/































































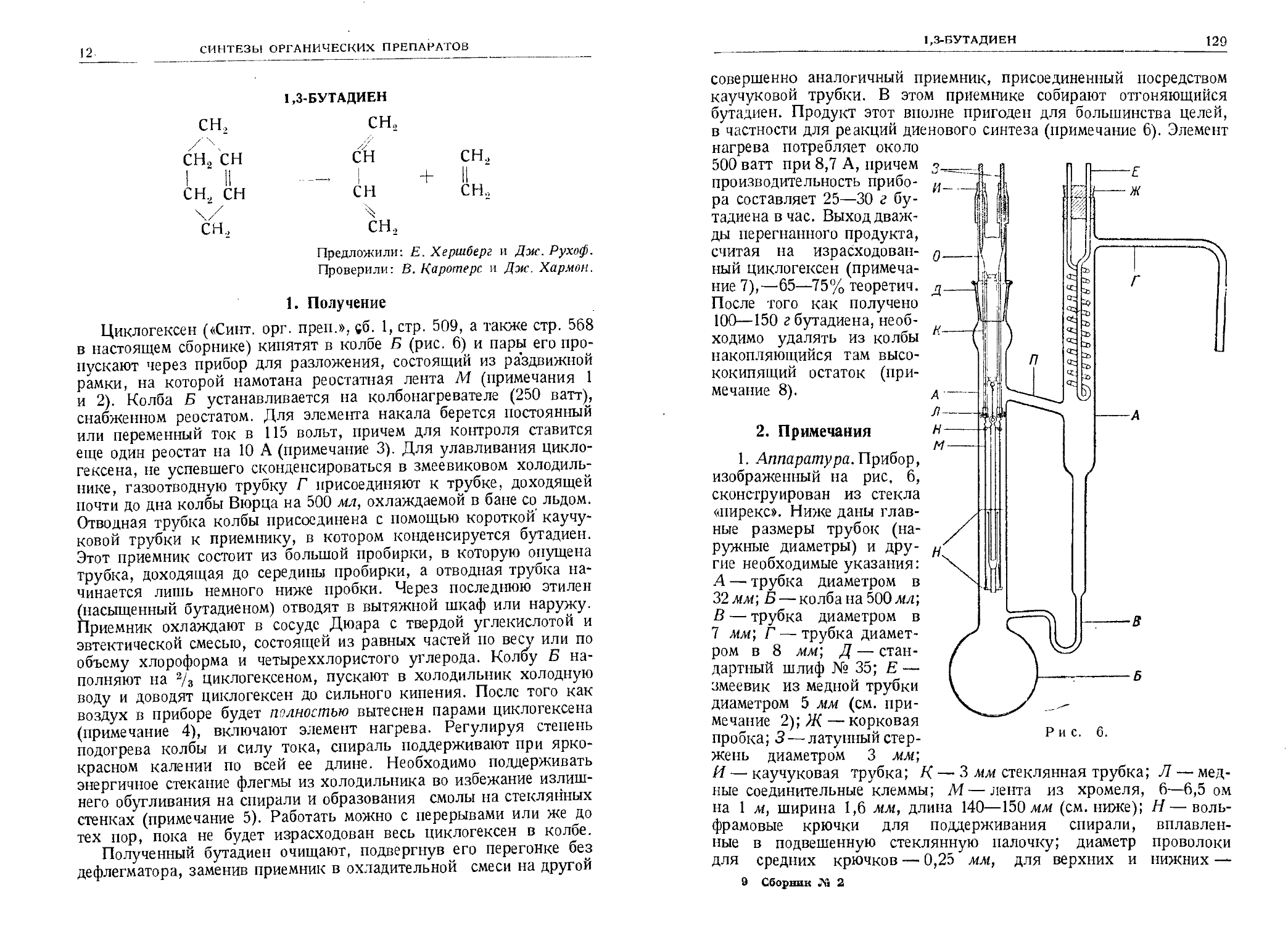












































































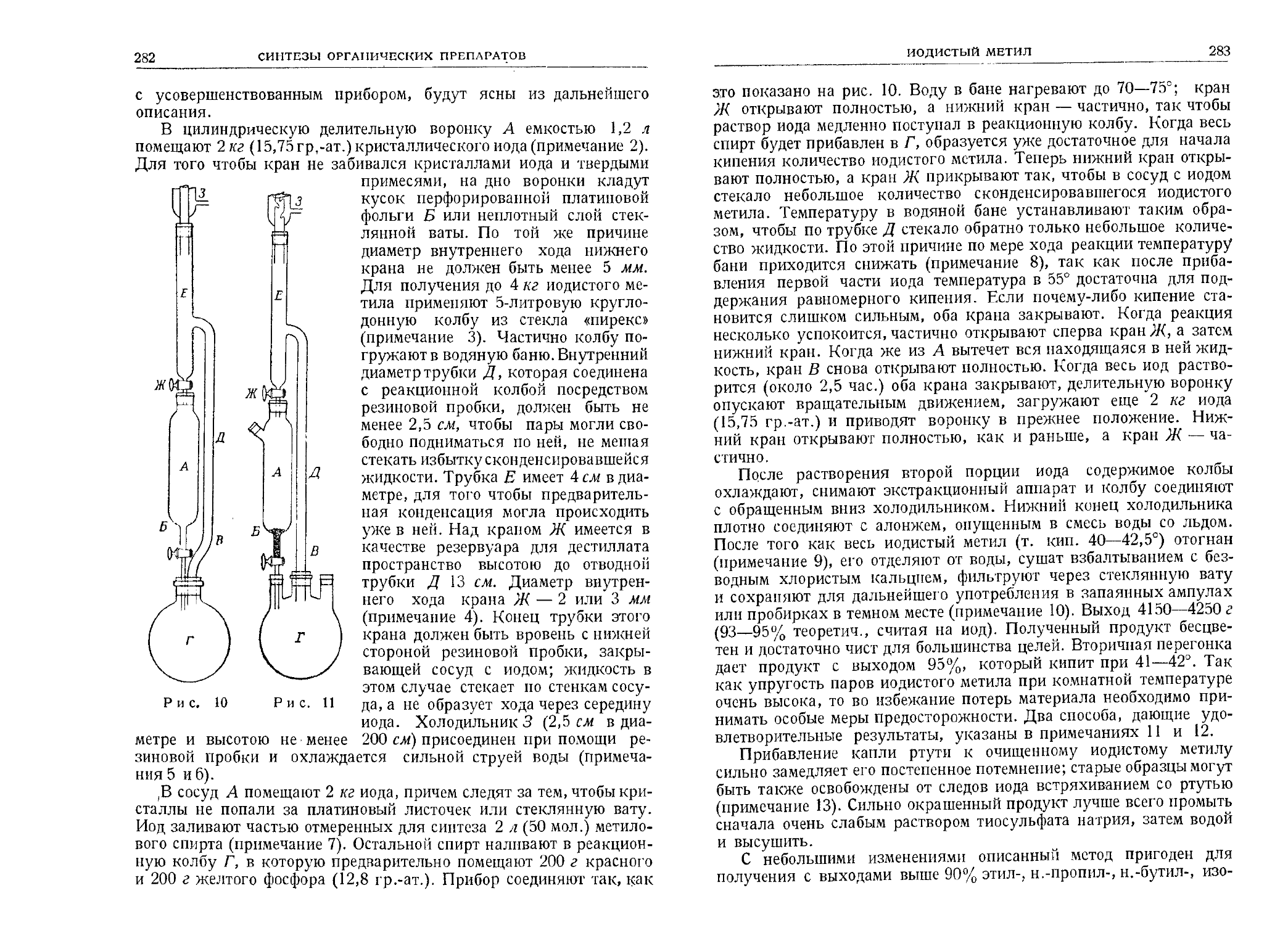













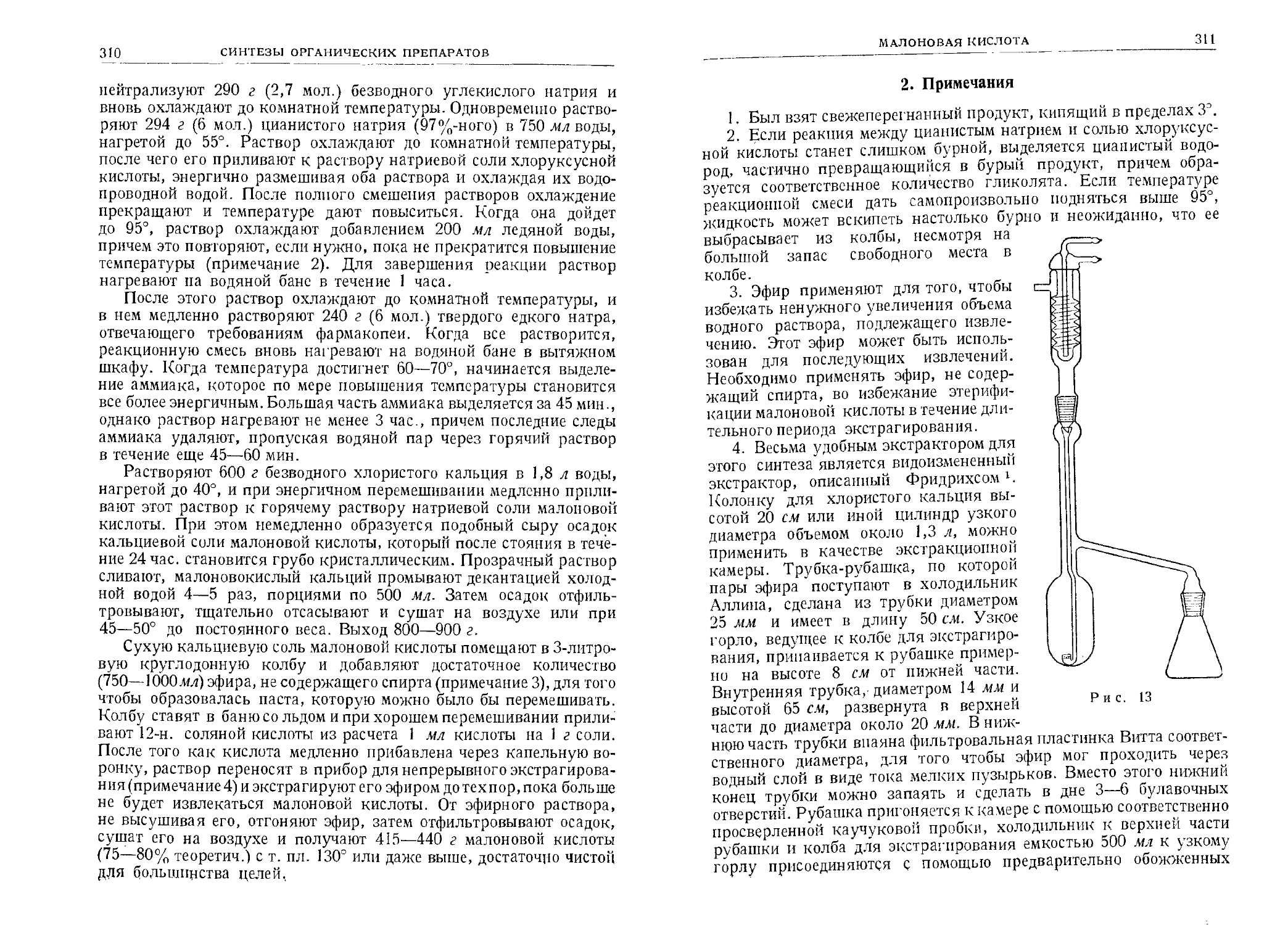
















































































































































































Текст
ORGANIC SYNTHESES
Collective Volume 2
A REVISED EDITION OF
ANNUAL VOLUMES X —XIX
Edited by A. Blatt, Secretary to the Board
Queens College, Flushing, N. Y.
EDITORIAL BOARD
R. ADAMS
H. ADKINS
С ALLEN
W. BACHMANN
W. CAROTHERS
H. CLARKE
J. CONANT
N. DRAKE
L. FIESER
R. FUSON
H. OILMAN
W. HARTMAN
J. JOHNSON
С MARVEL
С NOLLER
R. SHRINER
L. SMITH
F. WHITMORE
THIRD PRINTING
NEW YORK
СИНТЕЗЫ
ОРГАНИЧЕСКИХ
ПРЕПАРАТОВ
Сборник 2
РЕДАКТОРЫ АМЕРИКАНСКОГО ИЗДАНИЯ:
БЛЭТТ (общая редакция)
АДАМС
АДКИНС
АЛЛЕИ
БАХМАНН
ГИЛЬМАН
ДЖОНСОН
ДРЭК
КАРОТЕРС
КЛАРК
Перевод с
доктора хи
А. Ф.
КОНАНТ
МАРВЕЛ
НОЛЛЕР
СМИТ
УИТМОР
ФИЗЕР
ФЬЮЗОН
ХАРТМАН
ШРАЙНЕР
английского
иических наук
П Л АТЗ
Под редакцией академика
Б. А. КАЗАНСКОГО
19 4 9
ИЗДАТЕЛЬСТВО
ИНОСТРАННОЙ ЛИТЕРАТУРЫ
Москва
ПРЕДИСЛОВИЕ РЕДАКТОРА
В предисловии к сборнику № 1 «Синтезов органических пре-
паратов» отмечена огромная созидательная роль советских, а в про-
шлом российских, химиков-органиков в построении современной
органической химии.
За последнее время наша литература по синтезам в органической
химии обогатилась выпущенными в свет новыми произведениями
крупнейших советских ученых (например книгами В.М. Родионова,
Б. М. Богословского и А. М. Федоровой «Лабораторное руководство
по химии промежуточных продуктов и красителей», Н. Д. Пря-
нишникова «Практикум по органической химии»), отражающими не-
прерывный рост советской органической химии и все возрастающее
значение ее в технике, медицине, сельском хозяйстве. Задуманы
и готовятся новые книги-справочники, которые по широте
охвата материала и по целеустремленности, есть все основания
полагать, явятся исключительно ценными.
Американские сборники «Синтезы органических препара-
тов», 1-й из которых вышел в издании ИЛ в 1948г., выде-
ляются среди иностранной справочной литературы удачным под-
бором новых методов синтеза и сравнительно подробной характе-
ристикой их. В работе редакционной коллегии сборников участво-
вали такие известные органики, как Адаме, Уитмор, Гильман,
Каротерс и др.
2-й, настоящий, сборник «Синтезы органических препаратов»
представляет собой перевод, «Organic Syntheses, Collective Volume 2»,
вышедшего в США в 1946 г., в который вошли выпуски с X по
XIX, с исправлениями и дополнениями. Добавления, вошедшие в
этот том по сравнению с первоначальными выпусками, часто весьма
существенны, причем использована оригинальная литература и за
последние годы. Прописи синтеза некоторых препаратов претер-
пели настолько сильные изменения, что их можно считать соста-
вленными заново. В русском переводе (I—V тома, издания 1932—
1938 гг.) вышли американские выпуски I—XVII; таким образом
американские выпуски XVIII—XIX в русском переводе появляются
впервые.
Книги «Синтезы органических препаратов», предлагаемые вни-
манию советского читателя, никак не могут претендовать на исчер-
пывающий характер. В сборниках многого нет и многое едва ли будет
включено в будущие выпуски. К книгам нами, однако, не составлено
ПРЕДИСЛОВИЕ РЕДАКТОРА
дополнений, так как эти дополнения по объему заслуживали бы
отдельного издания. Подобный материал целесообразнее включить
в упомянутые выше подготавливаемые у нас произведения.
Примечания к тексту ограничиваются поэтому содержанием
текста и не указывают на недостаточно широкое освещение вопроса.
Приоритет советских ученых восстановлен в тех случаях, где
он не нашел отражения у американских составителей. В отношении
цитированных русских работ, опубликованных в русских жур-
налах, сделаны соответствующие дополнения и исправления.
Указатели к книге, как и для I тома, пополнены списком соста-
вителей прописей.
Кроме того исправлены явные ошибки и неточности американ-
ского издания.
Академик Б. А. Казанский.
ПРЕДИСЛОВИЕ
Сборник 2 «Синтезов органических препаратов» содержит в пере-
работанном виде материал, появлявшийся в ежегодных выпусках
Organic Syntheses X—XIX. Он является продолжением Сборника 1
и построен по одинаковому с ним общему плану. При обработке
материала для Сборника 2 были исправлены ошибки, допущенные
в ежегодных выпусках, проверены литературные ссылки и расчеты
исходных реагентов, внесены в текст изменения и улучшения мето-
дик, приводившиеся в ежегодных выпусках, и были добавлены один-
надцать новых и улучшенных проверенных методов. Новые методики
приведены для синтеза 2-карбэтоксициклопентанона, 1, 2-дибром-
циклогексана, этилового эфира адипиновой кислоты, этилового
эфира метилмалоновой кислоты, этилового эфира а-нафтойной кис-
лоты, 4-нитрофталимида, нитрозометилмочевины (два способа), пи-
мелиновой кислоты (два способа) и трифенилметилнатрия.
Раздел «Другие методы получения» по каждому синтезу был
проверен, и в него были включены методы, имеющие препаративное
значение и описанные в работах, рефераты которых были даны
в Chemical Abstracts, кончая 35 т. за 1941 г. Имеется также ряд
литературных ссылок на статьи, опубликованные в 1942 г., однако
за этот год литература исчерпывающим образом не просмотрена.
Для удобства читателей, желающих подобрать полную литературу
по данному препарату, в подзаголовке дано название препарата,
принятое в Chemical Abstracts, если это название отличается от
принятого названия препарата.
В тех случаях, когда описан синтез такого соединения, которое
может быть приобретено по цене не выше 5 долларов за 1 кг, у назва-
ния препарата поставлена звездочка 1. Возможно, что во многих
лабораториях изготовление такого препарата, если не учитывать
получаемых при этом экспериментальных навыков, нецелесооб-
разно.
Описание некоторых приборов и частей приборов, например,
таких, как видоизмененная колба Клайзена, ловушка для погло-
щения газов и т. д., упоминание о которых стало столь частым
в ежегодных выпусках «Синтезов органических препаратов», в тексте
Сборника 2 было давать нецелесообразно и только излишне загро-
моздило бы текст. Поэтому эти описания опущены. Для удобства
1 В переводе это обстоятельство, как не имеющее существенного значения
для советских химиков, не отмечалось. (Прим. ред.)
ПРЕДИСЛОВИЕ
тех читателей, которые сравнительно редко пользуются «Синтезами
органических препаратов», можно указать, что видоизмененные
колбы Клайзена описаны в настоящем сборнике на стр. 64 и в сбор-
нике 1 на стр. 142; ловушки для поглощения газов описаны в настоя-
щем сборнике на стр. 78 и в сборнике 1 на стр. 100. Опущено также
описание некоторых специальных реагентов. Так, например, если
для синтеза требуется абсолютный спирт, то не даются ссылки
на те несколько методов, которые описаны в сборнике для получе-
ния этого реагента; методы могут быть найдены по общему указа-
телю. Если, однако, для данного синтеза требуется абсолютный
спирт, приготовленный особым образом, то приводятся указания
для его получения.
Следует обратить внимание на следующее: вместо хлористого
кальция в осушительных трубках можно с успехом применять вату
(Org. Syn. 20, 9); вместо больших стаканов часто с успехом могут
быть применены эмалированные бани из нержавеющей стали;
мешалка с ртутным затвором часто может быть заменена более
простым устройством с резиновыми трубками (Org, Syn. 21, 40).
За эти указания приносим благодарность Ф. П. Пингерту, К. Ф. X.
Аллену и Л. П. Киридесу.
Редакторы выражают благодарность за многочисленные указа-
ния, исправления и улучшения, сделанные различными лицами.
Они будут приветствовать и в дальнейшем различные указания и
исправления, касающиеся новых томов «Синтезов органических
препаратов».
Благодарность выражается также Дж. Р. Крейцеру и Т. Герман
за содействие при выпуске настоящего сборника.
АЗЕЛАИНОВАЯ КИСЛОТА
Касторовое масло * СН,(СН.,M СНОНСНХН = СИ (СН2O СО2Н
Предложили: Док. Хилл и В. Мак-Ювен.
Проверили: Р. Фьюзон и Ч. Вудвард.
1. Получение
500 г касторового масла прибавляют к раствору 100 г едкого
кали в 1 л 95%-ного спирта. Смесь помещают в 3-литровую колбу,
соединенную с обратным холодильником, и кипятят в продолжение
3 часов. Затем раствор выливают в 3 л воды и подкисляют 100мл кон-
центрированной серной кислоты, разбавленной 300 мл воды. Выде-
лившуюся кислоту дважды промывают теплой водой, взбалтывают
с перерывами в течение 1 часа со 100 г безводного сернокислого
магния и затем отсасывают. Выход полученной таким образом сырой
рицинолеиновой кислоты составляет 480 г. Кислоту следует окис-
лять тотчас же (примечание 1) после получения.
240 г @,8 мол.) высушенной рицинолеиновой кислоты растворяют
в 1600 мл воды, содержащей 64 г едкого кали. В 12-литровую кру-
глодонную колбу, снабженную мощной механической мешалкой,
помещают 625 г C,5 мол.) перманганата калия и 7,5 л воды, нагре-
той до 35°. Для ускорения растворения перманганата смесь пере-
мешивают и, если нужно, подогревают настолько, чтобы темпера-
тура держалась около 35°. Когда перманганат полностью раство-
рится, к полученному раствору при энергичном перемешивании
прибавляют в один прием весь щелочной раствор рицинолеиновой
кислоты (примечание 2). Температура быстро поднимается до 75°.
Перемешивание продолжают еще полчаса, т. е. до тех пор, пока
в пробе, разбавленной водой, не исчезнет окраска перманганата.
После окисления смесь делят на две равные части и к каждой
части прибавляют раствор 200 г концентрированной серной кислоты
в 600 мл воды (примечание 3). Затем для коагуляции двуокиси
марганца смесь нагревают 15 мин. на водяной бане, после чего
осадок отфильтровывают, не допуская заметного охлаждения массы
(примечание 4). Отфильтрованную двуокись марганца помещают
в 4-литровый стакан и кипятят с 2 л воды для растворения азелаи-
10
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
новой кислоты, увлекаемой перекисью марганца. Жидкость филь-
труют горячей и фильтрат присоединяют к главной массе раствора.
Соединенные фильтраты и промывные воды, полученные в ре-
зультате обработки обеих частей окисленной смеси, упаривают
до объема приблизительно 4 л и раствор охлаждают в ледяной бане.
Выделившиеся кристаллы отсасывают, промывают один раз холод-
ной водой и сушат. Выход азелаиновой кислоты с т. пл. 95—105°
составляет 70—80 г.
Неочищенное вещество растворяют в 1200 мл кипящей воды,
раствор фильтруют с отсасыванием и оставляют охладиться.
Кристаллы отсасывают, промывают водой и сушат. Получают
48—55 г продукта C2—36% теоретического количества, считая на
неочищенную рицинолеиновую кислоту, взятую для окисления).
Температура плавления очищенной азелаиновой кислоты лежит
при 104—106°.
2. Примечания
1. При стоянии рицинолеиновая кислота полимеризуется. Со-
гласно Бекеру и Ингольду г при окислении заполимеризовавшейся
рицинолеиновой кислоты азотной кислотой получаются очень
плохие выходы.
2. В этот момент смесь сильно пенится, и, если перемешивание
недостаточно энергично, может произойти потеря продукта. В та-
ких случаях помогает прибавление небольших количеств эфира
или бензола. При хорошем перемешивании в этом нет необходи-
мости.
3. Во избежание быстрого выделения углекислоты и вспени-
вания кислоту надо прибавлять медленно и осторожно. Рекомен-
дуется, если это возможно, каждую из обрабатываемых двух частей
помещать в большой сосуд, например, в 12-литровую колбу.
4. Для этой цели рекомендуется взять три воронки Бюхнера
диаметром 20 см, вставленные в 2-литровые колбы для отсасывания.
3. Другие методы получения
Азелаиновая кислота может быть получена: окислением касто-
рового масла азотной кислотой 2; окислением рицинолеиновой кис-
лоты азотной кислотой * или щелочным раствором перманганата 3;
окислением метилового эфира олеиновой кислоты щелочным раство-
ром перманганата4; путем озонирования олеиновой кислоты и
последующего разложения озонида 5; путем озонирования метило-
вого эфира рицинолеиновой кислоты и последующего разложения
озонида6; действием углекислоты на димагниевое производное
1,7-дибромгептана 7; гидролизом 1,7-дициангептана 8, а также окис-
лением диоксистеариновой кислоты бихроматом и серной кисло-
АЗЛАКТОН а-СЕНЗОИЛАМИНО-З-АКРИЛОВОЙ КИСЛОТЫ
11
той, причем диоксикислоту получают действием перекиси водорода
на олеиновую кислоту9.
1 В а к е г, I п g о 1 d, J. Chem. Soc. 123, 128 A923); V е г к a d e,
Rec. trav. chim. 46, 137 A927).
2 А г р р е, Ann. 120, 288 A861); 124, 86 A862); Dale, Schorlem-
m e г, там же 199, 144 A879); Kiliani, Ber. 54, 469 A921); Day, Коп,
Stevenson, J. Chem. Soc. 117, 642 A920); Boeseken, Lutger-
h о г s t, Rec. trav. chim. 51, 164 A932).
3 Maquenne, Bull. soc. chim. C) 21, 1061 A899).
•Armstrong, Hilditch, J. Soc. Chem. Ind. 44, 43T A925).
5 Harries, Tank, Ber. 40, 4556 A907); R i e с h e, герм. пат. 565 168
[С. А. 27, 1008 A933)].
• Halle r, Brochet, Compt. rend. 150, 500 A910).
7 Braun, S о Ь е с к i, Ber. 44, 1926 A911).
8 Diontieau, Ann. chim. (9) 3, 249 A915).
9 Bennett, Gudgeon, J. Chem. Soc. 1938, 1679.
АЗЛАКТОН а-БЕНЗОИЛАМИНО-р-(ЗД-ДИМЕТОКСИФЕНИЛ)-
АКРИЛОВОЙ КИСЛОТЫ
[2-Фенш-4-вератрол-5D)-оксазолон, 2-фежл-4 C', 4'-диметоксибен-
залъ)-оксазолон]
сно
6H6CONHCH,CO;!H -f 2(CH3COJO
CH?,CO..Na
осн3
СН3О
сн
/—
%.
зО
>
1 = С
1.
/СО-О
\ 1
N==C-
Предложил
Проверили
Получение
¦ 4 СН3СО2Н
В 2-литровой конической колбе нагревают на электрической
плитке при постоянном взбалтывании смесь 160 г @,96 мол.) верат-
рового альдегида (примечание 1), 192 г A,07 мол.) растертой в по-
рошок сухой гиппуровой кислоты (см. стр. 158), 80 г @,98 мол.)
растертого свежесплавленного уксуснокислого натрия и 300 г
B78 мл; 2,9 мол.) высококачественного уксусного ангидрида.
Вначале смесь становится почти твердой, потом, по мере повышения
температуры, она постепенно превращается в жидкость и окраши-
вается в темножелтый цвет (примечание 2). Как только продукт
станет вполне жидким, колбу переносят на кипящую водяную баню
и продолжают нагревание в течение 2 часов. За это время часть
12
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
продукта выделяется в виде темножелтых кристаллов. Под конец
нагревания в колбу медленно приливают 400 мл спирта. Во время
прибавления спирта, ради ослабления бурного течения реакции,
колбу слегка охлаждают. Реакционную смесь оставляют стоять
на ночь и затем желтый кристаллический продукт отсасывают и
промывают непосредственно в воронке (порциями по 100 мл) сна-
чала два раза охлажденным до 0° спиртом, а затем — два раза ки-
пящей водой. После сушки продукт весит 205—215 г F9—73%
теоретического количества) и плавится при 149—150°. Продукт
достаточно чист для многих целей; его можно очистить перекристал-
лизацией из горячего бензола: из 1200 мл бензола получают 180—
190 г чистого азлактона с т. пл. 151—152Э.
2. Примечания
1. Вератровый альдегид, полученный метилированием ванилина
(стр. 142), может быть пущен в реакцию без дальнейшей очистки.
2. Смесь должна становиться совершенно жидкой при темпера-
туре около 110°. При перегревании продукт приобретает вместо
яркожелтой красную окраску.
3. Другие методы получения
Азлактоны а-бензоиламинокоричных кислот получались всегда
действием гиппуровой кислоты и уксусного ангидрида на аромати-
ческие альдегиды 1, обычно в присутствии уксуснокислого нат-
рия2. Метод, описанный выше, в основных чертах разработали
Кропп и Деккер 3.
1 Р 1 о с h I, Ber. 16, 2815 A883).
2 Erlenmeyer, Ann. 275, 3 A893).
8 К г о р р, Decker, Ber. 42, 1184 A909).
АЗОКСИБЕНЗОЛ
4 CfiHBNO2 + 3 As A -f-18 NaON *
2 C6H3N = NC6H6 + 6 Na3As04 4- 9 H2O
I
О
Предложили: Х. Бигелов и А. Пальмер.
Проверили: Г. Гильман и X. Ха.рвуд.
1. Получение
226 г A,1 мол.) истолченного в порошок мышьяковистого
ангидрида тщательно размешивают с небольшим количеством воды;
полученную тестообразную массу растворяют в растворе 275 г
(Ь,У мол.) едкого натра в 600 мл воды. Приготовленный таким
АЗОКСИБЕНЗОЛ
13
образом раствор мышьяковистокислого натрия разбавляют 600 мл
воды, помещают в 2-литровую трехгорлую колбу, снабженную об-
ратным холодильником и механической мешалкой, после чего к нему
прибавляют 150 г A25 мл; 1,2 мол.) свежеперегнанного нитробен-
зола (примечание 1).
Смесь кипятят с обратным холодильником на масляной бане
в течение 8 час. при непрерывном энергичном перемешивании
(примечание 2). Затем масляную баню удаляют и, продолжая пере-
мешивание, дают реакционной смеси охладиться приблизительно
до 80°, после чего переносят ее в делительную воронку, предвари-
тельно нагретую в сушильном шкафу примерно до той же темпера-
туры (примечание 3).
Верхний маслянистый слой отделяют (примечание 4), тотчас
же выливают в стакан и промывают водой, содержащей небольшое
количество соляной кислоты. Немедленно выпадают желтые кри-
сталлы азоксибензола (примечание 5). Выход продукта, плавящегося
при 35,5—36,5°, 102 г (85% теоретич.; примечания 6 и 7).
2. Примечания
1. Избыток мышьяковистокислого натрия и 8-часовое нагрева-
ние обеспечивают полное использование нитробензола. Благодаря
этому отпадает необходимость перегонки с паром или других про-
цессов, нужных для удаления не вошедшего в реакцию нитробен-
зола.
Можно взять неочищенный нитробензол, но лучше брать про-
дукт хорошего качества. При применении неочищенного нитро-
бензола обычно получается более темный продукт и с несколько
более низкой температурой плавления.
2. Температура внутри колбы должна быть около 104°, а тем-
пература бани не должна превышать намного 115°. Не следует
брать колбу меньшего размера, так как при случайной остановке
мешалки может произойти сильное вспенивание. Если реакцию
приходится почему-либо прервать или если прекратилось переме-
шивание, то масляную баню следует удалить. В противном случае
при возобновлении перемешивания реакционная масса может быть
выброшена через холодильник. Если принимать все указанные
меры предосторожности, то 8-часовое нагревание может и не быть
непрерывным.
Вопреки литературным данным, азоксибензол несколько летуч
с парами воды. Поэтому наличие маслянистых капель в холодиль-
нике под конец 8-часового кипячения не является признаком того,
что не весь нитробензол вошел в реакцию. Азоксибензол легко летуч
с водяным паром при 140—150°.
3. При таком способе работы отделение производится примерно
при 60°, и отпадает опасность охлаждения раствора до температуры,
14
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
при которой выделяется мышьяковокислый натрий. Если вслед-
ствие чрезмерного охлаждения он все же выпадет, то реакционную
смесь снова нагревают при перемешивании до полного растворения
выделившейся соли. Если смесь разбавить таким количеством воды,
которое оказывается достаточным для предотвращения выпадения
мышьяковокислого натрия при охлаждении раствора, то масло
оседает на дно, и его трудно отделить от водного слоя. Кроме того,
объем раствора при таком разбавлении сильно увеличивается, что
затрудняет работу.
4. При разбавлении раствора мышьяковокислого натрия рав-
ным объемом воды выпадает небольшое количество азоксибензола.
Его можно извлечь экстракцией бензолом, однако количество про-
дукта, получаемое этим путем, настолько ничтожно, что им можно
пренебречь.
5. Присутствие соляной кислоты ускоряет кристаллизацию.
Если кристаллизации не происходит, то рекомендуется внести в
масло кристаллик азоксибензола в качестве затравки.
6. При перекристаллизации из 50 мл горячего 95%-ного спирта
получается 72 г азоксибензола, плавящегося также при 35,5—36,5°.
Однако перекристаллизованный продукт получается более светлым.
7. Весьма удобен также следующий метод получения азокси-
бензола.
В 1-литровую трехгорлую колбу, снабженную обратным холо-
дильником и эффективной мешалкой (стр. 298), помещают 60 г
едкого натра, 200 мл воды и 41 г C4,2 мл; 0,33 мол.) нитробензола.
Колбу погружают в водяную баню, нагретую до 55—60°, после чего
в течение 1 часа и при работающей мешалке добавляют по частям
45 г @,23 мол.) декстрозы. Затем температуру бани повышают до
100° и поддерживают на этом уровне 2 часа. Горячую смесь выли-
вают в 2-литровую длинногорлую колбу и подвергают перегонке
с водяным паром для удаления нитробензола и анилина. На это тре-
буется около 20 мин., причем отгоняется около 2 л дестиллата. Когда
дестиллат станет прозрачным, остаток выливают в стакан и хорошо
охлаждают в бане со льдом. Затвердевший азоксибензол собирают,
куски его растирают в ступке, промывают водой и сушат. Выход
продукта с т. пл. 34—35° составляет 26—27 г G9—82% теоретич.).
После перекристаллизации из 15 мл метилового спирта возвращается
90% продукта с т. пл. 35—35,5° (Н. Ополоник, частное сообщение;
проверили Л. Ф. Физер и М. Физер).
3. Другие методы получения
Азоксибензол может быть получен восстановлением нитробен-
зола: спиртовым раствором едкого кали х, амальгамой натрия 2,
водородом в присутствии окиси свинца3, метиловым спиртом и
едким натром 4, метилатом натрия и метиловым спиртом 5, закисью
АКОНИТОВАЯ КИСЛОТА
15
свинца в щелочной суспензии 6, декстрозой в щелочной суспензии
(см. выше, примечание 7); электролитическим восстановлением7;
окислением азобензола хромовым ангидридом 8; окислением °-фе-
нклгидроксиламина: щелочным раствором марганцовокислого ка-
лия 9, нитробензолом10, минеральными кислотами" и меркура-
цетамидом 12; окислением анилина перекисью водорода 13 и кислым
раствором пермарганата калия в присутствии формальдегида14.
Метод, описанный выше, представляет собой незначительное видоиз-
менение одного из способов, описанных в литературе 15.
1 3 и н и н, J. prakt. Chcm. A) 36, 98 A845).
2 Алексеев, Bull. soc. chim. A) 1, 325 A864).
3 В г о w n, H e n k e, ам. пат. 1 451 489 [С. А. 17, 1969 A923)].
4 L а с h m a n, J. Am. Chem. Soc. 24, 1180 A902).
6 В г u h I, Ber. 37, 2076 A904).
6 Deutsche Gold- und Silber-Scheideanstalt vorm. Roessler; герм. пат. 486598
[С. А. 24, 1389 A930)].
'Lob, Ber. 33, 2332 A900); герм. пат. 116 467 (Chem. Zentr. 1901,4, 149>.
8 Wreden, Ber. 6, 557 A873).
9 R e i s s e r t, там же 29, 641 A896).
10 В a m b e r g e r, Renauld, там же 30, 2278 A897).
11 Bamberger, L a g u 11, там же 31, 1501 A898).
12 Forster, J. Chem. Soc.'73, 786 A898).
13 P r u d'h о m m e, Bull. soc. chim. C) 7, 622 A802).
14 Bamberger, Tschirner, Ber. 32, 342 A899).
15 Loesner, J. Prakt. Chem. B) 50, 564 A894).
снхо
1
С (ОН)
снхо
2н
со2
.н
АКОНИТОВАЯ
Н (H.so<)
КИСЛОТА
снсо.н
II
II
ССО,Н
снхо,н
нх>
Предложил: В. Брюс.
Проверили: Л. Физер и Д'. Фишер.
I. Получение
В 1-литровую круглодонную колбу, снабженную обратным холо-
дильником (примечание 1), помещают 210 г A мол.) измельченной
лимонной кислоты (с одной молекулой кристаллизационной воды)
и раствор 210 г A15 мл; 2 мол.) концентрированной серной кислоты
в 105 мл воды. Смесь в течение 7 час. нагревают на масляной бане,
температуру которой поддерживают при 140—145°. Светлобурый
раствор выливают в мелкую чашку и колбу ополаскивают 10 мл
горячей ледяной уксусной кислоты. Жидкости дают медленно охла-
диться до 41—42° (примечание 2), время от времени перемешивая
16
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
ее, чтобы разбить выделяющуюся массу твердой аконитовой кис-
лоты, и осадок отсасывают (примечание 3). Продукт тщательно
отжимают и отсасывают почти досуха, после чего его размешивают
в однородную пасту с 70 мл концентрированной соляной кислоты,
охлажденной в бане со льдом. Твердый продукт отсасывают (при-
мечание 3), промывают два раза холодной ледяной уксусной кис-
лотой порциями по 10 мл, тщательно отсасывают и для высушива-
ния раскладывают тонким слоем на пористой тарелке или на филь-
тровальной бумаге (примечание 4), Продукт этот практически
не содержит сульфата и для большинства целей является достаточно
чистым. Он бесцветен и в сухом виде весит 71—77 г D1—44% тео-
ретич.; примечание 5). Температура разложения в определенных
условиях (примечание 6) колеблется от 180 до 200°.
С целью очистки аконитовую кислоту перекристаллизовывают
из 150 мл ледяной уксусной кислоты, причем для горячего раствора
пользуются кислотоупорным фильтром (примечание 7). Аконитовая
кислота выпадает в виде небольших бесцветных игл. Выход соста-
вляет 50—60 г, причем упариванием маточного раствора в вакууме
до !/з первоначального объема можно выделить еще около 10 г.
Продукт сушат сперва на воздухе, а затем — в эксикаторе над
едким натром, для того чтобы удалить следы уксусной кислоты.
Одной перекристаллизации обычно бывает достаточно, для того
чтобы повысить температуру разложения до 198—199° (примеча-
ние 6).
2. Примечания
1. Желательно, чтобы холодильник был соединен с колбой по-
средством шлифа.
2. Если фильтровать при этой температуре, а не при более низ-
кой, то без значительных потерь аконитовой кислоты удается добиться
освобождения от небольшого количества низкоплавкой примеси.
J3. Фильтровать удобнее всего через воронку с пористой стеклян-
ной пластинкой или через кусок чистой шерстяной фланели, поло-
женной в воронку Бюхнера диаметром в 8 см.
4. В сырую погоду твердый продукт часто расплывается, и по-
тому сушить его приходится в эксикаторе. Продукт весьма упорно
удерживает уксусную кислоту и сушить его следует до тех пор,
пока запах растворителя не будет более заметен.
5. Определение по методу Пьючера, Виккери и Ливенуорзса 1
показывает, что в сернокислотном растворе остается 26 г лимонной
кислоты. Применять этот раствор вторично для получения акони-
товой кислоты не рекомендуется; накопившиеся вода и побочные
продукты значительно понижают как выход, так и качество полу-
чаемого продукта.
6. При нагревании в капилляре аконитовая кислота разлагается
сразу с энергичным выделением газа при температуре, в значитель-
АКОНИТОВАЯ КИСЛОТА
ной степени зависящей от скорости нагревания и от той температу-
ры, при которой введен в баню капилляр. В литературе 2 встре-
чаются указания на «температуру плавления», которая колеблется
от 182,5 до 194,5°. Если ввести неперекристаллизованную акони-
товую кислоту при 180° в небольшую баню с механическим пере-
мешиванием, нагреваемую со скоростью 2—3° в минуту, то разло-
жение кислоты наступает обычно при 189—190°. Однократно пере-
кристаллизованный продукт разлагается при 198—199°, если
ввести его в баню при 190°, если же его ввести при 195°, то он раз-
лагается при 204—205°. Определение с помощью блока Денниса 3
(метод, наиболее надежный для такого типа соединений) дает тем-
пературу разложения, равную 209°. Для получения хороших
результатов необходимо, чтобы проба была абсолютно сухой.
7. Горячий раствор разрушающе действует на фильтровальную
бумагу. Удобный фильтр можно получить, если в бюхнеровской
воронке диаметром в 6 см поместить 1—2 мм слой асбеста, покрыть
его 2—3 мм слоем активированного уГля, промыть с отсасыванием
и нагреть воронку вместе со склянкой для отсасывания до 120°
в сушильном шкафу. В сухом и горячем состоянии прибор готов
к употреблению.
3. Другие методы получения
Аконитовая кислота была получена из лимонной кислоты дей-
ствием серной кислоты 4, или хлористого водорода 5, или же нагре-
ванием ее 6. Она была получена также из метилового эфира ацетил-
лимонной кислоты7 и из ангидрида ацетиллимонной кислоты 8.
Описанный выше метод в основном заимствован у Гентшеля *.
Вместо серной можно взять фосфорную кислоту (85%), однако
в этом случае приходится гораздо строже следить за соблюдением
всех условий реакции, а выход получается лишь немногим лучше.
Квартароли и Бельфиори изучили влияние концентрации кислоты
и температуры на реакцию между серной и лимонной кислотами:
применение холодной пиросерной кислоты или горячей серной
кислоты концентрации не выше 94% приводит к образованию ако-
нитовой кислоты 9.
1 Pucher, Vickery, Leavenworth, Ind. Eng. Chem., Anal.
Ed. 6, 190 A934).
2Malachowski, Maslowski, Ber. 61, 2521 A928).
3 D e n n i s, S h e 1 t о n, J. Am. Chem. Soc. 52, 3128 A930).
4 H e n t s с h e 1, J. prakt. Chem. B) 35, 205 A887).
5 Hunaus, Ber. 9, 1751 A876).
6 Pawollcck, Ann. 178, 153 A875).
'Anschutz, Klingemann, Ber. 18, 1953 A885).
8 E a s t e r f i e 1 d, S e 1 1, J. Chem. S6c. 61, 1007 A892).
9 Qu a r t ar о 1 i, Belfiori, Ann. chim. applicata 28, 297 A938)
[C A. 33, 1669 A939)].
2 Сбор'шк JVs 2
18
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
АКРИДОН
o-C1C6H4C0.2H+C6H5NH2 T?~V o-C6H5NHC6H4CO,H
o-C6H5NHC6H4C02H
0
Предложили: К- Аллен и Дж. Мак-Ци.
Проверили: В. Хартман и А. Вейсбергер,
1. Получение
Л. N-Фенилантраниловая кислота. В 1-литровую круглодон-
ную колбу, снабженную обратным холодильником с воздушным
охлаждением, помещают смесь 155 г A,66 мол.) анилина, 41 г
@,26 мол.) о-хлорбензойной кислоты (примечание 1), 41 г @,3 мол.)
технического безводного поташа и 1 г окиси меди. Смесь кипятят
в течение 2 час. на масляной бане. Избыток анилина удаляют
перегонкой с водяным паром (на что требуется около 3 час), после
чего к оставшемуся бурому раствору прибавляют 20 г активирован-
ного угля (примечание 2) и смесь кипятят 15 мин. Массу фильтруют
с отсасыванием и фильтрат приливают при помешивании к смеси
'30 мл концентрированной соляной кислоты и 60 мл воды. По охла-
ждении выпавшую кислоту отсасывают и сушат на воздухе до
постоянного веса. Выход 46—52 г (82—93% теоретич.) почти бес-
цветного продукта. Температура плавления кислоты 179—181°,
причем несколько ниже этой температуры она начинает спадать
(примечания 3, 4 и 5).
Б. Акридон. В колбе емкостью 500 мл приготовляют раствор
42,7 г @,2 мол.) N-фенилантраниловой кислоты (примечание 6)
в 100 мл концентрированной серной кислоты (уд. в. 1,84) и раствор
нагревают на кипящей водяной .бане в течение 4 час, после чего
его выливают в 1 л кипящей воды. Чтобы избежать разбрызгивания,
раствор приливают по стенке сосуда. После кипячения в течение
5 мин. желтый осадок отфильтровывают и фильтрат сохраняют
(примечание 7). Влажный осадок кипятят в течение 5 мин. с раствором
30 г @,28 мол.) соды в 400 мл воды, после чего его отсасывают (при-
мечание 8) и' хорошо промывают водой. После высушивания на
воздухе вес неочищенного акридона составляет 35,5—37,5 г (91—
96% теоретич.); т. пл. продукта 344—346° (примечание 9). Для боль-
шинства целей продукт достаточно чист; его можно перекристал-
АКРИДОН
лизовать из смеси анилина и уксусной кислоты, причем следует
взять 10 мл анилина и 25 мл уксусной кислоты на каждые 2 г веще-
ства. Выход после перекристаллизации около 90%, температура
плавления перекристаллизованного продукта 348—352° (приме-
чание 10).
2. Примечания
1. 60 г технической [о-хлорбензойной кислоты растворяют
в 200 мл горячей воды, содержащей 20 г соды, добавляют Юг акти-
вированного угля, смесь кипятят в течение 10 мин. и фильтруют
с отсасыванием. Фильтрат приливают к хлористоводородной кис-
лоте, полученной разбавлением 31 мл концентрированной соляной
кислоты равным объемом воды. Вес высушенного на воздухе про-
дукта составляет 41 г, причем его можно использовать непосред-
ственно. Эта очистка необходима для того, чтобы получить хоро-
ший выход и чтобы продукт был хорошего качества. Если эту очистку
опустить, кислота получается окрашенной в темный цвет — от си-
него до черного, причем избавиться от такой окраски очень трудно.
Указания для получения о-хлорбензойной кислоты окислением
о-хлортолуола приведены на стр. 556.
2. Обычный животный уголь и уголь «дарко» дали одинаково
хорошие результаты.
3. N-Фенилантраниловая кислота медленно разлагается при
повышенной температуре. До достижения температуры плавления
наблюдается значительное предварительное спадание. Если поль-
зоваться методом погружения капилляра в нагретую баню, темпе-
ратура плавления достигает 182—187°. В литературе для чистой
кислоты приводятся т. пл. от 181 до 184°.
4. Эта кислота достаточно чиста для всех обычных целей. Тем-
пература плавления повышается лишь незначительно, если раство-
рить 5 г кислоты в 100 мл воды, в которой растворено 2,5 г соды,
добавить 2,5 г активированного угля, прокипятить 5 мин., профиль-
тровать и фильтрат подкислить. Выход 4,6 г. Если продукт полу-
чается окрашенным, то необходимо поступать так для того, чтобы
получить акридон более светлой окраски.
5. При перекристаллизации 5 г кислоты растворяют в 25 мл
кипящего спирта и добавляют 5 мл воды. Выход 4,8 г, температура
плавления 182—183°. Спирт можно заменить на уксусную кислоту
B мл на 1 г); это удобнее при перекристаллизации больших коли-
честв.
6. Можно применять и неперекристаллизованнуюТ^-фенилан-
траниловую кислоту, если только она была получена из очищенной
о-хлорбензойной кислоты. В противном случае неочищенную N-фе-
нилантраниловую кислоту следует обесцветить, как это описано
в примечании 4; иначе получается акридон зеленоватого цвета,
хотя с надлежащей температурой плавления.
2*
20
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
7. После стояния в течение ночи из фильтрата выпадает 1,6—
2 г весьма нечистого продукта с температурой плавления около 315°.
8. При подкислении фильтрата всегда выделяется немного
A,5—2 г) N-фенилантраииловой кислоты.
9. Неочищенный акридон спадается в капилляре при 330—335°
и расплавляется с образованием темноокрашенной жидкости при
344—346°.
10. Для перекристаллизации можно взять также изоамиловый
спирт. На 1 г акридона требуется 120 мл растворителя, причем
получается 0,75 г продукта с температурой плавления 354° при
определении температуры плавления по методу медного блока Берля-
Кульмана 1.
3. Другие методы получения
Практическим методом получения N-фенилантраниловой кис-
лоты является действие анилина на о-хлор- или о-бромбензойную
кислоту 2>3 или действие бромбензола на антраниловую кислоту *• 5>",
причем в обоих случаях пользуются солями меди или добавляют
окись меди.
Единственным методом, который имеет препаративное значе-
ние, является получение акридона замыканием цикла, исходя из
N-фенилантраниловой кислоты 7.
1 Berl, Kullman, Ber. 60, 811 A927); переработанное видоизменение
см. Wa I s h, Ind. Eng. Chem., Anal. Ей. 6, 468A934).
2 U 1 1 m a n n, Ber. 36, 2383 A903); Ann. 355, 322 A907).
a Meister, Lucius, Bruning; герм. пат. 145 189 (Chem. Zentr.
1903, II, 1097).
4 Goldberg, Ber. 39, 1691 A906).
5Houben, Brassert, там же 39, 3238 A906).
• Goldberg, UHmann, герм. пат. 173 523 (Chem. Zentr. 1906, II,
931); Goldberg; герм. пат. 187 870 (Chem. Zentr. 1907, II, 1465).
' Graebe, Lagodzinski, Ber. 25, 1734 A892); Ann. 276, 35 A893);
Mats urn ura, J. Am. Chem. Soc. 57, 1533 A935).
CH2CO4
I
CHXCK
Р- АЛАНИИ
($-Аминопропионовая кислота)
CH2NH2
СНХО,Н
>NH + KOBr+2KOH
KBr + KXO3
Предложили: Х. Кларк и Л. Бэр.
Проверили: В. Каротерс и В. Мак-Ювен.
1. Получение
К холодному @—5°) раствору 302 г едкого кали (в палочках)
в 2720 мл дестиллированной воды медленно, при помешивании, при-
бавляют 96,6 г C0,8 мл; 0,6 мол.) брома. Раствор охлаждают до
З-АЛАНИН
21
0э, после чего, перемешивая массу от руки, добавляют 59,4 г @,6 мол.)
сукцинимида (стр. 439). Смесь нагревают на водяной бане при 55—
60° до тех пор, пока она не обесцветится, после чего ее держат при
этой температуре еще два часа (примечание 1). После стояния в те-
чение ночи при комнатной температуре к смеси прибавляют концен-
трированной соляной кислоты до кислой реакции на конго красное
(около 380 мл, уд. в. 1,18; примечание 2) и выпаривают досуха на
водяной бане в вакууме. Остаток обрабатывают 1 л теплого 95%-ного
спирта; нерастворившийся бромистый калий отфильтровывают и
промывают 150—200 мл холодного спирта, приливая его неболь-
шими порциями. Фильтрат и промывную жидкость соединяют вместе
и выпаривают досуха при уменьшенном давлении, после чего оста-
ток экстрагируют 100 мл 95%-ного спирта. Полученный раствор
вновь выпаривают досуха и остаток экстрагируют 140 мл горячего
абсолютного спирта (примечание 3). Затем отгоняют основную
массу спирта, остаток разбавляют 200 мл дестиллированной воды
и взбалтывают его два раза с эфиром, причем каждый раз
берут эфира по 80 мл. Эфирные вытяжки отбрасывают (приме-
чание 4).
Водный раствор освобождают от эфира и спирта и кипятят с об-
ратным холодильником в течение 1—1,5 часа с целью омылить эфир
3-аланина, после чего упаривают в вакууме, чтобы удалить возможно
большую часть избытка соляной кислоты. Остаток растворяют
в воде, и объем раствора доводят точно до 1000 мл. Из этого раствора
берут 5 мл для определения суммарного количества галоидов.
К оставшейся части раствора прибавляют суспензию окиси серебра,
полученную из азотнокислого серебра, взятого с избытком на 10%
по сравнению с необходимым количеством (примечание 5), и смесь
хорошо перемешивают,-чтобы вызвать полное осаждение галоидных
солей. После стояния в течение ноии осадок отфильтровывают и
промывают водой. Фильтрат и промывные воды упаривают в ваку-
уме, пока объем не достигнет примерно 400 мл; затем раствор насы-
щают сероводородом и фильтруют через тонкий слой активирован-
ного угля, чтобы его обесцветить. Бесцветный фильтрат упаривают
до объема около 100 мл, если нужно обрабатывают обесцвечивающим
углем, концентрируют на водяной бане до начала кристаллизации
и охлаждают холодильной смесью. Кристаллы отсасывают, промы-
вают небольшим количеством холодного спирта и сушат. Если
упарить маточный раствор и вновь охладить его, то можно полу-
чить еще некоторое количество кристаллов (примечание 6). Обе
порции B8—30 г, т. пл. 189—192°) перекристаллизовывают из
воды, пользуясь той же методикой, причем получают 22—24 г
D1—45% теоретического количества) чистого р-аланина, который
плавится с разложением при 197—198° (с поправкой). Из
последнего маточника можно выделить еще около 2 г чистого
продукта,
22
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
2. Примечания
1. При этом чувствуется запах аммиака, что указывает на ча-
стичный гидролиз сукцинимида.
2. При подкислении иногда выделяется небольшое количество
брома, который быстро удаляется при последующем выпаривании.
3. При этом последнем экстрагировании удобно удалять часть,
нерастворимую в спирте, с помощью центрифуги.
4. При экстрагировании эфиром удаляются небольшие коли-
чества янтарной кислоты и ее сложных эфиров.
5. Окись серебра получают следующим образом: азотнокислое
серебро растворяют примерно в пяти частях холодной воды и доба-
вляют небольшой избыток 10%-ного раствора чистого едкого натра.
Осадок хорошо перемешивают, отделяют фильтрованием или центри-
фугированием и отмывают от солей натрия. Перед употреблением
его не следует сушить.
6. Последний маточный раствор представляет собой довольно
вязкую жидкость, содержащую некристаллизующиеся побочные
продукты.
3. Другие методы получения
Данные выше указания основаны на методике Гугеверфа и
Ван-Дорпа г с изменениями, которые внесли Хольм 2 и Хэль и
Хонан 3. [З-Аланин был также получен действием бромноватистых
солей на сукцинимид с последующим омылением получающейся
[3-уреидопропионовой кислоты4; действием аммиака на р-иодпро-
пионовую кислоту 5; омылением метилового эфира карбометокси-
C-аминопропионовой кислоты, полученной действием метилата на-
трия на сукцинбромимид в; восстановлением [3-нитрозопропионовой
кислоты7; нагреванием этилового эфира акриловой кислоты со
спиртовым раствором аммиака 8; из сложного эфира сукцинилгли-
цина через азид 9; действием жидкого аммиака на метиловый эфир
акриловой кислоты 10 и восстановлением цианоуксусной кислоты u
или ее этилового эфира с последующим омылением 12.
1 Hongewerff, Van Dorp, Rec. trav. chim. 10, 5 A891).
2 Holm, Arch. Pharm. 242, 597 A904).
3 Hale, Honan, J. Am. Chem. Soc. 41, 774 A919).
4 Weidel, R о i th n e r, Monatsh. 17, 172 A896).
5 H e i n t z, Ann. 156, 25 A870); Mulder, Ber. 9, 1902 A876); Abder-
h a 1 d e n, F о d о r, Z. physiol. Chem. 85, 114 A913).
0 Lengfeld, Stieglitz, Am. Chem. J. 15, 215, 504 A893).
'Pechmann, Ann. 264, 288 A891).
8 Wender, Gazz. chim. ital. 19, 437 A889).
"Curtius, Hech ten berg, J. prakt. Chem. B) 105, 289 A923).
10 Morsch, Monatsh. 63, 220 A933).
11 M e г с k; герм. пат. 597 305 [С. А. 28, 5078 A934I; Ruggli, Bu si n-
gcr, Helv. Chim. Acta 25, 35 A942).
12 We у g a n d, Ber. 74, 256 A941),
NH
1
CO
1
NH
CH;;CO2H^ Q
— CO
1
1
c~
и
II
— C-
o2 +
-NH4
-NH7
NH —
CO
NH —
>C0
H
C-
|
С
(
DH
АЛЛАНТОИН
АЛЛАНТОИН
Н„О 4 [О]
(KMnOs)
-NH
1
CO
-NH
NH-
i
CO
I
CO2K
1
1
-C
1
NH—С
NH
i
CO
1
NH
с
— NH
|
CO
1
— NH
)H
H
1
—
: — nhconh2
с
1
:l
0
Предложили: В. Хартман, Е. МоффеттнДж. Джки.
Проверили: В. Каротерс и В. Мак-Ювен.
1. Получение
100 г мочевой кислоты @,595 мол.) и 4,5 л горячей G0—85°)
воды помещают в 12-литровую круглодонную колбу, снабженную
механической мешалкой. Мешалку пускают в ход и в колбу прили-
вают раствор 80 г B мол.; примечание 1) технического едкого натра
в 120 мл воды. Перемешивание продолжают до тех пор, пока вся
мочевая кислота не перейдет в раствор (примечание 2), после чего
раствор охлаждают струей воды из водопровода. Когда температура
понизится до 25—30°, к энергично перемешиваемому раствору при-
бавляют сразу (примечание 3) 50 г @,32 мол.) перманганата калия
(примечание 4). Перемешивание продолжают еще 15—20 мин.
(примечание 5); затем смесь тотчас же фильтруют (примечание 6)
через воронку Бюхнера диаметром 19 см. Первая часть фильтрата
содержит небольшое количество двуокиси марганца. Эту часть
собирают отдельно и немедленно фильтруют вторично через ту же
воронку. Как только фильтрат станет прозрачным, его переносят
в 12-литровую круглодонную колбу, в которой находится 130 мл
A37 г; 2,2 мол.) ледяной уксусной кислоты. Затем раствор (он
Должен быть кислым на лакмус) упаривают 8 вакууме B0—30 мм)
24
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
на водяной бане до объема в 1,5—2 л. Полученный раствор оставляют
стоять на ночь в прохладном месте и выкристаллизовавшийся аллан-
тоин отфильтровывают с отсасыванием через воронку Бюхнера
диаметром 9 см (примечание 7). Продукт растворяют в 800—900 мл
кипящей воды, обрабатывают 5 г активированного березового угля
и раствор быстро фильтруют через складчатый фильтр на воронке,
обогреваемой паром. Фильтрат оставляют стоять на ночь в
прохладном месте (примечание 8) и на другой день отсасывают
выпавшие белые кристаллы аллантоина. Выход продукта с т. пл.
230—231° (примечание 9) составляет 60—71 г F4—75% теоре-
тич.). Если упарить полученный после процесса очистки маточ-
ный раствор до 100 мл, то можно получить еще 3—5 г аллан-
тоина.
2. Примечания
1. Если взять больше 80 г едкого натра, выход не увеличивается;
в этом случае, если нейтрализация не проведена тотчас же после
окончания реакции, избыток едкого натра вызывает разложение
некоторого количества аллантоина.
2. Очень важно, чтобы мочевая кислота совершенно раствори-
лась, иначе окисление ее пройдет не полностью. При охлаждении
раствора иногда выделяется небольшое количество белого осадка,
но на выход это не влияет.
3. Перманганат должен быть прибавлен быстро (в течение
1—5 мин,).
4. Количество перманганата может колебаться от 50 до 62 г.
@,32—0,39 мол.), не изменяя выхода.
5. Если продолжительность перемешивания уменьшить до 10
мин., часть мочевой кислоты останется неизмененной. Продолжитель-
ность перемешивания без вреда для выхода может быть увеличена
сверх 20 мин., но если перемешивание вести более часа, выход
значительно уменьшается.
6. Фильтрование следует закончить как можно быстрее, поэтому
необходимо брать большую воронку Бюхнера.
7. Фильтрат отбрасывают, так как в нем слишком мало аллан-
тоина, чтобы пытаться отделить его от других соединений, которые
имеются* там наряду с ним.
8. Кристаллизация может быть ускорена, если раствор охла-
ждать льдом при перемешивании.
9. Температура плавления, повидимому, зависит от скорости
нагревания. Вещество плавится при 228—230°, если капилляр
медленно нагревать в бане, начиная от комнатной температуры.
Если капилляр поместить в баню, уже нагретую до 228°, проба
плавится при 233—234°. С помощью медного блока получают еще
более высокие точки плавления.
АЛЛИЛАМИН
25
3. Другие методы получения
Аллантоин может быть получен окислением мочевой кислоты:
перманганатом калия \ перекисью свинца2, красной кровяной
солью 3, кислородом 4, перекисью марганца 5, озоном е, перекисью
водорода7 и при электролитическом окислении мочевокислого
лития 8. Он получается также при нагревании мочевины с глиокси-
ловой кислотой 9 или с любой другой двузамещенной уксусной
кислотой, например, дихлоруксусной ~~
10
1 С 1 a u s, Вег. 7, 226 A874); Sundwik, Z. physiol. Chem. 41, 343
A904); Behrend, Ann. 333, 141 A904); Biltz, Ber. 43, 1999A910); В i 1 t z,
G i e s 1 e г, там же 46, 3410 A913); Biltz, Max, там же 54, 2451 A921);
Neubauer, Ann. 99, 206 A856).
2 Wohler, Liebig, там же 26, 241 A838); Mulder, там же 159,
349 A871).
3 S с h 1 i e p e г, там же 67, 214 A848).
" Biltz, Max, Ber. 54, 2451 A921).
5 Wheeler, Zeit. fur Chem. 1866, 746.
6 Gorup-Besanez, Ann. 110, 94 A859).
7 Venable, J. Am. Chem. Soc. 40, 1099 A918).
8 F i с h t e r, Kern, Helv. Chim. Acta 9, 429 A926).
9 G r i m a u x, Ann. chim. phys. E) 11, 389 A877).
10 Merck & Co. Inc.; ам. пат. 2 158 098 [С. А. 33, 6350 A939)].
АЛЛИЛАМИН
CH, = CHCH.2NCS + H2O
CH3 = CHCH,NHa- HC1 + KOH
CH2 = CHCH2NH2.HC1 +COS
> CH2 = CHCHoNH, + KC1 + H2O
Предложил: М. Леффлер.
Проверили: В. Хартман и Е. Рарс.
1. Получение
В 5-литровую круглодонную колбу, снабженную обратным
холодильником, соединенным с ловушкой для газов (примечание 1),
помещают 2 л A2,1 мол.) 20%-ной соляной кислоты и 500 г E,05 мол.)
аллилизотиоцианата (примечание 2). Смесь кипятят на голом пла-
мени до полного исчезновения верхнего слоя аллилизотиоцианата.
Всего для гидролиза требуется около 15 час. По окончании реак-
ции жидкость выливают в 3-литровый стакан и упаривают на водя-
ной бане до тех пор, пока из горячего раствора не начнут выпадать
кристаллы. Это имеет место тогда, когда объем раствора достигнет
примерно 400 мл (примечание 3).
Еще теплый остаток разбавляют водой до объема в 500—550 мл
и раствор переливают в 2-литровую трехгорлую круглодонную
колбу, снабженную капельной воронкой емкостью 500 мл, мешалкой
26
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
с ртутным затвором и нисходящим холодильником. Нижний конец
холодильника присоединяют к приемнику, который представляет
собою склянку для отсасывания, емкостью 500 мл; боковой отвод
склянки присоединяют к обратному холодильнику (примечание 4).
Приемник погружают в баню со смесью из льда и соли, а трех-
горлую колбу помещают в водяную баню. Температуру воды в бане
доводят до 95—98°, после чего пускают в ход мешалку и из капель-
ной воронки добавляют по каплям 400 г G,1 мол.) едкого кали,
растворенного в 250 мл воды. После нейтрализации свободной
соляной кислоты начинает отгоняться амин. Скорость приливания
щелочи регулируют таким образом, чтобы аллиламин отгонялся
по каплям. После прибавления всего количества щелочи нагревание
и перемешивание продолжают до тех пор, пока не отгонится весь
амин.
Дестиллат сушат сперва 24 часа над твердым едким кали, а затем
над металлическим натрием (примечание 5). Сухой аллиламин
перегоняют на водяной бане, температуру которой поддерживают
при 70—78°, пользуясь дефлегматором высотой 30 см, и собирают
дестиллат, охлаждаемый льдом. Получают следующие две фракции:
до 54°/746 мм и 54—57°/746 мм. Первая фракция составляет 14—
16 г, и вторичной перегонкой из нее можно выделить 6—8 г чи-
стого вещества. Суммарный выход чистого аллиламина с т. кип.
54—57°/745 мм составляет 200—210 г G0—73% теоретич.).
2. Примечания
1. Для улавливания паров аллилизотиоцианата желательно
пользоваться ловушкой. Вполне подходящей является ловушка,
описанная на стр. 78.
2. Для этого синтеза был применен «практически чистый» *
аллилизотиоцианат (т. кип. 150—152°) фирмы Истмена.
3. Можно значительно увеличить скорость выпаривания, если
одновременно пользоваться вентилятором. Выгодно упарить рас-
твор как можно сильнее, чтобы удалить большую часть соляной
кислоты. Никакого вреда не будет, если упаривание вести до тех
пор, пока раствор не превратится в полутвердую массу кристаллов.
4. Необходимо тщательно следить за тем, чтобы не было потерь
вследствие летучести конечного продукта. Кроме того, пары аллил-
'амина не должны попадать в нос, так как они вызывают сильное
чихание.
* «Практически чистыми» — ('practical» — в американских прейскурантах
называются реактивы, по степени очистки занимающие промежуточное поло-
жение между теми, которые в СССР именуются «техническими» и «чистыми»
(например, перегоняющиеся в пределах 2—3').— Прим. ред.
АЛЬДЕГИДОФТАЛЕВАЯ КИСЛОТА
27
5. Во время сушки дестиллат необходимо поддерживать при
низкой температуре E—10°); кроме того, аллиламин перед сушкой
металлическим натрием необходимо отделить от едкого кали.
3. Другие методы получения
Аллиламин был получен гидролизом аллилизотиоцианата раз-
бавленной серной г или соляной 2 кислотой.
1 Н о f m a n п, Вег. 1, 183 A868).
2 Gabriel, Eschenbach, там же 30, 1124 A897).
АЛЬДЕГИДОФТАЛЕВАЯ КИСЛОТА
Ю]
(КМпО„)
/\COCOJH
со,н
(NaHSO,,)
(НС1)
/\сно
со,н
Предложили: Дж. Гарднер и К- Нэп лор мл.
Проверили: К- Ноллер и К- Лина.
1. Получение
В 3-литровую колбу с тремя горлами, снабженную механиче-
ской мешалкой с жидкостным затвором, обратным холодильником
и капельной воронкой,помещают500мл0,5-н. раствора едкого натра
и 32 г @,25 мол.) чистого нафталина. Смесь нагревают до кипения,
после чего в течение 1,5 часа при энергичном перемешивании (при-
мечание 1), добавляют маленькими порциями кипящий раствор
212 г A,34 мол.) КМпО4 в 1500 мл воды. После того как при-
бавлена последняя порция, смесь кипятят 30—45 мин., чтобы
завершить окисление. Не вступивший в реакцию перманганат
восстанавливают добавлением 20 мл спирта, колбу охлаждают,
чтобы дать затвердеть оставшемуся нафталину, и смесь фильтруют.
Фильтрат подкисляют 150 мл A,8 мол.) концентрированной
соляной кислоты (уд. в. 1,18), выпаривают до 500 мл, охлаждают
и фильтруют. Фильтрат нейтрализуют 30%-ным раствором едкого
натра A50—160 мл), добавляют 50 г @,48 мол.) бисульфита натрия
и смесь выпаривают досуха на паровой бане. Остаток размешивают
со 100 мл концентрированной соляной кислоты и выпаривают до-
суха на кипящей водяной бане. Обработку соляной кислотой и
выпаривание повторяют еще раз (примечание 2).
Остаток тщательно извлекают бензолом в большом экстракторе
Сокслета (примечание 3) и бензольные вытяжки выпаривают досуха.
Сырой продукт растворяют в 50 мл горячей воды, раствор филь-
труют, фильтрат охлаждают при помешивании в бане со льдом
(примечание 4). Кристаллы отсасывают и сушат на воздухе. Выход
28
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
слегка окрашенного продукта с т. пл. 94—95° составляет 15—15,5 г
D0—41% теоретич.).
Если этот продукт перекристаллизовать из 40 мл воды, добавив
1 г обесцвечивающего угля, и фильтрат охладить до 0°, то можно
получить 14—14,5 г белых кристаллов с т. пл. 96—96,5°.
2. Примечания
1. Во время приливания горячего раствора перманганата ни-
какого внешнего обогрева колбы не требуется. Время от времени
через рубашку обратного холодильника пропускают водяной пар,
чтобы возвратить возогнавшийся нафталин обратно в реакционную
колбу.
2. Обработку соляной кислотой и выпаривание повторяют для
того, чтобы добиться полного разложения бисульфитного соеди-
нения альдегидофталевой кислоты. Вегшейдер и Бонди х указывают
на то, что бисульфитное соединение необходимо нагревать на водя-
ной бане в течение нескольких дней с большим избытком соляной
кислоты, однако было установлено, что описанная выше обработка
вполне достаточна.
3. Можно воспользоваться или двумя экстракционными гиль-
зами размером 45 X 125 мм или четырьмя —размером 30x75 .ил.
Очень удобно воспользоваться видоизмененным экстрактором Сок-
слета типа Клаузницера2, в котором пары кипящего растворителя
обтекают экстракционную гильзу.
4. Весьма важно производить точный учет воды, которая берется
для перекристаллизации. В сырой кислоте содержится значительная
примесь фталевой кислоты, и недостаточное количество воды дает
грязный продукт. Слишком большое количество воды заметно умень-
шает выход.
3. Другие методу получения
Альдегидофталевая кислота была получена гидролизом 2-бром-
или 2-хлорфталвда3, о-трихлорметилбензальхлорида4, о-циан-
бензальхлоридаs и о-дихлорметилбензоилхлорнда6. Она была
также^получена действием озона на нафталин 7, окислением в ще-
лочной среде нафталина 8 или я-нитронафталина 9 с последующим
получением и разложением продукта конденсации фталоновой
кислоты с анилином 1и или же бисульфитного 1Д1 соединения фтало-
новой кислоты и действием углекислоты на продукт реакции о-хлор-
бензальдегида с натрием12. Подробные указания по получению
альдегидофталевой кислоты бромированием фталида и гидролизом
продукта бромирования приведены в Org. Syn., вып. 23.
1 Wegsch eider, Bondi, Monatsh. 26, 1055 A905).
3 Ho u ben, Weyl, Die Methoden der organischen Ctiemie, 3-е изд.,
Т. I, с, 565, Verlag Georg Thieme, Leipzig, 1925.
АМИД о-ТОЛУИЛОВОЙ КИСЛОТЫ
3 Racine, Ann. 239, 78 A887); Austin, Bousquet; ам. пат.
2 047 946 [С. А. 30, 6011 A936)].
4 Coulson, Gautier, Bull. soc. chim. B) 45, 507 A886).
6 Gabriel, Weise, Ber. 20, 3197 A887); Drory, там же 24, 2571
A891).
«Davies, Perkin, Clayton, J. Chem. Soc. 121, 2214 A922).
' Seekles, Rec. trav. chim. 43, 706 A923).
'Tcherniac; герм. пат. 79 693 [Frdl. 4, 162 A894—97)]; герм. пат.
86 914 [Frdl. 4, 163 A894—1897)].
9 Gardner, J. 'Am. Chem. Soc. 49, 1831 A927).
10 Soc. Chim. des Usines dn Rh6ne, герм. пат. 97 241 [Frdl. 5, 139 A897—
1900)]; Fu s о n, J. Am. Chem. Soc. 48, 1093 A926).
11 Graebe, Trump y, Ber. 31, 369 A898); Sidgwick, Clayton,
J. Chem. Soc. 121, 2263 A922).
"Morton, Le-Fevre, Hechenbleikner, J, Am. Chem. Soc.
58, 754 A936).
АМИД о-ТОЛУИЛОВОЙ КИСЛОТЫ
(о-Толуамид)
2 o-CH3CeH4CN + 2 Н.2О2 ~-а0Н— 2 o-CH8C,H4CONH!! + О2
Предложил: К. Ноллер.
Проверили: В. Хартман и Л. Смит.
1. Получение
В 2-литровую круглодонную колбу помещают 88 г @,75 мол.)
0-толунитрила («Синтезы орг. преп.», сб. 1, стр. 391), 300мл B,6 мол.)
30%-ной перекиси водорода, 400 мл 95%-ного спирта и 30 мл б-н.
раствора едкого натра (примечание 1). Из смеси начинает выде-
ляться кислород, причем смесь вскоре разогревается благодаря
теплоте, выделяющейся при реакции; внешним охлаждением тем-
пературу поддерживают при 40—50° (примечание 2). Приблизи-
тельно через час выделение тепла прекращается, после чего смесь
нагревают в течение 3 час. при 50°. По окончании нагревания еще
теплую смесь точно нейтрализуют по лакмусу 5%-ной серной кисло-
той и перегоняют с паром; отгоняют 1 л дестиллата, а остаток в коли-
честве около 600 мл (примечание 3) еще горячим переливают в лит-
ровый стакан и охлаждают до 20°. Выпавшие кристаллы отсасы-
вают, переносят в ступку и растирают в пасту со 100 мл холодной
воды, снова отсасывают и промывают на фильтре 100 мл холодной
воды.-Полученный таким образом амид о-толуиловой кислоты пред-
ставляет собою бесцветные кристаллы с т. пл. 141—141,5°. Выход
воздушно-сухого продукта: 91—93 г (90—92% теоретич.; приме-
чание 4). Продукт можно перекристаллизовать из воды A00 мл
на 10 г); выход при перекристаллизации 92%; точка плавления
при этом не изменяется (примечание 5).
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
2. Примечания
1. Для получения однородного раствора достаточно указанного
количества спирта.
2. Если температуре дать подняться значительно выше 50°,
то выделение кислорода становится настолько сильным, что смесь
вспенивается и может быть выброшена из колбы.
3. После отгонки главной массы спирта объем раствора в колбе
поддерживают постоянным, нагревая колбу на небольшом пламени.
4. По упаривании фильтрата можно получить еще 3—4 г низко-
плавкого продукта, но делать этого не имеет смысла.
5. По описанному методу, исходя из алифатических нитрилов,
можно получить и другие амиды с выходом 50—60%, из аромати-
ческих нитрилов — с выходом 80—95%. Приходится вносить лишь
небольшие изменения в условия проведения реакции и в способ
выделения амида, вызываемые различной растворимостью как
нитрилов, так и амидов. Если не считать трудно омыляемых нитри-
лов, например, е-замещенных ароматических нитрилов1, то лучшие
выходы получаются при применении эквивалентных количеств 6—
12%-ной перекиси водорода вместо 30%-ной.
3. Другие методы получения
Амид о-толуиловой кислоты был получен действием аммиака на
хлорангидрид о-толуиловой кислоты2 и действием спиртового
раствора едкого кали 3 или щелочного раствора перекиси водо-
рода 4>' на 0-толунитрил.
1 М с М a s t е г, N о 1 1 е г, Wash. Univ. Studies 13, 23 A925); I. Indian
Chem. Soc. 12, 652 A935) [C. A. 30, 1736 A936)].
2 Remsen, Reid, Am. Chem. I. 21, 289 A899).
3 Wei th, Ber. 6, 419A873).
i Kattwinkel, W о 1 f f e n s t e i n, там же 37, 3224 A904); Dub-
s k y, J. prakt. Chem. B) 93, 137 A916).
н.-АМИЛБЕНЗОЛ
C6H3CHX1 + Mg — CeH6CHJVlgCl;
CeHsCH.,MgCl + 2n-CH,CeH4SO8C4He
Cr>H3 (CH^CHs + C4H9C1 + (/r-CH3C6H4SO3JMg
Предложили: Г. Гильман и Дж. Робинсон.
Проверили: Н. Марвел и С, Россандер.
1. Получение
В 2-литровой круглодонной трехгорлой колбе приготовляют
раствор бензилмагнийхлорида из 24,3 г A rp-ат) магниевых стружек,
126,5 г A15 мл; 1 мол,) хлористого бензила и 500 мл абсолютного
н.-АМЙЛБЕНЗОЛ
31
эфира, по способу, описанному для получения н.-пропилбензола
(«Синтезы орг. преп.», сб. 1, стр. 364).
Раствор охлаждают проточной водой и через капельную воронку
прибавляют медленно и при помешивании 456 г B мол.) н.-бути-
лового эфира л-толуолсульфокислоты (примечание 1), растворенного
приблизительно в двойном количестве (по объему) абсолютного эфира,
с такой скоростью, чтобы эфир спокойно кипел (около 2 час). Скоро
выпадает белый твердый осадок и раствор принимает консистенцию
густых сливок. Перемешивание продолжают без охлаждения еще
около 2 час, после чего смесь выливают на измельченный лед,
к которому прибавлено 125 мл концентрированной соляной кислоты
(примечание 2).
Эфирный слой отделяют и водный слой экстрагируют 200 мл
эфира; соединенные эфирные вытяжки промывают один раз 100 мл
воды, а затем сушат, встряхивая в течение нескольких минут с 10 г
безводного поташа. После фильтрования эфир отгоняют на водяной
бане. Когда отгонится практически весь эфир, к остатку приба-
вляют около 5 г натрия, свеженарезанного тонкими ломтиками,
и кипятят в течение 2 час. (примечание 3). Жидкость деканти-
руют и перегоняют, пользуясь эффективной перегонной колонкой
и собирая фракцию, кипящую при 190—210°. При вторичной Пере-
гонке получается 74—88 г E0—59% теоретич.) н.-амилбензола
с т. кип. 198—202° (примечание 4).
2. Примечания
1. Метод получения н,-бутилового эфира л-толуолсульфокислоты
описан в «Синтезах орг. преп.», сб. 1, стр. 149, и в Org. Syn 20, 51.
2. Гидролиз удобно вести в 5-литровой конической колбе.
Магниевая соль л-толуолсульфокислоты плохо растворима в соля-
ной кислоте; поэтому полное растворение ее достигается последу-
ющим прибавлением приблизительно 2 л воды.
3. Металлический натрий при кипячении связывает небольшое
количество бензилового спирта, образующегося в результате окисле-
ния магнийорганического соединения кислородом воздуха.
4. Главная часть н.-амилбензола перегоняется при 199—201°.
Тщательной разгонкой фракции, переходящей около 75°, можно
получить 24 г B6% теоретич.) хлористого н.-бутила, кипящего при
76—80°.
3. Другие методы получения
н.-Амилбензол может быть получен: действием натрия на смесь
бромистого бензила и бромистого н.-бутилаа; взаимодействием
бензил-натрия с хлористым бутилом2; восстановлением н.-бутил-
фенилкетона муравьиной кислотой в присутствии меди при 300° 3
или цинком и соляной кислотой4; действием этилата натрия на
32
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
гидразон5 и ссмикарбазон6 н.-бутилфенилкетона и, наконец, описан-
ным здесь методом, основанным на данных Гильмана и Хека7,
а также Россандера и Марвела 8.
1 Schraram, Ann. 218, 388 A883).
2 Morton, Fallwell, Jr., J. Am. Chem. Soc. 60, 1429 A938).
3 Mailhe, deGodon, Bull. soc. chim. D) 21, 62 A917).
1 Stenzl, Fichter, Helv. Chim. Acta 17, 677 A934).
6 S с h m i d t, H о p p, S с li oe 1 1 e r, Ber. 72, 1893 A939).
6 Z i e g I e r, Dersch, Wollthan, Ann. 511, 38 A934).
7 Gilraan, Heck, J. Am. Chem. Soc. 50, 2223 A928).
8 Rossander, Marvel, там же 50, 1491 A928).
4-АМИНОВЕРАТРОЛ
C,4-Диметоксианилии)
2 (СН3ОJ C8H3CN + 2 Н2О2 — — ^ 2 (СН3ОJ C6H3CONHa + О2
(СНзО)^ C6HaCONH2 + NaOCl + 2 NaOH - ~
(СН36J C6H3NH2 + NaCl + Na2CO3 + H2O
Предложили: Док. Бек и В. Айди.
Проверили: Дж. Джонсон'и X. Стивенсон.
1. Получение
В 5-литровую колбу, снабженную механической мешалкой, по-
мещают 1,8 кг A,6 мол.) свежеприготовленного 3%-ного раствора
перекиси водорода, 100 г 25%-ного раствора едкого кали и 57 г
@,35 мол.) вератронитрила (стр. 145). Смесь при работающей ме-
шалке медленно нагревают до 45°, после чего источник нагревания
удаляют. Реакция протекает с выделением кислорода, и температура
продолжает повышаться (примечание 1). Вскоре начинает выделяться
амид; примерно через 50 мин. реакция заканчивается, и темпера-
тура начинает понижаться. Смесь охлаждают до 3—5° и оставляют
в бане со льдом на 1,5—2 часа. Белый кристаллический продукт
отсасывают и сушат па воздухе. Амид вератровой кислоты плавится
при 162,5—163,5°. Выход 55—58 г (87—92% теоретич.).
Щелочной раствор гипохлорита натрия получают пропусканием
хлора @,412 г на каждый грамм амида; примечание 2) через смесь,
состоящую из 300 г колотого льда и холодного раствора 80 г едкого
натра в 500 мл воды, находящуюся в 2-литровой круглодонной колбе.
К полученному раствору прибавляют в один прием все количе-
ство амида вератровой кислоты E5—58 г) и смесь медленно нагревают
на водяной бане при работающей мешалке. Вскоре масса начинает
темнеть, и при 50—55° (температура внутри колбы) начинается
выделение маслянистых капель. Смесь постепенно нагревают до
70° и поддерживают эту температуру в течение 1 часа. Затем
4-АМИНОВЕРАТРОЛ
33
медленно добавляют раствор 120 г едкого натра в 120 мл воды, темпе-
ратуру доводят до 80° и поддерживают ее на таком уровне еще 1 час.
По охлаждении смеси маслянистый слой амина застывает в крас-
ную кристаллическую массу. Сырой амин отсасывают, промывают
два раза ледяной водой порциями по 60 мл, хорошо отжимают и пере-
носят в обыкновенную колбу Клайзена на 125 мл. Фильтрат экстра-
гируют три раза бензолом, порциями по 60 мл, каждую вытяжку в
отдельности добавляют в колбу Клайзена и отгоняют бензол при
атмосферном давлении (примечание 3). Оставшийся амин перего-
няют в вакууме и собирают фракцию 172—174°/24 мм (примеча-
ние 4). Дестиллат быстро застывает в массу бесцветных кристаллов,
обладающих резкой точкой плавления 87,5—88° (примечание 5).
Из 58 г @,32 мол.) амида вератровой кислоты получают 39—40 г
(выход 80—82%) 4-аминовератрола.
2. Примечания
1. При этом происходит сильное вспенивание, и температура по-
вышается до 52—55°. Время от времени колбу снимают и взбал-
тывают от руки, чтобы привести в соприкосновение с раствором
частицы, увлеченные пеной.
2. Указанное количество хлора на 5% больше теоретического.
Хлор можно брать непосредственно из баллона, но небольшие ко-
личества его с большей точностью можно получать действием на
навеску перманганата калия избытка концентрированной соляной
кислоты @,367 г КМпО4 эквивалентны 0,412 г С12). На 55—58 г амида
вератровой кислоты следует взять 20,2—21,3 г перманганата.
С целью получения хлора отвешенное количество перманганата
калия помещают в колбу Вюрца на 500 мл, снабженную капельной
воронкой и укрепленную так, что ее можно взбалтывать. К кристал-
лам перманганата приливают по каплям концентрированную соля-
ную кислоту (всего требуется около 130 мл) и, по мерс того как идет
реакция, колбу подогревают. После прибавления всей кислоты
смесь осторожно кипятят в течение нескольких минут, чтобы выде-
лить остаток хлора, и затем быстро отъединяют прибор от сосуда с
раствором гипохлорита, чтобы избежать засасывания последнего.
Рекомендуется ставить предохранительную склянку между боковой
трубкой колбы Вюрца и трубкой, по которой хлор поступает в ще-
лочь.
3. Посредством такого метода отгонки бензола удаляется вода,
так что при последующей перегонке амина уже не образуется го-
ловной фракции, содержащей воду.
4. Принимая во внимание, что аминовератрол может застывать
уже в боковой трубке перегонной колбы, рекомендуется перегонку
вести быстро и поддерживать в бане температуру на 60° выше темпера-
туры отходящих паров. Следует принимать меры предосторожности,
3 Сборник Jfi 2
34
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
чтобы не загрязнить дестиллат небольшим количеством окрашен-
ных примесей, которые переходят в приемник, если перегонку
вести слишком глубоко.
5. Температура плавления 4-аминовератрола, полученного вос-
становлением 4-нитровератрола, согласно литературным данным,
равна 85—86°\ Амин под действием воздуха и света быстро приобре-
тает окраску; хранить его следует в запаянном сосуде из темного
стекла.
3. Другие методы получения
4-Аминовератрол был получен восстановлением 4-нитровера-
трола оловом1-2 или хлористым оловом3и соляной кислотой. При-
веденная выше методика основана на методе, применяемом для по-
лучения аминопиперола C,4-метилендиоксианилина) 4.
1 Moureu, Bull. soc. chim. C) 15, 647 A896).
2 S i mo n se n, Ra u, J. Chem.Soc. 113,28A918); Pollecoff, Robin-
son, там же 113, 645 (примечание) A918).
3 H e i n i s с h, Monatsh. 15, 232 A894).
« Rupe, Majewski, Ber. 33, 3401 A900).
а АМИНОИЗОМАСЛЯНАЯ КИСЛОТА
CH3COCH3 + NaCN + NH4C1 > (CH3J C(OH)CN + NaCl + NH3;
(CH3J C(OH) CN + NH3 - (CH3J C(NH2) CN + H2O;
(CH3J C(NH2) CN + 2 H2O + 2 HBr
(CH3J C(NH2 • HBr) CO.H + NH4Br;
(CH3), C(NH2 ¦ HBr) CO2H + C5H5N —->
(CH3J C(NH2) CO2H + C5H5N • HBr.
Предложили: X. Кларк и X. Бин,
Проверили: К. Марвел и Я. Бэйли.
1. Получение
В 1-лщровую круглодонную колбу помещают профильтрован-
ный раствор 200 г C,7 мол.)хлористого аммония в 500 лм воды; по-
гружением колбы в ледяную воду раствор охлаждают до 5—10°.
Затем прибавляют при перемешивании (примечание 1) раствор 175 г
C мол.) ацетона в 500 мл эфира, а за ним — раствор 160 г C,2 мол.)
цианистого натрия в 350 мл воды, также при перемешивании и с такой
скоростью, чтобы температура смеси не превышала 10° (примечание 2).
По прибавлении всего раствора цианистого натрия реакцион-
ную смесь продолжают перемешивать в течение одного часа, а затем
оставляют стоять на ночь. Эфирный слой отделяют, а водный рас-
твор экстрагируют шестью порциями эфира по 300 мл. Эфирные вы-
тяжки соединяют и отгоняют эфир. Остаток, состоящий главным
образом из циангидрина ацетона, растворяют в 800 мл метилового
ct-АМИНОИЗОМАСЛЯНАЯ КИСЛОТА
35
спирта; раствор охлаждают, насыщают газообразным аммиаком
(примечание 3), оставляют стоять в течение 2—3 дней (примечание 4),
а затем избыток аммиака вытесняют током воздуха. Метиловый
спирт отгоняют как можно тщательнее; к остатку прибавляют сперва
600 мл воды, а затем —¦ 1000 г 48%-ной бромистоводородной кисло-
ты, после чего смесь кипятят с обратным холодильником в течение
двух часов. *
Бромистоводородную кислоту отгоняют в вакууме на водяной
бане; к остатку прибавляют 400—500 мл воды и вневь концентри-
руют раствор в вакууме для того, чтобы возможно полнее удалить
бромистоводородную кислоту (примечание 5).
Остаток растворяют в 15—20-кратном количестве (по весу) ме-
тилового спирта (примечание 6), раствор фильтруют и смешивают
с избытком пиридина (примечание 7). После стояния в течение ночи
выпадает свободная аминокислота. Ее отсасывают, хорошо промы-
вают метиловым спиртом и сушат. Выход; 92—102 г C0—33% тео-
ретич.). Если нужно получить продукт, не содержащий пиридина,
то полученную по вышеописанному способу аминокислоту раство-
ряют в 200 мл теплой воды, раствор фильтруют, а фильтрат сме-
шивают с 2 л метилового спирта (примечание 8). В маточных рас-
творах остается менее 10 г аминокислоты, которую можно выделить
выпариванием раствора досуха, обработкой остатка метиловым
спиртом и повторным осаждением, как описано выше.
2. Примечания
1. Для получения хороших результатов необходимо энергичное
перемешивание.
2. Повышение температуры реакционной смеси до 15° не пони-
жает выхода. При 0J реакция идет очень медленно.
3. Для того, чтобы превратить образовавшийся в течение пер-
вой стадии процесса циангидрин ацетона в аминонитрил, необхо-
дим избыток аммиака.
4. В некоторых опытах смесь стояла только в течение 24 час,
однако выход понизился лишь незначительно.
5. После прибавления воды и последующего выпаривания по-
чти досуха полезно прибавить еще раз небольшое количество воды
B5—75 мл) и вновь выпарить досуха, чтобы обеспечить полное
удаление бромистоводородной кислоты. В случае необходимости
прибавление воды и выпаривание следует повторить несколько раз.
6. Количество метилового спирта не должно превышать 3 л: в
противном случае в растворе остается много аминокислоты. Для
полного растворения бромистоводородной соли аминокислоты смесь
необходимо долго перемешивать на холоду, хотя, повидимому, ни-
каких трудностей не встретится даже и в том случае, когда остаток
растворится не нацело.
Зб
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
7. Минимальное необходимое количество пиридина опреде-
ляется количеством бромистоводородной кислоты, содержащейся
в остатке. Избыток пиридина не причиняет вреда. Если пиридина
желательно израсходовать возможно меньше, его прибавляют неболь-
шими порциями до тех пор, пока раствор не будет нейтральным на
конго, а затем прибавляют еще 250 г.
8. Для увеличения выхода и получения дополнительного неболь-
шого количества аминокислоты маточный раствор упаривают до
небольшого объема на водяной бане; выпадающие при стоянии кри-
сталлы отсасывают и промывают метиловым спиртом.
3. Другие методы получения
Единственным удовлетворительным методом получения а-амино-
изомасляной кислоты является способ Штреккерах в том или дру-
гом его видоизменении2. Способ выделения аминокислоты из спирто-
вого раствора ее бромистоводородной соли с помощью пиридина в
основном копирует метод, предложенный для получения глицина3;
вместо пиридина можно применять анилин4.
1 Strecker, Ann. 75, 28 A850).
2 Tiemann, FriedUnder, Ber. 14, 1970 A881); Mnrckwald,
N e u m a r k, S t e 1 z n e г, там же 24, 3283 A891); Г у л е в и ч, там же 33,
1900 A900); Н е 1 1 s i n g, там же 37, 1923 A904); Г у л е d и ч и В а с м у с,
там же 39, 1184 A906); Зелинский, С т а д и и к о в, ЖРХО, 38, 722 A906);
Cocker, Lapworth, J. Chem. Soc. 1931, 1301.
3 Org. Syn. 4, 31 A925).
4 Benedict, J. Am. Chem. Soc. 51, 2277 A929).
е-АМИНОКАПРОНОВАЯ КИСЛОТА
/NH
(СН3)/ | + Н2О > NH2 (СН2)В СО2Н
Предложил: Дж. Экк.
Проверили: Л. Физер и Я- Фишер.
1. Получение
В круг лодонную колбу емкостью 500 мл помещают раствор 45 мл
концентрированной соляной кислоты (уд. в. 1,19) в 150 мл воды и
добавляют 50 г @,44 мол.) 2-кетогексаметиленимина (стр. 304). Рас-
твор кипятят около 1 часа, пока он не станет прозрачным (приме-
чание 1), и выпаривают досуха в вакууме на кипящей водяной бане.
Полученную хлористоводородную соль е-аминокапроновой ки-
слоты превращают в свободную кислоту так же, как это описано
при получении dZ-аланина («Синт. орг. преп.», сб. 1, стр. 20). Соль
растворяют в \л воды в стакане емкостью 1,5 л и последовательно
Г-АМИНОМАСЛЯНАЯ КИСЛОТА
37
обрабатывают 50 г измельченного глета, 25 г измельченного глета,
5 г свежеосаждснной гидроокиси свинца, 25 г измельченной окиси
серебра (примечание 2) и, наконец, сероводородом. При всех этих
операциях сохраняют первоначальный объем путем прибавления
небольших количеств воды.
После того как нацело удалены ионы галоида и металлов, рас-
твор упаривают до объема в 100 мл и добавляют 300 мл абсолютного
спирта. Затем осаждают аминокислоту, медленно добавляя к ней
при помешивании и охлаждении 500 мл эфира.
Полученную s-аминокапроновую кислоту отсасывают и сушат
в эксикаторе. Выход е-аминокапроновой кислоты с т. пл. 201—203°
составляет 52,5—53,5 г (90—92% теоретич.).
2. Примечания
1. Это указывает на то, что гидролиз закончен.
2. Точное количество окиси серебра, необходимое для реакции,
можно определить, оттитровав пробу раствора азотнокислым сере-
бром по Фольгарду.
3. Другие методы получения
е-Аминокапроновая кислота была получена омылением г-бен-
зоиламинокапронитрила \ омылением этилового эфира 8-фталимид-
бутилмалоновой кислоты2 и из циклогексаноноксима в результате
перегруппировки и гидролиза3.
iBraun, Steindorff, Ber. 38, 176 A905); В г a u n, там же 40,
1839 A907); Ruzicka, Hugoson, Helv. Chim. Acta 4, 479A921);
Marvel, MacCorquodale, Kendall, Lazier, J. Am. Chem.
Soc. 46, 2838 A924).
2 Gabriel, M a a s s, Ber. 32, 1266 A899).
3 W a 1 1 а с h, Ann. 312, 188 A900); Eck, Marve 1, J. Biol. Chem. 106,
387 A934).
'\
Y-АМИНОМАСЛЯНАЯ КИСЛОТА
,cox
C6H4<; /NK + Cl (CH2K CN -^ C6H4\ /N (CH2K CN -f KC1
/CO
C6H4<X j>N(CH4)aCN lHHs2o^ HaNCHXH2CHaCO2H
Предложил: К- ДеВитт.
Проверили: В. Хартман и А. Швадерер.
1. Получение
В 1-литровую круглодонную колбу, закрытую хорошей корко-
вой пробкой, через которую проходит воздушный холодильник,
38
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
помещают 100 г @,54 мол.) хорошо измельченного фталимида калия
(«Синт. орг. преп.», сб. 1, стр. 143) и 52 г @,5 мол.) у-хлорбутиро-
нитрила («Синт. орг. преп.», сб. 1, стр. 495). Колбу нагревают на
масляной бане, температуру которой поддерживают при 150—180°
в течение 1,5 часа (примечание 1), после чего ей дают охладиться.
Избыток фталимида калия вместе с образовавшимся хлористым
калием удаляют путем извлечения несколькими порциями дестил-
лированной воды до тех пор, пока промывные воды не дадут отри-
цательной пробы на хлор-ион. После этого колбу охлаждают, про-
дукту дают затвердеть и оставшуюся воду декантируют возможно
полнее. Осадок обрабатывают 140 мл концентрированной серной
кислоты и смесь осторожно нагревают на масляной бане с обрат-
ным холодильником до полного растворения -f-фталимидбутиро-
нитрила. Затем осторожно добавляют через обратный холодильник
200 мл дестиллированной воды и раствор энергично кипятят в те-
чение 3 час. Смесь охлаждают и оставляют на ночь, после чего от-
фильтровывают фталевую кислоту. Фильтрат переливают в боль-
шую фарфоровую чашку, приливают 1 л дестиллированной воды и
добавляют небольшими порциями избыток углекислого бария (около
550 г; примечание 2). Смесь выпаривают на водяной бане почти до-
суха, остаток хорошо размешивают с \л дестиллированиой воды
и вновь выпаривают (примечание 3). После этого опять прибавляют
1 л дестиллированной воды, массу хорошо размешивают и смесь
фильтруют через большую воронку Бюхнера. Осадок промывают
три раза горячей дестиллированной водой порциями по 200 мл, и
фильтрат вместе с промывными водами упаривают на кипящей
водяной бане до объема в 200 мл. Прибавляют 2 г активированного
угля, раствор фильтруют с отсасыванием через фильтр Уотмена
№ 42 и активированный уголь промывают несколько раз неболь-
шим количеством горячей дестиллированной воды. Фильтрат упа-
ривают на паровой бане до начала кристаллизации (около 75 мл) и
для того, чтобы осадить аминокислоту, добавляют 375—500 мл абсо-
лютного спирта. Массу хорошо перемешивают так, чтобы окрашен-
ные в желтый цвет примеси остались бы в растворе и по охлаждении
бесцветный кристаллический продукт отфильтровывают и промы-
вают абсолютным спиртом.
Спиртовой фильтрат упаривают до 50 мл и добавляют 50 г гидрата
окиси бария и 150 мл дестиллированной воды (примечание 4). Смесь
кипятят с обратным холодильником в течение 2 час. и избыток ги-
драта окиси бария осаждают углекислым газом. Углекислый барий
отфильтровывают и промывают горячей дестиллированной водой.
К фильтрату добавляют небольшой избыток серной кислоты для
того, чтобы выделить аминокислоту из ее бариевой соли, а для уда-
ления сульфат-иона добавляют избыток углекислого бария. Смесь
нагревают на паровой бане и перемешивают до тех пор, пока не
прекратится выделение газа, осадок отфильтровывают и промывают
Y-АМИНОМАСЛЯНАЯ КИСЛОТА
39
его горячей дестиллированной водой. Фильтрат и промывные воды
упаривают на водяной бане до объема в 100 мл, обесцвечивают рас-
твор 1 г активированного угля, фильтруют и упаривают до начала
кристаллизации (около 25 мл). Аминокислоту осаждают добавле-
нием 150 мл абсолютного спирта, осадок отфильтровывают и про-
мывают абсолютным спиртом.
Суммарный выход 24—32 г D7—62%, считая на взятый
в реакцию у-хлорбутиронитрил). Для перекристаллизации
аминокислоты ее растворяют в возможно меньшем количестве
дестиллированной воды и добавляют 5—7 объемов абсолютного
спирта.
2. Примечания
1. Через 45 мин. рекомендуется прервать нагревание и тщатель-
но перемешать пастообразную массу стеклянной палочкой. Более
длительное нагревание реакционной массы хотя и не является не-
обходимым, но не приносит вреда.
2. Углекислый барий нейтрализует серную кислоту и реаги-
рует с сернокислым аммонием, но не реагирует с аминокисло-
той.
3. Обычно в результате этих двух обработок аммиак бывает
удален нацело, однако, если он еще остался, перемешивание
осадка с водой и последующее выпаривание повторяют еще
раз.
4. В спиртовом фильтрате содержится заметное количество нир-
ролидона. Обработкой избытком гидроокиси бария можно превра-
тить его в бариевую соль аминокислоты 1>2.
3. Другие методы получения
у-Аминомасляная кислота была получена электролитическим
восстановлением сукцинимида в пирролидон с последующим гидро-
лизом его гидроокисью бария1; окислением пиперилуретана дымящей
азотной кислотой и обработкой полученного продукта концентриро-
ванной ""соляной кислотой в запаянных трубках при 100°; гидроли-
зом продукта конденсации 1М-(Р-бромэтил)-фталимида с натрий-
малоновым эфиром4 и с помощью метода, описанного выше,
который является незначительным видоизменением метода Габ-
риэля 2.
1 Т a f e I, Stern, Ber. 33, 2224 A900).
8 G a b r i e 1, там же 22, 3335 A889); 23, 1771 A890).
3 S с h о 11 e n, там же 16, 643 A883).
1 A s с h а п, там же 24, 2450 A891).
40
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
+ NaOH
2-АМИНО-4-МЕТИЛТИАЗОЛ
СН3 —СО HN
I + II
СН2С1 HS — С — NH2
СНЯ —С —N
II I!
НС С — NH3+ NaCl + 2 Н2О
Предложили: Дж. Байере и Дж. Дакки.
Проверили: Л. Физер и Б. Ригель.
1. Получение
В колбе емкостью 500 мл, снабженной обратным холодильником,
капельной воронкой и механической мешалкой, приготовляют сус-
пензию из 76 г A мол.) тиомочевины и 200 мл воды (примечание 1).
Пускают в ход мешалку и в течение 30 мин. прибавляют 92,5 г(80 мл,
1 мол.) хлорацетона (примечание 2). По мере того, как идет реакция,
тиомочевина растворяется и температура повышается. Желтый рас-
твор кипятят два часа. Затем его охлаждают и при не слишком сильном
перемешивании, чтобы избежать образования неприятной эмуль-
сии, добавляют 200 г твердого едкого натра, охлаждая при этом
колбу. Верхний маслянистый слой отделяют в делительной воронке,
а водный слой экстрагируют три раза эфиром, общее количество
которого составляет 300 мл (примечание 3). Тёмнокрасную масля-
нистую жидкость и эфирные вытяжки соединяют и раствор сушат
30 г твердого едкого натра, после чего его фильтруют без отсасы-
вания, чтобы удалить небольшое количество смолы. Эфир отгоняют
на водяной бане и остаток перегоняют в вакууме. После небольшой
головной фракции собирают 2-амино-4-метилтиазол при 117—
120°/8 мм или при 130—133°/18 мм. Выход продукта с т. пл. 44—45°
составляет 80—85,5 г G0—75% теоретич.).
2. Примечания
1. Реакцию можно проводить и без разбавителя, но в этом слу-
чае она может пойти очень бурно.
2. Продажный хлорацетон был подвергнут перегонке, причем
была взята фракция с температурой кипения 116—122°. Она почти
вся выкипала при 118—120°.
3. Если выпадает осадок и образуется эмульсия, добавляют
лед и воду до тех пор, пока она не растворится.
1-АМИНО-2-НАФТОЛ СОЛЯНОКИСЛЫЙ
41
3. Другие методы получения
В основном этот метод разработал Трауман 1. 2-Амино-4-метил-
тиазол был получен также из хлорацетона и тиоцианата аммо-
ния2; из хлорацетона и тиоцианата аммония в водном аммиаке3;
действием тиоцианата аммония на тиоцианоацетон 3; омыле-
нием и отщеплением углекислоты от циклического эфира, получен-
ного из этилового эфира у-бромацетоуксусной кислоты и тиомоче-
вины 4 и из тиоцианоацетона и аммиака в абсолютном эфире 5.
1 Tiaumann, Ann. 249, 37 A888).
2 Hantzsch, Traumann, Ber. 21, 938 A888); Hantzsch, We-
ber, тат же 20, 3118 A887).
3Tscherniac, Norton, там же 16, 345 A883); Tscherniac,
J. Chem. Soc. 115, 1071 A919).
1 S t e u d e, Ann. 261, 33 A891).
5 Hantzsch, Ber. 61, 1785 A928).
1 АМИНО 2 НАФТОЛ СОЛЯНОКИСЛЫЙ
I
NO
/чон
NO
NO
-\- NaOH
+ 4 [HI (Naas2°i),
ONa
\\ / \ /
NH,
ONa
+ 2HC1
H.,0
H2O
NaCl
Предложили: Дж. Конант и Б. Корсон.
Проверили: Ф. Уитмор и А. Остергоф.
1. Получение
В 8-литровый глиняный сосуд, снабженный механической мешал-
кой и трубкой для подвода пара, помещают 240 г A,39 мол.) нитрозо-
42
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
р-нафтола (из 200 г р-нафтола, см. «Синт. орг. преп.», сб. 1, стр. 300;
примечание 1), 1,5 л воды и 300 .мл 5-н. раствора едкого натра. Комки
разбивают палочкой и размешивают смесь в течение 30 мин. К концу
этого времени все нитрозосоединение практически переходит в рас-
твор; тогда прибавляют еще 1200 мл 5-н. раствора едкого натра.
Затем пропускают водяной пар до тех пор, пока температура смеси
не достигнет 35°, после чего доступ пара прекращают и прибавляют
к раствору, при перемешивании, 600 г технического гидросульфита
(минимум 85%-ного). Реакционную массу непрерывно перемешивают
в течение 5 мин., причем температура поднимается до 60—65°, и в
продолжение следующего получаса ведут перемешивание с 5-минут-
ными интервалами, каждый раз по одной минуте. Приблизи-
тельно через 15 мин. раствор становится прозрачным и светло-
желтым. На поверхности плавает небольшое количество черной
пены.
Когда перемешивание закончено, раствор охлаждают до 20°
прибавлением к нему около 1 кг льда и приливают при перемеши-
вании 500 мл технической концентрированной соляной кислоты
уд. в. 1,16. Эта последняя осаждает аминонафтол в виде объемистого,
почти белого осадка (примечание 2), который отсасывают на двух
20-сантиметровых воронках Бюхнера, быстро и как можно тщатель-
нее отжимают от маточного раствора и быстро же переносят (при-
мечание 3) в 8-литровый кувшин, содержащий 2,5 л воды и 250 мл
технической концентрированной соляной кислоты. Большие комки
разбивают палочкой, пропускают водяной пар и пускают в ход ме-
шалку. Пар впускают с такой скоростью, чтобы температура смеси
поднялась в течение 10 мин. до 85—90°, и продолжают перемеши-
вание при этой температуре еще в продолжение 1 часа. Затем горя-
чую смесь фильтруют с отсасыванием через 15-сантиметровую
воронку Бюхнера и охлаждают фильтрат до 35—40° в ледяной
бане.
Раствор цвета красного вина E200—5800 мл) фильтруют через
складчатый бумажный фильтр, положенный в 15-сантиметровую
воронку, в 1200 мл концентрированной соляной кислоты, находя-
щейся в 8-литровом сосуде. Тотчас же начинается выпадение соляно-
кислого аминонафтола. Смесь оставляют стоять не менее чем на
2 часа, время от времени перемешивая ее, чтобы ускорить полноту
осаждения. Выпавшую солянокислую соль 1-амино-2-нафтола отса-
сывают на 15-сантиметровой воронке Бюхнера, промывают после-
довательно тремя небольшими порциями 20%-ной соляной кислоты и
тремя порциями эфира по 50 мл (примечание 4) и сушат на воздухе,
разложив тонким слоем на фильтровальной бумаге. Выход 180—
200 г, считая на безводное вещество (примечание 5), т. е. 66—74%
теоретического количества, считая на [3-нафтол. Полученная соль
нестойка; она быстро разлагается в растворе, однако это разло-
жение можно в значительной мере предупредить прибавлением
1-АМИНО-2-НАФТОЛ СОЛЯНОКИСЛЫЙ
43
бисульфита натрия. Сухое твердое вещество разлагается медленнее,
но все же его следует применять в течение нескольких недель после
получения (примечание 6).
2. Примечания
1. Нет надобности сушить нитрозо-р-нафтол. Количество нитро-
зосоединения, приведенное в этом синтезе, соответствует 200 г
р-нафтола, и выход вычислен исходя из тех же 200 г. Нитрозо-°-наф-
тол растворяется в растворе 1 мол. едкого натра с образованием
зеленого раствора. В растворе остается в виде суспензии неболь-
шое количество аморфного бурого вещества, которое можно не
удалять.
2. Количества едкого натра, гидросульфита натрия и соля-
ной кислоты рассчитаны так, чтобы вызвать полное осаждение
аминонафтола. На всякий случай полноту осаждения следует
проверить прибавлением нескольких капель щелочи к одной
пробе фильтрата и небольшого количества кислоты — к другой
пробе.
3. Аминонафтол очень чувствителен к действию воздуха. Толь-
ко что осажденный аминонафтол — белого цвета, но на воздухе он
быстро становится пурпурным, и поэтому работать с ним следует
быстро. Чтобы уменьшить окисление, можно под конец смочить
аминонафтол слабым раствором бисульфита натрия, но обычно
этого можно не делать, так как небольшое количество бисульфита,
образовавшегося из гидросульфита, поглощается осадком и сохра-
няется в нем до конечного осаждения.
4. Промывание небольшим количеством эфира очень облег-
чает просушивание и несколько обесцвечивает продукт. Если веще-
ство будет применяться без сушки, то эфиром его можно не про-
мывать.
5. Для того, чтобы определить истинный выход, следует высу-
шить аликвотную пробу в вакууме, над едким натром. Вещество,
высушенное на воздухе и кажущееся совершенно сухим, содержит
от 10 до 15% влаги.
6. Свежеприготовленный солянокислый 1-амино-2-нафтол об-
ладает светлопурпурной окраской; при хранении темнеет. Его
можно очистить растворением в горячей воде, содержащей бисуль-
фит натрия, фильтрованием полученного раствора и повторным
осаждением соляной кислотой.
44
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
ЧЧ—он
II
+N2C6H4S(V + NaOH >
N = NC6H4SO3Na
I
N = NC6H,jSO3Na
"Voh
^'\
\
2 NaoS204 + 4 H20
+ 4 NaHSO3 + H2NC6H4SO3Na
Предложил: Л. Физер.
Проверили: К. Иоллер и В. Уайт.
1. Получение
А. Диазотирование сулъфаниловой кислоты. Смесь 105 г @,5 мол.)
сульфаниловой кислоты (с 2 мол. кристаллизационной воды), 26,5 г
@,25 мол.) безводного углекислого натрия и 500мл воды нагревают
и перемешивают до полного растворения сульфаниловой кислоты,
после чего раствор охлаждают в бане со льдом до 15° (при этой
температуре начинает кристаллизоваться натриевая соль сульфани-
ловой кислоты). Добавляют раствор 37 г @,54 мол,) азотистокис-
лого натрия в 100 мл воды и полученный раствор сразу же выли-
вают в смесь 106 мл A,25 мол.) (примечание 1) концентрированной
соляной кислоты (уд. в. 1,18) и 600 г льда, находящихся в 2-литро-
вом стакане. Раствор, из которого при перемешивании начинает
выпадать соль п-фенилдиазонийсульфокислоты, оставляют стоять
в бане со льдом на 15—25 мин. и за это время приготовляют раствор
нафтолята.
Б. Сочетание: оранж\\. В 5-литровой колбе растворяют ПО г
B,75 мол.) (примечание 2) едкого натра в 600 мл воды и в еще теплом
растворе растворяют 72 г р-нафтола @,5 мол.). Раствор охлаждают
1-АМИНО-2-НА.ФТОЛ СОЛЯНОКИСЛЫЙ
45
до 5°, для чего к нему добавляют 400 г льда. Затем добавляют сус-
пензию соли диазония, смесь хорошо перемешивают и оставляют
стоять, не охлаждая ее извне в течение 1 часа (примечание 3).
Из красного раствора вскоре выпадает азосоединение, образующее
в конце концов густую пасту.
В. Восстановление: 1,2-Аминонафтол солянокислый. Суспензию
оранжа II нагревают до 45—50° до тех пор, пока все не перейдет
в раствор с небольшим выделением газа. Затем осторожно прибавляют
около 0,1 части от 230 г (около 1,1 мол.) технического гидросуль-
фита натрия (примечание 4) и смесь перемешивают, пока не прекра-
тится образование пены, после чего незамедлительно прибавляют
остальное количество гидросульфита. Выделяющееся в начале жел-
тое вещество (вероятно, гидразосоединение) вскоре переходит в почти
бесцветный аминонафтол. Для того, чтобы завершить восстановле-
ние и получить хорошо фильтрующийся продукт, смесь сильно на-
гревают, пока она не начнет пениться; затем ее охлаждают до 25°
при перемешивании, поставив колбу в баню со льдом. Выпавший
розовый или кремового цвета продукт собирают и отмывают водой
от слегка желтоватого маточного раствора.
Сырой аминонафтол смывают в стакан, в котором находится на-
гретый до 30° раствор 2 г хлористого олова (с 2 мол. кристаллиза-
ционной воды) и 53 мл @,63 мол.) концентрированной соляной кис-
лоты в 1 л воды. При перемешивании амин вскоре растворяется,
и во взвешанном состоянии остается лишь небольшое количество
пушистого вещества, которое легко отличить от исходных кусков
(примечание 5). Для осветления раствора его перемешивают (без
нагревания) в течение 5 мин. с 10 г обесцвечивающего угля, после
чего раствор фильтруют с отсасыванием. Светложелтый раствор
обрабатывают 50 мл концентрированной соляной кислоты и нагре-
вают до кипения. По мере нагревания добавляют еще 50 мл кислоты.
Во время этой обработки окраска раствора несколько светлеет.
Сосуд с горячим раствором помещают в баню со льдом и содержимому
дают спокойно охладиться. Солянокислый 1-амино-2-нафтол выпа-
дает вскоре в виде больших совершенно бесцветных игл. Когда рас-
твор достаточно охладится, приливают 100 мл концентрированной
соляной кислоты и, прежде чем отфильтровывать продукт, массу
охлаждают до 0° (примечание б). Солянокислую соль промывают
холодным раствором 50 мл концентрированной соляной кислоты
в 200 мл воды и сушат ее на фильтровальной бумаге при температуре
не выше 30—35°. Выход 70—83 г G2—85% теоретич.). Если
продукт хранить без доступа света, то он остается совершенно
или почти совершенно бесцветным. Его свежий раствор в воде лишь
слабо окрашен, и при фильтровании остаются только следы приме-
си на фильтре.
Хотя продукт этот пригоден для большинства целей, его можно
подвергнуть очистке следующим образом. Его растворяют при нагре-
46
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
вании в растворе 2 г хлористого олова и 2 мл концентрированной
соляной кислоты в 1 л воды и горячий раствор осветляют фильтро-
ванием через Ь-мм слой обесцвечивающего угля (примечание 7).
Желтая или красная окраска, которая может при этом появиться,
исчезает, если раствор вновь нагреть до точки кипения. После
добавления 100 мл концентрированной соляной кислоты раствору
дают охладиться в бане со льдом, добавляют еще 100 .ил кислоты,
охлаждают до 0°, и продукт собирают и промывают, как и в первый
раз. Кристаллический продукт бесцветен, беззолен и может быть
признан чистым для анализа. Потери при перекристаллизации 80 г
составляют 5—10 г F—12%).
2. Примечания
1. Диазотирование может быть осуществлено и при применении
точно одного эквивалента кислоты @,5 мол.), однако раствор об-
разующейся таким образом диазокислоты NaO3SCfiH4N = NOH
значительно менее стоек, чем суспензия внутренней соли, образую-
щейся при применении большего количества кислоты.
2. Избыток щелочи требуется не для процесса сочетания, а ско-
рее для того, чтобы создать условия, подходящие для восстановле-
ния.
3. Выход не улучшается, если реакции дать итти большее коли-
чество времени. В указанных условиях смесь во время реакции
сочетания находится при температуре 5—10°.
4. Если гидросульфит плохого качества, его потребуется боль-
шее количество и едкого натра надо взять также дополнительное
количество.
5. Раствор сильно пересыщен, однако он может находиться
в таком состоянии, если только не дать ему стоять слишком большое
время. Ошибкой будет также добавить указанное количество кон-
центрированной соляной кислоты к суспензии аминонафтола, так
как это может вызвать кристаллизацию.
6. Другой метод кристаллизации заключается в том, что все
количество соляной кислоты B00 мл) добавляют к кипящему рас-
твору и дают ему медленно охладиться; при этом выпадают большие
толстые иглы. В присутствии хлористого олова можно не опасаться,
что раствор потемнеет вследствие окисления.
7. Этот метод следует предпочесть обычному при работе с веще-
ством, чувствительным к окислению воздухом.
3. Другие методы получения
1-Амино-2-нафтол был получен из р-нафтиламина 1 и, что имеет
большее практическое значение, из [3-нафтола через нитрозо- или
азосоединение. Нитрозо-р-нафтол был восстановлен в щелочном
1-АМИНО-2-НАфТОЛ| СОЛЯНОКИСЛЫЙ
47
растворе сероводородом 2-3 или гидросульфитом натрия4, однако,
прежние исследователи встречали затруднения при превращении
амина в его солянокислую соль без излишнего окисления. В ка-
честве антиоксиданта применялся сернистый ангидрид, который,
однако, оказался совершенно непригодным для этой цели. Восста-
новление в кислой среде, обычно хлористым оловом, дает более удо-
влетворительные результаты. Выделение соли амина с хлористым
оловом и утомительное разложение ее сероводородом 5 не нужны,
так как можно заставить кристаллизоваться солянокислую соль
амина без всякой примеси олова, если избегать избытка восстано-
вителя 2'8'7. Нитрозо-|5-нафтол был также восстановлен цинковой
пылью и серной кислотой 8, но пригодность такого продукта
для превращения в хинон находится под некоторым сомнением.
Применение нитрозосоединения в качестве промежуточного про-
дукта имеет тот недостаток, что приходится иметь дело с объеми-
стым осадком или большим объемом раствора; в этом случае может
иметь место частичное осмоление. Поэтому предпочтительнее ра-
ботать через стадию азосоединения. Технический оранж II был
восстановлен в нейтральной или щелочной среде сернистым натри-
ем 2 или гидросульфитом натрия9, причем сульфаниловая кислота
удалялась в виде растворимой соли. При работе с хлористым оло-
вом можно избежать необходимости выделения двойной соли амина
с хлористым оловом х, если взять точно вычисленное количество ре-
агента, а полученную смесь солянокислой соли амина и сульфанило-
вой кислоты разделить с помощью щелочного буфера2. Витт 10
нашел, что сульфаниловую кислоту можно удержать в растворе,
если он обладает достаточной кислотностью; воспользовавшись
этим усовершенствованием, Руссиг u разработал методику полу-
чения и восстановления оранжа II, которая, как указывалось,
дает прекрасные выходы. Однако, если судить по результа-
там превращения в хинон, эта методика дает продукт плохого
качества.
Оранж II был также восстановлен цинковой пылью и соля-
ной кислотой 12, электролитически 13 и каталитически 14. Ряд
других азокрасителей, производных ^-нафтола, был восстанов-
лен до 1,2-аминонафтола водородом в присутствии никеля
Рэнея 15:
Описанный выше метод является новым главным образом по-
тому, что в нем хлористое олово применяется в качестве антиокси-
данта при получении и перекристаллизации солянокислой соли
амина. С небольшими видоизменениями метод применим и к полу-
чению многих других аминофенолов 16.
1 Liebermann, Jacobson, Ann. 211, 49 A882).
2 Groves, J. Chem. Soc. 45, 294 A884); Stcnhouse, Groves,
Ann. 189, 153 A877).
48
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
3Lagodzinski, Hardine, Ber. 27, 3075 A894); В о е s е к е n,
Rec trav chim. 34, 272 A915); П о р а и - К о ш и ц, герм. пат. 463 519 (Chem.
Zentr. 1928, II, 1384).
4 См. стр. 41.
' Grandmougin, Michel, Ber. 25, 974 A892).
6 Z i n с к е, Ann. 268, 274 A892).
7 P a u 1, Z. Angew. Chem. 10, 48 A897).
8 Skit a, Rohrmann, Ber. 63, 1482 A930).
9 Grandmougin, там же 39, 3561 A906).
10 W i 11, там же 21, 3472 A888).
11 R u s s i g, J. Prakt. Chem. B) 62, 56 A900); В б e s e к e n, Rec. trav.
chim. 41, 780 A922).
12 Z i n с к e, Ann. 278, 188 A894).
«Boehringer и сыновья; герм. пат. 121 835 (Chem. Zentr. 1901,
II, 152); H u b b u с h, L о w у, С. А. 23, 343 A929).
11 Tetralin G. m. b. H.; герм. пат. 406 064 [Frdl. 14, 395 A921—25)].
15 Whitmore, Revukas, J. Am. Chem. Soc. 62, 1687 A940).
16 F i e s e r, F i e s e г, там же 57, 491 A935).
ОН
1,4-АМИНОНАФТОЛ СОЛЯНОКИСЛЫЙ
ОН
-f NaOH—*
\/\/
OH
+ 2 Na.SaO4 -f 4 H,0
N = NC6H4SO3Na
OH
/\
/ ч.
+ 4NaHS03
NH2
Предложил: Л.
Проверили: К-
+ H2NC(
Физер.
Ноллер и
н4
В.
SO3Na
Уайт.
1. Получение
Диазотируют 105 г @,5 мол.) сульфаниловой кислоты (с двумя
молекулами кристаллизационной воды) совершенно так же, как
это описано на стр. 44. Пока суспензия л-сульфофенилдиазония
1,4-АМИНОНАФТОЛ СОЛЯНОКИСЛЫЙ
49
охлаждается в бане со льдом, приготовляют раствор нафтолята,
для чего 72 г @,5 мол.) а-нафтола (примечание 1) вносят в теплый
раствор, полученный из 110 г B,75 мол.) едкого натра и 600 мл воды
и находящийся в 5-литровой колбе. Раствор нафтолята охлаждают
до 25°, добавляют 400 г льда, после чего приливают суспензию
соли диазония (примечание 2). Смесь хорошо перемешивают и оста-
вляют ее стоять на 1 час для завершения образования красителя
оранжа I.
Раствор оранжа I, окрашенный в темный пурпурово-красный
цвет, нагревают до 45—50°, осторожно добавляют около одной де-
сятой части от 230 г (около 1,1 мол.) технического гидросульфита
натрия (примечание 3) и смесь перемешивают до тех пор, пока не
прекратится вспенивание; после этого довольно быстро добавляют
остальной гидросульфит. Суспензию аминонафтола темнокремо-
вого цвета нагревают до 70°, чтобы вызвать достаточную коагу-
ляцию и тем самым облегчить фильтрование. Смесь быстро охла-
ждают до 25° в бане со льдом, непрерывно ее перемешивая, оса-
док отфильтровывают и промывают его свежим 1%-ным раствором
гидросульфита натрия.
Сырой аминонафтол быстро переносят в стакан, в котором при-
готовлен раствор 2 г хлористого олова (с двумя молекулами кристал-
лизационной воды) и 63 мл концентрированной соляной кислоты
в 800 мл воды, нагретый до 30°. Смесь перемешивают и нагревают,
причем аминонафтол растворяется. Раствор, обычно окрашенный
в тёмнокрасный цвет, фильтруют с отсасыванием; обрабатывать
его активированным углем не следует. Приливают 100 мл концен-
трированной соляной кислоты, раствор нагревают при температуре
кипения в течение 5—10 мин. и приливают еще 100 мл концентри-
рованной соляной кислоты. Во время нагревания окраска бледнеет
и становится светложелтой, а по охлаждении до 0° выпадают мел-
кие, почти бесцветные кристаллы.
Полученный продукт (вес в сухом состоянии 77—80 г) (приме-
чание 4) перекристаллизовывают из 700 мл воды, к которым до-
бавлено 2 г хлористого олова и 2 мл концентрированной соляной
кислоты. Горячий раствор осветляют, для чего его фильтруют че-
рез 5 мм слой активированного угля; к горячему фильтрату прили-
вают 100 мл концентрированной соляной кислоты. По охлаждении
в бане со льдом добавляют еще 100 мл концентрированной соляной
кислоты и охлаждают до 0°. Осадок отфильтровывают и промывают
холодным раствором 50 мл концентрированной соляной кислоты
в 200 мл воды. Хлористоводородная соль 1,4-аминонафтола выпа-
дает в виде мелких почти бесцветных игл, высокой степени чистоты.
Водный раствор соли окрашен в светлорозовый цвет, а кристаллы
при стоянии в течение нескольких недель также могут приобрести
светлорозовую окраску. Выход 70—73 г G2—75% теоретич.; при-
мечание 5).
4 Сбориик Лг 2
50
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
2. Примечания
1. а-Нафтол не должен содержать Ь-изомсра; если продукт
сильно окрашен, лучше подвергнуть его очистке путем перегонки
при атмосферном давлении. Удовлетворительным является продукт
с температурой плавления 95—96°.
2. При сочетании необходимо поддерживать низкую температуру,
чтобы избежать образования бис-азосоединения.
3. Если гидросульфит плохого качества, его следует взять боль-
ше; в этом случае нужно также взять добавочное количество едкого
натра.
4. Продукт этот желтоватого цвета и при сушке может стать
красным. Вероятно, он содержит следы 2,4-диамино-1-нафтола.
5. Приведенная здесь методика весьма близка к методике, опи-
санной на стр. 44 для получения хлористоводородной соли
1,2-аминонафтола. Эти методики следует сравнить: различия между
ними являются следствием лучшей растворимости и большей чув-
ствительности к окислению на воздухе 1,4-аминонафтола.
3. Другие методы получения
Обычным методом приготовления 1,4-аминонафтола является по-
лучение его из а-нафтола через азокраситель. Большинство исследова-
телей восстанавливали технический оранж I хлористым оловом 1-%-s-
*> •'•в; с помощью того же реагента был восстановлен и бензол-азо-а-на-
фтол7'8. Для того, чтобы можно было исходить из технического а-на-
фтола, был разработан метод 9 для получения бензолазосоединения,
отделения его от изомерного красителя, получающегося за счет при-
сутствующего р-нафтола, а также и от всякого иного бис-азосоедипе-
ния, посредством экстрагирования щелочью, а также метод вос-
становления азосоединения в щелочном растворе гидросульфитом
натрия. Процесс этот, однако, весьма длителен и дает нечистый
продукт.
1,4-Аминонафтол может быть получен восстановлением 1,4-ии-
трозонафтола, однако метод этот практического значения не имеет
ввиду малой доступности исходного продукта7. 1,4-Аминонафтол
может быть также получен с суммарным выходом в 60% восстано-
влением а-нитронафталина до нафтилгидроксиламина и перегруп-
пировкой последнего в аминонафтол 10.
'Licbermann, Jacobson, Ann. 211, 49 A882).
2 R u s s i g, J. prakt. Chem. B) 62, 50 A900); В о с s e k с n, Rec. trav.
chim. 41, 780 A922).
3Liebermann, Ann. 183, 247 A876).
* Lie. bermann, Ber. 14, 1796 A881).
5 S e i d e 1, там же 25, 423 A892).
• Z i n с k e, W i e g a n d, Ann. 288, 70 A895).
' Orandmougin,- Michel, Bcr. 25, 974 A892).
1-АМИНО-2-НАФТОЛ-4-СУЛЬФОКИСЛОТА
Si
s F u с h s, P i r a k, там же 59, 2456 A926).
¦' Conant, Lutz, Corson, «Синтезы орг. преп.», сб. 1, стр. 32.
"Neunhoeffer, Liebich, Ber. 71, 2247"A938).
11 F i e s e r, Fieser, J. Am. Chem. Soc. 57, 491 A935).
1-АМИНО-2-НАФТОЛ-4-СУЛБФОКИСЛОТА
NO NH,
•' Y он
-f 2 NaHSO3 + H2SO4
OH
-f 2 NaHSO4
SO4H
Предложил: Л. Физер.
Проверили: Ф. Уитмор и Д. Лодер.
1. Получение
Нитрозо-^-нафтол (см. «Синтезы орг. преп.», сб. 1, стр. 300;
примечание 1), полученный из 300 г ^-нафтола B,1 мол.), переносят
в 6-литровый сосуд, достаточно широкий, чтобы в него входила
30-сантиметровая воронка Бюхнера. Вещество, приставшее к стен-
кам воронки, смывают в сосуд холодным раствором из 600 г E,8 мол.)
бисульфита натрия и 100 мл б-н. раствора едкого натра в 2 л воды
(примечание 2). Смесь разбавляют водой до объема 4—4,5 л и пере-
мешивают в течение приблизительно 15 мин. до полного растворения
нитрозо-8-нафтола. Темный раствор сифонируют в большую ворон-
ку Бюхнера и фильтруют с отсасыванием для того, чтобы удалить
небольшое количество смолистого вещества, всегда содержащегося
в нитрозо-р-нафтоле. Прозрачный, желтовато-коричневый филь-
трат переливают в 8—10-литровый сосуд с широким горлом, раз-
бавляют водой до объема 7 л и при энергичном перемешивании
медленно приливают к нему 400 мл концентрированной серной кис-
лоты так, чтобы она стекала по стенкам сосуда; затем ставят смесь
в вытяжной шкаф и защищают ее от действия света (примечание 3).
Температура сразу повышается от 20—25° до 35—40°, и затем —
в течение 2 час. примерно до 50°; к этому времени реакция почти за-
кончена. После стояния, в общем в продолжение 5 час. или более
(примечание 4), выпавший плотный осадок отсасывают, переносят
в 1-литровый стакан, тщательно перемешивают с 200 мл воды, снова
отсасывают и промывают на фильтре 300 мл воды. Промытое и
отжатое вещество весит 700—800 г. Сушка до постоянного веса влаж-
ной 1-амино-2-нафтол-4-сульфокислоты при 120° не влечет за собой
сколько-нибудь заметного разложения. Продукт получается в виде
мелких, легких, серых игл. Выход: 410—420 г (82—84% теоретич,,
считая на р-нафтол; примечание 5).
52
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
2. Примечания
1. Для того чтобы приготовить нитрозо-р-нафтол в указанном
количестве, удобно взять 7—8-литровый сосуд с горлом диаметром
в 8—10 см, снабженный мешалкой из толстой стеклянной палочки
с 4 или 5 изгибами под прямыми углами, доходящими до горла со-
суда. Ширина изгибов должна быть максимальной, с учетом диа-
метра горла сосуда. Сосуд ставят в ведро с охлаждающей смесью
из льда и соли, которую время от времени перемешивают от руки.
При таком устройстве можно поддерживать температуру 0°, не
прибегая к внутреннему охлаждению.
2. Едкий натр прибавляется к раствору бисульфита для того,
чтобы нейтрализовать кислоту, оставшуюся в нитрозонафтоле и не
удаленную после промывки, так как свободная кислота выделит
из бисульфита сернистый ангидрид, который может частично вос-
становить нитрозонафтол еще до образования продукта присо-
единения. Взятый избыток щелочи способствует растворению
вещества.
3. Аминонафтолсульфокислота, особенно влажная, после дол-
гого стояния на свету становится розовой.
4. Весь синтез можно провести значительно быстрее, если от-
казаться от выделения нитрозо-р-нафтола. В этом случае смесь
бисульфита и щелочи прибавляют непосредственно к суспензии
сырого нитрозосоединения, что дает возможность избежать мед-
ленно протекающего отсасывания. Количество воды, прибавляе-
мой при последующих операциях, следует свести к минимуму. При
этом количество раствора едкого натра должно быть достаточным
для нейтрализации имеющегося избытка кислоты. Получаемая
таким путем аминонафтолсульфокислота — несколько худшего ка-
чества по сравнению с кислотой из отфильтрованного нитрозонаф-
тола; в этом случае выход понижается на 4—5%.
5. Этот серый продукт не совсем чист и содержит кристалли-
зационную воду. Таким образом приведенный здесь выход не точен.
Продукт лучшего качества может быть получен, если смесь нитрозо-
E-нафтола и бисульфита натрия энергично помешивать от руки
деревянной лопаточкой, в результате чего весь растворимый про-
дукт переходит в раствор через 3—5 минут. Суспензию фильтруют
возможно быстрее через две 15-сантиметровые воронки Бюхнера,
причем часто меняют бумажные фильтры. Прозрачный золотисто-
желтый фильтрат подкисляют немедленно по окончании фильтрова-
ния; в этом случае получают продукт светлосерого цвета. Напротив,
если подкисление производится не тотчас же, продукт приобретает
красную окраску и аминонафтолсульфокислота может оказаться
окрашенной в темный пурпурно-серый цвет. После фильтрования
и промывки водой продукт дополнительно промывают теплым
спиртом до тех пор, пока фильтрат не станет бесцветным, на
АНГИДРИД 3,4-ДИГИДРО-1,2-НАФТАЛЕВОЙ КИСЛОТЫ
53
что требуется 1,5—2 л спирта. Затем продукт дважды промывают
эфиром (порциями по 100 мл) и сушат до постоянного веса в темноте
при 60—80°; получают совершенно бесцветный продукт в виде мел-
кого порошка. Выход: 370—380 г G5—78% теоретического, считая
на ^-нафтол). В спирте, применяемом для промывки, растворяется
лишь незначительное количество аминонафтолсульфокислоты, по-
скольку упаривание тёмнокрасного фильтрата дает лишь 3—4 г
твердого остатка (Е. Л. Мартин и Л. Ф. Физер, частное сообщение).
3. Другие методы получения
1-Амино-2-нафтол-4-сульфокислота была получена: нагрева-
нием 2-нафтохинон-1-хлоримида с раствором бисульфита натрия1;
восстановлением 1-бензолазо-2-нафтол-4-сульфокислоты хлористым
оловом и соляной кислотой2; действием сульфита натрия на
солянокислый 1-амино-2-нафтол3; обработкой нитрозо-р-нафтола
бисульфитом натрия и соляной кислотой 4.
1 Friedlancler, Reinharclt, Ber. 27, 241 A894).
2 Marschalk, Bull. soc. chim. D) 45, 660 A929).
3 M a r s с h a 1 k, там же D) 45, 662 A929).
4 Schmidt, J. prakt. Chem. B) 44, 522 A891); В б n i g e r, Ber. 27,
23 A894).
АНГИДРИД 3,4-ДИГИДРО-1,2-НАФТАЛЕВОЙ КИСЛОТЫ
(Ангидрид 3,4-дигидро-1,2-нафталиндикарбоновой кислоты)
(А) С6Н5СН2СН2СН2СО2С2Н5 + (СО2С2Н5J <^НЛ>
(Б)
С6Н5СН2СН2СН (СО2СН5) СОСО2С,Н5 + CSHSOH
И./
СНСОаС.2Н-,
сна со
снй
о
СОСО2С2Н3
(H2SO4)
со
+2С,Н5ОН
Предложили: Е. Хершберг и Л. Физер.
Проверили: К. Номер и С. Кинсман.
1. Получение
А. Сложноэфирная конденсация. В 1-литровую круглодонную
колбу, снабженную обратным холодильником с хлоркальциевой
54
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
трубкой и капельной воронкой, помещают суспензию 6,1 г @,27 гр.-
атома) мелкораздробленного натрия (примечание 1) в 150 мл аб-
солютного эфира. Затем добавляют раствор 12,6 г @,27 мол.)
абсолютного этилового спирта (примечание 2) в 50 мл абсолютного
эфира и смесь оставляют на ночь для завершения реакции. К су-
спензии этилата натрия по частям добавляют 57 г @,39 мол.) этило-
вого эфира щавелевой кислоты («Синт. орг. преп.», сб. 1, стр. 562),
разбавленных 50 мл эфира. После того как закончится самопро-
извольная реакция, светложелтый раствор оставляют стоять
полчаса и затем приливают 50 г @,26 мол.) этилового эфира
у-фенилмасляной кислоты (примечание 3), разбавленных 50 мл
абсолютного эфира. Смесь осторожно кипятят 24 часа (приме-
чание 4).
Тёмнокрасный раствор охлаждают погружением колбы в ледя-
ную воду и нейтрализуют его при взбалтывании добавлением охла-
жденного до 0° раствора 15 мл концентрированной серной кисло-
ты в 200 мл воды. Эфирный слой отделяют, промывают водой и
сушат сульфатом натрия. Эфир отгоняют, прикапывая раствор из
капельной воронки, ножка которой доходит до дна эвакуирован-
ной колбы Клайзена, нагреваемой на водяной бане. Остаток
представляет собой светложелтое масло, состоящее из смеси
этилового эфира а-этоксалил-у-фенилмасляной кислоты и не про-
реагировавшего этилового эфира щавелевой кислоты (приме-
чание 5).
Б. Циклизация. Полученное масло медленно выливают в 500 мл
концентрированной серной кислоты, причем температуру поддер-
живают при 20—25°, охлаждая реакционную массу в бане со льдом.
После стояния в течение 1,5 часа при 20—25Э тёмнокрасный раствор
выливают в 3 л льда и воды. Ангидрид выпадает в виде светложел-
того осадка. Его отфильтровывают, хорошо промывают водой и
сушат в вакууме при 25°. Выход продукта с т. пл. 117—122°/40—
45 г. В результате перегонки в вакууме получается светложелтый
продукт с т. пл. 122—124°. Выход 38—42 г G3—81% теоретич.).
Продукт этот вполне пригоден для большинства целей. Если его
перекристаллизовать из 100 мл бензола с добавкой 75 мл лигроина
(т. кип. 60—80°), то можно получить 34—41 г светложелтых призм
с т. пл. 125—126° (примечание 6).
2. Примечания
1. Мелкораздробленный натрий, необходимый для этого син-
теза, можно получить по методу, описанному в «Синт. орг. преп.»,
сб. 1, стр. 548, или по методике, описанной ниже для калия, заменив
толуол ксилолом. (В случае эфиров некоторых других у-арил-
масляных кислот лучше применять калий.) Продажный металличе-
АНГИДРИД 3,4-ДИГМДРО-1,2-НАФТАЛЕВОЙ КИСЛОТЫ
55
ский калий очищают тем, что его расплавляют в толуоле. 10,4 г
@,27 гр-атома) металла и 150 мл сухого толуола помещают в 1-ли-
тровую колбу. Жидкость нагревают до кипения на электроплитке,
колбу удаляют и закупоривают притертой пробкой, к которой при-
паян кран. Необходимо иметь колбу с взаимно заменимыми шли-
фами. Один раз взбалтывают с открытым краном, чтобы дать сво-
бодный выход горячим парам, затем кран закрывают и колбу быстро
и энергично взбалтывают, чтобы раздробить металл. Смеси дают
спокойно охладиться и впускают азот. Пробку заменяют на насадку
для перегонки, соединенную с колбой на 500 мл, и в эту колбу
декантируют толуол. Раздробленный металл промывают несколько
раз абсолютным эфиром, затем покрывают слоем эфира A50 мл)
и с помощью 12,6гспирта, разбавленного 150 мл эфира, превращают
его в этилат. Следы калия в промывной жидкости вполне безопасно
разрушают путем кипячения со спиртом, разбавленным эфиром
с обратным холодильником.
2. Спирт был абсолютирован так, как это описано в «Синт. орг.
прел.», сб. 1, стр. 547, примечание 1.
3. Этиловый эфир у-фенилмасляной кислоты можно получить
с выходом 85—88% кипячением в течение трех часов смеси из 50 г
у-фенилмасляной кислоты, 150 мл спирта абсолютированного над
негашеной известью и 5 г концентрированной серной кислоты. Для
того, чтобы выделить эфир, отгоняют в вакууме па водяной бане
100 мл спирта, остаток разбавляют 200 мл воды, разделяют слои и
водный слой экстрагируют два раза порциями по 50 мл эфира.
Полученный ранее верхний слой и эфирный слой, содержащий
сложный эфир, сушат сернокислым натрием, эфир отгоняют и оста-
ток перегоняют в вакууме. Собирают фракцию, кипящую при
144—147с/19 мм.
4. В случае этилата калия реакция заканчивается через 12 ча-
сов.
5. Кетоэфир разлагается при перегонке, даже если она ведется
в вакууме.
6. Для циклизации кетоэфиров, полученных из эфиров у-на-
фтилмасляных кислот, лучше применять 80%-ную серную кислоту
и смесь нагревать при 70—80° и работающей мешалке в течение
30 мин.
3. Другие методы получения
Описанный здесь способх является видоизменением метода
Ауверса и Меллера -.
1 F 1 е s e r, Hershberg, J. Am. Chcm. Soc. 57, 1851 A935).
3 Auwers, Moller, J. prakt. Chem. B) 109, 137 A925).
56
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
АНГИДРИД ЯНТАРНОЙ КИСЛОТЫ
I
СНХО2Н СН.2СО
4- СН3СОС1 . ; /О + СН3СО2Н + НС1
снхо,н снхо
Предложили: Л. Физер и Е. Мартин.
Проверил: К. Ноллер.
1. Получение
В 1-литровую круглодонную колбу с обратным холодильником,
другой конец которого присоединен к газовой ловушке, помещают
118 г A мол.) янтарной кислоты и 215 мл B35 г, 3 мол.) хлористого
ацетила. Смесь осторожно кипятят на водяной бане до полного рас-
творения кислоты, на что требуется 1,5—2 часа. Раствору дают
спокойно охладиться, после чего его помещают в охладительную
смесь. Янтарный ангидрид, выпавший в виде прекрасных кристал-
лов, отфильтровывают через воронку Бюхнера, промывают два ра-
за эфиром, порциями по 75 мл, и сушат в вакуум-эксикаторе. Вы-
ход продукта с т. пл. 118—119° составляет 93—95 г (93—95% тео-
ретич.).
II
сн.,со,н
снхо,н
РОС13
снхо
снхо
НРО3 +ЗНС1
Предложили: Р. Шрайнер и X. Стрэк.
Проверили: К. Ноллер, Ф. Хилмер и Док. Пиккенс.
1. Получение
236 г B мол.) янтарной кислоты (примечание 1) и 153,5 г A мол.)
хлорокиси фосфора помещают в 1-литровую колбу Клайзсна, сое-
диненную с обратным холодильником. Второе горло, а также боко-
вой отвод колбы закрыты пробками. Для поглощения выделяюще-
гося хлористого водорода верхний конец холодильника соединяют
с ловушкой. Смесь нагревают (примечание 2) на голом огне в про-
должение 50 мин. до прекращения выделения хлористого водорода.
Затем холодильник отъединяют и колбу приспосабливают для от-
гонки. В качестве приемника берут бунзеновскую склянку или кол-
бу с боковым отводом, к которому для поглощения хлористого во-
дорода присоединяют каучуковую трубку и отводят трубку в кана-
лизацию. До 255°отгоняется лишь несколько миллилитров жидкости;
АНГИДРИД ЯНТАРНОЙ КИСЛОТЫ
57
при этой температуре приемник сменяют и ангидрид янтарной
кислоты собирают от 255 до 260° (примечание 3). Выход продукта с
т. пл. 118—120° равен 164—192 г (82—96%теоретич.). Неочищенный
ангидрид янтарной кислоты может быть перекристаллизован из
уксусного ангидрида. На 50 г ангидрида янтарной кислоты берут
35 мл уксусного ангидрида, нагревают до растворения и охлаждают
льдом. Кристаллы отсасывают, промывают двумя порциями по
20 мл холодного абсолютного эфира и быстро сушат на воздухе при
40°. Выход 43,5 г, т. пл. 119—120°.
2. Примечания
1. Для работы была взята янтарная кислота с температурой
плавления 189—190°. Менее чистая кислота дает продукт с более
низкой температурой плавления.
2. В начале реакции смесь сильно пенится, поэтому необходимо
медленное и осторожное нагревание. Лучше всего колбу нагревать
голым огнем, следя за тем, чтобы все части смеси нагревались рав-
номерно.
3. Смолистая масса, остающаяся в перегонной колбе, может быть
легко удалена теплым разбавленным раствором едкого натра.
3. Другие методы получения
Ангидрид янтарной кислоты может быть получен перегонкой
янтарной кислоты с пятихлористым фосфором 1'2, хлорокисью фос-
фора 2-3; пятииодистым фосфором 4, хлористым тионилом 5, хлори-
стым ацетилом 1> "¦ 7 или хлор ангидридом янтарной кислоты 8;
из бариевой или натриевой соли янтарной кислоты под действием
хлористого бензоила1, хлористого ацетила9, уксусного ангидрида 10,
бензофенондихлоридаи; из хлорангидрида янтарной кислоты и
щавелевой кислоты 6 или уксуснокислого натрия 9 и из этилового
эфира янтарной кислоты и хлористого бензоила 12.
Если метод, описанный в «Син. орг. преп.», сб. 1, стр. 96, для
получения ангидрида бензойной кислоты применить для синтеза
ангидрида янтарной кислоты, исходя из янтарной кислоты и ук-
сусного ангидрида, то ангидрид янтарной кислоты можно получить
с выходом 72% теоретич. Методика I обладает тем преимуществом,
что она удобнее, а II — тем, что она более экономична.
1 О е г h a r d t, Chiozza, Ann. 87, 292 A853).
2 V n 1 h a r d, там же 242, 150 A887).
3 Verkade, Hartman, Rcc. trav. chim. 52, 947 A933).
4 d'A г с e t, Ann. chim. phys. B) 58, 288 A835).
5 Meyer, Monatsh. 22, 420 A901).
6 Anschtitz, Ann. 226, 8 A884).
7 S с h u 1 z, Ber. 18, 2459 A885).
8 A n s с h ii t z, там же 10, 1883 A877).
58
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
9 Hei ntz, Jahresb. 1859, 279.
i° О d d о, M a n u e 1 1 i, Gazz. chim. ital. 26, A1) 482 A896).
11 Евлампиев, ЖОХ 7, 2934 A937).
12 Kraut, Ann. 137, 254 A866)
АНТРАХИНОН-а-СУЛЬФОКИСЛОТЫ КАЛИЕВАЯ СОЛЬ
A-Антрахинонсульфокислоты калиевая соль)
/СОЧ
г у\ / \Г Н 4- Н Я П <HSS°4)
^сск
С0
C6H3SO3H (а) + H2SO4
CeH
хо
>C6H3SO3H (а) + КС1 >
хсск
С6Н/ >C6H3SO3K (а) + НС1
чсск
, Предложили: В. Скотт и К. Аллен.
Проверили: Л. Фтер и Е. Хершберг.
1. Получение
Прибор состоит из трехгорлой колбы емкостью 500 мл, снаб-
женной механической мешалкой (примечание 1) и термометром;
колба установлена в вытяжном шкафу и наполовину погружена
в масляную баню. В колбу помещают 120 г 19—22%-ного олеума
и 1 г желтой окиси ртути, баню нагревают до 100° и через воронку
для сухих веществ, вставленную в колбу с помощью резиновой
трубки широкого диаметра, прибавляют 100 г @,48 мол.) антрахи-
нона (примечание 2). Смесь энергично перемешивают и нагревают
до 147'—152° в течение 45—60 мин., после чего масляную баню
удаляют, колбу снимают, а под мешалку устанавливают 2-литро-
вый стакан, в который налит 1 л горячей воды. Пускают в ход ме-
шалку и по внутренней стенке стакана осторожно приливают кис-
лотную смесь; затем, не останавливая мешалки, смесь кипятят еще
5 мин. Неизменившийся антрахинон E3—59 г; примечание 3) от-
фильтровывают с отсасыванием через воронку Бюхнера диаметром
20 см с фильтром из бумажной ткани и осадок промывают 200 мл
горячей соды. Светлобурый фильтрат вместе с промывными водами
нагревают до 90° и добавляют раствор 32 г хлористого калия в 250 мл
воды. По охлаждении до комнатной температуры (примечание 4)
калиевую соль, которая кристаллизуется в виде светложелтых
АНТРАХИНОН-а-СУЛЬФОКИСЛОТЫ КАЛИЕВАЯ СОЛЬ
59
листиков, отфильтровывают через большую воронку Бюхнера
(бумажный фильтр) и промывают 200 мл холодной воды. Выход про-
дукта, высушенного в вакууме при 100°, составляет 57—55 г G7—
86% теоретич., считая на прореагировавший антрахинон; приме-
чания б и 7).
2. Примечания
1. Необходима эффективная мешалка, приводимая в действие
сильным мотором. Приведенные здесь выходы были получены с
мешалкой Хершберга из хромелевой проволоки (стр. 117); с мешал-
ками других типов выход примерно на 10% ниже.
2. Применявшийся антрахинон имел температуру плавления
284,5—285,5° (исправл.). Его можно легко получить окислением
антрацена (стр. 546).
3. Полученный обратно продукт содержит ртуть и другие при-
меси и плавится при 265—275°. Если его использовать как таковой
в следующем синтезе, то дисульфокислоты образуется значительно
больше, чем при применении чистого антрахинона.
4. В указанных выше условиях реакция с образованием дисуль-
фокислоты имеет место лишь в малой степени, так что можно дать
охладиться смеси кристаллов и не бояться загрязнения продукта
дисульфонатами. При повторном применении антрахинона рекомен-
дуется собрать продукт после охлаждения смеси только до 60°,
и тогда дисульфонаты удерживаются в маточном растворе. Раство-
римость солей 1,5 -и 1,8-дисульфокислот антрахинона возрастает
с повышением температуры быстрее, чем растворимость соли а-моно-
сульфокислоты.
5. Выход в 57 г относится к тому случаю, когда обратно было
получено 53 г антрахинона; выход, выраженный в процентах, воз-
растает с увеличением количества выделенного обратно антра-
хинона.
6. Чистоту продукта можно проверить, превратив небольшое
количество его в а-хлорантрахинон (стр. 549) и определив его тем-
пературу плавления; продукт, полученный из описанной выше
соли, плавился при 158—160° (исправл.).
7. Условия, принятые в настоящей методике, благоприят-
ствуют образованию соли а-моносульфокислоты высокой степени
чистоты за счет уменьшения превращения антрахинона. Большей
степени превращения можно достигнуть, если сульфирование вести
при более высокой температуре или если взять большее количество
олеума, однако в обоих случаях возрастает количество образую-
щихся дисульфокислот. На степень сульфирования в jB-положение
температура влияет мало, однако на нее сильно влияет количество
ртутных солей в растворе. Приведенное выше количество отвечает
примерно пределу растворимости соли во взятой кислоте; при этом
60
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
образуется лишь малое количество р-сульфокислоты. Ввиду того,
что калиевая соль р-сульфокислоты легче растворима, чем соль
«-кислоты, следы ,?-изомера легко удалить при перекристаллизации,
3. Другие методы получения
Единственным практическим методом получения солей антра-
хинон-а-сульфокислоты является метод, основанный на открытии ',
что в присутствии небольшого количества ртутной соли антрахинон
сульфируется главным образом в а-положение, а не в ,?-положе-
ние. Подробные методики описали Фирц-Давид 2, Лауэр 3 и Грог-
гинс4; приведенные выше указания основаны главным образом
на наблюдениях Лауэра3.
1 Ильинский, Вег. 36, 4194A903); Жизнь, труды и изобретения. К
55-летию научной работы почетного академика М. А. Ильинского, изд А Н
СССР., 1938, М.—Л., стр. 121. Schmidt, Ber. 37, 66 A904).
2 F i e r z - D a v i d, Helv. Chim. Acta 10, 197 A927).
3 L a u e r, J. prakt. Chem. B) 130, 185 A931); там же B) 135, 164 A932)
4 G г о g g i n s, Unit Processes in Organic Synthesis, pp. 268—269, McGraw-
Hill Book Company, New York, 1935.
d-АРГИНИН СОЛЯНОКИСЛЫЙ
,,. гидролиз
Желатина * аминокислоты;
NHX (NH) NHCH.XHXHXH (NH2) CO.,H + CeHsCHO .
NHX (NH) NHCHaCH8CH2CH (NCHC,H5) CO,H • H.,O;
C13H18O,N4 • H2O + HC1 >
NHX (NH) NHCHXHXHXH (NH2HC1) CO2H + CeH5CHO.
Предложили: Бранд и Занберг.
Проверили: Кларк и С. Графф.
1. Получение
Бензилиденаргшин. Способ 1. К 500 г желатины (примечание
1) прибавляют 1500 мл концентрированной соляной кислоты уд,
веса 1,19 и смесь нагревают в течение 30 мин. на кипящей водяной
бане, а затем в продолжение 8—10 час. (примечание 2) кипятят на
голом огне с обратным холодильником, соединенным с ловушкой для
улавливания хлористого водорода. Полученный раствор упаривают
на водяной бане в вакууме (в аппарате, описанном в «Синтезах орг.
преп.», сб. 1, стр. 333) до 400 мл. Сиропообразный остаток разба-
вляют 500 мл дестиллированной воды и снова упаривают до 400 мл.
Этот процесс разбавления и упаривания повторяют еще дважды
(примечание 3). Полученный остаток растворяют в 500 мл горячей
дестиллированной воды и раствор обесцвечивают 10-минутным нагре-
d-АРГИНИН СОЛЯНОКИСЛЫЙ
61
ванием на кипящей водяной бане с 15 г обесцвечивающего угля. Жид-
кость фильтруют, фильтрат охлаждают смесью из льда и соли и под-
щелачивают 250—350 мл 40%-ного раствора едкого натра до слабо-
щелочной реакции на лакмус, поддерживая температуру ниже 10°.
Затем при температуре ниже +5° прибавляют еще 70 мл 40%-ного
раствора едкого натра и в четыре приема — 225 мл бензальдегида;
после прибавления каждой порции бензальдегида смесь энергично
взбалтывают, не допуская повышения температуры выше + 5е
(примечание 4). Прибавление бензальдегида отнимает около 10 минут.
Получившуюся эмульсию оставляют на ночь в холодильном
шкафу при 0—5°, Выпавший кристаллический осадок отсасывают;
его промывают сначала (в четыре приема) 80 мл ледяной воды, за-
тем— 50 мл смеси из двух объемов эфира и одного объема метилового
спирта и, наконец, эфиром (примечание 5) до тех пор, пока фильтрат
не станет бесцветным и не будет уже содержать бензальдегида.
Высушенный в вакуум-эксикаторе продукт весит 35—40 г
и плавится с разложением при 206—207° (исправл.).
Бензилиденаргинин. Способ 2. Гидролиз 500 г желатины и уда-
ление избытка соляной кислоты производят так же, как описано
выше. После обесцвечивания Животным углем A5 г) фильтрат раз-
бавляют до 2500 мл, нагревают почти до кипения, после чего к нему
добавляют раствор ПО г 2,4-динитро-1-нафтол-7-сульфокислоты
(«флавиановая кислота»; примечание б) в 400 мл горячей воды. Смесь
кипятят (примечание 7) в течение приблизительно 3 мин. и разба-
вляют кипящей водой до 4 л. Затем смесь быстро охлаждают до 45°;
потом ей дают медленно охладиться до комнатной температуры,
время от времени перемешивая ее и энергично потирая стеклянной
палочкой стенки сосуда. После подобной 2-часовой обработки при
комнатной температуре (примечание 8) практически весь арпшипди-
нитронафтолсульфонат (примечание 9) выделяется в кристалли-
ческой, легко отфильтровываемой форме. Продукт отсасывают и про-
мывают сначала тремя порциями (по 100лмH',5%-ного раствора ди-
нитронафтолсульфокислоты, а затем двумя порциями (по 25 мл)
95%-ного этилового спирта. Высушенный на воздухе продукт весит
113—118 г (примечание 10): при 245—265° он подвергается разло-
жению (примечание 11).
100 г @,21 мол.) мелкорастертого аргининдинитронафтолсульфо-
ната сразу вносят в 230 мл холодного 2-н. раствора едкого натра;
при перемешивании соль быстро растворяется. Затем без промедле-
ния (примечание 12) прибавляют в четыре приема при энергичном
взбалтывании 35 г @,33 мол.) бензальдегида; после внесения каж-
дой порции бензальдегида прибавляют по 75 мл ледяной воды. Бен-
зилиденаргинип выделяется в виде кристаллического осадка. Смесь
оставляют на i—2 часа при 15—20°, затем продукт отсасывают
(примечание 13) и промывают последовательно: сначала 2—4 раза
(каждый раз по 50 мл) ледяной водой, затем три раза (по 20 мл)
62
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
смесью из 20 мл метилового спирта и 40 мл эфира и наконец двумя
порциями (по 50 мл) эфира (примечание 5). Полученный осадок
сушат на воздухе. Выход: 39—43 г, что соответствует 44—48 г,
считая на 500 г желатины (примечание 10).
Аргинин солянокислый. Суспензию 50 г @,18 мол.) бензилиден-
аргинина в 39 мл 4-н. соляной кислоты нагревают в продолжение
45 мин. на кипящей водяной бане, время от времени взбалтывая
массу. Затем смеси дают охладиться, после чего ее освобождают от
бензальдегида, извлекая три раза эфиром (порциями по 100—150 мл).
Водный раствор, если нужно, фильтруют, обесцвечивают 3 г жи-
вотного угля, вновь фильтруют и упаривают при 70° на водяной
бане в вакууме до начала кристаллизации. Остаток извлекают из
колбы 25 мл горячего 70%-ного этилового спирта, после чего соляно-
кислый аргинин выделяют прибавлением 300 мл абсолютного спирта,
Продукт отфильтровывают; из маточного раствора можно получить
дополнительно небольшое количество кристаллической соли при-
бавлением 300 мл эфира. Общий выход (примечание 14): 33—34 г
(88—90% теоретич.). Продукт плавится при 220° (исправл.) и имеет
вращение Г«Н° = (+12,2) —(+12,3)° E%-ный раствор в воде).
2. Примечания
1. Техника испытания качества желатины основана на опре-
делении ее физических свойств; при этом различные образцы силь-
но отличаются по химическому составу. При проверке оказалось,
что лучшие выходы (9,7—10,3% от веса взятой желатины) бензили-
денаргинина дает чистая желатина, применяемая для микробиоло-
гических работ.
2. Проба на биуретовую реакцию через 5 час. дает обычно отри-
цательный результат.
3. Третий отгон содержит обычно только 1—2 г хлористого
водорода. При проверке этого способа в большом масштабе оказа-
лось удобным непрерывное прибавление воды под поверхность ки-
пящего сиропа; это изменение, которое является, собственно гово-
ря, перегонкой с паром в вакууме, приводит к более быстрому
удалению избытка соляной кислоты.
4. При температурах выше -f5° бензальдегид эмульсируется
с трудом.
5. Бензилиденарпшин нерастворим в эфире, но заметно раство-
рим в метиловом спирте и воде. Перекристаллизация из двух по-
следних растворителей дает продукт более низкого качества вслед-
ствие разложения, которое происходит при растворении. Загрязнен-
ные образцы можно очищать гидролизом с помощью горячей соля-
ной кислоты и повторным осаждением бензальдегидом после ней-
трализации.
d-АРГИНПН СОЛЯНОКИСЛЫЙ
6. Свободную диннтронафтолсульфокислоту можно легко по-
лучить из продажного нафтолового желтого S, обрабатывая на-
сыщенный профильтрованный раствор красителя тремя объемами
концентрированной соляной кислоты. Выделяющиеся кристаллы
промывают холодной 20%-ной соляной кислотой и сушат сначала
на воздухе, а затем—в вакуум-эксикаторе над твердым едким натром.
7. Кипячение препятствует выпадению аргининдифлавианата и
затрудняет выделение флавианатов других аминокислот.
8. Кристаллизация иногда задерживается, особенно при пер-
вых загрузках, когда в атмосфере еще нет следов аргининфлавианата
для заражения. В таких случаях раствор приходится охлаждать в
холодильном шкафу, время от времени сильно потирая стенки со-
суда стеклянной палочкой.
9. Из маточного раствора при долгом стоянии на холоду может
выделиться вторая фракция кристаллического осадка; повиди-
мому, он состоит главным образом из натриевой соли динитронафтол-
сульфокислоты; при дальнейшей обработке он не дает аргинина. Филь-
трат, полученный при работе по второму методу, может быть ис-
пользован для выделения других аминокислот, отличаясь тем са-
мым от маточника, получаемого но первому методу.
10. Этот выход был получен из партии желатины, из которой
по первому способу было получено 35—36 г бензилиденаргинина.
11. По литературным данным х чистый аргининдинитронафтол-
сульфонат плавится с разложением при 258—260°. Присутствие
влаги значительно снижает точку плавления. Если маточный
раствор оставить на несколько дней при 0—5", то из него может выкри-
сталлизоваться некоторое количество натриевой соли динитрона-
фтолсульфокислоты.
12. Бензальдегид необходимо прибавлять возможно быстрее,
так как иначе может выкристаллизоваться натриевая соль дини-
тронафтолсульфокислоты; воду прибавляют для задержки этой
кристаллизации. Присутствие избытка бензальдегида играет, по-
видимому, ту же роль.
13. Для отсасывания и промывания бензилиденаргинина удобны
воронки с впаянными пористыми стеклянными пластинками.
14. Количество общих потерь во всех стадиях видно из следу-
ющего опыта: 5 г азотнокислого аргинина были переведены через
дпнитронафтолсульфонат в бензилиденаргинин и обратно в азотно-
кислый аргинин; последнего было получено 4,2 г.
3. Другие методы получения
Аргинин был выделен: а) в виде серебряного производного2 при
рН равном 10; б) в виде динитропафтосульфоцата с последующим
расщеплением: 33%-ной серной кислотой \ совместным действием го-
рячей разбавленной серной кислоты и бутилового спирта3, холодной
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
концентрированной соляной кислотой с последующей обработкой
анилином4 или гидратом окиси бария 5; в) в виде бензилиденового
производного из растворов, подщелоченных гидратом окиси бария
или едким натром6; г) из белковых гидролизатов в условиях кон-
тролируемого рН 7.
Способ 1, описанный в настоящем руководстве, в главных чертах
тотже, что и разработанный Бергманом и Зервасом6. Способ 2 являет-
ся комбинацией методов (б) и (в); он дает сразу продукт высокой
степени чистоты, причем избегаются механические трудности,
с которыми приходится сталкиваться при полном удалении динитро-
нафтолсульфокислоты из ее соли с аргинином.
1 К о s s e I, Gross, Z. physiol. Chem. 135, 167 A924).
2 Kosscl, там же22, 176A896—7); 25, 165A898); Ко sse 1, Ku t s ch er,
там же 31, 165 A900); Vickery, Leavenworth, J. Biol. Chem. 72,
403 A927); 75, 115 A927).
3 Pratt, там же 67, 351 A926).
4 С о х, там же 78, 475 A928).
6 Felix, D i r r, Z. physiol. Chem. 176, 38 A928).
6 Bergman n, Zervas, там же 152, 282 A926); 172, 277 A927).
'Foster, Schmidt, J. Biol. Chem. 56, 545 A923); J. Am. Chem.
Soc. 48, 1709 A926); С о х, К i n g, В e r g. J. Biol. Chem. 81, 755 A929).
АЦЕТАЛЬ АКРОЛЕИНА
C1CHXHXH (OC2HOJ + KOH — CH3= CHCH (OC2H0J+KC1 +H2O.
Предложили: E. Вшпцеман, В. Эеанс, Г. Хасс. и Е. Шредер.
Проверили: Ф. Ушпмор н Г. Негер.
1. Получение
340 г (б мол.) сухого, истолченного в порошок едкого кали (при-
мечание 1) помещают в короткогорлую круглодонную колбу емк.
в 500 мл (примечание 2)' и туда же прибавляют
167 гA мол.) ацеталя [3-хлорпропионового аль-
дегида (стр. 67). Смесь энергично встряхивают
и немедленно соединяют колбу с трехшариковым
дефлегматором Глинского или другим подходя-
щим дефлегматором, который в свою очередь
соединен с нисходящим водяным холодильником
PlIC- 1 (примечание 3). Затем реакционную массу нагре-
вают на масляной бане при 210—220° до тех
пор, пока не прекратится отгонка (примечание 4). Дестиллат пере-
носят в делительную воронку и удаляют нижний водный слой. По-
лученный таким образом сырой ацеталь акролеина сушат 10 г пота-
ша, фильтруют и перегоняют из специальной колбы Клайзена (рис. 1)
собирая фракцию 122—126°. Выход: 98 г G5% теоретич.).
АЦЕТАЛЬ АКРОЛЕИНА
65
2. Примечания
1. Чистое едкое кали обезвоживают двухчасовым сплавлением
при 350° и по охлаждении возможно быстрее измельчают настолько,
чтобы полученный порошок проходил через сито с 24 отверстиями
на 1 см F0 меш). Рекомендуется применение дисковой мельницы с
диаметром 24 см и производительностью около 200 г в 1 мин. Очень
важно, чтобы едкое кали содержало возможно меньше воды, так как
в присутствии ее выход заметно понижается. При применении необез-
воженного едкого кали выход падает приблизительно на 60%.
2. Концентрированный раствор едкого кали, остающийся по
окончании реакции, быстро разъедает стекло. Если синтез повторя-
ют несколько раз подряд, то вместо колбы следует взять железную
реторту, сделанную из трубы диаметром в 10 см.
3. Реакция между ацеталем и щелочью протекает весьма бурно.
4. Большое количество головной низкокипящей фракции пока-
зывает, что взятое ед-кое кали содержало слишком много воды (при-
мечание 1).
3. Другие методы получения
Ацеталь акролеина может быть получен обработкой ацеталя
р-хлорпропионового альдегида сухим порошкообразным едким
кали1 и взаимодействием акролеина, ортомуравьиного эфира и
азотнокислого аммония в кипящем спирте 2.
1 W о h I, Ber. 31, 1798 A898); Witzemann, J. Am. Chem. Soc. 36,
1911 A914); Spoer, Young, Carnegie Inst. Washington Yearbook 25, 176
A925—1926); Expt. Sta. Record 57, 817 A927) [C. A. 22, 2368 A928)].
2 Fischer, Baer, Helv. Chim. Acta 18, 516 A935).
CH2 =
АЦЕТАЛЬ dl-ГЛИЦЕРИНОВОГО АЛЬДЕГИДА
CHCH (OC2H5J + H2O + [О]
НОСН2СНОНСН (ОС2Н8J
Предложили: Е. Витцеман, В. Эеанс, Г. Хасс и Е. Шредер-
Проверили: Ф. Уитмор и Г. Негер.
1. Получение
В 3-литровую открытую колбу, снабженную механической ме-
шалкой и термометром, помещают суспензию 65 г @,5 мол.) ацеталя
акролеина (стр. 64) в 600 мл воды. Жидкость охлаждают до 5° (при-
мечание 1) погружением колбы вводу со льдом, после чего к ней
прибавляют при перемешивании раствор 80 г @,5 мол.) марганцово.
5 Сборник Л1» 2
66
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
кислого калия в 1500 мл воды со скоростью около 25 л/л/мин.
В продолжение всего прибавления температуру смеси поддерживают
как можно ближе к 5°. По прибавлении всего марганцовокислого
калия, перемешивание прекращают. Реакционную массу оставляют
спокойно стоять в течение 2 час. (причем вскоре она превращается
в гель; примечание 2), а затем нагревают на водяной бане в течение
1 часа, а выпавшую перекись марганца отсасывают на большой во-
ропке Бюхнера C0 см), тщательно отжимают и промывают 150 мл
холодной воды. К фильтрату (около 2300 мл) прибавляют 1200 г
свежепрокаленного поташа, следя за тем, чтобы жидкость не на-
гревалась. Отслоившийся при этом ацеталь отделяют, и водный рас-
твор четыре раза извлекают эфиром порциями по 100 мл. Соединен-
ные эфирные вытяжки смешивают с ацеталем и эфирный раствор,
который может состоять из двух слоев (примечание 3), сушат 10 г
поташа. После отгонки эфира остаток перегоняют в вакууме. Выход
ацеталя rfl-глицеринового альдегида, кипящего при 120—12Г/8 мм;
55 г F7% теоретич.).
2. Примечания
1. Выход сильно зависит от температуры смеси во время окис-
ления. Наилучшие результаты получаются при 5°; даже небольшое
отклонение вызывает заметное понижение выхода.
2. Вскоре после прекращения перемешивания смесь должна пре-
вратиться в студнеобразную массу. Если этого не происходит, тс
выход обычно оказывается низким: при проведении реакции не был?
соблюдены температурные условия. .
3. Наблюдающееся иногда разделение раствора на два ело?
исчезает после прибавления поташа.
3. Другие методы получения
Ацеталь ^/-глицеринового альдегида может быть получен: на
греванием ацеталя оксихлорпропионового альдегида с растворол
углекислого калия1ш, обработкой глицеринового альдегида спирто
вым раствором хлористого водорода2 и окислением ацеталя акролеи
на марганцовокислым калием3.
1 Woh I, Ber. 31, 1799 A898).
2 W о h 1, N е u b e r g, там же 33, 3103 A900); W i t z e m a n n, J. Am
Chem. Soc. 36, 2229 A914).
й W о h 1, Ber. 31, 1799 A898); Evans, H a s s, J. Am. Chem. Soc. 48
2706 A926); W i t z e m a n n, там же 36, 1912 A914); Spoehr, Young
Carnegie Inst. Washington Yearbook 25, 177 A925—1926); Expt. Sta. Recon
57, 817 A927) [C. A. 22, 2368 A928)]; Fischer, Baer, Helv. Chim. Acta 18
516 A935).
АЦЕТАЛЬ Э-ХЛОРПРОПИО НОВОГО АЛЬДЕГИДА
67
АЦЕТАЛЬ 0-ХЛОРПРОПИОНОВОГО АЛЬДЕГИДА
СН2 = СНСНО + 2 С2НаОН 4- HC1 - С1СН2СН2СН (ОС2Н5J + Н2О
Предложили: Е. Витцеман, В. Эеанс, Г. Хасс и Е. Шредер.
Проверили: Ф. Уитмор и Г. Негер.
1. Получение
В 3-литровую круглодонную колбу, снабженную механической
мешалкой и газоприводной трубкой, помещают 200 г B53 мл) абсо-
лютного спирта (примечание 1). Колбу погружают в охладительную
смесь из льда и соли и спирт насыщают сухим хлористым водородом
при 0°. Пока идет насыщение, собира-
ют прибор для прибавления холодного
акролеина. Трубку делительной во-
ронки емк. в 200 мл вставляют в
отверстие пробки, запирающей корот-
кую обрезанную трубку большой во-
ронки (рис. 2). Затем промежуток
между обеими воронками заполняют
мелко истолченным льдом и водой и
в делительную воронку помещают
112 г B мол.) холодного акролеина
(«Синт. орг. преп.», сб. 1, стр. 17;
примечание 2).
По насыщении спирта хлористым
водородом каучуковую трубку, под-
водящую газ, отъединяют от газопри-
водной трубки; последнюю соединяют
с трубкой делительной воронки по-
средством каучуковой трубки, на ко-
торую надет винтовой зажим для ре-
гулирования тока жидкости. Затем к
спиртовому раствору хлористого водо-
рода медленно, при перемешивании,
прибавляют акролеин с такой ско-
ростью, чтобы температура смеси держалась около 0°, на что
требуется от 1 до 2 час. После того как образуются два слоя, ниж-
ний слой (ацеталь) отделяют и постепенно прибавляют к нему дву-
углекислую соду в порошке до полной нейтрализации всей ки-
слоты (примечание 3). Полученную смесь фильтруют с отсасыва-
нием; фильтрат промывают двумя порциями по 50 мл ледяной
воды (примечание 4), сушат 10 г поташа в течение 5—10 час, филь-
труют и перегоняют в вакууме. Выход ацеталя ^-хлорпропионового
альдегида, кипящего при 58—62°/8 мм; 112 г C4% теоретич.).
Рис. 2
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
2. Примечания
1. Следует применять спирт концентрации по крайней мере
не ниже 99,5% (см. «Синт. орг. преп.», сб. 1, стр. 555).
2. Работа с неохлажденным акролеином, ввиду раздражающего
действия его паров, крайне затруднительна и даже невыносима.
Делительную воронку следует закрыть пробкой, в которую вста-
влен тонкий стеклянный капилляр.
3. Вся кислота должна быть нейтрализована до промывки про-
дукта водой, так как разбавленная кислота очень легко омыляет
ацеталь.
4. Для промывки рекомендуется брать дестиллированную воду,
так как водопроводная вода иногда содержит такие примеси, кото-
рые ведут к разложению ацеталя (Ч. Ф. X. Аллен, частное сообще-
ние).
3. Другие методы получения
Ацеталь ^-хлорпропионового альдегида может быть получен:
действием спиртового раствора хлористого водорода на акролеин1
и из тех же реагентов в присутствии хлористого кальция 2.
1 Alsberg, Jahresber. 1864, 495; W о h 1, Ber. 31, 1797 A898); W о h 1,
Emmerich, там же 33, 2761 A900); Brabant, Z. physiol. Chem. 86,
208A913); Witzemann, J. Am. Chem. Soc. 36, 1909A914); S p о е h r,
Young, Carnegie Inst. Washington Yearbook 25, 176 A925—1926); Expt.
Sta. Record 57, 817 A927) [C. A. 22, 2368 A928)].
2 Crawford, Kenyon, J. Chem. Soc. 1927, 399.
АЦЕТИЛБЕН30ИН
( Бензоишцетат)
CeH5CHOHCOC6H5 + (CH3COJ 0
C6H6CH (OCOCH3) COC6H5 + CH3CO2H
Предложили: Б. Корсон и Н. Салиани.
Проверили: Ф. Уитмор и М. Уитмор.
1. Получение
К смеси 212 г A мол.) бензоина (примечание 1), 200 мл ледяной
уксусной кислоты и 200 мл B,1 мол.) уксусного ангидрида, нахо-
дящейся в стакане емк. в 1 л, снабженном механической мешалкой,
медленно прибавляют при перемешивании 20 мл концентрирован-
ной серной кислоты (х. ч.). Прибавление кислоты производят в те-
чение 5 мин.; за это время бензоин быстро растворяется и темпе-
ратура поднимается приблизительно до 50°. Стакан помещают на
20 мин. на кипящую водяную баню (примечание 2). Затем смеси
дают немного охладиться, переносят ее в большую делительную
воронку и медленно выливают в продолжение 30 мин. при энер-
АЦЕТИЛГЛИЦИН
69
гичном перемешивании в 2500 мл воды, находящейся в сосуде ем-
костью 4 л (примечание 3). Перемешивание продолжают в течение
часа. Осадок отсасывают возможно тщательнее на воронке Бюх-
нера диаметром 30 см и затем раскладывают на фильтровальной
бумаге. Приблизительно через 2 часа продукт переносят в стакан
емкостью 1 л и нагревают с 400 мл 95%-ного этилового спирта до
60°. Прозрачный раствор охлаждают при перемешивании до 5°
и выпавший продукт отсасывают. Воздушно-сухой ацетилбензоин,
обладающий слегка желтоватым оттенком, плавится при 80—82°.
Выход: 220—230 г (86—90% теоретич.). После второй перекристал-
лизации из 400 мл спирта получается вполне бесцветный продукт
с т. пл. 81,5—82,5°, причем теряется около 10 г.
2. Примечания
1. Бензоин («Синт, орг. преп.», сб. 1, стр. 95) можно не пере-
кристалл изо вывать.
2. Смесь не следует нагревать дольше или до более высокой
температуры.
3. Если продукт застывает в виде комков, то их надо вынуть,
растереть в большой ступке в пастообразную массу, переложить
обратно в стакан и затем снова начать перемешивание смеси.
3. Другие методы получения
Единственный способ, который представляет препаративный
интерес, состоит в ацетилировании бензоина хлористым ацетилом1
или уксусным ангидридом2. Точка плавления ацетилбензоин а по
Зинину—«ниже 100°», по Иена и Лимприхту 75°, позднейшие
исследователи приводят т. пл. 82—83°.
1 Зинин, Bull, de la classe physico-mathematique de l'Acad. Imper. des
Sciences de St.-Petersbourg, 15, N 18, 281 A857); Ann. 104, 120A857); Jena
L i m p r i с h t, там же 155, 92 A870); Papcke, Ber. 21, 1336 A888).
2 Francis, К e a n e, J. Chem. Soc. 99, 346 A911).
АЦЕТИЛГЛИЦИН
(Ацетуриноеая кислота)
H2NCH2CO2H + (CH3COJO CH3CONHCH2CO2H + CH3CO2H
Предложили: Р. Хербст и Д. Шемин.
Проверили: Р. Фьюзон и Е. Кливленд.
1. Получение
В 1-литровую коническую колбу, снабженную механической
мешалкой, помещают 75 г A мол.) глицина («Синт. орг. преп»»,
сб. 1, стр. 167) и 300 мл воды. Смесь энергично перемешивают до тех
70
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
пор, пока почти весь глицин не перейдет в раствор, после чего при-
ливают в один прием 215 г B мол.) 95%-ного уксусного ангидрида
(примечание 1). Энергичное перемешивание продолжают еще в
течение 15—20 мин., причем за это время раствор может разогреться
и ацетиглицин может начать выкристаллизовываться. Для за-
вершения кристаллизации раствор помещают на ночь в холодиль-
ный шкаф (примечание 2). На утро осадок отсасывают, промывают
на воронке ледяной водой и сушат при 100—110°. Вес полученного
продукта 75—85 г, т. пл. 207—208°. Фильтрат и промывные воды
соединяют и выпаривают досуха в вакууме на водяной бане при
50—60°. Остаток перекристаллизовывают из 75 мл кипящей воды
и полученную вторую порцию кристаллов также промывают ледя-
ной водой и сушат при 100—110°. Получают 20—30 г продукта с
т. пл. 207—208°. Упариванием маточного раствора можно получить
еще 4—б г несколько менее чистого продукта. Общий выход
104—108 г (89—92% теоретич.; примечание 3).
2. Примечания
1. Можно взять также эквивалентное количество 90%-ного
уксусного ангидрида.
2. Холодильный шкаф, в котором работали при проверке этого
синтеза, имел температуру 5—7°.
3. Этот метод можно применять для ацетилирования большин-
ства а-аминокислот с внесением лишь небольших изменений, кото-
рые зависят от растворимости индивидуальных кислот. При ацети-
лировании оптически деятельных аминокислот рацемизация не-
значительна или даже совсем не имеет места1.
3. Другие методы получения
Ацетилглицин был получен при взаимодействии хлористого аце-
тила и серебряной соли глицина в абсолютном эфире или бензо-
ле2-3; при действии уксусного ангидрида на суспензию глицина
в теплом бензоле3; при нагревании глицина с уксусным ангидри-
дом4; при действии кетена на водный раствор глицина или его нат-
риевой соли 5 и при действии уксусного ангидрида на водно-щелоч-
ной раствор глицина*.
1 В е h r, Clarke, J. Am. Chem. Soc. 54, 1631 A932).
3 Kraut, Hartmann, Ann. 133, 105 A865).
3 Curtius, Ber. 17, 1665 A884).
4 Radenhausen, J. prakt. Chem. B) 52, 437 A895).
5 Bergman n, Stern, Ber. 63, 437 A930).
6 С h a 11 a w a y, J. Chem. Soc. 1931, 2495.
АЦЕТИЛЕНДИКАРБОНОВАЯ КИСЛОТА
71
АЦЕТИЛЕНДИКАРБОНОВАЯ КИСЛОТА
HO3CCHBrCHBrCO2H + 4 КОН .
. KO2CC s= CCO2K + 2 KBr + 4Н2О;
К02СС=еССО2К + H2SO4 ¦— КО2СС = ССО.2Н +KHSO4;
КО2СС = ССОаН + H,SO4 (избыток) —- НО2СС = ССО2Н + KHSO4.
Предложили: Т. Абботт, Р. Арнольд и Р. Томпсон.
Проверили: Р. Фьюзон и В. Холленд.
1. Получение
В 2-литровой кругло донной колбе, снабженной обратным холо-
дильником, приготовляют раствор щелочи, для чего 122 г B,2 мол.—
1,5-кратное количество по сравнению с теоретическим) едкого
кали растворяют в 700 мл 95%-го метилового спирта (примечание 1).
К этому раствору щелочи добавляют 100 г @,36 мол.) а, р-ди-
бромянтарной кислоты (см. стр. 191) и смесь кипятят 1 час 15 мин.
на водяной бане. Реакционную смесь охлаждают и выпавший оса-
док отсасывают. Смесь солей промывают 200 мл метилового спирта
(примечание 2) и сушат между листами фильтровальной бумаги.
Вес сухой смеси составляет 144—150 г.
Смесь солей растворяют в 270 мл воды; добавлением 8 мл кон-
центрированной серной кислоты в 30 мл воды осаждают кислую
калиевую соль. После стояния в течение 3 час. или даже в течение
ночи смесь фильтруют с отсасыванием (примечание 3). Кислую соль
растворяют в 240 мл воды, к которой добавлено 60 мл концентри-
рованной серной кислоты. Полученный раствор извлекают пять
раз эфиром, порциями по 100 мл. Эфирные вытяжки упари-
вают досуха на водяной бане, причем остаток представляет
собой чистые кристаллы гидрата ацетилендикарбоновой кислоты.
После двухдневной сушки в вакуум-эксикаторе над концен-
трированной серной кислотой кристаллы плавятся с разложе-
нием точно при 175—176°. Выход: 30—36 г G3—88% теоретич.).
2. Примечания
1. В случае применения 95%-ного этилового спирта выход не-
сколько падает.
2. Эта смесь солеи состоит из бромистого калия и из калиевой
соли ацетилендикарбоновой кислоты.
3. Эта кислая соль практически не содержит брома и не ну-
ждается в дополнительном промывании.
72
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
3. Другие методы получения
Описанный метод принадлежит в основном Бандровскому1 и Байе-
ру2 с видоизменениями, внесенными Руггли 3. Тот же общий метод
применяли Бакер и ван дер Занден4, а также Мурё и Бонгран6.
1 Bandrowski, Ber. 10, 838 A877).
2 В а е у е г, там же 18, 677 A885).
3 R u g g I i, Helv. Chim. Acta 3, 5G4 A920).
1 Backer, van der Zanden, Rec. trav. chim. 47, 776 A928).
5 Moureu, Bongrand, Ann. chim. (9) 14, 9 A920).
«-АЦЕТОАМИНОКОРИЧНАЯ КИСЛОТА
C6HaCHO + CH2CO2H CeH5CH =
NHCOCH3
(CH3CO2Na)
N
со
I
о
CfiH,CH = C CO
N О + Н20
CH,
CeHaCH = CCO2H
CH.
NHCOCH3
Предложили: Р. Хербст и Д. Шемин.
Проверили: Р. Фьюзон и Е. Кливленд.
1. Получение
В 1-литровую коническую колбу помещают смесь 58,5 г @,5
мол.) ацетилглицина(стр. 69, примечание 1), 30 г @,37 мол.) безвод-
ного уксуснокислого натрия, 79 г @,74 мол.) свежеперегнанного
бензойного альдегида и 134 г A,25 мол.) 95%-ного уксусного ан-
гидрида. Колбу не плотно закрывают пробкой и нагревают на водя-
ной бане, время от времени помешивая содержимое до полного его
растворения A0—20 мин.). Полученный раствор кипятят в течение
1 часа с обратным холодильником, охлаждают и оставляют на ночь
в холодильном шкафу. Твердую массу желтых кристаллов обраба-
тывают 125 мл холодной воды и измельчают с помощью стеклянной
палочки. Кристаллы отсасывают через воронку Бюхнера, тщатель-
но промывают холодной водой (примечание 2) и сушат в вакуум-
эксикаторе над фосфорным ангидридом и едким кали. Выход не-
очищенного азлактона 69—72 г G4—77% теоретич.). Температура
а-АЦЕТОАМИНОКОРИЧНАЯ КИСЛОТА
плавления полученного продукта 148—150°; он является достаточно
чистым для препаративных целей (примечание 3).
В 1-литровой круглодонной колбе с коротким горлом раство-
ряют при кипячении 47 г @,25 мол.) неочищенного азлактона
а-ацетоаминокоричной кислоты в смеси 450 мл ацетона и 175 мл воды.
Чтобы завершить омыление, смесь кипятят с обратным холодиль-
ником 4 часа. Затем отгоняют большую часть ацетона при атмос-
ферном давлении на водяной бане. Оставшийся раствор разбавляют
400 мл воды, нагревают 5 мин. до кипения, чтобы обеспечить пол-
ное растворение ацётоаминокислоты и раствор фильтруют (примеча-
ния 4 и 5). Небольшое количество нерастворимой примеси, остаю-
щейся на фильтре @,2—0,5 г), промывают 50—75 мл кипящей воды.
Кристаллы, которые могут выпасть из фильтрата, вновь растворяют,
для чего раствор нагревают и кипятят его в течение 5 мин. с 10 г
активированного угля, а затем фильтруют с применением неболь-
шого вакуума почти при температуре кипения раствора (примеча-
ние 5). Уголь в воронке тщательно промывают четыре раза кипя-
щей водой порциями по 50 мл для того, чтобы удалить кристаллы,
которые могли выделиться при фильтровании, и промывные воды
присоединяют к основному фильтрату. После стояния в течение
ночи в холодильном шкафу выпадают бесцветные игольчатые кри-
сталлы, которые отсасывают (примечание 6), промывают 150—200 мл
ледяной воды и сушат в течение нескольких часов при 90—100°.
Выход 41—46 г (80—90% теоретич.) практически чистого продукта
с т. пл. 191—192° (примечание 7).
2. Примечания
1. Азлактон а-ацетоаминокоричной кислоты можно получить
также, если вместо ацетилглицина взять эквивалентное количество
глицина и увеличить количество уксусного ангидрида до 3 мол. на
1 мол. глицина. Однако в этом случае выход составляет только 45—
50% теоретического количества.
2. Если избыток бензойного альдегида не удален почти пол-
ностью повторной промывкой водой, то полезно под конец промыть
продукт 50—75 мл эфира, хотя это вызывает некоторую потерю аз-
лактона вследствие его растворимости в эфире.
3. Азлактон можно перекристаллизовывать из спирта, из четы-
реххлористого углерода или из уксусноэтилового эфира с добав-
кой петролейного эфира. Водных растворителей следует избегать,
так как вода легко расщепляет кольцо азлактона. Если применять
для перекристаллизации спирт, то можно опасаться, что кольцо
азлактона расщепится с образованием сложного эфира, особенно
при продолжительном нагревании раствора.
4. Раствор можно фильтровать или без отсасывания через
большой складчатый фильтр (лучше в воронке для горячего
74
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
фильтрования), или через воронку Бюхнера с легким отсасыва-
нием.
5. Растворимость а-ацетоаминокоричной кислоты в воде быстро
падает с понижением температуры ниже температуры кипения рас-
твора. Ввиду того, что раствор почти насыщен продуктом, значи-
тельное количество кислоты кристаллизуется в воронке во время
фильтрования, если раствору дать охладиться слишком сильно.
Учитывая это свойство продукта, нецелесообразно работать сразу
с большими его количествами.
6. Иногда после обработки активированным углем раствор ста-
новится зеленым в результате взаимодействия фенилпировиноград-
ной кислоты с следами железа. Если получающиеся при этом кри-
сталлы все еще желтого цвета, обработку активированным углем
следует повторить прежде чем собирать продукт на фильтре.
7. Если желательна дальнейшая очистка продукта, его можно
перекристаллизовать из 600 мл кипящей воды, причем потери со-
ставят около 5%. Эти потери частично обусловлены омылением
продукта с образованием фенилпировиноградной кислоты.
3. Другие методы получения
Азлактон а-ацетоаминокоричной кислоты был получен нагре-
ванием смеси глицина, бензойного альдегида, уксусного ангидрида
и безводного уксуснокислого натрия1'2, а также действием уксус-
ного ангидрида на N-хлорацетилфенилаланин2.
а-Ацетоаминокоричная кислота была получена из соответствен-
ного азлактона путем гидролиза либо водным раствором едкого
натра1, либо просто кипящей водой2.
1 Е г 1 е n m е у е г, Jr., Frustiick, Ann. 284, 48 A895).
2 В е г g m a n n, Stern, там же 448, 26 A926).
АЦЕТОЛ
A-Окси-2-пропанон)
НСОЙС2Н5 + КОН НСО2К + С2Н5ОН;
СН3СОСН2Вг + НСО2К —• СН3СОСН2ОСНО + КВг;
СН3СОСН2ОСНО + СН3ОН —¦ СН3СОСН2ОН + НСО2СН3.
Предложили: П. Левин и А. Уолти.
Проверили: Ф. Ушпмор и Дж. Холлингсхед.
1. Получение
В 3-литровую круглодонную колбу, снабженную обратным хо-
лодильником длиною 75 см, вливают раствор 210 г едкого кали
(очищенного спиртом) в 1500 мл безводного метилового спирта.
АЦЕТОЛ
75
Раствор охлаждают до температуры ниже 50° (примечание 1), при-
бавляют 300 г чистого этилового эфира муравьиной кислоты и ки-
пятят смесь с обратным холодильником в продолжение 2 час. (при-
мечания 2 и 3).
После этого прибавляют 410 г B51 мл; 3 мол.) бромацетона (стр.
103) и кипятят раствор в течение 16 час. с обратным холодильником
на водяной бане, нагретой до 95—97°. Выпавший бромистый калий
по охлаждении до 0° смесью льда с солью отсасывают на охлажден-
ной воронке Бюхнера, и фильтрат, после отгонки метилового спирта,
фракционируют в вакууме.
Фракция 25—35°/12 мм отбрасывается, так как она содержит
очень мало ацетола. Главная фракция кипит при 35—47°/12 мм
и весит 160 г. При вторичной перегонке выход ацетола, кипящего
при 40—43712 мм, составляет 120—130 г E4—58% теоретич.;
примечание 4).
2. Примечания
1^ Смесь необходимо охладить до температуры ниже 50°, чтобы
предотвратить улетучивание прибавляемого этилового эфира мура-
вьиной кислоты.
2. Технический этиловый эфир муравьиной кислоты очищается
следующим образом: его промывают сперва 3%-ным раствором
соды, затем холодной водой, сушат безводным сернокислым натрием,
фильтруют и фракционируют (ср. стр. 194). Чрезвычайно важно,
чтобы все вещества, применяемые для синтеза ацетола, были без-
водными, так как в присутствии небольших количеств влаги выход
сильно уменьшается, вследствие образования продуктов конден-
сации.
3. Если применять продажный муравьинокислый калий, то его
следует высушить в вакууме при 80°. На моль бромацетона берут
от 1,5 до 2 молей соли.
4. Ацетол легко полимеризуется при стоянии, но остается неиз-
мененным, если его растворить в равном объеме метилового спирта.
3. Другие методы получения
Ацетол обычно получался реакцией между бромацетоном и
муравьинокислым калием или натрием, или уксуснокислым калием
или натрием, с последующим гидролизом эфира метиловым спир-
том1»2. При действии на глицерин3 или на пропиленгликоль4
катализаторов дегидрогенизации при 200—300° также образуется
ацетол. Вместе с пировиноградной кислотой ацетол образуется и
при непосредственном окислении ацетона при помощи перекиси
ддетона (реактива Байера и ВиллигераM.
76
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
1 Nef, Ann. 335, 247, 260 A904).
2 Urion, Ann. chim. A1) 1, 78A934).
3 Holmes; англ. пат. 428, 462 [С. Л. 29, 6908 A935)].
4 Carbide and Carbon Chemicals Corporation, ам. пат. 2 143 383 fC. A. 33,
2914 A939)].
5 Pastureau, Bull. soc. chim. D) 5, 227 A909).
2-АЦЕТОТИЕНОН
(Метил-2-тиенил-кетон )
НС — CH
II II
НС CH
\/
s
CHoCOCl
(SnCJj)
НС —CH
II II
НС С — COCH3
\/
S
НС1
Предложили: Дж. Джонсон и Дж. Мзй.
Проверили: Р. Фьюзон и Е. Кливленд.
1. Получение
В круглодонную трехгорлую колбу емкостью 0,5 л, снабженную
термометром, капельной воронкой, мешалкой с жидкостным затво-
ром и хлоркальциевой трубкой, помещают 16,8 г @,2 мол.) тиофена
(см. стр. 458), 15,6 г A4 мл; 0,2 мол.) хлористого ацетила и 200 мл
сухого бензола. Раствор охлаждают до 0°, после чего по каплям,
при эффективно работающей мешалке, добавляют в течение 40 мин.
52 г B3 мл; 0,2 мол.) свежеперегнанного хлорного олова. При добав-
лении первых капель хлорного олова реакционная смесь окраши-
вается в пурпурный цвет и вскоре выпадает пурпурный осадок.
После добавления всего количества хлорного олова, охлаждаю-
щую баню удаляют и перемешивание продолжают еще 1 час. Про-
дукт присоединения гидролизуют медленным добавлением смеси
из 90 мл воды и 10 мл концентрированной соляной кислоты. Желтый
бензольный слой отделяют, промывают 25 мл воды и сушат над 5—
10 г безводного хлористого кальция. Бензол и не вступивший в реак-
цию тиофен (примечание 1) отгоняют на масляной бане с небольшим
дефлегматором и остаток перегоняют в вакууме. Выход 2-ацето-
тиенона с т. кип. 89—9 Г/9 мм составляет 20—21 г G9—83% теоре-
тич.; примечание 2).
2. Примечания
1. Если взболтать полученную смесь бензола и тиофена с рас-
твором 5,5 г хлорной ртути, 10 г ацетата натрия и 10 мл спирта в
80 мл воды, то оставшийся тиофен превратится в 2-хлор-ртуть-
тиофен (содержащий небольшую примесь хлористой ртути), из кото-
АЦЕТО-я-ЦИМОЛ
п
рого при действии соляной кислоты можно выделить тиофен. Коли-
чество выделенного обратно тиофена составляет 2—2,5 г.
2. 2-Ацетотиенон обладает следующими физическими констан-
тами: d420 = 1,168; Пд20 = 1,566. Т. пл. ссмикарбазона: 186—187°
(исправл.).
3. Другие методы получения
2-Ацетотиенон был получен действием на тиофен хлористого
ацетила в присутствии хлористого алюминия1 или хлорного олова2,
а также действием хлористого ацетила на 2-хлор-ртуть-тиофен3.
Настоящий метод основан на работе Стадникова и Гольдфарба2.
Для этой реакции следует предпочесть применение хлорного олова
в качестве катализатора, так как хлористый алюминий вызывает
полимеризацию тиофена.
1 Р е t е г, Вег. 17, 2643 A884); В i e d e r m a n п, там же 19, 636 A886).
2 Стадникоп, Гольдфарб, там же 61, 2341 A928).
3 V о 1 h a r d, Ann. 267, 178 A892); S t e i n k о р f, Bauraeister,
там же 403, 69 A914).
АЦЕТО-и-ЦИМОЛ
B-метил-5-изопропилацетофенон)
CH.,
+ СНХОС1
(AIC13)
CH (CH3J
—COCH,
HC1
CH (CH3J
Предложил: К- Аллеи.
Проверили: Р. Фьюзон и Ч. Вудвард.
1. Получение
1-литровая трехгорлая колба снабжается капельной воронкой,
мешалкой, термометром для низких температур (примечание 1) и
холодильником, к верхнему концу которого присоединена стеклян-
ная трубка для отвода выделяющегося хлористого водорода (при-
мечание 2). В колбу помещают смесь 200 мл сероуглерода и 180 г
A,35 мол.) безводного хлористого алюминия, после чего колбу ста-
вят в охладительную смесь и содержимое энергично перемешивают,
пока температура смеси не понизится до —5° или ниже. Тогда через
капельную воронку прибавляют 175 г A,3 мол.) и-цимола и ПО г
78
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
Газ
Вода
A00 мл, 1,4 мол.) хлористого ацетила с такой скоростью, чтобы
температура не поднималась выше 5°, на что требуется примерно
3 часа 20 мин. (примечание 3). Массу оставляют на ночь и затем
выливают в смесь 1 кг мелкого льда и 200 мл концентрированной
соляной кислоты. Смесь извлекают три раза порциями эфира по
700 мл. Эфирный раствор сушат безводным хлористым кальцием и
перегоняют при атмосферном давлении из снабженной елочным
дефлегматором колбы Клайзена, пока температура не достигнет
190°. Оставшийся в колбе продукт подвергают двукратной фракцио-
нированной перегонке при пониженном давлении.
Главная фракция представляет собой ацето-л-цимол,
светложелтую маслянистую жидкость, с т. кип. 124—
125°/12 мм; 155—157°/30 мм. Выход 115—125 г
E0—55% теоретич.; примечание 4). Около 50 г ци-
мола получают обратно (примечание 5); в колбе
остается небольшое количество A0—12 г) высококи-
пящих продуктов (примечание 6).
2. Примечания
1. Ввиду невозможности отсчитывать показания
термометра по той части шкалы, которая находится
в колбе, следует применять термометр, у которого
нуль расположен над пробкой. Рекомендуется поль-
зоваться термометром со шкалой от —50° до +50°.
2. Для улавливания хлористого водорода можно
пользоваться ловушкой, показанной на рис. 3.
26мм Другой тип ловушки описан в «Синт. орг. прсп.»,
-*/— сб.' 1, стр. 100.
Рис. 3 3. После прибавления около двух третей смеси
скорость приливания можно несколько увеличить.
Время, потребное для приливания, зависит от эффективности
охлаждения и перемешивания; последнее должно быть энергич-
ным. Если взять половину указанных выше количеств и реакцию
вести в колбе емкостью 500 мл, то время, необходимое для при-
ливания, составит примерно 1 час 20 мин., так как при таких
условиях легче регулировать температуру.
4. При первом фракционировании за сырой кетон принимается
фракция, кипящая в пределах 20°, иначе говоря, при 145—165°/28—
30 мм. Следует обратить внимание на то, что кетон имеет тенден-
цию перегреваться.
5. В случае применения хлористого ацетила получается лучший
выход и меньшее количество высококипящего остатка, чем в слу-
чае применения уксусного ангидрида.
6. Приведенная методика была успешно применена также и
для ацетилирования кумола и третичн. бутилбензола. Если рабо-
БАРБИТУРОВАЯ КИСЛОТА
79
тать при указанной выше низкой температуре, разложение бывает
очень незначительным, на что указывает малое количество высо-
кокипящего остатка.
3. Другие методы получения
Ацето-л-цимол может быть получен действием хлористого аце-
тила на л-цимол в присутствии безводного.хлористого алюминия х
или хлорного железа 2.
1 Lacourt, Bull. soc. chim. Belg. 38, 17 A929); С 1 a u s, Ber. 19, 232
A886); К 1 a ges, Lickroth, там же 32, 1563 A899); Ver ley, Bull. soc.
chim. C) 17, 910 A897).
! Meisscl, Ber. 32, 2421 A899).
БАРБИТУРОВАЯ КИСЛОТА
HN — CO
CH2 (CO2C2HSJ + CO (NH,J (C'H'0N!L CO CH, + 2 C.,H5OH
II"
HN —CO
Предложили: Дж. Дикки и А. Грей.
Проверили: Р. Фъюзон и В. Росс.
1. Получение
В 2-литровой круглодонной колбе с обратным холодильником,
запертым хлоркальциевой трубкой, растворяют 11,5 г @,5 г^ат)
мелко нарезанного натрия в 250 мл абсолютного спирта. К этому
раствору добавляют сперва 80 г. @,5 мол.) малонового эфира и
затем 30 г @,5 мол.) сухой мочевины, растворенной в 250 мл горя-
чего G0°) абсолютного спирта. Смесь энергично взбалтывают, после
чего ее кипятят 7 час. на масляной бане, нагретой до 110°; при этом
вскоре выпадает белый осадок. По окончании реакции добавляют
500 мл горячей E0°) воды, а затем такое количество соляной кислоты
(уд. в. 1,18), чтобы раствор стал кислым (около 45 мл.). Получен-
ный прозрачный раствор фильтруют и фильтрат оставляют на ночь
в бане со льдом. Выпавший белый продукт отсасывают, промывают
на воронке Бюхнера 50 мл холодной воды и сушат в сушильном
шкафу при 105—110° в течение 3—4 часов. Выход барбитуровой
кислоты: 46—50 г G2—78% теоретич.).
2. Другие методы получения
Барбитуровая кислота может быть получена: действием хлор-
окиси фосфора на малоновую кислоту и мочевину 1; действием уксус-
ного ангидрида на раствор мочевины и малоновой кислоты в уксус-
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
ной кислоте 2; из малонового эфира и мочевины, пользуясь в каче-
стве конденсирующего агента этилатом натрия 3, и из малонового
эфира и натриевого производного мочевины, полученного из моче-
вины и натрия в жидком аммиаке *. Настоящий синтез основан на
способе Михаэля 3.
1 Q r i m a u х, Compt. rend. 87, 752 A878);
Ber. 14, 1643 A881); Grimaux, Bull. soc. chim. B) 31
g n о n, Ann. chim. phys. F) 28, 289 A893).
2 В i 1 t z, W i t t e k, Ber. 54, 1035 A921).
3 M i с h a e 1, J. prakt. Chem. B) 35, 456 A887); T a f e 1, We i n s с h e n k,
Ber. 33, 3383 A900); Q a b r i e 1, С о 1 m a n, там же 37, 3657 A904).
1 J а с о b s о п, ам. пат. 2 190 594 [С. А. 31, 7068 A937)].
Conrad, Guthzeit,
146 A879); Mati-
БЕНЗАЛЬФТАЛИД
(З-Бензилиденфталид)
CfiH,CH,CO,H
Предложил: Р. Вейс.
Проверили: Дж. Джонсон и X. Снайдер.
1. Получение
В круглодонную колбу с коротким горлом (не длиннее 3 см)
емк. в 500 мл помещают 100 г @,67 мол.) фталевого ангидрида (при-
мечание) ПО г @,8 мол.) фенилуксусной кислоты («Синт. орг. преп.»,
сб. 1, стр. 440) и 2,6 г свежесплавленного уксуснокислого натрия.
К смеси прибавляют несколько кусочков неглазурованнои тарелки
и колбу закрывают пробкой, в которую вставлены термометр, дохо-
дящий почти до дна колбы, и широкая согнутая трубка, соединен-
ная с холодильником. Трубка должна кончаться как раз у нижнего
края пробки и не вдаваться в шейку колбы. Колбу погружают по
горло в песчаную баню и быстро нагревают до 230°, после чего тем-
пературу медленно поднимают до тех пор, пока через выводящую
трубку не отгонится полностью вода, образовавшаяся во время
реакции (а также некоторое количество органического вещества).
Воду собирают в небольшой сосуд, время от времени определяют
ее количество и таким образом судят о ходе реакции. Реакцию сле-
дует вести так, чтобы повышение температуры от 230 до 240° про-
должалось около 2 час. Температуру 240° поддерживают до тех
пор, пока не прекратится отгонка воды, что требует еще около часа.
Содержимое колбы превращается к этому времени в массу бу-
рого цвета, покрытую пленкой. Теперь пробку вынимают и при
ЁЕНЗАНТРОН
помощи стеклянной палочки берут пробу. Пробу помещают в про-
бирку или небольшой стаканчик, прибавляют к ней немного спирта
и нагревают до кипения. Если реакция прошла полностью, веще-
ство легко растворяется в горячем спирте и при охлаждении вы-
кристаллизовывается.
Если проба дает удовлетворительный результат, колбе дают
охладиться до Ш—95° и продукт растворяют в 400 мл кипящего
спирта. Раствор фильтруют от нерастворимого осадка и дают филь-
трату охладиться. Выпавшие желтые кристаллы бензальфталида
отсасывают и промывают 40—50 мл холодного спирта. Продукт
весит 115—116 г и плавится при 95—97°; с целью очистки его пере-
кристаллизовывают из 370—380 мл спирта. Выход чистого бензаль-
фталида с т. пл. 100—101° составляет 106— ПО г G1—74%теоретич.).
2. Примечание
Работу надо проводить с возопшшым фталевым ангидридом
хорошего качества (т. пл. 129—131°); если нельзя достать такой про-
дукт, можно очистить возгонкой обыкновенный фталевый ангид-
рид.
3. Другие методы получения
Бензальфталид был получен единственным путем — из фтале-
вого ангидрида и фснилуксусной кислоты в присутствии уксусно-
кислого натрия. Метод, описанный здесь, в основном разработан
Габриэлем1.
1 Gabriel, Ber. 18, 3470 A885).
БЕНЗАНТРОН
Си
н.,
глицерин
О
Предложили: Л. Маклеод и К- Аллен.
Проверили: Л. Физер и М. Тишлер.
1. Получение
В снабженной механической мешалкой и термометром 2-литро-
вой трехгорлой колбе растворяют при комнатной температуре 72 г
6 Сборник № 2
82
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
@,35 мол.) антрахинона (стр. 546) в 1060 г концентрированной сер-
ной кислоты и пускают в ход мешалку. Затем к окрашенному в
красный цвет раствору приливают 42 мл воды (примечание 1).
Колбу погружают до горла в масляную баню, после чего в течение
полутора часов добавляют 48 г @,76 гр-ат.) осажденной порошко-
образной меди (примечание 2). Температуру реакционной смеси
поддерживают при 38—42° до полного растворения всей меди, в
случае необходимости подогревая масляную баню; на растворение
требуется около трех часов (примечание 3).
Затем понемногу в течение 30 мин. прибавляют смесь из 96 г
A,04 мол.) глицерина (примечание 4) и 96 .мл воды; при этом темпе-
ратуре дают подняться до 85—90°. Вслед за этим смесь осторожно
нагревают до 120° в течение полутора часов так, чтобы темпера-
тура поднималась равномерно со скоростью Г в 3 мин. (приме-
чание 5). Смесь поддерживают при 118—120° еще в течение Зчас,
после чего массу охлаждают до 70—80° и при постоянном пере-
мешивании осторожно выливают в 4 л кипящей воды (примеча-
ние 6). Разбрызгивания избегают тем, что кислотную смесь при-
ливают по стенке стакана при одновременном перемешивании.
Получившуюся суспензию кипятят несколько минут, после чего
желательно до фильтрования оставить ее на несколько часов
стоять.
Темнозеленый безантрон отсасывают через большую воронку
Бюхнера, хорошо промывают водой и кипятят в течение 30—40
мин. с 1,2 л 1%-ного раствора едкого натра. Продукт отфильтровы-
вают, отмывают от темноокрашенной жидкости и сушат при 120°;
вес его — 67—71 г, содержание бензантрона — около 87%. Сырой
продукт кипятят с 500 мл тетрахлорэтана (технического), причем
все растворяется, за исключением примерно 8 г обуглившегося
остатка. Раствор в течение 15 мин. кипятят с обратным холодиль-
ником с 25 г активированного угля, фильтруют горячим через во-
ронку Бюхнера непосредственно в 2-литровую круглодонную колбу
с длинным горлом и остаток промывают горячим тетрахлорэтаном
A00—150 мл) до тех пор, пока фильтрат не станет бесцветным.
Добавляют 400—500 мл горячей воды и перегонкой с водяным па-
ром удаляют растворитель, что протекает очень быстро. Оставший-
ся в виде осадка бензантрон отфильтровывают и сушат при 120°.
Выход 56—60 г G0—75% теоретич.). Продукт имеет желтый цвет,
плавится при 168—170° и для большинства целей является доста-
точно чистым.
Для получения совершенно чистого препарата указанный выше
продукт растворяют при кипячении в 175 мл тетрахлорэтана и
раствор кипятят с обратным холодильником 15 мин. с 12 г активи-
рованного угля. Раствор фильтруют с отсасыванием в коническую
колбу и уголь промывают 50 мл горячего растворителя. Фильтрат
в горячем состоянии обрабатывают 750 мл кипящего спирта и остав-
БЕНЗАИТРОН
ляют кристаллизоваться. Бензантрон выделяется в виде желтых
игл с т. пл. 170—171°. Выход 48—52 г F0—55% теоретич.; приме-
чание 7).
2. Примечания
1. Растворение антрахинона происходит медленнее, если сперва
прибавляют воду.
2. Осажденную медь готовят, как указано на стр. 371, но берут
вдвое большие количества исходных продуктов.
3. Смесь приобретает желто-бурую окраску, причем выделяется
небольшое количество антранола; непрореагировавшую медь ясно
видно на дне колбы, если остановить на несколько минут мешалку.
4. Берется технический безводный глицерин.
5. Нагревание необходимо вести очень осторожно и температура
ни в коем случае не должна подниматься выше 120°. Более высокая
температура ведет к обугливанию и значительным потерям.
6. Получается крупнозернистый продукт, легче фильтрующийся,
чем в случае применения холодной воды.
7. При регенерации тетрахлорэтана перегонкой с водяным па-
ром маточного раствора выделяется еще некоторое количество про-
дукта E г), однако он темного цвета и плохого качества.
3. Другие методы получения
Бензантрон обычно получается нагреванием продукта восста-
новления антрахинона с серной кислотой и глицерином х или с
одним из производных глицерина2, или с акролеином3. Обычно
антрахинон восстанавливают в растворе серной кислоты непосред-
ственно перед реакцией с помощью сернокислого анилина 1б, же-
леза 1в или меди 1г. Однако имеются указания, что одновременно
проводимые восстановление и конденсация дают лучший выход 4.
Бензантрон был также получен дегидрогенизацией хлористым алю-
минием или хлорным железом 5 фенил-а-нафтил-кетона, дегидрата-
цией 1-фенилнафталин-8-карбоновой кислоты и нагреванием цин-
намалантрона со сплавом хлористого алюминия и хлористого на-
трия 7.
1 (a) Bally, Ber. 38, 194 A905); Badische Anilin- und Soda-Fabrik,
герм. пат. 176 018 [Frdl. 8,372 A905—7)]; (б) В а 1 1 у, S с h о 1 1, Вег. 44, 1665
A911); (в) Ильинский, сов. патент 18741 (Chem. Zentr. 1931, II,
1759; (г) С a s w e 1 1, Marshall, амер. пат. 626 392 [С. А. 21, 1992A927)];
(д) В а с h а г а с h, С a u I i f f, Jr., амер. пат. 1 893 575 [С. А. 27, 2163 A933)].
2 Badische Anilin- und Soda-Fabrik, герм. пат. 204 354 fFrdl. 9, 818
A908—10)].
3 С г о s s, P e r k i n, J. Chem. Soc. 1927, 1297.
4 Лукин, Пром. орг. хим., 4, 341 A937).
6 S с h о I 1, Seer, Ann. 394, 116 A912); Monatsh. 33,1 A912); S с h о 1 1,
герм. пат. 239 671 [Frdl. 10, 682 A910—12)].
6 Schaarschmidt, Ber. 50, 295 A917).
' I. G. Farbenind. A.-G.,repM. пат. 488 606 [Frdl. 16, 1438A927—29)].
84
СИНТЕЗЫ ОРГАНИЧЕСКИЙ ПРЕПАРАТОВ
ВЕНЗИЛФТАЛИМИД
(N-Бензилфталимад)
2С6Н4 (С0J NH + 2С6Н5СН2С1 + К2СО3
2С6Н4 (С02) NCH2C6H6 + С02 + 2КС1
Н2О
Предложил: Р. Манске.
Проверили: Г. Гилъмйн и X. Харвуд.
1. Получение
В тесной смеси 166 г A, 2 мол.) безводного поташа и 294 г
B мол.) фталимида (примечание 1) добавляют 506 гD мол.) хлори-
стого бензила (примечание 2) и. массу нагревают в продолжение
3 час. (примечание 3) с обратным холодильником на масляной бане
при 190°. Затем избыток хлористого бензила отгоняют из горячей
смеси с паром (примечание 4). Под конец отгонки бензилфталимид
закристаллизовывается. Смесь рекомендуется быстро охладить,
энергично перемешивая ее, с тем чтобы продукт выпал в возможно
более мелком состоянии. Осадок отсасывают на большой воронке
Бюхнера, тщательно промывают водой и возможно лучше отжи-
мают. Затем его промывают один раз 400 мл 60%-ного спирта и сно-
ва отжимают. Выход: 340—375 г G2—79%теоретич.). Температура
плавления 100—110°. Продукт хорошо очищается перекристалли-
зацией из горячей ледяной уксусной кислоты, причем выход пере-
кристаллизованного продукта составляет 80%. Температура плав-
ления чистого бензилфталимида: 116° (исправл.; примечание 5).
2. Примечания
1. Поташ легко обезвоживается нагреванием в большой чашке
над небольшим пламенем. Его необходимо растереть в очень мел-
кий порошок и смешать в ступке с фталимидом.
2. Для реакции применялся хлористый бензил, кипящий в пре-
делах 3°.
3. Механическое перемешивание не улучшает выхода.
4. Отогнанный избыток хлористого бензила отделяют от воды
и сушат хлористым кальцием. Таким образом возвращается обратно
200—300 г хлористого бензила.
5. По зтому же методу из бромистого триметилена можно полу-
чить ^-бромпропилфталимид и в качестве побочного продукта
а-, у-дифталимидпропан, а из бромистого р-фенилэтила — р-фенил-
этилфталимид.
3. Другие методы получения
Бензилфталимид может быть получен: из фталимид-калия и
хлористого бензила1; из фталимида, поташа и хлористого бензи-
ла 2 и из фталимида, этилата натрия и хлористого бензила3. В
БЕНЗИМИДАЗОЛ
85
обоих последних методах избегается необходимость получения фтал-
имид-калия. При кипячении с обратным холодильником смеси
фталевого ангидрида, бензиламина и ледяной уксусной кислоты
также образуется бензилфталимид4. Приведенный здесь метод
описан в литературе 2.
1 Gabriel, Bcr. 20, 2227 A887).
2 I n g, M a n s k e, J. Chem. Sac. 1926, 2348.
3Weisz, Lanyi, Magyar Chem. Folyoirat 39, 153 A933) [C. A. 28,
5815 A934)].
4 Vanags, Acta Univ. Latviensis, Kim. Fakultat, Ser. 4, No. 8, 405
A939) [C. A. 34, 1983A940)].
БЕНЗИМИДАЗОЛ
— NH»
— NH,
HCO2H >
CH +2H2O
Предложили: Е. Вйгнер и В. Миллетт.
Проверили: В. Хартманч Дж.Сауди.
1. Получение
В круглодонную колбу емкостью 500 мл помещают 54 г @,5 мол.)
