/




































































































































































































































































































































Автор: Фридрихсберг Д.А. Григоров О.Н. Карпова И.Ф. Козьмина З.П. Тихомолова К.П. Чернобережский Ю.М.
Теги: химия коллоидная химия
Год: 1964




Текст
О. Н. ГРИГОРОВ, И. Ф. КАРПОВА, 3, П. КОЗЬМИНА,
К. П. ТИХОМОЛОВА, Д. А. ФРИДРИХСБЕРГ,
Ю. М. ЧЕРНОБЕРЕЖСКИЙ
РУКОВОДСТВО
К ПРАКТИЧЕСКИМ РАБОТАМ
ПО КОЛЛОИДНОЙ ХИМИИ
Издаиие 2-е
Переработанное и дополненное
Допущено Министерством высшего и среднего
специального образования РСФСР
в качестве учебного пособия для университетов
ИЗДАТЕЛЬСТВО «ХИМИЙ»
Москва • 1964 . Ленинград-
ОГЛАВЛЕНИЕ
Предисловие........................................................ 3
Глава I
Дисперсность и структура коллоидных систем
Дисперсионный анализ суспензий, эмульсий и золей................... 5
Седиментационный анализ суспензий и эмульсий................... 9
Работа 1. Седиментационный анализ суспензий методом непре-
рывного взвешивания .......................................13
Работа 2. Пипеточный метод седиментационного анализа сус-
пензий ....................................................24
Работа 3. Седиментационный анализ суспензий в восходящем
потоке жидкости с разделением системы на фракции 26
Определение размеров и формы коллоидных частяц по рассеянию
ими света...................................................29
Работа 4. Определение величины коллоидных частиц при помо-
щи ультрамикроскопа .............36
Работа 5. Определение размеров частиц по интенсивности света,
рассеянного под углами Р = 45° и 135°............ 40
Методы определения размеров и формы частиц, основанные на из-
мерении интенсивности проходящего света..................40
Работа 6. Определение размеров коллоидных частиц по зависи-
мости коэффициента экстинкции от длины волны
света......................................................41
Работа 7. Определение размеров коллоидных частиц по харак-
теристической мутности системы.............................42
Работа 8. Определение размеров и формы анизодиаметрических
частиц по измерению интенсивности проходящего
света......................................................44
Структура капиллярных систем.....................................50
Работа 9. Определение общей пористости -мембран...........52
Работа 10. Определение среднего радиуса пор мембраны по
общей пористости и коэффициенту протекаемости. . 56
Работа 11. Определение среднего радиуса пор мембраны по ее
электросопротивлению и коэффициенту протекае-
мости ......................................................61
• 327
Работа 12. Определение максимального радиуса пор мембран 66
Удельная поверхность твердых дисперсоидов...........................71
Работа 13. Определение удельной поверхности порошков мето-
дом фильтрации жидкостей и газов. . •.....................73
Работа 14. Определение удельной поверхности порошков мето-
дом фильтрации разреженного газа.....................• ... 79
Литература .........................................-...............85
Глава II
Поверхностные явления и адсорбция
Поверхностное и пограничное натяжение...............................90
Работа .15. Измерение поверхностного натяжения методом сче-
та капель (метод сталагмометра)............................92
Работа 16. Измерение поверхностного натяжения методом наи-
большего давления пузырьков............................... 95
Работа 17. Измерение поверхностного натяжения методом ка-
пиллярного поднятия .......................................... $8
Работа 18. Измерение пограничного натяжения методом наи-
большего. давления капель.................................103
Адсорбция иа границах раздела жидкость — газ и твердое тело — жид-
кость ............................................................ 105
Адсорбция поверхностно-активных веществ из водных растворов . . 107
Работа 19. Исследование связи между поверхностным натяже-
нием и адсорбцией.........................................107
Работа 20. Применение уравнения' Фрейндлиха к адсорбции ор-
ганических кислот на твердых адсорбентах..........115
Адсорбция красителей..........................................120
Работа 21. Исследование .адсорбции красителей на угле. . . . 120
Обменная адсорбция..................... . ....... 123
Работа 22. Исследование обменной адсорбции ионов аналити-
ческим методом........................................... 127
Работа 23. Исследование обменной адсорбции ионов потенцио-
. метрическим методом ..............................130
Смачивание и флотация..................*...........................133
Работа 24. Измерение краевых углов и вычисление работы ад-
гезии . ..................................................133
Работа 25. Определение смачиваемости порошков по измере-
нию .давления вытеснения.........................141
Работа 26. Определение теплоты смачивания тонкоизмёльченных
веществ...................................................146
Работа 27. Исследование процесса флотации...................152
Эмульсии и пены . ;.............................................158
Эмульсии , , ......................................... 159
328
Работа 28. Приготовление концентрированных и предельно кон-
центрированных эмульсий и исследование их дисперс-
ности ..................................................... 159
Пены............................................................166
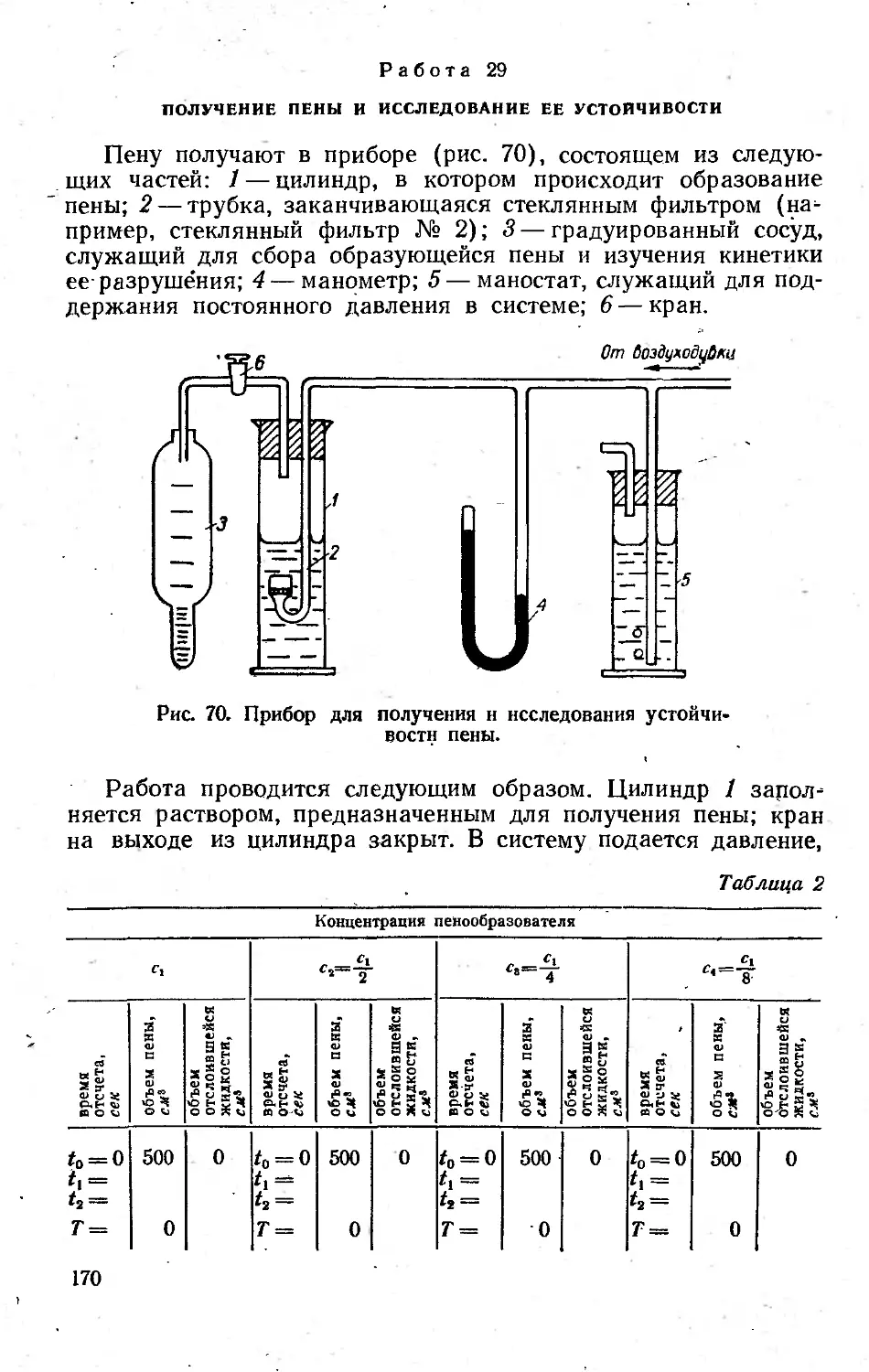
Работа 29. Получение пены и исследование ее устойчивости . . 169
Работа 30. Приготовление пены методом встряхивания раство-
ра пенообразователя в цилиндре..............................171
Работа 31. Оценка устойчивости пены по времени существова-
ния элементарной пленки ................................. 172
Литература ........................................................ 172
Глава III
Двойной электрический слой и электрокинетические явления
Электрокинетический потенциал.......................................176
Электроосмос ..........'........................... .'..........179
Работа 32. Определение электрокинетического потенциала диа-
фрагм методом электроосмоса.................................179
Потенциал протекания.............. ... ........................187
Работа 33. Определение электрокинетического потенциала диа-
фрагм методом потенциала протекания.........................187
- Электрофорез ....................... . 7"......................194
Работа 34. Определение электрокинетического потенциала золей
методом электрофореза (макроскопический метод) . . 196
Работа 35. Определение электрокинетического потенциала сус-
пензий методом электрофореза (микроскопический
метод) ................................................... 200
Электрокинетические свойства капиллярных систем.................' . . 204
Изменение чисел переноса ионов в мембранах..................... 205
Работа 36. Определение чисел переноса ионов в мембране ана-
.литическим методом........................ ... ... . .206
Работа 37. Определение чисел переноса ионов в мембране ме-
тодом диффузионного потенциала..............................210
Поверхностная проводимость ................................ ... 213
Работа 38. Определение поверхностной проводимости капилляр-
ных систем............................................. 213
Электродиализ...............'...........?.......................222
Работа 39. Очистка коллоидных растворов и суспензий от
' электролитов методом электродиализа ... . . 232
Литература ..................................................... 232
* 329
Глава IV
Устойчивость дисперсных систем
Работа 40. Исследование зон коагуляции (неправильные ряды) . 242
Работа 40а. Исследование зон коагуляции с помощью фото-
электроколориметра ........................................243
Работа 41. Взаимная коагуляция золей .....................245
Работа 42. Защитное действие высокомолекулярых соединений
(определение золотого числа) ................... 247
Работа 42а. Исследование зависимости числа прилипания ча-
'стиц кварца (стекла) от природы и концентрации
электролита............................................ 248
Литература .......................................................250
Г л а в а V
Структурообразование в дисперсных системах
Структурно-механические свойства дисперсных систем................253
Работа 43. Исследование упруго-пластических свойств структу-
рированных систем методом тангенциального смеще-
ния пластинки ............................................ 257
Работа 44. Определение напряжения сдвига н структурной вяз-
кости коллоидных систем методом капиллярной ви-
скозиметрии ............................................. 262
Фильтрационный анализ.............................................267
Работа 45. Исследование суспензий методом фильтрационного
анализа................................................... 267
Литература ...................................................... 278
Г лава VI
Свойства высокомолекулярных соединений и их растворов
Работа 46. Определение молекулярного веса высокомолекулярных
соединений методом осмотического давления...................281
Работа 47. Определение молекулярного веса высокомолекулярных
соединений по вязкости их растворов.........................288
Работа 48. Исследование процесса набухания твердых поли-
меров .....................................................295
Работа 49. Равновесие Доннана. Суспензионный и золь-кон-
центрационный эффекты...........................305
Литература .......................................................312
330
Глава VII
Указания для подготовки демонстрационных опытов к лекциям
Получение коллоидных растворов..................................... .313
Молекулярно-кинетические и оптические свойства дисперсных систем . . 314
Поверхностные явления и адсорбция....................................317
Явление смачивания и растекания...................................319
Эмульсии..........................................................321
Электрические явления на границе фаз................................ 322
Устойчивость суспензоидных золей и коагуляция.....................325
Структурно-механические свойства коллоидных систем . . . . .........325
Литература............,..............................................326
ПРЕДИСЛОВИЕ
Руководство к практическим занятиям по коллоидной химии
составлено преподавателями кафедры коллоидной химии хими-
ческого факультета Ленинградского ордена Ленина государ-
ственного университета имени А. А. Жданова на основе мате-
риала практических работ, выполняемых студентами-химиками.
Руководство содержит описание работ, входящих в общий
практикум по коллоидной химии для студентов-химиков всех
специальностей, а также работ, проводящихся студентами, спе-
циализирующимися по коллоидной химии. В настоящее время
в общем практикуме выполняются работы 1, 16, 19, 20, 21, 24,
32, 33, 39, 40, 40 а, 41, 42, 44 и 48. Остальные работы со-
ставляют содержание специальных практикумов на старших
курсах.
Многолетний опыт преподавания коллоидной химии в Ле-
нинградском университете показывает, что осуществление по-
следовательного прохождения всего учебного материала прак-
тикума вместе с читаемым курсом лекций встречает большие
затруднения. Поэтому в начале отдельных разделов и в описа-
нии каждой работы имеются краткие сведения по теории изу-
чаемого вопроса в объеме, необходимом для выполнения рабо-
ты, и в экспериментальной части даются методические указания
, к постановке эксперимента.
Данное руководство является вторым изданием, полностью
переработанным и значительно расширенным по сравнению с
первым изданием, выпущенным в 1955 году.
В данном руководстве наряду с применяемой в научных ис-
следованиях системой СГС в скобках и в .сносках указаны для
ряда формул значения в Международной системе единиц СИ
(ГОСТ 9867—61).
Формулы, включающие электрические величины, приводятся
также (обычно в сносках) в рационализированной системе СИ.
„ В этом случае уравнения для плоских систем приобретают
более простую форму путем исключения множителя 4к и вклю-
чают единицы, получившие широкое практическое распро-
странение (кулон, вольт, ампер, ом). Для этого в уравнение
3
вводится величина абсолютной диэлектрической проницаемости
в вакууме Do = 8,854 • 10-12 а- сек/в-м.
Материал между отдельными соавторами распределен сле-
дующим образом: работы 36 и 37 написаны О. Н. Григоровым;
работы 24, 26, 34, 35, 45, 46 и 48— И. Ф. Карповой; работы 1, 2,
3, 4, 9, 10, 11, 12, 13, 14, 20, 21, 39 и глава VII (указания к по-
становке демонстрационных опытов к лекциям) — 3. П. Козь-
миной; работы'5, 6, 7, 8, 17, 18 и 25 — К. П. Тихомоловой; ра-
боты 15, 16, 49, 22, 23, 32, 33, 38, 43, 44 и 47 — Д. А. Фридрих-
сбергом и работы 27, 28, 29, 30, 31, 40, 40а, 41, 42, 42а и 49 —
Ю. М. Чернобережским.
Общая редакция осуществлена проф. О. Н. Григоровым.
Авторы приносят глубокую благодарность за тщатель-
ный просмотр рукописи и сделанные критические замечания
акад. П. А. Ребйндеру, проф. С. С. Воюцкому, проф. Л. Я. Крем-
неву и сотрудникам кафедры коллоидной химии МГУ Е. П. Анд-
реевой, В. Н. Измайловой, О. И. Лукьяновой и Б. Я. Ямполь-
скому.
ГЛАВА I
ДИСПЕРСНОСТЬ И СТРУКТУРА коллоидных
СИСТЕМ
ДИСПЕРСИОННЫЙ АНАЛИЗ СУСПЕНЗИЙ, ЭМУЛЬСИЙ И ЗОЛЕЙ
Зависимость ряда физико-химических свойств вещества от
степени его дисперсности в настоящее время вполне установ-
лена. Имеется много общих закономерностей изменения свойств
Вещества в зависимости от размеров его частиц. Так, например,
^.уменьшением размера частиц вещества ниже определенного
редела растворимость его увеличивается. Скорость диффузии
Растворенных веществ также повышается с увеличением их дис-
Версности. Окраска многих коллоидных растворов изменяется
^зависимости от размеров частиц.
Большое значение имеет дисперсность веществ в различных
Ихнологических процессах. Например, качество цементов зави-
сит от размеров составляющих их частиц. 'В производстве ре-
зины качество вулканизата определяется в известной мере дис-
персностью-добавляемого порошка серы. Кроющая способность
краски зависит от степени размельчения пигмента. Интенсив-
ность. электроосмотического передвижения жидкости, широко
Применяющегося в последнее время для обезвоживания различ-
ных дисперсных систем, в частности грунтов, зависит также
от дисперсности объекта, подвергающегося осушке.
Приведенных примеров достаточно для заключения, что дис-
персность веществ имеет большое значение как при разрешении
Многих производственно-технических вопросов, так и для науч-
Й-исследовательскИх целей.
; Независимо от условий образования или способов получения
Дисперсных систем частицы их обычно разных размеров. Такие
Йрлидисперсные системы можно рассматривать как смесь моно-
Дисперсных частиц. Определение величины частиц и получение
Данных о распределении частип полидиспеосной системы по
вдределенным размерам и является задачей дисперсион-
5
него анализа. Различают качественный и количественный
дисперсионные анализы.
Качественный дисперсионный анализ устанавливает,
является ли исследуемая система грубодисперсной, коллоидно-
дисперсной или молекулярнодисперсной. К грубодисперсным си-
стемам относят системы с размером частиц больше 0,1 мк. Раз-
меры частиц от 6,1 мк до 1 ммк лежат в области коллоидной
дисперсности; частицы меньше 1 ммк относят к молекулярной
дисперсности. -
Количественный дисперсионный анализ заключается
в разделений исследуемого вещества на отдельные фракции,
содержащие частицы, размеры которых лежат внутри опреде-
ленного интервала, и установлении процентного содержания
отдельных.фракций в исследуемой дисперсной системе. Чем уже
выбран интервал размеров отдельных фракций, тем более пол-
ные данные получают для характеристики исследуемой поли-'
дисперсной системы по размерам частиц.
Дисперсионный анализ осуществляется различными мето-
дами. Для анализа систем грубодисперсных или систем, содер-
жащих некоторое количество частиц относительно больших
размеров, употребляют набор сит из тонкой проволоки или
шелка. Обычный набор сит дает возможность произвести раз-
деление частиц больше 0,25 мм. Набор стандартных проволоч-
ных сит позволяет разделить несколько более мелкие частицы.
Эти стандартные сита характеризуются числом отверстий —
хеш (meshe — петля) на линейный дюйм или сантиметр. Чем
больше число таких отверстий, тем меньше их сечение. ♦'
Приведем примеры поперечника отверстий наиболее тонких
сит (по системе Тайлера):
100 меш 0,147 мм
160 » 0,104 »
200 » 0,074 »
350 » 0,044 »
Набор таких сит дает возможность фракционировать из-
мельченные вещества, размеры частиц которых больше 44 мк.
Для дисперсных систем, содержащих частицы меньших раз-
меров, применяют другие методы дисперсионного анализа.
Для качественного определения степени однородности таких
систем пользуются микроскопическим методом, сущ-
ность которого заключается в непосредственном определении
размеров частиц с помощью обычного микроскопа. Можно про-
вести и количественный микроскопический анализ дисперсных
систем, размеры частиц которых больше разрешающей способ-
ности микроскопа, т. е. больше 0,2 мк. Однако количественное
определение микроскопическим методом затруднительно, так
как для полной характеристики дисперсности системы необхо-
дим<2 определить размеры очещ> брльщого числа частиц. Тем
6
объектива микроскопа и должна
Рис. 1. Измерительная шкала окуляр-
микрометра (а) и объектмикрометр (б).
|йё Менее этим методом приходится Пользоваться в тех случаях,
«когда другие методы анализа не могут быть применены.
При определении размеров частиц с помощью микроскопа
^применяют окулярный микрометр, представляющий собой оку-
ляр микроскопа со вставленной в него измерительной шкалой
>(рис. 1,а). Цена деления окулярной шкалы зависит от соотно-
Йшения увеличений окуляра и объектива микроскопа и должна
“быть определена предвари-
тельно. Для этого на пред-
метный столик микроскопа
'помещают предметный
«микрометр (объектмикро-
метр), представляющий со-
бой толстое предметное стек-
ло, на котором нанесена
шкала с определенной ценой
деления (рис. 1,6). Сфо-
^кусировав микроскоп на шкалу объектмикрометра, совмещают
какое-либо расстояние между штрихами обеих шкал и, отсчи-
тав число делений шкал на этом расстоянии, вычисляют цену
деления окулярной шкалы. Например, если а — число делений
шкалы объектмикрометра с ценой деления, равной Ь, соответ-
ствует при данном увеличении с делениям окулярной шкалы, то
Ц§на одного деления:
ab
Градуировку окулярной шкалы проводят при различных уве-
личениях, т. е. с различными объективами.
Для определения размеров частиц дисперсной системы не-
большое количество изучаемой суспензии или порошка, смочен-
ного жидкостью *, помещают на предметное стекло. Концен-
трация дисперсной фазы не должна быть большой, чтобы каж-
дая частица могла быть рассмотрена отдельно. Пробу сус-
пензии покрывают покровным стеклом и помещают на столик
микроскопа. Размеры частиц d определяются путем отсчета чис-
ла делений п окулярной шкалы на расстоянии, в котором укла-
дывается изображение частицы, и умножением его на цену деле-
ния при данном увеличении:
d = пх
Для проведения дисперсионного анализа системы необхо-
димо, кроме определения -размеров частиц, произвести подсчет
числа частиц различных размеров в поле зрения. Подсчеты
* Следует обращать внимание на выбор жидкости, чтобы предотвратить
коагуляцию частиц Обычно применяется дистиллированная вода, вазелино-
вое масло и др. Иногда добавляют пептизирующие вещества.
7
Следует производить При различных увеличениях на возможно
большем числе проб, меняя поле зрения в каждом опыте. Сде-
лав. относительно большое число измерений (1000—2000), полу-
чают достаточно точную характеристику распределения частиц
по размерам. Можно несколько облегчить проведение анализа,
фотографируя препараты под микроскопом и обрабатывая мик-
рофотографии.
Результаты микроскопического анализа представляют в виде
таблицы, содержащей относительное число частиц различных
размеров в процентах к общему числу*.
Наибольшее распространение получили методы седимен-
тационного анализа, основанные на определении скоро-
сти оседания частиц под действием силы тяжести. Применение
этих методов возможно для дисперсных систем, содержащих
Частицы, кинетическая энергия которых недостаточна для про-
тиводействия силе тяжести. При оседании полидисперсной сус-
пензии частицы большего размера будут иметь большую ско-
рость по сравнению с частицами меньшего размера. Мелкие
Частицы под действием силы Тяжести оседают очень медленно,
Что Ограничивает применение этих методов к высокодисперсным
системам.
Границы применения обычного седиментационного метода
анализа для высокодисперсных систем 'зависят как от величины
Частиц, так и от разности плотностей между частицей и диспер-
сионной средой. Для тяжелых частиц (например, металлических
с плотностью порядка 9—10 г) см?) практически нельзя Опреде-
лять радиусы меньше 50 ммк, а для частиц с Меньшей плот-
ностью эта граница еще больше Сдвигается в сторону крупных
Частиц. В большинстве случаев седиментационные методы ана-
лиза дают возможность охарактеризовать поЛидисперсные си-
стемы с размером частиц от 100 до 0,5 мк. Частицы больше
ДОС мк (г = 50 мк) предварительно отделяют, например отсеи-
ванием на ситах, и анализируют отдельно. Содержание в сус-
пензии частиц с размерами меньше 0,5 мк определяют суммарно
без разделения на фракции. В связи с этим большое вни-
мание было уделено разработке методов дисперсионного ана-
лиза, основанных на наблюдении за скоростью оседания частиц
под действием центробежной силы с применением ультрацен-
трифуг различной конструкции. Сведбергом были сконструиро-
ваны ультрацентрифуги, дающие ускорения, равные Ю5^ И
большие (g— ускорение силы тяжести). Таким методом можно
исследовать коллоидные системы высокой степени дисперсности
(например, с радиусом частиц до 2 ммк). Современные ультра-
* Результаты микроскопического анализа можно представить и в виде
графиков (стр.. 18),
8 ’
центрифуги дают возможность определять размеры частиц
вплоть до обычных молекул.
Размеры частиц, лежащих в коллоидной области диспепс-
ности, могут быть также определены с помощью ультрамикро-
скопа и электронного микроскопа, причем в последнем случае
имеется возможность судить и о форме частиц.
Седиментационный анализ суспензий и эмульсий
Большинство методов седиментационного анализа основано
на применении закона Стокса, согласно которому сила сопро-
тивления f движению шарообразной частицы в жидкости выра-
жается зависимостью:
f = &Г.Т;Г11 (1)
где т] — вязкость жидкости, г—радиус частицы, и — скорость
движения частицы.
Если частица оседает в поле земного тяготения, то силой,
вызывающей ее движение, является ее вес:
4 ЧЛ
Р = mg = -g лг3 dg
где tn — масса частицы, g — ускорение силы тяжести, d—плот-
ность частицы.
Потеря в весе частицы, .находящейся в жидкости, по закону
Архимеда составляет:
4
Pi ='§- nrbl'g
где d\ — плотность жидкости.
Следовательно, сила, под действием которой оседает частица
в жидкой среде:
4
/> = р — pl== — nr>(d — di)g (2)
Вначале движение частицы происходит с ускорением. Одна-
ко при возрастании скорости движения частийы, согласно урав-
нению (1), увеличивается сопротивление среды, т. е. ускорение
уменьшается и в некоторый момент времени становится равным
нулю. В этот момент сила сопротивления становится равной
силе, под действием которой происходит движение частицы,
и далее частица движется с постоянной скоростью, которую
легко найти из уравнений (1) и (2), так как:
/ = /*
или
4
бЛТЦГМ — ЛГ3 (d — rfj) g
9
Отсюда:
или
, / Оги ' ...
r~V 2(d-d,)g (4)
Из уравнения (3) следует, что скорость седиментации зави-
сит от радиуса частиц, вязкости среды и разности плотностей
диспергированного вещества d и дисперсионной среды dx. В том
случае, когда плотность частиц дисперсной фазы больше плот-
ности дисперсионной среды (d>dI), происходит седиментация
частиц с образованием осадка. Наоборот, при (d<dt)' диспер-
гированное вещество всплывает на поверхность, что часто встре-
чается при проведении дисперсионного анализа эмульсий.
Уравнение Стокса было выведено при соблюдении некото-
рых определенных условий, которым не всегда отвечают ре-
альные системы.
Разберем подробно эти условия и возможность применения
закона Стокса к оседанию частиц в жидкой среде:
1) частицы должны быть сферическими. Это условие выпол-
няется, например, в случае эмульсий с малой концентрацией
эмульгированной фазы и сферических частиц высокополимер-
ных соединений, полученных методом эмульсионной полимери-
зации. Частицы суспензий имеют обычно различную форму,
иногда сильно отличающуюся от сферической. Однако многими
авторами было показано, что в обычных измельченных веще-
ствах получающиеся отклонения вследствие нешарообразной
формы при оседании весьма невелики, за исключением тех слу-
чаев, когда мы имеем анизодиаметрические частицы (пластин-
чатые или палочкообразные), т. е. сильно отклоняющиеся от
сферической формы. Необходимо только иметь в виду, что
определение радиуса по уравнению (4) дает не действительную
величину, а лишь некоторый эффективный радиус, соответ-
ствующий радиусу сферической частицы того же вещества, осе-
дающей с той же* скоростью. Такой радиус называется экви-
валентным;
2) отсутствие взаимодействия между частицами. Взаимо-
действие между частицами нарушает оседание отдельных ча-
стиц и не учитывается уравнением Стокса. Однако взаимное
влияние частиц при малых концентрациях суспензии, когда рас-
стояние между частицами превышает их размеры не меньше
чем в 10 раз, очень мало и не отражается на получающихся
результатах. Поэтому седиментационный анализ суспензий сле-
дует проводить при небольших концентрациях (не выше 1—2%);
3) движущиеся частицы должны быть твердыми и гладки-
ми. Условие твердости связано с тем, что при седиментации
W
Жидких частиц (например, частиц эмульсий) внутри них могут
возникнуть микропотоки, способные изменить форму поверхно-
сти. Известны поправки к уравнению Стокса, связанные с невы-
полнением этого условия. Однако ими обычно не пользуются,
так как .было показано, что отклонения от закона Стокса в
большинстве случаев невелики.
При анализе суспензий условие твердости частиц всегда со-
блюдается; условие же гладкости частиц, т. е. отсутствия шеро-
ховатости на их поверхности, практически не выполнимо. В на-
стоящее время нет надежных методов оценки шероховатости
поверхности. Поэтому при определении размеров таких частиц
седиментационными методами пользуются эквивалентным ра-
диусом;
4) отсутствие скольжения между оседающей частицей и сре-
дой. В случае движения твердой частицы в жидкости, так же,
как и при движении жидкости вдоль неподвижной твердой
стенки, на пограничной поверхности образуется гидродинамиче-
ский слой, в котором существует некоторый градиент скорости
относительного передвижения жидкости. В условиях смачива-
ния молекулы жидкости, расположенные непосредственно на по-
верхности твердых частиц, движутся вместе с частицей. В случае
же движения жидкости вдоль твердой стенки молекулы жидко-
сти, расположенные на ее поверхности, остаются неподвижными.
Таким образом, скольжение происходит между слоями жидко-
сти, а не между жидкостью и твердой поверхностью. В связи с
этим в уравнении Стокса ц представляет собой коэффициент
вязкости жидкости. Одним из основных условий отсутствия
скольжения между оседающей частицей и средой является пол-
ная смачиваемость оседающих частиц жидкостью. Поэтому на,
выбор жидкости при седиментационных анализах следует обра-
щать большое внимание;
5) скорость оседания частиц не должна- превышать опреде-
ленного предела, что связано с возможностью возникновения
турбулентного движения в слое жидкости вблизи быстро осе-
дающих частиц. Это условие также указывает на известные
пределы применения уравнения Стокса в связи с размером
частиц и соотношением между плотностью диспергированного
вещества и среды, в которой происходит седиментация ча-
стиц;
6) постоянная скорость оседания частиц. Как указывалось
при выводе уравнения (4), частица в начале оседания движется
с некоторым ускорением и только после -того, как движущая
сила уравновешена силой сопротивления среды, частица оседает
с постоянной скоростью. Время, в течение которого частица до-
стигает постоянной скорости оседания, настолько мало, что не-
может оказать влияния на результаты седиментационного ана-
лиза. Например, время достижения постоянной скорости оседа-
11
ййя частиц кварца * с радиусом 50 жк в воде равно 3,4» 10-й сек,
для частиц с радиусом 1 мк оно равно 1,7 • 10-6 сек.
Кроме, рассмотренных условий применимости закона Стокса
к реальным системам, связанных с допущениями, сделанными
при выводе этого закона, следует учитывать и другие особен-
ности изучаемых объектов, а также влияние внешних факторов.
Та'к, суспензия должна быть устойчивой, не коагулировать в
процессе седиментации. Если частицы плохо смачиваются сре-
дой, то образуется неустойчивая суспензия, коагулирующая в
процессе оседания. В случае проведения седиментационного
анализа дисперсной системы, частицы которой плохо смачи-
ваются средой, необходимы добавки стабилизирующих веществ,
улучшающих смачивание. Оседание частиц должно происходить
в спокойной жидкости. Необходимо постоянство температуры
в условиях опыта. Все частицы должны иметь одинаковую плот-
ность, и п£>и малых размерах частиц следует учитывать наличие
сольватных и стабилизирующих слоев, так как сильное их раз-
витие, в особенности для частиц малых размеров, внесет неточ-
ность в результат определения. В дисперсной системе не дол-
жно быть пузырьков воздуха или другого газа, направление
движения которых противоположно оседающим частицам; по-
этому необходима тщательная подготовка образца для опыта.
Рекомендуется взятую навеску предварительно обработать не-
большими порциями жидкости при тщательном перемешивании,
иногда при подогреве, чтобы удалить адсорбированные на по-
верхности частиц газы.
Все методы седиментационного анализа можно разбить на
две группы.
К первой группе относятся методы, в которых анализ про-
водится с разделением дисперсной системы на отдельные фрак-
ции (отмучивание). Отмучивание может производиться в спо-
койной жидкости (метод отстаивания), а также в текущей струе
жидкости или воздуха.
- Ко второй группе относятся методы, в которых не произво-
дится непосредственного разделения дисперсной системы на
фракции. Седиментационный анализ, в котором не осуще-
ствляется непосредственное разделение дисперсной системы на
отдельные фракции, можно проводить, наблюдая за изменением
одной из следующих величин: 1) объема осадка, 2) концентра-
ции суспензии, 3) плотности суспензии, 4) гидростатического
давления столба суспензии и 5) веса осадка (весовой метод).
Дисперсионный анализ суспензий в поле центробежной силы
может проводиться также с разделением и без непосредствен-
ного разделения системы на фракции.
• Плотность кварца d = 2,7 г/см3 (2700 кг/м3).
12
Ниже рассматриваются два метода дисперсионного анализа
без разделения системы на фракции (метод непрерывного взве-
шивания осадка и пипеточный) и один метод с разделением
системы на фракции в восходящем потоке жидкости.
Работа 1
СЕДИМЕНТАЦИОННЫЙ АНАЛИЗ СУСПЕНЗИЙ МЕТОДОМ НЕПРЕРЫВНОГО
ВЗВЕШИВАНИЯ ОСАДКА
Этот метод основан на непосредственном определении увели-
чения веса осадка на какой-либо поверхности при седиментации.
Весовые методы вследствие своей чувствительности дают
возможность анализировать суспензии малой концентрации (до
0,001%). В литературе описано много разнообразных приборов,
позволяющих регистрировать увеличе-
ние веса осадка при седиментации,
х Однако только после работ Фигуров-
хского, предложившего чрезвычайно
простой прибор (микровесы), этот ме-
тод нашел широкое применение. В при-
боре Фигуровского использованы уп-
ругие свойства тонких кварцевых или
стеклянных палочек (шпицев), дефор-
мация которых при нагрузке в неко-
торых пределах точно следует закону
Гука.
Схема прибора Фигуровского при-
ведена на рис. 2. В штативе / закреп-
ляют стеклянную или кварцевую па-
лочку с оттянутым концом (шпиц) 2,
и на крючке подвешивают тонко-
стенную стеклянную чашечку 3 с за-
гнутыми краями. В центре чашечки
вплавлена упругая стеклянная нить с
крючком на конце, при помощи кото-
рого ее подвешивают к стеклянному
Рис. 2. Микровесы Фигу-
ровского.
шпицу, являющемуся коромыслом
микровесов. Хорошо оттянутый шпиц при нагрузке на конец и
при изгибе должен деформироваться по всей длине, И, как
указывалось выше, его деформация должна следовать закону
Гука.
Прибор Фигуровского применяют и для дисперсионного ана-
лиза эмульсий, но так как в большинстве случаев при их рас-
слоении дисперсная фаза не оседает, а всплывает на поверх-
ность, то чашечку приплавляют к стеклянной нити вверх дном
и погружают в эмульсию на небольшую глубину. По мере рас-
13
слоения эмульсии всплывающие капли собираются йод чашеч-
кой, в связи с чем появляется сила, выталкивающая ее. Вели-
чина этой силы пройорциональна гидростатическому весу
всплывающих капель эмульсии, вследствие чего этот метод ана-
логичен весовому методу седиментационного анализа суспензий.
В дальнейшем изложении мы рассмотрим весовой метод седи-
ментационного анализа суспензий, но следует отметить, что все
основные выводы и способы оформления экспериментальных
данных применимы и для седиментационного анализа эмуль-
сий [14].
Прежде чем приступить к седиментационному анализу, про-
веряют, происходит ли деформация коромысла по закону Гука.
Для этого чашечку подвешивают к коромыслу микровесов и
отмечают его деформацию при нагрузке от 0,01 до 0,5 г (поль-
зуясь отсчетным микроскопом или катетометром). Полученные
данные наносят на график, причем по оси ординат откладывают
величины деформации коромысла микровесов Д£, а по оси абс-
цисс- нагрузку р в г (рис. 3). В случае применимости закона
Гука зависимость деформации от
нагрузки должна быть прямоли-
нейной.
Седиментационный анализ
дисперсных систем может быть
проведен и при помощи торзион-
ных весов.
При седиментации суспензии
по мере накопления осадка на
чашечке отмечают смещение вниз
по вертикали конца стеклянного
шпица или делений микрошка-
лы, прикрепленной к стеклянной
нити * (а при использовании
торзионных весов — вес . осев-
наблюдения. Таким образом получают
строят кривую оседания, выражаю-
осадка р, пропорционального деформа-
на-
Рис. 3. Зависимость деформации
коромысла микровесов от
грузки.
ших частиц), и время
1 данные, по которым
щую зависимость веса
ции коромысла микровесов Д£, от времени седиментации t.
На основании кривой седиментации получают данные, характе-
ризующие распределение частиц по размерам.
Полученная таким образом седиментационная кривая для
реальных систем обычно имеет плавный ход и на ней уклады-
ваются все экспериментальные точки (рис. 4). Очевидно, что
полная кривая седиментации получится в том случае, когда
* Иногда применяют специальное оптическое устройство, освещая при-
крепленное к стеклянной нити зеркальце, которое отбрасывает зайчик на
отсчетную шкалу.
14
анализ доведен до конца, т. е. когда йдамые маленькие частицы
осели на чашечку микровесов. ОднакоЧнесмотря на то, что при
пользовании микровесами можно значительно сократить время
анализа путем уменьшения глубины оседания, все же часто не
удается довести анализ до конца, и тогда необходимо прежде
всего найти предел, к которому стремится кривая седиментации.
Известно несколько способов определения предела седимен-
тационной кривой, из которых разберем метод, предложенный •
Штыхновым. Этот метод состоит в том, что наряду с кривой
А л
седиментации строится кривая в координатах р, -j, где А —
любое целое число, t—время.
На рис. 4 изображена подобная кривая, которая характери-
зует скорость оседания частиц, причем участок, соответствую-
щий оседанию высокодисперсных фракций, оказывается прибли-
зительно прямолинейным. Через несколько точек этого участка
проводят прямую до пересечения с осью ординат. Точка пере-
сечения а соответствует весу всего осадка, так как при
А
t—хл, -->0. Прямая ab, проходящая через точку пересечения
и параллельная оси абсцисс, является пределом седиментацион-
ной кривой. Этот метод нахождения предела седиментационной
кривой является приближенным и рекомендуется в том случае,
Когда скорость оседания частиц к концу опыта очень мала и
15
выпала большая часть частиц суспензии, а также, когда в сус-
пензии относительно маЛо высокодисперсных фракций.
Разберем несколько подробнее кривую седиментации поли-
дисперсной суспензии. На рис. 5 изображен начальный участок
такой кривой ОА, выражающий зависимость веса осадка от
времени седиментации. В случае оседания полидисперсной сус-
пензии, в отличие от монодисперсной, концентрация частиц на
Рис. 5. Начальный участок седимен-
тационной кривой.
лись от чашечки на расстоянии,
этих частиц через s. Вес частиц,
составляет
определенной глубине изме-
няется с течением времени.
Так например, на чашечку, на-
ходящуюся на глубине h, через
некоторое время от начала
опыта ti полностью осядут все
частицы, скорость оседания ко-
торых
Обозначим вес таких час-
тиц через q. Частицы, скорость
оседания которых
осядут лишь частично, т. е.
осядут лишь те частицы, кото-
рые в начале опыта находи-
меньшем чем h. Обозначим вес
осевших к моменту времени t\,
P = q+s
и на рис. 5 представлен ординатой А\В\. Эквивалентный радиус
h
частиц и, оседающих со скоростью и—-*-, легко рассчитать
по уравнению (4). Частицы больше этого радиуса также полно-
стью осядут за время t\. Соответственно отрезок А2В2 дает вес
частиц, осевших за. время t2. Отрезки прямых СИд и С2А2 сбот-
ветствуют весу частиц, оставшихся еще в суспензии к моментам
времени tj и t2. Для определения из кривой седиментации вели-
чин q и s проведем касательную к кривой в точке А и продлим
ее до пересечения с осью ординат. Из точки пересечения D\
проведем прямую D\E\, параллельную, оси абсцисс. Скорость
оседания частиц в момент времени ti выразится произ-
водной:
16
; Эта производная определяет суммарную скорость седимен-
тации фракций, частично осевших к моменту времени fj:
dp _ dsx . ds2 . _ 'у' dsi
di ~~dt'~di' ~ £t~dT
' За период времени от 0 до t\ скорость оседания каждой из
?этих фракций была постоянной, соответствующей размерам ча-
'стиц. Следовательно, вес частиц этих фракций, выпавших к мо-
менту времени
I dt 1 dt
Вес частиц, полностью осевших к моменту времени t\, со-
ставляет:
, dp
? Из треугольника АхйхЕх очевидно, что
DxEx=tx, AxEx = DxExtga = tx ^- = s
следовательно
q = р — s = АХВХ — AXEX = EXBX — ODX
касательная к кривой седиментации отсекает по оси ординат
отрезок, отвечающий весу частиц, осевших полностью к мо-
менту времени, соответствующему ординате, проходящей через
точку касания Ах (в данном примере ti). Если провести каса-
тельную к точке А2, то отрезок OD2 соответствует весу частиц,
полностью осевших к моменту времени t2. Из рис; 5 также оче-
видно, что отрезок OD2—ODX соответствует весу фракции, со-
„ h
держащей частицы, оседающие со скоростью от «]=— до
h
и2~-7-, т. е. в пределах эквивалентных радиусов от t\ до г2.
Таким образом, при проведении ряда касательных к экспе-
риментально полученной кривой оседания можно разделить ис-1
следуемую полидисперсную систему на ряд фракций, размеры
частиц которых лежат в некоторых пределах. Чтобы найти пре-
дельные радиусы каждой фракции, через полученные точки
касания проводят ординаты, отсекающие по оси абсцисс отрез-
ки, соответствующие времени полного оседания различных
фракций. По найденным значениям t, пользуясь уравнением (4),
вычисляют эквивалентный радиус частиц. ‘Отрезки между каса-
тельными по оси ординат соответствуют весу отдельных фрак-
ций. Содержание в дисперсной системе отдельных фракций
можно выразить в процентах от общего количества вещества.
Для этого длину ординаты от 0 до предела кривой седимента-
ции принимают за 100%. Длины отрезков, отсеченных касатель-
17
Рис. 6. Интегральные кри-
вые распределения.
ними, отнесенные к общей длине ординаты, дают процентное
содержание отдельных фракций в системе.
Результаты седиментационного анализа могут быть пред-
ставлены в виде интегральной, или суммарной, кривой распре-
деления. При построении- такой кривой по оси ординат откла-
дывают ’ процентное содержание фракций с размером частиц
<гь <г2 и т. д., а по оси абсцисс, соответственно, rt, г2 и т. д.
(рис. 6, кривая /). Ордината .каждой
точки данной кривой дает процентное
содержание частиц с эквивалентными
радиусами, меньшими, чем соответ-
ствующий ей по оси абсцисс.
Суммарную кривую распределения
можно построить и таким образом,
что ордината каждой точки кривой
будет давать процентное содержание
частиц с большими эквивалентными
радиусами, чем соответствующий ей
на оси абсцисс. Для этого на оси
ординат откладывают процентное содержание частиц от гмакс
до гт, от Гмакс до гп и т. д., а по оси абсцисс значения радиусов
Гт, Гп И т. д. (рис. 6, кривая 2).
Результаты седиментационного анализа можно представить
более наглядно, введя понятие о функции распределения. Допу-
стим, что в начальный момент времени в исследуемой суспензии
содержится определенное количество
частиц (dQ) с эквивалентными радиу-
сами, лежащими в пределах от г до
г + dr, тогда функция распределения
а количество частиц этой фракции
dQ = /(r)dr
Рис. 7. Дифференциальная
кривая распределения.
Относительное количество дисперсной фазы [Q (г2)—Q (и)] с
’частицами, размеры которых лежат в пределах от и до г2, мож-
но выразить следующим соотношением:
г2 г2
Q(r2) — Q(r,)= J /(r)dr = J dr
Г, г,
На рис. 7 представлена графически найденная зависимость,
причем по оси абсцисс нанесены значения эквивалентных ра-
диусов, а по оси ординат = Вся площадь между кри-
вой и осью абсцисс дает общее количество частиц всех разме-
18
^ов, t. е. 100%, если полученные результаты анализа выражены
процентах. Процентное содержание отдельных фракций равно
;йлощади участка между ординатами соответствующих эквива-
лентных радиусов (от Г1 до г2 на рис. 7).
; Данные для построения дифференциальной кривой распре-
деления (рис. 7) могут быть получены из интегральной кривой
•(см. подробнее стр. 23).
Выполнение работы
В данной задаче седиментационный анализ проводится при
помощи торзионных весов* (рис. 8).
Прежде чем приступить к работе, устанавливают весы по
уровню посредством опорных виг
коромысло весов «3 передвиже-
нием вправо арретира 4 и уста-
навливают стрелку-указатель 5
на 0 с помощью рычага 6. Рав-
новесное состояние весов соот-
ветствует положению стрелки-
указателя равновесия 7 против
черты равновесия 8. Если при
передвижении стрелки-указателя
на 0 указатель равновесия не
находится против черты равно-
весия, то его приводят к этому
положению посредством винта 9.
Затем, повернув арретир 4 влево,
закрепляют коромысло.
Для приготовления суспензии
навеску исследуемого порошка
помещают в фарфоровую ступку
и перемешивают с водой, после
1, 2. Затем освобождают
Рис. 8. Установка для седимен-
тационного анализа суспензий
с торзионными весами.
чего переносят в стакан, который наполняют до метки дистил-
лированной водой**. Если порошок плохо смачивается жид-
костью и суспензия неустойчива, то по указанию преподавателя
„добавляют стабилизирующие вещества. Перед опытом произво-
дят перемешивание суспензии с помощью стеклянной палочки,
на конце которой прикреплен резиновый диск. Движением
стеклянной палочки вверх и вниз достигают равномерного рас-
пределения частиц суспензии по всему объему. Перемешивание
ведут в течение некоторого времени (3—5 мин). После оконча-
ния перемешивания суспензии ставят стакан у прибора (рис. 8)
* В случае применения для седиментационного анализа торзионных ве-
сов, проверка применимости закона Гука к деформации пружины весов не
производится.
** Метку на стакане ставят предварительно, отмечая объем в 1 дм\
19
и быстро погружают в него чашечку, подвешивая ее на крючок
коромысла 10. Одновременно с погружением чашечки включают
секундомер. Освободив коромысло 3 передвижением арретира 4,
замечают отклонение указателя равновесия от равновесного
положения. Первый отсчет производят через 10 сек. Для этого
посредством рычага натяжения 6. передвигают стрелку-указа-
тель влево до тех пор, пока указатель 7 не будет находиться
против черты равновесия. По мере накопления осадка на чашеч-
ке указатель равновесия смещается влево, и во время отсчетов
его возвращают снова в равновесное положение. После оконча-
ния анализа следует закрепить коромысло 3 посредством йрре-
тира 4, снять чашечку и установить стрелку-указатель 5 на 0.
Скорость процесса седиментации полидисперсной суспензии
бывает наибольшей в начале опыта. Поэтому в ^начале опыта
отсчеты производят через каждые 20 сек' по мере приближения
к концу опыта время между отсчетами постепенно увеличивают
да 10—15 мин. Опыт ведут в течение 2—3 ч.
За исходный отсчет в данной работе рекомендуется брать
вес чашечки в воде. Для этого перед опытом в стакан до метки
наливают воду и, погрузив чашечку, отмечают показания тор-
зионных весов, одновременно измеряя глубину погружения ча-
шечки (в см).
Данные опыта записывают в заранее заготовленную таблицу
(см. табл. 1).
Таблица 1
№ отсчета Время отсчета Вес чашечки с осевшими на ней частицами, мг Вес частиц, осевших на чашечке pt мг Время от начала опыта t, сек 1000 t
1 14 ч 30 мин *. 14 » 30 » 10 сек 314 10 10
2 14 » 30 >30 » 319 15 30
3 14 » 30 » 50 » 324 20 50
Примечание. Вес чашечки в воде 304 мг1, глубина погружения h 9 см.
* Время начала опыта (окончание перемешивания).
По данным таблицы 1 на миллиметровой бумаге вычерчи-
вают кривую, откладывая по оси ординат вес частиц, осевших
на чашечке, р в мг, а по оси абсцисс — время от начала опыта
t в сек. При построении графика необходимо выбрать масштаб,
удобный дл'я дальнейшего графического расчета *.
* Рекомендуется при построении седиментационных кривых размер бу-
маги 20 X 40 см,
20
Для нахождения предела седиментации по данным табл. 1 .
|строят кривую в координатах р, (А = 1000), подобную изо-
браженной на рис. 4. К полученной седиментационной кривой
^проводят несколько касательных (5—7), отсекающих на оси
^ординат близкие по величине отрезки, и из точек касания опу-
скают перпендикуляры на ось абсцисс.
| Следует обратить внимание на определение точки касания
кдля тех случаев, когда кривая и касательная на
|участке полностью сливаются, т.
рсогда некоторые участки кривой седи-
ментации являются прямолинейными,
fe«rro имеет место при
;стиц с определенным
^вычисления которого
^максимальное время,
Miee точке отрыва
^кривой.
' На рис. 9 приведен пример построе-
ния ординаты на прямолинейном уча-
«стке кривой оседания, причем пункти-
ром показано неправильное построе-
ние. Ординаты, проведенные из точек касания, отсекают на оси
абсцисс отрезки, соответствующие времени полного оседания
.•' различных фракций t. По уравнению (4) вычисляют эквивалент-
р ные радиусы частиц, полностью осевших к моментам времени,
соответствующим построенным ординатам.
( . При вычислении эквивалентных радиусов прежде всего опре-
L деляют константу уравнения (4):
е.
некотором .
оседании ча-
ра диусом, для
следует взять
соответствую-
касательной от
Рис. 9. Построение ординат
к кривой седиментации.
/<=
di)g
При выражении радиусов в микронах*:
(d — d\) g
Скорость оседания:
Тогда:
t
см(м)
(5)
/<=10«
♦ При вычислении эквивалентных радиусов можно пользоваться номо-
граммами [14, стр. 284],
21
Максимальный размер частиц порошка может быть опреде-
лен различными методами, например микроскопическим, путем
отбора определенных фракций при отмучивании и др.
Иногда для определения максимального размера частиц
пользуются графическим методом: проводят касательную к на-
чальному участку седиментационной кривой и по значению вре-
мени, соответствующему точке отрыва касательной от седимен-
тационной кривой (/Мин, рис. 4), рассчитывают максимальный
радиус по уравнению (5).
Следует, однако, отметить, что этот метод дает- приблизи-
тельное значение гмаКс и его нельзя применять при анализе сус-
пензий, содержащих небольшие количества грубых фракций *.
После вычисления эквивалентных радиусов измеряют длины
отрезков ординаты между касательными и выражают их в про-
центах от общей длины ординаты (от начала координат до пре-
дела седиментации. В табл. 2 ордината равна 177 мм).
• Таблица 2
Время оседания t, сек Радиус частиц г, мн Интервалы размеров частиц отдельных фракций, мк Длина отрезков между касатель- ными, мм Содержание фракций в системе, %
'2200 3,9 <3,9 21 11,9
3,9—5,2 29 16,4
1200 5,2 5,2—7,7 .— —
550 7,7 —. — —
Всего .... 177 100
Отрезок ординаты от начала координат до точки пересече-
ния первой касательной (отрезок ос на рис. 4), отнесенный к
общей длине, ординаты, дает процентное содержание частиц
в интервале между максимальным эквивалентным радиусом
и наибольшим, определенным по седиментационной кривой
Из величин отдельных отрезков., между касательными вычис-
ляют процентное содержание частиц фракции между соответ-
ствующими им эквивалентными радиусами. Отрезок от предела
седиментации до ближайшей к нему касательной (отрезок ad
на рис. 4) выражает относительное содержание частиц меныш
определенного из седиментационной кривой наименьшего экви
валентного радиуса (<гМин).
Полученные на основании седиментационной кривой данные
записывают в таблицу (см. табл. 2).
* В учебных лабораториях можно сообщать учащимся значение макси-
мального радиуса при выдаче задачи, предварительно определив его каким-
либо методом.
22
Таблица 3
Г Q А <? AQ Дг
2 5 5 2,5
ДО
Рис. 10. Дифференциальная кривая
распределения.
На основании данных таблицы строят суммарную кривую рас-
пределения: откладывают по оси ординат суммарное процентное
содержание фракций Q, начиная с наиболее мелких частиц; по
оси абсцисс — радиусы частиц, соот-
ветствующие большему значению
радиуса данной фракции. Напри-
мер, если в исследуемом порошке
наиболее мелкая фракция содержит
частицы с эквивалентными радиу-
• сами < 3,9 мк и относительное ко-
личество ее в дисперсной системе - _1
11,9%, то по оси ординат отклады-
вают 11,9%, а по оси абсцисс
3,9 мк; если же следующая фракция находится в пределах
3,9—5,2 мк и относительное ее количество 16,4%, то по оси
ординат откладывают суммарное процентное содержание обеих
фракций, т. е. 28,3%, а по оси абсцисс — 5,2 мк и т. д. Подоб-
ная суммарная кривая распределения представлена на рис. 6
(кривая 1). Любая точка этой кривой, как указывалось ранее,
показывает процентное содержание в системе частиц с мень-
шими эквивалентными радиу-
сами, чем соответствующий ей
по оси абсцисс. Суммарную
кривую распределения можно
также получить, начиная рас-
чет от наиболее крупной
фракции, причем по оси абс-
цисс откладывают эквивалент-
ные радиусы, соответствую-
щие меньшим значениям экви-
валентных радиусов отдельных
фракций, а по оси ординат —
суммарные количества фрак-
ций в системе.
Из суммарной кривой рас-
пределения находят величины
приращения процентного со-
держания частиц AQ через равные интервалы радиусов, для
чего может быть использована любая из двух полученных кривых.
Найденные результаты записывают в таблицу (см. табл. 3).
По данным табл. 3 строят дифференциальную кривую рас-
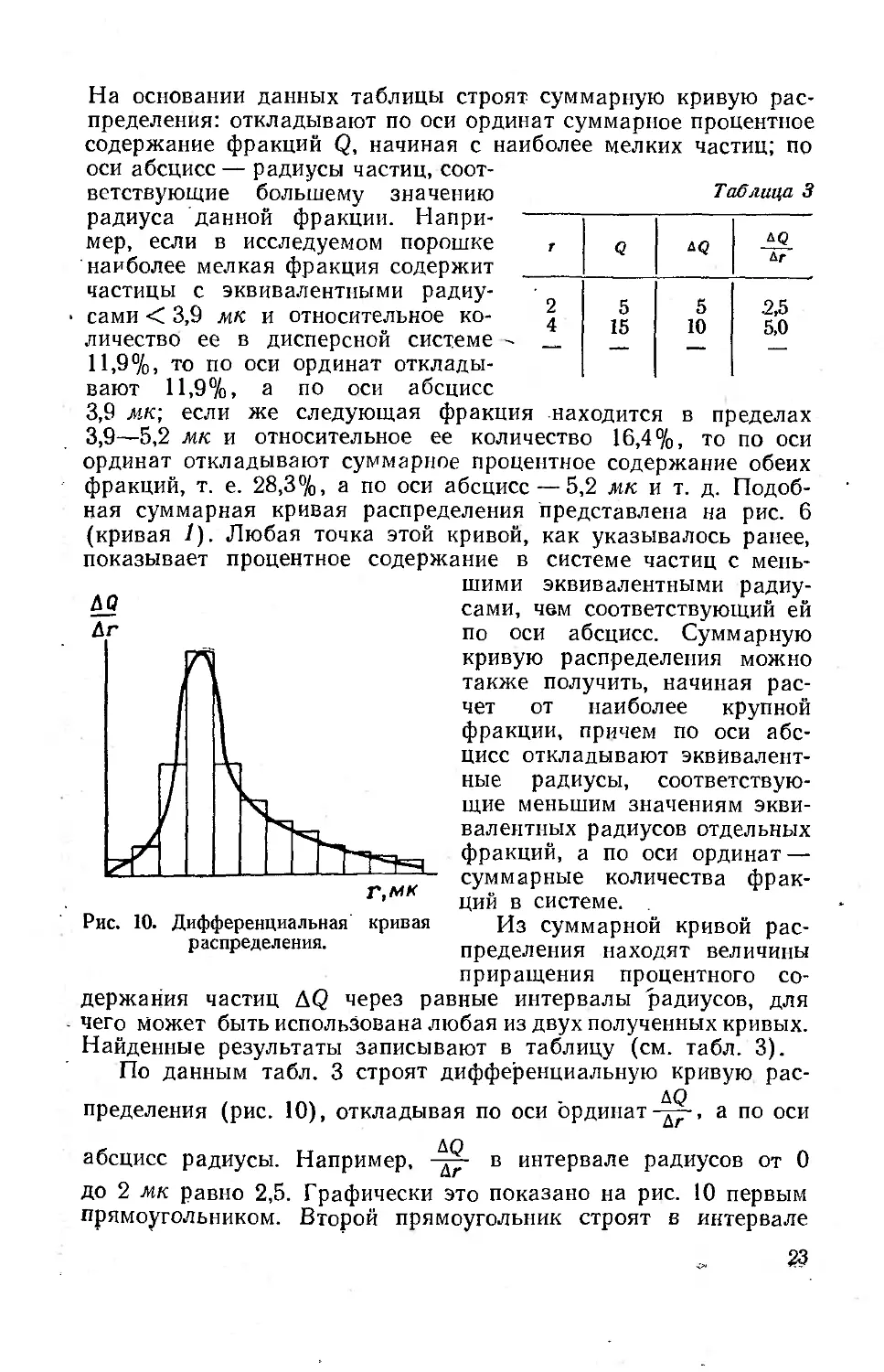
пределения (рис. 10), откладывая по оси ординат-^-, а по оси
абсцисс радиусы. Например, в интервале радиусов от 0
до 2 мк равно 2,5. Графически это показано на рис. 10 первым
прямоугольником. Второй прямоугольник строят в интервале
23
радиусов от 2 до 4 мк и т. д. — до максимального радиуса ча-
стиц. Соединив середины прямоугольников, получают плавную
кривую, подобную изображенной на рис. 10. Максимум на диф-
ференциальной кривой распределения соответствует наиболее
вероятному размеру частиц. Форма полученной дифференциаль-
ной кривой, в зависимости от характера дисперсности исследуе-
мой системы, может быть весьма различной.
Работа 2
ПИПЕТОЧНЫЙ МЕТОД СЕДИМЕНТАЦИОННОГО АНАЛИЗА СУСПЕНЗИИ
Этот метод является одним из основных методов седимента-
ционного анализа, широко применяемого в практике, особенно
при исследовании почв и грунтов. Сущность метода заключается
в определении изменения концентрации вещества на определен-
ной глубине суспензии, оседающей в спокойной жидкости.
При проведении седиментационного анализа этим методом
берут ряд проб с определенной глубины в суспензии через раз-
личные промежутки времени от начала опыта и определяют ве-
совую концентрацию вещества в каждой пробе. Время взятия
проб рассчитывают предварительно по уравнению (5) для ряда
выбранных значений'радиусов. При этом считают, что в пробу
. h
попадут только частицы, скорость оседания которых и -j
(h — глубина погружения, t — время от начала опыта); частицы
же, оседающие с большей скоростью, в пробе отсутствуют. Для
того чтобы не захватывать в пробу частицы из более глубоких
слоев, употребляют пипетки с боковыми отверстиями (нижний
конец заплавлен).
. Определение концентрации вещества в отдельных пробах
надо производить с большой тщательностью, так как неточ-
ность анализа сильно скажется на полученных результатах. Чем
больше концентрация суспензии, тем более надежные данные
получают при анализе проб. Однако, как указывалось выше,
седиментационный анализ не рекомендуется проводить при
больших концентрациях суспензии. Обычно при проведении се-
диментационного анализа пипеточным методом берут 1% сус-
пензию. Если производить отбор проб из такой суспензии на
определенной глубине через различные промежутки времени,
соответствующие времени полного оседания частиц определен-
ных размеров, то, зная первоначальную концентрацию суспен-
зии и ее объем, можно рассчитать весовую концентрацию в от-
дельных пробах.
Разность весовых концентраций между отдельными пробами
даст относительное содержание частиц соответствующих фрак-
ций в исследуемой суспензии. Вычислив процентное содержание
24
/Каждой фракции, можно оформить полученные результаты 6
> виде интегральной и. дифференциальной кривых распределения,
построение которых подробно рассмотрено в предыдущей ра-
боте.
'Выполнение работы
Прибор для седиментационного анализа по пипеточному ме*
тоду (рис. 11) состоит из цилиндра (емкостью в 1 дм3) с ме-
шалкой, буферной склянки с насосом и пипетки на 25 см3 для
отбора проб. Пипетка имеет наверху кран, нижний конец ее
удлинен и имеет внизу 4 отверстия,
расположенные сбоку, низ запаян.
С помощью насоса создается ва-
куум в буферной склянке; закрыв
выходной кран к насосу и на-
дев каучуковую трубку на пипетку,
можно путем открывания крана пи-
петки произвести отбор пробы сус-
пензии.
Перед опытом производят по
уравнению (5) расчет времени осе-
дания частиц различного радиуса
для исследуемого вещества. Затем
берут навеску порошка из расчета
приготовления 1% суспензии, т. е.
10 г на 1 дм3 суспензии, и приго-
тавливают суспензию, как указано
. на стр. 19. После перемешивания
суспензии осторожно устанавливают
пипетку, по соответствующей на
ней метке, На. определенную глу-
бину. Отсчет времени для первых
проб производят по секундомеру, а
затем По часам.
Рис. 11. Прибор для седимен*
тационного анализа пипеточ-
ным методом.
Соответственно рассчитанному
времени оседания частиц определен-
ного рддиуса производят отбор пробы в пипетку. Содержимое
пипетки выливают в фарфоровую чашку, предварительно взве-
шенную, и затем ополаскивают пипетку несколькими см3 дистил-
лированной воды в ту же чашку. Фарфоровую чашку Ставят
на Водяную баню и выпаривают содержимое досуха, после чего
высушивают остаток при 105° С в сушильном шкафу до постоян-
ного веса.
Суспензию в цилиндре снова перемешивают й по Истечений
соответствующего времени отбирают следующую пробу и т. д.
В отдельной пробе определяют влажность исследуемого по-
рошка высушиванием в сушильном шкафу при 105° С допостоян-
25
Кого веса. Так, если взята навеска р исследуемого Порошка (в г),
и отдельное определение влажности дало b % влаги, то
/>(100—6) л
• IQQ---- соответствует навеске абсолютно сухого порошка, а
, п/ . р (100 — 6) о
концентрация суспензии составляет (в и/о) —' 10q0—— с. Затем
вычисляют процентное содержание отдельных фракций. При
этом, как указывалось выше, считают, что если проба взята
через промежуток времени 1\, отвечающий времени полного осе-
дания на.данную глубину частиц с радиусом гь то в нее попа-
дут частицы с эквивалентным радиусом t\ и меньшим, в то
время как все частицы с эквивалентным радиусом большим г\
окажутся на большей глубине и в пробу не попадут. Если, на-
пример, взяты пробы по 25 см3 через промежутки времени, со-
ответствующие времени полного оседания частиц с эквивалент-
ными радиусами 50, 10, 5 и 1 мк, а величины сухих остатков в
последовательных пробах соответственно равнялись р50, рю, р5
и pt, то содержание частиц (в %), имеющих радиусы меньше
рассчитанных, определяют из следующих соотношений:
Радиусы частиц, мк Содержание частиц, %
<5о к =
С
<ю l = -'fog-’-122.
С
< 5
С
< ! /v=JAJ22.
С
Приняв все весовое количество порошка за 100%, получают
содержание частиц отдельных фракций:
Фракция, мк Содержание, %
>50 100 — К
50—10 К — L
10—5 L—M
5—1 M — N
< 1 N
Работа 3
СЕДИМЕНТАЦИОННЫЙ АНАЛИЗ СУСПЕНЗИЙ В ВОСХОДЯЩЕМ ПОТОКЕ
ЖИДКОСТИ С РАЗДЕЛЕНИЕМ СИСТЕМЫ НА ФРАКЦИИ
Этот метод основан на том, что в восходящем потоке жид-
кости могут оседать только те частицы, скорость оседания ко-
торых больше линейной скорости потока. Частицы же с мень-
шей скоростью оседания будут-уноситься потоком. Таким обра-
26
Рис. 12. Прибор для седимента-
ционного анализа суспензий в вос-
ходящем потоке жидкости.
зом, если жидкость со строго определенной скоростью будет
поступать на дно сосуда с суспензией и вытекать через спе-
циальные сливы из верхней его части, то через некоторое время
в нем не останется частиц, скорость оседания которых меньше
скорости потока жидкости, и в вытекающей жидкости не будет
взвешенных частиц. Так из полидисперсной системы можно вы-
делить фракцию, содержащую наиболее мелкие частицы.
Затем, повышая скорость потока жидкости, выделяют сле-
дующую фракцию с большими частицами. Когда в сосуде не
-останется частиц, скорость оседания которых меньше данной
скорости потока, последнюю снова увеличивают и т. д.
Меняя скорость потока жидкости, можно разделить полидис-
персную систему на фракции и определить их процентное содер-
жание в системе.
Расчет скорости оседания частиц с определенным эквива-
лентным радиусом, а следовательно, и линейной скорости вос-
ходящего потока жидкости обыч-
но проводят по уравнению (3):
2 г2р
Зная линейную скорость вос-
ходящего потока жидкости, мож-
но определить объемную ско-
рость, т. е. количество жидкости
vM, которое надо пропустить
в 1 мин через сосуд с суспен-
зией для достижения скорости и
восходящего потока:
= см31мин (6)
где и — скорость, вычисленная
по уравнению (3), см!сек\ q —
площадь поперечного сечения
сосуда, в котором проводится
анализ, см?.
Иногда для разделения поли-
дисперсной системы на отдель-
ные фракции пользуются прибором, изображенным на рис. 12.
Это несколько соединенных друг с другом сосудов разного диа-
метра. Суспензию помещают в самый маленький сосуд. Жид-
кость, поступающая в него со строго определенной скоростью,
перетекает в большие сосуды со все убывающей скоростью, и
Это дает возможность разделить полидисперсиую систему на
отдельные фракции. Разделение заканчивают после того, как
в вытекающей из последнего сосуда жидкости не будет взве-
шенных частиц. Прибор разбирают и определяют процентное
содержание отдельных фракций,
27
Выполнение работы
Рис. 13. Установка для седимен-
тационного анализа суспензий
в восходящем потоке жидкости.
В данной работе применяют прибор, схематически изобра-
женный на рис. 13. Вода поступает с определенной скоростью из
сосуда 1 в сосуд 3, в котором находится исследуемый порошок, 1
и вытекает через специальную сливную трубку 4 пьезометра 5,
который служит для фиксирова-
ния постоянства скорости про-
текания суспензии. Для того что-
бы жидкость поднялась, в вер-
тикальной трубке пьезометра,
употребляют сливные трубки с
оттянутым концом. В зависимо-
сти от скорости протекания сус-
пензии применяют сливные труб-
ки с разным диаметром оттяну-
того конца. Применение таких
трубок создает некоторое сопро-
тивление вытекающей суспензии,
в связи с .чем в вертикальной
трубке повышается уровень
жидкости, который остается по-
стоянным при определенной ско-
рости потока и фиксируется
по миллиметровой шкале 6 пьезо-
метра 5. Скорость потока жидко-
сти регулируют винтовым зажи-
мом 2.
Приступая к работе, прежде
всего определяют площадь по-
перечного сечения q сосуда 3, в
котором производится анализ.
Для этого наполняют его водой
до начала цилиндрической
части, вдоль которой наклеи-
вают полоску миллиметровой бу-
пипеткой воду и отмечают повы-
маги. Затем наливают в него
шение уровня. Зная объем прилитой воды и повышение уровня,
вычисляют площадь поперечного сечения сосуда. Добавляя сле-
дующие порции воды и отмечая каждый раз повышение уровня
жидкости, вычисляют площадь поперечного сечения сосуда в
различных участках и берут среднее значение.
Чтобы разделить исследуемый порошок на ряд фракций,
необходимо прежде всего определить значения линейной ско-
рости восходящего потока жидкости. Для этого вычисляют ско-
рость оседания частиц с предельными эквивалентными радиу-
сами каждой фракции по уравнению (3). (Предельные значе-
28
|сия эквивалентных радиусов частиц каждой фракции указы-
ваются преподавателем.) Затем по уравнению (6) вычисляют
Объемные скорости протекания жидкости, необходимые для до-
стижения вычисленных значений линейной скорости потока
даидкости.
ч Приступая к анализу, исследуемый порошок (навеска — по
наказанию преподавателя) сначала тщательно перемешивают
небольшим количеством воды (см. стр. 19), затем переносят
Ц сосуд 3, в который впускают воду. Установив объемную ско-
рость потока, равную наименьшей из рассчитанных, выделяют
Вз суспензии фракцию, содержащую самые мелкие частицы.
Йосле того как в вытекающей из сосуда 3 жидкости не будет
«взвешенных частиц, повышают скорость потока и собирают в
Отдельный сосуд следующую фракции? с частицами большего
размера и т. д.
Е - После разделения исследуемого порошка на фракции опре-
деляют вес последних и вычисляют процентное содержание
Важдой фракции (стр. 26). Полученные данные оформляют
ф виде таблицы:
Таблица 4
---------
| Эквивалент-
ное радиусы,
К мк
-----------
Линейная скорость
потока жидкости,
см(сек
Объемная скорость
потока жидкости,
см*!мин
Радиус
фракции,
мк
Вес
фракции
Содержание
отдельных
фракций, %
Определение размеров и формы коллоидных частиц
по рассеянию ими света
Согласно современной теории света, всякая точка среды, ко-
торой достиг фронт световой волны, становится источником
|йзйучения вторичных волн. Вторичные волны, посылаемые раз-
личными точками среды, интерферируют между собой, обуслов-
ливая ту или иную картину распределения интенсивности све-
тового потока в пространстве.
.Под интенсивностью светового потока следует
|йонимать количество энёргии, протекающее в единицу времени
рёрез единицу сечения и приходящееся на единичный телесный
•^гол.
%? В однородном веществе интерференция вторичных волн при-
водит к прямолинейному распространению света. Не так об-
стоит дело, если среда оптически неоднородна. Оптическая не-
однородность может быть вызвана различными причинами: флу
ртуациями плотности и концентрации, присутствием посторон-
29
них примесей. Среда, в которой число оптических неоднород-
ностей велико, а размеры их незначительны, называется мутной.
Распространение света в неоднородной среде не является пря-
молинейным. Свет наблюдается в большей или меньшей сте-
пени по всем направлениям, т. е. рассеивается.
Суммарный поток энергии, рассеянной частицей во всех на-
правлениях, отнесенный к единице интенсивности падающего
потока, называется коэффициентом рассеяния и обо-
значается символом /Ср. Рассеяние света характеризуется вели-
чиной интенсивности светового потока, рассеянного в различных
направлениях. Векторная диаграмма, показывающая распре-
деление интенсивности рассеянного света по всем направле-
ниям, называется индикатриссой рассеяния.
Изучение рассеяния света важно для суждения о величине
и форме частиц коллоидной дисперсности, которые слишком
малы для непосредственного исследования их с помощью обыч-
ного микроскопа. На явлении рассеяния света основан ряд ме-
тодов определения размера и формы частиц: с использованием
ультрамикроскопа, фотоэлектроколориметра, нефелометра и
поляриметра. В ультрамикроскопе каждая частица обнаружи-
вается в отдельности в виде светящейся точки или системы ди-
фракционных колец. В' остальных методах величина частицы
оценивается на основании измерений интенсивности светового
потока и степени поляризации в различных направлениях при
рассеянии света в мутной среде. В совокупности эти методы
дают возможность составить более или менее ясное представле-
ние и о форме частиц.
Распределение интенсивности рассеянного света в золях и
суспензиях. зависит от размера и формы частиц, длины волны
падающего света и от показателей преломления частиц и среды.
Зависимость степени поляризации рассеянного света от на-
правления рассеяния также определяется размером и формой
частиц.
Впервые теоретическое объяснение рассеянию света сфери-
ческими непроводящими частицами было дано Рэлеем. Согласно
теории Рэлея, интенсивность рассеяния естественного света опи-
сывается уравнением:
, , 9-2Ц2 I W2_ 1 12/ 1-|-COS2₽\
I I---2----) (1)
. где /0 — исходная интенсивность света;
V — объем частицы;
а —расстояние от центра частицы до точки наблюдения;
т — отношение показателя преломления частицы к показа-
телю преломления среды;
X — длина волны света в среде: Х = ^, где Хв — длина волны
света в вакууме, « — показатель преломления .среды; 0=180—0
3Q
Рис. 14. Схема хода лучей.
(где 6 — угол между направлением падающего луча И рассеян-
ного в данном направлении, — см. рис. 14).
Из формулы (1) следует, что интенсивность рассеянного
света обратно пропорциональна четвертой степени длины волны
падающего луча. Этим обьяс-
няется тот факт, что рассеянный
достаточно малыми частицами
свет при наблюдении сбоку
имеет голубоватый оттенок, а про-
ходящий — приобретает красно-
ватую окраску.
Интенсивности рассеяния све-
та в плоскостях координат, при
Направлении падающего луча
В отрицательном направлений
оси OZ (рис. 14), будут подчи-
няться следующим зависимо-
стям: в плоскости ХОУ, перпен-
дикулярной направлению падаю-
щего луча (J3 = 90°), I ~ const.
Индикатрисса рассеяния будет 'у
представлять собой окружность.
В плоскостях XOZ и YOZ:
I == 1 + cos2 р. Соответствующие
им индикатриссы Показаны на
рис. 15.
Таким образом, согласно теории Рэлея, индикатрисса рассея-
ния естественного света симметрична
в направлении распро-
Рис. 15. Индикатриссы рассеяния естествен-
ного света малой сферической частицей в пло-
скостях координат XOY, YOZ, и XOZ.
Страненйя падающего луча и в направлений, ему перпендику-
лярном.
Для вычисления коэффициента рассеяния кр необходимо,
Согласно определению, проинтегрировать индикатриссу рассея-
ния по всем направлениям и отнести полученную величину к
31
единице интенсивности падающего потока:
2л тс
Ар = j- J* dy JI sin ₽ d₽
° о о
(2)
где ф — угол, под которым наблюдается рассеяние света в плос-
кости XOY (см. рис. 14).
Подставив значения / из уравнения (1) и произведя необ-
ходимые вычисления, Получаем, что согласно теории Рэлея
_ 24лэуг I тг_1 12
X4 I «24-21
Введем следующие обозначения:
_ 3 I «2_1 |
а~ 4л [ «2.^2 |
(3)
(4)
т. е. ft — параметр, учитывающий соотношение между показа-
телями преломления частицы и среды;
8лг
г~~1~
Где г — радиус сферической частицы.
Тогда формула (3) примет вид:
Ар = 0,183г4лг2 [ a |s (6)
Теория Рэлея применима при следующих условиях:
а) должно соблюдаться определенное соотношение между
t размером частицы и длиной волны света, а именно — отношение
периметра частицы к длире волны не должно превышать 0,3.
Соотношение это обычно называется характеристиче-
ским параметром частицы и обозначается символом р:
б) частица не должна заметно поглощать Свет. Вследствие
Этого условия теория приложима к золям диэлектриков и не
может быть применима к золям металлов. j
Когда величина р становится больше, чем 0,3, интенсивность
рассеянного света Перестает быть пропорциональной V*. Зави-
симость интенсивности от уу- переходит постепенно в зависи-
мость Вследствие этого свет, рассеянный такими Золями в
боковом направлении, теряет голубоватый оттенок и переходит
в молочйо-белый. Зависимость интенсивности рассеянного света
от направления рассеяния приобретает более сложный харак-
тер. По мере укрупнения частиц индикатрисса рассеяния вее
32
5ольше теряет Свою Симметричность. Изменение индикатриссы
зассеяния по мере увеличения р изображено на рис. 16.
, Впоследствии теория Рэлея была распространена и на бо-
iee крупные частицы. На основании теории распространения
Электромагнитных волн в оптически неоднородной среде была
Рис. 16. Изменение формы индикатриссы рассеяния света
сферической частицей в плоскостях yoz и xoz по мере
ее укрупнения.
получена общая формула, Связывающая коэффициент рассея-
ния кр с размерами сферической частицы и применимая до
р == 5 [14]:
йр = яг2|а|2/?(г-) (8)
где /7(z) = -^-|-^- + 5s2 + (4z2—16)(ciz — Ins — C)—2zsinz-[-
co
+ 14(cosz—1)|; cis — — J dt
z
*C—постоянная Эйлера, равная 0,577...
Из сравнения формул (6) и (8) следует, что зависимость (8)
отличается от зависимости, требуемой, по уравнению Рэлея, на
величину функционального множителя, определяемого размером
частиц. В частном случае достаточно малых z (применимость
Теории Рэлея) сложная функция F (z) переходит в более про-
стую, а именно:
' Д (г) =0,183/
На рис. 17 приведены кривые kp = f(p), вычисленные с по-
мощью формул (6) и (8).
| Зависимость поляризации естественного света от направле-
;-ния также связана с размером частиц.
В случае достаточно малых частиц (применимость теории
1Рэлея) естественный свет будет полностью поляризован в на-
/правлении, перпендикулярном направлению падающего луча,
33
а в направлении распространения Луча останется деполяризо-
ванным. В промежуточных направлениях рассеянный свет ча-
стично поляризован. Зависимость степени поляризации от угла
наблюдения в этом случае выражается кривой, изображенной
на рис. 18.
При падении естественного света на систему, состоящую из
более крупных частиц (р>0,3), полной поляризации не наблю-
Лр
2,0
1,6 -
1,6 -
Г,4 •
1,2
1,0
о,в
0,6
0,4 -
0,2 -
° 1 ? 2 3 4 ~5
Р
Рис. 17.' Зависимость коэффи-
циента рассеяния света от вели-
чины характеристического пара--.
метра.
Кривая 1 построена по общей формуле;
кривая 2 получена, исходя из уравнения
Рэлея.
дается вообще. Область макси-
мальной поляризации смещается
в сторону источника света.
Нарушение сферичности фор-
мы, в свою очередь, приводит к
изменению зависимости как ин-
тенсивности рассеяния, так и
степени поляризации от направ-
ления рассеяния. Суммарная
световая энергия, рассеиваемая
стержневидной или дискообраз-
ной частицей, меньше, чем рас-
сеиваемая малой сферической ча-
Рис. 18. Зависимость степени поля-
ризации рассеянного света от угла
наблюдения.
стицей того же объема. Это отклонение от сферической формы,
т. е. анизодиаметричность частицы, может быть оценено введе-
нием некоторой функции q, зависящей от соотношения осей час-
тицы (анизодиаметрическая частица уподобляется эллипсоиду
вращения). Индикатрисса рассеяния такой частицы отличается
от индикатриссы сферических частиц. Большое значение при
этом имеет направление, в котором послан падающий луч. Если
падающий луч послан вдоль оси симметрии эллипсоида враще-
ния частицы, то в случае малой частицы (р < 1) индикатрисса
делается более узкой, но Не теряет симметричности. Для боль-
ших частиц в данных условиях индикатрисса вытягивается впе-
34
ред (так же как при укрупнении сферических частиц). Если па-
дающий свет направлен перпендикулярно оси симметрии, то
индикатрисса рассеяния зависит по иному от направления на-
блюдения. На рис. 19, а указаны индикатриссы рассеяния частиц
при направлении падающего
эллипсоида, на рис. 19,6—
перпендикулярно оси симмет-
рии в плоскости, включающей
ось симметрии и направление
падающего луча.
В реальных условиях, где
имеется большое число хаоти-
чески распределенных частиц,
«средняя» индикатрисса рас-
сеяния определяется следую-
щими закономерностями.
Отношение интенсивности
рассеяния света в данном на-
правлении к интенсивности па-
дающего света равно для
стержневидных частиц:
I 1 f sin IF / sin W\2
7o'_TF'J ~w~aw
о
(9)
луча вдоль оси симметрии
Рис. 19. Формы индикатрисе рассея-
ния света в зависимости от анизо-
диаметрии частиц и направления
падения луча:
а — луч послан вдоль оси симметрии частицы:
/ — частица сферическая; 5 —частица анизо-
диаметрическая (отношение осей 2 : 3); 5— ча-
стица анизодиаметрическая (отношение осей
1:10); б— луч послан перпендикулярно оси
симметрии частицы; частица анизодиаметри-
ческая (отношение осей 1 :10).
где W——у— sin, L — большая ось частицы; для сферических
частиц:
/- = •4-(sin О — О cos О)2 (10)
/0 и
-. 4т: г . ₽
где £7——^— sin
Имея построенные по формулам (9) и (10) кривые зависи-
мостей
(где (21 и 02 — углы, под которыми наблюдается рассеяние), и
получив экспериментальные значения -^Д- (при той же X), мож-
но определить отклонение формы частицы от сферической, т. е.
величину ее анизодиаметрии [12].
Величина анизодиаметрии может быть определена также на
основании зависимости степени поляризации от угла рассеяния
и сравнением поляризации рассеянных под углом 90° естествен-
ного и линейно поляризованного падающих лучей [10], [17].
3*
35
Для определения индикатриссы рассеяния на практике обыч-
но измеряют интенсивность рассеянного света при величине
угла ₽ = 0°, 45°, 90°, 135°.
Работа 4
ОПРЕДЕЛЕНИЕ ВЕЛИЧИНЫ КОЛЛОИДНЫХ ЧАСТИЦ ПРИ ПОМОЩИ
УЛЬТРАМИКРОСКОПА
В основе данного метода лежит явление рассеяния света кол-
лоидными частицами, называемое, я в л е н и е м Тиндаля и
наблюдаемое в том случае, когда размеры частиц меньше по-
ловины длины волны падающего света. На наблюдении явле-
ния Тиндаля под микроскопом основан принцип устройства
ультрамикроскопов различных систем. Коллоидный раствор
освещают сбоку на темном фоне сильным источником света и
наблюдения проводят с помощью обычного оптического микро-
скопа. Следует особо подчеркнуть, что в ультрамикроскопе не
видны отдельные частицы, но благодаря рассеянию ими света
обнаруживают в виде светящихся точек.
Кроме ультрамикроскопа могут быть использованы ультра-
конденсоры различной конструкции,. которые помещают под
столик микроскопа вместо обычного осветительного конденсора.
Принцип их устройства заключается в том, что свет, освещаю-
щий коллоидный раствор, не попадает в объектив микроскопа;
это дает возможность наблюдать рассеянный коллоидными ча-
стицами свет.
