/


























































































































































































































































































































































































































































































































Текст
с. с. воюцкий
КУРС
коллоидной
химии
2-Е ИЗДАНИЕ,
ПЕРЕРАБОТАННОЕ И ДОПОЛНЕННОЕ
Допущено Министерством высшего и среднего
специального образования СССР
в качестве учебника для студентов
химико-технологических специальностей вузов
ijHi. ыб-кДу
МОСКВА
ИЗДАТЕЛЬСТВО «ХИМИЯ» 1975
541
В 79
УДК 541.18(075.8)
Воюцкий С.С.
В 79 Курс коллоидной химии. Изд. 2-е, перераб. и
доп. М., «Химия», 1975.
512 с., табл. 22, рис. 169
Книга — второе переработанное и дополненное издание курса кол*
лоидной химии, являющегося учебником для химико-технологических
вузов (1-е издание вышло в 1964 г.). В ней изложены общие понятия и
законы коллоидной химии, опнсаиы свойства коллоидных систем, ме*
тоды их исследования и приложение коллоидной химии к решению
практических задач. Отдельная глава посвящена высокомолекулярным
веществам и их растворам. Наиболее переработаны введение, главы,,
посвященные адсорбции, и глава, в которой рассматривается устойчи*
вость и коагуляция коллоидных систем.
Книга может служить учебным пособием для студентов химиче»
ских факультетов университетов и аспирантов; она представляет ннте«
рес для научных работников, желающих ознакомиться с современным
состоянием коллоидной химии<
20503-017
050 (01)-75
17-75
541
© Издательство «Химия» 1975
СОДЕРЖАНИЕ
Предисловие .......................................................... 7
От автора.............................................................. 8
Глава I. Введение. Коллоидные системы и предмет коллоидной химии ... 9
1. Понятие о коллоидных системах и определение коллоидной химии как
науки................................................................... 9
2. Мера дисперсности...................................................15
3. Гетерогенность коллоидных систем как- основное отличие их от молеку-
лярных растворов........................................................18
4. Расклинивающее давление ............................................20
5. Влияние дисперсности на свойства дисперсных систем..................22
6. Классификации коллоидных и микрогетерогенных систем ....... 23
7. Значение коллоидных систем и коллоидных процессов в природе н тех-
нике .................................................................. 28
Глава II. Оптические свойства коллоидных систем . , . ..................33
1. Рассеяние света......................................................34
2. Абсорбция света......................................................39
3. Окраска коллоидных систем...........................................43
4. Оптические методы исследования коллоидных систем....................44
Глава 111. Молекулярно-кинетические свойства коллоидных систем .... 55
1. Тепловое движение молекул и броуновское движение . ............55
2. Диффузия в истинных растворах и в коллоидных системах..............58
3. Осмотическое давление..............................................66
4. Седиментационная устойчивость . . . ..............................оВ
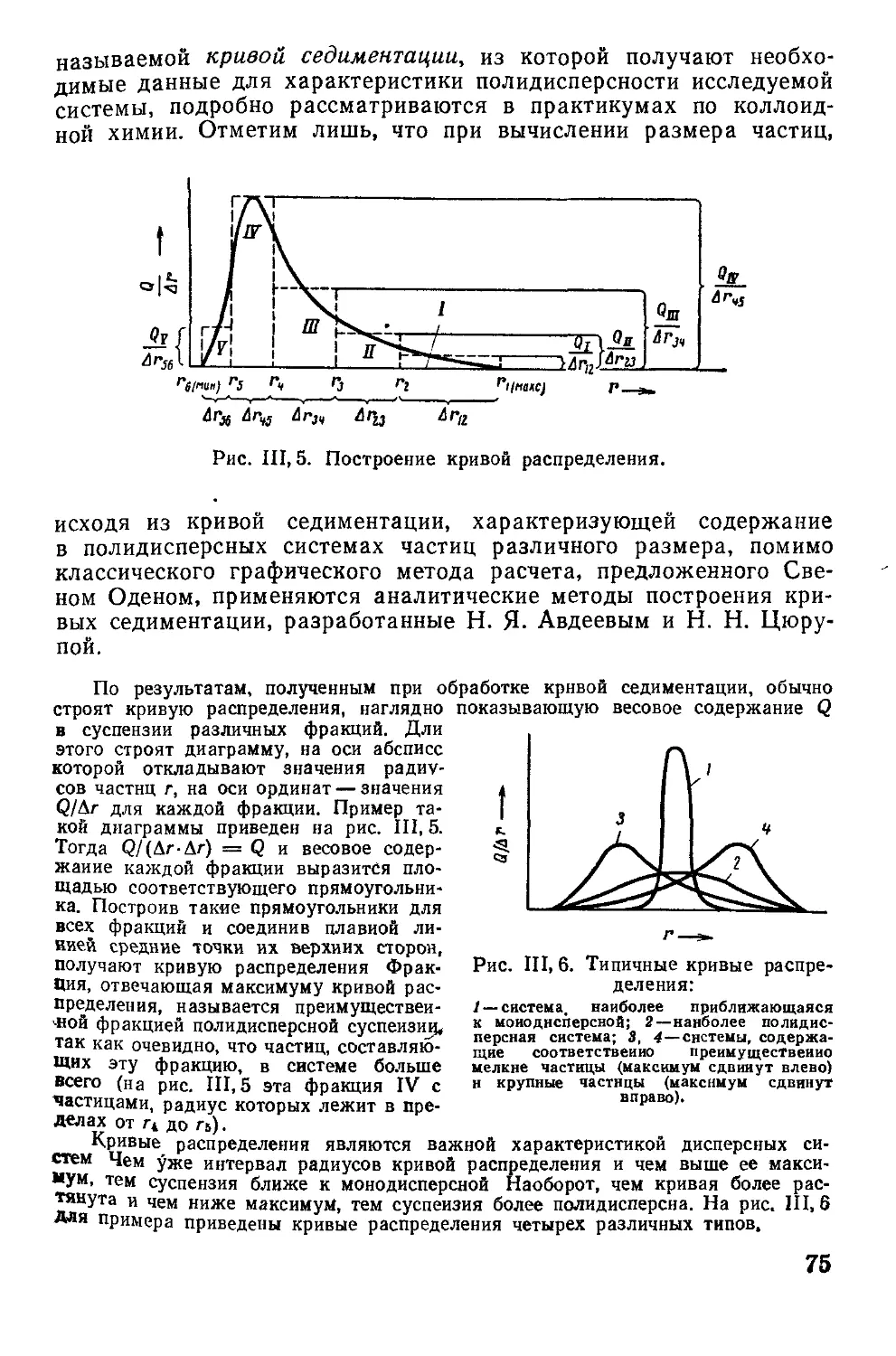
5. Седиментация и методы седиментационного анализа................... 73
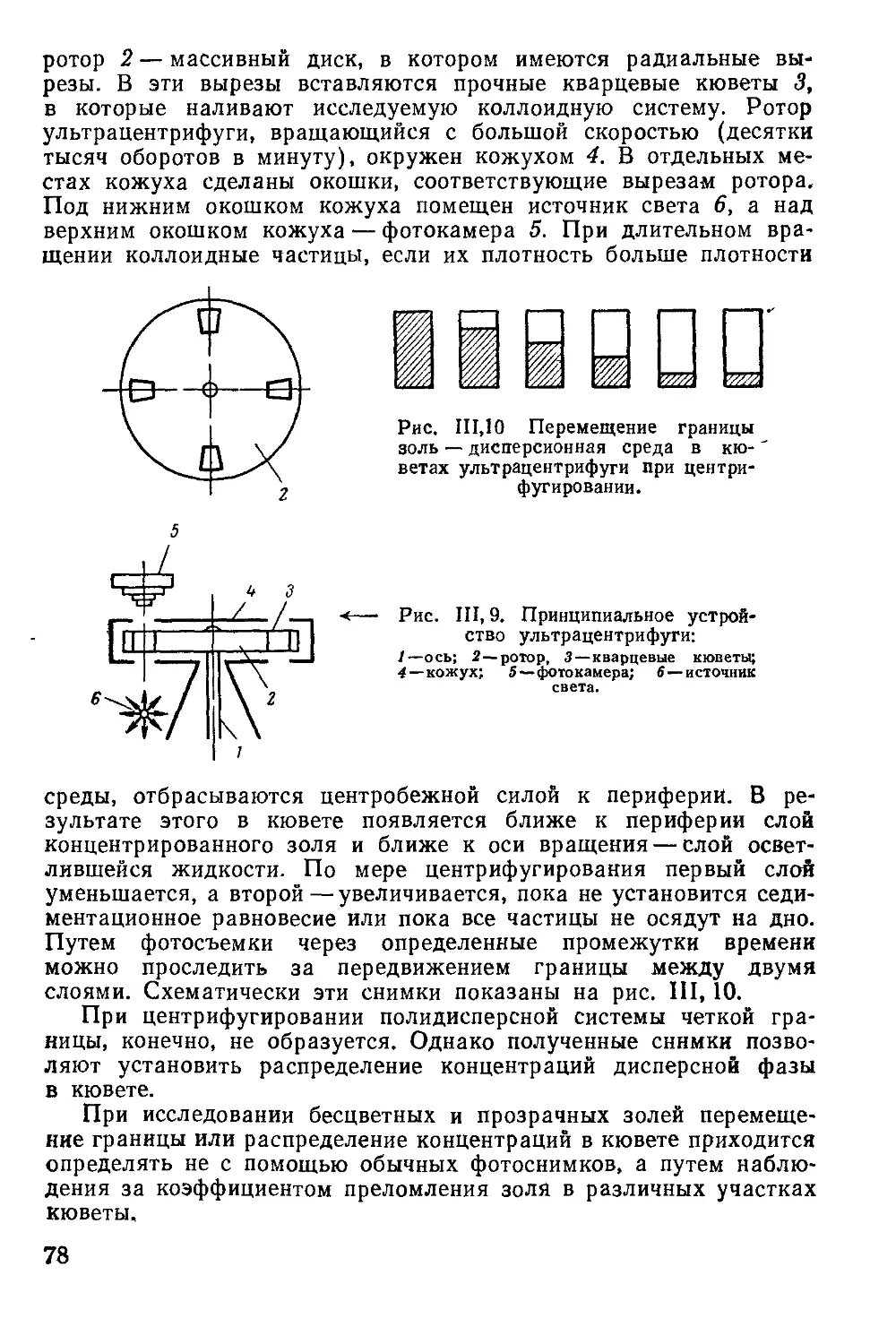
Ультрацентрифуга и ее применение для дисперсионного анализа ... 77
Глава IV. Ученйе об адсорбции. Адсорбция иа границе твердое тело — газ . 81
1. Понятие об адсорбции ............................................ 81
2. Природа адсорбционных сил..........................................85
3. Теория мономолекуляриой. адсорбции Ленгмюра....................... 88
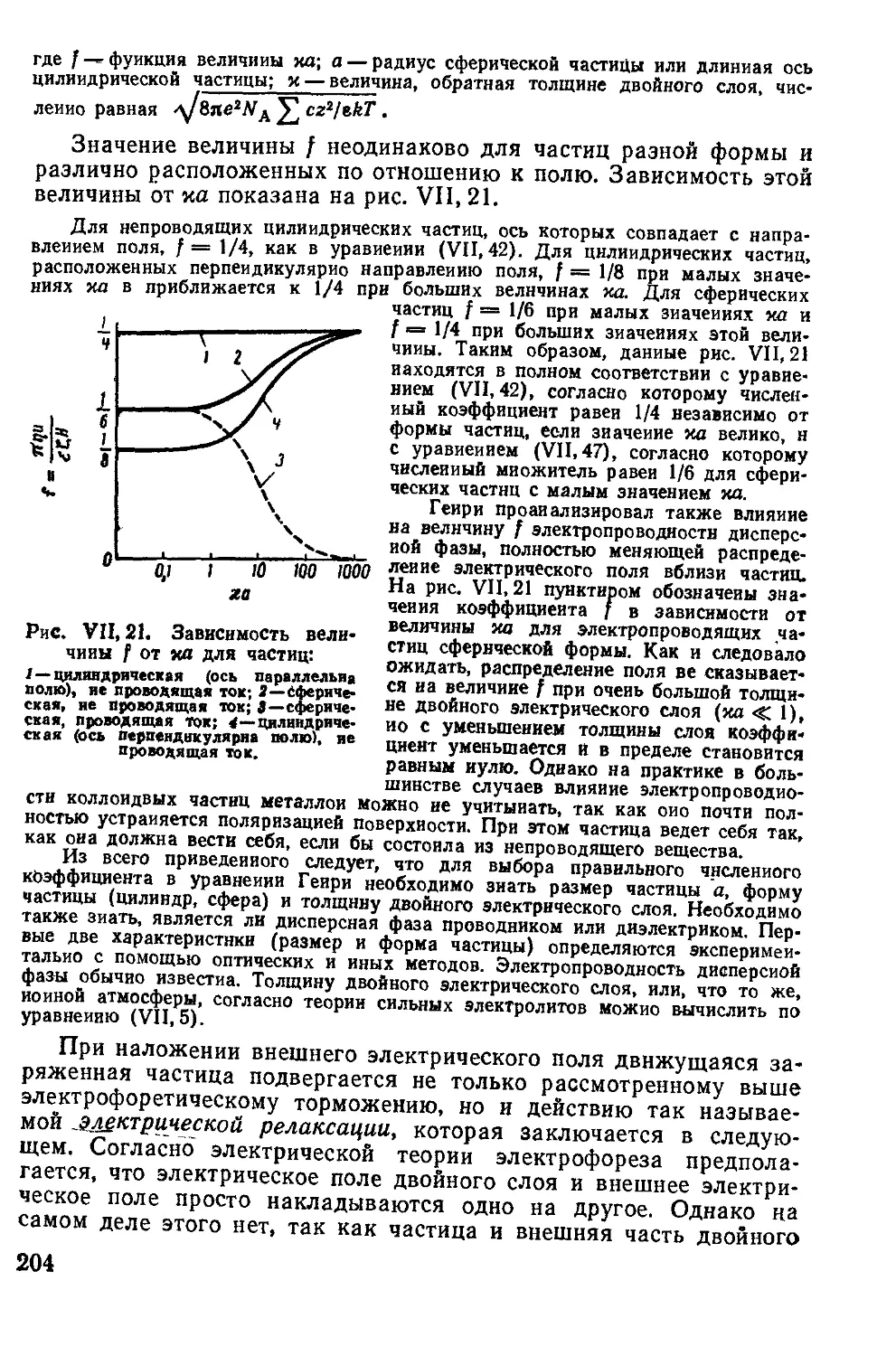
4. Теория полимолекулярной адсорбции Поляии и теория БЭТ ...... 93
5. Капиллярная конденсация........................................... 99
6. Химическая адсорбция ........................................... 103
1* 3
7. Теплота адсорбции.......................................... 105
8. Скорость адсорбции.............................................107
9. Влияние на адсорбцию свойств адсорбента и адсорбтива. Динамическая
адсорбция. Адсорбция из смесей газов .... ..................109
Глава V. Адсорбции на границе раствор — газ....................... 114
1. Поверхностное натяжение.......................... ... . . 114
2. Понятие о поверхностно-активных веществах . . ............117
3. Уравнение Гиббса...............................................120
4. Уравнение Шишковского. Переход от уравнения Гиббса к уравнению
Ленгмюра.......................................................... 124
5. Влияние на адсорбцию на границе раствор — газ строения и размера мо-
лекулы поверхностно-активного вещества. Правило Траубе ............126
6. Строение адсорбционного слоя на границе раствор — газ..........128
7. Весы Ленгмюра. Определение размера молекул поверхностно-активного
вещества ........................ . .......................132
Глава VI. Адсорбция иа границе твердое тело — раствор 137
1. Молекулярная адсорбция из растворов .......................... 137
2. Ионная адсорбция............................................. 146
3. Обменная адсорбция........................ . . ...............148
Обменная адсорбция на угле.....................................151
4. Явления смачивания ........................................... 153
5. Адгезия . . , ....................... 167
Глава VII. Электрические свойства коллоидных систем................169
4 Понятие об электрокинетических явлениях ........................ 169
Строение двойного электрического слоя...........................174
3. Влияние различных факторов на электрокинетический потенциал .... 191
i Электрофорез и электроосмос......................................197
Определение электрокинетического потенциала ................... 202
Электрофоретические методы .................................. 202
Электроосмотические методы ............. ....212
Сравнение значений ^-потенциала, найденных различными методами . . 217
в. Практическое значение электрокинетических явлений ............. 218
1. Некоторые другие электрические свойства коллоидных систем.......219
Г:лава VIII. Получение и очистка коллоидных систем. Строение коллоидных
мицелл............................................................ 223
1. Методы получения коллоидных систем............................ 223
2. Строение коллоидных мицелл......................................240
3. Примеры получения коллоидных систем........................... 245
4. Очистка коллоидных систем.......................................254
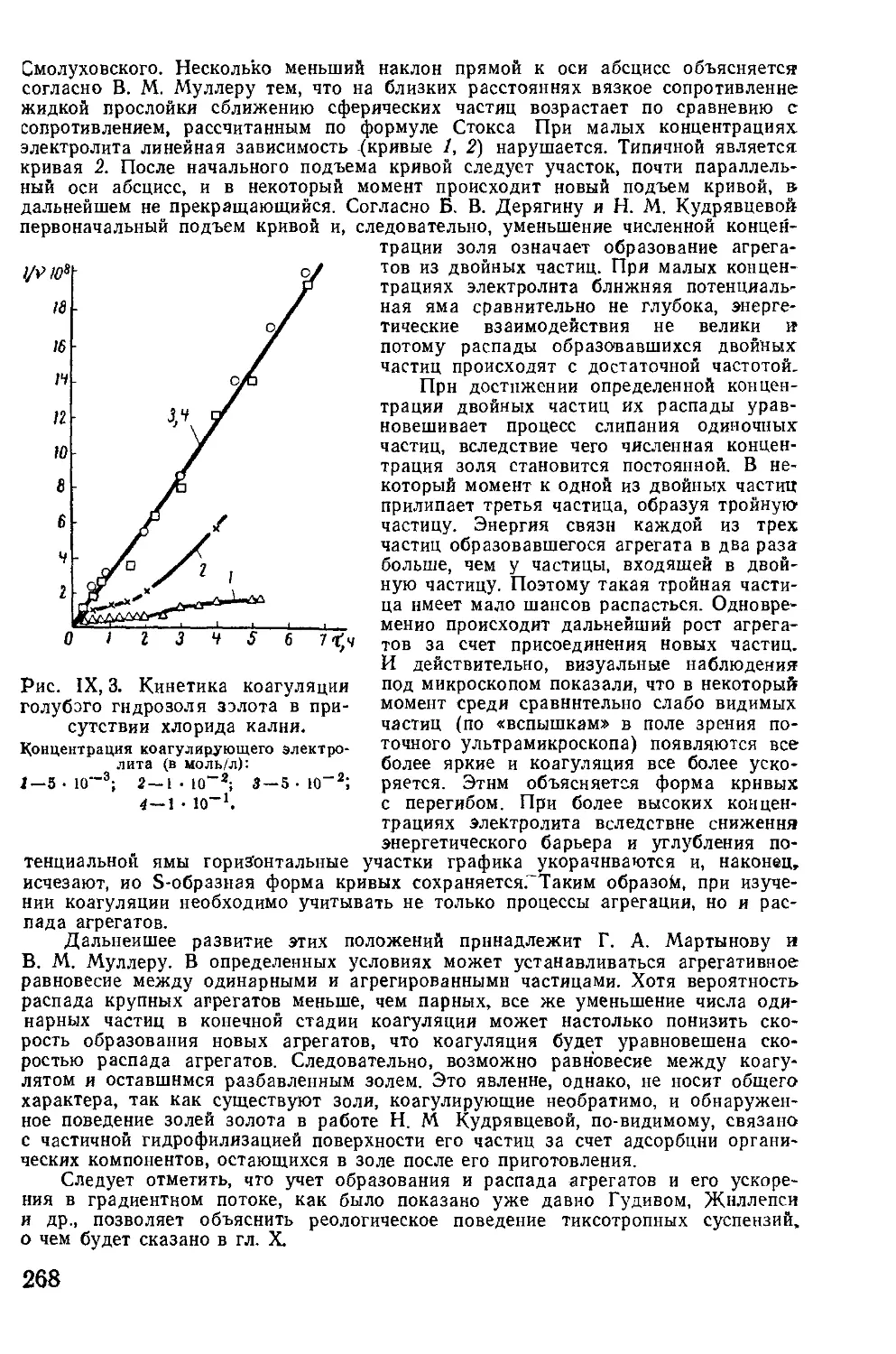
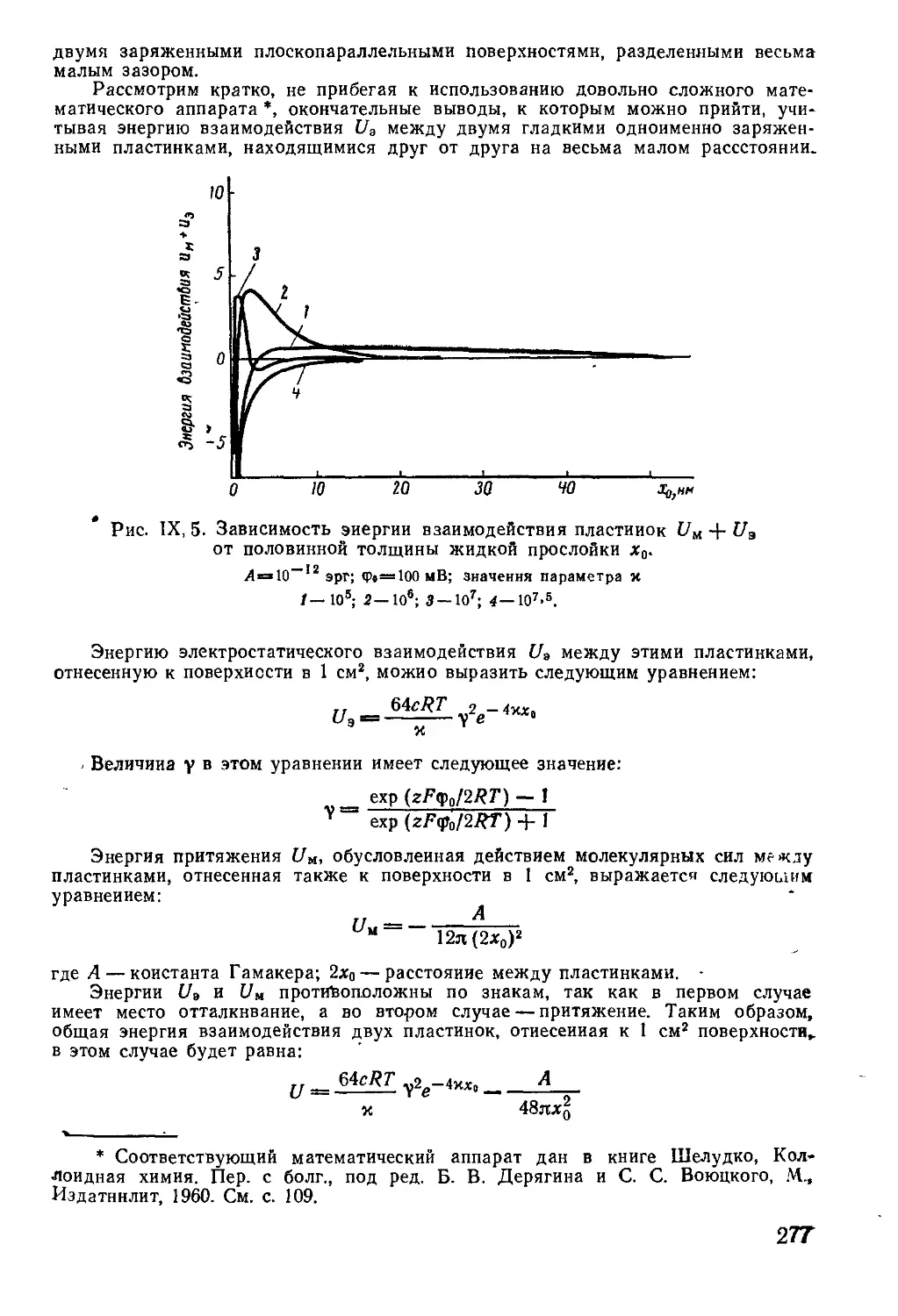
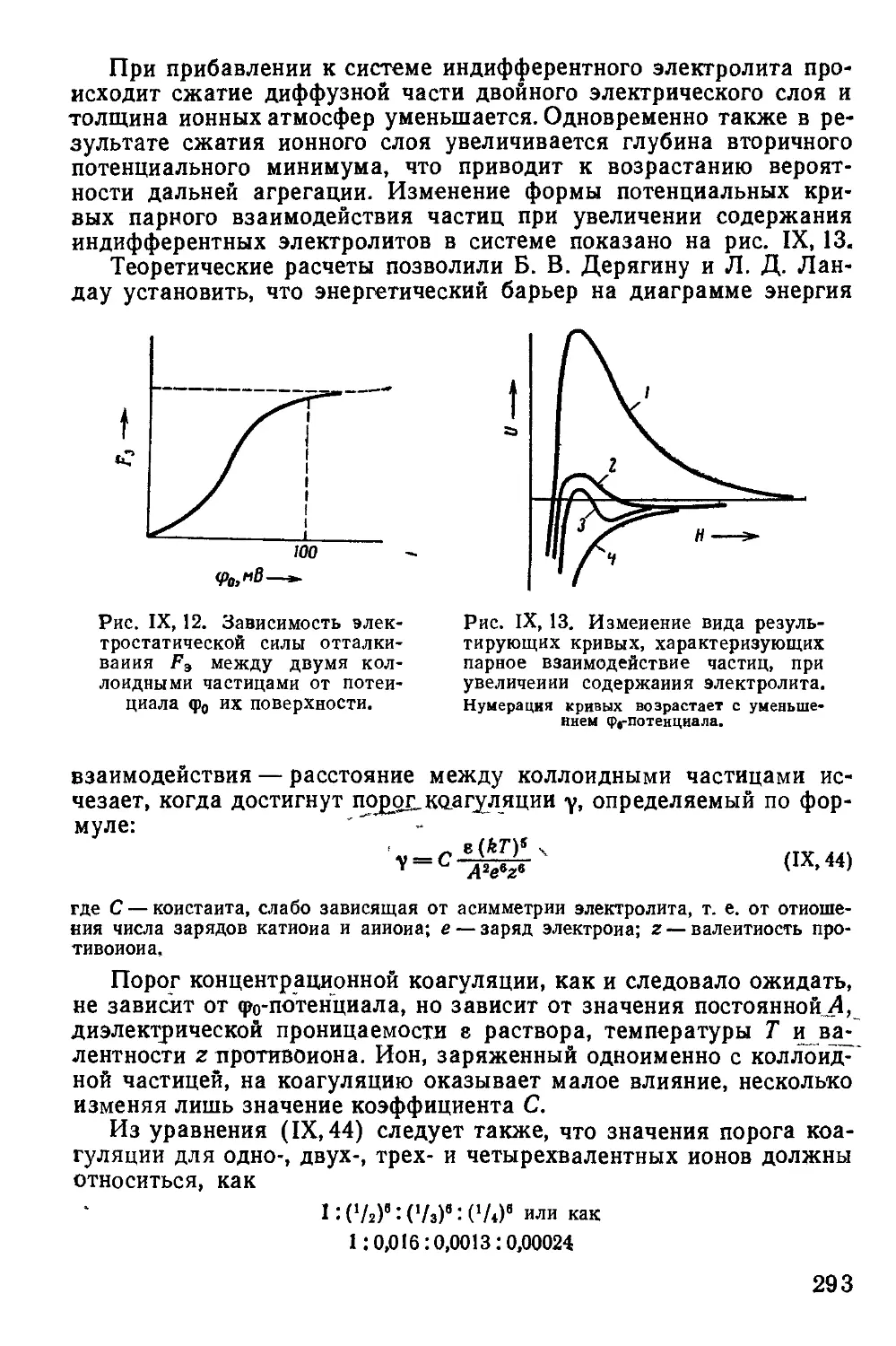
*1]лава IX. Устойчивость и коагуляция коллоидных систем............259
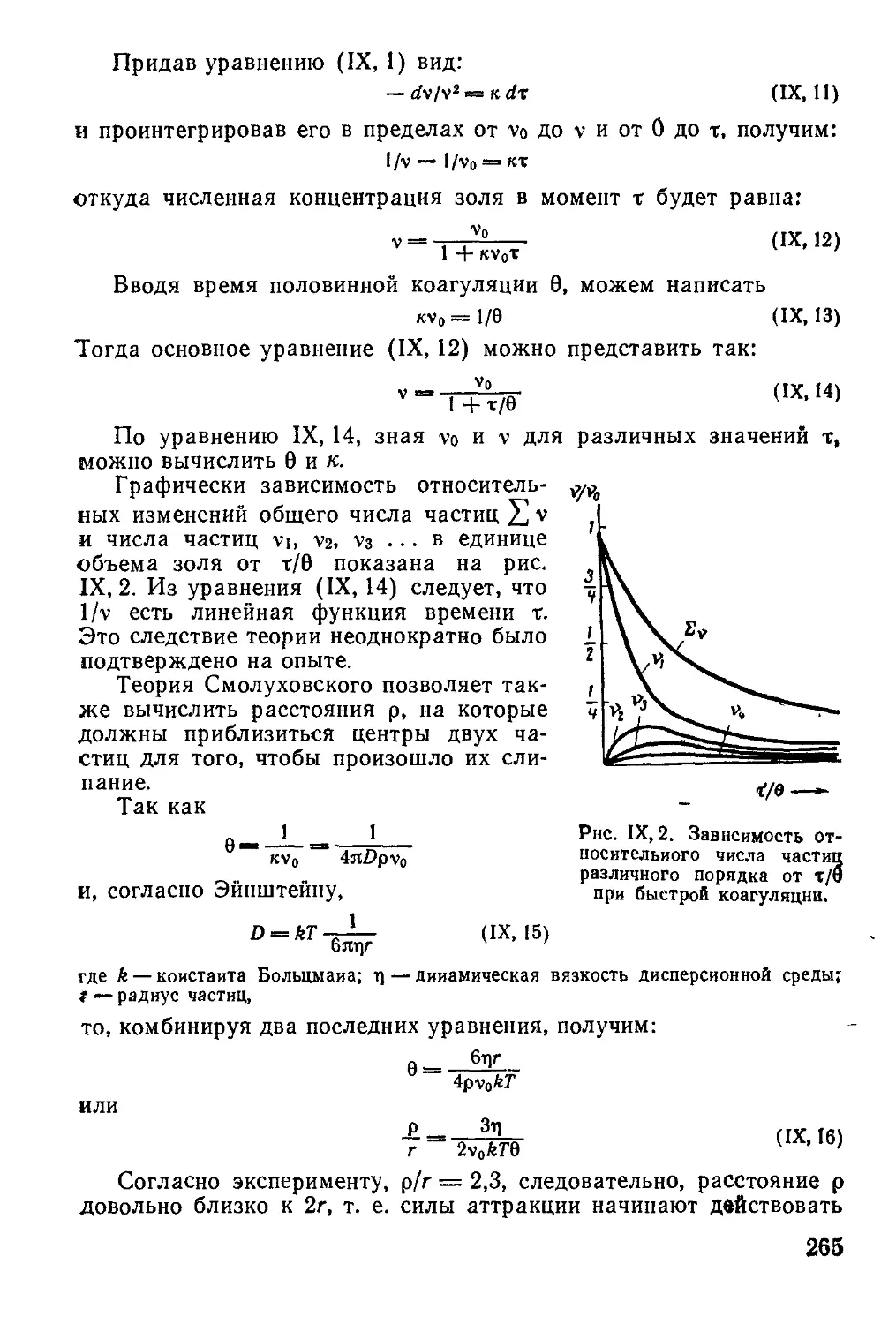
1. Кинетика коагуляции , , . , .................................. 261
2. Устойчивость тонких слоев жидкости и энергия взаимодействия между
поверхностью двух тел................'.............................269
3. Изменение энергии взаимодействия между мицеллами прн их сближении 278
4. Сольватация частиц, структурно-механический и энтропийный факторы
устойчивости ......................................................281
4
5. Правила коагуляции электролитами........................ 286
‘ 6. Теории коагуляции электролитами........................ ... 289
7. Влияние размера и концентрации частиц на их взаимодействие в дис-
персных системах........................................... 295
- 8. Значение адсорбционных явлений для коагуляции............. 296
9. Особые явления, наблюдающиеся при коагуляции электролитами . . . 300
Явления неправильных рядов ............... . . 300
Антагонизм и синергизм электролитов , .................. . , 301
Привыкание коллоидных систем ........................... . 303
Защита коллоидных частиц и сенсибилизация.............. ... 304
10. Коагуляция электролитами золей с неводной средой .........305
П. Гетерокоагуляция и гетероадагуляция коллоидных систем......307
12. Коагуляция под действием физических факторов .............308
Глава X. Структурно-механические свойства дисперсных систем...........313
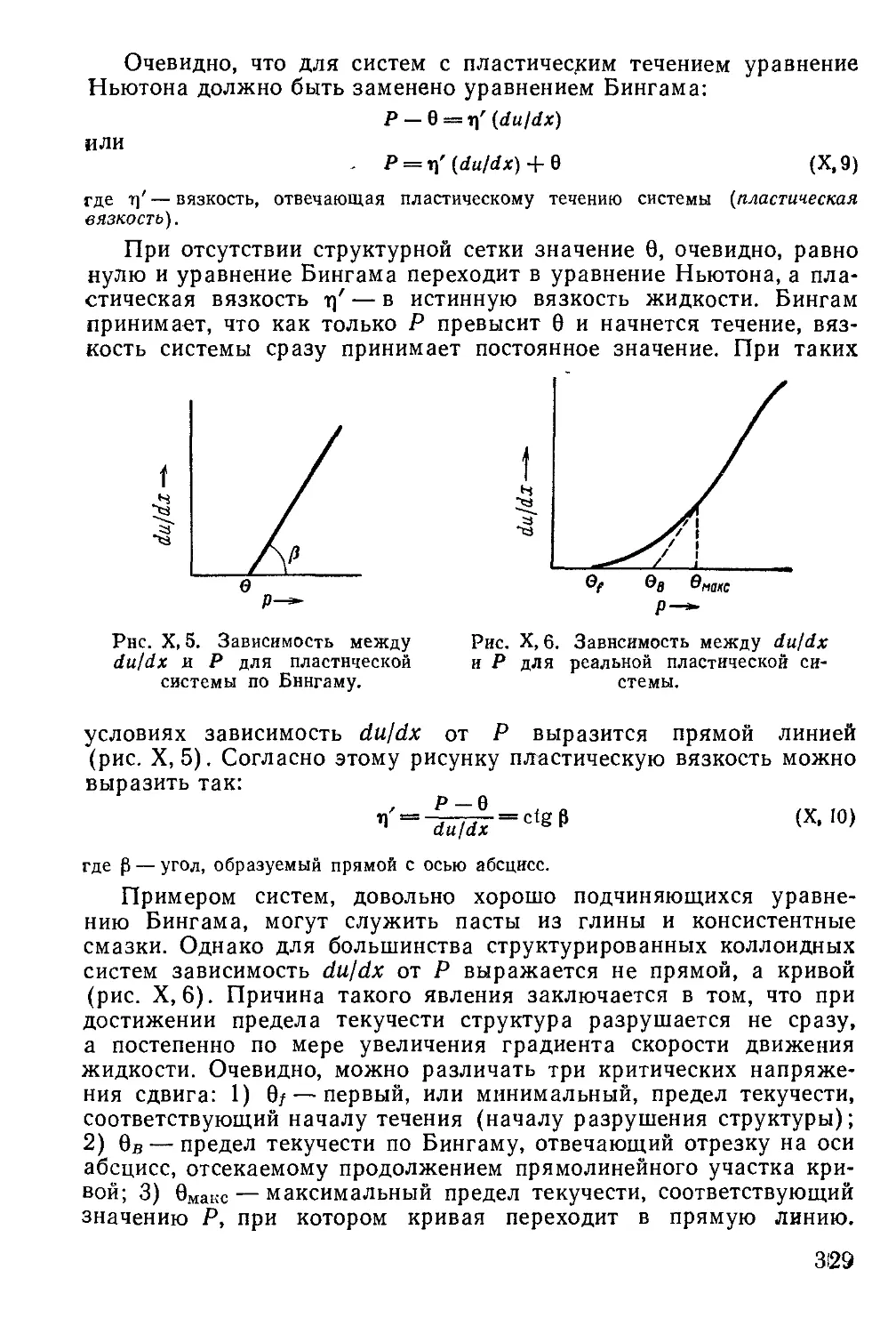
1. Возникновение и особенности структур в коллоидных системах .... 315
2. Вязкость истинных и коллоидных растворов...........................323
Определение вязкости жидкостей....................................325
Зависимость эффективной вязкости коллоидных систем от скорости те-
чения ............................................................326
3. Структурная вязкость..............................................328
4. Механические свойства коллоидных систем, проявляющих истинную упру-
гость . .......................................................... 331
Определение механических свойств коллоидных систем .... . . 334
5. Зависимость вязкости коллоидных систем от концентрации дисперсной
фазы................................................................ 335
Глава XL Системы с газовой дисперсионной средой........................340
1. Общая характеристика аэрозолей......................................340
2. Порошки и их свойства...............................................350
3. Методы получения аэрозолей..........................................356
4. Методы разрушения аэрозолей........................................360
5. Практическое значение аэрозолей.....................................364
Глава XII. Системы с жидкой и твердой дисперсной фазой..................366
1. Суспензии..........................................................366
2. Эмульсин .........................................................367
Классификация эмульсий.......................................... 368
Агрегативная устойчивость эмульсий и природа эмульгатора.....371
Методы получения и разрушения эмульсий...........................377
Обращение фаз эмульсий...........................................380
Практическое значение эмульсий и эмульгирования...............381
3. Латексы............................................................381
4. Пены..............................................................386
Устойчивость пен........................................... ' . . 387
Методы получения и разрушения пен. Практическое значение пен . . 394
5. Системы с твердой дисперсионной средой............................395
Глава XIII. Коллоидные поверхйостно-активные вещества...................399
1. Основные понятия и классификация коллоидных ПАВ....................400
2. Состояние ПАВ в растворе...........................................404
5
3. Стабилизующее действие ПАВ....................................... 410
"X Солюбилизация в растворах ПАВ.......................................412
5. Практическое значение растворов коллоидных ПАВ....................414
6. Танниды и красители............................................. 415
Глава XIV. Природа и некоторые свойства растворов высокомолекулярных
веществ..................................................416
1. Общие сведения о высокомолекулярных веществах....................417
Природные высокомолекулярные вещества...........................418
Синтетические высокомолекулярные вещества.......................419
Неорганические высокомолекулярные вещества . . . ..... 421
Промежуточные системы...........................................421
2. Полидисперсность и молекулярный вес высокомолекулярных веществ . . 423
Полидисперсность полимеров......................................423
Методы определения молекулярного веса высокомолекулярных веществ 424
Средний молекулярный вес........................................425
3. Строение макромолекул и структура высокомолекулярных веществ . . . 426
Гибкость линейных макромолекул . ...............................427
Фазовые состояния высокомолекулярных веществ ...................431
4. Теории растворов высокомолекулярных веществ .....................432
5. Термодинамика растворения высокомолекулярных веществ.............438
6. Набухание высокомолекулярных веществ _...........................442
7. Некоторые свойства растворов высокомолекулярных веществ..........451
Закон Рауля и кажущийся молекулярный вес высокомолекулярных ве-
ществ ..........................................................451
Осмотическое давление...........................................453
- Диффузия и седиментация........................................456
> Оптические свойства............................................457
Вязкость .......................................................459
Агрегативная устойчивость . , ..................................465
8. Полиэлектролиты..................................................468
Свойства растворов полиэлектролитов . ... 468
Применение полиэлектролитов ................................... 478
9. Студни..........................................................481
Застудневание........................••.........................482
Свойства студней .............................................. 486
Синерезис.......................................................490
Рекомендуемая литература по коллоидной химии 492
Предметный указатель...................................,. . . . 494
ПРЕДИСЛОВИЕ
«Курс коллоидной химии» С. С. Воюцкого в течение многих
лет являлся основным учебником для химико-технологических
институтов и химических факультетов университетов.
За это время коллоидная химия претерпела значительные из-
менения. Возникли новые проблемы и новые подходы к коллоид-
ным явлениям. Сюда следует отнести учение о расклинивающем
давлении, образование коллоидных систем в результате самодис-
пергирования, наличие периодических структур в коллоидных си-
стемах. Сильно расширилось учение о физико-химической меха-
нике. Все это заставило в значительной степени переработать
курс, введя в него новые представления, и одновременно изъять
из него устаревший материал.
Надеюсь, что учебник С. С. Воюцкого, написанный простым
и ясным языком на высоком научном уровне, будет полезен уча-
щимся химико-технологических вузов и всем желающим ознако-
миться с проблемами современной коллоидной химии.
Б. В. Дерягин
ОТ АВТОРА
Со времени выхода в свет первого издания этого учебника
прошло более десяти лет. За это время коллоидная химия в ре-
зультате трудов отечественных и зарубежных ученых претерпела
значительные изменения. Возник или сильно расширился ряд тео-
ретически интересных и практически важных разделов — учение
об устойчивости и коагуляции, физико-химическая механика, само-
произвольное диспергирование, физико-химия водных дисперсий
каучуков (латексов) и т. д.
Все это заставило автора подготовить второе переработанное
и дополненное издание учебника.
Как и в первом издании, автор стремился как можно более
приблизить излагаемый материал к практическим задачам, не
впадая, однако, в другую крайность — сделать книгу прописью
рецептов и описанием процессов различных областей технологии.
Автор старался излагать материал в понятной и ясной форме,
обращая главное внимание на физический смысл явлений.
Настоящий курс коллоидной химии соответствует программе
для химико-технологических специальностей высших учебных за-
ведений, утвержденной в 1974 г. Учебно-методическим управле-
нием Министерства высшего и среднего специального образова-
ния СССР. Кроме этого, в учебнике даны дополнительные сведе-
ния, набранные мелким шрифтом, которые будут полезны для
студентов химфаков университетов и аспирантов. В таком виде
книга, как мне представляется, будет полезна и для широких
кругов научных работников и инженеров, работающих в области
прикладной коллоидной химии.
Пользуясь случаем, автор выражает свою глубокую призна-
тельность сотрудникам кафедр коллоидной химии МГУ
им. М. В. Ломоносова, ЛГУ им. А. А. Жданова, ЛТИ им. Ленсо-
вета, МХТИ им. Д. И. Менделеева, МТИЛП, а также профессо-
рам Г. А. Мартынову, А. А. Трапезникову, Г. И. Фуксу и ряду
"других лиц за ценные советы при написании книги. Особую бла-
годарность автор выражает члену-корреспонденту АН СССР
проф. Б. В. Дерягину за его ценные указания и помощь при напи-
сании учебника.
С. С. Воюцкий
ГЛАВА I
ВВЕДЕНИЕ. КОЛЛОИДНЫЕ СИСТЕМЫ И ПРЕДМЕТ
КОЛЛОИДНОЙ ХИМИИ
1. ПОНЯТИЕ О КОЛЛОИДНЫХ СИСТЕМАХ И ОПРЕДЕЛЕНИЕ
КОЛЛОИДНОЙ ХИМИИ КАК НАУКИ
Коллоидная химия первоначально была лишь главой физи-
ческой химии. Со временем эта дисциплина чрезвычайно разрос-
лась и стала вполне самостоятельной наукой, со своим кругом
идей, лежащих в основе толкования экспериментальных фактов.
Были разработаны также специальные, вполне специфические
коллоидно-химические методы исследования — ультрамикроскопия,
электронная микроскопия, ультрацентрифугирование, электрофо-
рез и т. д. Практика показала огромное значение коллоидной
химии для современной техники. Сейчас невозможно указать от-
расль народного хозяйства, в которой в той или иной степени не
использовались бы коллоидные системы и коллоидные процессы
и не применялись бы их методы исследования. Все это и привело
к тому, что коллоидная химия выделилась в самостоятельную
дисциплину.
Чтобы ясно представить себе, чем занимается эта наука, надо
прежде всего ответить на вопрос, что такое коллоиды или кол-
лоидные системы. С коллоидными системами, встречающимися
в природе, человек имел дело с незапамятных времен. Однако
изучение этих систем началось сравнительно недавно.
Итальянский ученый Франческо Сельми в сороковых годах
XIX столетия обратил внимание на аномальные свойства некото-
рых растворов, являющихся, согласно современным представле-
ниям, типичными коллоидными системами. Эти растворы сильно
рассеивают свет; растворенные в них вещества выпадают в оса-
док от прибавления к ним даже весьма небольших количеств со-
лей, не взаимодействующих с растворенным веществом; переход
вещества в такой раствор и осаждение из него не сопровож-
даются изменением температуры и объема системы, что обычно
наблюдается при растворении кристаллических веществ. Сельми
назвал такие растворы, в отличие от обычных, «псевдораство-
рами». Позднее они получили название золей.
Английский химик Томас Грэм в начале второй половины про-
шлого века подробно исследовал свойства растворов, заинтересо-
вавших Сельми. Эти растворы, а также вещества, которые их
образуют, Грэм назвал коллоидами, так как думал, что клей,
£
называемый по гречески «колла», является типичным их предста-
вителем.
Остановимся на тех особенностях коллоидных растворов, ко-
торые были известны уже в шестидесятых годах XIX века.
1. Все коллоидные растворы способны рассеивать свет или,
как говорят, опалесцировать. Опалесценция становится осо-
бенно заметной, если, как это делал Тиндаль, через коллоидный
раствор пропускать пучок сходящихся лучей, поставив между ис-
точником света и кюветой с раствором линзу. При этих условиях
в коллоидном, растворе, наблюдаемом сбоку, виден ярко светя-
щийся конус (конус Тиндаля). Интенсивная опалесценция не
служит строгим доказательством наличия в системе межфазных
поверхностей раздела, но, безусловно, указывает на неоднород-
ность коллоидных растворов.
2. Диффузия частиц в коллоидных растворах протекает весьма
медленно.
3. Коллоидные растворы имеют очень малое осмотическое дав-
ление, которое часто даже трудно обнаружить.
Два последних свойства — замедленность диффузии и малое
осмотическое давление — указывают на то, что коллоидные рас-
творы содержат относительно крупные частицы растворенного ве-
щества. В самом деле, на диффузию влияет размер растворенных
частиц, так как с увеличением размера частицам все труднее пе-
редвигаться в среде молекул растворителя из-за возрастания тре-
ния. Осмотическое давление — это свойство коллигативное,- т. е.
зависящее при постоянной температуре только от числа частиц
в объеме, и его малое значение указывает на больший размер
частиц, так как при одной и той же весовой концентрации и оди-
наковой плотности растворенного вещества в растворе будет час-
тиц тем меньше, чем крупнее частицы.
4. Коллоидные растворы способны к диализу, т. е. с по-
мощью полупроницаемой перегородки (мембраны) могут быть от-
делены от растворенных в них примесей низкомолекулярных ве-
ществ. При диализе молекулы растворенного низкомолекулярного
вещества проходят через мембрану, а коллоидные частицы, не-
способные проникать через полупроницаемую перегородку (диа-
лизировать— по терминологии Грэма), остаются за ней в виде
очищенного коллоидного раствора. Способность к диализу также
указывает на то, что размер содержащихся в коллоидных раство-
рах частиц значительно больше размера молекул, до которых
раздроблено вещество в истинных растворах.
Следует отметить, что примеси, содержащиеся в коллоидных
растворах, могут играть весьма существенную роль. Еще
Д. И. Менделеев отметил, что ни один коллоидный раствор нельзя
получить без «подмеси» чужеродных веществ и что все попытки
получить совершенно чистые коллоидные растворы оказались
безуспешными. Позднее Д. И. Менделеев указывал, что некото-
рые вещества, считавшиеся ранее «подмесями», входят в ком-
10
плексный состав коллоидов. Значение таких примесей, служащих
стабилизаторами, т. е. веществами, без которых коллоидные рас-
творы не могут быть получены, было позднее вскрыто Н. П. Пес-
ковым.
• 5. В отличие от истинных растворов, являющихся вполне
устойчивыми (стабильными) системами, коллоидные растворы
агрегативно неустойчивы (лабильны), т. е. коллоидно растворен-
ное вещество способно сравнительно легко выделяться из рас-
твора (коагулировать) под влиянием незначительных внеш-
них воздействий. В результате в коллоидном растворе образуется
осадок (коагулят, коагулщм), представляющий собой агре-
гаты из слипшихся первичных частиц. Существенно, что агрега-
тивная неустойчивость коллоидных систем обычно проявляется
в тем большей степени, чем больше их концентрация. Поэтому
очень часто типичные коллоидные системы невозможно Получить
достаточно концентрированными.
К воздействиям, обусловливающим коагуляцию, относятся на-
гревание, замораживаниед интенсивное перемешивание и прежде
всего введение в раствор очень небольших количеств электроли-
тов (коагуляторов). При этом существенно, что коагуляция
под влиянием электролитов происходит и тогда, когда коагуля-
торы химически не взаимодействуют с коллоидно растворенным
веществом. Таким образом, коагуляция является не химическим,
а физическим процессом.
6. Коллоидные растворы обычно (но не всегда) обнаружи-
вают явление электрофореза, открытое Ф. Ф. Рейссом в Рос-
сии в 1808 г. Это явление заключается в переносе коллоидных
частиц в электрическом поле к тому или иному электроду. Следо-
вательно, частицы-коллоидно растворенного вещества, как и ионы,
могут обладать электрическим зарядом. Явление электрофореза
отличается от электролиза тем, что в последнем случае продукты
электролиза выделяются на электродах в эквивалентных количе-
ствах; при электрофорезе же происходит заметный перенос ве-
щества только в каком-нибудь одном направлении. Несоблюде-
ние при электрофорезе законов Фарадея, количественно характе-
ризующих электролиз, долгое время заставляло предполагать,
что между обоими явлениями нет прямой связи. На самом
Деле, как мы увидим - из дальнейшего, такой вывод был непра-
вильным.
Коллоидные_хистемы...могут-быть газообразными, жидкими
и твердыми. В начале настоящего курса будут рассмотрены глав-
ным образом коллоидные растворы, поскольку они наиболее изу-
чены и имеют чрезвычайно большое практическое значение.
И лишь в последующих главах мы ознакомимся с эмульсиями
и пенами, а также с газообразными «и твердыми коллоидными
системами.
В качестве примеров коллоидных систем можно привести
обычный водяной туман, дымы, коллоидные растворы металлов
И
(например, растворы платины, золота, серебра), коллоидные рас-
творы иодида серебра и сульфида мышьяка, растворы некоторых
органических красителей и мыл, эмульсии (например, молоко),
а также пемзу, рубиновое стекло, опал, чугун, некоторые сплавы
металлов.
Коллоидные свойства могут проявлять системы, состоящие не
только из неорганических, но и из органических веществ. Нако-
нец, коллоидные системы широко распространены в природе и мо-
гут быть получены в лаборатории. Следовательно, коллоидные
свойства системы не зависят от ее агрегатного состояния, ^имиче-
ской природы и происхождения. Чем же отличается всякая кол-
лоидная система от неколлоидной?
Грэм, по крайней мере в первое время, считал, что коллоиды
по своей природе отличны от обычных веществ (кристаллоидов).
Исходя из этого, он разделял все вещества на два мира — мир
кристаллоидов и мир коллоидов со своими особыми законами.
Однако это мнение Грэма оказалось неверным.
Еще современник Грэма И. Г. Борщев указывал на возмож-
ность кристаллического строения частиц, присутствующих в кол-
лоидных растворах. Позднее, в начале XX века русский ученый
П. П. Веймарн показал, что одно и то же вещество может в одних
условиях обладать свойствами кристаллоида, а в других условиях
давать коллоидные растворы. Так, канифоль при растворении
в спирте образует истинный раствор, а в воде — коллоидный рас-
твор. Наоборот, хлорид натрия в воде дает истинный раствор,
а в бензоле — коллоидный. Таким образом, правильнее говорить
не о коллоидном веществе, а о коллоидном состоянии ве-
щества.
Многочисленные исследования, проводившиеся на протяжении
многих десятилетий, показали, что коллоидное состояние веще-
ства— это высокодисперсное (сильно раздробленное) состояние,
в котором отдельные частицы являются не молекулами, а агре-
гатами, состоящими из множества молекул. Приняв это опреде-
ление коллоидного состояния (коллоидной системы), можно
сформулировать те принципиальные особенности, которые отли-
чают коллоидные системы от истинных растворов. Поскольку кол-
лоидные частицы состоят из множества молекул, то, очевидно, им
могут быть приписаны все термодинамические свойства фазы.
Равным образом молекулы среды, в которой диспергированы кол-
лоидные частицы, образуют другую фазу. Следовательно, всякий
коллоидный раствор является гетерогенной, многофазной (в про-
стейшем случае двухфазной) системой в отличие от истинных
растворов, которые являются гомогенными системами. Отсюда
же следует вывод, что поскольку всякий коллоидный раствор
представляет гетерогенную систему, условием ее образования яв-
ляется нерастворимость (или очень малая растворимость) веще-
ства одной фазы в веществе другой фазы, ибо только между такими
веществами могут существовать физические поверхности раздела.
12
В соответствии с тем, что число составляющих систему компо-
нентов равно двум или больше, коллоидные системы обычно яв-
ляются многокомпонентными. Однако в определенных условиях
могут образоваться и однокомпонентные коллоидные системы.
Примером этих систем, названных Во. Оствольдом изоколлоид-
ными, могут служить жидкости, в которых молекулы в резуль-
тате действия молекулярных сил образуют достаточно большие
агрегаты. Такие системы встречаются сравнительно редко и по-
этому в нашем курсе они не рассматриваются.
Принятое определение коллоидных систем подтверждается
рассмотренными выше характерными свойствами коллоидных
растворов. Именно такие гетерогенные системы должны сильно
рассеивать свет, обладать малой диффузионной способностью,
проявлять способность к диализу и быть агрегативно неустой-
чивыми.
Следует, однако, отметить, что твердые коллоидные системы не
обладают всеми перечисленными выше типичными коллоидными
свойствами. Так, все твердые коллоидные системы в обычных ус-
ловиях агрегативно устойчивы. Это объясняется только огром-
ной вязкостью этих систем, не позволяющей передвигаться час-
тицам растворенного вещества и образовывать более крупные аг-
регаты в результате слипания. При плавлении же этих систем
может проявляться их агрегативная неустойчивость. Металличе-
ские сплавы не обладают также опалесценцией. Но это обуслов-
ливается лишь непрозрачностью металла. Другие твердые кол-
лоидные системы, дисперсионная среда которых прозрачна (на-
пример, рубиновое стекло, опал), заметно опалесцируют. Недаром
явление опалесценции получило свое название от минерала
опала.
Поскольку из сказанного ясно, что всякая коллоидная система
представляет собою дисперсию одного тела (дисперсная
фаза) в другом (дисперсионная среда), то вообще более
правильно говорить не о коллоидах, 'а о коллоидных системах.
Таким образом, после всего изложенного можно дать опреде-
ление коллоидной химии как науки. Коллоидная химия — это
наука о свойствах гетерогенных высокодисперсных систем и
о протекающих в них процессах.
Следует подчеркнуть всю условность термина «коллоидная хи-
мия». Коллоидные системы представляют собою системы, содер-
жащие в виде дисперсных частиц не молекулы, а агрегаты моле-
кул. Наиболее типичный процесс для коллоидных систем — коа-
гуляция сводится к слипанию этих агрегатов в еще более
крупные под действием межмолекулярных, а не химических сил.
Другие процессы, характерные для коллоидных систем (физиче-
ская адсорбция, электрофорез и т. д.), также являются в основном
физическими или физико-химическими. Лишь при взаимодействии
Коагулятора со стабилизатором (веществом, находящимся в виде
адсорбционного слоя на поверхности коллоидных частиц и
13
обеспечивающим относительную агрегативную устойчивость си-
стемы) могут происходить химические реакции. Таким образом,
’ по существу коллоидная химия строится на основе двух областей
знания — физики h химии при явном преобладании первой, и толь-
ко по исторически сложившейся традиции учение о коллоидных
системах называют коллоидной химией, а не физической химией
гетерогенных высокодисперсных систем.
Рассматривая предмет коллоидной химии и коллоидные си-
стемы, необходимо указать на системы, которые, не являясь кол-
лоидными в прямом смысле этого слова, имели большое значение
для развития коллоидной науки.
Существует класс весьма важных веществ с очень большими
молекулами, так называемые высокомолекулярные соединения,
или полимеры. Сюда относятся белки, целлюлоза, каучук и ряд
синтетических продуктов. Размеры молекул этих веществ в от-
дельных случаях могут даже превышать размер коллоидных час-
тиц. Возникает вопрос, являются ли растворы этих веществ кол-
лоидными системами. Казалось бы, на этот вопрос следует отве-
тить положительно, так как эти растворы, содержащие гигантские
молекулы, обладают многими свойствами, характерными для кол-
лоидных растворов, например, способностью к диализу и малой
диффузией. Однако, как показали исследования последних деся-
тилетий, в достаточно разбавленных растворах высокомолекуляр -
ные соединения раздроблены до молекул и, следовательно, эти
растворы представляют собою гомогенные системы. Поэтому их
нельзя отнести к типичным коллоидным системам. Растворы бел-
ков, целлюлозы, каучука и других подобных веществ во избежа-
ние путаницы лучше называть не коллоидными растворами, как
это было принято раньше, а растворами высокомолекулярных ве-
ществ. Это название указывает, что данные системы, во-первых,
являются истинными растворами и, во-вторых, что в них содер-
жатся гигантские молекулы.
Несмотря на то, что растворы высокомолекулярных веществ
не являются коллоидными в точном смысле этого слова, описание
их свойств, как правило, включают в курс коллоидной химии, по-
скольку сходство ряда свойств коллоидных растворов и раство-
ров высокомолекулярных веществ позволяет рассматривать многие
проблемы одновременно для систем обоих типов. Помимо этого,
кроме типичных растворов высокомолекулярных веществ, в кото-
рых они существуют в виде больших, но не связанных друг с дру-
гом, обычно вытянутых или свернутых в весьма рыхлые клубки
молекул, известны растворы полимеров, по существу ничем не от-
личающиеся от коллоидных систем. Это растворы полимеров
в плохих растворителях; цепные молекулы в таких растворах
свернуты в компактный клубок с явно выраженной поверхностью,
на которой могут протекать явления адсорбции. Примером таких
систем являются натуральный и синтетические латексы, у кото-
рых сравнительно большие полимерные частицы находятся в вод-
14
ной среде и устойчивость которых обеспечивается благодаря ста-*
билизатору, адсорбированному на поверхности частиц. Таким об-
разом, между классическими коллоидными системами и раство-
рами полимеров не существует резкой границы.
В заключение необходимо упомянуть о коллоидных системах,
возникающих самопроизвольно (спонтанно), хотя, казалось бы,
это противоречит тому, что при образовании коллоидных систем
увеличивается межфазная поверхность, а значит, и свободная
энергия системы. Такие системы, имеющие вследствие больших
размеров частиц безусловно коллоидную природу, обнаружены
и исследованы в Советском Союзе П. А. Ребиндером и его школой
и в настоящее время привлекают пристальное внимание физико-
химиков, работающих в области коллоидной химии. Сюда сле-
дует отнести критические эмульсии, возникающие спонтанно при
температурах, близких к критической, эмульсии, представляющие
собой углеводороды с большим содержанием эмульгатора, неко-
торые неорганические дисперсные системы и т. д.
К рассмотрению спонтанного образования коллоидных си-
стем и природы растворов полимеров, имеющих явно выраженный
коллоидный характер, мы еще возвратимся в гл. VIII и XIV.
2. МЕРА ДИСПЕРСНОСТИ
Мерой раздробленности всякой дисперсной системы может
служить либо поперечный размер частиц а (для сферических
частиц — диаметр d, а частиц, имеющих форму куба, — ребро
куба /), либо обратная ему величина D = 1/а, называемая
обычно просто дисперсностью, либо, удельная поверхность зуд,
т. е. межфазная поверхность, приходящаяся на единицу объема
дисперсной фазы. Все эти величины взаимосвязаны. Чем меньше
размеры частиц, тем больше дисперсность или удельная поверх-
ность, и наоборот.
К коллоидным системам относятся системы, у которых значе-
ние а лежит в пределах 1—100 нм (10-7—10-5 см), а дисперс-
ность— в пределах 1—100 нм-1 (107—105 см-1). Верхний предел
дисперсности коллоидных систем обусловлен тем, что при даль-
нейшем дроблении вещества в растворе уже будут находиться
не агрегаты молекул, а отдельные молекулы, имеющие размер по-
рядка 0,1 нм. Нижний предел дисперсности коллоидных систем
определяется резким снижением интенсивности теплового движе-
ния частиц поперечным размером больше 100 нм. Несмотря на
установленный предел в 100 нм в курсе коллоидной химии рас-
сматриваются обычно и более грубодисперсные системы, размер
частиц которых может достигать несколько микрометров, а иногда
и значительно больше. Это целесообразно потому, что свойства
подобных систем, называемых микрогетерогенными, частицы ко-
торых хорошо видимы в микроскоп,' во многом совпадают со
свойствами коллоидных, или, иначе, ультрамикрогетерогенных
15
Рис. 1.1. Соотношение разме-
ров коллоидных частиц и моле
кул:
а—молекула водорода, d««0,l нм;
б—молекула хлороформа, J=0,8hm;
в—молекула гемоглобина, d=2,5 нм;
г, д, ж, з—частицы золотого золя,
соответственно им, 3 нм, 10 нм,
15 им н оседающие частицы.
а •
г»
«о
г □
систем, частицы которых уже не видны в микроскоп. К микрогете-
рогенным системам относятся порошки, суспензии, эмульсии, пены
и ряд других систем, имеющих огромное практическое значение.
На рис. 1,1 представлено соотношение размеров частиц золо-
того золя и некоторых молекул. Как видно из рисунка, мелкие
коллоидные частицы могут быть меньше молекул высокомолеку-
лярного вещества (например, гемоглобина) и лишь немногим
больше молекул низкомолекулярных
веществ (например, хлороформа).
Именно сравнительно малым раз-
мером коллоидных частиц опреде-
ляется сходство некоторых свойств
коллоидных систем и истинных рас-
творов. С другой стороны, относитель-
но большие размеры частиц коллоид-
ных систем объясняют их неспособ-
ность проникать через полупроницае-
мую мембрану, малую диффузионную
способность, способность оседать в до-
статочно мощном поле ультрацентри-
фуги. Более подробно особенности кол-
лоидных систем, связанные с разме-
рами частиц, будут рассмотрены в
гл. III, посвященной молекулярно-ки-
нетическим свойствам коллоидных си-
стем.
Говоря о размере частиц коллоидных систем, следует иметь
в виду два обстоятельства.
Во-первых, понятие «поперечный размер» имеет смысл для
сферических частиц и, пожалуй, еще для частиц, имеющих форму
куба. Если же частицы по форме сильно отличаются от шара, то
размер частицы зависит от направления, в котором проводят из-
мерение. Однако очень часто в коллоидной химии частицы при-
равнивают к сферическим, принимая, что эти сферические час-
тицы ведут себя в определенном отношении точно так- же, как
действительная частица. Диаметр такой условной шарообразной
частицы называют эквивалентным диаметром.
Во-вторых, в коллоидных системах частицы редко бывают од-
ного размера. Системы с частицами одинакового размера, назы-
ваемые монодисперсними системами, можно приготовить только
искусственно, пользуясь специальными приемами. Большинство
же коллоидных систем полидисперсно, т. е. содержит частицы раз-
ных размеров.
Удельная поверхность худ дисперсной системы выражается
уравнением
АС
5уд = $1, 2/^
(1,1)
где si, 2 — поверхность между фазами 1 и 2 (межфазная поверхность); V—сум-
марный объем дисперсной фазы,
16
Удельную поверхность дисперсной системы нетрудно вычис-
лить, если известны размер и форма частиц. Учитывая, что удель-
ная поверхность численно равна отношению поверхности частицы
sIt2 к ее объему tij, для системы, содержащей кубические частицы
с ребром I, имеем:
5уд = 51,2/О1 = 6/2//3==6// (1,2)
Для системы, содержащей сферические частицы радиусом г, по-
лучим:
Sya = s1,2/o1 = 4nr2/(73nr3) = 3/r = 6/d (1,3)
В общем случае:
Зуд ==«1, 2/«1 = k • 1/а = kD (1,4)
где k — коэффициент, зависящий от формы частиц.
Согласно уравнению (1,4) удельная поверхность прямо про-
порциональна дисперсности D и обратно пропорциональна раз-
меру частиц а.
С повышением дисперсности коллоидной системы ее удельная
поверхность резко возрастает. Это видно из табл. 1,1, в которой
показано изменение удельной поверхности 1 см3 вещества при
дроблении его на кубики меньших размеров.
Таблица 1,1. Изменение зуд при дроблении 1 см3 вещества
Z, см Число кубиков Объем кубика Up см3 Поверхность кубика 2» см2 5УД~5Ь2/°1
1 1 1 6 6
1 • 10“1 (1 мм) ыо3 1 • ю_3 6-10“2 6- ю1
1 • 10-2 1 • 106 1 • ю-6 6 • ю-4 6- ю2
1 • 10-3 1. 109 1 • ю-9 6 • ю-8 6 - 103
1 • 10-4 (1 мкм) ЫО12 1 • ю-12 6- ю-8 6-104
1-10-5 1.1015 ыо"18 6- ю-10 6- ю5
1 • ю~6 1 .1018 1 • ю-18 6- ю-12 6-10»
1 • 10 7 (1 нм) 1 • 1021 1 • ю-21 6- ю-14 6- ю7
По удельной поверхности коллоидные системы занимают осо-
бое положение среди дисперсных систем. В самом деле, удельная
поверхность в молекулярных системах, например в истинных рас-
творах, отсутствует, так как молекулы не обладают поверхностью
в обычном смысле слова. Вместе с тем удельная поверхность гру-
бодисперсных систем очень невелика. И лишь гетерогенные высо-
кодисперсные коллоидные системы имеют сильно развитую удель-
ную поверхность. Это наглядно показано на диаграмме (рис. 1,2),
изображающей изменение удельной поверхности с размером час-
тиц от грубодисперсных систем до систем молекулярной степени
17
дисперсности. Кривая sva = f(a) = k/a имеет вид равносторонней
гиперболы. Справа, в области грубодисперсных систем, кривая
асимптотически приближается к оси абсцисс. Слева она обры-
вается, когда коллоидные частицы достигают размеров молекул
и поверхность раздела между обеими фазами исчезает. Конечна,
Рис. 1,2. Зависимость удель-
ной поверхности системы от
размера частиц.
границу между коллоидной и молеку-
лярной степенью дисперсности нельзя
установить точно — для отдельных си-
стем она может быть сдвинута в ту
или иную сторону в зависимости от
химической природы дисперсной фазы
и дисперсионной среды.
Переход от грубодисперсных к мо-
лекулярно-дисперсным системам не-
прерывен, однако занимающие про-
межуточное положение коллоидные и
микрогетерогенные системы качествен-
но вполне специфичны. Благодаря
большой удельной поверхности этих
систем для них имеют огромное зна-
чение адсорбция и вообще поверх-
ностные явления, в то время как по-
ведение грубодисперсных и молекулярных систем определяется
в основном объемными свойствами.
3. ГЕТЕРОГЕННОСТЬ КОЛЛОИДНЫХ СИСТЕМ
КАК ОСНОВНОЕ ОТЛИЧИЕ
ИХ ОТ МОЛЕКУЛЯРНЫХ РАСТВОРОВ
Мы уже говорили о том, что агрегативная неустойчивость —>
специфическая особенность коллоидных систем. Это свойство кол-
лоидных систем имеет большое практическое значение. Не будет
преувеличением сказать, что основной задачей технолога произ-
водственного процесса, в котором имеют место коллоидные си-
стемы, является либо поддержание агрегативной устойчивости
системы, либо, наоборот, обеспечение известных условий коа-
гуляции.
Агрегативная неустойчивость является центральной проблемой
коллоидной химии, и уже в начале курса следует хотя бы в самом
общем виде рассмотреть, какие причины обусловливают агрега-
тивную неустойчивость коллоидных систем и почему многие кол-
лоидные системы, несмотря на их принципиальную агрегативную
неустойчивость, существуют весьма продолжительное время. При-
чины неустойчивости коллоидных систем могут быть объяснены
с двух точек зрения — термодинамической и кинетической.
Согласно термодинамике, агрегативная неустойчивость колло-
идных систем обусловлена достаточно большой и всегда положи-
тельной свободной поверхностной энергией, сосредоточенной на
18
межфазной поверхности системы. Поскольку поверхностная энер-
гия представляет свободную энергию и так как все системы, об-
ладающие избыточной свободной энергией, неустойчивы, это об-
условливает способность коллоидных систем коагулировать. При
коагуляции частицы слипаются, при этом межфазная поверхность
хотя бы частично исчезает и, таким образом, уменьшается сво-
бодная энергия системы. Впрочем, Смолуховский, а в последнее
время Г. А. Мартынов обратили внимание на то, что для умень-
шения свободной энергии системы непосредственный контакт час-
тиц не обязателен. Свободная энергия может уменьшаться
и тогда, когда частицы не входят в непосредственное соприкосно-
вение, а сближаются лишь на некоторое расстояние, позволяющее
им взаимодействовать через слой, разделяющий их среды.
В самом деле, пусть
F-U (1,5)
где F — свободная поверхностная энергия всей системы; st> 2 — межфазная по-
верхность; f— удельная свободная поверхностная энергия.
Величина f представляет собой сумму межфазной поверхностной энергии fa,
определяемой состоянием монослоя на границе фаз, и свободной энергии f®
вблизи поверхности, т. е. f = fa+ fv- Объемно-поверхностный вклад fv об-
условлен изменением состояния слоев жидкости вблизи поверхности раздела
фаз. Несмотря на то что вообще fa 3> fv, устойчивость системы ’в большинстве
случаев связана именно с изменением fv, так как при образовании агрегатов из
твердых частиц граница раздела фаз обычно не исчезает. Поэтому в ходе коа-
гуляции величина fa остается практически постоянной, а изменяется fv, причем
степень изменения зависит от уменьшения расстояния между частицами. Ко-
нечно, все это не относится к эмульсиям, где имеет место коалесценция, то
есть слияние частиц с полной ликвидацией первоначально разделяющей частицы
межфазной поверхности.
Поскольку коллоидные системы, обладающие большой удель-
ной поверхностью и большой свободной энергией, являются прин-
ципиально неравновесными системами, к ним неприложимо из-
вестное правило фаз. Такие системы, очевидно, всегда будут стре-
миться к равновесному состоянию, отвечающему разделению
системы на две сплошные фазы с минимальной межфазной по-
верхностью, хотя это равновесие практически может никогда и не
наступить. Термодинамическое толкование причин устойчивости
или неустойчивости коллоидных систем чрезвычайно просто. Од-
нако, как и всякая термодинамическая трактовка, это объяснение
формально, т. е. она не раскрывает сущности свойства агрега-
тивной неустойчивости. Кроме того, термодинамика не устанав-
ливает связи между свободной энергией системы и тем, как долго
система может пребывать в неравновесном состоянии. Поэтому
более полным в данном случае является объяснение агрегативной
неустойчивости или устойчивости коллоидных систем с позиций
физической кинетики.
Согласно кинетическим представлениям неустойчивость или
устойчивость коллоидной или микрогетерогенной системы опре-
деляется соотношением сил, действующих между отдельными ее
19
частицами. К таким силам относятся силы двух родов: силы сцеп-
ления, или аттракционные силы, стремящиеся сблизить частицы
и образовать из них агрегат, и силы отталкивания, препятствую-
щие коагуляции.
Силы сцепления имеют обычно ту же природу, что и межмо-
лекулярные (ван-дер-ваальсовы) силы. Существенно, что силы,
действующие между частицами, очень быстро возрастают при
сближении частиц.
Силами отталкивания могут являться электрические силы,
возникающие в результате избирательной адсорбции межфазной
поверхностью одного из ионов электролита, присутствующего
в системе. Поскольку частицы дисперсной фазы по своей природе
одинаковы и адсорбируют всегда определенный ион, все они при-
обретают электрический заряд одного и того же знака и испыты-
вают взаимное отталкивание, что препятствует сближению их на
такие расстояния, где уже могут действовать весьма значитель-
ные аттракционные силы. Другой причиной, препятствующей
сближению коллоидных частиц до расстояний, на которых начи-
нают превалировать силы сцепления, может явиться образование
на поверхности частиц сольватной оболочки из молекул среды.
Такая оболочка возникает в результате адсорбции дисперсной
фазой либо молекул среды, либо молекул или ионов третьего
компонента (стабилизатора) системы. Помимо этих двух факто-
ров существуют и другие факторы, обеспечивающие агрегатив-
ную устойчивость коллоидным системам. Подробно все факторы
устойчивости рассмотрены в гл. IX.
Таким образом, относительная устойчивость коллоидной си-
стемы определяется тем, достаточно ли велики силы отталкивания,
чтобы воспрепятствовать сближению частиц на близкие расстоя-
ния. Понятно, что такое объяснение не противоречит принципиаль-
ной неустойчивости огромного большинства коллоидных систем,
поскольку при непосредственной близости поверхностей частиц
силы сцепления, как правило, больше сил отталкивания и двум
отдельным частицам энергетически обычно выгодней образовать
агрегат. В дальнейшем мы увидим, что имеется много способов
уменьшения сил отталкивания, и в частности, одним из таких спо-
собов является введение в систему электролитов.
4. РАСКЛИНИВАЮЩЕЕ ДАВЛЕНИЕ*
При утоныпении прослойки жидкости, разделяющей поверх-
ности двух твердых тел или вообще двух любых адсорбировавших
ионы фаз, между поверхностями этих фаз возникают' силы взаи-
модействия двоякого рода. Во-первых, силы, зависящие от притя-
жения между молекулами обоих тел, между молекулами жидко-
сти и между молекулами жидкости и каждого тела (или фазы).
* Этот раздел главы иаписаи Б. В. Дерягиным.
20
Если оба тела одинаковы, то эти силы приводят к притяжению1
тел, стремящемуся утоньшить прослойку жидкости. Во-вторых,
в результате действия сил электрической природы между одина-
ковыми телами всегда возникает отталкивание, вызывающее
утолщение жидкой прослойки. Поэтому, чтобы толщина про-
слойки не изменялась и система в целом сохраняла термодина-
мическое и одновременно механическое равновесие, необходимо
приложить к поверхностям прослойки дополнительную силу,
в первом случае стремящуюся ее утолщить, во втором — утонь-
шить. Если обе фазы твердые, эти силы можно приложить к ним
непосредственно. Если фазы жидкие или газообразные, разделен-
ные жидкой перегородкой, равновесие можно поддержать, изме-
нив в них давление.
В обоих случаях эта дополнительная сила уравновесит давле-
ние в тонкой прослойке. Следовательно, равновесное значение
давления отличается от давления, которое было в прослойке до
утоньшения и которое сохраняется в объеме жидкой фазы. То из-
быточное давление, которое обнаруживает межфазная прослойка
жидкости при достаточном ее утоньшении, называется расклини-
вающим давлением и обозначается символом П(А). Понятие рас-
клинивающего давления было введено Б. В. Дерягиным в 1935 г.
Мерой величины П(Л) служит разность между давлением на про-
слойку, плоскопараллельную со стороны ограничивающих ее
фаз pi, и давлением р0 фазы, из которой образовалась прослойка
и с которой она сообщается по краям (периметру):
П (Л) = pi — р0
»
Функция П(А) имеет различный вид для разных фаз и про-
слоек, отражая закономерности изменения сил взаимодействия
между ними с изменением толщины прослойки. Следует подчерк-
нуть, что функцию П(А) можно вычислить теоретически только
для определенных случаев и лишь с ограниченной точностью. По-
этому особое значение имеют методы экспериментального опре-
деления величины П.
Закономерности расклинивающего давления были изучены
Б. В. Дерягиным с сотр. путем прямых измерений. Вначале им
было обнаружено и изучено расклинивающее давление для про-
слоек жидкостей между твердыми плоскими поверхностями, а
затем — для смачивающих пленок на различных поверхностях.
Позднее было обнаружено и исследовано расклинивающее дав-
ление пленок растворов мыл и других поверхностно-активных
веществ, помещенных между двумя газовыми средами (пузырь-
ками). Эти исследования были продолжены болгарским ученым
Шелудко, а затем английскими учеными Гайдоном и Коркилом,
американским ученым Майзельсом и др.
Вклад в расклинивающее давление могут вносить ван-дер-
ваальсовы силы (молекулярная слагающая расклинивающего
давления) и электрические силы (ионно-электростатическая
21
слагающая). Помимо этого Б. В. Дерягиным, Н. В. Чураевым,
3. М. Зориным и др. показано существование третьей «структур-
ной» слагающей расклинивающего давления. Она возникает, когда
толщина прослойки h (рис. 1,3) становится меньше суммы толщин
(hi + h2) ее поверхностных слоев, обладающих особым располо-
жением молекул, отличным от беспорядочного расположения мо-
лекул в объеме жидкости. Когда h < -ф h2, то, очевидно, часть
структурно измененных слоев, ограничивающих прослойку, должна
выдавиться в объем жидкости. При этом особая структура этих
П(П1=0 n(h) * n
Рис. I, 3. Схема, поясняющая возникновение расклинивающего да-
вления при перекрытии граничных слоев толщиной Л.
Стрелками обозначены силы расклинивающего давления для случая, когда оно
положительно.
граничных слоев разрушается, что требует затраты работы. Со-
гласно первому закону термодинамики затрата работы при утонь-
шении слоя должна быть связана с преодолением добавочного,
т. е. в данном случае расклинивающего давления.
Применяя первый закон термодинамики, мы предполагаем про-
цесс утоньшения прослойки настолько медленным, что диссипацией
энергии и затратой работы на преодоление вязкости жидкости
можно пренебречь. Расклинивающее давление никакого отношения
ни к вязкости, ни к другим механическим свойствам жидкости не
имеет. В заключение добавим, что расклинивающее давление мо-
жет быть как положительным, так и отрицательным.
5. ВЛИЯНИЕ ДИСПЕРСНОСТИ НА СВОЙСТВА ДИСПЕРСНЫХ СИСТЕМ
Многие свойства дисперсных систем весьма сильно зависят от
их дисперсности. Некоторые свойства проявляются сильнее при
переходе от грубодисперсных к высокодисперсным системам, на-
пример, способность к диффузии и осмотическое давление.