о-фенилендиамина (примечание 1) и добавляют 32 мл C4,6 г)
90%-ной муравьиной кислоты @,75 мол.; примечание 2). Смесь нагре-
вают на водяной бане при 100° в течение 2 час. По охлаждении к
ней медленно приливают 10%-ный раствор едкого натра, причем
содержимое колбы тщательно перемешивают, вращая колбу до
тех пор, пока смесь не станет чуть щелочной на лакмус. Сырой
бензимидазол отсасывают, пользуясь бюхнеровской воронкой диа-
метра 75 см. Для того, чтобы ополоснуть колбу, берут ледяную
воду. Сырой продукт тщательно отжимают на фильтре, промы-
вают примерно 50 мл холодной воды и затем подвергают очистке
без предварительной сушки (примечание 3).
Бензимидазол растворяют в 750 мл кипящей воды*в стакане
емкостью 1,5 л. Раствор нагревают 15 мин. с 2 г активированного
угля и быстро фильтруют через хорошо прогретую воронку (при-
мечание 4). Фильтрат охлаждают до 10—15° и бензимидазол от-
фильтровывают и промывают 50 мл холодной воды. Бесцветный
(примечание 5) продукт сушат при 100°. Т. пл. 170—172°, выход
49—50,5 г (83—85% теоретич.; примечания 6 и 7).
86
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
2. Примечания
11 Применялся продажный о-фенилендиамин хорошего каче-
ства с температурой плавления 99—ЮГ. Можно применять также
его солянокислую соль с добавлением или без добавления му-
равьинокислого натрия. Метод получения о-фенилендиамина приве-
ден на стр. 509.
2. Если уменьшить количество муравьиной кислоты почти до
теоретического количества, то это мало влияет на выход, но реко-
мендуется брать ее в избытке, достаточном для обеспечения исполь-
зования всего количества о-фенилендиамина. Бензимидазол обра-
зуется даже в том случае, если брать муравьиную кислоту концен-
трации значительно меньшей, чем 90%; хорошие выходы получались
даже с 40%-ной кислотой.
3. Вес неочищенного бензимидазола после высушивания при
100° составляет 57,5—59 г (97—99% теоретич.). Т. пл. 167—168°.
Продукт слегка окрашен в желтый цвет и обесцветить его можно
лишь с трудом. Окраска не исчезает даже после двух перекристал-
лизации (примечание 5).
4. Раствор почти насыщен при температуре кипения, и при ох-
лаждении немедленно начинается кристаллизация. Воронку для
фильтрования следует хорошо прогреть и фильтровать необходимо
быстро, так как иначе вещество начнет кристаллизоваться на
фильтре.
5. Если перекристаллизованный бензимидазол окрашен, то для
получения продукта хорошего качества поступают следующим обра-
зом. Бензимидазол растворяют в кипящей воде A3 мл на 1 г) и до-
бавляют концентрированный раствор перманганата калия до тех
пор, пока жидкость не станет непрозрачной в результате ьыпаде-
ния в осадок бурой двуокиси марганца. К горячей смеси добавляют
твердый бисульфит натрия до тех пор, пока раствор не просветлеет.
Затем добавляют активированный уголь, смесь нагревают 15 ми-
нут и фильтруют горячей. Выход 90—92%.
6. Если упарить маточный раствор до объема 30 мл, можно полу-
чить еще небольшое количество продукта B—2,5 г).
7. Этот метод является общим для получения бензимидазолов.
Если вместо муравьиной кислоты взять эквивалентное количество
уксусной кислоты D5 г), то с выходом 68% можно получить
2-метилбензимидазол с т. пл. 172—174°.
3. Другие методы получения
Бензимидазол был получен из о-фенилендиамина при действии
на него хлороформа и спиртового раствора едкого кали х и муравьи-
ной кислоты2, а также восстановлением о-нитроформанилида3.
Среди методов, которые практически менее удобны, можно упомя-
БЕНЗОГИДРОКСАМОВАЯ КИСЛОТА
87
нуть взаимодействие о-фенилендиамина с дихлорметилформами-
дином 4 или дифенилформамидином 5 или со смешанным ангидри-
дом муравьиной и уксусной кислот 6 и термическое декарбоксили-
рование бензимидазол-2-карбоновой кислоты7. Описанная выше
методика 8 в основном разработана на данных методики Вундта2.
Превращение алифатических кислот в 2-алкилбензимидазолы
путем нагревания их с о-фенилендиамином было предложено в
качестве общего метода получения их кристаллических производ-
ных для идентификации 9.
1 G r a s s i-C r i s t a I d i, L a m b a r d i, Gazz. chim. ital. 25, 225 A895).
2 Wundt, Ber. 11, 826A878); Heller, Kiihn, там же 37, 3116A904);
Paul у, Gundermann, там же 41, 4012 A908).
3 N i с me n to wsk i, там же 43, 3018 A910).
4 D a i n s, там же 35, 2503 A902).
5 Wagner, I. Org. Chem: 5, 136 A940).
« Be h a 1, герм. пат. 115 334 [Frdl. 6, 1280 A900-02)].
' Bistrzycki, Przeworski, Ber. 45, 3489 A912).
8 Wagner, Simons, J. Chem. Education 13, 267 A939).
9 S e k a, M it 1 I e r, Monatsh. 57, 97 A931); Pool, Harwood, Ral-
ston, J. Am. Chem. Soc. 59, 178 A937).
БЕНЗОГИДРОКСАМОВАЯ КИСЛОТА
C6H5CO2C2H5 + H2NOH + KOH
, C6H5CONHOK + C2H5OH + H2O
C6H3CONHOK + CH3CO2H C6HaC0NH0H + CH3CO.,K
Предложили: Ч. Хаузер и В. Ренфроу мл.
Проверили: К. Ноллер и М. Сайнерхолм.
1. Получение
А. Кплиееая соль бензогидроксамовой кислоты. Приготовляют
отдельно два раствора: 46,7 г @,67 мол.) хлористоводородного
гидроксиламина («Синт. орг. преп.», сб. 1, стр. 164) в 240 мл
метилового спирта и 56,1 г A мол.) едкого кали (х. ч.) в 140 мл
метилового спирта. Оба раствора готовят при температуре кипе-
ния растворителя, а затем охлаждают до 30—40° (примеча-
ние 1), после чего при взбалтывании раствор щелочи приливают
к раствору гидроксиламина. Повышение температуры при сме-
шении предотвращают, охлаждая периодически колбу в бане со
льдом. После того как прибавлена вся щелочь, смеси дают по-
стоять в бане со льдом в течение 5 мин., чтобы обеспечить полноту
осаждения хлористого калия. Затем при энергичном взбалтывании
добавляют 50 г @,33 мол.) этилового эфира бензойной кислоты и
смесь немедленно фильтруют с отсасыванием. Остаток в воронке
88
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
промывают небольшим количеством метилового спирта. Фильтрат
переливают в коническую колбу и оставляют стоять при комнат-
ной температуре. Примерно через 20 мин., а иногда и позднее, даже
через 3 часа, в зависимости от степени пересыщенности раствора,
начинают выпадать кристаллы. Черер 48 час. кристаллы отфиль-
тровывают, промывают небольшим количеством абсолютного эти-
лового спирта и сушат на воздухе. Выход 33—35 г E7—60% тео-
ретич.; примечания 2, 3 и 4).
Б. Бензогидроксамовая кислота. Смесь 35 г @,2 мол.) ка-
лиевой соли и 160 мл 1,25 н. уксусной кислоты перемешивают
и нагревают до получения прозрачного раствора, которому дают
охладиться до комнатной температуры, а под конец охла-
ждают в бане со льдом. При этом бензогидроксамовая кислота
выпадает в виде бесцветных кристаллов. После фильтрования и
высушивания выход продукта с т. пл.* 120—128° составляет 25—
26 г (91—95% теоретич.). Для очистки сырого продукта его раство-
ряют в 4,5-кратном количестве по весу горячего уксусноэтилового
эфира, отфильтровывают небольшое количество примеси и дают
раствору охладиться до комнатной температуры. Выпавшие бес-
цветные кристаллы отфильтровывают, промывают небольшим ко-
личеством бензола и сушат на воздухе. Из 26 г неочищенного про-
дукта получают 20 г перекристаллизованного с т. пл. 125—128°
G7%; примечание 5).
2. Примечания
1. Раствор хлористоводородного гидроксиламина не следует
охлаждать слишком быстро, чтобы кристаллизация не началась
до смешения. После смешения обоих растворов следует свести к
минимуму соприкосновение раствора с воздухом, чтобы" избежать
окисления свободного гидроксиламина.
2. Если упарить спиртовые маточные растворы, можно получить
еще 3—5 г калиевой соли.
3. Эту соль можно использовать для получения ацильных про-
изводных; например, если приготовить суспензию соли в диоксане
и подействовать хлористым бензоилом, можно получить с прекрас-
ным выходом дибензогидроксамовую кислоту.
4. Авторами настоящего синтеза были получены по той же мето-
дике и примерно с такими же выходами калиевые соли п-метил-
и п-метоксибензогидроксамовых кислот.
5. Число нейтрализации этого рродукта 137,5—138 (вычислено
137,1), если его определять следующим образом. Несколько капель
спиртового раствора 1,3,5-тринитробензола добавляют к 30—40 мл
воды, находящейся в колбе, и приливают 0,1 н. щелочь (около
0,5 мл) до появления розового окрашивания. Навеску (около 0,3 г)
бензогидроксамовой кислоты растворяют в этом растворе и тит-
руют ее титрованным раствором щелочи до появления той же розо-
е-БЕН30ИЛАМИН0-=с-БР0МКАПР0Н0ВАЯ КИСЛОТА
89
вой окраски. Количество пошедшей щелочи служит для вычисле-
ния числа нейтрализации.
3. Другие методы получения
Бензогидроксамовая кислота была получена действием гидро-
ксиламина на хлористый бензоил х, этиловый эфир бензойной кис-
лоты 2-3, или бензамид 4.
1 Lnssen, Ann. 161, 347 A872); J о n e s, H u r d, J. Am. Chem. Soc. 43,
2446 A921).
3 R e n f г о w, H a u s e г, там же 59, 2312 A937).
3 T i с m a n n, К r tiger, Ber. 18, 740 A885); Jeanrenaud, там же
22, 1272 A889).
4 H о f m a n n, там же 22, 2856 A889).
s-БЕНЗОИЛАМИНО-а-БРОМКАПРОНОВАЯ КИСЛОТА
(s-Бензамадо-а-бромкапроновая кислота )
3C6H,CONH (СН,M СО2Н + ЗВг2
— 3C6H5CONH (СН,L СНСОВг +
+ РВг3
Р(ОНK + 3 НВг
Вг
C6H5CONH (СН2L СНСОВг
Вг
C6H5CONH(CH2LCHCO2H
Вг
Предложили: Дж. Экк и К- Марвел.
Проверили: К. Ноллер и В. Мьюнич.
1. Получение
В 1-литровую трехгорлую колбу, снабженную делительной
воронкой, механической мешалкой (примечание 1) и воздушным
холодильником, который через хлоркальциевую трубку. присое-
динен к водяной ловушке, помещают тесную смесь 150 г @,64 мол.)
сухой s-бензоиламинокапроновой кислоты (стр.90) и 26,4г@,85г/?-ат.)
сухого красного фосфора. Реакционную колбу погружают в смесь
льда и соли, пускают в ход мешалку и из делительной воронки
по каплям приливают 408 г A31 мл, 2,55 мол.) сухого брома.
После того как весь бром прибавлен, охлаждающую баню удаляют
и смесь нагревают, сперва медленно, а затем на кипящей водяной
бане, при работающей мешалке, до практически полного исчезно-
вения паров брома. Горячую смесь медленно выливают в 400 мл
воды, налитой в 1-литровый стакан при перемешивании от руки.
Вязкий бромангидрид кислоты реагирует с водой с выделением
тепла и выпадает твердая кислота. Выпавшие куски измельчают,
смесь вновь помещают в ту же колбу и обрабатывают ее медленным
током сернистого газа, чтобы удалить избыток брома, Твердый
90
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
продукт отфильтровывают через воронку Бюхнера, промывают три
раза водой порциями по 50 мл и сушат на воздухе. Неочищенный
продукт растворяют в 250 мл горячего 95%-ного спирта, раствор
фильтруют и при перемешивании выливают в 1 л холодной воды
(примечание 2). Осадок отфильтровывают, сушат на воздухе и полу-
чают 130—180 г F4—89% теоретич.) кислоты с температурой плав-
ления 162—165°.
2. Примечания
1. Мешалка должна быть очень мощной, так как в реакционной
смеси выпадают куски, и в конце концов она становится очень вяз-
кой. Если на мешалке накапливаются комки и она перестает рабо-
тать, временно приходится вести перемешивание от руки до тех
пор, пока масса вновь не станет достаточно жидкой, чтобы можно
было возобновить работу механической мешалки. При проверке
синтеза была сделана попытка облегчить перемешивание, добавив
четыреххлористый углерод. Хотя этой цели и удалось достигнуть,
но выход оказался в два раза меньше, чем при работе без раствори-
теля.
2. Сырой продукт можно перекристаллизовать из этилового
спирта, однако температура плавления получается такой же, как
у продукта полученного осаждением водой, выход же значительно
ниже.
3. Другие методы получения
Приведенная выше методикаJ в основном заимствована у Брау-
на 2.
1 Е с к, М а г v e I, J. Biol. Chem. 106, 387 A934).
2 В г a u n, Ber. 42, 839 A909).
СН
s-БЕНЗОИЛАМИНОКАПРОНОВАЯ КИСЛОТА
(е-Бензамидокапроновая кислота)
/СН2 — СН2Ч ш,эол __ /СН2—СН2-
СО
—сн
>
СН2
H2N (СН2K СО2Н
ХСН2 — СН2 — NH
C6H-CONH (CH2M СО3Н
Предложили: Дж. Экк и К- Марвел.
Проверили: К. Ноллер и В. Мыонич.
1. Получение
А. Циклогексаноноксим. В 5-литровую колбу, снабженную
эффективной механической мешалкой и стеклянной трубкой для
ввода газа диаметром 8 мм, конец которой на 5 см не дохо-
дит до дна колбы, помещают 1,5 кг колотого льда и раствор
s-БЕНЗОИЛАМИНОКАПРОНОВАЯ КИСЛОТА
91
182 г B,5 мол.) технического азотистокислого натрия (92%-ного)
в 500 мл воды (примечание 1). Колбу помещают в баню со смесью
льда и соли, и к ней прибавляют холодный (—8°) раствор бисуль-
фита натрия, полученный насыщением сернистым газом раствора
143 г A,35 мол.) безводного углекислого натрия в 600 мл воды.
Поддерживая температуру ниже 0°, через смесь пропускают уме-
ренно сильный ток сернистого газа до кислой реакции на конго
ровно столько времени, чтобы уничтожить темную окраску,
которая появляется незадолго до того, как раствор станет
кислым.
К этому раствору прибавляют 196 г B мол.) технического цик-
логексанона и 500 мл 85%-ного этилового спирта; охлаждающую
баню заменяют на паровую, пускают в ход мешалку и смесь на-
гревают до 75°. Затем колбу окружают асбестом или любым
другим изоляционным материалом и содержимому колбы дают
медленно охладиться при эффективном перемешивании в течение
48 час. При комнатной температуре раствор точно нейтрализуют
на лакмус 50%-ным раствором едкого натра, охлаждая его и
перемешивая. На это требуется около 330 г раствора едкого
натра.
Маслянистый слой отделяют, а водный раствор извлекают два
раза эфиром, порциями по 200 мл. Маслянистую жидкость и эфир-
ные вытяжки соединяют вместе, эфир отгоняют и остаток перего-
няют из видоизмененной колбы Клайзена емкостью 500 мл с дефлег-
матором высотой 25 см. Фракция, кипящая при 95—100°/ 5 мм,
весит 170—190 г и плавится при 78—80°. Этот продукт переносят
в большую ступку, дают ему охладиться и растирают его с 120 мл
петролейного эфира (т. кип. 35—60°). Массу фильтруют с отсасы-
ванием и растворителю дают испариться с кристаллов. Получают
133—147 г циклогексаноноксима E9—65% теоретич.) с т. пл. 86—
88° (примечание 2).
Б. е-Бензоиламинокапроновая кислота. Реакцию перегруппи-
ровки 100 г @,88 мол.) чистого циклогексаноноксима (приме-
чание 3) осуществляют следующим образом. В 1-литровый ста-
кан помещают порцию в 10 г оксима и 20 мл 85%-ной серной
кислоты (уд. в. 1,783) (примечание 4). Стакан нагревают на
небольшом пламени и содержимое его перемешивают, придавая
стакану вращательное движение, до появления пузырьков. Немед-
ленно стакан снимают и дают пройти бурной реакции, которая про-
должается несколько секунд. Кислый раствор е-капролактама пе-
реносят в 5-литровую круглодонную колбу и в стакане проводят
перегруппировку новой порции оксима в 10 г с таким же, как и
в первом случае, количеством кислоты. Кислый раствор от 10 опы-
тов разбавляют 2,5 л воды и осторожно кипятят 30мин. с 5 г обес-
цвечивающего угля. Раствор фильтруют и точно нейтрализуют на
лакмус 50%-ным раствором едкого натра. Обычно на это требуется
92
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
около 510 г раствора едкого натра. Нейтральный раствор кипятят
30 мин. с 5 г обесцвечивающего угля и фильтруют.
Фильтрат помещают в 5-литровую колбу, снабженную механи-
ческой мешалкой, охлаждают колбу до 10° в бане со льдом и прибав-
ляют раствор 55 г A,37 мол.) едкого натра в 55 мл воды. Поддер-
живая температуру при 10°, из капельной воронки приливают по
каплям в течение 35—40 мин. 94 г @,67 мол.) хлористого бензоила
при энергичном перемешивании. Смесь перемешивают еще 1 час,
фильтруют и фильтрат переносят в 4-литровый стакан. Холод-
ный фильтрат медленно подкисляют на конго 10%-ной соляной
кислотой при перемешивании от руки (требуется около 450 мл
кислоты). Твердый осадок (примечание 5) отфильтровывают с от-
сасыванием, промывают водой и раскладывают слоем для высу-
шивания. Сухой продукт промывают два раза петролейным эфи-
ром (т. кип. 35—60°) порциями по 100 мл для удаления примеси
бензойной кислоты. Поглотившемуся растворителю дают испарить-
ся и окончательно сушат продукт в вакуум-эксикаторе над серной
кислотой. Выход очищенного продукта с температурой плавления
77—80° составляет 135—150 г F5—72% теоретич., считая на цик-
логексаноноксим).
2. Примечания
1. Можно добавить также твердый азотистокислый натрий к
2 кг льда, однако в этом случае необходимо предварительно вне
колбы тщательно смешать нитрит и лед во избежание спекания льда.
2. С лучшим выходом циклогексаноноксим можно получить,
исходя из кетона, хлористоводородного гидроксиламина («Синт.
орг. преп.», сб. 1, стр. 164) и углекислого натрия по методике,
приведенной на стр. 396. Однако синтез с применением хлористо-
водородного гидроксиламина обходится несколько дороже.
3. Перегруппировку оксима проводят порциями по 10 г и в
большом открытом стакане ввиду того, что реакция протекает весь-
ма бурно. Очень важно работать с оксимом хорошего качества,
иначе будет получен продукт значительно худший.
4. Кислоту такой концентрации можно приготовить, если сме-
шать 5 объемов концентрированной серной кислоты с 1 объемом
воды.
5. Если кислота выпадет в виде масла, надо ей дать постоять,
время от времени ее перемешивая, до тех пор пока она не затвер-
деет.
3. Другие методы получения
е-Бензоиламинокапроновая кислота была получена при дей-
ствии на бензоилпиперидин иятихлористого фосфора с образованием
е-бензоиламиноамилхлорида, превращением его в нитрил и омыле-
нием до кислоты 1, а также с помощью методики, описанной выше2.
БЕНЭОИЛЕНМОЧЕВИНА
93
Методика для перегруппировки циклогексаноноксима с целью
выделения е-капролактама и е-аминокапроновой кислоты приве-
дена на стр. 304 и 36.
1 Braun, Ber. 42, 839 A909).
2 Е с k, M a r v e I, J. Biol. Chem. 106, 387 A934).
БЕНЗОИЛЕНМОЧЕВИНА
[2,4 G,3)Хиназолиндион]
HCNO
со,н
СО2Н
NaOH
н
СО Hs_soj
н
ONa
C
0
Предложили: Н. Ланге и Ф. Шейбш.
Проверили: В. Хартман и Дж. Дикки.
1. Получение
В 3-литровом стакане приготовляют смесь из 20 г @,146 мол.)
антраниловой кислоты, 700 мл теплой воды C5°) и 11 мл A1,6 г,
0,19 мол.) ледяной уксусной кислоты. Все это перемешивают с
помощью механической мешалки и дают охладиться до комнатной
температуры. Затем при работающей мешалке по каплям прибав-
ляют свежеприготовленный раствор 15 г @,185 мол.) циаповокис-
лого калия (примечание 1) в 50 мл воды. Прибавление раствора
продолжается 15—20 мин. (примечание 2). Полученную пастооб-
разную массу перемешивают еще 20 мин., после чего медленно,
небольшими порциями добавляют 200 г E мол.) гранулированного
едкого натра (примечание 3). В течение всего этого времени темпе-
ратуру реакционной массы поддерживают ниже 40°, охлаждая
колбу холодной водой. На момент раствор становится прозрачным,
но вскоре в осадок выпадает в очень мелком состоянии гидрат одно-
натриевой соли бензоиленмочевины. Смесь охлаждают в течение ночи
в холодильном шкафу и выпавшую натриевую соль фильтруют через
94
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
воронку Бюхнера, пользуясь уплотненной фильтровальной бума-
гой (примечание 4). Бесцветную соль растворяют в 1 л горячей
воды (90—95°), раствор фильтруют и нагревают до кипения в 3-ли-
тровом стакане. Для того чтобы осадить бензоиленмочевину,
при энергичном перемешивании, добавляют разбавленной серной
кислоты A : 1) до кислой реакции на лакмус. Продукт выпадает
в виде гидрата, который образует мелкие блестящие бесцветные
иглы. Его собирают на воронке Бюхнера, промывают 200 мл воды
и сушат в сушильном шкафу при 100°. Выход 19,5—20,5 г (82—
87% теоретич.; примечание 5).
2. Примечания
1. Выход в значительной степени зависит от качества взятого
циановокислого калия, причем было найдено, что некоторые образ-
цы совершенно непригодны для этой цели. Указанный здесь выход
был получен при работе с обычным циановокислым калием фирмы
Истмсн.
2. Если прибавление ведется слишком быстро, то становится
очень сильным запах изоциановой кислоты (напоминающий запах
сернистого газа) и выход падает.
3. Продажный гранулированный едкий натр легко раство-
ряется и с ним удобно работать. Присутствие кремнезема не мешает,
так как его удаляют при фильтровании раствора натриевой соли,
перед осаждением бензоиленмочевины кислотой.
4. Удовлетворительны уплотненные фильтры Шлейхер и Шюль
(№ 575/9 см).
5. Т. пл. бензоиленмочевины лежит выше 350°.
3. Другие методы получения
Бензоиленмочевина была получена пропусканием циана через
спиртовой раствор антраниловой кислоты с последующим омыле-
нием образующегося 2-этокси-4-кетодигидрохиназолина г, сплав-
лением антраниловой кислоты с мочевиной 2>3 и действием водной1
циановой кислоты на антраниловую кислоту 2>3>*>5. Описанная
здесь методика заимствована у Богерта и Скечерда 5, причем вне-
сены некоторые изменения.
1 G r i e s s, Вег. 2, 415 A869).
2 G r i e s s, J. prakt. Chem. B) 5, 371 A872).
8 Bogert, Scatchard, J. Am. Chem. Soc. 41, 2056 A919).
4 Gabriel, Colman, Ber. 38, 3561 A905); Scott, Cohen J Chem
Soc. 119, 664A921).
6 Bogert, Scatchard, Am. Chem. Soc. 38, 1611 A916).
р-БЕНЗОИЛПРОПИОНОВАЯ КИСЛОТА
95
Р-БЕН30ИЛПР0ПИ0Н0ВАЯ КИСЛОТА
AICI3
сн2 — со
сн2 — со
)О + С6Н6
Предложили: Л. Сомервилл и К- Аллен.
Проверили: Р. Фьюзон и Д. Форман.
1. Получение
В 2-литровую трехгорлую круглодонную колбу, снабженную
механической мешалкой и двумя обратными холодильниками, по-
мещают 68 г @,68 мол.) ангидрида янтарной кислоты (стр. 56) и
350 г D,5 мол.) сухого, не содержащего тиофена, бензола (примеча-
ние 1). Мешалку пускают в ход и в колбу прибавляют за один прием
200 г A,5 мол.) растертого в порошок безводного хлористого алюми-
ния. Начинается выделение хлористого водорода и смесь разогре-
вается (примечание 2). При перемешивании смесь нагревают на
масляной бане до кипения еще в течение получаса (примечание 3);
затем смесь охлаждают холодной водой и через капельную воронку,
вставленную в верхнюю часть одного из обратных холодильников,
медленно приливают 300 мл воды (примечание 4). Избыток бензола
отгоняют с водяным паром и горячий раствор немедленно перели-
вают в 2-литровый стакан. По охлаждении жидкость сливают с
выделившегося осадка и подкисляют концентрированной соляной
кислотой (около 20 мл); выпадает от 5 до 15 г бензоилпропионовой
кислоты; ее отсасывают (примечание 5). Оставшуюся в стакане
суспензию кипятят в продолжение 5 час. с 1500 мл воды, содержа-
щей 360 г технической кальцинированной соды. Осадок отсасыва-
ют, промывают на фильтре горячей водой и фильтрат подкисляют
концентрированной соляной кислотой (около 300 мл) (примечание
б). Выпавшую бензоилпропионовую кислоту отсасывают и промы-
вают горячей водой (примечание 7). Высушенная в продолжение
одного дня кислота весит 95—100 г G2—82% теоретич,) и плавится
при 111—113° (примечание 8).
Если, как было указано, отделить первый окрашенный осадок
(вес 5—15 г), то остальную массу можно не очищать. Если нужен
вполне чистый препарат, то кислоту растворяют в разбавленном
растворе едкого натра и осаждают концентрированной соляной
кислотой, причем первую часть осадка отсасывают отдельно. Чи-
стая кислота плавится при 116° (примечание У).
2. Примечания
1. Технический бензол взбалтывают с концентрированной сер-
ной кислотой, промывают водой и сушат сначала хлористым каль-
цием, а затем металлическим натрием.
96
СИНТ ЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
2. Если реакция не начинается сразу, смесь слабо нагревают.
3. Выход не увеличивается от более длительного нагрева-
ния.
4. Начиная с этого момента метод может быть упрощен и улуч-
шен следующим образом. После добавления к комплексу с хлори-
стым алюминием воды приливают 100 мл концентрированной соля-
ной кислоты (уд. в. 1,18) и бензол отгоняют с водяным паром. Го-
рячую массу переносят в 1-литровый стакан, причем f-4-бензоилпро-
пионовая кислота выделяется в виде бесцветного масла, которое
вскоре затвердевает. После охлаждения до 0° твердый продукт
отделяют, промывают холодной смесью из 50 мл концентрирован-
ной соляной кислоты и 150 мл воды, а затем еще 200 мл холодной
воды. Неочищенную кислоту растворяют в растворе 75 г безвод-
ного углекислого натрия в 500 мл воды, для чего массу приходится
кипятить 15 мин. Раствор фильтруют с отсасыванием, небольшое
количество остающейся на фильтре гидроокиси алюминия дважды
промывают горячей водой, порциями по 50 мл. К горячему филь-
трату добавляют 4 г активированного угля, раствор перемешивают
3—4 мин. и фильтруют с отсасыванием. Прозрачный бесцветный
фильтрат переносят в 2-литровый стакан, охлаждают до 50—60°
и осторожно подкисляют 130 мл концентрированной соляной кис-
лоты. После охлаждения до 0° в бане со льдом и солью кислоту
отфильтровывают, тщательно промывают водой, сушат в течение
ночи при комнатной температуре, а затем до постоянного веса при
40—50°. Выход: ПО—115 г (92—95% теоретич,); т. пл.: 114—115°;
продукт не требует дальнейшей очистки (Е. Мартин и Л. Физер,
частное сообщение).
5. Количество выделяемой при этом кислоты зависит от того,
сколько времени раствор стоял до подкисления.
6. Первая часть осажденной кислоты обычно бывает окрашена;
лучше всего собирать ее отдельно. В этом случае остальная масс'а
кислоты — бесцветна и почти совсем чиста (т. пл.: 114—115°).
7. При упаривании фильтрата до небольшого объема и извле-
чении его эфиром получается еще 3 г кислоты.
8. Кислота упорно удерживает последние следы влаги, от чего
получаются преувеличенные выходы. Данные выше количества от-
носятся к продукту, который сушился в вакуум-эксикаторе в
течение ночи.
9. По этому способу может быть также получена у-бензоилмас-
ляная кислота. Ангидрид глутаровой кислоты @,68 мол.) раство-
ряют в части взятых для реакции 350 г бензола и раствор прибав-
ляют к остальному количеству бензола, в котором предварительно
суспендируют хлористый алюминий; температуру при этом под-
держивают в течение 1,5 часа (включая и время прибавления рас-
твора) ниже 15°; таким путем получают 80—85% теоретического
выхода "е-бензоилмасляной кислоты с т. пл. 125—126°.
З-БЕНЗПИНАКОЛИН
97
3. Другие методы получения
р-Бензоилпропионовая кислота может быть получена действием
ангидрида янтарной кислоты на бензол в присутствии безводного
хлористого алюминия1 или из янтарного ангидрида и броми-
стого магний-фенила2. Она была получена также продолжитель-
ным нагреванием смеси из коричного альдегида, синильной кисло-
ты, соляной кислоты и воды 3; восстановлением бензоилакриловой
кислоты4; кетонным расщеплением бензоилянтарного эфира5; на-
греванием фенилбромпараконовой, фенилбромизопараконовой или
7-фенил-|В,у-диброммасляной кислоты с водой в; гидролизом фена-
цилбензоилуксусного эфира7 и нагреванием фенацилмалоновой кис-
лоты 8.
1 В и г с k e r, Ann. chim. phys. E) 26, 435 A882); Kohler, Engel-
b г е с h t, J. Am. Chem. Soc. 41, 768 A919).
2 Komppa, Rohrmann, Ann. 509, 263 A934).
3 Ma ts mo to, Ber. 8, 1145A875); Peine, там же 17, 2114A884).
4 Pechmann, там же 15, 889 A882).
5 P e г к i n, J. Chem. Soc. 47, 276 A885).
8 F i t t i g, Leoni, Ann. 256, 81 A890); F i t t i g, О b e r m ii 1 1 e r,
S с h i f f e г, там же 268, 74 A892).
' К a p f, P a a 1, Ber. 21, 1487 A888).
8 К u e s, P a a 1, там же 18, 3325 A885).
(CeH5J С - С (С6Н6J
он он
р-БЕНЗПИНАКОЛИН
(I,)
С6Н5СОС (СвН3K + НаО
Предложил: В. Бахман.
Проверили: Дж. Джонсон и X. Снайдер.
1. Получение
В 1-литровую круглодонную колбу, снабженную обратным холо-
дильником, помещают раствор 1 г иода в 500 мл ледяной уксусной
кислоты. Затем добавляют 100 г @,27 мол.) бензпинакона (приме-
чание 1) и колбу нагревают на металлической сетке, встряхивая ее
время от времени, пока раствор не начнет слабо кипеть. Кипячение
продолжается 5 мин., в течение которых твердый бензпинакон со-
вершенно исчезает и образуется прозрачный красный раствор (при-
мечание 2). Раствор этот немедленно переливают в 1-литровый ста-
кан, где по охлаждении бензпинаколин выделяется в виде тонких
нитей. Продукт отсасывают, промывают 2—3 раза ледяной уксус-
ной кислотой, порциями по 60 мл, пока он не станет бесцветным,
и сушат. Фильтрат сохраняют для повторных синтезов. Выход
практически чистого бензпинаколина с т. пл. 178—179° составляет
90—91 г (95—96% теоретич.). Если желательно получить более
7 Сборник № 2
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
чистый продукт, сырой бензпинаколин растворяют в 450 мл горя-
чего бензола, фильтруют и к фильтрату добавляют 250 мл горячего
лигроина (т. кип. 90—100°). По охлаждении льдом бензпинаколин
отфильтровывают и сушат. Очищенный продукт весит 82—83 г
и плавится при 179—180°.
К фильтрату добавляют еще 100 г бензпинакона и реакцию
ведут аналогичным образом. Выход бензпинаколин а во втором и
в последующих опытах 94—94,5 г (98—99% теоретич.). Этот про-
цесс может быть повторен с одним и тем же фильтратом, пока пере-
группировке не будет подвергнуто 500 г пинакона.
2. Примечания
1. Бен^пинакон, полученный фотохимическим восстановлением
бензофенона (стр. 98), может быть применен без очистки.
2. Часто в течение последней минуты кипячения бензпинако-
лин начинает кристаллизоваться в кипящем растворе.
3. Другие методы получения
р-Бензпинаколин получается перегруппировкой бензпинакона.
Эту перегруппировку можно осуществить нагреванием бензпина-
кона с хлористым бензоилом \ с хлористым ацетилом 2, с уксус-
ной кислотой при 180—200°2, с разбавленной серной кислотой при
180—200°2 и с концентрированной соляной кислотой при 200°2.
Настоящий способ основан па методе, описанном Гомбергом и Бах-
маном 3.
1 Linnemann, Ann. 133, 28 A865).
2 Thorner, Zincke, Ber. 10, 1475 A877).
3 Gomberg, Bachmann, J. Am. Chem. Soc. 49, 246 A927).
БЕНЗПИНАКОН
(Тетрафенилгликоль)
2С6Н5СОС6Н5 + СН3СНОНСН3
(со—^е^ (СеНзJ с _ с (CgH5J + СНзС0СНз
ОН ОН
Предложил: В. Бахман.
Проверили: Дж. Джонсон и X. Снайдер.
1. Получение
В 1-литровую круглодонную колбу помещают 150 г @,82 мол.)
бензофенона (примечание 1), одну каплю ледяной уксусной кислоты
(примечание 2) и 665 г (850 мл, 11 мол.) изопропилового спирта
БЕНЗПИНАКОН
99
(примечание 3); смесь нагревают до 45°, затем плотно закрывают
корковой пробкой и последнюю укрепляют проволокой или бечев-
кой; колбу помещают горлом вниз на треножник и ее содержимое
подвергают прямому действию солнечных лучей. На ярком солнце
через 3—5 час. начинается появление кристаллов бензпинакона;
через 8—10 дней, в зависимости от интенсивности освещения (при-
мечание 4), колба оказывается заполненной кристаллами бензпина-
кона. Колбу охлаждают льдом, кристаллический продукт отсасы-
вают, промывают небольшим количеством изопропилового спирта
и сушат на воздухе. Фильтрат сохраняют для следующего восста-
новления (см. ниже). Выход практически чистого бензпинакона
с т. пл. 188—190° (примечание 5) составляет 141—142 г (93—94%
теоретич.). Для большинства целей продукт достаточно чист. Его
можно подвергнуть перекристаллизации, для чего продукт рас-
творяют в 1 л горячего бензола, фильтруют и к горячему фильтрату
добавляют 400 мл горячего лигроина (т. кип. 90—100°). После
охлаждения льдом кристаллы отфильтровывают и получают 129—
130 г очищенного продукта. В результате этой очистки точка пла-
вления не изменяется.
К фильтрату, состоящему в основном из изопропилового спирта,
добавляют следующую порцию бензофенона в 150 г и раствор под-
вергают действию солнечного света в таких же условиях, как и
при первом восстановлении. Выделившийся бензпинакон отфильтро-
вывают и сушат. Выход во втором и последующих опытах рав-
няется 142—143 г (94—95% теоретич.). Этот процесс можно повторять
с одним и тем же фильтратом б—7 раз, т. е. всего можно восстано-
вить 900—1050 г бензофенона.
2. Примечания
1. Для этого синтеза можно пользоваться продажным продук-
том; однако лучше взять бензофенон, предварительно перекристал-
лизованный из спирта. Метод получения бензофенона описан в
«Синт. орг. преп.», сб. 1, стр. 99.
2. Больше одной капли уксусной кислоты прибавлять не следует.
Кислота прибавляется для того, чтобы удалить следы щелочи, кото-
рая вызывает разложение пинакона на бензофенон и бензгидрол.
3. Если нет под руками изопропилового спирта, можно взять
абсолютный этиловый спирт. При применении этилового спирта
реакция протекает медленнее и получается желтый раствор; однако
кристаллы бензпинакона получаются бесцветными.
4. Для завершения восстановления необходимы пять безоблач-
ных солнечных дней. Реакцию можно прервать в любое время, от-
фильтровать кристаллы и фильтрат вновь подвергать действию света.
5. Ввиду того, что пинакон разлагается вблизи точки плавле-
ния, последняя может изменяться в зависимости от скорости
100
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
нагревания. Приведенные выше температуры плавления были полу-
чены при медленном нагревании; если же капилляр поместить в
баню при 150° и быстро ее нагревать, то наблюдаемая точка плав-
ления или разложения повысится до 193—195°.
3. Другие методы получения
Бензпинакон был получен действием бромистого фенилмагния
на^бензил или на метиловый эфир бензиловой кислоты 1. Обычно
он получается восстановлением бензофенона, причем в качестве
восстановителей применяется цинк и серная 2 или уксусная 3 кис-
лоты, амальгама алюминия4 и магний и йодистый магний5. Настоя-
щий метод основан на исследовании Когена в фотохимической реак-
ции, открытой Чиамичианом и Зильбером 7.
1 Асгее, Вег. 37, 2761 A904).
2 Linnemann, Ann. 133, 26 A865).
3 Загуменный, ЖРХО 12, 426 A880); Вег. 14, 1402 A881).
4 Cohen, Rec. trav. chim. 38, 75 A919).
6 Gomberg, Bachmann, J. Am. Chem. Soc. 49, 241 A927).
• Cohen, Rec. trav. chim. 39, 243 A920).
' Ciamician, Silber, Ber. 33, 2911 A900).
БЕТАИНГИДРАЗИД СОЛЯНОКИСЛЫЙ
(Реактив Жирара)
(СН3K N + С1СН.,СОХ,Н3 + H,NNH2
(СН3K NCH,CONHNH2 + С2Н5ОН
I
С1
Предложил:'А. Жирар.
Проверили: Л. Физер и Р. Джекобсен.
1. Получение
В 1-литровую трехгорлую колбу, снабженную мешалкой, тер-
мометром и змеевиковым холодильником, охлаждаемым льдом
(«Синт. орг. преп.», сб. 1, стр. 401), помещают раствор 98 г (84,5мл;
0,8 мол.) этилового эфира хлоруксусной кислоты (примечание 2 на
стр. 581) в 200 мл абсолютного спирта. Раствор охлаждают при
работающей мешалке до 0°, погрузив колбу в смесь соли со льдом,
после чего мешалку останавливают и добавляют сразу 74 мл D9 г;
0,83 мол.) триметиламина, который отмеряют после предваритель-
ного охлаждения до —5°. Экзотермическую реакцию контролируют,
погружая колбу в холодную воду, так, чтобы в течение 1 часа тем-
пература смеси поднялась до 60° (примечание 1). Когда выделение
БЕТАИНГИДРАЗИД СОЛЯНОКИСЛЫЙ
101
тепла прекратится, смесь оставляют стоять при комнатной темпе-
ратуре в течение 20 час. (не добавляя более льда в рубашку
холодильника).
После этого холодильник удаляют, термометр заменяют капель-
ной воронкой и при работающей мешалке добавляют в течение
10—15 мин. 40 г @,8 мол.) 100%-ного гидразина-гидрата (примеча-
ние 2). Через 45 мин., в течение которых мешалка работает, раствор
слегка охлаждают; если при этом кристаллизация продукта реак-
ции не начинается самопроизвольно, то ее вызывают потиранием
стенок колбы стеклянной палочкой (примечание 3). Продукт выпа-
дает в виде мелких бесцветных игл. По охлаждении колбы в бане
со льдом выпавшую чрезвычайно гигроскопичную соль быстро
отсасывают, промывают на воронке Бюхнера 150 мл холодного
абсолютного спирта и отжимают досуха с помощью резиновой про-
кладки. После сушки в вакуум-эксикаторе над концентрированной
серной кислотой продукт весит 100—108 г. Если от маточного рас-
твора и промывной жидкости отогнать 200—300 мл растворителя
в вакууме водоструйного насоса, то можно выделить еще некоторое
количество кристаллов. Суммарный выход соли с т. пл. 175—
180° (с разл.): 112—120 г (83,5—89,5% теоретич.; примечания
4, 5 и 6).
2. Примечания
1. Если не применять наружного охлаждения, температура по-
вышается примерно до 75°; при этом трудно избежать некоторой
потери амина.
2. Гидразин-гидрат может быть получен аммонолизом серно-
кислого гидразина («Синт. орг. преп.», сб. 1, стр. 158) по методу,
описанному в Org. Syn. 21, 70.
Разбавленный раствор гидразин-гидрата можно концентрировать
путем перегонки с ксилолом 1. Смесь 144 мл A50 г) 42%-ного рас-
твора и 230 мл ксилола подвергают перегонке из колбы емкостью
в 0,5 л с дефлегматором Гемпеля длиной 17 см. Пробка должна быть
обернута оловянной фольгой. Ксилол увлекает с собой около 85 мл
воды; остаток после перегонки дает 45—50г80—85%-ного гидразин-
гидрата. Этот продукт, после анализа титрованием кислотой с ме-
тилоранжем в качестве индикатора можно применять как таковой
или же подвергнуть дальнейшему концентрированию (см. приме-
чание 6).
3. В случае, применения более . низкопроцентного гидразин-
Гидрата кристаллизация протекает медленнее; однако до .того мо-
мента, как начнут появляться кристаллы, раствор не¦ рекомендуется
переохлаждать..
4. Хотя продукт этот и содержит небольшое количество; сим-
метричного дигидразида, освободиться от которого перекристалли-
102
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
зацией не легко, однако он вполне удовлетворителен в качестве
реактива для выделения кетонов. Более чистый продукт с т. пл.
192° (с разл.) может быть получен, если раствор, полученный из
этилового эфира монохлоруксусной кислоты и триметиламина,
приливать к спиртовому раствору, содержащему значительный
избыток гидразин-гидрата.
5. При хранении в сухом, герметически закупоренном сосуде
этот реактив может сохраняться длительное время без заметного
разложения (запах); однако отдельные порции, взятые через неко-
торое время, лучше все же перед употреблением перекристаллизо-
вывать из абсолютного спирта.
6. В случае применения 75- и 50%-ного гидразин-гидрата
лица, проверявшие этот синтез, получили выходы соответственно в
78 и 66%.
3. Другие методы получения
Описанный метод основан на работе Жирара и Сандулеско -.
1 Н u r d, В е n n e 11, J. Am. Chem. Soc. 51, 265 A929).
3 Girard, Sandulesco, Helv. Chim. Acta 19, 1095 A936).
БРОМАЛЬ
(CH3CHOK + 9Br, ЗСВГ3СНО -f 9HBr
Предложили: Ф. Лонг и Дж. Хоуард.
Проверили: В. Хартман и Дж. Бумер\
1. Получение
В 2-литровую круглодонную колбу с тремя горлами, снабжен-
ную механической мешалкой с жидкостным затвором, капельной
воронкой и эффективным обратным холодильником, помещают
720 г B30 мл, 4,5 мол.) брома (примечание 1) и 1,5 г серы (приме-
чание 2). Верхний конец обратного холодильника присоединен
посредством трубки к ловушке для улавливания выделяющегося
бромистого водорода. Затем медленно при работающей мешалке
добавляют в течение около 4 час. 69 г F9 мл, 0,52 мол.) безводного
паральдегида (примечание 1). Реакция идет за счет выделяющегося
при этом тепла в течение всего времени прибавления паральдегида;
после этого смесь нагревают еще 2 часа, поддерживая температуру
при 60—80°. Раствор подвергают перегонке и собирают фракцию
в пределах 155—175° (примечание 3).
• После вторичной перегонки в вакууме получают 220—240 г
E2—57% теоретич.) красновато-желтого бромаля с т. кип. 59—
6279 мм или 71—74°/18 мм.
БРОМАЦЕТОН
103
2. Примечания
1. Бром сушат, взбалтывая его с концентрированной серной
кислотой; паральдегид сушат хлористым кальцием.
2. Применение серы в качестве катализатора повышает выход
на 5—10% и не вызывает никаких затруднений при очистке.
3. Головная фракция составляет 90—-180 г и состоит главным
образом из брома, бромацетальдегида и дибромацетальдегида. Если
фракцию эту обработать еще раз небольшим количеством брома,
нагревать реакционную смесь до 60—80° в течение 2 час. и подверг-
нуть перегонке, как это указано выше, то можно выделить еще не-
которое количество бромаля.
3. Другие методы получения
Бромаль был получен бромированием раствора паральдегида
в уксусноэтиловом эфире ], пропусканием паров брома через аб-
солютный спирт 2, а также действием на хлораль бромида какого-
либо металла 3.
1 Pinner, Ann. 179, 67 A875).
2 S с h a f f e r, Ber. 4, 366 A871).
3 Mu I ler; ам. пат. 2 057 964 [С. А. 31, 112A937)].
БРОМАЦЕТОН
(I-Бром-2-пропанон)
CH3COCH3 + Br2 CH3COCH.Br
HBr
Предложил: П. Левин.
Проверили: Ф. Уитмор и Дж. Холлингсхед.
1. Получение
5-литровую трехгорлую круглодонную колбу снабжают мощной
механической мешалкой, 48-сантиметровым обратным холодильни-
ком Аллина, термометром и делительной воронкой емкостью 500 мл,
трубка которой доходит почти до дна колбы (примечание 1). Колбу
ставят на водяную баню.
Через делительную воронку вводят 1600 мл воды, 500 мл ацетона
(х. ч.) и 372 мл ледяной уксусной кислоты. Мешалку пускают
в ход и повышают температуру водяной бани до 70—80°, так что
смесь в колбе нагревается приблизительно до 65° (примечание 2).
Затем осторожно прибавляют через делительную воронку 354 мл
G,3 мол.) брома, регулируя прибавление таким образом, чтобы
в колбе не накоплялся не вступивший в реакцию бром (приме-
104
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
чание 3). Прибавление продолжается от 1 до 2 час. Обычно раствор
обесцвечивается примерно через 20 мин. после прибавления всего
брома. После того как раствор обесцветился,' его разбавляют 800 мл
холодной воды, охлаждают до 10°, нейтрализуют примерно 1000 г
твердой безводной соды (на конго), отделяют с помощью делитель-
ной воронки выделившееся масло и сушат его 80 г безводного хло-
ристого кальция. После просушки масло фракционируют и собирают
фракцию, кипящую при 38—48°/13 мм. Выход 470—480 г E0—51%
теоретич.). Полученное вещество можно применять без дальнейшей ¦
очистки для получения ацетола (стр.74). Если же хотят получить
более чистый продукт, то фракционируют полученное вещество еще
раз и собирают фракцию, кипящую при 40—42°/13 мм. Выход
400—410 г D3—44% теоретич.).
Высшие фракции содержат смесь изомерных дибромацетонов.
2. Примечания
1. Прибор следует установить в хорошо действующем вытяжном
шкапу, так как и бром и бромацетон действуют сильнораздражающе
на кожные покровы и слизистые оболочки. В качестве водяной
бани можно взять оцинкованное ведро.
2. Необходимо нагреть реакционную смесь до указанной темпе-
ратуры, чтобы обеспечить ровное течение реакции.
3. Не рекомендуется допускать скопления слишком большого
избытка брома, так как в этом случае реакция внезапно может
пойти слишком бурно.
3. Другие методы получения
Бромацетон может быть получен электролизом смеси ацетона и
бромистоводородной кислоты ], а также и более обычными методами
бромирования: добавлением брома к ацетону, разбавленному
десятикратным количеством воды (по весуJ; действием брома на
ацетон, в котором имеется взвесь мрамора3; действием брома на
смесь ацетона, воды и концентрированной соляной кислоты * и
введением паров брома в холодный ацетон с помощью тока воздуха'.
В литературе описан метод получения бромацетона, аналогич-
ный приведенному выше, за исключением того, что реакционную
смесь освещают сильным источником света в.
1 Richard, Compt. rend. 133, 879 A901).
2 Соколовский, ЖРХО 8, 279, 330 A876); Вег. 9, 1687 A876).
3 S с h о 1 1, М a 11 h a i о р о и 1 о s, там же 29, 1555 A896).
'Hughes, Watson, Yates, J. Chem. Soc. 1931, 3322.
8 Emmerllng, Wagner, Ann. 204, 29 A880).
•Gohr, Thiek6tter, Biochem. Z. 305, 374 A940).
л-БРОМБЕНЗАЛЬДЕГИД
105
л-БРОМБЕНЗАЛЬДЕГИД
л-Br C6H4CH3 + 2Br2 л-Br CeHjCHBro + 2HBr
л-Вг CeH4CHBr2 + H2O . /i-Br C6H4CHO + 2HBr
Предложили: Дж.Колеман и Дж. Хонейвел.
Проверили: В. Хартман и А, Швадерер.
1. Получение
В 1-литровую колбу с тремя горлами, снабженную механиче-
ской мешалкой, обратным холодильником, термометром и капель-
ной воронкой, помещают 100 г @,58 мол.) /i-бромтолуола («Синт.
орг. преп.», сб. 1, стр. 137). Трубка капельной воронки и термометр
должны доходить почти до дна колбы. Верхний конец холодиль-
ника соединен с ловушкой для поглощения газа. При работающей
мешалке колбу нагревают на масляной бане до тех пор, пока тем-
пература жидкости не достигнет 105°. Колбу освещают 150-ватт-
ной вольфрамовой электрической лампочкой (не матовой) и медленно
приливают из капельной воронки 197 г F1,8 мл, 1,23 мол.) брома
(примечание 1). Примерно половину всего брома приливают в те-
чение первого часа, причем температуру поддерживают при 105—
110°. Остальное количество прибавляется в продолжение 2 час,
причем температуру постепенно повышают до 135°. После того как
все количество брома прибавлено, температуру медленно повы-
шают до 150°.
Сырой продукт (примечание 2) переносят в 2-литровую колбу
и тщательно смешивают его с 200 г измельченного углекислого
кальция. Прибавляют 300 мл воды и смесь осторожно нагревают
(примечание 3), а затем кипятят с обратным холодильником в те-
чение 15 час, чтобы завершить гидролиз. Продукт подвергают пе-
регонке в быстром токе водяного пара (примечание 4), и дестиллат
собирают порциями по 500 мл, охлаждают, п-бромбензальдегид
отделяют и сушат в эксикаторе. Из первого литра дестиллата по-
лучают 50—60 г л-бромбензальдегида с т. пл. 55—57°; из следую-
щих 2 л дестиллата получают еще 15—20 г продукта с т. пл. 50—
56° (примечание 5). Эту порцию можно подвергнуть очистке, пере-
ведя ее через бисульфитное соединение (примечание 6), причем
получают 13—18 г продукта с т. пл. 55—57°. Суммарный выход
чистого альдегида 65—75 г F0—69% теоретич,).
2. Примечания
1. Скорость прибавления брома следует регулировать таким
образом, чтобы в реакционной смеси не накапливалось большое ко-
личество непрореагировавшего брома. О количестве наличного
106
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
брома можно приблизительно судить по окраске раствора и по
количеству паров брома, увлекаемых в холодильник.
2. л-Бромбензальбромид является лакриматором и, кроме того,
при попадании на кожу вызывает ощущение ожога. Облегчение
приносит промывание пострадавшего места спиртом.
3. Для того чтобы не разбить колбу, ее сперва нагревают на
водяной бане, а затем на сетке горелкой, причем до начала кипения
жидкости колбу непрерывно взбалтывают. После этого жидкость
может кипеть спокойно.
4. Трубка для подачи водяного пара должна доходить до дна
перегонной колбы. Для того чтобы обеспечить хорошее перемеши-
вание тяжелого осадка, удобно на конце этой трубки раздуть ша-
рик диаметром в 16 мм с четырьмя отверстиями по 0,8 мм. Если
масса перемешивается плохо, альдегид отгоняется очень медленно.
Целесообразно соединить колбу с холодильником посредством ка-
плеотбойника Гопкинса, чтобы избежать увлечения пены во время
перегонки.
5. Если оставшийся в перегонной колбе раствор подкислить,
то можно выделить 5—10 г сырой л-бромбензойной кислоты.
6. Продукт обрабатывают насыщенным раствором бисульфита
натрия B мл на 1 г), тщательно перемешивают и примерно через
3 часа пастообразную массу отсасывают. Продукт присоединения
промывают сперва абсолютным спиртом, а затем эфиром и перено-
сят в колбу, подготовленную для перегонки с водяным паром.
Прибавляют избыток раствора соды, и альдегид отгоняют вместе
с водяным паром.
3. Другие методы получения
л-Бромбензальдегид был получен окислением л-бромтолуола
хлористым хромилом х, омылением ацеталя, полученного из бро-
мистого л-бромфенилмагния, и ортомуравьиного эфира2, окисле-
нием этил-л-бромбензилового эфира азотной кислотой3, окисле-
нием бромистого л-бромбензила азотнокислым свинцом4; гидро-
лизом п-бромбензальбромида в присутствии углекислого кальция 5
и с помощью метода, приведенного на стр. 667 и заключающегося
в окислении л-бромтолуола хромовым ангидридом в присутствии
уксусного ангидрида, с последующим омылением образовавшегося
диацетата л-бромбензальдегида.
1 Worner, Ber. 29, 153 A896).
- Чичибабин. там же 37, 188 A904); Bodroux, Bull. soc. chim.
C) 31, 587 A904); G a 11 e r m a n n, Ann. 393, 223 A912).
3 E r r e r a, Gazz. chim. ital. 17, 206 A887).
* Jackson, White, Ber. 11, 1043 A878); Am. Chem. J. 3, 32 A881).
6 A d a m s, V о I 1 w e i I e r, J. Am. Chem. Soc. 40, 1738 A918).
а-БР0МИ30ВАЛЕРИАНОВАЯ КИСЛОТА
107
а-БРОМИЗОВАЛЕРИАНОВАЯ КИСЛОТА
(СН3J СНСН (СОХаН5); + 2КОН
(CO2K)S
2С3НООН
(СН3J СНСН (СО2К), + 2НС1 - (СН8)а СНСН (СО2Н), + 2КС1
(СН3J СНСН (СОоН), + Вг2 . (СН3), СНСВг (СО2НJ + НВг
(СН8J СНСВг (СО2НJ ЛнЛТ!!а.^!>, (СН3), СНСНВгСО,Н + СО2
Предложили: И.. Марвел и В. дю Випьо.
Проверили: Ф. Уитмор и А. Грисвольд.
1. Получение
Раствор 200 г C,6 мол.) едкого кали в 200 мл воды помещают в
2-литровую колбу, соединенную с обратным холодильником. Жид-
кость нагревают до 80° и прибавляют к ней через холодильник 202 г
A мол.) изопропилмалонового эфира (примечание 1) в течение при-
мерно 1 часа. Смесь следует часто и энергично взбалтывать, чтобы
увеличить поверхность соприкосновения эфира и раствора щелочи.
Омыление протекает быстро, и в результате получается однородная
прозрачная жидкость. Ее переливают в 20-сантиметровую чашку
для выпаривания и колбу ополаскивают 50 мл воды. Полученный
раствор изопропилмалоновокислого калия и избыток едкого кали
выпаривают досуха на паровой бане (примечание 2).
Остаток растворяют в 200 мл воды, раствор переносят в 1-лит-
ровую колбу и охлаждают до 0° смесью льда и соли. Затем медленно
прибавляют смесь 400 мл концентрированной соляной кислоты уд.
веса 1,19 и 200 г колотого льда до тех пор, пока реакционная масса
не станет кислой на конго. Во время подкисления температура ре-
акционной массы не должна превышать 10е (примечание 3). При
этом выделяется хлористый калий. Смесь экстрагируют не содер-
жащим спирта эфиром два раза порциями по 200 мл и четыре раза
по 100 мл (примечание 4), чтобы извлечь изопропилмалоновую
кислоту. Соединенные эфирные вытяжки (примечание 5) помещают
в колбу, соединенную с обратным холодильником, и прибавляют
постепенно 160 г A мол.) брома с такой скоростью, чтобы эфир слабо
кипел (примечание 6), на что требуется около 2 час. После того как
бромирование закончено, эфирный раствор промывают 100 мл воды,
чтобы удалить бромистоводородную кислоту, сушат 25 г хлори-
стого кальция и отгоняют эфир на водяной бане. Сырую изопро-
пилброммалоновую кислоту нагревают в перегонной колбе на мас-
ляной бане при 125—130° до тех пор, пока не прекратится выделе-
ние углекислого газа. Образовавшуюся а-бромизовалериановую
кислоту перегоняют в вакууме, собирая фракцию, кипящую при
140—160740 мм, и перегоняют ее вторично (примечание 7). Выход
108
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
продукта, кипящего при 148—153740 лш, 125—130715 жл*, 100—
120 г E5—66% теоретич.; примечание 8).
2. Примечания
1. Изопропшшапоновый эфир может быть получен по методу,
описанному при н.-бутилмалоновом эфире (см. «Синт. орг. преп.»,
сб. 1, стр. 546). Выход продукта, кипящего при 132—135744 мм
G0—75% теоретич.).
2. Если омыление прошло не полностью, конечный продукт бу-
дет содержать немного этилового эфира бромизовалериановой ки-
слоты, который находится в низкокипящих фракциях. Если не
удалить весь спирт, то после подкисления происходит частичная
этерификация.
3. Чем выше температура, тем легче изопропилмалоновая ки-
слота теряет углекислоту, превращаясь в изовалериановую ки-
слоту, которая не бромируется в описанных условиях.
4. Эфир для извлечения следует предварительно взболтать
с одной десятой объема насыщенного раствора хлористого кальция,
чтобы удалить спирт, в присутствии которого происходит частич-
ная этерификация кислоты.
5. Нет необходимости сушить эфирный раствор до бромирова-
ния, но следует очень тщательно отделить эфирный слой от водного.
6. Обычно бромирование начинается быстро, но иногда прихо-
дится нагревать смесь после того, как прибавлены первые несколько
капель брома. Чтобы закончить бромирование, иногда приходится
смесь нагревать.
7. При первой перегонке а-бромизовалериановая кислота не
имеет постоянной температуры кипения, так как она содержит не-
которое количество неразложившейся изопропилброммалоновой
кислоты, которая теряет углекислоту во время перегонки. Пред-
варительное нагревание, даже до температуры значительно более
высокой, чем указано в методике, не ведет к ее полному разложению,.
8. Этот метод является общим для получения а-бромзамещен~
ных кислот.- В тех же условиях можно получить а-бром-н.-капро-
новую кислоту из н.-бутилмалонового эфира (выход 65—70%);
а-бромизокапроновую кислоту из изобутилмалонового эфира (вы-
ход 65—70%) и а-бром-р-метилвалериановую кислоту из вторичн.
бутилмалонового эфира (выход 75—?0%).
3. Другие методы получения
а-Бромизовалериановая кислота может быть получена дейст-
вием брома на изовалериановую кислоту 1, из тех же реагентов
в присутствии фосфора 2 или треххлористого фосфора 3 и нагре.-
ванием изопропилброммалоновой кисловд.4..
БРОМИСТЫЙ ВОДОРОД
109
1 Ley, Popoff, Ann. 174, 63 A874).
3 S с h 1 е i с h e r, там же 267, 115 A892).
3 Ore. Syn. 20, 106.
4 Koenigs, Mylo, Ber. 41, 4437 A908).
БРОМИСТЫЙ ВОДОРОД (БЕЗВОДНЫЙ)
(Бромистоводородная кислота)
Вг2
2НВг
Предложили: Дж. Рухофф, Р. Бернетт и Э. Рид.
Проверили: В. Каротерс и В. Мак-Ювен.
1. Получение
А. Аппаратура. Колбу Вюрца емкостью 125 мл (Б на рис. 4;
примечание 1) снабжают резиновой пробкой с двумя отверстиями,
через которые проходят капельная воронка на 50 мл (примечание 2)
и стеклянная трубка диаметром около б мм, причем и та и другая
доходят до дна колбы. Колбу устанавливают в стакан емкостью
Водород
Рис. 4
800 мл, который служит в качестве водяной бани. Отводную трубку
колбы Вюрца соединяют с помощью небольшого куска каучуковой
трубки с узким концом (примечание 3) трубки для сожжения В из
стекла пирекс, внутренний диаметр которой равняется 20 мм,
а длина — 30 см. Трубку наполняют кусочками пористой тарелки,
расположенными по обе стороны двух сужений трубки, как это
видно на рисунке. Трубка укрепляется с обоих концов с помощью
двух зажимов на высоте 3—4 см над бунзеновской горелкой. От-
крытый конец трубки присоединяют с помощью пробок и треххо-
дового крана (примечание 4) к вертикальной трубке Г, диаметр
по
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
которой тот же, что и у трубки В, и длина которой равна 60 см.
Эту трубку наполняют медными стружками для удаления брома,
не вступившего в реакцию в трубке для сожжения. Перед прибо-
ром устанавливают предохранительную склянку А с водой или иной
подходящей жидкостью и с отводной трубкой, направленной в от-
душину вытяжного шкафа для отвода водорода в том случае, если
произойдет где-либо закупорка аппаратуры. Кроме того, эта пре-
дохранительная склянка служит для определения давления во-
дорода (примечание 5). Водород берут из баллона, снабженного '
редукционным вентилем.
Б. Методика работы. Трубку Г отъединяют от трубки для
сожжения, повернув соответственно трехходовый кран так, что
газы идут непосредственно в вытяжной шкаф. В колбу Б нали-
вают бром, водяную баню нагревают до 38° и все время поддержи-
вают ее при этой температуре (примечание 6). Через прибор пропу-
скают слабый ток водорода, и когда трубка для сожжения напол-
нится парами брома, под пустую часть трубки для сожжения по-
мещают горелку с небольшим пламенем. Вскоре внутри нагретой
части трубки появляется небольшое желтое пламя; после того как
пары брома будут полностью вытеснены из трубки для сожжения,
последнюю соединяют с трубкой Г (примечание 7). Пламя горелки
регулируют таким образом, чтобы нижняя часть трубки была на-
грета до тёмнокрасного каления; иногда бывает нужно увеличить
его или уменьшить в зависимости от скорости работы, причем ток
водорода регулируют так, чтобы получить желательный выход.
На этой аппаратуре можно легко получить 300 г бромистого водоро-
да в час(примечание 8).
2. Примечания
1. Отводную трубку колбы Вюрца удобно согнуть таким обра-
зом, чтобы она была перпендикулярна к горлу колбы.
2. Кран капельной воронки укрепляется с помощью резинового
колечка.
3. Перегонную колбу можно присоединить к трубке для сожже-
ния с помощью каучуковой пробки, однако удобнее припаять к труб-
ке для сожжения другую трубку диаметром в б мм и соединение
осуществить с помощью небольшого куска резиновой трубки. Даже
при таком устройстве необходимо следить за тем, чтобы трубка не
забивалась. Удобнее всего осуществить соединение колбы с трубкой
с помощью стеклянного шлифа.
4. Можно обойтись и без трехходового крана, однако он об-
легчает начало работы и дает возможность быстро отвести газ в том
случае, если реакция не пойдет должным образом, и тем самым из-
бежать порчи медных стружек.
БРОМИСТЫЙ ВОДОРОД
111
5. Если работа ведется таким образом, что образуется 300 г
бромистого водорода в час, уровень воды в трубке понижается при-
мерно на 12 см.
6. Следует поддерживать давление паров брома равным 0,5 am.
Упругость паров брома при 35° равняется 324 мм, а при 40° — 392 мм.
Если баня перегреется, ее следует немедленно
охладить льдом; в противном случае будет испа-
ряться больше брома и после реакции с водородом
будет оставаться непрореагировавший бром.
Более удобная форма прибора для испарения
брома показана на рис. 5. В принципе это тот же
прибор, что и описанный выше, только сосуд с
бромом нагревается не теплой водой, а парами ки-
пящего бромистого этила C8,4°). Таким образом
не требуется наблюдения за водяной баней.
7. Медные стружки удаляют малейшие следы
брома, превращая их в черную бромистую медь.
Если уровень почерневшей меди поднимается, зто
означает, что происходит увлечение паров брома.
Однако присутствие небольшого количества вла-
ги также вызывает некоторое потемнение всей
меди.
Бромистый водород можно также освободить
от примеси брома, если пропускать его через
раствор фенола в четыреххлористом углероде (Org.
Syn. 20, 65).
Если требуется особенно сухой бромистый водород, то на его
пути устанавливают небольшую ловушку, окруженную твердой
углекислотой. В ней будет собираться вода и небольшое количе-
ство бромистого водорода. Необходимо следить за тем, чтобы ловуш-
ка не забилась.
8. Согласно литературным данным \ главное затруднение при
получении бромистого водорода непосредственно из элементов за-
ключается в том, что либо реакция соединения их проходит со взры-
вом, или пламя самопроизвольно затухает. При повторной проверке
этого метода в течение многих часов на указанной выше аппаратуре
ни с одним из этих затруднений встретиться не пришлось.
3. Другие методы получения
Полный и сжатый обзор весьма обширной литературы по полу-
чению бромистого водорода имеется в руководстве Гмелина1. Наи-
более часто применяемые методы основаны на гидролизе некоторых
бромидов, в частности, следует указать на действие брома на крас-
ный фосфор и воду. Следует также упомянуть о действии брома на
углеводороды, в частности на тетралин, а также о непосредственном
Р II С.
112
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
соединении элементов. Действие брома на тетралин является удоб-
ным методом для получения бромистого водорода в малых масшта-
бах лаборатории 2. Метод непосредственного соединения элементов
имеет некоторые преимущества ввиду своей простоты и чистоты,
особенно в тех случаях, когда требуется получить большие коли-
чества бромистого водорода. В качестве катализаторов часто
применялись платина и активированный уголь, однако при доста-
точно высоких температурах можно обойтись и без катализа-
торов 3.
Подробные указания для получения бромистого водорода бро-
мированием тетралина и непосредственным синтезом из водорода
и брома приведены в соответственном руководстве 4.
1 G m e I i n, Handbuch der anorganischen Chemie, 8 изд., часть VII,
стр. 182, Verlag Chemie, Berlin, 1931.
2 Подробности см. Н о u b e n, Die Methoden der organischen Chemie, 3-е
изд., т. Ill, стр. 1156, Verlag Georg Thieme, Leipzig, 1930.
3 Ruhofi, Burnett, Reid, J. Am. Chem. Soc. 56, 2784 A934).
* Inorganic Syntheses, I, 149, McGraw-Hill, New York, 1939.
БРОМИСТЫЙ Н.-ДОДЕЦИЛ
A-Бромдодекан)
C12H25OH
HBr
H2O
С14Нг5Вг
Предложили: Э. Рид, Дж. Рухоф и Р. Бернетт.
Проверили: В. Каротерс и В. Мак-Ювен.
1. Получение
В колбу Вюрца емкостью 500 мл, снабженную термометром и
вводной трубкой, доходящей до дна колбы (примечание 1), поме-
щают 186 г A мол.) н.-додецилового спирта (примечание 2). К от-
водной трубке колбы присоединяют алонж, другой конец которого
погружен в 75 мл воды, налитой в коническую колбу, емкостью
125 мл. Прибор собирают на каучуковых пробках. Спирт нагревают
до 100° и при 100—120° (примечание 3) пропускают через него ток
сухого бромистого водорода (примечание 4) до тех пор, пока не
прекратится поглощение последнего (примечание 5). Сырой бромид
вместе с теми продуктами, которые были увлечены в приемник,
переливают в делительную воронку, отделяют от водной бромисто-
водородной кислоты, образовавшейся во время реакции, и взбал-
тывают с */3 частью по объему концентрированной серной кислоты
(примечание 6). Нижний кислотный слой отбрасывают (примеча-
ние 7). Оставшийся бромид смешивают с равным объемом 50%-ного
БРОМИСТЫЙ Н.-ДОДЕЦИЛ
из
метилового спирта (примечание 8), после чего прибавляют водный
аммиак, время от времени взбалтывая раствор до тех пор, пока он
не станет щелочным по фенолфталеину. Содержащий бромид ниж-
ний слой отделяют, промывают один раз равным объемом 50%-ного
метилового спирта, сушат над хлористым кальцием, фильтруют и
перегоняют. Выход продукта с т. кип. 199,5—201,5°/100 мм
win 134—135°/6 мм равняется 220 г (88% теоретич.; примеча-
ние 9).
2. Примечания
1. Для более полного поглощения бромистого водорода на конце
трубки, через которую он поступает, выдувают маленький шарик,
в котором делают ряд отверстий с помощью раскаленной вольфра-
мовой проволоки.
2. Применявшийся додециловый спирт кипел при 192,5—
193,5°/100 мм или при 151—152°/21 мм. В случае худшего качества
спирта выход несколько понижается. ^Додециловый спирт может
быть получен также по методу, описанному на стр. 306.
3. Теплоты реакции достаточно, чтобы поддерживать 100° до
конца процесса.
4. Бромистый водород весьма удобно получать непосредствен-
ным соединением водорода и брома (стр. 109). Следует избегать из-
бытка водорода, так как он ведет к потере продукта в результате
улетучивания.
5. На каждый моль спирта требуется около 1,5 мол. бромистого
водорода, из которых 1 мол. идет на превращение спирта в бромид
и около 0,5 мол. на насыщение воды, образующейся при реакции.
Скорость пропускания бромистого водорода регулируют таким об-
разом, чтобы насыщение продолжалось не менее полутора часов.
Коническую колбу, которая служит приемником, предварительно
взвешивают вместе с находящейся в ней водой. По окончании реак-
ции приемник быстро прибавляет в весе и нагревается за счет
теплоты растворения бромистого водорода в воде.
6. Сырой бромид необходимо энергично взбалтывать с серной
кислотой. Роль серной кислоты в данном случае, повидимому,
заключается в том, чтобы превратить оставшийся свободный доде-
циловый спирт в кислый эфир серной кислоты, который затем рас-
творяется в 50%-ной смеси метилового спирта и аммиака.
7. Необходимо следить за тем, чтобы разделение обоих слоев
как в этом, так и в последующих случаях было полным. Несоблюде-
ние этого требования обычно бывает причиной низких выходов.
8. Применение метилового спирта в значительной степени пре-
пятствует образованию затрудняющих работу эмульсий. При ком-
натной температуре в 100 мл 50%-ного метилового спирта раство-
ряется менее 0,1 г бромистого додецила.
8 Сборник .V 2
114
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
9. Авторы получили по этому методу целый ряд бромидов со
следующими выходами:
Бромид
Циклогекс.ил
н.-Гептил
Тетрадецил
Октадецил
Выход, %
Растворимость бромидов в метиловом спирте
72—75 j Менее 1 г в 100*.ил 65°/i)-H°ro метилового
спирта
87—90
90—91
„ 0,5 г в 100 мл 50°/0-ного метилового
спирта
0,1 г в 100 мл 50°/0-ного метилового
спирта
Практически нерастворим п 90°/0-ном метиловом
спирте
Совершенно очевидно, что в случае низших бромидов желатель-
но брать метилового спирта не более того количества, которое не-
обходимо, чтобы предотвратить образование эмульсии. Весьма удобно
воду, фенолфталеин и сырой бромид поместить в делительную во-
ронку и добавить столько аммиака, чтобы раствор стал розовым.
Затем небольшими порциями добавляют метиловый спирт до тех
пор, пока не разрушится эмульсия и после взбалтывания не обра-
зуется ясной границы между обоими слоями.
3. Другие методы получения
Описанный выше метод получения бромистого н.-додецила
(лаурила) в основном заимствован у Ружички 1 и был опубликован 2.
Этот метод проще и дает более чистый продукт по сравнению со
старым методом, по которому действуют водной бромистоводород-
ной кислотой на спирт в присутствии серной кислоты 3.
1Ruzicka,Stoii,Schinz) Helv. Chim. Acta 11, 685 A928).
2 Ruhoff, Burnett, Reid, J. Am. Chem. Soc. 56, 2784 A934).
s Kamm, Marve !,там же 42, 299A920); «Синт. орг. преи.», сб. 1, стр. 112.
БРОМИСТЫЙ ИЗОБУТИЛ
A-Бром-2-метилпропан)
3 (СН3J СНСН.ОН + РВг3 > 3 (СН3J СНСН2 Вг + Р (ОН),
Предложили: К- Ноллер и Р. Динсмор,
Проверили: Ф. Уитмор, Д. Ведертчер и А. Люкс.
1. Получение
В 2-литровую трехгорлую колбу, снабженную механической
мешалкой, термометром и капельной воронкой, помещают 518 г
F43 мл, 7 мол.) сухого изобутилового спирта (т. кип. 106—108°).
БРОМИСТЫЙ ИЗОБУТИЛ
115
Колбу помещают в смесь льда с солью и охлаждают спирт до —10°;
при этой температуре медленно прибавляют при перемешивании
695 г B44 мл, 2,56 мол.) трехбромистого фосфора (примечание 1).
Скорость прибавления регулируют таким образом, чтобы темпера-
тура не поднималась выше 0° (около 4 час). Охлаждение затем пре-
кращают, а перемешивание продолжают до тех нор, пока смесь не
примет комнатной температуры, после чего ее оставляют стоять на
ночь. На другой день удаляют мешалку, воронку и термометр, а
колбу соединяют с дефлегматором длиной 30 см и холодильником.
Полученный бромистый изобутил отгоняют в вакууме из реакционной
смеси приблизительно при 50° и 200 мм (примечание 2).
Дестиллат охлаждают примерно до 0° и промывают три раза
концентрированной серной кислотой, охлажденной до 0° порциями
по 50 мл. Затем продукт взбалтывают с безводным поташем до тех
пор, пока не исчезнет запах бромистоводородной кислоты, и пере-
гоняют его или при атмосферном давлении с 1-метровой колон-
кой, собирая фракцию, кипящую при 91—937760 мм (88,5—
90,5°/728 мм), или в вакууме на адиабатической колонке длиной
70 см и диаметром 2 см с головкой для регулируемого отбора дестил-
лата (примечание 3), собирая фракцию 41—43°/135 мм. Выход
525—570 г E5—60% теоретич.; примечание 4).
2. Примечания
1. Взятый для работы трехбромистый фосфор с т. кип. 171—
173°/760 мм; 168—1707725 мм был получен с выходом 90—95% при-
бавлением брома к перемешиваемой суспензии красного фосфора
в четыреххлористом углероде. Перегонку ведут с эффективной ко-
лонкой. Долго хранившийся красный фосфор, в котором содержатся
фосфорные кислоты, дает более плохой выход.
2. В некоторых опытах сырой бромид успешно перегонялся при
атмосферном давлении; но иногда при этой перегонке он бурно раз-
лагался. Если перегонка велась в вакууме, не встречалось никаких
затруднений. Работа проводилась с водоструйным насосом, а в ва-
куумной линии устанавливался тройной кран для спуска давления.
3. Перегонка велась с колонкой, которая описана Уитмором и
Люксом [J. Am. Chem. Soc. 54, 3451 A932)]. Продукт, полученный
фракционированием в вакууме при флегмовом числе 5:1, содержал
менее 1% бромистого третич.-бутила.
4. По описанному методу могут быть получены также следую-
щие бромиды при соответствующих выходах: бромистый еторично-
бутил, т. кип. 90—93°, выход 80%; бромистый пропил, т. кип.
70—73°, выход95%; бромистый изопропил, т. кип. 60—63°, выход68%.
По описанному здесь методу с трехбромистым фосфором все пе-
речисленные бромиды получались более чистыми и с лучшими вы-
ходами, чем по кислотному методу со смесью бромистоводородной
116
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
и серной кислот («Синт. орг. преп.», сб. 1, стр. 108). Метод с приме-
нением трехбромистого фосфора не пригоден для синтеза бромистого
третий.-бутила, который при этом трудно получить в чистом со-
стоянии.
3. Другие методы получения
Бромистый изобутил может быть получен из изобутилового
спирта при действии брома и фосфора '; водного раствора броми-
стоводородной кислоты 2 и газообразного бромистого водорода3;
из изобутилена и газообразного бромистого водорода 4 или раствора
бромистого водорода в ледяной уксусной кислоте 5-; изомеризацией
бромистого третич.-бутила при 210—220°6. Целый ряд бромидов,
в том числе и бромистый изобутил, был получен действием трех-
бромистого фосфора на спирты7. Описанный метод является видо-
изменением метода, применявшегося для получения бромистого
циклопентила 8.
1 Wurtz, Ann. 93, 114 A855).
2 N о г г i s, Am. Chem. J. 38, 640 A907).
8 F о u r n i e r, Bull. soc. chim. C) 35, 623 A906); Лонгинов, Л е р-
м а н, Хим. фарм. пром., 1933, 14.
4 Brunei, J. Am. Chem. Soc. 39, 1S78 A917).
5 Ogonowsky, Ber. 36, 1988 A903).
6 Фаворский, ЖРХО 39, 469 A907); Ann. 354, 343 A907).
7 Reynolds, Adltins, J. Am. Chem. Soc. 51, 280 A929); Tseng,
H о u, J. Chinese Chem. Soc. 2, 57 A934) [C. A. 28, 3711 A934)].
«Adams, N о 1 1 e r, J. Am. Chem. Soc. 48, 1084 A926).
БРОМИСТЫЙ ФЕНАЦИЛ
(а-Бромацетофгнон )
C6H5COCH3 + Br2 ^—^ C6HsCOCH2Br + HBr
Предложили: Р. Коупер и Л. Дэвидсон.
Проверили: Л. Смит и Е. Кайзер.
1. Получение
Раствор 50 г @,42 мол.) ацетофенона («Синт. орг. преп.», сб. 1,
стр. 106) в 50 мл абсолютного эфира (примечание 1) помещают в
сухую трехгорлую колбу, снабженную делительной воронкой, меха-
нической мешалкой и обратным холодильником (примечание 2).
Раствор охлаждают в бане со льдом и солью, прибавляют 0,5 г
безводного хлористого алюминия (примечание 3) и постепенно со
скоростью около 1 мл в минуту из делительной воронки при работаю-
щей мешалке приливают 67 г брома B1,5 мл, 0,42 мол.). Окраска
брома быстро исчезает, несмотря на то, что выделяется очень мало
бромистого водорода; к концу реакции раствор становится розовым.
После того как весь бром прибавлен, немедленно удаляют эфир
и бромистый водород (примечание 4) при пониженном давлении
БРОМИСТЫЙ ФЕНАЦИЛ
117
в слабом токе воздуха. Бромистый фенацил остается в колбе в виде
твердой массы коричневатожелтых кристаллов (примечание 5); окра-
ску удаляют взбалтыванием со смесью Юмл воды и 10 мл петролейного
эфира. Кристаллы отфильтровывают с отсасыванием и, если это тре-
буется, несколько раз промывают свежими порциями той же смеси
растворителей, пока не будет получен бесцветный продукт (приме-
чание 6). Вес сырого бромистогофенацила74—80г(88—96%теоретич.).
Т. пл. продукта 45—48°. Он достаточно чист для многих целей. При
желании получить продукт более высокой степени чистоты сырой
продукт можно перекристаллизовать из 25—30 мл метанола, при-
чем можно получить 54—55 г F4—66% теоретич.) белых кристал-
лов с т. пл. 49—5Г (примечание 7).
2. Примечания
1. В качестве растворителя можно применить сухой четырех-
хлористый углерод, однако он дает менее удовлетворительные
результаты, чем абсолютный эфир.
2. В один прием можно бромировать до 200 г ацетофенона с оди-
наково хорошими выходами, но делать это, как правило, не реко-
мендуется, если только не предполагается немедленно использовать
полученный продукт, так как при стоянии он приобретает окраску.
3. В отсутствие хлористого алюминия реакция протекает мед-
ленно и неполно.
4. Если эфир и бромистый водород не удалить немедленно, рас-
твор при стоянии чернеет, и получается более низкий выход менее
чистого продукта.
5. Бромистый фенацил является лакриматором, и при работе
с ним необходимо соблюдать осторожность, избегая попадания его
на кожу и не вдыхая его паров.
6. Вода удаляет желтую окраску, обусловленную остающимся
бромистым водородом, а петролейный эфир удаляет неизменив-
шийся ацетофенон или маслянистые побочные продукты. Поскольку
продукт совершенно нерастворим в воде и очень мало растворим
в холодном петролейном эфире, его можно промывать несколько раз,
не боясь потерь.
7. При проверке синтеза было замечено, что все образцы про-
дукта, даже после перекристаллизации, темнели и окрашивались
при стоянии в вакуум-эксикаторе над хлористым кальцием, не-
смотря на то, что вначале они были бесцветными.
3. Другие методы получения
Бромистый фенацил был получен бромированием ацетофенона
без растворителя1, в сероуглероде2'3, в уксусной кислоте3'4-5 и
в других органических растворителяхъ. Количественные резуль-
таты, получаемые при бромировании в различных растворителях,
118
СИШЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
исследовал Кренке 3. Применение эфира в описанной выше методике
основано на применении этого растворителя при бромировании
дезоксибензоина 8.
Бромистый фенацил — полезный реактив для идентификации ор-
ганических кислот путем превращения их в кристаллические фена-
циловые сложные эфиры4'7.
1 Е m m е г 1 i n g, E n g 1 е г, Вег. 4, 148 A871).
2 Н u n п i u s, там же 10, 2007 A877); S t a e d е I, К I e i n s с И m iid t,
т.ш же 13, 837 A880); Staedel, там же 16, 22 A883).
3 К г й h п k e, там же 69, 921 A936).
* R a t h e r, R e i d, J. Am. Chem. Soc. 41, 77 A919).
5 M ;i h 1 a u, Ber. 15, 2465 A882); L a z e n n e c, Bull. soc. chim. D) 5,
501 A909).
6 Limpricht, Schwa nert, Ann. 155, 68 A870).
7 Shrine r, Fuson, The Systematic Identification of Organic Compounds,
pp. 130—132, John Wiley & Sons, New York, 2-nd Ed., 1940.
БР0ММЕЗИТИЛЕН
сня
-bBr,
JCH,
сн3
HgC хч /CHS
HBr
Предложил: Л. Смит.
Проверили: Р. Адаме к X. Стерне.
1. Получение
В 3-литровую трехгорлую колбу, снабженную коротким об-
ратным холодильником, механической мешалкой и делительной
воронкой, помещают раствор 636 г E,3 мол.) мезитилена (примеча-
ние 1) в 375—440 мл четыреххлористого углерода. Колбу ставят
в охладительную смесь изо льда и соли и, когда температура рас-
твора опустится ниже 10°, к нему прибавляют при энергичном пере-
мешивании раствор 900 г B88 мл, 5,6 мол.) брома в 565 мл четырех-
хлористого углерода. Бромирование протекает очень легко; выде-
ляющийся бромистый водород отводят через холодильник в ловуш-
ку с водой. На прибавление всего раствора брома требуется около
3 час; в течение этого времени температуру реакционной массы
поддерживают при 10—15°.
После того как прибавление брома закончено, смесь оставляют
стоять при комнатной температуре 1 час. Полученный светложел-
тый раствор для удаления растворенной бромистоводородной кис-
лоты промывают последовательно водой, двумя порциями 20%-ного
раствора едкого натра, по 500 мл, а затем сушат хлористым каль-
БРОММЕЗИТИЛЕН
119
цием и фильтруют. Четыреххлористый углерод отгоняют, пользу-
ясь эффективной колонкой, до тех пор, пока температура в верхней
части колонки не достигнет 120°. После отгонки четыреххлористого
углерода маслянистая жидкость часто темнеет и дымит.
Остаток прибавляют к раствору 50 г натрия в 1 л 95%-ного спир-
та. Полученную смесь в течение часа кипятят с обратным холодиль-
ником (примечание 2) и затем оставляют стоять ночь. После этого
реакционную массу разбавляют б л воды и разделяют образовав-
шиеся слои. Водный слой экстрагируют тремя или четырьмя пор-
циями четыреххлористого углерода (примечание 3) по 500 мл каж-
дая, смешивают вытяжки с первоначально отделенным броммези-
тиленом, затем тщательно промывают этот раствор водой и по отде-
лении от воды сушат хлористым кальцием. Из сухого раствора
отгоняют четыреххлористый углерод на водяной бане и остаток тща-
тельно фракционируют в вакууме, из специальной колбы Клай-
зена. Броммезитилен собирают при 105—Ю7°/1б—17 мм (приме-
чание 4). Выход 840—870 г G9—82% теоретич.). Кроме того, полу-
чаются в небольшом количестве низкокипящая фракция (около
25 г) и высококипящий остаток. Полученный по этому способу
броммезитилен не дает осадка при стоянии в течение 24 час. со
спиртовым раствором азотнокислого серебра; т. пл. от —1° до
+1°.
2. Примечания
1. Мезитилен был получен по способу, описанному в «Синт.
орг. преп.», сб. 1, стр. 242; его т. кип. 58—59°/15 мм.
2. При обработке этилатом натрия полностью удаляются соеди-
нения с галоидом в боковой цепи. Нагревание, особенно вначале,
следует вести осторожно, так как раствор иногда вспенивается.
3. Для извлечения можно взять четыреххлористый углерод,
отогнанный от сырого броммезитилена. Общий объем раствора дол-
жен быть около 2 л.
4. Температура кипения броммезитилена при других давлениях:
132°/б2 мм; 139°/70 мм; 1577100 мм.
3. Другие методы получения
Броммезитилен всегда получают премированием мезитилена
в темноте х или при дневном свете 2 или действием бромистой серы
и азотной кислоты 3 или в присутствии металлического марганца
в отсутствие растворителя *.
1 S с h r a m m, Ber. 19, 212 A886).
2 Fittig, Store r, Ann. 147, 6 A868); Smith, MacDougall,
J. Am. Chem. Soc. 51, 3002 A929).
3 Kalle и. Ко, герм. пат. 123 746 [Frdl. 6, 53 A900—1902)].
4 Duke, Lewis, Dunbar, Proc. S. Dacota. Acad. Sci. 15, 21 A935)
[C. A. 30, 2556 A936)].
120
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
ОН
СО2Н
он
4-БРОМРЕЗОРЦИН
ОН
СО2Н
он
НВг
A00')
Вг
со„
со,н
Предложили: Р. Стдин и Р. Мак-Ни.
Проверили: В. Хартман и Дж. Дикки.
1. Получение
В 1-литровую колбу, снабженную механической мешалкой и
капельной воронкой, помещают 46,2 г @,3 мол.) 2,4-диоксибензой-
ной (^-резорциновой) кислоты (стр. 430) и 350 мл ледяной уксусной
кислоты. Пускают в ход мешалку и смесь нагревают до тех пор,
пока все не растворится D5°), после чего дают ей охладиться
до 35°. Затем при энергичном перемешивании через капельную
воронку прибавляют раствор 48 г A5 мл, 0,3 мол.) брома в 240 мл
ледяной уксусной кислоты. Прибавление брома продолжается около
часа, причем в продолжение реакции температуру реакционной
смеси все время поддерживают при 30—35=. После того как все
количество брома прибавлено, раствор выливают в 5 л воды, смесь
охлаждают до 0—5° и оставляют стоять на несколько часов. Мел-
кие белые кристаллы 2,4-диокси-5-бромбензойной кислоты соби-
рают в бюхнеровской воронке диаметром в 10 см и промывают
500 мл холодной воды. Сырой продукт после сушки на воздухе при
комнатной температуре плавится при 194—200°. Вес его 55—60 г.
С целью очистки его растворяют в 1,5 л кипящей воды, раствор кипя-
тят с обратным холодильником в течение часа (примечание 1), филь-
труют горячим и охлаждают в бане со льдом. Выкристаллизовавшийся
продукт собирают, промывают 100 мл холодной воды и сушат на
воздухе. Выход бесцветной 2,4-диокси-5-бромбензойной кислоты с
т.пл.206,5—208,5°(исправл.)составляет40—44гE7—63%теоретич.).
30 г очищенной 2,4-диокси-5-бромбензойной кислоты кипятят
с обратным холодильником в течение 24 час. с 375 мл воды. Полу-
ченный раствор фильтруют, охлаждают и экстрагируют сперва
400 мл, а затем 200 мл эфира. Эфир выпаривают и сушат 4-бром-
резорцин на паровой бане. Выход продукта с т. пл. 100—102° (при-
мечание 2) составляет 22—22,5 г (90—92% теоретич.).
о-БРОМФЕНОЛ
121
2. Примечания
1. Присутствующая обычно при этом 2,4-диокси-3,5-дибромбен-
зойная кислота превращается в хорошо растворимый 2,4-дибром-
резорцин, который и удаляется. Декарбоксилирование монобром-
кислоты протекает значительно медленнее.
2. Полученный в некоторых опытах продукт имел более низкую
температуру плавления — от 77 до 93°, однако при растворении
в хлороформе с последующим выпариванием растворителя всегда
получался продукт ст. пл. 100—102°. Считать это методом очистки
вряд ли представляется возможным.
3. Другие методы получения
4-Бромрезорцин был получен бромированием монобензойиого
эфира резорцина с последующим омылением *; из 2-бром-5-амино-
фенола через диазореакцию 2; действием на резорцин дихлормоче-
вины и бромистого калия 3 и бромированием 2,4-диоксибензойной
кислоты с последующим декарбоксилированием 4. Описанная выше
методика основана главным образом на наблюдениях Раиса 4.
1 Fries, Lindemann, Ann. 404, 61 A914).
2 Fries, Saftien, Ber. 59, 1254 A926).
3 Лихошерстов, ЖОХ З, 172 A933).
i Zehenter, Monatsh. 2, 480 A881); 8, 293 A887); Hemmelmayr,
там же 33, 977 A912); 34, 374 A913); Rice, J. Am. Chem. Soc. 48, 3125
A926); Davis, H a r r i n g t о п, там же 56, 129 A934).
о-БРОМФЕНОЛ
C6H5OH + 2H2SO4 С6Н3 (ОН) (SO,H)a (I, 2, 4) 4- 2Н2О
С6Н3 (ОН) (SO3HJ 4- 3NaOH
CeHs(ONa)(SOsNaJ+ Br2
С6Н3 (ONa) (SO3NaJ + ЗН2О
C6H2 (OH) (SO3NaJ (Br) + NaBr
CeH2 (OH) (SO3Na)a (Br) -f 2H,SO4
C6H2 (OH) (SO3H).2 (Br) + 2NaHSO4
OH
OH
Br
SO,H
Br
4- 2H2SO4
SO4H
Предложили: Р. Хустон и М. Баллард.
Проверили: Л. Физер и М. Тишлер.
1. Получение
В 3-литровую колбу с тремя горлами помещают смесь из 94 г
A мол.) фенола и 350 г A90 мл, 3,4 мол.) концентрированной серной
122
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
кислоты и смесь нагревают на кипящей водяной бане при непре-
рывном механическом перемешивании. После трехчасового нагре-
вания реакционную смесь охлаждают, для чего кипящую водяную
баню заменяют баней со льдом. Когда температура реакционной
смеси достигнет комнатной, ее подщелачивают, осторожно приба-
вляя раствор 280 г G мол.) едкого натра в 700 мл воды (примечание 1).
Прибавление щелочи ведут очень осторожно, при хорошем охла-
ждении, чтобы избежать вскипания. Выпадающая вначале твер-
дая соль при дальнейшем прибавлении щелочи почти полностью
растворяется.
Щелочной раствор охлаждают до комнатной температуры, опу-
скают в жидкость термометр и при все еще работающей мешалке
приливают в течение 20—30 мин. из капельной воронки 160 г A мол.)
брома. При этом дают температуре подняться до 40—50°. После
того, как весь бром прибавлен, перемешивание продолжают еще
полчаса. К концу прибавления брома раствор должен оставаться ще-
лочным и содержать лишь небольшое количество взвешенных частиц.
Затем раствор упаривают, нагревая колбу на масляной бане,
температуру которой доводят до 150—155°. По мере выделения осад-
ка содержимое колбы начинает сильно бросать, во избежание чего
через реакционную смесь пропускают довольно сильный ток воз-
духа. Это дает также и то преимущество, что продувание ускоряет
выпаривание (примечание 2). Нагревание продолжают до тех пор,
пока в колбе не останется густая пастообразная серая масса, для
чего требуется около 30—40 мин. Смеси дают охладиться и затем
делают ее сильно кислой, добавив 800 мл концентрированной серной
кислоты. Кислоту следует добавлять медленно в вытяжном шка-
фу, так как при этом обильно выделяется бромистый водород.
Затем колбу нагревают на масляной бане, температуру которой
поддерживают при 190—210°, и через реакционную смесь пропу-
скают струю водяного пара. При этом сульфогруппы отщепляются
и бромфенол перегоняется в виде тяжелого бесцветного или светло-
желтого масла. Приблизительно через час погон становится npoj
зрачным. Продукт извлекают эфиром, эфир отгоняют на водяной
бане и остаток перегоняют при атмосферном давлении (примеча-
ние 3). Фракция 194—200° представляет собой практически чистый
о-бромфенол. Выход 70—75 г D0—43% теоретич.; примечание 4).
о-Бромфенол — бесцветная жидкость характерного запаха, довольно
быстро разлагается при хранении, приобретая бурую или красную
окраску.
2. Примечания
1. Слишком большой избыток воды в реакционной смеси ведет,
повидимому, к образованию более высокобромированных продук-
тов. При недостаточном количестве воды масса во время бромиро-
вания затвердевает, что затрудняет перемешивание.
БРОМЦИАН
123
2. Небольшое количество образовавшегося трибромфенола уда-
ляетсяпри выпаривании, так как это вещество летуче с водяным паром.
3. Перегонку следует вести по возможности быстрее, так как
0-бромфенол при высокой температуре быстро разлагается. Было
найдено, что перегонка в вакууме особых преимуществ не дает.
4. В довольно большом высококипящем остатке содержатся,
по всей вероятности, продукты более глубокого бромирования фе-
нола.
3. Другие методы получения
о-Бромфенол был получен непосредственным бромированием
фенола в различных растворителях и под влиянием различных бро-
мирующих средств \ а также при высокой температуре в отсутст-
вие растворителя 2. Он был получен также декарбоксилированием
2-бром-З-оксибснзойной кислоты3, диазотированием о-броманили-
на 4 и из о-аминофенола посредством реакции Зандмейера 6. При-
веденный выше метод является видоизменением метода, разработан-
ного Такаги и Кутани6 для синтеза 2-хлорфенола. Внесенное
улучшение состоит в применении нитробензола в качестве раствори-
теля при бромировании фенолсульфокислоты 7.
1 Hubner, Brenken, Ber. 6, 171 A873); Dinwiddie, Kastle,
Am. Chem. J. 46, 502 A911); S k г a u b, В e i f u s s, Ber. 60, 1077 A927V
Лихошерстов, ЖРХО 61, 1019 A929).
2 E.Merc k; герм. пат. 76 597 [Frdl. 3, 845 A890—94I.
3 Lellman, Grothmann, Ber. 17, 2726 A884).
4 F i 11 i g, Mayer, там же 8, 362 A875).
6 Me nd о 1 a, Streatfeild, J. Chem. Soc. 73, 685 A898).
6Takagi,Kutani, J. Pharm. Soc. Japan, No. 517, 260 A925) ГС. А
20, 2669 A926)]. '
7 H u s to n, N e e 1 e y, J. Am. Chem. Soc. 57, 2176 A935).
БРОМЦИАН
NaCN + Br2 . BrCN -j- NaBr
Предложили: В. Хартман и Е. Дреджер.
Проверили: Р. Адаме и И. Озанн.
1. Получение
2-литровую круглодонную колбу, погруженную в воду со льдом
и снабженную мешалкой, делительной воронкой и отводной трубкой,
устанавливают в хорошо действующем вытяжном шкафу. В колбу
помещают 500 г A60 мл, 3,1 мол.) брома и 50 мл воды (примечание 1).
К этой смеси постепенно прибавляют при перемешивании раствор
170 г цианистого натрия C,5 мол.) в 1200 мл теплой воды. Темпе-
ратуру реакционной смеси поддерживают ниже 30°. По окончании
реакции (около 2 час. или меньше этого) бромциан отгоняют на
водяной бане в колбу емкостью 500 мл (примечание 2). Дестиллат
124
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
нагревают примерно со 100 г безводного хлористого кальция, филь-
труют и снова перегоняют, лучше всего без холодильника, непосред-
ственно из одной перегонной колбы в другую, горло которой соеди-
нено с отводной трубкой первой колбы. Бромциан кипит при 60—
62°. Его расплавляют в приемнике (примечание 3, тяга!) и выли-
вают в нагретую заранее взвешенную склянку. Выход продукта,
представляющего собой белое кристаллическое (примечание 4),
плавящееся при 49—51° вещество, 239—280 г G3—85% теоретич.).
2. Примечания
1. Воду прибавляют для того, чтобы уменьшить испарение
брома.
2. Желательно, чтобы отводная трубка непосредственно входила
в приемник, охлаждаемый снаружи струей воды, так как бромциан
вследствие близости температур кипения и плавления очень легко
закупоривает холодильник, если он слишком длинен или если диа-
метр его мал.
3. Бромциан очень ядовит, поэтому при переливании расплавлен-
ного продукта рекомендуется надевать противогаз.
4. Бромциан нестоек и иногда даже взрывается при стоянии.
Лучше всего получать его непосредственно перед применением.
3. Другие методы получения
Бромциан может быть получен из водного раствора цианистого
калия и брома при 0°1 действием брома на цианистый калий или
натрий в присутствии четыреххлористого углерода и уксусной
кислоты 3, действием брома на влажную цианистую ртуть3. В работе
Слотта г приведены подробные указания по получению бромциана
из брома и водного раствора цианистого калия3.
1 L a n g 1 и i s, Ann. cliim. phys. C) 61, 482 A861); Scholl, Ber. 29,
1823 A896); В a u m, там же 41, 523 A908); S 1 о 11 а, там же 67, 1028A934).
a National Aniline and Chemical Company, ам. пат. 1938 324 [С. А. 28,
1148 A934)].
3 S ё r u 1 1 a s, Ann. chim. phys. B) 34, 100 A827).
Р-БРОМЭТАНСУЛЬФОКИСЛОТЫ НАТРИЕВАЯ СОЛЬ
(Натриевая оль 2-бромэтапсульфокислоты)
BrCHXH^Br-f- NaaSO3 —¦ BrCHXH.SC^Na + NaBr
Предложили: К- Марвел и М. Спарберг.
Проверили: Ф. Уитмор и Д. Лодер.
1. Получение
В 5-литровую круглодонную колбу, снабженную обратным
холодильником, механической мешалкой и делительной воронкой,
;1-БРОМЭТАНСУЛЬФОКИСЛОТЫ НАТРИЕВАЯ СОЛЬ
125
помещают 615 г C,3 мол.) бромистого этилена (примечание 1),
1250 мл 95%-ного спирта и 450 мл воды (примечание 2). Мешалку
пускают в ход и нагревают смесь до кипения. К хорошо перемеши-
ваемой кипящей смеси прибавляют через делительную воронку
раствор 125 г A мол.) безводного сернистокислого натрия в 450 мл
воды, на что требуется около 2 час. После того как весь раствор сер-
нистокислого натрия прибавлен, смесь кипятят с обратным холо-
дильником 2 часа, затем колбу соединяют с обращенным вниз хо-
лодильником и отгоняют спирт и избыток бромистого этилена (при-
мечание 3). Оставшийся водный раствор выливают в большую
фарфоровую чашку и выпаривают его досуха на водяной бане. Из
полученной сухой смеси бромистого натрия, непрореагировавшего
сернистокислого натрия и натриевой соли ?-бромэтансульфоки-
слоты извлекают последнюю 2 л кипящего 95%-ного спирта. По
охлаждении раствора большая часть соли выкристаллизовывается;
маточным раствором вторично извлекают твердый остаток. Выход
165—190 г G8—90% теоретич.). Для окончательной очистки (при-
мечание 4) продукт можно перекристаллизовать из спирта и вы-
сушить при 110° (примечание 5). Выход при перекристаллизации
75—80%.
2. Примечания
1. Большой избыток бромистого этилена необходим для того,
чтобы понизить выход этандисульфокислоты.
2. Концентрация спирта играет, повидимому, важную роль;
при попытках изменить ее в ту или другую сторону были получены
более низкие выходы.
3. При разбавлении дестиллата, содержащего спирт, водою
A0 л) можно получить обратно около 400 г бромистого этилена.
4. Этот продукт может содержать от 2 до 5% бромистого натрия,
тем не менее он достаточно чист для получения таурина (стр. 441).
Очень чистый продукт можно получить в результате вторичной кри-
сталлизации из спирта.
5. Соль слегка гигроскопична.
3. Другие методы получения
Описанный метод получения основан на данных Колера х. На-
триевая соль р-бромэтансульфокислоты была получена также при
действии бисульфита натрия на окись этилена и превращением полу-
чаемой таким образом изэтионовой кислоты в бромкислоту дей-
ствием бромистоводородной кислоты 2.
1 К о h I e r, Am. Chem. J. 20, 692 A898); Marvel, Bailey, Spar-
ta e r g, J. Am. Chem. Soc. 49, 1835 A927).
2 Rumpf, Bull. soc. chim. E) 5, 879 A938).
126
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
0-БРОМЭТИЛАМИНА БРОМИСТОВОДОРОДНАЯ СОЛЬ
B-Бромэтиламина гидробромид)
HOCH.2CH,Nri, +2HBr BrCH2CH2NHsBr + Н2О
Предложил: Ф. Кортиз,
Проверили: К. Марвел п И- Флеминг.
1. Получение
В 12-литровую круглодонную колбу помещают 7 л (9,94 кг,
52 мол.) охлажденной до 0° бромистоводородной кислоты (уд. в.
1,421; примечание 1) и при работающей мешалке добавляют из капель-
ной воронки 1 кг. A6,4 мол.) охлажденного до 0° этаноламина (приме-
чание 2). К колбе присоединяют эффективный дефлегматор и отгоняют
1850 мл дестиллата. После этого уменьшают пламя горелки, так чтобы
жидкость перестала гнаться и дефлегматор служил бы обратным хо-
лодильником. Такое кипячение продолжают 1 час, после чеГо от-
гоняют еще 700 мл и раствор вновь кипятят с обратным холодиль-
ником 1 час. Таким путем последовательно отгоняют 600, 300, 250,
150, 100 и 50 мл дестиллата. Этот процесс может быть прерван в лю-
бое время. В конце концов раствор кипятят 3 часа, после чего от-
гоняют 2300 мл сырой бромистоводородной кислоты. Общий объем
дестиллата, включая и количество, отогнанное в промежутки между
периодами кипячения с обратным холодильником, должен быть не
менее 6270 мл и не более 6330 мл.
Темноокрашенный остаток делят на две примерно равные части
и каждую из этих частей выливают еще горячей в четырехлитро-
вый стакан. По охлаждении жидкости до 70°, к каждой порции до-
бавляют по 1650 мл ацетона. Смесь хорошо перемешивают, так
чтобы как можно большее количество темноокрашенного твердого
вещества пришло бы в соприкосновение с ацетоном. После стояния
в течение ночи в холодильном шкафу бромистоводородную соль
[i-бромэтиламина отсасывают в воронке Бюхнера, промывают аце-
тоном, пока соль не станет бесцветной (примечание 3) и сушат ее на
воздухе около 15 мин. Фильтраты соединяют вместе, упаривают до
объема в I ли охлаждают. После внесения затравки можно получить
вторую порцию почти чистого продукта. Если этот второй маточник
упарить вновь до сиропообразного состояния, охладить и внести
затравку, можно получить третью порцию слегка окрашенных кри-
сталлов (примечание 4). Выход около 2800 г (83% теоретич.; приме-
чание 5).
2. Примечания
1. Удельный вес бромистоводородной кислоты должен быть не
менее 1,42.
2. Продажный этаноламин фракционируют в стеклянной аппа-
ратуре и применяют фракцию с т. кип. 167—169°.
Э-БРОМЭТИЛАМИНА БРОМИСТОВОДОРОДНАЯ СОЛЬ
127
3. Рекомендуется для более эффективного промывания перенести
продукт из воронки в ступку и раздробить его.
4. Все три порции вполне пригодны для синтеза таурина (см.
стр. 441).
5. Эта методика была применена к получению гидробромидов
следующих Й-диалкиламиноэтилбромидов:
BrCH2CH2NHR2Br
Выход, % I Длительность периодон нагревании
83
80
55
20
5J
1—1,5 часа,
1
0,75 „
1
0,5 ,
2 часа
2 .
2 „
3 „
2 „
C,H'S
н.-С3Н7
н.-С4Нв
н.-С4Н„
Последняя перегонка не всегда проводилась настолько далеко,
как того требует методика Кортиза. Если дестиллат начинал окраши-
ваться в светлобурый или фиолетовый цвет, или же если начинали
выделяться белые пары, перегонку тотчас прерывали: дальнейшая
перегонка, повидимому, приводила к разложению. Однако обычно
окраска появлялась только к концу тех периодов нагрева, которые
указаны выше, и суммарный объем дестиллата никогда не был
менее чем 95% от указанного в прописи. Поскольку растворимость
в ацетоне получаемых продуктов с увеличением молекулярного веса
резко возрастает, при экстрагировании продуктов приходится при-
менять различные количества ацетона [частное сообщение Л. Амунд-
сена и К. Кранца; см. также J. Am. Chem. Soc, 63, 305A941)].
3. Другие методы получения
Бромистоводородная соль [3-бромэтиламина была получена:
действием калий-фталимида на бромистый этилен с последующим
гидролизом1; действием бромистого водорода на этиленимин2;
из этаноламина и бромистоводородной кислоты 3; и из этаноламина
и бромистого водорода при 140—190°4. Имеется указание на то, что
получение из этиленимина и бромистого водорода протекает успеш-
но только в том случае, если имин добавляют к кислоте 5.
1 Gabriel, Ber. 21,566A888).
2 Gabriel, там же 21, 1054 A888); Gabriel, Stelzner. там же
28, 2929 A895).
3 Gabriel, там же 50, 826 A917); С о г t e s е, !. Am. Chem. Soc. 58,
191 A936).
4 I. G. Farbenind. A.-G., англ. пат. 468 387 [С. А. 31, 8545 A937)].
5 Masters, BogertJ. Am. Chem. Soc. 64, 2710 A942).
12
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
сн2
сн2 сн
I II
сн2 сн
\/
сн.
1,3-БУТАДИЕН
сн,
сн
сн
сн.
сн2
Предложили: Е. Хершберг и Дж. Рухоф.
Проверили: В. Наротерс и Дж. Хармон.
1. Получение
Циклогексен («Синт. орг. преп.», ф. 1, стр. 509, а также стр. 568
в настоящем сборнике) кипятят в колбе Б (рис. 6) и пары его про-
пускают через прибор для разложения, состоящий из раздвижной
рамки, на которой намотана реостатная лента М (примечания 1
и 2). Колба Б устанавливается на колбонагревателе B50 ватт),
снабженном реостатом. Для элемента накала берется постоянный
или переменный ток в 115 вольт, причем для контроля ставится
еще один реостат на 10 А (примечание 3). Для улавливания цикло-
гексена, не успевшего сконденсироваться в змеевиковом холодиль-
нике, газоотводную трубку Г присоединяют к трубке, доходящей
почти до дна колбы Вюрца на 500 мл, охлаждаемой в бане со льдом.
Отводная трубка колбы присоединена с помощью короткой каучу-
ковой трубки к приемнику, в котором конденсируется бутадиен.
Этот приемник состоит из большой пробирки, в которую опущена
трубка, доходящая до середины пробирки, а отводная трубка на-
чинается лишь немного ниже пробки. Через последнюю этилен
(насыщенный бутадиеном) отводят в вытяжной шкаф или наружу.
Приемник охлаждают в сосуде Дюара с твердой углекислотой и
эвтектической смесью, состоящей из равных частей по весу или по
объему хлороформа и четыреххлористого углерода. Колбу Б на-
полняют на 2/з циклогексеном, пускают в холодильник холодную
воду и доводят циклогексен до сильного кипения. После того как
воздух в приборе будет полностью вытеснен парами циклогексена
(примечание 4), включают элемент нагрева. Регулируя степень
подогрева колбы и силу тока, спираль поддерживают при ярко-
красном калении по всей ее длине. Необходимо поддерживать
энергичное стекание флегмы из холодильника во избежание излиш-
него обугливания на спирали и образования смолы на стеклянных
стенках (примечание 5). Работать можно с перерывами или же до
тех пор, пока не будет израсходован весь циклогексен в колбе.
Полученный бутадиен очищают, подвергнув его перегонке без
дефлегматора, заменив приемник в охладительной смеси на другой
1,3-БУТАДИЕН
129
совершенно аналогичный приемник, присоединенный посредством
каучуковой трубки. В этом приемнике собирают отгоняющийся
бутадиен. Продукт этот вполне пригоден для большинства целей,
в частности для реакций диенового синтеза (примечание 6). Элемент
нагрева потребляет около
500 ватт при 8,7 А, причем
производительность прибо-
pa составляет 25—30 г бу-
тадиена в час. Выход дваж-
ды перегнанного продукта,
считая на израсходован-
ный циклогексен (примеча-
ние 7),—65—75% теоретич.
После того как получено
100—150 г бутадиена, необ-
ходимо удалять из колбы
накопляющийся там высо-
кокипящий остаток (при-
мечание 8).
2. Примечания
1. Аппаратура. Прибор,
изображенный на рис, б,
сконструирован из стекла
«пирекс». Ниже даны глав-
ные размеры трубок (на-
ружные диаметры) и дру-
гие необходимые указания:
А — трубка диаметром в
32 мм; Б — колба на 500 мл;
В — трубка диаметром в
7 мм; Г — трубка диамет-
ром в 8 мм; Д — стан-
дартный шлиф № 35; Е —
змеевик из медной трубки
диаметром 5 мм (см. при-
мечание 2); Ж — корковая
пробка; 3 — латунный стер-
жень диаметром 3 мм;
Я — каучуковая трубка; К — 3 мм стеклянная трубка; Л — мед-
ные соединительные клеммы; М — лента из хромеля, 6—6,5 ом
на 1 м, ширина 1,6 мм, длина 140—150 лш (см. ниже); Н — воль-
фрамовые крючки для поддерживания спирали, вплавлен-
ные в подвешенную стеклянную палочку; диаметр проволоки
для средних крючков — 0,25 мм, для верхних и нижних —
9 Сборник Кг 2
Рис.
130
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
0,38 мм; О — медная проволока; Л — трубка диаметром в
12 мм.
Лента М элемента нагрева пропущена через вольфрамовые
крючки Я, причем сверху и снизу имеется по пять крючков и во-
семь крючков в середине. Элемент нагрева подвешен с помощью
медной проволоки к стеклянной крестовине, как это показано на
рис. б. Нижний конец не закреплен, что даст ему возможность опу-
скаться по мере удлинения спирали — это предохраняет от корот-
кого замыкания в элементе.
Состав сплава, из которого сделана нить накала, оказывает
большое влияние как на выход продукта, так и на успешную работу
прибора. Хромоникелевые сплавы служат причиной значительного
обугливания и дают плохой выход. Значительно лучшие результаты
получаются при применении железохромоникелевых сплавов, так
называемого хромеля С и нихрома.
2. Для того чтобы змеевиковый холодильник работал хорошо
также и в жаркую погоду, следует увеличить число витков да
30—40. В том виде, как он изображен на чертеже, холодильник
работает хорошо при температуре водопроводной воды, равной
4—10°.
3. Ползунковый реостат на 2 А с проволокой, намотанной на
эмалированную железную трубку, может выдержать необходимую
нагрузку, если через трубку пропускать холодную воду.
4. Если в тот момент, когда накалится нить, в приборе будет
еще заметное количество воздуха, может произойти взрыв. Кроме то-
го, элемент накала немедленно же перегорит, если в приборе не
будет достаточного количества паров циклогексена, так как ток,
проходящий по проволоке, значительно превышает норму для воз-
душной среды.
5. Если будет иметь место слишком сильное обугливание, то
следует или усилить кипячение, или слегка понизить температуру
нити накала. Как правило, скорость паров должна быть возможно
больше, лишь бы не превышалась пропускная способность медного
холодильника.
6. Сырой продукт содержит заметные количества фракций
С2, С3 и Св. Содержание в нем бутадиена колеблется от 82 до 88%.
Если необходимо получить особо чистый продукт, бутадиен превра-
щают в тетрабромид, который перекристаллизовывают и вновь
превращают в углеводород с помощью цинка и спирта 1.
7. Циклогексен, который собирается в ловушке, охлаждаемой
льдом, обычно возвращается обратно в колбу. При определении вы-
хода продукт из ловушки вместе с остатком в колбе был под-
вергнут дробной перегонке, причем был выделен чистый исходный
продукт.
8. Такой прибор может быть также использован для получения
кетена из ацетона («Синт. орг. прен.», сб. 1, стр. 226).
Н.-БУТИЛНИТРИТ
131
3. Другие методы получения
Методы получения газообразных продуктов, содержащих боль-
шие или меньшие количества бутадиена, слишком многочисленны,
чтобы был смысл дать здесь их обзор. Это особенно справедливо
ввиду развития многочисленных промышленных процессов, мало
пригодных для работы в лаборатории. Наиболее удовлетворитель-
ным процессом для работы в лаборатории по получению бутадиена
является пиролиз циклогексена 2, который, как это было доказано,
дает продукт, в основном состоящий из бутадиена '>3. Для пиро-
лиза можно и применять как вышеописанный прибор, так и прибор
Уиллиамса и Херда4. Существуют также лабораторные методы
получения бутадиена, которые исходят из хлористого бути-
ла 5, 2,3-дибромбутана6, хлористого кротила6 и 1,3-бутиленгли-
коля 7.
1 Kistiakowsky, Ruhoff, Smith, Vaughan, I. Am. Chem.
Soc. 58, 146 A936).
2 Badische Anilin- und Soda-Fabrik: герм, пат 252 499 (Chem Zentr
1912, II, 1708);
3 Зелинский, Михайлов, Арбузов, ЖОХ 4, 856 A934)
1 Williams, H u r d, J. Org. Chem. 5, 122 A940).
6Muskat, Northrup, J. Am. Chem. Soc. 52, 4043 A930).
6 Harries, Ann. 383, 176 A911); Jacobson, J. Am. Chem. Soc.
7 N a ga i, J. Soc. Chem. Ind. Japan 44, Suppl. binding 64 A941) [C A.
35,3960A941)]. '
C4H9OH + MONO
н.-БУТИЛНИТРИТ
(H2SO4)
C4H9ONO + H2O
Предложил: В. Нойес.
Проверили: К- Ноллер и Б. Вилькоксон.
1. Получение
В 3-литровую кругло донную колбу с тремя горлами, снабжен-
ную механической мешалкой (примечание 1), капельной воронкой,
доходящей до дна колбы, и термометром, помещают 380 г E,5 мол.)
нитрита натрия (х. ч.) и 1,5 л воды. Колбу окружают смесью льда
и соли и раствор перемешивают до тех пор, пока температура не
понизится до 0°. Одновременно приготовляют смесь: 100 мл
воды, 136 мл B50 г, 2,5 мол.) концентрированной серной кислоты
(уд. в. 1,84; примечание 2) и 457 мл C70 г, 5 мол.) продажного н.-бу-
тилового спирта, охлаждают эту смесь до 0° и через капельную
воронку медленно вводят ее при работающей мешалке под поверх-
132
синтезьцорганических препаратов
ность раствора нитрита. Раствор спирта прибавляют так медленно,
что практически не имеет места выделение газа и температура
поддерживается при ±Г. Обычно для этого требуется 1,5—
2 часа.
Полученную массу оставляют стоять в холодильной смеси до
тех пор, пока не произойдет расслоение. Жидкие слои сливают
с сернокислого натрия в делительную воронку (примечание 3).
Нижний водный слой сливают, а слой бутилнитрита промывают
два раза порциями по 50 мл раствора, содержащего в 100 мл воды
2 г двууглекислой соды и 25 г хлористого натрия. Продукт сушат
20 г безводного сернокислого натрия и получают практически чи-
стый бутилнитрит в количестве 420—440 г (81—85% теоретич.;
примечания 4 и 5). При желании продукт можно подвергнуть пере-
гонке в вакууме, причем 98% его переходит при 24—27°/43 мм (при-
мечание 6). Бутилнитрит кипит при атмосферном давлении при
75° и при этом частично разлагается.
2. Примечания
1. Необходимо установить мощную мешалку с соответственным
мотором, которая могла бы приводить в движение и твердые части-
цы, так как к концу реакции выделяется большое количество серно-
кислого натрия.
Щ 2. Применяемая концентрация серной кислоты достаточна,
чтобы перевести в раствор бутиловый спирт, но недостаточна для
растворения бутилнитрита.
3. Если после первой декантации выделяется еще некоторое
количество бутилнитрита, декантируют вторично. При работе с
бутилнитритом необходимо соблюдать осторожность; вдыхание
паров его может повести к сильной головной боли и сердце-
биению.
4. Эта же методика вполне пригодна и для количеств в десять
раз меньших, чем здесь указано. При маленьких загрузках можно
обойтись без механического перемешивания, так как осторожное
вращение колбы от руки уже вызывает вполне удовлетворительное
перемешивание.
5. Изоамилнитрит может быть получен по тому же методу и при-
мерно с такими же выходами.
t&*fi. Бутилнитрит медленно разлагается при стоянии, поэтому
его следует хранить в холодном месте и пускать в дело через не-
сколько дней или, в крайнем случае, через несколько недель после
его получения. Образец, который хранился при жаркой погоде
в течение пяти месяцев, содержал только 20—25% нитрита. Про-
дукты разложения состоят из окислов азота, воды, бутилового
спирта и продуктов полимеризации масляного альдегида.
н.-БУТИЛОВЫЙ ЭФИР БОРНОЙ КИСЛОТЫ
133
3. Другие методы получения
Бутилнитрит всегда получался действием азотистой кислоты
на бутиловый спирт1. Описанная здесь методика2 представляет собою
видоизмененный метод Валлаха и Отто3 для получения этилнитрита.
1 В е г t о n i, Gazz. chim. ital. 18, 434 A888); A d a m s, К a m m, 1. Am.
Chem. Soc. 40, 1285 A918).
2 N о у е s, там же 55, 3888 A933).
3 W a 1 1 а с h, Otto, Ann. 253, 251 A889).
н.-БУТИЛОВЫЙ ЭФИР БОРНОЙ КИСЛОТЫ
3C4H9OH + В(ОНK > В(ОС4Н9K + ЗН2О
Предложили: Дж. Джонсон и С. Томпкинс.
Проверили: В. Хартмйн и Дж. Дички.
1. Получение
В 2-литровую круглодонную колбу, снабженную капельной
воронкой емкостью 200 мл и дефлегматором длиной 30 см, который
наполнен стеклянными бусами (примечание 1) и соединен с холо-
дильником длиной 40—50 см, помещают 124 г B мол.) борной кисло-
ты, 666 г (9 мол.) технического н.-бутилового спирта и несколько
кусочков неглазурованной глиняной тарелки. Реакционную смесь
нагревают до слабого кипения, причем нагревание регулируют
таким образом, чтобы в час отгонялось 90—100 мл дестиллата.
Температура паров у отводной трубки дефлегматора держится при
91° все время, пока перегоняется азеотропная смесь н.-бутилового
спирта и воды C—-3,5 часа; примечание 2). Через 2 часа отделяют
верхний слой дестиллата, состоящий из н.-бутилового спирта, су-
шат его небольшим количеством безводного поташа или сернокис-
лого магния и снова приливают через капельную воронку в реак-
ционную смесь. Таким же образом н.-бутиловый спирт отделяют,
сушат и возвращают в реакционную колбу после третьего часа
нагревания.
В течение третьего часа нагревания температура у отводной
трубки дефлегматора, по мере того как заканчивается отгонка воды,
медленно повышается. Через 4 часа, когда температура перегоняе-
мых паров достигнет 110—112°, нагревание прекращают и реакци-
онную смесь переносят в 2-литровую клайзеновскую колбу, при-
чем следят за тем, чтобы продукт был возможно меньше времени
в соприкосновении с влагой атмосферного воздуха (примечание 3).
Непрореагировавший н.-бутиловый спирт отгоняют в вакууме
(примечание 4) до тех пор, пока температура не начнет быстро под-
ниматься. Тогда приемник меняют, и главная фракция н,-бутило-
134
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
вого эфира борной кислоты перегоняется почти полностью при 103—
Ю5°/8 мм или 114—115°/15 мм. В перегонной колбе остается
незначительный остаток. Вес перегнанного н.-бутилового эфира
борной кислоты, загрязненного небольшим количеством н,-бутило-
вого спирта, — 410—435 г (89—94% теоретич.). Из водного дестил-
лата и головной фракции, полученной при перегонке в вакууме,
получают обратно 190—210 г н.-бутилового спирта.
Основную фракцию н.-бутилового эфира борной кислоты пере-
гоняют вторично из клайзеновской колбы с елочным дефлегматором
(примечание 5). Первые 4—6 мл дестиллата отбрасывают. Полу-
ченный очищенный продукте т, кип. 103—105°/8 мм или 114—
115°/15 мм весит 400—425 г (87—92%теоретич.; примечание 6).
2. Примечания
1. Можно работать с хорошим дефлегматором любого типа. При
проверке оказалось, что даже дефлегматоры длиннее 30 см наиболее
эффективных типов не дают результатов лучших, чем простой гем-
пелевский дефлегматор, наполненный стеклянными бусами или
битым стеклом.
2. Дестиллат, собранный при 9Г, разделяется на два слоя:
100 мл его содержат 72 мл сырого н.-бутилового спирта, находя-
щегося в верхнем слое.
3. Так как н.-бутиловый эфир борной кислоты быстро гидроли-
зуется влагой воздуха, необходимо при манипуляциях и перели-
ваниях по мере возможности уменьшить время соприкосновения
его с воздухом.
4. н.-Бутиловый эфир борной кислоты может быть очищен пере-
гонкой и при атмосферном давлении. Однако отделение от н.-бу-
тилового спирта идет легче в вакууме. По литературным данным1,
н.-бутиловый эфир борной кислоты кипит при 190° при 200 мм, а
при атмосферном давлении — при 230—235°.
5. Удобно работать с клайзеновской колбой, на отводную трубку
которой длиной 25—30 см надета муфта холодильника и которая
соединена с приспособлением, позволяющим собирать отдельные
фракции, не прекращая перегонки и не приводя продукт в сопри-
косновение с влажным воздухом.
6. По этому же методу может быть получен и н.-амиловый эфир
борной кислоты. Из 792 г (9 мол.) н,-амилового спирта и 124 гB мол.)
борной кислоты получается 510—525 г (93—96% теоретич.) н.-ами-
лового эфира борной кислоты с т. кип. 146—148°/16лш. При реак-
ции получается обратно 210—215 г н.-амилового спирта. Во время
реакции температура отгоняемых паров держится первые 2 часа
отгонки при 95°, и дестиллат содержит сравнительно много воды
A00 мл дестиллата содержат 56 мл воды и 44 мл н.-амилового спирта).
н.-БУТИЛСУЛЬФАТ
135
Через 2 часа температура медленно поднимается до 136—137°. Не
нужно и невыгодно возвращать полученный из дестиллата н.-ами-
ловый спирт обратно в реакционную смесь.
3. Другие методы получения
Вышеописанный способ приведен в главных чертах в патенте г.
н.-Бутиловый эфир борной кислоты может быть получен также при
действии н.-бутилового спирта на триацетилборную кислоту, или
на ангидрид борной кислоты.
1 W. J. В а п n i s t е г; ам. пат. 1 668 797 [С. А. 22, 2172 A928)].
н.-БУТИЛСУЛЬФАТ
(C4H9JSO3 + SO.2C12 > C4HeOSOaCl + С4Н9С1 + SO2
C4H9OSOaCl -t- (C4H9J SO3 - (C4H9J SO4 -r C4H9 Cl -{- SO2
Предложили: К- Сутер и X. Герхарт.
Проверили: К. Ноллер и М. Спйнерхолм,
1. Получение
В 2-литровую трехгорлую колбу, снабженную капельной во-
ронкой, механической мешалкой с ртутным затвором и холодиль-
ником, помещают 625 г C,2 мол.) н.-бутилсульфита (стр. 136).
Холодильник присоединяют к ловушке для поглощения газов и
в течение 30 мин. при энергичном перемешивании и охлаждении водо-
проводной водой в колбу приливают 217 г A31 мл, 1,6 мол.) хлори-
стого сульфурила (примечание 1). Затем капельную воронку заме-
няют термометром, шарик которого погружен в жидкость, и колбу
медленно нагревают при работающей мешалке до тех нор, пока
хлористый бутил не станет стекать сильной струей из обратного
холодильника (около 100—110°). При этом происходит обильное выде-
ление сернистого газа. Холодильник заменяют на елочный дефлег-
матор высотой 40 см и обращенный вниз холодильник; температуру
постепенно повышают до 130—135° и поддерживают ее до тех пор,
пока не прекратятся выделение сернистого газа и отгонка хлори-
стого бутила. Нагревание и перегонка продолжаются примерно
около 2 час. Остаток от перегонки, который представляет собой
сырой н.-бутилсульфат, охлаждают от комнатной температуры и
добавляют 100 мл насыщенного раствора углекислого натрия. Смесь
перемешивают 10мин., выливают ее в делительную воронку и оста-
вляют на 30 мин. для лучшего расслоения. Верхний слой сушат
хлористым кальцием по крайней мере в течение ночи, затем остав-
136
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
ляют стоять сутки с 15 г углекислого натрия или калия, время от
времени взбалтывая колбу. Продукт фильтруют в видоизмененную
колбу Клайзена емкостью 500 мл с дефлегматором высотой 25 см
и затем перегоняют с масляной бани. После первой перегонки полу-
чают 250—280 г G4—83% теоретич.) н.-бутилсульфата с т. кип.
ПО—114°/4 мм, обладающего резким запахом. Вторичная
перегонка дает чистый продукт со слабым запахом сложного эфира
ист. кип. 109—11174 мм, причем потери только механические
(примечание 2).
2. Примечания
1. Продажный хлористый сульфурил перегонялся, и была
использована фракция с т. кип. 69—70°.
2. Авторы, предложившие синтез, указывают, что аналогичным
путем может быть получен н.-пропилсульфат с выходом 66—70%.
В этом случае при вторичной перегонке берут короткий дефлег-
матор, для того чтобы удалить высококипящие примеси. Т. кип,
н.-пропилсульфата 88—9Г/4 мм.
3. Другие методы получения
н.-Бутилсульфат был получен при действии н.-бутилового
эфира хлорсульфоновой кислоты на н.-бутиловый эфир ортомуравьи-
ной кислоты или на н.-бутилсульфнт 1. Он был также получен окис-
лением н.-бутилсульфита перманганатом калия в растворе ледяной
уксусной кислоты2. Описанный выше метод, повидимому, дает
наиболее удовлетворительные результаты при работе в лабораторных
условиях.
1 L e v a i I I a n t, Compt. rend. 197, 648 A933); Barkenbus, Owen,
J. Am. Chem. Soc. 56, 1204 A934).
3 Evans, Ph. D. Dissertation, Northwestern University, 1935.
н.-БУТИЛСУЛЬФИТ
2C4H9OH + SOC12 > (C4H9J SOS'+ 2HC1
Предложили: К- Сутер и X. Герхарт.
Проверили: К- Ноллер и М. Сайнерхолм.
1. Получение
В 2-литровую трехгорлую колбу, снабженную механической
мешалкой с ртутным затвором, термометром, холодильником и
капельной воронкой, помещают 684 г (845 мл, 9,2 мол.) сухого
н.-БУТИЛСУЛЬФИТ
137
н.-бутилового спирта (примечание 1). Холодильник присоединяют
к ловушке для поглощения хлористого водорода и в течение 2 час.
при работающей мешалке приливают 500 г C05 мл, 4,2 мол.) хло-
ристого тионила (примечание 2). Во время прибавления первой
половины хлористого тионила температуру реакционной смеси
поддерживают при 35—45°, погружая колбу в ледяную воду (при-
мечание 3). Когда начнется выделение хлористого водорода, водя-
ную баню удаляют и ту же температуру поддерживают нагреванием
небольшим пламенем горелки. После того как весь хлористый тио-
нил прибавлен, для завершения реакции температуру постепенно
в течение 30 мин. повышают до кипения,что служит и для удаления
оставшегося количества хлористого водорода. Реакционную смесь
переносят в 1-литровую видоизмененную колбу Клайзена с дефлег-
матором высотой 25 см и подвергают ее дробной перегонке в вакууме.
После головной фракции, состоящей в основном из непрореагиро-
вавшего спирта, получают 625—689 г G7—84% теоретич.) н.-бутил-
сульфита с т. кип. 109—115715 мм. После повторной перегонки
получают 585—674 г G2—83%) продукта с т. кип. 109—112714 мм
(примечание 4).
2. Примечания
1. Продажный н.-бутиловый спирт с целью осушки был перегнан
с дефлегматором; головная фракция и небольшая фракция выше
117° были отброшены.
2. Продажный хлористый тионил перегонялся и была взята
фракция с т. кип. 78—80°.
3. До начала выделения газа происходит выделение значитель-
ного количества тепла, после чего реакция протекает с поглощением
тепла.
4. Авторы этого синтеза указывают, что с помощью той же
методики н.-пропилсульфит получается из н.-пропилового спирта,
но с несколько меньшим выходом.
3. Другие методы получения
н.-Бутилсульфит был получен из бутилового спирта и хлори-
стого тионила по указанному выше методу 1, а также с применением
абсолютного эфира в качестве растворителя и сухого пиридина для
поглощения выделяющегося хлористого водорода 2. Метод с пири-
дином был проверен, однако его преимущества не оправдывают
лишних затрат на эфир и пиридин. Бутилсульфит был так-
же получен действием хлористой серы на бутиловый спирт 3.
138
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
1 voss, Blank с, Ann. 485, 258 A931); Barkenbus, Owen,
I Am. Chem. Soc. 56, 1204 A934).
2 G e r r a r d, J. Chem. Soc. 1939, 99.
3 Bert, Compt. rend. 178, 1827 A924).
Н.-БУТИЛФОСФАТ
3C4H9OH + POC13+ 3C&H5N > PO(OC4H9K + 3C5H5NHCl
Предложили: Г. Дюттон и К. Ноллер.
Проверили: Дж. Джонсон и А. Хант.
1. Получение
В 2-литровую круглодонную колбу, снабженную обратным
холодильником, механической мешалкой с жидкостным затвором,
капельной воронкой (примечание 1) и термометром, помещают 222 г
B74 мл, 3 мол.) сухого н.-бутилового спирта, 260 г B65 мл, 3,3 мол.)
пиридина и 275 мл сухого бензола (примечание 2). Пускают в ход
мешалку и колбу охлаждают смесью льда и соли так, чтобы тем-
пература жидкости понизилась до —5°. При энергично работающей
мешалке прибавляют по каплям 153 г (91 мл, 1 мол.) хлорокиси
фосфора (т. кип. 106—107°) с такой скоростью, чтобы температура
не поднималась выше 10°. После того как все количество хлорокиси
прибавлено, реакционную смесь медленно нагревают до кипения
и эту температуру поддерживают в течение 2 час, затем смесь охла-
ждают до комнатной температуры и для растворения хлористово-
дородной соли пиридина добавляют 400—500 мл воды (примеча-
ние 4). Бензольный слой отделяют, промывают его 100—150 мл
воды (примечание 5) и сушат над 20 г безводного сернокислого
натрия.
Бензол и другие низкокипящие примеси отгоняют под давлением
в 40—50 мм до тех пор, пока температура в парах не достигнет
90°. Фракцию, содержащую н.-бутилфосфат, собирают при 160—
162715 мм или 143—14578 мм; выход ее 190—200 г G1—75%
теоретич.; примечание 6).
2. Примечания
1. Трубка капельной воронки должна оканчиваться достаточно
высоко над поверхностью реакционной смеси, чтобы избежать
кристаллизации в ней хлористоводородной соли пиридина. Целе-
сообразно пользоваться таким термометром, у которого шкала с де-
лениями выше — 5° находилась бы над пробкой. В противном слу-
чае пары в колбе и выкристаллизовывающаяся соль пиридина могут
затруднить чтение показаний термометра.
н.-БУТИЛФОСФАТ
139
2. Реагирующие вещества и растворитель высушивались по-
средством перегонки; были взяты фракции, кипящие в преде-
лах 1°.
3. Первые 10—15 мл хлорокиси фосфора следует прибавлять
очень медленно, чтобы избежать бурной реакции и перегрева.
Следует также избегать слишком низкой начальной температуры,
так как непрореагировавшая хлорокись фосфора может накапли-
ваться и затем вызвать бурную реакцию. Мешалка должна быть
таких размеров и работать с такой скоростью, чтобы теплота реак-
ции быстро и равномерно распределялась по всей массе, но чтобы
твердые частицы (и окклюдированные ими реагенты) не разбрыз-
гивались по верхней части стенок колбы.
4. Можно регенерировать около 50% взятого в реакцию пиридина,
если водный раствор упарить на паровой бане, обработать его кон-
центрированным раствором едкого натра и подвергнуть перегонке
выделившийся слой пиридина.
5. До перегонки раствор должен быть нейтральным. Присут-
ствие хлористого водорода ускоряет разложение сложных эфиров
фосфорной кислотых. Бензольный раствор не следует промы-
вать щелочными реагентами, например раствором соды, так как
щелочные реагенты также вызывают разложение при пере-
гонке.
6. Данный метод является общим для получения алкилфосфа-
тов. Пользуясь аналогичной методикой, можно получить из соот-
ветствующих спиртов н.-пропилфосфат с выходом 60—65%; вторич-
ный бутилфосфат — с выходом 40—45% и н.-амилфосфат — с вы-
ходом 60—65% а.
3. Другие методы получения
н.-Бутилфосфат был получен действием пятихлористого фосфо-
ра пли хлорокиси фосфора на н.-бутиловый спирт3, действием
хлорокиси фосфора на бушлат алюминия 4 или бутилат натрия 5
и окислением бутилфосфита6. Описанная выше методика2 ана-
логична той, которая применяется для получения алкилфос-
фитов7.
1 В а 1 а г с v, Z. anorg. allgem. Chem. 101, 227 A917).
2 N о 1 1 е г. D u t t о n, J. Am. Chem. Soc. 55, 424 A933).
3 N i с о 1 a i; ам. пат. 1 766 720 [С. А. 24, 4053 A930I; Celluloid Corpora-
tion; англ. пат. 455 014 [С. А. 31, 1427 A937I.
1 В a n n i s t e г; ам. пат. 1 799 349 [С. А. 25, 3014 A931)].
5Evans, Davies, Jones, J. Chem. Soc. 1930, 1310.
8 Chemische Fabrik von Heyden A.-G.; англ. пат. 398 659 [С. А 28,
13C2 A934)], и герм. пат. 605 174 fC. A. 31, 3066 A937)).
7 М i 1 о b e n il s k i, Sachnowski, Chernik Polski 15, 34 A917)
(C, A. 13, 2865 A919)].
140
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
БУТИРОИН
(Октанол-5-он-4)
2С3Н7СОАН, + 4Na
C3H7CONa
C3H-,CONa
2C.,H5ONa
H2SO.
C3H7CONa
C3H7COH C3H7C = О
C3H7CONa
СЯН,СОН
C3H7CHOH
Предложили: Дж. Снелл и С. Мак-Эльвен.
Проверили: Ц. Марвел и М. Лемйн.
1. Получение
В 3-литровую трехгорлую круглодонную колбу, снабженную
длинным обратным холодильником и эффективной механической
мешалкой, помещают 92 г D гр.-ат.) чистого металлического натрия
и около 150 мл ксилола. Натрий распыляют, нагревая ксилол до
плавления металла и охлаждая затем смесь при очень энергичном
перемешивании. Охлажденный ксилол декантируют, а распыленный
натрий тщательно промывают 4—5 раз сухим, не содержащим спирта,
эфиром. Затем приливают 1200 мл абсолютного эфира (примечание 1)
и колбу снабжают обратным холодильником, делительной воронкой
емкостью 250 мл и механической мешалкой (примечание 2).
Мешалку пускают в ход и через делительную воронку медленно
приливают 232 г B мол.) очищенного этилового эфира н.-масляной ки-
слоты (примечание 3). Рекомендуется сначала сразу прибавить 25 мл
эфира, при этом выделяющееся при реакции тепло доводит эфир
до кипения. Дальнейшее прибавление ведут с такой скоростью,
чтобы поддерживалось все время слабое кипение. Перемешивание
продолжают до тех пор, пока не кончится реакция и весь натрий
не перейдет в объемистый желто-белый осадок, который начинает
выпадать почти сразу, как только начнется реакция (примечание 4).
Реакционную колбу помещают в баню со льдом и при сильном
перемешивании к содержимому осторожно приливают через дели-
тельную воронку охлажденный раствор 210 г серной кислоты уд. в.
1,84, в 350 мл воды. Мешалку удаляют и колбу оставляют во льду
до тех пор, пока не закристаллизуется нижний слой, т. е. водный
сернокислый натрий Na2SOi-10H2O. Затем эфирный раствор декан-
тируют, а кристаллы сернокислого натрия промывают 100—200 мл
эфира.
Основной раствор соединяют с эфиром, полученным после про-
мывки, все взбшпывают со 100 мл 20%-ного раствора соды (приме-
чание 5) и сушат безводным поташом. Затем эфир и спирт быстро
БУТИРОИН
141
отгоняют, а остаток фракционируют в вакууме в видоизмененной
клайзеновской колбе емкостью 250 мл (примечание 6). Главная
фракция кипит при 80—86°/12 мм. Собирают также следующую
фракцию до температуры примерно на 15° выше температуры
кипения главной фракции. Выше и ниже кипящие фракции можно
фракционировать еще раз, чтобы получить дополнительно некоторое
количество бутироина. Общий выход 94—101 г F5—70% теоретич.).
Получаемый продукт окрашен следами дикетона в желтый цвет
(примечания 7 и 8).
2. Примечания
1. Реакцию можно вести и в бензоле, но она проходит значи-
тельно медленнее, чем в эфире.
2. Для распыления натрия лучше всего брать маленькую быстро-
действующую мешалку; для перемешивания реакционной смеси —
сравнительно большую, более медленно работающую мешалку.
3. Этиловый эфир масляной кислоты очищают следующим об-
разом: промывают дважды 10%-ным раствором соды, дважды рав-
ным объемом насыщенного раствора поваренной соли и сушат 24
часа над безводным поташом. Поташ отфильтровывают, а эфир
оставляют стоять на ночь над фосфорным ангидридом B% от веса
эфира). На другой день эфир отгоняют с дефлегматором над фосфор-
ным ангидридом и собирают фракцию, кипящую в пределах 2°
или даже более узко кипящую фракцию. При работе с менее тща-
тельно очищенным эфиром получают меньший выход бутироина.
4. На прибавление эфира требуется 1,5—2 часа, после чего
смесь нагревают еще 1 час.
5. В реакционной смеси имеются небольшие количества масля-
ной и дипропилгликолевой кислот.
6. Эфир следует отогнать как можно скорее и перегонку вести
не слишком медленно, так как продолжительное нагревание благо-
приятствует образованию высококипящего побочного продукта
неизвестного строения, который понижает выход бутироина.
7. Обычно количество дикетона очень незначительно. Дикетон
можно удалить, энергично взбалтывая в течение часа (с переры-
вами) бутироин со 100 мл насыщенного раствора бисульфита натрия.
Затем бутироин промывают концентрированным раствором поварен-
ной соли и опять перегоняют.
8. По этому же способу были получены:
пропионоин т. кип. 60—65712 мм, выход 50—55%
изобутироин » » 70—75714 мм, » 70—75%
пивалоин » пл. 80—81°; » 52—60%
т. кип. 85—95712 мм.
142
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
3. Другие методы получения
Алифатические ацилоины могут быть получены омылением про-
дукта реакции между натрием и влажными эфирными растворами
хлорангидридов кислот. Первичным продуктом в данном случае
является диэфир дизнольной формы ацилоина \ Больший пре-
паративный интерес представляет реакция между эфирными
растворами алифатических сложных эфиров и натрием2 или
калием 3.
1 К 1 i n g e r, S с h m i t z, Ber. 24, 1273 A891); Basse, KHneer,
там же 31, 1218 A898); Anderlini, Gazz. chim. ital. 25 (II) 51, 128A895):
Его p OB а, ЖРФХО 60, 1199A928).
о 2rB°oVeau ' t, Locqu i n, Bull. soc. chim. C) 35, 629A906); Fe i g 1,
™'»f' 2299 A925); Corson, Benson, Goodwin, 1. Am. Chem. Soc.
52, J98o A930).
"Scheibler, Emden, Ann. 434, 265 A923).
ВЕРАТРОВЫЙ АЛЬДЕГИД
C,4-Диметоксибензальдегид )
СНО
OCH4
(CH3), SO4 + NaOH
OH
СНО
/\
1
осн3
ОСН8
Предложил: Дж.
Проверили: Дж.
г- CH3SO,
Бек.
Джонсон
,Na +
и X.
Н20
Снайдер.
1. Получение
В 3-литровой трехгорлой колбе нагревают на водяной бане смесь
182 г A,2 мол.) ванилина (примечание 1) и 450 мл кипящей воды
Растворяют 150 г едкого натра (х. ч.) в 200—300 мл воды и затем
разбавляют раствор водой до 750 мл. Из этого раствора 360 мл
нагревают до 100° и сразу приливают к горячей смеси ванилина и
воды (примечание 2). Колбу снабжают обратным холодильником,
механической мешалкой и делительной воронкой емкостью 250 мл
. ВЕРАТРОВЫЙ АЛЬДЕГИД
143
Нагревание на водяной бане продолжают и к раствору из дели-
тельной воронки прибавляют 189 г A42 мл) диметилсульфата (при-
мечание 3) с такой скоростью, чтобы все время поддерживалось
слабое кипение, которое начинается после прибавления первых
10—15 мл (примечание 4). После прибавления диметилсульфата,
которое продолжается около 1 часа, смесь нагревают еще 45 мин.
и затем прибавляют с такой же скоростью, как и в первый раз,
еще 39 г C0 мл) диметилсульфата. К концу прибавления реакция
смеси должна быть кислой на лакмус (примечание 5). Нагревают
еще 10 мин. и слегка подщелачивают смесь, прибавляя 60 мл при-
готовленного ранее раствора едкого натра. Затем приливают еще
39 г диметилсульфата. Попеременное прибавление раствора едкого
натра и диметилсульфата (каждый раз по 39 г) повторяют еще два
раза, так что в общем прибавляют 345 г B,7 мол.) диметилсульфата.
Реакционную смесь после прибавления последней порции диметил-
сульфата делают сильно щелочной прибавлением 150 мл раствора
едкого натра и нагревают еще 20 мин. Затем быстро охлаждают
при постоянном перемешивании до 25° (примечание 6) и вератро-
вый альдегид экстрагируют три раза эфиром, порциями по
300 мл.
Соединенные эфирные вытяжки сушат безводным сернокислым
магнием и эфир отгоняют. После отгонки остается светложелтое,
быстро закристаллизовывающееся масло. Выход вератрового аль-
дегида, плавящегося при 43—44,5°, составляет 164—173 г (82—87%
теоретич.). Продукт достаточно чист для большинства целей, однако
может быть еще лучше очищен с небольшой потерей перегонкой
в вакууме. Из 164 г первоначального продукта получают 156 г
чистого вератрового альдегида, кипящего при 15378 мм и плавя-
щегося при 46° (примечание 7). Так как альдегид легко окисляется
на воздухе, его следует сохранять в плотно закупоренных или
запаянных сосудах.
2. Примечания
1. Для работы надо брать ванилин хорошего качества (т. пл.
8 1—82°).
2. Реагенты смешивают в горячем состоянии, для того, чтобы
в осадок не выпала натриевая соль ванилина.
3. Обычный технический диметилсульфат дает удовлетворитель-
ные результаты. Диметилсульфат очень ядовит, и необходимо тща-
тельно остерегаться, чтобы не вдыхать его паров или не распле-
скать его раствор. Специфическим противоядием служит аммиак.
Лучше всего проводить работу в хорошо действующем вытяжном
шкафу.
4. Если жидкость не кипит, выход обычно получается меньше.
5. Для того чтобы получить хороший выход, очень важно,
144
к СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
чтобы реакционная смесь несколько раз становилась кислой.
Поэтому последние 156 г диметилсульфата прибавляют в четыре
приема небольшими порциями и после каждого прибавления реак-
ционной смеси дают сделаться кислой. Удобнее всего определять
реакцию смеси, опуская в нее через трубку холодильника
стеклянную трубку, в конец которой вставлена лакмусовая
бумажка.
6. Если реакционную смесь слишком медленно охлаждать до
25° или охлаждать до температуры ниже 25°, то вератровый альде-
гид может закристаллизоваться во время охлаждения. В этом слу-
чае рекомендуется перед экстракцией эфиром нагреть смесь до
расплавления альдегида.
7. Вератровый альдегид достаточной степени чистоты, пригод-
ный для большинства синтетических целей, можно быстро и удобно
получить по методу Барджера и Зильбершмидта. [J. Chem. Soc,
133, 2924 A928)]. В 1-литровую трехгорлую круглодонную колбу
(или широкогорлую склянку), снабженную механической мешалкой,
обратным холодильником и двумя делительными воронками, поме-
щают 152 г A мол.) ванилина, который расплавляют, нагревая его
на водяной бане. Затем при энергичном перемешивании приливают
раствор 92 г A,5 мол.) 90%-ного едкого кали в 150 мл воды со ско-
ростью 2—3 капель в секунду. Через 20 сек. после начала прили-
вания щелочи прибавляют 160 г A20 мл, 1,25 мол.) диметилсуль-
фата примерно с такой же скоростью (непосредственно перед упо-
треблением диметилсульфат промывают равным количеством по
объему ледяной воды, затем 7з объема холодного насыщенного
раствора двууглекислой соды). Через несколько минут наружный
обогрев прекращают, и смесь продолжает кипеть за счет теплоты
реакции. Вскоре масса мутнеет, и после того как прибавлено при-
мерно половинное количество диметилсульфата, жидкость разде-
ляется на два слоя. Прибавление обоих реагентов должно быть
закончено примерно через 20 мин.
Окраска реакционной смеси пурпурно-бурая вначале к концу
реакции сразу становится желтой. Появление временной зеленовато-
желтой окраски в первой стадии реакции указывает на то, что
раствор стал кислым. В этом случае необходимо увеличить ско-
рость приливания щелочи. Конечная желтая окраска является
постоянной, и к концу реакции смесь дает щелочную реакцию
на лакмус.
Реакционную смесь немедленно переносят в большой стакан,
покрытый часовым стеклом, и дают ей охладиться в спокойном
состоянии, лучше всего оставив ее на ночь. Твердую кристалли-
ческую массу вератрового альдегида отделяют, растирают в ступке
с 300 мл ледяной воды, фильтруют с отсасыванием и сушат в ваку-
ум-эксикаторе. Выход 152—158 г (92—95% теоретич.). Продукт
плавится при 42,5—43,5° и дает оксим (т. пл. 89—90°) с выходом
ВЕРАТРОНИТРИЛ
145
90% и нитрил вератровой кислоты (т. пл. 65—66°) с выходом 68—
70% (ср. следующий синтез). Полученный таким образом
нитрил вполне пригоден для синтеза аминовератрола (стр. 32,
Дж. Джонсон и X. Стивенсон, частное сообщение).
3. Другие методы получения
Вератровый альдегид может быть получен действием на вера-
трол цианистого водорода в присутствии хлористого алюминия *;
конденсацией вератрола с формилпиперидином с последующим
омылением полученного продукта2; метилированием ванилина
йодистым метилом 3, диметилсульфатом 4, метиловым эфиром л-то-
луолсульфокислоты 5 или гидроокисью триметилфениламмония в.
1 Oattermann, Ann. 357, 367 A907).
2 Akabori, Senoh, Bull. Chem. Soc. Japan 14, 166 A939) [C. A. 33,
6270 A939)].
3 T i e m a n n, Ber. 8, 1135A875); 11, 663 A878); Ju 1 iu s be r g, там же
40, 119 A907).
* Kostanecki, Tambor, там же 39, 4022 A906).
5 Каневская, Arch. Pharm. 271, 462 A933).
6 Родионов, Bull. soc. chim. D) 45, 116A929).
CHO
n/.och8
OCH3
CH = NOH
7Ч.
ВЕРАТРОНИТРИЛ
C,4-Диметоксибензонитрил )
,ОН
(CH3COJO
0CH3
^ ОСН3
ОСН3
CN
ч/;осн3
осн3
1,0
2 СН3СО2Н
Предложили: Дж. Бек и В. Айди.
Проверили: Дж. Джонсон и Е. Амштутц.
1. Получение
В 1-литровой круглодонной колбе растворяют 83 г @,5 мол.)
вератрового альдегида (стр. 142) в 200 мл теплого 95%-ного спирта;
туда же добавляют теплый раствор 42 г @,6 мол.) солянокислого
10 Сборник № 2
146
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
гидроксиламина («Синт. орг. преп.», сб. 1, стр. 164) в 50 мл воды.
Оба раствора хорошо смешивают, после чего к смеси добавляют
раствор 30 г @,75 мол.) едкого натра в 40 мл воды. Смесь оставляют
стоять при комнатной температуре в течение 2,5 часа, затем доба-
вляют к ней 250 г измельченного льда и раствор насыщают углекис-
лым газом. Альдоксим выделяется в виде масла, которое затверде-
вает при стоянии в течение ночи в холодильном шкафу (примеча-
ние 1). Кристаллический оксим отсасывают, тщательно промывают
водой и сушат на воздухе. Выход оксима 88—89 г (97—98% теоре-
тич.).
Альдоксим вератрового альдегида помещают вместе со 100 г
уксусного ангидрида (94—96%-ного) в круглодонную колбу емкостью
300 мл с воздушным холодильником, присоединенным к колбе с по-
мощью шлифа (примечание 2), и массу осторожно нагревают,
Происходит бурная реакция, во время которой горелку оставляют.
После того как реакция несколько успокоится, раствор не сильно
кипятят в течение 20 мин. и затем при постоянном помешивании
осторожно выливают в 300 мл холодной воды. Перемешивание про-
должают, и, по мере охлаждения, нитрил выделяется в виде мелких
почти бесцветных кристаллов, которые отфильтровывают и сушат
на воздухе. Полученный таким образом вератронитрил является
вполне чистым; выход его 57—62 г G0—76% теоретич., считая на
вератровый альдегид). Плавится он при 66—67°,
2. Примечания
1. Иногда оксим после стояния в течение ночи не закристал-
лизовывается. В этом случае рекомендуется отделить маслянистый
слой, добавить к нему измельченного льда и вызвать кристаллиза-
цию потиранием стеклянной палочкой. Если для затравки имеется
кристалл оксима, то кристаллизация проходит без всяких затруд-
нений.
2. При применении корковой или каучуковой пробки продукт
обычно получается окрашенным.
3. Другие методы получения
Вератронитрил был получен из 4-аминовератрола диазотирова-
нием последнего с последующей обработкой цианистой закисью
меди 1, а также нагреванием вератрилглиоксиловой кислоты с гидро-
ксиламином2. Описанный выше общий метод был применен для
получения целого ряда замещенных ароматических нитрилов3.
1 М о u r e u, Bull. soc. chim. C) 15, 650 A896).
2 G а г е 1 1 i, Gazz. chim. ital. 20, 700 A890).
2 Marcus, Ber. 24,3650A891); Pschorr, Ann. 391,33A912).
ГАЛЛАЦЁТОФЁНОН
147
OH
ГАЛЛАЦЕТОФЕНОН
B, 3, 4-Триокшацетофеноп)
OH
OH
OH
+ (CH,CO)aO
ZnCls
+ CH,CO,H
.он
C0CH3
Предложили: И. Бадвар и К.. Вепкатараман.
Проверили: В. Хартман и Л. Ролл.
1. Получение
В круглодонной колбе емкостью 250 мл, снабженной обратным
холодильником с хлоркальциевой трубкой, растворяют 28 г @,21
мол.) свежесплавленного и хорошо измельченного хлористого цинка
(примечание 1) в 38 мл ледяной уксусной кислоты при нагревании
колбы на масляной бане, температура которой 135—140°. К прозрач-
ному светлокоричневому раствору сперва добавляют 40 г @,37 мол.)
95%-ного уксусного ангидрида, а затем в один прием 50 г @,4 мол.)
свежеперегнанного пирогаллола (примечание 2). Смесь нагревают при
140—145 (примечание 3) в течение 45 мин., причем во время нагре-
вания ее часто и энергично встряхивают. Не вошедший в реакцию
уксусный ангидрид и уксусную кислоту отгоняют при уменьшенном
давлении. Оставшуюся красно-бурую лепешку разбивают, приба-
вляя к ней 300 мл воды при механическом перемешивании в течение
нескольких минут. Смесь охлаждают ледяной водой, осадок отса-
сывают и промывают холодной водой. Сырой продукт в количестве
45—50 г перекристаллизовываютиз500 мл кипящей воды, насыщен-
ной сернистым газом, Выход соломенножелтых игл с т. пл. 171—
172° составляет 36—38 г E4—57%теоретич.). Если насытить маточ-
ный раствор солью и охладить его до 10°, можно выделить еще 4—
5 г сырого продукта, который после перекристаллизации дает 3—
4 г чистого продукта (примечание 4),
2. Примечания
1. Хлористый цинк должен быть хорошего качества, причем
перед работой его целесообразно сплавить.
2. Другие соотношения между количествами уксусной кислоты,
ангидрида и хлористого цинка не ведут к увеличению выходов.
3. Следует тщательно регулировать температуру, которая ни
в коем случае не должна подниматься выше 150°. Как при прове-
дении настоящего синтеза, так и вообще при получении других
кетонов по способу Ненцкого высокая температура ведет к образо-
ванию интенсивно окрашенного и осмолившегося продукта, в кото-
ром вероятно содержится небольшое количество дикетона.
148
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
4. Этот метод был применен также для получения других фено-
локетонов, как то: резацетофенона, 2-ацетил-1-нафтола 1, 2-фенил-
ацетил-1-нафтола и 2-фенилпропионил-1-нафтола 2.
3. Другие методы получения
Описанный выше метод представляет собою видоизмененный
метод Ненцкого и Зибера3. Галлацетофенон был также получен
действием хлористого ацетила на пирогаллол 4 и действием пере-
киси водорода и щелочи на 2-формил-4-ацетилрезорцин 5.
1 Witt, В г a u n, Вег. 47, 3227 A914).
2 Cheema, Venkataraman, J. Chcm. Soc. 1932, 919.
3Nencki, Sieber, J. prakt. Chem. B) 23, 151, 538 A881); Nencki,
Ber. 27, 2737 A894). См. также Crabtree, Robinson, J. Chem. Soc.
121, 1038 A922).
«Einliorn, Holland t, Ann. 301, 107A898); Fischer, Ber.
42, 1020 A909).
6 Nakazawa, J. Pharm. Soc. Japan 59, 297 A939) [C. A. 33, 8186 A939)].
н.-ГЕКСАДЕКАН
C16H33I + 2 [H] -(—ti?f!> ClfiH31HI
Предложил: П. Левин.
Проверили: В. Хартман, Л. Смита Дж. Дикки.
1. Получение
В 2-литровую круглодонную колбу, снабженную механической
мешалкой с жидкостным затвором, трубкой для подвода газа и труб-
кой для отвода хлористого водорода и паров уксусной кислоты
(примечание 1), помещают 915 мл ледяной уксусной кислоты,
327 г E гр.-ат.) цинковой пыли и 352 г A мол.) йодистого
цетила (т. пл. 20—22°, стр. 280). Смесь насыщают сухим хлористым
водородом, пускают в ход мешалку и массу нагревают на водяной
бане. После каждых 5 час. нагревания смесь вновь насыщают
хлористым водородом. Через 25 час. массе дают охладиться и отде-
ляют гексадекан, образующий верхний слой. Остаток выливают
в 3 л воды и фильтруют через бюхнеровскую воронку для удаления
цинковой пыли, которую промывают сперва 500 мл воды, а затем
250 мл эфира. Водные фильтраты соединяют вместе и экстрагируют
двумя порциями по 500 мл эфира. Эфирные вытяжки соединяют вместе,
добавляют их к гексадекану и полученный раствор промывают
сперва двумя порциями по 25 мл 20%-ного раствора едкого натра,
а затем водой до полного удаления щелочи. Эфирный раствор сушат
150 г безводного сернокислого натрия, фильтруют и перегоняют из
полулитровой видоизмененной колбы Клайзена с дефлегматором.
Выход н.-гексадекана с т. кип. 156—158°/14 мм и т. пл. 16——17°
составляет 192 г (85% теоретич.).
ГЕКСАМЕТИЛЕНГЛИКОЛЬ
149
2. Примечания
1. Если реакцию ведут в вытяжном шкафу, можно работать
в открытой колбе.
3. Другие методы получения
н.-Гексадекан был получен восстановлением йодистого цетила
цинком и соляной кислотой в спиртовом 1 или уксуснокислом 2>3
растворе; восстановлением цинк-медной парой 3, а также водородом
в присутствии палладиевого катализатора3. н.-Гексадекан был
получен также действием натрия на йодистый октил 4, нагреванием
диоктил-ртути как самой по себе, так и с цинковой пылью 3, нагре-
ванием пальмитиновой кислоты с иодистоводородной кислотой и
красным фосфором6 и восстановлением гексадецена-1 7.
н.-Гексадекан был получен в качестве побочного продукта при
синтезе бромистого октилмагния 8, а также при действии натрия на
смесь бромистого октила и бромистого этила9; кроме того, он
является одним из продуктов, образующихся при нагревании
стеарата натрия10 или цетилового эфира и.
1 S о г a b j i, J. Chem. Soc. 47, 38 A885).
2 Levene, West, van der Scheer, J. Biol. Chem. 20, 523 A915).
3 С a r e y, S m i t h, J. Chem. Soc. 1933, 346.
4 Z i n с k e, Ann. 152, 15 A869).
6 E i с h 1 e r, Ber. 12, 1882 A879).
6 К raff t, там же 15, 1701 A882).
7 Wibaut с сотрудниками, Rec. trav. chim. 58, 360 A936).
8 Braun, Deutsch, Schmatloch, Ber. 45, 1254 A912).
9 Lachowicz, Ann. 220, 180 A883).
10 Gran, Wir th, Ber. 53, 1310A920).
» О d d o, Gazz. chim. ital. 31 A) 346 A901).
ГЕКСАМЕТИЛЕНГЛИКОЛЬ
A,6-Гександиол)
(сисг.ад (Сн2)/СН2°Н +2C2HSOH
\сн2он
Предложили: В. Лэзир, Дж. Хилл и В. Аменд.
Проверили: Р. Шрайнер и Дж. Каплан.
1. Получение
В стальной реакционный сосуд (примечание 1) емкостью 400 мл
или даже больше, способный выдержать повышенное давление и
обладающий достаточным запасом прочности (примечание 2), поме-
150
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
щают 252 г A,25 мол.) этилового эфира адипиновой кислоты (т. кип.
144—145729 мм); (стр. 578) и 20 г меднохромового катализатора,
полученного с добавкой бария или беэ него (стр. 301). Реакционный
сосуд герметически закрывают и устанавливают на соответствую-
щее приспособление для взбалтывания. Сосуд соединяют с источ-
ником водорода и наполняют его водородом до давления в 150—200 am
(примечание 2).
Пускают в ход качалку и реакционную систему нагревают как
можно быстрее до 255°. При этой температуре (примечание 3) гидро-
генизацию продолжают до тех пор, пока поглощение водорода не пре-
кратится (примечание 4). Качалку останавливают, сосуд охлаждают
и давление спускают. Реакционную смесь переносят в стакан ем-
костью 600 мл с помощью четырех порций 95%-ного спирта по 25 мл.
Катализатор отделяют фильтрованием или с помощью центрифуги
и промывают его еще четырьмя порциями спирта по 25 мл (приме-
чание 5). К реакционному продукту (примечание 6) добавляют 50 мл
40%-ного раствора едкого натра и спиртовой раствор кипятят 2 часа
с обратным холодильником. Смесь переносят в 1-литровую перегон-
ную колбу и спирт отгоняют, пока термометр в парах не покажет
95°. Горячий остаток переносят в прибор для непрерывного экстра-
гирования жидкостей (стр. 478), колбу споласкивают 50 мл воды
и раствор исчерпывающим образом экстрагируют эфиром (приме-
чание 7). Эфир отгоняют и после удаления спирта и воды гликоль
перегоняют в вакууме из колбы Клайзена емкостью 250 мл. Выход
125—132 г (85—90% теоретич.). Гексаметиленгликоль кипит при
143—144° (температура бани 160°) при давлении 4 мм и плавится
при 41—42°.
2. Примечания
1. Подходящие реакционные сосуды (автоклавы) и приспособле-
ния для взбалтывания имеются в продаже или могут быть спе-
циально изготовлены 1.
2. Применяемое давление водорода зависит от имеющегося
оборудования. Водород в баллонах имеет максимальное давление
140 am. Существует также специальное оборудование для компри-
мирования водорода. Начальное давление водорода не должно
превышать 140 am, если максимальное рабочее давление, на которое
рассчитан прибор для гидрогенизации, составляет 350 am. Если
рабочее давление равно 700 am, начальное давление в реакционном
сосуде может достигать 210 am. Начальное давление не должно
равняться рабочему давлению, так как при нагревании сосуда да-
вление повышается. Так, при 255° давление будет в 1,8 раза больше,
чем при 20°. По мере того как идет гидрогенизация, давление па-
дает; по изменению давления можно следить за ходом реакции
Конец реакции заметен по тому, что давление становится постоянным.
ГЕКСАМЕТИЛЕНГЛИКОЛЬ
151
3. Регулировать температуру лучше всего с помощью автомати-
ческого терморегулятора, соединенного с реле, которое периоди-
чески включает и выключает ток.
4. Время F—12 час), необходимое для завершения реакции,
является функцией давления водорода, активности катализатора
и чистоты этилового эфира адипиновой кислоты. Если начальное
давление не очень высоко или объем реакционного сосуда менее
2 л, то во время реакции необходимо добавлять водород в реакцион-
ный сосуд. Во всяком случае, для того чтобы реакция гладко шла
до конца, необходимо поддерживать давление водорода не ниже
100 am.
5. Удобнее всего катализатор удалять с помощью центрифуги.
Если это почему-либо невозможно, его следует отфильтровать через
воронку иэ пористого стекла или через воронку Бюхнера.
6. На этой стадии количество сложного эфира можно опреде-
лить с помощью числа омыления реакционной массы, зная ее вес.
Это особенно важно в тех случаях, когда неизвестно состояние
прибора в отношении наличия каталитических ядов или когда при-
меняется новая порция катализатора. После того как партия ката-
лизатора испытана и аппаратура прокалибрирована, так что полнота
восстановления обеспечена, на этой стадии можно выделить гликоль
с помощью фракционной перегонки.
По данным Бэркса мл. и Адкинса [частное сообщение и статья
в J. Am. Chem. Soc. 62, 3300 A940)] гидрогенизация является обра-
тимой, и продукт реакции всегда содержит сложные эфиры. Для
получения чистого гексаметиленгликоля, не содержащего сложных
эфиров, рекомендуется более простая методика, чем описанная
здесь. 30 г сырого гликоля растворяют в 50 мл воды и извлекают
четыре раза бензолом порциями по 50 мл. Водный раствор подвер-
гают перегонке, пользуясь видоизмененной колонкой Видмера.
Выход гликоля, не содержащего сложных эфиров, 93%.
7. Время, необходимое для полного извлечения, колеблется
от 24 до 50 час. Оно зависит от конструкции прибора и от скорости
отгонки эфира. За экстракцией можно следить по уменьшению
объема водного слоя, содержащего гликоль. Экстракцию можно
считать законченной, когда после испарения небольшого коли-
чества эфира из верхнего слоя на часовом стекле не будет образо-
вываться остатка. Вместо эфира для экстракции можно взять бен-
зол.
3. Другие методы получения
Гексаметиленгликоль был получен действием на йодистый ге-
ксаметилсн уксуснокислого серебра и омылением полученного
ацетата 2, омылением бромида 3, восстановлением этилового зфира
адипиновой кислоты натрием и спиртом4 и методом, описанным
выше6. Каталитическая гидрогенизация в присутствии медно->
152
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
хромового катализатора карбэтокси-группы до гидро'ксильной
группы является весьма полезным общим методом для получения
спиртов и гликолсй1'6.
1 A d k i n s, Ind. Eng. Chem., Anal. Ed. 4, 342 A032); Reactions of Hydro-
gen with Organic Compounds over Copper-Chromium Oxide and Nickel Cata-
lysts, pp. 29—39, University of Wisconsin Press, Madison, Wisconsin, 1937.
2 H a m о n с t, Bull. soc. chim. C) 33, 538 A905).
3 Haworth, Perkin, J. Chem. Soc. 65, 598 A894).
4 Bo u ve a u 1 t, В la n c, Compt. rend. 137, 328 A903); Bull. soc.
chim. C) 31, 1203 A904).
6 L a z i e г, ам. пат. 2079 414 [С. А. 31, 4340 A937)].
• Adkins, Folkers, J. Am. Chem. Soc. 53, 1095 A931); 54, 1145
A932); W о j с i k, Ad к i n s, там же 55, 4939 A933).
ГЕПТАНОЛ-2
CH3C0 (СН3L СН3
JL^^—L CH3CH (ОН) (CH,L СН3
Предложили: Ф. Уитмор и Т. Оттербахер.
Проверили: Г. Гильман и X. Харвуд.
1. Получение
В 3-литровой круглодонной колбе, снабженной мощным обрат-
ным холодильником длиною в 100 и диаметром в 1 см, растворяют
228 г B мол.) метил-н.-амилкетона («Синт. орг. преп.», сб. 1, стр. 247)
в смеси 600 мл 95%-ного спирта и 200 мл воды. Через холодильник
прибавляют постепенно 130 г натрия E,6 гр.-ат.) в виде прово-
локи. Во время прибавления натрия колбу охлаждают струей
холодной воды (примечание 1), чтобы реакция не стала слишком
бурной (примечание 2).
После того, как натрий растворился (примечание 3), прибав-
ляют 2 л воды и охлаждают смесь до 15°, Затем отделяют верхний
маслянистый слой, промывают его последовательно смесью 25 мл
концентрированной соляной кислоты с 25 мл воды, затем 50 мл
воды, сушат 20 г безводного сернокислого натрия и перегоняют
с фракционирующей колонкой (примечание 4). После небольшой
головной фракции чистый гептанол перегоняется при 155—157,5°.
Выход 145—150 г F2—65% теоретич.).
2. Примечания
1. Если не охлаждать колбу, то образуются продукты конден-
сации, что значительно понижает выход.
2. Если содержимое колбы охлаждать льдом, то температуру
легко поддерживать ниже 30°. Подобное охлаждение, соединенное
с перемешиванием, чрезвычайно полезно во время начального при-
бавления натрия (в виде проволоки или маленьких кусочков).
яесии.и.-ГЕПТАХЛОРПРОПАН
153
При охлаждении и перемешивании жидкость кипит очень слабо,
и после прибавления около 60 г натрия реакция замедляется на-
столько, что можно прибавлять большие количества натрия сразу,
не опасаясь перегрева,
3. Время, нужное для прибавления натрия, можно значительно
сократить, если применять механическое размешивание. При раз-
мешивании выход не повышается, но зато масса не вспенивается,
благодаря чему натрий можно прибавлять быстрее.
4. Авторы применяли колонку Юнга с 20 дисками на расстоя-
нии 3 см друг от друга. Проверявшие синтез применяли дефлег-
матор Глинского с тремя шариками.
3. Другие методы получения
Гсптанол-2 может быть получен действием бромистого магний-
н.-амила на ацетальдегид х и восстановлением метил-н.-амилкетона
в спиртовом растворе натрием 2.
1 Henry, Rec. trav. chim. 28, 446 A909).
'Thorns, Mannich, Ber. 36, 2544 A903); Pickard, Kenyon,
J. Chem. Soc. 99, 58 A911).
яеси-И-И.-ГЕПТАХЛОРПРОПАН
A, 1, 1, 2, 2, 3, З'Гептахлорпропан)
2 = .CCl2+CHCl3 -™ CCl3CCl2CHCl2
Предложил: М. Фарлоу.
Проверили: Ф. Уитмор и Ф. Брейер.
1. Получение
В 1-литровую круглодонную колбу, снабженную обратным хо-
лодильником с хлоркальциевой трубкой, помещают 166 г A03 мл,
1,0 мол.) технического тетрахлорэтилена, 300 г B00 мл, 2,5 мол.)
сухого хлороформа и 27 г @,2 мол.) безводного хлористого алюми-
ния. Смесь не сильно кипятят на паровой бане в течение 15 час.
(примечание 1), охлаждают до комнатной температуры и выливают
в 1-литровую делительную воронку, наполовину наполненную
колотым льдом. Слой, содержащий органические продукты, несколь-
ко раз промывают водой и сушат хлористым кальцием или безвод-
ным сернокислым натрием. Путем фракционирования при атмосфер-
ном давлении с хорошим дефлегматором выделяют обратно 160—
165 г хлороформа. Фракцию, содержащую шгилш.-гептахлорпро-
пан, перегоняют в вакууме и выделяют продукт, кипящий при
1Ю—1137Ю мм или при 137—140732 мм, с т. пл. 29—30°. Выход
250—266 г (88—93% теоретич.; примечание 2).
154
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
2. Примечания
1. В начальной стадии реакции выделяется небольшое коли-
чество хлористого водорода.
2. Согласно данным Принса \ можно легко выделить гептахлор-
пропан, если реакционную смесь вылить в воду и удалить все непро-
реагировавшие продукты перегонкой с водяным паром. Перегонку
прекращают, как только начнет перегоняться сам продукт. По
охлаждении остатка получают бесцветное твердое вещество с над-
лежащей температурой плавления.
3. Другие методы получения
Этот метод был открыт Безекеном и Принсом2 и затем детальнее
исследован Принсом]3. Вместо тетрахлорзтилена можно пзять
пентахлорзтап, так как в присутствии хлористого алюминия послед-
ний превращается в непредельное соединение. нгашл.-Гептахлор-
пропан был получен также действием пятихлористого фосфора
на пентахлорацетон 4 и действием хлористого алюминия на дихлор-
ацетилхлорид 6.
1 Р г i n s, Rec. trav. chim. 54, 249 A935).
2 Boeseken, Prins, Chem. Zentr. 1911, I, 466.
3 Prins, J. prakt. Chem. B) 89, 414 A914).
* F r i t s с h, Ann. 297, 312 A897).
5 Boeseken, Rec. trav. chim. 29, 109 A910).
CH3 (CH2M CH = NOH + 4 [H]
н.-ГЕПТИЛАМИН
' CiH>0
CH3 (CH2N NH2 + H2O
Предложили: В. Латан, С. Пунтамбекер и К. Мареел.
Проверили: В. Царотерс и В. Мак-Юеен.
1. Получение
Раствор 258 г B мол.) оксима энаптола (стр. 394) в 4 л абсолют-
ного спирта (примечание 1) нагревают до кипения на водяной бане
в 12-литровой круглодонной колбе, запертой обратным холодиль-
ником, длиной 150 см, с очень широкой B,5 см) внутренней труб-
кой. Как только спирт начинает кипеть, баню удаляют и, поддер-
живая температуру кипения, прибавляют к раствору через холо-
дильник 500 г нарезанного длинными кусочками натрия. Приба-
вление следует вести как можно быстрее, чтобы раствор все время
кипел, но так, чтобы пары спирта успевали полностью конденси-
роваться в холодильнике (примечание 2). Последние 150 г натрия
плавятся в горячей смеси, и их можно прибавить очень быстро, без
потери спирта или амина через холодильник.
н.-ГЕПТИЛАМИН
155
Как только натрий растворится, реакционную массу охлаждают
и разбавляют 5 л воды. Колбу немедленно соединяют с обращенным
вниз холодильником и отгоняют смесь спирта, воды, гептиламина
и небольшого количества непрореагировавшего оксима энантола.
Нижний конец трубки холодильника до начала перегонки погру-
жают в смесь 300 мл концентрированной соляной кислоты и 300 мл
воды, находящуюся во второй 12-литровой колбе. Перегонку про-
должают до тех пор, пока проба дестиллата имеет щелочную реак-
цию. Если к концу отгонки начинается вспенивание, то в перегон-
ную колбу вливают еще 3 л воды. Общее количество дестиллата 8—9 л.
Спирт, воду и непрореагировавший оксим отгоняют в вакууме
при 20—30 мм на водяной или, лучше, на паровой бане. Затем колбу
с оставшимся в ней кристаллическим солянокислым гептиламином
соединяют с обратным холодильником и прибавляют через холо-
дильник 1 л 40%-ного раствора едкого .кали. Затем смесь взбалты-
вают, чтобы смыть со стенок колбы кристаллы, охлаждают и пере-
носят в делительную воронку. Нижний слой водной щелочи сли-
вают и оставшийся амин сушат в делительной воронке твердым ед-
ким кали. Образующийся во время сушки слой раствора едкого
кали время от времени удаляют и прибавляют свежие палочки ед-
кого кали до тех пор, пока не прекратится отделение водного слоя.
Чтобы окончательно высушить продукт, требуется 24—30 час.
Затем н.-гептиламин сливают через верх воронки в специальную
колбу Клайзена емкостью 250 мл и перегоняют при атмосферном
давлении, собирая фракцию 152—157°. Выход 140—170 г F0—73%
теоретич.; примечание 3).
2. Примечания
1. Если спирт обезвожен недостаточно, то получаются низкие
выходы. Абсолютный спирт, полученный перегонкой обыкновен-
ного абсолютного спирта над метилатом натрия, вполне пригоден
для этой цели («Синт. орг. преп.», сб. 1, стр. 545).
2. Чем быстрее проводят восстановление, тем лучше выход.
3. По этому же способу с выходом 50—60% были получены сле-
дующие амины: н.-бутиламин (т. кип. 75—80°) из оксима масляного
альдегида; вторичный бутиламин (т. кип. 59—65°) из оксима метил-
зтилкетона; циклогексиламин (т. кип. 133—135°) из циклогексанон-
оксима. Особенно тщательно следует сушить оба бутиламина.
3. Другие методы получения
н.-Гептиламин может быть получен восстановлением оксима энан-
тола амальгамой натрия и уксуснойкислотой1; амальгамой аммония2;
натрием и спиртом 3; каталитическим восстановлением 4; восстанов-
лением 1-нитрогептанажелезом и уксусной кислотой5; восстановле-
нием нитрила энантовой кислоты натрием и спиртом3-6 или его ката-
150
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
литическим восстановлением 7; восстановлением амида энантовой
кислоты натрием и спиртом 8 или его каталитическим восстановле-
нием 8; восстановлением фенилгидразона знантола амальгамой нат-
рия и уксусной кислотой10 и каталитическим восстановлением
знантола и аммиака в спиртовом растворе 11а. Образование вторич-
ного амина, что является серьезным затруднением при каталити-
ческом восстановлении нитрила знантовой кислоты, можно почти
полностью подавить, если восстановление вести в присутствии
большого избытка аммиака Иб. Среди других методов, также веду-
щих к образованию н.-гептиламина, следует упомянуть реакцию
между 1-бромгептаном и аммиаком12, гофмановскую перегруппи-
ровку амида каприловой кислоты 13 и бекмановскую перегруппи-
ровку оксима метил-н.-гептилкетона с последующим омылением ы.
1 Ooldschmidt, Ber. 20, 729 A887).
2 Т a k a k i, V е d a, J. Pharm. Soc. Japan 58, 276 A938) [С. А. 32, 5376
3 Suter, Moffett, J. Am. Chem. Soc. 56, 487 A934).
4 Mailhe, Compt. rend. 140, 1692 A905); Bull. soc. chim.
— ___ C) 33 963
A905); S a b a t'i e r, Mailhe, Ann. chim. phys. (8) 16, 102 A909); Paul.
Bull. soc. chim. E) 4, 1121 A937).
6 W о г s t a 1 1, Am. Chem. J. 21, 223 A899).
6 Forselles, Wahlforss, Ber. 25, 637 (Ref.) A892).
7 Mailhe, de Oodon, Bull. soc. chim. D) 23, 19 A918); M a i 1 h e,
Bellegarde, там же 25, 591 A919); Mailhe, Ann. chim. (9) 13, 203A920);
Schwoegler, Adkins, J. Am. Chem. Soc. 61, 3499 A939).
8 Scheuble, Loebl, Monatsh. 25, 1087 A904).
9 W о j с i k, Adkins, J. Am. Chem. Soc. 56, 2419 A934).
10 T a f e 1, Ber. 19, 1928 A886).
"aMignonac, Compt. rend. 172, 226 A921); E) Schwoegler,
Adkins, J. Am. Chem. Soc. 61, 3499 A939).
13 Davis, E 1 d e r f i e 1 d, там же 54, 1503 A932).
13 H о f m a n n, Ber. 15, 772 A882); Hoogewerff, van Dorp, Rec.
trav. chim. 6, 380 A887).
11 S о d e n, H e n 1 e, Pharm. Ztg. 46, 1026 A901) (Chem. Zentr. 1902,
I, 256).
«-ГИДРИНДОН
A-Инданон)
CHC1
i
CH2
CH./
ГО]
— CO
I
CH,
CH,
Предложили: Р. Пакауд и К- Аллен.
Проверили: Л. Физер и В. Кемпбелл.
1. Получение
В колбу емкостью 250 мл, снабженную доходящей до ее дна
трубкой для ввода газа и другой трубкой для вывода непрореагиро-
а-ГЙДРИНДОН
157
вавшего газа, а также термометром, помещают 80 г @,69 мол.)
свеже-перегнанного индена (примечание 1). Перед наполнением колба
погружается в баню с водой и льдом и, поддерживая температуру
жидкости при 5—10°, через нее с умеренной скоростью пропускают
сухой хлористый водород до тех пор, пока не поглотится 24—27 г
газа. Поглощение продолжается 8—10 час. и требует весьма незна-
чительного присмотра. Сырой а-хлоргидринден переносят в колбу
Клайзена на 250 мл и перегоняют его в вакууме. После небольшой го-
ловной фракции E—Юг), содержащей инден,перегоняется а-хлоргид-
ринден при 90— 103е/15 мм' выход составляет 80—90 г (примечание 2).
В трехгорлую колбу емкостью 500 мл, снабженную мешалкой,
термометром и капельной воронкой, помещают раствор 100 г A мол.)
хромового ангидрида в 100 мл воды и приливают 100 мл ледяной
уксусной кислоты. Затем, охлаждая колбу снаружи, через воронку
приливают а-хло-ргидринден с такой скоростью, чтобы температура
поддерживалась при 35—40°. На зто требуется около 1,5 часа.
Когдаприливаниеокончено,перемешивание продолжают еще 15 мин.,
после чего смесь выливают в большой стакан и разбавляют ее
300 мл воды. Для нейтрализации кислоты прибавляют твердый
углекислый натрий (примечание 3), после чего а-гидриндон отго-
няют от смеси с водяным паром, причем следят за тем, чтобы в начале
перегонки не перебросило пену. После того как отгонится около
2,5 л дестиллата, раствор опять начинает пениться. Тогда перегонку
прекращают, так как в дальнейшем она не дает заметного коли-
чества продукта.
а-Гидриндон обычно застывает при хорошем охлаждении дестил-
лата, причем образуется смесь бесцветных кристаллов и желтого
твердого вещества. Продукт отфильтровывают после тщательного
охлаждения смеси в бане со льдом. Еще влажный продукт раство-
ряют в 200 мл бензола и раствор подвергают перегонке до тех пор,
пока не отгонится вся вода. Затем бензол отгоняют в вакууме,
нагревая колбу на водяной бане, после чего перегоняют а-гидрин-
дон, который кипит при 125—126°/17 мм. Дестиллат представляет
собой светложелтое твердое вещество с т. пл. 39—41°. Выход 46—
55 г E0—60% теоретич., считая на взятый инден; примечания 4
и 5).
2. Примечания
1. Если применяется технический инден, то для этого синтеза
пригодна фракция с т. кип. 178—182°. При проверке синтеза при-
менялся инден с т. кип. 181,6—183,3°.
2. Большая часть продукта перегоняется при 100—103°/15 мм,
однако выход а-гидриндона увеличивается, если берется продукт,
выкипающий в более широких пределах.
3. Избыток углекислого натрия благоприятствует вспениванию
при перегонке, и поэтому его следует избегать. Ввиду того, что
158
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
с помощью индикаторной бумажки трудно определить момент
нейтрализации, соду добавляют все меньшими и меньшими порциями,
пока свежая порция не будет больше разлагаться с выделением газа.
4. Нецелесообразно высушивать продукт после перегонки с
водяным паром на воздухе ввиду того, что а-гидриндон обладает
настолько высокой упругостью паров, что потери весьма значи-
тельны. Сушить продукт можно в вакуум-эксикаторе, однако это
требует нескольких дней. В результате указанной здесь перегонки
получается совершенно сухой продукт и потери не превышают 2—5 г.
5. При перекристаллизации лучше всего а-гидриндон раство-
рить в спирте A мл на 1 г) при комнатной температуре, добавить
воды до начала появления твердой фазы и охладить раствор до 0°.
Вещество кристаллизуется либо в длинных пластинках, либо
в листочках.
3. Другие методы получения
а-Гидриндон был получен из а-хлоргидриндена 1, по методике,
описанной здесь^ из а-бромгидриндена нагреванием с разбавленной
серной кислотой2; циклизацией хлорангидрида гидрокоричной
кислоты хлорным железом3 или хлористым алюминием4; цикли-
зацией гидродсоричной кислоты фтористым водородом 5 или серной
кислотой е и взаимодействием хлорангидрида акриловой кислоты
с бензолом в присутствии хлористого алюминия 7.
1 Н ii с k e I, Sachs, Yantschulewitsch, Nerdel, Ann.
518f 17^ ( 1уЗо) .
3 Porter, Suter, J. Am. Chem. Soc. 57, 2022 A935).
3 We d e к i n d, Ami. 323, 255 A902)
* Kipping, J. Chem. Soc. 65, 484 A894).
'- Ficscr, Hershbcrg, J. Am. Chem. Soc. 61, 1278 A939).
'Price, Lewis, т.ш же 61, 2553 A939).
,„„ ' M°ureu> Bul1- s°c- chim. C) 9, 570 A893); Ann. chim. phys. G) 2,
198 A894); Kohler, Am. Chem. J. 42, 380 A909).
ГИППУРОВАЯ КИСЛОТА
C1CH2CO2H + 2 NH3 NH2CHXO,H + NH4C1
NHXHXO.H -f C6H5COC1 л. 2~NaOH
CeHaCONHCH2CO2Na + NaCl + 2 H2O
CeH3CONHCH,CO2Na + HC1 - C6HXONHCHXO,H + NaCl
Предложили: А. Ингерсолл и С. Бабкон.
Проверили: Р. Адаме н Б. Войцик.
1. Получение
К 3 л (около 45 мол.) концентрированного водного аммиака,
уд. в. 0,9 (примечание 1), находящегося в 5-литровой круглодонной
колбе, приливают при взбалтывании раствор 95 г A мол.) моно-
хлоруксусиой кислоты (примечание 2) в 100 мл воды. Колбу закры-
ГЙППУРОВАЯ КИСЛОТА
156
вают пробкой и оставляют стоять в течение четырех суток (приме-
чание 3). Затем колбу соединяют с обращенным вниз холодильни-
ком и раствор упаривают до 600—700 мл. Нижний конец холодиль-
ника соединен с широкой трубкой, которая доходит до дна 3-литро-
вой склянки, а в последнюю налито 1500 мл воды; зта склянка в свою
очередь соединена с другой меньшей склянкой, в которой также
имеется небольшое количество воды; благодаря этой системе избы-
ток аммиака улавливают во время отгонки. Склянки охлаждают
проточной водой. В первом уловителе регенерируется около 2 л
20—23%-ного аммиака (примечание 1).
Раствор после упаривания переливают из колбы в 2-литровый
стакан, прибавляют раствор 50 г A,25 мол,) едкого натра в 100 мл
воды и небольшое количество животного угля и смесь кипятят до
полного исчезновения запаха аммиака (примечание 4). Затем филь-
труют с отсасыванием, разбавляют водой до 500 мл и переносят
в 2-литровую круглодонную колбу, снабженную механической
мешалкой и охлаждаемую проточной водой. При постоянном пере-
мешивании и охлаждении ниже 30° к раствору приливают из двух
делительных воронок 150 г A,1 мол.) хлористого бензоила (приме-
чание 2) и холодный раствор 80 г B мол.) едкого натра в 200 мл воды.
Прибавление регулируют таким образом, чтобы реакция смеси
все время была слабощелочная. На прибавление реагентов уходит
около 1 часа, и затем смесь перемешивают еще в продолжение полу-
часа. Раствор выливают в 2-литровый стакан, где находится 125 мл
концентрированной соляной кислоты, и после охлаждения осадок
отфильтровывают и сушат. Получают 150—160 г сухого осадка,
который помещают в стакан; прибавляют 300 мл технического
четыреххлористого углерода, стакан покрывают часовым стеклом
и смесь слабо кипятят в продолжение 10 мин. После того, как смесь
несколько охладится, ее отсасывают, не давая полного разрежения,
и гиппуровую кислоту промывают на фильтре 50 мл четыреххло-
ристого углерода (примечание 5). Высушенный осадок весит 135—
140 г. Для окончательной очистки кислоту растворяют приблизи-
тельно в 2 л кипящей воды, фильтруют через воронку, обогреваемую
паром, и раствор оставляют для кристаллизации, причем не при-
меняют искусственного охлаждения. Кислота выделяется в виде
характерных белых игл с т. пл. 186—187° (не исправ.). Выход
115—122 г F4—68% теоретич., считая на взятую монохлоруксусную
кислоту). После упаривания маточного раствора до 200 мл полу-
чается еще 6—7 г гиппуровой кислоты, слегка окрашенной в корич-
невый цвет.
2. Примечания
1. В целом ряде опытов бралось эквивалентное количество реге-
нерированного водного аммиака уд. в. 0,93 и ниже, причем выходы
от этого не уменьшались.
100
СИНТЕЗУ ОРГАНИЧЕСКИХ Г1РЕПАРАТОВ
2. Применялись реактивы фирмы «Истмен» марки «практически
чистый» *.
3. Через два дня выход бывает немного меньше; через неделю
он не становится больше.
4. Во избежание образования бензамида необходимо удалить
аммиак полностью.
5. Четыреххлористый углерод можно получить обратно, для
чего следует фильтрат и промывные воды слабо подщелочить едким
натром, а затем кипятить их полчаса с обратным холодильником,
чтобы разрушить хлористый бензоил, и потом отогнать с паром.
Если профильтровать и подкислить водный остаток, можно получить
бензойную кислоту.
3. Другие методы получения
Гиппуровая кислота может быть получена из мочи травоядных
животных х; нагреванием бензамида с монохлоруксусной кисло-
той 2; нагреванием ангидрида бензойной кислоты с глицином3;
нагреванием бензойной кислоты с глицином1; нагреванием хлори-
стого бензоила с серебряной солью глицина, суспендированной
в бензоле 5, или с глицином и окисью цинка 6 и действием хлори-
стого бензоила на щелочной раствор глицина 7.
1 Gregory, Ann. 63, 125 A847); Н a I 1 о w а с h s, там же 106, 164
A858); Henneberg, Stohmann, Rau te n b e r g, там же 124, 200 A862).
2 Jazukowitsch, Zeit. Mr Chera. 1867, 460.
8 С u r t i u s, Ber. 17, 1662 A884).
4 Dessaignes, Jahresb. 1857, 367.
6'Curtius, J. prakt. Chem. B) 26, 145A883).
6 Dessaignes, Ann. 87, 325 A853).
' Batim, Ber. 19, 502 A886); Z. physiol. Chem. 9, 465 A885).
/гистидин солянокислый
Гидролиз протеинов НС===С — CHaCHNH2CO2H.HCl-H3O
крови
1
HN
1
N
н
Предложили: Дж. Фостер й Д. Шемин.
Проверили: Дж. Джонсон, X. Картер и П. Баррж.
1. Получение
В большую круглодонную колбу помещают 1,4 кг высушенной
пасты из кровяных телец (примечание 1) и 4,5 л концентрированной
соляной кислоты (уд. в. 1,18). Колбу нагревают на водяной бане
* См. примечание на стр. 26.
1-ГИСТИДИН СОЛЯНОКИСЛЫЙ
16!
до полного растворения белка и смесь осторожно кипятят с обратным
холодильником 18—20 час. Спирт от гидролизата отгоняют в вакууме
до консистенции густой пасты, после чего перегонку продолжают,
медленно приливая 1 л воды для удаления большей части избытка
соляной кислоты. Остаток растворяют в 8 л теплой воды, охлаждают
и нейтрализуют до рН 4,4—4,6 (по метилоранжу или бромкрезол
зеленому), добавляя концентрированный раствор едкого натра
(примечание 2). Смесь оставляют стоять в течение ночи, после чего
отфильтровывают выпавший пигмент через большую воронку Бю-
хнера с двумя слоями фильтровальной бумаги и 2-мм слоем инфузор-
ной земли. Фильтрат, окрашенный в тёмнокрасный цвет, обесцве-
чивают, для чего его нагревают и перемешивают 10 мин. с 60 г
активированного березового угля (норита).
Светложелтый фильтрат и промывные воды от промывки угля
разбавляют до объема в 25 л водопроводной водой и при переме-
шивании приливают раствор 600 г хлорной ртути в 2 л горячего
95%-ного спирта. Затем медленно добавляют при перемешивании
концентрированный раствор соды (отвечающий около 350 г без-
водного углекислого натрия), пока рН раствора не станет 7,0—7,5
(фенолрот или лакмус). Осадку дают осесть в течение нескольких
часов, лучше в течение ночи, раствор сифонируют и сосуд вновь
доливают водой до первоначального объема (примечание 3). Смеси
дают отстояться, промывную жидкость вновь сифонируют и такую
промывку повторяют еще дважды. После третьего раза промывную
жидкость сифонируют, а осадок отфильтровывают с отсасыванием
через большую воронку Бюхнера, в которую помещают два слоя
фильтровальной бумаги и 2-мм слой инфузорной земли.
Влажный комплекс гистидина с ртутью суспендируют в 5 л
воды и энергично перемешивают, пропуская при этом ток сероводо-
рода. Когда сернистая ртуть осадится полностью, суспензия стано-
вится равномерного черного цвета и при стоянии хорошо отстаи-
вается. Фильтрат и промывные воды после сернистой ртути (приме-
чание 4) упаривают в вакууме до объема около 1 л и осветляют
5 г активированного березового угля. Фильтрат и промывные воды
после угля упаривают далее до объема около 250 мл и смешивают
с тройным объемом 95%-ного спирта. Чтобы вызвать кристаллиза-
цию, раствор охлаждают и трут палочкой по стенке сосуда; хлористо-
водородная соль гистидина выпадает в виде пластинок. Выход
неочищенной хлористоводородной соли гистидина составляет 85—
90 г. Из фильтрата, после стояния в течение нескольких недель
в холодильном шкафу, обычно выпадают еще 4—5 г продукта.
Сырой продукт растворяют в пятикратном количестве по весу
воды и после осветления раствора небольшим количеством акти-
вированного угля раствор разбавляют 1,5-ным количеством по
объему 95%-ного спирта. Продукт выпадает в виде правильно
образованных белоснежных кристаллов, которые отфильтровывают
11 Сборник Л» 2
162
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
с отсасыванием после стояния в течение нескольких дней в холо-
дильном шкафу. Выход очищенной хлористоводородной соли гисти-
дина составляет 75—80 г (примечание 5). Соединение плавится при
251—252° с разложением. При применении указанной здесь мето-
дики не происходит рацемизации аминокислоты, и она обладает
свойственной ей оптической деятельностью, [a]?f= +8,0° в при-
сутствии трех молей хлористоводородной кислоты. Перекристал-
лизованный продукт обычно аналитически чист, и определение
азота по Ван-Сляйку дает правильные результаты. Иногда для
получения аналитически чистого продукта требуется еще одна пере-
кристаллизация.
2. Примечания
1. Для этого синтеза применялась продажная «сушеная паста
из кровяных телец». Эта паста содержит около 15% влаги и золы,
и 200 г ее содержат примерно столько сырого белка, сколько 1 л
свежей бычачьей крови A70 г белка на 1 л).
Если для синтеза применяется свежая кровь, удобно удалить
значительную часть воды следующим образом. 7 л бычачьей крови
обрабатывают в 12-литровой колбе 50 мл ледяной уксусной кислоты
и нагревают на водяной бане, время от времени перемешивая массу
до образования густой пасты. Около 4 л воды отгоняют в вакууме
на водяной бане, а остаток подвергают гидролизу, как описано выше:
2. Для нейтрализации требуется около 600 мл 50%-ного раствора
едкого натра. Следует избегать избытка щелочи.
3. Если, как это иногда бывает, комплекс гистидина с ртутью
оседает медленно, жидкость можно отсифонировать и профильтро-
вать. Небольшое количество вещества на фильтре присоединяют
к основной массе.
4. Сернистую ртуть можно сохранить и превратить в металличе-
скую ртуть или в сулему с помощью обычной методики.
5. Можно выделить еще 10 г сырой хлористоводородной соли
гистидина из маточного раствора, для чего его упаривают в вакууме
до 50—60 мл и приливают 1,5-кратное количество по объему 95%-ного
спирта. После перекристаллизации'получают 8—9 г чистого про-
дукта.
3. Другие методы получения
Получение гистидина гидролизом гемоглобина и осаждение
его хлорной ртутью в щелочном растворе впервые осуществил Френ-
кель 1>2'3. Гистидин может быть также осажден в виде серебряного
производного *.
1 F r a n k e I, Monatsh. 24, 229 A903).
2Abderhalden, Fleischmann, Irion, Fermentforschung
10, 447 A928) [С. А. 23, 2994 A929)].
3 О i 1 s о n, J. Biol. Chem. 124, 281 A938).
1 V i с к е г у, Leavenworth, там же 78, 627 A928).
di-ГЛИЦЕРИНОВЫЙ АЛЬДЕГИД
163
rf/ГЛИЦЕРИНОВЫЙ АЛЬДЕГИД
носн,снонсн(ос,н3)о+н2о ^so-
НОСН2СНОНСНО + 2 С2Н5ОН
Предложили: Е. Вицеман, У. Эванс, Г. Хасс и Е. Шредер.
Проверили: Ф. Уитмор и Г. Негер.
1. Получение
Смесь 50 г@,3 мол.) ацеталя ^//-глицеринового альдегида (стр. 65)
и 500 мл 0,1 н. серной кислоты оставляют стоять в продолжение
семи дней при температуре около 20° (примечание 1). К получен-
ному раствору прибавляют 30 мл ледяной уксусной кислоты, затем
тщательно нейтрализуют его раствором гидрата окиси бария (при-
мечание 2), перемешивают с 5 г животного угля, фильтруют и выпа-
ривают фильтрат в вакууме при 10 мм (примечание 3), до полного
прекращения отгонки воды. Для ускорения кристаллизации оста-
ток обрабатывают равным объемом абсолютного спирта, выпавшие
кристаллы отсасывают и сушат в вакуум-эксикаторе над натронной
известью н хлористым кальцием. Выход «//-глицеринового альдеги-
да, плавящегося при 137—139°, составляет 22 г (80% теоретич.).
2. Примечания
1. Для того чтобы глицериновый альдегид быстро закристалли-
зовался, омыление и выпаривание следует вести при температуре
ниже 30°.
2. Проба отфильтрованного раствора при прибавлении гидрата
окиси бария или серной кислоты должна оставаться прозрачной
или давать слабую опалесценцию.
3. При выпаривании под низким давлением A0 лш)получаю-
щийся глицериновый альдегид быстро кристаллизуется даже из
крепких растворов. Выпаривание при 20 мм дает сироп, который
часто кристаллизуется с большим трудом.
3. Другие методы получения
(//-Глицериновый альдегид может быть получен окислением
глицерина азотной кислотой х, бромом в присутствии соды 2 и пере-
кисью водорода в присутствии солей закиси железа3;при действии
ультрафиолетовых лучей на глицерин в нейтральном растворе 4;
при действии солнечного света на глицерин в присутствии серно-
кислого уранила 6; электролизом rfZ-эритроновой кислоты 6; омы-
лением ацеталя «//-глицеринового альдегида7; окислением акро-
леина 8; окислением циклического ацеталя бензальальдегида и
164
СИНТЕЗУ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
1,3-пропендиола с последующим омылением9 и щелочной конден-
сацией формальдегида 10.
1 К i I i a n i, Ber. 54, 467 A921); F i s с h е г, Т a f e 1, там же 20, 3385
A887).
2 Fischer, Taf el, там же 20, 3385 A887).
3 Fe n to n, Ja с k so n, Chem. News 78, 187 A898): J. Chem. Soc
75, 5 A899); W i t z e m a n n, J. Am. Chem. Soc. 36, 2227 A914).
4 Bierry, Henri, Ranc, Compt. rend. 152, 535 A911).
6 N e u b e r g, Biochem. Z. 13, 307 A908).
6 Neuberg, Scott, Lachmann, там же 24, 157 A910).
' W о h 1, Ber. 31, 1800, 2395 A898); W о h 1, Neuberg, там же 33,
3100 A900); Evans, H a s s, J. Am. Chem. Soc. 48, 2706 A926); W i t z e-
m a n n, там же 36, 1913 A914); Spoehr, Young, Carnegie Inst. Washing-
ton Yearbook 25, 177 A925—1926); Exp. Sta. Record 57, 817 A927) [C. A. 22,
2368 A928)].
8 Neuberg, Biochem. Z. 221, 492 A930); 255, 1 A932).
"Fischer, Ahlstrom, Richter, Ber. 64, 611 A931).
10 К у з и н, ЖОХ 8, 592 A938).
CH:lO<
ГОМОВЕРАТРОВАЯ КИСЛОТА
C,4-Диметоксифенилуксусная кислота)
.СО —О
С1 +ЗН2О
сн„о
(NaOH)
=с-с6н3
(СН3ОJ С6Н3СН2СОСО2Н -
(СН3ОJ С6Н3СН2СОСО2Н + Н2О2
NH3 -
(NaOH)
С6Н5СО,Н
сн.о/
сн2со2н
н2о + со3
СН3О
Предложили: X. Снайдер, Дж. Бек и В. Айди.
Проверили: Дж. Джонсон и П. Виттум.
1. Получение
А. Метиловый эфир гомовератровои кислоты. — В 3-литровую
круглодонную колбу помещают 1 л 10%-ного раствора едкого
натра и 200 г @,65 мол.) азлактона а-бензоиламино-ЩЗ,4-диме-
токсифенил)-акриловой кислоты ст. пл. 149—150° (см. стр. 11).
Колбу снабжают обратным холодильником и погружают в масля-
ную баню так, чтобы уровень жидкости в колбе был ниже
уровня масла в бане (примечание 1). Смесь не сильно кипя-
тят в течение б—7 час. до тех пор, пока не прекратится выде-
ление аммиака. Полученный раствор содержит натриевые соли
3,4-диметоксифенилпировиноградной кислоты (примечание 2) и
бензойной кислоты.
ГОМОВЕРАТРОВАЯ КИСЛОТА
165
Этот водный раствор переливают в 2-литровую широкогорлую
коническую колбу и к нему прибавляют 85 мл 40%-ного раствора
едкого натра. Устанавливают механическую мешалку и колбу охла-
ждают смесью льда и соли. Затем при перемешивании добавляют
75 мл 30%-ной перекиси водорода, разбавленной 75 мл воды, с та-
кой скоростью, чтобы температура не поднималась выше 15°. Массу
оставляют при комнатной температуре примерно на 10 час. (лучше
всего на ночь), после чего раствор подкисляют осторожным доба-
влением (примечание 3) 450 мл концентрированной соляной кислоты
(уд. в. 1,19). Теплый подкисленный раствор экстрагируют теплым
бензолом, причем первый раз берут 400 мл бензола и затем два раза
по 200 мл. Соединенные бензольные вытяжки сушат безводным серно-
кислым магнием и фильтруют через обыкновенную вату прямо в
3-литровую круглодонную колбу.
Бензол отгоняют, после чего добавляют 1 л метилового спирта
(примечание 4), содержащего 15 мл концентрированной серной кис-
лоты, и колбу снабжают эффективным обратным холодильником
с хлоркальциевой трубкой. После осторожного пятичасового кипя-
чения обратный холодильник меняют на холодильник, обращенный
вниз, и метиловый спирт отгоняют на водяной бане. Оставшуюся
жидкость охлаждают и взбалтывают с 500 мл холодной воды. Смесь
переносят в делительную воронку и экстрагируют бензолом, для
чего сперва берут 400 мл, а затем Две порции по 200 мл. Соединенные
вытяжки промывают двумя порциями по 100 мл 10%-ного раствора
соды и затем двумя порциями по 100 мл воды. Бензольный раствор
сушат безводным сернокислым магнием, переносят его в двухли-
тровую колбу и бензол отгоняют на водяной бане. Оставшуюся смесь
метиловых зфиров бензойной и гомовератровои кислот переносят
в колбу Клайзена на 250 мл и перегоняют в вакууме. Первая фрак-
ция до 100°/16 мм представляет собою метиловый эфир бензой-
ной кислоты (т. кип. 87°/16 мм) и весит около 75 г (85% теоретич.).
После небольшой промежуточной фракции, составляющей 2—3 г,
собирают чистый метиловый эфир гомовератровои кислоты при 176—
178716 мм или при 129—13171 мм. Выход 76—82 г E6—60%
теоретич., считая на азлактон).
Б. Гомовератровая кислота. В круглодонную колбу емкостью
500 мл помещают 250 мл 10%-ного раствора едкого натра
и 76 г @,36 мол.) метилового эфира гомовератровои кислоты. Колбу
снабжают обратным холодильником и смесь слабо кипятят. Омыле-
ние протекает быстро, и слой сложного эфира исчезает примерно
через 10 мин. Смесь слабо кипятят еще в течение 20 мин., после чего
раствор охлаждают в бане со льдом и затем медленно при поме-
шивании выливают в смесь 125 мл концентрированной соляной
кислоты и 350 г льда. Немедленно выпадают кристаллы гидрата
гомовератровои кислоты. После получасового стояния кристалли-
ческий продукт отфильтровывают с отсасыванием и на фильтре
166
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
промывают двумя порциями ледяной воды по 25 мл. Кристаллы
тщательно отжимают на фильтре, измельчают в порошок и оста-
вляют на ночь в вакуум-эксикаторе над натронной известью (для уда-
ления оставшейся соляной кислоты) и хлористым кальцием. Считая
на эфир, выход кислоты почти количественный и составляет 70 г
E5% теоретич. по исходному азлактону). Полученный продукт
плавится при 96—97° и содержит следы хлористого натрия. Для
очистки кислоту растворяют в 350 мл горячего бензола и раствор
фильтруют. К горячему фильтрату добавляют 150 мл горячего
лигроина (т. кип. 70—80°), раствор покрывают часовым стеклом
и дают медленно охладиться. После стояния в течение нескольких
часов (предпочтительно оставить па ночь) в холодном месте кри-
сталлы отсасывают и промывают холодной смесью 35 мл бензола и
15 мл лигроина, а затем 50 мл холодного петролейного эфира. Кри-
сталлы отжимают, по возможности полностью удаляя растворитель,
и сушат в вакуум-эксикаторе (примечание 5). Вес очищенной гомо-
вератровой кислоты 65 г E1% теоретич., считая на исходный азлак-
тон). Плавится она резко при 98°.
2. Примечания
1. Внутренний уровень должен быть ниже уровня масла, ибо
в противном случае раствор сильно бросает.
2. 3,4-Диметоксифенилпировиноградная кислота может быть
выделена из этого раствора следующим образом (Дж. С. Бэк и
В. С. Айди). Водный раствор натриевых солей насыщают сернистым
газом при температуре ниже 40°. В осадок выпадает бензойная
кислота, которую отсасывают и промывают небольшим количе-
ством воды. Фильтрат и промывные воды помещают в 3-литровую
круглодонную колбу, снабженную механической мешалкой, и
раствор нагревают до кипения. Затем при работающей мешалке
осторожно прибавляют концентрированную соляную кислоту до
ее явного избытка. Кислоту следует добавлять с осторожностью,
так как раствор может легко стать пересыщенным по отношению
к сернистому ангидриду, который в дальнейшем может выделиться
весьма бурно. Выпадает тяжелый осадок 3,4-диметоксифепилпиро-
виноградной кислоты, который по охлаждении отфильтровывают
с отсасыванием. Кислоту сушат и затем промывают двумя порциями
по 50 мл эфира. Выход 3,4-диметоксифенилпировиноградной
кислоты 110—116 г G6—80% теоретич.); т. пл. 181—184°. Дальней-
шая очистка может быть достигнута перекристаллизацией из ледя-
ной уксусной кислоты.
Другой метод получения гомовератровой кислоты (Дж. С. Бэк
и В. С. Айди) состоит в выделении 3,4-диметоксифенилпировиноград-
ной кислоты с последующим се окислением по методу, описанному
во второй части раздела А. Однако при проверке оказалось, что,
ДЕЗОКСИБЕНЗОИН
167
несмотря на исключение стадии этерификации, этот метод дает
худшие результаты по сравнению с описанным выше.
3. При прибавлении кислоты выделяется большое количество
углекислого газа.
4. Нет необходимости применять специально обезвоженный
метиловый спирт. Продажный метанол хорошего качества дает
вполне удовлетворительные результаты.
5. Ввиду того, что кислота образует гидрат, рекомендуется
не подвергать ее действию влаги воздуха.
3. Другие методы получения
Гомовератровая кислота была получена метилированием гомо-
протокатеховой кислоты1 и гомованилиновой кислоты2 йодистым
метилом, а также из вератрового альдегида через азлактон и 3,4-
диметоксифенилпировиноградную кислоту3. Гомовератровая кис-
лота была получена также омылением ее амида или нитрила: амид
был получен из хлорангидрида вератровой кислоты через диазо-
кетон * или из циангидрина вератрового альдегида через а-хлор-
гомовератрамид5; нитрил был получен каталитическим восстано-
влением ацильных производных циангидрина вератрового альде-
гида 6.
Приведенная выше методика основана на литературных данных
по получению гомовератровой кислоты 3 и п-метоксифенилуксусной
кислоты 7.
1 Р i с t e t, G a m s, Ber. 42, 2949 A909).
2 Tiemann, Matsmoto, там же 11, 143 A878).
3 Haworth, Perkin, Rankin, J. Chem. Soc. 125, 1693 A924).
1 Arndt, E i s t e r t, Ber. 68, 205 A935).
6 H a h n, S с h u 1 z, там же 72, 1302 A939).
«Kindle r, Peschke, Arch. Pharm. 271, 432 A933); К i n d 1 e r,
G e h 1 h a a r, там же 274, 377 A936).
' Cain, Simonsen, Smith, J. Chem. Soc. 103, 1036 A913).
ДЕЗОКСИБЕНЗОИН
ЗС6Н5СН2СО2Н + РС13 > ЗС6Н5СН2СОС1 + Н3РО3
С6Н5СН2СОС1 + С6Н6 -^^ C6H5CH2COC6HD + HC1
Предложили: К. Аллен и В. Барке р.
Проверили: К. Марвел и Це-Цинг-Чу.
1. Получение
В круглодонную 1-литровую колбу, соединенную с обратным
холодильником и системой для поглощения хлористого водорода,
помещают 68 г @,5 мол.) фенилуксусной кислоты, а затем приОа-
168
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
вляют 35 г @,25 мол.) треххлористого фосфора. Смесь нагревают
в течение 1 часа на кипящей водяной бане и затем, пока содержимое
колбы еще не остыло, добавляют 400 мл сухого бензола. Бензоль-
ный раствор хлорапгидрида фенилуксусной кислоты сливают с остаю-
щейся в колбе фосфористой кислоты на 75 г @,56 мол.) безводного
хлористого алюминия, находящегося в сухой круглодонной 1-лит-
ровой колбе, которую соединяют с тем же холодильником. Реакция
вначале идет бурно, почему необходимо охлаждение. После этого
смесь нагревают еще в течение 1 часа на водяной бане с обратным
холодильником, охлаждают ее и выливают на смесь, состоящую
из 500 г колотого льда и 200 г концентрированной соляной кислоты.
Бензольный слой отделяют, а водный извлекают один раз смесью
100 мл бензола и 100 мл эфира (примечание 2). Эфирно-бензольный
раствор промывают 100 мл воды (примечание 3) и сушат 40—50 г
хлористого кальция. Раствор фильтруют с отсасыванием (приме-
чание 4) в 1-литровую клайзеновскую колбу и растворитель
удаляют отгонкой в вакууме (примечание 5); остаток представ-
ляет собой коричневое масло, которое при охлаждении засты-
вает.
Сырой продукт (91—92 г) очищают перегонкой в вакууме из
клайзеновской колбы емкостью 250 мл (примечание 6). Продукт
перегоняется при 160°/5 мм; при 172°/15 мм; при 200°/30 мм в виде
бесцветного масла, застывающего при охлаждении. Выход 81—82 г
(82—83% теоретич., считая на взятую фенилуксусную кислоту);
т. пл. 53—54°. Продукт перекристаллизовывается из метило-
вого спирта, причем на каждый грамм берется 4 мл растворителя
(примечание 7). Выход перекристаллизованного продукта с темпе-
ратурой плавления 55—56° составляет 55—56 г. При охлажде-
нии фильтрата смесью льда с солью получают еще 7 г кристаллов
с т. пл. 55—56°. При дальнейшем охлаждении маточных растворов
выпадает около 5 г кристаллов с т. пл. 54—55°. Общий выход очи-
щенного продукта 67—70 г (примечание 8). Дальнейшая перекри-
сталлизация из метилового спирта не повышает точки плавления
продукта выше 55—56°.
2. Примечания
1. Метод получения фенилуксусной кислоты описан в «Синт.
орг. преп.», сб. 1, стр. 440. Фенилуксусная кислота высшего ка-
чества может быть приобретена также у поставщиков эфирных масел
и других препаратов для парфюмерии. В ряде случаев такой продаж-
ный продукт лучше полученного по методике, описанной в «Синт.
орг. преп.». Поскольку качество дезоксибензоина зависит от качества
взятой фенилуксусной кислоты, весьма важно применять кислоту
высшего качества.
ДЕЗОКСИБЕНЗОИН
169
2. Смесь бензола и эфира употребляется вместо эфира, потому
что таким образом достигается лучшее разделение слоев.
3. Промывание раствором едкого натра не является необходи-
мым и даже нежелательно, так как благодаря образованию эмульсии
выход продукта понижается на 8—10%, а качество его не улуч-
шается.
4. Даже если раствор прозрачен, лучше отделять хлористый
кальций фильтрацией, а не декантацией. Мелкие частицы хлори-
стого кальция и хлористого алюминия, оставшиеся в растворе,
ведут к толчкам и даже разложению во время перегонки.
5. Отгонка растворителя в вакууме на водяной бане делает
излишним фракционирование продукта при перегонке.
6'. Необходимо употреблять клайзеновскую колбу с широкой
отводной трубкой, так как дезоксибензоин может застыть и забить
аппарат. Перегонка в вакууме, если только следовать всем сделан-
ным выше указаниям, идет очень спокойно.
7. Метиловый спирт является лучшим растворителем для очистки.
При перекристаллизации из этилового спирта дезоксибензоин часто
выделяется в виде масла.
8. Дезоксибензоин неустойчив на свету, а потому сохранять
его следует в темных сосудах.
3. Другие методы пвлучения
Вследствие доступности исходных реагентов наиболее удобными
методами получения дезоксибензоина являются описанная выше
реакцияФриделя — Крафтса1 и восстановление бензоина2. Дезокси-
бензоин может быть также получен, причем часто с хорошим
выходом, нагреванием бромистого стильбена с водой в запаянной
трубке при 180—190°3; восстановлением бензила 4; действием цинка
и хлористого водорода на хлорбензил 5; при действии бензола на
фенилуксусную кислоту в присутствии фосфорного ангидрида °;
из хлористого бензоила и магний-галоидного производного натрие-
вой соли фенилуксусной кислоты7; из хлористого бензилмагния
и бензамида 8 и щелочным гидролизом дезилтиогликолевой кислоты9.
1 Graebe, Bungener, Ber. 12, 1080 A879).
2 К о h 1 е г, Am. Chem. J. 36, 182 A906); I r v i n e, W e i r, J. Chetn.
Soc. 91, 1388 A907); К о h 1 e r, N у g a a r d, J. Am. Chem. Soc. 52, 4133
A930); В a 1 1 a r d, D e h n, там же 54, 3970 A932).
3 Limpricht, Schwanert, Ann. 155, 60 A870).
4 J a p p, К 1 i n g e m a n n, J. Chem. Soc. 63, 770 A893).
5 T h i e 1 e, Straus, Ann. 319, 163 A901).
6 Z i n cl< e, Ber. 9, 1771 A876).
7 Ivanoff, Nicoloff, Bull. soc. chim. D) 51, 1331 A932).
8 Jenkins, J. Am. Chem. Soc. 55, 704 A933).
9 Behaghel, Schneider, Ber. 68, 1588 A935).
170
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
ДЕКАМЕТИЛЕНГЛИКОЛЬ
(Декандиол-1,10)
С,Н,0,С(СНЛ,СО,С1Н.+8[Н] <с.н.о„ + н«
НО (СН2I0 ОН + 2 С2Н5ОН
Предложил: Р. Манске.