При рассмотрении коллоидного раствора в ультрамикроскоп
можно сделать некоторые заключения о форме частиц. Напри-
мер, частицы, мало отличающиеся от сферических, светятся
равномерно, так как они рассеивают свет в одинаковой степени
при любом положении. При наблюдении в ультрамикроскоп
частиц, имеющих палочкообразную форму (золь V2O5) или лис-
точкообразную форму [золь Fe(OH)s] и тому подобную, видят
искрящиеся точки на темном фоне. Это связано с различной
интенсивностью рассеянного света в зависимости от положения,
в котором находятся частицы при броуновском движении по
отношению к падающим на них лучам.
Для определения размеров коллоидных частиц с помощью
ультрамикроскопа необходимо провести подсчет частиц в изве-
стном объеме, т. е. определить частичную концентрацию золя.
Если с — концентрация коллоидного раствора, т. е. число г
раздробленного вещества в единице объема, d — плотность ве-
щества, п — число частиц в объеме V см3, то масса отдельной
частицы;
eV
m=s~w an
36
Принимая форму частицы за куб с ребром I, можно выра-
зить ее массу уравнением:
m^l3d ' (12)
Из уравнения (11) и (12) получим: eV 13а = п
Отсюда: eV /=1/ (13) . г па
Если же считать частицу шарообразной с радиусом г, то:
4 _ eV
т = -тг пг3а =-
3 п
Откуда диаметр частицы:
з/ дту
£> = 2г = 21/ (14)
г 4я dn ' ’
Несмотря на некоторую условность сделанных предположе-
ний об определенной форме частиц и постоянстве плотности ве-
щества при дроблении (d — плотность вещества в массив-
ном состоянии), значения величины частиц, полученные уль-
трамикроскопическим путем, были подтверждены непосред-
ственным их определением при помощи электронного микро-
скопа.
Рассмотренный метод определения среднего размера кол-
лоидных частиц был усовершенствован Дерягиным и Власенко
[4], [5], [6], которые разработали метод поточной ультрамикро-
скопии и сконструировали на его принципе приборы — поточ-
ные ультрамикроскопы.
Идея этого метода заключается в подсчете частиц в непре-
рывном потоке золя, пересекающих за определенный промежу-
ток времени освещенную зону в направлении луча зрения. Поль-
зуясь этим методом, можно не только определить средний раз-
мер коллоидных частиц, но и провести дисперсионный анализ
исследуемой системы, ведя подсчет частиц при постоянной ско-
рости потока и постепенно уменьшающейся освещенности зоны
подсчета. При каждой освещенности глаз способен регистриро-
вать частицы с радиусом, большим определенной величины.
Поэтому, меняя освещенность и подсчитывая число частиц при
постоянной скорости потока, можно получить данные для по-
строения интегральной кривой распределения частиц по раз-
мерам.
37
Выполнение работы
В данной работе применяется щелевой ультрамикроскоп,
изображенный на рис. 20.
В металлической коробке 1 получают электрическую дугу,
пучок света от которой через отверстие 3 падает на линзу 4.
Рис. 20. Щелевой ультрамикроскоп.
Посредством линзы 4 изображение кратера дуги отбрасывается
на раздвижную прямоугольную щель 5. Далее свет проходит
через объектив и кювету с коллоидным раствором, где обра-
зует реальное изображение щели в виде тон-
кого луча прямоугольного сечения.
Подсчет светящихся частиц коллоидного
раствора производится при помощи микроско-
[^//^^///^2 па 8, расположенного перпендикулярно пучку
tZaMMBEl/ света, освещающего раствор. Определение
объема V, в котором производится подсчет
^^//////7/ частиц, проводят с помощью раздвижной
щели 5 (рис. 20, 21). На диске <3 (рис. 21),
имеющем в середине отверстие, закреплена
пластинка 1, над которой помещена подвиж-
ная пластинка 2, соединенная с микрометри-
ческим винтом 4. Вращая микрометриче-
ский винт, можно изменять расстояние между пластинками
1 и 2 и тем самым глубину пучка света, освещающего рас-
твор. Счетный объем V (обычно прямоугольный параллелепи-
пед) выделяется оптически из всего поля зрения, при помощи
Рис. 21. Схема
раздвижной щели
ультрамикроскопа.
окулярмикрометра с квадратной сеткой. Один из квадратов сет-
ки принимают за основание параллелепипеда. Высоту его, рав-
ную глубине пучка света, можно установить с помощью ми-
38
крометрического винта 4 (рис. 21) или путем поворота дис-
ка 3 на 90°. При таком повороте у пучка света прямоугольного
сечения, как это видно на рис. 22, глубина становится шири-
ной и может быть измерена. Тогда, если сторона квадрата /1,
а высота параллелепипеда h, то объем, взятый для подсчета
частиц:
Для определения V пользуются исходным неразбавленным
золем, так как при этом более резко выделяются границы све-
тового луча.
Определив глубину пучка света, поворачивают диск в исход-
ное положение и производят подсчет частиц, находящихся в
каждом квадрате. Так как кол-
лоидные частицы совершают ин-
тенсивное броуновское движение,
то рекомендуется производить
их подсчет при условии, чтобы в
счетном объеме в среднем со-
держалось не больше пяти час-
тиц. Вследствие того, что число
частиц в данном объеме все вре-
мя изменяется, необходимо про-
вести большое число подсчетов
(для получения точных резуль-
Рис. 22. Схема выделения счет-
ного объема.
татов — не менее ста) и взять
среднее значение для п. Определив V и п, вычисляют размбр
частиц по уравнениям (13) или (14).
Работу рекомендуется проводить в следующем порядке:
1) приготовляют коллоидный раствор по указанию препода-
вателя;
2) в кювету помещают неразбавленный коллоидный раствор
и включают электрическую дугу, причем электроды сближают
посредством винта 2 (рис. 20). Устанавливают микроскоп с
объективом малого увеличения (~Х10) так, чтобы отчетливо
были видны светящиеся частицы;
3) переведя микроскоп на объектив с большим увеличением
• (~Х40), устанавливают глубину пучка света, как указано
выше, посредством поворота диска 3 (рис. 21). Поворот диска
производят посредством рычага 6, но ни в коем случае нельзя
для этого пользоваться микрометрическим винтом 7 (рис. 20).
Устанавливают ширину светового пучка" в один квадрат сетки.
Учитывая, что при данном увеличении (~в 400 раз) сторона
квадрата равна определенному значению, вычисляют счеТный
. объем V. Размер стороны квадрата окулярной сетки определяют
. предварительным измерением по шкале объектмикрометра
(стр, 7);
89
4) повернув диск 3 (рис. 21) в исходное положение, напол-
няют кювету разбавленным раствором и подсчитывают число
частиц в поле зрения каждого квадрата;
5) сделав подсчеты частиц, вычисляют их размер по урав-
нению (14).
Работа 5
ОПРЕДЕЛЕНИЕ РАЗМЕРОВ ЧАСТИЦ ПО ИНТЕНСИВНОСТИ СВЕТА,
РАССЕЯННОГО ПОД УГЛАМИ fl - 45° И 135°
Выполнение работы
Приготовляется исследуемый золь. Измерения производят
С помощью нефелометров различного типа (например, НФМ).
Измеряют значения интенсивности света, рассеянного под уг-
лами р — 45° и р,== 135° (/45 и Л35). На основании полученных
данных вычисляют отношение Это отношение определяет
форму индикатриссы и яв'ляется характеристикой, позволяю-
щей определить размер частиц. Имеются таблицы, в которых
приведены значения г, соответствующих данным значениям
(таблица приведена ниже на стр. 44). По полученным экспери-
ментально значениям -у15- находят по таблице соответствующие
величины z и далее по формуле (5) рассчитывают величину г.
Таким методом могут быть исследованы, например, бесцвет-
ные золи различных окислов металлов, суспензии глин, бенто-
нита и т. п. --
Результаты записывают в виде таблицы.
Таблица 1.
spijcM^-ceK-cmep • ер^1см‘-сек-стер
* В системе СИ дж^-сек-стер, стер — единица телесного угла — стерадиан.
МЕТОДЫ ОПРЕДЕЛЕНИЯ РАЗМЕРОВ И ФОРМЫ ЧАСТИЦ,
ОСНОВАННЫЕ НА ИЗМЕРЕНИИ ИНТЕНСИВНОСТИ
ПРОХОДЯЩЕГО СВЕТА
Методы основаны на определении ослабления падающего
луча при прохождении им мутной среды. Измерения могут быть
проведены с помощью фотометров и фотоэлектроколориметров .
40
'(например, ФМ, ФМ-56, ФЭК-Н). Рассмотрим; случай, когда
ослабление света происходит только вследствие рассеяния, но
не поглощения — частицы заметно не поглощают свет. Это имеет
место, например, в случае бесцветных золей неметаллов,- Ин-
тенсивность падающего света ослабляется вследствие рассеяния
согласна следующему закону:
/ = /о(?"тх (15)
где х — толщина среды, через которую прошел луч, вследствие
чего интенсивность его снизилась от 70 до /; т — мутность среды,
или коэффициент экстинкции.
Из формулы (15) следует:
2,3031g
’ = - *----- (16)
Величина
1g ^-=О (17) .
называется оптической плотностью.
Величина т равна энергии, рассеиваемой 1 гм3 системы. Ее
можно рассматривать как величину, обратную расстоянию, ко-
торое поглощаемый луч должен пройти, чтобы быть ослаблен-
ным в е раз. Закономерность (15) имеет место только для до-
статочно разбавленных золей.
< Работав
ОПРЕДЕЛЕНИЕ РАЗМЕРОВ КОЛЛОИДНЫХ ЧАСТИЦ ПО ЗАВИСИМОСТИ
КОЭФФИЦИЕНТА ЭКСТИНКЦИИ ОТ ДЛИНЫ ВОЛНЫ СВЕТА
Зависимость коэффициента экстинкции т от длины волны
света Z, может быть представлена эмпирическим уравнением:
z = (18)
где b — коэффициент, не зависящий от размера частиц, их
формы и длины волны падающего света;
k — коэффициент, зависящий от размера и формы частиц
и не зависящий от длины волны света.
Из уравнения (18) следует:
AlgX
Величина k изменяется в пределах от 2 до 4 и достаточно
точно определяет размер частиц. Согласно зависимости г = f (k)
можно рассчитать величину г, зная k. Такие расчеты были про-
ведены. Имеются таблицы, дающие возможность определить ра-
диус частицы по экспериментально найденному значению k
(19)
41
(таблицы приведены ниже на стр. 44). В качестве ве-
личины, определяющей размер частиц в таблицах, дана вели-
чина z (5).
Коэффициент k находится экспериментально на' основании
измерения т при нескольких значениях X (19). Из таблицы по
полученному значению k определяется z. Затем по формуле (5)
по найденному значению z рассчитывается г. При расчете сле-
дует брать среднее значение длин волн, использованных в экс-
перименте при нахождении k.
Этот метод, так же как метод, изложенный в работе 5, имеет
то преимущество, что не требует измерения абсолютных величин
светорассеяния.
Выполнение работы
Приготовляют золь, подлежащий исследованию. На фото-
электроколориметре определяют значение оптической плотности
с красным и синим светофильтрами. По формуле (16) рассчи-
тывают величины т. Далее по формуле (19) определяют k, а по
таблице находят соответствующее полученной величине k зна-
чение z. По формуле (5) на основании найденного z вычис-
ляют г. Результаты записывают в виде таблицы.
Таблица 2
К СМ \р> см~
г, см
Работа 7
ОПРЕДЕЛЕНИЕ РАЗМЕРОВ КОЛЛОИДНЫХ ЧАСТИЦ ПО ХАРАКТЕРИСТИЧЕСКОЙ
МУТНОСТИ СИСТЕМЫ
Как уже указывалось, коэффициент рассеяния связан с раз-
мером частиц соотношением {см. формулу (8)]:
£р = яг21 “ I2 F (z)
Из определения мутности как суммарной энергии, рассеи-
ваемой .1 ши3 системы, следует:
j = kv (2°)
1 tar3 ' '
42
Здесь
с
Co6~~d
(21)
с — концентрация, г!см?-,
d — плотность частицы.
Решая уравнение (20)
ние kv из формулы (8) и выражая г через г (5), получаем:
г , t _ 6л I a I2 F (Z)
[ ] = —X--------Г-
относительно -— , подставляя зиаче-
соб
(22)
[т] называется характеристической мутностью.
6r.F(z) л , Ы?(г) . .
—принято обозначать через <p(z). —^-^-==<p(z).
Введя это обозначение в уравнение (22) и решая его относи-
тельно ф(г), получаем:
У И
(23)
Значения q>(z) протабулированы для различных г. Следова-
тельно, найдя <p(z), можно определить z и далее г.
Выполнение работы
Приготовляют золь определенной концентрации. На фото-
электроколориметре измеряют его оптическую плотность £>при
определенной длине волны (Z, в среде). Расчет размера частицы
производят следующим образом. По найденному значению D
рассчитывают мутность т [уравнение (16)]; концентрацию золя с,
выраженную в г/см:\ переводят в сОб [уравнение (21)]. Далее
из равенства [т] = — определяют характеристическую мутность
системы. По формуле (4) рассчитывают а. Теперь имеются все
величины для определения <₽ по формуле (23). Пользуясь таб-
лицей функций (стр. 44), по вычисленному таким образом зна-
чению ф(з) находят z и затем по формуле (5)—г. Результаты
записывают в виде таблицы.
43
Достаточно надежные измерения светорассеяния с помощью
методов, основанных на определении интенсивности проходя-
щего света, могут быть произведены лишь для систем со срав-
нительно большими величинами тСг^-Ю-2 сдг1).
Таблица значений функций ? (г), k и z для работ № 5, 6, 7
*135
Т(2) Ле k Z ¥(*) /<5 Лз5 k Z
3,260 — 1,00 241,6 2,676 2,960 5,00
Б,889 — — 1,20 289,0 3,371 2,807 5,50
8,768 — — 1,40 335,9 4,454 2,659 6,00
13,49 — — 1,60 344,3 — —_ 6,10
18,40 — — 1,80 353,3 .—. — 6,20
25,18 1,155 3,812 2,00 359,1 — — 6,30
32,64 — . — 2,20 371,1 — — 6,40
41,56 —_ — 2,40 381,2 6,191 2,533 6,50
- 46,30 1,254 3,686 2,50 425,5 8,957 2,457 7,00
51,52 — — 2,60 469,7 14,25 2,379 7,50
62,44 — — 2,80 511,6 25,47 2,329 8,00
74,33 1,401 3,573 3,00 598,0 .—. — 10,00
81,23 —- — 3,10 689,2 — — 11,0
84,16 —. - — 3,14 793,6 -—. 12,0
87,90 —— — 3,20 898,6 —. _— 13,0
95,06- — — 3,30 1002 — —х 14,0
102,3 .— 3,40 1102 — — 15,0
109,8 1,575 3,436 3,50 1201 — — 16,0
' 117,6 — — 3,60 1299 — -—. 17,0
125,6 — 3,70 1398 — — 18,0
133,7 — —_ 3,80 1496 — — 19,0
141,9 — — 3,90 1594 — — 20,0
150,5 1,826 3,284 4,00 1692 —
195,0 2,130 3,121 4,50 — — — —
Работа 8
ОПРЕДЕЛЕНИЕ РАЗМЕРОВ И ФОРМЫ АНИЗОДИАМЕТРИЧЕСКИХ ЧАСТИЦ ПО
ИЗМЕРЕНИЮ ИНТЕНСИВНОСТИ ПРОХОДЯЩЕГО СВЕТА
Этот метод применяется для определения размеров частиц,
длина которых близка к длине волны рассеиваемого света, а
ширина значительно меньше. С его помощью можно определить,
например, размеры вирусов, молекул высокополимеров и т. п.
Метод основан на завйсимости, эмпирически установленной
для стержневидных частиц:
AlgX * 7
(24)
где 7 величина, зависящая от формы частиц, определяющая
уменьшение рассеяния света при переходе от сферических ча-
44
стиц к анизодиаметрическим того же объема и называемая
показателем диссипации. Показатель диссипации 7
А т
однозначно определяется величинами -у- и q, где L — длина ча-
стицы, q — сложная функция от величины у.
Размеры частиц по этому методу определяют следующим об-
разом. С помощью уравнения Рэлея по интенсивности рассеян-
ного света вычисляют объем эквивалентной шарообразной ча-
стицы Рш. Затем измеряют т при нескольких длинах волн, по
формуле (24) находят показатель диссипации и по существую-
щим таблицам (3) по полученному 7 определяют у и q. Объем
анизодиаметрической частицы VaH = Рш<7-
Зная \РШ и L, нетрудно определить ширину частицы I. Для
определения L и I можно воспользоваться номограммами. В этом
случае достаточно знать значения -ц или отношение-ууj, т и тп.
Указания к пользованию номограммами даны на стр. 45.
Выполнение работы
Приготовляют золь определенной концентрации. По фор-
муле (21) определяют сОб. Затем измеряют D, по формуле (16)
вычисляют -г и переводят в [т] (22). По формуле (24) вычис-
ляют' 7. По полученным значениям [т] и 7 с помощью номограмм
находят искомые величины L и I.
Порядок пользования номограммами следующий: по номо-
грамме 1 по найденным значениям т, А и [т] определяют гэкв.
Затем по номограмме 2 по известным значениям гэкв, 7 и Л
определяют Lui. Результаты записывают в таблицу.
УКАЗАНИЯ К ПОЛЬЗОВАНИЮ НОМОГРАММАМИ
Номограмма 1
Точку, соответствующую найденному на опыте значению [т],
соединяют прямой с точкой, соответствующей данному значе-
нию т (прямая /). Точку пересечения прямой Z с прямой ab
45
[-r] t/CM'1
103^т-
s-
a
6-
5-
4-
3~
10*-
8Z
6-
s-
4-
f ikb(S fl)
400-
300-
200-
m
A-103(6A)r,1°
Рис. 23. Номограмма № 1. Определение эквивалентного
радиуса частицы по значениям т, h и [т]
Пример: [т] = 1 1 см~*; Л == 5• К)3 А; т — 1,20; гэ — 50А.
соединяют с точкой, соответствующей взятому в опыте значе-.
нию X— прямая 2. Точка пересечения прямой 2 с прямой, на
которой отложены значения гакв, даст искомое значение гзкв.
Номограмма 2
Рис. 24. Номограмма № 2. Определение характеристических размеров
частицы по известным значениям гэкв, ] и I
Пример: гв = 50А; -у = 0,22; Х=5-103А; £ = 1250А; / = 2бА.
Через точку, соответствующую определенному на опыте зна-
чению f или значению -j--, и точку, соответствующую найден-
ному с помощью номограммы 1 значению гэкв проводят прямую
до пересечения с прямой cd. Получают точку А. Через точку,
соответствующую используемому в опыте значению Л и найден-
ному значению -р проводят прямую до пересечения с прямой,
на которой отложены значения L. Точка пересечения этих пря-
мых дает искомое значение L. Соединяя точку, соответствующую
найденному значению L, с точкой А, находят искомое значение I.
На номограммах дан пример пользования ими.
Описание работы на приборе ФЭК-М. В основу измерения
положен принцип уравнивания интенсивностей двух световых
пучков с помощью переменной щелевой диафрагмы. Схема и
общий вид прибора даны на рис. 25.
Световые пучки от лампы л отражаются от зеркала 3( и 32.
Отраженные лучи проходят через светофильтры Ci и С2 и кю-
веты Ai и А2 и попадают на фотоэлементы. Уравнивание интен-
сивностей производится изменением ширины диафрагмы Д.
47
Перед началом работы рукоятку следует поставить в поло-
жение «открыто». С помощью рукоятки 4 стрелку гальванометра
установить на нуль. Наблюдение стрелки гальванометра произ-
водят через лупу 6. Измерение осуществляется двумя спосо-
бами:
1) Барабан 8 устанавливают на нулевом делении пропуска-
ния (устанавливать в направлении от меньших значений к
нулю). В правый световой пучок помещают кювету с исследуе-
мым раствором, в левый — с растворителем. Вращением'рукоят-
ки круговых фотометрических клиньев 2 и 3 стрелка гальвано-
метра устанавливается на нуль. Узел фотометрических клиньев
Рис. 25. Схема и общий вид фотоэлектроколориметра ФЭК-М.
состоит из рукоятки клина для грубой настройки 2 и рукоятки
клина для точной настройки 3. Установка на нуль производится
вначале при положении переключателя 12 на положении 1, за-
тем 2. Цифры на переключателе 12 означают: 0 — гальвано-
метр выключен; 1 — малая чувствительность; 2—большая чув-
ствительность. Переключатель 12 находится в положении 1 и 2
только при проведений измерений. ,
Затем в правый пучок света вводят кювету с растворителем;
При этом стрелка гальванометра отклоняется от нулевого поло-
жения. Вращением барабана 8 приводят ее в нулевое положе-
ние и определяют величину коэффициента светопропускания или
Оптической плотности .по левому отсчетному барабану 8. Отсчет-
ные барабаны имеют 2 шкалы: шкала коэффициентов светопро-
пускания (черная) н шкала оптической плотности (красная).
На левом барабане 100% по шкале светопропускания соответ-
ствует максимальному раскрытию диафрагмы, а 0%—полному
ее закрытию. На правом барабане 100% — щель раскрыта ми-
нимально, 30%—максимально.
48
Прибор имеет 4 светофильтра: нейтральный, зеленый, синий
и красный. Цифры 1, 2, 3, 4 на рукоятке 11 относятся к свето-
фильтрам: нейтральный—1, зеленый—.2, синий —3 и крас-
ный— 4.
По окончании работы стрелку необходимо арретировать, для
этого рукоятку поставить’в положение «закрыто».
2) В правый и левый пучки света помещают кюветы с рас-
творителем. Индекс правого барабана устанавливают на 0 деле-
ний шкалы оптической плотности. Щелевая диафрагма имеет
при этом минимальную ширину. Вращением рукоятки круговых
фотометрических клиньев устанавливают стрелку гальванометра .
на нуль. Затем в правый пучок вводят кювету с раствором. Вра-
щением измерительных барабанов устанавливают стрелку галь-
ванометра снова на нуль. Величина оптической плотности уста-
навливается по правому барабану 9. Пб окончании работы
стрелку гальванометра следует арретировать.
Оптическая плотность связана с коэффициентом пропуска-. -
ния следующим соотношением:
. 1
Оптическая плотность = In-гт-------------------
коэффициент светопропускания
Измерения следует производить через 15—20 мин после
включения прибора, так как в первые минуты прибор не дает
достаточно стабильных данных.
Приготовление золя серы. Для получения золя серы исполь-
зуется реакция между гипосульфитом натрия и серной кислотой:
NagSgOs HgSOj = Na2SO4 -J- HgO SO2 -f- S
Берут 11,5 см? охлажденной на льду концентрированной
серной кислоты (плотность 1,84). Отдельно приготовляют рас-
твор гипосульфита натрия. С этой целью 15 г гипосульфита рас-
творяют в 9 см3 дистиллированной воды при слабом нагрева-
нии (40—50° С). После того как весь гипосульфит растворится,
раствор охлаждают до комнатной температуры (охлаждение
следует производить при комнатной температуре). Затем рас-
твор гипосульфита по каплям, очень медленно, при интенсивном
перемешицании вливают в серную кислоту. Реакция проводится
на холоду (лед). В результате реакции'образуется густая жел-
товатая масса. К ней прибавляют 30 см3 дистиллированной
воды й колбочку помещают в ванну с горячей водой (60—70° С).
При этой температуре золь находится в течение получаса, после
чего охлаждается до комнатной температуры. Изредка его сле-
дует перемешивать. Полученный золь серы содержит большое
количество электролитов. Для удаления последних золь серы,
коагулируют 3 см3 насыщенного раствора NaCl, взбалтывают и
переносят на центрифугу. Центрифугирование производят в те-
чение 2—3 мин, скорость вращения 3 тыс. об)мин. После этого
49
ь
жидкость над осадком выливают, а осадок осторожно перено-
сят на фильтр и пептизируют 150 мл дистиллированной воды.
Полученный таким образом- золь содержит еще значительное
количество относительно крупных частиц. Для удаления по-
следних его центрифугируют 15—25 мин. Золь над осадком осто-
рожно отбирают пипеткой. Полученный таким образом золь раз-
бавляют в 2—3 раза и измеряют его светорассеяние. Золь мо-
жет быть разбавлен до центрифугирования. В зависимости от
порядка разбавления и центрифугирования получают золь с
различными размерами частиц. Для получения золей с еще бо-
лее мелкими частицами может быть проведена частичная коагу-
ляция их растворами NaCl различной концентрации. Так как в
первую очередь коагулируют более крупные частицы, при уве-
личении концентрации прибавленного к золю раствора NaCl и
последующем центрифугировании получаются золи, содержащие
все более мелкие частицы.
Приготовление золя сульфата бария. Сначала приготовляют
два раствора:
1) 1,1 г Na2SO4- ЮН2О растворяют в 50 см3 глицерина (I);
2) 2,45 г ВаС12-2Н2О растворяют также в 50 см3 глицери-
на (II).
Затем каждый раствор разбавляют спиртом и водой в опре-
деленном соотношении (см. ниже) и раствор, полученный из
раствора (I), осторожно приливают к раствору, полученному
из раствора (II), после чего растворы перемешивают. В зависи-
мости от соотношения спирта и воды при разбавлении получают
золи с разной величиной частиц. Рекомендуются следующие три
разбавления:
1) 1 см3 раствора (I) + 5 см3 воды + 4 см3 спирта
1 см3 (И) + 5 см3 воды + 4 см3 спирта
2) 1 см3 (I) + 2 см3 воды +3 см3 спирта
1 см3 (II) + 2 см3 воды + 3 см3 спирта
3) 1 см3 (I) + 1,5 см3 воды + 3,5 см3 спирта
1 см3 (II) + 1,5 см3 воды + 3,5 см3 спирта.
СТРУКТУРА КАПИЛЛЯРНЫХ СИСТЕМ
Капиллярными системами называются системы, образо-
ванные твердыми телами, пронизанными открытыми с обеих
сторон порами.
Твердое тело, образующее капиллярную систему, может быть
сплошным, тогда капиллярные системы называются жест-
кими. К жестким капиллярным системам относятся различ-
ные пористые тела (например, уголь, силикагель) и перегородки
(растительные и животные ткани и т. п.). В научной и технической
литературе различные пористые перегородки называют ди а»
50
фрагмами и мембранами, причем под диафрагмами
часто имеют в виду относительно грубопористые перегородки
значительной толщины, обладающие известной жесткостью (ке-
рамические, асбестовые и др.)» тогда как термин мембраны от-
носится преимущественно к эластичным тонким пленкам (кол-
лодиевые, желатиновые и др.).
В данном руководстве не проводится строгого разграничения
между терминами мембрана, и диафрагма, и они употребляются
как равнозначные.
. Капиллярные системы могут быть образованы и из отдель-
ных соприкасающихся между собой твердых частиц. Такие ка-
пиллярные системы называются порошковыми. В качестве
примера порошковых капиллярных систем можно привести при-
родные грунты, спрессованные тонкоизмельченные вещества
и т. п. В порошковых капиллярных системах могут быть два
рода пор: поры, образующиеся между отдельными частицами
при их соприкосновении, и более мелкие поры, пронизывающие
каждую частицу. Поры в капиллярных системах могут иметь
форму цилиндров, щелей или какую-либо иную и быть пря-
мыми или извилистыми, а также могут проходить отдельно друг
от друга или сообщаться между собой. Величина этих пор мо-
жет колебаться от микроскопических до коллоидных размеров.
Знание структуры капиллярных систем имеет большое зна-
чение при решении ряда теоретических и практических вопросов.
При оценке отдельных пористых сорбентов, обладающих внут-
ренней поверхностью, одним из существенных моментов является
представление о структуре сорбента. Исследование электро-
кинетических свойств капиллярных систем также не может про-
водиться без учета их структуры. Структура диафрагм имеет
большое значение для исследований, связанных с процессами
диализа, электродиализа, фильтрации, ультрафильтрации
и т. п. Большое значение структура мембран имеет также для
освещения многих биологических и биохимических вопро-
сов, связанных с проницаемостью растительных и животных
тканей для различных компонентов газовой или жидкой
фазы.
При определении структуры капиллярных систем вводят ряд
определенных предположений о форме поперечного сечения пор
и их расположения в капиллярной системе, так как действи-
тельная структура обычно нам неизвестна.
При исследовании структуры мембран в большинстве слу-
чаев считают, что они пронизаны порами цилиндрической
формы и в качестве основных структурных характеристик при-
нимают общую пористость и средний радиус пор.
Общей пористостью мембраны W называется от-
ношение объема пор Vp к общему объему Vn, т, е. объем пор
51
(1)
в единице объема мембраны:
Vp vn — v V
IF = — = —-----= 1_____.
v„ V„ vn
где V — объем твердого вещества или скелета мембраны.
Понятие среднего радиуса пор вводится вследствие того, что
диафрагмы обычно пронизаны порами различного радиуса. Для
более полной характеристики определяют максимальный радиус
пор и распределение пор мембраны по величине их радиусов.
Работа 9
ОПРЕДЕЛЕНИЕ ОБЩЕЙ ПОРИСТОСТИ МЕМБРАН
у Определение общей пористости можно произвести различ-
ными методами, в зависимости от характера образца пористого
тела. Весьма распространен упрощенный метод определения об'
щей пористости путем взвешивания сухого и пропитанного во-
дой образцов. Так, если т и тп, соответственно, массы сухого
и пропитанного водой образца, V — его объем, то общая пори-
стость:
™Н2О
где б?н2о — плотность воды, которая в подобных расчетах обычно
принимается за единицу.
Однако для многих объектов, напрймер коллодиевых, жела-
тиновых и т. п. мембран с небольшой толщиной, этот метод не-
пригоден вследствие того, что удаление с поверхности мембран
оставшихся капелек воды трудно провести сколько-нибудь удо-
влетворительно (например, фильтровальной бумагой), и по-
этому получается значительная ошибка при взвешивании. Луч-
шие результаты дает определение общей пористости из кажу-
щегося и истинного удельных весов. Под кажущимся
удельным весом понимают отношение веса сухой мем-
браны к общему объему образца вместе с порами:
1каж — tz
v п
. Выражая объем образца через кажущийся удельный вес
vn=>i~
7каж
а объем сухой мембраны через истинный удельный вес
у=-2-
Кист
52
и Подставляя найденные выражения в уравнение (1), получаем:
_ 1 Ткаж
‘ Тист
Для определения истинного удельного веса предварительно
взвешенный на воздухе образец пропитывают водой и затем
взвешивают на весах Вестфаля—Мора. Тогда, если р — вес об"
разца в воздухе и j?HsQ — вес его в воде, то:
Р7н,о
Тист — _
Р /Чо
Наибольшие затруднения возникают при определении кажу-
щегося удельного веса. Объем не очень тонких образцов (боль-
ше 0,5 мм) можно определять из геометрических размеров, из*
меряя толщину микрометром. Для определения общей порис-
тости более тонких мембран, толщина которых меньше 0,5 мм,
лучше пользоваться методом Манегольда, по которому объем
мембраны определяют посредством взвешивания ее в двух не-
смешивающихся жидкостях (например, в воде и четыреххлори-
стом углероде). Так, если Vn и рп— соответственно объем и вес
пропитанного водой образца в воздухе; Рпно и РпСС1—веса
его в воде и в четыреххлористом углероде; тНг0 и тссь— удель-
ные веса жидкостей, то:
^"н2о ~ рп ~ v«4o *
и
^"ссг4 ~Рп~ ^СС14
Откуда
Vn = -^Q~.<CI- (2)
•cci4 ’н2о
Определив сухой вес мембраны и учитывая, что отношение
Р Рпн&
------— равно объему V сухого вещества мембраны, можно
7н2о
вычислить общую пористость по уравнению:
У >-^н2о
* Это соотношение может быть легко получено из уравнения;
Рпнао =р~ %2о- так как V = - Ур. то рп^ = Р-(V„7H2O- V₽THjO)
или Рпнао в Р + v₽7Hao — ^н.о’Учитывая-что Р + г^н2о ~ Ра' 1аал^'№Ж‘
pnti^o"=Pn~V"(Hao
53
Выполнение работы
Для определения общей пористости, как это и следует из
уравнения (3), надо знать объем и вес в воде водонасыщенной
мембраны и ее сухой вес. Определение объема водонасыщенной
мембраны в данной работе проводится посредством взвешива-
ния образца мембраны в двух несмешивающихся жидкостях —
в воде и четыреххлористом углероде. Перед взвешиванием мем-
бран, содержащих воздух в порах, необходима предваритель-
ная пропитка образца водой. Пропитку обычно производят в
вакуум-эксикаторе, на дно которого
наливают теплую воду, а образец
подвешивают над нею. Эксикатор
соединяют с вакуумным насосом,
и некоторое время (около 10 мин)
мембрану оставляют в парах воды,
после чего ее толчком сбрасывают
в воду и выдерживают под ва-
куумом до прекращения выделе-
ния пузырьков воздуха. После про-
питки образца водой взвешивают
его в воде и четыреххлористом уг-
лероде на торзионных весах. Для
этого вначале взвешивают подвешен-
ную к крючку весов (см. рис. 8,
стр. 19) проволоку, свернутую в
Рис. 26. Взвешивание мембра- к0ль«0’ в котоРое потом помещают
ны в жидкости на весах Вест- мембрану типа керамической или
фаля — Мора. изогнутую в виде крючка, к которо-
му, затем подвешивают эластичную
мембрану типа коллодиевой. После взвешивания проволоки в
воде (обозначим ее вес через pi) прикрепляют к ней мембрану
и находят вес мембраны с проволокой р2- Разность весов р2—pi
даст вес мембраны в водеря .Затем мембрану переносят в че-
Н2О
тыреххлористый углерод. Следует отметить, что предварительно
обе жидкости должны быть взаимно насыщены.
В четыреххлористом углероде также сначала должна быть
взвешена подвесная проволока, а затем уже, проволока вместе
с мембраной. Если обозначить через р\ и р'2 соответственно вес
проволоки й вес проволоки с мембраной, то разность весов
р'2 — р’х даст вес мембраны в четыреххлористом углероде. Об-
разцы некоторых мембран относительно малого удельного веса
при взвешивании в четыреххлористом углероде всплывают.
В этом .случае следует употребить добавочный груз, который
прикрепляют к нижней части мембраны. Добавочный груз
вместе с проволокой предварительно взвешивают в четыреххло-
54
ристом углероде. Так как вес груза и проволоки р\ в этом слу-
чае будет больше веса проволоки, груза и мембраны р'2, т® в
формуле (2), выведенной для определения объема водонасы-
щенной мембраны, в числителе будет знак плюс.
Для определения веса образцов мембраны в жидкостях
можно использовать и весы Вестфаля—Мора. При этом вначале
подвешивают к нижнему крючку чашки весов проволоку и, по-
грузив ее в воду, уравновешивают весы, а затем взвешивают
проволоку с мембраной (рис. 26). Разность показаний весов
Вестфаля—Мора при взвешивании прово.локи р\ и проволоки
с мембраной р2 дает вес мембраны в воде Рпко- Затем произ-
водят взвешивание в четыреххлористом углероде и также по
разности показаний весов р[—р'2 находят вес мембраны в четы-
реххлористом углероде. При взвешивании мембран, всплываю-
щих в четыреххлористом углероде, применяют добавочный груз.
Для определения сухого веса р мембран образцы высуши-
вают в сушильном шкафу при 105° С до постоянного веса. Мем-
браны, которые нельзя нагревать до такой температуры (на-
пример, кодлодиевые), высушивают в вакуум-эксикаторе при
70—80° С*.
Рассчитав объем образца водонасыщенной мембраны по
уравнению (2) и зная ее сухой вес и вес в воде, вычисляют
Таблица 1
Вес, г Объем мембраны, пропитан- ной водой V, СМ9 Сухой вес Р, г Общая пори- стость W Площадь мембраны А, см2
в воде в четыреххлористом углероде
Pi Р2 Г12'-' Pi Р2 fl 2'-'
-
* Для студенческих работ можно рекомендовать коллодиевые мембра-
ны, которые могут иметь различную пористость, в зависимости от концен-
трации исходного раствора нитроклетчатки, времени высушивания и различ-
ных добавок. Мембраны получают следующим образом. В кристаллизатор
наливают раствор нитроклетчатки, который оставляют на воздухе в течение
некоторого времени и затем коагулируют водой. Для полного удаления рас-
творителя мембрану помещают на несколько суток в дистиллированную
воду, которую многократно сменяют. Варьируя время высушивания, можно
из обычного медицинского коллодия получить мембраны различной пори-
стости [2, стр. 43].
Кроме коллодиевых мембран в работах по определению структуры ка-
пиллярных. систем могут быть использованы любые пористые диафрагмы:
керамические, стеклянные и т. п.
55
общую пористость по уравнению (3), принимая tHj0 yt Опре-
деления проводят для нескольких образцов различного размера
и в каждом случае вычисляют общую пористость, затем берут
среднее значение. Полученные результаты записывают в таб-
лицу следующего вида (табл. 1). '
, Работа 10
ОПРЕДЕЛЕНИЕ СРЕДНЕГО РАДИУСА ПОР МЕМБРАНЫ ПО ОБЩЕЙ ПОРИСТОСТИ
) И КОЭФФИЦИЕНТУ ПРОТЕКАЕМОСТИ
(4)
- Среди различных методов определения среднего радиуса пор
мембран наибольшее распространение получил метод, основан-
ный на законе Пуазейля. Как известно, уравнение Пуазейля,
выведенное для отдельного капилляра, имеет вид:
V1 = —см3 (м3)
где V* — объем жидкости (или газа) с вязкостью т), протекаю-
щей под давлением Р за время t через капилляр радиуса г и
длины /.
Приложение закона Пуазейля к капиллярным системам ос-
новано на предположении, что он применим не только к капил-
лярам макроскопических размеров, но и к капиллярам, радиус
которых лежит в микроскопической и ультрамикроскопической
области. Кроме того,, так как действительная структура мембран
неизвестна, то приходится, как уже упоминалось выше,.вводить
ряд определенных предположений р форме поперечного сече-
ния пор и их расположении в мембране. Если предположить,
что все поры мембраны цилиндрической формы с одинаковым
поперечным сечением и расположены перпендикулярно поверх-
ности мембраны, то для мембраны с площадью А см2
Числом пор п на 1 см2 (1 м2) объем жидкости (или газа)
текающий через мембрану, согласно закону Пуазейля:
,, . itr*PtAn . ,
V = V'nA — —j—;— см3 (м3)
оца ' '
(Л12) И
К,.про-
(5)
где d— толщина мембраны.
Уравнение (5) содержит величину п, которую нельзя опреде-
лить экспериментально.' Однако число капилляров на единице
площади мембраны п можно выразить через общую пористость
и средний радиус пор. Действительно, если — объем от-
дельного капилляра, то w2dAn— общий объем всех пор мем-
бранвГ с площадью А. Следовательно, объем пор в единице
объема мембраны, или общая пористость:
w2dAn ,
dA
56
Откуда:
Подставляя значение п в уравнение (5), получим:
WPr2At
V ~
Отсюда:
г=1^й5тСЛ(Л) (7)
Такой вид имеет формула при условии, что все поры цилинд-
рической формы и расположены перпендикулярно поверхности
мембраны. Однако для реальных мёмбран трудно предположить
такое распределение пор и наиболее вероятно хаотическое их
расположение во всех направлениях. Манегольд, проведший ряд
работ по исследованию структуры различных мембран, доказал,
что такой хаотический характер распределения пор в отношении
скорости фильтрации равноценен системе прямолинейных пор,
пересекающихся в трех взаимно перпендикулярных направле-'
ниях. Если считать, что все поры равномерно распределены в
трех взаимно перпендикулярных направлениях, то жидкость бу-
дет проходить только через поры, пересекающие обе поверхно-
сти мембраны, т. е. через одну третью часть всего числа пор п.
Тогда число пор, проходящих через единицу площади мем-
браны:
И'=ЧИ
Вводя в данное равенство значение для п из уравнения (6),
имеем:
. 1 W
п — —<г
3 яг2
П'одставляя значение п' в уравнение (5) и произведя даль-
нейшие преобразования, получим выражение для среднего ра-
диуса пор:
см (л° . (8)
Количество жидкости (или газа), прошедшее через единицу
площади мембраны в единицу времени под давлением, равным
единице, Манегольд назвал коэффициентом протекав-
мости мембраны/).
у
D = см2 • сек/г (•** • сек/кг) (9)
где V — объем протекшей жидкости, см3 (м3); А г— площадь
мембраны, см2 (м2); Р — давление, дин1см2 (и/ж2); t— время, сек.
' . - ' ’ 57
Произведение коэффициента йротекаемости D, вязкости про-
текшей жидкости т] и толщины мембраны d называют абсо-
лютным коэффициентом протекаемости и обозна-
чают через Du- Следовательно:
DH = D-rfi см2 (м2) (10)
Вводя в уравнение (8) значение D из уравнения (9), полу-
чим формулу для вычисления среднего радиуса пор:
Г= у —СМ (м) (11)
или через абсолютный коэффициент протекаемости, учитывая
уравнение (10)
г = у ~W~ СМ
При выводе уравнения (11) был сделан ряд следующих
предположений:
1) прямолинейность пор. Если допустить, что поры не яв-
ляются прямолинейными, то это увеличит общую пористость W
и уменьшит коэффициент протекаемости D, тогда вычисленная
по уравнению (11) величина среднего радиуса пор г будет пре-
уменьшена;
2) постоянство сечения по всей длине отдельных пор. Если
принять, что это- сечение по всей длине неодинаково, то при
определении коэффициента протекаемости наибольшее значение
для количества протекающей жидкости будут иметь наиболее
узкие части пор;
3) равенство общего объема пустот мембраны фильтрующе-
' му объему. Обычно общий объем пустот не равен фильтрую-
щему объему, хотя бы вследствие наличия капилляров, не имею-
щих выхода на поверхность. Таким образом, величина общей
пористости W, вводимая в уравнение (11) для вычисления сред-
него радиуса пор г, будет преувеличена, и вычисленное значе-
ние для г меньше действительной величины;
4) равенство сечения всех пор мембраны. Обычно поры ре-
альных мембран не обладают одинаковыми сечениями. Таким
образом, средний радиус пор, определенный по уравнению (11),
является эквивалентным радиусом, т. е. таким радиусом, кото-
рым обладала бы мембрана с прямыми цилиндрическими пора-
ми, имеющая те же значения общей пористости и коэффициента
протекаемости; • что и исследуемая гетеропористая мембрана.
Действительно, всегда можно себе представить вместо реаль-
ной мембраны, обладающей различными по величине и форме
порами, гомопористую мембрану с прямыми цилиндрическими
порами, которая по протекаемости жидкости и общей пористо-;
сти эквивалентна реальной мембране.
58
Согласно уравнению (11) для нахождения среднего радиуса
пор мембраны необходимо определить общую пористость W,
коэффициент протекаемости D и толщину мембраны d. Методы
определения общей пористости описаны в работе (9).
Для определения коэффициента протекаемости D необходи-
мо измерить количество жидкости (воды), протекающей в неко-
торый промежуток времени через данную площадь мембраны
при заданном и постоянном в течение опыта давлении.
Толщина мембран может быть найдена или путем измерения
микрометром, или, лучше, посредством вычисления по формуле:
d = см (м) (12)
Л
где Vn и А — соответственно объем и площадь мембраны.
Выполнение работы
Прибор для определения коэффициента протекаемости изо-
бражен на рис. 27. Исследуемый образец зажимают в металли-
2
Рис. 27. Установка для определения коэффициента протекаемости мембран
ческие фланцы 3. Мембраны, которые могут деформироваться
под давлением, например, коллодиевые, желатиновые и т. п.,
помещают на металлической сетке. Между такой мембраной
и сеткой- должна быть прокладка из фильтровальной бумаги,
так как в противном случае протекание может происходить
59
не через всю поверхность мембраны. Радиус пор фильтроваль-
ной бумаги обычно значительно больше, чем исследуемого об-
разца, поэтому влияние его на протекаемость жидкости практи-
чески ничтожно. В случае неэластичных, жестких мембран типа
керамических, стеклянных и т. п. образец прикрепляют с по-
мощью менделеевской замазки или пицеина к эбонитовому коль-
цу, которое вставляют затем во фланец.
Зажав образец мембраны, наполняют прибор водой до кра-
ев (сначала одну половину) и закрывают пробкой с присоеди-
ненной к ней трубкой, посредством которой он соединяется с
остальной частью установки; затем наполняют водой другую
часть прибора с капиллярной пипеткой. Проследив, нет ли раз-
рывов водяного столба в системе, устанавливают мениск в
капиллярной пипетке 5 с помощью крана 4. Затем в буферную
бутыль 1 накачивают насосом воздух и дают давление в систе-
му.' После установления постоянной скорости течения деларт
ряд отсчетов мениска, пользуясь секундомером, и отмечают дав-
ление по манометру 2. Затем увеличивают давление и повто-
ряют отсчеты. Проведя ряд отсчетов, вычисляют величину коэф-
фициента протекаемости по уравнению*:
у
D = 13,& -981 (13)
Данные, полученные при определении коэффициента проте-
каемости, записывают в таблицу (табл. 2). ,
Таблица 2
№ образца мембраны Площадь образца А, см» Давление Р, см рт. ст. Объем протекшей жидкости V, CMS Время протекания жидкости tt сек Коэффициент протекае- мостн £>, см1-се к! г
/
Величину общей пористости мембраны, необходимую для
вычисления среднего радиуса пор, находят по уравнению- (3),
определив предварительно объем, вес в воде водонасыщенной
мембраны и ее сухой вес (стр. 54). Зная объем образца водо-
насыщенной мембраны и его площадь (стр. 59), вычисляют
толщину мембраны по уравнению (12).
Полученные данные при определении общей пористости за-
писывают в таблицу, подобную таблице 1 (стр. 55), дополнив
* В системе СИ: D = . 13 (здо. 9|8Т *
60
ее графой, содержащей значения толщины мембран, получен-
ные для каждого образца.
Определив средние значения коэффициента протекаемости,
общей пористости и толщины мембраны, вычисляют средний
радиус пор по уравнению (11).
Работа 11
ОПРЕДЕЛЕНИЕ СРЕДНЕГО РАДИУСА ПОР МЕМБРАНЫ ПО ЕЕ
ЭЛЕКТРОСОПРОТИВЛЕНИЮ И. КОЭФФИЦИЕНТУ ПРОТЕКАЕМОСТИ
Метод определения среднего радиуса пор мембраны по ее
электросопротивлению и коэффициенту протекаемости может
быть применен только к мембранам из непроводящего ток мате-
риала. Если такую мембрану пропитать раствором электролита
с концентрацией не ниже 0,1 н.*, то ее сопротивление выразится
соотношением:
4- (14)
где I — длина капилляров;
q— суммарная эффективная площадь сечения пор;
х— удельная электропроводность раствора.
Предположим, как и ранее, для упрощения, что поры мем-
браны расположены перпендикулярно ее поверхности и имеют
цилиндрическую форму, тогда:
l = d и q = пггпА
где d —'толщина мембраны;
г—радиус отдельного капилляра;
п — число капилляров на единицу площади мембраны;
А — площадь мембраны.
, Вводя в уравнение (14) значения I и q, получим выражение
Для сопротивления мембраны:
л 5)
“ 1мг2пА ' '
С другой стороны, коэффициент протекаемости такой мем-
браны (стр. 57): D <16>
Подставив в уравнение (16) значение объема жидкости, про-
текающей в единицу времени через мембрану [уравн. (5)
стр. 56], получим:
___________ ' <17>
* При более низких концентрациях уравнение (14) не будет справед-
ливым вследствие изменения электропроводности в порах мембраны.
61
Предполагая, что условия, определяющие прохождение элек-
трического тока аналогичны условиям потока жидкости через
мембрану, т. е. что значения d, А и п в уравнениях (15, 1'6, 17)
одинаковы, из уравнений (15) и (17) находим:
(18)
Таким образом, зная коэффициент протекаемости мембраны
D, ее площадь А, а также вязкость ц и электропроводность рас-
твора х и определив электросопротивление мембраны Rd, можно
найти средний радиус пор.
Если условия прохождения электрического тока и жидкости
через мембрану не одинаковы, например в случае определения
среднего радиуса пор порошковых мембран (стр. 64), то зна-
чения А и d различны. Тогда в уравнение (18) необходимо вве-
сти коэффициент, учитывающий это различие.
Можно избежать введения поправочного множителя в урав-
нение (18), определяя средний радиус пор по коэффициенту
протекаемости D и коэффициенту структурного со-
противления мембраны р, который показывает во
сколько раз увеличится сопротивление раствора между электро-
дами при введении мембраны:
₽ = (19)
где Rd—электросопротивление мембраны;
— электросопротивление слоя раствора, равного мембра-
не по площади и толщине.
Учитывая, что
С. Cv
где Cj и Cv — соответственно постоянные ячейки с мембраной
и без нее, получим:
₽ = -£*-
Величину Cv можно вычислить, зная толщину dt и пло-
щадь At мембраны:
___ dt
и ~ At
Тогда:
r —. CdAi Rd^At
p — dt ~ dt (20)
Из уравнений (15) и (20) найдем:
62
Предполагая, что число пор на единицу площади мембраны
одинаково при фильтрации жидкости и прохождении электриче-
ского тока, и введя значение п [уравнение (21)] в уравнение (17),
получим:
Г2
D-w <22>
Откуда:
г = / d$D (23)
Выполнение работы ,
Измерение электросопротивления мембран проводят обыч-
ным методом, как и при определении удельной электропровод-
ности раствора, при помощи мостика Кольрауша. Источником
тока может служить генератор звуковой частоты ЗГ-10 или
индукционная катушка. В качестве нуль-инструмента обычно
применяют низкоомный телефон. В зависимости от характера
исследуемого образца используют приборы различной кон-
струкции.
Электросопротивление жестких мембран, обладающих зна-
чительной толщиной (керамических, стеклянных и т. п.), можно
производить в приборе, изображенном на.рис. 28. Мембрану,
пропитанную 0,1 н. раствором
КС1, зажимают между фл-ан- 4
цами 4 прибора, состоящего . 5
из двух стеклянных сосу- : KF*)
дов 3, 5. Платиновые электро- ? /
ды 1, 2, вплавленные в стек-
лянные трубки, вставляют на ““
резиновых пробках в прибор. Рис. 28. Прибор для определения
Прибор наполняют 0,1 н. рас- электросопротивления мембран
твором КС1, после чего элек-
троды плотно прижимают к мембране и измеряют Сопротивле-
ние Rd- Определив электропроводность 0,1 н. раствора КС 1, коэф-
фициент протекаемости * и площадь мембраны (из геометриче-
ских размеров), вычисляют средний радиус пор по уравне-
нию (18).
Следует отметить, что этот метод определения среднего ра-
диуса пор жестких мембран нельзя применять к мембранам
неоднородным по толщине и с сильно шероховатой поверхно-
стью. В этом случае между электродами и диафрагмой будет
находиться слой раствора электролита, что внесет тем большую
ошибку в определение Rd, чем меньше толщина, диафрагмы.
Следует также учитывать, что эластичные мембраны (типа
* О методе определения коэффициента протекаемости см. стр. 59.
63
коллодиевых) могут деформироваться при приближении элек-
тродов к их поверхности, что также исказит величину Rd-,
Более точное значение среднего радиуса пор таких мембран
можно получить, используя уравнение (23). Метод определения
коэффициента структурного сопротивления жестких мембран
описан на стр. 217. Толщина мембран измеряется микрометри-
ческим винтом или вычисляется по уравнению (12) (стр. 59).
В отличие от жестких диафрагм выбор ячейки для измере-
ния электросопротивления порошковых капиллярных систем не
представляет затруднений, так
как порошком можно заполнить
сосуд любой формы. Поэтому в
зависимости от целей исследова-
ния структуры капиллярной си-
стемы можно использовать раз-
личные приборы.
В данной работе рекомен-
. дуется видоизмененный прибор
Самарцева и Остроумова (рис.
29). В этом приборе можно изме-.
рять как электросопротивление,
так и коэффициент протекаемо-
сти диафрагмы *, Прибор состоит
из трех частей: стеклянной труб-
ки 2 с вплавленными в нее двумя
платиновыми электродами 3 и 4,
плексигласового сосуда 1 с пер-
форированным дном и изогнутой трубки 5 с отводом, к кото-
рому присоединяется капиллярная пипетка.
При сборке прибора на перфорированное дно сосуда 1, со-
единенного каучуком с трубкой 5, кладут несколько слоев филь-
тровальной бумаги и наливают в прибор (через трубку 5) 0,1 н.
раствор КС1 так, чтобы фильтр был. покрыт небольшим слоем
жидкости. Затем в сосуд 1 на резиновой пробке вставляют
трубку 2 таким образом, чтобы нижний конец ее был плотно
прижат к фильтру; присоединяют прибор к бутыли (рис. 30)
и, наполнив его 0,1 н. раствором КС1, измеряют сопротивле-
ние Rh- Затем, закрыв кран 4 и убедившись в отсутствии пу-«
зырьков воздуха в приборе, приступают к формированию диа-
фрагмы. Для этого наполняют трубку 2 (рис. 29) суспензией ис-
следуемого порошка в 0,1 н. растворе КС1 **. После осаждения
* Этот же прибор применяют для определения ^-потенциала и коэф-
фициента эффективности порошковых -мембран (стр. 193, 217).
** Предварительно порошок необходимо обработать 0,1 н. раствором
КС1 для того, чтобы величина не изменялась во времени. Равновесие
обычно устанавливается через сутки при неоднократной смене раствора,
64
порошка, отобрав пипеткой раствор над порошком, снова на-
полняют трубку 2 суспензией. Эти операции повторяют до тех
пор, пока слой порошка не достигнет определенной высоты *.
Если частицы порошка малы и плохо оседают, то при формиро-
вании диафрагмы применяют отсасывание водоструйным насо-
сом через отвод трубки 5. После формирования диафрагмы,
заполнив обе части прибора 0,1 н. раствором КС1 и закрыв их
резиновыми пробками, открывают кран 4 (рис. 30) и фильтруют
Рис. 30. Установка для определения электросопротивления и
коэффициента протекаемости порошковых мембран.
раствор через порошок. Измерив ^сопротивление диафрагмы
вычисляют коэффициент структурного сопротивления (3 по урав-
нению (19).
Для определения коэффициента протекаемости D** изме-
ряют скорость протекания раствора через порошок при различ-
ных давлениях ***.
* В зависимости от размеров частиц порошка заполняют им трубку 2
на различную высоту, которую подбирают предварительными опытами так,
чтобы скорость фильтрации не была очень большой или, наоборот, очень
малой. Во всяком случае, слой порошка должен быть выше электродов не
менее, чем на 1 см.
** Подробнее см. стр. 59.
*** Если скорость фильтрации большая, то капилляр следует заменить
мерным цилиндром.
5 Зак. 254
65
Измерив толщину диафрагмы, через которую происходит
фильтрация жидкости, вычисляют средний радиус пор по урав-
нению (23).
Полученные данные в зависимости от выбранного метода
оформляют в виде одной из следующих таблиц (3 и 4):
Таблица 3
Образцы мембраны Площадь мембраны А, см? Коэффициент протекаемости D, см1 2 3-сек Сопротивление мембраны R&, Средний радиус пор г.
г ом мк
1.
2.
3.
Таблица 4
Образцы
мем-
браны
Толщина
мембраны
d, см
Коэффициент
протекаемости
D, ем2-сек!г
Сопротивление
мембраны,
R# ом
Сопротивление
слоя раствора
°м
Коэффициент
структурного
сопротивле-
ния р
Средний
радиус
пор г,
мк
1.
2.
3.
гсР!
Работа 12
определение максимального радиуса пор мембран
В ряде работ (например, при бактериологических исследова-
ниях) наиболее важной характеристикой структуры мембран,
применяющихся в качестве фильтров, является не средний ра-
диус пор, а их максимальный радиус. Расхождения же между
величинами среднего и максимального радиуса пор могут быть
довольно значительными (для некоторых коллодиевых мембран
они различаются между собой более чем в три раза).
Определение максимального радиуса пор мембран произво-
дят посредством продавливания воздуха или жидкости через
мембрану, пропитанную другой жидкостью. Если над мембра-
ной, пропитанной какой-либо жидкостью, поместить слой этой
жидкости и продавливать через нее снизу при постепенно повы-
шающемся давлении воздух .(или несмешивающуюся жидкость),
то при определенном давлении замечают над,поверхностью мем-
браны пузырьки воздуха (или капли жидкости). Образование
первых пузырьков на поверхности мембраны происходит над
66
порами, площадь сечения которых наибольшая. При дальней-
шем повышении давления число пузырьков воздуха (или ка?
пель жидкости) увеличивается, что связано с их образованием
над порами, площадь сечения которых меньше максимальной
величины. Наконец, замечают массовое выделение пузырьков
по всей поверхности мембраны.
Отметив давление, при котором на поверхности мембраны
образуются первые пузырьки воздуха (или капли жидкости),
можно вычислить максимальный радиус пор, используя урав-
нение Кантора:'
г = см (м) (24)
где г—радиус капилляра, см (и);
а—поверхностное натяжение на границе жидкость — воз-
дух или пограничное натяжение для двух несмешиваю-
щихся жидкостей, дин!см (н/м) ;
Р—давление, дин! см2 (н/м2).
Применение уравнения Кантора для определения максималь-
ного радиуса пор мембраны основано на ряде предположений,
упрощающих действительную структуру реальных мембран.
Так же, как и при определении среднего радиуса пор,
полагают, что мембрана пронизана цилиндрическими прямы-
ми порами, расположенными перпендикулярно ее поверхно-
сти, и что поверхность полностью смачивается наполняющей их
жидкостью.
Если радиус отдельных пор не постоянен на всем протяже-
нии, то этим методом определяют радиус узкой части капил-
ляра. Если поры не цилиндрической формы, а какой-либо иной,
то это также скажется на величине гмакс- Расположение пор
в мембране, не перпендикулярное к ее поверхности, может ока-
зать влияние лишь на время.образования видимого пузырька
воздуха или капли жидкости на поверхности мембраны. Если
жидкость не полностью смачивает капилляр, то в уравне-
ние (24) следует ввести поправку на краевой угол:
г = ~ cos 0 (25)
где S — краевой угол на границе стенка капилляра — жидкость
I — жидкость II.
Однако экспериментальное определение краевого угла для
капиллярных систем весьма сложно, и формулой Кантора поль-
зуются лишь в том случае, когда краевой угол равен нулю. По-
этому чрезвычайно важным является вопрос о выборе жидко-
сти, наполняющей поры мембраны’. При наполнении капилляров
хорошо смачивающей их поверхность жидкостью последняя по-
крывает слоем стенки капилляра, иг краевой угол 0 равен нулю.
5*
67
Наглядно это представлено на рис. 31, где а — мениск жидкости
в капилляре при неполном смачивании, а Ь—поток жидкости
в капилляре при полном смачивании.
Если мембрану пропитать водой и продавливать через нее
воздух *, то при давлении 760 мм рт. ст. можно определить
поры с радиусом около 1,5 мк. Если максимальный радиус пор
а 6
0<в<90е 0=0
Рис. 31. Схема движения жидкости в капилляре при
различном смачивании.
Рис. 32. Схема дви-
жения мениска жидко-
сти через капилляр.
исследуемой мембраны меньше этой величины, то лучше пользо-
ваться органическими жидкостями, например этиловым, бутило-
вым спиртами, толуолом, бензолом, для которых значения
поверхностного натяжения на границе с воздухом лежат в пре-
делах от 16 до 29 дин/см (16-10~3—29-10-3 н/м). Тогда при
разности давлений в 760 мм рт. ст. можно определить макси-
мальный радиус от 0,33 до 0,57 мк. Для определения меньших
радиусов пор можно или увеличить давление, или взять такие
две несмешивающиеся жидкости, погранич-
ное натяжение которых будет меньшей ве-
личиной.
Увеличение давления может привести к
деформации мембран и даже к образова-
нию трещин. Поэтому лучше применять две
• несмешивающиеся жидкости с малым по-
граничным натяжением, которое предвари-
тельно взаимно насыщают. Наиболее часто
употребляют систему изобутиловый спирт—
вода (о =1,8 дин/см) или бутиловый
спирт—вода (с =1,58 дин!см). Мембрану
пропитывают жидкостью, лучше смачивающей поверхность мем-
браны, и опыт проводят так, чтобы мембрана все время остава-
лась на границе двух жидкостей.
Рассмотрим движение жидкости через капилляр, наполнен-
ный другой жидкостью.
На рис. 32 изображен капилляр радиуса г и длины I, напол-
ненный жидкостью I с вязкостью т]1, через который должна
проходить жидкость II с вязкостью г]2. Пограничное натяжение
на границе жидкостей I и. II равно о. Когда приложенное дав-
* Поверхностное натяжение на границе вода — воздух при 20° равно
72,75 дин/см (72,75 • 10~3 н/м).
68.
ление достигает величины, равной -у—, мениск, находящийся
в капилляре, еще не движетоя. Чтобы преодолеть силы внутрен--
него трения для движения жидкости в капилляре, необходимо
несколько большее давление. Кроме того, чтобы жидкость про-
шла через капилляр под давлением АР, соответствующим дан-
ному радиусу г, необходимо определенное время t. Эго время
можно вычислить, если принять, что движение жидкости и тогда
подчиняется закону Пуазейля, когда в капилляре находится
граница раздела. До начала движения жидкости капилляр на-
полнен жидкостью с вязкостью т]1, в конце движения — жид-
костью с вязкостью г]2- На основании этого вместо т] в уравне-
ние Пуазейля вводят величину .
Тогда:
др= (26).'
(27)
(28)
Объем протекшей жидкости V в см3 равен объему капил-
ляра, а так как объем капилляра равен тг г2/, то:
др _ 4/2 (^i + Ча)
tw Г3
Откуда:
_ 4Z2(1Ji4-tj2)
lw~ . ДР/2
Следовательно, tw — то необходимое время ожидания, в те-
чение которого мениск жидкости дойдет до другого конца ка-
пилляра. Однако для того, чтобы на поверхности мембраны об-
разовалась видимая капля жидкости (или пузырек воздуха),
необходимо еще некоторое дополнительное время. В качестве
примера приведем время прохождения мениска жидкости через
капилляр с радиусом 1 мк и длиной 4 мм при употреблении в
качестве двух несмешивающихся жидкостей бутилового спирта
и воды, а также время, необходимое для образования видимого
пузырька жидкости на поверхности мембраны. Учитывая, что
вязкость бутилового спирта и воды соответственно равна 0,03
и 0,01 пз, найдем по. уравнению (28), что при давлении
10 см рт. ст. (и>=19,2 сек.
Образование же видимой капли жидкости на поверхности
мембраны требует значительно большего промежутка времени.
Рассчитаем по уравнению ПуаЗейля, каково время образования
капли воды радиусом 0,25 мм из рассматриваемого капилляра
при том же давлении. Принимая во внимание, что объем капли
4 о
равен у получим:
t = 32li<r* (29)
3 Apr4
69
Из приведенного вычисления следует, что время образования
капли'радиусом 0,25 мм равно 13 ч 52 мин 50 сек. Время обра-
зования капли радиусом 0,1 мм при тех же условиях равно
53 мин 12 сек. Таким образом, между установлением давления,
достаточного для продавливания капли жидкости (или пузырь-
ка воздуха) через мембрану, и образованием видимой капли
(или пузырька воздуха) на поверхности мембраны проходит
некоторый промежуток времени. Поэтому рекомендуется повы-
шать’давление постепенно, с паузами. Такое ступенчатое повы-
шение давления дает возможность ближе подойти к истинному
значению гмакс- Однако опыт в этом случае длится в течение
нескольких часов. Во много раз быстрее можно определить
максимальный радиус пор по методу чередования давлений,
заключающемуся в измерении давления в момент появления
и исчезновения пузырька.
Вначале, при повышении давления, отмечают давление, при
котором появляется пузырек. Затем, уменьшая давление, отме-
чают его величину при исчезновении пузырька. Проводят не-
сколько таких измерений. Отличающиеся в начале опыта от-
счеты давлений сближаются (до 2—3 мм при давлениях
400—700 мм рт. ст.) пли совпадают, если давление не превы-
шает 100 мм рт. ст. Сближение величин давления, при которых
появляется и исчезает пузырек, показывает, что это давление
отвечает искомой величине, соответствующей порам с макси-
мальной величиной радиуса.
Сравнение обоих методов определения максимального ра-
диуса пор дало одинаковые результаты.
Выполнение работы
Прибор для определения максимального радиуса пор изо-
бражен на рис. 33. Мембрану, пропитанную какой-либо жид-
костью, зажимают в металлические фланцы 1. Над мембраной
наливают слой этой жидкости. Необходимое давление получают
с помощью бомбы со сжатым воздухом. Над мембранами, кото-
рые могут деформироваться под давлением, помещают метал-
Таблица 5
№
счетов
hi см рт. ст. ,
в момент появления
пузырька
ft, см рт. ст.
в момент исчезновения
пузырька
70
Рис. 33. Прибор для опре-
деления максимального
радиуса пор:
/—фланцы с мембраной.
лическую сетку с большими отверстиями. Давление в момент
появления пузырька отмечают по ртутному манометру. Наблю-
дения за появлением пузырька проводят с помощью лупы. Этим
прибором можно пользоваться и при ра-
боте с двумя несмешивающимися
жидкостями.
В начале опыта дают на прибор не-
которое давление до появления пузырь-
ка воздуха на поверхности мембраны.
Затем уменьшают давление до исчезно-
вения ' пузырька. Отмечают показания
манометра в момент появления и исчез-
новения пузырька воздуха (йь /г2).
Полученные данные записывают в
следующую таблицу (табл: 5).
Сделав ряд отсчетов, добиваются по-
лучения близких по величине давлений
в момент появления и исчезновения пу-
зырька воздуха. Взяв среднее значение
величин давления из последнего измере-
• hi —|— Ло
ния п = —^-~—вычисляют радиус пор
по уравнению *:
гмакс —
2а
h -13,6 -981 СМ
(30)
УДЕЛЬНАЯ ПОВЕРХНОСТЬ ТВЕРДЫХ ДИСПЕРСОИДОВ
Характерным признаком высокодисперсных систем является
наличие большой поверхности раздела на границе фаз.
Степень дисперсности вещества можно характеризовать его
удельной поверхностью 50, т. е. отношением суммар-
ной поверхности частиц s к общему их объему V:
С___1
— V
При дроблении вещества резко возрастает его удельная
поверхность. Большой удельной поверхностью обладают также
различные пористые тела.
С увеличением поверхности возрастает свободная поверх-
ностная энергия системы, вследствие чего ряд свойств веществ,
находящихся в высокодисперсном состоянии, в основном опре-
деляется поверхностными свойствами. В частности, от величины
удельной поверхности адсорбента зависит количество адсорби-
2а
* В СИ гмакс — А 13600 g gl м.
71
рованного вещества как из газообразной, так и из жидкой сре-
ды. Однако в большинстве случаев, вследствие отсутствия дан-
ных о величине удельной поверхности, величины адсорбции,
так же как и значения теплоты адсорбции и смачивания, отно-
сят к единице массы или объема адсорбента. Величина удель-
ной поверхности имеет большое значение в гетерогенном ката-
лизе, при исследовании устойчивости высокодисперсных систем,
в капиллярных явлениях и ряде других.
Определение удельной поверхности пористых объектов — за-
дача довольно сложная. Существует ряд методов оценки удель-
ной поверхности: по адсорбции газов, паров или растворенных
веществ, по теплоте смачивания, по скорости растворения и др.
Широкое распространение получили методы определения
удельной поверхности по скорости фильтрации жидкости или
газа. В зависимости от целей исследования и характеристики
пористого объекта выбирают тот или иной метод определения
.удельной поверхности. Следует обратить внимание на то, что
само понятие удельной поверхности имеет относительное значе-
ние; например, определяя удельную поверхность пористого тела
по методу адсорбции, получают различные значения в зависи-
мости от его природы и структуры, а также свойств.вдсорбируе-
мого вещества. 1
Величина удельной поверхности пористого тела, определен-
ная по методу адсорбции, зависит от минимальных размеров
его пор, в которые может еще проникать адсорбируемое веще-
ство. Вследствие того, что размеры молекул газа изменяются
в небольших пределах, этот метод для различных газов дает
близкие величины. При определении удельной поверхности по
методу адсорбции из растворов получают данные, различающие-
ся иногда даже по порядку величин. Это можно объяснить тем,
что размеры частиц растворенных веществ, используемых в ад-
сорбционных опытах, изменяются от молекулярных и ионных
до коллоидных. С увеличением размеров частиц растворенного
вещества возрастает радиус пор, доступных для адсорбции,
и поверхность пор с меньшим радиусом окажется неучтенной.
Таким образом, различие в измеренных величинах удельной
поверхности по адсорбции растворенных веществ наиболее за-
метно для тонкопористых объектов.
Для одного и того же порошка величины удельной поверхно-
сти, полученные по методам адсорбции или фильтрации, могут
быть различными.
Методами, основанными на адсорбции, определяется вся ад-
сорбирующая поверхность, включая поры и трещины, не имею-
щие выхода на поверхность, методами, основанными на филь-
трации, только поверхность открытых пор, через которые проис-
ходит фильтрация жидкостей или теэа. Следует также иметь
в виду, что при выводе уравнения, связывающего скорость филь-
72
трации и удельную поверхность пористого тела, вводят ряд
предположений, упрощающих реальную структуру.
Кроме того, методы определения удельной Поверхности по
фильтрации жидкостей ие применимы к тонкодисперсным систе-
мам с большой поверхностью раздела. Скорость фильтрации
жидкости через ультратонкие поры, близкие к молекулярным
размерам, настолько мала, что затрудняет измерения. Следует
еще учесть, что свойства жидкостей (например, вязкость) могут
.изменяться в тонкокапиллярных системах и фильтрация жид-
кости через такие капилляры не будет следовать законам, уста-
новленным для течения жидкостей в сравнительно широких
капиллярах.
Имеются два основных метода определения удельной поверх-
ности по фильтрации воздуха или газа: по первому методу воз-
дух просасывается через пористую систему под давлением, близ-
ким к атмосферному; измеряется наружная поверхность частиц,
а поэтому метод пригоден для исследования более грубодисперс-
ных и менее пористых объектов. По второму методу протекаю-
щий газ * находится под большим разрежением (ниже
0,1 мм рт. ст.). Этот метод дает возможность измерять не толь-
ко наружную поверхность частиц порошка, но и поверхность
сквозных пор и трещин Метод пригоден для тонкопористых объ-
ектов. Результаты, полученные по этому методу для некоторых
образцов, приближаются к значениям, полученным по методу
адсорбции газов.
В случае наличия тупиковых капилляров в частицах порош-
ка этот метод дает меньшие значения удельной поверхности
по сравнению с методом адсорбции газов (например, для сили-
кагеля) .
Работа 13
ОПРЕДЕЛЕНИЕ УДЕЛЬНОЙ ПОВЕРХНОСТИ ПОРОШКОВ МЕТОДОМ
ФИЛЬТРАЦИИ жидкостей и газов •
Основной зависимостью, относящейся к течению жидкости
через пористую среду, является соотношение, установленное
Дарси, согласно которому скорость течения через пористую
среду пропорциональна площади поперечного сечения и гради-
енту давления.
Уравнение Дарси имеет вид:
« = - (1)
где v — объем жидкости, протекающей через пористую среду .
в единицу времени;
q — площадь поперечного сечения, перпендикулярного на-
правлению потока;
73
—----градиент давления, т. е. падение давления потока Д' Г
на единицу длины слоя /;
kx — коэффициент пропорциональности.
Уравнение Дарси, так же как и уравнение Пуазейля, спра
ведливо для ламинарного течения жидкости.
Как известно, всякая жидкость при определенных условиях
может течь ламинарным потоком; при изменении этих условий
можно превратить ламинарный поток в турбулентный.
Рейнольдс показал, что характер течения в трубах зависит
от средней скорости движения жидкости и, от диаметра тру-
бы D, от вязкости т] и плотности жидкости d. Соотношение
V
определяющее характер потока жидкости, получило название
числа Рейнольдса. Для цилиндрических труб ламинар-
ное течение жидкости переходит в турбулентное при значении
числа Рейнольдса порядка 2000.
Установлено, что при ламинарном течении жидкости для
числа Рейнольдса справедливо соотношение:
и Dd _ k' du? I .
~ 2 Dg№
где g — ускорение силы тяжести; k' — константа.
Из уравнения (2) получаем:
Если уравнение (3) применяют к прямому каналу нецилин-
дрической формы, то для характеристики его линейных разме-
ров пользуются понятием гидравлического радиуса R.
Гидравлическим радиусом R называют отношение
площади поперечного сечения к длине смоченного периметра
или отношение объема жидкости в канале к его поверхности.
В этом случае уравнение (3) принимает вид:
где k — константа.
Если уравнение (4) применяют в условиях течения жидкости
через пористую среду, то под R понимают объем пустот, делен-
ный на полную поверхность частиц:
где V—объем фильтрующего слоя;
Vr—объем частиц,-
s — поверхность частиц.
74
(6)
Одной из величин, характеризующих структуру пористого
тела, является общая пористость W, т. е. отношение
объема пор к общему объему образца:
V
Сравнивая уравнения (5) и (6), получаем WV — Rs — RS0Vr,
где So — удельная поверхность.
Так как из уравнения (6) следует, что Vr=V—WV, то
WV=RS0(V—WV), откуда
П ..
So (1 — №)
(7)
Подставляя в уравнение (4) значение R из уравнения (7),
получим следующее выражение для средней линейной скорости
течения жидкости через пористую среду:
£S2(1 — VT)2 7- I ()
Объемная скорость течения жидкости v через пористую сре-
ду равна произведению двух величин:— средней линейной ско-
рости и площади эффективного сечения. Последняя величина
выражается произведением площади сечения q, перпендикуляр-
ного направлению потока, и пористости фильтрующего слоя W.
Таким образом:
v uqW (9)
Вводя в уравнение (9) значение и, из уравнения (8), полу-
чаем
_____qW3g ЬР
~ Л5^(1 — IT)2ij ~Т
откуда:
gqW^p —
°~F — W)2l
Значение k, согласно экспериментальным данным ряда ис-
следователей, принимают равным 5, тогда:
, / qW3 кР
So = 14 у (J .см2/см3 — см 1 (10)
Если суммарную поверхность относят не к единице объема
пористого объекта,.а к единице массы, то' удельная поверхность:
14 / о IF3 ДР
s = ~d\ — См^г (м2/кг) О О
где d — плотность исследуемого вещества, г/см3.
75
Полученное уравнение устанавливает зависимость между
удельной поверхностью S и объемной скоростью течения жидко-
сти или газа через среду с пористостью W.
Величину общей пористости фильтрующего слоя можно вы-
числить по уравнению:
«"=—w- <12)
где V—объем фильтрующего слоя;
d — плотность вещества;
т — взятая навеска.
При определении удельной поверхности порошка по методу
фильтрации необходимо равномерное распределение частиц так,
чтобы жидкость или газ обтекали все частицы; кроме того, не-
обходимо достичь такого уплотнения, при котором поток воз-
духа не меняет положения частиц. Если порошок недостаточно
уплотнен, то положение частиц относительно друг друга может
быть различным, вследствие чего при одном и том же коэффи-
циенте пористости и прочих равных условиях скорость фильт-
рации будет различной, и определенные на опыте величины
удельной поверхности не будут одинаковы.
В случае переуплотненного слоя порошка частицы настолько
сближаются, что оказывают сопротивление фильтрации, подоб-
ное отдельным крупным частицам. -Поверхность же агрегата
всегда меньше суммарной поверхности составляющих его ча-
стиц. Поэтому при переуплотнении слоя получают значение
величины удёльной поверхности вещества ниже действительной
величины.
Таким образом, определение удельной поверхности следует
проводить при различных уплотнениях и максимальное значе-
ние из полученных величин считать наиболее близким к опреде-
ляемой величине.
Выполнение работы
В данной работе определение удельной поверхности порош-
ков проводится по методу фильтрации воздуха в приборе, изо-
браженном на рис. 34. Прибор состоит из следующих частей:
гильзы 5, .отдельные детали которой (стальная трубка 8, по-
лый внутри плунжер 7 и крышка 6) представлены на рисунке;
манометра 4 и аспиратора 2.
Исследуемый порошок помещают в стальную трубку 8 с пер-
форированным диском а, ниже которого имеется трубка для
присоединения гильзы к аспиратору. Плунжер служит для прес-
сования порошка в трубке, в нижней части его находится также
перфорированная пластинка б. Вводится плунжер в трубку 8
до соприкосновения упорного кольца в с верхней частью трубки.
76
Крышку 6 навинчивается на трубку так, чтобы плунжер был
прижат к трубке. Фиксирование плунжера в таком положении
позволяет производить опыты при одном и том же объеме спрес-
сованного порошка. Изменяя навеску порошка в гильзе, можно
проводить измерения удельной
поверхности при различной пори-
стости.
В верхней части аспиратора
(рис. 34) имеется воронка, через
которую наливается вода, и труб-
ка с краном для выхода воздуха
при наполнении.
В начале опыта определяют
площадь сечения гильзы q и объ-
ем той части, в которой поме-
щается слой порошка. Для этого
необходимо измерить внутренний
диаметр гильзы и высоту слоя
порошка I. Последняя вычисляет-
ся по разности расстояний от
верхнего края гильзы до пер-
форированного диска и от ниж-
ней плоскости плунжера до упор-
ного кольца.
Навеску исследуемого порош-
ка, взвешенную с точностью до
0,01 г, помещают в гильзу на
перфорированный диск, покры-
тый кружком фильтровальной бу-
маги. Для равномерного рас-
пределения порошка необходимо
слегка постучать по стенке гиль-
зы, после чего порошок прикры-
вают кружком фильтроваль-
Рис. 34. Установка для определе-
ния удельной поверхности порош-
ков по газопроницаемости.
ной бумаги и прессуют. Для этого плунжер нажимают рукой
так, чтобы его упорное кольцо соприкасалось с верхним краем
гильзы, после чего навинчивают на нее крышку до конца на-
резки.
Перед началом измерений необходимо проверить прибор на
герметичность. С этой целью каучуковую трубку 3 закрывают
стеклянной палочкой и открывают сливной кран /. Если прибор
герметичен, то начавшееся истечение воды вскоре прекращает-
ся. Только в этом случае можно приступить к измерениям. При-
соединив к прибору гильзу, открывают сливной кран /. Когда
величина разрежения, показываемая манометром, и скорость
истечения воды сделаются постоянными, отмечают показания
манометра и под аспиратор подставляют предварительно взве-
/7
/ •
шейный химический стакан, одновременно пуская в ход секун-
домер, Через некоторое время закрывают кран аспиратора и
взвешивают стакан с водой, после чего рассчитывают L'
скорость воздуха и, просасываемого через слой порошкк:
гьемную
где /И] — масса воды, вытекшей из аспиратора за время t. Из-
меняя время истечения жидкости и величины разрежения, полу-
Рис. 35. Пресс для уплотнения по-
рошка в гильзе.
чают 6—8 значений. После это-
го порошок из гильзы высы-
пают в фарфоровую чашку, бе-
рут другую навеску (по указа-
нию преподавателя), запол-
няют гильзу и, как указано
выше, определяют и. Для уп-
лотнения слоя порошка при
больших навесках гильзу с
введенным в нее плунжером
ставят под пресс (‘рис. 35).
Спрессование порошка прово-
дят до соприкосновения упор-
ного кольца плунжера с гиль-
зой. Несколько минут оста-
вляют в таком положении гиль-
зу под прессом, а затем навин-
чивают на нее крышку.
Проведя определение и при
различных уплотнениях (с не-
сколькими навесками), вы-
числяют значения удельной поверхности.
Для каждой серии измерений и с одной навеской расчет
ведут по формуле:
«1
Таблица I
Навеска
т, г
Пори-
стость W
Константа
k
Время
истече-
ния t,
сек
Масса
ВОДЫ mi
7771
WH2O
др,
см вод. ст.
см2
г
78
тле
14 f qW*
d V ij(l— Wyi
ДР —давление, см вод. ст.
Полученные данные записывают в виде таблицы (табл. 1).
Отмечают максимальное значение из полученных величин S.
Работа 14
ОПРЕДЕЛЕНИЕ УДЕЛЬНОЙ ПОВЕРХНОСТИ ПОРОШКОВ МЕТОДОМ ФИЛЬТРАЦИИ
РАЗРЕЖЕННОГО ГАЗА
При определении удельной поверхности тонкоизмельченных
веществ по методу фильтрации воздуха или другого газа при
давлениях, близких к атмосферному, получаются преуменьшен-
ные значения по сравнению с величинами, определенными дру-
гими методами. Это можно объяснить тем, что при выводе
уравнения, связывающего скорость фильтрации и удельную по-
верхность, предполагают отсутствие скольжения между твердой
стенкой и жидкостью или газом. Однако при течении.газа вдоль
твердой стенки всегда имеет место скольжение. на их границе.
Влияние скольжения газа на скорость фильтрации мало в том
случае, когда размеры пор велики по сравнению со средней
длиной пробега молекулы газа. При атмосферном давлении
средняя длина пробега молекулы меньше 0,1 мк, так что если
поперечное сечение пор равно нескольким десяткам микрон,
то для определения удельной поверхности можно вполне поль-
зоваться уравнением ‘(11).
Влиянием скольжения газа на скорость фильтрации в очень
узких порах пренебрегать нельзя. В данном случае сопротив-
ление слоя порошка заметно уменьшается за счет скольжения
газа по стенкам пор, и тем значительнее, чем больше величина
отношения длины свободного пробега молекул газа к среднему
сечению пор. Поэтому чем меньше сечение пор, т. е. чем более
диспергировано вещество, тем меньшие значения получаются
при определении удельной поверхности по методу газопрони-
цаемости. Уменьшить влияние скольжения газа на скорость
фильтрации можно путем проведения фильтрации при высоких
давлениях, так как при этом укорачивается длина свободного
пробега молекул. Однако проведение подобных опытов связано
с усложнением аппаратуры и не всегда может быть применено.
Дерягин разработал новый метод определения удельной по-
верхности по фильтраций разреженного газа, когда длина сво-
бодного пробега его молекул во много раз больше сечения наи-
более крупных пор. При фильтрации разреженного газа через
пористое тело молекулы газа в его порах соударяются друг
с другом во много раз реже, чем со стенками пор, вследствие
79
чего столкновениями молекул газа между собой мож1/о прене-
бречь. В таком случае движения отдельных молекул газа можно
рассматривать как взаимно независимые.
Течение газа при этих условиях детально исследовано Кнуд-
сеной, поэтому и получило название кнудсеновского по-
тока. Задолго до развития кинетической теории газов Грэм,
исследуя прохождение газов через пористые пластинки из гипса
(в настоящее время известно, что поры таких пластинок малы
по сравнению со средним свободным пробегом молекул газа),
установил, что количество прошедшего газа прямо пропорцио-
нально разнице давлений и обратно пропорционально корню
квадратному из молекулярного веса газа и температуры.
Была также экспериментально установлена зависимость
между количеством разреженного газа, проходящего через ка-
пилляр, и длиною последнего. Для объяснения этой зависимости
Кнудсен предположил, что молекулы после удара о стенку рас-
сеиваются во всех направлениях независимо от направления
удара. В результате некоторые молекулы после удара о стенку
будут двигаться в обратном направлении и не пройдут через
капилляр. Чем длиннее капилляр, тем больше таких молекул,
и поэтому количество газа, прошедшего через капилляр, умень-
шается с увеличением его длины. Согласно гипотезе Кнудсена,
число молекул, входящих в капилляр и движущихся после уда-
ра о стенку в обратном направлении, составляет определенную
долю от общего числа молекул, зависящую только от числа
столкновений молекул со стенкой. Приняв эту гипотезу Кнуд-
сена, при теоретическом рассмотрении явления фильтрации раз-
реженного газа через пористое тело Дерягин вывел следующую
математическую зависимость для количества газа, прошед-
шего через пористую систему:
, , IF2 1 dP
S( V MRT dl
(13)
где ч)п — число молей газа, протекающего через 1 см2 слоя ис-
следуемого вещества за 1 сек при градиенте давления
dP
вдоль потока
5] — удельная поверхность, см2! см? объема слоя;
W — пористость, которую можно рассчитать по уравнению
(12) предыдущей задачи, зная навеску и плотность
вещества;
М—молекулярный вес фильтруемого газа;
Т — абсолютная температура, °К;
R — универсальная газовая постоянная, эрг!моль -град.