Другие свойства, наоборот, становятся заметнее с переходом от
высокой дисперсности к низкой. К таким свойствам относится спо-
собность к седиментации (оседанию) частиц. Ряд свойств прояв-
ляется при промежуточных степенях дисперсности, отвечающих
частицам коллоидных размеров. Это — светорассеяние, интенсив-
ность окраски коллоидных систем, кроющая способность пигмен-
22
тов, действие наполнителей в каучуках и т. д. Весьма интересно,
что даже твердость сплавов, представляющих собою сложную си-
стему мельчайших кристалликов, максимальна при коллоидных
размерах этих кристалликов. Есть сведения, что и каталитическое
действие проявляется наиболее сильно у катализаторов, дисперс-
ность которых соответствует коллоидной степени раздробления.
Несмотря на бесспорную связь между размером частиц и свойствами дис-
персной системы, неверно все особенности дисперсной системы объяснять
только дисперсностью, как это делал, например, немецкий ученый Во. Ост-
вальд. Исходя из допущения о примате размера частиц над всеми остальными
свойствами, Во Оствальд даже предложил называть науку о коллоидных систе-
мах не коллоидной химией, а дисперсоидологией, т. е. учением о дисперсном
состоянии материи. Советскими учеными, и в первую очередь Н. П. Песковым,
было указано, что такой взгляд является односторонним и представляет собою
чисто механистический подход. Днсперсоидология, сводившая все только к умень-
шению или увеличению размера частиц, совершенно не учитывала сложного, в
большинстве случаев сопровождающегося адсорбцией, взаимодействия частиц
дисперсной фазы с дисперсионной средой, а также возможность чисто химиче-
ских взаимодействий при коагуляции А между тем эти явления играют весьма
важную роль в коллоидных системах. Кроме того, днсперсоидология, рассматри-
вая все дисперсные системы как качественно тождественные и отличающиеся
только размером частиц, не может объяснить особые свойства, которыми об-
ладают коллоидные системы и которые отличают их как от молекулярно-дис-
персных, так и грубодисперсных систем.
Наконец, несостоятельность дисперсоидологин особенно ясно выявилась по-
сле детального исследоваия природы растворов полимеров. Согласно Во Ост-
вальду и другим представителям дисперсоидологин, все коллоидные свойства
должны обязательно проявиться у систем, содержащих частицы коллоидных
размеров. Однако, как было уже показано, растворы высокомолекулярных ве-
ществ, молекулы которых отвечают коллоидным размерам, проявляют только
некоторые свойства, типичные для коллоидных систем (оптические, молекуляр-
но-кинетические свойства), в отношении же других свойств они имеют очень
мало общего с типичными коллоидными растворами
При объяснении свойств коллоидной системы необходимо учи-
тывать не только размер частиц, но и наличие межфазной поверх-
ности, обусловливающей возможность разнообразных адсорбцион-
ных явлений, а также различные химические реакции, которые
могут протекать на поверхности частиц.
6. КЛАССИФИКАЦИИ КОЛЛОИДНЫХ И МИКРОГЕТЕРОГЕННЫХ
СИСТЕМ
В коллоидной химии, как во всякой науке, имеющей дело со
множеством объектов, необходима классификация коллоидных
и микрогетерогенных систем, чтобы разобраться во всем их много-
образии. Однако несмотря на многочисленные попытки предло-
жить единую классификацию этих систем, такая классификация до
сих пор отсутствует. Причина этого заключается в том, что любая
—предложенная классификация принимает в качестве критерия не
в<;е свойства дисперсной системы, а только какое-нибудь одно из
них. В результате, как отметил еще Н. П. Песков, на каких бы
классификациях мы не останавливались, всегда найдутся такие
23
частные случаи, для которых принятая классификация окажется
спорной или даже неприложимой.
Рассмотрим кратко те классификации, которые получили наи-
большее распространение.
Классификация по дисперсности. Зндентопф н Знгмондн предложили части-
цы, видимые в обычный микроскоп, т. е с размерами больше его разрешающей
способности (0,2 мкм), называть микронами, коллоидные же частицы, которые
невидимы в микроскоп, — ультрамнкронами. Ультрамикроны эти авторы в свою
очередь разделяли на субмнкроны, т. е. частицы размером от 5 нм до 200 нм,
обнаруживаемые с помощью ультрамикроскопа, и на амикроны, т. е. частицы
с размером меньше 5 нм, не обнаруживаемые даже в ультрамикроскопе. Зиден-
топф н Знгмондн классифицировали собственно не дисперсные системы, а со-
держащиеся в них частицы. Это принципиально не вызывает никаких возраже-
ний, однако попытки использовать принцип, предложенный Зндентопфом н Зиг-
монди, для классификации коллоидных н мнкрогетерогенных систем оказались
не удачными.
В самом деле, как мы видели, дисперсность является только одним из фак-
торов, определяющих свойства системы, н, следовательно, характеристика дис-
персных систем по размеру содержащихся в них частиц будет неполной и одно-
сторонней. Кроме того, на практике очень редко встречаются монодисперсные
системы. К полнднсперсным же системам прилагать эту классификацию невоз-
можно. Наконец, в связи с явлениями агрегации, протекающими в коллоидных
системах, размер содержащихся в ннх частиц может меняться, н, таким образом,
одну и ту же систему в разное время ее существования придется относить к
различным классам.
Классификация по агрегатному состоянию дисперсной фазы
и дисперсионной среды была предложена Во. Оствальдом. Воз-
можны девять комбинаций дисперсной фазы и дисперсионной
среды в различных их состояниях (табл. 1,2). Однако практически
можно реализовать только восемь комбинаций, поскольку газы
в обычных условиях растворимы друг в друге и образуют гомо-
генную систему.
Таблица 1,2. Классификация дисперсных систем по агрегатному
состоянию дисперсной фазы и дисперсионной среды
Дисперсная фаза Дисперсионная среда Условное обозна: чение системы Название системы
Газ Газ г/г ‘ (Коллоидная система невозможна)
Жидкость Газ . ж/г Туманы
Твердое тело Газ т/г Дымы, пыль
Газ Жидкость г/ж Пены
Жидкость Жидкость Ж/Ж Эмульсин
Твердое тело Жидкость т/ж Коллоидные растворы, суспензии
Газ Твердое тело г/т Твердые пены, пористые тела
Жидкость Твердое тело ж/т Твердые эмульсии
Твердое тело Твердое тело т/т Твердые золн, сплавы
В коллоидной химии все системы, отвечающие коллоидной стет
пени дисперсности, принято называть золями. Поэтому системы
24
Ж/Г и Т/Г имеют общее название аэрозолей. Это название ус*
ловно, так как дисперсионной средой аэрозоля может быть не
только воздух, но и любой другой газ.
Системы с жидкой дисперсионной средой, обозначаемые Г/Ж,
Ж/Ж и Т/Ж, называют лиозолями (от греч. слова лиос — жид-
кость). В зависимости от природы дисперсионной среды лиозоли
делят на гидрозоли, алкозоли, этерозоли, бензозоли (дисперсион-
ной средой этих золей являются соответственно вода, спирт, эфир,
бензол). Коллоидные системы, дисперсионной средой которых яв-
ляется органическая жидкость, объединяют под одним названием
органозоли. Микрогетерогенные системы с твердой дисперсной фа-
зой и жидкой дисперсионной средой в коллоидной химии обычно
называются суспензиями.
Классификация по агрегатному состоянию дисперсной фазы
и дисперсионной среды весьма удобна для обобщения всего мно-
гообразия коллоидных систем и, пожалуй, в настоящее время яв-
ляется наиболее общепринятой. Этой классификации в известной
степени будем придерживаться и мы в нашем курсе. Однако эта
классификация обладает существенным недостатком: с уменьше-
нием размера частиц разница в агрегатном состоянии дисперсной
фазы в различных коллоидных системах постепенно сглаживается.
Говорить об агрегатном состоянии частиц с поперечным размером
в несколько ангстрем, состоящих из сравнительно небольшого
числа молекул, с точки зрения термодинамики едва ли можно.
С этим вполне согласуется и опыт, который показывает, что па
свойствам высокодисперсных систем невозможно отличить друг
от друга золи, для приготовления которых в качестве дис-
персной фазы были использованы вещества в жидком и твердом
состояниях.
Исходя из этого, Зигмонди упростил классификацию Во. Ост-
вальда, приняв в качестве классификационного признака лишь аг-
регатное состояние дисперсионной среды. Тогда восемь возмож-
ных классов Во. Оствальда сводятся всего к трем, а именно к си-
стемам с газовой, жидкой и твердой дисперсионной средой.
Классификация по взаимодействию дисперсной фазы и диспер-
сионной среды. Эта классификация пригодна только для систем
с жидкой дисперсионной средой. К системам с газовой или твер-
дой дисперсионной средой она, очевидно, неприложима.
Зигмонди предложил классифицировать коллоидные растворы
по способности сухого остатка, полученного в результате осторож-
ного выпаривания жидкости, растворяться в чистой дисперсионной
среде. Системы, сухой остаток которых не способен самопроиз-
вольно диспергироваться в дисперсионной среде, он назвал необ-
ратимыми. Сюда относятся типичные коллоидные растворы — лио-
золи металлов, гидрозоли иодида серебра и сульЛида мышьяка
и т. д. Обратимыми коллоидными системами он назвал системы,
сухой остаток которых при соприкосновении со средой обычно сна-
чала набухает, а затем самопроизвольно растворяется и снова
25
образует коллоидную систему. К таким системам относятся, на-
пример, раствор желатина в воде или каучука в бензоле.
Сравнение Систем, являющихся представителями этих классов,
показало, что они обладают и другими, отличающими их друг от
друга свойствами. Необратимые коллоидные системы имеют при-
знаки коллоидных растворов: их трудно получить с- высоким со-
держанием дисперсной фазы; они легко коагулируют при введе-
нии в них электролитов, образуя при этом компактные, содержа-
щие малое количество дисперсионной среды осадки. Обратимые
коллоидные системы, наоборот, можно получать достаточно высо-
кой концентрации; они гораздо менее чувствительны к электроли-
там, а осадки, которые все же могут выделяться при введении
в золи лишь большого количества коагулятора, весьма объемисты,
вязки и содержат много дисперсионной среды.
Фрейндлих высказал мнение, что обратимость и необратимость
коллоидной системы определяется взаимодействием дисперсной
фазы с диспереионной средой. Дисперсная фаза обратимых колло-
идов молекулярно взаимодействует с дисперсионной средой и по-
этому способна в ней растворяться. Исходя из этого, такие колло-
идные системы Фрейндлих предложил также называть лиофиль-
ными коллоидными системами (от греч. слова лиос — жидкость,
фило — люблю). Дисперсная фаза необратимых коллоидов неспо-
собна взаимодействовать с дисперсионной средой, а следовательно,
и растворяться в ней. Поэтому эти системы Фрейндлих назвал
лиофобными* (от греч. слова фобо — ненавижу). В том случае,
когда дисперсионной средой системы является вода, эти два класса
можно называть соответственно гидрофильными и гидрофобными
системами (от греч. слова гидра — вода).
Легко видеть, что в то время как необратимые, или лиофобные,
коллоидные растворы являются типичными коллоидными систе-
мами, обратимые, или лиофильные, системы представляют собою
не что иное, как растворы высокомолекулярных соединений. В са-
мом Деле, самопроизвольно растворяться в дисперсионной среде
и давать растворы с коллоидными свойствами способны только
вещества, распадающиеся в растворах на отдельные и притом
очень большие молекулы. Такими веществами как раз и являются
высокомолекулярные соединения. Самопроизвольное образование
типичных коллоидных систем с межфазной поверхностью раздела,
как правило, невозможно, так как это противоречит термоди-
намике.
Самопроизвольное образование коллоидных систем, а также
образование гетерогенных растворов полимеров будут рассмот-
рены в гл. VIII и XIV.-Зигмонди и Фрейндлих ошибочно отнесли
* В некоторых руководствах необратимые, или лиофобные, коллоидные си-
стемы называются также суспензоидами, а обратимые, или лиофильные, си-
стемы— эмульсоидами нз-за сходства некоторых свойств этих систем с сус-
пензиями или эмульсиями. Однако эта терминология малообоснована.
26
растворы полимеров к коллоидным, так как в то время еще не
было установлено, что эти растворы являются истинными.
Таким образом, классификация Зигмонди и Фрейндлиха по су-
ществу представляет собою классификацию не коллоидных си-
стем, а дисперсных систем, содержащих частицы, отвечающие кол-
лоидным размерам, причем в эту классификацию входят как ти-
пичные коллоидные системы, так и растворы полимеров.
Здесь уместно указать, что наряду с типичными необратимыми
и обратимыми системами, согласно классификации Зигмонди и
Фрейндлиха, существуют и промежуточные системы, которые труд-
но отнести к какому-нибудь одному из обоих классов. Это, напри-
мер, золи' гидроокисей некоторых металлов: А1(ОН)з, Бе(ОН)з,
Sn(OH)4. Исследование с помощью оптических методов указывает
на присутствие в этих системах коллоидных частиц (агрегатов мо-
лекул). Имеются и другие основания считать эти системы гетеро-
генными. Вместе с тем эти системы обратимы, могут быть полу-
чены с достаточно большой концентрацией дисперсной фазы и
менее чувствительны к электролитам, чем типичные лиофобные
системы. Такие свойства этих систем обычно объясняют исключи-
тельно большой гидратацией содержащихся в них частиц. Однако
в последнее время ряд исследователей стали считать, что в этих
системах в зависимости от способа получения дисперсная фаза мо-
жет находиться как в виде коллоидных частиц, так и в виде ма-
кромолекул. Природа этих растворов до сих пор окончательно не
ясна. К этому вопросу мы еще возвратимся в гл. IX и XIV.
В последнее время термину лиофильные коллоидные системы
некоторые ученые нашей страны стали придавать существенно
иной смысл по сравнению с тем, который придавали ему ранее.
Так, лиофильными золями, по предложению П. А. Ребиндера,
стали называть не растворы высокомолекулярных соединений,
а равновесные коллоидные системы, образующиеся при опреде-
ленных условиях в результате самопроизвольного раздробления
дисперсной фазы до частиц коллоидных размеров. Такое название
вполне обоснованно, так как частицы в таких системах имеют кол-
лоидные размеры и при том взаимодействуют с дисперсионной
средой, т. е. лиофильны по отношению к ней.
Некоторые ученые предлагают называть лиофильными золи,
приобретшие агрегативную устойчивость благодаря адсорбции на
поверхности их частиц неионогенных поверхностно-активных ве-
ществ. Такие золи отличаются от ионостабилизованных коллоид-
ных систем высокой устойчивостью к действию электролитов, ко-
торая обусловлена лиофильностью поверхности частиц дисперсной
фазы, приобретенной в результате адсорбции молекул неионоген-
ных поверхностно-активных веществ.
Классификация по взаимодействию между частицами. Согласно
этой классификации дисперсные системы разделяются на свобод-
нодисперсные и связнодисперсные.
27
к свободнодисперсным системам относятся бесструктурные си-
стемы, в которых частицы дисперсной фазы не связаны друг с дру-
гом в одну сплошную сетку и способны независимо перемещаться
в дисперсионной среде под влиянием броуновского движения или
силы тяжести. Такие системы не оказывают сопротивления сдви-
говому усилию, обладают текучестью и всеми остальными свой-
ствами, характерными для обычных жидкостей. К подобным си-
стемам относятся лиозоли, достаточно разбавленные суспензии
и эмульсии, а также аэрозоли. >
В связнодисперсных системах частицы связаны друг с другом
за счет межмолекулярных сил, образуя в дисперсионной среде
своеобразные пространственные сетки или каркасы (структуры).
Частицы, образующие структуру, очевидно, не способны к взаим-
ному перемещению и могут совершать лишь колебательные дви-
жения. К таким системам относятся гели, концентрированные сус-
пензии (пасты) и концентрированные эмульсии и пены, а также
порошки. Гели могут образоваться как в результате коагуляции
коллоидных систем и объединения в одно целое выпавшего осадка
(коагели), так и вследствие молекулярного сцепления в отдель-
ных местах частиц золя, образующих сравнительно рыхлые сетки
или каркасы (лиогели). В последнем случае в гелях сохраняется
внешняя однородность системы. Естественно, образованию геля
всегда способствует повышение концентрации дисперсной фазы
в системе. Переход золя в состояние геля называется гелеобразо-
ванием.
Связиодисперсные системы, обладающие в некоторой степени
свойствами твердого тела, не следует смешивать с системами
с твердой дисперсионной средой. У последних систем частицы
также неспособны перемещаться относительно друг друга, но при-
чина здесь существенно иная, а именно, огромная вязкость дис-
персионной среды.
Приведенная классификация приложима не только к коллоид-
ным системам, но и к системам, представляющим собою растворы
высокомолекулярных веществ.
7. ЗНАЧЕНИЕ КОЛЛОИДНЫХ СИСТЕМ И КОЛЛОИДНЫХ ПРОЦЕССОВ
В ПРИРОДЕ И ТЕХНИКЕ
Коллоидные системы чрезвычайно широко распространены
в природе. Огромное значение они имеют и в современной тех-
нике. Ниже приведены некоторые примеры, характеризующие роль
коллоидных систем и коллоидных процессов в окружающем нас
мире. Много других примеров, показывающих роль коллоидной
химии в практической деятельности человека, будет дано по ходу
изложения курса *.
* Ряд интересных примеров, в которых показана роль коллоидных систем
и процессов в природе и технике, можно найти в книге Во. Оствальда «Мир
обойденных величин» (Науч.-хнм.-техн. изд. Всехнмпром, ВСНХ СССР, 1930).
Конечно, эта книга сильно устарела.
28
Коллоидные системы и коллоидные явления наблюдаются дале-
ко за пределами Земли. Как известно, межзвездная материя состоит
главным образом из газов и пыли, причем с помощью наблюдений
над поглощением света было показано, что размер пылинок, со-
держащихся в космическом пространстве, обычно не превышает
3-10-5 см, т. е. пылинки-имеют типичные коллоидные размеры.
Кометы, являющиеся газово-пылевыми облаками, представляют
собою, таким образом, колоссальные коллоидные системы, а ха-
рактерное свечение комет, возникающее в результате освещения
мельчайших частиц лучами солнца, является не чем иным, как
проявлением светорассеяния. Остается пока неясным, чем обус-
ловлено длительное существование комет — огромной разрежен-
ностью космического газово-пылевого облака и малой частотой
встреч отдельных частиц друг с другом, или относительной агрега-
тивной устойчивостью системы, определяющейся каким-нибудь
фактором, например электрическим зарядом частиц, который мо-
жет возникать вследствие адсорбции пылевыми частицами ионов.
Как теперь установлено, в космическом пространстве содержатся
большие количества ионов, образующихся в результате действия
различных излучений на молекулы газов.
Проблема создания солнечной системы, или, по крайней мере,
проблема образования планет вокруг Солнца, также имеет прямое
отношение к коллоидным явлениям, как отмечал еще Аррениус.
По космогоническим представлениям Кейпера, Юри, В. Г. Фесен-
кова, солнечная система образовалась из газово-пылевого веще-
ства. Согласно одной из теорий образования планет, развитой
О. Ю. Шмидтом, планеты возникли из газово-пылевого облака, за-
хваченного Солнцем. Известно, что в пространствах нашей Га-
лактики существует множество таких облаков, и нет оснований
считать, что окружавшее Солнце допланетное облако материи, ка-
ково бы ни было его происхождение, существенно отличалось по
составу от галактических облаков.
Правильность своей теории О. Ю. Шмидт остроумно доказы-
вает тем, что планеты имеют почти круговые орбиты. Планеты
с такими орбитами могли образоваться только путем объединения
большого числа тел, содержащихся в газово-пылевом облаке, дви-
гавшихся до того по самостоятельным эллиптическим орбитам во-
круг Солнца. О. Ю. Шмидт не рассматривал детально механизм
объединения пылевых частиц, но можно думать, что при этом су-»
щественную роль играют те же факторы, что при слипании частиц
аэрозолей. Безусловно, на процесс образования агрегатов должны
влиять поверхностные силы, наличие у частиц электрического за-
ряда и т. д. Картина, конечно, сильно усложняется тем, что га-
зово-пылевое облако находится под интенсивным действием такого
мощного фактора, как солнечное излучение во всех его видах.
Коллоидные системы и процессы имеют огромное значение для
метеорологических явлений, при образовании горных пород и ми-
нералов, в сельском хозяйстве.
29
Облака и туманы представляют собою коллоидные системы
типа Ж/Г, причем очень часто их частицы несут электрический за-
ряд. Дождь, грозовые разряды и другие метеорологические явле-
ния должны рассматриваться как явления, связанные с коллоид-
ными процессами.
В настоящее время в результате всестороннего изучения
свойств аэрозолей Б. В. Дерягиным, Н. А. Фуксом, И. В. Петря-
новым, А. Г. Амелиным и другими советскими учеными разрабо-
таны способы борьбы с пылями, дымами и туманами, а также спо-
собы искусственного вызывания осадков, что чрезвычайно важно
для сельского хозяйства.
Образование дельт при впадении рек в море также является
в значительной мере коллоидным процессом. В пресной воде рек
обычно содержится огромное число взвешенных минеральных час-
тиц с размерами, близкими к коллоидным. Эти частицы обладают
электрическим зарядом, как и большинство коллоидных частиц.
При впадении рек в море в результате смешения речной воды
с морской, содержащей значительное количество электролитов,
взвешенные частицы теряют устойчивость, слипаются друг с дру-
гом и в виде агрегатов выпадают на дно, образуя отмели.
Огромное значение имеет коллоидная химия в земледелии.
Почва является сложнейшей коллоидной системой. Размер
и форма частиц почвы, наряду с их природой, определяют водо-
проницаемость и поглотительную способность почвы, которые
в свою очередь влияют на урожайность. Пески, обладающие не-
высокой дисперсностью, легко пропускают воду, высокодисперс-
ные же глины, наоборот, хорошо удерживают влагу. Присутствие
щелочей повышает дисперсность и гидрофильность почв. В проти-
воположность этому соли кальция коагулируют почву и понижают
ее гидрофильность. На этом основано известкование почвы, при-
меняемое Для того, чтобы понизить способность почвы удерживать
влагу. В последнее время широко применяются так называемые
структурирующие агенты на основе некоторых полимеров, внесе-
ние которых в почву устраняет эрозию и придает почве желатель-
ные свойства.
В народном хозяйстве нет ни одной отрасли промышленности,
которая в той или иной степени не имела бы дела с коллоидными
системами и коллоидными процессами. Например, задачей метал-
лурга является получение металла с оптимальной микро- и уль-
трамикроструктурой, что осуществляется введением в сплав
определенных присадок. В металлообрабатывающей промышлен-
ности такие процессы, как закалка, отжиг и прокатка, также
имеют целью изменение в нужном направлении микроструктуры
металла.
Керамическое производство теснейшим образом связано с кол-
лоидной химией, поскольку основное сырье этого производства —
глиняное тесто является концентрированной суспензией гидрати-
рованных силикатов алюминия. Существенно, что качество глины
30
определяется более физическими свойствами ее частиц (размер,
форма, состояние поверхности), нежели химическими свойствами.
Другое использование глин уже в качестве компонентов рас-
творов, используемых при бурении, катализаторов, носителей ка-
тализаторов, осушителей и осветлителей различных жидкостей,
начиная от вина и кончая продуктами нефтеперерабатывающей
промышленности, разрабатывается на Украине Ф. Д. Овчаренко
с сотр.
Весьма важны закономерности коллоидной химии при созда-
нии на базе минерального сырья новых строительных материалов.
Фундаментальные исследования П. А. Ребиндера и его школы
в области вяжущих средств привели к созданию конструкционных
материалов, обладающих повышенной прочностью и рядом дру-
гих ценных свойств.
С коллоидной химией связаны и производства, перерабатываю-
щие органическое сырье. Например, технология получения бумаги
включает процессы измельчения растительного волокна до высо-
кой степени дисперсности, приготовление дисперсий различных
проклеивающих агентов (канифоли, искусственных смол, каучука)
и отложение на поверхности измельченного волокна в результате
коагулирующего действия электролитов частиц этих дисперсий,
что придает бумаге ряд ценных свойств.
Крашение волокна и дубление кожи является также примером
технологий, где основную роль играют коллоидные процессы. Кра-
шение и дубление заключается в диффузии коллоидных частиц
красителя или дубителя в ткань или голье, в коагуляции этих час-
тиц при соприкосновении с элементарными волоконцами и в фик-
сации скоагулированных частиц на элементарных волоконцах.
В производстве синтетического каучука, резины и пластмасс
коллоидные процессы играют немаловажную роль. Так, эмульсион-
ная полимеризация, в результате которой получают дисперсии
синтетических каучуков (синтетические латексы), это процесс, про-
текающий в коллоидной системе. Резина и различные пластмассы
обычно содержат мельчайшие частицы минеральных наполните-
лей, придающие им нужные свойства, и поэтому должны рассмат-
риваться как коллоидные системы.
Основной операцией производства лаков и красок является из-
мельчение пигментов в соответствующих средах до возможно бо-
лее высокой степени дисперсности. Цвет и кроющая способность
лаков или красок в большой степени зависят от размера частиц
пигмента.
Многие основные операции в фармацевтической промышленно-
сти являются по существу коллоидными процессами Например,
изготовление эмульсий, кремов, мазей сводится к диспергирова-
нию нужных веществ в подходящих средах. Некоторые лекарства
применяются в коллоидной форме. Так, колларгол представляет
собою тончайшую дисперсию препарата серебра. Введение в ор-
ганизм лекарства в коллоидной форме, во-первых, локализует его
31
действие, а, во-вторых, увеличивает срок его действия на больной
орган, так как такое вещество выводится из тканей организма го-
раздо медленнее, чем если бы оно было введено в виде обычного
раствора.
Вспомогательные операции в ряде производств очень часто
представляют собою коллоидные процессы. Например, выделение
воды из нефти на нефтеперерабатывающих заводах, разрушение
эмульсий, образующихся в химических производствах при про-
мывке того или иного жидкого продукта водой, приготовление раз-
личных эмульсий в текстильном, кожевенном и ряде других про-
изводств. Типичными коллоидными процессами являются водо-
и газоочистка. Водоочистка сводится к коагуляции взвешенных
в воде мельчайших частиц электролитами или к извлечению из
воды примесей путем адсорбции. Один из современных способов
газоочистки заключается в придании содержащимся в газе или
дыме твердым или жидким частицам достаточно большого элек-
трического заряда и затем в отложении заряженных частиц на
противоположно заряженном электроде. Более подробно такой
способ газоочистки рассмотрен в гл. XI.
Мы рассмотрели здесь значение для человека коллоидных си-
стем и коллоидных процессов, но ничего не сказали о роли в при-
роде и технике высокомолекулярных соединений, растворы кото-
рых обладают многими коллоидными свойствами. Значение высо-
комолекулярных соединений в технике будет показано в гл. XIV
настоящего курса. Здесь же только укажем, что организмы расте-
ний и животных состоят из растворов и студней высокомолекуляр-
ных веществ. Поэтому биохимия и медицина теснейшим образом
связаны с коллоидной химией. Заметим также, что многие техно-
логические процессы пищевой промышленности по существу яв-
ляются коллоидными процессами. В хлебопекарной промышлен-
ности при приготовлении теста огромное значение имеют явления
набухания, а при выпекании хлеба — явления коагуляции. Приго-
товление маргарина, соусов и майонезов представляет собою не
что иное, как процесс эмульгирования. В молочной промышлен-
ности получение простокваши и сыра является процессом коагу-
ляции и синерезиса (явление, обратное набуханию). Наконец, за-
солка и варка мяса также сводятся к явлениям коагуляции или,
точнее, денатурации белков.
ГЛАВА II
ОПТИЧЕСКИЕ СВОЙСТВА КОЛЛОИДНЫХ СИСТЕМ
Учение об оптических свойствах коллоидных и микрогетеро-
генных систем является одним из основных разделов коллоидной
химии. Оптические свойства золя определяются свойствами кол-
лоидных частиц, поэтому, изучая оптические свойства системы,
можно установить размер, форму и строение частиц, не видимых
в обычный микроскоп. С помощью ультрамикроскопических на-
блюдений коллоидных систем удалось проверить основные моле-
кулярно-кинетические представления, долгое время носившие ги-
потетический характер; изучение оптических свойств способство-
вало количественному толкованию таких процессов, как диффузия,
броуновское движение, седиментация, коагуляция. Наконец, ввиду
того, что космическая пыль, туманы, облака и тончайшие взвеси
твердых частиц в морской и речной водах являются коллоидными
и микрогетерогенными системами, сведения об оптических свой-
ствах этих систем имеют и весьма важное практическое приложе-
ние в астрофизике, метеорологии, оптике моря. Вождение самоле-
тов и кораблей в тумане, фотографирование с помощью инфра-
красных лучей также имеют непосредственное отношение к оптике
коллоидных систем. Эта область науки сделала значительные ус-
пехи в последние годы в связи с развитием авиации, астронав-
тики и т. д.
К сожалению, в нашем курсе мы не имеем возможности по-
дробно остановиться на всех оптических свойствах коллоидных
систем из-за чрезвычайной сложности вопроса и сложности необ-
ходимого математического аппарата. Поэтому в данном курсе из-
ложены лишь главнейшие явления и закономерности, наблюдаю-
щиеся при падении светового луча на коллоидную систему, и ос-
новное внимание уделено приложению этих закономерностей к ре-
шению практических задач коллоидной химии.
При падении луча света на дисперсную систему могут наблю-
даться следующие явления:
1) прохождение света через систему;
2) преломление света частицами дисперсной фазы;
3) отражение света частицами дисперсной фазы;
4) рассеяние света (это явление проявляется в виде опалес-
ценции) ;
2 Зак. 664
33
5) абсорбция (поглощение) света дисперсной фазой с превра-
щением световой энергии в тепловую.
Прохождение света характерно для прозрачных систем моле-
кулярной или ионной степени дисперсности' (газы, большинство
индивидуальных жидкостей и истинных растворов, аморфные
и кристаллические тела). Преломление и отражение света всегда
наблюдаются у микрогетерогенных систем и находят свое выра-
жение в мутности относительно грубых суспензий и эмульсий и
дымов, наблюдаемой как в проходящем (прямом), так и отражен-
ном (боковом) свете. Для коллоидных систем наиболее харак-
терны рассеяние (дифракция) и абсорбция света. Далее рассмот-
рены только эти два явления, так как первые три подробно
изложены в курсе физики.
1. РАССЕЯНИЕ СВЕТА
На опалесценцию, обусловленную светорассеянием, обратил
внимание еще Фарадей (1857 г.), а затем Тиндаль (1869 г.), на-
блюдавший образование светящегося конуса при пропускании
пучка света через коллоидный раствор.
Светорассеяние наблюдается только тогда, когда длина свето-
вой волны больше размера частицы дисперсной фазы. Если длина
световой волны много меньше диаметра частицы, происходит от-
ражение света, проявляющееся в мутности, заметной визуально.
Следует отличать светорассеяние частицами, не проводящими
и проводящими электрический ток. Рассмотрим сначала первый,
более простой случай.
Рассеянный свет имеет ту особенность, что он распространяется
во всех направлениях. Интенсивность рассеянного света в разных
направлениях различна. Если частицы весьма малы по сравнению
с длиной волны, больше всего света рассеивается под углом в О
И. 180° к лучу, падающему на частицу. Если частицы сравнительно
велики (но все же меньше длины световой волны), максимальное
количество света рассеивается в направлении падающего луча
(вперед). Кроме того, рассеянный свет обычно поляризован. При
этом для малых частиц свет, рассеянный под углом -в 0 и 180°, не
поляризован вовсе, а свет, рассеянный под углом 90°, поляризован
полностью; для крупных частиц максимальная поляризация на-
блюдается при угле, отличном от 90°.
Картину рассеяния света удобно представлять в виде вектор-
ной диаграммы, предложенной Ми. Для получения такой диа-
грамма интенсивность неполяризованного и поляризованного
света, выраженную в каких-либо единицах, откладывают в виде
радиусов — векторов во всех направлениях от точки, изображаю-
щей частицу, и концы векторов соединяют непрерывной линией.
Диаграммы Ми, характеризующие рассеяние света весьма малой
и сравнительно крупной частицей, изображены на рис. II, 1 (стрел-
кой показано направление падающего на частицу света). Внешние
34
кривые на диаграммах соединяют концы радиусов — векторов, от-
вечающих общей интенсивности рассеянного света; внутренние
кривые ограничивают отрезки векторов, соответствующие интен-
сивности неполяризованного света. Таким образом, внешняя, за-
штрихованная часть диаграммы представляет собою поляризован-
ную часть рассеянного света, а вцутреняя, незаштрихованная —
неполяризованную часть света. Приведенные диаграммы относятся
Рис. II, 1. Диаграммы Ми, характеризующие рассеяние и поляри-
зацию света весьма малой частицей (а) и крупной частицей (б).
к рассеянию света сферическими частицами. "Позднее Ганс по-
дробно рассмотрел явление рассеяния света несферическими час-
тицами.
Для сферических частиц, не проводящих электрического тока,
малых по сравнению с длиной волны падающего света и отстоящих
друг от друга на достаточно большом расстоянии (разбавленная
система), Рэлей вывел следующее уравнение, связывающее интен-
сивность падающего света /0 с интенсивностью света, рассеянного
единицей объема системы /р:
/ „2 „2 .,„2
I ~~~ Ип 1 W
\ । / л
где n,i и по — показатели преломления дисперсной фазУ и дисперсионной среды;
v — численная концентрация *; о — объем одной частицы; Л — длина световой
волны
Уравнение Рэлея применимо для частиц, размер которых со-
ставляет не более 0,1 длины световой волны, т. е. для частиц не
больше 40—70 нм. Для частиц большего размера /р изменяется
обратно пропорционально не четвертый, а меньшей степени Л. Это,
конечно, способствует увеличению светорассеяния. Геллер детально
исследовал зависимость показателя степени при X от размера ча-
стиц в основном на примере моцодисперснйх латексов полисти-
рола, размер частиц которых определялся методом электронной
микроскопии. В своих работах (1946 г.) Геллер дал калибровочную
кривую в координатах радиуса частиц и показателя степени при X.
Число частиц, содержащихся в 1 см3 коллоидной системы, называется
^частичной» или «численной» концентрацией. Однако ввиду двусмысленности
S®oro термина («частичный» как противопоставление «полному») и дальней-
м мы будем пользоваться термином «численная» концентрация.
2* 35
Для золей различных полимеров показатель степени уменьшался
от 4 до 2,8.
Когда частицы становятся настолько велики, что их размер
значительно превышает Л, светорассеяние переходит в отражение
света, не зависящее от длины световой волны.
При увеличении частиц больше определенного размера отра-
жение света от частиц возрастает, что ведет к уменьшению интен-
сивности рассеянного света. Вместе с тем по мере уменьшения
размера частиц, как следует из уравнения Рэлея, интенсивность
светорассеяния также падает. Поэтому максимальным светорас-
сеянием обладают коллоидные системы.
Из уравнения Рэлея можно сделать следующие выводы.
1. Для частиц данного размера интенсивность рассеянного
света прямо пропорциональна концентрации золя. Это положение
можно использовать для определения концентрации дисперсной
фазы с помощью измерений светорассеяния золя. Однако следует
учесть, что при очень больших концентрациях возникает много-
кратное рассеяние и в уравнение Рэлея необходимо вводить соот-
ветствующие поправки.
2. Интенсивность рассеянного света пропорциональна квадрату
объема частицы или для сферических частиц шестой степени их
радиуса. В рэлеевской области уменьшение размера частиц при
сохранении весовой концентрации золя ве-
дет к соответствующему уменьшению све-
торассеяния.
Уравнение Рэлея можно представить в
виде:
/р = ftvtl2/0
Пусть при неизменной весовой концентра-
ции дисперсной фазы объем частиц в ре-
зультате дробления уменьшится в х раз.
Тогда численная концентрация увеличи-
вается в х раз, и будем иметь:
/р = kxv (а/*)2 /о ~ (frva2/*) Д>
Рис. 11,2. Рассеяние све-
та суспензией сульфата
(?ария в зависимости от
размера ее частиц.
т. е. светорассеяние уменьшится также в х раз. Это полностью Со-
впадает с результатами эксперимента, показывающими, что чем
выше дисперсность золя, тем он меньше рассеивает свет. При мо-
лекулярной степени раздробления опалесценция становится со-
вершенно визуально незаметной.
При увеличении частиц до размера, значительно превышаю-
щего длину световой волны, светорассеяние, как было указано
выше, переходит в отражение света и по мере увеличения частиц
интенсивность рассеянного света уменьшается. На рис. 11,2 пока-
зано выраженное в условных единицах рассеяние света суспензией
сульфата бария в зависимости от дисперсности системы (при по-
стоянной весовой концентрации). Светорассеяние характеризуется
начальной, восходящей частью кривой.
36
Поскольку коллоидной степени дисперсности системы отвечает
максимальное светорассеяние, становится понятным, почему на-
блюдение опалесценции является одним из чрезвычайно чувстви-
тельных методов обнаружения коллоидной природы системы
3. При опалесценции под действием белого света при боковом
освещении бесцветные коллоидные системы обнаруживают сине-
ватую окраску. Поскольку величина /р обратно пропорциональна
X4, рассеиваются главным образом синеватые (короткие) волны.
Наоборот, в проходящем свете эти коллоидные системы окрашены
в красноватый цвет, так как при прохождении через коллоидный
раствор из спектра в результате рассеяния выбывают лучи синего
света. При освещении системы монохроматическим светом описан-
ного явления,' естественно, не наблюдается, так как при этом рас-
сеянный свет может содержать только такую же волну, что и па-
дающий.
Следует заметить, что преимущественное рассеяние света с ма-
лой длиной волны объясняет цвет неба в различное время дня,
а также цвет морской воды. Причина голубого цвета неба днем
заключается в рассеивании коротких волн солнечного света атмо-
сферой Земли. Абсолютное значение интенсивности света, рассеян-
ного 1 см3 воздуха или воды, ничтожно, но оно становится замет-
ным благодаря огромной толщине земной атмосферы и флуктуаций
газовых молекул. Оранжевый или красный цвет неба при восходе
или заходе Солнца объясняется тем, что утром или вечером на-
блюдается, главным образом, свет, прошедший через атмосферу.
На зависимости светорассеяния от длины световой волны осно-
вано также применение синего света для светомаскировки и крас-
ного света для сигнализации. Лампы синего света применяют
когда хотят, чтобы они остались незамеченными с самолетов, так
как синие лучи при прохождении через достаточно толстый слой
воздуха, особенно если в нем содержатся частицы пыли или ту-
мана, полностью рассеиваются. Наоборот, когда хотят, чтобы свет
не рассеивался и был заметен в тумане, применяют фонари, светя-
щиеся красным светом.
4. Опалесценция золей (особенно, металлических) интенсивнее,
чем растворов высокомолекулярных соединений из-за большей
плотности, а, следовательно, большего показателя преломления
дисперсной фазы первых систем. Влияние соотношения показателей
преломления дисперсной фазы и дисперсной среды на светорас-
сеяние и мутность дисперсных систем очень удобно наблюдать на
эмульсиях. Как известно, эмульсии обычно сильно мутны. Однако
эмульсии глицерина в четыреххлористом углероде, стабилизован-
ные олеатом натрия, прозрачны. Это объясняется тем, что показа-
тели преломления глицерина и четыреххлористого углерода почти
одинаковы и, следовательно, множитель в уравнении Рэлея, в ко-
торый входят коэффициенты преломления, практически равен
нулю, т. е. эмульсия глицерина в четыреххлористом углероде
практически не рассеивает свет,
37
5. Опалесценция истинных растворов весьма незначительна, так
как вследствие малого объема частиц (молекул) выражение w2
в числителе уравнения Рэлея очень невелико. Однако светорассея-
ние в этих случаях может наблюдаться при применении лучей
с малой длиной волны, например рентгеновских лучей (длина
волны рентгеновских лучей равна 0,04—0,6 нм).
Индивидуальные жидкости и газы, о коэффициентах -прелом-
ления дисперсной фазы и дисперсионной среды которых говорить
бессмысленно, казалось бы, не должны рассеивать свет. Однако
они рассеивают свет из-за флуктуации плбтности: в результате
теплового движения молекул число их в том или ином ’микрообъ-
еме системы может случайно увеличиваться на весьма малое
время, при этом число молекул в другом микрообъеме умень-
шается, что приводит к разности плотностей вещества в микро-
объемах, а это, в свою очередь, обусловливает и разность в пока-
зателях преломления.
Для растворов помимо флуктуаций плотности наблюдаются
и флуктуации концентраций, которые, конечно, тоже могут яв-
ляться причиной рассеяния света. Совершенно очевидно, что у кол-
лоидных систем частицы дисперсной фазы формально также
можно рассматривать как флуктуации концентрации с существо-
ванием, затянувшимся на неопределенно долгое время. Благодаря
такой точке зрения возможен единый подход к объяснению свето-
рассеяния индивидуальными жидкостями, истинными растворами
и коллоидными системами и применение во всех случаях уравне-
ния Рэлея. К вопросу о флуктуациях мы возвратимся в следую-
щей главе.
Все сказанное относилось к рассеянию света бесцветными кол-
лоидными частицами, не проводящими электрического тока. При
специфическом поглощении каких-нибудь лучей зависимость ин-
тенсивности светорассеяния от %4 и v2, согласно уравнению Рэлея,
нарушается, меняется степень поляризации рассеянного света
и т. д. В частице, проводящей электричество, электромагнитное
поле световой волны индуцирует электродвижущую силу. В ре-
зультате в проводнике возникает переменный электрический ток,
как и в самом электромагнитном поле. Следствием этого является
преобразование электрической энергии в тепловую. В таких условиях
короткие электромагнитные волны (от 100 до 1000 нм) практически
полностью поглощаются. Это свойство проводников, к которым
относятся металлы, и является причиной их непрозрачности.
Светорассеяние металлическими сферическими частицами де-
тально изучено Ми и Гансом. Эксперимент показал, что при осве-
щении проводящих электрический ток частиц интенсивность опа-
лесценции с уменьшением длины волны света не возрастает, а про-
ходит через максимум, характерный для каждого металла. Кроме
того, максимум сдвигается в сторону длинных (красных) волн при
уменьшении степени дисперсности и в сторону коротких (синих)
волн при ее увеличении. Опыт также показал, что для золей мно-
38
гих металлов наблюдаются индивидуальные особенности опалес-
ценции.
В заключение отметим, что с опалесценцией внешне сходна
флуоресценция, характерная для истинных растворов некоторых
красителей, например флуоресцеина, эозина и др. Она заключается
в том, что раствор при наблюдении в отраженном свете имеет
иную окраску, чем в проходящем, и в нем можно видеть такой же
конус Тиндаля, что и в типичных коллоидных системах. Однако
это по существу совершенно различные явления. Опалесценция воз-
никает в результате рассеяния света, при этом длина волны рас-
сеянного света та же, что и падающего. Флуоресценция же пред-
ставляет собою внутримолекулярное явление, заключающееся
в селективном поглощении молекулой вещества светового луча
и в трансформировании его в световой луч с другой, большей дли-
ной волны. Существенно, что опалесценцию возбуждает любой
свет, в то время как флуоресценция обусловливается светом опре-
деленной длины волны, характерной для данного флуоресцирую-
щего вещества.
2. АБСОРБЦИЯ СВЕТА
В 1760 г. Ламберт, а еще ранее Бугер, изучая рассеяние света,
установили следующую зависимость -между интенсивностью про-
шедшего света и толщиной среды, через которую этот свет прошел:
= (П.2)
где 1а — интенсивность прошедшего света, 10 — интенсивность падающего света;
k — коэффициент поглощения, I — толщина поглощающего слоя
Согласно закону Бугера — Ламберта, если толщина слоя среды
растет в арифметической прогрессии, то интенсивность прошед-
шего света уменьшается в геометрической. Иначе говоря, поглоще-
ние во всех слоях, на которые мысленно можно разделить данную
среду, происходит таким образом, что каждый последующий слой
поглощает ту же долю проходящего света, что и предыдущий.
Бэр показал, что коэффициент поглощения растворов с абсо-
лютно бесцветным и прозрачным растворителем пропорционален
молярной концентрации с растворенного вещества:
k = ес
Вводя значение молярного коэффициента поглощения е в урав-
нение Бугера — Ламберта, получим закон Бугера — Ламберта —
Бэра:
/п = Л#ес/ (П,3)
устанавливающий зависимость интенсивности прошедшего света
iSr толщины слоя и концентрации растворенного вещества. Лога-
рифмируя уравнение (11,3), получим
1п (70//п) = ес/ (П,-4)
39
Левую часть этого уравнения называют оптической плотностью
раствора D или экстинкцией.
При работе с монохроматическим светом всегда следует ука-
зывать, при какой длине волны была определена оптическая плот-
ность, и обозначать ее через где индекс Л указывает на длину
волны света, примененного для определения.
Выражение /п/^о называют светопропусканием раствора или от-
носительной прозрачностью раствора.
Левую часть выражения:
*
А — At । _е— e.cl
Л)
принято называть относительным поглощением раствора.
Молйрный коэффициент поглощения, являющийся постоянной,
характерной для данного вещества величиной, можно легко опре-
делить, если с — 1 и I — 1. Тогда
е=1пф- (11,5)
Если е = 0, раствор не абсорбирует света, и в соответствии
с этим уравнение Бугера — Ламберта — Бэра примет вид:
/п = /0 (П,6)
т. е. интенсивность прошедшего света будет равна интенсивности
падающего.
Молярный коэффициент поглощения е зависит от длины волны
абсорбируемого света, температуры и природы растворенного ве-
щества и растворителя и, как правило, не зависит от концентра-
ции раствора. Однако возможны исключения, когда е изменяется
при разбавлении раствора. Это объясняется изменением химиче-
ских свойств системы — происходит гидролиз, образование гидра-
тов или ассоциация. Все это, конечно, может влиять на коэффи-
циент поглощения е.
Закон Бугера — Ламберта — Бэра, выведенный для гомоген-
ных систем, неоднократно пытались применить к коллоидным рас-
творам. Опыт показал, что дляузолей высокой дисперсности он
вполне приложим, если только слой жидкости не слишком толст,
а концентрация раствора не очень большая. Вопрос о приложении
этого закона к сравнительно низкодисперсным сильно опалесци-
рующим золям более сложен.
Размер частиц дисперсной фазы не входит в уравнение Бугера — Ламбер-
та — Бэра, и поэтому на первый взгляд кажется, что дисперсность золя не
должна влиять на его способность абсорбировать свет Однако влияние раз-
мера коллоидных частиц на абсорбцию света сказывается косвенно — через
светорассеяние Дело в том, что в результате светорассеяния проходящий белый
свет теряет часть излучения (главным образом, коротковолнового), что и вос-
принимается наблюдателем как абсорбция В отличие от истинной абсорбции
света, когда световая энергия абсорбируется системой и превращается в теп-
ловую, такая абсорбция, вызванная светорассеянием, называется фиктивной.
40
в этом случае уравнение (II, 3) принимает вид1
Zn = ехр [— (в + К'} cl]
(И. Л
где /('— коэффициент фиктивной абсорбции, обусловленной светорассеянием
Так как светорассеяние зависит от размера частиц, то, следовательно:
где г — радиус частиц
Если е = 0, т е золь белый, уравнение (11,7) с учетом фиктивной абсорб-
ции принимает вид
/п = /0 exp (— К’сГ) = Zo exp (— К"сЦМ)
(II, 8)
поскольку, по Рэлею, светорассеяние обратно пропорционально %4 При фиктив-
ной абсорбции золь, естественно, будет иметь в проходящем свете оранжевую
окраску, а в рассеянном—голубоватую В обоих случаях окраска, конечно, не
является собственно окраской вещества золя, а обусловлена рассеянием света
Опытная проверка уравнения (11,8) подтвердила его справедливость Ис-
следование зависимости фиктивной абсорбции от дисперсности золя показало,
что с увеличением размера частиц общее поглощение сначала растет, а затем,
достигнув максимума, начинает падать.
Металлические золи в отношении абсорбции света, так же как
и в отношении светорассеяния, обнаруживают аномальное поведе-
ние по сравнению с остальными коллоидными растворами. Как
и светорассеяние, абсорбция металли-
ческими золями достигает максимума
при определенных значениях длины
волны и радиуса частиц. На^рис. 11,3
показана зависимость абсорбции све-
та золями золота от длины волны па-
дающего света и от дисперсности золя.
Как видно, при увеличении степени
дисперсности золя максимум на кри-
вой светопоглощения сдвигается в сто-
рону коротких волн, причем значение
этого максимума сначала возрастает,
а затем падает.
Существрвание максимума на кри-
вой светопоглощения объясняется ча-
500 000 юо
Л, нм
Рис. 11,3. Зависимость абсорб-
ции света золотыми золями от
длины волны света и дисперс-
ности золя (дисперсность уве-
личивается в порядке возра-
стания номера кривых).
стично фиктивной абсорбцией, т. е.
рассеянием света, которая у металлических золей максимальна
при какой-то средней степени дисперсности,*»Однако одного свето-
рассеяния недостаточно, чтобы вызвать такой резкий эффект. То
обстоятельство, что с увеличением дисперсности абсорбция света
резко повышается, объясняется также исключительно большой спо-
собностью металлов поглощать свет. В результате уже тончайшие
металлические слои, толщина которых меньше длины световой
волны, не пропускают свет. Естественно, что при одной и той же
концентрации дисперсной фазы более высокодисперсные метал-
лические золи будут лучше экранировать свет.
41
Сказанное понятно из схем I и II, приведенных на рис. 11,4.
На схеме I показано прохождение световых лучей через неметал-
лический золь. В этом случае не имеет значения, будет ли дис-
персная фаза находиться в золе в виде крупных или мелких час-
тиц: благодаря ее прозрачности свет проходит как через те, так
и другие частицы. На схеме II показано прохождение лучей света
через металлический золь. Так как даже весьма мелкие металли-
ческие частицы обладают очень большой способностью поглощать
Рис. II, 4. Схема, поясняющая изменение экранирования при уве-
личении дисперсности неметаллического (I) и металлического (II)
золей:
а— низкодисперсные золн; б —высокодисперсные золи с тем же содержанием
дисперсной фазы.
Свет и с увеличением дисперсности степень экранирования возрас-
тает, при повышении дисперсности этого золя будет происходить
более интенсивное тушение света или, как говорят, экстинкция
возрастет. Понятно, что схема 11,4 является только грубым при-
ближением к действительности, так как в схеме не учтено «экра-
нирование» света за счет его рассеяния, зависящее от размера
тастиц дисперсной фазы.
Таким образом, для металлических золей уравнение светопо-
глощения должно учитывать дисперсность системы:
/п = 1а exp [— ecl/f (г3)]
(II, 9)
Интересно, что при дальнейшем росте дисперсности частицы
металлического золя становятся настолько малыми, что уже не
препятствуют прохождению света, и в этом случае золи ведут себя
<ак истинные растворы.
12
3. ОКРАСКА КОЛЛОИДНЫХ СИСТЕМ
Очень часто коллоидные системы окрашены. Окраска -драгоценных или по-
лудрагоценных камней обусловлена присутствием в них ничтожных количеств
тяжелых металлов и их окислов в состоянии коллоидной степени раздробления.
Например, в естественных рубинах такими примесями являются соединения же-
леза, в изумрудах — соединения хрома Так называемое рубиновое стекло, изго-
тавлявшееся еще М В. Ломоносовым, представляет собою стекло с весьма ма-
лой примесью коллоидного золота (0,0001%) Очень часто встречаются и окра-
шенные коллоидные системы с жидкой дисперсионной средой Особенно яркой
окраской обладают золи металлов Это объясняется большой разностью плотно-
стей, а следовательно, и показателей преломления дисперсной фазы н диспер-
сионной среды
Вообще следует сказать, что интенсивность окраски золей может быть весь-
ма высокой и значительно превышать интенсивность окраски молекулярных рас-
творов Например, при одинаковом содержании дисперсной фазы интенсивность
окраски золя золота превосходит интенсивность окраски раствора фуксина в
400 раз Интенсивная окраска коллоидных растворов позволяет определять
ничтожные количества коллоидно раздробленного вещества. Так, желтая окраска
коллоидного раствора сульфида мышьяка, которую визуально можно обнару-
жить при толщине слоя золя в 1 см, отвечает содержанию 1 ч As2Sj в 8-105 ч
воды, а красный цвет золя золота заметен в таких условиях при содержании
1 ч Au даже в 1 • 10s ч. воды. Это обстоятельство широко используется в ана-
литической химии.
Причины той или иной окраски данной коллоидной системы чрезвычайно
сложны На окраску коллоидных систем влияют не только природа дисперсной
фазы и дисперсионной среды, ио и дисперсность частиц, их форма и строение,
гак как эти факторы влияют на рассеяние и абсорбцию света Кроме того,
окраска коллоидных систем может зависеть от способа приготовления золя и от
условий его наблюдения (в проходящем и отраженном свете) Поэтому ниже
приведены только самые общие соображения, которые должны быть приняты
во внимание при рассмотрении причин той или иной окраски золя
Бесцветные (белые) неметаллические золи, не проявляющие избирательной
абсорбции света, в проходящем свете обычно обнаруживают оранжевую окра-
ску, а в отраженном свете — опалесцируют голубоватым светом Это явление,
как мы уже знаем, объясняется светорассеянием и полностью соответствует
закону Рэлея
Однако существует много золей с неметаллическими частицами, обладающих
специфической окраской, которая зависит от избирательной абсорбции световых
лучей их частицами Сюда, например, следует отнести золь берлинской лазури,
окрашенный в интенсивно синий цвет, красный золь сульфида сурьмы и т д.
Однако по мере понижения степени дисперсности, например при коагуляции,
интенсивность окраски золя снижается из-за быстрого возрастания светорассея-
ния Следует, впрочем, отметить, что заметные изменения в цвете золя при
коагуляции наступают лишь при тесном сближении частиц
Наиболее сложен вопрос об окраске золей, содержащих металлические ча-
стицы Цвет металлических золей, с одной стороны, обусловливается истинной
адсорбцией света металлическими частицами, в результате которой часть свето-
вой энергии переходит в тепло, с другой стороны, на цвет металлических золей
влияет и светорассеяние Благодаря тому, что абсорбция и светорассеяние с
увеличением размера частиц и длины волны света проходят через максимум,
золи одного и того же металла могут иметь разнообразную окраску Гак, гру-
<5одисперсиые золи золота, обладающие сравнительно малым истинным погло-
щением, сдвинутым в красную область спектра, и сильно рассеивающие свет
с максимумом в той же красной части спектра, обычно имеют голубой цвет
(в проходящем свете) и опалесцируют красным цветом (в рассеянном свете).
Высокодисперсиые золи золота, наоборот, обычно окрашены в красный цвет и
опалесцируют голубым цветом Это объясняется их способностью сильно абсор-
бировать свет с резким максимумом в желто-зеленой части спектра Интересно,
что при еще большей степени дисперсности золи золота приобретают желтый
43
цвет и приближаются по окраске к растворам хлорида золота, т. е. к окраске
молекулярно-дисперсных систем
Вопрос об окраске металлических золей во многих случаях усложняется
еще и тем, что помимо дисперсности системы на абсорбцию и рассеяние света
влияют форма и строение частиц Связь между этими факторами и окраской
металлических золей подробно рассмотрена в работах Ми и Ганса.
4. ОПТИЧЕСКИЕ МЕТОДЫ ИСЛЛЕДОВАНИЯ КОЛЛОИДНЫХ СИСТЕМ
В настоящее время оптические методы являются наиболее рас-
пространенными методами определения размера, формы и струк-
туры коллоидных частиц. Это объясняется не только быстротой
и удобством этих методов, но и точностью получаемых результа-
тов Грубые дисперсные системы (суспензии, эмульсии, пены, пыли)
обычно исследуют с помощью светового микроскопа. К наиболее
часто применяющимся методам исследования высокодисперсных
коллоидных систем относятся ультрамикроскопия, электронная
микроскопия, нефелометрия и турбидиметрия. Реже применяют
метод, основанный на определении двойного лучепреломления в
потоке, рентгенографию и электронографию для исследования вну-
тренней структуры и характера внешней поверхности частиц кол-
лоидной системы.
Ниже кратко освещены только принципы, на которых основаны
некоторые из этих методов. Устройство соответствующих приборов
и техника проведения определений здесь рассматриваться не бу-
дут, так как в этих вопросах учащийся сможет детально разо-
браться только на практических занятиях по коллоидной химии;х
Ультрамикроскопия явилась одним из первых оптических ме-
тодов исследования коллоидных систем. Наблюдение взвешенных
в воздухе частиц с помощью микроскопа на темном фоне при фо-
кусировании падающего на них сбоку света было описано еще
М. В. Ломоносовым. Однако лишь в 1903 г. Зидентопф и Зиг-
монди на основе этого явления предложили прибор — ультрамик-
роскоп, который был использован для исследования лиозолей. Не
будет ошибкой сказать, что это изобретение, давшее возможность
подтвердить реальность существования коллоидных частиц, поло-
жило начало бурному развитию коллоидной химии.
Теория показывает, что разрешающая способность микроскопа,
т. е. то наименьшее расстояние, при котором две точки можно еще
видеть отдельно друг от друга, составляет около половины длины
световой волны. Таким образом, при использовании обычного
света (длина волны 400—700 нм) в наилучший микроскоп видимы
частицы, размер которых составляет не менее 0,2 мкм. При ис-
пользовании ультрафиолетового света с помощью фотосъемки
можно получить изображение более мелких частиц, но с диамет-
ром все же не меньшим 0,1 мкм. Таким образом, коллоидные час-
тицы лежат за пределами видимости в обычном микроскопе.
Ультрамикроскоп позволяет констатировать присутствие кол-
лоидных частиц, подсчитывать и наблюдать их движение. Прин-
44
цип, на котором основан ультрамикроскоп, заключается в том, что
на коллоидную систему сбоку направляют сильный луч света
и с помощью обычного микроскопа наблюдают свет, рассеянный
отдельными частицами. По существу, принцип ультрамикроскопа
сводится к наблюдению под
микроскопом конуса Тин-
даля.
Различие в устройстве так на-
зываемого щелевого ультрамик-
роскопа от обычного микроскопа
легко можно уяснить из рис. 11,5.
Как можно видеть, в обычном
микроскопе наблюдение ведется
в проходящем свете. Частицы
при этом кажутся темными, так
как поглощают свет, а само поле
представляется светлым. При на-
блюдении в ультрамикроскоп, на-
оборот, поле зрения темное, так
как лучи от источника света не
попадают в глаз наблюдателя,
а коллоидные частицы представ-
ляются светлыми из-за их способ-
ности рассеивать свет. При этом,
поскольку размер коллоидных ча-
стиц обычно меньше половины
длины волны света, они воспри-
нимаются визуально в виде све-
тящихся точек. Частицы свобод-
нодисперсных систем, способные
совершать броуновское движение,
наблюдаются как точки, всегда
находящиеся в более или менее
оживленном движении.
При ультрамикроскопических
наблюдениях необходимо соблю-
дать следующие условия.
1. Золь должен быть доста-
точно разбавленным, чтобы рас-
стояние между частицами было
больше разрешающей способности
Рис. 11,5 Схема хода лучей в обыч-
ном микроскопе (а) и щелевом
ультрамикроскопе (б)
1—луч света, 2— зеркало, 3 — конденсор
Аббе, 4 — предметное стекло, 5 — исследуе-
мый препарат, 6“—покровное стекло,
7— объектив, 8 — окуляр, 9 — щелевая диа-
фрагма, 10—фокусирующая лииэа 11— кю-
вета с исследуемым золем
микроскопа. В противном слу-
чае отдельные точки будут сливаться друг с другом и наблюдение
за ними будет затруднено. /
2. Частицы не должны быть слишком малы или слишком ве-
лики. В первом случае их можно не увидеть из-за незначительной
интенсивности рассеиваемого ими света. Во втором — дифракцион-
ные кольца, образующиеся вокруг больших частиц, будут мешать
наблюдению.
45
3 Коэффициент преломления дисперсной фазы должен доста-
точно сильно отличаться от коэффициента преломления диспер-
сионной среды, иначе светорассеяние незначительно и частицы
будут мало заметными.
С помощью ультрамикроскопа можно наблюдать в металличе-
ских золях частицы размером не меньше 0,002—0,005 мкм. В не-
металлических золях, из-за меньшей разности в коэффициентах
преломления дисперсной фазы и дисперсионной среды, с помощью
ультрамикроскопа можно видеть частицы с диаметром не меньше
0,2 мкм. Наконец, для золей с органической дисперсной фазой
этот предел должен быть еще выше.
Вместо щелевых ультрамикроскопов в последнее время для ис-
следования коллоидных
Рис. II, 6. Схема хода лу-
чей в конденсоре темного
поля.
систем широко применяют так называе-
мые конденсоры темного поля, представ-
ляющие собою линзу со срезанным вер-
хом и посеребренной боковой поверх-
ностью. Принцип действия конденсора
темного поля заключается в том, что
проходящий через конденсор свет фоку-
сируется в точке, расположенной в поле
зрения микроскопа и в то же время на-
ходящейся в стеклянной кювете с иссле-
дуемой системой, помещенной сверху
конденсора. Благодаря специальному не-
прозрачному круглому экрану, установ-
ленному перед линзой, как и в щелевом
ультрамикроскопе, прямые лучи не по-
падают в окуляр, а поступают лишь лучи, рассеянные коллоидными
частицами. Схема хода лучей в конденсоре темного поля изо-
бражена на рис. II, 6. Конденсор темного поля чрезвычайно прост
и может быть приспособлен к любому обычному микроскопу.
С помощью ультрамикроскопа (или микроскопа с конденсором
темного поля) нельзя непосредственно определить размер кол-
лоидных частиц, однако это можно сделать косвенно. Из препарата
исследуемого золя тем или иным способом выделяют объем
в форме прямоугольного параллелепипеда, в котором подсчиты-
вают число коллоидных частиц.
Если высоту параллелепипеда обозначить через h, а через I
сторону квадрата, являющегося обычно основанием параллелепи-
педа, то объём, в котором производят подсчет частиц, будет равен
V = hl2. Так как коллоидные частицы обычно находятся в ожив-
ленном броуновском движении и число их в выделенном объеме
все время изменяется, приходится брать среднее значение из мно-
жества подсчетов, проведенных через определенные промежутки
времени. Объем, в котором подсчитывают число частиц, и числен-
ная концентрация не должны быть слишком большими для того,
чтобы наблюдатель мог сразу определить число находящихся
в объеме частиц.
46
Подсчитав среднее число частиц п в выделенном объеме V,
легко найти численную концентрацию v:
v = n/V (И, 10)
Если известны плотность дисперсной фазы р и весовая концен-
трация золя с, то, очевидно, должно соблюдаться равенство:
vpv = с (II, 11)
где v — средний объем частицы.
Отсюда
v = c/pv (II, 12)
Если частица шарообразна, то ее радиус легко найти по урав-
нению
г = ^/3ц/4л (11,13)
Если частица имеет кубическую форму, то размер ее ребра вы-
числяют по уравнению _
/ = -^о (П,14)
Наконец, наблюдая колдоидные системы в ультрамикроскоп,
можно не только определить средний размер частиц, но получить
и некоторое представление о форме частиц. Если1 частицы, види-
мые в темном поле, мерцают, то это признак их анизодиаметрич-
ности. Причина мерцания заключается в том, что несферическне
частицы, находящиеся в броуновском движении, поворачиваются
к световому лучу различными, неравными по площади плоскостями
и вследствие этого посылают в глаз наблюдателя в разное время
разное количество света. Если же частицы в темном поле светятся
спокойным, немерцающим светом, то это указывает на их при-
мерно изодиаметрическую форму.
Недостатком определения размера частиц с помощью ультра-
микроскопа является то, что найденное значение отвечает сред-
нему размеру частиц. Кроме того, такое определение очень утоми-
тельно (чтобы полученные результаты были достаточно достовер-
ными, приходится брать среднее из сотен и даже тысяч отдельных
определений).
Б. В. Дерягин и Г. Я. Власенко сконструировали специальный
поточный ультрамикроскоп, с помощью которого весьма быстро
можно определить число частиц в единице объема аэрозоля или
лиозоля. Схематическое устройство поточного микроскопа пока-
зано на рис. 11,7. Изучаемый лиозоль или аэрозоль наблюдается
в потоке, направленном вдоль канала кюветы 2, параллельного
оси тубуса микроскопа 5. Каждая частица, пересекая зону, осве-
щенную источником света 3, дает вспышку; общее число таких
вспышек может быть легко подсчитано наблюдателем. Разделив
число подсчитанных вспышек на измеряемый счетчиком 1 общий
объем аэрозоля, протекающего через контролируемую и вырезан-
ную окулярной диафрагмой 6 часть поля, легко найти численную
концентрацию.
47
При таком методе определения численной концентрации отпа-
дают почти все источники ошибок, возможные при классическом
ультрамикроскопическом методе счета, и, кроме того, значительно
сокращается время исследования. Так, при определении очень ма-
лых концентраций аэрозоля, остающегося после просасывания че-
рез аэрозольные фильтры, поточным методом время измерения со-
кращается в 100 и более раз при одновременном повышении точ-
ности определения.
При поточном методе ультрамикроскопического счета можно
автоматизировать счет частиц и, разбивая их на фракции или
Рис. II, 7. Схема поточного ультрамикроскопа:
Z —счетчик объема; 2—кювета; 3—источник света; 4 —линза; 5—тубус микро-
скопа; 6—окулярная диафрагма.
классы по различной яркости, вести счет частиц каждой фракции
в отдельности, т. е. изучать фракционный состав дисперсной фазы.
При автоматическом счете частиц свет поступает на катод фото-
умножителя. Каждая вспышка света от проходящей через кювету
частицы вызывает электрический импульс, который после усиле-
ния регистрируется счетчиком.
В настоящее время поточный ультрамикроскоп получил в Со-
ветском Союзе широкое распространение в ряде исследователь-
ских учреждений, а также в шахтах, в полевых и экспедиционных
условиях.
Электронная микроскопия. В электронном микроскопе вместо
световых лучей используется пучок быстрых электронов, что резко
увеличивает разрешающую способность микроскопа и дает воз-
можность непосредственно видеть или фотографировать коллоид-
ные частицы. Возможность применения в этом случае потока элек-
тронов обусловлена тем, что электроны обладают одновременно
как квантовой, так и волновой природой. Длина волны потока
электронов составляет всего 0,02—0,05 А, т. е. меньше размеров
48
атома, благодаря чему разрешающее расстояние с помощью элек-
тронного микроскопа может быть доведено до 5—10 А. Изображе-
ние, получаемое на флуоресцирующем экране, может быть сфото-
графировано, причем полученный снимок можно еще увеличить,
так что общее предельное увеличение современных электронных
микроскопов весьма большое.
Ход электронного пучка в электронном мик-
роскопе изображен на рис. 11,8. В общем он схо-
ден с ходом световых лучей в обычном микро-
скопе. Однако поскольку электроны легко рас-
сеиваются и поглощаются, для фокусировки пуч-
ка электронов применяют электромагнитные ка-
тушки, создающие электростатические или маг-
нитные поля. Для уменьшения рассеяния элек-
тронов внутри электронного микроскопа поддер-
живают высокий вакуум. Наконец, с той же
целью для исследования применяют объекты
очень малой толщины, нанесенные обычно на
тончайшую нитроцеллюлозную, кварцевую, угле-
родную или другие пленки, прозрачные для пуч-
ка электронов. Если последнее условие не будет
соблюдено, то под воздействием электронов мо-
жет процсходить нагревание и разрушение объ-
екта. Очень часто вместо самих объектов в элек-
тронном микроскопе наблюдают их отпечатки на
различных пленках. Такие пленки — отпечатки
(реплики) для придания им большей контраст-
ности обычно оттеняют с помощью напыления
каким-нибудь молекулярно-раздробленным ме-
таллом (например, хромом).
Недостатком электронной микроскопии яв-
ляется сложность подготовки объектов для ис-
следования и необходимость поддерживать в ми-
кроскопе высокий вакуум. Кроме того, посколь-
ку при наблюдении объект находится в вакууме,
в электронном микроскопе нельзя наблюдать
коллоидную систему как таковую, а можно ви-
деть лишь частицы, содержащиеся в ее сухом
остатке. Однако электронный микроскоп получает все более ши-
рокое применение в науке и технике, поскольку с его помощью
можно видеть мельчайшие частицы со всеми особенностями их
формы и строения. Благодаря его огромной разрешающей способ-
ности можно наблюдать даже отдельные большие молекулы (мо-
лекулы белков), вирусы.
На рис. 11,9 представлены фотоснимки, сделанные с помощью
электронного микроскопа.
Нефелометрия основана на способности коллоидных систем
рассеивать свет. Определяя интенсивность светорассеяния данной
Рис. II, 8. Схема
хода лучей в элек-
тронном микроско-
пе:
/ — источник быстрых
электронов (электрон-
ная пушка); 2—кон-
денсорная линза;
3 — объект исследова-
ния; 4—линза объек-
тива; 5—промежуточ-
ное изображение;
d —проекционная лин-
за; 7—конечное изо-
бражение на флуорес-
цирующем экране;
8—фотографическая
пластинка.
49
системой, можно определять размер частиц или концентрацию
дисперсной фазы, изучать явления коагуляции и т. д. Широкое
Рис. П,9. Электронные микрофотографии:
а—травленная поверхность алюминия (увеличение 6000); б—поверхность активного
угля; в—частицы, окиси цинка (увеличение 10000)
использовайие нефелометрии в коллоидной химии объясняется вы-
сокой чувствительностью метода, а также его простотой.
В осяове_ нефелрметрии лежит зависимость, вырджаемая^урав-
нением Рэлея, которое "можно представить в виде:
Ip = kvosIo = kvv • vlt> — kcrflo (П, 15)
где с — объемная концентрация дисперсной фазы в системе.
60
Зная все величины, входящие в уравнение Рэлея (11,1), объ-
емную концентрацию дисперсной фазы с и определив абсолютные
значения интенсивности падающего и рассеянного света (70 и /р),
можно вычислить средний объем частицы.
Абсолютные значения интенсивности падающего и рассеянного
света можно найти только с помодцью сложных приборов (тин-
дальметров), и полученные результаты требуют введения ряда по-
правок. Кроме того, при определении абсолютных значений ин-
монохромати-
тенсивности света надо пользоваться для
ческим светом. Поэтому гораздо боль-
шее распространение получили относи-
тельные методы нефелометрии, в кото-
рых эти трудности в значительной мере
отсутствуют.
При относительных измерениях опа-
лесценцию исследуемого раствора срав-
нивают с опалесценцией стандартного
раствора, размер частиц v" которого из-
вестен, и, пользуясь полученными данны-
ми, вычисляют размер частиц v' в иссле-
дуемой системе. Непременным условием
такого определения должна быть одина-1
ковая объемная концентрация дисперс-|
ной фазы 'в обоих растворах.
Для проведения измерений таким спо-
собом применяют чрезвычайно простые
приборы — нефелометры. Схема устрой-
ства простейшего из таких приборов —
визуального нефелометра Клейнманна,
1—источник света, 2, 3—подвиж-
ные экраны; 4, 5—цнлиндриге-
ские кюветы; 6, 7—стеклянные
цилиндрики, 5, Р—призма;
/0—окуляр
показана на рис. II, 10. Нефелометр имеет две совершенно одина-
ковые стеклянные цилиндрические кюветы 4 и 5, в первую из ко-
торых помещают стандартный раствор, а во вторую — испытуемый.
Свет от источника 1 (лежащего за плоскостью рисунка) равно-
мерно падает на обе кюветы Высоту освещенного столба жидкости
в каждой кювете можно регулировать, поднимая и опуская спе-
циальные экраны 2 и 3. Свет, рассеянный растворами, попадает
на сплошные стеклянные цилиндрики 6 и 7, погруженные на одну
и ту же глубину в растворы (эти цилиндрики применяют для того,
чтобы устранить отражение света менисками жидкостей). Из ци-
линдриков пучки рассеянного света с помощью специальных призм
8 и 9 направляются в окуляр 10, разделенный на две половины.
Каждая из его половин освещается за счет света, поступающего
из одной какой-нибудь кюветы.
При работе с нефелометром в кюветы наливают исследуемый
и стандартный растворы и, поднимая или опуская экраны у кювет,
Добиваются одинаковой освещенности обеих половин окуляра,
фчевидно, при таком условии интенсивность света, рассеянного ис-
Йяедуемым раствором /р, равна интенсивности света, рассеянного
51
стандартным раствором I". При этом должно соблюдаться и ра-
венство:
IQkv'ch' = I(ikv"ch" (II, 16)
так как интенсивность света, рассеянного каждой кюветой, про-
порциональна высоте ее освещенной части h.
Из уравнения (II, 16) следует:
v'h' = v"h" (II, 17)
или
v'lv"=-h"lh' (II, 18)
откуда
v' = v" (h"/hr) (II, I^-
no уравнению (11,19), зная размер частиц, содержащихся
в стандартном растворе, вычисляют размер частиц исследуемого-
золя. Конечно, следует помнить, что результаты таких вычислений
однозначны только тогда, когда коллоидные системы монодис-
персны. Кроме того, поскольку показатель степени при X в урав-
нении Рэлея не зависит от размера частиц только для высокодис-
персных золей, описанный способ можно применять для определе-
ния размеров сравнительно малых частиц.
Так как светорассеяние сильно зависит от размера частиц,
определение изменения интенсивности опалесценции может быть-
успешно применено для изучения протекающих в системе процес-
сов агрегации и дезагрегации. С этой целью целесообразно строить-
графики, на ординате которых откладывают значения светорас-
сеяния золя, а на абсциссе — время наблюдения.
Наконец, нетрудно видеть, что нефелометр можно использо-
вать для определения концентрации дисперсной фазы в системе.
При этом искомую концентрацию рассчитывают по формуле:
с' = с" (h"lh’) (II, 20)
вывод которой аналогичен выводу фбрмулы (11,19). Понятно, что-
стандартный и исследуемый растворы должны содержать частицы
одной и той же природы и одного и того же размера.
Помимо визуальных нефелометров широко применяют фотоэлектрические
нефелометры, в которых с помощью чувствительных микроамперметров опреде-
ляют силы фототоков i' и i", возникающих в фотоэлементах под действием
света, рассеянного стандартным и испытуемым растворами. При пользовании
фотоэлектрическими нефелометрами размер частиц и концентрацию дисперсной
фазы в золе определяют по уравнениям:
v' = v" (i'll") (П,21)
И
с' = с" (i'H") (11,22)
Для определения концентрации или размеров частиц золей,
слабо рассеивающих свет, иногда также можно использовать не-
фелометрические методы исследования. В этом случае следует
перейти от видимой части спектра к .ультрафиолетовым лучам.
52
Применяя соответствующий источник света, кварцевые линзы
и прибор, регистрирующий ток фотоэлемента, можно получить
данные для вычисления численной концентрации или размера
частиц.
Для определения размера частиц можно воспользоваться не
только способностью коллоидных систем рассеивать свет (нефе-
лометрия), но и их способностью ослаблять интенсивность про-
ходящего света в результате светорассеяния (турбидиметрия).
В этом случае измерения ведут с помощью обычных колориметров,
или спектрофотометров, позволяющих определять мутность. Ме-
тод турбидиметрии получил сейчас широкое распространение в
коллоидной химии; этот метод подробно описан в учебниках по
аналитической химии.
Наконец, размеры (а в некоторых случаях и форма) коллоид-
ных частиц могут бытЬ определены и по ряду других оптических
характеристик коллоидного раствора *. Однако рассмотрение всех
этих методов выходит за пределы настоящего курса.
В заключение отметим, что все методы определения размера
и формы коллоидных частиц, основанные на измерении рассеяния
света, пригодны в основном только для бесцветных (белых) зо-
лей. Для окрашенных золей и в особенности для металлических
золей эти методы без существенных коррективов применять
нельзя.
Рентгенография и электронография. Оба эти метода, основан-
ные на применении рентгеновских лучей или потока электронов,
подробно рассматриваются в курсе физической химии, и поэтому
мы не будем касаться здесь принципов, лежащих в их основе/От-
метим лишь, что методом рентгенографии можно получить инфор-
мацию о внутренней структуре коллоидных частиц. Вследствие
малого-размера этих частиц при исследовании коллоидных систем
с помощью рентгенографии получать диаграммы Лауэ затрудни-
тельно и приходится чаще всего ограничиваться получением и изу-
чением диаграмм Дебая —Шеррера.
Путем исследования диаграмм Дебая — Шеррера удалось уста-
новить кристаллическую структуру частиц многих золей. Особенна
хорошие результаты 'были получены при исследовании золей тя-
желых металлов и их соединений, так как способность рассеяния
лучей тяжелыми атомами весьма велика, а дисперсионная среда
здесь сравнительно мало мешает анализу. При этом было выяс-
нено, что структура дисперсной фазы сильно зависит от метода
приготовления и возраста золя. О работах В. А. Каргина
и 3. Я. Берестневой, показавших, что старение золей, как правило,
связано с кристаллизацией дисперсной фазы, будет сказано
в гл. VIII, посвященной синтезу золей.
* Исследование формы и строения коллоидных частиц, основанное на опре-
делении двойного лучепреломления в потоке, рассмотрено в первом издании
книги: С. С. Воюцкнй «Курс коллоидной химии», М., «Химия», 1964, 574 с. См.
53
Рентгенография имела огромное значение при исследовании
высокомолекулярных веществ, в частности при изучении струк-
туры природных и синтетических полимерных материалов, при вы-
яснении природы явлений набухания и т. д. Анализ диаграмм Де-
бая — Шеррера позволяет во многих случаях установить период
идентичности молекул полимеров и выяснить взаимное располо-
жение их структурных элементов в пространстве, хотя все это
требует чрезвычайно длительных и скурпулезных расчетов с при-
менением счетных машин. Именно методами рентгеноструктурного
анализа было установлено сложнейшее строение молекул таких
веществ, как пенициллин, витамин В12, гемоглобин и многих высо-
комолекулярных веществ.
Модификацией рентгенографической методики исследования
является определение среднего размера частиц путем рассеяния
рентгеновских лучей под малыми углами. Этим методом, были по-
лучены ценные сведения о размерах молекул белка и о степени их
гидратации.
Методы электронографии вследствие малой проникающей спо-
собности электронного пучка позволяют детально исследовать
только поверхность частиц дисперсной фазы коллоидных систем
и макромолекул высокомолекулярных веществ. Электронография
позволяет непосредственно определить расстояния между отдель-
ными атомами, лежащими на поверхности, на основании чего
можно найти другие параметры структуры вещества. Этот метод
исследования особенно пригоден для изучения адсорбционных
слоев.
Методы электронографии целесообразно сочетать с рентгено-
графическим анализом. При этом обычно удается получить доста-
точно сведений о внутренней структуре дисперсной фазы коллоид-
пых систем и растворов высокомолекулярных веществ, а также
об изменениях, наступающик в этой структуре в результате на*
тревания, деформации, набухания и тому подобных воздействий.
ГЛАВА III
МОЛЕКУЛЯРНО-КИНЕТИЧЪСКИЕ СВОЙСТВА
КОЛЛОИДНЫХ СИСТЕМ
1. ТЕПЛОВОЕ ДВИЖЕНИЕ МОЛЕКУЛ И БРОУНОВСКОЕ ДВИЖЕНИЕ
Согласно молекулярно-кинетическои теории газ представляет собою сово-
купность молекул или атомов, находящихся в хаотическом движении Средние
расстояния между молекулами в газах значительно превосходят линейные раз-
меры молекул, а суммарный объем, занимаемый собственно молекулами, ничто-
жен по сраинеиию с объемом газа Соударяясь друг с другом, молекулы газа
изменяют скорость и направление своего движения, однако их средняя кинети-
ческая энергия, зависящая только от температуры, остается всегда неизменной
и равной 4zkT (где k — постоянная Больцмана, Т—абсолютная температура).