Проверили: В. Каротерс и В. Мак-Ювен,
1. Получение
В 3-литровую круглодонную колбу, снабженную шариковым
холодильником длиною 60 см, который защищен от влаги воздуха
хлоркальциевой трубкой, помещают раствор 65 г @,25 мол.) диэти-
лового эфира себациновой кислоты (стр. 346) в 800 мл абсолютного
этилового спирта (примечание 1). Одновременно к этому раствору
прибавляют 70 г C гр.-ат.) металлического натрия в больших ку-
сках. Довольно бурную реакцию легко регулируют тем, что колбу
погружают в смесь мелкого льда и воды. Через некоторое время
реакция несколько успокаивается, колбу вынимают из охлаждаю-
щей смеси и реакции дают итти без внешнего охлаждения. Для
завершения восстановления смесь нагревают на водяной бане до
полного растворения натрия. Затем смеси дают немного охладиться,
добавляют 300 мл воды и нагревают на кипящей водяной бане до
тех пор, пока не перестанет отгоняться спирт. Оставшееся неболь-
шое количество спирта удаляют в слабом вакууме, пользуясь водо-
струйным насосом. Остаток разбавляют примерно 600 мл горячей
воды и смеси дают спокойно охладиться. Выделившийся масляни-
стый слой застывает, образуя твердую лепешку. Нижний водный
слой легко отделяют декантацией. Твердую массу промывают
один раз небольшим количеством холодной воды, воду удаляют
по возможности полно и вещество сушат, нагревая колбу на водяной
бане при пониженном давлении. Затем остаток экстрагируют че-
тыре раза горячим бензолом порциями по 250 мл. Соединенные
вытяжки обесцвечивают небольшим количеством активированного
угля, фильтруют и отгоняют большую часть, бензола. Остаток (около
60 мл) растворяют в спирте (около 200 мл), раствор опять фильтруют,
упаривают до небольшого объема (около 60 мл) и обрабатывают
равным объемом горячего бензола. При медленном охлаждении
смесь застывает, образуя сплошную массу больших кристаллов,
которые отсасывают и промывают эфиром. Выход гликоля с т. пл.
72—74° (испр.) составляет 32—33 г G3—75% теоретич.; примеча-
ния 2, 3 и 4).
2. Примечания
1. Спирт должен быть абсолютно сухим. Малейшие следы влаги
вызывают немедленно омыление зфира и соответственное уменьшение
выхода. Прекрасный метод получения действительно абсолютного
ДЕКАМЕТИЛЕНГЛИКОЛЬ
171
спирта состоит в том, что в 1 л 99,5%-ного спирта растворяют
7 г натрия, добавляют 27,5 г какого-нибудь высококипящего слож-
ного эфира, например, диэтилового эфира фталевой кислоты, кипя-
тят с обратным холодильником и спирт перегоняют г.
2. Если последовательно ставится несколько опытов получения
декаметиленгликоля или же если работают с большими количе-
ствами исходного вещества, соединенные маточные растворы можно
упарить, до полного удаления растворителя и остаток перегнать
в вакууме. Однако после перекристаллизации дестиллата из смеси
спирта и бензола получается только 5—7 г чистого гликоля на моль
себацинового эфира.
3. Общий метод Буво и Блана широко применяется для полу-
чения гликолей. Были предложены различные мелкие изменения
этой реакции, по большинство из них не имеет особого значения.
При проверке метода оказалось, что быстрое прибавление смеси
спирта и эфира к натрию дает примерно такие же результаты, как
и описанный выше способ, а механическое перемешивание реакцион-
ной смеси заметно понижает выход2. Выбор способа выделения того
или иного гликоля зависит от свойств последнего. Для малых
загрузок, описанных выше, метод кристаллизации является без-
условно наиболее подходящим в случае декаметиленгликоля. Низ-
шие члены этого рЗДа гликолей кристаллизуются не так легко.
8 частном сообщении К. Марвел указал на то, что для этих гли-
колей наилучшим методом является непрерывная экстракция эфи-
ром. В случае гексаметиленгликоля эта методика была с успехом
применена при проверке метода.
Пользуясь той же методикой, лица, проверявшие этот метод,
синтезировали следующие гликоли:
Выход, °/о
Гептаметиленгликоль т. кип. 143—146°/8 мм 88
Нонаметиленгликоль ь » 147—150°/2 мм 71
Ундекаметиленгликоль т. пл. 48—50° 57
Тридекаметиленгликоль » » 75—77° 88
Тетрадекаметиленгликоль » » 83—85° 61
Октадекаметиленгликоль » » 96—98° 54
Гептаметиленгликоль был выделен посредством непрерывной
экстракции эфиром щелочного раствора после восстановления.
Предварительно раствор был разбавлен водой и от него был ото-
гнан спирт. Нопаметилепгликоль был отделен от щелочной жидкости
декантацией (как описано выше) и подвергнут перегонке. Все дру-
гие гликоли были выделены кристаллизацией из бензола (без доба-
вления спирта). Такие же хорошие результаты были получены и при
больших загрузках (например 0,5 мол. сложного эфира).
4. Декаметиленгликоль может быть также получен с прекрас-
ным выходом восстановлением технического бутилового эфира себа-
циновой кислоты, пользуясь методикой, приведенной для синтеза
гексаметиленгликоля (стр. 149).
172
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
3. Другие методы получения
Декаметиленгликоль лучше всего получать восстановлением
эфиров себациновой кислоты натрием и спиртом 2 по методу Буво-
Блана, описанному выше, или же восстановлением водородом в при-
сутствии катализатора 3; такая методика описана на стр. 149. Оба
метода применимы для получения целого ряда других гликолем.
Декаметиленгликоль был также получен восстановлением амида
себациновой кислоты натрием и амиловым спиртом 4 и восстановле-
нием диметилового эфира себациновой кислоты натрием и жидким
аммиаком в абсолютном спирте 5.
1 S m i t h, J. Chem. Soc. 1927, 1288; Manske, J. Am. Chem. Soc 53,
1106 A931).
2 Bouveault, Blanc, Compt. rend. 137, 329A903); Bull soc chim
C) 31, 1205 A904); герм. пат. 164 294 [Frdl. 8, 1260 A905—7)]; F r a n k e,
Kienberger, Monatsh. 33, 1191 A912); Chuit, Helv. Chim. Acta
9,264A926); С а г о t h e r s, Hill, К i r b y, I а с о b s о n, I. Am
Chem. Soc. 52, 5287 A930).
3 F о 1 k e r s, A d k i n s, там же 54, 1146 A932).
1 Scheuble, Monatsh. 24, 623 A903); Scheuble, Loeble,
там же 25, 344 A904); Albert i, Smieciuszewski, там же 27, 411
A906).
5 С h a b 1 а у, Compt. rend. 156, 1021 A913); Ann.„chim. (9) 8, 216 A917).
ДИАЗОАМИНОБЕНЗОЛ
G,3-Дифенилтриазен)
C6H5NH3C1
NaNO2 -|- HCl
C6HbN2Cl + 2 H2O
NaCl
CeH3NH3Cl + CH3CO2Na > NaCl
C6H5NH2 + C6HflN2Cl + CH3CO2Na
C6H5N = NNHC6H
CH3CO.H + C,H3NH,
6H5
NaCl
CH3CO2H
Предложили: В. Хартман и Дж. Дикки.
Проверили: К. Ноллер и К- Кемп.
1. Получение
В 5-литровую колбу, снабженную механической мешалкой и
капельной воронкой, помещают 1 кг мелкого льда, 1,5 л воды,
279 г C мол.) технического анилина и 458 г C88 мл, 4,5 мол.) кон-
центрированной соляной кислоты (уд. в. 1,18). Пускают в ход
мешалку и в течение 15 мин. добавляют раствор 109 г A,5 мол.)
95%-ного нитрита натрия в 250 мл воды. Реакционную смесь пере-
мешивают 15 мин., после чего в течение 5 мин. добавляют раствор
422 г C,1 мол.) кристаллического уксуснокислого натрия в 800 мл
ДИАЗОАМИНОБЕНЗОЛ
173
воды. Немедленно начинается образование желтого осадка диазо-
аминобензола. Перемешивание продолжают еще 45 мин., причем
температура смеси должна быть ниже 20° (примечание 1), после
чего желтый диазоаминобензол отсасывают на воронке Бюхнера
диаметром 19 см (примечание 2), промывают его 5 л холодной воды,
отсасывают возможно лучше (примечание 3) и сушат на листе
фильтровальной бумаги. Полученный таким образом продукт
растворяют в 4 л кипящего бензина с т. кип. 60—90° (примеча-
ние 4), раствор фильтруют, дают ему охладиться до комнатной
температуры и оставляют на ночь. По окончании кристаллизации
желтые кристаллы отсасывают на воронке Бюхнера диаметром
19 см, промывают 500 мл холодного бензина с т. кип. 60—90° и
сушат при комнатной температуре. Выход желтых кристаллов
с т. пл. 92—94° составляет 242—251 г (82—85% теоретич.; при-
мечание 5). Если желательно получить еще более чистый про-
дукт, диазоаминобензол растворяют в 4 л кипящего бензина с
т. кип. 60—90° и подвергают кристаллизации, как это описано
выше. Выход перекристаллизованного диазоаминобензола с т. пл.
94—96° составляет 204—218 г F9—73% теоретич.; примеча-
ния 6 и 7).
2. Примечания
1. Является ли указанная выше температура максимальной,
при которой возможно вести реакцию, остается невыясненным.
2. Еще лучше воспользоваться центрифугой соответственных раз-
меров.
3. С целью удаления наибольшего количества воды воронку
Бюхнера покрывают резиновой пластиной, которую закрепляют
с помощью резинового кольца.
4. Длительное нагревание диазоаминобензола с бензином вызы-
вает разложение; поэтому прежде чем добавлять к бензину продукт,
подлежащий перекристаллизации, лигроин следует нагреть до
кипения. Растворение проводится по возможности быстро. Если
сырой диазоаминобензол недостаточно высушен, на дне колбы
выделяется слой воды. Его необходимо полностью отделить, прежде
чем фильтровать горячий бензиновый раствор.
5. Если маточные растворы упарить до объема в 1 л и охладить
их льдом, можно выделить еще 20—25 г кристаллов с т. пл. 79—
83°.
6. Можно вести синтез с половинным количеством исходных
реагентов. При этом получается около 125 г продукта.
7. Согласно литературным данным, чистый диазоаминобензол
представляет собой слегка желтоватые кристаллы с т. нл. 100°.
Двайер описал метод очистки диазоаминобензола, а также ряда
других аминоазосоединений \
174
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
3. Другие методы получения
Диазоаминобензол был получен действием нитрита натрия
на сернокислую соль анилина2;действием нитрита натрия на соляно-
кислую соль анилина 3; действием нитрита натрия и ацетата натрия
на солянокислый анилин4; действием нитрата аммония и серо-
водорода на солянокислый анилин в присутствии железа 5; дей-
ствием нитрита натрия и хромата или бихромата калия на анилин6;
действием амилнитрита на анилин 7.
Диазоаминобензол был получен также действием ангидрида
азотистой кислоты на анилин в спиртовом растворе 8; действием
нитрита серебра на солянокислый анилин 9, а также вместе с фенил-
мочевиной действием нитрозофенилмочевины на анилин в метило-
вом спирте 10. Ниментовский и Рожковский и исследовали диазо-
тирование анилина, солянокислой и сернокислой солей анилина
нитритом натрия и нитритом серебра. Описанный здесь метод
заимствован у Фишера 4.
1 Dwyer, J. Soc. Chcm. Ind. 56, 70T A937).
2 Staedel, Bauer, Ber. 19, 1952 A886).
3 Martius, Zeit. fur Chem. 1866, 381; Curtius, Ber. 23, 3033 A890);
V a u be 1, Chem. Ztg. 35, 1238 A94).
1 Fischer, Ber. 17, 641 A884).
5 Vaubel, Chem. Ztg. 37, 637 A913).
«Dwyer, Mellor, Trikojus, J. Proc. Roy. Soc. N. S. Wales,
66, 315 A932) [C. A. 27, 1331 A933)].
' Meyer, Ambuhl, Ber. 8, 1073 A875).
8 G r i e s s, Ann. 121, 257 A862).
"Niementowski, Roszkowski, Z. physik. Chem. 22, 158
A897).
10 Ha a ger, Monatsh. 32, 1089 A911).
uNiementowski, Roszkowski, Z. physik. Chem. 22, 145A897).
ДИАЗОМЕТАН
CH3N (NO) CONH2+ KOH CH2N2-f KCNO + 2 H2O
Предложил: Ф. Арндт.
Проверили: К. Ноллер и И. Бергштейнсон.
1. Получение
Диазометан чрезвычайно ядовит. При получении
диазометана а при работе с ним необходимо соблю-
дать сугубую осторожность.
В круглодонную колбу емкостью 500 мл помещают 60 мл 50%-
ного водного раствора едкого кали и 200 мл эфира. Смесь охлаждают
до 5°, после чего при взбалтывании прибавляют 20,6 г @,2 мол.)
нитрозометилмочевины (стр. 373). Колбу присоединяют к обращен-
ному вниз холодильнику, нижний конец которого снабжен алон-
ДИАЗОМЕТАН
175
жем, проходящим через резиновую пробку с двумя отверстиями
и погруженным в 40 мл эфира, находящихся в конической
колбе на 300 мл, охлаждаемой смесью льда и соли. Отходящие
газы проходят еще через одну ловушку с эфиром D0 мл), которая
также охлаждается до температуры ниже 0°. Реакционную колбу
погружают в водяную баню, нагретую до 50°, и эфир в колбе дово-
дят до кипения, причем время от времени колбу взбалтывают.
Отгонку эфира прекращают, как только переходящий дестиллат
сделается бесцветным. Обычно это наступает после того, как ото-
гнаны 2/3 эфира. Ни в коем случае не следует отгонять весь эфир.
В обоих приемниках с эфиром находится в растворенном состоя-
нии от 5,3 до 5,9 г диазометана F3—70% теоретич.; примечания 1
и 2), который для большинства целей является достаточно сухим.
Если требуется сухой (примечание 3) эфирный раствор, то
его оставляют стоять 3 часа над чистым гранулированным едким
кали (примечание 4). Для получения совершенно сухого раствора
его сушат натриевой проволокой.
2. Примечания
1. Для анализа определенный объем полученного раствора
(около Vso части) прибавляют при 0° к точной навеске чистой бен-
зойной кислоты (около 1,3 г), растворенной в 50 мл абсолютного
эфира. Бензойная кислота должна быть взята в избытке, о чем
можно заключить по полному обесцвечиванию раствора диазо-
метана. Не прореагировавшую бензойную кислоту оттитровывают
0,2 н. раствором щелочи.
2. Та же методика может быть применена и для получения
в два или три раза большего количества диазометана.
3. Полученный по этому способу раствор диазометана не содер-
жит ни аммиака, ни метилового спирта. В нем содержатся следы
метиламина, который образуется также и при получении диазо-
метана из нитрозометилуретана.
Если не требуется чистого безводного раствора, как это часто
случается при экспериментах с небольшим количеством материала,
можно воспользоваться упрощенной методикой. К 100 мл эфира
добавляют 30 мл 40%-ного едкого кали и смесь охлаждают до 5°.
Затем при непрерывном охлаждении и взбалтывании в течение
1—2 мин. добавляют маленькими порциями всего 10 г хорошо
измельченной нитрозометилмочевины. Темножелтый эфирный слой
легко отделяют декантацией; он содержит 2,8 г диазометана, а также
небольшое количество растворенных примесей и воды. Последнюю
можно удалить, высушив раствор гранулированным едким кали
в течение 3 час. Аналогичным образом можно получить раствор
диазометана в бензоле и других не смешивающихся с водой органи-
ческих растворителях.
176
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
4. Не следует применять едкое кали в виде разломанных пало-
чек, так как острые края облегчают разложение диазометана.
3. Другие методы получения
Для приготовления диазометана существуют четыре метода,
которые имеют практическое значение: дейазие спиртового раствора
едкого кали х или натрия, растворенного в гликоле 2, на нитрозо-
метилуретан; нагревание смеси едкого кали, хлороформа, гидра-
зингидрата и абсолютного спирта3; описанный выше метод —
действие едкого кали на нитрозометилмочевину4; действие алко-
голятов на нитрозо-производное р-метиламиноизобутилметилке-
тона 5. Обыкновенно выбор способа зависит от наличия того или
иного исходного материала. Способы приготовления исходных
материалов для первых трех методов описаны в настоящем сбор-
нике; способ приготовления нитрозопроизводного р-метиламино-
изобутилметилкетона, а из него — диазометана будет помещен в
следующем сборнике «Синт. орг. преп.».
Арндт, Леве и Аван 6, равно как и Эистерт7, в соответствующих
статьях рассмотрели различные способы приготовления диазо-
метана.
1 Pechmann, Ber. 27, 1888 A894); 28, 855 A895).
2 Meerwein, Burneleit, там же 61, 1845 A928).
3 S t a u d i n g e r, Kupfer, там же 45, 505 A912).
* Arndt, Amende, Angew. Chem. 43, 444 A930); Arndt, Scholz,
там же 46, 47 A933).
6 A d a m s о n, Kenner, J. Chem. Soc. 1935, 286; 1937, 1551.
6 Arndt, L о e w e, A v a n, Ber. 73, 606 A940).
' E i s t e r t, Angew. Chem. 54, 99, 124 A941).
сн3
NO.,
2,4-ДИАМИНОТОЛУ ОЛ
B,4-Толуилендиамин)
сн,
NIL
4H2O
Предложили: С. Мэхуд и Л. Шаффнер.
Проверили: Р. Адаме и П. Шильднек.
1. Получение
В трехгорлую колбу емкостью 500 мл, снабженную обратным
холодильником и механической мешалкой (примечание 1), поме-
щают 45,5 г @,25 мол.) 2,4-динитротолуола (примечание 2), 85 г
2,4-ДИАМИНОТОЛУОЛ
177
A,5 мол.) железа (примечание 3) и 100 мл 50%-ного (по весу) эти-
лового спирта (примечание 4). Смесь нагревают до кипения на водя-
ной бане, пускают в ход мешалку (примечание 5) и медленно при-
бавляют (примечание 6) раствор 5,2 мл @,06 мол.) концентрирован-
ной соляной кислоты в 25 мл 50%-ного (по весу) этилового спирта.
После того как вся кислота прибавлена, смесь кипятят с обратным
холодильником в продолжение 2 час. По истечении этого времени
холодильник отъединяют и горячую смесь слабо подщелачивают
на лакмус прибавлением вычисленного количества 15%-ного спир-
тового раствора едкого кали (примечание 7). Не давая смеси охла-
диться, отфильтровывают железо и споласкивают реакционную
колбу двумя порциями 95%-ного этилового спирта по 50 мл каждая;
этим же спиртом промывают остаток железа. К фильтрату прибав-
ляют 84 мл 6-н. серной кислоты, после чего выделяется средняя
сернокислая соль 2,4-диаминотолуола. Смесь охлаждают до 25°,
выпавшую соль отсасывают, промывают двумя порциями по 25 мл
95%-ного этилового спирта, сушат на воздухе в течение 2 час.
(примечание 8), а затем — до постоянного веса при 110°. Выход
49 г (89% теоретич.) сернокислого диаминотолуола, плавящегося
с разложением при 249—251° (примечание 9).
Раствор 20 г сернокислого 2,4-диаминотолуола в 200 мл воды,
нагретой до 60°, охлаждают до 40° (примечание 10) и прибавляют
к нему насыщенный раствор едкого натра (примечание 11) до щелоч-
ной реакции на лакмус. К полученному раствору прибавляют
еще 15 г сернокислого 2,4-диаминотолуола, растворяют его нагре-
ванием смеси до 55°, раствор охлаждают до 40° и снова слегка
подщелачивают на лакмус насыщенным раствором едкого натра.
Смесь охлаждают до 30° и отсасывают выпавший диаминотолуол.
Остаток сернокислого диаминотолуола A4 г) растворяют в филь-
трате при нагревании до 55°. Раствор охлаждают до 40° и опять
прибавляют насыщенный раствор едкого натра до щелочной реакции
на лакмус. Затем смесь охлаждают до 25° и отсасывают вторую
порцию диаминотолуола на воронке Бюхнера. Весь полученный
продукт сушат в эксикаторе над хлористым кальцием до постоян-
ного веса. Выход 26,5 г (95% теоретич., считая на сернокислый
диаминотолуол) продукта, плавящегося при 97—98,5°.
С целью очистки сырой диаминотолуол (примечание 12) раство-
ряют в 8-кратном количестве бензола B12 г) при 70°, и быстро
фильтруют раствор через нагретую воронку Бюхнера (примеча-
ние 13) с умеренным отсасыванием (примечание 14). Затем охла-
ждают фильтрат до 25° и декантируют маточный раствор с корич-
невых кристаллов. Маточный раствор упаривают до объема 25 мл,
отгоняя бензол в вакууме, охлаждают остаток до 25°, выпавшие
кристаллы диаминотолуола отделяют от жидкости декантацией,
присоединяют к первой порции и сушат весь продукт на воздухе.
Выход 22,5 г (81% теоретич., считая на сернокислый диамино-
12 Сборник № 2
178
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
толуол, и 74%, считая на динитротолуол) диаминотолуола, плавя
щегося при 98° (примечание 15).
2. Примечания
1. Чтобы предупредить спекание железа и местный перегрев
мешалка должна доходить почти до дна колбы.
2. 2,4-Динитротолуол не должен содержать маслообразнью
примесей и должен плавиться при 71°.
3. Железо должно быть или в виде порошка или в виде опилок
и в том и в другом случае для получения повторимых результате!
оно должно нацело проходить через сито в 100 меш. Если взяи
более крупные железные опилки, то восстановление может не пройть
до конца, хотя было достигнуто полное восстановление и с образцом
железа в порошке, содержащем опилки.
4. Концентрация спиртового раствора имеет большое значение.
С 95%-ным спиртом восстановление не проходит полностью, не про-
ходит оно до конца и с метанолом, как это предложил Уэст х. Реак-
ция идет до конца в водном растворе, но из реакционной смеси
трудно выделить диаминотолуол, и он получается недостаточно
чистым.
5. Мешалка должна вращаться с такой скоростью, чтобы железо
не оседало на дно, иначе железо спекается. Обычно хорошее пере-
мешивание достигается при скорости 750 об/мин,
6. Реакция между железом и соляной кислотой протекает очень
бурно, особенно в том случае, когда железо взято в порошке, и
поэтому соляную кислоту следует прибавлять сначала очень мед-
ленно. В течение первых 10 мин. следует прибавлять по одной капле
через каждые 10 сек., а затем можно прибавлять одну каплю уже
через каждые 4 сек. На все прибавление требуется около 30 мин.
7. Точное количество спиртового раствора едкого кали опре-
деляют предварительным титрованием 5,2 мл концентрированной
соляной кислоты спиртовым раствором едкого кали.
8. Большую часть спирта и альдегид следует удалить при ком-
натной температуре. При нагревании влажного вещества сразу
до 110° всегда образуются побочные продукты оранжевого цвета.
9. Тот же выход (в процентах) сернокислого 2,4-диаминотолуола
получается при увеличении загрузки в шесть раз.
10. Раствор охлаждают перед прибавлением щелочи, так как
нейтрализация сопровождается повышением температуры.
11. Следует пользоваться насыщенным раствором едкого натра,
так как надо по возможности избегать разбавления раствора диа-
минотолуола.
12. Диаминотолуол, выделенный из сернокислой соли, содержит
небольшие количества сернокислого диаминотолуола и, возможно,
сернокислого натрия, причем оба они в бензоле нерастворимы.
ДИБЕНЗАЛЬАЦЕТОН
179
13. Воронка Бюхнера должна быть горячей G0° и выше), так
как в противном случае кристаллы диаминотолуола могут выпасть
на воронке.
14. При сильном отсасывании бензол закипает и диаминотолуол
кристаллизуется в воронке.
15. При перегонке сухого перекристаллизованного диаминото-
луола под атмосферным давлением с коротким воздушным холодиль-
ником был получен желтый продукт с температурой кипения 292°.
При перегонке перекристаллизованного вещества в вакууме при
148—150°/8 мм был получен бесцветный диаминотолуол.
3. Другие методы получения
2,4-Диаминотолуол может быть получен из 2,4-динитротолуола
восстановлением железом и уксусной кислотой2, электролити-
ческим восстановлением 3 или восстановлением водородом в при-
сутствии никеля, приготовленного по Ренею 4, а также восстановле-
нием 4-нитро-о-толуидина или хлорангидрида 2,4-динитробензой-
ной кислоты 6 оловом и соляной кислотой 6.
1 We s t, J. Chem. Soc. 127, 494 A925).
2 Hoimann, Jahresber. 1861, 512.
3Hofer, Jakob, Ber. 41, 3192 A908).
4 A 1 b e r t, R i t с h i e, J. Proc. Roy. Soc. N. S. W a 1 e s 74, 74 A940)
[C. A. 34, 7286 A940)].
6 Nblting, Collin, Ber. 17, 268 A884).
«Красуский, ЖРХО 27, 337 A896).
ДИБЕНЗАЛЬАЦЕТОН
A,5-Дифенилпентанон-З)
2 C6H6CHO + СН3СОСН3
(NaOH)
С6Н3СН = СН — СО — СН = СНС6Н6 + 2 Н2О
Предложили: Ч. Конард и М. Доллиеер.
Проверили: X. Лозе и К- Ноллер.
1. Получение
Охлажденный раствор 100 г едкого натра в 1 л воды и 800 мл
спирта (примечание 1) помещают в 2-литровую широкогорлую
склянку, окруженную водой и снабженную механической мешалкой.
Температуру раствора поддерживают при 20—25° и энергично
его перемешивают (примечание 2), причем к раствору прибавляют
половину смеси, приготовленной из 106 г A мол.) бензальдегида
и 29 г @,5 мол.) ацетона (примечание 3). Через 2—3 мин. появляется
180
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
желтая муть, которая скоро переходит в хлопьевидный осадок.
Через 15 мин. прибавляют остаток смеси и сосуд, в котором она
находилась, споласкивают небольшим количеством спирта; послед-
ний также прибавляют к общей массе. Сильное перемешивание
продолжают еще в продолжение 30 мин., после чего выпавший
кашеобразный осадок отсасывают на большой воронке Бюхнера.
Продукт тщательно промывают дестиллированной водой (приме-
чание 4) и сушат до постоянного веса при комнатной температуре.
Выход 105—ПО г (90—94% теоретич.; примечание 5). Продукт
плавится при 104—107°.
Сырой дибензальацетон может быть перекристаллизован из
горячего этилового эфира уксусной кислоты, причем берется 100 мл
растворителя на каждые 40 г вещества. При перекристаллизации
получают около 80% продукта с температурой плавления ПО—
111*
2. Примечания
1. Спирт берут в количестве, достаточном для быстрого раство-
рения бензальдегида и для удержания в растворе бензальацетона,
пока он не прореагирует со второй молекулой альдегида. Более
слабые растворы щелочи замедляют образование дибензальацетона
и таким образом способствуют побочным реакциям и ведут к обра-
зованию липкого продукта. Крепкие растворы щелочи создают
затруднения при промывке. Указанные концентрации почти те же,
которые применяются при синтезе бензальацетофенона по методу,
описанному в «Синт. орг. преп.», сб. 1, стр. 77.
2. Были испытаны лишь температуры между 20 и 25°, так как
Предполагалось, что изменение температуры окажет то же действие,
что и при упомянутом выше получении бензальацетофенона.
Перемешивание очень важно, так как благодаря ему достигается
однородность продукта.
3. Употреблялся бензальдегид, промытый раствором соды и
перегнанный. Для работы был взят продажный ацетон марки
«х. чл. Для реакции берут теоретические количества, так как избы-
ток бензальдегида ведет к образованию липкого продукта, а избы-
ток ацетона способствует образованию бензальацетона. Чтобы
обеспечить прибавление эквивалентных количеств, смесь приго-
товляют непосредственно перед прибавлением.
4. Так как продукт практически не растворим в воде, то при
промывке можно брать ее в большом количестве. Соединения натрия,
по всей вероятности, являются главными примесями. Высушен-
ный продукт содержит некоторое количество соды, так как
отмыть едкий натр полностью трудно. Остаются также примеси,
которые не растворяются в воде. Тем не менее продукт доста-
точно чист для применения почти во всех реакциях.
1,4-ДИБЕНЗОИЛБУТАН
181
5. Если выпавший кашеобразный осадок оставить стоять не-
сколько часов, охладить и фильтровать холодным, получается
несколько больший выход, но делать это, однако, особого смысла
не имеет. Фильтрат можно применить в качестве раствора для вто-
рой операции, причем выход в этом случае будет около 93% теоретич.
Точка плавления этого второго продукта несколько ниже.
3. Другие методы получения
Дибензальацетон может быть получен конденсацией бензаль-
дегида с ацетоном, причем в качестве конденсирующих средств
были применены: сухой хлористый водород1, 10%-ный раствор
едкого натра 2 и ледяная уксусная кислота с серной кислотой 3.
Его получали также конденсацией бензальацетона с бензальдеги-
дом в присутствии разбавленного раствора едкого натра *. Штраус
и Экер 5 первые применили для кристаллизации продукта этило-
вый эфир уксусной кислоты.
1 Claiscn, Claparcde, Ber. 14, 350 A881).
2 S с h m i d t, тамже 14, 1460 A881); С 1 a i s e n, там же 14, 2470 A881);
Straus, С a s p a r i, там же 40, 2698 A907).
3 С 1 a i s e n, Claparede, тамже 14, 2460 A881).
4 Claisen, Ponder, Ann. 223, 141 A884).
5 Straus, Ecker, Ber. 39, 2988 A906).
CH2CH2CO2H
I -f
CH2CH2CO2H
CH2CH2COC1
CH-CH.COC1
1,4-ДИБЕНЗОИЛБУТАН
A,6-Дифенилгександион-1,6)
СН2СН2СОС1
2SOC12 | + 2SO2 + 2HC1
СН2СН2СОС1
WCI| СН2СН2СОС6На
+ 2С6Н6 — Л | + 2НС1
СН2СН2СОС6Н5
Предложили: Р. Фыозон и Дж. У онер.
Проверили: В. Каротерс и В. Мак-Ювен.
1. Получение
146 г A мол.) адипиновой кислоты («Синт. орг. преп.», сб. 1,
стр. 15), предварительно высушенной в вакууме над серной кисло-
той, помещают в ^литровую круглодонную колбу, соединенную
с обратным холодильником, и приливают сразу 357 г B17 мл,
3 мол.) хлористого тионила. Смесь слабо подогревают на водяной
бане, нагретой до 50—60°. Приблизительно через 4 час. наступает
полное растворение и выделение хлористого водорода прекра-
щается. Колбу соединяют с обращенным вниз холодильником и
182
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
нагревают в вакууме на водяной бане, чтобы отогнать избыток
хлористого тионила. Остающийся в колбе светложелтый хлор-
ангидрид адипиновой кислоты может быть сразу пущен в дальней-
шую обработку.
Смесь 300 г B,25 мол.) безводного хлористого алюминия и
1500 мл A7 мол.) бензола (высушенного и перегнанного над натрием,
помещают в 3-литровую трехгорлую колбу, снабженную мешалкой
с ртутным затвором, обратным холодильником и капельной ворон-
кой. Реакционную смесь охлаждают в бане с водой и льдом и при
энергичном перемешивании приливают через капельную воронку
хлорангидрид адипиновой кислоты. Прибавление хлорангидрида
ведут с равномерной скоростью в течение 45 мин.
(примечание 1). Реакционная смесь слегка темнеет, но не делается
черной. После того как прибавлено все количество хлорангидрида,
баню с водой и льдом отставляют и перемешивание продолжают
еще 2 часа при комнатной температуре (примечание 2).
Затем раствор медленно выливают при постоянном перемеши-
вании на смесь 1 кг колотого льда и 200 мл концентрированной
соляной кислоты, помещенной в 5-литровой колбе. Разложение
надо вести таким образом, чтобы по его окончании оставалось еще
некоторое количество льда. Когда весь лед растает, смесь из воды,
выпавшего дибензоилбутана и бензола делят на две равные части
и к каждой из них прибавляют по 1—1,25 л бензола. Осадок раство-
ряют при взбалтывании и слабом подогревании на водяной бане;
бензольные слои отделяют и промывают сначала равным объемом
слабого раствора соды, а затем водой.
Бензольный раствор помещают в 5-литровую колбу и отгоняют
3—3,5 л бензола, а оставшейся жидкости дают охладиться, ^ерез
несколько часов дибензоилбутан выкристаллизовывается и его
отсасывают. Продукт плавится без перекристаллизации при 104—
107° (примечание 3). К светлокоричневому фильтрату прибавляют
равный объем эфира и таким образом получают еще некоторое
количество кристаллов (примечание 4).
Выход неочищенного продукта 199—216 г G5—81% теоретич,).
Он может быть перекристаллизован растворением в 1 л горячего
95%-ного этилового спирта. После охлаждения выделяются кри-
сталлы с т. пл. 106—107°. Выход перекристаллизованного продукта
190—210 г.
2. Примечания
1. Очень существенно, чтобы во время прибавления хлоран-
гидрида адипиновой кислоты реакционная смесь оставалась холод-
ной, иначе может произойти заметное осмоление и продукт полу-
чится окрашенным.
2. Если реакционную смесь оставить стоять на некоторое время,
выход значительно понижается.
Y-ДИБРОМГИДРИН ГЛИЦЕРИНА
183
3. На точку плавления и окраску неочищенного продукта, пови-
димому, оказывает влияние качество хлористого алюминия. При
проверке брался очень чистый хлористый алюминий, причем
окраска реакционной смеси никогда не была темнее яркооранже-
вой, а неочищенный продукт получался чисто белого цвета и неиз-
менно плавился при 106—108°. В случае применения менее чистого
хлористого алюминия сырой продукт иногда получался бурого
цвета. От этой окраски можно полностью освободиться, промыв
окончательный продукт несколькими миллилитрами _ холодного
эфира, в котором дикетон почти нерастворим.
4. Во время проверки было установлено, что при прибавлении
эфира выделяется только 5—6 г вещества, поэтому едва ли имеет
смысл производить эту операцию.
3. Другие методы получения
Дибензоилбутан был получен при действии хлористого алюми-
ния на смесь бензола и хлорангидрида адипиновой кислоты * или
на смесь бензола и полимеризованного ангидрида адипиновой
кислоты2. Он был также получен из нитрила адипиновой кислоты
и бромистого фенилмагния 3 и в качестве побочного продукта при
действии цинка на а, Р-дибромпропиофенон * и а,а'-дибромбензоил-
бутан5. В литературе для дибензоилбутана указывались разные
температуры плавления между 102° х и 112° *.
1 Е ta i х, Ann. chim. phys. G) 9, 372 A896).
a H i 11, J. Am. Chem. Soc. 54, 4105 A932).
3 Compere, Bull. soc. chim. Belg. 44, 523 A935).
4 Ко h 1 e r, Am. Chem. J. 42, 384 A909).
5 F u s о n, F a r 1 о w, J. Am. Chem. Soc. 56, 1593 A934).
a, y-ДИБРОМГИДРИН ГЛИЦЕРИНА
A,3-Дибромпропанол-2)
3 HOCH2CHOHCH2OH + 2 P+ 3Br2
> 3BrCH2CHOHCH2Br+2P(OHK
Предложила: Г. Браун.
Проверили: Р. Фьюзон и С. Бабкок.
1. Получение
В 3-литровой круглодонной колбе, снабженной мощной мешал-
кой с глицериновым затвором, капельной воронкой и трубкой для
отвода выделяющихся газов, тщательно перемешивают 1,6 кг
A7,4 мол.) глицерина с 200 г F,5 гр.-ат.) красного фосфора (при-
мечание 1). Затем через капельную воронку приливают в течение
8 час. при энергичном перемешивании (примечание 2) 900 мл B808 г,
184
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
17,5 мол.) брома (примечание 3). Для того чтобы по возможности
уменьшить улетучивание брома, конец капельной воронки должен
доходить почти до дна колбы. Отходящие газы, образующиеся
в результате побочных реакций и состоящие главным образом из
бромистого водорода и некоторого количества брома, пропускают
через концентрированный раствор едкого натра или через газовую
ловушку. Реакция проходит с выделением тепла, и температура
быстро повышается до 80—100°; прибавление брома регулируют
таким образом, чтобы температура смеси держалась на этом уровне.
К концу прибавления брома колбу погружают в воду, нагретую
до 70—75°. После того как весь бром прибавлен, смесь оставляют
на ночь, а затем нагревают на водяной бане до полного поглощения
брома A—2 часа).
Смесь переносят в 3-литровую круглодонную колбу, снабжен-
ную резиновой пробкой с двумя отверстиями, через которые про-
ходят широкая отводная трубка и капилляр. Колбу помещают в
масляную баню и жидкость подвергают перегонке под уменьшенным
давлением, пользуясь водоструйным насосом. Приемник охлаждают
водой. Сперва переходит смесь бромистоводородной кислоты с водой;
затем перегоняется дибромгидрин. Температуру бани поднимают
настолько быстро, насколько это возможно, и доводят ее до 180°.
К концу перегонки необходимо тщательное наблюдение: ее пре-
рывают при первых признаках разложения. Последнее становится
заметным по обильному газообразованию, в результате которого
давление в перегонной установке повышается. К дестиллату,
окрашенному в соломенножелтый цвет, добавляют небольшой
избыток твердого бикарбоната натрия при энергичном взбалтыва-
нии, пока не прекратится выделение углекислоты. Неорганиче-
ские соли отсасывают и водный слой фильтрата отделяют от сырого
дибромгидрина. Последний очищают перегонкой в вакууме из
2-литровой колбы Клайзена. Перегонку ведут до тех пор, пока не
перестанет отгоняться вода и температура внутри колбы не повы-
сится до 100°. Дибромгидрин (в отгоне) отделяют от воды, сушат
безводным сульфатом натрия, фильтруют и выливают обратно
в перегонную колбу. Таким образом удается отделить главную
массу воды (примечание 4). Затем перегонку продолжают, и после
небольшой головной фракции собирают дибромгидрин при ПО—
112°/20 мм (примечание 5). Выход 2000—2050 г E2—54% теоретич.).
Дибромгидрин представляет собой тяжелую бесцветную жид-
кость с характерным запахом. При стоянии он постепенно желтеет.
Уд. в. его при 20° около 2,14.
2. Примечания
1. Красный фосфор следует тщательно перемешать с глицери-
ном, прежде чем начать прибавление брома. Бром не должен при-
2.6-ДИБРОМ-4-НИТРОФЕНОЛ
185
ходить в соприкосновение с сухим фосфором, иначе может произойти
бурная реакция.
2. Ввиду большой вязкости реакционной смеси требуется
мощная мешалка.
3. Для этого синтеза можно взять продажный 98%-ный глице-
рин и фармакопейный бром.
4. Вода образуется в результате химической реакции, проте-
кающей между фосфористой кислотой и глицерином или бром-
гидринами. Лучшие выходы получаются в тех случаях, когда
берется теоретическое количество брома, хотя, принимая во вни-
мание эту вторичную реакцию, и меньшее количество брома было бы
достаточным.
5. Сырой дибромгидрин перегоняется без всякого разложения
при давлении в 10—15 мм, если только температура масляной бани
не превышает 190°. Выше этой температуры начинается образова-
ние производных акролеина, присутствие которых придает дибром-
гидрину слезоточивые свойства.
3. Другие методы получения
г, у-Дибромгидрин глицерина был получен из глицерина и трех-
бромистого фосфора 1ш,
фосфора и брома 3>4'5.
из глицерина и орома ¦*; из глицерина,
phys. C) 48, 306 A856).
1 Berthelot, de Luc a, Ann. chim
2 В а г t h, Ann. 124, 349 A862).
3 A s с h a n, Ber. 23, 1826 A890).
4 Lespieau, Ann. chim. phys. G) 11, 236 A897).
5 В r a u n, J. Am. Chem. Soc. 52, 3172 A930).
2,6-ДИБРОМ-4-НИТРОФЕНОЛ
ОН ОН
ВгАвг
2 Br3
NO.
NO.,
2HBr
Предложили: В. Хартман и Дж. Дикки.
Проверили: Л. Физер и К- Фишер.
1. Получение
В 5-литровой кругло донной колбе, снабженной механической
мешалкой с жидкостным затвором, капельной воронкой и трубкой,
которая соединена с газовой ловушкой для отвода бромистого водо-
186
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
рода, растворяют 278 г B мол.) n-нитрофенола (т. пл. 112—113°)
в 830 мл ледяной уксусной кислоты. К этому раствору при комнат-
ной температуре и при работающей мешалке прибавляют по каплям
в течение 3 час. раствор 750 г B40 мл, 4,7 мол.) брома в 700 мл
ледяной уксусной кислоты. После того, как добавлен весь бром,
реакционную смесь перемешивают еще в течение получаса, а затем
в течение одного часа ее нагревают на водяной бане (температура
внутри колбы 85°), для того чтобы по возможности удалить избыток
брома. С целью удаления последних следов брома через реакцион-
ную смесь пропускают ток воздуха, после чего ее окраска стано-
вится желтой или коричневой. Смесь обрабатывают 1,1 л холодной
воды, перемешивают до тех пор, пока вся масса не охладится (при-
мечание 1), и оставляют на ночь на льду или в холодильном шкафу.
Светложелтый кристаллический продукт отсасывают на большой
(диам. 19 см) воронке Бюхнера и сначала промывйют 500 мл 50%-но-
го водного раствора уксусной кислоты, а затем тщательно водой.
Кристаллы сушат в сушильном шкафу при 40—60° или в__вакуум-
эксикаторе над едким натром. Выход 570—583г (96—98%теоретич.).
Продукт получается почти бесцветным и плавится с разложением
при 138—140° (примечание 2).
2. Примечания
1. Если во время охлаждения раствор перемешивать, то продукт
не образует корки на внутренней стороне колбы и потом лучше
промывается.
2. Продукт этот является достаточно чистым для большинства
целей, и качество его не намного улучшается при перекристаллиза-
ции из 50%-ной уксусной кислоты. Как очищенные, так и неочи-
щенные образцы 2,б-дибром-4-нитрофенола, полученные бромиро-
ванием нитрофенола, разлагаются при температуре плавления,
причем разложение до известной степени зависит от скорости
нагревания. Препарат, полученный нитрованием, плавится без
разложения при 144—145°г.
3. Другие методы получения
2,б-Дибром-4-нитрофенол был получен нитрованием 2,6-дибром-
фенолаг или дибромфенолсульфокислоты2, а также действием
азотной кислоты на 2,6-дибром-4-нитрозофенол 3 или 2,4,6-трибром-
фенол4. Он был также получен из соответственного этилового
эфира5 и действием брома на п-нитрозофенол *, 4,6-дибром-
2-нитрофенол7, 5-нитро-2-оксибензойную кислоту8 и 5-нитро-
2-оксибензосульфокислоту9. Введение двух атомов брома в моле-
кулу n-нитрофенола 10- u может быть осуществлено в сернокислот-
2,6-ДИБРОМХИНОН-4-ХЛОРИМИД
187
ной среде 1а и в присутствии хлористого алюминия 13. Описанный
здесь метод в основном заимствован у Мелау и Ульмана и.
1 Pope, Wood, J. Chem. Soc. 101, 1828 A912).
2 Armstrong, Brown, тамжс 25, 859A872); Contardi, Ciocca,
Gazz. chim. ital. 63, 878 A933).
3Kehrmann, Ber. 21, 3318 A888); Forster, Robertson,
J. Chem. Soc. 79, 688 A901).
* Raiford, Heyl, Am. Chem. J. 43, 395 A910).
'Jackson, Fiske, Ber. 35, 1132 A902); Am. Chem. J. 30, 60 A903).
«van Erp, Rec. trav. chim. 30, 290 A911).
' Armstrong, J. Chem. Soc. 28, 522 A875); Ling, там же 51,
147 A887).
8 L e 1 1 m a n n, Grothmann, Ber. 17, 2731 A884).
9 Post, Ann. 205, 94 A880).
"Brunck, Z. Chem. 1867, 204; там же 1868, 323; Vaubel, J.
prakt. Chem. B) 49, 544 A894).
"Mohlau, Uhlmann, Ann. 289, 94 A896).
12 Datta, Bhoumik, J. Am. Chem. Soc. 43, 310 A921).
13 Bodroux, Compt. rend. 126, 1285 A898); Bull. soc. chim. C) 19,
759 A898).
OH
2,6-ДИБР0МХИН0Н-4-ХЛ0РИМИД
B,6-Дибром-М-хлорхинонимин )
ОН
Br
5Sn + 14HCl-
NO,
Br
Br
SnCl6+4SnCl2
Br
OH
'4
NH3
О
Br
NH,
SnCl6+4Na0Cl~2
Br
Br
+SnCl4-T-4NaCH-4H2O
NCI
Предложили: В. Хартман, Дж. Дикки и Дж. Стемпфли.
Проверили: Л. Физер и К- Фишер.
1. Получение
А. 2,6-Дибром-4-аминофенол-хлоростаннат. В 5-литровую круг-
лодонную колбу помещают 148,5 г @,5 мол.) 2,б-дибром-4-нитро-
фенола (стр. 185), 300 мл C,7 мол.) концентрированной соляной
кислоты (уд. в. 1,19), 300 мл воды и 185 г A,56 гр.-ат.) волок-
188
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
нистого олова. Затем для того, чтобы уменьшить вспенивание,
прибавляют 3 мл октилового спирта и смесь нагревают, помеши-
вая, в открытой колбе на водяной бане до начала реакции. Ввиду
того, что реакция может начаться весьма бурно, рекомендуется
сперва нагревать массу очень осторожно. Время от времени добав-
ляют соляной кислоты и воды, причем вспенивание устраняют
добавлением воды. Всего прибавляют 520 мл F,4 мол.) концентри-
рованной соляной кислоты и 900 мл воды. После того как пройдет
бурная реакция, смесь энергично нагревают до полного растворения
дибромнитрофенола, горячий раствор (около 85°) фильтруют через
слой активированного березового угля, который помещают в нагре-
тую воронку Бюхнера. Фильтрат, который обычно бывает бесцвет-
ным, охлаждают до 0°, не прекращая перемешивания в течение
двух часов или же оставляют на ночь в холодном месте. Продукт,
выделившийся в виде бесцветных или окрашенных в светложелтый
цвет игл, отсасывают на воронке Бюхнера и промывают холодной
разбавленной соляной кислотой A объем концентрированной
кислоты на 1 объем воды). Препарат обыкновенно получается
бесцветным и его можно применить непосредственно для описывае-
мой ниже реакции (примечание 1). Вес оловянной соли после
сушки в сушильном шкафу при 50—60° или в вакуум-эксика-
торе над едким натром составляет 214—220 г. Теоретически, считая
на приведенную выше форму, должно было бы получиться 217 г.
Б. 2,6-Дибромхинон-4-хлоримид. Хлоростаннат удобно окис-
лять в двух порциях (примечание 2). В 3-литровую колбу помещают
раствор 115 г B,9 мол.) едкого натра в 175 мл воды и 1 кг измель-
ченного льда, после чего через смесь пропускают 108 г A,52 мол.)
хлора. При этом около 80% льда тает. Затем в 5-литровой колбе
растворяют ПО г @,127 мол.) оловянной соли 2,6-дибром-4-амино-
фенола в 1,2 л воды и 12 мл концентрированной соляной кислоты.
Для полного растворения смесь нагревают до 40—50°, после чего
ее охлаждают до 15—17°, добавляют 600 г льда и сразу при энергич-
ном перемешивании приливают раствор гипохлорита (работают
в вытяжном шкафу). Немедленно выпадает желтый осадок 2,6-
дибромхинон-4-хлоримида и выделяется хлор. После того как
раствор гипохлорита натрия хорошо смешался с остальной массой,
прибавляют 120 мл концентрированной соляной кислоты для того,
чтобы удержать в растворе соли олова (примечание 3). Мелкий
желтый осадок отсасывают в вытяжном шкафу на воронке Бюх-
нера (примечание 4) и промывают 1,5 л 5%-ной соляной кислоты
с целью удаления хлора и солей олова. Продукт сушат в стеклян-
ной чашке при 30—40° (примечание 5) или в вакуум-эксикаторе над
едким натром. Из двух таких порций получается всего 126—130 г
хлоримида (84—87% теоретич., считая на 2,6-дибром-4-нитрофенол,
взятый в реакцию в разделе .4). Температура плавления продукта
80—82°.
1,2-ДИБРОМЦИКЛОГЕКСАН
189
2. Примечания
1. Если продукт заметно окрашен в желтый цвет, его следует
перекристаллизовать из смеси 150 мл концентрированной соляной
кислоты и 375 мл воды, причем нагревать рекомендуется до 85—90°.
Выход перекристаллизованного продукта 180—190 г. При выпари-
вании маточного раствора до 1/3 части исходного объема при пони-
женном давлении можно получить еще 15—25 г оловянной соли.
2. Одновременно можно подвергнуть окислению до 700 г оло-
вянной соли, если в качестве реакционного сосуда взять глиняный
горшок на 70 л.
3. Если в этой стадии перемешивать массу 1 час, можно увели-
чить размер частиц хлоримида.
4. Первый литр фильтрата фильтруют вновь до тех пор, пока
он не станет прозрачным.
5. При сушке следует соблюдать осторожность, так как в одном
случае имело место бурное разложение значительной партии про-
дукта в открытой чашке при температуре около 60°.
3. Другие методы получения
2,6-Дибромхинон-4-хлоримид был получен действием гипохло-
рита натрия на 2,6-дибром-4-аминофенол, взятый в виде его хлор-
станната х или в виде солянокислой соли 2.
1 М о h 1 а и, Вег. 16, 2845 A883); F r i e d I a n d e r, S t a n g e, там же
26, 2262 A893); Mohlau, Uhlmann, Ann. 289, 94A896); Михайлов,
Труды Инст. чистых хим. реактивов № 16, 83 A939).
2 G i b b s, J. Biol. Chem. 72, 653 A927).
1,2-ДИБРОМЦИКЛОГЕКСАН
CH., CH2
Br2
CH2 CH
I II
CH.2 CH
CH2 CHBr
CH2 CHBr
CH,
CH.;
Предложили: Х. Снайдер и Л. Брукс.
Проверили: Л. Смит, Р. Арнольд и Дж. Райап.
1. Получение
В 2-литровую трехгорлую круглодонную колбу, снабженную
делительной воронкой емкостью 500 мл, механической мешалкой
и термометром, помещают раствор 123 г A,5 мол.) циклогексена
(примечание 1) в смеси 300 мл четыреххлористого углерода и 15 мл
190 СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
абсолютного спирта. Колбу охлаждают в бане со льдом и солью,
пускают в ход мешалку и, когда температура понизится до —5°, из
делительной воронки приливают раствор 210 г F7 мл, 1,3 мол.)
брома в 145 мл четыреххлористого углерода с такой скоростью,
чтобы температура реакционной смеси не превышала —Г (приме-
чание 2). На это требуется около 3 час.
Немедленно после прибавления всего брома содержимое колбы
переносят в 1-литровую видоизмененную колбу Клайзена емкостью
1 л и четыреххлористый углерод и избыток циклогексена отгоняют
с водяной бани (примечания 3, 4 и 5). Затем водяную баню заменяют
на масляную и продукт перегоняют в вакууме. После небольшой
головной фракции чистый дибромциклогексан перегоняется при
99—103°/16 мм A08—112°/25 мм). Выход 303 г (95% теоретич.;
примечания 6 и 7).
2. Примечания
1. Вполне удовлетворительным является циклогексен, выки-
пающий в пределах 2°. Методика получения циклогексена при-
ведена в «Синт. орг. преп.», сб. 1, стр. 509, а также в настоящем
сборнике на стр. 568.
2. Если не следить тщательно за температурой, то выход бывает
плохим вследствие реакций замещения. Даже при такой низкой
температуре замещение имеет место, если не взять избыток цикло-
гексена.
3. Трехгорную колбу можно ополоснуть небольшим количеством
четыреххлористого углерода.
4. При длительном соприкосновении с воздухом дибромид
разлагается и сильно темнеет. Поэтому продукт следует перегонять
немедленно,
5. Низкокипящая головная фракция содержит следы диброми-
да. Это видно по тому, что на воздухе она быстро темнеет.
6. Лучше всего продукт хранить в запаянных ампулах и как
можно меньше оставлять его в соприкосновении с воздухом.
7. Не темнеющий продукт может быть получен с помощью
следующего метода очистки. Сырой дибромид взбалтывают 5 мин.
с 7з по объему 20%-ного спиртового раствора едкого кали. Смесь
разбавляют равным ей объемом воды и слой, содержащий органи-
ческие продукты, отделяют, отмывают от щелочи, сушат и перего-
няют. Обработанный таким образом продукт может сохраняться
бесцветным неограниченно долгое время. Потери при очистке
составляют около 10% (В. Эггерс Дэринг и А. Дюрант мл.,
частное сообщение).
3. Другие методы получения
1,2-Дибромциклогексан был получен из циклогексена путем
присоединения брома в растворе хлороформа х, четыреххлористого
а, Э-ДИЁРОМЯНТАРНАЯ КИСЛОТА
углерода 2, эфира 3, ледяной уксусной кислоты *, в водном растворе
бромистого натрия 5, а также и в отсутствие растворителя 6. Бро-
мирование в растворе четыреххлористого углерода с добавлением
небольшого количества спирта дает более удовлетворительные
результаты, чем бромирование в растворе четыреххлористого
углерода, как это описано в одном из предыдущих выпусков 2.
1 В а е у е г, Ann. 278, 108 A894); Hofmann, Damm, Mitt, schlesischen
Kohlenforsch. Kaiser Wilhelm Ges. 2 97 A925) [C. A. 22, 1249 A928)];
Rothstein, Ann. chim. A0) 14, 542A930).
2 С о f f e y, Rec. trav. chim. 42, 398 A923); Greengard, Org. Syn.
12, 27.
3 F о r t e y, J. Chem. Soc. 73, 948 A898).
* Harries, Splawa-Neyman, Ber. 42, 695 A909).
5Марковников, ЖРХО 39, 174 A898); Ann. 302, 29A898);
S warts, Bull. soc. chim. Belg. 46, 13 A937).
« T r u f f a u 11, Bull. soc. chim. E) 1, 398 A934).
1. Получение
В 2-литровую трехгорлую круглодонную колбу, снабженную
механической мешалкой (примечание 1), капельной воронкой и
холодильником Фридрихса1 (примечание 2), помещают 200 г
A,7 мол.) фумаровой кислоты (примечание 3) и 400 г воды (приме-
чание 4). Все это тщательно смешивается, так чтобы фумаровая
кислота была смочена водой, и образовавшуюся густую вязкую
массу энергично перемешивают (примечание 5) и доводят до кипе-
ния, нагревая на сетке с помощью бунзеновской горелки (приме-
чание С).
Затем с возможной быстротой добавляют через капельную
воронку 276 г (94,3 мл, 1,7 мол.) брома (примечание 7). Скорость
прибавления регулируют таким образом, чтобы холодильник
Фридрихса был непрерывно заполнен флегмой примерно наполо-
вину (примечание 8). Прибавление брома продолжается около
1 часа (примечание 9). После того как прибавлено около 100 г
брома, быстро образуется дибромянтарная кислота, которая выпа-
дает в виде мелких белых игл. После окончания реакции должен
быть небольшой избыток брома, что видно по красной окраске
«, р-ДИБРОМЯНТАРНАЯ КИСЛОТА
192
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
раствора. В некоторых случаях приходится добавлять 5—10 г
брома, чтобы обеспечить его избыток.
Реакционную колбу погружают в воду со льдом и охлаждают
до 10°, не останавливая мешалки. Кристаллы отсасывают через
большую бюхнеровскую воронку и для удаления брома промывают
их холодной водой. Фильтрат можно отЗросить, так как в нем
содержатся только примеси. Полученный продукт сушат в течение
ночи при комнатной температуре. В перекристаллизации он не
нуждается. Выход 343—400 г G2—84% теоретич.).
2. Примечания
1. Для размешивания кристаллов, образующихся при реакции,
необходима массивная мешалка с возможно большей лопастью.
Можно обойтись без ртутного затвора, однако желательно, чтобы
направляющая трубка, в которой ходит мешалка, была погружена
в жидкость.
2. Следует применять стеклянные шлифы или каучуковые
пробки, так как горячий бром быстро разъедает корковые пробки.
3. Достаточно чистой для этого синтеза является продажная
фумаровая кислота. Синтез фумаровой кислоты описан на стр. 541.
4. Большее количество воды ведет к образованию монобромяб-
лочной кислоты, винной кислоты и других соединений неизвестного
строения 2.
5. Для получения хороших выходов необходимо энергичное
перемешивание.
6. Необходимо поддерживать кипение реакционной смеси во все
время течения реакции. Однако в период приливания брома следует
значительно уменьшить пламя, так как эта реакция экзотермична.
7. Прибор должен быть установлен в вытяжном шкафу или же
верх холодильника следует присоединить к ловушке для улавли-
вания паров брома, небольшое количество которых проскакивает
через холодильник.
8. При этом большая часть непрореагировавшего брома смы-
вается обратно в колбу, так что количество его, проскакивающее
через холодильник, сводится к минимуму.
9. Если бром прибавлять в течение большего количества вре-
мени, выход значительно уменьшается.
3. Другие методы получения
«,8-дибромянтарная кислота может быть получена нагреванием
янтарной кислоты с бромом и водой в запаянной трубке при 180°3;
нагреванием янтарной кислоты с красным фосфором и бромом в запа-
янной трубке при 140°4; нагреванием фумаровой кислоты с 2 молями
брома в растворе уксусной кислоты в запаянной трубке при 100J
ДЙ-н.-БУТИЛКАРБИН'ОЛ
в течение 7 часов Б; из фумаровой кислоты, брома и воды при 100°
под давлением в, а также методом, описанным выше.
1 Friedrichs, Z. angew. Chem. 33 (I) 30 A920).
2 К e k u 1 ё, Ann. Spl. Bd. 1, 338 A861).
3 К e k u 1 i, Ann. 117, 120 A861); Ann. Spl. Bd. 1, 338 A861); В о u r-
go i n, Bull. soc. chim. B) 19, 148 A873).
* Gorodetzky, Hell, Ber. 21, 1729 A888).
6 Michael, J. prakt. Chem. B) 52, 289 A895).
6 Kekule, Ann. Spl. Bd. 1, 129 A861); В а е у е r, Ber. 18, 674 A885).
ДИ-н.-БУТИЛКАРБИНОЛ
(Ношнол-5)
C4H9Br+Mg . C4H9MgBr
2 C4H9MgBr-f HCO2C2H6 > (C4H9J CHOMgBr-f C2H6OMgBr
2 (C4H9J CHOMgBr -f H2SO4 —> 2 (C4HeJ CHOH + MgBr2 + MgSO4
Предложили: Дж. Колеман и Крэг.
Проверили: Дж. Духонин и X. Стивенсон.
1. Получение
В 3-литровую трехгорлую круглодонную колбу, снабженную
делительной воронкой на 500 мл, механической мешалкой с жид-
костным затвором и обратным холодильником, помещают 36,5 г
A,5 гр.-ат.) магниевых стружек и 500 мл абсолютного эфира.
В делительную воронку наливают раствор 206 г A,5 мол.) бромистого
н.-бутила («Синт. орг. преп.», сб. 1, стр. 111) в 250 мл абсолютного
эфира. Пускают в ход мешалку, и из делительной воронки прили-
вают в колбу 10—15 мл раствора бромида, после чего через несколько
минут обычно начинается реакция (примечание 1). Как только
жидкость начнет энергично стекать обратно из холодильника,
колбу окружают водой со льдом, и прибавление бромида регули-
руют таким образом, чтобы обратное стекание жидкости из холо-
дильника происходило с умеренной скоростью. После того, как весь
раствор прибавлен, на что требуется 30—40 мин., баню со льдом
удаляют и перемешивание продолжают еще 15 мин. После этого
непрореагировавшего магния остается лишь незначительное коли-
чество.
Колбу снаружи охлаждают льдом и в делительную воронку
наливают раствор 55,5 г @,75 мол.) чистого этилового эфира муравьи-
ной кислоты (примечание 2) в 100 мл абсолютного эфира. Вновь
пускают в ход мешалку, и раствор этилового эфира муравьиной
кислоты прибавляют с такой скоростью, чтобы поддержать умерен-
ное кипение эфира. На это требуется около 30 мин. Баню со льдом
удаляют и перемешивание продолжают еще 10 мин.
13 Сборник Л» 2
194
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
. Далее при энергичном перемешивании (примечание 3) из дели-
тельной воронки прибавляют 100 мл воды с такой скоростью, чтобы
эфир кипел довольно энергично. Затем прибавляют холодный
раствор 85 г D6 мл, 0,85 мол.) концентрированной серной кислоты
в 400 мл воды. После того как кислота добавлена, оба слоя стано-
вятся прозрачными. Большую часть эфирного слоя сливают в 1-лит-
ровую круглодонную колбу, остаток вместе с водным слоем пере-
водят в делительную воронку. Оставшийся в колбе осадок промы-
вают два раза порциями по 25 мл эфира, и эти вытяжки добавляют
к содержимому делительной воронки. Эфирный слой отделяют
и присоединяют к декантированной части. Колбу соединяют с хоро-
шим дефлегматором и эфир отгоняют на водяной бане до тех пор,
пока температура паров не достигнет 50°. К оставшемуся загряз-
ненному карбинолу (примечание 4) добавляют 75 мл 15%-ного
водного раствора едкого кали и соединяют колбу с обратным холо-
дильником. Смесь энергично кипятят три часа, после чего очищен-
ный карбинол отгоняют с водяным паром, следя, чтобы в колбе
объем жидкости все время равнялся 250—300 мл. Дестиллат соби-
рают в делительную воронку, откуда время от времени спускают
нижний водяной слой. Перегонку можно считать законченной после
того, как соберется около 1,5 л воды.
Верхний слой, состоящий из ди-н.-бутилкарбинола, отделяют и
оставляют на один час над 10 г безводного поташа. Жидкость сли-
вают в колбу Клайзена на 500 мл, и оставшийся поташ промывают
три раза сухим эфиром порциями по 10 мл. Эфирный раствор при-
соединяют к веществу, находящемуся в колбе. После небольшой
головной фракции собирают 90—92 г (83—85% теоретич.) чистого
ди-н,-бутилкарбинола с т. кип. 97—98°/20 мм (примечание 5).
2. Примечания
1. Обычно реакция между магнием и эфирным раствором броми-
стого н.-бутила начинается самопроизвольно; в случае необходи-
мости можно для начала реакции прибавить небольшое количество
предварительно полученного реактива Гриньяра или же кристаллик
иода.
2. Лучше взять свежеперегнанный этиловый зфир муравьиной
кислоты, для очистки которого можно поступить следующим обра-
зом: к 100 г продажного этилового эфира муравьиной кислоты
добавить 15 г безводного поташа и смесь оставить стоять, взбалты-
вая ее время от времени. Затем эфир слить в сухую круглодонную
колбу на 200 мл, добавить 5 г фосфорного ангидрида, колбу соеди-
нить с хорошим дефлегматором и этиловый эфир муравьиной кислоты
перегнать, собирая дестиллат в приемник, защищенный от влаги
воздуха. Для данного синтеза была взята фракция, кипящая при
53—54°.
ДИГИДРОХОЛЕСТЕРИН
135
3. При прибавлении воды необходимо энергичное размешивание,
для того чтобы образующееся твердое вещество выпадало в осадок
в мелкораздробленном состоянии, а не в виде больших кусков.
4. В качестве примеси в сыром продукте содержится сложный
эфир н.-дибутилкарбинола и муравьиной кислоты, который омы-
ляется при кипячении с раствором едкого кали.
5. Ди-н.-бутилкарбинол можно перегонять при атмосферном
давлении без заметного разложения (т. кип. 193—1947743 мм),
однако предпочтительнее перегонять его в вакууме. В зависимости
от давления он кипит при следующих температурах: 97°/20 мм;
104730 мм; 109740 мм; 117760 'мм; 1307100 мм.
3. Другие методы получения
Ди-н.-бутилкарбинол был получен действием бромистого н.-бу-
тилмагния на н.-масляный альдегид 1 и на этиловый эфир муравьи-
ной кислоты х>2. Он был также получен каталитической гидро-
генизацией ди-н.-бутилкетона в присутствии платины 3.
1 Malengreau, Bull. acad. roy. Belg. cl. sci. 1906, 802 [C. A. 1, 1970
A907)].
! Dillon, Lucas, J. Am. Chem. Soc. 50, 1713 A928).
3Vavon, Ivanoff, Compt. rend. 177, 453 A923).
ДИГИДРОХОЛЕСТЕРИН
(?-Холестанол)
H.C
PtO
Предложили: В. Брюс и Дж. Ролле.
Проверили: Л. Физер, Р. Джекобсон и М. Ньюмен.
1. Получение
А. Из холестерина. 100 г @,26 мол.) продажного холесте-
рина перекристаллизовывают из 250 г ледяной уксусной кислоты,
причем в случае необходимости для обесцвечивания берут 1 г акти-
196
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
вированного угля. Очищенный продукт (примечание 1) без предва^
рительной сушки переносят в сосуд для гидрирования, который
снабжен термометром и приспособлением для нагрева (приме
чание 2). Добавляют 300 мл очищенной ледяной уксусной кислоть
(примечание 3) и 0,5 г окиси платины и гидрогенизацию проводят
при 65—75° при небольшом избыточном давлении. Все необходимое
количество водорода поглощается обычно за время от 2 до 4 час
(примечание 4). Водород вытесняют воздухом, раствор фильтруют
горячим и продукт выделяют путем кристаллизации и упариванш
фильтрата. Суммарный выход воздушносухого частично ацетил»
рованного дигидрохолестерина с т. пл. 130—135° составляет 85—
90 г.
Если не требуется специальной очистки (см. ниже), сыро?
продукт нагревают на паровой бане в течение 3 час. с 400 мл спирте
и раствором 25 г едкого натра в 100 л<л воды. По охлаждении продукт
отфильтровывают, промывают и перекристаллизовывают из 500 mj
спирта. Выход 75—80 г G5—80% теоретич.). Тщательно высушен-
ная проба (примечание 5) плавится при 140—141°.
Б. Из холестерилацетата (примечание 6). 5 г холестерилаце-
тата (примечание 7) и 0,1 г окиси платины суспендируют в 25 mj,
абсолютного эфира и 50 мл очищенной ледяной уксусной кислоть:
(примечание 3); гидрогенизацию проводят при комнатной темпера
туре и при небольшом избыточном давлении. Реакция заканчи-
вается обычно через 10—50 мин. Раствор фильтруют, добавляют
если это нужно, эфира для растворения выпавших кристалло!
и после отгонки растворителя в вакууме остаток либо омыляют
как это описано выше, либо подвергают очистке по следующем)
методу.
Очистка по методу Андерсона и Набенгауера Ч Раствор 20 ;
сырого, частично или нацело ацетилированного дигидрохолесте-
рина в 200 мл четыреххлористого углерода помещают в делительнук
воронку и взбалтывают со 100 мл уксусного ангидрида. Затеа
через трубку перевернутой воронки добавляют по каплям околс
5 мл концентрированной серной кислоты, причем воронку охла-
ждают и взбалтывают до тех пор, пока не будет больше измененш
в цвете. Обычно раствор приобретает голубую или зеленую окраску
интенсивность которой зависит от наличия в данном образце холе
стерина. Через 15—20 мин. при охлаждении и при осторожно»
встряхивании добавляют по каплям около 10 мл воды, так чтобь
образовалось два слоя. Раствор в четыреххлористом углерод;
(верхний слой) отделяют и отмывают от кислоты раствором хло
ристого натрия или соды (чистая вода образует эмульсию). Раство{
сушат сернокислым натрием, растворитель отгоняют в вакууме
остаток омыляют спиртовым раствором едкого кали так, .как эте
описано выше, и перекристаллизовывают из спирта. Вес чистогс
дигидрохолестерина составляет 12—14 г; температура плавленш
ДИГИДРОХОЛЕСТЕРИН
197
после тщательного высушивания (примечание 5) 142—143°. Про-
дукт дает слабую реакцию Либермана — Бурхарда (примечание 8)
только через 10—15 мин.
2. Примечания
1. Вес сухого продукта 90-—95 г. Для некоторых образцов тре-
буется повторная перекристаллизация.
2. Удобное приспособление для нагрева гидрогенизациоиного
сосуда см. «Синт. орг. преп.», сб. 1, стр. 46. Можно сконструиро-
вать и другой прибор: круглодонную колбу с длинным горлом
укрепляют за верхнюю часть с помощью зажима, состоящего из
двух лапок с ослабленным винтом; снизу колбу присоединяют
к эксцентрику и нагревают колбу, находящуюся в движении,
посредством неподвижной микрогорелки.
3. Ледяную уксусную кислоту кипятят с обратным холодиль-
ником в течение часа с 5 г переманганата калия на каждый литр
и подвергают перегонке.
4. Иногда катализатор теряет свою активность после поглоще-
ния приблизительно половины теоретического количества водорода,
в результате отравления примесями, которые не могли быть уда-
лены из продажного холестерина. В этом случае прибавление одной
или двух порций по 0,2 г катализатора обычно бывает достаточным,
чтобы довести реакцию практически до конца.
5. Стерин образует гидрат, из которого вода может быть уда-
лена только тщательной сушкой, например в вакууме при 100°.
6. Ацетильное производство восстанавливается легче, чем сво-
бодный стерин.
7. Холестерилацетат получается кипячением раствора 5 г
холестерина в 7,5 мл уксусного ангидрида в течение 1 часа с обрат-,
ным холодильником. Раствор охлаждают и кристаллический про-
дукт отфильтровывают и промывают холодным метанолом. Выход
5 г, т. пл. 114—115°.
8. Проба на холестерин: около 5 мг продукта растворяют
в 2 мл четыреххлористого углерода, добавляют 1 мл уксусного
ангидрида и 3—4 капли концентрированной серной кислоты. Холе-
стерин дает ряд последовательных изменений цвета.
3. Другие методы получения
Дигидрохолестерин был получен восстановлением холестенона
натрием в амиловом спирте 2, а также гидрогенизацией холесте-
рина. В присутствии платиновой черни или окиси платины были
получены выходы от 6,5 до 40% в растворе эфира3, ацетона4,
уксусноэтилового эфира 5 и уксусной кислоты в.
198
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
1 An derson, Nahenhauer, J. Am. Chem. Soc. 46, 1957 A924).
s D i e 1 s, Abderhalden, Ber. 39, 889 A906); D i e 1 s, S t a m m.
там же 45, 2230 A912); N e u b e r g, там же 39, 1155 A906).
3 W i 1 1 s t й 11 e r, M а у e г, там же 41, 2200 A908); D о г ё е, J. Chem.
Soc. 95, 044 A909); В о е h m. Biochem. Z. 33. 474 пот- w ; n л ¦> » ,
bull. soc. Chim. D) 53, 584 A933); Ruzic'ka, Brii
berger, Meyer, Helv. Chim. Acta 17, 1407 A934).
4 Nord, Biochem. Z. 99, 265 A919).
6 S h r i n с г, Ко, J. Biol. Cliem. 80, 6 A928).
• Ftirth, Felsenreich, Biochem. Z. 69, 420 A915).
2,6-ДИЙОД-л-НИТРОАНИЛИН
B,6-Д шод-4-ттроатлин )
NH2
n-NO2C6H4NH2
Предложили: Р. Сэндин, В. Дрэк и Ф. Jledotcep.
Проверили: Ф. Уитмор и М. Уитмор.
1. Получение
В 2-литровой трехгорлой круглодонной колбе, снабженной
механической мешалкой, обратным холодильником и капельной
воронкой, растворяют 138 г A мол.) л-нитроанилина (технического)
в 370 мл кипящей ледяной уксусной кислоты. После того как горелка
отставлена, через капельную воронку приливают смесь из 325 г
B мол.) хлористого иода (примечание 1)и 100 мл ледяной уксусной кис-
лоты. Приливание продолжается в течение получаса, причем раствор
быстро перемешивают. Происходит сильное разогревание. Затем
смесь нагревают в течение 2 час. на сильно кипящей водяной бане,
переносят в литровый стакан и охлаждают. Застывшую массу обра-
батывают 100 мл ледяной уксусной кислоты, и твердые комки раз-
давливают стеклянной пробкой (примечание 2). Смесь переносят
на большую воронку Бюхнера и отсасывают; оставшиеся в стакане
кристаллы смывают на воронку ледяной уксусной кислотой двумя
порциями по 25 мл. Осадок возможно лучше отсасывают от темного
маточного раствора. Кристаллы переносят обратно в стакан, хорошо
размешивают с 200 мл ледяной уксусной кислоты и снова отсасы-
вают на фильтре. Стакан смывают двумя порциями ледяной уксус-
ной кислоты по 25 мл каждая и осадок на фильтре отсасывают
по возможности досуха. Затем отсасывание прекращают и кристаллы
смачивают 50 мл эфира. Эфир удаляют также отсасыванием. Про-
дукт сушат на воздухе до постоянного веса (около 24 час). Выход
иесимм. ~ ДИМЕТИЛГИДРАЗИН СОЛЯНОКИСЛЫЙ
199
воздушно-сухого дииод-л-нитроанилина с т. пл. 243—245° состав-
ляет 220—250 г E6—64% теоретич.).
2. Примечания
1. Хлористый иод получают пропусканием сухого хлора в 254 г
A мол.) иода, находящегося в тарированной конической колбе емко-
стью 500 мл, до тех пор, пока привес не будет равен 71 г. При этом
необходимо частое встряхивание. Хлористый иод берется для реак-
ции без предварительной перегонки (ср. стр. 392).
2. Во время работы с ледяной уксусной кислотой рекомендуется
надевать резиновые перчатки.
3. Имеются указания, что выход увеличивается до 86% при
следующем видоизменении синтеза. Реакционную смесь кипятят
с обратным холодильником на масляной бане в течение 2 час, охла-
ждают ее и фильтруют с отсасыванием, стараясь возможно полнее
удалить растворитель. Полученную лепешку растирают в пасту
с 600 мл горячей воды, добавляют немного бисульфита натрия;
чтобы удалить избыток иода, продукт отсасывают и сушат. 2,6-
Дииод-4-нитроанилин можно перекристаллизовать из нитробен-
зола с последующей промывкой спиртом. После перекристаллиза-
ции получают продукт ст. пл. 249—250° [К. Ниман и К. Е. Редеман,
частное сообщение. См. также J. Am. Chem. Soc. 63, 1550 A941)J.
3. Другие методы получения
2,6-Дииод-п-нитроанилин был получен действием хлориода на
л-нитро анилин в хлороформе ином растворе 1 и в растворе ледяной
уксусной кислоты2.
1 Michael, Norton, Ber. 11, 113 A878).
2 Willgerodt, Arnold, там же 34, 3344 A901).
«есял*л*.-ДИМЕТИЛГИДРАЗИН СОЛЯНОКИСЛЫЙ
A,1-Диметилгидразингидрохлорид )
(CH3JNHHC1 + HONO > (CH3KNNO + HC1 + H2O
(CH3JNNO + 4[H] ^H^^^l (CHS)*NNH2+H2O
(CH3JNNH2+HCl > (CH3JNNH2-HCl
Предложил: X. Xamm.
Проверили: В. Харпшан и В. Петерсон.
1. Получение
А. Нитрозодиметиламин. В 2-литровую круглодонную колбу,
снабженную механической мешалкой, помещают 245 г C мол.)
солянокислого диметиламина, 120 мл воды и 10 мл приблизительно
2-н, соляной кислоты. Полученный раствор энергично размешивают
200
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
и поддерживают на водяной бане при температуре 70—75°, посте-
пенно прибавляя к нему из капельной воронки в течение 1 часа
суспензию 235 г C,23 мол.) 95%-ного азотистокислого натрия
в 150 мл воды. При этом довольно часто проверяют на лакмус
кислотность среды, поддерживая ее слегка кислой постепенным
(по 1 мл) прибавлением 2-н. соляной кислоты (всего требуется около
30—35 мл). После того как все количество нитрита прибавлено,
перемешивание и нагревание продолжают еще 2 часа.
Колбу приспособляют для перегонки и при небольшом вакууме
от реакционной смеси отгоняют воду и нитрозодиметиламин на
водяной бане до получения практически сухого остатка. К этому
остатку добавляют 100 мл воды и вновь в тех же условиях отгоняют
все досуха. Оба отгона соединяют вместе и насыщают поташом
(около 300 г); верхний слой нитрозодиметиламина удаляют, а вод-
ный слой экстрагируют три раза эфиром порциями по 140 мл.
Ншрозамин и эфирные вытяжки соединяют вместе, сушат безвод-
ным поташом и перегоняют с дефлегматором длиною в 30 см. Выход
продукта с т. кип. 149—150°/755лш составляет 195—200г(88—90%
теоретич.). Нитрозодиметиламин представляет собой маслянистую
жидкость желтого цвета, легко темнеющую на ярком свету.
Б. нешмм-Диметилгидразшгидрохлорид. В круглодонную 5-лит-
ровую колбу, снабженную механической мешалкой, капельной
воронкой и термометром, помещают 200 г B,7 мол.) нитрозоди-
метиламина, 3 л воды и цинковой пыли в количестве, которое
соответствует 650 г A0 гр.-ат.) 100%-ного цинка (примечание 1).
Пускают в ход мешалку, колбу погружают в водяную баню и, под-
держивая температуру при 25—30°, в течение 2 час. добавляют 1 л
A4 мол.) 85%-ной уксусной кислоты. После прибавления всего
количества кислоты температуру повышают до 60° и поддерживают
ее в течение часа. Затем реакционной массе дают охладиться, избы-
ток цинковой пыли отфильтровывают и промывают небольшим
количеством воды. Все водные растворы сливают вместе в 12-лптро-
вую колбу, которая подготовлена для перегонки с водяным паром.
Колбу снабжают капельной воронкой и перед пуском в колбу пара
ставят ловушку. Приемником служит большая склянка для отса-
сывания или колба Вюрца, причем отводную трубку приемника
присоединяют к двум поглотительным склянкам с соляной кислотой
A:1; примечание 2). Через капельную воронку прибавляют концен-
трированный раствор 1 кг едкого натра до явно щелочной реакции,
после чего жидкость подвергают перегонке с водяным паром до тех
пор, пока проба дестиллата не будет лишь очень слабо восстанавли-
вать фелингову жидкость. Обычно для удаления всего диметилгидра-
зина бывает достаточно отогнать 5—6 л дестиллата. Для того чтобы
ускорить перегонку, под перегонную колбу помещают горелку.
Водный отгон обрабатывают 650 мл концентрированной соляной
кислоты и упаривают его на водяной бане при пониженном давле-
иеснлш.-ДИМЕТИЛГИДРАЗИН СОЛЯНОКИСЛЫЙ
201
нии до тех пор, пока остаток не будет представлять собою сиропо-
образной массы (примечание 3). Для того чтобы еще более высушить
продукт, к нему приливают 150 мл абсолютного спирта и раствор
выпаривают в вакууме. Такую обработку повторяют два-три раза,
после чего твердый продукт бывает достаточно сухим и не пристает
к стенкам колбы. Сырой продукт сушат в вакуум-эксикаторе над
хлористым кальцием. Вес светложелтых сухих кристаллов 200—
215 г G7—83% теоретич.); с целью дальнейшей очистки его можно
растворить в равном весовом количестве кипящего абсолютного
этилового спирта и затем выморозить в бане со льдом. Выход чистого
бесцветного кристаллического продукта с т. пл. 81—82° составляет
180—190 г F7—73% теоретич.; примечание 4).
2. Примечания
1. Для того чтобы обеспечить полноту восстановления, необхо-
димо предварительно определить чистоту цинковой пыли.
2. При работе в вытяжном шкафу можно обойтись без промыв-
ных склянок. В начале перегонки с водяным паром происходит
выделение летучих азотистых оснований, состоящих главным обра-
зом из аммиака и метиламина.
3. Концентрированный водный раствор свободного гидразина
может быть получен следующим образом: сиропообразную массу
по каплям прибавляют к большому избытку твердого едкого натра
и получающееся основание отгоняют до тех пор, пока температура
не поднимется до 100э. Для того чтобы получить безводное основа-
ние, концентрированный водный раствор оставляют стоять над
едким кали, затем подвергают его перегонке, основание собирают
над окисью бария и после стояния в течение нескольких дней
вновь подвергают перегонке. Свободный гидразин кипит при 62—
65°/765 мм; он чрезвычайно гигроскопичен и разрушающе дей-
ствует на корковые и резиновые пробки.
4. Продукт представляет собою моногидрохлорид основания.
3. Другие методы получения
Получение несимметричного диметилгидразина восстановле-
нием нитрозодиметиламина описали Фишер1 и Ренуф2- Продукт
этот был также получен метилированием гидразина 3, восстановле-
нием питродиметиламина 4 и действием аминонадсерной кислоты
на диметиламин 5.
1 Fischer, Ber. 8, 1587 A875).
3 Renouf, там же 13, 2170 A880).
3 Harries, H a g а, там же 31, 56 A898).
'Franchimont, Rec. trav. chim. 3, 427 A884); Backer, там же
31, 150 A912).
5 § о ф m e r, S с h u 1 z; герм. пат. 338 609 [Frdl. 13, 203 A916—21)].
202
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
сижл.-ДИМЕТИЛГИДРАЗИН СОЛЯНОКИСЛЫЙ
CeH5CONHNHCOCeH5
SO, + 2NaOH >
2CeH5COCl + N2H4-H2SO4
CeH5CONHNHCOC6H5 + 2 (CH,J
СеН, CON (CHS) N (СНа) СОС6Н5 + 2 CHaSO4Na + 2 Н2О
C6H5CON (СН3) N (СН3) СОС6Н6 + 2 НС1 + 2 Н2О
CH3NHNHCHS • 2 НС1 + 2 С6Н5СО2Н
Предложил: X. Хатт.
Проверили: Р. Фъюзон и Э. Эллингбо.
1. Получение
А. Дибензоилгидразин. В 2-литровую колбу, снабженную механи-
ческой мешалкой (примечание 1) и охлаждаемую в бане с холодной
водой, помещают раствор 48 г A,2 мол.) едкого натра в 500 мл воды
и65г@,5мол.)гидразинсульфата («Синт. орг. преп.», сб. 1, стр. 158).
Затем пускают в ход мешалку и из двух капельных воронок мед-
ленно прибавляют 145 г A20 мл; 1,03 мол.) свежеперегнанного
хлористого бснзоила и 120 мл водного раствора, содержащего 45 г
A,1 мол.) едкого натра. Хлористый бензоил прибавляют в течение
1,5 часа; щелочь прибавляют немного быстрее. После того как все
количество прибавлено, перемешивание продолжают еще в течение
2 час. и затем раствор насыщают углекислым газом (примечание 2).
Дибензоилгидразин отсасывают, хорошо отжимают и растирают
в пасту с 50%-ным водным раствором ацетона. Эту пасту отсасы-
вают, промывают возможно лучше водой и хорошо отжимают.
Сырой продукт растворяют в 650 мл кипящей ледяной уксусной
кислоты, из которой по охлаждении дибензоилгидразин выделяется
в виде массы мелких белых игл. Кристаллы отсасывают, промы-
вают холодной водой и сушат, нагревая их в вакууме на водяной
бане в слабом токе воздуха. Первая порция с т. пл. 234—238° весит
80—90 г F6—75% теоретич.) и является практически чистой. Упари-
ванием маточного раствора можно получить еще небольшое коли-
чество менее чистого продукта.
Б. Дибензоилдиметилгидразин. Все описываемые ниже опера-
ции необходимо проводить в вытяжном шкафу. В 2-литровую трех-
горлую колбу, снабженную механической мешалкой, термометром
и двумя капельными воронками, помещают 80 г @,33 мол.) дибен-
зоилгидразина, 10 г @,25 мол.) едкого натра и 600 мл воды. С помощью
•водяной бани поддерживают температуру смеси около 90° (приме-
чание 3). Пуская в ход мешалку, прибавляют из обеих капельных
воронок 320 г B40 мл; 2,54 мол.) диметилсульфата (примечание 4)
и 250 мл водного раствора, содержащего 125 г C,1 мол.) едкого
натра. Диметилсульфат приливают порциями по 10 мл каждые.
симм.-ДИМЕТИЛГИДРАЗИН СОЛЯНОКИСЛЫЙ
203
5 мин., а раствор едкого натра с такой скоростью, чтобы реакция
все время была слегка щелочной. При явно щелочной реакции смесь
приобретает заметную желтую окраску; лучше поддерживать такую
степень щелочности, которая была бы несколько слабее и не вызы-
вала бы такой окраски.
Прибавление реагентов ведется около 2 час, причем дибензоил-
диметилгидразин выделяется в виде верхнего слоя (примечание 5);
полутвердый продукт, отбрасываемый мешалкой на стенки колбы,
необходимо время от времени смывать. Затем смесь нагревают еще
30 мин. и дают ей охладиться. Твердый дибензоилдиметилгидра-
зин собирают, растирают с водой, фильтруют и растворяют в 100 мл
хлороформа. Раствор фильтруют, чтобы освободиться от нераство-
римых примесей и сушат сернокислым натрием; хлороформ отго-
няют на водяной бане, в конце отгонки применяя вакуум. Твердый
остаток плавится при 77—84° и является достаточно чистым для
последующего употребления. Выход 77—83 г (86—93% теоретич.;
примечание 6).
В. Диметилгидразиндигидрохлорид. Смесь, состоящую из 67 г
@,25 мол.) дибензоилдиметилгидразина и 250 мл 32%-ной соляной
кислоты (уд. веса 1,16) не сильно кипятят в 2-литровой колбе
с обратным холодильником в течение 2 час. (в вытяжном шкафу).
Для удаления бензойной кислоты смесь подвергают перегонке
с водяным паром, причем собирают 10 л дестиллата (примечание 7);
раствор, оставшийся в колбе, выпаривают досуха в вакууме на
водяной бане. Кристаллический дигидрохлорид обрабатывают 25 мл
абсолютного этилового спирта и смесь выпаривают в вакууме
досуха. Обработку эту повторяют, после чего дигидрохлорид расти-
рают со смесью 25 мл абсолютного спирта и 2—3 мл концентриро-
ванной соляной кислоты; соль отфильтровывают и промывают
10—15 мл холодного абсолютного спирта. Эта первая порция
дигидрохлорида диметилгидразина после сушки в вакуум-экси-
каторе весит 22—23 г. Если выпарить маточные растворы и остаток
вновь обработать 5—6 мл абсолютного спирта и небольшим коли-
чеством соляной кислоты, то можно получить вторую порцию
кристаллов весом 3 г. Повторение этого процесса дает третью фрак-
цию количеством около 0,5 г (примечание 8). Суммарный выход
25—26 г G5—78% теоретич.).
2. Примечания
1. Можно работать без пробки и не в вытяжном шкафу, если
только в горло колбы неглубоко опустить стеклянную трубку,
соединенную с водоструйным насосом.
2. Большая часть дибензоилгидразина выделяется при реакции
в виде белого осадка; если смесь не насыщать углекислым газом,
ТО в растворе остается около 3 г продукта.
204
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
3. Температуру можно определять с помощью термометра,
находящегося в стеклянной трубке, проходящей через пробку и
погруженной в жидкость. Термометр удерживается с помощью
кусочка резиновой трубки, надетой на верхнюю часть его. Этим
приспособлением одновременно можно пользоваться и для опреде-
ления щелочности раствора.
4. Принимая во внимание сильную ядовитость диметилсуль-
фата, необходимо принимать меры предосторожности, чтобы не про-
лить жидкость и не дышать парами, которые выделяются реакцион-
ной смесью. Специфическим противоядием является аммиак.
5. Указанные количества диметилсульфата и едкого натра
являются только приблизительными. Диметилсульфат добавляют
с указанной выше скоростью до полного исчезновения твердого
дибензоилгидразина, после чего добавляют еще 25 мл. Щелочи
обычно требуется меньше указанного количества.
6. Дибензоилдиметилгидразин может быть получен в более
чистом виде, если к холодному профильтрованному хлороформен-
ному раствору прибавлять смесь эфира и петролеиного эфира
(т. кип. 40—60°), взятых в отношении 1 : 1, до тех пор пока не
начнется кристаллизация, а затем медленно добавить умеренный
избыток петролейного эфира. Гидразин выпадает в виде мелких
белых игл с т. пл. 84—87°.
7. Было найдено, что при такой обработке бензойная кислота
может быть удалена нацело. Вместо этого бензойную кислоту
можно экстрагировать смесью бензола и эфира A : 1).
8. Как моно-, так и дигидрохлориды весьма гигроскопичны,
поэтому их нельзя оставлять на воздухе. Растворы дигидрохлори-
да при выпаривании теряют соляную кислоту, и дигидрохлорид, по-
лученный из раствора посредством такой обработки, легко становит-
ся маслянистым. При перекристаллизации к спиртовому раствору до-
бавляют небольшое количество концентрированной солянойкислоты.
Для перекристаллизации дигидрохлорида его можно растворить
в кипящем абсолютном этиловом спирте (на 1 г требуется 20 мл
спирта), добавить небольшое количество концентрированной соля-
ной кислоты, слегка охладить раствор и прибавить около одной
пятой части (по объему) эфира. Диметилгидразиндигидрохлорид вы-
падает в виде белого кристаллического порошка ст. пл. 165—167°.
3. Другие методы получения
Симметричный диметилгидразин был получен нагреванием иод-
метилата 1-мётилпиразола с едким кали1. Обычный метод полу-
чения состоит в метилировании диформилгидразина с последующим
омылением соляной кислотой2. Описанный выше метод основан
на наблюдении Фольпмерса, который показал, что можно исходить
также и из дибензоилгидразина3.
ДИМЕТИЛГЛИОКСИМ
205
1 К п о г г, К о h 1 е г, Вег. 39, 3257 A906).
^ Harries К 1 a rr. t, там же 28, 503 A895); Harries, H a g a,
там же 31, 56 A898); Т h i e 1 е, там же 42, 2575 A909).
3 F о 1 р m e r s, Rec. trav. chim. 34, 34 A915).
ДИМЕТИЛГЛИОКСИМ
(Реактив Чугаева)
C2H5OH + HNO2
СН3СО
СН,С = NOH
3COCHXH3
+ NaO3SNHOH
C2H5ONO •
CH3COC (NOH) CH3 + C2H5OH
CH3C = NOH
—- +NaHSO4
CH3C = NOH
Предложили: R. Симон и В. Дамерелл.
Проверили: Г. Гилъмйн и Р. Фозерджилл.
1. Получение
А. Этиловый эфир азотистой кислоты. Раствор I. 620 г
(9 мол.) азотистокислого натрия F50 г технического, 95%-ного)
растворяют в воде, прибавляют 210 г D,6
мол.) спирта B85 мл 90%-ного денатуриро-
ванного или эквивалентное количество ректи-
фицированного) и доводят водой до объема
2500 мл. Раствори. 440 г серной кислоты
B55 мл уд. веса 1,84) смешивают с 210 г спирта
и разбавляют водой до объема 2500 мл. Эти-
ловый эфир азотистой кислоты можно полу-
чать непрерывно в виде газа, вливая рас-
твор II в раствор I.
Удобно пользоваться прибором, который
показан на рис.7. В нижний сосуд емкостью
б л наливают раствор I, в верхний емкостью
2,5 л — раствор II. Пробки берут каучуко-
вые и вставляют стеклянные трубки, как
показано на рисунке. Скорость приливания
раствора II к I регулируют винтовым зажи-
мом. Образующийся этиловый эфир азоти-
стой кислоты выходит через вторую трубку,
вставленную в пробку нижнего сосуда. Проб-
ки прикрепляют к горлам обоих сосудов
проволокой. Целесообразно снабдить нижний
сосуд механической мешалкой, которая способствует равно-
мерному выделению газа (примечание 1).
Ри с. 7
206
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
Б. Монооксим диацетила. В 2-литровую трехгорлую колбу,
снабженную холодильником, термометром, вводной трубкой для
впуска этилового эфира азотистой кислоты и приспособлением для
наружного охлаждения, помещают 620 г продажного метилэтил-
кетона, высушенного 75 г безводной сернокислой меди и отфиль-
трованного от нее. Затем прибавляют 40 мл соляной кислоты уд. в.
1,19 (примечание 2) и повышают температуру до 40°. После этого
начинают пропускать этиловый эфир азотистой кислоты, получен-
ный по вышеописанному способу, и поддерживают температуру
между 40 и 55°. После того как пропущен весь эфир (примечание 3),
из смеси отгоняют спирт, образовавшийся при реакции, до тех пор,
пока температура жидкости не достигнет 90°. Полученный сырой
продукт служит для получения-диметилглиоксима (примечание 4).
Если нужно получить чистый монооксим диацетила, то реакцион-
ную смесь нейтрализуют приблизительно 35 мл концентрирован-
ного водного аммиака и разбавляют половинным объемом воды.
Затем из раствора отгоняют спирт и избыток метилэтилкетона до
тех пор, пока дестиллат не потеряет способности воспламеняться.
Тогда приемник меняют, и смесь быстро перегоняют с перегретым
водяным паром. Почти весь монооксим диацетила переходит в пер-
вых 5 л дестиллата. Для выделения монооксима дестиллат насы-
щают 1—1,5 кг соли и затем охлаждают его до 0°. Твердый моно-
оксим диацетила выпадает в кристаллическом состоянии и отфиль-
тровывается. Выход 480—520 г. В случае надобности продукт
можно подвергнуть дальнейшей очистке перекристаллизацией из
воды (примечание 5).
В. Натриевая соль гидроксиламинсулъфокислоты. В 12-литровом
сосуде смешивают 5 кг молотого льда (примечание 6) с 569 г азоти-
стокислого натрия E94 г технического, 95%-ного). В эту смесь при-
бавляют при перемешивании суспензию кислого сернистокислого
натрия, содержащую 1 100 г активной сернистой кислоты (около
1 775 г технического бисульфита) в 750 мл воды. Затем прибавляют
при непрерывном размешивании 150 мл ледяной уксусной кислоты,
впуская ее через трубку, конец которой погружен в жидкость
(примечание 7), и вслед за нею — смесь 550 мл соляной кислоты
(уд. в. 1,19) с 400 г молотого льда (примечание 8). Температура
реакционной смеси неизменно должна быть ниже 0°; если заме-
чают, что температура начинает повышаться, то прибавляют еще
некоторое количество льда. После прибавления всей соляной
кислоты раствор должен обладать кислой реакцией на конго и
при правильном проведении реакции он содержит 6 или более мол.
натриевой соли гидроксиламиндисульфокислоты, которая быстро
гидролизуется в кислый раствор натриевой соли гидроксиламин-
моносульфокислоты.
Г. Диметилглиоксим. Сырой монооксим диацетила, получен-
ный после отгонки головной фракции до 90° и содержащий прибли-
ДИМЕТИЛГЛИОКСИМ
207
зительно 5 мол. монооксима диацетила, прибавляют к раствору
натриевой соли гидроксиламинсульфокислоты (отфильтрован-
ному от содержащегося в нем осадка), находящемуся в 15-литровой
колбе. Смесь нагревают до 70° и оставляют стоять при этой темпе-
ратуре, время от времени перемешивая ее, несколько часов (при-
мечание 9). Диметилглиоксим выпадает в виде кристаллов, которые
можно отфильтровать от раствора, как только он охладится (при-
мечание 10). Кристаллы промывают холодной водой до отрицатель-
ной реакции на сульфат-ион. Выход диметилглиоксима, плавя-
щегося при 238—240°, составляет 540—575 г.
Обычно получается чисто белый продукт без содержания смо-
листых примесей, и поэтому перекристаллизовывать его излишне
(примечание 5).
Д. Натриевая соль диметилглиоксима. К раствору 75 г едкого
натра в 300 мл воды прибавляют при перемешивании 100 г диметил-
глиоксима (примечание 11). Смесь нагревают, чтобы ускорить
растворение (примечание 12), и отфильтровывают от нераствори-
мых примесей. Еще горячий раствор вливают в 500 мл 95%-ного
спирта. Полученный раствор охлаждают при перемешивании
до 5°, отсасывают выпавшие кристаллы, суспендируют их в 150 мл
спирта, снова отсасывают и наконец сушат при 25°, пока кристаллы
не потеряют запаха спирта (примечание 13). Выход 213—230 г
октагидрата (81—88% теоретич.).
2. Примечания
1. Если не применять механической мешалки, то следует сле-
дить за тем, чтобы на дне сосуда с раствором I не образовался слой
раствора II, так как в этом случае даже слабое встряхивание вызо-
вет слишком бурное выделение газа.
Так как этиловый эфир азотистой кислоты ядовит, следует
вести реакцию в вытяжном шкафу или на открытом воздухе.
2. После прибавления соляной кислоты к метилэтилкетону
следует по возможности быстро начать пропускание этилового
эфира азотистой кислоты, так как соляная кислота вызывает кон-
денсацию кетона, вследствие чего понижается выход монооксима
диацетила.
3. Этиловый эфир азотистой кислоты можно впускать в раствор
очень быстро при условии, чтобы температура не превышала 55°.
Газ поглощается полностью, и на все прибавление требуется около
1,5 часа.
4. Необходимо следить за тем, чтобы отгонка спирта и избытка
метилэтилкетона велась до тех нор, пока температура жидкости
не достигнет точно 90°. Если отгонку прекращают слишком рано,
то неотогнанный спирт впоследствии повышает растворимость
диметилглиоксима; слишком высокая температура вызывает
208
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
образование смолистых продуктов. И в том и в другом случае
выход понижается.
5. После перекристаллизации получают 70—80% взятого моно-
оксима диацетила. Практически весь монооксим, оставшийся
в маточном растворе, можно выделить при помощи перегонки
с водяным паром. Перекристаллизованное и высушенное соедине-
ние плавится при 76,5°. Очистка перегонкой не рекомендуется.
Монооксим диацетила при хранении очень быстро принимает
коричневую окраску, поэтому его следует немедленно по получе-
нии прибавлять к моносульфонату гидроксиламина, если только
хотят получить бесцветный конечный продукт.
6. Молотый лед употребляют вместо толченого льда, так как он
охлаждает сильнее. Зимой рекомендуется применять также и снег.
7. Уксусная кислота оказывает буферное действие и поддержи-
вает низкую концентрацию водородных ионов в растворе, благо-
приятствующую получению хорошего выхода.
8. Кислоту можно прибавлять с любой скоростью при условии,
что температура смеси ниже 0° и не происходит выделения газа.
Все прибавление не должно продолжаться более 15 мин.
9. Смесь нагревают, чтобы ускорить гидролиз дисульфоната
в моносульфонат, а также чтобы повысить растворимость моно-
оксима диацетила в растворе моносульфоната.
10. Диметилглиоксим нельзя отсасывать, пока раствор не
охладился полностью, так как в теплом кислом растворе он
немного растворим.
11. Можно применять сырой или окрашенный диметилглиоксим,
так как загрязнения остаются в водно-спиртовом растворе.
12. Раствор диметилглиоксима в едком натре не следует кипя-
тить, так как продолжительное нагревание вызывает разложение.
13. Натриевую соль можно при желании подвергнуть дальнейшей
очистке растворением в воде и повторным осаждением спиртом. Боль-
шие кристаллы получаются при медленной кристаллизации из воды.
Слишком долгое просушивание или просушивание при слишком
высокой температуре ведет к частичной дегидратации соли, вслед-
ствие чего она растворяется значительно медленнее в воде и стано-
вится менее удобной для приготовления растворов для качествен-
ного и количественного анализов. Натриевая соль диметилглио-
ксима чрезвычайно легко растворима в воде. 3%-ный @,1 м.) водный
раствор может заменить 1%-ный спиртовой раствор, применяемый
в настоящее время для аналитических целей.
3. Другие методы получения
I Монооксим диацетила может быть получен действием изоамило-
вого эфира азотистой кислоты на метилэтилкетон в присутствии
едкого натра х или соляной кислоты 2 в качестве конденсирующих
2,4-ДИМЕТИЛ-3,5-ДИКАРБОЭТОКСИПИРРОЛ
209
средств и нагреванием до температуры плавления нитрозолевули-
новой кислоты3.
Диметилглиоксим может б^ть получен действием соляной
кислоты на монооксим ^ацетила 4; действием гидроксиламина на
диацетил5; действием гидроксиламина на монооксим диацетила6,
действием гидроксиламинмоносульфоната натрия на монооксим
диацетила7. Он образуется в небольших количествах вместе с этилни-
троловой кислотой при действии окислов азота на метилэтилкетон 8.
1 С laisen, Ber. 38, 696 A905).
2 D i е 1 s, F a r k a s, там же 43, 1957 A910); В i I t z, Z. anal. Chem.
48, 164 A909); Semon, Damerell, J. Am. Chem. Soc. 47, 2038 A925).
3 Th a 1, Ber. 25, 1720 A892).
4 S с h r a m m, там же 16, 180 A883); J о h 1 i n, J. Am. Chem. Soc.
36, 1218 A914).
5 F i 11 i g, Daimler, Keller, Ann. 249, 182 A888).
6 Ч у г а е в, Исследования в области комплексных соединений. Москва
1906, стр.64; Ber. 38, 2520 A905); Гандурин, J. prakt. Chem. B) 77, 414
A908); Biltz, Z. anal. Chem. 48, 164 A909); Adams, Kamm, J. Am.
Chem. Soc. 40, 1281 A918).
'Semon, D a m e r e II, там же 46, 1290 A924); 47, 2033 A925).
8 Behrend, Tryller, Ann. 283, 244 A894).
2,4-ДИМЕТИЛ-3,5-ДИКАРБОЭТОКСИПИРРОЛ
(Диэтиловый эфир 3,5-диметил-2,4-пирролдикарбоновой кислоты)
CH3COCH2CO2C2H5 + HNO2 > СН3СОСН (NO) CO2C2H5 + H2O
(NO)CO2C,H5 + 4[H] ^™^L
СН3СОСН (NHS) CO2C2H6 + Н20
НССО2С2Н3 СН8 ¦ СО2С2Н5
CHSCOH
(I
C,H,O,CCNH,
"Г
НОССНо
с,н5о2с
;сн3
+2Н2О
\N.'
н
Предложил: Г. Фишер.
Проверил: К. Ноллер,
1. Получение
В 3-литровую круглодонную колбу с тремя горлами, снабжен-
ную механической мешалкой с жидкостным затвором и делительной
воронкой, помещают 390 г C мол.) ацетоуксусного эфира («Синт.
орг. преп.», сб. 1, стр. 73) и 900 мл ледяной уксусной кислоты. Рас-
твор охлаждают льдом с солью до 5° и прибавляют по каплям при
энергичном перемешивании холодный раствор 107 г A,47 мол.)
95%-ного нитрита натрия в 150 мл воды с такой скоростью, чтобы
температура массы поддерживалась при 5—7°. Если охлаждение
достаточно эффективно, то для прибавления нитрита требуется
14 Сборник № 2
210
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
полчаса, после чего массу перемешивают еще в течение получаса.
Затем ее оставляют на 4 часа, и в течение этого времени ее температура
поднимается до комнатной.
Делительную воронку заменяют обратным холодильником с
широкой внутренней трубкой; третье горло колбы закрывают проб-
кой. Пускают в ход мешалку и через третье горло быстро прибавляют
по частям 196 г (Згр.-ат.) цинковой пыли (примечание 1). Прибавление
ведут таким образом, чтобы довести жидкость до кипения и чтобы
жидкость все время кипела (примечание 2). После того, как вся
цинковая пыль добавлена, реакционную смесь кипятят в течение
часа на горелке (примечания 3 и 4). Раствор в горячем состоянии
сливают с непрореагировавшего цинка в сосуд, куда предварительно
наливают 10 л воды. При этом раствор надо энергично перемешивать.
Остаток цинковой пыли промывают двумя порциями по 50 мл горя-
чей ледяной уксусной кислоты, которую также отделяют деканта-
цией, и приливают к водному раствору. Массу оставляют на ночь,
затем сырой продукт отсасывают, промывают его на фильтре двумя
порциями воды по 500 мл и сушат на воздухе до постоянного веса.
Выход 205—230 г E7—64% теоретич.). Продукт плавится при
126—130°. Если 50 г неочищенного продукта перекристаллизовать
из 100 мл 95%-ного спирта и промыть дважды порциями по 20 мл
холодного спирта, то получается 38,5 г желтых кристаллов с т. пл.
136—137° (примечание 5).
2. Примечания
1. Цинковая пыль должна быть не меньше чем 80% чистоты, при-
чем ее следует взять такое количество, которое соответствовало бы
196 г 100%-ного продукта. Избыток цинковой пыли в количестве
до 3,5 гр.-ат. не вызывал изменения выхода.
2. Чтобы довести жидкость до кипения, требуется примерно
четыре порции по 15 г, после чего оставшееся количество приба-
вляется порциями по 5 г в течение 45 мин. Необходимо тщательно
следить за тем, чтобы вначале цинковая пыль прибавлялась не в
слишком большом количестве, так как это может вызвать сильное
вспенивание. Чтобы быть в состоянии регулировать реакцию, если
она пойдет слишком бурно, необходимо иметь под рукой баню с
ледяной водой и мокрое полотенце.
3. По неизвестным причинам реакция идет не всегда одинаково.
Иногда, после того как прибавлено больше половины цинковой
пыли, вспенивание неожиданно прекращается. Тогда оставшееся
количество можно прибавлять с большей скоростью, что вызывает
энергичное кипение жидкости. При нагревании раствора смесь
иногда загустевает и становится клейкой. В этом случае, прежде
чем приступить к возобновлению перемешивания, необходимо до-
бавить около 300 мл уксусной кислоты. Наименьшие выходы были
получены именно в таких случаях.
2.4-ДИМЕТИЛ-5-КАРБОЭТОКСИПИРРОЛ
2П
4. Имеются указания, что выход может быть повышен до 75%,
если при восстановлении цинковой пылью добавить уксуснокис-
лый натрий. Последний образует комплексное соединение с уксус-
нокислым цинком* и тем самым повышает его растворимость. "Повы-
шение выхода возможно также, если добавлять раствор нитрозо-
соединения к смеси цинковой пыли, уксуснокислого натрия, ацето-
уксусного эфира и ледяной уксусной кислоты вместо того, чтобы
добавлять цинковую пыль*Ъ последнюю очередь1.
5. Сырой продукт под действием света окрашивается в розовый
цвет, тогда как перекристаллизованный продукт вполне стоек.
Можно получить несколько более светлый продукт, если при
перекристаллизации кипятить раствор с 2 г активированного угля,
но на температуру плавления это влияния не оказывает.
3. Другие методы получения
Описанный выше метод всегда применялся для получения 2,4-
диметил-3,5-дикарбоэтоксипиррола и в основном заимствован у
Кнорра42.
1Corwin, Quattlebaum, Jr., J. Am. Chem. Soc. 58, 1083 A936).
3 Knorr, Ann. 236, 318 A886).
2,4-ДИМЕТИЛ-5-КАРБОЭТОКСИПИРРОЛ
(Этиловый эфир 3,5-диметил-2-пирролкарбоновой кислоты)
И, СН3
+ C2H5MgBr > С,Н6 +
н
.
н,с
н
н,с
н
.N/
MgBr
сня
Вг
С1СО2С2Н5
н
н3с
н.с
1. Получение
Н
СН3
MgBr
сн3
СО2С2Н5
\N/
н
Предложил: Г. Фишер.
Проверил: К. Ноллер.
В 1-литровую круглодонную колбу с тремя горлами, снабжен,
ную эффективным обратным холодильником, мешалкой с жидкост.
212
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
ным затвором и капельной воронкой, помещают 13 г @453 гр.-ат.)
магниевых стружек, прибавляют несколько миллилитровчраствора
60 г D2 мл, 0,55 мол.) чистого бромистого этила в 50 мл абсолютного
эфира и пускают в ход мешалку (примечание 1). После того как нач-
нется реакция, приливают 200 мл абсолютного эфира и постепенно
прибавляют остальное количество раствора бромида с такой скоро-
стью, чтобы холодильник успевал конденсировать пары кипящего
эфира (около 30 мин.). Раствор перемешивают еще 15 мин., чтобы
завершить реакцию, и в течение 20 мин. приливают раствор 40 г
@,42 мол.) 2,4-диметилпиррола (стр. 216) в 100 мл абсолютного
эфира (примечание 2). После этого смесь кипятят 30 мин. на паровой
бане.
По охлаждении до комнатной температуры в течение 30 мин. до-
бавляют по каплям раствор 58 г E1 мл, 0,53 мол.) свежеперегнанного
этилового эфира хлормуравьиной кислоты (т. кип. 92,5—93,5°) в
100 мл абсолютного эфира (примечание 3). Смесь нагревают на паро-
вой бане еще 1,5 часа и оставляют на ночь при комнатной темпера-
туре.
Колбу погружают в смесь льда и соли и разлагают ее содержи-
йое, постепенно добавляя 300 мл насыщенного раствора хлористого
аммония и 100 мл воды (примечание 4). Водный слой отделяют в
делительной воронке емкостью 1,5 ли добавляют количество эфира,
достаточное для того, чтобы растворить желтый осадок. Суммарный
объем эфира равняется, примерно, 1 л. Эфирную вытяжку промы-
вают два раза водой порциями по 200 мл и все три водных слоя по-
следовательно извлекают 100 мл эфира. Соединенные эфирные вы-
тяжки сушат 30 г безводного сернокислого натрия, эфир отгоняют
на водяной бане, пока объем раствора не станет равным 200 мл, и
остаток охлаждают до комнатной температуры. Выкристаллизовав-
шийся продукт отсасывают и промывают два раза эфиром порциями
по 25 мл. Выход окрашенного в светложелтый цвет продукта с тем-
пературой плавления 122—123° составляет 35—38 г. От соединенных
фильтратов эфир нацело отгоняют на водяной бане и черный масля-
нистый продукт оставляют на ночь. Полутвердую массу фильтруют
с отсасыванием и промывают минимальным количеством холодного
эфира. Таким образом получают еще 6—7 г желтого продукта с
т. пл. 119—121°.
Весь полученный сырой продукт соединяют вместе и перекри-
сталлизовывают из 75 мл 95%-ного спирта. Получают 37—39 г
слегка окрашенного продукта с т. пл. 123—124°. После вторич-
ной перекристаллизации из спирта получают 34—36 г бес-
цветного продукта с той же температурой плавления. В
результате систематической переработки спиртовых маточных
растворов получают еще 5—6 г чистого продукта. Таким
образом суммарный выход равняется 40—41 г E7—58%
теоретич.).
2,4-ДИМЕТИЛ-5-КАРБОЭТОКСИПИРРОЛ
213
2. Примечания
1. Перемешивание не следует прерывать вплоть до того момента,
когда смесь оставляют на ночь.
2. Реакция эта не Экзотермична, однако ввиду большого объема
выделяющегося этана необходимо соблюдать осторожность при
прибавлении раствора диметилпиррола.
3. При прибавлении первых двух третей раствора имеет место
довольно значительное выделение тепла; в дальнейшем раствор
можно приливать быстрее.
4. Первую треть раствора хлористого аммония нужно прибавлять
очень медленно, часто и энергично взбалтывая колбу.
3. Другие методы получения
Наиболее удобным лабораторным методом получения 2,4-диме-
тил-5-карбоэтоксипиррола является только что описанный метод1.
Более дешевый метод его получения в больших количествах состоит
в частичном гидролизе 2,4-диметил-3,5-дикарбоэтоксипиррола серной
кислотой с последующим декарбоксилированием2. Этот сложный
эфир был также получен алкоголизом 5-трихлорацето-2,4-диметил-
пиррола в присутствии этилата натрия3. Свободная кислота была
получена из 1-[2,4-диметилпиррол-5]-2,4-диметилпиррол-5-карбоно-
вой кислоты 4 и из 2,4-диметилпиррол-5-альдегида 5.
1 F i s с h e r, Weiss, Schubert, Ber. 56, 1199 A923); I n g r a f-
f ia, Gazz. chim. ital. 63, 584 A933).
2 Fischer, Walach, Ber. 58, 2820 A925).
3 Houben, Fischer, там же 64, 2639 A931).
1 Magnanini, там же 22, 38 A889).
6 A 1 e s s a n d r i, Atti accad. Lincei 24, (II) 199 A915) [C. A. 10, 1350
A916)); Alessandri, Passerini, Gazz. chlm. ital . 51 (I), 277 A921).
214
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
СН3
!(CjHsJNH)
2С = 0
СН,
со2с2н5
/СН2Ч
С„Н5О2СС ССО2С2Н5
2,6-ДИМЕТИЛПИРИДИН
СН2О
С2Н5О2СС ССО2С2Н5
СН,С ССН3+Н2О
II
сн,с
ссн3
С2Н5О2С
сн:
CO2C2H
2^2" ХЪ
+ 2КОН
N
KO.Cf Х|СО„К
/ч.
СН3\^СН8
N
+ Са(ОН)а --
СН.
сня
2С2НаОН
СаСО3 + К2СО,
N
Предложили: А. Зингер и С. Мак-Элъеен.
Проверили: Р. Фьюзон и Ч. Вудвард.
1. Получение
В 1-литровую колбу помещают 500 г C,85 мол.) свежеперегнан-
ного ацетоуксусного эфира и колбу снаружи охлаждают льдом.
После этого добавляют 152 г B мол.) 40%-ного формалина и 20—
25 капель диэтиламина. Охлаждение реакционной смеси продол-
жают еще в течение 6 час, а затем смесь оставляют стоять при ком-
натной температуре 40—45 час. К концу этого времени образуются
два слоя: нижний слой—маслянистый, и верхний —водный. Оба
слоя разделяют и водный слой экстрагируют 50 мл эфира. Эфирный
раствор добавляют к маслянистому слою и полученный раствор
сушат 30 г хлористого кальция. Эфир отгоняют на водяной бане,
а остаток в количестве около 500 г разбавляют равным объемом спир-
та и тщательно охлаждают льдом. Затем через реакционную смесь
пропускают ток аммиака до насыщения. На это требуется от 4 до
2,6-ДИМЕТИЛПИРИДИН
215
8 час, причем все это время колбу снаружи охлаждают льдом. Амми-
ачно-спиртовой раствор оставляют стоять при комнатной темпера-
туре в течение 40—45 час, после чего большую часть спирта выпа-
ривают, остаток охлаждают и твердый 1,4-дигидро-3,5-дикарбоэтокси-
2,6-диметилпиридин отсасывают от оставшегося спирта. Сухой
сложный эфир плавится при 175—180°; выход его 410—435 г (84—
89% теоретич.).
200 г @,79 мол.) этого эфира помещают в 5-литровую колбу и
добавляют смесь 270 г воды, 72 г концентрированной азотной кис-
лоты (уд. в. 1,42) и 78 г концентрированной серной кислоты. Колбу
осторожно нагревают, одновременно придавая содержимому колбы
вращательное движение, медленно встряхивая ее рукой. Окисление
сопровождается сильным вспениванием, и если нагревание вести
слишком быстро, часть реакционной смесиможет выбросить из колбы.
После того как пена несколько спадет, реакционную смесь вновь
осторожно нагревают до тех пор, пока жидкость не приобретет тём-
нокрасную окраску. Весь процесс окисления занимает 10—15 мин.
После того, как прекратится кипение жидкости, ее обрабатывают
500 мл воды и 500 г мелкого льда. Полученный раствор сильно под-
щелачивают, постепенно добавляя водный аммиак (уд. в. 0,90). Вы-
павший в осадке 3,5-дикарбоэтокси-2,б-диметилпиридин отсасы-
вают, сушат на пористой тарелке и подвергают перегонке (приме-
чание 1). Выход продукта, кипящего при 170—172°/8 мм, — 115—
130 г, или 58—65% теоретич., считая на дигидроэфир.
Раствор 130 г @,52 мол.) этого эфира в 400 мл этилового спирта
помещают в двухгорлую 2-литровую колбу, снабженную капель-
ной воронкой и обратным холодильником, и нагревают его до ки-
пения. Затем из капельной воронки добавляют одну треть раствора
(примечание 2) 78, 5 г A,4 мол.) едкого кали в 40° мл спирта и спирто-
вый раствор кипятят до тех пор, пока он не сделается прозрачным.
После этого добавляют вторую треть раствора щелочи и реакцион-
ную смесь вновь кипятят до исчезновения осадка. Наконец добав-
ляют последнюю треть спиртового раствора едкого кали. Добавление
щелочи занимает около 20 мин. Реакционную смесь после этого кипя-
тят еще 40 мин.
Содержимое колбы в горячем состоянии выливают в чашку диа-
метром 30 см и спирт выпаривают на паровой бане. Сухую соль из-
мельчают, тщательно смешивают с 390 г окиси кальция и смесь по-
мещают в 2-литровую медную реторту (примечание 3), которую на-
гревают полным пламенем горелки Мекера. Дестиллат помещают
в перегонную колбу и нагревают его на паровой бане; часть, кипя-
щую до 90°, отбрасывают. Остатку дают стоять над твердым едким
кали в течение 12 час, после чего подвергают дробной перегонке.
Диметилпиридин перегоняется при 142—144°/743 мм. Выход 35—
36 г, или 63—65%теоретич., считаянаЗ,5-дикарбоэтокси-2,6-диметил-
пиридин, или 30—36%, считая на исходный ацетоуксусный эфир,
216
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
2. Примечания
1. Перед началом перегонки 3,5-дикарбоэтокси-2,6-диметилии-
ридин следует расплавить, погрузив колбу, в которой будет вестись
перегонка, в кипящую воду, а затем включить вакуум-насос. Если
этого не сделать, при перегонке происходит сильное вспенивание.
2. Если сразу добавить все количество щелочного раствора ед-
кого кали, то образуется бесцветный осадок, который не раство-
ряется даже при длительном нагревании.
3. Весьма желательно вести это разложение в металлической
реторте, так как стеклянные колбы размягчаются при той темпера-
туре, которая необходима для реакции.
3. Другие методы получения
2,6-Диметилпиридин был выделен из фракции каменноугольного
дегтя, содержащей основания1, а также из фракции 139—142° костя-
ного масла2; он был получен также из этилового эфира ацетопиро-
виноградной кислоты и этилового эфира р-аминокротоновой кислоты3.
В литературе описан весьма близкий метод окисления дигидроэфира,
а также омыления и декарбоксилирования полученного продукта*.
1 Lunge, Rosenberg, Ber. 20, 129 A887); Heap, Jones,
Speakman, J. Am. Chern. Soc. 43, 1936 A921); Komatsu, Mohri, J.
Chem. Soc. Japan 52, 722 A931) [C. A. 26, 4936 A932)].
2 Ladenberg, Roth, Ber. 18, 51 A885).
3 Mumm, Huneke, там же 50, 1568 A917).
•Опарина, Карасина, Смирн о в, ЖПХ 11, 965 A938).
2,4-ДИМЕТИЛПИРРОЛ
сн
с2н5о2с;
,СО,С2Н
2С2Н5
Н
+ 4КОН
сн,
н
\ /
\N/
н
1 X
СН3
+ 2С2Н50Н +
Предложил: Г
Проверил: К.
2 К2СОЭ
. Фишер
Ноллер.
1. Получение
В 3-литровой круглодонной колбе приготовляют раствор 270 г
D,8 мол.) едкого кали в 150 мл воды, прибавляют 120 г @,5 мол.)
2,4-ДИМЕТИЛПИРРОЛ
217
сырого 2,4-диметил-3,5-дикарбоэтоксипиррола (стр. 209) и щепотку
песка и все это хорошо смешивают при помощи взбалтывания. Колбу
снабжают обратным холодильником и смесь нагревают на масляной
бане при 130° в течение 2—3 час, время от времени взбалтывая ее
до тех пор, пока густая масса не станет несколько более жидкой
вследствие образования диметилпиррола.
Затем колбу соединяют с холодильником для перегонки с пере-
гретым паром и снабжают капельной воронкой для подачи воды в
середину колбы. В качестве приемника служит 3-литровая кругло-
донная колба, непосредственно соединенная с холодильником, ко-
торый ведет от перегонной колбы, а также со вторым холодильни-
ком, который стоит вертикально («Синт. орг. преп.», сб. 1, стр. 461).
Температуру масляной бани повышают до 160°, а затем в колбу про-
пускают перегретый пар с температурой 220—250°. Постепенно
температуру масляной бани повышают до 200°. Если масса чересчур
пенится, добавляют несколько капель воды из капельной воронки,
причем следят за тем, чтобы вода не попала на горячие стеклянные
стенки (примечание 1). Перегонку с паром ведут до тех пор, пока
не перегонится весь диметилпиррол. На это требуется 1—2 часа,
причем количество дестиллата достигает 2,5—Зл. Дестиллат извле-
кают один раз 200 мл эфира и три раза порциями по 100 мл эфира и
вытяжки сушат 2 часа 20 г безводного поташа. Эфир отгоняют
из видоизмененной колбы Клайзена на 100 мл с 15-сл* дефлегмато-
ром, причем раствор добавляют постепенно из капельной во-
ронки. После удаления эфира остаток перегоняют и собирают
фракцию 160—165°. Выход 27—30 г E7—63% теоретич.; примеча-
ния 2 и 3).
2. Примечания
1. Если содержимое колбы затвердеет или слишком загустеет,
температуру масляной бани следует понизить и скорость пропуска-
ния перегретого пара постепенно уменьшить.
2. Головной фракции почти не получается, однако если провести
несколько последовательных синтезов или работать с большими
количествами, то из вышекипящих фракций удается выделить около
2г продукта на каждый опыт. Если в качестве исходного вещества
брать перекристаллизованный продукт, диметилпиррол получается
в более чистом виде с меньшим количеством вышекипящих
продуктов, однако выход, считая на ацетоуксусный эфир, будет
меньше.
3. 2,4-Диметилпиррол очень легко окисляется воздухом с образо-
ванием красного смолообразного продукта. Если им не предполагают
воспользоваться немедленно, его следует хранить в атмосфере азота
или же в эвакуированной стеклянной ампуле.
218
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
3. Другие методы получения
2,4-Диметилпиррол был получен с помоцью целого ряда реак-
ций, однако метод Кнорра1 и конденсация а*Цетона с аминоацетоном2
являются единственными методами, имеющими препаративное зна-
чение. Описанный выше метод представляет собою видоизмененный
метод Кнорра. Согласно другой модификации этого метода эфир
пиррола и щелочь нагревают в стальной бомбе и продукт перего-
няют в атмосфере инертного газа; выход составляет 95%3.
1 К п о г г, Ann. 236, 326 A886).
2 Р i 1 о t у, К i r s с h, там же 395, 65 A913).
3 С о г vv i п, К г i e b I e, J. Am. Chem. Soc. 63, 1830 A941).
2,5-ДИМЕТИЛПИРРОЛ
Н
СН3СОСНаСНХОСН3+ NH3 .
Н2О
н
Предложили: Д. Юнг и К. Аллен.
Проверили: Цж. Джонсон и X. Стивенсон.
1. Получение
В коническую колбу емкостью 500 мл, снабженную обрат-
ным воздушным холодильником широкого диаметра (примечание 1),
помещают 100 г @,88 мол.) ацетонилацетона (примечание 2) и 200 г
A,75 мол.) углекислого аммония в кусках. Смесь нагревают на мас-
ляной бане при 100° до тех пор, пока не прекратится выделение пу-
зырьков газа, на что требуется 1—1,5 часа. Воздушный холодиль-
ник заменяют на обратный холодильник с водяным охлаждением,
также с широкой внутренней трубкой, и смесь осторожно нагревают
при 115° (температура бани) еще 30 мин. (примечания 1 иЗ). Затем
смесь охлаждают и отделяют верхний желтый слой, содержащий
диметилпиррол (примечание 4). Нижний слой извлекают 15 мл хло-
роформа, причем вытяжку добавляют к сырому диметилпирролу и
все вместе сушат безводным хлористым кальцием в плотно закупо-
ренном сосуде, из которого предварительно вытесняется воздух при
помощи азота.
Высушенный продукт переливают (примечание 4) в видоизменен-
ную колбу Клайзена с дефлегматором и хлороформ полностью от-
гоняют в вакууме (не улавливая паров). Диметилпиррол собирают
при 51—5378 мм или 78—807 25 мм; остаток в колбе очень незна-
2,5-ДИМЕТИЛПИРРОЛ
219
чителен D—5г, т. кип. 80—85725 мм). Выход совершенно чистого
продукта 68—72 г (81—86% теоретич.; примечание 5); хранить его
необходимо в атмосфере инертного газа в герметически закупоренном
сосуде из темного стекла.
2. Примечания
1. Необходимо следить за тем, чтобы возгоняющийся углекислый
аммоний не закупорил холодильник. Время от времени возогнав-
шуюся соль проталкивают стеклянной палочкой обратно в колбу.
2. Ацетонилацетон можно получить гидролизом 2,5-диметил-
фурана. Следующий метод получения сообщил Г. Бенсон. В колбу
емкостью 500мл помещают 125 г A,2мол.) 2,5-диметилфурана (т. кип.
93—96°), 60 г воды, 50 г ледяной уксусной кислоты и 3 мл 10%-ной
серной кислоты. Смесь осторожно кипятят в течение 36 час, доба-
вляют 1 г кристаллического уксуснокислого натрия, чтобы перевести
серную кислоту в сернокислый натрий, и подвергают перегонке
при атмосферном давлении, пользуясь дефлегматором. Отогнав фрак-
цию до 155°, остаток перегоняют в вакууме. Ацетонилацетон соби-
рают при 78—79715 мм или при 88—89725 мм. Выход 128—133 г
(86—90% теоретич.).
3. К концу первого и второго периодов нагревания целесообразно
растворить возогнавшийся углекислый аммоний, для чего в закры-
тый с одного конца холодильник вливают 5—10 мл горячей воды и,
закрыв холодильник с другого конца, переливают воду несколько
раз сверху вниз и обратно; полученный раствор приливают к реак-
ционной смеси.
4. Как при этой, так и при последующих операциях с диметил-
пирролом следует работать быстро, чтобы как можно меньше под-
вергать его действию атмосферного кислорода. Из прибора для пере-
гонки предварительно необходимо вытеснить воздух азотом, а по
окончании вакуум-перегонки прибор необходимо заполнить азотом,
а не воздухом.
5. Ввиду того, что 2,5-диметилпиррол не образует-твердых про-
изводных, в качестве критерия чистоты свежеперегнанного продукта
можно воспользоваться показателем преломления п%°—1,500.
При стоянии продукт постепенно окрашивается в красный цвет
и показатель преломления возрастает. Воздух и свет содействуют
ускорению этого процесса.
3. Другие методы получения
2,5-Диметилпиррол был получен нагреванием ацетонилацетона
с уксуснокислым аммонием в растворе ледяной уксусной кислоты *
или со спиртовым раствором аммиака при 150° в запаянной трубке2.
220
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
Он был получен гидролизом и декарбоксилированием 2,5-диме-
тил-3,4-дикарбоэтоксипиррола3, а также из монокарбоэтоксисое-
динения4.
1 A j е 1 I о, Cusmano, Gazz. chim. ital. 69, 2l6 A939).
2 Paal, Ber. 18, 2254 A885).
3 К n о г г, там же 18, 299, 1565 A885).
4Тимошевская, ЖОХ 9, 766 A939).
5,5-ДИМЕТИЛ-1,3-ЦИКЛОГЕКСАНДИОН
E,5-Диметилдигидрорезоруин; димедон)
ONa
С
(СН3J С = СНСОН3 С2Н5О2С С
NaOCsH6 I I
(СН3J С СО
сн2
о
с
+ СН2(СО
кон
а затем НС1
2^аНдJ
н
(СН3)
ОН
С
/ч
2С СН
2с сон
\ //
сн
Н2С СН.,
I I
(СН3JС СО
сн2
Предложили: Р. Шрайнер и X. Тодд.
Проверили: В. Кйротерс и В. Маи-Ювен.
1. Получение
В сухую 2-литровую круглодонную колбу с тремя горлами, снаб-
женную мешалкой с жидкостным затвором, капельной воронкой на
500л<ли эффективным обратным холодильником, защищенным хлор-
кальциевой трубкой от влаги воздуха, помещают 400 мл абсолютного
спирта. Через внутреннюю трубку холодильника добавляют 23 г
A гр.-ат.) чистого металлического натрия с такой скоростью, чтобы
раствор спокойно кипел. После того как натрий полностью раство-
рится, добавляют сперва 170 г A,06 мол.) диэтилового эфира мало-
новой кислоты, а затем медленно через капельную воронку 100 г
A,02 мол.) окиси мезитила (примечание 1). Раствор кипятят и пере-
мешивают два часа, после чего добавляют раствор 125 г B,2 м.ол.)
«&,5-ДИМЕТИЛ-1,3-ЦИКЛОГЕКСАНДИОН
22 f
едкого кали в 575 мл воды и смесь перемешивают и кипятят на водя-
ной бане еще 6 час.
Горячую массу нейтрализуют и добавлением разбавленной со-
ляной кислоты (уд. в. 1,055) A объем концентрированной кислоты на
два объема воды) делают чуть кислой по лакмусу. Для этого тре-
буется около 550 мл. К колбе присоединяют нисходящий холодиль-
ник и отгоняют на водяной бане возможно большее количество спирта
(около 550 мл).
Остаток в колбе кипятят с 15 г активированного березового
угля (примечание 2), фильтруют и вновь обрабатывают углем. Рас-
твор вновь нейтрализуют по лакмусу разбавленной соляной кислотой
(около 150 мл) и вновь кипятят с углем. Горячий, нейтральный или
щелочной желтый фильтрат подкисляют разбавленной соляной кис-
лотой E0—100 мл), так чтобы была ясно кислая реакция на метил-
оранж, кипятят несколько минут и дают охладиться, причем димедон
выкристаллизовывается. Продукт фильтруют с отсасыванием, про-
мывают ледяной водой и сушат на воздухе.'Выход 96—122 г F7—85%
теоретич.; примечание 3).
2. Примечания
1. Выход димедона зависит от чистоты окиси мезитила («Синт.
орг. преп.», сб. 1, стр. 319), которая должна быть свежеперегнанной,
причем в реакцию берется фракция 126—131°.
2. При прибавлении активированного угля следует соблюдать
осторожность, так как горячий кислый раствор сильно вспенивается
вследствие выделения углекислого газа. Следует работать в боль-
шой колбе и прибавлять уголь очень медленно.
3. Шрайнер и Тодд, которые предложили этот синтез, неизменно
получали выходы от 120 до 128 г продукта с т. пл. 145—147°. При
перекристаллизации из ацетона (около 1 л) получалось 100 г G0%)
чистого бесцветного продукта с т. пл. 147°. Каротерс и Мак-Ювен,
проверявшие этот синтез (пользуясь окисью мезитила, полученной
от фирмы «Истмен Кодак компани», которая перед работой перегоня-
лась), получали выходы от 96 до 112 г, причем продукт плавился при
147—148° и температура плавления после перекристаллизации не по-
вышалась. В литературе для димедона указывается т. пл. 148—150°.
3. Другие методы получения
5,5-Диметилдигидрорезорцин (метон, димедон) всегда полу-
чался из окиси мезитила и малонового эфира. Приведенный выше
метод заимствован у Форлендера Ч
*Vor lander, E r i g, Ann. 294, 314 A897); V о г 1 a n d e r, Z.
anal. Chem. 77, 245 A929); Z. angew. Chem. 42, 46 A929); Chavanne,
Miller, Cornet, Bull. soc. chim. Belg. 40, 673 A931).
222
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
ДИ-0-НАФТИЛРТУТЬ
2 C1OH7N2C1 • HgCl2 + б Си (C10H7JHg =f 2 NL, + 6 CuCl + Hg
Предложили:^А. Несмеянов и Э. Кем.
Проверили: Ф. Уатмор и Р. Витти.
1. Получение
В 2-литровую круглодонную колбу с мешалкой помещают 231 г
@,5 мол.) двойного соединения хлористого 8-нафтилдиазония и хлор-
ной ртути (стр. 557), 700 мл ацетона (т. кип. 55—57°) и 189 г C мол.)
порошка меди (примечание 1). Смесь быстро охлаждают до 20°, пере-
мешивают в течение 1 часа, затем добавляют 700 мл концентрирован-
ного водного аммиака (уд. в. 0,9), хорошо размешивают и оставляют
на ночь. Верхний слой жидкости декантируют, осадок отсасывают
на бюхнеровской воронке и промывают последовательно водой,
ацетоном и эфиром порциями по 25 мл. После того, как сырой про-
дукт высохнет на воздухе, его перекристаллизовывают из ксилола,
обесцветив активированным углем. Полученные слегка желтые
кристаллы (примечание 2) плавятся при 241,5—243,5°. Выход
51—55 г D5—48% теоретич., считая на взятый продукт присоеди-
нения; примечание 3).
2. Примечания
1. См. примечания при получении хлористой ,8-нафтилртути
(стр. 558).
2. Продукт реакции никогда не бывает чисто белого цвета.
Бесцветная ди-р-нафтилртуть может быть получена с хорошим вы-
ходом при действии на хлористую Р-нафтилртуть (стр. 557) йодистого
натрия согласно методу, приведенному в«Синт. орг. преп.», сб. 1,
стр. 201 для ди-л-толилртути.
3. Аналогичным образом из анилина, л-иоданилина и других
ароматических аминов получаются дифенилртуть, ди-л-иодфенил-
ртуть и т. д.
3. Другие методы получения
Ди-р-нафтилртуть была получена действием амальгамы натрия
на Р-бромнафталин2 и действием спиртового раствора йодистого нат-
рия на хлористую Р-нафтилртутьг.
lFrank С. Whitmore, R. J. Sobatzki. Частное сообщение.
2 С h a 11 a w а у, J. Chem. Soc. 65, 878 A894); М i с h a e 1 i s, Ber.
27, 251 A894).
2,4-ДИНИТРОАНИЛИН
223
2,4-ДИНИТРОАНИЛИН
(NOj),C,H,Cl + 2NH8 > (NO2JC6HSNH2
Предложили: Ф. Уэллс и К. Аллен.
Проверили: Р. Фьюзон и Чан Манн-Лу.
1. Получение
В широкогорлую колбу емкостью 250 мл (примечание 1) поме-
щают смесь 50 г @,25 мол.) технического 2,4-динитрохлорбензола
(примечание 2) и 18 г @,23 мол.) уксуснокислого аммония. Колбу
наполовину погружают в масляную баню и снабжают обратным
холодильником и стеклянной трубкой, диаметр нижнего конца ко-
торой не менее 2 см (во избежание засорения трубки), причем конец
ее почти доходит до поверхности реакционной смеси. Через эту
трубку подводят аммиак, который предварительно пропускают
через промывную склянку, служащую также счетчиком пузырьков.
В склянку наливают небольшое количество концентрированного
раствора едкого кали A2 г едкого кали на 10 мл воды).
Масляную баню нагревают до 170D, поддерживают эту темпера-
туру в течение б час. и в течение всего этого времени в колбу про-
пускают газообразный аммиак со скоростью 3—4 пузырьков в
секунду. После того как смесь охладится, твердую массу разби-
вают стеклянной палочкой, смешивают со 100 мл воды, смесь на-
гревают до кипения и фильтруют в горячем состоянии. Остаток рас-
творяют в 500 мл кипящего спирта и к раствору добавляют воды
(около 150 мл) до начала помутнения. Раствор вновь нагревают,
пока он не станет прозрачным, и дают ему охладиться. После стоя-
ния в течение ночи выпавшие кристаллы отфильтровывают и сушат.
Выход 2,4-динитроанилина с т. пл. 175—177° 31—35 г F8—76% тео-
ретич.; примечания 3 и 4). Для дальнейшей очистки продукт пере-
кристаллизовывают так же, как это описано выше, из спирта и воды,
причем на каждый грамм твердого вещества берут 20 мл спирта.
Из сырого продукта получают 90% чистого вещества, которое резко
плавится при 180°.
2. Примечания
1. Горло колбы должно быть достаточно широким, чтобы через
него можно было пропустить широкую вводную трубку. Весьма
удобной можно считать широкогорлую колбу для экстрагирования
емкостью 250 мл.
2. Применялся технический динитрохлорбензол с температурой
застывания 45°. В случае применения продукта перекристаллизован-
ного из спирта (т. пл. 48°), выход 2,4-динитроанилина соответствует
наибольшему указанному выше выходу.
3. В некоторых образцах технического дииитрохлорбензола со-
держались неизвестные примеси,, образующие двойные соединения
с динитроанилином. Соединения эти оставались в фильтрате, причем
224
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
не имело смысла извлекать оттуда те небольшие количества амина,
которые при этом терялись.
4. Ценность этого метода заключается в,том, что избегается
необходимость применения автоклава. В случаз, применения этого
способа кмононитрохлорбензолам полученные результаты оказались
неудовлетворительными.
3. Другие методы получения
2,4-Динитроанилин был получен нагреванием динитрохлорбен-
зола с аммиаком под давлением1: нагреванием 1,2,4-тринитробен-
зола с концентрированным раствором аммиака2; нагреванием дини-
трохлорбензола с мочевиной3 или уксуснокислым аммонием4;
омылением динитроацетанилида, полученного нагреванием динитро-
хлорбензола с ацетамидом при 200—210°5; нагреванием 2,4-динитро-
анизола или 2,4-динитроанисовой кислоты с водным или спиртовым
раствором аммиака под давлением 6; нагреванием 2,4-динитрофенола
с водным раствором аммиака под давлением7; самопроизвольной
перегруппировкой n-нитрофенилнитрамина8 или же перегруппи-
ровкой того же вещества с помощью концентрированной серной
кислоты при 0° 9.
1 С 1 е m m, J. prakt. Chem. B) 1, 170 A870); W i 1 1 g e г о d t, Ber. 9,
979 A876).
2 H e p p, Ann. 215, 362 A882).
• Rohm, Haas Company; ам. пат. 1 752 998 [С. А. 24, 2468 A930)].
* Soc. chim. de la Grande-Paroisse; англ. пат. 169 688 [С. А. 16, 721
A922)].
s Kym, Ber. 32, 3539 A899).
6 Salkowski, Ann. 174, 263 A874).
7 В а г r, Ber. 21, 1542 A888).
8 В a m b e r g e r, Dietrich, там же 30, 1253 A897).
« Hof t, Ann. 311, 98 A900).
2,4-ДИНИТРОБЕНЗАЛ ЬДЕГИД
CeH3N(CH3J + + 2
ONC6H4N (CH3J • HC1 + NaCl + H,0
ONC6H4N (CH3J • HC1 + CH3C6H3 (NO2J + NaCO3
(CH3J NC6H4N = CHC6H3 (NO2J + NaCl + NaHCO3 + H2O
(CH3JNC6H4N = CHC6H3 (NO2), + HaO + HC1
C6H3 (NO2J CHO + (CH3J NC6H4NH2 ¦ HC1
Предложили: Дж. Беннет и Е. Белл.
Проверили: Дж. Конант и Р. Шульц.
1. Получение
Раствор 300 г B,5 мол.) технического диметиланилина в 1050 мл
концентрированной соляной кислоты помещают в большую банку
2,4-ДИНИТРОБЕНЗАЛЬДЕГИД
225
или глиняный сосуд и охлаждают до 5° прибавлением мелко колотого
льда. К содержимому сосуда при механическом перемешивании
подливают медленно из делительной воронки, конец которой нахо-
дится под поверхностью жидкости, раствор 180гB,бмол.) азотисто-
кислого натрия в 300 мл воды. Прибавление ведут в продолжение
1 часа и все время поддерживают температуру ниже 8°, для чего в
случае надобности прибавляют лед. После того как весь нитрит при-
бавлен, смесь оставляют стоять еще час и затем фильтруют ее. Осадок
солянокислого л-нитрозодиметиланилина промывают сначала 400 мл
соляной кислоты A:1), а потом 100 мл спирта. После сушки навоз-
духе вес осадка 370—410 г (80—90% теоретич.; примечание 1).
В 3-литровой круглодонной колбе нагревают на кипящей водяной
бане в продолжение 30 мин. смесь 330 г A,8 мол.) воздушно-сухого
солянокислого нитрозодиметиланилина (примечание 2), 100 г без-
водной соды и 1,5 л 95%-ного спирта и затем отфильтровывают от
хлористого натрия и неизмененной соды. Фильтрат смешивают в
5-литровой круглодонной колбе, снабженной механической мешал-
кой и обратным холодильником, с 300г A,6 мол.) 2,4-динитротолуола
(т. пл. 70°) и смесь нагревают 5 час. на кипящей водяной бане. После
охлаждения продукт конденсации переносят на бюхнеровскую во-
ронку и хорошо просушивают отсасыванием, причем на поверхность
осадка накладывают пластинку мягкого каучука («rubber dam»).Кау-
чук присасывается к осадку и таким образом вытесняет жидкость.
Темиозеленый осадок переносят для промывания в большой стакан,
размешивают с 1 л 95%-ного спирта при нагревании в течение 30 мин.
на водяной бане, затем снова фильтруют и отжимают при помощи
каучука. Динитробензилиден-л-аминодиметиланилин в сыром со-
стоянии переносят в большую круглодонную колбу, в которой нахо-
дится 500 мл смеси из равных объемов концентрированной соляной
кислоты и воды, и пропускают в колбу водяной пар через трубку,
доходящую почти до дна. После того как температура жидкости
достигнет 105°, сильное пропускание пара продолжают еще 15 мин.
(примечание 3).
После охлаждения водный слой декантируют (или отфильтровы-
вают) от застывшего продукта, приливают еще 500 мл разбавленной
кислоты (примечание 4) и повторяют процесс нагревания паром.
Осадок отсасывают, промывают водой и сушат в вакууме. Коли-
чество полученного таким образом сырого динитробензальдегида
составляет 170—210 г, причем продукт плавится при 40—50°. Для
очистки его делят на две части; каждую из них нагревают с 8 л узко
кипящей бензиновой фракции (т. кип. 90—110°) в большой колбе
на водяной бане при механическом перемешивании в продолжение
3 час; раствор декантируют (температура жидкости 80°) и дают ему
закристаллизоваться. Выход 79—104 г B4—32% теоретич.) воз-
душно-сухого очищенного динитробензальдегида с т. пл. 69—71°
(примечание 5). Из маточных растворов при выпаривании или
15 Сборник Л» 2
226
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
отгонке с паром можно получить еще некоторое количество загряз-
ненного продукта; после кристаллизации ¦ по вышеописанному
способу получают еще 20 г альдегида с т. пл. 66Э.
2. Примечания
1. Имеются указания, что аналогичным способом может быть
получен и солянокислый л-нитрозодиэтиланилин, причем свободное
основание может быть получено из солянокислой соли следующим
образом: реакционную смесь, содержащую солянокислый л-нитро-
зодиэтиланилин, полученный из 50 г перегнанного один раз диэтил-
анилина, перемешивают и, поддерживая температуру при 5°, при-
бавляют к нему по каплям 580 мл 2-н, раствора углекислого натрия.
Прибавление соды продолжается около 2 час, после чего реакцион-
ная смесь окрашивается в темнозеленый цвет и выпадает темно-
зеленый осадок. Перемешивание продолжают еще 10 мин., затем
осадку дают осесть в течение 5 мин. и отсасывают его. Осадок про-
мывают сперва тремя порциями по 25 мл дестиллированной воды,
причем воду прибавляют по каплям из пипетки, затем спирто-эфир-
ной смесью A:1), тремя порциями по 20 мл, которые также прибав-
ляют по каплям. л-Нитрозодиэтиланилин сушат в эксикаторе над
хлористым кальцием. Выход 57 г (95% теоретич.). При желании
получить особо чистый продукт л-нитрозодиэтиланилин перекри-
сталлизовывают из петролейного эфира (М. Доджа и А. Мокит,
частное сообщение).
2. Попытки заменить свежеприготовленный материал техни-
ческим нитрозодиметиланилином оказались неудачными.
3. Очень важно, чтобы температура достигла максимума A05°)
и поддерживалась на этом уровне достаточно длительное время в те-
чение гидролиза.
4. Кислые водные растворы содержат л-аминодиметиланилин
и могут быть использованы для получения хинонимидных краси-
телей.
5. 2,4-Динитробензальдегид является ценным реактивом, даю-
щим хорошо кристаллизующиеся продукты конденсации с аминами
и с соединениями, имеющими реакционноспособную метиленовую
или метильную группу1.
3. Другие методы получения
2,4-Динитробензальдегид впервые был получен из динитротолу-
ола i-2-3. Он был также синтезирован из 2,4-диннтробензиланилина
окислением до бензилиденового соединения с последующим гидро-
лизом4'5 и окислением динитробензилового спирта 5.
л-ДЙНЙТРОБЕНЗОЛ
227
1 В е п п е 11, P r a 11, [. Ctiem. Soc. 1929, 1465.
2 Sachs, Kempf, Ber. 35, 1224 A902); Sachs; герм. пат. 121 745
[Ffdl. 6, 1047 A900—02)].
3 M Li 1 1 e r, Ber. 42| 3695 A909).
4 Sachs, E v e r d i n g, там же 35, 1237 A902).
6 С о h n, F r i e d 1 a n d e г, там же 35, 1266 A902).
тг-ДИНИТРОБЕНЗОЛ
n-NO2C,H4NH, + HNOa + HBF4 > n-NOXeH4N]1BF4 + 2 H2O
n-NOXeH4N2BF4 + NaNO, —-t „-C8H4 (NO2J + N2 + NaBF4
Предложил: Е. Старки.
Проверили: Л. Смит и X. Унгнаде.
1. Получение
В стакане на 400 мл растворяют 34 г @,25 мол.) л-нитроанилина
в 110 мл раствора борофтористоводородной кислоты (примечание 1).
Стакан помещают в баню со льдом и раствор перемешивают с по-
мощью эффективной механической мешалки, после чего по каплям
прибавляют холодный раствор 17 г @,25 мол.) азотистокислого на-
трия в 34 мл воды. Когда все количество нитрита добавлено, смесь
перемешивают еще несколько минут, а затем фильтруют с отса-
сыванием через пористый стеклянный фильтр. Твердый борофторид
диазония промывают один раз 25—30 мл холодной борофтористово-
дородной кислоты, дважды 95%-ным этиловым спиртом и несколько
раз эфиром (примечание 2). Выход продукта 56—59 г (95—99% тео-
ретич.).
В 2-литровом стакане растворяют 200 г твердого азотистокислого
натрия в 400 мл воды и добавляют 40 г медного порошка (примеча-
ние 3). Смесь перемешивают с помощью эффективной мешалки
(примечание 4) и к ней медленно прибавляют суспензию борофто-
рида диазония в 200 мл воды. При этом реакционная масса сильно
вспенивается, и для разрушения пены время от времени при-
бавляют 4—5 мл эфира. Реакция считается оконченной после того,
как прибавлено все количество соли диазония. Продукт отсасы-
вают, несколько раз промывают водой, дважды разбавленным рас-
твором едкого натра и вновь водой. Твердый продукт сушат в шкафу
при 110°, измельчают и извлекают кипящим бензолом порциями в
300, 200 и 150 мл. Бензол отгоняют на водяной бане, а остаток пере-
кристаллизовывают из 120—150 мл кипящей ледяной уксусной
кислоты. Выход красноватожелтых кристаллов с т. пл. 172—173° '
составляет 28—34,5 г F7—82%; примечание 5). После перекристал-
лизации из спирта получаются светложелтые кристаллы с темпера-
турой плавления 173°.
228
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
2. Примечания
1. Борофтористоводородную кислоту получают медленным до-
бавлением при непрерывном перемешивании 184 г борной кислоты к
450 г фтористоводородной кислоты D8—52%^ной), находящейся в
медном, свинцовом или покрытом серебром ^осуде, который охла-
ждается в бане со льдом.
Фтористоводородная кислота вызывает весьма болезненные ожо-
ги. При работе с нею открытые части тела должны быть защи-
щены. Ср. примечание 3 на стр. 539.
2. Для фильтрования следует пользоваться фильтром из пори-
стого стекла и борофторид диазония следует перед отсасыванием
хорошо перемешивать на фильтре. Это стойкий
продукт, который можно сушить в вакуум-
эксикаторе над фосфорным ангидридом.
3. Применялась металлическая медь, оса-
жденная в виде порошка.
4. При реакции разложения совершенно
необходимо эффективное перемешивание. Луч-
ше всего для этой цели применять не центри-
рованную мешалку. Реакция требует около
2 час. Менее эффективное перемешивание и
меньшая продолжительность реакции приво-
дят к образованию не чистого продукта, ко-
торый трудно очистить перекристаллизацией.
5. о-Динитробензол можно получить из
о-нитроанилина с помощью того же общего
метода. Из 34 г @,25 мол.) амина получается
38 г F3% теоретич.) сухой борофтористой соли о-нитробензол-
диазония. После реакции борофторида диазония с азотистокис-
лым натрием и медью, о-динитробензол выделяют не фильтрова-
нием и экстрагированием, а перегонкой с водяным паром. Чтобы
уменьшить весьма мешающее вспенивание и переброс жидкости при
перегонке, добавляют небольшое количество парафина. Кроме того,
с успехом может быть использована специальная колба, предназна-
ченная для подобной операции (примечание 6). После перегонки
с водяным паром кристаллический продукт отфильтровывают
и перекристаллизовывают из спирта. Выход о-динитробензола
с т. пл. 116—116,5° составляет 14—16 г C3—38% теоретич.).
6. В синтетических лабораториях «Истмен Кодак Компани» для
перегонки жидкостей, которые сильно вспениваются и перебрасы-
вают, применяют видоизмененную колбу Клайзена, изображенную
на рис. 8. Большой шарик, введенный в боковой отвод, должен по
своей емкости составлять около г]3 от объема перегонной колбы.
Во время перегонки колба устанавливается под углом, как это по-
казано на рисунке.
Рис. 8
2,4-ДИНИТРОФЕНИЛГИДРАЗИН
220
3. Другие методы получения
л-Динитробензол был получен из л-нитрозонитробензола при
действии азотной кислоты г, из п-нитроанилина реакцией Зандмейе-
ра2 и окислением л-нитроанилина концентрированной серной кисло-
той с персульфатом аммония3. о-Динитробензол был получен анало-
гичным образом из о-нитроанилина по реакции Зандмейера4 и окис-
лением персульфатом ам.мония3. Приведенная здесь методика опи-
сана в литературе5.
1 Bamberger, Hub пег, Вег. 36, 3808 A903).
2 Meisenheiraer, P a t г i g, там же 39, 2528 A906).
3 W i 11, К о р е t s с h n i, там же 45, 1134 A912).
4 Korner, Cnntardi, Atti accad. Lincei E) 23, I, 283 A914) [C. A.
8, 3020 A914)].
5 S ta r k e y, J. Am. Chem. Soc. 59, 1479 A937).
2,4-ДИНИТРОФЕНИЛГИДРАЗИН
C8H3 (NO2J Cl + 3 CH3CO2K + NH2NH2 • H2SO,j
C6H3 (NO2J NHNH3 + K2SO4 + KC1 + 3 CH3CO2H
Предложил: К- Аллен.
Проверили: Р. Фьюзон и М. Фарлоу.
1. Получение
35 г @,27 мол.) гидразинсульфата («Синт. орг. преп.», сб. 1,
стр. 158) суспендируют в 125 мл горячей воды, находящейся в ста-
кане емкостью 400 мл, и к смеси, перемешивая ее от руки, прибав-
ляют 85 г @,87 мол.) уксуснокислого калия (примечание 1), после
чего жидкость кипятят в продолжение 5 мин. и затем охлаждают
приблизительно до 70°. Прибавляют 75 мл спирта, осадок отсасы-
вают и промывают 75 мл горячего спирта. Профильтрованный рас-
твор гидразина используется в следующей стадии.
В 1-литровой колбе, снабженной мешалкой и обратным холо-
дильником, растворяют 50,5 г @,25 мол.) технического 2,4-динитро-
хлорбензола в 250 мл спирта; к полученному раствору прибавляют
раствор гидразина и смесь кипятят с обратным холодильником при
работающей мешалке в продолжение 1 часа. Главная масса продукта
выпадает в течение первых 10 мин. (примечание 2). По окончании
кипячения смесь хорошо охлаждают, осадок отсасывают и промы-
вают сначала 50 мл теплого F0°) спирта, чтобы удалить неизменен-
ное галоидное соединение, а затем 50 мл горячей воды. Продукт
плавится с выделением газа при 190—192° (примечание 3), весит 30 г
и достаточно чист для большинства целей. При отгонке половины
спирта от фильтрата получают вторую, менее чистую порцию дини-
230
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
1,4-ДИФЕНИЛБУТАДИЕН
231
трофенилгидразина, которую перекристаллизовывают из н.-бутило-
вого спирта C0 мл на I г продукта; примечание 4). Общий выход
40—42 г (81—85% теоретич.; примечания 5—,10).
2. Примечания
1. Вместо уксуснокислого калия можно взять эквивалентное
количество уксуснокислого натрия.
2. Во время выделения происходит сильное разогревание.
3. Точка плавления не резкая; вещество в капилляре спадает
при температуре на '10° ниже, чем расплавится полностью.
4. Лучшим растворителем для перекристаллизации, несмотря
на то, что его требуется большое количество, является н-бутиловый
спирт. Когда работа ведется с большими количествами, бутиловый
спирт можно заменить тстралином, пиридином или диоксаном A0 мл
на 1 г). К счастью, главная масса получающегося продукта не тре-
бует дальнейшей очистки.
5. При полном выпаривании фильтрата получается клейкий
остаток; количество содержащегося в нем динитрохлорбензола
настолько мало, что выделять его не имеет смысла.
6. Если вместо каждых 35 г уксуснокислого калия взять 10 г
едкого натра и раствор, прокипятив 5 мин., не отфильтровывать
от соли, получается 70%-ный выход динитрофенилгидразина; ка-
чество его хуже, чем у продукта, получаемого при работе с уксусно-
кислым калием.
7. Если для работы взять гидразингидрат, получается несколько
больший выход.
8. 2,4-Динитрофенилгидразин употребляют в качественном орга-
ническом анализе для получения кристаллических производных
карбонильных соединений г<2.
9. Этим же способом можно получить из 2,6-динитрохлорбензола
2,6-динитрофенилгидразин; хлористый пикрил дает 2,4,6-тринитро-
фенилгидразин.
10. Такие же хорошие результаты были получены при работе
по описанному способу с количествами в два и пять раз большими.
3. Другие методы получения
2,4-динитрофенилгидразин был получен из гидразингидрата и
2,4,-динитрохлорбензола2'3 или 2,4-динитробромбензола 4 и из тех
же галоидных соединений и уксуснокислого гидразина *.
1 А 1 1 е n, J. Am. Chem. Soc. 52, 2955 A930).
2 Brady, E Ism i e, Analyst 51, 77 A926); Brady, J. Chem. Soc.
1931» 757.
3 P u r go 11 i, Gazz. chim. ital. 24 (I) 555 A894).
¦ Curtius, Dedichen, J. prakt. Chem. B) 50, 258 A894).
1,4-ДИФЕНИЛБУТАДИЕН
(Бистирил)
C6H5CH = СНСНО —(
oo (CH3CO)SO
С6Н5СН,СО„Н
С6НаСН = СНСН = СНС6Н5 + СО2 + Н2о"
Предложил: Б. Корсон.
Проверили: К- Ноллер и Дж. Карсон.
1. Получение
В 1-литровую круглодонную колбу с обратным холодильником,
защищенным хлоркальциевой трубкой, помещают 150 г A,1 мол.)
фенилуксусной кислоты («Синт. орг. преп.», сб. 1, стр. 440), 147 г
A,1 мол.) свежеперегнанного коричного альдегида, 122 г свинцового
глета и 155 мл уксусного ангидрида. Смесь кипятят 5 час. (приме-
чание 1), выливают еще горячей в стакан и оставляют на ночь.
Полутвердую массу разбалтывают палочкой в кашицу, фильтруют с
отсасыванием через большую воронку Бюхнера и отжимают досуха.
Твердый продукт промывают в воронке два раза этиловым спиртом
порциями по 35 мл, хорошо перемешивая массу перед отсасыванием.
Твердый продукт переносят в стакан, размешивают с 50 мл спирта и
вновь фильтруют с отсасыванием. Аналогичным образом осадок
промывают еще раз 50 мл спирта (примечание 2). Продукт может
иметь различную окраску — от светложелтой до рыжеватокорич-
невой и весит 62—67 г B7—29% теоретич.; примечание 3). Темпе-
ратура плавления 149,5—153,5°.
С целью очистки продукт растворяют в 300 мл горячего бен-
зола, раствор кипятят в течение 3 мин. с 5 г обесцвечивающего угля,
фильтруют горячим через нагретую воронку Бюхнера с несильным
отсасыванием. Бензольный фильтрат обрабатывают 500 мл нагре-
того до кипения этилового спирта и затем охлаждают до 10° в бане
со льдом при взбалтывании. Кристаллы отсасывают, хорошо
отжимают и дают медленно просочиться через них 50 мл этилового
спирта, после чего вновь хорошо отсасывают. Вес продукта после
перекристаллизации 52—57 г B3—25% теоретич.; примечание 4).
Т. пл. 152,5—153,5°. Продукт представляет собою транс-транс-
форму диена.
2. Примечания
1. В течение первого получаса колбу следует нагревать осто-
рожно и неоднократно взбалтывать, чтобы облегчить растворение
окиси свинца.
2. Необходимо тщательное промывание.
3. Из маточного раствора можно выделить еще около 8 г сырого
углеводорода, однако выделение это весьма кропотливо и потому не
рекомендуется.
232
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
4. Если этот маточный раствор упарить до объема в 25 мл и
дать ему кристаллизоваться, то можно выделить еще. 5,5 г вещества.
3. Другие методы получения
Единственный метод, который имеет црепаративное значение,
состоит в конденсации фенилуксусной кислоты с коричным альдеги-
дом1. Описанная выше методика в основном взята у Куна и Винтер-
штейна2. Вистирил был также получен из стирилмагнийбромида и
хлорной меди3 или азобензола* и восстановлением р-бромстирола
гидразином в присутствии палладия3.
1 Thiele, Schleussner, Ann. 306, 198 A899).
2 Kuhn, Winterstein, Helv. Chim. Acta 11, 103 A928).
3 Sakellarios, Kyrirais, Ber. 57, 325A924); см. также Oilman,
Parker, J. Am. Chem. Soc. 46, 2827 A924).
4 Gilman, Pickens, там же 47, 2410 A925).
6 Busch, Weber, J. prakt. Chem. B) 146, 54 A936).
4,5-ДИФЕНИЛГЛИОКСАЛОН
D,5-Дифенил-2( 5 )-имидазолон )
С6Н5СНОНСОС8Н5 + NHX0NH2
C6H5CH —NH
С6Н5С = N
СО + 2Н,0
Предложили: Б. Корсон и Э. Фрибирн.
Проверили: Ф. Уитмор и М. Уитмор.
1. Получение
Смесь 212 г A мол.) бензоина (примечание 1), ПО г A,8 мол.) мо-
чевины и 800 мл ледяной уксусной кислоты нагревают в продолже-
ние 7 часов в 2-литровой круглодонной колбе с обратным холодиль-
ником (примечание 2). Горячий раствор быстро переливают в 2-ли-
тровый стакан (примечание 3) и оставляют стоять не менее 3 час.
Холодную смесь переносят большой деревянной ложкой или шпа-
телем на воронку Бюхнера диаметром 30 см, отсасывают и отжи-
мают с помощью резиновой прокладки возможно тщательнее. Филь-
трат выливают, а осадок переносят обратно в стакан, затем в тече-
ние 30 мин. размешивают при помощи механической мешалки с
500 мл эфира и снова отсасывают (примечание 4). Полученные кри-
сталлы раскладывают и дают им просохнуть в течение'ночи (приме-
чание 5). Затем продукт нагревают с'1 л ледяной уксусной кислоты
в 2-литровой круглодонной колбе с обратным холодильником (при-
мечание 6). Прозрачный раствор вливают при механическом пере-
мешивании в 2500 мл воды, находящейся в сосуде емкостью 4 л.
Перемешивание продолжают 30 мин. (примечание 7), осадок отса-
ДИФЕНИЛМЕТАН
233
сывают на воронке Бюхнера диаметром 30 см и возможно тщательнее
отжимают. Затем кристаллы переносят в 2-литровый стакан и раз-
мешивают при помощи механической мешалки в продолжение 10 мин.
с 1 л воды. После отсасывания промывку водой повторяют еще раз.
Затем кристаллы вновь помещают в стакан, размешивают с 750 мл
зфира и снова отсасывают. Продукт сушат на воздухе до постоянного
веса. Выход белых пушистых кристаллов дифенилглиоксалона с
температурой плавления 330—335° (исправл.; примечание 8) соста-
вляет 220—230 г (93—97% теоретич.).
2. Примечания
1. Бензоин («Синт. орг. преп.», сб. 1, стр. 95), для реакции
можно не перекристаллизовывать.
2. Первоначально красновато-оранжевая окраска затем перехо-
дит в темножелтую.
3. Если продукт оставить в колбе, он застынет настолько, что
позднее извлечь его будет трудно.
4. Непрореагировавший бензоин удаляют эфиром, в котором
дифенилглиоксалон растворяется с большим трудом.
5. Продукт трудно высушить досуха.
6. Операция бывает неудачной, если взято меньше 1 л ледяной
уксусной кислоты.
7. Вода удаляет непрореагировавшую мочевину.
8. Точку плавления удобно определять на поверхности ртути,
которую нагревают в пробирке с термометром, опущенным прямо в
ртуть.
3. Другие методы получения
Единственным способом, представляющим препаративный инте-
рес, является конденсация бензоина с мочевиной в уксуснокислом
растворе, которую описали Бильц1 и Четтэуэй2.
1 В i I tz, Ann. 368, 173 A909).
2 С h a 11 a w а у, С о u 1 s о n, J. Chem. Soc. 1928, 1363.
С6Н,СНХ1
C6H
ДИФЕНИЛМЕТАН
(Амальгама алюминия)
6^6
НС1
Предложили: В. Хартман и Р. Филлипс.
Проверили: Р. Фыозон и С. Бабкок.
1. Получение
В 5-литровую колбу, снабженную обратным холодильником и
S-образной трубкой, соединенной с капельной воронкой, помещают
10 г амальгамированной алюминиевой стружки (примечание 1) и 2 кг
B,3 л 25,6 мол.) бензола, предварительно обезвоженного перегон-
234
СИНТЕЗЫ ОРГАНИЧИХНИХ ПРЕПАРАТОВ
кой, при которой первый мутный погон был отброшен, ^ензол на-
тревают на паровой бане до кипения, затем доступ пара прекращают
и добавляют 500 г C,96 мол.) хлористого бензида с такой скоростью,
чтобы жидкость непрерывно кипела (примечание 2). Выделяющийся
хлористый водород поглощают водой или же|стводят его в тягу.
После того, как добавление хлористого бензила закончено (на что
требуется около 1 час), смесь нагревают в течение 10—15 мин. или
же до прекращения выделения хлористого водорода. По охлажде-
нии бензольный раствор дифенилметана отделяют декантацией от
небольшого количества смолы (примечание 3) и промывают сперва
5%-ным раствором едкого натра, а затем водой. После частичной
сушки хлористым кальцием бензол отгоняют на водяной бане, а
остаток перегоняют в вакууме. Первый погон собирают до \25°j10mm,
основную фракцию при 125—130°/10 мм и третью фракцию
до 1507Юлш (примечание 4). Повторная перегонка первой и третьей
фракций дает еще небольшое количество вещества,которое присоеди-
няют к основной фракции. Последнюю вымораживают и сливают
с кристаллов небольшое количество оставшегося масла. Выход
продукта с т. пл. 24—25° составляет 330—350 г D9,5—52,5% теоре-
тич.).
2. Примечания
1. Амальгаму алюминия приготовляют следующим образом:
алюминиевые стружки, предварительно промытые эфиром для обез-
жиривания, размешивают в течение нескольких минут с 5%-ным
раствором хлорной ртути, после чего их быстро промывают водой,
а затем метиловым спиртом. Амальгамированный алюминий надо
применять немедленно.
2. Иногда реакция долго не начинается. Сначала прибавляют
не более 50—60 г хлористого бензила и смесь нагревают до тех пор,
пока выделение хлористого водорода не укажет на начало реакции.
Если в колбе будет слишком много хлористого бензила, то реакция
может начаться столь бурно, что содержимое колбы может быть
выброшено.
3. В той же колбе, которая содержит алюминиевые стружки и
следы смолистого продукта, можно провести несколько синтезов,
не добавляя больше катализатора. При этом в последующих синте-
зах не наблюдается индукционного периода.
4. Остаток и высококипящая фракция могут быть частично пре-
вращены в дифенилметан нагреванием с одной третьей частью по
весу хлористого алюминия и с пятикратным весовым количеством
бензола.
3. Другие методы получения
Дифенилметан может быть получен из бензола и хлористого бен-
зила с применением в качестве конденсирующего агента хлористого
Р,Р-ДИФЕНИЛПРОПИОФЕНОН
235
алюминия г, фтористого водорода2, хлористого бериллия3, двойной
соли хлористого алюминия и хлористого натрия4, цинковой пыли8,
хлористого цинка 6 или амальгамы алюминия 7. Описанный выше
метод является небольшим видоизменением последнего способа.
Бензол и бензиловый спирт дают дифенилметан при действии
фтористого бора 8, фтористого водорода9 или хлористого бериллия3.
Дифенилметан был получен также из бензола, хлористого метилена
и хлористого алюминия10 и из бензола, формальдегида, этилового
спирта и концентрированной серной кислоты и. Восстановление бен-
зофенола до дифенилметана было осуществлено действием йодисто-
водородной кислотой и фосфором12, натрия и спирта13 и сплавлением
с хлористым цинком и хлористым натрием14. Конденсацию хлори-
стого бензилмагния с бензолом с образованием дифенилметана можно
осуществить добавлением небольших количеств магния и воды 1б.
1 Fried el, Balsohn, Bull. soc. chim. B) 33, 337 A880).
2 Simons, Archer, j. Am. Chem. Soc. 61, 1521 A939).
3Bredereck, Lehmann, Schonf eld, Fritzsche, Ber.
72, 1414 A939).
4 N 0 r r i s, Klcmka, J. Am. Chem. Soc. 62, 1432 A940).
5 Zincke, Ann. 159, 374 A871).
6 Friedel, Crafts, Ann. chim. phys. F) 1, 478 A884).
7 Hirst, Cohen, J. Chem. Soc. 67, 827 A895).
8 McKenna, Sow a, J. Am. Chem. Soc. 59, 470 A937).
9 Simons, A r с h e г, там же 62, 1623 A940).
10 F r i e d e 1, Crafts, Bull. soc. chim. B) 41, 324 A884); Schwarz,
Ber. 14, 1520 A881).
11 Blythe and Company Ltd., англ. пат. 446 450 [С. А. 30, 6760A936)].
12 Grae be, Ber. 7, 1624 A874).
13 К 1 a g e s, Allendorff, там же 31, 999 A898).
14 С 1 a r, там же 72, 1645 A939).
15 К h a r a s с h, G о 1 d b e r g, M а у о, J. Am. Chem. Soc. 60, 2004
A938).
0,0-ДИФЕНИЛПРОПИОФЕНОН
С6Н3СН = СНСОС6Н3 + С6Н6 —1 (С8Н5J СНСНХОС6Н5
Предложил: П. Шильднек.
Проверили: К- Шллер и К. Кемп.
1. Получение
В 3-литровую круглодонную колбу с тремя горлами, снабжен-
ную мешалкой с жидкостным затвором, термометром и делительной
воронкой емкостью 500 мл, помещают 1,7 л сухого бензола и 160 г
A,2 мол.) безводного хлористого алюминия в порошке (примечание 1).
Смесь охлаждают до 10°, погружая колбу в баню с водой и льдом, и
прибавляют раствор 120 г @,58 мол.) бензальацетофеиопа (приме-
236
СИНТЕЗЫ ОРГАНИЧИХ-КИХ ПРЕПАРАТОВ
чание 2) в 300 мл сухого бензола. Температуру реакционной смеси
поддерживают при 10—20°, причем вся операция прибавления про-
должается 30 мин, Охлаждающую баню удаляют и перемешивание
продолжают при комнатной температуре до тех пор, пока образо-
вавшийся вначале плотный желтый осадок не^ерейдет в раствор
(примечание 3). После этого перемешивание продолжают еще 1 час
и считают реакцию законченной.
Большую часть бензольного раствора, окрашенного в бурый
цвет, сливают в холодную смесь 100 мл концентрированной соляной
кислоты и 1,5 л воды, находящейся в 5-литровой круглодонной колбе.
Остаток фильтруют через бюхнеровскую воронку и кусочки хло-
ристого алюминия промывают два раза бензолом порциями по 100 мл.
Фильтраты присоединяют к главному раствору и все вместе тща-
тельно промывают разбавленной кислотой. Если жидкости плохо
расслаиваются, то смесь фильтруют при небольшом разряжении
и водный слой сифонируют, Раствор промывают два раза водой пор-
циями по 1,5 л и, если нужно, вновь фильтруют.
Прозрачный светложелтый раствор подвергают быстрой пере-
гонке с водяным паром (примечание 4) из той же 5-литровой колбы
и, после того как отгонится весь бензол, колбу охлаждают под водо-
проводным краном, не переставая взбалтывать. Оставшееся в колбе
масло застывает в виде светлокоричневых шариков. Их собирают,
возможно лучше отделяют от воды и растворяют в 2250 мл кипящего
спирта. Прибавляют 5 г обесцвечивающего угля, горячий раствор
фильтруют с отсасыванием и дают ему охладиться. Наилучшие
результаты получаются в том случае, если спиртовой раствор при
охлаждении его до комнатной температуры медленно перемешивать
с помощью механической мешалки. Перемешивание прекращают
тогда, когда масса станет полутвердой, после чего ее оставляют
стоять еще 24 часа. Мелкие бесцветные иглы отфильтровывают
через бюхнеровскую воронку диаметром в 15 см и как можно тща-
тельнее отжимают. Выход воздушно-сухого бесцветного продукта с
т. пл. 91—92° (примечание 5) составляет 125— 140г G6—85% теоре-
тич.; примечание 6).
2. Примечания
1. Пробные опыты показали, что для доведения реакции до
конца при комнатной температуре на каждый моль беизальацетофе-
иона требуется по крайней мере два моля безводного хлористого
алюминия. Если взять меньшее количество хлористого алюминия,
то желтый продукт присоединения не растворяется полностью даже
после перемешивания в течение 24 час, причем выход уменьшается.
2. Бензальацетофенон («Синт. орг. преп.», сб. 1, стр. 77) должен
быть совершенно чистым (т. пл. 55—56°) и, в частности, не должен
содержать бензальдегида.
ДИФЕНИЛСЁЛЕН
237
3. К концу реакции внешний вид смеси заметно изменяется.
Желтая окраска быстро переходит в темнобурую. Совершенно про-
зрачный раствор не образуется, так как хлористый алюминий
остается во взвешенном состоянии.
4. Если бензол отгонять с водяным паром непосредственно из
кислой смеси, то полученный сырой продукт окрашен в более тем-
ный цвет, труднее кристаллизуется и получается менее чистым после
кристаллизации.
5. Указанная в литературе1>2'3 температура плавления 3,8-дифе-
нилпропиофенона 96°. Продукт ст. пл.91—92° был подвергнут пере-
кристаллизации до постоянной температуры плавления из спирта и
из лигроина, однако температура плавления осталась равной 92°
(с поправкой). Было также на'йдено, что оксим и монобромид, по-
лученные по Колеру2, плавятся соответственно при 133° (с поправ-
кой) и при 166° (с поправкой).
6. Если маточный раствор упарить до объема в 400 мл, то можно
получить еще 12—16 г менее чистого продукта ст. пл. 88—9 Г.
3. Другие методы получения
р,Р-Дифенилпропиофенон был получен из бензальацетофенона
при реакции с бромистым фенилмагнием и с рядом фенильных про-
изводных других металлов2; конденсацией бензальацетофенона и
бензола посредством концентрированной серной кислоты3 или хло-
ристого алюминия4, а также при действии хлористого алюминия на
смесь бензола и солянокислого бензальацетофенона4. Описанная
выше методика в основном заимствована у Форлендера и Фрид-
берга 4.
1 К о h I e r, Am. Chem. J. 29, 352 A903).
2 G i 1 m a n, К i r b y, J. Am. Chem. Soc. 63, 2047 A941).
3 К oh ler, Am. Chem. J. 31, 642 A904).
a Vor lander, Friedberg, Ber. 56, 1144 A923).
ДИФЕНИЛСЁЛЕН
C6H5NH3 + NaNO2 + 2 HC1 - C6H5NX1 + NaC 1 + 2 H,0
2 CeH5NaCl + K.,Se.v > (C6H5)aSe + 2 KC1 + (x — 1) Se + 2 N,
Предложил: Г. Лейсестер.
Проверили: В. Хартман и Р. Буллард.
1. Получение
В стакан емкостью в 500 мл помещают смесь, предварительно
растертую в ступке и содержащую 360 г F,4 мол.) измельченного
едкого кали и 240 г C rp.-ат.) черного порошкообразного селенад
Смесь нагревают (примечание 1) на масляной бане при 140—150°
2"з§
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
до образования густой тёмнокрасной жидкости (примечание 2),
после чего ее добавляют (примечание 3) небольшими порциями
к 400 мл ледяной воды, находящейся в 5-литровой колбе. Раствор
сохраняют в бане со льдом.
- К 375 мл D,3 мол.) соляной кислоты (уд.-jp. 1,18) и 200 г льда
добавляют 139,6 г A,5 мол.) анилина. Полученный раствор диазо-
тируют раствором 103,5 г A,5 мол.) нитрит,натрия (х. ч.), причем
по мере необходимости к реакционной смеси добавляют лед, так
чтобы температура не поднималась выше 5°. Конечный объем диазо-
тированного раствора составляет около 1 л. Этот раствор прили-
вают тонкой струей из капельной воронки к раствору селенида
калия, который при этом энергично перемешивают механической
мешалкой. После того, как весь диазо-раствор добавлен, красный
водный раствор декантируют с образовавшегося темного масла
и нагревают до кипения (примечание 4). Затем его приливают
обратно к маслу, смесь хорошо перемешивают (примечание 5),
добавляют 200 мл хлороформа, селен отфильтровывают и на фильтре
промывают его еще небольшим количеством хлороформа (приме-
чание б). После отделения хлороформенного слоя водный слой
вновь экстрагируют 200 мл хлороформа. Соединенные вытяжки
перегоняют, при чем дифенилселен собирают от 300 до 315°. Выход
желтого масла с довольно неприятным запахом составляет 138—
150г G9—86% теоретич.; примечание 7). Если желательно получить
более чистый продукт, полученное вещество перегоняют в ва-
кууме; температура его кипения 165—167°/12 мм.
2. Примечания
1. Хотя при этой реакции никаких вредных газов и не выде-
ляется, однако смесь обладает довольно неприятным запахом,
так что лучше весь синтез проводить в вытяжном шкафу.
2. Если едкое кали и селен совершенно сухие, то образуется
густая паста. Добавление нескольких миллилитров воды ведет
к образованию упомянутой здесь тёмнокрасной жидкости.
3. Если массе дать охладиться и затвердеть, то становится
очень трудным измельчить ее и вновь растворить.
4. Водный слой необходимо нагревать отдельно, так как в про-
тивном случае стакан с вязкой маслянистой жидкостью неизменно
лопается даже в том случае, если вся масса хорошо перемешивается.
5. В результате этой обработки красная коллоидальная форма се-
лена переходит в черную модификацию, которая легче фильтруется.
6. Всего получается обратно 101—115 г селена.
7. При получении меньших количеств дифенилселена выход па-
дает до 70—75%. Этот метод может быть с успехом применен также
для получения соответственных производных селена из трех толуи-
динов с выходом от 50 до 70%.
ДИФЕНИЛСЕЛЕН ДВУХЛОРИСТЫЙ И ТРИФЕНИЛСЕЛЕН ХЛОРИСТЫЙ 239
3. Другие методы получения
Описанная здесь методика является видоизменением метода
Шеллера г. Дифенилселен может быть получен также из диазоти-
рованного анилина и моноселеиидов щелочных металлов2; реак-
цией Фриделя-Крафтса из бензола и четыреххлористого селена 8
или двуокиси селена d; из дифеиилсульфона и селена 5; из фенил-
магнийбромида и селена 6, двухлористого селена 7, хлорокиси
селена7 или двубромистого селена8; а также из селенофенолята
натрия и бромбензола9.
1 S с h о е 1 1 е г, Вег. 52, 1517 A919).
1 Lesser, Weiss, там же 47, 2521 A914).
3 К г a f f t, К a s с h a u, там же 29, 428 A896); В г a d t, Green, J.
Org. Chem. 1, 540 A937).
4 Lyons, Br a d t, Ber. 60, 60 A927).
5 К r a f f t, V о r s t с г, там же 26, 2817 A893); Krafft, Lyons,
там же 27, 1761 A894).
6 T a b о u г у, Ann. chim. (8) 15, 35 A908).
'Strecker, Willing, Ber. 48, 196 A915).
8Pieroni, Balduzzi, Gazz. chim. ital. 45, (II) 106A915).
«Foster, В г о w n, J. Am. Chem. Soc. 50, 1182 A928).
ДИФЕНИЛСЕЛЕН ДВУХЛОРИСТЫЙ И ТРИФЕНИЛСЕЛЕН ХЛОРИСТЫЙ
(А)
(С8Н5J Se
(Б) (CeHa), SeCl, + C6He
-НС^ (С6Н5J SeCl,
(CeHe)s SeCl + HC1
Предложил: Г. Лейсестер.
Проверили: В. Хартман и Р. Буллард.
1. Получение
А. Дифенилселен деухлористып. В стакан емкостью в 1,5 л
наливают 250 мл D мол.) азотной кислоты (уд. в. 1,42) и к ней
небольшими порциями приливают 125 г @,53 мол.) дифенилселена
(см. стр. 237), Затем добавляют соляной кислоты (уд. в. 1,18) до
полноты осаждения, на что требуется около 170 млB мол.) кислоты.
Смесь разбавляют 500 мл воды, желтый осадок отфильтровывают
и сушат на воздухе. Сырой продукт подвергают очистке, экстра-
гируя его 500 мл кипящего бензола. Выделяющиеся при охлажде-
нии кристаллы отсасывают, а фильтрат применяют для повторного
экстрагирования. Для перекристаллизации всего продукта необ-
ходимы три такие обработки. Выход желтых кристаллов, разла-
гающихся при 187—188°, составляет 137—141 г (85—87% теоретич.).
Б. Трифенилселен хлористый. К 87 г A00 мл, 1,1 мол.) бензола,
находящимся в 1-литровой трехгорлой колбе, снабженной меха-
нической мешалкой, добавляют 30 г @,22 мол.) безводного хлори-
240
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
ДИФЕНИЛСУЛЬФИД
241
стого алюминия. Суспензию охлаждают в бане со льдом и доба-
вляют к ней при работающей мешалке 40 г Ш, 13 мол.) двухлори-
стого дифенилселена порциями примерно по i г, всего в течение
25 мин. Перед добавлением каждой порции «температура должна
быть ниже 10°, чтобы избежать потемнения конечного продукта.
Когда все количество прибавлено, реакционную смесь оставляют
стоять 3 часа при комнатной температуре, после чего осторожно
добавляют 200 мл воды (примечание 1). Бензольный слой отделяют
и отбрасывают. Если водный слой получился окрашенным, то
с помощью нескольких экстрагирований бензолом можно удалить
большую часть окраски без ущерба для выхода. Затем водный слой
экстрагируют три раза хлороформом, порциями по 50 мл. Соеди-
ненные вытяжки упаривают до объема в 40 мл и обрабатывают
их 120 мл эфира (примечание 2). При этом выпадает желтое масло,
которое почти немедленно затвердевает в белый порошок. Хлори-
стый трифенилселен отфильтровывают и перекристаллизовывают из
300 мл метилэтилкетона, к которым добавлено 20 мл воды (приме-
чание 3). После сушки при 100° выход безводного продукта соста-
вляет 30 г F7% теоретич.; примечание 4).
2. Примечания
1. В начале гидролиза выделяется большое количество тепла.
2. Хлористый трифенилселен может быть осажден из водного
раствора в виде двойной соли с хлористым цинком г.
3. Хлористый трифенилселен плохо растворим в безводном
метилэтилкетоне B г в 300 мл). Если пользоваться смесью воды
и метилэтилкетона, то вещество кристаллизуется с двумя моле-
кулами воды. Кристаллизационную воду можно удалить нагре-
ванием в течение 30 мин. при 100°, однако за счет влажных раство-
рителей или влажного воздуха вода поглощается вновь.
4. Этот метод может быть применен также дли получения солей
л-толил- или смешанных фенил-л-толил-селенония.
3. Другие методы получения
Двухлористый дифенилселен может быть получен из дифенил-
селена 2>3 действием хлора или последовательной обработкой сперва
азотной, а затем соляной кислотой. Хлористый трифенилселен
был получен сплавлением дифенилртути с двухлористым дифенил-
селеном 4, а также действием двухлористого дифенилселена на бен-
зол в присутствии хлористого алюминия 3.
1 С г о w е 1 1, В г a d t, J. Am. Chem. Soc. 55, 1500 A933).
2 Foster, Brown, гам же 50, 1182 A928).
3 Leicester, Bergstrom, там же 51, 3587 A929).
«Leicester, Bergstrom, там же 53, 4428 A931); Leicester,
там же 57, 1901 A935).
ДИФЕНИЛСУЛЬФИД
(Феналсулъфид)
2 С6Н6 + S2C12 -^ C6HSSC6H5 + S + 2 НС1
Предложили: В. Хартман, Л. Смит и Дж. Дикки.
Проверили: Р. Фьюзоп и С. Бабкок.
1. Получение
В 5-литровую трехгорлую круглодонную колбу, снабженную
механической мешалкой, капельной воронкой и холодильником,
который соединен с ловушкой для улавливания хлористого водо-
рода, помещают 858 г (980 мл, 11 мол.) сухого бензола (примеча-
ние 1) и 464 г C,48 мол.) хлористого алюминия. Реакционную
смесь охлаждают льдом до 10°, после чего при работающей мешалке
прибавляют 405,1 г C мол.) технической хлористой серы, раство-
ренной в 390 г D50 мл, 5 мол.) бензола. Прибавление продолжается
примерно в течение часа, причем температуру все время поддер-
живают около 10°. Реакция начинается немедленно, что заметно
по выделению хлористого водорода и желтого вязкого комплекса
с хлористым алюминием. После того, как прибавлена вся хло-
ристая сера, реакционную смесь вынимают из бани со льдом и пере-
мешивание продолжают еще в течение двух часов при комнатной
температуре. Затем массу нагревают до 30°, пока практически
не прекратится выделение хлористого водорода A час). Смесь
выливают на 1 кг измельченного льда и по окончании гидролиза
бензольный слой отделяют от водного слоя с помощью делитель-
ной воронки. Бензол отгоняют на водяной бане, оставшееся темно-
окрашенное масло охлаждают до 0° и фильтруют через воронку
Бюхнера, чтобы отделить выделившуюся серу. Фильтрат раство-
ряют в 500 мл технического метилового спирта, раствор охла-
ждают до 0° и при этой температуре перемешивают три часа, после
чего выделившуюся серу удаляют так же, как и в первый раз.
Спирт отгоняют на водяной бане и остаток перегоняют из 1-литро-
вой видоизмененной колбы Клайзена, отводная трубка которой
служит одновременно приемником и охлаждается водой. Сперва
переходит небольшое количество низкокипящей фракции, а затем
получают 470—490 г (примечание 2) жидкости, окрашенной в жел-
тый цвет.и кипящей при 155—170°/18 мм. Полученный продукт
в течение часа нагревают на водяной бане при помешивании с
70 г цинковой пыли и 200 г 40%-ного раствора едкого натра (при-
мечание 3). Дифенилсульфид отделяют от раствора щелочи, про-
мывают два раза водой порциями по 500 мл, сушат над безводным
сернокислым натрием и перегоняют. Выход бесцветного дифенил-
сульфидас т. кип. 162— 163°/'18ла< составляет 450—464 г (81—83%
теоретич.).
16 Сборник № 2
242
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
2. Примечания
1. Бензол можно высушить, отогнав *г него на водяной бане
около 15%, пока дестиллат не станет прозргчньш.
2. При перегонке остатка переходит фракция ПО—200°/18 мм.
После перекристаллизации этого продукта из метилового спирта
получают 8—10 г тиантрена с т. пл. 155^-156°.
3. Для получения бесцветного препарата необходимо сырой
продукт подвергнуть указанной выше обработке.
3. Другие методы получения
Дифенилсульфид лучше всего получается при действии на бен-
зол в присутствии хлористого алюминия, хлористой серы1, двух-
хлористой серы 2 или серы 3. Наряду с дифенилсульфидом всегда
образуются следы тиофенола, а также различные количества тиан-
трена.
1 Boeseken, Rec. trav. chim. 24, 209 A905); Boeseken, Water-
man, там же 29, 319A910); В о e s e k e n, К о n i n g, там же 30, 116A911);
Genvresse, Bull. soc. chim. C) 15, 409 A896); Hartman, Smith,
Dickey, Ind. Eng. Chem. 24, 1317 A932).
2 Boeseken, Rec. trav. chim. 24, 217 A905); Boeseken, Koning,
там же 30, 116 A911).
3 F r i e d e I, Crafts, Ann. chim. phys. F) 14, 437 A888); Boese-
ken, Rec. trav. chim. 24, 17, 219 A905); Dougherty, Hammond,
J. Am. Chem. Soc. 57, 117 A935).
ДИФЕНИЛТРИКЕТОН J
(Дифешлпропаптрион)
C6H6COCH,COC6H5 + 2 Br,
C6HaCOCBrXOC6H5 + 2 HBr
C0H5COCBr2COC0H5 -f- 2 CHjCOaNa + 2 H3O
, C6HaCOC (OHKCOC6H3 -f- 2 NaBr -f- 2 CH3CO3H
с6н6сос (онJ сос6н5 -нагревание^ с6н5сососос6н5 -h h3o
Предложили: Л. Багелов и Р. Хатлик.
Проверили: В. Хартман и Л. Смит.
1. Получение
А. Дибензоилдабромметаи. В 1-литровую трехгорлую колбу,
снабженную механической мешалкой, капельной воронкой и термо-
метром, помещают 56 г @,25 мол.) дибензоилметана («Синт. орг.
преп.», сб. 1, стр. 186) и 14 мл хлороформа. Колбу окружают льдом,
мешалку пускают в ход и через капельную воронку медленно при-
ливают'в течение 30 мин. раствор 28,5 мл (88 г, 0,55 мол.) сухого
ДЙФЕНИЛТРИКЕТОМ
243
брома (примечание 1) в 230 мл хлороформа. Температура смеси
во время бромирования не должна быть выше 15е; для удаления
выделяющегося бромистого водорода над поверхностью жидкости
продувают слабый ток воздуха. После того, как весь бром прибав-
лен, перемешивание продолжают еще 15 мин. Раствор переносят
в колбу для перегонки и растворитель полностью отгоняют в ва-
кууме при комнатной температуре (примечание 2). Слабоокрашен-
ный остаток перекристаллизовывают из 125 мл горячего 95%-ного
этилового спирта. Дибензоилдибромметан получается в виде белых
кристаллов с температурой плавления 94—95е. Выход 72,4 г G6%
теоретич.; примечание 3).
Б. Дифенилтрикетонгидрат. В 1-литровой круглодонной колбе
приготовляют раствор 34,3 г @,42 мол.) плавленного уксуснокислого
натрия в 142 мл горячей ледяной уксусной кислоты. К раствору
прибавляют 72,4 г @,19 мол.) дибензоилдибромметана и смесь
нагревают с обратным холодильником до тех пор, пока не закон-
чится выпадение бромистого натрия A,5—2 часа). Смесь охлаждают
до комнатной температуры и, не переставая взбалтывать, разба-
вляют 150—200 мл воды, для того чтобы растворить неорганиче-
скую соль и высадить трикетонгидрат. Выпавший в виде белой
творожистой массы продукт (примечание 4) отсасывают, хорошо
промывают водой и сушат в сушильном шкафу при 60°. Точка
плавления колеблется от 65 до 90° в зависимости от того, насколько
продукт дегидратировался во время сушки. Выход 41,5 г (86% тео-
ретич., считая на дибензоилдибромметан).
В. Дифенилтршетон. Полученные 41,5 г @,16 мол.) трикетон-
гидрата перегоняют в вакууме из клайзеновской|'колбы, нагре-
ваемой на песчаной бане. В качестве приемника берут вюрцевскую
колбу; перегонку ведут без холодильника. Для того чтобы прибор
не забивался вследствие кристаллизации дестиллата, приходится
обогревать шейку колбы, служащей приемником. Безводный три-
кетон перегоняется при 174—176°/2 мм в виде красноватого масла,
застывающего в светложелтую кристаллическую массу. Продукт
растворяют в 70 мл горячего лигроина (т. кип. 90—120°). По охла-
ждении выпадают светложелтые иглы с т. пл. 68—70°. Выход 35 г
(91% теоретич., считая на трикетонгидрат, или 59% — считая на
дибензоилметан; примечание 5).
2. Примечания
1. Бром высушивают, промывая его концентрированной серной
кислотой.
2. Если отгонку растворителя вести при атмосферном давлении,
уменьшается выход и продукт получается менее чистый.
3. Если продукт не перекристаллизовывать, получается худ-
ший выход трикетонгидрата.
244
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
4. Если гидрат частично выделится в виде масла, его можно
растворить в небольшом количестве ледяной уксусной кислоты
и снова осадить равным объемом воды. •
5. Трикетон гигроскопичен и должен сохраняться в вакуум-
эксикаторе или в запаянной ампуле.
3. Другие методы получения
Известны два общих метода для получения дифенилтрикетона:
обработка дибензоилметана бромом1^ окислами азота2. Де-Нефвиль
и Пехманн, предложившие первый метод, рекомендуют1 переводить
сначала дикетон в дибензоилбромметан, получить ацетат последнего,
затем перевести его в дибензоилбромкарбинолацетат, который и
расщепляется, давая трикетон. Этот способ был с успехом применен
недавно Колером и Эриксоном 3 для получения трикетона с хоро-
шими выходами, но описанный нами выше метод, который был
упомянут, но не описан Де-Нефвилем и Пехманном1, значительно
проще.
iDe Neufville, Pechmann, Rer. 23, 3375, 3379 A890).
2 Wi eland, В 1 о с h, там же 37, 1524, 1531 A904).
3 К о h 1 е г, Е г i с k s о n, J. Am. Chem. Soc. 53, 2308 A931).
НС1 • H2N<
У/
F,BN,<
4,4-ДИФТОРДИФЕНИЛ
-/ /NH2 • HC1 +2HONO •
)-N2Cl +4H2O
3H2O
H3BO3 + 4HF > HBF4
Sn2C1 + 2 HBF4 -
>H,BF4
нагревание
4N2BF4 + 2 HC1
2 BF3
Предложили: Дж. Шиман и В. Винкельмюллер.
Проверили: В. Хартман, Дж. Байере и Дж. Дикки.
1. Получение
В 5-литровую круглодонную колбу помещают смесь 280 г
A,5 мол.) продажного бензидина и 880 мл A0,2 мол.) концентри-
рованной соляной кислоты (уд. в. 1,18); колбу нагревают на водя-
4,4'-ДИФТОРДИФЕНИЛ
245
ной бане 1—2 часа, время от времени взбалтывая ее, чтобы
получить двойную хлористоводородную соль. Затем к колбе при-
способляют механическую мешалку и капельную воронку и содер-
жимое колбы при работающей мешалке охлаждают до —10° в бане
со льдом и солью. По достижении этой температуры бензидин
диазотируют (обе аминогруппы), прибавляя в течение двух часов
раствор 232 г C,2 мол.) 95%-ного нитрита натрия в 800 мл воды,
до слабой реакции на азотистую кислоту при пробе на иодокрах-
мальную бумажку через 20 минут. В течение этой реакции темпе-
ратуру поддерживают ниже —5°.
В то время, пока ведется диазотирование, растворяют 104 г
A,68 мол.) борной кислоты в 222 г F,66 мол.) 60%-ной фтористо-
водородной кислоты. Раствор приготовляют в 1-литровом стакане,
покрытом с внутренней стороны парафином, и охлаждают его в воде
со льдом. Борную кислоту добавляют медленно, небольшими пор-
циями и смесь размешивают свинцовым стержнем. Необходимо
поддерживать температуру ниже 20—25°, чтобы теплота растворе-
ния не расплавила парафин со стенок стакана (примечание 1).
Охлажденный до 0° раствор борофтористоводородной кислоты
довольно быстро, при работающей мешалке, приливают к полу-
ченному тетразо-раствору, причем температуру поддерживают
ниже 10°. При этом образуется густая паста борофторида 4,4'-ди-
фенилен-#«с-диазония. Массу перемешивают при 10° еще 20—
30 минут. Затем кристаллы отфильтровывают через воронку Бюх-
нера диаметром 19 см и на воронке последовательно промывают
200 мл холодной воды, 200 мл холодного технического метилового
спирта и 200 мл продажного эфира. В промежутках между этими
промывками массу как можно лучше отжимают. После сушки в
вакуум-эксикаторе над серной кислотой (уд. веса 1,84) выход
сухого продукта составляет 393—400 г F8—69% теоретич.). Про-
дукт разлагается при 135—137°.
Разложение борофторида тетразония можно вести в 1-литро-
вой колбе Вюрца с широкой отводной трубкой. Непосредствен-
но к боковому отводу вюрцевской колбы присоединяют другую
колбу Вюрца на 500 мл, которую охлаждают проточной водой.
К боковому отводу приемника присоединяют каучуковую трубку,
которая заканчивается над поверхностью 2 л воды, налитой в
5-литровую колбу (примечание 2). Продукт, подлежащий разло-
жению (примечание 3), загружают в колбу и нагревают сверху
бунзеновской горелкой. Когда начнется выделение белых паров,
горелку удаляют и разложению предоставляют итти самопроиз-
вольно. По мере необходимости нагревание возобновляют. Чтобы
обеспечить полноту разложения, к концу реакции содержимое
колбы энергично нагревают. Небольшое количество 4,4'-дифтор-
дифенила собирается в приемнике, но большая часть его остается
в исходной колбе, откуда его извлекают, подвергая черную массу
246
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
ДИХЛОРУКСУСНАЯ КИСЛОТА
247
перегонке с паром. В результате вторичной перегонки с паром
получают чистый белый продукт, который после сушки в сушиль-
ном шкафу при 60—70° плавится при 88—89°. Если разложению
подвергаются 153 г борофторида тетразония, выход 4,4'-дифторди-
фенила составляет 61—62 г (80—81,5% теоретич., считая на боро-
фторид тетразония; 54—56%— считая на бензидин).
2. Примечания
1. Весьма удобно приготовлять раствор в небольшом свинцовом
горшке. Если сделать из свинца мешалку обычного типа, можно
применять механическое перемешивание. Мешалку следует про-
пустить через отверстие в свинцовой крышке достаточных размеров,
чтобы избежать разбрызгивания «фтористоводородной кислоты.
Фтористоводородная кислота вызывает чрезвычайно болезнен-
ные ожога. Во время работы части тела, подвергаемые ее действию,
должны быть защищены (ср. примечание 3 на стр. 536). Вместо того,
чтобы получать борофтористоводородную кислоту в лаборатории,
можно взять 355,5 г продажной 40%-ной.
2. Удобный прибор можно сконструировать, если соединить
согнутой трубкой большого диаметра B см) одну 1-литровую кругло-
донную колбу с другой такой же колбой, наполненной 500 мл воды.
Вторая колба снабжена выходной трубкой, через которую не раство-
рившиеся в воде газы отводятся в вытяжной шкаф.
3. Весьма важно, чтобы борофторид тетразония был хорошо
высушен. Если он высушен недостаточно, разложение будет проте-
кать очень бурно. При этом одновременно образуется продукт с
более высокой температурой плавления A60°), а также некоторое'
количество смолы. Хотя эти продукты и не летучи с водяным паром,
однако они значительно снижают выход 4,4'-дифтордифенила.
3. Другие методы получения
4,4'-Дифтордифенил может быть получен из 4,4'-дифенил-
бис-диазоний-пиперидида (полученного диазотированием бензидина
и сочетанием его с пиперидином) и концентрированной фтористо-
водородной кислоты х; действием натрия на л-фторбромбензол в
эфирной среде 2; из бензидина путем диаяотирования обеих амино-
групп и разложения полученной соли дифенил-бис-диазония кон-
центрированной фтористоводородной кислотой 3; описанным выше
методом, но в присутствии хлорного железа 4; и длительным кон-
тактом паров фторбензола с раскаленной докрасна проволокой 5.
Описанный здесь метод является улучшением метода Бальца и
Шимана 6 и в лабораторных условиях наиболее удобен.
1 W а 1 1 а с h, Ann. 235, 271 A886).
2 W a 1 1 а с h, H e u s I e r, там же 243, 244 A888).
3 Valentine г, Schwarz; герм. пат. 96 153 [Frdl. 5, 910 A897—
1900)|.
1 Valentine r, Schwarz; герм. пат. 186 005 [Frdl. 8, 1237 A905—
1907)].
5Meyer, Hofmann, Monatsh. 38, 149 A917).
6 В а 1 г, Schiemann, Ber. 60, 1189 A927).
ДИХЛОРУКСУСНАЯ КИСЛОТА
CNaCNI
2 СС13СН (ОНJ+2 СаСО3- (СНС1.ХО2J Са+2 СО2+2Н2О+СаС12
(СНС12СО,J Са+2 НС1 CaCU + 2 СНС12СО2Н
Предложили: А. Коп, Дж. Клйрк и Р. Коннор.
Проверили: Р. Шрайнер и Н. Мун.
1. Получение
В 3-литровую круглодонную колбу, снабженную обратным холо-
дильником и термометром, помещают раствор 250 г A,5 мол.) хло-
ральгидрата (фармакопейного) в 450 мл теплой воды E0—60°)
(примечание 1). Затем временно отсоединяют холодильник и при-
бавляют 152,5 г A,52 мол.) осажденного углекислого кальция,
после чего приливают 2 мл амилового спирта (примечание 2) и
раствор 10 г технического цианистого натрия в 25 мл воды. Не-
смотря на то, что реакция проходит с выделением тепла, смесь
нагревают небольшим пламенем горелки, так чтобы через 10 мин.
температура достигла 75°, после чего нагревание прекращают.
В течение 5—10 мин. температура продолжает повышаться и дости-
гает 80—85°, после чего она начинает падать. Как только начнется
понижение температуры, раствор нагревают до кипения и кипятят
20 мин. Затем смесь охлаждают до 0—5° в бане со льдом, подки-
сляют 215 мл концентрированной соляной кислоты (уд. в. 1,18) и
извлекают пять раз эфиром порциями по 100 мл (примечание 3).
Соединенные эфирные вытяжки сушат 20 г безводного сернокислого
натрия, эфир отгоняют на водяной бане, а остаток перегоняют
в вакууме из колбы Клайзена с дефлегматором (примечание 4).
Выход дихлоруксусной кислоты ст. кип. 99—104°/23 мм составляет
172—180 г (88—92% теоретич.; примечание 5).
2. Примечания
1. Количество выделяющегося цианистого водорода незначи-
тельно, поэтому реакцию можно вести в вытяжном шкафу без особого
приспособления для отвода газа. Применение механического пере-
мешивания не улучшает выход.
2. Амиловый спирт добавляют для того, чтобы уменьшить вспе-
нивание.
3. Для того, чтобы уничтожить эмульсию, часто образующуюся
при извлечении эфиром, раствор можно профильтровать через
складчатый фильтр или с отсасыоанием.
248
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
диэтилцинк
249
4. При перегонке при атмосферном давлении продукт разла-
гается.
5. Этот синтез был проведен и с удвоенными против указанных
в прописи количествами исходных веществ с одинаково хорошими
результатами.
3. Другие методы получения
Дихлоруксусная кислота была получена хлорированием уксус-
ной г или монохлоруксуснойг кислот, гидролизом пентахлор-
этана 3, а также из трихлоруксусной кислоты электролитическим
восстановлением 4 или действием на нее меди 5, и действием циа-
нистых солей щелочных металлов на хлоральгидрат в. Описанный
здесь метод в основном заимствован у Делепина 7.
1 М ii I I e r, Ann. 133, 159 A865); Dov/ Chemical Company; ам. пат.
1 921 717 [С. А. 27, 5084 A933)].
2 М a u m e n ё, Compt. rend. 59, 84 A864).
3 А 1 a i s, Froges, Cam argue, франц. пат. 773 623 [С. А. 29,
1437 A935)].
4 Brand, герм. пат. 246 661 [С. А. 6, 2496 A912)].
5 Doughty, Black, J. Am. Chem. Soc. 47, 1091 A925); Doughty,
D e r g e, там же 53, 1594 A931).
6 W a 11 а с h, Ann. 173, 288 A874); P u с h e r, J. Am. Chem. Soc. 42,
2251 A920); Chattaway, Irving, J. Chem. Soc. 1929, 1038.
7 Delepine, Bull. soc. chim. D) 45, 827 A929).
Р-ДИЭТИЛАМИНОЭТИЛОВЫЙ СПИРТ
B-Диэтиламшоэтанол )
(C2H5J NH + C1CHXH2OH + NaOH
" > (С2Н5J NCH2CH2OH + NaCl + H2O-
Предложил: В. Хартман.
Проверили: Б. Каротерс и В. Мак-Ювен.
1. Получение
В 2-литровую колбу, снабженную обратным холодильником
и капельной воронкой, помещают 380 г E,2 мол.) диэтиламина
(т. кип. 52—60°) и нагревают его на водяной бане до кипения, после
чего в течение часа приливают из капельной воронки 320 г D мол.)
этиленхлоргидрина. Реакционную смесь нагревают еще 8 час,
затем дают ей охладиться и довольно быстро при непрерывном
взбалтывании прибавляют раствор 230 г едкого натра в 350 мл
воды. При этом образуются два слоя и выпадает хлористый натрий.
Для растворения последнего добавляют 400 мл воды, затем 500 мл
бензола, пускают в ход механическую мешалку на 5 мин., после
чего бензольный слой отделяют, а водный — экстрагируют три раза
бензолом, порциями по 500 мл. Соединенные бензольные вытяжки
сушат 100 г твердого поташа при механическом перемешивании до
тех пор, пока раствор не станет прозрачным. Раствор подвергают
перегонке из 3-литровой колбы, снабженной колонкой длиною
50 см (набитой стеклом или карборундом) и термометром, опущен-
ным в жидкость. Перегонку ведут до тех пор, пока температура
жидкости не достигнет 100°, а температура паров в верхней части
колонки 85°. Остаток переливают в 1-литровую колбу Клайзена,
которая снабжена дефлегматором длиною 30 см, и перегоняют его
в вакууме. Отбирают следующие фракции: до 45°/20 мм, 45—
64°/18 мм и 64—65718 мм. "Вес последней фракции около 290 г.
Первые две фракции подвергают повторной перегонке, причем
получают дополнительное количество Р-диэтиламиноэтилового
спирта (примечание 1).Общий выход 320—330 г F8—70%теоретич.).
2. Примечания
1. Физические свойства р-диэтиламиноэтилового спирта по-
дробно описали Хедли, Коллетт и Лаззел *.
3. Другие методы получения
р-Диэтиламиноэтиловый спирт был получен восстановлением
диэтиламиноуксусного эфира натрием в спиртовом растворе2, дей-
ствием этиленхлоргидрина на диэтиламин 3, действием окиси эти-
лена на диэтиламин 1 и действием этилсульфата на этаноламин 4.
•Home, Schriner, J. Am. Chem. Soc. 54, 2928 A932); Headlee,
С о 1 1 e t t, L a z z e 1 1, там же 55, 1066 A933).
2 Q a u 1 t, Compt. rend. 145, 126 A907); Bull. soc. chim. D) 3, 369 A908).
3Ladenburg, Ber. 14, 1878 A881); Soderman, Johnson,
J. Am. Chem. Soc. 47, 1394 A925).
4 Carbide and Carbon Chemicals Corporation; франц. пат. 792 046 [С. А.
30, 4176 A936)].
ДИЭТИЛЦИНК
2 C,H3I + 2 C2H3Br + 4 Zn > 2 Zn (C2H5J + Znl2 + ZnBr2
Предложил: К- Ноллер.
Проверили: Г. Гилъман, Г. Саусгэт и К- Мелыпреттер.
1. Получение
В круглодонную колбу емкостью в 500 мл, снабженную обрат-
ным холодильником (примечание 1) и прочной мешалкой (приме-
чание 2), помещают 1.30 г (около 2 гр.-ат.) цинк-медной пары
250
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
(примечание 3). Затем прибавляют смесь 78 г @,5 мол.) йодистого
этила и 54,4 г @,5 мол.) бромистого этила (примечание 4). Мешалку
пускают в ход и смесь нагревают с обратным холодильником.
Реакция начинается через полчаса после начала нагревания (при-
мечание 5), что видно по усиливающемуся кипению. В этот момент
горелку отставляют.% Если реакция становится слишком бурной,
колбу охлаждают ледяной водой, но только так, чтобы реакция
лишь несколько ослабилась, но не прекратилась бы совсем (приме-
чание 6). Реакция обычно заканчивается через полчаса после того,
как отставлена горелка. Содержимому колбы дают охладиться до
комнатной температуры (примечание 7), удаляют обратный холо-
дильник, соединяют колбу с дефлегматором, холодильником, и
приемником и продукт перегоняют в вакууме (примечание 8).
Перегонку ведут прямо из реакционной колбы; приемником слу-
жит круглодонная колба емкостью 200 мл, которую погружают
в смесь льда с солью (примечание 9). Под конец перегонки через
прибор пропускают сухую углеки^шоту или очищенный азот (при-
мечание 10). Выход сырого продукта, достаточно чистого для боль-
шинства целей, 53—55 г (86—89% тсорстич.).
Дальнейшую очистку производят в следующем приборе: елоч-
ный дефлегматор длиною 30 см, соединяют с холодильником и
приемником, имеющим отводную трубку. Отводную трубку прием-
ника покрывают пробиркой, к которой припаяна небольшая тру-
бочка для впуска углекислоты; открытый конец пробирки неплотно
затыкают ватой (примечание 11). Прибор наполняют углекислотой
и круглодонную колбу с однократно перегнанным диэтилцинком
соединяют с дефлегматором. Диэтилцинк перегоняют затем при
атмосферном давлении. Выход продукта с температурой кипения
115—120° составляет 50—52 г (81—84% теоретич.; примечания 5
и 12).
2. Примечания
1. Для начала реакции очень важно, чтобы прибор был вполне
сух и защищен от атмосферной влаги. Для этой цели на конец
холодильника надевают хлоркальциевую трубку или соединяют
его с ловушкой, чтобы предохранить от влаги воздуха 1.
2. Для того чтобы весь цинк хорошо перемешивался, мешалка
должна вращаться медленно и подходить почти вплотную к стенкам
и дну колбы.
3. Цинк-медная пара может быть приготовлена по одному из
следующих способов:
а) Смесь из 120 г цинковой пыли и 10 г растертой в порошок
окиси меди осторожно нагревают на голом пламени горелки в
круглодонной колбе емкостью 200 мл. В колбу пропускают ток
водорода и продолжают нагревание при помешивании или встря-
диэтилцинк
251
хивании до тех пор, пока не будет восстановлена вся окись меди
и смесь не примет однородной серой окраски. Во время нагревания
температура нагревания должна быть немного ниже температуры
плавления цинка.
б) Сплав цинка и меди, содержащий 5—8% меди, приготовляют
сплавлением цинка с чистыми медными стружками. Полученному
сплаву придают форму палочек, которые для работы превращают
в мелкие опилки.
Если требуется приготовить лишь небольшие количества цинк-
алкилов, лучше применять метод (а). При получении больших
количеств считается более удобным работать со стружками сплава
цинка с медью (б). Цинк-медную пару, приготовленную по методу
(а), нужно или сейчас же пускать в реакцию, или сохранять в
атмосфере инертного газа, так как при доступе влажного воздуха
она быстро портится.
Метод, предложенный Нойесом2С, не дает такого активного
сплава, как вышеописанные способы. Для получения диэтилцинка
из чистых иодидов препарат Нойеса дает хорошие результаты, но
если для реакции берут смесь йодистых и бромистых алкилов, то
выходы ниже.
4. Употребление смеси йодистого и бромистого алкилов не
только дешевле, но и удобнее, так как реакция проходит менее
бурно, чем с одним йодистым алкилом. Методика для полу-
чения бромистого этила приведена в «Синт. орг. преп.», сб. 1,
стр. 113 и 117.
5. По наблюдению автора реакция начинается обычно через
20—40 мин. после начала кипения. При проверке, однако,
оказалось, что для начала реакции требуется 1—1,25 часа. Если
же не принять всех необходимых мер для удаления влаги, реакция
может не начаться в продолжение нескольких часов. В таких слу-
чаях, после того как смесь была нагрета некоторое время при
перемешивании, ее можно оставить стоять без перемешивания при
комнатной температуре. Реакция при таких условиях начиналась
затем обычно внезапно (иногда через 5 час.) и не требовала особого
наблюдения, так как кипение в таких случаях происходит не сильно
и применять охлаждение не приходится. Когда реакция начи-
нается не скоро, выход диэтилцинка несколько ниже.
6. Если слишком охладить смесь, реакция может прекратиться
и вызвать ее вновь затруднительно. В таких случаях выходы полу-
чаются значительно ниже.
7. При охлаждении реакционная масса застывает (вероятно,
благодаря перегруппировке RZnX) и может быть без опасения
приведена на короткое время в соприкосновение с воздухом. Твер-
дый материал менее способен к реакциям, чем диэтилцинк.
8. Давление должно быть ниже 30 мм ртутного столба, так как
иначе происходит разложение.
252
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
ДУРОЛ И ИЗОДУРОЛ
253
9. Употребляют или специальный алонж с боковым отводом для
соединения с насосом, или в пробку, соединяющую приемник
с холодильником, вставляют трубку, согнутую под прямым
углом.
10. Если перегонку ведут в вакууме, прибор можно не напол-
нять инертным газом. Qp всех остальных случаях для защиты от
доступа воздуха прибор наполняется углекислотой или азотбм.
При проверке была проведена перегонка на масляной бане
(около 200°) при 8 мм давления. Перегонка продолжалась около
1 часа. Колбу можно нагревать также непосредственно бунзенов-
ской горелкой.
11. Описанная система может быть заменена ртутным затвором.
Для этого соединяют верхнюю часть приемника со стеклянной
дважды согнутой под прямым углом трубкой, другой конец кото-
рой опущен в сосуд с ртутью. Трубка погружается в ртуть лишь
очень неглубоко. Если работать с ртутным затвором, после окон-
чания перегонки необходимо отсоединить приемник от прибора,
иначе при охлаждении прибора ртут! будет перетянута в прием-
ник.
12. При работе по описанному методу были получены также
высшие цинкалкилы со следующими выходами и температурами кипе-
ния: ди-н.-пропилцинк: выход 85—86%, т. кип. 39—40°/9 мм; ди-н.-
бутилцинк: выход 78—79%, т. кип. 81—82°/9 мм; ди-изоамилцинк:
выход 50—55%, т. кип. 100—103°/12 мм.
Высшие цинкалкилы следует перегонять всегда в вакууме.
3. Другие методы получения
Диэтилцинк был получен из цинка и диэтилртути 3 и из броми-
стого этила и цинк-медной пары в присутствии специального катали-
затора i. Обычно диэтилцинк получается действием йодистого
этила на цинк, обработанный предварительно различными спосо-
бами 5. Самым удобным из этих препаратов цинка является
пара цинк—медь 2. Приведенная выше методика описана в литера-
туре 6. Галоидные соединения цинкарилов и цинкдиалкилы и диа-
рилы могут быть получены действием реактива Гриньяра на без-
водный хлористый цинк в эфирном растворе 7. Улучшенный прибор
для получения, очистки и применения диэтилцинка опубликован
в литературе 8.
1 Oilman, Hewlett, Rec. trav. chim. 48, 1124 A929).
2 (a) G 1 a d s t о n e, Tribe, J. Chem. Soc. 26, 445, 678, 961 A873).
(b) L а с h m a n, Am. Chem. J. 19, 410 A897); 24, 31 A900).
(c) N о у e s, Organic Chemistry for the Laboratory, 3-rd Ed., p. 61, 1916
(d) R e n s h a w, Q r e e n 1 a w, J. Am, Chem. Soc. 42, 1472 A920)
3Frankland, Duppa, J. Chem. Soc. 17, 31 A864).
*J°b, Reich, Bull. soc. chim. D) 33, 1424 A923).
6 F r a n k 1 a n d, Ann. 85, 360 A853).
6 N о 1 1 e r, J. Am. Chem. Soc. 51, 594 A929).
7 Houben-Weyl, «Methoden der organischen Chemie», 2-е изд., 1924,
т. 4, стр. 754, 901; Blaise, Bull. soc. chim. D) 9, I—XXVI A911); G i 1-
m a n, Brown, j. Am. Chem. Soc. 52, 4482 (примечание 7) A930).
8McCleary, Degering, Proc. Indiana Acad. Sci. 43, 127 A934);
[C. A. 28, 7245 A934)].
ДУРОЛ И ИЗОДУРОЛ
А. ДУРОЛ A, 2, 4, 5-ТЕТРАМЕТИЛБЕНЗОЛ)
Побочные продукты — пента и гексаметилбензолы
СНЯ
С6Н4 (СН3), + 2 СН3С1
(А1С18)
сн„
+ 2НС1
сн.
Предложил: Л. Смит.
Проверили: Ф. Уптмор и Т. Холлингсхед.