Из уравнения (13) получим:
S, = 1,5-— • — смысле (14)
vnV MRT dl
80
см2!г
(15)
или
IT2 dP
dl
где 7каж — кажущийся удельный вес, т. е. вес образца, делен-
ный на объем слоя порошка вместе с порами.
Таким образом, уравнение (15) дает возможность опреде-
лить удельную поверхность вещества из измерения количества
газа, прошедшего через слой порошка, и градиента давления.
Выполнение работы
Удельная поверхность различных веществ в данной работе
определяется на установке (рис. 36), состоящей из следующих
Рис. 36. Установка для определения удельной, поверхности методом
фильтрации разреженного газа.
основных частей: реометра 1 с тремя капиллярами различного
диаметра для впуска воздуха в установку и манометра, напол-
ненного легкоподвижным нелетучим маслом; масляного 2 и
81
уравнению:
’ (16)
ртутного 3 манометров для измерения соответственно/малых и
больших перепадов давления; гильзы для порошка 4, закры-
того манометра 5 для оценки разрежения в системе.
Поскольку измерения удельной поверхности проводят при
достаточно больших разрежениях ( — 0,05 мм рт. ст.), все краны
установки должны быть вакуумными и хорошо притертыми.
Кран 6 служит для соединения одного из капилляров реометра
с установкой. Через кран 7 впускают воздух в установку. Трех-
ходовый кран 8 соединяет установку с манометрами 2 и 3. Кра-
ны 9 и 10 служат для отключения этих манометров от установ-
ки, кран 11 используется для создания перепада давления на
образце и кран 12 для соединения установки с буфером и
масляным насосом.
Количество газа, протекшего через исследуемое вещество,
определяют с помощью реометра и вычисляют по
ЛРС
Vn== -103
где Ар — показания реометра, см-,
С — константа реометра, равная объему газа
текающего через капилляр реометра за
разности уравнений в реометре в 1 см;
q — площадь сечения слоя порошка, см3;
Уо—-объем 1 моль газа-при нормальных условиях.
Прежде чем приступить к определению удельной поверхно-
сти исследуемого вещества, необходимо определить константу С
реометра. Для этого приключают к установке газовые часы че-
рез отверстие 13 и соединяют с установкой один из капилляров,
наиболее подходящий по диаметру для дальнейших измерений
(по указанию преподавателя). Осторожно (не до конца) откры-
вают кран 7 и пропускают в установку воздух, отмечая разность
уровней в реометре, количество прошедшего газа по газовым
часам и время протекания газа по секундомеру.
Константу реометра С вычисляют по уравнению:
с*
^Лр
где V—объем прошедшего газа, см3;
t — время протекания газа, сек;
йр — показания реометра.
Полученные данные оформляют в виде таблицы (табл. 2).
Из ряда определений константы реометра берут среднее зна-
чение. '
Определение удельной поверхности исследуемого вещества,
как и в предыдущей работе (стр. 76), проводят при различных
уплотнениях (навески по указанию преподавателя).
в см3, про-
1 сек при
82
Таблица 2
м н.п Показания реометра Лр, см Объем прошедшего газа V, см1 Время протекания газа t, сек • Константа реометра
/
Рис. 37. Схема гильзы.
На рис. 37 приведена схема гильзы, состоящей из стальной
трубки 6, в нижней части которой вставлен перфорированный
-диск 1, и полого внутри плунжера 3 с перфорированной пла-
стинкой 2. Навеску исследуемого вещества помещают в гильзу
на перфорированный диск, покрытый круж-' .
ком фильтровальной бумаги. Для равно-
мерного распределения порошка следует
слегка постучать по стенке гильзы, после
чего сверху покрыть кружком фильтроваль-
ной бумаги и спрессовать. Для этого вво-
дят плунжер в гильзу и завинчивают в
струбцину 4 так, чтобы упорное кольцо
плунжера плотно соприкасалось с резино-
вой прокладкой 5. Затем гильзу посред-
ством вакуумных каучуков присоединяют
к установке (рис. 36) и при закрытых кра-
нах 7 и 12 (все другие краны установки сле-
дует открыть) включают масляный насос,
после чего медленно открывают кран 12 и
производят откачку в течение 45—60 мин.
Перед впуском воздуха в установку
следует проверить ее герметичность. Если
при закрытом кране 11 не происходит поднятия масла в мано-
метре, то установка герметична, и можно приступить к измере-
ниям. Осторожно (не до конца) открывая кран 7, впускают воз-
дух в установку. Измеряют показания реометра йр и мано-
метра /iM.
При работе с тонкодисперсными порошками получают боль-
шие перепады давления на образце, которые измеряют с
помощью ртутного манометра, отключив предварительно
масляный манометр посредством кранов, 8 и 9. При работе с
грубодисперсными порошками перепад давления на образце из-
меряют с помощью масляного манометра.
Меняя скорость пропускания воздуха, что достигают изме-
нением положения крана 7, производят ряд отсчетов Лр и hM.
После окончания измерения закрывают кран 12 и, соединив
буферную склянку с воздухом, выключают масляный насос.
83
Затем, осторожно приоткрыв кран 7, впускают воздух в уста-
новку, после чего медленно открывают кран 12 и отсоединяют
гильзу. Порошок из гильзы высыпают, берут другую навеску и,
как указано выше, при различной скорости фильтрации воздуха
производят отсчеты hp и hM. Перед вычислением удельной по-
верхности исследуемого вещества необходимо проверить, доста-
точно ли было разрежение возду-
Рис. 38. График соотношения
между показаниями реометра
и манометра.
ха в системе, т. е. имел ли место
кнудсеновский поток.
Из уравнения (13) очевидно,
что количество прошедшего газа
должно быть пропорционально
перепаду давления на образце.
По имеющимся данным показа-
ний реометра и манометра для
различных скоростей фильтрации
воздуха строят график, отклады-
вая по оси ординат hM, а по оси
абсцисс hv.
Если получают прямолиней-
ную зависимость между величи-
нами hM и hp, подобно изображенной на рис. 38, то, следова-
тельно, разрежение воздуха достаточное. Если же прямолиней-
ной зависимости между Лм и hp не получают,— разрежение не-
достаточное и, следовательно, определение удельной поверхно-
сти проводить нельзя.
Перепад давления на образце вычисляют по соотношению:
ДР=Лмйм-981
(17)
где Ам — показания манометра;
dM—плотность жидкости, наполняющей манометр.
Вводя значения vn и ДР из уравнений (16) и (17) в уравне-
ние (15), получим:
s l,5W2V0<z-103-ftMdM-981
Ъя/ЖГАрС Ы
Объединив все постоянные в условиях опыта величины, по-
лучим:
Ткаж^р
где
1,5-Уо-1ОМм-981
• УТлйтсы
Полученные данные записывают в таблицу по следующей
форме (табл. 3).
84
Таблица 3
м
навески
Навеска
mi
Пористость
W
Кажущийся
удельный вес
^каж
S, смЧг
Для вычисления удельной поверхности исследуемого веще-
ства берут из табл. 3 среднее значение -г--
Лр
Проводя вычисление S для каждой навески, отмечают
максимальное значение как наиболее вероятное.
ЛИТЕРАТУРА
1. Ган Ф., Дисперсионный анализ, Госхимиздат, 1940.
2. Г р и г о р о в. О. Н„ Козьмина 3. П„ Маркович А. В., Фрид-
рихсверт Д. А., Электрокинетические свойства капиллярных си-
стем, Изд. АН СССР (1956).
3. Дерягин Б. В., ДАН СССР 53, № 7, 627 (1946).
4. Д е р я г и н Б. В., Власенко Г. Я., ДАН СССР, 63, № 2, 155
(1948).
5. Дерягин Б. В., Власенко Г. Я., Новые идеи в области изуче-
ния аэрозолей. Сб. статей, Изд. АН СССР, 1949.
6. Дерягин Б. В., Власенко Г. Я., Колл, ж., 13, В-4, 249 (1951).
7. Д е р я г и н Б. В., Крылов В. И., Ф р и д л я н д Р. М., ЖФХ 24
вып. 11, 1371 (1950).
8. Дерягин Б.В., Захаваева Н. Н., Талаев М. В., Филипов-
с к и й В. В., Определение удельной поверхности порошкообразных
тел по сопротивлению фильтрации разреженного воздуха, АН СССР
(1957).
9. 3 а в а р и ц к а я Т. А., Григоров О. Н., ДАН СССР 86, № 4, 757
(1952).
10. Крой т Г. Р., Наука о коллоидах, т. 1, гл. III, ИЛ, 1955.
11. Москвин Б. Н., Оптико-механ. пром., № 3 (107), 10 (1940).
12. С л он им И. Я., Оптика и спектроскопия 8, № 1, §8 (I960); 9, 2, 244
(1960).
13. Т о в а р о в В. В., Заводская лаборатория, 14, 68 (1948).
14. Фигуровский Н. А., . Седиментометрический анализ, Госхимиздат
(1940).
15. Физические методы органической химии, под ред. А. Вайсберга, т. IV,
ИЛ, 1955, стр. 682.
16. Фриш С. Э. и Тимо рева А. В., Курс общей физики, т. III, ГИТЛ
(1952).
17. 111 и ф р и н К. С., Рассеяние света в мутной среде, ГИТЛ (1951).
18. Doty Р., Steiner В. F., J. Chem. Phys. 18, 1211 (1950).
19. М a n е g о 1 d Е., Koll. Z., 79, 2, 156 (1937).
ГЛАВА II
ПОВЕРХНОСТНЫЕ ЯВЛЕНИЯ И АДСОРБЦИЯ
Коллоидное состояние характеризуется высокой степенью
дисперсности, а следовательно, значительная доля от всех моле-
кул или атомов, составляющих дисперсную фазу, находится
на поверхности раздела.
Понятие поверхности раздела в коллоидной химии
не ограничивается геометрической границей, разделяющей объ-
емные фазы, а охватывает реальный поверхностный слой, обла-
дающий определенной толщиной, в котором свойства вещества,
характеризуемые факторами интенсивности (например, концен-
трация, плотность энергии и др.), отличаются от их постоянных
значений в каждой из объемных фаз, непрерывно изменяясь по
нормали к поверхности. В большинстве случаев толщина такого
поверхностного слоя не превышает нескольких молекулярных
диаметров. Молекулы, находящиеся в поверхностном слое, от-
личаются от молекул в объеме по своему энергетическому со-
стоянию, вследствие асимметрии поля молекулярных сил у по-
верхности раздела. Так, молекула, находящаяся в глубине фазы,
испытывает притяжение одинаковых молекул, окружающих ее
равномерно со всех сторон, а следовательно, равнодействующая
молекулярных сил равна нулю. Для молекулы, находящейся
на поверхности раздела, равнодействующая не равна нулю и
направлена вглубь фазы по нормали к поверхности.
При диспергировании, связанном с образованием новой по-
верхности, часть молекул переходит из объема в поверхностный
слой, а следовательно, производится работа, направленная про-
тив молекулярных сил. Такая работа, производимая над систе-
мой, приводит к сгущению свободной энергии в межфазном
поверхностном слое, иначе говоря, к появлению свободной
поверхностной энергии F(s> *.
* Свободная поверхностная энергия VaK же как и другие термодина-
мические функции) определяется как ’ избыток свободной энергии в реаль-
ной системе (включающей поверхностный слой) по сравнению с такой
86
Высокодисперсные коллоидные системы обладают поэтому
Значительной свободной поверхностной энергией, в связи с
большой величиной удельной поверхности. Большинство явле-
ний, изучаемых в коллоидной химии, является следствием суще-
ствования поверхностной энергии, и самопроизвольные коллоид-
но-химические процессы направлены в сторону уменьшения этой
величины в соответствии со вторым началом термодинамики.
Явления адсорбции и смачивания, электрокинетические явле-
ния, процессы коагуляции, вопросы устойчивости коллоидных
систем и многие другие не могут быть поняты без изучения
свойств и особенностей поверхности раздела, в частности ее
энергетических характеристик. Поэтому учение о поверхностных
явлениях и адсорбции составляет центральный раздел коллоид-
ной химии.
Величина свободной поверхностной энергии Bs> выражается
соотношением:
F(S)=aS
где а — свободная энергия единицы поверхности (удельная
свободная поверхностная энергия);
S— площадь поверхности раздела.
Величина о является мерой напряженности молекулярно-сило-
вого поля в поверхностном слое.
Рассмотрим на примере границы раздела жидкость — газ
те закономерности изменения величины которые носят об-
щий характер и применимы в определенных условиях к другим
границам раздела.
Уменьшение величины F<s> в дисперсной системе при само-
произвольном ее изменении может идти двумя путями *, а имен-
но: путем уменьшения величины поверхности S и удельной по-
верхностной энергии о.
В чистой жидкости, состоящей из одинаковых молекул, поле
молекулярных сил (при данной температуре) является посто-
янным, а следовательно, величина о имеет вполне определенное
значение**. Поэтому чистая жидкость может уменьшить вели-
чину только за счет, уменьшения площади поверхности, пу-
тем изменения ее формы. Такие процессы, приводящие к обра-
зованию кривизны поверхности, лежат в основе капиллярных
явлений и явлений смачивания, которые имеют большое значение
идеальной системой, где плотность свободной энергии остается неизменной
вплоть до геометрической границы, разделяющей объемные фазы. Эти из-
быточные величины, характеризующие поверхностный слой, обозначаются
индексом (S) сверху.
* Конечно, возможны и весьма распространены более сложные процес-
сы, характеризуемые одновременным изменением обеих величин.
** Равновесная величина О, устанавливающаяся- в результате ориента-
ции молекул жидкости в поверхностном слое (в случае асимметричных мо-
лекул). .
8Т
в коллоидной химии, поскольку поверхность раздела в дисперс-
ных системах обычно обладает значительной кривизной.
Для растворов, состоящих из двух или более веществ, моле-
кулк которых различаются интенсивностью силовых полей меж-
молекулярного взаимодействия (полярностью), существует так-
же и другой путь понижения F<S), а именно: путь перераспреде-
ления молекул в растворе, приводящий к изменению состава
поверхностного слоя по сравнению с объемом. Действительно,
компонент менее полярный будет переходить из объема в по-
верхностный слой (а более полярный, наоборот, в объем), по-
скольку накопление менее полярного компонента приведет к
уменьшению напряженности молекулярно-силового поля, т. е.
свободной энергии в поверхностном слое.
Таким образом, в поверхностном слое происходит изменение
концентрации отдельных компонентов, называемое адсорб-
цией. Увеличение концентрации компонента, по сравнению
с объемной фазой, носит название положительной ад-
сорбции, уменьшение концентрации — отрицательной
адсорбции.
Процесс адсорбции идет в сторону уменьшения свободной
энергии поверхности о. Этот процесс, обусловленный молеку-
лярными силами, создает градиент концентрации у поверхности
и в предельном случае приводит к заполнению поверхностного
слоя тем компонентом, который обладает наименьшим значе-
нием о. Наоборот, тепловое движение молекул стремится вос-
становить равенство концентраций в объеме и в поверхностном
слое. Равновесие устанавливается тогда, когда процесс адсорб-
ции, приводящий к увеличению концентрации, компенсируется
процессом диффузии из поверхностного слоя вглубь раствора.
Это состояние равновесия, отвечающее минимуму свободной
энергии всей системы, описывается уравнением адсорбции Гиб-
бса, являющимся следствием второго начала термодинамики.
Таким образом, основная закономерность адсорбции заклю-
чается в том, что если величина свободной поверхностной энер-
гии для раствора меньше, чем для чистого растворителя, то
концентрация растворенного вещества в поверхностном слое
будет выше объемной. Такие вещества, понижающие при рас-
творении о на границе раздела, называются поверхностно-
активными. Если же растворенное вещество повышав? по-
верхностную энергию, оно будет удаляться из поверхностного
слоя в объем,— такие вещества называются поверхности о-
инактивными.
В обоих случаях процесс от исходного равномерного распре-
деления компонентов идет в сторону уменьшения свободной
энергии о.
Для того чтобы установить зависимость адсорбционного про-
цесса от химической природы адсорбируемого вещества, необ-
88
ходимо рассмотреть действие межмолекулярных сил. В водных
растворах поверхностно-активными являются растворимые в
воде органические вещества, например, спирты, кислоты, амины,
молекулы которых дифильны, т. е. имеют полярную группу
(связывающую их с водой) и неполярный радикал. Чем длин-
нее углеводородная цепь, тем большую работу нужно затра-
тить, чтобы ввести эту цепь в воду против когезионных сил
между диполями воды, тем больше будет, следовательно, умень-
шение свободной энергии при выходе неполярного радикала
из объема в поверхностный слой. Таким образом, с ростом дли-
ны цепи поверхностная активность увеличивается (правило
Дюкло — Траубе). Наоборот, компоненты более полярные, чем
растворитель, являются поверхностно-инактивными. В водных
растворах таковыми являются неорганические ионы. В растворе
сильного электролита величина о больше, чем в чистой воде,
так как для образования 1 см2 новой поверхности нужно затра-
тить дополнительную работу против кулоновских сил взаимо-
действия между .ионами в растворе сильного электролита. При
введении иона в объем раствора работа не затрачивается; на-
оборот, свободная энергия уменьшается за счет кулоновских
сил при взаимодействии ионов. Поэтому равновесная концентра-
ция электролита в поверхностном слое будет меньшей чем в
объеме, в соответствии с уравнением Гиббса.
Рассмотренные закономерности проявляются наиболее отчет-
ливо на границе раздела раствора с воздухом или паром, так
как в этом случае полем молекулярных сил в газовой фазе
можно практически пренебречь.
При адсорбции на границе раздела с твердым телом (адсор-
бентом) основную роль играют силы межфазного взаимодей-
ствия молекул жидкости и твердого тела, которые имеют ре-
шающее значение для процесса адсорбции. Еще более услож-
няется система в случае сосуществования трех границ раздела,
что имеет место при исследовании адсорбции в явлениях сма-
чивания.
Молекулярное поле на поверхности раздела твердого тела
может вызвать изменение концентрации не только раствора,
но и чистой жидкости, а особенно газа. Такой процесс увели-
чения концентрации (т. е. числа молекул в единице объема по-
верхностного слоя) приводит к уменьшению о, а следовательно,
также является положительной адсорбцией. Таким образом,
в общем случае адсорбцией называется изменение концентра-
ции вещества в поверхностном слое.
При всей сложности интерпретации тех явлений, в которых
наличие твердой фазы приводит к взаимодействию молекуляр-
ных полей различного рода, процесс физической адсорбции
всегда направлен в сторону уменьшения свободной энергии на той
поверхности раздела, где происходит изменение концентрации.
89
ПОВЕРХНОСТНОЕ И ПОГРАНИЧНОЕ (МЕЖФАЗНОЕ) НАТЯЖЕНИЕ
Свободная поверхностная энергия единицы площади * о на
границе жидкость — воздух обычно называется поверхност-
ным натяжением, на границе жидкость — жидкость назы-
вается пограничным или межфазным натяжением
(хотя эти термины, строго говоря, применимы для любой грани-
цы раздела).
Термин натяжение, возникший исторически, в связи с гипо-
тезой упругой пленки, выражает, представление о величине с
как о силе, действующей тангенциально (т. е. в плоскости, каса-
тельной к поверхности). Таким образом, величину о можно
определить равным образом как энергию и как силу, что легко
показать, рассматривая изотермический процесс образования
единицы новой поверхности. (
Работа расширения поверхности, равная adS, производится
в плоскости, касательной к поверхности. Следовательно,. в'сегда
есть возможность рассматривать ее как работу против силы,
действующей в этой плоскости в сторону сокращения поверх-
ности. Если при таком расширении линия периметра, ограничи-
вающего поверхность, длиной /, переместится на расстояние dx,
то работа против силы fl равна fldx (где f — сила, приходящая-
ся на единицу длины). Из сравнения двух выражений для ра-
боты видно, что cdS—fldx, т. е. свободная энергия единицы по-
верхности (1 см2) численно равна тангенциальной силе, прихо-
дящейся на 1 см длины контура, ограничивающего поверхность
раздела. Очевидно, что оба способа выражения должны быть
эквивалентны в отношении размерности. Действительно, энергия
единицы поверхности выражается в эрг/см2, т. е. в дин • см/см • см.
Сокращая, находим: дин/см или — н/м, т. е. раз-
мерность силы на единицу длины.
Таким образом, математически всегда возможна замена сво-
бодной энергии о величиной поверхностного натяжения, т. е.
силы, действующей тангенциально к поверхности. Следует пом-
нить, однако, что никаких особых сил, действующих только
в поверхностном слое, мы не знаем. Работа увеличения поверх-
ности совершается против тангенциальной составляющей тех же
вандерваальсовых межмолекулярных сил, равнодействующая
которых направлена нормально к поверхности в состояний
покоя. t
При любом способе выражения величина о определяется
работой, произведенной против сил молекулярного взаимодей-
ствия. Поэтому жидкости с более интенсивным полем молеку-
лярных сил, т. е. более полярные, характеризуются высокими
* Т. е. избыток свободной энергии, приходящийся на 1 см2 поверхност-
ного слоя.
90
Означениями а. Так, для сильно полярной жидкости — воды
= 72,75 эрг! см2 (72,75-10-3 дж/м2 или н/м) при 20°, для слабо
^Полярного гексана а — 18,43 эрг!см2 (18,43-10-3 дж!м2) при той
к’же температуре.
На границе двух жидкостей величина пограничного натяже-
ния определяется разностью напряженностей силовых полей
двух соприкасающихся фаз, и она' всегда будет меньшей, чем
i наибольшее из поверхностных натяжений этих жидкостей на
1 границе с воздухом.
Величина поверхностного натяжения уменьшается с ростом
температуры < oj вследствие ослабления сил молекуляр-
ного притяжения с увеличением среднего расстояния между
молекулами.
Применение уравнения Гиббса — Гельмгольца к поверхности
раздела
£ = ° ~~ Т (1)
показывает, что полная поверхностная энергия единицы поверх-
ности е больше ее свободной энергии о (так как < 0) и
слагается из двух величин а и T~ — q. Величина о равна мак-,
симальной работе процесса, в то время как q— (скрытая теп-
лота образования поверхности) показывает, какое количество
тепла поглощает система при изотермическом образовании 1 см2
поверхности для поддержания постоянной температуры.
Причина поглощения тепла, с молекулярно-кинетической точ-
ки зрения, заключается в том, что молекулы движутся к поверх-
ности, преодолевая молекулярное притяжение, направленное
вглубь фазы; при этом скорость их уменьшается, и поверхность
раздела образуется таким образом из молекул более «медлен-
ных», а следовательно, температура поверхностного слоя пони--
жается, если энергия в форме тепла не подводится извне.
Уменьшение о с температурой происходит приблизительно
= const! до температур, близких к критической,
где исчезает поверхность раздела фаз и о обращается в нуль.
Дифференцирование уравнения (1) по Т, при условии
да ,
-jy. == const приводит к выражению:
Таким образом, полная поверхностная энергия единицы по-
верхности е является температурным инвариантом и может быть
91
использована для сравнительной характеристики молекулярных
сил в жидкостях различного состава и строения. Для определе-
ния е по формуле (1) необходимо измерить е и найти темпера»
„ . . да
турныи коэффициент измеряя о при различных значени-
ях Т.
Среди большого числа методов измерения поверхностного
натяжения наиболее распространенными являются: 1) метод
капиллярного поднятия, 2) метод наибольшего давления пу-
зырьков, 3) метод отрыва кольца и 4) метод счета капель.
Работа 15
ИЗМЕРЕНИЕ ПОВЕРХНОСТНОГО НАТЯЖЕНИЯ МЕТОДОМ СЧЕТА КАПЕЛЬ
(МЕТОД СТАЛАГМОМЕТРА)
Метод сталагмометра является весьма распространенным,
хотя и уступает в отношении точности другим методам измере-
ния поверхностного натяжения. В основе метода лежит экспери-
ментально установленное положение, что вес капли, медленно
отрывающейся под действием силы тяжести от кончика верти-
кальной трубки, будет тем больше, чем больше поверхностное
натяжение жидкости на границе с воздухом. В первом прибли-
жении можно считать, что сила поверхностного натяжения, дей-
ствующая вертикально по окружности трубки и равная 2тгго,
поддерживает каплю, уравнове-
шивая ее вес р. В момент от-
рыва р = 2itro.
Это положение является спра-
ведливым в том случае, когда
отрыв капли совершается по
линии, равной внутреннему
периметру капиллярной трубки
радиуса г (обозначенной на
рис. 39,а). В действительности
же результаты киносъемки про-
цесса падения капли показы-
вают более сложную картину
(рис. 39), не позволяющую вы»
зависимость между величинами
Рис. 39. Схема отрыва капли от
капилляра.
вести строгую математическую
р, о и г; в особенности трудно учесть ту часть капли, которая
отрывается в виде маленькой капли или нескольких, следую-
щих за основной.
В результате ряда экспериментальных работ была установ-
лена зависимость:
^ = 27Е^(^)
92
'где f — функция от радиуса трубки г и объема капли w.
Обозначая -----через F, получаем:
2л/ -
\w ,я /
г
Для функции F в ряде справочников и монографий приве-
дены таблицы [1, стр. 484], однако практически они применяются
сравнительно редко ввиду трудностей определения радиуса ка-
пиллярной трубки. Обычно для измерения а пользуются стан-
дартной жидкостью с известным поверхностным натяжением о0.
В качестве стандартной применяют такую жидкость, которая
‘образует капли, близкие по объему к капТлям исследуемой жид-
кости, что позволяет’исключить функцию F. .Когда число капель
двух жидкостей, вытекающих из одного и того же объема V ста-
лагмометра, оказывается близким, значения функции F будут
одинаковы, поскольку F мало изменяется с w (при изменении w
в 1000 раз F изменяется от 0,17 до 0,26). Таким образом, для
исследуемой жидкости c = для стандартной с0= y-F0.
При F=F0 из двух уравнений получим:
, °о Ро
Отсюда:
а = =0 ~~ (2)
Ро
Для водных растворов неорганических веществ в качестве
стандартной жидкости применяют воду, так как в растворах
поверхностно-инактивных веществ о изменяется приблизительно
пропорционально концентрации и плотности, а следовательно,
условие приближенного равенства объемов может, быть выпол-
нено.
Для определения веса капли измеряют число капель и, вы-
текающих из объема V сталагмометра, а также плотность жид-
кости d, поскольку вес капли р может быть выражен формулой:
mg Vdg
? п п
(3)
где mi— масса жидкости в объеме V-
Ро
t VdBg
По
(4)
(5)
Подставляя (3) и (4) в (2), получим
а = а
drip
0 don
93
В другом способе вес капли р находят непосредственным
взвешиванием отсчитанного целого числа капель.
В случае летучих жидкостей измерения проводят в стеклян-
ной камере с насыщенным паром исследуемой жидкости. Суще-
ствуют также варианты метода, основанные на измерении фор-
мы и размеров висячей или лежачей капли [13].
Выполнение работы
Сталагмометр представляет собой стеклянную трубку (часто
коленчатую) с расширением в средней части и капилляром в
нижней (рис. 40);
Рис. 40. Сталагмо-
метр.
на трубке нанесены две круговые метки
(выше и ниже расширения). Целесообразно
применять трубки, градуированные вблизи
круговых меток. .^Капилляр предназначен
для ограничения скорости образования ка-
пель, которая не должна превышать 20 ка-
пель в 1 MiiH *; в зависимости от вязкости
жидкости применяются капилляры раз-
личного диаметра. Внизу трубка оканчи-
вается отшлифованной площадкой, в цент-
ре которой находится выходное отверстие
капилляра.
Перед началом работы сталагмометр
промывают хромовой смесью, свободной от
взвешенных частиц, затем тщательно про-
мывают водой. Если кончик засорен, нель-
зя прочищать его иглой, проволокой и т. д.,
а надо промыть хромовой смесью или дру-
гой жидкостью. Необходимо содержать
трубку и все части, соприкасающиеся с ис-
следуемой жидкостью, в полной чистоте;
совершенно недопустимо прикосновение
пальцем или каким-либо предметом к ниж-
ней площадке после очистки.
Для измерения сталагмометр закреп-
ляют в штатив, следя за'тем, чтобы нижняя
площадка находилась в горизонтальной
плоскости (вытекающие капли должны
иметь симметричную форму); надевая каучуковую трубку на
верхний конец, засасывают жидкость выше верхней метки. За-
тем отсчитывают число капель, вытекающих из объема V от
* При большей скорости вес образующейся капли быстро увеличивается
во времени и заметно изменяется за время отрыва. Поэтому вес оторвав-
шейся капли окажется преувеличенным. Кроме того, не успевают устано-
виться равновесные величины концентраций на поверхности капли. Кон-
струкция сталагмометра с замедленным истечением и автоматическим сче-
том капель разработана в ИФХ АН СССР,
94
^Верхней до нижней круговой метки. Необходимо избегать сотря-
сений, толчков, колебаний воздуха и изменения температуры во
время опыта.
Дальнейший ход измерений различается для капилляров с
градуировкой и без градуировки (имеющих только две метки).
Капилляры с градуировкой. Предварительно определяют,
скольким делениям соответствует одна капля. В начале опыта
(после того как уровень жидкости пройдет верхнюю круговую
метку) замечают деление, проходимое в момент отрыва капли.
Сразу же начинают счет капель; в конце отмечают деление, от-
вечающее моменту отрыва последней целой капли. Зная цену
"деления, вносят поправки к числу отсчитанных, капель и и п0.
^Измерения производят по 5—6 раз для каждой жидкости: для
^расчетов берут среднее значение.
f Измерения производят вначале со стандартной жидкостью.
Если таковой является вода, нет необходимости сушить сталаг-
мометр после промывки, а также при переходе от воды к вод-
ным растворам. Во всех случаях необходимо перед измерением
^промыть сталагмометр исследуемым раствором. При измерении
деерии растворов различной концентрации, вначале следует из-
мерять разбавленные растворы, переходя затем к более крнцен-
г,трированным. Плотность исследуемой жидкости d определяют
йри помощи пикнометра. Значения d0 и о0 берут из таблиц для
данной температуры. Для воды при 20° о0 = 72,75 эрг)см?-,
'(72,75• Ю*3 н/м), do = 0,998 г/см? (998 кг/м3). Вычисление ведут
go .формуле (5).
Капилляры без градуировки. Целое число капель стан-
дартной жидкости, ближе всего отвечающее объему V, заклю-
ченному между метками, собирают в бюкс, предварительно вы-
сушенный и взвешенный вместе с крышкой. Закрыв бюкс при-
шлифованной крышкой, взвешивают его вместе с жидкостью.
Не выливая жидкости, повторяют опыт несколько раз, собирая
..капли в тот же. бюкс и взвешивая. Определяют среднее значе-
ние веса Ро, деленного на число капель п0, т. е. р0 = —-. Таким
/г0
же образом производят определение для исследуемой жидкости
и вычисляют о по формуле (2).
Работа 16
ИЗМЕНЕНИЕ ПОВЕРХНОСТНОГО НАТЯЖЕНИЯ МЕТОДОМ НАИБОЛЬШЕГО
ДАВЛЕНИЯ ПУЗЫРЬКОВ
Давление, необходимое для отрыва пузырьков воздуха от
капиллярного кончика, погруженного в жидкость, прямо про-
порционально поверхностному натяжению жидкости, удержи-
вающему пузырек. Эта зависимость, вытекающая из физических
представлений, может быть доказана теоретически.
95
При выдувании пузырька из трубки в смачивающую жид-
кость необходимо приложить избыточное давление, чтобы пре-
одолеть капиллярное поднятие жидкости в трубку, обусловлен-
ное капиллярным давлением:
ДР = (6)
1\
где /? — радиус кривизны мениска.
Избыточное давление, необходимое для отрыва пузырька,
определяется тем максимальным значением капиллярного да-
вления ДРт, которое возникает в процессе образования пузырь-
ка. Величина ДРт является функцией радиуса капиллярной
трубки г. Действительно, если радиус трубки г достаточно мал,
то поверхность пузырька, выдуваемого из трубки в жидкость,
можно считать сферическим сегментом. Радиус кривизны R сег-
мента в момент начала образования пузырька велик, так как
поверхность близка к плоской; по мере выдувания R умень-
шается и становится наименьшим, когда пузырек примет форму
полусферы, а следовательйо R = г. '
Давление внутри пузырька возрастает, согласно (6), и в
этот момент становится наибольшим и равным ДРт. Для под-
держания равновесия необходимо, чтобы к газовой фазе внутри
пузырька было приложено избыточное (по сравнению с давле-
нием вне капилляра) внешнее давление, равное ДРт> так, чтобы
давление во всех точках газовой фазы внутри трубки было оди-
наковым (условие равновесия). За счет этой внешней разности
давлений и образуется пузырек. При дальнейшем росте пузырька
в равновесных условиях радиус кривизны, пройдя через полу-
сферу, начинает вновь увеличиваться. Это приводит к уменьше-
нию давления ДР внутри пузырька; воздух в трубке, находя-
щийся по-прежнему под внешним давлением ДРт, устремляется
в пузырек, й последний отрывается.
Максимальное давление ДРт отвечает условию: R = г. Под-
ставляя значение R в уравнение (6), находим:
Таким образом, максимальное внешнее давление равно тому
избыточному давлению &Рт, которое существует у сфериче-
ской поверхности данного радиуса. Это избыточное давление
определяется величиной о, а следовательно, и величина изме-
ренного максимального давления прямо пропорциональна по-
верхностному натяжению жидкости.
В приведенном выводе мы не учитывали гидростатического
давления столба жидкости, находящегося над пузырьком. Для
отрыва пузырька необходимо преодолеть и это давление, равное
96
Cgdhi (где hi — глубина погружения). Таким образом:
ДРи=-у- + ^^
При практических измерениях кончик капилляра обычно не
погружают дальше поверхности жидкости; в этом случае (hi 0)
. добавочный член исчезает:
О Л L Д-Р
Измеряя давление по разности уровней ДЛ=-^- в мано-
метре, соединенном с капиллярной трубкой, и определив по-
/стоянную для данного капилляра величину Л==—вычис-
ляют- поверхностное натяжение по формуле:
о — К Mtm (7)
Значение К обычно определяют посредством измерения мак*
< симальной Д/im для Стандартной" Жидкости (вода, бензол) с из-
вестным поверхностным натяжением со при данной температуре.':
i
Выполнение работы j
£ Для практических измерений целесообразно пользоваться»
^ Прибором, разработанным Ребиндером (рис. 41). Разность дав-
лений создается посредством разрежения над жидкостью вне
|капилляра; при этом происходит просасывание воздуха из
^трубки с капиллярным кончиком в жидкость с образованием
Р пузырьков.
f Перед началом работы трубку с капилляром и сосуд с боко-
вым отростком промывают хромовой смесью и водой; малейшие
загрязнения (например, прикосновение рукой) частей прибора,
соприкасающихся с изучаемой жидкостью, приводят к непра-
вильным результатам.
Ознакомившись с устройством прибора и подготовив термо-
стат, определяют константу К капилляра по воде. Для этого
наливают в сосуд 5—7 смг воды и вводят капиллярный кончик
таким образом, чтобы глубина его погружения в жидкость прак-
. тически равнялась нулю. Сосуд помещают в термостат, создают
‘ разрежение при помощи насоса, соединенного с буферной склян-
,кой и регулируют скорость образования пузырьков при помощи
;-крана. Число пузырьков не должно превышать 5—8 в 1 мин;
ф!ри большей скорости равновесные условия не достигаются и
\ .результаты оказываются неправильными.
97
Отсчет наибольшей величины Д/гт (отвечающей максималь-
ному давлению) по зеркальной шкале манометра производят
при помощи лупы таким, образом, чтобы отражение глаза на-
блюдателя в зеркале было на уровне мениска. Записывают по-
казания обоих колен манометра и берут их сумму. Вычисляют
Рис., 41. Установка для измерения поверхностного натяже-
ния методом наибольшего давления пузырьков.
константу К по формуле (8). Таким же образом проводят изме-
рения с исследуемой жидкостью, предварительно высушив сосуд
и капиллярную трубку. Вычисляют о по формуле (7).
Работа 17
ИЗМЕРЕНИЕ ПОВЕРХНОСТНОГО НАТЯЖЕНИЯ МЕТОДОМ КАПИЛЛЯРНОГО
ПОДНЯТИЯ
Следствием существования свободной поверхностной энергии
является факт появления разности давлений по обе стороны от
поверхности жидкости при ее искривлении. Эта разность дав-
лений носит название капиллярного давления. Градиент
капиллярного давления всегда направлен к центру кривизны
поверхности. 'Поэтому для жидкости с вогнутой поверхностью
капиллярное давление уменьшает внутреннее давление, а для
жидкости с выпуклой поверхностью увеличивает. В первом слу*
08
Чае капиллярное давление считается отрицательным, а во вто-
ром — положительным.
Величина капиллярного давления зависит от поверхностного
натяжения и кривизны поверхности:
где &Р—капиллярное давление;
о — поверхностное натяжение;
/?j и /?2—радиусы кривизны двух главных нормальных сече-
ний поверхности.
Это уравнение является основным в теории капиллярных яв-
лений и носит название формулы Лапласа. Оно дает зна-
чение капиллярного давления, вызываемого искривленной по-
верхностью жидкости любой формы.
Если поверхность сферическая, то /?i = R2 = R и
ДР = '7Г (6)
В капилляре поверхность жидкости, вследствие явления сма-
чивания, приобретает форму мениска. При смачивании капил-
ляра мениск — вогнутый, при несмачивании — выпуклый (см.
раздел «Смачивание»), Если капилляр поместить вертикально
таким образом, чтобы он пересекал границу раздела двух фаз,
то вследствие возникновения капиллярного давления равно-
весие в системе нарушается и граница раздела фаз начнет
перемещаться вверх или вниз в зависимости от условий избира-
тельного смачивания. Процесс перемещения границы внутри
капилляра будет продолжаться до тех пор, пока изменение
гидростатического давления не уравновесит капиллярное давле-
ние. Таким образом, в состоянии равновесия:
(10)
где dx и d2—плотности соприкасающихся фаз;
d2—плотность газовой фазы в случае границы раз-
дела жидкость — газ;
g—ускорение силы тяжести;
h — разность между уровнем границы фаз у плоской
поверхности и уровнем мениска в капилляре, на-
зываемая капиллярным поднятием.
На практике измерить радиус кривизны мениска практически
невозможно. Значительно проще пользоваться радиусом капил-
ляра г, равным r = .RcosS, где 6 — угол смачивания (рис. 42).
Величина капиллярного поднятия определится в этом случае
формулой:
. 1 2«-cosfl
Й = ---ТТ----------- (11)
• [di~~d2)g -г ' '
99
Как видно из формулы (II), величина капиллярного подня-
тия тем больше, чем лучше смачивание. При полном смачива-
нии угол 0 равен нулю, следовательно:
Рис. 42. Схема менис-
ка жидкости в капил-
ляре.
2а 1
’ gr
(12)
Формулы (11) и (12) могут быть ис-
пользованы для определения величины по-
верхностного натяжения с, .исходя из экспе-
риментально найденных Значений капил-
лярного поднятия. I
Метод капиллярного поднятия позволя-
ет получить наиболее точные по сравнению
с другими методами значения поверхност-
ного натяжения, что объясняется как про-
стотой теории, не требующей для своего
разрешения никаких допущений или приближений, так и воз-
можностью постановки точного эксперимента.
Выполнение работы
Определение поверхностного натяжения по методу капилляр-
ного поднятия производится в сосуде, указанном на рис. 43. Ос-
новную часть прибора составляют две трубки 2 и 1. Трубка 2—
капилляр, в котором наблюдается поднятие. Трубка 1 является
той частью прибора, где создается плоская поверхность жид-
кости, (т. е. трубка достаточно большого радиуса). С помощью
отвода 3 прибор может быть соединен с вакуумной установкой.
Важнейшие условия для получения точных значений поверх-
ностного натяжения по этому методу следующие.
I. Правильный подбор трубок прибора и точное определение
их радиуса. Трубки для прибора следует выбирать очень тща-'
тельно, чтобы ни внутренний, ни внешний диаметр не изменя-
лись по длине трубки. При изменении толщины стенок послед-
ние уподобляются призме, что обусловливает заметный сдвиг
изображения мениска. Эффект изменения видимой величины ка-
пиллярного поднятия будет наблюдаться и в том случае, если
трубки 2 и 1 (рис. 43) в приборе не параллельны, а наклонены,
хотя бы и немного, по отношению друг к другу. Внутреннее се-
чение капиллярных трубок должно представлять собой круг.
Трубки’ эллиптического сечения вызывают более высокое ка-
пиллярное поднятие, чем трубки с круговым сечением той же
площади. Величина радиуса трубок также имеет немаловаж-
ное значение. Капилляр 2 не должен быть слишком тонким, что-
бы можно было пренебречь изменением эффективного радиуса
капилляра, связанным с явлением адсорбции воздуха на стен-
ках капилляра. В то же время он должен быть достаточно ма-
100
Рис. 43. Прибор для опре-
деления поверхностного
натяжения методом ка-
пиллярного поднятия.
^'ого радиуса, чтобы обеспечить необходимую величину капил-
Йярного поднятия, позволяющую произвести точные измерения.
Црубка 1 должна быть достаточно широка, так как для точной
^фгнки поверхностного натяжения поверхность жидкости в ней
Должна быть абсолютно плоской. Существенную трудность пред-
ъявляет подбор однородных широких трубок. Поэтому, как
Давило, приходится ограничиваться
^рубками, радиус которых не больше 3—
см, а в случае особо точных опытов
вводить поправку на искривление ме-
даска.
Р; • Определение радиуса капилляра мо-
1СТ быть произведено либо измерением
капиллярного поднятия жидкости, точное
рачение поверхностного натяжения ко-
брой известно, либо калибровкой ка-
пилляра до его вмонтирования в прибор,
радиус большой трубки должен быть 2
^акже измерен до изготовления из
|tee прибора. Определение радиуса ка-
пилляра производится с точностью до
^А001 мм.
2. Обеспечение, условий полной сма-
чиваемости стенок сосуда, т. е. cos 6 = I.
Идя обеспечения условий полного сма-
йивания стенок жидкостью сосуд перед
ЩЙытом тщательно промывают хромовой
рмесью, ополаскивают дистиллированной
Водой и высушивают током воздуха,
Пропущенным над РгОв- Для получения
Наиболее точных результатов сосуд пе-
ред высушиванием помещают на 3—4 ч
над кипящей водяной баней и только после этого высушивают.
Затем в сосуд вливают необходимое количество исследуемой
деидкости, смачивают ею пробку, а шлиф тщательно притирают.
Верх пробки может быть залит парафином. Перед отсчетом
Жидкость несколько раз прогоняют через капилляр, и опыт ве-
дут с падающим .мениском.
3. Определение точной величины капиллярного поднятия.
Для получения истинной величины капиллярного поднятия тре-
буется отсчет определенного положения дна мениска и края
мениска, где он соприкасается со стеклом. Измерения могут
.быть произведены либо непосредственно с помощью катето-
метра, обеспечивающего точность до +Q,001 мм, либо установка
Заржет быть сфотографирована и положение мениска на снимке
уценено с помощью катетометра или компаратора. Измеряют
•Положение плоской поверхности широкой трубки, дна мениска
* *
и точки соприкосновения мениска со стенкой капилляра. Для
удобства наблюдения непосредственно за прибором помещают
темный экран в виде пластины, за экраном помещают лампу.
При измерениях положение экрана подбирают таким образом,
чтобы изображение мениска и плоской поверхности было тем-
ным и резко очерченным.
Таким образом определяют разность уровней между дном
мениска и плоской поверхностью /гР и между дном мениска и
его верхней точкой —ДЛ. Истинное значение поднятия жидкости
в капилляре выражается формулой:
h^h0 + ^ (13)
В ряде случаев оказывается необходимым учитывать искрив-
ленность поверхности жидкости в широкой трубке, Поправка
может быть введена с помощью уравнения:
0,8381+0,2789^+ |lg^ (14)
U iIq Г А/ Ч
где а — ’/2 hr;
h0 — видимый уровень поверхности;
Л'ч— «истинный» уровень плоской поверхности.
Для) получения более точных величин капиллярного подня-
тия следует провести ряд отсчетов положения мениска по раз-
личным-направлениям и взять среднюю величину.
4. Соблюдение постоянной температуры в течение опыта.
Для получения точных значений о необходимо, чтобы коле-
бания температуры не превышали 0,01° С. Для того чтобы не-
ровности стекла термостата не вызвали сдвига изображения
_ мениска или плоской поверхности, в термостат должно быть
вмонтировано оптическое стекло, через котороё производят на-
блюдение капиллярного поднятия.'
5. Проведение опыта в вакууме. Необходимость вакуумиро-
вания очевидна из формулы (12), с помощью которой рассчиты-
вают поверхностное натяжение по величине капиллярного под-
нятия, так как плотность d2 включает в себя плотность пара с
учетом воздуха. Разница в значениях капиллярного поднятия
в присутствии и отсутствии воздуха может достигать 0,5%- Для
удаления воздуха сосуд соединяют с вакуумной установкой, и
откачку производят до тех пор, пока жидкость в сосуде не за-
кипит. Через несколько минут отвод 3 закрывают, в вакуумную
установку отсоединяют.
Соблюдение указанных предосторожностей при постановке
опытов, измерении радиусов и величины капиллярного поднятия
с точностью до ±0,001 мм, колебаниях температуры около за-
данного значения в пределах ±0,01° дает возможность полу-
чить величины поверхностного натяжёния с точностью до
±0,01 эрг/см2 (10-Б дм/м2).. .
Ж2
Выполнение работы
Прежде всего сосуд промывают, высушивают, — как указано
выше, — и заполняют жидкостью с известной величиной поверх-
ностного натяжения. Далее прибор помещают в термостат, при-
чем трубки прибора должны находиться точно в вертикальном
положении, и сосуд вакуумируют (п. 5). Проверку вертикаль-
ного положения производят с помощью уровня. Высоту капил-
лярного поднятия измеряют как указано в п. 3. Радиус капил-
ляра рассчитывают по формуле (12). Плотности жидкости и
пара, входящие в формулу (12), могут быть либо измерены пик-
нометром, либо взяты из таблиц.
Затем сосуд промывают, высушивают и заполняют исследуе-
мой жидкостью. Измеряют величину максимального-капилляр-
ного поднятия и по формуле (12) определяют искомую вели-
чину поверхностного натяжения.
Работа 18
ИЗМЕРЕНИЕ ПОГРАНИЧНОГО НАТЯЖЕНИЯ МЕТОДОМ НАИБОЛЬШЕГО
ДАВЛЕНИЯ КАПЕЛЬ
Для измерения пограничного натяжения методом наиболь-
шего давления капель применимы в принципе те же положения,
которые были рассмотрены в работе 16.
Выполнение работы
Измерения в основном проводят по той же методике, что и
для поверхностного натяжения. Принципиальная схема уста-
новки дана на рис. 44.
В сосуд 1 наливают обе жидкости, на границе которых из-
меряют пограничное натяжение. В трубку 2 с капиллярным кон-
чиком засасывают ту из двух жидкостей, которая хуже смачи-
вает капилляр, так как при данной постановке опыта только в
этом случае возможно образование капли. Если более тяжелой
является сравнительно хорошо смачивающая жидкость, капил-
ляр имеет форму А, если хуже смачивающая, то трубка 2 на
конце изогнута (рис. 44, Б). Кончик капилляра должен подво-
диться возможно более точно к границе раздела жидкостей.
Избыточное давление &Рт (работа 16) будет в данном случае
определяться формулой:
2а
ДРт = “ + higdi — h2gd2
где с — пограничное натяжение;
h} — высота слоя более легкой жидкости вне трубки 2;
h2 — высота жидкости в трубке 2-г
103
fi?i — плотность более легкой жидкости;
d2— плотность хуже смачивающей капилляр жидкости;
g—ускорение силы тяжести.
Для упрощения расчетов эксперимент ставят так, чтобы гра-
ница жидкость — газ в трубке 2 и вне ее была на одном уровне,
т. е. при hi = й2. Тогда, если хуже смачивающая жидкость более
легкая, чем лучше смачивающая, то жидкость в трубке 2 и
верхняя жидкость в пробирке 1 одна и та же, т. е. di = d2, и вто-
рой и третий члены уравнения сокращаются. Если же более
Рис. 44. Установка для определения пограничного натяжения жидкость —
жидкость, методом наибольшего давления капель.
легкой является жидкость, лучше смачивающая Поверхность ка-
пиллярной трубки, то di + d2, и второй и третий члены уравне-
ния необходимо учитывать.
В том случае, когда di — d2, вышенаписанное уравнение мо-
жет быть представлено в виде:
o = = = (15)
где h — показания манометра;
d — плотность жидкости в манометре.
Для измерения пограничного натяжения следует пользо-
ваться не вертикальным манометром, как при определении по-
104
Ьерхностного натяжения, а наклонным манометром, заполнен-
'ным толуолом. Последний является более чувствительным, чем
вертикальный в раз (где а —угол наклона манометра),
г» Прежде всего определяется константа капилляра. Для этого
'Производят определение h на границе вода — бензол. Значение
*о на границе вода — бензол может быть взято из справочника.
/ При пользовании наклонным манометром следует учитывать,
что
<s = Kih (16)
причем
1 *•
Д' _ ZZ *
где К.1 — константа из формулы (15);
а-—угол наклона манометра;
х-—показание вертикального манометра, соответствующее •
100 делениям шкалы наклонного манометра.
— может быть вычислено по формуле (16).
Зная Кг, можно приступить к определению пограничных на-
ряжений на. различных границах раздела. Величину погранич-
ного натяжения вычисляют по формуле (16).
АДСОРБЦИЯ НА ГРАНИЦАХ РАЗДЕЛА ЖИДКОСТЬ-ГАЗ
И ТВЕРДОЕ ТЕЛО — ЖИДКОСТЬ
' Общие закономерности адсорбции на границе раздела жид-
кость— газ уже рассматривались выше. Процессы адсорбции .
из растворов на границе твердое тело (адсорбент) — жидкость
носят более сложный характер вследствие взаимодействия мо-
лекул твердого тела, растворителя и растворенных веществ, т е.
по меньшей мере трех компонентов. Условия адсорбции в такой
системе определяются интенсивностью молекулярных силовых
полей твердой и жидкой фаз на границе раздела.
Различие в интенсивности силовых полей, т. е. разность по-
лярностей двух фаз (растворителя и адсорбента), являются
Причиной возникновения свободной энергии о на границе, раз-
деляющей эти фазы. Чем больше разность полярностей, а сле-
довательно и величина с, тем сильнее выражена тенденция к ее
уменьшению. В этих условиях положительная адсорбция рас-
творенного вещества (поверхностно-активного для данной гра-
ницы раздела) будет наибольшей, растворителя — наименьшей.
Стремление к уменьшению разности полярностей и поверх-
ностной энергии в процессе адсорбции определяет характер
ориентации адсорбируемых молекул в поверхностном слое. Ди-
фильные молекулы ориентируются в поверхностном слое поляр
ной группой в сторону более полярной фазы, неполярным ради
105
калом — в сторону менее полярной, уменьшая тем самым раз-
ность полярностей двух ф.аз. Эти закономерности, подробно
изученные в работах Ребиндера, получили название п р а в и л-а
уравнивания полярностей.
Поэтому для адсорбции поверхностно-активных веществ, рас-
творенных в полярной жидкости, например в воде, применяют
неполярные гидрофобные адсорбенты (уголь), для адсорбции
же из неполярных жидкостей (СвН6, СС14 и др.) — полярные
гидрофильные адсорбенты (силикагель).
Рассмотрим основные принципы классификации процессов
адсорбции. Для твердых адсорбентов адсорбция имеет место
на всей поверхности капилляров, трещин, пор, обычно превы-
шающей во много раз величину видимой внешней поверхности.
При экспериментальном исследовании во многих случаях
трудно отделить адсорбцию от абсорбции — поглощения раство-
ренного вещества или газа всем объемом твердого тела. В тех
случаях, когда не удается установить механизм процесса или
же когда оба процесса происходят одновременно, применяется
общий термин — сорбция.
Второй принцип классификации исходит из природы сил
взаимодействия. Процесс изменения концентрации в поверхно-
стном слое, обусловленный молекулярными вандерваальсовыми
силами, называется физи-ческой адсорбцией. В том же
случае, когда происходит образование поверхностного соедине-
ния в результате действия химических сил, процесс носит на-
звание хемосорбции.
В настоящее время нельзя говорить о резком разграничении
физических и химических сил. Поэтому в случаях промежуточ-
ных (например, при сольватации) или же в тех случаях, когда
процесс хемосорбции сопровождается физической, адсорбцией
(например, Ог на угле), также применяют общий термин —
сорбция.
Тем не менее в большинстве случаев природу явления можно
устар'-'чить, исследовав величину теплового эффекта процесса.
При уменьшении свободной поверхностной энергии в процессе
адсорбции выделяется теплота адсорбции *. Очевидно, что в
процессе хемосорбции выделяется значительно большее коли-
чество тепла, чем в процессе физической адсорбции. В первом
случае теплота адсорбции по порядку величины близка к деп-
лоте химических реакций, во втором — к теплоте конденсации.
Существуют и другие, менее общие признаки различия, напри-
мер характер изотерм, кинетика процесса, его обратимость и др.
* В процессе адсорбции уменьшается не только свободная энергия, но
в большинстве случаев и энтропия, вследствие уменьшения степеней сво-
боды вещества в поверхностном слое. Следова1ельно, по уравнению Гибб-
са — Гельмгольца Д£ < О,
106
Процессы адсорбции из растворов подразделяются также в
зависимости от состояния адсорбируемого вещества. Раство-
ренное вещество может находиться в растворе в виде молекул,
ионов или коллоидных частиц. В соответствии с этим разли-
чают: 1) молекулярную адсорбцию (например, спиртов, жир-
ных кислот, сахара, иода и др.); 2) адсорбцию ионов (солей,ос-
нований, кислот); 3) адсорбцию из коллоидных растворов ча-
стиц дисперсной фазы (золь Au, As2Ss и др.).
Процессы адсорбции из растворов широко распространены
в природе и используются в промышленности, например, при
извлечении ценных примесей из природных и промышленных
вод, для очистки воды и различных жидкостей, для разделения
и анализа растворов сложного состава (хроматография) и т. д.
Адсорбция поверхностно-активных веществ из водных
растворов
Работа 19
ИССЛЕДОВАНИЕ СВЯЗИ МЕЖДУ ПОВЕРХНОСТНЫМ НАТЯЖЕНИЕМ
И АДСОРБЦИЕЙ
Задачей работы является: 1) исследование зависимости по-
верхностного натяжения от концентрации поверхностно-актив-
ного вещества в растворе, 2) определение адсорбции и вычис-
ление основньГх характеристик поверхностного слоя на границе
раствор — воздух, 3) изучение адсорбции на границе раствор —
твердый адсорбент с определением величины удельной поверх-
ности адсорбента.
Зависимость между изменением свободной поверхностной
энергии на поверхности раствора и концентрацией вещества в
поверхностном слое, т. е адсорбцией на границе раздела фаз,
выражена уравнением Гиббса:
_ а ( да \ , .
Т = ~РТ\да}тМ0Ль/СМ ‘
где Г — избыток вещества в моль на 1 см2 поверхности раздела;
, а — активность растворенного вещества в растворе;
У? — газовая постоянная, равная
8,31 • 107 эрг!моль-град(8,31 • 103 дж/кмоль-град)
Т — абсолютная температура.
Для разбавленных растворов можно принять, что вели-
чина а равна концентрации с, а следовательно:
Это фундаментальное уравнение, являясь приложением вто-
рого начала термодинамики к поверхностям раздела фаз, дает
107
количественное выражение для распределения, растворенного
вещества между объемом и поверхностным слоем в/результате
самопроизвольных процессов; приводящих к уменьшению сво-
бодной поверхностной энергии.
. Для веществ, понижающих свободную поверхностную энер-
д в ' ~
гию растворителя, т. е. поверхностно-активных, < 0, а следо-
вательно Г > 0, т. е. адсорбция положительна. Величина произ-
водной, взятая с отрицательным знаком, называется поверх-
ностной активностью и обозначается буквой g
да
& дс
Поверхностная активность является мерой способности рас-
творенного вещества понижать поверхностное натяжение и пе-
реходить из объема в поверхностный слой, т.. е. адсорбиро-
ваться. Из уравнения Гиббса видно, что адсорбция Г прямо
пропорциональна поверхностной активности g при данной тем?
пературе и крнцентрации веществ в растворе.
Уравнение Гиббса Позволяет построить кривую зависимости
Г от с на основании измерений поверхностного натяжения рас-
творов различной концентрации. Для. этого по данным опыта
- Строят кривую о—с и находят для отдельных значений с произ-
водные, проводя касательные и. измеряя тангенсы углов накло-
на. По этим данным вычисляют значения Г. Следует отметить,
что крутизна наклона характеризует поверхностную активность.
Другой путь, более распространенный, хотя и менее точный,
состоит в вычислении конечных разностей величин поверхност-
ного натяжения ci и а2, измеренных при двух различных, концен-
трациях Ci и с2. Эти разности о2— щ = До и с2 — = Дс под-
ставляют в уравнение Гиббса:
______£_ _Ао_
RT Лс
(в качестве с в этом случае подставляют среднее арифметиче-
ское -. = fcp I.
Определение разностей и ход зависимости вычисленных зна-
чений Г от с показывает рис. 45.
Уравнение Гиббса Г = f(o, с) не даёт однозначного выраже-
ния для функции r = f(c), т. е. для изотермы адсорбции, так
как термодинамическое описание системы, включающей поверх-
ность раздела, содержит не менее 3 переменных (в данном слу-
чае о, с, Г) по условию равновесия, выражаемому уравнением -
Гиббса. Для исключения одной из независимых переменных (на-
пример, а) необходимо наложить дополнительное условие, кото-
рое может быть получено при помощи молекулярной теории.
Таким условием является, например, то или иное представление
(2)
108
Й5> Строении Поверхностного Слоя. В первом приближении можно
считать; чтр на границе раствор — воздух и во многих случаях
жа границе раствор — твердое тело поверхностный слой яв-
ляется мономолекулярным, так как поле молекулярных сил
^твердого тела>в основном экранируется первым слоем молекул.
А - В поверхностном слое полярная часть молекулы поверхно-
стно-активного '.вещества обращена в сторону более полярной
^фазы, неполярная — в сторону менее полярной. При малых кон-
центрациях адсорбированные молекулы движутся по поверх-
ности раздела независимо друг от друга, наподобие двухмер-
ного газа; оси молекул при вытянутой их форме расположены
.тангенциально или под ма-
лым углом к поверхности. С
ростом концентрации поверх-
ностный слой приобретает свой-
ства двухмерной жидкости:
молекулы тесно соприкасаются
но ориентация отсутствует. С
дальнейшим ростом концентра-
ции молекулы принимают вер-
тикальную ориентацию, зани-
мая минимальную площадь в
поверхностном слое, приобре-
тающем свойства двухмерной
Ориентированной структуры
Анизотропной жидкости или
Рис. 45. Кривые зависимости поверх-
ностного натяжения (а) н адсорб-
ции (Г) от равновесной концентра-
ции раствора.
твердого тела.
Дальнейшее увеличение чис-
ла молекул в поверхностном
слое невозможно, следователь-
но, с ростом концентрации ве-
щества в растворе наступает
предел адсорбции. Предельное число молей вещества, адсорби-
рованных на 1 см2 поверхности раздела,'обозначают Г»,. Исходя
из представления о мономолекулярности слоя, Ленгмюр вывел
следующее выражение для изотермы адсорбции:
г==г
со
kc
1-1- kc
(3)
В этом уравнении величины Г и Ги обозначают не избыточ-
ное, а полное количество повёрхностио--активного компонента в
1 см2 поверхностного слоя. Однако для разбавленных растворов
этим различием можно пренебречь. Уравнение содержит две
константы Гоо и k. Константа k является константой равнове-
сия; в рамках молекулярно-кинетической теории она предста-
вляет собой отношение констант скоростей процессов адсорбции
109
/л •
й десорбции, термодинамически онй характеризует работу ад-
сорбции, равную RT In k. Константа зависит от приррды раство-
ренного вещества и характеризует его поверхностную активность.
Уравнение Лэнгмюра хорошо согласуется с данными опыта
и правильно отражает ход кривой Г = [(с), а именно: при м'а-
лых концентрациях (&с<С1) уравнение дает прямую пропор-
циональность между адсорбцией Г и концентрацией с; при
больших концентрациях (kc^>>\) уравнение приводит к пре-
дельному значению адсорбции Г = Гоо. Уравнение (3) позво-
ляет вычислить константы, в частности величину Гет, являю-
щуюся важнейшей характеристикой, позволяющей (стр. 111) вы-
числить размеры адсорбируемых молекул. Для нахождения кон-
стант можно решить систему уравнений с двумя неизвестными,
написанную для двух растворов различной концентрации
(ci и с2):
р _р kCl . р р ^2
1~ со1+ЛС1> ^2 — J-co
Разделив одно уравнение на другое, получим:
Г1 (1 + kci) с2 = Г2 (1 kci) Ci
Решая подобные системы для различных пар *, находят зна-
чения k, берут среднее и, подставляя его во все исходные урав-
нения, получают значения Г^,, из которых вычисляют среднее
арифметическое.
Вместо указанного способа часто пользуются графическим
методом нахождения констант. Для этого преобразуют урав-
нение (3) так, чтобы получить уравнение прямой, например, де-
лят с на обе части уравнения. После сокращения получают
с 1,1
г - + гга с
т. е. уравнение типа у = а+Ьх. В координатах (у-, с) уравне-
ние отвечает прямой, не проходящей через начало координат.
Величину Гю определяют по углу наклона прямой, а именно:
Гса=="Д(с/Ту ==ctga
Построив график -р-----с, находят разности ’ величин Дс и
Д(т')’ отвечающие отрезкам на графике (рис. 46), и вычис-
ляют Гто. По отрезку -р-Ц- отсекаемому на оси ординат, можно,
определив Гоо, найти вторую константу k.
* Например, для пяти растворов различной концентрации можно соста-
5«4
вить -j -g- = 10 различных пар.
110
Пользуясь изложенными представлениями о плотнейшей упа-
ковке молркул в монослое при предельной адсорбции, можно
вычислить Площадь, занимаемую одной молекулой, т. е. попереч-
ное сечение молекулы. Для этого надо разделить величину пло*
щади на число молекул; на площади 1 см2, согласно определе-
нию величины Г, адсорбируются Г молей или [W молекул (где
N— число АвоУадро). Следовательно, площадь занимаемая од-
ной молекулой:
Можно также вычислить толщину поверхностного слоя 8,
иначе говоря, осевую длину ориентированной молекулы. Массу
вещества, адсорбированного на
1 см2, с одной стороны, можно оп-
ределить, как произведение объема
s5 = 18 на плотность d, а с дру-
гой, — как число молей Гоо, умно-
женное на молекулярный вес
ы=ттМ
а следовательно:
d
Определение поперечного сече-
ния и длины молекулы позволило
впервые установить размеры и фор-
му различных органических моле-
кул. Вместе с исследованием по-
верхностных пленок нерастворимых
веществ этот метод и в настоящее
чение, как для теории строения, так и для определения струк-
туры ориентированных слоев.
Величины адсорбции Г на поверхности раствор.— воздух
всегда вычисляют рассмотренным выше способом — из изме-
ренных значений а, ввиду значительных трудностей прямого экс-
периментального измерения количества адсорбированного ве-
щества. Действительно, определить изменение концентрации в
объеме раствора невозможно в обычных условиях, так как
вследствие малой площади поверхности раздела раствор — воз-
дух процесс адсорбции практически не изменяет концентрации
в объеме. Для границы раздела раствор — твердая фаза приме-
нение адсорбентов с большой удельной поверхностью приводит
к адсорбции столь значительной, что изменение концентрации ве-
щества в объеме может быть определено аналитически. В этом
случае величину адсорбции х, отнесенную к единице массы т
HI
адсорбента (поверхность которого обычно неизвестна/, находят
по разности концентраций: /
~ = $С°.!Г£Х). V. моль/г {моль/кг) / (4)
где с0 — концентрация раствора до адсорбции/
сх — концентрация после адсорбции; /
V — объем раствора, дм3. ,
Измерение концентраций может бы^ь выполнено любым ме-
тодом количественного анализа. Для растворов поверхностно-
активных 'веществ таким методом может быть измерение по-
верхностного натяжения.
Измеряя поверхностное натяжение тех же растворов после
адсорбции ах, можно
по кривой о—с, построенной ранее, наити
графически значения сх и вычислить х
по уравнению (4). В некоторых случаях
целесообразно нанести значения сх после
адсорбции на тот же график, сохраняя
прежние значения абсцисс, и по ним по-
строить вспомогательную кривую *
(рис. 47). При таком способе построения
величина Со — сх, т. е. разность абсцисс
точек айв основной кривой, отвечаю-
щих значениям со и сх, может быть най-
дена графически по расстоянию между
двумя точками, лежащими на разных
кривых при одной ординате (точки а и
d). Таким способом из двух кривых мы
можем получать значения с0—сх, а сле-
довательно, и х не только для измерен-
ных растворов, но и для любых проме-
Рис. 47. График для на-
хождения равновесной
концентрации раствора
после адсорбции по ве-
личине поверхностного
натяжения.
х
жуточных значений. сх, т. е. построить непрерывную кривую за-
висимости адсорбции ~ (рис. 48) от равновесной концентра*
ции сх (изотерму адсорбции).
Различные способы выражения величины адсорбции связа-
ны соотношением:
v —=rs
т
где 5 —удельная поверхность адсорбента, т. е. суммарная пло-
щадь поверхности, отнесенная к единице массы адсорбента
(1 г).
* Эта кривая не отвечает условиям равновесия, так как выражает за-
висимость величин о после адсорбции от исходной концентрации (до ад-
сорбции).
Для
предельной
адсорбции:
~ = Г S
т 00
Рнс. 48. Кривая зависи-
мости адсорбции на границе
твердое тело — жидкость от
равновесной концентрации
раствора.
Это выражение позволяет найти величину удельной поверх-
ности, если известны и Го,.
В предельном случае, когда адсорбированные молекулы об-
разуют монослбц с плотнейшей упаковкой, число молей Гоо
Помещающихся на единице площади, будет определяться только
эффективным поперечным сечением молекулы и не зависит от
Природы поверхности; поэтому для
вычисления S можно использовать ве-
личину Гоо, найденную ранее для гра-
ницы раствор—воздух. В этом слу-
чае:
S == см2/г (м2[кг) (5)
В реальных условиях на границе
адсорбент — раствор молекулы веще*
ства могут иногда покрывать поверх-
ность не сплошным слоем, адсорби-
руясь лишь на отдельных активных
Центрах; тогда соотношение (5) даст
Преуменьшенные значения S. В других
Случаях может образоваться несколько
рлоев, вплоть до объемного заполне-
ния части пор, тогда соотношение (5) даст преувеличенные
Значения S. Тем не менее сравнение с данными, полученными
при помощи независимых методов,' показало, что часто метод
Дает правильные результаты. К сожалению, определение удель-
ной поверхности — этой важнейшей коллоидно-химической ха-
рактеристики системы — связано с большими трудностями как
методического, так и принципиального характера. Поэтому из-
ложенный метод, даюший правильную ориентировочную оценку,
величины удельной поверхности, с успехом применяется на
практике, например для сравнительной оценки активности раз-
личных адсорбентов и катализаторов.
Выполнение работы
Иэ раствора, полученного у преподавателя (см. стр. 115)
приготовляют серию разбавленных растворов. Из каждого при-
готовленного раствора отбирают определенный объем V (25 или
50 см3) в хорошо промытые и высушенные колбы. В колбы
помещают равные навески т адсорбента (уголь), закрывают
пробками и оставляют стоять в течение 2—3 ч, периодически
Ш
встряхивая. Оставшиеся порции растворов использует для из-
мерения поверхностного натяжения методом наибольшего дав-
ления пузырьков (работа 16). Измерения о произцбдят сначала
для воды, затем для всех растворов, переходя от меньшей кон-
центрации’ к большей. Наносят точки на график и вычерчивают
кривую а—с. Вычисляют значения Г, располагая данные и вы-
числения по следующей форме (табл. 1).
Таблица 1
С рт— -|-Л2 СТ Дс Дст Дст сср г.1010 сср _7 Ю
0,00 36,4 72,75
0,0103 —7,35 —714 0,0051 1,50 3,41
0,0103 32,7 65,4
0,0103 —5,6 —544 0,0154 3,44 4,48
0,0206 29,9 59,8
же графике по результатам вычислений
наносят
том
Таблица 2
На
кривую Г—сср. Находят величину предельной адсорбции Гет
графическим методом из кривой у— сср или расчетным мето-
дом (во втором случае последний столбец таблицы не запол-
няют). Вычисляют площадь, занимаемую молекулой в поверх-
ностном слое А и толщину слоя 8.
Затем переходят к изучению адсорбции на поверхности твер-
дого адсорбента. Содержимое колб с углем фильтруют, отбра-
сывая первые порции
фильтрата. Измеряют ве-
личины поверхностного
натяжения сх после сорб-
ции для всех растворов.
Данные и вычисления
записывают по следующей
форме (табл. 2).
Значения сх находят
графически. В табл. 2
оставляют место для вы-
числения промежуточных значений по вспомогательной кривой,
С
о
Со
т
согласно указанию преподавателя. Вычисляют величины —.
Вычерчивают график изотермы адсорбции-^- —сх. Если пре-
дел адсорбции не достигнут *, принимают в качестве предель-
* Это может быть в случае высокопористых адсорбентов или при ма-
лой растворимости адсорбируемого вещества,
114
него наибольшее значение — . Вычисляют величину дей-
ствующей удельной поверхности 5.
Для работы могут быть рекомендованы следующие исход-
ные растворы: 2М раствор этилового спирта; 1,5 М раствор изо-
пропилового спирта; 0,5 М растворн-бутилового спирта, 0,6 М
раствор изобутилового спирта; 0,1 М раствор йзоамилового
спирта; 0,5 М раствор масляной кислоты; 0,25 М раствор изо-
валериановой кислоты.
Из каждого раствора приготовляют, путем разбавления
вдвое, еще 5 растворов (V2, ’Л, Vs, Vie, V32 от исходного). Таким
образом каждая серия составляется из 6 растворов.
Работа 20
ПРИМЕНЕНИЕ УРАВНЕНИЯ ФРЕЙНДЛИХА К АДСОРБЦИИ ОРГАНИЧЕСКИХ
КИСЛОТ НА ТВЕРДЫХ АДСОРБЕНТАХ
В настоящее время нет общей теории адсорбции из раство-
ров на твердых адсорбентах. В этом случае вопрос более сло-
жен, чем при адсорбции газов на твердых адсорбентах, так как
следует учитывать адсорбцию растворителя и взаимодействие
молекул растворенного вещества не только с адсорбентом, но
и с растворителем.
Определяя количество поглощенного вещества, как функцию
равновесной концентрации при постоянной температуре, можно
судить о характере процесса.
Рассмотрим некоторые простей-
шие случаи, в которых процесс
поглощения вещества носит оп-
ределенный характер.
Если происходит абсорбция,
т. е. растворенное вещество рас-
пределяется в объеме твердой
фазы, то, согласно закону Генри
или правилу распределения веще-
ства между двумя гомогенными
фазами, количество поглощенно-
го вещества пропорционально
концентрации, что и представле-
но прямой 1 на рис. 49, где по
оси ординат нанесены- значения
Рис. 49.. Изотермы сорбции раз-
личного типа.
количества поглощенного веще-
ства х на единицу массы т, а по оси абсцисс концентрации
вещества в растворе после установившегося равновесия с.
В том случае, когда растворенное вещество ассоцииро-
вано, наблюдается отклонение от закона Генри, и зависимость
1Ц>
поглощенного вещества от равновесной концентрации имеет вид
вогнутой-к оси ординат кривой, подобно кривой 2.
В случае образования химического соединения между рас-
творенным веществом и твердым телом (процесс хемосорбции)
зависимость от с представлена графически в виде кривой 3.
Точка b соответствует некоторой минимальной концентра-
ции, необходимой для образования определенного химическо-
го соединения. В дальнейшем пока идет реакция, равновес-
ная концентрация остается постоянной. По окончании реак-
ции растворенное вещество уже не поглощается твердым те-
лом.
Если на поверхности твердого тела происходит адсорбция
растворенного вещества, то зависимость адсорбированного ве-
щества от равновесной концентрации при постоянной темпера-
туре (изотерма адсорбции) изобразится плавной - кривой
(рис. 50). Из изотермы адсорбции
' <* слеДУет, что при малых концентра-
циях растворенного вещества (уча-
сток оа) наблюдается прямолиней-
/ ная зависимость количества адсор-
ц/ бированного вещества от концент-
Г рации. При сравнительно больших
/ концентрациях (участок Ьс) адсор-
Г бированное количество остается по-'
----------------------стоянным, независимо от возраста-
ния концентрации, т. е. достигается
Рис. 50. Изотерма адсорбции, предел адсорбции. В области сред-
них концентраций (участок ab) в
довольно широком интервале количество адсорбированного ве-
щества возрастает пропорционально дробной степени кон-
центрации. Для этого учацтка изотермы применимо эмпириче-
ское уравнение:
1
4=/<с" (6)
m
где —количество адсорбированного вещества на единицу
массы адсорбента;
с — равновесная концентрация;
К—константа, которая представляет собой количество ад-
сорбированного вещества при равновесной концентра-
ции, равной единице (величина К может изменяться
в широких пределах и зависит от способа выражения
концентрации);
^-—константа, значение которой равно правильной
дроби.
116
, . Приведенное ныше эмпирическое уравнение адсорбции из-
-вестно как уравнение Фрейндлиха, который широко пользовался
им в своих исследованиях.
Следует отметить, что адсорбция зависит от величины по-
|верхности адсорбента, и поэтому было бы правильнее количе-
ство адсорбированного вещества относить не к единице массы,
fea к единице поверхности. Однако определение величины поверх-
ности адсорбентов довольно сложная задача, поэтому количе-
ство адсорбированного вещества в большинстве случаев отно-
сят к единице массы адсорбента.
Определение констант уравнения Фрейндлиха дает возмож-
)Сть характеризовать количественно процесс адсорбции, а так-
е сравнивать адсорбционную активность различных адсорбен-
тов по отношению к отдельным растворенным веществам.
Появление дробного показателя в уравнении адсорбции
Фрейндлиха для средних значений концентрации можно интер-
претировать следующим образом.
V. Представим себе поверхность адсорбента в соприкосновении
раствором. Молекулы растворенного вещества адсорбируются
Цри попадании на незанятые места поверхности. При введении
$£ раствор первых, очень небольших, порций вещества, когда
ройерхность адсорбента свободна, молекулы легко попадают
|;на эти участки. Величина адсорбции зависит только от средней
брродолжительности пребывания молекул на поверхности. Таким
Образом, в результате введения малого количества вещества
столько небольшая часть поверхности окажется занятой в каж-
Ййлй момент времени. Поэтому адсорбция из отдельных вводи-
мых затем небольших порций практически не зависит от адсорб-
ции предыдущего количества. Это .и обусловливает пропорцио-
нальность величины адсорбции и равновесной концентрации.
С дальнейшим увеличением концентрации все большая
‘часть поверхности оказывается занятой, вероятность попадания
^молекул на незанятые активные места значительно уменьшает-
. ся, а это приводит к тому, что адсорбция растет пропорциональ-
но некоторой дробной степени концентрации.
Наконец, постоянство величины адсорбции при больших кон-
центрациях легко объяснить тем, что вся поверхность адсор-
бента занята молекулами растворенного вещества.
Следовательно, уравнение Фрейндлиха не характеризует все-
то процесса адсорбции. Действительно, при малых концентра-
циях количество адсорбированного вещества пропорционально
-равновесной концентрации, а при больших концентрациях до-
стигает постоянной величин^, т. е. в первом случае—= 1, во
втором случае -i- = 0, в то время как в уравнении Фрейндлиха
является постоянной величиной.
117
Так как приближение к пределу насыщения поверхности при-
1
водит к резкому уменьшению —, следует ожидать, что чем бы-
стрее (т. е. при меньших концентрациях при прочих равных ус-
ловиях— навеска, температура и т. д.) достигается-предел на-
сыщения, тем меньше должны быть значения —. Подтвержде-
ние этого положения мы находим в адсорбции жирных кислот,
которая, согласно Траубе, возрастает в гомологическом ряду.
„ 1
Так, например, для ряда уксусная — валериановая кислоты —
уменьшается от 0,41 до 0,23.
Очевидно, что константа К, напротив, будет возрастать в
гомологическом ряду, так как возрастает адсорбционная способ-
ность. Действительно, в том же ряду уксусная — валериановая
кислоты значение К увеличивается от 0,1 до 0,34.
С увеличением поверхности адсорбента при адсорбции од-
ного й того же вещества (например, уксусной кислоты углем)
общая величина адсорбции возрастает, что находит выражение
и в возрастании К.. Но с увеличением площади поверхности воз-
растает число незанятых активных мест, а это ведет к повы-
1
шению величины —.
п
С повышением температуры адсорбция понижается, так как
увеличивается кинетическая энергия молекул и уменьшается
средняя продолжительность пребывания их. в поверхностном
слое. Константа К поэтому уменьшается. Так как с повыше-
нием температуры оказывается занятой все меньшая часть ак-
1
тивных мест поверхности, то — возрастает.
Процесс адсорбции протекает обычно с очень большой ско-
ростью. Однако диффузия растворенного вещества в раство-
рах, посредством которой восполняется убыль концентрации
у поверхности адсорбента, происходит весьма медленно, что
замедляет установление адсорбционного равновесия. Для
более быстрого установления равновесия рекомендуется
перемешивать или встряхивать растворы с адсорбентом. В боль-
шинстве случаев равновесие достигается в течение несколь-
ких минут; только при мелкопористых сорбентах и больших
молекулах адсорбированного вещества процесс длится несколь-
ко суток.
Исследование адсорбции из растворов твердыми сорбентами
ведут при определенной температуре, учитывая изменение кон-
центрации раствора после сорбции. Поэтому всегда необходимо
оперировать с растворами определенного объема и известной
концентрации.
118
Выполнение работы
Прежде всего определяют концентрацию данной для опыта
кислоты посредством титрования раствором NaOH.
Разультаты титрования записывают в табл. 3.
Концентрацию кислоты
Nk вычисляют по известно-
му соотношению: УкА^к =
= А^аОнУкаОН, В КОТОРОМ
Ук и Укаон—соответственно
объем взятой кислоты и объ-
ем раствора NaOH, пошед-
шего на титрование. А^асн—
концентрация раствора
NaOH в г-экв/дм3.
Из исходного раствора
кислоты приготовляют рас-
творы различной концентрации посредством разбавления вдвое.
Для этого в 5 сухих ко'лб наливают пипеткой по 50 см3 дестил-
лированной воды. Затем 50 см3 исходного раствора добавляют
в первую колбу, хорошо взбалтывают и, несколько раз набирая
раствор в пипетку и'выливая его обратно в колбу, споласки-
вают пипетку приготовленным раствором. После этого отбирают
.этой же пипеткой 50 см3 приготовленного раствора и добавляют
его в следующую колбу, хорошо перемешивают содержимое
колбы, как при получении первого растцора и споласкивают
пипетку. Потом берут 50 см3 для приготовления следующего
раствора и т. д. — до тех пор, пока не получат шесть растворов,
включая исходный.
К 50 см3 каждого полученного раствора добавляют по 1 г
адсорбента (угля). Встряхивают в течение некоторого времени
(обычно несколько минут) в зависимости от природы адсорбен-
та и концентрации раствора, после чего отфильтровывают рас-
твор, отбрасывая первые порции фильтрата. Остальное употреб-
ляют для титрования (по три порции). Полученные при титро-
Таблица 3
Проба
Взято
кислоты,
см^
На титро-
вание
пошло
NaOH,
Среднее
значение
1
2
3
10
10
10
Таблица 4
№ опыта Количество вещества в 50 г .и 8 первоначального раствора а, . ммоль Количе- ство угля т, г Количеств в фи л равновесная концентрация с о вещества ьтрате в 50 см* раствора F, ммоль Сорбированное количество кислоты на 1 г угля, a — F ш tn
- *
П9
вании данные, так же как при титровании исходного раствора,
записывают в протокол опыта. Вычисляют количество вещества
(в миллимолях), находящегося в 50 см3 до и после адсорбции.
Следует заметить, что до адсорбции от-
Таблица 5 титровывают только исходный раствор, и
№
опыта
содержание вещества в более разбав-
ленных растворах вычисляют посредст-
вом уменьшения в два раза количества-
вещества, содержащегося в 50 см3 пре-
дыдущего раствора.
Полученные данные записывают в
таблицу (табл. 4).
Эти данные используют также для
построения изотермы адсорбции, подоб-
но изображенной на рис. 50, откладывая на оси абсцисс значения
равновесной концентрации, а по оси
ординат — количество вещества, адсор-
бированное 1 г угля.
Для нахождения констант К. и -
логарифмируют уравнение Фрейндли-
ха (6):
Выписывают в виде таблицы значе-
ния lg-^- и 1g с (табл. 5). Нанося значе-
Рис. 51. График для
определения констант
в уравнении Фрейндлиха.
ния 1g по оси ординат, а по оси абсцисс — 1g с, получают
прямую, подобную изображенной на рис. 51. Отрезок ОА, отсе-
каемый прямой на ординате, дает значение 1g К, а тангенс угла
наклона прямой к оси абсцисс tga — величину—. Находят зна*
„ 1
чение констант ли—.
Адсорбция красителей
Работа 21
ИССЛЕДОВАНИЕ АДСОРБЦИИ КРАСИТЕЛЕЙ НА УГЛЕ
В настоящее время адсорбцией красителей, в частности ме-
тиленовой сини, пользуются для определения удельной поверх-
ности различных веществ, что может быть объяснено сравни-
тельно простой техникой эксперимента, а также тем, что в ряде
случаев, например на производствах, необходима сравнительная
120
оценка поверхностей одного й того же или подобных мате*
"риалов.
". Такая сравнительная оценка величины поверхностей может
'быть проведена методом адсорбции красителей. Однако следует
иметь в виду, что для абсолютных определений удельной по-
верхности этот метод весьма неточен и часто совершенно непри-
годен.
В зависимости от природы красителя адсорбция его может
протекать различно. Известно, что одни адсорбенты адсорби-
руют только кислые красители, другие — преимущественно
основные. Кроме того, наиболее тонкие поры могут быть недо-
й'ступны для молекул красителей, так что поверхность их не бу-
!Дет учтена при соответствующих определениях.
? Количество адсорбированного из раствора красител-я может
:быть определено с помощью фотоэлектрического колориметра.
Как известно, интенсивность света уменьшается при прохож-
дении его через какую-либо материальную среду. Согласно за-
кону Ламберта —- Беера, это уменьшение интенсивности света
зависит от толщины слоя среды и концентрации в нем погло-
щающего вещества. Математическое выражение закона Ламбер-
1та —Б^ера следующее:
I = IoeK cd
где /—интенсивность прошедшего через среду света;
£ /0 — интенсивность падающего света;
id и с — соответственно толщина слоя и концентрация в нем
i поглощающего вещества;
К— константа, зависящая от свойств среды и света;
е — основание натуральных логарифмов.
Из приведенного уравнения очевидно, что если толщина слоя
^постоянна, то изменение интенсивности прошедшего через него
света будет зависеть только от' концентрации Поглощающего
вёщества.
; Применение закона Ламберта —Беера к растворам крася-
щих веществ, а также к коллоидным растворам основывается
На положении, что как растворитель, так и дисперсионная сре-
да в случае коллоидных растворов практически не поглощают
Света.