В твердых кристаллических телах молекулы, атомы или ионы расположены
в определенном порядке, отвечающем наименьшему значению свободной энер-
гии, и колеблются около положений равновесия — узлов кристаллической ре-
шетки
Жидкости по своим свойствам занимают промежуточное положение между
газами и твердыми телами По плотности они близки к твердым телам — рас-
стояния между молекулами в жидкости почти так же малы, как в кристаллах,
и близки к размерам самих молекул Однако молекулы жидкости, как и мо-
лекулы газа, способны менять свои места, правда, не выходя из сферы влияния
молекулярных сил соседних молекул
Ранее считалось, что молекулы в жидкости расположены беспорядочно по
отношению друг к другу Однако рентгенографические исследования показали,
что в весьма малых областях жидкости имеется определенный порядок распо-
ложения молекул Принято считать, что структура жидкости характеризуется
ближним порядком в отличие от кристаллов, которым свойственен дальний по-
рядок При этом следует учитывать, что области с квазикристаллическим по-
рядком в жидкости во времени не постоянны, — возникнув в одном месте и
просуществовав очень недолго, они распадаются и образуются в другом месте.
Согласно современной теории жидкостей, продложенной Я И Френкелем
н независимо от него Эйрингом, передвижение молекул в жидкости совершается
таким образом, что когда какая-нибудь молекула меняет свое место, происходит
перегруппировка соседних молекул и эта перегруппировка продолжается до тех
пор, пока каждая молекула снова не займет положение, наиболее выгодное в
энергетическом отношении Рассмотрим представления Я И Френкеля несколько
подробнее
Наличие ближнего и отсутствие дальнего порядка в жидкости указывает
на то, что в ее молекулярной упаковке имеются пустоты, или дырки По-
вышение температуры способствует увеличению числа дырок и уменьшению ква-
знкристаллической упорядоченности Наличие дырок и обусловливает способность
жидкости к течению и увеличению объема при плавлении
Многие молекулы жидкости могут оказаться расположенными по соседству
с дырками, как изображено на рис III, 1 В энергетическом отношении такие
молекулы находятся в потенциальной яме и отделены от других возможных
равновесных положений энергетическим барьером Однако каждая из этих мо-
лекул, если она обладает достаточной энергией, может совершить скачок и за-
Яять находящуюся рядом дырку, перейдя в новое положение равновесия Есте-
ственно, что в результате такого скачка молекула оставит после себя дырку,
55»
в которую в свою очередь может перескочить другая молекула. Число возможных
•скачков будет тем больше, чем больше число дырок и чем меньше высота энер-
гетического барьера, который должна преодолеть молекула при переходе из
одного положения равновесия в другое.
Такое перескакивание молекул, очевидно, определяет возможность их теп-
лового перемещения в жидкости, или самоднффузнн. При увеличении тем-
пературы скорость самоднффузнн возрастает, так как прн этом молекула, рас-
положенная рядом с дыркой, имеет большую вероятность приобрести энергию,
необходимую для преодоления энергетического барьера Помимо этого скорость
диффузии растет с температурой нз-за увеличения числа дырок в результате
термического расширения жидкости Минимальная энергия, необходимая для
того, чтобы молекула жидкости могла переместиться нз одного временного по-
ложения равновесия в другое, называется энергией активации диффузии. Эта
величина зависит от компактности упаковки молекул, их
Рис. Ш, 1. Схема
расположения мо-
лекул вблизи дыр-
ки.
размера н от межмолекуляриых сил.
В рассмотреинном выше случае самоднффузнн прн от-
сутствии виешиого силового поля скачкн молекул одина-
ково часты во всех направлениях, н поэтому подобный про-
цесс не приводит к переносу вещества в каком-инбудь пре-
имущественно одном направлении. Прн иалнчнн внешнего
силового поля, например напряжения сдвига, более ве-
роятен скачок в направлении действия поля, так как не-
необходимая для этого энергия активации будет меньше,
чем для скачка в противоположном направлении. В ре-
зультате произойдет вынужденная диффузия, обусловли-
вающая вязкое течение жидкости (последнее рассмотрено
в гл. X).
Аналогично объясняется и. диффузия молекул веще-
ства, растворенного в жидкости. Молекулы вещества в рас-
творе, совершая скачкн примерно таким же образом, как н молекулы раствори-
теля, в силу беспорядочности движения не могут оставаться в каком-либо опре-
деленном месте, а распределяются в среднем равномерно по всему объему жид-
кости. Согласно термодинамике это соответствует увеличению эитропнн си-
стемы.
Можно сказать, что тепловое движение каждой молекулы жидкости состоит
частично нз колебательных движений около положения равновесия н частично
нз небольших поступательных движений, когда под влиянием особо сильного
удара соседней молекулы нлн иесколькнх случайных ударов в близких друг
другу направлениях молекула настолько далеко удаляется от равновесного по-
ложения, что оказывается ближе к некоторому новому положению равновесия.
Таким образом, все молекулы жидкости как бы ведут кочевую жнзиь, причем
кратковременные «переезды» (скачкн нз одного положения равновесия в дру-
гое) сменяются относительно продолжительными периодами «оседлой жизни»
(колебание вокруг положения равновесия). Именно сравнительно длительными
периодами «оседлого» существования молекул в жидкостях объясняется, что
диффузия в жидкостях происходит гораздо медленнее, чем в газах.
Важно отметить, что при высоких температурах жидкости по своим свой-
ствам приближаются к газам В критическом состоянии различие между жидко-
стью и газом исчезает, а прн температурах выше критической жидкость пре-
вращается в газ. Наоборот, при низких температурах, близких к температурам
кристаллизации, жидкости по своим свойствам приближаются к кристаллам-
Однако переход жидкости в кристаллическое состояние всегда происходит скач-
кообразно Когда жидкости по тем или иным причинам не могут перейти в кри-
сталлическое состояние, они с понижением температуры переходят в стеклооб-
разное (аморфное) состояние
Тепловое движение частиц в коллоидных и микрогетерогенных
•системах получило название броуновского движения в честь анг-
лийского ботаника Р. Броуна, обнаружившего его в 1827 г, при
56
наблюдении под микроскопом водной суспензии цветочнойг
пыльцы. Броун не смог объяснить природу этого явления, но уста-
новил, что* движение частиц пыльцы не ослабевает со временем,
не зависит от внешних источников энергии (света, сотрясений)
и тем интенсивнее, чем выше температура. Позднее разные иссле-
дователи пытались объяснить броуновское движение жизнедея-
тельностью пыльцы, наличием конвекционных токов в системе,
электрическими явлениями, неравномерным смачиванием частиц
средой. Однако первое предположение пришлось отбросить, так
как броуновское движение оказалось свойственным не только сус-
пензии пыльцы, но и вообще всем суспензиям, в том числе и сус-
пензиям неорганических веществ. Объяснение броуновского дви-
жения существованием в жидкости микроскопических конвекцион-
ных токов оказалось несостоятельным, так как при этом соседние
частицы должны были бы передвигаться с одинаковой скоростью
и параллельно друг другу. На самом же деле микроскопическое
наблюдение показывает, что соседние частицы движутся с раз-
личными скоростями по траекториям, пересекающимся под раз-
ными углами. Несостоятельными оказались и объяснения броу-
новского движения с помощью электрических сил и неодинакового
смачивания поверхности частиц дисперсионной средой.
Гуи (1888 г.) и Экснер (1900 г.) предположили, что броунов-
ское движение имеет молекулярно-кинетическую природу, т. е. яв-
ляется следствием теплового движения. Правильность этой точки
зрения была подтверждена теоретическими расчетами Эйнштейна
и Смолуховского и экспериментальными работами Перрена, Свед-
берга и ряда других исследователей. Теперь точно установлено,
что движение коллоидных частиц является следствием беспоря-
дочных ударов, наносимых им молекулами среды, находящимися
в тепловом движении. Если частица достаточно мала, то число
ударов на нее приходящихся с разных сторон обычно неодина-
ково и частица получает периодические импульсы, заставляющие-
ее двигаться в разных направлениях по очень сложной траекто-
рии. С увеличением размера и массы частицы вероятность ком-
пенсации ударов возрастает, а инерция частицы становится
больше. Это приводит к тому, что большие частицы, порядка
5 мкм, совершают движения, воспринимаемые нами как колеба-
ния около некоторого центра. При диаметре частицы больше 5 мкм
броуновское движение практически прекращается.
Следует указать, что кроме поступательного движения малые
частицы вследствие ударов молекул претерпевают и вращательное
броуновское движение.
В результате огромного числа ударов, которые наносят моле-
кулы среды коллоидной частице, последняя меняет свое направ-
ление и скорость весьма часто. За одну секунду коллоиДная час-
тица может изменить свое направление свыше 1020 раз. В таких
условиях определить истинный путь коллоидной частицы невоз-
можно, но легко определить среднее расстояние, на которое она
57
Рис. Ш, 2. Схема броуновского
движения частицы.
смещается в единицу времени. На рис. III, 2 схематически пред-
' ставлена проекция движения частицы на плоскость. Эту проекцию
можно найти, наблюдая броуновское движение одной частицы
в микроскопе и регистрируя ее положение в поле зрения через
равные промежутки времени. При этом движение частицы от од-
ного зарегистрированного положения до другого только условно
изображено прямой линией. На самом деле оно также имеет
' весьма сложную зигзагообразную траекторию в пространстве. По-
скольку обычно интересуются пере-
движением частицы в каком-нибудь
определенном направлении (напри-
мер, в направлении, в котором идет
диффузия), то для количественных
расчетов, о которых речь будет идти
ниже, берут не само смещение, или
сдвиг, а величину А, представляю-
щую среднее квадратичное значение
проекции смещения частицы на
ось х, параллельную выбранному
направлению. Среднеарифметиче-
ским значением проекции смеще-
как вследствие равной вероятности
ния пользоваться нельзя, так
всех направлений движения частицы оно равно нулю. Среднее
квадратичное значение проекции смещения находится из урав-
нения:
Д? + Д* + Д1+ ...
(Ill, I)
n
где Д1, Дг, Дз...— отдельные проекции смещения частицы на ось х; п — число
•таких проекций, взятых для расчета
2. ДИФФУЗИЯ В ИСТИННЫХ РАСТВОРАХ И
В КОЛЛОИДНЫХ СИСТЕМАХ
Диффузией называют самопроизвольно протекающий в системе
процесс выравнивания концентрации*молекул, ионов или коллоид-
йыхчастиц под влиянием их теплового хаотического движения. Та-
ким образом, диффузия является макроскопическим проявлением
теплового движения молекул и поэтому всегда идет тем быстрее,
чем выше температура. Явление диффузии необратимо, она проте-
кает до полного выравнивания концентраций, так как хаотиче-
скому распределению частиц отвечает максимальная энтропия
системы.
Част<^ за причину диффузии принимают осмотическое давле-
ние. Это представление было развито Нернстом (1885 г.). Так как
осмотическое давление может проявляться лишь при наличии по-
лупроницаемой перегородки (мембраны), то это давление бес-
смысленно рассматривать как какую-то реальную силу, суще-
58
ствующую вне связи с мембраной. Тем не менее осмотическое дав-
ление, являющееся также результатом хаотического движения
молекул, иногда действительно очень удобно принимать за при-
чину диффузии. Правомерность такого подхода подтверждается
соответствием теоретических данных, полученных, исходя из этой
точки зрения, с экспериментальными.
Перенос массы в результате диффузии формально сходен с за-
кономерностями переноса тепла или электричества. Воспользовав-
шись такой аналогией, Фик (1855 г.) сформулировал первый за-
кон диффузии:
dm = - D ~ s dr (Ш, 2)
dx
где т— количество продиффундировавшего вещества; D — коэффициент диффу-
зии, зависящий от свойств диффундирующих частиц и среды, dc/dx — градиент
концентрации; 5 — площадь, через которую идет диффузия; т — продолжитель-
ность диффузии.
Знак минус перед правой частью равенства поставлен потому,
что производная dcldx имеет отрицательное значение, так как
с увеличением значений х величина с уменьшается.
Уравнение (111,2) можно представить также в виде:
> 1Д= I/s (dm/dr) = — D dc/dx (III, 3>
Величину /д принято называть удельным потоком диффузии, так
как она характеризует количество вещества, переносимое в ре-
зультате диффузии за единицу времени через сечение, равное еди-
нице площади. —
Диффузионный поток в общем случае является функцией от х
и т, так как от этих величин зависит градиет концентрации dc/dx.
Только если поддерживать градиент концентрации постоянным,
диффузионный поток не изменяется с течением времени и в си-
стеме устанавливается стационарный процесс диффузии. Для этого-
случая, очевидно, можно написать:
tic
m = —D-~sr (111,4)-
dx
Принимая dc)dx = —1, s = 1 и т = 1, получим
D = m (111,5)-
т. е. коэффициент диффузии D численно равен количеству веще-
ства, продиффундировавшего через единицу площади в единицу
времени при градиенте концентрации, равном единице. Из уравне-
ния (III, 4) также следует, что коэффициент диффузии измеряется
в см2/с. Однако очень часто при рассмотрении явлений диффузия
в связи с медленностью процесса за единицу времени берут не се-
кунду, а сутки.
Эйнштейн (1908 г.) вывел уравнение, связывающее коэффи-
циент диффузии D с абсолютной температурой системы Т, вяз-
костью дисперсионной среды т] и радиусом частиц дисперсной
5&
фазы г. Для вывода этого уравнения, имеющего большое практи-
ческое и теоретическое значение, представим себе изображенную
на рис. III, 3 трубку с сечением в s см2, заполненную раствором,
причем примем, что концентрация растворенного вещества равно-
мерно убывает слева направо. Выделим мысленно в этой трубке
слой раствора бесконечно малой толщины, ограниченный вообра-
жаемыми плоскостями xlx'i и х^, объем этого элементарного
слоя V будет равен sdx. Обозначим концентрацию раствора
и осмотическое давление слева от этого слоя через Ci - и ла,
а справа от него — через с2 и л2. Очевидно, а > с2 и л, > л2. При
таких условиях в трубке происходит
диффузия растворенного вещества,
причем перенос вещества будет идти
слева направо, как указано на рисун-
ке стрелкой. Как известно, диффузия
обусловлена хаотическим тепловым
движением частиц, когда частицы дви-
жутся в отдельные моменты с различ-
ной скоростью и в разных направле-
Рис. III, 3. Схема, поясняющая ниях. Однако, рассматривая диффу-
лывод уравнения Эйнштейна. зию в целом как направленный поток
частиц, можно говорить о некоторой
эффективной постоянной скорости. Такой скоростью должны об-
ладать все частицы, чтобы, двигаясь прямолинейно, перенести че-
рез единицу сечения за единицу времени при данном градиенте
концентрации определенное количество вещества.
Постоянная скорость диффундирующих частиц может быть
объяснена только тем, что движущая сила f, действующая на от-
дельную частицу, уравновешивается обратно направленной силой
трения f', испытываемой этой частицей, т. е. что
(Ш,6}
Сила трения f' равна произведению коэффициента трения В между
частицей и средой на скорость движения частицы и:
f = Ви
(Ш,7)
Коэффициент В для сферических частиц равен по Стоксу 6лт]Г
(где л — динамическая вязкость среды и г — радиус частицы).
Для вычисления движущей силы, действующей на одну час-
тицу, определим сначала движущую силу F, приходящуюся на
•единицу объема элементарного слоя. Очевидно
Jtt — л2 dn
sdx sdx sdx
Тогда, если молярную концентрацию растворенного вещества в эле-
ментарном слое обозначить через с, то движущую силу f, прихо-
€0
дящуюся на одну частицу, можно представить следующим об-
разом:
RT de
scN« dx
f_______dn
' scdxN
(Ш, 8)
тде В— универсальная газовая постоянная; Т — абсолютная температура; Уа—
число Авогадро.
Вставляя найденные выражения для f' и f в уравнение (III, 6),
лслучаем:
de
dx
о ВТ
Bu.~----jt-
зсУ,
(IH, 9)
Это уравнение можно переписать следующим образом:
RT de 1
sJV л dx В
uc = —
Но ис есть не что иное, как количество вещества т, продиффун-
дировавшее в единицу времени через единицу площади. Поэтому
RT de 1
sN\ dx В
(HI, 10)
tn — —
Согласно уравнению Фика, для тех же условий
dx
(111,11)
Приравнивая правые части уравнений (111,10) и (111,11), на-,
ходим:
RT 1 kT
N.' В
(III, 12)
1 kT
блцг
(HI, 13)
Это и есть уравнение Эйнштейна. Для частиц, по форме близких
к сферическим, В = 6лт]г и
Уд 6лг|г
Как можно видеть, коэффициент диффузии прямо пропорциона-
лен абсолютной температуре и обратно пропорционален вязкости
среды и радиусу частиц. Поскольку размеры коллоидных частиц
очень велики по сравнению с размерами обычных молекул, коэф-
фициент диффузии в коллоидных системах мал.
Поэтому Грэм пришел в свое время к неправильному заключе-
нию, что диффузия в коллоидных системах отсутствует.
,, Пользуясь уравнением Эйнштейна, можно легко определить
Массу 1 моля вещества, если известны значения D, Т и т]. В самом
Деле, из уравнения (III, 13) следует, что
r = kT
(Ш, 14)
61
Определив по уравнению (III, 14) радиус частиц, можно вычис-
лить массу 1 моля вещества М:
Л1 = 4/3лг3рУл (III, 15>
где р — плотность дисперсной фазы, которая обычно известна.
Таким образом, измеряя коэффициент диффузии, можно опре-
делить молекулярный вес вещества (численно равный массе 1 моля
вещества).
Перрен и Герцог на основании определения коэффициента диффузии в вод-
ных растворах вычислили молекулярные веса некоторых углеводов, допустии,-
что молекулы их имеют сферическую форму и они настолько малы, что воду
можно рассматривать как непрерывную среду и что увеличения радиуса мо-
лекул, а следовательно, и коэффициента В вследствие сольватации растворен-
ного вещества не происходит. Полученные ими результаты приведены в
табл. Ill, 1.
Таблица III. I. Молекулярный вес разлччных углеводов, вычисленный
по коэффвциенту диффузии
Углевод D, см2/суткн р, Г/см3 М
опытное значение теоретическое значение
Арабиноза 0,540 1,6182 120 ieo
Тростниковый сахар 0,384 1,5873 332 342
Молочный сахар 0,377 1,5245 331 342
Мальтоза 0,373 1,4992 336 342
Рафиноза 0,316 1,5017 553 504
Совпадение экспериментальных результатов с рассчитанными
на основании данных о химическом строении этих веществ доста-
точно удовлетворительно, несмотря на сделанные допущения.
Для экспериментального определения коэффициента диффузии
предложено много методов. Все они основаны на том, что раствор^
диффундирующего вещества приводят в контакт с растворителем
таким образом, чтобы между ними образовалась возможно более
четкая граница раздела. Полученную систему выдерживают неко-
торое время при постоянной температуре в условиях, полностью'
исключающих сотрясения и тепловую конвекцию, и затем опре-
деляют наступившее в результате диффузии новое распределение
концентрации растворенного вещества в системе. По полученный
результатам, обычно с помощью специальных таблиц, вычисляют
коэффициент диффузии.
Различные модификации этого способа отличаются только ап-
паратурным оформлением и способом определения распределения
концентрации вещества после диффузии. Распределение концен-
трации может быть установлено с помощью осторожного отбора
проб и их анализа, а также путем определения характера измене-
ния окраски, абсорбции света или коэффициента преломления рас-
твора по высоте столба жидкости. Экспериментальные методы
62
П
Д
л
2
&
N
определения коэффициента диффузии подробно описаны в руко-
водствах к практическим занятиям по физической и коллоидной
химии.
Причина диффузии в ястинных-растверах,- -как выше указано,
заключается в. тепловом движении молекул. Аналогично в кол-
лоидных системах причиной диффузии дисперсной фазы является
броуновское движение частиц. фс'ЛИ существует связь между броу-
новским движением и диффузией, то должна существовать связь
между средним квадратичным значением проекции смещения час-
тицы Д и коэффициентом диффузии D.
Эта связь была установлена Эйнштей-
ном (1905 г.) и независимо от него
Смолухрвским (1906 г.).
Для установления зависимости
Д = /:(/)) воспользуемся несколько
упрощенными рассуждениями Эйн-
штейна, приведенными в одной из его
работ. Кал и при выводе уравнения
Эйнштейна, представим себе изобра-
женную на рис. III,4 трубку с попе-
речным селением в s см2, наполненную
коллоидным раствором, концентрация
которого равномерно уменьшается сле-
ва направо. В этом же направлении,
конечно, будет идти и диффузия кол-
лоидных частиц. Выделим мысленно в
твора 1 и 2 со средними концентрациями растворенного вещества
Ci и с2, отвечающими концентрации вещества в центре выделен-
ных слоев. Пусть Д — среднее квадратичное значение проекции
смещения частицы на ось, параллельную направлению диффузии,
за время т.
Вследствие хаотичности броуновского движения перенос любой
частицы растворенного вещества равновероятен вправо и влево.
Масса вещества, перенесенного за время т через воображаемую
плоскость MN вправо, будет mi = Ci Дз/2, а влево т2 = с2Дз/2.
Очевидно, в результате обоих этих процессов количество веще-
ства, продиффундировавшее через плоскость MN слева направо,
выразится уравнением:
т — — т2 = ’Дн As — V2C2 = '/2^1 — сг} «
т2= у сД с
Рис. III, 4. Схема, поясняющая
зависимость Д = Ц£>).
этой трубке два слоя рас-
(III, 16)
Но из рис. III, 4 следует, что
или С] — сг de Д dx (HI, 17)
— de ‘ dx (1П, 18)
63
Подставив уравнение (111,18) в уравнение (111,16), получим:
1 — de
m = -±.^2^-s (Ш, 19)
2 dx
Объединяя уравнение (111,19) и уравнение Фика т ——Dstds/dx,
имеем: _
D = Д2/(2т) (111,20)
или _
Д==У2Р^ (Ш,21)
Из этого следует важный вывод, что величина А, а следова-
тельно, и само смещение пропорциональны не т, а V т.
Вставляя в уравнение (111,21) значение D из уравнения Эйн-
штейна (III, 13), можно получить:
/ kTx
'V Злдг
RT2x
N дблдг
(Ш,22)
Это уравнение обычно называют уравнением Эйнштейна — Смолу-
ховского.
Сведберг проверил правильность уравнения Эйнштейна — Смо-
луховского, исследуя под ультрамикроскопом золи золота. Он
определял значения А через разные промежутки времени и вычис-
лял теоретические значения А по формуле A = V&iT> где kx =
= &77(3лт]г). Ниже приведены полученные им результаты для
золя золота с частицами диаметром 0,044 мкм:
Время наблюдения, с.........? 1 2 3 4
Значение Д, мкм
экспериментальное............ 4,3 5,8 6,6 8,3
вычисленное............... 4,1 5,8 7,6 8,2
Совпадение результатов оказалось вполне удовлетворительным.
Подобные же результаты были получены Сведбергом на других
объектах, а также и другими исследователями.
Помимо этого для проверки правильности уравнения Эйн-
штейна — Смолуховского Сведберг определял зависимость А от
вязкости дисперсионной среды коллоидной системы. В этом слу-
чае для вычисления теоретического значения А он пользовался
формулой A = VWn, где k2 = кТг/(Злг).
Наконец, Зеддиг проверил правильность уравнения Эйнштей-
на — Смолуховского, определяя зависимость А от температуры.
При этом теоретические значения А он вычислял по уравнению
где &з = &т/(Злг). В правой части этого равенства
фигурирует величина т], поскольку вязкость дисперсионной среды
меняется с температурой. И в этом случае опыт подтвердил
теорию.
Пользуясь уравнением Эйнштейна — Смолуховского и зная зна-
чение А и всех остальных величин, можно вычислить число Аво-
64
гадро. Это проделал Перрен, проводивший опыты с суспензией
гуммигута. Вычисленное им число Авогадро равнялось 6,8-1023,
что близко к значениям NA, определенным другими методами
(6,02-1023).
Интересно, что Перрен нашел значение числа Авогадро, наблюдая не только
поступательное, но и вращательное движение микроскопически видимых частиц.
Как было указано в предыдущем разделе, под влиянием ударов, сообщаемых
частице молекулами среды, она начинает вращаться. Это вращательное движе-
ние, как показал Эйнштейн, подчиняется уравнению:
V Аа 4my3
(III, 23)
где 0 — среднеквадратичное угловое смещение частицы за время т.
Перрен, наблюдая под микроскопом вращение относительно больших частиц
суспензии мастики и пользуясь уравнением (111,23), получил для числа Аво-
гадро значение, равное 6,5-1023.
Исследование броуновского движения и диффузии в коллоид-
ных системах не только дало многое для понимания природы дис-
персных систем и установления общности молекулярно-кинетиче-
ских свойств этих систем и систем молекулярной дисперсности, но
и явилось доказательством правильности молекулярно-кинетиче-
ской теории в целом. Теория броуновского движения, созданная
Эйнштейном и Смолуховским, подтвердила реальное существова-
ние молекул как раз в то время, когда по этому вопросу разверну-
лась ожесточенная дискуссия, поднятая Вильгельмом Оствальдом
и другими представителями энергетической школы, советовавшими
избегать пользоваться понятиями атома и молекулы, поскольку,
по их мнению, за этими словами не кроется объективная ре-
альность.
Таким образом, значение теории броуновского движения выхо-
дит далеко за пределы коллоидной химии, в которой она, кстати
говоря, явилась первой количественной теорией. Теория броунов-
ского движения, согласно которой движение коллоидных частиц —
прямое следствие теплового движения молекул, приобрела огром-
ное значение в физической химии, физике и философии, явившись
убедительным обоснованием правильности материалистического
мировоззрения. Исследование броуновского движения привело
к созданию теории флуктуаций и способствовало развитию стати-
стической физики.
Флуктуации представляют собой спонтанные отклонения ка-
кого-нибудь параметра от среднего равновесного значения в до-
статочно малых объемах системы. Флуктуациям подвержены
физические величины (например, плотность, концентрация), био-
логические (рост, продолжительность жизни), социально-экономи-
ческие (плотность населения в данном месте, цена товара)
И Т- д. Основы общей теории флуктуаций заложены американским
ученым Гиббсом (1902).
3 Зак 664 ЙЦ
Простейшим способом наблюдения флуктуаций в коллоидной
системе является определение через равные промежутки времени
числа частиц, находящихся в микроскопически малом объеме. На-
пример, при подсчете таким образом числа частиц, находящихся
в 1000 мкм3 золотого золя, Сведбергом был получен следующий
ряд значений: 1, 2, 0, 0, 2, 0, 0, 1, 3, 2, 4, 1, 1, 2, 3, 1, 1, 1, 1, 3, 1,
1, 2, 5, 1, 1, 2, 3. Всего им было сделано 518 наблюдений. В сред-
нем число частиц в выделенном объеме составило 1,545, но в от-
дельные моменты оно падало от 0 и повышалось до 7.
Несоответствие найденных чисел среднему значению можно
объяснить только тем, что хаотическое движение частиц приводит
к случайному попаданию в выделенный микрообъем то большего,
то меньшего числа частиц. Понятно, что если бы вместо микро-
объема был выделен для наблюдения достаточно большой объем,
содержащий огромное число частиц, то в результате компенсации
избытка частиц в одних его участках недостатком частиц в дру-
гих участках средняя численная концентрация в таком макрообъ-
еме оказалась бы постоянной.
Из сказанного следует, что флуктуации представляют собою
явление как бы обратное явлению диффузии, хотя оба они обус-
ловлены тепловым движением. При диффузии происходит вырав-
нивание концентраций в макрообъемах, а флуктуации представ-
ляют собою спонтанное отклонение концентрации от среднего его
значения в микрообъемах.
В нашем курсе мы не можем рассматривать теорию флуктуа-
ций. Отметим лишь, что применительно к коллоидным системам
эта теория позволяет вычислить вероятность различных числен-
ных концентраций в микрообъемах, а также найти тот промежуток
времени, через который данная концентрация снова повторится
в выделенном объеме. Это время обычно весьма резко возрастает
с увеличением отклонения концентрации от среднего значения.
Так, расчеты показывают, что в 1 мл газа при нормальных усло-
виях отклонение численной концентрации на 1% может произойти
через 10180 лет, т. е. практически невозможно Однако абсолютной
невозможности осуществления подобного события все же нет, что
характерно для второго начала термодинамики, носящего стати-
стический характер.
3. ОСМОТИЧЕСКОЕ ДАВЛЕНИЕ
Осмотическое давление л достаточно разбавленных коллоидных
растворов можно найти по уравнению:
я=-^обд/^. /?T = v H- = vfeT (III, 24)
где т<>бщ — масса растворечиого вещества, т — масса частицы; V — объем си-
стемы, а — число Авогадро, Т — абсолютная температура, v — численная кон-
центрация.
66
Это уравнение аналогично известному уравнению Вант-Гоффа для
осмотического давления истинных растворов:
Я = ,?общ/Л£ (III, 25)
где д/ — масса 1 моля растворенного вещества, с — весовая концентрация.
Следует заметить, что молекулярно-кинетические уравнения,
пригодные для истинных растворов, приложимы и к коллоидным
растворам с той только разницей, что масса моля вещества заме-
няется в них массой частицы.
Особенностями осмотического давления лиозолей по сравнению
с истинными растворами является его малое значение и непо-
стоянство. Причина низкого осмотического давления у лиозолей
становится понятной из следующего пояснения.
Для двух систем с осмотическими давлениями ла и Л2, нахо-
дящихся при одной и той же температуре, можно написать:
л1Я=У1(/?Г/УА) (111,26)
л2 = v2 (7?Г/Уд) (Ш, 27)
Разделив уравнение (111,26) на уравнение (111,27), получаем:
«1/«2='V1/V2 (111,28)
Из этого уравнения следует, что осмотическое давление дисперсной
системы определяется только численной концентрацией и не зави-
сит от природы и размера частиц. Это же уравнение объясняет и
малое осмотическое давление любой коллоидной системы, так как
благодаря большой массе коллоидных частиц при Одной и той же
весовой концентрации численная концентрация коллоидной си-
стемы всегда значительно меньше, чем у истинного раствора.
Весьма малое осмотическое давление у лиозолей было причиной
ошибки Томаса Грэма, не располагавшего чувствительными осмо-
метрами и считавшего, что у коллоидных систем осмотическое дав-
ление отсутствует вовсе.
Вторая особенность осмотического давления лиозолей — его не-
постоянство— объясняется явлением агрегации, характерным для
коллоидных систем. Для двух коллоидных систем с дисперсной
фазой одной и той же природы и с одинаковой весовой концен-
трацией с, но различающихся размером частиц, можно написать:
_ У. VQ/^р) _ Г23
«2 V2 с/(‘/з^р) Г? 1 ’ '
где г, и г2 — радиусы частиц первой и второй системы; р — плотность дисперс-
ной фазы
Согласно этому уравнению отношение осмотических давлений
Двух таких систем обратно пропорционально отношению куба ра-
диусов их частиц. Отсюда следует, что даже небольшое изменение
3* 67
радиуса частицы вызывает большое изменение осмотического да-
вления. Ясно, что при такой зависимости с увеличением среднего
радиуса частиц системы в результате их слипания и образования
агрегатов должно весьма сильно падать осмотическое давление.
Наоборот, при распаде агрегатов на первичные частицы осмотиче-
ское давление должно сильно повышаться. Поскольку явления
агрегации и дезагрегации в коллоидных системах весьма легко
наступают под действием иногда даже очень слабых внешних воз-
действий, становится понятным непостоянство осмотического дав-
ления лиозолей и зависимость его от предыстории раствора.
Малое значение и непостоянство осмотического давления лио-
золей являются причиной того, что осмометрия, а также эбулио-
скопия и криоскопия не применяются для определения численной
концентрации или размера коллоидных частиц. Следует, впрочем,
заметить, что осмометрические, эбулиоскопические и криоскопиче-
ские методы нельзя использовать для определения размера кол-
лоидных частиц не только вследствие указанных причин, но и
из-за обычного присутствия в лиозолях электролитов. При очистке
лиозолей, например диализом, вместе с посторонними электроли-
тами может удаляться и стабилизующий электролит, что приводит
к нарушению агрегативной устойчивости системы, укрупнению ча-
стиц и, следовательно, к получению неправильных значений осмо-
тического давления. Кроме того, на результатах осмометрических
определений сильно сказывается так называемое «мембранное
равновесие», или равновесие Доннана. Это равновесие устанав-
ливается в результате сложного распределения ионов между
коллоидным раствором в осмотической ячейке и внешним раство-
ром, о чем подробно сказано в гл. XIV.
Однако осмометрия вполне применима для определения мо-
лекулярного веса высокомолекулярных веществ, образующих
истинные растворы и не требующих для стабилизации растворов
присутствия в них электролитов. Такое определение возможно
благодаря тому, что растворы высокомолекулярных веществ мог.ут
быть получены достаточно высокой концентрации, как правило,
вполне агрегативно устойчивы и обычно хорошо выдерживают
операции очистки.
4. СЕДИМЕНТАЦИОННАЯ УСТОЙЧИВОСТЬ
При рассмотрении диффузии мы не принимали во внимание
влияние гравитационного поля (земного притяжения) на систему.
Между тем обязательно следует учитывать это влияние на частицы
достаточно большой массы, так как такие частицы под действием
гравитационного поля будут оседать, или седиментировать. В ре-
зультате этого в системе установится определенное равновесное
распределение частиц по высоте либо, если частицы достаточно
тяжелы, все они выпадут в осадок.
Способность дисперсной системы сохранять равномерное рас-
пределение частиц по всему объему принято называть седимента*
68
ционной, или кинетической, устойчивостью системы. Очевидно,
о седиментационной устойчивости или неустойчивости имеет смысл
говорить только при рассмотрении свободнодисперсных систем,
когда каждая частица свободна и находится в тепловом движе-
нии.
Грубодисперсные системы (например, пыль или суспензия песка
в воде) седиментационно неустойчивы и оседают, так как частицы
их тяжелы и практически не могут осуществлять теплового (броу-
новского) движения. Наоборот, высокодисперсные системы (газы,
истинные растворы) обладают высокой кинетической устойчи-
востью, так как им свойственны тепловое движение и способность
к диффузии. Коллоидные системы (аэрозоли, лиозоли) по устой-
чивости занимают промежуточное положение.
Распределение одинаковых по размеру частиц, видимых в микроскоп или
ультрамикроскоп, по высоте можно исследовать двумя методами. В первом слу-
чае микроскоп располагают горизонтально и при исследовании системы пере-
двигают его по высоте. Тогда сразу видно, что число частиц убывает с высотой.
Однако для выявления зависимости убывания частиц с высотой обычно поль-
зуются вторым методом Согласно этому методу микроскоп при исследовании
устанавливают вертикально, при этом видны только частицы, находящиеся в
слое, на который фокусирован микроскоп. Толщина этого слоя в опытах Пер-
рена, работавшего с моиодисперсным золем гуммигута, составляла ~ 1 мкм
Поднимая или опуская тубус, микроскоп можно было фокусировать на слои,
которые лежали выше или ниже начального В одной из серий опытов Перрена
при общем числе частиц 13 000 и диаметре их в 0,212 мкм соотношение числа
частиц в слоях, отстоявших от дна кюветы на расстояниях 5, 35, 65 и 95 мкм,
составляло 100 : 47 : 22,6:12. Как можно видеть, через каждые 30 мкм число
частиц в поле зрения микроскопа убывало вдвое. Таким образом, при возраста-
нии высоты в арифметической прогрессии число частиц в поле зрения микро-
скопа уменьшалось в геометрической прогрессии Следовательно, как и пред-
полагал Перрен, взвешенные в жидкости частицы распределяются по высоте
в гравитационном поле по той же барометрической формуле, что и молекулы
газа. За эти опыты, увенчавшиеся окончательной победой атомизма и отличав-
шиеся исключительной точностью, остроумием и простотой, Перрену в 1926 г.
была присуждена Нобелевская премия.
Удобным методом рассмотрения того, как влияет диффузия
или седиментация на кинетическую устойчивость дисперсной си-
стемы, является сравнение диффузионного потока с противонаправ-
ленным ему седиментационном потоком.
Удельный диффузионный поток 1Д, как было показано ранее,
выражается уравнением (III, 3):
г’д = — D dc/dx
Удельный седиментационный поток ic, учитывая, что для одной
частицы, оседающей с постоянной скоростью, сила трения Ви
равна движущей силе (силе тяжести) mg, можно представить
уравнением:
г'с = ис = (mg/В) с (111,30)
Где и скорость седиментации; с—концентрация; m — эффективная масса ча-
ицы; g — ускорение свободного падения; В — коэффициент трения между кол-
ядной частицей и дисперсионной средой.
69
Разделив уравнение (III, 30) на уравнение (III, 3) и приняв во
внимание уравнение (III, 12), получим:
4 mg с р(р — p0)g с (III ЗП
(д = kT dcldx kT dc/dx '
где v — объем частицы; р и ро — плотность дисперсной фазы и дисперсионной
среды.
При i'c/г'д 1 можно принимать во внимание только седимен-
тацию, при ic/1'д 1—только диффузию, а при 1с/»д 1, т. е.
когда ic « 1д, необходимо учитывать оба процесса. В последнем
случае в системе устанавливается определенное распределение
дисперсной фазы по высоте.
Из уравнения (111,31) вытекает также, что если рассматривать
устойчивость системы, в которой дисперсная фаза равномерно
распределена по объему (например, в результате предваритель-
ного перемешивания), то в первое время всегда преобладает
седиментация, поскольку вначале dc/dx = 0. Однако со временем
равномерное распределение вещества в системе нарушается и про-
изводная dc/dx приобретает все возрастающие значения. Массопе-
ренос будет продолжаться до тех пор, пока поток »д не станет
равным t'c, т. е. пока не будет соблюдено условие 1сЛд = 1 и в
системе установится равновесие. Учитывая, что градиент концен-
трации изменяется по высоте, и заменяя х на h, можно написать
уравнение (111,31) в виде
mg с
kT ' dc/dh
Это уравнение можно написать и в таком виде:
- —(Ш,32)
с kT
Интегрируя от с0 до и соответственно от 0 до h, получим:
in (c0/cft) = mgh/kT — mNAghjRT (III, 33)
ИЛИ
= (И1.34)
Если в уравнении (III, 33) величину с заменить пропорциональной
ей величиной давления р, то получим хорошо известную бароме-
трическую формулу:
^{PolPh),s=m8hlkT (111,35)
Это так называемый гипсометрический закон, которому подчи-
няется распределение молекул газа по высоте.
Поскольку концентрация дисперсйби фазы с пропорциональна
численной концентрации v, уравнение (III, 33) можно представить
также в виде:
In (v0/vA) == mgh/kT (111,36)
7Q
Это уравнение можно переписать следующим образом:
kT In (vn/v.)
h =-----(Ш, 37)
mg
С помощью этой формулы удобно вычислять для любой сво-
боднодисперсной системы величину й, представляющую собою вы-
соту, на которую надо подняться, чтобы численная (или, соответ-
ственно, весовая) концентрация уменьшилась с vo до v*, или в voM
раз.
В табл. III, 2 приведены вычисленные по уравнению (III, 37)
величины й при (v0/vft) = 2 и (v0/vA) = 106.
Таблица Ш,2. Снижение концентрации вещества с высотой в различных
дисперсных системах
Система Диаметр частиц, нм Высота h (в см), на которой концентрация убывает
в 2 раза в 106 раз
Кислород 0,27 500 000 (5 км) 10 000 000 (100 км)
Золь золота 1,86 215 4 300
То же 8,35 2,5 50
» » 186 2 • 10“s (0,2 мкм) 4 • 10“4 (4 мкм)
Суспензия гуммигута 230 3 • 10“3 (30 мкм) 6 • 10“2 (600 мкм)
Как видно из данных табл. III, 2, значение й резко падает с уве-
личением массы (диаметра) частиц. Более высокое значение й для
суспензии гуммигута с частицами диаметром в 230 нм, чем для
золя золота с частицами 186 нм в диаметре, объясняется тем, что
плотность золота во много раз больше плотности гуммигута.
Понятно, что распределение частиц по высоте, подчиняющееся
гипсометрическому закону, осуществляется только в монодисперс-
ных системах. В случае полидисперсных систем картина распре-
деления гораздо более сложная. Ниже приведены данные о влия-
нии броуновского движения и седиментации на скорость передви-
жения частиц в полидисперсном гидрозоле серебра:
Диаметр частиц, мкм.............. 0,1
Расстояние, проходимое частицей
за 1 с, мкм
в результате броуновского дви-
жения ...........................10,0
в результате седиментации . . 0,0676
1 10
3,1 1,0
6,75 676,0
Из сравнения скорости броуновского движения и седиментации
можно видеть, что частицы серебра диаметром меньше 0,1 мкм
будут более или менее равномерно распределены по всему объему
системы, частицы серебра диаметром более 10 мкм будут нахо-
диться в основном в осадке, частицы же промежуточных размеров
71
будут распределены по высоте согласно гипсометрическому закону,
причем для каждой фракции будет устанавливаться свое равно-
ьесие.
Несмотря на то, что для золей, согласно гипсометрическому за-
кону, концентрация должна уменьшаться очень быстро с высотой,
весьма часто дисперсные системы практически обнаруживают одну
и ту же концентрацию по всей высоте столба. Особенно это харак-
терно для высокодисперсных золей. Такое явление объясняется
тем, что с уменьшением размера частиц сила тяжести, обусловли-
вающая оседание, уменьшается-гораздо быстрее, чем сила трения,
поскольку сила тяжести для частиц сферической формы пропор-
циональна третьей степени радиуса частиц, а сила трения пропор-
циональна только первой степени радиуса. В результате этого
установление равновесия, при котором приложим гипсометриче-
ский закон, достигается у высокодисперсных золей чрезвычайно
медленно, и в большинстве случаев мы имеем дело с системами,
в которых распределение частиц по высоте очень далеко от пред-
писываемого теорией.
В качестве иллюстрации влияния размера частиц на скорость
оседания ниже приведены скорости седиментации сферических ча-
стиц кварца в воде в зависимости от их радиуса (для SiO2
р = 2,7 г/см3; для Н2О т] — 0,015 П):
Радиус частиц, см 10~3 10-4 1Q-5 10~e 10-7
Скорость седи-
ментации, см/с 3,2 • 10~2 3,2 • I0-4 3,2 -10 3,2-10 3,2 • 10-!О
Время, необходи-
мое для оседа-
ния частиц на
расстояние в
1 см........ 31 с 51,7 мин 86,2 ч 359 дней 100 лет
На равновесное распределение частиц в системе влияют самые
незначительные толчки и сотрясения, а также неодинаковая тем-
пература в различных участках золя, что приводит к образованию
в системе конвекционных потоков. Расчеты показывают, что доста-
точно, например, колебаний температуры на 0,001 °C в 1 ч, чтобы
седиментация в высокодисперсном золе золота была полностью
исключена.
Тем не менее в условиях достижения равновесного распределе-
ния частиц в системе гипсометрический закон для лиозолей соблю-
дается достаточно точно. Доказательством этому служит то об-
стоятельство, что Перрен, исходя из установленного им с помощью
микроскопа равновесного распределения по высоте относительно
больших частиц монодисперсной суспензии гуммигута, смог вы-
числить число Авогадро. Найденное таким образом значение
числа АД оказалось равным 6,82- 1023, что довольно близко к зна-
чению, найденному с помощью других методов Вестгрен, работая
с золями золота, получил еще более точное значение числа Аво-
72
гаДр0 — 6,05-1023. Интересно, что Вестгрен достигал равновесия
как при седиментации вещества, так и при его диффузии со дна
сосуда.
5. СЕДИМЕНТАЦИЯ И МЕТОДЫ СЕДИМЕНТАЦИОННОГО АНАЛИЗА
Наблюдение за скоростью седиментации в суспензиях, т. е.
в дисперсных системах с достаточно большими частицами, обла-
дающих практически полной кинетической неустойчивостью, по-
зволяет сравнительно легко и удобно определять размер частиц.
Применяющиеся при этом методы получили название методов се-
диментационного анализа.
Рассмотрим, как оседает в жидкости отдельная частица такой
суспензии.
Оседание частицы, очевидно, происходит под действием силы
тяжести f, которая с учетом на потерю в весе, по закону Архи-
меда, составляет:
f = v(p —p0)g
где v — объем частицы; р — плотность вещества частицы; ро — плотность среды;
g — ускорение свободного падения
Оседанию противодействует сила трения f'i
f' = Ви
где В— коэффициент трения между частицей и средой; и — скорость седимен-
тации частицы
Вначале частица движется ускоренно, так как при малых ско-
ростях сила тяжести превышает силу трения. По мере увеличения
скорости движения сила трения возрастает и в некоторый момент
уравновешивает силу тяжести, вследствие чего частица начинает
двигаться с постоянной скоростью. При стационарном режиме осе-
дания, очевидно, должно соблюдаться равенство:
v(p — Po)g = fi« (Ш, 38)
Применительно к сферическим частицам это уравнение принимает
вид
4/3№ (р — р0) g = блтуи (Ш, 39)
Из уравнения (111,39) легко найти скорость седиментации ча-
стицы:
2 г2
и = -д^-(р —Po)g (Ш,40)
Согласно уравнению (111,40) скорость седиментации частицы
прямо пропорциональна квадрату радиуса (или диаметра) ча-
стицы, обратно пропорциональна вязкости среды и зависит от раз-
вести р — р0 так, что при р 2> р0 происходит оседание, а при
Р < ро (например, суспензия парафина в воде) всплывание ча-
стиц— обратная седиментация.
73
(П1.41)
имею-
частиц
Из уравнения (111,39) легко также найти радиус частицы,
зная скорость ее оседания и значение величин т], р и ро:
V9r|«
2(р — Ро) g
Уравнение (111,39) справедливо для водных суспензий,
щих частицы размером от 0,1 до 100 мкм, так как для этих
время нарастания скорости оседания до постоянного значения на-
столько мало, что не оказывает влияния на результаты седимен-
тационного анализа. Например, время достижения постоянной
скорости оседания частиц кварца радиусом 50 мкм в воде состав-
ляет 3,4-10-3 с, а для частиц радиусом 1 мкм оно равно всего
1,7-10~6 с.
К частицам радиусом больше 100 мкм, в обычных условиях
оседающим ускоренно, и к частицам радиусом меньше 0,1 мкм,
содержащимся в кинетически устойчивых системах, уравнение
(111,39) неприложимо. Поэтому обычный седиментационный ана-
лиз в этом случае непригоден.
Рассмотрим теперь седиментацию дисперсных систем, состоя-
щих из множества частиц. При этом примем, что частицы в таких
суспензиях оседают совершенно независимо друг от друга.
В монодисперсной системе, поскольку скорость оседания одина-
ковых по размеру частиц одинакова, отстаивание будет происхо-
дить равномерно (высота слоя осветленной жидкости пропорцио-
нальна времени оседания т). При этом граница раздела отстояв-
шейся концентрированной суспензии и прозрачной среды будет
смещаться на некоторое расстояние. Тогда скорость оседания и
выразится уравнением:
и = Щх ' (Ш, 42)
а радиус частиц уравнением:
r = k^Hix (HI, 43)
где __________
k
Л/ 2 (р — p0)g
По уравнению (III, 43) легко вычислить радиус частиц суспен-
зии по результатам наблюдения за ее оседанием визуально. Ско-
рость седиментации монодисперсной суспензии можно определить,
наблюдая за оседанием какой-нибудь одной из ее частиц в микро-
скоп.
При отстаивании полидисперсной суспензии в ртличие от моно-
дисперсной граница оседающего слоя оказывается размытой, так
как частицы, имеющие различные радиусы, проходят за одно и то
же время различные пути. Поэтому седиментационный анализ
полидисперсной системы сводится к определению скорости накоп-
ления осадка.
Способы установления зависимости между массой выпавшего
осадка т и временем оседания т, графически изображаемые так
74
называемой кривой седиментации, из которой получают необхо-
димые данные для характеристики полидисперсности исследуемой
системы, подробно рассматриваются в практикумах по коллоид-
ной химии. Отметим лишь, что при вычислении размера частиц,
Рис. III, 5. Построение кривой распределения.
исходя из кривой седиментации, характеризующей содержание
в полидисперсных системах частиц различного размера, помимо
классического графического метода расчета, предложенного Све-
ном Оденом, применяются аналитические методы построения кри-
вых седиментации, разработанные Н. Я. Авдеевым и Н. Н. Цюру-
пой.
По результатам, полученным при обработке кривой седиментации, обычно
строят кривую распределения, наглядно показывающую весовое содержание Q
в суспензии различных фракций. Дли
этого строят диаграмму, на оси абсписс
которой откладывают значения радиу-
сов частиц г, на оси ординат — значения
Q/Ar для каждой фракции. Пример та-
кой диаграммы приведен на рис. III, 5.
Тогда Q/(Ar-Ar) = Q и весовое содер-
жание каждой фракции выразится пло-
щадью соответствующего прямоугольни-
ка. Построив такие прямоугольники для
всех фракций и соединив плавной ли-
нией средние точки их верхних сторон,
получают кривую распределения Фрак-
ция, отвечающая максимуму кривой рас-
г—
Рис. III, 6. Типичные кривые распре-
деления:
1 — система, наиболее приближающаяся
к монодисперсной; 2—наиболее полидис-
персная система; 3, 4—системы, содержа-
щие соответствеиио преимущественно
мелкие частицы (максимум сдвинут влево)
и крупные частицы (максимум сдвинут
вправо).
пределения, называется преимуществен-
ной фракцией полидисперсной суспензии,
так как очевидно, что частиц, составляю-
щих эту фракцию, в системе больше
всего (на рис. III, 5 эта фракция IV с
частицами, радиус которых лежит в пре-
делах от г4 до г5).
Кривые распределения являются важной характеристикой дисперсных си-
стем Чем уже интервал радиусов кривой распределения и чем выше ее макси-
мум, тем суспензия ближе к монодисперсной Наоборот, чем кривая более рас-
тянута и чем ниже максимум, тем суспензия более полидисперсна. На рис. 111,6
Для примера приведены кривые распределения четырех различных типов.
75
Остановимся теперь кратко на экспериментальных приемах, ис-
пользуемых при седиментационном анализе полидисперсных си-
стем.
Весовая модификация седиментационного анализа заключается
в определении скорости накопления осадка на чашке весов. Для
этой цели были предложены седиментационные весы самых раз-
нообразных конструкций. Наиболее распространенными седимен-
тационными весами является прибор Н. А. Фигуровского, предло-
(рис. Ш, 7) представляет собою
закрепленный в горизонталь-
ном положении в держателе
штатива J гибкий стеклянный
или кварцевый шпиц 2, на ко-
нец которого подвешивают на
стеклянной нити тонкостенную
стеклянную чашечк^ <?• Рас-
стояние между уровнем жид-
кости и подвешенной чашечкой
обозначено через Н. При по-
гружении чашечки в цилиндр
с испытуемой суспензией на
чашечке постепенно накапли-
вается осадок и шпиц проги-
бается. За прогибом шпица
женный им в 1936 г. Этот прибор
Рис. III, 7. Седиментометр Фигурон-
ского:
1—держатель штатива; 2—шпиц; 3 — чашечка.
следят при помощи отсчетного
микроскопа. Отмечая во време-
ни перемещения конца шпица
по микрошкале, строят график
изменения величины прогиба шпица во времени. Так как при про-
гибе шпица имеет место подчинение закону Гука, то полученный
график выражает зависимость массы осадка т от времени т, т. е.
m = f(r). Вместо того, чтобы подвешивать чашечку к концу гиб-
кого шпица, ее можно укрепить 'к коромыслу торзионных весов.
При этом за накоплением осадка следят по перемещению стрелки
на циферблате весов.
‘ Другой метод наблюдения за ходом седиментации полидисперсных систем
, предложен Вигнером в 1918 г. Этот метод основан на измерении гидростатиче-
/ ского давления столба суспензии при выделении из нее дисперсной фазы в ре-
зультате седиментации. Седиментация по этому методу проводится в специаль-
ном приборе — седиментометре Вигнера (рис. III, 8). При закрытом кране, со-
единяющем оба колена прибора, в широкую трубку 1 заливают суспензию, а в
узкую трубку 2 вводят дисперсионную среду, затем кран открывают. Вначале
уровень жидкости в трубке 2 выше, чем в трубке 1, так как средняя плотность
суспензии обычно больше, чем плотность среды. Однако по мере выпадения
дисперсной фазы суспензии и накопления осадка на дне трубки 1, плотность сус-
пензии будет постепенно приближаться к плотности среды и разность уровней
жидкости h в обоих коленах будет уменьшаться. Зависимость h от времени
седиментации можно использовать для построения кривой седиментации.
Весьма интересный метод седиментационного анализа, хотя и требующий
довольно сложного оборудования, разработали Н. А. Фигуровский и Т. Б. Гав-
76
рилова. Эти авторы применили для оседания дисперсной фазы сконструирован-
ные нми автоматические центробежные приборы, позволяющие определять раз-
меры частиц высокоднсперсных систем (до 0,05 мкм прн плотности дисперсной
фазы 2—4 г/см3). С помощью этих приборов можно изучать ки-
нетику оседания частиц и затем получать кривую распределения
частиц по размерам.
В заключение следует отметить ряд условий, огра-
ничивающих применимость седиментационного ана-
лиза. Во-первых, основное уравнение седиментацион-
ного анализа (111,39) пригодно только для расчета
размера сферических частиц. Для частиц, отличаю-
щихся по форме от сферических, уравнение (111,39)
позволяет определить только так называемый эф-
фективный, или эквивалентный радиус, т. е. радиус
воображаемых сферических частиц, обладающих той
же плотностью и оседающих с той же скоростью,
что и частицы суспензии. Во-вторых, при седимента-
ционном анализе с использованием уравнения (111,39)
можно получить правильные результаты только в том
случае, если частицы не сольватированы. Понятно,
что влияние сольватации будет сказываться в тем
большей степени, чем меньше размер частиц. Нако-
нец, в-третьих, седиментационный анализ можно
применять только тогда, когда частицы оседают раз-
дельно друг от друга (когда концентрация системы
не слишком велика) и когда они не образуют агре-
гатов.
Рис. 111,8.
Седименто-
метр Вагне-
ра:
1 — трубка для
суспензии,
2 — трубка для
дисперсионной
среды.
Ультрацентрифуга и ее применение для дисперсионного
анализа
Как уже указывалось, под действием гравитационного поля осе-
дают только достаточно крупные частицы. Коллоидные частицы
под действием силы тяжести не седиментирует или седиментируют
чрезвычайно медленно. Так, частицы кварца радиусом 0,1 мкм
проходят при оседании путь в 1 см за 86,2 ч. Однако, заменяя
гравитационное поле действием центрифуги с гораздо большим
ускорением, в сотни тысяч раз превышающим ускорение свобод-
ного падения, можно заставить оседать достаточно быстро и кол-
лоидные частицы. В частности, в центробежном поле с ускорением
105 g та же суспензия кварца должна оседать на 1 см всего за 3 с.
Использовать ультрацентрифугу для определения размера кол-
лоидных частиц впервые в 1910 г. предложил А. В. Думанский.
Шведский ученый Сведберг широко использовал эту идею, он раз-
работал. ряд конструкций ультрацентрифуг для определения раз-
мера коллоидных частиц и молекул высокомолекулярных веществ,
например белков.
Принципиальное устройство ультрацентрифуги показано на
рис. 111,9. На приводимую в движение ось центрифуги 1 надет
77
ротор 2 — массивный диск, в котором имеются радиальные вы-
резы. В эти вырезы вставляются прочные кварцевые кюветы 3,
в которые наливают исследуемую коллоидную систему. Ротор
ультрацентрифуги, вращающийся с большой скоростью (десятки
тысяч оборотов в минуту), окружен кожухом 4. В отдельных ме-
стах кожуха сделаны окошки, соответствующие вырезам ротора.
Под нижним окошком кожуха помещен источник света 6, а над
верхним окошком кожуха — фотокамера 5. При длительном вра-
щении коллоидные частицы, если их плотность больше плотности
Рис. 111,10 Перемещение границы
золь — дисперсионная среда в кю-
ветах ультрацентрифуги при центри-
фугировании.
Рис. III, 9. Принципиальное устрой-
ство ультрацентрифуги:
1—ось; 2—ротор, 3—кварцевые кюветы;
4—кожух; 5*-фотокамера; б—источник
света.
среды, отбрасываются центробежной силой к периферии. В ре-
зультате этого в кювете появляется ближе к периферии слой
концентрированного золя и ближе к оси вращения — слой освет-
лившейся жидкости. По мере центрифугирования первый слой
уменьшается, а второй—увеличивается, пока не установится седи-
ментационное равновесие или пока все частицы не осядут на дно.
Путем фотосъемки через определенные промежутки времени
можно проследить за передвижением границы между двумя
слоями. Схематически эти снимки показаны на рис. III, 10.
При центрифугировании полидисперсной системы четкой гра-
ницы, конечно, не образуется. Однако полученные сннмкн позво-
ляют установить распределение концентраций дисперсной фазы
в кювете.
При исследовании бесцветных и прозрачных золей перемеще-
ние границы или распределение концентраций в кювете приходится
определять не с помощью обычных фотоснимков, а путем наблю-
дения за коэффициентом преломления золя в различных участках
кюветы.
78
По данным, полученным тем или иным способом, можно вычис-
лить скорость седиментации или найти седиментационное равно-
весие. На основании этого, в свою очередь, можно рассчитать мо-
лекулярный вес или размер частиц, подвергающихся седимен-
тации.
Рассмотрим более подробно определение размера частиц по
скорости седиментации в ультрацентрифуге. Для расчетов приме-
нимо уравнение, в общем сходное с обычным седиментационным
уравнением (111,38). Однако поскольку/.при центрифугировании
частицы, постепенно удаляясь от оси вращения, двигаются с пе-
ременной все возрастающей скоростью, в уравнении величина и
должна быть заменена на dx/dx (где х — расстояние частицы от
оси вращения). В то же время из механики известно, что ускоре-
ние в поле центрифуги равно а2х (где со — угловая скорость).
Тогда, очевидно, уравнение (111,38) в применении к ультрацентри-
фуге можно написать следующим образом:
В dxfdx — v (р — ро) ®2х (111,44)
Разделяя переменные и интегрируя от Xi до х2 и соответственно
от 0 до т, можно получить:
Хг X
С , , v (р — Ро)®2 f ,
\ dxfx = —----------------\ dx
Xi О
In (x2/xi) = V (р — р0) а2х/В
(111,45)
Принимая, что частицы имеют сферическую форму, и подста-
вив соответствующее значение для В, получим:
In (х2М1) = 2г2 (р — ро) <о2т/(9т]) (III, 46)
Решая это уравнение относительно г, можно написать:
)Т| In (ХгД1)
(р — р0)®2т
(III, 47)
Пользуясь уравнением (111,47), можно рассчитать численный
или молекулярный вес по перемещению в поле ультрацентрифуги
сферических частиц на расстояние х2— Xi за время т. Понятно,
что для этого необходимо также знать величины т), р, ро и со.
Уравнение (111,45) можно представить таким образом, что
в правой части его будут находиться все постоянные величины, ха-
рактеризующие исследуемую систему:
(Р - р°1,4 д const
по2 В В
(Ш, 48)
Выражение ln(x2/xi)/(w2) называют константой седиментации
И обозначают через S. Константа седиментации может служить
мерой «седиментируемости» данной системы.
Если известен коэффициент диффузии, то из соотношений
& ~ kT/B для диффузии и уравнения S = пг/В можно исключить
79
В и найти массу т, а следовательно, и численный или молекуляр-
ный вес. Такой способ определения численного или молекулярного
веса особенно пригоден для систем, содержащих частицы, сильно
отличающиеся по своей форме от сферической или сильно сольва-
тированные. Это связано с тем, что величины S и D одинаково за-
висят от формы и степени сольватации частиц.
Размер коллоидных частиц, как уже указывалось, можно
найти не только по скорости седиментации в ультрацентрифуге,
но и определяя седиментационное равновесие. Для этой цели
применяют центрифугирование при не слишком больших частотах
вращения (обычно около 20 000 об/мин), так как иначе превали-
ровала бы седиментация и равновесие не устанавливалось. Чис-
ленный или молекулярный вес, найденный по седиментационному
равновесию, отвечает равновесному распределению частиц в си-
стеме, он не зависит от способа достижения этого распределения,
и, следовательно, на результатах анализа не может сказываться
форма частиц и их сольватация.
Комбинируя определение скорости седиментации с определе-
нием седиментационного равновесия, можно найти и кривую рас-
пределения частиц, если центрифугированию подвергается поли-
дисперсная система. Сравнение результатов седиментации в уль-
трацентрифуге по обоим методам позволяет также судить и
о форме частиц.
Современная ультрацентрифуга представляет собою сложный
прибор, конструкция которого обеспечивает равномерное враще-
ние ротора и отсутствие вибраций, исключает малейшие темпера-
турные колебания в кювете и т. д. В новейших ультрацентрифугах
ротор диаметром всего в несколько сантиметров, изготовленный
обычно из хромоникелевой стали, вращается в токе разреженного
водорода. Водород, обладающий высокой теплопроводностью, обес-
печивает быстрый отвод тепла, выделяющегося вследствие трения,
и таким образом уменьшает возможность тепловой конвекции
в кювете. Такие ультрацентрифуги приводятся во вращение с по-
мощью масляных турбин. Существуют и воздушные ультрацентри-
фуги, ротор которых приводится во вращение и поддерживается
во взвешенном состоянии потоком воздуха.
Следует отметить, что в последнее время ультрацентрифуги на-
чинают использоваться не только для определения численного или
молекулярного веса дисперсных систем, но и для чисто препара-
тивных целей, например, для разделения смесей высокомолекуляр-
ных соединений в растворах на отдельные фракции, отличающиеся
размером молекул.
ГЛАВА IV
УЧЕНИЕ ОБ АДСОРБЦИИ.
АДСОРБЦИЯ НА ГРАНИЦЕ ТВЕРДОЕ ТЕЛО —ГАЗ
t. ПОНЯТИЕ ОБ АДСОРБЦИИ
Адсорбцией называется сгущение газообразного или растворен-
ного вещества на поверхности раздела фаз. Газ или растворенное
вещество принято называть в этом случае адсорбтивом *, а жид-
кость или твердое тело, адсорбировавшее их, — адсорбентом.
Так как адсорбция является поверхностным явлением, то, есте-
ственно, она имеет огромное значение для коллоидных систем,
обладающих большой поверхностью. Коагуляция лйозолей, пепти-
зация коллоидных осадков, изменение знака заряда частиц и тому
подобные явления теснейшим образом связаны с адсорбцией.
Адсорбцию газов углем наблюдал еще в XVIII в. Шееле. На
явление адсорбции веществ из раствора впервые обратил внима-
ние в 1785 г. русский академик Т. Е. Ловиц. Французский ученый
Соссюр в 1814 г. нашел, что все пористые тела, т. е. тела с боль-
шой поверхностью, способны адсорбировать газы и что при этом
обычно выделяется тепло. Соссюр сделал также очень важное на-
блюдение, что адсорбция идет тем лучше, чем легче сжижается
газ В конце XIX в. Гиббс разработал общую термодинамическую
теорию адсорбции. В XX в. явление адсорбции подробно исследо-
вали Ленгмюр, Поляни, Брунауэр, Де Бур, а в Советском Союзе —
Л. Г. Гурвич, Н. А. Шилов, М. М. Дубинин, А. В. Киселев и дру-
гие ученые.
Различают физическую, или ван-дер-ваальсову, ад-
сорбцию и химическую адсорбцию, или хемосорбцию.
В первом случае адсорбционные силы имеют ту же природу, что
и межмолекулярные, или ван-дер-ваальсовы, силы. Физическая ад-
сорбция всегда обратима. При химической адсорбции адсорбцион-
ные силы имеют химическую природу. Хемосорбция обычно необ-
ратима. В нашем курсе мы будем рассматривать, главным обра-
зом, физическую адсорбцию и лишь в соответствующем месте
укажем на принципиальное различие между обоими видами ад-
сорбции
Иногда адсорбированное вещество называют адсорбатом. Однако это
может повести к недоразумениям, так как некоторые ученые под адсорбатом
понимают адсорбент с адсорбированным на нем адсорбтивом (Н. П. Песков,
81
При химической адсорбции молекулы адсорбтива, связанные
с адсорбентом прочными химическими силами, естественно не мо-
гут перемещаться по поверхности последнего. В отличие от этого
при физической адсорбции могут иметь место как нелокализо-
ванная адсорбция, когда молекулы адсорбтива способны пере-
двигаться по поверхности адсорбента, так и локализованная
адсорбция, когда молекулы адсорбтива не могут перемещаться по
поверхности. Локализованная физическая адсорбция объясняется
тем, что поверхность адсорбента состоит из различных атомов,
ионов или молекул, по разному взаимодействующих с молекулами
адсорбента. Чтобы молекулы адсорбтива могли передвигаться по
поверхности адсорбента, очевидно, они должны преодолевать оп-
ределенные потенциальные барьеры. Однако очень часто преодо-
ление таких барьеров, если они достаточно велики, невозможно.
Понятно, что с повышением температуры локализованная физиче-
ская адсорбция может переходить в нелокализованную вследствие
возрастания кинетической энергии молекул и их способности пре-
одолевать потенциальный барьер.
Физическая адсорбция протекает самопроизвольно. Адсорбтив
стремится занять всю поверхность адсорбента, но этому препят-
ствует процесс, противоположный адсорбции — десорбция, вызван-
ная, как и диффузия, стремлением к равномерному распределению
вещества вследствие теплового движения."Для каждой концентра-
ции адсорбтива в окружающей среде существует состояние ад-
сорбционного равновесия, аналогичное равновесию между конден-
сацией и испарением. Понятно, что чем выше концентрация
адсорбтива, тем больше адсорбция. Также ясно, что чем выше
температура, тем меньше физическая адсорбция. Для каждой
температуры также существует свое состояние равновесия. Влия-
ние температуры на физическую адсорбцию вполне согласуется
с принципом Ле Шателье —Брауна, поскольку десорбция как про-
цесс, обратный адсорбции, сопровождается поглощением тепла.
Для определения количества адсорбированного вещества необ-
ходимо экспериментально найти давление газа или концентрацию
адсорбтива в сосуде, в котором происходит адсорбцйя, до и после
адсорбции. Очень часто количество адсорбированного вещества
определяют по привесу адсорбента. Следует заметить, что опреде-
ление .количества адсорбированного вещества вызывает часта
большие трудности, всегда имеющие место, когда малая искомая
величина является разностью двух больших измеряемых величин.
Чтобы уменьшить ошибку измерений, определение обычно прово-
дят, применяя в качестве адсорбента пористые тела с большой
удельной поверхностью, связывающие поэтому большое количе-
ство адсорбтива. Однако это в свою очередь имеет тот недоста-
ток, что на адсорбции может сказываться диаметр пор адсорбента.
Сравнительно большие молекулы адсорбтива не смогут проникать
в узкие капилляры адсорбента и достигнутый предел адсорбций
будет являться фиктивной величиной, не характеризующей адсорб-
82
ционное взаимодействие адсорбтива с адсорбентом. При малых
радиусах пор может происходить сжижение адсорбтива (капил-
лярная конденсация) и получаются завышенные значения коли-
чества адсорбированного вещества.
Количественно адсорбция может быть выражена с помощью
нескольких величин:
1. Величиною а, представляющей собою количество адсорб-
тива, находящегося в объеме адсорбционного слоя, отвечающего
единице массы адсорбента. Эту величину обычно измеряют
в моль/г.
2. Величиною а, показывающей количество адсорбированного
вещества, приходящегося на единицу поверхности адсорбента. Эта
величина представляет собою нечто иное, как поверхностную кон-
центрацию адсорбтива. Единицами измерения ее являются моль/м2
и ммоль/см2.
3. Введенной Гиббсом величиной Г, представляющей собою из-
быток числа молей адсорбтива в объеме поверхностного слоя пло-
щадью 1 см2 по сравнению с числом его молей в том же объеме,
если бы у межфазной границы не происходило изменения концен-
трации адсорбтива. При малых концентрациях адсорбтива гибб-
совская адсорбция Г близка к поверхностной концентрации а, при
больших концентрациях адсорбтива величина Г отличается от а.
В случаях, когда по тем или иным причинам концентрация адсорб-
тива в поверхностном слое меньше его концентрации в объеме,
величина Г отрицательна, а само явление называется отрицатель-
ной адсорбцией.
Адсорбцию можно характеризовать:
1) зависимостью количества адсорбированного вещества а от
температуры при постоянных равновесных давлении р или кон-
центрации с; графики а = f(T) при р = const называются изоба-
рами, а при с = const — изопикнами адсорбции;
2) зависимостью равновесного давления (или концентрации)
от температуры при постоянном количестве адсорбированного ве-
щества; графики р = f(T) и с = f(T) при а = const называются
изостерами;
3) зависимостью количества адсорбированного вещества а от
равновесного давления (или концентрации) при постоянной тем-
пературе; графики а — f(p) или а = f(c) при Т = const назы-
ваются изотермами адсорбции. Изотермы имеют особенно боль-
шое значение при изучении адсорбции, и поэтому в дальнейшем
мы ограничимся в основном рассмотрением этой зависимости. Схе-
матически вид обычных изотерм адсорбции изображен на
рис. IV, 1. Как можно видеть, изотермы имеют три участка. На-
чальный круто поднимающийся вверх почти прямолинейный уча-
сток кривой показывает, что при малых давлениях или концен-
трациях адсорбция практически пропорциональна этим величинам,
“то отвечает в значительной степени еще свободной поверхности
адсорбента. Почти горизонтальный участок, соответствующий
83
большим давлениям или концентрациям, отвечает поверхности ад-
сорбента, полностью насыщенной адсорбтивом. В этих условиях,
если на поверхности адсорбента может образоваться лишь моно-
молекулярный слой адсорбтива, количество адсорбированного ве-
щества перестает практически зависеть от давления или концен-
трации. Средний участок кривой соответствует промежуточным
степеням заполнения поверхности.
Крутой подъем изотермы при малых равновесных концентра-
циях хорошо согласуется с тем, что последние количества адсорб-
тива десорбируются с поверхности адсорбента с большим трудом.
PpallH (сра6н) *"
Ряс. IV, 1. Обычный вид изо-
терм адсорбция при различ-
ных температурах:
1 — изотерма, отвечающая темпера-
туре Тг, 2—изотерма,, отвечающая
температуре
Так, для удаления следов газа, адсор-
бировавшихся на внутренней поверх-
ности приборов, приходится произво-
дить длительное вакуумирование, ино-
гда при повышенной температуре. Су-
щественно, что на идеальной изотерме
физической адсорбции нет никаких
резких перегибов, характерных иногда
для изотерм адсорбции, сопровождаю-
щейся химическим взаимодействием.
Это указывает на то, что при физиче-
ской адсорбции не образуется опре-
деленных соединений между адсорбен-
том и адсорбтивом.
Из сказанного ранее о зависимости
адсорбции от температуры совершенно
очевидно, что с повышением темпера-
туры величина равновесной адсорбции
будет уменьшаться и вследствие этого изотермы для высоких
температур лежат ниже изотерм для низких температур (см.
рис. IV, 1). Однако при повышении температуры не должен изме-
няться предел адсорбции, т. е. количество адсорбтива, приходя-
щегося на единицу поверхности при предельно плотной упаковке
его молекул в мономолекулярном слое. Предел адсорбции прак-
тически не зависит от температуры и должен определяться только
размерами молекул адсорбтива. Следовательно, изотермы, отве-
чающие разным температурам, с повышением равновесного дав-
ления или концентрации в пределе должны были бы слиться
в одну. Однако этого обычно не наблюдается, так как при высоких
температурах предел адсорбции соответствовал бы очень высокому
равновесному давлению или концентрации.
Изотерма адсорбции по виду напоминает параболу. Поэтому
Бедекер, а затем Фрейндлих для ее аналитического выражения
предложили эмпирическое уравнение вида:
а = х/ш = Кр'!п
(IV, 1)
где х — количество адсорбированного вещества; m — масса адсорбента; р — рав-
новесное давление газа в системе; К и 1/п — константы.
84
Для адсорбции из раствора уравнение Фрейндлиха, как пра-
вило, пишется в таком виде:
а = х!т = 0с^"В11 (IV, 2>
где р — константа, соответствующая константе К. предыдущего уравнения; с —
равновесная концентрация.
Константа 0 уравнения (IV, 2) обычно колеблется в широких
пределах. Физический смысл ее становится ясным, если принять
с= 1, тогда р представит собой величину адсорбции при равно-
весной концентрации адсорбтива, равной 1 моль/л. Показатель 1/п,
имеющийся в обоих уравнениях, является правильной дробью и
характеризует степень приближения
занного об изменении формы изо-
термы под влиянием температуры
легко видеть, что с повышением
температуры коэффициенты Д' и р
должны уменьшаться, а 1/п — уве-
личиваться.
Константы уравнения Фрейндлиха лег-
ко найти графически по изотерме, построен-
ной в логарифмических координатах. Так,
для адсорбции из раствора имеем:
, lg а = 1g р + 1/п 1g Сравн (IV, 3)
изотермы к прямой. Из ска-
Это уравнение прямой. Логарифмируя Рис. IV, 2. Изотерма адсорбции
экспериментально найденные значения а в логарифмических координатах^
иен откладывая на осях координат 1g а
н 1g с, получают график, изображенный
на рнс. IV, 2. Отрезок, отсекаемый прямой на оси ординат, равен 1g 0, а тан-
генс угла наклона 0 прямой к осн абсцисс равен 1/п. Следует заметить, что
при логарифмировании уравнения (IV, 2) принято выражать а в ммоль/г, а
Срази — В МОЛь/л.
Как указано выше, уравнение Фрейндлиха является эмпирическим уравне-
нием Кроме того, это уравнение, представляющее собою уравнение параболы,
не может объяснить почти прямолинейного нарастания адсорбции, не завися-
щего от концентрации. Очевидно, почти прямолинейный участок изотермы, от-
вечающий малым давлениям нлн концентрациям, может быть получен с по-
мощью уравнения Фрейндлиха только в том случае, если 1/п — 1. Точно так же
горизонтальный прямолинейный участок изотермы, отвечающий высоким дав-
лениям нлн концентрациям, может быть получен только прн 1/п — 0. Таким
образом, показатель 1/п по существу должен являться сам функцией р или с.
Поскольку 1/п принимается постоянным н лежащим в пределах 0,2—1 для ад-
сорбции нз газовой среды н 0,1—0,5 для адсорбции нз растворов, уравнение
Фрейндлиха пригодно только для интервала средних давлений или концентра-
ций. Аналитически адсорбционные изотермы в целом гораздо лучше описываются
известным уравнением Ленгмюра, которое подробно рассматривается далее.
2. ПРИРОДА АДСОРБЦИОННЫХ СИЛ
Взаимодействие молекул адсорбтива с поверхностью адсор-
бента при физической адсорбции может обусловливаться различ-
ными причинами. Рассмотрим кратко эти причины, так как
Подробное рассмотрение адсорбционных сил выходит за рамки
85
коллоидной химии и сведения о них можно найти в специальных
монографиях, посвященных адсорбции, или в фундаментальных
курсах физической химии *.
Наиболее простым случаем является адсорбция неполярной
молекулы, также неполярным адсорбентом, когда действуют толь-
ко дисперсионные силы притяжения и борновские силы отталки-
вания.
Потенциал б, определяющий взаимодействие одной молекулы
адсорбента с одним атомом неполярного адсорбтива, приближенно
можно выразить, например, уравнением Леннарда — Джонса:
e = -Cr-e+ Bz~12 (IV, 4)
где г — расстояние между центрами частиц; С — константа дисперсионного при-
тяжения, В — константа, характеризующая энергию сил отталкивания.
Константа С для многоэлектронных атомов может быть выра-
жена через электрические и магнитные свойства взаимодействую-
щих молекул с помощью различных уравнений, основанных иа
квантовомеханических представлениях, которые здесь не будут
рассматриваться. Существенно, что, как это показывается в физи-
ческой химии, не только энергия дисперсионных, но и индукцион-
ных, и ориентационных сил притяжения зависят от расстояния г
одинаковым образом, а именно — обратно пропорционально ше-
стой степени расстояния.
Выражение Вг~12 введено в уравнение (IV,4) как удачное эм-
пирическое приближение. Совершенно очевидно, что на сравни-
тельно далеких расстояниях должны превалировать силы притя-
жения, а на весьма малых расстояниях — силы отталкивания.
Ясно также, что на определенном расстоянии эти силы должны
быть равны, так как это соответствует минимуму свободной энер-
гии системы.
Однако поскольку при адсорбции дисперсионные силы дей-
ствуют одновременно между каждой неполярной частицей, напри-
мер атомами аргона, и всеми близлежащими атомами адсорбента,
суммарный потенциал Ф адсорбционных сил приближенно может
быть получен суммированием потенциала парных взаимодействий
атома аргона с каждым отдельным атомом адсорбента по всем его
атомам с учетом потенциала сил отталкивания:
Ф = £ег = -с£<-6 + в £гг12 (iv,5>
В этом уравнении — С У, гг6 — суммарный потенциал диспер-
сионных сил притяжения Z?Xr~12 — суммарный потенциал сил
отталкивания, а rf— расстояния от центра атома аргона до цен-
тров различных атомов адсорбента. Поскольку энергия взаимодей-
* Например. Курс физической химии Под общей редакцией Я. И. Гераси-
мова, Т. I, М., «Химия», 1969. См. с, 460,
36
ствия частиц быстро убывает с расстоянием, то практически до-
статочно произвести суммирование по 100—200 ближайшим ато-
мам адсорбента. Существенно, что при адсорбции сложных непо-
лярных молекул потенциальную энергию взаимодействия можно
приближенно вычислить как сумму потенциальных энергий ад-
сорбции ее звеньев (силовых центров).
Если адсорбент состоит не из атомов, а из ионов, то к дей-
ствию дисперсионных сил добавляется действие индукционных сил
притяжения диполя, индуцированного в молекуле адсорбтива элек-
трическим полем, создаваемым ионами решетки адсорбента. При
этом доля индукционных сил в адсорбционном взаимодействии
пропорциональна поляризуемости молекулы адсорбтива и квад-
рату напряженности электростатического поля над поверхностью
адсорбента.
Если на неполярном адсорбенте адсорбируются полярные мо-
лекулы адсорбтива, то постоянные диполи молекул адсорбтива
поляризуют атомы адсорбента, т. е. индуцируют в них электриче-
ские моменты. Вследствие этого возникает индукционное притя-
жение, которое добавляется к дисперсионному. Индукционное
взаимодействие обычно невелико и в зависимости от диполя мо-
лекулы адсорбтива и поляризуемости адсорбента может достигать
не более нескольких ккал/моль.
Наконец, если полярные молекулы адсорбируются на адсор-
бенте, имеющем на поверхности ионы или диполи, то возникает
взаимодействие ионов или диполей адсорбтива с электростатиче-
ским полем адсорбента. При этом молекулы адсорбтива могут
ориентироваться в электростатическом поле адсорбента, т. е. про-
исходит ориентационное кулоновское взаимодействие.
Следует заметить, что если суммарное дисперсионное 'взаимо-
действие молекулы адсорбтива с адсорбентом всегда больше взаи-
модействия ее с одним активным центром адсорбента, то суммар-
ное электростатическое взаимодействие молекулы адсорбтива
может быть и меньше ее электростатического взаимодействия с
одним центром адсорбента. Такое явление можно объяснить тем,
что отрицательный полюс диполя молекулы адсорбтива, притяги-
ваемый катионом решетки адсорбента, одновременно испытывает
отталкивание со стороны соседних с этим катионом анионов, обра-
зующих вместе с катионами знакопеременную поверхность адсор-
бента.