1. Получение
Колбу емкостью 5 л, установленную на паровой бане, снабжают
широкой (примечание 1) газоприводиой трубкой, доходящей до
дна колбы, и обратным холодильником, верхний конец которого
закрыт пробкой. В последнюю вставлена изогнутая стеклянная
трубка, оканчивающаяся на дне толстостенного стеклянного ци-
линдра, в который на высоту около 10 см налита ртуть. Так как
реакция ведется при несколько повышенном давлении, все соеди-
нения прибора должны быть вполне герметичными. Пробки сле-
дует хорошо пригнать и укрепить проволокой, соединительные
каучуки должны быть также укреплены проволокой. В колбу
вносят 3180 г C100 мл, 30 мол.) ксилола (примечание 2) и 1000 г
безводного хлористого алюминия (примечание 2). Вводную трубку
колбы соединяют с баллоном с хлористым метилом или с прибором
для его получения (примечание 3); баню нагревают до кипения и в
смесь пропускают довольно быстрый ток хлористого метила (при-
мечание 4). Сначала происходит быстрое поглощение хлористого
метила, и скорость газа следует регулировать таким образом, чтобы
ртуть не засасывалась в отводную трубку. Образующийся хлори-
стый водород удобно поглощать в ловушке. После того, как реак-
ция замедляется, давление в приборе начинает постепенно увели-
чиваться, и вскоре хлористый водород и хлористый метил начинают
проходить через ртуть. Тогда скорость впускаемого газа следует
уменьшить, чтобы предупредить бесполезную потерю хлористого
254
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
ДУРОЛ И ИЗОДУ РОЛ
255
метила. Всего на проведение реакции требуется около 100 час.
(примечание 5).
Когда реакция закончена, баню охлаждают, колбу отъединяют
от баллона и холодильника, не плотно закрывают пробкой и оста-
вляют стоять на ночь. Црсле этого реакционную смесь медленно
при помешивании выливают на 5 кг колотого льда. Зеленоватый
маслянистый слой, всплывающий над водным слое», отделяют,
сушат хлористым кальцием, фильтруют и дважды фракционируют
из круглодонной колбы, снабженной эффективным дефлегматором
и воздушным холодильником (примечание 6).
Собирают следующие фракции:
Фрак-
ция
I и II
Ш
IV
V
Темп., °С
до 150
150—180
180—205
Выше 205
Фракция содержит
Бензол, ксилол
Триметилбензолы
Тстраметилбензолы
Пентаметилбензол
(главным образом)
Выход, г
Немного
570
2 075
815
В таблице приведены средние цифры. В различных опытах
выходы колеблются в пределах 10—20% в зависимости от неболь-
ших изменений в деталях проведения реакции и чистоты применяе-
мых материалов, особенно хлористого алюминия. Чем этот послед-
ний активнее, тем больше получается высших фракций.
Фракция тетраметилбензола (IV) содержит много дурола, кото-
рый можно выморозить и отфильтровать, так как он обладает
сравнительно высокой температурой плавления (80°). Для этого
фракцию IV охлаждают смесью льда и хлористого кальция, отса-
сывают с хорошим разрежением выкристаллизовавшийся дурол
через охлаждаемую воронку (примечание 7) и осадок тщательно
отжимают. Когда отсасывание закончено и через охлаждаемую
воронку более не капает жидкости, воронке дают дойти до комнатной
температуры (примечание 7) и продолжают отсасывать до тех пор,
пока полностью не прекратится стекание жидкости в колбу, после
чего твердый продукт собирают. Выход 540—610 г.
Фракцию III можно метилировать до тетрамстплбензола нагре-
ванием на кипящей водяной бане со 100 г безводного хлористого
алюминия и пропусканием в смесь 225 г хлористого метила. Из
фильтратов от дурола, содержащих его изомеры, также можно
получить значительное количество дурола, если нагревать их на
кипящей водяной бане с 50 г хлористого алюминия. Продукт реак-
ции обрабатывают обычным путем, т. е выливают на двойное
весовое количество колотого льда, отделяют от водного слоя,
фракционируют дважды и вымораживают дурол, как описано.
После однократного метилирования триметилбензольнои фракции и
однократной обработки фильтратов дурола общий выход сырого ду-
рола достигает в среднем 1000—1400г B5—35% теоретич., считая
на 30 мол. ксилола).
Для того чтобы очистить дурол, помещают 200 г сырого веще-
ства в 1-литровую круглодонную колбу, снабженную обратным
холодильником, и расплавляют его на водяной бане при 95°; затем
прибавляют через верхний конец холодильника 200 мл теплого
E0q) 95%-ного этилового спирта и осторожно нагревают смесь,
пока она не станет однородной. Раствор фильтруют через воронку
с обогревом, оставляют его плотно закрытым (примечание 8) стоять
в теплом месте C5°) на ночь, потом охлаждают до 0°, и выпавшие
кристаллы отсасывают. Продукт (около 169 г) плавится при 74—78°.
Вторая перекристаллизация дает 149 г дурола с температурой пла-
вления 77—79 . Третья кристаллизация дает 140 г с точкой плавления
79—80°. Из маточных растворов спирт отгоняют с дефлегматором
и остаток присоединяют к фильтратам от вымороженного дурола.
Пентаметилбензол. Фракцию V, а также смесь, метилированную
вышететраметилбензолов, можно переработать в пентаметилбензол.
Если из ксилола хотят получить пентаметилбензол непосредственно,
то хлористого метила нужно пропустить на один моль больше, и
поэтому метилирование следует вести на 10 час. дольше, чем в слу-
чае получения дурола, т. е. всего ПО час. В остальном методика
точно такая же. Реакционную массу выливают на лед и выделен-
ную смесь углеводородов фракционируют обычным путем. Фракция
выше 205° (фракция V) в результате разгонки дает следующие три
фракции:
Фракция
VI
VII
VIII
Теми., ГС
205-215
215—235
Остаток выше 235
Фракция содержит
Тетра- и пентаметилбензолы
Главным образом пентаметилбензол
Гексаметилбензол и смолы
Фракция VII представляет собой довольно чистый пентаметил-
бензол. Если его перекристаллизовать один раз из 95%-ного спирта
или из смеси равных объемов спирта и бензола, то получается
белоснежный продукт, плавящийся, однако, в широких преде-
лах и потому не пригодный для большинства целей. Чтобы полу-
чить вполне чистый пентаметилбензол, фракцию VII фракциони-
руют в вакууме, собирая погон 123—133°/22 мм (большая часть
переходит при 127—129°). Затем застывающий при охлаждении
дестиллат перекристаллизовывают так, как это описано ниже, и
получают продукт с температурой плавления 52° (исправл. 53°).
256
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
ДУРОЛ И И30ДУР0Л
257
600 г неочищенного пентаметилбензола нагревают до 100° и
медленно при перемешивании приливают к 1000 мл 95%-ного эти-
лового спирта, нагретого до 70° в 2-литровом стакане.
Полученный раствор оставляют стоять для кристаллизации на
ночь при 30°. Выпавшие кристаллы тщательно отсасывают и сушат
в течение ночи при комнатной температуре на пористой пластинке.
Выход продукта около 250 г (примечание 9).
Гексаметилбензол. Фракцию VIII перегоняют порциями по
250 мл из колбы Клайзена при 20 мм давления и собирают следую-
щие погоны:
Фракция
IX
X
XI
XII
Темп., СС
Фракция содержит
§0—110
110—135
135—170
Остаток выше
170
Главным образом тетра- и пентаметил-
бензолы
пентаметилбензол
Гексаметилбензол
Гексаметилбензол и смолы
Эту фракционированную перегонку можно производить в обык-
новенной клайзеновской колбе, но тогда трудно поддерживать
нужное давление, так как углеводороды легко растворяют каучук
пробок. Чтобы предупредить последние от разрушительного дей-
ствия паров полиметилбензолов, берут клайзеновскую колбу с
очень длинными горлами и широкой боковой трубкой. Перегонку
следует вести настолько быстро, чтобы дестиллат не затвердевал
в дефлегматоре и отводной трубке колбы. Кроме того, при
продолжительном нагревании гексаметилбензол отчасти осмо-
ляется.
Для получения больших количеств гексаметилбензола быстро
метилируют пентаметилбензол или фильтраты от дурола. Смесь
378 г пентаметилбензола и 200 г безводного хлористого алюминия
нагревают на масляной бане при 190—200° и пропускают через
нее быстрый ток сухого хлористого метила в продолжение.3—4 час,
пользуясь тем же прибором, что и при получении дурола, после
чего смесь оставляют стоять на ночь при комнатной температуре.
Затем прибавляют 1 л горячего ксилола, чтобы растворить застывшее
вещество, и выливают полученный раствор на 3 кг колотого льда.
Образующийся маслянистый слон отделяют и отгоняют ксилол и
другие низкокипящие вещества в вакууме (примечание 10). Фрак-
ции разделяют, как было указано выше. После двух дробных пере-
гонок и двух перекристаллизации (примечание 11) получают 98—
121 г белых кристаллов, плавящихся при 157—161° (примеча-
ние 12).
2. Примечания
1. В качестве газоприводной трубки удобно пользоваться вну-
тренней трубкой холодильника. Ее следует поместить так, чтобы
широкий конец был внутри большой колбы.
2. Ксилол должен быть бесцветным, хорошего качества и кипеть
при 135—140°. Для удаления воды его перегоняют и отбрасывают
первые 10% дестиллата. Следует применять хлористый алюминий
только высшего качества, так как с плохими сортами метилирование
идет очень плохо. Хлористый алюминий нужно разбить на малень-
кие кусочки, но не следует толочь его в порошок.
3. Хлористый метил получают в 5-литровой колбе, установлен-
ной на песчаной бане и соединенной с обратным холодильником,
в верхнее отверстие которого вставляется пробка с газоотводной
трубкой. По выходе из холодильника газ пропускают последова-
тельно через семь промывных склянок, из которых первая, четвер-
тая и седьмая — пустые (предохранительные), вторая и третья
(по направлению движения газа) наполнены водой, и пятая и ше-
стая — серной кислотой. Чтобы получить приблизительно 45 мол.
(теоретич.) хлористого метила, берут смесь 200 г воды с 2200 г
A200 мл) концентрированной серной кислоты, вливают ее в колбу
и прибавляют 1400 г A760 мл) метилового спирта, при охлаждении,
с такой скоростью, чтобы температура не поднималась выше 70°.
Затем прибавляют 2400 г хлористого натрия, плотно соединяют все
части прибора и нагревают колбу на песчаной бане настолько
сильно, чтобы газ выделялся с большой скоростью. Путем- опыта
было установлено, что при применении продажных материалов выход
хлористого метила составляет приблизительно 55—65% теоретиче-
ского количества, так что следует брать приблизительно удвоен-
ные количества по сравнению с рассчитанными. Следовательно,
для того, чтобы превратить 30 мол. ксилола в тетраметилбензол,
прибор должен быть загружен три раза. Если имеется баллон с
хлористым метилом, то на то же самое количество ксилола берут
65—70 мол. хлористого метила. Чтобы иметь возможность контро-
лировать количество взятого в реакцию хлористого метила, бал-
лон взвешивают перед началом опыта и реакцию заканчивают после
того, как вес баллона уменьшится на соответствующую вели-
чину.
4. Опыты показали, что быстрый ток хлористого метила вызы-
вает достаточное перемешивание.
5. Норму времени в 100 час. можно сократить, если взять больше
хлористого алюминия. Однако в этом случае продукт реакции полу-
чается очень вязким, что, особенно при работе с большими коли-
чествами, сильно затрудняет дальнейшую переработку.
6. Для того, чтобы достичь хорошего разделения, абсолютно
необходимо произвести две систематические фракционировки (не
17 Сборник № 2
258
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
повторные перегонки) с эффективной колонкой. Чем колонка
эффективнее, тем лучше бывают результаты.
7. Вещество следует отсасывать на большой бюхнеровской во-
ронке, которая охлаждается снаружи смесью льда и хлористого
кальция до тех пор, пока в приемник капает жидкость. По литера-
турным данным, первый фильтрат, полученный при отсасывании
с охлаждением, состоит главным образом из изодурола A, 2, 3, 5-),
а фильтрат, полученный при отсасывании при комнатной темпера-
туре, состоит из псевдодурола или пренитола A, 2, 3, 4-) с т. пл. —4°.
8. Дурол очень летуч, а потому его не следует оставлять на
воздухе дольше, чем это вызывается необходимостью. С парами
спирт^ он также очень летуч, и поэтому отогнанный от маточных
растворов спирт всегда содержит некоторое количество дурола.
Чтобы не терять вещество, отгоны применяют для последующих
перекристаллизации, а остаток после отгонки нагревают с хлори-
стым алюминием для изомеризации изодурола и пренитола в дурол.
9. Сырой пентаметилбензол загрязнен главным образом гекса-
метилбензолом, который не может быть удален при помощи кри-
сталлизации. Поэтому», перекристаллизация очень мало повышает
температуру плавления пентаметилбензола. Единственный метод
для отделения гексаметилбензола — это фракционированная пере-
гонка.
10. При продолжительном нагревании гексаметилбензола до
высокой температуры он разлагается. Поэтому достигают лучших
результатов, если производят перегонку в вакууме.
11. Небольшие количества примесей сильно понижают темпе-
ратуру плавления гексаметилбензола. Для того, чтобы полечить
резко плавящийся продукт, следует произвести несколько пере-
кристаллизации фракции, кипящей в указанных пределах.
12. Небольшое количество B5 г или меньше) более или менее
чистого гексаметилбензола удобнее всего перекристаллизовывать
из этилового спирта. В 600 мл кипящего спирта растворяется
около 25 г вещества, и выпадает по охлаждении 20 г. Эфир и бензол
растворяют гексаметилбензол значительно легче, и поэтому боль-
шие количества вещества удобнее перекристаллизовывать из одного
из этих растворителей или из смеси одного из них со спиртом.
125 г тщательно отфракционированного гексаметилбензола распла-
вляют и медленно выливают при помешивании в 1500 мл 95%-ного
спирта. Небольшую нерастворившуюся часть переводят в раствор
прибавлением около 300 мл горячего бензола, при нагревании на
водяной бане и непрерывном помешивании (до полного растворения).
Раствор оставляют стоять на ночь при температуре около 25°.
Выпавшие кристаллы отсасывают и промывают 95%-ным спиртом
в количестве, достаточном, чтобы хорошо их смочить (приблизи-
тельно 25 мл). Высушенный продукт весит около 112 г и плавится
при 155—159°.
ДУРОЛ И ИЗОДУРОЛ
3. Другие методы получения
Дурол, пентаметилбензол и гексаметилбензол обычно полу-
чаются из бензола или из его метилпроизводных по методу Фриделя-
Крафтса !. Дурол был получен из бромпроизводных метилированных
бензолов по методу Фиттига 2. Его получили также с выходом 20%
пропусканием паров метилового спирта и ацетона через нагретую
окись алюминия 3, и с выходом 45% хлорметилированием ксилола
и восстановлением хлорметилированного продукта 4. Гексаметил-
бензол был получен действием хлористого цинка на метиловый
спирт 5 или ацетон 6. Метод, приведенный выше, описан в лите-
ратуре 7.
Б. ИЗОДУРОЛ A, 2, 3, 5-ТЕТРАМЕТИЛБЕНЗОЛ)
Br MgBr
СН,/
сн.
сн3
MgBr
/ч
СН,
сн3
сн3
Mg
(Эфир) СН31
сня
f 2 (СН3J SO4
сня
СН3Вг + (CH3OSO2OJ Mg
Предложил: Л. Смит.
Проверили: Р, Адаме и В. Мойер.
1. Получение
В 3-литровую трехгорлую колбу, снабженную обратным холо-
дильником с хлоркальциевой трубкой, делительной воронкой и
механической мешалкой и установленную на паровой бане, поме-
щают 48г Bгр.-ат.) магниевых стружек, 150 мл абсолютного эфира
и 100 г броммезитилена (стр. 118). Реакция начинается не сразу,
и иногда бывает необходимо прибавить немного иода или ката-
лизатора Гильмана (примечание 1). После того, как реакция
началась и протекает гладко, к реакционной смеси прибавляют
260
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
ДУРОЛ И ИЗОДУРОЛ
261
еще 298 г броммезитилена (в общем 2 мол.) примерно в 700 мл
абсолютного эфира с такой скоростью, чтобы эфир все время энер-
гично кипел. После того, как прибавлен весь броммезитилен, смесь
нагревают на водяной бане до тех пор, пока практически весь
магний не растворится (примечание 2).
Раствор гриньярова реактива охлаждают до 10° и прибавляют
при энергичном перемешивании раствор 600 г D,8 мол.) диметил-
сульфата (примечание 3) в 500 мл абсолютного эфира, в течение
2—3 час. Реакция идет очень бурно, и может произойти сильное
вскипание вследствие выпадения нерастворимых соединений маг-
ния. После Прибавления диметилсульфата, реакционную смесь
оставляют стоять (примечание 4) в течение около 24 час. и затем
комплекс разлагают, прибавляя через делительную воронку раз-
бавленную соляную кислоту. Как только масса станет достаточно
жидкой, начинают перемешивание. Когда магниевые соли полно-
стью перейдут в раствор, перемешивание прекращают, дают смеси
отстояться, сливают водный слой и промывают эфирный раствор
три раза водой. По удалении магниевых солей эфир отгоняют на
водяной бане и остаток медленно приливают к раствору 30 г натрия
в 500 мл абсолютного спирта (примечание-5), кипятят смесь в тече-
ние 30 мин. с обратным холодильником, охлаждают, смешивают со
150—200 мл эфира и удаляют щелочь и спирт тщательным промы-
ванием водой (примечание 6). Эфирный раствор сушат хлористым
кальцием, отгоняют эфир, нагревают остаток на водяной бане в
течение 3—4 часов с 25—30 г металлического натрия (примечание 7),
полученную смесь фильтруют и фильтрат тщательно фракциони-
руют в вакууме из специальной колбы Клайзена. Фракция до
85718 мм весит около 50 г и состоит главным образом из мезити-
лена: изодурол переходит при 85—87°/18 мм. Выход 140—160 г
E3—61%теоретич.), продукт имеет температуру плавления —24,2°.
2. Примечания
1. Катализатор Гильмана 8 легко приготовить нагреванием в
• эвакуированной колбе сплава магния, содержащего 12,75% меди
и примерно 20% (по весу) иода. Чтобы вызвать реакцию с галоидным
соединением, достаточно всего около 0,25 г этого продукта. При
применении катализатора Гильмана к реакционной смеси приба-
вляют обыкновенные магниевые стружки, как только начнется
реакция. Начало реакции можно вызвать также прибавлением
небольшого количества эфирного раствора какого-либо магний-
органического соединения, например, бромистого этилмагния.
2. При применении катализатора Гильмана к концу реакции
всегда остается избыток магния.
3. Диметилсульфат нужно тщательно перегнать в вакууме,
собирая фракцию, кипящую в пределах 1°.
Диметилсульфат чрезвычайно ядовит, поэтому следует вести
работу с ним очень осторожно и ни в коем случае не вдыхать его
паров и не разбрызгивать жидкость на руки и платье. Специфиче-
ским противоядием является аммиак.
4. Иногда реакционная смесь становится почти твердой, в этом
случае перемешивание бесполезно.
5. Избыток диметилсульфата не удаляется полностью при
обработке продукта водным раствором щелочи. Реакция между
диметилсульфатом и этилатом натрия бывает иногда очень бурной,
поэтому смешивание растворов следует производить с осторож-
ностью. Если имеется большой избыток диметилсульфата, то для
того, чтобы раствор оставался щелочным, может понадобиться
большее количество этилата натрия.
6. Образующуюся эмульсию можно разрушить подкислением
смеси.
7. Обработка натрием обеспечивает получение вещества, не
содержащего галоида*
3. Другие методы получения
Изодурол может быть получен из броммезитилена, йодистого
метила и натрия9; из мезитилена, хлористого метила и хлористого
алюминия 10; из мезитилена, йодистого метила, хлористого алюми-
ния и сероуглерода11; из 1, 3, 4, 5-тетраметилбензонитрила с
хлористым водородом при 250°12; действием хлористого цинка или
иода на камфору 13; в небольших количествах действием концен-
трированной серной кислоты на ацетон и. Приведенный выше ме-
тод получения описан в литературе 15.
Ни один способ с хлористым алюминием не дает чистого про-
дукта, так как в его присутствии происходит изомеризация изо-
дурола с образованием всех трех тетраметилбензолов, а из этой
смеси нельзя выделить в чистом состоянии изодурол. (Ср. приме-
чание 7, стр. 258.)
1 A d о г, R i 1 1 i e t, Ber, 12, 331 A879); Friedel, Crafts, Compt.
rend. 91, 257 A880); Ann. chim. phys. F) 1, 449 A884); Jacobsen, Ber.
14, 2629 A881); 18, 338 A885); 20, 896 A887); A n s с h it t z, Ann. 235, 185
A886); Claus, Foecking, Ber. 20, 3097 A887); Beaurepaire, Bull,
soc. chim. B) 50, 676 A888).
2 Jannasch, Fittig, Z. Chem. 13, 161 A870); I a n n a s с h, Ber.
7,~692 A874); 10, 1354 A877).
g 8 R e с k 1 e b e n, S с h e i b e г, там же 46, 2363 A913).
4 v. В r a u n, N e 1 1 e s, там же 67, 1098 A934).
5LeBel, Greene, Compt. rend. 87, 260 A878).
6 G r e e n e, там же 87, 931 A878).
'Smith, D о b г о v о I n у, J. Am. Chem. Soc. 48, 1413 A026).
sGilman, Peterson, Schulze, Rec. trav. chim. 47, 19 A928).
9 Jannasch, Ber. 8, 356 A875); В i e 1 e f e 1 d t, Ann. 198, 380 A879);
Jannasch, Weiler, Ber. 27, 3442 A894).
262
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
ДУРОХИНОН
263
10 Jacobsen, там же 14, 2629 A881).
11 С 1 a u s, F о е с к i n g, там же 20, 3097 A887).
" Hofmann, там же 17, 1915 A884).
"Armstrong, Miller, там же 16, 2259 A883).
14 О г n d о г f f, Young, Am. Chem. J. 15, 267 A893).
"Smith, MacDougall, j. Am. Chem. Soc. 51, 3003 A929).
H3C,
2 C6 (CH3L (NO2J
ДУРОХИНОН
^ (HNO3)
Ce (CH3L (NO.,),
СН3
- [Ce (CH3L (NH2 ¦ НС1L]а • SnCl4
О
V'[C6
), (NH2 • НС1J]2 ¦ SnCl4 —- 2
H3C
H,C
,сн3
CH3
0
Предложил: Л. Смит.
Проверили: Ф. Уитмор и Т. Холлингсхед.
1. Получение
А. Динитродурол. В стакан емкостью 800 мл, снабженный тер-
мометром и эффективной механической мешалкой, наливают 75 мл
концентрированной серной кислоты и к ней прибавляют раствор
13,4г @,1 мол.; примечания 1 и 2) дурола (стр. 253) в 100 мл хлоро-
форма. Реакционную массу охлаждают до 10°, прибавляют по
каплям при перемешивании 16 г A0,7 мл) дымящей азотной кис-
лоты (уд. в. 1,5) (примечание 3) из делительной воронки емкостью
125 мл, с такой скоростью, чтобы температура не превышала 50°
(все прибавление длится приблизительно 15 мин., причем снаружи
смесь охлаждается смесью льда и соли). Как только вся кислота
прибавлена, смесь переносят в делительную воронку, удаляют слой
серной кислоты и немедленно (примечание 4) выливают верхний
хлороформенный слой в 500 мл 10%-ного раствора соды. Слой
серной кислоты отбрасывают, так как он содержит очень мало
динитродурола. Весь процесс проводят с самого начала еще три
раза, соединенные хлороформенные растворы промывают дважды
2,5%-ным раствором соды, сушат в течение ночи 30 г безводного
хлористого кальция, фильтруют и отгоняют хлороформ до тех
пор, пока не начнут выпадать кристаллы динитродурола. Тогда
прибавляют к раствору четырехкратный объем горячего 95%-ного.
этилового спирта (около 500 мл) и охлаждают полученную смесь
до 10°. Выпавший кристаллический осадок отсасывают и промы-
вают дважды 50 мл холодного A0°) 95%-ного этилового спирта.
Выход, полученный от четырех нитрований, 82,5—84 г (92—94% тео-
ретич.) продукта, плавящегося при 207—208° (примечание 5).
Б. Восстановление динитродурола. Раствор 90 г динитродурола
в 1 л ледяной уксусной кислоты кипятят в колбе емкостью 12 л
(примечание 6). Одновременно растворяют 700 г хлористого олова
в 800 мл концентрированной соляной кислоты и раствор нагревают
до кипения. Раствор нитрососдинения в уксусной кислоте пере-
стают нагревать и очень осторожно (в течение 10 мин.) вли-
вают в него раствор хлористого олова. Реакция заканчивается в
15 мин., и по мере охлаждения раствора начинают выпадать кри-
сталлы двойного соединения хлорного олова и солянокислого
диаминодурола. Реакционную смесь охлаждают холодной водой
со льдом до 10°, выпавшие кристаллы отсасывают, промывают
дважды 50 мл 95%-ного этилового спирта и дважды 50 мл эфира и
сушат. Фильтраты содержат очень небольшое количество диамино-
дурола и поэтому ими можно пренебречь. Состав полученного
соединения: [Ce(CH3L (NH2- HC1J]2 SnCl4; оно кристаллизуется из
реакционной смеси в виде мелких блестящих, почти бесцветных
пластинок. Выход 145 г (97% теоретич.).
В. Дурохинон. 100 г двойной оловянной соли суспендируют в
растворе 300 г кристаллического хлорного железа в 150 мл воды,
к которой добавлены 20 мл концентрированной соляной кислоты;
суспензию оставляют стоять на ночь при температуре около 30°
и затем отсасывают. Продукт растворяют в 150 мл горячего 95%-ного
этилового спирта, раствор фильтруют и оставляют стоять ночь
при 30°. Выход дурохинона 40 г (90% теоретич.; т. пл. 109—110°).
2. Примечания
1. Дурол лучше нитровать небольшими порциями, так как
хороший выход и чистый продукт получаются только в том случае,
если реакция производится очень быстро и время соприкосновения
азотной кислоты с динитродуролом, минимально;
2. Хорошие результаты можно получить только с чистым дуро-
лом. Его следует перекристаллизовывать из метилового спирта
до тех пор, пока он не будет плавиться при 79—80°.
3. Большой избыток азотной кислоты не желателен, так как
он понижает выход. Концентрация азотной кислоты также играет
роль; для получения хороших результатов надо брать кислоту
уд. в. 1,5 или больше.
4. Необходимо вылить хлороформенный слой в раствор соды как
можно скорее, так как продолжительное стояние даже с небольшим
количеством кислоты приводит к образованию значительного коли-
264-
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
ИЗОНИТРОЗОПРОПИОФЕНОН
265
чества красного смолистого вещества, что сильно затрудняет после-
дующую очистку нитросоединепия.
5. При нитровании дурола мононитросоединение не образуется;
получается или динитродурол, или неизмененное вещество, и
продукты окисления.
6. Большая колба необходима, потому что восстановление идет
очень бурно и реакционная смесь при вскипании заполняет колбу
указанного размера полностью.
3. Другие методы получения
Дурохинон был получен действием щелочей на 2,3-дикетопен-
танх йЯи 3,3-дихлор-2-пентанон 2; из дуренола сочетанием его
с диазотированной сульфаниловой кислотой, с последующим вос-
становлением азокрасителя и окислением полученного аминофе-
нола до хинона3; и из дурола посредством ряда реакций, опи-
санных выше4, основой для которых послужил способ Нефа5.
Методика нитрования, применяемая для получения динитродурола,
представляет собой видоизменение способа нитрования, введен-
ного впервые Вильштеттером и Кубли 6.
1 Pechmann, Ber. 21, 1420 A888).
2 Фаворский, ЖРХО26, 559A804); J. prakt. Chcm. B) 51, 538 A895).
3 Smith, Opie, Wawzonek, Prichard, J. Org. Chem. 4,
318 AS39).
•'Smith, Dobrovolny, J. Am. Chem. Soc. 48, 1420 A926).
5 Nef, Ber. 18, 2806 A885);'Ann. 237, 5 A887).
• Wills tatter, Kubli, Ber. 42, 4151 A90S).
ИЗОНИТРОЗОПРОПИОФЕНОН
B-Оксим '/-фенил-1,2-пропандиона)
CeH5COCH2CH3
CHgONO — ~ C6H5COCCH3
NOH
CHgOH
Предложили: В. Хартунг и Ф. Нроссяей,
Проверили: Р. Фьюзон и Р. Петерсон.
1. Получение
3-литровую круглодонную колбу с тремя горлами (рис. 9, А)
снабжают обратным холодильником, механической мешалкой с
жидкостным затвором и двумя трубками для впуска газа Т1 и Т2,
погруженными возможно глубже в колбу. Метнлнитрит получают
в 2-литровой конической колбе Б, снабженной капельной ворон-
кой В емкостью 500 мл и соединенной с колбой А посредством
трубки Tj. Сухой хлористый водород поступает через трубку Т2.
Весь прибор желательно собрать в хорошо действующем вытяж-
ном шкафу.
В колбу А помещают раствор 469 г C,5 мол.) пропиофенона
(примечание 1) в 2,3 л обыкновенного этилового эфира, а в колбу
Б— смесь из 290 г D,0 мол.) 95%-ного азотистокислого натрия,
180мл A42 г, 4,5 мол.) метилового спирта и 170 мл воды. В капель-
ную воронку В наливают 455 мл холодной разбавленной серной
кислоты (полученной прибавлением одного объема концентрирован-
ной кислоты к двум объемам
воды).
Пускают в ход мешалку и
через трубку Т2 со скоростью 6—
10 пузырьков в секунду начи-
нают пропускать хлористый во-
дород. Кислоту из воронки В
прибавляют по каплям к со-
держимому колбы Б, и газооб-
разный метилнитрит (примеча-
ние 2) по трубке Tj поступает
в реакционную смесь. Раствор
в А окрашивается в коричнево-
красный цвет, и примерно через
10 мин. эфир начинает спокойно
кипеть(примечание 3). Скорость
выделения метплнитрита регу-
лируют таким образом, чтобы эфир непрерывно, но не сильно
кипел. Время, необходимое для прибавления метилнитрита, рав-
няется примерно 4 час. Перемешивание и пропускание хлористого
водорода продолжают после этого еще 30 мин. К концу этого
времени раствор перестает кипеть м приобретает светложелтую
окраску.
Реакционную смесь оставляют стоять на несколько часов (пред-
почтительно на ночь), после чего ее многократно экстрагируют
10%-ным раствором едкого натра порциями по 500 мл до тех пор,
пока щелочная жидкость не будет более окрашиваться после взбал-
тывания ее с эфирным раствором (примечание 4). Обычно требуется
пятикратное извлечение. Соединенные щелочные вытяжки мед-
ленно и при помешивании выливают в смесь, состоящую из 700—
750 мл концентрированной соляной кислоты и 1 кг льда. Кристаллы
изонитрозопропиофенона отсасывают и сушат. Вес полученного
продукта — 370—390 г F5—68% теоретич.); его т. пл. 111—113°.
Продукт этот может быть перекристаллизован из толуола (около
550 мл), причем получается 315—335 г белоснежных кристаллов
с т. пл. 112—113° (примечание 5).
Рис. 9
266
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
ИЗОПРОПИЛОВЫЙ ЭФИР МОЛОЧНОЙ КИСЛОТЫ
267
2. Примечания
1. Пропиофенон может быть получен с выходом в 70—80% из
бензола и хлористого пропионила или пропионового ангидрида в
присутствии хлористого алюминия.
2. Для получения малых количеств изонитрозокетона удобнее
применять более высококипящий алкилнитрит, например бутил-
нитрит (стр. 131), который можно добавлять непосредственно к
реакционной смеси, заменив трубку 7\ на капельную воронку.
Бутилнитрит должен быть свежеполученным или перегнанным
перед самым употреблением.
3. Скорость перемешивания должна быть постоянной, так как
неожиданное увеличение скорости может вызвать чересчур энер-
гичное кипение эфира.
4. Эфирный раствор, оставшийся после извлечения щелочью,
содержит непрореагировавший прошюфенон, который может быть
выделен, если отогнать эфир и остаток подвергнуть дробной пере-
гонке. Количество выделенного пропиофенона, с т. кип. 210—
216°, колеблется от 80 до ПО г.
5. Из толуольного маточного раствора можно выделить еще
25—30 г продукта, если раствор извлечь щелочью и вновь осадить
изонитрозопропиофенон кислотой.
3. Другие методы получения
Изонитрозопропиофенон был получен из сложных эфиров <х-бен-
зоилпропионовой кислоты путем омыления, нитрозирования и
декарбоксилирования *; из 1 -фенил-1,2-пропандиона действием гид-
роксиламина 2, из пропиофенона действием на него амилнитрита 3,
метилнитрита 4 или бутилнитрита 5.
1Pechmann, Muller, Ber. 21, 2119 A888).
2 Ко lb, Ann. 291, 292 A896).
3 Claisen, Manasse, Ber. 22, 529 A889).
1 S I a te r, J. Chem. Soc. 117, 590 A920).
5 H a r t u n g, Munch, J. Am. Chem. Soc. 51, 2264 A929).
ИЗОПРОПИЛОВЫЙ ЭФИР МОЛОЧНОЙ КИСЛОТЫ
СН3СНОНСО2Н + (СН3J СНОН (-H-lS-? CH3CHOHCO2CH(CH3K+ Н,0
Предложил: Ф. Мак-Дермот.
Проверил: К. Ноллер.
1. Получение
В 3-литровой круглодонной колбе (примечание 1), соединенной
с колонкой для фракционирования длиной в 1 м1, смешивают
450 г G,5 мол.) безводного изоиропилового спирта (примечание 2),
212 г B мол.) 85%-ной фармакопейной молочной кислоты, 1 л бензола
и 5 мл концентрированной серной кислоты. Колбу ставят в воздушную
баню на электрическую плитку (примечание 3) и медленно отгоняют
в течение б—7 час. тройную смесь бензол — изопропиловый спирт—
вода, которая кипит при 66,5°. Отгонку продолжают до тех пор, пока
температура паров в верхней части колонки не повысится и не
остановится на 71—72° (бинарная смесь изопропиловый спирт — бен-
зол); отделение воды при этом прекращается. Тогда к смеси приба-
вляют 10 г осажденного углекислого кальция и продолжают пере-
гонку до тех пор, пока температура не достигнет 80° для того,
чтобы удалить большую часть бензола и избыток изопронилового
спирта (примечания 4 и 5). После этого содержимое колбы фильтруют
в специальную колбу Клайзсна и перегоняют фильтрат в вакууме,
собирая фракции до 60°, 60—75°, 75—80° и 80—100° при 32 мм.
Фракция, кипящая при 75—80°/32 мм, представляет собой изо-
пропиловый эфир молочной кислоты, и количество ее составляет
130—160 г. Повторной перегонкой высших и низших фракций
получают еще 30—60 г эфира. Общий выход 160—180 г F0—68%
теоретич.). При атмосферном давлении эфир кипит при 166—168°
с небольшим разложением.
2. Примечания
1. Для больших количеств целесообразно взять колбу с двумя
горлами; второе горло служит для прибавления к смеси углекислого
кальция.
2. Пригоден технический изопропиловый спирт. Изопропило-
вый спирт очень трудно хорошо высушить. Бинарная смесь изо-
пропиловый спирт—вода, кипящая при 80,35°, содержит 12,1%
воды по весу. Тройная смесь бензол — изопропиловый спирт — вода,
кипящая при 66,5°, содержит 73,8% бензола, 18,7% изопропило-
вого спирта и 7,5% воды. Следовательно, после прибавления 120 г
сухого бензола к 100 г смеси и после отгонки до температуры 82°
в"колбе остается 55—60 г почти сухого изопропилового спирта.
3. Можно применять также водяную, паровую или масляную баню.
4. До прибавления углекислого кальция температура паров не
должна превышать 72°. Если отогнать слишком много спирта
до того, как кислота нейтрализована, то происходит обугливание
и осмоление, что понижает выход эфира.
5. Полученные обратно бензол и избыточный изопропиловый
спирт можно высушить перегонкой и применять для последующих
синтезов.
3. Другие методы получения
Изопропиловый эфир молочной кислоты был получен нагрева-
нием изопропилового спирта и молочной кислоты в запаянной
трубке при 170°2 и из молочнокислого серебра и йодистого изопро-
268
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
пила, вместе с изопропиловым эфиром а-изопропоксипропионовой
кислоты3. При непосредственной этерификации молочной кис-
лоты изопропиловым спиртом в присутствии серной кислоты
нельзя получить выход более 20%, и полученный продукт менее
чист.
1 Clarke, Rahrs, Ind. Eng. Chem. 15, 349 A923).
2 S i 1 v a, Bull. soc. chim. B) 17, 97 A872).
3Purdie, Lander, J. Chem. Soc. 73, 298 A898).
ИЗОПРОПИЛТИОЦИАНАТ
(Изопропиловый эфир роданистой кислоты)
(СН3J СНВг + NaSCN (СН3J CHSCN + NaBr
Предложил: Р. Шрайнер.
Проверил: К- Ноллер.
1. Получение
В 3-литровую круглодонную колбу, снабженную весьма эффек-
тивной механической мешалкой (примечание 1), обратным холо-
дильником и делительной воронкой емкостью 500 мл, помещают
445 г E,5 мол.) роданистого натрия (примечание 2) и 1250 мл
90%-ного этилового спирта. Мешалку пускают в ход, нагревают
смесь до кипения и в течение 1 часа медленно прибавляют 615 г
E мол.) бромистого изопропила (стр. 115; «Синт. орг. преп.», сб. 1,
стр. 118). После этого, не прекращая перемешивания, продолжают
кипячение еще 6 час. Затем выпавший бромистый натрий отсасы-
вают и промывают 250 мл 95%-ного спирта. Фильтрат упаривают
на водяной бане насколько это возможно, остаток смешивают
с 500 мл воды и отделяют верхний слой изопропилтиоцианата.
Водный слой экстрагируют двумя порциями эфира по 100 мл (при-
мечание 3), эфирные вытяжки прибавляют к ранее отделенному
тиоцианату, и полученный эфирный раствор сушат безводным
сернокислым натрием (примечание 4). Высушенный раствор два
раза фракционируют из специальной колбы Клайзена с 25-санти-
метровым дефлегматором, собирая следующие фракции: до 60°;
60—100°; 100—130°; 130—146° и 146—151°. Последняя фракция
представляет собой чистый изопропилтиоцианат. Выход 320—
345 г F3—68% теоретич.). Спиртовой дестиллат, отогнанный ранее
на водяной бане, подвергают вторичной перегонке с эффективной
колонкой (примечание 5), до тех пор, пока не отгонят весь спирт
(примечание 6), и остаток После отделения воды перегоняют, как
указано выше. Таким образом получают дополнительно 55—65 г
продукта, кипящего при 146—151°. Общий выход 385—400 г G6—
ИМИНОДИФЕНИЛМЕТАН СОЛЯНОКИСЛЫЙ
269
79% теоретич.). При вторичной перегонке соединенных фракций,
кипевших при 146—151°, практически весь продукт переходит
при 149—151°.
2. Примечания
1. Необходимо очень энергичное механическое перемешивание,
так как в противном случае оседающий на^дно бромистый натрий
вызывает сильные толчки.
2. Пригоден технический роданистый натрий. Роданистый ка-
лий не представляет по сравнению с солью натрия никаких пре-
имуществ.
3. Если водный слой экстрагировать бензолом, то для получе-
ния того же выхода необходимо произвести три дробные перегонки.
4. Сернокислый натрий удаляет не всю воду, поэтому при по-
следующей фракционировке как только появится водный слой,
его следует удалять с помощью делительной воронки.
5. Пригоден 8-шариковый дефлегматор типа, описанного Клар-
ком и Рарсом !.
6. Перегонку продолжают до тех пор, пока в самом нижнем
шарике не появится вода.
3. Другие методы получения
К"; Изопропилтиоцианат был получен действием йодистого изо-
пропила на роданистый калий 2.
1 Clarke, Rahrs, Ind. Eng. Chem. 18, 1092 A926).
2 Henry, Ber. 2, 496 A869); Geislich, Ann. 178, 80 A875).
ИМИНОДИФЕНИЛМЕТАН СОЛЯНОКИСЛЫЙ
(Бензгидрилидгнимин солянокислый )
8 (С6Н5J С = NOH >
4 (С6Н3), СО + 4 (СвН5K С = NH + N, -4, 2 Н2О + 2 N0
(С6Н^J С = NH + НС1 —.- (С„Н8L С = NH • НС1
Предложил: А. Лахман.
Проверил: К. Ноллер.
1. Получение
Кусок стеклянной трубки длиной 80 см с внутренним диамет-
ром 2 см запаивают с одного конца и наполняют неплотно 49 г
@,25 мол.) бензофеноноксима (стр. 381). Трубку укрепляют почти
в горизонтальном положении с легким наклоном вниз к закрытому
концу и соединяют с маленькой колбой для отсасывания при
270
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
л-ИОДАНИЛИН
271
помощи каучуковой пробки и стеклянной трубки, согнутой под пря-
мым углом. Затем отсасывают воздух водоструйным насосом, впус-
кают сухой углекислый газ, снова выкачивают его и опять напол-
няют углекислым газом. После этого трубку нагревают на голом
пламени, причем начинают нагревать от верхнего конца и нагре-
вают одно место до тех пор, пока не наступит разложение, а затем
уже переходят дальше (примечание 1). После того, как весь оксим
разложился, в течение короткого времени сильно нагревают смесь,
скопившуюся в закрытом конце трубки (примечание 2), для того
чтобы закончить разложение, и продукт охлаждают. Затем из трубки
снова отсасывают воздух и отгоняют сконденсировавшуюся на стен-
ках трубки воду слабым нагреванием. Жидкость переливают в
маленькую перегонную колбу и перегоняют при давлении около
20 мм. Дестиллат, состоящий из смеси бензофенона и иминодифенил-
метана, растворяют в 400 мл нефтяной фракции F0—90°) и осаждают
солянокислый имин пропусканием сухого хлористого водорода. Соль
отсасывают (примечание 3), промывают небольшим количеством той
же нефтяной фракции, сушат и сохраняют, защищая от влажности
(примечание 4). Она возгоняется без разложения при 230—250° (при-
мечание 5). Выход 16—18 г E9—66% теоретич.; примечание 6).
2. Примечания
1. Если нагревать осторожно, то газы не увлекают с собой ве-
щества из трубки.
2. Необходимо следить за тем, чтобы капли воды, сконденси-
ровавшиеся в холодной части трубки, не стекали обратно и не сме-
шивались с горячей жидкостью.
3. Из фильтрата можно выделить бензофенон.
4. На влажном воздухе хлоргидрат превращается в смесь бен-
зофенона и хлористого аммония. Свободное основание при стоя-
нии на воздухе выделяет аммиак, причем постепенно выделяется
кристаллический бензофенон.
5. По способу Ганча и Крафта соль имина можно перевести в
свободный имин, обрабатывая раствор соли в хлороформе сухим
аммиаком.
6. Выход зависит в значительной степени от качества оксима
бензофенона. Если последний был влажным или находился в со-
прикосновении с влажным воздухом, особенно в закрытом сосуде,
то выход заметно понижается.
3. Другие методы получения
Иминодифенилметан (или его хлоргидрат) может быть получен
нагреванием дифенилдихлорметана с уретаном при 130°1; действием
аммиака на дифенилдибромметан2; обработкой бензофепонхлор-
имида пятихлористым фосфором в эфирном растворе 3 или сухим
хлористым водородом в растворе лигроина4; действием бромистого
фенилмагния на N-бромбензамид2, бензонитрил5, бромистый
циан 6, хлористый циан 7 и на арилтиоцианаты 8. Он был получен
также пропусканием аммиака и паров бензофенона над окисью
тория при 380—390°9; каталитическим восстановлением оксима бен-
зофенона водородом в присутствии никелевого катализатора в рас-
творе абсолютного спирта при комнатной температуре и атмосферном
давлении10; пропусканием смеси водорода и паров оксима бен-
зофенона над восстановленной медью при 200° и; наконец, дей-
ствием этилата натрия на N-монохлордифенилметиламин в спирто-
вом растворе 12. Метод, описанный здесь, опубликован ранее
Лахмаиом 13.
1Hantzsch, Kraft, Ber. 24, 3516 A891).
2 М о о г е, там же 43, 564 A910).
3 V о s b u r g Ь, J. Am. Chem. Soc. 38, 2095 A916).
* Peterson, Am. Chem. J. 46, 331 A911).
5Moureu, Mignonac, Compt. rend. 156, 1806 A913); Ann. chim.
(9) 14, 336 A920).
" Orignard, В e 1 1 e t, Courtot, там же (9) 4, 34 A915).
7 Q r i g n a r d, В e 1 1 e t, С о u r t о t, там же (9) 12, 379 A919).
«Adams, Bramlet, Tendick, J. Am. Chem. Soc. 42, 2372
A920).
9 Mignonac, Compt. rend. 169, 239 A919).
10 M i g n о п а с, там же 170, 938 A920).
"Yamaguchi, Bull. Chem. Soc. Japan 1, 35 A926) [C,. A. 21, 75
A927)].
12 H e 1 1 e r m a n, S a n d e r s, J. Am. Chem. Soc. 49, 1742 A927).
13 L а с h m a n , там же 46, 1477 A924).
л-ИОДАНИЛИН
C6H5NH2 + l, + NaHCOg —¦- n-IC6H4NH2 + Nal + H2O -f- CO2
Предложил: P. Брюстер.
Проверили: В. Каротерс и В. Мак-Ювен.
1. Получение
В 3-литровый стакан помещают ПО г A,2 мол.) анилина, 150 г
A,8 мол.) бикарбоната натрия и 1 л воды и охлаждают смесь до 12—
15° прибавлением небольшого количества льда. Затем стакан снаб-
жают эффективной механической мешалкой и погружают в жид-
кость большой фарфоровый шпатель (плоскостью по радиусу ста-
кана), который, создавая препятствие вращательному движению,
улучшает перемешивание. Мешалку пускают в ход, прибавляют к
272
СИНТЕЗЫ ОРГАНИЧЕСКИХ «ПРЕПАРАТОВ
реакционной смеси 254 г (I мол.) истолченного в порошок иода
порциями по 15—20г через промежутки в 2—3 мин., таким образом,
чтобы все прибавление закончить в полчаса, и затем продолжают
перемешивание в течение еще 20—30 мин. За это время реакция
заканчивается, и окраска, обусловленная иодом, практически
исчезает. Сырой л-иоданилин, выпавший в виде темной кристалли-
ческой массы, собирают на воронке Бюхнсра, как можно тщательнее
отжимают и сушат на воздухе. Фильтрат можно сохранить с целью
выделения иода (примечание 1).
Сырой продукт очищают, нагревая его с 1 л бензина (примеча-
ние 2) с обратным воздушным холодильником в 2-литровой колбе
на водяной бане при 75—80° (примечание 3). Смесь часто взбалты-
вают в течение 15 мин., затем горячий раствор осторожно сливают
с осадка в стакан, охлаждаемый смесью льда и соли при непрерыв-
ном перемешивании. л-Иоданилин немедленно выпадает в виде
почти бесцветных игол; его отсасывают и сушат на воздухе (приме-
чания 4 и 5). Фильтрат выливают обратно в колбу, смесь нагревают,
раствор сливают с осадка, охлаждают и т. д. (примечание 6). Выход
165—185 г G5—84% теоретич») л-иоданилина, плавящегося при
62—63°.
2. Примечания
il. Для того, чтобы превратить в иод йодистый натрий, находя-
щийся в водном растворе, поступают следующим образом. К вод-
ному фильтрату от л-иоданилина прибавляют 100 мл концентриро-
ванной серной кислоты и раствор 200 г двухромовокислого натрия
в 200 мл воды. Выпавшему иоду дают осесть, промывают его три
раза водой посредством декантации, отсасывают и сушат на часо-
вом стекле. Выход неочищенного иода 167—179 г.
2. Обыкновенный бензин перегоняют, собирая фракцию 70—
150°. При применении неперегнанного бензина иоданилин обычно
получается загрязненным высококипящими углеводородами, кото-
рые очень трудно удалить.
3. При нагревании до более высокой температуры иногда про-
исходит заметное осмоление, вследствие чего понижается выход.
4. Если охлаждать недостаточно быстро, то продукт часто вы-
падает в виде масла.
5. Удобно сушить п-иоданилин в токе теплого воздуха, поль-
зуясь фэном, который применяется для сушки волос.
6. Обычно достаточно двух экстракций, но если остается еще
значительное количество нерастворившегося вещества, то следует
произвести третью экстракцию. Первая порция практически бес-
цветна, вторая и третья окрашены в светлокоричневый цвет, если
только раствор не обесцветить небольшим количеством животного
угля.
5-ИОДА.НТРАНИЛОВАЯ КИСЛОТА
Й73
3. Другие методы получения
/т-Иоданилин может быть получен восстановлением п-нитроиод-
бензола *; омылением л-иодацетанилида, полученного действием
хлористого иода на ацетанилид 2; непосредственным иодированием
анилина 3. Вышеописанный метод представляет собою переработан-
ный способ иодирования толуидинов Уилера 4 и Ханна и Берли-
нера 5.
1 G г i e s s, Zeit. fur Chem. 1866, 218; К е k u 1 ё", там же 687; К 6 г п е г,
We n d e r, Gazz. chim. ital. 17, 489 A887); В а е у е г, Вег. 38, 2762 A905);
Montagne, там же 51, 1490 A918).
2 Michael, Norton, там же 11, 108 A878); Chattaway, Con-
stable, J. Chem. Soc. 105, 125 A914).
3 H о f m a n n, Ann. 67, 65 A848); Bradfield, Orton, Roberts,
J. Chem. Soc. 1928, 783; M i 1 i t z e r, Smith, Evans, J. Am. Chem.
Soc. 63, 436A941).
'Wheeler, Liddle, . Am. Chem. J. 42, 501 A909); Wheeler,
там же 44, 128, 500 A910).
6 H a n n, В e r 1 i n e r, J. Am. Chem. Soc. 47, 1710 A925).
5-ИОДАНТРАНИЛОВАЯ КИСЛОТА
NH,
+ IC1
V
COaH
NH9
HC1
Предложили: В. Уоллингфорд и П. Крейгер.
Проверили: Ф. Уитмор и Л. Сюзерленд.
1. Получение
В 3-литровом стакане растворяют ПО г @,8 мол.) антраниловой
кислоты (примечание 1)в 1 л воды и 80 .мл химически чистой концен-
трированной соляной кислоты (уд. в. 1,19); раствор охлаждают до20°.
В 2-литровом стакане приготовляют раствор однохлористого иода
в соляной кислоте путем разбавления 140 Мл концентрированной
соляной кислоты (х. ч.) 500 мл холодной воды, после чего добавляют
достаточное количество льда, чтобы температура понизилась до 5°,
и в течение 2 мин. при перемешивании добавляют 131 г @,8 мол.)
однохлористого иода (стр. 199).
Раствор однохлористого иода, охлажденный до 5°, при переме-
шивании быстро приливают к раствору антраниловой кислоты,
IS Сборник JVi 2
274
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
температура которого 20°. Почти немедленно выпадает 5-иодан-
траниловая кислота в виде гранулированного осадка рыжеватоко-
ричневого цвета или даже фиолетового и температура повышается до
18—22°. Смесь перемешивают в течение 1 часа, пока она доходит до
комнатной температуры, затем ее фильтруют через бюхнеровскую
воронку диаметром 13 см. Кислоту хорошо отжимают, промывают
три раза холодной водой порциями по 100 мл и затем сушат при
90—100°. Выход кислоты 185—189 г (88—90% теоретич.). Окраска
кристаллов от бурой до пурпурной, температура плавления 185—
190° с разложением (примечание 2).
5-Иодантраниловую кислоту лучше всего очистить путем пере-
кристаллизации ее аммонийной соли. Для этого в стакан емкостью
400 мл помещают 100 г кислоты, добавляют 200 мл горячей воды и
кислоту растворяют в концентрированном аммиаке (уд. в. 0,9) при
60°, перемешивая до полного растворения и так, чтобы был неболь-
шой избыток аммиака. На это требуется около 40 мл концентриро-
ванного аммиака. Добавляют гидросульфит натрия порциями по
1 г до тех пор, пока больше не будет заметно белящего действия,
затем добавляют 5 г обесцвечивающего угля, перемешивают 3 мин.
и смесь фильтруют с отсасыванием через предварительно нагретую
бюхнеровскую воронку в нагретую колбу для отсасывания. Фильтр
промывают 10 мл кипящей воды.
Фильтрат и промывные воды переносят в стакан на 400 мл и
раствору дают охладиться медленно без перемешивания, пока не
прекратится образование кристаллов, после чего охлаждают до 5°.
Кристаллическую аммонийную соль отфильтровывают с отса-
сыванием через большую бюхнеровскую воронку, кристаллы про-
мывают 15 мл ледяной воды, хорошо отсасывают, отжимают, рас-
кладывают тонким слоем на эмалированном или стеклянном про-
тивне и сушат на воздухе при 35—50°. Выход аммонийной соли,
окрашенной в желтый до фиолетового цвет, составляет 80—89 г
или 76—84% (примечания 3 и 4).
Аммонийную соль растворяют в трех частях горячей воды, если
нужно, то добавляют аммиак, чтобы завершить растворение, при
60°, раствор вновь обрабатывают гидросульфитом натрия и 3—4 г
обесцвечивающего угля, фильтруют горячим и вновь осаждают
5-иодантраниловую кислоту, для чего добавляют соляную кислоту
(х. ч.) порциями по 3—5 мл и тщательно перемешивают после ка-
ждого прибавления до тех пор, пока реакционная смесь не будет
давать слабо кислую реакцию на конго. Затем добавляют лед,
пока температура не понизится до 20° и выпавшую кислоту отфиль-
тровывают с отсасыванием, обильно промывают холодной водой и
сушат при 100—110° (примечание 5). 5-Иодантраниловая кислота
почти количественно осаждается из ее аммонийной соли и полу-
чается в виде желтого порошка с температурой плавления 190—195°
(с разложением; примечание б).
6-ИОДАНТРАНИЛОВАЯ КИСЛОТА
275
2. Примечания
1. Вполне пригодна продажная сублимированная антранило-
вая кислота.
2. Точка разложения бывает различной в зависимости от метода
определения. Указанные здесь величины были получены следую-
щим образом: образец помещался в капилляр, капилляр помещался
в баню, предварительно нагретую до температуры на 25° ниже
точки разложения, и нагревался со скоростью 2—3° в минуту.
3. В условиях сушки аммиачная соль является нестойкой. Сухой
продукт представляет собой смесь 5-иодантраниловой кислоты и ее
аммонийной соли. При соприкосновении с воздухом окраска ее
может измениться от желтой до фиолетовой.
4. Для того, чтобы выделить 5-иодантраниловую кислоту из
маточных растворов их чуть подкисляют на конго с помощью пла-
стинки для взятия проб концентрированной соляной кислотой,
охлаждают до 20°, отфильтровывают, обильно промывают холод-
ной водой и сушат при 90—110°. Эту порцию кислоты можно при-
бавить к главной порции при дальнейших перекристаллизациях
или переработать отдельно.
5. Сушильный шкаф должен быть с хорошей циркуляцией воз-
духа и с хорошо отрегулированной температурой: сушка при 120°
вызывает выделение значительного количества свободного иода.
6. Этот продукт достаточно чист для превращения его в м-иод-
бензойную кислоту (стр. 276). При желании получить продукт
большей степени чистоты аммонийную соль можно перекристалли-
зовать из двухкратного по весу количества воды, при температуре
от 45° до 5° с выходом 85—90%. Если после высушивания соль
остается почти белой, выделенная щз соли кислота плавится с раз-
ложением при 210°. Для этого могут потребоваться 4—5 перекри-
сталлизации.
3. Другие методы получения
5-Иодантраниловая кислота была получена восстановлением
2-нитро-5-иодбензойной кислотыг, обработкой антраниловой ки-
слоты иодом в растворе едкого кали 2, обработкой ангидрида 5-ок-
симеркурантраниловой кислоты иодом в водном растворе йодистого
калия 3 и иодированием антраниловой кислоты в растворе ледяной
уксусной кислоты однохлористым иодом 4 или в растворе разбав-
ленной уксусной кислоты иодом 5.
1 Ого the, J. prakt. Chem. B) 18, 326 A878).
2 Wheeler, Johns, Am. Chem. J. 43, 403 A910); К 1 e m m e, Hun-
ter, J. Org. Chem. 5, 230 A940).
3Schocller, Hue ter, Ber. 47, 1938 A914).
* Borsch e, Weussmann, Fritzsche, там же 57, 1774 A924).
5Militzer, Smith, E v a n s, J. Am. Chem. Soc. 63, 436 A941).
276
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
со,н
^ NHO
СО2Н
/4NX1
л*-И0ДБЕН30ЙНАЯ КИСЛОТА
со.2н
NaNO2 + 2 MCI /^ N,Cl + NaCl + 2 Н2О
СО2Н
С,Н5ОН —
(Си) /\
СН3СНО + НС1
Предложили: В. Уоллингфорд и П. Крюгер.
Проверили: Ф. Уитмор и Л. Сюзерленд.
1. Получение
В 1-литровой колбе растворяют 132 г @,5 мол.) 5-иодантранило-
вой кислоты (стр. 273) и 35 г @,5 мол.) азотистокмслого натрия в
смеси 500 мл теплой воды и 60 мл 30%-ного раствора едкого натра.
По охлаждении до 20° раствор приливают из капельной воронки
в течение 15—20 мин. к хорошо перемешиваемой смеси 250 мл
концентрированной соляной кислоты (уд. в. 1,18) и 250 г льда,
находящейся в 2-литровом стакане. Если нужно, добавляют еще
льда, чтобы температура была ниже 20°. Нерастворимая желтая
соль диазония выпадает еще до окончания диазотирования. После
того как весь раствор прибавлен из капельной воронки, реакцион-
ную массу перемешивают еще 5 мин. и проверяют на иодокрахмаль-
ную бумажку, имеется ли избыток азотистой кислоты. Если тре-
буется, добавляют еще небольшое количество твердого азотисто-
кислого натрия и пробу повторяют каждые 3 мин. до тех пор, пока
не будет твердо установлено наличие небольшого избытка азоти-
стой кислоты. Соли диазония дают осесть и сливают возможно боль-
шее количество жидкости с пастообразной массы.
В 3-литровый стакан помещают 750 мл 95%-ного спирта и 1,5 г
хорошо измельченной сернокислой меди, после чего смесь нагре-
вают на электроплитке или на водяной бане до 70°. Диазониевую
пасту прибавляют порциями по 30 мл к спирту при хорошем пере-
мешивании. Температуру при этом поддерживают при 60—70° и
перед прибавлением новой порции выжидают, пока прекратится
выделение азота. Последние следы соли диазония смывают неболь-
шими порциями ранее декантированного раствора, остаток кото-
рого приливают к спирту порциями по 100 мл. Реакционную смесь
нагревают и перемешивают 30 мин. при 65—70J, после чего без переме-
шивания охлаждают до 5°. Выпавшую .м-иодбензойную кислоту филь-
Л-И0ДБЕН30ЙНАЯ КИСЛОТА
277
труют с отсасыванием через_воронку Бюхнера, осадок промывают три
раза холодной водой порциями по 50 мл и сушат при 90—110°.
Выход сырой .и-иодбензойной кислоты, окрашенной в коричневый
цвет, составляет 107—116 г (86—93% теоретич.; примечание 1).
Сырую .м-иодбензойную кислоту очищают путем перекристалли-
зации ее аммонийной соли. К 100 г кислоты в стакане емкостью
250 мл добавляют 75 мл горячей воды и кислоту частично нейтрали-
зуют 24 мл концентрированного аммиака (уд. в. 0,9). Массу пере-
мешивают при 80° до тех пор, пока не прекратится растворение
кислоты. Нейтрализацию завершают добавлением 2—5 мл аммиака
до полного растворения кислоты. Раствор нагревают до 90°, приба-
вляют 1 г обесцвечивающего угля, смесь фильтруют с отсасыванием,
пользуясь предварительно нагретыми воронкой Бюхиера и отса-
сывающей склянкой. Остаток на фильтре промывают 15 мл кипя-
щей воды. Фильтрат и промывные воды переносят в стакан на
250 мл и дают охладиться без "перемешивания до 25—35°, после
чего любым способом охлаждают до 5°. Кристаллы аммонийной
соли л-иодбензойной кислоты отфильтровывают, отжимают, как
можно лучше на воронке Бюхнера, раскладывают тонким слоем
на стеклянной или эмалированной чашке и сушат при температуре
не выше 60°. Получают 86—90 г призматических кристаллов,
окрашенных в светложелтый или рыжеватокоричневый цвет. Выход
аммонийной соли составляет 80—84% (примечание 2):
Полученную таким образом аммонийную соль перекристалли-
зовывают до тех пор, пока она не станет бесцветной, для чего ее
растворяют в равном весовом количестве воды при 80° и раствор
охлаждают до 5°. Выход после такой очистки составляет 75—85%
(примечание 3).
.м-Иодбензойную кислоту с т. пл. 187—188° получают, раство-
ряя чистую аммонийную соль ее в четырехкратном по весу коли-
честве горячей воды и подкисляя раствор на конго красное концен-
трированной соляной кислоты с добавлением льда, чтобы понизить
температуру до 20°. Выделившийся осадок отфильтровывают
с отсасыванием, кислоту промывают значительным количеством
холодной воды и сушат при 90—110° (примечание 4).
2. Примечания
1. Если маточный раствор выпарить до появления мути и охла-
дить до 5°, то можно получить несколько граммов кислоты, однако
весьма грязной и осмолившейся.
2. При таких условиях сушки аммонийная соль льиодбензойной
кислоты оказывается недостаточно стойкой. Продукт содержит
некоторое количество свободной кислоты.
3. В некоторых опытах даже после пяти перекристаллизации
не удавалось получить бесцветные кристаллы аммонийной соли,
278
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
однако в этих опытах переосаждение кислоты после 3—4 перекри-
сталлизации давало продукт с нужной температурой плавления.
Для переработки маточных растворов после этих перекристал-
лизации лучше всего их подкислить концентрированной соляной
кислотой до кислой реакции на конго, осадок отфильтровать, про-
мыть, высушить и переработать выделенную кислоту.
4. Суммарный выход чистой кислоты из неочищенной зависит
от количества взятой сырой кислоты, от того, какие меры были при-
няты для избежания потерь, и от числа перекристаллизации, ока-
завшихся необходимыми для достижения необходимой чистоты.
Если не выделять кислоты из маточных растворов, то из 100 г не-
очищенной кислоты, после превращения ее в аммонийную соль и
после трех перекристаллизации этой соли, можно получить 40—45 г
чистой кислоты. Суммарный выход чистой кислоты из 100 г неочи-
щенной составляет 50 г чистой кислоты с т. пл. 187—188°, 10 г
менее чистой кислоты с т. пл. 184—186° и около 10 г осмолившегося
твердого остатка.
3. Другие методы получения
.м-Иодбснзойная кислота была получена диазотированием .м-ами-
нобензойной кислоты с последующим действием йодистого калия
на кислый раствор диазония 1>а>3 и действием концентрированной
азотной кислоты на раствор иода и бензойной кислоты в ледяной
уксусной кислоте 4.
1 Q r i e s s, Ann. 113, 336 A860).
! Cohen, R а р е г, J. Chem. Soc. 85, 1273 A904).
3 С a 11 e 1 a i n, Bull. soc. chim. D) 41, 1546 A927).
' Datta, Chatterjee, J. Am. Chem. Soc. 41, 294 A919).
ИОДБЕНЗОЛ
C6H5NH2 + NaNO2 + 2 HC1 ¦ C6H6N2C1 + 2 H2O + NaCl
CeH5NaCl.+ KI —* C6H6I + N2 + KC1
Предложили: X. Люкас и Е. Кеннеди.
Проверили: Дж. Джонсон и П. Барршс,
1. Получение
В глиняный сосуд емкостью 10—15 л помещают 950 мл (ИЗО г,
11,7 мол.) концентрированной соляной кислоты (уд. в. 1,19), 950 мл
воды, 200 г A96 мл, 2,15 мол.) анилина и 2кг льда (примечание 1).
Смесь перемешивают с помощью механической мешалки, и как
только температура понизится до 5°, довольно быстро приливают
из делительной воронки, ножка которой опущена ниже поверхно-
сти жидкости, охлажденный раствор 156 г B,26 мол.) азотистокис
ИОДБЕНЗОЛ
279
лого натрия в определенном объеме G00—1000 мл) воды. Прили-
вание раствора не должно быть столь быстрым, чтобы температура
поднялась выше 10° или чтобы имело место выделение окислов
азота. Последние 5% раствора нитрита приливают медленнее и
время от времени берут пробу на иодокрахмальную бумажку до
тех пор, пока не будет обнаружен избыток свободной азотистой
кислоты.
Перемешивание продолжают еще 10 мин., и, если это нужно,
раствор быстро фильтруют через простую вату в большой воронке.
К раствору приливают водный раствор 358 г B,16 мол.) йодистого
калия и реакционную смесь оставляют на ночь. Затем реакцион-
ную массу переносят в большую колбу (или в две меньшие колбы)
и нагревают на водяной бане с воздушным холодильником, пока
не прекратится выделение газов, после чего жидкости дают спокой-
но охладиться до полного осаждения тяжелого органического слоя-
Большую часть верхнего водного слоя сифонируют и отбрасывают
(примечание 2). Остаток подщелачивают осторожным прибавлением
концентрированного раствора едкого натра (обычно требуется 100—
125 г технического твердого едкого натра) и все немедленно подвер-
гают перегонке с водяным паром. Последнюю х/3 дестиллата,получен-
ного при перегонке с водяным паром, собирают отдельно и соеди-
няют вместе с водным слоем, полученным от предыдущих порций
дестиллата. Смесь подкисляют 5—10 мл концентрированной сер-
ной кислоты и вновь перегоняют с водяным паром. Полученный
при этом иодбензол присоединяют к главной порции и сушат 10—
15 г хлористого кальция (примечания 3 и 4). После перегонки в
вакууме получают 327—335 г G4—76% теоретич.) иодбензола с
т. кип. 77—78°/20 мм или 63—64°/8 мм (примечание 5).
2. Примечания
1. Если взять большее количество льда, часть его не растает
по окончании диазотирования.
2. При хорошем разделении слоев теряется с верхним слоем
не более 1—2 г иодбензола.
3. Твердый хлористый кальций удерживает довольно значи-
тельное количество иодбензола. Если применявшийся для сушки
хлористый кальций обработать водой, можно выделить 8—12 г
иодбензола.
4. Вес сырого иодбензола 350—355 г (80% теоретич.). Он доста-
точно чист для большинства целей без перегонки.
5. При слишком глубокой отгонке дестиллат будет окрашен.
3. Другие методы получения
В предыдущем сборнике г приведено получение иодбензола
иодированием бензола иодом и азотной кислотой, а также обзор
280
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ.
ЙОДИСТЫЙ МЕТИЛ
281
препаративных методов получения. Приведенная здесь методика,
основанная на методе Гаттермана 2, дает более чистый продукт.
1 «Сипт. орг. преп.», сб. 1, стр. 220.
2 Гаттермаи-Виланд, Практические работы по органической
химии, 5 изд., Госхимиздат, 1948, стр. 323.
3. В начале перегонки выделяются следы иода, и поэтому пер-
вая фракция обычно интенсивно окрашена. Если перегонку пре-
рвать, то по возобновлении ее вновь в первых каплях дестиллата
появляются следы иода. Почти бесцветный дестиллат получается
в том случае, если фракции отбирают, не прерывая перегонки.
ЙОДИСТЫЙ н.-ГЕКСАДЕЦИЛ
A-Иодгексадекан, цетил йодистый)
3 С16Н33ОН + PI,
3 C16H33I + H3PO3
Предложили: В. Хартман, Дж. Байере и Дж. Дикки.
Проверили: В. Каротерс и В. Мак-Ювен.
1. Получение
В 3-литровую круглодонную колбу помещают 242 г (I мол.)
цетилового спирта (примечание 1), Юг @,32 rp.-ат.) красного фос-
фора и 134 г A,06 rp.-ат.) дважды возогнанного иода и смесь нагре-
вают на масляной бане до тех пор, пока спирт не расплавится.
После этого колбу снабжают обратным холодильником и механи-
ческой мешалкой с жидкостным затвором и при работающей ме-
шалке смесь нагревают до 145—150° (температура масляной бани)
в течение 5 час. По охлаждении реакционной смеси йодистый це-
тил извлекают один раз 250 мл эфира и дважды порциями по 125 мл.
Соединенные эфирные вытяжки фильтруют, чтобы отделить фосфор,
и промывают сперва 500 мл холодной воды, затем 250 мл 5%-ного
раствора едкого натра и вновь 500 мл воды. Эфирный раствор сушат
безводным хлористым кальцием, эфир отгоняют на водяной бане
и йодистый цетил перегоняют в вакууме. Главную фракцию соби-
рают при 220—225°/22 мм или 210—215°/12 мм. Вес ее 300 г (85%
теоретич.) и т. пл. 18—20° (примечание 2). После вторичной пере-
гонки получают 275 г G8% теоретич.) вещества с т. кип. 220—
223722 мм или 203—205°/9 мм ист. пл. 20—22° (примечание 3).
2. Примечания
1. Цетиловый спирт получают согласно методике, приведен-
ной на стр. 307. Вполне приго'ден продукт с т. пл. 48—49°. При при-
менении цетилового спирта худшего качества выход может пони-
зиться до 70%.
2. Этот продукт, повидимому, является достаточно чистым для
большинства целей. В литературе имеются указания и на более
высокую температуру плавления, а именно 25Э.
3. Другие методы получения
Вышеописанный метод в основном заимствован у Смита 1. Це-
лый ряд исследователей пользовался аналогичным методом2. Йоди-
стый цетил был получен также нагреванием цетилового спирта
с желтым фосфором и иодом в сероуглеродном растворе 3; повтор-
ным пропусканием сухого йодистого водорода через расплавлен-
ный цетиловый спирт с некоторыми перерывами между отдельными
опытами 4, а также нагреванием цетилового спирта или цетилстеа-
рата с 55%-ной иодистоводородной кислотой при 120° в течение
2 час. 5.
1 S m i t h, J. Cliem. Soc. 1932, 738.
2 Fridau, Ann. 83, 9 A852); Levene, West, van der Scheer,
J. Biol. Chem. 20, 523 A915); D e 1 с о u r t, Bull. soc. chim. Belg. 40, 284 A931);
[C. A. 25, 5661 A931)].
3 G a s с а г d, Ann. chim. (9) 15, 372 A921).
4 К r a f f t, Ber. 19, 2219 A886).
'Guyer, Bieler, H a r d m e i e r, Helv.. Chim. Acta 20, 1466 A937).
ЙОДИСТЫЙ МЕТИЛ
(Подметан)
А. ИЗ МЕТИЛОВОГО СПИРТА
3 СН3ОН + PI, > 3 СН31 + Н3РО3
Предложил: Г. Кинг.
Проверил: Л. Физер.
1. Получение
Смесь метилового спирта и фосфора нагревают до кипения в
колбе прибора, изображенного на рис. 10. Пары конденсируются
в холодильнике, а стекающая жидкость растворяет иод и поступает
обратно в реакционную колбу. Прибор, в котором ведется реакция
(рис. 10), может быть сделан более удобным, если взять трехгорлую
реакционную колбу (рис. 11); дальнейшим усовершенствованием
является боковая трубка в цилиндрической делительной воронке
А, через которую можно загружать иод (примечание 1). Описывае-
мый синтез был проведен с более простым прибором, изображен-
ным на рис. 10; изменения, которые приходится ввести при работе
282
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
ЙОДИСТЫЙ МЕТИЛ
283
Ж
с усовершенствованным прибором, будут ясны из дальнейшего
описания.
В цилиндрическую делительную воронку А емкостью 1,2 л
помещают 2кг A5,75гр,-ат.)кристаллического иода (примечание 2).
Для того чтобы кран не забивался кристаллами иода и твердыми
примесями, на дно воронки кладут
кусок перфорированной платиновой
фольги Б или неплотный слой стек-
лянной ваты. По той же причине
диаметр внутреннего хода нижнего
крана не должен быть менее 5 мм.
Для получения до 4 кг йодистого ме-
тила применяют 5-литровую кругло-
донную колбу из стекла «пирекс»
(примечание 3). Частично колбу по-
гружают в водяную баню. Внутренний
диаметр трубки Д, которая соединена
с реакционной колбой посредством
резиновой пробки, должен быть не
менее 2,5 см, чтобы пары могли сво-
бодно подниматься по ней, не мешая
стекать избытку сконденсировавшейся
жидкости. Трубка Е имеет А см в диа-
метре, для того чтобы предваритель-
ная конденсация могла происходить
уже в ней. Над краном Ж имеется в
качестве резервуара для дестиллата
пространство высотою до отводной
трубки Д 13 см. Диаметр внутрен-
него хода крана Ж — 2 или 3 мм
(примечание 4). Конец трубки этого
крана должен быть вровень с нижней
стороной резиновой пробки, закры-
вающей сосуд с иодом; жидкость в
этом случае стекает но стенкам сосу-
да, а не образует хода через середину
иода. Холодильник 3 B,5 см в диа-
метре и высотою не менее 200 см) присоединен при помощи ре-
зиновой пробки и охлаждается сильной струей воды (примеча-
ния 5 и б).
,В сосуд А помещают 2 кг иода, причем следят за тем, чтобы кри-
сталлы не попали за платиновый листочек или стеклянную вату.
Иод заливают частью отмеренных для синтеза 2 л E0 мол.) метило-
вого спирта (примечание 7). Остальной спирт наливают в реакцион-
ную колбу Г, в которую предварительно помещают 200 г красного
и 200 г желтого фосфора A2,8 гр.-ат.). Прибор соединяют так, как
Рис. 10
Рис. 11
это показано на рис. 10. Воду в бане нагревают до 70—75°; кран
Ж открывают полностью, а нижний кран — частично, так чтобы
раствор иода медленно поступал в реакционную колбу. Когда весь
спирт будет прибавлен в Г, образуется уже достаточное для начала
кипения количество йодистого метила. Теперь нижний кран откры-
вают полностью, а кран Ж прикрывают так, чтобы в сосуд с иодом
стекало небольшое количество сконденсировавшегося йодистого
метила. Температуру в водяной бане устанавливают таким обра-
зом, чтобы по трубке Д стекало обратно только небольшое количе-
ство жидкости. По этой причине по мере хода реакции температуру
бани приходится снижать (примечание 8), так как после приба-
вления первой части иода температура в 55° достаточна для под-
держания равномерного кипения. Если почему-либо кипение ста-
новится слишком сильным, оба крана закрывают. Когда реакция
несколько успокоится, частично открывают сперва кран Ж, а затем
нижний кран. Когда же из А вытечет вся находящаяся в ней жид-
кость, кран В снова открывают полностью. Когда весь иод раство-
рится (около 2,5 час.) оба крана закрывают, делительную воронку
опускают вращательным движением, загружают еще 2 кг иода
A5,75 гр.-ат.) и приводят воронку в прежнее положение. Ниж-
ний кран открывают полностью, как и раньше, а кран Ж — ча-
стично.
После растворения второй порции иода содержимое колбы
охлаждают, снимают экстракционный аппарат и колбу соединяют
с обращенным вниз холодильником. Нижний конец холодильника
плотно соединяют с алонжем, опущенным в смесь воды со льдом.
После того как весь йодистый метил (т. кип. 40—42,5°) отогнан
(примечание 9), его отделяют от воды, сушат взбалтыванием с без-
водным хлористым кальцием, фильтруют через стеклянную вату
и сохраняют для дальнейшего употребления в запаянных ампулах
или пробирках в темном месте (примечание 10). Выход 4150—4250 г
(93—95% теоретич., считая на иод). Полученный продукт бесцве-
тен и достаточно чист для большинства целей. Вторичная перегонка
дает продукт с выходом 95%, который кипит при 41—42°. Так
как упругость паров йодистого метила при комнатной температуре
очень высока, то во избежание потерь материала необходимо при-
нимать особые меры предосторожности. Два способа, дающие удо-
влетворительные результаты, указаны в примечаниях 11 и 12.
Прибавление капли ртути к очищенному йодистому метилу
сильно замедляет его постепенное потемнение; старые образцы могут
быть также освобождены от следов иода встряхиванием со ртутью
(примечание 13). Сильно окрашенный продукт лучше всего промыть
сначала очень слабым раствором тиосульфата натрия, затем водой
и высушить.
С небольшими изменениями описанный метод пригоден для
получения с выходами выше 90% этил-, н.-пропил-, н.-бутил-, изо-
284
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
ИОДИСТЫЙ^МЕТИЛ
285
бутил-, н.-амил- и изоамилиодидов. Вторичный бутилиодид полу-
чается с несколько худшим выходом вследствие отщепления йоди-
стого водорода.
2. Примечания
1. Верхнее отверстие воронки А (рис. 11) должно быть такого
же размера, как и отверстие трубки Е, так чтобы воронка могла
служить приемником во время описываемой ниже перегонки (при-
мечание 12).
2. Указанные объем и форма воронки не обязательны, но были
найдены наиболее удобными. Даже если работать с шарообразной
делительной воронкой, стекающая жидкость все же смачивает ее
стенки.
3. Для больших количеств можно брать колбу емкостью до 12 л.
4. Для этого годится кран от маленькой разбитой делительной
воронки.
5. В жаркую погоду обычно происходит потеря некоторого
количества йодистого метила через холодильник. Испаряющийся
йодистый метил можно уловить, если отвести газы, выделяющиеся
из верхнего конца холодильника, под поверхность смеси воды со
льдом. Для этой цели удобна пипетка на 50 мл, верхней трубке
которой придают U-образную форму и соединяют с концом холо-
дильника, а нижнюю погружают в смесь воды со льдом.
6. При проверке синтеза вместо длинного холодильника был
взят измененный холодильник Уэста с вдавленными неправиль-
ными зубцами (рис. 11). Длина его муфты — 90 см, внутренний
диаметр внутренней трубки 2,2 см, внутренний диаметр верхнего
отверстия 3,3 см. К верху этого холодильника присоединяют
шариковый холодильник высотой 30 см, который можно охлаждать
ледяной водой. При работе с водопроводной водой, температура
которой равнялась 5°, было легко достичь полной конденсации
и не приходилось пользоваться ловушкой, описанной в примеча-
нии 5. В жаркую погоду, по всей вероятности, пришлось бы
воду для холодильника предварительно пропускать через большие
банки со льдом.
7. Метанол можно брать не абсолютный. В одном из опытов
он был разбавлен 10% воды (по весу). Выход йодистого метила
в этом опыте был тот же, что и в проверочном опыте с абсолют-
ным метанолом.
Кипение начинается спокойнее, если в колбу предварительно
поступает раствор иода. В этом случае сразу образуется количество
йодистого метила достаточное для того, чтобы кипение началось
при сравнительно низкой температуре.
8. Увеличение скорости реакции зависит главным образом от
того, что иод легче растворим в йодистом метиле, чем в метаноле,
и поэтому по мере хода реакции скорость поступления иода
в реакционную колбу увеличивается. Реакция идет с выделе-
нием тепла.
9. Имеются указания на то, что этот дестиллат представляет
собою, вероятно, постоянно кипящую смесь йодистого метила с
метиловым спиртом. Смесь эта состоит из 93% (по весу) йодистого
метила и кипит при 39°. Для выделения чистого йодистого метила
вполне пригодна приведенная выше методика отделения его от
ледяной воды (Р. Динерштейн, частное сообщение).
10. Остаток в реакционной колбе содержит желтый фосфор и
потому с ним следует обращаться с осторожностью. Необходимо
немедленно остатки заливать водой.
11. К концу внутренней трубки холодильника припаивают конец
стеклянного змеевика (диаметр 2 мм). Другой конец змеевика при-
паивают к трубке, которая вставлена в пробку, закрывающую при-
емник; для предохранения от влаги воздуха в эту же пробку встав-
лен бунзеновский затвор; как змеевик, так и приемник охлаждают
льдом с солью или твердой углекислотой.
12. Удобное приспособление для перегонки получают также
при работе с холодильником, изображенным на рис. 11, и делитель-
ной воронкой А. Йодистый метил отгоняют из 3-литровой колбы,
причем холодильник устанавливают в вертикальном положении
и соединяют с колбой посредством перевернутой U-образной трубки
B2 мм). Делительная воронка служит приемником и позволяет
легко собирать отдельные фракции. Боковое отверстие делительной
воронки соединяют с небольшим холодильником, закрытым хлор-
кальциевой трубкой. Простой и хороший способ для определения
температуры кипения состоит в том, что в перевернутую U-образ-
ную трубку вставляют длинный термометр; шарик его упирается
в небольшую пробку, помещенную на дне колбы.
И, 13. Предостережение. Йодистый метил при соприкосновении
со ртутью на солнечном свету очень легко образует йодистую
метил-ртуть, которая чрезвычайно ядовита.
Б. ИЗ ДИМЕТИЛСУЛЬФАТА
KI + (CH3JSO4 ---- СН31 + CH3SO4K
Предложил: В. Хартман.
Проверили: Дж. Джонсон и Х.Пассино.
1. Получение
Реакция проводится в 3-литровой трехгорлой колбе, снабжен-
ной термометром, механической мешалкой с жидкостным затвором
и небольшим дефлегматором, присоединенным к обращенному вниз
холодильнику. Сосуд, который служит в качестве приемника,
охлаждают ледяной водой (примечание 1). В реакционную колбу
286
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
2-ЙОДТИОфЕН
28?
помещают раствор 800 г D,8 мол.) фармакопейного йодистого калия
в 430 мл воды, добавляют 60 г углекислого кальция и при работаю-
щей мешалке смесь нагревают до 60—65°.
Поддерживая температуру 60—65°, постепенно через делитель-
ную воронку приливают 630 г D73 мл, 5 мол.) «практического»
(см. сноску на стр. 26) диметилсульфата (примечание 2). Скорость
приливания должна быть такой, чтобы быстро отгонялся йодистый
метил, причем вся операция прибавления занимает около 2 час.
После того, как прибавлено все количество диметилсульфата,
температуру повышают до 65—70° и поддерживают ее 40 мин. для
того, чтобы завершить перегонку-'иодистого метила. Продукт отде-
ляют от небольшого количества воды, тщательно сушат над безвод-
ным хлористым кальцием и сливают в сухую перегонную колбу.
Добавляют несколько кристаллов йодистого калия и продукт
перегоняют на водяной бане. Выход йодистого метила с т. кип.
41—43° составляет 615—640 г (90—94% теоретич.; примечание 3).
2. Примечания
1. Весьма важно предотвратить потери легко летучего йоди-
стого метила. По этому поводу см. примечания в методике (А).
2. Диметилсульфат очень ядовит. Следует избегать попадания
жидкости на кожу или вдыхание паров. Специфическим противо-
ядием является аммиак.
3. Такой же процентный выход может быть получен и при
загрузках в 10 раз больше приведенных выше.
3. Другие методы получения
Йодистый метил был получен действием диметилсульфата на
водный раствор йодистого калия 1; медленной перегонкой смеси
метилового спирта с большим избытком иодистоводородной кислоты
с постоянной температурой кипения 2; электролизом водного рас-
твора уксуснокислого калия в присутствии иода или йодистого
калия 3; действием водного раствора йодистого калия на метиловый
эфир я-толуолсульфокислоты4; и действием метилового спирта
на раствор пятииодистого фосфора в йодистом метиле 5. Наиболее
широко применяемым методом получения йодистого метила являет-
ся взаимодействие метилового спирта с трехиодистым фосфором
(или со смесью иода и фосфора, безразлично красного или желтого
или смесью обоих) 6. Были предложены различные видоизменения
этого метода, причем предлагались также некоторые варианты для
получения высших йодистых алкилов, которые вполне применимы
для получения йодистого метила 7.
Методика, описанная в (А), является видоизменением метода
Уокера 8. Метод постепенного введения в реакционную колбу рас-
творимого компонента, описанный выше, может быть применен и
к другим реакциям, например, к получению различных диалкиль-
ных производных ртути 9.
При работе по методике (Б) в качестве иодсодержащего исход-
ного вещества с успехом может быть использован йодоформ, если
его предварительно нагреть со спиртовой щелочью 10.
1Weinland, Schmid, Ber. 38, 2327 A905); герм. пат. 175 209
[Frdl. 8, 17 A905—1907I.
3 N о г г i s, Am. Chem. J. 38, 639 A907).
8Kaufler, Herzog, Ber. 42, 3860 A909).
4 P e а с о с k, Menon, Quart. J. Indian Chem. Soc. 2, 240 A925);
Родионов, Bull. soc. chim. D) 39, 323 A926).
5 Walker, Johnson, J. Chem. Soc. 87, 1595 A905).
«Dumas, Peligot, Ann. 15, 20 A835); ЖРХО 27, I, 364 A895)
[Ber. 29, (R) 90 A896)]; J. prakt. Chem. B) 53, 275 A896); С г i s m e r, Ber.
17, 652 A884); Walker, Johnson, J. Chem. Soc. 87, 1592 A905); Hay-
wood, там же 121, 1911 A922).
' Hunt, там же 117, 1592 A920); В о g e r t, S 1 о с u m, J. Am. Chem.
Soc. 46, 763 A924).
8 W a 1 k e r, J. Chem. Soc. 61, 717 A892); N a g a i, J. Pharm. Soc. Japan
1916, No 407, 1;[C. A. 10,987 A916)]; Adams, Voorhees, J. Am. Chem.
Soc. 41, 796 A919); Adams, Kamm, Marvel, Organic Chemical Rea-
gents 1, Univ. Illinois Bull. 16, D3) 19 A919); Reynolds, Adkins,
J. Am. Chem. Soc. 51, 280 A929); King, Proc. Trans. Nova Scotian Inst.
Sci. 16, B) 87 A924).
8 G i 1 m a n, Brown, J. Am. Chem. Soc. 51, 928 A929).
10 К i m b a 1 1, J. Chem. Education 10, 747 A933).
НС CH
2-ИОДТИОФЕН
HC CH
2 о,
+ 2 I2 + HgO > 2
,CH
Hgla
НСЧ XI
Предложил: В. Миннис.
Проверили: Р. Адаме и X. Коган.
1. Получение
В широкогорлую склянку с притертой пробкой, охлаждаемую
водой со льдом, помещают 35г @,42 мол.) тиофена (стр. 458) и 50 мл
бензола (примечание 1). Постоянно взбалтывая эту смесь, а если
надо, то и охлаждая, к ней прибавляют в продолжение 15—20 мин.
попеременно и небольшими порциями 75 г @,35 мол.) желтой окиси
ртути и 109 г @,43 мол.) иода. Желтая окись ртути постепенно
переходит в красную йодную ртуть. Осадок отфильтровывают и
промывают три раза эфиром, порциями по 25 мл. Эфирно-бензоль-
ный раствор для удаления избытка иода взбалтывают с разбавлен-
ным раствором тиосульфата, затем сушат 5 г хлористого кальция
и фильтруют. Эфир и бензол отгоняют на водяной бане (примеча-
288
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
Л-ИОДФЕНОЛ
289
ние 3), а остаток фракционируют в вакууме. 2-Иодтиофен перего-
няется при 73715 мм, 80—81720 мм и 90—947 34—38 мм (примеча-
ние 4). Выход 63—66 г G2—75% теоретич.; примечание 5). Если иод-
тиофен все еще окрашен следами иода, окраску можно уничтожить
встряхиванием с небольшим количеством окиси ртути.
2. Примечания
1. Бензол можно заменить нефтяной фракцией (т. кип. 100—120°).
2. При энергичном встряхивании от руки выходы получаются
лучше, чем при перемешивании механической мешалкой. Обыч-
ная механическая мешалка не в состоянии поддерживать окись
ртути во взвешенном состоянии.
3. Непрореагировавший тиофен может быть получен обратно
из эфирно-бензольного отгона, если его обработать окисью ртути
и разбавленной уксусной кислотой, затем отсосать выпавший
белый осадок JC4H2S(Hg0C0CH3)Hg0H] на бюхнеровскои воронке
и разложить его концентрированной соляной кислотой 1. В прове-
рочном опыте непрореагировавшего тиофена было слишком мало
для того, чтобы его имело смысл регенерировать.
4. При этой реакции образуется небольшое количество 2,5-ди-
иодтиофена. Можно выделить около 4 г кристаллического дииод-
тиофена, с т. пл. 40—41°, из остатка после отгона 2-иодтиофена
(т. кип. 65,5—66,579 мм; О. И. Ли, частное сообщение).
5. Иодтиофен реагирует с магнием, давая гриньяров реактив,
и потому может быть употреблен для получения других производ-
ных тиофека.
3. Другие методы получения
Иодтиофен был получен только действием иода и окиси ртути
на тиофен 2.
1 D i m r о t h, Ber. 32, 759 A899).
2 Meyer, К r e 1 s, там же 17, 1558 A884); Т h у s s e n, J. prakt. Chem.
B) 65, 5 A902).
гс-ИОДФЕНОЛ
2rc-HOC6H4NH2 + 3H2SO4 + 2NaNO2 >
2 iz-HOC,H4N3SO4H + Na2SO4 + 4~H3O
r-HOC6H4N3SO4H + KI —— rc-HOC6H4I + N3 + KHSO4
Предложили: Ф. Дэйис и Ф. Эберли.
Проверили: Р. Фьюзон и X. Хуллп.
1. Получение
109 гA мол.) л-аминофенола (примечание 1) растворяют в смеси
500 г льда, 500 мл воды пЬЬ мл(\20 г, 1,2 мол.) концентрирован-
ной серной кислоты (уд. веса 1,84). Раствор этот охлаждают холо-
дильной смесью до 0° и в течение 1 часа добавляют к нему при не-
прерывном помешивании раствор 72 г A мол.) 95%-ного азотисто-
кислого натрия в 150 мл воды. Перемешивание продолжают еще
20 мин., после чего .добавляют 20 мл C7 г, 0,37 мол.) концентриро-
ванной серной кислоты.
Реакционную смесь выливают в охлажденный до 0° раствор
200 г A,2 мол.) йодистого калия в 200 мл воды. Через несколько
минут добавляют 1 г медной бронзы (примечание 2) при непрерыв-
ном перемешивании и раствор медленно нагревают на водяной
бане. Температуру поддерживают при 75—80° до тех пор, пока
не прекратится выделение азота. Иодфенол при этом выделяется
в виде тяжелого темноокрашенного масла. По охлаждении до ком-
натной температуры реакционную смесь извлекают три раза пор-
циями по 165 мл хлороформа и соединенные вытяжки промывают
разбавленным раствором тиосульфата. Растворитель отгоняют на
водяном бане, а остаток перегоняют в вакууме, причем л-иодфе-
нол собирают при 138—140°/5 мм. Однократная перекристаллиза-
ция из 2 л нефтяной фракции (т. кип. 90—110°) дает бесцветный
продукт с резкой температурой плавления 94°. Выход продукта
после перекристаллизации 153—159 г F9—72% теоретич.).
2. Примечания
1. Применялся продажный я-аминофенол, который плавился
при 182—183° с разложением.
2. Некоторые образцы продажной бронзы, применяемые для
бронзовых красок, покрыты пленкой из стеариновой кислоты. Для
химических операций следует брать медную бронзу, которая не
подвергалась предварительной обработке такого рода.
3. Другие методы получения
«-Иодфенол впервые был получен в качестве побочного продукта
при действии иода на салициловую кислоту в щелочном растворе
или же нагреванием иодсалициловой кислоты 1. Он был получен
также действием иода на фенол в щелочном растворе 2 или в присут-
ствии окиси ртути 3, а также действием однохлористого иода 4. Лучше
всего он получается диазотированием «-аминофенола с последую-
щей заменой группы диазония на иод 5, хотя он был получен также
и из л-иоданилина диазотированием с последующей заменой
группы диазония на гидроксил 6.
1 Lautemann, Ann. 120, 299 A861); К е k u 1 ё, там же 131, 221
A864).
3 Н о 1 1 е m a n, R i n k e s, Rec. trav. chim. 30, 96 A911).
3 H 1 a s i w e t z, Weselsky, Ber. 2, 523 A869).
t9 Сборник № 2
290
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
«Schiitzenberger, Sengenwald, Jahresb. 1862, 413.
6 N б 1 t i n g, Wrzcsinski, Ber. 8, 820 A875); Nolting, Stri-
eker, там же 20, 3021 A887); Neumann, Ann. 241, 74 A887).
• G r i e s s, Zeit. fur Chem. 1865, 427; Holleman, Rinkes, Rec.
trav. chim. 30, 95 A911).
ИТАКОНОВЫЙ АНГИДРИД И ИТАКОНОВАЯ КИСЛОТА
СН2 С (ОН) — СН2
СО2Н СО2Н СО,Н
сн, = с сн„
сн2 = с сн3
О = С\ /С = О
хох
сн, = с -
2 Н2О + СО3
сн.
н2о
SO'
СО2Н СО,Н
Предложили: Р. Шрайнер, С. Форд и Л. Ролл.
Проверил: К- Ноллер.
1. Получение
А. Итаконовый ангидрид. Колбу Кьельдаля из стекла «пирекс»
соединяют с помощью изогнутой стеклянной трубки, диаметром
12 мм, с обращенным вниз для отгонки водяным холодильником
длиною 100 см; внутренняя трубка холодильника, также из
стекла пирекс, имеет местами вдавленности (примечание 1). В каче-
стве приемников берут две колбы Вюрца с длинными горлами
емкостью по 250 мл, соединенные последовательно. Пары поступают
в центр каждой колбы, проходя в обоих случаях через алонж, и
которому присоединена стеклянная трубка. Во время перегоню
приемники охлаждают ледяной водой.
200 г @,95 мол.) кристаллической лимонной кислоты, содер-
жащей 1 мол. воды, помещают в реакционную колбу (примечание 2]
и нагревают ее на голом пламени, пока кислота не расплавится
После этого колбу очень быстро нагревают большой горелкой Me-
кера, стремясь окончить перегонку как можно быстрее A0—12 мин.)
но тщательно избегая перегрева (примечание 3). Дестиллат состоит
из воды и итаконового ангидрида, большая часть которого перего-
няется при 175—190°. Весь отгон немедленно переносят в делитель-
ную воронку, и отделяют нижний слой итаконового ангидриде
(примечание 4). Выход ангидрида 40—50 г C7—47% теоретич.)
Продукт достаточно чист для получения цитраконового ангидриде
(стр. 570, примечания 5 и 6).
Б. Итаконовая кислота. 40 г итаконового ангидрида кипятяг
с обратным холодильником со 100 мл воды в течение 1 часа. Потол
раствору дают охладиться сперва до комнатной температуры, а зател
ИТАКОНОВЫЙ АНГИДРИД И ИТАКОНОВАЯ КИСЛОТА
291
его охлаждают в ледяной воде. Выпавшие кристаллы итаконовой
кислоты отсасывают и сушат. Выход 11—18 г B4—39% теоретич.)
продукта с температурой плавления 162—165°. Концентрированием
маточного раствора до одной трети первоначального объема можно
получить еще некоторое количество кислоты с более низкой темпе-
ратурой плавления (примечания 7 и 8).
2. Примечания
1. Трубка из простого стекла легко может лопнуть при быстрой
перегонке. Рекомендуется взять трубку из стекла «пирекс», раз-
мягчить отдельные места ее пламенем паяльной горелки и, применяя
вакуум, несколько вдавить их внутрь.
2. Для каждой порции лимонной кислоты необходимо брать
чистую колбу, так как в присутствии остатка от предыдущей опера-
ции в начале перегонки происходит сильное вспенивание. Легче
всего вымыть колбу, влив в нее 25%-ный раствор едкого натра, пока
остаток от перегонки еще не затвердел. Последующее нагревание
вызывает полное растворение массы.
3. Перегревание увеличивает превращение в цитраконовый
ангидрид. Колбу следует нагревать со всех сторон так, чтобы пла-
мя охватывало значительную поверхность. Перегонку следует
прекратить, как только в реакционной колбе появятся желтые пары.
4. Быстрое отделение ангидрида от водного слоя сводит к ми-
нимуму гидролиз ангидрида. Упариванием водного слоя можно
получить смесь итаконовой и цитраконовой кислот.
• ' 5. Чистота получаемого итаконового ангидрида, повидимому,
сильно зависит от условий реакции. Из сырого продукта при стоя-
нии всегда выпадают кристаллы итаконовой кислоты, вероятно,
вследствие гидролиза ангидрида растворенной или взвешенной
в нем водой. О чистоте ангидрида можно судить до известной сте-
пени по количеству полученной из него итаконовой кислоты. Если
перегонку вести с указанной скоростью, то получается чистый
ангидрид с температурой плавления 67—68°.
6. Если требуется большее количество итаконового ангидрида,
лучше провести несколько опытов с порциями по 200 г лимонной
кислоты, а не подвергать разложению сразу большую порцию
кислоты. Обычно с увеличением загрузки выход итаконового
ангидрида падает.
7. Иногда итаконовая кислота не кристаллизуется. Это проис-
ходит, повидимому, в тех случаях, когда перегонку лимонной
кислоты вели недостаточно быстро, в результате чего итаконовый
ангидрид содержит большое количество цитраконового ангидрида.
8. Для получения значительных количеств итаконовой кислоты
удобнее воспользоваться методикой, приведенной ниже, которая
дает значительно больший выход. Быстро (за 4—б мин.) перегоняют
девять порций по 120 г лимонной кислоты из колб Кьельдаля
292
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
емкостью 300 мл и все дестиллаты собирают в один и тот же прием-
ник. Дестиллат, который обычно не состоит из двух слоев, пере-
ливают в чашку для выпаривания, добавляют 50 мл воды и смесь
держат 3 часа на кипящей водяной бане. По охлаждении продукт
застывает в полутвердую массу, которую отфильтровывают и про-
мывают 150 мл воды. Остаток представляет собою совершенно белые
кристаллы с температурой плавления 165°. Общий вес кристаллов
составляет 138 г, а упариванием фильтрата можно получить еще
42 г продукта с т. пл. 157—165°. Суммарный выход 26—27% тео-
ретич. (К. Уильсон и К' Аллен, частное сообщение).
3. Другие методы получения
Итаконовый ангидрид был получен нагреванием итаконовой
Кислоты и перегонкой лимонной кислоты г.
Итаконовая кислота была получена перегонкой лимонной кис-
лоты 2, аконитовой кислоты3 и итамалевой кислоты4; нагрева-
нием лимонной кислоты с разбавленной серной кислотой в запаян-
ных трубках 5; действием воды на аконитовую кислоту при 180°;
нагреванием цитраконовой кислоты с едким натром 7; нагреванием
цитраконового ангидрида с водой при 150° 8; нагреванием концен-
трированного раствора цитраконовой кислоты при 120—130° в
запаянных трубках9 и действием грибка Aspergillus itaconicus
на тростниковый сахар 10.
Смесь цитраконовой и итаконовой кислот получается, если
прилить концентрированный водный раствор лимонной кислоты
в нагретый эвакуированный сосуд, перегнать в вакууме смесь
образовавшихся ангидридов и дать смеси прореагировать с водой и.
1 Anschiitz, Ber. 13, 1541 A880).
2 В а и р, Ann. 19, 29 A836).
8 Crass о, там же 34, 63 A840).
4 Schwartz, Zeit. fur Chem. 1867, 649.
5 M a p к о в н и к о в, П у р г о л д. там же 1867, 264.
6 Р е b a I, Ann. 98, 94 A856).
' D е 1 i s I e, там же 269, 86 A892).
8 F i 11 i g, L a n d о 1 t, там же 188, 72 A877).
8 W i 1 m, там же 141, 29 A867).
10 Kinoshita, Acta Phytochim. 5, 271 A931) [C. A. 26, 966 A932)].
11 Boehringer Sohn A.-G.; анг. пат. 452 460 [С. А. 31, 1045, A937)].
КАЗЕИН
Предложили: Е. Кон и Дж. Хендри.
Проверили: X. Кларк и В. Кеннан.
1. Получение
К 1 л молока, с которого почти полностью сняты сливки (приме-
чание 1), прибавляют медленно и при перемешивании 0,05 м. соля-
КАЗЕИН
293
ную кислоту через капиллярную трубку, доходящую до дна сосуда.
Соляную кислоту прибавляют до тех пор, пока рН раствора не
достигнет 4,6 (примечание 2). Чтобы установить этот момент, берут
5 мл раствора, разбавляют до 50 мл, прибавляют метилрот и сра-
внивают окраску с эталоном ряда буферных растворов (примеча-
ние 3). Требуется около 1000 мл кислоты; после прибавления
этого количества отделение казеина практически закончено. Тогда
прибавляют 3 л воды, прекращают перемешивание и ставят смесь
в холодильный шкаф, чтобы дать осесть хлопьевидному осадку ка-
зеина, на что требуется от 12 до 24 час. Прозрачный верхний слой,
содержащий растворимые белки и соли, удаляется сифонированием
возможно более тщательно; осадок переносят на бюхнеровскую
воронку и промывают холодной дестиллированной водой до тех
пор, пока промывная вода не будет свободна от кальция (проба с
щавелевокислым аммонием).
Казеин, загрязненный фосфорнокислым кальцием и жирами,
отжимают до наименьшего объема (около 500 мл) и помещают в
двухлитровый стакан. Затем его обрабатывают 0,1 м. едким натром,
причем щелочь прибавляют медленно через капилляр, который
доходит до дна сосуда (примечание 4) и при постоянном помеши-
вании. Щелочь прибавляют до тех пор, пока рН смеси не достигнет
6,3 (примечание 5); количество требующейся щелочи — 100—•
150 мл. Момент окончания прибавления устанавливают сравнением
окраски пробы раствора с окраской эталонов буферного ряда (при-
мечание 6) в присутствии индикатора дибром-о-крезолсульфофта-
леина («бромкрезолпурпур»). При таком рН казеин полностью пере-
ходит в раствор в виде натриевой соли; жиры, фосфорнокислый
кальций и казеиновокислый кальций остаются нерастворенными.
Следует тщательно следить за тем, чтобы не прибавить щелочи
больше, чем нужно для того, чтобы рН достигло указанной вели-
чины (примечание 4). Молочный раствор фильтруют через толстый
слой A0—15 мм) фильтровальной бумажной массы, плотно
уложенной в воронке для отсасывания. Фильтрат может
слегка опалесцировать; если он недостаточно прозрачен, то его
снова фильтруют через свежий слой фильтровальной бумажной
массы.
К фильтрату прибавляют 0,05 м. соляную кислоту до тех пор,
пока его рН не достигнет 4,6. Обычно расходуется от 220 до 250 мл
0,05 м. кислоты. Количество кислоты, необходимое для второго
осаждения, определяют так же, как и при первом осаждении,
титруя аликвотную часть раствора, разбавленную в 5 раз, 0,01 м.
соляной кислотой до окраски соответственного эталона буферного
ряда. По мере осаждения казеина уменьшают скорость прибавле-
ния кислоты для того, чтобы предупредить забивание выпадающим
осадком конца капиллярной трубки; энергичное механическое
помешивание совершенно необходимо. После того, как обработка
294
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
КАПРОНОВЫЙ АЛЬДЕГИД
295
кислотой закончена, прибавляют 5 л холодной дестиллированной
воды, смесь ставят в ледяной шкаф и дают хлопьевидному осадку
осесть. Прозрачный раствор сливают с осадка при помощи сифона,
казеин переносят на воронку для отсасывания с плотной бумагой,
промывают его холодной дестиллированной водой до полного уда-
ления хлоридов, отжимают по возможности досуха и сушат над
хлористым кальцием в вакуум-эксикаторе. Выход — 23—29 г
бесцветного плотного продукта, который можно легко измельчить
в ступке.
2. Примечания
1. Для того, чтобы удалить сливки, достаточно оставить молоко
стоять ночь в холодильном шкафу, а затем отделить нижний слой
при помощи сифона.
2. Казеин содержится в молоке в виде кальциевой соли; рН
4,6 — изоэлектрическая точка свободного казеина, который рас-
творим в воде в количестве только 0,11 г на литр1.
3. Для этого ряда можно приготовить следующие буферные
растворы:
0,1 м. уксусная
кислота
7,35 мл
6,3 »
5,1 „
4,0 „
2,95 „
0,1 м. уксуснокислый
натрии
2,65 мл
3,7 „
4,9 „
6,0 „
7,05 „
рН
4,2
4,4
4,6
4,8
5,0
4. Необходимо избегать местного избытка щелочи, так как это
может денатурировать казеин а.
5. При этом рН казеиновокислый натрий переходит почти пол-
ностью в раствор, тогда как казеиновокислый кальций остается
в основном нерастворенным 3.
6. Удобно пользоваться следующим буферным рядом:
Vis M- динатрий-
фосфат
0,78 мл
1,2 .
1,85 ,
2,65 ,
3,75 ,
5,0 „
Vis M- монокалий-
фосфат
9,22 мл
8,8 ,
8,15 ,
7,35 ,
6,25 „
5,0 „
РН
5,8
6,0
6,2
6,4
6,6
6,8
3. Другие методы получения
Все методы получения основаны на осаждении казеина из мо-
лока прибавлением той или иной кислоты. Тем не менее они зна-
чительно разнятся в отношении способов дальнейшей очистки.
По способу Хаммарстена * точно прибавляется такое количество
щелочи, которое растворяет казеин полностью. При этом способе
щелочность раствора оказывает некоторое влияние на физические
свойства получаемого казеина, но, повидимому, не влияет на его
состав. При работе по способу Ван-Сляйка и Босворта 5 последние
следы кальция удаляются прибавлением соли щавелевой кислоты
к аммиачному раствору казеина. Однако Ван-Сляйк и Бэкер дока-
зали, что такая обработка является лишней 6.
Описанный выше метод в большей своей части основан на спо-
собе Ван-Сляйка и Бэкера. Внесенные изменения обоснованы тем
фактом, что в нейтральных растворах казеин образует значительно
более растворимые соединения с одновалентными основаниями,
чем с двухвалентными.
1 Cohn, I. Gen. Physiol. 4, 697 A922).
2 С о h n, Hendry, там же 5, 521 A923).
3 L о с b, там же 3, 547 A920—21).
4 Hammarsten, Textbook of Physiological Chemistry, перевод 7 изд.,
стр. 619, John Wiley & Sons, 1911.
6 Van Slyke, Bosworth, J. Biol. Chem. 14, 211 A913).
6 V a n Slyke, Baker, там же 35, 127 A918).
КАПРОНОВЫЙ АЛЬДЕГИД
(н-Гексальдегид)
CsHnMgBr + СН (OC2He)s . СВНЦСН (ОС2Н5J + C.2H5OMgBr
С5НПСН (ОС2Н5)а + Н2О 1^-1 СНцСНО + 2 С,Н3ОН
Предложил: Дне. Бахман.
Проверили: К- Ноллер и В. Вун.
1. Получение
В 2-литровую трехгорлую круглодонную колбу, снабженную ме-
ханической мешалкой с жидкостным затвором, капельной воронкой
емкостью 250 мл и обратным холодильником, снабженным хлоркаль-
циевой трубкой, помещают ЗОг A,25 гр, -ат.) магниевых стружек, 50 мл
абсолютного эфира и небольшой кристаллик иода. Пускают в ход
мешалку и добавляют 5 мл F г) бромистого н.-амила (примечание 1).
Как только начнется реакция, добавляют 300 мл абсолютного эфира
и затем медленно приливают раствор 183 г (вместе с раньше при-
бавленными 189 г, т. е. 1,25 мол.) бромистого н.-амила в 150 мл
296
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
2-КАРБЭТОКСИЦИКЛОПЕНТАНОН
297
абсолютного -эфира. Если обеспечено наружное охлаждение, то все
количество галоидалкила может быть прибавлено в течение 30 мин.,
после чего раствор осторожно кипятят еще 30 мин. для завершения
реакции. Удалив источник нагревания, колбу охлаждают до 50°
и в течение 15—20 мин. добавляют 148 г A мол.) ортомуравьиного
эфира («Синт. орг. преп.», сб. 1, стр. 554). Смесь кипятят в течение
б час. (примечание 2); к концу этого времени обратный холодиль-
ник заменяют на нисходящий и эфир полностью отгоняют на паро-
вой бане.
Реакционную смесь охлаждают и осторожно прибавляют 750 мл
хорошо охлажденной 6%-ной соляной кислоты. Чтобы содер-
жимое колбы оставалось холодным, вместе с кислотой добавляют
время от времени в колбу лед. Как только все перейдет в раствор
(примечание 3), отделяют верхний маслянистый слой, содержащий
ацеталь н.-гексальдегида. Ацеталь омыляют, подвергая его пере-
гонке с раствором 100 г E5 мл) концентрированной серной кислоты
в 700 мл воды. Свободный альдегид отгоняется быстро, и перегонку
можно считать законченной, когда проба свежего дестиллата со-
держит не более 5% не смешивающегося с водой масла. Дестиллат
собирают в раствор 100 г A мол.) бисульфита натрия в 300 мл воды.
Смесь энергично взбалтывают в течение нескольких минут; масля-
нистый слой, не растворившийся в растворе бисульфита, в основ-
ном представляет собой н.-амиловый спирт, который отбрасывают.
Чтобы удалить последние следы н.-амилового спирта и других
примесей, бисульфитный раствор подвергают перегонке с водяным
паром, причем собирают 200 мл дестиллата.
Оставшийся раствор бисульфита ого соединения альдегида ох-
лаждают до 40—50°, осторожно прибавляют суспензию 80 г бикар-
боната натрия в 200 мл воды и свободный альдегид отгоняют с
водяным паром. Верхний слой дестиллата отделяют, промывают
тремя порциями воды по 50 мл (примечание 4), сушат 20 г безвод-
ного сернокислого натрия и перегоняют, пользуясь дефлегматором
высотою в 20 см. Выход капронового альдегида с т. кип. 126—129°
составляет 45—50 г D5—50% теоретич.).
2. Примечания
1. Бромистый н.-амил был получен обычным способом («Синт.
орг. преп.», сб. 1, стр. 108) и перегнан в пределах 127^-129°.
2. В некоторых случаях немедленно начинается образование
белого осадка, однако чаще осадок появляется только после 20—
30-минутного кипячения. Если этот период нагревания заметно
сократить, то после' отгонки эфира имеет место неожиданная экзо-
термическая реакция, что ведет к значительному уменьшению
выхода. Более длительное нагревание не ведет к увеличению вы-
хода н.-гексальдегида.
3. Растворение протекает медленно и может быть значительно
ускорено применением механического перемешивания.
4. Растворенный в промывных водах альдегид может быть вы-
делен перегонкой с водяным паром, однако делать это не имеет
особого смысла, так как капроновый альдегид довольно плохо
растворим в воде.
3. Другие методы получения
Капроновый альдегид был получен из капроновой кислоты про-
пусканием ее над цинковой пылью при 300°1, реакцией с амиленом
при 300° в присутствии окиси тория 2, пропусканием ее над закисью
марганца при 300—360° с двумя объемами муравьиной кислоты 3
и перегонкой ее кальциевой соли с формиатом кальция 4. Он был
также получен нагреванием а-оксигептановой кислоты или еще
лучше а-ацетоксигептановой кислоты 5, а также по методике, опи-
санной выше 6.
1 М a i I h e, Chem. Ztg. 33, 243 A909).
2 М a i 1 h e, там же 34, 1174 A910).
8Sabatier, Mailhe, Compt. rend. 158, 986 A914).
1 Lieben, Janecek, Ann. 187, 130 A877).
5 В a gard, Bull. soc. chim. D) 1, 313 A907).
6 Bachman, J. Am. Chem. Soc. 55, 4281 A933).
2-КАРБЭТОКСИЦИКЛОПЕНТАНОН
(Этиловый эфир 2-оксициклопентанкарбоновой кислоты)
О
(СН2L<
,COX2HS
со,с,н5
н.,с
,сох*н,
нх-
н
+
сн,
Предложил: П. Пиннни.
Проверили: Л. Физер и Т. Джекобе.
1. Получение
Прибор состоит из 3-литровой трехгорлой круглодонной колбы,
снабженной механической мешалкой с ртутным затвором (приме-
чание 1), капельной воронкой емкостью 250 мл и обратным холо-
дильником, защищенным от влаги воздуха хлоркальциевой трубкой.
В колбу помещают 23 г A гр.-ат.) натрия и 250 мл сухого толуола
(примечание 2). Пускают в ход мешалку и из капельной воронки
298
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
приливают 202 г A мол.) этилового эфира адипиновой кислоты
(стр. 578) с такой скоростью, чтобы все ириливание заняло около
2 час. Обычно реакция начинается немедленно после прибавления
этилового эфира адипиновой кислоты. Температуру масляной бани
поддерживают при 100—115°, как во время прибавления эфира,
так и еще в течение около 5 час. после этого. Время от времени
через холодильник приливают сухой толуол для того, чтобы реак-
ционная смесь оставалась достаточно жидкой и было возможно
эффективное перемешивание (примечание 3). Таким путем прибав-
ляют 750—1000 мл толуола.
Реакционную смесь охлаждают в бане со льдом и медленно
выливают ее в 1 л 10%-ной уксусной кислоты, охлажденной до 0°
в смеси льда и соли. Толуольный слой отделяют, промывают один
раз водой, два раза охлажденным 7%-ным раствором соды и вновь
водой. Толуол отгоняют при атмосферном давлении, а остаток пе-
регоняют в вакууме. Выход продукта с т. кип. 83—88°/5 мм. или
79—84°/3 мм составляет 115—127 г G4—81% теоретич.; примеча-
ния 4 и 5).
2. Примечания
1. Мешалка Гершберга, часть которой изображена на рис. 12,
обеспечивает эффективное перемешивание как в данном случае,
так и вообще в случае
пастообразных смесей.
К оси мешалки с одно-
го конца припаяно два
стеклянных кольца под
прямым углом друг к
другу и через каждое
кольцо пропущена хро-
мелевая или иихромо-
вая проволока № 18 (на
рис. 12 проволока по-
казана только для ниж-
него кольца; верхняя
проволока имеет толь-
ко кольца и не имеет
нижней соединяющей
части). Мешалка легко
вводится и выводится
через узкое отверстие и
при работе ходит по
внутренней поверхности
колбы. Удобно ось мешалки делать из стеклянной трубки и
надеть на нее два небольших шарикоподшипника, поверх коротких
2-КАРБЭТОКСИЦИКЛОПЕНТАНОН
299
Рис. 12
кусочков каучуковой трубки (один из таких подшипников пока-
зан на рисунке).
Указанные выше выходы были получены с такой мешалкой.
Применяя различные другие типы мешалок, редко удавалось полу-
чать повторимые результаты.
2. Для того, чтобы высушить толуол, его перегоняют над нат-
рием.
3. Если реакционной смеси дать слишком загустеть и переме-
шивание при этом будет затруднено, или если температуру масля-
ной бани поднять выше 115—120°, твердое натриевое производное
будет спекаться на стенках колбы. Это затрудняет полное удале-
ние из колбы реакционной массы и ее разложение.
4. Согласно литературным данным 2> 3, полученный таким об-
разом продукт может содержать этиловый эфир адипиновой кислоты.
Чтобы его удалить, продукт охлаждают до 0° л медленно прили-
вают к 600 мл 10%-ного раствора едкого кали, температуру кото-
рого поддерживают при 0° с помощью бани со смесью льда и соли.
Затем добавляют воды до тех пор, пока выпавшая соль не раство-
рится, и холодный щелочной раствор извлекают два раза эфиром
порциями по 200 мл. Щелочной раствор, температуру которого
поддерживают при 0°, медленно при перемешивании приливают
к 900 мл 10%-ной уксусной кислоты, причем температуру под-
держивают ниже 1° (пользуются смесью льда и соли). Выделившийся
маслянистый слой извлекают 400 мл эфира, водный слой извлекают
еще четыре раза эфиром, порциями по 250 мл. Эфирные вытяжки
промывают два раза холодным 7%-ным раствором соды и сушат
над сернокислым натрием. После отгонки эфира остаток перего-
няют в вакууме при 79—81°/3 мм. Выход составляет только 80—
85%, и в случае хорошо проведенного синтеза количество этило-
вого эфира адипиновой кислоты, которое При этом удаляется, не
превышает 1% от общего количества продукта. За исключением
тех случаев, когда синтез прошел неудачно, такую кропотливую
очистку лучше опустить.
Если нужен продукт, совершенно не содержащий следов этило-
вого эфира адипиновой кислоты, то можно сэкономить время и
материалы, опустив первую перегонку (наблюдение проверяв-
ших синтез). Толуольный раствор неочищенного 2-карбэтокси-
циклопетанона охлаждают до 0° и медленно приливают при пере-
мешивании к 300 мл 10%-ного раствора едкого кали, температуру
которого поддерживают ниже 1°. Добавляют воды до полного рас-
творения плохо растворимой калиевой соли. Толуольный слой
отделяют и дважды промывают холодным 10%-ным раствором
едкого кали порциями по 150 мл. После каждой промывки добав-
ляют холодной/воды для растворения выпадающего осадка. Толу-
ольный раствор, который при этом становится светложелтым,
окончательно промывают два раза холодной водой, порциями по
300
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
КАТАЛИЗАТОР МЕДНО-ХРОМОВЫЙ
301
150 мл- Водные растворы соединяют вместе, экстрагируют 250 мл
эфира и обрабатывают, как это описано выше для щелочного рас-
твора. Выход составляет 100—115 г F4—74%).
5. Следующая методика дает несколько лучшие результаты.
В 3-лнтровую круглодонную колбу, снабженную обратным
холодильником, защищенным от влаги воздуха хлоркальциевой
трубкой, помещают 50 г «молекулярного» натрия и добавляют
1250 мл бензола, высушенного перегонкой над натрием. Затем
сразу приливают 303 г A,5 мол.) этилового эфира адипиновой кис-
лоты, после чего добавляют 3 мл абсолютного спирта. Колбу на-
гревают на водяной бане до тех пор, пока не начнется бурная реак-
ция, что происходит через несколько минут, и не станет выделяться
лепешка натриевого соединения. На этой стадии колбу хорошо
взбалтывают от руки. После того, как бурная реакция несколько
успокоится, смесь нагревают на водяной бане в течение ночи, и
затем охлаждают в бане со льдом. Продукт разлагают льдом и
б н. соляной кислотой, причем кислоту добавляют до синей окраски
бумажки конго. Бензольный слой отделяют и водный слой извле-
кают один раз 200 мл бензола. Объединенные бензольные растворы
промывают 200 мл 5%-ного раствора соды, а затем 300 мл воды.
Раствор переливают в 3-литровую колбу Вюрца и бензол и воду
отгоняют при атмосферном давлении. Остаток фракционируют
в вакууме. Выход продукта с т. пл. 108—11 Г/15 мм составляет
185—192 г G9—82% теоретич.). После повторной •перегонки про-
дукт кипит при 102°/11 мм.
Отличительными чертами этой методики является добавление
спирта, в результате чего совершенно исчезает или значительно
сокращается индукционный период, и во-вторых, избыток натрия,
что способствует полноте реакции. Бензол в качестве растворителя
можно заменить петролейным эфиром (т. кип. 60—80°).
Карбэтоксициклопентанон, перегнанный один раз, достаточно
чист для обычных синтетических целей; он дает С-метильное про-
изводное с выходом 93%. (Р. Линстед и Е. Мид, частное сообще-
ние 5. Проверили Р. Шрайнер и Н. Мун).
3. Другие методы получения
2-Карбэтоксициклопентанон был получен из этилового эфира
адипиновой кислоты при действии натрия *¦ 2- 3- 5, натрийамида 6
и этилата натрия 7. Приведенный выше метод основан на работе
Корнюбера и Борреля 2.
1 Hershberg, Ind. Eng. Chem., Anal. Ed. 8, 313 A936).
'Cornubert, Borrel, Bull. soc. chim. D) 47, 301 A930).
3Bouveault, там же (З) 21, 1019 A899); Зелинский,
Ушаков, ЖРХО 56, 67 A925); Bull. soc. chim. D) 35, 484 A924).
'van Rysselberge, Bull. acad. rcy. Belg. (Sci.) E) 12, 171 A926);
(Chem. Zentr. 1926, II, 1846).
5 L i n s t e a d, Meade, J. Chem. Soc. 1934, 940.
"Bouveault, Locqin, Bull. soc. chim. D) 3, 440 A908).
'Wislicenus, Schwanhausser, Ann. 297, 112 A897).
КАТАЛИЗАТОР МЕДНО-ХРОМОВЫЙ
прокаливание CuCr2O4
> комплексные хроматы > ВаСг„О4
(Примечание 1)
Предложили: В. Лэзир и X. Арнольд.
Проверили: Р. Шрайнер и Дж. Каплан.
fcSa(INO3).>
(NH4)a CrO4
1. Получение
Смесь 26 г @,1 мол.) азотнокислого бария и 800 мл дестиллиро-
ванной воды нагревают до 70° и после полного растворения соли
добавляют 218 г @,9 мол.) химически чистой азотнокислой меди
с 3 мол. кристаллизационной воды. Затем смесь перемешивают при
70° до тех нор, пока не будет получен прозрачный раствор (приме-
чание 2).
Раствор хромовокислого аммония приготовляют растворением
126 г @,5 мол.) двухромовокислого аммония (х. ч.) в 600 мл дестил-
лированной воды с добавлением 150 мл 28%-ного водного аммиака
(уд. в. 0,9; примечание 3). Теплый раствор нитратов перемешивают
(можно от руки) и к нему приливают тонкой струей раствор хро-
мовокислого аммония. Продолжают перемешивание еще в течение
нескольких минут, после чего красноватобурый осадок двойной
хромовокислой соли меди и бария отсасывают (примечание 4) через
воронку Бюхнера диаметром 16 см, осадок хорошо отжимают и
сушат при 110°. Сухой осадок помещают в никелевую чашку, не
плотно покрытую крышкой (примечание 5) или в один или два фар-
форовых сосуда, покрытых часовыми стеклами, и нагревают его
в муфельной печи в течение 1 часа при 350—450° (примечание 6).
На этой стадии выход хромита должен быть около 160 г. Остаток
после прокаливания измельчают в ступке, чтобы разбить комки,
которые могут образоваться (примечание 7), и высыпают его в
2-литровый стакан, куда предварительно налито 1,2 л 10%-ной
уксусной кислоты. Перемешивают 10 мин., после чего смеси дают
отстояться. Примерно через 10 мин. 2/3 или даже больше отрабо-
танного раствора кислоты сливают и заменяют 1,2 л свежей 10%-
ной уксусной кислоты и извлечение повторяют. Остаток промывают,
повторно экстрагируя его четыре раза дестиллированной водой,
которой каждый раз берут по 1,2 а (примечание 8). Нерастворимую
зш
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
часть отсасывают через воронку Бюхнера, сушат при 110° и расти-
рают в ступке, так чтобы получился мелкий черный порошок (при-
мечание 9). Выход 130—140 г (примечания 10 и 11).
2. Примечания
1. Протекающую в данном случае реакцию нельзя выразить
количественно простым уравнением. Процесс был исследован Гре-
гером х.
2. Азотнокислый барий плохо растворим в холодной воде и
еще хуже в растворе азотнокислой меди. Поэтому для того, чтобы
обе соли перевести в раствор, необходимо смесь нагреть.
3. Теплый, свежеприготовленный раствор хромовокислого аммо-
ния можно применять для осаждения немедленно или же спер-
ва дать ему охладиться до комнатной температуры. Однако рас-
твор, полученный так, как это здесь указано, пересыщен при ком-
натной температуре, и при стоянии из него выделяются кристаллы.
Если необходимо сохранить раствор хромовокислого аммония до
следующего дня, то часть аммиака нужно сохранить и добавлять
немедленно перед осаждением. Для получения хромовокислого
аммония можно применять также хромовую кислоту, если только
раствор кислоты хранить на холоду и достаточное количество
аммиака добавлять под поверхность кислоты.
4. Количество взятого аммиака вычисляется таким Тобразом,
чтобы получился маточник, нейтральный на лакмус с учетом
основного характера осадка. Хотя отклонение в любую сторону
отрицательно влияет на выход, однако лучше не пытаться устанав-
ливать точно конец реакции после осаждения, а просто внести по-
правку в то количество аммиака, которое нужно взять в следую-
щем опыте. Маточный раствор сам служит индикатором: кислый
раствор — желтого цвета, щелочной — темнозеленого.
5. Удобный размер противня: глубина 6 см, длина 19 см и ши-
рина 10 см. Он может быть сделан из листового никеля толщиной
0,8 мм.
6. Прокаливание смешанной хромовокислой соли меди и ам-
мония вызывает самопроизвольную экзотермическую реакцию,
протекающую с быстрым выделением газа. Хотя количество одно-
временно прокаливаемой соли и не имеет значения, однако сосуд
не должен быть слишком полным, в противном случае будут иметь
место значительные механические потери продукта. Также не реко-
мендуется измельчать куски сухого осадка, так как если осадок
находится в виде кусочков, то это уменьшает разбрасывание хро-
мита при прокаливании.
7. До экстрагирования катализатор должен представлять собой
голубовато-черный хрупкий порошок. Это вполне удовлетвори-
тельный катализатор для дегидрогенизации спиртов и для таких
КАТАЛИЗАТОР МЕДНО-ХРбМОВЫЙ
303
легче протекающих реакций гидрогенизации, как восстановление
нитросоединений. Смесь хромита меди и окиси меди несколько
менее активна и легче восстанавливается до металлической меди
по сравнению с катализатором, из которого окись меди удалена
экстрагированием кислотой.
8. Если промывать слишком длительное время, катализатор
может перейти в коллоидальное состояние, и тогда его трудно
выделить путем декантации или фильтрования.
9. Никаких особых предосторожностей при работе или при
хранении меднохромового катализатора не требуется, так как дей-
ствие воздуха или влаги не оказывают па него вредного влияния.
10. Барий вводится в качестве одного из компонентов катализа-
тора, потому что он оказывает защитное действие против отравляю-
щего действия сульфатов; имеются также указания на то, что он
оказывает стабилизирующее действие на катализатор в отношении
его восстановления. Приведенную здесь методику можно применить
также и к получению медно-хромового катализатора не содержа-
щего бария. В этом случае не берут совсем азотнокислого бария,
а азотнокислой меди берут 242 г A мол.). Все остальные детали
синтеза сохраняются.
11. Другой метод описывает Адкинс 2. Этот метод заключается
в следующем: 31 г азотнокислого бария растворяют в 820 лмдестил-
лированной воды, предварительно нагретой до 80°. К горячему
раствору добавляют 260 г кристаллической азотнокислой меди
C мол. Н2О) и смесь перемешивают и нагревают до полного раство-
рения. Одновременно растворяют 151 г двухромовокислого аммо-
ния в 600 мл дестиллированной воды и к раствору добавляют 225 мл
28%-ного водного аммиака. Горячий раствор азотнокислых солей
приливают тонкой струей при перемешивании к раствору хромо-
вокислого аммония. Оранжевый осадок отфильтровывают, хорошо
отжимают и отсасывают по возможности досуха. Осадок сушат
в шкафу при 75—80° в течение 12 час, хорошо измельчают и раз-
деляют на три порции. Каждую порцию разлагают в отдельности
в большой фарфоровой кастрюле (диаметр 15 см), нагревая ее на
голом пламени. Массу необходимо нагревать настолько, чтобы
разложение протекало при возможно более низкой температуре.
Во время разложения порошок непрерывно перемешивают сталь-
ным шпателем и нагревание регулируют таким образом, чтобы раз-
ложение протекало не слишком бурно. Для этого нагревают только
одну сторону кастрюли и перемешивание усиливают, как только
начнется разложение для того, чтобы распространить его по всей
массе. Во время этого процесса окраска порошка меняется от оран-
жевой до бурой и в конце концов до черной. Когда вся масса ста-
нет черной, выделение газа прекращается, порошок вынимают из
кастрюли и дают ему охладиться. После этого все количество про-
дукта перемешивают 30 мин. с 600 мл 10%-ного раствора уксусной
304
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
2-КЕТОГЕКСАМЕТИЛЕНИМИН
305
кислоты, осадок отфильтровывают, с целью промывки суспенди-
руют шесть раз в воде (каждый раз берут по 100 мл), сушат 12 ча-
сов при 125° и измельчают. Выход катализатора 170 г.
3. Другие методы получения
Хромит меди был получен прокаливанием основной хромово-
кислой меди при красном калении и термическим разложением
двойной хромовокислой соли меди и аммония х. Приведенная здесь
методика является видоизменением последнего метода 3, согласно
которому добавляется также двойная хромовокислая соль бария
и аммония 4. Медно-хромовые окисные катализаторы гидрогениза-
ции были также получены измельчением или совместным нагре-
ванием окиси меди и окислов хрома, разложением карбонатов
меди, аммония и хрома или нитратов меди и хрома 2>4, а также
низкотемпературным прокаливанием двойных хромовокислых со-
лей меди и аммония в.
1 Gro ger, Z. anorg. Chem. 58, 412 A908); 76, 30 A912).
2 Adkins, Reactions of Hydrogen with Organic Compounds over Copper-
Chromium Oxide and Nickel Catalyst, p. 12, University ot Wisconsin Press,
Madison, 1937.
3 L a z i e г; ам. пат. 1 746 782 и 1 746 783 [С. А. 24, 1649 A930)]; ам. пат.
1 964 000 [С. А. 28, 5075 A934)].
'Connor, F о 1 k e r s, A d к i n s, J. Am. Chem. Soc. 53, 2012 A931);
54, 1138 A932).
5 С а 1 i n g a e r t, Edgar, Ind. Eng. Chem. 26, 878 A934).
2-КЕТОГЕКСАМЕТИЛЕНИМИН
B-Оксогексаметилешмин; е-капролактам)
(CH2M С = NOH
„NH
Предложили: К- Марвел и Дж. Экк.
Проверили: Л. Физер и К. Фишер.
1. Получение
Перегруппировку 120 г A,06 мол.) чистого оксима циклогек-
санона (примечание 1) осуществляют с двенадцатью 10-граммовыми
порциями так, как это описано в первых восьми строках раздела
Б на стр. 91. Кислые растворы е-каиролактама от двенадцати
опытов соединяют вместе в 3-литровой кругло донной колбе, сна б.
женной механической мешалкой и делительной воронкой, поме-
щенной в охладительную смесь. Раствор охлаждают до 0° и осто-
рожно подщелачивают на лакмус 24%-ным раствором едкого кали,
который добавляют очень медленно E—б час.) при хорошем охла-
ждении (примечание 2). Обычно расходуется около 1550 мл щелоч-
ного раствора. Для того чтобы избежать гидролиза в этой стадии
работы, температуру поддерживают ниже 10°.
Выпавший в осадок сернокислый калий отфильтровывают и
промывают два раза хлороформом порциями по 100 мл. Слабоще-
лочной раствор экстрагируют около пяти раз хлороформом пор-
циями по 200 мл (примечание 3), и соединенные хлороформные
растворы промывают один раз 50 мл воды, чтобы удалить щелочь.
Хлороформ отгоняют и остаток подвергают фракционированной
перегонке в вакууме. Выход 2-кетогексаметиленимина с т. кип.
127—133°/7 мм и т. пл. 65—68° составляет 71—78 г E9—65% теоре-
тич.; примечание 4).
2. Примечания
1. Для работы необходимо взять чистый гексаноноксим с т. пл,
86—88° (стр. 90 и 396), так как при действии серной кислоты на
продукт худшего качества имеет место значительное осмоление.
2. В случае едкого кали результаты получаются лучше, чем
в случае едкого натра, так как при применении последнего из рас-
твора выпадает большое количество кристаллического сернокислого
натрия, что мешает хорошему охлаждению.
3. Экстрагирование заканчивают тогда, когда хлороформный
слой уже не содержит более заметного количества продукта.
4. В литературе указана т. кип. 139—140°/12 мм. Для темпе-
ратуры плавления указаны различные величины от 65 до 70°.
3. Другие методы получения
2-Кетогексаметиленимин был получен нагреванием е-амино-
капроновой кислоты или ее этилового эфира 1, а также перегруп-
пировкой циклогексаноноксима 2>3'4. Описанный выше метод
основан на указаниях Валлаха 2 относительно перегруппировки
оксима с видоизменениями, внесенными Ружичкой 3. Эта перегруп-
пировка может быть осуществлена также в виде непрерывного про-
цесса 5.
1Gabriel, Maass, Ber. 32, 1271 A899); В г a u n, там же
40, 1840 A907); Carothers, Berchet, J. Am. Chem. Soc. 52, 5289 A930).
2 W a 1 1 а с h, Ann 312, 187 A900); 343, 43 A905).
3 R u z i с k a, S e i d e 1, H u g о s о n, "Helv. Chim. Acta 4, 477 A921).
i Eck, Marvel, J. Biol. Chem. 106, 387 A934).
5 E. I. du Pont de Nemours and Company; ам. пат. 2 234 566 [С. А. 35, 3650
A941)]; I. О. Farbenind. A.-O.; ам. пат. 2 249 177 [С. А. 35, 6599 A941)J.
20 Сбораик № 2
306
Синтезы органических препаратов
ЛАУРИЛОВЫЙ СПИРТ
(Додециловый спирт)
Предложили: С. Форд и К- Марвел.
Проверили: Ф. Уитмпр и Д. Лодер.
1. Получение
5-литровую трехгорлую круглодонную колбу снабжают механи-
ческой мешалкой с ртутным затвором, делительной воронкой и
большим обратным холодильником длиной около 2 м, с внутрен-
ней трубкой 2,5 см в диаметре (примечание 1), который соединен
с колбой посредством куска толстой резиновой трубки.
В колбу помещают 70 г (Згр.-ат.) натрия и 200 мл сухого толуола
(примечание 2) и нагревают ее на масляной бане, пока натрий
не расплавится. Затем пускают в ход мешалку, и когда натрий
разобьется в мелкие шарики, масляную баню удаляют и смеси
дают охладиться. Во время охлаждения продолжают размешива-
ние для того, чтобы натрий оставался в размельченном состоянии.
Когда смесь охладится до 60°, к ней прибавляют через делитель-
ную воронку сперва раствор 114 г @,5 мол.) этилового эфира лаури-
новой кислоты (примечание 3) в 150 мл абсолютного спирта (приме-
чание 4) и затем еще 500 мл спирта с наиболее возможной быстротой
(примечание 5), без потери вещества через холодильник. На при-
бавление эфирного раствора и спирта требуется менее 5 мин. (обычно
2—3 мин.). Когда реакция успокоится, колбу нагревают на водяной
бане, пока весь натрий не растворится (примечание 6). Затем смесь
перегоняют с водяным паром, чтобы отделить толуол и этиловый
спирт. После отгонки содержимое колбы переливают в горячем
состоянии в делительную воронку и промывают три раза порциями
горячей воды по 200 мл для того, чтобы извлечь лауриловокислый
натрий (примечание 7). По охлаждении лауриловый спирт извле-
кают эфиром. Соединенные эфирные вытяжки промывают водой,
раствором соды и вновь водой и сушат безводным сернокислым
магнием. Эфир отгоняют и перегоняют лауриловый спирт в вакууме.
Выход 60—70 г F5—75% теоретич.) продукта, кипящего при
143—146° /18 мм или 198—200°/ 135 мм (примечание 8).
2. Примечания
1. Реакция идет очень бурно, и поэтому, если применять холо-
дильник с меньшими длиной и диаметром внутренней трубки,
возможно, что смесь будет выброшена через верхний конец холо-
дильника и вследствие присутствия измельченного натрия может
легко воспламениться. Внутреннюю трубку холодильника целе-
сообразно сделать из латуни или меди.
ЛАУРИЛОВЫЙ СПИРТ
307
2. Толуол сушат посредством перегонки, причем первые 10%
дестиллата отбр'асывают. Перегнанный толуол хранят над натрием.
3. Этиловый эфир лауриновой кислоты был получен алко-
голизом кокосового масла и фракционированием полученных
эфиров. Вещество кипит при 127—132° /5 мм [См. Org. Syh. 20,69,
а также Organic Chemical Reagents III. Univ. Illinois Bull. 19,
F) 62 A921)].
4. Качество абсолютного спирта, взятого для восстановления,
играет очень важную роль. Спирт, высушенный над известью, дает
очень плохие выходы. Спирт, применявшийся в описанных опытах,
был высушен метилатом магния (см. «Синт. орг. преп.», сб. 1,
стр. 545).
5. Наивысшие выходы получаются, когда восстановление про-
водят быстро. Если реакция идет слишком бурно, то останавливают
мешалку и некоторое время смесь охлаждают льдом.
6. Если надо произвести несколько восстановлений, то можно
сэкономить время, перелив смесь в другую колбу, чтобы освобо-
дить прибор для следующей операции.
7. Лауриновокислыйнатрий следует извлечь очень тщательно, так
как в противном случае он вызывает образование стойких эмульсий.
8. Таким же способом этиловый эфир ундециловой кислоты может
быть восстановлен в ундециловый спирт (т. кип. 123—125°/6 мм)
с выходом 70%; этиловый эфир миристиновой кислоты —¦ в ми-
ристиловый спирт (т. кип. 170—173°/20 мм; т. пл. 39—39,5°)
с выходом 70—80%, и этиловый эфир пальмитиновой кислоты —
в цетиловый спирт (т. кип. 178—182°/12 мм; т. пл. 48,5—49,5°)
с выходом 70—78% теоретич.
3. Другие методы получения
Лауриловый спирт может быть получен восстановлением лаури-
нового альдегида 1, восстановлением эфиров лауриновой кислоты
натрием и абсолютным спиртом 2, или натрием, жидким аммиаком
и абсолютным спиртом 3 или каталитическим восстановлением4;
восстановлением амида лауриновой кислоты натрием и амиловым
спиртом 5. Описанный выше способ в основном разработали Левин
и Аллен 2.
1 Кг aff t, Ber. 16, 1718 A883); Сивков, Новикова, ЖПХ 13,
1272 A940).
2 Bouveault, Blanc, Bull. soc. chim. C) 31, 674 A904); герм. пат.
164 294 [Frdl. 8, 1260 A905-07)]; L с v e n e, Allen, J. Biol. Chem. 27,
443 A916); Marvel, Tanenbaum, J. Am. Chem. Soc. 44, 2649 A922);
Adams, Marvel, Org. Chem. Reagents IV, Univ. Illinois Bull. 20. (8)
54 A922). '
3 С h a b 1 a y, tompt. rend. 156, 1021 A913); Ann. chim. (9) 8, 215 A917).
1 Ad kins, Folkers, J. Am. Chem. Soc. 53, 1095 A931); Bohme
A.-G.; англ. пат. 356 606 [С. А. 26, 5573 A932)].
1 Scheuble, Loebl, Monatsh. 25, 348 A904).
308
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
МАЛОНОВАЯ КИСЛОТА
309
dZ-ЛИЗИНА ХЛОРИСТОВОДОРОДНЫЕ СОЛИ
C6H5CONH (СН,LСНСО,Н -™ C6H3CONH (СН2L СНСО2Н
I I
Br NH,
-^±^L cinh3(ch,Lchco2h .™=l cinh3 (сн2Lснсо2н
NH3C1 NH3
Предложили: Дж. Экк и К. Марвел.
Проверили: К- Ноллер и В. Мьюнич.
1. Получение
A. dl-z-Бензоиллизин. В 5-литровую колбу фильтруют раствор
180 г @,57 мол.) е-бензоиламино-а-бромкапроновой кислоты
(стр. 89) в 2 л водного аммиака (уд. в. 0,9) и раствор оставляют
стоять на двое суток. Кристаллы, которые могут образоваться
за это время, отфильтровывают, и фильтрат упаривают на паровой
бане в вакууме до объема около 1 л. Кристаллы отфильтровывают,
присоединяют к первой порции и промывают сперва 100 мл спирта,
а затем 100 мл эфира. Водный фильтрат упаривают в вакууме
досуха; остаток промывают два раза водой порциями по 100 мл,
чтобы удалить бромистый аммоний, а затем последовательно 50 мл
спирта и 50 мл эфира. Суммарный выход г-бензоиллизина, с тем-
пературой плавления 265—270°, составляет 100—116 г G0—81%
теоретич.).
Б.й1-Лизина двухлористоводороджясоль. Раствор 100г@,4мол.)
бензоиллизина в смеси 600 мл соляной кислоты (уд. веса 1,18)
и 400 мл воды кипятят 10 час. с обратным холодильником. Затем
смесь охлаждают и бензойную кислоту отфильтровывают. Филь-
трат упаривают па водяной бане в вакууме до образования густого
сиропа, который переносят в стакан емкостью 1,5 л с помощью
четырех порций (около 400 мл) горячего абсолютного спирта. Если
нужно, то раствор фильтруют. Раствор охлаждают до 15—20°
и медленно, при перемешивании, приливают 500 мл эфира. Осадок
отфильтровывают и сушат. Продукт плавится при 187—189°;
выход 67—75 г G6—85% теоретич.) аналитически чистой дву-
хлористоводородной соли лизина (примечание 1).
B. dl-Лшина монохлористоводородная соль. К раствору 55 г
@,25 мол.) двухлористоводородной соли лизина в 1 л кипящего
95%-ного спирта (примечание 2) добавляют при перемешивании
раствор 25 г @,32 мол.) пиридина в 40 мл горячего 95%-ного спирта.
Немедленно выпадает бесцветная кристаллическая монохлористо-
водородная соль. После стояния в течение ночи в холодильном
шкафу кристаллы отфильтровывают и промывают два раза холод-
ным абсолютным спиртом, порциями по 50 мл. После высушива-
ния продукт плавится при 260—263°; выход его 42—43 г (91—94%
теоретич.).
G целью дальнейшей очистки и удаления хлористоводородной
соли пиридина полученный продукт растворяют в 85 мл кипящей
воды и при перемешивании добавляют 650 мл кипящего 95%-ного
этилового спирта. По охлаждении в течение ночи в холодильном
шкафу твердый осадок отфильтровывают и промывают один раз
20 мл холодного абсолютного спирта. Выход монохлористоводород-
ной соли с"т. пл. 263—264° (исправл.) составляет40—42г (95—97%).
2. Примечания
1. Если получается продукт с более низкой температурой плав-
ления, то его можно очистить, для чего его растворяют в 1 л горя-
чего 95%-ного спирта, если нужно, раствор фильтруют, охлаждают
и, не отделяя того вещества, которое могло выкристаллизоваться,
медленно приливают 1,5 л эфира. Если продукт выпадает в виде
масла, то при стоянии он вскоре кристаллизуется. При проверке
было установлено, что из одной порции G5 г), которая плавилась
при 173—178°, было получено после такой обработки 67 г (89%)
продукта ст. пл. 187—189°.
2. Если раствор непрозрачен, то до добавления пиридина его
следует профильтровать.
3. Другие методы получения
Приведенная выше методика получения dZ-бензоиллизина и
хлористоводородных солей dZ-лизина является видоизменениемг
метода, опубликованного Брауном2.
iEck, Marvel, J. Biol. Chem. 106, 387 A934).
3 В г a u n, Ber. 42, 844 A909).
МАЛОНОВАЯ КИСЛОТА
C1CH,CO,H —— ClCH2CO2Na —-1 CNCH,COaNa —
NaOH
CaCI2
r CH2(CO2NaJ --- CH2(CO2JCa
HCl
СН2(СО,НJ
Предложил: Н. Вейнер.
Проверили: f(. Нолпер и М. Сайнерхолм.
1. Получение
В 5-литровой круглодонной колбе растворяют 500 г E,3 мол.)
хлоруксусной кислоты в 700 мл воды; раствор нагревают до 50°,
310
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
нейтрализуют 290 г B,7 мол.) безводного углекислого натрия и
вновь охлаждают до комнатной температуры. Одновременно раство-
ряют 294 г F мол.) цианистого натрия (97%-ного) в 750 мл воды,
нагретой до 55°. Раствор охлаждают до комнатной температуры,
после чего его приливают к раствору натриевой соли хлоруксусной
кислоты, энергично размешивая оба раствора и охлаждая их водо-
проводной водой. После полного смешения растворов охлаждение
прекращают и температуре дают повыситься. Когда она дойдет
до 95°, раствор охлаждают добавлением 200 мл ледяной воды,
причем это повторяют, если нужно, пока не прекратится повышение
температуры (примечание 2). Для завершения реакции раствор
нагревают на водяной бане в течение 1 часа.
После этого раствор охлаждают до комнатной температуры, и
в нем медленно растворяют 240 г F мол.) твердого едкого натра,
отвечающего требованиям фармакопеи. Когда все растворится,
реакционную смесь вновь нагревают на водяной бане в вытяжном
шкафу. Когда температура достигнет 60—70°, начинается выделе-
ние аммиака, которое по мере повышения температуры становится
все более энергичным. Большая часть аммиака выделяется за 45 мин.,
однако раствор нагревают не менее 3 час, причем последние следы
аммиака удаляют, пропуская водяной пар через горячий раствор
в течение еще 45—60 мин.
Растворяют 600 г безводного хлористого кальция в 1,8 л воды,
нагретой до 40°, и при энергичном перемешивании медленно прили-
вают этот раствор к горячему раствору натриевой соли малоновой
кислоты. При этом немедленно образуется подобный сыру осадок
кальциевой соли малоновой кислоты, который после стояния в тече-
ние 24 час. становится грубо кристаллическим. Прозрачный раствор
сливают, малоновокислый кальций промывают декантацией холод-
ной водой 4—5 раз, порциями по 500 мл. Затем осадок отфиль-
тровывают, тщательно отсасывают и сушат на воздухе или при
45—50° до постоянного веса. Выход 800—900 г.
Сухую кальциевую соль малоновой кислоты помещают в 3-литро-
вую круглодонную колбу и добавляют достаточное количество
G50—1000мл) эфира, не содержащего спирта (примечание 3), для того
чтобы образовалась паста, которую можно было бы перемешивать.
Колбу ставят в баню со льдом и при хорошем перемешивании прили-
вают 12-н. соляной кислоты из расчета 1 мл кислоты на 1 г соли.
После того как кислота медленно прибавлена через капельную во-
ронку, раствор переносят в прибор для непрерывного экстрагирова-
ния (примечание 4) и экстрагируют его эфиром до техпор, пока больше
не будет извлекаться малоновой кислоты. От эфирного раствора,
не высушивая его, отгоняют эфир, затем отфильтровывают осадок,
сушат его на воздухе и получают 415—440 г малоновой кислоты
G5—80% теоретич.) с т. пл. 130° или даже выше, достаточно чистой
для большинства целей,
МАЛОНОВАЯ КИСЛОТА
311
2. Примечания
1. Был взят свежеперегнанный продукт, кипящий в пределах Зэ.
2. Если реакция между цианистым натрием и солью хлоруксус-
ной кислоты станет слишком бурной, выделяется цианистый водо-
род, частично превращающийся в бурый продукт, причем обра-
зуется соответственное количество гликолята. Если температуре
реакционной смеси дать самопроизвольно подняться выше 95°,
жидкость может вскипеть настолько бурно и неожиданно, что ее
выбрасывает из колбы, несмотря на
большой запас свободного места в
колбе.
3. Эфир применяют для того, чтобы
избежать ненужного увеличения объема
водного раствора, подлежащего извле-
чению. Этот эфир может быть исполь-
зован для последующих извлечений.
Необходимо применять эфир, не содер-
жащий спирта, во избежание этерифи-
кации малоновой кислоты в течение дли-
тельного периода экстрагирования.
4. Весьма удобным экстрактором для
этого синтеза является видоизмененный
экстрактор, описанный Фридрихсом Ч
Колонку для хлористого кальция вы-
сотой 20 см или иной цилиндр узкого
диаметра объемом около 1,3 л, можно
применить в качестве экстракционной
камеры. Трубка-рубашка, по которой
пары эфира поступают в холодильник
Аллина, сделана из трубки диаметром
25 мм и имеет в длину 50 см. Узкое
горло, ведущее к колбе для экстрагиро-
вания, припаивается к рубашке пример-
но на высоте 8 см от нижней части.
Внутренняя трубка,-диаметром 14 мм и
высотой 65 см, развернута в верхней
части до диаметра около 20 мм. В ниж-
нюю часть трубки впаяна фильтровальная пластинка Витта соответ-
ственного диаметра, для того чтобы эфир мог проходить через
водный слой в виде тока мелких пузырьков. Вместо этого нижний
конец трубки можно запаять и сделать в дне 3—б булавочных
отверстий. Рубашка пригоняется к камере с помощью соответственно
просверленной каучуковой пробки, холодильник к верхней части
рубашки и колба для экстрагирования емкостью 500 мл к узкому
горлу присоединяются с помощью предварительно обожженных
Рис. 13
312
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
МЕТИЛБЕНЗИЛКЕТОН
313
корковых пробок. С помощью этого прибора 395—400 г малоновой
кислоты экстрагировались за 72 часа, причем эфир менялся каждые
24 часа, а последние следы экстрагировались еще 24 часа. Экстрак-
тор Фридрихса изображен на рис. 13.
3. Другие методы получения
Малоновая кислота была получена омылением ее нитрила кон-
центрированной соляной кислотой 2, гидратацией недокиси угле-
рода 3, а также действием цианистого калия или натрия на бром-
уксусный эфир 4, хлоруксусный эфир 5 или хлоруксусную кислоту 6
с последующим омылением. Наиболее удобным и выгодным методом
является действие цианистого натрия на хлоруксусную кислоту.
1 Friedrichs, Chem. Fabrik 1, 91 A928).
- Henry, Compt. rend. 102, 1396 A886).
3 D i e 1 s, Wolf, Ber. 39, 696 A906).
4 Franchimont, там же 7, 216 A874).
5 К о 1 b e, Ann. 131, 349A864); M il 1 1 e г, там же 131, 352 A864); П е т-
p и е в, ЖРХО, 10, 64 A878).
«Miller, J. prakt. Chem. B) 19, 326 A879); Qrimaux, Tcher-
n i a k, Bull. soc. chim. B) 31, 338 A879); В о u r g о i n, там же B) 33, 574
A880); Conrad, Ann. 204, 126 A880)
CHgC
МЕЗАКОНОВАЯ КИСЛОТА
(HNO3)
О
H,0
СН С^=(
СН3ССО2Н
ноххн
Предложили: Р. Шрайнер, С. Форд и Л. Ролл.
Проверил: К. Ноллер.
1. Получение
Смесь 100 г @,89 мол.) цитраконового ангидрида (примечание 1),
100 мл воды и 150 мл разбавленной азотной кислоты A часть по
объему концентрированной азотной кислоты на четыре части по
объему воды) выпаривают до появления красных паров в кониче-
ской колбе емкостью 500 мл (примечание 2). Раствор охлаждают
и выкристаллизовавшуюся мезаконовую кислоту отсасывают. Маточ-
ный раствор упаривают до объема 150 мл и вновь отсасывают выпав-
шую кислоту. Дальнейшее упаривание маточного раствора до 50 мл
дает еще некоторое количество вещества (примечание 3). Все порции
мезаконовои кислоты соединяют и перекристаллизовывают из 100 мл
воды. Выход продукта, плавящегося при 203—205°, равен 50—60 г
D3—52% теоретич.).
2. Примечания
1. Цитраконовый ангидрид можно заменить цитраконовой кис-
лотой. Методика получения цитраконовой кислоты и цитраконового
ангидрида приведена на стр. 570.
2. Чтобы произошла изомеризация, необходимо вести выпари-
вание до того момента, когда появятся красные пары. При этом
обычно объем раствора равен примерно 250 мл.
3. Упаривание производится в несколько приемов, чтобы пол-
нее выделить мезаконовую кислоту.
3. Другие методы получения
Мезаконовая кислота была получена нагреванием цитраконовой
кислоты с разбавленной азотной кислотой 1, с иодистоводородной
кислотой 2, с концентрированным раствором едкого натра 3; нагре-
ванием концентрированного водного раствора итаконовой или
цитраконовой кислоты до 180—200°4; обработкой цитрадибром-
пировинной кислоты и мезодибромпировинной кислоты йодистым
калием и медью при 150° °; нагреванием цитраконового ангидрида
с азотной кислотой 6.
1 Gottlieb, Ann. 77, 268 A851).
2 К е k u 1 с, Ann. Spl. 2, 94 A862).
3 De lis le, Ann. 269, 82 A892).
4 S w a r t s, Jahresb. 1873, 579.
5 S w a r t s, Zeit. fur Chem. 1868, 259.
6 F i t t i g, Landolt, Ann. 188, 73 A877).
МЕТИЛБЕНЗИЛКЕТОН
(/-Фенил-2-nponaiwH )
А. ИЗ ФЕНИЛУКСУСНОЙ И УКСУСНОЙ КИСЛОТ
с6н5сн2ссш + сн3со2н <т-
свн,хн2сосн
со2 + н2о
,хн2сосн.
Предложили: Р. Хербст и Р. Минске.
Проверили: К- Ноллер и К- Лов.
1. Получение
Реакция проводится в приборе, приведенном на рис. 14. Л — А
трубка для сожжения из стекла «пмрекс», длиной 90 см и диаметром
2 см. К трубке с помощью специального тройника Б, с боковым
отводом В, присоединена делительная воронка Д. Середина трубки,
по длине в 60 см, заполняется катализатором двуокисью тория
(примечание 1), который в нижней части трубки удерживается
с помощью вдавливаний в стекле. Горячий конец Г пирометра
314
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
соприкасается с серединой трубки, а часть трубки, наполненная
катализатором, обернута тонким слоем асбестовой бумаги.
Трубку помещают в цилиндрическую электропечь, длиной 60 см,
установленную с небольшим наклоном к горизонтали. Кольцевое
пространство между трубкой и печью по концам набивают асбестом,
чтобы обеспечить равномерную температуру по длине печи и чтобы
удерживать каталитическую трубку в неподвижном состоянии.
Нижний конец реакционной трубки присоединен с помощью алонжа
Ё к вертикальной стеклянной трубке Ж, длиной 40 см, с диаметром
2 см, наполненной стеклянными бусами, которая служит холо-
дильником. Нижний конец трубки Ж присоединен к конической
колбе или к какому-либо другому приемнику.
Г
— 50 см
— 90 СМ
Рис. 14
Печь нагревают до 430—450° и через трубку пропускают ток
углекислого газа, поступающего через боковой отвод В. Угле-
кислый газ предварительно пропускается через промывную склянку
с концентрированной серной кислотой с целью осушки и для опре-
деления скорости пропускания газа. В делительную воронку
помещают раствор 136 г A мол.) фенилуксусной кислоты с т. пл.
77—79° («Синт. орг. преп.», сб. 1, стр. 440) в 120 млA20 г, 2 мол.)
ледяной уксусной кислоты и этот раствор приливают со скоростью
12—15 капель в 1 мин. Прибавление всего количества раствора
занимает 12—15 час. При этом через каталитическую трубку про-
пускают очень слабый ток углекислого газа A пузырек в 1 сек.),
для того чтобы поддерживать газы в движении. После того как
весь раствор прибавлен, в делительную воронку наливают 10 мл
ледяной уксусной кислоты, которые также пропускают через
трубку, для того чтобы вытеснить продукт реакции. Дестиллат
Состоит из слегка флуоресцирующей светлобурой маслянистой
МЕТИЛБЕНЗИЛКЕТОН
315
жидкости и водного слоя; оба слоя обрабатывают 300 г смеси льда
и воды и подщелачивают на лакмус небольшим избытком 50%-ного
раствора едкого натра (примечание 2).
Маслянистый слой отделяют (примечание 3), а водный слой
извлекают два раза бензолом порциями по 50 мл. Бензольные
вытяжки соединяют вместе с маслянистым слоем и растворитель
отгоняют. После дробной перегонки в вакууме получают 80—95 г
фракции, отвечающей метилбензилкетону и кипящей при 110—¦
120° /21—22 мм; остаток представляет собой дибензилкетон (при-
мечание 4). После повторной перегонки главной фракции полу-
чают 74—87 г E5—65% теоретич.) метилбензилкетона с т. кип.
110—115° /21—22 мм (примечания 5, 6 и 7).
2. Примечания
1. Катализатор приготовляют следующим образом: кусочки
пемзы, размером с горошину, в количестве, достаточном для напол-
нения трубки, обрабатывают некоторое время горячей концентри-
рованной азотной кислотой, а затем тщательно промывают горячей
дестиллированной водой. В фарфоровой чашке пемзу смешивают
с раствором 40 г кристаллического азотнокислого тория [Th (NO3L+
+ 12 Н2О] в 100 мл воды и раствор выпаривают досуха при частом
перемешивании, для того чтобы обеспечить равномерное распре-
деление соли. Пропитанную пемзу прокаливают на горелке Бун-
зена до полного разложения нитрата. На пемзе откладывается
около 15 г двуокиси тория.
2. Из щелочного раствора можно выделить около 10—15%
фенилуксусной кислоты, если его подкислить серной кислотой.
Кислота выделяется в виде масла, которое при охлаждении медленно
кристаллизуется.
3. Чтобы облегчить разделение, можно добавить соли.
4. Если проводится несколько опытов, то остатки можно соеди-
нить вместе и перегнать в вакууме. Фракция, кипящая при 190—
210°/20 мм, составляет около 19 г на опыт и представляет собой
главным образом дибензилкетон.
5. С целью дальнейшей очистки продукта кетон можно превра-
тить в его бисульфитное соединение, последнее промыть эфиром,
разложить бикарбонатом натрия и перегнать продукт с водяным
паром.
6. Если предполагается провести несколько опытов, катализа-
тор следует регенерировать после каждого опыта, для чего через
реакционную трубку пропускают в течение 3 час. воздух, посте-
пенно повышая при этом температуру до 550°. В первом опыте
пыход может оказаться несколько меньшим, особенно если не все
окислы азота были удалены из реакционной трубки.
316
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
7. Пользуясь той же методикой, примерно с тем же выходом,
можно получить следующие кетоны: этилбензилкетон из фенил-
уксусной кислоты и пропионовой кислоты, метил-^-фенилзтил-
кетон из гидрокоричной и уксусной кислот и этил-р-фенилэтил-
кетон из гидрокоричной и пропионовой кислот.
р-МЕТИЛВАЛЕРИАНОВАЯ КИСЛОТА
317
Б. ИЗ а-ФЕНИЛАЦЕТОАЦЕТОНИТРИЛА
С6Н5СН (CN) СОСН3
Н2О i"^! С6Н5СН2СОСН
3TNH3+CO2
Предложили: Л. Джулиан и Дж. Оливер.
Проверил: К- Ноллер.
1. Получение
В 3-литровую колбу наливают 350 мл концентрированной
серной кислоты и охлаждают ее до —10°. Медленно, при взбалты-
вании, прибавляют все количество первой порции влажного а-фенил-
ацетоацетонитрила, полученного, согласно методике, описанной
на стр. 503 (что соответствует 188—206 г или J ,2-1,3 мол. сухого
продукта), причем температуру поддерживают ниже 20° (приме-
чание 1). После того как все добавлено, колбу нагревают на паровой
бане до полного растворения компонентов и еще в течение 5 мин. Рас-
твор охлаждают до 0°, быстро приливают 1750 мл воды и колбу ставят
в энергично кипящую водяную баню, на которой ее нагревают 2 часа,
временами взбалтывая. Образуется слой кетона, который по охлажде-
нии отделяют, а кислотный слой извлекают 600 мл эфира. Масляни-
стый слой и эфирные вытяжки промывают последовательно 100 мл
воды, затем эфирный раствор присоединяют к маслянистой жид-
кости, и все вместе сушат 20 г безводного сернокислого натрия.
Раствор фильтруют, сернокислый натрий промывают эфиром, после
чего его выбрасывают. От фильтратов отгоняют эфир и остаток
перегоняют в вакууме из видоизмененной колбы Клайзена с дефлег-
матором высотой 25 см. Собирают фракцию с т. кип. 109—112°/24 мм.
Выход 125—150 г G7—86% теоретич.; примечание 2).
2. Примечания
1. Если применяется чистый сухой а-фенилацетоацетонитрил, то
к серной кислоте необходимо прибавить половинное количество
воды от веса нитрила, в противном случае при нагревании на
паровой бане будет иметь место обугливание.
2. Обычно почти весь продукт перегоняется в этих пределах;
головная фракция и остаток отсутствуют. Однако иногда полу-
чается до 30 г высококипящего остатка, главным образом неизме-
нившегося нитрила. Когда это имеет место, выход соответственно
уменьшается.
3. Другие методы получения
Метилбензилкетон был получен перегонкой смеси бариевых 1
или кальциевых2 солей фенилуксусной и уксусной кислот; про-
пусканием паров этих кислот над нагретой окисью тория, взятой в ка-
честве катализатора 3>4, и нагреванием фенилуксусной кислоты,
уксусно-кислого натрия и уксусного ангидрида 5. Метилбензилкетон
был также получен из хлористого фенилацетила и метилцинка 8, из
хлористого ацетила и дибензилкадмия 7, перегруппировкой окиси
я-фенил-^-метилэтилена 8, нагреванием а-фенил-р-метилэтиленгли-
коля с разбавленной серной кислотой9, нагреванием продукта
присоединения хлорацетона и бромистого фенилмагнияs, дей-
ствием на смесь хлорацетона и бензола хлористым алюминием 1П
и омылением а-фенилацетоуксусного эфирап или фенацилмало-
нового эфира 12.
1 Radziszewski, Ber. 3, 198 A870).
2 Y о u n g, I. Chem. Soc. 59, 621 A891).
3 S e n d e r e n s, Ann. chim. phys. (8) 28, 318 A913).
• Pickard, Kenyon, J. Chem. Soc. 105, 1124 A914).
5 Магидсон, Га р куша, ЖОХ 11, 339 A941).
"Попов, ЖРХО 4, 213 A872); Ber. 5, 500 AS72).
' Oilman, Nelson, Rec. trav. chim. 55, 518 A936).
8Fourneau, Tiffeneau, Compt. rend. 141, 663 A905).
9 T i i f e n e a u, Ann. chim. phys. (8) 10, 345, 368 A907).
10 Mason, Terry, J. Am. Chem. Soc. 62, 1622 A940).
n Beckh, Ber, 31, 3163 A898).
12 M e t г n e r, Ann. 298, 378 A897).
0-МЕТИЛВАЛЕРИАНОВАЯ КИСЛОТА
(З-Метилпентановая кислота)
CH3CH2CH (СНа) СН (СО2С2Н5), + 2 КОН >
СН3СН2СН (СН3) СН (СО,КJ + 2 С2Н5ОН
СН3СН3СН (СН8) СН (СО2КK + H2SO4 >
СН3СН2СН (СН3) СНоСО.Н + СОа + K.SO^
Предложили: Е. Влиет, К- Марвел и К. Юзуех.
Проверили: Г. Гильман и Р. Браун.
1. Получение
В 2-литровую круглодонную колбу, снабженную обратным
холодильником, механической мешалкой и делительной воронкой,
помещают раствор 200 г C,6 мол.) едкого кали (примечание 1)
318
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
в 200 мл воды. Мешалку пускают в ход и медленно прибавляют
к горячему раствору 200 г @,92 мол.) этилового эфира вторично-
бутилмалоновой кислоты (примечание 2); при этом благодаря
выделяющейся теплоте омыления раствор слабо кипит. После при-
бавления всего эфира раствор нагревают так, чтобы он слабо кипел
в течение 2 час. Затем реакционную массу разбавляют 200 мл воды
и отгоняют 200 мл дестиллата, для того, чтобы удалить весь спирт,
образовавшийся во время омыления (примечание 3).
Раствору дают охладиться и прибавляют к нему через делитель-
ную воронку холодную смесь 320 г C,3 мол.) концентрированной
серной кислоты (примечание 4) и 450 мл воды. Подкисление сле-
дует вести медленно и при перемешивании, во избежание вспени-
вания; при этом раствор разогревается и может самопроизвольно
начать кипеть. После того как прибавлена вся серная кислота,
раствор кипятят с обратным холодильником в течение около 3 час.
Когда метилвалериановая кислота выделится в виде маслянистого
слоя, обратный холодильник заменяют автоматическим отдели-
телем (примечание 5) и раствор перегоняют до тех пор, пока вся
органическая кислота практически не будет отделена, причем вод-
ная часть стекает обратно в колбу, на что требуется от 10 до 15 час.
По окончании отгонки воду, находящуюся в самом отделителе
(около 100 мл), взбалтывают с эфиром, чтобы извлечь растворен-
ную метилвалериановую кислоту (примечание 6). Эфир отгоняют,
остаток присоединяют к главной массе сырой кислоты, смешивают
ее с равным объемом сухого бензола (примечание 7) и перегоняют
смесь из видоизмененной колбы Клайзена с дефлегматором. Сна-
чала переходят бензол и вода, а затем при 193—196°/743 мм 8-метил-
валериановая кислота. Выход 66—69 г F2—65% теоретич.; при-
мечание 8).
2. Примечания
1. Едкий натр непригоден для омыления, так как при работе
с ним выпадает натриевая соль e/ло/шчно-бутилмалоновой кислоты
и получается полутвердая масса.
2. Этиловый эфир ешерично-бутилмалоновой кислоты удобно
получать из бромистого еторично-бутила и малонового эфира по
общему способу, описанному в «Синт. орг. преп,», сб. 1, стр. 546.
Выход эфира, кипящего при 124—132°/28 мм,—80—81% теоретич.
Указанный выше выход р-метилвалериановой кислоты был получен
при применении полученного данным способом этилового эфира
еш/?«чнс-бутилмалоновой кислоты.
3. После омыления весь спирт необходимо удалить. Если оста-
вить в растворе некоторое количество спирта, то образуется этило-
вый эфир |В-метилвалериановой кислоты и конечный продукт будет
содержать значительное количество низкокипящих веществ.
МЕТИЛГИДРАЗИН СЕРНОКИСЛЫЙ
319
4. Если взять соляную кислоту, то она перегоняется вместе
с водой и метилвалериановой кислотой и затрудняет очистку про-
дукта .
5. Весьма эффективным оказалось приспособление, описанное
в «Синт. орг. преп.», сб. 1, стр. 523, рис. 25. Для извлечения кис-
лоты были испробованы и другие методы, причем они дали мен<?е
удовлетворительные результаты. При экстракции растворителями
выход понижается приблизительно на 10%, и продукт получается
загрязненным смолистыми примесями.
6. Эфиром извлекается всего 1—2 г кислоты.
7. Кислоту можно высушить и иначе, но описанный метод
является наиболее удобным.
8. По этому методу можно получить и другие одноосновные
жирные кислоты: например н,-капроновую кислоту из этилового
зфира н.-бутилмалоновой кислоты с выходом 75%.
3. Другие методы получения
^-Метилвалериановая кислота может быть получена отщеп-
лением углекислоты от emo/шчн.-бутилмалоновой кислоты * и
действием бромистого этилмагния на N-метиланилид кротоновой
кислоты с последующим омылением 2.
'Van Romburgh, Rec. trav. chim. 6, 153 A887); К u 1 i s с h,
Monatsh. 14, 559 A893); В e n 11 e y, J. Chem. Soc. 67, 267 A895); Olivier,
Rec. trav. Chim. 55, 1032 A936).
s Maxim, I о a n i d, Bull. Soc. Chim. Romania 12, 28 A930) [C. A. 25,
488A931)].
МЕТИЛГИДРАЗИН СЕРНОКИСЛЫЙ
2C6H5CHO + N.2H4 • H,SO4
C6H5CH = N — N = CHC6H5 + (CH3),SO4 + 3 H2O
. 2 C6H5CHO + CH3NHNH2 • HaSO4 + CH3OH
Предложил: X. Xamm.
Проверили: В. Хартман и В. Петерсон.
1. Получение
А. Бензалъазип. В 5-литровую круглодонную колбу, снабжен-
ную прочной стеклянной механической мешалкой, помещают 240 г
A,85 мол.) измельченного гидразинсульфата («Синт. орг. преп.»,
сб. 1., стр. 158), 1,8 л воды и 230 мл B07 г, 3,4 мол.) 28%-ного
водного аммиака (уд. в. 0,90). Смесь перемешивают и, после
того, как весь гидразинсульфат растворится, с помощью делитель-
ной воронки прибавляют в течение 4—5 час. 440 мл D60 г, 4,35 мол.)
320
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
МЕТИЛИЗОПРОПИЛКАРБИНОЛ
321
бензальдегида (примечания 1 и 2). Затем массу перемешивают
еще 2 часа и выпавший бензальазин отсасывают через бюхнеров-
скую воронку, промывают водой и хорошо отжимают. Продукт
растворяют в 800 мл кипящего 95%-ного этилового спирта, и, по
охлаждении, азин выделяется в виде желтых игл с т. пл. 92—93°.
Выход 350—360 г (91—94% теоретич.); кроме того, из маточного
раствора можно выделить еще 10—15 г менее чистого продукта.
Для того чтобы удалить этиловый спирт из азина, его сушат в ваку-
ум-эксикаторе над хлористым кальцием.
Б. Сернокислый метилгидразин. В 3-литровой круглодонной
колбе, снабженной обратным холодильником с хлоркальциевой
трубкой, смешивают 200 г @,96 мол.) беизальазина, 350 мл сухого
бензола, не содержащего тиофена, и 100 .ил A33 г, 1,05 мол.) диметил-
сульфата (примечание 3). Смесь непрерывно нагревают на водяной
бане в течение 5 час, время от времени взбалтывая ее. Затем смесь
охлаждают и твердый продукт присоединения разлагают добавле-
нием 600 мл воды, причем взбалтывание продолжают до тех пор,
пока не исчезнет весь осадок. Бензол и бензальдегид удаляют
перегонкой с водяным паром; оставшуюся жидкость, по охлажде-
нии, обрабатывают 15—20 мл бензальдегида и оставляют на ночь.
Смолу и бензальазин отфильтровывают (примечание 4).
Фильтрат выпаривают в вакууме на водяной бане до тех пор,
пока вся масса не станет полукристаллической. С целью дальней-
шей осушки ее дважды упаривают при уменьшенном давлении
с абсолютным этиловым спиртом, причем каждый раз берут по 50 мл.
Полученную кристаллическую массу растирают с 50 мл абсолют-
ного этилового спирта, фильтруют и этот процесс повторяют еще
раз. Белый кристаллический продукт представляет собою почти
чистый метилгидразинсульфат и содержит очень мало гидразин-
сульфата. После сушки в вакуум-эксикаторе над хлористым каль-
цием выход составляет 105—ПО г G6—80% теоретич.). С целью
очистки сульфат растворяют примерно в 250 мл кипящего 80%-ного
этилового спирта и нерастворившийся осадок (главным образом
гидразинсульфат) отфильтровывают. По охлаждении метилгидразин-
сульфат выпадает в виде белых пластинок, которые отсасывают
и промывают небольшим количеством абсолютного спирта. После
сушки над хлористым кальцием первая порция имеет т. пл. 141 —
142° и весит 70—75 г E1—54% теоретич.; примечание 5).
2. Примечания
1. Бензальдегид не должен содержать бензойной кислоты, для
чего предварительно его взбалтывают с водным раствором соды.
2. Во время реакции смесь энергично перемешивают и в колбе
укрепляют одну или две толстые стеклянные палочки, которые
служат для разбивания комков бензальазина.
3. Учитывая, чрезвычайную ядовитость диметилсульфата, при
работе с ним необходимо соблюдать осторожность, не проливать
его и не вдыхать паров реакционной смеси. Специфическим противо-
ядием от диметилсульфата служит аммиак.
4. Для того, чтобы удалить непрореагировавший гидразин-
сульфат, его превращают в бензальазин. Если фильтрат смешать
с 5 Мл бензальдегида и оставить стоять на 4 часа, то заметного обра-
зования осадка не должно быть.
5. Из маточного раствора можно выделить еще 12 г менее чистого
продукта с т. пл. 133—136°.
3. Другие методы получения
Описанная выше методика в основном заимствована у Тиле Ч
Метилгидразин был также получен восстановлением с последующим
гидролизом нитрозометилмочевины 2, нитрометилуретана 3 и нитро-
зометиламиносульфокислоты 4, а также метилированием гидразин-
гидрата йодистым метилом 5 или диазометаном 8.
1 Т h i e I e, Ann. 376, 244 A910).
а В г u n i n g, там же 253, 7 A889).
3 Backer, Rec. trav. chim. 31, 193 A912).
•Traube, Brehmer, Ber. 52, 1286 A919).
5 H a r r i e s, H a g а, там же 31, 56 A898).
«Staudinger, Kupfer, там же 45, 501 A912).
МЕТИЛИЗОПРОПИЛКАРБИНОЛ
(З-Метилбутанол-2)
(СН3J СНВг + Mg
(СН3J CHMgBr + СН3СНО
(СН3J СНСН (OMgBr) CHg +
> (CH3J CHMgBr
> (СН8)Я СНСН (OMgBr) CH3
H2O (СН3J СНСНОНСН3
Предложили: Н. Дрэк и Дж. Кук.
Проверили: К- Мареел и Б. Войцик.
1. Получение
В 3-литровую трехгорлую колбу, снабженную механической
мешалкой, делительной воронкой и обратным холодильником,
закрытым сверху хлоркальциевой трубкой, помещают 146 г
F гр.-ат.) сухих магниевых стружек (примечание 1) и около 250 мл
абсолютного эфира (примечание 2).
Через делительную воронку прибавляют раствор 600 г D,9 мол.)
бромистого изопропила в 300 мл абсолютного эфира (примечание 3).
Реакция начинается после того, как прибавлено около 15 мл раствора
(примечание 4). Прибавление раствора ведут с такой скоростью,
21 Сборник № 2
322
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
чтобы реакционная смесь все время слабо кипела. Если кипение
становится слишком сильным, колбу охлаждают проточной водой.
Прибавление раствора бромистого изопропила занимает от 3,5
до 4 час, после чего реакционную смесь кипятят 40 мин, с обратным
холодильником на водяной бане.
Затем продукт реакции охлаждают до —5° (примечание 5)
и при этой температуре в течение 1 часа приливают раствор 200 г
D,5 мол.) уксусного альдегида (примечание 6) в 250 мл абсолют-
ного эфира.
После прибавления раствора уксусного альдегида продукт
разлагают, выливая реакционную смесь на 2 кг колотого льда. От
избытка магния освобождаются декантацией. Основную галоидную
соль магния растворяют прибавлением около 1 л 15%-ной серной
кислоты. Эфирный раствор отделяют и водный слой экстрагируют
четыре раза эфиром порциями по 150 мл. Эфирные растворы соеди-
няют, сушат 25 г прокаленного поташа, фильтруют и фракциони-
руют с коротким дефлегматором. Метилизопропилкарбинол кипит
при 110—111,5°. Фракцию, перегоняющуюся при 37—109°, надо
снова высушить и вторично фракционировать. Общий выход 210—
215 г E3—54% теоретич.).
2. Примечания
1. Избыток магния берется для того, чтобы повысить выход
гриньярова реактива.
2. Если во время реакции теряется эфир, можно прибавить его
еще некоторое количество. Эфир для этой реакции надо сушить над
чистой натриевой проволокой.
3. Бромистый изопропил с т. кип. 59—60° применялся без всякой
предварительной очистки. Метод получения бромистого изопропила
описан в «Синт. орг. преп.», сб. 1, стр. 118, а также в настоящем
сборнике на стр. 114.
4. Если вся аппаратура и реактивы совершенно сухие, то для
того, чтобы началась реакция, нет необходимости подогревать
смесь на водяной бане.
5. Температуру необходимо поддерживать не выше —5°.
6. Уксусный альдегид можно удобно получить деполимериза-
цией чистого сухого паральдегида при помощи толуолсульфокислоты
в качестве катализатора. Для данной реакции надо брать уксус-
ный альдегид, кипящий при 20,5—21°. Применение газообразного
уксусного альдегида не ведет к повышению выхода.
3. Другие методы получения
Метилизопропилкарбинол был получен восстановлением метил-
изопропилкетона амальгамой натрия1 и натрием2; обработкой
хлорангидрида хлоруксусной кислоты цинкметилом 3; обработкой
МЕТИЛИЗОПРОПИЛКЕТОН
323
бромангидрида бромуксусной кислоты цинкметилом4; действием
изомасляного альдегида на бромистый метилмагний 5 и как побоч-
ный продукт при реакции между хлорацетоном и йодистым метил-
магнием 6.
1 Munch, Ann. 180, 339 A876).
з Michael, Zeidler, Ann. 385, 262 A911).
3 Богомолец, ЖРХО, 13, 396 A881); Ann. 209, 86 A881).
4 В и н о г р а д о в, там же 191, 128 A878).
5 Henry, Compt. rend. 145, 22 A907).
6 Fo ur n e a u, T i f f e n e a u;i там же 145, 438 A907).
МЕТИЛИЗОПРОПИЛКЕТОН
C-M em илбутанон-2)
(СН3J С (ОН) СН2СН3 + Вг2 > (СН3JСВгСНВгСН3
(СН3J СВгСНВгСНз + Н20 (СН3J CHCOCHg + 2 НВг
Предложили: Ф. Уитмор, В. Звери и X. Ротрок.
Проверили: В. Хартман и Л. Ролл.
Н2О
1. Получение
В круглодонную колбу из стекла «пирекс» емкостью 1,5 л,
снабженную капельной воронкой, эффективной механической мешал-
кой и термометром, помещают 176 г B мол.) дважды перегнанного,
кипящего в пределах 0,5°, /яреяшчн.-амилового спирта. Колбу
погружают в водяную баню и в нагретый до 50—60° спирт медленно
вливают при перемешивании в течение 2 час. 320 г A03 мл, 2 мол.)
брома (примечание 1). Окончив прибавление брома, перемешивание
продолжают еще несколько минут до исчезновения окраски брома
(примечание 2).
В реакционную колбу с сырым триметилэтилендибромидом
прибавляют затем 540 мл воды и колбу соединяют с длинным обрат-
ным холодильником и мешалкой с ртутным затвором (примечание 3).
Смесь кипятят при перемешивании до тех пор, пока гидролиз прак-
тически не пройдет полностью, на что требуется от 3 до 5 час. (при-
мечание 4); обратный холодильник заменяют на обращенный вниз,
и полученный метилизопропилкетон отгоняют при перемешива-
нии из реакционной смеси. Отгонку ведут до тех пор, пока темпе-
ратура не начнет повышаться (примечание 5) и не перестанет отго-
няться масло (около 1,5 часа) или пока перегоняющееся масло не
станет тяжелее воды. Остаток в колбе может быть перегнан для
получения около 380 мл постоянно кипящей бромистоводородной
кислоты.
К дестиллату, состоящему из желтого масла и небольшого слоя
воды, прибавляют соду в порошке (около 10 г) и смесь взбалтывают
324
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
до тех пор, пока водный слой не станет щелочным и почти насыщен-
ным (примечание 6). Слои разделяют и масло кипятят около 16 час.
с обратным холодильником с 20 г порошкообразного поташа и 5 мл
воды (примечание 7). Масляный слой вновь отделяют и сушат
6 г безводного хлористого кальция или поташа (примечание 8).
Затем масло переносят в колбу с 2 г сухой соды и перегоняют с хоро-
шим дефлегматором или перегонной колонкой. Выход метилизо-
пропилкетона, кипящего при 92—94° (примечание 9), составляет
около 102 г E9% теоретич.). Продукт слегка окрашен в желтый
цвет.
2. Примечания
1. Бром прибавляют с такой скоростью, чтобы в реакционной
смеси все время был лишь небольшой избыток его, заметный по
красно-оранжевой окраске. Скорость реакции зависит от темпера-
туры и от эффективности перемешивания. Температура реакцион-
ной смеси все время держится на несколько градусов выше темпе-
ратуры водяной бани.
После того, как прибавлено около половины брома, смесь
благодаря выделению воды становится мутной, и для того, чтобы
бром вступал в реакцию, достаточно уже более низкой температуры
D0—45°).
2. Сырой триметилэтилендибромид может быть очищен промыв-
кой раствором соды и воды, а затем перегонкой высушенного про-
дукта в вакууме; т. кип. 49—51°/11 мм; выход 60—70%. При гидро-
лизе очищенного дибромида выход кетона составляет 78%. Однако
потери при очистке дибромида ведут к некоторому понижению
суммарного выхода кетона, так что очистка дибромида нецелесооб-
разна.
3. Для гидролиза дибромида и переведения всего брома в по-
стоянно кипящую D7,3%) бромистоводородную кислоту берут коли-
чество воды на 50% больше теоретического. Можно брать и меньше
воды, но при этом реакционная смесь темнеет сильнее и выход
кетона несколько понижается.
4. Эффективное перемешивание уменьшает время гидролиза,
и при этом не происходит потери материала, которая вызывается
неравномерным, внезапным вскипанием. Чтобы быть уверенным
в полноте гидролиза, нагревание следует продолжать по меньшей
мере еще 1 час после того, как удельный вес маслянистого слоя
станет меньше удельного веса водного слоя.
5. Как дестиллат, так и остаток вызывают слезоточивость и
с ними надо работать в хорошо действующем вытяжном шкафу.
6. Кетон немного растворим в воде, и сода служит не только
для нейтрализации бромистоводородной кислоты, но и для его
высаливания.
МЕТИЛИЗОПРОПИЛКЕТОН
325
7. Неочищенный кетон содержит небольшие количества бро-
мидов и темнеет, если его сначала не прокипятить над поташом,
даже после нескольких перегонок.
8. Кетон необходимо хорошо высушить, так как иначе он пере-
гоняется с водой около 78°. Для того, чтобы получить кетон обратно,
фракцию эту следует высушить.
9. Хорошие результаты получены с колонкой высотой 55 см,
набитой стеклянными трубочками длиной и диаметром по б мм.
Если работа проводилась с хорошими холодильниками, то всегда
получалось несколько граммов триметилэтилена с т. кип. 34—
36°/735 мм. Присутствие небольшого количества соды во время
последней перегонки препятствует разложению продукта.
3. Другие методы получения
Триметилэтилендибромид был получен присоединением брома
к триметилэтилену х и к третичному амиловому спирту2.
Метилизопропилкетон был получен гидратацией изопропилаце-
тилена 3; из бромистого изопропилмагния и уксусного ангидрида 4;
из хлористого изопропилмагния и уксусноэтилового эфира5;
омылением этилового эфира диметилацетоуксусной кислоты8;
каталитическим путем — пропусканием смеси паров изомасляной
и уксусной кислот над окисью тория 7, а также из бутана или
изобутана и окиси углерода под действием хлористого алюминия 8.
Метилизопропилкетон может быть получен также перегруппиров-
кой метилизопропенилкарбинола 9 или окиси триметилена i°; нагре-
ванием триметилэтиленгликоля с разбавленной соляной кисло-
той п; нагреванием триметилэтиленхлоргидрина B-метил-2-окси-
3-хлорбутана) с водой12 и гидролизом триметилэтилендибромида13.
'Михайленко, ЖРХО 27, 56 A895); F г о е b e, H ос hs tetter,
Monatsh. 23, 1075 A902).
2 J. prakt. Chem. B) 53, 266 A896).
8Флавицкий, Крылов, ЖРХО 10, 347 A878); Кучеров,
Ber. 42, 2761 A909).
4 Sweet, Marvel, J. Am. Chem. Soc. 54, 1188 A932).
5 Ivanoff, Spassoff, Bull, soc' chim. E) 2, 816 A935).
6 Schryver, J. Chem. Soc. 63, 1336 A893).
'Senderens, Compt. rend. 149, 996 A909).
8Hopff, Nenitzescu, Isacescu, Cantuniari, Ber.
69, 2244 A936).
9 Кондаков, ЖРХО 17, 300 A885); N. V. de Bataafsche Petroleum
Maatschappij, франц. пат. 777 032 [С. А. 29, 3686 A935)].
"Леонтович, ЖРХО 35, 606 A903); Ber. 36, 2018 A903); Shell
Development Company; ам. пат. 2 106 347 [С. А. 32, 2542 A938)].
11 Bauer, Ann. 115, 91 A860).
12 Красуский, ЖРХО 34, 287 A902).
13 ЖРХО 27, 359 A895); Froebe, Hochstetter, Monatsh. 23,
1075 A902); С о 1 о n g e, Bull. soc. chim. E) 3, 501 A936).
326
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
S-МЕТИЛИЗОТИОМОЧЕВИНА СЕРНОКИСЛАЯ
B-Метил-2-тиопсевдомочевиш сернокислая)
SCH3
2(H3NKCS+(CH3JSO,
, (HN = С — NH2J;- H2SO4
Предложили: П. Шильднек и У. У индус.
Проверили: Г. Гильман и В. Шульц.
1. Получение
В 2-литровой круглодонной колбе смешивают 152" г B мол.)
мелкорастертои тиомочевины и 70 мл воды и к смеси прибавляют
138 г A,1 мол.) технического диметилсульфата (примечание 1).
Колбу немедленно присоединяют к длинному обратному холодиль-
нику, который в свою очередь соединен с ловушкой. Реакции дают
итти самопроизвольно (примечание 2), применяя охлаждение лишь
в том случае, если она пойдет слишком бурно и колба наполнится
парами. Когда первоначально бурно идущая реакция закончится,
смесь нагревают еще 1 час с обратным холодильником, причем
в это время она закристаллизовывается (примечание 3). Ей дают
охладиться, в колбу прибавляют 200 мл 95%-ного этилового спирта,
продукт отсасывают, дважды промывают порциями по 100 мл
95%-ного спирта и сушат на воздухе. Выход вещества, плавящегося
с разложением при 235°, составляет 190 г. Из спиртового фильтрата,
если его упарить до кашеобразной консистенции и по охлаждении
смешать со 120 мл 95%-ного спирта, можно получить еще 43 г кри-
сталлов с т. пл. 230°. Общий выход 220—233 г G9—84% теоретич.).
2. Примечания
1. Если взятый для синтеза технический диметилсульфат не
окрашен в темнокоричневый цвет, то перед реакцией его можно
не перегонять.
2. Если смесь охладить слишком сильно ледяной водой, само-
произвольно идущая реакция почти прекращается и требуется
слабое подогревание, чтобы она началась снова. Если смесь совсем
не охлаждать, то начавшаяся реакция идет настолько бурно, что
могут произойти потери материала через холодильник. Этого
следует остерегаться, так как диметилсульфат очень ядовит. Необ-
ходимо иметь наготове раствор аммиака, который является специ-
фическим противоядием при отравлениях диметилсульфатом.
При проверке настоящей работы реакция не началась самопроиз-
вольно и, чтобы пустить ее в ход, надо было и колбу и ее содержи-
мое нагреть на^небольшом пламени горелки, придавая ей от руки
МЕТИЛИМИНОДИУКСУСНАЯ КИСЛОТА
327
вращательное движение. Когда реакция начинается, пламя уда-
ляют. Следует иметь наготове сосуд с ледяной водой, чтобы в случае
необходимости замедлить слишком бурную реакцию, особенно,
если длина холодильника менее 125 см.
3. Окончание бурной реакции показывает, что половина тиомо-
чевины прометилирована и что диметилсульфат перешел в моно-
метилсульфат. Для завершения метилирования необходимо силь-
ное нагревание.
3. Другие методы получения
Описанный метод в основном разработан Арндтом1.
1 А г nd t, Ber. 54, 2236 A921).
МЕТИЛИМИНОДИУКСУСНАЯ КИСЛОТА
(NaOH)
2С1СН2СО2Н +CHSNH3
CH3N(CH.2CO2H),+2HC1
Предложил: Дж. Берчет.
Проверили: Дж. Джонсон и П. Баррик.
1. Получение
В 2-литровую колбу, снабженную механической мешалкой,
делительной воронкой и термометром, помещают 189 г B мол.)
хлоруксусной кислоты и 150 мл воды. Колбу охлаждают ледяной
водой и при работающей мешалке приливают холодный раствор
160 г D мол.) едкого натра в 500 мл воды с такой скоростью, чтобы
температура не поднималась выше 30° (примечание 1). После того,
как вся щелочь прибавлена, охлаждающую баню удаляют и мед-
ленно приливают водный раствор (примечание 2), содержащий
31 г A мол.) метиламина. Реакция эта идет с выделением тепла и
температуру поддерживают ниже 50°, погружая колбу время от
времени в ледяную воду. После того как весь метиламин добавлен,
для завершения реакции раствор оставляют стоять в течение 2 час.
Затем к реакционной смеси прибавляют раствор 257 г A,05 мол.)
хлористого бария (ВаС12. 2 Н2О) примерно в 500 мл горячей воды
при энергичном взбалтывании и смесь нагревают на водяной бане
в течение 30 мин. Немедленно выпадает осадок бариевой соли амино-
кислоты, которую по охлаждении до комнатной температуры отфиль-
тровывают с отсасыванием, переносят в стакан и промывают горя-
чей водой (80°) два раза порциями по 250 мл. Бариевую соль сушат
при 100°, выход ее 225—230 г (80—82% теоретич.).
Сухую бариевую соль помещают в 2-литровую колбу, снабжен-
ную механической мешалкой, добавляют 600 мл воды и смесь
нагревают до кипения. Затем из делительной воронки в течение
328
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
МЕТИЛНИТРАТ
329
1 часа постепенно прибавляют к хорошо перемешиваемой смеси
вычисленное количество 5-н. серной кислоты (примечание 3). Смесь
подвергают центрифугированию или фильтруют с отсасыванием
(примечание 4) через тонкий слой фуллеровой земли. Осадок серно-
кислого бария переносят в стакан и извлекают его два раза кипящей
водой порциями по 250 мл. Фильтрат и промывные воды переносят
в колбу Вюрца и, нагревая ее на водяной бане, упаривают в ваку-
уме, до объема 175—200 мл (примечание 5). Сиропообразный оста-
ток выливают в большой стакан и обрабатывают 500 мл абсолют-
ного метанола. Немедленно появляются кристаллы кислоты. Смесь
оставляют стоять на 3—4 часа в бане с льдом, чтобы завершить
осаждение, и кристаллы фильтруют с отсасыванием. Их промывают
два раза метанолом, порциями по 75 мл и сушат при 100°. Метил-
иминодиуксусная кислота образует мелкие бесцветные кристаллы
с т. пл. 215°. Выход 92—105 г F3—71% теоретич.).
При желании получить особенно чистый продукт кислоту можно
переосадить, растворив ее в равном весовом количестве воды и доба-
вив три объема метанола. Потери при очистке составляют 4—5%.
2. Примечания
1. Температуру необходимо регулировать, чтобы избежать обра-
зования гликолевой кислоты. Половины щелочи достаточно для
нейтрализации кислоты; остальное количество можно приливать
быстро, не боясь, что повысится температура.
2. Можно применить технический водный раствор метиламина
B8—33%). Содержание амина следует предварительно определить
титрованием стандартным раствором кислоты.
3. Для выделения кислоты из бариевой соли требуется 1,416 мл
5-н. серной кислоты на 1 г. Перед употреблением кислоту следует
оттитровать.
4. Если в растворе останется неизменившаяся бариевая соль
метилиминодиуксусной кислоты, то она пептизирует сернокислый
барий, что может затруднить фильтрование. Если образуется
коллоидальный сернокислый барий, то рекомендуется добавить
небольшой избыток (менее 1 %) серной кислоты и продолжить нагре-
вание на 20 мин. дольше. Если нужно, следы серной кислоты в ко-
нечном продукте можно удалить переосаждением кислоты мета-
нолом.
5. Остаток должен иметь консистенцию сиропа, однако доста-
точно жидкого, чтобы его можно было свободно вылить из колбы.
3. Другие методы получения
Метилиминодиуксусная кислота до настоящего времени полу-
чалась действием метиламина на циангидрин формальдегида с по-
следующим омылением образующегося динитрила *. Приведенный
выше метод, как было найдено автором, дает значительно более
удовлетворительные результаты.
1 Esch we i 1 е г, Ann. 279, 39A894); I. G. Farbenind. A.-G.; франц.
пат. 804 497 [С. А. 31, 3505 A937)].
МЕТИЛНИТРАТ
CH3OH + HONO2 -"—— CH3ONO2 + H2O
Предложили: А. Блж и Ф. Беберс.
Проверили: Дж. Джонсон и X. Стивенсон.
1. Получение
В колбе, охлаждаемой в бане со льдом, смешивают 425 г C00 мл,
4,7 мол.) химически чистой азотной кислоты (уд. в. 1,42), не содер-
жащей азотистой кислоты (примечание 1), и 550 г C00 мл) хими-
чески чистой серной кислоты (уд. в. 1,84). Во второй колбе, также
охлаждаемой в бане со льдом, к 119 г A50 мл, 3,7 мол.) чистого
метилового спирта (примечание 2) приливают 92 г E0 мл) хими-
чески чистой серной кислоты, поддерживая температуру ниже 10°.
Холодную смесь азотной и серной кислот разливают поровну
в три конические колбы емкостью 500 мл (примечание 3) и каждую
порцию в отдельности обрабатывают х/3 частью смеси метилового
спирта и серной кислоты при непрерывном взбалтывании и тща-
тельном перемешивании (примечание 4). Температуре дают под-
няться довольно быстро до 40° и с помощью внешнего охлаждения
поддерживают эту температуру. При прибавлении смеси метилового
спирта и серной кислоты большая часть метилнитрата выделяется
в виде почти бесцветного маслянистого слоя над кислотным слоем.
Время, необходимое для завершения реакции, составляет 2—3 мин.
для каждой колбы. Реакционную смесь оставляют стоять на холоду
еще на 15 мин., но не больше. Затем быстро отделяют нижний
слой отработанной кислоты и сразу выливают его в большой объем
холодной воды (около 1 л на каждую порцию), чтобы избежать
разложения, которое быстро наступает с обильным выделением
окислов азота.
Оба эфирных слоя соединяют и промывают два раза охлажден-
ным до 0° раствором соли (уд. в. 1,17; примечание 5), порциями
по 25 мл. Ко второй порции промывной жидкости прибавляют
немного (8—10 капель) концентрированного раствора едкого натра
до слабо щелочной реакции на лакмус. Сложный эфир отмывают
от щелочи охлажденным до 0° раствором соли и под конец промы-
вают два раза водой порциями по 15 мл (примечание 6). Продукт
обрабатывают 10—15 г безводного хлористого кальция и оставляют
330
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
стоять над ним, время от времени взбалты"вая его, в течение 1 часа
при 0°. Затем его сливают на свежую порцию хлористого кальция
E г) и после стояния в течение 30 мин. отфильтровывают. Без
дальнейшей очистки сырой эфир (примечание 7) можно применять
для большинства синтетических целей непосредственно, например
для синтеза фенилнитрометана (стр. 513). Выход 190—230 г F6—
80% теоретич.). Неочищенный эфир нельзя хранить — его следует
быстро использовать.
2. Примечания
1. Если азотная кислота окрашена, ее можно обработать неболь-
шим количеством мочевины (около 1—2 г на 100 мл), однако это
следует делать только в том случае, если кислота заметно окрашена.
2. Применялся продажный метанол высокой степени чистоты
без дальнейшей очистки. Для этого синтеза синтетический мета-
нол лучше, чем метиловый спирт древесного происхождения.
3. Необходимо отметить, что при смешении реагирующих
веществ наблюдается сокращение объема. Общий объем смеси кислот
составляет 585 мл (вместо 600 мл), а объем смеси метанола с серной
кислотой — 182 мл (вместо 200 мл).
4. Обработка метанола смесью концентрированных азотной и
серной кислот не лишена некоторых элементов опасности, и поэтому
необходимо принять соответствующие предохранительные меры.
Однако авторы, предложившие настоящую методику, указывают,
что ими было проведено более ста синтезов без единого взрыва или
бурного разложения.
5. Это соответствует 22%-ному раствору хлористого натрия.
Было найдено, что раствор такой концентрации дает удовлетвори-
тельное разделение и не образует эмульсии.
6. Следы кислоты, остающейся в эфире, способствуют разложе-
нию. При нагревании таких препаратов может иметь место сильный
взрыв.
7. Метилнитрат может быть перегнан, если соблюдать должные
меры предосторожности. Продукт нельзя нагревать сразу, и он
не должен содержать свободной кислоты. Однако не рекомендуется
перегонять метилнитрат, поскольку сырой эфир (после промывки
и сушки) в большинстве синтетических реакций дает такие же хо-
рошие выходы, как и перегнанный продукт. Потери при перегонке
не велики, и чистый продукт кипит при 64,5—65°. Нельзя пере-
гревать остаток в перегонной колбе.
3. Другие методы получения
Метилнитрат был получен перегонкой смеси метанола и азотной
кислоты, к которой по каплям прибавлялась смесь метанола с сер-
ной кислотой 1, а также по методике вполне похожей на приведен-
МЕТИЛОВЫЙ ЭФИР ЩАВЕЛЕВОЙ КИСЛОТЫ
331
ную выше, но с разбавленной азотной кислотой2. Ни один из
этих методов не дает удовлетворительных результатов, и часто
имеют место взрывы.
1 Lea, Am. J. Sci. B) 33, 227 A862); Chem. Zentr. 1862, 602.
2 D e 1 ё р i n e, Bull. soc. chim. C) 13, 1044 A895).
МЕТИЛОВЫЙ ЭФИР ЩАВЕЛЕВОЙ КИСЛОТЫ
(СО2НJ + 2 СН3ОН -^1 (СО,СН3J
2 Н2О
Предложил: Э. Боуден.
Проверили: К- Марвел и А. Койд.
1. Получение
В колбу из стекла «пирекс» емкостью 500 мл, закрытую пробкой,
в которую свободно вставлена стеклянная механическая мешалка
и делительная воронка, помещают 90 г A мол.) безводной щавелевой
кислоты («Синт. орг. преп.», сб. 1, стр. 522) и 100 мл G9 г, 2,5 мол.)
метанола (примечание 1). Энергично перемешивая смесь, к ней
медленно прибавляют через делительную воронку 35 мл чистой
концентрированной серной кислоты (примечание 2). Затем смесь
нагревают, если надо (примечание 3), почти до кипения и по воз-
можности быстро фильтруют через 15-сантиметровый фильтр, вло-
женный в слабо нагретую стеклянную воронку; фильтрат собирают
в широкогорлую коническую колбу емкостью 500 мл. Реакцион-
ную колбу споласкивают 40 мл горячего метанола и жидкостью
промывают фильтр. После стояния при 15° в течение 24 час.
(примечание 4) выпавшие кристаллы отсасывают по возможности
досуха, отжимают между листами фильтровальной бумаги и сушат
на воздухе в течение нескольких минут. Фильтрат охлаждают
до —10°, быстро отсасывают выкристаллизовавшийся продукт и
сушат его, как указано выше. Всего получается 100—115 г щаве-
левого эфира,'слегка загрязненного серной кислотой и плавящегося
при 50—52°.
Для очистки полученное вещество растворяют в 100 мл горя-
чего перегнанного метилового спирта, фильтруют раствор через
теплую воронку и оставляют кристаллизоваться. По истечении
нескольких часов кристаллы отсасывают, а фильтрат охлаждают
и обрабатывают,,как указано выше. В общем получается 80—90 г
метилового эфира щавелевой кислоты F8—76% теоретич.), пла-
вящегося при 52,5—53,5° (примечание 5).
332
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
1-МЕТИЛ-2-ПИРИДОН
333
2. Примечания
1. Для реакции применяют продажный метанол, в котором
почти не содержится ацетона. Для перекристаллизации метилового
эфира щавелевой кислоты его предварительно перегоняют.
2. Если взять меньшее количество серной кислоты, чем указано,
то получается меньший выход, в то время как при применении боль-
ших количеств иногда получаются трудно фильтрующиеся растворы.
Важно прибавлять кислоту медленно и при энергичном перемеши-
вании, иначе происходит местное перегревание, раствор темнеет
и конечный продукт получается окрашенным. Некоторые продаж-
ные сорта метанола сильно темнеют в присутствии серной кислоты.
Метанол, применяемый для настоящего синтеза, не должен стано-
виться бурым в присутствии серной кислоты; получаемые филь-
траты окрашены в светложелтый цвет.
3. Растворение щавелевой кислоты вызывает резкое понижение
температуры, в то время как растворение серной кислоты ведет
к повышению температуры.
4. В основном реакция заканчивается в несколько минут, но для
завершения кристаллизации необходимо несколько часов.
5. При больших загрузках получают такой же выход. Если
синтез повторяют несколько раз, то маточный раствор, получен-
ный после первой перекристаллизации, берут вместо исходного
спирта для следующего синтеза и т. д. При таком способе выход
несколько повышается.
3. Другие методы получения
Метиловый эфир щавелевой кислоты может быть получен пере-
гонкой смеси щавелевой кислоты, метилового спирта и серной
кислоты х; растворением безводной щавелевой кислоты в горячем
метаноле 2; этерификацией щавелевой кислоты метиловым спиртом
в присутствии безводного хлористого водорода в качестве катали-
затора3; переэтерификацией этилового эфира щавелевой кислоты4;
пропусканием паров безводного метилового спирта через водную
щавелевую кислоту до удаления воды5; отгонкой смеси метилового
спирта и воды от смеси метилового спирта и водной щавелевой
кислоты и пропусканием полученного дестиллата через слой без-
водного поташа с автоматическим возвращением обезвоженного
отгона в реакционную колбу8. Описанный здесь метод проще
всех вышеперечисленных и дает вполне удовлетворительные выходы,
1 Dumas, Peligot, Ann. chim. phys. B) 58, 44 A835); Ann. 15,
32 A835); WOhler, там же 81, 376 A852).
2 E г 1 e n m e у е г, Jahresb. 1874, 572.
3 R i s i n g, S t i e g 1 i t z, J. Am. Chem. Soc. 40, 726 A918).
1 Pfantil, Monatsh. 31, 316A910); Reimer, Downes, J. Am.
Chem. Soc. 43, 950 A921).
5 Dutt, J. Chem. Soc. 123, 2714 A923).
6 Kenyon, «Синт. орг. преп.». сб. 1, стр. 565.
1-МЕТИЛ-2-ПИРИД0Н
C5H5N +(CH3JSO4 (C5H5NCH3) SO4CH3
(C5H5NCH3) SO4CH3 +3NaOH +2K3Fe (CN),
CH
/4
HC CH
II I +Na(CH3)SO4+2K3NaFe(CNN+2H.)O
HC CO
N
CH
s Предложили: Е. Прилл и С. Мак-Элъвен.
Проверили: К. Марвел и С. Бабкок.
1. Получение
В 5-литровую круглодонную колбу, снабженную делительной
воронкой и обратным холодильником, помещают 145 г A,83 мол.)
сухого пиридина (примечание 1) и из делительной воронки при-
бавляют по каплям 231 г A,83 мол.) диметилсульфата. После добав-
ления всего количества диметилсульфата колбу нагревают на кипя-
щей водяной бане в течение 2 час. для завершения реакции.
От колбы, содержащей сырую соль пиридиния, отъединяют
холодильник и соль растворяют в 400 мл воды. Колбу оборудуют
механической мешалкой, эффективность которой не должна зави-
сеть от объема жидкости в колбе, и раствор охлаждают до 0° смесью
льда и соли. Отдельно приготовляют раствор 1,2 кг C,65 мол.)
железосинеродистого калия в 2,4 л воды и отдельно — раствор
300 г G,5 мол.) едкого натра в 500 мл воды и оба эти раствора при-
бавляют по каплям из двух делительных воронок к энергично
перемешиваемому раствору соли пиридиния с такой скоростью,
чтобы температура реакционной смеси не поднималась выше 10°.
Скорость прибавления обоих растворов регулируется таким обра-
зом, чтобы раствор едкого натра был добавлен к реакционной
смеси одновременно с половиной раствора железосинеродистого
калия, на что обычно требуется около 1 часа. Вторую половину
раствора железосинеродистого калия добавляют еще в течение
1 часа, после чего реакционную массу оставляют стоять 5 час,
причем за это время ее температура поднимается до комнатной.
1-Метил-2-пиридон выделяют из реакционной смеси высалива-
нием, для чего при энергичном перемешивании добавляют 400—500 г
безводного углекислого натрия. После того, как углекислый натрий
перестанет растворяться, перемешивание прекращают и от водного
раствора отделяют желтый или бурый маслянистый слой, содер-
334
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
жащий большую часть метил-пиридона, а также и некоторое коли-
чество непрореагировавшей соли пиридиния, воды и неорганических
солей. Водный слой фильтруют (примечание 2), чтобы отделить
избыток углекислого натрия и выпавший в осадок железистосине-
родистый калий или натрий. Фильтрат разделяют на три части
и каждую часть экстрагируют два раза порциями по 200 мл техни-
ческого изоамилового спирта (примечание 3). Спирт после вторич-
ного извлечения первой водной порции может быть употреблен
для первичного извлечения второй порции, и т. д., так что всего
берется 800 мл изоамилового спирта. Вытяжки соединяют вместе
и добавляют их к маслянистому слою, предварительно отделенному
от реакционной смеси. При этом обычно появляется водный слой,
который отделяют и извлекают 100 мл изоамилового спирта.
Соединенные вытяжки подвергают перегонке под уменьшенным
давлением из видоизмененной колбы Клайзена на водяной бане.
Весь растворитель при зтом возвращается обратно и его можно
вновь применить при следующем синтезе.
Остаток переносят в видоизмененную колбу Клайзена емкостью
250 мл и подвергают его перегонке в вакууме на масляной бане.
Если изоамиловый спирт был тщательно удален, низкокипящей
фракции почти не получается. Выход продукта с т. кип. 122—
124°/11 мм составляет 130—140 г F5—70% теоретич.; примеча-
ния 4 и 5). После того как отгонится весь пиридон, в колбе
остается небольшое количество черного твердого вещества.
2. Примечания
1. Был взят без дополнительной очистки продажный пиридин.
2. Удобнее всего фильтровать через тампон из стеклянной ваты,
заложенный в воронку. Жидкость в воронке следует перемешивать
так, чтобы тяжелый осадок не оседал и не вызывал засорения
фильтра.
3. Если в качестве растворителя для экстрагирования метил-
пиридона из водного раствора взять бензол или эфир, извлечение
проходит очень медленно. Сухой 1-метил-2-пиридон хорошо раство-
рим в этиловом эфире, бензоле и в большинстве других органи-
ческих растворителей. В петролейном эфире и в лигроине метил-
пиридон практически не растворим. Если взболтать смесь воды,
бензола и небольшого количества пиридона, то весь пиридон ока-
жется в водном слое. Если метилпиридон высаливать из водного
раствора достаточным количеством поташа, то маслянистый слой
не растворяется при прибавлении бензола или этилового эфира,
а образуются три слоя. Повидимому, метилпиридон можно извлечь
эфиром или бензолом только, если водный раствор насыщен едким
натром или едким кали, но в этом случае происходит образование
стойкой эмульсии, что крайне затрудняет разделение слоев. '
6-МЕТИЛУРАЦИЛ
335
Если взболтать равные количества воды и технического изоами-
лового спирта с небольшим количеством метилпиридона, то метил-
пиридон распределяется примерно поровну между обоими раство-
рителями. Для извлечения метилпиридона из водного раствора
можно также с успехом применять хлороформ.
4. 1-Метил-2-пиридон в чистом состоянии представляет собой
жидкость без цвета и запаха. Если его не сохранять в запаянной
ампуле, при стоянии он окрашивается в темный цвет.
5. Фишер и Хур * приводят следующие температуры кипения
при различных давлениях: 250°/740 мм; 130°/14,5 мм; 126°/12,5 мм
и 1217Ю мм.
3. Другие методы получения
1-Метил-2-пиридон был получен метилированием 2-пиридона г,
окислением йодистого 1-метилпиридиния 2 и 1-метилпиридиний-
метилсульфата 3 железносинеродистой солью, а также электролити-
ческим окислением 1-метилпиридинийметилсульфата *.
1 Pechmann, Baltzer, Ber. 24, 3144 A891).
2 Decker, J. prakt. Chem. B) 47, 28 A893).
'Decker, Kaufman n, там же B) 84,435A911); Fargher, Fur-
ness, J. Chem. Soc. 107, 690 A915).
«Fisher, Neund linger, Ber. 46, 2544A913); Neundlin-
g e r, Chur, J. prakt. Chem. B) 89, 466 A914); Fisher, С h u г, там же
B) 93, 363 A916).
6-МЕТИЛУРАЦИЛ
NH2CONH
CH3C (NHCONH2)
NHCONH2
CH3COCH2CO2C,H5
CH3C (NHCONH2) == СНСО2С2Н3 -f H,,0
CHCO2C2HS -f NaOH
CH3C (NHCONH2) = CHCO2Na + C2H5OH
NH—CO—NH
CH3C = CHCO2Na + HC1 CH3C = CH — CO + H2O + NaCl
Предложили: Дж. Донливи и М. Кайз.
Проверили: Р. Фьюзон и В. Росс.
1. Получение
В большом кристаллизаторе (диаметром 12,7 см) тщательно
размешивают 80 г A,33 мол.) мочевины в порошке со смесью 160 г
A55 мл, 1,23 мол.) ацетоуксусного эфира (примечание 1), 25 мл
абсолютного спирта (примечание 2) и 10 капель концентрированной
соляной кислоты. Кристаллизатор неплотно покрывают часовым
336
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
стеклом и ставят в вакуум-эксикатор над концентрированной сер-
ной кислотой. Эксикатор непрерывно эвакуируют водоструйным
насосом до тех пор, пока смесь не станет сухой (примечание 3),
на что обычно требуется от 5 до 7 дней (примечание 4). Вес совер-
шенно сухого неочищенного [3-ураминокротонового эфира составляет
200—205 г.
Сухой тщательно измельченный неочищенный р-ураминокрото-
новый эфир размешивают с раствором 80 г B мол.) едкого натра
в 1,2 л воды при 95°. Прозрачный раствор охлаждают до 65° и при
помешивании осторожно подкисляют, медленно добавляя концентри-
рованную соляную кислоту. Почти немедленно выпадает 6-метил-
урацил. По охлаждении смеси продукт отфильтровывают, промы-
вают холодной водой, спиртом и эфиром и сушат на воздухе. Веще-
ство получается в виде бесцветного порошка высокой степени
чистоты. Выход ПО—120 г G1—77% теорстич.). С целью дальней-
шей очистки пиримидин можно подвергать перекристаллизации
из ледяной уксусной кислоты. 6-Метилурацил разлагается выше
300°.
2. Примечания
1. Удовлетворительные результаты получаются даже при при-
менении продажного ацетоуксусного эфира. Метод получения ацето-
уксусного эфира описан в «Синт. орг. преп.», сб. 1, стр. 73.
2. Большее количество спирта только увеличивает период
сушки, не увеличивая выхода. Если спирта не брать, то конден-
сация протекает медленно и выход падает.
3. Если продукту конденсации не дать высохнуть, то в следую-
щей стадии при подкислении выделяется большое количество угле-
кислогб газа, что указывает на неполное использование ацетоуксус-
ного эфира.
4. Рекомендуется часто менять серную кислоту, не реже одного
раза в сутки. Во время сушки образующиеся комочки необходимо
периодически растирать, чтобы ускорить процесс сушки.
3. Другие методы получения
Синтез 6-метилурацила из ацетоуксусного эфира и мочевины
впервые описал Беренд1. Вещество это также было получено
действием гидроокиси свинца на метилтиоурацил в щелочной
среде2; кипячением бензаль-2-D-окси-6-метил)-пиримидилгидра-
зина с соляной кислотой 8 и из мочевины и дикетена 4.
1 В е h r e n d, Ann. 229, 5 A885); Behrend, Roosen, там же 251,
238 A889). См. также В i I t z, H е у п, там же 413, 109 A917).
2 List, там же 236, 23 A886).
3 Т h i е 1 е, В i h a n, там же 302, 308 A898).
1 Carbide and Carbon Chemicals Corporation; ам. пат. 2 138 756 [С. А. 33,
2152 A939)]; Standard Oil Development Company; ам. пат. 2 174 239 [С. А. 34,
450 A940)]; Boese, Ind. Eng. Chem. 32, 16 A940).
й-МЕТИЛ-а-ФЕНЙЛГИДРАЗИН
33?
а-МЕТИЛ-а-ФЕНИЛГИДРАЗИН
( 7 -Метил- У -фенилгидразин )
C6HaN (NO) CH3 + 4 [HI -— - C6H5N (NH2) CH3 + H3O
Предложили: В. Хартман и Л. Ролл.
Проверили: Л. Физер и Дж. Уокер.
1. Получение
В 2-литровую трехгорлую колбу, снабженную эффективной
механической мешалкой, термометром и капельной воронкой,
помещают смесь 200 г C,1 rp.-ат.) цинковой пыли и 300 мл воды. При
энергичном перемешивании к суспензии медленно прибавляют
раствор 100 г @,73 мол.) N-нитрозометиланилина (стр. 372) в 200 мл
B10г, 3,5 мол,) ледяной уксусной кислоты. Поддерживают темпе-
ратуру между Ю и 20° путем внешнего охлаждения или внесения
внутрь мелко наколотого льда. После прибавления всего кислого
раствора (от 1,5 до 2 час. в зависимости от охлаждения) смесь
перемешивают еще 1 час при комнатной температуре, а потом
нагревают до 80° на водяной бане. Горячий раствор отфильтровы-
вают от непрореагировавшего цинка, который промывают три раза,
порциями по 100 мл раствором теплой 5%-ной соляной кислоты.
Фильтрат и промывные воды соединяют, охлаждают и затем при-
бавляют 40%-ный раствор едкого натра в количестве, достаточном
для того, чтобы вновь растворить выпадающий вначале гидрат
окиси цинка (около 1,2 л). Маслянистый слой отделяют, а водный
извлекают два или три раза эфиром порциями по 100 мл. Масло
и эфирные вытяжки соединяют и эфир отгоняют на водяной бане;
остаток перегоняют в вакууме. Выход бесцветного (примечание 1)
а-метил-а-фенилгидразина, кипящего при 106—109713 мм, состав-
ляет 46—50 г E2—56% теоретич.).
2. Примечание
1. а-Метил-а-фенилгидразин при стоянии темнеет.
3. Другие методы получения
а-Метил-а-фенилгидразин был получен восстановлением нитро-
зометиланилина цинком и уксусной кислотой1 или электролитиче-
ским восстановлением2; восстановлением фенилгидразона N-метил-
нитроформальдегида цинком и уксусной кислотой3; нагреванием
метилбензои'лфенилгидразина с концентрированной соляной кисло-
той 4; и метилированием фенилгидразина йодистым метилом в при-
сутствии амида натрия5.
1 Fischer, Ann. 190, 152 A878); 236, 198 A886).
2 Wells, Babcock, France, J. Am. Chem. Soc. 58, 2630 A936).
Bamberger, Schmidt, Ber. 34,
T a i e 1 там же 18, 1744 A885).