Таким образом, если между источником света и фотоэлемен-
том поставить кювету с раствором красителя, то в зависимости
"от концентрации исследуемого раствора произойдет определен-
ное уменьшение интенсивности падающегр на фотоэлемент све-
та. С помощью фотоэлектрического коло'риметра можно опре-
делить светопропускание или оптическую плотность исследуе-
мого раствора £> —Ig-j- (см. стр. 41). Если провести подобные
фотоэлектрические измерения растворов до адсорбции, то, зная
121
концентрации растворов с и показания прибора, можно постро-
ить градуировочную кривую, нанося на ось ординат значения
оптической плотности, а на ось абсцисс — концентрации иссле-
дуемых растворов. Проведя фотоэлектрические измерения рас-
творов после адсорбции по градуировочной кривой, находят зна-
чения равновесных концентраций растворов. По изменению кон-
центрации растворов до и после адсорбции определяют
количество адсорбированного вещества.
Выполнение работы
В работе применяют фотоэлектрический колориметр (модель
ФЭК-М), принципиальная схема которого приведена на стр. 48.
В начале работы по заданию преподавателя приготовляют
6—8 растворов красителя различной концентрации, после чего
приступают к измерению оптической плотности приготовленных
растворов (по первому способу см. стр. 48). Следует иметь
в виду, что при исследовании темных (сильно окрашенных) рас-
творов надо пользоваться кюветами.с малой толщиной слоя
раствора (1—3 мм). Напротив, для прозрачных растворов вы-
бирают кювету с большей толщиной слоя (30—50 мм). Необхо-
димо также выбрать светофильтр, с которым в дальнейшем про-
водят всю работу. С этой целью для двух растворов (исходного
и наиболее разбавленного) производят измерения оптической
плотности с тремя светофильтрами. Выбирают тот светофильтр,
для которого получают наибольшую разницу в оптической плот-
ности этих двух растворов. Если значение оптической плотности
концентрированного раствора лежит в интервале от 0,7 до 1,
то все дальнейшие измерения проводят с этой кюветой. В про-
тивном случае выбирают другую кювету, причем при больших
значениях берут кювету с меньшей толщиной слоя раствора.
При значениях оптической плотности 0,4—0,7 выбирают кювету
с большей толщиной слоя раствора. В тех'случаях, когда, поль-
зуясь кюветой с максимальной толщиной слоя, получают для
раствора наибольшей концентрации значение оптической плот-
ности меньше 0,5, следует все измерения проводить по второму
способу (см. стр. 49).
Экспериментальные данные, полученные при выборе свето-
фильтра, записывают в таблицу 5.
Выбрав светофильтр и кювету, производят измерения опти-
ческой плотности приготовленных растворов, после чего строят
градуировочную кривую.
Затем к 50 см3 каждого раствора добавляют уголь (навеска
по указанию преподавателя) и в течение 20 мин встряхивают
растворы, после чего растворы центрифугируют. При пользова-
нии центрифугой необходимо иметь в виду, что пробирки, нахо-
дящиеся друг против друга, должны быть уравновешены (взве-
122
Таблица 5
Фильтр Концентрация растворов Оптическая плотность О,-О2
*1 с, р. О,
Зеленый Синий Красный
шивание производят на технических весах). После центрифуги-
рования осторожно отбирают растворы пипеткой, переносят в
кювету и определяют оптическую плотность, пользуясь той же
кюветой и тем же способом измерения, что и при получении
данных для градуировочной кривой. Зная оптическую плотность,
находят по градуировочной кривой концентрации растворов
после адсорбции и вычисляют количество адсорбированного ве-
щества.
Полученные данные записывают в таблицу по следующей
форме (табл. 6).
Таблица 6
Приготовление растворов Оптическая плот- ность раство ов Количество коасителя. г Со—с % адсорб- ции Примеча- ние
раствор красителя, см? вода,' см? до ад- сорбции после адсорб- ции до .ад- сорбции Со по~ле адсорб- ции с
В примечании отмечают навеску адсорбента, выбранный
фильтр и толщину слоя раствора в кювете.
Обменная адсорбция
Адсорбция ионов характерна для гетерополярных адсорбен-
тов как естественного (почвы, глины, глауконит, белки и др.),
так и искусственного (силикагель, синтетические смолы) проис-
хождения и носит обменный характер.
На границе раздела твердой фазы с водой и водными рас-
творами практически всегда возникает двойной электрический
слой в результате диссоциаций ионогенных групп, находящихся
на поверхности твердой фазы, или же вследствие избирательной
адсорбции одного , из ионов, носящей специфический характер
123
(см. стр. 174). Ионы внешней обкладки, т. е. .противоионы, не
входящие в состав твердой фазы и связанные с ней электроста-
тическими силами, находясь в динамическом равновесии с иона-
ми раствора, обмениваются в эквивалентном количестве на
ионы того же знака, находящиеся в растворе.
Таким образом, адсорбция ионов представляет собой эквива-
лентный обмен ионов одного знака, противоположного знаку
заряда твердой фазы. Поэтому данное явление называют обмен-
ной адсорбцией или обменом ионов.
Обмен ионов характерен также для высокомолекулярных
полиэлектролитов и в первую очередь для ионообменных смол,
представляющих собой сплошную пространственную сетку (кар-
кас) полимера, в узлах которой равномерно закреплены ионы
одного знака (аналогичные ионам внутренней обкладки); по-
движные противоионы находятся в растворе внутри сетки и
являются обменными. Сетка полимера, заполненная раство-
ром, рассматривается в настоящее время как одна гомогенная
фаза; поэтому представления о границе раздела фаз и ад-
сорбции в этих системах теряют физический смысл. Тем не ме-
нее законы ионного обмена являются общими для таких поли-
электролитов и для типичных гетерогенных адсорбентов. По-
этому все поглотители, для которых характерен процесс экви-
валентного обмена подвижных ионов, в то время, как ионы дру-
гого знака закреплены в структуре, носят общее название
ионитов.
В дальнейшем изложении термины: адсорбент и ионит (а
также: адсорбция и обмен ионов) будут применяться для ион-
ных поглотителей как гетерогенного, так и гомогенного типа.
Обмен ионов на границе с раствором может быть в общем
виде выражен следующим образом:
v IIMj + М2 ПМ2 + Mj
где П — обозначает твердое тело, являющееся поглоти-
телем;
М1,_И М2 — ионы, участвующие в процессе обмена.
Развитие учения об адсорбции ионов тесно связано с почво-
ведением и агрохимией, где вопросы обмена ионов имеют ог-
ромное значение для агротехники, а также при изучении гене-
зиса почв и природных вод.
Мировую известность приобрело учение Гедройца об обме-
не ионов почвами; им было впервые введено понятие о поч-
венном поглощающем комплексе, сыгравшее боль-
шую роль в развитии современных представлений-о структуре
почв.
Развитие теории привело к установлению количественных за-
кономерностей процесса обмена ионов. Обобщенный термодина-
мический вывод был дан Никольским. Для обмена двух ионов
124
НМ было получено следующее уравнение:
хУг> а*'*'
1 17 1
*2 «2
'тде Xi и х2— равновесные количества адсорбированных ионов
на единицу массы адсорбента;
Zj и z2 — их валентности; •
г?! и а2 — активности в равновесном растворе;
К—константа обмена.
§цля одновалентных ионов, например при обмене К+ и Na+
‘имеем:
XNa+ % gNa+
хк+ ак+
В настоящее время процессы обменной адсорбции находят
‘все возрастающее применение, в частности для умягчения и
Опреснения жестких вод, для извлечения ценных веществ из
разбавленных растворов, для разделения ионов, в хроматогра-
фическом анализе, для изготовления электрохимически актив-
ных мембран, используемых в электродиализе, в качестве ката-
лизаторов, и в ряде других областей.
В связи с этим разработаны методы получения новых синте-
тических ионитов на основе высокомолекулярных соединений.
'Поглотители, содержащие кислые группы, сульфогруппы, кар-
боксильные и другие, адсорбируют катионы и называются ка-
тионитами.
' Анионитами являются поглотители, содержащие основ-
ные группы (например, аминогруппы).
В зависимости от степени диссоциации ионогенных групп
различают сильные и слабые иониты. Для каждого ионита раз-
личают Н+-форму, Иа+-форму и т. д., в зависимости от природы
противоиона. Большое значение имеет предв.арйтельна'я обра-
ботка, позволяющая получить поглотитель в нужной ионной
.форме.
- Важнейшей характеристикой ионита является количество по-
глощенных им ионов. Эта способность поглощать определенное
количество ионов зависит от природы поглотителя и называется-
обменной способностью. Обменная способность может
меняться в широких пределах в зависимости от концентрации
адсорбируемого иона в растворе и от величины pH.
Количественное выражение этой зависимости для случая ад-
сорбции из водного раствора соли, установленное Никольским,
имеет следующий вид:
' k
ти г 1 z ° м 1
125
где хм — количество металлического катиона, поглощенного
единицей массы адсорбента;
z — его валентность;
ам — его активность в растворе;
k — емкость двойного слоя *;
А — константа интегрирования.
Так, увеличение концентрации одновалентных ионов в рас-
творе в 10 раз вызывает такое же изменение адсорбированного
количества %м как увеличение pH на единицу.
Указанная зависимость х = f (а, pH) имеет общий характер
и связана с тем, что происходит конкуренция за места в адсорб-
ционном слое между катионом и ионом Н, всегда присутствую-
щим в воде. Несмотря на малые количества Н+ (~10-7 г-экв!л
в нейтральном растворе), во многих случаях адсорбционная
способность Н+ весьма велика, и участие его в обмене оказы-
вается значительным. Это наблюдается для всех слабых катио-
нитов (в почвах, силикатах и др.), где Н-ион образует прочную
связь, т. е. слабо диссоциирующую кислоту, в то время как соли
этой кислоты хорошо диссоциированы.
Адсорбционная способность различных ионов неодинакова:
чем больше заряд иона и чем меньше его гидродинамический
радиус, тем больше вероятность перехода из свободного раство-
ра и диффузного слоя в адсорбционный слой и вытеснения им
одноименно заряженного иона.
Таким образом, по адсорбционной способности ионы обра-
зуют тот же ряд, который характерен для коагулирующей спо-
собности, а именно — для катионов:
А13+ > Ва2+ > Са2+ > Mg2+ > К+ > Na+ > Li+
Поэтому величина обменной адсорбции будет различной для
отдельных ионов.
В связи с тем, что величина обменной адсорбции зависит
от ряда условий, для сравнительной оценки различных погло-
тителей были выработаны определенные стандартные условия;
величина обменной способности, отвечающая стандартным ус-
ловиям, называется условной емкостью обмена.
В качестве условной емкости обмена обычно принимают ко-
личество эквивалентов иона бария, поглощенное 1 кг адсорбента
при pH, равном 6,5 и концентрации Ва2+ в растворе, равной
0,1 н. Эти условия являются наиболее удовлетворительными.
* Емкость выражена в «химических» единицах, т. е. в миллиэквивален-
тах на 1 г адсорбента и на единицу pH, а именно:
. 2,3/г'ЯГ
А =
где k' — емкость в единицах CGSE или в фарадах,
126
В некоторых случаях применяются другие условия, однако
всегда значения pH. концентрации и природы иона должны
быть точно установлены и оговорены.
Для исследования обменной адсорбции применяются два
метода — аналитический и потенциометрический. В первом ме-
тоде обменная адсорбция изучается в динамических условиях,
во втором — в статических.
Работа 22
ИССЛЕДОВАНИЕ ОБМЕННОЙ АДСОРБЦИИ ИОНОВ
АНАЛИТИЧЕСКИМ МЕТОДОМ
Принцип метода заключается в обработке навески адсор-
бента буферным раствором соли до установления адсорбцион-
ного равновесия при постоянных величинах концентрации вы-
тесняющего иона и pH раствора. После этого адсорбированный
ион вытесняется другим ионом и анализируется в фильтрате.
Рассмотрим метод применительно к адсорбции Ва2+ на ка-
тионите.
Необходимость применения буферных растворов, особенно
для слабых ионитов, обусловлена тем, что даже ничтожное
количество вытесненного Н+ в случае небуферного раствора на-
столько резко изменит pH, что адсорбционное равновесие сильно
сместится в пользу Н+, а следовательно, вытеснение Н-иона
ионом Ва будет незначительным. По расчету, чтобы изменить
равновесное значение pH от 5,5 до 6,5 для почвы при навеске
в несколько граммов, потребуется несколько сот литров небу-
ферного 0,1 н. раствора хлористого бария.
Наступление адсорбционного равновесия обычно определяет-
ся по равенству pH поступающего и вытекающего (равновес-
ного) раствора.
Такой способ определения возможен лишь в том случае, •
когда pH изменяется в процессе обмена, например, когда адсор-
бент находился первоначально в Н+-форме. Для этого в некото-
рых случаях рекомендуется предварительная промывка адсор-
бента слабым раствором кислоты.
После достижения равновесия перед вытеснением адсорби-
рованного Ва2+ необходимо учесть количество ионов бария, на-
ходящихся в растворе, пропитывающем навеску; этот механи-
чески задержанный барий при ориентировочной оценке величи-
ны обменной адсорбции обычно отмывают водой. Однако при
этом ионы Н+, содержащиеся в воде, вытесняют некоторое коли-
чество адсорбированного бария, что приводит к преуменьшен-
ным результатам. Более точным является учет механически
задержанного бария; для этого определяют вес раствора извест-
ной концентрации, пропитывающего адсорбент.
127
Вытеснение ионов бария после установления равновесия
обычно производится подкисленным раствором хлористого ам-
мония. Вытеснение ведут, до отсутствия реакции на Ва2+ в
фильтрате.
Обработка адсорбента растворами обычно проводится на
Рис. 52. Установка для измерения
обменной адсорбции.
аналитических воронках; целег
сообразно применение усовер-
шенствованной аппаратуры,
например автоматического
аппарата (рис. 52), в котором
навеску адсорбента помещают
в патрон между Двумя стек-
лянными фильтрами. Нижний
фильтр 1 вплавлен в патрон,
а верхний 3 удерживается кон-
цом отводной трубки 4 , за-
крепленной в пробке. Раствор
протекает через адсорбент
снизу вверх из бутыли 5, снаб-
женной трубкой 6 для защиты
от СОг, под напором гидроста-
тического столба жидкости, вы-
сотой 1—2 м.
При определении обменной
адсорбции в условиях рН>6,5
необходимо проводить обра-
ботку навески адсорбента без
доступа воздуха, чтобы устра-
нить возможность образования
ВаСО8 и его осаждения на ад-
сорбенте. Точно так же при
исследовании щелочных образцов необходима их предваритель-
ная обработка кислотой.
Выполнение работы
Приготовляют буферные растворы, для этого к раствору
ХЛорисТого бария добавляют уксусную или малеиновую кисло-
ту*, раствор разбавляют водой до 0,1 н. по барию й добавляют
0,1 н. раствор гидроокиси бария до требуемого значения pH**
согласно указанию преподавателя.
Навески адсорбента помещаются на бумажные фильтры в
аналитические воронки или в колонки. Необходимо ставить
* Так, чтобы после разбавления раствор был порядка 0,01 н. по кис-
лоте.
** Таким же образом поступают при изучении'адсорбции других ионов,
128
r*' •
2—3 параллельных опыта. Воронки или колонки должны
выть предварительно высушены и взвешены вместе с филь-
трами.
Проводят предварительную обработку навески адсорбен-
та небольшим количеством 0,01 и. НС1 до отчетливо красной
реакции фильтрата по метиловому оранжевому (если
изучается Н+-форма адсорбента, то такая обработка не тре-
буется).
v; Далее навески обрабатывают буферным раствором ВаСЬ-
Скорость протекания раствора через колонки не должна превы-
;Шать 1 см^мин. При работе с воронками необходимо приливать
Даствор отдельными порциями. Для обработки одной навески
'Обычно требуется 0,3—0,8 дл3 раствора. В конце обработки во-
•ронки необходимо прикрывать часовыми стеклами. После того
£как значение pH вытекающего раствора будет равно-pH исход-
/ного, обработку прекращают*. После этого избыток раствора
^слегка отсасывают, не допуская просасывания воздуха через
‘•навеску, и быстро взвешивают воронку с навеской и часовым
: Стеклом. Отнимая вес воронки с сухим адсорбентом, фильтром
И стеклом, находят вес механически задержанного раствора.
:Дная содержание Ва2+ в 1 г исходного раствора вычисляют
' количество механически задержанного Ва2+, которое нужно вы-
честь из общего количества найденного бария. После взвеши-
вания вытесняют Ва2+ посредством дальнейшей обработки рас-
твором, содержащим 0,5 моль NH4C1 и 0,05 моль НС1 в 1 Л.
Фильтрат собирают. Вытеснение ведут до исчезновения реакции
'На Ва2+, что проверяется в отдельной пробе.
После окончания вытеснения фильтрат упаривают др объема
~200 мл и определяют барий весовым методом. Параллельно
анализируют навеску исходного буферного раствора для опре-
Таблица 7
Пробы На- веска, е Вес воронкн с навеской и фильтром, г Вес механи- чески задержан- ного раствора, е Вес BaSO4, г Ва2+, лг-экв Емкость мг*экв{г
сухйй влажный Всего механи- чески задер- жанного адсорби- рован- ного
•
* Определение pH исходного раствора и контроль величины pH выте-
кающих растворов производят в отдельной пробе электрометрическим
путем.
129
деления содержания бария в 1 г раствора. Из найденного коли-
чества бария вычитают- механически задержанный. Разность
дает величину обменной адсорбции, которую вычисляют в
мг-экв!г адсорбента.
Данные опыта и результаты вычислений записывают по
форме табл. 7.
Работа 23
ИССЛЕДОВАНИЕ ОБМЕННОЙ АДСОРБЦИИ ИОНОВ ПОТЕНЦИОМЕТРИЧЕСКИМ
МЕТОДОМ
Аналитическим методом обычно пользуются для характери-
стики обменной адсорбции адсорбентов при определенном зна-
чении pH. Потенциометрический метод позволяет провести
быстро измерения при различных концентрациях Н+, и таким
образом установить зависимость обменной адсорбции от вели-
чины pH.
Изучение кривых х — pH имеет большое значение для харак-
теристики свойств функциональных (ионогенных) групп погло-
тителя. В соответствии с этим различают 4 типа ионитов
(рис. 53).
Рисунок приведен для катионитов; для анионитов получаются
те же соотношения, если по оси абсцисс откладывать рОН.
Кривая типа I характерна для монофункциональных силь-
ных ионитов; ионогенная группа представляет собой сильнук? ки-
слоту, например R—БОз Н+. Уже при низких значениях pH
(~2) кислота полностью диссо-
циирована, и величина адсорбции
металлического катиона почти до-
стигает предельной величины для
данного адсорбента.
Кривую II дают иониты типа
слабых кислот, у которых диссо-
циация ионогенных групп отве-
чает высоким значениям pH (8—
2 4 6 в 70 pH 9 для карбоксильных, 10—11 для'
Рис. 53. Кривые зависимости ад- фенольных). Кривая III харак-
сорбции от pH раствора для иоии- терна для бифункциональных
тов различного типа. ионитов с сильной и слабой кис-
лотной группой; по отношению
ординат горизонтальных участков можно определить долю ад-
сорбции, приходящуюся на каждую группу.
Кривая IV типична для полифункциональных адсорбентов,
для почв, алюмосиликатов и др. Увеличение концентрации ОН~
приводит к диссоциации все более слабых кислотных групп, по-
следовательно увеличивая величину адсорбции.
130
Сущность определения обменной способности по потенцио-
метрическому методу заключается в построении двух кривых
титрования раствора соли щелочью и кислотой в отсутствии и
при наличии адсорбента. Если адсорбент находится в Н+-фор-
ме, то при титровании до того же значения pH раствора во вто-
ром случае потребуется больше, щелочи, так как часть щелочи
пойдет на нейтрализацию Н-ионов,
вытесненных из адсорбционного
слоя.
Таким образом на рис. J54 кри-
вая титрования в присутствии адсор-
бента (II) будет сдвинута вправо,
.в сторону больших количеств щело-
чи по сравнению с кривой титрова-
ния I без адсорбента.
Разность абсцисс точек, лежа-
щих на двух кривых при одном
значении pH, показывает количе-
ство адсорбированных Н-ионов,
нейтрализованных при этом значе-
нии pH, а следовательно, количест-
см3 NaOH
во хм металлических катионов, по- Рис к титрованиЯ:
ступивших из раствора на их место. , „ „
Измеряя ЭТИ разности ДЛЯ различ- адсорбента.
ных значений pH, находят величи-
ны хм для каждого заданного значения (например, через
каждые 0,5 единиц pH).
c(nt — По)
--------т
(7)
где «1 — число см3, NaOH при титровании с адсорбентом;
п0 — то же без адсорбента;
с—концентрация раствора щелочи в мг-экв!дм3’,
т— навеска адсорбента.
Непосредственное титрование в присутствии навески адсор-
бента обычно затруднительно, ввиду медленности наступления
равновесия, особенно в. почвах. Поэтому поступают следующим
образом: берут ряд навесок в колбочки с раствором соли, до-
бавляют различные количества щелочи и после наступления
равновесия измеряют pH в каждой, находя таким образом от-
дельные точки кривой II. Соль и щелочь .добавляют в таких ко-
личествах, чтобы равновесные концентрации катиона во всех
колбах были одинаковыми.
Для изучения адсорбции в сильно кислой области в некото-
рых случаях' приходится добавлять не щелочь, а кислоту. .
В щелочной области необходимо применять растворы, сво-
бодные от СОг и исключить доступ ее иа-воздуха.
131
Выполнение работы
Предварительная подготовка заключается в переводе адсор-
бента в Н+-форму *. Для этого адсорбент обрабатывают на во-
ронке Бюхнера раствором кислоты (например 0,05 н. НС1) до
наступления равновесия, т. е. до тех пор, пока концентрация ис-
ходного раствора и фильтрата ‘не станут одинаковыми (прове-
ряют титрованием). Применяется также метод электродиализа
(см, работу) в котором адсорбент с водой помещают в среднюю
камеру между двумя отрицательно заряженными (например
целлофановыми) мембранами. Процесс ведут со сменой воды
в крайних камерах до отсутствия металлических катионов в ка-
тодной камере (например по сухому остатку). Обработанный
кислотой адсорбент тщательно отмывают водой до отсутствия
в пробах фильтрата кислой реакции по метиловому красному.
Полученный в Н+-форме адсорбент высушивают и берут навески,
по указанию преподавателя, в сухие склянки (8—10 штук).
В склянки с навесками наливают по 50 см3 0,2 н. NaCl и раз-
личные количества 0,1 н. NaOH (например, 1 см3 во вторую,
2 см3 в третью и т. д. в порядке возрастания). Затем добавляют
воды так, чтобы общий объем жидкости составлял 100 см3. Рав-
новесная концентрация Na-ионов во всех склянках будет равна
0,1 н., так как часть (отвечающая количеству добавленного
NaOH) перейдет в адсорбционный слой. Все сосуды закрывают
пробками, помещают в аппарат для встряхивания на 2—3 ч и
затем оставляют стоять до наступления равновесия (обычно
2—3 суток) при периодическом взбалтывании.
После этого определяют pH растворов, находящихся в рав-
новесии с адсорбентом. Берут несколько параллельных проб из
каждого сосуда. Растворы не фильтруют, так как присутствие
адсорбента (например, в виде взвеси) при измерении pH обе-
спечивает получение более точных результатов.
Данные опыта наносят на график; по оси абсцисс отклады-
вают П1 — число см3 NaOH, по оси’ординат--полученные зна-
чения pH.
* Для анионита в ОН-форму.
132
s Для получения кривой в отсутствии адсорбента берут 50 мл
0,2 н. NaCl, добавляют 5О‘си3 HjO и проводят потенциометри-
ческое титрование раствора, добавляя по каплям 0,1 н. NaOH.
уДля построения той же кривой в кислой области такой же ра-
‘•Створ соли титруют по каплям 0,1 н. НС1.
' . Наносят обе кривые на один график. По графику измеряют
фазности абсцисс точек, имеющих одинаковую ординату; орди-
наты берут при различных значениях pH, например через ка-
ждые 0,5 единиц.
По разности абсцисс (ni — n0) вычисляют значения хм Для
^различных значений pH согласно уравнению (7). Запись ведут
|jao следующей схеме (табл. 9)..
Таблица 9 •
pH Ло «1—Ло ХМ
с Значения хм выражают в мг-экв!г сухого вещества адсор-
бента. Полученные результаты выражают в виде графика
•Хм — pH.
СМАЧИВАНИЕ И ФЛОТАЦИЯ
Среди явлений, происходящих на границе раздела трех фаз,
j?t₽iue всего встречаются и имеют большое практическое значе-
•ние явления смачивания и растекания. Условия смачивания по-
верхности твердого тела жидкостью, характеризующие молеку-
лярное взаимодействие различных фаз, играют большую роль в
процессах проникновения жидкости, и в частности воды,, в ка-
пиллярные системы — различного рода пористые тела, грунты и
почвы. Возможность изменения условий смачивания исполь-
зуется при приготовлении составов для борьбы с вредителями
•растений, для придания водонепроницаемости тканям, стенным
^(покрытиям и т. д. Особо, важное значение имеют условия смаг
чивания для осуществления процесса флотации, широко при-
; меняющегося при добыче полезных ископаемых. Количественная
оценка смачиваемости может быть осуществлена различными
методами.
. Работа 24
V ИЗМЕРЕНИЕ КРАЕВЫХ УГЛОВ И ВЫЧИСЛЕНИЕ РАБОТЫ АДГЕЗИИ
Капля жидкости, находящаяся на поверхности другой, несэде-
Шивающейся с ней жидкости или на поверхности твердого тела,
133
может растекаться в тонкую пленку или остаться на поверхно-
сти в виде линзы.
Условия растекания или нерастекания капли могут быть оп-
ределены энергетическими соотношениями в системе, т. е. велйг
чинами свободной энергии на межфазных границах и их соот-
ношением, выраженным посредством адгезии и когезии.
Под адгезией или прилипанием понимают силу сцепления
между двумя, приведенными в соприкосновение разнородными
телами.
Когезией называется сила притяжения между одинаковы-
ми молекулами.
Адгезию часто характеризуют работой, которую надо затра-
тить, чтобы разделить две фазы, имеющие поверхность сопри-
косновения 1 см2.
В этом случае, вместо одной границы раздела между двумя
фазами образуются две поверхности каждой фазы, граничащие
с воздухом. Соответственно этому получится два значения сво-
бодной энергии поверхности.
Для поверхности раздела твердое тело — жидкость работа
адгезии выразится следующим соотношением, установленным
Дюпре:
Ж ~ °Т—Г °Ж-Г-°т-ж (1)
в котором индексы т, ж и г соответствуют твердому телу, жидко-
сти и газовой фазе.
Таким образом, работа адгезии будет равна разности между
суммой величин, образовавшихся новых свободных поверхност-
ных энергий и межфазной свободной поверхностной энергии.
Под работой когезии понимают работу разделения какой-
нибудь однофазной системы, например столба жидкости с по-
перечным сечением в \ .см2, на два столба.
Применив уравнение Дюпре к отдельной жидкости, видим*
что член ат-ж исчезает и уравнение для работы когезии полу-
чится следующим:
W't-jk = 2«ж_г
Работа когезии (сцепления молекул) может рассматриваться,
как работа адгезии на воображаемой поверхности, площадью
в сл«2, внутри самой жидкости.
Чем больше работа адгезии жидкости к твердому телу и чем
меньше работа когезии жидкости, а следовательно и ее поверх-
ностное натяжение, тем лучше жидкость смачивает поверхность
твердого тела. Поэтому, жидкости .с низким поверхностным на-
тяжением хорошо смачивают различные твердые поверхности.
Так, масла и углеводороды, у которых поверхностное натяже-
ние на границе с воздухом колеблется в пределах от 17—
25 эр^рм,2 (17; lQ-з—25 .10 3 dyclM2}смачивают все известные
тела. Вода, для которой поверхностное натяжение На границе
с роздухом равно 73 эрг/см? (73-Ю-3 дж]м2) (при 20°), хорошо
смачивает стекло, кварц и ряд других твердых тел; ртуть, имею-
щая поверхностное натяжение 460 эрг/слг2, (460 • 10*3 дж1м2)
смачивает только некоторые твердые тела.
Рассмотрим условия равновесия при смачивании твердого
тела жидкостью (рис. 55).
На рис. 55 дается поперечный разрез капли жидкости (ж),
помещенной на поверхности твердого тела (т), в атмосфере га-
за (г).
Рис. 55. Краевые углы: 1 — смачивание (6 < 90°); 2—переходный слу-
чай (0 = 90°); 3 — иеСмачивание (0 > 90°).
Соответственно трём соприкасающимся фазам, мы имеем в
данном случае, три поверхности раздела: твердое тело — газ т—г,
твердое тело — жидкость т — ж и жидкость — газ ж — г.
Линейная граница, по которой соприкасается поверхность
раздела т—же поверхностью раздела т — г, называется пери-
метром смачивания.
Рассматривая поверхностные натяжения, как силы, дей-
ствующие тангенциально к поверхности, что вполне допустимо
для данного случая, имеем три силы поверхностного натяже-
ния, действующие в точке соприкосновения трех фаз ст_г;
Ст—ж! Сж—г-
На границе раздела твердое тело — газ действует сила вдоль
поверхности раздела ст-г. Эта сила стремится растянуть каплю
вдоль границы твердого тела. Сила, действующая на границе
раздела твердое тело—жидкость ст-ж стремится стянуть ее. Си-
ла, действующая на границе раздела жидкость—газ с)И .г, на-
правлена по касательной к поверхности капли. Эта касательная
образует с поверхностью твердого тела в точке соприкосновения
трех фаз угол 9, условно измеряемый в сторону жидкости и на-
зываемый углом смачивания или краевым углом
(рис. 55).
Величина этого угла определяется условием механиче-
ского равновесия. Согласно уравнению Лапласа, величина
cos 9 при равновесии связана с межфазными поверхностными
135
натяжениями следующим соотношением:
ат_г = &т—ж Ч~ °ж —г cos 6 ИЛИ cos6 = -^b=i ат~ — (2)
аж—г
Из уравнения Лапласа видно, что краевой угол или величина
его косинуса зависит от молекулярной природы поверхности
раздела и не зависит от разменов капли.
Величина краевого угла или величина cos 9 является количе-
ственной оценкой смачиваемости и служит мерой смачивания.
Если свободная поверхностная энергия на границе твердое
тело — газ имеет большее значение, чем на границе твердое те-
ло— жидкость, то твердое тело будет смачиваться этой жидко-
стью. Тогда равновесный угол 0 будет меньше 90°, т. е. краевой
угол будет острый, a cos 9 будет >0 (рис. 55,/). При 9 = 0 и
cos 9 = + 1 будет происходить полное смачивание.
Условие смачиваемости определяется, таким образом, нера-
венством:
°т-г > ат-ж
В обратном случае, когда свободная поверхностная энергия
на границе твердое тело —жидкость имеет большее значение,
чем на границе твердое тело— газ, поверхность твердого тела
будет противодействовать растеканию на ней жидкости. Это бу-
дет определяться следующим неравенствам:
°т-г < °т—ж
В этом случае угол 6 будет больше 90°, а обе 9 < 0 (рис. 55,5),
капля не смачивает поверхности твердого тела.
' Полной несмачиваемОсти отвечает условие:
0 = 180°; cosO = — 1
Условия сМаЧйваеМосТи и смысл краевого угла, Как указы-
валось ранее, становятся более понятными при рассмотрении их
0 точки зрения работы адгезий между Твердым телом и жидко-
стью.
Поскольку достаточно точных методов измерения и оценки
Величин поверхностного натяжения на границе с твердым те-
лом нет, то для определения работы адгезий на границе Твердое
тело — жидкость Пользуются следующим уравнением Юйга, в
котором .поверхностное натяжение твердого ТеЛа исключено С
помощью уравнения Дюпре (1) и Лапласа (2):
^Т-Ж = 0Ж-Г (1 4“ CDS 6) (3)
/ Уравнение (3) дает возможность, зная краевой угол И поверх-
Постное натяжение на границе жидкость — газ, вычислить ве-
личину работы адгезии между твердым телом и жидкостью.
Как указывалось выше при условии полного смачивания
краевой угол отсутствует или его значение равно нулю. В этом
136
случае по уравнению
^т-ж “= 2о>к—г
т. е. работа когезии жидкости равна работе адгезии между твер-
дым телом и жидкостью.
Краевой угол, равный 90°, означает, что адгезия между твер-
дым телом и жидкостью вдвое меньше ее когезии:
^г-ж — г
При краевых углах, близких к 180°, имеет место полное не-
смачивание. Адгезия в этом случае должна отсутствовать. Од-
нако практически угол в 180° невозможен, так как в. какой-то
степени адгезия всегда существует. Краевой угол характеризует
«избирательность смачивания» для тех случаев, когда с поверх-
ностью твердого тела соприкасаются две жидких фазы несме-
шивающихся жидкостей.
• Если жидкость — вода, можно количественно характеризо-
вать степень гидрофильности или гидрофобности поверхности
твердого тела, придавая некоторую условность этим терминам.
Из рис. 55 видно, что краевой угол у гидрофильных поверх-
ностей острый, величина его меньше 90°, а величина cos 6 изме-
няется в пределах от 0 до +1.
У гидрофобных поверхностей величина краевого угла колеб-
лется в пределах от 90° до 180°, а величина cos 6 изменяется в
пределах от 0 до —1 (рис. 55,5).
Поверхности твердых тел переходного типа (рис. 55,2)
имеют краевой угол ~90°.
В качестве примера можно указать, что к гидрофильным
. веществам относятся карбонаты, сульфаты, окислы, многие си-
ликаты, слюда и др., т. е. вещества,^имеющие полярный характер.
Примерами гидрофобных веществ являются: уголь, сажа, се-
ра, парафин, металлы, т. е. неполярные вещества, поверхности
которых хорошо смачиваются неполярными, углеводородными
жидкостями.
Следует отметить, что многие твердые тела, являясь по су-
ществу гидрофобными, благодаря легкой окисляемое™, при из-
мерении краевого угла показывают некоторую гидрофильность.
Так, металлы по своей молекулярной структуре, имея гомеопо-
лярную атомную решетку — типично гидрофобные тела, однако
поверхность металлов почти всегда покрыта тончайшей плен-
кой окислов, благодаря чему поверхность металла в той или
иной степени оказывается гидрофильной. Для того, чтобы уда-
лить окисную пленку, необходимо путем обработки (шлифовки)
обнажить свежую металлическую поверхность.
Наиболее надежным способом получения чистой, неизменен-
ной твердой поверхности является раскалывание кристалла.
437
Природу твердой поверхности можно изменить и превратить
гидрофильную поверхность в гидрофобную, а гидрофобную в
гидрофильную. Для этого нужно создать на поверхности смачи-
ваемого твердого тела адсорбционный слой. Так, если мы возь-
мем пластинку алюминия, у которой поверхность покрыта плен-
кой окиси алюминия, то поверхность этой пластинки будет уже
не гидрофобной, а гидрофильной, т. е. будет довольно хорошо
смачиваться водой.
С другой стороны, бентонит гидрофильный в обычном состоя-
нии становится гидрофобным при прибавлений к воде неболь-
ших количеств аминов.
Прибавление поверхностио-активных веществ к воде, в кото-
рой находится гидрофобная поверхность, приводит к тому, что
на поверхности твердого тела образуется адсорбционный слой
ориентированных дифильных молекул поверхностно-активных
веществ, причем неполярная группа молекул (углеводородные
радикалы) обращена к поверхности, а полярная группа (ОН,
СООН, NH2) направлена в сторону полярной жидкости — воды.
Такая ориентация молекул связана со стремлением системы
уменьшить свободную поверхностную энергию (правило уравни-
вания полярностей Ребиндера).
Благодаря ориентации молекул в адсорбционном слое, на
поверхности происходит обращение угла смачивания или так на-
зываемая инверсия смачивания.
Возможность изменять природу поверхности твердого тела и
придавать ей в зависимости от ряда условий гидрофобность или
гидрофильность имеет большое практическое значение в самых
различных областях промышленности.
Выполнение работы
Для измерения краевых углов было предложено довольно
много методов, так как точное измерение встречает ряд трудно-
стей. Эти трудности связаны, главным образом, с явлением
статического и динамического гистерезиса сма-
чивания. Гистерезисом смачивания называется откло-
нение величины краевого угла от значения его, соответствую-
щего состоянию равновесия капли.
Статический гистерезис вызывается статическим
трением по периметру капли, препятствующим ее растеканию.
При натекании капли жидкости на смачиваемую поверхность
краевой угол значительно больше, чем при оттекании. Разность
краевых углов, лежащих в пределах между минимальным уг-
лом оттекания и максимальным углом натекания и будет ста-
тическим гистерезисом.
Гистерезис, зависящий от времени, которое необходимо для
того, чтобы капля жидкости, нанесенная на поверхность твер-
138
дого тела, приняла равновесное положение, Называется дина-
мическим гистерезисом.
Из методов определения углов смачивавия наибольшее рас-
пространение получил «метод пузырьков», который заключается
iB томг что испытуемый образец помешается в исследуемую
жидкость, а под поверхность образца подводится пузырек воз-
духа. В этом случае ошибки, происходящие за счет гистерезиса,
меньше, чем в случае, когда на твердую поверхность наносят
каплю жидкости.
Определение угла смачивания по указанному методу произ-
водится путем проектирования пузырька воздуха на экран у
данной поверхности твердого тела. Для этой цели образец в
виде небольшой тонкой пластинки, имеющей гладкую поверх-
ность*, предварительно хорошо промытой водой, приклеивают
(с помощью парафина, сургуча или 3%-ного раствора’киноплен-
ки в ацетоне) к металлической лапке держателя. Л^ожно поль-
зоваться проволочным держателем, куда образец вставляется
без клея.
Закрепленный образец помещают в стеклянную кювету с ис-
следуемой жидкостью. Кювета склеивается из плоско-парал-
лельных, прозрачных пластин стекла. Перед употреблением кю-
вета должна быть хорошо вымыта и. сполоснута исследуемой
жидкостью. Образец не следует погружать слишком глубоко в
жидкость во избежание влияния гидростатического давления на
форму пузырька. Пузырек воздуха подводится с помощью ми-
кропипетки с загнутым концом. Можно пользоваться медицин-
ским шприцем, в который вставляют вместо обычной иголки
загнутый стеклянный капилляр. В обоих случаях перед употреб-
лением кончик капилляра должен быть хорошо промыт. Уста-
новка для определения краевых углов изображена на рис. 56.
В качестве источника освещения удобно пользоваться проек-
ционным фонарем или фильмоскопом. Включив источник осве-
щения, проектируют пузырек воздуха на экран. Проекция об-
разца должна быть достаточно отчетливой, края пузырька дол-
жны быть резкими и неразмытыми, что достигается установкой
на резкость изображения перемещением линзы, проектора и
экрана. Во избежание влияния гидростатического давления пу-
зырек не должен быть велик.
Контур пузырька аккуратно и точно обрисовывается остро
отточенным карандашом на листе миллиметровой бумаги, при-
крепленном к экрану, а еше лучше фотографируется.
Для вычисления краевого угла 5 и 'его cos можно пользо-
ваться двумя методами.
По первому методу к контуру пузырька проводят касатель-
ную в точке соприкосновения трех фаз—-у основания пузырька.
* В случае шероховатой поверхности образец может быть отшлифован.
139
Полученный при этом угол, направленный в сторону жидкости,
и будет краевым углом 6.
Рис. 56. Установка для измерения краевых углов.
Касательную в некоторых случаях удобнее проводить, поль-
зуясь зеркалом следующим образом: небольшое прямоугольное
зеркало ставят ребром в точку 2 кри-
4вой (рис. 57), к которой следует
—s. провести касательную. Поворачивая
{ \ s' зеркало около этой точки (точка со-
( \3прикосновения трех фаз), достигают
\ такого его положения, когда отраже-
2\ ние контура кривой в зеркале составит
\ не ломаную, а непрерывную кривую.
\ Очертив на рисунке положение ребра
\ зеркала в виде прямой линии 7, полу-
\ ‘ чают направление нормали к кривой
\ в данной точке. Проведенный к ней
' перпендикуляр и будет касательной,
Рис. 57. Схема определения определяющей краевой угол 0. Числен-
касательной с помощью • ное значение величин краевого угла
зеркала. определяют, пользуясь транспортиром.
Соответствующие значения cos 6 на-
ходят по таблицам тригонометрических величин. Данные запи-
сывают в таблицу 1, приводя значения для всех измерений.
В каждом случае следует измерить углы для 3—4 пузырьков,
140
записав в таблицу 1 значения краевых углов ДЛЯ Каждого пу-
зырька, и подсчитать среднее значение краевого угла.
Таблица 1
Исследуемое твердое тело Состав и концен- трация раствора Углы смачивания Среднее значение cos в W т-ж
изме- рение 1 изме рение 2 изме- рение 3
Стекло Вода 33 35 34 34 4-0,82 132
Бакелит . . . Вода 84 84 81 83 4-о,12 81,7
При исследовании влияния поверхностно-активных веществ
при различных концентрациях измерения краевого угла произ-
водят сначала в чистой воде, а затем в
активного вещества (начиная с малых
концентраций). По полученным дан-
ным вычерчивают изотерму смачива-
ния, откладывая по оси абсцисс кон-
центрации поверхностно-активного ве-
щества, а по оси ординат значение
cos 6, нанося вверх от нуля положи-
тельные значения cos 6, вниз — отрица-
тельные (рис. 58).
Работу адгезии рассчитывают,
пользуясь уравнением (3), подставляя
в уравнение среднее значение cos б
^т-ж = =ж-г(1 + со8 0)
Значения для поверхностного натя-
растворе поверхностно-
Рис. 58. Ход кривой инвер-
сии смачивания.
жения на границе жидкость — газ при
температуре опыта берут из справочных таблиц. Поверхностное
натяжение для воды на границе с воздухом при / = 20° С рав-
но 72, 75 эрг/см2 (72, 75- 10-3 дж/м2).
Работа 25
ОПРЕДЕЛЕНИЕ СМАЧИВАЕМОСТИ ПОРОШКОВ ПО ИЗМЕРЕНИЮ ДАВЛЕНИЯ
ВЫТЕСНЕНИЯ
. Смачиваемость твердого тела жидкостью определяется соот-
ношением между когезией жидкости и. адгезией ее к твердому
телу. Указанное соотношение может быть охарактеризовано ве-
личиной адгезионного натяжения, под которым пони-
мается следующее выражение:
- ' ' Ат~ж “ — аж-г (4)
141
Где Лт-ж — адгезионное натяжение;
W'.,-.,, — работа адгезии жидкости к твердому телу;
о.ж_,—поверхностное натяжение на границе жидкость —
газ, равное, как известно, половине работы когезии.
В том случае, когда соприкасающимися фазами являются
твердое тело, жидкость и газ, связь между /1т-ж и углом сма-
чивания может быть получена, исходя из уравнения (3) (обо-
значения соответствуют принятым , в работе № 24):
Ат_ж = °ж—г cos 6 (5)
Уравнение (5) формально неразрешимо, если 6 = 0. Угол
6 = 0 не только в случае, когда адгезия жидкости к твердому
телу равна когезии жидкости, но и когда адгезия превышает
когезию. Во втором случае уравнение (3) перестает быть спра-
ведливым, вследствие чего и уравнение (5) также не может
быть использовано для определения адгезионного натяжения
как произведения ож_г cos 6.
В том случае, когда соприкасающимися фазами являются
твердое тело и две не смешивающиеся между собой жидкости,
связь между величинами адгезионных натяжений и углом сма-
чивания может быть определена следующим образом.
Согласно уравнению Лапласа можно написать:
°жг—г ’ COS 6жг = °т—г °т—ж«
_ (6)
°жн- г ' cos “жи — ст—г ат-жн
°ЖС-ЖИ cos ^жс-жн ~ °Т-ЖН °т-жс
где 6И( —угол смачивания на границе раздела твердое тело,
воздух, смачивающая жидкость;
6и<н — угол смачивания на границе раздела твердое тело —
воздух — несмачивающая жидкость;
6жс-жн—угол смачивания на границе раздела твердое тело —
смачивающая — несмачивающая жидкость, измерен-
ный в сторону смачивающей жидкости.
Термины «смачивающая жидкость» (индекс «с») и «несмачи-
вающая жидкость» (индекс «н») имеют условный характер: в
действительности первая смачивает лучше второй.
Соотношение между величинами адгезионных натяжений и
углом смачивания определится совместным решением приведен-
ной системы уравнений (6), с учетом, что
•^Жс —Т ~ аЖс — Г COS fljKg
•^Жй -Т — °ЖН—Г COS 0>кн
Таким образом:
4жс-т — -4жн-т = 5ж^-жн'СОЗ 6Жс_Жн (7)
142
В капилляре вследствие наличия мениска на границе фаз, су-
шествует определенное капиллярное давление, величина
рого зависит от угла смачивания (см. стр. 99).
Под влиянием капиллярного давления жидкость,
смачивающая капилляр, вытесняет несмачиваю-
щую. Чтобы воспрепятствовать перемещению
фаз, необходимо, приложить к системе давление,
равное капиллярному по величине, но направ-
ленное в противоположную сторону. Это проти-
водавление называется давлением вытес-
нения. Очевидно, что давление вытеснения свя-
зано с адгезионным натяжением.
Порошковые диафрагмы являются капилляр-
ными системами и, следовательно, определение
смачиваемости порошка по измерению давления
вытеснения может быть использовано для опреде-
ления их смачиваемости различными жидкостями.
Выполнение работы
кото-
Для измерения давления вытеснения употреб-
ляется прибор, показанный на рис. 59.
Прибор состоит из следующих деталей: стек-
лянной цилиндрической трубки /, с впаянной в
нее перфорированной стеклянной пластинкой 3,
отводами 4, 5, 6 и краном 2; стеклянной цилинд-
рической трубки 7 с краном 3; градуированного
капилляра 9; перфорированной пластинки 10;
вставляемой в трубку 1.
Рекомендуется следующий порядок работы
Прежде всего определяется эффективный радиус
пор капилляров порошковой диафрагмы. С этой
целью измеряется давление вытеснения воздуха
жидкостью, полностью смачивающей поверх-
ность порошка (cos0= 1). В случае силикатов
в качестве такой жидкости можно взять воду.
Заполнение прибора и измерения производятся
следующим образом. Часть прибора — трубка 1
устанавливается в вертикальном положении.
Фильтр 1(7 вынут из прибора. Отводы 4 и 5 за-
крыты. Пластинка 3 покрывается кружком филь-
тровальной бумаги. Затем на пластинке форми-
Рис. 59. Прибор для определения смачиваемости порош-
ков по давлению вытеснения.
7
руется диафрагма из сухого порошка. Для этого порошок вводится
в трубку небольшими порциями и после введения каждой порций
143
тщательно утрамбовывается стеклянной палочной. Формирование
диафрагмы производится до тех пор, пока верх мембраны не бу-
дет лишь немного ниже отвода б. Сверху диафрагма плотно при-
” жимается фильтром 10. К прибору присоединяется часть 7 и все
свободное пространство над диафрагмой и в части 7 запол-
няется данной жидкостью с помощью отвода 6, после чего по-
следний закрывается. Краны 2 и 8 открываются. Очевидно, что
жидкость, находящаяся в верхней части прибора начнет пропи-
тывать диафрагму. Пропитка производится до отвода 5. Окон-
чание пропитки легко определить визуально. Затем краны 2 и 8
закрываются. Прибор устанавливается в горизонтальное поло-
жение, к нему присоединяется капилляр 9, заполненный данной
жидкостью, и производится измерение давления вытеснения в
системе твердое тело — воздух — жидкость. Для этой цели при-
бор соединяется через кран 2 с манометрической установкой,
оба крана открываются и на систему подается давление. Давле-
ние вытеснения измеряется тем предельным давлением, которого
еще недостаточно, чтобы мениск в капилляре начал двигаться
в сторону, противоположную самопроизвольному вытеснению.
Очевидно, что величина этого давления определяется соотноше-
нием величин адгезии между твердым телом и соприкасающи-
мися с ним фазами. Для получения точных данных показания
следует снимать не тотчас после подачи давления, а чёрез неко-
торое время, когда установится стационарное состояние. Радиус
капилляров диафрагмы вычисляется по формуле (стр. 99):
2а COS 0Ж„
и с I
-dgr . '
где г — эффективный радиус капилляров диафрагмы;
о — поверхностное натяжение на границе жидкость — газ;
А — разность высот в манометре;
d — плотность жидкости в манометре;
g — ускорение силы тяжести;
6—угол смачивания.
В данном случае cos 6= 1.
Следующей стадией работы является определение адгезион-
ного натяжения. Для проведения необходимых измерений при-
бор после определения эффективного радиуса пор диафрагмы
-разбирают, освобождают от порошка, моют и тщательно высу-
шивают. Прибор заполняют таким же образом, как при опреде-
лении г, но в качестве жидкой фазы берут исследуемую жидкость.
Таким же образом, как и раньше измеряют давление вы-
, теснения. Зная из предыдущего опыта г, легко вычислить вели-
чину сж-г • cos 6 = В (8). Полученная величина В может быть
приравнена адгезионному натяжению только в том случае, если
cos 9 I. Следовательно, чтобы судить о том, можно ли вели-
чину В считать равной величине адгезионного натялсения, сле-
Н4 .
(8)
дует определить cps0. С этой целью в отдельном опыте изме-
ряют Сж-г и определяю-? cos 6:
' о В
COS и =2=--------
°ж-г
Еслисозб =£ 1, то В = Дт_ж. Если же cos 0= 1, т. е. исследуемая
жидкость полностью смачивает порошок, определить адгезион-
ное натяжение по результатам этого опыта невозможно. По-
этому, в данном частном случае, для определения величины ад-
гезионного натяжения следует поступать следующим образом:
сначала подбирается любая жидкость, которая не полностью
^смачивает поверхность порошка (cos 6 =/= 1) и не растворима в
•исследуемой жидкости.
”. Для силиката в качестве такой жидкости можно взять а
бром-нафталин. Измерив по вышеописанной методике давление
вытеснения в системе твердое тело — воздух—несмачивающая
жидкость, по формуле (8) определяют величину Дкн-т — адге-
зионное натяжение несмачивающей жидкости к твердому телу.
Затем прибор разгружают; промывают, тщательно высуши-
вают и заполняют снова, но по несколько иной методике. Сна-
чала формируют диафрагму до отвода 5 (рис. 59) т. е. часть
пространства трубки 1, обозначенная б. Формирование диафраг-
: мы производится так же, как описано выше. Часть диафраг-
мы б пропитывается несмачивающей жидкостью, т. е. той, с ко-
торой проводился предыдущий опыт. После того, как пропитка
данной части диафрагмы закончена, часть в через отвод 4 за-
полняется этой, не полностью смачивающей порошок, жидко-
стью; кран 2 и отвод 4 герметически закрывают. Лишняя жид-
кость, находящаяся в верхней части прибора над диафрагмой,
удаляется и верхняя часть прибора тщательно высушиваете?
фильтровальной бумагой. Затем'на сформированной части ди-
афрагмы формируют новую часть а до тех пор, пока, ее верх
не будет лишь несколько ниже отвода б. Сверху диафрагма
плотно прижимается фильтром 10, к прибору присоединяют
часть 7 и производят пропитку этой части диафрагмы исследуе-
мой жидкостью. С этой целью верхнюю часть пространства над
диафрагмой и часть 7 заполняется исследуемой жидкостью (с
‘помощью отвода 6 и крана 8) и отвод 6 закрывают. Пропитка
производится при открытых кране 8 и отводе 5. Убыль жидко-
сти в верхней части прибора, происходящая за счет пропитки,
должна быть восполнена. Прописка части а производится до
тех пор, пока верхняя жидкость не дойдет до границы порошка,
пропитанного несмачивающей жидкостью. Воздух, находящийся
первоначально в порах диафрагмы а вытесняется через отвод 5.
Окончание пропитки легко определить как визуально, так и по
появлению исследуемой жидкости в отводе 5. По окончании про-
питки отвод 5 и кран 8 закрывают и к прибору присоединяют
145
капилляр 9. Прибор устанавливают в горизонтальном положении
и производят измерение давления вытеснения системы твер-
дое тело — несмачивающая жидкость — смачивающая жидкость.
Методика измерения давления вытеснения та же, что и в преды-
дущих опытах. Зная давление вытеснения данной системы, г и
Аки-т, легко определить искомую величину адгезионного натя-
жения. Действительно, подставив найденные значения h и г в
формулу (8), можно определить величину °жс-жн • со5 0Жс-жн- Со-
гласно формуле (7) искомая величина адгезионного натяжения
может быть найдена теперь по соотношению:
^жс-т Акн-т + °жс-жн cos ®жс-жн
Результаты измерения записываются в виде следующей таб-
лицы.
Таблица 2
А.,, °,,. -cos В А ,
Жц— Т — Жд '“с—т
apzjcM1 эрг!см* эрг[см2
Рассмотренный выше метод определения смачиваемости по-
рошков по измерению давления вытеснения не может претендо-
вать на большую точность. Для получения точных значений ад-
гезионного натяжения должен быть учтен гистерезис смачивания.
Последний особенно резко выражен в случае образования на
твердой поверхности адсорбционного слоя растворенных ве-
ществ. Однако, если исследуется избирательное смачивание
твердых поверхностей чистыми жидкостями; не показывающими
значительного гистерезиса смачивания, метод дает возможность
судить о величинах адгезионного натяжения между разного ро-
да жидкостями и твердым телом.
Работа 26
ОПРЕДЕЛЕНИЕ ТЕПЛОТЫ СМАЧИВАНИЯ ТОНКОИЗМЕЛЬЧЕННЫХ ВЕЩЕСТВ
Степень смачиваемости порошкообразных и пористых адсор-
бентов можно определить, измеряя (калориметрически) теплоту
смачивания.. Теп л отой смачивания называется то коли-
чество тепла, которое выделяется при ьогружении 1 е адсор-
бента в смачивающую жидкость.
Можно определять интегральную (полную) теплоту
смачивания, под которой понимают общее количество те-
145
ила, выделенное при смачивании 1 а измельченного вещества
избытком жидкости и дифференциальную теплоту
смачивания, выражающую собой тепловой эффект, отнесен-
ный к последовательно добавляемым к адсорбенту порциям
смачивающей жидкости.
Между величиной теплового эффекта и химической природой
твердого вещества и жидкости существует ясно выраженная
связь. При данной степени дисперсности теплота смачивания
тем больше, чем больше сродство жидкости к данной поверх-
ности, т. е. чем меньше разность полярностей между дисперсной
фазой и жидкостью.
Поэтому полярные (гидрофильные) порошки имеют больший
тепловой эффект в полярных жидкостях, например в воде. Те-
пловой эффект смачивания таких порошков в углеврдородах
сравнительно невелик (см. табл. 3).
Таблица 3
Жидкость Тепловой ффект, кал!г *
глина кварц уголь
Вода . . . .- Четыреххлористый уг- 12,6 15,6 5,9
лерод ........ 1,8 8,1 20,0
* Для перевода в систему СИ (дж[г) величину в калориях
надо умножить на 4,186.
Большим недостатком обычных измерений теплоты смачива-
ния является то, что этот параметр относят к единице массы
твердых адсорбентов, тогда как следовало бы относить его к
единице поверхности. Чтобы получить сравнимые величины ну-
жно знать степень дисперсности и удельную поверхность (для
исключения влияния величины удельной поверхности). Ребинде-
ром был предложен коэффициент р, дающий термическую ха-
рактеристику гидрофильности поверхности твердого тела. Коэф-
фициент р является отношением значений теплоть?смачивания од-
ного и того же твердого вещества в воде Qi и в углеводороде Qz".
' Для гидрофильных порошков этот коэффициент больше еди-
ницы, для гидрофобных — меньше. Величиной 3 удобно пользо-
ваться, так как она не зависит от дисперсности порошка. Если
5 — общая поверхность дисперсной системы, (в кал) — тепло-
та смачивания 1 см2 поверхности порошка водой, (в кал) —
147
теплота смачивания 1 см2 бензолом, то:
Qi = Sqi и Q2 = Sq2
Разделив одно выражение на другое, получаем
(11)
V2 42
т. е. отношение величин теплот смачивания 1 см2 поверхно-
сти порошка не зависит от дисперсности.
Процесс выделения тепла вызывается тем, что при смачива-
нии происходит замена поверхности раздела твердое тело —
воздух с высоким значением полной поверхностной энергии
(уравн. стр. 91) поверхностью твердое тело — жидкость с отно-
сительно низким значением поверхностной энергии.
Если удельная поверхность твердого тела S см21г, ет-г — пол-
ная поверхностная энергия на границе твердое тело — газ (на
1 си2), ет-ж — полная поверхностная энергия на границе твердое
тело — жидкость, то теплоту смачивания можно выразить с по-
мощью уравнения:
Q = S (ет_г ет—ж)
В некоторых случаях можно вычислить теплоту смачивания,
пользуясь значением поверхностной энергии, т. к. точные значе-
ния е неизвестны.
Если предположить, что имеет место полное смачивание (т. е.
6 = 0 и cos6= 1), то уравнение 2 на стр. 136 перейдет в урав-
нение Ст—г = Ст—ж "Ь Сж—г*
. Для этого случая можно заменить ст-г — ст-ж в этом урав-
нении величиной-сж-г на границе раздела жидкость — воздух.
Переходя от свободной к полной поверхностной энергии, мо-
жно принять, что:
с Q .
ет—г ет—ж — еж—г ИЛИ о — — ,
6ж—г
Q — бж-г • •S
Для водьк полная поверхностная энергия еж-г равна
.117 эрг/см2 (0,117 дж/м2). Зная теплоту смачивания, можно вы-
числить удельную поверхность. .
Несмотря на трудности в пбстроении полной теории теплоты
смачивания, экспериментальные исследования в этой области
представляют большую ценность, давая непосредственное пред-
ставление об энергетике' взаимодействия жидкости с твердым
телом, характеризуя порошки с точки зрения их гидрофильно-
сти или гидрофобности и величины адгезии.
Измерение теплот смачивания имеет поэтому большое зна-
чение в различных отраслях промышленности.
148
Определение теплоты смачивания порошков с большой удель-
ной поверхностью (десятки и сотни м2/г) может быть осуще-
ствлено в калориметрах сравнительно простой конструкции. Из-
мерение теплот смачивания грубодисперсных систем с малой
удельной поверхностью требует применения высокочувствитель-
ных микрокалориметров.
Выполнение работы
Для измерения теплоты смачивания пользуются калоримет-
рами самых разнообразных конструкций. В лаборатории кол-
лоидной химии ЛГУ применяют весьма
^простой прибор типа калориметра Шот*
тки. Калориметр (рис. 60) состоит из со*
суда — кожуха 7 (дюаровский сосуд), по*
решенного в термостат 8. В сосуд вста*
Является второй сосуд 6 с капилляром и
^шкалой 1 и боковым отростком 3 с кра-
ном и воронкой. Во внутреннем сосуде
^имеется вплавленная пробирка. Внутрен-
ний сосуд наполняется жидкостью с
большим коэффициентом термического
расширения (толуол или хлороформ),
а сверху водным раствором какого-ни-
будь красителя, для облегчения наблю-
дений по капиллярной шкале.
Шкалу градуируют по теплоте ней-
трализации для того, чтобы получить
цену одного деления шкалы в калориях.
В пробирку 5, находящуюся во внутрен-
нем сосуде \6, вставляют вторую пробир-
ку 4 с возможно, малым зазором между
стенками — так, чтобы она свободно
скользила внутри вплавленной про-
бирки. Определив истинный температур-
ный ход калориметра, как это обычно
делают при калориметрических измере-
ниях, определяют теплоту смачивания
Рис. 60. Прибор для опре-
деления теплоты смачи-
вания порошков.
испытуемого вещества, быстро всыпая
навеску в жидкость, помещенную ,в.о
внутреннюю пробирку 4, и перемешивая
порошок с помощью ручной мешалки 2.
Вследствие теплового эффекта окра-
шенный раствор поднимается по капилляру, что дает возмож-
ность, наблюдая за положением мениска в капилляре и учиты-
вая тепловой ход прибора, определить теплоту смачивания 1 г
порошка.
149
Рис. 61. График для определения скачка
температуры.
Измерение теплоты смачивания удобно проводить в следую-
щем порядке: оставляют подготовленную к опыту жидкость во
внутренней пробирке 4 на 2—3 ч при определенной температуре
для того, чтобы в приборе установилось температурное равно-
весие.
Наблюдают температурный ход прибора, т. е. изменение по-
казаний термометрической шкалы во времени. Определение тем-
пературного хода необходи-
мо, так как избежать пол-
ностью теплообмена калори-
метра с окружающей средой
невозможно.
Истинный скачок темпе-
ратуры можно найти графи-
чески, путем экстраполяции
линейного хода температур-
ной кривой до и после изу-
чаемого процесса.
Наблюдаемые во время
опыта отсчеты по термоме-
трической шкале делятся на
три периода: первый период
до смачивания — ~ 5 мин,
второй — основной период,
во время которого выде-
ляется теплота смачивания, .
и . третий — после смачи-
вания— также ~5 мин
(рис. 61).
Для определения значе-
ния одного деления шка^ы
(в калориях) температурную шкалу градуируют по теплоте
нейтрализации.
При нейтрализации пользуются 0,1 н. НС1 и 0,1 н. NaOH с то-
чно установленным титром. Известно, что нейтрализация 1 г-экв
любой сильной кислоты любым сильным основанием в разбав-
ленных растворах сопровождается одним и тем же тепловым
эффектом, равным 13700 кал!моль*\ 5 см3 0,1 н.НС1 наливают
в пробирку 4 (рис. 60), а в отдельную запасную пробирку на-
ливают 0,1 н. NaOH с таким расчетом, чтобы в результате ней-
трализации 5 см3 0,1 н. НС1, вся кислота была нейтрализована
и остался небольшой избыток щелочи. Растворы кислоты и ще-
лочи следует до опыта выдержать некоторое время при опреде-
ленной температуре в термостате. Нейтрализацию кислоты про-
изводят, приливая Щелочь к кислоте и отмечая показания шка-
* В системе СИ 57 348 дж[моль.
150
лы во времени. Время наблюдают по секундомеру, определив
предварительно температурный ход прибора.
Установив по шкале высоту поднятия, соответствующую те-
плоте, выделенной при нейтрализации, зная значение теплоты
нейтрализации кислоты щелочью (13 700 кал/моль*) и учиты-
вая температурный ход до нейтрализации и после, определяют
цену одного деления шкалы в калориях.
Для определения теплоты смачивания порошка в чисто вы-
мытую и высушенную пробирку наливают жидкость (10 см3),
смачивающую порошок, например дистиллированную воду, и
выжидают (2—3 ч), пока система придет в равновесие.
Одновременно в отдельную пробирку помещают навеску вы.-
сушенного до постоянного веса при 110° С порошка. После на-
ступления равновесия и повторного определения температурного
хода порошок быстро высыпают в пробирку с жидкостью, произ-
водя перемешивание мешалкой и наблюдая каждые 30 сек за
поднятием жидкости в капилляре. Наблюдения производят до
тех пор. пока жидкость в капилляре не начнет равномерно опу-
скаться.
Определив графически истинный температурный скачок, вы-
числяют теплоту смачивания, отнесенную к 1 а порошка.
Подобные же измерения производят с неполярной жидко-
стью, например с бензолом.
Поправка на теплообмен, происходящий между калоримет-
ром и окружающим пространством, делается следующим об-
разом.
- На график (рис. 61) наносят значения показаний термомет-
рической шкалы до опыта и во время опыта. По оси ординат
откладывают показания шкалы, по оси абсцисс — время в мину-
тах. На графике определяют момент конца равномерного изме-
нения температуры до введения навески и начала постоянного
хода после смешения. Промежуток времени между этими момен-
тами делят пополам и через полученную точку проводят прямую
перпендикулярно оси абсцисс. Экстраполируя участки равно-
мерного хода температуры до и после смешения до пересечения
с прямой, получают отрезок At (в делениях условной шкалы),
отвечающий истинному температурному скачку.
Таблица 4
Исследуемое вещество Теплота смачивания водой, кал!г Теплота смачивания неполярной жидкостью, кал(г Коэффициент Ь*12
SiO2 9,1 4,4 2,1
Уголь ....... 5,9 16,0 0,37
151
Рассчйтав теплоту смачивания в калориях на 1 а испытуе-
мрго порошка в воде, а затем в неполярной жидкости (напри-
мер, бензоле), характеризуют смачиваемость порошка коэффи-
циентом р12 [11].
Полученные данные записывают в виде таблицы (табл. 4).
Работа 27
ИССЛЕДОВАНИЕ ПРОЦЕССА ФЛОТАЦИИ: ВЛИЯНИЕ ФЛОТОРЕАГЕНТОВ НА
ПРОЦЕСС ФЛОТАЦИИ; СВЯЗЬ МЕЖДУ ВЕЛИЧИНОЙ КРАЕВОГО УГЛА
И ФЛОТИРУЕМОСТЬЮ
Флбтация представляет собой процесс обогащения полез-
ных ископаемых, основанный на избирательном прилипании
тонкоизмельченных частиц минералов к поверхности раздела во-
да — воздух, вода — масло или вода — твердое тело. Этот про-
цесс получил широкое распространение в промышленности и в
настоящее время используется не только для обогащения прак-
тически всех добываемых минералов, но и при очистке воды, со-
держащей небольшие количества целлюлозы из отходов бумаж-
ного производства, при очистке сточных вод, а также для извле-
чения органических смол из растительных продуктов.
Различают четыре вида флотации:
1. При пенной флотации минеральные частицы опреде-
ленного состава прилипают к пузырькам воздуха, поднимаю-
щимся в водной суспензии данного минерала (пульпе). Прилип-
шие частицы уносятся в поверхностный слой пульпы, где обра-
зующаяся пена с частицами минерала снимается и разрушается;
частицы другого состава не прилипают к пузырькам и остаются
в пульпе.
2. При пленочной флотации минеральные частицы
тонким слоем наносятся на движущуюся поверхность воды. Ча-
стицы, хорошо смачиваемые водой, тонут,, а другие, хуже смачи-
ваемые, остаются на поверхности воды и собираются.
•3. В процессе масляной флотации частицы минерала,
смачиваемые маслом, прилипают к капелькам масла и выно-
сятся наверх, тогда.как частицы, смачиваемые водой, остаются
в пульпе.
4. Четвертый вид флотации — так называемая флотация
твердой стенкой. В пульпу вводится твердая поверхность,
обработанная специальными реагентами, к которой избиратель-
но прилипают частицы определенного минерального состава, то-
гда как другие частицы остаются в пульпе.
Следует отметить, что в настоящее время наибольшее рас-
пространение получила пенная флотация.
Так как включения ценных минералов в порода ,тёсно свя-
заны друг с другом и с минералами породы, то перед разд еде-
153
пенной флотации необходимо,
воду с поверхности минерала;
Рис. 62. Соотношение поверхно-
стных сил для системы твердое
тело — жидкость — газ.
нием всю массу руды подвергают измельчению в мельницах,‘вы-
деляют в классификаторах частицы определенного размера, ко-
торые затем и поступают во флотационные машины.
Флотация, как и большинство других методов разделений ве-
ществ (кристаллизация, перегонка), основанных на различии в
физических свойствах, не дает возможности при практическом
осуществлении полного (100%) извлечения ценного минерала.
Поэтому обычно применяется фракционная флотация, при кото-
рой концентраты, получаемые при флотации, подвергаются допол-
нительной флотации, дающей концентрат более высокого качества.
' Для осуществления процесса
чтобы воздух частично вытеснял
для процесса масляной флотации
необходимо, чтобы масло вы-
тесняло воду; пленочная флота-
ция возможна в том случае, если
Вода , не может полностью вытес-
нить воздух с минеральной по-
верхности.
Рассмотрим случай вытесне-
ния воды с поверхности твердого
тела воздухом, т. е. случай,
имеющий место при пенной фло-
тации (рис. 62). Работа адгезии
или иначе прилипания пузырька воздуха к твердой поверхности
равняется:
^Т-Г — °Т-Ж ~Ь °Ж — г °т—Г (12)
Решая это уравнение совместно с уравнением Лапласа (2)
°т—г ~ °т—ж ~Ь °ж—г cos ®
Получаем:
Гт_г = аж_г(1-СО8в) (13)
Этим же выражением определяется величина работы, кото-
рая должна быть совершена для отрыва пузырька воздуха от
Поверхности минерала. Уравнение (13) показывает, что Способ-
ность прилипать к пузырьку воздуха, и флотироваться зависит
от величины краевого угла В.'Если 6=0, то 1^т-г = 0, тогда тен-
денция к прилипанию и флотация отсутствуют. Прн 6 = 180е
прочность прилипания достигает максимума, равного 2сж-г. Сле-
довательно, минерал может обладать флотируемостью при лю-
бом 6, отличном от 0°> Однако, чем больше величина 6, тем
более ярко выражена флотируемость минерала. Отсюда стано-
вится понятной -важность определения краевых углов для про-
цессов флотаций.
В случае пленочной флотации частица минерала при Доста*
Точно малой ее величине остается на поверхности жидкости
153
и не Тонет. Рассмотрим условия, необходимые для осуществле-
ния этого процесса. Разделим процесс перехода твердого тела
из газообразной фазы в жидкую на четыре стадии (рис. 63).
Условие само' роизвольного погружения тела в жидкость
ст_г>ст_Н(. Однако это условие является необходимым, но не
достаточным, так как твердое тело должно пройти границу
раздела жидкость—газ.
Условия, необходимые для протекания каждой стадии со-
стоят в следующем:
от стадии I до стадии II
от стадии II до стадии III
от стадии III до стадии IV
°т—г + °ж-г > °т—ж
°т—г > °т—ж
°т—г > °ж—г4~ат-ж
Отсюда следует, что если возможен переход от стадии III
до стадии IV, то возможен весь процесс погружения тела. Сле-
Рис. 63. Схема процесса перехода твердого тела из газообразной среды
в жидкую.
довательно, существенным условием погружения тела является:
°Т —Г > °Ж —Г *Р °Т—Ж ИЛИ °Т —Г °т-ж > °ж— г
Твердое тело будет плавать на поверхности и не утонет в
том случае, если
°т—г “ °т—ж < °ж—Г
ИЛИ
8т-г“~ °т-ж < |
аж-г
Учитывая, что левая часть неравенства Соответствует cosS,
окончательно получаем:
cos в < 1
Следовательно, условие пленочной флотации твердого тела,
так же как и пенной флотации — наличие конечной величины
краевого угла 6, отличной от нуля.
Приведенный расчет не учитывает гравитационных сил.
В случае, если 6>0, но рассматриваемая частица имеет значи-
тельный вес, она может утонуть в воде, однако условие 6 > О
154
дает принципиальную возможность нахождения твердого тела
на поверхности жидкости.
Из вышесказанного следует, что все факторы, способствую-
щие увеличению краевого угла минералов будут положительно
сказываться на их флотации.
Минералы, подвергнутые измельчению, могут обладать есте-
ственной флотируемостью, либо она может быть вызвана искус-
ственной гидрофобизацией говерхности.
На практике процесс флотации производят в присутствии
флотореагентов, которые по своему воздействию на процесс
флотации могут быть разделены на следующие четыре большие
группы:
1) вспениватели — вещества, способствующие образова-
нию устойчивых пузырьков и пены в пульпе;
2) собиратели — Вещества, которые повышают краевой
угол 6 и тем самым гидрофобизируют поверхность минерала;
3) активаторы — вещества, способствующие закрепле-
нию собирателя на поверхности минерала;
4) депрессоры — вещества, которые в противополож-
ность активаторам препятствуют закреплению собирателя на
поверхности минерала и тем самым ухудшают флотируемость
данного минерала.
Следует отметить, что часто вещество, являющееся актива-
тором в одном конкретном случае, может быть депрессором
в другом случае. Поэтому часто две последние группы объеди-
няются в одну под общим названием регуляторов (модифика-
торов) .
Для флотации большое значение имеет степень гидратиро-
ванности минеральных. частиц. Чем более гидратирована ча-
стица и чем более устойчива гидратная оболочка, тем менее
флотируется частица.
Собирателями обычно являются вещества, молекулы кото-
рых содержат полярную и неполярную группы. Аналогичное
строение имеют и молекулы пенообразователя. Наиболее часто
употребляемыми собирателями являются ксантогенаты с общей
формулой
/OR
C\sm
дитиокарбаматы
/NR2
'^SM
дитиофосфаты
/OR
- S = P—OR
- - \>M
85
»
алкилсульфаты RSO4M и алкилсульфонаты RSO3M, жирные
кислоты, а также амины. (В приведенных формулах R-—орга-
нический радикал, М—металл).
Пенообразователи, применяемые во флотации, обычно
представляют собой масла, полученные при сухой перегонке
дерева, листьев или каменного угля. Они не являются индиви-
дуальными соединениями, а представляют собой смесь терпе-
нов, терпеновых спиртов, камфор.
Благодаря своему дифильному характеру, молекулы собира-
теля располагаются так, что их полярные части направлены
к поверхности минерала, а неполярные в сторону жидкости
(рис. 64,а). Этим и достигается гидрофобизация поверхности.
Рис. 64. Схема закрепления молекул собирателя на поверх-
ности минерала а и молекул пенообразователя на поверх-
ности пузырька б. —
Молекулы пенообразователя, адсорбируясь на поверхности
пузырьков (рис. 64,6), гидрофилизируют поверхность послед-
них и тем самым повышают их устойчивость и степень дисперс-
ности.
В качестве регуляторов обычно применяются соли-тяжелых
металлов, цианиды, сульфид натрия, сода, известь, жидкое стек-
ло и т< п. В одних условиях они действуют как активаторы,
в других являются депрессорами.
Выполнение работы
При исследовании процесса флотации минералов в лабораг
торных условиях обычно используются флотационные аппараты
малых размеров, моделирующие процесс флотации, происходя-
* щий в промышленных условиях. Однако для ускорения и углуб-
ления изучения процесса часто используются способы, не моде*
лирующие полностью промышленный процесс, но позволяющие
выяснить основные закономерности и подробно изучить отдель-
ные факторы, оказывающее, влияние на процесс флотации,
1&6
К ним относится работа с аппаратом Халлимонда, который и
используется в лаборатории коллоидной химии ЛГУ. Аппарат
изготовляется из обычного или органического стекла, прост
по конструкции (рис. 65) и требует для проведения опыта всего
0,2—1 г минерала. Ток воздуха создается за счет жидкости,
перетекающей из бутыли 2 в бутыль 3, воздух проходит через
склянку с раствором NaOH (для очистки от СО2) и затем попа-
вдает в трубку Халлимонда.
Работа производится следующим образом. На аналитиче-
;1ских весах берут 4—5 навесок исследуемого минерала * (по
С0,5—1 г). Затем готовят 4—5 растворов (по 100 см3), отличаю;
?.щихся друг от друга концентрацией собирателя или величиной
pH раствор’а, при постоянной концентрации собирателя и т. п.
Навески минерала переносятся в приготовленные растворы и
в течение 15—20 мин приводятся (при перемешивании) в равно-
весие с растворами й затем поочередно помещаются во флота-
ционный аппарат и подвергаются флотации. .
Частицы минерала перемешиваются в трубке магнитной ме-
‘шалкой 5. Воздух вводится в прибор по капилляру 1 через стек-
лянный фильтр Шотта (№ 2) 4. Частицы минерала прилипают
к поднимающимся пузырькам воздуха и выносятся на поверх-
* В качестве минералов могут быть взяты кварц, барит, кальцит, тальк
и др. Размер частиц минерала ~ 100 мк. Наиболее доступным минералом
.является кварц. Для него условия проведения флотации могут быть сле-
дующие: собиратель — уксуснокислый лауриламин 1—10 мг/дм3, время фло-
тации — 10—20 мин. Процесс активации может быть изучен на системе кварц
См 10—50 мг/дм3 FeCl3, собиратель в этом случае — олеат натрия (1—
/ .10 мг/дм3},
5 157
ность 7. Достигнув поверхности, пузырьки лопаются, а освобо-
дившиеся минеральные частицы собираются в отростке 6. По
окончании опыта (время флотации указывается преподавателем)
отфлотированный минерал, собравшийся в 6 и называемый кон-
центратом и остальную часть, называемую хвостами, собирают
в отдельные, предварительно взвешенные бюксы и высушивают
в сушильном шкафу до постоянного веса. На основании резуль-
татов взвешиваний определяют относительное количество мине-
рала, перешедшего в концентрат (7, %).
7 =
количество перешедшего в концентрат минерала
навеска минерала
100
Аналогичная работа проделывается со следующими навес-
ками минералов. Полученные результаты записывают в таблицу
(табл. 5) и строят на графике зависимость величины 7 от кон-
центрации собирателя, pH или от другого изучаемого фактора.
Таблица 5
№ опыта Условия опыта (концентрация собирателя, pH, время опыта> ' Навеска минерала, мг Вес концен- трата, мг Вес хвостов, мг 1 % Краевой угол 6 или cos 6
После проведения опытов по флотации определяют величи-
ны краевого угла 6 (по методике, описанной в работе 24) на
шлифах исследованного минерала в применявшихся ранее рас-
творах. Полученные значения 6 сравниваются с величинами 7.
ЭМУЛЬСИИ И ПЕНЫ
Эмульсии — дисперсные системы, образованные двумя не
смешивающимися между собой жидкостями, и пены, в кото-
рых дисперсная фаза — газ — распределена в жидкой дисперси-
онной среде, по ряду свойств весьма близки друг к другу,
вследствие чего эти два вида дисперсных систем обычно рас-
сматриваются совместно. Общность многих свойств эмульсий
и пен находит свое отражение и в применяющейся терминоло-
гии: часто название эмульсия применяется только для разбав-
ленных систем, независимо от того будут ли частицы дисперс-
ной фазы являться жидкостью или газом (газовые эмульсии).
Эмульсии и пены широко распространены в природе и нахо-
дят широкое применение в промышленности, например в тек-
стильной, кожевенной, металлообрабатывающей, фармацевти-
158
чёской, Нйщевой, а Также b сельском хозяйстве' й Дорожном
строительстве. Этим обусловливается необходимость количе-
ственной оценки ряда важных технологических характеристик
этих систем. Ниже приводится методика исследования некото-
рых свойств эмульсий и пен.
Эмульсии
Работа 28
ПРИГОТОВЛЕНИЕ КОНЦЕНТРИРОВАННЫХ И ПРЕДЕЛЬНО КОНЦЕНТРИРОВАННЫХ
ЭМУЛЬСИЙ И ИССЛЕДОВАНИЕ ИХ ДИСПЕРСНОСТИ
Основными проблемами изучения эмульсий являются усло-
вия их образования, устойчивости и разрушения.
Эмульсии, подобно другим дисперсным системам, могут
быть получены двумя путями: конденсацией паров одной жид-
кости в другой или диспергированием. Наибольшее распростра-
нение получил второй способ, при котором диспергирование
жидкости производится размешиванием, встряхиванием или
растиранием. Хороший способ получения высокодисперсных
эмульсий — диспергирование с помощью ультразвука.
Если дисперсной фазой является не смешивающаяся с водой
органическая или другая жидкость, которую обычно называют
маслом, система носит название прямой эмульсии и ус-
ловно обозначается М/В. Если дисперсной фазой служит вода,
а дисперсионной средой — масло, система называется обрат-
ной эмульсией и обозначается В/М.
В зависимости от условий получения могут образоваться
эмульсии двух типов: прямые или обратные. При изменении
условий один тип эмульсии может переходить в другой. Такое
явление носит название обращения фаз эмульсии.
Эмульсии могут быть получены почти с любым содержа-
нием дисперсной фазы. При малых концентрациях эмульсии
капли могут свободно перемещаться в среде, тогда как при
очень высоких концентрациях (до 99 объемн. %) жидкость,
составляющая дисперсионную среду, практически превращается
в очень тонкие прослойки и отличается по своим свойствам
от обычных жидкостей в объеме.
Полученные эмульсии могут быть устойчивыми или разру-
шаться во времени путем слияния (коалесценции) отдельных
капель. Слияние капель эмульсии уменьшает степень дисперс-
ности частиц и в пределе приводит к полному расслаиванию
двухфазной системы. Отсюда вытекает важная проблема устой-
чивости и разрушения эмульсий.
. Обычно устойчивые эмульсии получают в присутствии, специ-
альных стабилизирующих веществ, называемых эмульгаторами,
159
образующих на поверхности капель адсорбционные защитные
слои, препятствующие слиянию капель.
Разбавленные эмульсии с содержанием дисперсной фазы
от 0,01 до 0,1% по объему могут быть практически весьма
устойчивыми,.так как возможность коалесценции капель вслед-
ствие малой вероятности их столкновения очень небольшая.
Стабилизирующим фактором в этом случае может явиться на-
личие двойного электрического слоя на поверхности раздела
масло — вода. Значительная толщина двойного электрического
слоя (высокие значения ^-потенциала) выполняет роль стабили-
зирующего фактора, являющегося вполне достаточным для
обеспечения устойчивости разбавленных эмульсий. Такие си-
стемы могут быть устойчивыми и при значительных величинах
пограничного натяжения.
В концентрированных эмульсиях, содержание дисперсной
фазы в которых достигает 99% по объему,, прорыв тонких про-
слоек дисперсионнрй среды и коалесценция капель при недоста-
точной стабилизации происходит очень быстро.
Устойчивые высокодисперсные, концентрированные эмульсии
получают понижением пограничного натяжения почти до нуля,
что ведет к образованию самопроизвольных или, как их иногда
называют, критических эмульсий. Получающиеся эмуль-
сии относятся к классу лиофильных коллоидных систем и яв-
ляются равновесными, термодинамически устойяивыми. Про-
исходящее в процессе самопроизвольного диспергирования не-
которое возрастание поверхностной энергии компенсируется
.увеличением энтропии системы в связи с более равномерным
распределением в ней вещества дисперсной фазы.
На практике такие системы получаются при введении боль-
Щого количества (десятки процентов) поверхностно-активных
веществ, снижающих пограничное натяжение до 0,1——
0,01 эрг) см? (10-4—10-5 дж!м?). Примером таких систем яв-
ляются растворимые в воде масла, получающиеся при добавле-
нии к минеральным маслам до 40% натриевых мыл.
В отличие от лиофильных систем, лиофобные дисперсные
системы являются термодинамически неустойчивыми и требуют
специальной стабилизации. В настоящее время различают три
фактора устойчивости лиофобных дисперсных систем:
1) наличие двойного электрического слоя иа поверхности
дисперсных частиц, приводящее к возникновению энергетиче-
ского барьера, препятствующего сближению частиц до расстоя-
ний, на которых заметно проявляются силы молекулярного
притяжения;
2) образование на поверхности частиц значительного соль-
ватного слоя, препятствующего их сближению;
3) образование на. поверхности частиц со стороны диспер-
сионной среды стабилизирующей адсорбционной пленки, меха-
160
нически препятствующей агрегированию, в особенности коалес-
ценции капелек.
Более детальное обсуждение первых двух факторов при-
ведено в главе IV. Здесь же мы остановимся на действии треть-
его фактора — механической прочности стабилизирующей плен-
ки, препятствующей уменьшению ее толщины и коалесценции
капель. Вязкость в таком слое постепенно нарастает от нор-
мальной вязкости дисперсионной среды до максимальных зна-
чений непосредственно вблизи поверхности капель. Такие слои
•могут быть образованы либо молекулярными коллоидами —
высокомолекулярными соединениями типа желатины и каучука,
либо полуколлоидами типа мыл. Адсорбируясь в поверхностном
слое, эти вещества образуют лиогель, обладающий значитель-
ной прочностью,,
, Следует отметить, что с изменением концентрации эмульга-
тора. наступает оптимум структурно-механических свойств, выше
которого система становится снова неустойчивой (работы Тра-
пезникова). Наличие такого оптимума прочности стабилизирую-
щих пленок связано с необходимостью существования некото-
рой подвижности адсорбционного слоя, которая будет приво-
дить к «самозалечиванию» случайных разрывов в пленке.