Обычно энергии индукционного и ориентационного взаимодей-
ствия гораздо меньше энергии дисперсионного взаимодействия, и
поэтому часто принимают, что энергия межмолекулярного притя-
жения определяется энергией дисперсионного притяжения.
Причиной адсорбции, близкой к физической, может быть
также образование водородной связи. В частности, такая связь
возникает при адсорбции на адсорбентах, содержащих на поверх-
ности гидроксильные группы таких молекул, как молекулы воды,
спиртов, аммиака и аминов. Например, при адсорбции воды на
87
гидроксилированной поверхности силикагеля водородная связь мо-
жет возникать по следующей схеме:
н н
НН НН
''b o' 'о
—Si— о— Si— + Н2о —> —Si—О—Si—
II II
При образовании водородной связи энергия взаимодействия
адсорбтива с адсорбентом довольно велика, и поэтому теплота,
выделяющаяся при такой адсорбции, обычно значительно больше
теплоты адсорбции веществ, сходных по геометрической форме и
размеру молекул, но не образующих водородной связи.
Существенный вклад в раскрытие сущности адсорбционных яв-
лений дало использование для их изучения инфракрасной спек-
троскопии. В частности, А. Н. Терениным было показано, что при
адсорбции на поверхности кремнезема происходит возмущение
гидроксильных групп, причем в инфракрасном спектре это прояв-
ляется в виде смещения частоты валентных колебаний гидроксиль-
ных групп поверхности в более длинноволновую область спектра.
Это смещение оказалось не одинаковым для различных адсорби-
рованных поверхностью молекул.
3. ТЕОРИЯ МОНОМОЛЕКУЛЯРНОЙ АДСОРБЦИИ ЛЕНГМЮРА
Различают адсорбцию газа на твердом теле, адсорбцию рас-
творенного вещества на границе раствор — газ и, наконец, адсорб-
цию растворенного вещества на границе твердое тело — раствор.
Адсорбция газа на твердом теле является простейшим слу-
чаем адсорбции, так как система газ — твердое тело состоит всего
из двух компонентов. Поэтому адсорбция газа твердым телом осо-
бенно удобна для теоретического рассмотрения явления адсорб-
ции.
Имеется несколько теорий физической адсорбции, из которых
рассмотрим прежде всего теорию мономолекулярной ад-
сорбции. Теория эта была предложена в 1915 г. американским
ученым Ленгмюром, но, как отмечает сам Ленгмюр, им использо-
ваны представления об адсорбционных силах, впервые выдвинутые
русским ученым Л. Г. Гурвичем.
При разработке теории Ленгмюр исходил из следующих по-
ложений.
1. Адсорбция является локализованной и вызывается силами,
близкими к химическим. Однако следует отметить, что Ленгмюр
считал химическими все силы, обуславливающие когезионную
прочность вещества, а также проявляющиеся в явлениях испаре-
ния, кристаллизации, поверхностного натяжения и т. д.
£8
2. Адсорбция молекул адсорбтива происходит на активных
центрах, всегда существующих на поверхности адсорбента. Такими
центрами могут быть пики и возвышения, имеющиеся на любой,
даже самой гладкой поверхности. Так, на поверхности кристаллов
известкового шпата имеются выступы высотой в 10~4—10~5 см,
даже тонко отполированные зеркала имеют на поверхности вы-
ступы до 3-10-7 см.
В результате большой ненасыщенности силового поля около
таких пиковки выступов эти участки обладают способностью удер-
живать налетающие молекулы газа, причем центр тем более ак-
тивен, чем ненасыщеннее молекулы адсорбента на его поверх-
ности.
Примером поверхности с центрами различной активности мо-
жет служить поверхность восстановленного никеля, для которой
Тейлор дает следующую схему строения:
I
—Ni—
—Ni— — Ni— —Ni—
II II I II
—Ni—Ni— —Ni—Ni— —Ni— —Ni—Ni—
I I I I I I I I I I I
—Ni—Ni—Ni—Ni—Ni—Ni—Ni—Ni—Ni—Ni—Ni—
I I I I I I I I I I I
—N i—N i—Ni—N i—Ni—N i—N i—N i—Ni—Ni—Ni—
I I I I I I I I I I I
Атомы никеля, связанные с поверхностным слоем одной связью,
благодаря большой ненасыщенности адсорбируют молекулы ад-
сорбтива сильнее или в большем количестве, чем атомы никеля,
связанные с поверхностью двумя связями. В свою очередь, на
последних адсорбция проходит интенсивнее, чем на атомах никеля,
связанных с поверхностью большим числом связей.
Ряд исследователей считает также, что активными центрами
являются ребра и углы кристаллов и границы зерен в микроне-
однородном или,-как говорят, микрогетерогенном адсорбенте, около
которых образуются особенно интенсивные силовые поля. То, что
ребра и углы кристаллов обладают повышенной способностью ад-
сорбировать молекулы адсорбтива, можно показать на следующем
примере. Если поместить кристалл медного купороса в спиртовый
раствор сероводорода, то почернение кристалла из-за образования
сульфида меди всегда начинается с углов и ребер.
Обычно принимают, что активные центры занимают лишь не-
значительную часть поверхности адсорбента. Эту точку зрения
подтверждает то обстоятельство, что количество яда, отравляю-
щего катализатор, как правило, гораздо меньше того количества,
Которое потребовалось бы для покрытия ядом всей адсорбционной
поверхности.
3. Вследствие малого радиуса действия адсорбционных сил,
имеющих природу, близкую к химической, и способности их
к насыщению каждый активный центр, адсорбируя молекулу ад-
сорбтива, становится уже неспособным к дальнейшей адсорбции.
В результате этого на поверхности адсорбента может образоваться
только мономолекулярный слой адсорбтива.
4. Адсорбированные молекулы удерживаются активными цен-
трами только в течение определенного промежутка времени. Через
некоторое время, в результате флуктуации кинетической энергии,
молекулы отрываются от поверхности адсорбента и переходят
в газовую фазу. Взамен этих молекул активные центры могут ад-
сорбировать новые молекулы, которые в свою очередь десорби-
руются, и т. д.
Время пребывания молекулы в адсорбированном состоянии на
активном центре в значительной степени зависит от температуры.
При низких температурах это время может быть очень большим.
При высоких температурах порядка 1000—2000 °C время пребы-
вания молекулы в адсорбированном состоянии может равняться
всего миллионным долям секунды.
5. Ленгмюр не учитывал силы взаимодействия между адсор-
бированными молекулами. Другими словами, согласно Ленгмюру,
время пребывания молекул газа на активных центрах не зависит
от того, Заняты молекулами газа соседние активные центры
или нет.
Исходя из приведенных выше положений, Ленгмюр смог дать
общее уравнение изотермы локализованной адсорбции, пригодное
как для описания адсорбции газов, так и растворенных веществ.
Для вывода этого уравнения применительно к адсорбции газа
представим локализованную адсорбцию как квазихимическую ре-
акцию между молекулой газа и активным центром адсорбента,
в результате которой образуется адсорбционный комплекс, т. е.
молекула адсорбтива, адсорбированная адсорбентом:
молекула газа + активный центр адсорбента адсорбционный комплекс
Согласно этой реакции и принимая во внимание, что число моле-
кул газа, ударяющихся о поверхность адсорбента, пропорцио-
нально давлению газа, можно написать:
a = fepa0 (IV, 6)
где а — поверхностная концентрация адсорбтива; ао — концентрация свободных
активных центров на поверхности адсорбента; р — давление газа; k — константа,
не зависящая от концентрации и постоянная при данной температуре.
При установившемся адсорбционном равновесии:
k = а/(раа)
В этом случае k является константой равновесия.
Принимая во внимание, что каждый активный центр может
быть занят одной молекулой, можно написать:
а 4- а0 = Омаке (IV, 7)
где обмане — поверхностная концентрация адсорбтива при заполнении им всех
активных центров.
90
Уравнение (IV, 7) можно переписать в виде:
ао в амакс °
Подставим это уравнение в уравнение (IV, 6):
a = kp (амакс — а)
Решая уравнение (IV, 9) относительно а, получим:
а = ИмаксбрЛ 1 + kP)
(IV, 8)
(IV, 9)
(IV, 10)
Это и есть уравнение изотермы адсорбции Ленгмюра. Иногда
уравнение Ленгмюра пишут в несколько ином виде;
a амаксР _ ачаксР
" 1/6 + Р А + р
где А — величина, обратная константе равновесия.
Поскольку величина адсорбции а пропорциональна а, уравне-
ние Ленгмюра можно представить и в таком виде:
а = амакс6р/(1 + 6р) = амакср/(Д + р) (IV, 11)
В этом уравнении аМакс— количество адсорбтива (в молях), ад-
сорбированное единицей массы адсорбента (1г), соответствующее
полному заполнению всех активных центров. Таким образом, в
уравнении Ленгмюра обе константы амакс (или амаьс) и k имеют
определенный физический смысл.
Уравнение Ленгмюра хорошо описывает изотерму адсорбции,
давая при малых и больших значениях р приблизительно прямо-
линейные участки на графике, чего не дает уравнение Фрейндлиха.
В самом деле, при малых давлениях в знаменателе уравнения
(IV, 11) можнб пренебречь членом kp, весьма малым по сравнению
с единицей, и уравнение Ленгмюра принимает вид, тождественный
с уравнением Генри, которому подчиняется распределение веще-
ства между двумя фазами: а = аМакс kp = Kp. Таким образом,
при малых р адсорбированное количество вещества прямо про-
порционально равновесному давлению. Этот случай соответствует
начальному почти прямолинейному участку изотермы. При боль-
ших значениях р в знаменателе уравнения Ленгмюра можно,
наоборот, пренебречь единицей, и тогда уравнение примет вид
а == амакс, т. е. адсорбированное количество вещества перестает
зависеть от давления, что отвечает относительно прямолинейному
Участку изотермы, идущему почти параллельно оси давлений.
Для графического решения ураинение Ленгмюра приводят к виду:
Р 1 । Р
Я Ямаксб Ямакс
(IV, 12)
Полученное уравнение есть уравнение прямой в координатах p/а и р. Наглядно
эт° видно из рис. IV, 3. Очевидно, Омане = ctg 0, где 0 — угол наклона прямой
91
к абсциссе, а 1/(амак<?^)—отрезок, отсекаемый прямой на оси ординат. Зная
1/(амакс&) и Ямакс, легко вычислить k*
Как известно, константа равновесия k всякой химической реакции связана
с теплотой реакции <2 уравнением
k = geiIRT (IV, 13)
где g — постоянный множитель
Так как при физической адсорбции всегда выделяется тепло, т е <2 > 0, то
из уравнения (IV, 13) следует, что с ростом температуры константа равновесия
должна уменьшаться Это происходит из-за увеличения кинетической энергии
молекул и возрастания вследствие этого десорбции Отсюда становится понят-
ным, почему адсорбция газов при высоких температурах и не слишком высоких
давлениях незначительна, а предел адсорбции в этих условиях не достигается
(см рис IV,1)
Уравнение изотермы адсорбции Ленгмюра хорошо приложимо,
«ели адсорбция вызывается силами, близкими по своей природе
Рис. IV, 3. Решение уравнения
Ленгмюра путем его преобра-
зования в уравнение прямой.
Рис. IV, 4. Изотерма ступен-
чатой .адсорбции.
к химическим силам, и если адсорбция не осложняется рядом по-
бочных явлений, например диссоциацией молекул адсорбирован-
ного газа на поверхности.
Представления, развитые Ленгмюром, позволяют объяснить так
называемую ступенчатую адсорбцию. В этом случае изотерма ад-
сорбции носит своеобразный ступенчатый характер, как показано
на рис. IV, 4. Это явление легко объяснить, если допустить, что на
поверхности адсорбента существуют группы активных центров,
резко отличающиеся по активности. Первая ступень изотермы,
-очевидно, отвечает заполнению группы наиболее активных центров,
происходящему уже при малых давлениях, вторая — заполнению
группы активных центров с меньшей активностью, что требует
более высоких давлений и т. д.
Однако некоторые случаи физической адсорбции не могут быть
объяснены положениями теории мономолекулярной адсорбции и
требуют принципиально иного подхода с позиций полимолекуляр-
ной адсорбции.
* Другой способ графического решения уравнения Ленгмюра, пожалуй, не-
-сколько менее точный, можно найти в 1-ом издании «Курса коллоидной химии»,
€ С Воюцкого М, «Химия», 1964 См с. ПО,
92
4. ТЕОРИЯ ПОЛИМОЛЕКУЛЯРНОЙ АДСОРБЦИИ ПОЛЯНИ
И ТЕОРИЯ БЭТ
Опыт показал, что наряду с изотермами адсорбции, вид кото-
рых показан на рис. IV, 1, довольно часто на практике встречаются
изотермы, не имеющие второго участка, почти параллельного оси
давлений и отвечающего насыщению поверхности адсорбента мо-
лекулами адсорбтива. Вид таких изотерм изображен на рис. IV, 5.
Как можно видеть, в точке А изотерма Ленгмюра круто подни-
мается кверху. Очевидно, связывание адсорбтива адсорбентом не
прекращается после образования мономолекулярного слоя, а про-
должается дальше. Форму подобных изотерм нельзя объяснить
Рис. IV, 5. Вид изотермы, ха-
рактерной для полимолекуляр-
иой адсорбции.
Рис. IV, 6. Схема адсорбционного объема
по представлениям полимолекулярной
теории адсорбции.
как следствие капиллярной конденсации, так как такая изотерма
наблюдается и у непористых адсорбентов, когда капиллярная
конденсация невозможна.
Для объяснения этого явления Поляни в 1915 г. предложил
теорию полимолекулярной адсорбции, называемую
также потенциальной. Рассмотрим кратко исходные положе-
ния этой теории, особенно пригодной в случае адсорбции паров на
твердом теле.
1. Адсорбция обусловливается чисто физическими силами.
2. На поверхности адсорбента нет активных центров, а адсорб-
ционные силы действуют вблизи от поверхности адсорбента и об-
разуют около этой поверхности со стороны газовой фазы непре-
рывное силовое поле.
3 Адсорбционные силы действуют на сравнительно большие
расстояния, во всяком случае большие, чем размеры отдельных
молекул адсорбтива, и поэтому можно говорить о существовании
У поверхности адсорбента адсорбционного объема, кото-
рый заполняется при адсорбции молекулами адсорбтива.
4. Действие адсорбционных сил по мере удаления от поверх-
ности уменьшается и на некотором расстоянии практически стано-
вится равным нулю.
93
5. Притяжение данной молекулы поверхностью адсорбента не
зависит от наличия в адсорбционном пространстве других моле-
кул, вследствие чего возможна полимолекулярная адсорбция.
6. Адсорбционные силы не зависят от температуры, и, следо-
вательно, с изменением температуры адсорбционный объем не
изменяется Это не противоречит тому, что с повышением темпе-
ратуры адсорбция уменьшается; в этом случае снижение адсорб-
ции обусловливается не уменьшением адсорбционных сил, а уве-
личением в результате нагревания интенсивности теплового дви-
жения адсорбированных молекул, что приводит к увеличению
десорбции.
Из рассмотрения положений теории полимолекулярной адсорб-
ции можно видеть, что силовое поле, возникающее у поверхности
адсорбента, во многом сходно с гравитационным полем (ненасы-
щаемость поля молекулами адсорбтива, находящимися непосред-
ственно на поверхности адсорбента; независимость сил, действую-
щих в поле, от температуры).
На рис. IV, 6 схематически показан разрез адсорбционного
объема, отвечающий представлению теории полимолекулярной ад-
сорбции. Как и во всяком силовом поле, в адсорбционном поле
можно представить себе эквипотенциальные поверхности, т е. по-
верхности с одним и тем же адсорбционным потенциалом. На ри-
сунке они обозначены пунктирными линиями. Под адсорбционным
потенциалом 0 следует понимать работу, совершаемую против ад-
сорбционных сил при перемещении 1 моля адсорбтива (пара) из
данный точки поля в газовую фазу. Очевидно, максимальный ад-
сорбционный потенциал должен существовать на границе адсор-
бент — адсорбционный объем. На границе адсорбционный объем —
газовая фаза, т. е. там, где кончается действие адсорбционных сил,
потенциал 0 должен быть равным нулю.
Изменение адсорбционного потенциала обычно выражается в теории поли-
молекулярной адсорбции не как функция расстояния I от поверхности адсор-
бента, а как функция адсорбционного объема V. Это обусловлено тем, что
функция 0 = f{V) экспериментально сравнительно легко определяется, в то
время как нахождение функции 0 = ((/) вызывает подчас иеопреодолимые труд-
ности В самом деле, адсорбционный объем легко найти, если работа ведется,
например, при достаточно низких температурах Тогда адсорбтив находится в
адсорбционном пространстве в виде жидкости. В этом случае заполненный жид-
костью адсорбционный объем Уж, отвечающий 1 г адсорбента, выражается
следующим уравнением
— aV мол (IV, 14)
где а — количество адсорбтива (в молях), адсорбированного 1 г адсорбента!
Умол — мольный объем конденсата.
Толщину слоя жидкости в адсорбционном объеме можно было бы иайтя
по уравнению-
= (IV, 15)
где s — поверхность адсорбента
К сожалению, из-за сложности определения истинной поверхности адсор-
бента эту величину часто очень трудно определить, поэтому Поляни и предло-
жил выражать 0 не как функцию I, а как функцию пропорциональной вели-
чины V.
94
Изменение адсорбционного потенциала 0 с изменением адсорб-
ционного объема V, как это представлял Поляни, показано на
рис. IV, 7. Подобные кривые, не зависящие от температуры и ха-
рактерные для каждого данного адсорбента, Поляни называет ха-
рактеристическими кривыми.
Теория полимолекулярной адсорбции принимает, что для ад-
сорбционного объема применимо уравнение состояния газа. По-
этому изотермы, характеризующие зависимость плотности адсорб-
тива р от адсорбционного объема V для разных температур, напо-
минают изотермы р, V.
В определенных условиях, например при достаточно низкой
температуре, адсорбционные силы на поверхности могут вызвать
конденсацию пара в жидкость плотностью р®. При температурах
значительно более низких, чем крити-
ческая, в результате конденсации весь
адсорбционный объем будет заполнен
жидкостью, так что У® = Умакс- В этом
случае кривая p = f(V) на участке,
соответствующем адсорбционному
объему, идет почти параллельно оси
абсцисс в связи с малой сжимаемостью
жидкости. Далее на границе адсорб-
ционный объем — газовая фаза кри-
вая резко опускается вниз И ПЛОТ- Рис. IV, 7. Типичный вид ха-
ность адсорбтива достигает значения рактеристической кривой,
плотности газовой фазы ро.
При температурах значительно более высоких, чем критиче-
ская, адсорбтив будет вести себя как идеальный газ и график за-
висимости р от V выразится кривой, сходной с изотермой для
идеального газа pV = RT. В этом случае адсорбированный газ бу-
дет обладать максимальной плотностью у поверхности адсорбента,
минимальной — в непосредственной близости от газовой фазы.
Плотность адсорбтива в адсорбционном объеме здесь нигде не
достигает плотности жидкости.
Наконец, когда температура близка к критической, зависи-
мость р от V выразится, очевидно, кривой, близкой по своему виду
к изотерме, описываемой уравнением Ван-дер-Ваальса. В этом
случае часть адсорбированного вещества будет находиться в ад-
сорбционном'объеме в жидком состоянии, а часть—в газообраз-
ном. Понятно, наиболее резко кривая снижается в участке, отве-
чающем переходу от жидкости к газу.
Исходя из приведенных выше представлений, Поляни указал путь для по-
строения характеристической кривой. Потенциал 0«, отвечающий точке i, нахо-
дящейся на поверхности раздела жидкость — газ в адсорбционном объеме, мож-
но представить как работу сжатия 1 моля газа при температуре Г;
Р<
(IV, 16)
Р»
95
где р, — давление насыщенного пара над поверхностью жидкости, р0 — давление
газа в газовой фазе
Так как V = RT/p, значение О, можно вычислить по уравнению:
₽Z ₽i d
0,= V dp^RT —- = RT In — (IV, 17)
Ра P-
Значение p, для большинства веществ известно; значение р0 определяется из
опыта. Для нанесения соответствующей точки на диаграмму, выражающую зави-
симость адсорбционного потенциала от адсорбционного объема, необходимо
знать, какому значению V, соответствует найденное значение 0,. Для этого
пользуются вышеприведенным уравнением (IV, 14).
Определив V, и 0, для ряда значений р0 при температуре значительно бо-
лее низкой, чем критическая, когда адсорбтив находится в адсорбционном объ-
еме в основном в виде жидкости, можно построить график 0 = f(V). Наличие
в правой части уравнения (IV, 17) множителя Г не указывает на то, что зави-
симость 0, от V, должна меняться с температурой; а лишь показывает, что при
другой температуре, но при том же значении р0 адсорбционный потенциал, соот-
ветствующий границе жидкость — газ, будет иным
М М. Дубинин показал, что потенциальная теория адсорбции дает воз-
можность вычислить изотермы адсорбции различных паров на одном и том же
адсорбенте по характеристической кривой, полученной из изотерм адсорбции
одного пара, так как соотношение адсорбционных потенциалов различных паров
практически не зависит от адсорбционного объема. Из этого следует, что коор-
динаты точек характеристических кривых для разных адсорбтивов в случае
одного и того же адсорбента при всех значениях адсорбционного объема нахо-
дятся в постоянном отношении р, т. е. эти кривые являются афинными. Отно-
шение р называется коэффициентом афинности характеристических кривых. От-
сюда следует, что построив характеристическую кривую по экспериментальной
изотерме адсорбции одного адсорбтива и зная соответствующий коэффициент
афинности для какого-нибудь другого адсорбтива, можно найти изотерму ад-
сорбции для этого второго адсорбтива.
Мы рассмотрели кратко две теории адсорбции — теорию моно-
молекулярной адсорбции Ленгмюра и теорию полимолекулярной
адсорбции Поляни, на первый взгляд исключающие друг друга.
Возникает вопрос, какая из этих теорий более правильна? На это
следует ответить, что обе теории ограничены в применении. В за-
висимости от природы адсорбента и адсорбтива и в особенности
от условий адсорбции в одних случаях приложима одна, а в дру-
гих— другая теория адсорбции. Теория Поляни применима только
к явлениям чисто физической адсорбции. Теория Ленгмюра охва-
тывает с известными ограничениями явления как физической, так
и химической адсорбции. Однако теория Ленгмюра не может быть
применена для объяснения адсорбции на тонкопористых адсорбен-
тах, имеющих сужающиеся поры. В местах сужения, вследствие
аддитивности дисперсионных сил, адсорбционный потенциал более
высок и в таких местах происходит более интенсивная адсорбция.
Это особенно заметно при температурах ниже критической темпе-
ратуры адсорбтива, т. е. при адсорбции паров, которые в этом
случае заполняют наиболее узкие места капилляров в виде жидко-
сти. Применение уравнения Ленгмюра к адсорбции тонкопори-
стыми адсорбентами, имеющими поры с сужениями, затрудни-
тельно. Конечно, формально, уравнением Ленгмюра можно описать
96
адсорбцию такими адсорбентами. Однако константа амакс в этом
случае теряет смысл емкости плотного монослоя, и использование
этой константы для определения удельной поверхности таких ад-
сорбентов уже не дает правильных результатов.
Делались попытки обобщить представления Ленгмюра и По-
ляни и описать изотермы различной формы с помощью одного
уравнения. В частности, такая обобщенная теория была развита
Брунауэром, Эмметом и Теллером в 1935—1940 гг. применительно
к адсорбции паров. Их теория получила название теории БЭТ по
первым буквам имен авторов. Основные положения теории БЭТ
следующие.
1. На поверхности адсорбента имеется определенное число рав-
ноценных в энергетическом отношении активных центров, способ-
ных удерживать молекулы адсорбтива.
Рис. IV, 8. Схема полимолекулярной адсорбции, принятая по тео-
рии БЭТ.
2. Для упрощения взаимодействием соседних адсорбированных
молекул в первом и последующих слоях пренебрегают.
Оба эти допущения соответствуют ленгмюровской адсорбции
на однородной поверхности без взаимодействия адсорбированных
молекул.
3. Каждая молекула первого слоя представляет собой возмож-
ный центр для адсорбции и образования второго адсорбционного
слоя; каждая молекула второго слоя является возможным центром
адсорбции в третьем и т. д.
4. Предполагается, что все молекулы во втором и более дале-
ких слоях имеют такую же сумму статистических состояний, как
в жидком состоянии (в общем отличающуюся от суммы состояний
первого слоя).
Таким образом, адсорбированная фаза может быть представ-
лена как совокупность адсорбционных комплексов — молекуляр-
ных цепочек, начинающихся молекулами первого слоя, непосред-
ственно связанных с поверхностью адсорбента. При этом цепочки
энергетически не взаимодействуют друг с другом. Схема строения
адсорбционного слоя по теории БЭТ показана на рис. IV, 8.
Все эти предпосылки достаточно условны. В частности, со-
гласно теории БЭТ принимается, что каждая молекула жидкости
имеет только двух ближних соседей (т. е. сверху и снизу в це-
почке), в то время как у молекулы реальной жидкости гораздо
больше ближайших соседей. Тем не менее эта теория оказалась
в некоторых отношениях весьма плодотворной,
4 Зак, 664 д?
Теория Поляни не позволяет вывести уравнения изотермы ад-
сорбции. Теория БЭТ, как и теория Ленгмюра, дает аналитическое
уравнение для изотермы, которая в этом случае имеет S-образную
форму (см. рис. IV, 5). Не приводя достаточно сложного вывода
уравнения изотермы адсорбции Брунауэра, Эммета и Теллера,
остановимся лишь на некоторых положениях, лежащих в основе
этого вывода, и приведем само уравнение изотермы.
Брунауэр, Эммет и Теллер при выводе уравнения рассматри-
вают адсорбцию молекул пара как серию квазихимических реак-
ций образования единичных и кратных адсорбционных комплексов:
пар + свободная поверхность единичные комплексы
пар + единичные комплексы двойные комплексы
пар + двойные комплексы тройные комплексы и т. д.
При этом теплота адсорбции первого слоя молекул Qi, т. е. теп-
лота образования единичных комплексов, гораздо больше, чем для
всех последующих слоев. Теплоты адсорбции всех последующих
слоев приблизительно одинаковы и равны теплоте объемной кон-
денсации L.
Для константы равновесия полимолекулярной адсорбции С можно написать
следующее уравнение:
C = g'exp(^~) (IV, 18)
(Qi — L)—так называемая чистая теплота адсорбции
На основе этих представлений Брунауэр, Эммет и Теллер вы-
вели следующие уравнения изотермы адсорбции паров:
где рз — давление насыщенного пара при данной температуре, р/рз— относи-
тельное давление пара, а, аМакс, а, амакс, р— имеют те же значения, что и в
уравнении Ленгмюра
При значениях р, далеких от р, и С>1, адсорбция приводит
к образованию мономолекулярного слоя и уравнения (IV, 19) и
(IV, 20) переходят в уравнения адсорбции Ленгмюра (IV, 10) и
(IV, 11). По мере приближения р к ps число свободных активных
центров сокращается и кратность комплексов растет. При р = pt
происходит объемная конденсация пара.
Уравнение изотермы полимолекулярной адсорбции БЭТ легко привести
к линейной форме
pips _ 1 , С — 1
_ \ л Г< г» г»
О (1 — p/ps) Омакс^- Ямакс^
(IV, 21)
По наклону этой линии и отсекаемому ею отрезку на оси ординат можно найти
значения констант Омане и С.
98
Теория БЭТ, так же как и теория Ленгмюра, указывает путь
для определения удельной поверхности адсорбента. Найдя амакс
для паров простых веществ (N2, Аг, Кг) при низких температурах
и зная площадь $о, занимаемую молекулой адсорбтива, легко вы-
числить удельную поверхность адсорбента $уд:
5уд = аМаксЛ/А50 (IV, 22)
где Na — число Авогадро.
Легко решить и обратную задачу. Для этого определяют с по-
мощью адсорбции при низких температурах стандартного пара
поверхность $уд данного адсорбента, после чего, найдя экспери-
ментально изотерму адсорбции пара исследуемого адсорбтива и
значение емкости монослоя амакс, вычисляют значение $0:
-'э = '\л/(“.маИЛ) (IV, 23)
Опыты по определению удельной поверхности одного и того же
адсорбента при адсорбции различных паров дали близкие резуль-
таты, в общем совпадающие с результатами определения удельной
поверхности другими, не адсорбционными методами. Это под-
тверждает правильность толкования формы изотермы с помощью
теории БЭТ. Несмотря на ряд недостатков, теория БЭТ является
в настоящее время лучшей и наиболее полезной теорией физиче-
ской адсорбции.
5. КАПИЛЛЯРНАЯ КОНДЕНСАЦИЯ
Как мы уже отмечали, полимолекулярная адсорбция характе-
ризуется S-образной изотермой адсорбции, изображенной на
рис. IV. 5. Однако следует иметь в виду, что кривую аналогичной
формы можно получить при адсорбции, осложненной капилляр-
ной конденсацией. Рассмотрим это явление более подробно.
Капиллярная конденсация обусловлена наличием у адсорбента
мелких пор. Пары адсорбтива конденсируются в таких порах при
давлениях, меньших давления насыщенного пара над плоской по-
верхностью вследствие образования в капиллярах вогнутых ме-
нисков. Возникновение этих менисков следует представлять как
результат слияния жидких слоев, образовавшихся на стенках ка-
пилляра вследствие адсорбции паров. Понятно, что возникновение
вогнутых менисков возможно только в том случае, если образо-
вавшаяся жидкость смачивает стенки капилляра.
Связь между радиусом г шаровидного мениска, образовавшегося в капил-
ляре, опущенном в смачивающую его стенки жидкость, и давлением насыщен-
ного пара р над мениском дается известным уравнением В. Томсона (Кельвина!,
вывод которого приводится в курсах физики:
р = Рзехр(--?У“0л) (IV, 24)
где р„ — давление насыщенного пара над плоской поверхностью жидкости; а —
поверхностное натяжение жидкости; КМол— мольный объем жидкости; R— га-
зовая постоянная; Т — абсолютная температура.
4*
99.
Следует отметить, что уравнение (IV, 24) выведено для вогнутого шаровид-
ного мениска Для цилиндрического мениска, у которого одно из главных зна-
чений кривизны равно нулю, оно принимает вид
pa = psexp(— а^°л ) (IV, 25)
Таким образом, давление пара иад цилиндрическим мениском больше, чем
иад шаровидным мениском у капилляра с тем же радиусом, т е рц > р Это
обстоятельство играет существенную роль при капиллярной конденсации.
Уравнение В Томсоиа является основным при расчетах, связанных с явле-
ниями капиллярной конденсации Если известны давление пара жидкости р, и
радиус капилляров адсорбента *, то по уравнению В Томсона можно вычислить
давление пара ph, выше которого в капиллярах начинается конденсация Если
заданы р, и ph, то, пользуясь уравнением В. Томсоиа, можно вычислить макси-
мальный радиус капилляров, в которых будет происходить конденсация (что
нужно знать для правильного подбора адсорбента).
Явление конденсации не следует смешивать с физической ад-
сорбцией. Элементарная теория капиллярной конденсации не
учитывает специфического действия поверхностных сил. Доказа-
тельством различия капиллярной конденсации и полимолекуляр-
ной физической адсорбции служит и тот факт, что полимолеку-
лярная адсорбция может происходить на плоских поверхностях,
тогда как капиллярная конденсация в таких условиях невозможна.
Б В Дерягин и 3 М Зорин, изучавиГие механизм капиллярной конденсации
оптическим методом на гладкой поверхности стекла, показали, что крутой
подъем изотермы, характеризующей капиллярную конденсацию, начинается для
неполярных веществ (СС14 и др) при р/р, около 0,98, а для полярных веществ
(вода, спирты, нитробензол)—при несколько меньших относительных давлениях.
Этн исследователи также нашли, что при сорбции полярных веществ переход
ОТ адсорбционного слоя к объемной жидкой фазе совершается скачкообразно.
Этот основной вывод подтверждает одновременное существование линзообразных
зародышей — островков новой жидкой фазы и полимолекуляриых адсорбцион-
ных слоев равномерной толщины Объемная жидкая фаза образуется путем ро-
ста этих островков, причем при этом процессе окружающий их адсорбционный
слой заметно не изменяется В противоположность этому объемная фаза при
поверхностной конденсации неполярных реществ, по-вндимому, появляется в ре-
зультате непрерывного утолщения адсорбционного слоя, причем эта конденсация
протекает вполне обратимо
Приведенные данные позволяют считать, что для полярных веществ раз-
личие между адсорбционным полимолекулярным слоем и объемом жидкости но-
сит характер фазовых различий Это дает право считать полимолекулярные ад-
сорбционные слои как особые граничные фазы Наоборот, адсорбционный слой
паров неполярных веществ нельзя рассматривать как особую фазу, отличную
от объемной, поскольку между ними возможен непрерывный переход и невоз-
можно их сосуществование.
При адсорбции, сопровождающейся капиллярной конденсацией,
часто наблюдается явление гистерезиса, когда изотермы адсорб-
ции и десорбции не совпадают Это явление подробно изучали
Ван-Беммелен и Зигмонди на примере адсорбции воды силикаге-
лем. Результаты их опытов представлены схематически в виде
* Радиус капилляра R, как это следует из молекулярной физики, связан
с радиусом кривизны шаровидного мениска г смачивающей стенки капилляра
жидкости простым уравнением R — г соз 0, где 0 — краевой угол
100
диаграммы на рис. IV, 9. На диаграмме по оси ординат нанесены
масса поглощенной силикагелем воды т, а на оси абсцисс — рав-
новесные значения давления пара р.
При р — 0 силикагель еще содержит немного воды, что харак-
теризуется отрезком ОА. Это кристаллизационная вода, которая
может быть удалена только прокаливанием. Изотерма адсорбции
обратима лишь на участке АВ. От точки В изотерма становится
необратимой — одной и той же массе влаги rtii при поглощении
отвечает давление пара а при обезвоживании — рг, причем
pi > Р2- Это становится ясным, если
провести параллельную абсциссе ли-
нию, пересекающую гистерезисную
петлю, и из точек пересечения опу-
стить перпендикуляры на ось давле-
ний. Зигмонди объяснил подобное яв-
ление тем, что на участке BED проис-
ходит капиллярная конденсация, а на
участке BCD — испарение воды из ка-
пилляров. Воздух, адсорбированный
сухими стенками капилляров, препят-
ствует их смачиванию при оводнении
силикагеля. Очевидно, вследствие это-
го краевые углы, образуемые жид- Рис. IV, 9. Гистерезис при ка-
костью со стенками капилляров при пиллярной конденсации,
оводнении силикагеля, будут всегда
больше соответствующих углов при испарении, когда стенки пол-
ностью смочены водой. В результате мениски жидкости, заполняю-
щей капилляры, в первом случае также всегда будут менее во-
гнуты, чем во втором, и давление пара, отвечающее одному и тому
же количесту поглощенной силикагелем жйдкости, при оводнении
будет больше, чем при обезвоживании
Кривые BCD и BED имеют определенный наклон к оси давле-
ний. Это объясняется тем, что капилляры силикагеля, имеющие
разные радиусы, заполняются или опустошаются последовательно.
Заполнение конденсатом узких капилляров происходит уже при
малых давлениях, в то время как заполнение широких капилля-
ров требует значительно более высоких давлений. При испарении
воды из капилляров, понятно, наблюдается обратная зависимость.
Предварительное тщательное удаление воздуха из пористого адсорбента
обычно очень сильно уменьшает гистерезис Это как будто подтверждает пра-
вильность объяснения гистерезиса адсорбцией воздуха на стенках капилляров
Есть, однако, и другие объяснения этого сложного явления. В частности, гисте-
резис при капиллярной конденсации может быть объяснен, исходя из формы
пор ^с°Р®ента Представим, что адсорбент содержит поры, изображенные на
При конусообразной форме в порах (см рис, IV, 10а) образуется адсорб-
ционная пленка с вогнутой поверхностью, причем шаровидная поверхность с
максимальной кривизной наблюдается в наиболее узкой части поры При
Р Р> ехр[—2аУмол/{г RT)] пар будет насыщенным по отношению этой поверх-
ности и начнет конденсироваться. Это приведет к продвижению жидкости и в
101
более широкую часть поры, что, конечно, вызовет увеличение г. Для того чтобы
пар продолжал конденсироваться, давление р должно возрастать (см. изотерму
на рис. IV, 10а). При уменьшении р жидкость со стен капилляра десорбируется
и изотерма пойдет в обратном направлении таким же путем, т. е. капиллярная
конденсация в конусообразных порах полностью обратима.
В порах цилиндрической формы, закрытых с одного конца, т. е. имеющих
форму пробирки (см. рис. IV, 106), у закрытого конца при адсорбции обра-
зуется шаровидный мениск. При р — р, ехр [—2аУмол/(г ЛГ)! происходит ка-
пиллярная конденсация, и в результате этого поры заполняются жидкостью.
Однако в отличие от предыдущего случая радиус мениска при этом будет по-
стоянным, и поэтому заполнение пор происходит при постоянном значении р,
чему соответствует вертикальная часть
изотермы капиллярной конденсации
(см. изотерму на рис. IV, 106). Про-
цесс десорбции пойдет в обратном
направлении таким же путем, т. е.
капиллярная конденсация в цилин-
дрических капиллярах с одним закры-
тым концом также вполне обратима.
Наконец, в цилиндрических порах,
открытых с обоих концов (см. рис.
IV, 10в), шаровидный мениск при ад-
сорбции не может образоваться, и кон-
денсация начнется на внутреннем ци-
линдрическом мениске пленки, покры-
вающей стенки капилляра, при дав-
лении рц = psexp[—а^мол/(г/?Т)].
В результате конденсации толщина
пленки жидкости увеличивается, а ра-
диус поры уменьшается, и поэтому
Рис. IV, 10. Схема капиллярной конден-
сации в порах различной формы:
а—конусообразной; б — цилиндрической, за-
крытой у одного конца; в — цилиндрической,
открытой с обоих концов.
она заполняется жидкостью при дав-
лении р. Изотерма капиллярной кон-
денсации, как и в предыдущем слу-
чае, имеет вертикальный участок (см.
изотерму на рис. IV, 10в). Однако
вследствие меньшей кривизны цилин-
дрической поверхности мениска по
сравнению с кривизной шаровой поверхности (при одном и том же радиусе
капилляра) вертикальный участок на изотерме соответствует большим зна-
чениям давления пара. После заполнения поры на обоих ее концах возникнут
шаровидные мениски, кривизна которых с повышением давления пара умень-
шается. При десорбции процесс вначале пойдет обратимо — при испарении не-
больших количеств жидкости в устья капилляров будут вдавливаться шаровид-
ные мениски со все возрастающей кривизной. Однако при р =
= psexp[—сУмолЦг RT)] эти шаровидные мениски прорваться еще не могут и
капилляр при этом давлении останется еще заполненным. Только при сниже-
нии давления пара до р = ра ехр [—2<тИмол/(г/?Т)] радиус шаровидного ме-
ниска станет равным радиусу адсорбционной пленки в цилиндрическом капил-
ляре и вся жидкость, заполнявшая капилляр, испарится. Все это обусловит то,
что десорбционная ветвь разойдется с адсорбционной, т. е. получится характер-
ная петля капиллярно-конденсационного гистерезиса.
Реальные адсорбенты не обладают порами какого-нибудь одного размера
к какой-нибудь одной формы. Их поры заполняются или опустошаются не одно-
временно. Это является причиной того, что ветви гистерезисной петли обычно
наклонены к оси абсцисс.
На явлениях адсорбции и, главным образом, капиллярной кон-
денсации основана рекуперация (возвращение в производство)
летучих растворителей, теряющихся при технологических процесса^.
102
Рассмотрим для примера процесс рекуперации растворителя из клеев, при-
меняемых в резиновом производстве. При изготовлении прорезиненных тканей
на один рулон из 300 м ткани расходуется обычно около 180 кг каучукового
клея, содержащего около 85% высокосортного бензина. Весь бензин при высу-
шивании тканн после покрытия ее клеем улетучивается и смешивается с возду-
хом. Таким образом, при отсутствии рекуперации огромные количества дефи-
цитного и дорогостоящего бензина теряются.
Для рекуперации летучего растворителя смешанные с воздухом пары бен-
зина отсасывают при сушке ткани из сушилок и с помощью воздушных насосов
подают в рекуперационную установку, состоящую из двух адсорберов. Пары
бензина поступают в один заполненный активным углем адсорбер. Другой ад-
сорбер в это время отключен. В первом адсорбере, куда поступила паро-воз-
душная смесь, происходит сначала адсорбция, а затем и капиллярная конден-
сация паров бензина до полного насыщения адсорбента летучим растворителем,
что легко установить по проскоку паров бензина через слой угля. После дости-
жения насыщения первый адсорбер отключают от подающей трубы и подклю-
чают к ней второй адсорбер. В отключенный адсорбер подают горячий водяной
пар для испарения и десорбции бензина. Пары бензина н воды подают в холо-
дильник, а затем в сепаратор, где сконденсированные бензин и вода отделяются
друг от друга путем простого расслаивания этих несмешивающихся жидкостей.
За это время второй адсорбер поглотил достаточное количество бензина. Теперь
от подающей трубы отделяют его для проведения процесса десорбции, а к трубе
присоединяют снова первый адсорбер. Так осуществляется непрерывный произ-
водственный процесс рекуперации летучего растворителя.
6. ХИМИЧЕСКАЯ АДСОРБЦИЯ
Наряду с типичной физической адсорбцией часто на практике
имеет место химическая адсорбция, или хемосорбция, т. е. адсорб-
ция, осуществляющаяся за счет химических сил. Однако провести
резкую границу между обоими видами адсорбции невозможно. Ад-
сорбция одного и того же адсорбтива на одном и том же адсор-
бенте в одних условиях может быть физической, а в других усло-
виях —- химической. Очень часто физическая адсорбция предше-
ствует химической — адсорбтив, адсорбированный под действием
физических сил, затем связывается с адсорбентом уже химиче-
следую-
и мало-
обычно
скими силами.
Физическая и химическая адсорбция различаются по
щим признакам. Физическая адсорбция вполне обратима
специфична. Теплота физической адсорбции составляет
всего 2—8 ккал/моль и соизмерима с теплотой конденсации. Теп-
лота химической адсорбции может достигать 200 ккал/моль, т. е.
имеет порядок теплот химических реакций. Химическая адсорбция
обычно необратима.
Особенно характерно различное действие температуры на фи-
аическую и химическую адсорбцию. Повышенная температура
уменьшает физическую адсорбцию и, наоборот, способствует хемо-
сорбции. В последнем случае это объясняется тем, что хемосорб-
ция является химическим процессом, обычно требующим значи-
тельной энергии активации (10—30 ккал/моль). Именно поэтому
Хемосорбция, как правило, является активированной адсорбцией.
Поскольку химическую адсорбцию обусловливают химические
Рмы, десорбция протекает с большим трудом и почти всегда
103
вместо адсорбированного вещества десорбируется другое. По су-
ществу десорбция осуществляется здесь не за счет ухода молекулы
с поверхности адсорбента, а в результате разложения образовав-
шегося при хемосорбции поверхностного соединения. Хемосорбция,
как и химическая реакция, вполне специфична, т. е. с адсорбентом
могут взаимодействовать лишь определенные адсорбтивы. Суще-
ственно, что энергия активации возрастает с повышением степени
покрытия поверхности хемосорбированными молекулами. Это
можно объяснить лишь существованием активных центров с раз-
личными энергиями активации.
. Если химическая адсорбция обратима, зависимость количества
адсорбированного вещества от температуры может приобретать
довольно сложный характер.
На рис. IV, 11 показана изо-
бара адсорбции СО палла-
дием. При низких температу-
рах наблюдается только физи-
ческая адсорбция, которая бы-
стро падает с повышением
температуры. Однако при до-
стижении определенной темпе-
-100 -юо о юо zoo ратуры вступает в действие
*°с активированная адсорбция, и
Рис. IV, 11. Изобара адсорбции окиси количество адсорбированного
углерода палладием. вещества начинает возрастать
при увеличении температуры.
При этом практически протекает только хемосорбция, а физиче-
ская адсорбция становится очень незначительной. В области вы-
соких температур наблюдается вновь уменьшение количества ад-
сорбированного вещества в результате его разложения.
Типичным примером хемосорбции может служить адсорбция
кислорода на угле. На поверхности угля имеются, очевидно, атомы
углерода со свободными валентностями, что можно представить
схематически так:
Благодаря ненасыщенности атомов
угля может адсорбироваться кислород:
углерода на поверхности.
104
При попытке десорбировать адсорбтив путем нагревания с по-
верхности удаляется не кислород, а окислы углерода. Это указы-
вает на то, что связь между атомами кислорода и углерода проч-
нее, чем связь между атомами углерода.
Образующиеся при химической адсорбции мономолекулярные
слои новых соединений — поверхностные соединения, как их на-
звал Н. А. Шилов, нельзя рассматривать как новую фазу, новое
вещество. В самом деле, между адсорбированными молекулами
адсорбтива и атомами (молекулами) адсорбента возникает хими-
ческая связь, но в то же время поверхностные атомы адсорбента
сохраняют связь с остальными его атомами. Энергия образования
химической связи между молекулами адсорбтива и адсорбента,
очевидно, недостаточна для отрыва поверхностных атомов адсор-
бента от кристаллической решетки. При подводе энергии извне,
например при повышении температуры, такой отрыв может на-
ступить, в результате чего поверхностная реакция превращается
в обычную гетерогенную реакцию и образуется новая фаза.
Интересно, что теплота поверхностных реакций обычно значи-
тельно больше, чем теплота образования химического соединения.
Так, теплота адсорбции кислорода на угле примерно в два раза
больше теплоты сгорания твердого углерода.
Как физическая, так и химическая адсорбция играют огромную
роль в гетерогенном катализе, так как на поверхности катализа-
тора молекулы адсорбтива реагируют друг с другом гораздо быст-
рее. Разные исследователи высказывали различные мнения о при-
чине этого явления. Адсорбент, сгущая и ориентируя на своей по-
верхности молекулы участвующих в реакции компонентов, способ-
ствует тем самым протеканию реакции, в конечном счете сам в нее
не вступая. Ускорение реакции на поверхности адсорбента (ката-
лизатора) может являться результатом и того, что поверхностные
силы обусловливают диссоциацию молекул адсорбтива на более
реакционноспособные атомы или, по крайней мере, вызывают ос-
лабление связи между атомами молекулы. Роль адсорбции в гете-
рогенном катализе подробно рассматривается в специальных кур-
сах физической химии газовых реакций и катализа.
7. ТЕПЛОТА АДСОРБЦИИ
Как уже было указано, физическая адсорбция является экзо-
термическим процессом. Количество тепла, выделяющегося при
адсорбции, можно измерить с помощью либо изотермического, либо
адиабатического калориметра. При использовании изотермического
калориметра выделившаяся теплота адсорбции определяется, на-
пример, по количеству льда, превратившегося в воду. Температура
системы при этом остается постоянной, и теплота расходуется
исключительно на фазовое превращение. При использовании адиа-
батического калориметра количество выделившегося тепла опре-
деляется по повышению температуры в калориметре. Следует
105
заметить, что экспериментально определенные с помощью калори-
метра значения теплоты адсорбции часто недостаточно точны.
Причина этого заключается в том, что адсорбция, а следовательно,
и выделение тепла растягиваются на очень длительный промежу-
ток времени. Последнее неизбежно приводит к потерям тепла.
Различают два способа выражения теплоты адсорбции — ин-
тегральную и дифференциальную. Рассмотрим эти два
понятия несколько подробнее.
Интегральная теплота адсорбции дш,т представляет собой об-
щее количество выделившегося при адсорбции тепла, отнесенное
к 1 г адсорбента. Очевидно
<7hht = Q/'W (IV, 26)
где Q — общее количество выделившегося тепла, кал; т — масса адсорбента, г.
Единицами измерения интегральной теплоты адсорбции в со-
ответствии с уравнением (IV, 26) являются кал/г (адсорбента).
Рис. IV, 12. Зависимость интеграль-
ной теплоты адсорбции </инт от коли-
чества адсорбированного вещества а:
1 — при ’условии пропорциональности <7ИНТ
величине а; 2—экспериментально получен-
ная зависимость.
Рис. IV, 13. Зависимость диффе-
ренциальной теплоты адсорб-
ции *7диф от количества адсорби-
рованного вещества а.
На первый взгляд может показаться, что теплота ^инт должна
быть прямо пропорциональна количеству адсорбированного веще-
ства а. Однако на самом деле этого нет (рис. IV, 12).. Это объяс-
няется тем, что в начале процесса газ адсорбируется на наиболее
активных центрах с максимальным тепловым эффектом, а в конце
процесса вступают в действие менее активные центры, при адсорб-
ции на которых выделяется сравнительно мало тепла.
Дифференциальная теплота адсорбции Допустим, что к дан-
ному моменту адсорбировалось на поверхности адсорбента п мо-
лей адсорбтива и при этом выделилось Q калорий тепла. Допу-
стим далее, что после этого адсорбировалось при той же темпера-
туре еще дополнительно dn молей адсорбтива и выделилось dQ
калорий тепла. Дифференциальная теплота адсорбции ^диф пред-
ставляет собой отношение этого дополнительно выделившегося
106
тепла к дополнительно адсорбированному количеству адсорбтива,
т. е.
‘}пиф = dQ/dn (IV, 27)
Иными словами, дифференциальной теплотой адсорбции назы-
вается тепло, выделившееся при дополнительной адсорбции малого
количества адсорбтива и пересчитанное на 1 его моль. Единицами
измерения дифференциальной теплоты адсорбции в соответствии
с уравнением (IV, 27) являются кал/моль (адсорбтива).
Зависимость дифференциальной теплоты адсорбции от количе-
ства адсорбированного вещества можно схематически выразить
графиком, изображенным на рис. IV, 13. Дифференциальная теп-
лота адсорбции уменьшается в ходе процесса вследствие того, что
молекулы адсорбтива, по мере насыщения наиболее активных цен-
тров, адсорбируются все менее и менее активными участками по-
верхности.
Исследование теплот адсорбции различных газов разными по-
верхностями внесло большой вклад как в понимание природы
адсорбционных процессов и явлений гетерогенного катализа, так
и в решение ряда практических задач, например при подборе ка-
тализаторов.
8. СКОРОСТЬ АДСОРБЦИИ
Скорость физической адсорбции на непористых адсорбентах
обычно весьма велика, и поэтому часто ее измерить очень сложно.
Во многих случаях адсорбционное равновесие достигается за 10—
20 с, причем 90—95% адсорбтива связываются адсорбентом уже
за 1—2 с. Практически принимают, что скорость адсорбции опре-
деляется скоростью, с которой адсорбтив достигает поверхности
адсорбента, т. е. скоростью диффузии.
Причину наблюдающейся иногда замедленной физической ад-
сорбции следует искать в строении адсорбента. Адсорбенты часто
являются пористыми, и проникновение молекул адсорбтива в поры
требует иногда длительного времени. Иногда причина замедленной
адсорбции заключается в том, что физическая адсорбция сопро-
вождается хемосорбцией, требующей более длительного времени.
Наконец, причиной замедленной адсорбции может быть присут-
ствие на поверхности адсорбента адсорбированного воздуха или
паров воды.
Типичные кинетические кривые адсорбции изображены на
рис. IV, 14. Форма этих кривых имеет: то же объяснение, что и
форма изотермы адсорбции. Вначале величина адсорбции практи-
чески пропорциональна времени, так как поверхность адсорбента
свободна от адсорбтива. После установления равновесия адсорб-
ции последняя перестает зависеть от времени и соответствующие
участки кривых идут почти параллельно оси времени.
Уравнение скорости адсорбции обычно имеет вид:
dajdx = k (аравн — «г) (I V, 27)
107
Рис. IV, 14. Зависимость коли-
чества адсорбированного веще-
ства от времени адсорбции т при
различных температурах:
1—кривая, отвечающая температуре
2—кривая, отвечающая температуре
(Т1 < Гр.
где Дравв — количество адсорбированного вещества, отвечающее адсорбционному
равновесию при данных условиях; ах—количество вещества, адсорбированного
ко времени т; k — константа уравнения.
Физический смысл этого уравнения вполне понятен: чем ближе
система к равновесию или чем больше насыщенность поверхности,
характеризуемая множителем (аравн — ах), тем медленнее идет
адсорбция. Константа k зависит от размера адсорбирующей по-
верхности и от коэффициента диффузии адсорбтива.
Иногда в качестве уравнения скорости адсорбции принимают
параболическое уравнение, напоминающее по виду уравнение
Фрейндлиха:
а = Лт1/п (IV, 28)
Недостатки этого уравнения те же, что и уравнения Фрейндлиха.
Экспериментально скорость адсорбции исследуют либо путем
определения в намеченные моменты времени количества газа,
адсорбированным, либо по привесу
адсорбента. Особенно широкое при-
менение для исследования кинети-
ки адсорбции получили весы Мак-
Бена, представляющие собой мини-
атюрную чашечку, подвешенную на
очень чувствительной пружинке. На
чашечку кладут кусочек адсорбен-
та и пружинку вместе с чашечкой
помещают в сосуд с адсорбтивом.
В результате адсорбции масса это-
го кусочка возрастает, и растяже-
ние пружинки соответственно уве-
личивается. По растяжению заранее
откалиброванной пружинки во вре-
мени можно судить о кинетике ад-
сорбции количества адсорбирован-
ного вещества.
Интересно влияние температуры на скорость адсорбции. С по-
вышением температуры скорость адсорбции возрастает, так как
нагревание всегда способствует ускорению установления равнове-
сия в системе. С другой стороны, при повышении температуры ад-
сорбция, отвечающая равновесному состоянию, падает. Таким об-
разом, кинетические кривые адсорбции при разных температурах
должны пересекать друг друга, как это и показано на рис. IV, 14.
Температурный коэффициент скорости физической адсорбции
невелик. Это связано с тем, что энергия активации физической ад-
сорбции близка к нулю, и ускорение адсорбции с повышением тем-
пературы обусловлено, главным образом, только увеличением ско-
рости подвода адсорбтива к поверхности адсорбента в результате
роста скорости диффузии. При хемосорбции температурный коэф-
фициент скорости адсорбции имеет тот же порядок, что и при хи-
108
мических процессах, так как энергия активации хемосорбции до-
статочно велика и температура значительно ускоряет химическое
взаимодействие.
9. ВЛИЯНИЕ НА АДСОРБЦИЮ СВОЙСТВ АДСОРБЕНТА И АДСОРБТИВА.
ДИНАМИЧЕСКАЯ АДСОРБЦИЯ. АДСОРБЦИЯ ИЗ СМЕСЕЦ ГАЗОВ
Влияние свойств адсорбента. Как следует из рассмотренных
теорий адсорбции, на способность того или иного адсорбента ад-
сорбировать газы сильно влияет его пористость, а также его фи-
зическое состояние. Аморфные адсорбенты обычно гораздо лучше
адсорбируют газ, чем кристаллические. Это объясняется, очевидно,
тем, что поверхность аморфного адсорбента шероховата, в то
время как поверхность кристалла, за исключением ребер и углов,
гладкая.
Непористые адсорбенты, получаемые в результате химических
реакций в растворе и последующего осаждения (например, суль-
фат бария), а также путем размельчения твердых тел, обладают
обычно сравнительно небольшой удельной поверхностью (1—
10 м2/г) и поэтому имеют довольно ограниченное применение. Бо-
лее высокодисперсные адсорбенты с непористыми частицами можно
получить при неполном сгорании органических соединений (угле-
родные, или черные сажи) или кремнийорганических соединений
(белые сажи), а также в результате гидролиза галогенидов крем-
ния (SiCl4, S1F4) в парах воды (аэросилы). Получаемые порошки
с удельной поверхностью порядка сотен м2/г применяют в каче-
стве найолнителей полимеров, лаков и смазок.
В качестве поглотителей, осушителей, катализаторов или но-
сителей каталитически активных веществ обычно используют вы-
сокодисперсные пористые адсорбенты. Такие адсорбенты приме-
няют, как правило, не в виде порошка, а в форме достаточно
механически прочных гранул или таблеток. Это обеспечивает зна-
чительные удобства при их применении (отсутствие пыления) и
уменьшает сопротивление потоку газа или жидкости, из которого
проводится адсорбция.
В качестве тонкопористых адсорбентов наиболее часто приме-
няют древесный уголь, животный (костный) уголь, силикагель,
различные природные силикаты, алюмогель и алюмосиликагель.
Из древесных углей для адсорбции применяют уголь, полученный
из твердых древесных пород, так как уголь, полученный из мягких
пород, например из сосновой древесины, весьма непрочен и легко
рассыпается. Лучшие сорта угля для адсорбции получают из скор-
лупы кокосовых орехов и абрикосовых косточек. Кроме того, для
адсорбции обычно применяется активный уголь.
Обычный уголь (уголь-сырец) имеет сравнительно небольшую
адсорбционную способность, так как его удельная поверхность
сравнительно невелика и поры в значительной степени заполнены
смолами и продуктами неполного сгорания, образующимися при
109
-получении угля. Активирование угля заключается в термической
обработке, в результате которой его удельная поверхность увели-
чивается, при этом продукты неполного сгорания частично сгорают,
частичо улетучиваются. Термическую обработку угля, во избежа-
ние больших потерь в результате сгорания, проводят в атмосфере
водяного пара или двуокиси углерода (при 750—950°C). Содер-
жащиеся в угле органические вещества, а частично и сам уголь
реагируют с водяным паром и двуокисью углерода с образова-
нием СО и Н2. Поскольку эти процессы эндотермические, активи-
рование в атмосфере пара или двуокиси углерода легко остановить
в тот момент, когда сгорят ненужные органические вещества и
не будет еще затронута основная часть угля. Изменение струк-
туры угля при активировании показано на рис. IV, 15. (области,
Рис IV, 15. Постепенное изменение структуры угля при его активи-
ровании.
занятые смолами и продуктами неполного сгорания, на рисунке
заштрихованы). Удельная поверхность активного угля колеблется
в пределах от 300 до 1000 м2/г, а диаметр микропор — от 30 до
90 А.
Уголь как адсорбент применяется для заполнения противога-
зов, рекуперации растворителей, рафинирования сахара, обесцве-
чивания многих жидкостей, очистки воздуха в промышленных
предприятиях, а также используется в медицине. Адсорбцию актив-
ным углем не следует смешивать с активированной адсорбцией.
Другим адсорбентом, часто применяющимся на практике, яв-
ляется силикагель — гидратированная двуокись кремния, приго-
товленная в виде очень пористого тела или порошка. Силикагель
обычно получают, вводя раствор силиката натрия при сильном пе-
ремешивании в 5—10%-ный раствор хлористоводородной кис-
лоты. Образовавшийся пористый силикагель измельчают и про-
мывают водой. Затем кусочки силикагеля сушат при температуре
около 500 °C, дробят до частиц нужных размеров и для удаления
пыли отсеивают. Удельная поверхность приготовленного таким об-
разом силикагеля составляет 400—500 м2/г. Капилляры силикагеля
несколько шире, чем капилляры активного угля, и более одно-
родны по размерам.
Силикагель отличается от угля меньшей адсорбционной спо-
собностью при очень низких давлениях и способностью избира-
ло
тельно поглощать пары воды. Поэтому его обычно применяют для
осушки газов.
В последнее время широкое применение в качестве адсорбентов
получили молекулярные сита. Примером таких сит являются цео-
литы, кристаллы которых построены из чередующихся кремне- и
алюмокислородных тетраэдров и содержат поры с диаметром от
4 до 7,5 А в зависимости от типа цеолита. Рыхлые пространствен-
ные решетки цеолитов способны поглощать и удерживать доста-
точно малые молекулы, в то время как большие молекулы в эти
решетки проникнуть не могут. На этом и основано молекулярно-
ситовое действие цеолитов, используемых для осушки, разделения
смесей паров и выделения растворенного вещества из растворов.
В частности, осушка органических растворителей с помощью цео-
литов основана на том, что молекулы воды (диаметр 2,75 А) легко
проникают в узкие поры кристаллов цеолита, в то время как боль-
шие по размерам молекулы растворителя в такие поры не попа-
дают.
Существенным преимуществом пористых кристаллов является
высокая однородность их пор по размерам.
Влияние свойств адсорбтива. Как уже было указано, Соссюр
установил, что газ адсорбируется тем лучше, чем легче он сжи-
жается, чем выше его критическая температура. Позднее было
установлено, что адсорбция газа тем больше, чем выше темпера-
тура кипения вещества. Установлена также связь между адсорб-
цией и теплотой испарения газа. Наконец, Аррениус нашел, что
количество адсорбированного газа увеличивается с возрастанием
константы а в известном уравнении Ван-дер-Ваальса.
В табл. IV, 1 приведены данные, характеризующие связь ме-
жду адсорбцией и физическими свойствами некоторых газов.
Таблица IV,1. Зависимость адсорбции газов на древесном угле
от их физических свойств
Газ Мол. вес Температура кипения, К Критическая температура, К Объем газа (в см3), адсорбированного 1 г угля при 15 °C
so2 64 263 430 379,7
NH3 17 240 405 180,9
N2O 44 183 309 54,2
с2н2 26 189 308 48,9
со2 44 195 242 47,6
со 28 81 134 9,3
n2 28 77 126 8,0
Н2 2 20 33 4,7
Из приведенных в табл. IV, 1 данных видно, что температура
кипения, критическая температура вещества, а также адсорбция
связаны друг с другом. Так как температура кипения со-
ставляет обычно 2/з от критической, то, естественно, что адсорбция,
111
возрастающая с ростом критической температуры, должна увели*
чиваться и с возрастанием температуры кипения. Связь между
физическими свойствами газов и адсорбцией вполне объяснима,
так как силы, действующие прн сжижении газов и их адсорбции,
близки по своей природе.
Связь между адсорбцией и указанными физическими свой-
ствами газов обнаруживается только при физической адсорбции.
При хемосорбции, в результате ее специфичности, подобная связь
обычно отсутствует.
Динамическая адсорбция. Все, что говорилось ранее об ад-
сорбции, относилось к случаю, когда адсорбция’протекает в ста-~
тических условиях, т. е. когда адсорбент адсорбирует мо-
лекулы из одного и того же объема газа вплоть до установления
адсорбционного равновесия. Однако на практике большое значение
имеет динамическая адсорбция, когда адсорбирующийся газ или
смесь его с воздухом пропускают через слой адсорбента. Такие
условия имеют место при работе установок, на которых проводится
рекуперация паров растворителей из смеси с воздухом, при работе
противогаза и т. д.
Динамическая активность адсорбента существенно отличается
от его статической активности. Статическая адсорбционная актив-
ность при данной температуре и концентрации газа определяется
количеством адсорбтива, поглощенного единицей массы адсорбента
при установлении равновесия. Динамическая активность адсор-
бента характеризуется длительностью пропускания газа или его
смеси с воздухом через слой адсорбента до момента обнаружения
первых следов газа за слоем адсорбента. Эта величина зависит
как от статической активности адсорбента, так и от других факто-
ров, а именно от соотношения между толщиной и площадью слоя
адсорбента, диаметра зерен адсорбента, концентрации газа и ско-
рости его протекания. Поэтому динамическую адсорбцию можно
характеризовать только временем, протекающим до проскока газа
через слой адсорбента при данных условиях процесса, и нельзя
характеризовать количеством газа, адсорбированным единицей
массы или объема адсорбента. Динамическая активность адсор-
бента подробно изучалась в Советском Союзе Н. А. Шиловым,
а затем М М. Дубининым и другими учеными.
Адсорбция газов из их смесей. Адсорбция газов из их смесей
имеет большое практическое значение, так как условия, при кото-
рых адсорбент окружен атмосферой одного какого-нибудь газа, на
практике встречаются крайне редко. С адсорбцией газов из их
смесей с воздухом приходится иметь дело при рекуперации раство-
рителей, при кондиционировании воздуха, при очистке двуокиси
углерода, водорода, аммиака.
При адсорбции из газовой смеси каждый отдельный газ адсор-
бируется всегда в меньшем количестве, чем если бы происходила
адсорбция одного этого газа. При этом чем лучше газ адсорби-
руется, тем лучше адсорбируется он и из смеси. Опыт показывает,
112
что адсорбция одного газа может настолько превосходить адсорб-
цию другого, смешанного с ним газа, что адсорбция второго газа
практически не происходит. В частности, это наблюдается при ад-
сорбции активным углем из воздуха паров органических раствори-
телей, когда влияние воздуха на адсорбцию паров растворителей
ничтожно.
В заключение отметим, что, применяя адсорбент, никогда не
следует забывать, что он мог ранее адсорбировать на себе тот или
иной газ. То, что мы принимаем за адсорбцию, часто является
лишь вытеснением с поверхности адсорбента другого газа.
ГЛАВА V
АДСОРБЦИЯ *НА ГРАНИЦЕ РАСТВОР —ГАЗ
Для рассмотрения явлений адсорбции растворенного вещества
на границе раствор — газ молекулярно-кинетические представле-
ния, которыми мы широко пользовались в предыдущей главе, мало
пригодны. Здесь гораздо целесообразнее рассматривать явления
с термодинамических позиций и связывать адсорбцию растворен-
ного вещества с изменением свободной энергии поверхности или ее
поверхностного натяжения.
Из-за однородности и гладкости поверхности всякой жидкости
при изучении адсорбции на ее поверхности совершенно неприло-
жимы обычные представления об активных центрах. Очевидно,
из-за равноценности всех участков поверхности жидкости и тепло-
вого движения ее молекул нельзя также говорить о каком-нибудь
закреплении молекул адсорбтива в определенных местах. Моле-
кулы адсорбтива, если только они не связаны друг с другом мо-
лекулярными силами, способны свободно перемещаться на поверх-
ности и, как правило, находятся в тепловом движении.
1. ПОВЕРХНОСТНОЕ НАТЯЖЕНИЕ
Учение о поверхностном натяжении и методы его определения подробно
рассматриваются в курсе физики. Здесь только напомним некоторые сведения
о поверхностном натяжении жидкостей и твердых тел.
Поверхностное натяжение является следствием существования внутреннего
давления — силы, втягивающей молекулы внутрь жидкости и направленной пер-
пендикулярно поверхности. Внутреннее давление тем выше, чем полярней веще-
ство, так как причиной его является действие молекулярных сил. Например,
внутреннее давление воды составляет 14 800 атм, а бензола — только 3800 атм.
Огромные значения внутреннего давления объясняют, почему жидкости мало-
сжимаемы под действием внешних давлений, обычно применяемых на практике^
Внутреннее давление втягивает молекулы, расположенные на поверхности
жидкости, внутрь н тем самым стремится уменьшить поверхность до минималь-
ной при данных условиях. Сила, действующаи на единицу длины границы раз-
дела и обусловливающая сокращение поверхности жидкости, называется силой
поверхностного натяжения, или, просто, поверхностным натяжением. Единицы
измерения этой величины дин/см. Эта сила всегда направлена тангенциально к
поверхности жидкости
Заметим, что для увеличения поверхности жидкости необходимо затратить
работу, связанную с преодолением сил, обусловливающих внутреннее давление.
Работа при обратимом и изотермическом процессе, необходимая для создания
единицы поверхности, равна удельной свободной энергии поверхности. Очевидно,
114
единицы измерения этой величины эрг/см2. Помня, что эрг = дип-см, можно полу-
чить для удельной свободной поверхностной энергии, или работы создания 1 см2
поверхности, те же единицы измерения, что и для поверхностного натяжения,
т. е. дин/см. Удельная свободная поверхностная энергия численно равна поверх-
ностному натяжению; обе эти величины обозначают символом ст. Следует иметь
д виду, что свободная поверхностная энергия равна поверхностному натяжению
только в случае однокомпонентной жидкости.
Под влиянием поверхностного натяжения жидкость при отсутствии внешних
рил всегда стремится принять форму шара, так как поверхность шара — это
наименьшая поверхность, ограничивающая объем, и, следовательно, поверхност-
ная энергия системы при этом будет минимальной.
В табл. V, 1 приведены значения ст некоторых веществ в жидком состоянии
на границе с воздухом или паром этой же жидкости.
Таблица V, I. Поверхностное натяжение некоторых веществ
в жидком состоянии на границе с воздухом или паром (по И. И. Жукову)
Жидкость Температура измерения, °C а, эрг/см2 Жидкость Температура измерения, °C (Т,ЭрГ/См2
Ртуть 20 485 Гексан 20 18,5
Вода 20 72,75 Золото 1200 1120
Глицерин 20 66,0 Олово 900 510
Этиленгликоль 20 46,7 Хлорид натрия 811 113
Анилин 20 42,9 Кислород — 198 17
Бензол 20 28,9 Водород -252 2
Хлороформ 20 27,1 Гелин —270 0,24
Этиловый спирт 20 21,6
Поверхностное натяжение пропорционально внутреннему давлению, и, следо-
вательно, чем сильнее межмолекулярные взаимодействия, тем выше поверхност-
ное натяжение
Значение поверхностного натяжения при образовании поверхности для ин-
дивидуальной жидкости устанавливается практически мгновенно, примерно в
течение 0,001 с.
Как известно, при повышении температуры поверхностное натяжение жидко-
стей понижается приближенно по прямолинейному закону. Это значит, что тем-
пературный коэффициент —da/dT имеет почти постоянное отрицательное значение
вплоть до температур, близких к критической. При критической температуре
исчезает различие между граничащими фазами и поверхностное натяжение
становится равным нулю. Это относится не только к системе жидкость — пар,
но и к системе жидкость — жидкость, когда поверхностное натяжение исчезает
при критической температуре растворимости
Для определения поверхностного натяжения на границе жидкость — газ или
жидкость — пар применяют методы капиллярного поднятия, взвешивания или
счета капель, наибольшего давления пузырьков, отрыва кольца и ряд других
описанных в курсах физики и в практикумах по коллоидной химии.
Выше мы рассматривали поверхностное натяжение на границе жидкость —
газ или жидкость — пар Для этих систем вследствие большей разряженности
газа или пара взаимодействием между молекулами жидкости и газа или пара
можно пренебречь. Этого, конечно, нельзя сделать в случае поверхностного на-
тяжения на границе жидкость — жидкость. Наличие над слоем первой жидкости
слоя другой, несмешивающейся с нею жидкости приводит к понижению меж-
фазного поверхностного натяжения, поскольку молекулы второй жидкости при-
•Тягивают к себе молекулы первой ч таким образом уменьшают действие не-
компенсированных сил на поверхности первой жидкости. Понижение межфазного
115
поверхностного натяжения, очевидно, тем значительнее, чем меньше различие
в полярностях обеих жидкостей. Жидкости, близкие по полярности смеши-
ваются друг с другом во всех отношениях, и поэтому поверхностное натяжение
между ними должно равняться нулю. Если жидкости ограниченно растворимы
друг в друге, то как Показал Г. Н. Антонов еще в 1907 г., поверхностное натя-
жение на границе жидкость — жидкость приближенно равно разности между
поверхностными натяжениями взаимно насыщенных жидкостей на границе их'
с воздухом (правило Антонова), В табл. V,2 приведены данные, пока-
зывающие, насколько правило Антонова отвечает действительности, если одной
из жидкостей является вода.
Таблица V,2. Поверхностное натяжение на границе взаимно насыщенных
жидкостей (жидкость—вода) и на границе этих жидкостей
с воздухом (по И. И. Жукову)
Жидкость Темпера- тура измере- ния, °C <7 на границе с воз- духом, эрг/см2 <г наДгранице жидкость — вода, эрг/см2
ВОДНЫЙ слой СЛОЙ органи- ческой жидкости вычислен- ное значение экспери- менталь- ное значение
Бензол 19 63,2 28,8 34,4 34,4
Анилин 26 46,4 42,2 4,2 4,8 •
Хлороформ 18 59,8 26,4 33,4 33,8
Четыреххлористый углерод 17 70,2 26,7 43,5 43,8
Амиловый спирт 18 26,3 21,5 4,8 4,8
Крезол 18 37,8 34,3 3,5 3,9
Поверхиостиое иатяжеиие иа границе жидкость — жидкость можно опреде-
лять методами, во многом аналогичными методам, применяемым для нахожде-
ния поверхностного натяжения на границе жидкость — газ.
Говоря о поверхностном натяжении твердых тел, следует прежде всего от-
метить, что для его определения существуют лишь косвенные методы. В табл. V, 3
Таблица V,3. Поверхиостиое иатяжеиие некоторых кристаллов
(по И. И. Жукову)
Кристаллы Температура измерения, °C О, ЭРГ/СМ2
CaF2 30 2500
Sr SO 4 30 1400
BaSO4 25 1250
PbF2 25 900
A?CrO4 26 575
CaSO4 • 2H2O 30 270
Pbl2 30 130
приведены значения поверхностного натяжения некоторых кристаллов, найден-
ные с помощью метода растворимости, Этот метод основан иа законе В. ТоиН
116
соиа (Кельвина), связывающем растворимость вещества с размером его малых
частиц и поверхностным натяжением.
Следует заметить, что когда говорят о поверхностном натяжении кристал-
лов вообще, допускают известное усреднение, потому что в зависимости от
плотности заполнения граней кристалла атомами или ионами и, следовательно,
в зависимости от интенсивности действия молекулярных сил разным граням
кристалла обычно соответствует различное поверхностное натяжение.
2. ПОНЯТИЕ О ПОВЕРХНОСТНО-АКТИВНЫХ ВЕЩЕСТВАХ
Выше были рассмотрены представления о поверхностном натя-
жении индивидуальных жидкостей. На поверхностном натяжении
растворов сильно сказывается явление адсорбции. .
Все растворимые вещества по их способности адсорбироваться
на границе жидкость — воздух можно разделить на две группы:
поверхностно-активные вещества и поверхностно-инактивные ве-
щества.
Поверхностно-активные вещества способны накапливаться
в поверхностном слое, и, следовательно, при этом должна происхо-
дить положительная адсорбция, т. е. Г > 0. Поверхност-
но-активные вещества должны обладать поверхностным натяже-
нием, меньшим поверхностного натяжения растворителя (иначе
накопление вещества в поверхностном слое термодинамически не-
выгодно), и сравнительно малой растворимостью; при хорошей
растворимости они стремились бы уйти с поверхности в глубь
жидкости. Иначе говоря, взаимодействие между молекулами по-
верхностно-активного вещества (в целом) и молекулами раство-
рителя всегда меньше взаимодействия между молекулами раство-
рителя. Поэтому поверхностно-активные вещества будут преиму-
щественно выталкиваться из объема раствора на поверхность, т. е.
в этом случае Г > 0. В результате накопления на поверхности рас-
твора молекул этих веществ, слабо взаимодействующих друг
с другом, межмолекулярное взаимодействие в поверхностном слое
уменьшается и поверхностное натяжение падает.
Поверхностно-активными веществами относительно воды яв-
ляются многие органические соединения, а именно жирные кислоты
с достаточно большим углеводородным радикалом, соли этих жир-
ных кислот (мыла), сульфокислоты и их соли, спирты, амины. Ха-
рактерной особенностью строения молекул большинства поверх-
ностно-активных веществ является их дифильность, т. е. строение
молекулы из двух частей — полярной группы и неполярного угле-
водородного радикала. Обладающая значительным дипольным мо-
ментом и хорошо гидратирующаяся полярная группа обусловли-
вает сродство поверхностно-активного вещества к воде. Гидрофоб-
ный углеводородный радикал является причиной пониженной
растворимости этих соединений. Наименьшее значение поверхност-
ного натяжения водного раствора поверхностно-активных веществ
Может достигать 25 эрг/см2, т. е. почти равняться поверхностному
натяжению углеводородов.
117
В дальнейшем дифильные молекулы поверхностно-активного
вещества мы будем изображать общепринятым символом:
...—О
где кружок обозначает полярную группу, а черточка — неполярный радикал.
Поверхностно-инактивные вещества стремятся уйти с поверх-
ности жидкости в объем, в результате чего происходит отрица-
тельная адсорбция, т. е. Г < 0. Поверхностно-инактивные
вещества обладают поверхностным натяжением, большим поверх-
ностного натяжения растворителя (иначе они самопроизвольно на-
капливались бы в поверхностном слое), и обычно высокой раство-
римостью, что способствует их стремлению уйти с поверхности в
объем. Другими словами, взаимодействие между молекулами по-
верхностно-инактивных веществ и растворителя всегда больше, чем
взаимодействие между самими молекулами растворителя, поэтому
поверхностно-инактивные вещества стремятся перейти в объем
раствора.
Поверхностно-инактивными веществами в отношении воды яв-
ляются все неорганические электролиты — кислоты, щелочи, соли.
Молекулы этих веществ не имеют гидрофобной части и распа-
даются в воде на хорошо гидратирующиеся ионы. Одновалентные
ионы вызывают сравнительно небольшое повышение поверхност-
ного натяжения.
Двухвалентные ионы эффективнее одновалентных в эквимоляр-
ных растворах.
Из органических веществ поверхностно-инактивными относи-
тельно воды являются лишь ионизирующиеся вещества, у которых
неполярная часть молекулы отсутствует или очень мала. К таким
веществам относятся, например, муравьиная и аминоуксусная кис-
лоты.
В неводных растворителях неорганические электролиты также
повышают поверхностное натяжение, причем величина этого эф-
фекта зависит от природы растворителя. Так, при введении иодида
натрия в метиловый спирт сильно повышается поверхностное на-
тяжение, у этилового спирта поверхностное натяжение повышается
примерно вдвое меньше, в спиртах большего молекулярного веса
эффект еще меньше.
Вещества, не влияющие на поверхностное натяжение раствори-
теля, распределяются равномерно между поверхностным слоем и
объемом раствора и, следовательно, для них Г = 0. Такие вещества
обладают поверхностным натяжением, близким к поверхностному
натяжению растворителя. Примером веществ, весьма малоактив-
ных в отношении воды, могут служить сахара — при растворении
сахаров в воде ее поверхностное натяжение почти не изменяется.
Однако следует заметить, что сахара, не изменяя заметно поверх-
ностного натяжения на границе водный раствор — воздух, часто
118
оказываются поверхностно-активными на границе водный рас-
твор — твердая фаза или другая жидкость.
На рис. V, 1 приведены изотермы поверхностного натяжения —
кривые, характеризующие изменение поверхностного натяжения
при повышении концентрации вещества. Как можно видеть, при
повышении концентрации поверхностно-активного вещества (кри-
вая 7) изотерма сначала круто падает почти по прямой. Наличие
этого участка на изотерме определяется тем, что вначале вся по-
верхность раздела жидкость — воздух свободна от поверхностно-
активного вещества и небольшие его количества, присутствующие
Рис. V, 1. Зависимость поверхност-
ного натяжения ст от концентрации
раствора:
1— изотерма для поверхностно-активного
вещества, 2 —то же для поверхностно-ин-
активного вещества ; 3 — то же для ве-
щества, ие влияющего на поверхностное
натяжение растворителя.
Рис. V, 2. Зависимость поверхност-
ного натяжения а от температуры:
1 — индивидуальная жидкость; 2— раствор
поверхностно-активного вещества.
в растворе, почти целиком уходят на поверхность. Затем следует
криволинейный участок изотермы, отвечающий средним концентра-
циям поверхностно-активного вещества. В этих условиях значи-
тельная часть поверхности уже занята молекулами поверхностно-
активного вещества, что снижает дальнейшую его 'адсорбцию на
границе раздела. Наконец, большим концентрациям поверхностно-
активного вещества на изотерме отвечает почти горизонтальный
участок, показывающий, что поверхностное натяжение мало зави-
сит от концентрации. При этих условиях на поверхности жидкости
образуется сплошной мономолекулярный слой поверхностно-актив-
ного вещества и дальнейшая адсорбция уже невозможна.