3 Bamberger, Schmidt, Ber. 34, 591 A901).
4 T a i e 1, там же 18, 1744 A885).
6 Orammaticakis, Compt. rend. 210, 303 A940).
22 Сборник JV« 2
338
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
5-МЕТИЛФУРФУРОЛ
E-Метил-2-фуралъдегид )
Левулеза (из сахарозы) -\- НС1 С1СН2 — С4
НС —СН
II !
С1СН2—С С —CHO+SnCU + HCl
о
— CHO+4H2O
НС —СН
II II
СН3 — С С —CHO+SnCl4
о
Предложил: И. Ринкес.
Проверили: Дж. Джонсон и А. Бломквист.
1. Получение
В 12-литровую круглодонную колбу, снабженную пробкой,
через которую проходят термометр и широкая согнутая стеклян-
ная трубка, наливают б л 32%-ной соляной кислотьГ(уд. в. 1,163;
примечание 1). Кислоту нагревают до 50° и в ней растворяют при
взбалтывании 1 кг B,9 мол.) измельченного сахара (примечание 2).
Темноокрашенный раствор быстро нагревают до 70—72°, поддер-
живают эту температуру в течение 10 мин. и сразу выливают в боль-
шой глиняный сосуд, в который предварительно помещают 3 кг
колотого льда (рекомендуется работать в вытяжном шкафу; при-
мечание 3). По охлаждении до комнатной температуры к смеси
добавляют 600 г B,67 мол.) продажного кристаллического хло-
ристого олова (SnCl, ¦ 2Н2О). Реакционную смесь энергично пере-
мешивают в течение 10 мин., после чего оставляют стоять двадцать
четыре часа.
На следующий день кислую жидкость фильтруют с отсасыванием
через большую бюхнеровскую воронку, чтобы удалить большие
количества образовавшихся смолистых веществ. Осадок на фильтре
промывают двумя порциями по 350 мл воды и затем двумя порциями
по 300 мл бензола. Объем фильтрата и промывных вод составляет
примерно 10 л. 5-Метилфурфурол извлекают из водного слоя бен-
золом, для чего требуются три порции растворителя по 300 мл
на каждые 2 л жидкости (примечание 4). Соединенные бензольные
вытяжки (около 5 л) разделяют на две или три части и каждую часть
промывают двумя порциями по 150 мл 5%-ного раствора соды и
двумя порциями по 100 мл воды. Бензольный раствор сушат 100—
150 г безводного сернокислого магния (или сернокислого натрия)
5-МЕТИЛ<1*УРФУРОЛ
339
и бензол отгоняют из 1-литровой колбы, снабженной коротким
дефлегматором и капельной воронкой для непрерывного добавле-
ния раствора. Перегонку прекращают, когда температура отходя-
щих паров достигнет 85°.
Остаток переносят в колбу Клайзена на 250 мл, причем послед-
ние капли жидкости в колбе споласкивают двумя небольшими пор-
циями E—10 мл) бензола. Последние следы бензола удаляют
осторожным нагреванием при пониженном давлении, после чего
собирают 5-метилфурфурол при 83—85°/15 мм (примечание 5).
Выход 63—70 г B0—22% теоретич., считая па содержащуюся
в сахаре левулезу; примечание 6).
2. Примечания
1. Техническая соляная кислота дает такие же удовлетвори-
тельные результаты, как и химически чистая. Концентрация соляной
кислоты не должна превышать 32% (уд. в. 1,163), так как в случае
более концентрированной кислоты при нагревании происходит
вспенивание и выход несколько падает E6—57 г).
2. Применялся обычный сахарный песок, содержащий около 3%
крахмала; последний не оказывает вредного действия на реакцию.
3. Более длительное нагревание или повышение температуры
выше 72° является безусловно вредным.
4. Извлечение бензолом должно производиться немедленно после
фильтрования, так как при стоянии вновь выпадают смолистые
вещества.
5. 5-Метилфурфурол при стоянии быстро темнеет и через неко-
торое время становится совсем черным. Обычные антиоксиданты
не замедляют в заметной степени это изменение.
6. 5-Метилфурфурол может быть получен по несколько видоиз-
мененному методу, который ведет к более быстрым, но несколько
худшим результатам 1. Раствор 800 г сахарозы в 1 л горячей воды
медленно приливают к кипящему раствору 500 г кристалличе-
ского хлористого олова и 2 кг хлористого натрия в 4 л 12%-ной
серной кислоты, находящемуся в 12-литровой колбе. Альдегид
отгоняется по мере его образования, дестиллат подщелачивают
содой и подвергают перегонке с паром. Этот второй дестиллат
экстрагируют бензолом и бензольную вытяжку после отгонки бен-
зола перегоняют под уменьшенным давлением. Выход 27—35 г
A0—13% теоретич.).
3. Другие методы получения
5-Метилфурфурол был получен перегонкой рамнозы с разбав-
ленными минеральными кислотами2, а также восстановлением
5-бром- и 5-хлорметилфурфурола хлористым оловом а. Описанный
340
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
выше метод, в котором в качестве исходного сырья применяется
сахароза, был опубликован Ринкесом 4.
1 Scott, Johnson, J. Am. Chem. Soc. 54, 2553 A932).
2 Maquenne, Ann. chim. phys. F) 22, 91 A891); Runde, Scott
Johnson, J. Am. Chem. Soc. 52, 1288 A930).
3 Fenton, Gostling, J. Chem. Soc. 79, 811 A901).
* Rinkes, Rec. trav. chim. 49, 1123 A930); 52, 337 A933).
df-МЕТИОНИН
: N — С (Na) (CO2C2H5J + C1CH2CH,SCH3
CO'
/
C°
/
I! \
IN
! r 1..1 \ NaOH
I
CH.,CH,SCHS
HC1
)N-C(CO2C2H5J
CH.,CH2SCH3
CO3H
ч / CONHC (CO2HJ
CH2CH2SCH3
CONHC (CO2HJ—- CH3SCH2CH,CH (NH,) CO2H
CH2CH2SCH3
Предложили: Дж. Барджер и Т. Вейксслъбаум.
Проверили: X. Кларк и С. Гурин.
1. Получение
А. Натриевое производное этилового эфира фталимидмалоновой
кислоты. К раствору 9,2г@,4гр.-ат.) металлического натрия вЗООлм
абсолютного спирта при 60° прибавляют при энергичном перемеши-
вании 126 г @,41 мол.) этилового эфира фталимидмалоновой кис-
лоты («Синт. орг. преп.», сб. 1, стр. 558). Смесь быстро охлаждают
до 0° и кристаллический продукт немедленно отсасывают и промы-
вают последовательно двумя порциями по 200 мл абсолютного спирта
и двумя порциями по 200 мл эфира. Затем кристаллы сушат сперва
(«-МЕТИОНИН
341
в вакуум-эксикаторе, а затем в течение 8 час. нагревают при пони-
женном давлении при 15 мм в колбе, погруженной в масляную баню,
температура которой поддерживается при 145—155° (примечание 1).
Выход 108—111 г (82—85% теоретич.).
Б. Диэтиловый эфир 1-метилтиол-3-фталимидопропан-3,3-ди-
карбоновой кислоты. В 1-литровую трехгорлую колбу, снабженную
холодильником, термометром и стеклянной трубкой для отбора
пробы (трубка закрывается пробкой), помещают смесь 85 г @,26 мол.)
натриевого производного этилового эфира фталимидмалоновой кис-
лоты и 43 г @,39 мол.) |3-хлорэтилметилсульфида (стр. 562) и все
это нагревают на масляной бане до 160—165°. Через полтора-два
часа смесь уже не дает щелочной реакции на лакмус. Избыток хлор-
этилметилсульфида отгоняют при пониженном давлении (приме-
чание 2), маслянистый остаток обрабатывают 150 мл теплой воды
и получившуюся смесь переносят в стакан, помещенный в охлади-
тельную смесь. Выпавшие кристаллы отсасывают, промывают
100 мл холодной воды и перекристаллизовывают из возможно мень-
шего количества теплого абсолютного спирта. Выход чистого про-
дукта с т. пл. 66—67° составляет 75—79 г G6—80% теоретич.).
В. 1-Метилтиол-3-фталамидпропан-3,3-дикарбоновая кислота.
Раствор 25 г @,066 мол.) полученного выше эфира в 30 мл 95%-ного
спирта нагревают в круглодонной колбе емкостью 200 мл на водяной
бане и прибавляют 70 мл 5-н. раствора едкого натра. Мутную жид-
кость нагревают до тех пор, пока проба при разбавлении водой не
станет прозрачной, для чего требуется около 2 час. Раствор охла-
ждают до 0° и осторожно нейтрализуют по конго 0,2-н. раствором
соляной кислоты, после чего медленно добавляют еще 75 мл 5-н.
соляной кислоты, причем температуру все время поддерживают при
0°. Кислота выпадает в виде бесцветных кристаллов. Для заверше-
ния этого выпадения медленно добавляют 60 мл концентрированной
соляной кислоты (уд. в. 1,19). Полученный продукт фильтруют
с отсасыванием и отмывают от минеральных солей небольшими пор-
циями ледяной воды. После сушки в вакууме получают 21,5—22 г
продукта с т. пл. 141 —143°, что составляет 95,5—98% теоретич.
Г. Метионин. На кипящей водяной бане нагревают суспензию
21,5 г @,063 мол.) полученной выше трикарбоновои кислоты в 350 мл
горячей воды и приливают 40 мл концентрированной соляной кис-
лоты (уд. в. 1,19). При этом выделяется углекислый газ, и вещество
переходит в раствор. После полуторачасового нагревания доба-
вляют еще 200 мл концентрированной соляной кислоты и нагрева-
ние продолжают еще 45 мин. По охлаждении из раствора выпадает
фталевая кислота, которую отфильтровывают и промывают два раза
водой порциями по 50 мл (примечание 3). Фильтрат и промывные
воды соединяют вместе и упаривают досуха на водяной бане при
уменьшенном давлении. Сухой остаток растворяют в 50 мл горячей
воды, раствор обрабатывают 18 мл пиридина и выливают в 150 мл
342
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
горячего абсолютного спирта. При этом метионин быстро выкристал-
лизовывается. По охлаждении его отфильтровывают, промывают
спиртом и сушат. Вес первой порции кристаллов около 6,9 г. Маточ-
ный раствор упаривают досуха, остаток извлекают 15 мл горячей
воды и раствор выливают в 50 мл абсолютного спирта, в резуль-
тате чего получают еще 1,3 г. Полученные 8,2 г почти чистого метио-
нина суспендируют в 200 мл кипящего абсолютного эфира, филь-
труют и сушат. Выход метионина с т. пл. 279—280° (исправл.)
7,9—8,0 г (84—85% теоретич.).
2. Примечания
1. Натриевое производное этилового эфира фталимидмалоновой
кислоты кристаллизуется с 1,5 молекулами спирта, которые можно
удалить только нагреванием в вакууме выше 140°.
2. Если дестиллат подвергнуть вторичной перегонке, можно
получить 10—12 г чистого продукта.
3. Полученная таким образом фталевая кислота весит около
7,8 г G5% теоретич.) и плавится при 188°. Если в этот момент не
удалить большую часть фталевой кислоты, то вместе с метионином
может выделиться пиридинфталат, что осложнит дальнейшую
очистку.
3. Другие методы получения
^/-Метионин был впервые синтезирован по методу Штрекера,
но с очень малым выходом х. Описанная выше методика, основанная
на указаниях опубликованных авторами настоящего синтеза 2,
имеет то преимущество по сравнению с методом Виндуса и Мар-
вела -\ что выход значительно выше E4—60% против 13—19%,
считая на взятый [3-хлорэтилметилсульфид).
Метионин был получен также и без применения |3-хлорэтилметил-
сульфида, исходя из я-бензоиламино-у-бутиролактона 4 и из а-аце-
тил-у-бутиролактона s. В первом из этих синтезов сперва полу-
чается этиловый эфир а-бензоиламино-у-хлормасляной кислоты,
затем продукт его взаимодействия с метилмеркаптаном, который
затем омыляется. Согласно второму способу, получается а-окси-
ашшо-у-бутиролактон, который восстанавливают до а-амино-у-бу-
тиролактона и последний превращают в 3,6-б«с(|?-оксиэтил)-
2,5-дикетопиперазин. Диоксидикетопиперазин превращают в соответ-
ственное дихлорэтиловое соединение, из которого после реакции с
метилмеркаптаном и последующего гидролиза получается метионин.
1 Barge r, Coyne, Biochem. J. 22, 1417 A928).
2 В а г g e r, Weichselbaum, там же 25, 997 A931).
3 Windus, Marvel, J. Am. Chem. Soc. 52, 2575 A930).
4 Hill, R о b s о n, Biochem. J. 30, 248 A936).
6Snyder, Andre en, Cannon, Peters, J. Am. Chem. Soc,
64, 2082 A942).
МЕТОКСИАЦЕТОНИТРИЛ
343
МЕТОКСИАЦЕТОНИТРИЛ
NaCN -f- CH.,0 -f- HaO HOCH2CN -\- NaOH
HOCH,CN -f- (CH3)a SO4 -f- NaOH
. CH3OCH2CN -f- CH3NaSO4 -f H.,0
Предложили: Дж. Скарроу и Я. Аллен.
Проверили: Р. Фьюзон и Ч. Вудеард.
1. Получение
Приготовление метоксиацетонитрила надо про-
изводить в вытяжном шкафу с хорошо работающей
тягой. Диметилсульфат очень ядовит, и, несмотря
на высокую точку кипения, при обычной темпера-
туре давление его паров высоко. Обычным противоя~
дием для диметилсульфата служит аммиак, ко-
торый необходимо все время иметь под рукой,
чтобы разрушать случайно пролитый диметил-
сульфат. Рекомендуется во время работы часто
обмывать руки слабым раствором аммиака.
В 1-литровую трехгорлую круглодонную колбу, снабженную
мешалкой, термометром для отсчета низких температур (примеча-
ние 1) и капельной воронкой, помещают 98 г B мол.) растертого
в порошок цианистого натрия (примечание 2) и 200 мл воды. Ме-
шалку пускают в ход и начинают прибавлять к раствору неболь-
шими количествами 60 г B мол.) параформальдегида (примечание 3).
Когда температура поднимется до 20—25° и цианистый натрий пе-
рейдет в раствор, колбу помещают в охладительную смесь и осталь-
ное количество параформальдегнда прибавляют при температуре
ниже 25е.
В капельную воронку помещают 200 мл B70 г, 2,1 мол.) техни-
ческого диметилсульфата и, когда температура внутри колбы упа-
дет до 13°, к реакционной смеси прибавляют 20—30 мл диметилсуль-
фата. При этом должна начаться экзотермическая реакция; иногда
приходится, чтобы дать реакции начаться, удалить баню со льдом
(примечание 4). Когда температура начнет понижаться, прибавляют
остальной диметилсульфат с такой скоростью, чтобы температура
держалась между 12 и 15°: обычно на это требуется не менее 20 мин.
После окончания прибавления перемешивание продолжают еще
40 мин., за это время температ.ура обычно падает до 5°. Мешалку
останавливают и немедленно отделяют верхний маслянистый слой
(примечание 5). Нижний водный слой переносят опять в колбу и
снова метилируют так же, как и первый раз, свежей порцией 200 мл,
диметилсульфата (примечание б).
344
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
Маслянистый верхний слой сушат 10 г безводного сернокислого
натрия (примечание 7) и перегоняют в вакууме. Перегонку ведут
с хорошим дефлегматором (примечание 8). Фракция, кипящая при
15 мм ниже 70°, в главной массе является метоксиацетонитрилом;
его получается 60—70 г. Верхний слой, полученный после второго
метилирования, обрабатывают точно так же, причем метоксиацето-
нитрила получается 55—60 г. В колбе после отгонки остается диме-
тилсульфат, который можно снова применять для дальнейшей
работы (примечание 9).
Полученные фракции соединяют и перегоняют при атмосферном
давлении с хорошим дефлегматором; при 118—122° перегоняется
в виде бесцветной жидкости около 95% продукта. Суммарный вы-
ход метоксиацетонитрила 100—110 г G0—77% теоретич.).
2. Примечания
1. Удобнее всего работать с термометром длиной 30—35 см,
градуированным от — 50° до +50°; нужные температуры на шкале
будут находиться в этом случае вне колбы.
2. Цианистый калий дает худшие результаты.
3. Точно такие же хорошие выходы получают и при работе с про-
дажным раствором формалина; необходимо лишь учитывать коли-
чество вводимой вместе с ним воды — общий объем не должен пре-
вышать 200 мл.
4. Реакция обычно начинается сразу, иногда же лишь после
того, как смесь нагреется.
5. Если смесь оставить стоять, то температура поднимается до
40—50°, появляется красная окраска и выход получается ничтожный.
6. Полученный обратно диметилсульфат можно использовать
в дальнейшей работе без предварительной очистки.
7. Ни одно из применяемых обычно для сушки веществ не яв-
ляется вполне удовлетворительным: все же лучшим можно считать
сернокислый натрий.
8. По мнению авторов, лучше всего для этой перегонки брать
дефлегматор Глинского. Для того, чтобы кипение шло без толчков,
в пробку колбы .вставляют капиллярную трубку, которая доходит
почти до дна колбы и через которую поступает воздух. При второй
перегонке достаточно для этой цели кусочка пористой глиняной
тарелки.
9. Удельные веса жидкости, остающейся в колбе B опыта),
1,31 и 1,32. Удельный вес чистого диметилсульфата 1,35.
3. Другие методы получения
Метоксиацетонитрил получается при метилировании оксиаце-
тонитрила диметилсульфатрм х при действии цианистой меди или
МОНОЭТИЛОВЫЙ ЭФИР СЕБАЦИНОВОЙ КИСЛОТЫ
345
цианистой ртути на хлорметиловый эфир2 и при дегидратации
метоксиацетамида фосфорным ангидридом 3. Описанный выше ме-
тод является видоизменением метода Польсторфа и Мейера х.
IPolstorff, Meyer, Ber. 45, 1011 A912); S 1 a t с г, S t с р h e n,
J. Chem. Soc 117, 312 A920); M a 1 k i n, Robinson, там же 127, 372 A925).
3 G a u t h i e r, Ann. chim. phys. (8) 16, 302, 306 A909).
3 К i 1 p i, Z. physik. Chem. 86, 671 A914).
МОНОЭТИЛОВЫЙ ЭФИР СЕБАЦИНОВОЙ КИСЛОТЫ
(Кислый этиловый эфир себациновой кислоты)
(НС1)
НО2С (СН2)8 СО2Н + С.2Н5ОН
С2Н4ОХ (СН2)8 СОоН + Н2О
Предложили: Ш. Сванн мл., Р. Элер и Р. Вусвелль.
Проверили: Л. Смит и Дж. Клегг.
1. Получение
В 1-литровую видоизмененную колбу Клайзена (примечание 1),
боковой отвод которой закрыт корковой пробкой, помещают смесь
202 г A мол.) себациновой кислоты, 150 г @,58 мол.) диэтилового
эфира себациновой кислоты (примечание 2), 50 млщ-н.-бутилового
эфира (примечание 3) и 30 г B5 мл) концентрированной соляной
кислоты (уд. в. 1,19). В верхней части колбы устанавливают обрат-
ный холодильник.
Колбу нагревают на металлической бане (сплав Вуда) до 160—
170°, пока смесь не станет совершенно гомогенной. Тогда темпера-
туру бани понижают до 120—130° и через холодильник приливают
60 мл A мол.) 95%-ного этилового спирта. Смесь кипятят в течение
2 час, к концу этого времени приливают дополнительно 20 мл
этилового спирта и раствор кипятят еще 2 часа.
Металлической бане дают охладиться до 75° и реакционную смесь
подвергаютперегонке в вакууме водоструйного насоса (примечание 4).
Температуру бани медленно повышают и перегонку с водоструйным
насосом продолжают, пока температура бани не достигает 125°.
Тогда баню вновь охлаждают до 75—80J и перегонку продолжают
при меньшем давлении, пользуясь масляным насосом.
Первые фракции содержат небольшое количество спирта, воду
и дн-н.-бутиловый эфир (примечание 5). Следующая фракция с
т. кип. 156—158°/6 мм является дизтиловым эфиром себацнновой
кислоты (примечание 6). Моноэтиловый эфир себациновон кислоты
собирают при 183—18776 мм. Продукт плавится при 34—36°, и
вес его 114—124 г E0—54% вычисленного количества, считая на
использованную себашшовую кислоту). Повторная перегонка го-
ловной фракции (т. кип. 175—18376 мм) и хвостовой фракции
346
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
1,2-НАФТАЛЕВЫЙ АНГИДРИД
347
(т. кип. 187—195°/6 мм) дает еще 24—26 г чистого моноэфира.
Суммарный выход 138—150 г F0—65% теоретич.; примечания
7 и 8).
2. Примечания
1. Дефлегматор колбы должен быть длиной не менее 35 см и
хорошо изолирован. Его можно обмотать 10 мм асбестовым шнуром.
2. Добавление диэтилового эфира себациновой кислоты в на-
чале реакции уменьшает его образование из реагирующих веществ,
так что главным продуктом реакции становится моиоэфир.
Диэтиловый эфир себациновой кислоты удобно получать кипя-
чением с обратным холодильником 130 г @,65 мол.) себациновой
кислоты, 250 г этилового спирта и 25 мл концентрированной серной
кислоты. Выход составляет 90% и продукт кипит при 156—158°/6 мм.
См. также примечание 8, стр. 579.
3. н.-Бутиловый эфир применяется потому, что по сравнению
с другими возможными соединениями он допускает образование
гомогенной реакционной смеси.
4. В начале перегонки жидкость в колбе очень сильно пенится.
Поэтому желательно понижать давление постепенно и не применять
слишком глубокого вакуума, пока вспенивание не прекратится.
5. н.-Бутиловый эфир может быть вновь выделен в чистом со-
стоянии простой перегонкой после отделения от него воды.
6. После того как отогнан диэтиловый эфир себациновой кислоты,
воду из холодильника следует спустить, для того чтобы монозфир
не застывал до попадания в приемник. Полученный диэтиловый
эфир весит 150—175 г и может быть непосредственно использован
в последующих синтезах.
7. В последующих синтезах остаток от перегонки оставляют
в колбе. Этим путем при загрузках в 1 или 2 мол. выход увеличи-
вается до 70—77"%-
8. Авторы, предложившие синтез, указывают, что при загрузке
в 1 мол. выход моноэтилового эфира адипиновой кислоты с т. кип.
155—157°/7 мм по этой методике составил 71—75%.
3. Другие методы получения
Моноэтиловый эфир себациновой кислоты был получен непосред-
ственной этерификацией себациновой кислоты этиловым спир-
том ', полуомылением диэтилового эфира себациновой кислоты2
и нагреванием эквимолекулярных количеств себацииовой кислоты
и диэтилового эфира себациновой кислоты в течение нескольких
часов 3.
1 N е i s о n, J. Chem. Soc. 29, 310 A876).
3 Walker, там же 61, 713 A892).
3 Fourneau, Sabetay, Bull., soc. chim. D) 43, 859 A928); D) 45,
1,2-НАФТАЛЕВЫЙ АНГИДРИД
A,2-Нафталиндикарбоноеой кислоты ангидрид)
СО,
i
СО
о
-СО
о
>—со
+ H2S
Предложили: Е. Хершберг и Л. Физер.
Проверили: К- Ноллер и Киноман.
1. Получение
В колбу Клайзена емкостью 50 мл, к которой в качестве прием-
ника припаяна колба Вюрца, помещают 20 г @,1 мол.) 3,4-дигидро-
1,2-нафталевого ангидрида (стр.53) и 3,2г @,1 rp.-ат.) серы. Колбу
погружают в баню (примечание I), предварительно нагретую до
230—235°, и взбалтывают ее до тех пор, пока комочки серы не рас-
творятся A5—20 мин.). Затем температуру повышают до 250°
и поддерживают ее в течение 30 мин. (примечание 2). Остаток пере-
гойяют в вакууме (примечание 3) и дестиллат перекристаллизовы-
вают из 150 мл бензола, к которому добавлено 50 мл лигроина
(т. кип. 60—80°) при температуре кипения. Выход 15—18 г G6—
91% теоретич.) светложелтых игл с температурой плавления
166—167°.
2. Примечания
1. Можно взять баню из сплава Вуда (т. пл. около 150°) или
из смеси 10 частей азотнокислого калия и 7,5 частей азотистокис-
лого натрия.
2. Если нагревание при 250° продолжить до прекращения выде-
ления сероводорода (около 10 час), то после перекристаллизации
получается продукт, который светлее по цвету и в меньшей степени
спадается перед расплавлением.
3. Продукт перегоняется при 210—215°/12—13 мм, при темпе-
ратуре бани 260°.
3. Другие методы получения
1,2-Нафталевый ангидрид был получен омылением динитрила
1,2-нафталевой кислоты1, окислением соответственно замещенных
348
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
а-НАФТОЙНАЯ КИСЛОТА
349
углеводородов или кетонов 2, и дегидрогенизацией соответствен-
ного 3,4-дигидро-соединения бромом 3 или серой 4.
1 С 1 е v е, Вег. 25, 2475 A892); Waliimann, J. prakt. Chem. B) 127,
197 A930); Cook, J. Chem. Soc. 1932, 462.
2 F r e u n d, Fleischer, Ann. 399, 186, 210 A913); Kruber,
Sch a de, Ber. 68, 11 A935).
3 A u w e r s, M о 1 1 e r, J. prakt. Chem. B) 109, 141 A925).
4 Fieser, Hershberg, J. Am. Chem. Soc. 57, 1853 A935).
а-НАФТОЙНАЯ КИСЛОТА
A-Нафтойная кислота)
Br
•Mg > C10H7MgBr
C10H7MgBr + CO.,
2 C10H,CO2MgBr + H,SO4 2
C10H7CO2MgBr
CO,H
MgBt\,+MgSO4
Предложили: Г. Гильман, Н. Сент-Джон и Ф. Шульце.
Проверил: К- Иоллер.
1. Получение
¦В 2-литровую трехгорлую колбу, снабженную механической
мешалкой, обратным холодильником и делительной воронкой,
помещают 24,3 г A rp.-ат.) магниевых стружек (примечание 1). Маг-
ний обливают 100 мл абсолютного эфира, прибавляют Ю мл A5 г,
0,07 мол.) гх-бромнафталина (примечание 2) и один или два кристал-
лика иода (примечание 3), для того чтобы вызвать начало реакции.
Под колбу ставят теплую водяную баню D5° или выше) и удаляют
ее, как только начнется реакция. Мешалку пускают в ход и при-
бавляют к реакционной смеси 192 г @,93 мол.) «-бромнафталина
в 500 мл абсолютного эфира с такой скоростью, чтобы реакция шла
энергично, но не очень бурно, на что требуется от 1,5 до 3 час.
Когда прибавление а-бромнафталина закончено, под колбу снова
ставят теплую водяную баню и продолжают перемешивание и кипя-
чение с обратным холодильником в продолжение 30 мин.; после
этого магнийорганическое соединение, которое собирается в виде
тяжелого масла на дне колбы, растворяют прибавлением 533 мл
сухого бензола (примечание 4).
Затем реакционную смесь охлаждают смесью льда и соли. Дели-
тельную воронку заменяют каучуковой пробкой с двумя отверстиями,
в одно из которых вставлен термометр, так что его шарик погружен
в реакционную смесь, а в другое — короткая стеклянная трубка,
вытянутая снаружи в тонкий капилляр (примечание 5). Когда тем-
пература реакционной смеси достигнет —7°, холодильник заме-
няют газоприводной трубкой диаметром 10 мм, укрепленной таким
образом, чтобы ее конец находился приблизительно на 50 мм над
поверхностью реакционной смеси (примечание б). Смесь перемеши-
вают и впускают через газоприводную трубку сухую углекислоту
(примечание 7). Скорость тока газа регулируют таким образом,
чтобы температура смеси не превышала —2°. После того как
реакция закончится, что обычно происходит через Р/4—*/а часа,
температура падает ниже — 7е и не повышается при ускорении
тока углекислого газа.
Когда это достигнуто, к реакционной смеси, не прекращая охла-
ждения и перемешивания, медленно прибавляют 25%-ную серную
кислоту до тех пор, пока не прекратится реакция и весь избыток
магния не растворится (примечание 8). Затем маслянистый слой
отделяют, а водный слой экстрагируют двумя порциями эфира по
100 мл. Соединенные эфирно-бензольные вытяжки (примечание 9)
извлекают в делительной воронке тремя порциями 25%-ного едкого
натра по 100 мл каждая.
Каждую щелочную вытяжку экстрагируют последовательно
100 мл эфира, затем их смешивают и нагревают до 100°, для того
чтобы удалить летучие примеси.
Полученный щелочный раствор охлаждают, сильно подкисляют
50%-ной серной кислотой, выпавшую а-нафтойную кислоту отса-
сывают, промывают до удаления сернокислых солей и сушат. Вы-
ход неочищенного продукта, который плавится при 142—155°,
равен 130—135 г. С целью очистки кислоту растворяют d 400 мл
горячего толуола, прибавляют к раствору небольшое количество
инфузорной земли и раствор фильтруют с отсасыванием через горя-
чую бюхнеропскую воронку. Фильтрат охлаждают ледяной водой,
выпавшую а-нафтойную кислоту отсасывают, промывают холодным
толуолом до тех пор, пока фильтрат не станет бесцветным, и сушат.
Таким образом получается светлоокрашенная а-нафтойная кислота,
плавящаяся при 159—161°. Выход 118—121 г F8—70% теоретич.;
примечание 10).
2. Примечания
1. Применялся магний в виде мелких стружек. После того как
реакция начиналась, она шла гладко и с любым сортом стружек.
2. а-Бромнафталин («Синт. орг. преп.», сб. 1, стр. 127) следует
перегнать в вакууме, собирая фракцию 144— 147°/1б мм.
350
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
р-НАФТОЙНАЯ КИСЛОТА
351
3. При проверке было найдено, что реакция начинается быстро,
после прибавления 0,5 г иода.
4. Необходимо растворить гриньяров реактив в бензоле, чтобы
предотвратить его затвердение при охлаждении раствора. Бензол
следует прибавлять к реакционной смеси до охлаждения, так как
после охлаждения магнийорганическое соединение затвердевает
и растворяется с большим трудом.
5. Капилляр предназначен для того, чтобы обеспечить медлен-
ный выход избытка углекислого газа и чтобы давление в колбе не
было чрезмерным.
6. Если газоприводную трубку опустить ниже, то она может
закупориться.
7. Углекислый газ из баллона надо высушить пропусканием
через две промывные склянки с серной кислотой.
8. Внешнее охлаждение льдом дает возможность быстро про-
вести разложение без опасности потерять часть продукта в резуль-
тате нагрева массы до кипения.
9. Если эфирно-бензольный раствор не вполне прозрачен, то
перед извлечением едким натром его следует профильтровать.
10. Вторая перекристаллизация из толуола дает почти белый
продукт с т. пл. 160,5—162°.
3. Другие методы получения
а-Нафтойная кислота может быть получена из бромистого а-наф-
тилмагния и газообразной J или твердой 2 углекислоты; омылением
этилового эфира а-нафтойной кислоты, в свою очередь также полу-
ченного из бромистого а-нафтилмагния3; омылением цианистого
а-нафтила * и из метил-а-нафтилкетона и хлорноватистокислого
калия2. Кислота была также получена сплавлением натриевой
соли а-нафталинсульфокислоты с муравьинокислым натрием 5 и из
нафталина и хлорангидрида карбаминовой кислоты в присутствии
хлористого алюминия с последующим омылением образовавшегося
амида а-нафтойной кислоты 6. Методы получения а-нафтойной кис-
лоты обсуждены в работе Валя, Гедкоопа и Геберлейна7.
Вышеописанный способ 8 разработан на основе методов Экри х,
Блайках и Уитмора и Фокса*.
1 А с г е е, Вег. 37, 625 A904); В 1 i с k e, J. Am. Chem. Soc. 49, 2846
A927); Whitmore, Fox, там же 51, 3363 A929).
2 F i e s e r, Holmes, Newman, там же 58, 1055 A936).
i Loder Whitmore, там же 57, 2727 A935); см. также стр. 592.
4 Merz, Muhlhaiiscr, Ber. 3, 712 A870).
6 Meyer, там же 3, 364 A870).
°Gattermann, Schmidt, там же 20, 860 A887).
'Wahl, Goedkoop, Heberlein, Bull. soc. chim. E) 6, 533
A939).
«Oilman, St. John, St. John, Rec. trav. chim. 48, 594 A929).
COCH,
Р-НАФТОЙНАЯ КИСЛОТА
3NaOCl —
Na
"+CHCl3+2Na0H
C]eH7CO2Na + HC1
. C10H7CO2H + NaCl
Предложили: М. Ньюмен и X. Холъмс.
Проверили: В. Хартман и Ф. Джонс.
1. Получение
В 3-литровой колбе приготовляют раствор 184 г D,6 мол.) едкого
натра в 300—400 мл воды и добавляют достаточное количество льда,
чтобы общий объем равнялся 1,5 л. Через раствор пропускают хлор,
поддерживая температуру ниже 0э погружением колбы в баню
с охладительной смесью. Хлор пропускают до нейтральной реакции
на лакмус, после чего приливают раствор 34 г едкого натра в 50мл
воды (примечания 1 и 2). Колбу зажимают в лапке и снабжают
термометром и эффективной мешалкой. Раствор нагревают до 55°
и добавляют 85 г @,5 мол.) метил-^-нафтилкетона (примечание 3).
Смесь энергично перемешивают и, после того как начнется экзотер-
мическая реакция, температуру поддерживают при 60—70° (приме-
чание 4), часто погружая колбу в баню со льдом до тех пор, пока
температура не перестанет подниматься. На это требуется от 30 до
40 мин. Раствор перемешивают еще 30 мин., после чего избыток гипо-
хлорита разрушают добавлением раствора 50 г бисульфита натрия
в 200 мл воды (примечание 5). По охлаждении до комнатной темпе-
ратуры реакционную смесь переносят в 4-литровый стакан и осто-
рожно подкисляют 200 мл концентрированной соляной кислоты.
Сырую бесцветную кислоту собирают на воронке Бюхнера, промы-
вают водой и возможно лучше отсасывают, пользуясь резиновой
пластинкой. Кислоту сушат и перекристаллизовывают(примечание 6)
из 600 мл 95%-ного спирта. Получают 75—76 г (87—88% теоре-
тич.) 8-нафтойной кислоты с т. пл. 184—185° (исправлен.). Если
отогнать от маточного раствора 450 мл растворителя, то можно
получить еще 9 г A0% теоретич.) кислоты с т. пл. 181—183° (ис-
правлен.; примечание 7).
2. Примечания
1. Имеются указания, что, приготовляя раствор гипохлорита
натрия пропусканием хлора через раствор едкого натра, очень
трудно определить момент нейтрализации вследствие мгновенного
обесцвечивания индикатора. В случае избытка хлора, даже при
щелочной реакции Конечного раствора, достигнутой дополнительным
добавлением едкого натра, окисление метил-р-нафтилкетона в наф.
352
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
1,2-НАФТОХИНОН
353
тойную кислоту все же не будет иметь места. Поэтому рекомендуется
следующий видоизмененный метод получения раствора гипохлорита.
Раствор 218 г E,45 мол.) едкого натра в 300 мл воды охлаждают
в 3-литровой колбе до комнатной температуры водопроводной водой.
Добавляют 1250 г льда и быстро пропускают хлор, пока привес
не составит 161 г D,5 мол.). Охлаждать колбу снаружи не требуется,
так как добавленного количества льда достаточно для того, чтобы
по окончании поглощения хлора температура раствора равнялась 0°.
Начиная с этого момента, методика та же, что и описанная на
стр. 351 (частное сообщение Э. К. Стерлинга).
2. Раствор гипохлорита может быть также с успехом пригото-
влен из продажного гипохлорита кальция, содержащего не менее
65% активного гипохлорита кальция.
В 3-литровой круглодонной колбе растворяют 250 г продажного
гипохлорита кальция в 1 л теплой воды и туда же добавляют те-
плый раствор 175 г поташа и 50 г едкого кали в 500 мл воды. Колбу
закупоривают и энергично взбалтывают до тех пор, пока образо-
вавшийся вначале полутвердый гель не превратится в жидкость.
Взвешенный твердый осадок отфильтровывают через большую во-
ронку Бюхнера, промывают 200 мл воды и возможно лучше отса-
сывают с применением резиновой пластинки и хорошо работаю-
щего насоса. Фильтрат, объем которого составляет приблизительно
1,5 л, переливают в 3-литровую круглодонную колбу, после чего
к нему можно прибавлять метил-р-нафтилкетон так, как это опи-
сано выше.
Такой раствор содержит около 200 г B,3 мол.) гипохлорита
калия. Можно пользоваться гипохлоритом натрия или калия; гипо-
хлорит кальция непригоден ввиду плохой растворимости кальцие-
вой соли [j-нафтойной кислоты.
3. Был взят продукт фирмы «Истмен» с т. пл. 53—55°.
4. Если смесь не охлаждать, то реакция протекает очень бурно,
происходит быстрое выделение хлороформа, и некоторое количество
кетона может отогнаться с водяным паром.
5. Рекомендуется после приливания раствора бисульфита про-
верить реакционную массу раствором йодистого калия, чтобы уве-
риться в том, что весь гипохлорит разрушен. Если гипохлорит
остался, то хлор, выделяющийся при подкислении раствора, обра-
зует какую-то примесь с высокой температурой плавления.
6. Влажную кислоту можно перекристаллизовать и без предва-
рительной сушки, но в этом случае приходится брать большее коли-
чество спирта для того, чтобы перевести весь продукт в раствор.
7. Методика эта может быть использована и для получения
больших количеств кислоты. В случае двадцатикратной загрузки
выход достигает 87%. Этот метод может быть использован также
для получения других ароматических кислот в тех случаях, когда
соответственные кетоны являются доступными.
3. Другие методы получения
[З-Нафтойная кислота получается главным образом омылением
?-нафтонитрила 1. Выход при получении ее из (З-нафтиламина, на-
триевой соли |3-нафталинсульфокислоты и из кальциевой соли
^-нафталинсульфокислоты равняется (приблизительно) 20, 21 и
50% соответственно 2. Кислота эта была также получена действием
углекислого газа на гриньяровский реактив, который получается
из менее доступного р-бромпроизводного 3, хлорированием (з-метил-
нафталина с последующим гидролизом и окислением 4 и по мето-
дике, приведенной вышеs. Обзор различных методов получения
,3-нафтойной кислоты приведен в работе Валя, Гедкоопа и Гебер-
лейна ".
1 Baeyer, Besemfelder, Ann. 266, 187 A891).
2 D e r i с k, Kamiti, J. Am. Chem. Soc. 38, 408 A916).
3 Oilman, St. John, Rec. trav. chim, 48, 743 A929).
« Wahl, Goedkoop, Heberlein, Bull. soc. chim. E) 6, 533
n\
A939).
Fieser, Newman, Holmes, J. Am. Chem. Soc. 58, 1055 A936).
1,2-НАФТОХИНОН
(Нафтохинон)
О
NHX1
О
[О]
(FeCl8)
Предложил: Л. Физер.
Проверили: К. Ноллер и В. Уайт.
1. Получение
Чтобы получить хорошие результаты, необходимо работать
быстро. Вся нужная посуда и реактивы должны быть пригото-
влены заранее. Окисляющий раствор получают растворением 240 г
@,89 мол.) хлорного железа (с б мол. воды) в смеси, состоящей из
90 мл концентрированной соляной кислоты и 200 мл воды, при
нагревании; затем раствор охлаждают до комнатной температуры,
добавляя в него 200—300 г льда, и фильтруют с отсасыванием.
В 5-литровую круглодонную колбу помещают 80 г @,41 мол.)
хлористоводородной соли 1,2-аминонафтола высокой степени чи-
стоты (примечание 1) и добавляют раствор 5 мл концентрированной
соляной кислоты в 3 л воды, предварительно нагретый до 35°.
23 Сборник № 2
354
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
НИТРИЛ МАЛОНОВОЙ КИСЛОТЫ
355
При взбалтывании все быстро растворяется A—2 мин.); раствор
быстро фильтруют с отсасыванием, чтобы освободиться от неболь-
шого количества нерастворившейся примеси, и переносят его в чи-
стую 5-литровую колбу (примечание 2). Сразу прибавляют весь
окисляющий раствор, причем колбу энергично вращают, чтобы оба
раствора хорошо перемешались. Хинон выпадает немедленно в виде
объемистого микрокристаллического желтого осадка. Продукт со-
бирают на воронке Бюхнера и хорошо промывают водой (примеча-
ние 3). Затем для лучшего промывания осадок переносят в большой
стакан, размешивают несколько минут с 2 л нагретой до 30° воды и
вновь фильтруют. Полученную лепешку разрезают на ломтики и
сушат на фильтровальной бумаге при комнатной температуре в атмо-
сфере, не содержащей кислотных паров. Выход 60—61 г (93—94%
теоретич.).
Полученный продукт окрашен в золотистожелтый цвет и пла-
вится с разложением при 145—147°, несколько размягчаясь при
температуре около 140°; в нем совершенно отсутствует черный плохо
растворимый динафтилдихингидрон, и продукт нацело растворяется
в спирте или в бензоле. Под микроскопом видно, что продукт со-
стоит из массы правильно образованных мелких игл; посредством
перекристаллизации из спирта или бензола его можно получить
в виде красивых ораижевокрасных игл, однако такая очистка не
может считаться надежной, так как она ведет к значительным по-
терям, и температура разложения понижается на 10—20°. Таким
образом, целесообразнее хранить и употреблять хинон в том виде,
в каком он получается: в этом состоянии он является вполне чистым
и может сохраняться неопределенно долгое время. Его не следует
измельчать в порошок, так как он сильно наэлектризовы-
вается.
2. Примечания
1. Качество нафтохинона в значительной степени зависит от
чистоты исходного материала. Кристаллический продукт, описан-
ный на стр. 45, может быть использован без перекристаллизации
и даже без предварительной сушки. Рекомендованный в рецептуре
избыток окислителя достаточен для количества аминонафтола, не-
сколько большего, чем это указано там; если выход амино-
нафтола получается меньше, то избыток хлорного железа не
вредит.
2. На этой стадии раствор должен быть прозрачным, и в толще
он может иметь заметную оранжевожелтую окраску, однако он
не должен быть пурпурового цвета.
3. Желтая окраска промывной воды объясняется частичной
растворимостью хинона.
3. Другие методы получения
Единственным удовлетворительным методом получения 1,2-нафто-
хинона (Р-нафтохинона) является окисление ^^-аминонафтола
в кислом растворе. Основной проблемой является получение этого
промежуточного соединения с хорошим выходом и в достаточно
чистом состоянии. Эта проблема и относящаяся к ней литература
рассмотрены на стр. 46. В большинстве работ, касающихся полу-
чения аминонафтола, содержатся также и данные об его окислении;
однако единственно ценным является наблюдение, что хлорное
железо представляет собою лучший окислитель, чем хромовая
кислота, так как при низкой температуре, даже будучи в избытке,
оно не разрушает хинона х.
Другим методом синтеза р-нафтохинона является получение его
из 1,2-бромнафтола через кетонитробромид2. Хотя соответственный
хинон сам по себе является настолько чувствительным соединением,
что полученный таким образом продукт разлагается через несколько
часов, метод этот представляет значительный интерес для получе-
ния некоторых замещенных р-нафтохинонов 3.
1 Groves, J. Chem. Soc. 45, 298 A884).
2 Fries, Ann. 389, 315 A912).
s Fries, Schimmelschmidt, там же 484, 245 A930).
НИТРИЛ МАЛОНОВОЙ КИСЛОТЫ
CNCH2CONH2+PC15
2CNCrI,CONH2-f POC13
CH2(CNJ + POCI3+2HC1
2CH2(CNJ+HPO3+3HC1
Предложили: Б. Корсон, Р. Скотт и /{. Возе.
Проверили: К- Марвел и ДжУГармон.
1. Получение
150 г A,8 мол.) чистого цианацетамида («Синт. орг. преп.к сб. 1,
стр. 498) тщательно смешивают в большой (диаметр 20 см) ступке
со 150 г @,7 мол.) пятихлористого фосфора (примечание 1). Смесь
переносят (примечание 2) как можно быстрее в 1-литровую колбу
Клайзена, снабженную термометром на 360° и трубкой для впуска
воздуха (примечание 3). Колбу Клайзена соединяют при помощи
воздушного холодильника удвоенной длины со склянкой для от-
сасывания емкостью 250 мл, которая соединена в свою очередь с во-
доструйным насосом (примечание 4) и манометром.
Прибор эвакуируют примерно до 30 мм ртутного столба и затем
ставят колбу Клайзена на кипящую водяную баню. Смесь плавится
и принимает оранжевую окраску. Кипение начинается нриблизи-
356
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
НИТРИЛ МАЛОНОВОЙ КИСЛОТЫ
357
тельно через 15 мин., еще до того, как твердый осадок расплавится
полностью, и давление повышается примерно до 150 мм вследствие
выделения хлористого водорода и хлорокиси фосфора (примеча-
ние 5). Когда через 30—35 мин. выделение газа замедлится, что
заметно по понижению давления и менее бурному кипению реак-
ционной смеси, приемник меняют. Перегонную колбу снимают с ки-
пящей водяной бани, вытирают досуха и погружают в масляную
баню, нагретую до 140° (примечание 6), а свежий приемник ставят
в ледяную воду.
Нитрил малоновой кислоты начинает перегоняться при 113°/ЗО мм
A25°/50 мм). Температуру масляной бани медленно повышают
в течение 25 мин. до 180° (примечание 7) и собирают нитрил между
113° и 125°. Когда перегонка почти закончилась, масляную баню
удаляют, чтобы предупредить изменение окраски получаемого
продукта (примечание 8). Выход сырого нитрила 80—95 г F7—80%
теоретич.; примечание 9). Нитрил можно очистить перегонкой в ва-
кууме (примечание 10) с потерей около 10%; его собирают при
113—12О°/ЗО мм. Одна перегонка дает жидкость, прозрачную, как
вода, которая быстро застывает и становится твердой, напоминая по
виду лед (примечание 11), и плавится при 28—30°. Вещество обла-
дает слабым запахом, который имеет сходство с запахом ацетамида.
2. Примечания
1. Относительные количества реагентов соответствуют отноше-
нию 5 молекул амида на 2 молекулы пятихлористого фосфора. Если
взять больше пятихлористого фосфора, то выход понижается1.
Пятихлористый фосфор должен быть хорошего качества — сухим
и твердым.
2. Перенесение смеси в колбу очень облегчается, если взять
15-сантиметровую стеклянную воронку с обрезанной ножкой и от-
верстием, расширенным почти до размеров горла колбы Клайзена.
Смесь высыпают в воронку и проталкивают через широкое отверстие
стеклянной палочкой. При этом следует работать в противогазе и
быть очень осторожным, чтобы не вдохнуть испарений, так как они
вызывают резкий кашель, который может продолжаться несколько
дней.
3. Во избежание закупорки приводящая воздух трубка не
должна быть вытянута в капилляр.
4. Масляный насос применять нельзя, так как выделяющиеся
пары вызывают его коррозию.
Имеются указания, что один водоструйный насос обычно не
может справиться достаточно быстро с выделяющимися газами,
в результате чего вещество механически уносится с пеной. Это за-
труднение можно преодолеть, если работать с двумя параллельно
присоединенными насосами (К. Аллен, частное сообщение).
5. Если не хотят выделять образующуюся хлорокись фосфора,
то рекомендуется ее отсасывать. Это легко выполнимо, если прием-
ник не охлаждать.
6. Если первоначальная температура масляной бани ниже 140°,
нитрил перегоняется слишком медленно (что соответственно пони-
жает выход); если температура выше 140°, то нитрил переходит
слишком быстро. В последнем случае некоторое количество нитрила
конденсируется в резиновых трубках, ведущих к насосу, и может
их закупорить.
Колбу необходимо погрузить в масло как можно глубже (при-
мерно до глубины 100 мм от верха колбы). Весьма удобна в каче-
стве масляной бани эмалированная кастрюля на 3,5 л. Обычный
паяный сосуд не выдерживает такой высокой температуры.
Т. Температура масляной бани не должна намного превышать
180°, иначе дестиллат окрасится и может произойти сильное вски-
пание и перебрасывание. Нужно следить за тем, чтобы дестиллат
не затвердел в холодильнике, что часто случается во время первой
части перегонки. Затвердевшее в холодильнике вещество легко рас-
плавить нагреванием на маленьком пламени.
8. Остаток от перегонки в колбе Клайзена можно размягчить
добавлением воды и разбить при помощи палочки. Небольшое
оставшееся количество легко смыть концентрированной азотной
или, лучше, концентрированной серной кислотой.
9. Выход зависит всецело от быстроты работы. Нитрил мало-
новой кислоты очень реакционноспособен, а потому его следует
выделять из реакционной смеси как можно быстрее. После одно-
или двукратного повторения синтеза выходы превысят 67% бла-
годаря приобретенному опыту.
10. Нитрил малоновой кислоты можно перегонять небольшими
порциями E0 мл или около этого количества) при обычном давлении;
температура кипения — около 220е. Более продолжительное нагре-
вание, необходимое при больших количествах, может вызвать бур-
ное разложение. В этом случае жидкость темнеет, внезапно заки-
пает и под конец выбрасывается из колбы в виде облака белого
пара и горячей жидкости, которая по охлаждении частично затвер-
девает в хрупкое, твердое вещество красного цвета.
11. По определению азота нитрил получается 99,5%-ный. Если
его сохранять в коричневых склянках, защищенных от света, он
остается бесцветным по меньшей мере в течение года. Если же он
будет сохраняться в обыкновенных склянках и под действием света,
то он быстро темнеет.
3. Другие методы получения
Нитрил малоновой кислоты был получен действием пятихлори-
стого фосфора на цианацетамид х и действием пятиокиси фосфора на
358
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
амид малоновой кислоты2 или на цианацетамид8. Применение
пятиокиси фосфора дает значительно менее удовлетворительные
результаты по сравнению с методом, приведенным выше.
1 Hesse, Am. Chem. J. 18, 726 A896).
2 Henry, Compt. rend. 102, 1394 A886).
3 H e n г у, там же 102, 1395 A886).
НИТРИЛ «-ФЕНИЛ-р-БЕНЗОИЛПРОПИОНОВОЙ КИСЛОТЫ
(а-Фенацил-а-толунитрил)
CeH5CH = CHCOCeHs + HCN -—> CeH5CH (CN) СН2СОС6Н5
Предложили: Я. Аллен и Р. Кимболл.
Проверили: Дж. Конант и Е. О'Бриен.
1. Получение
Все операции, включая отсасывание сырого продукта и промы-
вание его водой, следует производить в вытяжном шкафу с хорошей
тягой.
В 5-литровую колбу или склянку, поставленную на водяную
баню и снабженную мешалкой, термометром и делительной ворон-
кой, помещают. 208 г A мол.) бензальацетофенона (примечание 1),
3,5 л 95%-ного этилового спирта (примечание 2) и 60 г A мол.)
ледяной уксусной кислоты (примечание 3). Смесь нагревают при
перемешивании до 35° и прибавляют через делительную воронку
раствор 130 г B мол.) цианистого калия в 375 мл воды в течение
15 мин. После того как вся уксусная кислота войдет в реакцию,
раствор становится щелочным (примечание 3) и первоначальная
зеленоватая окраска смеси переходит в желтую. Перемешивание
продолжают в течение 3 час., причем поддерживают температуру 35°.
В течение этого времени около половины нитрила выкристаллизо-
вывается. После этого колбу неплотно закрывают пробкой и оста-
вляют на 50 час. в холодном месте. (При холодной погоде удобно
оставить колбу на открытом воздухе, но так, чтобы на нее не падал
солнечный свет.) Затем отфильтровывают кристаллический осадок
и промывают его, сначала 500 мл холодного 50%-ного спирта, а потом
водой до тех пор, пока фильтрат не будет свободен от цианистого
калия (проба с азотнокислым серебром). Выход высушенного на
воздухе вещества 220—227 г (93—96% теоретич.), т. пл. 125°.
Полученный нитрил достаточно чист для большинства целей, хотя
он содержит следы высокоплавкой примеси. Если желательно полу-
чить продукт с более высокой температурой плавления, то сырой
нитрил можно перекристаллизовать из 1 л 95%-ного спирта или
375 мл ацетона. Чистый нитрил а-фенил-3-бензоилпропионовой
кислоты плавится при 127°.
1-НИТРО-2-АЦЕТИЛНАФТИЛАМИН
359
2. Примечания
1. Вполне пригоден сырой бензальацетофенон, не содержащий
щелочи и высушенный на воздухе («Синт. орг. преп.», сб. 1, стр. 77).
2. Реакцию можно вести в более концентрированном растворе
A000 мл спирта), причем выход остается тем же, но получается
менее чистое вещество. В этом случае следует после прибавления
всего цианистого калия производить размешивание в течение 15 мин.
при 50°, а затем охладить реакционную смесь водопроводной водой.
Так как из раствора такой концентрации вещество выпадает в виде
масла> следует внести в качестве затравки кристаллик нитрила.
3. Если раствор станет слишком щелочным, то образовавшийся
нитрил присоединяется ко второй молекуле непредельного кетона
настолько легко, что конечный продукт состоит почти полностью
из высокоплавкого вещества B84—286°). По этой причине необхо-
димо отмерять уксусную кислоту точно; если взять ее слишком
много, то не происходит присоединения синильной кислоты.
3. Другие методы получения
Нитрил а-фенил-р-бензоилпропионовой кислоты был получен
действием цианистых натрия или калия на р-хлорбензилацетофе-
нон х или на дибромид бензальацетофенона2; присоединением си-
нильной кислоты к бензальацетофенону в присутствии цианистых
натрия или калия 3.
1 Anschutz, Montfort, Ann. 284, 2 A895); Rupe, Schnei-
der, Ber. 28, 960 A895).
2 Dodwadmath, Wheeler, Proc. Indian Acad. Sci. 2A, 438 A935)
[C. A. 30, 1771 A936)].
3Hann, Lapworth, J. Chem. Soc. 85, 1358A904); Lapworth,
W e с h s 1 e г, там же 97, 41 A910).
//
1-НИТРО-2-АЦЕТИ ЛНАФТИ ЛАМИН
[N-A-Нитро-2-нафтил )-ацетамид]
NO2
NHCOCH3
HNO3 >
NHCOCH3
+н2о
Предложили: В. Хартман и Л. Смит.
Проверили: Л. Физер и Дж. Уокер.
1. Получение
В 2-литровую круглодонную колбу, снабженную механической
мешалкой, термометром и капельной воронкой и укрепленную на
штативе таким образом, чтобы по мере надобности ее можно было
360
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
погружать в баню со льдом и водой, помещают 300 г A,62 мол.)
ацетил-|3-нафтиламина (т.пл. 131—132°) и 500 мл ледяной уксусной
кислоты. Мешалку пускают в ход и при комнатной температуре
начинают прибавлять по каплям 200 г A43 мл, 2,1 мол.) концентри-
рованной азотной кислоты (уд. в. 1,4). Прибавление кислоты продол-
жается около 45 мин., и колбу время от времени погружают в охла-
дительную смесь, чтобы не дать температуре подняться выше 40°.
Когда прибавлено около х/ю части азотной кислоты, перемешивание
смеси делается очень затруднительным, так как смесь становится
густой. Тогда прибавление кислоты временно прерывают. Очень
скоро (через 3—5 мин.) масса становится снова жидкой, и тогда
опять начинают прибавлять азотную кислоту. После прибавления
примерно одной четверти азотной кислоты все твердое вещество
переходит в раствор. Происходит сильное разогревание, и необхо-
димо хорошее охлаждение, чтобы температура не поднялась выше
40°; остальную кислоту прибавляют при непрерывном охлаждении
смеси. После внесения всей кислоты перемешивание продолжают
еще 10 мин.
Колбу закрывают пробкой и охлаждают водой со льдом в про-
должение 3 час; продукт реакции выделяется при этом в виде жел-
той, кристаллической каши (примечания 1 и 2). Кристаллы отсасы-
вают на бюхнеровской воронке диаметром 19 см и промывают сна-
чала 200 мл 50%-ной уксусной кислоты и затем 400 мл этилового
эфира. Неочищенный сухой продукт B70—290 г) помещают в 3-лит-
ровую колбу и кипятят с обратным холодильником с 1,7 л бензола
в течение 20 минут. Смеси дают охладиться до 40—45° и фильтруют
с отсасыванием на бюхнеровской воронке (диаметр 19 см). Осадок
состоит из смеси труднорастворимых изомеров главным образом
5- и 8-нитро-2-ацетилнафтиламина. При охлаждении профильтро-
ванного раствора получают 190—200 г 1-нитро-2-ацетилнафтиламина
с температурой плавления 117—119°. Продукт перекристаллизо-
вывают из 500 мл горячего 95%-ного этилового спирта. Получаются
мелкие желтые кристаллы, плавящиеся при 123—124°. Выход
175—182 г D7—49% теорётич.).
2. Примечания
1. Продукт иногда выкристаллизовывается медленно; в таком
случае рекомендуется тереть о стенки сосуда стеклянной палочкой.
Если же и это не помогает, можно заразить раствор кристаллами,
полученными при разбавлении небольшой порции раствора водой.
2. Через 3 часа кристаллизация закончена, но не полностью.
Если маточный раствор оставить стоять на пять дней, выпадает еще
некоторое количество вещества, но при его очистке получается
только 3 г чистого 1-нитро-2-ацетилнафтиламина.
лс-НИТРОАЦЕТОФЕНОН
361
3. Другие методы получения
Описанный способ разработан на основании имеющихся в лите-
ратуре данных о нитровании 2-ацетил-р-нафтиламина 1.
1 Jacobson, Ber. 14, 803 A881); Liebermann, Jacobson,
Ann, 211, 44 A882); Friedlaender, Littner, Ber. 48, 328 A915);
S с h i e m a n n, L e у, там же 69, 963 A936).
М-П ИТРО АЦЕТОФ EH ОН
CeH4COCHs+HNO3 ^-NO2CeH4COCHs+H2O
Предложили: Б. Корсон и Р. Хазен.
Проверили: Р. Адаме и В. Мопер.
1. Получение
В 1-литровую широкогорлую коническую колбу, поставлен-
ную в 8-литровый глиняный сосуд, который наполнен смесью
льда и соли, вливают 150 мл концентрированной серной кислоты.
Колба снабжается мощной механической мешалкой, маленькой ка-
пельной воронкой и термометром, доходящим почти до дна колбы
(примечание 1). Мешалку пускают в ход и, после того как серная
кислота охладится до 0° или ниже, к ней по каплям медленно при-
бавляют через капельную воронку 60 г @,5 мол.) чистого ацетофе-
нона с такой скоростью, чтобы температура не поднималась выше 5°
(приблизительно в течение 10 мин.; примечание 2). После того,
как реакционная смесь охладится, но на этот раз до —7°, через ка-
пельную воронку прибавляют охлажденную A5—20°) нитрующую
смесь, состоящую из 40 мл @,65 мол.) азотной кислоты (уд. в. 1,42
при 15,5°; примечание 3) и 60 мл концентрированной серной кис-
лоты, с такой скоростью A00—120 капель в минуту), чтобы темпе-
ратура реакционной массы не поднималась выше 0° (примечание 4).
После прибавления нитрующей смеси продолжают размешива-
ние в течение 10 мин. и затем выливают (примечание 5) содержимое
колбы при сильном размешивании от руки в смесь 750 г толченого
льда и 1500 мл воды. Продукт выпадает в виде желтого хлопьевид-
ного осадка.
После того, как лед растаял, продукт отсасывают и несколько
клейкую массу тщательно отжимают. Затем осадок переносят
в ступку и растирают последовательно с тремя порциями холодной
воды по 300 мл (для удаления кислоты), а затем перемешивают
в кашицу с двумя порциями по 25 мл охлажденного до 0° этилового
спирта (для удаления маслянистых примесей). После каждой из
пяти промывок осадок тщательно отсасывают и возможно сильнее
отжимают. Затем продукт отжимают на неглазурованной глиняной
362
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
тарелке и, когда он станет достаточно сухим (приблизительно через
1 час), его растворяют в 100—120 мл горячего спирта (примечание б).
Темный раствор быстро фильтруют с отсасыванием через малень-
кую воронку (примечание 7) и медленно выливают горячий филь-
трат в 1000 мл холодной воды, при энергичном перемешивании (при-
мечание 8) стеклянной палочкой в продолжение всего времени
прибавления и еще несколько минут после того, как весь раствор
уже прибавлен. Смесь оставляют несколько минут стоять, затем
отсасывают желтый твердый осадок, промывают его 200 мл холод-
ной воды и отжимают досуха, сначала на воронке, а потом на пори-
стой глиняной тарелке.
Когда вторично осажденный л/-нитроацетофенон E0—55 г) высу-
шен, его растворяют в 100 мл горячего спирта в конической колбе
емкостью 300 мл. Колбу ставят затем в ледяную баню и сильно
встряхивают все время, пока идет кристаллизация. Особенно энер-
гичное встряхивание необходимо в то время, когда температура
массы понижается с 60° до 50°, так как в этом интервале сильно
уменьшается растворимость ^-нитроацетофенона в спирте. Если
размешивание во время кристаллизации производилось недоста-
точно сильно, то вместо более чистой и легче высыхающей кристал-
лической кашицы образуются большие комки кристаллов. После
того как температура достигнет 20° (приблизительно через 1 час)
смесь отсасывают (примечание 9). Твердый осадок (примечание 10)
промывают 10 мл охлажденного до 0° спирта и отжимают досуха на
глиняной тарелке. Светложелтый продукт размягчается при 74°
и плавится при 76—78°. Общий выход 45 г E5% теоретич.; приме-
чание 11).
2. Примечания
1. Наиболее подходящим для проведения этой реакции сосудом
является однолитровая широкогорлая коническая колба. Боль-
шая поверхность плоского дна такой колбы обеспечивает быстрое
охлаждение. Мешалка-пропеллер с длинными широкими лопастями
размешивает вязкую жидкость значительно лучше, чем мешалка
типа центрифуги. Лопасти должны быть настолько длинны, на-
сколько это допускает ширина горла колбы. Термометр, показываю-
щий температуру реакционной смеси, должен быть опущен в колбу
под углом и доходить почти до дна колбы, поскольку количество
жидкости невелико. Нулевая точка термометра должна находиться,
самое меньшее, на 15 см от шарика, для того чтобы облегчить отсчет
температуры. Следить за температурой необходимо в продолжение
всего опыта. Применять баню с охладительной смесью меньшего
размера, чем указано (около 8 л), нецелесообразно.
2. Серную кислоту, ацетофенон и нитрующую смесь можно
прибавлять через одну и ту же капельную воронку, не промывая ее.
м- НИТРОАЦЕТОФЕНОН
363
При быстром размешивании ацетофенон можно без труда прибавить
в течение 7 мин. так, чтобы температура реакционной смеси не
поднималась выше 3°.
3. Азотная кислота с удельным весом меньше чем 1,42 при 15,5°
дает нечистый продукт. Концентрацию обыкновенной крепкой
азотной кислоты обычно приходится увеличить до нужной величины
прибавлением дымящей азотной кислоты.
4. Хорошие результаты получаются при соблюдении двух усло-
вий: низкая температура @° или ниже) и быстрота прибавления
нитрующей смеси (не более 45 мин.). При эффективном охлаждении
можно поддерживать температуру между — 5° и 0°. Если темпера-
тура поднимется один или два раза до 3°, то это не отражается на
выходе, при условии, что реакционная смесь будет немедленно и
быстро охлаждена до указанного предела. Для того чтобы избежать
местного перегрева, следует направить ножку капельной воронки
таким образом, чтобы нитрующая смесь смешивалась с реакционной
массой в месте наиболее сильного перемешивания.
Перемешивание должно быть очень энергичным. Наиболее благо-
приятная скорость различна для разных типов мешалок и зависит
от формы и размера лопастей. Скорость описанной здесь мешалки
1600 об/мин. Во время прибавления ацетофенона большая скорость
не нужна; фактически ее даже нельзя поддерживать из-за сильного
разбрызгивания. Однако во время прибавления нитрующей смеси,
когда реакционная смесь становится более густой, можно и нужно
вести перемешивание с большой скоростью. Смесь льда и соли
следует неоднократно размешивать палкой и прибавлять к ней
время от времени еще лед и соль. Температура охлаждающей бани
должна быть около — 16°, баня должна содержать достаточное
количество жидкости, чтобы нижняя половина колбы была погру-
жена в нее. Если не соблюдать этих условий тщательно, то приба-
вление нитрующей смеси занимает от 2 до 3 час. (поскольку темпе-
ратура реакционной массы не должна подниматься выше 0°). Такое
продолжительное действие смеси азотной и серной кислот на ацето-
фенон так же вредно, как повышение температуры: продукт полу-
чается плохого качества, а выход падает с 55% до 15% и даже ниже.
Если колбу охлаждать твердой углекислотой и ее же приба-
влять в реагирующую смесь, то при нитровании можно поддержи-
вать температуру — 20° Ч Согласно частному сообщению В. В.
Хартмана, такое применение твердой углекислоты дает возмож-
ность вести синтез в большем масштабе.
5. Перерыв в работе возможен только после того, как продукт
вылит в воду.
6. Для того чтобы растворить л/-нитроацетофенон в указанном
количестве спирта, достаточно нагреть раствор до 55°, но чтобы
предупредить выпадение кристаллов во время фильтрования, жела-
тельно довести спиртовой раствор до кипения.
364
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
7. Чрезмерное вспенивание в склянке для отсасывания легко
приостановить, зажимая время от времени каучуковую трубку,
ведущую к насосу.
8. Сильное перемешивание и медленное выливание совершенно
необходимы. Если этого не делать, то продукт выделится не в виде
хлопьев, а в виде комков.
9. Так как кривая растворимости при температурах, близких
к комнатной, идет полого, то нет необходимости в очень сильном
охлаждении перед фильтрованием. Разность в растворимости между
20° и 10° составляет только 1 г на 100 мл раствора. Растворимость
л/-нитроацетофенона в 96%-ном спирте изменяется следующим
образом:
НИТРОБАРБИТУРОВАЯ КИСЛОТА
365
Темп.,
8
17
22
23
27
28
32
39
42
СС
Количество
(в 10 мл
раствора)
в г
0,16
0,25
0,31
0,34
| 0,41
0,45
0,94
0,97
1,22
Темп.,
48
50
52
53,5
55
57
5Э
60
°С
Количество
(в 10 мл
раствора)
в г
1,83
2,38
3,03
4,05
5,46
8,05
11,00
15,60
10. Из маточного раствора можно получить еще небольшое коли-
чество продукта (около 5 г) концентрированием его до 20 мл и по-
следующим охлаждением льдом.
3. Другие методы получения
jw-Нитроацетофенон обычно получается нитрованием ацетофе-
нона 1>2; он был получен также кетонным расщеплением .м-нитро-
бензоилацетоуксусного эфира 3. Приведенная здесь методика была
видоизменена Моргеном и Уотсоном4; авторы указывают, что по
видоизмененному методу выход ^-нитроацетофенона возрастает до
83% теоретич.
1 Barkenbus, Clements, J. Am. Chem. Soc. 56, 1369A934).
2Emmerling, Engler, Ber. 3, 886 A870); В u с h k а, там же
10, 1714 A877); Engler, тэд же 18, 2238 A885); Kostanecki, Tarn-
b о г, там же 34, 1691 A901); Rupe, Braun, Z e m b r u s k i, там же 34,
3522 A901); Camps, Arch. Pharm. 240, 5 A902).
3 Gevekoht, Ann. 221, 334 A883). „
4 Morgan, Watson, J. Soc. Chem. Ind. 55, 29T A936).
НИТРОБАРБИТУРОВАЯ КИСЛОТА
E-Нитробарбитуроеая кислота)
NH — CO
СО СН2 • 2 Н2О
NH — СО
HNO3
дымящая
NH — СО
СО CHNOo • 3 Н.,0
II"'
NH —СО
Предложили: В. Хартман и О. Шеппард.
Проверили: К. Марвел и Б. Войцик.
1. Получение
В 2-литровую колбу, снабженную механической мешалкой и
находящуюся в ледяной бане, помещают 143 мл дымящей азотной
кислоты (уд. в. 1,52). При постоянном перемешивании прибавляют
в течение 2 час. 100 г @,61 мол.) барбитуровой кислоты (стр. 79);
при этом поддерживают температуру ниже 40°. После того, как вся
барбитуровая кислота прибавлена, смесь перемешивают еще 1 час,
а затем, продолжая перемешивание, прибавляют 430 мл воды и
раствор охлаждают до 10°. Осадок отсасывают, промывают холодной
водой и сушат при 60—80° (примечание 1) на стеклянном противне.
Полученную таким образом нитробарбитуровую кислоту раство-
ряют в 860 мл кипящей воды в 2-литровой колбе, нагревая раствор
на кипящей водяной бане и пропуская в смесь острый пар до пол-
ного растворения кислоты (примечание 2). Раствор фильтруют и
дают ему охладиться за ночь; выпавшие кристаллы отсасывают,
промывают холодной водой и сушат на противнях в сушильном
шкафу 2—3 часа при 90—95°. При быстром нагревании продукт
плавится с разложением при 181—183°. Выход 139—141 г (приме-
чание 3). Если нитробарбитуровую кислоту сушить 2—3 часа при
110—115°, то получается 90—94 г безводного продукта (85—90%
теоретич.). Этот безводный продукт плавится с разложением при
176°.
2. Примечания
1. Если продукт не высушить до перекристаллизации, то всю
азотную кислоту удалить трудно, и окончательный продукт будет
сильно пахнуть азотной кислотой.
2. Если не получается светложелтого раствора, то до фильтро-
вания надо прибавить активированного угля.
3. Выход этот несколько больше теоретического A39 г), что
объясняется, вероятно, большей степенью гидратации, чем указано
в формуле.
366
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
rt-НИТРОБЕНЗАЛЬДЕГИД
367
3. Другие методы получения
Нитробарбитуровая кислота была получена при окислении виолу-
ровой кислотыг и обработкой барбитуровой кислоты дымящей
азотной кислотой 2 или концентрированной азотной кислотой 3.
1 С е г е s о 1 е, Вег. 16, 1134 A883).
2 В а е у е г, Ann. 130, 140 A864).
s F r e d h о 1 m, Z. anal. Chem. 104, 400 A936).
И-НИТРОБЕНЗАЛЬДЕГИД
(A) n-OoNC6H4CH3+2(CH3COK O+2 [
n-O2NC6H4CH (OCOCH3J + 2 CH8CO2H
(Б) n-O2NCeH4CH (OCOCH3K+ H2O J^iL
n-O2NCeH4CHO + 2 CH8CO2H
Предложили: С. Либерман и Р. Коннор.
Проверили: Дж. Джонсон и Е. Кливленд.
1. Получение
А. п-Нитробензальдшщтат. В 2-литровую трехгорлую кругло-
донную колбу, снабженную механической мешалкой и термометром
и погруженную в смесь льда и соли, помещают 570 мл F00 г) ледяной
уксусной кислоты, 565 мл F12 г, 6 мол.) уксусного ангидрида (при-
мечание 1) и 50 г @,36 мол.) л-нитротолуола (примечание 2). К этому
раствору медленно при перемешивании прибавляют 85 мл A,5 мол.)
концентрированной серной кислоты (примечание 3). После охла-
ждения смеси до 5° прибавляют небольшими порциями 100 г
A мол.) хромового ангидрида (примечание 2) с такой скоростью,
чтобы температура не поднималась выше 10° (примечание 4). После
прибавления всего количества хромового ангидрида перемешивание
продолжают еще 10 мин. Содержимое колбы выливают в два 3-лит-
ровых стакана, на 2/3 наполненных колотым льдом, после чего до-
ливают холодной воды, так чтобы общий объем составил 5—6 л.
Осадок отфильтровывают с отсасыванием и промывают холодной
водой до тех пор, пока промывная вода не станет бесцветной. Про-
дукт суспендируют в 500 мл холодного 2%-ного раствора соды и
перемешивают с помощью механической мешалки. После тщатель-
ного перемешивания твердый продукт отсасывают (примечание 5)
и промывают сперва холодной водой, а затем 20 мл холодного спирта.
Выход продукта, высушенного в вакуум-эксикаторе, составляет
44—49 г D8—54% теоретич.). Его температура плавления 120—
122° (примечание 6).
Сырой продукт вполне может быть применен для омыления,
а также использован для других реакций без дальнейшей очистки.
Чистый диацетат можно получить перекристаллизацией из 150 мл
горячего спирта. Горячий раствор фильтруют через складчатый
фильтр для удаления нерастворимых примесей. Выход чистого
л-нитробензальдиацетата с т. пл. 125—126° составляет 43—46 г
D7—50% теоретич.).
Б. п-Нитробензальдегид. Смесь 45г @,18 мол.) сырого п-нитробен-
зальдиацетата, ЮОл/лводы, ЮОллспиртаи 10 мл концентрированной
серной кислоты кипятят с обратным холодильником 30 мин., филь-
труют через складчатый фильтр и фильтрат охлаждают в бане
с льдом. Кристаллы отфильтровывают с отсасыванием, промывают
холодной водой и сушат в вакуум-эксикаторе. Вес первой порции
22—24 г (82—89% теоретич.), т. пл. 106—106,5°. Если фильтрат
разбавить примерно 300 мл воды, можно получить вторую порцию
кристаллов в количестве 2—3 г. Общий выход 24—25,5 г (89—94%
теоретич.; примечание 6).
2. Примечания
1. «Практически чистый»* уксусный ангидрид хорошего ка-
чества (95%-ный) дал такой же хороший выход, как и 99—100%-ный
уксусный ангидрид.
2. Был взят «-нитротолуол «практически чистый», с т. пл. 50—51°.
Хромовый ангидрид удовлетворял требованиям фармакопеи и был
98%-ной чистоты.
3. Если серную кислоту приливать слишком быстро, то имеет
место обугливание.
4. Весьма важно поддерживать температуру реакционной смеси
ниже 10°. Если окислитель прибавлять настолько быстро, что тем-
пература поднимется выше 10°, то выход заметно понижается. Если
пользоваться хорошей баней со льдом и солью, то на прибавление
окислителя нужно затратить 45—60 мин.
5. Если подкислить промывную жидкость после промывания
содой, можно получить 7—10 г n-нитробензойной кислоты с т. пл.
242—243°.
6. Повидимому, эта методика применима к получению таких
замещенных бензальдегидов, в которых заместитель не затраги-
вается при реакции окисления. Так, авторы метода указывают, что
n-бромбензальдегид может быть получен по той же методике, если
вместо я-нитротолуола взять 62 г @,37 мол.) п-бромтолуола, а окис-
ление и выделение проводить совершенно аналогичным способом.
Выход сырого п-бромбензальдиацетата 51—64 г D8—60% теоретич.),
т. пл. 90—92°. Чистый диацетат получается, если растворить сырой
продукт в 150 мл горячего спирта, профильтровать раствор через
складчатый фильтр и охладить его. После фильтрования получают
* См. сноску на стр. 26.
368
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
39—52 г чистого продукта с т. пл. 94—95°. Вторую порцию кристал-
лов можно получить разбавлением фильтрата. Общий выход 47—
56 г D6—54% теоретич., считая на бромтолуол). Сырой продукт
подвергают гидролизу до л-бромбензальдегида кипячением 45 г
@,157 мол.) продукта с 100 мл воды, 150 мл спирта и 10 мл концен-
трированной серной кислоты. Раствор фильтруют через складчатый
фильтр, охлаждают и кристаллы отсасывают. Вторую порцию по-
лучают разбавлением фильтрата 300 мл воды. Общий выход продукта
с т. пл. 55—57° составляет 24—28 г (83—96% теоретич.).
Имеются также указания, что л-циантолуол дает л-цианбензаль-
дегид, выход в этом случае такой же, как и в случае л-нитробензаль-
дегида (Л. Вейслер, частное сообщение).
3. Другие методы получения
л-Нитробензальдегид был получен из л-нитротолуола при дей-
ствии на него изоамилнитрита в присутствии метилата натрия г
или окислением хлористым хромилом2, двуокисью церия3 или
хромовым ангидридом в присутствии уксусного ангидрида4. Он
может быть также получен окислением хлористого л-нитробензилаs,
n-нитробензилового спиртав или сложных эфиров л-нитрокоричной
кислоты 7.
1 Angel i, Angelic о, Atti accad. Lincei E) 8, И, 28 A899) (Chem.
Zentr. 1899, II, 371).
2 R i с h t e r, Ber. 19, 1060 A886); Law, P e r k i n, J. Chem. Soc. 93,
1635 A908).
8 Farbw. vorm. Meister, Lucius, Briining, герм. пат. 174 238 [Frdl. 8,
150 A905—07I.
* T h i e 1 e, Winter, Ann. 311, 353 A900).
5 Fischer, Greiff, Ber. 13, 669 A880); Schmidt, герм, пат,
15 881 (Frdl. 1, 60 A877—87)].
•Cohen, Harrison, J. Chem. Soc. 71, 1057 A897); Walter-
герм, пат. 118 567 [Frdl. 6, 131 A900—02)].
' В а е у е г; герм. пат. 15 743 [Frdl. 1, 60 A877—87)].
Я-НИТР0БЕНЗИЛБР0МИД
(а-Бром-п-ттротолуол)
n-NOsCeH4CHa -f- Br2
n-NO2C6H4CH,Br +
Предложили: Дж. Колеман и Дж. Хонейвел.
Проверили: В. Каротерс и В. Мак-Ювен.
1. Получение
В 1-литровую трехгорлую колбу помещают 300 г B,2 О
технического л-нитротолуола (т. пл. 51—52 ). КолЬу снаьжают
мешалкой с жидкостным затвором, обратным холодильником и
П-НИТРОБЕНЗИЛБРОМИД
369
делительной воронкой, установленной таким образом, что трубка
ее почти достигает дна колбы. Другой конец холодильника при-
соединен к газовой ловушке. Колбу нагревают на масляной бане
до температуры 145—150° (примечание 1) и в течение 2 час. доба-
вляют по каплям 368 г A18 мл, 2,3 мол.) брома (примечание 2).
После того как все количество брома прибавлено, нагревание
и перемешивание продолжают 10 мин. и содержимое колбы выли-
вают еще в жидком состоянии (примечание 3) в 5-литровую кругло-
донную колбу, в которую уже налито 4 л горячего лигроина (т. кип.
90—100°). Туда же добавляют 15 г обесцвечивающего угля. Колбу
нагревают на электрической плитке, пока все не растворится, кипя-
тят 10 мин. и быстро фильтруют с отсасыванием (примечание 4).
По охлаждении до 20° выпавшие кристаллы отсасывают, хорошо
отжимают и промывают два раза холодным лигроином порциями
по 50 мл. Вес сырого продукта с т. пл. 94—97° составляет 280—315 г
E9—66% теоретич.). Продукт этот достаточно чист для ряда целей.
Для дальнейшей очистки его растворяют в 3—3,5 л горячего
лигроина, кипятят с 10—15 г обесцвечивающего угля и фильтруют
с отсасыванием (примечание 4). Фильтрат охлаждают льдом, кри-
сталлы отсасывают, отжимают и промывают два раза холодным
лигроином порциями по 25 мл. Светложелтый продукт плавится при
97,5—99° и весит 250—280 г E3—59% теоретич.; примечания 5 и 6).
2. Примечания
1. Для получения хороших результатов необходимо строго
придерживаться указанных температурных пределов.
, 2. Бром следует прибавлять в течение 2 час, даже если некото-
рое количество его при этом будет потеряно через холодильник.
3. При работе с л-нитробензилбромидом и его растворами необ-
ходимо соблюдать осторожность. При попадании его на кожу необ-
ходимо немедленно смазывать пораженное место спиртом.
4. Метод обратного фильтрования Боста и Констэбля (стр. 479)
особенно удобен для фильтрования горячих растворов нитробен-
зилбромида, так как в этом случае не только уменьшается опасность
пожара, но и облегчается работа с растворами, которые обладают
лакриматорным действием. Чтобы не забивалась трубка, соединяю-
щая склянки для фильтрования, диаметр ее должен быть равен
8—10 мм. Из маточного раствора можно выделить обратно около
3,5 л лигроина.
5. л-Нитробензилбромид применяется как реактив для иденти-
фикации многих кислот х и фенолов 2 путем превращения их в л-ни-
тробензильные простые и сложные эфиры.
6. Имеются данные, что бромирование по методике Брюстера 3
проще и удобнее, чем по методу, приведенному выше, если в качестве
источника искусственного света применять лампу с вольфрамовой
24 Сборник J* 2
370
Синтезы органических препаратов
нитью накала. Для бромирования 300 г «-нитротолуола достаточен
свет от двух вольфрамовых ламп по 300 ватт. Выход я-нитробензил-
бромида составляет 60—70% теоретич. (Л. Вейслер и Д. Пирлман,
частное сообщение).
3. Другие методы получения
n-Нитробензилбромид обычно получался бромированием п-нитро-
толуола *> 3. Он был также получен реакцией между п-нитробензи-
ловым спиртом и бромистоводородной кислотой * и нитрованием
бромистого бензила 5.
1 R e i d, J. Am. Chem. Soc. 39, 126 A917); L у о n s, R e i d, там же 39,
1728 A917).
2 R e i d, там же 39, 304 A917); L у m a n, R e i d, там же 42, 615A920).
3 Brewster, там же 40, 406 A918).
* N о г г i s, Watt, Thomas, тамже 38, 1077 A916).
* Moureu, Brown, Bull. soc. chim. D) 29, 1008 A921).
И-НИТРОДИФЕНИЛОВЫЙ ЭФИР
(п-Нитрофенилфениловый эфир)
n-NO2C6H4OC6H6+KCl + H.2O
Предложили: Р. Брюстер и Т. Гренинг.
Проверили: В. Хартман и Дж. Дикки.
1. Получение
В 2-литровую колбу помещают 160 г A,7 мол.) фенола хорошего
качества и 80 г A,43 мол.) едкого кали и смесь нагревают при 130—
140° до полного растворения щелочи. Образовавшийся фенолят
калия охлаждают до 100—110°, добавляют к нему 0,5 г медного
катализатора (примечание 1) и 78,8 г @,5 мол.) п-нитрохлорбензола.
Колбу снабжают механической мешалкой, термометром и обратным
холодильником, пускают в ход мешалку и содержимое колбы нагре-
вают с помощью бунзеновской горелки до 150—-160°. При этой тем-
пературе начинается энергичная реакция, вся масса вскипает и
происходит выделение хлористого калия. В этой стадии реакции
горелку удаляют. Кипение заканчивается примерно через 5—7 мин.,
после чего добавляют 78,8 г @,5 мол.) n-нитрохлорбензола. Смесь
вновь нагревают до тех пор, пока не начнется вторая энергичная
реакция, которая продолжается также около 5 мин. без нагревания
извне. Когда кипение, вызванное экзотермической реакцией, пре-
кратится, вновь подставляют горелку и поддерживают температуру
150—160° в течение еще 30 мин. Темноокрашенный плав выливают
Л-НИТРОДИФЕНИЛОВЫЙ ЭФИР
371
в 1,5 л ледяной воды, содержащей 50 г едкого натра и массу хорошо
перемешивают, чтобы удалить избыток фенола. Сырой ?г-нитроди-
фениловый эфир выделяется в виде темнобурой кристаллической
массы, которой дают осесть. Продукт отсасывают на воронке Бюх-
нера, промывают 2 л воды и отжимают по возможности тщательнее
от воды. Кристаллы сушат на воздухе и перегоняют из колбы
Клайзена емкостью 500 мл. Небольшую фракцию, содержащую
л-нитрохлорбензол и кипящую до 170°/8 мм, отбрасывают. Соби-
рают фракцию, кипящую при 170—188°/8 мм; вес ее 14 г (примеча-
ние 2). Главная фракция кипит при 188—193°/8 мм и при вторичной
перегонке почти нацело переходит при 188—190°/8 мм, п-Нитроди-
фениловый эфир застывает по охлаждении в массу кристаллов, по-
хожих на алмазы, с т. пл. 56—58°. Выход 173—177 г (80—82%
теоретич.; примечание 3).
2. Примечания
1. Активный медный катализатор можно получить из сернокис-
лой меди. Для этого 100 г @,4 мол.) сернокислой меди (CuSO4-5H2O)
растворяют в 350 мл горячей воды в 1-литровом стакане. По охла-
ждении до комнатной температуры постепенно добавляют 35 г
@,53 гр.-ат.) цинковой пыли (а если нужно, то и большее количество)
до полного обесцвечивания раствора. Выпавшую медь промывают
декантированием водой. К осадку добавляют разбавленную соляную
кислоту E%), чтобы удалить избыток цинка, и массу перемешивают
до тех пор, пока не прекратится выделение водорода. Порошко-
образную медь отфильтровывают, промывают водой и сохраняют во
влажном состоянии в хорошо закупоренной склянке.
2. Из фракции, кипящей при 170—188°/8 мм, при вторичной
перегонке можно получить 4 г fi-нитродифенилового эфира.
3. Если взять 0-нитрохлорбензол, получается 0-нитродифенило-
вый эфир с т. кип. 183—185°/8 мм с выходом в 84%- Для получения
л-нитродифенилового эфира лучшим методом является метод Улль-
мана и Шпонагеля 1, по которому исходят из .м-бромнитробензола,
так как ^-хлорнитробензол образует большое количество смоли-
стых продуктов.
3. Другие методы получения
я-Нитродифениловый эфир получается нитрованием дифенило-
вого эфира 2 и нагреванием я-нитрохлорбензола 3 или нитрофтор-
бензола 4 с фенолятом калия и фенолом.
1 Ullmann, Sponage'l, Ber. 38, 2211 A905).
2 Su ter, J. Am. Chem. Soc. 51, 2583 A929).
3 Haeussermann, Teichmann, Ber. 29, 1446 A896).
* R a r i с k, Brewster, D a i n s, J. Am. Chem. Soc. 55, 1289 A933).
372
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
НИТРОЗОМЕТИЛМОЧЕВИНА
373
N-НИТРОЗОМЕТИЛАНИЛИН
( N-MemuA-N-нитрозоанилин )
CeH5NHCH3 + NaNO2 + НС1 > CeH5N (NO) CH3 + NaCl + H2O
Предложили: В. Хартман и Л. Ролл.
Проверили: Л. Физер и Дж. Уокер.
1. Получение
В 3-литровую колбу, снабженную механической мешалкой, по-
мещают смесь 107 г A мол.) метиланилина (примечание 1), 145 мл
концентрированной соляной кислоты и 400 г льда. Смесь энергично
перемешивают и при температуре 10° или ниже (если надо, приба-
вляют еще льда) в течение 5—10 мин. приливают раствор 70 г A мол.)
азотистокислого натрия в 250 мл воды. Перемешивание продолжают
еще 1 час. Маслянистый слой отделяют, а водную часть извлекают
два раза бензолом порциями по 100 мл. Бензол отгоняют при атмо-
сферном давлении, а остаток перегоняют в вакууме. Главная фрак-
ция нитрозометиланилина перегоняется в виде светложелтой жид-
кости при 135—137°/13 мм. Выход (примечание 1) 118—127 г (87—
93% теоретич.).
2. Примечание
1. Выход зависит от качества взятого метиланилина. Самый
лучший выход получен с чистым метиланилином, кипящим при
81—82714*м.
3. Другие методы получения
N-Нитрозометиланилин впервые был получен действием азоти-
стой кислоты на метиланилин Ч Он получается также действием
йодистого метила на натриевую соль фенилнитрамина и последую-
щим восстановлением 2; при обработке диметиланилина тетранитро-
метаном 3 или фенилнитрокарбинолом4; при кислотном гидролизе
нитрозофенилглицина 5 и при окислении диметилдифенилгидразина
окислами азота *.
1 Нерр, Вег. 10, 329 A877).
2 В a m b e r g e г, там же 27, 373 A894).
s S с h m i d t, F i s с h e г, там же 53, 1538 A920).
4 Cohen, Calvert, J. Chcm. Soc. 73, 164 A898).
5 Fischer, Ber. 32, 249 A899).
e Wieland, Fressel, Ann. 392, 148 A912).
НИТРОЗОМЕТИЛМОЧЕВИНА
С а-Метил-а-нитрозомочевина)
I. ИЗ ХЛОРИСТОВОДОРОДНОЙ СОЛИ МЕТИЛАМИНА
CH3NH3C1+H2NCONH2 -
CHSNHCONH2+HNO, --
- CH3NHCONH2+NH4C1
CH3N (NO) CONH2+ HaO
Предложил: Ф. Арндт.
Проверили: К. Ноллер i
С. Либерман.
1. Получение
Во взвешенную 1-литровую колбу наливают 200 г A,5 мол.)
24%-ного водного раствора метиламина (примечание 1) и добавляют
концентрированной соляной кислоты, пока раствор не станет кис-
лым на метилрот, на что требуется около 155 мл кислоты. Затем
доливают столько воды, чтобы общий вес достиг 500 г, и прибавляют
300 г E мол.) мочевины, после чего раствор осторожно кипятят с об-
ратным холодильником 2 часа 45 мин., а после этого энергично кипя-
тят еще 15 мин. Раствор охлаждают до комнатной температуры и
растворяют в нем 1ЮгA,5мол.) 95%-ного азотистокислого натрия
и охлаждают его до 0°. В 3-литровом стакане приготовляют смесь
бООгльда и 100гAмол.) концентрированной серной кислоты, охла-
ждая стакан эффективно действующей смесью льда и соли, и к этому
раствору при перемешивании механической мешалкой медленно при-
ливают холодный раствор метилмочевины и нитрита с такой ско-
ростью, чтобы температура не поднималась выше 0° (примечание 2).
Нитрозометилмочевина всплывает на поверхность в виде кристал-
лического пенистого осадка, который немедленно отфильтровывают
с отсасыванием и на фильтре хорошо отжимают. Кристаллы раз-
мешивают до образования пасты с 50 мл холодной воды, по возмож-
ности лучше отсасывают, отжимают (примечание 3) и сушат в ва-
куум-эксикаторе до постоянного веса. Выход 105—115 г F6—72%
теоретич.; примечание 4).
2. Примечания
1. Содержание метиламина в продажном водном растворе опре-
делялось титрованием стандартным раствором кислоты с метилро-
том в качестве индикатора. Если раствор содержит иное количество
метиламина, берется эквивалентное его количество. '
2. Удобно держать раствор метилмочевины и нитрита в бане
со льдом и солью и сливать его с помощью сифона в раствор кис-
лоты, так чтобы конец сифона был погружен в жидкость. Это при-
бавление продолжается около 1 часа.
3. Проба влажного продукта должна нацело растворяться в ки-
пящем метаноле. Если остается заметный остаток, что бывает редко,
374
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
НИТРОЗОМЕТИЛМОЧЕВИНА
375
промывку повторяют. Каждая последующая промывка несколько
понижает выход.
4. Полученный таким образом препарат можно хранить в холо-
дильном шкафу неопределенно долгое время. При температуре
выше 20° его не следует хранить более, чем в течение нескольких
часов. При температурах около 30° нитрозометилмочевина может
претерпеть неожиданное разложение без взрыва, но с выделением
паров, вызывающих раздражение. Имеются указания на то, что
стойкость препарата возрастает, если прибавить к нему несколько
капель уксусной кислоты.
II, ИЗ АЦЕТАМИДА
2CH3CONH2-f Bra + 2NaOH
CH3NHCONHCOCH3 + 2 NaBr-|-2 H2O
CH3NHCONHCOCHS + H2O ~l CH3NHCONH2 + CH3CO,H
CH3NHCONH2+HONO > CH3N (NO) CONH,+ H2O "
Предложили: Е. Амштутц и Р. Майерс.
Проверили: К. Аллен и Дж. Дек.
1. Получение
А. Ацетилметилмочевиш. В 4-литровом стакане приготовляют
раствор 59 г A мол.) ацетамида в 88 г @,55 мол.) брома (примечание 1)
и к нему прибавляют по каплям при перемешивании от руки рас-
твор 40 г A мол.) едкого натра в 160 мл воды. Полученную желтую
реакционную смесь нагревают на водяной 5ане до начала реакции
(примечание 2), после чего нагревание продолжают еще 2—3 мин.
Обычно из раствора, окрашенного в желтый или даже красный цвет,
немедленно начинают выпадать кристаллы (примечание 3), и для
завершения кристаллизации раствор охлаждают в течение 1 часа
в бане со льдом. После фильтрования и высушивания на воздухе
получают 49—52 г бесцветного кристаллического ацетильного про-
изводного метилмочевины (84—90% теоретич.; примечания 4 и 5).
Его температура плавления 169—170°.
Б. Нитрозометилмочевина. Смесь 49 г @,42 мол.) ацетильного
производного метилмочевины (примечание 6) и 50 мл концентри-
рованной соляной кислоты нагревают при перемешивании от руки
на паровой бане до тех пор, пока не будет ясно, что твердое вещество
больше не растворяется (примечание 5). После этого нагревание
продолжают еще 3—4 мин. (всего нагревание на паровой бане про-
должается 8j—12 мин.), и раствор разбавляют равным объемом воды
и охлаждают ниже 10° в бане со льдом. Затем при перемешивании
медленно приливают холодный насыщенный раствор 38 г @,55 мол.)
азотистокислого натрия в 55 мл воды. Смесь оставляют стоять в бане
со льдом в течение нескольких минут, после чего нитрозометилмо-
чевину отфильтровывают и промывают 8—10 мл воды, предвари-
тельно охлажденной до 0°. После высушивания на воздухе полу-
чают 33—36 г G6—82% теоретич.) нитрозометилмочевины в виде
бледножелтых кристаллов с т. пл. 123—124°.
2. Примечания
1. Осторожное нагревание на водяной бане способствует раство-
рению ацетамида. Однако необходимо соблюдать осторожность,
чтобы во время нагревания терялось лишь минимальное количество
брома.
2. Иногда реакция начинается очень бурно, поэтому ее проводят
в большом сосуде.
3. Если на этой стадии раствор совершенно бесцветен, то про-
дукт обычно кристаллизуется медленнее, содержит большее коли-
чество бромистого натрия, и выход получается ниже. По этим при-
чинам и желателен небольшой избыток брома.
4. Если полученный выход отвечает наименьшему из приведен-
ных чисел, оставшееся количество продукта может быть выделено
при длительном охлаждении фильтрата на льду.
5. Неочищенное ацетильное производное метилмочевины содер-
жит некоторое количество бромистого натрия в виде белого кристал-
лического продукта, нерастворимого в концентрированной соляной
кислоте в стадии (Б). Бромистый натрий растворяется при после-
дующем разбавлении раствора и не оказывает влияния на дальней-
шую обработку азотистокислым натрием.
6. Ацетильное производное метилмочевины, полученное в стадии
(А), может быть применено без высушивания.
3. Другие методы получения
Нитрозометилмочевина всегда получается нитрозированием ме-
тилмочевины. Метилмочевина, в свою очередь, может быть получена
из а) хлористоводородной соли метиламина и цианистого калия 1ш,
б) диметилсульфата, аммиака и цианистого калия 1; в) хлористово-
дородной соли метиламина и мочевины 2 и г) ацетамида, брома и
щелочи3. Проверенные указания для получения метилмочевины
с целью ее последующего нитрозирования по способам (а) и (б)
описаны на стр. 48 «Org. Syn.», т. 15. Получение метилмочевины и ее
последующее нитрозирование по способам (в) и (г) приведены выше,
в методиках I и II. Методы, основанные на применении цианистого
калия, обладают тем недостатком, что этот реактив дорог и мало
376
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
доступен. Арндт, Леве и Аван 2 рассмотрели преимущества различ-
ных методов получения нитрозометилмочевины.
1 А г n d t, Amende, Z. angew. Chem. 43, 444 A930); A r n d t,
S с h о 1 z, там же 46, 47 A933).
! Arndt, Loewe, Avan, Ber. 73, 606 A940); E i s t e r t, Angew.
Chem. 54, 124 A941).
3 Owen, Ramage, Simonsen, J. Chem. Soc. 1938, 1213.
НИТРОЗОМЕТИЛУРЕТАН
(Этиловый эфир N-mmpo3o-N-метилкарбаминовой кислоты)
CH3NHCO2C2H5 + HNO2 CH3N (NO) CO2C2H3 + H2O
Предложили: В. Хартман и Р. Филлипс.
Проверили: Л. Физер и Дж. Уокер.
1. Получение
К 206 г B мол.) этилового эфира N-метилкарбаминовой кислоты
(стр. 587) и 600 мл диэтилового эфира, находящимся в 5-литровой
колбе, прибавляют одновременно 200 г льда и 650 г (9 мол.) 96%-ного
азотистокислого натрия, растворенного в 1 л холодной воды (при-
мечание 1). Колбу закрывают пробкой, в которую вставлены тер-
мометр, трубка для выхода выделяющихся окислов азота и дели-
тельная воронка, нижний конец которой доходит до дна колбы. Через
воронку осторожно прибавляют в течение 1,5 часа 1,2 г F,7 мол.)
раствора холодной 35%-ной азотной кислоты, приготовленной
смешением 600 г D26 мл) концентрированной азотной кислоты с 600 г
льда. Колбу время от времени взбалтывают, придавая ее содержи-
мому вращательное движение, но перемешивание производят глав-
ным образом выделяющиеся газы. Температура не должна быть
выше 15°; в случае надобности к смеси прибавляют лед. Эфирный
слой становится сначала розовым, а потом постепенно сине-зеленым.
Как только окраска станет зеленой, эфирный слой отделяют (при-
мечание 2) и дважды промывают сначала холодной водой, а потом
холодным раствором поташа до тех пор, пока не прекратится выде-
ление углекислоты. Раствор сушат твердым поташом и эфир отго-
няют на водяной бане из 1-литровой колбы для вакуум-перегонки
с дефлегматором C0 см длины). Как только отгонится большая
часть эфира, прибор присоединяют к вакууму и слабо нагревают,
так чтобы температура жидкости была не выше 45—50° (примеча-
ние 3), пока давление не упадет до 20 мм. Выход нитрозометилуре-
тана, кипящего при 59—61°/10 мм, составляет 200 г G6%теоретич.).
Удельный вес продукта 1,133 при 20°.
НИТРОМЕЗИТИЛЕН
377
2. Примечания
1. Чтобы получить хороший выход, необходим большой избыток
нитрита. Вероятно, это является следствием реакции, протекающей
согласно уравнениям:
2 HNO2 NOa + NO + Н2О
CH3NHCO2C2H5 +2 NO., CH3N(NO) CO2C2H5 + HN3O
CH3NHCO2C2H6 + 4HNO,- CH3N(NO)CO2C,H5 + 2 NO + HNO3+
+ 2 HaO
Таким образом, окись азота (NO) не входит в реакцию. Не рекомен-
дуется стремиться использовать ее, окисляя кислородом, во избе-
жание взрыва, или воздухом, так как в этом случае благодаря уве-
личивающемуся испарению могут произойти потери.
2. Нитрозометилуретан вызывает раздражение кожи.
3. По литературным данным, при перегонке под атмосферным
давлением нитрозометилуретан взрывается.
3. Другие методы получения
Нитрозометилуретан получается обработкой этилового эфира
метилкарбаминовой кислоты азотистокислым натрием и серной
кислотой г и при пропускании газов, выделяющихся при действии
мышьяковистого ангидрида на азотную кислоту, в эфирный раствор
этилового эфира метилкарбаминовой кислоты 2.
1 К 1 о b b i e, Rec. trav. chim. 9, 139 A890).
2 Pechmann, Ber. 28, 856 A895); Schmidt, там же 36, 2477
A903); В r a h 1, там же 36, 3635 A903).
НИТРОМЕЗИТИЛЕН
B-Нитромезитилен)
СН,
НСЧ
СН,
HNOo
н2о
Предложили: Г. Поуэлл и Ф. Джонсон.
Проверили: В. Хартман и Дж. Дакки.
1. Получение
В трехгорлую круглодонную колбу емкостью 500 мл, снабжен-
ную механической мешалкой, капельной воронкой и гильзой для
термометра, помещают 40 г @,333 мол.) мезитилена («Синт. орг.
преп.», сб. 1, стр. 242) и 60 г E5,5 мл) уксусного ангидрида (приме-
чание 1). Колбу помещают в баню с водой и льдом и по охлаждении
реакционной смеси ниже 10° начинают приливать смесь 31,5 г B0,8 мл,
378
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
1-НИТРО-2-НАФТОЛ
379
0,5 мол.) дымящей азотной кислоты (уд. в. 1,51), 20 г A9,1 мл)
ледяной уксусной кислоты и 20 г A8,5 мл) уксусного ангидрида
(примечание 2). Нитрующую смесь приливают при перемешивании
в течение 40 мин., поддерживая температуру между 15 и 20°.
После того, как добавлена вся азотная кислота, колбу с реакцион-
ной смесью вынимают из охлаждающего раствора и оставляют ее
стоять при комнатной температуре в течение 2 час. Затем колбу
нагревают при взбалтывании до 50° на водяной бане (примечание 3)
и поддерживают эту температуру в течение 10 мин. Смесь охлаждают
и медленно выливают в 800 мл ледяной воды при энергичном пере-
мешивании. Затем добавляют около 40 г хлористого натрия, водный
слой декантируют и экстрагируют 200—250 мл эфира. Эфирную
вытяжку добавляют к оставшемуся нитромезитилену и эфирный
раствор промывают 3 или 4 порциями по 30 мл 10—15%-ного рас-
твора едкого натра до тех пор, пока водная вытяжка не станет
явно щелочной. Эфир отгоняют, пользуясь водяной баней, а к остатку
приливают около 150 мл 10%-ного раствора едкого натра и подвер-
гают его перегонке с водяным паром. Перегонку с водяным паром
продолжают до тех пор, пока взятая проба дестиллата не станет про-
зрачной и не будет содержать масляных капель. Перегонка продол-
жается около 3 час, причем собирается около 1,5 л дестиллата.
Нитромезитилен собирается на дне сосуда. Воду декантируют через
бумажный фильтр, а остаток растворяют в 30 мл эфира. Бумажный
фильтр, если на нем собрались твердые частицы, промывают 10 мл
эфира, оба раствора соединяют вместе и помещают в перегонную
колбу на 150 мл, присоединенную к приемнику с водяным охла-
ждением. После отгонки эфира на водяной бане желтый остаток
перегоняют на голом огне. Почти весь продукт переходит в преде-
лах 243—250° и по охлаждении затвердевает^ причем остаток соста-
вляет 0,5—1,5 г. Вес полученного желтого кристаллического про-
дукта — 47 г. Для очистки его растворяют в 25 мл продажного
метилового спирта и при помешивании охлаждают смесью льда и
соли. Кристаллы отсасывают через маленькую бюхнеровскую во-
ронку и дважды промывают холодным метиловым спиртом порциями
по 5 мл. Из промывной жидкости с помощью той же методики выде-
ляют 6—8 г сырого продукта с т. кип. 243—249°, который при
перекристаллизации дает 4 г чистого нитромезитилена. Чистый
нитромезитилен светложелтого цвета, плавится при 43—44°. Выход
41—42 г G4—76% теоретич.).
2. Примечания
1. Можно взять технический продукт с содержанием 85% уксус-
ного ангидрида.
2. Азотную кислоту осторожно приливают к смеси уксусной
кислоты и уксусного ангидрида, поддерживая все время темпера-
туру ниже 20° с помощью бани со льдом и солью. Если азотную
кислоту приливать слишком быстро, то происходит реакция со
взрывом.
3. Повышать температуру выше 50° не рекомендуется.
3. Другие методы получения
Нитромезитилен был получен непосредственным нитрованием
мезитилена концентрированной азотной кислотой 1, а также дей-
ствием бензоилнитрата на мезитилен в растворе четыреххлористого
углерода при низкой температуре 2 и из динитромезитилена восста-
новлением одной нитрогруппы и заменой образовавшейся амино-
группы на водород 3.
1 F i t t i g, S t о r e r, Ann. 147, 1 A868); Biedermann, Lcdoux,
Ber. 8, 58A875); Sch u 1 t z, там же 17, 477 A884); Bamberger, Ri-
sing, там же 33, 3625 A900).
2 F r a n с i s, там же 39, 3801 A906).
3 L a d e n b u r g, там же 7, 1135 A874).
1-НИТРО-2-НАФТОЛ
N02
NHCOCHg,
NO2
//\/\
-2NaOH
N02
'SoNa
lONa
NO.,
V
CH,CO.,H
¦CH3CO2
CH,CO.,Na
Предложили: В. Хартман, Дж. Байере и Дж. Дшски.
Проверили: Л. Физер и Дж. У оке р.
1. Получение
В 3-литровую круглодонную колбу, соединенную с обратным
холодильником, помещают 100 г @,435 мол.) 1-нитро-2-ацетилнаф-
тиламина (стр. 359) и раствор 112 г B,8 мол.) едкого натра в 2,7 л
воды (примечание 1). Смесь кипятят до тех пор, пока не прекра-
тится выделение аммиака (от 6 до 7 час). Полученный раствор
окрашен в тёмнокрасный цвет, и в нем взвешены кристаллы нитро-
нафтолята, которые растворяют прибавлением 1 л горячей воды.
Затем раствор отфильтровывают от небольшого количества нерас-
творимого вещества; осадок промывают горячей водой до тех пор
пока промывные воды не станут бесцветными, и отбрасывают его,
380
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
хотя он и содержит немного нитронафтиламина. Фильтрат соединяют
с промывными водами и подкисляют 500 мл ледяной уксусной
кислоты. Нитронафтол выпадает в виде мелких яркожелтых кри-
сталлов, которые отсасывают на воронке Бюхнера диаметром 10 см,
промывают водой и сушат. Выход вещества с т. пл. 101—103° со-
ставляет 76—81 г (92—98% теоретич.).
Продукт очищают перекристаллизацией из 500 мл метилового
спирта, содержащего 5 мл концентрированной соляной кислоты.
Получают 60 г кристаллов с т. пл. 103—Ю4°. При упаривании
маточного раствора до 150 мл выпадает еще 12—13 г. Общий выход
перекристаллизованного продукта 72—73 г (88—89% теоретич.).
2. Примечание
1. Данная концентрация щелочи была найдена наиболее удо-
влетворительной. Если брать более крепкую щелочь, гидролиз
сначала идет очень быстро, однако скоро выпадает натриевая соль,
образующая густую пасту и вызывающая толчки при кипении.
Слабая щелочь требует для окончания гидролиза гораздо больше
времени.
3. Другие методы получения
1-Нитро-2-нафтол был получен нитрованием этилового эфира
Э-нафтола г>2; окислением 1-нитрозо-2-нафтолая и действием ацетил-
азотной кислоты на |3-нафтол4. Он получается также действием
3 мол. азотистокислого натрия на °-нафтиламин в присутствии
избыточного количества кислоты 5; при разложении азотной кисло-
той бензолазо-3-нафтола6; при нагревании в вакууме нитрата псевдо-
кумолазо-°-нафтола6; при обработке 1-бром-2-нафтола азотной
кислотой в среде уксусной кислоты 7 и при реакции между двуокисью
азота и Р-нафтолом 7. Он был получен также из 1-нитро-Ьбром-
2-кетодигидронафталина действием едкой щелочи 8 и сплавлением
а-нитронафталина с едким натром 9.
Описанный выше способ основан на данных Андреони и Бидер-
манна и других исследователей 10.
1 W i 11 k a m p f, Ber. 17, 393 A884).
2 G a e s s, J. prakt. Chem. B) 43, 22 A891).
3Stenhouse, Groves, Ann. 189, 151A877); Fierz-David,
I s с h e r, Helv. Chim. Acta 21, 680 A938).
• P i с t e t, Krijanowski, Arch. sci. phys. nat. Geneve D) 16,
191 A903) (Chem. Zentr. 1903, II, 1109).
5 D e n i n ge r, J. prakt. Chem. B) 40, 300 A889).
eCharrier, Ferrer i, Gazz. chim. ital. 44, A) 176 A914).
'Armstrong, Rossiter, Ber. 24 (R), 720 A891); J. Chem. Soc.
Proc. 1891, 87, 89.
8 Fries, Ann. 389, 317 A912).
' W о h 1, герм. пат. 116 790 [Frdl. 6, 114 A900—1902)].
10 A n d г е о li i, Biedermann, Ber. 6, 342 A873); Jacobson,
там же 14, 806 A881); Liebermann, Jacobson, Ann. 211, 46A882).
2-НИТРОТИОФЕН
381
CH — CH
2-НИТРОТИОФЕН
CH —CH
CH CH+HNO, - — CH
Предложил: В. Бабасинян.
Проверили: Р. Адаме и А. Кнауф.
1. Получение
Приготовляют два раствора: 84 г A мол.) тиофена (стр. 458)
растворяют в 340 мл уксусного ангидрида и 80 г A,2 мол.) дымя-
щей азотной кислоты (уд. в. 1,51) растворяют в 600 мл ледяной
уксусной кислоты (примечание 1). Каждый раствор разделяют на
две равные части. Половину раствора азотной кислоты помещают
в 2-литровую трехгорлую круглодонную колбу, снабженную тер-
мометром, механической мешалкой и капельной воронкой, и охла-
ждают до 10е. Затем при несильном перемешивании, по каплям
прибавляют половину раствора тиофена с такой скоростью, чтобы
температура реакционной смеси была ниже комнатной. Особенно
быстро повышается температура при прибавлении первой части
тиофенового раствора. В холодную погоду температуру регули-
руют таким образом, что колбу, в которой проводится нитрование,
погружают в баню с холодной водопроводной водой. Слишком силь-
ное охлаждение не является необходимым, но следует избегать
перегрева реакционной смеси (примечание 2). После того как при-
бавлена первая половина раствора тиофена, температуру реакцион-
ной смеси понижают до 10°, в колбу быстро вливают оставшуюся
часть раствора азотной кислоты и продолжают постепенное приба-
вление тиофена. Во все время нитрования раствор должен быть
светлобурого цвета. Появление розового или тёмнокрасного окра-
шивания указывает на то, что имеет место окисление. Продукт
оставляют стоять при комнатной температуре в течение 2 час,
после чего его обрабатывают равным количеством (по весу) коло-
того льда при энергичном взбалтывании. Мононитротиофен выде-
ляется в виде светложелтых кристаллов. При стоянии в холодиль-
ном шкафу в течение 24 час. или даже больше выпадает дополни-
тельное количество кристаллов. Их отсасывают (примечание 3)
на воронке Бюхнера или на воронке с пористым стеклянным филь-
тром при низкой температуре, тщательно промывают ледяной во-
дой, отжимают и сушат в эксикаторе из темного стекла или же в от-
сутствии света (примечание 4).
Фильтрат и промывная жидкость содержат в растворенном виде
некоторое количество продукта, которое может быть выделено
382
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
П-НИТРОФЕНИЛИЗОЦИАНАТ
383
перегонкой с водяным паром. Кислый дестиллат состоит из бело-
снежных кристаллов (если они защищены от действия света) нитро-
тиофена и из его раствора. Продукт отсасывают и промывают,
фильтрат нейтрализуют содой и экстрагируют эфиром. Эфирную
вытяжку сушат, эфир отгоняют и получают мононитротиофен
с примесью некоторого количества динитротиофена (примечание 5).
Выход продукта с т. пл. 44—45° составляет 90—110 г G0—85%
теоретич.). Если его подвергнуть очистке путем перегонки с водя-
ным паром и повторной перекристаллизации из петролейного
эфира (т. кип. 20—40°), то получают бесцветные кристаллы с тем-
пературой плавления 45,Ъ° (примечания б и 7).
2. Примечания
1. Обе кислоты следует смешивать постепенно, добавляя азот-
ную кислоту к уксусной и взбалтывая при этом колбу. Часто при-
ходится охлаждать смесь.
2. Успех в значительной степени зависит от правильного регу-
лирования температуры. Обычно все протекает нормально, если
реакционная смесь хорошо охлаждается холодной водой.
3. Мононитротиофен является сильным ядом '. При попадании
эфирного раствора на кожу образуются болезненные нарывы. При
несчастном случае пострадавшую часть кожи промывают спиртом.
4. Уже давно замечено, что это соединение чувствительно
к свету2.
5. Желтая окраска нитротиофена обусловлена наличием следов
динитротиофена и других примесей. Для того, чтобы обнаружить
присутствие динитротиофена, растворяют несколько кристаллов
в спирте и добавляют одну каплю слабого спиртового раствора
едкого кали. Немедленно появляется розовое или тёмнокрасное
окрашивание. Избыток едкого кали разрушает окраску3.
6. Мейер и Штадлер2 указывают, что нитротиофен перегоняется
без разложения при 224—225°.
7. Раньше для кристаллизации применяли эфир, спирт, бензол
и другие растворители. Как правило из этих растворителей нельзя
получить белоснежный продукт. В настоящей работе было найдено,
что петролейный эфир (т. кип. 20—40°) обладает несомненными
преимуществами, так как при длительном кипячении с обратным
холодильником он извлекает мононитротиофен, но лишь с трудом
растворяет примеси. При применении петролейного эфира были
получены белоснежные кристаллы в виде игл длиною от Ю до 20 см.
3. Другие методы получения
Нитротиофен был получен наряду с динитротиофеном при про-
садывании сильного тока воздуха, насыщенного парами тиофена,
через красную дымящую азотную кислоту s. Он был получен также
нитрованием тиофена при 0—5° смесью уксусного ангидрида и
дымящей азотной кислоты 4.
1 М е у е г, Вег. 18, 1772 A885).
2 Meyer, S t a d 1 е г, там же 17, 2648 A884).
3 Meyer, S t a d I e r, там же 17, 2780 A884).
*Steinkopf, Liitzkendorf, Ann. 403, 27 A914); герм. пат.
255 394 [Frdl. 11, 144 A912—14)].
Я-НИТР0ФЕНИЛИ30ЦИАНАТ
(п-Нитрофениловый эфир изоцшновой кислоты)
n-O2NC6H4NHa+COCl2 n-O2NC6H4NHCOCl +
n-O2NCeH4NHCOCl n-OsNC6H4NCO + HCl
Предложили: P. Шрайнер, В. Хорне и Р.
Кокс.
Проверили: В. Хартман и Дж. Дикки.
1. Получение
Прибор для проведения этого синтеза изображен на рис. 15.
С одного конца прибора вводится фосген, а другой конец соединен
с водоструйным насосом для создания небольшого разрежения.
В 5-литровой колбе А насыщают при комнатной температуре фо-
сгеном 500 мл сухого уксусноэтилового эфира (примечание 1). Фо-
Рис.
сген предварительно очищают, пропуская его через промывалку Б
с хлопковым маслом для удаления хлора, а затем через промывалку
В с концентрированной серной кислотой, как это показано на ри-
сунке (примечание 2). Затем через капельную воронку Г медленно
приливают раствор 150 г A,09 мол.) л-нитроанилина в 1,5 л сухого
уксусноэтилового эфира в течение 3—4 час. Добавление раствора
л-нитроанилина ведется с такой скоростью, чтобы образующийся
сперва осадок солянокислого л-нитроанилина успевал растворяться
и чтобы он не накапливался (примечание 3). Все это время через
384
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
раствор пропускают ровный ток фосгена, чтобы обеспечить его
избыток (примечание 4). К концу реакции раствор осторожно кипя-
тят с помощью бунзеновской горелки, для того чтобы способство-
вать растворению комков солянокислого л-нитроанилина, которые
иначе растворяются очень медленно.
После того как добавлен весь л-нитроанилин, пропускание
фосгена продолжают еще 5 мин,, затем пламя усиливают и отгоняют
уксусноэтиловый эфир. Следует быть осторожным, чтобы к концу
перегонки не перегреть остаток. Бурый остаток (примечание 5)
обрабатывают 800 мл горячего сухого четыреххлористого углерода,
а нерастворимый остаток (двузамещенную мочевину) отфильтро-
вывают.
Около двух третей четыреххлористого углерода отгоняют, после
чего раствор охлаждают и по возможности быстро отфильтровывают
кристаллы л-нитрофенилизоцианата таким образом, чтобы избе-
жать длительного действия на это соединение влаги воздуха. Кон-
центрированием маточного раствора получают еще некоторое коли-
чество кристаллов. Полученный продукт перекристаллизовывают
из сухого четыреххлористого углерода; получаются светложелтые
иглы с т. пл. 56—57° (примечание 6). Выход после однократной пере-
кристаллизации 152—170 г (85—95% теоретич.; примечание 7).
2. Примечания
1. Уксусноэтиловый эфир, не содержащий этилового спирта
сушат безводным сернокислым магнием.
2. Такого типа прибор дает возможность спокойно и безопасно
вести реакцию с фосгеном при наличии хорошего вытяжного шкафа
с хорошим вентилятором. Во всей системе поддерживают небольшое
разрежение. Избыток фосгена поглощают 20%-ным раствором
едкого натра в склянке Д.
Хотя обычно фосген содержится в стальных баллонах, иногда
он встречается и в стеклянных ампулах. Если для этого синтеза
берется ампула с фосгеном, то следует взять только одну третью
часть по весу л-нитроанилина, отвечающего данному количеству
фосгена. При этих условиях избыток фосгена обеспечен: ср. приме-
чание 4. Однако применение стеклянных ампул с фосгеном не реко-
мендуется, ввиду связанной с этим опасности.
3. Для растворения солянокислого л-нитроанилина, если это
потребуется, смесь можно слегка нагреть.
4. Избыток фосгена замедляет следующую реакцию:
264 263
. n-NO2C6H4NHCONHC6H4NO,-n+HCl
5. Неочищенный остаток представляет собой смесь солянокис-
лого л-нитроанилина, л-нитрофенилкарбамилхлорида, л-нитро-
2-НИТРОФЛУОРЕН И 2-АМИНОФЛУОРЕН
385
фенилизоцианата и л, л'-динитрофенилмочевины. л-Нитрофенил-
карбамилхлорид превращается в л-нитрофенилизоцианат при пере-
кристаллизации из горячего четыреххлористого углерода.
6. Свежеполученный продукт плавится при 56—57°, но при
хранении температура плавления вскоре понижается до 54°, осо-
бенно в том случае, если склянку, в которой хранится продукт,
иногда открывают. Под действием влаги воздуха образуется
л, л'-динитродифенилмочевина (т. пл. 360°). Продукт лучше сохра-
няется при хранении в запаянных сосудах.
7. л-Нитрофенилизоцианат перегоняется без разложения при
160—162718 мм.
3. Другие методы получения
л-Нитрофенилизоцианат был получен нагреванием л-нитрофенил-
карбамилхлорида. Последний получается действием фосгена на
л-нитроанилин в бензольно-толуольных растворах г, а также дей-
ствием пятихлористого фосфора на метил-л-нитрофенилкарбамат 2.
Описанная выше методика основана на данных, опубликованных
ее авторами3.
xVittenet, Bull. soc. chim. C) 21, 586 A899); Van Hoogstra-
ten, Rec. trav. chim. 51, 418 A932).
2 S w a r t z, Am. Chem. J. 19, 318 A897).
3 S h r i n e r, Cox, J. Am. Chem. Soc. 53, 1603 A931); Home, Shri-
n e г, там же 53, 3186 A931).
2-НИТРОФЛУОРЕН И 2-АМИНОФЛУОРЕН
|+HNO8
сн„
сн,
NOo
3Zn-4-H,O-
/
сн2
NO,
¦Jnh,
+н2о
-3ZnO
сн,
Предложил: В. Кун.
Проверили: Л. Физер и Дж. Уокер.
1. Получение
А 2-Нитрофлуорен. В 1-литровой трехгорлой колбе, снабжен-
ной термометром, механической мешалкой и капельной воронкой
и помещенной в водяную баню, растворяют 60 г @,36 мол.) флуо-
рена (примечание 1) в 500 мл теплой ледяной уксусной кислоты.
25 Сборник JV; 2
386
СИНТЕЗУ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
Температуру поднимают до 50° и при перемешивании прибавляют
в течение 15 мин. 80 мл A,3 мол.) концентрированной азотной кис-
лоты, уд. в. 1,42. Во время прибавления кислоты раствор слегка
желтеет и выпадает небольшой осадок. Температуру водяной бани
медленно повышают до 60—65°; осадок растворяется, и раствор
темнеет. Перемешивание продолжают и смесь постепенно нагревают
до 80° (примечание 2). Через 5 мин. водяную баню удаляют и смеси,
состоящей из полутвердой массы мелких желтых игол, дают охла-
диться в течение 2 час. до комнатной температуры. Продукт отса-
сывают на воронке Бюхнера самым тщательным образом и промы-
вают двумя порциями по 25 мл холодной ледяной уксусной кислоты,
содержащей 0,5 г уксуснокислого калия. Затем промывают еще
несколько раз водой и сушат. Полученный 2-нитрофлуорен плавится
при 155—156° и достаточно чист для большинства целей. Выход
60 г G9% теоретич.).
Если требуется более чистый продукт, полученный 2-нитрофлуо-
рен можно пере кристаллизовать из 200 мл ледяной уксусной кис-
лоты. Получается 56 г очищенного продукта с температурой
плавления 157°.
Б. 2-Аминофлуорен. В 2-литровой круглодонной колбе разме-
шивают в жидкую пасту 30 г @,14 мол.) высушенного и растертого
2-нитрофлуорена с 1 л 78%-ного спирта (820 мл 95%-ного спирта и
180 мл воды). К суспензии прибавляют раствор 10 г хлористого
кальция в 15 мл воды вместе с 300 г цинковой пыли и смесь хорошо
перемешивают. Колбу соединяют с эффективным обратным холодиль-
ником и реакционную массу кипятят в продолжение 2 часов.
Осадок, который состоит из цинковой пыли и окиси цинка,
отсасывают от кипящего раствора и экстрагируют (примечание 3)
50 мл кипящего 78%-ного спирта. Соединенные фильтраты выли-
вают в 2 л воды, причем выделяется белый хлопьевидный осадок,
который отсасывают и перекристаллизовывают из 400 мл горячего
50%-ного спирта. Очищенный 2-аминофлуорен кристаллизуется
в иглах с т. пл. 127,5°. Выход 20—21 г G8—82% теоретич.).
2. Примечания
1. Для работы был взят флуорен с т. пл. 113—114°.
2. В этой стадии реакция идет с выделением тепла и темпера-
тура реакционной смеси может подняться на 10—15° выше, чем
в бане. Если температура поднимается выше 85°, получится сильно-
окрашенный и нечистый 2-нитрофлуорен.
3. Осадок цинковой пыли и окиси цинка фильтруют горячим
через предварительно нагретую воронку. Если раствор не будет
нагрет до кипения, то часть продукта выкристаллизуется при филь-
тровании. Если часть цинковой пыли пройдет через фильтроваль-
4-НИТРОФТАЛЕВАЯ КИСЛОТА
387
ную бумагу, рекомендуется фильтрат нагреть до кипения и профиль-
тровать второй раз без отсасывания через складчатый фильтр на
воронке для горячего фильтрования.
3. Другие методы получения
Описанный способ в основном разработан Дильсом 1. !2-Нитро-
флуорен был получен также при пропускании окислов азота через
раствор флуорена в бензоле 2.
1 D i e I s, Вег. 34, 1758 A901).
2 Monti, Martello, Valente, Oazz. chim. ital. 66, 31 A936).
4-НИТРОФТАЛЕВАЯ КИСЛОТА
>NH+
2H2O
o,N
(hno3)
Предложили: E. Хентресс, E. Шлос мл. и П. Эрлих.
Проверили: в. Хартман и Дж. Сауди.
1. Получение
К раствору, содержащему 26,6 г @,66 мол.) едкого натра в 240 мл
воды, добавляют 80 г @,416 мол.) 4-нитрофталимида (стр. 389;
примечание 1). Смесь быстро доводят до кипения и не сильно кипя-
тят в течение 10 мин. Раствор слегка подкисляют на лакмус концен-
трированной азотной кислотой (уд. в. 1,42); для этого после того,
как реакция станет нейтральной, добавляют еще 70 мл A00 г, 1,1 мол,)
азотной кислоты (примечание 2). Раствор вновь кипятят 3 мин.,
затем охлаждают до температуры ниже комнатной, переливают его
в делительную воронку емкостью в 1 л и два раза экстрагируют
несодержащим спирта эфиром, порциями по 300 мл (примечание 3).
Прежде чем разделять слои, содержимое воронки необходимо энер-
гично взболтать. Эфирные вытяжки сушат безводным сернокислым
натрием и эфир отгоняют до появления твердой массы. Концентри-
рованный эфирный раствор выливают в фарфоровую чашку и остав-
шемуся растворителю дают испариться в вытяжном шкафу (приме-
чание 4). Температура плавления белых кристаллов 4-нитрофтале-
вой кислоты равна 163—164°. Кислотное число равно 105,5 (вычисл.
105,5). Выход 85—87 г (95—99% теоретич.).
388
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
2. Примечания
1. Если требуется омылить большое количество 4-нитрофтал-
имида, то целесообразнее провести ряд опытов с количествами, ука-
занными выше.
2. Взятого вначале количества щелочи достаточно для того,
чтобы нейтрализовать нитроимид, и для того, чтобы щелочность
полученного раствора была бы около 1-н. При нейтрализации крас-
ная окраска раствора переходит в грязнобурую, которая при под-
кислении превращается в бледножелтую. Дополнительных 70 мл
концентрированной азотной кислоты достаточно для того, чтобы
выделить в свободном виде всю 4-нитрофталевую кислоту, и при
этом не остается большого избытка кислоты, который затруднял бы
извлечение эфиром.
Целесообразнее применять азотную кислоту, а не соляную или
серную. Хотя выходы получаются и одинаковые, но при примене-
нии азотной кислоты продукт получается чистого белого цвета
и в лучшем физическом состоянии.
3. Необходимо избегать одновременного присутствия в эфирной
вытяжке азотной кислоты и спирта, так как при испарении эфира
может иметь место окислительный процесс, подобный взрыву. Кроме
того, присутствие спирта может повести к загрязнению продукта
следами сложного эфира.
4. Выпаривание последней порции эфира протекает медленно;
мягкие и рыхлые вначале кристаллы кислоты постепенно становятся
твердыми и плотными. Оставшаяся азотная кислота не причиняет
затруднений, если только эфир был свободен от спирта и было взято
указанное количество азотной кислоты.
3. Другие методы получения
4-Нитрофталевая кислота получалась обычно нитрованием фта-
левой кислоты * или фталевого ангидрида 2-3 с последующим отде-
лением ее от сопутствующей 3-нитрофталевой кислоты 4>5'в. Она
была также приготовлена из 6-нитро-2-нафтол-4-сульфокислоты
(полученной из технического внутреннего ангидрида диазотирован-
ной б-нитро-1-амино-2-нафтол-4-сульфокислотыO. Описанный выше
метод удобнее, чем все ранее предложенные.
1 Miller, Ann. 208, 223 A881); Huisinga, Rec. trav. chim. 27,
261 A908).
2 Levy, Stephen, J. Chem. Soc. 1931, 79.
8 Oulhane, Woodward, «Синт. орг. npen.», сб. 1, стр. 316.
* Bogert, Boroschek, J. Am. Chem. Soc. 23, 752 A901).
5 Cohen, Woodroffe, Anderson, J. Chem. Soc. 109, 232 A916).
• L a w r a n с e, J. Am. Chem. Soc. 42, 1872 A920).
' R u g g 1 i, Knapp, Merz, Zimmerman, Helv. Chim. Acta
12, 1043 A929).
4-H ИТРОФТАЛ ИМИД
389
4-НИТРОФТАЛИМИД
CO4
NH+HN03
(H,SO4)
V\Co
Предложили: Е. Хентресс и Р. Шрайнер.
Проверили: Ф Уитмор и К. Криммел.
1. Получение
В 3-литровый стакан наливают 1,4 л концентрированной серной
кислоты (х. ч.; уд. в. 1,84) и к ней добавляют 240 мл E,7 мол.)
дымящей азотной кислоты (х. ч.; уд. в. 1,50). Смесь охлаждают
в бане со льдом и, как только температура достигнет 12°, при пере-
мешивании, возможно более быстро прибавляют 200 г A,36 мол.)
продажного фталимида, поддерживая температуру при 10—15°.
Реакционной смеси дают нагреться до комнатной температуры по
мере того, как лед в бане тает, и оставляют ее на ночь.
Прозрачный светложелтый раствор, при энергичном перемеши-
вании, медленно выливают на 4,5 кг колотого льда; при этом тем-
пература смеси не должна подняться выше 20°. Сырой продукт
нитрования отфильтровывают с отсасыванием, причем в бюхнеров-
скую воронку кладут полотняный фильтр, и массу по возможности
хорошо отжимают. Полученную лепешку хорошо размешивают
с 2 л ледяной воды. Осадок отсасывают и вновь размешивают его
с 2 л ледяной воды. Такую промывку повторяют четыре раза. Неочи-
щенный продукт после высушивания на воздухе плавится при 185—
190°; выход его 165—174 г F3—66% теоретич.). С целью очистки
продукт перекристаллизовывают из 3—3,2 л 95%-ного этилового
спирта. При этом получается 136—140 г E2—53% теоретич.) 4-нитро-
фталимида с т. пл. 198°.
2. Другие методы получения
4-Нитрофталимид был получен из 4-нитрофталевой кислоты 1,
а также нитрованием фталимида 2. Описанная здесь методика яв-
ляется видоизменением способа Леви и Стефена2.
1 S e i d e I, Ber. 34, 4351 A901); S e i de I, Bittner, Monatsh! 23,
420 A902); Bogert, Renshaw, J. Am.' Chem. Soc. 30, 1137 A908).
2 Levy, Stephen, J. Chem. Soc. 1931, 79.
390
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
он
2
п-ОКСИБЕНЗОЙНАЯ КИСЛОТА
ок
+С6Н5ОН +
СО.2К (нагревание)
B30°)
л-КОС6Н4СО2К +
СО2К
. п-нос6н4со2н+:
Предложили: К. Бюлер и В. Кэт.
Проверили: Дж. Джонсон и К- Никольс.
1. Получение
В фарфоровую чашку диаметром 20 см помещают смесь из 100 г
@,725 мол.) салициловой кислоты и 150 мл воды и медленно, при
перемешивании, добавляют 60 г @,43 мол.) углекислого калия (при-
мечание 1). Раствор выпаривают на кипящей водяной бане до обра-
зования густой пастообразной массы, которую разбивают на мелкие
куски и сушат в сушильном шкафу при 105—110° в течение 2 час.
Сухой продукт измельчают в тонкий порошок, который сушат еще
два часа при 105—110° и затем вновь растирают в порошок.
Хорошо измельченную смесь калиевой соли салициловой кис-
лоты и углекислого калия помещают в круглодонную колбу ем-
костью 500 мл, которую погружают в масляную баню так, чтобы
из бани немного выделялась только верхняя часть горла колбы
(примечание 2). Баню нагревают до 240° (примечание 3) и эту тем-
пературу поддерживают полтора часа. При этом время от времени
содержимое колбы перемешивают с помощью изогнутой стеклянной
палочки со сплющенным концом.
По завершении реакции (примечание 4) продукт переносят еще
в горячем состоянии и по возможности полностью в 2-литровую
колбу, в которую предварительно налит 1 л горячей воды. Реак-
ционную колбу споласкивают несколько раз этим горячим раство-
ром. Щелочной раствор подкисляют концентрированной соляной
кислотой (требуется около 75 мл), нагревают его почти до кипения
и обрабатывают 5—б г активированного угля. Горячий раствор
фильтруют, чтобы освободиться от небольшого количества бурой
смолы. Фильтрат охлаждают под краном водопроводной водой и
выпавший сырой кристаллический продукт коричневого цвета отса-
сывают. Фильтрат упаривают до объема около 300 мл и вновь охла-
ждают. Вторую порцию неочищенной кислоты также отсасывают
и присоединяют к первой порции. Общий выход неочищенной
л-оксибензойной кислоты с т. пл. 208—211° достигает 40—45 г.
П-ОКСИБЕНЗОЙНАЯ КИСЛОТА
391
Неочищенную кислоту растворяют в 300 мл горячей воды, рас-
твор кипятят в течение нескольких минут с 4—5 г активированного
древесного угля и фильтруют. После тщательного охлаждения под
водопроводным краном очищенный продукт отсасывают и промы-
вают 10—15 мл холодной воды. Вес очищенной кислоты 35—40 г
G0—80% теоретич.); т. пл. 211—212°.
2. Примечания
1. Углекислый калий берется в избытке, так как это предохра-
няет массу от спекания при последующем нагревании. Хотя холод-
ная смесь имеет сильно щелочную реакцию, однако прозрачный
раствор получается только после нагревания смеси.
2. При такой методике работы образующийся при реакции фенол
отгоняется от смеси. Операцию эту следует вести в вытяжном шкафу.
3. Указывается температура масляной бани; температура внутри
колбы около 230°. Необходимо внимательно следить за температу-
рой, так как при более высокой температуре наступает заметное
разложение.
4. Окончание реакции может быть грубо определено следующим
образом: небольшую пробу обрабатывают 3—4 мл горячей воды и
подкисляют концентрированной соляной кислотой. Так как п-окси-
бензойная кислота растворима значительно лучше, чем салицило-
вая кислота, отсутствие осадка в теплом растворе указывает на то,
что реакция в основном закончена.
3. Другие методы получения
п-Оксибензойная кислота была получена нагреванием фенолята
калия в токе углекислого газа * или с четыреххлористым углеро-
дом 3, и нагреванием п-крезола с щелочами и окислами различных
металлов 3. Описанная выше методика близка к описанной в лите-
ратуре несколько десятков лет тому назад 4. Имеются указания, что
если обезвоживать двукалиевую соль салициловой кислоты путем
нагревания ее в вакууме, а затем нагреть в атмосфере углекислого
газа, то имеет место почти полное превращение салициловой кислоты
в л-оксибензойную5.
1 К о 1 b e, J. prakt. Chem. B) 10, 100 A874); Hartmann, там же
B) 16, 39 A877); Ost, там же B) 20, 208 A879).
aReimer, Tiemann, Ber. 9, 1285 A876); Н a s s e, там же 10,
2186 A877).
» Graebe, Kraft, Ber. 39, 797 A906); Friedlander, Low-
Beer; герм. пат. 170 230 [Frdl. 8, 158 A905—07)].
* Ко 1 be, J. prakt. Chem. B) 11, 24 A875); H e у d e n; герм. пат.
48 356 [Frdl. 2, 132 A887—90)].
* Dow Chemical Company, ам. пат. 1937 477 [С. А. 28, 1056 A934)].
392
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
ОКСИМ БЕНЗОФЕНОНА
393
2-ОКСИ-3,5-ДИИОДБЕНЗОЙНАЯ КИСЛОТА
C,5-Дииодсалщиловая кислота)
СОаН СО„Н
ОН /V
2IC1
ОН
I
2НС1
Предложили: Г. Вуллетт и В. Джонсон.
Проверили: В. Хартман и Е. Рарс.
1. Получение
В 2-литровом стакане с механической мешалкой растворяют
25 г @,18 мол.) салициловой кислоты (т. пл. 159—160°) в 225 мл
ледяной уксусной кислоты (примечание 1). При работающей ме-
шалке прибавляют сначала раствор 62 г @,38 мол.) хлористого иода
(примечание 2) в 165 мл ледяной уксусной кислоты, а затем 725 мл
воды. При этом выпадает желтый осадок дииодсалициловой кислоты.
Продолжая перемешивание, реакционную смесь постепенно нагре-
вают на электрической плитке до 80° и эту температуру поддержи-
вают в течение 20 мин. Весь процесс нагревания продолжается
около 40 мин. К концу реакции масса перемешивается с трудом,
так как выпадает объемистый осадок. По охлаждении до комнатной
температуры (примечание 3) осадок отсасывают на воронке Бюхнера
и промывают сперва уксусной кислотой, а затем водой. Массу хо-
рошо отжимают и полученный твердый остаток G5 г) растворяют
в 100 мл теплого ацетона и фильтруют без отсасывания. К филь-
трату медленно добавляют при взбалтывании 400 мл воды. Мелкий
хлопьевидный осадок отсасывают, промывают водой и сушат.
Выход дииодсалициловой кислоты с т. пл. 235—236° составляет
64—64,5 г (91—92% теоретич.; примечание 4).
2. Примечания
1. Взятое количество ледяной уксусной кислоты может ока-
заться недостаточным для полного растворения салициловой кис-
лоты. Растворение завершается после прибавления раствора хло-
ристого иода.
2. Достаточно чистый для этого синтеза хлористый иод можно
получить следующим образом: сухой хлор пропускают через 127 г
A rp.-ат.) иода, которые находятся в перегонной колбе емкостью
125 мл, до тех пор, пока увеличение веса не составит 34,5 г @,97
rp.-ат.). Трубка, через которую впускают хлор, должна доходить
почти до дна колбы, причем колбу все время осторожно встряхи-
вают. По достижении указанного выше привеса хлористый иод
перегоняют обычным образом, причем в качестве приемника слу-
жит колба Вюрца, защищенная от влаги воздуха хлоркальциевой
трубкой. Собирают фракцию 97—105°. Выход 142 г (87% теоретич.).
Хлористый иод можно хранить в сухой склянке с притертой проб-
кой. В этом синтезе необходимо брать избыток иода.
3. Если имеется свобо'дный иод, его удаляют 5%-ным раствором
серноватистокислого натрия.
4. При проверке данного синтеза было найдено, что 4-окси-
3,5-дииодбензойная кислота может быть получена из 4-оксибензой-
ной кислоты по этому же самому методу, однако в этом случае
продукт не перекристаллизовывают из ацетона, в котором он плохо
растворим. Выход 4-окси-3,5-дииодбензойной кислоты с т. пл.
278—279° (с разложением) 59 г (84% теоретич.).
3. Другие методы получения
Описанный выше метод основан на работе Кофмана 1. Дииод-
салициловая кислота была получена нагреванием салициловой
кислоты с иодом в спиртовом растворе 2; из тех же реагентов, но
с добавлением окиси ртути 3; действием иода на салициловую кис-
лоту в присутствии щелочи 3-4 и действием на салициловую кислоту
иода и йодноватой кислоты 6. Однако, повидимому, ни один из этих
методов не дает хорошего выхода чистого продукта.
1 Cofman, Gazz. chitn. ital. 50 (II) 297 A920).
2 Lautemann, Ann. 120, 300 A861).
3 W e s e 1 s k у, там же 174, 103 A874).
4 К e k u 1 ё, там же 131, 226 A864).
5 L i e с h t i, там же Spl. 7, 133, 141 A870).
ОКСИМ БЕНЗОФЕНОНА
(C,HB), CO+H,NOH • HCl + NaOH >
(CeH6)a С = NOH+2H2O+NaCl
Предложил: А. Лйхман.
Проверил: К. Ноллер.
1. Получение
100 г @,55 мол.) бензофенона («Синт. орг. преп.», сб. 1, 99),
60 г @,86 мол.) солянокислого гидроксиламина («Синт. орг. преп.»,
сб. 1, стр. 164), 200 мл 95%-ного этилового спирта и 40 мл воды
смешивают в 2-литровой круглодонной колбе. К смеси прибавляют
постепенно при встряхивании ПО г B,75 мол.) истолченного в по-
рошок едкого натра. Если реакция становится слишком бурной,
колбу охлаждают водопроводной водой. После того, как весь едкий
натр прибавлен, колбу соединяют с обратным холодильником и
реакционную массу кипятят в течение 5 мин. После охлаждения
смесь выливают в раствор 300 мл концентрированной соляной кис-
лоты в 2 л воды. Выпавший твердый осадок отсасывают, тщательно
394
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
ОКСИМ ЭНАНТОЛА
промывают водой и сушат (примечание 1). Выход 106—107 г (98—
99% теоретич.) вещества, плавящегося при 141—142°. При перекри-
сталлизации 20 г из 80 мл метанола получается 13 г кристаллов
с той же температурой плавления (примечание 2).
2. Примечания
1. Этот сырой продукт после сушки в течение ночи при темпе-
ратуре около 40° практически чист и, если применять его сразу, то
он вполне пригоден для получения солянокислого иминодифенил-
метана (стр. 269).
2. В присутствии кислорода и следов влаги оксим бензофенона
превращается постепенно в смесь бензофенона и азотной кислотых.
Чтобы продукт не разлагался при хранении, его сушат в вакуум-
эксикаторе, затем последний наполняют чистым углекислым газом,
опять эвакуируют и вновь наполняют углекислотой. Обработанный
таким образом оксим сохраняют в герметически закупоренных
склянках, также наполненных углекислотой.
3. Другие методы получения
Оксим бензофенона получается в больших количествах обработ-
кой водно-спиртового раствора бензофенона и солянокислого гидро-
ксиламина соляной кислотой3, содой 3, спиртовым раствором едкого
кали4 или водным раствором едкого натраь. Кроме того, он был
получен обработкой ?ш>нитрозилбензгидрила спиртовым раствором
едкого кали 6 и окислением а-аминодифенилметана раствором пер-
сульфата магния 7.
1 Hollemann, Rec. trav. chim. 13, 429 A894); L а с h m a n, J. Am.
Chem. Soc. 46, 1478 A924).
2 Beckmann, Ber. 19, 989 A886).
3 J a n n у, там же 15, 2782 A882).
4 Derick, Bornmann, J. Am. Chem. Soc. 35, 1287 A913).
6 L а с h m a n, там же 46, 1481 A924); 47, 262 A925).
e Behrend, Platner, Ann. 278, 369 A894).
' Bamberger, Seligman, Ber. 36, 704 A903).
ОКСИМ ЭНАНТОЛА
(Гептальдоксим)
2CH3(CH2)SCHO+2NH2OH • HCl + Na2CO3
2 CH3 (CH2M CH = NOH + 2 NaCl + CO2+ 3 H2O
Предложил: Е. Бускет.
Проверили: В. Каротерс и В. Мак-Ювен.
1. Получение
В 5-литровую двугорлую колбу, снабженную механической
мешалкой, обратным холодильником, термометром и делительной
воронкой, помещают водный раствор 348 г E мол.) солянокислого
305
гидроксиламина («Синт. орг. преп.», сб. 1, стр. 164) в 600 мл холод-
ной воды и 460 г D мол.) энантола (примечание 1). Мешалку пускают
в ход (примечание 2) и прибавляют к реакционной массе раствор
265 г B,5 мол.) безводного химически чистого углекислого натрия
в 500 мл воды с такой скоростью, чтобы температура смеси не пре-
вышала 45°. После того как прибавление раствора соды закончено,
перемешивание продолжают еще в течение 1 часа при комнатной
температуре.
Затем отделяют верхний маслянистый слой и промывают его
двумя порциями воды по 100 мл (примечание 3). Промытый продукт
переносят в специальную колбу Клайзена емкостью 1,5 л и перего-
няют в вакууме на масляной бане. Первая фракция состоит из нит-
рила энантовой кислоты, оксима энантола и очень небольшого
количества воды. Оксим собирают при 103—Ю7°/б мм (температура
масляной бани 140—147°, примечание 4). Выход 420-—480 г (81—
93% теоретич.). По охлаждении дестиллат медленно затвердевает
и плавится при 44—46°; его можно применять непосредственно
без дополнительной очистки для получения н.-гептиламина (стр. 154).
С целью очистки 25 г перегнанного продукта перекристалли-
зовывают из 70 мл 60%-ного этилового спирта. После одной пере-
кристаллизации (примечание 5) оксим энантола получается в виде
бесцветных листочков, с т. пл. 53—55° (примечания б и 7). Выход
перекристаллизованного продукта из одного опыта 315—320 г.
2. Примечания
1. Температура кипения взятого в реакцию энантола была 54—
59°/16 мм.
2. Для получения хорошего выхода необходимо быстрое и энер-
гичное перемешивание, так как энантол не растворим в воде и с вод-
ным раствором солянокислого гидроксиламина образует гетероген-
ную смесь.
Чтобы получить гомогенный раствор, к реакционной смеси можно
прибавить этиловый спирт, но при этом выход несколько понизится,
вследствие образования примесей с более высокой температурой
кипения.
3. Продукт реакции так плохо растворим в воде, что для выде-
ления его из водного раствора не нужно обрабатывать последний
эфиром, если для разделения слоев имеется достаточно времени.
4. Температура масляной бани во время перегонки имеет большое
значение. Первую фракцию кончают собирать, как только будет
достигнута постоянная температура кипения. Это наступает тем
скорее, чем лучше отрегулирована до начала перегонки температура
масляной бани. Первой фракции должно получиться не более 50 мл,
из которых около 10 мл воды. Если тщательно регулировать тем-
пературу бани, то практически весь продукт перегоняется при по-
стоянной температуре.
396
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
5. Оксим растворяют при осторожном нагревании и затем рас-
твор в продолжение нескольких часов охлаждают до 0° или ниже.
Вещество, оставшееся в маточном растворе (около 30% всего коли-
чества), можно выделить после отгонки спирта в виде не чистого,
трудно кристаллизующегося масла.
6. Температура плавления оксима энантола в капилляре E3—
55°) зависит от скорости нагревания. По различным литературным
данным оксим плавится в пределах от 50° до 58°.
7. По описанному способу из циклогексанона можно получить
с таким же выходом оксим циклогексанона. Так как этот оксим
плавится выше оксима энантола, то он затвердевает еще до окон-
чания прибавления раствора соды. Чтобы довести реакцию до конца,
через реакционную массу пропускают струю водяного пара до пол-
ного расплавления оксима и затем четыре раза сильно взбалтывают
смесь с 5-минутными перерывами. Оксим циклогексанона кипит
при Ю0—1057Ю—12 мм и плавится при 87—88°.
3. Другие методы получения
Оксим энантола был получен только действием энантола на
водный раствор солянокислого гидроксиламина в присутствии
щелочи *; вышеописанный метод представляет собой видоизменение
способа Вестенбергера '.
1 Westenberger, Вег. 16, 2992 A883); Golcischmidt, Z а-
п о 1 i, там же 25, 2593 A892); Bourgeois, Dambmann, там же
26, 2860 A893).
g-ОКСИЭТИЛМЕТИЛ СУЛЬФИД
B-Метилмеркаптозтанол )
SCH3
(HN = C — NH2JH2SO4+2NaOH
NH
Р-ОКСИЭТИЛМЕТИЛСУЛЬФИД
397
H2N — С — NHCN+ 2CH3SH+ Na2SO4+ 2 HgO
CH3SH + Na0C2H5
CH3SNa+CH3OHCH2Cl
CH3SNa+C2H5OH
CH3SCH2CH2OH+NaCl
Предложили: У. Уиндус и Я.' Шильднек.
Проверили: X. Кларк и С. Гурин.
1. Получение
Прибор состоит из 2-литровой трехгорлой колбы, снабженной
капельной воронкой, краном и длинным холодильником. После
холодильника установлена ловушка, состоящая из пустой промыв-
ной склянки для газа, соединенной с другой такой же. дклянкой.
в которую наливают 100 мл разбавленной серной кислоты A объем
концентрированной серной кислоты на 2 объема воды). Затем газ
проходит через колонку (высотой около 30 см) с хлористым каль-
цием, через пустую 2-литровую колбу, которая служит ловушкой,
и, наконец, через 2-литровую колбу с поглотительной смесью.
Последняя состоит из раствора 80,5 г C,5 гр.-ат.) чистого натрия
в 1,5 л абсолютного спирта. Отводную трубку из колбы с поглоти-
тельной смесью присоединяют к пустой 1-литровой колбе, которая
в свою очередь соединена с литровой колбой, содержащей 500 мл
насыщенного раствора уксуснокислого свинца (примечание 1). Отвод-
ная трубка последнего сосуда присоединена к водоструйному насосу.
В трехгорлую колбу помещают 556 г B мол.) сернокислой S-ме-
тилизотиомочевины (стр. 326), после чего через прибор просасывают
очень слабый ток воздуха с помощью водоструйного насоса и из
капельной воронки добавляют к сернокислой S-метилизотиомочевине
800 мл холодного 5-н. раствора едкого натра. Смесь медленно нагре-
вают для того, чтобы получить метилмеркаптан (примечание 2).
По мере того как реакция близится к концу (примерно через два
часа), смесь нагревают более энергично в течение 30 мин. (примеча-
ние 3).
Раствор натрийметилсульфида в абсолютном спирте переносят
в трехгорлую колбу емкостью 3 л, установленную на водяной бане
и снабженную капельной воронкой, обратным холодильником и меха-
нической мешалкой. Раствор нагревают до начала кипения спирта,
нагревание прекращают и по каплям добавляют 302 г C,75 мол.)
этиленхлоргидрина (примечание 4) при энергично работающей
мешалке в течение примерно двух часов (примечание 5). Реакцион-
ную смесь концентрируют, отгоняя по возможности большее коли-
чество спирта на водяной бане. Затем смеси дают охладиться и от-
фильтровывают выпавший хлористый натрий. Колбу ополаскивают
и хлористый натрий промывают три раза 95%-ным спиртом порциями
по 100 мл. Фильтрат и промывные жидкости соединяют вместе и
упаривают в вакууме на водяной бане, пока не прекратится отгон
дестиллата. Остаток переносят в видоизмененную колбу Клайзена
и подвергают его дробной перегонке в вакууме. Выход 238—265 г
G4—82% теоретич., считая на взятый в реакцию натрий). Продукт
кипит при 68—70°/2О мм (примечание 6).
2. Примечания
1. Уксуснокислый свинец связывает не вступивший в реакцию
метилмеркаптан, осаждая его в виде свинцовой соли.
2. Скорость выделения метилмеркаптана регулируют степенью
нагревания. После того как начнется быстрое выделение газа,
реакционную смесь следует нагревать очень осторожно. Получению
ровного тока газа способствует создание слабого вакуума.
398
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
ОКТААЦЕТАТ а-ЦЕЛЛОБИОЗЫ
399
3. Выделение метилмеркаптана в основном заканчивается через
1,5—2 часа. Продолжительное энергичное нагревание увеличивает
количество выделяющегося аммиака.
4. Этиленхлоргидрин следует предварительно перегнать и в реак-
цию взять фракцию, кипящую при 126—128°.
5. Реакция обычно заканчивается немедленно после добавления
этиле нхлоргидрина, так что последующее кипячение с обратным
холодильником является излишним. Реакцию можно считать закон-
ченной, когда раствор имеет нейтральную реакцию на лакмус.
6. Можно взять пятикратные загрузки по сравнению с указан-
ными выше, что не вызывает уменьшения выхода. В этом случае
удобнее отфильтровать хлористый натрий до упаривания раствора.
3. Другие методы получения
Методы получения метилмеркаптана и р-оксиэтиленметилсуль-
фида в основном разработаны соответственно Арндтомх и Кир-
нером 2.
1 А г n d t, Ber. 54, 2238 A921).
2 К i r n е г, J. Am. Chem. Soc. 50, 2451 A928).
ОКТААЦЕТАТ а-ЦЕЛЛОБИОЗЫ
(С12Н20О10)я+ л Н2О+ 8 п (СН3СО2) О .
, п сиНмОи (СОСН3)8 + 8 п СН3СО2Н
Предложила: Г. Браун.
Проверили: Р. Фьюзон, В. Росс и В. Кемпбелл.
1. Получение
В 1-литровую склянку с широким горлом и с притертой пробкой
наливают 400 мл уксусного ангидрида, который охлаждают до —10°
в охладительной смеси, после чего при взбалтывании сразу доба-
вляют 36 мл концентрированной серной кислоты. Температура
раствора повышается до 20°. Склянку вынимают из охладительной
смеси и в жидкость сразу погружают при помощи толстой стеклян-
ной палочки 20 г гигроскопической ваты (примечание 1). При непре-
рывном перемешивании склянку нагревают на водяной бане, тем-
пературу которой поддерживают при 60°, до тех пор, пока темпе-
ратура смеси не достигнет 45° (около 10 мин.), затем склянку вы-
нимают и температуре не дают подняться выше 55°, охлаждая
склянку, когда это нужно, проточной водой. Перемешивание
ведется непрерывно (примечание 2). Примерно через 20 мин. (от
начала реакции) смесь становится жидкой, ее охлаждают до 50° и,
продолжая перемешивание, прибавляют вторую порцию ваты
в 20 г, причем температуре вновь не дают подняться выше 55°.
Процесс охлаждения до 50° и прибавления по 20 г ваты продол-
жают с интервалами в 10 мин. до тех пор, пока не будет прибавлено
всего 100 г ваты. После прибавления последней порции перемеши-
вание продолжают до тех пор, пока масса не станет жидкой (около
10 мин.), склянку закупоривают и нагревают на бане, температуру
которой поддерживают при 50° в течение 1 часа. За это время вата
нацело растворяется, образуя жидкий, светлобурый сироп. После
этого закупоренную склянку держат в сушильном шкафу при 35°
в течение 7 дней.
Раствор темнеет, приобретает цвет темного красного вина и
уже на второй день начинает выкристаллизовываться октаацетат
а-целлобиозы (примечание 3). После семидневного стояния при 35°
полукристаллическую массу прибавляют при помешивании к 20 л
холодной воды. После энергичного перемешивания хлопьевидный
осадок октаацетата а-целлобиозы и ацетатов целлюлозы и декстрина
становится кристаллическим и через 1—2 часа его собирают на
воронке Бюхнера диаметром в 12,5 см, отмывают холодной водой
от кислоты и тщательно отжимают. Влажный продукт весом около
250 г размешивают с 250 мл теплого метилового спирта и по охла-
ждении до комнатной температуры нерастворившуюся часть соби-
рают на воронке Бюхнера диаметром в 7 см, промывают Зраза мети-
ловым спиртом порциями по 50 мл и сушат при 40°. Выход почти
чистого октаацетата а-целлобиозы составляет 69—74 г; с целью
очистки его растворяют в 300 мл хлороформа и раствор фильтруют
с отсасыванием в сухой приемник через слой угля, предварительно
промытый спиртом и находящийся в бюхнеровской воронке. Хлоро-
форменный раствор фильтруют еще тогда, когда слой угля все еще
смочен спиртом, и фильтр немедленно промывают 100 мл хлоро-
форма, не прерывая фильтрования. Бесцветный фильтрат упари-
вают в вакууме до начала кристаллизации ацетата (около 250 мл),
нагреванием кристаллы вновь переводят в раствор и все выливают
в 750 мл теплого метилового спирта. Немедленно начинает выкри-
сталлизовываться ацетат в виде мелких игл, образующих в конце
концов густую пасту. Смесь при помешивании охлаждают до 0°,
и, примерно через час, продукт отфильтровывают, промывают
100 мл метилового спирта и сушат при 40°. Выход бесцветного
октаацетата а-целлобиозы с т. пл. 220—222°, имеющего [а]^0" =
+ 41,6°, составляет 65—69 г C5—37% теоретич., если считать,
что влажность ваты равна 10%; примечания 4, 5 и 6).
С целью дальнейшей очистки раствор этого продукта в 350 мл
хлороформа, если нужно, осветляют и выливают в 750 мл метило-
вого спирта, после чего смесь охлаждают до 0°. Выход чистого аце-
тата с т. пл. 225—226° и удельным вращением [и]2?° + 42,5°
составляет 61—65 г. При повторной кристаллизации константы его
не изменяются.
400
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
ОЛЕИНОВЫЙ СПИРТ
401
2. Примечания
1. Можно взять продажную хлопковую вату или фильтроваль-
ную бумагу. Материал этот обычно содержит 7—10% влаги.
2. Для того чтобы получить хороший выход, необходимо тща-
тельно следить за температурой и энергично перемешивать реак-
ционную массу. Если за температурой не следить, она быстро
может подняться выше 100°, что поведет к значительному разло-
жению.
3. Для ускорения кристаллизации можно потереть стенки со-
суда стеклянной палочкой или же внести затравку. Эту затравку
легко получить, выливая небольшое количество реакционной смеси
в большое количество воды. Осадок отфильтровывают, обрабаты-
вают спиртом и сушат при 40°.
4. В более ранних опытах во время прибавления ваты поддер-
живали температуру при 30—40°, в результате чего на эту операцию
требовалось больше времени. Ацетолиз протекал в течение шести
дней при комнатной температуре. Выход продукта с т. пл. 220—
222° составлял 50—56 г, но мог быть увеличен и до 62—70 г, если
операция продолжалась 8 дней.
5. Указанные выше количества можно удвоить, не вводя изме-
нений в методику и получая тот же выход.
6. Метод выделения ацетата по Припсгейму1 дает такие же хоро-
шие результаты. Вместо того, чтобы после ацетолиза смесь выли-
вать в воду, ее держат при 0° в течение 1—2 дней, кристалличе-
ский продукт собирают на большой бюхнеровской воронке и про-
мывают 50 мл уксусного ангидрида, предварительно охлажденного
до 0°. Ацетаты целлюлозы и декстрина вместе с другими побочными
продуктами переходят в фильтрат. Сырую массу обрабатывают
200 мл теплого метилового спирта, отфильтровывают и перекристал-
лизовывают из смеси хлороформа и спирта так, как это описано
выше. Выход 65—68 г, т. пл. 220—222°.
3. Другие методы получения
Получение октаацетата а-целлобиозы ацетолизом целлюлозы было
открыто Франшимоном 2, и этот процесс был исследован подробно
целым рядом ученых ъз,*,ьш При разработке данной методики осо-
бенно ценными оказались наблюдения Фрейденберга 4 и Клейна 5.
1 Pringslieira, Merkatz, Z. physiol. Chem. 105, 173A919).
я Franchimont, Ber. 12, 1941 A879).
Sk г я u n, Ко ni g, Monatsh. 22, 1011 A901); Maquenne, Good-
c. chim. C) 31, 854 A904); Schliemann, Ann. 378,
:, там же 398, 332 A913).
3 Sk г аи р,
Bull, soc
win, Bull. soc. chim. C) 31, 854 A904)
366 A911); О s t, там же 398, 332 A913).
4 Freudenberg, Ber. 54, 767 A921).
5 К le i n, Z. angew. Chem. 25, 1409 A912).
ОЛЕИНОВЫЙ СПИРТ
C17H33CO2C4H9+ 4 Na+ 3 C4H9OH — C17H33CH2OH+ 4 C4H9ONa
Предложили: Е. Рид, Ф. Кокерилл, Дж. Мепер, В. Кокс мл. и Дж. Рухов.
Проверили: К. Ноллер и Р. Баннерот.
1. Получение
В 5-литровую круглодонную колбу, снабженную широким рога-
тым форштоссом и двумя обратными холодильниками с широкими
внутренними трубками, помещают 3 л безводного бутилового спирта
(примечание 1) и 507 г A,5 мол.) бутилового эфира олеиновой кислоты
(примечание 2), затем сразу прибавляют 180 г G,8 rp.-ат.) чистого
натрия, нарезанного кубиками (ребро кубика 2,5 см), и колбу со-
единяют с холодильниками. Сперва реакция проходит довольно
медленно, так что для того, чтобы достигнуть температуры кипения
бутилового спирта, требуется около получаса (примечание 3),
однако в дальнейшем она протекает очень бурно. При наличии двух
обратных холодильников потери обычно не имеют места, однако,
если реакция пойдет чересчур бурно или произойдет сильное вспе-
нивание, колбу следует обернуть мокрым полотенцем до тех пор,
пока реакция не успокоится (примечание 4). К концу реакции колбу
помещают на нагретую песчаную баню и поддерживают равномер-
ное кипение жидкости до тех пор, пока не прореагирует весь нат-
рий. После этого на время нагревание прекращают, постепенно
добавляют через холодильник 160 мл воды и раствор снова не сильно
кипятят в течение часа (примечание 5). Нагревание прекращают
и добавляют 1,2 л воды. После энергичного взбалтывания смеси
дают разделиться на два слоя. Нижний водный слой, содержащий
едкий натр, сифонируют и отбрасывают (примечание 6 и 11).
К содержимому колбы добавляют 200 г твердого хлористого
натрия (примечание 7) и бутиловый спирт отгоняют с водяным па-
ром (примечание 8). Маслянистый слой отделяют еще в горячем
состоянии (примечание 9), переливают в 1-литровый стакан и нагре-
вают на горячей плитке при помешивании до тех пор, пока темпе-
ратура не поднимется до 160°. К этому времени олеиновый спирт
не содержит уже воды и вспенивание прекращается (примечание
10). Горячую жидкость переносят в 1-литровую колбу Клайзена
с дефлегматором длиною 25 см и перегоняют ее в вакууме при
давлении в 3 мм. После небольшой головной фракции E—10 г)
главную фракцию собирают при 177—183°/3 мм. Выход олеино-
вого спирта 330—340 г (82—84% теоретич,: примечание 11).
2. Примечания
1. Для обезвоживания продажный бутиловый спирт перегоняют,
пользуясь колонкой длиною 1 м, и собирают фракцию 117,5—118,5°.
Обычный продажный спирт является достаточно чистым, так что
26 Сборник № 2
402
Синтезы органических препаратов
после того как температура паров достигнет 117°, все оставшееся
в колбе количество можно непосредственно использовать для реак-
ции восстановления без перегонки.
2. Бутиловый эфир олеиновой кислоты был получен алкоголи-
зом 3 кг вымороженного оливкового масла кипячением его с обратным
холодильником с 7 л бутилового спирта и 150 г концентрированной
серной кислоты в течение 20 час. Смесь промывалась три раз?
насыщенным раствором хлористого натрия, порциями по 2,5 л.
При последней промывке к раствору прибавлялся метилоранж и
углекислый натрий в количестве, достаточном для нейтрализации
оставшейся кислоты. Избыток бутилового спирта отгонялся и оста-
ток тщательно фракционировался, для чего применялась колба
Клайзена емкостью 2 л с дефлегматором длиною 25 см. Всего после
перегонки было получено 3075 г сложного эфира, из которых 2422 г
(около 70%, считая на взятое в реакцию оливковое масло) перего-
нялось в пределах 204—208°/3 мм, причем йодное число этой фрак-
ции было близко к теоретическому. Полученный продукт хотя и
содержал некоторое количество предельных сложных эфиров,
однако был все же значительно чище продукта, получаемого из
продажной олеиновой кислоты, и являлся вполне удовлетвори-
тельным для большинства целей.
3. При желании сократить это время реакционную массу можно
нагревать извне, пока она не достигнет температуры кипения.
В одном из опытов, когда бутиловый спирт нагревался до того, как
добавлялся натрий, реакция началась более бурно, но выход прак-
тически не изменился.
4. Реакционную смесь не следует охлаждать ниже температуры
кипения бутилового спирта, так как непрерывное и бурное проте-
кание реакции является необходимым условием для получения
Хороших выходов.
5. Это делается с целью омыления непрореагировавшего слож-
ного эфира.
6. От начала синтеза до этого момента обыкновенно требуется
около 3,5 часа, причем работу надо вести непрерывно.
7. Хлористый натрий препятствует образованию эмульсии при
перегонке с водяным паром.
8. Бутиловый спирт может быть легко регенерирован и высушен
с очень небольшими потерями.
9. Мыла, а возможно и высококипящие примеси, при охлаждении
вызывают застывание даже жидких спиртов с образованием студня.
10. Этим мероприятием в значительной степени устраняются
осложнения, которые вызывает вспенивание в начале перегонки
в вакууме.
П. Описанная выше методика вообще применима для восстано-
вления сложных эфиров в спирты, с прекрасными выходами. При
получении твердых нормальных предельных спиртов методика при
ОРТАНИЛОВАЯ КИСЛОТА
403
желании может быть изменена для регенерации кислоты из не
восстановленного сложного эфира. После того как удалена щелочь,
спиртовой слой промывают два раза 20%-ным раствором соли,
причем промывные растворы отбрасывают. Ни крепкая щелочь,
ни раствор соли не извлекают в этих условиях заметных количеств
органической кислоты. К раствору в бутиловом спирте добавляют
раствор 50 г хлористого кальция в 150 мл воды, смесь подвергают
перегонке с водяным паром до полного удаления бутилового спирта
и колбе с ее содержимым дают охладиться. В застывшей корке твер-
дого спирта делают отверстие, через которое удаляют водный слой.
Прибавляют 2 л толуола и колбу нагревают и взбалтывают до тех пор,
пока весь спирт не растворится и останутся только мелкие кристаллы
кальциевой соли невосстановленной кислоты. Раствор охлаждают
до 35° и фильтруют с отсасыванием. Кальциевое мыло удаляют
с фильтра, нагревают его с 500 мл толуола, охлаждают, фильтруют
и промывают небольшим количеством толуола. Соединенные толуоль-
ные растворы можно концентрировать и затем дать спирту выкри-
сталлизоваться или же толуол можно отогнать до конца и остаток
перегнать в вакууме. Нерастворимое кальциевое мыло можно раз-
ложить, вновь этерифицировать и использовать для следующего
восстановления.
3. Другие методы получения
Олеиновый спирт был получен восстановлением этилового эфира
олеиновой кислоты водородом в присутствии цинк-хромового ката-
лизатора 1 или же натрием в среде абсолютного этилового спирта2,
1 S a u e r, A d k i n s, J. Am. Chem. Soc. 59, 1 A937).
! Bouveault, Blanc, Bull. soc. chim. C) 31, 1210A904). См.
приготовление лаурилового спирта — на стр. 306.
ОРТАНИЛОВАЯ КИСЛОТА
( Аналин-о-сульфокислота; о-Аминобензолсульфокислота )
0-O2NC6H4SSC6H4NO2-0+4[O]+Cl2 <!™2 o-02NC6H4S02Cl
"o-02NC6H4S02Cl-f-H20 > o-02NCeH4S03H+HCl
o-02NC6H4S03H+ б [Н] <ре + сн^нЛ 0-H2NCeH4SO3H + 2 Н2О
Предложил: Е. Вертхейм.
Проверили: Р. Фьюзон и Р. Шрейбер,
1. Получение
А. о-Нитробензолсулъфожлхлорид. 3-литровую круглодонную
колбу с тремя горлами снабжают мощной мешалкой с жидкостным
затвором, обратным холодильником и трубкой для подачи хлора,
404
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
опущенной глубоко в жидкость. Стеклянная трубка, ведущая от
обратного холодильника, отведена в вытяжной шкаф. В колбу
помещают 200 г @,65 мол.) ди-о-нитрофенилдисульфида («Синт.
орг. преп.», сб. 1, стр. 200), 1 л концентрированной соляной кислоты
(уд. в. 1,18) и 200 мл концентрированной азотной кислоты (уд. в.
1,42). Через реакционную массу пропускают хлор со скоростью
около двух пузырьков в секунду, нагревая ее при этом на водяной
бане до 70°. Примерно через 30 мин. дисульфид расплавляется, и
раствор окрашивается в оранжево-красный цвет. После того как
весь дисульфид расплавится, нагревание и пропускание хлора
продолжают еще 1 час, после чего сульфонилхлорид немедленно
отделяют декантацией от верхнего слоя жидкости, промывают его
два раза теплой водой G0°) порциями по 300 мл и дают ему затвер-
деть, причем воду отделяют как можно тщательнее и отжимают
твердый продукт.
Промытый хлорангидрид растворяют при 50—60° в 140 мл ледя-
ной уксусной кислоты и раствор быстро фильтруют с отсасыванием.
Фильтрат охлаждают, погружая колбу в холодную воду, и при этом
массу энергично перемешивают для того, чтобы сульфонилхлорид
выделился в виде мелких кристаллов. Затем массу тщательно раз-
мешивают с 1 л холодной воды и декантируют водный слой через
большую воронку Бюхнера. Эту операцию повторяют два раза.
После этого к смеси добавляют 1 л холодной воды, затем при поме-
шивании приливают 10 мл концентрированного водного аммиака
(уд. в. 0,90). Массу немедленно фильтруют, на фильтре промывают
200 мл воды и сушат на воздухе. Выход 240г(84%теоретич.). Про-
дукт светло желто го цвета с т. пл. 64—65° и может быть использо-
ван без дальнейшей очистки (и без сушки) для получения ортанило-
вой кислоты.
Б. Ортапиловая кислота. В 3-литровую колбу, снабженную
обратным холодильником и мешалкой с жидкостным затвором, поме-
щают смесь из 200 г @,90 мол.) о-нитробензолсульфонилхлорида,
100 г безводного углекислого натрия и 600 мл воды. Смесь нагревают
на электрической плитке до кипения и непрерывно перемешивают,
чтобыускорить омыление, которое обычно заканчивается через 45мин.
после того, как хлорангидрид расплавится. Оранжево-красный
раствор фильтруют и фильтрат точно подкисляют на лакмус уксус-
ной кислотой, на что требуется ее около 25 мл. Раствор переносят
в 3-литровую круглодонную колбу с тремя горлами, снабженнук
обратным холодильником и эффективной мешалкой с жидкостным
затвором. Раствор нагревают на электроплитке до кипения и npi'
энергичном перемешивании добавляют железные опилки (размеров
около 20 меш) со скоростью около 25 г каждые 15мин. Всегоопилоь
прибавляют 350 г. Через несколько минут смесь приобретает темно-
бурую окраску и имеет склонность к вспениванию. После четырех-
часового перемешивания проба при фильтровании должна дат!
ОРТ АН ИЛОВАЯ КИСЛОТА
405
почти бесцветный фильтрат; если фильтрат продолжает оставаться
красного или оранжевого цвета, перемешивание и нагревание сле-
дует продолжить. После того как получится светлоокрашенный
фильтрат, добавляют 2 г активированного угля, горячую смесь
фильтруют с отсасыванием и остаток промывают несколько раз
небольшим количеством горячей воды, причем промывные воды
прибавляют к фильтрату. Фильтрат охлаждают до 15° и медленно
приливают 95 мл концентрированной соляной кислоты. Ортанило-
вая кислота выпадает в виде бесцветных кристаллов, имеющих под
микроскопом форму гексагональных пластинок (примечание 1).
После того как температура вновь понизится до 15°, всю массу
фильтруют и осадок промывают сперва водой, а затем этиловым
спиртом. Если к фильтрату добавить еще 20 мл концентрированной
соляной кислоты, то после стояния в течение нескольких часов выпа-
дает еще около 1 г вещества. Выход 89 г E7% теоретич.) вещества
97—100%-й чистоты; для ряда целей оно не нуждается в перекристал-
лизации. Вполне чистый продукт (ч. д. а.) может быть получен в ре-
зультате однократной перекристаллизации из горячей воды. Т. разл,
около 325° (блок Макенна).
2. Примечание
1. При охлаждении растворов ортаниловой кислоты ниже 13,5°
выпадает кристаллогидрат кислоты, который кристаллизуется в виде
игл (см. фотографии в работе Фирц-Давида и др.)х.
3. Другие методы получения
Ортаниловая кислота впервые была получена восстановлением
нитробензолсульфокислоты сернистым аммонием2. Это восстано-
вление было осуществлено также электролитически и посредством
железа или цинка 3. Кислота эта была получена кроме того перегруп-
пировкой фенилсульфаминовой кислоты4; действием бромновати-
стого натрия на калиевую соль о-карбаминобензолсульфокислоты 5;
восстановлением смеси нитробензолсульфокислот с последующим
разделением изомеров 6; действием метилового спирта на о-нитро-
фенилсульфохлорид7; действием кислоты на диацетилдифенилсульф-
амид 8; отнятием брома от n-броманилин-о-сульфокислоты 9; вос-
становлением 1,2,6,-аминотиофенолсульфокислоты 10 и омылением
и восстановлением л-нитробензолсульфонилхлорида, полученного
из ди-о-нитрофенилдисульфида ц.
1Fierz-David, Schlittler. Waldemann, Helv. Chitn.
Acta 12, 663 A929).
2 L i m p r i с h t, Ann. 177, 79, 98 A875).
3Wohlfahrt, J. prakt. Chem. B) 66, 556 A902); Goldschmidt,
Eckardt, Z. physik.' Chem. 56, 411 A906); Holleman, P о 1 a k, Rec.
trav. chim. 29, 419 A910); Шарвин, Арбузов и Варшавский,
ЖХП 6, 1409 A929).
406
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
1 Baraberger, Hindermann, Ber. 30, 654 A897); В a m b e r-
g е г, К u n z, там же 30, 2276 A897); Bretschneider, J. prakt. Chem.
B) 55, 286 A897); Bamberger, Rising, Ber. 34, 249 A901); В a u m-
g a r t e n, там же 59, 1976 A926).
5 В r a d s h a w, Am. Chem. J. 35, 339 A906).
6 В a h 1 m a n n, Ann. 186, 307 A877); Franklin, Am. Chem. J.
20, 457 A898); О b e r m i 1 1 e г, герм. пат. 281 176 [Frdl. 12, 125 A914—16)J.
7 Z i n с к e, F a r r, Ann. 391, 59—66 A912).
8 W О h I, Koch, Ber. 43, 3301 A910).
9 Thomas, Ann. 186, 128 A877); Basclcr Chem. Fabrik Bindschedler,
герм. пат. 84 141 [Frdl. 4, 90 A894—97)]; К г е i s, Ann. 286,377 A895); Brad-
shaw, Am. Chem. J. 35, 340 A906); Boyle, J. Chem. Soc. 95, 1698 A909);
Scott, Cohen, тамже 121, 2042 A922).
10 Rasso w, Do h 1 e, J. prakt. Chem. B) 93, 188, 203 A916).
11 E 1 ge r s m a, Rec. trav. chim. 48, 752 A929).
ПЕЛАРГОНОВАЯ КИСЛОТА
(н.-Нонановая кислота)
СН2 (СОаС2Н5J+ C,H16Br+ C4H9ONa
. С,Н15СН (СО2С2Н5
С,Н15СН(СО2С2Н5J+2Н2О —"! С7Н15СН(СО2НJ+2С2Н5ОН
С7Н15СН (СО2НJ > С,Н15СН2СО2
Предложили: Е. Рид и Дж. Рухоф.
Проверили: В. Хартман и Дж. Сауди,
1. Получение
В 5-литровую колбу с тремя горлами, снабженную механиче-
ской мешалкой с жидкостным затвором, обратным холодильником,
капельной воронкой и термометром, помещают 2,5 л абсолютного
бутилового спирта (примечание 1) и сразу прибавляют 115 г
E гр.-ат.) чистого, блестящего мелконарезанного металлического
натрия. Для того, чтобы облегчить растворение натрия, можно
пустить в ход мешалку, однако нагревать раствор не следует. Когда
натрий растворится полностью, раствор охлаждают до 70—80°,
после чего быстро, при помешивании, прибавляют 800 г E мол.)
свежеперегнанного малонового эфира (т. кип. 135—136°/100 мм).
Раствор нагревают до 80—90° и прибавляют 913 г E,1 мол.) чистого
бромистого гептила (стр. 114, т. кип. 179—180°). Сперва, пока не
начнется выделение бромистого натрия, бромид следует прибавлять
медленно. Затем его прибавление ведут с такой скоростью, чтобы
бутиловый спирт спокойно кипел. Обычно на прибавление бромистого
гептила требуется около 1 часа, после чего смесь осторожно кипятят
до тех пор, пока она не станет нейтральной на лакмус (около 1 часа).
Все содержимое колбы вместе с выпавшим в осадок бромистым
натрием переносят в 12-литровую колбу. Туда же выливают и неболь-
ПЕЛАРГОНОВАЯ КИСЛОТА
407
шое количество воды, которой ополаскивают реакционную колбу.
Затем медленно при взбалтывании прибавляют раствор 775 г
A2,5 мол.) 90%-ного едкого кали в равном весовом количестве
воды. Смесь осторожно нагревают, время от времени ее взбалтывая
до начала кипения (примечание 2). Кипячение продолжают до конца
реакции омыления (около 4—5 час), после чего колбу сейчас же при-
способляют для перегонки с водяным паром (<<Синт. орг. преп.»,
сб. 1, стр. 461) и смесь перегоняют до тех пор, пока не перестанет
переходить бутиловый спирт (примечание 3). К остатку добавляют
осторожно при взбалтывании 1350 мл A5,5 мол.) концентрированной
соляной кислоты (уд. в. 1,18) и смесь кипятят еще 1 час (примеча-
ние 4). По охлаждении водный слой сливают посредством сифона
и выбрасывают его (примечание 5).
Полученную маслянистую жидкость переносят в 3-литровую
круглодонную колбу и нагревают с обратным воздушным холодиль-
ником на масляной бане при температуре около 180°. После того
как прекратится выделение углекислого газа (примерно через
2 часа), маслянистый слой сливают с небольшого осадка. Этот
осадок обрабатывают 200—300 мл концентрированной соляной
кислоты, причем получают еще некоторое количество маслянистой
жидкости, которую добавляют к основному продукту.
Сырую пеларгоновую кислоту перегоняют в вакууме из видоиз-
мененной колбы Клайзена с дефлегматором, причем собирают
фракцию 140—142712 мм A88— 190о/Ю0 мм). Выход 525—590 г
F6—75%теоретич.). Т.пл. чистой кислоты 12—12,5° (примечание 6),
2. Примечания
1. Продажный бутиловый спирт был высушен твердым поташом
и перегнан с елочным дефлегматором высотой 90 см. Была взята
фракция с т. кип. 117—118°.
2. Сперва образуются два слоя, однако по мере того, как про-
текает омыление, раствор становится гомогенным. В колбу необ-
ходимо положить кусочки битой пеглазурованной тарелки, и,
особенно вначале, нагревание следует вести осторожно, время от
времени взбалтывая колбу, так как реакция может пойти очень бурно.
3. Между омылением и перегонкой не следует давать охла-
ждаться колбе. Рекомендуется колбу нагревать, чтобы не очень
увеличивался объем дестиллата. Обычно собирают около 7 л дестил-
лата, из которого можно выделить бутиловый спирт.
4. Когда прекратится увеличение маслянистого^ слоя, можно
считать разложение гептилмалоната калия законченным. На дне
колбы иногда собирается слой соли, поэтому при нагревании колбы
следует соблюдать осторожность, чтобы колба не лопнула.
5. Извлечение водного слоя органическим растворителем не
имеет смысла.
408
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
6. Пользуясь тем же методом и примерно с одинаковым выходом
можно получить н.-капроновую кислоту, исходя из бромистого
н.-бутила (см. также стр. 319). В этом случае частичное разложение
замещенной малоновой кислоты вызывают кипячением водного
раствора в 12-литровой колбе непосредственно после прибавления
соляной кислоты. Бутилмалоновая кислота заметно растворима
в воде, и расслоение с образованием маслянистого слоя имеет место
только после того, как она уже в значительной степени разложи-
лась с образованием капроновой кислоты. Необходимое для этого
время колеблется от 8 до 10 час. Рекомендуется нагревать кислот-
ный слой с обратным воздушным холодильником так же, как и
в случае пеларгоновой кислоты.
3. Другие методы получения
Пеларгоновая кислота была получена окислением олеиновой
кислоты ', а также омылением цианистого октила 2 или гептилацето-
уксусного эфира 3.
1 Redtenbacher, Ann. 59, 52 A846); Harries, Thieme,
там же 343, 355 A905); Harries, Turk, Ber. 39, 3737 A906); M о 1 i n a r i,
S и п с i n i, там же 39, 2739 A900); Molinari, Barozi, там же 41,
2795 A908); Егоров, ]. prakt. Chem. B) 86, 531 A912).
2 Zincke, Franc himont, Ann. 164, 333 A872).
3 Joufdan, там же 200, 107 A880).
н.-ПЕНТАН
CH3 (СНД, CHBrCH3+ Mg . СН3 (СН,J СН (MgBr) СН3
2 СН3 (СН2J СН (MgBr) CH3+ H.SOj >
2 СН3 (СН3K СН3+MgBra + MgSO,
Предложил: /<. Ноллер.
Проверили: Р. Адаме и Л. Ролл.
1. Получение
В 5-литровую колбу, установленную в водяной бане и снабжен-
ную механической мешалкой, делительной воронкой, термометром
в гильзе (примечание 1) и хлоркальциевой трубкой, вносят после-
довательно 182 г G,5 гр.-ат.) магниевых стружек, кристалл иода
и 100 мл смеси 1133 г G,5 мол.) 2-бромпентана (примечание 2) и
750 г ди-н.-бутилового эфира (примечание 3). Мешалку пускают
в ход и нагревают колбу на водяной бане до тех пор, пока не начнется
реакция, на что может потребоваться от 15 мин. до 1 часа. В это время
за процессом следует непрерывно наблюдать, так как реакция,
начавшись, идет очень бурно с выделением большого количества
тепла. Как только реакция начнется, в колбу приливают 750 г
ди-н.-бутилового эфира, а затем смесь 2-бромпентана с ди-н.-бутило-
вым эфиром с такой скоростью, чтобы температура реакционной массы
н.-ПЕНТАН
409
держалась в пределах 50—60°. Чтобы ускорить прибавление 2-бром-
пентана, реакционную массу охлаждают холодной водой. После того
как прибавление окончено, на что требуется около 3 час, смесь
нагревают на кипящей водяной бане в течение 1 часа.
Одновременно в 12-литровую колбу, снабженную мешалкой,
делительной воронкой и мощным, обращенным вниз холодильни-
ком с ледяной водой, помещают раствор 450 мл концентрированной
серной кислоты в 3 л воды. Мешалку пускают в ход и к кислоте при-
бавляют полученный выше раствор магнийорганического соеди-
нения. Посредством охлаждения извне температуру регулируют
таким образом, чтобы смесь несколько нагрелась, но не кипела.
После того, как прибавление магнийорганического соединения
закончено, реакционную массу нагревают на водяной бане до тех
пор, пока не прекратится отгонка пентана. Остаток в колбе охла-
ждают, отделяют слой дибутилового эфира, переносят его в 5-литро-
вую колбу, соединенную с обращенным вниз холодильником, и на-
гревают голым пламенем до тех пор, пока не будет достигнута тем-
пература кипения дибутилового эфира (примечание 4). Оба отгона
соединяют вместе, отделяют от небольшого количества воды, про-
мывают дважды холодной концентрированной серной кислотой пор-
циями по 125 мл и оставляют стоять ночь с безводным поташом.
Поташ отфильтровывают, и н.-пентан дважды фракционируют с эф-
фективной колонкой (высотою в 100см). Выход 270—290 г E0—53%
теоретич.) продукта, кипящего при 35,5—36,5°.
2. Примечания
1. В качестве гильзы для термометра удобна наполненная
ртутью запаянная с одного конца стеклянная трубка подходящего
диаметра. Конец трубки должен быть погружен в реакционную
смесь, а шарик термометра — в ртуть.
2. 2-Бромпентан удобно готовить из вто/я/чш-амилового алко-
голя (метил-н,-пропилкарбинола) действием бромистоводородной
кислоты. Если исходный продажный амиловый спирт содержит
изомеры, то н.-пентан получается менее чистый, с т. кип. 33—35°
и содержит значительное количество изопептана.
3.' Дибутиловый эфир следует перегнать, собирая фракцию 142—
144°. Этиловый эфир применять нельзя, так как его температура
кипения близка к температуре кипения пентана.
4. Перегонкой остатка легко получить обратно около 70% дибу-
тилового зфира.
3. Другие методы получения
н.-Пентан получался многими способами, но реакции, имеющие
препаративный интерес, немногочисленны. К ним относятся; вос-
становление 2-бромпентана цинком и соляной кислотой х; восстано-
вление 3-иодпентана цинком и иодистоводородной кислотой 2;
410
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
каталитическое восстановление пиридина при 50°3; восстановление
н.-амилена 4; разложение бромистого вторично-шплмагния хлори-
стым аммонием5. Применение н.-бутилового эфира в описанном выше
способе, чем избегается вопрос о разделении н.-пентана и этило-
вого эфира5, заимствовано из работы Марвела, Бломквиста и Бона6.
1 Clarke, Talbot, неопубликованные данные.
aKarvonen, Acta Chem. Fennica 3, 101 A930) [С. А. 25, 2412 A931I.
3 S k i t а, В r u n n e r, Ber. 49, 1598 A916).
4 Fettindustrieges. m. b. H., герм. пат. 329 471 [Frdl. 13, 178 A916—21I;
W i ba u t и сотрудники, Rec. trav. chim. 58, 337 A939).
. 'Fischer, К 1 e m m, Z. physik. Chem. A 147, 275 A930).
•Marvel, Blomquist, Vaughn, J. Am. Chem. Soc. 50, 2810
A928).
CH2
/\
сн2 сн2
! I
CH2 CO
\/
CH,
ПИМЕЛИНОВАЯ КИСЛОТА
А. ИЗ ЦИКЛОГЕКСАНОНА
+ C3H5O3CCO,C2H5
(NaOCsH3)
CH3
CH., CHCOCOX.H»
CH, CO
C2H5OH
CH,
сн2 снсосо2с2н5
сн2 со
нагревание
(Fe -j- битое
стекло)
сн2
сн2
/\
СН„ СНСОаС.Н6
I " I
сн2 со
2Н2О -^^
2 (HCI)
сн.,
сн2
/\
СН2 СНСО2С2Н5
сн., со
\/
сн2
НОХ (СНа)в С(
+ СО
Н6ОН
сн0
Предложили: X. Снайдер, Л. Брукс и С. Шапиро.
Проверили: Л. Смит, Р. Арнольд и Дж. Моран.
1. Получение
В 2-литровой трехгорлой колбе, снабженной капельной ворон-
кой, механической мешалкой с ртутным затвором и обратным
ПИМЕЛИНОВАЯ КИСЛОТА
411
холодильником, закрытым пробкой с хлоркальциевой трубкой,
приготовляют раствор этилата натрия, для чего осторожно раство-
ряют 4бгBгр.-ат.) чистого натрия в 600 мл абсолютного этилового
спирта (примечание 1). Колбу погружают в баню со льдом и солью
и пускают в ход мешалку. Когда температура раствора понизится
до 10° (примечание 2), из капельной воронки в течение 15 мин.
приливают предварительно охлажденный до 0° раствор 196 г B мол.)
циклогексанона (примечание 3) в 292 г B мол.) этилового эфира
щавелевой кислоты (примечание 4). Чтобы избежать полного затвер-
девания реакционной смеси, необходимо энергичное перемешивание
(примечание 5). После того как все прибавлено, продолжают еще
охлаждать смесь льдом в течение 1 часа, после чего продолжают
перемешивать ее при комнатной температуре около б час.
После этого реакционную смесь разлагают, осторожно приба-
вляя охлажденный до 0° разбавленный раствор серной кислоты,
полученный добавлением 56 мл концентрированной серной кислоты
(уд. в. 1,84) к 435 г льда. При нейтрализации температуру смеси
поддерживают при 5—10°, охлаждая колбу в бане со льдом и солью.
Раствор после этого должен иметь кислую реакцию на конго^ и его
разбавляют холодной водой, доводя объем до 4 л. Этиловый эфир
2-кетоциклогексилглиоксалевой кислоты выделяется * в виде тяже-
лой маслянистой жидкости, которую отделяют. Водный раствор
извлекают четыре раза бензолом порциями по 500 мл. Сырой про-
дукт присоединяют к бензольным вытяжкам, и полученный раствор
промывают два раза водой порциями по 200 мл. Для лучшего отде-
ления от воды бензольный раствор оставляют стоять в делительной
воронке в течение нескольких минут.
Бензольный раствор порциями по 500 мл переносят, не высуши-
вая его, в 1-литровую колбу Клайзена, присоединенную к водяному
холодильнику. Колбу нагревают на водяной бане до тех пор, пока
не прекратится отгонка бензола. Тогда водяную баню заменяют
на масляную и систему постепенно эвакуируют до остаточного
давления 10—12 мм, поддерживая температуру масляной бани
при 90°. После того как бензол, непрореагировавший сложный
эфир и кетон отгонятся (примечание 6), температуру масляной
бани повышают. Когда температура паров достигнет 105°/10—12 мм,
меняют приемник. Температуру масляной бани немедленно повы-
шают до 175° и собирают весь продукт, отгоняющийся между 105°
и 165° при 10—15 мм, на что требуется от 30 мин. до 1 часа. За это
время температуру бани постепенно повышают до 200°, для того
чтобы получить последние порции дестиллата. Выход 250 265 г
F3—67% теоретич.). Дестиллат переносят в видоизмененную колбу
Клайзена емкостью 500 мл и добавляют незначительное количество
(следы) железного порошка и немного мелко растертого легкоплав-
кого стекла (примечание 7). Смесь перегоняют при давлении 40 мм,
поддерживая температуру-'бани при 165—175°л. (примечание 8).
412
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
При этом выделяется окись углерода; дестиллат собирают в пределах
125—140°. Пиролитическое разложение продолжается 1,5—2 часа.
Выход этилового эфира 2-кетогексагидробензойной кислоты соста-
вляет 200—210 г E9—62% теоретич., считая на циклогексанон).
Этот продукт, имеющий показатель преломления Щ 1,476—1,479,
достаточно чистый для того, чтобы его можно было применить
в следующей стадии без дополнительной перегонки.
В 2-литровую трехгорлую колбу, снабженную капельной ворон-
кой, обратным холодильником и специальной мешалкой (приме-
чание 9), помещают 100 г B,5 мол.) едкого натра и 300 мл безводного
метанола. Пускают в ход мешалку и смесь нагревают в течение
1 часа на масляной бане, температуру которой поддерживают при
120° для того, чтобы растворить большую часть едкого натра.
Перемешивание и нагревание продолжают еще в течение 2 час,
в течение которых прибавляют 100 г @,59 мол.) этилового эфира
2-кетогексагидробензойной кислоты. Полученную смесь нагревают
в течение еще одного часа, поддерживая температуру бани при 120°.
Затем смесь разбавляют 600 мл воды и обратный холодильник заме-
няют на обращенный вниз. Метанол отгоняют до тех пор, пока тер-
мометр, погруженный в кипящий раствор, не покажет 98—100°.
Оставшийся водный раствор энергично перемешивают и к нему
осторожно, по каплям прибавляют точно 210 мл концентрирован-
ной соляной кислоты (уд. в. 1,18; примечание 10). Горячий кислый
раствор обрабатывают 2—4 г активированного угля и филь-
труют его через нагретую бюхнеровскую воронку. Фильтрат охла-
ждают в бане со льдом. Пимелиновую кислоту отфильтровывают
через воронку Бюхнера и перекристаллизовывают из кипящей воды,
беря на каждые 45 г кислоты 100 мл воды. После высушивания на
воздухе кислота плавится при 103,5—104°; выход ее составляет
65—73 г. Маточные растворы после гидролиза и перекристаллиза-
ции соединяют вместе и выпаривают досуха на водяной бане. Полу-
ченный твердый остаток два раза извлекают ацетоном порциями
по 500 мл. Ацетон отгоняют на водяной бане и оставшуюся сырую
пимелиновую кислоту перекристаллизовывают из минимального
количества бензола. Это дает еще 10—12 г чистого продукта, так
что суммарный выход составляет 75—83 г (80—88%теоретич., счи-
тая на этиловый эфир 2-кетогексагидробензойной кислоты, или
47—54%, считая на циклогексанон).
2. Примечания
1. Такие же хорошие результаты дает и сухой мётилат натрия.
Вместо раствора этилата натрия берут 108 г B мол.) метилата нат-
рия и 400 мл абсолютного этилового спирта.
2. На этой стадии реакции капельную воронку можно заменить
на пробку, в которую вставлен термометр.
ПИМЕЛИНОВАЯ КИСЛОТА
413
3. Продажный циклогексанон был перегнан с колонкой высотой
50 см с насадкой из карборунда. Вполне пригодна фракция с т. кип.
1547746 мм.
4. Продажный этиловый эфир щавелевой кислоты был перегнан
из видоизмененной колбы Клайзена, причем была использована
фракция с т. кип. 83—86725 мм.
5. Примерно через 1—2 часа после
прибавления реагентов обыкновенно вы-
падает осадок. Если раствор окрашен в
оранжевый цвет и осадок не выпадает,
его все же можно подвергнуть дальней-
шей переработке и получить удовлетво-
рительные результаты.
6. Последние порции этого дестил-
лата проверяют на реакцию с хлорным
железом. Красная или фиолетовая ок-
раска указывает на присутствие этило-
вого эфира 2-кетогексагидробензойной
кислоты.
7. Если взять более 1 мг железа,
то хотя скорость пиролиза и возрастает,
однако получается твердый остаток, ко-
торый трудно удалить из колбы. Обыкно-
венное легкоплавкое стекло растирают
в ступке; достаточно взять 0,5—1 г.
8. Если температура бани слишком
высока, то будет отгоняться не подверг-
нувшийся пиролизу эфир. Высокий по-
казатель преломления указывает на при-
сутствие неизменившегося эфира.
9. Мешалку делают следующим об-
разом (рис. 16). Вокруг изогнутой полу-
кругом стеклянной палочки наматывают
двойным слоем медную проволоку для
электромагнита. На определенных про-
межутках из проволоки делают петли,
которые закручивают так, чтобы длина
конца была около 5 см от стеклянной
палочки. Мешалку вводят в центральное
горло трехгорлой колбы. Концы про-
волоки должны быть такими, чтобы при
вращении мешалки они касались стенки колбы. Концам придают
нужное положение с помощью стальной или стеклянной палочки,
введенной через одно из боковых горл. Медная проволока скребет по
стенкам колбы, что препятствует отложению на них соли, а это, в
свою очередь, умеряет толчки при кипении. В случае применения
р и с. 16
414
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
ПИМЕЛИНОВАЯ КИСЛОТА
415
на этой стадии обычной мешалки выход уменьшается не менее чем
на 10%.
10. Если добавлено достаточное количество кислоты, раствор
становится почти совершенно прозрачным.
Б. ИЗ САЛИЦИЛОВОЙ КИСЛОТЫ
o-HOCeH4C02H+H20+4[H]
1iol) HO2C (СН2KСО2Н
Предложил: А. Мюллер.
Проверили: Р. Фьюзон, Дж. Литтл и У. Фъюгэт.
1. Получение
В 5-литровой двугорлой колбе, снабженной капельной воронкой
и обратным холодильником, помещают 400 мл свежеперегнанного
изоамилового спирта (т. кип. 128—132°) и нагревают его до 90—100°
на масляной бане. Добавляют 240гA0,4rp.-ат.) чистого натрия и
температуру масляной бани быстро повышают так, чтобы спирт
энергично кипел (примечание 1). В колбу приливают раствор 100 г
(О, 73 мол.) салициловой кислоты в 2 л изоамилового спирта со ско-
ростью 100 мл каждые 4 мин., так что на все приливание требуется
1 час 20 мин., после чего капельную воронку ополаскивают 100 мл
изоамилового спирта. Сперва раствор в колбе прозрачный, но по
мере прибавления салициловой кислоты он становится мутным.
Температуру масляной бани регулируют таким образом, что в те-
чение всего опыта спирт быстро стекает из обратного холодиль-
ника. Примерно через 7—8 час. натрий полностью переходит в
раствор.
Колбе дают охладиться до 100° и при энергичном взбалтыва-
нии приливают 800 мл горячей воды (примечание 2). Горячую смесь
переносят в 5-литровую делительную воронку и колбу ополаски-
вают 200 мл горячей воды (примечание 3). Смесь энергично взбал-
тывают и образовавшиеся слои разделяют. Раствор в изоамиловом
спирте извлекают четыре-пять раз водой, нагретой почти до кипе-
ния, порциями по 200 мл (примечание 4). Объединенные водные
растворы подвергают перегонке с водяным паром, чтобы освободить
водный раствор натриевой соли от изоамилового спирта. Собирают
около 500 мл дестиллата (примечание 5).
Колбе дают охладиться и приливают 920 мл соляной кислоты
(уд. в. 1,19). Не вошедшую в реакцию салициловую кислоту отго-
няют с водяным паром. Колбу при этом сильно нагревают так, что
реакционная масса упаривается и к концу перегонки начинает вы-
падать в осадок хлористый натрий. Удаление салициловой кислоты
бывает практически полным после того, как отогнано 10—12 л
дестиллата, хотя дестиллат продолжает еще давать положительную
реакцию на салициловую кислоту с хлорным железом. Раствор в
колбе охлаждается в течение ночи, после чего его сильно охлаждают
в бане со льдом.
Кристаллический осадок, представляющий собой смесь хлори-
стого натрия и пимелиновой кислоты, отфильтровывают через во-
ронку Бюхнера с отсасыванием, но не промывают. Продукт сушат
в фарфоровой чашке на водяной бане в течение нескольких часов,
часто его перемешивая, причем бурая масса частично расплавляется.
По охлаждении эту массу переносят в бумажный патрон аппарата
Сокслета на 500 мл. Аппарат Сокслета нагревают на электроплитке
и осадок извлекают 650 мл бензола. О полноте извлечения судят
по тому, что жидкость в сифонной трубке перестает быть мутной
(примечание 6). Экстракция закончена, когда после испарения не-
большой пробы раствора из экстрактора не остается остатка B—
Зчаса). По завершении извлечения бензольный растйор упаривают
до объема около 300 мл и пимелиновой кислоте дают выкристал-
лизоваться. Кристаллы отфильтровывают, промывают холодным бен-
золом и сушат на воздухе. Продукт плавится при 104—105°. Вы-
ход его 50—58 г D3—50% теоретич.).
2. Примечания
1. Выход пимелиновой кислоты заметно понижается, если
на этой стадии спирт не будет энергично кипеть,
2. Воду следует приливать медленно и в начальной стадии кол-
бу энергично взбалтывать, так как в смеси могут еще быть следы
непрореагировавшего натрия.
3. Извлекают обязательно горячей водой (85—90°), иначе об-
разуются весьма трудно разделяемые эмульсии. Если эмульсия все
же образуется, ее разрушают, пропуская острый пар через раствор
прямо в делительной воронке.
4. Значительную часть изоамилового спирта можно выделить
обратно и использовать в последующих синтезах. Влажный спирт
после извлечения водой непосредственно подвергают перегонке и
собирают фракцию с т, кип. 128—135°. В головной фракции отде-
ляют слой спирта, который можно подвергнуть перегонке вместе
с последующей порцией влажного спирта.
5. Этиловый эфир пимелиновой кислоты может быть получен
следующим образом. Вместо того, чтобы подвергать соединенные
водные вытяжки перегонке с водяным паром, их упаривают на во-
дяной бане до образования густой кристаллической массы, которая
лишь слабо пахнет изоамиловым спиртом. Массу растворяют в 600 мл
воды и добавляют 800 мл концентрированной соляной кислоты.
Смесь охлаждают до комнатной температуры, фильтруют и к филь-
трату приливают 170 мл концентрированной соляной кислоты.
416
СИНТЕЗЫ ^ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
Кислую смесь извлекают три раза эфиром порциями по 400 мл;
с успехом может быть применен экстрактор для непрерывного из-
влечения, например, описанный в «Синт. орг. преп.»,сб. 1, стр. 352,
и от полученных вытяжек отгоняют зфир. По охлаждении остатка
в бане со льдом выпадают кристаллы. После тщательного охлажде-
ния их отфильтровывают через воронку Бюхнера и отжимают на
пористой тарелке. К фильтрату добавляют несколько кусочков по-
ристой тарелки и оставляют его стоять ночь в вакуум-эксикаторе
над концентриропанной серной кислотой. Выпавшие кристаллы
отфильтровывают, отжимают на пористой тарелке и присоединяют
к основной порции. Всего получают 60—70 г.
Сырой продукт, представляющий собой смесь пимелиновой и
салициловой кислот, подвергают этерификации, для чего его кипятят
4 часа с 520 мл абсолютного этилового спирта и 6 мл концентрирован-
ной серной кислоты. Затем 2/3 спирта отгоняют, а к остатку добав-
ляют 600 мл воды и 400 мл зфира, смесь взбалтывают и водный
слой отделяют. Эфирный раствор дважды взбалтывают с 2 н. рас-
твором едкого натра, порциями по 200 мл, для того, чтобы удалить
этиловый эфир салициловой кислоты, а затем с водой до исчезно-
вения щелочной реакции. Эфир отгоняют, а остаток перегоняют
в вакууме. Этиловый эфир пимелиновой кислоты кипит при 153—
156°/24 лш; 148—152°/22 мм. Выход 54—60 г C5—38% теоретич.,
считая на салициловую кислоту).
При этой методике весьма важно удалить бурую густую массу,
которая образуется при частичном подкислении щелочного раствора
натриевых солей. Если всю соляную кислоту прибавить сразу, то
извлечение эфиром всего осадка является неприятной операцией,
ввиду медленного разделения эфирного и водного слоев. Очень
важно также тщательно промывать эфирный раствор этиловых
эфиров пимелиновой и салициловой кислот раствором едкого натра,
чтобы избежать потери продукта при гидролизе. (Частное сообще-
ние А. Мюллера и Э. Рэльца. Проверили В. Каротерс и В. Мак-Ювен.)
6. В некоторых опытах пимелиновая кислота начинает кри-
сталлизоваться в сифонной трубке. Чтобы по возможности пре-
одолеть это затруднение, прибор утепляют асбестом. Иногда сифон-
ную трубку приходится обогревать острым паром.
3. Другие методы получения
Пимелиновая кислота была получена в качестве побочного
продукта при взаимодействии бромистого триметилена с натрий-
циануксусным эфиром \ при действии углекислоты на бромистый
пентаметилен-1,5-димагний2, при гидролизе цианистого пентаме-
тилена 3, при действии натрия и амилового спирта на салицило-
вую кислоту, гваяколкарбоновую кислоту * или антраниловую
кислоту 5 и из 2-цианциклогексанона 6.
пиг!ерониловая кислота
417
Получение пимелиновой кислоты из циклогексанона, описанное
п части А, и из салициловой кислоты, описанное в части Б 7, пред-
ставляют собой новые методики в <<Синт. орг. преп.». Старая
методика для получения этилового зфира пимелиновой кислоты 8
приведена в примечании 5 на стр. 415.
1 Carpenter, Perkin, J. Chem. Ssc. 75, 933 A899).
2 G r i g n a r d, V i g n о n, Compt. rend. 144, 1359 A907).
3 Ha mo net, там же 139, 60 A904); Bull. soc. chim. [31] 33, 532
A905); Braun, Ber. 37, 3591 A904).
i Einhorn, Lumsden, Ann. 286, 259, 266 A895); W a 1 k e r, L u m-
s d e n, J. Chem. Soc. 79, 1198 A901).
5 Einhorn, Meyenberg, Ber. 27, 2467 A894).
6 Meyer, Helv. Chim. Acta 16, 1293 A933).
7 M ii 1 1 er, Monatsh. 65, 18 A935).
8 M u 1 1 e r, R б 1 z, там же 48, 734 A927); Org. Syn. 11, 42.
CH,-
0
ПИПЕРОНИЛОВАЯ КИСЛОТА
CH,
0
o-
CHO
[0]
(KM11O4)
0—-
CO,H
Предложили: Р. Шрайнер и Е. Клейдсрер.
Проверили: X. Кларк и С. Графф.
1. Получение
60 г @,4 мол.) пиперонала (примечание 1) и 1 500 мл поды по-
мещают в 5-литровую колбу, снабженную эффективной механиче-
ской мешалкой. Колбу ставят на водяную баню, нагревают смесь
до 70—80° и пускают в ход мешалку (примечание 2). Затем в эмуль-
сию из пиперонала и воды вливают раствор 90 г @,56 мол.) перман-
ганата калия в 1 800 мл воды, на что требуется 40—45 мин. (приме-
чание 3). Перемешивание и нагревание продолжают после этого
еще в продолжение 1 часа; к концу этого времени перманганат пол-
ностью раскисляется. К реакционной массе прибавляют 10%-ный
раствор едкого кали в таком количестве, чтобы она стала щелочной.
Затем горячую смесь фильтруют с отсасыванием, промывают пе-
рекись марганца тремя порциями горячей воды по 200 мл каждая
и соединенные фильтраты и промывные воды охлаждают. Выпадаю-
щий при этом непрореагировавший пиперонал следует отфильтро-
вать (примечание 4). Затем раствор подкисляют соляной кислотой,
прибавляя ее до тех пор, пока не прекратится образование осадка.
Полученную пиперониловую кислоту отсасывают, промывают
27 Сборник Л» 2
418
СИНТЕЗЫ ОРГАНИЧЕСКИХЦЧРЕПАРАТОВ
холодной водой до полного удаления хлоридов и сушат. Выход 60—
64 г (90—96% теоретич.) бесцветного вещества, плавящегося при
224—225° (исправл.). Для большинства целей сырой продукт
достаточно чист; его можно, однако, перекристаллизовать из 10-
кратного весового количества 95%-ного этилового спирта, причем
получают 52—56 г G8—84% теоретич.) игольчатых кристаллов,
плавящихся при 227—228° (исправл.).
2. Примечания
1. Можно применять без очистки продажный пиперонал с тем-
пературой плавления 36°.
2. Эффективное механическое перемешивание необходимо для
toro, чтобы расплавленный пиперонал образовал с водой эмульсию.
3. Обратный порядок прибавления понижает выход до 50%.
4. Если поддерживать температуру реакционной смеси при
70—80° и прибавлять перманганат с указанной скоростью, то пи-
перонал окисляется полностью.
3. Другие методы получения
Пиперониловая кислота была получена раскислением пипери-
новой кислоты 1, пиперонала1, сафрола 2 и изосафрола 2 перман-
ганатом калия. Она была получена также действием йодистого
метилена на протокатеховую кислоту3 в присутствии щелочи.
1 F i 11 i g, M i e 1 с k, Ann. 152, 40 A869).
2 Ciamician, Silber, Ber. 23, 1160 A890).
8 Fittig, Remsen, Ann. 168, 94 A873).
ПИРОМЕЛЛИТОВАЯ КИСЛОТА
Древесный уголь
HO2C
НОХ
?О,Н
Jco.
н
Предложили: Е. Филиппы и Р. Телен.
Проверили: X. Кларк и С. Графф.
1. Получение
В снабженную термометром 5-литровую круглодонную колбу
вносят 100 г истолченного в мелкий порошок соснового или ело-
вого древесного угля (примечание 1), 650 мл 82—88%-ной серной
кислоты уд. в. 1,76—1,80 (примечание 2) и небольшую каплю ртути
(примечание 3). Колбу ставят на проволочную сетку в вытяжном
шкафу п нагревают на маленьком пламени так, чтобы температура
реакционной массы поднялась за 4 часа до 250°. В течение следую-
ПИРОМЕЛЛИТОВАЯ КИСЛОТА
419
щего получаса температуру повышают до 290°, причем серная кис-
лота начинает испаряться, а смесь вспенивается и сильно увеличи-
вается в объеме (примечание 4). В продолжение следующего часа
температуру поднимают до 300°, а по истечении еще одного часа —
до 315°. За это время смесь несколько сгущается. Затем внезапно
начинается очень бурное кипение, и в горле колбы появляются тон-
кие белые иглы ангидрида пиромеллитовой кислоты (примечание 5).
Тогда прибавляют 50 мл серной кислоты, обмывая ею стенки колбы,
нагревают сиропообразную смесь в течение нескольких минут до
250° и переливают горячую жидкость в реторту с тубусом емкостью
1000 мл (примечание 6). Колбу споласкивают минимальным коли-
чеством воды и сливают этот раствор также в реторту.
Сначала на голом пламени отгоняют воду, затем прибавляют
к смеси 30 г кислого сернокислого калия и перегонку продолжают.
Сперва переходит почти бесцветная серная кислота. Как только
в шейке реторты начинают появляться кристаллы ангидрида пиро-
меллитовой кислоты, приемник заменяют другим, содержащим
50 мл воды. Нагревание продолжают до тех пор, пока перегонка не
прекратится. Реторту споласкивают 100 мл воды, раствор фильтруют
и выпаривают на водяной бане приблизительно до 25 мл (при-
мечание 7). Последний отгон также выпаривают до объема 25 мл.
По охлаждении кристаллизуется пиромеллитовая кислота. Про-
дукт, полученный из обеих порций, переносят на воронку для от-
сасывания с плотным фильтром, промывают двумя порциями ле-
дяной воды по 10 мл (примечание 8) и перекристаллизовывают из
четырех частей кипящей воды. Пиромеллитовую кислоту (кристал-
лизуется с 2 мол. воды) сушат при 105°. Т. пл. 271° B62° ненс-
правл.). Выход б—8 г (примечание 9).
2. Примечания
1. Выход пиромеллитовой кислоты зависит в значительной
степени от сорта взятого древесного угля. Из ивового угля пиро-
меллитовой кислоты не получается.
2. Удельный вес кислоты не должен выходить из указанных пре-
делов.
3. Полезное действие сернокислой ртути доказано рядом опытов.
4. Если сократить время первого периода нагревания (т. ниже
250°), то вспенивание в этом периоде усиливается и конечный выход
понижается.
5. Окончание реакции легко установить по начинающемуся
бурному вспениванию смеси, наступающему взамен спокойного
кипения. Если смесь недостаточно густа, то в следующих опытах
следует взять меньшее количество серной кислоты. Если же она
затвердеет, то надо взять больше кислоты.
6. Смесь следует перенести п реторту до того, как она застынет.
420
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
Тубус реторты (если только она не из стекла «пирекс» с притертой
пробкой) лучше всего закупорить стеклянной пробкой, диаметр
которой меньше диаметра тубуса. Пробку следует обернуть асбе-
стовой бумагой, смоченной раствором кремнекислого натрия.
7. Если в шейке реторты скапливается значительное количе-
ство ангидрида пиромеллитовой кислоты, то рекомендуется рас-
твор от ополаскивания реторты сливать в чистый сосуд, а не в при-
емник, в котором находится сравнительно большое количество
серной кислоты.
8. При выделении пиромеллитовой кислоты из маточного рас-
твора рекомендуется сначала удалить серную кислоту прибавле-
нием небольшого избытка гидроокиси бария и затем подкислить
раствор на конго соляной кислотой.
9. Ангидрид пиромеллитовой кислоты можно получить из сухой
безводной кислоты кипячением ее с уксусным ангидридом.
3. Другие методы получения
Пиромеллитовая кислота может быть получена окислением
гомологов бензола, содержащих органические заместители в 1,2,
4- и 5- положениях, например окислением дурола азотной кислотой1;
конденсацией бензола с хлорангидридом диэтилмалоновой кислоты,
восстановлением полученного продукта в углеводород, вторичной
конденсацией по тому же способу и, наконец, окислением получен-
ного тетраэтилбензодигидриндендиона 2; из ^-ксилола аналогич-
ным синтезом, заключающимся в конденсации его с хлорангидридом
уксусной кислоты, восстановлении и окислении3; окислением
тетрагидро-5,б-бензиндан-1-она 4.
Пиромеллитовую кислоту можно получить декарбоксилирова-
нием меллитовой кислоты простым нагреванием 5 или нагреванием
в присутствии серной кислоты в.
Окисление древесного угля серной кислотой дает в результате
меллитовую кислоту и продукты ее декарбоксилирования 7; можно
применять также азотную кислоту 8. Пиромеллитовая кислота была
получена также электролитическим окислением графита в щелоч-
ной среде 9.
1 J а с о b s e п, Всг. 17, 2516 A884).
2 Freund, Fleischer, Goffer je, Ann. 414, 26 A918).
3 P h i 1 i p p i, S e k a, F г о e s с h 1, там же 428, 300 A922).
'Daruns, Levy, Compt. rend. 201, 904 A935).
5 E r d m a n n, Ann. 80, 281 A851); В а е у е r, Ann. Suppl. 7, 36 A870).
6 E r d m a n n, Ann. 80, 282 A851); Silberrad, J. Chem. Soc. 89,
1795 A906).
7 Verneuil, Cumpt. rend. 118, 195 A894); 132, 1340 A901); P h i-
1 i p p i, T h e 1 e n, Ann. 428, 296 A922).
8 Silberrad, герм. пат. 214 252 fFrdl. 9,173A908—10I. P h i 1 i p p i,
Rie, Ann. 428, 287 A922).
9 Bar to li, Papasogli, Gazz. chim. ital. 12, 113 A882); 13, 37A883).
г-ПРОПИЛЕНГЛИКОЛЬ
421
/-ПРОПИЛЕНГЛИКОЛЬ
A-1,2-Пропандшл)
^ СН3СНОНСН2ОН
СН3СОСН2ОН+2[Н]
Предложили; П. Левин и Д. Уолти.
Проверили: Ф. Уитмор и Дж. Холлингсхед.
1. Получение
Раствор 1 кг сахарозы в 9 л воды вливают в 20-литровую бутыль,
снабженную счетчиком пузырьков. 1 кг пекарских дрожжей раз-
ламывают на кусочки и размешивают в 1 л воды в тестовидную
массу. Затем прибавляют это тесто к сахарному раствору и оста-
вляют смесь стоять при комнатной температуре, пока не начнется
энергичное выделение газа (от 1 до 3 час). К сильно бродящему
раствору прибавляют 100 г свежеприготовленного ацетола (стр. 74)
и снова оставляют смесь стоять при комнатной температуре до тех
пор, пока реакция не успокоится (примечание 1). Затем сосуд ста-
вят в нагретый до 32° термостат, где брожение начинается вновь.
Реакция обыкновенно заканчивается к концу третьего дня; при
испытании фелинговой жидкостью в растворе обнаруживается
лишь незначительное количество восстанавливающих 4 Сахаров.
По окончании брожения к смеси прибавляют 20—30 г коротко-
волокнистой стеклянной ваты или асбеста и отсасывают дрожжи.
Фильтрат выпаривают на водяной бане в вакууме до густоты си-
ропа при температуре не выше 40° (примечание 2). Остаток (около
200 мл) взбалтывают со смесью 400 мл абсолютного спирта и
100 мл абсолютного эфира (примечание 3). Образовавшийся оса-
док удаляют центрифугированием, верхний эфирно-спиртовой
слой сливают, а сиропообразный остаток экстрагируют смесью
200 мл 98,5%-ного спирта и 100 мл абсолютного эфира (примеча-
ние 4). Соединенные спиртово-эфирные растворы упаривают в ва-
кууме при 35—40° до получения густого сиропа. Остаток снова
взбалтывают со смесью 400 мл 98,5%-ного спирта и 100 мл абсолют-
ного эфира и центрифугируют (примечание 4). Верхний эфирно-
спиртовой слой отделяют, упаривают его в вакууме и перегоняют
остаток из специальной колбы Клайзена. Выход сырого продукта,
кипящего при 86—91°/12 мм, около 100 г. Сырой продукт опять
перегоняют и собирают фракцию 88—90°/12 мм или 187—
189°/760 мм. Конечный продукт (примечание 3) представляет собою
бесцветную жидкость с уд. в. 1,04 и удельным вращением [а]^°° =
= — 15,0°. Выход 50—60 г D9—58% теоретич.; примечание 5).