При описанной стабилизации понижение поверхностного
натяжения хотя и происходит в той или иной степени, однако
не имеет определяющего значения. Для подтверждения этого
положения можно привести пример стабилизации эмульсий так
называемыми бронирующими твердыми эмульгаторами. Стаби-
лизация такими порошковыми - эмульгаторами (глины, сажа
и т: п.) имеет широкое распространение.
Процесс закрепления частиц порошка на границе раздела
капля дисперсной фазы — дисперсионная среда происходит по
причине, аналогичной закреплению частиц на пузырьках воз-
духа в процессе флотации (работа 27). Для получения устой-
чивой прямой эмульсии М/В частицы твердого эмульгатора дол-
жны хорошо смачиваться водой, однако полного смачивания
быть не должно, иначе они перейдут целиком в водную фазу.
Необходимым условием закрепления частиц на границе раздела
масло — вода с преимущественной ориентацией в водную фазу
является выполнение соотношения 0<cos6< 1, т. е. сводится
к условию образования частицей с поверхностью капель конеч-
ного краевого угла меньше 90°, считая всегда краевые углы во
внешней среде. Это означает, что для образования эмульсии
прямого типа частицы твердого эмульгатора должны быть гид-
рофильными (глина) и, наоборот, для образования обратной
эмульсии — гидрофобными (сажа). На рис. 66 изображено бро-
нирование капельки частицами твердого эмульгатора.
.Под эмульгирующей способностью эмульга-
тора часто понимают то максимальное количество, дисперс-
161
ной фазы, которое можно диспергировать в данном объеме
эмульгатора при некоторых определенных условиях. Количе-
ственно эмульгирующую способность можно выразить в объ-
емных процентах дисперсной фазы
Уф
_-—_—- . 100 — объемн. %
’ кф~г »*с
или в величинах кратности эмульсии:
Гф
-рг- = кратность эмульсии
где Уф — объем дисперсной фазы;
Vc — объем дисперсионной среды.
Эмульсии с максимальным содержанием дисперсной фазы
Умакс — носят название предельных.
Следует отметить, однако, что величины эмульгирующей
способности, выраженной в объемных процентах или в числах
Рис. 66. Закрепление частиц твер-
дого эмульгатора на поверхности
капли:
а—гидрофильный эмульгатор; 6—гидрофоб-
ный эмульгатор.
кратности, имеют постоянные
значения для данных эмульга-
торов только при вполне Опре-
деленном режиме процесса
эмульгирования.
Более правильным методом
оценки эмульгирующей способ-
ности эмульгатора, учитываю-
щей строение адсорбционно-
сольватных слоев, является
метод, предложенный Крем-
невым [14], [15]. Кремнев при-
нимает за меру эмульгирую-
щей способности предельную
величину поверхности стабили-
зирующего слоя, образован-
ную 1 сма раствора эмульгатора в предельно концентрирован-
ных эмульсиях при достижении слоем критической толщины 8кр,
независимо от конечного содержания Умакс.
Попытка * дальнейшего увеличения поверхности стабилизи-
рующих слоев ниже 8кр невозможна. Следовательно, условия
8 < 8кр не существует. Кремнев считает, что сольватация эмуль-
гатора происходит как со стороны внешней, так и внутренней
фазы, в связи с этим существуют две величины 8кр — для масля-
* Устойчивые эмульсии чистых масел [25 а] могут быть получены при
условии образования гидрофильным твердым эмульгатором (например, бенто-
нитом) в водной фазе объемной коагуляционной структуры, а одночастичный
слой может стабилизировать эмульсии в сочетании с образованием в по-
верхностном слое структурированных металлических мыл основного харак-
тера.
162
ной фазы &кр и для водной В'р. Молекулы эмульгатора, являю-
щиеся дифильными, скрепляют жидкости в эмульсии. Это
скрепление осуществляется одновременным связыванием при
помощи антиполярных частей молекул эмульгатора известных
количеств обеих жидкостей с образованием двух слоев с ано-
мальными свойствами на поверхности капель и внутри них.
Соотношение т = 5кр/&кр имеет решающее значение для ста-
билизации эмульсий того или иного типа. В случае, если тол-
щина сольватного слоя со стороны воды Вкр будет больше тол-
щины сольватного слоя со стороны масла 8кр. т. е. р> 1, обра-
зуется прямая эмульсия; при обратном соотношении устойчивой
является обратная эмульсия.
Выполнение работы
Рис. 67. Ци-
линдр и спи-
раль для по-
лучения
эмульсий по
методу
Кремнева.
Целью настоящей работы является приготовление концен-.
трированных и предельно концентрированных эмульсий и ис-
следование их степени дисперсности. Данные дисперсионного
анализа позволяют вычислить предельную поверх-
ность стабилизирующего слоя S«> и его критиче-
скую толщину 8кр.
Для приготовления таких эмульсий мы поль-
зуемся методикой, предложенной Кремневым, кото-
рая состоит в следующем. В открытый градуиро-
ванный цилиндр на 50—100 см3 микропипеткой вво-
дят 0,5—1 см3 раствора 'эмульгатора и опускают
в него проволочную спираль, с помощью которой
производят эмульгирование. Спираль имеет 3-—
5 витков, причем диаметр наибольшего витка не-
сколько меньше диаметра цилиндра, в котором го-
товится эмульсия (рис. 67).
Последний виток спирали переходит в держа-
тель, длина которого несколько больше длины ци-
линдра. Спираль должна хорошо прилегать ко дну
цилиндра и находиться с самого начала опыта в
растворе эмульгатора, что является необходи-
мым условием успешной работы и легко выполнимо, так как
эластичная проволока может быть изогнута по форме дна ци-
линдра. Эмульгируемую жидкость из бюретки выпускают кап-
лями с определенной скоростью так, чтобы она стекала по стен-
кам ци'линдра при одновременном плавном, и непрерывном дви-
жении спирали по оси цилиндра. Скорость движения спирали
должна находиться в соответствии со скоростью подачи‘эмуль-
гируемой жидкости из бюретки таким образом, чтобы послед- '
няя успевала непрерывно диспергироваться и не накапливалась
значительными объемами на поверхности эмульсии. При дости-
163
жении определенного содержания дисперсной фазы Уф (по
условиям задания) поступление жидкости из бюретки прекра-
щается и одновременно прекращают движение спирали. Таким
образом получаются концентрированные эмульсии. Полученная
концентрированная эмульсия подвергается дисперсионному ана-
лизу под микроскопом (описание методики анализа смотри
ниже).
Далее проводится получение новой порции концентрирован-
ной эмульсии по описанной выше методике с соблюдением по-
стоянными—Уф, скорости подачи эмульгируемой жидкости и
частоты движения спирали, однако время диспергирования /2
увеличивается. Полученную эмульсию анализируют под микро-
скопом. Аналогично получают еще более концентрированные
эмульсии, увеличив время диспергирования до A, h /5 *. Степень
дисперсности каждой эмульсии определяется микроскопически
сразу же после ее получения.
Свидетельством получения предельно концентрированной
эмульсии может служить появление первых крупных капель
дисперсной фазы, образующихся при непрерывном диспергиро-
вании эмульсии. Постоянство величин S и 8, вычисленных по
данным дисперсионного анализа при разных значениях t, яв-
ляется доказательством того, что полученная эмульсия — пре-
дельно концентрированная.
Методика микроскопического анализа состоит в следующем.
Препарат для микроскопических измерений готовится путем
осторожного перемешивания небольшого количества свежепри-
готовленной эмульсии со значительным объемом нагретого на
водяной бане до 30—40° С 1% раствора желатины (с целью
сведения до минимума возможной коалесценции капель). Капля
приготовленной таким образом разбавленной эмульсии вносит-
ся в лунку предметного стекла и сверху накрывается покров-
ным стеклом так, чтобы под ним не оставалось пузырька воз-
духа. Для каждой исследуемой эмульсии готовится 2—3 препа-
рата, в которых в различных местах рассматривается 10—
20 полей зрения **. Всего в каждой эмульсии измеряется около
200 капель, размер которых вначале определяется в условных
единицах (по числу делений окулярмикрометрической сетки,
занимаемых одной каплей). Все измеренные капли разбиваются
на классы: 0—1; 1—3; 3—6; 6—9 делений и т. д. Капли каждого
класса характеризуются средним диаметром di, равным сред-
нему арифметическому из значений для границ промежутка:
di = 0,5; d2 = 2; d2 = 4,5; 7,5 делений и т. д.
* По описываемой методике может быть получена, например, эмульсия
керосина в воде. Эмульгатором является 0,5—1,0% раствор желатины, на-
гретой на водяной бане до 30—40°, V® = 10 см3, ti, t?, h, ft и равны co<
ответственно 3, 5, 7, 9, 11 мин.
** Измерения ведутся при увеличении 20X40.
164
Обработка данных дисперсионного анализа производится
следующим образом. На основании полученных данных состав-
ляется таблица, функций распределения капель по размерам
N(d) (табл. 1)
jVz = _^_.100%
I
где п{ — число капель класса I с диаметром (Ц;
_ 2 — общее число капель всех классов;
Nt— относительная концентрация частиц, %'.
рованной в 1 см3 раствора эмульгатора, определяется по сле-
дующей формуле:
__ i_______________КФ ______ji_____
. ~ ‘ Ve ~ V.a'^d^Ni
i i
где a — цена одного деления окулярмикрометрической сеточки.
Учитывая, что S-8 = 1, мы можем записать:
т. е.
0 =-----------
-
i
Для предельно концентрированных эмульсий таким образом
могут быть рассчитаны предельная величина поверхности ста-
билизующего слоя So, и критическая толщина слоя 8Кр.
165
Пены
Пенами являются дисперсные системы, _в которых дис-
. персная фаза — газ — распределена в жидкой дисперсионной
среде, причем среда представлена в виде тонких прослоек жидко-.
ста. Таким образом, пены могут быть определены как высоко-
концентрированные эмульсии газа в жидкости,
подобно обычной высококонцентрированной эмульсии, где
имеется вырождение дисперсионной среды в тонкие прослойки,
находящиеся между капельками жидкости.
Пены, так же как и эмульсии, находят широкое практическое
применение. Они используются при огнетушении, в процессах
флотации (пенная флотация), при получении ячеисто-пористых
тепло- и звукоизоляционных материалов (пенобетоны, пено-
пласты, пеностекло), в пищевой промышленности (пастила,
мусс) и т. д.
Пена может быть получена встряхиванием ра'створа пенооб-
разователя в цилиндре или пропусканием воздуха через порис-
тый фильтр, помещенный в раствор пенообразователя. Третий
способ получения пен из растворов, дающих высокоустойчивые
пены, состоит в том, что струя раствора с определенной высоты
падает на поверхность того же раствора, находящегося в ци-
линдре.
Пена образуется в том случае, когда скорость образования
и подхода пузырьков газа к поверхности жидкости оказывается
больше скорости их разрушения. Чем больше скорость образо-
вания пузырьков и чем меньше скорость их разрушения, тем
более обильная пена может быть получена.
Пены, как и все дисперсные системы, являются термодинами-
чески неустойчивыми по причине высокой удельной поверхности
и, следовательно, большой величины свободной поверхностной
энергии системы.
Для характеристики устойчивости пены обычно при-
нимают время существования пены с момента ее возникновения
до полного разрушения или- время, за которое высота столба
пены убывает в 2 раза. »
Устойчивость пены также может быть оценена по времени
жизни отдельного пузыры?Э,. образованного на поверхности
жидкости, или по времени жизни элементарной пленки,
образованной данной жидкостью в проволочном (платиновом)
кольце. Оба метода могут дать представление об устойчи-
вости пены, причем, как правило,- устойчивость элементарной
пены изменяется параллельно устойчивости пены в целом,
хотя в последнем случае могут иметь место более сложные
соотношения.
Устойчивость имеет кинетический характер. Рассмотрим, ка-
кие же силы обусловливают устойчивость пены', т. е. способ-
166
Рис. 68. Схема .кинетики
разрушения пены: а, б,
в — последовательные
стадии разрушения пены.
ность системы определенное время сохранять свое первоначаль-
ное состояние.
Многими авторами было показано, что чистые жидкости не
дают устойчивой пены, только добавка к ним второго компо-
нента может привести к. ее образованию. Дистиллированная
вода не дает устойчивой пены. Прибавление к ней поверхностно-
: активных веществ приводит к появлению пены, причем с рос-
том концентрации поверхностно-активного вещества интенсив-
ность пенообразования увеличивается, проходит через максимум
снова падает для концентрированных растворов. Однако, как
|?иоказано Ребиндером, такого падения устойчивости с ростом
^концентрации поверхностно-активного вещества не наблюдается
хдля- высокомолекулярных поверхностно-
Кактивных веществ (сапонин, белки),
’•когда устойчивость монотонно возра-
стает с ростом концентрации пенообразо-
вателя.
f Молекулы пенообразователя адсорби-
руются на границе раздела фаз таким
.^образом, что их гидрофобная часть на-
правлена в сторону газовой фазы, а гид-
рофильная— в воду. Гидрофильная часть
молекулы гидратируется водой, образуя
^гидратные слои определенной тол-
идины, (как и в случае эмульсий), за-
щищающие пузырьки воздуха от коалес-
ценции.
Рассмотрим кинетику разрушения пены (рис. 68). Свеже-
приготовленная пена имеет прослойки жидкости (рис. 68,а),
обладающие значительной толщиной и находящиеся между пу-
. зырьками газа, покрытыми гидратированными молекулами
поверхностно-активного вещества. Толщина гидратных слоев 8
составляет часть от толщины прослоек между пузырьками. Вода,
заключенная между пузырьками, начинает стекать вниз — тол-
щина прослойки уменьшается до тех пор, пока гидратные слои
двух поверхностей не придут в' соприкосновение (рис. 68, б).
/Вторым этапом разрушения пены является утоньшение гидрат-
ных слоев и, наконец, достижение прослойкой такой толщины,
при которой она становится неустойчивой, что приводит к коа-
лесценции пузырьков (рис. 68,в).
1 Наиболее интересным и важным этапом в кинетике разру-
шения пены является II этап — разрушение гидратных слоев.
Одна из причин устойчивости таких гидратных слоев — на-
личие расклинивающего давления в двухсторонних пленках
(гл. IV).
Другая возможная причина устойчивости таких слоев — эф-
фект Марангони — Гиббса, состоящий в следующем.
167
Рис. 69. Схема строения пены.
Рассмотрим строение пены (рис. 69). Пена состоит из ячеек,
построеннйх аналогично пчелиным сотам. В местах, где соеди-
няются стенки пузырьков, образуются утолщения, в которых
жидкость имеет сильно искривленную (вогнутую) поверхность.
Жидкость в вогнутых участках пленки находится под гидроста-
тическим давлением Pi, пониженным по сравнению с давле-
нием Ро в плоских участках на величину капиллярного давления
ДР5= — (где г—радиус кривизны данного участка пленки) и,
следовательно, при возникновении пены возникает ток жидкости
из плоских участков в углы
пленок. Поток жидкости
включает в себя и поверх-
ностный слой, обогащенный
поверхностно-активным ком-
понентом, который заменяет-
ся раствором из внутрен-
них слоев жидкости, менее
богатым поверхностно-ак-
тивным компонентом. Это
приводит к увеличению о на
плоской поверхности по
сравнению с вогнутой ча-
стью, т. е. создается гра-
диент с, а также поверх-
ностной концентрации по-
верхностно-активного веще-
ства между плоской и
искривленной поверхностью
пузырька. Возникает поверхностный противоток жидкости, под-
питывающий пленку и мешающий ее дальнейшему утоньшению.
Таким образом, вытекание жидкости из прослойки аналогично
протеканию ее в капилляре; чем более тонкой становится плен-
ка, тем медленнее происходит протекание жидкости. Чтобы вся
жидкость смогла вытечь из слоя между пузырьками, необхо-
димо определенное время.
Другая причина устойчивости пузырька может лежать в
его эластичности. Происхождение ее объясняется следующим:
любая деформация сферического пузырька, приводит к уве-
личению его поверхности и, следовательно, к уменьшению
концентрации поверхностно-активного вещества на поверх-
ности и увеличению с. Поэтому деформированный участок стре-
мится вернуться к своей исходной форме и тем самым умень-
шить в.
Разрушение пены может происходить за счет роста крупных
пузырьков пены и исчезновения мелких. Этот процесс объяс-
няется диффузией газа через пленку из более мелких пузырьков
168
в более крупные и связан с разностью давлений в различных по
величине пузырьках.
Наконец, наиболее сильным фактором устойчивости пен, как
это следует из работ Ребиндера, является механическая проч-
ность слоев, образованных вокруг пузырьков пены. В пределе
такие слои могут обладать свойствами твердого тела, а сама
система будет беспредельно устойчивой (пенопласты).-
Зная причины устойчивости пены, можно объяснить отсут-
ствие устойчивой пены для чистых жидкостей.
Дистиллированная вода не дает пены, так как в отсутствии
поверхностно-активных веществ не могут образоваться гидрат-
ные слои вокруг пузырьков воздуха, и, следовательно, нет ника-
ких сил, препятствующих их слиянию.
Экстремальная зависимость пенообразования от концентра-
ции низкомолекулярных поверхностно-активных веществ может
быть объяснена следующим образом. При больших концентра-
циях поверхностно-активного вещества, т. е. при предельном на-
сыщении поверхностного слоя молекулами поверхностно-актив-
ного вещества, градиент концентрации между поверхностным
слоем и объемом раствора наименьший. Таким образом, растя-
жение поверхности пузырька или отток жидкости к углам в
эффекте Марангони — Гиббса не приводит к значительному
увеличению о, а следовательно, отсутствует сила, препятствую-
щая утончению поверхностного слоя жидкости. Наоборот, при
меньших концентрациях, когда поверхностный слой еще не
является насыщенным и существует значительный градиент
концентрации поверхностно-активного вещества между поверх-
ностью и объемом жидкости, отток жидкости приводит к более
резкому возрастанию о на поверхности раздела пузырек—рас-
твор, а следовательно, появляется сила, препятствующая такому
оттеканию.
Отсутствие экстремальной зависимости устойчивости пены от
концентрации поверхностно-активного вещества для высокомо-
лекулярных поверхностно-активных веществ говорит о другой
природе стабилизирующего фактора, которая связана, по-види-
мому, с образованием гелеобразных структурированных за-
щитных слоев на поверхности пузырька газа. Такие струк-
турированные слои обеспечивают значительную устойчивость
пен.
Как и в случае эмульсий, стабилизатор, обладающий значи-
тельными структурообразующими свойствами, но небольшой
поверхностной активностью, может быть вытеснен более силь-
ным поверхностно-активным веществом, адсорбционные слои
которого не обладают достаточными механическими,свойствами.
Этот путь является наиболее простым и удобным при необхо-
димости разрушения устойчивых пен.
169
Работа 29
ПОЛУЧЕНИЕ ПЕНЫ И ИССЛЕДОВАНИЕ ЕЕ УСТОЙЧИВОСТИ
Пену получают в приборе (рис. 70), состоящем из следую-
щих частей: 1 — цилиндр, в котором происходит образование
пены; 2 — трубка, заканчивающаяся стеклянным фильтром (на-
пример, стеклянный фильтр № 2); 3 — градуированный сосуд,
служащий для сбора образующейся пены и изучения кинетики
ее- разруше'ния; 4 — манометр; 5 — маностат, служащий для под-
держания постоянного давления в системе; 6 — кран.
От боздуходуйки
Рис. 70. Прибор для получения н исследования устойчи-
вости пены.
Работа проводится следующим образом. Цилиндр 1 запол-
няется раствором, предназначенным для получения пены; кран
на выходе из цилиндра закрыт. В систему подается давление,
Таблица 2
Концентрация пенообразователя
Су 2 с са- 4 г -£1 С,-Т
время отсчета, сек объем пены, С.И8 объем отслоившейся жидкости, см» время отсчета, сек объем пены, ел® объем отслоившейся жидкости, I сж3 I rt № Н ЗБ -в* £ £ « Си н В О tj объем пены, СМ» объем отслоившейся жидкости, см» время отсчета, сек объем пены, см» объем отслоившейся жидкости, см»
"S ю*»-*©* II II II II О 500 0 0 "S о** II II II- II О 500 0 с ° 11 II II о — еч с с-* 500 0 0 II II II II о 500 0 0
170
и когда оно достигает постоянной величины, открывается
кран 6.
Открытие крана приводит к бурному выделению пузырьков
и образованию пены, которая поднимается по трубке и проходит
через кран. Первые порции пены отбрасываются. При установ-
лении стационарного потока пены она подается в приемник 3
«собирается в нем до определенного уровня (например500 см3).
Сразу же после заполнения приемника пеной включается’секун-
домер и производятся измерения высоты (объема) пены и
объема отслоившейся жидкости, которая собирается в так назы-
ваемом «пальце», находящемся в нижней части приемника. Из-
мерения проводятся через определенные промежутки времени
в зависимости от устойчивости получающейся пены. Измерения
чзрекращаются при исчезновении столба пены — при появлении
«зеркала» (свободной от пены центральной части поверхности
жидкости в сосуде).
Время с момента получения пены до момента ее полного ’
разрушения (появление зеркала) будем называть временем
жизнипеныТ.
Полученные результаты записываются в таблицу и изобра-
жаются в виде следующих графиков: 1) изменение объема пены
во времени (Vn—t); 2) изменение объема отслоившейся жид-
кости во времени (Уж — t).
Работа 30
ПРИГОТОВЛЕНИЕ ПЕНЫ МЕТОДОМ ВСТРЯХИВАНИЯ РАСТВОРА
ПЕНООБРАЗОВАТЕЛЯ В ЦИЛИНДРЕ
Пена может быть получена н более простым способом —•
встряхиванием раствора пенообразователя в цилиндре с при-
тертой пробкой.
Таблица 3
Концентрация пенообразователя
Cl с «•2- 2 fl
время, сек объем пены И, см9 время, сек объем пены V, см3 время, сек объем пены V, смг время, сек объем пены V, см*
° IIII • -
Работа проводится следующим образом. В цилиндр с при-
тертой пробкой (емкостью 50—100 см3) наливают определённый
171
объем (20—50 см3) раствора пенообразователя и встряхивают
в течение 15 сек. После прекращения встряхивания включается
секундомер и одновременно отмечается объем образовавшейся
пены. Далее отмечают изменения объема пены во времени.
Аналогичные опыты проводят с растворами пенообразователя,
разбавленными в 2, 4, 8, 16 раз и т. д. (до тех концентра-
ций, пока пенообразователь устойчивой пены давать не будет —
получающаяся пена мгновенно разрушается). Полученные ре-
зультаты заносят в таблицу и строят следующие графические
зависимости: 1) максимальный объем образовавшейся пены,
как функция концентрации пенообразователя; 2) кинетика раз-
рушения пены (Уп—t).
Работа 31
ОЦЕНКА УСТОЙЧИВОСТИ ПЕНЫ ПО ВРЕМЕНИ СУЩЕСТВОВАНИЯ
ЭЛЕМЕНТАРНОЙ ПЛЕНКИ
Рис. .71. Опре-
деление устой-
чивости пены
по времени су-
ществования
элементарной
пленки.
Устойчивость пены может быть оценена по времени жизни
элементарной пленки. Работа проводится следующим образом.
Готовится ряд растворов исследуемого пенообразователя с по-
стоянно уменьшающейся концентрацией (как и в предыдущей
работе с = 1, 1/2, 1/4, 1/8 и т. д.). Исследуемый
раствор помещается в стаканчик на 50 см3 и в
него погружается платиновое проволочное коль-
цо диаметром 15—20 мм (рис. 71). Кольцо
осторожно вынимается из жидкости и по секун-
домеру фиксируется время с момента образова-
ния пленки до ее разрушения. В каждом рас-
творе производится 20—30 отсчетов времени
жизни элементарной пленки (в секундах). Полу-
ченные результаты усредняются. Аналогичные
опыты проводятся со всеми исследуемыми кон-
центрациями.
Результаты опытов изображаются графиче-
ски в виде зависимости: время жизни элемен-
тарной пленки — концентрация пенообразова-
теля. Полученная зависимость сравнивается с зависимостью, по-
лученной в предыдущей работе (график 1).
ЛИТЕРАТУРА
1. Адам Н. К-, Физика н химия поверхностей, ОГИЗ, 1947.
2. Богданов О. С., Вопросы теории и практики флотации, 1959.
3. Богданов О. С., Труды Ин-та Механобр, вып. 124 (1959).
4. Бромберг А. В., Колл, ж., 8, № 3, 117 (1946).
5. Венстрем Е. К, Ре б ин дер П. А., ЖФХ, 2, № 6, 754 (1931),
172
\
\
6. ВоЛцкий С. С., Усп. хим., 30, 1237 (1961).
7. Гедройц К. К., Химический анализ почв, Сельхозгиз, 1932.
8. Гельфер их Ф., Иониты. Основы ионного обмена, ИЛ, 1962.
9. Ерчн^овский Г. О., Образование флотационной пены, ГОНТИ,
1939.
10. Ж у к о в И. И., Коллоидная химия, т. I, 1949.
И. Классен В. И., Мо к р о у с о в В. А., Введение в теорию флотации,
Госгортехиздат, 1959.
12. КлейтЬн В., Эмульсии. Их теория и техническое применение, ИЛ,
13. К о ш е в и и к Л. Ю., Кусаков М. М., Л у б м а н Н. М., ЖФХ, 27,
1887 (1958).
'4. Кремнев Л. Я., Со.скин С. А., ЖОХ, 16, № 12, 2000 (1946).
15. Кремнев Л. Я., Колл, ж., 13, 15, 394 (1951).
16, Матерова Е. А., Парамонова В. И., Уч. зап. ЛГУ, 163, № 12,
112 (1953).
17. Н и к о л ь ск и й Б. П., Парамонова В. И., Усп. хим., 8, вып. 10-
(1939).
18. Н и к о л ь с к и й Б. П., сб. «Довременные методы исследования физико-
химических свойств почв», 4, вып. 3, Изд. АН СССР, 1947.
19. Р а б и н е р со н А. И., Проблемы коллоидной химии, ОНТИ, 1937.
20. Ребиндер П. А., Липец М. Е., Римская М. М., Тауб-
м а н А. Б., Физико-химия флотационных процессов, Металлургиздат,
1933.
21. Р е б и н д е р П. А., Исследования в области поверхностных явлений,
ОНТИ, 1936.
22. Р е б и н д е р П. А., Колл, ж., 8, № 3, 157 (1946).
23. Ребиндер П. А., Конспект общего курса коллоидной химии, Изд.
МГУ, 1949.
24. С а з е р л е н д К- Л., У о^к И. В., Принципы флотации, Металлург-
издат, 1958.
25. Талмуд Д. Л., Суховольская С. Д., ЖФХ, 2, № 1, 31 (1931).
25а. Таубман А. Б., К о р е цк ий А. Ф-, ДАН СССР, 120, № 1, 126 (1958);
124, № 2, 358 (1959).
26. Хроматография. Сб. материалов совещания, под ред. Б. П. Николь-
ского, Изд. ЛГУ, 1956.
27. Шелудко А., Коллоидная химия, ИЛ, 1960.
28. В i k е г m а п I. I., Surface chemistry, 1958.
29. Bartell F. E., Osterhof H. Y., Ind. Eng. Chem., 19, № 11, 1277
(1927).
30. Bartell F. E., Miller F. L., Ind. Eng. Chem., 20, № 7, 738 (1928).
31. Harkins W. D., Brown F. E„ J. Am. Chem. Soc., 41, I, 499 (1919).
32. R i c h a r d s T. W., Coombs L. W., J. Am. Chem. Soc., 37, II, 1656
• (1915).
33. Richards T. W., Carver E. K. J., Am. Chem. Soc., 43, I, 827 (1921).
ГЛАВА III
' ДВОЙНОЙ ЭЛЕКТРИЧЕСКИЙ СЛОЙ И
ЭЛЕКТРОКИНЕТИЧЕСКИЕ ЯВЛЕНИЯ
На границе раздела фаз, как правило, возникает двойной
электрический слой; его происхождение можно объяснить сле-
дующим образом.
В гомогенной фазе химический потенциал ц любого’ компо-
нента в равновесном состоянии имеет постоянную величину во
всем объеме фазы. Вместе с тем величина ц для данного ком-
понента в разных фазах имеет различные значения. Поэтому
при соприкосновении двух фаз на границе раздела происходит
переход компонентов из фазы с более высоким значением хими-
ческого потенциала в фазу с меньшей величиной ц. Переход за-
ряженных компонентов (ионов, электронов) приводит к образо-
ванию заряда в одной фазе и равного, но противоположного по
знаку, заряда в другой. Эти противоположные заряды, благо-
даря взаимному притяжению, остаются на границе раздела, об-
разуя двойной электрический слой.
Дальнейший переход вещества из одной фазы в другую бу-
дет задерживаться возникающей разностью потенциалов Дф и,
наконец, прекратится при достижении максимальной равновес-
ной величины Дф,
В состоянии равновесия работа переноса 1 моль против элек-
трических сил ZiF\q (где Zi — валентность z-го компонента, F —
число Фарадея), должна быть’равна уменьшению химического
потенциала компонента Др, а следовательно:
Дщ -f- г1? = О*
Такие процессы на границе проводников с растворами изу-
чаются в электрохимии. Коллоидная химия изучает наряду с
проводниками системы с непроводящей твердой фазой, где
двойной электрический слой возникает за счет перехода ионов
из дисперсной фазы в дисперсионную среду или же в обратном на-
правлении.
* В общем случае раздельное экспериментальное определение Д<р и Др
не представляется возможным.
174
Первый случай имеет место при диссоциации молекул, соста-
вляющих^ поверхностный слой твердой фазы (например, поли-
кислот). 'При этом один из ионов (например, Н+) выходит в
жидкую фазу, в то время как другой ион, связанный более проч-
но (обычнА силами главных валентностей), остается в твердой
•:фазе. Во втором случае двойной электрический слой возникает
в результате преимущественной адсорбции одного из ионов рас-
•твора, обычно иона общего или изоморфного с ионами твердой
фазы (например, Ag+ на AgCl).
Таким образом, в результате диссоциации или адсорбции
’дэдна фаза приобретает положительный заряд, другая — отрица-
|тельный. Поэтому наложение внешнего электрического поля
^приводит к относительному перемещению двух фаз, каждая из
^которых будет двигаться, если она не закреплена, к противопо-
ложному полюсу. Наоборот, относительное движение фаз под
.Действием внешнего механического усилия приводит к смеще-
нию зарядов, а следовательно, к возникновению градиента по-
тенциала в направлении смещения.
Явления относительного движения фаз вдоль поверхности
раздела, обусловленные внешним электрическим полем или при-
водящие к возникновению электрического поля, называются
электрокинетическими явлениями..
Движение под действием внешнего электрического поля
д) свободных частиц дисперсной фазы (суспензии, эмульсии,
г золи) в дисперсионной среде называется электрофорезом
’^катафорезом), б) жидкости относительно неподвижной твер-
,Дой фазы (капиллярные системы, гели) называется электро-
космосом.
Возникновение' электрического поля при механическом дви-
жении а) свободных частиц дисперсной фазы в дисперсионной
среде (обычно под действием силы тяжести) называется п о -
тенциалом седиментаций (эффект Дорна), б) жид-
кости относительно неподвижной твердой фазы (обычно под
действием внешнего давления) называется потенциалом
протекания (течения).
Изучение этих явлений имеет большое и разностороннее зна-
чение для теории процессов, происходящих в поверхностном
слое (в частности, в вопросах устойчивости дисперсных систем),
и широко используется при решении различных практических
задач.
Наряду с рассмотренными явлениями наличие двойного элек-
трического слоя изменяет условия прохождения тока в рас-
творе, находящемся в равновесии с твердой фазой, а именно, из-
меняет числа переноса ионов и электропроводность в жидкости,
заполняющей поры капиллярных систем.
Эти свойства, получившие название электрокинетиче-
ских свойств дисперсных систем, тесно связаны с
175
электрокинетическими явлениями и играют большую роль при
практическом применении капиллярных систем, в частности при
использовании мембран в процессе электродиализа.
ЭЛЕКТРОКИНЕТИЧЕСКИИ (S) ПОТЕНЦИАЛ /
/
Представление о двойном электрическом слое, кай о плоском
конденсаторе, развитое в классических работах Гельмгольца,
получило дальнейшее развитие в трудах Смолуховского, Гуи,
Чэпмена, Штерна и других ученых.
Рассмотрим современное представление о строении двойного
электрического слоя ионов на границе твердой фазы с водой
или с разбавленным водным раствором электролита (например,
на границе стекло — раствор NaCl) (рис. 72а).
Ионы, входящие в состав твердой фазы (например, анионы
поликремневых кислот п SiOj • SiOf-, возникающие в результате
диссоциации), образуют внутреннюю обкладку двойного слоя.
Ионы противоположного знака (катионы Na+, отделившиеся при
диссоциации от твердой фазы), т. е. противоионы, образуют
внешнюю обкладку двойного слоя, обращенную к жидкой
фазе.
Ионы внешней обкладки, притянутые электрическими силами
вплотную к внутренней обкладке, образуют плотный * слой вне-
шней обкладки.
Наряду с этим, благодаря молекулярному тепловому движе-
нию, часть ионов внешней обкладки, обладающих большей ки-
нетической энергией, преодолевает потенциальную энергию по-
ля и уходит в объем раствора.
, В результате устанавливается динамическое равновесие, в
котором скорость диффузии ионов из поверхностного слоя в
объем раствора, равна скорости обратного процесса, обуслов-
ленного полем электростатических сил. Это равновесие анало-
гично распределению молекул газа в гравитационном поле и
подчиняется закону Больцмана. Действие электрического поля
приводит к увеличению концентрации ионов противоположного
знака (с+) и к уменьшению концентрации одноименно заряжен-
ных ионов в растворе (с_), вблизи поверхности (рис. 72,в).
Расчеты, сделанные по уравнениям теории Гуи, показывают,
что плотность заряда, обусловленная избытком противоионов
больше плотности, связанной с недостатком одноименных ионов;
поэтому увеличение с+ больше уменьшения с-.
Процесс диффузии, наоборот, стремится выравнять концен-
трации ионов по всему объему. В результате в жидкой фазе со-
* Называется также: адсорбционный слой, слой Гельмгольца, слой
Штерна.
176
здается объемный заряд, плотность которого постепенно убы-
вает с удалением от поверхности раздела. СЛой, в котором
диффузно распределен объемный за-
ряд (противоположный по знаку заря-
ду поверхности), носит название
диффузного слоя.
Согласий теории Штерна, часть
противоионов находится в непосред-
ственном соприкосновении с ионами
твердой фазы, образуя плотный слой;
другая часть противоионов составляет
диффузный слой. Обычное схемати-
ческое изображение распределения
этих ионов приведено на рис. 72, б, где,
в отличие от 72, а, показаны не все
ионы, содержащиеся в растворе, а
только избыточные.
Система в целом всегда является
электронейтральной, то есть число за-
рядов внутренней обкладки должно
быть равно числу зарядов противо-
ионов (плотный слой + избыток в диф-
фузном слое).
Толщина плотного слоя имеет по-
рядок ионного диаметра. Толщина
диффузного слоя зависит, от темпера-
туры и концентрации. Чем выше кон-
центрация ионов в растворе, тем силь-
нее смещается равновесие в сторону
перехода ионов из объема раствора в
поверхностный слой, т. е. из диффуз-
ного слоя в плотный. Таким образом
с ростом концентрации происходит
сжатие диффузного слоя.
Согласно теории Гуи, приведен-
ная толщина диффузного
с л о я 8 (т. е. расстояние от поверхно-
сти до центра тяжести электрических
зарядов) обратно пропорциональна
корню квадратному из величины кон-
центрации. Для 10-2 н. раствора одно-
валентного электролита 8 = 3 Мц, для
10“* н. 8 = 30 тир, т. е. во много раз
больше толщины плотного слоя.
Рис. 72. Схема двойного
электрического слоя:
а и б—распределение избыточ-
ных ионов <в двойном электриче-
ском слое; в —изменение концен-
траций ионов в зависимости от
расстояния; г—изменение потен-
циала в зависимости от расстоя-
ния.
Падение потенциала в плотном слое, по аналогии с плоским
конденсатором, принимается линейным, а градиент потенциа-
ла — постоянным.
177
В диффузн£М слое градиент потенциала постепенно
уменьшается, по мере удаления от поверхности, с уменьшением-
плотности объемного заряда р, согласно уравнению Пуассона
дг<р 4тч .
-jp- =---g- р для плоской поверхности. , •
Ход падения потенциала <р с расстоянием от твердой фазы
представлен на рис. 72, г. 1
При относительном перемещении фаз ионы диффузной части
двойного слоя участвуют в движении жидкой фазы, в то время
как .ионы плотного слоя остаются вместе с твердой фазой. По-
этому участие ионов диффузного слоя в относительном движе-
нии жидкости характеризуется не всем межфазным потенциа-
лом е, а лишь той частью его, которая представляет собой паде-
ние потенциала в области диффузного слоя. Эта часть падения
потенциала, точнее разность потенциалов между границей сколь-
жения жидкости и глубиной раствора, т. е. областью, где объем-
ный заряд равен нулю,.называется эле’ктрокинетическим
или ^-потенциалом.
Таким образом, g-потенциал характеризует объемный заряд
жидкой фазы, а следовательно, и электрокинетические явления,
обусловленные ионами диффузного слоя. Граница скольжения
жидкости в разбавленных растворах обычно совпадает с грани-
цей между плотным и диффузным слоем. В концентрированных
растворах, где потенциал быстро изменяется с расстоянием, эти
границы могут не совпадать, поэтому в общем случае g-потен-
циал не всегда может быть отождествлен с падением потенциа-
ла в диффузном слое (обозначаемым обычно через ф1).
Увеличение концентрации ионов в растворе приводит, как
было уже отмечено, к переходу ионов диффузного слоя в плот-
ный, а следовательно, к уменьшению g-потенциала.
. При рассмотрении реальных капиллярных систем следует
учитывать, что внутри каждого капилляра возникает двойной
электрический слой. Распределение плотности заряда в поверх-
ностном слое, а следовательно, и величина g-потенциала одно-
значно определяется (при данной температуре) составом фаз, а
именно: химической природой твердой фазы, составом раствора
и его концентрацией. Таким образом, величина g-потенциала по
физическому смыслу не должна зависеть от структурных пара-
метров, т. е. от размеров капилляра, что подтверждается и эк-
спериментально. Точно так же, в коллоидных растворах, напри-
мер в суспензиях, величина g-потенциала у частиц дисперсной
фазы не должна зависеть от их размеров *.
* Сказанное справедливо для поверхностей, у которых, радиус кривиз-
ны' значительно превышает толщину диффузного слоя.- В тех случаях, ко-
гда эти величины являются соизмеримыми, распределение плотности за-
ряда, а следовательно и реличипа g-потенциала, могут изменяться с кри-
визной поверхности.
178
Таким образом, отсутствие зависимости от структурных па-
раметров позволяет считать величину ^-потенциала однознач-
ной характеристикой поверхности раздела твердой фазы в рав-
новесии с раствором данного состава и концентрации.
Электроосмос
Работа 32
^ОПРЕДЕЛЕНИЕ ЭЛЕКТРОКИНЕТИЧЕСКОГО ПОТЕНЦИАЛА ДИАФРАГМ МЕТОДОМ
i ЭЛЕКТРООСМОСА
Электроосмосом называется движение жидкости через капил-
ляр или капиллярную систему (диафрагму) под действием при-
йложенной внешней электродвижущей силы.
; ' Механизм электроосмотического переноса жидкости можно
^Представить себе следующим образом (рис. 73). При наложении
^внешнего электрического поля вдоль находящейся в растворе
{системы капилляров, подвижные ибны диффузного слоя, пере-
гдвигаются к противоположному полюсу.
'' Таким образом, в каждом капилляре создается цилиндриче-
ская (для капилляров круглого сечения) оболочка ионов диф-
фузного слоя, передвигающаяся к определенному полюсу и
увлекающая за. собой, вследствие молекулярного сцепления и
..трения, всю массу жидкости в капиллярах. После достижения
‘ стационарного состояния процесса вся жидкость движется, как
одно целое.
Из изложенного очевидно, что при уменьшении избытка ио-
нов в диффузном слое, например при переходе их в плотный
слой,, ионы, ушедшие из диффузного слоя, перестают участво-
вать в электроосмосе. Очевидно, что такой переход, связанный
с уменьшением эффективного заряда и ^-потенциала, приводит
к уменьшению, движущей силы в данном электрическом поле,
а следовательно, к уменьшению перенесенного количества
жидкости.
Наоборот с увеличением избытка ионов в диффузной части,
с увеличением ^-потенциала возрастает количество перенесенной
жидкости в единицу времени, т. е. объемная скорость электро-
осмоса V. Таким образом, v пропорциональна £.
Эта связь и является основой для количественного определе-
ния величины ’^-потенциала по методу электроосмоса. Гельм-
гольц и Смолуховский разработали теорию электроосмоса и по-
лучили выражение для ^-потенциала, смысл которого можно по-
нять при помощи следующих рассуждений.
При наличии диффузного слоя ионов непосредственной при-
чиной электроосмотического движения жидкости является гра-
диент потенциала Н на диафрагме, обусловленный внешним
179
электрическим полем и действующий вдоль границы раздела
фаз
н Е
где Е — разность потенциалов между концами капилляра или
сторонами диафрагмы,
I — длина капилляра или системы капилляров.
Рис. 73. Схема движения раствора в капиллярах (порах)
мембраны при электроосмосе.
Градиенту потенциала пропорциональна линейная скорость
v * л
жидкости w = -j, где v— объемная скорость, А— суммарная
эффективная площадь поперечного сечения капиллярной систе-
мы. Следовательно, объемная скорость v ~ wA пропорциональ-
на НА = ~ А. Очевидно также, что скорость потока обратно
180
(1)
равный где D — диэлектриче-
пропорциональна коэффициенту вязкости жидкости т). Объеди-
няя все сказанное, находим, что
_ tilv
v ~ — • х, -Д—
' /vj АЕ
Посредством применения уравнения Пуассона, связывающего
потенциал с зарядом V2cj> =-jy Р*, был найден коэффициент
пропорциональности в (1),
ская проницаемость.
Таким образом
Avr\lv*
DEA
£
(2)
для одиночных капилляров, редко
вывести из приве-
вместо /? его вы-
раствора х
Эта формула, применяемая
используется для капиллярных систем ввиду трудности опреде-
ления геометрических структурных параметров А и I. Обычно
пользуются другой формулой, которую легко
денной выше. По закону Ома:
Я
где 7? — сопротивление системы. Подставляя
ражение через удельную электропроводность
К 1. А
отсюда значение Е в (2),получим
не содержащее структурных па-
Е- И
находим: с — , подставляя
выражение для ^-потенциала,
раметров **:
С
(3)
~ТБ~
Величина ^-потенциала, являясь функцией распределения ио-
нов между плотным и диффузным слоями, не должна зависеть
от структурных характеристик.
Действительно, на основании многочисленных эксперимен-
тальных данных было установлено, что отношение v[l, а следо-
вательно, и ^-потенциал являются постоянными для данной гра-
ницы раздела и не зависят от толщины диафрагмы (длины
* В рационализированной системе (см. стр. 3) =
цИ)
“ DD0EA •
** В рационализированной системе (см. стр. 3)
г . V-v
~ IDD0
и С =
(За)
181
капилляров), от площади ее поперечного сечения (числа пор) и
от радиуса отдельных капилляров *.
Таким образом, объем перенесенной жидкости V, рассчитан-
ный на единицу количества электричества q, должен быть вели-
чиной постоянной для границы раздела между фазами опреде-
ленного состава
V V v
- = 7F = -f= const
В случае же расчета объема перенесенной жидкости на
1 в)сек по формуле (2), т. е. — const, необходимо
учесть структурные характеристики I и А, так как они опреде-
ли ы Е
ляют градиент потенциала на диафрагме /7 = -у и пропорцио-
о V
нальную ему величину линеинои скорости жидкости w =
Таким образом, градиент потенциала, вызывающий электро-
осмотический перенос жидкости, входит в неявном виде в
оба выражения для ^-потенциала; в уравнении (3) он вы-
ражен через отношение плотности тока к электропроводности
раствора.
Движущие электроосмотические силы в пористых объектах
могут быть охарактеризованы с помощью максимального
электроосмотического поднятия. Если жидкость, пе-
редвигающуюся вследствие электроосмоса, заставить подни-
маться в одной из частей прибора, то возникает гидростатиче-
ское давление, направленное в сторону противоположную элек-
троосмотическому переносу. Очевидно, по мере подъема жидко-
сти скорость электроосмотического переноса будет замедляться.
В равновесных условиях гидростатическое давление полностью
уравновесит электроосмотический перенос и движения жидко-
сти наблюдаться не будет.
Величина гидростатического давления, уравновешивающая
электроосмотический перенос жидкости, называется максималь-
ным электроосмотическим поднятием и определяется форму-
лой**: •
. 2DZE ...
“макс — W
* Завиимость £ от радиуса капилляра может иметь место при очень
малых радиусах, соизмеримых с толщиной двойного слоя, .вследствие не-
полного развития диффузных слоев. При очень больших радиусах, когда дви-
> жение ионов вдоль стенки не может увлечь за собой всей массы жидкости
в капилляре значения v будут преуменьшенными, а следовательно и ?-по-
тенциал, вычисленный по формуле (3), будет меньше истинного значения,
8 DD ^F
** В рационализированной системе (см. стр. 3) ЛМакс в—•
182
где ^макс — величина максимального электроосмотического под-
нятия;
С — электрокинетический потенциал;
Е — приложенное напряжение;
г — эффективный радиус пор диафрагмы.
Выполнение работы
Для практического определения .величины ^-потенциала ме-
тодом электроосмоса, в зависимости от характера исследуемого
Объекта, могут быть применены различного типа приборы.
Рис. 74. Прибор для измерения электроосмоса в жестких
мембранах.
Если исследуемый образец представляет собой жесткую диа-
фрагму, обладающую известной механической прочностью и ма-
лой протекаемостью (например, целлофан, коллодиевая мембра-
на, керамическая диафрагма), его зажимают во фланцах ме-
жду двумя сосудами, наполненными исследуемым раствором.
При исследовании порошков капиллярная система образуется
путем формирования порошка между двумя перфорированными
пластинками.
Измерение электроосмоса в жестких мембранах. Измерение
объемной скорости электроосмоса производится в приборе, скон-
струированном в лаборатории коллоидной химии ЛГУ (рис. 74).
Диафрагма зажимается между резиновыми прокладками во
фланцах 2 и 3, изготовленных из плексигласа. В отверстия флан-
цев вклеены сосуды 4, заполняемые раствором электролита. Со-
суду придается изогнутая форма для предотвращения диффузии
к диафрагме из электродного пространства за время опыта.
183
Неполяризующиеся электроды 5, вводимые на пробках, пред-
ставляют собой трубки, заполненные в верхней части раствором
CuSO4, в нижней — 3% студнем агара, содержащим электролит.
В раствор погружена медная проволока, проходящая через проб-
ку, закрывающую трубку. Отсчетные капилляры 1, оканчиваю-
щиеся чашечками, вводятся на шлифах или пробках в ближай-
шие к фланцам отверстия сосудов.
Отсчетные капилляры, на которые нанесены деления, пред-
варительно градуируют для определения цены деления в едини-
цах объема (см3). Такую градуировку легко произвести с по-
мощью ртути. Для этого в тщательно вымытый и высушенный
капилляр втягивают небольшое количество ртути. Замечают,
скольким делениям на капилляре отвечает данная навеска. За-
тем столбик ртути осторожно перемещают по длине капилляра
и делают ряд отсчетов. Из полученных значений берут среднюю
величину. Ртуть переносят в бюкс и взвешивают. Объем капил-
т
ляра в пределах отмеченных делении равен где т — масса
ртути, d—плотность ртути, равная 13,6 г1см?. Зная число деле-
ний N, отвечающих данной навеске, находят объем а, соответ-
„ т
ствующии одному делению капилляра а = .
Собранный прибор с открытыми верхними отверстиями на-
полняют до уровня отверстий. При заполнении удаляют пузырь-
ки воздуха путем покачивания прибора, вводят отсчетные капил-
ляры, заполняют раствором полностью электродные простран-
ства и вставляют электроды. Затем, при помощи кранов в ниж-
ней части прибора, медленно выпускают часть раствора так,
чтобы уровни менисков в отсчетных капиллярах установились
вблизи середины шкалы.
Прибор для работы устанавливают так, чтобы капилляры на-
ходились в горизонтальном положении и на одном уровне во
избежание фильтрации жидкости из одной половины в другую.
После заполнения прибор проверяют на отсутствие пузырьков
воздуха и на герметичность. Если положение менисков не из-
меняется в течение .3—5 мин, это показывает, что прибор собран
правильно и можно включать ток. После включения тока пре-
жде всего определяют знак заряда исследуемой. диафрагмы.
Знак заряда определяют по направлению движения жидкости
через диафрагму по отношению к полюсам источника тока. На-
правление определяется по перемещению менисков в отсчетных
капиллярах.
После этого устанавливают величину объемной скорости
электроосмотического переноса жидкости v.
Отмечают начальные положения менисков по шкале для пер-
вого и второго NH капилляра в покое. Затем включают од-
новременно ток и секундомер, и отмечают конечные положения
184
N'k и Nk- Разность NK— NH^=N3 дает число делений, пройден-
ных за время t. Меняя направление тока при помощи переклю-
чателя, производят ряд отсчетов, отмечая каждый раз силу то-
ка I. Полученные данные записывают в виде таблицы.
в уравнение (3). Значения D и т] берут из справочных таблиц
для чистой жидкости, так как небольшие количества электро-
лита существенно не влияют на эти величины. Для воды и вод-
ных растворов*/) = 81; т] = 0,01 пз при 20° С.
Величина удельной электропроводности х Данного раствора
определяется в обычной ячейке.
Для получения величины электрокинетического ^-потенциала
в вольтах необходимо ввести переводный множитель, так как
все величины, входящие в уравнение (3), были выражены в аб-
солютных единицах CGSE **.
Таким образом, если значения I мы выразим в а, х— в
ом~1 • смг1, a g — в в, то в формулу (3) необходимо ввести пере-
счетные коэффициенты
С 4лт;-л 9 • 10%
D/-3-10»
или
С=^.9.Ю4в
Результат вычисления должен содержать величину и знак
^-потенциала, соответствующий знаку заряда твердой фазы, т. е.
внутренней обкладки двойного слоя.
Для работы могут быть рекомендованы коллодиевые мем-
браны (приготовление см. стр. 55) или керамические диафраг-
мы (например, выточенные из слабо обожженных фарфоровых
пластинок, применяемых в органическом анализе), вклеенные в-
* Время t выражают в секундах. Если измерения проводились в оди-
наковые отрезки времени и сила тока была постоянной, можно вычислить
среднее значение Na и, пользуясь им, найти v/1.
** В рационализированной системе (см. стр. 3) t, вычисляется непо-
средственно по формуле (За) (г] — н сек/м2, v — м3/сек, I — а, и —
ОМ'1 • At'1).
185
обойму из оргстекла (зажимаемую между фланцами). Рабочими
растворами могут служить 0,005 —0,01 н. растворы КС1, NaCl,
А1С13. Перед работой диафрагмы выдерживаются в растворе
1—2 суток.
Измерение электроосмоса в порошковых диафрагмах. Для
измерения электроосмоса в порошковых системах может быть
рекомендован прибор, схематически представленный на рис. 75.
Основной частью прибора является U-образная трубка 4.
В нижнюю часть одного из колен трубки впаян стеклянный
крупнопористый фильтр 6, при формировании диафрагмы его
закрывают кружком фильтровальной бумаги; трубка над фильт-
ром заполняется суспензией порошка. Путем седиментации по-
рошка или, в случае медленно оседающих частиц, осторожно от-
сасывая жидкость на водоструйном насосе, получают столбик
порошка необходимой толщины.
Рис. 75. Прибор для измерения электроосмоса
Ь порошковых мембранах.
Как было указано ранее, для поверхности определенной хи-
мической природы при определенном составе и концентрации
прилегающего к ней раствора, электрокинетический потенциал —
величина постоянная. Однако практически требуется соблюде-
ние ряда условий, чтобы полученная величина скорости движе-
ния жидкости через капиллярную систему была действительной
характеристикой электроосмотической скорости. В системе дол-
жны быть созданы все условия для установления ламинарного
стационарного потока жидкости. На это положение в особенно-
сти следует обращать внимание при работе с такими сложными
капиллярными системами, как порошковые, где для установле-
ния ламинарного стационарного потока жидкости необходимо
использовать диафрагмы достаточно большой толщины. Толщи-
на диафрагмы не должна быть ниже определенного критического
значения. Это значение не является постоянным, оно увеличи-
вается с увеличением радиуса капилляров пор диафрагмы и с
уменьшением £-потенциал а. Для .обычно применяемых в работе
186
порошков критическое значение имеет порядок нескольких см-
ири меньшей толщине движение ионов двойного слоя не сможет
увлечь всей массы жидкости и вычисленные из объемной скоро-
сти значения £ окажутся преуменьшенными.
Фильтр 5 должен быть закреплен в приборе как можно бо-
лее плотно. Поэтому сверху между фильтром 5 и пробкой, удер-
живающей электролитический ключ 3, рекомендуется помещать
стеклянную трубку^ плотно прижимающую фильтр и не мешаю-
щую свободному продвижению жидкости. После того как мем-
брана сформирована и фильтр 5 закреплен, прибор заполняется
^исследуемым раствором и к нему присоединяются электролити-
ческие ключи и отсчетйые капилляры /, с помощью которых
Определяется объем перенесенной жидкости. С помощью элек-
йгролитических ключей 3 и 7, опущенных в стаканчики с раство-
Цом CuSO4, система соединяется с полюсами источников тока.
(Градуировка капилляров для измерения и расчеты описаны вы-
Sjttie (стр. 184).
t Измерение максимального электроосмотического поднятия.
$Для измерения максимального электроосмотического поднятия
^используются приборы того же типа как и для определения ско-
рости электроосмотического переноса ^-потенциала. Заполнение
Приборов, определение их герметичности и горизонтальности по-
ложения капилляров производится так же, как в предыдущей
работе. Для определения величины поднятия один из капилля-
ров присоединяется к манометрической установке. При включе-
нии тока мениск в отсчетном капилляре начинает двигаться. На
^систему подается давление до тех пор, пока движение мениска
йне прекратится. Величина •приложенного давления, очевидно,
равна величине максимального электроосмотического поднятия.
В случае крупнопористых объектов величины электроосмоти-
ческого поднятия относительно малы и тогда более точные зна-
чения последней в данном случае могут быть получены с помо-
щью несколько иной методики, а именно, отсчетные капилляры
1 устанавливаются вертикально и манометрической установкой
не пользуются. Величина максимального электроосмотического
-поднятия определяется по- разности уровней в отсчетных капил-
лярах.
Потенциал протекания
Р а б о т а 33
ОПРЕДЕЛЕНИЕ ЭЛЕКТРОКИНЕТИЧЕСКОГО ПОТЕНЦИАЛА ДИАФРАГМ МЕТОДОМ
ПОТЕНЦИАЛА ПРОТЕКАНИЯ
Потенциалом протекания Е называется разность
потенциалов, возникающая между концами капилляра или си-
.стемы капилляров при протекании раствора под действием внеш-
него давления.
187
Измерение потенциала протекания является одним из наибо-
лее надежных методов определения величины ^-потенциала в
капиллярных системах.
Поток жидкости, протекая через систему капилляров, увле-
кает за собой ионы диффузной части двойного электрического
Рис. 76. Схема возникновения потенциала протекания при движении
жидкости через поры мембраны.
слоя. Это движение ионов, являясь поверхностным током, и при-
водит к возникновению разности потенциалов между объемами
раствора, разделенными капиллярной системой (рис. 76).
По мере увеличения разности потенциалов, движение ионов,
находящихся в избытке (например, катионов при отрицательном
заряде твердой фазы), все более замедляется, так как оно на-
188
правлено против возникающего одноименного объемного заряда
(распределяющегося равномерно и практически мгновенно по
всему объему, вследствие значительной электропроводности
воды и водных растворов); в то же время ускоряется движение
ионов другого знака, так как возникший градиент потенциала
способствует их движению. Эти процессы эквивалентны возник-
новению встречного объемного тока, обусловленного потенциа-
лом Е. В результате установится стационарное состояние с по-
стоянной величиной Е, при котором поверхностный ток скомпен-
сирован объемным, и через капилляр будут проходить с раство-
ром эквивалентные количества анионов и катионов.
Гельмгольц и Смолуховский разработали теорию потенциала
рротекания и получили для стационарного состояния выражение,
Связывающее потенциал протекания Е с величиной электрокине-
тического потенциала Смысл этого выражения можно уяснить
цз следующих рассуждений. Величина потенциала протекания
Сбудет тем выше, чем больше ионов диффузного слоя будет вы-
йесено из капилляра в единицу времени. Количество этих ионов
йропорционально, с одной стороны, ^-потенциалу, с другой —
объемной скорости жидкости, тем большей, чем больше прило-
женное давление Р и чем меньше коэффициент вязкости жидко-
сти Т].
Наряду с этим Е будет тем выше, чем меньше встречный
объемный ток, пропорциональный удельной'электропроводности
раствора х.
. Объединяя сказанное, получим:
СР V.E
Коэффициент пропорциональности, определяемый уравнени-
ем Пуассона, равен (работа 32), где D — диэлектрическая
проницаемость.
Таким образом, мы приходим к уравнению Гельмгольца —
Смолуховского для потенциала протекания *:
г__ 4пур.Е
Q DP~
Величина ^-потенциала не должна зависеть от структурных
характеристик диафрагм. Действительно, на основании много-
численного экспериментального материала установлено, что
(5)
* В рационализироваииой системе (см. стр. 3):
г . _
t ~ DDoP
= DDqP (V +
(5а)
(6а)
189
отношение -р , а следовательно и £, не изменяется с изменением
радиуса капилляров *, их числа и длины (толщины диафраг-
мы). Таким образом, как и следовало ожидать (см. стр. 178),
величина ^-потенциала является определенной электрокинетиче-
ской характеристикой твердой фазы, соприкасающейся с раство-
ром данного состава и концентрации.
Поскольку фактическая величина удельной электропроводно-
сти в порах х отличается от таковой в свободном растворе
zv, в уравнение вводится поправка на поверхностную проводи-
мость xs:
x = xv + xs
£=-^(v+%s) (6)
или
С = аСэ
х. -|-'х '
где а==—------— неисправленное, L, — исправленное значе-
7v
ние электрокинетического потенциала.
Таким образом, поправочным множителем является коэф -
фициент эффективности а, определяемый эксперимен-
тально** (работа 38). Величина xv определяется при данной
температуре' в обычном сосуде для электропроводности. Из
уравнений (5) и (6) следует, что величина потенциала протека*
ния прямо пропорциональна приложенному давлению, иначе го-
воря, график Е— Р должен давать прямую, проходящую через
начало координат. Проверка этого положения служит контролем
примененной методики по определению потенциала протекания.
В работе поставлены следующие задачи: 1) измерение потен-
циала протекания коллодиевой или керамической (см. электро-
осмос) диафрагмы в водном 0,001—0,003 н. растворе электро-
лита при различных давлениях (15—ЗО.смрт. ст.) и построение
графика Е— Р; 2) вычисление ^-потенциала диафрагмы и ис-
правление этой величины посредством введения поправки на
поверхностную проводимость **.
Выполнение работы
Измерение потенциала протекания в жестких мембранах. На
жестких мембранах и диафрагмах в растворах хлоридов потен-
* За исключением очень узких капилляров, радиус которых соизмерим
с толщиной двойного электрического слоя.
** В работах общего практикума величина « не определяется и испра-
вление величины £ не производится.
190
циал протекания измеряется как разность потенциалов между
двумя хлоросеребряными электродами 2 и 4, помещенными
в раствор с двух сторон диафрагмы (рис. 77). Прибор для изме-.
рения соединен посредством переключателя 7 с потенциоме-
трической установкой 10, а также с баллоном 1 и манометром 6
посредством трехходового крана 5.
Рис. 77. Схема установки для измерения потенциала протекания
в жестких мембранах.
Коллодиевая или керамическая диафрагма *, предваритель-
;но пропитанная раствором электролита путем выдерживания
д нем в течение суток, нажимается между резиновыми проклад-
ками во фланцах прибора 5. Прибор наполняют тем же раство-
ром засасыванием через нижние отростки. Измеряется разность
Потенциалов между электродами без давления ко, т. е. потен-
циал асимметрии. Измерив ко, проверяют его устойчивость во
времени (3—5 мин). После этого соединяют одну половину
Прибора с манометром и включают давление посредством пово-
t' * Керамические диафрагмы удобнее всего вклеить при помощи замаз-
ки в обойму из плексигласа.
191
рота крана 5. Разность потенциалов кл измеряют через 3 мин
после включения давления и затем повторяют измерение, чтобы
убедиться в постоянстве величины Ki во времени. Определяют
знак заряда твердой фазы. Для этого устанавливают направле-
ние возникающей э. д. с., (по клеммам потенциометра при ком-
пенсации), в зависимости от направления давления, а следова-
тельно и знак заряда твердой фазы в соответствии со схемой
(рис. 77). После этого отключают давление, снимают каучуко-
вую трубку и вновь измеряют ко. Затем включают давление на
другую половину прибора и опять измеряют потенциал. Вели-
чина £ вычисляется как разность ki—ко- Если значения ki или
ко несколько изменяются, для расчета берут величину ко, изме-
ренную непосредственно после снятия давления.
Для удобства записи необходимо условиться относительно
знаков, например считать положительными давление, а также
э.*д. с., если они направлены слева направо (если положитель-
ным является левый электрод).
Измерения для каждой стороны проводят 2—3 раза, после
чего вычисляют среднюю величину Е при данном давлении. Опи-
санную серию измерений повторяют при различных величинах
давления (например, 15, 20, 25, 30 см рт. ст.), после чего строят
график Е — Р и вычисляют ^-потенциал.
Таблица 2
Диафрагма коллодиевая № , 0,001 н. КС1
t от начала опыта, р, 7Г1, 1Г0, Е, дур,
сек см рт. ст. Мв мв мв мв[см рт. ст.
0 0 4-0,3 — , IT
3 0 4-о,з — —_
3 4-15,0 —3,7 ——
6 15,0 —3,9 —— —
9 15,0 —3,9 —4,3 —0,29
9 0 4-0,4 — —
12 0 4-0,4 — —
12 —15,0 —
15 —15,0 4-4,9 — —
18 —15,0 4-5,0 4-4,5 —0,30
18 0 4-0,5 —— —
и т. д.
Запись опыта ведут по следующей схеме (табл. 2). Для вы-
числения £ по уравнению (5) все величины должны быть выра-
жены в системе CGSE *. Поэтому, при подстановке величин Р,
* В рационализированной. системе (см. стр. 3) £ вычисляется непо-
средственно по уравнениям (5а, 6а) (ц—н-сек!мг, Р — н/м2, £ и Е — в).
192
g, Ё и х в практических единицах необходимо ввести числовой
коэффициент, а именно:
С 4лт) Е %-9-Ю’1
300-1000 ~ D ’ 300-1000' Р-981-13,6
или
С_в,75.№.«
где Р — в см рт. ст., g и Е — в мв и х — в ом 1 • см~\
Е
Определяют графически среднюю величину -р и, подставляя
ее в формулу, вычисляют -g-потенциал. Результат вычислений
должен содержать величину и
знак g-потенциала, равный знаку
заряда стенки.
Измерение потенциала проте-
кания в порошковых диафраг-
мах. Опыт проводят в приборе,
предназначенном для одновре-
менного измерения потенциала
протекания, поверхностной про-
водимости (стр. 218) и коэффи-
циента протекаемости (стр. 57),
изображенном на рис. 78.
Порошковая диафрагма 2
формируется в трубке 5 путем
многократного наполнения ее
суспензией порошка в воде или
разбавленном растворе электро-
лита * с последующим отсасыва-
нием (например, водоструйным
насосом). Для измерения потен-
циала протекания служат хлоро-
серебряные электроды 6 и 7,
подключаемые к клеммам потен-
циометра. Измерения произво-
дятся при одном направлении да-
Рис. 78. Ячейка для измерения по-
тенциала протекания и поверх-
ностной проводимости в порош-
ковых мембранах.
вления, в том же порядке, что и для жестких мембран
(стр. 190). Раствор фильтруют через диафрагму в направлении
сверху вниз до установления равновесия, характеризуемого по-
стоянством величины Е/Р.
При работе необходимо следить за тем, чтобы при формиро-
вании или во время работы с диафрагмой в нее не попадал воз-
дух, а также за отсутствием воздушных пузырьков в трубке под
основанием (для их удаления наклоняют прибор, заполненный
* Во избежание образования рыхлой коагуляционной структуры,
193
раствором). Запись ведут по схеме табл. 2; указывается знак
электрода 6, стоящего перед диафрагмой, давление обозначается
как положительное.
В литературе описаны также приборы, в которых порошко-
вая диафрагма запрессовывается между дисковыми перфориро-
ванными платиновыми электродами. Преимущество этих прибо-
ров заключается в уменьшении сопротивления, а следовательно
в повышении чувствительности, недостаток — в том, что потен-
циалы на платине менее воспроизводимы и медленно устанавли-
ваются вследствие поляризации. Покрытие платины слоем
Ag + AgCl не дает, положительных результатов, так как AgCl
легко стирается при запрессовке, загрязняя диафрагму. Кроме
того, наличие переменного сопротивления этого слоя затрудняет
измерение поверхностной проводимости.
Электрофорез
Движение заряженных частиц, взвешенных в неподвижной
жидкости, под влиянием электрического поля, называется элек-
трофорезом.
В зависимости от знака заряда частицы передвигаются
к аноду или катоду, поэтому, наблюдая за направлением пере-
движения частиц, можно
определить знак их за-
ряда.
Движение частицы в
жидкости, в электриче-
ском поле можно пред-
ставить себе следующим
образом.
Рис. 79. Схема электрофореза. Как это показано на
рис. 79,а, частица в жидко-
сти окружена двойным электрическим слоем ионов. При прило-
жении электрического поля (рис. 79, б) распределение ионов в
диффузном слое нарушается: иоиы диффузного слоя начинают
смещаться по отношению к частице и происходит непрерывный
обмен ионными атмосферами вокруг частиц, тогда как сама
частица с частью противоионов движется по направлению про-
тивоположного заряженного полюса. Так, отрицательно заря-
женная частица двигается к положительному полюсу, положи-
тельно заряженная — к отрицательному полюсу.
Измерив скорость движения частиц и зная градиент потен-
циала приложенного электрического поля, можно вычислить
электрофоретическую подвижность частиц.
Электрофоретической подвижностью U назы-
вают путь, который проходят частицы в секунду, при градиенте
потенциала 1 в/см.
194
Электрофоретическая подвижность рассчитывается по урав-
нению:
JT — — . w Е
U-~ГН—Н' Н^~Т
гдё Л —путь, пройденный частицами, сл;
I — время, сек\
„ Е
п — -----градиент потенциала внешнего электрического поля;
' Е — разность потенциалов, в;
I—расстояние между электродами, см.
Согласно уравнению Гельмгольца — Смолуховского, электро-
форетическая подвижность прямо пропорциональна электроки-
jнетическому потенциалу частиц £ и обратно пропорциональна
коэффициенту вязкости жидкости ц
.._ CD [ см/сек ]
4ят]
в/см
гдё D — диэлектрическая постоянная
: или
(7)
____ 4ят)(7’
С D~
Зная электрофоретическую -подвижность можно вычислить
величину ^-потенциала, Так, например, в воде (при 18°) ц = 0,01,
.0 = 81; при градиенте электрического поля Н = 1 в/см (1 е==
1 . „ 4.3,14-0,01
==’300 абсолютных электростатических единиц), (. = —.^300"
абсолютных единиц. Тогда величина электрокинетичёского по-
тенциала в вольтах будет
4 3,14 • 0,01 • 300 4-3,14-0,01 ..
---8Г-1/300 ‘ ~-------81-----(300) -U
С
или
H = 14OU в*.
При электрофоретической подвижности частиц, равной
С = 14 мв
1 мк!сек (т ц)- 4 см/сек \
в/см \ ' ‘ в/см )’
Опыт показывает1, что в воде частицы самой различной хими-
ческой природы (живые клетки, различные суспензии, эмульсии)
сравнительно мало отличаются друг от друга по электрофорети-
ческой подвижности.
* В рационализированной системе (см. стр. 3) С = (7а) или
10-3
С = 81-8,854. Ю"18-^ = 1,4 ’ltW ГДО U м1сек1в1м-
195
Электрофоретическая подвижность частиц обычно лежит
в пределах от 2 до 4 мк/сек.
Дебай и Гюккель вводят в уравнение Смолуховского число-
вой коэффициент К, который зависит от формы частиц. Если
принять, что коллоидные частицы имеют сферическую форму и
применить уравнение Стокса, то электрофоретическая скорость
должна быть равна:
Согласно Генри, исследовавшему этот вопрос, размеры и
форма частиц влияют на электрофоретическую подвижность
в том случае, когда толщина двойного слоя сравнима с величи-
ной частиц, т. е. в случае очень маленьких частиц; в остальных
случаях уравнение Гельмгольца — Смолуховского остается пра-
вильным.
Измерение электрофоретической подвижности можно про-
изводить двумя методами: макроскопическим и микроскопи-
ческим.
Работа 34
ОПРЕДЕЛЕНИЕ ЭЛЕКТРОКИНЕТИЧЕСКОГО ПОТЕНЦИАЛА ЗОЛЕЙ МЕТОДОМ
ЭЛЕКТРОФОРЕЗА (МАКРОСКОПИЧЕСКИЙ МЕТОД)
Среди ряда макроскопических методов электрофореза наибо-
лее распространены методы с подвижной границей.
Для измерения электрофоретической скорости в этом случае
наблюдают за перемещением границы между золем и наслоен-
ной на него жидкостью в электрическом поле. Наблюдения за
скоростью передвижения границы золь — боковая жидкость про-
изводят в различных приборах, в зависимости от характера ис-
пытуемого золя.
На рис. 80 изображен прибор Бэртона, сконструированный
в виде U-образной трубки с ответвлением, снабженным ворон-
кой 1 и краном 2. Электроды 3 и 4 погружены в боковую жид-
кость, которая наслаивается на золь.
Кён ввел некоторые усовершенствования в прибор Бэртона,
заключающиеся в том, что боковые трубки градуированы и
имеются два боковых крана (рис. 81).
Дальнейшее видоизменение сделано Рабиновичем и Фодиман
(рис. 82).
Градуировка боковых трубок нанесена по обе стороны кра-
нов, что представляет удобство для работы с такими золями, ко-
торые имеют меньшую плотность, чем боковая жидкость. В этом
случае боковая жидкость наливается также в нижнюю часть
основной трубки прибора. Электроды отведены в специальные
196
боковые ответвления, которые могут быть заполнены агаровым
студнем.
Простым и удобным прибором является прибор, предложен-
ный Чайковским, использующийся в лаборатории кафедры кол-
лоидной химии ЛГУ (рис. 83).
Прибор состоит из трубки 1 с двумя градуированными коле-
нами, в которые вставлены агаровые
%бор с электродами. В среднее от-
ветвление 2 вставляется на пробке
пипетка с исследуемым золем так,
чтобы кончик пипетки отстоял от
стенки трубки на I—2 мм.
Исследуемый золь набирают в
пипетку, на конец которой надет
кусок резиновой трубки, зажатый
с помощью винтового зажима. Пи-
петку вставляют в трубку, напол-
ненную до половины боковой жидко-
ключи, соединяющие при-
Рис. 81. Прибор Кёна
для электрофореза.
Рис. 80. Прибор Бэртона
для электрофореза.
стью, свободной от дисперсной фазы. При заполнении прибора,
очень осторожно и медленно открывают зажим, при этом золь
постепенно вытесняет боковую жидкость в градуированных
коленах, образуя резкую границу, необходимую для наблюде-
ния. Когда граница раздела золя в обоих коленах дойдет при-
мерно до середины боковых трубок, зажим закрывают и, убе-
дившись в герметичности прибора, отмечают уровень границы
раздела в обоих коленах.
197
В качестве электродов лучше пользоваться Обратимыми (в
частности медными в растворе, CuSO<) в отдельных сосудах
(стаканчиках) и агаровыми ключами, вставленными в колена
боковых трубок. Прибор включают последовательно с миллиам-
перметром в цепь постоянного тока с напряжением — 100 в.
Отмечают направление и скорость передвижения границы
золь — боковая жидкость с помощью секундомера.
Сделав ряд определений в обоих коленах, с помощью пере-'
ключателя производят перемену полюсов и повторяют измере-
ния. Для расчета берут средйее значение скорости перемещения
границы. Для определения величины градиента потенциала Н
необходимо определить разность потенциалов на электродах и
расстояние между ними. Лучшие резуль-
таты получаются при определении удель-
ной электропроводности золя <х, силы
тока I н площади сечения А баковых ко-
лен трубки 1.
В этом случае, градиент потенциала
Н будет определяться следующим соот-
ношением:
Рие. 82. Прибор Рабино-
вича и Фодиман для
где I — сила тока, в а;
х — удельная электропроводность
жидкости, ом~1 • см~';
А — площадь сечения трубки, см2 (м2).
Способ измерения электрофоретиче-
электрофореза. ской подвижности по перемещению гра-
ницы раздела золь — жидкость связан
с некоторыми затруднениями и необходимостью выполнять ряд
условий, нарушение которых приводит к ошибкам.
Весьма существенным является вопрос о составе боковой
жидкости, в которую погружены электроды.
Для получения более резкой границы между золем и боко-
вой жидкостью в течение опыта нужно, чтобы электропровод-
ность боковой жидкости была равной или немного больше, чем
электропроводность золя. Равенство электропроводности золя и
боковой жидкости важно еще и потому, что тогда при расчетах
градиента потенциала (входящего в формулу для вычисления
электрокинетического потенциала) не вносится ошибки за счет
неравномерного падения потенциала по расстоянию между элек-
тродами.
Наилучшие результаты получаются при применении в каче-
стве боковой жидкости ультрафильтрата золя, содержащего те
же ионы, что и золь. Вместо ультрафильтрата можно пользо-
198
ваться раствором после центрофугирования золя. Полученные
данные записывают в таблицу.
. Таблица 3
Сила тока, а Время наблюдения it сек Путь, пройденный частицами,. см Градиент потенциала Н, в/см Электро- форетическая скорость U, мк]сек'в ^потенциал, мв
*-
Из средних значений скорости U вычисляют величину ^-по-
тенциала по уравнению (7). Значения вязкости г) для чистой
жидкости (в пз) при t опыта берут из справочных таблиц; для
воды и разбавленных водных растворов т] = 0,01 пз, D для воды
и водных растворов равна 81. Удельную электропроводность бо-
ковой жидкости и золя определяют по обычной схеме Коль-
рауша.
199
Работа 35
ОПРЕДЕЛЕНИЕ ЭЛЕКТРОКИНЕТИЧЕСКОГО ПОТЕНЦИАЛА СУСПЕНЗИЙ МЕТОДОМ
ЭЛЕКТРОФОРЕЗА (МИКРОСКОПИЧЕСКИЙ МЕТОД)
Метод микроэлектрофореза состоит в наблюдении за пере-
движением частиц непосредственно в микроскоп. Суспензию ви-
димых в микроскоп частиц помещают в маленькую стеклянную
камеру, прилагают разность потенциалов между концами кю-
веты, после чего фиксируют положение отдельной частицы и
измеряют путь, который она проходит за определенный проме-
жуток времени.
Метод микроэлектрофореза, являясь более чувствительным,
чем макрометод, имеет целый ряд и других преимуществ. Метод
этот дает возможность непосредственно определять размеры и
форму движущихся частиц, если это представляет интерес.
Исследования могут производиться в растворах солей как очень
малых, так и относительно больших концентраций. Свойства
дисперсионной среды не изменяются заметным образом в тече-
ние опыта. Отдельные измерения занимают мало времени.
Недостатком метода микроэлектрофореза является то, что на
электрофоретическую скорость частиц может налагаться ско-
рость электроосмотического потока дисперсионной среды, дости-
гающей заметной величины вследствие малого сечения капил-
ляра микрокамеры. При наличии электроосмоса наблюдаемая
скорость передвижения частиц в электрическом поле изменяется
по глубине камеры, так как она слагается из истинной электро-
форетической скорости частиц и скорости движения жидкости.
Поскольку электрофоретическая микрокамера герметически
закрыта, то возникший электроосмотический поток жидкости на
границе со стеикой камеры, направленный к одному полюсу
источника тока, обусловливает обратный ток жидкости в цен-
тральной части камеры. Если дисперсионная среда — вода или
водный раствор, то для стеклянной, плоской микрокамеры на
границе со стеклом, несущим отрицательный заряд, как со сто-
роны верхней части, так и у дна камеры, жидкость будет пере-
мещаться по направлению к катоду, оттекая в середине камеры
к аноДу. Поэтому, если частицы суспензии отрицательно заря-
жены и движутся к аноду, то в середине камеры движение ча-
стиц будет ускоренным, а у поверхности и у дна — замедленным.
В идеально плоской камере наблюдаемая скорость передви-
жения частиц изменяется по параболе с изменением уровня —
по сечению камеры.
Наблюдаемая скорость передвижения частицы нНабл предста-
вляет собой алгебраическую сумму истинной скорости и скоро-
сти движения жидкости вследствие электроосмоса uw:
^набл = И |~ Пщ, (8)
200
Таким образом, истинная скорость, т. е. скорость движения
частицы относительно жидкости, не меняется с глубиной, а ка-
жущаяся или наблюдаемая в зависимости от глубины изме-
няется.
Для закрытой плоской камеры имеет место условие:
d
~ J uwdx = О
о
где d— толщина камеры;
х — расстояние от стенки.
Откуда:
d
U = ^набл dx
О
Измеряя Ннабл на различных уровнях, можно найти истинную
скорость и посредством графического интегрирования.
Более удобные условия, исключающие электроосмотическую
скорость, предложил Смолуховский, который вывел уравнение,,
дающее возможность вычислить истинную скорость электрофо-
реза, если кажущаяся величина скорости измерена на двух глу-
бинах камеры.
На глубине х скорость переноса жидкости uw выражается
уравнением:
где w — электроосмотическая скорость;
d — толщина камеры.
Вблизи стенки камеры х = 0, uw — и; следовательно:
«набл = “ + 'г (10)
Из уравнения (9) можно определить значение при ко-
тором 0. Решая квадратное уравнение (9) относительно .
находим, что нш = 0 при значении -—=0,211 и 0,789; следова-
тельно, измерения, сделанные на глубины, дают истинное зна-
чение электрофоретической скорости, что было подтверждено
на плоской камере закрытого типа.
Поэтому электрофоретические измерения в плоской камере
можно производить, наблюдая за скоростью' движения частиц
на определенной глубине и пренебрегая движением жидкости.
Для определения электрофоретической подвижности поль-
зуются следующим уравнением:
U — Дизм. см/сек (11)
Н в/см ' '
201
где:
—
А -X
Отсюда величина ^-потенциала *
c=jM« 300S
HU
(12)
где Н—градиент потенциала, I — сила тока а,
х — удельная электропроводность жидкости, ом~’ • см~’,
А — площадь поперечного сечения камеры, см2, остальные
обозначения даны в начале раздела.
Выполнение работы
Рис. 84. Микрокамера Абрамсона — Дорф-
мана для электрофореза.
Для количественных измерений электрофоретической под-
вижности пригодны только герметически закрытые плоские ка-
меры. Одной из лучших микрокамер такого типа является ка-
мера Абрамсона, видоизмененная Дорфманом (рис. 84). Прибор
состоит из стеклянной
плоскопараллельной ка-
меры б высотой 0,5—1мм,
шириной 7—12 мм и дли-
ной 30—40 мм, вставлен-
ной на шлифах в боко-
вые отростки 5 и 7 трех-
ходовых кранов. Послед-
ние дают возможность
наполнить камеру суспен-
зией, помещенцой в во-
ронку 8, или соединить
камеру со вставленными
в нижние трубки 2, 10 неполяризуюшимися электродами 1 и И.
Наполнение прибора и его промывка производятся при помощи
воронки 8, отростка 12 и каучуковой трубки 4, снабженной за-
жимом. В качестве обратимых электродов употребляются мед-
ные в растворе CuSO4; для предохранения камеры от проникно-
вения в нее продуктов электролиза из электродных пространств
верхняя часть трубок 3 и 9 заполняется небольшим количеством
агарового студня. Краны для герметичности смазываются ва-
куумной смазкой,или чистым (прокипяченным в дистиллирован-
ной воде) вазелином. В качестве источника тока пользуются
аккумуляторами или сухими батареями на 100 в. Сила тока из-
меряется миллиамперметром. Путь, пройденный частицами, от-
* В рационализированной системе С = и , где %, олг1 • лг1; Н, в/м,
п D Un
I, а.
202
мечается с помощью калиброванного окулярного микрометра.
Время отмечается секундомером. Направление тока меняется
с помощью переключателя.
Прежде чем приступить к измерению электрофоретической
скорости, надо найти 1/5 глубины камеры, для чего определяют
высоту камеры. Определение высоты производится следующим
образом. С помощью туши на верхней и нижней стенках ка-
меры, приблизительно в центре, делаются маленькие отметки а
и щ (рис. 85). В камеру засасывается тонкоизмельченная сус-
пензия графита, частицы которого оседают на дно камеры.
Микроскоп фокусируют на верхнюю метку а. Далее с помо-
щью. микрометрического винта микроскопа с известной ценой
деления фокусируют микроскоп на частицы графита, осевшие на
дно камеры, и определяют расстояние
от точки а до точки би а также от
точки а до точки а.\.
После этого камеру переворачи-
вают меткой а\ кверху, фокусируют
микроскоп на метку и определяют
.расстояние от точки ai до Ь и от точки
01 до а: Зная расстояние от ах до а и
толщину стенки аб и а^би можно оп-
б
б,
а.
Рис. 85. Схема определения
высоты микрокамеры.
ределить высоту камеры. Площадь поперечного сечения камеры,
необходимую для расчета градиента потенциала И, находят из
средних значений ширины и высоты камеры.
Высоту камеры определяют, как указывалось выше. Значе-
ние ширины камеры удобно находить, пользуясь микроскопом
Брюнеля. Другой метод определения поперечного сечения, даю-
щий лучшие результаты, состоит в измерении электрофоретиче-
ской скорости частиц, имеющих определенную, постоянную под-
вижность U.
Определив скорость иИЗм—у* можно найти величину гра-
диента И, так как Зная силу тока и удельную'
электропроводность золя х, можно вычислить площадь попереч-
ного сечения А:
Путь, пройденный частицей за время t, определяют, наблю-
дая за частицей, пересекающей черту линейки окулярмикроме-
тра, и отмечая число пройденных частицей делений. Делается
20—30 отсчетов в обе стороны с переключением полюсов тока.
В качестве результата берут среднее значение. При значениях
градиента потенциала больше 12 в/см электроосмотический по-
ток жидкости в камере может иметь турбулентный характер,
203
Поэтому все измерения надо проводить при более Низких значе-
ниях. .
Определив согласно уравнению (11) электрофоретическую
Подвижность, вычисляют величину ^-потенциала. Полученные
.Данные записывают в следующие таблицы (табл. 4 и 5):
Таблица 4
Расстояние от а до «1 Расстояние от а до б\ Расстояние от ai до б Высота , камеры Площадь попе- речного сечения
Путь, пройденный частицами
(число делений)
Таблица 5
Из средних значений U вычисляют величину ^-потенциала по
уравнению (12).