При повышении концентрации поверхностно-инактивного веще-
ства в растворе (кривая 2) изотерма полого поднимается. Это
объясняется тем, что поверхностно-инактивные вещества благодаря
большому поверхностному натяжению и хорошей растворимости
уходят в объем, а на границе раздела жидкость — воздух имеется
лишь сравнительно небольшая часть этого вещества, попадающая
туда в результате диффузии из объема раствора.
Наконец, при повышении концентрации веществ, не влияющих
на поверхностное натяжение (кривая 3), изотерма, естественно,
представляет собой прямую, параллельную оси концентраций.
119
В заключение следует отметить, что поверхностная активность
вещества зависит не только от его природы, но и от свойств рас-
творителя (среды). Если растворитель обладает большим поверх-
ностным натяжением, данное вещество может проявлять значи-
тельную поверхностную активность, если же растворитель имеет
малое поверхностное натяжение, те же вещества могут стать по-
верхностно-инактивными. Вода, наиболее часто применяющийся
растворитель, обладает большим поверхностным натяжением, и
поэтому по отношению к ней многие вещества проявляют поверх-
ностную активность. Спирты обладают значительно меньшим по-
верхностным натяжением, чем вода. Поэтому некоторые вещества
поверхностно-активные в отношении воды, являются инактивными
в отношении спиртов.
Установление равновесного значения поверхностного натяже-
ния. Баланс молекулярных сил на вновь образованной поверхности
индивидуальных жидкостей, а следовательно, и равновесное значе-
ние поверхностного натяжения устанавливаются практически мгно-
венно. На поверхности же растворов поверхностно-активных ве-
ществ должна установиться равновесная концентрация поверх-
ностно-активного вещества, что осуществляется в результате
диффузии. Поэтому, если молекулы поверхностно-активного веще-
ства большие, медленно диффундирующие, например молекулы
высших жирных кислот и их солей, равновесное значение поверх-
ностного натяжения на границе раствор — воздух может устанав-
ливаться довольно долго.
Зависимость поверхностного натяжения от температуры.
Весьма своеобразна зависимость поверхностного натяжения рас-
творов поверхностно-активных веществ от температуры, изученная
П. А. Ребиндером. Поверхностное натяжение индивидуальных
жидкостей, как уже было сказано, монотонно уменьшается с тем-
пературой. Поверхностное натяжение растворов некоторых поверх-
ностно-активных веществ с повышением температуры может изме-
няться по кривой с максимумом. На рис. V, 2 схематически изо-
бражена температурная зависимость поверхностного натяжения
для индивидуальных жидкостей (/) и растворов таких поверх-
ностно-активных веществ (2). Максимум на кривой 2 объясняется
десорбцией поверхностно-активного вещества в определенном ин-
тервале температур, что приводит к увеличению поверхностного
натяжения. При более высоких температурах, после окончания
процесса десорбции, поверхностное натяжение снова начинает сни-
жаться.
3. УРАВНЕНИЕ ГИББСА
Для интерпретаций явлений адсорбции на границе раствор —
газ весьма существенно установить связь между избытком адсор-
бированного вещества в поверхностном слое Г, концентрацией по-
верхностно-активного вещества в растворе с- и поверхностным
натяжением а на границе раствор — газ. Эта связь для разбавлен-
120
ных растворов дается известным уравнением адсорбции Гиббса,
выведенным им в 1876 г.:
Приведем упрощенный вывод уравнения Гиббса, данный Во,
Оствальдом.
Представим себе раствор поверхностно-активного вещества
с поверхностью со и поверхностным натяжением а. Пусть поверх-
ностный слой этого раствора содержит избыток в 1 моль раство-
ренного вещества. Очевидно, в этом случае можно написать:
Г = 1/<о (V,2>
Допустим, что в объеме поверхностно-активного раствора со-
держится тоже 1 моль поверхностно-активного вещества. Перене-
сем мысленно весьма малое количество поверхностно-активного
вещества из объема в поверхностный слой. При этом поверхностное
натяжение изменится на величину da.
Так как система вначале находилась в равновесии, то перенос
вещества из объема в поверхностный слой потребует затраты не-
которой работы на преодоление осмотического давления Vdn.
(где V — объем раствора; dn. — разность осмотических давлений
до и после переноса).
Поскольку изменение поверхностной энергии должно быть
равно значению затраченной осмотической работы с обратным зна-
ком, можно написать:
coda = — Vdn
Но так как V — RT/я и поскольку для достаточно разбавленных,
растворов л пропорционально концентрации с, последнее уравне-
ние можно представить следующим образом:
, RT ал RT de ...
coda ----------------- (V, 3>
ле
Подставляя в это уравнение значение со, найденное из уравне-
ния (V, 2), и решая его относительно Г, получим уравнение
Гиббса:
г______с da
1 — . RT de
В случае, когда имеют дело с достаточно концентрированными
растворами, концентрацию с в уравнении Гиббса следует заменить
на активность вещества а.
Приведем еще один более строгий вывод уравнения Гиббса, основанный на.
применении химического потенциала.
Как известно из термодинамики, для системы, состоящей из двух компонен-
тов, связь между изобарно-изотермическим потенциалом G и химическими по-
тенциалами щ и Ца обоих компонентов выражается уравнением:
G = + ргПг
121
где и п2 — число молей первого и второго компонентов, например раствори-
теля и растворенного вещества.
Это уравнение не учитывает возможности изменения поверхности системы. Для
реального раствора, обладающего поверхностью, в уравнение следует ввести
член as (где ст — поверхностное натяжение, a s— площадь поверхности). Тогда
приведенное выше уравнение можно написать следующим образом:
G = as + + Ц2П2
После дифференцирования имеем:
dG — a ds + s do + dn, + n, dp1 + p2 dn2 + n2 dp2 (V, 4)
С другой стороны, из термодинамики известно, что
dG = — S dT + V dp + dnt + p2 dn2
где S — энтропия; V — объем; p — давление.
Это уравнение также не учитывает возможности изменения поверхности. При
введении соответствующей поправки оно принимает вид:
dG = -SdT+Vdp + <Jds+pldnl+p.2dn2 (V.5)
Приравнивая правые части равенств (V, 4) и (V, 5), получаем:
S dT — V dp + s do + nj dpt + n2 dp2 =0
При постоянной температуре и давлении это уравнение упрощается:
s do + nj dpi + n2 dp2 = 0 (V, 6)
Рассматриваемый раствор можно представить состоящим из двух частей —
части, испытывающей действие поверхностных сил (поверхностная фаза), и ча-
сти, не находящейся под действием этих сил (объемная фаза). Обозначим через
п® и и® число молей первого и второго компонентов в объемной фазе,
п2 — число молей тех же компонентов в поверхностной фазе.
Очевидно, для поверхностной фазы пригодно уравнение (V, 6). Для
ной же фазы аналогичным уравнением будет:
n°ldnl + п^р2 = 0
Решим уравнение (V, 7) относительно dpt:
„о
^2
' ^Р1 =------
«1
Подставив найденное значение dpt в уравнение (V, 6), получим:
/ «1«]\
s do + I п2---г— I dp2 = 0
к «17
или
do п2 — n^l nJ
dp2 ~ s
где пг — число молей растворенного вещества, связанное с и, молями
рителя в поверхностной фазе; ~ число молей растворенного вещества,
связанное с nt молями растворителя в объемной фазе.
Правая часть уравнения (V, 8) представляет собой избыток количества рас-
творенного вещества на единице поверхности, т. е. величину адсорбции Г. Тогда
уравнение (V, 8) примет вид:
r = -da/dp2 (V,9)
а nt и
объем-
(V.7)
(V,8)
122
с увеличением с, т. е. если
срабн —*
Рис. V, 3. Построение изотер-
мы адсорбции по изотерме по-
верхностного натяжения.
Так как для разбавленных растворов р.2 = р.® +/?У In с, т° при постоянной
температуре dp.2 = RT d In с. Подставляя это значение dp2 в уравнение (V, 9),
получаем уравнение Гнббса:
г=_____с da
RT ' de
Из уравнения Гиббса ясно, что если поверхностное натяжение
увеличивается с концентрацией с, т. е. если dofde > 0, то Г < О,
иначе говоря, концентрация растворенного вещества в поверхност-
ном слое меньше, чем в объеме раствора (отрицательная адсорб-
ция). Наоборот, если о уменьшается
da Ide <0, то Г > Q и концентрация
вещества в поверхностном слое боль-
ше, чем во всем объеме (положитель-
ная адсорбция). Наконец, если о не
зависит от с, то концентрация раство-
ренного вещества в поверхностном
Слое и в объеме раствора одинакова
(адсорбции не наблюдается).
Величина — dal de называется по-
верхностной активностью. Взятая на
практически прямолинейном участке
изотермы поверхностного натяжения,
когда концентрация растворенного ве-
щества невелика и ее значение по-
стоянно, она может служить мерой по-
верхностной активности данного ве-
щества.
В честь Гиббса величину —'da/de обозначают через G и называют гиббсом.
Уравнение Гиббса в этом случае принимает очень простую форму:
/»
Г = ^гС (V,10)
откуда
G = --^- = RT — (V, 11)
de с ' 7
Как следует из уравнения (V, 11), единицы измерения G эрг-см/моль.
Уравнение Гиббса было выведено на основании термодинами-
ческих представлений. Экспериментальная проверка этого уравне-
ния весьма затруднительна в связи со сложностью определения
концентрации растворенного вещества в поверхностном слое. Тем
не менее опыты Мак-Бена, в которых с поверхности раствора с по-
мощью прибора, напоминающего микротом, срезался очень тонкий
слой жидкости, показали, что во всех исследованных случаях экс-
периментально найденные значения адсорбции в пределах ошибки
опыта совпадали со значениями, вычисленными по уравнению
Гиббса. Правильность уравнения Гиббса была подтверждена
также опытами А. Н. Фрумкина.
123
Пользуясь уравнением Гиббса, по изотерме поверхностного на-
тяжения для поверхностно-активного вещества легко построить
соответствующую изотерму адсорбции следующим образом. Возь-
мем какую-нибудь точку на изотерме поверхностного натяжения и
проведем через нее касательную и прямые, параллельные осям
координат, как это показано на рис. V, 3. Отрезок, отсекаемый на
оси ординат касательной и прямой, параллельной оси концентра-
ций, деленный на отрезок абсциссы, отсекаемый на ней прямой,
проведенной через точку параллельно оси ординат, равен —delete,
т. е
z do
с de
Подставив значение, найденное для —defde, в уравнение Гиббса,
получим:
______с do __ г
1 “ RT "dc~ RT
Определив для ряда точек изотермы поверхностного натяже-
ния соответствующие значения величины Г, легко построить изо-
терму адсорбции.
4. УРАВНЕНИЕ ШИШКОВСКОГО. ПЕРЕХОД ОТ УРАВНЕНИЯ ГИББСА
К УРАВНЕНИЮ ЛЕНГМЮРА
При малых концентрациях поверхностно-активного вещества
поверхностное натяжение раствора о уменьшается прямо пропор-
ционально концентрации с, т. е.:
Д = а0 — o = kc (V, 12)
где Д — уменьшение поверхностного натяжения; Оо — поверхностное натяжение
чистого растворителя; k — константа.
При сравнительно больших концентрациях снижение во вре-
мени о с возрастанием с описывается эмпирическим уравнением
Б. А. Шишковского, предложенным им в 1908 г.:
Д — по — о = о0В 1п -Ь 1) (V, 13)
где В — константа, мало зависящая от природы поверхностно-активного веще-
ства и равная 0,2 при температуре ~20°С; 1/А — константа, называемая удель-
мой капиллярной постоянной, характерная для каждого поверхностно-активного
вещества
Уравнение Шишковского хорошо применимо для вычисления
поверхностного натяжения жирных кислот с не слишком большим
числом атомов углерода в молекуле (до Се). ।
Как было показано Ленгмюром в 1917 г , пользуясь уравнением
Шишковского в дифференцированном виде, можно перейти от
уравнения Гиббса к уравнению Ленгмюра. В самом деле, уравне-
ние Шишковского можно записать так:
<т0 — о = оаВ In -|- — а0В In — °оВ In (с + А) — о0В In А
124
Дифференцируя это уравнение, получаем:
, а0В de а0В de
- do = aaBd In (c + 4) =
или
da Ba0
de A + c
(V, 14)
(V, 15)
Подставим уравнение Шишковского в дифференцированном
виде в уравнение Гиббса:
„ с da с Ва0 Ва0 с/А
1 — RT ' de ~ RT ’ А + с ~ RT ' 1 + с/А
Обозначая Ba0/RT через аМакс, 1/Л через k и учитывая, что при
малых концентрациях Г практически равно а, имеем:
а = амакс£с/(1 + kc) (V, 16)
Поскольку величина адсорбции а пропорциональна а, можно
также записать:
а — Пмакс^с/( 1 + ^с)
Таким образом, мы пришли к уравнению Ленгмюра. При этом
существенно, что константа 1/А, входящая в уравнение Шишков-
ского и служащая мерой капиллярной активности вещества, ока-
залась равной постоянной k в уравнении Ленгмюра. Следова-
тельно, уравнение Шишковского является тем переходным мостом,
который соединяет уравнение Гиббса, выведенное исходя из чисто
термодинамических представлений, и уравнение Ленгмюра, выве-
денное на основе молекулярно-кинетических положений.
Пользуясь уравнением Шишковского в дифференцированном виде, можно
также найти связь между поверхностной активностью —da/dc и удельной ка-
пиллярной постоянной 1/4. В самом деле, при концентрациях поверхностно-ак-
тивного вещества с <С 4 уравнение (V, 15) принимает вид-
= = <v-17)
Наконец, пользуясь уравнением (V, 15), можно установить смысл эмпири-
ческой постоянной В в уравнении Шишковского При достаточно большой кон-
центрации, когда 4 <С с, величиной 4 можно пренебречь и уравнение (V, 15)
принимает вид:
da Ва0
de с
Подставив это уравнение в уравнение Гиббса, в котором при больших значениях
с величина Г = ГмаКс = const, получим:
Гмакс = Ва$/КТ
откуда
В = ГМакс^П°го (V, 18)
Так как значения Гмакс одинаковы для всех членов любого гомологического
ряда и мало различаются для обычных поверхностно-активных веществ (жирные
кислоты, спирты и т. д), становится понятным, почему В является постоянной
величиной.
125
5. ВЛИЯНИЕ НА АДСОРБЦИЮ НА ГРАНИЦЕ РАСТВОР—ГАЗ
СТРОЕНИЯ И РАЗМЕРА МОЛЕКУЛЫ ПОВЕРХНОСТНО-АКТИВНОГО
ВЕЩЕСТВА. ПРАВИЛО ТРАУБЕ
Как мы уже отмечали, молекулы поверхностно-активных ве-
ществ обычно дифильны, т. е. имеют полярную и неполярную
часть. Полярной частью молекул поверхностно-активного вещества
могут быть группы, обладающие достаточно большим дипольным
моментом: —СООН; —ОН; —NH2; —SH; —CN; —NO2; —NCS;
—СНО; —SO3H. Неполярной частью молекулы поверхностно-ак-
тивного вещества обычно являются алифатические или ароматиче-
ские радикалы. Длина углеводородного радикала сильно сказы-
вается на поверхностной активности молекулы. Дюкло, а затем
Траубе, изучая поверхностное натяжение водных растворов гомо-
логического ряда предельных жирных кислот, нашли, что поверх-
ностная активность этих веществ на границе раствор — воздух тем
выше, чем больше длина углеводородного радикала, причем
в среднем она увеличивается в 3,2 раза на каждую группу СН2.
Это легко доказывается тем, что изотермы поверхностного натя-
жения для гомологического ряда жирных кислот почти полностью
совмещаются, если при переходе от одного члена ряда к следую-
щему изменить масштаб значений на оси концентраций в 3,2 раза.
Другая формулировка правила Дюкло — Траубе сводится
к тому, что когда длина цепи жирной кислоты возрастает в ариф-
метической прогрессии, поверхностная активность увеличивается
в геометрической прогрессии. Подобное же отношение должно
соблюдаться при удлинении молекулы и для величины 1/А, по-
скольку поверхностная активность веществ при достаточно малых
концентрациях пропорциональна удельной капиллярной постоян-
ной.
Причина зависимости, установленной сначала Дюкло, а затем
в более общем виде Траубе, заключается в том, что с увеличением
длины углеводородной цепи уменьшается растворимость жирной
кислоты и тем самым увеличивается стремление ее молекул пе-
рейти из объема в поверхностный слой. Например, если масляная
кислота смешивается с водой во всех отношениях, то уже валериа-
новая кислота образует только 4%-ный раствор, все же другие кис-
лоты с более высоким молекулярным весом еще менее растворимы
в воде.
Согласно Ленгмюру, правило Дюкло — Траубе можно обосновать следую-
щим образом Примем, что толщина поверхностного слоя равна б Тогда сред-
няя коицеитрация в этом слое будет Г/б. Из термодинамики известно, что ма-
ксимальная работа А, требующаяся для сжатия газа от объема до объема V2,
может быть представлена как
Л = /?Пп(Г1/72) (V, 19)
Для двух идеальных растворов с концентрацией С; и с2 соответственно,
когда == ЛгУг или (Vi/V2)= (Лг/лО, можно написать:
А = RT 1п (я2/Л1) = RT In (cVcJ (V, 20)
где л — осмотическое давление.
126
Тогда работа адсорбции, т е. работа обратимого переноса одного моля
растворенного вещества из объема в поверхностный слой, будет равна:
А = RTln = /?Г1п (V.21)
с f>c ’
Обозначая через Ап и An-i максимальную работу адсорбции для двух
смежных членов гомологического ряда, имеем:
Ап - An-t = RT In (г^С)п t = ЯГ In 3 = 640 кал/моль (V, 22)
Из этого уравнения видно, что работа адсорбции должна увеличиваться на
постоянную величину при удлинении углеводородной цепи на группу СН2. Это
значит, что при небольших концентрациях, при которых только и соблюдается
правило Дюкло — Траубе, все группы СН2 в цепи занимают одинаковое поло-
жение по отношению к поверхности, что возможно лишь, когда цепи располо-
жены параллельно поверхности, т. е. лежат на ней. К вопросу об ориентации
молекул поверхностно-активного вещества в поверхностном слое мы возвратимся
ниже в этом разделе.
Насколько соблюдается правило Дюкло — Траубе для гомоло-
гического ряда жирных кислот, можно видеть из данных табл. V, 4.
Правило Дюкло — Траубе соблюдается не только для жирных кис-
лот, но и для других гомологических рядов — спиртов, аминов
и т. д.
Таблица V, 4. Зависимость G и 1/4 от длины молекул жирных кислот
(по И. И. Жукову)
Кислота <?, эрг «см моль °П + 1 1/А <[М)п+1 (1/Л)га
Уксусная СНзСООН — 2,84 3,1
Пропионовая С2Н5СООН 8,5 • 10* 3,4 8,93 2,2
н-Масляпая С3Н7СООН 2,9 • 105 3,1 19,6 3,5
н-Валериаповая С4Н9СООН 8,9 • 105 2,7 68,5 3,5
н-Капроновая С5НПСООН 2,4 • 106 3,1 233 2,4
н-Гептиловая С6Н13СООН 7,6 • 106 — 555 4,0
я-Октиловая С7Н15СООН — — 2222 3,2
н-Нониловая С8Н17СООН — — 7140 —
Правило Дюкло — Траубе так, как оно сформулировано выше,
выполняется при температурах, близких к комнатной. При более
высоких температурах отношение 3,2 уменьшается, стремясь к еди-
нице, так как с повышением температуры поверхностная актив-
ность снижается в результате десорбции молекул и различие ме-
жду поверхностной активностью гомологов сглаживается.
Следует заметить также, что правило Дюкло — Траубе соблю-
дается лишь для водных растворов поверхностно-активных ве-
ществ. Для растворов этих же веществ в неполярных растворите-
лях правило Дюкло — Траубе обращается, так как с увеличением
127
углеводородной цепи растворимость поверхностно-активного веще-
ства в неполярных жидкостях возрастает и вещество стремится пе-
рейти из поверхностного слоя в раствор.
6. СТРОЕНИЕ АДСОРБЦИОННОГО СЛОЯ НА ГРАНИЦЕ РАСТВОР-ГАЗ
Выше мы рассматривали адсорбцию на поверхности раздела
раствор — газ в основном с термодинамической точки зрения. Рас-
смотрим теперь ориентацию молекул поверхностно-активных ве-
ществ на границе раствор — газ и строение адсорбционного слоя.
Чтобы получить представление о строении адсорбционного слоя
на поверхности раздела, рассмотрим результаты исследования
строения пленок, образуемых при нанесении на поверхность воды
различных жидкостей, мало растворимых в воде. Небольшое ко-
личество такой жидкости, нанесенное на неограниченно большую
поверхность воды, либо образует линзообразную каплю, когда при-
тяжение молекул жидкости больше друг к другу, чем к молекулам
воды, либо растекается по воде, образуя тончайший мономолеку-
лярный слой, когда притяжение молекул жидкости к воде больше,
чем друг к другу. Образование капель наблюдается в том случае,
когда наносимая на поверхность воды жидкость неполярна. Расте-
кание и образование мономолекулярного слоя происходит тогда,
когда молекулы жидкости дифильны. Растекающимися на поверх-
ности воды, но практически не растворяющимися в ней являются
высшие жирные кислоты, спирты, амины.
Мономолекулярные пленки на поверхности воды могут нахо-
диться в трех состояниях: газообразном, жидком и твердом.
Жидкие и твердые поверхностные пленки называются также кон-
денсированными пленками. Агрегатное состояние мономолекуляр-
ной пленки определяется молекулярными силами, действующими
между молекулами пленки.
Газообразные поверхностные плеики. Если силы, действующие
между молекулами в пленке, сравнительно невелики, то моле-
кулы поверхностно-активного вещества при достаточно большой
предоставленной им поверхности воды стремятся рассеяться по
ней и удалиться друг от друга на возможно большие расстояния.
В результате теплового движения молекулы все время переме-
щаются по поверхности воды независимо друг от друга, что обус-
ловливает поверхностное давление, действующее в направлении,
противоположном поверхностному натяжению. Такую пленку *
с полным основанием можно считать двухмерным газом, по-
скольку молекулы этого газа не могут оторваться от воды и вы-
нуждены двигаться только в двух измерениях.
* Термин <пленки» в данном случае применяется условно На самом деле
молекулы двухмерного газа не образуют на поверхности воды сплошную пленку,
а обладают тепловым движением, сходным с тепловым движением молекул
трехмерного (обычного) газа. Иногда такие пленки называют слоями поверх-
ностно-активных веществ, хотя и это название не совсем удачно.
128
Полярная группа дифильной молекулу поверхностно-активного
вещества благодаря гидрофильности и способности гидратиро-
ваться будет погружена в воду, а углеводородный радикал будет
лежать «плашмя» на воде, так как между радикалом и молеку-
лами воды существуют силы притяжения. (Следует помнить, что
выталкивание неполярного радикала на поверхность происходит
не под действием сил отталкивания между радикалом и молеку-
лами воды, а потому что притяжение между молекулами воды друг
к другу больше, чем притяже-
ние между молекулами воды и
радикалом.) а
Расположение молекул
двухмерного газа на поверхно-
сти воды схематически пока-
зано на рис. V, 4а.
Веществами, образующими
на воде газообразные пленки,
И
Рис. V, 4. Ориентация молекул поверх-
иостно-активных веществ в адсорбцион-
ном слое:
а—двухмерный газ; б — конденсированная
пленка.
являются органические соеди-
нения с дифильными молеку-
лами, имеющими не слишком
большой и не слишком малый
углеводородный радикал, на-
пример жирные кислоты с чис-
лом углеродных атомов в цепи
не меньшим 12 и не большим 20—22, алифатические спирты и
амины с не слишком большим молекулярным весом.
Аналогия между трехмерным и двухмерным газом не ограни-
чивается существованием в том и другом случае давления газа.
Существенно, что и для того и для другого газа может быть при-
менено уравнение состояния одного и того же вида. Это легко по-
казать следующим образом.
Если содержание поверхностно-активного вещества в системе
мало, т. е. когда на поверхности жидкости находится двухмерный
газ, можно написать:
<т0 — в = А = fee
Дифференцируя это уравнение, находим:
— do = d\ = k de (V.23)
Подставляя уравнение (V, 23) в уравнение Гиббса (V, 1), полу-
чим:
г______С do с db ck de _ Ь .
RT de RT de RT de RT
Обозначим площадь, которую занимает 1 моль поверхностно-
активного вещества в виде двухмерного газа, через со. Тогда
Г=1/а
5 Зак. 664
129
Подставим это значение Г в уравнение (V, 24) г
(V, 25)
ИЛИ
Дсо —(V,26)
Это уравнение аналогично уравнению Менделеева — Клапей-
рона, отнесенное к одному молю газа:
pV = RT
В самом деле, в уравнении Менделеева — Клапейрона V —
объем трехмерного газа, а в уравнении (V, 26) ® — «объем» двух-
мерного газа, т. е. его поверхность. В уравнении Менделеева —
Клапейрона р — давление, а в уравнении (V, 26) А — уменьшение
поверхностного натяжения в результате действия поверхностного
давления, т. е. величина, равная давлению двухмерного газа.
Состояние реального трехмерного газа описывается известным
уравнением Ван-дер-Ваальса:
(р + a/V2) (V - b) = RT
Для двухмерного газа в соответствующих условиях, как показал
А. Н. Фрумкин, необходимо применять аналогичное уравнение:
(А + a/со2) (<о - 0) = RT (V, 27) '
В уравнении (V, 27) величины аи0 отвечают постоянным а и b
в уравнении Ван-дер-Ваальса. При этом поправка а/ы2, обуслов-
ленная существованием сил притяжения между углеводородными
радикалами молекул, начинает сказываться только при их доста-
точной длине.
Конденсированные (жидкие и твердые) пленки. Если танген-
циально действующие силы между углеводородными радикалами
молекул поверхностно-активных веществ в поверхностной пленке
велики, то молекулы слипаются и образуют крупные конденсиро-
ванные «острова», в которых тепловое движение молекул сильно
затруднено. В таких «островах» молекулы обычно ориентируются
параллельно друг другу и перпендикулярно поверхности воды, об-
разуя своеобразный «частокол». Впрочем, согласно высказываниям
некоторых исследователей, молекулы в такой пленке могут быть
ориентированы и под некоторым углом к поверхности. Отдельные
молекулы, конечно, могут отрываться от этих «островов» и запол-
нять поверхность между ними в виде разреженной газообразной
пленки. Такое явление аналогично испарению жидкости или суб-
лимации твердого тела. Строение конденсированных пленок пока-
зано схематически на рис. V, 4 б.
Веществами, образующими в воде в обычных условиях конден-
сированные пленки, являются органические соединения с дифиль-
ными молекулами, имеющими длинные углеводородные радикалы,
так как удлинение цепи, естественно, увеличивает силы взаимодей-
ствия между молекулами. Следует, однако, заметить, что во мно-
130
гих случаях при изменении условий, например при повышении тем-
пературы, конденсированные поверхностные пленки способны пе-
реходить в газообразные.
Конденсированные пленки обычно жидкие и молекулы в них
перемещаются довольно свободно. Однако если действующие ме-
жду радикалами молекул силы настолько велики, что молекулы не
могут перемещаться, то конденсированные пленки можно рассма-
тривать как твердые. Это имеет место при относительно очень
длинных углеводородных радикалах дифильных молекул, содержа-
щих больше 20—24 атомов углерода. О наличии у конденсирован-
ных пленок свойств твердого тела можно убедиться, нанося на них
легкий порошок. Если пленка твердая, то при осторожном сдува-
нии порошок остается неподвижным. Если пленка жидкая, поро-
шок передвигается по поверхности. Другой метод определения
агрегатного состояния пленки состоит в том, что в жидкость напо-
ловину погружают маленький стеклянный диск, подвешенный на
кварцевой нити к горизонтально вращающейся головке. Если
пленка твердая, то при вращении головки образуется некоторый
угол закручивания, прежде чем диск, разорвав пленку, последует
за головкой. Если же пленка жидкая, диск следует за закручивае-
мой головкой без образования угла закручивания.
Следует указать, что помимо описанных выше типов пленок
существуют еще так называемые растянутые пленки, проме-
жуточные по свойствам между газообразными и конденсирован-
ными пленками. Такие пленки образуются, например, из конден-
сированных пленок при повышении температуры. В определенном
интервале температур конденсированные пленки расширяются,
после чего площадь, занимаемая пленкой, уже почти не зависит от
температуры. Полагают, что в растянутых пленках углеводородные
цепи молекул не ориентированы параллельно, а до известной сте-
пени переплетены между собой или, по крайней мере, энергети-
чески взаимодействуют друг с другом (слипаются), лежа «плашмя»
на воде, что и препятствует неограниченному растеканию пленки.
Полярные же группы молекул в таких пленках относительно сво-
бодно движутся в приповерхностном слое воды. Измерения, прове-
денные с помощью весов Ленгмюра (которые описаны ниже), по-
казали, что в растянутых пленках площадь, занимаемая молеку-
лой, имеет промежуточное значение, между площадями молекул,
ориентированных по нормали к поверхности воды и лежащих на
ней «плашмя».
Причина образования растянутых пленок заключается в уве-
личении с возрастанием температуры кинетической энергии угле-
водородных радикалов молекул, образующих конденсированную
пленку. Интересно, что одинаковое уменьшение длины углеводо-
родной цепи всегда вызывает одинаковое понижение температуры,
при которой происходит растяжение пленки. Вместе с тем эта тем-
пература не зависит от природы полярной группы. Способность
к образованию растянутых пленок увеличивается при наличии
5*
131
в середине алифатического радикала дифильных молекул двойной
связи, что объясняется, очевидно, большим притяжением к воде
двойной связи. Опыт также показал, что на способность поверх-
ностной пленки находиться в растянутом состоянии влияет состоя-
ние полярных групп. Так, жирные кислоты при определенных
температурах образуют растянутые пленки на нейтральных и кис-
лых растворах, т. е. при практически недиссоциированных поляр-
ных группах.. На щелочных же растворах жирные кислоты в тех
же условиях дают газообразные пленки, что обусловлено оттал-
киванием одноименных зарядов соседних полярных групп, появив-
шихся в результате их диссоциации.
7. ВЕСЫ ЛЕНГМЮРА. ОПРЕДЕЛЕНИЕ РАЗМЕРА МОЛЕКУЛ
ПОВЕРХНОСТНО-АКТИВНОГО ВЕЩЕСТВА
Существование газообразных и конденсированных поверхност-
ных пленок было блестяще доказано Ленгмюром при помощи скон-
струированных им специальных весов/ позволяющих определять
Рис. V, 5. Весы Ленгмюра:
/—кювета; 2, 3—левый и правый барьер; 4—ко-
ромысло; 5—чашечка.
Рис. V, 6. Зависимость поверхност-
ного давления р от площади, зани-
маемой молекулой кислоты s0:
7 —изотерма для лауриновой кислоты;
2—то же для миристиновой кислоты;
3—то же для пальмитиновой кислоты.
поверхностное давление, под действием которого находится пленка,
и устанавливать зависимость этого давления от площади, занимае-
мой пленкой.
Устройство весов Ленгмюра в первоначальном их виде схема-
тически показано на рис. V, 5.
Измерение с помощью таких весов проводят следующим обра-
зом. На поверхность воды, налитой вровень с краями кюветы 1 и
ограниченной слева и справа барьерами 2 и 3 из полосок пропи-
танной парафином бумаги, помещают небольшое количество лету-
чего растворителя, содержащего нерастворимое вещество, способ-
ное образовывать поверхностную пленку. Растворитель быстро
132
улетучивается, а дифильные молекулы вещества распространяются
по поверхности воды. Приближая левый подвижный барьер 2
к правому 3, связанному с коромыслом 4, уменьшают простран-
ство, занятое пленкой. При этом под действием возросшего поверх-
ностного давления пленки барьер 3 передвигается вправо и таким
образом выводит из равновесия связанное с ним коромысло 4. По-
мещая нужные разновесы на чашечку 5, можно привести весы
к нулевому положению и таким образом измерить давление, под
которым находится пленка.
Проделав эту операцию несколько раз, можно получить данные
для построения изотермы, связывающей давление, под действием
которого находится пленка, с площадью, занятой известным коли-
чеством исследуемого вещества, или с площадью, занимаемой од-
ной молекулой этого вещества. Последнюю легко вычислить, зная
площадь, занятую всем веществом, его массу и молекуляр-
ный вес.
На рис. V, 6 представлены такие изотермы, полученные Адамом
для различных жирных кислот при температуре 12,5—14,5 °C. Кри-
вая 1 относится к лауриновой кислоте (СцНгзСООН), которая
благодаря сравнительно короткому углеводородному радикалу
образует только газообразные поверхностные пленки. Как можно
видеть, эта изотерма по своей форме напоминает изотерму
pV = const для идеального трехмерного газа. Кривая 2 была по-
лучена для миристиновой кислоты С13Н27СООН, которая способна
давать и газообразные и конденсированные пленки. В этом случае
после достижения известной степени сжатия пленки (определен-
ной площади, приходящейся на одну молекулу) давление пере-
стает увеличиваться и кривая на некотором участке идет парал-
лельно оси абсцисс. Наличие этого участка, аналогичного подоб-
ному участку на изотерме р, V для реального трехмерного газа,
указывает на конденсацию двухмерного газа и отвечает наличию
равновесия между двумя поверхностными фазами — газообразной
пленкой и островками конденсированной пленки.
Интересно отметить, что наблюдаемое максимальное давление
двухмерного Газа при его конденсации, равное примерно 0,4 дин/см,
может показаться весьма малым. Однако это не так. Следует
учесть, что молекулы двухмерного газа лежат «плашмя» и тол-
щина поверхностной пленки составляет всего около 4,5 А. Соот-
ветствующий расчет показывает, что поверхностное давление
в 0,4 дин/см примерно соответствует 8,7-Ю6 дин/см2, или 8,5 атм.
Это значение уже довольно близко к критическому давлению для
обычных жидкостей.
При дальнейшем сжатии пленки, когда «островки» прибли-
жаются вплотную друг к другу и двухмерный газ между ними
исчезает, давление снова резко увеличивается. Наконец, при срав-
нительно высоких давлениях оно достигает определенного предела
и уже не изменяется (на рис. V, 6 этот участок не показав). Этот
предел соответствует давлению, при котором пленка разрушается.
133"
При этом отдельные обломки пленки наползают друг на друга, как
льдины во время ледохода, и на поверхности воды образуется уже
полимолекулярный слой поверхностно-активного вещества.
Кривая 3 относится к пальмитиновой кислоте С15Н31СООН,
обладающей большим молекулярным весом. Эта кислота из-за
сильного взаимодействия между длинными углеводородными ра-
дикалами при обычной температуре образует только конденсиро-
ванные пленки. Если такой пленке предоставлена достаточно
большая поверхность, то на остальной поверхности жидкости, не
занятой пленкой, конечно, находится двухмерный газ, но давление
его настолько мало, что его трудно измерить. При достаточном
сжатии подобных пленок наблюдается внезапное резкое возраста-
ние давления, отвечающее тому, что вся поверхность воды покры-
вается конденсированной пленкой. Дальнейшее сжатие пленки
приводит к ее разрушению аналогично разрушению пленки из ми-
ристиновой кислоты.
Установление зависимости между поверхностным давлением и
площадью, занимаемой молекулой, позволяет не только найти
связь между природой поверхностно-активного вещества и харак-
тером образующейся пленки, но и выяснить влияние температуры
на строение пленки. Опыт показал, что по мере повышения темпе-
ратуры сначала в пленке преодолеваются молекулярные силы ме-
жду углеводородными радикалами и пленка может стать растяну-
той, затем пленка превращается в газообразную.
Все сказанное об ориентации молекул поверхностно-активного
вещества на границе вода — воздух относилось к пленкам, полу-
ченным из дифильных молекул, очень мало растворимых в воде.
Однако это же относится и к поверхностным пленкам растворимых
веществ, образованных в результате адсорбции их молекул. Необ-,
ходимо лишь помнить, что между количеством веществ на поверх-
ности и в объеме всегда устанавливается известное равновесие.
Кроме того, следует учесть, что поверхностные пленки раствори-
мых поверхностно-активных веществ, благодаря сравнительно^ не-
большой длине углеводородных радикалов их молекул, почти всёгда
бывают газообразными.
По зависимости поверхностного давления от площади пленки
в кювете весов Ленгмюра можно определить размер и форму моле-
кул, образующих’пленку. В некоторых случаях подобные исследо-
вания позволили уточнить строение молекул, до этого остававшееся
неясным. Именно с помощью этого метода выяснено строение ряда
эфиров высокомолекулярных спиртов и других органических соеди-
нений.
Рассмотрим, как можно определить указанным способом раз-
мер и форму дифильных молекул нерастворимых в воде веществ.
Согласно сказанному выше, резкий подъем изотермы давление —
площадь пленки отвечает образованию плотно упакованного слоя
молекул, ориентированных перпендикулярно поверхности воды. Со-
ответствующий перегиб кривой позволяет найти площадь s0, при-
134
холящуюся на одну молекулу, расположенную в вертикальном (по
отношению к плош сти воды) положении:
___ S 1 __ 1 J W S
NAm/M = NAm/MS- ГмаксДГА '
где s — площадь всей пленки, равная произведению расстояния между барье-
рами на ширину кюветы весов Ленгмюра; т — масса взятого для определения
вещества, г; М — молекулярный вес вещества; Аа— число Авогадро.
Значение s0, найденное по этому методу для всех жирных кис-
лот, имеющих в цепи 14 и больше атомов углерода, оказалось
равным 20,5 А2. Тот факт, что площадь, занимаемая молекулой
в сплошном мономолекулярном слое, не зависит от длины цепи,
подтверждает характер расположения молекул, а именно, цепи не
лежат на поверхности воды, а ориентированы более или менее вер-
тикально. В результате рентгенографических исследований было
установлено, что в кристаллах площадь сечения параллельных
плотно упакованных углеводородных цепей составляет 18,4 А2.
Соответствие значению 20,5 А2 довольно хорошее. Несколько боль-
шие значения s0 для молекул жирных кислот в пленках можно
объяснить либо меньшей плотностью их упаковки, либо тем, что
они не вполне перпендикулярны к поверхности, а образуют с нею
некоторый угол.
Опыты также показали, что s0, не изменяясь с увеличением
длины углеводородного радикала, зависит от полярной группы мо-
лекулы. Так, s0 для жирных кислот равно 20,5 А2, для сложных
эфиров предельных кислот — 22 А2 и для спиртов — 21,6 А2. Это
указывает, что s0 является молекулярной константой, которая ха-
рактеризует полярную группу дифильной молекулы. Кроме того,
опыты показали, что s0 сравнительно мало зависит от природы по-
верхности, на которой образовалась пленка. Так, было найдено,
что площадь, занимаемая молекулой n-толуидина на границе вод-
ный раствор — воздух, равна 25,4 А2, на границе вода — гексан —
26,2 А2 и на границе вода — бензол — 25,8 А2. Поэтому, зная ГмаКо
и число молей п вещества, адсорбированного поверхностью в усло-
виях, отвечающих образованию сплошного мономолекулярного
слоя, можно вычислять поверхность адсорбента s, пользуясь про-
стым уравнением:
5 — Имакс/Гмакс (V,29)
Этот метод широко применяется для определения удельной по-
верхности различных адсорбентов и дисперсных систем. Для этого
можно воспользоваться следующим уравнением:
5уд = s/m (v> 3°)
Толщину поверхностной пленки 6, или, что то же, длину мо-
лекулы вещества при допущении вертикального ее расположения
на поверхности, также можно вычислить. Объем поверхностного
слоя, приходящийся на единицу поверхности, очевидно, численно
135
равен 8. С другой стороны, объем этого же слоя равен ГмаксУмол
(где Умол — мольный объем). Поэтому
в = Гмакс^мол и Г макс-М/ р ( V, 31)
где р — плотность адсорбированного вещества.
Ясно, что значение 6 в гомологическом ряду должно правильно
возрастать с увеличением длины цепи. И действительно, опыт по-
казал, что при удлинении алифатической цепи на группу СНг б
возрастает на 1,43 А. Рентгенографически установленное расстоя-
ние между двумя атомами углерода в углеводородной цепи, где
валентные связи расположены под углом 109°28', равно по прямой,
соединяющей оба атома, 1,54 А, а по вертикали—1,26 А.
Несовпадение последнего значения со значением 1,43 А объяс-
няется, вероятно, теми же причинами, что и несовпадение значе-
ний So, определенных с помощью весов Ленгмюра и рентгеногра-
фически.
ГЛАВА VI
АДСОРБЦИЯ НА ГРАНИЦЕ ТВЕРДОЕ ТЕЛО —РАСТВОР
Адсорбция из раствора на твердой поверхности, пожалуй, наи-
более важна для коллоидной химии, так как именно она лежит
в основе таких важнейших явлений, как образование и разрушение
лиозолей, а также их устойчивость.
Адсорбция на границе твердое тело — раствор в общем сходна
с адсорбцией на поверхности твердое тело — газ, но в первом слу-
чае явление сильно усложняется наличием третьего компонента —
среды (растворителя), молекулы которого могут также адсорбиро-
ваться на поверхности адсорбента и, следовательно, являться кон-
курентами молекул адсорбтива. Таким образом, адсорбция этого
вида является всегда адсорбцией из смеси. Кроме того, адсорбция
на границе раздела твердое тело — раствор всегда усложняется
взаимодействием молекул адсорбтива с молекулами среды.
При рассмотрении адсорбции из раствора на твердом теле при-
лито различать два случая: адсорбцию неэлектролитов, когда ад-
сорбируются молекулы адсорбтива, и адсорбцию электролитов,
когда избирательно адсорбируется один из ионов электролита.
1. МОЛЕКУЛЯРНАЯ АДСОРБЦИЯ ИЗ РАСТВОРОВ
Количество вещества а, молекулярно адсорбированное 1 г ад-
сорбента из раствора, обычно вычисляют по уравнению;
а = (с° ~ С1) V 1000 (VI, 1)
т
где с0 и Ci — начальная и равновесная концентрации адсорбтива, моль/л; V —
объем раствора, из которого происходит адсорбция, л; т — масса адсорбента, rj
1000 — переводной миожитель (для перевода измеряемой величины в ммоль/г).
Иногда, когда известна удельная поверхность адсорбента, ве-
личину адсорбции относят к единице поверхности (обычно к 1 см2).
Зависимость молекулярной равновесной адсорбции из раствора
на твердом теле от концентрации адсорбтива характеризуется
обычной изотермой адсорбции, и для достаточно разбавленных
растворов в этом случае адсорбция хорошо описывается эмпири-
ческим уравнением Фрейндлиха (IV, 2) или уравнением Ленгмюра
i(IV, 10), Конечно, для описания адсорбции из раствора на твердом
137
теле применимо и уравнение Гиббса, но, к сожалению, сложность
определения поверхностного натяжения на границе твердое тело-*
раствор не позволяет непосредственно использовать его в этом
случае.
Тем не менее величину гиббсовской адсорбции легко определить
экспериментально, измеряя изменение мольной концентрации ад-
сорбтива в растворе в результате адсорбции. Если общее число
молей вещества в растворе равно п, а мольная доля адсорбтива до
адсорбции составляла в растворе No и при равновесии с адсорбен-
том N, то
Г = п_(К„_-Е) (VI, 2)
™-5уд
где т — масса адсорбента; эУд — удельная поверхность адсорбента.
Влияние природы среды. Поскольку, как уже было указано,
при адсорбции из раствора молекулы адсорбтива и среды являются
конкурентами, очевидно, чем хуже адсорбируется среда на ад-
сорбенте, тем лучше будет происходить адсорбция растворенного
вещества. Исходя из того, что поверхностно-активные вещества
обладают малым поверхностным натяжением, можно считать, что
чем больше поверхностное натяжение самой среды, тем меньше ее
молекулы способны к адсорбции на твердом теле и тем лучше на
нем адсорбируется растворенное вещество. Именно поэтому ад-
сорбция на твердом теле обычно хорошо идет из водных растворов
и гораздо хуже из растворов в углеводородах, спиртах и других
органических жидкостях со сравнительно малым поверхностным
натяжением.
Другим критерием пригодности растворителя в качестве среды для адсорб-
ции является теплота смачивания этим растворителем адсорбента. При введе-
нии адсорбента в жидкость выделяется теплота смачивания Q, равная разности
полных поверхностных энергий адсорбента *:
(VI’3)
где Зуд — удельная поверхность адсорбента; Et и Ег — полная поверхностная
энергия на границе адсорбент — воздух и адсорбент — жидкость соответственно.
Разность полярностей поверхностей на второй границе раздела всегда мень-
ше, чем на первой, а поэтому Et > Е2 и Q > 0. Теплота смачивания обычно
колеблется в пределах 1—20 кал на 1 г адсорбента и, понятно, зависит как от
значений Et и Ez, так и от пористости или дисперсности адсорбента.
Чем больше тепла выделяется при смачивании, тем интенсивнее энергети-
ческое взаимодействие растворителя с адсорбентом и тем, следовательно, худ-
шей средой для адсорбции является данная жидкость. В табл IV, 1 приведены
* Полная энергия единицы поверхности Е является суммой свободной энер-
гии поверхности о и связанной энергии Qs:
Е = о +
Величина Qs представляет собой теплоту, необходимую для поддержания по-
стоянной температуры прн изотермическом растяжении поверхности на единицу
площади Более подробно см. Н. К Ада м. Физика и химия поверхности. М.,
ОГИЗ, 1947, с. 25.
138
значения теплот смачивания флоридина различными растворителями и количе-
ство бензойной кислоты, адсорбированной флоридином из этих растворителей,
подтверждающие приведенное положение.
Таблица VI, 1. Адсорбция на флоридине бензойной кислоты
из 10/0-ных растворов
Растворитель Т еплота смачивания, к а л/г- Количество адсорбированной кислоты, ммоль/г
Ацетон 27,3 0
Этилацетат 18,5 0,22
Хлороформ 8,4 3,50
Бензол 5,6 3,64
Четыреххлористый угле- 4,6 3,95
род Лигроин (фракция от 60 до 80 °C) 4,2 4,01
Вещества, состоящие из полярных молекул, дают большой тепловой э'ффект
при смачивании полярными жидкостями (например, водой); вещества, состоя-
щие пз неполярных молекул, выделяют больше тепла при смачивании неполяр-
ными жидкостями (например, углеводородами). Поскольку на теплоту смачи-
вания сильно влияет значение удельной поверхности, П. А. Ребиндер предложил
в качестве критерия взаимодействия полярной среды (воды) с адсорбентом
величину а — отношение теплоты смачивания порошкообразного тела водой (Qi)
и углеводородом (Qa):
a=Ql/Q2 (VI,4)
Диэлектрическая проницаемость до некоторой степени является
мерой полярности вещества; следовательно, существует известная
связь, с одной стороны, между диэлектрической проницаемостью
жидкостей и с другой — адсорбционной способностью в этих жид-
костях различных адсорбтивов.
В первом приближении можно также принять, что чем лучше
среда растворяет адсорбтив, тем хуже идет в этой среде адсорб-
ция. Это положение является одной из причин обращения правила
Дюкло — Траубе. Так, когда адсорбция жирной кислоты происхо-
дит на гидрофильном адсорбенте (например, силикагеле) из угле-
водородной среды (например, из бензола), адсорбция с увеличе-
нием молекулярного веса кислоты не возрастает, как это следовало
бы из правила Дюкло — Траубе, а уменьшается, так как высшие
жирные кислоты лучше растворимы в неполярной среде.
Влияние свойств адсорбента и адсорбтива. На адсорбцию из
растворов сильно влияют полярность и пористость адсорбента. Не-
полярные адсорбенты, как правило, лучше адсорбируют неполяр-
ные адсорбтивы, а цоляриые-адсорбенты— полярные адсорбтивы.
Влияние пористости адсорбента зависит от соотношения раз-
меров пор адсорбента и молекул адсорбтива. При увеличении по-
ристости адсорбента адсорбция малых молекул адсорбтива из
растворов обычно возрастает, так как мелкопористые адсорбенты,
139
как правило, обладают большим избирательным действием и влия-
ние химической природы поверхности у них повышено. Однако эта
зависимость соблюдается лишь в том случае, когда молекулы ад-
сорбтива достаточно малы и могут легко проникать в поры. Круп-
ные молекулы адсорбтива не могут попасть в узкие поры адсор-
бента, и адсорбция уменьшается или, во всяком случае, чрезвы-
чайно сильно замедляется. Результатом этого может явиться также
обращение правила Дюкло — Траубе, т. е. с увеличением размера
молекул адсорбтива адсорбция на адсорбентах с узкими порами
не возрастает, а падает. В табл. VI, 2 приведены данные, характе-
ризующие это положение, причем за характеристику адсорбцион-
ной способности взят коэффициент р из уравнения Фрейндлиха.
Таблица VI, 2. Адсорбция жирных кислот из водных
растворов иа адсорбенте с узкими порами
Кислота Число атомов С з цепи Коэффициент Р
Муравьивая 0 0,546
Уксусная 1 0,238
Пропионовая 2 0,111
Масляная 3 0,056
Валериановая 4 0,032
Капроновая 5 0,020
Понятно, что на непористых, с гладкими поверхностями адсор-
бентах такого обращения правила Дюкло — Траубе наблюдаться
не может. ~
Рассматривая (влияние химической природы; адсорбтива на его
способность адсорбироваться на твердом теле, трудно сделгггь ка-
кие-нибудь обобщения, так как адсорбируемость одного и того же
адсорбтива сильно зависит от полярности адсорбента и среды. Все
же, определяя влияние на адсорбцию свойств самого адсорбтива,
можно исходить из правила уравнивания полярности, сформули-
рованного П. А. (Ребиндером. Согласно этому правилу вещество С
может адсорбироваться на поверхности раздела фаз Л и В в том
случае, если наличие вещества С в поверхностном слое приводит
к уравниванию разности полярностей этих фаз. Иначе говоря, ад-
сорбция будет идти, если значение полярности вещества С, харак-
теризуемой, например диэлектрической проницаемостью е, лежит
между значениями полярностей веществ А и В, т. е. если будет
соблюдаться условие:
ел>ес>8в или 8л<ес<ев
Из правила уравнивания полярностей также вытекает, что дем
больше разность полярностей между растворимым веществом и
раствором, т\_е._чем меньше растворимость вещества, тем лучше
оно будет адсорбироваться.
140
Из правила П. А. Ребиндера также следует, что дифильные мо-
лекулы поверхностно-активного вещества должны ориентироваться
на границе раздела адсорбент — среда таким образом, чтобы по-
лярная часть молекулы была обращена к полярной фазе, а непо-
лярная часть — к неполярной. Воздух, если он является одной из
фаз, можно считать неполярной фазой. Ориентацию молекул ди-
фильных веществ, например, молекул жирной кислоты, на границе
раздела между различными фазами поясняет схема, представлен-
ная на рис. VI, 1.
Исходя из сказанного, можно сделать заключение, что все по-
лярные гидрофильные поверхности должны хорошо адсорбировать
поверхностно-активные вещества из неполярных или слабополяр-
ных жидкостей и, наоборот, неполярные гидрофобные поверхности
! ---------------------------------------—
Уголь Силикагель вода
Рис. VI, I. Схема ориентации дифильиых молекул на границе раз-
дела фаз разной природы.
хорошо адсорбируют поверхностно-активные вещества изХщоляр-
ных жидкостей (например, из водных растворов). Именно на этом
основано практическое применение полярных адсорбентов (сили-
кагель, глины, флоридин) для адсорбции поверхностно-активных
веществ из неполярных сред и неполярных адсорбентов (уголь)
для адсорбции из полярных сред.
Помимо общих положений о влиянии природы адсорбтива на
адсорбцию имеется и ряд частных правил. Так, с увеличением мо-
лекулярного веса способность вещества адсорбироваться возрас-
тает. Именно поэтому алкалоиды, а также красители, обладающие
обычно высокими молекулярными весами, хорошо адсорбируются.
Замечено также, что ароматические соединения вообще адсорби-
руются лучше, чем алифатические, а непредельные соединения
лучше, чем насыщенные. Наконец, так же как и при адсорбции на
границе раствор — воздух, при адсорбции жирных кислот и спир-
тов на твердых веществах качественно соблюдается известное пра-
вило Траубе.
Влияние времени, температуры и концентрации раствора. Ад-
сорбция вещества из раствора идет медленнее адсорбции газа, так
как уменьшение концентрации в граничном слое может воспол-
няться только путем диффузии, происходящей в жидкости в общем
довольно медленно. Для ускорения установления адсорбционного
равновесия при этом часто применяют перемешивание системы.
Особенно медленно происходит адсорбция больших молекул на
адсорбенте с достаточно узкими капиллярами. В этих условиях.
141
как мы уже указывали, равновесие устанавливается чрезвычайно
медленно или вовсе не наступает. В табл. VI, 3 приведены данные,
Таблица VI, 3. Зависимость времени, необходимого для установления
адсорбционного равновесия, от размера молекул жирных кислот
и размера пор угля
—1 - Характеристика пор Вррмя установления адсорбционного равновесия для
пропионовой кислоты гептиловой кислоты
Сравнительно широкие Среднего диаметра Узкие Весьма узкие < 1 ч 3 суток 10 суток >30 суток 2 ч ~ 32 суток Равновесие практически не наступает То же
Рис. VI, 2. Зависимость Г от
.мольной доли N адсорбтива;
1 — сильная адсорбция адсорбтива
if слабая адсорбция растворителя;
2—слабая адсорбция адсорбтива и
сильная адсорбция растворителя;
Л—слабая адсорбция компонентов.
характеризующие скорость адсорбции двух жирных кислот различ-
ного молекулярного веса на сахарном угле, имеющем поры раз-
личного диаметра. Как можно видеть, с увеличением молекуляр-
ного веса кислоты время наступления адсорбционного равновесия
сильно возрастает.
При повышении температуры адсорбция из раствора умень-
шается, однако обычно в меньшей степени, чем адсорбция газов.
Однако, когда растворимость малорас-
творимого адсорбтива в растворителе.
с увеличением температуры повышает-,
ся, адсорбция также может возрастать,
вследствие достижения значительно
более высоких концентраций разновес-'
ного раствора. Такое явление наблю-
дается, например, при адсорбции наф-.
талина на гидроксилированной поверх-
ности кремнезема из раствора в «-геп-
тане.
На рис. VI, 2 схематически показа-
но изменение величины гиббсовской
адсорбции Г в зависимости от моль-
ной доли N адсорбтива в равновесном
растворе. При сильной адсорбции ад-
сорбтива и слабой адсорбции раство-
рителя кривая 1 вначале круто подни-
мается вверх, проходит через максимум и затем падает до нуля
почти прямолинейно. Снижение величины адсорбции Г объяс-
няется тем, что при больших значениях N концентрации адсорб-
тива в поверхностном слое и в объеме раствора становятся очень
большими и притом одинаковыми. Изотерма 2 характеризует от-
рицательную адсорбцию, абсолютное значение которой возрастает
142
сначала линейно, потом достигает максимума и, наконец, при очень
высокой концентрации адсорбтива в поверхностном слое и в объе-
ме падает до нуля. Изотерма 3 характеризует сначала положитель-
ную гиббсовскую адсорбцию, которая затем меняет знак, переходя
через нуль, и становится отрицательной. Отрицательная адсорбция
с увеличением V также достигает нуля. В точке пересечения изо-
термы с осью абсцисс концентрации поверхностного и объемного
растворов одинаковы, так что данный адсорбент не может разде-
лить смеси. Это явление называют адсорбционной азеотропией.
Следует заметить, что адсорбция из раствора определяется не
полным значением потенциальной энергии системы молекула ад-
сорбтива — адсорбент, как это имеет место при адсорбции газа
при малом давлении, а разностью потенциальных энергий моле-
кулы адсорбтива по отношению к адсорбенту и по отношению
к растворителю. Поэтому теплота адсорбции адсорбтива из рас-
твора обычно в несколько раз меньше его адсорбции на том же
адсорбенте из газовой фазы.
В заключение отметим, что адсорбция жирных кислот и ряда других по-
верхностно-активных веществ из их растворов в неполярных жидкостях может
приводить к формированию на поверхности твердых тел граничных полимоле-
кулярных слоев толщиною 0,05—0,5 мкм. Как показали Б. В Дерягин с сотр. и
Г И Фукс с сотр.. механические свойстве таких слоев отличаются от свойств
объемных слоев раствора и зависят от структуры и молекулярного веса моле-
кул поверхностно-активного вещества Было также показано, что толщина гра-
ничного слоя растворов жирных кислот к гексане или бензоле является линей-
ной функцией длины углеводородного радикала, а температура «плавления»
этого слоя (снижение механических свойств до значения свойств объема рас-
твора) зависит от температуры плавления соответствующих поверхностно-актив-
ных веществ Граничные слои обеспечивают устойчивость дисперсных систем
в неполярных жидкостях и играют важную роль в действии смазочных масел.
Значение адсорбции из растворов в природе и технике. Адсорб-
ция из растворов имеет огромное значение для большинства фи-
зико-химических процессов, происходящих в растительных и
животных организмах. Явления химических превращений при
усвоении питательных веществ обычно начинаются с накопления
реагирующих веществ на поверхности природных катализаторов —
ферментов. Проникновение Веществ в организм через полупрони-
цаемые перегородки также обычно начинается с явления адсорб-
ции, происходящего на поверхности раздела.
В технике молекулярная адсорбция из растворов получила
очень широкое применение. Т. Е. Ловиц впервые применил ад-
сорбцию еще в XVIII в. для очистки древесным углем растворов
от различных примесей. В настоящее время обычный способ освет-
ления сахарных сиропов осуществляется обработкой их активным
углем. Смазочные масла также очищают с помощью специальных
гяин, действующих в качестве адсорбента.
Путем адсорбции извлекают малые количества веществ, рас-
творенных в больших объемах жидкости. Этим, например, поль-
зуются в технологии редких элементов.
143
заключается в
Рис. VI, 3. Хро-
матограмма
хлорофилла:
1—бесцветный
слой; 2—ксанто-
филл 3 (желтый).
3 — хлорофиллин р
(желто-зеленый),
4—хлоро филлин а
(зелеио-синий),
5—ксантофилл
(желтый), 6—-ксан-
тофилл а' (жел-
тый), 7—ксанто-
филл а (желтый),
8-—хлорофиллии
(серо-стальной).
Очень широко адсорбцию применяют в аналитической химии
для разделения трудно разделяемых соединений. На процессах
адсорбции основана хроматография, впервые предложенная в
1903 г. русским ученым М. С. Цветом. Этим методом М. С. Цвет
смог разделить сложный растительный пигмент хлорофилл на со-
ставные части.
Метод адсорбционной хроматографии в простейшем его виде
том, что через трубку (адсорбционную колонку)',
наполненную каким-нибудь адсорбентом (А12О3,
MgO, СаО, СаСО3, фуллерова земля, флоридин)',
в течение длительного времени медленно пропу-
скают раствор смеси веществ, которые необходи-
мо разделить. При этом на адсорбенте в верхней
части трубки отлагаются сильно адсорбирующиеся
компоненты смеси, менее сильно адсорбирующие-
ся компоненты отлагаются в средних частях труб-
ки и наименее адсорбирующиеся компоненты от-
лагаются на адсорбенте в самом низу трубки.
Мало адсорбируемые вещества не могут задержи-
ваться в верхних слоях адсорбента, так как при
прохождении через эту зону новых порций рас-
твора вещества, обладающие высокой адсорбцион-
ной способностью, вытесняют с поверхности ад-
сорбента менее адсорбционноспособные соедине-
ния.
В результате по длине столба адсорбента об-
разуются зоны, насыщенные отдельными компо-
нентами смеси. Если отдельные компоненты имеют
различную окраску, то эти зоны легко отличить по
цвету. Если же компоненты бесцветны, но способны
к люминесценции, то для определения границ ме-
жду этими зонами слой адсорбента исследуют в
ультрафиолетовом свете.
Для выделения отдельных компонентов столбик
адсорбента осторожно выдавливают ч из трубки и
разделяют на ряд коротких столбиков (зон), из
которых затем можно извлечь адсорбированный
компонент подходящим растворителем.
На рис. VI, 3 показана хроматограмма хлорофилла, состоя-
щего, как известно, из нескольких близких по строению, но раз-
лично окрашенных веществ, что не позволяет их разделить хи-
мическими методами, но дает возможность различить их по окра-
ске, применяя метод хроматографии.
Для увеличения резкости границы между отдельными зонами, в которых ад-
сорбировано то или иное вещество, приходится обычно прибегать к дополнитель-
ному «проявлению» хроматограммы. Для этого через колонку пропускают срав-
нительно небольшой объем исследуемой смеси, растворенной в подходящем
растворителе. После этого через колонку пропускают чистый растворитель (или
144
последовательно ряд растворителей) и исследуют состав выходящего раствора,
в котором компоненты появляются один за другим по мере увеличения их спо-
собности адсорбироваться адсорбентом. Этот метод называется проявительным.
На рис. VI, 4 приведена типичная дифференциальная хроматограмма, получен-
ная при разделении смеси аминокислот на колонке с крахмалом. Как можно
видеть, почти каждый компонент дает на кривой самостоятельный пик и может
быть собран в отдельную фракцию.
Вместо растворителя через колонку можно пропускать раствор какого-ни-
будь вещества, которое адсорбируется сильнее, чем любой из компонентов ис-
следуемой смеси. В этом случае хроматограмма имеет также ряд пиков, распо-
ложенных в порядке возрастания адсорбируемости веществ, и заканчивается
Объем бы текшей жидкости, мл
Рнс. VI, 4. Элюционное разделение аминокислот на крахмальной колонке (по
Штейну и Муру):
растворители: н-бутаиол, н-пропанол, 0,1 и. НС1 (1 :2 : 1) и, после аспарагиновой кислоты,
н-пропанол, 0,5 и. НС1 (2 : 1)„
выходом чистого вытесняющего вещества. Этот метод хроматографии назы-
вается вытеснительным.
Мы кратко остановились на некоторых методах жидкостной хроматографии.
Но существует еще много других методов хроматографии — осадочная, газовая,
газо-жидкостная и т. д. Все эти методы подробно рассматриваются в специаль-
ных разделах аналитической химии.
Для хроматографии первичный акт адсорбции может иметь
в зависимости от адсорбента и разделяемых веществ характер
молекулярной адсорбции или ионного обмена. Первичные про-
цессы молекулярной адсорбции при хроматографии подчиняются
уравнениям Ленгмюра или Фрейдлиха, а первичные процессы при
ионном обмене — уравнению Б. П. Никольского, о котором будет
сказано ниже.
Хроматография получила очень широкое применение при раз-
делении и очистке лекарственных веществ, витаминов, пигментов,
энзимов, протеинов и алкалоидов.
Интересно, что хроматография сыграла очень большую роль
при открытии новых, искусственно приготовленных трансурановых
элементов. Именно с помощью этого метода были разделены эле-
менты № 99 эйнштейний (Es), № 100 фермий (Fm) и № 101 мен-
делевий (Md).
145
2. ИОННАЯ АДСОРБЦИЯ
Адсорбция электролитов не укладывается в рамки учения о мо-
лекулярной адсорбции и требует специального рассмотрения, по-
скольку адсорбент может по различному адсорбировать ионы, на
которые распадается молекула электролита в растворе. Так как
наибольшее практическое значение имеют водные растворы элек-
тролитов, рассмотрим лишь адсорбцию электролитов из таких
растворов.
На адсорбции ионов существенным образом сказывается при-
рода адсорбента. Ионы, способные поляризоваться, адсорбируются
обычно только на поверхностях, состоящих из полярных молекул
или из ионов. Поэтому ионную адсорбцию часто называют также
» ।___1----1 А
О 1 I з
Рис. VI, 5. Соотношение между истинными радиусами одновалент-
ных катионов и их радиусами в гидратированном состоянии.
полярной адсорбцией. Микроучастки поверхности, несущие опре-
деленный заряд, адсорбируют противоположно заряженные ионы.
При этом ионы электролита, несущие противоположный знак, не-
посредственно не адсорбируются, но под; действием сил электро-
статического притяжения остаются вблизи адсорбированных
ионов, образуя с ними на поверхности адсорбента так называемый
двойной электрический слой. Строение этого слоя подробно рас-
смотрено в гл. VII, посвященной электрическим свойствам кол-
лоидных частиц.
Радиус ионов сильно влияет на их способность адсорбиро-
ваться. Из ионов одинаковой валентности максимальную адсорб-
ционную способность проявляют ионы наибольшего радиуса. При-
чина этого явления, с одной стороны, заключается в большой
поляризуемости таких ионов и, следовательно, их способности
притягиваться поверхностью, состоящей из ионов или полярных
молекул, с другой стороны, в меньшей гидратации ионов (чем
больше радиус иона, тем меньше при одном и том же заряде его
гидратация). Гидратация вообще препятствует адсорбции ионов,
так как наличие гидратной оболочки уменьшает электрическое
взаимодействие.
Различие в гидратации одновалентных катионов иллюстри-
рует схема, приведенная на рис VI, 5. Сплошная линия обозна-
чает границу самого иона, а пунктирная — границу его гидратной
оболочки. Из схемы видно, что ион лития гораздо более гидрати-
146
рован, чем ион цезия. Ряды ионов, составленные в порядке умень-
шения их способности связывать среду, называются лиотропными
рядами, или рядами Гофмейстера. Одновалентные катионы можно
поставить в следующий ряд по возрастающей способности адсор-
бироваться:
Li+<Na+<K+<Rb+ < Cs+
Для двухвалентных катионов это будет следующий ряд-
Mg2+< Са2+ <Sr2+ <Ва2 +
Одновалентные анионы по их возрастающей способности адсор-
бироваться располагаются в такой последовательности:
СГ < Вг- < NO3~ < 1“ < NCS-
Адсорбционная способность ионов весьма сильно зависит также
от их валентности. Чем больше валентность иона, тем сильнее он
4- + +• +
@ @
О-A/ 0-К+ «-Г
Рис. VI, 6. Достройка кристалла иодида серебра в рас-
творе иодида калия.
притягивается противоположно заряженными микроучастками по-
верхности. Катионы различной валентности по их возрастающей
адсорбционной способности можно расположить в следующий ряд:
К+ < Са2+ < А13+ < Th4+
Особый интерес для коллоидной химии представляет адсорб-
ция ионов поверхностью кристалла, в состав которого входят ионы
той же природы. При этом адсорбцию можно рассматривать как
кристаллизацию, т. е. как достройку кристаллической решетки
способным адсорбироваться ионом. Согласно Л анету и Фаяису,
кристаллы достраиваются лишь теми ионами или атомами, кото-
рые входят в их состав. Например, кристаллы Agl, внесенные в
раствор К1, адсорбируют на поверхности иодид-ионы. Если же
147-
кристаллы Agl внести в раствор AgNOs, то происходит адсорбция
ионов серебра. Понятно, что силы, под влиянием которых проис-
ходит такая достройка, являются химическими и одновременно
электростатическими силами, и ионы, достраивающие кристалл,
адсорбируются в этом случае особенно прочно. На рис. VI, 6 по-
казана схема достройки кристалла иодида серебра в растворе
иодида калия (первоначальная граница кристалла Agl обозна-
чена на схеме пунктиром).
Существенно, что достраивать кристаллическую решетку спо-
собны не только ионы, входящие в состав решетки, но и изоморф-
ные с ними. Важно также, что образовать прочную связь с по-
верхностью кристалла могут не только ионы, входящие в кри-
сталлическую решетку, но и вообще атомные группы, близкие
к атомным группам, находящимся на поверхности. Так, уголь
прочно удерживает органические радикалы, а окиси и гидраты
окисей алюминия и железа прочно связывают группы, содержащие
кислород.
В заключение отметим, что на поверхности адсорбента могут
адсорбироваться как ионы, так и молекулы, из которых они об-
разовались, причем между первыми и последними существует
адсорбционное равновесие. Наличие молекулярной формы адсор-
бированного электролита на поверхности коллоидных частиц было
показано в работах В. А. Каргина на примере ферри- и алюмо-
золей.
3. ОБМЕННАЯ АДСОРБЦИЯ
Если на поверхности адсорбента уже адсорбирован электпо-
лит, то при контакте этого сорбента с другим электролитом почти
всегда в той или иной степени наблюдается обменная адсорбция,
или, правильнее, обмен ионов между - двойным электрическим
слоем адсорбента и средой. При обменной адсорбции адсорбент,
поглощая определенное количество каких-либо ионов, одновре-
менно выделяет в раствор эквивалентное количество других ионов
того же знака, вытесненных с поверхности.
К обмену способны не только чужеродные ионы с адсорбиро-
ванными адсорбентами, но и ионы, образующиеся из самого ад-
сорбента в результате диссоциации его молекул. При этом по-
верхностное явление, каким является адсорбция, может перехо-
дить в объемное явление, т. е. в обмене могут участвовать ионы,
расположенные в глубинных слоях адсорбента, если только к ним
возможен доступ раствора. В частности, это происходит при об-
мене ионов на пермутите натрия и ионообменных смолах, о кото-
рых будет сказано ниже. Понятно, что вещества, способные к объ-
емному обмену ионов, обладают особенно высокой емкостью по-
глощения соответствующего иона из раствора.
Обменная адсорбция имеет ряд особенностей.
Во-первых, обменная адсорбция специфична, т. е. к обмену
способны только определенные ионы. Иными словами, на обмен-
148
ную адсорбцию сильно влияет как природа твердой фазы и имею-
щегося на ней двойного электрического слоя, так и природа ад-
сорбируемого иона. В зависимости от химической природы ионов,
которые могут обмениваться с ионами, содержащимися в адсор-
бентах, различают кислотные и основные адсорбенты. Кислотные
адсорбенты ведут себя подобно кислоте и способны обменивать
с растворами катионы; основные адсорбенты сходны по свойствам
с основаниями и обменивают анионы. Впрочем, существует много
амфотерных адсорбентов, которые в одних условиях обменивают
катионы, а в других — анионы. Специфичность обменной адсорб-
ции указывает на то, что по своей природе этот процесс прибли-
жается к химическим явлениям.
Во-вторых, обменная адсорбция не всегда обратима.
В-третьих, обменная адсорбция, как правило, протекает более
медленно, чем молекулярная адсорбция. Особенно медленно она
пр'отёкает, когда происходит обмен ионов, находящихся в глубине
адсорбента. Очевидно, в данном случае время необходимо для
того, чтобы ионы из раствора продиффундировали в глубь ад-
сорбента и вытеснили оттуда ионы, которые в свою очередь должны
перейти в раствор.
Наконец, в-четвертых, при обменной адсорбции может изме-
няться pH среды. Это наблюдается в том случае, когда ионом,
обмениваемым адсорбентом, является водородный или гидроксиль-
ный ион. Если адсорбент заменяет на какой-нибудь катион водо-
родный ион, то последний, поступая в раствор, уменьшает pH
среды, при этом адсорбент ведет себя подобно кислоте. Если ад-
сорбент меняет на какой-нибудь анион гидроксильный ион, то pH
раствора, наоборот, увеличивается, причем адсорбент ведет себя
как основание. Наглядно обмен ионов в обоих этих случаях можно
изобразить следующими схемами:
| адсорбент- | Н+ + Na* + Cl- —► [ адсорбент- | Na+ + Н+ -|- С1-
| адсорбент* | ОН + Na* + Cl —► | адсорбент* | С1~ + Na* + ОН
Б. П. Никольский (1939 г.) предложил уравнение, количе-
ственно характеризующее обмен ионов 1 и 2 на твердой поверх-
ности:
(VI, 5)
gj/2‘ a\lz'
где gi> g2 — содержание обменивающихся ионов в адсорбенте, г-экв/г; at, а2 — ак-
тивности обменивающихся ионов в растворе; z», zz — валентность ионов; k —•
константа.
При z, = z2 = 1 уравнение упрощается:
gilgz = k' (ai/a2) (VI, 6)
При небольших концентрациях электролита вместо активно-
сти а можно пользоваться значением концентрации. В этом случае
149
уравнение (VI, 6), связывающее адсорбционную способность ионов
1 и 2 с их концентрацией в растворе, принимает вид:
gi/g2 = fe'(c1/c2) . (VI,7)
Обменная адсорбция имеет большое значение в земледелии,
биологии и технике. Почва способна поглощать и удерживать
определенные ионы, например катионы К+ и NHJ, содержащиеся
в удобрениях и необходимые для питания растений. Взамен этих
катионов почва выделяет эквивалентные количества других катио-
нов, например Са2+ и Mg2h. Анионы, как, например, СГ, NO3, SOi",
почти не поглощаются почвой. Согласно К. К. Гедройцу (1933 г.),
детально исследовавшему явление обмена ионов в почве, погло-
щать основания способен так называемый поглощающий ком-
плекс— высокодисперсная смесь нерастворимых алюмосиликатов
и органоминеральных соединений. От природы поглощенных ионов
в значительной мере зависят физические и агротехнические свой-
ства почвы.
В технике обменная адсорбция имеет существенное значение.
Например, при крашении растительного волокна оно адсорбирует
из среды окрашенные катионы красителя, выделяя эквивалентное
количество ионов кальция, всегда присутствующих в техническом
Волокне. Здесь следует отметить, что очень часто явление обмена
обусловлено не самим веществом адсорбента, а содержащимися
в адсорбенте незначительными примесями.
Обменная адсорбция широко применяется при умягчении воды.
Как известно, наличие в воде больших количеств солей жесткости
(ионов Са2+ и Mg2+) очень часто затрудняет применение такой
воды в технике. Мыла в жесткой воде переходят в форму нерас-
творимых кальциевых и магниевых мыл и теряют свое моющее
и стабилизующее действие. Применение жесткой воды в паровых
котлах приводит к образованию на их стенках накипи, понижаю-
щей теплопроводность и увеличивающей потери тепла, а в отдель-
ных случаях может явиться причиной взрыва котла (из-за мест-
ного перегревания и постепенного изменения структуры металла).
Пища, сваренная в жесткой воде, обычно безвкусная и твердая.
Для умягчения жесткой воды Ганс предложил применять алюмо-
силикатный поглотитель, названный им пермутитом, состав кото-
рого можно выразить следующей формулой:
Na2O • А12О3 • 3SiO2 • 2Н2О
В настоящее время вместо относительно дорогого пермутита
натрия для умягчения воды применяют также природные мине-
ралы — глаукониты.
Умягчение жесткой воды с помощью пермутита мржно пояс-
нить следующей схемой:
| пермутит2- | 2Na+ -f- Са2+ + SO|~ -—> | пермутит2- | Са2+ -|- 2Na+ -у
150
Пермутит, обменявший Na+ на Са2+, в согласии с законом дей-
ствия масс, можно снова легко регенерировать путем обработки
концентрированным раствором поваренной соли. При этом ионы
Na+ вытесняют ионы Са2+ в раствор, и после промывки пермутит
снова пригоден для применения. Нетрудно видеть, что обработка
жесткой воды пермутитом ведет к ее умягчению, но не к демине-
рализации.
В последние годы вместо пермутита и глауконитов для умяг-
чения воды широко применяют ионообменные смолы, которые по-
дробнее будут описаны в гл. XIV.
Обменная адсорбция используется также для улавливания цен-
ных веществ из чрезвычайно разбавленных растворов, из которых
выделять эти вещества другими методами нерентабельно. Таким
образом, можно регенерировать, например, медь из рудничных
вод и сточных вод производства искусственного медноаммиачного
шелка; серебро из сточных вод фабрик, изготовляющих кино-
пленку; хром из электролитических хромовых ванн и т. д. Обмен-
ная адсорбция применяется при извлечении из растворов радио-
активных элементов.
Наконец, на обменной адсорбции основано также точное установление экви-
валентной точки при титровании" растворов в аналитической химии. Например,
если раствор хлорида натрия титровать в присутствии флуоресцеина нитратом
серебра, то пока в растворе имеется хотя бы небольшой избыток хлорида нат-
рия, иа поверхности образующихся кристаллов AgCl будет возникать двойной
электрический слой, состоящий из ионов С1* и ионов Na+. В результате этого
выделяющийся осадок будет белым, а раствор имеет желто-зеленую окраску.
Однако как только в растворе окажется небольшой избыток нитрата серебра, на
поверхности кристаллов AgCl образуется уже двойной слой из ионов Ag+ и
NOJ. Так как окрашенный анион флуоресцеина обладает большой адсорбцион-
ной способностью, он вытеснит из двойного электрического слоя ион NO3 и в
результате этого осадок окрасится в желто-зеленый цвет, раствор же станет
бесцветным. Такое изменение окраски наступает весьма резко, что позволяет
легко устанавливать эквивалентную точку при титровании.
Обменная адсорбия на угле
Уголь, не обладающий полярностью, казалось бы, не может
адсорбировать ионы сильных электролитов. Однако опыт показы-
вает, что уголь не только способен избирательно адсорбировать
ионы электролитов, но на нем могут протекать и явления обменной
адсорбции. Уголь является практически наиболее важным адсор-
бентом, поэтому подробно рассмотрим причины обменной адсорб-
ции на угле.
Прежде всего следует отметить, что древесный или животный
угли могут проявлять способность к обменной адсорбции вслед-
ствие содержания в них небольшого количества неорганических
веществ. Так, уголь адсорбирует из раствора метиленовой сини
окрашенные катионы красителя и отдает взамен их в раствор
Неокрашенные ионы Са2+. Однако, как показал опыт, к обменной
151
адсорбции способны и угли, совершенно лишенные неорганических
компонентов. В этом случае обменную адсорбцию можно объяс-
нить на основе чисто электрохимических представлений, развитых
А. Н. Фрумкиным, и представлений Н. А. Шилова и его школы
о поверхностных соединениях.
Согласно А. Н. Фрумкину, уголь может вести себя как газовый
электрод, напоминающий водородный электрод, получаемый в ре-
зультате насыщения платиновой черни газообразным водородом.
Поверхность угля может адсорбировать водород, который обра-
зуется в процессах получения угля и при его активации. С другой
стороны, уголь может поглощать кислород из воздуха при полу-
чении, активации и хранении угля. В зависимости от того, чем
насыщена поверхность угля, он может играть роль водородного
или кислородного электрода.
На поверхности угля, адсорбировавшего водород, при контакте
с водой образуется ион водорода, причем атом водорода является
донором, а уголь — акцептором электронов:
уголь | Н2 —► [ уголь2-! 2Н+
Такой уголь в растворе электролита способен обменивать ионы
водорода на любые другие катионы. Например, если в растворе
содержится хлорид натрия, произойдет следующий обменный
процесс:
| уголь- | Н+ + Na+ + С1— —► | уголь-1 Na+ + Н+ -|- Cl-
При этом уголь ведет себя как кислотный адсорбент, и раствор
после обмена подкисляется.
Уголь, на поверхности которого адсорбировался кислород, в
воде является донором электронов, а атомы кислорода — акцепто-
рами электронов:
| уголь | О2 —► | уголь4 + | 2О2-
Весьма неустойчивые ионы О2- тотчас взаимодействуют с водой
с образованием гидроксильных ионов:
| уголь24" | О2- + Н2О —► [ уголь24" 20Н-
В растворе электролита такой уголь ведет себя как основной
адсорбент, обменивающий гидроксильные ионы на любые анионы:
| уголь4" | ОН- + Na4" + Cl- —► | уголь4" | Cl- -|- Na4" + ОН-
При этом раствор нейтрального электролита подщелачивается.
Правильность теории А. Н. Фрумкина подтверждается тем, что
уголь, с поверхности которого тщательным эвакуированием уда-
лены адсорбированные газы, теряет способность поглощать из
раствора ионы электролитов.