2. Примечания
1. Прибавление ацетола может на некоторое время замедлить
реакцию.
422
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
н.-ПРОПИЛСУЛЬФИД
423
2. Выпаривание следует производить при возможно более низ-
кой температуре. Аппарат для выпаривания в вакууме больших
количеств жидкости описан в «Синт. орг. преп.», сб. 1, стр. 333.
3. Продукт имеет слабо кислую реакцию. Если нужно получить
вполне нейтральный /-пропиленгликоль, то следует нейтрали-
зовать первый (по порядку получения) сироп раствором метилата
натрия в метаноле, снова упарить его и остаток экстрагировать по
вышеописанному способу.
4. Если не имеется центрифуги, то аналогичного результата
можно достигнуть, если к раствору прибавить около 15 г коротко-
волокнистой стеклянной ваты или асбеста, размешать механической
мешалкой или сильно встряхивать в течение 5 мин., а затем отсосать.
5. Оптически активные гликоли являются подходящим исход-
ным материалом для получения оптически активных карбинолов,
оксикислот и т. д. Биологический метод асимметрического восста-
новления — единственный практически удовлетворительный спо-
соб для получения этих гликолей. В общих чертах другие опти-
чески активные гликоли получаются так же, как и /-пропиленгли-
коль,— из ацетона. В некоторых случаях целесообразно окислить
хлоргидрин в соответствующий хлоркетон, а в остальном процесс
вести так же, как и в случае синтеза /-пропиленгликоля.
3. Другие методы получения
/-Пропиленгликоль был получен из оптически неактивного гли-
коля действием микроорганизмовх и дробной кристаллизацией
соединений со стрихнином 2. Описанный синтез основан на методе
Фербера, Норда и Нейберга 3.
1 Le Be I, Jahresb. 1881, 512.
2 Griin, Ber. 52, 260 A919).
3 Farber, Nord, Neuberg, Biochgm. Z. 112, 313 A920).
Н.-ПРОПИЛСУЛЬФИД
C2HBONa+H2S > NaSH+C2H6OH
NaSH + CaHsONa > Na2S+C2H3OH
Na2S + 2CH3CH2CH2Br > (СН3СНаСН2K S-f 2 NaBr
Предложили: P. Бост а М. Конн.
Проверили: Р. Фьюзон и Ч. Вудеард.
1. Получение
В 2-литровую круглодонную колбу, снабженную обратным
холодильником и стеклянной трубкой диаметром 6 мм, доходящей
до дна колбы и с другой стороны закрытой резиновой трубкой и
зажимом, помещают 800 мл абсолютного спирта и 50,6 г B,2гр,-ат.)
чистого натрия, нарезанного маленькими кусочками. После того
как натрий полностью растворится, половину раствора переносят
в 2-литровую круглодонную колбу с тремя горлами, снабженную
капельной воронкой, механической мешалкой с жидкостным за-
твором и обратным холодильником с хлоркальциевой трубкой. Кол-
бу, в которой находится другая половина раствора, вновь соеди-
няют с обратным холодильником и раствор зтилата натрия насы-
щают из баллона сероводородом со скоростью двух пузырьков в се-
кунду до полного насыщения раствора (на что требуется около
6 час). Этот раствор сулфгидрата натрия добавляют к раствору
этилата натрия, находящемуся в трехгорлой колбе, и в течение
1 часа эту смесь кипятят с обратным холодильником. По охлажде-
нии до комнатной температуры добавляют абсолютный спирт в ко-
личестве, которое необходимо для растворения всего сернистого
натрия (около 200 мл; примечание 1).
К этому раствору сернистого натрия по каплям добавляют при
перемешивании 246 г B мол.) бромистого н.-пропила («Синт. орг.
преп.», сб. 1, стр. 118). После добавления всего количества бромида
колбу нагревают на паровом конусе (воронке) 8 час. (примечание 2).
При этом массу перемешивают, но не очень энергично. Затем колбу
охлаждают и ее содержимое добавляют к 2 л 25%-ного водного
раствора хлористого натрия, находящегося в делительной воронке.
Смесь энергично взбалтывают, дают расслоиться, отделяют верх-
ний маслянистый слой пропилсульфида и сушат его безводным серно-
кислым натрием. Нижний слой экстрагируют петролейным эфиром
(т. кип. 24—45°; примечание 3) пять раз порциями по 200 мл. Вы-
тяжки сушат 20 г сернокислого натрия и петролейный эфир отго-
няют с дефлегматором длиною 60 см, пока температура паров не
достигнет 60°. Остаток добавляют к отделенному пропилсульфиду
и все вместе подвергают перегонке. Выход продукта с т. кип. 140—
143°—80—100 г F8—85% теоретич,; примечание 4).
2. Примечания
1. Желательно, чтобы сернистый натрий полностью перешел
в раствор, прежде чем будет добавляться бромид. Однако последую-
щее экстрагирование облегчается, если раствор имеет возможно
меньший объем.
2. Для того чтобы быть уверенным, что при кипячении не про-
исходит потери пропилсульфида, обратный холодильник следует
соединить с ловушкой, в которую налит водный раствор хлорной
ртути. Желательно, чтобы вода, которая поступает в холодильник,
имела температуру 5—10°.
3. Чтобы избежать загрязнения продукта, петролейный эфир
до употребления следует перегнать. Этиловый эфир в качестве эк-
страгирующего средства не пригоден,
424
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
ПРОПИОНОВЫЙ АЛЬДЕГИД
425
4. Аналогичным образом могут быть получены н.-бутил и вто-
ричн. -амилсульфиды.
3. Другие методы получения
н.-Пропилсульфид всегда получался действием спиртового рас-
твора сернистой щелочи на н.-пропилгалогенид 1.
1 С a h о u r s, Compt. rend. 76, 133 A873); W j n s s i n ge r, Bull, soc,
chim. B) 48, 109 A887).
ПРОПИОНОВЫЙ АЛЬДЕГИД
3CH3CH2CH3OH+K2CrA
323+2A + 2
3 CH3CH2CHO+ K2SO4+ Cr2 (S0J.
H2O
Предложили: Ч. Херд и Р. Мейнерт.
Проверили: В. Мак-Ювен и В. Каротерс.
1. Получение
100 г A25 мл, 1,7 мол.)н,-пропилового спирта с т. кип. 96—96,6°
помещают в 2-литровую трехгорлую колбу, снабженную мешалкой
с ртутным затвором (примечание 1), капельной воронкой и шари-
ковым холодильником длиной 60 см (примечание 2), поставленным
под углом в 45°. Через холодильник пропускают воду, нагретую
до 60°. Другой холодильник, поставленный наклонно для отгонки,
присоединен к верху первого холодильника. Холодная вода цир-
кулирует через этот второй холодильник. Конец последнего соеди-
нен при помощи алонжа с приемником, который охлаждается ле-
дяной водой.
Спирт в колбе нагревают до кипения и через капельную воронку
приливают при перемешивании смесь из 164 г @,56 мол.) двухро-
мовокислого калия, 120 мл концентрированной серной кислоты
B,2 мол.) и 1 л воды. Во время прибавления, продолжающегося
30 мин., поддерживают бурное кипение. После прибавления всей
окислительной смеси содержимое колбы кипятят еще в продолжение
15 мин., чтобы отогнать последние следы альдегида. Пропионовый
альдегид, полученный в приемнике, сушат 5 г безводного сернокис-
лого натрия и фракционируют. Выход пропионового альдегида
с т. кип. 48—55° и показателем преломления 1,364 (примечание 3)
составляет 44—47 г D5—49% теоретич.).
2. Примечания
1. Выход пропионового альдегида зависит главным образом
от того, насколько эффективно работает мешалка.
2. Первый холодильник имеет целью не давать отгоняться
пропиловому спирту и возвращать его обратно в колбу.
3. По литературным данным, показатель преломления Яд°° =
= 1,3636.
3. Другие методы получения
Пропионовый альдегид был получен при пропускании паров
пропилового спирта над тонким порошком нагретой восстановлен-
ной меди г; при пропускании смеси пропилового спирта и воздуха
над горячей платиновой спиралью 2 или серебряным катализатором
с небольшой добавкой окиси самария3; при пропускании смеси
водяного пара и окиси пропилена над силикагелем при 300°4;
при прибавлении пропилового спирта к хромовой смеси 5 или при
добавлении хромовой смеси к пропиловому спирту6; при нагре-
вании пропиленгликоля до 500°7; при нагревании смеси кальцие-
вых солей муравьиной и пропионовой кислот 8; при действии йо-
дистого этилмагния на амиловый эфир муравьиной кислоты9;
при каталитическом восстановлении акролеина10; при электро-
лизе растворов хлористого кальция или слабой серной кислоты
в пропиловом спирте и; при пропускании паров пропилового спир-
та над окисью кадмия при 325° 12; при пропускании паров пропио-
новой и муравьиной кислот над окисью титана при 250—300° 13;
при окислении пропилового спирта током воздуха в присутствии
медной бронзы, нитробензола и хинолина u и при действии этило-
вого эфира ортомуравьиной кислоты на бромистый этилмагний 1а.
1 Sabaticr, Senderens, Compt. rend. 136, 923 A903).
2 T r i 1 ! a t, Bull. soc. chim. C) 29, 38 A903).
3 D a y, E i s n e r, J. Phys. Chem. 36, 1912 A932).
i I. G. Farbenind. A.-G.; герм. пат. 618 972 [С. А. 30, 1066 A930)].
5Lieben, Zeisel, Monatsh. 4, 14 A883); Л а г е р е в, ЖОХ, 5, 515
A935).
iHurd, Meinert, Spence, J. Am. Chem. Soc. 52, 1140 A930).
' N e f, Ann. 335, 203 A904).
8 L i n n e m a n, там же 161, 21 A872).
9 Bayer und Сотр., герм. пат. 157 573 [Frdl. 8, 156 A905—07)].
10 Sab a tie r, Senderens, Ann. chim. phys. (8) 4,398A905);
(8) 16, 72 A909); S k i t a, Bcr. 45, 3316 A912).
11 R e i 11 j n g e r, Z. Elektrochem. 20, 261 A914); F e у е г, там же 25,
142A919).
12 S a b a t i e r, M a i 1 h e, Ann. chim. phys. (8) 20, 303 A910).
13 S a b a t i e r, Mailhe, Compt. rend. 154, 563 A912).
14 Rosen m und, Zetzsche, Ber. 54, 2034 A921).
is W о о d, С о m 1 e y, J. Soc. Chem. Ind. 42, 431 T A923).
426
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
0-ПРОПИОФЕНОЛ И п-ПРОПИОФЕНОЛ
427
о-ПРОПИОФЕНОЛ И й-ПРОПИОФЕНОЛ
(о-Оксипропиофенон и п-оксипропиофенон)
с.нвосос,н
2Н5
^г* о-и" л-СвН4 (ОН) СОСаНв
Предложили: Э. Миллер и В. Хартунг.
Проверили: В. Хартман и Л. Ролл.
1. Получение
В 2-литровую трехгорлую круглодонную колбу, снабженную
обратным холодильником, механической мешалкой (примечание 1)
и капельной воронкой емкостью 100 мл, помещают 374 г B,8 мол.)
безводного хлористого алюминия и 400 мл сероуглерода (примеча-
ние 2). К суспензии при перемешивании медленно прибавляют че-
рез капельную воронку 375 г B,5 мол.) фенилового эфира пропио-
новой кислоты (примечание 3). Почти сейчас же начинается реак-
ция с выделением хлористого водорода (примечание 4) и сероугле-
род начинает кипеть благодаря теплоте, выделяемой при реакции
(примечание 5). После прибавления всего пропионата (около 1,5 часа)
смесь нагревают на водяной бане так, чтобы она спокойно кипела
до тех пор, пока выделение хлористого водорода практически не
закончится (около 2 час). Обратный холодильник заменяют на
обращенный вниз и сероуглерод отгоняют. Затем водяную баню
удаляют и смесь нагревают на масляной бане в течение 3 час. при
140—150° (примечание б). При этом снова происходит выделение
хлористого водорода; смесь постепенно густеет и наконец засты-
вает в коричневую тягучую массу. Перемешивание продолжают,
пока это возможно (примечание 7).
Массе дают охладиться и комплексное алюминиевое соединение
разлагают, медленно прибавляя к нему сначала смесь из 300 мл
концентрированной соляной кислоты и 300 мл воды, а затем 500 мл
воды (примечание 8); при этом на поверхности собирается черный
маслянистый продукт. Смесь оставляют стоять на ночь в холодиль-
ном шкафу; большая часть маслянистого слоя при этом закристал-
лизовывается и ее можно отсосать. Твердый продукт (п-пропиофе-
нол) перекристаллизовывают из 400 мл метилового спирта. Выход
продукта, окрашенного в свстложелтый цвет и плавящегося при
145—147°, составляет 129—148 г C4—39% теоретич.). После второй
перекристаллизации т. пл. повышается до 147—148°.
Маслянистый фильтрат соединяют с упаренными маточными рас-
творами, полученными после перекристаллизации, растворяют в
500 мл 10%-ного раствора едкого натра и экстрагируют два раза
эфиром (по 100 мл), для того чтобы удалить все нейтральные про-
дукты (не фенольного характера). Щелочной раствор подкисляют
соляной кислотой и маслянистый слой отделяют, сушат безводным
сернокислым магнием и перегоняют. о-Пропиофенол кипит при
110—11576 мм. Выход 120—132 г C2—35% теоретич.). Во время
перегонки получается также около 40 г л-пропиофенола с т. кип.
135—150°/11 мм. Общий выход неочищенного n-пропиофенола до-
стигает таким образом 169—188 г D5—50% теоретич.; примечание 9).
2. Примечания
1. Для того чтобы мешалка могла работать, когда масса стано-
вится густой, лучше всего сделать ее из толстой стеклянной палочки,
которую следует согнуть, как согнут конец клюшки для игры в гольф.
2. Если в качестве растворителя берут нитробензол, всегда
получается много смолы.
3. Сырой неперегнанный фениловый эфир пропионовой кислоты,
полученный осторожным нагреванием смеси эквивалентных коли-
честв фенола и хлористого пропионила до тех пор, пока не прекра-
тится выделение хлористого водорода, вполне пригоден для данной
перегруппировки. Эфир можно также легко получить, если к смеси
фенола и пропионовой кислоты (каждого по 1,05 мол.) медленно
прибавить 1 мол. хлористого тионила, удалить выделяющийся
хлористый водород и сернистый ангидрид и перегнать получен-
ный продукт.
4. Рекомендуется проводить работу в хорошо действующем вы-
тяжном шкафу, так как выделяется большое количество хлористого
водорода. Если же вытяжного шкафа не имеется, можно восполь-
зоваться ловушкой, которая описана на стр. 78.
5. Скорость прибавления фенилпропионата регулируют так,
чтобы смесь слабо кипела.
6. Температура реакционной смеси обычно держится на 10°
ниже температуры бани, а именно 130—135°. Количественное соот-
ношение изомеров зависит от температуры реакции. Более высокая
температура A60—170°) способствует образованию ортоизомера.
7. Для того чтобы облегчить выход выделяющемуся при нагре-
вании хлористому водороду, необходимо густеющую массу непре-
рывно перемешивать, иначе она может быстро подняться и забить
выходные отверстия колбы.
8. При разложении происходит выделение большого количества
тепла, поэтому разбавленный раствор кислоты надо приливать очень
медленно.
9. Описанный способ оказался пригодным для получения це-
лого ряда гомологов о- и л-пропиофенола, например ацетофенола,
бутирофенола и капрофенола.
3. Другие методы получения
о- и п-Пропиофенолы были получены конденсацией пропионовой
кислоты и фенола в присутствии хлористого цинка1 и из фенола
и хлористого пропионилаа. Приведенный здесь способ является
428
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
видоизменением метода, описанного Коксом3 и Гатунгом, Ман-
чем, Миллером и Кросслеем 4. Согласно другой методике, фенило-
вый эфир пропионовой кислоты добавляют непосредственно к хло-
ристому алюминию, без растворителя 5.
1Goldzweig, Kaiser, J. prakt. Chem. B) 43, 86 A891).
2 Perk i n, J. Chem. Soc. 55, 547 A889).
3 Cox, J. Am. Chem. Soc. 49, 1028 A927).
4 Hartung, Munch, Miller, С г о s s 1 e у, там же 53, 4153 A931).
6 F a r i n h о 11, H a r d e n, T w i s s, там же 55, 3386 A933).
ПротоКатеховый альдегид
429
О CH2
ПРОТОКАТЕХОВЫЙ АЛЬДЕГИД
О СС12
ЗРС1а
—о
сно
СНС1,
О СС1,
// V
-о
2Н„О
+ РОС13+2РС13-[-2НС1
О СО
к I
—о
+ 4НС1
СНС12
О СО
У~ о
сно
Н.0
сно
он
l4
;/\--он
сно
Предложили: Док. Бэк и Ф. Циммерман.
Проверили: Р. Фыозон и В. Росс.
1. Получение
В 3-литровую круглодонную колбу помещают 108 г @,72 мол.)
пиперонала и порциями по 20—30 г добавляют 454 г B,18 мол.)
свежеприготовленного пятихлористого фосфора. Вначале реак-
ция протекает бурно и колбу охлаждают льдом; содержимое колбы
предохраняют от действия влаги. После того, как добавлена при-
мерно половина пятихлористого фосфора, реакция замедляется
и колбу перестают охлаждать. Прибавление всего количества пяти-
хлористого фосфора занимает около 30 мин. Получившуюся жид-
кость зеленого или голубого цвета, содержащую не растворившийся
пятихлористый фосфор, весьма осторожно нагревают на голом пла-
мени горелки в течение 1 часа для удаления хлористого водорода.
Из образовавшейся мутной светлобурои жидкости летучие продукты
удаляют при нагревании на водяной бане в вакууме водоструйного
насоса, что занимает около 30 мин. После этого содержимое колбы
выливают в 5 л холодной воды, налитой в 12-литровую круглодон-
ную колбу (примечание 1). Образуется молокообразное масло,
которое подымается и опускается в воде и примерно через 30 мин.
затвердевает. После стояния в течение ночи смесь осторожно ки-
пятят 3 часа, бурый раствор, содержащий небольшое количество
смолообразных примесей, осветляют активированным углем и упа-
ривают в вакууме до 700 мл. При этом начинает выпадать альдегид.
Раствор оставляют стоять на ночь при температуре около 0°; вы-
падает большое количество кристаллов, которые отфильтровы-
вают и промывают небольшим количеством воды. С целью очистки
продукт перекристаллизовывают из тройного по весу количества
воды. Выход вещества с т. пл. 153—154° (с разложением) составляет
61 г F1% теоретич.).
2. Примечания
1. Выливать следует осторожно, так как оставшийся пяти-
хлористый фосфор бурно реагирует с водой.
3. Другие методы получения
Протокатеховый альдегид был получен многими разнообразными
методами, однако обычно его получают из пирокатехина по реак-
ции Реймера-Тимана \ из ваниллииа 2 или вератрового альдегида 3
реакцией деметилирования и из пиперонала действием хлористого
алюминия4 или пятихлористого фосфора с последующим омыле-
нием 5.
1 R е i m е г, Т i e m a n п, Вег. 9, 1268 A876); Tiemann, Koppe,
там же 14, 2015 A881).
2 Tiemann, Haarmann, там же 7, 620 A874).
3 Dreyfus, герм. пат. 193 958 [Frdl. 9, 161 A908—10)].
4 Givaudan-Delawanna, Inc.; ам. пат. 2 027 148 [С. А. 30, 1395 A936)].
5 F i 11 i g, Remsen, Ann. 159, 144A871); Pauly, Ber. 40, 3096
A907); В a r g e r, J. Chem. Soc. 93, 563 A908); Hoering, Baum, Ber.
41, 1914 A908).
430
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
Й-РОДАНДИМЕТИЛАНИЛИН
431
Р-РЕЗОРЦИЛОВАЯ КИСЛОТА
(ОНJ
СО2Н
iOH
ОН
Предложили: М. Ниренштейн и Д. Нлиббенс.
Проверили: Р. Адаме и Ф, Н.ендалл.
1. Получение
Раствор 200 г A,8 мол.) резорцина и 1 кг (9,9 мол.) кислого угле-
кислого калия (примечание 1) в 2 л воды (примечание 2) медленно
нагревают на водяной бане в течение 4 час. в 5-литровой колбе,
соединенной с обратным холодильником. Затем колбу нагревают
на сетке и смесь энергично кипятят 30 мин.; Во время кипячения
через раствор пропускают сильную струю углекислого газа.
Горячий раствор подкисляют 900 мл концентрированной соля-
ной кислоты, уд. в. 1,19, прибавляя последнюю через делительную
воронку, ножка которой доходит до дна реакционной колбы, для
того чтобы предупредить образование слоя кислоты над не нейтра-
лизованным еще раствором. После подкисления раствору дают
охладиться до комнатной температуры, а затем охлаждают его
в бане со льдом. При этом выпадают почти бесцветные призмати-
ческие кристаллы резорциловой кислоты. На воздухе продукт
постепенно становится розовым, что объясняется примесью не-
большого количества резорцина. Выход сырой кислоты 225 г.
Извлекая маточный раствор несколько раз эфиром, можно получить
еще 35 г резорциловой кислоты и небольшое количество непрореа-
гировавшего резорцина. Резорциловую кислоту извлекают из эфира
взбалтыванием с водным раствором двууглекислого натрия. Водный
раствор подкисляют соляной кислотой и снова извлекают эфиром.
По отгонке эфира остается обычно сильно окрашенная резорцило-
вая кислота. Для обесцвечивания ее нужно несколько раз перекри-
сталлизовать из кипящей воды с добавлением активированного
березового угля.
С целью очистки всю полученную сырую кислоту B60—270 г)
растворяют в 1 л горячей воды, кипятят раствор приблизительно
с 25 г угля, фильтруют его через нагретую воронку и выкристал-
лизовывают кислоту охлаждением раствора в холодильной смеси
при сильном перемешивании. Таким образом получают мелко кри-
сталлический бесцветный продукт. Если дают кристаллам выпадать
медленно, то получают слегка окрашенное вещество. Выход чистой
резорциловой кислоты, плавящейся при 216—217°, равен 160—'
170 г E7—60% теоретич.; примечание 3).
2. Примечания
1. Вместо кислого углекислого калия можно взять кислый
углекислый натрий в соответствующем количестве.
2. Если взять на 1 часть резорцина менее 10 частей воды, выход
понижается.
3. Высушенные на воздухе кристаллы отдают при 110° воду
в количестве, соответствующем половине молекулы кристалли-
зационной воды.
3. Другие методы получения
Описанный метод получения 1 [3-резорциловой кислоты пред-
ставляет собою видоизмененный метод Быстржицкого и Костанец-
кого 2. Был также описан более быстрый, но менее эффективный
вариант этого метода 3.
1 Clibbens, Nierenstein, J. Chem. Soc. 107, 1494 A915).
2 Bistrycki, Kostanecki, Ber. 18, Ш84 A885).
3 Couturier, Ann. chim. A1) 10, 570 A938).
Л-РОДАНДИМЕТИЛАНИЛИН
(N, N-Диметил-п-тиоцшнанилин)
C6HfiN(CH3)s,+ Br2+
-—-> 2 NH4Br-f n-NCSC6H4N (CH3J-f-HSCN
Предложили: P. Брюстср и У. Шредер.
Проверил: В. Каротерс.
1. Получение
В 1-литровом стакане приготовляют раствор 60,5 г @,5 мол.)
диметиланилина и 80 г A,05 мол.) роданистого аммония в 250 мл
ледяной уксусной кислоты и раствор охлаждают до 10—20° в бане
с водой и льдом. Раствор перемешивают с помощью механической
мешалки в течение 20—30 мин. и по каплям приливают к нему рас-
твор' 80 г B5,7 мл, 0,5 мол.) брома в 100 мл ледяной уксусной ки-
слоты, причем температуру поддерживают ниже 20° (примечания
1 и 2). После того как весь бром прибавлен, реакционную смесь
вынимают из охлаждающей бани и после стояния при комнатной
температуре в течение 10 мин. выливают ее в 5—6 л воды. Боль-
шая часть п-родандиметиланилина выпадает в виде светложелтого
твердого вещества (примечание 3), которое отфильтровывают с от-
сасыванием и промывают водой. После высушивания на воздухе
получают 50—55 г вещества с т. пл. 71—73°. Еще 10—15 г менее
чистого продукта можно получить, если фильтрат сделать щелоч-
432
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
СОЛЬ РЕЙНЕКЕ
433
ным на лакмус, на что требуется около 1250 мл 20%-ного едкого натра.
Оба продукта соединяют вместе, растворяют примерно в 1,2 л ки-
пящего лигроина (т. кип. 90—100°) и быстро фильтруют через боль-
шой складчатый фильтр, находящийся в нагретой воронке. Из
фильтрата продукт выпадает в виде длинных желтых игл, причем
кристаллизацию завершают, тщательно охлаждая сосуд с веще-
ством. Температура плавления очищенного продукта 73—74°,
суммарный выход 56—60 г F3—67% тёоретич.; примечание 4).
2. Примечания
1. При более высокой температуре образуется значительное
количество желтого полимера родана, который загрязняет продукт.
2. К концу приливания раствора брома на стенках стакана на-
чинает накапливаться тяжёлый осадок, который время от времени
следует удалять с помощью шпателя.
3. п-Родандиметиланилин слабое основание, и соли его легко
гидролизуются.
4. Если выпарить маточный раствор, можно получить еще 4—5 г
низкоплавкого продукта.
3. Другие методы получения
я-Родандиметиланилин был получен из диметиланилина и ро-
дана г, хлортиоциана 2, роданистого аммония и брома 3>4 и рода-
нистого свинца и фенилиодидхлорида в. Роданирование аромати-
ческих аминов было осуществлено также электролитически 4i6.
»S6derback, Ann. 419, 275 A919).
8 Lecher, Joseph, Ber. 59, 2603 A926).
3Лихошерстов, Петров, ЖОХ 3, 183 A933).
1 Helwig, ам. пат. 1 810 848 [С. А. 25, 5355 A931I.
5 Neu, Ber. 72, 1505 A939).
6 F i с h t e r, S с h б п m a n n, Helv. Chira. Acta 19, 1411 A936).
СОЛЬ РЕЙНЕКЕ
NH4 [Cr (NH3J (SCNL] • H2O
Предложил: Х. Джин.
Проверили: X. Кларк н Р. Ингольс.
1. Получение
В белой эмалированной кастрюле емкостью около 4 л осторожно
нагревают 800 г A0,5 мол.) роданистого аммония тремя горелками
с небольшим пламенем (примечание 1). Массу перемешивают тер-
мометром, заключенным в стеклянную трубку, до тех пор, пока
соль частично не расплавится и температура не поднимется до 145—•
150°. Когда такое состояние достигнуто, прибавляют тесную смесь
170 г @,675 мол.) хорошо измельченного двухромовокислого аммо-
ния и 200 г B,6 мол.) роданистого аммония, порциями по 10—12 г
при постоянном помешивании. После того как прибавлено около
десяти таких порций, начинается довольно бурная реакция с вы-
делением аммиака, причем температура поднимается до 160°. Го-
релки гасят и оставшуюся часть смеси прибавляют с такой ско-
ростью, чтобы за счет теплоты реакции поддерживалась температура
160° (примечание 2). В то время как масса охлаждается, ее продол-
жают перемешивать, причем одновременно соскабливают куски,
приставшие к боковым стенкам сосуда (примечание 3).
Еще в горячем состоянии (примечание 4) продукт хорошо из-
мельчают и размешивают с 750 мл ледяной воды, налитой в большой
стакан. Через 15 мин. нерастворившуюся часть отфильтровывают
с отсасыванием, отжимают по возможности лучше от маточного
раствора, но не промывают (примечание 5). Осадок размешивают
с 2,5 л воды, которую предварительно нагревают до 65°. Затем тем-
пературу быстро повышают до 60° (примечание б), раствор немед-
ленно фильтруют через воронку с горячим обогревом и фильтрат
на ночь ставят в холодильный шкаф.
Выпавшие кристаллы отсасывают, а маточник применяют для
вторичного экстрагирования осадка при 60°. Это дает дополнитель-
ное количество кристаллической соли Рейнеке. Затем маточный
раствор упаривают при 40—50° в вакууме до объема 250—300 мл
и при этом получают еще небольшое количество кристаллов A2—
13 г). Общий выход воздушно-сухих кристаллов 250—275 г E2—57%
тсорстич.; примечание 7).
Нерастворимый остаток от вторичного экстрагирования состоит
главным образом из соли Морланда (гуанидиновая соль кислоты
Рейнеке) и составляет 130;—135г C3—34%тёоретич.; примечание 8).
2. Примечания
1. Необходимо по возможности равномерное нагревание.
2. Добавление смеси ведут в течение 5—7 мин.
3. Продукт отделяют от стенок во время охлаждения, так как
в холодном состоянии отделить его очень трудно.
4. Продукт следует измельчать еще в теплом состоянии, чтобы
он не успел поглотить влагу из воздуха.
5. Фильтрат, состоящий главным образом из неизменившегося
роданистого аммония и продуктов его разложения, содержит слиш-
ком мало соли Рейнеке, чтобы имело смысл извлекать ее оттуда.
6. Соль Рейнеке разлагается в водном растворе, причем раствор
синеет и выделяется свободный цианистый водород. При комнат-
28 Сборник № 2
434
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
ной температуре для этого разложения требуется примерно 2 неде-
ли; выше 65° оно протекает весьма быстро. Аналогичное разложе-
ние происходит и в кипящем спирте.
7. Соль Рейнеке служит реактивом для осаждения первичных
и вторичных аминов, пролина и оксипролина и некоторых амино-
кислот \
8. Соль Морланда растворима в ацетоне и содержит небольшое
количество бесцветного сернистого соединения, нерастворимого
в горячей воде. Частично она может быть превращена в соль Рей-
неке, если на нее подействовать в слабоаммиачном растворе боль-
шим избытком хлористого аммония. Однако получающиеся выходы
настолько малы, что такая операция невыгодна.
3. Другие методы получения
Соль Рейнеке была получена действием двухромовокислого ка-
лия 2 или аммония 1>3 на расплавленный роданистый аммоний.
1 Kapfhammer, Ее k, Z. physiol. Chem. 170, 310 A927); Grass-
mann,Lang, Biochem. Z. 269, 223 A934).
2 R e i n e с к e, Ann. 126, 113 A863); Christensen, J. prakt. Chem
B) 45, 213 A892); Zeletiy, Qor tner, J. Biol. Chem. 90, 430 A931).
3 Werner, Z. anorg.'Chem. 15, 260 A897); Ann. 406, 276 A914).
СОЧЕТАНИЕ о-ТОЛИДИНА И ЧИКАГР-КИСЛОТЫ
(Получение азокраски, не содержащей минеральных солей)
ОН NH3
СОЧЕТАНИЕ О-ТОЛИДИНА" И ЧИКАГО-КИСЛОТЫ
435
C!N,f
+ 2
СНЯ
NH» ОН
СН3
SO3Na
OH NH,
Na.OsS'
N ¦= NV
CH,
CH,
NaO3S
SO3Na
Предложили; Дж. Хартвел и Л. Физер.
Проверили: В. Хартман и Дж. Бумер.
1. Получение
В 1-литровом стакане приготовляют жидкую пасту' из 21,2 г
@,1 мол.) о-толидина и 300 мл воды, которую затем переводят в рас-
твор прибавлением 20 мл B3,6 г, 0,23 мол.) концентрированной
соляной кислоты (уд. в. 1,18); в случае необходимости массу слегка
подогревают. Раствор охлаждают льдом до 10°, пускают в ход мешал-
ку и добавляют еще 21 мл B5 г, 0,24 мол.) концентрированной со-
ляной кислоты (примечание 1), что вызывает частичное выделение
хлористоводородной соли о-толидина. Поддерживая температуру
при 10—15°, быстро приливают раствор 14,5 г @,2 мол.) 95%-ного
нитрита натрия в 40 мл воды, причем последние 10% раствора до-
бавляют медленно и только до тех пор, пока не получится четкая
положительная реакция на азотистую кислоту по иодокрахмальнои
бумажке. Избыток азотистой кислоты должен сохраняться в те-
чение получаса. В течение всего процесса диазотирования необ-
ходим избыток соляной кислоты, который бы давал сильно кислую
реакцию на конговую бумажку. Для точного завершения реакции
по окончании диазотирования избыток азотистой кислоты уда-
ляют осторожным прибавлением небольшого количества раствора
хлористоводородной соли о-толидина, пользуясь иодокрахмальнои
бумажкой (примечание 2).
В 3-литровом стакане приготовляют пасту из 82 г @,21 мол.)
88%-ной технической чикаго-кислоты A-амино-8-нафтол-2,4-ди-
сульфокислоты; примечание 3) и 500 мл воды. Чикаго-кислоту
переводят в раствор добавлением 8 г @,2 мол.) едкого натра в 30 мл
воды, причем реакцию раствора контролируют по лакмусу так,
чтобы она оставалась слегка кислой (примечание 4). Добавлением
льда раствор охлаждают до 18° и перед самым сочетанием приба-
вляют 35 г @,33 мол.) безводного бикарбоната натрия.
При энергично работающей мешалке довольно быстро прилива-
ют раствор соли диазония к раствору чикаго-кислоты. Сначала вы-
деляется голубой краситель, который при дальнейшем перемеши-
вании постепенно растворяется. Через 30 мин. раствор проверяют
на щелочность (примечание 5) и на полноту сочетания (примеча-
ния 6 и 7).
После того как смесь перемешивается в общей сложности уже
в течение 2 час, ее нагревают до 85°, добавляют 15 г обесцвечиваю-
щего угля, перемешивают еще 15 мин. и фильтруют; фильтрат вновь
нагревают до 85° при работающей мешалке и на-глаз определяют
объем раствора. На каждые 100 мл раствора медленно и при энер-
гичном перемешивании прибавляют 27 г кристаллического уксус-
нокислого натрия (обычно требуется 450—500 г). Соль при-
бавляется в четыре или пять приемов (примечание 8). В этих
условиях краситель выпадает в легко фильтрующемся виде. Если
с помощью стеклянной палочки перенести каплю реакционной мас-
сы на фильтровальную бумагу, краситель должен образовать си-
нюю массу в центре красного или фиолетового круга (примечание 9).
В еще теплом состоянии смесь фильтруют с отсасыванием через во-
ронку Бюхнера, диаметром в 20 см, причем по возможности полнее
удаляют маточный раствор (примечание 10). Твердую массу рас-
436
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
СОЧЕТАНИЕ 0-ТОЛИДИНА И ЧИКАГО-КИСЛОТЫ
437
творяют в 1,2 л воды при 85° и вновь высаливают краситель, приба-
вляя примерно 300 г ацетата натрия (в несколько приемов), или до
тех пор, пока почти весь краситель не будет высолен. В пробе на
вытек на фильтровальной бумаге края должны быть слабо окра-
шены в красный цвет. Продукт выделяют, вновь растворяют
в 1,2 л воды при 85° и вновь высаливают. На этот раз берут столько
ацетата натрия, чтобы уже при пробе на вытек образовался неболь-
шой ободок, на котором не были бы заметны красные примеси (при-
мечание 11). Продукт выделяют, сушат в шкафу при 110° в течение
не менее чем 24 часа, измельчают и просеивают через сито в 40 меш.
Чтобы удалить уксуснокислый натрий, просеянный краситель
обрабатывают 10—15 мин. кипящим 95%-ным этиловым спиртом, по-
следовательно, порциями по 500 мл, причем после каждой такой об-
работки краситель отсасывают и на фильтре промывают небольшим
количеством спирта (примечание 12). После четырех таких опера-
ций фильтрат должен дать отрицательную пробу на содержание
ацетата. Продукт, меняющий свой цвет при такой обработке от
бронзового до зеленого, сушат на воздухе при комнатной темпера-
туре. Выход красителя, не содержащего неорганических солей
и органических примесей, составляет 80—81 г (83—84% теоретич.;
примечание 13).
2. Примечания
1. Если всю соляную кислоту прибавить сразу, а не в два при-
ема, как здесь указано, то выпадет осадок, состоящий из о-толиди-
на, покрытого его солью с двумя молекулами НС1, и диазотирование
протекает очень медленно.
2. Если при этом не получается прозрачного раствора, то все
нерастворимые частицы можно отфильтровать.
3. Ввиду того, что этот промежуточный продукт является про-
дуктом техническим и был выделен путем высаливания, необходимо
выяснить степень его чистоты. Чикаго-кислота находится в этом
продукте в виде кислой натриевой соли (молекулярный вес 341).
Быстрый метод анализа, достаточно точный для данной цели, состоит
в том, что навеску чикаго-кислоты в разбавленном сильно кислом
растворе титруют 0,5-н. раствором нитрита натрия, титр которого
установлен по сульфаниловой кислоте, причем конец реакции опре-
деляется на иодокрахмальную бумажку.
Некоторые образцы технической чикаго-кислоты могут содержать
такое количество примеси, ведущей к образованию красного кра-
сителя, что удаление этого красного красителя путем повторного
высаливания, как это рекомендуется (примечание 9), представляет
значительные трудности. В таких случаях целесообразнее очистить
чикаго-кислоту следующим образом. Берут 700 г чикаго-кислоты
в пасте, в которой, по данным анализа, содержится 26,6% 1-амино-
8-пафтол-2,4-дисульфокислоты, и при нагревании растворяют в
2 л воды; раствор фильтруют, нагревают до 70° и высаливают добавле-
нием хлористого натрия F00—650 г) до образования густой пасты.
Осадок отфильтровывают при 40°, хорошо отжимают при помощи
резиновой пластины, растворяют в 2 л воды и вновь высаливают
совершенно так же, как и в первый раз. После высушивания в те-
чение 36 часов в вакуум-шкафу при 75°, получают почти белый про-
дукт в количестве 150—155 г, который содержит 78% активного
реагента. Краситель («толидин-1824»), полученный из этого про-
дукта при пробе на вытек после второго высаливания, не содержал
красной примеси (Л. Физер и М. Гросс, частное сообщение).
4. Аминонафтолы быстро темнеют в щелочном растворе вслед-
ствие окисления. Поэтому до самого момента сочетания раствор
оставляют слегка кислым.
5. Эту пробу осуществляют следующим образом: несколько ка-
пель диазо-раствора переносят с помощью стеклянной палочки
в углубление в белой фарфоровой пластинке, краситель высаливают,
перемешивая его с небольшим количеством химически чистого хло-
ристого натрия, и пробуют массу на лакмусовую бумажку.
6. Проба на полноту сочетания состоит в испытании на избыток
соли диазония (а) и на избыток чикаго-кислоты (б).
а) Одну или две капли реакционной смеси растворяют в про-
бирке примерно в 20 мл воды и раствор делят на две равные части,
которые наливают в две пробирки. К одной пробирке добавляют не-
сколько капель 10%-ного раствора соли Шеффера (соль 2-нафтол-
б-сульфокислоты) или Н-соли(соль2-нафтол-3, 6-дисульфокислоты),
полученных из 0,5 г кислоты, 5 мл воды и раствора соды, достаточ-
ного для полного растворения продукта. Вторая пробирка служит
для холостого опыта. Обе пробирки быстро доводят почти до кипе-
ния и сравнивают их окраску. Если окраска слишком темная, обе
пробирки разбавляют одинаковым количеством воды до тех пор, пока
растворы не станут светлоголубого цвета. Окраску можно сравнить,
вылив раствор из каждой пробирки на фильтровальную бумагу.
Если окраска раствора в первой пробирке оказалась хотя немного
краснее, чем в контрольной пробирке, то это означало бы, что в азо-
смеси имеется избыток соли диазония.
б) Несколько капель азосмеси высаливают так, как это ука-
зано в примечании 5, массу переносят на фильтровальную бумагу
и около этого пятна, с обратной стороны, помещают каплю раствора
диазотированной сульфаниловой кислоты. Красное или красно-
фиолетовое окрашивание на границе между обоими пятнами ука-
зывает на наличие избытка чикаго-кислоты.
7. В продолжение всей реакции необходимо поддерживать
щелочную реакцию и в случае необходимости прибавлять соду.
Если в смеси обнаружен избыток соли диазония и одновременно
избыток чикаго-кислоты, то это означает, что прошло еще недоста-
438
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
СУКЦИНИМИД
439
точно времени для полного сочетания. Если же обнаружен недо-
статок чикаго-кислоты, следует добавить небольшое количество ее
(в виде щелочного раствора).
8. Физическое состояние красителя и его способность хорошо
фильтроваться зависят от температуры высаливания, скорости
прибавления соли, скорости и продолжительности перемешивания
и от щелочности раствора. Описанный выше метод дает хорошо
фильтрующийся продукт.
9. Примесь красного цвета представляет собой другой краси-
тель, полученный сочетанием соли диазония с продуктом окисления,
который содержится в технической чикаго-кислоте. Обычно легче
очистить голубой краситель от этой красной примеси путем повтор-
ного высаливания, чем очистить исходную чикаго-кислоту. При
синтезе же трипанового голубого (продукт сочетания толидина
с двумя молекулами Н-кислоты) освободиться от красной примеси
посредством высаливания невозможно. Эту примесь можно удалить
полностью путем экстрагирования спиртом, в котором она раство-
рима лучше, чем голубая компонента. С этой целью лучше всего
осадить горячий насыщенный водный раствор красителя не менее,
чем шестикратным количеством (по объему) спирта.
10. Как при этом, так и при последующем фильтрованиях це-
лесообразно пользоваться резиновой пластинкой, закрепленной
при помощи резиновых колечек. Применение резиновой пластины
особенно выгодно в случае медленного фильтрования, продолжаю-
щегося в течение ночи.
11. Наиболее чувствительной пробой на чистоту красителя
является капиллярная пробаг, основанная на том, что красная
примесь растворима в воде лучше, чем голубое соединение, а также
хуже адсорбируется фильтровальной бумагой. С этой целью берут
кусочек пасты, соответствующий примерно, 0,05 г сухого красите-
ля, растворяют ею в стакане емкостью 400 мл в 200 мл воды, до-
бавляют 0,5 г поваренной соли и повышают температуру до 95°.
В жидкость погружают полоску фильтровальной бумаги размером
2 х 15 см, подвешенную на стеклянной палочке, которая лежит на
краях стакана. Температуру поддерживают при 95° в течение
15 мин., после чего полоску фильтровальной бумаги удаляют.
Наличие красной примеси совершенно ясно обнаруживается выше
голубой окраски. Если окажется, что продукт недостаточно очи-
щен, его вновь подвергают высаливанию до получения совершенно
чистого продукта.
12. За удалением уксуснокислого натрия можно следить сле-
дующим образом: к 10 мл спиртового фильтрата в пробирке доба-
вляют 3 капли концентрированной серной кислоты. Если немедлен-
но не образуется белый осадок сернокислого натрия, пробирку
охлаждают льдом. Осадок образуется уже при наличии 0,002 г
уксуснокислого натрия в 10 мл спирта".
13. Применяя ту же методику, можно диазотировать и другие
диамины, например, бензидин и дианизидин, и сочетать их с дру-
гими аминонафтолами, такими, например, как S-кислота A-амино-
8-нафтол-4-сульфокислота), I-кислота B-амино-5-нафтол-7-суль-
фокислота), ^кислота B-амино-8-нафтол-6-сульфокислота) и
Нгкислота A-амино-8-нафтол-3,б-дисульфокислота), или с про-
стыми нафтолами, как, например, кислота Невиль-Винтера
A-нафтол-4-сульфокислота), кислота Шеффера B-нафтол-6-сульфо-
кислота) и R-кислота B-нафтол-3,б-дисульфокислота). Различие
будет заключаться только в способе высаливания красителей, так
как для каждого из них имеются определенные оптимальные усло-
вия (примечание 8). В тех случаях, когда щелочная реакция ук-
суснокислого натрия ведет к заметному увеличению растворимости
красителя, вместо него можно применить бромистый аммоний. По-
следний легко экстрагируется горячим этиловым, а еще лучше го-
рячим метиловым спиртом.
Пользуясь этим методом, в качестве второй компоненты нельзя
ввести нафтиламины, так как в этом случае сочетание проводится
в кислом растворе.
3. Другие методы получения
Красители, получение которых описано выше, а также те, кото-
рые упомянуты в примечании 13, описаны в патентной литературе 2.
Обычные методы получения дают продукты, содержащие неоргани-
ческие соли и другие примеси.
1 Mu I I i k e n , Identification of the Commercial Dyestuffs, pp. 10—11,
John Wiley & Sons, New York, 1910.
2 Bayer und Ко.; герм, патенты 35 341 и 38 802 [Frdl. 1, 469, 488 A877—
87I; Cas'sella und Ко; герм. пат. 3949 [Frdl. 3, 685 A890—94)]; Badische Ani-
lin- und Soda-Fabrik; герм, патенты 57 327 и 75 469 [Frdl. 3, 687, 690
A890—94)].
СУКЦИНИМИД
енхо
CHXO,NH4
нагревание
NH + NH. + 2 H.,0
енхо
Предложили: X. Кларк, и Л. Бер.
Проверили: В. Каротерс и В. Мак-Ювен.
1. Получение
В 1-литровую колбу Вюрца с боковым отводом длиною 40 см
и внутренним диаметром не менее 10 мм (примечание 1) помещают
236 г B мол.) янтарной кислоты и при охлаждении и взбалтывании
медленно приливают 270 мл B43 г, 4 мол.) 28%-ного водного амми-
ака (уд. в. 0,90). При этом большая часть кислоты растворяется,
440
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
образуя прозрачный раствор. Колбу устанавливают для перегонки
и к боковому отводу ее присоединяют в качестве приемника колбу
Вюрца емкостью 500 мл, которую охлаждают водой. Можно принять
также меры для улавливания аммиака, выделяющегося через боко-
вой отвод приемника. Смесь осторожно нагревают голым пламе-
нем, причем быстро наступает растворение и небольшое количество
не вступившего в реакцию аммиака переходит вместе с первыми
порциями дестиллата. Температура паров повышается до 100°
и остается постоянной до тех пор, пока не отгонится около 200 мл
воды. Пламя увеличивают, и аммонийная соль янтарной кислоты
начинает разлагаться с выделением аммиака; при перегонке сле-
дующих 30 мл температура паров падает до 97°. После того, как
температура вновь поднимется до 102°, меняют приемник и собирают
промежуточную фракцию до 275°. Затем собирают сукцинимид.
в пределах 275—289°, причем главная масса переходит в пределах
285—289°. При разложении в незначительной степени образуется
черная смола; перегонку прекращают, как только осмелившийся
остаток начнет разлагаться с выделением желтых паров.
Количество целиком затвердевшего сырого сукцинимида соста-
вляет около 168 г. Промежуточную фракцию вновь подвергают пере-
гонке из меньшей колбы, причем в пределах 275—289° собирают еще
около 10 г. Обе порции неочищенного сукцинимида перекристалли-
зовывают из 95%-ного этилового спирта, причем на 1 г продукта
берут 1 мл растворителя. Если раствор охлаждать до 0° в течение
нескольких часов до фильтрования и затем для промывки взять
около 25 мл холодного спирта, то вес первой порции составляет
163—164 г (82—83% теоретич.). Если маточный раствор упарить
до 7з первоначального объема, то можно выделить еще 4—5 г
(примечание 2). Продукт не содержит кристаллизационной воды
и плавится при 123—125°.
2. Примечания
1. Боковая отводная трубка ни в коем случае не должна быть
меньшего диаметра, иначе при перегонке она может легко забиться
кристаллами сукцинимида.
2. Если темный маточный раствор упарить досуха и остаток
перекристаллизовать из свежего 95%-ного спирта, то можно полу-
чить еще небольшое количество менее чистого продукта.
3. Другие методы получения
Сукцинимид обычно получается нагреванием янтарнойкислоты в то-
ке аммиака1'2 или перегонкой аммонийной соли янтарнойкислоты1'3.
1 F e h I i n g, Ann. 49, 198 A844).
- Franchimont, Friedman n, Rec. trav. chim. 25, 79 A906).
3 Bu n ge, Ann. Suppl. 7,118A870); M e н ш у т к и н, Ann. 162, 166 A872).
ТАУРИН
441
ТАУРИН
('^-Аминоэтансулъфокислота )
1
BrCHXH,S03Na + NH3 > NH2CH2CH2SO3H -f NaBr
Предложили: К- Мареел и К. Бэйли.
Проверили: Ф. Уитмор и Д. Лодер.
1. Получение
Раствор ПО г @,52 мол.) натриевой соли р-бромэтансульфокис-
лоты (стр. 124) примерно в 2 л B8 мол.) концентрированного вод-
ного аммиака (уд. в. 0,9) оставляют стоять 5—7 дней (примечание 1),
а затем выпаривают досуха. Последние следы воды удаляют нагре-
ванием на паровой бане. Остаток растворяют в минимальном
количестве горячей воды (около 500 мл) и в случае необходимости
раствор обесцвечивают 5 г активированного березового угля. Бес-
цветный раствор упаривают до 65—70 мл и прибавляют к нему 250л*л
95%-ного спирта. Через короткий промежуток времени выпадает
таурин в смеси с некоторым количеством бромистого натрия. По
окончании кристаллизации сырой таурин отсасывают и перекри-
сталлизовывают. Для этого его растворяют в 100 мл горячей воды
и затем к раствору прибавляют такое количество 95%-ного этило-
вого спирта (около 500 мл), чтобы конечная концентрация спирта
была 80%. Выпадающий в этих условиях таурин обычно не содер-
жит бромидов. Однако иногда приходится перекристаллизовывать
его четыре или пять раз, чтобы получить продукт, не содержащий
бромистого натрия. Выход чистого таурина (примечание 2) 31—
36 г D8—55% теоретич.).
2. Примечания
1Г Реакция проходит приблизительно на 25% через 5 час, на
60% через 30 час. и на 90% через 5 дней, что было установлено тит-
рованием бром-иона.
2. Чистота получаемого этим способом таурина была устано-
влена анализом.
11
BrCH,CH2NH3Br + Na2S03
2 NaBr
Предложили: Ф. Кортиз.
Проверили: К- Марзел и К- Флеминг,
1. Получение
Раствор 615 г C мол.) бромистоводородного р-бромэтиламина
(стр. 127) и 416 г C,3 мол.) безводного сернистокислого натрия.
-442
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
^примечание 1) в 2,4 л воды упаривают на водяной бане до минималь-
ного объема, на что требуется 36—48 час. По охлаждении смеси
холодную влажную лепешку тщательно растирают с 1,5 л концен-
трированной соляной кислоты. После этого массу фильтруют с отса-
сыванием через асбестовый фильтр, помещенный в воронке Бюх-
нера. Осадок десять раз промывают концентрированной соляной
кислотой порциями по 150 мл. Фильтрат хорошо размешивают,
если это нужно, отделяют декантацией от выпавших солей и упари-
вают на голом пламени горелки до объема в 600 мл.
К горячему остатку приливают при энергичном помешиваний
2,4 л 95%-ного этилового спирта и через 15 мин. выпавший осадок
отфильтровывают и на фильтре промывают 95%-ным этиловым спир-
том до тех пор, пока он не станет бесцветным, после чего его сушат
ла воздухе. Сырой продукт подвергают очистке, для чего его рас-
творяют в четырехкратном количестве по весу горячей воды, при-
бавляют активированного березового угля, фильтруют и к горя-
чему фильтрату добавляют пятикратное количество (по объему)
95%-ного этилового спирта.
Полученный продукт представляет собой практически чистый
таурин; он разлагается при 305—310° (блок Макенна). Выход
255—275 г F8—73% тсоретич.).
2. Примечание
1. Можно взять также эквивалентное количество (831 г) более
дорогого кристаллического сернистокислого натрия.
3. Другие методы получения
Таурин обычно получают из бычачьей желчи х или из большого
мускула моллюска абалона 2. Он был получен также окислением
цистамина3 и декарбоксилированием цистеиновой кислоты4; из
этиленимина и сернистого ангидрида 6; из хлорэтансульфокис-
лоты и аммиака 6; из 2-меркаптотиазолина окислением бромной
водой "'; из уксусного альдегида путем сложного ряда реакций, в
том числе сульфирования, образования альдегид-аммиака и ими-
досульфокислоты и восстановления 8. Обычно таурин синтезируют
из бромэтансульфокислоты и аммиака 9 по методу (I) или из °-бром-
этиламина бромистоводородного и сернистокислого натрия10 по
методу (II).
1 Hammarsten, Z. physiol. Chem. 32, 456 A901); Т a u b e г, Beitr.
¦сhem. Physiol. Path. 4, 324 A904).
2 Schmidt, Watson, I. Biol. Chem. 33, 499 A918).
s Schoberl, Z. physiol. Chem. 216, 193 A933).
4 Fried mann, Beitr. chem. Physiol. Path. 3, 1 A903): White.
Pishman. |. Biol. Chem. 116, 457 A936).
ТЕТРАБРОМ- И ТЕТРАИОДНЕОПЕНТАН
443
5 Gabriel, Ber. 21, 2667 A888).
6 К о 1 b e, Ann. 122, 42 A862); Anscliutz, там же 415, 97 A918).
7 Gabriel, Ber. 22, 1154 A889).
s A u z i e s, Rev. gen. chim. 14, 278 (Chem. Zentr. 1911, 11, 1433).
9 Marvel, Bailey, Sparberg, J. Am. Chem. Soc. 49, 1836A927).
10 R e у с h 1 e r, Bull. soc. chim. Belg. 32, 247 A923); С о r t e s e, J. Am.
Chem. Soc. 58, 191 A936).
ТЕТРАБРОМ- И ТЕТРАИОДНЕОПЕНТАН
[Тетрабром- и тетраиодгидрин пентаэритрита, тетра-{бромметил)
метан и тетра-('иодметил)-метан]
3 С (СН2ОНL + 4 РВг3 > 3 С (СН2ВгL + 4 Н3Р03
С (СН2ВгL + 4 Nal — С(СН,1L +4 NaBr
Предложил: X. Шуринк.
Проверили: В. Каротерс и В. Мак-Ювен.
1. Получение
А. Тетрабромнеопентан. В круглодониую колбу емкостью
500 мл, снабженную воздушным обратным холодильником, к верх-
нему концу которого присоединена капельная воронка с длинной
трубкой и согнутая стеклянная трубка, помещают 125 г @,92 мол.)
сухого пентаэритрита («Синт. орг. преп.», сб. 1, стр. 333). Согну-
тую трубку соединяют с соответствующей ловушкой для поглоще-
ния выделяющегося в большом количестве бромистого водо-
рода. Колбу нагревают на паровой бане и из капельной воронки
осторожно приливают 500 г A75 мл, 1, 85 мол.) свежеперегнанного
трехбромистого фосфора (стр.115). После прибавления всего коли-
чества паровую баню заменяют масляной и температуру постепенно
повышают до 170—180° (примечание 1). При этой температуре нагре-
вание продолжают 20 час, после чего оранжевокрасную реакцион-
ную смесь выливают в стакан, в который предварительно налит 1л хо-
лодной воды. Смесь энергично перемешивают для измельчения про-
дукта реакции. Красный хлопьевидный продукт фильтруют с от-
сасыванием и несколько раз промывают горячей водой, после чего
тщательно промывают 2 раза холодным 95%-ным этиловым спиртом
порциями по 200 мл (примечание 2). Продукт сушат и переносят
в большой экстрактор Сокслета, где его тщательно экстрагируют
95%-ным спиртом (примечание 3). По охлаждении из спирта вы-
падает тетрабромнеопентан, который отсасывают. Выход неочищен-
ного продукта с т. пл. 158—160° составляет 245—270 г F9—76%
теоретич.). Продукт этот достаточно чцст для превращения его в
иодид (примечание 4). С целью очистки сырой продукт можно под-
вергнуть перекристаллизации из 95%-ного спирта; на 1 г продукта
идет 30 мл растворителя. Температура плавления при этом повы-
шается до 163°, выход около 85%.
444
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
ТЕТРАГИДРОФУРАН
445
Б. Тетраиоднеопентан. В круглодонную колбу емкостью
500 мл с обратным холодильником помещают 100 г @,67 мол.) вы-
сушенного при 120° йодистого натрия, 300 мл B40 г) метилэтил-
кетона (примечание 5) и 50 г @,13 мол.) неочищенного тетрабромнео-
пентана (т. пл. 158—160°). Смесь кипятят на паровой бане 48 час,
меняют обратный холодильник на обращенный вниз, отгоняют рас-
творитель и остаток смывают горячей водой в нагретую бюхнеров-
скую воронку. Продукт в воронке тщательно промывают кипящей
водой, хорошо отжимают и переносят в большой экстрактор Сокс-
лета. С целью очистить продукт от примесей его экстрагируют ки-
пящим 95%-ным спиртом до тех пор, пока проба, взятая из экстрак-
ционного стакана, не будет плавиться при 233° (примечание 6).
Продукт вынимают из стакана и сушат; выход 66—73 г (89—98%
теоретич.). Неочищенный продукт может быть подвергнут перекри-
сталлизации из горячего бензола; на 1 г продукта требуется 18 мл
растворителя. Выход около 80%, причем температура плавления
не изменяется.
2. Примечания
1. Температуру следует повышать медленно, чтобы избежать
образования самопроизвольно воспламеняющихся гидридов фос-
фора, которые могут загореться и уничтожить весь препарат. Ана-
логичный результат получается и в том случае, если трехброми-
стый фосфор прибавляют при 170°.
2. Посредством этой промывки удаляют образующиеся в ка-
честве промежуточных соединений бромгидрины.
3. Так как экстрагирование протекает медленно, рекомендуется
разделить продукт на несколько порций и работать одновременно
с несколькими экстракторами. В данном случае применялся экстрак-
тор Сокслета видоизмененного типа (стр. 28, примечание 3), в ко-
тором трубка экстрактора обогревается парами растворителя.
4. Неочищенный продукт обладает неприятным запахом, веро-
ятно, вследствие присутствия какого-то соединения фосфора. От
этого запаха можно избавиться, если продукт нагреть до 120° и
затем несколько раз его перекристаллизовать.
5. Вместо метилэтилкетона в качестве растворителя можно взять
ацетон, однако в этом случае реакцию надо вести в запаянном
сосуде при 95—100° в течение 30—36 час.
6. Экстракция продолжается от 12 до 16 час. в зависимости от
быстроты стекания конденсирующихся паров спирта.
3. Другие методы получения
Тетрабромгидрин пентаэритрита был получен действием трех-
бромистого фосфора на пентаэритрит 1>2, а также действием рас-
твора бромистого водорода в уксусной кислоте на тетраацетат пен-
таэритрита 3. Иодид был получен действием красного фосфора и
иодистоводородной кислоты на пентаэритрит 4, а также действием
йодистого натрия на бромид в растворе ацетона 2.
1 R a v e, Toilers, Ann. 276, 61 A893); Густавсон, J. prakt.
Chem. B) 54, 98 A896).
3 Backer, Schurink, Rec. trav. chim. 50, 924 A931).
3 Perkin, S i m о n s e n, J. Chem. Soc. 87, 860 A905).
4 Tollens, Wigand, Ann. 265, 331 A891).
ТЕТРАГИДРОФУРАН
(Окись тетраметилена; 1,4-этоксибутан)
СН
II
СН
СН
||
СН+2Н,
(PdO — Pd)
сн2
1
СН,
-СН,,
S
СН.,
о
о
Предложили: Д. Старр и Р. Хиксон.
Проверили: Дж. Джонсон и X. Стивенсон.
1. Получение
.4. Получение окиси палладия. В кастрюле емкостью 350 мл
растворяют 2,2 г @,02 гр.-ат.) металлического палладия в неболь-
шом количестве царской водки и раствор (примечание 1) обрабаты-
вают 55 г азотнокислого натрия (х. ч.) и достаточным количеством
дестиллированной воды, чтобы образовалась густая паста. Веще-
ства тщательно перемешивают и осторожно нагревают, чтобы уда-
лить воду. Нагревание усиливают до расплавления смеси (около
270—280°), после чего осторожно продолжают нагревание. Не-
сколько выше температуры плавления смесь перемешивают и с
осторожностью нагревают, так как происходит выделение окислов
азота и имеет место вспучивание. После того, как выделение газов
почти закончится (примерно через 5 мин.), массу нагревают пол-
ным пламенем бунзеновской горелки в течение 10 мин. Всего на-
гревание продолжается около 30 мин. По мере того, как масса охла-
ждается, кастрюлю вращают с той целью, чтобы плав застыл по
стенкам сосуда. После обработки дестиллированной водой (около
200 мл) до полного растворения натриевых солей темнокоричневыи
осадок окиси палладия отфильтровывают и тщательно промывают
1%-ным раствором азотнокислого натрия (примечание 2). Окись
не следует промывать чистой водой, так как она легко переходит
в коллоидальное состояние. Выход окиси палладия после сушки
в вакуум-эксикаторе составляет 2,3—2,4 г (91—95% теоретич.;
примечание 3).
446
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
ТЕТРАГИДРОФУРАН
447
Б. Тетрагадрофуран. В сосуд для каталитического гидрирова-
ния под давлением (примечание 4) помещают 10 г чистого фурана
(примечание 5) и 0,2 г окиси палладия. Воздух из сосуда вытесняют
водородом (примечание б) и создают начальное давление, равное при-
близительно 7 am (примечание 7). После 10-минутного индукцион-
ного периода гидрирование протекает гладко, и примерно через час
поглощается теоретическое количество водорода. Реакция заметно
зкзотермична. После того как реакция закончена, добавляют 20 г
фурана и 0,2 г окиси палладия (примечание б), воздух вновь выте-
сняют водородом и давление водорода доводят до 7 am. После этого
индукционный период бывает незначительным, и реакция протекает
даже несколько скорее, чем в первый раз; температура повышается
до 40—50°. Когда реакция почти закончится, добавляют 30 г фу-
рана и 0,2 г окиси палладия и восстановление продолжают. Таким
образом продолжают прибавлять порции по 30 г фурана и по 0,2 г
окиси палладия до тех пор, пока сосуд не наполнится на 2/3. После
этого для полноты восстановления добавляют еще порцию палла-
дия, и смесь взбалтывают до тех пор, пока не прекратится погло-
щение водорода. Катализатору дают осесть (примечание 8) и тет-
рагидрофуран декантируют через фильтр в перегонную колбу.
Продукт восстановления нацело перегоняется при 64—66°.
Для восстановления 120 г A28 мл, 1,76 мол.) фурана требуется
15—20 час. в зависимости от чистоты фурана и от активности ката-
лизатора (примечание 9). Выход перегнанного тетрагидрофурана
составляет 114—118 г (90—93% теоретич.). Так как восстановление
протекает количественно, выход определяется аккуратностью
работы с летучими фураном и тетрагидрофураном.
2. Примечания
1. Можно также взять водный раствор эквивалентного коли-
чества химически чистого хлористого палладия.
2. Фильтраты должны быть чистыми и бесцветными. Если они
приобретают желто-оранжевую опалесценцию, это значит, что часть
окиси перешла в коллоидальное состояние. Палладий может быть
выделен в виде окиси 1, если фильтраты выпарить досуха и вновь
подвергнуть сплавлению, или же в виде палладиевой черни, если
их слегка подщелочить содой и нагреть с формалином.
3. Небольшое количество окиси палладия пристает к кастрю-
ле, и его нельзя удалить обычными способами. Царская водка рас-
творяет ее лишь с трудом, по ее легко растворить кипячением
с 48%-ной бромистоводородной кислотой.
4. Если предполагают работать при первоначальном давлении,
равном б—7 am, то обычный аппарат для каталитического восста-
новления («Скнт. орг. преп.», сб. 1, стр. 46) следует несколько видо-
изменить, заменив резиновую трубку, соединяющую сосуд с
водородом и сосуд для восстановления, гибкой медной трубкой.
Чтобы избежать несчастных случаев и потери материалов, сосуд,
для восстановления всегда должен быть окружен металлической сет-
кой, а сам сосуд необходимо предварительно проверить на макси-
мальное применяемое давление. Пробку сосуда укрепляют с по-
мощью медного зажима. Резиновую пробку предварительно обра-
батывают щелочью, тщательно промывают и сушат.
5. Как и во всех случаях каталитического гидрирования, чи-
стота исходных материалов имеет здесь первостепенное значение.
Вполне удовлетворительным является свежеперегнанный фуран с
т. кип. 31—32°, полученный по методу Уильсона («Синт. орг. преп.»,
сб. 1, стр. 449). Имеются указания на то, что фуран, полученный
по методу Гильмана и Луизипьяна 2, следует высушить хлористым
кальцием и подвергнуть тщательной фракционировке. Рекомен-
дуется перегнать фуран незадолго до употребления и избегать кон-
такта с резиновыми пробками.
6. Ввиду большой летучести фурана и тетрагидрофурана, колбу
перед наполнением водородом не эвакуируют как обычно, а вытес-
няют воздух водородом. По этой же причине необходимо охлаждать
сосуд для гидрогенизации, прежде чем спускать давление перед при-
бавлением новых порций фурана икатализатора во избежание боль-
ших потерь. Весьма удобное и быстрое охлаждение достигается
тем, что на сосуд для гидрогенизации (при работающей качалке)
направляют струю эфира из промывалки.
7. Гидрогенизация фурана может производиться и при первона-
чальном давлении в 3 am, однако в этом случае восстановление про-
текает медленнее.
8. Катализатор можно отделить, высушить в вакуум-эксика-
торе над серной кислотой и использовать вновь. Обычно второе вос-
становление протекает значительно медленнее, так что целесооб-
разнее применять наряду со старым катализатором 1—2 порции
свежего. Использованный катализатор можно регенерировать, пре-
вращая его в окись так, как это описано в части (А).
9. В приборе Адкинса для гидрогенизации при высоком давлении
можно прогндрировать сразу 120 г фурана с 10 г никелевого ката-
лизатора Ренея4. При работе под давлением в 100—150am и в тем-
пературных пределах от 100 до 150° восстановление протекает
чрезвычайно быстро и является сильно экзотермичным. Катализа-
тор окись платины — платиновая чернь не является удовлетвори-
тельным катализатором для восстановления фурана 3.
3. Другие методы получения
Получение катализатора окись палладия — палладиевая чернь
м применение его для восстановления органических соединений
изучали ШраГшер и Адаме1. Палладиевая чернь и коллоидальный
448
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
а-ТЕТРАЛОН
449
палладий широко применяются в качестве катализаторов гидро-
генизации 5.
Тетрасидрофуран был получен восстановлением фурана в паро-
вой фазе над никелевым катализатором при 170° 6; в растворе бути-
лового спирта при 50° с никелевым катализатором Ренея 7; с ката-
лизатором палладий — палладиевая чернь в отсутствии раствори-
теля 3; и с катализатором осмий на асбесте 8, а также восстановле-
нием эфиров янтарной кислоты с медно-хромовым катализатором 9
и дегидратацией 1,4-бутиленгликоля 10.
1 Shrine r, Adams, J. Am. Chem. Soc. 46, 1684A924); К e r n, S h r i-
n с г, Adams, там же 47, 1147 A925).
2 Gilman, Louisinian, Rec. trav. chim. 52, 156 A933).
3 S t a r r, H i x о n, J. Am. Chem. Soc. 56, 1595 A934).
"Covert, A d k i n s, там же 54, 4116 A932).
5 Сабатье. Катализ в органической химии. Госхимтехиздат, Л. 1932.
6 Bourguignon, Bull. soc. chim. Belg. 22, 88 A908); Ш у й к и н,
Бунина, ЖОХ 8, 669 A938).
7 С 1 о к е, А у е г s, J. Am. Chem. Soc. 56, 2144 A934).
s Шуйкин, Чиликина, ЖОХ 6, 279 A936).
9 Е. I. du Pont de Nemours and Company, ам. пат. 2 130 501 [С. А. 32,
9101 A938)].
10 Струков, Хим.-фарм. пром. 1935, № 1,35; General Aniline and Film
Corporation; ам. пат. 2 251 292 [С. А. 35, 6982 A941)]; I. Q. Farbenind. A.-G..
герм. пат. 700 036 [С. А. 35, 6982 A941)].
C6HS (CH2K CO.2H + SOC12
Cl —CO
«-ТЕТРАЛОН
[3А-Ди.гидро-1'- B )-шфталинон]
—+ CeH5 (CH,K С0С1 4- НС1 -I- SO.
0
CH..
(A1C1-)
CH,
CH.,
CH.,
С Ho
4- HCi
Предложили: Е. Мартин и Л. Физер.
Проверил: /(. Ноллер.
1. Получение
В круглодонную колбу емкостью 500 мл, снабженную обратным
холодильником, который с помощью стеклянной трубки соединен
с ловушкой для поглощения газов, помещают 32,8г@,2мол.O-фе-
нилмасляной кислоты (стр. 511) и 20 мл C2 г, 0,27 мол.) хлористого
тионила (примечание 1). Смесь осторожно нагревают на водяной
бане до полного расплавления кислоты, после чего реакции дают итти
без нагревания извне. Через 25—30 мин. выделение хлористого
водорода прекращается, и смесь нагревают в течение 10 мин. на водя-
ной бане. После этого колбу присоединяют к водоструйному на-
сосу и после создания вакуума нагревают ее 10 мин. на водяной бане,
а под конец — 2—3 мин. на небольшом пламени горелки, чтобы
удалить избыток хлористого тионила. Получаемый в результате этой
реакции хлорангидрид представляет собой почти бесцветную жид-
кость и не нуждается в дополнительной очистке. Колбу охлаждают,
добавляют МЬмл сероуглерода и раствор охлаждают снаружи льдом.
Затем быстро и сразу прибавляют 30 г @,23 мол.) хлористого алю-
миния и колбу присоединяют немедленно к обратному холодиль-
нику. Через несколько минут энергичное выделение хлористого водо-
рода прекращается, и смесь медленно нагревают на водяной бане
до кипения. После нагревания и взбалтывания в течение 10 мин.
реакцию можно считать законченной. Реакционную смесь охла-
ждают до 0° и комплекс с хлористым алюминием разлагают, осто-
рожно добавляя при взбалтывании 100 г льда. Затем добавляют
25 мл концентрированной Соляной кислоты и смесь переносят в
2-литровую круглодонную колбу и подвергают ее перегонке с водя-
ным паром (примечание 2). Сперва отгоняется сероуглерод (при-
мечание 3), после чего наблюдается резкий перелом в перегонке,
и продукт реакции нацело переходит с последующими 2 л дестил-
лата. Маслянистый слой отделяют, а водный слой экстрагируют
тремя порциями бензола по 100 мл. Маслянистую жидкость и бен-
зольную вытяжку соединяют вместе, растворитель отгоняют и
остаток перегоняют в вакууме. Выход а-тетралона с т. кип.
105—107°/2 мм составляет 21,5—26,5 г G4—91% теоретич., считая
на 7-фенилмасляную кислоту).
2. Примечания
1. Хлористый тионил был очищен следующим образом: 50 г
продажного хлористого тионила были перегнаны сперва в 10 мл
хинолина, а затем с 20 мл кипяченого льняного масла в приборе,
который был защищен от влаги воздуха.
2. Перегонку следует вести сильной струей пара и поставить
мощный холодильник (см. «Синт. орг. преп.», сб. 1, стр. 461), так
как а-тетралон лишь умеренно летуч с водяным паром.
3. При регенерации сероуглерода остается лишь весьма незна-
чительный остаток.
3. Другие методы получения
а-Тетралон был получен каталитической гидрогенизацией
а-нафтола ь, из хлорангидрида -у-фенилмасляной кислоты и хло-
ристого алюминия2; из f-фенилмасляной кислоты и концентри-
29 Сборник № 2
450
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
рованной серной кислоты3 и окислением тетралина хромовым
ангидридом4 или кислородом воздуха5. Подробные указания для
получения а-тетралона по этому последнему методу приведены
в Org. Syn. 20, 94.
1 Schroeter; герм. пат. 352 720 [С. А. 17, 1245 AS23)].
3 Kipping, Hill, J. Chem. Soc. 75, 147 A899); A m a g a t, BuIS.
soc. chim. D) 41, S40 A927).
3 Krollpfeiffer, Schafer, Ber. 56, 624 A923); Home, S h r i-
n e r, J. Am. Chem. Soc. 55, 4652A933); Cook, Hewett, J. Chem. Soc. 1934,
373
* Schroeter; герм. пат. 346 S48 [Frdi. 14, 491 A921—25)].
6 I. G. Farbenind. A.-G.; герм. нат. 539 476 [С. А. 26, 1614A932)];
Cook, J. Chem. Soc. 1938, 1778; Hock, Susemihi, Ber. 66, 61 AS33);
Brown, Widiger, Letang, J. Am. Chem. Soc. 61, 2601 AS3S).
CH,
CH,
ТЕТРАМЕТИЛЕНХЛОРГИДРИН
D-Хлорбутанол-1)
¦CH2
CH2
HC1
—. ClCHsCHaCHaCH,OH
0
Предложили: Д. Старр и Р. Хиксон.
Проверили: Дж. Джонсон и X. Стивенсон.
1. Получение
В трехгорлую колбу емкостью 500 мл помещают 114 г A,58 мол.)
тетрагидрофурана (стр. 445) и колбу снабжают обратным холо-
дильником, погруженным в жидкость термометром и согнутой
стеклянной трубкой для ввода газообразного хлористого водорода
(примечание 1), которая доходит почти до дна колбы. Верхний
конец обратного холодильника присоединяют к колбе Вюрца на
150 мл, которую охлаждают смесью льда и соли и которая служит
для улавливания продуктов, увлекаемых хлористым водородом.
Тетрагидрофуран нагревают до температуры его кипения F4—
65°) и через жидкость пропускают медленный ток хлористого
водорода. По мере того, как протекает реакция, температура кипя-
щей жидкости повышается сперва медленно, а затем более быстро,
пока не превысит 100° (примерно после 4 час. нагревания). Прибли-
зительно через 5 час. температура становится практически постоян-
ной в пределах 103,5—105,5°, и реакцию прекращают. Светлобурую
жидкость охлаждают, переносят в колбу Клайзена емкостью 250 мл
ТЕТРАМЕТИЛЕНХЛОРГИДРИН
451
с дефлегматором длиною 20 см и жидкость перегоняют в вакууме
с водоструйным насосом. Вначале выделяется большое количество
хлористого водорода, и только через некоторое время устанавли-
вается нужное разрежение. На протяжении всей перегонки необ-
ходимо применять ловушку, охлаждаемую до—15° смесью льда и
соли для улавливания тетрагидрофурана.
После небольшого головного погона главную фракцию собирают
в пределах 80—90°/14 мм или 65—75°/7 мм (примечание 2). Вес
этой фракции 95—100 г. В колбе остается немного E—10 г) высоко-
кипящего остатка. Вторичное фракционирование сырого продукта
дает 93—98 г E4—57% теоретич.) чистого тетраметиленхлоргидрина,
выкипающего в пределах одного градуса при 81—82°/14 мм или
70—7 Г/7 мм (примечания 3 и 4).
2. Примечания
1. Хлористый водород, получаемый прибавлением по каплям
концентрированной серной кислоты к смеси хлористого натрия и
концентрированной соляной кислоты, может быть употреблен непо-
средственно, без предварительной сушки.
2. Имеются указания х на то, что тетраметиленхлоргидрин в слу-
чае перегонки его при давлениях, заметно выше 15 мм, претерпе-
вает отщепление хлористого водорода. Если главную фракцию
перегоняют с масляным насосом, то последний необходимо защи-
тить от действия хлористого водорода колонками с натронной
известью.
3. Для того чтобы выделить тетрагидрофуран, конденсат из
ловушек и головные погоны, полученные при дробной перегонке,
соединяют вместе, охлаждают в бане со льдом и осторожно обраба-
тывают 15—20 мл 40%-ной щелочи. Верхний слой отделяют, сушат
небольшим количеством хлористого кальция и перегоняют. Вес
выделенного тетрагидрофурана с т. кип. 64—67° составляет 20—
22 г A7—19% исходного количества). Остаток A2—14 г) после
отгонки тетрагидрофурана перегоняется при 43—45°/10 мм и пред-
ставляет собою тетраметилендихлорид.
4. Тетраметиленхлоргидрин может быть превращен действием
трехбромистого фосфора в хлорбромид с прекрасным выходом2.
3. Другие методы получения
Тетраметиленхлоргидрин был получен действием хлористого
тионила на тетраметиленгликоль в присутствии пиридинах, а также
с помощью метода, описанного выше 2.
1 Kirner, Richter, J. Am. Chem. Soc. 51, 2503 A929).
2 S t a r r, H i x о n, там же 56, 1595 A934).
452
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
р-тиодигликоль
453
ТИОБЕНЗОФЕНОН
CH8CH2ONa +
С6Н5СС12С6Н4 + 2 NaSH
H2S
> С2Н6ОН + NaSH
C6H6CSCeH6 + 2 NaCl
HaS
Предложили: X. Штаудингери X. Фрейденбергер.
Проверили: Р. Адаме и Е. Вудруф.
1. Получение
Приготовляют спиртовой раствор сульфгидрата натрия • насы-
щением сухим сероводородом раствора 150 мл абсолютного спирта,
в которых растворены 4,6 г @,2 гр.-ат.) натрия. В 3-литровую трех-
горлую колбу, снабженную капельной воронкой и трубкой для
подачи сухого углекислого газа, механической мешалкой и обрат-
ным холодильником, помещают 25 г @,11 мол.) дифенилдихлор-
метана (примечание 1). Колбу наполняют углекислым газом и затем
медленно прибавляют спиртовой раствор сульфгидрата натрия
(примечание 2). Бурно протекающую реакцию несколько умеряют
погружением колбы в холодную воду, причем реакционная смесь
окрашивается в синий цвет.
После того, как сульфгидрат весь прибавлен, реакционную
массу оставляют стоять в течение получаса, затем прибавляют
воды и смесь экстрагируют эфиром. Эфирный раствор сушат хло-
ристым кальцием, отгоняют эфир и остаток перегоняют в вакууме
в атмосфере углекислого газа (примечание 3). Тиобензофенон
кипит при 174°/14 мм; он переходит в виде синего масла, которое,
если оно достаточно чисто и высушено, по охлаждении застывает
в красивые синие кристаллы. Выход сырого продукта 10—12,5 г
E0—63% теоретич.; примечание 4).
Сырой продукт, содержащий около 75% тиобензофенона, очи-
щают перекристаллизацией из петролеиного эфира (фракция с
т. кип. -70—90°). Выход очищенного продукта с т. пл. 53—54°.
8,4—9,9 г D2—50% теоретич.).
Тиобензофенон хранят в темноте в запаянных стеклянных ампу-
лах, наполненных сухим углекислым газом.
2. Примечания
1. Дифенилдихлорметан (дихлорид бензофенона) получается на-
греванием эквимолекулярных количеств бензофенона и пятихлори-
стого фосфора при 145—150° в течение 2 час. и последующей дроб-
ной перегонкой смеси в вакууме. Т. кип. 201—202°/35 мм.
2. Необходимо, чтобы все время был избыток дифенилдихлор-
метана, потому что сульфгидрат легко восстанавливает тиокетон
в соответственный дисульфид. Если прибавляют хлорид к раствору
сульфгидрата натрия, то получается с выходом 70% чистый дибенз-
гидрилдисульфид, а тиокетон не образуется.
3. Все операции, связанные с очисткой, следует проводить
очень быстро и по возможности без доступа воздуха.
4. При увеличении загрузки дифенилдихлорметана до 100 г
получается тот же выход тиобензофенона (в процентах).
3. Другие методы получения
Тиобензофенон был получен действием тиофосгена на бензол в
присутствии хлористого алюминияа; действием пятисернистого
фосфора2 или этилового эфира тиоацетоуксусной кислотыs на
бензофенон и действием спиртового раствора сернистого калия *
или сульфгидрата натрия 6 или тиоуксусной кислоты в на дифенил
дихлорметан.
f В е г g г е е п, В е г. 21, 341 A888).
2 G a 11 e r m a n п, там же 28, 2877 A895).
3 М i t r a, J. Indian Chem. Soc. 9, 637 A932) [С. А. 27, 3922 A933)].
* Gattermann, Schulze, Ber. 29, 2944 A896).
5 Staudinger, Freudenberger, там же 61, 1577 A928).
«Schonberg, Schutz, Nickel, там же 61, 1378 A928).
g-тиодигликоль
B,2'-Тиодиэтанол)
2 HOCH2CH2C1 + Na2S HOCH2CH,SCH2CH2OH + 2 NaCl
Предложили: Е. Фабер и Дж. Миллер.
Проверили: В. Мак-Ювен и В. Каротерс,
1. Получение
В 3-литровую круглодонную колбу, снабженную механической
мешалкой, помещают 1,5 кг C,7 мол.) 20%-ного раствора этилен-
хлоргидрина (примечание 1) и 750 г воды. Затем колбу ставят в
пустую чашку такого размера, чтобы она могла служить баней
на случай, если понадобится охлаждение. Пускают в ход мешалку
и к раствору прибавляют 493 г B,05 мол.) кристаллического сер-
нистого натрия с 9 частицами кристаллизационной воды; приба-
вление ведут с такой скоростью, чтобы температура раствора была
30—35°, на что обычно требуется 40—60 мин. После того как весь
сернистый натрий прибавлен, перемешивание раствора продол-
жают еще в течение 30 мин.
Затем мешалку убирают, колбу соединяют с обратным холо-
дильником и в пробку вставляют термометр так, чтобы его шарик
был погружен в жидкость. Колбу нагревают на водяной бане до
тех пор, пока температура жидкости не поднимется до 90° и затем
в течение 45 мин. поддерживают температуру при 90—95°. Раствор
454
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
ТИОСАЛИЦИЛОВАЯ КИСЛОТА
455
охлаждают до 25° и нейтрализуют прибавлением по каплям кон-
центрированной соляной кислоты до реакции на куркумовую
бумажку (примечание 2). После фильтрования раствор помещают
в колбу для упаривания в вакууме.
Колбу соединяют с коротким дефлегматором и обращенным вниз
холодильником. Для предотвращения толчков при кипении вста-
вляют капилляр. Затем воду отгоняют при 30—40 мм; колбу поме-
щают на водяную баню, которую постепенно нагревают до кипе-
ния с такой скоростью, чтобы перегонка шла все время равномерно
и гладко. Остаток в колбе, состоящий из хлористого натрия и
тиодигликоля, извлекают два раза порциями по 500 мл горячего
абсолютного спирта для того, чтобы растворить сульфид. После
второго извлечения соль переносят на воронку Бюхнера и промы-
вают небольшим количеством горячего спирта (примечание 3).
Спиртовые вытяжки и спирт после промывки помещают снова
в перегонную колбу и спирт отгоняют в вакууме. Когда практиче-
ски весь спирт отогнан, температуру бани повышают до 100° и
остаток нагревают в продолжение 3 час. при давлении в 30 мм.
Сырой бесцветный или бледножелтый продукт весит 200—215 г.
Он имеет т. кип. 164—1бб°/20 мм и может быть очищен перегонкой
в вакууме (примечание 5). Выход чистого продукта 180—195 г
G9—86% теоретич.).
2. Примечания
1. Для синтеза тиодигликоля могут быть использованы водные
растворы этиленхлоргидрина с содержанием его от 18 до 40%;
особенно удобен 20%-ный раствор, так как при его применении
реакция идет гладко и ее легко регулировать.
В том случае, если хлоргидрин имеется в растворе более слабом,
чем 20%, его следует сконцентрировать перегонкой. Хлоргидрин
и вода дают постояннокипящую смесь. Содержание в ней хлор-
гидрина равно 42,5% и кипит она при 95,8°/735 мм.
2. Под конец реакции жидкость становится щелочной и ее
необходимо нейтрализовать, иначе при отгонке происходит зна-
чительное разложение. Необходимо следить за тем, чтобы нейтра-
лизация была точной, иначе возможно образование в небольшом
количестве горчичного газа. Кроме того, в случае избытка кислоты
при выпаривании в вакууме имеет место осмоление, и выход дестил-
лата падает до 50%. Лакмусовая бумага при нейтрализации непри-
годна.
3. Если для извлечения берут 95%-ный спирт, то вместе с тио-
дигликолем растворяется и часть соли. Эту соль можно легко
отфильтровать после того, как спирт отогнан.
4. Продолжительность сушки зависит от того, при каком давле-
нии она происходит; при 20 мм вода и спирт удаляются за 1 час.
Если вода не была полностью удалена перед экстракцией спиртом,
в веществе может остаться некоторое количество соли после того,
как отогнан спирт. От соли можно избавиться или декантируя
продукт с осадка, или фильтруя его через стеклянную вату.
5. Если при опыте были взяты реактивы хорошего качества,
то сырой продукт практически белого цвета и для большинства
целей достаточно чист. Вполне чистый продукт можно получить
перегонкой в вакууме.
3. Другие методы получения
Метод, описанный выше, является видоизменением метода, пер-
воначально описанного Мейером х. Ирвин 2 доказал, что этот общий
метод пригоден и для работ в заводском масштабе. Р-Тиодигликоль
может быть получен также из окиси этилена и сероводорода, при-
чем "этот процесс удается только в том случае, если в качестве
растворителя для реагирующих веществ применять некоторое
количество готового тиодигликоля 3.
1 М е у е г, Вег. 19, 3260 A886); С I a r k e, J. Chem. Soc. 101, 1583 A912);
G о m b e r g, J. Am. Chem. Soc. 41, 1414 A919).
2 Irvine, английский военный отчет.
3 Чичибабин, франц. пат. 769 216 [С А. 29,481A935)]; Чичиба-
бин, Бестужев, Compt. rend. 200, 242 A935); Nenitzescu, Scar-
1 atescu, Вег. 68,587A935); Oth me r, Kern, Ind. Eng. Chem. 32,160A940).
ТИОСАЛИЦИЛОВАЯ КИСЛОТА
(о-Меркаптобензойная кислота)
iCO2H
NH2
__L o-HO2CC2H4N,Cl
NaN0 "
2o-HO,CC6H1N2Cl ~S^ o-H02CC6H4SSC6H4C02H-o'
rz ¦ сн со m f^'Y°*H
o-HOoCCeHjSSCeH.CO.H-o' i—L—i-i-L 2
\/SH
Предложили: /(. Алмн и Д. Мак-Кей.
Проверили: Р. Адаме и А. Кнауф.
1. Получение
200 лы воды нагревают до кипения в 4-литровом стакане и при
нагревании и перемешивании в ней растворяют 260 г A,1 мол.)
456
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
ТИОСАЛИЦИЛОВАЯ КИСЛОТА
457
кристаллического сернистого натрия (Na2S-9H2O) и 34 г мелко
растертой серы. Затем прибавляют раствор 40 г едкого натра в
100 мл воды и смесь охлаждают сначала холодной водой, а затем
смесью льда и соли.
В 2-литровый стакан, находящийся в охладительной смеси и
снабженный мешалкой и термометром для отсчета температур около
0°, помещают 500 мл воды, 137 г A мол.) антраниловой кислоты
и 200 мл концентрированной соляной кислоты. Мешалку пускают в
ход и смесь охлаждают до 6°. Отдельно растворяют 69 г Aмол.)
азотистокислого натрия в 280 мл горячей воды и полученный рас-
твор охлаждают льдом. Этот раствор помещают небольшими пор-
циями в делительную воронку подходящего размера. Воронка уста-
навливается таким образом, чтобы ее нижний конец был опущен
под поверхность раствора антраниловой кислоты. Когда темпе-
ратура понизится до 5°, начинают приливать нитрит и одновре-
менно постепенно прибавляют около 500 г колотого льда, чтобы
температура не поднималась выше 5°. Это прибавление занимает
около 10 мин. (примечание 1). По окончании диазотирования капля
раствора должна сразу вызывать синее окрашивание на иодкрах-
мальной бумаге.
Мешалку и термометр затем переносят в щелочной раствор
сульфида, температура которого должна быть ниже 5°. Диазорас-
твор прибавляют к сульфиду в течение 20—30 мин. вместе с 950 г
льда так, чтобы температура не поднималась выше 5°. Когда при-
бавление закончено, баню со льдом отставляют и дают смеси на-
греться до комнатной температуры; через 2 часа выделение азота
заканчивается (примечание 2). Затем к раствору прибавляют около
180 мл концентрированной соляной кислоты до кислой реакции
на конго и осадок дитиосалициловой кислоты отфильтровывают и
промывают водой.
Для удаления избытка серы осадок растворяют при кипячении
в растворе 60 г безводной (кальцинированной) соды в 2 л воды,,
смесь фильтруют горячей, делят фильтрат на 5 равных частей
(примечание 3) и дитиосалициловую кислоту осаждают, как и в
первый раз, концентрированной соляной кислотой. Дитиосалици-
ловую кислоту по возможности тщательно отсасывают.
Влажный осадок смешивают в 1-литровой круглодонной колбе
с 27 г цинковой пыли и 300 мл ледяной уксусной кислоты. Затем
смесь энергично кипятят около 4 час. с обратным холодильником
(примечание 4). После окончания восстановления смесь охлаждают
и фильтруют с отсасыванием. Осадок на фильтре промывают один
раз водой, переносят его в 1-литровый стакан и размешивают с
200 мл воды. Суспензию нагревают затем до кипения. Горячий
раствор подщелачивают до сильно щелочной реакции прибавле-
нием около 40 мл 33%-ного водного раствора едкого натра, кипятят
около 20 мин. для полного извлечения продукта из осадка и
отфильтровывают от нерастворимой части (примечание 5). Затем
выделяют тиосалициловую кислоту из раствора прибавлением кон-
центрированной соляной кислоты до кислой реакции на конго.
Продукт отсасывают, промывают один раз водой и сушат в сушиль-
ном шкафу при 100—110°. Выход продукта с т. пл. 162—163° соста-
вляет 110—130 г G1—84% теоретич., считая на антраниловую-
кислоту).
Продукт достаточно чист для большинства целей (примечание 6).
Для перекристаллизации 5 г вещества растворяют в 20 мл горя-
чего 95%-ного спирта, к раствору прибавляют 40 мл воды, кипятят
с небольшим количеством активированного угля, фильтруют горя-
чим и дают охладиться. Продукт кристаллизуется в виде желтых
хлопьев. Выход перекристаллизованной тиосалициловой кислоты
с т. пл. 163—164° составляет 4,7 г.
2. Примечания
1. При этом способе диазотирования процесс протекает гораздо,
быстрее, чем если применять только внешнее охлаждение («Синт.
орг. преп.», сб. 1, стр. 262). Общий объем раствора не играет роли,
так как дитиосалициловая кислота очень трудно растворима в воде
и легко отсасывается.
2. Иногда при выделении азота раствор очень сильно пенится.
Прибавление время от времени нескольких миллилитров эфира
уничтожает пену.
3. Дитиосалициловую кислоту можно выделить также всю одно-
временно и провести восстановление сразу всего количества. В этом
случае восстановление следует вести в 5-литровой колбе с хорошей
мешалкой. Смесь кипятят с обратным холодильником на кольце-
вой горелке в продолжение 10 час. При работе в лаборатории удоб-
нее делить материал и восстанавливать его небольшими порциями.
При восстановлении всего количества сразу выход практически не
понижается.
4. Восстановление не всегда проходит гладко, иногда цинк
собирается комками и становится неактивным. В таких случаях:
приходится еще добавлять цинка. Для определения конца восстано-
вления берут пробу, охлаждают и фильтруют. Осадок кипятят
с крепким раствором едкого натра, фильтруют и подкисляют соля-
ной кислотой. Если восстановление закончено, выпавший осадок
плавится при 164° или ниже; если восстановление не закончено,
осадок плавится выше 164°. В таком случае главную массу кипятят
еще с обратным холодильником и, если нужно, прибавляют цинка,
пока проба не покажет, что реакция прошла полностью.
При определении точки плавления капилляр с пробой следует
опускать в баню, которую предварительно нагревают до 163—
164°.
458
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
5. Если восстановление ведут в пяти частях, то для каждой
порции достаточно одной экстракции едким натром. Если же вос-
становление ведут сразу, приходится производить несколько экс-
тракций; в том случае лучше всего остаток после первой обработки
щелочью прокипятить с соляной кислотой, профильтровать и только
потом снова обрабатывать щелочью.
6. Тиосалициловая кислота применяется при получении окситио-
нафтена и многих тиоиндигоидных красителей.
3. Другие методы получения
Из многих методов, описанных для получения тиосалициловой
кислоты, лишь следующие имеют препаративный интерес: нагре-
вание ортогалоидозамещенных бензойных кислот с щелочным
гидросульфидом при 150—200° в присутствии меди или медных
солей >'2 или при действии сернистого натрия при 200°3; восстано-
вление дитиосалициловой кислоты глюкозой 4 или металлами 5-2
в щелочном растворе. Дитилосалициловая кислота получается при
обработке диазотированной антраниловой кислоты двусернистым
натрием в щелочном растворе 6.
1 (a) Cassella und Ко, герм. пат. 189200 [С. А. 2,607 A908)]; (b) Cain,
Intermediate Products for Dyes, p. 151.
2 Chem. Age 21, Dyestuffs Suppl. p. 11 A929).
3 Cassella und Ко; герм. пат. 193 290 [С. А. 2, 1514A908I; Ref. I (b).
* С 1 a a s z, Ber. 45, 2427 A912).
5 Kalle und Ко, герм. пат. 204 450 [С. А. 3, 1695 A909)]. Ref. I (b).
ТИОФЕН
CH2CO2Na НС СН
p2s3
ТИОФЕН
459
СН,ССШа
НС СН
Предложил: Р. Филлипс.
Проверили: Р. Адаме и X. Коган.
1. Получение
В 3-литровую круглодонную колбу помещают возможно более
тесную смесь 486 г C мол.) мелко растертой безводной натриевой
соли янтарной кислоты (примечание 1) и 648 г D,1 мол.) мелко
растертого трехсернистого фосфора (примечание 2). Колбу соеди-
няют с обращенным вниз холодильником длиной 100 см (примеча-
ние 3) и, кроме того, через пробку вставляют трубку, которая дохо-
дит до середины колбы, для ввода углекислоты. Холодильник
соединяют с 2-литровой колбой, охлаждаемой смесью льда с солью.
Несконденсировавшиеся газы пропускают через две 2-литровые
колбы, соединенные последовательно с приемником. В каждую
колбу помещают по 1 кг колотого льда и 200 мл 40%-ного рас-
твора едкого натра. Обе колбы снаружи охлаждают смесью льда
и соли.
Реакционную колбу наполняют углекислотой (примечание 4),
причем ее вращают, чтобы удалить воздух, удерживаемый растер-
той реакционной смесью, затем соединяют с холодильником и при
пропускании слабого тока углекислоты смесь нагревают сначала
в течение 30 мин. на слабом огне горелки Бунзена (примечание 5),
а затем полным пламенем до прекращения выделения желтых паров
(около 30 мин.). Для того, чтобы иметь уверенность в том, что весь
тиофен отогнан, во время второго периода реакции углекислоту
пропускают быстрее (примечание б). Содержимое приемника и
обоих поглотителей соединяют и перегоняют с водяным паром из
5-литровой колбы до тех пор, пока не перестанут перегоняться
капли масла. Тиофеновый слой в дестиллате отделяют от воды,
сушат сначала твердым едким натром, затем металлическим натрием
и фракционируют. Выход продукта, кипящего при 83—86°, соста-
вляет 63—75 г B5—30% теоретич.; примечания 7 и 8).
2. Примечания
1. Натриевая соль янтарной кислоты получалась из янтарной
кислоты и сушилась в продолжение нескольких дней в плоских
чашках на водяной бане.
2. Если трехсернистый фосфор нельзя получить в готовом виде,
то его можно приготовить по разработанному А. Грисуолдом
методу: однородную смесь из вычисленных количеств мелко растер-
той серы и красного фосфора помещают в глиняный цветочный
горшок, отверстие в дне которого закрыто пробкой. Горшок вста-
вляют в ведро с песком и над верхом ведра держат наготове тяже-
лую крышку. В смесь бросают зажженную спичку, крышку быстро
закрывают, а щели засыпают песком. Реакцию хорошо вести под
открытым небом, так как она проходит очень бурно, часто происхо-
дят небольшие взрывы и выбрасывает пламя. После полного охла-
ждения цветочный горшок разламывают, содержимое его собирают
и сохраняют в банках с хорошо притертыми пробками. Желательно
некоторое время сохранять приготовленное вещество не размель-
чая, так как оно еще не вполне кристаллично и с большим трудом
поддается растиранию.
460
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
3. Благодаря быстрому выделению большого объема газов
трубка, соединяющая колбу с холодильником, должна иметь 2 см
в диаметре, и все пробки должны быть хорошо подобраны и гер-
метичны.
4. Если до нагревания воздух не полностью вытеснен из колбы,
реакция может протекать с силой взрыва.
5. При проверке работы оказалось, что на этой стадии реак-
ции необходимо непрерывное нагревание.
6. До полного охлаждения реакционной колбы не следует от-
крывать пробки, так как иначе при соприкосновении с воздухом
избыток трехсернистого фосфора может воспламениться, Остаю-
щуюся в колбе твердую массу можно размягчить обработкой горя-
чей водой, после чего ее можно раздробить и вытряхнуть.
7. Из свежеприготовленного продукта при стоянии часто выпа-
дает небольшое количество коричневого осадка. Если его отфиль-
тровать, то больше осадка не образуется.
8. Опыты в больших масштабах были проведены с удовлетвори-
тельным результатом, но при этом имеется опасность, что реакция
пойдет слишком бурно.
3. Другие методы получения
Тиофен имеется в небольших количествах в каменноугольном
газе и бензоле Ч Он был получен пропусканием этилена или ацети-
лена в кипящую серу2; при пропускании этилсульфида через
горячую трубку 8; при пропускании этилена или светильного газа
над горячими пиритами4; при нагревании ангидрида янтарной
кислоты с пятисернистым фосфором5; при обработке эритрита
пятисернистым фосфором6; при обработке альдегида янтарной кис-
лоты трехсернистым фосфором7; при пропускании ацетилена над
пиритом при 300° 8; при обработке натриевой соли янтарной кис-
лоты трехсернистым фосфором9 и при пропускании ацетилена
и сероводорода над бокситом при 320° или над гидратом окиси
никеля при 300°10, а также при пропускании фурана и сероводорода
над нагретой окисью алюминия и.
1 Meyer, Ber. 16, 1471 A883); 17, 2642 A884).
2 Meyer, Sandmeyer, там же 16, 2176 A883).
3 К е k u 1 ё, там же 18, 217 A885).
4 N a h n s e п, там же 18, 217 A885).
6 Volhard, Erdmann, там же 18, 454 A885).
• Р а а 1, Т a f е 1, там же 18, 689 A885).
' Harris, там же 34, 1496 A901).
e Steinkopf, Kirchoff, Ann. 403, 5 A914); герм. пат. 252 375
iC. A. 7, 538 A913)]; S t e i n к о р f, Chem. Ztg. 35, 1098 A911).
0 Volhard, Erdmann, Ber. 18, 454 A885); Friedburg, J. Am.
Chem. Soc. 12, 85 A890).
10 S t u e r, G г о b; ам. пат. 1 421 743 [С. А. 16, 3093 A922)].
11 Юрьев, ЖОХ 6, 972 A936); Ber. 69, 440 AS36).
n-ТОЛИЛКАРБИНОЛ
461
и-ТОЛИЛКАРБИНОЛ
(п-Метилбензиловый спирт)
л-СН3С6Н4СНО +СН2О+КОН —п-СН3СвН4СН2ОН +НСО2К
Предложили: Д. Даеидсон и М. Вейсс.
Проверили: Р. Фьюзон и Е. Кливленд.
1. Получение
Прибор состоит из 3-литровой трехгорлой колбы, снабженной
механической мешалкой с ртутным затвором, обратным холодиль-
ником, капельной воронкой и термометром, доходящим почти до
дна колбы. В колбу помещают 500 г G,6 мол.) едкого кали в грану-
лах (85%-ного) и 750 мл продажного абсолютного метанола (не со-
держащего ацетона) и пускают в ход мешалку. Большая часть
щелочи растворяется через несколько минут с выделением тепла.
Колбу помещают в большую баню с холодной водой и, когда тем-
пература внутри колбы упадет до 60°, добавляют смесь 360 г C53 мл,
3 мол.) п-толуилового альдегида (примечание 1), 300 мл формалина
C,9 мол.; примечание 2) и 300 мл абсолютного метанола с такой
скоростью, чтобы температура внутри колбы поддерживалась при
60—70°. Прибавление продолжается около 15мин. Затем температуру
внутри колбы поддерживают при 60—70° в течение 3 час, после чего
обратный холодильник заменяют на обращенный вниз и метанол отго-
няют с бани с солевым раствором до тех пор, пока температура внутри
колбы не повысится до ЮГ. Тогда к теплому раствору приливают
900 мл холодной воды и смесь охлаждают. Образовавшиеся два
слоя немедленно разделяют (примечание 3) и водный слой извле-
кают три раза бензолом, порциями по 200 мл. Маслянистую жид-
кость и вытяжки соединяют вместе, промывают пять или шесть
раз водой, порциями по 50 мл (примечание 4), а промывные воды
извлекают 50 мл бензола и бензольные вытяжки присоединяют к
промытым вытяжкам. Бензольный раствор осветляют взбалтыва-
нием с несколькими граммами безводного сернокислого натрия и
перегоняют его при уменьшенном давлении. После отгонки бен-
зола получают 331 г (90% теоретич.) п-толилкарбинола (т. кип.
116—118°/20 мм). В приемнике продукт затвердевает в массу про-
питанных маслянистой жидкостью кристаллов с т. пл. 54—55°.
После перекристаллизации из равного по весу количества продаж-
ного гептана (т. кип. 90—100°) получают с выходом 80% длинные
иглы с т. пл. 61°. Если упарить маточный раствор, то можно полу-
чить еще 8% продукта (примечания 5 и 6).
2. Примечания
1. Удовлетворительные результаты были получены с техниче-
ским п-толуиловым альдегидом. Метод получения п-толуилового
альдегида описан на стр. 464.
462
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
2. Содержание формальдегида в растворе определяется анализом
(стр. 480).
3. После стояния верхний слой затвердевает.
4. В результате этой промывки удаляется калиевая соль л-то-
луиловой кислоты. Если ее не удалить, то затрудняется перегонка
продукта.
5. Конечный остаток от маточного раствора представляет собою
маслянистую жидкость, которая не застывает в охладительной
смеси и, повидимому, является смесью п- и л*-толилкарбинолов.
При окислении ее перманганатом были получены лишь следы
о-фталевой кислоты (реакция на образование фенолфталеина);
часть масла, лучше растворимая в воде, дает фенилуретан, сме-
шанная проба которого с фенилуретаном как п-толилкарбинола,
так и бензилового спирта имеет пониженную температуру плав-
ления.
6. В аналогичных условиях бензальдегид дает бензиловый спирт
с выходом 80%, а пиперонал — пиперониловый спирт с выходом
86%.
3. Другие методы получения
л-Толилкарбинол был получен из п-толуилового альдегида дей-
ствием спиртового раствора едкого кали г, электролитическим вос-
становлением 2 и при восстановительном действии реактива
Гриньяра 3, а также электролитическим восстановлением л-толуи-
ловой кислоты 4. Описанная выше методика является применением
общего метода восстановления ароматических альдегидов в соот-
ветственные спирты 5.
1 Cannizzaro, Ann. 124, 252 A862).
2 Law, J. Chem. Soc. 91, 755 A907).
3 Oddo, Gazz. chim. ital. 41, A) 285A911).
* M e 11 1 e r, Ber. 39, 2933 A906).
5 Davidson, Bogert, J. Am. Chem. Soc. 57, 905 A935).
CHS
О-ТОЛУИЛОВАЯ КИСЛОТА
сн3
/Ч
2H2O+H2SO, >
СО.2Н
-j- NH4HSO4
Предложили: X. Кларк и Е. Тзйлор.
Проверили: К. Марвел и В. Мойер.
1. Получение
В 5-литровую колбу, снабженную механической мешалкой,
обратным холодильником и делительной воронкой, помещают 3 кг
75%-ной серной кислоты уд. в. 1,67, нагревают ее примерно до
о-ТОЛУИЛОИАЯ КИСЛОТА
463
150°, пускают в ход мешалку и прибавляют через делительную-
воронку в продолжение 2 час. 1 кг (8,54 мол.) о-толунитрила («Синт..
орг. преп.», сб. 1, стр. 391). Перемешивание при 150—160° продол-
жают еще в течение 2 час. после того, как весь нитрил прибавлен.
Затем температуру повышают до 190° и массу перемешивают при
этой температуре еще 1 час. Обыкновенно под конец нагревания в
холодильнике появляется некоторое количество кристаллического
вещества. Реакционную смесь охлаждают, выливают в ледяную
воду и выпавшую толуиловую кислоту отсасывают. С целью очистки
сырой продукт растворяют в избытке 10%-ного раствора едкого
натра (примечание 1), раствор фильтруют горячим и подкисляют
фильтрат разбавленной серной кислотой. Выделившуюся о-толуи-
ловую кислоту отсасывают, сушат и перекристаллизовывают из
3 л бензола (примечание 2). Получается 930—1030 г (80—89%
теоретич.) о-толуиловой кислоты, плавящейся при 102—103° (при-
мечание 3).
2. Примечания
1. Нерастворимое в щелочи вещество представляет собою амид
толуиловой кислоты, который может быть выделен. Если его обра-
зуется много, то это доказывает, что нагревание велось недоста-
точно долго или же что температура была недостаточно высока.
Если сократить время нагревания, то можно получить значи-
тельное количество амида.Сырой амид очищают перекристаллизацией
из воды; температура плавления чистого продукта 139—140°.
2. Еще некоторое количество чистой о-толуиловой кислоты
можно получить после отгонки большей части бензола от бензоль-
ного маточного раствора и охлаждения остатка.
3. и-Толуиловую кислоту (т. пл. 178°) можно получить из
л-толунитрила по указанному же способу и с тем же выходом.
Эта кислота труднее растворима в бензоле, и поэтому для пере-
кристаллизации ее требуется около 9 л бензола.
3. Другие методы получения
о-Толуиловая кислота была получена нагреванием 1,3-нафталин-
дисульфокислоты, 1,3-диоксинафталина, 1-нафтол-З-сульфокислоты
или 1-нафтиламин-З-сульфокислоты с едким натром*; восстано-
влением фталида иодистоводородной кислотой и фосфором2; электро-
литическим окислением о-ксилола3; окислением о-ксилола раз-
бавленной азотной кислотой *; каталитическим восстановлением
фгалевого ангидрида 5; омылением о-толунитрила 75%-ной серной
кислотой 6 и действием углекислоты на эфирный раствор продукта
реакции бутиллития и о-бромтолуола 7.
» Kalle und Ко; герм. пат. 79 028 [Frdl. 4, 147A894—97)]; Fri e d I a e n-
der, Rudt, Ber. 29, 1611 A896).
-464
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
я-ТОЛУИЛОВЫЙ .'АЛЬДЕГИД
465
2 Н е s s e r t, там же 11, 238 A878); Racine, Ann. 239, 72 A887).
3Fichter, Rinderspracher, Helv. Chim. Acta 10, 41 A927)
* F i 11 i g, Bieber, Ann. 156, 242 A870).
6 Willstatter, Jaquet, Ber. 51, 771 A918).
« Cahn, Ann. 240, 280 A887).
'Oilman, Langham, Moore, J. Am. Chem. Soc. 62, 2330 A940).
я-ТОЛУИЛОВЫЙ АЛЬДЕГИД
CO+HC1
>_ HCOC1
CH3CeH5
HCOC1 -(AIC^U «-CH3CeH4CHO + HCI
Предложили: Дж. Колеман и Д. Крзг.
Проверили: Г. Гилъман и Дне. Дикки.
1. Получение
Прибор собирают в вытяжном шкафу. Его расположение видно
да рис. 17. Узкую широкогорлую реакционную банку А емкостью
500 мл снабжают эффек-
тивной механической ме-
шалкой с ртутным за-
твором, трубкой Б для
введения смеси газов и
выводящей трубкой, со-
единенной с промывной
склянкой Д. В банку А,
поставленную в нагре-
тую до 20° водяную баню.
помещают 200 г B,17
мол,) сухого толуола и
затем при хорошем пе-
ремешивании быстро
прибавляют 30 г @,3
мол.) полухлористой ме-
ди (примечание 1) и 267 г
B мол.) мелко растер-
того безводного хлори-
стого алюминия.
Смесь хлористого во-
дорода и окиси углерода
(примечание 2) пропу-
скают до дна реакцион-
ного сосуда через трубку
Б. Пропускание газов ведут 7 час, причем количество пропу-
скаемой окиси углерода должно быть вдвое больше количества
Рис. 17
хлористого водорода. Скорость притока газов можно сравнивать
по пузырькам во время их прохождения через промывные склянки В
и Г. О поглощении можно судить по количеству отходящих
газов, проходящих через промывную склянку Д. Окись угле-
рода в начале реакции поглощается почти количественно, но но
мере того, как смесь густеет, поглощение становится менее
полным.
Через 7 час. смесь медленно при взбалтывании приливают
(примечание 3) к 1,5 кг колотого льда, находящегося в 3-литровой
колбе. Полученную смесь перегоняют с паром до тех пор, пока
не будет отогнан полностью весь альдегид и неизмененный толуол.
После прибавления к дёстиллату 50 мл эфира слои разделяют;
водный слой извлекают 150 мл эфира, который присоединяют к пер-
вому неводному слою (примечание 4). Эфирный раствор высушивают
хлористым кальцием, эфир отгоняют и альдегид перегоняют из колбы
емкостью 500 мл с дефлегматором (примечание 5). Выход л-толуило-
вого альдегида, перегоняющегося при 201—205°, составляет 121—
132 г D6—51% теоретич., считая на толуол). Вторичная перегонка
сопряжена с очень небольшой потерей и дает почти бесцветный
продукт, кипящий при 203—205° (примечание 6).
2. Примечания
1. Приготовление полухлористой меди описано в «Синт. орг.
преп.», сб. 1, стр. 491. Осадок полухлористой меди несколько раз
промывают декантацией раствором сернистой кислоты, отсасывают
на большой воронке Бюхнера, промывают ледяной уксусной
кислотой и сушат в выпарительной чашке до исчезновения
запаха уксусной кислоты. Полученная полухлористая медь
чисто белого цвета. Препарат должен сохраняться без доступа
воздуха. Другой метод получения полухлористой меди описан на
стр. 552.
2. Окись углерода можно вытеснить водой из газгольдера ем-
костью в 75 л. Газ сушат, пропуская его через две промывные
склянки с концентрированной серной кислотой.
Вместо того, чтобы брать окись углерода из большого газголь-
дера, удобнее получать ее следующим образом: 170 г A41 мл;
3,69 мол.) чистой муравьиной кислоты (уд. в. 1,2) прибавляют
постепенно в 1-литровую колбу Вюрца к 250 г A35,8 мл) концен-
трированной серной кислоты, нагретой на масляной бане до 70—
80°. Отводную трубку колбы соединяют с промывными склянками
с серной кислотой. Чтобы обеспечить постоянный и ровный ток
окиси углерода, муравьиную кислоту помещают в капельную во-
ронку, которая вставлена в резиновую пробку с двумя отверстиями,
закрывающую горло. Другое отверстие этой пробки соединяют при
30 Сборник JN» 2
466
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
помощи резиновой трубки с пробкой, закрывающей капельную
воронку и имеющей одно отверстие. При этих условиях вспенивания
смеси не наблюдается; если же оно все-таки случится, рекомен-
дуется к серной кислоте в реакционной колбе прибавить неболь-
шое количество парафина.
3. Часто смесь становится настолько густой, что ее нельзя
вылить и приходится перекладывать при помощи шпателя.
4. Отделение альдегида от неизмененного толуола, при помощи
бисульфита натрия в этой стадии работы не повышает заметно
чистоты продукта.
5. Перегонку ведут с обыкновенным водяным холодильником,
а когда температура поднимется до 150°, этот холодильник заме-
няется воздушным.
6. При хранении л-толуилового альдегида рекомендуется при-
бавлять к нему несколько кристаллов гидрохинона.
3. Другие методы получения
Описанный метод представляет собою видоизмененный метод
Гаттермана и Коха1. Для получения л-толуилового альдегида
существуют еще следующие способы: действие синильной кислоты
и хлористого водорода на толуол в присутствии хлористого алю-
миния 2; действие карбонила никеля на толуол в присутствии
хлористого алюминия3; восстановление нитрила л-толуиловой
кислоты хлористым оловом с последующим гидролизом образую-
щегося сначала альдимина 4; .окисление «-ксилола, особенно окис-
ление его хлористым хромилом 5; действие на бромистый л-толил-
магний фенилгидразона формальдегида 6, ортомуравьиного эфира,
метилформанилида-, этоксиметиленанилина или сероуглерода. Син-
тезы, в которых исходят из гриньярова реактива, за исключением
первого из упомянутых методов, были рассмотрены и сравне-
ны Смитом и Бейлисом7 и Смитом и Никольсом'; они реко-
мендуют применять этоксиметиленанилин или ортомуравьиный
эфир.
1 Oattermann, Koch, Ber. 30, 1622 A897).
2 Bayer und Ко, герм. пат. 99 568 [Frdl. 5, 98 A897—1900)]; Н i n k e 1,
Ay ling, Morgan, J. Chem. Soc. 1932, 2790; H i n k e 1, А у 1 i n g,
В е у п о п, там же 1935, 677.
3 D e w a r, Jones, J. Chem. Soc. 85, 216 A904).
4 Stephen, там же 127, 1874A925); Williams, J Am. Chem
Soc. 61, 2248 A939).
5 L a w, P e r k i n, J. Chem. Soc. 91, 258 A907).
6 Grammaticakis, Compt. rend. 210, 303 A940).
7 Smith, В а у 1 i s s, J. Org. Chem. 6, 437 A941); Smith, Nichols,
там же 6, 489 A941).
С1ШЯ.-ТРИБРОМБЕНЗОЛ
467
силг-и.-ТРИБРОМБЕНЗОЛ
A,3, 5-Трибромбензол)
C6HaBrsNH2 • H2SO4
C6H6NH2 + 3 Br2 C6H2Br3NH3 -f 3 HBr
HNO2 CeHsjBrgN.HSOj + 2 H2O
C6H2Br3N2HSO4 + C2HaOH
l,3,5-C6H3Br3 + CH3CHO + N2 + H2SOU
Предложили: Дж. Колеман и В. Талбот.
Проверили: Р. Фъюзон и Ч. Вудвард.
1. Получение
Прибор составляют из 12-литровой круглодонной колбы и колбы
для отсасывания емкостью 250 мл. Колбы соединяют между собой,
как показано на рис. 18, посредством пробок и стеклянных трубок.
После загрузки колбу А помещают в баню со льдом, а Б — в водя-
ную баню, нагретую до 40—50°. В колбу А загружают 100 г
A,1 мол.) анилина, 1 л воды и 100 мл A,2 мол.) концентрированной
соляной кислоты; после растворения анилина добавляют еще воды
до общего объема 5 л. В колбу Б загружают 577 г A85 мл; 3,6 мол.)
брома. Затем колбы погружают в бани и, соединив В с насосом,
через Aw Б, пропускают быстрый ток воздуха,
насыщаемого в ?парами брома (примечания 1 и 2).
Пропускание брома продолжают до тех пор,
пока раствор в Л не примет ясножелтой окраски,
на что требуется приблизительно 3—4 часа,
после чего реакцию можно считать законченной.
Триброманилин отсасывают на бюхнеровской
воронке, тщательно промывают водой от броми-
стоводородной кислоты, возможно тщательнее
отжимают и без дальнейшей сушки используют
в следующей стадии реакции.
Влажный триброманилин помещают вместе с
2,1 л 95%-ного спирта и 525 мл бензола в 5-литровую двухгор-
лую колбу. Одно отверстие колбы соединяют с обратным холо-
дильником, другое закрывают пробкой, которую можно легко
открывать для прибавления нужных реактивов. Триброманилин
растворяют, нагревая колбу на водяной бане. К раствору при-
бавляют 140 мл концентрированной серной кислоты, а затем как
можно быстрее, насколько только позволяет бурная реакция,
140 г B,03 мол.) растертого азотистокислого натрия. Когда пер-
воначальная реакция несколько успокоится, раствор доводят до
кипения и кипятят, пока не закончится выделение газа, после
чего его оставляют стоять в теплом месте 3 часа.
Рис. 18.
468
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
1,2,3-ТРИИ ОД-5-НИТРОБЕНЗОЛ
460
После охлаждения смеси (лучше всего в бане со льдом) маточ-
ный раствор отделяют от твердого осадка декантацией через воронку
Бюхнера; чтобы осадок остался в колбе, на горло колбы можно
надеть проволочную сетку. К осадку затем прибавляют раствор
150 мл концентрированной серной кислоты в 1,5 л воды. После
разложения избытка азотистокислого натрия осадок переносят на
воронку Бюхнера и промывают сначала водой, а потом небольшим
количеством спирта. Выход сухого неочищенного трибромбензола
250—260 г G4—77% теоретич.). Красновато-коричневый продукт
плавится при 112—116°.
Для дальнейшей очистки каждые 100 г сырого продукта раство-
ряют в кипящей смеси 1560 мл ледяной уксусной кислоты и 350 мл
воды; раствор кипятят несколько минут с 25 г активированного
угля, фильтруют горячим и дают охладиться. Выпавшие кристаллы
отсасывают и промывают в бюхнеровской воронке небольшим коли-
чеством холодного 95%-ного спирта для удаления уксусной кис-
лоты. Таким образом получают 216—240 гF4—71% теоретич.) слабо
окрашенного трибромбензола с т. пл. 121,5—122,5° (исправлен.).
При дополнительной обработке маточных растворов выход
неочищенного продукта может быть увеличен еще на 50—60 г.
Таким образом общий выход будет равен 300—320 г (89—95% тео-
ретич.). Спиртовые растворы и спирт после промывки разбавляют
до б л и водный слой отделяют от тяжелого масла, выпадающего
на дно. Затем бензол отгоняют от масла из колбы Вюрца, причем
надо следить за тем, чтобы не перегреть продукт после того, как
будет отогнан бензол. Полученный таким образом трибромбензол
можно пере кристаллизовать, как описано выше.
2. Примечания
1. При реакции выделяется значительное количество тепла и
для избежания потерь брома необходимо колбу А охлаждать. Для
того, чтобы воздух был сильно насыщен парами брома, колбу
Б надо погрузить в баню, нагретую до 40—50°. Если воздух будет
насыщен парами брома при более низкой температуре, то благо-
даря сильному перемешиванию, которое производится воздухом,
в реакционной смеси образуется легкая пена, причем она легко
может быть выброшена из колбы.
2. Между реакционной колбой и насосом целесообразно поставить
предохранительную склянку. В нее наливают некоторое количество
воды, через которую и проходит воздух, после того, как из него
поглощены пары брома. Это приспособление преследует две цели:
по скорости, с которой воздух проходит через воду, можно судить
о скорости просасывания воздуха и, кроме того, можно следить
за полнотой поглощения брома в реакционной колбе, так как даже
небольшое количество непоглощенного брома окрашивает воду в
ясножелтый цвет. В этом случае временно прекращают отсасывание,
в результате чего вода из предохранительной склянки пересасы-
вается в реакционную колбу, и тем самым бром возвращается в
реакционную смесь.
3. Другие методы получения
симм.-Трибромбензол был получен из 3,5-диброманилина заме-
ной аминогруппы на бром *; из бромацетилена при действии света 2;
при разложении 2,4,6-трибромфенилгидразина 3, при восстановле-
нии сернокислого 2,4,6-трибромбензолдиазония 4>s и как побочный
продукт при получении 2,4,6-трибромбензонитрила в.
1 Ко г пег, Gazz. chim. ital. 4, 410 A874).
2 Сабанеев, ЖРХО 17, 1, 176 A885).
3 Chat taw ay, Vonderwahl, J. Chem. Soc. 107, 1508 A915).
1 Jackson, Moore, Am. Chem. J. 12, 167 A890).
6 J а с k s о п, В e n 11 e у, там же 14, 335 A892).
6 Montagne, Rec. trav. chim. 27, 347 A908).
1,2,3-ТРИИОД-5-НИТРОБЕНЗОЛ
NOa NO,
NO,
NaNO.
"~h^so7
KI
NH,
I
N,HSO,
1
Предложили: Р. Сэндин и Т. КэрНС.
Проверили: Ф. Уитмор и Л. Сузерленд.
1. Получение
В 1-литровой колбе с двумя или тремя горлами, снабженной
механической мешалкой, растворяют 50 г @,13 мол.) 2,6-дииод-4-
нитроанилина (стр. 198) в 200 мл концентрированной серной кис-
лоты (уд. в. 1,84). Раствор охлаждают до 5° смесью льда и соли
и при работающей мешалке добавляют к нему смесь 100 мл концен-
трированной серной кислоты и 12 г @,17 мол.) азотистокислого
натрия, также охлажденного до 5° (примечание 1). Чтобы выделить
азотистую кислоту из нитрозилсерной кислоты, медленно прили-
вают из капельной воронки при энергично работающей мешалке
200 мл 85%-ной фосфорной кислоты A51 мл фармакопейной фосфор-
ной кислоты, разбавленной до 200 мл). Во время приливания кислоты
температуру поддерживают ниже 10°. Смесь вынимают из охлади-
тельной бани и продолжают перемешивать до тех пор, пока не будет
закончено диазотирование A—2 часа; примечание 2). К концу этого
времени раствор выливают при помешивании в 2 л смеси колотого
470
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
ТРИКАРБОМЕТОКСИМЕТАИ
471
льда и воды, находящихся в 4-литровом стакане. Чтобы разрушить
избыток азотистой кислоты, добавляют небольшими порциями при
помешивании около 15 г мочевины до тех пор, пока продолжается
образование газа. Если смесь непрозрачна, то ее фильтруют и
затем постепенно обрабатывают раствором 30 г @,18 мол.) йоди-
стого калия в 200 мл воды. Для завершения реакции смесь нагре-
вают до прекращения выделения газа. Если выделился свободный
иод, то его удаляют бисульфитом натрия, после чего смесь филь-
труют через бюхнеровскую воронку, отмывают от серной кислоты и
неорганических солей и как можно лучше отжимают. Продукт
сушат на воздухе до постоянного веса. Выход светлобурого сырого
продукта с т. пл. 160—162° составляет 60—61 г (94—95% теоретич.).
Чистый продукт может быть получен, если растворить сырой про-
дукт в 200 мл кипящего бензола, профильтровать его и фильтрат
охладить до 10°. Выход желтых кристаллов с т. пл. 161—162°
составляет 40—42 г F5—70% от сырого продукта; примечание 3).
2. Примечания
1. Азотистокислый натрий должен быть хорошо измельчен и его
следует прибавлять медленно при энергичном перемешивании к
серной кислоте, температуру которой поддерживают при 5° охла-
ждением в бане со льдом и солью. Следует избегать выделения
окислов азота при прибавлении нитрита.
2. За это время температура смеси постепенно повышается до
комнатной. Когда диазотирование закончено, капля смеси дает
прозрачный желтый раствор при прибавлении ее к 10 мл холодной
воды.
3. Имеются указания в литературе, что описанной здесь мето-
дике следует предпочесть методику, которую описали Ходжсон и
Уокер \ и что сырой трииоднитробензол лучше всего очищать пере-
кристаллизацией из целлосольва [К. Ниманн и К. Е. Редеман,
частное сообщение и J. Am. Chem, Soc. 63, 1550 A941)].
3. Другие методы получения
1,2,3-Трииод-5-нитробензол был получен диазотированием
2,6-дииод-4-нитроанилина (без применения фосфорной кислоты) с
последующей обработкой йодистым калием г. Настоящая методика
является примером общего метода, разработанного Шоутиссеном2
для диазотирования слабо основных аминов, например 2,6-дига-
лоидо-производных л-нитроанилина.
1 W i I I g е г о d t, Arnold, Ber. 34, 3343 A901); Kalb, Schwei-
zer, Zellner, Berhold, там же 59,1866 A926); Harington,
В a r g e r, Biochem. J. 21, 175 A927); Hodgson, Walker, J. Chem. Soc.
1933, 1620.
2 Schoutissen, J. Am. Chem. Soc. 55, 4531 A933).
ТРИКАРБОМЕТОКСИМЕТАН
(Триметиловый эфир метантрикарбоновой кислоты)
2'СН3 (СО2СН3), + 2 Na > 2 NaCH (C0aCH3J + Н2
NaCH (СО2СН3J"+ С1СО,СН3 > СН "
NaCl
Предложили: В. Корсон и Дж. Сэйр.
Проверили: Р. Адаме и А. Кнауф.
1. Получение
В 2-литровую трехгорлую колбу, снабженную обратным холо-
дильником, делительной воронкой и мешалкой с ртутным затво-
ром, помещают 400 мл сухого ксилола и 13 г @,56 rp.-ат.) натрия.
Колбу нагревают на масляной бане до расплавления натрия и смесь
перемешивают до тех пор, пока натрий не разобьется на мелкие
капли. Затем в течение 5—10 мин. прибавляют 69 г @,57 мол.)
диметилового эфира малоновой кислоты (примечание 1).
Смесь охлаждают при непрерывном перемешивании и, когда
температура опустится приблизительно до 65°, прибавляют в тече-
ние 5—10 мин. 57 г @,6 мол.) метилового эфира хлоругольной кис-
лоты. Затем смесь медленно нагревают в течение 15—20 мин. до
кипения и нагревание и перемешивание продолжают еще 5 час.
Смесь охлаждают до комнатной температуры, колбу на две трети
наполняют водой и перемешивают 5 мин. Ксилольный раствор
отделяют, промывают водой, сушат хлористым кальцием, филь-
труют и перегоняют в вакууме. После отгонки растворителя при
128—142°/1?> мм перегоняется трикарбометоксиметан. Выход неочи-
щенного продукта 50—51 г E0—51% теоретич.). При охлаждении
продукт становится полутвердым.
Для очистки полученного продукта его растворяют в равном
объеме метилового спирта и раствор охлаждают в охладительной
смеси до тех пор, пока при охлаждении маточного раствора не будут
больше выделяться кристаллы. Кристаллы отсасывают и маточный
раствор снова охлаждают в охладительной смеси. В случае необ-
ходимости эту операцию повторяют в третий раз или до тех пор,
пока при охлаждении маточного раствора не прекратится выделение
новой порции кристаллов. Кристаллы переносят с воронки в ста-
кан, размешивают с 70 мл петролейного эфира (т. кип. 32—45°),
фильтруют, отсасывают досуха и промывают небольшим количе-
ством петролейного эфира. Выход мелких белоснежных кристал-
лов, плавящихся при 43—45°, около 40—42 г D0—42% теоретич.;
примечание 2).
2. Примечания
1. Происходит энергичное выделение водорода, и натриевое
соединение диметилового эфира малоновой кислоты выпадает в
472
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
ТРЦКЛРБЭТОКСИМЕТАН
473
виде густой пасты. Для того, чтобы не дать натриевой соли превра-
титься в комки, необходимо хорошее перемешивание как во время
прибавления малонового эфира, так и во время последующего
нагревания.
2. Трикарбометоксимстан очень легко растворим в метиловом
спирте, и только при многократном охлаждении и отсасывании можно
добиться хорошего выхода. Благодаря чрезвычайно большой раство-
римости при перекристаллизации происходит большая кажущаяся
потеря. Однако значительную часть теряющегося продукта можно
получить обратно из спиртового маточного раствора.
3. Другие методы получения
Описанный метод очень похож на метод, который опубликовали
Адикес, Бруннерт и Люкер 1 для получения триэтилового эфира.
Согласно другим способам, натриевую соль диметилового эфира
малоновой кислоты выделяют 2. Ср. также «Другие методы полу-
чения» трикарбэтоксиметана (стр. 474).
1 A d i с k e s, Brunnert, Lticker, J. prakt. Chem. B) 130, 163
A931).
- Scholl.Egerer, Ann. 397, 355 A913); Philippi, Hanusch,
W а с e k, Ber. 54, 901 A921); Backer, Lolkelma, Rec. trav. chim. 57,
1237 A938).
ТРИКАРБЭТОКСИМЕТАН
(Этиловый эфир метантрикарбоновой кислоты)
СНа (СО2С2Н5J + Mg + С2Н5ОН - C3H5OMgCH (СО2СаН5J + Н,
C2HBOMg СН (СО2СаН5J + С1СО2С2Н8- ClMgC (CO2C2H6K + С,Н5ОН
CIMgC (СО,С3Н5)з + СН3СО2Н СН (СО2С2Н3)з + СН3СО2 MgCl
Предложили: X. Лунд и А. Фойгт.
Проверили: В. Каротерс и В. Мак-Ювен.
1. Получение
В 1-литровую круглодонную колбу, снабженную эффективным
обратным холодильником с неслишком узкой внутренней трубкой,
помещают 25 г A,03 гр.-ат.) магниевых стружек, подготовленных
как для реакции гриньяра, 25 мл абсолютного спирта (примечание 1),
1 мл четыреххлористого углерода (примечание 2) и 30 мл смеси,
состоящей из 160 г A51 мл, 1 мол.) малонового эфира и 80 мл абсо-
лютного спирта. Подготовляют холодную воду для охлаждения
колбы, если это понадобится, и смесь осторожно нагревают до
начала выделения водорода. Реакция может пойти настолько бурно,
что будет необходимо произвести охлаждение колбы снаружи. Через
холодильник постепенно приливают малоновый эфир, причем
следят за тем, чтобы реакция протекала энергично, но все же подда-
валась регулированию. Когда реакция становится более умерен-
ной, колбу охлаждают и добавляют через холодильник 300 мл
эфира, который предварительно простоял 24 часа над хлористым
калбцием. Колбу осторожно нагревают, выпавшие сперва кристаллы
растворяются, и некоторое время вновь выделяется водород без
внешнего подогревания. Реакцию заканчивают на паровой бане,
затем баню удаляют и через холодильник приливают с помощью
капельной воронки 100 мл A,05 мол.) хлормуравьиного эфира и
100 мл абсолютного эфира с такой скоростью, чтобы жидкость
энергично кипела в течение всего времени прибавления смеси
(примечание 3). Колбу нагревают 15 мин. на паровой бане, после
чего реакцию считают законченной.
Образовавшееся вязкое магнийорганическое соединение осто-
рожно разлагают разбавленной уксусной кислотой G5 мл в 300 мл
воды), причем колбу охлаждают под краном проточной водой.
Образуются два прозрачных слоя, по разделении которых водный
слой экстрагируют 100 мл эфира; соединенные эфирные растворы
промывают водой, сушат сернокислым натрием, эфир отгоняют
на паровой бане и остаток перегоняют в вакууме. После неболь-
шого головного погона температура быстро повышается до 130°
при 10 мм и начинает перегоняться чистый трикарбэтоксиметан.
Выход продукта, который выкипает в пределах 5°, составляет
204—215 г (88—93% теоретич.). Продукт затвердевает при 25°.
Т. пл. чистого вещества 28—29Э.
2. Примечания
1. Предпочтительнее работать с совершенно безводным спир-
том, однако допустимо и применение высокосортного продажного
абсолютного спирта, что не ведет к заметному уменьшению выхода.
Авторы пользовались спиртом, который был обезвожен магнием 1.
2. Четыреххлористый углерод (так же как и ряд других галоид-
ных соединений) значительно ускоряет реакцию между магнием и
спиртом. Если применять безводный спирт, то реакция начинается
через некоторое время и без нагревания, если же работать с
99,5%-ным спиртом, его необходимо нагреть почти до температуры
кипения, прежде чем выделение водорода станет достаточно энер-
гичным.
3. К концу реакции магниевое соединение этилового эфира
метантрикарбоновой кислоты выпадает в виде вязкой массы, кото-
рая мешает реакции между оставшимся магниймалоновым эфиром
и хлормуравьиным эфиром. Энергично кипятя смесь, мешают
образованию компактной массы. В случае работы с большими
количествами в этой стадии процесса желательно применять пере-
мешивание. .
474
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
3. Другие лгетоды получения
Трикарбэтоксиметан был получен действием этилового эфира
хлормуравьиной кислоты на натриймалоновый эфир в бензоль-
ной 2. толуольной или ксилольной3 суспензии; перегонкой эти-
лового эфира этоксалилмалоновой кислоты 4, из этилового эфира
угольной кислоты и малонового эфира 5 и по методике, которая
описана выше в.
По данным Беккера и Лолкельма, ароматические эфиры метан-
трикарбоновой кислоты, а также этиловый эфир этой кислоты
лучше всего получаются по методу Лунда, в котором применяется
магний; другие алифатические эфиры метантрикарбоновой кислоты
получаются лучше, если применять натрий (ср. синтез трикарбоме-
токсиметана), причем в качестве среды для реакции применяют
толуол или ксилол.
1 Lund, В j e r r u m, Ber. 64, 210 A931).
2 Conrad, Quthzeit, Ann. 214, 32 A882).
3 Backer, Lolkelma, Rec. trav. chim. 57, 1237A938).
4 В о u v e a u 1 t, Bull. soc. chim. C) 19, 79 A898); Scholl, Egerer
Ann. 397, 353 A913).
5 Wallingford, Homeyer, Jones, J. Am. Chem. Soc. 63, 2056
A941).
«Lund, Ber. 67, 938 A934).
/-ТРИПТОФАН
(Побочный продукт: l-тирозин)
Гидролиз казеина >
(-ТРИПТОФАН
475
ССНХН (NH2) ССШ и
сн
N
I
Н
я-НОСвН4СН2СН (NHa) CO2H
Предложили: Дж. Кокс и Г. Кинг.
Проверили: X. Кларк и Дж. Леланд.
1. Получение
А. Триптофан. В 8-литровую бутыль помещают 600 г продаж-
ного казеина (в виде крупного порошка) и 3,2 л водопроводной
воды, нагретой до 37° (примечание 1). Бутыль встряхивают до
тех пор, пока весь казеин не увлажнится. После этого к смеси
прибавляют сперва раствор 60 г безводной соды (примечание 2)
и б г фтористого натрия (примечание 3) в 1 л воды C7°), а затем
полужидкую пасту из 20 г продажного панкреатина и 100 мл воды
C7°). Смесь покрывают слоем толуола (80 мл), разбавляют до б л.
закупоривают, хорошо встряхивают и ставят в теплую комнату
или в термостат при 37°.
Через 4—5 дней, в продолжение которых бутыль ежедневно
встряхивают, большая часть казеина переходит в раствор и начи-
нает выделяться тирозин. По истечении 5 дней прибавляют вторую
порцию смеси из 20 г панкреатина и 100 мл воды. Через 12 дней
бутыль ставят на ночь в ледяной шкаф для охлаждения, после
чего отфильтровывают осадок (примечание 4) и сохраняют его для
получения тирозина.
Фильтрат F,9—7 л) отмеряют в 16-литровый глиняный сосуд
и прибавляют на каждый литр 163 мл разбавленной холодной серной
кислоты (один объем 95%-ной серной кислоты на один объем воды).
Первую порцию кислоты следует прибавлять осторожно, так как
при этом выделяется углекислота.
Затем триптофан осаждают прибавлением раствора 200 г серно-
кислой окиси ртути (примечание 5) в смеси I860 мл воды и 140 мл
концентрированной 95%-ной серной кислоты. Реакционную массу
оставляют стоять в течение 24—48 часов, после чего при помощи
сифона удаляют прозрачную жидкость, отфильтровывают желтый
осадок и промывают его (примечание 6) раствором 100 мл концен-
трированной серной кислоты и 20 г сернокислой ртути в 1,9 л дестил-
лированной воды до тех пор, пока фильтрат не станет бесцветным
и не перестанет давать характерной окраски с реактивом Милло-
на (примечание 7). Для этого необходимо около 1,5 л раствора.
Затем осадок промывают последовательно тремя порциями дестил-
лированной воды по 500 мл для удаления большей части серной
кислоты.
Влажный остаток A20—130 г) суспендируют посредством меха-
нического перемешивания в 1,2—1,3 л дестиллированной воды и
прибавляют горячий 20%-ный водный раствор гидрата окиси
бария до тех пор, пока смесь не станет и не будет оставаться щелоч-
ной цо фенолфталеину (на что требуется около 120 мл). Затем при
перемешивании пропускают быстрый ток сероводорода, пока ртуть
полностью не будет осаждена (примечание 8). Осадок отфильтровы-
вают и промывают водой до тех пор, пока проба фильтрата не пере-
станет давать с бромной водой реакцию на триптофан (примеча-
ние 9). Из соединенных фильтратов удаляют барий прибавлением
точно эквивалентного количества разбавленной серной кислоты (при-
мечание 10) и отфильтровыванием выпавшего сернокислого бария.
Затем фильтрат выпаривают в вакууме приблизительно до 80 мл.
Для того, чтобы извлечь триптофан из водного раствора, послед-'
ний несколько раз взбалтывают в делительной воронке с н.-бути-
ловым спиртом, которого берут каждый раз по 25 мл, при этом
время от времени прибавляют воду, чтобы объем водного раствора
оставался приблизительно постоянным (примечание 11). Спиртовые
вытяжки упаривают в вакууме. После того, как вся вода отогнана,
476
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
/-ТРИПТОФАН
477
в перегонной колбе из раствора выпадает триптофан, что может
вызвать бурное вскипание. Когда вода полностью удалена, что
устанавливают по прекращению образования капель воды в трубке
холодильника, перегонку прекращают; по охлаждении триптофан
отсасывают и промывают небольшим количеством свежего бутило-
вого спирта. Извлечение бутиловым спиртом и перегонку повторяют
многократно до тех пор, пока выделяемое количество триптофана
не будет настолько незначительным, что им можно пренебречь
(примечание 11).
Приготовленный таким способом триптофан (всего 7—8 г) не
всегда получается одинакового качества. Для очистки его раство-
ряют в 60 мл разбавленного спирта (два объема 95%-ного спирта
на один объем воды), отфильтровывают из горячего раствора зна-
чительное количество нерастворимой примеси, осадок вновь извле-
кают 10 мл кипящего разбавленного спирта и соединенные филь-
траты обесцвечивают 1 г активированного угля. Полученный рас-
твор фильтруют и оставляют стоять в ледяном шкафу. Выпавшие
серебристые листочки триптофана отфильтровывают и промывают
последовательно холодным 70, 80 и 95%-ным спиртом и наконец
небольшим количеством эфира. При каждой перекристаллиза-
ции получают менее половины взятого триптофана (примечание 12).
Выход чистого триптофана (примечание 13) 4,0—4,1 г; кроме того
получается меньше 0,1 г продукта, содержащего примеси.
Б. Тирозин. Нерастворимое вещество A60—170 г), полученное
при фильтровании гидролизованиого раствора казеина (стр. 475),
суспендируют в 320 мл воды с добавлением 80 мл концентрирован-
ной 36%-ной соляной кислоты и смесь осторожно кипятят в тече-
ние 30 мин. (примечание 14) и фильтруют через плотную ткань.
Фильтрат обесцвечивают б г активированного угля (примечание 15)
и снова фильтруют горячим (примечание 16). Теплый F0—70°)
раствор взбалтывают с тремя порциями бензола по 20 мл (приме-
чание 17) и затем нагревают до кипения (примечание 18). После
этого осторожно прибавляют небольшой избыток A20—150 мл)
28%-ного аммиака и оставляют раствор в ледяном шкафу на ночь.
Выпавший кристаллический продукт отфильтровывают и промы-
вают тремя порциями ледяной воды по 40 мл. Вес высушенного
продукта около 23 г. Маточный раствор и промывные воды выпа-
ривают приблизительно до 200 мл и получают еще около 1 г веще-
ства.
С целью очистки сырой тирозин суспендируют в 400 мл воды и
.переводят его в раствор, прибавляя раствор 8 г едкого натра в
20—30 мл воды (примечание 19). Полученный раствор обесцвечи-
вают 2 г активированного березового угля, фильтруют и промы-
вают уголь на воронке 20—30 мл горячей дестиллированной воды.
Соединенные фильтраты нагревают до кипения (примечание 18)
и прибавляют 13 мл соляной кислоты (примечание 20); при этом
обыкновенно начинается кристаллизация. Затем раствор подкисляют
(на лакмус) уксусной кислотой (примечание 21) и оставляют стоять
на ночь в ледяном шкафу. Выпавший тирозин отфильтровывают и
промывают охлажденной до 0° дестиллированной водой A30—150 мл)
дб тех пор, пока промывные воды не перестанут давать реакцию на
ион хлора. Промытый и отжатый продукт сушат на воздухе или
в вакуумсушилке. Выход 17,0—18,2 г бесцветных шелковистых игл
тирозина. Дополнительное количество (около 0,5 г) несколько
менее чистого продукта можно получить концентрированием маточ-
ного раствора до объема приблизительно в 120 мм.
2. Примечания
1. Трипсин действует быстрее, если вода нагрета до 37°. Дестил-
лированная вода в данном случае не нужна.
2. Сода взята с большим избытком, можно взять и меньшее коли-
чество .
3. Фтористый натрий, повидимому, препятствует действию
оксида з.
4. Это фильтрование может протекать медленно. Удобнее всего
вести фильтрование в воронке Бюхнера диаметром 20 см. Осадок
отсасывают досуха после каждого наполнения и затем меняют филь-
тровальную бумагу.
5. Требуется приблизительно такое количество сернокислой
окиси ртути, чтобы полностью осадить триптофан, что устанавли-
вается пробой с глиоксиловой кислотой по Гипкинсу-Колу.
6. Промывание производят для того, чтобы удалить тирозин,
осажденный в виде ртутного соединения, которое несколько более
растворимо, чем соединение триптофана. Добавление сернокислой
окиси ртути ведет к уменьшению растворимости триптофана.
7. Фильтраты даже после полного удаления тирозина продол-
жают давать красную окраску с реактивом Миллона, но по оттенку
она заметно отличается от первоначальной, вызванной тирозином.
8. Раствор после некоторого стояния должен содержать избыток
сероводорода. Проба фильтрата должна давать после подкисления
уксусной кислотой обильный черный осадок с уксуснокислым
свинцом.
9. Реакция с бромной водой в данных условиях более чувстви-
тельна, чем с глиоксиловой кислотой. Так как бром реагирует
с сероводородом с выделением серы, его предварительно следует
удалить кипячением. Испытываемый раствор должен быть под-
кислен уксусной кислотой.
10. Это количество лучше всего определить титрованием алик-
вотной части раствора B0 мл) 2%-ной титрованной серной кислоты.
11. При проверке оказалось целесообразным экстрагировать
водный раствор в непрерывно действующем аппарате (рис. 19).
478
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
riuui.-ТРИТИАН
479
Экстрагирование продолжают до тех пор, пока жидкость в колбе
не начнет кипеть толчками вследствие выпадения твердого осадка;
тогда берут новую порцию н.-бутилового спирта; около пяти порций
достаточно для полного извлечения. Этот процесс повторяют до
тех пор, пока остаток после 3 часов экстрагирования не перестанет
давать с бромной водой красного окрашивания, характерного для
триптофана. Длительность экстрагирования, естественно, зависит
от интенсивности кипячения; при про-
Jc= насосу верке способа на эту операцию затра-
ш чивалось 28—30 час.
12. Перекристаллизация сырого трип-
тофана является очень трудной опера-
цией. Приходится удалять не только
менее растворимый побочный продукт,
но и значительные количества более
растворимых клейких примесей, остаю-
щихся в маточных растворах. Каждый
раз после отсасывания триптофана ма-
точный раствор следует выпарить до
небольшого объема в вакууме на водя-
ной бане и остаток обработать двойным
объемом спирта. Этот процесс повторя-
ют до тех пор, пока не будут больше
получаться кристаллы, а станет выпа-
дать лишь одна клейкая масса.
13. Чистота триптофана была провере-
на определением вращения ([а]с=от
—28 до — 33°) и определением аминного
азота F,8—6,9) по методу Ван-Сляйка.
Рис. 19.
14. Кипящий раствор кислоты гидролизует протеины, присут-
ствие которых весьма замедляет фильтрование.
15. Активированный древесный уголь не обесцвечивает раствора
полностью, но благодаря этой обработке конечный продукт полу-
чается бесцветным.
16. Фильтрование при очистке тирозина во всех случаях, за
исключением, быть может, последнего, лучше всего производить
в воронке Бюхнера (диаметр 20 см). При применении активирован-
ного древесного угля для получения прозрачного фильтрата перед
фильтрованием можно прибавлять кизельгур.
17. Бензол извлекает следы вещества (по всей вероятности,
жирной кислоты), которое замедляет фильтрование и присут-
ствие которого сильно ухудшает качество конечного про-
дукта.
18. Тирозин кристаллизуется в виде длинных шелковистых
иголок, легче отсасывающихся, если раствор нейтрализовать при
температуре кипения.
19. Небольшое количество хлопьевидных примесей остается
нерастворимым.
20. Осаждение ведут соляной кислотой, чтобы по присутствию
ионов хлора в промывных водах судить о степени промывки.
21. Тирозин очень мало растворим в водных растворах уксус-
ной кислоты, и потому избыток ее не ведет к уменьшению выхода.
3. Другие методы получения
Вышеописанный способ получения триптофана основан на
методах Хопкинса и Кола г, Дэкина 2 и Онслоу 3.
Тирозин в качестве первичного продукта можно легко полу-
чить гидролизом шелка соляной кислотой, нейтрализацией полу-
ченного раствора едким натром и последующим подкислением уксус-
ной кислотой.
1 Н о р k i n s, С о 1 е, J. Physiol. 27, 418 A902).
2 D а к i n, Biochem. J. 12, 302 A918).
3 О n s 1 о w, там же 15, 392 A921).
симм. -ТРИТИАН
( Т ритиоформальдегид )
/СГ1о Оч
3 СН2О
3H,S -<Н^ S
3 Н2О
"СН,-
Предложили: Р. Бост и Е. Констэбль.
Проверили: Р. Фъюзон и Ч. Вудвард.
1. Получение
Смесь, состоящую из 326 г C,9 мол.) 36%-ного формалина (при-
мечание 1) и 700 мл концентрированной соляной кислоты (уд. в.
1,18), помещают в высокий стеклянный
цилиндр (примечание 2) и через раствор
пропускают сероводород до тех пор, пока
не прекратится образование осадка. С
целью облегчения процесса образующийся
осадок время от времени отфильтровывают.
Необходимое для завершения реакции вре-
мя колеблется от 12 до 24 час. Получают
176 г (98% теоретич.) сырого продукта,
представляющего собой почти бесцветные
иглы с т. пл. 210—213°.
Продукт подвергают очистке с помощью
метода обратного фильтрования. Прибор
показан на рис. 20. 2-литровая круглодонная колба снабжена
обратным холодильником и согнутой стеклянной трубкой диаметром
Рис.
480
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
ТРИФЕНИЛМЕТИЛНАТРИЙ
481
8—10 мм (примечание 3)- К нижнему концу этой трубки при-
соединен с помощью корковой пробки наконечник диаметром
25 мм, сделанный из гильзы для аппарата Сокслета и наполненный
стеклянной ватой. В качестве приемника для горячего фильтрата
служит 2-литровая коническая колба.
Сырой продукт помещают в круглодонную колбу, добавляют
1 л бензола и колбу нагревают до энергичного кипения раствори-
теля. Через несколько минут источник нагревания удаляют и
смеси дают успокоиться. Фильтровальную гильзу, которая до
этого момента находилась в верхней части колбы (рис. 20), опускают
в нормальное положение и присоединяют коническую колбу. Путем
приложения небольшого вакуума жидкость пересасывается в кони-
ческую колбу. Горячий раствор удаляют, дают ему охладиться и
фильтруют. В это время процесс экстрагирования повторяют со
второй 1-литровой порцией бензола. Обе порции бензола приме-
няют попеременно так, как это описано выше, до тех пор, пока
не будет перекристаллизован весь сырой продукт. Обычно бывает
достаточно 10 экстрагирований; таким образом каждая 1-литровая
порция бензола оборачивается 5 раз.
Выход чистого продукта ст. пл. 214—215°составляет 165—169 г
(92—94% теоретич.).
2. Примечания
I. Принимая во внимание, что продажный формалин содержит
метиловый спирт, количество формальдегида, определенное по
таблицам удельных весов, неточно (ср. «Синт. орг. преп.», сб. 1,
стр. 279). Содержание формальдегида в формалине определяют
посредством анализа. Рекомендуется воспользоваться для этого
иодометрическим методом Боргстрома и Хорша х. Выход вычисляют
на основе действительного содержания формальдегида согласно ана-
лизу.
2. Высокий цилиндрический сосуд обеспечивает хорошее сопри-
косновение раствора с пробулькивающим через него газом.
Сероводород брался из баллона.
3. Для успешного применения метода обратного фильтрования
необходимо обратить внимание на следующие детали: а) соедини-
тельная трубка должна быть достаточно широкой; б) длина трубки
между колбами должна быть как можно меньше; в) горячий раствор
должен пересасываться быстро; г) следует избегать слишком боль-
шого вакуума.
Прибор для обратного фильтрования весьма удобен для про-
стых перекристаллизации, а также для повторных экстрагирований.
Он особенно удобен для работы с летучими и легко воспламеняю-
щимися растворителями, а также с растворами, которые обладают
лакриматорным действием.
3. Другие методы получения
сллш.-Тритиан был получен действием цинка и соляной кис-
лоты на сероуглерод2, этиловый эфир изотиоциановой кислоты3
или калиевую соль тиоциановой кислоты 3, нагреванием йодистого
метилена со спиртовым раствором кислого сернистого натрия *
и действием на водный раствор формальдегида сероводорода и кон-
центрированной соляной кислоты6.
1 В о г е s t г о in, Horsch, J. Am. Chem. Soc. 45, 1493 A923).
2 Oirard, Compt. rend. 43, 396 A856); Ann. 100, 306 A856).
3 Hofmann, Ber. 1, 176, 179 A868).
« H u s e m a n n, Ann. 126, 293 A863).
Б Hofmann, там же 145, 360 A868); В a u m a n n, Ber. 23, 67 A890).
ТРИФЕНИЛМЕТИЛНАТРИЙ
(CeH3K CC1 + 2 NaHg > Na [C (C6H5K] 4- NaCl 4- 2 Hg
Предложили: В. Ренфроу мл. и Ч. Хаузер.
Проверили: Л. Смит и Е. Баллард.
1. Получение
В 2-литровой склянке с хорошо притертой пробкой пригото-
вляют раствор 63 г @,226 мол.) чистого трифенилхлорметана (при-
мечание 1) в 1,5 л чистого абсолютного эфира и к раствору доба-
вляют 1150 г свежеприготовленной 1%-ной амальгамы натрия
@,5 гр.-ат. натрия; примечание 2). Пробку смазывают небольшим
количеством вакуумной смазки и склянку закупоривают герме-
тично. Затем склянку помещают на качалку для взбалтывания,
хорошо ее укрепляют и энергично взбалтывают. Реакция весьма
экзотермична, и во время взбалтывания склянку охлаждают
мокрыми полотенцами. Вскоре, обычно через 10 мин., появляется
постоянная кроваво-красная окраска. После трехчасового взбал-
тывания склянку охлеакдают до комнатной температуры, снимают
с качалки и оставляют спокойно стоять до тех пор, пока не
осядет хлористый натрий (примечание 3).
Эфирный раствор трифенилметилнатрия отделяют от хлори-
стого натрия и от амальгамы следующим образом. Пробку выни-
мают из реакционной склянки и немедленно заменяют ее плотно при-
гнанной пробкой с двумя отверстиями, в которые вставлены короткая
стеклянная трубка, формы перевернутой U-образной трубки. Корот-
кую трубку присоединяют через осушительную систему к баллону
с азотом. Один конец U-образной трубки оканчивается па 4 ел выше
дна склянки, другой конец заканчивается как раз под пробкой
с двумя отверстиями, вставленной плотно в 2-литровую кониче-
31 Сборник № 2
482
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
скую колбу. Колбу наполняют азотом и она служит приемником
для декантируемого раствора. Во второе отверстие в пробке кониче-
ской колбы вставляют делительную воронку с короткой ножкой,
которая служит для выпуска азота во время декантации, а затем
может быть использована для последующего введения реагентов
(примечание 4). Пробки покрывают слоем парафина. Кран дели-
тельной воронки слегка приоткрывают, и эфирный раствор трифе-
иилметилнатрия медленно и непрерывно передавливают в колбу,
наполненную азотом, давая небольшое давление азота из баллона.
Если стеклянная трубка установлена на правильной высоте над
поверхностью хлористого натрия и амальгамы, из склянки можно
передавить все количество эфирного раствора за исключением 50—
75 мл.
В случае применения трифенилхлорметана хорошего качества
и свежеприготовленной амальгамы, выход трифенилметилнатрия
почти количественный. Раствор можно проанализировать прибли-
женно следующим образом: из известного объема берут 50 мл и
приливают их к 25 мл воды, налитой в делительную воронку. Вод-
ный слой сливают и эфирный раствор промывают три раза водой
порциями по 25 мл. Водные растворы соединяют вместе, кипятят
для удаления эфира и титруют 0,2-н. серной кислотой, применяя
в качестве индикатора метилрот.
2. Примечания
1. Следует взять трифенилхлорметан хорошего качества с
т. пл. 112—113°. Продажный продукт можно перекристаллизовать
из смеси 5 частей лигроина (т. кип. 90—110°) и 1 части хлористого
ацетила, применяя около 1,8 г растворителя на 1 г продукта. Ука-
зания для получения трифенилхлорметана можно найти в 23 вы-
пуске Org. Syn.
2. Для приготовления 1%-ной амальгамы натрия 11,5 г
@,5 rp.-ат.) натрия нарезают небольшими кубиками высотою в
5 мм. и растворяют его в 1150 г очищенной ртути, прибавляя по
одному кусочку натрия. Ртуть находится в широкогорлой кониче-
ской колбе емкостью 500 мл, а каждый кусочек натрия накалы-
вается на острый конец длинной стеклянной палочки и погружается
в ртуть, под ее поверхность. В результате бурно протекающей
реакции маленькие кусочки натрия могут разбрызгиваться. По-
этому стеклянную палочку пропускают через кусок толстого кар-
тона, который во время реакции покрывает горло колбы.
3. Если применять твердую 3%-ную амальгаму натрия, то
можно получить растворы, содержащие почти 0,67 мол. трифенил-
метилиатрия на 1 л, т. е. примерно в пять раз более концентриро-
ванные, чем получаемые согласно приведенной выше методике.
ТРИФЕНИЛЭТИЛЕН
483
1,7 кг 3%-ной амальгамы натрия B,2 гр.-ат. натрия) получают
прибавлением 1649 г ртути к 51 г натрия расплавленного под мине-
ральным маслом (ср. «Синт. орг. преп.», сб. 1, стр. 235, примеча-
ние 1). В еще горячем состоянии амальгаму выливают в большую
фарфоровую чашку и дают ей охладиться. Затем ее переносят в
большую железную ступку, измельчают на куски размером 1 мм,
отмывают от масла лигроином и помещают в 2-литровую склянку
с притертой пробкой. Прибавляют 1,5 л абсолютного эфира и 278 г
A мол.) трифенилхлорметана, склянку закупоривают стеклянной
пробкой, смазанной вакуумной смазкой, и склянку взбалтывают
на качалке до тех пор, пока не останется более твердой амальгамы,
а затем еще в течение 2 час. Если ход качалки 10—12 см и качалка
делает 3—4 качания в секунду, суммарное время взбалтывания
на ^качалке составляет 5—8 час. При менее эффективно работаю-
щей качалке требуется больше времени. Реакция протекает доста-
точно медленно, так что охлаждения мокрыми полотенцами не
требуется. Далее продукт реакции обрабатывается так, как это
описано на стр. 481 (частное сообщение Ч. Хаузера и Б. Худ-
зона мл. Проверили Л. Смит и Р. Лиггетт).
4. Трифенилметилнатрий полезный реактив для получения на-
триевых производных очень слабых кислот (алифатических слож-
ных эфиров, ангидридов кислот и т. п.). Применение этой методики
на примере этилового эфира изомасляной кислоты описано на
стр. 582.
3. Другие методы получения
Описанная выше методика является видоизменением метода
Шленка и Окса *.
'Schlenk, Ochs, Ber. 49, 608A916).
ТРИФЕНИЛЭТИЛЕН
CeH5CH#gCl + (СвН5J СО
(СвН5J С (OMgCl) СН2СвН6
(С6Н5JС (OMg С1) СН2С6Н5 + Н2О
(СвН5J С (ОН) CH2CeH5 + Mg (ОН) С1
(С6Н5J С (ОН) CH2C6HS -^4-t (C6H5JC = CHC6H5 + Н2О
Предложили: Г. Адкинс и В. Цартман.
Проверили: К- Ноллер и Ф. Мак-Миллан.
1. Получение
В 3-литровую колбу с тремя горлами, снабженную механической
мешалкой, обратным холодильником и делительной воронкой,
помещают 24,3 г A rp.-ат.) магниевых стружек, 500 мл абсолютного
эфира, кристаллик иода и небольшую часть E—10 мл) от общего
484
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
количества 126,5 г A15 мл, 1 мол.) свежеперегнанного хлористого
бензила (т. кип. 177—179°). Реакция начинается через несколько
минут (примечание 1), причем в случае надобности ее регулируют,
охлаждая колбу мокрым полотенцем. Пускают в ход мешалку
и приливают остальное количество хлористого бензила с макси-
мальной скоростью, какую только допускает стекание эфира из
обратного холодильника. На прибавление хлористого бензила
требуется от 1 до 2 час, после чего смесь кипятят на паровой бане,
при работающей мешалке, еще 3 часа и, не останавливая мешалки,
приливают 182 г A мол.) бензофенона («Синт. орг. преп.», сб. 1,
стр. 99), растворенных в 500 мл абсолютного эфира с такой ско-
ростью, что смесь энергично кипит. На это требуется около 20 мин.,
после чего реакционную смесь оставляют стоять 2 часа (примеча-
ние 2).
Колбу помещают в баню со льдом, добавляют внутрь 700 г
колотого льда и, чтобы растворить гидроокись магния, приливают
500 мл холодной 20%-ной серной кислоты. Эфирный слой отделяют,
водный слой экстрагируют два раза эфиром порциями по 200 мл.
Эфирные вытяжки и раствор соединяют вместе, эфир отгоняют и
остаток кипятят в течение 2 час. с обратным холодильником со
100 мл 20%-ной серной кислоты для того, чтобы дегидратировать
карбинол. Продукт реакции отделяют от водного слоя и подвер-
гают его перегонке в вакууме (примечание 3). Собирают фракцию
215—225°/15 мм, которая весит 160—170 г. Продукт этот плавится
при 60—68° (примечание 4). Путем перекристаллизации из 900 мл
горячего 95%-ного спирта с последующим охлаждением до 0°
получают 140—150 г углеводорода с т. пл. 68—69° E4—59% тсо-
ретич.). Если маточный раствор упарить до 150 мл и охладить, то
можно получить еще 5—10 г вещества с т. пл. 65—67,5° (приме-
чание 5).
2. Примечания
1. Если через 30 мин. реакция все еще не началась, то при
работающей мешалке колбу начинают нагревать на водяной
бане.
2. Если реакционную смесь оставить стоять на ночь, то полу-
чаются несколько лучшие результаты.
3. Продукт не следует промывать водой, так как, повидимому,
присутствие следов серной кислоты при перегонке необходимо для
того, чтобы завершить дегидратацию.
4. Для того чтобы вызвать кристаллизацию, обыкновенно при-
ходится внести затравку.
5. Пользуясь той же методикой, можно получить стильбен с
выходом 25—35%, исходя из бензальдегида и хлористого бензил-
магния.
ТРИХЛОРЭТИЛОВЫЙ СПИРТ
485
3. Другие методы получения
Трифенилэтилен был получен реакцией бромистого фенилмаг-
ния с дезокеибензоином или с этиловым эфиром фенилуксусной
кислоты 1, а также реакцией дифенилкетенхинолина с бензальде-
гидом2. Описанная выше методика заимствована в основному
Гелля и Виганда 3. Образующийся в качестве промежуточного про-
дукта дифенилбензилкарбинол можно с успехом дегидратировать,
если его нагреть с бисульфитом калия или же если его раствор
кипятить в ледяной уксусной кислоте 4.
lKlages, Heilmann, Ber. 37, 1455 A904).
sStaudinger, Kon, Ann. 384, 89 A911).
3Hell, Wiegandt, Ber. 37, 1429 A904).
•Van de Kamp, Sletzinger, J. Am. Chem. Soc. 63, 1880
A941).
ТРИХЛОРЭТИЛОВЫЙ СПИРТ
( 2-Трихлорэтанол)
3CC13CHO + Al (OCH2CH3K
2 Al (OCH2CC13)S + 3 H2SO4
3CH3 CHO
Ala (SO4K
Al (OCH2CC18)8
6CC13 CH,OH
Предложил: У. Чалмерс.
Проверили: Р. Фьтон и X. Хулпи.
1. Получение
В погруженную в масляную баню 2-литровую колбу с тремя
горлами помещают 250 г A,7 мол.) безводного хлораля, 650 мл
абсолютного спирта (примечание 1)и75г @,46 мол.) чистого этилата
алюминия (примечание 2). В центральном горле колбы устанав-
ливают на корковой пробке эффективный дефлегматор (примеча-
ние 3), а в одно из боковых отверстий вставляют трубку, доходя-
щую до дна колбы, для подачи сухого азота (примечание 4). Третье
горло герметично закрывают пробкой; оно служит для отбора проб
жидкости.
К отводной трубке дефлегматора присоединяют U-образную
трубку и склянку Тищенко емкостью по 100 мл каждая. U-образную
трубку погружают в охладительную смесь, а в склянку Тищенко
наливают насыщенный раствор бисульфита натрия. После того
как все это сделано, масляную баню нагревают до 135° и начи-
нают пропускать слабый ток газа. Смесь энергично кипит, при этом
спирт стекает обратно в колбу, а ацетальдегид, образующийся
в результате реакции, собирается в охладительной смеси и в склянке
Тищенко.
486
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
Для определения конца реакции отбирают в пробирку несколько
капель реакционной смеси и обрабатывают их водой. После
осаждения гидрата окиси алюминия прозрачную жидкость декан-
тируют и добавляют к ней несколько капель желтого сернистого
аммония. Если остались еще хотя бы следы хлораля, то при нагрева-
нии до начала кипения появится темнобурое окрашивание. Если
удалять жидкость, накопляющуюся в и-образной трубке через
каждые несколько часов, то конец реакции можно легко заметить по
уменьшению количества собирающегося в ней ацетальдегида.
После 23—24-часового нагревания (на протяжении 2—3 дней)
реакция обычно заканчивается. Температуре в бане дают понизиться
до 120° и спирт отгоняют через обычный холодильник, который
присоединяют вместо дефлегматора. После того как остаток три-
хлорэтилата алюминия станет почти сухим (примечание 5), колбу
удаляют с масляной бани, твердую массу обрабатывают 250 мл
20%-ной серной кислоты и тщательно перемешивают для того,
чтобы обеспечить полное разложение.
Смесь подвергают перегонке с водяным паром до тех пор, пока
не перейдет весь трихлорэтиловый спирт. При этом получают около
4 л дестиллата (примечание 6). Маслянистую жидкость отделяют
отводного слоя, последнийнасыщают сернокислым натрием и извле-
кают тремя порциями эфира по 200 мл. Эфирный раствор добавляют
к главной порции спирта и всё вместе сушат безводным сернокислым
натрием.
Эфир отгоняют и продукт подвергают перегонке в вакууме
(примечание 7). При этом получают 215 г (84% теоретич.)
трихлорэтилового спирта с т. кип. 94—97°/ 125 мм и с т. пл. 16—17°
(примечание 8). Для получения более чистого препарата его вто-
рично перегоняют в вакууме и кристаллы отжимают на охлажденной
пористой тарелке. Температура плавления чистого трихлорэтило-
вого спирта 19° (примечание 9).
2. Примечания
1. Действие небольшого количества этилата алюминия на чистый
хлораль или на хлораль, разбавленный бензолом, ведет к обра-
зованию трихлорэтилтрихлор ацетата совершенно аналогично
тому, как этилацетат образуется из ацетальдегида под действием
этилата алюминия, однако выходы бывают очень небольшими.
Несколько лучшие выходы получаются в том случае, если исход-
ные вещества берутся в молекулярных отношениях, однако хорошие
выходы получаются только в присутствии большого количества
спирта.
2. В настоящее время этилат алюминия производится в промыш-
ленном масштабе. Автор рекомендует для его получения1 следую-
щий проверенный на практике метод:
ТРИХЛОРЭТИЛОВЫЙ СПИРТ
487
в колбу с обратным холодильником помещают 27 частей алю-
миния в стружках или зернах, которые обрабатывают 276 частями
абсолютного спирта, 0,1—'0,25 части хлорной ртути и несколькими
кристаллами иода. Через несколько минут происходит бурное
выделение водорода и после нагревания в течение нескольких часов
на водяной бане этилат получается в виде сероватого порошка.
Не вошедший в реакцию спирт отгоняют на масляной бане до
тех пор, пока оставшаяся масса не расплавится с образованием тем-
ноокрашенной жидкости, которую переливают в колбу Клайзена
и перегоняют в вакууме с коротким воздушным холодильником,
причем в качестве приемника служит склянка для отсасывания из
стекла «пирекс». Ввиду того,что этот этилат алюминия довольно легко
возгоняется, между приемником и вакуум-линией помещают там-
пон из стеклянной ваты, для того чтобы не произошло закупорки.
Перегонку ведут на голом огне, и притом достаточно быстро. Дестил-
лат еще в жидком состоянии переливают в колбу из стекла «пирекс»,
где ему дают охладиться. Он образует твердую белую массу, которую
необходимо сохранять в хорошо закупоренной колбе во избежание
поглощения водяных паров. Выход достигает 90% теоретич.
Для описанной выше реакции, при которой следы воды не меша-
ют, некоторое количество этилата можно расплавить, вылить в ступ-
ку, покрыть часовым стеклом и по охлаждении измельчить и взвесить.
Автор нашел, что по описанному выше методу может быть полу-
чено большое количество этилата даже с 95%-ным спиртом, однако
лучшие результаты получаются, если спирт предварительно проки-
пятить и дважды перегнать над негашеной известью.
3. Этот дефлегматор должен быть таким, чтобы он мог доста-
точно хорошо отделять ацетальдегид от этилового спирта. Весьма
удобным является дефлегматор Гана а, в котором постоянная тем-
пература поддерживается кипящим в ере дней трубе метиловым спир-
том, тогда как пары перегоняющегося вещества идут по внешнему
кольцевому пространству. Необходимо, чтобы нижняя часть приме-
няемого дефлегматора была достаточно широка (диаметр около
2 см), так как в противном случае в дефлегматоре возможно
накопление флегмы.
Колонку, построенную по тому же принципу, можно сделать из
двух лабораторных холодильников. Нижнее отверстие внешней ру-
башки холодильника, присоединенного к колбе, закрывают и рубашку
наполняют на две трети ее высоты метиловым спиртом. Для того
чтобы жидкость не выбрасывало, в нее вводят несколько кусочков
пемзы. Верхний отросток рубашки первого" холодильника соеди-
няют со вторым обратным холодильником для конденсации паров
метилового спирта. Внутреннюю трубку дефлегматора наполняют
стеклянными бусами, которые удерживаются с помощью зубцов,
сделанных в нижней части трубки. Верхний конец этой трубки при-
соединяют к U-образной трубке, погруженной в холодильную смесь.
488
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
Должны быть приняты все меры, чтобы обеспечить полное раз-
деление альдегида и спирта, так как даже небольшая, но непрерыв-
ная потеря растворителя ведет в результате длительного нагревания
к большим потерям. Температура кипения хлораля лежит только на
двадцать градусов выше температуры кипения этилового спирта
и всякая потеря последнего поведет к потере некоторого количества
хлораля.
4. Азот берется из баллона и предварительно сушится, для чего
его пропускают через хлоркальциевую трубку и через трубку с фос-
форным ангидридом, смешанным со стеклянной ватой или бусами.
Между трубкой с фосфорным ангидридом и реакционной колбой
помещают трубку со стеклянной ватой, для того чтобы в случае
сильного тока азота фосфорный ангидрид не попал в реакционную
смесь.
5. Следует соблюдать меры предосторожности, чтобы не пере-
греть остаток, так как в противном случае он может стать очень
плотным и частично осмолиться, а это значительно замедляет дей-
ствие на него кислоты.
6. Из остатка после перегонки с паром можно извлечь эфиром
небольшое количество трихлоралдоля, СС1аСНОНСН2СНО.
7. Продукт может быть перегнан и при обыкновенном давлении
(т. кип. 151°), однако ввиду частичного разложения дестиллат
получается бурого цвета.
8. Даже при простом кипячении с обратным холодильником
без отделения альдегида можно получить выход, равный 65% 3. Реак-
ция между зтилатом алюминия и хлоралем, с одной стороны, и три-
хлорэтилатом алюминия и ацетальдегидом— с другой, является рав-
новесной. Некоторая часть ацетальдегида удаляется из сферы
реакции, конденсируясь в уксусноэтиловый эфир в результате
каталитического действия этилата алюминия, однако в данной
смеси эти изменения протекают медленно. Большое количество
альдегида превращается в паральдегид и ацеталь, но превращения
эти являются обратимыми.
9. Применение этилата алюминия в присутствии спирта являет-
ся удобным методом для восстановления целого ряда соединений,
которые не могут быть восстановлены обычными способами. Меер-
вейн и его сотрудники применили этот метод для превращения бро-
маля в трибромэтиловый спирт и получили также коричный спирт
и различные галоидозамещенные коричные и кротоиовые спирты из
соответственных альдегидов 4>5. Во всех этих случаях получаются
прекрасные выходы.
3. Другие методы получения
Описаный выше метод в основном заимствован у Меервейна и его
сотрудников Шмидта5 и Бока3. Теорию этой реакции и ее
применение рассматривали также Дворжак 4 и Верлей 6. Имеется
ТРИЭТИЛКАРБИНОЛ
489
целый ряд патентов, в которых предлагается использование этих
реакций; в некоторых из них вместо этилового спирта применяется
вторичный спирт, например изопропиловый7. Трихлорэтиловый
спирт является одним из продуктов хлорирования этилового спирта
и содержится в высококипящих фракциях, получаемых при про-
изводстве хлораля 8. Гарцаролли-Турнлак9 и Делакр 10 полу-
чили его действием диэтилцинка на хлораль.
lMeister, Lucius, Bruning, герм. пат. 286 596 [Frril. 12, 29
A914—1916)].
2 Н a h n, Ber. 43, 419 A910): Измененный вид этого прибора описан в
Org. Synt. 20, 27.
8 v. В о с k, Dissertation, Albertus-Universitat, KOnigsberg Pr., Германия,
1926.
1 Dworzak, Monatsh. 47, 11 A926).
5Meerwein, Schmidt, Ann. 444, 233 A925).
6 V e r 1 e y, Bull.' soc. chim. D) 37, 537 A9-5).
7 Bayer & Ко., англ. пат. 235 584 [С. А. 20, 917 A926)]; I. Q. Farbenind.
А.-О., англ. пат. 286 797 [С. А. 23, 395 A929)]; Winthrop Chemical Company;
ам. пат. 1 725 054 [С. А. 23, 4709 A929)].
8 А 1 t s с h u I, Meyer, Bcr. 26, 2758 A893).
9 Garzarolli-Thurnlackh, Ann. 210, 63 A881).
10 D e 1 а с г е, Bull. soc. chim. B) 48, 784 A887).
ТРИЭТИЛКАРБИНОЛ
(З-Этилпентанол-3)
C2H5Br + Mg
3C2H5MgBr + (С2Н5JСО3
* (С,Н5K COMgBr + Н2О
> C2H5MgBr
> (С2Н5K COMgBr + 2C2H3OMgBr
> (С2Н5K СОН + Mg (ОН) Вг
Предложили: В. Мойер и К. Марвел.
Проверили: Ф. Уитмор и Д. Лодер.
1. Получение
В 3-литровую трехгорлую колбу, снабженную механической
мешалкой, делительной воронкой емкостью 500 мл и эффективным
обратным холодильником, закрытым пробкой с хлоркальциевой
трубкой, помещают 107г D,4гр.-ат.) магния в стружках и 800 мл
абсолютного эфира. Начало реакции вызывают прибавлением 5 мл
G г, 0,06 мол.) бромистого этила (примечание 1) без перемешивания.
Затем мешалку пускают в ход и прибавляют раствор 480 г D,4 мол.)
бромистого этила в 1 л абсолютного эфира с такой скоростью, чтобы
эфир сильно кипел, на что требуется около 2час. (примечание 2). После
того как весь бромистый этил прибавлен, реакция практически окон-
чена, но перемешивание следует продолжать еще в течение 15 мин.
400
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
К полученному раствору магнийорганического соединения
прибавляют при энергичном перемешивании в течение около 3 час.
раствор 156 г A,32 мол.) этилового эфира угольной кислоты (приме-
чание 3) в 200 мл эфира. Реакция идет очень бурно, и эфир непре-
рывно кипит. После того как весь эфир угольной кислоты прибав-
лен, реакционную массу при перемешивании нагревают на водяной
бане в течение еще 1 часа.
Затем ее выливают при частом взбалтывании в смесь 1,5 кг коло-
того льда и раствора 300 г хлористого аммония в 600 мл воды,
находящуюся в 5-литровой круглодонной колбе. Отделяют эфир-
ный слой и водный слой экстрагируют двумя порциями эфира
по 500 мл (примечание 4).
От соединенных вытяжек отгоняют эфир, остаток сушат 10 г
безводного поташа и перегоняют при атмосферном давлении, соби-
рая фракцию, кипящую при 139—142 ° (80—90 г). Низкокипящий
дестиллат сушат 5 г безводного поташа, фильтруют и вторично
перегоняют, причем получается вторая порция (около 25 г) три-
этилкарбинола, кипящего при 139—142°. Эту операцию повторяют
еще раз, а в случае необходимости — два раза, и собирают еще 20 г
(примечание 5) алкоголя. Общий выход 125—135 г (82—88% теоре-
тич.; примечание 6). Триэтилкарбинол представляет собою вязкую
жидкость с резким запахом, напоминающим запах камфары.
2. Примечания
1. Бромистый этил сушат хлористым кальцием, а затем перего-
няют над фосфорным ангидридом, собирая фракцию, кипящую при
38—39°. Метод получения бромистого этила приведен в «Синт. орг.
преп.», сб. 1, стр. 113, 117.
2. Время прибавления можно сократить, если охлаждать колбу
снаружи. Вокруг колбы, выше уровня эфира, обматывают сложен-
ное в узкую полосу полотенце и накладывают на верхнюю часть
колбы колотый лед. При таком способе охлаждения значительное
количество паров эфира конденсируется на стенках колбы, без того,
чтобы реакционная смесь сколько-нибудь заметно охлаждалась.
3. Продажный 99%-ный этиловый эфир угольной кислоты
подвергают очистке так, как это описано в примечании 2 на стр. 593.
4. Для экстрагирования можно взять эфир, отогнанный от рас-
твора триэтилкарбинола.
5. При проверке синтеза первое просушивание продолжалось в
течение 15 час. Первая фракционировка дала 122 г карбинола,
вторая — 10 г, а третья не дала ничего.
6. Заменяя бромистый этил соответственным галоидалкилом, по
этому способу можно получить следующие триалкилкарбинолы:
три-н.-пропилкарбинол, т. кип. 89—92°/ 20 мм, выход 75%; три-
н.-бутилкарбинол, т. кип. 129—131°/ 20 мм, выход 84%; три-н.-
УРАМИЛ
491
амилкарбинол, т. кип. 160—164°/ 19 мм, выход 75% и три-н.-геп-
тилкарбинол, температура кипения 195—2007 б мм, выход 72%.
3. Другие методы получения
Триэтилкарбинол был получен действием цинка на смесь йоди-
стого этила и диэтилкетонаг; действием магния на бромистый
этил в растворе диэтилкетона 2; действием натрия и бромистого
этила на диэтилкетон или на этиловый эфир пропионовой кислоты 3;
как побочный продукт при реакции между бромистым этилмагнием
и сероокисью углерода4; действием бромистого этилмагиия на
этиловый эфир пропионовой5, хлормуравьиной6, цианмуравьи-
ной 7 или угольной кислот 8.
'Баратаев, Зайцев, J. prakt. Chem. B) 34, 463 A886).
2 D a v i e s, К i p p i n g, J. Chem. Soc. 99, 298 A911).
'Morton, Stevens, J. Am. Chem. Soc. 53, 2247 A931).
* Weigert, Ber. 36, 1009 A903).
5 Schreiner, J. prakt. Chem. B) 82, 295 A910); Boeseken, Wild-
s с h u t, Rec. trav. chim. 51, 169 A932).
6 Мазуревич, ЖРХО 42, 1582 A910).
7 В г и у 1 a n t s, Bull. soc. chim. Belg. 33, 529 A924).
8Whitmore, Badertscher, J. Am. Chem. Soc. 55, 1560 A933).
УРАМИЛ
(Аминобарбитуровая кислота)
NH —CO
CO CHNO, • 3H2O+6HC1+3Sn
NH-CO
NH — CO
I I
CO CHNH, + 5 H,0 + 3 SnCl,
II"
NH —CO
Предложили: В. Хартман и О. Шегшард.
Проверили: К. Марвел и Б. Войцик.
1. Получение
В 5-литровую колбу помещают 100 г @,44 мол.) нитробарбиту-
ровой кислоты (стр. 365) и 600 мл концентрированной соляной
кислоты и смесь нагревают на кипящей водяной бане. К горячей
смеси в течение 30 мин. прибавляют 250г B,1 rp.-ат.) губчатого олова
и 400 мл соляной кислоты и нагревание продолжают до тех пор,
пока не исчезнет желтая окраска (примечание 1). Затем добавляют
492
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
еще около 3 л концентрированной соляной кислоты и нагревают
до полного растворения белого осадка. Прибавляют активированный
березовый уголь и горячую смесь фильтруют через стеклянный пори-
стый фильтр (примечание 2). Фильтрат оставляют на ночь в холо-
дильном шкафу, на другой день отсасывают (примечание 3) ура-
мил и промывают, сначала большим количеством разбавленной соля-
ной кислоты и под конец водой (примечание 4). Фильтрат упаривают
в вакууме до 1 л и охлаждают в течение ночи. Урамил, выпавший
из фильтрата, отсасывают на бюхнеровской воронке и присоединяют
к первоначально полученному продукту (примечание 5). Урамил
сушат в эксикаторе сначала над концентрированной серной кисло-
той, а под конец для удаления соляной кислоты — над 40%-ным
едким натром (примечание 6).
Урамил представляет собою тонкий белый порошок, при стоянии
сначала розовеющий, а потом краснеющий. Выход продукта,
плавящегося выше 400°, составляет40—46г F3—73% теоретич.).
2. Примечания
1. Водный раствор нитр о барбитуровой кислоты окрашен в жел-
тый цвет, в присутствии концентрированной соляной кислоты
окраска ослабевает. В конце реакции раствор должен быть вполне
бесцветным.
2. Если не имеется воронки с пористым стеклянным фильтром,
раствор можно профильтровать через слой активированного угля
толщиной в 1 см, положенный на двойном слое фильтровальной
бумаги. После того как урамил растворен в концентрированной
соляной кислоте, он лишь очень медленно выделяется из раствора,
и если фильтрование проводить быстро, раствор можно предвари-
тельно охладить до 60—80° с очень небольшой потерей продукта.
3. В некоторых случаях урамил при этом не выкристаллизо-
вывается, тогда раствор надо упарить и снова охладить.
4. Если продукт не промыт как следует водой, он будет содер-
жать соли олова.
5. Для качественной пробы на урамил испытуемый раствор
кипятят при доступе воздуха с аммиаком. В присутствии урамила
появляется розовая, постепенно темнеющая окраска. Реакция идет
быстрее в присутствии окиси ртути.
6. Если продукт сушится на воздухе или в сушильном шкафу, он
почти всегда получается розовым. Если в воздухе имеется аммиак
или амины, розовая окраска появляется быстрее.
3. Другие методы получения
Урамил был получен при кипячении аллоксантина с хлористым
аммонием 1; при восстановлении нитрозобарбитуровой или нитро-
ФЕНАНТРЕН 2- И 3-СУЛЬФОКИСЛОТЫ
493
барбитуровой кислот йодистым водородом 2 и при восстановлении
фенилгидразона аллоксана оловом и соляной кислотой 3.
1 W б h I e r, Liebig, Ann. 26, 309 A838).
2 В а е у е г, там же 127, 223 A863).
3 Ktthling, Ber. 31, 1973 A898).
ФЕНАНТРЕН 2- И 3-СУЛЬФОКИСЛОТЫ
Ч
"Ч
Предложил: Л. Физер.
Проверили: В. Хартман и Дж. Бумер.
1. Получение
Сульфирование проводится в 2-литровой круглодонной колбе с
тремя горлами, снабженной термометром, капельной воронкой
и мешалкой (примечание 1). В колбу помещают 500 г B,8 мол.) чис-
того фенантрена (примечание 2) и расплавляют его, поместив колбу
в масляную баню, нагретую до 110°. Пускают в ход мешалку и доба-
вляют 327 мл F00 г, 5,8 мол.) концентрированной серной кислоты
с такой скоростью, чтобы температура внутри колбы не поднима-
лась выше 120° A0—15 мин.), Реакционную смесь перемешивают
и поддерживают при температуре 120—125° в течение 3,5 час,
после чего проба реакционной смеси должна почти нацело раство-
ряться в воде. Реакция эта экзотермична, так что баню следует под-
держивать при температуре на 5—10° ниже температуры смеси.
Одновременно выделяется некоторое количество сернистого газа,
и реакционная масса становится зеленой.
Вязкий раствор еще в горячем состоянии выливают в 4 л воды и
по растворении добавляют раствор 400 г едкого натра в 600—800 мл
воды. После тщательного охлаждения в бане со льдом выпавшую
натриевую соль отсасывают через большую воронку, хорошо отжи-
мают и тщательно промывают 1 л полунасыщенного раствора хло-
ристого натрия (около 180 г на 1 л). Осадок состоит главным обра-
зом из натриевых солей 2- и 3-сульфокислот. Фильтрат содер-
жит смесь натриевых солей дисульфокислот и его отбрасывают
(примечание 3). Для того чтобы предварительно сконцентрировать
менее растворимую соль 2-сульфокислоты, смесь натриевых солей
растворяют в 7—8 л кипящей воды, содержащей 100 мл концентри-
рованной соляной кислоты (примечание 4), раствор фильтруют, филь-
трат нейтрализуют едким натром и дают ему кристаллизоваться. По
отделении маточного раствора (А), который сохраняют, кристаллы
(состоящие главным образом из натриевой соли 2-сульфокислоты)
494 СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
растворяют приблизительно в 8 л кипящей воды (примечание 5) и к
горячему раствору добавляют 100 г твердого двухводного хлори-
стого бария. Выпавший мелкий белый осадок бариевой соли
фенантрен-2-сульфокислоты недолго кипятят и отфильтровывают
через воронку Бюхнера диаметром в 20 см, которую предварительно
нагревают на паровой бане. Маточный раствор (Б) сохраняют.
Выпавшая бариевая соль 2-сульфокислоты содержит еще некото-
рое количество соли 3-сульфокислоты, которую необходимо извлечь
горячей водой. Осадок обрабатывают при температуре кипения
6-литровыми порциями кипящей воды до тех пор, пока не будет
найдено, что оставшаяся соль не содержит изомеров. Чистота опреде-
ляется на основании температуры плавления n-толуидиновой соли
по методике, описанной ниже. Обычно двух-трех таких промыва-
ний бывает достаточно. Маточный раствор (Б) от бариевой соли
2-сульфокислоты соединяют с промывными водами и все вместе
упаривают до б л. Для осаждения бария прибавляют серную кис-
лоту B5—30 мл концентрированной кислоты, разбавленной водой);
фильтрат (примечание б) упаривают до 2 л и нейтрализуют едким
кали, для осаждения смеси калиевых солей сульфокислот (В),
Фильтрат от калиевых солей можно еще более упарить и обработать
хлористым калием, чтобы обеспечить полноту выделения калиевых
солей сульфокислот. Оставшийся фильтрат отбрасывают. Смесь
калиевых солей, полученных в результате всех этих операций,
сохраняют (В).
Маточный раствор (.4) после кристаллизации натриевой соли
упаривают до объема в 2—3 лик горячему раствору добавляют
200 г хлористого калия. Выделившуюся по охлаждении смесь кали-
евых солей собирают и присоединяют к ранее полученным калиевым
солям (В); фильтрат отбрасывают. Полученную смесь растворяют
в минимальном количестве горячей воды, раствор фильтруют,
нагревают до кипения и дают спокойно охладиться. При этом выпа-
дает большое количество калиевой соли 3-сульфокислоты в очень чи-
стом виде A80—210 г). Маточный раствор, содержащий калиевую
соль 3-сульфокислоты с примесью соли 2-сульфокислоты, упари-
вают до небольшого объема, продукт высаливают хлористым калием
и отмывают от сульфат-иона раствором хлористого калия. Горя-
чий водный раствор этого продукта обрабатывают 10 г кристалли-
ческого хлористого бария и выпавшую в осадок бариевую соль
2-сульфокислоты E—10 г) отмывают от изомеров. Первый фильтрат
сохраняют, но промывные воды выливают. Из этого фильтрата
путем упаривания, осаждения бария серной кислотой, нейтрализа-
ции едким кали и кристаллизации продукта получают еще некото-
рое количество чистой калиевой соли 3-сульфокислоты E0—70 г).
Конечные выходы следующие: бариевой соли фенантрен-2-суль-
фокислоты 150—200 г A7—21%); калиевой соли фенантрен-3-суль-
фокислоты 200—220 г B4—26%; примечания 7 и 8).
ФЕНАНТРЕН 2- И 3-СУЛЬФОКИСЛОТЫ
495
Идентификация и проба на чистоту г состоит в получении и
исследовании пробы л-толуидиновой соли сульфокислоты (приме-
чание 9). Водный раствор сульфокислоты (или ее натриевой или
калиевой соли) обрабатывают избытком л-толуидина и соляной кис-
лоты, добавляют достаточное количество воды, чтобы все перевести
в раствор при температуре кипения и оставляют кристаллизоваться.
Выпавшие кристаллы хорошо промывают водой. В случае бариевой
соли ее кипятят с разбавленной серной кислотой, добавляют неболь-
шое количество обесцвечивающего угля и профильтрованный рас-
твор обрабатывают я-толуидином. Если соль амина выпадает в виде
масла, следует поскрести стенки сосуда палочкой, так как л-толуи-
диноваясоль,особенновслучае 3-сульфокислоты, даже в почти чистом
состоянии может некоторое время оставаться в виде масла. С другой
стороны, нечистая кислота дает п-толуидиновую соль, которая
может пребывать в виде масла неопределенно долгое время. Это
обстоятельство указывает на наличие смеси изомеров почти с такой же
точностью, как и депрессия точки плавления, хотя эта депрессия
и значительна. Для определения температуры плавления соли
амина ее можно высушить, отжав на фильтровальной бумаге,
однако капилляр необходимо поместить в баню при температуре
ниже 130°. Предварительное нагревание является вполне доста-
точным для того, чтобы вполне высушить продукт. Не вполне высу-
шенная соль плавится на 20—30° ниже истинной температуры
плавления.
л-Толуидиновая соль фенантрен-2-сульфокислоты образует пло-
ские иглы или пластинки с т. пл. 282° B91° исправ.), соль 3-сульфо-
кислоты образует толстые иглы с т. пл. 217° B22° исправл.).
2. Примечания
1. Можно получить удобную мешалку, если согнуть стеклянную
палочку под углом в 45° на 4 см от конца. С помощью такой мешалки
можно отделять от стенок приставшее к ним твердое вещество.
2. Следует пользоваться фенантреном хорошего качества. Техни-
ческий 70%-ный фенантрен может быть подвергнут очистке по
методу Коена и Кормье 2. Весьма важно отметить, что наличие
в фенантрене более 2% антрацена повышает его температуру плав-
ления.
3. Считается, что при этом получается не менее 12 изомерных
дисульфокислот. Полезных продуктов из такой смеси выделить не
удалось.
4. Растворимость солей сульфокислот заметно возрастает в при-
сутствии минеральной кислоты.
5. Удобным, хотя и не очень прочным, сосудом для кипячения
и выпаривания больших объемов растворов может служить эмали-
рованное ведро емкостью в 10 л, которое нагревают кольцевой
496
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
^-З-ФЕНИЛАЛАНИН
497
горелкой. Содержимое ведра можно вычерпывать с помощью
ковша.
6. Прибавление Небольшого количества обесцвечивающего угля
в значительной степени облегчает коагуляцию сернокислого бария
и способствует удержанию его на фильтре.
7. Низкий выход моносульфокислот зависит в значительной
степени оттого, что наряду с монокислотами неизбежно образуются
также и дисульфокислоты.
8. Для некоторых реакций, например, для щелочного плава,
бариевая соль является удобной формой применения фенантрен-2-
сульфокислоты. Для превращения ее в калиевую соль весьма целе-
сообразно брать бариевую соль в том влажном и мелкораздроб-
ленном состоянии, в котором она первоначально получается. Так
как реакция эта протекает медленно, то для осаждения всего бария
следует брать несколько свежих порций серной кислоты.
9. Этот метод может быть рекомендован как общий метод для
идентификации сульфокислот. По экономии времени и материала он
более выгоден, чем получение свободной кислоты, хлорангидрида,
сложного эфира, амида или фенола. Пользуясь этим методом, можно
быстро идентифицировать несколько милиграммов кислоты или
любой из ее солей с металлами, независимо от того, находятся
они в твердом состоянии или в растворе.
3. Другие методы получения
Сульфирование фенантрена изучали Вернер и его ученики 3, а
также Зандквист4, Физер г и Иоффе 5.
1 F i e s e r, J. Am. Chem. Soc. 51, 2460, 2471 A929).
2 Cohen, Cormier, там же 52, 4363 A930).
3 Werner и др., Ann. 321, 248 A902).
*Sandquist, там же 392, 76 A912).
6 Иоффе, ЖОХ 3, 448 A933).
df-p-ФЕНИЛАЛАНИН
А. ИЗ АЗЛАКТОНА а-БЕН30ИЛАМИН0К0РИЧН0Й КИСЛОТЫ
.со —о
сйнйсн = с
(Р + HI)
\n-=c
с6н5
со.,н
2Н2О +2[Н]
С„Н6СОгН
Предложили: X. Джиллеспи и X. Снайдер.
Проверили: В. Хартман и Дж. Дикки.
1. Получение
В 1-литровую трехгорлую круглодонную колбу, снабженную
обратным холодильником, механической мешалкой и капельной
воронкой (примечание 1), помещают 25 г @,1 мол.) азлактона а-бен-
зоиламинокоричной кислоты (примечания 2 и 3), 20 г @,64 гр.-ат.)
красного фосфора и 135 г A25 мл) уксусного ангидрида. Затем в
течение примерно одного часа прибавляют при работающей мешалке
195 г A25 мл, 0,76 мол.) 50%-ной иодистоводородной кислоты (уд. в.
1,56; примечание 4). Смесь кипятят с обратным холодильником
3—4 часа, затем охлаждают и фильтруют с отсасыванием. Не
вступивший в реакцию фосфор промывают на фильтре двумя пор-
циями по 5 мл ледяной уксусной кислоты и отбрасывают его,
Фильтрат и промывную жидкость отгоняют досуха при понижен-
ном давлении из колбы Клайзена емкостью 500 мл, нагреваемой
на водяной бане. В качестве приемника служит колба Вюрца на
250 мл, охлаждаемая льдом. Дестиллат сохраняют для повторения
синтеза (примечание 5).
К сухому остатку в клайзеновской колбе добавляют 100 мл
воды и вновь выпаривают все досуха. Этот дестиллат отбрасывают.
К остатку в колбе приливают 150 мл воды и 150 мл эфира и смесь
взбалтывают до полного растворения. Водный слой отделяют и
экстрагируют три раза эфиром порциями по 100 мл. Эфирные вы-
тяжки отбрасывают, водный раствор нагревают на кипящей водяной
бане с 2—Зг активированного березового угля и со следами сульфита
натрия до тех пор, пока не будет удален весь растворенный эфир.
Раствор фильтруют, фильтрат нагревают до кипения и нейтрали-
зуют 15%-ным водным аммиаком (уд. в. 0,94), применяя в качестве
индикатора конго красный. Обычно требуется около 25 мл аммиака.
Фенилаланин выделяется в виде бесцветных пластинок, которые по
охлаждении раствора отфильтровывают и на фильтре тщательно
промывают двумя порциями по 30 мл холодной воды. Выход 10,5—
11 г F3,6—67% теоретич.). При определении температуры плавле-
ния продукт разлагается при 284—288° (исправл.; примечание 6).
2. Примечания
1. Следует пользоваться чистыми корковыми пробками, оберну-
тыми оловянной фольгой.
2. Этот азлактон легко получается из бензальдегида по методу,
описанному для азлактона а-оензоиламино-[ЦЗ,4-диметоксифенил)-
акриловой кислоты (стр. 11). Из 53 г @,5 мол.) бензальдегида,
89,5 г @,5 мол.) гиппуровой кислоты (стр. 158), 41 г плавленого
уксуснокислого натрия и 153 г уксусного ангидрида получают
78—80 г F2—64% теоретич.) почти чистого продукта с т. пл.
165—166° (исправл.). Полученный продукт достаточно чист для
получения из него фенилаланина. После перекристаллизации из
150 мл бензола получают продукт с т. пл. 167—168° (исправл.).
3. Восстановление можно вести таким же образом, исходя из
а-бензоиламинокоричной кислоты; при этом получаются более
32 Сборник № 2
498
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
высокие выходы. Азлактон может быть превращен в свободную
кислоту следующим образом:
В колбе емкостью 12 л, снабженной механической мешалкой,
суспендируют 62,3 г @,25 мол.) азлактоиа в 6 л воды, после чего
добавляют 11 г @,275 мол.) едкого натра в виде 10%-ного раствора.
Смесь нагревают на водяной бане при работающей мешалке до тех
пор, пока все не растворится, на что требуется от 3 до 4 час. Горя-
чий раствор фильтруют и подкисляют соляной кислотой. а-Бензоил-
аминокоричная кислота выделяется из горячего раствора в виде
белых призм и по охлаждении отфильтровывается. Выход 55,5—
64,5 г (83—96,5% теоретич.). Продукт достаточно чист и плавится
с разложением в пределах 2° между 224 и 236° (исправл.). Сырую
кислоту можно перекристаллизовать из спирта, однако темпера-
тура плавления при этом не изменяется.
4. При прибавлении иодистоводородной кислоты реакционная
смесь может затвердеть. Если это произошло, мешалку останав-
ливают и при помешивании стеклянной палочкой прибавляют один
или два раза по 5 мл раствора иодистоводородной кислоты. После
этого масса становится достаточно жидкой, чтобы вновь можно было
пустить мешалку.
5. При повторной операции дестиллат помещают в 1-литровую
колбу и добавляют 4 мл воды, 25 г азлактона и 20 г красного фос-
фора. Смесь нагревают с обратным холодильником 3—4 часа, после
чего поступают так же, как описано выше. Выход получается прак-
тически такой же, как и в первом опыте.
6. Температура разложения может быть весьма различной и
зависит от скорости нагревания. Приведенные выше результаты
достигаются в том случае, если капилляр с веществом помещают в
нагретую до 200° баню и после этого быстро повышают температуру.
Б. ИЗ а-АЦЕТОАМИНОКОРИЧНОЙ КИСЛОТЫ
dl-3-ФЕНИЛАЛАНИН
499
С6Н5СН = ССО2Н
NHCOCH3
свн6сн2снсо2н
NHCOCH3
(Pt)
н»о
(HCI)
СвН5СН2СНСО3Н
NHCOCH3
CeH5CH2CH(NH2)C(XH
Предложили: Р. Хербст и Д. Шемин.
Проверили: Р. Фьюзон и Е. Кливленд.
1. Получение
В склянку прибора для восстановления Бурге сса-Парра помещают
раствор 20,5 г @,1 мол.) а-ацетоаминокоричной кислоты (стр. 72)
в 150 мл ледяной уксусной кислоты (примечание 1), добавляют 0,5 г
катализатора окиси платины («Синт. орг. преп.», сб. 1, стр. 357) и
смесь взбалтывают в атмосфере водорода при начальном давлении
около 3 am до тех пор, пока не будет поглощено вычисленное коли-
чество водорода; обычно на это требуется около 2 час. (примеча-
ния 2 и 3). Когда восстановление закончено, катализатор отфиль-
тровывают с отсасыванием и промывают небольшим количеством
воды. Фильтрат и промывные воды соединяют вместе и упаривают
досуха в вакууме на водяной бане.
Кристаллический остаток (примечание 4) растворяют в 400 мл
1-н. соляной кислоты, переносят в 1-литровую колбу, снабженную
обратным холодильником, и кипятят 10 час. (примечание 5). Получен-
ный раствор упаривают в вакууме на водяной бане досуха; к остатку
медленно приливают из капельной воронки 100 мл воды примерно
с той же скоростью, с которой она отгоняется, для того чтобы по
возможности полнее удалить избыток соляной кислоты. Остаток
растворяют в 30—40 мл кипящей воды и, осторожно добавляя кон-
центрированный аммиак и уксусную кислоту, доводят рН раствора
до щелочной реакции на конго красное, но следя затем, чтобы реак-
ция еще была кислой на лакмус (примечание 6). Затем, чтобы способ-
ствовать выделению фенилаланина, приливают двойной объем
95%-ного спирта. Смесь на сутки помещают в холодильный шкаф,
после чего продукт переносят в воронку Бюхнера, где его промы-
вают сперва три раза ледяной водой порциями по 25 мл, а затем
спиртом. Выход 14,5 г. Фильтрат упаривают досуха в вакууме
на водяной бане и остаток извлекают 70 мл ледяной воды, в 3—4 пор-
ции. Нерастворимый продукт промывают 95%-ным спиртом и при-
бавляют его к главной порции фенилаланина. Общий выход 16 г
(примечание 7).
Обе порции, весом около 16 г, растворяют в минимальном коли-
честве кипящей воды (примечание 8), приливают двойной объем
95%-ного спирта и колбу помещают на ночь в холодильный шкаф,
чтобы завершить кристаллизацию. Фенилаланин переносят в во-
ронку Бюхнера, несколько раз промывают небольшими порциями
ледяной воды, а затем спиртом. Выход 10,5—11 г. Если фильтрат и
промывные воды вновь упарить, можно получить еще 3—3,5 г про-
дукта. Общий выход 14—14,3 г (85—86% теоретич.) аналитически
чистого фенилаланина.
2. Примечания
1.'Иногда приходится нагреть смесь для того, чтобы полностью
растворить ацетоаминокоричную кислоту в этом количестве уксус-
ной кислоты. В этом случае раствор необходимо охладить до ком-
натной температуры, прежде чем помещать его в прибор для восста-
новления.
500
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
ФЕНИЛАРСИНОВАЯ КИСЛОТА
501
2. После того как поглощено вычисленное количество водорода,
катализатор перестает быть коллоидным и скорость поглощения
водорода становится очень малой.
3. В случае свежеприготовленного и влажного катализатора
может также восстановиться бензольное ядро с образованием
N-ацетилгексагидрофенилаланина. Когда это имеет место, поглоще-
ние водорода продолжается с большой скоростью даже после того,
как поглотилось количество его, необходимое для восстановления
двойной связи. После перекристаллизации из воды или разбавлен-
ного спирта гексагидросоединение образует иглы с температурой
плавления 178°.
'4. Чистый N-ацетилфенилаланин можно получить, если перекри-
сталлизовать остаток из горячей воды или из горячего разбавлен-
ного спирта. При этом получаются бесцветные иглы с т. пл, 150—151°.
5. Омыление 1-н. соляной кислотой бывает не полным, если
нагревание ведется менее 10 час. При кислоте более высокой концен-
трации омыление может быть завершено быстрее.
6. Если раствор будет только щелочным на конго красное, то
продукт выделяется в виде почти твердой массы; прибавление спирта
облегчает проверку рН, так как масса при этом измельчается,
а также уменьшается растворимость продукта.
7. Этот продукт содержит около 2,5% хлористого аммония; если
это учесть, то выход фенилаланина составляет 94%. Если не тре-
буется абсолютно чистый фенилаланин, последующую кристаллиза-
цию можно опустить.
8. Фенилаланин растворяется довольно медленно в кипящей
воде. Поэтому удобнее начинать с избытка воды и упаривать раствор
на голом пламени до тех пор, пока из горячего раствора не начнут
выпадать кристаллы.
3. Другие методы получения
^/-Фенилаланин был получен действием аммиака и цианистого
водорода на фенилацетальдегид1; восстановлением оксима2 или
фенилгидразона 3 фенилпировиноградной кислоты; восстановлением
пировиноградной кислоты в спиртово-аммиачном растворе4; вос-
становлением а-аминокоричной кислоты или ее производных5 и
действием аммиака на а-бром-^-фенилпировиноградную кислоту6;
детальные указания для работы по этой методике приведены в Org.
Synt., 21, 99.
'Erlenmeyer, Lipp, Ann. 219, 194 A883).
2 Erlenmeyer, там же 271, !69 A892); Knoop, Hoessli, Ber.
39, 1479 A906); Shem in, Her bs t, J. Am. Chem. Soc. 60, 1951 A938).
8 Феофилактов, Виноградова, ДАН 24, 759 A939); ЖОХ 10,
255 A940).
'Knoop, Oe s te r 1 i n, Z. physiol. Chem. 148, 311 A925).
6 P 1 6 с h 1, Ber. 17, 1623 A884); Erlenmeyer, Ann. 275, 15 A893);
В e r a m a n n Stern, Witte, там ше 449, 280 (примечание) A926);
Наг? JTgton. М с С о г t n с у, Biochem. J. 21, 854 A927); Lamb.Rob-
son, там же 25, 1234 A931).
« Fischer, Ber. 37, 3064 A904).
CeH6NHa
ФЕНИЛАРСИНОВАЯ КИСЛОТА
(Фенилмышьяковая кислота)
2НС1 + NaNO2 C6H5NX1
2 Н2О + NaCl
CeH5N2Cl
CeH6As03Na3
Na3As03 -^°- C6H5As O3Na2 4- N3 4- NaCl
2НС1
C6H5As03H2 + 2 NaCl
Предложили: Р. Буллард и Дж. Дикки.
Проверили: В. Каротерс и В. Мак-Ювен.
1. Получение
В крутлодонную колбу емкостью 12 л, снабженную механической
мешалкой, наливают 1 л воды. Воду нагревают до кипения, после
чего прибавляют 500 г D,7 мол.) безводного углекислого натрия.
Как только он растворится, добавляют при работающей мешалке
250 г A,26 мол.) трехокиси мышьяка и 11 г кристаллической серно-
кислой меди. После полного растворения всех реагентов раствор
при перемешивании охлаждают до 15° в струе водопроводной воды.
Одновременно с получением раствора мышьяковистокислого
натрия приготовляют раствор хлористого фенилдиазония. Для этого
к хорошо перемешиваемой смеси 186 г B,0 мол.) технического
анилина, 400 мл D,8 мол.) концентрированной соляной кислоты
(уд. в. 1,19), 1 л воды и достаточного количества колотого льда, так
чтобы суммарный объем равнялся 3 л, медленно приливают раствор
145 г B,0 мол.) 95%-ного нитрита натрия в 500 мл воды. На это тре-
буется от 30 до 40 мин.
Раствор хлористого фенилдиазония добавляют при перемеши-
вании в течение 1 часа к суспензии мышьяковистокислого натрия,
предварительно охлажденной с помощью бани со льдом и солью
до 0°. Температуру при этой реакции поддерживают ниже 5° (приме-
чание 1). При этом вследствие выделения азота происходит вспени-
вание, которое умеряют тем, что время от времени добавляют
небольшие количества бензола. После того как прибавлено все коли-
чество раствора хлористого фенилдиазония, перемешивание про-,
должают еще 1 час и смесь фильтруют, чтобы отделить все твердые
частицы. Осадок промывают 500 мл холодной воды, фильтрат и
промывные воды соединяют вместе и упаривают на голом огне до
объема 1,5 л (примечание 2).
К горячему концентрированному раствору, окрашенному в тем-
нобурый цвет, добавляют концентрированной соляной кислоты
502
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
до тех пор, пока не прекратится выделение смолистого продукта
(примечание 3). Смолу отфильтровывают, после чего добавляют
еще соляной кислоты. Если снова выделяется смола, ее отфильтро-
вывают. При этом должен получиться прозрачный бледножелтый
раствор. Весьма -важно всю смолу отделить именно в этот момент;
в противном случае даже при повторной перекристаллизации нельзя
будет получить бесцветный продукт. Из полученного раствора фенил-
арсиновую кислоту осаждают добавлением 250 мл концентрирован-
ной соляной кислоты (уд. в. 1,19: примечание 4). После того как
смесь охладится (лучше всего после стояния в течение ночи), фенил-
арсиновую кислоту отсасывают на воронке Бюхнера и промывают
200 мл холодной воды. Светложелтые кристаллы растворяют в
500 мл кипящей воды, добавляют 20 г активированного березового
угля, раствор фильтруют горячим и фильтрату дают охладиться.
Выпавшие белые кристаллы отфильтровывают и сушат. Фениларси-
новая кислота плавится с разложением при 154—158° и переходит
при этом в ангидрид, CeH6As02. Выход 160—182 г C9—45% теоре-
тич.; примечание 5).
2. Примечания
1. Эта температура является оптимальной как с точки зрения
максимального выхода, так и в отношении легкости дальнейшей
очистки продукта. Однако хорошие результаты получаются и при
температурах до 15°.
2. Раствор упаривают при атмосферном давлении, так как при
упаривании в вакууме происходит сильное вспенивание.
3. Всего требуется около 100 мл соляной кислоты. Необходимо
следить за тем, чтобы в осадок не выпала фениларсиновая кислота.
4. Слишком большой избыток соляной кислоты ведет к раство-
рению части продукта и к понижению выхода.
5. В некоторых случаях выход достигал 57%, однако причины,
обусловливающие такой высокий выход, неизвестны.
3. Другие методы получения
Фениларсиновая кислота была получена окислением фенилди-
хлорарсина 1 или фенилдииодарсина 2 хлором в водной среде; окис-
лением фениларсина азотной кислотой или воздухом3; разложе-
нием водой фениларсинтетрахлорида или фениларсиноксихлорида4,
нагреванием иодбензола или бромбензола с мышьяковистокислым
калием5; диазотированием п-арсаниловой кислоты с последующим
разложением в растворе гидросульфита натрия и соляной кислоты6;
действием мышьяковистокислого калия на калиевую соль бензол-
изодиазотата7; действием хлористого фенилдиазония на мышьяко-
вистокислый натрий в присутствии соединений меди 8 или хлористого
магния и порошкообразной меди9 и действием нейтральной или
а-ФЕНИЛАЦЕТОАЦЕТОНИТРИЛ
503
щелочной смеси, содержащей трехокись мышьяка, медную соль и
восстановитель, на хлористый феиилдиазоний.
Приведенная выше подробная методика в основном заимство-
вана у Пальмера и Адамса". Имеются указания на то, что приме-
нение буферных растворов с целью обеспечить постоянство рН ведет
к увеличению выхода фениларсиновой кислоты в этой реакции п.
1 Michaelis, Loesner, Ber. 27, 265 A894); Rosenheim, Bi-
le с k i, там же 46, 551 A913); R о e d с г, В 1 a s i, там же 47, 2752 A914).
2 В е г t h e i m, там же 47, 274 A914).
3 Palmer, Dehn, там же 34, 3599 A901); D e h n, Am. Chem. J. 33,
149 A905).
1 Michaelis, Ber. 10, 625 A877); LaCoste, Michaelis, Ann.
'5 Dehn, Am. Chem. J. 33, 140 A905); Rosenmund, Ber. 54, 438
A921).
•Bertheim, шже41, 1853 A908).
' Bart; герм. пат. 250 264 [Frdl. 10, 1254 A910—12)].
8 Chem Fabrik von Heyden A.-G., герм. пат. 264 924 [Frdl. 11, 1030
A912—14I; Palmer, Adams, J. Am. Chem. Soc. 44, 1361A922); N о г г i s,
I Ind Ene Chem. 11, 825 A919); Schmidt, Ann. 421, 169 A920).
9 Bar t; герм пат. 254 092 [Frdl. 11, 1030 A912—14)].
i» M о u n e у r a t; англ. пат. 142 947 [С. А. 14, 2802 A920)].
" Bias, Genie civil 115, 448 A939) [C. A. 34, 2342 A940)].
CeH5CH2CN + CH3COX2H
a^2ll5
а-ФЕНИЛАЦЕТОАЦЕТОНИТРИЛ
(а-Ацетил-а.-толунитрил)
(C2H5ONa)
~ С6Н5СН (CN) СОСН3 + С3Н6ОН
Предложили: П. Джулиан, Дж. Оливер, Р. Кимболл,
А. Пайи. и Дж. Джефферсон.
Проверили: К- Ноллер и Сайнерхолм.
1. Получение
В 2-литровой круглодонной колбе с обратным холодильником
приготовляют раствор этилата натрия из 60 г B,6 rp.-ат.) чистого
натрия и 700 мл абсолютного спирта. К горячему раствору прилива-
ют смесь 234 г B мол.) чистого цианистого бензила (примечание 2)
и 264 г C мол.) сухого уксусноэтилового эфира (примечание 3)^
Смесь энергично встряхивают, холодильник закрывают пробкой
с хлоркальциевой трубкой и раствор нагревают на водяной бане
2 часа, после чего оставляют его стоять на ночь^(примечание 4).
На утро смесь перемешивают деревянной палочкой, чтобы разбить
куски, охлаждают в охладительной смеси до —10° и поддерживают
при этой температуре 2 часа. Натриевую соль отфильтровывают через
воронку Бюхнера диаметром 15 см и на воронке промывают четыре
раза эфиром, порциями до 250 мл.. Отфильтрованная лепешка вдчтд
504
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
а-ФЕНИЛАЦЕТОАЦЕТОНИТРИЛ
505
бесцветна и соответствует 250—275 г сухой натриевой соли, или
69—76% теоретич. Фильтраты охлаждают в охладительной смеси
прежде чем перерабатывать их, как описано ниже.
Натриевую соль, еще смоченную эфиром, растворяют в 1,3 л
дестиллированной воды при комнатной температуре, раствор охла-
ждают до 0°'и нитрил осаждают медленным приливанием, при энер-
гичном взбалтывании, 90 мл ледяной уксусной кислоты, поддержи-
вая при этом температуру ниже 10°. Осадок отфильтровывают с отса-
сыванием и на воронке промывают четыре раза водой порциями по
250 мл. Влажная лепешка, весом около 300 г (примечание 5), соот-
ветствует 188—206 г 59—64% сухого бесцветного а-фенилацето-
ацетонитрила с т. пл. 87—89", пригодного для большинства целей.
При желании сырой продукт можно перекристаллизовать, для
чего влажную лепешку растворяют в 100 мл горячего метилового
спирта, раствор фильтруют и при перемешивании охлаждают до
10°. Кристаллы отфильтровывают с отсасыванием и один раз про-
мывают на фильтре 40 мл охлажденного до —10° метанола. Выход
продукта после высушивания 173—191 гE4—60%); т. пл. 88,5—89.5°.
Фильтраты и промывные растворы после выделения натриевой
соли переливают в 5-литровую колбу и разбавляют ледяной водой
до тех пор, пока колба не будет полной. Нижний слой сифонируют,
большую часть эфира декантируют, а оставшееся количество раз-
деляют с помощью делительной воронки. Водный слой таким же
образом экстрагируют два раза эфиром порциями по 500 мл и эфир-
ные вытяжки отбрасывают. Эфир, оставшийся в водном слое, уда-
ляют при пониженном давлении, просасывая воздух через раствор
с помощью водоструйного насоса в течение 1 часа и о-фенилацето-
ацетонитрил осаждают приливанием 60 мл ледяной уксусной кис-
лоты. Если при этом выпадает масло, колбу помещают в баню со
льдом, пока масло не закристаллизуется. Кристаллы отсасывают и
на фильтре промывают четыре раза водой порциями по 50 мл. После
высушивания вес продукта рыжеватокоричневого цвета 50—55 г,
т. пл. 83—86 г. Его растворяют в метанольном маточном растворе от
первой перекристаллизации. Раствор кипятят с небольшим количе-
ством активированного угля, фильтруют его и охлаждают до—10°.
Образовавшиеся кристаллы отфильтровывают, промывают 10 мл
холодного метанола и сушат. Получают 43—48 г продукта, окрашен-
ного в бледносоломенный цвет, с т. пл. 87—89°. Его перекристал-
лизовывают из 25 мл чистого метанола и промывают 10 мл холод-
ного метанола. При этом получают 37—41 г с т. пл. 88,5—89,5°.
Таким образом выход чистого продукта составляет в общем 210—
232 г F6—73% теоретич.; примечания 6 и 7).
2. Примечания
1. Абсолютный спирт можно получить, если 95%-ный спирт
дважды высушить над негашеной известью или один раз над нега-
шеной известью и один раз над натрием, согласно указаниям при-
мечания 1, «Синт. орг. преп.», сб. 1, стр. 547. Можно также взять
продажный абсолютный спирт и перед употреблением высушить его
один раз негашеной известью или натрием.
2. Цианистый бензил был получен согласно методике, описанной
в <<Синт. орг. преп.», сб. 1, стр. 502, включая очистку концентри-
рованной серной кислотой.
3. Продажный абсолютный уксусноэтиловый эфир кипятился
с обратным холодильником в течение 30 мин. над фосфорным анги-
дридом и перегонялся непосредственно перед употреблением.