ЭЛЕКТРОКИНЕТИЧЕСКИЕ СВОЙСТВА КАПИЛЛЯРНЫХ СИСТЕМ
Электрокинет ическими свойствами капил-
лярных систем называются свойства, которые проявляются
как следствие наличия двойного электрического слоя ионов на
границе раздела твердое тело — жидкость. К таким электроки-
нетическим свойствам капиллярных систем относятся электроки-
нетический потенциал, поверхностная проводимость и изменение
числа переноса ионов в порах капиллярной системы—мембраны.
Наличие поверхностной проводимости и избирательной про-
ницаемости капиллярных систем по отношению к различным
ионам электролитов, находящимся в растворе, соприкасающим:
ся с мембраной, обусловливает ряд особенностей, которые необ-
ходимо учитывать при рассмотрении многих процессов, проте-
кающих в природе, а также при использовании мембран для
технических Целей.
В качестве рримера той значительной роли, которую играют
электрокинетические свойства капиллярных систем — мембран
при их использовании в технике, можно указать на электродиа-
204
лиз, в настоящее время широко применяющийся в лабораторной
технике и промышленности для очистки различных дисперсных
систем и растворов от электролитов.
Изменение чисел переноса ионов в мембранах
Среди электрокинетических свойств капиллярных систем —
мембран и диафрагм существенную роль играет изменение
чисел переноса ионов в порах мембраны по сравнению со сво-
бодным раствором. Рассмотрим сущность данного явления.
Представим себе капилляр в продольном разрезе, наполненный
раствором электролита с двойным электрическим слоем ионов
на внутренней поверхности, при отрицательном заряде стенки
(рис. 86). В объеме АБВГ, где ионы сохраняют подвижность
при наложении электриче-
ского поля, концентрация
катионов больше, чем
анионов, вследствие того,
что сюда входят избыточ-
ные катионы диффузного
слоя. Этот объем всегда
можно рассматривать как
сумму объемов централь-
ной части капилляра —
д.. £. *. +..+.. + £ + + ±. Т + ±1*5
я.-Т.. ,г.Э-....У. ,±....Г...*. ...Т....Т..
+ ± 4- +4-+ + + ±. +
Рис. 86. Схема переноса ионов в широком
капилляре.
цилиндра с радиусом
А*— 8, где R — радиус капилляра, 8 — толщина диффузного
ионного слоя и объема цилиндрической оболочки тс/?2/—
— тг(/?— Б)2/, где I — длина капилляра.
В центральной части капилляра, вне пределов двойного элек-
трического слоя, числа переноса будут такие же, как и в сво-
бодном растворе без мембраны, так как подвижности и
концентрации ионов раствора, наполняющего капилляр в цен-
тральной части и в свободном растворе, одинаковы. В цилиндри-
ческой оболочке, входящей в двойной слой, вследствие влияния
электростатических сил поверхности, подвижности и концентра-
ции находящихся там ионов будут отличаться от свободного
раствора и поэтому числа переноса в этом слое будут иные,
чем в свободном растворе. Очевидно, что при больших радиусах
капилляра объем центральной его части, вне пределов двойного
слоя, будет составлять подавляющую часть общего объема ка-
пилляра, и поэтому то изменение, которое вносится ионами диф-
фузного слоя, ничтожно, и суммарное значение числа переноса
по всему сечению капилляра не изменяется по сравнению со сво-
бодным раствором.
Если же мы перейдем к капилляру, величина радиуса кото-
рого и толщина двойного электрического слоя близки, по разме-
рам, то нетрудно видеть, что в этом случае (рис. 87) в объеме
205
•А’Б’В’Г* будут' преобладать ионы двойного Слоя й Поэтому
числа переноса в этом-случае будут определяться подвижностью
и количеством ионов в двойном электрическом слое. При отри-
цательно заряженной стенке капилляра в объеме А’Б’В’Г’ число
переноса катиона будет приближаться к единице, тогда как чис-
ло переноса аниона будет близко к нулю.
. При положительно заряженной поверхности капилляра кар-
тина будет обратной, так как в диффузной части двойного слоя
будут преобладать анионы. Поэтому с уменьшением радиуса ка-
пилляра до пределов толщины двойного слоя, чи’сло переноса
аниона становится близким к единице, а число переноса ка-
тиона приближается к нулю.
Основываясь на приведенном механизме изменений чисел
переноса, приходим к. заключению, что с изменением радиуса
..i:.....i...+.. ±. +
Рис. 87. Схема переноса ионов в узком капилляре.
капилляров мембраны в сторону уменьшения мы должны на-
блюдать соответствующее увеличение числа переноса противо-
иона и уменьшение для иона одноименного по знаку заряда
с мембраной.
Изменение чисел переноса ионов должно зависеть от концен-
трации раствора электролита. С уменьшением концентрации
раствора доля, вносимая поверхностным сгущением ионов одного
Знака, будет оказывать большее влияние на соотношение /ионов,
проходящих через мембрану в электрическом поле, и изменение
чисел переноса должно возрастать.
Для измерения чисел переноса ионов через мембраны суще-
ствуют два основных метода. Первый — метод непосредствен-
ного аналитического определения концентрационных изменений
в растворе, вносимых мембраной, второй — методика диффузи-
онного потенциала.
Работа 36
ОПРЕДЕЛЕНИЕ ЧИСЕЛ ПЕРЕНОСА ИОНОВ В МЕМБРАНЕ
АНАЛИТИЧЕСКИМ МЕТОДОМ
Изменение концентрации растворов, омывающих мембрану,
является -мерой изменения чисел переноса. Рассмотрим отрица-
тельно заряженную мембрану (рис. 88) в цепи постоянного тока
в растворе. На рис. 88 стрелками показано направление движе-
206
ния ионов, и число делений отвечает соотношению подвижностей
ионов в мембране и в растворе, принимая подвижности. в сво-
бодном растворе одинаковыми. На катодной стороне мембраны
приход катионов после прохождения некоторого числа кулонов
q должен составлять пк ~ и расход п%~^, где пйк и /гд соответ-
ственно числа переноса катиона в свободном растворе и в мем-
бране и F—-число Фарадея. Результативное изменение концен-
трации отдельных ионов по сторонам мембраны будет опреде-
ляться разностью чисел переноса в мембране и в растворе, и
Рис. 88. Схема переноса ионов через диафрагму.
баланс катионов на катодной стороне Dk будет положительным
(пк > п°) и составит:
Д"
*
Точно так же положительным окажется на катодной стороне
и баланс анионов
Da==4<na— «а> = 4
а Г “ а' г
Следовательно
D* = Da = DK
Последнее равенство очевидно, так как в силу сохранения
электронейтральности раствора повышенное поступление катио-
нов из капилляров мембраны на катодную сторону должно быть
уравновешено задержкой анионов перед капиллярами на этой
стороне. На анодной стороне мембраны повышенный расход
катионов компенсируется переносом анионов от мембраны к по-
южительному полюсу. Таким образом, на анодной стороне
мембраны имеет место общий отрицательный баланс, т. е. пони-
жение концентрации электролита.
Для катионов:
Dk==4("°~"«)==~4 Д"
Для анионов:
^ = Т-(«а-«а) = -4 Д”
207
Аналогично, в силу сохранения электронейтральности рас-
твора:
£>“==£>“ = D'"
Среднее изменение составит'
откуда
Ди = ^ . (1)
Я
Для отрицательно заряженной мембраны искомое число
переноса катиона через мембрану «к будет, очевидно, равно
пк = «к + Дп <2>
а число переноса аниона
пя = п® — Ди
или, поскольку пк + па = 1. то:
Лд = 1
Для положительно заряженной мембраны, увеличивающей
число переноса аниона по уравнению со свободным раствором,
или, что то же, уменьшающей число переноса катиона, концен-
трационные соотношения будут обратные и очевидно:
«а = аа + Дп (3)
Выполнение работы
Прибор, в котором производится определение, изображен
схематически на рис. 89.
Как видно на рисунке, прибор состоит из центральной
части — двух широких трубок 2 и 9, снабженных внизу кранами.
Трубки имеют боковые отростки, входящие во фланцы 1, между
которыми (на резиновых прокладках) зажимается исследуемая
мембрана. В верхней части трубок на шлифах вставляются бо-
ковые части 3 и 6, заканчивающиеся отростками 4 и 8, куда
вставляются трубки 5 и 7 на пробках, наполненные агаровым
студнем и в верхней своей части имеющие медные электроды
в растворе CuSO4.
Прибор наполняют исследуемым раствором определенной
концентрации (обычно 0,01 н. раствором КС1) и включают в
цепь постоянного тока. Количество прошедшего электричества
учитывается с помощью включаемого в цепь вольтаметра Ки-
сти ковско го.
208
Рис. 89. Прибор для определения
чисел переноса ионов через
мембраны по аналитической ме-
тодике.
По окончании опыта, который проводят обычно около двух
часов, выпускают раствор через краны: сначала из отделений 4
и 8 и затем из пространств Зи6и2и9в отдельные сосуды.
Растворы из отделений 4 и 8 отбрасываются, остальные подвер-
гаются анализу. Количества раствора из пространств 2 и 9 учи-
тываются отдельно путем взвешивания на технических весах или
измеряются с помощью мерного цилиндра. В случае изменений
концентрации раствора в контрольных пространствах 3 и 6 опыт
бракуется. В растворах определяют концентрацию иона хлора
титрованием раствором AgNOs .(титрование проводят в присут-
ствии флюоресцеина или по Фольгардту, или потенциометриче-
ским методом [7] ряда проб определенного объема (50 см3).
Разницу в концентрации исход-
ного раствора и после опыта
пересчитывают на весь объем ка-
мер 2 и 9, суммируют, и это со-
ставляет общее изменение коли-
чества эквивалентов иона хлора
по обеим сторонам мембраны
£>к _|_ /)а Разделив на 2, по-
лучают среднее изменение в эк-
вивалентах D, входящее в рас-
четную формулу (1)‘для вычис-
ления величины изменения числа
переноса Дм.
Количество прошедшего элек-
тричества в кулонах получают
путем анализа раствора из воль-
таметра. Содержимое вольта-
метра титруют после опыта рас-
створом KCNS или NH4CNS, используя в качестве индикатора
железо-аммиачные квасцы.
Для вольтаметра пользуются 15% раствором KNO3 свобод-
ного от Cl-ионов, которым наполняется немного более половины
трубки вольтаметра. В нижнюю часть трубки помещается се-
ребряный электрод. На раствор KNO3 осторожно наслаивают
0,5 н. раствор HNO3 высотой 3—5 см. В раствор азотной кис-
лоты помещают вверху платиновый электрод, являющийся като-
дом. Серебряный электрод, погруженный в раствор KNO3, яв-
ляется анодом.
Значение числа переноса определяемого иона в свободном
растворе, необходимое для расчета числа переноса в мембране
по формулам (2) и (3), берут из справочных таблиц (для
Cl-иона в 0,01 н, растворе КО, п°С1 = 0,504). В качестве мембран
удобно использовать коллодиевые, керамические, целлофан, же-
латину (нанесенную на бумагу или ткань и дубленую раство-
ром формалина), а также ионообменные мембраны.
209
Работа 37
ОПРЕДЕЛЕНИЕ ЧИСЕЛ ПЕРЕНОСА ИОНОВ В МЕМБРАНЕ МЕТОДОМ
ДИФФУЗИОННОГО ПОТЕНЦИАЛА
Второй метод определения чисел переноса через мембраны
основывается на испрльзовании диффузионного потенциала.
Диффузионный потенциал возникает, как известно, при сопри-
косновении двух растворов электролитов различной концентра-
ции, вследствие разной скорости диффузии отдельных ионов
разного знака заряда. При диффузии ионов электролита в сто-
рону более разбавленного раствора, если катион обладает боль-
шей подвижностью по сравнению с, анионом, то более разбав-
ленный раствор приобретает положительный заряд. При боль-
шей относительной подвижности аниона более разбавленный
раствор получает отрицательный заряд. Величина диффузион-
ного потенциала зависит от соотношения подвижностей катиона
и аниона. По Нернсту величина диффузионного потенциала я
связана с подвижностью катиона и и аниона v следующим соот-
ношением
nF и -|- v у2С2
(4)
где — концентрация более концентрированного раствора;
с2 — концентрация более разбавленного раствора;
Ti и Та — соответствующие коэффициенты активности.
Полная электродвижущая сила концентрационной цепи типа
М | М+ (ct) | (с2) М+1 М
Ei * 5s
будет
Е = ?i — $2 4-71 (5)
Здесь g, и g2— потенциалы употребляемых электродов и
я — диффузионный потенциал.»
Если мы возьмем два раствора хлористого калия различной
концентрации, то величина диффузионного потенциала на гра-
нице этих растворов будет ничтожной вследствие того, что по-
движности К+ и С1~ близки между собой. Однако, если мы раз-
делим эти два раствора мембраной, изменяющей соотношение
подвижностей катиона и аниона, или, что то же самое, изме-
няющей числа переноса по сравнению со свободным раствором,
то между этими растворами возникает разность потенциалов.
Величина этой разности потенциалов будет возрастать соответ-
ственно изменению чисел переноса мембраной. Эта зависимость
и составляет основу для измерения чисел переноса путем диф-
фузионного потенциала,
210
Для измерения получающейся разности потенциалов исполь-
зуются каломелевые или хлоросеребряные электроды. Употреб-
ление хлоросеребряных электродов дает, по данным ряда иссле-
дователей, лучшие результаты.
Для этого случая формулу для расчета числа переноса мож-
£10 легко вывести из уравнений (4) и (5)
t -----4-lln-I^-
Г U-f-V 12^2 F \«+*' U~\-V) f2C2
Обозначим
fc,. и v
к * —,— = n+; =— = n_,
a-f-v + u-}-v
^гогда
h K = (n+ —
Г + F Wi
tio уравнению (5)
Г £=sI — ь+ъ
Г |2c2 .
Следовательно
E = fl + n+ - „_) ln-I£- = 2л+ ln-I£-
r I2C2 r I2C2
Кугкуда
Выполнение работы
Измерение возникающей на мембране разности потенциалов
црризводится в приборе, изображенном на рис. 90.
Рис. 90. Прибор для определения чисел переноса ионов
через мембраны по диффузионному потенциалу.
Мембрана зажимается во фланцы 3. Боковые пространства 2
и 4 наполняются растворами КС1 различной концентрации. Со*
этношение концентраций не должно быть велико, вследствие
имеющейся зависимости чисел переноса ионов в мембране от
концентрации электролита (обычно это соотношение берут
211
равное 2, т. е. например, 0,01 н./0,02 н.). Мембрану предвари-
тельно выдерживают в растворе меньшей концентрации, так как
при направленности процесса диффузии в сторону малой кон-
центрации это обеспечивает совпадение границы между раство-
рами различной концентрации с разделяющей их мембраной во
время опыта.
Разность потенциалов, возникающая на мембране, опреде-
ляется с помощью потенциометрической установки. Предвари-
тельно определяют потенциал асимметрии, образуя цепь из двух
хлоросеребряных электродов, погруженных в раствор КС1 опре-
деленной концентрации без мембраны (можно в отдельном ста-
кане) *. Электроды должны показывать потенциал асимметрии
не больше 0,5 мв. Потенциал асимметрии проверяется перед
каждым опытом.
За окончательную величину потенциала на мембране при-
нимают значение, не меняющееся в течение 5 мин. Расчет числа
переноса ведется по формуле (6), т. е.
Е
где пк—число переноса катиона в мембране;
£—.полученная величина разности потенциалов на мем-
бране;
и с2 — концентрации растворов по сторонам мембраны;
Т1 и 72 — соответствующие коэффициенты активности электро-
дита, равные для растворов КС1:
Молярность ' раствора Коэффициент активности
0,001 0,965
0,002 0,951
0,005 0,926
0,01 0,889
0,02 0,865
0,05 0,809
0,1 0,762
0,2 0,715
Следует иметь в виду, что метод диффузионного потенциала
дает менее надежные и точные результаты по сравнению с ана-
литической. методикой. Необходимость использования двух рас-
Для опытов пользуются хлоросеребряными электродами, которые при-
готовляют следующим образом. Платиновые электроды тщательно очищают,
промывают дистиллированной водой и серебрят в специальном растворе
(35 г AgNO? + 40 г KCN на 1 дм3 дистиллированной воды) в течение 24 ч
при плотности тока 1—2 ма/см2. После серебрения электроды многократно
ополаскивают дистиллированной водой и хлорируют в растворе 0,1 н. НС1
при плотности тока 5—6 ма/см2 (в этом случае электрод делают анодом).
После тщательной промывки электрод может быть использован для ра-
боты.
212
творов различной концентрации по сторонам мембраны вносит
неопределенность в значение числа переноса через мембрану п,
которое зависит от концентрации раствора электролита. В ана-
литической методике концентрация исходного раствора одина-
кова по обеим сторонам мембраны и воспроизводимость парал-
лельных опытов значительно лучше, чем по методике диффу-
зионного потенциала.
Поверхностная проводимость
Работа 38
ОПРЕДЕЛЕНИЕ ПОВЕРХНОСТНОЙ ПРОВОДИМОСТИ КАПИЛЛЯРНЫХ СИСТЕМ
Явление поверхностной проводимости наблюдается в узких
капиллярах и, особенно отчетливо, в капиллярных системах.
'Явление заключается в том, что раствор электролита, содержа-
щийся в капилляре, или в порах мембраны, обладает большей
удельной электропроводностью, нежели окружающий равновес-
ный раствор.
Эта добавочная электропроводность обусловливается прово-
димостью избыточных ионов, а именно ионов двойного электри-
ческого слоя. Таким образом, проводимость раствора в порах х
слагается из удельной электропроводности свободного раствора
Ху и поверхностной проводимости Xs
''•=7-y+7-s
Следует иметь ввиду, что величина Xs является не удельной
электропроводностью поверхностного слоя, а той, распределен-
ной равномерно по всему объему раствора в порах, добавкой
к ху, которая обусловлена избытком подвижных ионов.
Изучение поверхностной проводимости имеет существенное
значение, поскольку эта величина является одной из важнейших
электрокинетических характеристик поверхности раздела. Так,
определение поверхностной проводимости позволяет оценить ве-
личину фактического электросопротивления диафрагм и мем-
бран, что весьма важно,’например, при рациональном выборе
диафрагм для электродиализа, при' исследовании электриче-
ского сопротивления живых тканей, для определения пористо-
сти грунтов методом электропроводности, для характеристики
ионообменных адсорбентов и т. д.
Наряду с этим поверхностную проводимость необходимо
учитывать при вычислении ^-потенциала. Так, в уравнении
Гельмгольца — Смолуховского для потенциала протекания
, 4r.rtE
u — Dp *у
213
Пходит величина удельной электропроводности раствора; оче-
видно, что для получения правильных значений £ в уравнение
необходимо подставлять фактическую величину электропровод-
ности раствора в порах мембраны, т. е.
_ 4ят)£
С ~ DP (V + х$)
Эта поправка может быть выражена посредством множителя
V
называемого коэффициентом эффективности мем-
браны,
(П
а
С = а£°
Таким образом, для получения исправленного значения £-по-
тенциала, необходимо величину £°, вычисленную, исходя из
удельной электропроводности свободного раствора Ху, умно-
жить на коэффициент эффективности а.
Проводимость поверхностного слоя иоцов, характеризуемую
значениями Ху и а, целесообразно относиТь к единице поверх-
ности в тех случаях, когда величина поверхности известна.
Величина поверхностной проводимости, отнесенная к 1 см2
поверхностного слоя, называется удельной поверхност-
ной проводимостью.
Удельная поверхностная проводимость Ks по физическому
смыслу не зависит от величины поверхности, а следовательно,
и от радиуса капилляров, или иначе/от степени дисперсности
системы. Изменение концентрации раствора, как показали экс-
периментальные данные, мало влияет на величину KS- ‘По-
скольку с ростом концентрации ионы диффузного слоя перехо-
дят в плотный слой, отсутствие заметных изменений ве-
личины Ks свидетельствует о »том, что ионы в плотном
слое обладают подвижностью. Отсюда следует, что поверхност-
ная проводимость обусловлена всеми ионами внешней обкладки
двойного слоя. Вёличина Ks связана с плотностью заряда и в
первом приближении равна:
KS^\U1 + \U2
где vjj — поверхностная плотность ионов плотного слоя,
Т?2 — диффузного слоя,
Ux и U2 — подвижности ионов.
Таким образом, не изменяясь со степенью дисперсности ка-
пиллярной системы и с концентрацией ионов в растворе, удель-
ная поверхностная проводимость Ks характеризует ионную при-
роду поверхности раздела.
В связи с этим можно следующим образом охарактеризо-
вать рассмотренные нами величины xs и а. Поверхностная про-
214
водимость для всего
боковая поверхность,
Для цилиндрического
;а следовательно:
капилляра равна KsS или xsQ, где 5 —-
Q — объем капилляра
капилляра с радиусом
(на 1 см длины).
г, Ks • 2w = xsW2,
2/G
х =—*
$ г
следует, что величина
(2)
2К5
xs = для дан-
Из уравнения (2)
фой капиллярной системы (г = const) пропорциональна Кв, Я
^следовательно, так же мало изменяется с изменением концен-
трации. Однако в отличие от Ks, величина xs связана обратной
пропорциональностью с радиусом капилляра г. Чем меньше ра-
здиус капилляра, тем больше избыточных ионов двойного слоя
Додержится в единице объема раствора, пропитывающего" капил-
лярную систему и тем больше,' следовательно, поверхностная
^проводимость.
К Коэффициент эффективности а также может быть выражен
|через удельную поверхностную проводимость Ks посредством
^формул (1) и (2):
U '
£
(3)
а ~
2KS
t Приведенное выражение показывает, что коэффициент эф-
фективности а увеличивается при уменьшении радиуса пор г,
также с уменьшением концентрации раствора (пропорцио-
нальной электропроводности ху). Действительно, в обоих слу-
чаях общее количество ионов в объеме капилляра уменьшается
Гораздо быстрее, чем число ионов двойного слоя. Поэтому от-
носительная доля поверхностных ионов будет возрастать с
^уменьшением г и ху. Таким образом, коэффициент эффектив-
ности а, подобно изменению чисел переноса в диафрагме (ра*
Иота 36), характеризует долю участия поверхности раздела, т. е.
ионов двойного слоя, в общем переносе электричества через ка-
пиллярную систему.
При исследовании проводимости капиллярных систем имеет
^значение и другой коэффициент {В, названный коэффициен-
те м структурного сопротивления и показывающий,
Йо сколько раз уменьшается суммарное эффективное сечение
Нор, а следовательно и электропроводность капиллярной си-
стемы за счет наличия непроводящего скелета твердой фазы.
' Соотношением между величинами а и ₽ определяется явление
капиллярной сверхпроводимости, состоящее в том,
что при внесении в раствор между электродами диафрагмы,
пропитанной тем же раствором, сопротивление раствора в ряде
случаев не увеличивается, а уменьшается. Происходит это по-
тому, что при замене эквивалентного слоя раствора мембраной,
его сопротивление Rh увеличивается в р раз за счет непроводя-
215
щего скелета и в а раз уменьшается за счет поверхностной про-
водимости в порах. Следовательно:
Rh~=Rd или 7^ = 4 <3а)
где Rd— сопротивление мембраны
Rh— сопротивление эквивалентного слоя раствора, равного
мембране по площади и толщине.
Очевидно, что если а. > J3, то Rh> Rd-
Поскольку (3 с концентрацией не меняется, а а с разбавле-
нием растет, условие а > (3 удовлетворяется в разбавленных рас-
творах, и мы можем наблюдать явление «капиллярной сверх-
проводимости».
Количественное изучение этого явления основано на сравне-
нии измеренных на опыте величин сопротивления мембраны
(Rd) и эквивалентного слоя (Rh) в одном и том же растворе.
. Для измерения удельной электропроводности раствора, со-
держащегося в порах х, необходимо знать постоянную сопротив-
ления капиллярной системы Cd = Rd*- Величину Cd можно опре-
делить, применяя раствор с известной величиной х. Таковым
может быть раствор' с высокой концентрацией электролита.
В этих условиях в порах мембраны ионы поверхностного слоя
составляют незначительную долю от общего количества ионов и
xsCxy, а следовательно, х « xv. В качестве такого раствора,
называемого контрольным, обычно применяют 0,1 н. раствор
КС1, где х° « xv * и тогда
Г'О_
Таким образом, для определения величин xs или а необхо-
димо измерить сопротивление мембраны дважды: а) в исследуе-
мом растворе (Rd) и б) в контрольном (0,1 н.) растворе (Rd)
для определения постоянной сопротивления мембраны Cd-
Измерив эти величины, а также сопротивление растворов —
исследуемого Rv и контрольного Ry, в обычной ячейке с по-
стоянной сопротивления Су, производят дальнейшие вычисления
* следующим образом:
z4) хЧ)
— D0 • Gd — Ка*у, ~ D
KV Kd
(4)
Для вычисления коэффициента (3 измеряют сопротивление
раствора Rh, равного дйафрагме по площади и толщине.
* Индексом (0) обозначаются данные, относящиеся к контрольному
0,1 н. раствору.
216
Для этого замещают диафрагму раствором, не изменяя поло-
жения электродов. Зная электропроводность этого раствора,
вычисляют постоянную СЛ:
С. = х../?.
h V h
Величину Ch можно назвать постоянной ячейки без
диафрагмы. Очевидно, что величина Ch, измеренная в любом
растворе, будет одинаковой так же, как и Cv. Например, для
контрольного раствора
*X=X = Q (5)
по определению:
Rd
₽ = • • (6)
Kh
Из соотношений (4), (5), (6) находим также
Следует отметить, что из уравнений (4) — (7) можно легко
получить приведенное ранее соотношение (За).
Выполнение работы
Измерительная схема не отличается от обычной схемы, при-
меняемой для измерения удельной электропроводности раство-
ров, и представляет собой мост Кольрауша, состоящий из четы-
рех сопротивлений; в качестве одного из сопротивлений вклю-
чается измерительная ячейка. Источником переменного высоко-
частотного тока служит генератор звуковой частоты ЗГ-10 или
индукционная катушка. В качестве нуль-инструмента обычно
применяют низкоомный телефон (с сопротивлением катушек
порядка 100 ом) или осциллограф. Для более точных измере-
ний (необходимых в исследовательской работе) в схему вклю-
чается магазин переменной емкости с для компенсации емкости
измерительной ячейки. При этом получается более отчетливый
минимум звука. Для устранения утечек тока рекомендуется
ввести дополнительную ветвь моста, состоящую из сопротивле-
ний Дб, Де (по 300 ом) и /?? (150 ом), как указано на схеме
рис. 91.
Подробное описание схемы имеется у Глесстона. Конструк-
ция измерительной ячейки зависит от характера исследуемого
объекта.
Измерение поверхностной проводимости порошковых капил-
лярных систем. Для измерения поверхностной проводимости
капиллярных систем, образованных суспензиями, порошками
217
и др., удобно применять ячейку конструкции Самарцева и Ост-
роумова (рис. 78), описанную на стр. 193.
До формирования диафрагмы в ячейке измеряют сопротив-
ление Rh свободных растворов различной концентрации (на-
пример, 0,001 н., 0,01 н. и 0,1 н.) и находят величину Ch —
= xv • Rn- Эта величина должна быть по-
стоянной для данной ячейки. После того,
как диафрагма сформирована, через нее
пропускают контрольный (0,1 н.) раствор
до установления постоянной величины
сопротивления Rd. по которому находят
константу ячейки с диафрагмой Cd- За-
тем отмывают диафрагму водой и обра-
батывают исследуемым раствором. Если
контрольным служил раствор КС1, а ис-
следование проводится в другом электро-
лите, необходимо предварительно обра-
ботать диафрагму 0,1 н. раствором
исследуемого электролита (для вытесне-
ния ионов К+ или С1_ из двойного слоя),
а затем отмывать водой.
Определяют установившиеся величи-
ны сопротивления диафрагмы Rd для
всех исследуемых растворов, приготов-
ленных по указанию преподавателя. Из-
меряют сопротивление всех растворов Rv
в обычном сосуде.
Из полученных данных вычисляют xv,
и, а затем xg, а и [3 по вышеприведенным'
формулам.
Запись ведут по следующей схеме
(табл. 1).
Измерение поверхностной проводимости жестких диафрагм
в разъемной ячейке. В случае капиллярных систем, обла-
Рис. 91. Электрическая
схема измерения сопро-
тивления:
/^—сопротивление ячейки;
/?2~ ^ — сопротивление плеч
основного и дополнительного
моста; ЗГ—генератор звуко-
вой частоты; С—магазин ем-
костей; Г—телефон; Я —пере-
ключатель.
218
дающих определенной формой (не способных заполнять весь
рбъем ячейки), например коллодиевых и керамических мембран,
измерение поверхностной проводимости усложняется вследствие
трудности заполнения капиллярной системой всего пространства
между электродами. Для диафрагм, обладающих значительной
толщиной, приближенные измерения можно производить непо-
средственным приложением плоских электродов к двум сторо-
жам диафрагмы, пропитанной раствором. Для мембран тонких,
также неоднородных по толщине, такой метод неприменим,
|гак как между электродами, наряду с мембраной, часть про-
странства занимают прослойки свободного раствора. Конструк-
ция ячейки, разработанная в лаборатории коллоидной химии
ИГУ, позволяет учесть и ис-
ключить сопротивление этих
|рослоек.
Для этого измеряют сум-
марное сопротивление R мем-
браны и слоев свободного рас-
твора между электродами и
мембраной. Затем мембрану
^янимают и измеряют сопроти-
вление тех же слоев раствора
|ез мембраны Rv- По разности
находят сопротивление caMoif
мембраны Rd = R—Rv.
Разъемная ячейка состоит
|з двух пластин, изготовлен-
иях из плексигласа (рис. 92),
^Ириной 3 см и высотой 3 см
| нижней части. Два платино-
Йях электрода вклеены в уг-
лубления нижней части пластин 6; вводы к ним изолированы
Й проходят внутри пластин между склеенными слоями плекси-
гласа. При соединении пластин три направляющие шпильки 3
рбеспечивают параллельное расположение пластин, а резиновое
Кольцо 4 — воспроизводимость постоянных размеров ячейки
расстояния между электродами).
£. Во время измерений ячейку, погруженную в небольшой ста-
канчик, помещают в термостат. Измерение производят в сле-
дующем порядке: из мембраны вырезают- образцы размером
3X4 см, а также рамки, т. е. образцы такого же размера, но
е отверстиями 1,5 X 2 см в середине. Два образца мембран и
два образца рамок помещают в исследуемые разбавленные
растворы и в контрольный 0,1 н. КС1 на 2 суток со сменой рас-
твора. Рамки можно помещать не во все стаканы, так как ве-
йичина Ch одинакова для всех растворов.
Рнс. 92. Разъемная ячейка для изме-
рения поверхностной проводимости
в жестких мембранах.
219
На образцах,- помешенных в один сосуд, делают какую-
нибудь отметку, например срезают уголки. Прибор в стакан-
чике с исследуемым раствором помещают перед опытом в водя-
ной термостат и выдерживают 1—2 ч. Перед измерениями рас-
твор в стаканчике заменяют свежим, нагретым до температуры
термостата. Раскрывают прибор, удаляют пузырьки воздуха из
нижней части, собирают ячейку без мембраны посредством вве-
дения направляющих шпилек и. измеряют сопротивление свобод-
ного раствора. Такая ячейка без мембраны с известной Cv мо-
жет служить одновременно в качестве сосуда для определе-
ния zv- Измерения повторяют несколько раз, раскрывая и вновь
собирая ячейку. Затем вкладывают образец мембраны (№ 1),
измеряют сопротивление R = Rd + Rv. Повторяют измерения
несколько раз, каждый раз раскрывая ячейку. Схема II пред-
ставляет раскрытую ячейку, схема III — собранную ячейку с
мембраной.
Вынимают мембрану и вновь измеряют Rv (2—3 раза)..За-
тем вкладывают рамку (№ 2) и измеряют несколько раз сум-
марное сопротивление R' = Rn + Rv Те же измерения прово-
дят с параллельными образцами (№ 3 и № 4). Подсчитывают
средние величины для каждого образца. Из средних величин
R и R' вычитают средние значения сопротивлений Rv, измерен-
ных до и после измерения образца, т. е. вычисляют Rd — R—Rv
и Rh = R'—Rv.
Запись опыта для каждого раствора ведут по следующей
форме (табл. 2).
Таблица 2
№ образца 4v R (О R> (2) R R (3) *v R' (4) *v
1 1360 2060 1370 2290 1380 2110 1370 2300 1400
2 1350 2100 1390 2270 1400 2080 1350 2310 1410
3 1380 2050 1390 2280 1390 2080 1390 2280 1370
Среднее 1363 2070 1383 2280 1390 2090 1370 2297 1393
Среднее Rv 1373 1387 1380 1382
Rd- = 697 Rh- = 893 Rd- = 710 Rh- = 915
Вычисляют средние значения Rd и Rh. Повторяют тот же
цикл измерений для другой концентрации и в конце — для кон-
трольного 0,1 н. раствора. Вычисляют удельные электропровод-
ке
ности растворов хк=-р~ • исходя из известной величины Cv-
Kv
220
Данные опыта и вычисления записывают по той же форме,
как и в случае порошковых диафрагм (табл. 1). Вычисляют
Cd, Ch, затем х, xg, аир. Для работы применяются коллодие-
вые или целлофановые мембраны в 5-10~4—2-Ю-3 н.растворах
КС1, NaCl, NH4C1.
Измерение поверхностной проводимости жестких мембран в
щелевой вращающейся ячейке. Для более точных измерений
сопротивления тонких мембран применяется вращающаяся
ячейка (рис. 93). Ячейка состоит из двух дисков 4 диаметром
.36 мм, изготовленных из плексигласа, скрепленных на высту-
пах 1 и 2 латунными винтами так, что между дисками остается
щель 7 толщиной 0,5 мм *. С внутренней стороны дисков вклеены
Рис. 93. Щелевая ячейка для измерения поверхностной проводимости
в жестких мембранах.
круглые платиновые электроды 5 (можно заменить медными
платинированными) так, что диаметр открытой поверхности
электрода равен 15 мм. Электроды углублены по отношению
к плоскостям, образующим щель и после скрепления дисков
отстоят один от другого на 1,5 мм. В щель между ними, за-
полненную раствором, вводится при помощи стеклянного крючка
и палочки мембрана 8, вырезанная как показано на рис. 93 и
.снабженная ушком из серебряной проволоки. Для облегчения
введения мембраны (под раствором) в щель служат два вы-
ступа 3 на нижнем диске вместе с небольшим расширением на
верхнем. Изолированные вводы 6 к дисковым электродам, изго-
товленные из толстой медной проволоки, изогнуты под прямым
углом к плоскости дисков и направлены вверх вдоль непрово-
дящего эбонитового стержня к двум контактным латунным
кольцам, закрепленным на стержне на разных уровнях при по-
мощи дисков из полистирола. На верхней поверхности каждого
кольца имеются желобки с ртутью, куда погружены проволочки,
ведущие к измерительной схеме, описанной выше. Ячейка
* Если нужно расширить щель, между выступами вставляют тонкие
пластинки из оргстекла.
221
закрепляется на штативе вертикально, посредством подшипника;
верхний конец стержня соединяется с мотором Уоррена, обеспе-
чивающим равномерное вращение ячейки со скоростью 1 об/сек
в двухлитровом стакане с исследуемым раствором, помещенном
в .водяном термостате. При вращении центробежная сила обес-
печивает омывание мембраны раствором, устраняющее погреш-
ности, связанные с нагреванием, изменением концентрации,
электролизом и пр. .
В этой ячейке измеряется сопротивление ячейки с мембраной
R и без мембраны Rv. Разность этих величин Д7? = R—Rv
равна сопротивлению мембраны Rd за вычетом сопротивления
слоя раствора, эквивалентного мембране Rh; &R = Rd — Rh, я
следовательно:
Rd = bR+Rh (8)
В этом методе Rh определяют, исходя из измеренного Rv,
расстояния между электродами а и толщины мембраны d:
Величину d определяют микрометром или по методике, опи-
санной на стр. 59, величину а — из постоянной сопротивления
ячейки Cv = nvRv = ; a — nvRvA, где А — рабочая площадь
электрода.
Подставляя значение Rh в уравнение (8) находим:
Значения nv находят из Rv и Cv. Величина Cv определяется
по измерению Rv эталонного раствора, для которого и известна
из таблиц (например, 0,01 и., КС1). Величины и, хв, а и р вы-
числяют из значений Rd, Rh, Kv по формулам, приведенным
выше.
Электродиализ
Работа 39
ОЧИСТКА КОЛЛОИДНЫХ РАСТВОРОВ И СУСПЕНЗИИ ОТ ЭЛЕКТРОЛИТОВ
МЕТОДОМ ЭЛЕКТРОДИАЛИЗА
Очистка коллоидных растворов или суспензий от находя-
щихся в них электролитов может быть достигнута посредством
двух основных методов: диализа и электродиализа. Для прове-
дения диализа между коллоидным раствором и водой поме-
щают полупроницаемую перегородку — мембрану (например,
222
целлофановую, коллодиевую и т. п.), поры которой проницаемы
для ионов электролита, но не проницаемы для коллоидных час-
.тиц. Через такую мембрану, вследствие градиента концентра'
ций, будет происходить диффузия растворенного электролита в
• воду и воды в коллоидный раствор до уравнивания концентра-
1ции электролита по обе стороны мембраны. Сменяя воду, можно
^довести очистку коллоидного раствора до-желаемой степени.
|Рднако следует иметь в виду, что скорость диализа пропорцио-
нальна разности концентраций диализуемого вещества по сто-
ронам мембраны. Вследствие этого по мере удаления электро-
лита скорость диализа будет уменьшаться и полная очистка
коллоидного раствора от растворенных электролитов практи-
чески невозможна.
Диализ можно значительно ускорить путем применения по-
стоянного электрического тока, т. е. соединяя диализ с электро-
|лизом. Такой комбинированный процесс называют электродиа-
лизом. Преимущество электродиализа по сравнению с диализом
^заключается не только в быстроте удаления электр.олитов,
Йю и в возможности более полной очистки коллоидного рас-
твора.
кг Для проведения электродиализа применяют различной кон-
струкции аппараты, называемые электродиализаторами. Осно-
;.даой таких аппаратов является трехкамерная ячейка, среднее
Пространство которой отделено от крайних электродных камер
^Мембранами. Подлежащий очистке коллоидный раствор поме-
чают в среднюю камеру, в то время как крайние камеры напол-
няют водой. Мембрана, расположенная у отрицательного элек-
трода называется — катодной, а у положительного — анодной.
^Следует обращать большое внимание на выбор материала для
|анода, чтобы избежать анодного растворения и переноса ионов
^металла через анодную мембрану в среднюю камеру. В связи с
йэтим в качестве анода обычно употребляют платину или графит.
jjB качестве катода могут служить различные металлы — железо,
шинель, медь.
Е Легко показать, что только введение двух мембран, разде-
ляющих электродиализатор на три части, дает принципиальную
^возможность очистки, коллоидного раствора, находящегося в
Передней камере. Так, на рис. 94,а схематично представлен про-
цесс электролиза раствора сернокислого натрия, причем обра-
зующиеся у электродов кислота и щелочь могут свободно диф-
фундировать вглубь, образуя вновь раствор. Na2SO4. Введение
одной мембраны, проницаемой для ионов Na и SO4, будет за-
труднять диффузию продуктов электролиза и приведет к разло-
жению раствора Na2SO4 на кислоту и щелочь (рис. 94,6).
С введением двух мембран продукты разложения — кислота
и щелочь — остаются в электродных пространствах, и коллоид-
ный раствор, помещенный "в среднем пространстве, может
223
быть очищен от содержащихся в нем электролитов, например
сернокислого натрия (рис. 94,в).
Применение мембран при электродиализе обусловливает ряд
явлений, осложняющих процесс электролиза. Во-первых, числа
переноса ионов электролита в мембране могут отличаться от их
значений в свободном растворе. Эффективность электродиализа,
как увидим ниже, зависит от природы мембран и их расположе-
ния в электродиализаторе; во-вторых, в процессе электродиали-
за может не только уменьшиться концентрация раствора элек-
тролита в средней камере, но и измениться его состав, вслед-
ствие различной скорости удаления ионов. Например, при очист-
ке какого-либо коллоидного раствора или суспензии от Na2SO4
в средней камере может образоваться H2SO4 (стр. 228);
Рис. 94. Схема, поясняющая различие между электролизом и электро-
диализом.
в-третьих, может наблюдаться электроосмос через мембраны.
Направление движения жидкости при электроосмосе зависит
от знака заряда мембран и расположения их по отношению
к электродам в электродиализаторе. Поэтому электроосмотиче-
ский перенос жидкости может быть направлен как из средней
камеры в электродные, так и наоборот. В результате может
значительно изменяться объем раствора в средней камере. Если
жидкость движется из электродных камер, где в процессе элек-
тродиализа образуются кислота и щелочь, в среднюю камеру,
то вследствие этого там также может произойти изменение со-
става электролита.
Кроме того при электродиализе дисперсных систем происхо-
дит электрофорез. Движущиеся при , электрофорезе частицы
могут осаждаться на мембране и тем самым изменять ее свой-
ства.
В ряде работ кафедры коллоидной химии ЛГУ было пока-
зано, что изменение чисел переноса ионов в мембране является
основной электрохимической характеристикой мембран, приме»
224
няющихся при электродиализе. Как известно, изменение чисел
переноса ионов в мембране зависит от знака заряда мембраны
и от величины среднего радиуса пор.
По влиянию на числа переноса ионов мембраны можно раз-
делить на три группы:
1) мембраны электрохимически неактивные, которые не из-
меняют чисел переноса ионов по сравнению с их значением
в свободном растворе. Такими мембранами являются относи-
тельно грубопористые мембраны, радиус пор которых велик
по сравнению с толщиной двойного электрического слоя;
2) мембраны электрохимически активные, т. е. изменяющие
числа переноса ионов по сравнению с их значениями в свобод-
ном растворе. В таких мембранах в зависимости от знака их
заряда повышается число переноса катиона (отрицательно заря-
женные мембраны) или аниона (положительно заряженные
мембраны) по сравнению с их значением в свободном растворе.
Радиус пор этой группы мембран значительно меньше, чем мем-
бран электрохимически неактивных. С уменьшением радиуса
пор мембраны увеличивается изменение чисел переноса ионов;
3) мембраны -идеально электрохимически активные, т. е.
проницаемые лишь -для ионов одного знака заряда. В зависи-
мости от знака заряда поверхности число переноса ионов одного
знака заряда в таких мембранах равно единице, а другого —
нулю. В таких мембранах в растворе нет одноименно заряжен-
ных с поверхностью ионов и ток переносится только противо-
ионами.
Такое разделение мембран, применяющихся при электродиа-
лизе, является относительным вследствие того, что изменение
чисел переноса ионов в мембране зависит также от природы
электролита и концентрации раствора, которая изменяется в
процессе электродиализа. С уменьшением концентрации элек-
тролита электрохимическая активность мембран возрастает, и,
кроме того, мембраны электрохимически неактивные в концен-
трированных растворах могут' оказаться электрохимически ак-
тивными в разбавленных растворах.
Рассмотрим характер процесса’ электродиализа с мембра-
нами различного типа. Чтобы оценить значение, природы мем-
браны в процессе электродиализа, представим себе, что во всех
трех камерах электродиализатора находится, один и тот же
раство’р электролита, например- сернокислого натрия, причем
в электродных камерах при условии непрерывного протекания
раствора Na2SO4 концентрация его не изменяется. При такой по-
становке опыта в случае применения электрохимически неактив-
ных мембран в средней камере не может происходить никаких
изменений в составе и концентрации электролита. Действитель-
но, вследствие равенства чисел переноса ионов в порах мем-
браны и в свободном растворе взамен ионов, уходящих через
225
соответствующие Мембраны из средней камеры к электродам, из
электродных камер через вторые мембраны будут поступать тс
же ионы в тех же соотношениях. Если в крайних камерах не
поддерживать искусственно концентрацию растворов, равную
исходной, то равенство концентрации растворов, во всех каме-
рах электродиализатора^ может быть только в начале, опыта,
когда нет продуктов разложения у электродов.
При проведении электродиализа в реальных условиях край-
ние камеры наполняются дистиллированной водой, — тогда пр
мере накопления кислоты и щелочи в электродных простран-
ствах вместо ионов электролита в среднюю камеру будут по-
ступать ионы водорода и гидроксила, образуя -воду. Таким об-
разом, уменьшение концентрации электролита в средней камере
при применении электрохимических неактивных мембран проис-
ходит только вследствие ухода ионов электролита с заменой их
ионами водорода и гидроксила из электродных камер.
При использовании электрохимически активных мембран
возможно изменение концентрации растворов в средней камере
и в том случае, когда во всех трех камерах электродиализатора
находится раствор одного и того же электролита. Было установ-
лено, что изменение концентрации раствора при электродиализе
зависит только от значений чисел переноса ионов в мембране
и не зависит от чисел переноса ионов в свободном растворе.
Следует обратить внимание на то, что в этом случае чрезвы-
чайно важно расположение мембран по отношению к полюсам
источника тока. Уменьшение концентрации электролита может
происходить только в том случае, когда число переноса катиона
в катодной мембране больше, чем число переноса катиона в
анодной мембране. Очевидно, что в этом случае число переноса
аниона в катодной мембране будет меньше, чем в анодной! мем-
бране. При таком расположении мембран катионы будут в от-
носительно большем количестве уходить из средней камеры че-
рез катодную мембрану, чем поступать в нее из анодного про-
странства. Соответственно и анионы также в относительно
большем количестве будут удаляться из средней камеры через
анодную мембрану, чем поступать в нее через катодную мем-
брану. Следовательно, при электродиализе с мембранами раз-
ного знака заряда положительно заряженную мембрану надо
поставить на анод, а отрицательно заряженную — на катод.
При обратном расположении мембран, т. е. когда число пере-
носа катиона больше в анодной! мембране, чем в катодной, в сред-
ней камере концентрация электролита будет увеличиваться.
При электродиализе коллоидного раствора или суспензии
с применением электрохимически активных мембран в обычных
условиях, когда в крайних камерах образуются соответственно
растворы, кислоты и щелочи, изменение концентрации электро-
лита в средней камере происходит как вследствие замены ионов
226
электролита ионами водорода и гидроксила из электродных
пространств, так и вследствие различия в числах переноса ионов
в порах анодной и катодной мембран.
С увеличением разности чисел переноса ионов в анодной
и катодной мембранах возрастает скорость изменения концен-
трации электролита в средней камере. Можно повысить ско-
рость электродиализа, применяя мембраны одного знака заря-
да, но разной электрохимической активности. При этом, если
мембраны приготовлены из одного и того же материала и име-
ют отрицательный заряд поверхности, то мембрану с большим
средним радиусом пор ставят на анод. В случае двух положи-
тельно заряженных мембран анодная мембрана должна иметь
меньший радиус пор по сравнению с катодной. Наиболее эффек-
тивно процесс электродиализа будет идти с идеально электро-
химически активными мембранами разного знака заряда.
В этом случае разность чисел переноса ионов электролита в.
анодной и катодной мембранах достигает максимальной вели-
чины, т. е. единицы.
При проведении электродиализа до недавнего времени при-
меняли различные пористые мембраны. Отрицательно заряжен-
ных мембран известно много, например керамические, перга-
ментные, целлофановые, коллодиевые и др. Выбор положитель-
но заряженных мембран был весьма ограничен.
В настоящее время при проведении электродиализа широко
применяют ионитовые, мембраны, т. е. мембраны из ионообмен-
ных смол (стр. 125). Такие мембраны по свойствам прибли-
жаются к идеально электрохимически активным и. обладают
малым электросопротивлением. Из ионообменных смол можно
готовить как положительно, так и отрицательно заряженные
мембраны. Для приготовления отрицательно заряженных мем-
бран используют катиониты; положительно заряженные мем-
браны готовят из анионитов.
Ионообменные смолы применяют и для поглощения приме-
сей, удаляемых из средней камеры. Для этого устанавливают
постоянную циркуляцию промывных вод из боковых камер че-
рез колонки, наполненные ионообменными смолами. По окон-
чании опыта поглощенные смолой примеси можно извлечь и
проанализировать.
Эффективность применения тех или иных мембран при элек-
тродиализе оценивается затратами электроэнергии. На рис. 95
представлен пример теоретического подсчета выхода по току
при электродиализе раствора Na2SO4 с-электрохимически неак-
тивными мембранами. Вследствие большей подвижности ионов
И и ОН в порах катодной и анодной мембран при электродиа-
лизе находятся соответственно растворы щёлочи и кислоты.
Следовательно, числа переноса ионов Na и ОН в порах ка-
тодной мембраны равны их значениям в растворе католита,
227
т. e. NaOH(nNa+=0,19; nOH- =0,81), а числа переноса ионов
Н и SO4 в анодной мембране равны их значениям в растворе
анолита, т. е. H2SO4(nH+ = 0,82; nso- = 0,18). После прохожде-
ния одного фарадея электричества из средней камеры уйдет
0,19 г-экв Na+ и 0,18 г-экв SO4~.ho в то же время в среднюю ка-
меру поступит 0,82 г-экв Н+ и 0,81 г-экв ОН- или 0,81 г-экв Н2О
и 0,01 г-экв Н+, т. е. в средней камере появится избыток кисло-
ты в 0,01 г-экв H2SO4. Таким образом, вычисленная величина
выхода по току для ионов SO4 и Na оказывается соответственно
равной 18 и 19%.
H2so4
Na2SO4
NaOH
J.
0,62г-эк»
SO4 -*•
0,16 г-эка
он-
0,81 г -эк»
-Na+
0,19г-экв
+Q«?-ja»H2SO4
-Ц/9г-з«Ка2ЗО4
+O,0fe-wH2SO4
+0,19г-экМаОН
Рис, 95. Схема расчета теоретического значения выхода
по току при электродиализе (с электрохимически,неактив-
ными мембранами).
Применение электрохимически активных мембран при элек-
тродиализе значительно увеличивает выход по току.
- С увеличением чисел переноса катиона в катодной и аниона
в анодной мембране выход по току будет'возрастать и при при-
менении идеально электрохимически активных мембран дости-
гает 100%. Вычисленную из значений чисел переноса ионов ве-
личину выхода по току обычно принято называть истинным
выходом по току.
На вычисленную величину выхода по току будет влиять диа-
лиз, а при малых концентрациях электролита также и электро-
осмос. Направление этих процессов зависит для диализа от со-
отношения концентраций электролита в камерах электродиали-
затора, а для электроосмоса — от знака заряда мембран.
В случае, если эти процессы направлены из средней камеры
в электродные, они увеличат вычисленную величину выхода по
току; обратное направление этих процессов приведет к ее умень-
шению. Поэтому для характеристики использования тока при
228
электродиализе введено понятие кажущегося -выхода по
току, которым называют выраженное в процентах отношение
количества электричества, необходимого по закону Фарадея для
удаления катиона или аниона электролита, к количеству элек-
тричества, фактически затраченному для удаления того же ко-
личества ионов.
Кажущийся выход по току может быть вычислен на основа-
нии изменений концентрации электролита в средней камере в
Рис. 96.. Схема процесса электродиализа в многокамерном электро-
диализаторе с ионообменными мембранами.
зависимости от количества прошедшего электричества по сле-
дующему уравнению:
т _ (со - с/) V-96 500
Чкаж — ——— • IUU
где ^каж — кажущийся выход по току;
Cq и ct— соответственно концентрации данного иона до и после
электродиализа, г • экв/дм3;
V—объем раствора, дм3;
I — сила тока, а; ’
t — длительность электродиализа, -сек.
Если количество прошедшего электричества Q выражать
в а-ч, объем раствора V — в дм3, концентрацию с-—
в мг-экв!дм3; то:
_ _ (Co-Q) V-96500.100 _ (Co-C/)V
’Ыж 1000.3600-Q — 4Ы> Q (H)
229
Чем больше величина кажущегося выхода по току, тем эф-
фективнее процесс электродиализа.
Расход электроэнергии можно значительно уменьшить, про-
водя электродиализ в многокамерном аппарате и используя
ионитовые мембраны. В таком аппарате между двумя электро-
дами попеременно чередуются большое число катионитовых и
анионитовых мембран. При электродиализе во всех четных ка-
мерах (независимо от их числа) произойдет очистка раствора,
так как анионы легко пройдут через расположенные на их пути
анионитовые мембраны, а катионы — через катионитовые. В не-
четных камерах, наоборот, произойдет концентрирование ионов
растворенных солей, вследствие обратного расположения мем-
бран в этих камерах (рис. 96).
Большей скорости и высокой степени очистки достигают
проведением электродиализа при высоком напряжении (200—
400 в!см).
Выполнение работы
Электродиализатор, применяющийся в данной работе, изо-
бражен на рис. 97 *. Он состоит из П-образных рам и двух
сплошных крайних пластин, скрепленных болтами. Мембраны
Рис. 97. Трехкамерный лабораторный
электродиализатор.
вставляют между резиновыми
прокладками, вырезанными по
форме рам. Катодом служит
никелевая пластина, анодом —
графит. Среднюю камеру
наполняют раствором, под-
лежащим очистке, электрод-
ные камеры — дистиллирован-
ной водой, затем пропускают
постоянный ток.
Через определенные проме-
жутки времени, в зависимости
от концентрации раствора и объема средней камеры, производят
отбор проб пипеткой'из средней камеры, отмечая одновременно
показания амперметра. Обычно пробы берут по 5—10 см5 через
10—20 мин.
За ходом электродиализа следят в зависимости от получен-
ного задания по изменению электропроводности или концентра-
* Пластины и рамы прибора могут быть изготовлены из плексигласа,
эбонита, а также из дерева. Деревянные части прибора необходимо пара-
финировать, для этого погружают их в расплавленный парафин и оста-
вляют в нем до окончания выделения пузырьков воздуха. Размеры прибора
могут быть различными. Для студенческих работ можно изготовить рамы
из квадратных пластин, стороны которых равны 15 см. Толщина пластин
3—4 см.
230
цни раствора, определяемой аналитически *. В первом случае
определяют сопротивление раствора в каждой пробе и, вычис-
лив значения удельной электропроводности, оформляют полу-
ченные данные в виде следующей таблицы (табл. 1).
Таблица I
Длительность электродиа- лиза, ч Сила тока /, а Средняя сила тока /ср, а Количеств чества за время А/ о электри- Q, а-ч за все время опыта Сопротивле- ние раствора, ом Удельная электропро- водность, ом~ 1 2-см'~1 [
*
Для вычисления прошедшего количества электричества
в а • ч берут среднюю силу тока, соответствующую промежуткам
времени между соседними измерениями 7Ср, и умножают на
время Д7 в часах, т. е. в случае проб, берущихся через каждые
15 мин, на 0,25.
Строят график, на котором по оси абсцисс откладывают ко-
личество ампер-часов, прошедших от начала опыта; по оси
ординат значения электропроводности растворов, причем на оси
ординат отмечают значение электропроводности исходного рас-
твора.
При наблюдении за изменением концентрации растворов,
определяемой аналитически, анализ проб производится по
одному из трех ниже указанных методов.
Анализ на ион CI. Раствор (5 с.и3), взятый из среднего про-
странства, разбавляют 15 см3 воды и титруют раствором AgNOs
в присутствии индикатора КгСгО4 (метод Мора).
Анализ на ион SO4. Пробу отбирают пипеткой на 10 см3 и
при перемешивании на холоду вливают в стакан, содержащий
15 см3 раствора хлоргидрата бензидина. При этом выпадает
мелкокристаллический осадок, который отфильтровывают под
вакуумом, промывают водой, отсасывают и вместе с фильтром
переносят в эрленмейеровскую колбу. Добавляют 30 см3 воды,
закрывают пробкой и сильно встряхивают до тех пор, пока
* Для очистки методом электродиализа в учебных лабораториях можно
рекомендовать следующие системы:
.1.0,1% раствор агар-агара или 1 %-ная-суспензия глины, приготовлен-
ные на 0,01—0,02 н. растворах электролита (ДС1, NaCl, NaaSOi, CaClj
и др.).
2. Золь берлинской лазури (стр. 314). За ходом электродиализа в этом
случае следует следить по изменению электропроводности золя.
231
фильтр не превратится в мелкие волокна. Затем смывают пробку
и шейку колбы водой, добавляют одну каплю фенолфталеина,
нагревают до кипения и титруют 0,1 н. раствором NaOH (метод
Рашига).
Анализ на ион Са. Раствор, взятый из. средней камеры
(10 см3), вливают в стакан, добавляют 5 см3 NH4C1 и2—3капли
аммиака, нагревают и осаждают кипящим раствором
(NH4)2C2O4. Дают постоять на водяной' бане 2 ч, после чего
отфильтровывают осадок, промывают его теплой водой и еще
влажный смывают в стакан с водой. В этот же стакан через
фильтр пропускают теплую разбавленную серную кислоту
(20 см3), чтобы разложить еще оставшиеся следы щавелево-
кислого кальция," и титруют 0,1 н. раствором КМпО4.
Данные опыта заносят в таблицу по следующей форме
(табл. 2).
Таблица 2
Длительность электродиа- лиза, ч Сила тока /, а Средняя сила тока /Ср, а Количество электри- чества Q, а‘Ч Концентрация раствора в средней камере, мг~экв!дмъ
за время А/ за все время опыта
Строят график: по оси абсцисс откладывают количество про-
шедшего электричества от начала опыта (в а • ч), а по оси орди-
нат— концентрацию раствора в средней камере в момент взятия
проб.
Кажущийся выход по току т)каж вычйсляют по уравнению (9),
причем если сила тока не сильно изменилась в процессе опыта,
то значение Q можно взять из таблицы 1 или 2. Для более
точного определения цкаж в цепь необходимо ввести вольтаметр,
по которому и находят количество электричества, прошедшее за
время опыта.
ЛИТЕРАТУРА
1. Глесстон С., Введение в электрохимию, Госхимиздат, 1951.
2. Г р и г о р о в О. Н., Козьмина 3. П., Маркович А. В., Фрид-
р и х с б е р г Д. А., Электрокинетические свойства капиллярных си-
стем, Изд. АН СССР, 1956.
3. Григоров О. Н„ Уч. зап. ЛГУ, Сер. ест. наук, № 68, (1944).
4. Жуков И. И., Коллоидная химия, т. I, 1949.
5. Коагуляция коллоидов, Сб, под ред. А. И. Рабиновича, ОНТИ, 1931,
232
6. К а р п о в а И. Ф., Уч. зап. ЛГУ, Сер. ест. наук, № 150 (1951).
7. Кольтгоф И.,_ Количественный анализ, Химиздат, 1938.
8. К р о й т Г. Р., Наука о коллоидах, ИЛ, 1955.
9 Марк ович А. В., Изв. естеств.-ваучн. ин-та им. П. Ф. Лесгафта, XXIV
(1951).
10. С а м а р ц е в А. Г., О с т р о у м о в В. В., Колл. ж. 12, 186 (1950).
11. Стендер В. В., Диафрагмы для электролиза водных растворов, Гос-
химиздат, 1948. . .
12. Л аскор ин Б. Н., С пир ков а Н. М., Рактмаи М К., Иовооб-
менные мембраны и их применение, Госатомиздат, 1961.
13. Каргин В. А., М а т в ее в а Т. А., ДАН СССР, 105, № 2, 294 (1955),
14. Abramson Н. A., Electrophoresis of proteins, 1942.
15. Shmid G„ Shwartz H., Z. Electrochem., 55, № 4, 295 (1951).
ГЛАВА IV
УСТОЙЧИВОСТЬ ДИСПЕРСНЫХ СИСТЕМ
Под устойчивостью дисперсных систем понимают спо-
собность дисперсной фазы сохранять исходную степень дисперс-
ности частиц, а также равномерное их распределение в диспер-
сионной среде.
Можно рассматривать седиментационную (кинетическую) и
агрегативную устойчивость: седиментационная устой-
чивость, количественно выражающаяся гипсометрическим
законом распределения частиц по высоте, определяется броунов-
ским движением и силой тяжести частиц. Если частицы дисперс-
.ной системы достаточно малы, они удерживаются в растворе
благодаря броуновскому движению, несмотря на действие силы
тяжести. Такие системы называются седиментационно устойчи-
выми. Агрегативной устойчивостью называется
способность частиц системы сохранить степень дисперсности,
т. е. не слипаться и не давать агрегатов под влиянием различ-
ных воздействий.
Нарушение агрегативной устойчивости, происходящее вслед-
ствие слипания отдельных, первичных частиц в более крупные
агрегаты, называется коагуляцией. Следствием нарушения
устойчивости является выпадение дисперсной фазы в осадок.
Золи обладают высокой седиментационной устойчивостью,
тогда как их агрегативная устойчивость обычно мала. С увели-
'чением размеров частиц седиментационная устойчивость золей
падает.
Коллоидная химия изучает устойчивость не только золей, но
также эмульсий и суспензий, являющихся системами седимента-
ционно малоустойчивыми.
Изучение устойчивости эмульсий и суспензий представляет
большой интерес благодаря большому их значению для про-
мышленности, техники и сельского хозяйства.
Различные факторы влияют на седиментационную и агрега-
тивную устойчивость по-разному. Например, седиментационная
устойчивость, определяющаяся броуновским движением,
234
увеличивается с повышением температуры, тогда как агрега-
тивная устойчивость, наоборот, падает, так как с ростом темпе-
ратуры увеличивается вероятность столкновения частиц между
собою.
Устойчивость, обусловленная в основном зарядом на поверх-
ности в виде двойного электрического слоя и, как следствие
этого электрокинетическим потенциалом, характеризующим эф-
фективный заряд поверхности, присуща, главным образом,
суспензоидным золям типа металлов, окислов металлов, сульфи-
дов и т. д.
Электрокинетический или ^-потенциал, не являющийся, со-
гласно современным воззрениям, единственной причиной устой-
чивости коллоидных систем, часто используется, однако, в каче-
стве относительной меры устойчивости суспензоидных золей.
Поскольку устойчивость суспензоидных золей объясняется
наличием заряда частиц, то состав и концентрация раствора
электролита имеют большое значение. Суспензоидные золи по-
этому очень чувствительны к прибавлению электролитов и
коагулируют при добавлении их в небольшом количестве. Во
многих случаях концентрация, необходимая для почти мгновен-
ной коагуляции, ничтожно мала.
Многочисленными исследованиями было установлено, что
явление коагуляции наступает не только в тех случаях, когда
величина ^-потенциала становится равной нулю, но также при
всех значениях, лежащих ниже некоторой величины ^-потенциа-
ла, называемой критическим потенциалом (величина
эта имеет порядок ~ 30 мв).
Наименьшая концентрация электролита, вызывающая коагу-
ляцию за определенный промежуток времени и выраженная
в миллимолях на дм3, называется порогом коагуляции
золя Ск данным электролитом.
Величина, обратная порогу коагуляции, называется коагу-
лирующей способностью; она йзмеряется количеством
дл«3 золя, которое может быть скоагулировано одним миллимо-
лем данного электролита.
Поскольку коагуляция представляет собой процесс, идущий
во времени, изучение кинетики коагуляции очень существенно
для понимания механизма процесса. .
Скорость процесса коагуляции определяется двумя факто-
рами: 1) вероятностью достаточного сближения частиц под
влиянием броуновского движения, 2) вероятностью того, что
при таком сближении эти частицы действительно образуют
агрегат. Поэтому рассматривая процесс коагуляции при доба-
влении электролита, различают медленную коагуляцию,
отвечающую слипанию наиболее быстро движущихся частиц при
уменьшении сил отталкивания, и быструю коагуляцию,
когда каждое столкновение частиц приводит к их слипанию.
235
Теория кинетики быстрой коагуляции была детально разрабо-
тана Смолуховским.
При медленной коагуляции скорость коагуляции зависит от
природы и количества добавленного электролита. При достиже-
нии быстрой коагуляций скорость коагуляции уже не зависит от
количества дополнительно добавляемого электролита, так как
все соударения частиц являются эффективными.
Принимая во внимание, что процесс коагуляции происходит
во времени, для изучения явлений коагуляции лучше пользо-
ваться методикой, связанной с кинетикой процесса. Для этого
можно оставлять концентрацию прибавленного электролита по-
стоянной и измерять время, необходимое для появления види-
мой мути или окраски. Можно также оставлять постоянным
время и определять концентрацию электролита, которая вызы-
вает коагуляцию.
Исследование коагуляции можно проводить прямыми и кос-
венными методами. К первым относится ультрамикроскопиче-
ский метод счета частиц золя (поточный ультрамикроскоп) ч
К косвенным методам относятся все методы, основанные на из-'
мерении вторичных эффектов (мутность золя, изменение окра-
ски, вязкости и др.).
Коагулирующее действие различных электролитов резко
различается, что особенно сказывается на скорости коагуляции.
Согласно правилу Шульце-Гарди, при коагуляции золя
электролитами коагулирующий ион всегда имеет заряд, проти-
воположный заряду коллоидной частицы; порог коагуляции при
этом тем меньше, чем выше 'валентность коагулирующего иона.
Так, отрицательно заряженные золи золота и As2S3 коагулируют
под влиянием катиона; положительно заряженные золи, подобно
Fe(OH)3, коагулируют под действием аниона.
Коагулирующее действие электролита с увеличением валент-
ности коагулирующего иона выражается следующими средними
цифрами:
Следует отметить, что ионы тяжелых металлов вызывают
коагуляцию отрицательных золей не соответственно своей ва-
лентности. Замечено также; что многие ионы органических со-
единений обладают чрезвычайно большой коагулирующей спо-
собностью независимо от валентности.
. Коагулирующая способность катионов и анионов одной ва-
лентности также неодинакова. Так, катионы щелочных метал-
лов, связанные с одним и тем же анионом (например, при
анионе NO3), по коагулирующей способности можно располо-
жить в так называемый лиотропный ряд:
Cs > Rb > К > Na > Li
236
Такое различное действие отдельных ионов связано с их
энергией и степенью гидратации, которые зависят в свою оче-
редь от величины радиуса и поляризуемости ионов, т. е. от де-
формируемости электронных оболочек атомов. Если в негидра-
тированном состоянии радиус ионов возрастает от Li к Cs, то
в гидратированном состоянии наблюдается обратная зависи-
мость.
Многовалентные катионы (Al3+, Fe3+, Th4+ и др.) обладают
способностью не только уменьшать g-потенциал и тем вызывать
коагуляцию, но могут, кроме того, изменять знак ^-потенциала
на противоположный.
Такое явление наблюдается, например, при коагуляции
суспензоидных золей при прибавлении многовалентных 'катио-
нов (в «неправильных» рядах, или «чере-
довании зон коагуляции»).
Кривая зависимости ^-потенциала от
концентрации прибавленного А1С1з пред-
ставлена на рис. 98.
Как видно из рисунка, вначале имеет
место резкое падение отрицательного
значения g-потенциала, исчезновение g-по-
тенциала в нулевой точке, а затем изме-
нение знака заряда, сопровождающееся
сначала повышением положительного
значения g-потенциала, а затем пони-.,
жением при дальнейшем увеличении кон-
центрации электролита.
Изменение знака эффективного заря-
Рис. 98. Зависимость v-
потенциала от концен-
трации электролита.
да объясняется способностью многова-
лентных ионов к специфической адсорб-
ции. Силы специфической адсорбции ха-
рактеризуются адсорбционным потенциа-
лом, величина которого зависит от индивидуальных свойств
иона.
Ион, обладающий большим адсорбционным потенциалом, мо-
жет адсорбироваться в сверхэквивалентном количестве, т. е.
в количестве, большем, чем это требуется для нейтрализации
зарядов ионов внутренней обкладки двойного электрического
слоя. Избыточные ионы притягивают электростатически ионы
противоположного знака, располагающиеся диффузно, чем и
обусловливается перемена знака, электрокинетического потен-,
циала.
Если имеется исходный отрицательно заряженный золь, то
при прибавлении многовалентного катиона, соответственно
уменьшению и в дальнейшем перемене знака электрокинетиче-
ского потенциала, золь показывает сначала — отсутствие
коагуляции (отрицательный потенциал выше критического),
237
после чего наступает первая зона коагуляции —I заштрихован-
ная область (отрицательный потенциал приближается к нулю).
Затем следует зона, в которой коагуляция снова отсутствует
(золь имеет положительный g-потенциал выше критического) и,
наконец, вторая зона коагуляции — II заштрихованная об-
ласть, связанная со сжатием диффузного слоя, состоящего в
этом случае из анионов, при увеличивающейся концентрации
электролита.
Если многовалентные катионы дают неправильные ряды
с отрицательно заряженным золем, то в отношении положитель-
но заряженных золей такую же роль играют многовалентные
анионы. Так, при действии лимоннокислого натрия на золь
Ее(ОН)з наблюдаются две зоны отсутствия коагуляции и две
зоны коагуляции в той же последовательности, что и в случае
неправильных рядов у отрицательных золей.
При взаимодействии двух противоположно заряженных зо-
лей наблюдается явление взаимной коагуляции. На-
пример, отрицательно заряженный золь сернистой сурьмы Sb2S3
коагулирует при прибавлении положительно заряженного золя
гидрата окиси железа.
Опыты показывают, что если мы будем рассматривать со-
стояние смешанного золя по обе стороны от зоны коагуляции,
то частицы золя несут знак заряда того золя, который нахо-
дится в .избытке.
В основе механизма явления взаимной коагуляции лежит
электростатическое притяжение противоположно заряженных
частиц и взаимодействие ионов, образующих двойные слои.
Явление взаимной коагуляции играет большую роль в поч-
венных процессах: часть содержащихся в почвах коллоидов
образуется в результате взаимной коагуляции положительно
заряженных золей Fe(OH)s, А1(ОН)з и отрицательно заряжен-
ных золей кремневой кислоты, а также гуминовых веществ.
Явление взаимной коагуляции используется при очистке воды
от органических веществ.
Коагуляция суспензоидных золей может быть вызвана при-
бавлением неэлектролитов, поверхностно-активных веществ, из-
менением температуры, длительным диализом и т. п.
‘ Растворы высокомолекулярных соединений типа желатины,
белков, крахмала гораздо более устойчивы, чем суспензоиды.
Это связано с тем, что высокомолекулярные соединения в под-
ходящих растворителях являются истинными молекулярными
растворами, т. е. образуют гомогенную систему, в которой не
существует границы раздела между фазами. Растворы высоко-
молекулярных соединений мало чувствительны к прибавлению
электролитов.
На устойчивости растворов высокомолекулярных соединений
основано явление з а щи т ы,. заключающееся в том, что при при-
238
бавлении к суспензоидам высокомолекулярных соединений
в очень небольших концентрациях суспензоиды приобретают
в отношении устойчивости свойства, сходные с растворами высо-
комолекулярных соединений.
Вещества, вызывающие повышение устойчивости суспензоид-
ных золей, называются защитными коллоидами.
Золь, чувствительность которого к электролитам понижена,
называется защищенным. Защитное действие имеет специ-
фический характер. Вещества, дающие наилучшую защиту для
одних суспензоидных золей, могут оказаться сравнительно менее
действенными по отношению к другим. Защитное действие за-
висит не только от природы защищающего и защищаемого кол-
лоида, но также и от степени дисперсности частиц суспензоид-
ного золя, от присутствия в золе примесей, от знака заряда и
pH среды.
Механизм защитного действия сводится к образованию ад-
сорбционного слоя защитного коллоида на поверхности частиц,
гидрофобного золя, сообщающего им свойства самого защитного
коллоида. Частицы суспензоидного золя оказываются покры-
тыми тонкой пленкой, имеющей большое сродство к раствори-
телю и поэтому приобретают в значительной степени свойства
молекулярного коллоида. Защищенные золи ведут себя по отно-
шению к электролитам так же, как сами защитные коллоиды.
Правило Шульце — Гарди здесь оказывается уже непримени-
мым, валентность коагулирующего иона оказывает малое влия-
ние на количество электролита, которое требуется для коагу-
ляции. .
Защитное действие имеет очень большое значение для мно-
гих процессов, рассматриваемых в биологии и медицине, а так-
же широко используется в различных отраслях промышленности.
Рассмотрим более подробно причины, обусловливающие
устойчивость и коагуляцию дисперсных систем.
Эффект взаимодействия двух суспензоидных частиц, имею-
щих гидрофобную поверхность, рассмотрен в ряде работ Деря-
гина [1]. При приближении двух одинаковых частиц друг к дру-
гу и взаимном перекрытии ионных атмосфер, окружающих ча-
стицы, происходит перераспределение ионов в ионных атмосфе-
рах.
Учет электростатических и осмотических сил, возникающих
при этом, приводит к экспоненциальной зависимости энергии от-
талкивания Е двух частиц от расстояния между ними Н (рис. 99,
кривая 1). Кроме сил отталкивания при сближении частиц дей-
ствуют силы притяжения, имеющие природу сил Ван-дер-Вааль-
са. Наиболее универсальным типом этих сил являются лондо-
новские силы, возникновение которых связано с частично син-
хронизованным движением электронов во взаимодействующих
молекулах. Энергия взаимодействия всех молекул приводит к
239
энергии притяжения, изменяющейся обратно пропорционально
квадрату расстояния между частицами (рис. 99, кривая 2), Пол-
ная энергия взаимодействия двух частиц получается суммирова-
нием энергий отталкивания и притяжения (рис. 99,' кривая 3).
На рис. 99 положительная величина Е соответствует увеличению
энергии системы.
•' На больших расстояниях между частицами преобладают си-
лы притяжения (вследствие более медленного падения сил при-
тяжения с расстоянием между ча-
Рис. 99. Зависимость энергии
взаимодействия двух частиц
от расстояния между ними.
стицами по сравнению с силами от-
талкивания), однако при уменьше-
нии. Н начинают действовать силы
электростатического отталкивания,
что приводит к появлению энерге-
тического барьера, для преодоления
* которого необходима определенная
кинетическая энергия сближающих-
ся частиц. При еще меньших Н
энергия притяжения оказывается
- большей по сравнению с энергией
электростатического отталкивания,
частицы начинают самопроизвольно
сближаться и в конце концов коагу-
лируют.
Таким образом, величина энерге-
тического барьера Es является от-
ветственной за устойчивость колло-
идной системы — чем выше величи-
на Et, тем более устойчивой являет-
ся коллоидная система. Снижение
величины Е$ должно' приводить к
уменьшению устойчивости системы,
и, наконец, когда величина ста-
новится равной или меньшей энергии броуновского движения
частиц, наступает быстрая коагуляция.
На величину Е%оказывает влияние как потенциал-поверхно-
сти* частиц ф, так и толщина двойного электрического слоя d.
Устойчивое состояние, при постоянном значении ф, может быть
получено за счет увеличения d. Уменьшение величины ф, при по-
стоянном значении d должно приводить к потере устойчивости
системы. Таким образом, уменьшение устойчивости системы мо-
жет происходить либо за счет уменьшения потенциала поверх-
ности ф*, либо за счет уменьшения толщины двойного электри-
ческого слоя.
* Приведенные выше рассуждения связывают устойчивость системы
с величинами ф и d. Одиако ввиду большей простоты в определении £-по-
240
Расчеты, проведенные Дерягиным и Ландау, позволили вычи-
слить условие исчезновения энергетического барьера на диаг-
рамме энергия — расстояние между коллоидными частицами.
Это условие связывает порог коагуляции ск электролитом с
валентностью коагулирующего иона и имеет следующий вид:.
const
ск---
где z — валентность коагулирующего иона.
Приведенное выражение хорошо объясняет эмпирическое
правило Шульце—Гарди. Действительно, для противоионов,
имеющих разные валентности (I—II—III), получаем следую-
щий ряд для ск:
Следовательно, для коагулирующих концентраций можем за-
писать следующий ряд:
730:11:1
т. е. получаем ряд, очень близкий к эмпирически установлен-
ному ряду ШуЛьце—Гарди.
Выше рассматривался вопрос взаимодействия двух одина-
ковых частиц, при котором перекрытие ионных атмосфер приво-
дит к отталкиванию частиц и отсутствию коагуляции системы.
Рост концентрации электролита должен способствовать коагуля-
ции (слипанию частиц).
Однако экспериментально на моделях Дерягиным с сотруд-
никами • было показано, что даже значительное увеличение
концентрации электролита (до 1 н. NaCl) не уничтожает пол-
ностью энергетического барьера между частицами и на неболь-
ших расстояниях между ними (~100А) существуют значитель-
ные силы отталкивания. Так как в этом случае невозможно
говорить о силах электростатического отталкивания (отсутствует
диффузная часть двойного электрического слоя), то Дерягиным
введено представление о силах иной природы, пр-видимому, свя-
занных с сильной сольватацией поверхности и особой структу-
рой образующихся сольватных слоев. Оба вида сил, как элек-
тростатического; так и сольватационного характера Дерягин
объединяет под общим названием расклинивающего да-
вления.
Согласно взглядам Ребиндера, вышеназванный фактор ус-
стойчивости действует только в случае разбавленных дисперсных
теициала по сравнению *с ip-потенциалом, а также часто наблюдающимся
параллелизмом этих величин, обычно на практике устойчивость дисперсных
систем связывают не. с ^ потенциалом (что было бы более правильным),
а с ^-потенциалом.
241
систем. В случае концентрированных систем с большим со-
держанием дисперсной фазы и в предельно концентрированных
системах (например, предельно концентрированные эмульсии)
решающую роль играет другой фактор устойчивости — структур-
но-механические свойства адсорбционных слоев, образованных
на границе раздела дисперсная фаза — дисперсионная среда. Та-
кие адсорбционные слои могут обладать значительной струк-
турной вязкостью, превышающей вязкость дисперсионной среды
на несколько порядков, и в пределе обладать свойствами твер-
дого тела.
Наличие структурно-механического барьера, как отмечает
Ребиндер, становится достаточным только тогда, когда на на-
ружной поверхности такого адсорбционного слоя поверхностная
энергия достаточно мала. В обратном случае, при наличии хотя
и структурированной, но не лиофильной, а лиофобной оболочки
коагуляции — вторичная флоккуляция — происходит путем сцеп-
ления оболочек наружными поверхностями.
В свете современных воззрений причины устойчивости дис-
персных систем могут быть разделены на три группы:
1) электростатическое отталкивание заряженных частиц,
возникающее при перекрытии ионных атмосфер;
2) силы неэлектростатического характера, связанные с нали-
чием на поверхности частиц особых сольватных слоев, состоя-
щих из молекул дисперсионной среды;
3) образование на поверхности частиц адсорбционной геле-
образной оболочки, обладающей высокой механической прочно-
стью.
Р а б о т а 40
ИССЛЕДОВАНИЕ ЗОН КОАГУЛЯЦИИ (НЕПРАВИЛЬНЫЕ РЯДЫ)
Для проведения опыта в мензурку берут 10 см3 молярного
раствора А1С1з. В 22 чисто вымытые пробирки наливают по
5 см3 золя канифоли. Из мензурки с А1С1з отбирают пипеткой
5 см3 раствора и приливают в пробирку с золем, на которой
восковым карандашом делается отметка: 22. Полученную смесь
хорошо встряхивают. Мензурку с А1С1з доливают водой до
10 см3, в результате чего раствор разбавляется вдвое. Полови-
ну этого раствора приливают в пробирку с золем, на которой
делают отметку: 21. Мензурку снова доливают водой до 10 см3
и т. д. Таким образом получают 22 пробирки с золем канифоли
и прибавленным раствором А1С13. Пробирки устанавливают в
штатив и оставляют в покое на сутки. Через сутки наблюдают
происшедшие в пробирках изменения и записывают результаты
по'следующей схеме, начиная запись с пробирки № 1, т. е. с са-
мой малой концентрации (табл. 1).
242
Для получения правильных результатов необходимо пользо-
ваться чистой посудой.
Таблица 1
№ пробирок Концентрация раствора Результаты наблюдений через 24 ч
1 ~ Не коагулировало
2 — Слабо коагулировало и
3 —- т. д.
Для работы приготовляют золь канифоли следующим обра-
зом: 1 г .канифоли растворяют в 25 см3 спирта. Полученный ра-
створ канифоли вливают небольшими порциями в колбу с
600 см3 дистиллированной воды. Канифоль, растворяющаяся в
спирте, практически не растворяется в воде, вследствие чего по-
лучается молочно-белый опалесцирующий золь ’ канифоли.
При получении грубодисперсных частиц золь отфильтровы-
вают через фильтр?»
'Работа 40а
ИССЛЕДОВАНИЕ ЗОН КОАГУЛЯЦИИ С ПОМОЩЬЮ ФОТОЭЛЕКТРОКОЛОРИМЕТРА
Коагулированный золь обладает большим светорассеянием
(большей мутностью) по сравнению с некоагулированным зо-
лем. Это может быть использова-
но для определения зон коагуля-
ции оптическим методом с по-
мощью фотоэлектроколориметра.
В этом случае работа по опреде-
лению зон коагуляции имеет
более количественный характер
(по сравнению с работой 40) и
проводится за более короткое
время (4—5 ч).
Зависимость мутности золя т
от концентрации коагулирую-
щего электролита (иона) изоб-
ражена на рис. 100 (приведенная
зависимость соответствует коагу- Рис 1(Х) Зависимость мутности
ляции золя AgJ, полученного при 30ля от ЛОгарифма концентрации
сливании равных объемов 1 X коагулирующего иона.
X Ю"4 М раствора AgNO3 и 4 X
Х10"4 М раствора KJ раствором A1(NO3)3). В зоне I золь ус-
тойчив и величина т имеет небольшое значение. Увеличение
концентрации коагулирующего иона приводит к коагуляции
243
золя (И.зона), однако дальнейший рост концентрации коагули-
рующего электролита приводит к перезарядке золя и появлению
новой зоны устойчивости золя (III зона*). Наконец, при ещё
больших концентрациях коагулирующего иона наступает вторая
зона коагуляции (IV зона), связанная со сжатием двойного элек-
трического слоя, существующего вокруг коллоидных частиц.
Определение т производится с помощью фотоэлектроколори-
метра по одной из двух методик (стр. 48). С помощью данной
методики могут быть исследованы неправильные ряды, полу-
чающиеся при коагуляции отрицательно заряженных золей Agl,
AgBr, AgCl ионами Al3+, Zr4+,Th4+, положительно заряженных
золей галогенидов серебра цитрат-ионами и др.
Выполнение работы
Исследуют- неправильные ряды на примере коагуляции золя
Agl раствором Al(NO3)s. Методика работы состоит в следую-
щем [8]. Приготавливаются растворы: 4 • 10-4 М раствор KJ,'
1 • 10-4 М раствор AgNO3, 2,1 • 10-4 М, 2,1 • 10“3 и 2,1 • 10~2 М рас-
Рис. 101. Изменение оптиче-
ской плотности коагулирую-
щего золя во времени.
творы А1(МО3)з. Изучают коагуля-
цию золя AgJ раствором A1(NO3)3b
области концентраций 1 • 10~6— 1 X
X1 • Ю~3 М. Золь получают непосред-
ственно в кювете, в которой в даль-
нейшем производится измерение оп-
тической плотности D. В кювету на-
ливается необходимое количество
см3 Н2О, A1(NO3)3 (табл. 1а), 10см3
AgNO3 и затем 10 см3 КД (объем
получающегося золя в кювете со-
ставляет 21 см3). Включают секун-
домер, содержимое кюветы тщатель-
но перемешивают в течение 0,5 мин, продувая воздух через пи-
петку. Кювета помещается в фотоэлектроколориметр и произво-
дятся измерения оптической плотности D через 3, 5, 8 и 10 мин
после смешения растворов **. После проведенных измерений
золь выливается, кювета тщательно ополаскивается дистиллиро-
ванной водой и аналогичные эксперименты проводятся с боль-
шими количествами коагулирующего электролита. Полученные
величины D наносятся на график (рис. 101) в виде зависимости
D = f(t), где t — время, отсчитываемое с момента сливания
* Согласно работам Матиевича, перезарядка отрицательно заряженных
золей связана с адсорбцией на коллоидных частицах-, ие многовалентных
ионов, например,, Al3+, Th4+, а их продуктов гидролиза (например, для А13+
продукты гидролиза имеют общую формулу А1л (ОН)+<3п-т’).