152
Н. А. Шилов объяснял обменную адсорбцию на обеззоленном
угле с совершенно иных позиций. Согласно Н. А. Шилову, при по-
лучении или активировании угля на его поверхности возникают
тончайшие слои окислов, не являющиеся новой фазой и прочно
связанные с кристаллической решеткой адсорбента. Эти окислы»
образующиеся на поверхности угля, полученного в одних усло-
виях, реагируя с водой, могут давать карбоксильные группы, спо-
собные обменивать водород на катион. Иначе говоря, такой уголь
будет вести себя как кислота. При получении угля в других усло-
виях на его поверхности могут образоваться при взаимодействии
с водой гидроксильные группы. Такой уголь способен обменивать
гидроксил на анион, и, таким образом, он является основанием.
Если получение или активирование угля проводить при промежу-
точном режиме, когда на его поверхности образуется примерно
одинаковое число групп первого и второго типа, то уголь будет < .
обладать амфотерными свойствами и изменения pH раствора не
наблюдается.
Обеззоленный уголь, полученный специальной очисткой или и»
чистых веществ (например, сахара), как показал опыт, адсорби-
рует из водных растворов кислоты, а к щелочам остается индиф-
ферентным. Такой уголь адсорбирует также анионы из растворов
неорганических нейтральных солей (NaCl, KCI, KNO3, K2SO4
и т. д.). Поскольку на поверхности угля всегда находится некото-
рое число групп — С—ОН, гидроксилы этих групп в виде ионов
ОН- поступают взамен анионов в раствор, т. е. идет обменная
адсорбция, изменяющая pH системы. Такую адсорбцию принято
называть гидролитической.
При детальном рассмотрении теории обменной адсорбции на
угле А. Н. Фрумкина и Н. А. Шилова оказывается, что эти теории
не противоречат друг другу, а являются толкованиями одного и
того же явления с различных точек зрения. К этому выводу легко
прийти, если учесть, что поверхностные соединения Н. А. Шилова
способны в воде ионизироваться и образовывать двойной электри-
ческий слой газового электрода.
4. ЯВЛЕНИЯ СМАЧИВАНИЯ
К явлению адсорбции близки явления смачивания, также опре-
деляющиеся интенсивностью взаимодействия между молекулами '
различных веществ. Рассмотрим явления смачивания на примере
капли жидкости, нанесенной на поверхность твердого тела, хотя»
конечно, можно говорить и о смачивании жидкости жидкостью.
Смачивание и краевой угол. Если молекулы жидкости взаимо-
действуют с молекулами твердого тела сильнее, чем между собою»
то жидкость растекается по поверхности или, как говорят, смачи-
вает ее. Растекание происходит до тех пор, пока жидкость не по-
153
кроет всю поверхность твердого тела или пока слой жидкости не
станет мономолекулярным. Такой случай называется полным
смачиванием. Он наблюдается, например, при нанесении капли
воды на поверхность чистого стекла.
Если молекулы жидкости взаимодействуют друг с другом зна-
чительно сильнее, чем с молекулами твердого тела, растекания
не произойдет. Наоборот, жидкость соберется на поверхности в
каплю, которая, если бы не действовала сила тяжести, имела бы
почти сферическую форму. Случай, близкий к этому, наблюдается
при нанесении капли ртути на любую неметаллическую поверх-
ность.
Между этими двумя крайними случаями в зависимости от со-
отношения интенсивности молекулярных сил, действующих, с од-
ной стороны, между молекулами жидкости и, с другой — между
_________ е А
а б б
Рис. VI, 7. Различные случаи неполного смачивания:
а —9 <90°; 6—9=90°; в—9 >90°.
молекулами жидкости и твердого тела, возможны переходные слу-
чаи неполного смачивания, когда капля образует с поверхностью
твердого тела определенный равновесный угол, называемый крае-
вым углом, или углом смачивания.
На рис. VI, 7 изображены капли, образующие на твердой по-
верхности острый краевой угол (0<9О°), краевой угол, равный
90°, и тупой краевой угол (0>9О°). Следует иметь в виду, что
краевой угол, образуемый каплей на поверхности твердого тела,
всегда измеряют со стороны жидкости. Полного несмачивания,
т. е. случая, когда краевой угол равен 180°, практически никогда
не наблюдается, так как между жидкостью и твердым телом все-
гда действуют силы притяжения, хотя бы и очень малые.
Ниже приведены значения краевого угла, образуемого водой
на поверхности различных твердых тел в атмосфере воздуха:
Кварц Малахит Галенит Графит Тальк Сера Парафйн
0° 17° 47° 55—60° 69° 78° 106°
Смачивание жидкостью твердого тела можно объяснить как
результат действия сил поверхностного натяжения. Рассмотрим
пример неполного смачивания, изображенный на рис. VI, 8. Оче-
видно, периметр смачивания, или окружность, капли является гра-
ницей взаимодействия трех сред — жидкости 1, воздуха 2 и твер-
дого тела 3. Эти среды имеют разграничивающие их поверхности:
поверхность жидкость — воздух с поверхностным натяжением ai>2,
поверхность воздух — твердое тело с поверхностным натяжением
<Т2,з и поверхность жидкость — твердое тело с поверхностным на-
тяжением oii3. Таким образом, на единице длины периметра сма-
154
чивания действуют три силы ощ 2, 02,3 и ощз, обозначенные на
рисунке соответствующими векторами и направленные перпенди-
кулярно к отдельным элементам окружности и касательно к соот-
ветствующей поверхности раздела. При образовании равновесного
краевого угла 0 все три силы должны уравновешивать друг друга.
Силы 02, з и Qi, з действуют в плоскости поверхности твердого тела.
Сила же 2 направлена к поверхности под углом 0. Однако по-
скольку точка приложения трех сил может только передвигаться
1
, /\в | \
д,3 d,2-cos0 3
Рис. VI, 8. Зависимость между краевым углом и поверхностными
натяжениями о112, с2,з и 04,3:
1 — жидкость; 2—воздух; 3—твердое тело.
по поверхности, но не может от нее оторваться, вместо силы ai>2
следует взять ее проекцию на плоскость, т. е. щ, 2cos 0. Тогда усло-
вие равновесия сил выразится уравнением Юнга:
<т2> з = щ, з + щ, г соз0 (VI, 8)
Решив это уравнение относительно cos 0, получим:
°2, з — <-'1, з
‘ cos 0 ------------------ (VI, 9)
°1, 2
Помимо приведенного выше «силового» вывода, уравнение Юнга можно
вывести и из термодинамических соображений. Приводим этот вывод пъ Адам-»
сону.
Представим себе, что жидкость продвинулась по поверхности на весьма
малое расстояние и покрыла поверхность твердого тела, равную As. Учитывая,
что удельные межфазные энергии численно равны соответствующим значениям
поверхностного натяжения, изменение свободной поверхностной энергии ДА а
этом случае будет равно:
ДА = Де (щ, з — О2) 3) + Asoh 2 cos (0 — Д0)
или
i\F/hs — <т1( з — о2, з + 01,2 cos (0 — А0)
При равновесии lim kF/ks = О
As>0
Тогда
О'], з — з “В Oi, 2 cos 0 = 0
Откуда получаем:
02,3 = 01, 3 + О], 2 COS 0
„ °2, 3 — С1, 3
cos 0 =------------
<h, 2
Величину cos 0, характеризующую способность жидкости сма-
чивать поверхность, принято называть смачиванием и обозначать
155п
через В. Очевидно, что при полном смачивании (0 = 0) В — +1»
а гипотетическому случаю полного несмачивания (0 = 180°) отве-
чает В — —1. Поскольку жидкость тем лучше смачивает твердое
тело, чем меньше взаимодействие между ее молекулами, неполяр-
ные жидкости с малым поверхностным натяжением обычно хо-
рошо смачивают поверхность. Например, углеводороды с поверх-
ностным натяжением порядка 20—30 эрг/см2 практически смачи-
вают все твердые тела; вода с поверхностным натяжением
72,75 эрг/см2 (при 20 °C) смачивает лишь некоторые тела, напри-
мер стекло, кварц, неорганические соли; ртуть с поверхностным
натяжением 475 эрг/см2 смачивает только некоторые металлы.
Смачивание является процессом, при котором в системе из трех соприка-
сающихся фаз происходит уменьшение свободной энергии. В самом деле, пусть
очень тонкий слой жидкости 1 растекается по поверхности твердого тела 3, ко-
торое находится в среде 2, и пусть площадь, покрытая жидкостью, в результате
этого увеличивается на As При этом:
1) поверхность твердого тела, соприкасающегося со средой 2, уменьшится
па As, в результате чего поверхиостиая энергия системы уменьшится на аг, gAs;
2) поверхность, которой жидкость 1 соприкасается со средой 2, увеличится
на As, благодаря чему поверхностная энергия возрастает на щ, jAs,
3) площадь межфазной поверхности между жидкостью 1 и твердым телом 3
возрастает на As, вследствие чего поверхностная энергия системы увеличится
ий Gt, sAs.
Таким образом, общее изменение свободной поверхностной энергии F при
растекании жидкости будет:
AF = (— Os, а + Of, 2 + Oj, з) As
отсюда
AF/hs === <т>, з -}• Oj, 2 — <?2, з (71,10)
Но из уравнения (VI, 8) следует, что при растекании жидкости, полностью сма-
чивающей поверхность (cos0 = 1), должно соблюдаться условие
<Ъ. 3>°’l, 3 + Cl, 2
Или
С1>з + С1,2 — С2,3<0 (VI, 11)
Сравнивая уравнения (VI, 10) и (VI, 11), можно сделать заключение, что
жидкость растекается и смачивает поверхность, если при этом энергия системы
уменьшается.
Исходя из общего условия минимальной поверхностной энер-
гии при равновесии соприкасающихся сред, за меру смачивания
тела жидкостью можно принять убыль свободной поверхностной
энергии при образовании межфазной поверхности между жид-
костью и твердым телом. Отсюда следует, что из двух жидкостей
лучше смачивает данную поверхность та, при растекании которой
поверхностная энергия системы уменьшается на большую вели-
чину. Поскольку смачивание сопровождается уменьшение^ по-
верхностной энергии, в процессе смачивания выделяется тепло.
Теплота смачивания 1 см2 поверхности обычно колеблется от 10~3
До 10-5 кал. Теплота смачивания может служить характеристикой
способности жидкости смайивать поверхность твердого тела, если
нельзя определить краевой угол смачивания, например при сма-
чивании жидкостью порошков.
156
Явление смачивания можно наблюдать и тогда, когда вместо
воздуха взята вторая жидкость, не смешивающаяся с первой и
имеющая меньшую плотность. Если каждая из двух жидкостей мо-
жет смачивать поверхность, то, очевидно, между ними будет про-
исходить конкуренция, аналогичная конкуренции при адсорбции
двух адсорбтивов. Исходя из того, что смачивание определяется
соотношением молекулярных сил, действующих между молекулами
каждой отдельной жидкости, с одной стороны, и между молеку-
лами жидкостей и молекулами твердого тела, с другой стороны,
нетрудно видеть, что из двух жидкостей смачивать поверхность
будет та, значение полярности которой ближе к полярности твер-
дого тела. О жидкости, лучше смачивающей поверхности, говорят,.
что она обладает большим избирательным смачиванием по отно-
шению к данной поверхности.
Избирательное смачивание было исследовано П. А. Ребинде-
ром. Рассмотрим это явление более подробно. Представим себе,
что поверхность твердого тела находится в соприкосновении с по-
лярной водой и каким-нибудь неполярным углеводородом. Тогда,
если вода избирательно смачивает поверхность, т. е. если краевой
угол 0, образуемый со стороны воды, <90°, а значение В =
— cos 0 > 0, поверхность называют гидрофильной. Согласно урав-
нению (VI, 9) этот случай имеет место при условии, когда стг, з>
>О1,з или когда 02, з— Ш, з > 0- Избирательное смачивание во-
дой наблюдается обычно,‘когда разность полярностей между водой
и твердым веществом меньше, чем между неполярным углеводоро-
дом и твердым веществом. К веществам с гидрофильной поверх-
ностью относятся вещества с сильно выраженным межмолекулярным
взаимодействием, например кварц, стекло, корунд, гипс, малахит,
т. е. силикаты, карбонаты, сульфаты, а также окиси и гидраты
окисей металлов. Из органических веществ в качестве веществ
с гидрофильной поверхностью можно указать целлюлозу.
Если же твердое тело лучше смачивается неполярным углево-
дородом, т. е. если для воды 0 > 90°, а значение В = cos 0 < О,
поверхность называют гидрофобной или олеофильной. Согласно
уравнению (VI, 9) этот случай может наблюдаться лишь тогда,
когда Стг, з<<Т1,з или когда огг, з — «й, з < О- Избирательное сма-
чивание неполярным углеводородом наблюдается тогда, когда раз-
ность полярностей между углеводородом и твердым веществом
меньше, чем между водой и этим веществом. К веществам с гид-
рофобной поверхностью относятся все углеводороды и другие орга-
нические соединения с большими углеводородными радикалами.
Из неорганических соединений сюда надо отнести сульфиды тяже- .
лых металлов, тальк, графит, серу.
При краевом угле, равном 90°, очевидно, будет наблюдаться
промежуточный случай, когда избирательное смачивание отсут-
ствует.
Интересно, что если все минералы разместить в ряд соответ-
ственно возрастанию их твердости, то этот ряд, как указал
157
П. А. Ребиндер, совпадает с рядом тех же минералов, располо-
женных по их избирательной смачиваемости водой. Минералы вы-
сокой твердости — кварц, корунд, алмаз — обладают очень высо-
кой гидрофильностью.
Следует заметить, что во многих учебниках и монографиях
гидрофильной или гидрофобной поверхностью называется поверх-
ность, на которой вода образует, соответственно, острый или тупой
краевой угол на границе с воздухом. Так определять гидрофиль-
ность или гидрофобность неправильно, поскольку большинство по-
верхностей дают с водой в атмосфере воздуха острые краевые
углы. Иначе обстоит дело при избирательном смачивании поверх-
ности водой в присутствии неполярной жидкости. В этом случае
вода далеко не всегда образует острый краевой угол. Гидрофиль-
ные поверхности в этих условиях будут избирательно смачиваться
водой, при этом краевые углы 0 < 90°; однако если поверхности
гидрофобные, углеводород частично вытесняет воду и для воды
6 > 90 °C.
Определение краевого угла. Для измерения краевого угла, об-
разуемого жидкостью на твердом теле, на его поверхность наносят
небольшую каплю жидкости и с помощью пучка света, направлен-
ного параллельно поверхности, проектируют боковое изображение
капли на экран, затем на экране очерчивают контур капли, сидя-
щей на поверхности твердого тела, и через точку, в которой со-
прикасаются все три фазы, проводят касательную к контуру капли.
Угол наклона этой касательной и есть краевой угол.
Для измерения краевого угла, образуемого двумя несмешиваю-
щимися жидкостями на поверхности твердого тела, пластинку из
исследуемого вещества погружают в стеклянную кйвету с более
легкой жидкостью и затем на поверхность пластинки осторожно
с помощью пипетки наносят каплю более тяжелой жидкости. Да-
лее поступают так же, как и в предыдущем случае.
Помимо этого простейшего, но не очень точного метода опре-
деления краевого угла есть и другие, более точные, но и более
сложные методы, которые рассматриваются в практикумах по кол-
лоидной химии.
Следует заметить, что определить истинное значение краевого
угла часто затруднительно по следующим причинам.
1. На краевой угол могут влиять следы веществ, загрязняющих
поверхность. Резкое изменение смачивания происходит уже при
образовании на поверхности мономолекулярного слоя, загрязняю-
щего вещества, для чего нужны ничтожные его количества. По
данным некоторых исследователей, на смачивание поверхности мо-
жет сказываться даже контакт этой поверхности в течение некото-
рого времени с воздухом промышленных городов, всегда содержа-
щим следы углеводородов.
2. Многие вещества, например металлы, хорошо окисляются и
на их поверхности образуется тончайшая, трудно обнаруживаемая
пленка окислов. Эта пленка также может сильно влиять на сма-
158
чивание. Поэтому для определения краевого угла надо брать не-
загрязненные и неокислившиеся поверхности.
3. Поверхности обычно хорошо адсорбируют воздух. Естествен-
но, что адсорбированный воздух замедляет процесс растекания
жидкости по твердому телу, так как для вытеснения воздуха с по-
верхности и установления равновесного краевого угла требуется
определенное время. Подобное замедление установления равновес-
ного краевого угла называется гистерезисом смачивания. Во мно-
гих случаях равновесное значение краевого угла из-за гистерезиса
может и не достигаться вовсе.
Явление гистерезиса наглядно проявляется в том, что краевой
угол очень часто зависит от условия его образования. А именно,
если каплю жидкости прижать к поверхности твердого тела, то
краевой угол будет меньше, чем в том случае, если каплю осто-
рожно нанести на поверхность. В капиллярах с неполностью сма-
чиваемыми стенками вследствие гистерезиса жидкость подни-
мается на различную высоту в зависимости от того, поднимается
ли жидкость свободно по капилляру, или же сначала заполняют
капилляр и затем дают стечь жидкости до достижения равновес-
ного положения.
Гистерезис является также причиной того, что краевой угол,
образуемый при натекании жидкости, обычно гораздо больше, чем
при оттекании. Последнее явление можно наблюдать, когда капли
дождя стекают не по слишком чистому оконному стеклу, при этом
капли как бы задерживаются и снизу образуют гораздо больший
краевой угол, чем сверху,
С целью устранения гистерезиса, происходящего в результате
адсорбции воздуха, методику определения краевого угла можно
видоизменить следующим образом. Пластинку твердого вещества
вводят в стеклянную кювету с жидкостью и после полного сма-
чивания ее поверхности устанавливают в горизонтальном положе-
нии. Затем под пластинку, не вынимая ее из жидкости, с помощью
пипетки с загнутым концом вводят пузырек воздуха. Далее опре-
деление краевого угла, который и в этом случае измеряют со сто-
роны жидкости, проводят так,же, как было описано выше*.
4. На смачивание твердого тела может влиять шероховатость
поверхности, причем чем больше шероховатость, тем резче про-
являются свойства поверхности, обуславливающие притяжение
или отталкивание воды. Подобное влияние можно объяснить тем,
что при 0 < 90° (для гладкой поверхности) жидкость проникает
в углубления поверхности подобно тому, как она всасывается в
смачиваемые ею капилляры.’ Понятно, что это улучшает смачива-
ние шероховатой поверхности. Если 0 > 90°, то жидкость но
* Более подробные сведения о гистерезисе смачивания можно найти в ра-
ботах: П. А. Р е б и н д е р и др. Исследования в области поверхностных явле-
ний. М„ ОНТИ, 1936, 299 с. '
159
проникает в углубления, что ухудшает смачивание шероховатой
поверхности. Иными словами, гидрофильной поверхности шерохо-
ватость придает как бы еще большую гидрофильность, а шерохо-
ватость гидрофобной поверхности делает ее еще более гидрофоб-
ной. Для исключения влияния микрорельефа поверхность иссле-
дуемого твердого тела должна быть возможно более гладкой.
Венцель первый указал путь, позволяющий учитывать влияние шерохова-
тости поверхности на ее смачивание жидкостью. Для этого входящие в расчет-
ное уравнение (уравнение Юнга) поверхностные натяжения вг, з и щ, з следует
умножить на так называемый фактор шероховатости, т. е. на отношение факти-
ческой поверхности раздела к поверхности твердого тела, если бы она была
гладкой *. В результате вместо уравнения Юнга надо написать:
Г (<Т2, 3 — 01, з) = Щ, 2 COS 0
5. На краевой угол могут влиять условия образования поверх-
ности. Так, поверхность стеариновой кислоты, полученная охла-
ждением ее расплава на воздухе, гидрофобна. Поверхность же
стеариновой кислоты, полученная охлаждением ее расплава на
границе со стеклом, оказывается гидрофильной. Это явление мож-
но объяснить тем, что в первом случае наружу слоя кислоты
(в воздух) обращены, главным образом, гидрофобные углеводо-
родные радикалы стеариновой кислоты, а во втором случае, бла-
годаря действию поверхности полярного стекла на расплав, на-
ружу обращены полярные гидрофильные карбоксильные группы.
Вообще благодаря определенной ориентации молекул вещества
в поверхностном слое можно говорить о поверхностной и
объемной гидрофильности или гидрофобности вещества.
В. А. Пчелиным было показано, что поверхность животного во-
лоса гидрофобна, в то время как само вещество волоса гидро-
фильно.
На практике наиболее важен случай, когда смачивающей жид-
костью является вода. Плохо смачиваются водой неполярные ве-
щества, например поверхности, покрытые углеводородами. Для
улучшения смачивания водой обычно применяют смачиватели —
растворимые, хорошо адсорбирующиеся, поверхностно-активные
вещества, понижающие поверхностное натяжение на границе
твердое тело — жидкость и одновременно на границе жидкость —
воздух. При нанесении на поверхность смачивателя краевой угол,
являющийся мерой смачивания, согласно уравнению (VT, 9),
уменьшается в результате снижения значений oi, 3 и <ji, 2, а сма-
чивание соответственно возрастает.
Обычно молекулы смачивателей дифильны, что характерно для
всякого поверхностно-активного вещества, и адсорбируются на
поверхности воды, ориентируясь углеводородными цепями наружу
в воздух, благодаря чему на поверхности воды создается как бы
* Более подробно о гистерезисе см. С. С. Воюций, Физико-химические
основы пропитывания и импрегнироваиия волокнистых материалов дисперсиями
полимеров. Л., «Химия», 1969, с. 13.
160
пленка углеводорода. Этим и объясняется понижение поверхност-
ного натяжения раствора и повышение смачивающей способности
до значений, соответствующих поверхностным натяжениям орга-
нических неполярных жидкостей.
Интересным свойством монослоев поверхностно-активных ве-
ществ является их способность переходить из раствора на твердую
поверхность и образовывать на этой поверхности полимолекуляр-
ные слои. Это свойство было обнаружено еще Блоджетт на сле-
дующем опыте.
В мономолекулярный плотно упакованный слой молекул по-
верхностно-активного вещества, образованный на поверхности
воды, осторожно вводят в вертикальном,положении стеклянную
пластинку. При погружении этой пластинки гидрофильная поверх-
ность ее покрывается мономолекулярный слоем поверхностно-
активного вещества, причем полярная часть молекул вещества,
естественно, будет обращена к полярной поверхности стекла и
поверхность пластинки, таким образом, станет гидрофобной. После
этого пластинку вместе с адсорбированным на ней слоем поверх-
ностно-активного вещества осторожно извлекают из раствора. При
этом на пластинке, уже адсорбировавшей мономолекулярный слой
поверхностно-активного вещества, адсорбируется второй слой того
же вещества, причем неполярные радикалы молекул этого слоя
обращены к неполярным радикалам первого слоя, т. е. по направ-
лению к пластинке. В результате на поверхности пластинки обра-
зуется бимолекулярный слой молекул, обращенных друг к другу
неполярными радикалами, а поверхность пластинки станет снова
гидрофильной. Неоднократно повторяя эту операцию, можно пе-
ренести на пластинку значительное число слоев поверхностно-
активного вещества, делая поочередно поверхность пластинки то
гидрофобной, то гидрофильной.
Практическое значение смачивания. Смачивание имеет большое
значение для успешного проведения ряда важнейших технологиче-
ских процессов. Например, в текстильной технологии хорошее сма-
чивание волокна или тканей является важным условием для кра-
шения, беления, расшлихтовки, пропитки, стирки и т. д. Совер-
шенно понятна роль смачивания для эффективного применения
инсектофунгисидов, поскольку листья растений и шерстяной по-
кров животных всегда в той или иной степени гидрофобны. Боль-
шое значение имеет смачивание и в типографском деле. Смачива-
ние соответствующими жидкостями металлов и неметаллических
тел ускоряет и облегчает иХ механическую обработку (резание,
сверление, шлифовку, полировку). Бурение нефтяных скважин в
горных породах также облегчается, если применять специальные
бурильные растворы, содержащие смачиватели. При лужении,
спайке, сварке металлов, а также склеивании различных твердых
тел необходимо прежде всего хорошее смачивание их поверхности.
Наконец, на явлениях избирательного смачивания основано обо-
гащение руд — флотация. Рассмотрим в качестве примера роль
6 Зак. 664 161
г
смачивания в таких процессах, как стирка, пропитка и фло-
тация.
Целью стирки является удаление с поверхности твердого Teyia,
обычно волокна или ткани, прилипших к ней загрязнений — шер-
стяного жира, шлихты, сажи, пыли, белковых веществ и т. д. При
стирке поверхность раздела волокно — загрязнение заменяется на
поверхности раздела волокно — раствор моющего вещества и за-
грязнение— раствор моющего вещества. Отличие процессов, ле-
жащих в основе стирки, от избирательного смачивания заклю-
чается только в том, что роль первой жидкости играет твердое или
полутвердое загрязнение.
Рассмотрим процесс стирки на примере удаления жидкого угле-
водорода (жира) с поверхности ткани с помощью обычного мыла.
Задача технолога при проведении этого процесса — как можно
Рис. VI, 9. Схема отмывания углеводорода при стирке:
а—жидкость, смачивающая поверхность; б—жидкость, не смачивающая по-
верхность.
1—мыльный раствор; 2— углеводород; 3 — ткань; 4 — гидрофильный слой мою-
щего вещества.
больше уменьшить поверхностное натяжение на границах раздела
моющий раствор — загрязнение и моющий раствор — волокно. По-
верхностные натяжения этих поверхностей уменьшаются в резуль-
тате адсорбции мыла на поверхности жира и ткани, причем ад-
сорбция происходит, конечно, таким образом, что молекулы мыла
обращаются полярной частью в воду, неполярной — к загрязнению
или волокну. Тогда под влиянием неизменившегося поверхностного
натяжения на границе твердое тело — загрязнение жир соберется
в каплю, которую легко можно удалить с поверхности волокна
путем даже незначительного механического воздействия. Происхо-
дит отмывание, заключающееся в резком увеличении В = cos 0
(для воды), т. е. в уменьшении краевого угла. Это наглядно пока-
зано на рис. VI, 9.
При стирке моющее вещество, адсорбируясь как на поверхно-
сти ткани 3, так и на поверхности загрязняющей ткань частицы
углеводорода 2, образует на них гидрофильный слой мыла 4. Этот
слой способствует отделению частицы загрязнения от ткани, так
как улучшение смачиваемости отмываемой поверхности водой в
условиях избирательного смачивания означает понижение смачи-
вания ее углеводородом. Кроме того, образовавшийся на капель-
ках углеводорода слой гидратированных частично ионизированных
162
молекул мыла придает устойчивость возникшей эмульсии и этим
способствует удалению загрязнения вместе с моющим раствором
в конце стирки.
Следует заметить, что на поверхности отбеленной гидрофиль-
ной ткани, благодаря полярности целлюлозы, адсорбирующиеся
молекулы мыла могут ориентироваться полярными группами к
волокну и неполярными углеводородными радикалами — в мою-
щий раствор. Казалось бы, в этом случае должна происходить
гидрофобизация поверхности, понижающая эффект стирки. Од-
нако имеются данные, что при использовании достаточно концен-
трированных растворов мыла после возникновения первого слоя
молекул на ткани образуется второй слой мыла, молекулы кото-
рого ориентированы противоположным образом, т. е. полярной
группой в моющий раствор. Таким образом, и в этом случае на-
блюдается гидрофилизация поверхности ткани.
П. А. Ребиндер считает, что обязательным условием для мою-
щего действия является также достаточная механическая проч-
ность и вязкость гидратированных адсорбционных слоев моющего
вещества. С одной стороны, такие слои на границе моющий рас-
твор— воздух способствуют пенообразованию, играющему при
стирке важную роль. С другой стороны, образование прочных гид-
ратированных адсорбционных слоев вокруг частиц жира обеспе-
чивает эмульгирование отмытых загрязнений и препятствует их
вторичному оседанию на волокне *.
Процессом, в известной степени обратным стирке, является пропитка тканей
с целью повысить их водонепроницаемость при сохранении воздухопроницаемо-
сти (так называемая пористая водоотталкивающая пропитка). Задача технолога
при проведении этого процесса заключается в образовании на поверхности от-
дельных волоконец ткани тонких пленок, на которых вода образует большой
краевой угол. С этой целью ткани пропитывают растворами или дисперсиями
гидрофобных, так называемых водоотталкивающих веществ. В качестве таких
веществ можно использовать ацетат алюминия, мыла поливалентных металлов,
парафин, асфальт, нефтяные остатки, кремнийорганические соединения и смеси
этих веществ. Иногда пропитку тканей с целью повышения их водонепроницае-
мости проводят в два приема. Например, ткань пропитывают сначала диспер-
сией парафина, содержащей мыло в качестве эмульгатора, а затем раствором
ацетата алюминия, при этом частицы парафина отлагаются на волокне в ре-
зультате коагуляции.
После пропитки ткань высушивают, и при этом на волоконцах материала
образуются пленки водоотталкивающих веществ. При пропитке ткани диспер-
сией частицы водоотталкивающего вещества отлагаются на поверхности волоко-
нец дискретно. Однако в условиях последующей сушки при повышенной темпе-
ратуре гидрофобное вещество обычно плавится и растекается по волокну.
В результате пропитки поверхность капилляров ткани становится гидро-
фобной, несмачиваемой водой. Продвижению воды по такому капилляру ме-
шает большое поверхностное натяжение на границе вода — гидрофобизированная
поверхность, которое стремится уменьшить поверхность соприкосновения между
водой и стенкой капилляра и не позволяет воде течь по стенкам капилляра.
Поэтому вода, проникая в устье капилляра, не смачивает его стенки, а образует
выпуклый мениск, как это показано на рис. VI, 10. Искривленная поверхность
* Более подробно о механизме стирки см. П. А. Ребиндер и др. Физико-
химия моющего действия. М., Пищепромиздат, 1935, 178 с.
6*
163
этого мениска под действием поверхностного натяжения стремится стянуться
и стать плоской, при этом возникает направленное вверх давление. Это давле-
ние уравновешивает давление воды, стремящейся проникнуть в капилляр. По-
нятно, что чем выше поверхностное натяжение и чем уже капилляр, тем больше
должно быть давление, которое может протолкнуть воду через капилляр. Следо-
вательно, для получения ткани с высокой водонепроницаемостью нужно, чтобы
гидрофобизируемая ткань была достаточно
плотной и чтобы в ней не содержалось поверх-
ностно-активных веществ (остатков эмульгато-
ров эмульсии, которой производилась пропит-
ка)*.
Рис. VI, 10. Несмачивание во-
дой капилляра гидрофобизиро-
ванной ткани.
На явлениях смачивания и несма-
чивания, как уже указывалось, ос-
нован и процесс флотации — метод
обогащения полезных ископаемых, по-
лучивший в настоящее время исклю-
чительно широкое применение. В осно-
ве этого метода лежит использование
различий в смачивании разделяемых
частиц водой. Чтобы уяснить себе принцип, на котором основана
флотация, рассмотрим поведение достаточно малых гидрофобных
и гидрофильных минеральных частиц на границе раздела вода —
воздух или вода — масло.
Гидрофильные частицы смачиваются водой и под действием
поверхностного натяжения на границе вода — воздух или вода —
масло частица втягивается в воду. Наряду с силой поверхностного
РиС. VI, 11. Разделение гидрофильных (а) и гидрофобных (б)
частиц путем флотации:
/—водная фаза; 2 — воздух илн масло; 3—твердое тело.
натяжения на частицу действует и сила тяжести. В результате
частица, смачиваемая водой, целиком перейдет в водную фазу и
потонет.
Гидрофобные частицы вода не смачивает, и под действием' по-
верхностного натяжения на границе вода — воздух или вода —
масло частица может остаться на границе раздела, если только
она не слишком велика и сила тяжести не превысит флотационную
силу. Схематически эти оба случая изображены на рис. VI, 11.
Понятно, что измельчая минерал до нужной дисперсности, всегда
* Подробнее о механизме пропитки см. С. С. В о ю ц и й, Физико-химиче-
ские основы пропитывания и импрегнирования волокнистых ыатеоиалов диспер-
сиями полимеров. Л., «Химия», 1969, 236 с,
1Q4
можно подобрать размер частиц, при котором частицы будут оста-
ваться на поверхности, поскольку при дроблении сила тяжести
уменьшается прямо пропорционально кубу радиуса частицы, а
флотационная сила снижается прямо пропорционально первой сте-
пени радиуса. Однако слишком высокая дисперсность может ока-
заться вредной — мелкие частицы подвержены интенсивному броу-
новскому движению, что уменьшает вероятность их закрепления
на границе вода — воздух или вода — масло.
Флотационное обогащение руд полезных ископаемых основано
на том, что сернистые соединения, в виде которых металлы обычно
находятся в руде, обладают большей гидрофобностью, чем пустая
порода, например кварц. Практически флотационное разделение
руды никогда не проводят простым введением измельченной руды
в воду, поверхность - которой граничит с воздухом или маслом.
В таком виде флотационный процесс слишком неэффективен. В на-
стоящее время широкое применение получила так называемая
пенная флотация. Она заключается в том, что в суспензию мине-
рала— флотационную пульпу — тем или иным способом вводят пу-
зырьки воздуха. При всплывании пузырьки собирают по своей по-
верхности те частицы руды, на' которых вода образует большой
краевой угол. В результате на поверхности пульпы образуется
минерализованная пена. Эту пену самотеком или с помощью спе-
циальных гребков удаляют с поверхности пульпы в виде концен-
трата. руды. Хорошо смачиваемые водой частицы пустой породы
не прилипают к пузырькам, оседают на дно и образуют отходы
флотации, так называемые «хвосты».
Оптимальный размер зерен минерала при флотации 0,15—
0,01 мм. Однако при флотации каменного угля и самородной серы
частицы минерала могут быть и более крупными (0,5—1 мм). Ве-
совое отношение руды к воде в пульпе обычно лежит в пределах
от 1 : 4 до 1 : 2.
Рассмотрим в самых общих чертах термодинамику пеиной флотации. Для
этого вычислим измеиеиие свободной энергии системы при пениой флотации.
До флотации для свободной межфазной энергии Fo системы можно написать
следующее уравнение:
Fq = 2s1.2 + <T1, з$1,з (VI, 12)
где <Ti. 2 и (Т1 ,з — удельная поверхностная энергия (численно равная межфаз-
ному натяжению) на границе водная среда — воздух и водная среда — флоти-
руемое вещество соответственно; st, 2 и Si, з — соответствующие межфазные по-
верхности. В результате флотации свободная энергия в расчете на единицу по-
верхности контакта флотируемой частицы с воздухом равна:
Fj = <Т1, 2 ($1, 2 — 1) + <Т1, 3 (®1, 3 — 1 ) + О2, 3 (VI, 13)
Отсюда изменение свободной энергии вследствие флотации составит:
— AF = F0— Ft = (Tt> 2 + (Ti. з — Оз, з (VI, 14)
Согласно уравнению (VI,8)-
Ot, з — 02, з = — 01, 2 cos 0 (VI, 15)
165'
Подставляя это уравнение в уравнение (IV, 14), получим:
— ДР = О], 2 — О1. 2 COS0 = О], 2 (1 — COS 0) = 01,2 (1 — В) (VI, 16)
Вероятность флотации W связана с изменением свободной энергии следующим
образом:
F~exp(-A/W) = exp [<Т1,2(1 - B)/RT] (VI, 17)
Следовательно, вероятность флотации частиц возрастает с увеличением поверх-
ностного натяжения на границе вода — воздух и с уменьшением смачивания
водной фазой флотируемых частиц.
Обычно разность в гидрофобности поверхности частиц ценного
минерала и пустой породы сравнительно невелика. Поэтому для
повышения эффективности флотации почти всегда применяют так
называемые коллекторы, или собиратели. В качестве коллекторов
используют органические вещества с дифильной молекулой, спо-
собные адсорбироваться на поверхности частиц ценного минерала
таким образом, что полярная часть молекулы обращается к ад-
сорбенту, а углеводородный радикал — наружу. В результате этого
гидрофобность частиц минерала возрастает и флотационный про-
цесс протекает интенсивнее. Наиболее часто в качестве коллекто-
ров применяют ксантогенаты R0—(где R— углеводород-
\SM
ный радикал, М — щелочной металл). Имеются данные, что ксан-
тогенаты не просто адсорбируются поверхностью частиц сернистых
металлов, но вступают с ними в химическое взаимодействие.
При флотации несульфидных минералов в качестве коллекто-
ров обычно применяют жирные кислоты и их мыла. Ионогенные
группы этих коллекторов всегда обращены к твердой фазе (Solid),
поэтому эти группы принято называть солидофильными. Особенно
пригодны такие коллекторы для солеобразных минералов, в со-
став которых входят катионы щелочноземельных металлов Са2+,
Mg2+, Sr2+. В кристаллических решетках этих минералов преобла-
дает ионная связь, и их катионы активно взаимодействуют с хи-
мически адсорбирующимися поверхностно-активными ионами
RCOO- кислоты или мыла. На закреплении коллекторов на по-
верхности ' флотируемых частиц сказывается также и влияние
длины углеводородного радикала, а именно, взаимодействие угле-
водородных цепей друг с другом способствует образованию' ад-
сорбционной пленки и чем сильнее такое взаимодействие, тем
прочнее закрепляются адсорбционные слои коллектора на поверх-
ности минерала.
Для неполярных неметаллических минералов (графит, сера,
тальк, ископаемый уголь), которые плохо смачиваются водой, нет
необходимости в сильных коллекторах. Для них обычно исполь-
зуют мало растворимые в воде коллекторы, например масла. При
флотации расходуется сравнительно мало коллектора — на 1 т
руды берут обычно только сотни граммов реагентов. Это указывает
166
на то, что для улучшения флотации на поверхности минеральных
частиц достаточно образования тончайшего мономолекулярного
слоя, иногда даже ненасыщенного.
Помимо коллекторов при флотации применяют пенообразова-
тели и регуляторы.
Пенообразователи нужны для получения достаточно (но не слишком) стой-
кой минерализованной пены. Пенообразователи не должны заметно изменять
смачивание флотируемых частиц, а должны адсорбироваться на границе пузы-
рек воздуха — водная фаза. Этими свойствами обладают алифатические спирты,
так как группа —ОН обычно слабо закрепляется на поверхности минеральных
частиц и в то же время хорошо адсорбируется на границе раствор — газ, бла-
годаря поверхностной активности спирта, способствуя тем самым вспениванию.
При выборе пенообразователей следует избегать веществ, сильно понижаю-
щих поверхностное натяжение, так как иначе пена окажется пустой, т. е. не
содержащей ценного минерала или содержащей его в очень малом количестве.
Регуляторы применяют для регулирования избирательного действия коллек-
торов. В одних случаях регулятор, действуя непосредственно на поверхность
минерала, облегчает взаимодействие с ним коллектора и тем улучшает флота-
цию. Такой регулятор является активатором флотации. В других случаях, наобо-
рот, регулятор затрудняет взаимодействие минерала с коллектором, что по-
давляет флотацию этого минерала. В этом случае регулятор можно назвать де-
прессором. Примером активатора при флотации является сульфид натрия, улуч-
шающий флотацию сернистых металлов с помощью ксантогенатов. В качестве
депрессора может служить жидкое стекло, ухудшающее флотацию силикатных
минералов.
Флотацию можно применять не только для выделения из по-
лезного ископаемого какого-нибудь одного продукта, но и для
разделения ряда продуктов. Такая флотация называется селектив-
ной, она осуществляется обычно с помощью регуляторов. Приме-
ром селективной флотации может служить флотация свинцово-
цинковых руд. При этом сначала получают свинцовый концентрат,
вводя для этого в пульпу депрессоры, предотвращающие флота-
цию цинковых минералов, затем с помощью активаторов улучшают
флотационную способность цинковых минералов и добиваются
перехода их в пену, получая таким образом цинковый концентрат.
Применение флотации для обогащения полезных ископаемых
непрерывно расширяется. Флотация используется для обогащения
сульфидных и ряда руд цветных металлов, например свинцово-
цинковых, медных, медно-цинковых, молибденовых, железных, оло-
вянных и руд редких металлов. Флотация применяется для обо-
гащения таких ископаемых, как сера, графит, уголь, а также руд,
содержащих апатит, плавиковый шпат, барит и т. д. Значение
флотации особенно возрастает вследствие того, что она позволяет
использовать тонко вкрапленные в горные породы руды, запасы
которых неисчерпаемы.
5. АДГЕЗИЯ
К явлениям адсорбции и смачивания довольно близки также явления адге-
зии, когда две взаимно нерастворимые жидкости, либо жидкость и твердое тело,
либо, наконец, два твердых тела приводятся в тесный контакт друг с другом
167
и под действием межмолекулярных или иных (например, электрических) сил
прочно прилипают друг к другу так, что для их разделения нужно приложить
известное усилие нли произвести работу.
Различают три случая адгезии.
1. Адгезия между двумя жидкостями. Мысленно отделим друг от друга в
среде 2 две жидкости 1 и 3, соприкасающиеся по площади s, как это показано
на рис. VI, 12. Тогда образуются две поверхности: жидкости 1 с поверхностным
натяжением оч, 2 и жидкости 3 с поверхностным натяжением оъ, з, а поверх-
ность раздела с поверхностным натяжением оч, 3 исчезнет. Таким образом, ра-
бота адгезии, т. е. работа, которая нужна для разделения единицы поверхности
одной жидкости от другой, будет равна:
А/а == оч, 2 + <Тг, з “ сП, з (VI, 18)
Это уравнение впервые дал Дюпре.
2. Адгезия между жидкостью и твердым телом. Допустим, что на поверх?
ности твердого тела 3 находится капля жидкости 1 в среде 2. Работу адгезии
в этом случае по уравнению (VI, 18) вычислить нельзя, так как поверхностные
натяжения оч, э и оч,э обычно неизвестны.
Однако ffi, з и <т2, з можно исключить
из уравнения (VI, 18) с помощью форму-
лы (VI, 8). Тогда для вычисления ра-
боты адгезии можно использовать урав-
нение:
A/s = oj, 2 + <Х|, 2 cos 0==
= <т.,2(1 +cose) (VI, 19)
3. Адгезии между твердыми телами.
Работу адгезии в этом случае невоз-
Рнс. VI, 12. Схема, показывающая
изменение межфазных поверхностей
при отделении
жидкостей 1
несмешицающихся
и 3 в среде 2.
много раз превышает
можно определить, исходя из значений
поверхностного натяжения, так как по-
верхностное натяжение на границе твер-
дое тело— воздух обычно неизвестно.
Кроме того, Б. В. Дерягиным и Н. А. Кро-
товой показано, что работа адгезии
работу, вычисленную на основании кос-
в этом случае во
венных теоретических предположений, и что она зависит от скорости разруше-
ния адгезионного соединения. Это указывает на неравновесный характер про-
цесса разрушения адгезионного соединения между твердыми телами. Для объ-
яснения адгезии твердого тела к твердому телу в разное время был выдвинут
ряд теорий — молекулярная, часто называемая не совсем правильно адсорбцион-
ной теорией (Дебройн, Мак-Ларен н другие зарубежные ученые), электрическая
теория, затем развившаяся в электронную (Б. В. Дерягин, Н. А. Кротова и
В. П. Смилга), так называемая диффузионная теория, приложимая к частному
случаю—адгезии полимера к полимеру (С. С. Воюцкий). и др. Вероятней всего
универсальной теории адгезии твердого тела к твердому’ телу вообще не суще-
ствует. В зависимости от природы твердых тел н условий образования адгезион-
ного соединения адгезию в том или ином случае можно объяснить, исходя из
разлитых теорий *.
* Теория адгезии изложены в следующих монографиях (приводятся только
главнейшие); Б. В. Дерягин, Н. А. Кротова, Адгезия. М. — Л. Изд. АН
СССР, 1949. с. 244; Адгезия, цементы, прнпон. Сборник под ред. Н. Дебройна
и Р. Гувинка. Пер. с англ. М., Издатинлит, 1954, 584 с.; С. С. Воюцяий.
Адгезия. Энциклопедия полимеров, т. 1. М., «Советская энциклопедия», 1972. См.
с 22; С. С. Воюцкий. Аутогезия и адгезия высокополимеров. М, Ростехиздат,
1960. с. 244; Б. В. Дерягин, Н. А. Кротова, В. П. Смилга. Адгезия твер-
дых тел. М., «Наука», 1973, с. 270; А. А. Берлин, В. Е. Басин. Основы,
адгезии полимеров. М., «Химия», 1974, с. 392.
168
ГЛАВА VII
ЭЛЕКТРИЧЕСКИЕ СВОЙСТВА КОЛЛОИДНЫХ СИСТЕМ
В этой главе рассмотрены электрические свойства высокодис-
персных коллоидных систем с твердой дисперсной фазой и жидкой
дисперсионной средой. Об электрических свойствах аэрозолей,
эмульсий, а также растворов коллоидных поверхностно-активных
веществ сказано в главах, посвященных этим системам.
1. ПОНЯТИЕ ОБ ЭЛЕКТРОКИНЕТИЧЕСКИХ ЯВЛЕНИЯХ
Наличие у частиц дисперсных систем электрического заряда
было открыто еще в 1808 г. профессором Московского универси-
тета Ф. Ф. Рейссом. Он показал, что при наложении разности
электрических потенциалов на электроды, опущенные в заполнен-
ные водой стеклянные трубки, воткнутые в кусок сырой глины,
как это схематически показано на рис. VII, 1, жидкость в трубке
с положительным полюсом мутнела, а в трубке с отрицательным
полюсом вода оставалась прозрачной. Это указывало на то, что
частицы глины переносятся в электрическом поле к положитель-
ному полюсу. Более поздними исследованиями было установлено,
что частицы переносятся в электрическом поле с постоянной ско-
ростью. Эта скорость тем больше, чем выше приложенная разность
потенциалов и диэлектрическая проницаемость среды, и тем мень-
ше, чем больше вязкость среды. Перенос частиц в электрическом
поле получил название электрофорез^или Катафореза.
Рейсс заметил также, что если тонкий кварцевый песок по-
местить в среднюю часть U-образной трубки так, чтобы он обра-
зовал как бы пористую диафрагму, затем заполнить трубку водой
и приложить электрический ток к электродам, помещенным в оба
колена трубки (рис. VII, 2), то уровень воды в колене с отрица-
тельным электродом будет повышаться до тех пор, пока разность
уровней в обоих коленах не достигнет определенного значения.
Подобно электрофорезу этот процесс идет с постоянной скоростью,
и количество перенесенной жидкости прямо пропорционально при-
ложенной разности потенциалов и диэлектрической проницаемости
и обратно пропорционально вязкости среды. Исследованиями Ви-
демана, проведенными в 1852 г., было установлено, что количество
жидкости, прошедшей через капилляры пористой диафрагмы,
169
пропорционально силе тока и при постоянной силе тока не зависит
от площади сечения или толщины диафрагмы. Это явление было
названо~электроосмосом, или электроэндоосмосом.
Причина обоих явлений, обнаруженных Ф. Ф. Рейссом, одна
и тажё— наличие разноименных зарядов у твердой и жидкой
фазы. При электрофорезе в результате возникновения электриче-
ского поля между электродами, благодаря малому размеру частиц
глины, происходит перенос отрицательно заряженной дисцерсной
фазы к положительному электроду. При электроосмосе ввиду того,
что частицы песка слишком тяжелы, под влиянием электрического
поля по капиллярам, имеющимся в слое песка, к отрицательному
электроду передвигается положительно заряженная жидкость.
Рис. VII, 1. Схематическое изо-
бражение опыта Рейсса по элек-
трофорезу.
Рис. VII, 2. Схематическое изо-
бражение опыта Рейсса по
электроосмосу.
'В дальнейшем были обнаружены два явления, как бы противо-
положные электрофорезу и электроосмосу. Дорн в 1878 г.-обна-
ружил, что при оседании каких-либо частиц в жидкости, например
песка в воде, возникает электродвижущая сила между двумя элек-
тродами, введенными в разные места столба Жидкости. Это явле-
ние, противоположное электрофорезу, получило название эффекта
Дорна, или потенциала седиментации.
При продавливании жидкости через пористую перегородку, по
обеим сторонам которой находятся электроды, также было обна-
ружено возникновение разности потенциалов. Явление это, откры-
тое Квинке в 1859 г. и обратное электроосмосу, было названо по-
тенциалом протекания, или потенциалом течения.
Все четыре указанных'явления, поскольку" "в них происходит
передвижение частиц или жидкости при приложении разности по-
тенциалов или, наоборот, возникает разность потенциалов при
передвижении частиц или жидкости, получили общее название
электрокинетических явлений. Эти явления, будучи связанными
с наличием межфазной поверхности, проявляются легче всего
в высокодисперсных системах с большой удельной поверхностью.
Рассмотрим подробнее электрофорез и электроосмос, так как
эти явления весьма важны для изучения и понимания электриче-
ских свойств коллоидных систем.
170
Совершенно очевидно, что причина всех электрокинетических
явлений заключена в противоположности знаков заряда твердой
фазы и жидкости. Это положение было принято еще Квинке и
Гельмгольцем во второй половине XIX столетия. Однако вопрос,
почему возникают эти заряды на межфазной границе, оказался
гораздо более сложным.
Причиной возиикиовения заряда коллоидных частиц вначале считали пере-
ход электронов из одной фазы в другую при контакте двух фаз. Однако если
бы эта точка зрения была правильной, то при электрофорезе должно было бы
соблюдаться известное правило Кёна, согласно которому тела с большей ди-
электрической проницаемостью должны заряжаться положительно, а с мень-
шей— отрицательно, поскольку первые обычно являююя донорами, а вторые —
акцепторами электронов. Для некоторых коллоидных систем, например для гид-
розолей серы или эмульсий масла в воде, это правило как будто соблюдается.
Однако для большого класса коллоидных систем, а именно для коллоидных рас-
творов металлов и их окислов в воде, оно оказалось^ совершенно неприемле-
мым Частицы металлов, обладающих бесконечно большой диэлектрической про-
ницаемостью, как правило, несут отрицательный заряд, тогда как вода, имеющая
по сравнению с ними небольшую диэлектрическую проницаемость, оказывается
заряженной положительно. Кроме того, опыт показал, что знак заряда колло-
идной частицы может меняться на обратный под действием весьма небольших
количеств некоторых электролитов, не влияющих сколько-нибудь заметно на ди-
электрическую проницаемость среды. Эти наблюдения показали несостоятель-
ность теории, связывающей возникновение заряда с контактом двух фаз.
С современной точки зрения заряд на коллоидных частицах
лиозолей, проявляющийся при электрофорезе, обусловлен нали-
чием на их поверхности двойного электрического слоя из ионов’,
возникающего либо в результате избирательной адсорбции одного
из ионов электролита, находящегося в растворе, либо за счет
ионизации поверхностных молекул веществ. Правильность такой
точки зрения подтверждают опыты, показавшие, что электрокине-
тические явления не наблюдаются или почти не наблюдаются в
жидких средах с очень малой диэлектрической проницаемостью,
в которых не происходит заметной диссоциации электролитов.
К таким жидкостям относятся хлороформ, петролейный эфир, се-
роуглерод. В то же время электрокинетические явления наблю-
даются в нитробензоле в таких слабо полярных жидкостях, как
ацетон, этиловый и метиловый спирты, и в особенности — в воде.
Образование двойного электрического слоя в результате изби-
рательной адсорбции одного из ионов рассмотрено в гл. VI. На
рис. VII, 3 в самом общем виде изображен двойной электрический
слой, возникающий на кристаллах иодида серебра, находящихся
в слабом растворе иодида калия *. Иодид-ионы (потенциалопре-
деляющие ионы) достраивают кристаллическую решетку иодида
* Кристалл Agl, имеющий кубическую форму, условно изображен на
рис. VII, 3, в виде кружка К этому приему, ради простоты, будем прибегать
в аналогичных случаях и в дальнейшем. Потенциалопределяющие ионы пока-
заны на рис. VII. 3 с внешней стороны частицы. В некоторых руководствах эти
ионы помещают непосредственно в ее поверхностном слое. Каждый способ раз-
мещения потенциалопределяющих ионов имеет свои преимущества Первый спо-
соб подчеркивает то важное обстоятельство, что потеициалопределяющий ион
171
Рис. VII, 3. Схема образова-
ния двойного Электрического
слоя вокруг частицы иодида
серебра, находящейся в рас-
творе иодида калия.
серебра и тем самым придают частицам отрицательный заряд,
а ионы калия (противоионы) находятся в растворе вблизи меж-
фазной поверхности. В целом, однако, весь комплекс, называемый
мицеллой, остается электронейтральным.
Впрочем, образование двойного электрического слоя в резуль-
тате избирательной адсорбции одного из ионов, присутствующих
в дисперсионной среде, может происходить и тогда, когда до-
стройки кристаллической решетки нет. Например, двойной элек-
трический слой образуется на частицах парафина, диспергирован-
ного в слабом растворе щелочи, за
счет избирательной адсорбции гидр-
оксильного иона, который в данных
условиях проявляет лучшую адсорби-
руемость, чем ион щелочного металла.
Возникновение двойного электри-
ческого слоя за счет ионизации мож-
но проиллюстрировать образованием
двойного электрического слоя на ча-
стицах водного золя двуокиси крем-
ния. Молекулы SiOa, находящиеся на
поверхности таких частиц, взаимодей-
ствуют с дисперсионной средой, гидра-
тируются и образуют кремневую кис-
лоту, способную ионизироваться:
H2SiO3 SiO|“ + 2Н+
При этом силикатные ионы SiOl"
остаются на поверхности частицы, об-1
условливая ее отрицательный заряд, а ионы водорода переходят
в раствор. Схематически двойной электрический слой на поверх-
ности частиц SiC>2 можно изобразить так, как это показано на
рис. VII, 4. Понятно, что и в этом случае весь комплекс в целом
будет электронейтрален. Конечно, вполне возможно, что не все
молекулы НгЗЮз, находящиеся на поверхности, ионизируются.
Кроме того, возможна- и одноступенчатая диссоциация на ионы
SiOsH- и Н+. Но это не меняет сути дела.
Более подробно образование двойного электрического слоя на
коллоидных частицах рассмотрено в следующей главе.
При действии электрического поля на частицы, несущие двой-
ной электрический слой, происходит явление, напоминающее элек-
тролиз. Если дисперсная фаза заряжена отрицательно, коллоид-
ные частицы вместе с адсорбированными на них отрицательными
в большинстве случаев адсорбируется частицей из внешней среды. Второй способ
указывает на то, что адсорбированный потенциалобразующий ион достроил ча-
стицу и составляет с ней одно целое. Учитывая, что ни один из этих способов
размещения ионов не имеет явных преимуществ, в дальнейшем будем прибе-
гать к первому способу, поскольку он используется в большинстве отечествен-
ных учебников и был применен в первом издании этой книги,
172
потенциалопределяющими ионами движутся к аноду, а положи-
тельно заряженные противоионы — к катоду. Если дисперсная
фаза заряжена положительно, направление движения частиц и
ионов меняется на обратное. Следует отметить, что к электроду,
имеющему заряд, одноименный с заряженными частицами, дви-
жется только часть противоионов. Другая часть противоионов, на-
ходящихся весьма близко от поверхности дисперсной фазы, под
действием сравнительно значительных электрических и адсорб-
ционных сил оказывается связанной с частицами и вынуждена
двигаться вместе с ними. Явление электрофореза можно пояснить
Рис. VII, 5. Схема движения кол-
лоидной частицы н противоионов при
электрофорезе.
Рис. VII, 4. Схема образования двой-
ного электрического слоя вокруг
частицы двуокиси кремния, находя-
щейся н воде.
схемой, изображенной на рис. VII, 5. При этом совершенно про-
извольно принято, что потенциалопределяющие ионы заряжены
отрицательно, а противоионы — положительно. Конечно, электро-
форез надо рассматривать не как простой перенос заряженных ча-
стиц и противоионов к соответствующим электродам, а как пере-
нос, сопровождающийся постоянным взаимным обменом между
противоионами соседних коллоидных частиц. В этом отношении
электрофорез полностью сходен с электролизом, который осу-
ществляется путем обмена ионов соседних молекул.
Изложенному представлению о существе электрофореза, ка-
залось бы, противоречат сделанные ранее наблюдения об одно-
сторонности этого явления, т. е. наблюдения, показавшие, что при
электрофорезе переносится только коллоидное вещество, но не
происходит переноса ионов. Однако противоречие здесь только
кажущееся, так как для образования на частицах двойного элек-
трического слоя требуется ничтожно малое количество электро-
лита, которое очень трудно определить количественно. Так, было
найдено, что при получении золя сульфида мышьяка, для кото-
рого стабилизатором являются молекулы сероводорода, на 0,67 г
AS2S3, выделившегося на аноде, приходилось всего IO75 г водо-
рода, выделяющегося на катоде. Понятно, что такое количество
водорода с помощью обычных аналитических методов определить
178
невозможно, что и привело к возникновению в свое время непра-
вильных представлений об односторонности процесса электрофо-
реза.
Аналогичное объяснение имеет и явление электроосмоса. Двой-
ной электрический слой в этом случае образуется на внутренней
поверхности капилляров пористого тела либо в результате изби-
рательной адсорбции одного из ионов электролита, присутствую-
щего в жидкости, заполняющей капилляр, либо вследствие иони-
зации молекул вещества, из которого состоят стенки капилляра,
либо, наконец, в результате адсорбции на поверхности капилляра
ионов ОН- или Н+, всегда присутствующих в воде.
На рис. VII, 6 изображен двойной электрический слой, образо-
вавшийся в капилляре, причем условно принято, что потенциал-
определяющие ионы заряжены отрицательно, а противоионы — по-
ложительно. При наложе-
нии на капилляр электриче-
+ + +4-4- ++ ++4-4- + + 4-
+
_+_ +_+_+ _+_+_+. + _+_+ +_+_+ +_
777777Z7777Z777777777z77i
Рис. VII, 6. Схема движения жидкости
противоионов при электроосмосе
ского поля путем введения
в сосуды, которые он соеди-
няет, электродов слой про-
тивоионов в капилляре бу-
дет смещаться параллельно
и неподвижному слою потен-
циалопределяющих ионов к
катоду, что вызовет и пере-
мещение к катоду всей жидкости, заполняющей капилляр, под дей-
ствием сил трения и молекулярного сцепления. Освободившиеся
места противоионов немедленно занимают катионы, находящиеся
в объеме жидкости, заполняющей капилляр, а соответствующие
этим катионам ионы направляются к аноду (на рис. VII, 6 ионы,
находящиеся в объеме жидкости, заполняющей капилляр, не изо-
бражены) .
К явлениям электрофореза и электроосмоса мы еще вернемся
в конце этой главы, при рассмотрении количественной стороны
обоих явлений.
2. СТРОЕНИЕ ДВОЙНОГО ЭЛЕКТРИЧЕСКОГО СЛОЯ
Для упрощения изложения будем рассматривать, в основном,
плоский двойной электрический слой, хотя в коллоидных раство-
рах и тонкопористых телах такой слой практически и не встре-
чается. Подобное упрощение допустимо, когда толщина двойного
слоя мала по сравнению с радиусом кривизны поверхности кол-
лоидных частиц или капилляров.
Кроме того, при рассмотрении двойного электрического слоя
примем ряд следующих общих положений, из которых исходили
все авторы теорий его строения. Двойной электрический слой со-
стоит из ионов одного знака, относительно прочно связанных с
дисперсной в случае лиозолей твердой фазой (потенциалопреде-
ллющие ионы), и эквивалентного количества противоположно за-
174
6
Рис. VII, 7. Двой-
ной электрический
слой по Гельм-
гольцу — Перрену
и соответствую-
щий скачок потен-
циала.
ряженных ионов, находящихся в жидкой дисперсионной среде
вблизи межфазной поверхности (противоионы). Заряд на поверх-
ности твердой фазы в первом приближении рассматривается как
поверхностный заряд, равномерно распределенный по всей поверх-
ности. Между противоионами и свободными (не входящими в
двойной электрический слой) ионами того же знака, находящи-
мися в жидкости, существует динамическое равновесие. Диспер-
сионная среда представляется всегда как непрерывная фаза, влия-
ние которой на двойной электрический слой оп-
ределяется лишь ее диэлектрической проницае-
мостью.
При таких предпосылках отличие между тео-
риями строения двойного электрического слоя
заключается, в основном, только в различном
толковании структуры слоя противоионов.
Теория Гельмгольца — Перрена. Согласно
этой теории двойной слой представляется как бы
плоским конденсатором, одна обкладка которого
связана непосредственно с поверхностью твер-
дого тела (стенкой), а другая обкладка, несу-
щая противоположный заряд, находится в жид-
кости на очень малом расстоянии от первой. По-
тенциалов таком двойном слое, равно как и
потенциал в плоском конденсаторе, очевидно,
должен падать весьма круто (по прямой), а зна-
чение поверхностного заряда а будет опреде-
ляться известной из физики формулой:
е
° ~ 4лб ф0
Где е — абсолютная диэлектрическая проницаемость среды,
заполняющей пространство между обкладками конденса-
тора, фо — разность потенциалов между дисперсной фазой
и раствором; б — расстояние между обкладками
(VII, 1)
На рис. VII, 7 дана схема строения такого двойного электри-
ческого слоя. На этом рисунке заштрихованная часть представ-
ляет твердую фазу, а незаштрихованная — раствор; положитель-
ные и отрицательные ионы, образующие двойной слой, обозначены
соответственно через и —; свободные ионы электролита, всегда
присутствующие в жидкости, чтобы не усложнять схему, не пока-
заны. Рис. VII, 7 иллюстрирует также падение потенциала с уве-
личением расстояния х от поверхности твердого тела, причем об-
щий скачок потенциала в таком двойном слое является в то же
время и скачком потенциала между твердой фазой и раствором.
Приведенная схема строения двойного электрического слоя не
объясняет ряд особенностей электрокинетических явлений. В на-
стоящее время она представляет для коллоидной химии только
исторический интерес. Основным недостатком этой схемы является
175
то обстоятельство, что толщина двойного слоя Гельмгольца — Пер-
рена очень мала и приближается к молекулярным размерам. В то
же время в результате гидродинамических исследований было
установлено, что место разрыва (плоскость или граница скольже-
ния) при перемещении твердой и жидкой фаз относительно друг
друга всегда находится в жидкой фазе на сравнительно большом
расстоянии От межфазной границы. Толщина слоя жидкости, «при-
липающего» в этих условиях к твердой поверхности, во всяком
случае больше толщины двойного электрического слоя Гельм-
гольца — Перрена.
Если бы теория Гельмгольца — Перрена была правильной, то
при оседании коллоидных частиц в жидкости или при продавли-
вании жидкости через капилляр вообще не должен был бы наблю-
даться эффект Дорна или потенциал протекания, а явления элек-
трофореза и электроосмоса были бы невозможны. Однако если
даже допустить, как это принималось ранее, что поверхность сколь-
жения проходит между двумя обкладками двойного электрического
слоя, то и в этом случае представления Гельмгольца — Перрена
приводят к противоречию. В самом деле, при таком допущении
электрокинетический потенциал, т. е. потенциал, обнаруживаемый
при электрофорезе или электроосмосе, должен был бы соответство-
вать разности между всеми потенциалопределяющими ионами и
всеми противоионами, т. е. равняться общему скачку потенциала.
Однако опыты показали, что электрокинетический потенциал не
только, как правило, меньше общего скачка потенциала, но изме-
няется под влиянием различных факторов совсем иначе. Например,
общий скачок потенциала не зависит сколько-нибудь существен-
ным образом от индифферентных' электролитов, ие содержащих
ионов, способных достраивать, кристаллическую решетку, в то же
время такие электролиты сильно влияют на электрокинетический
потенциал.
Все изложенное показывает, что общий скачок потенциала и
электрокинетический потенциал являются различными величинами
и, следовательно, представления Гельмгольца и Перрена о пло-
ском электрическом слое недостаточны для объяснения электроки-
нетических явлений.
Теория Гуи — Чэпмена. Значительным шагом вперед явилась
теория двойного электрического слоя с диффузным слоем проти-
воионов, предложенная независимо друг от друга Гуи (1910 г.) и
Чэпменом (1913 г.). Эта теория в значительной мере устранила
недостатки теории Гельмгольца — Перрена. По теории Гуи — Чэп-
мена противоионы не могут быть сосредоточены только у межфаз-
ной поверхности и образовывать моноионный слой, а рассеяны
в жидкой фазе на некотором расстоянии от границы раздела. Та-
кая структура двойного слоя определяется, с одной стороны, элек-
трическим полем у твердой фазы, стремящимся притянуть эквива-
лентное количество противоположно заряженных ионов возможно
ближе к стенке, а с другой стороны, тепл<-ь »м движением ионов,
176
вследствие которого противоионы стремятся рассеяться во всем
объеме жидкой фазы.
В непосредственной близости от межфазной границы преобла-
дает действие электрического поля. С удалением от межфазной
границы сила этого поля постепенно ослабевает и проявляется все
сильнее рассеивание противоионов двойного слоя в результате
теплового движения, вследствие чего концентрация противоионов
падает и становится равной концентрации тех же ионов, находя-
щихся в глубине жидкой фазы. Таким образом возникает равно-
весный диффузный слой противоионов, связанных с твердой фазой.
Понятно, что равновесие этого диффузного слоя динамическое.
Рис. VII, 8. Зависимость концентра-
ции С противоионов (кривая /) и
потенциалопределяющих ионов (кри-
вая 2) от расстояния х от плоской
стенки (1/х — толщина двойного элек-
трического слоя).
Рис. VII, 9. Двойной электри-
ческий слой по Гуи — Чэпмеиу
и падение в нем потенциала.
С другой стороны, находящиеся в жидкости ионы того же
знака, что и адсорбированные стенкой потенциалопределяющие
ионы, отталкиваются электрическими силами от твердой фазы и
уходят в глубь раствора. Это обусловливает распределение потен-
циалопределяющих иоиов и противоионов в диффузной части
двойного электрического слоя, что иллюстрирует рис. VII, 8.
Строение двойного электрического слоя по Гуи — Чэпмену и
падение потенциала в этом слое схематически изображены на
рис. VII, 9.-Потенциал на этой схеме падает не по прямой, а по
кривой в связи с тем, что компенсирующие заряд стенки противо-
ионы распределены неравномерно. Падение кривой круче в тех
местах, где больше компенсирующих противоионов, и, наоборот,
кривая более полога там, где компенсирующих противоионов мало.
Поскольку диффузность слоя противоионов определяется теп-
ловым разбрасыванием, то при температуре абсолютного нуля все
противоионы должны были бы находиться у твердой поверхности.
177
Следовательно, теоретические представления Гельмгольца — Пер-
рена оказываются частным случаем теории Гуи — Чэпмена.
Равновесное распределение ионов, которое устанавливается
у твердой стенки, аналогично равновесному распределению моле-
кул газа в атмосфере под влиянием силы тяжести с тем лишь раз-
личием, что гравитационное поле не зависит от распределения мо-
лекул, а электрическое поле в случае двойного электрического слоя
само является функцией распределения заряженных ионов. Число
противоионов, находящихся у заряженной поверхности твердой
фазы, по мере увеличения расстояния от границы раздела по на-
правлению внутрь раствора, уменьшается по закону распределения
Больцмана, а число потенциалопределяющих ионов увеличивается
согласно тому же закону. Отсюда следует, что если концентрацию
положительных и отрицательных ионов в точке, потенциал которой
равен фж, соответственно обозначить через с+ и с_ (в молях на
единицу объема), то для расстояния х — оо
с+=с_ = с0о (VII, 2)
а для не слишком большого расстояния х
с+=Сооехр(—^гфх//??) (VII, 3)
с- = Coo exp (Гz^xIRT) ( VII, 4)
где с» — концентрация электролита на бесконечно большом расстоянии от твер-
дой фазы при фоо = 0; F—число Фарадея (96 540 Кл); г— валентность иона;
R — газовая постоянная; Т — абсолютная температура.
Согласно уравнению распределения Больцмана произведение
Г2фх в экспоненциальном множителе уравнений (VII, 3) и (VII, 4)
представляет собой.электрическую работу переноса одного моля
соответствующего вида ионов из объема раствора (где ф = 0) до
точки с потенциалом фж.
Ясно, что при х = оо фж = фоо = 0, а при х = 0 фж = фо, т. е.
на бесконечно большом расстоянии от границы раздела потенциал
равен нулю, а на границе раздела — общему скачку потенциала
поверхности.
Представления, развитые Гуи и Чэпменом, позволяют объяс-
нить некоторые электрокинетические явления. Так, поскольку пло-
скость скольжения АВ при перемещении твердой и жидкой фаз
относительно друг друга лежит в жидкости на некотором малом
расстоянии Д от межфазной границы, где потенциал еще не сни-
жается до потенциала жидкой фазы (см. рис. VII, 9), то разность
между ним и потенциалом внутри жидкой фазы в этом месте со-
ответствует заряду этой части диффузного слоя. Этот потенциал
и будет определять перемещение фаз при наложении электриче-
ского поля, т. е. обусловливать явления электрофореза или элек-
троосмоса. Ясно, что электрокинетический потенциал, как его часто
называют ^-потенциал, является частью общего скачка потен-
циала ф0. Таким образом, становится понятным, почему электро-
178
кинетический потенциал отличен от нуля, но не равен общему
скачку потенциала. Более того, схема строения двойного электри-
ческого слоя, предложенная Гуи и Чэпменом, позволяет понять,
почему различные факторы влияют на оба потенциала по-раз-
ному.
Рассмотрим, например, как влияет на оба эти потенциала вве-
дение в систему индифферентного электролита. В этом случае
общий скачок потенциала почти не изменяется. Совсем иначе бу-
дет обстоять дело с электрокинетическим потенциалом. С повы-
шением концентрации вводимого электролдда вследствие того, что
для компенсации потенциалопределяющих ионов требуется всегда
одно и то же (эквивалентное)
число зарядов противоположного
знака, толщина диффузного слоя
уменьшается. Как принято го-
ворить, двойной электрический
слой сжимается. В результате
этого меняется и распределение
в нем потенциала. Будет менять-
ся и ^-потенциал, отвечающий
плоскости скольжения жидкости
при электрофорезе или электро-
осмосе.
Рис. VII, 10. Влияние индифферент-
ного электролита на толщину двой-
ного электрического слоя и электро-
кннетический потенциал (количество
электролита увеличивается от кри-
вой 1 к кривой 4).
Наглядно изменение £-потен-
циала при введении в систему
все увеличивающихся количеств
индифферентного электролита
показано на рис. VII, 10. При до-
статочно больших концентрациях
электролита диффузный слой может сжаться до моноионного слоя,
и двойной электрический слой, таким образом, превратится в слой
Гельмгольца — Перрена. Понятно, что поскольку этот слой будет
находиться ближе к стенке, чем плоскость скольжения, g-потен-
циал будет равен нулю.
Следует заметить, что в основе теории Гуи — Чэпмена лежат
те же физические представления, что и в основе известной теории
сильных электролитов Дебая—Гюккеля, причем, кстати заметим,
первая возникла раньше второй.
Как известно, толщина ионной атмосферы 1/х по теории силь-
ных электролитов определяется следующим уравнением:
2
z
efer
8ле2АА ci2i
«.RT
£ eft
еЯТ
8nF2 £ erf
(VII, 5)
где е — абсолютная диэлектрическая проницаемость жидкости; k — постоянная
Больцмана; ~Т — абсолютная температура; е — заряд электрона; Na — число Аво-
гадро; Ci — концентрация ионов различной природы; Zt — валентность ионов;
R — газовая постоянная; F—число Фарадея,
179
Уравнение VII, 5, связывающее 1/х, с, и z„ можно представить
в упрощенном виде:
— = fe—
* VT7?
где k — константа уравнения; — ионная сила раствора.
Из приведенного уравнения вытекает, что ионная атмосфера
должна быть тем меньше, чем выше концентрация ионов в рас-
творе и чем больше их валентность. Естественно, что аналогичное
соотношение соблюдается и тогда, когда ионная атмосфера обра-
зуется не вокруг иона, а вблизи границы раздела фаз. Однако
только при малых потенциалах поверхности значение ^-потенциала
определяется одной толщиной ионной атмосферы и только в этих
условиях на нее могут одинаково влиять как заряды противоионов,
так и заряды побочных ионов, присутствующих в системе. При вы-
соких потенциалах поверхности (больше 50 мВ), как показывает
теория, на снижение ^-потенциала во много раз сильнее влияет
заряд противоиона, в особенности когда он велик. Физический
смысл этого явления заключается в том, что сильно заряженный
противоион электролита притягивается к поверхности и сильно ее
экранирует. Возрастание способности противоиона снижать ^-по-
тенциал с ростом его валентности наглядно видно нз следующих
опытных данных:
Валентность противоиона IV III II I
СС................ 1 10-20 60-100 500-1000
. Здесь значения eg представляют собой относительную концен-
трацию противоиона, необходимую для снижения электрокинети-
ческого потенциала до одного .и того же значения, например
50 мВ. Как видно, этот ряд не дает каких-нибудь точных отноше-
ний. Этого, впрочем, и нельзя было ожидать, так как способность
иона,сжимать двойной электрический слой зависит не только от
его валентности, определяющей электрическое взаимодействие со
стенкой, но и от его размера, поляризуемости, способности гидра-
тироваться и т. д. Мюллер, исходя из представлений Гуи и Чэп-
мена, чисто математическим путем показать что способность про-
тивоионов понижать ^-потенциал действительно должна быстро
расти с их валентностью, причем для случая плоской поверхности
раздела его вычисления дали такой ряд:
Валентность противоина IV III II I
сс............... 1 7,5 57 540
Это достаточно хорошо согласуется с экспериментальными дан-
ными.
Рассмотрим теперь количественную сторону теории Гуи — Чэпмена.
Одним из основных исходных уравнений в этой теории является уравнение
Пуассона, предусматривающее связь межу объемной плотностью заряда р и по-
180
тенциалом <р Это уравнение имеет вид:
V<p= —4лр/е (VII, 6)
где е — абсолютная диэлектрическая проницаемость; V — сокращенное обозначе-
ние оператора Лапласа:
V<p = (<Э 2ф/«Э х2) + (^2ф/<Э i/2) + (д2ф/<Эгг) (VII, 7)
При плоской поверхности твердой фазы потенциал изменяется только в на-
правлении по нормали к поверхности, и производные по у и г, таким образом,
равны пулю В этом частном случае зависимость потенциала от расстояния до
стенки х выражается следующим простым уравнением:
d2qldx2 = — 4лр/е (VII, 8)
Очевидно, при х = оо
Ф = 0; d<p/dx = 0 и р = 0
а при х = 0
Ф = Фо и dyfdx = — 4ла/в
где фо — потенциал межфазной поверхности; а — заряд (поверхностная плот-
ность электричества) той же поверхности
Объемную плотность заряда можно представить так:
p = Fz(c+-c_) (VII, 9)
где F — число Фарадея; г — валентность ионов; с+ и с- — концентрация поло-
жительных и отрицательных ионов, моль/л
Формула (VII, 9) пригодна, очевидно, только для бинарного симметричного
электролита. В других случаях формула (VII, 9) должна иметь вид алгебраиче-
ской суммы:
p=2>zzcz (VII, 10)
Однако этот более сложный случай мы здесь рассматривать не будем
Подставив в уравнение (VII,9) уравнения (VII,3) и (VII,4), имеем-
р = FzcM [exp (— Fzq/RT) — exp (Fzrp/RT)] (VII, 11)
Если найденное значение p подставить в уравнение (VII,8), можно получить:
"dx? = ~ Т Fzc°° [ехр (“ ~RT~) “ еХр (~/?Г)] (VII> 12)
Для того чтобы провести первое интегрирование и получить производную
dtf/dx, преобразуем полученное уравнение следующим образом:
[ир (-Z3L) _ „„ (^.)]
h (- » - “р (»] % *
М£)—h (- » - (»] “>
181
Проинтегрируем
<ftp/dx->0, имеем
полученное выражение от 0 до <р. Учитывая, что при <р -> О
ч>
о
<р
О
Ш [“> (т?)+>М=
«ЛИ _____
d(p / SnRTc^ Г /Лгф\ f Fzy Y1
dt = ± V-~ Lexp ( 2RT ) - eXP (- w)j
Так как при увеличении х величина ф уменьшается, то производная d<p]dx
имеёт отрицатёльйбе значение, и окончательно имеем:
Это дифференциальное уравнение позволяет сделать ряд интересных выво-
дов. Если электролит, концентрация’ которого в растворе равна с», является ин-
дифферентным, т. е. не содержит потеициалопределяющие ионы, то и потен-
циал фо, равный общему скачку потенциала, не зависит от. с„. Следовательно,
при изменении с», согласно формуле (VII, 13), может изменяться только наклон
кривой ф = f(x), а значение ф0 должно оставаться постоянным. На рис. VII, 10
лриведеио несколько кривых <p = f(x), отвечающих одному и тому же значе-
нию фо, ио при разных с«,, для которых dq/dx, в соответствии с уравнением
(VII, 13), возрастает от кривой 1 к кривым 2, 3, 4. Так как плоскость скольже-
ния на рис. VII, 10 соответствует линии АВ, то понятно, что начальный электро-
кинетический потенциал Ji с возрастанием концентрации электролита будет
уменьшаться, принимая значения и т. д. При валентности противоиона,
большей единицы, производная d<p/dx, учитывая уравнение (VII, 13), с возрас-
танием концентрации должна увеличиваться еще сильнее, т е. электрокииетиче-
-скин потенциал будет падать еще более резко
Таким образом, теория Гуи — Чэпмена хорошо объясняет падение ^-потен-
циала при увеличении концентрации противоиона и возрастании его валентности.
При малых Фо и.соответственно при значениях Ехф0/(2/?Г) 1 можно вос-
пользоваться приближением е“= 1 +а и е~а = 1 —а, что дает еа — е~а~2а.
Тогда вместо уравнения (VII, 13) получим приближенное уравнение:
dtp___
dx
V8nRTcx Fz<p __________ / 8nF2z2coo
в RT ~~ '\f eRT
ф = — Хф
где
8nF2z2Coo
eRT
8ne2z2N Ac,
IkT
(VII, 14)
Интегрируя уравнение (VII, 14) и учитывая, что при х = 0 Ф = <ц>, полу-
чаем:
<р = фое-их (VII, 15)
182
Уравнение (VII, 15) показывает, что потенциал уменьшается в е — 2,72 раз
на расстоянии порядка 1/х*.
Уравнение (VII, 13) позволяет установить соотношение между плотностью
поверхностного заряда а на межфазной границе и потенциалом поверхности <ро-
Из условия электронейтральности двойного электрического слоя следует, что
поверхностный заряд по абсолютному значению равен общему объемному за-
ряду в растворе, т. е.
СО
(T = -^pdx (VII, 16)
о
С помощью уравнения Пуассона (VII, 8) это уравнение можно проинтегри-
с др. ду ЮТТТ и м обпНЗОМ1
pODSTb
е -JL
4л dx2 4л \ dx /х=о
(VII, 17)
Заменяя dq/dx найденным для этой производной значением из уравнения
(VII, 13), получаем:
Пользуясь упрощением, аналогичным тому, которое было применено при вы-
воде формулы (VII, 14), из уравнения (VII, 18) в случае малых значений фо и
Fz^a/^RT можно получить следующее приближенное выражение:”
VzRTcx Fz<f0 _ в / 8nF2z2coo ex .q,
-----RT~ 15Г Л/ —RT~ ’фо ф° ( VI1’ '9>
где __________ __________________
V8nF2z2cco / 8ле2х2Удсоо
eRT = V IkT
В этом случае заряд и потенциал на границе раздела пропорциональны друг
другу и двойной электрический слой ведет себя как плоский конденсатор, рас-
стояние между пластинами которого равно 1/х « б. То, что расстояние между
обкладками конденсатора, эквивалентное двойному слою, равно примерно 1/х,
оправдывает обычный способ выражения: «толщина двойного электрического
слоя равна 1/х».