4. Если позволяет время, переработку можно продолжить, не
давая смеси стоять в течение ночи. Если сушка спирта и уксусно-
этилового эфира начаты с утра, именно этот момент удобен для того,
чтобы сделать перерыв.
5. Если продукт предназначается для получения метилбензил-
кетона (стр. 313), его не следует сушить.
6. Не имеет смысла пытаться выделить более чистый продукт
упариванием маточных растворов.
7. Число стадий можно сократить, если опустить выделение
натриевой соли. В этом случае реакционную смесь, после стояния
в течение ночи, разбавляют в 5-литровой колбе 2 у^воды и содер-
жимое колбы взбалтывают до растворения натриевой соли. Приба-
вляют 1 л колотого льда и смесь экстрагируют два раза эфиром
порциями в 1 л и в 500 мл. Водный раствор после извлечения осво-
бождают от эфира, как описано выше, и осаждают раствором 150 мл
ледяной уксусной кислоты и 400 мл воды, осадок отфильтровывают
и промывают водой. Продукт получается окрашенным и имеет более
низкую температуру плавления, чем полученный из очищенной на-
триевой соли; его следует дважды перекристаллизовать из метанола,
чтобы т. пл. стала 88,5—89,5°. Суммарный выход продукта с такой
температурой плавления несколько меньше, чем приведенный в
основной методике.
3. Другие методы получения
а-Фенилацетоацетонитрил был получен конденсацией уксусно-
этилового эфира с натриевым производным цианистого бензила, полу-
ченного из цианистого бензила и амида натрия в эфире 1, и конден-
сацией уксусноэтилового эфира и цианистого бензила с помощью
сухого2 этилата натрия или его спиртового раствора 3.
1 Bodroux, Compt. rend. 151, 234A910); Bull. soc. chim. D) 7, 848
"- W a 1 t h e r, S с h i с k 1 e r, J. prakt. Chem. B) 55, 343 A897).
•' В е с k h, Ber. 31, 3160 A898); Post, M icha le k, J. Am. Chem. Soc.
52, 4358 A930).
506 СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
ФЕНИЛБЕН30ИЛДИА30МЕТАН
(Азибензил, а-диазо-а-фенилацетофенон)
°\С = N — NH2 + HgO > " 5Ч-С<^ ||
г н сс\/ г н со^ \n
ФЕНИЛГЛИОКСАЛЬ
507
Hg + Н2О
Предложили: К. Нетщеску и Е. Соломоника.
Проверили: Л. Физер и Р. Темпл.
1. Получение
В ступке смешивают 30 г @,134 мол.) бензилгидразона (приме-
чание 1) с 60 г @,28 мол.) желтой окиси ртути и с 15 г безводного серно-
кислого натрия (примечание 2). Смесь переносят в склянку на 500 мл
с притертой пробкой и поверх наливают 200 мл абсолютного эфира
(примечание 3). В качестве катализатора добавляют 4 мл холодного
насыщенного спиртового раствора едкого кали (примечание 4) и
смесь взбалтывают 10—15 мин. Раствор фильтруют без отсасывания
через тонкую фильтровальную бумагу и остаток промывают не-
сколько раз эфиром до тех пор, пока фильтрат не станет лишь слабо
окрашенным. Соединенные эфирные вытяжки выпаривают досуха в
вакууме водоструйного насоса, причем колбу нагревают на водяной
бане до температуры не выше 40° (примечание 5). Желтый кристалли-
ческий продукт сушат на пористой тарелке и перекристаллизовывают
из абсолютного эфира. Выход азибензила, плавящегося с разложе-
нием при 79°, составляет 26—28 г(87—94%теоретич.; примечание 6).
2. Примечания
1. Бензилгидразон1 может быть получен следующим образом2:
смесь, состоящую из 52 г @,4 мол.) сернокислого гидразина («Синт.
орг. преп.», сб. 1, стр. 158), ПО г @,8 мол.) уксуснокислого натрия
и 250 г воды кипятят в течение 5 мин., охлаждают до 50° и добавляют
225 мл метилового спирта. Выпавший в осадок сернокислый натрий
отфильтровывают и промывают небольшим количеством спирта.
Затем приготовляют горячий раствор 50 г @,24 мол.) бензила
(«Синт. орг. преп»., сб. 1, стр. 83) в 75 мл метилового спирта, к
которому добавляют полученный выше раствор, предварительно
нагретый до 60°. Большая часть бензилгидразона выделяется не-
медленно, но выход увеличивается, если смесь кипятить в течение
1 часа с обратным холодильником. По охлаждении гидразон отфиль-
тровывают и промывают небольшим количеством эфира, чтобы уда-
лить желтую окраску. Выход 50,5 г (94% теоретич.). Продукт пла-
вится с разложением при 147—1513.
Метод получения бензилгидразона из бензила и гидразин-
гидрата описан в Org. Synt. 20, 48.
2. Сернокислый натрий поглощает воду, образующуюся в резуль-
тате реакции.
3. Применение эфира вместо бензола или петролейного эфира,
как это рекомендуется в более старых методах, облегчает удаление
растворителя после окончания реакции.
4. Без катализатора окисление может продолжаться несколько
часов и выходы бывают весьма различными и в значительной степени
зависят от качества окиси ртути.
5. Если упаривание вести на водяной бане при атмосферном
давлении, то может произойти взрыв.
6. Этой же методикой можно воспользоваться и для получения
диазофлуорена3.
3. Другие методы получения
Фенилбензоилдиазометан (азибензил) был получен окислением
бензилгидразона окисью ртути* с применением в качестве раство-
рителя бензола или петролейного эфира, а также и без указанного
выше катализатора.
1 С u r t i u s, T h u n, J. prakt. Chem. B) 44, 176 A891).
2 Частное сообщение К. Ф. X. Аллена.
3Staudinger, Kupfer, Ber. 44, 2207 A911); Staudinger,
Gaul e, там же 49, 1955 A916).
4 С и г ti u s, T h u n, J. prakt. Chem. B) 44, 182 A891).
C6H5COCH3
ФЕНИЛГЛИОКСАЛЬ
SeO3 > CeH5COCHO + Se + HaO
Предложили: Х. Рилей и А. Грей.
Проверили: Л. Физер и К. Фишер.
1. Получение
В 1-литровую трехгорлую круглодонную колбу, снабженную
мешалкой с жидкостным затвором и обратным холодильником,
помещают 600 мл диоксана (примечание 1), 111 г A мол.) двуокиси
селена (примечание 2) и 20 мл воды (примечание 3). Смесь нагре-
вают до 50—55°, перемешивают до тех пор, пока все не перейдет в
раствор, сразу добавляют 120 г A мол.) ацетофенона и полученную
смесь кипятят при непрерывном перемешивании в течение 4 час.
(примечание 4). Горячий раствор отделяют декантацией от выпав-
шего селена, а диоксан и воду отгоняют с небольшим дефлегматором.
Фенилглиоксаль перегоняют в вакууме из колбы Клайзена емкостью
250 мл (примечание 5), причем собирают фракцию 95—97°/25 мм
(примечание 6). Выход 93—96г F9—72% теоретич.; примечание 7),
508
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
О-ФЕНИЛЕНДИАМИН
500
Альдегид при стоянии застывает в студенообразную массу, что,
вероятно, объясняется полимеризацией. При перегонке происходит
деполимеризация, не сопровождающаяся значительными потерями.
Фенилглиоксаль можно хранить также в виде его гидрата, который
легко получить, если растворить желтую жидкость в 3,5—4 объемах
горячей воды и дать продукту кристаллизоваться (примечание 8),
2. Примечания
1. В качестве растворителя можно взять также этиловый спирт
(95%). Кроме того реакцию можно вести с избытком ацетофенона
B мол.) в качестве растворителя, однако результаты при этом полу-
чаются менее удовлетворительные. Диоксан может быть регенери-
рован и вновь использован при последующих синтезах.
2. Для получения двуокиси селена 200 г A41 мл) концентриро-
ванной азотной кислоты нагревают в 3-литровом стакане на электри-
ческой плитке в хорошо действующем вытяжном шкафу и добавляют
100 г селена отдельными порциями по 5—Ю г. Стеклянная меха-
ническая мешалка сбивает пену и ускоряет процесс окисления. По-
лученный раствор переливают под тягой в большую чашку для выпа-
ривания и нагревания на электрической плитке при температуре не
выше 200°, пока не произойдет полное обезвоживание селенистой
кислоты. Для очистки сырого продукта его подвергают возгонке. Для
этого 50 г окиси переносят в 1-см фарфоровый тигель, покрывают его
колбой для фильтрования емкостью 250 мл, через которую пропус-
кают ток холодной воды из-под крана. Тигель предохраняют асбестом
и нагревают на небольшом пламени, до тех пор пока все не возго-
нится B0—30 мин.). По охлаждении тигля возогнанную двуокись
селена очищают с колбы, которая служит холодильником. При ра-
боте с двуокисью селена необходимо соблюдать сугубую осторож-
ность, так как двуокись селена очень ядовита.
Другой метод окисления селена в двуокись селена описали Ган и
Шалее \
3. Вместо смеси двуокиси селена с водой можно взять продаж-
ную селенистую кислоту A29 г, 1 мол.).
4. Примерно через два часа раствор становится прозрачным и
дальнейшее осаждение селена почти незаметно.
5. Можно получить несколько граммов гидрата, если головную
фракцию добавить к равному объему теплой воды и оставить про-
дукт кристаллизоваться.
6. В литературе приведена т. кип. 120°/'50 мм и 142°/125 мм.
7. Вместо ацетофенона можно взять фенилацетальдегид. Анало-
гичным образом из пропиофгнона получается фенилметилглиоксаль;
кроме того в целом ряде случаев соединения, содержащие метиле-
новую группу рядом с карбонильной группой, могут быть окислены
двуокисью селена в соответственный я-кетоальдегид или а-дикетон 2.
Для окисления лепидина и хинальдина в соответственные аль-
дегиды ряда хинолина необходимо применять свежеприготовленную
двуокись селена 3.
8. В литературе указано, что растворимость гидрата при 20°
равняется 1 части в 35 частях воды. Температура плавления приво-
дится от 73 до 91°; при этом указывается, что различия в темпера-
туре плавления зависят от различной степени влажности образцов4.
Гидрат хорошо кристаллизуется из воды; в качестве растворителей
могут быть взяты также хлороформ, сероуглерод, спирт или смесь
эфира с лигроином. Фенилглиоксаль может быть выделен из гидрата
перегонкой в вакууме.
3. Другие методы получения
Фенилглиоксаль был получен из изонитрозоацетофенона через
бисульфитное соединение4-5 или действием нитрозилсерной или азоти-
стой кислоты7. Он был получен также окислением бензоилкарбинола
уксуснокислой медью 3, нагреванием бромфенацилацетата9 и окисле-
нием ацетофенона двуокисью селена2.
1 Hahn, Schales, Ber. 67, 1823 A934).
2 R i 1 е у, М о г 1 е у, F r i с n d, J. Chem. Soc. 1932, 1875; англ. пат.
354 798 [С. А. 26, 3804 A932)]; ам. пат. 1 955 890 [С. А. 28, 4067 A934)].
3 Kaplan, J. Am. Chem. Soc. 63, 2654 A941).
i Pinner, Ber. 35, 4132 A902); там же 38, 1532 A905).
6 Pechmann, там же 20, 2904 A887); Miiller, Pechmann,
там же 22, 2556 A889); S m e d 1 e у, J. Chem. Soc. 95, 218 A909).
«Neuberg, Hofmann, Biocliem. Z. 229, 443 A930).
7 Neuberg, Hofmann, там же 239, 495 A931); Cusma no, Gazz.
chim. ital. 68, 130 A938).
«Net, Ann. 335, 271 A904); H e n z e, Z. physiol. Chem. 198, 83 A931);
там же 200, 232 A931).
9 Madelung, Oberwegner, Ber. 65, 935 A932).
О-ФЕНИЛЕНДИАМИН
/f\
NH2
NO,
+ 3Zn + H.,0 —
(NaOH)
;nh.
+ 3ZnO
Предложил: Е. Мартин.
Проверили: В. Хартман и С. Фирке.
1. Получение
В 1-литровую трехгорлую круглодонную колбу, снабженную ме-
ханической мешалкой с жидкостным затвором и обратным холо-
дильником, помещают 69 г @,5 мол.) о-нитроанилина («Синт. орг.
преп.», сб. 1, стр. 291), 40 мл 20%-ного раствора едкого натра и
200 мл 95%-ного этилового спирта. Смесь энергично перемешивают
510
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
у-ФЕНИЛМАСЛЯНАЯ КИСЛОТА
511
и нагревают на водяной бане до начала кипения. Затем нагревание
прекращают и прибавляют 130 г B гр.-ат.; примечание 1) цинковой
пыли порциями по 10 г, так чтобы раствор непрерывно кипел (при-
мечания 2 и 3). После того как цинковая пыль прибавлена, смесь
перемешивают и кипятят еще 1 час; окраска раствора изменяется от
тёмнокрасной до почти бесцветной. Горячую смесь фильтруют с отса-
сыванием, остаток цинковой пыли вновь возвращают в колбу и
извлекают два раза горячим спиртом порциями по 150 мл. Филь-
траты соединяют вместе, добавляют к ним 2—3 г гидросульфита
натрия и раствор упаривают в вакууме водоструйного насоса на
водяной бане до объема 125—150 мл. После тщательного охлажде-
ния в смеси льда и соли выпавшие светложелтые кристаллы отса-
сывают, промывают один раз небольшим количеством ледяной воды
и сушат в вакуум-эксикаторе. Выход неочищенного о-фенилендиами-
на с т. пл. 97—100° составляет 46—50 г (85—93% теоретич.). При
Желании получить более чистый продукт полученные кристаллы
растворяют в 150—175 мл горячей воды, к которой добавлено 1—2 г
гидросульфита натрия, и раствор обрабатывают обесцвечивающим
углем. После тщательного охлаждения смесью льда и соли бесцвет-
ные кристаллы отфильтровывают с отсасыванием и промывают 10—
15 мл ледяной воды. Выход очищенного о-фенилендиамина состав-
ляет 40-—46 г G4—85% теоретич.). Т. пл. 99—101° (примечания 4 и 5).
2. Примечания
1. Цинковая пыль должна быть не ниже 80%-ной чистоты, при-
чем взятое количество должно отвечать 130 г 100%-ного продукта.
Опыты, поставленные с большим избытком цинковой пыли, не при-
вели к повышению выхода.
2. Необходимо соблюдать осторожность и вначале не прибавить
слишком много цинковой пыли, так как в этом случае реакция про-
текает очень бурно. Хорошо иметь под рукой баню со льдом и
мокрые полотенца, для того чтобы регулировать реакцию, если она
пойдет слишком бурно.
3. Иногда реакция неожиданно приостанавливается, и для того,
чтобы она вновь пошла, необходимо добавить еще 10 мл 20%-ного
раствора едкого натра.
4. Продукт можно также очистить перегонкой в вакууме в атмо-
сфере инертного газа. Однако только в том случае, если исходный про-
дукт не достаточно чист, имеет место значительное разложение и пе-
регнанный продукт быстро темнеет при соприкосновении с воздухом.
5. Свободный диамин может быть также превращен в двойную
хлористоводородную соль, а соль очищена следующим образом.
Сырой о-фзнилендиамлн растворяют в смеси 90—100 мл концентри-
рованной соляной кислоты (уд. в. 1,19) и 50—60 мл воды, к которой
добавлено 2—3 г хлористого олова. Горячий раствор обрабатывают
обесцвечивающим углем, раствор фильтруют и приливают 150 мл
концентрированной соляной кислоты. Смесь хорошо охлаждают
смесью льда и соли, бесцветные кристаллы отфильтровывают с отса-
сыванием, промывают небольшим количеством холодной концен-
трированной соляной кислоты и сушат их в вакуум-эксикаторе над
твердым едким натром. Выход двухлористоводородной соли о-фени-
лендиамина 77—81 г (85—90% теоретич., считая на весовое коли-
чество взятого о-нитроанилина).
3. Другие методы получения
о-Фенилендиамии был получен восстановлением о-нитроанилина
оловом и соляной кислотой1, хлористым оловом и соляной кислотой2,
станнитом натрия3, цинковой пылью в присутствии воды4, гидро-
сульфитом натрия и едким натром5 и цинковой пылью и спиртовым
раствором щелочи6; электролитическим восстановлением в растворе
водного спирта в присутствии уксуснокислого натрия7 и из о-хлор-
бензола или о-хлоранилина действием на них водного аммиака при
150° под давлением в присутствии меди8. Описанная выше методика
является видоизменением метода Гинсберга и Кенига 6.
о-Фенилендиамин был предложен в качестве реактива для иден-
тификации жирных кислот, путем превращения их в кристалличе-
ские 2-алкилбензимидазолы (стр. 85).
iZincke, Sintenis, Ber. 6, 123 A873); К о е г n e r, Gazz. chim.
ital. 4, 320 A874); Hubner, Ann. 209, 361 A881).
2 Goldschmidt, Ingebrochtsen, Z. physik. Chem. 48, 448
A904); Goldschmidt, Sunde, там же 56, 23 A906).
8 Goldschmidt, E с k a r d t, там же 56, 400 A906).
4 Bamberger, Bcr. 28, 250 A895).
5 Borsche, Chem. Zentr. 1909, II, 1550.
"Hinsberg, Konig, Ber. 28, 2947 A895).
' Rohde, Z. Elektrochem. 7, 339 A900).
8 Soc. pour l'ind. chim. a Bale; франц. пат. 788 348 [С. А. 30, 1395 A936)].
Y-ФЕНИЛМАСЛЯНАЯ КИСЛОТА
+ 4 [Н]
свн6снаснаснасо2н + н2о
Предложил: Е. Мартин.
Проверили: К. Ноллер и Ф. Мак-Миллан.
1. Получение
Для получения амальгамированного цинка смесь 120 г цинковой
шерсти, 12гхлористой ртути, 200 мл воды и 5—б мл концентрирован-
ной соляной кислоты взбалтывают в 1-литровой круглодонной колбе
в течение 5 мин. Раствор сливают и в колбу прибавляют следующие
512
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
ФЕНИЛНЙТРОМЕТАН
513
реактивы в указанном ниже порядке: 75 мл воды, 175 мл концентри-
рованной соляной кислоты, 100 мл толуола и 50 г @,28 мол.) 2-бен-
зоилпропионовой кислоты (стр. 95). Колбу закрывают пробкой,
в которую вставлен обратный холодильник, верхний конец которого
присоединен к газовой ловушке (примечание 1). Реакционную смесь
энергично кипятят 25—30 час. (примечание 2), причем каждые б час.
приливают по 50 мл концентрированной соляной кислоты. По охла-
ждении до комнатной температуры слои разделяют и водный слой
разбавляют 200 мл воды и затем извлекают эфиром три раза порция-
ми по 75 мл. Толуольный слой и эфирные вытяжки соединяют, про-
мывают водой и сушат хлористым кальцием. Растворители отгоняют
в вакууме на водяной бане, после чего у-фенилмасляную кислоту
также перегоняют при 178—181°/19 мм A48—154°/8— 10 мм, 125—
130°/3 мм). Выход кислоты с т. пл. 46—48° (примечание 3) соста-
вляет 38—41 г (82—89% теоретич.; примечание 4).
2. Примечания
1. В начале нагревания выделяется значительное количество
хлористого водорода, так что казалось бы целесообразным вместо
концентрированной соляной кислоты брать кислоту с постоянной
точкой кипения. Однако в этом случае получается продукт с т. пл.
40—44°, и выход несколько понижается.
2. Если по какой-либо причине приходится прерывать кипя-
чение, следует принять особые меры предосторожности при возобно-
влении нагревания, для избежания сильного вспенивания. Время от
времени можно прогревать верхнюю часть колбы горелкой от руки.
После того как оба слоя хорошо смешались друг с другом, кипе-
ние протекает спокойно.
3. В литературе указана т. пл. от 47 до 5Г. Кислоту можно пере-
кристаллизовать из горячей воды G5 мл на 1 г), однако выход при
этой операции не превышает 50%. До сих пор для перекристалли-
зации не найдены более подходящие растворители или смесь рас
творителей. Вторичная перегонка ведет к повышению т. пл. до
47—48° и сопровождается только механическими потерями.
4. Приведенная здесь методика отличается от описанной в
Org. Syn. 15, 64 тем, что к реакционной смеси добавляют толуол.
В присутствии толуола концентрация органических веществ в вод-
ном слое чрезвычайно мала, и полимолекулярные реакции проте-
кают в меньшей степени, чем при работе по первоначальной методике.
Вследствие этого выход чистого продукта возрастает.
Если эту видоизмененную методику применить к получению
соединений с более высокой температурой плавления, например, у-на-
фтилмасляных кислот, то слои разделяют после того, как реакцион-
ная смесь охладится до 50—60°; для извлечения применяют бен-
зол и бензольно-толуольный раствор осветляют активированным
березовым углем до сушки, после чего его несколько упаривают и
оставляют кристаллизоваться. При получении метоксилированных
кислот, например i'-анизил- или "-вератрилмасляных кислот, имеет
место частичное деметилирование. В этом случае толуольный слой
и вытяжки смешивают с избытком разбавленного раствора едкого
натра и органические растворители отгоняют перегонкой с водя-
ным паром. Щелочной раствор обрабатывают при 80° избытком
диметилсульфата, раствор осветляют активированным древесным
углем, охлаждают и подкисляют. При этом продукт выпадает в
чистом виде.
3. Другие методы получения
Из многочисленных методов получения у-фенилмасляной кислоты
препаративное значение имеют следующие методы; отщепление
углекислоты от у-фенилэтилмалоновой кислоты1; карбоксилирова-
ние бромистого у-фенилпропилмагния2 и восстановление В-бензоил-
пропионовой кислоты амальгамированным цинком и соляной
кислотой3 или восстановление ее гидразона этилатом натрия4.
Применение толуола при восстановлении по Клемменсену C-бензоил-
пропионовой кислоты и более десятка других аналогичных соеди-
нений описано Мартином5.
1Fischer, Schmitz, Ber. 39, 2212 A906).
2 G г i g п а г d, Compt. rend. 138, 1049 A904); R u p e, P г о s k e, Ber.
43, 1233 A910).
3 К г о 1 1 p f e i f f e r, Schafer, там же 56, 620 A923).
4 Staudinger, M ii 1 1 e г, там же 56, 713 A923).
6 M a r t i n, J. Am. Chem. Soc. 58, 1438 A936).
ФЕНИЛНЙТРОМЕТАН
(а-Нитротолуол)
CeHsCH.,CN + CH3ONO2 + NaOC2H5 >
C6H5C = NOaNa + CH3OH + C2H5OH
CN
~~~ CRHX=NO,Na
CN
CO,Na
HCl
C6H6CH = N02H + CO2
C6H3CH ==¦ NO2H > C6H5CH2NO2
Предложили: А. Блек и Ф. Бебсрс.
Проверили: Дж. Джонсон и X. Стивенсон.
1. Получение
Л. Натриевая соль феншацинитроацетонитрила. В 2-литро-
вую круглодонную колбу с эффективным обратным холодильником
33 Сборник К: 2
5D
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
наливают 400 мл абсолютного спирта и через внутреннюю трубку
холодильника прибавляют с максимальной быстротой 46 г B гр.-ат,)
свеженарезанного металлического натрия, после чего колбу нагре-
вают на масляной бане. Примерно через 0,5 часа остается только
небольшой шарик расплавленного натрия @,5—1,0 г) и начинает
выпадать этилат натрия. Тогда приливают 100 мл абсолютного спирта
и смесь охлаждают до 0°. Затем поверх твердой лепешки этилата
натрия в колбе приливают еще 100 мл абсолютного спирта (примеча-
ние 1). Вместо обратного холодильника колбу закрывают пробкой,
в которую вставлена делительная воронка и хлоркальциевая трубка.
При непрерывном взбалтывании прибавляют охлажденную до 0° смесь
234 г B мол.) свежеперегнанного цианистого бензила («Синт. орг.
преп.», сб. 1, стр. 502) и 216 гA80лл, 2,8 мол.) метилнитрата (приме-
чание 2) с такой скоростью, чтобы температура поддерживалась при
4—8°. После того как прибавление окончено, на что требуется около
1 часа, реакционную смесь оставляют стоять при 4—8° еще в течение
1 часа, время от времени взбалтывая содержимое колбы. Затем колбу
закрывают пробкой, в которую вставлен бунзеновский клапан, и кол-
бу помещают на 24 часа в охладительную смесь. Выпавшую натрие-
вую соль аци-нитросоединения отфильтровывают с отсасываниемчерез
воронку Бюхнера, тщательно промывают абсолютным эфиром (при-
мечание 3) и еушат на воздухе. Вес первой порции кристаллов со-
ставляет 215—275 г E8—75% теоретич.). Маточный раствор и эфир-
ную промывную жидкость соединяют вместе и упаривают до объ-
ема около 150 мл в вакууме. Последовательно отфильтровывают с
отсасыванием выпадающие порции натриевой соли и промывают их
абсолютным эфиром. Суммарный выход неочищенной натриевой
соли составляет 275—300 г G5—82% теоретич.). Этот продукт
обычно применяют без дальнейшей очистки.
Б. Фенилнитрометан. В 4-литровом стакане растворяют 300 г
едкого натра в 1,2 л воды. Стакан помещают в эмалированную каст-
рюлю (в качестве предосторожности, чтобы он не треснул) и раствор
нагревают до кипения. К кипящей щелочи в течение 1 часа неболь-
шими порциями прибавляют высушенную на воздухе неочищенную
натриевую соль фенилнитроацетонитрила B75—300 г). Кипячение
продолжают до тех пор, пока не прекратится выделение аммиака
(около 3 час); время от времени приливают горячую воду, чтобы
объем раствора оставался примерно постоянным (примечание 4).
Горячий щелочной раствор выливают в плоскую фарфоровую чашку,
в которой по охлаждении он застывает в воскообразную массу.
Лепешку сырой натриевой соли фенилнитроуксусной кислоты
разбивают ложкой, переносят в 4-литровый стакан и размешивают
с 500 г льда. Стакан помещают в большой глиняный сосуд, наполнен-
ный смесью льда и соли, и в стакане устанавливают механическую
мешалку. Когда раствор в стакане охладится до —5", через капель-
ную воронку медленно приливают при энергичноработающеимешалке
ФЕНИЛНИТРОМЁТАН
515
концентрированную соляную кислоту до слабо кислой реакции на
конго. Во время приливания кислоты температура не должна под-
ниматься выше —5° (примечание 5). Обычно требуется около 900 мл
кислоты, и приливание продолжается около 2 час. Холодный раствор
экстрагируют один раз 500 мл эфира и два раза по 250 мл эфира.
Эфирные вытяжки соединяют и промывают ледяным насыщенным
раствором двууглекислой соды до тех пор, пока промывная жид-
кость не станет бесцветной или лишь слабожелтой (обычно бывает
достаточно взять две порции по 100 мл). Затем эфирный раствор
промывают 250 мл ледяной воды, к которой добавлено 2 капли со-
ляной кислоты, и, наконец, еще тремя порциями по 50 мл ледяной
воды. Эфирный раствор сушат безводным сернокислым натрием и
оставляют стоять в течение 3—4 дней, чтобы завершилась изомериза-
ция лабильной аци-формы. Раствор фильтруют, эфир отгоняют при
15—20° в вакууме и оставшееся масло перегоняют при давлении
3 мм из обыкновенной колбы Клайзена. Фенилнитрометан получают
в виде светложелтого масла с т. кип. 90—92°/3 мм (примечание 6).
Выход 135—150 г E0—55% теоретич., считая на цианистый бензил;
примечание 7).
2. Примечания
1. Верхний слой спирта препятствует непосредственному сопри-
косновению прибавляемых веществ с этилатом натрия и вызы-
вает местный перегрев.
2. Большой избыток метилнитрата заметно увеличивает выход.
Свежеприготовленный метилнитрат (стр, 329) был высушен и приме-
нялся непосредственно без перегонки. Весьма удобно использовать
все количество его (обычно 210—230 г), полученное из 120 г метило-
вого спирта.
3. Можно применить обыкновенный эфир, постоявший несколько
дней над безводным хлористым кальцием.
4. За реакцией необходимо тщательно наблюдать. Иногда имеет
место сильное вспенивание, что требует прибавления небольших
количеств воды из промывалки.
,»* 5. При —5° аци-нитросоединение выпадает в виде серого пасто-
образного твердого вещества. Когда раствор становится кислым,
коллоидальный осадок стремится коагулировать.
6. При перегонке сырого продукта весьма важно поддерживать
низкое давление и избегать перегрева. Если перегонку вести слиш-
ком далеко, то происходит разложение и дестиллат быстро окра-
шивается при стоянии. Если перегонку вести не достаточно осто-
рожно, то может произойти бурное разложение, в результате
которого обычно срываются все каучуки.
Перегнанный продукт разлагается при хранении и его сле-
дует быстро использовать. Фенилнитрометан нельзя хранить
516
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
в сосуде с притертой стеклянной пробкой, так как пробку
легко заедает и попытки открыть склянку могут повести к
взрыву.
7. Описанная здесь методика может быть значительно сокращена
и упрощена, если работать при несколько более высоких температу-
рах, чем указано выше. Так, было найдено, что выгодно добавлять
раствор цианистого бензила и метилнитрата к этилату натрия при
взбалтывании и при температуре 5—15°. При этой температуре
реакция протекает гладко и с постоянной скоростью. Общий
выход натриевой соли удовлетворительного качества составляет
300—320 г.
Омыление нитрила и подкисление лучше всего проводить в том
же 4-литровом стакане; при этом избегается необходимость пере-
ливания. Для растворения едкого натра, необходимого для омыле-
ния, было взято 1,5 л воды. Как только прекратится выделение амми-
ака (лакмус) стакан помещают в смесь льда и соли и смесь энергично
перемешивают с помощью механической стеклянной мешалки. Когда
температура понизится до 30°, добавляют 500 г льда. Подкисление
соляной кислотой (800—850 мл) проводят, при 0—10° при непре-
рывном перемешивании. Реакционную массу оставляют на ночь и
на следующее утро извлекают эфиром. Эфирные растворы достаточно
оставить стоять над сернокислым натрием на 20—24 часа перед пере-
гонкой. При соблюдении этих условий выход составляет 153—163 г
светлоокрашенного продукта с т. кип. 92—95°/4 мм (температура
бани 135°). От остатка при более высокой температуре можно ото-
гнать еще 10—15 г более темного продукта (Л. Физер и Е. Берли-
нер, частное сообщение).
3. Другие методы получения
Фенилнитрометан был получен нитрованием толуола разбавлен-
ной азотной кислотой в запаянной трубке1, взаимодействием между
хлористым фенилдиазонием и нитрометаном в щелочном растворе2;
действием нитрита серебра на хлористый бензил3 или йодистый бен-
зил4 и конденсацией этилнитрата с цианистым бензилом с после-
дующим омылением6. Применение метилнитрата, который может
быть получен с большими удобствами и с меньшей опасностью, имеет
определенные преимущества.
1 Коновалов, ЖРХО 27, ч. II, 54 A895); Вег. 28, I860 A895).
а Bamberger, Schmidt, Levinstein, там же 33, 2053 A900).
3 Hollemann, Rec. trav. chim. 13, 405 A894).
4 Hantzsch, Schultze, Ber. 29, 700 A896).
6 Wislicenus, Endres, там же 35, 1755 A902); Гатте р ман—
В и л а н д, Практические работы по органической химии, 5 изд., Госхим-
техиздат, М. — Л., 1948, стр. 293.
2-ФЕНИЛПИРИДИН
517
2-ФЕНИЛПИРИДИН
С6Н5Вг + 2 Li = C6H5Li
C6H5Li + C5H5N
C6H6
H
¦ LiH +C6H5
N
Li
N
Предложили: Дж. Эванс и Н. Аллен.
Проверили: Р. Фъюзон, В. Росс и Е. Кливленд,
1. Получение
В 1-литровую трехгорлую колбу, снабженную капельной ворон-
кой, термометром, механической мешалкой и обратным холо-
дильником, защищенным от влаги воздуха (примечание 1), после
промывки колбы сухим азотом помещают 3,5 г @,5 гр.-ат.) лития, на-
резанного кусочками размером с горошину, и 100 мл абсолютного
эфира. Пускают в ход мешалку и из капельной воронки приливают
около 10 мл смеси 40 г бромбензола @,25 мол.) в ЬО мл абсолютного
эфира. Обычно при этом протекает бурная реакция (примечание 2).
Остальное количество смеси приливают постепенно в течение 30 мин.,
после чего лития почти не остается (примечание 3).
Затем из капельной воронки медленно приливают при работаю-
щей мешалке раствор 40 г @,5 мол.) сухого пиридина (примечание 4)
в 100 мл сухого толуола. Эфир отгоняют (примечание 5) и оставшуюся
суспензию перемешивают при 110° (температура внутри колбы) в те-
чение 8 час. Реакционную массу охлаждают до 40°, осторожно
приливают 35 мл воды и, если нужно, жидкости фильтруют (приме-
чание 6). Нижний слой отделяют и отбрасывают. Толуольный слой
сушат в течение 1 часа 20 г измельченного едкого кали и осторожно
перегоняют, пользуясь видоизмененной колбой Клайзена с дефлег-
матором. Головную фракцию до 150° отгоняют при атмосферном
давлении, а остаток перегоняют в вакууме; после двух фракциони-
рованных перегонок выход 2-фенилпиридина с т. кип. 140°/12 мм
составляет 15,5—19 г D0—49% теоретич.).
2. Примечания
1. Прибор и реактивы должны быть высушены, столь же тща-
тельно как для магний-органического синтеза.
2. Иногда реакция не начинается без нагревания; однако, как
только реакция начнется, источник нагрева следует удалить.
3. Выход фениллития около 75%. Этот выход можно определить,
если фениллитий заставить прореагировать с избытком бензофенона
518
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
ФЕНИЛПИРОВИНОГРАДНАЯ КИСЛОТА
519
и взвесить количество образовавшегося трифенилкарбинола, пред-
полагая, что выход карбинола количественный1.
4. Успех синтеза зависит от сухости пиридина. Пиридин кипя-
тился 8 час. с обратным холодильником со свежей негашеной
известью, перегонялся, после чего подвергался такой же обработке,
но в присутствии порошкообразного едкого кали. Ни один из сортов
окиси бария таких хороших результатов не давал. Наиболее высо-
кие выходы были получены с пиридином для медицинских целей.
5. Это легко осуществить, если спустить воду из холодильника
при нагревании до 110°.
6. Обычно остающиеся небольшие частицы не вошедшего в реак-
цию металла мешают расслаиванию.
3. Другие методы получения
2-Фенилпиридин был получен из пиридина и фениллития 2 или
хлористого фенилдиазония3. В указанной выше методике при
замене эфира в качестве растворителя толуолом избегается необ-
ходимость работать в запаянной трубке 4.
1 G i I m a n, Zoellner, Selby, J. Am. Chem. Soc. 54, 1957 A932).
2 Ziegler, Zeiser, Ber. 63, 1847 A930).
3 Haworth, Heilbron, Hey, J. Chem. Soc. 1940, 352.
* Walters, McElvain, J. Am. Chem. Soc. 55, 4625 A933).
ФЕНИЛПИРОВИНОРРАДНАЯ КИСЛОТА
CBH,CH = CCO,H
NHCOCH,
4- 2 Н20 + НС1 >
С6НЙСН2СОСО,Н -f CHgCO.H + NH4C1
Предложили: Р. Хербст и Д. Шемин.
Проверили: Р. Фьюзон и Е. Кливленд.
1. Получение
В колбу емкостью 500 мл, к которой на шлифу присоединен
обратный холодильник, помещают Юг@,05мол.) а-ацетоаминокорич-
ной кислоты (стр. 72) и 200 мл 1-н. соляной кислоты (примечание 1).
Омыление заканчивается после кипячения в течение 3 час. Из кипя-
щего раствора могут выделиться несколько капелек светлозеленого
масла, которые отфильтровывают. По охлаждении из фильтрата
выпадают кристаллы фенилпировиноградной кислоты, которые
отсасывают (примечание 2) и в воронке Бюхнера промывают
небольшим количеством ледяной воды. Фильтрат и промывные
воды соединяют и извлекают четыре раза эфиром порциями по
50 мл. Эфир удаляют выпариванием при комнатной температуре,
под конец в вакуум-эксикаторе (примечание 3). Остаток присоеди-
няют к первой порции кристаллов и продукт сушат в вакуум-экси-
каторе над хлористым кальцием и едким кали. Выход 7,2—7,7 г (88—
94%теоретич.), причем т. пл. 150—154° (примечания 4 и 5).
2. Примечания
1. Можно получить и большие количества фенилпировиноград-
ной кислоты, если соответственно увеличить количество исходных
продуктов. Однако так рекомендуется поступать только в том слу-
чае, если продукт предполагают использовать немедленно, так как
фенилпировиноградная кислота при стоянии начинает разлагаться
уже через несколько дней.
2. Количество фенилпировиноградной кислоты, выпадающее из
фильтрата, увеличивается, если до фильтрования раствору дать
постоять в течение нескольких дней в холодильном шкафу. Если
продукт находится в виде взвеси в холодной разбавленной кислоте,
разложения не наблюдается.
3. Выпаривание при комнатной температуре можно удобно осу-
ществить, если над поверхностью раствора продувать ток сухого
воздуха, а над раствором установить стеклянный колпак.
4. Температура плавления в значительной степени зависит от
скорости нагревания образца.
5. Фенилпировиноградную кислоту можно перекристаллизо-
вать из дихлорэтана, бензола или хлороформа, однако потери при
этом вследствие нестойкости продукта довольно значительны.
3. Другие методы получения
Фенилпировиноградная кислота была получена омылением я-бен-
зоиламинокоричной кислоты щелочами или кислотами1'2; омыле-
нием кислотами этилового эфира фенилщавелевоуксусной кислоты 3;
омылением кислотами этилового эфира фенилцианпировиноградной
кислоты2-4; дегидратацией р-фенилглицериновой кислоты сер-
ной кислотой5 и щелочным гидролизом «-ацетоаминокоричной
кислотыв.
1 Р loch I, Ber. 16, 2817 A883).
г Erlenmeyer, Jr., Ann. 271, 165, 173 A892).
3 W i s 1 i с е n u s, Ber. 20, 592 A887).
1 Erlenmeyer, Jr., Arbenz, Ann. 333,228A904); Hemmerle,
Ann. chim. (9) 7, 229 A917).
5 Dieckmann, Ber. 43, 1034 A910).
«Bergmann, Stern, Ann, 448, 27 A926).
520
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
ФЕНИЛПРОПИОЛОВАЯ КИСЛОТА
С6Н5СНВгСНВгСО2С2Н5 + ЗКОН
СвН5С = ССО2К + С2Н6ОН + 2 КВг +
С6Н3С = ССО2К + H2SO4 > С6Н5С не ССО2Н + KHSO4
2 Н2О
Предложил: Т. Абботт.
Проверили: Г. Гильман и Дж. Райт
1. Получение
В 3-литровой круглодонной колбе, снабженной обратным холо-
дильником и нагреваемой на водяной бане, приготовляют раствор
252,5 г D,5 мол.) едкого кали (примечание 1) в 1,2 л 95%-ного
спирта. К щелочному раствору, охлажденному до 40—50°, приба-
вляют 336 г A мол.) неочищенного этилового эфира а,[3-дибром-
р-фенилпропионовой кислоты (стр. 586). Когда первоначально бур-
ная реакция прекратится, содержимое колбы нагревают в продол-
жение 5 час. с обратным холодильником на кипящей водяной
бане.
Затем реакционную смесь охлаждают и выделившиеся соли
отсасывают. К фильтрату добавляют до нейтральной реакции на
лакмус (примечание 2) концентрированную соляную кислоту и
выпавшие соли снова отсасывают. Затем фильтрат концентрируют
отгонкой до тех нор, пока температура паров не достигнет 95°.-
Остаток и ранее отфильтрованные соли соединяют, растворяют
в 800 мл воды и охлаждают прибавлением колотого льда. Лед при-
бавляют в таком количестве, чтобы общий объем был равен 1,8 л
(примечание 3). Охлажденный раствор помещают в баню с ледяной
водой и при механическом перемешивании к нему приливают20%-ный
раствор серной кислоты до сильно кислой реакции на лакмус. Пере-
мешивание ведут 20 мин. и затем фенилпропиоловую кислоту отса-
сывают и промывают четыре раза порциями по 30 мл 2%-ного
раствора серной кислоты.
Полученную в виде светлокоричневых зерен кислоту растворяют
в 1 л 5%-ного раствора соды, прибавляют к раствору 20 г активи-
рованного угля и в течение 30 мин. нагревают на водяной бане, пере-
мешивая время от времени раствор. Затем его фильтруют, охлаждают
извне, а внутрь прибавляют 200 г колотого льда. При механическом
перемешивании к раствору медленно приливают 20%-ный раствор
серной кислоты. Выпавшую кислоту отсасывают, промывают сначала
50 мл 2%-ной серной кислоты, затем небольшим количеством воды и
сушат на воздухе. Выход кислоты, плавящейся между 115 и
125°, составляет 112—118 г G7—81% теоретич.).
ФЕНИЛПРОПИОЛОВАЯ КИСЛОТА
521
100 г полученной кислоты можно очистить перекристаллизацией
из 200—300 мл четыреххлористого углерода; получается 70 г
фенилпропиоловой кислоты с т. пл. 135—136°.
2. Примечания
1. Лучшие выходы получаются, если брать 50% избыток едкого
кали. Концентрация щелочи почти не влияет на выход.
2. Спирт лучше отгонять от нейтральных, а не от щелочных
растворов.
3. Чтобы не произошло отщепления углекислоты, температуру
надо поддерживать как можно ниже. Если этой предосторожности
не соблюдать, то выход понижается и продукт получается менее
чистый.
3. Другие методы получения
В основном описанный способ разработал Перкин х. Фенилпро-
пиоловая кислота может быть получена также из эфирных раство-
ров ^-бромстирола 2 и ^-хлорстирола 3 действием натрия и углекис-
лого' газа; из ^-бромстирола и бугиллития в эфирном растворе 4;
действием спиртового раствора щелочи на а-бромкоричную кислоту2,
[3-бромкоричную кислоту5 или этиловый эфир а-бромкоричной
кислоты 6, а также действием углекислого газа на натрий-фенил-
ацетилен 2'7.
Получение фенилпропиоловой кислоты действием щелочи на
«, р-дибромкоричную кислоту, т.е. еще более прямой путь, чем из
сложного эфира, применялось мало вследствие трудностей, связан-
ных с получением дибромкислоты. Однако в литературе имеется
указание, что «, °-дибромкоричная кислота может быть легко полу-
чена с выходом в 95% присоединением брома к коричной кислоте
в кипящем четыреххлористом углероде и что неочищенный продукт
может быть использован для синтеза фенилпропиоловой кислоты 8.
В этой же статье описан упрощенный способ получения небольших
количеств фенилпропиоловой. кислоты, исходя из а, р-дибромкорич-
ной кислоты.
1 Р е г k i n, J. Chem. Soc. 45, 172A884); Liebermann, Sachs с,
Ber. 24, 4113 A891).
2 G laser, Ann. 154, 140, 162 A870).
3 Erlenmeyer, Ber. 16, 152 A883).
i G i 1 m a n, L a n g h a m, M о о r e, J. Am. Chem. Soc. 62, 2328 A940).
6 В a r i s с h, J. prakt. Chem. B) 20, 180 A879).
«Michael, Ber. 34, 3647 A901).
' E. I. du Pont de Nemours and Company; ам. пат. 2 194 363 [С. А. 34, 4745
A940)].
s R e i m e r, J. Am. Chem. Soc. 64, 2510 A9-12).
522
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
а-ФЕНИЛЭТИЛАМИН
523
НС — СН
ФЕНИЛТИЕНИЛКЕТОН
(Фенил-2-тиенилкетон)
НС —СН
НС СН + С6Н6СОС1
\/
S
А1С13
НС С — СОС6Н3 + НС1
Предложил: В. Миннис.
Проверили: Р. Адаме и X. Коган.
1. Получение
В 1-литровую трехгорлую колбу, снабженную механической
мешалкой, обратным холодильником и термометром (шарик опущен
в жидкость), помещают 100 г @,75 мол.) безводного хлористого алю-
миния и 300 г сероуглерода (примечание 1). Суспензию охлаждают
до 15—25° и через холодильник в течение 3,5 часа при перемешива-
нии приливают раствор 60 г @,71 мол.) тиофена (стр. 458) и 105 г
@,75 мол.) хлористого бензоила в 225 г сероуглерода (примечание 2).
Раствору дают нагреться до комнатной температуры, перемешивание
продолжают еще 3 часа и затем реакционную смесь оставляют стоять
на ночь. На другой день смесь кипятят с обратным холодильником
на водяной бане в продолжение 3,5 часа, охлаждают, выливают на лед
и экстрагируют эфиром. Эфирный раствор промывают сначала рас-
твором соды, потом водой и сушат хлористым кальцием. Эфир отго-
няют на водяной бане, а остаток перегоняют в вакууме. Выход
продукта, кипящего при 200—209730—40 мм, составляет 117—
120 г (88—90% теоретич.). При перекристаллизации из 1 л петро-
лейного эфира (т. кип. 65—110°) получается ПО—112 г продукта
с т. пл. 52°. Вторичная кристаллизация из петролейного эфира
дает продукт, плавящийся при 55—56°. Потеря при второй кристал-
лизации около 10%.
2. Примечания
1. Сероуглерод сушился хлористым кальцием.
2. Тиофен и хлористый алюминий, суспендированный в серо-
углероде, реагируют бурно. Если затем хлористый бензоил в рас-
творе сероуглерода прибавлять к тиофену, происходит осмоление
и получается небольшой выход кетона.
3. Другие методы получения
Фенилтиенилкетон был получен действием хлористого бензоила
на хлористую тиенилртуть 1; действием хлористого бензоила на тио-
фен в присутствии хлористой тиенилртути2, фосфорного ангид-
рида 3, хлорного олова 4 и хлористого алюминия Б. Он был получен
также из йодистого тиенилмагния и бензонитрила 6.
1 V о 1 h a r d, Ann. 267, 179 A892); Steinkopf, Killingstad,
там же 532, 288 A937).
2 Steinkopf, Bauermeister, там же 403, 70 A914).
3 S t e i n k о р f, там же 413, 349 A917).
4Стадников, Раковский, Ber. 61, 269 A928); Гольдфарб,
ЖРХО 62, 1073 A930).
6 Comey, Ber. 17, 790 A884).
•Thomas, Couderc, Bull. soc. chim. D) 23, 289 A918).
а-ФЕНИЛЭТИЛАМИН
(а-Метилбензиламин)
C6H5COCH3 + 2 HCO2NH4
C6H5CH (NHCHO) CH3 + 2 H2O + NH3 + СО,
С6Н5СН (NHCHO) CH3 + Н3О + НС1 ¦-.
С6Н5СН (NH3C1) СН3 + НСО2Н
CeH6CH (NH8C1) СН3 + NaOH — С6Н5СН (NH,) СН3 + NaCl + Н2О
Предложил: А. Ингерсолл.
Проверили: Р. Фьюзон и В. Росс.
1. Получение
В видоизмененную колбу Клайзена емкостью 500 мл помещают
250 г D мол.) муравьинокислого аммония (примечание 1), 150 г
A,25 мол.) ацетофенона (примечание 2) и несколько кусочков не-
глазурованной тарелки. Колбу снабжают пробкой, через которую
проходит термометр, доходящий почти до дна, а ее боковой отвод
присоединяют к небольшому обращенному вниз холодильнику.
Колбу нагревают на небольшом пламени, смесь сперва распла-
вляется, образуя при этом 2 слоя, и начинается перегонка. При 150—
155° смесь становится однородной и начинается реакция, причем
имеет место некоторое вспенивание. Нагревание продолжают в слу-
чае нужды более медленно, пока температура не достигнет 185°.
В течение около трех часов отгоняются вода, ацетофенон и углекис-
лый аммоний; процесс не требует тщательного наблюдения. При
185° нагревание прекращают, верхяий слой ацетофенона отделяют
от остальной части дестиллата и без предварительной сушки вли-
вают его обратно в реакционную колбу. После этого смесь нагре-
вают 3 часа при 180—185®. Дестиллат экстрагируют 25—30 мл
бензола, чтобы извлечь ацетофенон (примечание 3), а водную часть
выливают.
Реакционную смесь охлаждают и взбалтывают в делительной во-
ронке емкостью 500 мл со 150—200 мл воды для того, чтобы удалить
524
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
муравьинокислый аммоний и формамид. Неочищенный а-фенил-
этилформамид вновь сливают в ту же колбу, а водный слой экстра-
гируют два раза бензолом порциями по 30 мл, после чего его выли-
вают. Бензольные вытяжки соединяют вместе с основной массой и
добавляют 150 мл концентрированной соляной кислоты и несколько
кусочков неглазурованной тарелки. Смесь осторожно нагревают,
пока не отгонится весь бензол, после чего ее кипятят еще 40—50 мин.
Гидролиз протекает быстро, и смесь становится однородной, если
не считать небольшого слоя ацетофенона и других нейтральных
примесей. Смесь охлаждают и экстрагируют сперва 50 мл, а затем
тремя или четырьмя порциями по 25 мл бензола. Вытяжки эти со-
храняют для того, чтобы выделить из них обратно ацетофенон (при-
мечание 3).
Водный кислый слой переносят в 1-литровую круглодонную
колбу, снабженную делительной воронкой и подготовленную для
перегонки с водяным паром. Через воронку приливают раствор
125 г едкого натра в 250 мл воды, и смесь подвергают перегонке
с водяным паром (примечание 4). Большая часть амина содержится
в первом литре дестиллата, однако, дестиллат следует собирать до
тех пор, пока он не станет уже слабощелочным. В колбе остается
небольшое количество ди-(д-фенилэтил)-амина и нейтральных
веществ, которые можно отбросить.
Дестиллат экстрагируют 5 раз бензолом порциями по 50 мл,
бензольный раствор тщательно сушат измельченным в порошок
едким натром и перегоняют (примечание 5). Большая часть амина
переходит при 184—186°, однако для большинства целей достаточно
чистой является более широкая фракция 180—190° (примечание 6).
Выход этой фракции составляет 80—88 г. Если соединить вместе
головную бензольную фракцию и остаток от перегонки, экстраги-
ровать их разбавленной кислотой и выделить вновь амин так, как
это указано выше, можно получить еще 10—12 г продукта (приме-
чание 7), так что суммарный выход составляет 90—100 г F0—66%
теоретич., считая на взятый в реакцию ацетофенон; примечание 8).
2. Примечания
1. Муравьинокислый аммоний можно получить в большом коли-
честве действием на тьердый углекислый аммоний небольшим избыт-
ком продажной 85%-юой муравьиной кислоты и упариванием рас-
твора по частям на паровой бане в вакууме. Слегка влажный про-
дукт, получаемый после отсасывания, вполне пригоден для этого
синтеза. ?
2. Применялся ацетофенон с т, пл. 16—20°. Метод получения
ацетофенона приведен в «Синт. орг. преп.», сб. 1, стр. 106.
3. Бензольный раствор промывают разбавленной щелочью,
сушат и перегоняют, причем собирают фракцию 198—207°.
а-ФЕНЙЛЭТИЛАМиН
525
4. При перегонке с водяным паром рекомендуется непосредственно
нагревать перегонную колбу, чтобы объем раствора оставался почти
неизменным.
5. Амин действует разрушающе на корковые и каучуковые
пробки и поглощает из воздуха углекислоту. Лучше всего перего-
нять его из колбы, боковой отвод которой входит на небольшое рас-
стояние внутрь горла колбы, и собирать в приемник, защищенный
трубкой с натронной известью.
6. Если желают получить амин в очень чистом виде, то описан-
ный выше продукт растворяют вместе с 1,04 частями кристалличе-
ской щавелевой кислоты в 8 частях горячей воды. После обесцве-
чивания углем из профильтрованного раствора по охлаждении вы-
падают кристаллы щавелевокислой соли. В каждых 100 мл маточ-
ного раствора остается около 5 г соли; большая часть ее может
быть выделена путем выпаривания и дополнительной кристалли-
зации. Амин выделяется из чистого оксалата с помощью едкого
кали. Его отгоняют с водяным паром и очищают так, как это опи-
сано выше. Когда надо получить определенное количество амина
в водном растворе (например для расщепления на оптические анти-
поды), берут взвешенное количество (безводного) оксалата, раз-
лагают его и амин количественно перегоняют с водяным
паром.
7. Когда проводится последовательно несколько опытов, кислый
раствор амина можно присоединить к следующей порции до пере-
гонки с водяным паром.
8. Описанный выше метод представляет собою общий метод.
С соответственными изменениями в части, которая касается очистки
амина, по этому методу могут быть получены из соответствующих
кетонов: а-п-толилэтиламин G2%); а-п-хлорфенилэтиламин F5%);
а-л-бромфенилэтиламин F3%); а-л-ксилилэтиламин F6%) и а-
(?-нафтил)-этиламин (84%).
3. Другие методы получения
Настоящая методика была разработана по данным работ Вал-
лаха х и Фрейлона 2, основанных на общем методе, который открыт
Лейкартом 3. а-Фенилэтиламин может быть также получен с удо-
влетворительными результатами восстановлением оксима ацето-
фенона натрием в абсолютном спирте 4, или амальгамой натрия 5,
или амальгамой аммония6, или электролитическим восстановле-
нием7. Амин этот был также получен восстановлением фенилгидра-
зона ацетофенона амальгамой натрия и уксусной кислотой 8, из
бромистого а-фенилэтила и гексаметилентетраминаэ, действием
йодистого метилмагния на гидробензамид 10 и каталитическим вос-
становлением ацетофенона водородом в присутствии аммиака и
526
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
d- и f-к-ФЕНИЛЭТИЛАМИН
527
никелевого катализаторап. Подробные указания для работ по
этому методу приведены в Org. Syn. 23.
1 W a 1 1 а с h, Ann. 343, 60 A905).
2 F г е у 1 о n, Ann. chim. phys. (8) 15, 141 A908).
3 Leuckart, Ber. 22, 1413 A889).
* Mo h r, J. prakt. Chem. B) 71, 317 A905).
6 Kraft, Ber. 23, 2783 A890).
6 T a k a k i, U e d a, J. Pharm. Soc. Japan 58, 276 A938) [C. A. 32, 5376
A938)].
7 Tafel, Pfeffermann, Ber. 35, 1515 A902).
8 T a f e 1, там же 19, 1929 A886); 22, 1856 A889).
9 Andre, Vernier, Compt. rend. 193, 1192 A931).
10 В u s с h, Leefhelm, J. prakt. Chem. B) 77, 5 A908).
11 Couturier, Ann. chim. A1) 10, 610 A938); Schwoegler, Ad-
kin s, J. Am. Chem. Soc. 61, 3499 A939).
d- и /-а-ФЕНИЛЭТИЛАМИН
(d- и 1-а.-Метилбензиламж)
Предложил: А. Ингерсолл.
Проверили: Р. Фьюзон и В. Росс.
1. Получение
d-ai-Фенилэтиламин. Раствор 100 г @,75 мол.; примечание 1)
/-яблочной кислоты в 500 мл дестиллированной воды смешивают
со 120 г A мол.) dZ-a-фенилэтшгамина (стр. 523); полученный рас-
твор недолго нагревают на паровой бане, фильтруют в 1-литровый
стакан и дают медленно охладиться. Через несколько часов отса-
сывают выкристаллизовавшуюся соль d-a-фенилэтиламина и /-яблоч-
ной кислоты и на фильтре промывают ее 25 мл ледяной воды.
Фильтрат и промывные воды упаривают на паровой бане примерно
до двух третей объема фильтрата и по охлаждении отсасывают вто-
рую порцию кристаллов (примечания 2 и 3). Повторяя этот про-
цесс, можно получить третью, а иногда и четвертую порцию кри-
сталлов, после чего маточный раствор становится настолько вязким,
что получить удовлетворительные результаты при кристаллизации
невозможно. Оставшийся маточный раствор сохраняют.
Последовательно полученные порции кристаллов подвергают
систематической перекристаллизации следующим образом, применяя
в случае необходимости активированный уголь. Около двух третей
первой порции кристаллов растворяют примерно в 3-х частях воды
и горячему раствору дают кристаллизоваться при медленном
охлаждении (примечание 3). Жидкость фильтруют или деканти-
руют, растворяют в ней оставшееся от первой порции количество и
процесс кристаллизации повторяют. Остальные порции перекри-
сталлизовывают последовательно таким же образом из той же
жидкости, причем перед каждой кристаллизацией раствор упаривают
до соответственного объема. Последний маточный раствор упаривают
по стадиям и вязкий остаток присоединяют к остатку от первона-
чальной кристаллизации. Все эти порции кристаллов систематически
перекристаллизовывают из свежей воды до тех пор, пока продукт
не станет совершенно чистым (примечание 4). Таким образом можно
получить 80—-90г F3—70% теоретич.) чистой соли d-основания и
/-кислоты (безводной кислой соли).
Чистую соль яблочной кислоты (молекулярный вес 255) разла-
гают нагреванием с несколько большим количеством (примечание 5),
чем 2 эквивалента, приблизительно, 2-н. едкого натра. По охла-
ждении амин экстрагируют 3—4 порциями чистого бензола по 25 мл,
раствор тщательно сушат измельченным в порошок едким натром и
чистый амин с т. кип. 184—185° и уд. вращением [«J^5° от -\- 39,2°
до + 39,7° (без растворителя) получают перегонкой (примечание 6).
Небольшое количество амина перегоняется вместе с бензолом. Выход
35—40 г (92—94% теоретич., считая на чистую соль /-яблочной
кислоты).
Маточные растворы как от первой, так и от последующих кри-
сталлизации обрабатывают аналогичным образом и растворы на-
триевой соли яблочной кислоты соединяют вместе и сохраняют,
чтобы выделить /-яблочную кислоту (примечание 7). Количество
выделенного обратно амина составляет 75—80 г, причем он содержит
40—50% избыточного /-амина.
l-a-Феиилэтиламин. Полученный обратно амин превращают
в кислый тартрат. Работают в водном растворе, причем на каждый
грамм амина берут 1,25 г d-винной кислоты и 4 мл воды. Если это
бывает нужным, раствор кипятят с обесцвечивающим углем и дают
ему медленно охладиться в спокойном состоянии (примечание 3).
Выпадает плотная масса грубых кристаллов неочищенной соли
/-a-фенилэтиламина и d-винной кислоты. Раствор декантируют и
упаривают примерно до двух третей первоначального объема и полу-
чают вторую порцию кристаллов тем же способом, который опи-
сан выше. Эти операции повторяют еще один или два раза, после
чего обычно уже нельзя получить грубых кристаллов, а получается
только смесь иглоподобных кристаллов (смешанных солей; приме-
чание 8). Одновременно первую порцию кристаллов перекристал-
лизовывают из двухкратного по весу количества воды. Примерно
две трети всего количества сырой соли получают в чистом виде;
маточный раствор служит в качестве растворителя для второй пор-
ции и т. д. Последний маточный раствор присоединяют к остатку
от первой кристаллизации и сохраняют для того, чтобы выделить
только частично разложенный на оптические антиподы амин.
Выход чистой соли /-основания и d-кислоты (безводной кислой
соли) составляет 75—100 г, что соответствует несколько большему
количеству, чем избыток /-амина в исходной смеси. Чистая соль
528
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
ФЁНОКСТИН
529
обладает удельным вращением в 8%-ном водном растворе от
+ 13,0 до +13,2°.
Чистую соль (молекулярный вес 271) растворяют в четырех
частях воды; амин выделяют избытком 20—25%-ного раствора ед-
кого натра, экстрагируют бензолом и подвергают очистке так, как
это описано для rf-амина. Константы совпадают с константами, при-
веденными для rf-амина, выход 32—42 г (94—96% теоретич., считая
на чистую соль винной кислоты или 53—70%, считая на весь /-амин,
содержащийся в исходном продукте).
Амин, выделенный из маточных растворов таким же путем, как
это описано выше, составляет 40—50 г. В нем содержится небольшой
избыток rf-амина. Его удобно использовать в последующих опытах
вместо rfZ-амина.
2. Примечания
1. Количество яблочной кислоты теоретически достаточно для
того, чтобы половину амина превратить в кислую соль, а вторую
половину в нейтральную соль.
2. Небольшое количество летучего амина теряется в результате
гидролиза при выпаривании и перекристаллизации соли.
3. Дробную кристаллизацию можно облегчить, если внести в теп-
лый раствор затравку того кристалла, который должен выделиться,
и дать продукту кристаллизоваться медленно в спокойном состоянии.
4. Чистоту соли rf-a-фенилэтиламина и /-яблочной кислоты не
легко определить по температуре плавления или по удельному вра-
щению. Лучше ее определять по форме крупных кристаллов и по
растворимости. Кислые и нейтральные соли /-основания и /-кис-
лоты растворимы значительно лучше и обычно совсем не выкристал-
лизовываются.
5. Если предполагают выделить обратно яблочную кислоту, то
следует избегать избытка щелочи, так как /-яблочная кислота легко
претерпевает рацемизацию при нагревании с концентрированной
щелочью.
6. Если предполагается использовать амин в водном растворе, то
можно подвергнуть разложению щелочью взвешенное количество
чистой соли, основание отогнать количественно с водяным паром и
использовать весь дестиллат.
7. Раствор, содержащий натриевую соль яблочной кислоты,
нейтрализуют уксусной кислотой, разбавляют таким образом, чтобы
в нем содержалось 5% натриевой соли яблочной кислоты и при
температуре кипения обрабатывают 10%-ным раствором уксусно-
кислого свинца до тех пор, пока не перестанет выпадать в осадок
свинцовая соль яблочной кислоты. По охлаждении эту соль соби-
рают и промывают, растирая ее с кипящей водой. Затем соль расти-
рают с дестиллированной водой в жидкую пасту и разлагают серо-
водородом B дня). Сернистый свинец отфильтровывают и раствор
яблочной кислоты упаривают до нужного объема. Определяют содер-
жание кислоты титрованием пробы раствора и этим раствором поль-
зуются в следующих опытах вместо чистой кислоты. Выход 70—80%.
8. Соль /-основания и d-кислоты не может быть получена в чи-
стом состоянии, если в растворе содержится равное количество со-
лей й- и /-аминов. По этой же причине нельзя расщеплять исходный
rfZ-амип непосредственно с помощью rf-винной кислоты.
3. Другие методы получения
Настоящий метод заимствован у Ловена 1. Расщепление на опти-
ческие антиподы было осуществлено rf-a-бромкамфара-л-сульфо-
кислотой (/-форма);2-3; /- и dZ-яблочными кислотами {й- и /-фор-
мами) 4; /-хинной и d-винной кислотой (й- и /-формами)Б, а также
й- и /-б^-динитродифеновыми кислотами (d- и /-формамиN. Методы,
в которых применялись d-бензилметилацетилхлорид 7, rf-оксимети-
ленкамфара 8, /-хинная кислота 9 и камфарный ангидрид 10 имеют
только теоретический интерес. rfZ-Амин нельзя расщепить на опти-
ческие антиподы с помощью камфар- 10-сульфокислоты п или мин-
дальных кислот 12.
1 L о v e" n, I. prakt. Chem. 72, B) 307 A905); см. также Loven, Ber. 29,
2313 A896).
2 Hunter, Kipping, J. Chem. Soc. 83, 1147 A903).
3 Ingold, Wilson, там же 1933, 1502.
1 Inge r soil, J. Am. Chem. Soc. 47, 1168A925).
5 Andre, Vernier, Compt. rend. 193, 1192 A931).
6 I n g e r s о 1 1, L i 11 1 e, J. Am. Chem. Soc. 56, 2123 A934).
7 К i p p i n g, S a 1 w a y, J. Chem. Soc. 85, 444 A904).
'Pope, Read, там же 95, 171 A909).
9 Marckwald, Meth, Ber. 38, 801 A905).
10 Freylon, Ann. chim. phys. (8) 15, 140 A908).
"Pope, Harvey, J. Chem. Soc. 75, 1110A899).
12 I n ge r s о 1 1, Babcock, Burns, J. Am. Chem. Soc. 55,411 A933).
ФЕНОКСТИН
(Феноксатиин )
,-Os
(CeH5),0 + 2S
(A1CI3)
+
Предложили: К. Сутер и Ч. Максвелл.
Проверили: Р. Фьюзон и Е. Кливленд.
1. Получение
В 5-литровую колбу помещают 1886 г A1 мол.) дифенилового
эфира (примечание 1), 256 г (8 гр.-ат.) серы (серного цвета), и 510 г
C,8 мол.) безводного хлористого алюминия. Колбу энергично взбал-
34 Сборник № 2
530
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОЙ
тывают, чтобы хорошо перемешать реагенты; смесь при этом окра-
шивается в пурпурный цвет. Колбу снабжают обратным холодиль-
ником с водяным охлаждением (примечание 2) и нагревают на водя-
ной бане в хорошо действующем вытяжном шкафу. Бурное вначале
выделение сероводорода примерно через 1,5 часа замедляется. Колбу
нагревают в общей сложности 4 часа, время от времени взбалтывая,
после чего реакционную смесь при помешивании медленно выливают
в 4-литровый стакан, наполовину наполненный льдом, к которому
добавлено 250 мл концентрированной соляной кислоты. Если нужно,
то добавляют еще льда. Колбу ополаскивают водой и эту воду при-
соединяют к главному продукту. После разделения обоих слоев
водный слой отбрасывают, а слой, содержащий фениловый эфир и
фенокстин, сушат в течение ночи над хлористым кальцием, после
чего смесь перегоняют при остаточном давлении 5 мм из специ-
альной 3-литровой колбы Клайзена с эффективным дефлегматором
высотой 45 см. После отгонки фенилового эфира собирают фрак-
цию 140—1бО°/5 мм, главная часть которой переходит при 150—152°,
и эту фракцию считают за фенокстин (примечание 3). Выход 700 г
(87i% теоретич.). Этот продукт, который слегка окрашен и обладает
сильным запахом, очищают перекристаллизацией из 1,2—1,5 л
кипящего метанола; раствор следует быстро охладить и при этом
хорошо перемешивать, чтобы воспрепятствовать выделению вещества
в виде масла. Потеря при перекристаллизации составляет около
3%, и высушенный продукт плавится при 56—57° (примечание 4).
После вторичной перекристаллизации получается продукт, который
плавится еще на Г выше.
ФЛОРОАЦЕТОФЕНОН
531
4-хлорбензойной кислоты х, из фенокстеллурина и серы 2, и дей-
ствием серы и хлористого алюминия на дифениловый эфир3.
1 Mauthner, Ber. 39, 1340A906).
2 D r e w, J. Chem. Soc. 1928, 519.
3 F B h
, J ,
F e r r a r i о, Bull. soc. chim. D)9, 536A911); Ackermann, герм. пат.
234 743 [Frdl. 10, 153 A910— 12)J; S u t e r, McKenzie, Maxwell,
J. Am. Chem. Soc. 58, 717 A936); Bennett, Lesslie, Turner, J
Chem. Soc. 1937, 444; Suter, Green, J. Am. Chem. Soc. 59, 2578 A937).
НО
ОН
\/он
он
но>
он
ФЛОРОАЦЕТОФЕНОН
B,4,6-Триоксиацетофенон )
ОН
CH3CN —L
3 (ZnCI.)
но хх/он
С (= NH • НС1) СН3
он
• нс!)сн3
н„о —
но
ф NH4C1
он
Предложили: К- Гулати, С. Сет и К. Венкатараман.
Проверили: Дж. Джонсон и М. Буш.
2. Примечания
1. Удовлетворительным, является продажный дифенилоксид.
2. Из холодильника стекает обратно небольшое количество фени-
лового эфира, который в противном случае увлекался бы сероводо-
родом.
3. Головную фракцию дифенилового эфира с т. кип. 98—Ю1°/5мм
можно использовать в последующих синтезах. Температуры кипе-
ния компонентов смеси следующие: дифенилового эфира —134—
137°/15 мм, 259—262°/745 мм; фенокстина— 180—183715 мм,
31Г/745 мм. При фракционировании при 15 мм выход фенокстина
меньше, и в колбе остается большее количество смолистого остатка,
чем при фракционировании при 5 мм.
4. Чистый образец плавится При 57,5—58°.
3. Другие методы получения
Фенокстин был получен с помощью ряда реакций с использо-
ванием в качестве исходных продуктов тиокатехина и 3,5-динитро-
1. Получение
В склянку для отсасывания емкостью 250 мл, снабженную хлор-
кальциевой трубкой и резиновой пробкой, через которую проходит
трубка для подачи хлористого водорода (примечание 1), помещают
20 г @,16 мол.) хорошо высушенного флороглюцина (примечание 2),
13 г @,32 мол.) безводного ацетонитрила, 80 мл абсолютного эфира
и 4 г хорошо измельченного сплавленного хлористого цинка. Склянку
охлаждают в смеси льда и соли и время от времени взбалтывают,
пропуская при этом через раствор в течение двух часов сильный ток
сухого хлористого водорода. Склянку оставляют на 24 часа в холо-
дильном шкафу, после чего через раствор, который уже приобрел
светлооранжевую окраску, еще раз пропускают в течение двух
часов хлористый водород. Колбу закрывают пробкой и оставляют
в холодильном шкафу на 3 дня.
Объемистый оранжевожелтый осадок солянокислого кетимина
освобождают от эфира декантацией и дважды промывают абсолют-
ным эфиром, порциями по 20 мл. Осадок переносят в 2-литровую
круглодонную колбу, куда налит 1 л горячей воды. Колбу снаб-
532
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
ФТАЛИД
533
жают обратным холодильником и желтый раствор энергично кипя-
тят на проволочной сетке 2 часа. Затем добавляют небольшое ко*
личество C—4 г) активированного березового угля, раствор кипя-
тят 5 мин. и еще горячим фильтруют с отсасыванием. Уголь
экстрагируют двумя порциями кипящей воды по 100мл и этот филь-
трат добавляют к главной массе.
После стояния в течение ночи бесцветные или окрашенные в
бледножелтый цвет иглы флороацетофенона отсасывают и сушат
в сушильном шкафу при 120° (примечание 3). Выход 20—23,5 г
G4—87% теоретич.). Продукт плавится при 217—219° (исправл.)
и вполне чист, так что его можно непосредственно применять для
большинства целей. Пербкристаллизовать его можно из 35-кратного
количества (по весу) горячей воды, причем потеря составляет около
5%. После перекристаллизации продукт плавится при 218—219°
(исправл.).
2. Примечания
1. Для подачи хлористого водорода необходимо взять трубку
с расширением на конце, чтобы избежать закупорки вследствие вы-
деления твердого солянокислого кетимина.
2. Все реактивы должны быть тщательно высушены. Обыкновен-
ный флороглюцин («Синт. орг. преп.», сб. 1, стр. 446) содержит две
молекулы кристаллизационной воды, которые могут быть удалены
сушкой в течение ночи при 120°. Взятые в реакцию ацетонитрил и
эфир были свеже перегнаны над фосфорным ангидридом.
3. Флороацетофенон кристаллизуется из водных растворов с од-
ной молекулой воды Ч Высушенные в сушильном шкафу кристаллы
при лежании на воздухе легко поглощают влагу. Флороацетофенон
с хлорным железом дает красное окрашивание, напоминающее
красное вино, в отличие от флороглюцина, который дает фиолето-
вое окрашивание 2.
3. Другие методы получения
Наиболее удовлетворительным методом получения флороацето-
фенона является реакция Геша 3. Описанная выше методика при-
надлежит Робинсону и Венкатараману4. Флороацетофенон был
получен также действием хлористого ацетила на флороглюцин в при-
сутствии хлористого алюминия 2.
1Gulati, Seth, Venkataraman, J. Chem. Soc. 1934, 1766.
2 Shriner, Kleiderer, J. Am. Chem. Soc. 51, 1269 A929).
3 Hoesch, Ber. 48, 1129 A915).
4 Robinson, Venkataraman, J. Chem. Soc. 1926, 2347.
ФТАЛИД
/CO2Na
--- С6Н4^
(HCI)
>o
СН2ОН N
Предложили: Дж. Гарднер и Я. Нэйлор мл.
Проверили: X. Кларк и Д. Блюмеыталъ.
1. Получение
В 2-литровой круглодонной колбе, снабженной механической
мешалкой, приготовляют путем размешивания густую пасту из
180 г B,75 гр.-ат.) цинковой пыли и раствора 1 г сернокислой меди
в 35 мл воды (примечание 1), после чего к ней добавляют 400 г
C27 мл) 20%-ного водного раствора едкого натра. Содержимое колбы
охлаждают до 5° путем погружения ее в баню со льдом и добавляют
маленькими порциями при работающей мешалке 147 г A,0 мол.)
фталимида («Синт. орг. преп.», сб. 1, стр. 448) с такой скоростью,
чтобы температура не поднималась выше 8°, на что требуется около
30 мин. После того, как все количество фталимида прибавлено, пере-
мешивание продолжают еще 30 мин. Смесь разбавляют 400 мл воды,
нагревают на паровой бане до тех пор, пока не прекратится выделе-
ние аммиака (около 3 час.) и упаривают в вакууме примерно до
объема в 400 мл. Продукт фильтруют и фильтрат подкисляют на
конго концентрированной соляной кислотой (около 150 мл). Смесь,
в которой фталид уже выделился в виде масла, кипятят в течение
1 часа, чтобы завершить лактонизацию оксиметилбензойной кислоты
и в горячем состоянии переносят ее в стакан. По охлаждении масля-
нистый продукт застывает, образуя красно-бурую лепешку. Смесь
оставляют на ночь в холодильном шкафу, причем из водного слоя
выделяется еще некоторое количество кристаллов. Холодную массу
фильтруют с отсасыванием (примечание 2). Сырой фталид, содержа-
щий значительное количество хлористого натрия, пepeкpиcтaллизoJ
вывают порциями по 20 г из 1,5 л воды. Маточный раствор от первой
порции применяют для перекристаллизации следующих порций.
Каждая порция фильтруется в горячем виде, затем кристаллы от-
фильтровываются при температуре ниже 5° и промываются неболь-
шим количеством ледяной воды (примечание 3). Фталид кристалли-
зуется в виде прозрачных пластинок с т. пл. 72—73°. Выход
90—95 г F7—71% теоретич.: примечание 4).
2. Примечания
1. При проверке этого синтеза неоднократно бывали полные
неудачи, даже в тех случаях, когда применялась хорошая продажная
цинковая пыль. Только при активировании цинка сернокислой
медью имел место процесс восстановления.
534
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
П-ФТОРБЕНЗОЙНАЯ КИСЛОТА
535
2. Маточный раствор при упаривании его даже меньше чем до
половинного объема не выделяет больше кристаллов при охлаждении.
3. В начале кристаллизации при охлаждении вместе со фталидом
выделяется незначительное количество какой-то желтоватой при-
меси, которая, очевидно, не может быть удалена кипячением с акти-
вированным углем. Однако количество этой примеси столь незна-
чительно, что она не влияет на температуру плавления продукта.
4. Упаривание последнего маточного раствора до объема в 500 мл
дает еще очень небольшое количество фталида, однако эта операция
едва ли оправдывается.
3. Другие методы получения
Фталид был получен бромированием о-толуиловой кислоты с по-
следующим гидролизом г; восстановлением фталевого ангидрида 2,
аммонийной соли фталевой кислоты3 или моноэтилового эфира
фталевой кислоты4, а также по методу, описанному выше 5.
1 Н j e I t, Ber. 19, 412 A886); 3 а л к и н д, С е м е н о в, ЖРХО 46, 512
A914); D a v i е s, Р е г k i п, J. Chem. Soc. 121, 2207 A922).
2 W i s 1 i с e n u s, Ber. 17, 2181 A884); Godchot, Bull. soc. chim.
D) 1, 830 A907); Sabatier, Kubota, Compt. rend. 172, 736 A921); An s-
t i n, Bosquet, Lazier, J. Am. Chem. Soc. 59, 864 A937).
3 De If ino , Somlo, IX Congr. intern, quim. pura applicada 4, 360
A934); [C. A. 30, 2855 A936)].
4 E. I. du Pont de Nemours and Company, ам. пат. 2 114 696 [С. А. 32,
4607 A938)].
5 R e i s s e r t, Ber. 46, 1489 A913); Kalle und Ко., А.-О.; герм. пат.
267 596 [Frdl. 11, 196A912—14)].
я-ФТОРБЕНЗОЙНАЯ КИСЛОТА
C2H5O2CC6H4NH2 + NaNO2 + 2 НС1 >
> C2H5OXC6H4NX1 + NaCl + 2 H2O
C3H5OXC6H4NX1 + HBF4 C2H6OXC6H4N,BF4 + HC1
C.HACCeH^BF, . C2H5OXCeH4F + N.2 + BF3
H2O > FC6H4CO2H + C2H5OH
Предложили: Дж. Шиман и В. Винкельмюллер.
Проверили: В. Хартман, Дж. Байере и Дж. Джки.
C2H5OXC6H4F
1. Получение
В 5-литровую круглодонную колбу помещают 165 г A мол.)
этилового эфира л-аминобензойной кислоты (примечание 1), 300 мл
воды и 204 мл B,5 мол.) концентрированной соляной кислоты, уд.
в, 1,19. Смесь в течение 1 часа нагревают на кипящей водяной бане,
время от времени взбалтывая. В колбе получается солянокислый
этиловый эфир л-аминобензойной кислоты в виде белой пасты, а за-
тем колбу вместе с этой пастой помещают в баню со смесью льда и
соли и охлаждают до 0°. При механическом перемешивании к смеси
медленно приливают при температуре не выше 7° раствор 72,6 г
A мол.) 95%-ного азотистокислого натрия в возможно меньшем
количестве воды. Диазотирование считается законченным, когда
через 10 мин. после прибавления раствора нитрита иодо-крахмаль-
ная бумага будет давать слабую реакцию на азотистую кислоту.
Пока происходит диазотирование, растворяют 68 г A,1 мол.)
борной кислоты в 133 г D мол.) 60%-ной фтористоводородной кис-
лоты (примечание 2), находящейся в стакане, который покрыт тон-
ким слоем парафина. Для того, чтобы не расплавился парафин,
температуре не дают подниматься выше 25°, и после прибавления
всего количества реакционную массу охлаждают в бане со льдом.
Холодный раствор борофтористоводородной кислоты быстро
прибавляют при перемешивании к раствору диазония; температура
во время прибавления должна быть ниже 10°. Выпадает густой оса-
док борофтористого карбэтоксифенилдиазония; перемешивание про-
должают еще 20—30 мин. Осадок отсасывают на воронке Бюхнера
диаметром 18,5 см и промывают последовательно 300 мл холодной
воды, 300 мл технического метилового спирта и 200 мл обыкновен-
ного серного эфира. После каждой промывки осадок возможно тща-
тельнее отсасывают. Затем борофторид высушивают в вакуум-
эксикаторе над концентрированной серной кислотой, уд. в. 1,84
(примечание 3). Выход сухого борофторида 198—205 г G5—78%
теоретич.); температура разложения 93—94°.
_ Термическое разложение удобно вести в 2-литровой колбе.
Вторая 1-литровая колба Вюрца соединяется непосредственно с
отводной трубкой первой колбы и служит приемником. Отводную
трубку приемника соединяют при помощи каучуковой трубки
с 5-литровой круглодонной колбой, в которую налито 2 л воды.
Выделяющиеся при разложении газы проходят над поверхностью
воды; трехфтористый бор растворяется, а остальные газы отводят
в хорошо действующий вытяжной шкаф (примечание 4). Борофторид
«-карбэтоксифенилдиазония помещают в колбу для разложе-
ния и верхнюю часть колбы, рядом с поверхностью вещества, нагре-
вают бузеновской горелкой. Когда появятся белые пары трехфтори-
стого бора, пламя удаляют и предоставляют реакции итти само-
произвольно. Если нужно, колбу время от времени подогревают.
К концу реакции для полного разложения и расплавления продукта»
реакционную массу сильно нагревают. Часть этилового эфира л-фтор-
бензойной кислоты (т. кип. 105—106°/ 25 мм) во время разложе-
ния уносится газами и собирается в приемнике, но большая часть
остаетеяв колбе, в которой проводилось разложение. Продукт извле-
кают из колбы и приемника этиловым эфиром, который затем отгоняют
536
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
на водяной бане. Остаток кипятят в продолжение 1 часа с обратным
холодильником с раствором 56 г A мол.) едкого кали в 80 мл
95%-ного этилового спирта и 120 мл воды. Горячий раствор филь-
труют и гс-фторбензойную кислоту, осаждают из горячего фильтрата
прибавлением концентрированной соляной кислоты до кислой реак-
ции на конго. По охлаждении осадок отсасывают и высушивают.
Для очистки л-фторбензойную кислоту растворяют в горячем рас-
творе углекислого калия D0 г К2СО3 в 400 мл Н2О). Раствор обра-
батывают активированным углем и фильтруют горячим. Для оса-
ждения фторбензойной кислоты к фильтрату при перемешивании
прибавляют соляную кислоту. По охлаждении выпавшую кислоту
отсасывают, промывают водой и сушат.
При разложении 85 г @,32 мол.) борофторида л-карбэтокси-
фенилдиазония получается 38—40 г л-фторбензойной кислоты (84—
89% теоретич., считая на эфир л-аминобензойной кислоты). Т. пл.
очищенной кислоты 186°. До очистки кислота плавится при 183—
184°.
2. Примечания
1. Этиловый эфир л-аминобензойной кислоты можно получить
восстановлением соответственного нитросоединения по методу, опи- •
санному в «Синт. орг. преп.», сб. 1, стр. 533, или же непосредствен-
ной этерификацией л-аминобензойной кислоты, для чего 274 г
B мол.) л-аминобензойной кислоты вносят в 1,4 кг этилового спирта,
насыщенного сухим хлористым водородом, и нагревают 24 часа
с обратным холодильником. Затем реакционную смесь выливают
в воду, нейтрализуют содой и нерастворимый в воде эфир отсасы-
вают. Выход 80% теоретич.
2. Фтористоводородная кислота причиняет чрезвычайно болез-
ненные ожоги. При работе с фтористоводородной кислотой необ-
ходимо защищать все части тела, которые могут подвергнуться
ее действию. Ср. примечание 3, стр. 539.
Вместо стакана, покрытого парафином, при получении борофто-
ристоводородной кислоты удобнее работать со свинцовой чашкой.
Вместо перемешивания от руки можно также установить свинцовую
механическую мешалку обычной конструкции. Проводить мешалку
следует через отверстие в свинцовой крышке итак, чтобы предотвра-
тить разбрызгивание фтористоводородной кислоты.
Борофтористоводородная кислота D0%) имеется теперь в про-
даже. Для работы по описанному способу нужно брать 200 г такой
кислоты.
3. Очень важно, чтобы борофторид был сухой. Если осадок еще
влажен, разложение идет очень бурно, происходит осмоление и
выход уменьшается.
4. Более простым прибором для разложения может служить
2-литровая круглодонная колба с коротким горлом, соединенная
ФТОРБЕНЗОЛ
537
посредством широкой B см) изогнутой трубки с 1-литровой колбой,
в которую налито 500 мл воды. Газы из второй колбы отводятся
в вытяжной шкаф с хорошей тягой.
3. Другие методы получения
л-Фторбензойная кислота была получена окислением л-фтор-
толуола хромовой кислотой в слабой серной кислоте при 160°1;
окислением л-фтортолуола псрманганатом калия 2; электролитиче-
ским окислением л-фтортолуола 3; нагреванием хлористого л-карб-
оксифенилдиазония с дымящей фтористоводородной кислотой4;
окислением л-фторбензальдегида Б; окислением л, л'-дифторстиль-
бена перманганатом калия в; окислением 4,4'-дифторбифенила азот-
ной кислотой' и окислением 4,4'-дифторбифенила хромовой кис-
лотой 8. Описанный метод заимствован у Бальца и Шиманна9.
1 W а 1 1 а с h, Ann. 235, 263 A886).
2 Slothouwer, Rec. trav. chim. 33, 324A914); Holleman, тамже
25, 332 A906); Holleman, Slothouwer, Proc. К. Акай. Wetensch.
Amsterdam 19, 497, 500 A910) (Chem. Zentr. 1911, I, 74); К о о р а 1, Rec. trav.
chim. 34, 152 A915); Meyer, Hub, Monatsh. 31, 933 A910).
3 Fichtcr, Rosenzweig, Helv. Chim. Acta 16, 1154 A933).
4Schmitt, Gehren, J. prakt. Chem. B) 1, 394 A870); P a t e r n o,
О 1 i v e r i, Oazz. chim. ital. 12, 87 A882).
5 R i n к е s, Chem. Weekblad 16, 206 A919).
«Meyer, Hoffmann, Monatsh. 38, 154 A917).
' Hove, Bull. acad. roy. Belg. E) 12, 801 A926) (Chem. Zentr. 1927,
I, 884).
8Schiemann, Roselius, Ber. 62, 1813 A929).
» Balz, S с h i e m a n n, там же 60, 1186 A927).
ФТОРБЕНЗОЛ
б^, • HC1 + HNO, CeH5N2Cl + 2 H2O
C,H5NaCl + HBF4 -—> C6H5N2BF., + HC1 "
C6H5N2BF4 CeH5F + N2 + BF2
Предложил: Д. Флоод.
Проверили: В. Хартман и Дж. Байере.
1. Получение
В большую стеклянную или глиняную банку C0 X 30 см)
емкостью 40 л помещают смесь 1350мл воды и 1650 мл B0 мол.) кон-
центрированной соляной кислоты, уд. в. 1,19; жидкость энергично
перемешивают при помощи механической мешалки (примечание 1)
и хорошо охлаждают смесью льда и соли. Пока раствор охлаждается,
отвешивают 2075 г A6 мол.) солянокислого анилина (примечание 2)
и готовят раствор 1,2 кг A7 мол.) азотистокислого натрия в 1,5 л
538
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
воды. Когда температура кислоты опустится до 5° или ниже, к ней
прибавляют около трети всего количества солянокислого анилина и
при температуре не выше 7° начинают диазотировать, медленно при-
ливая раствор нитрита. Время от времени добавляют солянокислый
анилин в таких количествах, чтобы в реакционной смеси все время
имелся избыток кристаллов. Весь анилин должен быть прибавлен
к тому времени, когда прилита половина раствора нитрита. Диазо-
тирование заканчивают, когда иодокрахмальная бумага даст реак-
цию на присутствие свободной азотистой кислоты. Обычно для этого
приходится прибавить почти весь раствор нитрита.
Пока производится диазотирование, приготовляют раствор боро-
фтористоводородной кислоты: 1 кг A6,2 мол.) борной кислоты (кри-
сталлической фармакопейной) вносят небольшими частями в 2150 г
F,5 мол.) 60%-ной фтористоводородной кислоты (примечание 3).
Получение кислоты ведут в двух 3-литровых покрытых изнутри
парафином колбах, охлаждая их ледяной водой и взбалтывая (при-
мечание 4). Температура получаемой кислоты не должна быть
выше 20—25°.
Приготовленный и охлажденный до 0° раствор борофтористоводо-
родной кислоты вливают в раствор диазония, охлажденный до тем-
пературы ниже 0°. Во время прибавления кислоты, которое ведут
довольно быстро, температура не должна подниматься выше 10°.
Для того, чтобы хорошо размешать образующуюся густую массу,
необходимо энергичное перемешивание (примечание 1). Затем про-
должают перемешивание еще 20—30 мин. и окрашенную в коричне-
вый цвет массу отсасывают на двух воронках Бюхнера диаметром
24 см. Желтоватый кристаллический осадок промывают последова-
тельно ледяной водой (около 800 мл), метиловым спиртом (около
800 мл) и этиловым эфиром (900 мл). После каждой промывки сле-
дует как можно лучше отсасывать жидкость. Хорошая промывка
имеет большее значение, так как увеличивает стабильность продукта
(примечание 5).
Светлокоричневую объемистую соль раскладывают на фильтро-
вальной бумаге и на ночь оставляют на сквозном ветру (лучше всего
класть соль на стол, который находится вблизи вытяжного шкафа).
Затем соль переносят в 12-литровую колбу (примечание 6). При
помощи изогнутой трубки колбу соединяют с длинным холодильни-
ком с достаточно широким диаметром внутренней трубки, причем
холодильник в свою очередь соединяют с тремя 2-литровыми кони-
ческими колбами, установленными последовательно и погруженными
в смесь льда с солью. Для удаления большого количества паров
трехфтористого бора отводную трубку последней колбы отводят в хо-
рошо действующий вытяжной шкаф или соединяют с системой уло-
вителей, наполненных льдом с водой или раствором соды (примеча-
ние 7). Осадок осторожно разлагают, нагревая слабым пламенем
одно место колбы, недалеко от верхней поверхности осадка, до тех
ФТОРБЕНЗОЛ
539
пор, пока не начнется реакция. Тогда пламя удаляют и реакции
. дают продолжаться самостоятельно до конца. Если реакция стано-
вится слишком бурной, то колбу надо охлаждать, прикасаясь к ней
куском льда (примечание 8). Когда реакция приостанавливается,
смесь следует осторожно подогревать. К концу реакции колбу сильно
нагревают до тех пор, пока не прекратится выделение паров трех-
фтористого бора. Последние следы фторбензола можно удалить из
колбы при помощи легкого отсасывания, соединяя приемник с насосом.
Полученные дестиллаты соединяют и отделяют выделившийся
фенол, затем их промывают 4—5 раз 10%-ным раствором едкого
натра, пока промывные воды не станут почти бесцветными, а по-
следнюю промывку производят один раз водой (примечание 9). За-
тем сушат взбалтыванием с измельченным хлористым кальцием и
быстро перегоняют из 2-литровой колбы с коротким дефлегматором.
Иногда первая часть отгона содержит немного воды, и тогда ее снова
сушат. Продукт представляет собою бесцветную жидкость, по запаху
напоминающую бензол. Выход фторбензола, кипящего при 84—
85°, составляет 780—870 г E1—57% теоретич.; примечание 10).
2. Примечания
1. Так как во время процесса приходится несколько раз иметь
дело с большими количествами суспендированного твердого про-
дукта, то лучше всего брать для работы медленно вращающуюся
весельную мешалку с несколькими лопастями. Одинаково пригодны
мешалки как деревянные, так и металлические, защищенные от
действия кислот краской, хотя краска обыкновенно быстро отстает.
Можно работать также с мешалками, покрытыми слоем резины.
2. Применялся технический солянокислый анилин. Азотисто-
кислый натрий, соляная кислота и фтористоводородная кислота
также могут быть не химически чистыми продуктами, а техниче-
скими. Вместо солянокислого анилина, кислоты и воды можно взять
анилин A485 г) и соляную кислоту C л), не прибавляя воды. Но
в таких случаях образующийся солянокислый анилин часто выпа-
дает на стенках сосуда, образуя твердый кристаллический слой,
очень затрудняющий перемешивание смеси и ее охлаждение.
3. Можно взять эквивалентное количество 48%-ной или 52%-ной
фтористоводородной кислоты. При работе с указанными количе-
ствами имеется избыток фтористоводородной кислоты. Можно брать
и больший избыток, но выход при этом значительно не изменяется.
Борофтористоводородная кислота D0%-ная) имеется теперь в про-
даже в готовом виде.
Фтористоводородная кислота при соприкосновении с кожей при-
чиняет чрезвычайно болезненные ожоги. Поэтому необходимо при-
нимать все возможные предосторожности для защиты открытых ча-
стей тела, особенно рук и глаз. Следует надевать длинные кислото-
устойчивые резиновые перчатки и очки. При ожоге кислотой обож-
540
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
женное место белеет. Ожог надо держать под струей воды до тех
пор, пока тело снова не примет обычного цвета. Чтобы ожоги не
разболелись, рекомендуется быстро применитьмазь, приготовленную
из окиси магния и глицерина (Fredenhagen и. Wellmann, Angew.
С hem, 45, 537, 1932).
4. Борофтористоводородную кислоту удобнее приготовлять в
свинцовом сосуде с механической мешалкой. Такой сосуд легко
можно сделать из листового свинца. Лист сгибают, придают ему
нужную форму и швы спаивают. В качестве мешалки может слу-
жить железный прут, вставленный в узкую свинцовую трубку с при-
паянной на конце полосой свинца. Крышку можно сделать из дере-
вянной доски, которую с нижней стороны выкладывают свинцом.
Постепенно кислота разъедает спайку, а потому для защиты спайку
надо покрывать кислотоупорной смазкой. Полученную борофтористо-
водородную кислоту сифонируют из сосуда через резиновую трубку.
5. Присутствие влаги уменьшает стойкость борофторида фе-
нилдиазония. Если оставить стоять влажный плотно слежавшийся
продукт, может произойти бурное разложение. Если в сырую погоду
продукт разложить на бумаге, он темнеет и медленно разлагается.
То же с ним происходит, если борофторид фенилдиазония плохо
промыт.
6. Разложение можно вести в два приема из двух 5-литровых
колб, причем в этом случае реакцию легче регулировать.
7. Так как при разложении выделяются большие количества газа,
все соединения должны быть сделаны при помощи широких трубок.
8. Обычно разложение протекает гладко, причем приходится
время от времени подогревать колбу. Но если соль была взята влаж-
ная, реакция может пойти более бурно, а при недостаточном охла-
ждении настолько бурно, что регулировать ее будет уже невозможно
(примечание 5).
9. Удельный вес фторбензола около 1,025, поэтому при промывке
необходимо для получения хорошего разделения слоев брать раствор
едкого натра указанной концентрации. Перед промывкой водой
едкий натр следует полностью отделить.
10. Получение фторбензола было успешно проведено и с количе-
ствами в два и три раза большими, чем указанные выше; выходы
получались почти пропорциональные. При работе с большим коли-
чеством материала хорошо пользоваться, кроме описанного выше
свинцового сосуда (примечание 4), также керамиковым нутчем.
3. Другие методы получения
Описанный выше метод получения фторбензола в основном раз-
работали Больц и Шиманн1. Рекомендуется осаждать борофторид фе-
нилдиазония из раствора, в котором суммарная концентрация водо-
родного иона не превышает 2 г на литр 2. Борофторид диазония был
получен также из солянокислого анилина и нитрозилборофторида3,.
фумаровая кислота
541
Фторбензол был получен с выходом 50% из бензолдиазопипери-
дида и фтористоводородной кислоты4. Эта реакция проходит очень
бурно, и сразу нельзя брать больше 10—15 г пиперидида. Все осталь-
ные методы основаны на получении фтористого фенилдиазония и его
разложения на фторобензол при нагревании 5'в-7. Автору не уда-
лось при проверке подтвердить сообщение Валентинера и Шварца 5
о том, что реакцию можно вести в водном растворе.
1 В а 1 г, S с h i e m a n п, Вег. 60, 1188 A927).
2 Е. 1. du Pont de Nemours and Company, ам. пат. 1 916 327 [С. А. 27, 4539
A933)].
'Курский, ЖОХ 8, 524 A938).
* W a 1 1 а с h, Ann. 235, 258 A886); W a 1 1 а с h, H e u s 1 с г, там жг
243, 219 A888).
6Valentiner, Schwarz, герм. пат. 96 153 [Frdl. 5, 910 A897—
1900); Chem. Zentr. 1898, I, 1224].
¦Holleman, Beekman, Rec. trav. chim. 23, 225 A904).
7 S w a r t s, там же 27, 120 A908).
ФУМАРОВАЯ КИСЛОТА
НС —СН
НС ССНО
\/
о
4 [О]
(NaClO3)
нохен
со,
нссо,н
Предложил: Н. Милас.
ПроверЬли: Р. Адаме и К- Амштутц.
1. Получение
В 5-литровую плоскодонную колбу, снабженную длинным (80—
90 мм) с широкой внутренней трубкой холодильником, делительной
воронкой и механической мешалкой (примечание 1) и закрепленную
на высоте в 10 см над электрической плиткой, помещают 450 г
D,2 мол.) хлорноватокислого натрия, 2 г пятиокиси ванадия (при-
мечание 2) и 1 л воды. Пускают в ход мешалку, смесь нагревают до
70—75° (примечание 3) и затем прибавляют около 5—10 мл из от-
вешенных заранее 200 г B,06 мол.) фурфурола (примечание 4).
Как только начнется бурная реакция, прибавляют оставшееся ко-
личество фурфурола с такой скоростью, чтобы реакция не затихала
(примечание 5). Все прибавление занимает 70—80 мин. После того,
как прибавление фурфурола закончено, продолжают перемешивание
смеси при 70—75° в течение 10—11 часов и оставляют ее при ком-
натной температуре на ночь. Выпавшую сырую фумаровую кислоту
отсасывают и сушат на воздухе. Выход 155—170 гF5—72%теоретич.;
примечание 6).
Еще некоторое количество фумаровой кислоты можно получить
из фильтрата нагреванием его на водяной бане с 50 мл концентри-
542
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
рованной соляной кислоты; к концу реакции жидкость обычно
окрашивается в синий цвет. Раствор упаривают приблизительно
до объема в 700 мл и затем охлаждают проточной водой. Выпавшую
фумаровую кислоту отсасывают и сушат на воздухе. Выход 10—
15 г продукта, плавящегося при 282—284° в запаянном капилляре.
Сырой продукт очищают перекристаллизацией примерно из
1 250 мл 1-н. соляной кислоты. Выход 100—ПО г чистой фумаро-
вой кислоты, плавящейся при 282—284° в запаянном капилляре.
Еще некоторое количество кислоты можно получить упариванием
фильтрата до небольшого объема на водяной бане. Общий выход
чистой фумаровой кислоты 120—138 г E0—58% теоретич.).
2. Примечания
1. Если не применить механического перемешивания, то выход
понижается.
2. Катализатор приготовляют следующим образом: 20 г хими-
чески-чистого метаванадата аммония взмучивают в 200 мл во-
ды и к смеси медленно прибавляют 30 мл концентрированной
соляной кислоты уд. в. 1,19. Образовавшийся красновато-коричне-
вый полуколлоидальный осадок промывают несколько раз водой
посредством декантации, затем суспендируют его в 300 мл воды
и оставляют стоять при комнатной температуре в течение трех дней.
Получающийся таким образом легко фильтрующийся зернистый
осадок отсасывают и промывают несколько раз водой, чтобы уда-
лить соляную кислоту. После промывки пятиокись ванадия сушат
при 120° в течение 12 час, растирают в мелкий порошок и снова
сушат в течение 12 час. при 120°.
3. Для того, чтобы иметь возможность следить за температурой,
в колбу можно подвесить термометр через внутреннюю трубку холо-
дильника. Сначала смесь можно нагреть до 70—75° на голом пламени.
4. Пригоден продажный 99%-ный фурфурол. Можно взять
также сырой фурфурол, полученный по методу, описанному в «Синт.
орг. преп.», сб. 1, стр. 454.
5. Повидимому, реакция начинается не сразу после прибавления
первой порции фурфурола. Когда начинается бурная реакция, тем-
пература повышается примерно до 105° и на этом уровне поддер-
живается в течение некоторого времени. Выход фумаровой кислоты,
повидимому, зависит до некоторой степени от скорости протекания
реакции на этой стадии. Поэтому необходимо регулировать приба-
вление фурфурола таким образом, чтобы реакция проходила доста-
точно бурно.
6. Сырая фумаровая кислота содержит 74—78% чистой кислоты,
что было определено титрованием щелочью. Кроме неорганических
солей продукт загрязнен только кислым малеиновокислым натрием,
который разлагается при кристаллизации из соляной кислоты.
кЕЛИДОНОВАЯ КИСЛОТА
543
3. Другие методы получения
'Фумаровая кислота была получена из бромянтарной кислоты
нагреванием с водой \ с разбавленной бромистоводородной кисло-
той 2 и нагреванием кислоты выше температуры плавления 3. Она
была также получена нагреванием яблочной кислоты 4, изомериза-
цией малеиновой кислоты5 и восстановлением винной кислоты
фосфором и иодом 6. Описанный выше способ является наиболее
удобным лабораторным методом и представляет незначительное
видоизменение способа, описанного в литературе '.
1 V о 1 h a r d, Ann. 268, 256 A892); Miiller, Sucker t, Ber. 37,
2598 A904).
2 Fi tti g, Ann. 188, 90 A877).
3 К e k u 1 ё, там же 130, 22 A864); V о 1 h a r d, там же 242, 158 A887).
«Lassaigne, Ann. chim. phys. B) 11, 93 A819); P e 1 о u z e, Ann.
11, 265 A834); W i s 1 i с е n u s, там же 246, 91 A888); Michael, J. prakt.
Chem. B) 46, 231 A892); Jungfleisch, Bull. soc. chim. B) 30, 147 A878).
6 W i s 1 i с e nu s, Ber. 29, 1080 (реф.) A896); С i a m i с i a n, S i 1-
b e г, там же 36, 4267 A903); S k r a u p, Monatsh. 12, 107 A891); Weiss,
Downs, J. Am. Chem. Soc. 44, 1119 A922); Terry, Eichelberger,
там же 47, 1402 A925).
« P h e 1 p s, герм. пат. 254 420 [Frdl. 11, 99 A912—14)]; Org. Chem. Rea-
gents II, Univ. Illinois Bull. 18, F), 35A920).
' M i 1 a s, J. Am. Chem. Soc. 49, 2007 A927); В у л ы г и н а, Маслобойно-
жировое дело, 1934, 4, 43.
ХЕЛИДОНОВАЯ КИСЛОТА
(•(-Пирон-2,6-дшарбоновая кислота)
СО2С2Н5
СН3СОСН3
о
(NaOC2H5)
о
СН.СН.
Н5С„О2ССО
н2о
НС
II
НО2СС
СН
II
ССО2Н
\
о
.Предложили: Е. Ригель и Ф. Цвильгмейер'
Проверили: Р. Фьюзон и В. Росс.
1. Получение
В 1-литровой круглодонной колбе, снабженной обратным холо-
дильником, который защищен хлоркальциевой трубкой, растворяют
544
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕг1аРАТОЙ
хинон
545
46 г B гр.-ат.) натрия в 600 мл абсолютного спирта (примечание 1).
Прибавление натрия продолжается около одного часа, а для пол-
ного его растворения необходим еще один час. К концу реакции
колбу можно осторожно нагревать небольшим коптящим пламе-
нем. Пока натрий растворяется, в сухих склянках с притертыми
пробками отвешивают следующие реактивы: 58 г A мол.) сухо-
го ацетона (примечание 2), 150 г A,03 мол.) свежеперегнан-
ного этилового эфира щавелевой кислоты («Синт. орг. преп.»,
сб. 1, стр. 562) и 160 г A,1 мол.) этилового эфира щавелевой
кислоты.
Примерно половину раствора этилата натрия выливают в 3-ли-
тровую круглодонную колбу с тремя горлами, снабженную мешал-
кой с жидкостным затвором и обратным холодильником; другую
половину поддерживают в теплом состоянии с помощью небольшого
пламени. Первой половине алкоголята дают несколько охладиться,
пока он не начинает частично затвердевать, затем сразу прибавляют
смесь 58 г сухого ацетона и 150 г этилового эфира щавелевой кислоты,
после чего пускают в ход мешалку. При этом происходит выделение
тепла, жидкость буреет, но остается прозрачной. Как только рас-
твор начнет мутнеть, прибавляют единовременно вторую половину
горячего раствора этилата натрия и 160 г щавелевого эфира, причем
обе жидкости сливают таким образом, что они смешиваются еще до
попадания в колбу. Первоначально жидкость остается прозрачной
и окрашенной в темнокоричневый цвет, но примерно после 30 мин.
перемешивания масса становится фактически твердой. Тогда обрат-
ный холодильник заменяют на нисходящий, колбу нагревают на
масляной бане до 110° и отгоняют 150 мл спирта. Колбу защищают
хлоркальциевой трубкой и реакционную смесь охлаждают до 20°.
Натриевое производное переносят с помощью стеклянной палочки
в 3-литровый стакан и обрабатывают его смесью из 300 мл кон-
центрированной соляной кислоты (уд. в. 1,19) и 800 г колотого льда
(примечание 3). Все кусочки тщательно растирают и кремово-жел-
тую суспензию этилового эфира хелидоновой кислоты собирают на
воронке Бюхнера диаметром в 15 см. Сложный эфир удаляют с во-
ронки, размешивают его со 100 мл ледяной воды и вновь собирают
на воронке (примечание 4). Для того, чтобы его омылить, сырой
продукт нагревают в 5-литровой колбе (примечание 5) с 300 мл
концентрированной соляной кислоты на паровой бане в течение
20 час. По охлаждении до 20° твердый кристаллогидрат кислоты
отфильтровывают на воронке Бюхнера диаметром в 10 см, два раза
промывают ледяной водой порциями по 50 мл и сушат сперва
при 100° в течение 2 час, а затем при 160° до постоянного
веса, чтобы удалить кристаллизационную воду. Выход продукта
с т. разл. 257° (исправл.) составляет 140—145 г G6—79% теоре-
тич.).
2. Примечания
1. Необходим высококачественный абсолютный спирт («Синт.
орг. преп.», сб. 1, стр. 555).
2. Ацетон сушат несколько дней над хлористым кальцием, филь-
труют и перегоняют. При этом некоторое количество ацетона
теряется в результате образования продукта присоединения к хло-
ристому кальцию г.
3. Во время нейтрализации следует поддерживать возможно
более низкую температуру, так как повышение ее ведет к окраши-
ванию продукта в темный цвет.
4. Если этот неочищенный сложный эфир промыть еще раз, а за-
тем высушить в вакуум-эксикаторе над серной кислотой, то полу-
чается 220 г (85% теоретич.) продукта с т. пл. 98—100°.
5. Надо брать большую колбу, так как вначале происходит
сильное вспенивание. Если пена чересчур мешает, ее можно сбить,
прибавив немного эфира.
3. Другие методы получения
Природную хелидоновую кислоту получают из травы чистотела
(Chelidonium majus). Синтез ее из этилоксалата и ацетона впервые
был описан Клайзеном а. Значительные упрощения внесли Виль-
штеттер и Пуммерер 3, дальнейшие улучшения сделали Ружичка и
Форназир 4. Настоящая методика основана именно на работе этих
авторов.
1 В a gs ter, J. Chem. Soc. Ill, 494A917).
2 С 1 a i se n, Ber. 24, 111 A891).
3 W i 1 1 s t a 11 e r, Pummerer, там же 37, 3744 A904).
'Ruzicka, Fornasir, Helv. Chim. Acta 3, 811 A920).
3 n-HOC6H4OH -f NaClOa
ХИНОН
(Бензохинон)
(V2O6)
хн = снх
30 = СС >С-0 + NaCl + 3 Н20
Чн = сн/
Предложили: X. Ундереуд мл. и В. Уолш.
Проверили: Л. Физер и Д. Поттер.
1. Получение
В 2-литровую круглодонную колбу, снабженную механической
мешалкой, помещают 1 л 2%-ной серной кислоты, 0,5 г пятиокиси
ванадия (примечание 1), ПО г A мол.) гидрохинона и 60 г @,56 мол.)
хлорноватокислого натрия. Смесь энергично перемешивают при-
мерно в течение 3 час. до тех пор, пока первоначально образующийся
зеленый хингидрон не перейдет в желтый хинон. Температура реак-
35 Сборник Я: 2
546
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОЙ
ХЛОРАНГИДРИД ФТАЛЕВОЙ КИСЛОТЫ самн. и несамм.
547
ционной смеси повышается примерно до 40° (примечание 2). Конец
реакции наступает обычно через 3,5—4 часа. Колбу отъединяют,
охлаждают проточной водой под краном, смесь фильтруют с отса-
сыванием и хинон промывают один раз 100 мл холодной воды. Про-
дукт сушат в эксикаторе над хлористым кальцием, причем полу-
чается 86—90 г вещества с т. пл. 110—112°. Для большинства целей
продукт достаточно чист. Фильтрат и промывные воды извлекают
четырьмя порциями бензола по 100 мл и получают еще 12—14 г
хинона. Общий выход 99—104 г (92—96% теоретич.; примечание 3).
Очень чистый хинон может быть получен или возгонкой в ва-
кууме или перекристаллизацией из кипящего лигроина (т. кип. 90—
120°). На 100 г неочищенного хинона для перекристаллизации тре-
буется около 1,2 л лигроина, причем получается 92—97 г яркожел-
того продукта с т. пл. 111—113°.
2. Примечания
1. Пятиокись ванадия может быть взята в готовом виде или же
получена из метаванадата аммония по методике, описанной в при-
мечании 2 на стр. 542.
2. Если в реакцию берется больше одного моля гидрохинона,
температуре не следует давать подниматься выше 40°.
3. Путем подбора подходящего органического растворителя тот
же окислитель может быть применен и для получения антрахинона.
Смесь, состоящую из 90 г @,51 мол.) хорошо измельченного чистого
антрацена, 0,5 г пятиокиси ванадия, 76 г хлорноватокислого
натрия, 1 л ледяной уксусной кислоты и 200*1 2%-ной серной кис-
лоты, нагревают с обратным холодильником до начала бурной реак-
ции. Источник нагревания удаляют и реакции дают итти около
20 мин., затем смесь вновь кипятят с обратным холодильником в те-
чение 1 часа, после чего колбу охлаждают в бане со льдом. Светло-
желтое вещество отсасывают, хорошо промывают водой и сушат при
110°. Выход 92—96 г (88—91% теоретич.). Продукт плавится при
273—275°.
Применяемый в этом случае окислитель (хлорноватокислый
натрий в присутствии пятиокиси ванадия) не является сильным
окислителем и хотя он легко окисляет весьма реакционноспособныи
антрацен, однако его нельзя применить для превращения углеводо-
родов ряда нафталина и фенантрена в соответствующие хиноны или
же для окисления аценафтена или флуорена (наблюдения проверяв-
ших этот синтез Физера и Поттера).
3. Другие методы получения
Окисление гидрохинона бихроматом натрия в сернокислотном
растворе, а также ссылки на другие методы получения были приве-
дены раньше \
1 «Синт. орг. преп.», сб. *1, стр. 463.
ХЛОРАНГИДРИД ФТАЛЕВОЙ КИСЛОТЫ самм. и несамм.
(Фталилхлорид)
>0 + РС15
)
,С0С1
,С0С1
!С0С1
+ poci3
,1
Ic
A1CI,
oci
со
О • А1С1,
/\со
х >СС1.2
со
О + Al (OH)S + 3HC1
- А1С1а + ЗНаО -
Предложил: Э, Отт.
Проверили: Г. Гилъман и Ф. Прохаска.
1. Получение
А. Симметричный хлорангидрид о-фталевой кислоты. Смесь
148 г A мол.) фталевого ангидрида (примечание 1) и 220 г A,06 мол.)
пятихлористого фосфора (примечание 2) помещают в колбу Клайзена
емкостью 500 мл, соединенную с обратным воздушным холодиль-
ником, ^верхний конец которого закрыт пробкой с хлоркальциезой
трубкой; отводную трубку колбы закупоривают пробкой. Колбу
ставят немного наклонно так, чтобы хлорокись фосфора, конденси-
рующаяся в отводной трубке, стекала бы обратно в колбу. После
12-часового нагревания на масляной бане при 150° воздушный холо-
дильник удаляют, вынимают пробку, которая закрывает отводную
трубку, и соединяют колбу с обращенным вниз водяным холодиль-
ником. Затем отгоняют большую часть хлорокиси фосфора, посте-
пенно повышая температуру масляной бани до 250°. Жидкий оста-
ток перегоняют в вакууме; сначала переходит небольшое количество
хлорокиси фосфора, а затем —при 131—133°/9—10 мм — симм.-
хлорангидрид о-фталевой кислоты. Полученный таким образом
продукт содержит небольшую примесь фталевого ангидрида; он
затвердевает при охлаждении смесью льда и соли и плавится при
11—12° (примечание 3). Выход— 187 г (92% теоретич.).
Б. Несимм. хлорангидрид о-фталевой кислоты. Смесь 105 г
симметричного хлорангидрида о-фталевой кислоты и 75 г мелко
548
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
растертого хлористого алюминия (примечание 4) нагревают на паро-
вой бане в течение 8—10 час. в колбе, защищенной от доступа влаги
хлоркальциевой трубкой. Смесь следует часто перемешивать до
тех пор, пока весь хлористый алюминий не перейдет в раствор.
По охлаждении образуется твердая масса, которую еще теплой
разбивают на маленькие куски. После полного охлаждения плав
небольшими порциями растирают в ступке с кусочками льда.
Образующийся белый осадок отсасывают и немедленно растворяют
приблизительно в 300 мл теплого бензола D0—50°). Бензольный
раствор отделяют от небольшого водного слоя, сушат хлористым
кальцием в течение 8 час. и фильтруют. Бензол отгоняют в вакууме
на водяной бане при 30—40°. Кристаллический осадок экстраги-
руют в аппарате Сокслета петролейным эфиром (с т. кип. 20—40°)
до тех пор, пока остаток в гильзе не будет состоять почти целиком
из чистого фталевого ангидрида (примечание 5). Затем петролейный
эфир отгоняют и оставшийся сырой несгмш.-хлорангидрид фтале-
вой кислоты очищают дробной кристаллизацией из петролейного
эфира (т. кип. 20—50°). Очищенный хлорангидрид плавится при
87—89°. Точка плавления не резкая, так как несимметричный хлоран-
гидрид легко изомеризуется в симметричный. Выход чистого несим-
метричного хлорангидрида фталевой кислоты составляет около 76 г
G2% теоретич.) и зависит от качества взятого хлористого алюминия.
2. Примечания
1. Следует взять чистый возогнанный фталевый ангидрид (с т.- пл.
128—129°). Обыкновенный технический фталевый ангидрид можно
очистить возгонкой.
2. Пятихлористый фосфор не должен содержать ни треххлори-
стого фосфора, ни хлорокиси фосфора. С целью очистки обычный
пятихлористый фосфор помещают в колбу, соединенную с прием-
ником, охлаждаемым льдом, и нагревают колбу на водяной бане в
возможно более высоком вакууме водоструйного насоса. Между насо-
сом и приемником следует поставить трубку с хлористым кальцием.
3. Вполне чистый симметричный хлорангидрид о-фталевой кис-
лоты нельзя получить даже перекристаллизацией из чедыреххло-
ристого углерода. При перегонке несимм.-хлорангидрида фталевой
кислоты при атмосферном давлении получается силш.-ангидрид,
плавящийся при 16°.
4. Хлористый алюминий должен быть хорошего качества. Если
его нет под руками, то он может быть получен нагреванием высушен-
ного зерненого алюминия в токе чистого, сухого хлористого водо-
рода. Газ тщательно сушат, пропуская его через трубку, напол-
ненную пятиокисью фосфора, нанесенной на стеклянную вату. Во
время взвешивания и растирания в порошок хлористый алюминий
следует как можно тщательнее оберегать от доступа влаги.
а-ХЛОРАНТРАХИНОН
549
5. На извлечение требуется от 8 до 10 час. Остаток должен
состоять из практически чистого фталевого ангидрида (т. пл. 126°).
3. Другие методы получения
силш.-Хлорангидрид о-фталевой кислоты был получен нагрева-
нием фталевого ангидрида с пятихлористым фосфором 1, хлористым
тионилом или бензотрихлоридом. В случае двух последних реаген-
тов в качестве катализатора применялся взятый в небольшом коли-
честве хлористый цинк2.
Чистый силш.-хлорангидрид о-фталевой кислоты пытались полу-
чать повторным нагреванием сырого содержащего фталевую кис-
лоту хлорангидрида с небольшим количеством пятихлористого
фосфора s.
Несимметричный изомер получается с плохим выходом при на-
гревании симметричного изомера с хлорным оловом 4.
1 С I a u s, Hoch, Ber. 19, 1187A886).
2 К у г i d e s, J. Am. Chem. Soc. 59, 206 A937).
3 В r ii h 1, Ann. 235, 13 A886).
•Csanyi, Ber. 52, 1792A919).
s-ХЛОРАНТРАХИНОН
A-Хлорантрахинон)
3C6H
6H4
C6H3SO3K + NaC103 + 3 HCl
3C6H4
c0
C6H3C1 (a) + 3KHSO4 -| NaCl
Предложили: В. Скотт и К- Аллен.
Проверили: Л. Физер и Е. Хершберг.
1. Получение
В вытяжном шкафу собирают прибор, состоящий из 2-литровой
трехгорлой колбы, снабженной мешалкой (примечания 1 и 2),
обратным холодильником и капельной воронкой (примечание 3).
В колбу помещают 20 г @,061 мол.) калиевой соли антрахинон-а-
сульфокислоты (стр. 58), 500 мл воды и 85 мл A мол.) концентриро-
ванной соляной кислоты. Раствор нагревают до кипения, пускают
в ход мешалку и по каплям, в течение. 3, час, приливают раствор
20 г @,19 мол.) хлорноватокислого натрия (примечание 4) в 100 мл
воды (примечание 5). Смесь осторожно кипятят еще один час, после
чего выпавший в.осадок «-хлорантрахинон отсасывают.и отмывают.
550
СИНТЕЗЫ ОРГАНИЧЕСХИХ ПРЕПАРАТОВ
от кислоты горячей водой (около 350 мл). Яркожелтый продукт
после сушки в вакууме при 100° имеет т. пл. 158— 160° (исправл.).
Выход 14,6—14,7 г (97—98% теоретич.; примечания б и 7).
2. Примечания
1. Ввиду того что к концу реакции наблюдается тенденция
к вспениванию, необходимо пользоваться эффективной мешалкой.
При проверке этого синтеза с успехом была применена мешалка
Хершберга (см. стр. 298), сделанная из тан-
таловой проволоки. Металл этот заметно не
разъедался даже при повторных синтезах.
2. Для укрепления мешалки можно ис-
пользовать обычный стеклянный затвор, од-
нако лучше пользоваться затвором, описан-
ным в «Синт. орг. преп.», сб. 1, стр. 116,
рис. 5А, в котором в качестве запорной жид-
кости применяется не ртуть, а вода.
3. Обыкновенная капельная воронка обла-
дает тем недостатком, что за ней необходимо
непрерывно следить. Поэтому при проверке
описываемого способа было применено устрой-
ство, предложенное Хершбергом и изобра-
д,Чмм. жешюе па рис. 21.
Вольфрамовая 4. Калиевая соль хлорноватой кислоты
менее пригодна вследствие своей плохой
растворимости.
5. Если приливание вести слишком быстро
или же кипятить смесь слишком энергично,
имеет место значительная потеря хлори-
рующих газов через холодильник.
6. При перекристаллизации продукта из 200 мл н,-бутилового
спирта получается 13,4 г продукта в виде желтых игл с т. пл. 161—
162° (исправл.). Большие количества лучше всего перекристалли-
зовывать из толуола из расчета 2 мл толуола на 1 г вещества.
7. Температура плавления тщательно очищенного а-хлорантра-
хинона равна 162,5° (исправл.). Возможными примесями являются
р-изомер, а также 1,5- и 1,8-дихлорантрахиноны. Все эти вещества
хотя и имеют более высокую температуру плавления, однако сни-
жают температуру плавления «-хлорантрахинона.
проволока
— 0,5 ммх8см.
Капилляр
Рис. 21.
3. Другие методы получения
а-Хлорантрахинон может быть получен: из а-аминоантрахинона
посредством диазо-реакции 1, действием хлористого тионила на
jk-ХЛОРБЕНЗАЛЬДЕГИД
551
калиевую соль антрахинон-а-сульфокислоты под давлением а и по
описанному выше способу 3.
1 Bayer und Ко; герм. пат. 131 538[Frdl. 6, 311 A900-02)]; G г о g g i п s,
Unit Processes in Organic Syntheses, p. 175, McGraw-Hill Book Company, New
2> M e is t e r, Luc ius, Br ii n i ng; герм. пат. 267 544 (Chem. Zentr.
19Нз Baver'und Ко; герм. пат. 205 195 (Chem. Zentr. 1909, I, 414); Badische
Anilin- und Soda-Fabrik, герм. пат. 228 876 (Chem. Zentr. 1911, 1, 102); U 1 1-
mann, Ochsner, Ann. 381, I A911).
.м-ХЛОРБЕНЗАЛЬДЕГИД
jn-NOXeHXHO + 3 SnCl, + б НС1
> jK-NHXeHXHO + 3 SnCl4 + 2 H2O
*-NHXeHXHO + NaNO2 + 2 HC1
> jK-ClN2CeH4CHO + NaCl + 2 H2O
лс-(ЖХвНХНО
N2
Предложили: И. Бек и В. Айди.
Проверили: Дж. Джонсон и П. Виттум.
1. Получение
Раствор 450 г B мол.) кристаллического хлористого олова (при-
мечание 1) в 600 мл концентрированной соля'ной кислоты помещают
в 3-литровый стакан, снабженный эффективной механической
мешалкой, и охлаждают в ледяной бане. Когда температура пони-
зится до + 5°, в раствор вносят в один прием 100 г @,66 мол.)
л-нитробензальдегида (примечание 2). Температура в течение пер-
вых 5 мин. медленно поднимается до 25—30°, а затем быстро дохо-
дит до 100°. Необходимо энергичное перемешивание, иначе реакцион-
ную смесь может выбросить из стакана (примечание 3). Во время
реакции нитробензальдегид растворяется и получается почти про-
зрачный красный раствор; его охлаждают смесью льда и соли до
тех пор, пока температура не понизится до +2°. Во время охлажде-ч
ния выделяются оранжево-красные кристаллы и получается густая
суспензия.
Над стаканом устанавливают делительную воронку на 250 мл
так, чтобы конец ее был опущен под поверхность пастообразной
суспензии. В воронку помещают раствор 46 г @,67 мол.) азотисто-
кислого натрия в 150 мл воды и при энергичном перемешивании
552
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
медленно приливают его к смеси в стакане до тех пор, пока иодо-
крахмальная бумага не даст реакции на азотистую кислоту. Во
время прибавления раствора нитрита, которое занимает около
90 мин., температуру смеси поддерживают между 0 и + 5° (при-
мечание 4). Обыкновенно приходится прибавить почти весь раствор
нитрита, за исключением 5—8 'мл, и лишь тогда иодокрахмальная
бумага показывает избыток азотистой кислоты.
Под конец диазотирования аминобензальдегида подготовляют
горячий раствор полухлористой меди: 189 г @,75 мол.) растертых
в порошок кристаллов сернокислой меди, и 161 г хлористого натрия
растворяют в 600 мл горячей воды в 5-литровой круглодонной
колбе; к этому раствору прибавляют раствор 41 г @,22 моль) мета-
бисульфита натрия Na2S2O5 и 27 г @,67 мол.) едкого натра в 300 мл
воды. К концу реакции температура полученного раствора полу-
хлористой меди должна быть около 75°,
Раствор диазония приливают к горячему раствору полу-
хлористой меди, причем содержимое колбы взбалтывают от руки,
но не охлаждают. Хорошо перемешав растворы, прибавляют к смеси
840 мл концентрированной соляной кислоты и оставляют стоять на
ночь. На другой день для выделения ж-хлорбензальдегида реакцион-
ную смесь перегоняют с водяным паром; практически ,и-хлорбен-
зальдегид отгоняется с первыми 1,5 л дестиллата. Из водного
дестиллата ж-хлорбензальдегид извлекают два раза эфиром пор-
циями по 150 мл, эфирный раствор сушат 10—15 г безводного хло-
ристого кальция и затем сливают с него. Эфир отгоняют, а остаток
перегоняют в вакууме. ж-Хлорбензальдегид кипит при 84—86°/8мм;
107—109°/26 мм (примечание 5). Выход 70—74 г G5—79% тео-
ретич.; примечание 6).
2. Примечания
1. При работе было взято кристаллическое химически чистое
хлористое олово SnCl2 • 2Н2О. При работе с техническим хлори-
стым оловом выходы получаются ниже.
2. Для работы был взят л«-нитробензальдегид обычного каче-
ства с т. пл. 52—55°.
3. Во время бурной реакции рекомендуется хорошо перемеши-
вать как реакционную смесь, так и охладительную баню. Если
лг-нитробензальдегид вносить несколькими порциями для того,
чтобы реакция протекала спокойнее, то выходы получаются хуже.
4. При температурах ниже 0° скорость диазотирования значи-
тельно замедляется. Выше + 5° происходит некоторое разложение
диазония.
5. Так как ж-хлорбензальдегид легко окисляется кислородом
воздуха, его следует сохранять в плотно закупоренных сосудах или
в запаянных ампулах.
.М-ХЛОРБЕНЗАЛЬДЕГИД
553
6. По данным авторов, предложивших настоящий синтез,
ж-бромбензальдегид можно получить по тому же методу, если вместо
полухлористой меди взять раствор полубромистой меди, который при-
готовляется из 189 г сернокислой меди, 91 г бромистого натрия, 41 г
метабисульфита натрия и 27 г едкого натра. Вместо 840 мл концен-
трированной соляной кислоты к смеси растворов диазония и броми-
стой меди прибавляют 200 мл 48%-ной бромистоводородной кислоты.
ж-Бромбензальдегид кипит при 93—98°/8 мм. Выход л*-бромбенз-
альдегида 80 г, т. е. 65% теоретич.
Однако, согласно имеющимся данным, полученный таким обра-
зом л<-бромбензальдегид может содержать до 20% ж-хлорбензаль-
дегида. Чтобы избежать этой примеси, в качестве восстановителя
следует взять бромистое олово.
Раствор бромистого олова получают нагреванием 119 г A гр.-ат.)
волокнистого олова с 705 г D мол.) 46%-ной бромистоводородной
кислоты на водяной бане в течение 2 час. с перемешиванием механи-
ческой мешалкой. Раствор охлаждают до 40° и сразу при работаю-
щей мешалке прибавляют 50 г @,33 мол.) л*-нитробензальдегида.
В результате выделяющегося при реакции тепла температура смеси
повышается и доходит приблизительно до 105°. Затем смесь нагре-
вают еще 30 мин. на водяной бане, после чего ее охлаждают до 0°
и амииобензальдегид диазотируют, для чего постепенно добавляют
раствор 23 г@,33мол.) азотистокислого натрия в 75 мл воды. Раствор
диазония приливают к горячей суспензии полубромистой меди,
добавляют при помешивании 100 мл 46%-ной бромистоводородной
кислоты и смесь оставляют стоять на ночь. Затем реакционную массу
подвергают перегонке с водяным паром, после чего ж-бромбензаль-
дегид извлекают эфиром и после отгонки растворителя перегоняют
в вакууме. Т. кип. 90—92°/4 мм; выход 41 г F7% теоретич.;
Ф. Тайзон, частное сообщение).
3. Другие методы получения
л*-Хлорбензальдегид был получен хлорированием бензальде-
гида 1 и окислением л<-хлорбензилового спирта 2 и ж-хлортолуола 3.
Удобнее всего получать его из л«-нитробензальдегида через ж-амино-
бензальдегид реакцией диазотирования4. Описанный метод в
основных чертах тот же, что и имеющийся в патентной лите-
ратуре *.
1 Ми Пег; герм. пат. 30 329; 33 064 [Frdl. 1, 143, 146 A877—1887I.
2 Mettle r, Ber. 38, 2812A905).
3 L a w, Р е г k i п, J. Chem. Soc. 93, 1636 A908).
* М е i s t e r, Lucius, Briining, герм. пат. 31 842 [Frdl. 1, 144
A877—1887)]; Erdmann, Schwechten, Ann. 260, 59 A890); E i с h e n-
g r u n, Ein^horn, там же 262, 135 A891).
554
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
л-С1С6Н4СН3
п-Cl С6Н4СНС12
я-ХЛОРБЕНЗАЛЬДЕГИД
2С1, > л-С1С,Н4СНСЦ + 2НС1
Н20 > л-С1СвН4СНО + 2НС1
Предложил: В. Мак-Ювен.
Проверили: Г.Тильман и Ч. Лиу.
1. Получение
Круглодонную колбу емкостью 500 мл с двумя горлами соеди-
няют с обратным воздушным холодильником B см в диаметре"),
наполненным на протяжении 60 см стеклянными бусами или коль-
цами диаметром 5—6 мм (примечание 1). Хлор вводят в колбу через
стеклянную трубку диаметром 4 мм, вставленную в пробку, кото-
рая закрывает одно из отверстий колбы; трубка доходит почти до
дна колбы и оканчивается небольшим перфорированным шариком,
для того чтобы ток газа разбивался на мелкие пузырьки. От боль-
ших количеств образующегося хлористого водорода можно изба-
виться с помощью ловушек.
Во взвешенную колбу помещают 126,5 г A мол.) п-хлортолуола
(«Синт. орг. преп.», сб. 1, стр. 491) и 3,8 г пятихлористого фосфора.
Колбу нагревают на бане, температуру которой поддерживают при
160—170° (примечание 2), и при освещении прямым солнечным све-
том или 100-ваттной вольфрамовой лампой пропускают сильный
ток хлора прямо из баллона до тех пор, пока вес'колбы не увели-
чится на 55—66 г (примечание 3).
Продукт, окрашенный в слабожелтый или желтозеленый цвет,
переносят в широкогорлую 4-литровую склянку, в которой нахо-
дится 400 мл концентрированной серной кислоты, и смесь сильно
перемешивают в продолжение 5 часов (тяга; примечание 4). Затем
вязкую жидкость переводят в делительную воронку и оставляют на
ночь, после чего нижний слой (примечание 5) медленно выливают
при помешивании в стакан емкостью 3 л, наполненный на три чет-
верти колотым льдом. После того, как лед растает, полученный оса-
док кремового цвета отсасывают, промывают водой, отжимают до-
суха на воронке и делят на три равные части. Затем каждую часть
растворяют в возможно меньшем количестве эфира и эфирный рас-
твор несколько раз встряхивают с 2%-ным раствором едкого натра
до тех пор, пока при подкислении пробы промывной воды не пере-
станет выпадать осадок п-хлорбензойной кислоты (примеча-
ние 6).
Эфир отгоняют на водяной бане, а остаток перегоняют в ваку-
уме из колбы Клайзена. Выход л-хлорбензальдегида с т. кип.
108—111725 мм и т. нл. 46—47° составляет 76—84 г E4—60%
теоретич.).
п-ХЛОРБЕНЗАЛЬДЕГИД
55 5
2. Примечания
1. Стеклянная насадка холодильника уменьшает возможность
увлечения п-хлортолуола током хлористого водорода в виде мел-
ких капелек. Если надо провести только одну операцию, то годятся
обыкновенные хорошие пробки. Если же требуется провести не-
сколько операций, рекомендуется пропитать пробки раствором жид-
кого стекла, чтобы защитить их от разъедания хлористым водоро-
дом и хлором.
2. Баня наполняется маслом или графитом.
3. Для этого требуется 4,5 часа. При работе с большими исход-
ными количествами G50 г п-хлортолуола и 23 г пятихлористого
фосфора), по данным автора, для достижения прироста в весе
330—360 г потребовалось 6—10 час.
4. Энергичное перемешивание необходимо для того, чтобы избе-
жать сильного вспенивания. Большая часть хлористого водорода
выделяется в начале перемешивания.
5. Верхний воскоподобный слой выбрасывают.
6. Выход л-хлорбензойной кислоты около 20 г. При больших
загрузках G50 г п-хлортолуола) выход кислоты в среднем 260 г.
3. Другие методы получения
п-Хлорбензальдегид может быть получен: из п-хлор- или п-бром-
бензилхлорида при действии водного раствора азотнокислого
свинца г; из /г-хлортолуола и хлористого хромила 3 путем гидро-
лиза хлористого л-хлорбензальхлорида 3; из п-аминобензальдегида
диазотированием и последующей обработкой полухлористой медью4;
из бромистого п-хлорфенилмагния и этилового эфира ортомуравьи-
ной кислоты5; из п-хлорбензилхлорида и гексаметилентетрамина
с последующим гидролизом 6; из п-хлорбензонитрила через имино-
хлорид с последующим гидролизом7; при действии окиси углерода
и хлористого алюминия на хлорбензол 8 и действием углекислого
натрия на 2,5-дихлорбензолсульфонил-4-хлорбензоилгидразин9.
iBeilstein, Kuhlberg, Ann. 147, 352 A867); Jackson,
White, Am. Chem. J. 3, 30 A881).
2 L a w, P e r k i n, J. Chem. Soc. 93, 1636 A908).
sErdmann, Kirchhoff, Ann. 247, 368 A888); E r d m a n n,
Sen we с h ten, там же 260, 63 A8S0); К a e s wu r m, Ber. 19, 742 A886).
* W a 1 t h e r, R a e t z e, J. prakt. Chem. B) 65, 258 AS02).
6 В о d г о u x, Bull. soc. chim. C) 31, 587 A904).
«Mayer, English, Ann. 417,78A918).
'Stephen, J. Chem. Soc. 127, 1874 A925).
8 Boehringer und Sohne. герм. пат. 281 212 (Chem. Zentr. 1915, I, 178).
3 М с F a d у e n, Stevens, J. Chem. Soc. 1936, 584.
556
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
ХЛОРИСТАЯ Р-НАФТИЛРТУТЬ
557
о-ХЛОРБЕНЗОЙНАЯ КИСЛОТА
С1С6Н4СН3 + 2 КМпО4 > С1С6Н4СО2К + 2 МпОа + КОН + Н2О
Предложили: X. Кларк и Е. Тэйлор.
Проверили: Г. Гильман и Дж. Мак-Глемфи.
1. Получение
В 12-литровой колбе, снабженной мешалкой и обратным холо-
дильником, смешивают 600 г C,8 мол.) марганцовокислого калия,
7 л воды и 200 г A,6 мол.) о-хлортолуола («Синт. орг. преп.», сб. 1,
стр. 491). Смесь медленно нагревают до кипения (примечание 1)
при непрерывном размешивании, пока не исчезнет окраска перман-
ганата, на что требуется от 3 до 4 час. После этого колбу соединяют
с обращенным вниз холодильником и смесь кипятят при перемеши-
вании до тех пор, пока с водой не перестанет отгоняться масло.
Количество полученного таким образом не вошедшего в реакцию
о-хлортолуола достигает 25—30 г. Горячую смесь отсасывают и
осадок перекиси марганца промывают дважды горячей водой пор-
циями по 500 мл. Соединенные фильтраты упаривают (примечание 2)
до объема 3,5 л; если раствор не вполне прозрачен, то его можно
осветлить при помощи 1—2 г угля. Затем горячий еще раствор
осторожно подкисляют, прибавляя к нему при постоянном разме-
шивании 250 мл концентрированной соляной кислоты, уд. в. 1,19.
После того как смесь охладится, белый осадок о-хлорбензойной
кислоты отсасывают и промывают холодной водой. После высуши-
вания получается 163—167 г G6—78% теоретич., считая на вошед-
ший в реакцию о-хлортолуол) почти совсем чистой (примечание 3)
о-хлорбензойной кислоты с т. пл. 137—138°. Для полной очистки
ее можно перекристаллизовать из 600 мл толуола; при этом полу-
чается 135—140 г продукта, плавящегося при 139—140°. Некоторое
количество вещества можно получить упариванием маточного
раствора.
2. Примечания
1. Если нагревать смесь слишком быстро, то реакция может
пойти бурно с самого начала. В этом случае течение реакции уме-
ряют, накладывая мокрые тряпки на верхнюю часть колбы.
2. Удобнее всего упаривать на водяной бане в вакууме (см.
«Синт. орг. преп.», сб. 1, стр. 333).
3. Для описанного метода получения следует брать чистый
о-хлортолуол, в противном случае о-хлорбензойная кислота может
оказаться загрязненной изомерными кислотами, которые чрезвы-
чайно трудно удалить. Поэтому о-хлортолуол следует получать
из чистого о-толуидина или из о-хлортолуолсульфокислоты..
Метод очистки технической 0-хлорбензойной кислоты описан
на стр. 19.
3. Другие методы получения
Единственными практическими методами получения о-хлор-
бензойной кислоты является окисление 0-хлортолуола и замещение
аминогруппы антраниловой кислоты атомом хлора. Оба этих метода
были детально исследованы Гребе *; для получения относительно
больших количеств он рекомендует первый метод. Окисление
0-хлортолуола перманганатом впервые описал Эммерлинг 2.
1 О г а е b e, Ann. 276, 54 A893).
2 Е m m e r I i n g, Ber. 8, 880 A875).
ХЛОРИСТАЯ 0-НАФТИЛРТУТЬ
B-Хлормеркурнафталин )
C10H7NH2 + NaNO, + 2 НС1 -f HgCl2
C1OH7N2C1 • HgCl2 + NaCl + 2 H2O
C1OH7N2C1 • HgCl2 + 2Cu > C10H7HgCl + N3 + 2CuCl
Предложил: А. Несмеянов.
Проверили: Ф. Уитмор и Р. Витти.
1. Получение
В 4-литровый глиняный сосуд, снабженный эффективной мешал-
кой, помещают 450 мл концентрированной соляной кислоты, уд. в.
1,19, и 500 мл воды, и затем прибавляют 143 г A мол.) [3-иафтил-
амина. Суспензию солянокислого амина охлаждают прибавлением
500 г колотого льда. Когда температура понизится до 5°, прибавляют
твердый азотистокислый натрий (около 69 г) до избытка на иодо-
крахмальную бумажку. Во время диазотирования постепенно при-
бавляют еще 600 г колотого льда с такой скоростью, чтобы поддер-
живать температуру при 5°. Холодный раствор соли диазоиия от-
фильтровывают от незначительного осадка и снова помещают
в тот же сосуд. Раствор 271 г A мол.) хлорной ртути в 300 мл кон-
центрированной соляной кислоты смешивают с 300 г льда и медленно
приливают к быстро перемешиваемому раствору диазония. Выпадает
тяжелый желтый осадок двойной соли хлористого р-нафтилдиазо-
ния и сулемы. Для того, чтобы реакция прошла полностью, пере-
мешивание продолжают еще в продолжение получаса. Осадок
отсасывают возможно тщательнее на воронке Бюхнера диаметром
20 см и промывают двумя порциями воды по 400 мл и двумя пор-
циями ацетона по 150 мл (примечание 1). Осадок сушат на воздухе
при температуре около 20° (примечание 2) до постоянного веса.
Выход 380—390 г (82—84% теоретич.; примечание 3).
558
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
ХЛОРИСТЫЙ ДЕЗИЛ
559
В 2-литровую круглодонную колбу из стекла ширекс», снаб-
женную мешалкой, помещают 139 г @,3 мол.) продукта присоеди-
нения, 700 мл ацетона (примечание 4), 38 г (О,бгр.-ат.) порошка меди
(примечание 5). Смесь сразу же охлаждают до 20°, перемешивают
в течение 1 часа и оставляют стоять на ночь. Нерастворившуюся
часть отсасывают на воронке Бюхнера (примечание 6). Получаю-
щийся осадок экстрагируют Ъл кипящего продажного ксилола
и раствор фильтруют через воронку с обогревом. По охлаждении
фильтрата выпадают кристаллы хлористой |3-нафтилртути (приме-
чание 7). Выход продукта с т. пл, 266—267° составляет 52—64 г
D0—49% теоретич., считая на взятый амин; примечания 8 и 9).
2. Примечания
1. Промывка большими количествами растворителей необхо-
дима для удаления незначительных количеств сильно окрашенных
примесей. Сам продукт практически нерастворим в холодных
жидкостях, употребляемых для промывки.
2. При нагревании двойная соль сильно взрывает. Для синтеза
ртуть-органических соединений двойная соль может быть и не
вполне сухой.
3. Хранить двойные соли сулемы и хлористых диазониев в закры-
той посуде небезопасно. Применять двойную соль следует по воз-
можности в день синтеза. Если возникает необходимость ее хранить,
рекомендуется рассыпать ее тонким слоем в большой открытой
чашке или в неплотно закрытом сосуде.
В ряду большого числа таких синтезов однажды около 960 г
двойной соли сулемы с хлористым фенилдиазонием были высушены
на воздухе и помещены в большую склянку с навинчивающейся
крышкой. Примерно через 4 часа комплекс разложился с большой
силой, причем в течение этого времени он не подвергался ни нагре-
ванию, ни действию сильного света (Г. Гильман, частное сообще-
ние).
4. Применявшийся ацетон кипел при 55—57°. Замена ацетона
этиловым или метиловым спиртом несколько понижает выход.
5. Порошок меди приготовляют из цинковой пыли и избытка
раствора сернокислой меди при температуре ниже 70°.
6. Ацетоновый фильтрат содержит лишь следы продукта.
7. Маточный раствор после отделения кристаллов может быть
вновь употреблен для извлечения следующего сырого осадка, так
как продукт практически нерастворим в холодном ксилоле.
8. Если взять бромистоводородную кислоту и бромную ртуть,
то получается бромистая [З-нафтилртугь.
9. Подобным же образом можно перевести анилин, все три
толуидина и аминофенолы в соответствующие хлористые ртутные
соединения с выходом около 40%.
3. Другие методы получения
Хлористая -Э-нафтилртуть была получена действием хлорной
ртути в амиловом спирте на ди-,3-нафтилртуть 1; из ,6-иафталии-
сульфиновой кислоты и хлорной ртути 2; из р-нафтилборной кислоты
и хлорной ртути 3 и вышеописанным способом.
1 М i с h a e I i s, Ber. 27, 251 A8S4).
2Whitmore, Howe, Organic Compounds of Mercury, by Frank
С Whitmore, The Chemical Catalog Company, New York, 1921, p. 203.
3 К о n i g, S с h a r r n b e с k, J. prakt. Chem. B) 128, 168 A930).
ХЛОРИСТЫЙ ДЕЗИЛ
(а-Хлор-а-фенилацетофенон)
C6H6CHOHCOC6H5 + SOC12
C5H5N
C6H5CHC1COC6H3 + SO2 +HC1
6H5
Предложил: А. У орд.
Проверили: Я. Марвел и Це-Цинг Чу.
1. Получение
В 1-литровый стакан помещают 100 г @,47 мол.) бензоина («Синт,
орг. преп.», сб. 1, стр. 95) и 50 г E7 мл) пиридина. Смесь нагре-
вают до получения однородного раствора и затем охлаждают льдом
до тех пор, пока она не застынет. Застывшую массу растирают
(не очень мелко) и к ней медленно приливают при энергичном пере-
мешивании и охлаждении водой 75 г D6 мл; 0,63 мол.) хлористого
тионила. После каждого прибавления хлористого тионила реак-
ционная смесь сильно разогревается, и выделяются значительные
количества сернистого газа и хлористого водорода. Масса сначала
размягчается, но вскоре застывает, причем получается твердый
продукт, окрашенный в светложелтый цвет. Примерно через 1 час
прибавляют воды, твердую массу растирают и отсасывают. Затем
продукт еще дважды хорошо растирают с водой, отсасывают и как
можно -лучше отжимают. Полученный белый порошок сушат до
'постоянного веса над серной кислотой или хлористым кальцием.
Выход сырого продукта около 125 г. Полученное соединение раство-
ряют в 450 мл кипящего 95%-ного спирта (примечание 1), раствор
фильтруют и фильтрат охлаждают водопроводной водой. Полу-
чают 77 г бесцветных кристаллов, которые после сушки на воздухе
плавятся при 66—67°. Охлаждая маточный раствор смесью льда
с солью, получают еще 9 г кристаллов с т. пл. 65—66°. Дальнейшее
охлаждение фильтрата уже не дает больше кристаллов. Общий
выход 80—86 г G4—79% теоретич.; примечания 2 и 3).
560
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
S-ХЛОР-о-НИТРОТИОФЕНОЛ
561
2. Примечания
1. Продукт можно нерекристаллизовывать из петролейного
эфира (т. кип. 40—60°), но этот растворитель менее удобен при
работе с большими количествами продукта.
2. Если опыт вести в количествах, равных х/5 указанных выше,
выход получается лишь 70% теоретич.
3. Хлористый дезил разлагается и темнеет на солнечном свете,
но при хранении в темных склянках вполне устойчив.
3. Другие методы получения
Хлористый дезил был получен действием хлористого тионила
на бензоин 1 и /-бензоин2. Его получали также действием хло-
ристого водорода на азибензил 3.
1 S с h г о е t e г, Вег. 42, 2348 A909).
2 М с К е n z i e, Wren, J. Chem. Soc. 97, 481 A910).
3 С u r t i u s, Lang, J. prakt. Chem. B) 44, 547 A891).
S-ХЛОР-о-НИТРОТИОФЕНОЛ
(Хлористая о-нитрофенилсера, о-нитрофенилсульфотя хлорид)
NO2C6H4SSC6H4NO2 + CLj * 2 NO2C6H4SC1
Предложил: М. Хубачер.
Проверили: Л. Физер и Д. Поттер.
1. Получение
В 1-литровую трехгорлую колбу, снабженную термометром,
обратным холодильником и трубкой для ввода газа с оттянутым
концом, доходящей до дна колбы, помещают 600 мл сухого четырех-
хлористого углерода, 154 г @,5 мол.) ди-о-нитрофенилдисульфида,
с т. кип. 193—195° («Синт. орг. преп.», сб. 1, стр. 200), и 0,25 г иода.
К верхнему концу обратного холодильника присоединяют стеклян-
ную трубку, другой конец которой погружен в пробирку с неболь-
шим количеством четыреххлористого углерода. Через реакционную
смесь, температуру которой поддерживают при 50—60°, пропу-
скают ток хлора, предварительно осушаемого серной кислотой.
Скорость пропускания хлора A6—17 г в час) регулируют таким
образом, чтобы газ совсем или почти совсем не проходил через
ловушку с четыреххлористым углеродом. Постепенно желтый
ди-о-нитрофенилдисульфид исчезает, и через 2—2,5 часа получают
гомогенный темножёлтый раствор (примечание 1). Для того, чтобы
освободиться от небольшого количества темного осадка, теплый
раствор фильтруют через обогреваемую воронку Бюхнера, причем
как колбу, так и фильтр ополаскивают 30 мл теплого четыреххло-
ристого углерода. Желтый фильтрат (примечание 2) охлаждают
до 5° и оставляют кристаллизоваться. Сростки кристаллов разби-
вают стеклянной палочкой, собирают на бюхнеровской воронке,
хорошо, отсасывают и отжимают. Продукт быстро сушат B часа)
при 50° и хранят в закупоренной склянке (примечание 3).
Т. пл. этого продукта 73—74,5°, а вес его 126—135 г F6—71%
теоретич.). Можно получить еще некоторое количество кристаллов,
если отогнать растворитель от маточного раствора на водяной
бане (примечание 4) и оставшееся темноокрашенное масло вылить
в фарфоровую чашку. Последние следы четыреххлористого угле-
рода удаляют сушкой при 50°, причем масло закристаллизовывается.
Полученный продукт плавится при 67—72° и весит 48—58 г. Он
обладает достаточной чистотой для большинства целей (примеча-
ние 5). Общий выход 183—184 г (96—97% теоретич.; примечания б
и 7).
2. Примечания
1. Избыток хлора не вредит.
2. Для большинства целей можно без дальнейшей очистки при-
менять этот фильтрованный раствор, если предварительно отогнать
около 100 мл растворителя с целью удаления избытка хлора. Выход
продукта, находящегося в растворе, составляет около 98% теоретич.
3. Чистый продукт плавится при 74,5—75°. В контакте с влаж-
ным воздухом S-хлор-о-нитротиофенол разлагается в течение
нескольких дней, отщепляя хлористый водород. В герметично
закупоренной склянке из темного стекла с притертой пробкой его
можно хранить в течение нескольких месяцев.
4. Ввиду того, что S-хлор-о-нитротиофенол самопроизвольно
разлагается при нагревании выше 170°, рекомендуется для удаления
растворителя пользоваться водяной баней.
5. Продукт можно перекристаллизовать из горячего четырех-
хлористого углерода B мл на 1 г), охладив затем профильтрован-
ный раствор до 5°. Получается 75% вещества, считая на сырой
продукт, с т. пл. 70—73°.
6. Тот же метод может быть применен для получения S-хлор-
о-нитро-п-хлортиофенола (т. пл. 95—97°); хлорирование дисуль-
фида (т. пл. 212—213) в этом случае протекает значительно медлен-
нее. 8-Хлор-2,4-динитротиофенол может быть получен хлориро-
ванием соответственного дисульфида, суспендированного в нитро-
бензоле, при 120—130°. Ввиду того, что хлорид обладает взрывча-
тыми свойствами, растворитель следует отгонять в вакууме при 130°.
Сырой продукт плавится при 89—92°. После перекристаллизации
из четыреххлористого углерода т. пл. повышается до 94—95°.
7. Арилтиохлориды применяются для введения группы ArS-,
причем по своим реакциям они напоминают хлорангидриды кислот.
36 Сборник Хг 2
562
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
3. Другие методы получения
S-Хлор-о-нитротиофенол всегда получался хлорированием соот-
ветственного дисульфида '.
1 Z i n с к е, Вег. 44, 770 A911); Z i n с к е, F а г г, Ann. 391, 63 A912)..
Р-ХЛОРЭТИЛМЕТИЛСУЛЬФИД
CH3SCH2CH2OH 4- SOC12 > CH3SCH2CHX1 + SO2 + HC1
Предложили: В. Кирнер и В. Виндус.
Проверили: X. Кларк и С. Гурин.
1. Получение
В 1-литровой трехгорлой колбе смешивают 150 г A,63 мол.)
|3-оксиэтилметилсульфида (стр. 396; примечание 1) и 200 г сухого
хлороформа (примечание 2). Колбу помещают на водяную баню
и снабжают ее капельной воронкой, механической мешалкой и холо-
дильником. К холодильнику присоединяют ловушку для улавли-
вания хлористого водорода и сернистого газа. Затем к раствору
р-оксиэтилметилсульфида по каплям прибавляют раствор 204 г
A,7 мол.; примечание 3) хлористого тионила в 135 мл сухого хлоро-
форма в течение примерно двух часов (примечание 4). Реакционную
смесь энергично перемешивают как при этом прибавлении, так и по
окончании его еще в течение 4 час. Хлороформ отгоняют на водяной
бане, а остаток перегоняют в вакууме. Выход продукта, кипящего-
при 55—56°/30 мм, составляет 135—153 г G5—85% теоретич.;
примечание 5).
2. Примечания
1. Можно увеличить загрузку в семь раз, и это не вызовет
уменьшения выхода в процентах.
2. Хлороформ сушат перегонкой, причем применяют фракцию
с т. кип. 60—61°.
3. Хлористый тионил перегоняют и берут фракцию, кипящую
в пределах 2°.
4. Реакционную смесь подогревают после того, как добавлено
половинное количество хлористого тионила, так чтобы хлороформ
спокойно кипел. Нагревание после прибавления всего количества
хлористого тионила не рекомендуется.
5. [З-Хлорэтилметилсульфид вызывает на коже нарывы, почему
обращение с ним требует осторожности. При атмосферном давлении
он кипит при 140°.
ХОЛЕСТАНОН
3. Другие методы получения
Приведенный выше метод в основных чертах тот же самый»,
который описан в литературе *.
1 К i г п е г, J. Am. Chem. Soc. 50, 2452 A928).
ХОЛЕСТАНОН
НХ
НХ
нх
с8н17
СгОя
нх
/\
о
Предложил: У. Брюс.
Проверили: Л. Физер и Р. Джекобсеп..
1. Получение
Раствор 50 г @,13 мол.) дигидрохолестерина (примечание 1>
в 500 мл беизола медленно при охлаждении (примечание 2) добав-
ляют к раствору 68 г @,23 мол.) кристаллического двухромово-
кислого натрия, 50 мл ледяной уксусной кислоты и 90 мл концен-
трированной серной кислоты в 300 мл воды, находящемуся в 3-литро^
вой колбе. Смесь энергично перемешивают, или поставив колбуг
на качалку, или же при помощи эффективной мешалки (примеча-
ние 3), в течение 6 час, при 25—30° (примечание 4).
После этого бензольный раствор отделяют и дважды промывают
100 мл воды, один раз 200 мл 5%-ного раствора едкого кали и вновь
два раза водой. Если раствор не стал еще бесцветным, его обесцве-
чивают 1 г угля. Бензол отгоняют и полученную сиропообразную*
массу растворяют при нагревании в 300 мл спирта. По охлаждении
холестанон выпадает из раствора в виде хорошо образованных игл.
Выход промытого и воздушно-сухого продукта с т. пл. 129—130°"
составляет 41,5—42 г (83—84% теоретич.). Если к фильтрату при-
бавить 80 мл воды, то можно получить еще 2 г продукта с т. пл.
125—126°.
2. Примечания
1. Пригодным для синтеза является продукт с т. пл. 140—14F
(стр. 195). Присутствие следов холестерина не оказывает вредного»
564
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
ЦЕЛЛ0БИ03А
565
действия, так как он при окислении превращается в продукты кисло-
го характера, от которых освобождаются в процессе очистки.
2. Если растворы смешивать без охлаждения, то температура
поднимается до 60° и выход несколько падает.
3. При проверке этого синтеза с успехом была применена мешалка
Гершберга (стр. 298).
4. Если перемешивание вести в два раза дольше, выход заметно
не изменяется. Период в 6 час. является минимальным для переме-
шивания при данной загрузке.
3. Другие методы получения
Холестанон был получен окислением дигидрохолестерина хро-
мовым ангидридом в растворе уксусной кислоты 1. Выход иногда
бывает несколько меньшим в результате частичного ацетилирова-
ния стерина.
1Diels, Abderhalden, Ber. 39, 884 A906); W i 1 1 s t a 11 e г,
Mayer, там же 41, 2199 A908); Vavon, Jakubowicz, Bull. soc.
chim. D) 53, 584A933).
ЦЕЛЛОБИОЗА
C12Hi4Ou (C0CH3)s + 8 CH3OH -
CiaH22Ou + 8 CH3CO2CH3
Предложил: Г. Браун.
Проверили: Р. Фыозон, В. Росс и В. Кемпбелл.
1. Получение
В трехгорлой колбе емкостью 500 мл, снабженной мешалкой
с ртутным затвором и хлоркальциевой трубкой, получают суспен-
зию 68 г @,1 мол.) октаацетата о-целлобиозы с т. пл. 220—222°
(стр. 398) в 300 мл абсолютного метилового спирта. Затем прибав-
ляют раствор, полученный внесением 0,25 г @,01 гр.-ат.) натрия
в 50 мл метилового спирта, и смесь энергично перемешивают при
комнатной температуре в течение одного часа (примечание 1).
По мере того как протекает гидролиз, смесь становится все более
жидкой, а растворитель слегка окрашивается. По истечении ука-
занного времени кристаллический осадок отсасывают, промывают
четыре раза метиловым спиртом порциями по 25 мл и сушат при 40?.
Вес неочищенной и почти бесцветной целлобиозы соответствует
теоретическому количеству C4 г). С целью очистки ее растворяют
в 125 мл горячей воды, к которой прибавлено несколько капель
ледяной уксусной кислоты, раствор просветляют 1—2 г угля и
фильтруют с отсасыванием. Бесцветный фильтрат упаривают
в вакууме до небольшого объема. Выпаривание прекращают, когда
большая часть целлобиозы выкристаллизуется, после чего кристал-
лическую массу смывают в коническую колбу 100 мл метило-
вого спирта. Смесь хорошо перемешивают и оставляют стоять
в течение нескольких часов, чтобы закончилась кристаллизация.
Кристаллы собирают на бюхнеровской воронке, промывают 25 мл
метилового спирта и сушат при 40°. Выход чистой целлобиозы,
имеющей [о]2о° + 34,8° (в 6%-ном водном растворе), составляет
31 г (91% теоретич.). Если упарить маточный раствор до неболь-
шого объема и вновь прибавить спирта, то можно получить еще 1 г
такого же чистого продукта. Таким образом, суммарный выход
составляет 94% теоретич. (примечание 2).
2. Примечания
1. С одинаковым успехом реакцию можно вести также при меха-
ническом встряхивании в закупоренной склянке.
2. При работе по старому методу 1, согласно которому раствор
октаацетата в хлороформе обрабатывали раствором метилата
натрия, а затем водой, выход чистой целлобиозы достигал тольк©
67—79% теоретич.
3. Другие методы получения
Целлобиозу впервые получили Скрауп и Кениг2 омылением
октаацетата спиртовым раствором едкого кали. Позднее этот метод
усовершенствовали Прингсгейм и Меркац3. Для этой цели приме-
нялся также водный гидрат окиси бария 4. Для омыления ацетатов
углеводов был широко использован раствор аммиака в метиловом
спирте. Метод каталитического омыления небольшим количеством
метилата натрия впервые предложил Цемплен 1, который рассмат-
ривал механизм этой реакции, как присоединение реагента к карбо-
нилам сложноэфирных групп ацетата Сахаров с последующим
разложением продукта присоединения в результате реакции со
спиртом8. Настоящая методика, взятая из работы Цемплена,
Гереча и Гадаши 6, обладает значительными преимуществами по
сравнению с первоначальной г (см. примечание 2).
1 Z e m p 1 ё п, Вег. 59, 1254 A926).
3 S k г а и р, К о n i ?, там же 34, 1115 A901).
3Pringsheim, Merkatz, Z. physiol. Chetn. 105, 173 A919).
•Abderhalden, Zemplen, там же 72, 58 A911).
'Zemplcn, Kunz, Ber. 56,1705A923).
«Zemplen, Oerecs, Hadacsy, там же 69, 1827 A936).
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
ЦИАНГИДРИН АЦЕТОНА
(а-Оксиизобутиронитрил)
{(СН3J СО + NaCN + H2SO4 (СН3J С (ОН) CN + NaHSO4
Предложили: Р. Кокс и Р. Стормонт.
Проверили: Р. Фьюзон и М. Хэнт,
1. Получение
В 5-литровую круглодонную колбу с тремя горлами, снабжен-
ную эффективной мешалкой (примечание 1), капельной воронкой
и гильзой для термометра, помещают раствор 500 г (9,7 мол.) измель-
ченного 95%-ного цианистого натрия в 1,2 л воды и 900 мл G13 г,
12,3 мол.) ацетона. Колбу помещают в баню со льдом и раствор
энергично перемешивают. Когда температура понизится до 15°,
добавляют в течение 3 час. 2,1 л (8,5 мол.) 40%-ной серной кислоты
{примечание 2), причем во время прибавления температура должна
поддерживаться между 10 и 20°. После того как все количество
кислоты прибавлено, перемешивание продолжают еще 15 мин.,
затем мешалку останавливают и дают осесть образовавшейся соли.
Обычно образуется слой циангидрина ацетона, который сливают
и отделяют от водного слоя. Бисульфат натрия отсасывают и про-
мывают тремя порциями ацетона по 50 мл каждая. Фильтрат, про-
мывную жидкость и водный слой соединяют вместе и трижды экстра-
гируют эфиром порциями по 250 мл (примечание 3). Вытяжки
присоединяют к отделенному ранее циангидрину и все вместе сушат
¦безводным сернокислым натрием. Эфир и ацетон отгоняют на водяной
«бане, а остаток перегоняют в вакууме. Низкокипящую фракцию
отбрасывают и собирают циангидрин ацетона при 78—82°/15 мм.
Выход 640—650 г G7—78% теоретич.; примечание 4). •
2. Примечания
1. Целесообразно применять тяжелую металлическую мешалку,
так как к концу реакции масса становится весьма вязкой. Ввиду
того, что реакционная смесь выделяет небольшое количество циа-
нистого водорода, пробку, через которую проходит мешалка, сле-
дует снабдить трубкой для отвода выделяющихся газов или же
вести реакцию в вытяжном шкафу.
2. Для получения 40%-ной серной кислоты 650 мл концентри-
рованной серной кислоты (уд. в. 1,84) прибавляют к 1,8 л воды.
3. Экстрагирование и перегонку следует начать немедленно по
окончании реакции; перегонку следует вести как можно быстрее,
чтобы избежать разложения.
ЦИКЛОГЕКСИЛБЕНЗОЛ
567
4. Метод получения циангидрина ацетона из цианистого калия
и бисульфитного соединения ацетона описан в Org. Syn. 20, 43.
Методика, которая описана выше, дает менее чистый циангидрин,
пригодный, однако, для ряда синтетических целей.
3. Другие методы получения
Циангидрин ацетона был получен из ацетона и безводного
цианистого водорода в присутствии катализаторов, имеющих
основной характер, а именно в присутствии углекислого калия,
•едкого кали или цианистого калия *; кроме того, он был получен
действием цианистого калия на продукт присоединения бисульфита
натрия к ацетону 2, а также действием цианистого водорода, полу-
чаемого непосредственно в реакционной смеси, на водный раствор
ацетона3. В более современных промышленных процессах приме-
няют ацетон, цианистый водород и катализатор основного харак-
тера 4.
'Urech, Ann. 164, 256A872); U 1 t ё е, Ber. 39, 1857A906); Rec. trav.
«him. 28, 7 A909).
2Bucherer, Grolee, Ber. 39, 1225 A906); герм. пат. 141 509 (Ckem.
Zentr. 1903, I, 1244); Org. Syn. 20, 43.
3 W e 1 с h, Clemo, J. Chem. Soc. 1928, 2629.
* Triplex Safety Glass Company Ltd.; англ. пат. 416007 [С. А. 29, 814A935)]
и 452 285 [С. А. 31, 417 A937)]; I. G. Farbenind. A.-Q.; франц. пат. 804 124
{С. А. 31, 2615 A937)]; Deutsche Gold- und Silber-Scheideanstalt vortn. Roes-
sler; франц. пат. 812 366 [С. А. 32, 858A938)].
-\
,CHL —I
CH, — I
ЦИКЛОГЕКСИЛБЕНЗОЛ
( Фенилу иклогексан )
Предложил: Б. Корсон.
Проверили: Дж. Джонсон и Е. Кливленд.
1. Получение
В 1-литровую трехгорлую колбу, снабженную механической
мешалкой, капельной воронкой и термометром, помещают 468 г
E30 мл, 6 мол.) бензола и 92 г E0 мл) концентрированной серной
кислоты (уд. в. 1,84). Смесь охлаждают в бане со льдом и при рабо-
тающей мешалке в течение 1,5 часа приливают 164 г B03 мл, 2 мол.)
циклогексена (примечание 1). Температуру при этом поддержи-
вают при 5—10°. После того как прибавлен весь циклогексен, пере-
мешивание продолжают еще в течение 1 часа.
568
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
Отделяют углеводородный слой, охлаждают его льдом и про-
мывают четыре раза холодной концентрированной серной кислотой,
порциями по 50 мл (примечание 2). Затем продукт дважды промы-
вают теплой E0°) водой, дважды 3%-ным раствором едкого натра
и дважды чистой водой (примечание. 3). Смесь углеводородов сушат
безводным хлористым кальцием и дважды подвергают дробной
перегонке, пользуясь елочным дефлегматором длиной 30 см или
каким-либо аналогичным дефлегматором. Циклогексилбензол соби-
рают в пределах 238—243° (примечания 5 и 6). Выход 210—220 г
F5—68% теоретич.).
2. Примечания
1. Для получения сравнительно небольших количеств цикло-
гексена весьма удобно дегидратировать циклогексанол 85%-ной
фосфорной кислотой по методике Дена и Джексона [J. Am. Chem.
Soc. 55, 4285 A933)]. При этом имеет место незначительное обугли-
вание в отличие от сернокислотного метода дегидратации (См.
«Синт. орг. преп.», сб. 1, стр. 509), при котором имеет место значи-
тельное обугливание и продукт получается загрязненным сернистым
ангидридом.
В 2-литровую трехгорлую колбу, снабженную делительной
воронкой и трехшариковым дефлегматором Вюрца, наполненным
стеклянными трубками, наливают 200 г 85%-ной фосфорной
кислоты. После дефлегматора присоединяется эффективно
действующий холодильник, который ведет к приемнику, охлаждае-
мому льдом. Колбу нагревают на масляной бане до 165—170°.
Из делительной воронки по каплям в течение 4—5 час. прибавляют
1 кг A0 мол.) технического циклогексанола. После того как все
количество циклогексанола прибавлено, температуру бани посте-
пенно повышают до 200° и эту температуру поддерживают 30 мин.
В течение всего этого времени температура в верхней части дефлег-
матора не превышает 90°. Верхний слой дестиллата отделяют
(можно добавить соли, чтобы уничтожить эмульсию и добиться
лучшего разделения слоев) и сушат безводным сернокислым маг-
нием. Нижний водный слой можно сохранить для дальнейшей
переработки. Отработанное осушающее средство можно обработать
водой для выделения удерживаемого им циклогексена. Сырой
циклогексен перегоняют с хорошим дефлегматором и собирают
фракцию с т. кип. 81—83°. Выход 660—690 г G9—84% теоретич.).
Остаток состоит главным образом из циклогексанола и может быть
подвергнут повторной обработке, как это описано ниже.
Можно получить дополнительно 25—30 г циклогексена, если
соединить вместе остаток от перегонки сырого циклогексена и вод-
ный слой от первоначального дестиллата и подвергнуть их пере-
гонке с 25 г 85%-ной фосфорной кислоты. Полученный дестиллат
присоединяют к низкокипящей головной фракции, полученной при
ЦИКЛОГЕКСИЛБЕНЗОЛ
569"
перегонке сырого циклогексена, отделяют нижний водный слой,
а верхний слой сушат безводным сернокислым магнием и перегоняют,
как это описано выше.
Фосфорная кислота может быть регенерирована, если ее разба-
вить4 водой, профильтровать, а затем упарить с небольшим коли-
чеством азотной кислоты до нужной концентрации.
С помощью той же методики, исходя из 86 г A мол.), циклопен-
тапола и 15 мл 85%-ной фосфорной кислоты, было получено 55 г
(81% теоретич.) циклопентена с т. кип. 44—45°. Попытки выделить
циклопентанол сделано не было (Оливер Груммит и Дж. Джон-
сон, частное сообщение).
2. Серной кислотой промывают для превращения дициклогек-
силсульфата в кислый циклогексилсульфат, который в дальней-
шем может быть отмыт.
3. Для того, чтобы по возможности избежать образования эмуль-
сии, целесообразнее пользоваться теплой водой, а не холодной и
разбавленной щелочью и не концентрированной. Несмотря на то,
что промывные воды имеют молочный вид, связанные с промыва-
нием водою потери весьма незначительны.
4. Полезно дать отстояться взвешенной воде, для чего продукт
оставляют на ночь, прежде чем добавлять осушитель.
5. В типичном опыте при второй перегонке были собраны сле-
дующие фракции: 78—85°, 296 г; 85—235°, 2 г; 235—238°, 2 г;
238—243°, 215 г; 243—265°, 2 г; остаток выше 265°, 46 г.
6. Остаток от перегонки по охлаждении становится полутвер-
дым вследствие выделения 1,4-дициклогексилбензола, который
можно выделить, Для этого твердое вещество отфильтровывают
с отсасыванием, промывают метиловым спиртом и перекристалли-
зовывают из ацетона D мл ацетона на 1 г неочищенного вещества).
Выход очищенного дициклогексилбензола с т. пл. 100—ЮГ состав-
ляет 15—24 г.
3. Другие методы получения
Циклогексилбензол был получен гидрогенизацией бифенила 1
и циклогексинилбензола 2, реакцией между хлористым 3 или бро-
мистым * циклогексилом и бензолом в присутствии хлористого
алюминия и из бензола и циклогексена или циклогексанола в при-
сутствии хлористого алюминия 5, серной кислоты 6, или галоидных
соединений бора 7.
1 Е i j k m a n, Chem. Weekblad 1, 7 A903).
2Sabatier, Murat, Compt. rend. 154, 1390 A912); Alder, R i-
cker t, Ber. 71, 379 A938).
3 Курсаиов, ЖРХО 33, 685 A901); Ann. 318, 309 A901).
4 Br au n, Ber. 60, 1180 A927).
6 В о d г о u x, Ann. chitn. A0) II, 511 A929); Berry, R e i d, J. Am.
Chem. Soc. 49, 3142 A927); Corson, там же 59, 645 A937); Наметкин к
570
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
Покровская, ЖОХ 7, 962 A937); Цукерваичк и Сидорова,
ЖОХ, 7, 641 A937).
» Deschauer, герм. пат. 515 177 (Chem. Zentr. 1931, I, 1829); Truf-
f au It, Compt. rend. 202, 1286 A936); С о r s о n, J. Am. Chem. Soc. 59,
645 П937).
7 H о f m a n n, Wulff, англ. пат. 307 802 (Chem. Zentr. 1929, П,
2101); McKenna, Sow a, J. Am. Chem. Soc. 59, 470 A937).
ЦИТРАКОНОВЫЙ АНГИДРИД И ЦИТРАКОНОВАЯ КИСЛОТА
сн., = с — с = о сн3с — с = о
¦ | >о — | >о
СН, — С = О НС С = О
СН3С
I
НС
с = о
^> о
сн,с
н2о
со2н
НС
Предложили: Р. Шрайнер, С. Форд и Л. Роля.
Проверил: К- Номер.
1. Получение
А. Цитраконовый ангидрид. 250 г итаконового ангидрида (при-
мечание 1) быстро перегоняют при атмосферном давлении из спе-
циальной колбы Клайзена емкостью 500 мл с 15-сантиметровым
дефлегматором (примечание 2). Приемники для дестиллата следует
менять, не прерывая перегонки. Дестиллат, переходящий при тем-
пературе ниже 200°, состоит из воды и других продуктов разложе-
ния. Фракция, кипящая при 200—215°, состоит из цитраконового
ангидрида, и ее собирают отдельно. Продукта с т. пл. 5,5—6°
получается 170—180 г F8—72% теоретич.) продукта. Повторная
перегонка в вакууме дает 155—165 г цитраконового ангидрида, кипя-
щего при 105—110°/22 мм и плавящегося при 7—8° (примечание 3),
т. е. 62—66% теоретич.
Б. Цитраконовая кислота. К 22,4 г @,2 мол.) чистого цитрако-
нового ангидрида, находящегося в стакане емкостью 100 мл, при-
бавляют из пипетки ровно 4 мл @,22 мол.) дестиллированной воды.
Смесь перемешивают при слабом нагревании на электрической
плитке, пока не образуется гомогенный раствор, затем накрывают
стакан часовым стеклом и оставляют стоять иа 48 час. К концу
указанного времени масса полностью затвердевает. Получается
26 г цитраконовой кислоты с т. пл. 87—89°. Для дальнейшей очистки
ее тщательно растирают в ступке, промывают 50 мл холодного
бензола, отсасывают, сушат на воздухе и затем в течение 24 час.
сушат в вакууме-эксикаторе над фосфорным ангидридом. Полу-
чается 24,4 г (94% теоретич.) продукта, плавящегося при 92—93°.
ЭНАНТОВАЯ КИСЛОТА
571
2. Примечания
1. Пригоден сырой итаконовый ангидрид (стр. 290), вместо кото-
рого можно взять также итаконовую кислоту.
2. Успешность работы зависит от быстроты перегонки. Не сле-
дует прекращать нагревание при смене приемников. Наилучшие
выходы получаются в том случае, если период нагревания длится
недолго.
3. Сырой цитраконовый ангидрид содержит небольшое коли-
чество воды, ацетона и цитраконовой кислоты, которые отделяются
при перегонке в вакууме без сколько-нибудь значительного по-
нижения выхода.
3. Другие методы получения
Цитраконовый ангидрид был получен перегонкой цитраконовой
и лимонной кислот 1.
Цитраконовая кислота была получена перегонкой лимонной
кислоты 2, молочной кислоты 3 и оксипировинной кислоты 4, а также
действием иодистоводородной кислоты на лимонную кислоту5.
Смесь цитраконовой и итаконовой кислот была получена при при-
ливании концентрированного водного раствора лимонной кислоты
в нагретый эвакуированный сосуд, после чего смесь образовавшихся
ангидридов перегонялась в вакууме и обрабатывалась водой6.
1 Anschutz, Ber. 14, 2788 A881).
2 С г a s s о, Ann. 34, 68A840); К е k u 1 ё, Lehrbuch der organischen
Chemie, 2, 317A866); Wilm, Ann. 141, 28 A867).
3 E n g e 1 h a r d t, там же 70, 243, 246 A849).
1 D e m a r 9 a y, Ber. 9, 963 A876).
'Kammerer, Ann. 139,269A866).
6 Boehringer Sohn A.-G.; англ. пат. 452 460 [С. А, 31, 1045A937).
ЭНАНТОВАЯ КИСЛОТА
(н.-Гептановая кислота)
3 СвН13СНО
вН13
3 С6Н13СО2Н
2 KMnO4
K2SO4
H2SO4
2MnOa
Предложил: Дж. Рухоф.
Проверили: К- Ноллер и
Н,0
М. Патт.
1. Получение
В 5-литровую колбу, снабженную механической мешалкой и
охлаждаемую в бане со льдом, помещают 2,7 л воды и 350 мл F44 г)
концентрированной серной кислоты (уд. в. 1,84). Когда темпера-
тура упадет до 15°, добавляют 342 г D03 мл, 3 мол.) энантола (н.-гепт-
альдегида; примечание 1) и затем 340 г B,15 мол.) перманганата
572
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
калия порциями nol 5 г. Перманганат прибавляют с такой скоростью,
чтобы температура не поднималась выше 20° (примечание 2). После
того как все количество перманганата прибавлено, через раствор
пропускают сернистый газ до тех пор, пока раствор не станет про-
зрачным (примечание 3). Маслянистый слой отделяют, промывают
его один раз водой и подвергают перегонке из видоизмененной
колбы Клайзена с дефлегматором высотою 30 см. Головные фракции
отделяют от воды и вновь перегоняют; высококииящие фрак-
ции подвергают повторной перегонке. Выход продукта, кипящего
при 159—16Г/Ш0 мм, составляет 296—305 г G6—78% теоретич.;
примечание 4). Продукт этот является достаточно чистым для боль-
шинства целей; титрование показывает, что он содержит 95—97%.
энантовой кислоты.
Для дальнейшей очистки продукт вносят в раствор 140 г C,5
мол.) едкого натра в 700 мл воды и подвергают его перегонке с водя-
ным паром из 2-литровой колбы до тех пор, пока в дестиллат не
перестанет переходить маслянистый продукт. Оставшийся в колбе
раствор охлаждают до комнатной температуры и подкисляют
375 мл D,5 мол.) концентрированной соляной кислоты. Энантовую
кислоту отделяют и перегоняют из видоизмененной колбы Клай-
зена с дефлегматором. Выход кислоты с т. кип. 155—157°/80 мм
составляет 85—90% от веса взятого нечистого продукта. Титрова-
ние показывает, что это продукт 100%-ной чистоты.
2. Примечания
1. Был взят свежеперегнанный энантол с т. кип. 85,5—87,5°/
90 мм.
2. Перемешивание должно быть весьма энергичным. Время,
необходимое для прибавления перманганата, равняется примерно
2 час.
3. Сернистый газ в присутствии серной кислоты восстанавли-
вает выпавшую в осадок двуокись марганца в растворимый сульфат;
удаление большого количества этого хлопьевидного продукта зна-
чительно облегчает выделение энантовой кислоты. Пропускание
сернистого газа продолжается около 2 час, необходимо избегать
избытка его. В случае отсутствия сернистого газа можно воспользо-
ваться бисульфитом натрия.
4. Иногда энантовая кислота получается окрашенной в желтый
цвет. При перегонке эта окраска не исчезает.
3. Другие методы получения
Энантовая кислота была получена окислением энантола азот-
ной кислотой !, перманганатом калия в щелочном водном растворе 2
или в растворе ацетона3 или бихроматом калия с серной кислотой4,
ЭПИХЛОРГИДРИН И ЭПИБРОМГИДРИН
573
а также при действии углекислоты на продукт реакции между
натрием и 1-хлоргексаном.
iTillev Ann 67, 107 A848); М е h 1 i s, там же 185, 360 A877);
Krafft, Ber. 15, 1717A882)
a
Г58, 754A936).
ЭПИХЛОРГИДРИН И ЭПИБРОМГИДРИН
2ХСН2СНОНСН2Х + Са(ОН)а ¦
— 2 СН, — СН — СН2Х + СаХ2 + 2 Н2О
О
Предложил: Г. Браун.
Проверили: В. Хартман и Дж. Бумер.
1. Получение
А. Эпихлоргидрин. В 5-литровую круглодонную колбу поме-
щают 1350 г (988 мл, 10,5 мол.) a.Y-дихлоргидрина глицерина
(«Синт. орг. преп.», сб. 1, стр. 213), 840 г A0 мол.) хорошо измель-
ченной технической гашеной извести (88%-ной) и 840 мл воды
B0°) и все это энергично взбалтывают в течение 15 мин. (приме-
чание 1). При этом вначале образуется густая паста, но вскоре
затем эпихлоргидрин отделяется от солей кальция в виде подвиж-
ной жидкости. Колбу закрывают резиновой пробкой с широкой
отводной трубкой и смесь перегоняют с водяной бани вначале при
давлении 40—50 мм. Затем давление понижают до 10 мм и темпе-
ратуру постепенно повышают до 95—100° (примечание 2). Для
того, чтобы обеспечить максимальный выход, необходимо хорошо
охлаждать приемник смесью льда и соли до —5° или даже ниже.
Дестиллат переносят в делительную воронку, отделяют водный
(верхний) слой, сливают его обратно в колбу и повторяют пере-
гонку. Третья перегонка, проведенная аналогичным образом, дает
еще небольшое количество эпихлоргидрина (примечание 3). Ниж-
ние слои, получаемые при всех этих перегонках, соединяют вместе
и подвергают перегонке с дефлегматором при пониженном давлении.
Фракцию эпихлоргидрина собирают до 75°/5О мм, а остаток (около
160—180 мл), содержащий большое количество дихлоргидрина,
сливают оОратно в реакционную колбу, куда прибавляют еще
150 мл воды. После новой перегонки в вакууме, как это описано
574
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
ЭРУКОВАЯ КИСЛОТА
575
выше, нижний слой дестиллата присоединяют к основному коли-
честву эпихлоргидрина. Сырой продукт подвергают перегонке при
атмосферном давлении до тех пор, пока температура паров не
поднимется до 115°; тогда перегонку прерывают и отделяют
в дестиллате водный слой. Нижний слой дестиллата сушат
безводным сернокислым натрием и выливают обратно в перегон-
ную колбу. После небольшого головного погона эпихлоргид-
рин перегоняется при 115—117°. Выход 650—700 г F7—72%
теоретич.).
Б. Эпибромгидрин. В 5-литровой круглодонной колбе суспен-
дируют 2140 г A л, 9,8 мол.) а,у-дибромгидрина глицерина (стр. 183)
в 1,5 л воды, после чего в течение 15 мин. при взбалтывании посте-
пенно прибавляют 400 г измельченной технической гашеной извести
(88%-ной). Затем сразу добавляют еще 400 г гашеной извести
(всего 9,5 мол.) и эпибромгидрин отгоняют в вакууме так, как это
описано для эпихлоргидрина (примечание 2). Нижние слои от двух
таких перегонок (около 750 мл) соединяют вместе, сушат безвод-
ным сернокислым натрием и подвергают дробной перегонке при
атмосферном или пониженном давлении. Выход эпибромгидрина
с т. кип. 134—136° или 61—62°/50 мм составляет 1130—1200 г
(84—89% теоретич.).
2. Примечания
1. Необходимо строго придерживаться указанного количества
воды; большее количество воды вызывает вспенивание. Реакция
идет без выделения тепла.
2. Эпихлоргидрин кипит при 30—32°/10 мм, эпибромгидрин —
при 61—62°/50 мм. Эти обе жидкости летучи с водяным паром при
пониженном давлении.
3. Объем слоя эпихлоргидрина, получаемого при последователь-
ных перегонках, приблизительно равняется: 500 мл, 200 мл
20 мл.
3. Другие методы получения
Эпихлоргидрин т и эпибромгидрин 2 были получены действием
на дихлор- и дибромгидрины глицерина щелочей в различных
условиях. Описанная здесь методика представляет собой лабора-
торное применение метода Грисгейма 3.
1 «Сннт. орг. преп.», сб. 1, стр. 527.
2 В е г t h e 1 о t, Luc a, Ann. chitn. C) 48, 306, 311 A856); R e b о u 1,
там >кеC) 60, 32A860).
3 Chemische Fabrik Oriesheim-Elektron; герм. пат. 246 242 [Frdl. 10, 22
A910—12)]; R r a u n, J. Am. Chem. Soc. 54, 1248A932).
ЭРУКОВАЯ КИСЛОТА
CH3 (CH2O СН = СН (СНа)и СО.,Н
(Гидролиз сурепного масла)
Предложили: К- Ноллер и Р. Талбот.
Проверили: X. Кларк и Е. Тэйлор.
1. Получение
В соединенную с обратным холодильником 5-литровую колбу
помещают 2,5 л 95%-ного этилового спирта и 340 г D,5 мол!)
технического G3—75%-ного) едкого кали. Смесь осторожно
взбалтывают, пока не растворится едкое кали, а затем при
взбалтывании прибавляют 1 330 г A,5 л, примерно 4 эквива-
лента) сурепного масла и смесь кипятят е обратным холо-
дильником на водяной бане в течение 25—30 час. (примеча-
ние 1).
Горячую смесь выливают при перемешивании в 15 л теплой
воды E0—60°), прибавляют к полученному раствору 700 мл
(8,2 мол.) концентрированной 36%-ной соляной кислоты (при-
мечание 2) и оставляют стоять до расслаивания (от 10 до
15 мин.). Затем сифонируют как можно тщательнее нижний
слон и промывают масло двумя порциями теплой воды па 1 л.
Полученную таким образом смесь кислот в количестве 1460—
1600 мл растворяют в тройном объеме 95%-ного этилового спирта
и раствор охлаждают до температуры ¦— 10°—0°, причем кристал-
лизуется эруковая кислота (примечание 3). После 6—8-часового
охлаждения сырую эруковую кислоту отделяют от маточного
раствора на центрифуге (примечание 4). Из фильтрата по глубоком
охлаждении выделяется еще некоторое количество эруковой кис-
лоты. Всего получается 800—1100 г сырого продукта, который
вследствие присутствия олеиновой кислоты плавится частично
или весь при комнатной температуре. С целью очистки его раство-
ряют в равном объеме спирта, раствор охлаждают в течение 6 час.
до 0°, вновь центрифугируют и получают вещество в виде хорошо
образованных кристаллов. Вторая порция, получающаяся по упа-
ривании маточного раствора, по чистоте соответствует сырой (один
раз кристаллизованной) эруковой кислоте и поэтому должна быть
перекристаллизована вторично. Отцентрифугированнын продукт
кристаллизуют еще раз из равного объема 95%-ного этилового
спирта. Перекристаллизованная кислота содержит спирт, который
удаляют нагреванием вещества до постоянного веса на водяной бане
в вакууме. Всего получается 260—360 г кислоты (примечание 5)
с т. пл. 31—32° (примечание 6).
.376
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
2. Примечания
1. Если смешивать раствор по указанному способу, то сурепное
масло при вливании в щелочь образует эмульсию и кипячение
с обратным холодильником начинается более равномерно.
2. Если прибавить кислоту раньше воды, то образуется много
эфиров, вследствие чего выход сильно понижается (менее 200 г).
3. Лучше всего производить кристаллизацию охлаждением
до 0° в течение нескольких часов. При охлаждении смесью соли
и льда кристаллизация идет значительно быстрее, но получаю-
щаяся при этом эруковая кислота содержит больше олеиновой
кислоты.
4. Эруковую кислоту легко отфильтровать, пользуясь центри-
фугой. Если ее отделение от маточного раствора происходит не-
достаточно быстро, она плавится и выделить ее становится невоз-
можным. Если центрифугу применить нельзя, кислоту следует
отфильтровать при 0°, что весьма неудобно во все времена года,
кроме зимы.
5. Маточные растворы от перекристаллизации можно соединить
вместе, отогнать из них спирт и остаток перегнать в вакууме,
собирая при этом две равные фракции. Первая фракция B00—220°/
5 мм) состоит преимущественно из олеиновой кислоты; высшая
B20—230°/ 5 мм) затвердевает при 20° и после перекристаллизации
дает добавочное количество эруковой кислоты.
6. Полученная кислота содержит небольшую примесь арахино-
вой кислоты и других насыщенных жирных кислот и имеет йодное
число 66,9 (вместо 75). Если продукт не вполне бесцветен, то его
можно перегнать в вакууме (т. кип. 241— 243°/5 мм; 252—254°/12 мм).
При перегонке практически не происходит потерь; только ничтож-
ное количество высококипящих примесей остается в перегонной
колбе.
3. Другие методы получения
Эруковая кислота входит в состав различных природных масел,
однако всего удобнее получать ее из сурепного масла. Описанный
здесь процесс в главных чертах разработали Реймер и Билль х.
Были предложены методы получения чистой эруковой кислоты из
насыщенных кислот2, но они очень сложны, состоят из длительных
процессов дробного осаждения и перекристаллизации и неизбежно
дают небольшие выходы. Продукт, получаемый по описанному
выше способу, пригоден для большинства целей.
1 R е i m е г, Will, Вег. 19, 3320A886); М и Мег, R 6 1 ъ, Wiener,
там же 67, 296A934).
2 Н о I d e, W i I k e, Z. angew. Chetn. 35, 105, 186, 289 A922); Т а и f e I,
Bauschinger, там же 41, 157 A928).
ЭТИЛОВОГО ЭФИРА ГЛИЦИНА ХЛОРИСТОВОДОРОДНАЯ СОЛЬ
577
ЭТИЛОВОГО ЭФИРА ГЛИЦИНА ХЛОРИСТОВОДОРОДНАЯ СОЛЬ
CH2 = NCH,CN + С2Н5ОН + 2НС1 + 2Н2О
- НС1 ¦ NH2CH2CO2C2H6 + СН2О + NH4C1
Предложил: К- Марвел.
Проверили: Л. Физер и С. Джудкинс.
1. Получение
В 3-литровую круглодонную колбу помещают 500 мл D00 г,
8,7 мол.) абсолютного спирта, насыщенного на холоду хлористым
водородом (примечание 1), 870 мл F80 г, 14,8 мол.) 96%-ного спирта
(примечание 2) и 70 г A,03 мол.) метиленаминоацетонитрила (при-
мечание 3). Смесь нагревают с обратным холодильником на кипя-
щей водяной бане в течение 3 час. (примечание 4), причем выде-
ляется хлористый аммоний. После того как реакция закончена,
горячий спиртовой раствор фильтруют с отсасыванием и фильтрат
охлаждают, причем хлористоводородная соль этилового эфира
глицина выпадает в виде мелких белых игл. Продукт отсасывают,
отжимают возможно тщательнее на фильтре и сушат на воздухе.
Выход около 110 г. Спирт от фильтрата отгоняют (примечание 5)
до тех пор, пока в колбе не останется одна третья часть по объему.
Остаток опять охлаждают и выделяют вторую порцию кристаллов.
Общий выход продукта с т. пл. 142—143° составляет 125—129 г
(87—90% теоретич.). Если требуется особенно чистый препарат,
сырой продукт можно перекристаллизовать из абсолютного спирта.
2. Примечания
1. 500 мл абсолютного спирта охлаждают льдом и пропускают
через него ток сухого хлористого водорода, пока увеличение веса
не достигнет 163 г. Это количество достаточно для получения насы-
щенного раствора. Раствор должен быть защищен от влаги воздуха
хлоркальциевой трубкой.
2. Если желательно добиться лучших выходов, следует пользо-
ваться спиртом точно указанной крепости. Рекомендуется перед
работой проверить концентрацию спирта спиртометром. В 870 мл
96%-ного спирта содержится как раз достаточное количество воды
для гидролиза. Поэтому, если взять спирт меньшей концентрации,
хлористоводородная соль эфира глицина не образуется с такой
легкостью и не отделяется так легко от раствора. Опыты с приме-
нением 96%-ного спирта, насыщенного хлористым водородом и
870 мл 96%-ного спирта, дали выход на 8—10 г меньше указанного
выше. Применение спирта, концентрация которого меньше 96%,
дает продукт худшего качества и еще меньший выход.
3. Можно пользоваться неочищенным продуктом, синтез кото-
рого см. «Синт. орг. преп.», сб. 1, стр. 256.
37 Сборник № 2
578
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
ЭТИЛОВЫЙ ЭФИР АДИПИНОВОЙ КИСЛОТЫ
579
4. При кипячении с обратным холодильником надо пользо-
ваться корковой, а не резиновой пробкой, так как последняя ведет
к окрашиванию получаемого продукта.
5. Следует принять меры предосторожности против того, чтобы
в спирт не попала вода, так как хлористоводородная соль эфира
глицина довольно хорошо растворима в воде. Концентрирование
фильтрата на водяной бане не следует вести в открытом сосуде,
так как в этом случае спирт будет поглощать влагу и продукт
не будет кристаллизоваться.
3. Другие методы получения
Хлористоводородная соль эфира глицина была получена дей-
ствием абсолютного спирта и хлористого водорода на глицин *; из
хлорангидрида глицина и спирта 2; действием аммиака 3 или гекса-
метилентетрамина4 на монохлороуксусную кислоту с последующим
омылением спиртовым раствором соляной кислоты; действием
хлористого водорода на метиленаминоацетонитрил 6 и восстановле-
нием этилового эфира цианмуравьиной кислоты 6 или хлористо-
водородной соли соответственных имидоэфиров 7.
1 С u r t i u s, О о e b e 1, J. prakt. Chem. B) 37, 159 A888); Harries,
Weiss, Ann. 327, 365 A903).
2 Fischer, Ber. 38, 2916 A905).
3Hantzsch, Silberrad, там же 33, 70 A900); Hantzsch,
Metcalf, там же 29, 1681 A896).
4 A u g e r, Bull. soc. chim. C) 21, 6 A899); L о с q u i n, там же 23, 662
A900).
5 Jay, Curtius, Ber 27, 60 A894); К 1 a g e s, там же 36, 1508 A903).
6 Oes. fur Kohlentechnik m. b. H., герм. пат. 594 219 [С. А. 28, 3417
A934)]; 638 577 [С. А. 31,3066 A937)]; E. Merck, герм. пат. 597 305 [С. А.
28, 5078 A934)].
7 Oes. fur Kohlentechnik in. b. H., герм. пат. 604 277 [С. А. 29, 812A935)].
ЭТИЛОВЫЙ ЭФИР АДИПИНОВОЙ КИСЛОТЫ
НО., С (СН,L СОЛ
2 Н2О
(толуол)
С2Н5О,С (СН.L СО2С2Н3
Предложил: В. Мшович.
Проверили: Р. Фьюзон и Е. Кливленд.
1. Получение
В 3-литровую колбу Вюрца помещают 438 г (Змол.) адипиновой
кислоты, 1080 мл (9 мол.) абсолютного этилового спирта, 540 мл
толуола и 2,5 мл концентрированной серной кислоты (примеча-
ние 1). Колбу присоединяют к обращенному вниз холодильнику
и нагревают ее на масляной бане (примечание 2). При 75° начинает
отгоняться азеотропная смесь спирта, толуола и воды. Перегонку
продолжают до тех пор, пока термометр в горле колбы не покажет
78°, после чего нагревание прерывают.
Дестиллат собирают в 2-литровую колбу, в которую предвари-
тельно помещают 450 г безводного поташа (примечание 3). Колбу
хорошо взбалтывают, содержимое ее фильтруют через бюхнеров-
скую воронку и дестиллат возвращают в колбу Вюрца (примеча-
ние 4). Колбу вновь нагревают, пока температура в парах не достиг-
нет 78—80°, после чего перегонку прекращают (примечание 5).
Оставшуюся жидкость (примечание 6) выливают в 1-литровую колбу,
причем большую колбу ополаскивают небольшим количеством
спирта, и смесь подвергают перегонке в вакууме. Сперва переходят
спирт и толуол; затем температура повышается и при 138°/20 мм
отгоняется этиловый эфир адипиновой кислоты (примечание 7).
Выход 580—588 г (95—97% теоретич.; примечания 8 и 9).
2. Примечания
1. При этерификации по этому методу берется тройное коли-
чество абсолютного этилового спирта по сравнению с теоретическим.
Если взять только двойное по сравнению с теоретическим коли-
чество, то этерификация будет не полной. Необходимое количество
серной кислоты составляет 1% от веса взятой адипиновой кислоты.
При меньших количествах органической кислоты достаточно
нескольких капель серной кислоты.
2. Следует поддерживать температуру бани около 115° до
растворения смеси и до начала отгонки. Позднее температуру
можно несколько снизить и температура 100—110° является вполне
достаточной.
3. На 1 мол. адипиновой кислоты, подлежащей этерификации,
требуется 150 г безводного поташа.
4. Если работать с малыми количествами, то можно фильтро-
вать через складчатый фильтр непосредственно в колбу Вюрца.
5. Дестиллат, содержащий спирт, толуол и воду, можно высу-
шить, перегнать и вновь использовать для этерификации, добавив
необходимое количество абсолютного спирта. Можно также доба-
вить воды и выделить один только толуол, высушить его над хло-
ристым кальцием и перегнать.
6. Если раствору дать охладиться, выпадают мелкие кристаллы
свободной кислоты. Количество ее ничтожно.
7. К концу перегонки температура поднимается на несколько
градусов, однако перегонку следует продолжить, так как при
повторной перегонке эфир перегоняется без остатка.
8. Автор метода указывает, что с помощью той же методики
с выходом 94—98% могут быть получены и другие этиловые эфиры
двухосновных органических кислот от щавелевой до себациновои.
560
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
9. Этиловый эфир адипиновой кислоты можно также получить1,
если кипятить с обратным холодильником 175 г A,2 мол.) адипи-
новой кислоты, 175 г B22 мл) этилового спирта, 450 мл бензола и
80 г D3,5 мл) концентрированной серной кислоты в течение 5 час.
на водяной бане. Выход сложного эфира с т. кип. 136—137719 мм
составляет 218 г (90% теоретич.; частное сообщение П. С. Пинкни.
Проверили Л. Ф. Физер и Т. Л. Джекобе).
3. Другие методы получения
Этиловый зфир адипиновой кислоты получается при кипячении
адипиновой кислоты, этилового спирта, бензола и серной кислоты 1;
из адипиновой кислоты, спирта и хлористого водорода 2; из адипи-
новой кислоты, абсолютного спирта и серной кислоты 3; при пере-
гонке смеси этилового спирта, толуола и адипиновой кислоты
с добавлением небольшого количества хлористоводородной кислоты,
действующей в качестве катализатора 4 и по методике, описанной
выше 5.
!Van Rysselberge, Bull. soc. chim. Belg. 35, 312 A926).
2 А г p p e, J. prakt. Chem. A) 95, 208 A865).
3 С u г t i u s, там же B) 91, 4 A915); Bouveault, Locquin, Bull,
soc. chim. DK, 439A908).
4 L о с q u i n, E 1 g h о z у, там же D) 41, 445 A927).
5 M i i о v i с, там же E) 4, 1661 A937).
ЭТИЛОВЫЙ ЭФИР АЦЕТОЯНТАРНОЙ КИСЛОТЫ
СН3СОСНХОХ2Н5 + NaOC2H6
СН3С (ONa) = СНСОХ2Н3 + С2Н5ОН
СН3С (ONa) = СНСОХ2Н5 + С1СНХОХ2Н5
СНХОСН (СО.Х2Н5) СНХОХ2Н5 + NaCl
Предложили: Г. Адкинс, Н. Исбелл и Б. Войцик.
Проверили: Дж. Джонсон и X. Снайдер.
1. Получение
В 3-литровую круглодонную колбу, снабженную механической
мешалкой, обратным холодильником и капельной воронкой, поме-
щают 400 мл абсолютного спирта (примечание 1), после чего через
форштосс холодильника медленно прибавляют 23 г A rp.-ат.) чистого
металлического натрия, нарезанного тонкими ломтиками. Чтобы
ускорить завершение реакции, колбу нагревают на водяной бане.
После того как натрий полностью растворится, медленно добавляют
143 г A,1 мол.) ацетоуксусного эфира («Синт. орг. преп.», сб. 1,
стр. 73). Затем пускают в ход мешалку и в течение часа медленно
добавляют 123 г A мол.) этилового эфира монохлоруксуснои кис-
лоты (примечание 2), после чего смесь кипятят в течение 5—6 час.
ЭТИЛОВЫЙ ЭФИР АЦЕТОЯНТАРНОЙ КИСЛОТЫ
581
По истечении этого времени реакционная смесь не должна показы-
вать щелочной реакции на влажную лакмусовую бумажку.
По охлаждении отсасывают выпавший хлористый натрий и затем
промывают его два раза абсолютным спиртом порциями по 50 мл.
Спирт отгоняют с небольшим дефлегматором на водяной бане.
Остаток фильтруют, переносят в круглодонную колбу и подвер-
гают фракционированной перегонке в вакууме, пользуясь колон-
кой Видмера со спиралью длиной 8 см (примечание 3). Собирают
фракцию 121—12475 мм. Выход 121—134 г E6—62% теоретич.;
примечание 4).
2. Примечания
1. Требуется хорошо абсолютированный спирт. Для его полу-
чения обычный абсолютный спирт сушат небольшим количеством
металлического натрия, добавляют несколько миллилитров этило-
вого эфира янтарной кислоты и перегоняют, собирая дестиллат
прямо в реакционную колбу (см. примечание 1, стр. 170 и «Синт.
орг. преп.», сб. 1, стр. 555).
2. Применялся этиловый эфир монохлоруксуснои кислоты
с т. кип. 142—145°. Этот эфир можно легко получить, если кипя-
тить в течение б час. с обратным холодильником смесь из 200 г
хлоруксусной кислоты, 120 г абсолютного спирта и 25 г концентри-
рованной серной кислотых. Полученный продукт очищают, как
обычно, причем выход равняется 185 г G0% теоретич.).
3. Для этой фракционированной перегонки выгодно пользо-
ваться колонкой с электрическим обогревом. Главным побочным
продуктом реакции является этиловый эфир Р-ацетотрикарбалли-
ловой кислоты2 (т. кип. 190°/16 мм), образующийся при дальней-
шем действии этилового эфира монохлоруксуснои кислоты на этило-
вый эфир ацетоянтарной- кислоты.
4- Аналогичным образом может быть получен этиловый эфир
а-ацетоглутаровой кислоты, для чего вместо этилового эфира
монохлоруксуснои кислоты следует взять 181 г A мол.) этилового
эфира р-бромпропионовой кислоты («Синт. орг. преп.я, сб. 1,
стр. 543). В этом случае продукт собирают при 132—134°/4 мм
и выход составляет 120 г E2% теоретич.).
3. Другие методы получения
Этиловый эфир ацетоянтарной кислоты был получен при взаимо-
действии натрийацетоуксусного эфира с этиловым эфиром моно-
хлоруксуснои 1 или монобромуксусной кислоты 2. Описанный выше
метод является видоизменением 3 метода, описанного Конрадом г.
1 Con г ad, Ann. 188,218A877).
'Emery, Ber. 23, 3755 A890); Fichter, Pfister, там же 37, 1997
A904).
Ms be 11, Wojcik, Adkins, J. Am. Chem. Soc. 54, 3685 A932).
582
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
ЭТИЛОВЫЙ ЭФИР БЕНЗОИЛДИМЕТИЛУКСУСНОЙ КИСЛОТЫ
(Этиловый эфир я-бензоилизомасляной кислоты)
(СН3J CHCOSC,H5 + NaC (C6H5K
. Na [(СН8JССОаС2Н5] + НС (С6Н5K
С6Н5 СОС1 + Na [ (СН3J ССО,С2Н5]
С6Н3 СОС (СН3J COsCaH5 + NaCl
Предложили: Ч. Хаузер и В. Ренфрю мл.
Проверили: Л. Смит и Е. Баллард.
1. Получение
К свежеприготовленному раствору 0,187 мол. трифенилметил-
натрия (стр. 481), находящемуся в колбе, в которую он был перене-
сен после получения, добавляют 21,7 г B5 мл, 0,187 мол.) чистого
этилового эфира изомасляной кислоты (примечание 1). Смесь
взбалтывают, и, после того как она 10 мин. постоит при комнатной
температуре, к ней прибавляют, продолжая взбалтывание, раствор
26 г B1,5 мл, 0,186 мол.) чистого хлористого бензоила в 50 мл абсо-
лютного эфира. Смесь разогревается, и немедленно выпадает белый
осадок хлористого натрия. После стояния при комнатной темпе-
ратуре в течение нескольких часов смесь нагревают на водяной
бане и эфир отгоняют, пока объем не уменьшится до 300—400 мл.
Приливают раствор 5 мл уксусной кислоты в 300 мл воды и смесь
взбалтывают в делительной воронке до тех пор, пока при стоянии
жидкость не будет расслаиваться на два гомогенных слоя. Водный
слой спускают и отбрасывают; эфирный слой взбалтывают с 10%-ным
раствором соды и сушат над хлористым кальцием или «драйе-
ритом» *. Раствор фильтруют и перегоняют на водяной бане до тех
пор, пока не отгонится почти весь эфир. Остаток охлаждают в холо-
дильном шкафу, выкристаллизовавшийся трифенилметан отфиль-
тровывают с отсасыванием и промывают несколькими небольшими
порциями абсолютного эфира. Фильтрат после отгонки эфира пере-
гоняют в вакууме и собирают все, что отгонится, до 180° при 15 мм.
Дестиллат вновь перегоняют и фракцию, отгоняющуюся до 160°
при 15 мм, подвергают еще раз перегонке. При этом получают
20,5—22,5 г E0—55% теоретич.) этилового эфира бензоилдиметил-
уксусной кислоты с т. кип. 146—148°/15 мм или 133—135°/9 мм
(примечания 2 и 3).
2. Примечания
1. Весьма важно, чтобы исходные реактивы были чистыми.
Продажный этиловый эфир изомасляной кислоты можно вполне
* безводным хлорнокислым магнием. — Прим. ред.
ЭТИЛОВЫЙ ЭФИР БЕНЗОИЛДИМЕТИЛУКСУСНОЙ КИСЛОТЫ
583
удовлетворительно очистить, если его промыть 10%-ным раствором
соды, высушить в течение нескольких дней над драйеритом и под-
вергнуть дробной перегонке с хорошим дефлегматором. Для настоя-
щего синтеза следует взять фракцию, кипящую в пределах 1°
A10—111°). Хлористый бензоил очищается перегонкой в вакууме,
причем собирается фракция, кипящая в пределах менее 2°.
2. Если применять более концентрированный раствор трифе-
нилметилнатрия, полученный так, как это описано в примечании 3
на стр. 482, то можно получить ,в одном опыте около 120 г этило-
вого эфира бензоилдиметилуксусной кислоты.
Склянку с эфирным раствором 0,85 мол. трифенилметилнатрия
по/ружают на 2/3 ее высоты в баню со льдом. Когда склянка хорошо
охладится, пробку вынимают, и при этом у горла склянки держат
трубку, через которую пропускают быстрый ток сухого азота.
В склянку быстро вставляют каучуковую пробку с тремя отвер-
стиями, в которые вставлены механическая мешалка (с открытой
осью), капельная воронка и согнутая трубка, оканчивающаяся
в склянке на 1 см ниже горла склянки. Через эту трубку пропу-
скают медленный ток сухого азота; азот подают также в верхнюю
часть капельной воронки. Содержимое склянки энергично перемеши-
вают и быстро приливают 102 г @,875 мол.) этилового эфира изомасля-
ной кислоты. Через 5 мин. с помощью капельной воронки приливают
123 г A01 мл, 0,875 мол.) хлористого бензоила с такой скоростью,
чтобы смесь кипела не слишком бурно (прибавление продолжается
около 5 мин.). После того как все добавлено, перемешивание про-
должат еще 2—3 мин. Склянку вынимают из бани со льдом, закупо-
ривают ее, взбалтывают и оставляют стоять на 30 мин. Реакцион-
ную смесь переносят в делительную воронку емкостью 2,5 л и ртуть
сливают. Оставшуюся смесь экстрагируют 300 мл воды, причем,
если образуется густая масса, ее собирают отдельно. Эту массу
извлекают небольшим количеством эфира и эфирные вытяжки при-
бавляют к эфирному слою в делительной воронке. Эфирный раствор
промывают два раза 10%-ным раствором соды порциями по 100 мл,
для высушивания взбалтывают с 50 г безводного сернокислого
натрия или магния и оставляют стоять в закупоренной 2-литровой
конической колбе над 50 г «драйерита». Раствор фильтруют
в 2-литровую круглодоппую колбу и эфир отгоняют. Остаток
переносят в 1-литровую колбу Клайзена и перегоняют его в вакууме
на металлической или масляной бане4. При этом собирают все,
перегоняющееся до 175° при 15 мм. Дестиллат переносят в кругло-
донную колбу емкостью 200 мл и вновь перегоняют его с металли-
ческой бани, пользуясь дефлегматором Видмера высотой 15 см.
Выход этилового эфира бензоилдиметилуксусной кислоты с т. кип.
146—148715 мм составляет 115—125 г F1—67% теоретич., считая
на трифенилметилнатрий). (Ч. Хаузер и Б. Худзон мл., частное
сообщение.)
584
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
ЭТИЛОВЫЙ ЭФИР БЕНЗОИЛУКСУСНОЙ КИСЛОТЫ
585
3. Авторы, предложившие синтез, указывают, что аналогич-
ным образом можно получить этиловый эфир а, а, у, ^-тетраметил-
ацетоуксусной кислоты, если вместо хлористого бензоила взять
хлорангидрид изомасляной кислоты. Выход сложного эфира с т. кип.
94,5—95,5°/18 мм составляет около 55% теоретич. Ряд других
примеров ацилирования сложных эфиров по этому методу можно
найти в работе Худзона мл. и Хаузера *.
3. Другие методы получения
Этиловый эфир бензоилдиметилуксусной кислоты был получен
конденсацией этилового эфира изомасляной кислоты с этиловым
эфиром бензойной кислоты 2, фениловым эфиром бензойной кисло-
ты 3, бензойным ангидридом или хлористым бензоилом 3. Описан-
ная здесь методика является видоизменением способа Шейблера
и Штейна *.
1 Hud son, Jr., H a u s e r, J. Am. Chem. Soc. 63,3159A941).
2 Re nf row, Jr., Ha user, там же 60,464A938).
"Hudson, Jr., Dick, Hauser, там же 60,1961A938).
4Scheibler, Stein, J. prakt. Chem. B) 139, 111 A934).
ЭТИЛОВЫЙ ЭФИР БЕНЗОИЛУКСУСНОЙ КИСЛОТЫ
(АJСН3СОСНХО2С2Н5 + 2CeHsCOCl + 2Na
2CeHsCOCl
2СН3СОСНСОоС2Н3
2NaCl
СОС6Н5
(Б) CH3COCHCO2C2HS + НаО + NH3
сос6н3
CeH5COCHXOX2H5 + CHgCt
Предложили: Р. Шрайнер, А. Шмидт и Л. Ролл.
Проверили; Я. Ноллер и И. Бергштейнсон.
1. Получение
А. Получение этилового эфира бензоилацетоуксусной кислоты.
В 5-литровую трехгорлую колбу, снабженную механической мешал-
кой с жидкостным затвором и обратным холодильником, помещают
3,4 л сухого бензола (примечание 1), 195 г A,5 мол.) ацетоуксус-
ного эфира («Синт. орг. преп.», сб. 1, стр. 73) и 34,5 г A,5гр.-ат.)
чистого натрия. Смесь нагревают при перемешивании на паровой
бане и дают ей спокойно кипеть 24 часа. После того как суспен-
зия натрийацетоуксусного эфира несколько охладится, приливают
в течение 3 час. 263 г B18 мл, 1,9 мол.) хлористого бензоила. Смесь
перемешивают и кипятят еще 8 час, после чего ее охлаждают до
комнатной температуры и прибавляют 375 г колотого льда. После
тщательного взбалтывания отделяют бензольный слой, содержа-
щий этиловый эфир бензоилацетоуксусной кислоты, промывают
его 75 мл 5%-ного раствора бикарбоната натрия, сушат сернокис-
лым натрием и перегоняют (примечание 2). Остаток перегоняют
в вакууме из видоизмененной колбы Клайзена емкостью 500 мл
с дефлегматором высотой 50 см. После небольшого головного погона,
содержащего хлористый бензоил, собирают фракцию с т. кип.
142—14876 мм или при 177—181°/20 мм. Выход 223—263 г F3—
75% теоретич.).
Б. Омыление этилового эфира бензоилацетоуксусной кислоты.
В конической колбе емкостью 500 мл растворяют 32 г @,6 мол.)
хлористого аммония в 150 мл (8,3 мол.) воды и добавляют 10 мл
@,15 мол.) аммиака (уд. в. 0,9), Раствор нагревают до 42°, быстро
добавляют 58,5 г @,25 мол.) этилового эфира бензоилацетоуксусной
кислоты, температура которого 20°, и смесь взбалтывают (примеча-
ние 3). Колбу ставят в водяную баню, нагретую до 42° точно на
10 мин., после чего ее быстро охлаждают, погрузив в баню со льдом.
Раствор дважды извлекают эфиром порциями по 100 мл и эфирные
вытяжки сушат безводным сернокислым магнием. Эфир отгоняют
и остаток перегоняют в вакууме; выход этилового эфира бензоил-
уксусной кислоты с т. кип. 132—137°/4 мм или 165—169°/20 мм
составляет 37,0—37,5 г G7—78% теоретич.).
2. Примечания
1. Чтобы получить сухой бензол, его перегоняют и головную
фракцию отбрасывают.
2. Весьма важно, чтобы эти стадии при выделении продукта
проводились как можно быстрее.
3. Если работать с большими количествами, то выход бывает
меньше. Необходимо точно придерживаться условий реакции
в отношении времени, температуры и количеств реагентов. Синтез
должен проводиться без перерыва.
3. Другие методы получения
Этиловый эфир бензоилуксусной кислоты был получен конден-
сацией (в присутствии этилата натрия) уксусноэтилового эфира
с бензойноэтиловым эфиромг, ацетофенона с угольным эфиром2
и ацетофенона с щавелевым эфиром с последующим нагреванием 3;
действием концентрированной серной кислоты на этиловый эфир
фенилпропиоловой кислоты4 или на а-бромкоричную кислоту8,
взаимодействием диазоуксусного эфира с бензальдегидомв, кон-
денсацией бензола с моноэтиловым эфиром хлорангидрида мало-
новой кислоты в присутствии хлористого алюминия7, или
586
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
ЭТИЛОВЫЙ ЭФИР N-МЕТИЛКАРБАМИНОВОЙ КИСЛОТЫ
587
хлористого бензоила с продуктом реакции между магнием и этило-
вым эфиром хлоруксусной кислоты в эфирном растворе 8, действием
спирта на хлористоводородную соль бензоилацетиминоэтилового
эфира 9 и омылением этилового эфира бензоилацетоуксусной кис-
лоты 10.
1Claisen, Lowman, Ber. 20, 651 A887).
- С 1 a i s e n, там же 20, 655 A887).
3Wislicetius, там же 28, 811 A895).
4 В а у е г, там же 15, 2705 A882).
5MichaeI, Browne, там же 19, 1392 A886).
8 В и с h II с г, Curtius, там же 18, 2371 A885).
7 М а г g u е г у, Bull. soc. chim. C) 33, 549 A905).
«Meyer, Togel, Ann. 347, 76 A906).
9 H a 11 e r, Bull. soc. chim. B) 48, 23 A887).
10 С 1 a i s e n, Ann. 291,71A896); S h r i n e r, Schmidt, J. Am.
Chem. Soc. 51, 3636 A929).
ЭТИЛОВЫЙ ЭФИР «,?-ДИБРОМ-3-ФЕНИЛПРОПИОНОВОЙ КИСЛОТЫ
CfiH5CH=CHCO2C2H5 + Br2 C6H5CHBrCHBrCO3C.,H3
Предложили: Т. Эбботт и Д. Алыпхаузен.
Проверили: Г. Гильман и Дж. Райт.
1. Получение
В 1-литровую круглодонную колбу, закрытую пробкой с двумя
отверстиями, в которые вставлены капельная воронка и отводная
трубка, помещают 176,2 г A мол.) этилового эфира коричной кис-
лоты («Синт. орг. преп.», сб. 1, стр. 548), растворенных в 100 мл четы-
реххлористого углерода (примечание 1). Колбу ставят в лед и при
частом перемешивании приливают небольшими порциями 159,8 г
E1,2 мл, 1 мол.) брома (примечание 2). ,
Через час раствор выливают в большую плоскую чашку и дают
улетучиться избытку брома и четыреххлористому углероду (при-
мечание 3). Дибромэфир выделяется в виде больших кристаллов,
образующих на дне чашки плотные сростки (примечание 4). Осадок
разминают, раскладывают тонким слоем на большой воронке
Бюхнера, отсасывают до полного исчезновения следов брома и затем
сушат, отжимая между большими листами фильтровальной бумаги.
Выход сырого дибромэфира 280—285 г (83—85% теоретич.).
Т. пл. 65—71°.
Если требуется чистый продукт, его получают перекристалли-
зацией из петролейного эфира (с температурой кипения 70—90°).
Из 100 г сырого продукта получается 80—85 г этилового эфира
а,Э-дибром-3-фенилпропионовой кислоты с температурой плавле-
ния 74—75°.
2. Примечания
1. Четыреххлористый углерод берется вместо эфира потому,
что при употреблении последнего образуется соединение, вызы-
вающее слезотечение.
2. Присоединение брома в начале реакции происходит очень
быстро, под конец оно замедляется. Выделения бромистого водо-
рода не происходит. Присоединение протекает в течение 20—25 мин.
3. Этот процесс идет довольно медленно; его можно ускорить,
если над поверхностью чашки поместить опрокинутую большую
воронку, соединенную с насосом. В этом случае кристаллический
осадок выделяется приблизительно через 2 часа.,
4. Если реакцию вести тщательно, то маточного раствора прак-
тически не остается. Если же остается раствор, то при его испаре-
нии можно получить загрязненные кристаллы, которые перед
употреблением следует очистить перекристаллизацией.
3. Другие методы получения
Этиловый эфир а, !3-дибром-|3-фенилпропионовой кислоты полу-
чается присоединением брома к этиловому эфиру коричной кисло-
ты *.