.** С целью получения более высоких значений D при измерениях при-
меняется синий светофильтр.
244
всех растворов. Из найденных величин Dl0 (величина D при t=
= 10 мин) вычисляются значения тю по формуле тю ------j--,
где I — толщина слоя среды в см, через который проходит луч в
кювете.
На основании полученных результатов строится окончатель-
ная зависимость тю от логарифма концентрации коагулирую-
щего иона (рис. 100). Полученные результаты оформляются трк-
же в виде таблицы 1а. 1
Таблица 1а
№ Количество и концен- трация исходного А1 (ЫОз)з Количе- ство н2о, см5 Концентра- ция Al (NO3)S в золе, молъ!дм* t D Т10
V, см9 с, молъ/дм’ 3 мин 5 мин 8 мин 10 мин
1 0,0 2,1 • 10~4 1,0 0
. 2 0,1 2,1 • 10- 0,9 1 10~в
3 0,2 2,К-10~4 0,8 2-Ю-6
4 0,3 2,1 • 10j 0,7 3-10-®
5 0,5 2,1 10~4 0,5 5-Ю"3
6 0,8 2,1 10~4 0,2 8-Ю-’
7 1,0 2,1-10 4 0,0 1 10~6
8 0,2 2,1 • 10“? 0,8 2 -10“®
9 0,3 2,1 • 10~3 0,7 З-Ю-6
10 0,5 2,1 10“’ 0,5 5-10-®
11 0,8 2,1-ю-3 0,2 8-10~s
12 1,0 2,1 • Ю"8 0,0 1 • IO'4
13 0,2 2,1 10“* 0,8 2-10~4
14 0,3 2,1 Ю 0,7 3-10“4
15 0,5 2,1 10“’ 0,5 5 IO"
16 0,8 2,1 W , 0,2 8 • Ю-4
17 1,0 2,1 • 10~2 0,0 1 • 10-3
Работа 41
ВЗАИМНАЯ КОАГУЛЯЦИЯ ЗОЛЕЙ
. Опыты по взаимной коагуляции производят следующим об-
разом. Восковым карандашом помечают номера десяти проби-
рок. В пробирки наливают золь. Fe (ОН)з в убывающем количе-
стве по порядку (табл 2). В каждую пробирку приливают с по-
мощью бюретки золь МпО2 в количестве, отмеченном в табл. 2.
Опыты по взаимной коагуляции можно также проводить,
пользуясь Ре(ОН)з и отрицательно заряженным золем Аз25з
(табл. 3).
По окончании опытов отмечают номер пробирки, в которой
наблюдается максимум коагуляции.
245
Приготовление положительно Заряженного золя Fe(OH)3.
Золь гидрата окиси железа получают методом гидролиза сле-
дующим образом: 95 см3 дистиллированной воды нагревают до
кипения. К кипящей воде прибавляют небольшими порциями
5 см3 10% раствора FeCls-
. Таблица 2
№ пробирок Fe (OHU см* МпО2, см* Результаты смешения
1 9,9 0,1 Без изменений
2 9,7 0,3 Небольшая муть и т. д.
3 9,5 0,5
4 9,0 1,0
5 7,0 3,0
6 5,0 5,0
7 3,0 7,0
8 1,0 9,0
9 0,5 9,5
10 0,2 9,8
После несколЬких минут кипячения получается красно-кори-
чневый золь гидроокиси железа, заряженный положительно.
Золь лучше предварительно подвергнуть диализу (стр. 314),
Таблица 3
№ пробирок Fe (OH)S, смъ As2S3> см* Результаты смешения
1 9,0 1 Коагуляции не наблю-
дается
2 7,0 3 Слабая муть и т. д.
3 5,0 5
4 2,0 8
5 1,0 9
6 0,5 9,5
7 0,3 9,7
8 0,2 9,8
9 0,1 9,9
Приготовление отрицательно заряженного золя МпО2. К 50 см3
0,1 н. КМпО4 при тщательном перемешивании стеклянной па-
лочкой приливают по каплям 1% раствор перекиси водорода до-
тех пор, пока капля образующегося золя, взятая стеклянной па-
лочкой и нанесенная на фильтровальную бумагу, не перестанет
окрашивать бумагу в розовый цвет. Получается темно-коричне-
вый отрицательно заряженный золь МпО2. Полученный золь
разбавляют в три раза.
246
Получение золя сернистого мышьяка. При кипячении полу-
чают насыщенный раствор H3AsO3; 50 см3 этого раствора разбав-
ляют до 200 см3 и в течение 5 мин пропускают через раствор
сероводород. Образуется желтый опалесцирующий золь серни-
стого мышьяка. Золь отфильтровывают от грубых частиц и диа-
лизуют до отсутствия реакции на SH . Избыток сероводорода
можно удалить, пропуская через золь ток водорода.
Работа 42
ЗАЩИТНОЕ ДЕЙСТВИЕ ВЫСОКОМОЛЕКУЛЯРНЫХ СОЕДИНЕНИЙ
(ОПРЕДЕЛЕНИЕ «ЗОЛОТОГО ЧИСЛА»)
Мерой защитного действия различных высокомолекулярных
соединений является золотое число. Золотым числом назы-
вают то минимальное количество сухого защитного коллоида
в мг, которое способно воспрепятствовать перемене окраски
10 см3 красного золя золота
(приготовленного по одному
из стандартных методов) в
синий при добавлении к нему
1 см3 10% раствора NaCl. 1
Опыт проводят следующим
образом. В 10 чистых, сухих
пронумерованных пробирок
наливают по 10 см3 красного
золя золота, полученного од-
ним из нижеуказанных мето-
дов. В каждую пробирку до-
бавляют по 1 см3 раствора же-
латины из соответствующих по
номерам пробирок с раствором
желатины (табл. 4). Содержимое
вают и приливают в каждую по 1 см3 10% раствора NaCl.
Так как защитное действие проявляется не мгновенно, то по-
сле перемешивания выжидают 2—3 мин и отмечают номер про-
бирки, в которой наблюдают изменение красной окраски в си-
нюю. Для вычисления золотого- числа берут среднее значение
рассчитанного веса желатины в мг для двух смежных пробирок,
в одной из которых коагуляция обнаружена, а в другой отсут-
ствует.
Получение золя золота. Красный золь-золота можно полу-
чить одним из следующих методов.
Получение золя с формальдегидом. 120 см3 дистиллирован-
ной воды нагревают в колбе, предварительно хорошо вымытой.
Во время нагревания прибавляют 2,5 см3 0,6% раствора НАиСЦ
и 2 см3 0,2 н. КгСО3. Смесь доводят до кипения, после чего,
Табли (Д 4
' №
пробирок
Раствор
желатины,
см9
Н2О, cjks
1
2
3
4
5
6
7
8
' 9
1
2
3
4
5
6
7
8
9
9
8
7
6
5
4
3
2
1
пробирок
хорошо
взбалты-
247
сняв коЛбу с плитки, быстро приливают небольшими порциями
(при энергичном перемешивании) 3—5 см3 разбавленного фор-
мальдегида. Через 1—2 мин после прибавления жидкость розо-
веет, а затем окрашивается в темйо-красный цвет.
Реакция получения золя происходит по следующей схеме:
НАиС14 + 2К2СО3 + Н2О = Au (ОН)3 + 2СО2 -|- 4КС1
2Аи (ОН)3 + К2СО3 =-2КАиО2 + ЗН2О 4-СО2
2КАиО2 + ЗНСОН + К2СО3 = 2Аи %- ЗНСООК + КНСО3 + Н2О
Полученный золь золота имеет размеры частиц 20—40 ммк.
Получение красного золя золота со спиртом. К 100 см3 дис-
тиллированной воды приливают 2 см3 0,6% раствора НАиС1<
2 см3 0,2 н. раствора К2СО3 и нагревают до кипения. Сняв колбу
с плитки, прибавляют небольшими порциями 2—5 см3 спирта
при тщательном перемешивании. Продолжают нагревание, пока
появившаяся розовая окраска не станет вишнево-красной.
Полученный, золь золота очень чувствителен к прибавлению
электролитов.
Приготовлений раствора желатины. 1 см3 0,1% раствора же-
латины разбавляют до 100 см3. Полученный раствор нали-
вают в десять пронумерованных пробирок,, как это указано
в табл. 4.
Работа 42а
ИССЛЕДОВАНИЕ ЗАВИСИМОСТИ ЧИСЛА ПРИЛИПАНИЯ ЧАСТИЦ КВАРЦА
(СТЕКЛА) ОТ ПРИРОДЫ И КОНЦЕНТРАЦИИ ЭЛЕКТРОЛИТА
В основе процессов коагуляции лежат силы адгезии (прили-
пания), возникающие при сближении частиц дисперсной фазы.
Поэтому исследования сил прилипания представляют большой
теоретический и практический интерес. Рядом советских и ино-
странных ученых были разработаны различные методы, позво-
ляющие изучать слипание'частиц, наблюдая непосредственно за
взаимодействием частиц с поверхностью.
В лаборатории коллоидной химии ЛГУ используется метод,
предложенный Бузагом. Бузаг принимает прилипание частиц
суспензии к твердой пластинке из того же материала, что и ча-
стицы, за модель коагуляции, считая, что между взаимным при-
липанием частиц и их прилипанием к твердой стенке имеется
полный параллелизм.
Принцип Метода заключается в том, что частицы при-
водятся в соприкосновение с пластинкой и что силе тяжести
противодействует сила взаимодействия между частицей и
стенкой.
248
Исследуемую монодисперсную суспензию кварца или сте-
кла помещают в закрытую микрокамеру (рис. 102). Микрока-
мера представляет собой стеклянную трубку длиной приблизи-
тельно 20 см, с внутренним диаметром 5—5,5 мм, снабженную
по краям двумя кранами. В середине камеры имеется шарооб-
разное вздутие 1, обрезанное с нижней и верхней сторон, с
параллельно отшлифованными краями, к которым с помощью
канадского бальзама приклеены стеклянные пластинки (напри-
мер, микроскопические покровные стекла).
Микрокамеру заполняют исследуемой суспензией засасыва-
нием. Опыты проводятся с разбавленными суспензиями (по воз-
можности монодисперсными), i
0,02—0,05%. Микрокамера ус-
танабливается на столике
микроскопа. После оседания
всех частиц на определенной
площади поверхности дна ка-
меры с помощью окулярной
сетки определяется среднее
число осевших частиц. Для
удобства наведения на фокус
и облегчения подсчета частиц
на поверхности пластинки, при-
крывающей камеру, делают
тонкую отметку тушью. Через
определенный отрезок време-
ни после осаждения частиц
(5—10 мин) камеру осторож-
ным движением (без сотря-
сения) переворачивают вверх
дном, так что нижняя пластин-
концентрацию частиц
Рис. 102. Схема микрокамеры и при-
бора для измерения чисел прилипания
микроскопических частиц.
ка оказывается сверху, затем (через определенное время) под-
считывают число прилипших к поверхности частиц на той же
площади поверхности, на которой подсчитывалось число осев-
ших частиц. Число прилипших частиц, выраженное в процен-
тах к общему числу осевших частиц, называется' числом
прилипания и принимается за количественную меру коагу-
ляции.
Опыты по адгезии частиц производят в воде и в рдстворах
электролитов (NaCl, КС1, MgCl2, СаС12 и др.) различной кон-
центрации (1 • 10~3— 1 • 10-s М). В работе необходимо соблюдать
максимальную чистоту, микрокамера должна быть хорошо про-
мыта перед каждым опытом. При заполнении камеры надо сле-
* Для работы могут быть использованы фракции с размером частиц:
I—5 мк, 5—10 мк, 10—20 мк, 20—30 мк. Выделение отдельных фракций
производится по методу восходящей струи (стр. 26),
249
дить за тем, чтобы в камеру не попадали пузырьки воздуха.
Для проверки чистоты камеры необходимо проводить контроль-
ный опыт со стандартной суспензией в чистой воде.
ЛИТЕРАТУРА .
1. Дерягин Б. В., Труды III Всесоюзной конференции по коллоидной хи-
мии, 225, 1956.
2. Дискуссия. Колл, ж., 23, № 3, 353 (1961).
3. Коагуляция коллоидов, Сб. под ред. А. И. Рабиновича, ОНТИ, 1931.
4. Кройт- Г. Р., Наука о коллоидах, ИЛ, 1955.
5. Л е в и ч В. Г., ДАН СССР, 103, We 3, 453 (1955).
6. Р е б и н д е р П. А., Колл, ж., 20, № 5, 527 (1958).
7. Matijevic Е., Abramson М., Shulz К., Kerker М., J. Phys. Chem.,
64, 1157 (1960).
8. Matijevic Е., Mathai К-> Ottewill R., Kerker M., J. Phys. Chem.,
65, 826 (1961).
9. Matijevic E., Mathai K., Kerker M., J. Phys. Chem., 66, 1799
(1962).
ГЛАВА V
СТРУКТУРООБРАЗОВАНИЕ В ДИСПЕРСНЫХ СИСТЕМАХ
Наличие дисперсной фазы (часто в относительно малом ко-
личестве) может существенно изменить структурно-механиче-
ские свойства системы по сравнению с чистой дисперсионной
средой. Возможность изменения механических свойств жидкой
дисперсионной среды зависит от химической природы веществ,
образующих дисперсную систему, и определяется молекулярны-
ми силами сцепления между частицами дйсперсной фазы и
взаимодействием их с дисперсионной средой.
В тех случаях, когда такое взаимодействие между частицами
дисперсной фазы и дисперсионной средой слабо выражено, ме-
ханические свойства подобных систем являются качественно те-
ми же, что и чистой дисперсионной среды.
Поэтому все дисперсные системы могут быть разделены на
две группы: 1) бесструктурные системы и 2) струк-
турированные системы, обладающие структурой, охва-
тывающей весь занимаемый ими объем.
Частицы дисперсной фазы бесструктурной системы не связа-
ны между собой, способны перемещаться независимо друг от
друга в среде молекул дисперсионной среды под действием
внешних сил. В качестве примера бесструктурных дисперсных
систем могут быть указаны обычные суспензоидные золи или
разбавленные эмульсии. Подобные суспензоидные системы про-
являют свойства истинно вязких жидкостей и не обладают ме-
ханической прочностью. Отличие свойств этих систем от свойств
дисперсионной среды носит количественный характер и заклю-
чается в том, что вязкость их больше, чем вязкость дисперсион-
ной среды (за счет заполнения части объема частицами дисперс-
ной фазы). Такое относительное повышение вязкости в бесструк-
турных системах, как это было теоретически установлено Эйн-
штейном, пропорционально отношению объема частиц дисперс-
ной фазы к общему объему системы:
251
В ЭТОМ, уравнении Эйнштейна т]— вязкость дисперсной си-
стемы, цо —вязкость дисперсионной среды, Vx — объем частиц
дисперсной фазы, Vo — общий объем системы и k — коэффи-
циент, зависящий от формы частиц (для сферических частиц
k = 2,5).
Из уравнения следует, что вязкость дисперсной системы дол-
жна быть линейной функцией концентрации дисперсной фазы,
что было подтверждено для суспензоидов многочисленными
экспериментальными данными.
Для второй группы, т. е. структурированных дисперсных си-
стем, характерным является развитие в той или иной степени
упруго-пластических свойств, связанных с образованием струк- ,
туры и возможностью изменения агрегатного состояния системы,
с переходом в твердое тело. Частицы дисперсной фазы в таких
системах связаны межмолекулярными силами в одну общую
структуру, распространяющуюся на весь объем, занимаемый
дисперсной системой.
Возможность проявления сил молекулярного сцепления ме-
жду частицами, необходимых для образования сплошной про-
странственной сетки, значительно повышается при условии
достаточно высокой дисперсности и при частицах анизодиаметри-
ческой формы, т. е. с резко различными размерами по отдель-
ным направлениям (пластинчатых или палочкообразных, вытя-
нутых частицах). Предполагая для анизодиаметрических частиц
различную толщину адсорбционного сольватного слоя и воз-
можность его утоньшения и прорыва в местах наибольшей кри-
визны — углах и ребрах, можно прийти к заключению о наличии
условий, благоприятствующих сцеплению и агрегированию ча-
стиц. В этих случаях достаточно весьма малоё объемное содер-
жание дисперсной фазы- для того, чтобы частицы могли войти
в соприкосновение друг с другом концами или ребрами и обра-
зовать сплошную' пространственную сетку, обладающую извест-
ной механической прочностью. Такой процесс часто называется
лиофильной коагуляцией, чем подчеркивается коагуля-
ционный механизм образования таких рыхлых скелетов струк-
тур, в отличие от компактных структур, образующихся при лио-
фобной коагуляции, а также при осаждении первичных,
не агрегированных частиц.
Дисйерсные системы, принадлежащие к этой группе, не под-
чиняются уравнению Эйнштейна, и вязкость их возрастает с кон-
центрацией больше, чем это следует по линейному закону.
Процесс структурообразования может привести к тому, что
все частицы дисперсной фазы окажутся более или менее прочно
связанными между собой, полностью утратив свою подвижность,
и вся дисперсионная среда окажется заключенной в промежут-
ках между частицами. Такая система, утратившая основное
свойство жидкости — текучесть, носит название гель.
252
Таким образом, гель состоит из двух фаз: первая составляет
его скелет, образованный частицами дисперсной фазы, обладаю-
щий механической прочностью и придающий всей системе свой-
ства твердого тела, и вторая—-жидкость, заполняющая все про-
межутки этого скелета.
В зависимости от свойств структурных элементов, образую-
щих гель, и прочности их сцепления могут бнть получены твер-
дые системы с «жестким» сцеплением частиц, обладающие ча-
сто значительной твердостью и хрупкостью, как, например, твер-
дые гели кремневой кислоты или алюмогели.
Особенно благоприятные условия для процесса структурооб-
разования имеют растворы высокомолекулярных и высокополи-
мерных органических соединений (гл: VI).
Сильно выраженное взаимодействие между макромолекула-
ми этих соединений и дисперсионной средой в специфических
растворителях, а также особенности строения макромолекул об-
разующих нитеобразные и разветвленные .формы — все это со-
здает условия, способствующие образованию сплошной про-
странственной сетки по объему и цереходу в твердое тело.
Получающиеся при этом процессе системы, в отличие от ге-
лей (являющихся гетерогенными образованиями), оказываются
гомогенными, однофазными и носят название — стущни, сам
процесс — застудневания. Студни, в противоположность
хрупким гелям, обладают хорошо выраженными упруго эласти-
ческими свойствами, что дало основание употреблять для их
описания термйн эластические гели, имевший некоторое
распространение.
СТРУКТУРНО-МЕХАНИЧЕСКИЕ СВОЙСТВА ДИСПЕРСНЫХ СИСТЕМ
Структурированные дисперсные системы весьма распростра-
нены и исследование их свойств имеет большое значение для
развития ряда отраслей народного хозяйства, в частности строи-,
тельства (цементы, бетоны), инженерной геологии (грунты), ке-
рамической промышленности (пасты, шликеры), при нефтяном
бурении (глинистые суспензии), при' изготовлении консистент-
ных смазок (олеофильные системы), синтетических волокон и
пластических масс.
Важнейшей характеристикой этих систем, сочетающих в себе
свойства твердого тела и жидкости, является комплекс механи-
ческих свойств — прочности, упругости, пластичности, вязкости. <
Совокупность этих свойств решает вопросы практического ис-
пользования таких систем (например, в качестве строительных
и других материалов).
Механические свойства рассматриваемых систем опреде-
ляются их структурой — природой сил, связывающих частицы
дисперсной фазы, дисперсностью частиц, характером их
253
взаиморасположения и движения *, Вот почему эти свойства на-
зываются структурно-механическими.
Для разработки способов модификации структуры с целью
получения нужных для практики свойств необходимо прежде
всего установить количественные характеристики механических
свойств структурированных систем. Среди них наибольшее зна-
чение имеют упруго-пластические (реологические)" свойству,
подробно изученные в работах Ребиндера и его школы, пред-
ставляющих собой основы физико-химической механики дис-
персных систем.
Упруго-пластические свойства определяются характером де-
формации (т. е. изменения формы) системы под действием при-
ложенной внешней силы, обычно называемой нагрузкой. Отно-
шение силы к площади, к
которой она приложена, называется
, напряжением. Различные ви-
ды деформаций могут быть све-
дены к двум элементарным: рас-
тяжению и сдвигу. Растяжение
, (или сжатие) вызывается напря-
жением, нормальным к поверх-
ности (т. е. давлением), а
сдвиг — тангенциальным. Наибо-
Рис. 103. Схема тангенциального лее полную характеристику струк-
сдвига. турированных систем дает иссле-
дование деформации сдвига. Под
влиянием тангенциальной си-
лы f (рис. 103) слои тела сдвигаются и каждое нормальное се-
чение тела поворачивается на некоторый угол ф; при малых
ЬЬ' ~ л
значениях ф имеем: ф = -у-- 1 аким образом относительная
деформация сдвига характеризуется величиной угла сдви-
га (выраженной в радианах). Деформации делятся на обрати-
мые и остаточные (пластические). Обратимые деформации ис-
чезают после прекращения действия силы. Тела, обратимо вос-
станавливающие первоначальную форму после снятия нагрузки,
называются упругими телами.
Идеально упругие тела подчиняются закону Гука, согласно
которому величина относительной деформации е пропорциональ-
на приложенному напряжению Р
Р—Ег
(1)
* Истинно-твердые тела (кристаллы) по современным воззрениям, так-
же обладают свойствами коллоидных систем, вследствие существования
дефектов кристаллической решетки различного рода в реальных телах; сред-
ние расстояния между дефектами близки к размерам коллоидных частиц
и механические свойства реальных кристаллов определяются в значитель-
ной’ степени структурой дефектов.
254
(2)
Коэффициент Е, называемый модулем упругости, характеризует
жесткость тела. При напряжениях, превышающих так назы-
ваемый предел упругости Ph (стр. 260), пропорциональ-
ность нарушается: происходит либо разрушение структуры, ха-
рактерное для хрупких тел, предел прочности которых Рт бли-
зок к пределу упругости, либо возникают остаточные (пласти-
ческие) деформации, не исчезающие после снятия нагрузки. Те-
ла, обнаруживающие остаточную деформацию при напряжениях,
превышающих предел упругости, называются пластичными
телами. Одним из видов остаточной деформации является
течение, характерное для вязких жидкостей, при котором вели-
чина деформации непрерывно увеличивается при постоянно дей-
, ствующем напряжении. Вязким называется тело, изменяющее
форму при любом, сколь угодно малом напряжении (Р/{ = 0).
Идеально вязкие тела — жидкости — подчиняются закону Нью-
тона, согласно которому градиент скорости сдвига или, иначе
говоря, скорость относительной деформации сдвига пропорци-
ональна приложенному напряжению
г, dt
p^~dF
где т] — коэффициент вязкости.
Преобладание обратимых деформаций приближает свойства
системы к твердому телу, остаточных — к жидкости. Однако по
современным представлениям различие
жидким состояниями не является резким и определяется лишь'
скоростью рассасывания (спада) приложенного к телу напря-
жения, т. е. периодом релаксации т, равным отношению
двух констант: вязкости и жесткости *
Величина т, имеющая размерность времени и показывающая,
за какое время начальное напряжение, приложенное к телу,
уменьшается в е раз, является основной константой, объеди-
няющей свойства твердого тела и жидкости. Если время воз-
действия значительно больше т — данное тело ведет себя как
жидкость, если меньше — как твердое тело, подвергаясь хруп-
кому разрыву при напряжениях, превышающих прочность
структуры.
между твердым и
(3)
Р dt Р
* Из (1) dP = Edt-, из (2) =-------, следовательно dP = — Р dt.
4
т] dP dt С (* dt
Обозначая = получим: I dlnP= I — (где Ро — началь-
но о
, р
ное напряжение), откуда Р = Рйе 1,1, а следовательно, при t — т; р = —° .
е
255
Так, для кристаллов льда £=9-1010 г^м-сек2:,
т] = 1,2 • Ю14 г/см-сек, а следовательно т 13000 сек. Поэтому
при быстрых воздействиях лед ведет себя как хрупкое твердое
тело, но при длительных — проявляет текучесть (ледники). Для
жидкой воды - величина Е имеёт тот же порядок, но вязкость
rj = 10~z г/см-сек и т=Ю-13 сек. Поэтому вода течет, но при
очень быстрых ударах (например, при простреле пулей) струя
воды подвергается хрупкому разрушению; упругость воды про-
является также при мгновенном касании камня. Высоковязкие
жидкости, например асфальт, при низких температурах обла-
дают большими tj и <г и ведут себя как упругие тела; с ростом
температуры значения г] и t уменьшаются на несколько поряд-
ков и асфальт становится пластичным (остаточные деформа-
ции) хотя й разрывается при быстрых ударах.
Таким образом между твердым телом и жидкостью суще-
ствует непрерывный ряд переходов, осуществляемых структу-
рированными системами, сочетающими в себе свойства обоих
состояний. Так, в твёрдообразных упругих системах (например,
в бентонитовых гелях) при малых, но длительных напряжениях
наблюдается очень медленное течение, называемое ползу-
ч е с т ь ю. При этом структурная сетка, разрушаясь, успевает
обратимо восстанавливаться. При дальнейшем увеличении Р
наступает лавинное разрушение структуры, вязкость умень-
шается скачкообразно на несколько порядков и система с раз-
рушенной структурой течет далее как обычная жидкость. Чем
резче выражено это уменьшение вязкости, тем более твердооб-
разным явл'яется тело.
Приведенные примеры показывают, что место структуриро-
ванной системы при переходе от твердого состояния к жидкому
определяется, с одной стороны, прочностью структуры, с дру-
гой— отношением времени воздействия к периоду релаксации,
выражаемому величинами т] и Е.
В структурированных системах, в отличие от обычных (нью-
тоновых) жидкостей, вязкость не является постоянной величи-
ной, а зависит от напряжения сдвига, уменьшаясь с ростом Р
в связи с разрушением структуры. Поэтому для решения ряда
задач теории пластичности и характеристики пластических
свойств системы вместо эффективной (структурной) вязко-
сти г](Р) вводится ряд постоянных значений т], отвечающих
различным стадиям разрушения структуры (стр. 260).
Таким образом, измерения реологических параметров — мо-
дулей упругости, граничных напряжений и вязкостей — позво-
ляют характеризовать упруго-пластично-вязкие свойства реаль-
ных структурированных дисперсных систем.
Методы, применяемые для исследования этих свойств, мож-
но разделить на две группы: методы первой группы применяют-
ся для исследования сильно структурированных, тиксотропных,
256
гелеобразных систем. Их основу составляет измерение дефор-
мации сдвига. Сюда относятся методы коаксиальных цилин-
дров [5], конического пластометра, смещения пластинки и т. д.
Вторая группа методов применяется для изучения относи-
тельно мало структурированных коллоидных систем — золей и
основывается на измерении скорости их течения через капил-
лярную трубку, в зависимости от приложенного давления (вис-
козиметрические методы).
Работа 43
ИССЛЕДОВАНИЕ УПРУГО-ПЛАСТИЧЕСКИХ СВОЙСТВ СТРУКТУРИРОВАННЫХ
СИСТЕМ МЕТОДОМ ТАНГЕНЦИАЛЬНОГО СМЕЩЕНИЯ ПЛАСТИНКИ
Метод тангенциального смещения заключается в том, что
в исследуемую систему погружается рифленая пластинка. Пла
стинка» посредством жесткой нити (металлической или стеклян
ной), соединенной с микрофо-
тошкалой, подвешивается к
кварцевой или стеклянной спи-
ральной пружинке (метод Вей-
лера и Ребиндера).
Вместо пружинки можно
применять коромысло обыкно-
венных аналитических весов
(рис. 104). Сообщаемое усилие
р (нагрузка на чашку весов
или опускание столика вниз в
случае растяжения пружинки)
вызывает смещение пластин-
ки I, регистрируемое наблюде- Рис 104 Прибор
для измерения на-
нием положения крючка ни- пряжения сдвига методом танген-
ти 1 в микроскоп 2 с окуляр- циального смещения пластинки,
микрометром. Груз 3 уравно-
вешивает чашку весов 4. Результаты опыта обычно выражают
графически в координатах р—е и е*— t', это позволяет вычислить
величины, характеризующие упруго-пластические свойства кол-
лоидной системы.
Рассмотрим кривые р—е, полученные при помощи растяже-
ния спиральной пружинки (рис. 105) или опускания чашки ве-
сов (рис. 106). Кривые показывают наличие трех типов струк-
туры. Кривая / характерна для структур с упругими свойствами
(например, 10% суспензия природного -бентонита). Начальный
прямолинейный участок ОА отвечает упругой деформации и по-
зволяет вычислить модуль упругости по уравнению (1).
С ростом нагрузки при достижении рн происходит хрупкий
разрыв структуры: пластинка резко вырывается вверх, и даль-
нейшая пластическая деформация после разрушения структуры
257
(участок ВС) может происходить п{Ти нагрузке р\ меньшей,
чем ph (это уменьшенная величина устанавливается автомати-
чески, вследствие сокращения пружинки, рис. 105. При приме-
нении весов кривая с максимумом могла бы получиться лишь
при снятии части груза с чашки в момент деформации. Практи-
чески, при постоянной нагрузке, кривая 7 пойдет так, как пока-
зано на рис. 106.
Значение pi отражает величину того остаточного структуро»
образования, при котором происходит дальнейшая пластическая
деформация.
Рис. 105. Кривые напряжение —
деформация (метод Вейдера —
Ребиндера).
Рис. 106. Кривые напряже-
ние — деформация (метод
пластинки с коромыслом).
Величина предельного напряжения pk, при котором проис-
ходит разрушение структуры (начало течения), отнесенная к
единице площади поверхности скольжения, представляет собой
предельное статическое напряжение сдвига 6S (сов-
падающее в данном случае с величиной Р^):
где S — площадь поверхности пластинки.
Кривые типа 77 получены для систем с пластическим разру-
шением структуры (например, диализованная 10% суспензия
бентонита).
В этом случае после прямолинейного участка упругой дефор-
мации ОА продолжается рост р при значительной деформации е
(участок AD). Этот участок отражает состояние суспензии, в
.которой начинается преобладание пластических деформаций
над остаточными упругими деформациями, переходящих далее
в пластическое течение. В этом случае разрушение структуры
начинается в точке отрыва кривой от прямолинейного участ-
ка А, отвечающей пределу текучести, тогда как прочность ха-
рактеризуется значением рт в точке D.
258
Кривая Ш характерна для структуры с заметным преобла-
данием пластических свойств над упругими (например, 10%
суспензия бентонита в Ыа+-форме); напряжение сдвига с ростом
деформации достигает некоторого предельного постоянного зна-
чения pi=pm, отвечающего пластическому течению.
Исследование упруго-пластических свойств производится
также посредством анализа кривых зависимости деформации е
от времени t при постоянной нагрузке, т. е. кривых е—t. Кривая
для упругой системы, в том случае, когда величина нагрузки
не превышает предела упругости, показана на рис. 107. В мо-
мент приложения нагрузки (f=0) возникает упругая деформа-
ция £о, исчезающая после снятия
нагрузки со скоростью звука в
данной системе. Она характери-
зуется величиной условно-мгно-
венного модуля упругости
Е-^4- (1а)
где Р — напряжение, т. е. нагруз-
ка р, приходящаяся на единицу
площади пластинки. При сохра-
нении постоянной нагрузки в те-
Рис. 107. Кривая деформация —
чение нескольких минут проис- время для упругого тела.
ходит эластическая деформа-
ция (ет—е0), представляющая собой процесс упругого после-
действия. Тела, обладающие значительной деформацией упру-
гого последействия, называются эластичными и характеризуются
величиной модуля эластичности
£2 = —
Ет~ Е0
(16)
Низкие значения Е2 характерны для эластомеров (каучука,
резины). Эластичность их объясняется растягиванием свернутых
в клубки макромолекул, возвращающихся в исходное, более
вероятное, состояние после снятия нагрузки. Таким образом,
эластическая деформация является механически обратимой *,
как и упругая. При снятии нагрузки (р=0), как видно из
рис. 107, процесс идет в обратном направлении, и система воз-
вращается полностью к исходному состоянию.
Если нагрузка превышает предел упругости, происходит
либо упругий разрыв, либо возникает пластическая деформа-
ция. Типичная кривая кинетики деформации реальной упруго-
пластической системы (например, 30% глинистой суспензии)
представлена на рис. 108.
* Но необратимой термодинамически вследствие рассеяния свободной
энергии.
- 259
В этом случае после мгновенной упругой деформации е0,
наряду с затухающим упругим последействием на участке АВ,
происходит постепенное нарастание пластической деформации.
Дальнейший прямолинейный участок ВС обусловлен постоян-
ной величиной пластической деформации (течения). По углу
наклона этого участка можно вычислить, в соответствии с фор-
мулой (2), весьма важную характеристику пластического тече-
ния, а именно наименьшую пластическую вяз-
кость т)1 системы с разрушенной структурой; при этом надо
учесть, что часть напряжения Рь
(предел упругости) затрачивает-
ся на упругую деформацию, а
следовательно, напряжение, вы-
зывающее пластическое течение,
равно Р—Рь- Из формулы (2)
находим, обозначая Де = е — ет:
Рис. 108. Кривая деформация —
время
для упруго-пластичиого
тела.
(5)
Де
Предел упругости Рь, являю-
щийся также пределом текучести,
i определяется как величина на-
пряжения сдвига, при которой
кривая е—t без течения (рис. 107)
переходит в кривую с течением
(рис. 108). Независимым крите-
Pk является инвариантность вели-
правильности выбора
т]1, вычисленной по (5) для разных значений Р. При сня-
рием
чины
тии нагрузки (р = 0 при t — t\) система не возвращается к ис-
ходному состоянию. Конечное состояние отличается от началь-
ного на величину
графика следует,
ствия нагрузки t\
а следовательно:
остаточной пластической деформации ер Из
что отношение е к продолжительности дей-
равно отношению разностей в уравнении (5),
’ll
P~Pk
(6)
Разбор кривых е—t
пластических свойств
ЕО и* £1 *.
dz/dt Е]
показывает, что соотношение упругих и
может быть выражено величинами
* На основании кривых
вязкость эластичности Т]2
развития эластической
е — t может быть найдена также условная
по начальной (наибольшей) скорости
л. ' (dz\
деформации < из которой вычтена постоянная
скорость течения
(d»/dOe —(de/dOi
260
Рассмотренные методы позволяют определить важнейшие
параметры (Еь Е2, Ph, т)ь Ла), количественно характеризующие
упруго-пластические свойства структурированных коллоидных
систем. Например, исследование изменения этих величин в за-
висимости от времени, прошедшего с момента формирования
системы, позволяет провести количественную оценку тиксотроп-
ных свойств структурированных систем, имеющих большое зна-
чение для практики.
Выполнение работы
Приготовляют структурированные системы по указанию пре-
подавателя за сутки до опыта (если нет других указаний). Пе-
ред опытом проверяют нулевую точку весов (рис. 104).-Опреде-
ляют вес и размеры пластинки. Подбирают набор гирь, уравно-
вешивающих пластинку, погруженную в коллоидную систему
Производят калибровку окулярмикрометра по эталону. Про-
веряют правильность установки прямоугольной кюветы квадрат-
ного сечения. Центр квадрата должен находиться на продол-
жении отвесного направления нити с крючком, идущей от коро-
мысла, при горизонтальном его положении. Закрепляют коро-
мысло в этом положении посредством арретира. Фокусируют
микроскоп на конец крючка. Сильно встряхивают исследуемую
дисперсную систему (например, 10% суспензию бентонита), раз-
рушая структуру, выливают в кювету и немедленно погружают
в нее пластинку, надевая петлю нити на крючок. На чашку
весов помещают компенсирующий груз и освобождают арретир
Оставляют систему в покое в течение указанного времени (на-
пример, 10 мин), проверяя правильность фокусировки микроско-
па в конце срока.
Последовательно нагружая чашку весов, записывают пока-
зания шкалы окулярмикрометра п при различных нагрузках р.
Опыт ведут до появления пластического течения, т. е. до той
нагрузки, при которой равновесие не устанавливается. Вычисля-
ют относительную деформацию е, как разность1 между равновес-
ным п и исходным п0 показаниями на шкале, деленную на рас-
стояние до стенки. Опыт повторяют с пластинкой, имеющей
другую площадь S. Результаты опыта выражают графически в
координатах е — р. Определяют рь (конец прямолинейного
участка), рт и вычисляют 6S по формуле (4). Вычисляют Е из
угла наклона прямой ОА по формуле (3).
После этого изучают систему при нескольких постоянных
нагрузка^, больших и меньших рт. Для этого, после окончания
периода покоя, помещают на чашку весов сразу всю нагрузку.
Одновременно включают секундомер, записывают, величину
мгновенного смещения s0 и отмечают показания шкалы через
различные промежутки времени. Снимают нагрузку, отметив
261
время ti> и продолжают наблюдать смещение во времени. Для
удобства снятия нагрузки к гирькам предварительно прикреп-
ляются тонкие ниточки. Результаты выражают графически в
координатах е—t. Вычисляют величину модулей £7 и Е% по (1а,
16) и наименьшей пластической вязкости по формуле (6). При
изучении тиксотропных свойств измерения производят после
различных промежутков покоя.
Работа 44
ОПРЕДЕЛЕНИЕ НАПРЯЖЕНИЯ СДВИГА И СТРУКТУРНОЙ ВЯЗКОСТИ
КОЛЛОИДНЫХ СИСТЕМ МЕТОДОМ КАПИЛЛЯРНОЙ ВИСКОЗИМЕТРИИ
Методы капиллярной вискозиметрии основаны на измерении
объемной скорости течения структурированных коллоидных рас-
творов через капиллярные трубки при различных градиентах
давления.
В условиях ламинарного течения применение уравнения
Ньютона приводит к следующему соотношению между объем-
ной скоростью v (т. е. объемом V жидкости, протекшей за еди-
ницу времени), приложенным давлением Р и вязкостью жидко-
сти (или газа) tj:
Постоянная k определяется геометрическими параметрами
системы и для случая цилиндрического капилляра к = §/-• Под-
становка этой величины в формулу приводит к известному урав-
нению Пуазейля
где I—длина;
г—радиус капилляра.
Обычные жидкости (например, вода, спирт, глицерин) под-
чиняются закону Ньютона и уравнениям (7) и (8). Для этих
жидкостей, называемых также истинно вязкими или ньютоно-
выми, коэффициент вязкости т] является величиной постоянной,
а следовательно, построение графика v—Р или f—P (при по-
стоянном объеме) должно дать прямую, проходящую через на-
чало координат (рис. 109,1).
Структурированные системы не подчиняются указанным за-
конам; зависимость v—Р для таких систем обычно выражается
кривой Ц. -При малых давлениях жидкость вообще не течет,
ввиду наличия упругого сопротивления структуры. В точке А, где
усилие, развиваемое давлением, достигает величины предель-
ного статического напряжения сдвига (работа 43), начинается
262
разрушение структуры и медленное пластическое течение с вы-
„ [ dv
СОКОИ ВЯЗКОСТЬЮ -77,
\ dP
тически неразрушенной структуре. С ростом давления и скоро-
сти происходит разрушение структуры во все большей и боль-
шей степени, которое, однако, никогда не доходит до конца, так
как некоторая доля связей, образующих пространственную
структуру, успевает обрятимо восстанавливаться в потоке даже
при больших скоростях течения. ~
структуре характеризуется
молинейным отрезком ВС
сывается уравнением
k \
— очень мало) типа ползучести в прак-
Такое течение
пря-
опи-
ь
v=-^(P-Pd)
разрушенной
в
Рис. 109. Кривые течения для
ньютоновой жидкости (I) и струк-
турированной жидкости (II).
и
(Ю)
(9)
где Pd — отрезок, отсекаемый на
оси Р продолжением
прямой ВС.
Физический смысл величины
Pd очевиден из изложенного;
Pd — то добавочное давление, ко-
торое необходимо для разруше-
ния структуры. Таким образом,
объемная скорость v пропорцио-
нальна. тому действующему да-
влению Р—Pd, под которым течет
жидкость. Величина Pd, отнесен-
ная к единице поверхности, назы-
вается предельным динамическим напряжением
..сдвига (динамическим сопротивлением сдвигу):
я - / . р^г2 п г
- d S 2т.г1 d' 21
Статическое напряжение сдвига вычисляется таким же обра-
зом по давлению Ps, отвечающему началу текучести (точка А):
= <П)
Величина 6S характеризует усилие, необходимое для началь-
ного разрушения структуры (начало текучести), в то время как
величина 6^ представляет собой то усилие, которое затрачивает-
ся на разрушение структуры в текущей жидкости.
Системы с преобладающими пластическими свойствами ха-
рактеризуются условием 6d>6s в то время , как для упругих си-
стем с хрупким разрывом структуры типа I (рис. 105) характер-
но обратное соотношение, а именно: 68>6а, выражаемое крц
вой II (рис. 110).
263
Возвращаясь к рис. 109, отметим, что изменение характера
кривой выше точки С отвечает переходу ламинарного течения
в турбулентное. Этот переход наступает для структурированных
жидкостей при меньших значениях числа Рейнольдса по сравне-
нию с ньютоновыми жидкостями, поскольку наличие структуры
нарушает послойное скольжение.
Из графика v—Р можно найти коэффициент вязкости тр,
отвечающий практически разрушенной структуре. Дифференци-
руя (9), получаем:
Рис. 110. Кривые течения для
ньютоновой жидкости (I) и
упруго-пластичного тела (II).
Производную -^р находят по углу наклона касательной; ве-
личина r)=£-ctga, уменьшаясь с ростом давления, остается в
значительном интервале давлений постоянной (отрезок ВС).
Эта величина называется наи-
меньшей пластической вяз-
костью*. Величину, необходимую
для расчета k, находят из графи-
ка v—Р для истинно'вязкой жидко-
сти с известным значением т), на-
пример для воды. Практически из-
меряют обычно не v, а пропорцио-
1
нальную ей величину у , где г —вре-
мя истечения постоянного объема
V жидкости. В этом случае
= k'. d (1/0 (12)
где k' = -y.
Для установившегося течения
(отрезок ВС) величина структурной
вязкости может быть выражена в конечных разностях, находи-
мых графически
лр
= тд/о =*'ctga (13)
В большинстве случаев ограничиваются определением отно-
TJoth == - 7Д1
'отн Мо
сительнои вязкости
где т;Н2О — вязкость воды.
* Из этих же графиков по углу наклона касательной AD может быть
найдена величина наибольшей пластической вязкости си-
стемы с неразрушенной структурой:
264
Эту величину можно найти непосредственно из соотноше-
ния углов наклона прямолинейных отрезков кривых I (для
воды) и Иг
г, : - ctEa
Ч0ТН ЧЩО Ctg“0
Для вискозиметрических определений применяют капилляр-
ный вискозиметр Уббелоде (рис. 111); постепенное повышение
давления производится по-
средством баллона через
буферную склянку • или же
подъемом маностата на бло-
ке. Для регистрации начала
текучести (определение 6S)
удобнее всего применять
капиллярный пластометр,
т. е. горизонтальную градуи-
рованную капиллярную труб-
ку с краном (рис. 112). В
обоих' случаях необходимо
учитывать, что величины,
характеризующие образова-
ние и разрушение структу-
Рис. 111. Вискозиметр Уббелоде.
ры, в некоторых случаях зависят от диаметра капилляра, а сле-
довательно, значения 6 и тц оказываются инвариантными лишь
в определенной области.
Рис. 112, Установка для измерения упруго-пластических свойств
методом капиллярного пластометра.
На основании исследований Серб-Сербиной следует считать
целесообразным применение капилляров диаметром от 1,5 до
2,2 мм и проведение параллельных опытов в капиллярах
различного диаметра. ’
265
Для сильно структурированных, тиксотропных систем труд-
но установить инвариантную область; кроме того, в этих слу-
чаях может иметь место сплошное скольжение столба раствора,
как целого, вдоль поверхности стекла, искажающее результаты
измерений 6S. Поэтому такие гелеобразные системы (например,
цементные пасты) целесообразнее исследовать при помощи пер-
вой группы методов (смещение пластинки). Наоборот, для вы-
сокодисперсных пластичных систем с небольшими величинами
Рт и т] применяются методы капиллярной вискозиметрии.
Выполнение работы
Приготавливают серию коллоидных растворов (по указанию
преподавателя) в соответствии с теми факторами, влияние ко-
торых на коллоидно-химические свойства подлежит исследова-
нию. Так может изучаться влияние концентрации дисперсной
фазы, состава или концентрации добавленного электролита,
влияние pH и др., а также сравнение объектов (например, глин)
различной природы.
Растворы должны быть приготовлены за сутки до измере-
ний, если нет специальных указаний.
Предварительно определяют радиус капилляров в вискози-
метре (рис. 111) и в пластометре (рис. 112).
Для этого через отверстие 1 Дрис. 111) левого колена высу-
шенного вискозиметра вводят навеску ртути т в капилляр 2 и,
переводя прибор в горизонтальное положение, измеряют дли-
ну I в капилляре при Помощи металлической линейки. Измере-
ния повторяют 2—3 раза с разными навесками. Вычисляют
радиус капилляра г ___
v.r'Hd —т\ г= \/
V ~ld
где d — плотность ртути (13,6 г[см3).
Таким же образом находят г для капиллярного пластометра.
Далее устанавливают константу вискозиметра по воде. Через
отверстие 1 заполняют вискозиметр водой до нижней части рас-
ширений (при открытом кране). После этого, закрыв кран, за-
полняют водой левое колено выше метки 3. Создают давление
в системе и отмечают его по манометру. Открыв кран 5, вклю-
чают секундомер в момент прохождения мениском метки 3 и
определяют время протекания объема жидкости V (между мет-
ками 3 и 4) из левого колена в правое, выключив секундомер
при достижении метки 4. Производят измерения t при различ-
ных величинах давления Р.
Полученные данные наносят на график —Р, по которому,
эная вязкость воды (т) — 0,01 пз при 20°С), находят k' из урав-
нения (13),
266
Такие же измерения производят для серии растворов при
различных давлениях. Полученные данные также выражают
графически и вычисляют щ по формуле (13) и ба по (10). Зна-
чения вязкости выражают в пз, значения ба — обычно в динасы?.
Если значения Р измерены в см вод. ст., то:
Pd • 981. г!см сек2
где Pd — отрезок на оси Р, отсекаемый при экстраполяции пря-
молинейного участка.
Для определения 6s исследуемый раствор засасывают в пла-
стометр 1 (рис. 112), переводят его в горизонтальное положение
и соединяют с буферной склянкой 2.
Медленно и плавно повышая давление, отмечают по мано-
метру 3 высоту Ps, отвечающую .началу движения столбика
жидкости; опыт повторяют несколько раз. Вычисляют 6g по
формуле:
es = -r-Ps.98i
Следует отметить, что в тиксотропных системах все вели-
чины изменяются в зависимости от времени, прошедшего с мо-
мента их формирования. Поэтому все измерения должны про-
изводиться через одинаковые (или определенные) промежутки
времени после заполнения прибора.
Полученные данные выражают графически в виде зависимо-
сти б<2, б« и т]! от величины, вызывающей изменения системы
(например, от концентрации дисперсной фазы).
ФИЛЬТРАЦИОННЫЙ АНАЛИЗ
ч
Работа 45
ИССЛЕДОВАНИЕ СУСПЕНЗИИ МЕТОДОМ ФИЛЬТРАЦИОННОГО АНАЛИЗА
Сущность процесса фильтрации заключается в отделении
твердых частиц от жидкости при прохождении последней через
пористую среду, называемую фильтром.
Фильтрация является наиболее распространенным процес-
сом на предприятиях химической, горнорудной, нефтеобраба-
тывающей, пищевой и других отраслей промышленности.
Из обширной области применения' фильтрации в промыш-
ленности можно еще упомянуть производство соды, гипса, као-
лина, минеральных удобрений, лаков, красок и'искусственного
волокна.
Фильтрация воды при получении химически чистых продук-
тов, очистка воды на водопроводных станциях имеют громадное
19* 267
значение так же, как и фильтрация грунтовых почвенных вод
сквозь осадочные горные породы. '
Принцип метода фильтрационного анализа заключается в
том, что осадок твердой фазы, непрерывно образующейся при
фильтровании суспензии, со своей стороны действует как фильтр.
Это явление автофильтрации, связанное со степенью дис-
персности частиц, их гидрофильностью или гидрофобностью,
а также характером коагуляционной структуры осадка, может
быть использовано для качественно-количественной характери-
стики состояния суспензий, золей и структурированных коллоид-
ных систем.
Пользуясь методом фильтрационного анализа, можно изу-
чать такие явления, как коагуляция, пептизация, набухание,
структурообразование и ряд других, имеющих большое значе-
ние не только в коллоидной химии, но также и в технике и,
в частности, в химической промышленности, использующей
фильтрацию с какой-либо специальной целью.
Следует отметить также, что метод фильтрационного ана-
лиза, широко применяющийся в почвоведении, даёт возмож-
ность изучать диспергирующее действие различных катионов,
поглощенных почвой. Установлено, что изменение дисперсности
почвы связано с природой поглощенного катиона, в частности
со степенью его гидратации.
Поглощенные катионы по своей диспергирующей способно-
сти располагаются в лиотропный ряд, согласно уменьшению
их гидратации:
Na+ > К+ > КН4+ > Mg2+ > Са2+ > Ва2+ > Н+
Метод фильтрационного анализа дает возможность опреде-
лить состав поглощенных оснований в почве, а также солонце-
ватость почв.
Поскольку фильтрационный анализ основан на фильтрации
суспензии сквозь образующийся осадок, рассмотрим те законо-
мерности, которые наблюдаются в капиллярных системах, не
имеющих жесткого закрепленного скелета, а состоящих из от-
дельных соприкасающихся между собой частиц.
• Процесс фильтрации с образованием осадка осуществляется
обычно на промышленных фильтрах (такая фильтрация назы-
вается также фильтрацией через шламм).
Фильтрация дисперсионной среды, происходящая через уже
сформировавшийся осадок, называется стационарной
фильтрацией. Фильтрация дисперсионной среды из суспен-
зий при непрерывно накопляющемся осадке называется не-
стационарной фильтрацией.
Специфической особенностью фильтрации суспензий, отли-
чающей ее от фильтрации однородных жидкостей, является не
только изменение концентрации суспендированных частиц в
268.
жидкости при движении ее через фильтрующий слой, но также
изменение гидродинамических условий движения жидкости по
мере накопления вещества в объеме осадка.
Вопросы, связанные с изменением гидродинамических уело*
вий движения воды через осадок, являются предметом гидро-
динамики фильтрации суспензий.
Вопросы, связанные с эффектом изменения концентрации
суспензии при фильтрации, с влиянием различных компонентов
состава дисперсионной среды и свойств дисперсной фазы на
кинетику фильтрации и характер структурных изменений филь-'
трующего осадка, относятся к коллоидной химии.
Исследование структуры осадка, образующегося на фильтре
при различных условиях, представляет большой интерес. Одна-
ко дать количественную характеристику структуры такой капил-
лярном системы, состоящей
формы и различного разме-
ра, невозможно; поэтому
приходится подходить к ним,
исходя из ряда предположе-
ний, упрощающих реальную
структуру.
При гидродинамических
исследованиях фильтрации
через грунт исходят из
предположений, что все по-
ры грунта имеют цилиндри-
ческую форму и расположе-
обычно из частиц неправильной
Рис. 113. Схема основного элемента
модели фиктивного грунта.
ны параллельно друг другу или считают, что грунт состоит из
одинаковых .по размеру шарообразны^ частиц. Первый грунт
называют идеальным, второй — фиктивным.
Исходя из предположения о сферической форме частиц,
можно дать количественную характеристику структуры капил-
лярных систем, состоящих из отдельных соприкасающихся меж-
ду собой твердых частиц.
Слихтер путем ряда геометрических построений определил
пористость фиктивного грунта. Рассматривая систему из восьми
одинаковых шаров и предполагая симметричное расположение
остальных шаров, Слихтер пришел к заключению, что пори-
стость системы зависит от упаковки шаров. Им было показано,
что все возможные расположения шаров могут колебаться меж-
ду крайними конфигурациями: самой плотной и самой свобод-
ной упаковкой йаров. Так как все шары имеют одинаковый
диаметр, то расстояние между центрами двух любых соприка-
сающихся шаров равно сумме их радиусов, т. е. диаметру. Сле-
довательно, центры каждых восьми соприкасающихся шаров
расположены в вершине ромбоэдра, каждая грань которого
представляет собой ромб (рис. 113),
269
Как это видно на рис. 114, проекции сечения поры имеют
максимальную площадь, когда углы ромба становятся равны-
ми 90°, что соответствует самой свободной укладке шаров.
В этом случае ромбоэдр переходит в куб.
Когда же угол равен 60°, имеет место самая тесная упаковка
шаров (рис. 115). Каждый шар при тесной упаковке имеет
12 точек соприкосновения с соседними шарами и 6 — при сво-
бодной упаковке. Пористость такого фиктивного грунта W, т, е.
Рис. 114. Схема рас-
положения шаров при
наиболее свободной
укладке (.0 = 90°)-
Рис. 115. Схема распо-
ложения шаров при наи-
более плотной укладке
(0 = 60°).
отношение свободного пространства ко всему объему грунта,
определяется соотношением:
6(1 — cos 0) + 2 cos О
где 6 — угол ромба.
Таким образом, пористость фиктивного грунта, состоящего
из одинаковых шарообразных частиц, не зависит от их диамет-
ра, а зависит только от их относительной упаковки, обуслов-
ленной величиной угла 0.
Для двух рассматриваемых крайних случаев упаковки вели-
чина общей пористости лежит в пределах 0,4764 — для наибо-
лее свободной упаковки и 0,2595 — для наиболее тесной.
Геометрические соотношения в реальных капиллярных си-
стемах, как уже упоминалось выше, очень сложны. Вероятно,
в этом случае имеет место сочетание плотной и свободной упа-
ковки. Основываясь на предположении, что поры в системе
из сферических частиц имеют форму трубок с трехгранным
сечением, Слихтер вывел уравнение для фильтрации, подтвер-
дившееся экспериментально для фильтра, образованного шаро-
образными зернами приблизительно одинаковой величины.
Одним из важнейших факторов, влияющих на фильтрацию,
является давление, под которым жидкость проходит через
фильтр,
270
Й
Связь между Перепадом давления и скоростью фильтрации
при ламинарном течении жидкости была установлена Дарси.
Согласно закону Дарси, скорость фильтрации определяется со-
отношением:
V=K~h
где К—константа, называемая нроницаемостью фильтра;
h — толщина слоя; <-
ДР
—----градиент давления в слое.
Скорость фильтрации v измеряется количеством жидкости V,
протекающей в единицу времени через единицу площади
фильтра:
V
v~~t
При непрерывно нарастающей толщине фильтрующего сЛоЯ
и увеличении общего сопротивления потоку скорость фильтра-
ции может приниматься постоянной лишь для бесконечно ма-
лого промежутка времени и должна быть выражена в диффе-:
ренциальной форме:
Таким образом, объемная скорость фильтрации (фильтра-
ция через площадь А в единицу времени) равна:
dV 1 ДР
где т] — вязкость среды.
Согласно уравнению Дарси, скорость фильтрации при по-
стоянной толщине фильтрующего осадка пропорциональна по-
тере напора Р или при изменяющейся толщине осадка пропор-
ДР
циональна градиенту давления
В данном случае измерение фильтрации сводится к опреде-
лению коэффициента проницаемости К, связанного с пори-
стостью осадка.
При одной и той же пористости проницаемость будет тем
меньше, чем меньше размер частиц, из которых состоит осадок,
а следовательно, чем медленнее происходит фильтрация. На-
оборот, при одном и том же размере частиц, из которых состоит
фильтрующий слой, проницаемость зависит от пористости —
возрастая вместе с ней.
Так как для капиллярной системы, образованной отдельны-
ми частицами, эквивалентный диаметр капилляра нельзя не-
посредственно измерить и он не имеет реального физического
значения, предложен ряд уравнений, где средняя величина пор
271
выражается через пропорциональный ей размер самих частиц
или их удельную поверхность.
Уравнение Дарси обычно действительно только при неболь-
ших давлениях; при больших значениях давления скорость
фильтрации жидкости оказывается не пропорциональной дав-
лению.
Ребиндером предложена .следующая количественная харак-
теристика для процесса фильтрации суспензий. Для случая ста-
ционарной фильтрации суспензии, принимая структуру осадка
ДР .
неизменной и —— const, получаем:
dV 1 ДР0 .
—— = -----^ = Л0 = const
dt h0
где ДР0 — перепад гидростатического давления при стацио-
нарной фильтрации;
Ло—постоянная высота слоя осадка;
kQ — тангенс угла наклона прямой к оси t.
В системе координат t и V этот закон выражается прямыми,
проходящими через начало координат и различающимися тан-
Рис. 116. Зависимость объема филь-
трата (v) от времени (f) для. про-
цесса стационарной фильтрации.
генсами углов наклона k0, оп-
ределяющими скорость стацио-
нарной фильтрации (рис. 116).
В том случае, если происхо-
дит нестационарная фильтра-
ция дисперсионной среды из
суспензии с непрерывно накоп-
ляющимся осадком, скорость
фильтрации будет непрерывно
уменьшаться во времени, соот-
ветственно увеличению осадка.
В простейшем случае можно
допустить, что концентрация
дисперсной фазы с в суспен-
зии не меняется (с = const),
что плотность слоя осадка
постоянна и не зависит от его
высоты h; тогда высота слоя массы т определяется следующим
соотношением: h — ат, где а=-------коэффициент рых-
лости осадка.
Считая, что масса дисперсной фазы, входящая в осадок, про-
порциональна объему отфильтровавшейся жидкости, т. е. что
п = eV, скорость фильтрации выразится следующим образом:
dt acv А' 1J
(2)
272
или
V^KJ£-.LA
at ас т]
Вводим обозначение -J-Д тогда
ас ij
dV *
Г—= В; VdV=Bdt
dt-
(3)
Рис. 117. Зависимость lg v от lg t
для условий нестационарной филь-
При В = const путем интегрирования (3) от t = 0 до t на*
ходим:
V2
-^=Bt или V2 = 2Bt
откуда
V=V2Bt
Если
V2B^a
ТО
V=^at'!* (4)
Логарифмируя полученное уравнение для малых концентра-
ций, находим;
lg V=lge-}-0,51gf (5)
lg V линейно возрастает с ростом lg t, как это видно из пред-
ставленного графика ряда экспериментальных данных (рис. 117).
Наклон кривых для самых раз-
личных суспензий почти одина-
ков, и тангенс угла наклона бли-
зок к величине 0,5. *
Предложенная Ребиидером
схема- процесса для стационар-
ной и нестационарной фильтра-
'ции представляет несомненный
интерес при применении метода
фильтрационного анализа.
Как показали исследования,
проведенные в лаборатории кол-
лоидной химии ЛГУ, ряд раз-
личных факторов влияет на ско-
рость фильтрации в зависимости
от того, происходит ли фильтра-
ция в стационарных или неста-
ционарных условиях.
Экспериментальные данные ранних исследователей, приме-
нявших метод фильтрационного анализа, показали, что скорость
фильтрации в большой степени зависит от концентрации при-
бавляемого электролита и валентности входящих ионов, подчи-
няясь при прочих одинаковых условиях правилу Шульце-Гарди.
.273
Как по коагулирующему действию, так и по скорости фильт-
рации действителен ряд:
NaOH < Na2CO3 = Н2О < Na2SO4 = NaCl == КС1 < MgCl2 =
= CuSO4 = CuCl2 = BaCl2 < A12(SO4)3 < Th(NO3)4
Однако в вопросе, касающемся влияния электрокинетиче-
ского потенциала на фильтрационную способность капил-
лярных систем, до последнего времени не было достаточной
ясности.
Работами кафедры коллоидной химии ЛГУ установлено, что
влияние прибавляемого электролита с изменением значения
электрокинетического потенциала не сказывается на скорости
фильтрации при прохождении жидкости через капиллярную си-
стему, состоящую из относительно крупных частиц.
Этот факт объясняется тем, что структура подобной системы
не изменяется при прибавлении электролита вследствие отсут-
ствия процесса агрегации у крупных частиц при фильтровании
через осадок. Такие системы аналогичны жёстким закреплен-
ным системам.
В том случае, когда фильтрующий осадок сформирован в
воде и образован первичными частицами, а не агрегатами, из-
менение состава и концентрации раствора без механического
нарушения структуры осадка также не производит структурных
изменений осадка, и, следовательно, скорость стационарной
фильтрации не изменяется при прибавлении различных элек-
тролитов, несмотря на значительные изменения величины элек-
трокинетического потенциала.
В том случае, когда происходит процесс нестационарной
фильтрации тонкодисперсной суспензии с возможным измене-
нием структуры осадка вследствие происходящей агрегации
частиц, на скорость фильтрации влияет концентрация прибав-
ляемого электролита и его валентность (при соответствующем
изменении величины ^-потенциала).
. Структурообразование осадка можно представить следую-
щим образом: вначале идет агрегирование первичных частиц,
тем более беспорядочное, чем выше концентрация электролита,
что далее приводит к оседанию образующихся агрегатов.
Структурные свойства осадков-коагулятов несомненно зависят
от природы коагулируемых частиц, от степени их гидрофобности
или гидрофильности, а также от формы частиц.
Возрастание концентрации электролита приводит к увели-
чению агрегации частиц, вследствие чего осадок разрыхляется
и занимает больший объем. ’Чем меньше концентрация, тем
'больше независимость отдельных' частиц осадка, чем больше
плотность упаковки частиц, тем меньше тенденция к образова-
нию хлопьевидных, рыхлых осадков.
274 '
Вполне Понятно, Ито с увеличением объема осадка и степени
его рыхлости скорость фильтрации увеличивается. Поэтому при
исследовании процесса нестационарной фильтрации представ-
ляют несомненный интерес наблюдения за изменением объема
осадка, как величины, характерной для исследуемых дисперс-
ных систем и связанной со структурой осадка. Такие определе-
ния производят обычно в каком-нибудь градуированном сосуде.
На основании изменения величины объема осадка W можно
вычислить: 1) степень уплотнения осадка во времени t:
. _ Wk
“ Wt
где/j —степень уплотнения осадка;
IFK — конечный, неизменяемый объем осадка;
Wt — объем осадка через время t\
2) скорость изменения объема осадка во времени Ь:
значения b находят графически по tg угла, образуемого каса-
тельной к кривой оседания.
Рост V и W свидетельствует о развитии коагуляции.
Выполнение работы
Прибор для фильтрационного анализа (рис. 118) состоит из
бюретки 1, в верхний конец которой плотно вставляется воронка
Бюхнера 2 емкостью 75—100 см3, с отводной трубкой, соеди-
няющей бюретку с открытым водяным манометром 4 и мано-
статом 5. Маностат свободно перемещается вверх и вниз на
блоке, чем достигается точная и быстрая регулировка давления.
Бюретка, манометр и маностат посредством стеклянной трубки
соединяются с водоструйным насосом. Между прибором и на-
сосом помещается предохранительная буферная склянка 6, на
20 дм3. Между манометром и бюреткой помещается трехходо-
вой кран, чем достигается возможность соединения бюретки
попеременно с атмосферой или манометром.
Маностат 5 во время опыта лучше держать наполненным
до половины водой.
Употребляемый для фильтрационного анализа материал (пе-
сок, кварц, стекло), дробится на шаровой мельнице, после чего
для удаления железа, если это необходимо, например, при ис-
пользовании стальных шаров в мельнице, отмывается 10% сер-
ной кислотой и многократно промывается водой до удаления
SOl-. Промытый и высушенный порошок фракционируется с
275
помощью набора стандартных Сит до желаемой степени дисперс-
ности.
Для получения более мелких фракций часть, прошедшая
сквозь самое мелкое сито (350 меш=50 мк), отмучивается в
воде.
При приготовлении суспензии определенной концентрации
перемешивание порошка с водой лучше производить не со всем
количеством воды, а сначала только с частью (20—30%) с по-
следующим растиранием в ступке до образования однородного
густого теста. Затем приливают остальную порцию воды, взбал-
Рис. 118. Установка для фильтрационного анализа.
тывая полученную суспензию в течение 10—20 мин. Таким обра^
зом получают более однородную и стабильную суспензию.
Во всех сравниваемых опытах должны быть одинаковы:
1) объем фильтрующейся суспензии (50 см3);
2) концентрация суспензии в зависимости от исследуемого
порошка (10—25%);
3) давление (20—50 см вод. ст.);
4) сорт и число слоев фильтровальной бумаги, помещаемой
в качестве подложки в воронку Бюхнера.
Опыт по фильтрационному анализу производят следующим
образом. При помощи трехходового крана выключают бюрет-
ку 1, пускают в ход водоструйный насос и создают необходимое
разрежение поднятием или опусканием баллона маностата 5.
Фильтруемую суспензию быстро наливают на бумажный фильтр
воронки 2, предварительно смоченный исследуемым раствором,
и в течение определенного времени (обычно 1—2 мин) остав-
276
ляют систему в покое. Осевшая суспензия образует слой,
через- который идет фильтрование. Затем с помощью трехходо-
вого крана 3 соединяют бюретку с манометром и, пуская в ход
секундомер, производят отсчеты объема фильтрата через опре-
деленные промежутки времени. Объем фильтрующейся жид-
кости первые 10—15 мин отмечают через каждую минуту, а
затем в течение 1 или 2 ч (в зависимости от условий) сначала
через каждые 5 мин, а потом через 10 мин. Кроме того, следует
отметить время, в течение которого отфильтровывается 25 см3
жидкости, а также продолжительность всего про-
цесса фильтрации и общий объем полученного
фильтрата.
Все полученные значения записывают в таб-
лицу результатов исследования. .
Строят график зависимости объема отфиль-
тровавшейся жидкости V от времени t. Получен-
ные данные представляют в логарифмическом
виде, откладывая по оси абсцисс 1g /, по оси ор-
динат — lg V. Находят tg угла наклона получен-
ных прямых.
Определение изменений объема осадка про-
изводят Вг сосуде с градуированной нижней
частью (рис. 119). Исследуемую суспензию на-
ливают в сосуд и после интенсивного взбалтыва-
ния отмечают объем осадка Wt, время t и конеч-
ный неизменяемый объем осадка WK.
Далее вычисляют степень уплотнения осад-
ка t, исходя из соотношения:
Рис. 119. Сосуд
для измерения
объема осадка.
В заключение находят скорость изменения' объема осадка
во времени. Скорость изменения объема находят графически,
откладывая по оси ординат Wt, а по оси абсцисс — t. Проводят
касательные к кривой оседания Wt=f (t). Тангенсы углов, со-
ставленные касательными, равны скорости изменения объема
осадка
В качестве объектов исследования можно рекомендовать
глину, кварц, песок, стекло.
По указанию преподавателя делают одну из следующих
задач:
1. Определение влияния электролитов на скорость фильтра-
ции и объем осадка. Для этой цели пользуются растворами
электролитов различной концентрации: КС1, ВаСЬ, А1С13,
NaOH. Для сравнения берут чистую воду.
277
2. Определение влияния степени дисперсности порошка на
скорость фильтрации. В зависимости от характера исследуемого
порошка пользуются несколькими фракциями, например:
Фракция < 3 мк Фракция 10—25 мк
» 3—10 мк » 40—80 мк
Сравнивают скорость фильтрации исследуемого порошка раз-
личных фракций в чистой воде и в растворе 0,01 н. КС1, а также
соответствующие изменения объема осадка во времени.
Полученные данные записывают в таблицы (табл. 1 и 2):
Таблица 1
Концентрация
суспензии,
%
Давление,
см
вод. ст.
Время,
мин
V, см9
dV
dt
Ig V
Ig t
lg a
ЛИТЕРАТУРА
1. Антипов-Каратаев И. H., Применение фильтрационного метода
к изучению факторов дисперсности почв. Труды лаборатории ин-та
агропочвоведения, вып. II (1930).
2. Бейлер С. Я., Ребиндер П. А., ДАН СССР, 49, № 5 41945).
3. Л е й б е н з о н Л. С., Движение природных жидкостей и газов в пори-
стой среде, Гостехиздат, 1947.
4. Михайлов Н. В., Ребиндер П. А., Колл, ж., 17, № 2, 107 (1955).
5. Путилова И. Н., Руководство к практическим занятиям по коллоид-
ной химии, Изд. Высшей школы, 1961.
6. Ребиндер П. А. Конспект общего курса коллоидной химии, Изд.
МГУ, 1949.
7. Ребиндер П. А. Физико-химическая механика, сер. IV, № 39, 40, Изд.
«Знание», 1958.
8. Р е б и н д е р П. А., Сб. «Новые методы физико-химических исследова-
ний поверхностных явлений», Труды Ин-та физ. химии, 1, 5, 1950.
9. Сега лова Е. Е., Ребиндер П. А., Колл, ж., 10, № 3, 228 (1948).
10. Се р б - С е р б и н а Н. Н., Ребиндер П. А., Колл, ж., 9, № 5, 381
’ (1947).
11. Совещание по вязкости жидкостей и коллоидных растворов, Изд. АН
СССР, 1941.
12. Ч е р н е н к о Л. Е., Р е б и н д е р П. А., Колл, ж., 12, № 5, 386, (1950)'.
ГЛАВА VI
СВОЙСТВА ВЫСОКОМОЛЕКУЛЯРНЫХ СОЕДИНЕНИЙ
И ИХ РАСТВОРОВ
К высокомолекулярным соединениям относятся вещества, ко-
торые включают в состав своих молекул сотни и тысячи отдель-
ных атомов, связанных друг с другом силами главных валент-
ностей. Такие огромные молекулы часто называют макро-
молекулами. Кроме большого количества различных
органических соединений, составляющих подавляющее боль-
шинство веществ этой группы, к высокомолекулярным, или вы-
сокополимерным соединениям относятся и некоторые неоргани-
ческие вещества.
Большой молекулярный вес высокополимеров обусловливает
ряд свойств, которые отсутствуют у низкомолекулярных соеди-
нений: растворением высокополимерных соединений в специфи-
ческих растворителях можно получить высоковязк^е рас-
творы, а в твердом состоянии многие из них характеризуются
высокой прочностью и эластичностью, чем и обусловливается
широкое и все возрастающее применение этих материалов в
технике.
С возрастанием молекулярного веса веществ, в пределах
определенного класса органических соединений, повышается
вязкость их растворов, увеличивается прочность и эластичность
нитей и пленок, повышается температура плавления, умень-
шается растворимость. Поэтому молекулярный вес высокополи-
мера является одной из важнейших характеристик и для его
определения применяется большое количество различных ме-
тодов.
Следует иметь в виду, что природные и синтетические высо-
кополимерные соединения представляют собой смесь молекул
различной величины, т. е. являются полидисперсными
веществами, которые обычно характеризуются средними вели-
чинами молекулярных весов и для получения более полных све-
279
дений 6 распределении молекул по величине приходится прибе-
гать к разделению исходного полимера на отдельные фракции.
Применение физико-химических методов определения моле*
кулярных весов связано с использованием растворов высоко-
молекулярных соединений. Свойства получающегося раствора
высокомолекулярного соединения зависят от химической при-
роды растворителя и высокополимера. В специфических раство-
\ рйтелях образуются растворы, термодинамически устойчивые,
имеющие свойства истинных растворов. Это обусловливает воз*
можиость применения методов, основанных на молекулярно-
кинетических свойствах растворов высокополимёрных соедине-
ний, ,поскольку число частиц в единице объема, несмотря нй
относительную крупность кинетической единицы по сравнению
с обычными низкомолекулярными соединениями оказывается
достаточно большим для количественного учета измеряемых в
опыте величин.
Наиболее распространенными методами определения моле-
кулярных весов являются методы, основанные на измерении
осмотического давления и вязкости растворов высокополимеров
(включенные в данное руководство).
Для высокомолекулярных соединений характерным является
самопроизвольное проникновение растворителя в сухой поли-
мер, сопровождающееся часто значительным увеличением его
объема с образованием студня. Этот процесс, предшествующий
растворению высокополимера и растворителя, называется на-
буханием. В настоящее время процесс набухания рассматри-
вается как процесс, аналогичный смешению жидкостей, проте-
кающий односторонне вследствие значительной разницы в вели-
чине молекул высокополимера и растворителя.
В начальной стадии набухания полимеров в большинстве
случаев происходит выделение тепла, которое может быть зна-
чительным. Большой тепловой эффект свидетельствует о силь-
ном энергетическом взаимодействии между молекулами поли-
. Мера и низкомолекулярной жидкости, которое приводит к со-
стоянию большей упорядоченности в расположении молекул
системы в. целом и, следовательно, к уменьшению энтропии.
Дальнейшее поглощение жидкости происходит без заметного
теплового эффекта или связано с поглощением тепла при возра-
стании энтропии. Возрастание энтропии в процессе смешения
разнородных молекул при дальнейшем переходе в раствор свя-
зано с увеличением термодинамической вероятности, т. е. увели-
чением числа. способов распределения гибких макромолекул
полимера в растворе при разбавлении.
Количественная оценка процесса набухания, помимо теоре-
тического значения, является весьма важной для оценки воз-
можности применения высокополимерных материалов в техни-
ческих целях.
28Q
Работа 46
определение молекулярного веса высокомолекулярных соединении
МЕТОДОМ осмотического давления
Одним из основных и распространенных методов определе-
ния молекулярного веса высокомолекулярных соединений яв-
ляется метод определения осмотического давления разбавлен-
ных растворов.
- При растворении высокомолекулярных соединений в соот-
ветствующих растворителях, в которых они самопроизвольно
растворяются и обратимо осаждаются, получаются растворы
устойчивые в своих свойствах во времени и не отличающиеся
во многих отношениях от истинных растворов низкомолекуляр-
ных веществ. Подобного рода растворы высокомолекулярных
соединений устойчивы, имеют значительно большую величину
осмотического давления, чем суспензоидные золй, и могут быть
хорошо очищены от низкомолекулярных примесей. Для таких
растворов высокомолекулярных соединений осмотический ме-
тод определения молекулярного веса получил большое распро-
странение.
В тех случаях, когда высокомолекулярные соединения яв-
ляются электролитами, например белки, следует учитывать рав-
новесие Доннана и влияние pH среды.
Осмометрия имеет преимущества перед другими методами,
например эбулиоскопией и криоскопией, являясь более точным
методом в. тех случаях, когда приходится определить молеку-
лярный вес полимеров, превышающий 20 000. Осмотическим ме-
тодом можно определять молекулярные веса, лежащие в пре-
делах 25000—1500000.
Нижний определяемый предел молекулярных весов обуслов-
лен возможностью изготовления пригодных полупроницаемых
мембран. Верхний предел зависит от точности определения
высоты поднятия столба раствора.
Величина осмотического давления в разбавленных растворах
неэлектролитов определяется уравнением Вант-Гоффа:
п V = п • RT (1)
где it — осмотическое давление;
V—объем раствора;
п — число Молей вещества в единице объема;
R — газовая постоянная;
Т — абсолютная температура.
г- ' CV
Если заменить п через -др где с — концентрация, выражен-
ная в г/см3 раствора, а М — молекулярный вес,
-281
то уравнение (1) принимает следующий вид:
— —RTc
с ~ М * Л ~ М
(2)
• Следует указать, что даже в области разбавленных раство-
ров могут иметь место отклонения от уравнения Вант-Гоффа,
которые для низкомолекулярных соединений, а также для изо*
диаметрических молекул незначительны, однако для линейных
и разветвленных полимеров становятся заметными.
Тогда, согласно Галлеру, можно воспользоваться следую-
щим уравнением:
RT 10 2 /-П
" = "лГ с +Вс
где В — коэффициент, учитывающий взаимодействие, между мо-
лекулами. Разделив обе части уравнения (3) на с, получаем
следующее соотношение, отвечающее уравнению прямой линии:
Измерив величину осмотического давления для различных
концентраций, строят график в осях — и с (рис. 120), полу-
чают прямую линию и, экстраполируя на нулевое значение кон-
Рис. 120.. График к опреде-
.лению молекулярного веса
осмотическим методом.
центрации с, получают на оси ординат
RT
отрезок, равный
Осмотическое давление можно оп-
ределять двумя основными методами:
статическим и динамическим. В этих
методах используются полупроницае-
мые мембраны, качество которых име-
ет большое значение для правиль-
ного определения молекулярного ве-
са. Выбор наиболее идеальной полу-
проницаемой мембраны, т. е. мембра-
ны, хорошо пропускающей раствори-
тель и не пропускающей растворенный
полимер, должен проводиться экспериментально.
При выборе мембран, в основном, следует руководствоваться
следующими принципами:
1) мембрана не должна набухать в тех растворителях, в ко-
торых определяют осмотическое давление;
2) мембрана должна обеспечивать достаточно большую
скорость прохождения растворителя и быть достаточно мелко-
пористой .для того, чтобы не пропускать макромолекулы раство-
ренного полимера;
282
3) мембрана не должна быть хрупкой и должна выдержи-
вать значительное давление и сравнительно высокую темпера-
туру.
Механизм действия полупроницаемых мембран недостаточно
выяснен. В какой-то степени мембрана действует подобно так
называемому молекулярному ситу, поэтому
бран следует характеризовать ее со сто-
роны ее структуры, определив средний
радиус пор мембраны (см. работу 10).
Проницаемость мембраны в отношении
растворенного вещества лучше определить
ультрафильтрацией раствора через ис-
пытуемую мембрану в приборе Зейца (или
другом).
Чаще всего употребляются мембраны
из ацетилцеллюлозы, целлофана разных
марок, из целлюлозы, нитроцеллюлозы, де-
нитрированного коллодия, пористого стекла,
ферроцианида меди, осажденного на колло-
диевой мембране.
Статический метод определения
осмотического давления основан на том, что
осмотическое давление раствора уравнове-
шивается давлением столба жидкости, воз-
никающем в результате проникновения
растворителя в раствор.
Преимущества статического метода за-
ключаются в простоте аппаратуры. Кроме
того, благодаря тому, что равновесие уста-
при подборе мем-
Рис. 121. Осмометр
Шульце.
на результаты из-s
навливается длительное время, оставшиеся
в растворе низкомолекулярные примеси,
свободно распределяясь по обе стороны
мембраны, не оказывают заметного влияния
мерений.
С другой стороны, длительность достижения равновесного
состояния, продолжающегося иногда в течение нескольких не'
дель, может исказить результаты благодаря адсорбции раство-
ренного вещества мембраной, а также возможности изменения
некоторых веществ во времени (гидролиз).
Наиболее прост и имеет широкое распространение для ста'
тического определения осмотического давления осмометр, пред-
ложенный Шульце, ,(рис. 121).
Осмометр состоит из камеры 2 емкостью ~ 10 см3 из стекла
или хромированной латуни. Камера присоединяется с по-
мощью винтов к пластинке, в середине которой имеются отвер-
стия (диаметром 1 мм). Нижняя сторона камеры плотно прижи-
мает мембрану к пластинке — сетке, толщина которой должна
283
быть не больше 0,5 мм. Часть осмометра, в которой находится
раствор, называется осмотической ячейкой. Раствор наливается
в, ячейку через верхнее отверстие /, куда для отсчета величины
давления вставляется пришлифованный градуированный капил-
ляр диаметром 1 мм и длиной 50 см. Нижняя пришлифованная
часть капилляра входит в камеру на 0,5—1 мм для предохране-
ния камеры от пузырьков воздуха при заполнении.
Камера с раствором вставляется в сосуд 3, наполненный чи-
стым растворителем. Сосуд закрывается .пришлифованной
крышкой во избежание испарения растворителя. При измере-
нии осмотического давления осмометр помещают в термостат.
Динамический метод, предложенный Ван-Кампеном,
основан на том, что осмотическое давление компенсируется на-
ложенным на раствор переменным противодавлением. Величина
осмотического давления вы-
Рис. 122. Компенсационный осмометр
Думанского.
числяется на основании из-
мерения скорости проникно-
вения растворителя через
мембрану. Преимущество
динамического метода за-
ключается в быстроте изме-
рений. Кроме того, ввиду
малого времени соприкосно-
вения раствора с мембра-
ной, можно не учитывать
адсорбцию растворенного
вещества мембраной.
Следует указать, что
этот метод требует очень
точного термостатирования,
а также тщательной очистки раствора от низкомолекулярных
примесей, так как вследствие быстроты проводимых измерений
низкомолекулярные примеси не успевают равномерно распре-
делиться по обе стороны мембраны.
На практике употребляют осмометры самых разнообразных»
конструкций. В настоящее время предложено ~50 различных
типов осмометров. • »
При выборе типа осмометра следует учитывать следующее:
1) объем осмотической ячейки должен быть возможно ма-
лым, так как количество раствора и растворителя должно быть
небольшим;
2) необходимо тщательное термостатирование прибора при
проведении опытов;
3) измерения должны происходить быстро;
4) . необходимо следить за тем, чтобы не происходило утечки
растворителя через краны и не попадали пузырьки воздуха в ос-
мометр;
284
5) полупроницаемая мембрана должна быть хорошо Закреп-
лена на осмометре, чтобы при измерении она не прогибалась.
Простым и довольно распространенным осмометром является
осмометр, предложенный Думанским, позволяющий быстро про-
водить измерения, не дожидаясь установления равновесия в си-
стеме.
На рис. 122 представлен компенсационный осмометр Думан-
ского.
Осмотическая ячейка 6 соединяется с мембраной 7, аспира-
тором 1 и манометром 3. Ячейку и внешний сосуд помещают в
термостат 5. Поднятием верхней части аспиратора регулируется
внешнее давление таким образом, чтобы оно было больше или
меньше осмотического давления.
В первом случае мениск в капилляре 4 опускается вниз со
скоростью Vi, пропорциональной-избыточному давлению Р\—it.
Во втором случае, мениск поднимается со скоростью У2, про-,
порциональной разности it—Р2, тогда отношение скоростей пере-
мещения растворителя по капилляру Vi/V2, будет равно отно-
шению разностей давлений, обусловливающих поднятие или
опускание мениска в капилляре, т. е.:
К. А-*
у2 л—Рг
и отсюда
VM+Vf,
V. +
Выполнение работы
Динамический метод Ван-Кампена можно несколько видо-
изменить, применяя осмометр типа Эршлера, изображенный на
рис. 123.
Осмометр Эршлера состоит из стакана 1 емкостью ~ 1 дм3,
который заполняется растворителем. В стакан вставляется ци-
линдрический сосуд 2 емкостью 40—50 см3. В нижней своей
части этот сосуд имеет фЛанец 5 для укрепления мембраны. Со
Стороны растворителя под мембрану подкладывается сетка с от-
верстиями для предотвращения прогиба мембраны.
В верхней суженной части цилиндрического сосуда имеется
так называемое горло 3, шлифованное внутри, в которое встав-
ляется полая внутри и открытая снизу пробка 4, переходящая
в стеклянную трубку с двумя боковыми отростками. Отростки
трубки снабжены пришлифованными кранами. Горизонтальный
отросток с краном 6, отросток, направленный вверх, 7.
Над краном 7 расположена стеклянная воронка 9, закры-
вающаяся пришлифованной пробкой, через которую наполняют
ячейку исследуемым раствором.
285
Четвёртый отросток переходит в калибров энную капилляр-
ную пипетку 10, с помощью которой измеряют объем раствори-
теля, проходящего через мембрану при различных давлениях.
Пипетка 10 заканчивается трехходовым краном 11, при по-
мощи которого осмотическая ячейка может быть соединена с ат-
мосферным и заданным давлением.
Постоянство давления можно регулировать ртутным регуля-
тором давления и маностатом. Величина давления регистри-
руется ртутным манометром и может быть изменена поднятием
и опусканием трубки регулятора давления, погружаемой на оп-
ределенную глубину в ртуть.
В этом случае кран 11 поворачивается таким образом, чтобы
осмотическая ячейка была отсоединена от манометра.
Рис. 123. Осмометр Эршлера.