Важным свойством двойного электрического слоя, которое легко охаракте-
ризовать количественно, является его дифференциальная и интегральная емкость:
<Эсг/дф0 и сг/фо (VII, 20)
Для небольших потенциалов дифференциальная и интегральная емкости равны
одной и той же величине С, которую можно вычислить по уравнению:
С = о/ф0 = ex/(4rt) (VII, 21)
Для более высоких потенциалов емкость двойного электрического слоя стано-
вится больше этой величины.
* Здесь следует заметить, что величину 1/х, получившую в теории сильных
электролитов название радиуса ионной атмосферы (или дебаевского радиуса)»
нельзя понимать геометрически, как радиус сферы, за которой действие поля
рассматриваемого иона становится равным нулю. Физическая величина радиуса
ионной атмосферы, строго говоря, неограничена. Одиако потенциал поля убывает
с удалением от рассматриваемого иона экспоненциально и иа расстоянии, много
большем 1/х, влиянием этого потенциала можно пренебречь.
183
При комнатной температуре и г = 1 значение х равно приблизительно
3-107 Vc" (где с — концентрация электролита, моль/л) Отсюда в 1 н водном
растворе электролита с одновалентными катионом и анионом вычисленная ем-
кость двойного электрического слои равна 3-107-80/4 = 200 мкФ/см2 Емкости,
определенные экспериментально, равны всего одной десятой этого значения
Несостоятельность теории Гуи—Чэпмена может быть показана и другим
путем Если раствор электролита не очень разбавлен (например, 0,1 н) и по-
тенциал у стенки высок (например, 200 мВ), то для того, чтобы теоретическое
и экспериментально найденное значения емкости совпадали, концентрация про-
тивоиона около стенки должна быть равна 300 н , что совершенно невозможно
Причина расхождения экспериментально и теоретически найденных значений
емкости двойного электрического слоя заключается в том, что теория Гун —
Чэпмена не принимает во внимание размера ионов, рассматривая их как то-
чечные заряды, которые могут сколь угодно близко подойти к стенке, что и
обусловливает более высокие значения расчетных величин
Другой недостаток теории Гуи — Чэпмена заключается в том,
что она не объясняет так называемого явления перезарядки — пе-
ремены знака электрокинетического потенциала при введении
в систему электролита с многовалентным ионом, заряд которого
противоположен по знаку заряду дисперсной фазы.
Далее теория Гуи—Чэпмена не объясняет различного дей-
ствия разных по природе противоионов одной и той же валентно-
сти на двойной электрический слой. Согласно этой теории введе-
ние эквивалентного количества разных противоионов одинаковой
валентности должно сжимать двойной электрический слой и по-
нижать ^-потенциал в одинаковой степени. Однако опыт показы-
вает, что это не так. Эффективность действия ионов одной и той
же валентности на двойной электрический слой возрастает с уве-
личением радиуса иона.
Наконец, теория Гуи — Чэпмена, относительно хорошо прило-
жимая в случае достаточно разбавленных коллоидных растворов,
оказывается неприемлемой для более концентрированных.
Все эти затруднения в значительной мере преодолены в тео-
рии строения двойного электрического слоя, предложенной Штер-
ном
Теория Штерна. В 1924 г. Штерн предложил схему строения
двойного электрического слоя, в которой он объединил схемы
Гельмгольца — Перрена и Гуи — Чэпмена. Разрабатывая теорию
двойного электрического слоя, Штерн исходил из двух предпосы-
лок. Во-первых, он принял, что ионы имеют конечные,, вполне оп-
ределенные размеры и, следовательно, центры ионов не могут на-
ходиться к поверхности твердой фазы ближе, чем на расстоянии
ионного радиуса. Во-вторых, Штерн учел специфическое, не элек-
трическое взаимодействие ионов с поверхностью твердой фазы.
Это взаимодействие обусловлено наличием на некотором малом
расстоянии от поверхности поля молекулярных (адсорбционных)
сил. Как будет показано при обсуждении причин устойчивости и
коагуляции коллоидных систем, молекулярные силы, действующие
между телами, состоящими из множества молекул, вследствие
своей аддитивности являются относительно дальнодействующими.
184
Рис. УП, 11. Двойной электри-
ческий слой по Штерну и па-
дение в нем потенциала.
Однако в отличие от электрических сил их действие быстро умень-
шается с расстоянием, и поэтому их действие в данном случае
следует учитывать только у самой поверхности твердого тела на
расстоянии порядка нескольких ангстрем.
Согласно Штерну, первый слой или даже несколько первых
слоев противоионов притягиваются к стенке под влиянием как
электростатических, так и адсорбционных сил. В результате этого
часть противоионов удерживается поверхностью на очень близком
расстоянии, порядка 1—2 молекул, образуя плоский конденсатор
толщиной 6, предусмотренный теори
Этот слой, в котором, естественно, на-
блюдается резкое падение электриче-
ского потенциала, одни авторы на-
зывают гельмгольцевским, другие —
штерновским, третьи — адсорбционным
слоем. Остальные противоионы, нуж-
ные для компенсации потенциалопре-
деляющих ионов, в результате тепло-
вого разбрасывания образуют диффуз-
ную часть двойного слоя, в которой
они распределены согласно тем же
законам, что и в диффузном слое
Гуи — Чэпмена. Эту часть двойного
слоя, в которой потенциал падает от-
носительно постепенно, иногда назы-
вают слоем Гуи. Схема двойного элек-
трического слоя по Штерну и падение
в нем электрического потенциала по-
казаны на рис. VII, 11.
Из схемы можно видеть, что полное падение потенциала <р0
слагается из падения потенциала <р6 в диффузной части двойного
слоя и разности потенциалов (<р0 — <р6) между обкладками кон-
денсатора. Место границы скольжения в таком слое остается до
сих пор неясным. Некоторые авторы принимают, что она совпа-
дает с границей между слоем Гельмгольца и слоем Гуи. Однако
в общем случае ее можно представить себе находящейся в слое
Гуи, как это изображено на рис. VII, 11 (граница скольжения обо-
значена пунктирной линией АВ). Таким образом, потенциал на
границе слоя Гельмгольца и слоя Гуи не обязательно должен быть
равен ^-потенциалу.
Понятно, что с введением электролитов в систему диффузный
слой будет сжиматься и все большее и большее число противо-
ионов будет попадать в адсорбционный слой. Двойной электри-
ческий слой, согласно-взглядам Штерна, при этом все больше при-
ближается к слою, предусмотренному в теории Гельмгольца —
Перрена, а ^-потенциал уменьшается, постепенно *.приближаясь
к нулю При разбавлении системы, наоборот, диффузный слой рас-
ширяется и ^-потенциал возрастает.
185
На распределение ионов в двойном электрическом слое по тео-
рии Штерна сильно влияет природа противоионов. Если противо-
ионы обладают различной валентностью, то толщина диффузного
слоя и число противоионов в адсорбционном слое определяются,
главным образом, валентностью ионов и, следовательно, обуслов-
ливаются электростатическими силами. Понятно, диффузный слой
тем тоньше и ^-потенциал тем ниже, чем больше валентность Про-
тивоионов. При этом надо принимать во внимание те же сообра-
жения, что и при объяснении влияния валентности противоиона на
^-потенциал по теории Гуи — Чэпмена.
Если противоионы имеют одинаковую валентность, толщина
двойного электрического слоя и число противоионов в диффузном
слое определяются специфической адсорбционной способностью
ионов, обусловленной, как показано в гл. VI, их поляризуемостью
и гидратацией. Эти свойства ионов определяются их истинным ра-
диусом или, что то же, положением соответствующих элементов
в таблице Д. И. Менделеева.
Большая поляризуемость иона, конечно, должна способствовать
уменьшению толщины двойного электрического слоя, поскольку
при этом возникают дополнительные адсорбционные силы между
твердой фазой и индуцированным диполем и, кроме того, ион
может ближе подойти к поверхности. Так как деформируемость
иона увеличивается с его размерами и поскольку радиусы анионов
вообще значительно больше радиусов катионов, поляризуемость
анионов обычно больше, чем катионов:
Катион а1024, см3 Анион alo24, cm'
Li* 0,03 F- 0,96
Na+ 0,19 ci- 3,60
К+ ’0,89 Br- 5,00
Rb+ 1,50 Г 7,60
Cs+ 2,60
В этом, между прочим, кроется объяснение того, почему отри-
цательный заряд поверхности встречается в природе гораздо чаще,
чем положительный.
Гидратация ионов, как известно, уменьшается с увеличением
истинного радиуса ионов. Снижение гидратации ионов должно спо-
собствовать сжатию двойного электрического слоя, так как гидрат-
ная оболочка уменьшает электростатическое взаимодействие ме-
жду противоионами и поверхностью твердой фазы.
Из всего приведенного становится понятным, почему способ-
ность сжимать двойной электрический слой и уменьшать ^-потен-
циал возрастает в ряду катионов от Li+ к Cs+, а в ряду анионов
от F" к Г. На рис. VII, 12 приведена найденная Енни и Рейтемейе-
ром зависимость ^-потенциала отрицательно заряженных частиц
водной суспензии глины, насыщенной различными одно- и двух-
валентными противоионами, от размера ионов. Как можно видеть,
1S6
характер зависимости ^-потенциала от радиуса двухвалентных ка-
тионов такой же, что и для одновалентных катионов.
Рассмотрим теперь представления Штерна, с количественной стороны.
По абсолютной величине заряд а на твердой поверхности, согласно теории
Штерна, равен сумме заряда ионов, находящихся в адсорбционном слое си, и
заряда диффузной части двойного слоя <т2:
a = ai+a2 (VII, 22}
Для вычисления си Штерн использовал метод, аналогичный тому, который
применяется для вывода изотермы адсорбции Ленгмюра. Для симметричного
электролита (у которого оба иона имеют одни и тот же заряд) Штерн получил
следующие уравнения, характеризующие
число адсорбированных единицей поверхно-
сти молей ионов различного знака:
+ “ макс 1 + 1/Сооехрце+ + Г2фб)//?г]
(VII, 23)
г =г _____________________I_______________
иакс j + 1/Со. ехр ц0_ _ Г2фб)//?Г]
(VII, 24)
где 0+ и 0- — специфические адсорбцион-
ные потенциалы для положительных и от-
рицательных ионов.
В уравнениях (VII, 23) и (VII, 24) чис-
литель экспоненты представляет общий ад-
сорбционный потенциал ионов, т. е. работу
Рис. VII, 12. Зависимость g-по-
тенциала отрицательно заряжен-
ных частиц глины от радиуса
противоионов г.
переноса одного моля ионов из объема раствора на поверхность адсорбента.
Эта сложная величина, представляющая алгебраическую сумму специфического
адсорбционного потенциала 0+ или 0- и электрического адсорбционного потен-
циала fztpj. Для положительных иоиов общий адсорбционный потенциал, оче-
видно, будет 0++ fzq>6, для отрицательных он равен 0- — fzcpj.
Заряд адсорбционного слоя си представляет собой разность отрицательных
и положительных зарядов, приходящихся на единицу адсорбционной поверхно-
сти. Тогда
<r,_Fz(r+ Г—) ДзГиакс{ ( + 1/Ссо ехр [(0+ + Fz4>syRT]
1 ---Б---(VII, 25)
-Л2ф6)//?Г] J
1 + 1/Со» ехр [(0_
На практике очень часто достаточно принимать в расчет только одно из слагае-
мых в уравнении (VII, 25), так как или положительные, или отрицательные ионы
полностью вытесняются из молекулярного конденсатора. Другое упрощение сво-
дится к тому, что единицу в знаменателе обоих уравнений можно отбросить,
если концентрация электролита не слишком высока.
Заряд диффузного слоя <т2 непосредственно «связан с потенциалом в этом
слое фа и вычисляется согласно теории Гуи — Чэпмена по уравнению (VII, 18)
(т. е. в этом случае действие адсорбционных сил не учитывается):
« - Vt [“₽ - “Р(- ^)] (VIC25>
187
Подставляя уравнения (VII, 25) и (VII, 26) в уравнение (VII, 22), получим:1
° = /^Гмакс { , + 1/Соо ехр [(0+ + /7гфв)/2/?Г] ~
_________________1_____________] +
1 + 1/см ехр [(0_ — Fz<p6)/2RT] ] ‘
<™.2ч
Так как значение первого члена выражения в фигурных скобках в правой
части равенства меняется с концентрацией сильнее, чем значение второго, то,
очевидно, при разбавлении раствора Hi уменьшается быстрее, чем <т2, и струк-
тура слоя приближается к модели Гуи — Чэпмеиа, при увеличении же концен-
трации структура двойного электрического слоя приближается к модели Гельм-
гольца Таким образом, анализ уравнения (VII, 27) приводит к тем же выводам,
к которым мы пришли ранее иа основании общих положений.
Первый член суммы в правой части уравнения от (см. VII, 27) представляет
интерес еще и потому, что даже тогда, когда 0+ и 0- равны нулю, т. е. при
отсутствии специфической адсорбции, величина от все же не равна нулю Это
значит, что противоионы, даже если они не адсорбируются специфически, могут
втягиваться в адсорбционный слой под действием электростатических сил.
Емкость полного двойного слоя С определяется как емкость двух последо-
вательно расположенных конденсаторов по известной формуле:
г М1СЛ (VII, 28)
Ьмт Сд
где Си—емкость молекулярного конденсатора; Сд— емкость диффузного еЛоя.
Емкость молекулярного слоя См равна отношению заряда стенки а к разности
потенциалов <p0 — ф^:
си = ^/('₽о-'₽в) (VII, 29)
Штерн принимал емкость молекулярного конденсатора постоянной. Однако
эта емкость, безусловно, должна зависеть от специфических свойств иоиов, на-
ходящихся в адсорбционном слое. Так как величина См почти постоянна, а ве-
личина Сд сильно зависит от концентрации электролита, характер падения по-
тенциала в двойном электрическом слое также зависит от количества находяще-
гося в системе электролита. Таким образом, если даже потенциал <р0 является
постоянным, то фй, значение которого определяется состоянием диффузного слоя,
снижается с уменьшением 1/х.
Если емкость одного из двух конденсаторов значительно больше емкости
другого, то полная емкость практически равна меньшей составляющей. Так,
в разбавленных растворах С ~ Сд, а в концентрированных С « См.
Значения емкости двойного электрического слоя, вычисленные
по теории Штерна с учетом радиусов ионов, оказались близкими
к экспериментально найденным, и, таким образом, эта теория пре-
одолела один из недостатков, присущий теории Гуи — Чэпмена.
Далее, в отличие от теории Гуи — Чэпмена, теория Штерна может
Объяснить причину изменения знака электрокинетического потен-
циала при введении в систему многовалентных ионов, заряд кото-
рых противоположен по знаку заряду дисперсной фазы. Такие мно:
говалентные ионы втягиваются в адсорбционный слой как из-за
сильных электростатических взаимодействий, так и из-за большой
адсорбируемости, связанной с поляризуемостью таких ионов. Ионы
188
-V
Рис. VII, 13. Изменение падения
потенциала двойного электриче-
ского слоя при перезарядке с по-
мощью сильно адсорбирующихся
ионов:
/—падение потенциала до перезарядки;
2—падение потенциала после переза-
рядки.
могут адсорбироваться^ таком количестве, что не только нейтра-
лизуют заряд твердойпбверхности, но и перезаряжают частицу.
В результате, как это можно видеть из схемы рис. VII, 13, характер
падения потенциала в двойном электрическом слое изменится ко-
ренным образом, а <р6- и ^-потенциалы, имевшие ранее тот же знак,
что и <р-потенциал (на схеме ф61 и
£1), меняют знак на обратный (на
схеме ф62 и g2)- При этом, конечно,
потенциал ф0 остается постоянным,
так как чужеродные ноны не спо-
собны достраивать кристаллическую
решетку твердой фазы. Таким об-
разом, создается положение, когда
Фо- и ^-потенциалы будут иметь раз-
личные знаки. ПоняРтнр, что даль-
нейшее повышение концентрации
электролита в'системе должшг-нри-
водить к сжатию-диффузного слоя
и падеййю ^-потенциала.
Для перезарядки частиц, имею-
щих отрицательный g-потенциал,
особенно часто применяют многова-
лентные ионы алюминия и тория.
Впрочем, перезарядку способны вы-
зывать и одновалентные ионы, если
они обладают * большим адсорб-
ционным потенциалом. Сюда отно-
сятся ионы многих алкалоидов, на-
пример стрихнина и хинина, и ос-
новных красителей (кристалличе-
ский фиолетовый, метиленовый го-
лубой и др.). Это объясняется тем,
что такие крупные ионы не только
способны поляризоваться, но и яв-
ляются постоянными диполями.
Из изложенного выше видно, что теория Штерна соответствует
результатам экспериментальных наблюдений лучше, чем теория
Гуи — Чэпмена. Благодаря уточнению роли размера ионов и вве-
дения представления об адсорбционном потенциале, она может
объяснить ряд специфических особенностей действия тех или иных
электролитов на двойной электрический слой и электрокинетиче-
ский потенциал. Однако необходимо указать, что и эта теория не
является совершенной, поскольку она исходит из ряда допущений
и в ней имеется много неопределенностей, например, допущение
о независимости адсорбционного потенциала от концентрации, что
едва ли вероятно. Следует также заметить, что представления
р плоскости скольжения в двойном электрическом слое весьма ус-
ловны. По существу, плоскости скольжения так, как ее представ-
189
ном электрическом слое, но и
о I
лял Штерн, очевидно, нет, а имеет место скольжение жидкости
от межфазной поверхности в глубь раствора, начинающееся на
определенном расстоянии от поверхности твердой фазы. Таким
образом, значение ^-потенциала, весьма важной характеристики
всякой коллоидной системы, определяется при электрокинетиче-
ских явлениях не только характером падения потенциала в двой-
характером движения жидкости
вблизи твердой пбверхности, за-
висящим от ее реологических
свойств.
В дальнейшем был проведен ряд ис-
следований с целью уточнить предполо-
женное Штерном строение двойного
электрического слоя. Из этих исследова-
ний наибольшее значение имеет работа
Грехема, согласно которой слой Гельм-
гольца состоит из внешней н внутрен-
ней частей. Во внешней части находятся
гидратированные противоионы, которые
удерживаются у межфазной поверхности
электростатическими силами. Во внут-
ренней части находятся хемосорбирован-
ные твердой поверхностью ионы, одно-
именно заряженные с твердой фазой и
с частично разрушенными гидратными
оболочками.
слоя у сферических частиц. Для сфери-
ческих частиц при сравнительно небольших значениях потенциала <р0 прибли-
женное выражение для падения потенциала с расстоянием было дано Дебаем
и Гюккелем:
<Р = (фог/а) ехр [— и (а — г)] (VII, 30)
1
Рис. VII, 14. Схема строения двой-
ного ионного слоя обычной н ди-
польной коллоидной частицы (по
И. Ф. Ефремову):
а—коллоидная частица, не являющаяся
диполем, б—дипольная коллоидная ча-
стица.
Строение двойного электрического
где г -— радиус частицы; а — расстояние от ее центра. Заряд частицы Q прн этом
зависит от потенциала <р0 следующим образом:
Q = re (1 + яг) <р0 (VII, 31)
Нахождение подобных зависимостей для больших значений потенциала <р0
является пока весьма сложной задачей в связи с возникающими при этом прин-
ципиальными н математическими трудностями. Однако имеются специальные
таблицы, которые составили Лбб, Овербек и Вирзема, позволяющие находить
связь между зарядом и потенциалом сферических частиц.
Строение двойного электрического слои у частиц с постоянным дипольным
моментом. Н. А. Толстой с сотр. показали, что существуют коллоидные частицы
с электрической дипольной структурой, образующиеся вследствие самопроизволь-
ной униполярной ориентации адсорбированных на их поверхности диполей дис-
персионной среды (например, Н2О, ОН- н т. д) или вследствие ориентации
полярных групп самого вещества частиц. Подобные частицы, как показали раз-
личные электрооптические методы исследования, обладают жестким большим
электрическим моментом (тысячи и миллионы дебаев). Так, перманентная ди-
польная структура обнаружена у пятиокнсн ванадия, у частиц суспензий глины,
гуминовых золей, суспензий ряда красителей н некоторых бактерий и вирусов.
Можно с достаточной уверенностью сказать, что подобные дипольные струк-
туры, привлекшие в последнее время особое внимание исследователей, широко
распространены в коллоидных и биологических системах.
Вследствие наличия постоянных диполей частицы могут взаимодействовать
друг с другом и образовывать в золе сложные структуры.
190
Электрическую структуру коллоидной частицы, имеющую постоянный ди-
поль, можно в первом приближении представить как взаимодействие двойного
электрического слоя Гуи — Чэпмена и дипольной структуры (рис. VII, 14).
Аналогичные взаимодействия .наблюдаются и у магнитных дисперсных ча-
стиц
3. ВЛИЯНИЕ РАЗЛИЧНЫХ ФАКТОРОВ
НА ЭЛЕКТРОКИНЕТИЧЕСКИЙ ПОТЕНЦИАЛ
Поскольку ^-потенциал в некоторых случаях является величи-
ной, характеризующей устойчивость коллоидной системы, весьма
важно рассмотреть влияние на него таких факторов, как введение
в систему электролитов, изменение pH, концентрации раствора,
температуры и т. д.
Влияние индифферентных электролитов. Рассмотрение влияния
различных факторов на ^-потенциал целесообразно начать с наи-
более простого и практически чрезвычайно важного случая — вве-
дения в систему индифферентных электролитов, т. е. электролитов,
не имеющих ионов, способных' достраивать кристаллическую
решетку коллоидной частицы. Следует указать, что именно введе-
нием индифферентных электролитов на практике чаще всего поль-
зуются для коагуляции коллоидных систем.
Как уже было показано, индифферентные электролиты не мо-
гут сколько-нибудь существенно изменить общий скачок потен-
циала коллоидных частиц, а электрокинетический потенциал такие
электролиты в общем случае снижают в результате увеличения
концентрации противоионов и сжатия двойного электрического
слоя.
При введении индифферентных электролитов следует различать
два случая: 1) в систему вводится электролит, один из ионов ко-
торого одинаков с противоионами, 2) в систему вводится электро-
лит, не имеющий общих ионов с электролитом — стабилизатором.
Первый случай рассмотрен при обсуждении теорий Гуи — Чэп-
мена и Штерна. Очевидно, по мере увеличения содержания в си-
стеме такого электролита толщина двойного электрического слоя
стремится стать равной толщине адсорбционного слоя за счет
сжатия диффузного слоя. В результате ^-потенциал понижается,
пока не станет равным нулю, что будет отвечать так называемому
изоэлектрическому состоянию систему.
Второй случай, когда в систему вводится электролит, не со-
держащий обоих ионов с электролитом — стабилизатором, отли-
чается от первого только тем, что здесь имеет место явление об-
мена противоионов коллоидной частицы на эквивалентное число
одинаковых по знаку ионов введенного электролита. Наиболее
простой обмен ионов происходит, когда на поверхности твердой
фазы имеется двойной электрической слой типа Гуи — Чэпмена,
т. е. когда можно пренебречь специфическим адсорбционным по-
тенциалом ионов. Очевидно, при этом обмен будет определяться
только валентностью ионов. Например, если отрицательно заря-
женная дисперсная фаза находится в растворе, содержащем два
191
вида одновалентных катионов, то отношение концентраций этих
ионов в любой точке двойного сдоя должно быть тем же, что и их
отношение в глубине раствора. Ионный обмен, очевидно, будет
подчиняться закономерности, аналогичной уравнению Б. П- Ни-
кольского (VI, 6):
g1/g2 = fe(c1/c2) (VII,32)
где gi и g2 — концентрация ионов 1 и 2 электролита в двойном слое; С] и с2 —
концентрация этих ионов в растворе.
Если валентность противоионов неодинакова, то равновесие при
обмене значительно смещено в сторону катиона с более высокой
валентностью, который в большем количестве накапливается в ме-
Рис. VII, 15. Влияние на ^-потенциал
межфазной поверхности стекла сле-
дующих электролитов:
1 — KC1; 2—Ca(NOi),; 3—АЦИОЛ; 4—кри-
сталлический фиолетовой; 5—Th(NO,)«.
стах с отрицательным потенциа-
лом. ‘
При наличии специфической
адсорбции ионов ионный обмен
сильно усложняется. При этом
ионЬ1 одной и той же валентности
по своей способности переходить
в двойной электрический слой
располагаются в те же ряды, ко-
торые уже были приведены при
обсуждении зависимости толщи-
ны двойного электрического слоя
от природы образующих его
ионов.
Предложено большое число уравне-
ний для количественного описания ион-
ного обмена, учитывающих специфиче-
скую адсорбцию ионов. Наиболее общим
из таких уравнений, приложимым толь-
ко к обмену иоиов одинаковой валентно-
сти, является следующее:
gi/g2 = f(ci/c2) (VII, 33)
В этом уравнении коэффициент f является величиной, обусловленной раз-
личием специфических адсорбционных потенциалов 0] и 02 обоих иоиов:
f^expj8^;02] (VII,34)
При очень большом адсорбционном потенциале ионы, заряд ко-
торых по знаку противоположен заряду дисперсной фазы, могут
вызвать перезарядку коллоидных частиц. Это явление было рас-
смотрено при изложении теории Штерна.
На рис. VII, 15 в качестве примера приведены кривые, характе-
ризующие изменение ^-потенциала стекла при введении в кол-
лоидный раствор различных электролитов * (данные заимство-
* На рис. VII, 15 по оси ординат вверх от нуля отложены отрицательны?
значения ^-потенциала, а вниз от нуля — его положительные значения. Тако*
прием графического изображения часто применяется в коллоидной химии в свя-
зи с тем, что в большинстве коллоидных систем частицы заряжены отрицательно^.
192
ваны из работы Ратджерса и Де Смета). Как можно видеть, элек-
тролиты с одно- и двухвалентными катионами (кривые 1, 2)
только понижают ^-потенциал отрицательно заряженной стеклян-
ной поверхности, в то время как электролиты с трех- и четырехва-
лентным катионом вызывают перезарядку (кривые 3, 4, 5).
Рассматривая влияние индифферентных электролитов, мы принимали, что на
электрокинетический потенциал оказывают влияние ионы, заряд которых проти-
воположен по знаку заряду коллоидной частицы и одинаков с зарядом противо-,
ионов Возникает вопрос, могут ли влиять на S-потенциал ионы вводимого ин-
дифферентного электролита, заряженные одноименно с коллоидной частицей
(так называемые сопутствующие или побочные ионы). На этот вопрос иссле-
дователя отвечают по-разному, но во всяком случае, если эти ионы и влияют
на электрокинетический потенциал, то незначительно. К этому вопросу мы воз-
вратимся в гл. IX.
Рис. VII, 17. Зависимость потен-
циала фр (кривая /) и ^-потенциала
(кривая 2) частиц золя Agl от ло-
гарифма концентрации иодид-иоиов
в растворе.
Рис. VII, 16. Влияние иеиндиффе-
рентиого электролита на ф0- и £-
потенциалы:
1—кривая падения потенциала в двойном
электрическом слое до введения электро-
лита; 2—то же после введения иеиидиффе-
реитиого электролита; 3—то же после вве-
дения значительного количества иепидиф-
фереитиого электролита.
Влияние неиндифферентных электролитов. Влияние электро-
лита, один из ионов которого способен достраивать кристалличе-
скую решетку дисперсной фазы, заключается в том, что потенциал-
определяющий ион этого электролита может повышать потенциал
wo, а находящийся с ним в паре ион, одноименный с зарядом про-
тивоиона, способен сжимать двойной электрический слой. При ма-
лых концентрациях неиндифферентного электролита проявляется,,
в основном, первая тенденция, связанная с поверхностным дей-
ствием иона, способного достраивать кристаллическую решетку.
При больших концентрациях, когда достройка кристаллической
решетки завершена, превалирует вторая тенденция. Поэтому при
введении в систему все возрастающих количеств неиндифферент-
ного электролита ^-потенциал сначала возрастает, а потом падает,
проходя через максимум. Это видно из схемы, представленной на
Рис. VII, 16.
7 Зак. 664 193>
На рис. VII, 17 приведены данные Трельстра и Кройта, харак-
теризующие изменение фо- и ^-потенциалов частиц золя иодида се-
ребра в зависимости от логарифма концентрации иодид-ионов
(lg Cj-) в растворе. Характер изменения обоих потенциалов с из-
Рис. VII, 18. Изменение ф0- и
^-потенциалов при перезарядке
с помощью неиндифферентного
электролита:
/—падение потенциалов до перезаряд-
ки; 2— падение потенциалов после пере-
зарядки.
менением концентрации потенциал-
определяющих ионов вполне согла-
суется со всем сказанным выше.
При введении неиндифферентно-
го электролита возможна и переза-
рядка коллоидных частиц. Сущ-
пость такой перезарядки поясним
на следующем примере. Рассмо-
трим, что будет происходить при
введении в золь иодида серебра,
стабилизованный нитратом серебра,
раствора иодида калия. До введения
электролита потенциалопределяю-
щим ионом в золе, очевидно, будет
ион Ag+, а противоионом — ион NOj.
Частицы такого золя заряжены по-
ложительно. После введения в си-
стему избытка иодида калия потен-
циалопределяющим ионом станет
ион I", а противоионом — ион К+-
Сами же частицы золя приобретут
отрицательный заряд. Кроме того,
нитрат серебра, содержащийся в
дисперсионной среде, вступит в ре-
акцию с введенным иодидом калия,
в результате чёго в системе обра-
зуется некоторое дополнительное
количество отрицательно заряженной дисперсной фазы. Этот слу-
чай перезарядки отличается от уже рассмотренного случая переза-
рядки с помощью чужеродных ионов тем, что здесь происходит из-
менение не только но и фо-потенциала. Схема, поясняющая пе-
резарядку дисперсной фазы с помощью неиндифферентного элек-
тролита, показана на рис. VII, 18.
Влияние pH среды. Значение pH дисперсионной среды может
сильно сказываться на ^-потенциале коллоидных частиц, так как
водородные и гидроксильные ионы обладают высокой способностью
адсорбироваться; первые — благодаря' малому радиусу, что по-
зволяет им близко подходить к поверхности твердой фазы; вто-
рые — из-за большого дипольного момента.
Особое значение приобретает влияние pH для лизолей, у котрых дисперс-
ная фаза является амфотерным соединением, например золи А1(ОН)з, Ре(ОН)з,
Sn(OH)4, Th(OH)4. У этих золей с изменением pH дисперсионной среды может
происходить перезарядка частиц вследствие изменения характера ионизации ве-
194
щества дисперсной фазы. Рассмотрим перезарядку этого типа на примере золя
гидроокиси алюминия.
В не слишком кислой среде ионизация молекул А1(ОН)3, находящихся иа
поверхности коллоидных частиц, идет следующим образом:
А1(ОН)3 А1(ОН)++ОН-
Гидроокись ведет себя здесь как основание, так как избыток водородных иоиов
подавляет ионизацию по кислотному типу. В результате потеициалопределяющим
ионом в этом случае является иои АЦОН)*, а противоионом — ОН’. Коллоид-
ная частица при этом будет заряжена положительно. В средах с более низким
pH образуются ионы А134:
А1(ОН)^" + ОН- ч > А13+ + ЗОН-
Сначала это приведет к большему увеличению общего положительного заряда
поверхности, а затем гидроокись алюминия растворится в кислоте и, следова-
тельно, коллоидный раствор перейдет в истинный.
В щелочной среде ионизация по основному типу невозможна из-за избытка
гидроксильных ионов. В щелочной среде гидроокись алюминия ведет себя как
кислота, и ионизация А1(ОН)3 идет по следующей схеме:
А1(ОН)3 А1(0Н)20Ч-Н+
В результате этого потеициалопределяющим ионом уже будет ион А1(ОН)2О~,
а противоионом — Н+. Коллоидная частица окажется заряженной отрицательно.
В средах с более высоким pH происходит образование иона AlOg по схеме:
А1(ОН)2О" + Н+ A1O2' + H+ + H2O
При еще большей концентрации гидроксильных ионов гидроокись алюминия
растворится в растворе щелочи с образованием алюмината и коллоидный рас-
твор также перейдет в истинный.
Естественно, что при изменении потенциала <р0 у коллоидных частиц будет
изменяться и С-потенциал. В кислой среде ^-потенциал, как и <р0-потенциал,
имеет положительный знак, в щелочной среде — отрицательный. Очевидно так-
же, что должно существовать такое значение pH, при котором ^-потенциал
равен нулю и система окажется в так называемом изоэлектрическом состоянии.
При этом состоянии число положительных и отрицательных зарядов на поверх-
ности одинаково.
Приведенный пример перезарядки дисперсной фазы отличается от ранее
рассмотренного случая перемены знака С-потенциала при введении неиндиффе-
рентного электролита только тем, что ион, определяющий потенциал после пере-
зарядки, не вводится в систему, а образуется из вещества самой дисперсной
фазы в результате изменения характера ионизации.
Влияние концентрации коллоидной системы. Исходя из самых
общих представлений, можно предполагать, что при разбавлении
всякой коллоидной системы ^-потенциал должен возрастать, так
как толщина двойного электрического слоя увеличивается в ре-
зультате уменьшения концентрации противоионов в растворе. Вме-
сте с тем при разбавлении может наблюдаться десор'бция потен-
циалопределяющего иона с поверхности дисперсной фазы, что
должно приводить к падению фо-потенциала и соответственно £-по-
тенциала. Концентрирование коллоидной системы обуславливает,
конечно, прямо противоположное действие. В каком направлении
в итоге изменяется g-потенциал при изменении концентрации кол-
лоидной системы, очевидно, определяется тем, влияние какого из
7*
195
двух факторов — утолщения (сжатия) двойного электрического
слоя или десорбции (адсорбции) потенциалопределяющих ионов —
в данном конкретном случае окажется сильнее.
Влияние температуры. Аналогично концентрации на ^-потенциал
действует и температура. С повышением температуры ^-потенциал
должен расти вследствие возрастания интенсивности теплового
движения противоионов н увеличения ТОЛЦЩНЫ двойнога элек-
трического слоя. Однако одновременно может возрастать и десорб-
ция потенциалопределяющих ионов, и при этом фа- и ^-потенциалы
уменьшаются. При понижении температуры должна наблюдаться
обратная зависимость. Вопрос о том, как будет изменяться ^-по-
тенциал с изменением температуры, очевидно; должен решаться
отдельно для каждой коллоидной системы с учетом ее индивиду-
альных особенностей.
Понятно, что вышеприведенные рассуждения приложимы
только к тому случаю, когда разбавление (нагревание) или кон-
центрирование (охлаждение) системы существенно не влияют на
растворимость стабилизующего электролита и на переход его из
молекулярной в ионизированную форму.
Следует отметить, что, к сожалению, экспериментальных дан-
ных о влиянии на ^-потенциал частиц различных лиозолей как
концентрации дисперсной фазы, так и температуры очень мало.
Влияние природы дисперсионной среды. Как отмечалось в на-
чале этой главы, электрокинетические явления, а следовательно,
и наличие двойного электрического слоя на межфазной границе
характерны для систем с, полярными дисперсионными средами.
Большое число проведенных исследований* показало, что ^-потен-
циал дисперсной фазы тем больше, чем больше полярность рас-
творителя. В табл. VII, 1 приведены результаты определения ско-
Таблица VII. 1. Скорость электроосмотического переноса
различных жидкостей в стеклянных капиллярах
Жидкость Диэлектри- ческая проницаемость Дипольный момент молекулы р-‘1О18» эл.-ст. ед. Скорость электро- осмотического пере- носа ыо’Ю5» смЗ/С’В
Вода 81 1,7 16,16
Нитробензол 36 3,9 5,53
Пропиловый спирт 22 1,75 1,95
Анилин 7,2 1,51 1,15
Хлороформ 5,2 1,06 0,63
Бензол 2,25 0 0
Четыреххлористый углерод 2,23 0 0
ростей электроосмотического переноса некоторых чистых жидко-
стей в стеклянных капиллярах, полученные в опытах Файербра-
зера и Балкина. Как следует из данных таблицы, чем больше по-
лярность жидкости, характеризуемая ее диэлектрической прони-
196
цаемостью и дипольным моментом молекулы, тем больше скорость
электроосмотического переноса.
Влияние неравновесных электроповерхностных сил. Выше были рассмотрены
равновесные поверхностные силы, действующие у межфазной границы и способ-
ные препятствовать сближению двух одноименно заряженных частиц. В послед-
ние годы Б. В. Дерягин и С. С. Духин проанализировали действие электропо-
верхностиых сил в системах, в которых имеют место нарушения термодинамиче-
ского равновесия. Они установили, что деформация двойного электрического
слоя, вызванная внешним электрическим полем или конвективным движением
жидкости, приводит к образованию такого электрического поля, радиус дей-
ствия которого часто на несколько порядков превосходит радиус действия не-
деформиров энного слоя в тех же условиях.
Вопросу поляризации двойного электрического слоя посвящены также ра-
боты Овербека, Буутса, Ньюмеиа и других исследователей. В этих работа ав-
торы пришли к выводу, что осмотическим влиянием можно пренебречь при ма-
лом значении С-потеициала (t, < 25 мВ) или же при очень малой толщине
двойного электрического слоя.
Теория неравновесных поверхностных сил диффузионной природы, развитая
Б В. Дерягиным и С. С. Духииым, имеет существенное значение при рассмотре-
нии закономерностей электрокииетических явлений и взаимодействия поляризо-
ванных частиц. Учет диффузии и поляризации двойного слоя позволил Б В. Де-
рягину и С. С. Духину предсказать новое явление, родственное электрофорезу,—
диффузиофорез, заключающееся в движении дисперсных частиц при отсутствии
внешнего электрического поля под влиянием только перепада концентрации ионов
Роль электроповерхностных неравновесных сил в различных процессах, ве-
роятно, весьма значительна. Деформация двойного электрического слоя может
происходить не только под действием внешнего электрического поля (этот случай
будет рассмотрен в разд. 5 настоящей главы), ио и при действии конвективных
потоков жидкой среды, гравитационного поля, поля центробежных сил, ультра-
звукового поля, механических вибраций, броуновского движения. В частности,
было обнаружено влияние электрического поля, возникающего при оседании мел-
ких частиц, на скорость седиментации. В. Г. Левнчем и А Н. Фрумкиным было
указано, что вблизи поверхности капли, движущейся в жидкой среде, может
возникать электрическое поле диффузионного происхождения. Поляризация ион-
ных слоев, наступающая вследствие деформации двойного электрического слоя,
обусловливает проявление дальнодействующих сил притяжения между индуци-
рованными диполями. Наконец, Штауф наблюдал • образование периодических
структур из непроводящих коллоидных частиц, находящихся в переменном элек-
трическом поле. Некоторые из этих эффектов более подробно рассмотрены в
гл. IX.
4. ЭЛЕКТРОФОРЕЗ И ЭЛЕКТРООСМОС
В начале этой главы описаны явления электрофореза и элек-
троосмоса в самом общем виде. Рассмотрим элементарную теорию
электрокииетических явлений и применяемые на практике методы
Определения электрофоретической подвижности и скорости элек-
троосмотического переноса более подробно, поскольку эти вели-
чины позволяют вычислить весьма важную характеристику кол-
лоидных систем — ^-потенциал.
Квинке объяснял Электрокинетические явления возникновением
у межфазной границы двойного электрического слоя. Гельмгольц
развил идеи Квинке и попытался количественно подойти к объяс-
нению электрокииетических явлений. При рассмотрении электро-
кинетических явлений Гельмгольц исходил из следующих поло-
жений.
197
1. Электрические заряды поверхностей жидкости и твердой
фазы противоположны по знаку и расположены параллельно друг
другу, в результате чего образуется двойной электрический слой.
2. Толщина двойного электрического слоя имеет размеры,
близкие к молекулярным.
3. Прн электрокииетических явлениях слой жидкости, непо-
средственно прилегающий к поверхности твердой фазы, остается
неподвижным, тогда как остальная жидкость, находящаяся вблизи
этой поверхности, подвижна и к ней приложим закон трения, при-
меняемый к нормальным жидкостям.
4. Течение жидкости в двойном электрическом слое при элек-
трокинетических явлениях происходит ламинарно и выражается
обычными гидродинамическими уравнениями.
+___1.1________+ , ~Г +__________+...t _
± ±_______± —______±±_______—
’ 6
Рис. VII, 19. Поведение двойного электрического слоя прн электро-
кинетических явлениях:
а—схема двойного слоя по Гельмгольцу — Перрену; б—схема двойного слоя
с учетом диффузного слоя Гуи и адсорбционного слоя Штерна.
5. Двойной электрический слой можно рассматривать как пло-
скопараллельный конденсатор.
6. Распределение зарядов в двойном слое не зависит от на-
пряженности прилагаемого электрического поля, и внешняя раз-
ность потенциалов просто накладывается на поле двойного элек-
трического слоя.
7. Твердая фаза является диэлектриком, жидкость же проводит
электрический ток.
Исходя из этих положений, выведем уравнение, связывающее
g-потенциал со скоростью электрофореза или электроосмотиче-
ского переноса. Для этого представим себе у твердой поверхности
двойной электрический слой, находящийся под действием разно-
сти электрических потенциалов, приложенной тангенциально
к межфазной границе. Такой слой изображен на рис. VII, 19а.
Находящиеся в жидкости ионы (противоионы) под влиянием внеш-
него электрического поля стремятся передвинуться вправо к по-
люсу, несущему противоположный заряд (в данном случае к ка-
тоду). Понятно, что вблизи твердой поверхности вместе с ионами
стремится передвинуться вся жидкость, в которой находятся эти
ионы. Наоборот, под влиянием этого же поля твердая поверхность
с закрепленными на ней ионами (потенциалопределяющими ио-
198
1
нами) стремится передвинуться в противоположную сторону
(в данном случае к аноду). В зависимости от того, что является
неподвижным — жидкость или твердая поверхность, — наблю-
дается передвижение твердой фазы (электрофорез) или жидкости
(электроосмос).
Если не прилагать к двойному электрическому слою разности
потенциалов, а смещать одну фазу относительно другой, то проис-
ходит перенос электрических зарядов, связанных с фазой, и соот-
ветственно возникает электрический ток, а значит, и разность по-
тенциалов. В зависимости от того, передвигается ли жидкость
относительно неподвижной твердой стенки или передвигаются
твердые частицы в жидкости, наблюдается либо потенциал тече-
ния, либо потенциал Дорна-
Допустим, что расстояние между ионами, связанными с твер-
дой фазой (потенциалопределяющими ионами), и ионами, находя-
щимися в жидкости, равно 6. Примем далее, что величина элемен-
тарного электрического заряда, умноженная на число единичных
электрических зарядов, приходящихся на единицу поверхности
твердой фазы, равна ст. Эта величина представляет, очевидно, не
что иное, как поверхностную плотность электричества. Ввиду элек-
тронейтральности системы поверхностная плотность заряда в жид-
кости также должна равняться величине ст, но с обратным знаком.
Это сгущение электрической энергии около межфазной границы
эквивалентно заряду конденсатора и соответствует разности по-
тенциалов £ между его обкладками или соответственно между фа-
зами. Так как б очень малая величина, то кривизной межфазной
границы можно пренебречь, считая, таким образом, что двойной
электрический слой представляет собой плоский конденсатор. Из-
вестно, что между величинами 6, ст и £ для плоскопараллельного
конденсатора имеется следующая зависимость:
£ = 4лба/е (VII, 35)
где е — абсолютная диэлектрическая проницаемость вещества между обклад-
ками конденсатора
Это же уравнение можно переписать так:
ог = 8£/(4зтд) (VII, 33)
Если тангенциально к двойному слою ионов приложена раз-
ность потенциалов Е, а расстояние между электродами состав-
ляет L, то, очевидно, градиент потенциала внешнего поля Н будет
равен:
H = E/L (VII, 37)
Электрическая сила, действующая тангенциально на единицу
межфазной поверхности, когда последняя находится в электриче-
ском поле с градиентом потенциала Н, и стремящаяся сдвинуть
Две фазы относительно друг друга, равна:
Fl = aH (VII, 33)
199
Как уже отмечалось, после короткого периода установления
стационарного режима как электрофорез, так и электроосмос идут
с постоянной скоростью. Это можно объяснить только тем, что
сила трения уравновешивает электрическую силу, обусловливаю-
щую относительное перемещение фаз. Согласно закону Ньютона
сила трения, приходящаяся на единицу поверхности раздела фаз,
составит:
F 2 == г) (du/dl) (VII, 39)
где г) — динамическая вязкость жидкости; и — скорость передвижения жидкости
относительно твердой стенки; I — толщина слоя жидкости, в котором меняется
ее скорость, равная при принятой схеме двойного электрического слоя расстоя-
нию между обкладками конденсатора д.
Принимая, что скорость течения жидкости в двойном электриче-
ском слое линейно изменяется с расстоянием и учитывая, что
I = 5, получим:
F2 = n(«/d) (VII, 40)
Так как при постоянной скорости электрофореза или электро-
осмоса электрическая сила, обусловливающая движение, и сила
треиия равлы, т. е. Fi = Fz, то
<rff = n(u/0) (VII, 41)
Подставив в уравнение (VII, 41) значение о из уравнения (VII, 36),
произведя необходимые сокращения и решив его относительно а,
получим известное уравнение Гельмгольца — Смолуховского:
и = 8£&/(4лп) (VII, 42)
Скорость электрофоретического переноса очень часто относят
к единице градиента приложенного потенциала, для чего обе части
уравнения (VII, 42) делят на Я:
и0 = и)Н = в£/(4лп) (VII, 43)
Величина и0 называется электрофоретической подвижностью и
обычно служит для сравнения способности к электрофорезу раз-
личных коллоидных систем.
Решая уравнение (VI 1,42) относительно ^-потенциала, полу-
чим:
I = 4лт)и/(еЯ) (VII, 44)
С помощью формулы (VII, 44) можно вычислить ^-потенциал при
электрофорезе или электроосмосе, если известна величина и.
При пользовании уравнением (VII, 44) необходимо помнить,
что если приложенная извне разность потенциалов Е выражена,
как обычно, в В, a L в см, то Н = E/L (В/см) = E/L-300 (эл.-ст.
ед/см). Отсюда потенциал, выраженный в абсолютных эл.-ст. ед.,
можно найти с помощью уравнения:
I = 4лци/(еЯ) = 4лт)и£ • 300/(е£) (VII, 45)
200
Однако ^-потенциал, как и всякий другой электростатический по-
тенциал, принято выражать в вольтах. Для этого вычисленное по
уравнению (VII, 45) значение потенциала надо умножить еще раз
на 300. Уравнение для вычисления электрокинетического потен-
циала в вольтах имеет следующий вид:
£ = 4лт]«Ь-3002/(е£) (VII, 46)
Следует заметить, что при выводе уравнений (VII, 42) и
(VII, 44) был сделан ряд упрощений и не вполне обоснованных
допущений. Прежде всего, как уже было указано при рассмотре-
нии строения двойного электрического слоя, схему, из которой мы
исходили, нельзя считать удовлетворительной. Двойной электри-
ческий слой, согласно новейшим представлениям, надо представ-
лять не плоскопараллельным конденсатором, а конденсатором,
одна из обкладок которого состоит из диффузно распределенных
ионов. Часть этих ионов находится в приповерхностном слое и от-
стоит от твердой поверхности на меньшем расстоянии, чем пло-
скость скольжения. В результате этого электрокинетический по-
тенциал соответствует не всему заряду на поверхности стенки,
а разности между общим поверхностным зарядом и зарядом всех
противоионов, находящихся в приповерхностном слое. Поведение
такого слоя при электрофорезе или электроосмосе следует пред-
ставлять себе так, как это показано на рис. VII, 19 б. Правда, та-
кое представление о двойном электрическом слое не обесценивает
приведенный вывод, так как этот слой по-прежнему можно рас-
сматривать как электрический конденсатор. Возникает лишь во-
прос о том, насколько допустимо при количественных выводах при-
равнивать расстояние I, на котором происходит изменение скоро-
сти течения жидкости в двойном слое, к усредненному расстоянию
между обеими обкладками электрического конденсатора с размы-
той внешней обкладкой.
Далее, поскольку скольжение происходит не в плоскости,
а в слое, имеющем малую, но все же конечную толщину и содер-
жащем некоторое число противоионов, изменение скорости течения
жидкости в этом слое в зависимости от расстояния происходит не
по строго прямолинейному закону. Следовательно, допущение, что
du/dl — и/1 в некоторой степени условно.
Наконец, принимая значения диэлектрической порницаемости
и коэффициента вязкости жидкости в двойном электрическом слое
равными значению соответствующих- характеристик раствора, мы
допускаем также некоторую ошибку, поскольку из-за повышенной
концентрации ионов значения е и т] в двойном электрическом слое
могут быть иными, чем в дисперсионной среде. На диэлектрическую
проницаемость может влиять также поле высокого напряжения,
возникающее в двойном электрическом слое. Вязкость у поверх-
ности твердой фазы может быть повышена за счет изменения
структуры приповерхностного слоя жидкости, вызванного дей-
ствием молекулярных сил. На существенное повышение вязкости
201
жидкости у поверхности твердой фазы указывалось в работах
Б. В. Дерягина. Кроме того, и диэлектрическая проницаемость и
вязкость являются макроконстантами и применение их при вычис-
лении ^-потенциала, где они должны характеризовать слои жидко-
сти молекулярной толщины, является условным.
Отличие значений диэлектрической проницаемости и вязкости в двойном
электрическом слое жидкости от значений этих характеристик в объеме диспер-
сионной среды может приводить не только к получению неточных абсолютных
значений ^-потенциала, но и к изменению харак-
Рис. VII, 20. Влияние кон-
центрации с электролита
в растворе ка электрокине-
тические свойства системы:
1 — изменение диэлектрической
проницаемости е; 2 — изменение
^-потенциала, вычисленного без
учета изменения е; 3 — изменение
£-потенциала, вычисленного с уче-
том изменения е; 4—изменение
электрофоретической подвиж-
ности и.
тера зависимости ^-потенциала от какого-нибудь
влияющего на него фактора. Для иллюстрации
этого положения приведем следующий пример.
Известно, что при возрастании концентрации
электролита диэлектрическая проницаемость рас-
твора изменяется, проходя через минимум. В ре-
зультате этого ^-потенциал, вычисленный по урав-
нению Гельмгольца — Смолу ховского с учетом
изменения величины е, будет изменяться по кри-
вой с максимумом, если даже электрофоретиче-
ская подвижность изменяется с увеличением кон-
центрации электролитов по прямолинейному за-
кону. Сказанное становится более понятным из
схемы, приведенной на рис. VII, 20.
материал указывает на
При определении значений ^-потен-
циала с помощью электрофореза и элек-
троосмоса приходится встречаться с ря-
дом частных экспериментальных и теоре-
тических трудностей, о которых будет
сказано ниже. Тем не менее не следует
делать вывода о недостоверности полу-
чаемых из опыта величин ^-потенциала.
Наоборот, огромный экспериментальный
то, что если даже абсолютное значение
электрокинетического потенциала определяется и не совсем точно,
то все же всегда можно получить правильную картину его изме-
нения под влиянием различных факторов.
5. ОПРЕДЕЛЕНИЕ ЭЛЕКТРОКИНЕТИЧЕСКОГО ПОТЕНЦИАЛА
Электрофоретические методы
Значение ^-потенциала можно вычислить по уравнению
(VII, 44). Для этого необходимо измерить скорость электрофоре-
тического передвижения частиц в системе при известном градиенте
потенциала прилагаемого напряжения и знать значения вязкости и
диэлектрической проницаемости жидкости.
Однако прежде чем рассматривать экспериментальные методы
определения скорости электрофоретического переноса, остановимся
на различного рода ограничениях, которые надо иметь в виду при
применении уравнений (VII, 42) и (VII, 44).
202
Согласно исходным положениям, электрофорез представляет
собой явление, близкое электроосмосу. И для электрофореза, и
для электроосмоса, как мы приняли ранее, перемещение жидкости
по отношению к поверхности твердой фазы определяется силами,
действующими на двойной электрический слой. Именно исходя из
этих предпосылок нами и было выведено уравнение Гельмгольца —
Смолуховского, выражающее зависимость скорости электрофореза
от градиента потенциала внешнего поля. Однако применение урав-
нения (VII, 42) для описания электрофоретических явлений огра-
ничено следующими условиями. Во-первых, толщина двойного
слоя (обычно характеризуемая величиной 1/х) должна быть мала
по сравнению с размером частицы. Во-вторых, вещество частицы
не должно проводить электричества, а поверхностная проводимость
на межфазной границе должна быть настолько малой, чтобы она
практически не влияла на распределение внешнего электрического
поля.
Длительное время уравнение Гельмгольца — Смолуховского
считали справедливым во всех случаях, независимо от того, выпол-
няются ли вышеприведенные условия или нет. Но в 1924 г. Гюк-
кель, основываясь на новой теории сильных 'электролитов, дал
другое уравнение, отличающееся от уравнения (VII, 42) числен-
ным множителем 2/з:
и — е£Н/(6ж\) (VII, 47)
При выводе этого уравнения коллоидная частица принята экви-
валентной сферической частице и введена поправка на так назы-
ваемое электрофоретическое запаздывание (торможение), вызван-
ное влиянием внешнего поля на двойной электрический слой. Под
действием этого поля противоионы передвигаются в направлении,
противоположном движению частицы, сообщая этим самым дви-
жение окружающей жидкости в том же направлении. Это приво-
дит к тому, что частица перемещается не в покоящейся, а в дви-
жущейся жидкости, в результате чего электрофоретическая
скорость уменьшается.
Генри в 1931 г. показал, что уравнения (VII, 42) и (VII, 47)
справедливы каждое в отдельном случае. При выводе уравнения
(VII, 42) учитывалась деформация электрического поля, вызван-
ная непроводящей частицей, а при выводе уравнения (VII, 47)
допускалось, что непроводящие частицы не влияют на поле в двой-
ном слое и объеме жидкости. Последнее предположение справед-
ливо лишь тогда, когда электропроводность частиц равна электро-
проводности среды или размеры частиц так малы по сравнению
с толщиной двойного слоя, что деформация в большей части двой-
ного слоя становится несущественной.
Тщательно рассматривая вопрос с математической точки зрения
и принимая во внимание деформацию внешнего поля, Генри пока-
зал, что скорость электрофореза во всех случаях может быть вы-
ражена уравнением
« = /е^ЯДлп)
(VII, 48)
203
где Jфункция величины ха; а — радиус сферической частицы или длинная ось
цилиндрической частицы; х — величина, обратная толщине двойного слоя, чис-
ленно равная д/8яе2АГА У, сг2/ъкТ .
Значение величины f неодинаково для частиц разной формы и
различно расположенных по отношению к полю. Зависимость этой
величины от иа показана на рис. VII, 21.
Для непроводящих цилиндрических частиц, ось которых совпадает с напра-
влением поля, / = 1/4, как в уравнении (VII, 42). Для цилиндрических частиц,
расположенных перпендикулярно направлению поля, f — 1/8 при малых значе-
ниях ' ‘ п~~ ...
«
Рис.
иа в приближается к 1/4 при больших величинах иа. Для сферических
частиц f = 1/6 при малых значениях иа и
f = 1/4 при больших значениях этой вели-
чины. Таким образом, данные рис. VII, 21
находятся в полном соответствии с уравне-
нием (VII, 42), согласно которому числен-
ный коэффициент равен 1/4 независимо от
формы частиц, если значение иа велико, и
с уравнением (VII, 47), согласно которому
численный множитель равен 1/6 для сфери-
ческих частиц с малым значением иа.
Геири проанализировал также влияние
на величину f электропроводности дисперс-
ной фазы, полностью меняющей распреде-
ление электрического поля вблизи частиц.
На рис. VII, 21 пунктиром обозначены зна-
чения коэффициента f в зависимости от
величины иа для электропроводящих ча-
стиц сферической формы. Как и следовало
ожидать, распределение поля ве сказывает-
ся иа величине f при очень большой толщи-
не двойного электрического слоя (иа С 1),
ио с уменьшением толщины слоя коэффи-
циент уменьшается И в пределе становится
равным нулю. Однако на практике в боль-
шинстве случаев влияние электропроводно-
жа
VII, 21. Зависимость вели-
чины f от иа для частиц:
1—цилиндрическая (ось параллельна
полю), не проводящая ток; 2—Сфериче-
ская, яе проводящая ток; J—сфериче-
ская, проводящая ток; <—цилиндриче-
ская (ось перпендикулярна полю), яе
проводящая ток.
сти коллоидвых частиц металлов можно не учитывать, так как оно почти пол-
ностью устраняется поляризацией поверхности. При этом частица ведет себя так,
как она должна вести себя, если бы состояла из непроводящего вещества.
Из всего приведенного следует, что для выбора правильного численного
коэффициента в уравнении Геири необходимо знать размер частицы а, форму
частицы (цилиндр, сфера) и толщину двойного электрического слоя. Необходимо
также знать, является ли дисперсная фаза проводником или диэлектриком. Пер-
вые две характеристики (размер и форма частицы) определяются эксперимен-
тально с помощью оптических и иных методов. Электропроводность дисперсной
фазы обычно известна. Толщину двойного электрического слоя, или, что то же,
ионной атмосферы, согласно теории сильных электролитов можно вычислить по
уравнению (VII, 5).
При наложении внешнего электрического поля движущаяся за-
ряженная частица подвергается не только рассмотренному выше
электрофоретическому торможению, но и действию так называе-
мой электрической релаксации, которая заключается в следую-
щем. Согласно электрической теории электрофореза предпола-
гается, что электрическое поле двойного слоя и внешнее электри-
ческое поле просто накладываются одно на другое. Однако на
самом деле этого нет, так как частица и внешняя часть двойного
204
слоя движутся в противоположных направлениях, и первоначаль-
ная симметрия двойного слоя относительно частицы нарушается.
Вследствие электропроводности и диффузии двойной слой стре-
мится восстановить симметрию, но это восстановление требует вре-
мени (времени релаксации), и поэтому внешняя часть двойного
слоя все же остается позади движущейся частицы. В результате
этого возникает добавочное электрическое поле, которое действует
на частицу, стремясь двигать ее в обратном направлении, и тем
самым влияет на скорость электрофореза.
в|_______________________________,
Ч0' °,' I Ю ЮО 1000
ха
Рис. VII, 22. Влияние релаксации и различных типов электролитов
на электрофорез коллоидных частиц с отрицательным J-потеициа-
лом, равным 50 мВ.
Тип электролита обозначен цифрами у кривых: первая цифра характеризует
валентность катиона, вторая—валентность аниона.
Не рассматривая подробно влияние электрической релаксации на скорость
электрофореза, отметим лишь, что, согласно Овербеку, эффект релаксации зави-
сит от С-потенциала, величины ха и от валентности ионов электролитов, присут-
ствующих в системе. На рис. VII, 22 в качестве иллюстрации показано влияние
электрической релаксации для сферических коллоидных частиц с отрицательным
^-потенциалом, равным 50 мВ, и различных типов электролитов. На оси абсцисс
отложены значения ха, а на оси ординат — зваченвя величины f, ва которую
следует умножить скорость электрофоретического переноса, вычисленную по
уравнению Гюккеля (VII, 47), чтобы получить правильные результаты. Пунктир-
ной линией, показана кривая, характеризующая изменение скорости, вычисленной
по уравнению Геири без учета релаксации.
По данным рис. VII, 22 можно сделать вывод о незначительных релаксацион-
ных эффектах при малых и больших значениях ха. Характерно различное распо-
ложение кривых в зависимости от электролитов различного типа. Электрофорез
отрвцательно заряженных частиц замедляется симметричными электролитами и
электролитами, имеющими поливалентные катионы. Наоборот, поливалентные
анноны ускоряют электрофорез отрицательно заряженных частиц. .
С увеличением электрокинетнческого потенциала и валентности ионов по-
правка на релаксацию быстро возрастает. Но при значениях электрокинетиче-
ского потенциала, меньших 25 мВ, или при использовании электролитов с одно-
валентными катионами и анионами релаксационный эффект мал и поправку на
205
релаксацию в уравнение Генри можно не вводить, так она не превышает в
этом случае 3%.
Помимо явлений электрофоретического запаздывания и электрической ре-
лаксации на скорость электрофореза может влиять и агрегатное состояние дис-
персной фазы. Так, скорость электрофоретического переноса жидких частиц при
всех прочих одинаковых условиях электрофореза равна подвижности твердых
частиц лишь в частном случае, когда в результате адсорбции поверхностно-
активных веществ поверхность капли становится неподвижной, что делает жид-
кую частицу похожей на твердую. В общем же случае жидкие частицы, обла-
дающие достаточно высокой проводимостью, движутся при электрофорезе
значительно быстрее, чем твердые. Это объясняется следующими причинами. Во-
первых, трение о поверхность жидкой частицы всегда меньше, чем трение о по-
верхность твердого шарика таких же размеров, так как капли жидкости могут
деформироваться при движении среды Во-вторых, двойной электрический слой
Рис. VII, 23. Движение жидко-
сти в капельке ртути при элек-
трофорезе.
Рьс. VII, 24. Схема -прибора
Кёна для электрофореза.
на границе двух жидкостей возникает в обеих фазах, и его часть, приходящая
на каплю, играет существенную роль, заставляя циркулировать жидкость внутри
капли, что сказывается на скорости электрофореза.
Следует упомянуть о работах А. Н. Фрумкина, определявшего электрофо-
ретическую подвижность капель ртути. А. Н. Фрумкин показал, что капля ртути
полностью поляризована, так что ее поведение подобно поведению непроводника.
Однако поляризация изменяет поверхностное натяжение на полюсах капли, вы-
зывая движение ртути вдоль поверхности капли. Если капля заряжена положи-
тельно (обычный случай), то заряд на поверхности капли у полюса, обращенного
к положительному электроду, уменьшается и поверхностное натяжение в этом
месте возрастает, тогда как на другом полюсе капли происходит обратное яв-
ление. В результате разницы в поверхностных натяжениях внутри капли возни-
кает движение ртути, что схематически представлено на рис. VII, 23. Стрелки
внутри капли показывают движение ртути, стрелки снаружи капли — направле-
ние движения дисперсионной среды. Большая стрелка внизу рисунка обозначает
направление движения всей капли. Не трудно понять, что это движение ртути
должно ускорять перенос частицы к отрицательному электроду. Такие круговые
движения могут увеличивать скорость переноса капли на несколько порядков по
сравнению с обычными скоростями электрофореза
Рассмотрим теперь кратко основные методы определения ско-'
рости электрофореза, уделяя внимание главным образом их прин-
ципиальным особенностям, так как технические подробности экс-
периментальных методик изложены в соответствующих руковод-
ствах к практическим занятиям по коллоидной химии. Поскольку
206
электрофорез во многих отношениях сходен с электролизом, его
скорость может быть измерена во многих случаях теми же спосо-
бами, какими определяется подвижность обычных ионов, напри-
мер путем определения чисел переноса, измерения скорости пере-
мещения границы раздела, аналитическими методами, а также
с помощью микроскопа или ультрамикроскопа.
Метод Гитторфа. Этот метод аналогичен методу, применяемому для опреде-
ления чисел переноса ионов. Через коллоидный раствор, помещенный в специаль-
ный сосуд, пропускают в течение некоторого времени электрический ток и затем
в пробах раствора, отобранных из разных мест, аналитически определяют коли-
чество дисперсной фазы, переместившейся к одному из электродов. Очевидно,
это количество т прямо пропорционально скорости электрофоретического пере-
носа и, концентрации дисперсной фазы с, силе тока / и времени пропускания
тока t и обратно пропорционально электропроводности жидкости у, т. е.
т = uclxly (VII, 49)
Из этого уравнения можно определить скорость электрофореза:
и = ущ/(с/т) (VII, 50)
Этот метод в применении к коллоидным системам особенно точен благодаря
большой массе дисперсной фазы, приходящейся на единицу заряда Однако
этот способ применяется на практике довольно редко. Таттье использовал метод
Гитторфа не только для определения электрофоретической скорости, но и для
одновременного определения-подвижности противоионов.
Метод подвижной границы. Этот метод получил особенно ши-
рокое распространение на практике благодаря относительной про-
стоте. Принцип его основан на наблюдении за скоростью передви-
жения под влиянием электрического поля границы между обычно
мутным или окрашенным коллоидным растврщш—и прозрачной
бесцветной специальной «боковой жидкостью». Для исследования
прозрачного бесцветного золя используют специальные приемы,
способствующие проявлению границы, например освещение уль-
трафиолетовым светом.
В разное время исследователями было предложено много приборов для осу-
ществления метода подвижной границы. Одним из таких приборов, очень про-
стым по устройству, является прибор Кёна, изображенный на рис. VII, 24. Этот
прибор представляет собой широкую стеклянную U-образную трубку, каждое
колено которой имеет внизу стеклянный кран с диаметром отверстия, равным
внутреннему диаметру трубки. Верхние части обоих колен трубки отградуиро-
ваны, причем большое деление равно 1 см. В оба колена U-образной трубки
вставляются платиновые электроды. Коллоидный раствор можно вводить в при-
бор с помощью специальной воронки, соединенной с нижней частью U-образной
трубки узкой стеклянной трубкой с краном.
Прибор собирают для работы следующим образом. В нижнюю часть U-об-
разной трубки при открытых кранах с помощью воронки вводят исследуемый
коллоидный раствор в таком количестве, чтобы его уровень оказался несколько
выше кранов. Затем закрывают краны на обоих коленах U-образной трубки и
кран на трубке, соединяющий прибор с воронкой. С помощью пипетки (или
наклоняя весь прибор) из обоих колен удаляют жидкость, находящуюся над
кранами. После этого в оба колена с помощью пипетки вводят определенные
количества «боковой жидкости» так, чтобы уровни ее в коленах U-образпой
трубки находились на одной высоте и были на 3—4 см выше градуировки. Да-
лее в каждое колено вставляют укрепленный на резиновой пробке электрод и
осторожно открывают крапы в обоих коленах. В таком виде прибор готов .для
работы.
207
Определение сводится к измерению времени (с помощью секундомера), за
которое в одном из колен трубки после включения постоянного тока определен-
ной силы граница раздела коллоидная система — боковая жидкость передви-
нется на высоту 1 см. Проводят несколько таких определений, не выключая
тока, н для вычисления скорости электрофореза берут нз них среднеарифметиче-
ское значение. Одновременно с измерением времени с помещью вольтметра,
включенного параллельно в электрическую цепь, отмечают напряжение тока на
электродах. В конце работы необходимо измерить также расстояние между
'обоими электродами по длине трубки; это расстояние нужно для вычисления
значения градиента потенциала внешнего поля.
В более совершенных приборах боковые колена градуированы как над кра-
нами, так н под ннмн, что позволяет работать с коллоидными системами менее
плотными, чем боковая жидкость. Это особенно важно прн работе с эмульсиями,
у которых плотность дисперсной фазы (обычно углеводорода), как правило,
меньше плотности дисперсионной среды (воды). При этом боковая жидкость по-
мещается внизу, а коллоидный раствор — вверху.
Для устранения влияния поляризации электродов применяют неполярнзую-
щнеся электроды. Для того чтобы в систему не проникали продукты электро-
лиза, электроды обычно помещают в отдельные широкие сосуды, соединяемые
с коленами прибора прн помощи стеклянных трубок, заполненных агаровым
студнем, содержащим хлорид калия, как это принято в электрохимии прн опре-
делении pH.
Для большей точности передвижение границы системы коллоидный рас-
твор — боковая жидкость можно наблюдать с помощью короткофокусной опти-
ческой трубы, укрепленной на катетометре.
При выборе боковой жидкости следует руководствоваться не-
которыми требованиями к ее составу и свойствам. Помимо того,
что в отличие от коллоидного раствора она должна быть прозрач-
ной или бесцветной, она должна также не влиять на ^-потенциал
переходящих в нее из золя коллоидных частиц и обладать элек-
тропроводностью, равной или немного большей электропровод-
ности коллоидной системы. Это обычно обеспечивает образование
резкой границы между золем -и боковой жидкостью и позволяет
обходиться без введения поправок на их различную электропро-
водность при вычислении градиента внешнего потенциала.
В качестве боковой жидкости часто применяют ультрафильтрат
золя или дисперсионную среду, полученную коагуляцией коллоид-
ной системы путем замораживания. Однако если исследуют отно-
сительно концентрированные коллоидные растворы с небольшим
содержанием электролитов, приготовленная таким способом боко-
вая жидкость обладает все же несколько иной электропровод-
ностью по сравнению с золем. В этом случае при вычислении ско-
рости электрофореза необходимо- вводить поправки на распреде-
ление напряженности в электрическом поле, что подчас бывает
трудно.
Метод Тизелиуса. Этот метод в принципе сводится к методу подвижной
границы, однако в нем много специфических особенностей. Измерения по этому
методу проводят в приборе, который представляет собою усовершенствованный
аппарат Кёиа. Для создания резкой границы между коллоидным раствором и
боковой жидкостью в приборе Тизелиуса используют не краны, а сдвиг пришли-
фованных частей U-образной трубки относительно друг друга. На рис. VII, 25
дан разрез основной части прибора, соответствующей U-образной трубке при-
бора Кёна. Нижняй часть трубки в момент наполнения верхней части боковой
208
жидкостью должна быть уже заполнена испытуемым золем. Поперечное сечение
U-образнон трубки имеет не круглую, а прямоугольную форму, что облегчает
наблюдение за передвижением границы. Кроме того, в таких условиях жидкость
быстрее принимает температуру водяного термостата, являющегося необходимой
частью прибора Определения проводят, как правило, прн +4 °C, что соответ-
ствует максимальной плотности воды. В этих условиях прн небольших темпе-
ратурных колебаниях плотность жидкости изменяется весьма мало, поэтому
Рис. VII, 25. Внд (сбоку) U-образной трубки в приборе Тизелиуса:
а—рабочее положение, б — положение в момент заполнения верхней части
трубки.
исключается образование конвекционных токов н размывание границы раздела
незначительное Электроды, с помощью которых создается внешнее электриче-
ское поле, расположены в боковых широких сосудах, благодаря чему продукты
электролиза не попадают в золь На рнс VII, 26 показан прибор Тизелиуса в
собранном виде. Передвижение границы
определяют по различию в коэффициен-
те преломления исследуемого раствора
и его ультрафильтрата, который обычно
используют в качестве боковой жид-
кости.
Очень остроумная оптическая систе-
ма Фнльпота — Свенссона позволяет ха-
рактеризовать распределение градиента
коэффициента преломления в области
подвижной границы графическим спосо-
бом Этим методом, прн котором фото-
графируют нли наблюдают плоский све-
товой луч, проходящий через U-образную
трубку, можно регистрировать все не-
равномерности распределения коллоид-
ного вещества в столбике жидкости. Если
градиент коэффициента преломления от-
Рнс. VII, 26. Схема прибора Тнзе-
сутствует, световой луч благодаря осо-
бому устройству линз дает на фотопла-
стннке одну сплошную вертикальную лн- лнуса в собранном виде.
нию Если имеется такой градиент, то на
фотопластинке ему будет соответствовать пнк. По характеру этого пика можно
судить о положении границы раздела в столбике жидкости, о резкости границы
и о разности концентраций по обе стороны от границы раздела.
Метод Тизелиуса особенно шнрокс-применяется прн исследовании белков и
других высокомолекулярных электролитов, поскольку с его помощью можно не
только определить скорость электрофореза, но и разделить смесь высокомолеку-
лярных веществ на отдельные компоненты. В самом деле, если исследуемый
раствор содержит нескблько компонентов с различными электрофоретическими
подвижностями, то фронт передвижения более подвижных компонентов будет
обгонять фронт движения компонентов, движущихся медленнее, и образуется
столько границ, сколько компонентов в смеси.
209
Электрофорез на бумаге. Прибор Тизелиуса очень сложен. В последнее
время широкое распространение для исследования белков получил другой метод,
предложенный также Тизелиусом, но осуществляемый проще. Это метод элек-
трофореза на бумаге.
Он выполняется следующим образом. На середину полоски плотной, гомо-
генной фильтровальной (хроматографической) бумаги, пропитанной буферным
раствором с определенным значением pH, наносят каплю исследуемого коллоид-
ного раствора. Затем на полоску бумаги накладывают разность потенциалов.
Под влиянием образующегося электрического поля отдельные компоненты, со-
держащиеся в капле, обладающие разными электрофоретическими подвижностя-
ми, передвигаются по полоске с различными скоростями. Через некоторое время
компоненты распределяются на бумаге в виде стольких зон, различно удаленных
от исходной точки, сколько компонентов содержалось в растворе. Полоску вы-
сушивают и прогревают для денатурации и фиксации находящихся на ней бел-
ков и после этого окрашивают подходящими красителями. В результате прояв-
ляется распределение компонентов по длине полоски. Роль бумаги в этом методе
сводится к устранению диффузионного и конвекционного перемешивания белков
при электрофорезе.
Методом электрофореза на бумаге можно не только исследовать состав
смесей высокомолекулярных веществ, но и выделить отдельные компоненты.
С этой целью полоску бумаги, не прогревая, разрезают на части и из них
экстрагируют отдельные, уже разделенные компоненты. Теория этого метода,
при котором существенную роль играют поверхностные и адсорбционные явле-
ния, еще мало разработана.
Микроскопический и ультрамикроскопический методы. Эти ме-
тоды определения электрофоретической подвижности заключаются
в определении скорости передвижения индивидуальных коллоид-
ных частиц в электрическом поле при помощи микроскопа или
ультрамикроскопа. Преимущество этого метода перед методом по-
движной границы состоит в том, что при исследовании с помощью
микроскопа частицы находятся в одной и той же окружающей их
среде и отсутствует поверхность раздела между коллоидной си-
стемой и боковой жидкостью. Другое преимущество этого метода
заключается в том, что для определения достаточно очень малое
количество раствора. Недостаток этого метода тот, что нельзя ис-
следовать электрофоретическую подвижность частиц в растворах
с более или менее значительной концентрацией дисперсной фазы,
так как в таких растворах наблюдение за перемещением отдель-
ной частицы невозможно. Разбавление же системы чужеродной
жидкостью всегда влияет на ^-потенциал.
При микроскопическом или ультрамикроскопическом методе испытуемый
коллоидный раствор помещают в снабженную электродами закрытую кювету
с прямоугольным или цилиндрическим сечением и измеряют время, необходимое
для того, чтобы выбранная частица передвинулась на определенное расстояние.
Так как частицы находятся в броуновском движении, то необходимо брать сред-
нее арифметическое значение из многих таких определений. Кроме того, следует
учитывать, что жидкость в кювете находится в движении в результате электро-
осмотического перемещения вдоль стенок кюветы. Однако вследствие того, что
кювета закрыта, в центре кюветы устанавливается обратный ток жидкости, и,
таким образом, перенос жидкости в кювете в целом равен нулю. Г.кодцс.ть. дни-
жения жидкости в кювете налагается на электрофоретическую скорость движе-
ния частиц, и найденное значение представляет алгебраическую сумму этих ско-
ростей. Понятно, что значение этой суммы сильно зависит от местонахождения
частицы в кювете. Примером этому могут служить результаты работы Эллиса,
210
определившего зависимость между скоростью движения масляных капель в кю-
вете с прямоугольным сечением и высотой, на которой находились капельки
относительно дна кюветы. Эти данные представлены в виде точек на рис. VII, 27
(парабола на рисунке соответствует теоретически вычисленной скорости элек-
трофореза).
Для вычисления истинной электрофоретической скорости можно взять сред-
нее значение наблюдаемых скоростей по всей плоскости поперечного сечения
кюветы. Однако это сложно, и практически проще измерять электрофоретическую
скорость в том месте поперечного сечения кюветы, где скорости электроосмоса
и обратных потоков равны друг другу и где вследствие этого жидкость нахо-
дится в покое Теория, с помощью которой находятся такие места в кювете,
дана Смолуховским, она приводится в практикумах по коллоидной химии. Здесь
Рис. VII, 27. Наблюдаемая
(точки) и вычисленная (кривая)
скэрости электрофоретического
переноса масляных капель на
различной глубине кюветы.
Рис. VII, 28. Схема прибора, предло-
женного Абрамсоном для микроэлек-
трофореза:
1 — воронка для исследуемого раствора;
2—трехходовые краны; 3 — деполяризую-
щиеся электроды; 4 — объектив микроскопа;
5 — конденсор; 6 — кювета; 7 — трубка для
отвода раствора.
же укажем лишь, что для кюветы с круглым сечением слой, где скорость дви-
жения жидкости равна нулю, лежит на расстоянии 0,148 диаметра от стенки.
Для плоской кюветы большой ширины таких слоев два, причем каждый из них
находится на расстоянии, равном 0,212 от ее высоты. Иначе говоря, эти слои
соответствуют примерно 1/5 и 4/5 высоты кюветы.
Для микроскопического и ультрамикроскопического определе-
ния электрофоретической скорости предложен ряд приспособлений
и приборов. Простейшие из них представляют собой кюветы, со-
стоящие из предметного и покровного стекла, между которыми
помещают две тонкие платиновые пластинки, приклеенные к обоим
стеклам и служащие электродами. После введения золя в эту кю-
вету боковые щели ее замазывают вазелином. Понятно, что при
работе с такими кюветами не исключается поляризация электро-
дов и переход продуктов электролиза в золь.
На рис. VII,28 дана схема прибора для микроэлектрофореза, предложенного
Абрамсоном. Этот прибор состоит из плоской стеклянной кюветы 6, переходя-
щей в стеклянные трубки, заканчивающиеся трехходовыми кранами 2. Один из
этих кранов соединяет кювету с воронкой I. Пользуясь воронкой и кранами,
вначале кювету промывают, а затем наполняют исследуемым коллоидным
211
раствором. Непосредственно перед измерением, поворачивай краны, кювету от-
соединяют от воронки и сливиой трубки 7 и соедиииют с иеполиризующимиси
электродами 3 цинковыми палочками в насыщенном растворе сульфата циика.
Как правило, электрофорез проводят при постоянном токе, причем в элек-
трическую цепь прибора вводят переключатель для перемены направления тока.
Интересно, что Сведберг еще в 1919 г. использовал для определении электро-
форетической скорости переменный ток, при котором частицы совершают коле-
бания и видны в ультрамикроскопе с темным полем как светлые линии опреде-
ленной длины. Зная частоту тока и измерив длину светлой линии, можно вы-
числить электрофоретическую скорость.
В заключение следует указать, что описанный метод определе-
ния электрофоретической подвижности можно применять и к рас-
творам высокомолекулярных соединений, отдельные молекулы ко-
торых в ультрамикроскопе не видимы. Для этого в раствор вводят
малые частицы кварца или угля, которые адсорбируют на себе
высокомолекулярное вещество. Как показали многие экспери-
менты, электрофоретическая подвижность таких частиц такая же,
как и подвижность свободных макромолекул. Это становится по-
нятным, если учесть, что электрофоретическая скорость, согласно
уравнению Гельмгольца — Смолуховского, не зависит от размера
частиц. Однако всегда следует помнить, что ^-потенциал, вычис-
ленный по результатам таких измерений, является в некоторой сте-
пени фиктивной величиной, так как в этом случае довольно трудно
представить себе наличие двойного слоя с более или менее по-
стоянным потенциалом.
Электроосмотические методы
Как мы уже зиаем, С-потеициал можно вычислить ие только по скорости
электрофореза, но и по электроосмотической скорости передвижении. Хотя пря-
мое определение электроосмотической скорости возможно, гораздо удобнее для
вычислении С-потенциала измерять объем жидкости, проходящий при электро-
осмосе через капиллир или пористую мембрану, или давление, развивающееся
в результате электроосмотического движения жидкости.