»Anschiitz, Kinnicutt, Ber. 11, 1220 A878); А г о n s t e i n,
H-o lie ma n n, там же 22, 1181 A889); L e i g h t о n, Am. Chem. J. 20, 136
A898); Sudborough, Thompson, J. Chem. Soc. 83, 671 A903). .
ЭТИЛОВЫЙ ЭФИР N-МЕТИЛКАРБАМИНОВОЙ КИСЛОТЫ
(N-Метилу ретан)
С1СО2С2Н5 + CH3NH2 + NaOH > CH3NHCO2C2H5 + NaCl + H2O
Предложили: В. Хпртман и М. Брезен.
Проверили: Дж. Конант и К. Бзйли.
1. Получение
В 2-литровую круглодонную колбу, снабженную мешалкой и
охлаждаемую смесью льда с солью, помещают 300 мл эфира и 186 г
B мол.) 33%-ного водного раствора метиламина. К перемешивае-
мой смеси при т. 5° приливают 217 г B мол.) хлоругольного эфира
(примечание 1), причем температура не должна подниматься выше 5°.
После того, как прибавлена почти половина хлоругольного эфира,
начинают одновременно прибавлять холодный раствор 80 г B мол.)
чистого едкого натра в 120 мл воды с такой скоростью, чтобы при-
ливание обоих растворов закончилось одновременно. Необходимо,
чтобы во все время этой операции работала мешалка (примечание 2).
588
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
ЭТИЛОВЫЙ ЭФИР МЕТИЛМАЛОНОВОЙ КИСЛОТЫ
589
Через 15 мин. после окончания прибавления эфирный слой отде-
ляют, а водный раствор извлекают 100 мл эфира. Соединенные
эфирные растворы быстро высушивают встряхиванием с 8 г поташа,
которые прибавляют в две порции. Эфир отгоняют, а остаток пере-
гоняют в вакууме, причем собирают фракцию, кипящую при 55—
60°/12 мм. Выход бесцветного маслянистого продукта 182—185 г
(88—90% теоретич.).
2. Примечания
1. Был взят технический хлоругольный эфир.
2. Скорость прибавления зависит от того, насколько хорошо
охлаждается реакционная смесь. Если охлаждение вести извне
смесью льда и соли, то на прибавление эфира требуется около
5 час.
3. Другие методы получения
Этиловый эфир N-метилкарбаминовой кислоты был получен
при прибавлении водного раствора метиламина к хлоругольному
эфиру ! и из метилкарбамилхлорида и этилового спирта 2.
'Schreiner, J. prakt. Chem. B) 21, 124A880); Pechmann, Ber.
28, 855 A895); E i s t e r t, Angew. Chem. 54, 124 A941).
2 G a 11 e r m a n n, Ann. 244, 35 A888).
ЭТИЛОВЫЙ ЭФИР МЕТИЛМАЛОНОВОЙ КИСЛОТЫ
(Диэтиловый эфир метилмалоновой кислоты)
А. ИЗ ЭТОКСАЛИЛПРОПИОНОВОГО ЭФИРА
СН3СНСО2С2Н5
СОСО2С2Н5
(нагревание)
— СН3СН (СО2С2Н5J + СО
Предложили: Р. Цоке и С. Мак-Элыт.
Проверили: Р. Фьюзон и В. Росс.
1. Получение
В круглодонную колбу подходящей величины с обратным холо-
дильником и термометром, который подвешен в холодильнике и
погружен в жидкость, помещают 345 г A,7 мол.) этилового эфира
этоксалилпропионовой кислоты ст. кип. 114—1167Ю ш« (стр. 604)
и нагревают его до начала бурного выделения окиси углерода
A30—150°). Температуру жидкости постепенно повышают по мере
того, как замедляется выделение газа, и под конец жидкость кипятят
до тех пор; пока не прекратится выделение газа. Этиловый эфир
метилмалоновой кислоты перегоняют. Он кипит при 194—
19б°/745 мм, причем выход составляет 288 г (97% теоретич.).
Б. ИЗ МАЛОНОВОГО ЭФИРА
СН3Вг + Na [СН (СО2С2Н5J] > СН3СН (СО2С2Н5J
Предложил: Н. Вейнер.
NaBr
Проверили: Р. Фьюзон и Е. Кливленд.
1. Получение
В 2-литровую трехгорлую колбу, снабженную механической
мешалкой и обратным холодильником, снабженным хлоркальциевой
трубкой, помещают 1 л абсолютного этилового спирта (примеча-
ние 1) и добавляют 46 г B гр.-ат.) натрия, нарезанного небольшими
кусочками. Когда весь натрий перейдет в раствор, добавляют
320 г B мол.) малонового эфира (примечание 2), после чего через
перемешиваемый раствор пропускают с помощью стеклянной трубки
(диаметром не менее 4 мм), вставленной в третье горло колбы и
доходящей до дна колбы, газообразный бромистый метил в ко-
личестве 200 г B,1 мол.; примечание 3),
Пропускание бромистого метила занимает около 4 час. Реак-
ция протекает гладко; в результате выделения тепла температура
смеси иногда достигает точки кипения. Во время реакции выпадает
бромистый натрий. Когда добавление бромистого метила закончено,
раствор становится светлооранжевого цвета и имеет слабо щелоч-
ную реакцию. Его кипятят еще 30 мин., после чего нейтрализуют
ледяной уксусной кислотой и охлаждают. Бромистый натрий отса-
сывают и на воронке промывают небольшим количеством холодного
спирта. Большую часть спирта отгоняют при атмосферном дав-
лении (примечание 4). Бромистый натрий растворяют в 600—700 мл
воды, содержащей 10 мл концентрированной соляной кислоты.
Полученный раствор (примечание 5) добавляют к остатку от пере-
гонки и смесь хорошо взбалтывают. Водный слой отделяют от слоя
сложного эфира и дважды извлекают его эфиром. Сложный эфир
и эфирные вытяжки соединяют вместе и высушивают путем недол-
гого взбалтывания с хлористым кальцием, после чего раствор
немедленно фильтруют. Эфир отгоняют, а сложный эфир взбалтывают
ровно 1 мин. с холодным раствором 10 г едкого натра в 30 мл воды
(примечание 6). Щелочной слой отделяют, сложный эфир промывают
разбавленной кислотой и сушат, как и в первый раз, хлористым
кальцием. Его перегоняют в вакууме и собирают фракцию с т. кип.
96716 мм.Нрм этом почти не бывает ни головного погона, ни остатка.
Выход 275—290 г G9—83% теоретич.).
590
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
ЭТИЛОВЫЙ ЭФИР МЕТИЛМЛЛОНОВОЙ КИСЛОТЫ
591
2. Примечания
1. Большое значение имеет качество спирта, так как даже следы
воды заметно понижают выход. Обычный абсолютный спирт пере-
гоняют над 2—3% натрия и отбрасывают головную фракцию и
25% остатка.
2. Было найдено, что малоновый эфир даже лучшего качества
содержит достаточное количество примесей, которое ведет к умень-
шению выхода. Однако уже однократная перегонка в вакууме даже
технического малонового эфира дает продукт достаточно чистый
для настоящего синтеза.
3. Ампулы или баллончики с бромистым метилом охлаждают
в бане со льдом, вскрывают, с помощью обыкновенной резиновой
трубки присоединяют к трубке, через которую бромистый метил
подается в колбу, и после этого дают нагреться до комнатной тем-
пературы. Испарение жидкости поддерживает ее в достаточно
холодном состоянии, и газ поступает в раствор с нужной скоростью.
С целью осушки газ лучше всего пропускать через колонку с гра-
нулированным едким кали.
Если нет бромистого метила, то он может быть получен следую-
щим образом: 500 г 95%-ной серной кислоты медленно прибавляют
при взбалтывании к 400 г обычного метилового спирта, охлажден-
ного в бане со льдом. Затем в 1-литровой круглодонной колбе,
закрытой пробкой с двумя отверстиями, в которые вставлены
капельная воронка и отводная трубка, приготовляют суспензию из
половины ранее приготовленной смеси серной кислоты и метило-
вого спирта и 600 г бромистого натрия. Для того, чтобы началось
выделение бромистого метила, колбу нагревают на водяной бане
до 50°, после чего из воронки медленно приливают оставшуюся
смесь метилового спирта и серной кислоты по мере уменьшения
объема смеси в колбе. По мере того, как скорость выделения газа
падает, температуру постепенно повышают до тех пор, пока выде-
ление бромистого метила не прекратится и содержимое колбы не
затвердеет. В период выделения газа время от времени колбу встря-
хивают для лучшего смешения компонентов. Выделяющийся газ
сушат, пропуская его через осушительную колонку с гранулиро-
ванным едким кали.
4. Для того, чтобы удалить спирт при атмосферном давлении,
лучше всего фильтрат слить обратно в колбу, в которой сохранена
механическая мешалка, но обратный холодильник заменен на
отводную трубку и обращенный вниз холодильник. По мере отгонки
спирта раствор перемешивают, в результате чего избегаются толчки
при перегонке, которые в противном случае имеют место вследствие
выделения бромистого натрия из концентрированного раствора.
5. Такого рода использование бромистого натрия одновременно
служит нескольким целям: выделяется сложный эфир, который мог
поглотиться водным раствором, понижается растворимость слож-
ного эфира в воде и повышается плотность водного слоя, что бла-
гоприятствует рассаливанию.
6. Такого рода обработка служит для удаления неизменивше-
гося малонового эфира. Михаэль 1 показал, что такая обработка пол-
ностью удаляет неизменившийся малоновый эфир и почти совсем
не затрагивает этиловый эфир метилмалоновой кислоты. В случае
применения бромистого метила в качестве метилирующего агента
этилового эфира диметилмалоновой кислоты не образуется.
3. Другие методы получения
Наиболее широко распространенный метод получения этилового
эфира метилмалоновой кислоты состоит в алкилировании малоно-
вого эфира йодистым метилом 2> *, бромистым метилом 3 или диме-
тилсульфатом4. Отделение конечного продукта от следов не из-
менившегося исходного продукта и от этилового эфира диметилма-
лоновой кислоты не может быть достигнуто перегонкой, так как
точки кипения всех трех сложных эфиров лежат в пределах 3,5°.
Михаэль * нашел, что не вступивший в реакцию малоновый эфир
может быть полностью отделен, если воспользоваться тем, что он
легче омыляется щелочью, а Гэн и Ингольд5 получили чистый
продукт путем омыления, перекристаллизации метилмалоновой
кислоты и последующей этерификацией. На основании опытов
Сальковского мл. 6 с ацетоуксусным эфиром можно сделать заклю-
чение, что в случае применения бромистого метила в качестве
алкилирующего агента образование диметильного производного не
имеет места. Методика Б, основанная на работе Михаэля 1, описана
в литературе 7.
Чистый этиловый эфир метилмалоновой кислоты может быть
также получен восстановлением метилиденмалонового эфира в при-
сутствии никеля, приготовленного по Рэнею 8; из этилового
эфира а-бромпропионового эфира через нитрил9 (этот процесс
следует признать дорогостоящим, учитывая стоимость исходных
реагентов и низкие выходы) и пиролитическим разложением эти-
лового эфира этоксалилпропионовой кислоты. Этот последний метод,
лежащий в основе методики (А), впервые описал Вислиценус 10.
1 М i с h a e I, J. prakt. Chem. B) 72, 537 A905).
2 Z u b 1 i n, Ber. 12, 1112 A879); Co n r ad, В i s с h*o f f, Ann. 204, 146
A880); Herzig, Wenzel, Monatsh. 24, 115A903).
'Lucas, Young, J. Am. Chem. Soc. 51, 2536 A929).
4 N e f, Ann. 309, 188 A899).
6 G a n e, I n g о 1 d, J. Chem. Soc. 1926, 14.
«Salkowski, Jr., J. prakt. Chem. B) 106, 256 A923).
' F i e s e r, N о v e 1 1 o, J. Am. Chem. Soc. 62, 1856 A940).
8 W о j с i k, A d k i n s, там же 56, 2424 A934).
9 Зелинский, ЖРХО 20, 659 A888); Ber. 21, 3162 A888); S t e e 1 е, J.
Am. Chem. Soc. 53, 286 A931).
"Wislicenus, Ber. 27, 796 A894).
592
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
ЭТИЛОВЫЙ ЭФИР а-НАФТОЙНОЙ КИСЛОТЫ
(Этиловый эфир 1-нафтойной кислоты)
C10H7Br + Mg > C10H7MgBr
C10H7MgBr + (С2Н5J СО3 С1ОН7СО2С2Нб + C2H6OMgBr
Предложили: Ф. Уигпмор и Д. Лодер.
Проверили: Р. Фъюзон и '/. Вудвард.
1. Получение
Приготовляют раствор бромистого а-нафтилмагния из 24,3 г
A гр.-ат.) магния и 207 гA мол.) а-бромнафталина по методике, опи-
санной на стр. 348 (примечание 1), но только берут большую
делительную воронку и 3-литровую трехгорлую колбу. Гриньяров
реактив переносят в делительную воронку и в колбу помещают
177 г A,5 мол.) этилового эфира угольной кислоты (примечание 2)
и 100 мл абсолютного эфира. Пускают в ход мешалку и приливают
раствор бромистого а-нафтилмагния с такой скоростью, с какой
только позволяет стекание эфира из обратного холодильника.
После того как все добавлено, перемешивание продолжают еще
30 мин. и смесь оставляют стоять на ночь.
Чтобы осуществить гидролиз, реакционную смесь выливают при
перемешивании в 5-литровую колбу, в которую предварительно
помещено 1,2—1,5 кг колотого льда. Выпавший основной бромистый
магний растворяют постепенным добавлением 145 мл холодной
30%-ной серной кислоты C0 мл концентрированной кислоты и 120 мл
воды). Эфирный слой отделяют, а водный слой извлекают 100 мл
эфира. От объединенных эфирных растворов эфир отгоняют на
водяной бане, пока объем раствора не станет равным 400 мл. Оста-
ток промывают два раза 5%-ным раствором соды, порциями по 40 мл
(примечание 3), и сушат 20 г безводного хлористого кальция.
Раствор фильтруют для отделения хлористого кальция и эфир
отгоняют на водяной бане, Остаток переносят в колбу Вюрца на
500 мл и перегоняют. Фракцию от 287° до 307° собирают и счи-
тают неочищенным этиловым эфиром а-нафтойной кислоты. Сырой
продукт перегоняют из видоизмененной колбы Клайзена емкостью
250 мл. Выход чистого эфира с т. кип. 143—144,5°/3 мм составляет
136—147 г F8—73% теоретич., примечание 4).
2. Примечания
1. Если к реагенту не прибавлять бензола, то выпадает осадок
бромистого а-нафтилмагния и тогда надо пользоваться делитель-
ной воронкой, у которой отверстие крана имеет достаточно широ-
кий диаметр, чтобы его не забило во время цриливания реагента
к сложному эфиру.
ЭТИЛОВЫЙ ЭФИР н.-ТРИДЕКАНОВОЙ КИСЛОТЫ
593
2. Для очистки этилового эфира угольной кислоты 200 мл его
промывают 40 мл 10%-ного раствора соды, затем 40 мл насыщенного
раствора хлористого кальция и, наконец, 60 мл воды. После стоя-
ния в течение 2 час. над 10 г хлористого кальция эфир перегоняют
и собирают фракцию с т. кип. 125—126°. Не следует оставлять
стоять этиловый эфир угольной кислоты над безводным хлористым
кальцием более чем 1 сутки, так как эфир образует с солью ком-
плексное соединение.
3. При подкислении содового раствора можно получить 2—3 г
а-нафтойной кислоты.
4. Этот синтез был успешно проведен также с количествами,
в пять раз превышающими приведенные здесь.
3. Другие методы получения
Этиловый эфир а-нафтойной кислоты был получен в результате
взаимодействия хлористого а-нафтоила с абсолютным этиловым
спиртом *, при нагревании смеси а-нафтойной кислоты с этиловым
спиртом в присутствии серной кислоты 2 и по методике, описанной
выше 3.
1 Hot mann, Ber. 1, 42 A868).
2 Per kin, J. Chem. Soc. 69, 1178 A896).
3Loder, Whitmore, J. Am. Chem. Soc. 57, 2727 A935).
ЭТИЛОВЫЙ ЭФИР н.-ТРИДЕКАНОВОЙ КИСЛОТЫ
н.-СиН26Вг + KCN h.-C12H25CN + KBr '
h.-C12H25CN + 2H,0 _(—L, н.-С12Н25СО2Н + NH3
н.-С18На5СО,Н + C,HfiOH -—°~ н.-СиНюСОаСяН6 + H2O
Предложил: Дж. Рухоф.
Проверили: В. Каротерс и В. Мак-Ювен.
1. Получение
В 12-литровую колбу, снабженную обратным холодильником и
эффективной мешалкой с жидкостным затвором (примечание 1),
помещают 2,5 л 95%-ного этилового спирта, 872 г C,5 мол.) чистого
бромистого н.-додецила («Синт. орг. преп.», сб. 1, стр. 112) и 278 г
C,85 мол.) хорошо измельченного 90%-ного цианистого калия
(примечание 2). Смесь кипятят с обратным холодильником 15 час.
при работающей мешалке на водяной бане (примечание 3). К концу
этого времени прибавляют еще 278 г цианистого калия и смесь
кипятят, не прекращая перемешивания еще 15 час.
Колбе дают охладиться и добавляют раствор 670 г 90%-ного
едкого кали в 1 л воды (примечание 4). Раствор кипятят с обратным
холодильником при работающей мешалке 30 час. Затем колбу соеди-
няют с насадкой для перегонки с водяным паром (примечание 5)
38 Сборник ."Nt 2
594
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
и пропускают пар до тех пор, пока вспенивание не заставит прекра-
тить перегонку. После того как колба несколько охладится, трубку
для ввода пара заменяют на капельную воронку, ножка которой
доходит до дна колбы, и через нее добавляют 1,5 л концентрирован-
ной соляной кислоты (уд. в. 1,18) или эквивалентное количество
более разбавленной кислоты. Колбу время от времени взбалтывают,
чтобы предотвратить отслаивание соляной кислоты. Капельную
воронку заменяют вновь трубкой для ввода пара; в течение 30 мин.
пропускают водяной пар. К концу этого времени с помощью
сифона отделяют водный слой и отбрасывают его (примечание 6).
Сырую кислоту еще в теплом и жидком состоянии обрабатывают
815 мл A4 мол.) 95%-ного этилового спирта, содержащего 20—30 г
C—5% по весу) безводного хлористого водорода, и фильтрованием
отделяют раствор от нерастворимых примесей. Фильтрат помещают
в 3-литровую колбу, снабженную обратным холодильником, доба-
вляют 160 г безводного хлористого кальция и смесь кипятят
с обратным холодильником в течение 24 час. (примечание 7).
С помощью сифона отделяют нижний слой и разбавляют его двух-
кратным объемом воды, чтобы выделить растворившийся сложный
эфир, который присоединяют к главной порции. Ко всему коли-
честву добавляют 408 мл спирта с содержанием 3—5% безводного
хлористого водорода и 80 г хлористого кальция, после чего смесь
кипятят еще 24 часа. Нижний слой вновь сифонируют и выделяют
из него растворенный эфир так, как это описано выше. Верхний слой
дважды промывают равным объемом воды C0—40°) и переносят
в 2-литровую делительную воронку. Объем маслянистой жидкости
составляет примерно 1 л. В ту же воронку приливают 500 мл теп-
лой воды, несколько капель фенолфталеина и постепенно, при
взбалтываний, добавляют водный раствор аммиака (концентриро-
ванный водный аммиак, разбавленный двадцатью объемами воды)
до тех пор, пока образовавшаяся эмульсия не станет розовой (на
это требуется 50—100 мл). После этого добавляют спирт порциями
по 100 мл, для того чтобы разрушить эмульсию и добиться быстрого
разделения слоев; обычно бывает достаточно прибавить 2—3 пор-
ции спирта. Нижний слой спускают и сохраняют (примечание 8).
Маслянистый слой промывают три раза теплой водой, причем,
если это нужно, добавляют спирт для того, чтобы уничтожить
эмульсию. Сложный эфир сушат безводным хлористым кальцием,
фильтруют и перегоняют в вакууме. Выход этилового эфира три-
декановой кислоты с т. кип. 163—16575 мм A78—180°/20 мм;
197—198°/60 мм) составляет 615—660 г G3—78%теоретич,). Путем
дробной перегонки головной и остаточной фракций можно полу-
чить еще 50—70 г продукта; 15—30 г эфира и кислоты выделяют из
аммиачных промывных вод и из хлористого кальция, которым су-
шилась главная порция. Общий выход 685—710 г (81—84% теоре-
тич.; примечания 9 и 10).
ЭТИЛОВЫЙ ЭФИР н.-ТРИДЕКАНОВОЙ КИСЛОТЫ
595
2. Примечания
1. Можно пользоваться мешалкой с ртутным затвором, но пред-
почтительнее вазелиновый затвор.
2. Цианистый калий дает лучшие результаты, чем цианистый
натрий. Необходимым условием является его тщательное измель-
чение.
3. Работать необходимо в вытяжном шкафу. В качестве водяной
бани удобно использовать небольшую ванночку, поставленную
на скамейку, и обогревать ее сбоку бунзеновской горелкой.
4. Это соответствует избытку в 3,85 мол. едкого кали. Этого
количества достаточно для того, чтобы по окончании гидролиза
в растворе осталось 10% щелочи. Если брать меньшее количество
щелочи, то возрастает количество неомыленного нитрила.
5. Холодильник должен вести к 2-литровой склянке для отса-
сывания, боковой отвод которой соединен непосредственно с тягой
вытяжного шкафа. Спирт пахнет синильной кислотой, и его
выбрасывают.
6. Для некоторых целей пригодна сырая кислота, однако в ней
содержится небольшое количество неомыленного нитрила, а также
примесь продукта с весьма неприятным запахом (возможно — изо-
нитрил) и обычно небольшое количество синего осадка (комплекс-
ный цианид железа). При получении сложного эфира осадок
удаляется фильтрованием спиртового раствора кислоты, так как
присутствие его мешает удалению свободной тридекановой кислоты
после этерификации.
7. Чтобы избежать толчков при кипении, добавляют несколько
кусочков неглазурованной тарелки.
8. Если промывную жидкость подкислить, можно выделить еще
15—30 г сложного эфира и кислоты.
9. В случае еще ббльших загрузок методика остается неизмен-
ной и выходы получаются несколько более высокие. Так, например,
автор при загрузках в два раза больших получал выход равный
88—89%.
10. Аналогичным способом из бромистого н.-тетрадецила (стр.114)
были получены пентадекановая кислота и ее этиловый эфир примерно
с тем же выходом.
3. Другие методы получения
Тридекановая кислота была получена синтезом при помощи ма-
лонового эфира, исходя из йодистого ундецила1, окислением
а-оксимиристиновой кислоты в ацетоновом растворе перманганатом
калия 2, действием бромистого додецила на цианистый калий с по-
следующим омылением 3, а также из йодистого ундецила и этило-
.96
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
вого эфира циануксусной кислоты4. Приведенная выше методика
является видоизменением способа, который описали Ружичка,
Штоль и Шинц 3.
1 L e v e n e, West, Allen, van der Scheer, I, Biol. Chem
23, 73 A915).
2 L e v e n e, West, там же 18, 465 A914).
'Ruzicka, Stoll, Schinz, Helv. Chim. Acta 11, 685 A928).
* R о b i n s о n, J. Chem. Soc. 125, 230 A924).
ЭТИЛОВЫЙ ЭФИР а-ФЕНИЛАЦЕТОУКСУСНОЙ КИСЛОТЫ
(Этиловый эфир а-ацетил-а-толуиловой кислоты)
СН8СОСН (С6Н6) CN + С,Н5ОН -—- СН3СОСН (С6Н5) С — ОС2Н5
СН8С0СН (С6Н5) С — ОС2Н3 + H2SO4
II
Н,0
NH
II
NH
СН3С0СН (С6Н6) СО2С2Н5
NH4HSO4
Предложили: Р. Кимболл, Д. Джефферсон и А. Пайк.
Проверил: К. Ноллер.
1. Получение
Прибор состоит из 1-литровой трехгорлой колбы, снабженной
механической мешалкой с ртутным затвором и трубкой для ввода
газа, диаметром б мм, доходящей до дна колбы. В третье горло
вставлена корковая пробка с термометром для низких температур
и трубкой с фосфорным ангидридом, нанесенным на стеклянную
вату, и хлористым кальцием. Трубка для ввода газа присоединена
к трем осушительным колонкам высотой 20 см, из которых две на-
полнены фосфорным ангидридом, нанесенным на стеклянную вату,
и одна хлористым кальцием. Последняя колонка в свою очередь
соединена с 2-литровой колбой Флоренса, снабженной промывной
склянкой и предохранительной склянкой, в которую налит 1 л кон-
центрированной серной кислоты. Колбу Флоренса присоединяют
к генератору хлористого водорода, описанному в «Синт. орг. преп.»,
сб. 1, стр. 214, в который Загружают 1,5 л концентрированной
серной кислоты и 800 мл соляной кислоты, что достаточно для
данного синтеза. Между колбой генератора и воронкой должен
находиться прибор для уравнивания давления.
Трубку для ввода газа вынимают, в реакционную колбу нали-
вают 400 мл абсолютного спирта (примечание 1) и добавляют 161 г
A мол.) сухого а-фенилацетоацетонитрила (т. пл. 88,5—89,5°;
стр. 503). Горло на время закрывают пробкой и нитрил растворяют
при нагревании и перемешивании. Затем колбу погружают в
ЭТИЛОВЫЙ ЭФИР я-ФЕНИЛАЦЕТОУКСУСНОЙ КИСЛОТЫ
597
охладительную смесь и раствор энергично перемешивают так, что
кристаллизующийся нитрил выпадает в мелко дисперсном состоя-
нии. Когда температура достигнет —10°, вставляют трубку для
ввода газа и при умеренном перемешивании пропускают ток сухого
хлористого водорода с такой скоростью, чтобы можно было счи-
тать пузырьки в промывной склянке с серной кислотой. Пропуска-
ние хлористого водорода продолжается 5—8 час, до насыщения
смеси (примечание 2). Тогда баню со льдом удаляют и перемешива-
ние продолжают до растворения твердого осадка (около 1 часа),
после чего колбу оставляют стоять на ночь (примечание 3).
Большую часть избытка хлористого водорода удаляют, добавив
в колбу кусочки пористой тарелки и эвакуируя колбу в течение
30 мин. с помощью водоструйного насоса, одновременно поставив
ее в водяную баню, нагретую до 40°. В 5-литровой колбе раство-
ряют 200 г углекислого натрия в 1,2 л воды и добавляют 2 л колотого
льда. К этому раствору приливают тонкой струей при энергичном
перемешивании реакционную смесь и немедленно раствор экстра-
гируют три раза эфиром, порциями по 500 мл. Эфирные вытяжки
поочередно промывают четыре раза ледяным 5%-ным раствором
хлористого натрия порциями по 250 мл, затем их соединяют вместе
и помещают в 3-литровую колбу, охлаждаемую в бане со льдом.
В 5-литровой колбе приготовляют раствор 100 г концентрирован-
ной серной кислоты (х. ч.) в 700 мл воды, добавляют 1,5 л колотого
льда и смесь взбалтывают до тех пор, пока снаружи колбы не
образуется корка льда. Примерно половину этого растворд прили-
вают через воронку, на которой задерживается лед, к холодному
эфирному раствору иминоэфира, смесь взбалтывают точно 15 сек.
(примечание 4), дают ей расслоиться и нижний слой отделяют.
Оставшуюся кислоту приливают к эфирному раствору в две пор-
ции, причем каждый раз смесь взбалтывают 15 сек. и слои разде-
ляют. Поскольку эфирный раствор, хотя и не содержит более ими-
ноэфира, все еще содержит небольшое количество этилового эфира
фенилацетоуксусной кислоты, его сохраняют, чтобы позднее при-
соединить к главной порции.
Раствор сернокислой соли иминоэфира в серной кислоте быстро
становится мутным вследствие выделения этилового эфира а-фенил-
ацетоуксусной кислоты. Для завершения гидролиза смесь нагре-
вают на водяной бане в течение 30 мин. при температуре, когда
эфир едва кипит (около 50°; примечание 5). Затем раствор охла-
ждают, эфирный слой отделяют и кислоту извлекают один раз 250мл
эфира. Эфирный раствор промывают один раз 100 мл воды и вод-
ную вытяжку присоединяют к кислоте. Кислый раствор вновь
помещают на водяную баню и нагревают 45 мин. после того, как
температура достигнет 80—90°. По охлаждении раствора его вновь
экстрагируют, как ив первый раз, а-затем все эфирные вытяжки,
включая и первую, из которой был удален иминоэфир,- соединяют
598
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
вместе и промывают один раз 250 мл 5%-ного раствора бикарбоната
натрия, один раз 250 мл воды и сушат над 20 г безводного серно-
кислого натрия (примечание 6). Сернокислый натрий отфильтро-
вывают и промывают его эфиром. Эфир от всех фильтратов отгоняют
и остаток перегоняют в вакууме из видоизмененной колбы Клай-
зена емкостью 250 мл с дефлегматором высотой 25 см. Главная фрак-
ция кипит при 139—143°/12 мм или при 130—134°/5 мм и весит
103—167 г E0—81% теоретич.). В результате дробной перегонки
головной и главной фракций, а также остатка можно получить
продукт, выкипающий в пределах 1—2°, причем выход остается
прежним (примечание 7).
2. Примечания
1. Спирт был высушен один раз негашеной известью и один раз
натрием, как это указано в примечании 1, «Синт. орг. преп.», сб. 1,
стр. 547.
2. При более быстром насыщении выход заметно понижается.
3. На дно оседает мелкий осадок, вероятно, хлористый аммоний.
В наиболее удачных опытах этого осадка не было или он был незна-
чителен.
4. Всякая задержка на этой стадии ведет к омылению части
иминоэфира в конечный продукт, который остается в эфирном
растворе.
5. Необходимо соблюдать осторожность при вынимании колбы
из баниГ так как даже при небольшом смешивании эфир закипает
и его может выбросить из колбы.
6. Удаление продукта время от времени при омылении, повиди-
мому, ведет к повышению выхода.
7. Жидкий сложный эфир представляет собой равновесную
смесь, содержание энола в которой увеличивается при перегонке,
после чего при стоянии оно медленно падает до 30% *. Т. кип. 145—
147°/11 мм, приведенная в литературе, выше чем температура,
полученная в настоящей работе.
3. Другие методы получения
Этиловый эфир а-фенилацетоуксусной кислоты может быть полу-
чен омылением а-фенилацетоацетонитрила в абсолютном спирте
сухим хлористым водородом 1. Настоящий метод отличается тем,
что хлористый водород нейтрализуется углекислым натрием и что
гидролиз иминоэфира проводится разбавленной серной кислотой,
так что продукт выделяется немедленно по образовании. Это пре-
дохраняет сложный эфир от дальнейшего разложения, что заметно
повышает выход.
lBeckh, Ber. 31, 3160 A898); Post, M i с h a I e k, I. Am. Chem.
Soc. 52,4358A930).
ЭТИЛОВЫЙ ЭФИР ФЕНИЛМДЛОНОВОЙ КИСЛОТЫ
ЭТИЛОВЫЙ ЭФИР ФЕНИЛМАЛОНОВОЙ КИСЛОТЫ
С6Н5СН2СОАН5 + (СО2С,Н5)., <Ei^N!>
С6Н6СН (СО,С2Н8) СОСОАН5 + С2Н5ОН
С6Н3СН (СО2С2Н5) СОСОХ,Н3 <нагреван-1. СвН5СН (СО2С2НбJ + СО
Предложили: Л. Левин и Дж. Мейер.
Проверили: Л. Физер и К. Фишер.
1. Получение
В 2-литровую колбу с тремя горлами, снабженную мешалкой
с ртутным затвором, обратным холодильником и капельной ворон-
кой, помещают 500 мл абсолютного этилового спирта (примечание 1)
и постепенно добавляют 23 г(\ гр.-ат.) металлического натрия, наре-
занного кусочками. После того как весь натрий растворится,
раствор охлаждают до 60° и при энергичном помешивании добавляют
из капельной воронки непрерывной струей 146 г A мол.) этилового
эфира щавелевой кислоты (примечание 2); остатки его смывают
небольшим количеством абсолютного спирта, после чего немед-
ленно приливают 175 г A,06 мол.) этилового эфира фенилуксусной
кислоты. Затем сразу же прекращают перемешивание, реакцион-
ную колбу опускают так, чтобы мешалка была вынута из раствора,
и одновременно подготовляют 2-литровый стакан. Примерно через
4—6 мин. после прибавления этилового эфира фенилуксусной
кислоты начинается кристаллизация. При первых признаках кри-
сталлизации содержимое колбы выливают в стакан. Кристаллиза-
ция протекает почти мгновенно.
Почти твердой массе натриевого производного дают охладиться
до комнатной температуры, после чего ее тщательно перемешивают
с 800 мл абсолютного эфира. Осадок отсасывают и несколько раз
промывают абсолютным эфиром. Этиловый эфир фенилщавелево-
уксусной кислоты выделяют из его натриевой соли, разбавленной
серной кислотой B9 мл концентрированной серной кислоты в 500 мл
воды). Почти бесцветный маслянистый слой отделяют, а водный
слой экстрагируют тремя порциями эфира по 100 мл- Эфирные
вытяжки присоединяют к основному продукту и сушат эфирный
раствор над безводным сернокислым натрием. Эфир отгоняют, а
остаток помещают в видоизмененную колбу Клайзена с дефлегма-
тором и нагревают под давлением около 15 мм на бане из сплава
Вуда. Температуру бани постепенно доводят до 175° и поддержи-
вают эту температуру до тех пор, пока не прекратится выделение
окиси углерода. В случае временного повышения давления в
течение этого процесса нагревание прерывают. К концу реакции
(через 5—6 час.) перегнанную маслянистую жидкость вливают
обратно в колбу и этиловый эфир фенилмалоновой кислоты пере-
гоняют в вакууме. Собирают фракцию 158—162°/10 мм. Выход
189—201 г (80—85% теоретич.).
GOO
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
2. Примечания
1. Необходимо пользоваться абсолютным спиртом хорошего
качества. К обыкновенному «абсолютному» спирту прибавляют 5%
по весу металлического натрия и перегоняют его непосредственно
в реакционную колбу.
2. Для того, чтобы все реактивы были действительно сухими
и нейтральными, этиловые эфиры щавелевой («Синт. орг. преп.»,
сб. 1, стр. 562) и фенилуксусной кислот («Синт. орг. преп.», сб. 1,
стр. 557) предварительно взбалтывают с безводным поташом и пе-
регоняют под уменьшенным давлением, причем до перегонки оба
продукта нагревают при атмосферном давлении до тех пор, пока
не будут достигнуты температуры их кипения.
3. Другие методы получения
Описанная выше методика основана на стандартном методе
Вислиценуса *. Этиловый эфир фенилмалоновой кислоты был полу-
чен также из цианистого бензила и этилугольного эфира2. Фенил-
малоновая кислота была также получена действием углекислого
газа на энолят фенилуксусной кислоты 3.
1 W i s I i с е n u s, Ber. 27, 1091 A894); Ruhemann, J. Chem. Soc.
81, 1214 A902); Р i с k a r d, Y a t e s, там же 95, 1015 A909); F о г s t e г,
Ми Пег, там же 97,135A910); Baker, In go Id, там же 1927,835;
Blum-Bergman n, Ber. 65, 115 A932).
sNelson, Cretcher, J. Am. Chem. Soc. 50, 2760 A928).
Mvanoff, Spassoff, Bull. soc. chim. D) 49, 20 A931).
ЭТИЛОВЫЙ ЭФИР ФЕНИЛЦИАНПИРОВИНОГРАДНОЙ КИСЛОТЫ
C6H5CH2CN + СО2С2Н5 + C2H5ONa
со2с2н3
С6Н5С = С (ONa) СО2С2Н5 + 2С2Н5ОН
CN
СвН5С = С (ONa) СО2С2Н3 + НС1 CeH5CHCOCOaC2H5 + NaCl
CN
CN
Предложили: Р. Адаме и X. Калвери.
Проверили: Дж. Нонант и Д. Блюменталь.
1. Получение
В 3-литровую круглодонную колбу, соединенную с обратным
холодильником, наливают 650 мл абсолютного спирта (примеча-
ние 1) и прибавляют к нему через холодильник как можно быстрее,
ЭТИЛОВЫЙ ЭФИР ЭТИЛЕНТЕТРАКАРБОНОВОЙ КИСЛОТЫ
601
но так, чтобы не было потерь, 46 г B гр.-ат.) натрия. Если весь нат-
рий не растворится, то смесь нагревают. К горячему раствору эти-
лата натрия приливают как можно быстрее 312 г B,1 мол.) этило-
вого эфира щавелевой кислоты («Синт. орг. преп.», сб. 1, стр. 562),
после чего немедленно прибавляют 234 г B мол.) цианистого бен-
зила («Синт. орг. преп.», сб. 1, стр. 502) и реакционную смесь оста-
вляют стоять на ночь. Полученный раствор переносят в 3-литро-
вый стакан, прибавляют к нему 250—300 мл воды (примечание 2),
нагревают до 35° и приливают, при механическом перемешивании,
концентрированную соляную кислоту до сильно кислой реакции на
лакмус. По охлаждении раствора до комнатной температуры выпа-
дает этиловый эфир фенилцианпировиноградной кислоты в виде ли-
монножелтых кристаллов. Выход 360—385 г, т. пл. 126—128°. После
перекристаллизации из 60%-ного спирта сложный эфир плавится
при 130°. Выход чистого продукта 300—325 г F9—75% теоретич.).
2. Примечания
1. Вполне пригоден абсолютный спирт, получаемый по методи-
ке, указанной в «Синт. орг. преп.», сб. 1, стр. 555.
2. Указанное количество воды разбавляет спирт настолько,
что сложный эфир хорошо кристаллизуется. Если взять больше
воды, то эфир выпадает немедленно в виде масла, которое посте-
пенно затвердевает.
3. Другие методы получения
В литературе описан только изложенный выше способ г.
1Erlenmeyer, Ann. 271, 173 A892).
ЭТИЛОВЫЙ ЭФИР ЭТИЛЕНТЕТРАКАРБОНОВОЙ КИСЛОТЫ
2 СНВг (СО2С2Н5J + Na2CO3
2 NaBr + Н2О + СО2 + (С2Н5О2СJ С = С (СО2С2Н5J
Предложили: Б- Корсон и В. Бенсон.
Проверили: К- Марвел и X. Мунро.
1. Получение
Смесь 200 г A,9 мол.) безводного углекислого натрия (примеча-
ние 1) и 300 г A,25 мол.) этилового эфира броммалоновой кислоты
(«Синт. орг. преп.», сб. 1, стр. 541) нагревают в продолжение 3 час.
в 1-литровой колбе (примечание 2) на масляной бане при 150—160°
(примечание 3). По истечении указанного времени к горячей еще
массе (примечание 4) прибавляют 300 мл ксилола (примечание 5).
602
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
Твердую массу тщательно раздробляют палочкой, смесь переносят
в 2-литровый стакан и колбу споласкивают смесью 100 мл ксилола
и 100 мл воды. Эту смесь переливают в 2-литровый стакан и туда же
прибавляют еще 600 мл воды; при перемешивании твердая масса
легко растворяется. Жидкую смесь переливают в делительную
воронку, хорошо взбалтывают и дают отстояться (примечание 6).
Нижний водный слой отбрасывают (примечание 7), а ксилольный
слой переносят в 1-литровую перегонную колбу и отгоняют ксилол
до тех пор, пока температура жидкости не достигнет 170°. Жидкий
остаток переливают в колбу Клайзена емкостью 500 мл и перего-
няют в вакууме. Первый отгон, кипящий до 170э/15 мм, отбрасы-
вают; собранный при 170—230°/15 мм (примечания 8 и 9) продукт
примерно через 15 мин. затвердевает. Выход 150—160 г G5—80%
теоретич.).
С целью очистки сырой продукт растворяют в 75 мл 95%-ного
спирта при 40"; этой температуры вполне достаточно для полного
растворения. Раствор охлаждают до 12° (примечания 10 и И) и
выпавший эфир отсасывают. Выход высушенного на воздухе бес-
цветного продукта, плавящегося при 52,5—53,5°, составляет 95—
110 г D7—55% теоретич.). Отгонкой спирта от маточного раствора,
перегонкой остатка в вакууме и перекристаллизацией затвердев-
шего дестиллата можно повысить выход до 110—115 г E5—57% тео-
ретич.).
2. Примечания
1. Углекислый натрий должен нацело проходить через сито
в 100 меш.
2. Смесь нагревают в открытой колбе. Поскольку этиловый
эфир броммалоновой кислоты и этиловый эфир этилентетракарбо-
новой кислоты обладают высокой температурой кипения, потери
вследствие испарения ничтожны. При нагревании смеси в колбе
с обратным холодильником выход понижается, вероятно, потому,
что вода, образующаяся во время реакции, остается в реакционной
смеси и вызывает омыление одного или обоих эфиров.
3. Колбу ставят в холодную баню; нагревание следует вести
3 часа, считая от того момента, когда температура бани достигнет
150°.
4. Если дать смеси охладиться, то она затвердевает, и тогда ее
очень трудно извлечь из колбы.
5. Вместо ксилола можно взять толуол или бензол, но работа
с первым удобнее, так как он кипит выше.
6. Полное разделение слоев наступает через 10 минут.
7. Опыт показал, что содержащееся в водном слое количество
продукта ничтожно.
8. Этиловый эфир этилентетракарбоновой кислоты кипит при
19778 мм; 203713 мм; 210722 мм; 22 Г/33 мм; 234748 мм.
ЭТИЛОВЫЙ ЭФИР ЭТИЛЕНТЕТРАКАРБОНОВОЙ КИСЛОТЫ
601!
9. В колбе остается очень небольшой остаток. Перегонку сле-
дует прекратить, как только капли переходящего дестиллата станут
темножелтыми.
10. По охлаждении раствор превращается в сплошную массу
кристаллов, пропитанных маточным раствором, которая обладает
низкой теплопроводностью. Поэтому, чтобы понизить температуру
всей массы до 12°, требуется довольно продолжительное время.
Температуру необходимо контролировать термометром, так как
кривая растворимости эфира начинает резко подниматься выше
12°. Охлаждение кристаллизующейся массы ниже 12° не дает ни-
каких преимуществ.
11. Ниже приведена растворимость этилового эфира этиленте-
тракарбоновой кислоты в 100 мл 95%-ного этилового спирта:
2,0 г при 0°
9 К П"
!o „
8,0 „
9,7 „
23°
26е
16,0
19,0
28,0
35,0
61,0
при 30°
„ зг
„ 33е
„ 34°
„ 36,5°
3. Другие методы получения
Этиловый эфир этилентетракарбоновой кислоты был получен
отщеплением элементов галоидоводородной кислоты от монохлор-
и моноброммалонового эфира действием натрия х, этилата натрия 2,
уксуснокислого калия3, углекислого калия *, уретаннатрия5, форм-
анилиднатрия и ацетанилиднатрия 6- Он был получен также дей-
ствием брома 7 или иода 8 на динатриевое производное этилового
эфира этан-1,1, 2,2-тетракарбоновой кислоты; действием на дибром-
малоновый эфир натрия9 или этилата натрия 10 или натриймало-
нового эфира (с последующим действием поташаI1 и действием
иода12 на динатриймалоновый эфир. Имеются указания, что при-
менение влажного бензола в качестве среды при реакции бромма-
лонового эфира с углекислым калием ведет к повышению выхода
этилового эфира этилентетракарбоновой кислоты до 80% 13-
1 С о n r a d, G u t h г е i t, Ber. 16, 2631 A883).
2 С о n r a d, Guthzeit, Ann. 214, 76 A882).
¦Conrad, Bruckner, Ber. 24, 2998 A891).
'Blank, Samson, там же 32, 860 A899).
6 D i e 1 s, H e i n t z e 1, там же 38, 303 A905).
6 P a a 1, О 11 с п, там же 23, 2591 A890).
' К о t z, S t a 1 m a n n, I. prakt. Chem. B) 68, 163 A903).
8Bischoff, Hausdorfer, Ann. 239, 130 A887).
"Conrad, Bruckner, Ber. 24, 3004 A891).
"Curtiss, Am. Chem. J. 19, 699 A897).
ii Ad i с k e s, J. prakt. Chem. B) 145, 236 A936).
«Bischoff, Rach, Ber. 17, 2781 A884).
13Malachowski, Sienkiewiczowa, там же 68, 33 A935).
604
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
1Ч-ЭТИЛ-.И-ТОЛУИДИН
605
ЭТИЛОВЫЙ ЭФИР ЭТОКСАЛИЛПРОПИОНОВОЙ КИСЛОТЫ
СН3СНХОХоН5 + СОХ2Н5 ^l^-t СН3ССОХ2Н5
I " .1
СОХ2Н5 C(ONa)COX2H5
^^ снхнсох,н
2~а* "о
СОСОХ2Н5
Предложили: Р. Кикс и С. Мак-Эльв ен.
Проверили: Р. Фьюзон и В. Росс.
1. Получение
В 3-литровой колбе с тремя горлами распыляют в ксилоле 69 г
C rp.-ат.) металлического натрия. Смесь охлаждают, ксилол сливают
и натрий промывают два раза небольшими порциями абсолютного
эфира. Затем к мелкораздробленному натрию приливают 1 л аб-
солютного эфира. Колбу снабжают мешалкой с ртутным затвором,
эффективным обратным холодильником и капельной воронкой, при-
чем . холодильник и воронку защищают от влаги воздуха хлор-
кальциевыми трубками. Из воронки добавляют по каплям 138 г
A75 мл, 3 мол.) абсолютного этилового спирта (примечание 1).
После того как весь спирт прибавлен и не осталось больше непро-
реагировавшего натрия (это видно по прекращению кипения жид-
кости), колбу погружают в баню с водой и льдом и через капель-
ную воронку медленно прибавляют смесь 306 г C мол.) этилового
эфира пропионовой кислоты и 438 г D04 мл, 3 мол.) этилового
эфира щавелевой кислоты («Синт. орг. преп.», сб. 1, стр. 562; приме-
чание 2).
После того как смесь сложных эфиров прибавлена, мешалку
убирают и обратный холодильник меняют на обращенный вниз.
Образовавшиеся при реакции спирт и эфир отгоняют на водяной
бане (примечание 3). Остаток, который по охлаждении обычно
затвердевает, обрабатывают 600 мл холодной 33%-ной уксусной
кислоты. Смесь оставляют стоять на несколько часов, время от
времени взбалтывая колбу, чтобы полностью разложить натриевое
производное, и продукт экстрагируют четыре раза эфиром пор-
циями по 500 мл. Эфирную вытяжку промывают 1 л воды, 2 раза
10%-ным раствором бикарбоната натрия порциями по 500 мл и,
наконец, 1 л воды. Эфир отгоняют на водяной бане, а остаток под-
вергают дробной перегонке в вакууме с хорошим дефлегматором
(примечание 4). Собирают фракцию 114—116°/10 мм. Выход 363—
425 г F0—70% теоретич,).
2. Примечания
1. Прибавление спирта занимает от 4 до 6 час. в зависимости от
эффективности холодильника и мешалки.
2. Прибавление смеси сложных эфиров следует производить
достаточно медленно, чтобы эфир не кипел. На прибавление эфир-
ной смеси требуется от 2 до 3 час.
3. После того как большая часть спирта отогнана, на поверхно-
сти красной вязкой жидкости образуется желтая пена, в этот момент
перегонку прекращают. По охлаждении раствора натриевое про-
изводное кристаллизуется, причем происходит значительное уве-
личение в объеме.
4. Если перегонку вести при 10 мм, то заметного разложения
этоксалилового эфира с образованием этилового эфира метилмалоно-
вой кислоты не происходит. Чтобы избежать перегрева, рекомен-
дуется нагревать колбу на масляной бане и обогревать дефлегматор.
3. Другие методы получения
Этиловый эфир этоксалилпропионовой кислоты был получен
конденсацией по Клайзену щавелевого эфира с этиловым эфиром
пропионовой кислоты ! так, как это описано выше, или же алкили-
рованием этилового эфира этоксалилуксусной кислоты 1.
1 W i s I i с е n u s, Arnold, Ann. 246, 329, 336 A888).
jM-CHsC,H4NH
Л-ЭТИЛ-.И-ТОЛУИДИН
Ь С2Н5Вг > Af-CH8C,H4NHCaH5 • НВг
+ HNO2 —— Jn-CH8CeH4N (NO) C2H5 + Н2О
m-CH3C6H4N (NO) C,H5 -r 6 [H] >
^-CH3C6H4NHC3H5
NH3
HX>
Предложили: Дж. Бек и К. Ферри.
Проверили: Дж. Джонсон и П. Баррик.
1. Получение
Берут две обыкновенные узкогорлые склянки емкостью 250 мл
и в каждую из них помещают по 32,1 г @,3 мол.) л<-толуидина и по
33 г B3 мл, 0,3 мол.) бромистого этила (примечание 1). Склянки
закупоривают каучуковыми пробками, укрепляют пробки про-
волокой и оставляют стоять при комнатной температуре в течение
24 час. в 2-литровом стакане, наполненном водой (примечание 2).
В каждой склянке белую кристаллическую массу раздробляют и
выделяют амин взбалтыванием с 150 мл 10%-ного раствора едкого
606
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
натра и 50 мл эфира. Содержимое обоих склянок соединяют вместе,
нижний водный слой отделяют и отбрасывают, а эфирный раствор
амина промывают 150 'мл воды. Эфир отгоняют на водяной бане и
получают чистый амин (90—92 г).
Сырой амин при охлаждении прибавляют к раствору 100 мл
концентрированной соляной кислоты (уд. в. 1,18) в 350 мл воды.
Раствор хлористоводородной соли охлаждают в бане со льдом и при
энергичном перемешивании медленно приливают раствор 41,5 г
@,6 мол.) азотистокислого натрия в 150 мл воды. Во время приба-
вления температуре не дают подниматься выше 12°. После того как
весь нитрит добавлен, смесь оставляют стоять в течение 10 мин.;
после чего ее извлекают три раза эфиром порциями по 100 мл. Эфир
испаряют, осторожно нагревая эфирный раствор на водяной бане
и продувая ток воздуха над его поверхностью. Необходимо следить
за тем, чтобы во время испарения эфира температура была воз-
можно более низкой (примечание 3).
Сырое нитрозосоединение постепенно прибавляют при непрерыв-
ном перемешивании к раствору 407 г A,8 мол.) кристаллического
хлористого олова B мол. Н20) в 420 /*лD,8мол.) концентрированной
соляной кислоты (уд. в. 1,18) в 3-литровой колбе. Реакция эта экзо-
термичная, и колбу необходимо охлаждать для того, чтобы под-
держивать температуру ниже 60°. После стояния в течение не менее
1 часа (примечание 4) смесь сильно подщелачивают осторожным
прибавлением холодного раствора 520 г A3 мол.) едкого натра в
800 мл воды. Во время приливания щелочи смесь энергично переме-
шивают и охлаждают проточной водой.
Полученную молочную суспензию подвергают перегонке с во-
дяным паром и собирают около 2 л дестиллата, который насыщают
хлористым натрием и извлекают три раза бензолом, цорциями по
100 мл. Вытяжки тщательно сушат в течение ночи гранулирован-
ным едким кали и сливают с осушителя декантацией. После отгонки
растворителя амин перегоняют в вакууме. Практически весь про-
дукт переходит при 111—112°/20 мм или при 115,5—117°/26 мм.
Чистый амин представляет собой практически бесцветную жид-
кость, с высоким показателем преломления. Выход 51—53 г F3—
66% теоретич.; примечание 5). При стоянии продукт довольно
быстро окрашивается.
2. Примечания
1. Применялся ти-толуидин высокого качества. Бромистый этил
обыкновенного качества дает удовлетворительные результаты.
2. Если в начальной стадии реакции дать итти слишком быстро,
в склянках может создаться большое давление. С целью пре-
дохранения от взрыва склянки следует защитить проволочной
сеткой.
ЭТОКСИУКСУСНАЯ К-ТА И ЭТИЛОВЫЙ ЭФИР ЭТОКСИУКСУСН. К-ТЫ 607
3. Нитрозосоединение разлагается при нагревании или при стоя-
нии. Его не следует сохранять, а рекомендуется немедленно обраба-
тывать восстановителем.
4. Никакого вреда не будет, если смесь оставить стоять и на
больший срок (на ночь). Часто выпадает гранулированный осадок
комплексной соли олова и амина.
5. Практически по той же методике могут быть получены и дру-
гие N-алкил-л-толуидины. Авторы синтеза указывают, что н.-про-
пил-, изопропил- и н.-бутилпроизводные могут быть легко получены
из л-толуидина и соответственных йодистых (лучше чем бромистых)
алкилов. С этими галогенидами алкилирование осуществляют таким
образом, что закупоренную склянку помещают в стакан с водой,
предварительно нагретой до 70—80° и держат ее в теплом помеще-
нии до окончания реакции, что обычно занимает несколько дней.
3. Другие методы получения
N-Этил-л-толуидин был получен при пропускании ж-толуидина
и этилового спирта над катализатором при высокой температуре г,
а также алкнлированием л<-толуидина с помощью этилового эфира
и-толуолсульфокислоты2. Настоящий метод очистки является видо-
изменением общего метода очистки вторичных аминов, разработан-
ного Дипольдером 3.
1 М a i h I e, d e G о cl о n, Compt. rend. 172, 1417A921); М a i 1 h e;
франц. пат. 23 891 (Chem. Zentr. I922, IV, 760).
2 Finzi, Ann. chim. applicata 15, 41 A925) [C. A. 19, 2647 A925)].
1 D i e p о 1 d e r, Ber. 32, 3514 A899).
ЭТОКСИУКСУСНАЯ КИСЛОТА И ЭТИЛОВЫЙ ЭФИР ЭТОКСИУКСУСНОЙ
кислоты
С1СН2СО2Н + 2C2H5ONa
C2H5OCH2CO2Na + HC1
с,н5осн2со,н
C,HsOCH2CO2Na + NaCl +C2HSOH
— - C2H~OCH2CO2H + NaCl
с.н5он -<HC- с,н5осн2со2с,н5 + н2о
Предложили: Р. Фыозон и В. Войцик.
Проверили: К. Ноллер и Дж. Гордон.
1. Получение
А. Этоксиуксусная кислота. 1250 мл абсолютного этилового
спирта (примечание 1) помещают в 2-литровую круглодонную
колбу, соединенную с обратным холодильником длиною 70—80 см.
Через трубку холодильника в колбу прибавляют 69 г C гр.-ат.)
металлического натрия с такой скоростью, чтобы спирт все время
608
СИНТЕЗЫ ОРГАНИЧЕСКИХ ПРЕПАРАТОВ
ЭТОКСИУКСУСНАЯ К-ТА И ЭТИЛОВЫЙ ЭФИР ЭТОКСИУКСУСНОЙ К-ТЫ 609
слабо кипел. Когда натрий полностью растворится, к раствору эти-
лата натрия медленно прибавляют порциями по 20 мл раствор 142 г
A,5 мол.) хлоруксусной кислоты в 180 мл абсолютного спирта (при-
мечание 2). После прибавления всей кислоты смесь слабо нагре-
вают в течение 10 мин. Затем избыток спирта удаляют полностью,
сначала отгонкой на водяной бане, а под конец отгонкой с водяным
паром. Водный раствор охлаждают, и к нему прибавляют 140 мл
A,7 мол.) концентрированной соляной кислоты, уд. в. 1,19. Выпав-
ший хлористый натрий отсасывают и промывают два раза порциями
но 50 мл эфира. Водный фильтрат насыщают безводным серно-
кислым натрием C0—35 г) и извлекают эфиром, которым раньше
промыли хлористый натрий, с добавкой еще 100 мл чистого эфира.
Эфир отделяют от водного слоя, который затем экстрагируют еще
4 раза чистым эфиром порциями по 100 мл. Эфирные вытяжки
соединяют, эфир отгоняют на водяной бане, а остаток перегоняют
в вакууме из колбы Клайзена емкостью 500 мл, соединенной с при-
емником такой же емкости. Кислота перегоняется при 109—111°/
17—18 мм. Выход 115—116 г кислоты G4% теоретич.). При повтор-
ной пер*егонке низшей фракции собирают фракцию 150—210°, кото-
рой получается 7—10 г и которая в основной массе представляет
собою этоксиуксусную кислоту и может быть присоединена к глав-
ной массе для перевода в эфир.
Б. Этиловый эфир этоксиуксуспой кислоты. Этоксиуксусную
кислоту, которой должно быть около 125 г A,2 мол.), помещают
в коническую колбу емкостью 750 мл. В этой колбе находится 230 мл
C,9 мол.) абсолютного этилового спирта. Колбу погружают в ведро
с холодной водой (примечание 3) и в смесь пропускают сухой хлори-
стый водород. После насыщения (примечание 4) смесь оставляют
стоять 24 часа, чтобы реакция закончилась при комнатной темпе-
ратуре (примечание 5). Затем раствор охлаждают и, помешивая,
чтобы избежать сильного вспенивания, к нему осторожно при-
бавляют насыщенный раствор соды. Соду прибавляют до слабо-
щелочной реакции на лакмус; избытка соды следует избегать, так
как он понижает выход. Полученный эфир извлекают четыре раза
порциями по 100 мл обыкновенного эфира; вытяжку сушат 25 г
безводного поташа и эфир отгоняют в водяной бане. Остаток пере-
гоняют при атмосферном давлении. Выход эфира, кипящего при
153—155°, составляет 110—115 г E5—58% теоретич., считая на
этоксиуксусную кислоту).
3. Во время реакции происходит разогревание, и если колбу
не охлаждать холодной водой, то поглотится недостаточное коли-
чество хлористого водорода, что ведет к уменьшению выхода.
4. Требуется значительное количество сухого хлористого водо-
рода. Газ надо пропускать через смесь по крайней мере 5 час.
5. Выход, повидимому, ограничен равновесием, устанавливаю-
щимся между кислотой и ее эфиром. Для достижения такого равно-
весия требуется не менее 24 час.
3. Другие методы получения
Этоксиуксусную кислоту впервые получил Гейнтцх взаимо-
действием хлоруксусной кислоты с этилатом натрия. Описанный
выше способ в основном разработан Соммеле2. Этоксиуксусная
кислота была получена также омылением этоксиацетонитрила кон-
центрированной соляной кислотой3 и при действии избытка эти-
лата натрия на 1,1,1,2-тетрахлорэтан и на а,р-дихлорвинилэтило-
вый эфир *. В одном патенте описывается синтез этой кислоты,
исходя из диэтилового эфира и углекислоты5 при высоком давлении.
Этиловый эфир этоксиуксусной кислоты был получен действием
йодистого этила на натриевую соль этоксиуксусной кислоты6,
действием этилата натрия на этиловый эфир хлоруксусной кислоты7,
действием спирта на неочищенный диазоуксусный эфир 8 и при
алкоголизе этоксиацетонитрила спиртовым раствором хлористого
водорода 3'9.
1 Н е i n t z, Ann. Physik 109, 331 (I860); 111, 555A860); R о t h s te i n,
Bull. soc. chim. D) 51, 838 A932).
2 S о m m e I e t, Ann. chim. phys. (8) 9, 489 A906).
3 G a u t h i e г, там же (8) 16, 304 A909).
'Geuther, Brockhoff, J. prakt. Chem. B) 7, 113 A873).
'Dreyfus, франц. пат. 671 103 [С. А. 24, 1867 A930)].
«Heintz, Ann. 129, 40 A864).
'Henry, Ber. 4, 706 A871).
8 С u r t i u s, J. prakt. Chem. B) 38, 424 A888).
•Soramelet, Ann. chim. phys. (8) 9, 501 A906).
2. Примечания
1. Для работы пригоден спирт, абсолютированный негашеной
известью.
2. Хлоруксусную кислоту надо прибавлять довольно быстро,
так, чтобы раствор кипел.
39 Сборник
УКАЗАТЕЛЬ СИНТЕЗОВ ПО ТИПАМ РЕАКЦИЙ
УКАЗАТЕЛЬ СИНТЕЗОВ ПО ТИПАМ РЕАКЦИЙ
В этом указателе большинство препаратов, которые входят в состав настоя-
щего сборника, отнесено к какому-либо определенному типу реакций; в указатель
включены только такие препараты, которые можно с достаточной определен-
ностью классифицировать по намеченным рубрикам. Как типы реакций, так
и препараты в пределах каждого типа расположены в алфавитном порядке.
АЛКИЛИРОВАНИЕ (см. также
Гринъяра реакция, замещение, кон-
денсация, Фриделя-Крафтса реак-
ция).
Вензилфталимид 84
Вератровый альдегид 142
симм. - Дибензои лдиметилгидра-
зин 203
Дифенилметан 223
Метилгидразин сернокислый
319
S-Метилизотиомочевина серно-
кислая 326
Метоксиацетонитрил 343
Пеларгоновая кислота 406
н.-Пропилсульфид 422
Этиловый эфир а-ацетоглутаро-
вой кислоты 581
Этиловый эфир ацетоянтарной
кислоты 580
Этиловый эфир вторично-бутил-
малоновой кислоты 318
Этиловый эфир изопропилма-
лоновой кислоты 108
Этиловый эфир метилмалоновой
кислоты 588
N-Этил-.м-толуидин 605
АЛКОГОЛИЗ.
Бутиловый эфир олеиновой кис-
лоты 402
АРИЛИРОВАНИЕ.
о-Нитродифениловый эфир 371
л-Нитродифениловый эфир 370
2-Фенилпиридин 517
АЦИЛИРОВАНИЕ (см. также заме-
щение металлов на другие группы, К).
Ацетилбензоин 68
Ацетилглицин 69
Вензгидроксамовая кислота 87
е-Бензоиламинокапроновая
кислота 90
Галлацетофенон 147
Гиппуровая кислота 158
силш.-Дибензоилгидразин 202
Октаацетат а-целлобиозы 398
Холестерилацетат 196
БУВО-ВЛАНА ВОССТАНОВЛЕ-
НИЕ (см. восстановление, Е).
CO2R—CH2OH[Na + ROH].
ВОССТАНОВЛЕНИЕ (см. также за-
мещение галоида на водород, Д).
Восстановитель заключен в
скобки перед названием пре-
парата.
А. СН = СН — СН2СН2
[Каталитическое, Pt] N-Ацетил-
гексагидрофенилаланин 500
[Каталитическое, Pt] N-Ацетил-
феиилаланин 500
[Каталитическое, Pt] Дигидро-
холестерин 195
[Каталитическое, Pd] Тетра-
гидрофуран 445
[Р + Ш каталитическое, PtJ
di-p-Фенилаланин
Б. СНО-*СН2ОН
[СН2О — КОН]
спирт 462
[СН.,0 — КОН] Пипероииловый
спирт 462
[СН2О — КОН] - ^~~-—~«»
пол 461
[А!(ОС2Н5K]
спирт 485
В. С = О — СНОН
[Na + C2H5OH] 2-Гептанол 152
611
л-Толилкарби-
Трихлорэтиловый
OH]
М1
[Редуктаза] i-Пропиленгликоль
421
Миристиловый
Нонаметилен-
Г. С = О — CH2
[Zn 4- HCl] -у-Фенилмасляная
кислота 511
[Zn = Си 4- NaOH] Фталид 533
Д. CO2R — СН2ОН
[Na + С2Н5ОН] Гепта.четилен-
гликоль 170
[Na 4- С2Н5ОН] Декаметилен-
гликоль 170
[Na 4- С2Н5ОН] Цетиловый спирт
307
[Каталитическое, хромит меди]
Гексаметиленгликоль 149
[Na 4- С2Н5ОН] Лауриловый
спирт 306
[Na 4- С2Н5ОН]
спирт 307
[Na 4- С2Н5ОН]
гликоль 171
[Na 4- С2Н5ОН] Октадекамети-
ленгликоль 171
[Na 4- CdHeOH] Олеиловый спирт
401
[Na 4- С2Н5ОН] Тетрадекамети-
ленгликоль 111
[Na 4- С2Н5ОН] Тридекамети-
ленгликоль 171
[Na 4- С2Н6ОН] Ундекаметилен-
гликоль 171
[Na + С2Н5ОН] Ундецилсниловый
спирт 307
Е. C=NOH-~CH2NH2
[Na 4- С2Н5ОН] н.-Бутиламин 155
[Na + С2Н5ОН] вторичио-Бутил-
амин 155
[Na 4- С2Н5ОН] н.-Гептиламин
154
[Na 4- СаН5ОН]
амии 155
Циклогексил-
Ж. N =
[Na2S2O4] 1-Амино-4-нафтол со»
лянокислый 48
3. N = 0
NH,
со-
Бензиловый
[Na2S2O4] 1-Амино-2-нафтол со-
лянокислый 41
[Na2S2O4] 1-Амино-2-нафтол
лянокислый 41
[Zn 4- СН3СО2Н] Диметилгидра-
зин солянокислый 199
[Zn 4- СН3СО2Н] 2,4-Диметил-
3,5-дикарбоэтоксппиррол 209
[Zn 4- СН3СО2Н]з-Метил-а-фенил-
гидразин 337
И. N02— NH2
[Zn 4- Н2б] 2-Аминофлуорен 385.
[SnBr2 4- НВг] л-Вромбеизаль-
дегид 552
[SnCl2 4- НС1] Диаминодурол-
хлоростаннат 263
[Fe + HC1] 2,4-Диаминотолуол;
176
[Sn 4- НС1] 2,6-Дибром-4-амино-
фенол-хлоростаннат 186
[Fe 4- СН3СО Н] Ортаниловая;
кислота 403
[Sn 4- НС1] Урамил 491
[Zn4- NaOH]fl-Фeнилeндиaмин511
[SnCl2 4- HCl] л-Хлорбензаль-
дегид 551
К. Другие реакции восстановления
[Na3As03] [Декстроза] Азоксибен-
зол 12
[СН3СНОНСН3] Вензопинакон 96.
[Na 4- u30-C5HjjOH] Пимелино-
вая кислота 410
[Zn 4- СН3СОгН] Тиосалицило-
вая кислота 455
[SnCl2 4- HCl] N-Этил-л-толуи-
дин 605
ГАЛОГЕНИРОВАНИЕ.
А. Хлорирование
о-Нитробензолсульфонилхлорид
404 ''
а-Хлорантрахинои 549
л-Хлорбензальдегид 551
8-Хлор-2,4-динитротиофенол
561
S-Хлор-о-нитро-п-хлортиофенол
561
S-Хлор-о-нитротиофенол 560
Б. Бромирование
с-Бензоиламино-а-бромкапроно-
вая кислота 89
612
УКАЗАТЕЛЬ СИНТЕЗОВ ПО ТИПАМ РЕАКЦИЙ
УКАЗАТЕЛЬ СИНТЕЗОВ ПО ТИПАМ РЕАКЦИЙ
Бромаль 102
Бромацетон 103
л-Бромбензальдегид 105
а-Бромизовалериановая кисло-
та 107
tx-Бромизокапроновая кислота
108
а-Бром-и.-капроновая кислота
108
Броммезитилен 118
¦а - Бром - fi - метилвалериановая
кислота 108
о-Бромфенол 121
БрОмциан 123
.Дибензоилдибромметан 242
2,6-Дибром-4-нитрофенол 185
.2,4 - Диокси - 5 - бромбензойная
кислота 120
л-НитробензилбрОмид 368
силш.-ТрибрОмбензол 467
Триметилэтилендибромид 324
Фенацилбромид 116
SB. Иодирование
2,6-Дииод-л-нитрОанилин 198
л-Иоданилин 271
5-Иодантраниловая кислота 273
2-Иодтиофен 287
2-Окси-3,5-дииодбензойная кис-
лота 392
4-Окси-3,5-дииодбензойная кис-
лота 393
ГАЛОФОРМЕННАЯ РЕАКЦИЯ
fi-Нафтойная кислота 351
ГЕША РЕАКЦИЯ.
Флороацетофенон 531
ГИДРОЛИЗ. Подзаголовки иллю-
стрируют типы соединений, подвер-
гаемых гидролизу. Гидролизующий
агент заключен в скобки перед на-
званием препарата.
А. Азлактоиы
а-Ацетоамииокоричная кислота
72
[NaOH] а-Бензоиламинокоричная
кислота 497
[NaOH] 3,4-Диметоксифенилпи-
ровиноградная кислота 166
<<П-,Ч-Фенилаланин 496
S. Амиды кислот
[НС1] df-Лизина хлористоводород-
ная соль 308
[НС1] <П-Метионин 340
[HC1] dZ-fi-Фенилаланин 496
[HC1] а-Фенилэтиламин 523
В. Амииы
[NaOH] 1-Нитро-2-нафтол 379
[НС1] Фенилпировиноградная кис-
лота 518
Г. Ангидриды кислот
Итаконовая кислота 290
Мезаконовая кислота 312
Цитракоиовая кислота 570
Д. Белковые вещества
[НС1] d-Аргинин солянокислый 60
г-Гистидин солянокислый 160
[Панкреатин] /-Тирозин 476
[Панкреатин] ^-Триптофан 474
Е. Двугалондные производные
л-Бромбензальдегид 105
[H2SO4] л-ХлОрбензальдегид 554
Ж. Имиды
(Н,8О4]]--Аминомасляная кислота
37
[NaOH] 1-Метилтиол-З-фталимид-
пропан-3,3-дикарбоновая кис-
лота 341
[NaOH] 4-Нитрофталевая кислота
387
3. Имииы
Флороацетофенон 531
[H2SO4] Этиловый эфир а-фепил-
ацетоуксусной кислоты 596
И. Лактамы
[НС1] е-Аминокапроновая кислота
36
[H2SO4] г - Бензоиламинокапр'оио-
вая кислота 90
К. Нитрилы
[NaOH—Н2О2] Амид вератровой
кислоты 32
[NaOH—Н202]Амид-о-толуиловой
кислоты 20
[Н28О4]т-Амин0масляная кисло-
та 37
NaOH] Малоновая кислота 309
H2SO4] Метилбензилкетон 313
H2SOi] о-Толуиловая кислота
462
[H2SO4] л-Толуиловая кислота
463
[NaOH] Феиилнитрометан 513
[НС1] Этилового эфира глицина
хлористоводородная соль 577
[КОН] Этиловый эфир н.-триде-
циловой кислоты 593
Л. Сложные эфнры
[H2SO4] л-Бромбензальдегид 552
[КОН] а-Бромизовалериановая
кислота 107
[КОН] а-Бромизокапроновая кис-
лота 108
[КОН] а-Бром-н-капроновая кис-
лота 108
[КОН] а-Бром-р-метилвалериано-
вая кислота 108
[NaOH] Гомовератровая кислота
[КОН] 2,6-Диметилпиридин 214
[КОН] 2,4-Диметилпиррол 216
[КОН] н.-Капроновая кислота
319, 408
[КОН] р-Метилвалериановая кис-
лота 317
NaOH] 6-Метилурацил 335
КОН] Пеларгоновая кислота -106
NaOH] Пимелиновая кислота 410
НС1] Хелидоновая кислота 543
КОН] Эруковая кислота 575
NH4OH] Этиловый эфир бензоил-
уксусной кислоты 584
М. Углеводы
[H2SO4-(CH3COJO] Октаацетат
целлобиозы 398
- [NaOCH3] Целлобиоза 564
Н. Шиффа основания
[НС!] d-Аргинин солянокислый 60
[НС1] 2,4-Динитробензальдегид
224
[НС1] Этилового эфира глицина
хлористоводородная соль 577
О. Другие реакции гидролиза
[НС1] Аллиламин 25
[H2SO4] Ацетонилацетон 219
[H2SOJ о-Бромфенол 121
[H2SO4] (П-Глицериновый альде-
гид 163
[НС1] енлш.-Диметилгидразин-
дигидрохлорид 202
[Na2CO3] Ортамиловая кислота
403
ГРИНЬЯРА РЕАКЦИЯ.
н.-Амилбензол 30
н.-Гексальдеги^ 295
Ди-н.-бутилкарбинол 193 .
2,4-Диметил-5-карбоэтоксипир-
рол 211
Изодурол 259
Метилизопропилкарбинол 321
а-Нафтойная кислота 348
н.-Пептап 408
Три-н.-амилкарбинол 490
Три-н.-бутилкарбинол 490
Три-н.-пропилкарбинол 490
Трифенилэтилен 483
Этиловый эфира-нафтойиой кис-
лоты 592
ДЕГИДРАТАЦИЯ. Применяемые ре-
агенты заключены в скобки перед
названиями препаратов.
[H2SOJ Аконитовая кислота 15
2-Амино-4-метилтиазол 40
Бензальазин 319
Бензилгидразон 506
Бензилиденаргинин 60
Бензимидазол 85
Бемзоиленмочевина 93
Г(СН3СОJО] Вератронитрил 145
2,5-ДиметилпиррОл 218
2,4-Динитробензальдегид 224
4,5-Дифенилглиоксалон 232
[нагревание] Итаконовый ан-
гидрид 290
2-Метилбензимидазол 80
[НС1] 6-Метилурацил 335
Нитрил малоновой кислоты 355
Оксим бензофенона 369, 393
Оксим циклогексанона 90, 396
Оксим энантола 394
[НС1] симм.-Тритиан 479 '
[H2SO4] Трифенилэтилен 483
Хелидоновая кислота 543
[Н3РО4] Циклогексен 567
[Н3РО4] Циклопентен 569
[СН3СОС1] [РОС13] Янтарный
ангидрид 56
ДЕГИДРОГЕНИЗАЦИЯ (см. также
окисление). Применяемые реагенты
заключены в скобки перед назва-
нием препарата.
[S] 1,2-Нафталевый ангидрид 347
[HgO] Фенилбе'нзоилдиазометан
506
[S] Фенокстии 529
ДЕКАРБОКСИЛИРОВАНИЕ.
tz-Бромизовалериановая кисло-
та 107
сс-Бромизокапроновая кислота
108
614
УКАЗАТЕЛЬ СИНТЕЗОВ ПО ТИПАМ РЕАКЦИЙ
УКАЗАТЕЛЬ СИНТЕЗОВ ПО ТИПАМ РЕАКЦИЙ
615
сс-Бром-н.-капроновая кислота
108
а - Бром - Э - метилвалериановая
кислота 108
4-Бромрезорцин 120
2,6-Диметилпиридин 214
2,4-Диметилпиррол 216
к.таконовый ангидрид 290
н.-Капроновая кислота 319, 408
Метилбензилкетон 313
В-Метилвалериановая кислота
317
di-Метионип 340
Пеларгоновая кислота 406
Фенилнитрометан 513
ДИАЗОТИРОВАНИЕ.
A. Получение соли диазония
Двойная соль хлористого р-на-
фтилдиазония и сулемы 558
4,4-Дифенил-бис-диазоиия бо-
рофторид 245
о-Нитробензолдиазония боро-
фторид 228
л-Нитробензолдиазония боро-
фторид 227
Б. Реакция сочетания
1-Амино-2-нафтол солянокис-
лый 41
1-Амино-4-нафтол солянокис-
лый 48
Диазоаминобензол 172
Сочетание о-толидина и Чикаго-
кислоты 434
B. Восстановление азогруппы
1-Амино-2-нафтол солянокис-
лый 41
1-Амино-4-нафтол солянокис-
лый 48
Г. Замещение группы диазония
1. На Вг
.м-Бромбеизальдегид 552
2. На С1
jw-Хлорбензальдегид 551
3. На F
4,4'-Дифтордифенил 244
Фторбензол 537
л-Фторбензойная кислота 534
4. На Н
л-Иодоензойная кислота 276
шлш.-Трибромбензол 467
5. На [
Иодбензол 278
л-Иодфенол 288
1, 2,3-Трииод-5-нитробензол
469
6. На. NO,
о-Динитробензол 228
п-Динитробензол 227
7. На Se
Дифенилселен 237
Ди-.м-толилселеи 239
Ди-о-толилселен 240
Ди-п-толилселен 240
8. Прочие реакции замещения
группы диазония
Ди-р-нафтилртуть 222
Тиосалициловая кислота 455
Фениларсиновая кислота 501
Хлористая ji-нафтилртуть 557
ЗАМЕЩЕНИЕ (см. также Гриньяра
реакция, дшзопшровате). Подзаго-
ловки иллюстрируют типы реакции
замещения, причем X представляет
собою галоид.
A. ОН на Вг
р-Вромдибутиламиноэтила бро-
мистоводородная соль 127
Р-Бромдипропиламипозтила
бромистоводородная соль 127
р-Бромдиэтиламиноэтила броми-
стоводородная соль 127
Бромистый вторично-бутл 115
Бромистый н.-геитил 114
Бромистый н.-додецил 112
Бромистый изобутил 114
Бромистый изопропнл 115
Бромистый октадецил 114
Бромистый иентаэритрит 443
Бромистый н.-пропил 115
Бромистый тетрадецил 114
Бромистый циклогексил 114
fi-Бромэтиламина бромистово-
дородная соль 126
а,г-Дибромгидрнн глицерина
183
Б. ОН на С1
Хлористый дезил 559
{S-Хлорэтилметилсульфид 562
B. ОН на I
Йодистый н.-амил 284
Йодистый н.-бутил 284
Йодистый н.-гексадецил 280
Йодистый изоаиил 284
Йодистый изобутил 289
Йодистый метил 281
Йодистый н.-пропил 284
Йодистый этил 284
Г. X на NH, или на замещенные
аминогруппы
Гиппуровая кислота 158
di-s-Дибензоиллизин 308
2,4-Динитроанилин 223
2,4-Динитрофенилгидразин 229
Я-Диэтиламиноэтиловьш спирт
248
Метилиминодиуксусная кислота
327
Таурин 441
N-Фенилантраниловая кислота
18
Этиловый эфир N-метилкар-
баминовой кислоты 587
Д. X на CN
Малоновая кислота 309
Этиловый эфир н.-тридециловой
Кислоты 593
Е. X на Н
н.-Гексадекан 148
5-Метилфурфурол 338
Ж. X ва металлы
Трифенилметклиатрий 481
Цинкди-н.-бутил 252
Цинкдиизоамил 252
Цинкди-н.-пропил 252
Цинкдиэтил 249
3. X на SO3H
fi-Бромэтансульфокислоты на-
триевая соль 124
Таурин 441
И. X на другие группы
Ацетол 74
Дифенилметаи 233
Дифенилтрикетон 242
Изопропилтиоцианат 268
р-Оксиэтилметнлсульфед 396
Тиодигликоль 453
К' Металлои иа другие группы
¦f-Амнномасляная кислота 37
Диэтиловый эфир 1-метилтиол-
3 - фталимидопропан - 3,3 - ди-
карбоиовой кислоты 341
Йодистый метил 281
(З-Оксиэтнлметилсульфид 396
Трикарбометоксиметан 471
2-Фепилпиридин 517
Этиловый эфир бензоилдиметн л-
уксусной кислоты 582
Этиловый эфир бензоилуксус-
ной кислоты 584
Этиловый эфир а, а, -[^-тетраме-
тилацетоуксусной кислоты
584
КАРБОКСИЛИРОВАНИЕ.
р-Резорциловая кислота 430
КАННИЦЦАРО РЕАКЦИЯ.
(см. восстановление, Б. СНО-.
-СН3ОН [СН2О + КОН]).
КЛЕММЕНСЕНА ВОССТАНОВЛЕ-
НИЕ (см. восстановление, Г).
С==О — СН2 [Zn + HC1]
КНОРРА СИНТЕЗ ПИРРОЛА.
2,4-Диметил-3,5-дикарбоэтокси-
пиррол 209
КОНДЕНСАЦИЯ (см. также а.тили-
рование, восстановление, Гриньяра
реакция, дегидратация, диазотиро-
вание, перегруппировки, присоеди-
нение, Фридем-Крафшса реакция).
Термин «конденсация» применяется
здесь в ограничительном смысле и
относится к тем реакциям, интрамо-
лекулярным или интермолекуляр-
ным, которые ведут к образованию
связи углерод-углерод, обычно в
результате отщепления простейшей
неорганической молекулы.
Подзаголовки иллюстрируют
типы образующихся соединений.
Применяемые реагенты заключены
в скобки перед названием препа-
рата.
А. Гетероциклические соединения
[CH3CO,Na-(CH3COJO] Азлактон
а-ацетоамииокоричной кислоты
72
[CH3CO2Na-(CH3COJO] Азлактон
о-бензоиламино-?-C,4-диметок-
сифенил)-акриловой кислоты 11
[CH3CO2Na-(CH3COJO] Азлактон
сс-бензоиламинокоричной кис-
лоты 497
[H,SOt] Акридон 18
[(C2H5JNH1 1,4-Дигидро-3,5-ди-
карбоэтокси-2,6-диметилпири-
дин
[Н„804]3,4-Дигидро-1,2-нафтале-
вой кислоты ангидрид 53
2,4-Диметил-3,5-днкзрбоэтокси-
пиррол 209
616
УКАЗАТЕЛЬ СИНТЕЗОВ ПО ТИПАМ РЕАКЦИЙ
УКАЗАТЕЛЬ СИНТЕЗОВ ПО ТИПАМ РЕАКЦИЙ
617
Б. Кстоепирты
Na
Na'
Na
Na
Бутироин 140
Изобутиронн 141
Пивалоин 141
Пропионоин 141
В. Кстоэфиры
[NaOC2H5] 5,5-Диметил-1,3-цик-
логександион 220
[Nal 2-Карбоэтоксициклопента-
нон 297
[NaOC2H5] Этиловый эфир 2-кето-
циклогексилглиоксиловой кис-
лоты 411
[NaOC2H5] Этиловый эфир фенил-
малоновой кислоты 599
[NaOCjHj] Этиловый эфир фенил-
цианпировинограднон кислоты
600
[NaOCaH5] Этиловый эфир хели-
доповой кислоты 544
[NaOCsH5] Этиловый эфир этокса-
лилпропионовой кислоты 604
Г. Другие реакции конденсации
fCH3COoNa] Бензальфталид 80
[Н2804]"Вснзантрон 81
[NaOH] Дибензальацетон 179
[(СН3СОJО — РЬО] 1,4-Дифенил-
бутадиен 231
[Си2С13 — А1С1„] л-Толуиловый
альдегид 464
[NaOC2H5] а-Фенилацетоацето-
нитрил 503
[Na2CO3] Этиловый эфир этилен-
тетракарбоновой кислоты 601
ЛЕУКАРТА РЕАКЦИЯ.
сс-л-Бромфенилэтиламин 525
а-л-Ксенилэтиламин 525
а-(Р-Нафтил)-этиламин 525
а-л-Толуилэтиламин 525
а-Фенилэтиламин 523
сс-л-Хлорфенилэтиламин 525
НЕНЦКОГО РЕАКЦИЯ.
Галлацетофенон 147
НИТРОВАНИЕ.
Динитродурол 262
1-Нитро-2-ацетилнафтиламин
.м-Нитроацетофенон 361
Нитробарбитуровая кислота
365
Нитромезитилен 377
2-Нитротиофен 381
2-Нитрофлуорен 385
4-Нитрофталимид 389
НИТРОЗИРОВАНИЕ.
2,4 - Диметил - 3,5 - дикарбоэто-
ксипиррол 209
Монооксим диацетила 206
Нитрозодиметиламин 199
л-Нитрозодиметиланилин соля-
нокислый 225
л-Нитрозодиэтиланилин 226
N-Нитрозометиланилии 372
Нитрозометилмочевина 373
Нитрозометилуретан 376
N-Этил-ж-толуидин 605
ОКИСЛЕНИЕ (см. также дегидрогени-
зация). Подзаголовки иллюстри-
руют тип окисления. Окислитель
заключен в скобки перед названием,
препарата.
A. СН3— СНО
СгО3] л-Бромбензальдегид 105
СгО3] л-НитробенЗальдегид 365
SeO2] Фенилглиоксаль 507
Б. СН3-*СО2Н
[КМпО4] Хлорбензойная кислота
B. СН2ОН — СНО
[К2Сг20-] Пропионовый альдегид.
424
Г. СНОН — С = О
[K3Fe(CNN] 1-Метил-2-пиридО№
[СгО3] Холестанон 563
Д. СНО — СО2Н
[КМпО4] н.-Гептановая кислота
571
[КМпО4] Пиперониловая кислота
417
Е. С=С—С02Н
[КМпО4] Азелаиновая кислота 9
Ж. Другие реакции окисления
КМпО4] Аллантоин 23
NaCIO3] Антрахинон 547
КМпО4] Ацеталь й/-глицерино-
вого альдегида 65
[СгО3] сс-Гидриндон 156
[Н202]Гомовератровая кислота 164
[NaOCl] 2,6-Дибромхинон-4-хлор-
имид 187
[HNO3] 3,5-Дикарбоэтокси-2,6-
диметилпиридин 215
FeCl.,] Дурохинон 262
NaOCllp-Нафтойная кислота 348
FeCL] 1,2-Нафтохинон 353
H»SO4] Пиромеллитовая кислота
418
[КМпО4] Фтальальдегидная кис-
лота 27
[NaClO3] Фумаровая кислота 541
NaC103j Хинон 545
[HNO3—HC1] 8-Хлор-2,5-ди-
нитротиофеиол 569
ОТЩЕПЛЕНИЕ НХ. Реагент, приме-
няемый для отщепления, заключен
в скобки перед названием препа-
рата.
[КОН] Ацеталь акролеина 64
[ КОН] Ацетилендикарбоновая
кислота 71
[КОН] Фенилпропиоловая кисло-
та 520
[Са(ОНJ] Эпибромгидрин 573
[Са(ОНJ] Эпихлоргидрин 574
ПЕРЕГРУППИРОВКИ.
р-Аланин 20
Аллантоин 23
4-Аминовератрол 32
Ацетилметилмочевина 374
е-Бензоиламипокапроновая
кислота 90
р-Бензопинаколин 97
2-Кетогексаметиленимин 306
Мезаконовая кислота 312
Мвтилизопропилкетон 321
1-Метил-2-пиридон 333
п-0ксибензойная кислота 390
о- и л-ПропиофенОл 426
несилш.-Хлорангидрид-о-фтале-
вой кислоты 547
Цитраконовый ангидрид 570
ПИРОЛИЗ.
1,3-Бутадиен 128
Метилбензилкетон 313
Сукцинимид 430
Этиловый эфир 2-кетогексагид-
робензойной кислоты 412
Этиловый эфир метилмалоновой
кислоты 588
Этиловый эфир фенилмалоноиой
кислоты 599
ПРИСОЕДИНЕНИЕ (см. также вос-
становление, гидролиз, Гринъяра
реакция). Реагенты, которые при-
соединяются, заключены в скобки
перед названием препарата.
A. к С = С
[Cl—CHC1J несилш.-Гептахлор-
пропан 153
[Вг— Вг] 1,2-Дибромциклогексан
189
[Вг—Вг] а,р-Дибромянтарная
кислота 191
[Cl—CI] а-Хлоргидринден 157
[Н—С6Н6] Циклогексилбензол
576
[Вг—Вг] Этиловый эфир,а,Р-ди-
бром-р-фенилпропиновой кис-
лоты 586
Б. к С = N
[СН3 — SO4CH3] 1 -Метил-2-пири-
дон 333
B. к С = О
[Н — CN1 Метоксиацетонитрил'
343
[Н = CN] Циангидрин ацетона 566»
Г. к С = С-С = О
[Н — СН(СО2С2Н6J] 5,5-Диметил-
1,3-циклогександиОн 220
[Н—СбН6] р, р-Дифенилпропио-
фенон 235
[Н — CN] а-Фенил-3-бензоилпро-
пионитрил 358
Д. к C=N
[Н — С6Н2(О Н)8 ] Ф лороацетофе-
нон 531
[Н — ОС2Н5] Этиловый эфир а-фе-
нилацетоуксусной кислоты 596-
Е. к N = C = O
[Н — NHCeH5] Бензоиленмоче-
вина 93
РАЗДЕЛЕНИЕ НА ОПТИЧЕСКИЕ
ИЗОМЕРЫ. J
й- и /-сс-Фенилэтиламин 526
СУЛЬФИРОВАНИЕ.
Антрахинонсульфокислоты ка-
лиевая соль 58
о-Бромфсиол 121
Фенантрен-2-сульфокислоты ба-
риевая соль 496
Фенантрен-3-сульфокислоты ка-
лиевая соль 494
ТИОЦИАНИРОВАНИЕ.
л-Тиоциандиметиланилин 431
618
УКАЗАТЕЛЬ СИНТЕЗОВ ПО ТИПАМ РЕАКЦИЙ
•ФРИДЕЛЯ-КРАФТСА РЕАКЦИЯ.
2-Ацетотиенон 76
Ацето-тг-цимол 77
7-Бензоилмасляная кислота 96
[i-Бензоилпропионовая кислота
95
Гексаметилбензол 256
Дезоксибенэоин 167
1,4-Дибензоилбутан 181
Дифенилсульфид 214
Дурол 253
Пентаметилбензол 254
а-Тетралон 448
Фенилтиенилкетон 522
Хлористый трифенилселен 239
ШТРЕКЕРА СИНТЕЗ.
а-Аминоизомасляная кислота
34
ЭТЕРИФИКАЦИЯ (см. также ацили-
рование).
н.-Амиловый эфир борной кис-
лоты 134
н .-Амилфосфат 139
н.-Бутилнитрит 131
н.-Бутиловый эфир борной кис-
лоты 133
н.-Бутнлсульфат 135
.н.-Бутилсульфит 136
н.-Бутилфосфат 138
Изопропиловый эфир молочной
кислоты 266
Метилнитрат 329
Метиловый эфир гомовератро-
вой кислоты 164
Метиловый эфир щавелевой кис-
лоты 331
Моноэтиловый эфир адипиновой
кислоты 346
Моноэтиловый эфир себацино-
вой кислоты 345
н.-Пропилсульфат 136
н ,-Пропилфосфат 139
Этиловый эфир адипиновой кис-
лоты 578
Этиловый эфир л-аминобензой-
ной кислоты 534
Этиловый эфир пимелиновой
кислоты 415
Этиловый эфир себациновой
кислоты 345
Этиловый эфир н.-тридецило-
вой кислоты 593
Этиловый эфир -[-фенилмасля-
ной кислоты 54
Этиловый эфир хлоруксусной
кислоты 580
Этиловый эфир этоксиуксусной
кислоты 608
УКАЗАТЕЛЬ СИНТЕЗОВ ПО ТИПАМ СОЕДИНЕНИЙ
Каждый препарат отнесен к тому или иному типу в зависимости от того,
какая группа вводится в описанном синтезе. Так, например, .и-бромнитро-
бензол, получаемый бромированием нитробензола, был бы помещен в разделе
галоидопроизводиых, а не в разделе нитросоединений. Если нет особых указа-
ний, двойная углерод-углеродная связь не рассматривается как замести-
тель. Соли помещены при соответственных кислотах и основаниях.
АЗОКСИСОЕДИНЕНИЯ.
Азоксибензол 12
АЗОСО ЕДИНЕНИЯ.
Диазоаминобензол 172
Продукт сочетания о-толидина
и Чикаго-кислоты 434
АЛЬДЕГИДЫ.
А. Незамещенные альдегиды
н.-Капроновый альдегид 295
Пропионовый альдегид 424
л-Толуиловый альдегид 464
Б. Замещенные альдегиды
л-Бромбензальдегид 105
(//-Глицериновый альдегид 163
2,4-Динитробензальдегид 224
л-Нитробензальдегид 366
л-Хлорбензальдегид 554
АМИДЫ.
Амид о-толуиловой кислоты 20
4-Аминовератрол 32
Ацетилглицин 69
АМИНОКИСЛОТЫ (см. кислоты).
АМИНЫ.
А. Незамещенные амнны
Аллиламин 25
2-Аминофлуорен 385
н.-Бутиламин 155
вторичио-Вутлампн 155
н.-Гептиламин 154
Диаминодурол хлоростаннат263
2,4-Диаминотолуол 176
а-л-Ксенилэтиламин 525
а-(Р-Нафтил)-этиламин 525
сс-л-толилэтиламин 525
о-фенилендиамин 511
й- и i-cc-фенилэтиламин 526
(П-а-фенилэтиламин 523
Циклогексиламин 155
N-Этил-л-толундин 605
Б. Замещенные амины
4-Аминовератрол 32
1-Амино-2-нафтол солянокис-
лый 41
1-Амино-4-нафтол солянокис-
лый 48
(П-е-Бснзоиллизин 308
сс-л-Бромфенилэтиламин 525
2,6-Дибром-4-аминофенол хло-
ростаинат 187
2,4-Динитроанилин 223
р-Диэтиламиноэтиловый спирт
248
<П-Лизин 308
й/-Лизина хлористоводородная
соль 308
Метилиминодиуксусная кис-
лота 327
Ортаниловая кислота 403
Урамил 491
а, л-Хлорофенилэтиламин 525
АНГИДРИДЫ КИСЛОТ.
Ангидрид янтарной кислоты 56
3,4-Дигидро-1,2-нафталевой ки-
слоты ангидрид 53
Итаконовый ангидрид 290
620
УКАЗАТЕЛЬ СИНТЕЗОР ПО ТИПАМ СОЕДИНЕНИЙ
1,2-Нафталевый ангидрид 347
Цитраконовый ангидрид 570
АЦЕТАЛИ.
Ацеталь акролеина 64
Ацеталь fi-хлорпропионового
альдегида 67
БЕЛКИ.
Казеин 292
ГАЛОИДОАНГИДРИДЫ КИСЛОТ.
Хлорангидрид о-нитробензол-
сульфокислоты 404
Хлорангидрид фталевой кис-
лоты сими, и несимм. 547
ГАЛОИДОПРОИЗВОДНЫЕ УГЛЕ-
ВОДОРОДОВ.
A. Моногалоидные алкилы
1. Хлориды
ос-Хлоргиндриндон 157
2. Бромиды
вторично-Бромистый бутил 115
Бромистый н.-гептил 114
Бромистый н.-додеци л 112
Бромистый изобутил 114
Бромистый изопропил 115
Бромистый октадецил 114
Бромистый н.-пропил 115
Бромистый тетрадецил 114
Бромистый циклогексил 114
3. Иодиды
Йодистый н.-амил 284
Йодистый н.-бутил 284
Йодистый н.-гексадецил 280 }
Йодистый изоамил 284
Йодистый изобутил 289
Йодистый метил 281
Йодистый н.-пропил 284
Йодистый этил 284
Б. Моногалоидные арилы
Броммезитилен 118
Иодбеизол 278
Фторбензол 537
B. Полигалоидные производные
Бромистый пентаэритрит 443
несилш.-Гептахлорпропан 153
1,2-Дибромциклогексан 189
Дифенилдихлорметан 452
4,4'-Дифторбифенил 244
Йодистый пентаэритрит 444
енлш.-Трибромбензол 467
Триметилэтилендибромид 324
ГАЛОИДОПРОИЗВОДНЫЕ ДРУ-
ГИХ КЛАССОВ.
A. Галоидоальдегиды
Бромаль 102
л-Бромбензальдегид 552
,/И-Хлорбензальдегид 551
Б. Галоидоамины
,3-Бромдибутиламиноэтила бро-
мистоводородная соль 127
р-Бромдиметиламииоэтила бро-
мистоводородная соль 127
jB-Бромдипропиламиноэтила
бромистоводородная соль 127
[З-Бромдиэтиламиноэтила бро-
мистоводородная соль 126
[З-Бромэтиламина бромисто-
водородная соль 126
л-Иоданилин 271
B. Галоидокетоиы
Бромацетон 103
Бромистый фенацил 116
Дезил хлористый 559
Дибензоилдибромметан 242
Г. Галондонитросоедииеиия
л-Нитробензилбромид 368
1,2,3-Трииод-5-нитробензол 469"
Д. Галоидоспирты игалоидофенолы
Бромрезорцин 120
о-Бромфеиол 121
а у-Дибромгидрин глицерина
183
гг-Иодфенол 288
Тетраметиленхлоргидрин 4501
Е. Прочие галоидопроизводные
а-Бромизовалериановая кис-
лота 107
а-Бромизокапроновая кислота
108
а-Бром-н.-капроновая Кислота
108
а-Бром-;!-метилвалериановая
кислота 108
2,6-Дибром-4-иитрофенол 185
а,р-Дибромянтарная кислота
191
2,6-Дииод-л-нитроанилин 198
2,4-Диокси-5-брОмбензойнаяки-
слота 120
5-Иодантраниловая кислота
273
2-Иодтиофен 278
2-Окси-3,5-дииодбензойная кис-
лота 392
УКАЗАТЕЛЬ СИНТЕЗОВ ПО ТИПАМ СОЕДИНЕНИЙ
621
л-Фторбензойная кислота 534
р-Хлорантрахинон 549
Р-Хлорэтилметилсульфид 562
Этиловый эфир а.Э-дибром-?-
фснилпропионовой кислоты
586
ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИ-
НЕНИЯ.
A. Азотсодержащие гетероциклы
Акридон 18
Аллантоин 23
Барбитуровая кислота 79
Бензимидазол 85
Бензоиленмочевииа 93
1,4-Дигидро-3,5-дикарбоэтокси-
2,6-диметилпиридин 215
3,5-Дикарбоэтокси-2,6-диметил-
пиридин 215
2,4- Димети л-3,5-дикарбоэтокси-
пиррол 209
-2,4-Дииетил-5- карбоэтоксипир-
. рол 211
2,6-Диметилпиридин 214
2,4-Диметилпиррол 216
2,5-Диметилпиррол 218
4,5-Дифенилглиоксалон 232
2-Кетогексаметиленимин 306
2-Метилбензимидазол 86
1-Метил-2-пиридон 333
6-Метилурацил 335
2-Фенилпиридин 517
S. Кислородсодержащие гетеро-
циклы
Бензальфталид 80
5-Метилфурфурол 338
Тетрагидрофуран 445
Фталид 533
Хелидоновая кислота 543
Эпибромгидрин 573
Эпихлоргидрин 574
B. Серусодержащие гетероциклы
Тиофеи 458
снлш.-Тритиан 479
Г. Гетероциклы, содержащие два
различных гетероатома
Азлактон а-ацето- аминокорич-
ной кислоты 72
Азлактон а-бензоиламино-fi-
C,4-диметоксифенил)-акр ило-
вой кислоты 11
Азлактон ос-бензоиламинокорич-
ной кислоты 497
2-Амино-4-метилтиазол 40
Фенокстин 529
ГИДРАЗИН И ЕГО ПРОИЗВОД-
НЫЕ.
Бензальазин 319
Бензилгидразои 506
Бетаингидразид солянокислый
100
силш.-Дибензоилгидразин 202
симм .-Дибензоилдиметилгид-
разин 203
нгсилш.-Диметилгидразин 199
сил<л<.-Диметилгидразин диги-
дрохлорид 202
несимм. -Диметилгидразин со-
лянокислый 199
2,4-Динитрофенилгидразин 229
Метилгидразии сернокислый
319
а-Метил-з-фенилгидразин 337
ГИДРОКСИЛАМИН И ЕГО ПРОИЗ-
ВОДНЫЕ.
Бензогидроксамовая кислота
87
Бензогидроксамовой кислоты
калиевая соль 87
Вератральдоксим 146
Диметилглиоксим 205
Изонитрозопропиофенон 264
Моноксим диацетила 206
Оксим бензофенона 393
Оксим циклогексана 90
Оксим энантола 394
ДИАЗОНИЯ СОЛИ.
Двойная соль хлористого C-
нафтилдиазония н сулемы 558
4,4' - Дифенилен-бис-диазония
борофторид 245
гг-Карбоэтоксифенилдиазония
борофторид 535
о-Нитробензолдиазония боро-
фторид 228
л-Нитробеизолдиазония боро-
фторид 227
ДИАЗОСО ЕДИНЕНИЯ.
Диазометан 174
Фенилбензоилдиазометан 506
ИЗОЦИАНАТЫ.
л-Нитрофенилизоцианат 383
ИМИДЫ.
Бензилфталимид 84
622
УКАЗАТЕЛЬ СИНТЕЗОВ ПО ТИПАМ СОЕДИНЕНИЙ
2,6-Дибромхинон-4-хлоримид
187
Сукцинимид 430
имины.
Бензилиденаргинин 60
Дифенилметаиимингидрохлоиид
269
КЕТОНЫ.
A. Монокетоны
Ацето-л-цимол 77
Бензантрон 81
?-Бензопинаколин 97
а-Гидриндон 156
Дезоксибензоии 167
р^-Дифенилпропиофенон 235
Метилбензилкетон 313
Метилизопропилкетон 329
а-Тетралон 448
Холестанон 563
Б. Поликетоны
Ацетонилацетон 219
1,4-Дибензоилбутан 181
5,5-Диметил- 1,3-циклогексан-
дион 220
Дифенилтрикетон 242
Дифенилтрикетонгидрат 242
B. Замещенные кетоны
Оксикетоны
Галлацетофенон 147
о- и л-Пропиофенол
Флороацетофеноы
Прочие замещенные кетоны
2-Ацетотиенон 76
Дибензальацетон 179
а-Фенилацетоацетонитрил 503
Фенилтиенилкетон 522
КИСЛОТЫ.
А. Незамещенные кислоты
1. Одноосновные
и.-Капроновая кислота 319
В-Метилвалериановая кислота
317
а-Нафтойная кислота 348
ti-Нафтойная кислота 315
Пеларгоновая кислота 406
о-Толуиловая кислота 462
л-Толуиловая кислота 463
у-Фенилмасляная кислота 511
Фенилпропиоловая кислота 520
Энантовая кислота 571
Эруковая кислота 575
2. Миогоосновные
Азелаиновая кислота 9
Аконитовая кислота 15
Ацетилендикарбоновая кисло-
та 71
Итаконовая кислота 290
Малоновая кислота 309
Малоновой кис-юты кальцие-
вая соль 310
Мезаконовая кислота 312
Пимелиновая кислота 410
Пиромеллитовая кислота 418
Фумаровая кислота 191
Цитраконовая кислота 570
Б. Замещенные кислоты
1. Аминокислоты
р-Аланин 20
а-Аминоизомасляная кислота
34
?-Аминокапроновая кислота
36
7-Аминомасляная кислота 37
d-Аргинин гидрохлорид 60
N-Ацетилгексагидрофенилала-
нин 500
N-Ацетилфенилаланин 500
а-Ацетоаминокоричная кисло-
та 72
Е-Бензоиламинокапроновая ки-
слота 90
а-Бензоиламинокоричная кис-
лота 497
Гиппуровая кислота 158
/-Гистидина хлористоводород-
ная соль 160
Метилиминодиацетиловая кис-
лота 327.
dZ-Метионин 340
Таурин 441
/-Тирозин 476
/-Триптофан 474
й/-р-Фенилаланин 496
N-Фенилантраниловая кислота
18
2. Галоидокислоты
Е-Бензоиламино-а-бромкапро-
новая кислота 89
Дихлоруксусная кислота 247
.м-Иодбензойная кислота 276
о-Хлорбензойная кислота 556
3. Кстокислоты
у-Бензоилмасляная кислота 96
fJ-Беизоилпропионовая кисло-
та 95
Фенилпировиноградная кис-
лота 51 &
УКАЗАТЕЛЬ СИНТЕЗОВ ПО ТИПАМ СОЕДИНЕНИЙ
623
4. Оксикислоты
л-Оксибензойная кислота 390
fj-Резорциловая кислота 440
5. Прочие замещенные кислоты
Альдегидофталевая кислота 27
Гомовератровая кислота 164
3,4 - Диметоксифенилпировино-
градная кислота 166
4-Нитрофталевая кислота 387
Пиперониловая кислота 417
Эгоксиуксусная кислота 607
МОЧЕВИНЫ ПРОИЗВОДНЫЕ.
Ацетилметилмочевина 374
МЫШЬЯК-ОРГАНИЧЕСКИЕ СО-
ЕДИНЕНИЯ.
Фениларсиновая кислота 501
НАТРИЙ-ОРГАНИЧЕСКИЕ СО-
ЕДИНЕНИЯ.
Натриевая соль фенилациии-
троацетонитрила 513
Натриевое производное этило-
вого эфира фталимидмалоно-
вой кислоты 340
Трифенилметилнатрий 481
НИТРИЛЫ.
Вератронитрил 145
Метоксиацетонитрил 343
Нитрил малоиовой кислоты 355
а-Фенил-3-бензоилпропионит-
рил 358
Циангидрин ацетона 566
НИТРОЗОСОЕДИН ЕНИЯ.
Нитрозодиметиламин 199
л-Нитрозодиметиланилин соля-
нокислый 225
л-Нитрозодиэтиланилин 226
N-Нитрозометиланилин 372
Нитрозометилмочевина 373
Нитрозометилуретан 376
НИТРОСОЕДИНЕНИЯ.
А. Незамещенные
о-Динитробензол 228
л-Динитробензол 227
Динитродурол 262
Нитромезитилеи 377
2-Нитрофлуорен 385
Фенилнитрометан 513
Б. Замещенные
1-Нитро-2-ацетипаминонафталин
359
.м-Нитроацетофенон 361
Нитробарбитуровая кислота
365
2-Нитротиофен 381
4-Нитрофталимид 389
ПРОСТЫЕ ЭФИРЫ
Вератровый альдегид 142
Метоксиацетонитрил 343
о-Нитродифениловый эфир 37Г
л-Ннтродифениловый эфир 370
ПРОТЕИНЫ (см. белки).
СЕЛЕН-ОРГАНИЧЕСКИЕ СОЕДИ-
НЕНИЯ.
Ди-.и-толилселен 239
Дифенилселен 237
Дифенилселен двухлористый:
239
Трифенилселен хлористый 239-
СЕРУСОДЕРЖАЩИЕ СОЕДИНЕ-
НИЯ (см. также гетероцикличе-
ские соединения).
1-Амино-2-нафтол-4-сульфокис-
лота 51
Антрахинонсульфокислоты ка-
лиевая соль 58
Р-Бромэтансульфокислоты на-
триевая соль 124
Дибензогидрилдисульфид 453
Дифенилсульфид 241
Диэтиловый эфир 1-метилтиол-
3-фталимидопропан-3,3-ди-
карбоновой кислоты 341
S-Метилизотиомочевина серно^
кислая 326
й/-Метионин 340
Морланда соль 443
,Ь-Оксиэтилметилсульфид 396
н.-Пропилсульфид 136
Рейнеке соль 342
Таурин 441
Тиобензофенон 452
р-Тиодигликоль 453
Тиосалициловая кислота 455
Фенантрен-2-сульфокислоты ба-
риевая соль 494
Фенантрен-3-сульфокислоты
калиевая соль 494
8-хлор-2,4-динитротиофенол 561
S-Хлор-о-нитротиофенол 560
СЛОЖНЫЕ ЭФИРЫ НЕОРГАНИ-
ЧЕСКИХ КИСЛОТ.
н.-Амиловый эфир борной кис-
лоты 134
.624
УКАЗАТЕЛЬ СИНТЕЗОВ ПО ТИПАМ СОЕДИНЕНИЙ
н.-Амилфосфат 139
н.-Бутилнитрит 131
н.-Бутиловый эфир борной кис-
лоты 133
н.-Бутилсульфат 135
н.-Бутилсульфит 136
«тлорично-Бутилфосфат 139
н.-Бутилфосфат 138
Изопронилтиоцианат 268
Метилнитрат 329
Метилнитрит 265
н.-Пропилсульфат 136
н.-Пропилфосфат 139
Этилнитрит 205
СЛОЖНЫЕ ЭФИРЫ ОРГАНИЧЕ-
СКИХ КИСЛОТ.
А. Незамещенные сложные эфиры
1. Одноосновных кислот
и.-Бутиловый эфир олеиновой
кислоты 402
Холестерилацетат 196
Этиловый эфир а-нафтойной кис-
лоты 592
Этиловый эфир н.-тридекановой
кислоты 539
Этиловый эфир -/-фенилмасля-
ной кислоты 55
2. Многоосновных кислот
Метиловый эфир щавелевой кис-
лоты 331
Трикарбометокснметан 471
Трикарбоэтоксиметан 472
Этиловый эфир адипиновой Кис-
лоты 578
Этиловый эфир вторито-бутк-
малоновой кислоты 31,8
Этиловый эфир изопропилмало-
новой кислоты 108
Этиловый эфир метилмалоновой
кислоты 588
Этиловый эфир пимелнновой
кислоты 455
Этиловый эфир себациновой
кислоты 17
Этиловый эфир фенилмалоновой
кислоты 599
Этиловый эфир этнлентетра-
карбоновой кислоты 601
•Б. Замещенные сложные эфиры
1. Галоидоэфиры
Бромбензальдиацетат 367
Этиловый эфир монохлоруксус-
ной кислоты 100
2. Кетоэфиры
Ацетилбензоин 68
2-Карбоэтоксициклопентанон
297
Этиловый эфир ацетоянтарной
кислоты 581
Этиловый эфир бензоилацето-
уксусной кислоты 584
Этиловый эфир бензоилуксуе-
ной кислоты 584
Этиловый эфнр 2-кетогексагид-
робензойной кислоты 412
Этиловый эфир монохлоруксус-
ной кислоты 100
Этиловый эфир а,яд,7-тетра-
метилацетоуксусной кислоты
584
Этиловый эфир а -фенилацето-
уксусной кислоты 596
Этиловыйэфир хелидоновой кис-
Лоты 544
Этиловый эфир этоксалилпро-
пионовой кислоты 604
3. Прочие сложные эфиры
Изопроииловый эфир молочной
кислоты 226
Метиловый эфир гомовератро-
вой кислоты 164
Нитробензальдиацетат 366
Этилового эфира глицина хло-
ристоводородная соль 577
Этиловый эфнр л-аминобензой-
ной кислоты 534
Этиловый эфир N-метилкарб-
аминовой кислоты 587
Этиловый эфир фенилциашшро-
виноградной кислоты 600
Этиловый эфир этоксиуксусной
кислоты 608
СПИРТЫ.
А. Незамещенные спирты
1. Первичные.
Бензиловый спирт 462
Гексаметиленгликоль 149
Гептаметиленгликоль 170
Декаметиленгликоль 170
Лауриловый спирт 306
Миристиловый спирт 307
Нонаметиленгликоль 171
Октадекаметиленгликоль 171
Олеиловый спирт 401
Тетрадекаметиленгликоль 111
л-Толилкарбинол 461
Тридекаметиленгликоль 171
Ундекаметиленгликоль 171
Ундециленовый спирт 307
Цетиловый Спирт 307
УКАЗАТЕЛЬ СИНТЕЗОВ ПО ТИПАМ СОЕДИНЕНИЙ
625
2. Первично-вторичные
/-Пропиленглпколь 421
3. Вторичные
Ди-н.-бутилкариинил 193
2-Гептанол 152
Дигидрохолестерии 195
Метилизопроггнлкарбннол 321
4. Третичные
Бензопинакон 96
Три-н.-амилкарбинол 490
Трй-н.-бутилкарбннол 490
Три-н.-гептилкарбинол 490
Три-н.-пропилкарбииол 490
Триэтилкарбинол 489
Б. Замещенные спирты
1. Кетоспирты
Ацетол 74
Бутироин 140
Изобутироин 141
Пиналоин 141
Пропионоин 141
2. Прочие замещенные спирты
Ацеталь (//-глицеринового аль-
дегида 65
Пиперониловый спирт 462
Трихлорэтиловый спирт 485
РТУТЬ-ОРГАНИЧЕСКИЕ СОЕДИ-
НЕНИЯ.
Ди-^-нафтилртуть 222
Хлористая fi-нафтилртуть 557
УГЛЕВОДЫ И ИХ ПРОИЗВОДНЫЕ
Октаацетат п-целлобиозы 398
Целлобиоза 564
УГЛЕВОДОРОДЫ.
А. Алифатические и алицикли-
ческие
1,3-Бутадиен 129
н.-Гексадекан 148
Пентан 408
Циклогексен 128
Циклопентен 569
Б. Ароматические
н.-Амилбензол 30
Гексаметилбензол 256
1,4-Дифенилбутадиен 231
Дифенилметан 233
Дурол 253
Изодурол 259
Пентаметилбензол 254
Трифенилэтилен 483
Циклогексилбензол 567
ФЕНОЛЫ.
1-Нитро-2-нафтол 379
Протокатеховый альдегид 428
хиноны.
АнТрахинон 547
Дурохинон 262
1,2-Нафтохинон 353
Хинон 545
ЦИНК-ОРГАНИЧЕСКИЕ СОЕДИ-
НЕНИЯ.
Ди-н.-бутилцинк 252
Диизоамилцинк 252
Ди-н.-пропилцинк 252
Диэтилцинк 249
40 Сборнш; № 2
УКАЗАТЕЛЬ ВЕЩЕСТВ ПО ФОРМУЛАМ
В этом указателе перечислены все препараты. Принятая в указателе
система та же, что и в Chemical Abstracts. Основные принципы се заключа-
ются в следующем: 1) все обозначения химических элементов в формулах
расположены в алфавитном порядке за исключением того, что в органических
соединениях на первом месте всегда стоит С, а непосредственно за ним Н,
если соединение содержит также и водород; 2) сами формулы расположены
по возрастающему числу атомов углерода; при равном числе атомов углеро-
да — по возрастающему числу атомов водорода; при равном числе атомов
углерода и водорода — в алфавитном порядке остальных входящих в фор-
мулу элементов; 3) соединения, имеющие одинаковую формулу, расположены
в алфавитном порядке названий изомеров; 4) неорганические соли органических
кислот и продукты присоединения неорганических соединений к органическим
расположены при формуле тех соединений, производными которых они явля-
ются; 5) кристаллизационная вода не входит в состав приводимых формул.
CBrN, Бромциан, 123
CHeN2, диазометан 174
СН31, йодистый метил 281
CH3NO2, метилнитрит 265
CH3NO3, метилнитрат 329
CH4S, метилмеркаптан 397
CH6N2, метилгидразин сернокислый
319
С2НВг30, бромаль 102
С,Н2С1„О2, дихлоруксусная кислота
W
С2Н3С13О,
485
трихлорэтиловый спирт
С Н 5 BrO3S, fi-бромэтансульфокислоты
"натриевая соль 124
С2Н51, йодистый этил 284
C2H5N3O2, нитрозометилмочевииа 373
C2H6BrN" р-бромэтиламина бромисто-
"водородная соль 126
C2HeN3O, нитрозодиметиламин 199
C2HeN2S, S-метилизотиомочевина сер-
нокислая 326
C2H7NO3S, таурин 441
C2H8N2, не?1шл«.-диметилгидразин 199
» ?цлш.-диметилгидразин диги-
дрохлорид 202
C2HeN2, нешмм.-диметилгидразин ги-
дрохлорид 199
С3НС17, несшим.-гептяхлорпропан 153
C3H2N2, нитрил малоиовой кислоты
С3Н4О4, малоновая кислота 309
С3Н4О4, малоновой кислоты кальцие-
вая соль 310
CjHjBrO, бромацетон 103
» эпибромгидрин 573
С3Н5С1О, эпихлоргидрин 574
C3H5NO, метоксиацетонитрил 343
С3НвВг,О, я,у-дибромгидрин глице-
рина" 183
С3НвО, пропионовый альдегид 424
С3Н8О2, ацетол 74
C3HSO3, ^/-глицериновый альдегид
164
C3HeS3, симм.-тритиан 479
С3Н,Вг, бромистый изопропил 115
С3Н,Вг, бромистый н.-пропил 115
C3H7CIS, р-хлорэтилметилсульфид 562
С3Н,I, йодистый н.-пропил 284
C3H7N, аллиламин 25
C3H,NO2, р-аланин 20
C3H8OS, р-оксиэтилметилсульфид 3&6
С3Н3О2, /-пропиленгликоль 421
С4Н2О4, ацетилендикарбоновая кис-
лота 71
C4H3IS, 2-иодтиофен 287
C4H3NO2S, 2-нитротиофен 381
C4H3N3O5, нитробарбитуровая кис-
лота 365
С4Н4Вг204, а,^-дибромянтарная кис-
лота 191
C4H4N2O3, барбитуровая кислота 79
УКАЗАТЕЛЬ ВЕЩЕСТВ ПО ФОРМУЛАМ
627
С4Н4О3, ангидрид янтарной кислоты
56
С4Н4О4, фумаровая кислота 541
C4H4S, тиофен 458
C4H5N0o, сукцинимид 430
C4H5N3O3, урамил 491
С4Нв, 1,3-бутадиен 128
C4H6N2S, 2-амино-4-метилтиазОл 40
C4H6N4O3, аллантоин 23
С4Н6О4, метиловый эфир щавелевой
кислоты 331
С4Н7СЮ2, этиловый эфир монохлор-
уксусной кислоты 580
C4H7NO, циангидрин ацетона 566
C4H7NO2, монооксим диацетила 206
C4H,NO3, ацетилглицин 69
C4H7NS, изопропилтиоцианат 268
C4H8N2O2, ацетилметилмочевина 374
» диметилглиоксим 205
,> диметилглиоксима натрие
вая соль 207
C4H8N2O3, нитрозометилуретан 376
С4Н8О, тетрагидрофуран 445
С4Н3О3, этоксиуксусная кислота 608
С.Н.Вг бромистый вторично-бутил
115
С4Н9Вг, бромистый изобутил 114
С4Н9С1О, тетраметиленхлоргидрии
450
С4Н„1, йодистый н.-бутил 284
I, йодистый изобутил 289
C4H9NO2, 7-аминомасляыая кислота
о I
» а-аминоизомасляная ки-
слота 34
» н.-бутилнитрит 131
)) этилового эфира глицина
хлористоводородная соль 577
C4H9NO2, этиловый эфир N-метилкар-
баминовой кислоты 578
C4Hl0BrN, р-бромэтиламина броми-
стоводородная соль 126
C4Hl0CrN7S4, соль Рейнеке 432
C4Hl0O2S, fi-тиодигликоль 453
C4Hl0Zn, диэтилцинк 249
C4HUN, н.-бутиламин 155
» вторично-бутнламин 155
С5Н4О3, цитраконовый ангидрид 570
» итаконовый ангидрид 290
C5HeN2O2, 6-метилурацил 335
С5Н6О4, итакоповая кислота 290
С5Н6О4, мезаконовая кислота 312
» цитраконовая кислота 570
С5Н8Вг4, тетрабромнеопентан 443
С5Н814, тетраиоднеопентаи 444
С5Н9Вг02, а-бром-изовалериановая
кислота 107
C5H9NO4, метилиминодиуксусная кис-
лота 327
С5Н1ОВг„, триметилэтилендибромид
'324
С5Н10О, метилизопропилкетон 329
С5Н1г1, йодистый и.-амил 284
» йодистый изоампл 284
CjHuNO^S, (//-метионин 340
С5Н12, нГ-пентан 408
C5H12CrH9S4, соль Морланда 443
С5Н12О, метилизопропилкарбинол 321
C5H14C1N3O, бетаингидразид соляно-
кислый 100
С6Н2Вг2С1Г>Ю,2,6-Дибромхинон-4-хлор-
имид
CeH2I3N0o, 1,2,3-трииод-5-нитробен-
зол 469"
C6H3Br2NO3, 2,6-дибром-4-нитрофенол
185
СвН3Вг3, «иаг.-трибромбензол 467
C,H3C1NO2S, S-хлор-о-нитро-п-хлор-
тиофенол 561
C6H3C1N2O^S, 5-хлор-2,4-динитротио-
фенол 561
CeH4BF4N3O2, о-нитробензолдиазония
борофторид 228
CeH4BF4N3O2, л-нитробензолдиазопия
борофторид 227
C6H4C1NO2S, S-хлор-о-нитротиофенол
560
С4Н 4C1NO4S, о-ннтробензолсульфонил-
хлорид 404
C6H4ION2O2, дииод-п-нитроанилин 198
CeH4N2O4, о-динитробензол 228
"> л-динитробензол 227
СеН4О„, хинон 545
С6Н6Вг0, о-бромфенол 121
СвН6Вг02, бромрезорцин 120
CeH5Br2NO, 2,6-дибром-4-аминофенол-
хлоростаннат 187
CeH6F, фторбензол 537
СвН61, иодбензол 278
СвН5Ю, л-иодфенол 288
C6H5N3O4, 2,4-динитроянилин 223
C6HeIN, л-иоданилин 271
CH,NtO,, 2,4-динитрофенплгидра-
зии 229
C6H6OS, 2-ацетотиенон 76
СвН6О„, 5-метилфурфурол 338
C6H6Oj, аконитовая кислота 15
CeH7As03, фениларсиновая кислота
501
CeH7NO, 1-метил-2-пиридон 333
CeH7NO3S, ортаниловая кислота 403
CeH8N2, о-фенилендиамин 511
i> " в-фенилендиамин дигидрохло-
рид 510
628
УКАЗАТЕЛЬ ВЕЩЕСТВ ПО ФОРМУЛАМ
C6H9N, 2,4-диметилпиррол 216
» 2,5-диметилпиррол 218
C6H9N3O2, /-гистидина хлористоводо-
родная соль
СрНщ, циклогексен 567
СеН1()Вг2, 1,2-дибромциклогексан 189
CpHjuOj, ацетонилацетон 219
С6НпВг, циклогексилбромид 114
С6НпВгО2, а-бром-н.-капроновая кис-
лота 108
С6Н11ВгО2, а-бромизокапроновая кис-
лота 108
СсНпВгО2, а-бром-р-метилвалериано-
вая кислота 108
СвНп, оксим циклогексанона 90
>> 2-кетогексаметиленимин 306
С6Н12О, н.-капроновый альдегид 295
С.Н12О2, н.-капроновая кислота 319,
408
л fi-метилвалериановая кис-
лота 317
* пропионоин 141
СвН12О3, изопропиловый эфир молоч-
ной кислоты 266
СвН12О3, этиловый эфир этоксиуксус-
ной кислоты 608
CeH13N, циклогексиламип 155
C6H13NO2, е-аминокапроновая кисло-
та 36
C8H14BrN, fi-диэтиламиноэтилбромида
гидробромид 127
C6H14N2O2, dZ-лизина хлористоводо-
родная соль 308
CeH14N2O2, d/-лизина хлористоводо-
родная "соль 308
CeH14N4O2, d-аргинин солянокислый
60
С6Н14О2, гексаметилснгликоль 149
C6H14O4S, н.-пропилсульфат 136
CeH14S, и.-пропилсульфид 422
CeH14Zn, ди-н.-пропилцинк 252
CjHj 5NO, fi-диэтиламиноэтиловый
спирт 248
C7H4I2OS, 2-окси-3,5-дииодбензойная
кислота 392
С,Н412О3, 4-окси-3,5-дииодбензойная
кислота 393
C7H4N2O3, л-нитрофенилизоцианат 383
C.H4N2O5, 2,4-динитробензальдегид
224
C7H4OS, хелидоновая кислота 543
С7Н5Вг0, л-бромбензальдегид 552
С7Н5Вг0, л-бромбензальдегид 105
С7Н5Вг04, 2,4-диокси-5-бром-бензой-
ная кислота 120
С7Н5С1О, л-хлорбензальдегид 551
» п-хлорбензальдегид 554
С,Н5СЮ2, о-хлорбензойная кислота
556
C7H5FO2, л-фторбензойная кислота 534
С7Н5Ю0, л-иодбензойная кислота 276
C,H5NO3, л-нитробензальдегид 366
C7HeBrNO2, л-нитробензилбромид 368
C,HeINO2, 5-иодантраниловая кисло-
та 273
C,HeN2, бензимидазол 85
C7HeO2S, тиосалициловая кислота 455
С,Н6О3, л-оксибензойная кислота 390
» протокатеховый альдегид
428
С7НвО4, fi-резорциловая кислота 430
C7H7NO2, бензогидроксамовая кисло-
та 87
C,H7NO2, фенилнитрометан 513
» бензогидроксамовой кисло-
ты калиевая соль 87
C,H8N2O, N-нитрозометиланилин 372
С7Н8О, бензиловый спирт 462
C,H9N, 2,6-диметилпиридин 214
C,H10N2, 2,4-диаминтолуол 176
» 2,4-диаминтолуол сернокис-
лый 177
C7H10N2, а-метил-а-фенилгидразин 337
С7Н10Ов, трикарбометокснметан 471
C7H1SO4, пимелиновая кислота 410
С,Н14О2, ацеталь акролеина 64
» энантовая кислота 571
С,Н15В4, бромистый н.-гептил 114
С7Н15С1О2, ацеталь й-хлорпропионо-
вого альдегида 67
C,H15NO, оксим энантола 394
С7Н1бО, 2-гептанол 152
>> трпэтилкарбинол 489
С7Н1вО,, гептаметиленгликоль 170
С7Н1еО4, диэтиловый ацеталь dl-гли-
церинового альдегида 65
C,H17N, н.-гептиламин 154
С8Н4С12О2, хлорангидрид фталевой
кислоты симм, н несимм. 547
C8H4N2Od, 4-нитрофталимид 389
C8H5NO6, 4-нитрофталевая кислота 387
C8HeN2O2, бензоиленмочевина 03
» фенилацинитроацетонитри-
ла натриевая соль 513
С8НвО12 фенилглиоксаль 507
» фталид 533
С8НвО3, альдегидофталевая кислота 27
С8Н6О4, пиперониловая кислота 417
С8Н,ВгО, бромистый фенацил 116
C8H7NOa, л-нитроацстофеноп 361
C8H8N2, 2-метилбеизимидазол
C8HSO, п-толуиловый альдегид 464
С8Н8О„, о-толуиловая кислота 462
» " п-толуиловая кислота 463
УКАЗАТЕЛЬ ВЕЩЕСТВ ПО ФОРМУЛАМ
623
С8Н8О3, пиперониловый спирт 462
С8Н8О4, галлацетофенон 147
» флороацетофенон 531
C8HeNO, амид о-толуиловой кислоты
20
C8Hi0BrN, а-л-бромфеиилэтиламин
525
C8H,OC1N, а-л-хлорфенилэтиламин
525
C8HieN2O, л-иитрозодиметиланилин
солянокислый 225
С8Н10О, л-толилкарбинол 461
C8HnN, ^/-ж-фенилэтиламин 526
» d-ct-фенилэтиламин 523
» Z-з-фенилэтиламин 526
C8HnNO2, 4-аминовератрол 32
С8Н12О2, 5,5-диметил-1,3-циклогексан-
дион 220
С8Н12О3, 2-карбоэтоксициклопента-
нон 297
С8Н14О4, этиловый эфир л-амипобен-
зойной кислоты 534
С8Н14О4, этиловый эфир метилмалоно-
вой кислоты 588
С8Н16О2, бутироин 140
>> изобутироин 141
C8H18BrN, ^-бромдипропиламиноэтил-
гидробромид 127
C8H18O0S, н.-бутилсульфат 135
CSH18O4S, н.-бутилсульфит 136
ClsH18Zn, ди-н.-бутилцинк 252
С9НеО2, фенилпропиоловая кислота
520
С8Н8О, а-гидриндон 156
С9Н8О3, фенилпировиноградная кис-
лота 518
C9H9BF4N2O,, и-карбоэтоксифенил-
диазония борофторид 535
С9Н9С1, а-хлоргидринден 157
C9H9NOS, изонитрозопропиофенон 264
» " вератронитрил 145
CjHjNOa, гиппуровая кислота 158
С 9HlcN,S, л-тиоциандиметиланилин
431
С9Н10О, метилбензилкетон 313
С9Н10О2, о- и л-пропиофенол 426
С9Н10О3, вератровый альдегид 142
С9Н,0Вг, броммезитилен 118
C9HnNO2, этиловый эфир л-аминобен-
зойной кислоты 534
CJHJJNO2, нитромезитилен 377
d ^/-|5-фенилаланин 496
C9HnNOc, амид вератровой кислоты
32
C9HUNO3, /-тирозин (/-триптофан) 476
C9H13N, а-л-толилэтиламин 525
» К-этил-л<-толуидин 605
CeH13NO2, 2,4-диметил-карбоэтокси-
пиррол 211
С9Н14О„ этиловый эфир 2-кетогекса-
гидробензойной кислоты 412
СвН14О6) этиловый эфир этоксалилпро-
пионовой кислоты 604
С9Н1вО4, азелаиновая кислота 9
С9Н18О2, пеларгоновая кислота 406
С9Н26О, ди-н.-бутилкарбинол 193
С„Н2СО2, нонаметиленгликоль 171
С„Н21О4Р, н.-пропилфосфат 139
С10НвО2, 1,2-иафтохинон 353
С10Н6О8, пиромеллитовая кислота 418
ClCH,BrHg, бромистая f-нафтилртуть
DO о
C10H7ClHg, хлористая р-нафтилртуть
Cl0H,Cl3HgN2, двойная соль хлори-
стого ^-нафтилдиазония и сулемы
558
C1CH,NO3, 1-нитро-2-нафтол 379
ClCH9NO, 1-амино-2-нафтол соляно-
кислый 41
Cl0H9NO, 1-амино-4-нафтол соляно-
кислый 48
(С10Н 9NO), а-фенилацетоацетонитрил
С10Н 9NO4S, 1 -амино-2-нафтол-4-суль-
фокислота 51
С1СН10О, а-тетралон 448
Cl0Hl0O3, р-бензоилпропионовая кис-
лота 95
Cl0H12N2O4, динитродурол 262
С10Н12О2, дурохинон 262
»" 7-Фенилмасляная кислота
511
Cl0Hj,O4, гомовератровая кислота 164
С10Н14, дурол 253
» изодурол 259
C10H14N2O, л-нитрозодиэтиланилин226
CjoHjjOj, этиловый эфир 2-кетоцикло-
гексилглиоксалевой кислоты 411
C,OH1SN2 диаминодуролхлоростаннат
263
C10HlsO5, этиловый эфир ацетоянтар-
ной кислоты 580
C10HlsOe, трикарбоэтоксиметан 472
Cl0H18O3, этиловый эфир а,а,7,у-
тстраметилацетоуксуспой кислоты
584
С10Н18О4, этиловый эфир адипиновой
кисло 1ы 578
С10Н18О4, этиловый эфир изопропил-
малоновой кислоты 108
С10Н2СО2, пивалоин 141
Cl0H22BrN, ^-бромди-н.-б}'тиламшю-
этйлгидробромид 127
630
УКАЗАТЕЛЬ ВЕЩЕСТВ ПО ФОРМУЛАМ
УКАЗАТЕЛЬ ВЕЩЕСТВ ПО ФОРМУЛАМ
631
Cl0H32O, три-и.-пропилкарбинол 490
Cl0H22O,, декаметиленгликоль 170
Cl0H22Zn, диизоамилцинк 252
CUHSOS, фенилтионилкетон 522
CjiHjOj, а-нафтойная кислота 348
» р-нафтойиая кислота 351
CUH,N, 2-фенилпиридин 517
CUH9NO2, азлактон а-ацетоамшюко-
ричной кислоты 72
¦О4, л-бромбензальдиацетат
^Н^МО^а-ацетоаминокоричная кис-
лота 72
C.jHjjNO., л-нитробензальдиацетат
366
СпН12Вг2О2, этиловый эфир а,р-ди-
бром-^-фенилпропионовой кислоты
586
СПН12О3, /-триптофан 474
» f-бензоилмасляная кислота 96
CjjH^Oj, этиловый эфир бензоилук-
сусной кислоты 584
CUH12O5, 3,4-диметоксифенилпирови-
ноградная кислота 166
CUH13NO3, N-ацетилфенилаланин 500
CjjH14O4, метиловый эфир гомоверат-
ровой кислоты 164
СиН14О7, этиловый эфир ацгтондикар-
боиовой кислоты 543
CuHle> н.-амилбензол 30
» пентаметилбензол 254
СаН13О5, этиловый эфир а-ацетоглу-
таровой кислоты 581
CjjHj 9NO3, N-ацетилгексагидрофенил-
алании 500
CjjHjoOj, этиловый эфир вторично-
бутилмалоновой кислоты 318
CUH21)O4, этиловый эфир пимелиновой
кислоты 415
CUHS2O, ундециловый спирт 307
CjjH24Oa, ундекаметилеигликоль 171
^i2HeO3, 1,2-нафталевый ангидрид 347
С12Н8В2РвГЧ4,4,4'-дифенилеи-бц?-диа-
зония борофторид 245
C12H8F,, 4,4'-дифтордифеннл 244
C12HBOS, фенокстин 529
^12Нв0з, ангидрид 3,4-дигидро-1,2-иа-
фталевой кислоты 53
C12H9NO3> о-иитродифениловый эфир
C12H9NO3, л-нитродифениловый эфир
C12HiaCl2Se, дифенилселен двухлорп-
стый 239
C12Hl0NsO, азоксибензол 12
C12Hl0N;O3, 1-мнтро-2-ацетилиафтил-
ам.ш '359
C12Hj0S, дифенилсульфид 241
C12H10Se, дифенилселен 237 .
C12HnNO3, этиловый эфир фенилциан-
пировиноградной кислоты 600
Ci2HnN3, диазоаминобепзол 172
C12H12N, а-(,5-нафтил)-этиламин 525
^чЛА). этиловый эфир а-фенилаце-
тоуксусной кислоты 596
С12Н1в, циклогексилбензол 567
С12Н16О, ацето-л-цимол 77
С12Н18О2, этиловый эфир у-фенилмас-
ляной кислоты 55
C12H1;NO4, 2,4-диметил-3,5-дикарбо-
этоксипиррол 209
C12H17NOS, соль d-2-фенилэтиламина и
/-яблочной кислоты 526
CJ2H17NOe, соль /-з-фснилэтиламина и
tf-винной кислоты 527
С12Н1В, гексаметилбензол 256
С12Н22О4, моноэтиловый эфир себа-
цицовой кислоты 345
С12Н2„ОИ, целлобиоза 564
С12Н25Вг, бромистый н.-додецил 112
С12Н28О, лауриловый спирт 306
С12Н27ВО3) н.-бутиловый эфир борной
кислоты 133
С12Н27О4Р, н.-бутилфосфат 135
» еторцч/ю-бутилфцсфат 139
Cl3H9NO, акридон 18
^13Н9^О2, 2-интрофлуорен 385
С13Н10С1а, дифенилдихлорметаи (кето-
хлорид бензофенона) 452
CinHl0S. тиобензофенон 452
C13HUN, 2-аминофлуорен 385
» иминодифенилметан соляно-
кислый 269
C13HUNO, оксим бензофенона 96, 393
CjjHjjNOj, N-фенилантраниловая кис-
лота 18
С13Н12, дифенилметан 233
ci3Hi2Na°> беизилгидразон 506
С13Н12О2, этиловый эфир а-нафтойной
кислоты 592
Ci3Hl4O4, этиловый эфир бензоилук-
сусной кислоты 584
C13HieBrNO3, е-бензоиламино-а-бром-
капроновая кислота 89
С12Н16О3, этиловый эфир бензоилди-
метилуксусной кислоты 582
С13Н1вО4, этиловый эфир фенилмало-
новой кислоты 599
QsHi7NO3, е-бензоиламинокапроновай
кислота 90
C13HJ7NO4, 3,5-дикарбоэтокси-2,6-дн-
метилпиридин 215
Cj3HlsN2Oj, ^/-^-бензоиллизин 308
*--i34j8N4O2( бензилиденаргинин 60
Ci3HieNO4, 1,4-дигидро-3,5-дикарбо-
этокси-2,6-диметилпиридин 215
С13Н2ВО, три-н.-бутилкарбинол 490
^laHgsOj, тридекаметиленгликоль 171
С14Н?С1О„, й-хлорантрахинон 549
С14Н3О2, антрахиион 547
Cl4H8O5S, антрахиноисульфокислоты
калиевая соль 58
Cj jHl0N.O, фенилбеизоилдиазометаи
506
C14Hl0O3S, фенантрен-2-сульфокислоты
бариевая соль 494
Cl4Hj0O3S, фенантрен-3-сульфокислоты
калиевая соль 494
С14НПСЮ, дезил хлористый 559
CjjHjjNj, бензальазин 319.
С14Н ,N2O2 дибензоилгидразин 202
C14Hj20, дезоксибензоин 167
C14H14Se, ди-ж-толуилселен 239
» дн-о-толуилселен 240
» ди-л-толуилселен 240
Cl4H15N, а-п-ксилилэтиламин 525
C14Hj 5NO7S, 1 -метилтиол-3-фталимид-
пропан-3,3-карбоиовая кислота 341
С14Н„„О8, этиловый эфир этилеитетра-
карбоновой кислоты 601
С14Н2вО4, диэтиловый эфир себацино-
вой кислоты 345
CJ4H29Br, бромистый тетрадецил 114
С14Н30О, миристиловый спирт 307
С14Н30О2, тетрадекаметиленгликоль
Ci5Hj(,Br202, дибензоилдибромметан
242
С15Н10О2, бензальфталид 80
С15Н10О3, дифенилтрикетон 242
» дифенилтрикетон гидрат
242
CjsHuNCXj, бензилфталимид 84
Cl5H12N2O, 4,5-дифенилглиоксалои
232
C15HJ4NOeNa, этилового эфира фтал-
имидмалоновой кислоты натриевое
производное 340
С16Н30О2, этиловый эфир н.-тридека-
иовой кислоты 593
С16Н33О3, н.-амиловый эфир борной
кислоты 134
С1бН33О4Р, н.-амилфосфат 139
CleHjjNO2, азлактон а-бенэоиламино-
коричной кислоты 497
C1(,H13NO, нитрил-а-фенил-р-бензоил-
пропионовой кислоты 358
C1(.HJ3NO3, с-бензоиламинокоричная
кислота 497
CleHjj, 1,4-дифенилбутадиеи 231
CJ(iHjjO3, ацетилбензоин 68
C1(.H](.N2O2, дибензоилдиметилГидра-
зин 203
CipH^N^joS, аргининдинитронафтол-
сульфонат 61
С16Н331, йодистый и.-гексадецил 280
С16Н34, и.-гексадекан 148
С^Н^О, цетиловый спирт 307
» три-н.-амилкарбинол 490
С17Н10О, бензантрон 81
С15Н14О, дибензальацетон 179
C.gH.jCISe, хлористый трифенилселен
239
C,SH15NO4, азлактон а-бензоиламино-
р-C,4-диметилоксифенил)-акрило-
вой кислоты 11
С13Н1ВО„, 1,4-дибензоилбутан
ClsHolN06S, диэтиловый эфир 1-ме-
тилтиол-3-фталимидопропан-3,3-дн-
карбоновой кислоты 341
С1ВНзвО, олеиловый спирт 401
С18Н„7Вг, бромистый октадецил 114
C18HgB02, октадекаметиленгликоль 171
C19H]5Na, трифенилметнлнатрий 481
C20H14Hg, ди-р-нафтилртуть 222
С20Н1в, трифенилэтилен 483
С .Н1аО, ^fi-дифенилпропиофенои
235
.jH.NOsS, п-толуидииа и 2-фенан-
"трен-2-сульфокислоты соль 495
С22Н42О2, бутиловый эфир олеиновой
кислоты 402
С2„Н42О2, эруковая кислота 575
С„2Н4вО, три-н.-гептилкарбинол 490
С26Н22О2, бензопинакои 98
» [-{-бензпинаколин 97
CoeH22S2, дибеизгидролдисульфид 453
С27Н46О, холестанон 563
С27Н48О, дигидрохолестерин 195
С;'Н38О19, октаацетат а-целлобиозы
'398
СНщО,, холестерилацетат 196
c;4H23NeO14S4, продукт сочетания
о-толидина и Чикаго-кислоты 434
НВг, бромистый водород 109
IC1, хлориод 199, 393.
УКАЗАТЕЛЬ РИСУНКОВ
Аппаратура
для получения ацеталя у-хлорпро-
пионового альдегида 67
для получения изонитрозопропио-
фенона 265
для получения йодистого метила
282
для получения п-нитрофенилизо-
цианата 383
для получения п-толуилового аль-
дегида 464
дляполучениятриброманилина 467
Выпариватель для брома 111
Перегонная колба 64, 228
Капельная воронка 550
Экстрактор 311, 478
Фильтрование, перевернутое 479
Ловушка для поглощения газа 78
Генератор газа
Этилнитрит 205
Метилнитрит 265
Пирогенетические реакции
Бутадиен 129
Бромистый водород 109
Метилбснжлкетон 314
Мешалка, механическая 298, 413
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Адипииовая кислота 181, 578
Азелаиновая кислота 9
Азлактон а-ацетоамииокоричиой кис-
лоты 72
Азлактон а-беизоиламино-р-3,4-диме-
токсифенил-акриловой кислоты 11,
164
Азлактон а-бензоиламинокоричио й
кислоты 497
Азоксибензол 12
Азотная кислота 215, 239, 319, 329,
360, 361, 376, 386, 404, 508
Азотная кислота дымящая 262, 365.
376, 381, 389
Авотиокислая медь с 3 мол. кристал.
воды 301
Азотнокислый барий 301
Аконитовая кислота 15
Акридои 18
Акролеин 64, 65, 67
р-Аланин 20
Аллантоии 23
Аллиламин 25
Аллилизотиоциаиат 25
Альдегид бензойный 61, 72, 179, 180,
181, 320, 321, 497
Альдегид вератровон кислоты 11,
142, 144, 145
Альдегид коричный 231
Альдегидофталевая кислота 27
Ачьдегид уксусный 322
Алюминий 486
Алюминий хлористый 77, 95, 116,
117, 153, 154, 168, 182, 240, 241,
242, 253, 254, 257, 261, 426, 449,
465, 522, 529, 547
Алюминия этилат 485, 486
Амальгама алюминия 233, 234
Амальгама натрия 1%'ная 482, 482
Амальгама натрия 3°/0-пая 483
Амальгама цинка 511
Амид вератровой кислоты 32
Амид о-толуиловой кислоты 20, 463
н.-Амилбензол 30
н.-Амил бромистый 295, 296
н.-Амил йодистый 284
и.-Амиловый спирт 134
трет.-Амиловый спирт 329
и.-Амиловый эфир борной кислоты
134
втярцчед-АмилСульфид 424
н.-Амилфосфат 139
4-Амииовератрол 32
а-Амииоизомасляная кислота 34
s-Амииокапроиовая кислота 36
7-Аминомасляиая кислота 37
2-Амиио-4-метилтиазол 40
1 -Амино-З-иафтол-2,4-дисульфокие-
лота 435
1 -Амино-8-нафтол-3,6-дисульфокиС ло-
та 437
1-Амиио-2-нафтол солянокислый 41,
355
1-Амино-4-иафтол солянокислый 48
1 -Амино-2-иафтол-4-сульфокислота 51
!-Амнно-8-иафтол-4-сульфокислота
439
2-Амино-5-нафтол-7 сульфокислота 439
2-Амнио-8-нафтол-6-сульфокислота
439
л-Амипофеиол 288
2-Амииофлуорен 385
Аммиак 35, 214, 223
Аммиак водный 158, 301, 308, 319,
439, 441, 585
Аммоний двухромовокислый 301, 433
Аммоний муравьинокислый 523
Аммоний роданистый 431, 432
Аммоний углекислый 218
Аммоний уксуснокислый 223
Аммоний хлористый 34, 585
Ангидрид глутаровой кислоты 96
Ангидрид итаконовый 290, 570
Ангидрид мышьяковистый 12
Ангидрид уксусный 11, 68, 72, 147.
196, 231, 366, 378, 381, 398, 497
Ангидрид цитраконовый 312, 570
Ангидрид янтарный 56, 95
634
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Анилин 18, 172, 238, 271, 278, 467,
501
Анилин солянокислый 537
Антиоксиданты 45, 49, 147
Антраниловая кислота 93, 273, 45^.
Антрахинон 59, 82, 547
Антрахиноисульфокислоты калиевая
соль 58
Антрацен 546
Аргининдинитронафтолсульфонат 61
rf-АрГинии солянокислый 60
Ацеталь-d/-глицеринового альдегида
65, 163
Ацеталь капронового альдегида 296
Ацеталь fi-хлорпропиондвого альде-
гида 64, 67
Ацеталь акролеина 64
Ацетамид 374
Ацетилбензоин 68
N-Ацетилгексагидрофенилалашш 500
Ацетилглицин 69, 72
Ацетнлендикарбоновая кислота 71
Ацетилметилмочевина 374
fi-Ацетилнафтиламин 360
N-Ацетилфенилаланин 500
Ацетил хлористый 56, 76, 58
а-Ацетоаминокоричная кислота 72
Ацетол 74, 421
Ацетон 34, 103, 179, 545, 556
Ацетонилацетон 219
Ацетоиитрил 531
Ацетотиенон 76
Ацето-гс-цимол 77
Барбитуровая кислота 79, 365
Барий азотнокислый 301
Барий хлористый 494
Бария гидрат окиси 39
Бензальазин 319
Бензальацетофенон 236, 358
Бензальфталид 89
Бензантрон 81
Бензидин 245, 439
Бензил 506
Бензилгидразон 506
Бензилиденаргинин 60
Бензиловый спирт 462
Бензилфталимид 84
Бензил хлористый 30, 84, 234, 480
Бензил цианистый 503, 514, 601
Беизимидазол 85
Бензогидроксамовая кислота 87
Бензогидроксамовой кислоты калие-
вая соль 87
Вензоин 68, 232, 559
оензойнып альдегид 61, 72, 179, 320,
497
?-Бензоиламино-я-бромкапроновая ки-
слота 89, 308
е-Бензоиламинокапроновая кислота
89, 93
а-Вензоиламинокоричная кислота 497
Бензоиленмочевина 93
<//-г-Бензоиллизин 308
¦/-Бензоилмасляная кислота 96
Ь-Бензоилпропионовая кислота 95
Бензоил хлористый 92, 160, 202, 522,
582, 584
Бензол 95, 168, 182, 233, 235, 239,
241, 267, 567
/З-Бекзопикаколин 97
Бензопинакон 96, 97
Бензофенон 96, 393, 452, 484
Бепзофенона дихлорид 452
Бензофенона оксим 96, 393
Бетаннгидразин солянокислый 100
Бисульфита продукт присоединения
106
Борная кислота 133, 228, 245, 535, 538
Бром 20, 89, 102, 103, 104, 105, 107,
ПО, 116, 118, 120, 122, 123, 153,
182, 186, 190, 196, 243, 323, 369,
375, 431, 467
Бромаль 102
Бромацетон 75, 103
.м-Бромбензальдегид 552
п-Бромбензальдегид 105, 368
л-Бромбензальдиацетат 367
Бромбензойная кислота 106
Бромбензол 517
fs-Бромдибутиламиноэтила бромисто-
водородная соль 127
р-Бромдиметиламиноэтила бромисто-
водородная соль 127
р-Бромдипропилакиноэтила бромисто-
водородная соль 127
р-Бромдиэтиламиноэтила бромистово-
дородная соль 127
а-Вромизовалериановая кислота 107
а-Вромизокапроновая кислота 108
Бромистая медь 553
Бромистая р-нафтилртуть 558
Бромистоводородная кислота 109
Бромистый н.-амил 295, 296
Бромистый н.-бутил 193
Бромистый «торично-бутил 115
Бромистый водород 108, 112
Бромистый н.-гептил 114
Бромистый н.-додецил 112, 593
Бромистый изобутил 114
Бромистый изопропил 115
Бромистый лаурил 112
Бромистый метил 589, 590
Бромистый натрий 553
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
635
Бромистый октадецил 114
Бромистый н.-пропил 115
Бромистый тетрадецил 114
Бромистый фенацил 116
Бромистый циклогексил 114
а-Бром-н.-капроновая кислота 108
Броммезитилен 118, 529
а-Бром-'з-метилвалериаиовая кислота
108
а-Бромнафталин 292, 348
2-Бромпентан 408
7-Бромпропилфталимид 84
Бромрезорцин 120
п-Бромтолуол 105
а-тг-Бромфеиилэтиламин 525
¦о-Бромфенол 121
Бромциан 123
р-Бромэтансульфокислоты натриевая
соль 124, 441
§-Бромэтиламина бромистоводородная
соль 126, 441
1,3-Бутадиен 128
н.-Бутиламин 155
вторично-Бутпжпн 155
н.-Бутил бромистый 193
вторично-Вугнл бромистый 115
н.-Бутилнитрит 131
и.-Бутиловый спирт 131, 133, 138
и -Бутиловый спирт безводный 137,
401, 402, 406, 407
н.-Бугиловый эфир 346, 408
н -Бутиловый эфир борной кислоты
133
н -Бутиловый эфир олеиновой кислоты
402
н.-Бутиловый эфир л-толуолсульфо-
кислоты 31
я.-Бутилсульфат 135
и.-Бутилсульфид 424
II .-Бутилсульфит 136
М-Бутил-л«-толуидин 607
н.-Бутилфосфат 138
«торцчно-Бутилфосфат 139
Бутироин 140
Ванилин 142
Вата 398
Вератральдоксим 146
Вератровый амид 32
Вератронитрил 44, 145
d-Виниая кислота 527
Водород 150, 196
Водород бромистый 109, ПО, 112, 446,
499
Водород хлористый 67, 148, 157, 265,
270, 450, 464, 531, 577, 594, 596,
608
Вспенивание, устранение его 187, 347
Галлацетофенон 147
Гашеная известь 573
н.-Гексадекап 148
н.-Гексадецил йодистый 148, 280
н.-Гексальдегид 295
Гексаметилбензол 256
Гексаметиленгликоль 149
Гептальдегид 395, 571
Гептаметиленгликоль 170
н.-Гептановая (эиантовая) кислота 571
2-Гептанол 152
песимм. -Гептахлорпропан 153
н.-Гептиламин 154
н.-Гептил бромистый 114
Гершберга мешалка 298
Гидразингидрат 101
Гидразинсульфат 202, 229, 319, 506
а-Гидриндон 156
Гидроксиламин солянокислый 87, 145,
393, 395
Гидроксиламинсульфокислоты натрие-
вая соль 206
Гидроокись свинца 37
Гидросульфит 42, 49
Гидрохинон 545
Гилылана катализатор 259, 260
Гиппуровая кислота 158, 497
/-Гистидина хлористоводородная соль
160
Глет 231
Глицерин 82, 183
Глицерина а^-дибромгидрин 183,574
Глицерина а,у-дихлоргидрин 573
^//-Глицериновый альдегид 163
Глиции 69
Глицина этилового эфира хлористово-
дородная соль 577
Гомовератровая кислота 164
Двуокись селена 507, 508
Двойная соль хлористого {3-нафтил-
диазония и сулемы 222, 558
Дезил хлористый 559
Дезоксибензоин 167
Декаметиленгликоль 170
Декстроза 15
Диазоаминобензол 172
Диазометан 174
Диаминодурол хлоростаннат 263
2,4-Диаминотолуол 176
2,4-Диаминотолуол сернокислый 177
Диаиизидин 439
Дибензальацетон 179
Дибензгидролдисульфид 453
1,4-Дибензоилбутан 181
Дибензоилгидразин 202
636
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Дибензоилдибромметан 242
Дибензоилдиметилгидразин 203
Дибензоилметан 242
2,6-Дибром-4-аминофенол-хлоростан-
нат 187
ад-Дибромгидрин глицерина 183, 574
2,6-Дибром-4-нитрофенол 185, 187
2,6-Дибромхинон-4-хлоримид 187
1,2-Дибромциклогексан 189
а,^-Дибромянтарная кислота 71, 191
fS-Дибутиламиноэтила бромистоводо-
родная соль 127
Ди-н.-бутилкарбинол 193
Ди-н.-бутилцинк 252
1,4-Дигидро-3,5-дикарбОс»токси-2,б-дн-
метилпиридии 215
3,4-Дигидро-1,2-нафта левой кислоты
ангидрид 53, 347
Дигдрохолестерин 195, 563
Диизоамилцинк 252
2,6-Дииод-л-нитроанилин 198, 469
2,5-Дииодтиофен 288
3,5-Дикарбоэтокси-2,6-диметилпири-
дин 215
Димедон 220
Диметиламин солянокислый 199
Диметиланилин 224, 431
несимм. -Диметилгидразин 199
симм.-Диметилгидразин дигидрохло-
рид 202
несимм.-Диметилгидразин солянокис-
лый 199
Диметилглиоксим 205
2,4-Диметил-3,5-дикарбоэтоксипиррол
209
2,4-Диметил-5-карбоэтоксипиррол 211
Диметилнитрозоамин 199
Диметиловый эфир малоновой кислоты
471
2,6-Диметилпиридин 214
2,4-Диметилпиррол 212, 216
2,5-Диметилпиррол 218, 220
Диметилсульфат 143, 203, 260, 286,
320, 326, 333, 343
5,5-Диметил-1,3-циклогександион 220
3,4-Диметоксифснилпировиноградная
кислота 164, 166
Ди-^-нафтилртуть 222
2,4-Динитроанилин 223
2,4-Динитробензальдегид 224
2,4-Динитробензилидеи-л-аминодиме-
тиланилин 225
о-Динитробензол 228
л-Динитробензол 227
Динитродурол 262
2,4-Динитро-1-нафтол-7-сульфокисло-
та (флавиановая кислота) 63
2,4-Дииитротолуол 176, 225
2,4-Динитрофепилгидразин 229
2,6-Динитрофенилгидразин 231
Ди-о-нитрофеиилдисульфид 404, 560
2,4-Динитрохлорбензол 223, 229
2,4-Диоксибензойная кислота 430
2,4-Диокси-5-бромбензойная кислота
120
Ди-н.-пропилцинк 252
Дитиосалициловая кислота 456
Ди-ж-толилселен 239
Ди-о-толилселен 240
Ди-л-толилселен 240
4,4'-Дифенил-бнс-диазонияборофторид
245
1,4-Дифенилбутадиен 231
4,5-Дифенилглиоксалои 232
Дифенилдихлорметан 452
Дифенмлметан 233
?,р-Дифенилпрониофенон 235
Дифенилселен 229, 237
Дифенилселен двухлористый 239
Дифенилсульфид 241
Дифенилтрикетон 242
Дифенилтрикетонгидрат 242
а,у-Дифталимидпропан 84
4,4'-Дифторбифенил 244
ад-Дихлоргидрин глицерина 573
Дихлоруксусная кислота 247
1,4-Дициклогексилбензол 569
Диэтйламин 214, 248
S-Диэтиламиноэтнловый спирт 248
Диэтиловый эфир-1 -метилтиол-3-фта~
лимидопропан-3,3-дикарбоновой
кислоты 341
Диэтиловый эфир фталевой кислоты
171
Диэтилцинк 249
н.-Додециловый спирт 112, 306
Древесный уголь еловый 418
Древесный уголь сосновый 418
Дрожжи 421
Дурол 253
Дурохинон 262
Желатина 60
Железо хлорное 263, 353
Жерара реактив 100
Изоамиловый спирт 414
Изобутилоный спирт 114
Изобутироин 141
Изодурол 259
Изонитрозопропиифенон 264
Изопропиловый спирт 98
Изопропиловый спирт безводный 266
Изопропиловый эфир молочной кис-
лоты 266
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
637
Изопропилтиоцианат 268
N-Изопропил-л-толуидин 606
Иминодифенил.метан солянокислый
269
Инден 157
Иод 97, 271, 280, 281, 287, 392
Иод, его обратное получение из йоди-
стого натрия 272
л-Иоданилин 271
5-Иодантраниловая кислота 273
л-Иодбензойная кислота 276
Иодбензол 278
Йодистая метилртуть 285
Иодистоводородная кислота 497
Йодистый н.-амил 284
Йодистый н.-бутил 284
Йодистый н.-гексадецил 280
Йодистый изоамил 284
Йодистый изобутил 289
Йодистый метил 281
Йодистый н.-пропил 284
Йодистый этил 284
Иод однохлористый 198, 199, 273, 392
2-Иодтиофен 287
л-Иодфенол 288
Итаконовая кислота 290
Итаконовый ангидрид 290
Казеин 292
Калий двухромовокислыи 424
Калий железосинеродистый 333
Калий йодистый 279, 286, 289, 470
Калий углекислый 18, 84, 390
Калий уксуснокислый 229
Калий хлористый 494
Калий цианистый 93
Калий цианивокислый 358, 593
Калия гидроокись 11, 20, 32, 64, 71,
74, 87, 107, 174, 194, 215, 210, 221,
237, 317, 370, 406, 461, 506, 520,
570, 576, 593
Калия перманганат 9, 23, 27, 65,
417, 556, 571
Кальций углекислый 247
Кальций хлористый 310
Кальция гидроокись 573
Кальция гипохлорит 352
Кальция окись 215
н.-Капроновая кислота 319, 408
л-Карбоэтоксифенилдиазония боро-
фторид 535
2-Карбоэтокснциклопеитанон 297
Касторовое масло 9
Катализатор Гильмана 260
Катализатор медно-хромовый 301
Катализатор окись тория 313, 315
Катализатор палладиевый 445
Катализатор платиновый 196, 499
2-Кетогексаметиленимин 34, 306
Кислота бромистоводородная 109
» 3,4-диметоксифенилпирови-
ноградная 164, 166
Кислота иодистоводородная 497
» Невиля-Винтера 439
» ортаниловая 403
» |5-резорциловая 430
» рицннолеиновая 9
» салициловая 339, 392, 414
» себациповая 445
>> флавиановая 61
» фтористоводородная 228,245,
535, 538
Кислота хлористоводородная 25, 41,
49, 60, 160, 172, 178, 187, 200, 203,
224, 238, 239 244 263 276 278,
292, 309, 335, 338, 341, 434, 456,
479, 491, 501, 510, 511, 518, 524,
534, 537, 549, 551, 605
Кислота щавелевая 331
» /-яблочная 526
» /-яблочная, выделение ее
обратно 528
Кислота янтарная 56, 439, 459
» у 439
» Н 439
)> I 439
» Невиля-Винтера 439
» S 439
Клайзена колба специальная 64, 228
Коричный альдегид 232
я-л-Ксенилэтиламин 525
Ксилол 253
Лауриловый спирт 306
й /-Лизина хлористоводородная соль
308
Лимонная кислота с 1 мол. кристал,
воды 12, 290
Литий 517
Ловушка для улавливания газа 78
Магний 30, 193, 211, 259, 295, 321,
348, 408, 472, 483, 489
Малоновая кислота 309
Малоновой кислоты кальциевая соль
310
Меди окись 18
Медно-хромовый катализатор 149,301
Медный порошок 81, 222, 227, 330,
558
Медь полухлористая 552
Мезаконовая кислота 312
Мезитилен 118, 377
Метил-н.-амилкетон 152
638
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Метиламин 325, 373, 578
Метиланилин 372
Метилбснзилкстон 313
2-Метилбензимидазол 86
л-Метилбензогидроксамовой кислоты
калиевая соль 88 s
[З-Метилвалериановая кислота 317
Метилгидразин сернокислый 319
Метиленаминоацетонитрил 577
Метилизопропилкарбинол 321
Метилизопропилкетон 329
S-Метилизотиомочевина сернокислая
326, 397
Метилиминодиуксусная кислота 327
Метил йодистый 281
Метилмеркаптан 397
Метил-р-нафтилкетон 351
Метилнитрат 329, 514
Метилнитрит 265
Метиловый спирт 165, 265, 281, 329,
331
Метиловый спирт абсолютный 564
Метиловый эфир гомовератровой кис-
лоты 164
Метиловый эфир хлормуравьиной кис-
лоты 473
Метиловый эфир хлоругольной кис-
лоты 471
Метиловый эфир щавелевой кислоты
331
1-Метил-2-пиридон 333
1-Метилтиол-3-фталимид-3,3-дикарбо-
новая кислота 341
6-Метилурацил 335
а-Метил-а-фенилгидразин 337
Метил-р-фенилэтилкетон 316
5-Метилфурфурол 338
Метил хлористый 253, 257
Метилэтилкетон 208
сМ-Метионин 340
Метоксиацетонитрил 343
л-Метоксибензогидроксамовой кис-
лоты калиевая соль 88
Мешалка Гершберга 298
Миристиловый спирт 307
Молоко 292
Моиооксим диацетила 206
Моноэтиловый эфир адипиновой кис-
лоты 346
Моноэтиловый эфир себационовой кис-
лоты 345
Морлаида соль 443
Муравьиная кислота 85
Мышьяковистый ангидрид 12, 501
Натриевая соль диметилглиоксима
207
Натрий 79, 152, 154, 170, 220, 292,
306, 340, 396, 401, 406, 410, 414,
422, 452, 471, 503, 513, 543, 564,
580, 584, 588, 599, 600, 607
Натрий азотистокислый 44, 92, 131,
172, 200, 205, 209, 225, 227, 238,
245, 276, 278, 289, 372, 373, 435,
456, 467, 469, 501, 535, 537, 551,
557, 606
Натрий азотнокислый 445
Натрий бромистый 558
Натрий двухромовокислый 563
Натрий йодистый 444
Натрий роданистый 268
Натрий сернистокислый 453, 455
Натрий сернистый 125, 441, 453, 455
Натрий углекислый 91, 144, 225, 310,
395, 435, 501, 601
Натрий уксуснокислый И, 72, 80,
172, 243, 438, 497, 506
Натрий фтористый 474
Натрий хлорноватокислый 445, 541,
549
Натрий цианистый 34, 123, 247, 310,
343, 566
Натрия бикарбонат 271
Натрия бисульфит 42, 49, 61
Натрия гидроокись 12, 23, 29, 33, 41,
49, 60, 91, S3, 142, 144, 164, 179,
188, 195, 202, 207, 242, 248, 292,
309, 327, 333, 341, 351, 352; 374,
384, 387, 412, 458, 498, 509, 514,
533, 552, 587.
Натрия метабисульфит 552
1,2-Нафталевый ангидрид 347
Нафталин 27
р-Нафтиламин 557
р-Нафтилртуть бромистая 558
р-Нафтилртуть хлористая 557
а-(р-Нафтил)-этиламин 525
а-Нафтойная кислота 348
р-Нафтойная кислота 351
а-Нафтол 49
Р-Нафтол 42
2-Нафтол-3,6-дисульфокислота 347
1-Наотол-4-сульфокислота 439
2-Нафтол-6-сульфокислота 437
1,2-Нафтохинон 353
Невиля-Винтера кислота 439
Нитрил малоновой кислоты 355
Нитрил-а-фенол-^-бензоилпропионо-
вой кислоты 358
в-Нитроанилин 228, 509
л-Нитроанилин 198, 227, 383
1-Нитро-2-ацетилнафтиламин 359, 379
.м-Нитроацетофенон 361
Нитробарбитуровая кислота 365, 491
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
639
л-Нитробензальдегид 551
л-Нитробензальдегид 366
л-Нитробензальдиацетат 366
л-Нитробензилбромид 368
Нитробензол 13
о-Нитробензолдиазония борофторид
228
л-Нитробензолдиазония борофторид
227
в-Нитробензолсульфонилхлорид 404
e-Нитродифениловый эфир 371
л-Нитродифениловый эфир 370
Нитрозодиметиламин 199
л-Нитрозодиметиланилин солянокис-
лый 225
л-Нитрозодиэтиланилин 226
л-Нитрозодиэтиланилии солянокис-
лый 226
N-Нитрозометиланилин 372
Нитрозометилмочевина 174, 373
Нитрозометилуретан 376
Нитрозо-р-нафтол 43, 51
Нитромезитилен 377
1-Нитро-2-нафтол 379
2-Нитротиофен 381
л-Нитротолуол 366, 368
л-Нитрофенилизоцианат 383
л-Нитрофенол 186
2-Нитрофлуорен 385
4-Нитрофталевая кислота 387
4-Нитрофталимид 387, 389
в-Нитрохлорбензол 371
Нонаметиленгликоль 171
Окись меди 18
Окись палладия, катализатор 445
Окись платины, катализатор 196, 4Р9
Окись ртути 58, 287, 506
Окись серебра 21, 37
Окись тория 313, 315
Окись углерода 464, 465
л-Оксибензойная кислота 390
2-Окси-3,5-дииодбензойная кислота
392
4-Окси-3,5-дииодбензойная кислота
393
Оксим бензофенона 369, 393
Оксим вератрового альдегида 146
Оксим циклогексанона 90, 396, 304
Осим энантола 154, 394
р-Оксиэтилметилсульфид 396, 562
Октаацетат а-целлобиозы 398, 564
Окгадекаметиленгликоль 171
Олеиловый спирт 401
Оливковое масло 402
Олово хлористое 45, 49, 263, 338,
551, 606
Олово хлорное 76
Оранж I 49
Оранж II 44
Ортаниловая кислота 403
Ортомуравьиный эфир 296
Палладий 445
Палладия окись, катализатор 445
Панкреатин 474
Паральдегид 102
Параформальдегид 343
Паста кровяных телец 160
Пеларгоновая кислота 406
Пентаметилбензол 254
н.-Пентан 408
Пентаэритрит 443
Пивалоин 141
Пимелиновая кислота 410
Пиперонал 417, 429
Пиперониловая кислота 417
Пиперониловый спирт 462
Пиридин 35, 138, 309, 333, 518, 559
Пирогаллол 147
Пиромеллитовая кислота 418
Пиромеллитовый ангидрид 430
Платины окись, катализатор 196,499'
/-Пропилепгликоль 421
н.-Пропиловый спирт 424
н.-Пропилсульфат 136
н.-Пропилсульфид 422 •
н.-Пропилсульфит 137
N-Пропил-л-толуидин 607
н.-Пропилфосфат 139
Пропионовый альдегид 424
Пропионоин 141
о- и л-Пропиофенол 426
Пропиофенон 264, 266
Протокатеховый альдегид 428
В-Резорциловая кислота 120, 430
Резорцин 430
Рейнеке соль 432
Рицинолеиновая кислота 9
R-соль 437
Ртути окись 58, 287, 506
Ртуть 418
Ртуть хлорная 161, 487, 511, 557
Сааициловая кислота 330, 392, 41-5
Сахар 338, 421
Сахар в порошке (пудра) 338
Сахароза 421
Себациновая кислота 345
Селен 237, 507
Селена окись 507
Сера 102, 166, 572
640
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Сера хлористая 241
Серная кислота 15, 18, 38, 51, 54,
68, 81, 91, 121, 131, 148, 164, 176,
205, 219, 264, 266, 288, 296, 304,
315, 329, 331, 346, 361, 366, 380,
398, 402, 416, 418, 425, 462, 463,
467, 469, 554, 56"; 567. 571, 578,
581, 597
Серная кислота дымящая 58
Сернистый газ 91, 166, 572
Сернокислая медь 276, 371, 501, 533,
552
Сероводород 37, 161, 423, 452, 479
Сероуглерод 426, 522
Соли тг-толуидина и фенантрен-2- н
3-сульфокислоты 495
Соль /-а-фенилэтиламина и ^-винной
кислоты 527
Соль(/-а-фенилэтиламина и /-яблочной
кислоты 526
Соль Шеффера 439
Сочетание о-толидина и Чикаго-кис-
лоты 434
Сукцинимид 430
Сульфаниловая кислота 44, 48
Сульфокислоты, их идентификация
495
Сульфурил хлористый 135
Таурин 441
Тетрабромнеопентаи 443
Тетрагидрофуран 445, 450
Тетрадекаметиленгликоль 111
Тетраиоднеопентан 444
а-Тетралон 448
Тстраметиленхлоргидрии 450
Тетрахлорэтилен 153
Тнантрсн 242
Тиобензофенон 452
р-Тиодиглнколь 453
Тиомочевипа 40, 320
Тионил хлористый 137 181, 449, 559
562
Тионнл хлористый, его очистка 449
Тиосалицмловая кислота 455
Тиофен 76, 287, 381, 458, 522
Тиофен, его выделение 288
л-Тиоциандиметиланилин 431
/-Тирозин 476
о-Толидин 434
л-Толилкарбинол 461
а-л-Толилэтиламин 525
л-Толуидип 605
о-Толуиловая кислота 462
л-Толуилопая кислота 463
л-Толуиловый альдегид 464
о-Толунитрил 29, 463
Толуол 465, 512, 578
Три-н.-амилкарбинол 490
симм.-Трибромбензол 467
Три-н.-бутилкарбинол 490
Три-н.-гептилкарбинол 490
Тридекаметиленгликоль 171
1,2,3-Трииод-5-нитробензол 469
Трикарбометоксиметан 471
Трикарбоэтоксиметан 472
Триметиламин 100
Триметилэтилендибромид 324
2,4,6-Тринитрофенилгидразин 230
Трипановый голубой 438
Три-н.-пропилкарбинол 490
/-Триптофан 474
симм.-Тритиан 479
Трифеиилметилнатрин 481
Трифенилселен хлористый 239
Трифенилхлорметан 483
Трифенглэтилен 483
Трихлорэтиловый спирт 485
Триэтилкарбинол 4S9
Углекислота 314, 349, 430
Уксусная кислота 87, 89, 93, 191,
198, 209, 314, 337, 358, 381, 404,
456
Уксусный альдегид 322
Уксусный ангидрид 11, 68, 69, 72,
147, 196, 231, 336, 377, 381, 398,
497
Ундекаметпленгликоль 171
Ундециловый спирт 307
Урамил 491
Фенацтрен 493
Фенантреи-2- и 3-сульфокпслоты 493
Фенантрсн-2-сульфокислоты бариевая
соль 494
Фенантрен-3-сульфокислоты калиевая
соль 494
Фенацилбромид 116
dZ-fi-Фенилаланин 496
N-Феннлантраниловая кислота 18
Фениларсиновая кислота 501
а-Фенилацетоацетонитрил 316, 503,
596
Фенилацинитроацетоиитрила натрие-
вая соль 513
Фенилбензоилдиазометан 506
Фенилглиоксаль 507
Фенилдиазония борофторид 227
о-Фснилепдиамин 83, 85, 511
о-Фепилендиамина двухлористоводо-
родная соль 510
Фениллитий 517
у-Фепплмасляпая кислота 54, 449, 511
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
641
Фенилнитрометан 513
Фениловый эфир 529
Фениловый эфир пропноновой кисло-
ты 426, 427
2-Фенилппридин 517
Фенилпировиноградная кислота 518
Фенилпропиоловая кислота 520
Фснилтиенилкетон 522
Фенилуксуспая кислота 81, 167, 231,
314
а-Фенилэтиламин 523, 526
d- и /-з-Фепилэтиламин 526
р-Фенилэтилфталимид 84
Фенокстин 529
Фенол 427
Фильтрование обратное 479
Фильтр с асбестом и углем 17
Фильтр с мягкой фланелью 16
Флавиаповая кислота 61
Флороацетофенон B,4,6-триоксиацето-
фенон) 531
Флороглюцин 531
Флуорен 385
Формалин 214, 461, 479
Формальдегид 214, 461, 480
Фосген 383
Фосфора хлорокись 56, 138
Фосфор желтый 281
Фосфор красный 89, 183, 280, 281, 497
Фосоорная кислота 469, 569
Фосоор пятисернистый 460
Фосфор пятихлористый 355, 428, 452,
547, 554
Фосфор трехбромистый 115, 116, 443
Фосфор трехсернистый 458, 459
Фталевый ангидрид 80, 547
Фталид 533
Фталимид 84, 389, 533
Фталимидкалий 37
л-Фторбензойная кислота 534
Фторбензол 537
Фтористоводородная кислота 227, 228,
245, 535, 538
Фумаровая кислота 191, 541
Фурян 446
Фурфурол 541
Хелидоновая кислота 543
Хинон 545
Хлор 32, 188, 351, 392, 403, 554, 560
4-8-хлор-2,4-динитротиофенол 561
5-8-Хлор-о-нитротиофенол 560
S-Хлор-о-нитро-л-хлортиофенол 561
Хлораль 485
Хлоральгидрат 247
симм.-и несимм. Хлорангпдрпд о-фта-
левой кислоты 547
а-Хлораптрахинон 549
Хлорацстон 40
.м-Хлорбензальдегид 551
л-Хлорбепзальдешд 554
«-Хлорбензойная кислота 18, 556
л-Хлорбензойпая кислота 554
7-Хлорбутиронитрил 37
я-Хлоргпдрипден 157
Хлористая .S-нафтилртуть 557
Хлористый ацетил 56. 76, 78
Хлористый бензил 30, 84, 234, 484
Хлористый оензонл 92, 159 202 522,
582, 584
Хлористый дезил 559
Хлористый метил 253. 256
Хлористый сульфурил 135
Хлорная ртуть 162, 487, 511, 557
Хлорное железо 263, 353
Хлороформ 153, 262
2-Хлорртутьтиофен 76
о-Хлортолуол 556
л-Хлортолуол 554
Хлоругольный эфир 212, 473, 587
Хлоруксусная кислота 158, 309, 327,
581, 608
Хлорэтилметилсульфид 341, 562
Хлорфенилэтиламин 525
а,л-Хлорфенилэтиламин 525
Холестанон 563
Холестерилацетат 196, 197
Холестерин 195
Хромовый ангидрид 157, 336
Царская водка 445
Целлобноза 564
Цетпл йодистый 148, 280
Цетиловый спирт 280, 307
Циангидрин ацетона 566
Цианистый бензил 6, 503, 514
Цианоацетамид 355
л-Цианобензальдегид 368
Циклогексанол 568
Циклогексанон 91, 411
Цпклогексаноноксим 90, 304, 396
Циклогексен 128, 187, 567
Цнклогексиламин 155
Циклогексилбензол 567
Циклогексил бромистый 114
Цпклопентанол 569
Циклопсптен 569
л-Цимол 77
Цинк 148, 200, 210, 241, 337, 371,
386, 456, 510, 533
Цинк-медная пара 249, 250
Цинк хлористый 147, 531
Цитраконовая кислота 570
Цитраконовый ангидрид 570
41 Сборник № 2
642
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
643
Чикаго-кислота 435
Чикаго-кислота, ее очистка 436
Чикаго-кислота, сочетание с е-толн-
дином 434
Шеффера соль 439
Щавелевая кислота 331
Энантовая кислота 571
Эиантол 571
Эпибромгндрип 573
Эпихлоргидрнн 574
Эруковая кислота 575
Этаноламии 126
Этил бромистый 212, 250, 251, 489,
605
Этил иодистый 250, 251, 284
N-Этил-л-толуидин 605
Этил-^-фенилэтилкетон 316
Этилат алюминия 485, 486
Этилбензилкетон 316
Этилендибромид 124
Этиленхлоргидрин 248, 397
Этилнитрнт 205
Этилового эфира глицина хлористо-
водородная соль 577
Этилового эфира фталимидмалоновой
кислоты натриевое производное
340
Этиловый спирт 152, 205, 276, 345,
467, 577, 593
Этиловый спирт абсолютный 53, 67,
79, 100, 154, 170, 171, 220, 296,
307, 340, 373, 384, 396, 410, 416,
422, 452, 472, 485, 503, 513, 543,
580, 578, 588, 589, 599, 600
Этиловый эфир 30, 53, 193, 211, 259,
295, 310, 321, 348, 481, 483, 489,
592
Этиловый эфир абсолютный 30, 53,
193, 212, 259, 295, 310, 321, 348, 481,
483, 489, 592
Этиловый эфир адипииовой кислоты
578
» » азотистой кислоты
205
» » л-алщнобензойной ки-
слоты 534
» » а-ацетоглутаровой
кислоты 581
» » fj-ацетотрнкарбаллн-
ловой кислоты 581
>> >> ацетоуксусной кис-
лоты 209, 214, 335,
580, 584
>> » ацетоянтарной ки-
слоты 580, 581
Этиловый эфир бензонлацетоуксус-
ной кислоты 584
* » бензоилдиметилуксу-
сной кислоты 582
» » бензоилуксусной ки-
слоты 584
>> » бензойной кислоты 87
>> » броммалоновой ки-
слоты 601
>> » бромпроппоновой ки-
слоты 581
» » вторично-бутлпа-
лоновой кислоты 31 &
» » а,3-дибром->фенил-
пропионовой кис-
лоты 586
» » изомасляной кисло-
ты 582
>> » изопропилмалоновой
кислоты 107, 108
» » 2-кетогексагидробен-
зойной кислоты 412
» >> 2-кетоциклогексил-
глиоксалевой кис-
лоты 411
» » коричной кислоты
586
» » лауриновой кислоты
306
» >> малоновой кислоты 79,
221, 406, 472, 580
» >> и.-масляной кислоты
140
» » N-метилкарбамино-
вой кислоты 587
» » метплмалоновой ки-
слоты 288, 588
» » монохлоруксусиой
кислоты 100 580,
581
» >> муравьиной кислоты
75
>> » муравьиной кислоты
(его очистка) 75, 332
» » а-нафтойной кислоты
592
» » пентадецпловоп кис-
лоты 595
>> » пнме.'шновой кислоты
415
» » пропионовой кислоты
604
>> |) себациновой кислоты
170, 345, 346
» » а, а, ¦;, у-тетраметил-
ацетоуксусной ки-
слоты 584
Этиловый эфир н.-тридецнловой ки-
слоты 593
» » угольной кислоты
490, 592
» » угольной кислоты
(его очистка) 593
>> |) fl-ураминокротоповой
кислоты 336
» » а-фенилацетоуксус-
ной кислоты 317,
596
» . » фенилмалоновоп ки-
слоты 599
» » у-фенилмасляной ки-
слоты 54, 55
» >> фенилуксусной кис-
лоты 600
» >> феннлцианпнровино-
градной кислоты
600
» » фталимидмалоновой
кислоты 340
Этиловый зфир хелидоновой кисло-
ты 544
» » щавелевой кислоты
54, 411, 544, 601,
604
» » этилентетракарбоно-
вон кислоты 601
>> >> этоксалилпроппоно-
вой кислоты 604
» •> а-этоксалил-у-фенил-
масляной кислоты
504
» :> зтоксиуксусной ки-
слоты 608
Этоксиуксусная кислота 607
Эфир (см. Этиловый эфир)
Эфир ортомуравьиный 296
Эфир уксусноэтиловын 503
Янтарная кислота 56
Янтарный ангидрид 56, 95
УКАЗАТЕЛЬ СОСТАВИТЕЛЕЙ РЕЦЕПТУРЫ
Абботт Т. (Abbott Т. W.) 71, 520, 586
Адаме P. (Adams Roger) 118, 123, 158,
176 259, 287, 361, 381, 408, 452,
455, 458, 471, 522, 541, GOO
Адкипе Г (Adkins Homer) 483, 580
Айяи В. (lde W. S.) 11, 32, 145, 164,
551
Аллен К. (Allen С. F. H.) 18, 58, 77,
81, 95, 156, 167, 218, 223, 229, 343,
358, 374, 455, 517, 549
Альтхаузен Д. (Althousen Darrell) 586
Аменд В. (Amend W. J.) 149
Амштутц Е. (Amstutz E. D.) 145, 374
Амштутц К. (Amstutz К. L.) 541
Арндт Ф. (Arndt F.) 174, 373
Арнольд P. (Arnold Richard T.) 71,
189, 410
Арнольд X. (Arnold H. R.) 301
Бабасинян В. (Babasinian V. S.) 381
Бабкок С. (Babcock S. Н ) 158, 183,
233, 241, 333
Вадвар И. (Badhwar I. С.) 147
Вайерс Дж. (Byers J. R.) 40, 244, 280,
379, 534, 537
Валлард М. (Ballard Murel M.) 121
Валлард Е. (Ballard E. С.) 481, 582
Баннерот P. (Bannerot R.) 401
Барджер Дж. (Barger G.) 340
Варкер В. (Barker W. Е.) 167
Баррик П. (Barrick P. L.) 160, 278,
327, 605
Бахман В. (Bachmann W. Е.) 97, 08
Бахман Дж. (Bachman G. В.) 295
Беберс Ф. (Babers Frank H.) 329, 513
Бедертчер Д. (Badcrtscher D. Е.) 114
Бек Дж. (Buck Johannes S.) 11, 32,
142 145, 164, 428, 551, 605
Велл Е. (Bell E. V.) 224
Беннет Дж. (Bennett G. М.) 224
Бенсон В. (Benson W. L.) 601
Бергштейнсон И. (Bergsteinsson 1.)
174, 584
Вернетт P. (Burnett Robert E.) 109,
112
Берчет Дж. (Berchet G. J.) 327
Бигелов Л. (Bigelow Lucius A.) 242
Бигелов X. (Bigelow Н. Е ) 12
Бин X. (Bean H. J.) 34
Битти P. (Beattie R. W.) 222, 557
Блолжвист A. (Blomquist А. Т.) 338
Блюменталь Д. (Blumenthal D.) 533,
600
Блэк A. (Black Alvin P.) 329, 513
Бост P. (Bost R. W.) 422, 479
Боуден Э. (Bowden Everett) 331
Бранд Е. (Brand E.) 60
Браун Г. (Braun Geza) 183, 398, 564,
573
Браун P. (Brown R. E.) 317
Брезен M. (Brethen M. R.) 587
Брейер Ф. (Breuer F. W.) 153
Брукс Л. (Brooks L. A.) 189, 410
Брюс В (Bruce Williams F.) 15, 195,
563
Брюстер P. (Brewster R. Q.) 271, 370,
431
Буллард P (Bullard R. H.) 237, 239,
501
Бумер Дж. (Boomer G. L.) 102, 434,
493, 573
Бусвелль P. (Buswell R. J.) 345
Бускет E. (Bousquet E. W.) 394
Буш M. (Bush M T ) 531
Бэйли К. (Bailey С. F.) 34, 441, 587
Бэр Л. (Behr Letha Davies) 20, 439
Бюлер К. (Buehler С. А.) 390
Вагнер E. (Wagner E. C.) 85
Вейксельбаум Т. (Weichselbauin Т.Е.)
340
Вейнер Н. (Weiner Nahtan) 309, 589
Вейс Р. (Weis Richard) 80
Вейсбергер A. (Weissberger A.) 18
Вейсс М. (Weiss Marvin) 461
Венкатараман К. (Venkataraman К.)
147 531
Вертхейм E. (Wertheim E.) 403
Вилькоксон Т. (Wjlcoxon В. Н.) 131
Виндус В. (Windus Wallace) 562
УКАЗАТЕЛЬ СОСТАВИТЕЛЕЙ РЕЦЕПТУРЫ
645
Винкельмюллер В fWinkelmuller W )
244, 534
Виньо, дю (V. clti Via;neaud) 107
Butt К. (Witt de С. С.) 37
Виттум П. (Vittum P. W.) 164, 551
Витцеман Е. (Witzemann E. J.) 64,
65, 67, 163
Влиет Е. (Vliet E. В.) 317
Возе К (Vose С. Е.) 355
Войцик Б. (Wojcik В. Н.) 158, 321
365, 491, 589, 607
Вуднард Ч. (Woodward Charles F.)
9, 77, 214, 343, 422, 467, 592
Вудруф Е. (Woodruff E. H.) 452
Вуллётт Г. (WooIIett G. Н.) 392
Вун В. (Woon W. S.) 295
Гарднер Дж. (Gardner J. H.) 27, 533
Гармон Дж. (Harmon J.) 355
Гархарт X. (Gerhart H. L.) 135, 136
Гильман Г. (Giiman Henry) 12 30
84, 152, 205, 249, 317, 326, 348, 464,'
520, 547, 554, 556, 586
Гордон Дж. (Gordon j. J.) 607
Графф С. (Graff S.) 60, 417 418
Грей A. (Gray A. R.) 79, 507
Тренинг Т. (Groenjng Theodore) 370
Грисвольд A. (Griswold A. M.) 107
Гулати К. (Gulati К. С.) 531
Турин С. (Gurin S.) 340, 306, 562
Давидсон Д. (Davidson David) 461
Дамерелл В. (Damerell V. R.) 205
Дек Дж. (Dec J.) 374
Джекобе Т. (Jacobs T. L.) 297
Джекобсен P.(Jacobsen Robert P.) 105,
563
Джефферсон Дж. (Jefferson George D.)
503, 596
Джиллеспи X. (Gillespie H. В.) 496
Джонс Ф. (Jones F. W.) 351
Джонсон Дж. (Johnson John R) 11,
32, 76, 80, 97—98, 133, 138, 142,
145, 160, 164, 193, 218, 278, 285,
327, 338, 366, 390, 445, 450, 513
531, 551, 567, 580, 605
Джонсон Ф. (Johnson F. R.) 377
Джонсон В. (Johnson W. W.) 392
Джудкинс С. (Judkins S. L.) 577
Джулиан П. (Julian Percy L.) 316, 503
Дикки Дж. (Dickey J. В ) 23, 40, 70,
93, 120, 133, 148, 172, 185, 187,
241, 244. 280, 370, 377, 379 383, 464,
496, 501, 534
Дпнсмор Р. (Dinsmore R.) 114
Долливер М. (Dolliver Morris A.) 179
Дипливи Дж. (Donleavy John J.) 335
Дреджер E. (Dreger E. E.) 123
Дрэк В. (Drake W. V.) 198
Дрэк Н. (Drake Nathan L.) 321
Дэвидсон Л. (Davidson L. H.) 116
Дэйнс Ф. (Dains F. B.) 228 .
Дэкин X. (Dakin H. D.) 432
Дюттон Г. (Dutton G. R.) 138
Жирар A. (Girard Andre) 100
Запберг М. (Sandberg M.) 60
Зингер A. (Singer Alvin) 214
Ингерсолл A. (Ingersoll A. W.) 158,
523, 526
Ингольс Р. (Ingols R. S.) 432
Исбелл Н. (Isbell Neville) 580
Кайз М (Kise Mearl A.) 335
Кайзер E. (Kaiser E. W.) 116
Калвери X. (Calvery H. O.) 600
Кан Э. 222
Каплан Дж. (Kaplan J. F.) 149, 301
Каротерс В (Carothers W. H.) 20, 23,
109, 112, 128, 154, 170, 181, 220,
248, 271, 280 368, 394, 424, 431,
439, 443, 453, 472, 501, 593
Карсон Дж. (Carson J. F.) 231
Картер X. (Carter H. E.) 160
Кемп К. (Kemp С. R.) 172, 235
Кемпбелл' В (Campbell W P.) 156,
398 564
Кендалл Ф. (Kendall F. E.) 430
Кеннеди E. (Kennedy E. R.) 278
Кеннаи В. (Kennan W. M.) 292
Кимболл Р (Kimboll R. R.) 358, 503,
596
Кинг Г. (King Harold S.) 281, 474
Киноман С. (Kinsman S.) 53, 347
Кирнер В. (Kirner W. R.) 562
Кларк Дж. (Clark John R.) 247
Кларк X. (Clarke H. T.) 20, 34, 60,
292, 340 396, 417—418 432, 439,
462, 474, 533, 556, 562, 575
Клегг Дж. (Clegg J. W.) 345
Клейдерер E. (Kleiderer E. C.) 417
Клиббенс D. (Clibbens D. A.) 430
Кливленд Е. (Kleveland E. A.) 60. 72,
76, 366, 46f, 498, 517, 518, 529, 567,
589
Киауф A. (Knauf A E ) 381, 455, 471
Коган X. (Cogan H. D.) 287 '58, 522
Коид A. (Koide A. T.) 331
Кокернлл Ф. (Cockcrille F. O.) 401
Кокс Дж. (Сох Gerald J.) 474
Кокс В. .мл. (Cox W. M.) 401
64G
УКАЗАТЕЛЬ СОСТАВИТЕЛЕЙ РЕЦЕПТУРЫ
Кокс Р (Сох R. F. В.) 383, 566, 588,
604
Котеман Дж. (Coleman G. Н.) 105,
193, 368, 464, 467
Кон Е. (Cohn E. J.) 292
Конант Дж. (Conant J. В.) 41, 224,
358, 587, 600
Конард Ч. (Conard Charles R.) 179
Кони М. (Conn M. W.) 422
Коннор P. (Connor Ralph) 247, 366
Констэбль Е. (Constable E. W.) 479
Кои А. (Соре Arthur С.) 247
Корсон Б. (Corson В. В.) 41, 68, 231,
232/ 355, 361, 471, 567, 601
Кортиз Ф. (Cortese Frank) 126, 441
Коунер P. (Cowper R. M.) 116
Крейгер П. (Krueger Paul A.) 273,
276
Криммель К. (Krimmel С. R.) 389
Кросслей Ф. (Crossley Frank) 264
Крэг Д. (Craig David) 193, 464
Кук Дж. (Cooke Giles В.) 321
Кун В. (Kuhn W. E.) 385
К:>рнс Т. (Cairns T. L.) 469
Кэт В. (Cate W. Е.) 390
Лайкан В. (Lycan W. Н.) 154
Ланге Н. (Lange N. А.) 93
Лахман A. (Lachman Arthur) 269, 393
Левин П. (Levene P. А.) 74, 103, 148,
421, 599
Леджер Ф. (Leger Frank) 198
Лейсестер Г. (Leicester Henry M.) 237,
239
Леланд Дж. (Leland Jessica P.) 474
Леман М. (Lehman M. R.) 140
Леффлер М. (Leffler M. Т.) 25
Лпберман С. (Ljebermann S. V.) 366,
373
Лини К. (Linn Carl) 27
Литтл Дж. (Little |. R.) 414
Лиу Ч. (Liu Chuan) 554
Лов К. (Love С. F ) 313
ЛодерД. (Loder D. J.) 51, 124, 306,
441, 489, 592
Лозе X. (Lohse H.) 179
Лонг Ф. (Long F. А.) 102
Лунд X. (Lund Hakon) 472
Лэзир В. (Lazier W. А.) 149, 301
Люкас X. (Lucas H. J.) 278
Люкс A. (Lux A. R.) 114
Майерс P. (Myers R. R.) 374
Мак-Глемфи Дж. (McOIumphy J. H.)
556
Мак-Дер.мот Ф. (McDermott F. А ) 260
Мак-Кен Д. (MacKay D. D.) 455
Мак-Ки Г. (McKee G. Н. W.) 18
Мак-Ки P. (McKee R. А.) 120
Маклеод Л. (Macleod L. С.) 81
Мак-Миллан Ф. (McMillan F М ) 483,
511
Максвелл Ч. (Maxwell Charles F.) 529
Мак-Эльвеи С. (McEIvain S M ) 140,
214, 333, 588, 604
Мак-тЮвен В. (McEwen W. L.) 9, 20,
23, 109, 112, 154, 170, 181, 220, 248,
271, 280, 368, 394, 424, 439, 443
453, 472, 501, 554, 593
Манске P. (Manske Richard H. F.) 84
170, 313
Марвел К. (Marvel С. S.) 30, 34, 89,
90, 107, 124, 126 140, 154, 167 304
306, 308, 317, 321 331, 333, 355,
365 441, 462, 489^ 491, 559, 577,
601
Мартин Е (Martin E L.) 56, 448, 509,
511
Мейер Дж. (Meyer G. М.) 401, 599
Мейнерт P. (Meinert R. N.) 424
Мельтреттер К. (Mehltretter С.) 249
Милас Н. (Milas Nicholas A.) 541
Миллер Э. (Miller Ellis) 426
Миллетт В. (Millett W. Н.) 85
Миннис В. (Minnis Wesley) 287, 522
Мичовнч В. (Micovic V. M.) 578
Мойер В. (Moyer W. W.) 259, 361,
462, 489
Моран Дж. (Moran John) 410
Моффетт Е. (Moffett E. W.) 23
Мун Н. (Moon Neil S.) 247
Муиро X. (Munro H. E.) 601
Мюллер A. (Muller Adolf) 414
Мэй Дж. (May G. Е.) 76
Мэхуд С. (Mahood S. А.) 176
Мьюнич В (Munich William) 89, 90.
308
Негер Г. (Neher Harry T.) 04, 65, 67,
163
Нейлор К. мл (Naylor С. A. jr.) 27,
533
Нешщеску К. (Nenitzescu Costin D.)
506
Несмеянов А. 222, 557
Никольс К. (Nichols С. Р.) 390
Ниренштейн М. (Nierenstein M.) 430
Нойес В. (Noyes W. А.) 131
Ноллер К. (Noller С. R.) 27, 29, 44,
48, 53, 56, 87, 89, 90, 114, 131, 135,
136, 138 172 174, 179, 209, 211,
216, 231, 235, 249 266, 268, 269,
290 295 308 309' 312 313 316
347, 348^ 353, 373, 393, 401, 408,
УКАЗАТЕЛЬ СОСТАВИТЕЛЕЙ РЕЦЕПТУРЫ
047
448, 483, 503, 511, 570, 571, 575,
584, 596, 607
Ньюмен М. (Newman M. S.) 195, 351
О'Бриен Е. (O'Brien Helen) 358
Озанн (Ozanne 1. L.) 123
Оливер Дж. (Oliver John J.) 316, 503
Остергоф A (Osterhof A. L.) 41
Отт Э. (Ott Erwin) 547
Оттербахер Т. (Otterbacher Т.) 152
Пайк A. (Pike Arthur B.) 503, 59G
Пакауд Р. (Pacaud R. A.) 156
Пальмер A. (Palmer Albert) 12
Пассино X. (Passino H. J.) 285
Патт М (Patt M.) 571
Петерсон В. (Peterson W. D.) 199, 319
Летерсон Р. (Peterson R. F.) 264
Пиикни П. (Pinkney P. S.) 297
Поттер Д. (Potter D. J.) 545, 560
Поуэлл Г. (Powell Garfield) 377
Прилл Е. (Prill E. A.) 333
Прохзска Ф. (Prochaska F\ J.) 547
Пуитамбекер С. (Puntambeker S. V.)
154
Райан Дж. (Ryan [ohn) 189
Райт Дж (Wright G. F.) 520, 586
Papc E. Дж. (Rahrs E. J.) 25, 392
Рейнсмит Г. (Rhinesmith Herbert S.)
191
Ренфроу В. мл (Renfrow W. В. Jr.)
87, 481, 582
Ригель В. (Riegel Byron) 40
Ригель E. (Riegel E. Raymond) 543
Рид Э. (Ried E. Emmet) 109, 112, 401,
406
Рилей X. (Riley H. A.) 507
Ринкес И. (Rinkes I. J.) 338
Робинсон Дж. (Robinson J.) 30
Ролл Л. (Roll L. J.) 147, 290, 312,
323, 337, 408, 426, 570, 584
Ролле Дж. (Rails J. O.) 105
Росс В (Ross W. E.) 79, 191, 335, 398,
428, 507, 523, 526, 543, 564, 588,
604
Россаидер С. (Rossander S. S.) 30
Ротрок X. (Rothroek H. S.) 323
Рухофф Дж. (Ruhoff John R.) 109,
112, 128,401, 406, 571,593
Сайнерхолм М. (Synerholm M.) 87,
135, 136, 309, 503
Салианн Н. (Saliani N. A.) 68
Сауди Дж (Sawdey G. W.) 85, 387,
406
Саусгэт Г. (Southgate Harriet A.) 249
Сванн Ш. мл. (Swann Sherlock) 345
Сент Джон Н (St. John Nina B.) 348
Сет С. (Seth S. R.) 531 .
Симон В. (Semon W. L.) 205
Скорроу Дж. (Scarrow J. A.) 343
Скотт В. (Scott W. J.) 58, 549
Скотт Р. (Scott R. W.) 355
Смит Л. (Smith Lee Irvin) 29, 116, 118,
148, 189, 227, 241, 242 253, 259,
2G2, 345, 359, 410, 481 582
Снайдер X. (Snyder H. R.) 11, 80, 97,
98, 142, 164, 189, 410, 496, 580
Снелл Дж. (Snell John M.) 140
Соломоника Е (Solomonica Eugen)
506
Сомервилл Л. (Somerville L. F.) 95
Спарберг M. (Sparberg M. S.) 124
Старки E. (Starkev E. B.) 227
Старр Д. (Starr Donald) 445, 450
Стемпфли Дж. (Stampfli J. G.) 187
Стерне X. (Stearns H. A.) 118
Стивенсон X. (Stevenson H В K2, 193,
218, 329, 445, 450, 513
Стормонт P. (Stormont R. T.) 566
Сутер К. (Suter С. М.) 135—136, 529
Сэйр Дж. (Sayre J. L.) 471
Сэндин P. (Sandin R. B.) 120, 198, 469
Сюзерленд Л. (Sutherland L. H.) 273,
276, 469
Талбот В. (Talbot William F.) 467
Талбот P. (Talbot R. H.) 575 -
Телен Р. (Thelen R.L18
Темпл Р. (Temple Ralph S.) 506
Тишлер М (Tishler Max) 81, 121
Тодд X. (Todd H. R.) 220
Томпкинс С. (Tompkins S. W.) 133
Томпсон Р. (Thompson Ralph B.) 71
Тэйлор E. (Taylor E. R.) 462, 556, 575
Уайт В. (White W. R.) 44, 48, 353
Уиндус У. (Windus Wallace) 326, 396
Уитмор М (Whitmore Marion M.) 68,
198, 232
Уитмор Ф. (Whitmore Frank C.) 41,
51, 64, 65, 67, 68, 74, 103, 107, 114,
124 152, 153, 162, 198, 222, 223,
253, 262, 273, 276, 306, 323, 389,
421, 441, 469, 489, 557, 592
Унгнаде X. (Ungnade H. E.) 227
Ундервуд X. мл. (Underwood H. W.
Jr.) 545
Уоллингфорд В. (Wallingford V. H.)
273, 276
Уолти A. (Walti A.) 74, 421
Уолш В. (Walsh W. L.) 545
648
УКАЗАТЕЛЬ СОСТАВИТЕЛЕЙ РЕЦЕПТУРЫ
УКАЗАТЕЛЬ СОСТАВИТЕЛЕЙ РЕЦЕПТУРЫ
649
Уокер Дж. (Walker Joseph T.) 181,
337, 359, 372, 376, 379, 385
Уорд A. (Ward A. M.) 559
Уэллс Ф. (Wells F. В.) 223
Фабер Е. (Faber E. М.) 453
Фарлоу М. (Farlow Mark W.) 153, 229
Ферри'К. (Ferry Clayton W.) 605
Физер Л. (Fieser Louis F.) 15, 36, 40,
44, 48, 51, 53, 56, 58, 81, 100 121,
156, 185, 187, 195, 281, 297, 304,
337, 347, 353, 359, 372, 376, 379,
385, 434, 448, 493, 506, 507, 545,
549, 560, 563, 577, 599
Филиппи E. (Philippi E ) 418
Филлипс Р. (Phillips Ross) 233, 376,
Фирке С. (Fierke S. S.) 509
Фишер К. (Fisher С. Н.) 15, 36, 185,
187, 304, 507, 599
Фишер Г. (Fisher Hans) 209, 211, 216
Флеминг К. (Fleming) 126, 441
Флоод Д. (Flood D. Т.)
Фозерджилл P. (Fothergill R.) 205
Фойгт A. (Voigt Axel) 472
Форд С. (Ford S. G.) 290, 306, 312,
570
Форман Д. (Forman Don. В.) 95
Фостер Дж. (Foster G. L.) 160
Фрейденбергер X. (Freudenberger H.)
452
Фриборн Э. (Freeborn Emeline) 232
Фьюгэт У. (Fugate Wesley) 414
Фыозон P. (Fuson Reynold'С.) 9, 69,
71—72, 76—77, 79, 95, 181, 183,
191, 202, 214, 229, 233, 241, 264, 288,
335, 343, 398, 403, 414, 422 428,
461, 467, 479, 485, 498, 517—518,
523, 526, 529, 543, 564, 566, 578,
588—589, 592, 604, 607
Хазен P. (Hazen R. К.) 361
Ханслик P. (Hanslick Roy S.) 242
Хант A. (Hunt Anthony) 138
Харвуд X. (Harwood H. J.) 12, 84,152
Хармон Дж. (Harmon J.) 128
Хартвел Дж. (Hartwell J. L.) 434
Хартман В. (Hartman W. W.) 18 23,
25, 29, 37, 85, 93, 102, 105, 120,
123, 133, 147—148, 172, 185, 187,
199, 233, 237, 239 241—242, 244,
248, 280, 285, 319, 323, 337, 351,
359, 365, 370, 372, 376, 377, 379,
383, 387, 392, 406, 426. 434, 4S3, 491,
493, 496, 509, 534, 537, 573, 587
Хартупг В. (Hartung Walter H.) 264,
420
Хасс Г. (Hass Henry) 64, 65, 67, 163
Хатт X. (Hatt H. H.) 199, 202, 31«
Хаузер Ч. (Hauser C. R.) 87, 481, 582
Хентресс К. (Huntress E H ) 387, 389
Хербст P. (Herbst R. M.) 69, 72, 313,
498, 518
Херд Ч. (Hurd Charles D.) 424
Хершберг E. (Hershberg E. B.) 53, 58,
128, 347, 549
Хиксон P. (Hixon R. M.) 445, 450
Хилл Дж. (Hill J. W.) 9, 149
Холленд В. (Holland W. E.) 71
Холлингсхед Дж. (Hollingshead J.
Pauline) 74, 103, 421
ХоллингсхедТ.(Hollingshead Thos. E.)
253, 262
Хольмс X. (Holmes H. L.) 351
Хонейвел Дж. (Honeywell G. E.) 105,
368
Хорне В. (Home W. H.) 383
Хоуард Дж. (Howard J. W.) 102
Хубачер М. (Hubacher Max H.) 560
Хулли X. (Hully H. H.) 288, 485
Хустон P. (Huston Ralph C.) 121
Хэнт М. (Hunt Madison) 566
Цвильгмейер Ф. (Zwilgmeyer F.) 543
Це-Цинг Чу (Tse-Tsing Chu) 167, 559
Циммерман Ф. (Zimmermann F. J.)
428
Чалмерс У. (Chalmers William) 485
Чан-Манн Лу (Chan Mann Lu) 223
Шапиро С. (Shapiro S. H.) 410
Шаффнер П. (Schaffner P, V. L.) 176
Швадерер A. (Schwaderer A. J.) 37,
105
Шейбпи Ф. (Sheibley P. E.) 93
Шемин Д. (Shemin D.) 69, 72, 160,
498, 518
Шеппард О. (Sheppard О. E.) 36o, 491
Шильднек П. (Shildncck P. R.) 176,
235, 326, 396
Шиман Дж. (Schiemann G.) 244, 534
Шлос Е. мл (Shloss E. L. Jr.) 387
Шмидт A. (Schmidt A. G.) 584
Шрайнер Р. (Shriner R. L.) 149, 220,
247 268 290, 301, 312, 383, 389,
417, 570, 584
Шредер E. (Schroeder E. F.) 64, 65,.
07, 103
Шредер У. (Schroeder Wesley) 43
Шрепбер Р. (Schreiber R. S.) 403
Штаудппгер X. (Staudinger H.) 45
Шулыд В. (Schulz W. F.) 326
Шульц P. (Schultz R. E.) 224
Шульце Ф. (Schulze F.) 348
Шуринк X. (Schurink H.'B.) 443
Эберли Ф. (Eberly Floyd) 288
Эванс В. (Evans Wm. Lloyd) 64, 65,
67, 163
Эванс Дж. (Evans J. С W.) 517
Эвери В. (Evers W. L.) 323
Экк Дж. (Eck J. C.) 36, 89, 90, 304,
308
Элер Р. (Oehler Rene) 345
Эллингбо Э. (EUingboe Ellsworth) 202
Эрлих П. (Ehrlich P.) 387
Юзуех К. (Hsueh С. М.) 317
Юнг Д. (Young D. M.J18
СОДЕРЖАНИЕ
Стр..
Предисловие редактора 5
Предисловие 7
Азелаиновая кислота 9
Азлактон а-беизоилам1шо-^C,4-Д1шетоксифеиил)-акриловой кислоты . . 11
Азоксибензол 12
Аконитовая кислота ; . 15
Акридон 18
м-Аланин 20
Аллантоин 23
Аллиламин 25
Альдегидофталевая кислота 27
Амид о-толуиловой кислоты 29
н.-Амилбензол 30
4-Аминовератрол 32
а-Амнионзомасляпая кислота 34
е-Аминокапроновая кислота 36
у-Аминомасляиая кислота 37
2-Амино-4-метилтиазил 40
1-Амино-2-нафтол солянокислые 41
1,4-Аминонафтол солянокислый 48
1-Амино-2-нафтол-4-сульфокислота 51
Ангидрид 3,4-дигидро-1,2-нафталевой кислоты 53
Ангидрид янтарной Кислоты 56
Антрахинон-а-сульфокислоты калиевая соль 58
d-Аргииин солянокислый 60
Ацеталь акролеина 64
Ацеталь d/-глицеринового альдегида 65
Ацеталь р-хлорпропионового альдегида 67
Ацетилбензоин 68
Ацетилглицин 69
Ацетилендикарбоновая кислота 71
а-Ацетоаминокоричная кислота ¦ 72
Ацетол 74
2-Ацетотиенон 76
Ацето-л-цимол 77
Барбитуровая кислота 79
Бензальфталид • 80
Бензантрон 81
Бензилфталимид 84
Бензимидазол 85
Бензогидроксамовая кислота 87
s-Бензоиламино-а-бромкапроновая кислота 86
Е-Бензоиламинокапроновая кислота 90
Бензоиленмочевина 93
fi-Бензоилпропионовая кислота 95
fl-Бензпинаколин 97
Бензпинакон 98
СОДЕРЖАНИЕ
651
Стр.
Бетаингидразид солянокислый ЮО
Бромаль 102
Бромацетон 103
л-Бромбензальдегид 105
а-Бромизовалернаповая кислота 107
Бромистый водород (безводный) 109
Бромистый н.-додецил 112
Бромистый нзобутил 114
Бромистый фенацил 116
Броммезитилен 118
4-Бромрезорции 120
о-Бромфенол 121
Бромциан 123
З-Бромэтансульфокислоты натриевая соль 124
З-Бромэтилампна бромистоводородная соль 126
'1,3-Бутадиен 128
н.-Бутилнитрит 131
н.-Бутиловый эфир борной кислоты 133
н.-Бутилсульфат ]35
н.-Бутилсульфит ]36
н.-Бутилфосфат 138
Бутироин 140
Вератровый альдегид 142
Вератронитрил М5
Галлацетофенон 147
я.-Гексадекан |48
Гексаметиленгликоль , 149
Гептанол-2 152
леомш.-Гептахлорпропан 153
н.-Гептиламин 154
а-Гидриндон 156
Гиппуровая кислота 158
/-Гистидин солянокислый 1бо
^/-Глицериновый альдегид юз
Гомовератровая кислота 164
Дезоксибензоин 167
Декаметиленгликоль 170
Диазоаминобензол 172
Диазометан 174
2,4-Диаминотолуол 176
Дибензальацетон 179
1,4-Дибензоилбутан 181
ад-Дибромгидрин глицерина 183
2,6-Дибром-4-нитрофенол 185
2,6-Дибромхинон-4-хлоримид 187
1,2-Дибромциклогексан , 189
а,р-Дибромянтарная кислота 191
Ди-н.-бутилкарбинол 193
Дигидрохолестерин 195
2,6-Дииод-л-нитроанилин 198
яесилш.-Диметилгидразин солянокислый 199
симл.-Диметилгидразин солянокислый 202
Диметилглиоксим 205
2,4-Диметил-3,5-дпкарбоэтокатиррол 209
2',4-Дчметил'каРб0ЭТ0КС1ШНРР0Л 211
652
СОДЕРЖАНИЕ
Стр.
2,6-Диметилпиридни 214
2,4-Диметилпиррол 216
2,5-Диметилпиррол 218
5,5-Диметил-1,3-циклогександнон 220
Дг ^-нафтилртуть 222
2,4-Динитроанилин ' 223
2,4-Динитробензальдегид 224
«-Динитробензол . . . , 227
2,4-Динитрофенилгидразнп 22&
1,4-Дифенилбутадиен 231
4,5-Дифенилглиоксалон 232.
Дифеиилметан 233
?,5-Дифенилпропиофенон 235
Дифенплселен 237
Дифенилселен двухлористый и трифепилселен хлористый 239
Дифенилсульфид . . . ф 241
Дифенилтрикетон 242
4,4'-Дифтордифенил 244
Дихлоруксусная кислота" 247
(З-Диэтиламиноэтиловый спирт 248
Диэтилцинк 249
Дурол и изодурол ." 253
Дурохиноп 262
Изонитрозопропиофенсн 264
Изопропиловый эфир молочной кислоты 266
Изопропилтиоцианат 268
Имииодифенилметан солянокислый 26ft
я-Иоданилин 271
5-Иодаптраниловая кислота 273
л-Иодбензойная кислота 27&
Иодбензол 278
Йодистый н.-гексадецил 280
Йодистый метил 281
2-Иодтиофен 287
я-Иодфеиол 288
Итаконовый .ангидрид и итаконовая кислота 290
Казеин 292
Капроновый альдегид 295
2-Карбоэтоксициклопентанон 297
Катализатор медно-хромовый 301
2-КетОгексаметилеиимин 304
Лауриловый спирт 306
dl-Лизина хлористоводородные соли 308
Малоиовая кислота _ _ 309-
Мезаконовая кислота 312
Метилбензилкетон 313-
ji-Метилвалериановая кислота 3!7
Метилгидразин сернокислый 319
Метилизопропилкарбинол 321
Метилизопропилкетон 323
S-Метилизотиомочевина сернокислая '. 32о
Метилимнноднуксусная кислота 327
Метилнитрат ' ' ' ' ' . ' ' 320
Метиловый эфир щавелевой кислоты ........... . ' . . . . . 33 >
1-Метил-2-пнрндон оо--1
СОДЕРЖАНИЕ
653
Стр.
6-Метилурацил 335
а-Метил-а-фенилгидразин 337
5-Метилфурфурол 338
Й/-Метионин 340
Метоксиацетоиитрил 343
Моноэтиловый эфир себаииновой кислоты 345
1,2-Нафталевый ангидрид 347
а-Нафтойная кислота 348
З-Нафтойная кислота 351
1,2-Нафтохинон 353
Нитрил .малоновой кислоты 355
Нитрил а-феиил-р-бензоилпропионовой кислоты 358
1-Нитро-2-ацетшшафтцла,чнн 359
.«-Нитроацетофенон 361
Нитробарбитуровая кислота 365
.п-Нитробензальдегид 366
л-Нитробеизилбромид 368
л-Нитродифениловый эфир 370
N-Нитрозометиланилин 372
Нитрозометиллючевина 373
¦.Нитрозометилуретан 376
Нитромезитилен 377
1-Нитро-2-нафтол 379
2-Нитротиофен 381
л-Нитрофенилизоцианат 383
2-Нитрофлуорен и 2-аминофлуорен 385
4-Нитрофталевая кислота : 387
4-Нитрофталимид 389
л-Оксибензойная кислота 390
2-Окси-3,5-дииодбензойная кислота ' 392
Оксим бензофенона 393
¦Оксим энантола 394
i-Оксиэтилметилсульфид 396
Ъктаацетат а-целлобиозы 398
Олеиновый спирт 401
Ортаниловая кислота 403
Пеларгоновая кислота 406
н.-Пентаи 408
Пимелиновая кислота 410
Пиперониловая кислота 417
Пиромеллитовая кислота 418
/-Пропиленгликоль 421
н.-Пропилсульфид 422
Пропионовый альдегид 424
о-Пропнофенол и я-пропиофенол - . . 426
Протокатеховый альдегид 428
З-Резорциловая кислота 430
л-Роданди.метиланилин 431
Соль Рейиеке 432
Сочетание о-толидина и Чикаго-кислоты 434
Сукцинимид 439
Таурии 441
Тетрабром- и тетраиоднеогшитан 443
Тетрагидрофуран 445
654
СОДЕРЖАНИЕ
Стр.
а-Тетралон 448-
Тетраметиленхлоргидрнн 450
Тиобензофеноп 452
2-1 иодигликоль 453
Тиосалициловая кислота 455
Тиофен 458-
л-Толилкарбинол 461
о-Толуиловая кислота . . ' 462
л-Толуиловый альдегид 464
силш.-Трибромбензол 467
1,2,3-Трииод-5-11итробепзол 469
Трикарбометокси.метан 47!
Трикарбоэтоксиметан 472
/-Триптофан 474
силш.-Тритнан 479
Трифенилметилпатрпй 481
Трифенилэтилен 483
Трихлорэтиловый спирт 485
Триэтилкарбинол 489
Урамил 491
Фенантрен-2- и 3-сульфокпслоты 493
^/-^-Фенилалании 496
Фепиларсиновая кислота 501
а-Фенилацетоацетоннтрил 503
Фепнлбепзоилдиазомстап 506
Фенилглиоксаль 507
о-Феиилендпамин 509
7-Феиилмасляная кислота 511
Феиилнитрометаи 513
2-Фенилпирпдин 517
Фенилпировшюгрлдпая кислота 518
Фснилпропиоловая Кислота 520
Фенилтиенилкетон 522
а-Фенилэтпламин 523
й- и /-а-Фенилэтпламин 526
Феиокстин 529
Флороацетофенон 531
Фталид 533
тт-ФторбеизоГшая кислота 534
Фторбензол 537
Фумаровая кислота 541
Хелпдоновая кислота 543
Хшюн 545s
Хлорангидрид фталевой кислоты симм. и несшим 547
а-Хлорантрахиион 549
Л4-Хлорбеизальдегид 551
л-Хлорбензальдегид 554
о-Хлорбензойная кислота 556
Хлористая i'i-нафтилртуть 557
Хлористый дезил 559
З-Хлор-о-нитротпофеноч 560
р-Хлорэтнлметнлсульфид 562
Холестанон 563
Целлобкоза - 564
Циаигпдрин ацетона 566
СОДЕРЖАНИЕ
655
Циклогексилбепзол . .
Цитраконовый ангидрид
и цитраконовая кислота
Энантовая кислота
Эпихлоргидрип и эпибромгидрип
Эруковая кислота
Этилового эфира глицина хлористоводородная
Этиловый эфир адипииовой Кислоты
аиетоянтарной кислоты
бензоилдиметилуксусной кислоты
бензоилуксусной кислоты
этоксалилпропионовой кислоты
Этоксиуксусиая кислота и этиловый эфир этоксиуксусной кислоты
Стр.
507
570
571
573
575
577
578
580
582
584
586
а^-дмбром-^-фепилпропионовой кислоты оои
N-метилкарбаминовой кислоты 587
метилмалоновой кислоты 588
а-нафтойиой кислоты
н.-тридекановой кислоты ......
а-фенилацетоуксусной кислоты . . .
фенилмалоновой кислоты
фенилиианпнровиноградной кислоты
этилентетракарбоиовой кислоты wi
605
607
592
593
596
599
600
601
610
619
Указатель синтезов по типам реакции
Указатель синтезов по типам соединений
Указатель веществ по формулам - 626
Указатель рисунков
Предметный указатель
Указатель составителей рецептуры 644
632
633