Вода из водопроводного крана, поступающая в регулятор
давления и в маностат, сжимает-воздух и создает давление, со-
ответствующее глубине погруженной в ртуть трубки регулятора
давления.
. При равенстве напора воды и создаваемого давления вода
перестает поступать в маностат. Избыток воды сливается через
боковой отросток регулятора давления, после чего водопровод-
ный кран прикрывается до минимума. Положение мениска ре-
гулируется кранами 7 и 8.
После заполнения ячейки, помещенной в термостат, измере-
ния производят через некоторое время, для того чтобы система
пришла в равновесие.
Заполнение ячейки производится следующим образом: ячей-
ка 2 заполняется исследуемым раствором, раствор через горло 3
засасывается в трубку 4. Краны 7 и 8 должны быть при этом
закрыты. После заполнения трубки 4 кран 7 открывается,
286
Рис. 124. График скорости про-
текания растворителя через
мембрану.
кран 6 закрывается для заполнения раствором капиллярной
части трубки, на которой нанесены деления для отсчета.
В начале опыта внешний сосуд (стакан) и осмотическая
ячейка заполняются дистиллированной водой или другим рас-
творителем. После этого задаются различные величины давле-
ния (4—5 значений) и с помощью секундомера отмечается время
прохождения определенного количества жидкости (например,
0,2 см3) через мембрану по отсчетному капилляру. Мениск жид-
кости в капилляре осмометра лучше рассматривать с помощью
/ наведенного микроскопа или катетометра. Измерения произво-
дятся для каждого заданного давления не менее 4-х раз и для
расчета скорости протекания берет-
ся среднее значение из всех измере-
ний. На основании полученных дан-
ных вычерчивается кривая скорости
протекания растворителя через мем-
брану, кривая CD (рис. 124).
Обычно, вследствие сопротивле-
ния, возникающего при протекании
жидкости через мембрану и капил-
лярных сил в отсчетном капилляре,
кривая CD не проходит через нуль.
Это, учитывается далее в опытах с
раствором исследуемого вещества.
Затем осмотическая ячейка осво-
бождается от растворителя, пропо-
ласкивается исследуемым растворо:
твором. Накладывая на раствор последовательно разные да-
вления, вычерчивают кривую скорости прохождения раствора
через мембрану при разных противодавлениях.
Когда разность между уровнями раствора и растворителя
равна осмотическому давлению или на раствор накладывается
давление водяного столба, равное осмотическому давлению рас-
твора, то скорость проникновения растворителя через полупро-
ницаемую мембрану равна нулю. Измерив скорость прохожде-
ния растворителя при нескольких значениях давления водяного
столба, можно измерить осмотическое давление экстраполяцией
на нулевую скорость. (Метод нулевой скорости). (Рис. 124, пря-
мая АВ).
Из полученного экстраполяцией значения осмотического да-
вления следует вычесть величину отрезка, полученного для опы-
тов с чистым растворителем.
Для определения молекулярного веса осмотическим путем
выбор мембраны имеет большое значение. Трудно получить
мембрану, способную пропускать молекулы растворителя и за-
держивать молекулы исследуемого вещества с молекулярным
весом ниже 20000.
287
и заполняется этим
*В качестве полупроницаемых мембран используют гидрат-
целлюлозную пленку, получаемую омылением нитроцеллюлоз-
ной пленки, мембрану из целлофана, мембраны из синтетиче-
ских полимеров и другие. Полупроницаемая мембрана не долж-
на пропускать молекул исследуемого вещества, не должна взаи-
модействовать химически с растворителем, не должна раство-
ряться в растворителе и должна пропускать растворитель
с большой скоростью, для того чтобы осмотическое равновесие
наступало в короткое время.
Сделав ряд определений для нескольких концентраций, нахо-
дят графически величину и определяют молекулярный вес
по уравнению Вант-Гоффа (2).
Для приобретения навыков в работе с осмометром, можно
предварительно провести измерение осмотического давления
(рассчитав в дальнейшем молекулярный вес) растворов саха-
розы или растворов конго красного.
Теоретическое значение молекулярного веса сахарозы рав-
но 342.
Теоретический молекулярный вес конго красного равен 696.
Анализ на сахар в растворителе (воде) производится с по- i
мощью цветной реакции с а-нафтолом. Для этой цели 2—3 см3
исследуемого на сахар раствора наливают в пробирку. Сверху
осторожно приливают 2—3 см3 концентрированной H2SO4- и
1—2 капли а-нафтола. Малейшие следы сахара показывают фио-
летовое кольцо.
Для определения осмотического давления сахарозы следует
пользоваться мембранрй, имеющей средний радиус пор ~ 1 ммк.
Работа 47
ОПРЕДЕЛЕНИЕ МОЛЕКУЛЯРНОГО ВЕСА ВЫСОКОМОЛЕКУЛЯРНЫХ СОЕДИНЕНИИ
ПО ВЯЗКОСТИ ИХ РАСТВОРОВ
Растворы высокомолекулярных соединений характеризуются
весьма высокой вязкостью даже при малых концентрациях-рас-
творенного вещества. К ним неприменимо уравнение Эйнштейна
(стр. 251), выведенное для частиц сферической формы, а именно:
V
Иуд =U k (1)
И О
где.т]уД — удельная вязкость, т. е. относительное прира-
щение вязкости растворителя цо при введении в него полимера:
г Чуд=2Н^='^“1==1101«~1
288
Называется относительной вяз-
вязкостей).
для растворов полимеров величина
не только от концентрацию раство-
и от молекулярного веса М. При
(3)
Рис. 125. Кривые зависимости
приведенной вязкости от кон- -
центрации раствора полимера.
Величина г]Отн
костью (или отношением
Было установлено, что
удельной вязкости зависит
Vx
ренного вещества -у, но
одинаковой весовой (или объемной) концентрации величина т]уд
тем больше, чем больше М. Штаудингер на основании большого
экспериментального материала вывел формулу:
•п =~1(Мс
'уд > «
где с — концентрация полимера;
Км — константа, которую Штаудингер считал постоянной для
данного полимергомологического ряда и не 'зависящей
от природы растворителя.
Однако'позднейшие исследования не подтвердили этой про- .
стой зависимости; было показано, что уравнение (3) может быть
получено теоретически как предель-
ный случай при следующих усло-
виях: а) отсутствие взаимодействия
между макромолекулами; б) пре-
дельное выпрямление цепи макро-
молекулы в растворе.
Первое из этих условий может
быть осуществлено при очень малых
с. Поэтому применяется экстраполя-
Чуд
ция величины-у, называемой при-
веденной вязкостью (а так-
же числом вязкости) к нуле-
вой концентрации. Согласно урав-
Чуд
нению (3) величина -у должна
быть постоянной, однако практиче-
ски вследствие увеличения взаимо-
действия между макромолекулами с ростом с приведенная вяз-
кость увеличивается приблизительно линейно (рис. 125).
Графическая экстраполяция величины -у до нулевой кон-
центрации дает величину:
Эта величина называется характеристической вяз-
костью (а также предельным числом вязкости) и
обозначается буквой т] в квадратных скобках. Благодаря эк-
страполяции величина [т]] не включает взаимодействия между
289.
Вчуд
тех случаях, когда зависимость —-------с
оказывается не линейной, целесообразно на том же графике от-
ln rjrl0 ~
ложить зависимость —от с. Разлагая эту функцию в ряд
7]уд 1П TjTfl
по степеням ——, получим —-—
с2
X -5- — • • •, откуда видно, что
О
^УД I ^уд \2 С , I Чуд \3
с ~\~с~/
lim
с->0
Этот прием облегчает экстраполяцию, т. к. функция
меньше изменяется с концентрацией; кроме того, обе кривые
должны пересекаться на оси ординат (рис. 125).
Таким образом, введение величины [rj] учитывает первое из
указанных выше условий применимости уравнения Штаудин-
гера. Второе условие, а именно предельное выпрямление макро-
молекулы, вообще не реализуется, поэтому необходимо введение
поправки, учитывающей конфигурацию молекулы. Вытянутые
цепи оказывают гидродинамическое сопротивление течению
жидкости, молекулы которой, огибая цепи, вынуждены замед-
лять движение. При полном выпрямлении цепи сопротивление
ее было бы тем больше, чем длиннее цепь; отсюда понятна пря-
мая пропорциональность между т] и М в уравнении (3). Однако
в действительности цепные макромолекулы в растворе свернуты
в той или иной степени в клубки и оказывают меньшее сопро-
тивление потоку. Если М- и длйна цепи возрастает, например в
2 раза, очевидно, что сопротивление увеличится меньше, чем
вдвое. Статистический расчет для совокупности макромолекул,
каждая из которых обладает полной свободой вращения звеньев
и свернута в беспорядочный клубок, дает следующее выра-
жение:
Однако этот расчет не учитывает взаимодействия макромо-
лекулы с молекулами растворителя. Это. взаимодействие приво-
дит к ограничению свободы вращения звеньев и к растягива-
нию цепи, т. е. к увеличению среднего расстояния между кон-
цами цепи. Если это увеличение выразить при помощи пара-
^•2 _—.
метра «2 = —, где г2 — средний квадрат расстояния между
'о „
концами цепи при взаимодействии, — то же без взаимодей-
ствия, то теория, развитая Флори и Фоксом, дает следующее
выражение:
h ] =
290
Для плохих растворителей (почти не взаимодействующих с
полимером) а « 1, для хороших — а приблизительно пропорцио-
нален М0’1, следовательно frj] = КМ^.
Таким образом, влияние конфигурации макромолекулы в
растворе может быть выражено посредством возведения вели-
чины молекулярного веса в степень а, постоянную для данной
системы полимер — растворитель и лежащую в пределах
0,5—0,8 в зависимости от взаимодействия макромолекул с рас-
творителем:
[>;] = КМа (4)
Это уравнение Марка — Хувинка применяется в настоящее
время для определения молекулярного веса высокополимеров.
Для нахождения констант К и а были измерены величины^],
а также молекулярные веса М (независимым методом) для не-
скольких низших членов гомологического ряда в данном рас-
творителе. На основании этих данных были найдены значения
К и а графически путем применения логарифмических коор-
динат
lg N = Al (5)
В этих координатах отрезок на оси ординат дает 1g К,
a tg угла наклона — величину а. Найденные таким путем значе-
ния К и а для ряда систем полимер — растворитель приводятся
в литературе в виде таблиц. В табл. 1 приводятся данные для
некоторых систем.
Пользуясь данными этой таблицы, можно вычислить. моле-
кулярный вес полимера М по уравнению (5), измерив вязкость
при разных концентрациях полимера.
Таблица I
Полимер Растворитель л °C K-W- а Область применения м-ю-6
Целлюлоза Медно-аммиач- 25 0,85 0,81 —
Ацетилцеллюлоза ный раствор Ацетон 25 1,49 0,82 0,3—3,9
Полиизобутилен Толуол — 2,6 0,64 —
» Диизобутилен 20 3,6 0,64 0,05—13
Полиизопрен (НК) Толуол -— 5,03 0,67 0,4—15
» Бензол 20 4,9 0,66 —
Дивинилстирольный Толуол 30 5,4 0,66 0,25—9,2
каучук Полистирол » 30 . 3,7 0,62 2—18
Полиметилметакрилат Хлороформ -— 60 0,79 —
» Бензол 25 0,94 0,76 0,5—9,8
Поливинилацетат Ацетон 50 2,8 0,67 0,8—8,5
Поливиниловый спирт Вода 50 5,9 0,67 0,4—1,1
Найлон Муравьиная ки- — 11,0 0,72 0,05—0,25
Диметилсилоксан слота Толуол — 2 0,66 0,002—0,12
291
0
6
5
Рис. 126. Вискози-
метр с переменной
высотой столба
жидкости.
Измерение вязкости осложнено тем, что растворы некото-
рых полимеров не являются ньютоновскими жидкостями, т. е.
для них величина т] не является постоянной, а уменьшается
с ростом градиента скорости течения раствора в капилляре. При
значительных концентрациях это изменение обусловлено нали-
чием структуры, образованной взаимодействием макромолекул
между собой (см. работу 44).
В области малых концентраций, недостаточных для струк-
турообразования, эта зависимость объясняется тем, что длинно-
цепочечные молекулы деформируются и
ориентируются вдоль потока, оказывая
меньшее сопротивление течению жидкости,
а следовательно т] становится меньше. По-
скольку изложенная выше теория исходит
из наиболее вероятных конфигураций мак-
ромолекул в растворе без учета их дефор-
мации в потоке, естественно, что наимень-
шие отклонения от теории будут при низ-
ких градиентах скорости. Поэтому обычно
измеряют величины (т]—т]о)/т|о при различ-
ных градиентах, скорости, а затем экстра-
полируют к нулевому градиенту. Эти изме-
рения можно проводить в вискозиметре при
разных давлениях (см. работу 44) или же
применить специальный вискозиметр, изо-
браженный на' рис. 126, в котором изме-
нение градиента скорости достигается
установлением различной высоты столба
жидкости. Измеряя последовательно время
протекания жидкости между метками 5 и 4,
4 и 3, 3 и 2, 2 и 1 под действием силы тяже-
сти, можно в одном опыте получить серию
данных, отвечающих различным градиентам скорЬсти.
. В точных работах по измерению вязкости жидкостей необ-
ходимо учитывать, что в уравнение Пуазейля:
РпгЧ
где Р— перепад давления;'
г — радиус капилляра;
/ — его длина;
V—объем;
t — время; нужно ввести две существенные поправки.
Одна из них обусловлена тем, что в результате искривления
линий потока жидкости на концах капилляра происходит рас-
сеяние энергии. Этот концевой эффект может быть учтен до-
бавлением слагаемого Д к длине капилляра I.
292
Вторая поправка связана с тем, что часть перепада давле-
ния ДР расходуется не на преодоление вязких сил, а на совер-
шение работы, переходящей в кинетическую энергию истечения.
Можно показать, что:
\ / mdV2
т2гЧ2 1
!
где d — плотность жидкости;
т — коэффициент; близкий к единице.
Введение обеих поправок в уравнение Пуазейля дает:
md V „
8V (/+/,) W
Если давление, под которым течет жидкость, обусловлено си-
лой тяжести, то:
P=hdg
где h — средняя высота столба жидкости.
Подставляя это значение Р в уравнение (6) и объединяя по-
стоянные величины, получим:
где А и В — постоянные для данного вискозиметра и фиксиро-
ванной средней высоты столба жидкости. Для фиксации вы-
соты столба непосредственно под капилляром делается расши-
рение, разрывающее столб жидкости. Таким образом нижний
уровень устанавливается автоматически на одном и том же
месте, независимо от объема жидкости в вискозиметре.
Для нахождения констант вискозиметра А и В измеряют
время истечения нескольких жидкостей с известными величи-
нами т], например глицеринововодных смесей, или же время ис-
течения воды при разных температурах для каждого участка
вискозиметра.
Из уравнения (7) следует:
<8>
Нанося на график как функцию , находят константы Л
(отрезок на оси ординат) и В (tg угла наклона). Следует отме-
тить, что в тех случаях, когда поправка на кинетическую энер-
гию ничтожно мала, по уравнению (6) т] = Adt, а следователь-
но В ~ 0, и график должен дать горизонтальную прямую.
Для работы с сильно летучими растворителями применяют
насадки различных конструкций, описанные в литературе.
293
Выполнение работы
Измерение вязкости в вискозиметрах с внешним давлением
описано в работе 44; т]0Тв определяется как отношение ctg углов
наклона касательных. Измерение в вискозиметре, изображен-
ном на рис. 126, производится следующим образом.
В широкое колено 7 вводят объем жидкости, достаточный
для заполнения средней трубки 6 с расширениями.
Затем давлением воздуха из баллона или груши переводят
жидкость в трубку 6, закрывая пальцем трубку 8. Жидкость
должна подняться выше метки 5. Затем, отключив давление,
дают жидкости вытекать через капилляр. Прохождение меток
верхним уровнем жидкости фиксируется посредством поочеред-
ных отсчетов на двух секундомерах.
Вначале производится калибровка прибора водой при раз-
ных температурах для нахождения констант капилляра 7 и 6
(для каждого расширения отдельно). Запись производится по
схеме табл. 2. Измерения повторяют 3—4 раза при каждой тем-
пературе, для расчета берут среднее.
Таблица 2
В студенческой работе константы А и В могут быть даны
преподавателем без измерения.
После калибровки производят измерения с чистым раствори-
телем, повторяя их 3—4 раза. '
Далее производят такие же измерения с каждым из серии
растворов полимера, приготовленных по указанию преподава-
теля. Плотность растворов определяют пикнометрически. Дан-
ные опытов оформляют в виде следующей таблицы (см. Табл, 3,
стр. 295).
Вычисляют значения т] и т]о по уравнению (7) для каждого
интервала между метками. Если величины т] для раствора дан-
ной концентрации оказываются практически постоянными, бе-
рут среднее значение т]. Если же эти значения различаются, не-
обходимо экстраполировать на нулевую скорость сдвига. По-
скольку скорость сдвига пропорциональна средней высоте
столба жидкости h, необходимо построить график т]—h для рас-
294
Таблица 3
находят как разность между верхним уровнем жидкости в мо-
мент, отвечающий половинному времени вытекания //2 и ниж-
ним концом капилляра. Отсчет делают по линейке с миллимет-
ровыми делениями *.
Полученные значения—где с—концентрация раствора,
е/100 см3, наносятся на график по оси ординат. По оси абсцисс
откладывают значения с.
•Величину т] находят посредством экстраполяции к с = 0.
Для вычисления М применяют уравнение (5), подставляя зна«
чения констант К и а из таблицы.
Работа 48
Исследование процесса набухания твердых полимеров
Избирательное поглощение жидкости сухим образцом поли-
мера сопровождается сильным увеличением его объема. Это яв-
ление называется набуханием. Избирательная способность
набухания связана с сольватацией цепей макромолекул, обра-
зующих остов полимера при проникновении в него молекул
жидкости.
Хрупкие, неэластичные гели, к которым относятся главным
образом неорганические гели типа SiO2, SnO2, TiO2, предста-
вляют собой пористые тела, впитывающие в силу капиллярно-
сти любую смачивающую их жидкость независимо от химиче-
ского состава. При этом объем этих гелей практически почти не
изменяется.
Высокомолекулярные соединения, такие, как желатин, агар-
агар, каучук, иначе относятся к растворителям; не все смачи-
вающие жидкости поглощаются данным высокополимером, а
* Высокой точности отсчета не требуется, поскольку величина т] обычно
мало изменяется с ft,
295
только некоторые: желатин поглощает воду, но не поглощает
бензол; каучук, напротив, поглощает бензол и не поглощает
воду. Высокомолекулярное вещество набухает с образованием
студня в жидкостях, близких ему по химическому строению.
Высокомолекулярные соединения, поглощая жидкость, уве-
личиваются в объеме, развивая при этом-значительную механи-
ческую силу, способную достигать нескольких сотен килограм-
мов на 1 см2 площади студня. Переменное изменение объема
в процессе набухания и отбухания при высыхании студня может
быть повторено неограниченное число раз в том случае, когда
имеет место ограниченное набухание, т. е. такое набухание, при
котором высокомолекулярное вещество при обычной темпера-
туре набухает до известного предела (желатина, агар-агар в
воде при обычной температуре).
Свойство эластичности студня связано с гибкостью цепей
молекул, составляющих остов студня.
Гибкость молекул зависит, в свою очередь, от природы ато-
мов, входящих в состав макромолекулы, от характера распре-
деления этих атомов, от длины цепи, величины межмолекуляр-
ных сил и от температуры.
Комплекс процессов, сопровождающих набухание, является
весьма сложным, поэтому механизм набухания не вполне ясен
до настоящего времени. Несомненно, однако, что набухание свя-
зано с процессом растворения.
Низкомолекулярные соединения могут переходить в раствор
непосредственно, тогда как растворению высокомолекулярных
веществ, цепи которых имеют большие размеры по сравнению
с молекулами растворителя, должно предшествовать набухание.
Процесс набухания можно- себе представйть как проникно-
вение молекул низкомолекулярной жидкости в пространство
между молекулами высокомолекулярного соединения. Такое
проникновение происходит благодаря тому, что звенья гибких
цепей полимера не подходят друг к другу вплотную и образуют
свободные пространства, в которые проникают молекулы низко-
молекулярной жидкости. Когда свободные пространства запол-
няются жидкостью, молекулы начинают раздвигать звенья це-
пей полимера, образуя новые пустоты, которые снова запол-
няются низкополимерной жидкостью.
Если набухший студень при той же температуре далее само-
произвольно переходит в раствор, происходит неограниченное
набухание. Так, натуральный и дивиниловый'каучуки в бен-
золе, нитроцеллюлоза в ацетоне, гуммиарабик в воде сначала
набухают,- а затем переходят в раствор, образуя золь. В этом
случае происходит полное, раздвигание цепей полимера и отрыв
’макромолекул друг от друга. При ограниченном ( набухании
раздвигание цепей происходит только в некоторых участках,
остальные части цепи остаются связанными между собой.
296
Повышение температуры часто приводит к тому, что неко-
торые ограниченно набухающие полимеры начинают* раство-
ряться в жидкостях, в которых они набухали. Так, желатина
растворяется в воде при нагревании.
В ряде случаев мы встречаемся с ограниченным набуханием,
не переходящим в неограниченное при повышении температуры
(вулканизированный каучук, задубленная желатина). Это
объясняется тем, что молекулы полимера образуют между со-
бой мостичные связи, препятствующие текучести и затрудняю-
щие переход молекул в раствор.
При большом числе мостичных связей, например у эбонита,
пространственная сетка полимера делается жесткой, цепи утра-
чивают свою гибкость, и полимер теряет способность к набу-
ханию.
Начальная стадия набухания характеризуется обычно поло-
жительным тепловым эффектом. В дальнейшем набухание про-
' текает почти без выделения тепла и с повышением температуры
увеличивается.
Количество тепла в калориях, которое выделяется в течение
всего процесса набухания 1 г сухого полимера в условиях из-
бытка жидкости, называется интегральной теплотой
набухания.
Количество тепла, выделяющееся при поглощении полиме-
ром 1 г жидкости, в различных состояниях набухающего студня,
называется дифференциальной теплотой набу-
хания. Наибольшее значение дифференциальная теплота на-
бухания имеет в начальный момент процесса набухания.
Теплота набухания объясняется взаимодействием раствори-
теля и молекул полимера с образованием адсорбционно-соль-
ватного слоя и характеризует это взаимодействие с энергетиче-
ской стороны. Часть растворителя связывается набухающим по-
лимером. В случае водного раствора образуется связанная вода,
молекулы которой ориентированы по отношению к макромоле-
кулам полимера и имеют более плотную упаковку, чем свобод-
ная вода. Поэтому связанная вода отличается от свободной
воды по ряду физико-химических свойств: большей плотностью,
неспособностью растворять, меньшей диэлектрической постоян-
ной, пониженной температурой замерзания и др.
Следствием относительно большей плотности ориентирован-
ных молекул связанной воды является сжатие системы сту-
день— жидкость, характеризующееся тем, что сумма объёмов
сухого вещества и поглощенной им жидкости больше, чем объем
набухшего полимера.
Это явление называется контракцией и наблюдается,
главным образом, у высокомолекулярных соединений, набухаю-
щих в воде: желатины, агар-агара, крахмала. Набухание каучу-
ка в углеводородах происходит без заметной контракции.
297
Для количественного учета контракции можно пользоваться
следующим соотношением:
где —контракция;
Vo — объем сухого вещества;
1^! — объем поглощенной жидкости;
V2 — объем набухшего полимера.
Для удобства сравнения результатов отдельных опытов по
набуханию введено понятие о степени набухания, под которым
разумеется количество жидкости,
поглощенное 1 г сухого вещества.
Предельное количество жидко-
сти, поглощенное 1 г полимера, на-
зывается набухаемостью.
Количество жидкости, поглощен-
ной при ограниченном набухании,
может быть очень велико, объем
поглощенной жидкости может более
чем в десять раз превышать. объем
взятого для набухания сухого по-
лимера. Процесс набухания проис-
ходит во времени с определенной
природы набухающего полимера й
Рис. 127. Зависимость степени
набухания (W) от времени (Z).
скоростью, зависящей от
растворителя.
Процесс набухания протекает как реакция первого порядка:
(1)
где Л — постоянная, зависящая от природы полимера;
W—количество поглощенной при набухании жидкости на
1 г сухого вещества;
t — время набухания;
Wco — предельное количество поглощенной при набухании
жидкости.
Интегрируя уравнение (1), получаем:
Л 1 I
t W^ — W
(2)
Приведенное выше' уравнение для скорости набухания мож-
но решить графическим путем. Для этого строят кривую за-
висимости степени набухания W от времени t и для различных
величин W находят скорости набухания проведением касатель-
ных (рис/127).
, Тангенсы углов, составляемых касательными, равны скорос-
- 4 dW т
тям набухания = при различных величинах 1и.,
298
Уравнение (1) можно представить в виде:
С ТО7 dw 1
если w и -ц- величины переменные, а д- величина постоян-
ная, то это уравнение прямой. Поэтому, нанеся, на координаты
значения W и —^-.получаем прямую (рис. 128), где отрезок по
оси ординат ОК равен:
при ~ = 0
Отрезок OD по оси абсцисс равен A W’oo, поскольку при
W = О
°0 A dt
Таким образом, можно определить постоянные Woo и А гра-
фическим путем. Постоянство А не соблюдается в начале про-
Рис. 128. График для
расчета постоянных А
и 1Г».
Рис. 129. Кривые набуха-
ния для ограиичеино набу-
хающих веществ.
цесса набухания. Приведенное уравнение скорости набухания
было подтверждено опытами многих* исследователей.
Графически кинетика набухания выражается кривой зави-
симости степени набухания W от времени (рис. 129). На рис. 129
представлены данные для ограниченно набухающих образцов
двух веществ (кривые I и II), правильную оценку дает макси-
мальная степень набухания, так как ход кривых I, II может
быть различен в начальные периоды’ времени.
Для неограниченно набухающих полимеров с переходом
студня в раствор, кривая зависимости степени набухания от вре-
мени показывает максимум, после чего резко снижается
(рис. 130).
Большую группу высокомолекулярных соединений, обладаю-
щих рядом особенностей в отношении процесса набухания.
299
представляют полиэлектролиты, в частности белковые вещества
в воде и водных растворах. В этих условиях при измерении сте-
пени набухания необходимо учитывать pH среды. При некото-
рой концентрации водородных ионов, характерной для белка
определенной природы, белок отщепляет как кислотные, так и
Рис. 130. Кривая набухания
для неограниченно набухаю-
щего вещества.
основные группы в равной степени,
т. е. является электрически нейт-
ральным. Такое состояние белка на-
зывается изоэлектрическим
состоянием1.
Изоэлектрической точ-
кой называется такое значение pH
среды, при котором частицы белка
не .передвигаются в электрическом
поле и, следовательно, суммарный
заряд их равен нулю.
При повышении концентрации
Н-ионов среды, т. е, при сдвиге
в кислую сторону от изоэлектрической точки белки заряжаются
Изоэлектрическая рн
точка
Рис. 131. Кривая зависимости
степени набухания от pH рас-
твора.
положительно.
Наоборот, при сдвиге в щелочную сторону частицы белка
будут находиться в состоянии анионов и будут поэтому нести
отрицательный заряд.
Изоэлектрическая точка белков не совпадает с нейтральной
реакцией раствора (pH = 7), вследствие неравенства щелочной
и кислотной констант диссоциации
у белков. Обычно она сдвинута в
кислую сторону.
Приводим для примера несколь-
ко значений pH изоэлектрической
точки у различных белков:
4,7
4,55
4,7
6,7
степени не-
обычно ми-
студней от pH раствора,
Желатина ....
Яичный белок . .
Казеин........
Гемоглобин . . .
.Кривая зависимости
бухания от pH имеет
нимум в изоэлектрической точке, и
по обе стороны от этой точки сте-
пень набухания возрастает, как это
показано на рис. 131.
Причина зависимости набухания
т. е. влияние водородных и гидроксильных ионов на набухание,
заключается в изменении отталкивательных сил между цепями
макромолекул, приводящего к изменению структуры студня.
В изоэлектрическом состоянии происходит более уплотнен-
ная агрегация макромолекул с образованием более сложных
300
комплексов. Этот процесс связан с некоторой десольватацией
в изоэлектрическом состоянии.
Благодаря наличию у белков ионогенных групп на набуха-
ние также оказывают влияние нейтральные соли. До'известной
степени влияние это зависит и от pH раствора.
В сильно кислой среде • все соли уменьшают набухание;
вблизи изоэлектрического состояния соли могут как понижать,
так и повышать степень набухания, причем анионы оказывают
более сильное влияние, чем "катионы.
Более сильно гидратированные ионы С1 вызывают меньшее
набухание, чем менее гидратированные ионы Вг и J. По влия-
нию на процесс набухания анионы располагаются в следующий,
гак называемый лиотропный ряд (Гофмейстера):
CNS- > J" > Вг' > NO3- > С1О3- >
> ацетат"> цитрат > сульфат
Изучение процесса набухания высокомолекулярного соеди-
нения дает весьма ценные данные для его физико-химической
характеристики.
Выполнение работы
Определение степени набухания можно производить весовым
и объемным путем.
В первом случае определяют вес сухого вещества и вес в
набухшем состоянии. По разности находят привес в граммах на
1 г сухого вещества.
При объемном методе измеряют увеличение объема набу-
хающего студня, происходящее при поглощении им жидкости,
или учитывают уменьшение объема жидкости, в которой проис-
ходит процесс набухания.
В последнем случае, удобно пользоваться прибором, изобра-
женным на рис. 132.
Прибор наполняют жидкостью, в которой определяют набу-
хание. Прибор закрепляют вертикально в штатив, устанавливая
21 Зак. 254
301
его резервуаром / кверху. Отмечают уровень жидкости в изме-
рительной трубке 3, после чего поворачивают прибор в наклон-
ное положение так, чтобы жидкость покрыла всю навеску поли-
мера, помещенную в резервуар 1.
Через определенные промежутки времени (15, 30, 45, 60,
120 мин), в, зависимости от условий опыта, определяют объем
поглощенной жидкости. Для этого жидкость переливают в ре-
зервуар 2 и, поставив прибор в вертикальное положение, выжи-
дают, чтобы вся жидкость стекла из резервуара 1, после чего
отмечают положение мениска в трубке 3. Уменьшение объема
жидкости соответствует количеству поглощенной жидкости. По-
лученные данные записывают в таблицу. Рассчитывают степень
набухания для каждого взятого отрезка времени и строят гра-
фик зависимости степени набухания от времени. По оси орди-
нат откладывают степень набухания, по оси абсцисс — время в
минутах. На полученных кривых набухания W = f(t) проводят
касательные для различных величин W.
Найдя тангенсы углов, составляемые касательными tg<p —
dW .w dW ~
2^-, строят кривую зависимости между w и• Определяют
постоянные №оо и А.
Методом Пржеборовского и Тиле [8] можно исследовать ско-
рость набухания одновременно весовым и объемным путем, поль-
зуясь при этом одной навеской. Метод основан на взвешивании,
исследуемого образца в двух жидкостях, в которых данный вы-
сокополимер не набухает.
Взвешивание производят на торзионных весах. В начале ис-
следования определяют вес ро сухого образца в воздухе, под-
вешивая образец на тонкой проволоке *.
После этого для определения объема сухого образца его
взвешивают в жидкости 1, в которой данное вещество не набу-
хает (для желатины пользуются бензолом), а затем в жид-
кости 2 (желатина в СС14), предварительно сполоснув образец
этой жидкостью. При- взвешивании в СС14 пользуются дополни-
тельным грузиком для того, чтобы образец не всплывал из-за
большого удельного веса СС14. Проволоку, на которой предва1
рительно взвешивают образец, взвешивают в СбН6 и в СС14, а
грузик — только в СС14. Объем сухого вещества Уо рассчиты-
вают по приведенному ниже уравнению (6).
Определив вес и объем образца, помещают его в сосуд с жид-
костью, в которой происходит набухание. Через определенный
промежуток времени (например, 15 мин), образец вынимают из
сосуда, освобождают от прилипшей жидкости осторожным при-
* Можно пользоваться чашечкой, сделанной из легкого материала, на-
пример из алюминиевой фольги. На дне чашечки делают отверстия. Ча-
шечку подвешивают на тонкой проволоке к весам. В этой. же чашечке мо-
жет происходить набухание.
302
косновением фильтровальной бумаги и взвешивают на торзион-
ных весах сначала в жидкости 1, затем в жидкости 2. Каждый
раз образец споласкивают жидкостью, в которой производят
взвешивание.
Взвешенный образец промывают дистиллированной водой и
снова погружают в сосуд для набухания, повторяя взвешивание
через некоторое время. В зависимости от условий опыта набу-
хание происходит в течение 2—3 ч.
Таким образом, получают следующие данные:
р0 — вес сухого образца в воздухе;
рг — вес сухого образца в бензоле (жидкость 1) ;
р2 — вес сухого образца в четыреххлористом углероде (жид-
кость 2);
Pl — вес набухшего образца в жидкости 1 через время /;
Тг — удельный вес жидкости 1; для бензола при 20° С
71 = 0,879 г/сж3(8620 н/м3);
72 — удельный вес жидкости 2; для четыреххлористого угле-
рода при 20°С 72= 1,595 а/сл13(15646 н/мг).
На основании этих данных можно вычислить вес набухшего
(через время t) полимера р и объем сухого Vo и набухшего
(через время t) образца Vt-
Для веса образца в жидкости I (через время t):
Pt = P—Vfp
где р — вес набухшего образца;
Vt — объем набухшего образца.
Для веса образца в жидкости 2:
Pt=P— VZ72
При условии 72 > 71 получаем:
Уравнением (4) пользуются для вычисления объема набух-
шего полимера. Поскольку р = pt + Vt7i и р = Pt 4- V/72 для
веса набухшего полимера, получаем:
(л + Х) + (п + 12)^ ...
р =---------------------- (5)
Для вычисления объема сухого образца имеем аналогично:
Pi — Ро Ц) ' Ti> Pi —Ро Vo ' Т2
откуда:
(6)
12 —71
303
Зная вес и объем^сухого образца, можно рассчитать коли-
чество поглощенной жидкости и определить степень набу-
хания.
Полученные данные наносят на график, откладывая по
оси абсцисс время в минутах, по оси ординат — степень набу-
хания.
Проводят ряд касательных к полученной кривой набухания
и находят тангенсы углов. Графически определяют константы
И А.
По указанию преподавателя делают одну из следующих за-,
дач:
1. Влияние анионов на скорость набухания желатины. Для
опыта берут несколько образцов желатины и набухание проис-
ходит в растворах солей KCNS, KBr, KNO3 и в чистой воде.
' 2. Влияние pH на скорость набухания желатина. Для опытов
пользуются растворами ‘электролитов различной концентрации:
НС1, H2SO4, NaOH.
3. Определение контракции желатина. Для этой цели поль-
зуются прибором, изображенным на рис. 132.
4. Определение скорости набухания каучука в различных
растворителях. Набухание каучука проводят в керосине, бен-
зине, бензоле, толуоле и ацетоне. Полученные данные записы-
вают в таблицу 2'при работе первым методом, в таблицу 3 —
при работе вторым.
Таблица 2
Вес образца Ро Объем взятой для опыта жидкости Объем поглощен- ной жидкости vt Время набухания it мин Степень , набухания W Скорость набухания dW dt U7 со А
*
Таблица 3
Ро t, мин Pl Pi Pt Pt p Vo Vt' w № dt w co A
»
<304
Работа 49
РАВНОВЕСИЕ ДОННАНА, СУСПЕНЗИОННЫЙ И ЗОЛЬ-КОНЦЕНТРАЦИОННЫИ
ЭФФЕКТЫ
Если два раствора электролита разделены мембраной, непро-
ницаемой хотя бы для одного из ионов (обычно это ион кол-
лоида), то все остальные ионы распределяются по обе стороны
мембраны неравномерно. Это сказывается на величине измеряе-
мого осмотического давления коллоидного раствора, а также
проявляется в обнаружении разности потенциалов между кол-
лоидным раствором и равновесной с ней жидкостью. Данное
явление было открыто в 1911 г. Доннаном и получило назва-
ние мембранного равновесия или равновесия Доннана. Очень
близко связаны с этим явлением так называемый суспен-
зионный и золь-концентрационный эффекты.
Рассмотрим наиболее простой случай равновесия Доннана
для вещества типа коллоидного электролита состава NaR. Пусть
по одну сторону мембраны вначале имеем раствор NaR (I), а по
другую — NaCl (II); предположим, что мембрана проницаема
для всех ионов кроме R”.
I и
Na+ Na+
R” СГ
NaCl будет диффундировать из II в I и при равновесии:
I п
Na+ Na+
R”
СГ СГ
Рассмотрим следующее, бесконечно малое (dn моль), изо-
термическое обратимое изменение системы:
dn моль Na+ II —> I
dn моль СГ II —> 1
Равновесие будет достигнуто в том случае, когда работа изо-
термического переноса dn моль Na* из II в I, равна работе, не-
обходимой для переноса dn моль С1 из II в I, т. е. при равно-
весии справедливо следующее равенство:
[Na+]n [СГК, .
^rlnRJ5< + dn/?7’ln7ci=t=0
где [Na+]n, [С1~]п, [Na+]i и [Cl~]i — концентрации ионов Na+ и Cl”
в камерах II и I.
Следовательно [Na+]n[Cl~]n — [Na+]i[CI”]i, т. е. равновесие до-
стигается, когда произведения концентраций свободно диффун-
дирующих ионов по обе стороны мембраны' становятся одина-
ковыми.
305
Учитывая условия электронейтральности растворов по обеим
сторонам мембраны можно записать:
[Na+]n=[Crjn и [Na+]j= [С!-],-)-[R"],
Следовательно
[СГ]у = [Na+Jj [С1-],
так как
[Na+J, > [СГ],
то
[cr^xcrij
Аналогично можно получить, что [Na+]n < [Na+]j.
Полученные выражения говорят о неравномерном распреде-
лении электролита, в частности отдельных ионов, по обеим сто-
ронам мембраны, в том случае, если в одной из двух камер си-
стемы присутствуют заряженные частицы, неспособные по ка-
ким-либо причинам к диффузии.
Для более ясного представления сущности процесса прове-
дем некоторые расчеты. С целью упрощения рассмотрения вве-
дем следующие два предположения: 1) полная диссоциация
NaR и NaCl, 2) объемы жидкости по обе стороны мембраны
одинаковы.
Получающееся равновесие можно представить следующим
образом:
Na+ R~ СГ Na+ СГ
Cj X Сц—X Сп — X
где Cj и сп — исходные концентрации коллоида и электролита;
х— количество электролита, прошедшего в камеру (I)
из камеры (II). Тогда из условия равновесия мож-
но записать:
(с1-|-л)л = (с11—л)2
• Решая полученное Уравнение, находим величину х:
си
Х fi + 2cn
Тогда — есть распределительная пропорция электролита
(NaCl) между пространствами II и 1 при равновесии. Легко по-
казать:
си х _ ci ~Ь сц
х ~ сп
Если Сц мало по сравнению с cj, тогда можно пренебречь
в числителе, правой части равенства величиной Сп, тогда
306
€11—-X . Cl
——» т. e. чем больше и меньше еп, тем меньшее от-
носительное количество электролита перейдет в камеру I. Если
еп Су тогда ———== 1, т. е. распределение электролита ока-
зывается равномерным по обе стороны мембраны, приближаясь
к случаю отсутствия-коллоида.
Как указывалось выше, неравномерность распределения
электролита оказывает влияние на величину измеряемого осмо-
тического давления коллоидного раствора.
Действительное осмотическое давление чистого коллоидного
электролита равно P0 — 2ciRT. Обозначим через Р противодав-
ление, возникающее за счет неравномерного распределения
электролита между камерами I и II. Тогда Р = 2(сп — х)РТ.
Наблюдаемое осмотическое давление Рп равняется
Рн==2/?7'(с[4-л) —2/?Г(сп —jc) = 2/?7'(cj —с([ 4-2х)
Откуда следует, что
Рк 2РТ(с} — сп + 2х) _ с, — сп + 2х
~Р-О- 2C.RT ~ - с,
Подставляя значение х, окончательно получаем:
Рн _ ci + сп
. р0 С1+2СП
Если Q-sCc,,, то Рн = уР0 и
если С|^>с(1, то /эн = По-
следовательно, истинное значение осмотического давления
коллоида получается только в том случае, когда концентрация
его много больше концентрации присутствующего электролита.
) Вследствие неравномерного распределения ионов при равно-
весии между растворами, должна существовать разность потен-
циалов (так называемый «мембранный потенциал»), которая
может быть вычислена ио формуле:
RT [Na+], RT [СГ]П
р__ in 1 -Ч in -41
F [Na+]„ ~ F ln [С1],
Приведенную формулу мы можем переписать иначе:
Подставляя значение для х, получаем:
RT
ci + cii
с.»
307
Если ci<On, то Е«=0. Этот случай соответствует отсутствию
коллоида и равномерному .распределению электролита по обе
стороны мембраны. В случае, если сг^>сп, получаем, что
Следовательно, величина мембранного потенциала зависит
от концентрации коллоида, возрастая с ростом концентрации
последнего. Наоборот, рост концентрации электролита (при
постоянной концентрации коллоида) приводит к уменьшению
величины мембранного потенциала.
Мембранный потенциал может быть легко обнаружен с по-
мощью двух каломельных электродов, помещенных в равновес-
ные жидкости, находящиеся по обе стороны от мембраны.
В случае положительно заряженного коллоида, потенциал»кало-
мельного электрода, погруженного ‘в камеру с коллоидом I,
всегда оказывается более положительным, чем потенциал кало-
мельного электрода, опущенного в равновесный раствор, не со-
держащий коллоида. ’
Рассмотрим связь между мембранным потенциалом в равно-
весии Доннана и так называемыми суспензионным или золь-
концентр ационным эффектами.
Сущность суспензионного эффекта состоит в том, что pH
суспензии и выделенного из нее центрифугата или ультрафиль-
трата в общем случае не одинаковы. При этом оказывается,
что в суспензии из отрицательно заряженных частиц pH ниже,
чем pH центрифугата (ультрафильтрату). Положительно заря-
женные частицы дают обратный эффект — pH суспензии выше
чем pH центрифугата.
На это явление впервые обратили внимание почвоведы, изу-
чавшие концентрацию водородных ионов в почвенных суспен-
зиях й вытяжках из почв. Они нашли, что pH суспензии зависит
от взятого для измерения соотношения почва — вода, хотя ранее
казалось, что раствор, насыщенный растворимыми компонен-
тами почвы, должен показывать постоянное значение pH неза-
висимо от содержания почвы в воде. •
• Большое количество работ, проведенных по измерению сус-
пензионного эффекта, в частности работы, проводившиеся на
кафедре коллоидной химии ЛГУ, показали, что суспензионный
эффект является достаточно общим явлением и увеличивается
с увеличением концентрации дисперсной фазы. В случае отрица-
тельно заряженных частиц суспензия оказывается более кислой
по сравнению с равновесной жидкостью, для положительно за-
ряженных частиц — эффект имеет обратное направление. Уве-
личение дисперсности частиц и уменьшение концентрации элек-
тролита приводит к увеличению суспензионного эффекта, кото-
рый можно характеризовать как разницу в электрометрически
308
измеренных величинах:
pHcycn рНр. Ж ДрН
Рис. 133. Зависимость С-по-
тенциала и суспензионного
эффекта (Д pH) А12О3 от
pH раствора.
Было показано, что суспензии веществ, обладающих амфо-
терными свойствами, в зависимости от pH раствора, могут пока-
зывать как кислый (суспензия более кислая, по сравнению с
равновесной .жидкостью), так и щелочной суспензионные эф-
фекты. Для примера на рис. 133 приведена зависимость суспен-
зионного эффекта (кривая. II) и С-потенциала (кривая I) для
суспензий AI2O3. Слева от изоточки,
^-потенциал положителен, и суспен-
зии оказываются более щелочными
по сравнению с равновесным рас-
твором. Справа от изоточки части-
цы А12О3 несут ^отрицательный заряд,
^-потенциал отрицателен, и суспензии
А12О3 оказываются более -кислыми
по сравнению с равновесным рас-
твором.
В случае электрометрического из-
мерения эффекта в золях он носит на-
звание зо л ь-концентрацион не-
го эффект а. Таким образом нет
принципиального различия между сус-
пензионным и золь-концентрационным
эффектами.
Следует отметить, что эти эффек-
ты могут измеряться не только отно-
сительно Н-ионов, но также и по отно-
шению к любому иону, находящемуся в
J-, С1~, SOt и т. д.
Большинство авторов для объяснения суспензионного эффек-
та привлекает теорию равновесия Доннана. Рассмотрим более
подробно измерение мембранного потенциала в цепи Доннана,
а также измерение суспензионного эффекта и покажем их иден-
тичность.
Для того чтобы измерить величину мембранного потенциала,
составляется следующая цепь:
системе; например Ag+,
Hg Hg2Cl2 КС1нас ( равновесный раствор суспензия или коллоидный КС1нас Hg2Cl2 Hg
раствор
М
Цепь состоит из двух каломельных электродов, солевые мо-
стики которых погружены в равновесный раствор и в дисперс-
ную систему, разделенные друг от друга мембраной М.
309
В обычных определениях pH со стеклянным электродом цепь
имеет следующий вид:
Ag AgCl НС1
стекло
исследуемый
раствор
КС1нас Hg2Cl2 Hg
т. е. цепь состоит йз одного каломельного и одного стеклянного
электродов.
На рис. 134 представлены схематически суспензия и находя-
щаяся с ней в равновесии жидкость, которые отделены друг
от друга мембраной. В каждую жидкость помещены обратимый
электрод Н (например, стеклянный электрод) и солевой мо-
стик s с каломельным электродов. Тогда величина мембранного
потенциала может быть измерена между двумя каломельными
электродами (солевыми мостиками) (Es)i— (ДОг, а величина
суспензионного эффекта равна
рНсуСП РНР. )К = [(£\) 1 — (Ен) 1] [(ДО 2 (Еп) 2],
т. е. ДрН=(Д)1—(Д)2—(Дт)1+(£ц)2. Поскольку два обра-
тимых электрода имеют одинаковый потенциал в жидкостях, на-
ходящихся в истинном термодинамическом равновесии (т. е.
Рис. 134. Схема, показывающая
идентичность мембранного потен-
циала и золь концентрационного
(суспензионного) эффекта.
(£н)1=(Дг)2), ТО ДрН = (Д)1-
— (Д)2. (В данном случае мы
величину ДрН выражали не в
единицах pH, а в мв, однако для
данного рассмотрения это не име-
ет принципиального значения, так
как от мв мы легко можем пе-
рейти к pH).
Важным выводом из приведен-
ного рассмотрения является то,
что величина мембранного потен-
циала точно соответствует ДрН,
т. е. эти два эффекта являются
идентичными.
Приведенное термодинамиче-
ское рассмотрение указывает на
идентичность ДрН и мембранного
потенциала в системе, находящейся в истинном термодинами-
ческом равновесии, однако,- механизм возникновения мембран-
ного потенциала в данном случае не рассматривается. Такой
причиной может быть или равновесие Доннана, или какая-
либо другая причина.
Большинство авторов считает, что причиной суспензионного,
эффекта является равновесие Доннана с его неравномерным
распределением ионов между коллоидом и равновесным рас-
твором. Однако за последнее время появились работы, в кото-
310
pbix указывается возможность возникновения потенциала меж-
ду двумя каломельными электродами и появления ДрН не за
счет неравномерного распределения ионов между суспензией
и равновесным раствором, а за счет наличия диффузионного
потенциала на границе КС1 мостик — суспензия.
Действительно, в том случае, когда КС1 — мостик опущен
в равновесный раствор, диффузионным потенциалом соединения
можно пренебречь из-за незначительного различия в числах пе-
реноса ионов К и С1. Однако в том случае, когда КС1 — мостик
опускается в суспензию, состоящую из значительного числа за-
ряженных частиц, диффузия КС1 из мостика в суспензию может
привести к значительной величине диффузионного потенциала,
возникающего за счёт изменения чисел переноса ионов К и С1
в суспензии. (Суспензия из некоагулирующих взвешенных ча-
.стиц может быть рассмотрена как мембрана с некоторыми рас-
стояниями между частицами, аналогичными порам «жесткой
мембраны»).
Так как мы не можем без произвольности разделить э. д. с.
цепи Доннана на истинный мембранный потенциал и два диф-
фузионных потенциала КС1 солевых мостиков, то обычно рас-
сматривают э. д. с. всей цепи Доннана, оставляя в стороне без
рассмотрения вопрос о точном значении разности потенциалов
между суспензией и равновесным с ней раствором.
Выполнение работы
Целью работы является измерение э. д. с. цепи Доннана
и золь-концентрационного эффекта (ДрН), а также демонстра-
ция их идентичности.
Работа проводится следующим образом. Приготовляется
1,5 дм3 раствора электролита (например НС1) определенной
концентрации (по указанию преподавателя). На полученном
растворе готовится 40—50 см3 1%-ного раствора желатины. Да-
лее коллодиевый мешочек, закрытый резиновой пробкой, вме-
стимостью 40—50 см3, наполняется раствором желатины, после
чего в пробку вставляется капилляр (для этой .цели можно ис-
пользовать капиллярную пипетку на 1 см3). Мешочек поме-
щается в стакан объемом 1 Ди3, до уровня пробки (рис. 135).
Стакан заполняется раствором приготовленного электролита.
Система оставляется в покое на 1—2 дня для достижения рав-
новесия, что может быть установлено по постоянству высоты
мениска жидкости в капилляре, характеризующей величину
осмотического давления. После достижения равновесия капил-
ляр удаляется и вместо него в отверстие пробки вставляется
маленькая воронка. Путем сдавливания мешочка можно до-
биться поднятия жидкости в воронку, после чего производится
измерение э, д. с, с помощью двух каломельных электродов.
311
Предварительно определяется потенциал асимметрии каломель-
ных электродов, который учитывается затем при вычислении
э. д. с. цепи Доннана. Один. КС1 солевой мостик, опускается
в равновесный раствор, а второй в воронку с раствором жела-
тины. Получающаяся э. д. с. измеряется с помощью потенцио-
метра. Измеренная величина э. д. с. (с учетом потенциала асим-
метрии каломельных электродов) является э. д. с. цепи Донна-
на. Затем производятся измерения pH равновесного раствбра
Рис. 135. Схема установки для измере-
ния мембранного потенциала.
ктрод, соединенный с коллоидным
и раствора желатины с по-
мощью водородного или сте-
клянного электродов. Вели-
чины э. д. с. цепи Доннана
и Д£, полученные при изме-
рениях pH, должны быть
равны. Различие этих вели- w
чин говорит о том, что си-
стема не достигла равнове-
сия.
Следует отметить, что в
том случае, когда частицы
коллоида заряжены поло-
жительно, каломельный эле-
раствором, оказывается бо-
лее положительным, по сравнению с каломельным электродом,
соединенным с равновесной жидкостью. В этом случае pH кол-
лоидного раствора оказывается выше, чем pH равновесного
раствора. При отрицательно заряженном коллоиде наблюдаются
обратные закономерности. В случае нахождения коллоида в
изоэлектрической точке оба эффекта должны отсутствовать, и
таким путем может быть определена изоэлектрическая точка
исследуемого вещества.
ЛИТЕРАТУРА
1. В о л ж и н с к и й .И. А., Л ь во в В. Н., Рейхсфельд В. О., Практи-
кум по синтетическим каучукам, Госхимиздат, 1955.
2. - Д у м а н с к и й А. В., Учение о коллоидах, Госхимиздат, 1948.
3. К о р ш а к В. В., Р а ф й к о в С. Р., Введение к изучению высокомоле-
кулярных соединений, Изд. АН СССР, 1946.
4. Кройт Г. Р., Наука о коллоидах, т. 1, ИЛ, 1955.
5. Сб. «Методы‘исследования полимеров», ИЛ, 1961.
6. -Никольский Б. П., Почвоведение, 9, 138 (1939).
7. Пасы некий А. Г., Коллоидная химия, Госхимиздат, 1961.
8. П р же б о р ов с к и й Я- С., Тиле В. Е., Колл, ж., 16, № 4, 304 (1954).
9. Тагер А. А., Физико-химия полимеров, Госхимиздат, 1963.
ГЛАВА VII
;, наполненный водой,
Рис. 136. "Прибор для
получения золей метал-
лов методом.электриче-
ского распыления.
УКАЗАНИЯ ДЛЯ ПОДГОТОВКИ ДЕМОНСТРАЦИОННЫХ
ОПЫТОВ к ЛЕКЦИЯМ
ПОЛУЧЕНИЕ КОЛЛОИДНЫХ РАСТВОРОВ
Лекции по получению коллоидных систем могут сопровож-
даться демонстрацией следующих опытов.
Получение коллоидного раствора свинца методом электриче-
ского распыления. Получение коллоидного раствора свинца ме-
тодом распыления удобно демонстрировать в приборе, изобра-
женном на рис. 136. В стеклянный сосу
вставляют на пробке два свинцовых
стержня, служащие электродами. При-
бор через реостат включают в электро-
сеть. Вращением винта I сближают
электроды до соприкосновения и разво-
дят так, чтобы между ними образо-
валась электрическая дуга. Сразу же
вокруг электрической дуги образуется
коллоидный раствор.
Получение высокодисперсного (крас-
ногб) золя золота путем восстановления
таннином. В предварительно тщательно
вымытую эрленмейеровскую колбу нали-
вают 400 см? дистиллированной воды,
4 см? 0,5%-ного раствора HAuCU и 4см3
1 %-кого раствора К2СО3, хорошо взбал-
тывают и нагревают до кипения. -Затем,
не прекращая кипячения, добавляют
при встряхивании по каплям 1 % раствор таннина (приготов-
ленный перед лекцией) до появления- интенсивной красной
окраски.
Получение грубодисперсного (голубого) золя золота.
К 400 см3 дистиллированной воды добавляют 6 см3 0,5%-ного
раствора НАиСЦ и 2 см3 1%-ного раствора солянокислого
313
гидразина. Сразу же образуется коллоидный раствор золота,
прозрачный, голубого цвета в проходящем свете ш сильно опа-
лесцирующий и имеющий коричневатый оттенок в отраженном.
Получение золя канифоли! К 4'00 см3 дистиллированной воды
добавляют по каплям при встряхивании 5% Спиртовой раствор
канифоли. Немедленно образуется молочно-белый коллоидный
раствор ка.нифоли.
Получение золя гидрата окиси железа. Нагревают до кипе-
ния 400 см3 дистиллированной воды и прибавляют отдельными
порциями ~20 см3 предварительно (перед лекцией) приготов-
ленного на холоду 2%-ного раствора FeCl3. После нескольких
минут кипячения получают красно-коричневый золь гидроокиси
железа.
Получение золя берлинской лазури. К 400 см3 дистиллиро-
ванной воды добавляют 4 см3 1%-ного раствора K4Fe(CN)6 и
приливают по каплям 2% раствор FeCl3, свежеприготовленный
на холоду. Сразу же получается золь берлинской лазури.
МОЛЕКУЛЯРНО-КИНЕТИЧЕСКИЕ И ОПТИЧЕСКИЕ СВОЙСТВА
ДИСПЕРСНЫХ СИСТЕМ
Диффузия. Различие в скорости диффузии истинных и кол-
лоидных растворов можно показать на диффузии их в гелях.
Для этого приготовляют 3% раствор желатины следующим об-
разом: сначала навеску желатины оставляют на 1 ч в холодной
воде, после чего сосуд с желатиной ставят на водяную баню,
затем еще горячий раствор разливают в 4 цилиндра (по 200 см3)
и оставляют застывать при комнатной температуре.
Сверху застывшего раствора желатины наливают по 20 см3
золя золота, золя Fe(OH)3, раствора метиленового голубого и
насыщенного раствора C11SO4. Через 1—2 дня обнаруживают
ясную диффузию раствора CuSO4 и метиленового голубого, в
то время как частицы коллоидных растворов практически в
гель не диффундируют.
Диализ истинных и коллоидных растворов. Для демонстра-
ции диализа истинных и коллоидных растворов можно исполь-
зовать заранее приготовленные коллодиевые мешочки. Если
взять два одинаковых мешочка, наполнив наполовину один —
насыщенным раствором CuSO4, а другой — голубым золем золо-
та, опустить в стакан с водой, то через несколько минут можно
наблюдать появление окраски в стакане с CuSO4, тогда как
жидкость во втором стакане останется без изменений.
Для получения коллодиевого мешочка берут чистую сухую
колбу и наполняют обычным медицинским коллодием, представ-
ляющим собой 4% раствор нитроклетчатки (коллоксилина) в
смеси спирта и эфира (1:3 по объему). Затем, медленно вра-
щая колбу, выливают раствор, следя за тем, чтобы оставшимся
314
коллодием была равномерно покрыта вся внутренняя поверх-
ность колбы, и сушат некоторое время (5—10 мин) на воздухе.
После образования на поверхности твердой пленки споласки-
вают колбу несколько раз водой. Потом, слегка отделив пленку
от края колбы, наливают воду между пленкой и стенкой колбы
и осторожно вынимают мешочек. Для получения более толстых
и прочных коллодиевых мешочков после образования твердой
пленки колбу еще раз наполняют коллодием и уже после вто-
ричного высушивания споласкивают водой.
Явление Тиндаля. Для демонстрации на лекции явления
Тиндаля пропускают луч света от электрической дуги, включен-
ной с реостатом в электрическую сеть, через конденсор в сосуд
с плоскопараллельными стенками. Во время демонстрации этого
опыта электрическая дуга покрывается металлическим кожу-
хом — покрышкой.
Вначале в кювету наливают дистиллированную воду (лучше
накануне демонстрации) и включают ток. При освещении ее на-
блюдают очень слабый, чуть заметный конус Тиндаля. Добав-
ляют некоторое количество раствора CuSO4 и при этом не на-
блюдают заметных изменений в интенсивности конуса Тиндаля.
При добавлении небольшого количества какого-либо коллоид-
ного раствора, например золя золота, интенсивность конуса
сразу же резко увеличивается.
Затем пропускают луч света от электрической дуги через ряд
коллоидных растворов, полученных ранее (золи золота, золь
гидрата окиси железа, канифоли, свинца, берлинской лазури),
и во всех случаях наблюдают яркий конус Тиндаля.
Следует отметить, что при подготовке этого опыта для де-
монстрации необходимо тщательно проверить на конус Тиндаля
воду и добавляемый к ней раствор CuSO4.
Если в воде и растворе CuSO4 будут загрязнения (пылинки)
и пузырьки воздуха, то они также могут дать заметный конус
Тиндаля.
Дисперсность коллоидных систем и их окраска. Зависимость
окраски коллоидных систем от размеров частиц можно демон-
стрировать на золях серебра, полученных методом восстановле-
ния. Проводя реакцию в щелочной среде и меняя количествен-
ные соотношения гидрохинона и лимоннокислого натрия, можно
получить коллоидные растворы серебра различной окраски (от
желтой до синей).
Необходим небольшой избыток щелочи, который устанавли-
вают предварительно. Для этого к 2 см3 0,01 н. раствора AgNO3
добавляют 2 см3 0,005 н. раствора гидрохинона (свежеприготов-
ленного), 4 см3 0,01 н. лимоннокислого натрия и встряхивают.
Если раствор через 10—15 сек не начнет желтеть, то к 100 см3
раствора лимоннокислого натрия необходимо добавить одну
каплю разбавленного раствора аммиака (1:3) и повторить
•15
опыт. Если и в этом случае жидкость не желтеет, то к раствору
лимоннокислого натрия добавляют еще одну каплю аммиачного
раствора и снова повторяют опыт, и так до появления окраски.
Следует избегать избытка аммиака, так как это может вызвать
быструю коагуляцию растворов.
Приготовив раствор лимоннокислого натрия, смешивают его
с раствором гидрохинона, в следующих количествах:
№ колбы................... 1 2
Лимоннокислый натрий, см3 80* 55
Гидрохинон, см3 .......... 0,25 0,35
3 4 5 6 7 8
40 28 20- 14 7 5
0,5 0,7 5 7 14 20
Затем в 8 цилиндров .емкостью 100 см3 (с притертыми
пробками) наливают по 20 см3 0,01 н. раствора AgNO3 и, до-
бавив соответственно в каждый цилиндр смешанные растворы
гидрохинона и лимоннокислого натрия, хорошо взбалтывают.
В цилиндр 8 добавляют каплю аммиачного раствора. Если в
первых двух-трех цилиндрах не наблюдается появление окраски
через 15—20 сек, то к ним также добавляют по капле аммиач-
ного раствора. Через несколько минут отмечают различную
окраску золей. Можно во все цилиндры добавить воды до
100 см3.
Если почему-либо неудобно получать-растворы на лекции,
то для демонстрации можно пользоваться заранее полученными
молярными растворами лимоннокислого натрия и гидрохинона,
так как при такой концентрации растворов получаются более
устойчивые золи серебра. Раствор AgNO3 и количественныё со-
отношения растворов лимоннокислого, натрия и гидрохинона,
а также ход опыта остаются без изменений. В связи с тем, что
окраска растворов в этом случае появляется не так быстро,
опыт лучше проводить накануне лекций.
Ультрамикроскопическое наблюдение броуновского движе-
ния. Наблюдение броуновского движения коллоидных частиц
с помощью ультрамикроскопа можно проводить как на раство-
рах суспензоидных золей, так и на аэрозолях.
Устройство ультрамикроскопа описано в работе 4. Для на-
блюдения броуновского движения частиц коллоидного раствора
пользуются, как и в вышеуказанной работе, стеклянной кюветой.
Наблюдение броуновского движения частиц аэрозолей с по-
мощью ультрамикроскопа — более эффектное для демонстра-
ции— проводится в металлической камере (рис. 137) с двумя
стеклянными пластинками, через одну из которых проходит
освещающий золь луч света, а через вторую ведут наблюдения
за частицами. Впустив через каучук табачный дым в камеру
и-надев каучуковую трубку на металлическую отводную трубку,
укрепляют камеру на столике микроскопа. Затем получают
электрическую дугу и наблюдают броуновское движение частиц.
316
Наблюдение под ультрамикроскопом коллоидных растворов,,
содержащих частицы различной формы. Для наблюдения под
ультрамикроскопом коллоидных частиц различной формы мож-
но взять голубой золь золота, например, приготовленный по ре-
цепту, указанному на стр. 313, и золь V2O5. При рассмотрении
золя золота^ в ультрамикроскоп видны равномерно светящиеся7
точки, тогда как палочкообразные частицы золя V2O5 представ-
ляются в виде искрящихся точек.
Золь V2Os готовят по следующему рецепту: 0,5 г аммонийной
соли ванадиевой кислоты растирают в ступке с несколькими
каплями разбавленной соляной
кислоты (1 :5).Полученный крас- »
ный осадок пятиокиси ванадия
переносят на воронку с обычным
бумажным фильтром и промы-
вают его водой до тех пор, пока
не начнет протекать темнокрас-
ный коллоидный раствор пяти-
окиси ванадия. После этого оса-
док сливают в эрленмейеровскую
колбу и доливают 200 сл3 ди-
стиллированной воды. Через несколько часов получается про-
зрачный раствор темно-красного цвета в проходящем свете.
При старении в течение нескольких дней иЬлучают золи, даю-
щие при перемешивании характерные шелковистые струи,
которые можно обнаружить при наблюдении невооружённым
глазом.
Рис. 137. Камера для демонстра-
ции броуновского движения в аэро-
золях.
ПОВЕРХНОСТНЫЕ ЯВЛЕНИЯ И АДСОРБЦИЯ
Влияние добавок поверхностно-активных веществ на поверх-
ностное натяжение воды. Влияние добавок поверхностно-актив-
ных веществ на поверхностное натяжение воды можно демон-
стрировать с помощью сталагмометра (работа 15). Для этой
цели лучше брать сталагмометр небольшого размера. В малень-
кий химический стакан наливают воды и, наполнив сталагмо-
метр, как указано в работе 15, дают ей свободно вытекать, под-
считывая число капель. Затем вносят в стакан небольшое коли-
чество (стеклянной палочкой) какого-либо поверхностно-актив-
ного вещества (например, амилового или гептилового спирта).
Наполняют полученным раствором сталагмометр и снова, при
вытекании из сталагмометра, подсчитывают число капель. По-
лучают значительно большее количество капель раствора по
сравнению с числом капель воды, вытекающей из того же объ-
ема сталагмометра.
При демонстрации этого опыта перед большой аудиторией
целесообразно пользоваться проекционным фонарем.
317
Адсорбция красителей на угле. Большую воронку с бумаж-
ным фильтром наполняют углем и фильтруют через него рас-
твор метиленового голубого. Концентрацию раствора (в зависи-
мости от адсорбционных свойств взятого угля) подбирают пред-
варительными опытами, чтобы весь краситель поглощался
углем.
Адсорбция красителя на стекле. 5% раствор метиленового
голубого наливают в колбу и взбалтывают, чтобы раствор об-
текал стенки колбы; затем выливают раствор, споласкивают
колбу водой до тех пор, пока вода не будет совершенно бесцвет-
ной; после этого в колбу приливают несколько см3 концентри-
рованной уксусной кислоты и отмечают синюю окраску.
Адсорбция брома на силикагеле и кварцевом песке. К склян-
ке 1 (рис. 138), дно которой покрыто небольшим количеством
брома, присоединяют через тройной кран 2 стеклянную труб-
ку 3, предварительно наполненную слоем силикагелй I, затем
кварца II и снова силикагеля III. Другой конец трубки при-
соединяют к водоструйному насосу. В зависимости от положе-
ния крана через трубку можно пропускать пары брома или воз-
дух. Пары брома, проходя через трубку, сильно адсорбируются
слоями силикагеля, изменяя его окраску до красно-коричневой
в то время, как кварц остается без изменений.
Если после паров брома пропустить через трубку воздух,
то вследствие десорбции брома сначала исчезнет окраска пер-
вого, а затем и второго слоя силикагеля.
Таким образом, этим опытом можно демонстрировать влия-
ние величины поверхности химически однотипных адсорбентов
на их адсорбционные свойства, а также обратимость адсорбции.
Хроматографическое разделение ионов. Демонстрацию хро-
матографического разделения ионов можно проводить, исполь-
зуя окись алюминия (для хроматографии) и растворы азотно-
кислых солей меди и кобальта.
' Смешав равные объемы 1 н. растворов азотнокислых солей
меди и кобальта, выливают полученную смесь фиолетового цве-
та в заранее приготовленную адсорбционную колонку и соеди-
318
няют с вакуумом. Сразу же появляется голубая окраска в верх-
ней части колонки и розовая — внизу. По мере протекания рас-
твора окрашенные зоны увеличиваются, розовая окраска
перемещается вниз и может быть совсем вытесненной.
Для адсорбционной колонки можно использовать трубку с
оттянутым концом (внутренний диаметр 1,5—2 см), в нижнюю
часть которой помещают кусок ваты. Приготовив концентриро-
ванную суспензию окиси алюминия, выливают ее в трубку,
которую соединяют с вакуумом (водоструйный насос). После
удаления воды колонка готова к употреблению.
ЯВЛЕНИЯ СМАЧИВАНИЯ И РАСТЕКАНИЯ
Парафинированное сито. Предварительно из медной или ла-
тунной сетки с отверстиями около 1 мм сгибают две прямо-
угольные коробки, углы которых можно пропаять оловом. Одну
из этих коробок парафинируют, опуская в расплавленный пара-
фин. Если, вынув коробку из парафина и охладив ее, обнару-
живают, что отверстия полностью покрыты парафином, то для
их освобождения снова слегка подогревают дно коробки и силь-
но дуют в нее.
На лекции наливают воду в обе коробки, демонстрируя, что
парафинированная коробка не пропускает воду. Можно поме-
стить обе коробки на поверхность воды, налитой в большой кри-
сталлизатор. Парафинированная коробка будет плавать, вто-
рая — опустится на дно. Перевернув парафинированную короб-
ку вв^рх дном, показывают, что жидкость в нее не проникает.
Плавание тяжелых тел. Алюминиевый диск толщиною
0,8—1 мм осторожно кладут на поверхность воды, налитой в
кристаллизатор. Пластинка опускается на дно. Достают ее, хо-
рошо вытирают и смазывают вазелином. После этого пластинка
не только плавает на воде, но и может быть нагружена. Так,
если диаметр диска ~30 см, то на него можно положить груз
(гирьки) ~ 100 г.
Диск можно изготовить и из другого материала, причем сле-
дует учитывать разницу в удельных весах. Например, толщина
цинкового диска должна быть приблизительно в три раза мень-
ше алюминиевого.
Чем больше диаметр диска, тем большую нагрузку он вы-
держит.
Свойства поверхностных пленок. Образование и некоторые
свойства поверхностных пленок можно демонстрировать следую-
щими опытами.
Опыт с тальком и олеиновой кислотой. Кристаллизатор, на-
полненный водой, ставят на черную подкладку. На поверхность
воды насыпают равномерно порошок талька, который не смачи-
вается водой, а плавает на поверхности, резко выделяясь на
319
темном фоне. Концом стеклянной палочки, смоченной олеиновой
кислотой, прикасаются к поверхности воды. Кислота растекает-
ся, образуя тонкую пленку, а частицы талька сдвигаются к кра-
ям кристаллизатора. Если внесенного количества кислоты не-
достаточно для образования пленки по всей поверхности, то
в месте соприкосновения палочки с водой образуется черное
круглое пятно, вокруг которого рас-
положены частицы талька.
—'"'7 Наличие поверхностного давле-
У—• [ ния при образовании поверхностных
/ __пленок можно демонстрировать сле-
дующими опытами.
Опыт Дево (лодочка .с камфо-
Рис. 139. «Лодочка» с камфо- рой). Из тонкой алюминиевой или
рой. оловянной фольги вырезают пла-
стинку (рис. 139). В угол выреза
кладут кусочек камфоры. В середину пластинки можно парафи-
ном или воском прикрепить соломинку с легким бумажным
флажком. Осторожно взяв за стерженек, переносят пластинку
на поверхность воды, налитой в большой кристаллизатор. Пла-
стинка немедленно начнет дви-
гаться по поверхности воды. Мож- х—
но, изогнув ее края, добиться,
чтобы она кружилась по воде. 11 1 ~
«Танец» камфоры. На поверх-
ность воды, налитой в чистый
кристаллизатор, высыпают мел- < -- ____
кие кусочки камфоры, не прика- жЧ ‘Х>
саясь к ним пальцами. Можно _______________ ____________<
взять кусочек камфоры за один
конец И перочинным ножом СО- Рис. 140. «Двухмерная пушка»,
скоблить ее с другого конца в
воду. Наблюдают движение камфоры, которое будет тем энер-
гичнее, чем мельче куски.
«Двухмерная пушка». Из тонкой пластинки слюды вырезают
модель двухмерной орудийной трубки и «снаряд» (рис. 140)
и помещают их на поверхность воды так, чтобы между «снаря-
дом» и «стволом пушки» был зазор в 1—2 мм. Затем в
этот зазор наносят каплю масляной кислоты. Мгновенно
снаряд выскакивает из трубки, а трубка отходит в другом на-
правлении.
«Двухмерное колесо». Из тонкой пластинки слюды выре-
зают изогнутую полоску с выемками на концах и отверстием
в середине. Затем ее парафинируют, в вырезы / и2 (рис. 141)
кладут по кусочку камфоры и, вставив проволоку в середину,
закрепляют ее в кристаллизаторе с помощью воска, парафина
или пластилина. После чего осторожно наполняют кристаллиза-
320
тор водой. При соприкосновении поверхности воды с камфорой
полоска начинает вращаться, напоминая вращение колеса.
Можно не закреплять полоску на проволоке, а положить ее
на поверхность воды. Она будет вращаться, перемещаясь по
поверхности воды в кристаллизаторе.
«Живые» капли ртути. На дно кюветы с горизонтальным
дном, наполненной ра’створом азотной кислоты (на 100 объемов
воды 20 объемов концентрированной кислоты), выкапывают из
пипетки несколько капель ртути разного размера. Затем вблизи
каждой капли кладут по кристаллику двухромовокислого ка-
лия. При растворении кристалликов окружающая их жидкость
окрашивается в оранжевый цвет. Как только окрашенный рас-.
твор дойдет до капли ртути, она
приходит в оживленное движе-
ние, причем большие по сравне-
нию с кристаллом капли движут-
ся вперед, толкая кристалл перед
собой; маленькие капли придви-
гаются к поверхности кристалла
и непрерывно дрожат.
Масляная флотация. Берут
три закрытых пробками цилинд-
ра (на 250 см3), в первые два на-
ливают поровну бензин и воду, в
Рис. 141. «Двухмерное колесо»,
третий — воду и хлороформ.
В первый цилиндр добавляют 0,5 г угля, во второй 0,5 г глины,
в третий 0,25 г глины и 0,25 г угля. Хорошо взбалтывают со-
держимое цилиндров.
В первом цилиндре уголь переходит в верхний слой (бен-
зин), во втором цилиндре частицы глины находятся в нижнем
слое — в воде, в третьем — внизу в хлороформе будет уголь,
а . наверху в воде —глина.
ЭМУЛЬСИИ
Образование эмульсин (самопроизвольное). Предварительно
приготавливают раствор следующего состава: 3 см3 касторового
масла, 25 см3 95% этилового спирта, 0,1 г олеата натрия, 0,1 г
камфоры.
На лекции из пипетки по каплям выливают раствор на
поверхность воды. Образующиеся при этом капельки масла
распределяются в • объеме, и получается молочно-белая
эмульсия масла в воде. Отбирают пипеткой небольшое количе-
ство эмульсии и рассматривают под микроскопом. Приготов-
ленная таким образом эмульсия сохраняется в течение несколь-
ких дней. ' 1
Влияние эмульгатора на устойчивость эмульсий.. Берут два
цилиндра (на 500 сл3), наливают 200 см3 воды и 50 см3 подсол-
321
нечного масла или бензола. В один из цилиндров добавляют
1 см3 10%-ного раствора сапонина и оба цилиндра встряхивают.
В цилиндре без эмульгатора эмульсия быстро разрушается, в
то время как в цилиндре с добавленным эмульгатором полу-
чается стойкая эмульсия.
Получение эмульсий типа М/В и В/М (масло в воде и вода
в масле). Берут в пробирку 4 см3 хлопкового масла'или рыбь-
его жира и добавляют к нему при нагревании очень не-
много (на кончике шпателя) красителя, Судана II или III.
После полного растворения красителя охлаждают масло и раз-
ливают в две пробирки поровну. Приливают в одну пробирку
2 см3 0,1 н. раствора NaOH, а в другую — 2 см3 0,1 и. раство-
ра СаС12. Энергично встряхивают, особенно вторую пробирку.
Затем наносят по капле на предметное стекло и, положив на
каплю покровное стекло, рассматривают в микроскоп. Эмуль-
сия М/В видна в виде красных шариков на белом фоне, тогда
как в случае эмульсии В/М видят белые шарики на красном
фоне.
Образование защитных слоев на поверхности ртути. Покрыв
дно двух кристаллизаторов чистой ртутью, наливают в один
из них воды,, в другой 1% раствор сапонина или 0,4% рас-
твор желатина (толщина слоя жидкости над ртутью должна
быть примерно в два раза больше толщины слоя ртути). За-
тем шпателем медленно разрезают ртуть, касаясь дна кри-
сталлизатора. Ртуть под водой сразу же соединяется. В кри-
сталлизаторе же с раствором ее можно разделить на отдель-
ные части.
ЭЛЕКТРИЧЕСКИЕ ЯВЛЕНИЯ НА ГРАНИЦЕ ФАЗ
Электрокапиллярные явления. «Ртутное сердце». Изменение
величины поверхностного натяжения в зависимости от заряда
поверхности можно демонстрировать на ртути. Для этого на
большое часовое стекло наливают ртуть (3—5 см диаметром),
раствор серной кислоты с двухромовокислым калием (серную
кислоту разбавляют до 20% по объему 0,1%-ным раствором
двухромовокислого калия). Затем в штативе укрепляют метал-
лический стержень с острым концом. При прикосновении метал-
лического острия стержня капля разрядится, вследствие увели-
чения поверхностного натяжения она сожмется и на некоторое
время отойдет от проводника. Затем в растворе снова заря-
дится, поверхностное натяжение уменьшится — капля расплы-
вется, подойдет к проводнику, где, разрядившись, сожмется,
и т. д. Такое уменьшение и увеличение поверхности капли про-
исходит ритмически* и продолжается довольно долго, почему
и получило название «ртутного сердца»,
322
Электроосмос (максимальное электроосмотическое подня-
тие). Для демонстрации явления электроосмотического подня-
тия жидкости очень удобен прибор, изображенный на рис. 142.
Керамическую или стеклянную диафрагму вклеивают в обойму
из плексигласа и зажимают между металлическими фланца-
ми 2. Прибор наполняют 0,001 н. раствором NaOH. В колена Г
Рис. 142. Прибор для демонстра-
ции максимального электроосмо-
тического поднятия.
ми 2. Прибор наполняют 0,001 н.
и 3 вставляют агар-агаровые клю-
чи; другой конец ключей погру-
жают в стакан с насыщенным
раствором CL1SO4, где находятся
и медные электроды. Краны слу-
жат для удобства наполнения
прибора. Капиллярные трубки
(~1,5 м длиной) вставляют в
прибор на резиновых пробках и
поддерживают в лапках на шта-
тивах. Концы капиллярных тру-
бок загнуты и к ним подвешены
маленькие стеклянные пробирки.
При наполнении прибора раствор
через краны продавливается в
капиллярные трубки примерно на
половину их длины. Ток подается
от кенотрона с напряжением
~400 в. В цепь включают мил-
лиамперметр и переключатель,
чтобы менять направление тока
в цепи.
При включении тока жидкость
в одной из трубок поднимается,
доходит до конца трубки и вы-
текает в подвешенную пробирку.
Во втором капилляре жидкость
ление тока, наблюдают поднятие жидкости в другом капил-
ляре.
Для того, чтобы лучше была видна жидкость в капилляре,
можно перед тем, как вставлять в прибор капиллярные
трубки, добавить к раствору несколько капель фенолфталеина-
и за прибором поставить лист белой бумаги.
Электрофорез. Для демонстрации явления электрофореза
можно пользоваться прибором Бэртона (рис. 143), представляю-
щим собой U-образную трубку с ответвлением, снабженным
воронкой и краном.
Прибор наполняют через воронку 1 коллоидным раствором
и вставляют на пробках -электроды. Электроды представляют
собой медные пластинки или спирали из толстой медной прово-
локи. Ток подается от кенотрона с напряжением ~ 100 в.
опускается. Изменив направ-
323
В качестве коллоидного раствора для демонстрационных
опытов в данном случае можно применить золи AgBr различ-
ного знака заряда, приготовленные следующим образом:
1) 3 см3 0,1 н. КВг разбавляют до 10 см3 водой и медленно
при встряхивании вливают в раствор AgNO3, полученный путем
разбавления 5 см3 0,1 н. AgNO3 до 40 см3;
2) 5 см3 0,1 н, КВг разбавляют водой до 40 см3 и получен-
ный раствор по каплям добавляют к раствору AgNO3, приготов-
ленному путем разведения водой 4 см3 0,1 н. AgNO3 до 15 см3.
Рис. 143. Прибор для
демонстрации электро-
фореза.
Рис. 144. Лопатка
для демонстрации
применения элек-
троосмоса в борь-
бе с прилипанием
грунтов.
Через несколько минут после включения тока наблюдают
скопление коллоидных частиц у одного из электродов.
Применение электроосмоса в технике. Можно демонстриро-
вать применение электроосмоса в борьбе с прилипанием грунтов
к металлическим частям различных механизмов. Для этого не-
обходимо изготовить металлическую лопатку (рис. 144), состоя-
щую из 3-х металлических пластинок, положенных на изоляци-
онных прокладках друг на друга. Большая по размеру ’ соеди-
няется с положительным, а две других с отрицательным, пол юсом
источника тока (20—30 в). Перемешивая лопаткой заранее при-
324
готовленную глинистую пасту, наблюдают прилипание глины
к металлической лопатке. Ведя перемешивание при пропускании
тока, отмечают легкое отставание глины от поверхности лопатки
вследствие электроосмоса.
УСТОЙЧИВОСТЬ СУСПЕНЗОИДНЫХ коллоидов И КОАГУЛЯЦИЯ
Защитное действие высокомолекулярных веществ. Для де-
монстрации защитного действия высокомолекулярного соедине-
ния ставят два цилиндра (на 200 см3) с раствором красного
золя золота; в один из цилиндров добавляют 15 см3 10%-ного
раствора желатины и перемешивают раствор. Затем из пробир-
ки с концентрированной серной кислотой стеклянной палочкой
вносят некоторое количество кислоты в цилиндр с незащищен-
ным золем золота. Сразу же при перемешивании раствора про-
исходит изменение цвета — раствор синеет.
В то же время неоднократное внесение кислоты из пробирки
к защищенному желатиной золю, не приводит к изменению
окраски." Можно даже добавить некоторое количество кислоты
прямо из пробирки, при этом цвет не меняется.
• Для лучшего наблюдения изменения окраски золя золота
можно за цилиндрами поместить лист белой бумаги.
Коагуляция цветных вод, содержащих органические веще-
ства. В бутыль емкостью 10—20 дм3 наливают воды и добав-
ляют алюминиевые квасцы или сернокислый алюминий из рас-
чета 60—100 мг на 1 дм3 (в зависимости от состава цветной
воды следует путем предварительных опытов установить необ-
ходимое для коагуляции количества квасцов). Затем раствор
перемешивают и наблюдают через 1—3 мин помутнение и осе- ’
дание хлопьев.
СТРУКТУРНО-МЕХАНИЧЕСКИЕ СВОЙСТВА КОЛЛОИДНЫХ СИСТЕМ
Образование геля (получение твердого спирта). К спирту
добавляют постепенно при перемешивании палочкой, насыщен-
ный раствор уксуснокислого кальция. Образуется гель. Если
вырезать из него кубик и поднести его к пламени горелки, то
произойдет вспышка.
Тиксотропия. Явление тиксотропии можно демонстрировать
на суспензии бентоцита. Для этого мелко размельчают бенто-
нит и, перемешивая сначала с небольшим количеством воды
при растирании в фарфоровой ступке, готовят 15—20%-ную
суспензию. Концентрация суспензии, при которой можно
наблюдать явление тиксотропии, зависит от свойств бентонита
и должна быть установлена предварительными опытами. При-
готовленную суспензию помещают в цилиндр (на 200 см3), за-
крывают пробкой и демонстрируют на лекции, периодически
встряхивая.
325
ЛИТЕРАТУРА
1. Д у м а н с к и й А. В., Учение о коллоидах, Госхимиздат, 1948.
2. Жуков И. И., Химия коллоидов, Изд. ЛГУ, 1948.
3. Млодзеевский А. Б., Лекционные демонстрации по физике, Моле-
кулярная физика и термодинамика, ОГИЗ, Гостехиздат, 1948.
4. О с т в а л ь д В., Краткое практическое руководство по коллоидной хи-
мии, ГНТИ, 1931.
5. Холмс Г., Лабораторные' работы по коллоидной химии, ОНТИ, Хим-
теорет, 1936.
6. Manegold Е„ Allgem. u. Angewan. Kolloidkunde, В. 1, 1956.
7. W е i d i п g е г Н. G., Z. f. Chem., 12, 363, 1962.
Проект- ОТКРЫТЫЙ ДОСТУП
Над оцифровкой данной книги работали:
Ружинский С.И. rygmskifaiaport.ги
Ружинский Ю.И.
Раенко А.С.
август 2005, г. Харьков, Украина
г.Харьков, ул. Чкалова 1
МП «Городок»
Популяризация применения химических добавок и оригинальных технологий в строительной индустрии.
ryginski@aport.ru
+38(057)315-32-63
Здесь может быть Ваша реклама!
Закажи книгу по бетоноведению или строительству на оцифровку и размести в ней свою рекламу.
Дополнительная информация: ryginski@aport.ru