Определение по объему электроосмотически перенесенной жидкости. Дли
объема жидкости V, протекающего через одиночный капиллир радиуса г в еди-
ницу времени, очевидно, справедливо выражение:
РУФ (VII, 51)
. 4ят) 4т) ' '
Следует помнить, что при электроосмотическом течении жидкости через
капилляр движущая сила имеет электрическую природу и она действует на
периферические части цилиндра жидкости, заполняющей капилляр, где сосредо-
точены свободные противоионы. В результате этого при наложении электриче-
ского поля скорость движения жидкости в капилляре будет сначала максималь-
ной у стенки капиллира и минимальной у его оси. Затем, вследствие трения
между слоями жидкости, скорости выравниваются и при стационарном режиме
течения жидкость движется практически с одинаковой скоростью по всему тече-
нию капилляра. Схема, иллюстрирующая установление стационарного течения
при электроосмосе, приведена иа рис. VII, 29а.
При электроосмосе характер движении жидкости прямо противоположен
тому, который наблюдалси бы при движении жидкости под влиянием гидроста-
тического давления, когда в начале течения скорости жидкости всюду равны.
Одиако в последнем случае постепенно начинает сказыватьси трение у стенки,
212
скорость течения периферийных слоев становится меньше, чем центральных, и,
наконец, эпюра скоростей при стационарном режиме течения принимает форму
параболы (рис. VII, 296).
Пористую мембрану, согласно Смолухонскому, можно рассматривать как
сумму капилляров с общей площадью сечения s. Тогда объем жидкости, элек-
троосмотически протекающей в единицу времени через мембрану, будет выра-
жаться следующим уравнением:
(VII, 52}
Таким образом, объем жидкости, электроосмотически перенесенной через мем-
брану, прямо пропорционален поперечному сечению капилляров, диэлектрической
проницаемости, ^-потенциалу, приложенной электро-
движущей силе и обратно пропорционален вязкости
жидкости.
Согласно закону Ома:
£ = //?
(VII, 53)
где Е— приложенная разность потенциалов; I — сила
тока; R — электрическое сопротивление системы.
Электрическое сопротивление раствора электролита
равно:
£
Y«
(VII, 54)
где L — толщина слоя раствора; а —площадь сече*
ния; у — удельная электропроводность раствора. Под-
ставляя в уравнение (VII, 53) приведенное выраже-
ние для R, получим:
e^ir^-~-
Vs
(VII, 55)
i
Рис. VII, 29. Схема, пояс-
няющая установление
стационарного режима
течения жидкости в ка-
пилляре:
о—при электроосмосе; б—под
действием гидростатического
давления.
Пользуясь этим выражением для Е, градиент внешнего
зить следующим образом:
потенциала
можно выра-
„ Е IL I
L syL ys
Подставляя найденное значение градиента в уравнение (VII,52),
v st&H seg/ eg/
4лт) 4лт)уа 4лт|у
(VII, 56>
имеем:
(VII, 57}
Так как в уравнение (VII, 57) не входит ии L, ни а, то, очевидно, при постоян-
ной силе тока объем жидкости, прошедший сквозь мембрану, не зависит от попе-
речного сечения и длины капилляров. Правильность уравнения (VII, 57) была
подтверждена опытами Видемана.
Из уравнения (VII, 57) легко найти значение С-потеициала:
g = ±^K (VII, 58)
По аналогии с уравнением (VII, 46) правую часть этого уравнения надо умно-
жить на 3002 для того, чтобы g-потеициал был выражен в вольтах. Тогда урав-
нение (VII, 58) будет иметь вид:
4лт)у7300!
-----Vi----
(VII, 59)
Таким образом, для того чтобы определить значение g-потенциала по элек-
троосмосу, следует экспериментально определить объем жидкости, перенесенной
21»
•через капилляр или пористую мембрану, удельную электропроводность раствора
и силу тока, при которой проводился электроосмос. Остальные величины берут
-обцчно из справочников.
При использовании уравнения (VII, 58) необходимо учитывать возможность
явления так называемой поверхностной проводимости. Это явление сводится к
тому, что находящиеся около межфазной границы ионы изменяют состав среды,
я следовательно, и удельную электропроводность раствора у межфазной гра-
ницы. Если радиус капилляра достаточно велик по сравнению с толщиной слоя
у стенки, где проявляется поверхностная проводимость, средняя электропровод-
ность раствора остается приблизительно той же, что и в его объеме, и вели-
чина у в уравнении (VII, 58) является удельной электропроводностью раствора.
Рис. VII, 30. Зависимость ^-потенциала от радиуса капилляра г
с учетом (/) н без учета (2) поверхностной проводимости:
А—мембрана из кварца; S—мембрана нз корунда.
Если же капилляры тонкие, это условие не соблюдается и у становится уже
функцией радиуса капилляра. В этом случае вместо уравнения (VII, 58) следует
•пользоваться уравнением:
^(r±OVs/s).y (Vn60)
•тде О — длина окружности капилляра; з — его сечение; у4 — поверхностная про-
водимость.
Множитель (у + Oy>/s) в числителе уравнения (VII, 60) ставится вместо
величины у, исходя из следующих соображений. Электропроводность жидкости,
заполняющей капилляр, у равна сумме электропроводностей (обратных сопро-
тивлений) объемной части жидкости и приповерхностного слоя:
1/^=1//?+l//?s (VII,61)
где \/R = ys/L, \/R — ys/L и l/R, = ys O/L, причем принимается, что толщина
слоя с поверхностной проводимостью мала по сравнению с радиусом капилляра.
Отсюда общая электропроводность у равна
V = Y + (VII, 62)
Величина Oys/s, очевидно, представляет собой увеличение электропроводно-
•сти жидкости в капилляре в результате поверхностной проводимости. Так как
O/s ~ \/г, то поправка на поверхностную проводимость не нужна при капил-
лярах достаточно большого радиуса.
Сравнивая уравнения (VII, 58) и (VII, 60) и учитывая, что величина Oy„/s
всегда положительна, можно видеть, что вследствие поверхностной проводимо-
сти истинные значения С-потенциала при достаточно тонких капиллярах всегда
выше вычисленных без введения поправки ча поверхностную проводимость.
Эксперименты подтвердили это положение. На рис. VII, 30 приведены резуль-
^14
тэты опытов И. И. Жукова по определению С-потенциала протекания, вычислен-
ные с учетом (кривые /) и без учета (кривые 2) поверхностной проводимости.
В качестве пористых мембран были взяты диафрагмы из кварца и корунда,
в качестве жидкости — слабый раствор хлорида калия. По характеру кривых
видно, что значения С-потенциала без учета поверхностной проводимости сильно
уменьшаются при переходе к диафрагмам с порами малого сечения, а при вве-
дении поправки иа поверхностную проводимость получаются почти одни и те же-
значения электрокинетического потенциала во всем испытанном диапазоне радиу-
сов капилляров. Следовательно, при определении С-потенциала электроосмоти-
ческим путем нли методом протекания необходимо всегда учитывать радиус пор-
капилляров.
Приведенная выше поправка характеризует поверхностную проводимость
только отдельного капилляра, тогда как на практике в большинстве случаев-
реальными объектами являются поричые тела. Для определения поправки на
Рис. VII, 31. Схема прибора Перрена для электроосмоса:
1 — порошок исследуемого вещества; 2 — электроды; 3 — вороика; 4— кран;
5 — прокалиброванная трубка.
поверхностную проводимость пористых тел измеряют электросопротивление-
мембраны в исследуемом и стандартном растворах и по этим данным вычисляют
величину Ys-
С поверхностной проводимостью связано явление так называемой капилляр-
ной сверхпроводимости. Это явление, открытое Д. А. Фридрихсбергом, заклю-
чается в следующем. Если трубку с проводящим ток раствором перегородить
пористой диафрагмой из непроводящего материала, то электрическое сопротив-
ление системы должно возрасти вследствие того, что часть сечения трубки будет
занята диэлектриком. Однако если у поверхности капилляров диафрагмы обра-
зуется слой, обладающий поверхностной проводимостью, то за счет этой прово-
димости для весьма тонкокапиллярных пористых тел в разбавленных растворах
электролитов может наблюдаться повышение общей проводимости. Будет иметь
место на первый взгляд парадоксальный эффект понижения электрического
сопротивления при введении в трубку диафрагмы из диэлектрика.
Остановимся теперь кратко на приборах, применяемых для определения
С-потенциала по объему электроосмотически перенесенной жидкости. Во всех
приборах этого типа электроосмос проводится не с одиночным капилляром, а с
помощью пористой диафрагмы из твердого вещества.
На рис. VII, 31 приведена схема прибора для электроосмоса, предложенного
еще Перреном. В среднюю часть стеклянной U-образной трубки, присоединяемой
к боковым коленам с помощью шлифов, набивают достаточно плотно порошок 1
исследуемого вещества. В оба боковых колена вставлены платиновые электро-
ды 2, питаемые постоянным током, напряжение которого должно составлять не
менее 40—50В. В правом боковом колене имеется воронка 3 с краном для за-
полнения прибора дисперсионной средой, а в левом колене — кран 4 для слива
215-
жидкости из прибора. Кроме того, к правому колену трубки присоединена ка-
пиллярная горизонтальная заранее прокалиброванная трубка 5, служащая для
наблюдения за перемещением в ней жидкости.
Перед определением прибор заполняют жидкостью так, чтобы в порах диа-
фрагмы 1 не оставалось воздуха и установилось постоянное положение мениска
жидкости в левой части капиллярной трубки 5. После этого включают ток в та-
ком направлении, чтобы мениск в капилляре 5 передвигался слева направо, и
отсчитывают скорость его перемещения по секундомеру. Одновременно с по-
мощью миллиамперметра, включенного в электрическую цепь прибора, измеряют
силу тока. Недостатком прибора является поляризация электродов и образова-
ние продуктов электролиза, которые могут проникать в капилляры диафрагмы
и этим самым вносить ошибку в результаты измерения.
В' более совершенных приборах применяют иеполяризующиеся электроды,
достаточно удаленные от диафрагмы или соединенные с ней с помощью агаро-
вых мостиков. В приборе для электроосмоса, предложенном Уметсу, диафрагма
из порошка располагается в вертикальной стеклянной трубке со стеклянной ре-
шеткой в нижней ее части.
Диафрагму необходимой высоты получают, наполняя трубку суспензией по-
рошка н отсасывая затем жидкость водоструйным насосом. После этого прибор
заполняют раствором, вставляют агаровые мостики для соединения диафрагмы
с неполяризующимися электродами и включают постоянный электрический ток.
Скорость электроосмоса определяют с помощью калиброванного стеклянного
капилляра, при этом начальное положение мениска жидкости устанавливают,
приоткрывая специальный боковой кран.
Определение по электроосмотическому давлению. При этом способе опреде-
ляют не объем жидкости, протекшей при электроосмосе через капилляр или
пористую мембрану в единицу времени, а давление, возникающее в результате
электроосмотического движения жидкости. Для этого то колено электроосмо-
тического прибора, в которое поступает жидкость, должно заканчиваться верти-
кальной или наклонной трубкой, с помощью которой можно установить высоту
столба жидкости, уравновешивающего электроосмотическую силу. Давление этого
столба заставляет жидкость течь через капилляр или пористую мембрану в об-
ратном направлении, пока не установится равновесие между-силой тяжести и
осмотическим давлением.
Уравнение, с помощью которого можно вычислить С-потенциал, выводят,
исходя из того, что в равновесном состоянии объем жидкости Уь поступающей
в отдельный капилляр в единицу времени под влиянием электроосмотической
силы, равен объему жидкости V!t вытекающему за то же время из капилляра
под влиянием гидростатического давления Р. Объем V-, выражается уравне-
нием (VII, 51):
4П
Для Vj, согласно уравнению Пуазейля, можно написать:
где I — длина капилляра.
В равновесном состоянии Vt = V2 и, следовательно:
Откуда
raeg/7 пРг>
4т) 8т]1
2е£/П
№
(VII, 63)
(VII, 64)
Еще Квинке и Видеман нашли, что электроосмотическое давление Р про-
порционально градиенту внешнего потенциала Н и обратно пропорционально г*,
что полностью соответствует уравнению (VII, 64).
216
Из уравнения (VII, 64) легко найти значение С-потеициала по давлению Р
или высоте поднятия жидкости в трубке.
Подобный метод можно применить (в отсутствие поверхностной проводи-
мости) также и при электроосмосе через пористую мембрану. Одиако он не
получил широкого распространения на практике.
Сравнение значений {-потенциала, найденных '
различными методами
Из изложенного выше ясно, что определение электрокинетиче-
ского потенциала сопряжено со многими трудностями, связанными
как с постановкой эксперимента, так и с вычислением {-потенциала
Рис. VII, 32. Зависимость {-потенциала
стенки капилляра из стекла пирекс от
концентрации электролита:
/—данные, вычисленные по результатам элек-
троосмотических определении; 2—данные, по-
лученные при определении потенциалов тече-
ния.
по экспериментальным . дан-
ным. Еще сравнительно недав-
но исследователи не учитыва-
ли влияния поверхностной про-
водимости, электрофоретиче-
ского запаздывания и электри-
ческой релаксации, и поэтому
нз большого числа исследова-
ний в области электрокннети-
ческих явлений лишь немногие
давали достоверные значения
{-потенциала.
Нанлучшим доказатель-
ством точности значений ^-по-
тенциала, определяемых с уче-
том нужных поправок, может
служит)» идентичность его зна-
чений, найденных при различ-
ных электрокииетических яв-
лениях.
Опыты, проведенные Рат-
джерсом, а также Вьигом с
целью сравнения значений {-потенциала, определенного методом
протекания и по электроосмосу, показали одинаковость этих зна-
чений. На рис. VII, 32 предбТавлена зависимость между ^-потен-
циалом стенки стеклянного капилляра и концентрацией раствора
электролита, заполняющего капилляр. Как можно видеть, значения
{-потенциала, вычисленные из электроосмотических данных и из
данных, полученных при определении потенциала протекания, ло-
жатся на одну и ту же кривую. Расхождения данных для KNO3
объясняются другой обработкой капилляра, примененного в этом
опыте.
Эквивалентность значений {-потенциала, найденных с помощью
электрофореза и электроосмоса, также неоднократно проверялась.
В частности, Абрамсон в своих тщательных опытах по исследова-
нию электроосмоса и микроэлектрофореза покрытых белком ча-
стиц при учете всех необходимых поправок установил равенство
217
значений ^-потенциала, найденных обоими способами. Наконец,
^-потенциал, определенный по методу протекания, оказался рав-
ным ^-потенциалу, вычисленному из электрофоретических данных.
Таким образом, хотя экспериментальный материал не очень
велик и не всегда является абсолютно доказательным, тем не ме-
нее можно утверждать, что все три электрокинетические явления
эквивалентны и значения ^-потенциала, определенные этими тремя
методами, одинаковы.
6. ПРАКТИЧЕСКОЕ ЗНАЧЕНИЕ ЭЛЕКТРОКИНЕТИЧЕСКИХ ЯВЛЕНИЙ
Электрокинетические явления имеют большое практическое
значение. Используя их, можно определять весьма важную харак-
теристику дисперсных систем — ^-потенциал, а с помощью электро-
фореза можно разделять на фракции и характеризовать такие
сложные смеси, какими являются природные белки и другие вы-
сокомолекулярные электролиты.
Электрокинетические явления широко используются не только
при научных исследованиях, но и в технике. В частности, электро-
форез применяют для нанесения тонкого слоя частиц коллоидных
размеров на поверхность проводящего материала. Этим способом
лолучают весьма однородные покрытия, толщину которых легко
регулировать. Электроотложение можно проводить в таких сре-
дах, как спирт, ацетон и других, что исключает выделение газов
на электродах даже при большой силе тока и малой электропро-
водности жидкости. Для нанесения токопроводящих покрытий
электрофорез используют при производстве изолированных нагре-
вательных спиралей и активированных катодов для радиоламп,
представляющих собой металлическую проволоку, покрытую тон-
ким слоем окисла щелочноземельного металла.
Электрофорез применяют также для покрытия металлических
деталей каучуком путем отложения на их поверхности частиц кау-
чука, содержащихся в латексах (водных дисперсиях каучука).
В этом процессе отрицательно заряженные частицы латекса дви-
жутся к аноду, которым служит подлежащий покрытию предмет,
и осаждаются на нем, образуя более или менее толстую пленку.
В латекс предварительно можно вводить усиливающие каучук на-
полнители и вулканизирующие агенты. Благодаря этому на дета-
лях получают резиновые покрытия, обладающие высоким каче-
ством.
Б. В. Дерягиным, С. С. Духиным и А. А. Коротковой был раскрыт механизм
образования латексных пленок при их получении- с помощью так называемого
«ионного отложения», широко применяющегося в технологии переработки ла-
тексов. Этот способ сводится к тому, что на форму, обычно стеклянную, фарфо-
ровую или металлическую, наносят слой фиксатора, содержащего соль с поли-
валентным катионом, способный астабилизовать обычно отрицательно заряжен-
ные частицы латекса; форму погружают в латекс, в результате чего на ией
образуется латексная пленка нужной формы.
Отложение пленок латекса на форме обычно объясняется желатинированием
латекса под действием ионов, диффундирующих из слоя фиксатора в глубь
218
латекса. Однако остается неясным, почему в ходе процесса значительно повы-
шается концентрация латекса у поверхности формы. Авторы указывают, что
источником сил, способных перемещать частицы латекса по направлению к фор-
ме, являются градиенты электрического и химического потенциалов, сопровож-
дающие диффузию ионов содержащегося в фиксаторе электролита в глубь рас-
твора. Иначе говоря, движение взвешенных частиц объясняется действием
электрического поля, образуемого при диффузии электролита.
Поскольку С-потенциал глобул латекса обычно отрицателен, то для хлорида
кальция, который обычно применяют при ионном отложении н для которого
коэффициент диффузии положительного нона меньше коэффициента диффузии
отрицательного, движение латексных частиц всегда направлено к форме. Само
явление переноса заряженных частиц под действием электрического поля, обра-
зуемого при диффузии электролита, как уже было указано, авторы назвали
диффузиофорезом.
О значении электрофореза для разрушения аэрозолей сказано
в гл. XI.
Электроосмос используют при обезвоживании пористых мате-
риалов. Для этого влажную массу помещают между электродами,
при этом вода в образовавшемся электрическом поле передви-
гается к одному из них, обычно катоду, где она и стекает. Элек-
троосмос можно применять также для обезвоживания (в комбина-
ции с давлением) при фильтровании. Фильтр-прессы, применяемые-
для такого обезвоживания, называются в технике электроосмо-
тическими фильтр-прессами. Было много попыток использовать-
электроосмос для обезвоживания торфа, однако до сих пор эта
проблема еще не решена из-за сравнительно большого расхода
энергии.
Интересно, хотя пока практически и не осуществлено, предло-
жение Квинке об использовании потенциала течения для получе-
ния электрической энергии.
7. НЕКОТОРЫЕ ДРУГИЕ ЭЛЕКТРИЧЕСКИЕ СВОЙСТВА
КОЛЛОИДНЫХ СИСТЕМ
В этом разделе кратко рассмотрим электропроводность и ди-
электрическую проницаемость лиозолей, так как эти свойства
тесно связаны с электрокинетическими явлениями, устойчивостью-
и коагуляцией коллоидных систем.
Электропроводность лиозолей. Электропроводность лиозоля
слагается из электропроводности, обусловленной коллоидными ча-
стицами, и электропроводности ионов, присутствующих в системе.
Поэтому электропроводность коллоидного раствора зависит от за-
ряда, числа и подвижности коллоидных частиц и ионов, находя-
щихся в золе.
Нетрудно представить себе соотношение между электропроводностью, обус-
ловленной коллоидными частицами, и электропроводностью за счет ионов, при-
сутствующих в золе. Примем, что в золе содержится 1 объемн.% дисперсией
фазы, радиус частиц г = 50 А и С-потеициал составляет 100 мВ в дисперсиенней
среде, для которой х = 105. Для заряда частиц, по аналогии с уравнением
(VII, 31), можно написать уравнение:
Q = reg (I + хг) (VII, 65>
219
Если ие учитывать электрическую релаксацию, то электрофоретическую по*
движность частиц можно выразить соотношением:
«о
eg
бЯТ)
Число частиц v, содержащихси в единице объема коллоидного раствора, со-
ставляет:
v =
0,01
4/з№
Тогда удельная электропроводность уИОлл,
цами, будет равна:
г, . 0,01
Yk<wi=Qvu0 = reg(1 + кг} -^—3
обусловленная коллоидными части-
eg в 0,01еаеа(1Я-хг)
бят] ™ 8я!Г!Т]
(VII, 66)
Если в последнее уравнение подставить принятые выше значения к и г,
значения а и г] для воды, а удельную электропроводность выразить в практи-
ческих единицах, то получим:
Уколл 4 • 10“5 Ом-1 • см-1
Электропроводность же дисперсионной среды, для которой к — 105, состав-
ляет примерно 10~® Ом~1-см~|. Таким образом, электропроводность, обусловлен-
ная присутствием в золе коллоидных частиц, вполне измерима, если только ча-
стицы не слишком велики и концентрация золя не слишком мала.
Паули, а затем и другие исследователи измеряли электропро-
водность гидрозолей с помощью кондуктометрического титрования
Рис. VII, 33. Результаты
кондуктометрического ти-
трования золя сульфида
мышьяка:
/ — кривая титрования с КОН;
2—кривая титрования с ВаС12.
для определения ионных компонентов
системы. С помощью кондуктометриче-
ского титрования, как показали А. И. Ра-
бинович и его сотр., не только можно
определить число противоионов, а сле-
довательно, и заряд коллоидных частиц,
но также получить сведения относитель-
но локализации и фиксации противо-
ионов в двойном слое. Поясним это на
следующем примере.
На рис. VII, 33 в виде схематических
кривых приведены результаты кондукто-
метрического титрования растворами
гидроокиси калия и хлорида бария гид-
розоля сульфида мышьяка, для которого
потенциалопределяющим ионом является ион HS-, а противоио-
ном — ион Н+. Как можно видеть, кривая 1 титрования с помрщью
КОН имеет минимум. Существование этого минимума объясняется
следующим образом. При добавлении первых порций щелочи про-
исходит замещение в адсорбционном слое мицеллы противоиона
Н+ на ион К+, и это приводит к образованию из ионов Н+ и OIH
почти неионизированной молекулы Н2О. В результате уменьшения
содержания ионов водорода, обладающих исключительно высокой
подвижностью, электропроводность системы падает. -Однако после
замещения всех ионов водорода при дальнейшем добавлении ще-
лочи электропроводность золя увеличивается. По количеству ще*
220
лочи, отвечающему минимуму на кривой, можно судить о количе-
стве в золе связанных ионов водорода.
Кривая 2 титрования раствором ВаС1г не имеет максимума и
состоит из криволинейного и прямолинейного участков. Так как
при титровании ВаС1г вода не образуется, кривая не может иметь
минимума, и по мере титрования происходит постепенное увеличе-
ние электропроводности. Благодаря большей подвижности ионов
Н+ по сравнению с ионами Ва2+ электропроводность золя в начале
титрования растет больше, чем это соответствует количеству до-
бавленного электролита, что и обусловливает характерную форму
этого участка кривой. Точка перехода криволинейного участка
в прямолинейный, соответствующая минимуму на кривой титрова-
ния щелочью, очевидно, отвечает полному замещению ионов во-
дорода ионами бария. Дальнейшее введение раствора хлорида ба-
рия, конечно, может приводить только к линейному увеличению
электропроводности.
При титровании как КОН, так и BaCh получаются в общем
совпадающие результаты. Ввиду большого адсорбционного потен-
циала ионов бария из мицеллы вытесняются все ионы Н+, находя-
щиеся в адсорбционном слое.
При работе с другими гидрозолями и электролитами результаты
могут быть иными. Так, опыты показали, что при титровании золя
мастики электропроводность с самого начала прямо пропорцио-
нальна количеству добавленного электролита. Прямолинейная
форма графика кондуктометрического титрования указывает на то,
что в этом случае не происходит вытеснения иона водорода из
адсорбционного слоя.
Таким образом, путем кондуктометрического титрования можно
установить характер сил, за счет которых противоионы удержи-
ваются в адсорбционном слое. Если результаты титрования•ука-
зывают на способность к обмену всех противоионов мицеллы, т. е.
свидетельствуют об их способности вытесняться как из диффуз-
ного, так и адсорбционного слоя, то противоионы удерживаются
в мицелле только в результате действия физических сил (электри-
ческих, адсорбционных). Если, наоборот, часть противоионов из
двойного слоя не вытесняется или вытесняется с большим трудом,
то следует полагать, что эти ионы удерживаются в мицелле си-
лами, близкими по своей природе к химическим силам.
Высокая частота и высокое напряжение тока влияют на электропроводность
гидрозолей аналогично тому, как они влияют на электропроводность обычных
электролитов. Однако для коллоидных систем это влияние сказывается более
резко, поскольку оно связано с электрофоретическим запаздыванием н электри-
ческой релаксацией, эффект которых проявляется особенно сильно у частиц кол-
лоидных размеров.
Прн измерении электропроводности в поле высокой частоты колебания ча-
стиц настолько быстры, что полная асимметрия двойного слоя не успевает раз-
виться. В результате этого эффект релаксации уменьшается, а электропровод-
ность соответственно возрастает.
В поле очень высокого напряжения (105 В/см) скорость частиц настолько
увеличивается, что они вырываются из окружающей их атмосферы ионов.
221
Следствием этого является исчезновение эффекта электрофоретического тормо-
жения в электрической релаксации.
Диэлектрическая проницаемость лиозолей. Диэлектрическая проницаемость
лиозолей отличается от диэлектрической проницаемости дисперсионной среды.
Это объясняется следующим.
1. Частицы золя имеют обычно другую и, как правило, меньшую диэлектри-
ческую проницаемость, чем дисперсионная среда, в результате чего общая ди-
электрическая проницаемость золя снижается. Этот эффект, очевидно, является
простым объемным эффектом.
2. Частицы золя могут иметь постоянный дипольный момент. При определе-
нии диэлектрической проницаемости в электрическом поле диполи ориенти-
руются, вследствие чего увеличивается поляризация и возрастает значение е.
Этот эффект обычно незначителен у типичных коллоидных систем, частицы ко-
торых, как правило, не имеют постоянного дипольного момента. У растворов
же высокомолекулярных соединений, молекулы которых могут обладать по-
стоянным дипольным моменотм, этот эффект может приводить к значительному
увеличению диэлектрической проницаемости
3. Частицы золя могут приобретать дипольные моменты, противоположно
направленные внешнему электрическому полю вследствие деформации двойного
электрического слоя в этом поле. Очевидно, при этом центры тяжести положи-
тельных и отрицательных зарядов частицы смещаются относительно друг друга,
т. е. частицы поляризуются, что приводит к возрастанию диэлектрической прони-
цаемости. Подобный эффект характерен для всех коллоидных систем и раство-
ров высокомолекулярных электролитов.
4. Частицы золя могут гидратироваться или, в общем случае, сольватиро-
ваться Молекулы среды в сольватном слое адсорбированы и ориентированы
под действием значительных адсорбционных сил. Такая фиксация молекул среды
приводит к уменьшению поляризации, а следовательно, и диэлектрической про-
ницаемости системы
Из-за электропроводности лиозолей диэлектрическую проницаемость изме-
рять необходимо с помощью переменного тока. Однако при этом надо помнить,
что полученные значения зависят от частоты переменного тока. При не слишком
больших частотах значение диэлектрической проницаемости не отличается су-
щественно от тех значений, которые можно было бы найти в статическом поле,
так как частицы успевают полностью поляризоваться в промежуток времени
меньший, чем продолжительность одного колебания поля. Однако при больших
частотах последнее условие уже не выполняется, и в растворе обнаруживается
дисперсия (рассеяние) диэлектрической проницаемости, характер которой зави-
сит от того, какой фактор обусловливает ее особенности для дайной системы.
В первом случае, когда отклонение диэлектрической проницаемости об-
условлено простым объемным эффектом, дисперсии этой величины не наблю-
дается Во втором случае дисперсия происходит при такой частоте, когда ди-
поли уже не могут следовать за изменением направления поля. В третьем случае
дисперсия наблюдается при частоте, уже не вызывающей асимметрии двойного
слоя, т. е. при частоте, отвечающей увеличению электропроводности. Что ка-
сается того, влияет ли на дисперсию сольватация частиц, то этот вопрос до сих
пор неясен. Имеющиеся экспериментальные данные об увеличении диэлектриче-
ской проницаемости растворов желатина и агара с возрастанием частоты можно
объяснить ие только изменением гидратации макромолекул, но и действием
ряда других факторов — влиянием частоты на двойной слой, иа поведение по-
стоянных диполей и т. д.
ГЛАВА VIII
ПОЛУЧЕНИЕ И ОЧИСТКА КОЛЛОИДНЫХ СИСТЕМ.
СТРОЕНИЕ КОЛЛОИДНЫХ МИЦЕЛЛ
Как известно, золи по размеру частиц дисперсной фазы зани-
мают промежуточное положение между истинными растворами и
суспензиями, поэтому, естественно, они могут быть получены либо
путем соединения отдельных молекул или ионов растворенного ве-
щества в агрегаты, либо в результате диспергирования сравни-
тельно больших частиц. В соответствии с этим Сведберг делит
методы синтеза коллоидных систем на конденсационные и
диспергационные. Особо от этих методов стоит метод пеп-
тизации, который заключается в переводе в коллоидный рас-
твор осадков, первичные частицы которых уже имеют коллоидные
размеры. Наконец, в некоторых случах коллоидные системы могут
образоваться путем самопроизвольного диспергиро-
вания дисперсной фазы в дисперсионной среде.
Основными двумя условиями получения коллоидных систем,
независимо от применяемых методов синтеза, являются нераство-
римость или достаточно малая растворимость дисперсной фазы
в дисперсионной среде и наличие в системе, в которой образуются
частицы, веществ, способных стабилизировать эти частицы, а в
случае конденсационных методов и замедлять (приостанавливать)
их рост. Такими веществами могут быть как чужеродные вещества,
специально вводимые в систему, так и соединения, образующиеся
при взаимодействии дисперсной фазы с дисперсионной средой.
1. МЕТОДЫ ПОЛУЧЕНИЯ КОЛЛОИДНЫХ СИСТЕМ
Конденсационный метод. Обычно считается, что образование
коллоидных систем в результате конденсации является не чем
иным, как процессом кристаллизации, а образовавшиеся частицы
представляют собой мельчайшие кристаллики. Таких взглядов
придерживался, наприм'ер, русский ученый П. П. Веймарн, один
из первых исследователей, детально изучавших конденсационные
методы образования лиозолей.
Образование обычных кристалликов протекает в две стадии:
1) возникновение зародышей (центров кристаллизации) в пе-
ресыщенном растворе, причем пересыщение может быть вызвано
223
химической реакцией, приводящей к получению малорастворимого
соединения, уменьшением растворимости соединения при замене
лучшего растворителя худшим, охлаждением раствора и другими
причинами;
2) рост зародышей, что приводит к образованию достаточно
крупных кристаллов.
Долгое время принимали, что образование зародышей проис-
ходит самопроизвольно (спонтанно). Такую точку зрения развивал
Тамман. Он считал, что в некоторых участках пересыщенного рас-
твора, находящегося в метастабильном состоянии, молекулы или
ионы растворенного вещества сами по себе без участия каких-ни-
будь посторонних взвешенных частиц могут располагаться в кри-
сталлическом порядке, образуя мельчайшие зародыши, способные
к дальнейшему росту.
Согласно этой точке зрения скорость образования зароды-
шей «1 пропорциональна относительному пересыщению и может
быть выражена уравнением:
с?~Ся (VIII,!)
св
где k — коэффициент пропорциональности; св — концентрация пересыщенного
раствора; ся — концентрация насыщенного раствора (обычная растворимость ве-
щества).
Разность сп — Сн представляет собой избыток вещества, спо-
собного образовывать кристаллы, и, следовательно, в известной
степени может служить мерой скорости выделения вещества из
раствора. Величина сн характеризует взаимодействие растворен-
ного вещества с растворителем и, таким образом, -является мерой
сопротивления выделению вещества из раствора. Считают, что ^ем
больше разность сп — си и меньше величина сн, тем скорее обра-
зуются зародыши, тем больше возникает центров кристаллизации
и тем меньше по размеру окажутся коллоидные частицы, так как
все количество способного выделиться вещества распределится ме-
жду большим числом образовавшихся центров кристаллизации.
Однако уже довольно скоро было экспериментально установ-
лено, что зародыши кристаллизации образуются, как правило, не
путем удачного столкновения молекул или ионов в растворе вслед-
ствие флуктуаций концентрации, а в результате осаждения рас-
творенного вещества на чужеродных мельчайших пылинках, слу-
чайно оказавшихся в системе. Например, было найдено, что в рас- г
творах, тщательно очищенных от посторонних взвешенных частиц,
в течение долгого времени даже при значительном пересыщении
не образуется кристаллов. Наоборот, при введении в такие рас- !
творы чужеродных зародышей или кристаллов растворенного ве- »
щества в них немедленно начиналась кристаллизация. Все это *,
привело к тому, что стали считать вероятным образование зароды- J
шей (кристаллизационных центров) на уже готовых поверхностях '
раздела, j
224 1
Теория образования новой дисперсной фазы зародилась в ис-
следованиях Гиббса (1878 г.) по термодинамике поверхностных
явлений и получила развитие в двадцатых годах нашего столетия
(школа Фольмера) в экспериментальных и теоретических исследо-
ваниях конденсации пересыщенного пара. Взгляды Фольмера на
образование зародышей в пересыщенном паре детально рассмо-
трены в гл. XI, посвященной аэрозолям. Здесь же лишь укажем,
что растворимость или давление насыщенного пара малых частиц
любой фазы, как это следует из термодинамики, больше, чем
у крупных частиц (закон В. Томсона). Иначе говоря, увеличение
дисперсности фазы повышает ее растворимость в окружающей
среде, или способность вещества к выходу из данной фазы. По-
этому раствор, насыщенный относительно крупных кристаллов,
является еще ненасыщенным относительно мелких кристаллов того
же вещества. В таких условиях самопроизвольное образование
достаточно крупных кристаллических зародышей мало вероятно,
а очень мелкие зародыши, возникающие в результате флуктуаций,
не могут вызвать кристаллизацию, так как по отношению к ним
раствор не является пересыщенным. Очевидно, зародыши новой
фазы могут образовываться лишь при очень высоких степенях пе-
ресыщения, когда возникновение сравнительно больших зароды-
шей статистически более вероятно.
Все только что изложенное можно объяснить и несколько
иначе. Процесс образования микрокристаллических коллоидных ча-
стиц представляет собой не что иное, как переход метастабильной
фазы в стабильную, сопровождающийся уменьшением свободной
энергии системы. Этот процесс самопроизвольный, за исключе-
нием стадии образования зародышей. Для того чтобы метаста-
бильная фаза перешла в стабильную, необходимо образование до-
статочного числа зародышей. Однако для возникновения большего
числа зародышей затрачивается энергия на создание новой по-
верхности раздела фаз — стабильной и метастабильной *. По-
скольку процесс перехода метастабильной фазы в стабильную на
первой зародышевой стадии всегда сопровождается не уменьше-
нием, а увеличением свободной энергии вследствие образования
новой поверхности, он не может происходить самопроизвольно до
тех пор, пока зародыши, образующиеся в системе, не достигнут
определенного размера. После этого переход совершается само-
произвольно.
Совершенно иначе обстоит дело, если в системе присутствуют
чужеродные зародыши или в нее введены кристаллики стабильной
* Метастабильным состоянием в термодинамике называют такое состояние
системы, которое не отвечает устойчивому равновесию в данных условиях, но
сохраняется во времени. Таково, например, состояние пересыщенного пара или
раствора, переохлажденной илн перегретой жидкости, малоустойчивой кристал-
лической модификации. МетастабилЬиое состояние способно переходить под
влиянием сравнительно слабых внешних воздействий или при внесений в систему
зародышей более устойчивой фазы в устойчивое, стабильное состояние.
8 Зак, 664
225
фазы, полученные отдельно. При этом образование новой фазы
облегчено наличием в системе межфазной поверхности, на кото-
рой процессы конденсации протекают особенно легко.
Следует отметить, что введение чужеродных зародышей лежит
в основе получения золей с заранее заданной дисперсностью, так
как в пересыщенный раствор можно вводить любое число посто-
ронних зародышей, между которыми и распределяется вещество,
выделяющееся в виде кристаллической фазы. Более подробно об
этом сказано далее, при описании методов синтеза отдельных кол-
лоидных систем.
Из изложенного следует, что ценность формулы (VIII, 1) весьма
ограниченна. Тем не менее она указывает путь регулирования дис-
персности при образовании коллоидной системы. Для повышения
степени дисперсности, согласно этой формуле, необходимо увели-
чить относительное пересыщение, т. е. увеличить значение са или
уменьшить сн, или, наконец, одновременно увеличить сп и пони-
зить св.
Рассмотрим вторую стадию образования коллоидной системы —
рост зародышей кристаллизации в результате отложения на них
вещества из пересыщенного раствора. Для объяснения роста кри-
сталлов в разное время было предложено много теорий.
Гиббс, Кюри, а впоследствии русский ученый Г. В. Вульф при
интерпретации явлений, связанных с ростом кристаллов, исходили
из связи между формой кристалла и поверхностной энергией всех
его граней. Согласно диффузионным теориям процесс образования
кристаллической грани протекает с бесконечно большой скоростью
и поэтому зависит только от скорости подвода вещества к кри-
сталлу из раствора, т. е. от скорости диффузии. В двадцатых го-
дах нынешнего столетия для объяснения роста кристаллов Фоль-
мер предложил адсорбционную теорию, согласно которой частицы
кристаллизующегося вещества при достижении поверхности обра-
зуют своеобразный адсорбционный слой — двумерное кристалли-
ческое образование, присоединяющееся затем к грани кристалла.
Странский считает вероятным возможность образования на расту-
щем кристалле ионных рядов или слоев, сходных с двумерными
кристаллическими образованиями Фольмера.
Если принять диффузионный механизм роста кристаллов, то
скорость их роста м2 можно представить уравнением:
«2 = ^(сп-сн) (VIII, 2)'
где D — коэффициент диффузии; s — поверхность кристалла; 6 — толщина слоя
раствора, через который происходит диффузия (в этом слое концентрация ве-
щества растет от сн на поверхности до сп в объеме пересыщенного раствора).
В результате роста зародыша степень пересыщения раствора
уменьшается за счет перехода растворенного вещества в кристал-.
лическую фазу и одновременно снижается растворимость частиц
из-за увеличения их размеров. Уравнение (VIII, 2) только в пер*
226
вом приближении описывает рост кристалликов и далеко не для
всех условий. Часто связь между скоростью роста и степенью пе-
ресыщения весьма сложна, многие детали процесса еще не ясны.
Есть основания полагать, например, что в некоторых случаях ско-
рость роста кристалликов все же определяется не диффузией,
а скоростью отложения молекул вещества на гранях кристалла.
Помимо этого надо помнить, что процесс роста относительно хо-
рошо изучен только для крупных кристаллов с хорошо сформиро-
ванными гранями. В процессе же образования мельчайших кол-
лоидных частиц существенны только начальные стадии роста.
При получении коллоидной системы скорость образования за-
родышей «1 должна быть велика, а скорость роста кристаллика «2
мала, так как лишь в этом случае образуется множество кристал-
ликов, каждый из которых соответствует коллоидным размерам.
Наоборот, если скорость ut мала, а скорость «2 велика, то все вы-
делившееся вещество отложится на небольшом числе зародышей и
в результате образуется сравнительно небольшое количество круп-
ных кристаллов.
Важно отметить, что в первом случае будут образовываться
сравнительно монодисперсные золи, а во втором — полидисперс-
ные. Действительно, при малой скорости образования зародышей
и большой скорости их роста в начале процесса образования золей
возникает небольшое число зародышей, которые -к концу процесса
вырастут до кристалликов больших размеров, в то время как кри-
сталлики, образующиеся на зародышах, возникших в конце про-
цесса, останутся маленькими.
Очень существенное значение для получения коллоидных систем имеет кон-
центрация реагирующих растворов. В результате химических реакций, приводя-
щих к образованию плохо растворимых веществ, при малых концентрациях ре-
агирующих веществ получаются золи, при больших концентрациях — осадки и
прн весьма больших концентрациях — гели. Это хорошо можно проследить иа
примере реакции желтой кровяной соли K«[Fe(CN)6] и хлорида железа FeCl3,
в результате которой образуется берлинская лазурь Fe,[Fe(CN)6]3. Если быстро
смешать в эквивалентных количествах концентрированные растворы хлорида
железа и желтой кровяной соли, то берлинская лазурь выделяется в виде гу-
стого геля. Небольшое количество этого геля при размешивании в большом
объеме воды дает стойкий золь Если вместо концентрированных растворов
исходных веществ взять 10-кратио разбавленные растворы, то в результате
реакции образуется осадок, ие способный переходить в золь, сколько бы его ие
размешивали. Наконец, если растворы хлорида железа и желтой кровяной соли
разбавить очень сильно и затем смешать, то получится устойчивый золь берлин-
ской лазури.
Все эти явления можно объяснить следующим образом. В сильно разбав-
ленных растворах скорость кристаллизации невелика, так как для роста кри-
сталликов к ним сначала должно продиффундировать растворенное вещество.
В этих случаях за время, нужное для перехода всего вещества в кристалличе-
скую фазу, образуется достаточно большое число зародышей. В очень концен-
трированных растворах скорость образования зародышей велика потому, что
кристаллизация ие может привести к 'быстрому использованию большого коли-
чества вещества и прекращению образования новых зародышей. В растворах
средней концентрации скорости возникновения зародышей и кристаллизации при-
мерно одинаковы, в результате чего и происходит образование относительно
больших частиц.
8*
227
Вводя в систему во время образования коллоидного раствора
различные специально подобранные вещества, препятствующие
возникновению зародышей или тормозящие их рост, Хиге еще
в 1914 г. изучил раздельно роль скорости возникновения центров
кристаллизации и скорости укрупнения микрокристалликов. Ока-
залось, что к веществам, предотвращающим или замедляющим
образование зародышей, относятся Кз[Ре(СМ)6] и K4[Fe(CN)6], а
к веществам, тормозящим рост кристалликов, — КВг и KI. При
введении первых веществ кристаллики из пересыщенного раствора
совсем не образуются, однако процесс кристаллизации легко мож-
но вызвать, если в систему ввести чужеродные зародыши. При до-
бавлении веществ, тормозящих рост кристалликов, можно полу-
чить золи с мельчайшими, субмикроскопическими частицами.
Действие веществ, препятствующих возникновению зародышей,
вероятно, объясняется чисто химическими явлениями, происходя-
щими в растворе. Действие веществ,- задерживающих рост заро-
дышей, вызвано адсорбцией их на зародышевых кристалликах и
образованием на поверхности кристалликов тончайшего чужерод-
ного слоя, препятствующего дальнейшей достройке кристаллика.
Это объяснение подтверждается тем, что зависимость скорости
роста кристалликов от равновесной концентрации вещества, пре-
пятствующего росту, может быть выражена уравнением, по виду
сходным с известным адсорбционным уравнением Фрейндлиха.
Интересно, что под влиянием специальных добавок может по-
разному меняться скорость роста отдельных граней кристалликов.
Так, при введении метилового фиолетового в процессе получения
золя иодида серебра коренным образом меняется форма кристал-
ликов Agl. Это объясняется, очевидно, тем, что молекулы мети-
лового фиолетового адсорбируются преимущественно на определен-
ных гранях кристаллика (на гранях с наибольшей поверхностной
энергией), что и тормозит рост кристалла в направлении, перпен-
дикулярном данной грани.
Наряду с теорией, рассматривающей образование коллоидных
систем как процесс кристаллизации, уже сравнительно давно су-
ществовали взгляды, согласно которым при быстром осаждении
вещества из раствора могут быть получены коллоидные системы
с аморфными частицами, лишь впоследствии приобретающими
кристаллическое строение. Например, Габер еще в 1-922 г. считал,
что характер новой фазы зависит от скорости двух процессов —
скорости упорядочения и скорости агрегирования молекул. Если
скорости первого процесса больше, то могут получаться кристал-
лические частицы. Если, наоборот, быстрее протекает второй про-
цесс, то возникает аморфная фаза. Аналогично высказывался и
А. В. Думанский.
Детальные исследования В. А. Каргина и 3. Я. Берестневой по-
казали, что образование аморфных частиц при синтезе коллоид-
ных систем чаще является правилом, чем исключением. Эти ис-
следования, сводившиеся к прямому наблюдению образовавшихся
228
частиц с помощью электронного микроскопа, позволили устано-
вить, что при получении золей двуокиси тцтана, двуокиси крем-
ния, сульфида мышьяка, гидроокиси алюминия, пятиокиси ванадия
и золота сначала возникают круглые или бесформенные аморфные
частицы. На рис. VIII, 1а приведена электронная микрофотогра-
фия частиц свежеполученного золя двуокиси титана. Как можно
видеть, частицы имеют размер от 0,1 до 0,8 мкм и образуют це-
почечные агрегаты. Аморфность этих частиц видна из рис. VIII,2а,
представляющего собой электронограмму того же препарата.
Рис. VIII, 1. Электронные микрофотографии частиц золя двуокиси
титана:
а—свежеприготовленны* золь; б —Золь через 1—2 ч после приготовления.
Однако спустя некоторое время после получения внутри частиц
всех вышеперечисленных золей происходят явления упорядочения
и кристаллизации, приводящие к распадению частиц на отдельные
кристаллики. В качестве примера на рис. VIII, 16 приведена элек-
тронная микрофотография препарата, изготовленного из постарев-
шего золя двуокиси титана. Как можно видеть, внутри первона-
чальных аморфных сфероидальных частиц образовалось множе-
ство мелких кристаллических по форме частиц. Одновременно на
электронограммах, как это показано на рис. VIII, 26, появились
кольца из точечных рефлексов, что подтверждает кристаллическую
структуру образовавшихся мелких частиц.
Продолжительность существования аморфных частиц весьма
различна для разных коллоидных систем. Так, кристаллизация
частиц золя золота происходит через 3—5 мин после его приго-
товления, золя пятиокиси ванадия — через 1ч, золя двуокиси ти-
тана— через 1—2 -ч, золя гидроокиси алюминия — через сутки,
.229
золя кремневой кислоты — через 2 года. Можно предполагать, что
скорость кристаллизации определяется быстротой образования
упорядоченных участков и их числом.
Очень сильное влияние на скорость кристаллизации первона-
чально образовавшихся аморфных частиц оказывает температура.
Например, частицы золей гидроокиси алюминия и двуокиси ти-
тана, приготовленных при 80—90 °C, сразу дают на электроно-
граммах картину, характерную для мелкокристаллических частиц.
Очевидно, у этих золей процесс кристаллизации при повышенных
Рис. VIII, 2. Электронограммы частиц золя двуокиси титана:
а— свежеприготовленный золь; 6х— золь через 1-42 ч после приготовления*
температурах протекает настолько быстро, что практически невоз-
можно получить электронограмму первоначально образующихся
аморфных частиц.
Дальнейший процесс старения перечисленных коллоидных си-
стем сопровождается как кристаллизацией частиц, так и образо-
ванием агрегатов, чаще всего в виде цепочечных или сетчатых
структур. Возникновение таких структур свидетельствует о неод-
нородности коллоидных частиц. Но даже и первичные сферические
аморфные частицы образуют цепочки. Это свидетельствует о том,
что и на шарообразных частицах имеются более активные участки,
по которым происходит их слипание. -
В. А. Каргин -и 3. Я. Берестнева на основании наблюдавшихся
ими явлений считают, что возникновение в растворах кристалли-
ческих образований в качестве первого акта синтеза коллоидных
систем маловероятно, так как трудно допустить, что при соударе-
нии молекул, атомов или ионов они соединялись сразу в порядке,
соответствующем их кристаллической решетке, тем более, что
коллоидные системы получаются из сильно пересыщенных рас-
творов. Более вероятно, что в пересыщенном растворе каждое со-
230
ударение приводит к слипанию частиц. Поскольку соударения ме-
жду молекулами, атомами или ионами равновероятны во всех
направлениях, первичные частицы новой фазы имеют обычно ша-
рообразную форму и аморфную структуру. Различная дисперс-
ность образовавшихся коллоидных частиц обусловливается, по-ви-
димому, теми же факторами, что и различная скорость роста
отдельных плоскостей кристаллов в присутствии примесей, кото-
рые могут либо ускорять, либо замедлять рост.
Вследствие захвата молекул дисперсионной среды при образо-
вании аморфных частиц и рыхлости их упаковки создаются усло-
вия, при которых молекулы, атомы или ионы, вошедшие в состав
аморфной частицы, сохраняют достаточную подвижность внутри
этих частиц. Так как образовавшаяся система неравновесна, ча-
стицы будут кристаллизоваться, что приведет к уменьшению сво-
бодной энергии системы. Благодаря возникновению кристалличе-
ских образований внутри аморфной частицы в ней создаются
напряжения, и частица распадается на множество отдельных мел-
ких, но уже кристаллических частичек. Таким образом, размер
•образовавшихся кристаллических частиц связан не с условием
роста их из раствора, как предполагалось ранее, а с кристаллиза-
цией при распаде первичных аморфных частиц. Весьма вероятно,
что описанный механизм образования новой кристаллической фазы
в коллоидных системах имеет очень широкое распространение.
Точку зрения В. А. Каргина и 3. Я. Берестневой на механизм
образования коллоидных систем подтвердила М. Ф. Талина, изу-
чавшая получение золей гидрата окиси железа по методу Грэма
путем взаимодействия карбоната аммония с хлоридом железа.
Однако ее электронно-микроскопические и рентгенографические
исследования частиц золя Fe(OH)3, полученного с помощью гид-
ролиза, показали иную картину — коллоидные частицы в этом слу-
чае имели аморфное строение.
Таким образом, из вышеизложенного напрашивается вывод, что
образование частиц в золях может происходить по различным ме-
ханизмам.
В рассмотренных выше теориях образования коллоидных си-
стем в результате процессов конденсации основное внимание
уделялось образованию достаточно малых частиц как условию,
обеспечивающему коллоидной системе седиментационную устойчи-
вость. Однако для получения длительно существующих коллоид-
пых систем одного этого условия недостаточно, системе необхо-
димо еще придать агрегативную устойчивость. На это особое
внимание обратил еще Н. П. Песков. Он правильно указал на не-
состоятельность взглядов некоторых исследователей, считавших,'
что для получения золя необходимо лишь раздробить дисперсную
фазу в дисперсионной среде до частиц, отвечающих коллоидным
размерам.
Мы уже знаем, что агрегативная устойчивость коллоидных
систем может определяться одноименным зарядом коллоидных
231
i
частиц вследствие избирательной адсорбции на их поверхности од-
ного из ионов присутствующего в системе стабилизующего элек-
тролита. Существует и ряд других объяснений агрегативной устой-
чивости. Однако, каковы бы ни были взгляды на причины агрега-
тивной устойчивости гидрофобных коллоидных систем, все они
предусматривают адсорбцию присутствующего в системе стабили-
затора на поверхности частиц, что и обусловливает взаимодей-
ствие между дисперсной фазой и инертной к этой фазе дисперси-
онной средой. Более подробно вопрос о стабилизации коллоидных
частиц рассмотрен в разделе этой главы, посвященном строению
мицелл различных золей, а также в гл. IX.
Одной из разновидностей метода конденсации является поли-
меризация, с помощью которой получают синтетические латексы,
представляющие собой водные дисперсии полимеров, т. е. типич-
ные коллоидные системы. Однако рассматривать механизм полу-
чения синтетических латексов мы здесь не будем: этот вопрос
освещен в курсах химии и физики полимеров.
Диспергационный метод. Прежде чем говорить о получении
коллоидных систем путем диспергцров'ания грубых частиц, необ-
ходимо хотя бы кратко рассмотреть сущность процесса дисперги-
рования вообще
Диспергированием называют такое измельчение твердых
или жидких тел в инертной (не взаимодействующей с измельчае-
мым веществом) среде, при котором, резко повышается дисперс-
ность и образуется дисперсная система, обладающая значительной
удельной межфазной поверхностью. В противоположность раство-
рению диспергирование происходит, как правило, не самопроиз-
вольно, а с затратой внешней работы, расходуемой на преодоле-
ние межмолекулярных сил при дроблении вещества. >
Процесс диспергирования имеет большое практическое значе-
ние в ряде производств и технологических процессов: при полу-
чении высокодисперсных порошков, служащих активными напол-
нителями для полимеров и пигментами для красок, при изготовле-
нии суспензии графита для смазок, при измельчении руд полезных
ископаемых перед их обогащением, при изготовлении муки и дру-
гих пищевых продуктов и т. д.
В результате изучения механизма диспергирования твердых
тел было установлено, что при деформации твердого тела на его
поверхности образуются микротрещины. Работы А. Ф. Иоффе и
его школы показали, что именно образование микротрещин и осо-
бенно поверхностных микротрещин служит главной причиной рез-
ко пониженной прочности твердых тел по сравнению с теоретиче-
ски возможной прочностью, вычисленной на основании данных об
их строении.
Микротрещины образуются обычно в слабых местах кристал-
лической решетки. Все твердые тела обладают дефектами струк-
туры, распределенными в объеме так, что участки твердого тела
между ними имеют в среднем размеры порядка 10-6 см. Иначе
232
говоря, один дефект встречается в среднем через 100 правильных
межмолекулярных или межатомных расстояний. «Слабыми ме-
стами» могут являться границы между отдельными кристалликами,
если тело состоит из микрокристалликов, и любые неоднородности.
При снятии нагрузки, если не было достигнуто разрушения тела,
образовавшиеся микротрещины, по П. А. Ребиндеру, смыкаются
и исчезают, как бы «залечиваются». В случае же нагрузок, пре-
вышающих предел прочности, разрушение тела в основном идет по
этим микротрещинам.
П. А. Ребиндер, Е. Д. Щукин и др. в своих работах показали,
что развитие микрощелей под действием внешних деформирующих
сил может происходить значительно легче при адсорбции различ-
ных веществ из среды, в которой ведется диспергирование. Адсор-
бироваться могут как ионы электролитов, так
и молекулы поверхностно-активных веществ.
Образуя на адсорбировавшей их поверхности
двухмерный газ в результате нелокализован-
ной адсорбции, они под давлением этого газа
проникают в устья возникших микрощелей
и стремятся раздвинуть каждую микрощель,
содействуя таким образом внешним дефор-
Рис. VIII, 3. Схема
развития микрощелей
под давлением двух-
мирующим силам и способствуя диспергиро-
ванию.
На рис. VIII, 3 схематически изображено
развитие микрощелей под влиянием двухмер- мерного газа.
ного газа, образовавшегося в результате ад-
сорбции вещества на поверхности частиц. Следует отметить, что
диспергирование облегчается не только благодаря давлению двух-
мерного газа, но и вследствие экранирования сил сцепления, дей-
ствующих между противоположными поверхностями щели, при
попадании в образующиеся микрощели постороннего вещества.
Облегчение диспергирования под влиянием адсорбции полу-
чило название эффекта Ребиндера или адсорбционного понижения
твердости, а вещества, повышающие эффективность диспергирова-
ния, называются понизителями твердости.
Адсорбционное понижение твердости имеет большое практиче-
ское значение. Оно используется не только при получении дисперс-
ных систем, но и при достаточно грубом разрушении и деформи-
ровании различных материалов, например при бурении горных
пород, при обработке металлов на металлорежущих станках и т. д.
Благодаря адсорбционному понижению твердости эти процессы
ускоряются, снижаются энергетические затраты и удлиняется
срок работы режущего инструмента.
Переходя непосредственно к получению коллоидных систем ме-
тодом диспергирования, следует указать, что при простом меха-
ническом дроблении или растирании образуются обычно порошки,
размер частиц которых не меньше нескольких микрометров. Этот
предел обусловен тем, что при механическом измельчении
233
происходит также и слипание частиц. Однако еще П. П. Веймарн
в 1912 г. заметил, что если при растирании в обычной ступке не-
растворимых окислов, сульфидов и хлоридов металлов добавлять
к растираемому веществу сахар или другие органические соеди-
нения, то дисперсность продукта значительно увеличивается. Ана-
логичные результаты были получены в лаборатории Сведберга.
При растирании серы с мочевиной и растворении полученной
смеси в воде образовывались довольно стойкие суспензии серы.
Подобное действие третьего компонента объясняется адсорбцион-
ным понижением твердости. Кроме того, способствующее диспер-
гированию вещество может являться стабилизатором.
Тем не менее методы диспергирования обычно значительно
уступают методам конденсации по дисперсности полученных си-
стем. Способом диспергирования даже в присутствии стабилиза-
тора редко удается получить системы, у которых частицы были бы
меньше 1 мкм. А
Метод пептизации. Пептизацией называют переход в кол-
лоидный раствор осадков, образовавшихся при коагуляции. Тер-
мин пептизация был введен еще Грэмом на основании чисто внеш-
него сходства процесса пептизации с растворением белков под.
влиянием пепсина. Пептизация может происходить в результате
промывания осадка или под действием специальных веществ —
пептизаторов. При этом из осадка удаляются коагулирующие
ионы или пептизатор адсорбируется коллоидными частицами осад-
ка, что ведет к образованию двойных электрических слоев или
сольватных оболочек вокруг коллоидных, частиц и к преодолению
благодаря ним сил сцепления между частицами. Ставшие свобод-
ными частицы под влиянием теплового движения распределяются
равномерно во всем предоставляемом им объеме жидкости. Таким
образом, пептизация является процессом, как бы обратным ко-
агуляции.
Следует заметить, что пептизировать осадок удается далеко
не всегда. Пептизации препятствуют явления рекристаллизации и
старения, приводящие к сращиванию частиц друг с другом. Очень
трудно также осуществить пептизацию осадка, полученного путем
коагуляции золя поливалентными ионами, весьма прочно удержи-
вающимися на поверхности адсорбировавших их частиц.
При пептизации, как и при коагуляции, не наблюдается сте-
хиометрических отношений между количествами пептизатора и
пептизированного осадка. Для пептизации осадка и получения
лиозоля не требуется, чтобы вся поверхность частиц была покрыта
слоем адсорбированного пептизатора. Так, Фаянс установил, что'
для получения устойчивого золя бромида серебра частицы его
должны быть покрыты всего на */4—7ю часть от всей поверхности
пептизатором, которым в этом случае будет электролит, содержа-
щий бромид-ионы. Однако от количества пептизатора зависит дис-„
персность частиц в полученном золе. При малом содержании вве-
денного пептизатора образуются частицы высших порядков, со-
234
стоящие из нескольких первичных частиц, при достаточно боль-
шом — отдельные первичные частицы.
Пептизация протекает с определенной скоростью, причем при
достаточном количестве пептизатора процесс вначале идет весьма
быстро, а затем постепенно замедляется. Для скорости пептизации,
понятно, имеет значение перемешивание системы. При хорошем
перемешивании ускоряется проникновение пептизатора внутрь
агрегатов, что способствует отрыву частиц друг от друга и пере-
ходу их в раствор. Скорость пептизации, как правило, возрастает
с повышением температуры.
При пептизации наблюдается весьма характерная зависимость
между количествами пептизированного вещества, взятого осадка и
пептизатора. Эта закономерность, называемая иногда правилом
Взятого для ростборепия
Рис. VIII, 4. Зависимость количества
осадка, перешедшего в коллоидный
раствор, от количества осадка, взя-
того для растворения (прн постоян-
ном содержании пептизатора).
Рис. VIII, 5. Зависимость количе-
ства осадка, перешедшего в коллоид-
ный раствор, от концентрации вве-
денного пептизатора с (при постоян-
ном количестве осадка, взятого для
растворевия).
осадка и изученная Во. Оствальдом и Бузагом, заключается в том,
что при постоянном содержании пептизатора с возрастанием ко-
личества взятого для пептизации осадка количество осадка, пе-
решедшего в раствор, сначала увеличивается, а затем уменьшается
(рис. VIII, 4).
Объяснение правила осадка заключается в следующем. Для
пептизации одной частицы осадка требуется некоторое минималь-
ное количество пептизатора. Поэтому при введении первых порций
осадка, когда в системе пептизатора много, а осадка еще мало,
последний легко переходит в золь. Однако по мере добавления
осадка на одну его частицу будет приходиться все меньше и
меньше пептизатора. Это приведет сначала к снижению коллоид-
ного растворения. Затем, когда пептизируемого вещества в системе
станет много, осадок не только перестанет растворяться, но даже
выпадет уже растворившийся осадок, так как вследствие перерас-
пределения пептизатора между коллоидными частицами его уже
не будет хватать для того, чтобы эти частицы находились в рас-
творе.
235
Как следует из зависимости между количествами коллоидно
растворенного осадка и взятого для растворения, пептизация резко
отличается от обычного растворения, в ротором после достижения
насыщения содержание растворенного вещества перестает зави-
сеть от количества вещества, взятого для растворения. Такое раз-
личие объясняется, конечно, тем, что для коллоидного растворения
требуется пептизатор, в то время как для истинного растворения
никакой третий компонент не нужен.
Если брать для растворения одинаковые количества осадка и
пептизировать их различными, все увеличивающимися количе-
ствами пептизатора, то начнется пептизация, которая быстро воз-
растает с увеличением количества вводимого пептизатора, и, нако-
нец, наступит полная пептизация осадка (рис. VIII, 5). Подобную
зависимость также легко объяснить тем, что для коллоидного рас-
творения осадка необходимо определенное количество пептиза-
тора.
Явление пептизации имеет большое значение в технике при
переведении различных осадков в коллоидные растворы, а также
в препаративной коллоидной химии при получении золей. Однйко
часто пептизация может играть и отрицательную роль. Например,
при извлечении сахара из свеклы диффузией возможна пептиза-
ция пектина и других веществ, содержащихся в растительных
тканях.
Самопроизвольное диспергирование. Весьма интересно явление
самопроизвольного образования равновесных и устойчивых кол-
лоидных систем. Опыт показывает, что в отдельных случаях твер-
дое тело или жидкость могут самопроизвольно диспергироваться
в жидкой среде с образованием двухфазной, но термодинамически
устойчивой коллоидной или микрогетерогенной системы. Такие
системы, полученные в результате самопроизвольного диспергиро-
вания, П. А. Ребиндер, как уже мы указывали в гл. I, предлагает
называть лиофильными коллоидными системами, поскольку
при этом достаточно сильно выражено взаимодействие между ве-
ществом дисперсной фазы и средой.
К таким системам относятся критические эмульсии, возникаю-
щие спонтанно при температурах, близких к критическим; высоко-
дисперсные золи парафина в углеводородах; водные растворы
эмульсолов — углеводородов с большим содержанием (10—40%);
мыл или мылоподобных поверхностно-активных веществ (эмуль-
солы применяют в качестве смазочнорежущих жидкостей при хо-'.
лодной обработке металлов) и т. д. Эти системы могут существо-;
вать длительное время без заметных изменений.
О причинах возникновения и существования подобных систем в последние
годы велась оживленная дискуссия (П. А. Ребиндер, Г. И. Фукс, Е. Д Щукин
и др), которая до настоящего времени не привела к окончательному решению
вопроса Однако учитывая большое принципиальное значение самопроизвольного
возникновения термодинамически равновесных двухфазных систем, этот процесс
следует рассмотреть более подробно, исходя из представлений, развиваемы^
П. А. Ребиндером.
236
Как для классических лиофобных, так и для лиофильных систем в новом
смысле этого слова характерно существование межфазной груницы, разделяю-
щей фазы 1 и 2 (рис VIII, 6а). Однако фактически межфазная граница не:
является геометрической плоскостью, а имеет некоторую толщину Л;,2, хотя и-
весьма малую вследствие резкого снижения действия молекулярных сил с рас-
стоянием (см. рис. VIII, 66) В этом случае правильнее говорить о пограничном
межфазном слое.
Свободная энергия системы F, включая
и межфазный слой, может быть выражена
уравнением:
> F = Ft + F2 + Flt 2 =
= /^1+/2V2 + ff1,2S1,2 (VIII, 3)
где Ft и F2 — свободные энергии первой и <
второй фаз, ft и f2 — плотности свободной $
энергии этих фаз; Vi и V2 — объемы фаз; “ *
F,,2- свободная энергия пограничного слоя; Рис VIII 6> Межфазная граница:
Oi,2 — сгущение свободной энергии в погра- „ .
.. _ J „ а—идеальной системы; б—реальной
НЯЧНОМ слое С площадью St,2. . системы, s, , — поверхность границы
В объеме фаз плотности энергий fl и f2 раздела; Л. 2—общая толщина погра-
постоянны. Однако вблизи поверхности раз- личного слоя, равная сумме толщин hi
дела фаз плотность энергии возрастает и й> приповерхностных слоев первой
с приближением к поверхности раздела под н второй фазы,
действием энергии взаимодействия фаз или
некомпеисированности межмолекулириых сил в поверхностном слое. Таким об-
разом, переход ог энергетического уровня первой фазы ft к энергетическому
уровню второй фазы f2 совершается через энергетический барьер, отвечающий
сгущению свободной энергии в поверхностном слое. Это слой на рис. VIII, 7
показан в .виде заштрихованного «языка». Площадь «языка» соответствует сгу-
щению свободной энергии в поверхностном слое или для слоя площадью в
1 см2 — межфазному поверхностному иатяжеияю CFj,2-
В типичных лиофобных коллоидных системах межфазный слой характери-
зуется большим увеличением плотности свободной энергии и четкой границей
Рис. VIII, 7. Изменение плот-
ности свободной энергии f у меж-
фазной границы лиофобных кол-
лоидных систем.
Ряс. VIII, 8. Изменение плот-
ности свободной энергии f у меж-
фазной границы лиофильных кол-
лоидных систем.
раздела фаз. В этом случае Ot,2 оНр, где оНр — критическое значение меж-
фазного поверхностного натяжения, при котором возможно образование термо-
динамически равновесной коллоидной системы, т. е имеет место самопроизволь-
ное диспергирование в результате теплового движения вещества. Поэтому
в лиофобных системах (при О;, 2 оНр) самопроизвольно могут идти только про-
цессы коагуляции, структурообразоваиия, рекристаллизации и т. д.
В лиофильных коллоидных системах межфазный слой характеризуется ма-
лым увеличением плотности свободной энергии и ие имеет четкой границы. На
237
рис. VIII, 8 даиа схема, характеризующая сгущение энергии в поверхностном
слое частиц лиофильного золя.
Разность (fi — ft), как можно видеть из этого рисунка, очень мала и энер-
гетический барьер в этом случае практически отсутствует. Поэтому лиофильные
системы (в ионом смысле этого слова) способны к самопроизвольному диспер-
гированию под влиянием теплового движения.
Изменение свободной энергии AF при самопроизвольном дис-
пергировании можно представить уравнением:
AF = АС/+ Дзь 2ffb 2 - Т AS (VIII, 4)
где U — внутренняя- энергия; S — энтропия.
В данном случае Д(7 близко к нулю. Тогда условие самопроиз-
вольного диспергирования очевидно будет выражаться неравен-
ством:
Asl,2<rl,2-I'AS<0 (VIII,5)
Первый член левой части этого неравенства представляет собой
возрастание свободной энергии при ,диспергировании, а второй
член левой части характеризует убыль свободной энергии вслед-
ствие равномерного распределения дисперсной фазы в объеме. При
достаточно малой величине <ц, 2 энтропийный член может превали-
ровать, в результате чего и происходит самодиспергирование.
Из уравнения (VIII, 5) можно получить следующее:
2 < nykT (VIII, 6)
где п — число частиц; а — размер частиц; k — постоянная Больцмана; у — без-
размерный коэффициент.
Величина па2 пропорциональна изменению поверхности, а ве-
личина nyk — изменению энтропии системы при диспергировании.
Константа у при диспергировании до частиц коллоидных размеров
имеет значение около 10.
Из уравнения (VIII, 6) можно получить критическое значение
поверхностного натяжения:
°гкР = -^т (VIII, 7)
Для частиц коллоидных размеров ааИО-6 см, а Окр~0,01 эрг/см2.
Иначе говоря, самопроизвольное диспергирование вЬзможно для
получения коллоидных систем, у которых О], 2 < Окр ~ 0,01 эрг/см2.
При самопроизвольном диспергировании образуются дисперс-
ные системы, характеризующиеся нормальными кривыми распре-
деления с-некоторым наиболее вероятным радиусом частиц. Это
значит, что существует какая-то оптимальная, характерная для
данной системы дисперсность. Дальнейшее самодиспергирование
вплоть до молекул П. А. Ребиндер с сотр. считают невозможным.
Согласно их точке зрения термодинамическая устойчивость двух-
фазных дисперсных систем определяется двумя условиями: доста-
точно низким межфазным поверхностным натяжением и быстрым
его повышением с уменьшением радиуса частиц. Однако причины
238
повышения межфазного натяжения с уменьшением размера частиц
трудно объяснить.
Можно дать и другие объяснения, почему явление самодиспер-
гирования не идет до молекул. Так, при разбавлении водой эмуль-
солов отдельные молекулы углеводорода не могут существовать
в системе из-за нерастворимости углеводорода в воде. По этой
причине образовавшиеся молекулы углеводорода тотчас слипаются
друг с другом в капельки, а присутствующее в системе поверхност-
но-активное вещество адсорбируется на этих капельках и пони-
жает 01,2 До значений, меньших Окр. Таким образом, при раство-
рении эмульсола одновременно протекают два противоположно
направленных процесса — растворение эмульсола до молекул и
слипание молекул углеводорода в агрегаты, адсорбирующие по-
верхностно-активное вещество. При установлении равновесия
между обоими процессами получается устойчивая равновесная
эмульсия. Размеры капелек этой эмульсии определяются количе-
ственным соотношением углеводород : поверхностно-активное веще-
ство, природой обоих компонентов, температурой и т. д.
В случае образования критических эмульсий при нагревании
двух несмешивающихся при обычной температуре жидкостей не-
возможность растворения вплоть до молекул объясняется двумя
одновременно протекающими, но противоположно направленными
процессами. С одной стороны, идет диспергирование вплоть до мо-
лекул одной из фаз, а с другой — одновременно происходит ко-
алесценция капелек этой фазы, в результате чего устанавливается
динамическое равновесие, которое тотчас нарушается с изменением
температуры. Конечно, состояние критической эмульсии также со-
ответствует минимальной свободной энергии системы.
V В заключение обобщим кратко те термодинамические положе-
ния, которые необходимо иметь в виду при рассмотрении различ-
ных методов синтеза коллоидных систем.
При получении коллоидных систем методом диспергирования
работа, затрачиваемая на преодоление межмолекулярных сил при
дроблении дисперсной фазы, запасается системой в виде свободной
энергии на межфазной поверхности. Избыток свободной энергии
делает систему термодинамически неустойчивой. Для придания
системе агрегативной устойчивости избыток свободной энергии
должен быть уменьшен посредством адсорбции. Однако практиче-
ски в результате адсорбции никогда не удается избавиться от сво-
бодной поверхностной энергии полностью, и поэтому устойчивость
типичных коллоидных систем носит обычно временный характер.
-При дроблении вещества, понятно, увеличивается энтропия си-
стемы. Однако увеличение энтропии благодаря сравнительно боль-
шим размерам частиц не сказывается сколько-нибудь заметно на
устойчивости коллоидного раствора. Только при очень малых меж-
фазных поверхностных натяжениях увеличение энтропии может
приводить к самопроизвольному диспергированию и образованию
равновесных коллоидных систем.
239
Для пептизации внешней энергии на перевод осадка в раствор
не требуется, так как свежий осадок представляет собой первич-
ные частицы, очень непрочно слипшиеся друг с другом только
в отдельных местах. Для преодоления сил сцепления в систему
достаточно ввести пептизатор, диффундирующий к поверхности
частиц и образующий на ней двойной электрический слой или соль-
ватную оболочку.
Фактором, обусловливающим при этом равномерное распреде-
ление частиц по всему объему жидкости, ставших свободными и
устойчивыми, является броуновское движение.
Получение коллоидных систем путем самопроизвольного дис-
пергирования близко по своему существу к пептизации. В этом
случае работа диспергирования мала благодаря небольшой меж-
фазной свободной энергии. При этом работа диспергирования
настолько невелика, что для коллоидного растворения достаточно
одного теплового движения. Возрастание энтропии системы в ре-
зультате более равномерного распределения диспергированного
вещества с избытком компенсирует увеличение свободной поверх-
ностной энергии вследствие возрастания поверхности раздела фаз.
Наиболее сложными кажутся на первый взгляд термодинами-
ческие условия получения коллоидных систем методом конденса-
ции. Может даже показаться, что золи, синтезированные, напри-
мер, в результате химической реакции, образуются самопроиз-
вольно и, следовательно, их получение сопровождается уменьше-
нием свободной энергии системы. Однако не следует забывать, что
при химической реакции свободную энергию системы следует
сравнивать не со свободной энергией растворов исходных компо-
нентов реакции, а со свободной энергией полученной системы с
выкристаллизовавшейся дисперсной фазой. При этом причины не-
устойчивости коллоидных растворов, полученных методом конден-
сации, становятся совершенно ясными.
2. СТРОЕНИЕ КОЛЛОИДНЫХ МИЦЕЛЛ
Ознакомившись со строением двойного электрического слоя,
составляющего внешнюю оболочку мицеллы, и с физико-химиче-
скими основами'синтеза коллоидных систем, можно перейти к
рассмотрению строения коллоидных мицелл в целом.
Строение мицелл в золях интересовало ученых уже давно, и по этому во-
просу имеется множество мнений, часть из которых представляет теперь лишь
исторический интерес
Иордис в 1902 г. при изучении химических методов получения различных
золей пришел к выводу, что состав коллоидных мицелл не соответствует тем
веществам, которые должны образоваться в результате предполагаемой реакции.
Иордис одни из первых отметил, что дисперсная фаза золя всегда содержит в
качестве примеси вещества, из которых она была получена При удалении этих
веществ, например, путем диализа, золь теряет устойчивость. На основании этого
Иордис правильно считал, что примеси не безразличны для коллоидной системы.
Согласно Иордису, коллоидная частица представляет собой комплексное соеди-
нение сложного состава.
240
Аналогичных взглядов придерживался и известный французский ученый
Дюкло Он, как и Иордис, считал весьма существенным для свойств коллоидных
систем присутствие в них небольших количеств исходных веществ и полагал, что
эти примеси входят в состав коллоидных частиц. Например, состав частиц золя
As2S3, получающегося в результате взаимодействия As2O3 и H2S и всегда содер-
жащего небольшие количества H2S, Дюкло изображал формулой
As2S3 • nH2S
С помощью буквенного коэффициента п в этой формуле Дюкло хотел под-
черкнуть возможность широкого изменения содержания H2S в коллоидной ча-
стице. Для подобной сложной частицы Дюкло первый предложил название «ми-
целла». Небольшое же количество стабилизатора в мицелле он назвал активной
частью и указал, что именно эта часть обусловливает движение частицы в элек-
трическом поле и ее присутствием объясняется поведение золя при введении в
него электролитов
Паули расширил представления Иордиса и Дюкло. Он тоже считал, что
мицелла состоит из сравнительно инертного ядра и способной к ионизации ак-
тивной части Эту способную к ионизации часть мицеллы он назвал ионогенным
комплексом Паули рассматривал этот комплекс как настоящее комплексное со-
единение по теории Вернера и поэтому выражал строение мицеллы, например
золя сульфида мышьяка, следующей формулой:
xAs2S3 • j/As2S4H~ • j/H+
Эта схема объясняет как заряд частиц золя сульфида мышьяка, так и поведение
золя при. введении в него электролитов. Однако схема ничего не говорит о том,
каким образом обеспечивается связь между ионогенным комплексом и неактив-
ной частью мицеллы. Эта схема не может объяснить и того, почему коллоидные
частицы характеризуются как общим скачком потенциала на границе двух фаз,
так и особым ^-потенциалом, обнаруживающимся только при электрокннетиче-
ских явлениях.
Наряду с рассмотренными выше представлениями, которые
можно назвать химическими гипотезами строения коллоидных ми-
целл, существовал и другой, физический, или адсорбционный, под-
ход к объяснению структуры мицеллы. В 1914 г. Панет показал,
что кристаллы некоторых нерастворимых в воде солей особенно
энергично адсорбируют из окружающего раствора ионы, образую-
щие с противоположно заряженными ионами кристаллической по-
верхности малорастворимые соединения. Причина этого явления
заключается в том, что при образовании малорастворимой соли
энергия гидратации всегда меньше энергии кристаллической ре-
шетки.
Дальнейшее развитие этой концепции и приложение ее к объ-
яснению образования коллоидных систем принадлежит Фаянсу.
Фаянс считал, что коллоидные частицы являются ультрамикроско-
пическими кристалликами и что, например, кристаллики Agl,
полученные в результате реакции КД, взятого в избытке с AgNOs
и находящиеся в растворе иодида калия, испытывают тенденцию
к росту. Поскольку из ионов К+ и I", присутствующих в растворе,
нерастворимое соединение с ионами Ag+ и I-, находящимися на
поверхности кристалла, способен давать только ион I-, он один
и способен достраивать кристалл. Понятно, что достройка кри-
сталла ионом I- может продолжаться только до тех пор, пока
этим ионом не будут закрыты на кристаллической поверхности
241
все ионы Ag+, которые только и могут взаимодействовать с ионом
I- вследствие разноименное™ зарядов.
Достройка кристалла Agl имеет место и тогда, когда кристалл
находится в растворе AgNO3. В этом случае ионом, достраиваю-
щим поверхность, будет ион Ag+, который придаст кристаллу уже
положительный заряд.
Достройка кристалла Agl ионами I- и Ag+ происходит не
только потому, что они образуют нерастворимое соединение на
кристаллической поверхности, но и потому, что эти ионы иден-
тичны по размерам и свойствам с ионами I" и Ag+, уже имеющи-
мися в кристалле, и, следовательно, наиболее подходят к данной
кристаллической решетке. Правильность такой точки зрения под-
тверждается тем, что ионами, достраивающими решетку, могут
быть не только ионы, общие с ионами кристалла, но и изоморф-
ные с ними. Так, кристалл Agl может достраивать не только-
ион I-, но и изоморфные с ним ионы Вг_ или С1_.
Легко видеть, что в результате достройки на поверхности
кристалла образуется двойной электрический слой. При этом, ко-
гда кристалл Agl находится в растворе KI, потенциалопределяю-
щим ионом будет ион I-, а противоионом — ион К+; когда же кри-
сталлик находится в растворе AgNO3, потеициалопределяющим
ионом будет ион Ag+, а противоионом — ион NO3.
Существенно, что как химическая, так и физическая теории
строения мицеллы приводят к одним и тем же выводам, а именно-
к тому, что ионы электролита — стабилизатора препятствуют даль-
нейшему росту кристаллика, сообщают ему заряд и тем самым
способствуют агрегативной устойчивости коллоидной системы.
Следует, однако, заметить, что химические и приведенные выше
адсорбционные представления приложимы далеко не всегда. На-
пример, при получении водцых эмульсий углеводородов с приме-
нением в качестве стабилизатора обычных мыл также образуется
двойной электрический слой на поверхности капелек. При этом
потенциалопределяющими ионами служат анионы жирной кис-
лоты со сравнительно длинным углеводородным радикалом, а про-
тивоионами— катионы щелочного металла. Понятно, что никакого
комплексообразования или достройки кристаллической решетки в
этом случае не может быть, так как капельки углеводорода хи-
мически инертны и аморфны. Однако существенно то, что в этом
случае капельки углеводорода адсорбируют ионы, в состав кото-
рых входят углеводородные радикалы.
Таким образом, обобщая приведенное выше* можно прийти к
выводу, что заряд коллоидной частицы всегда определяется род-
ственными ионами стабилизатора, способными достраивать кри-
сталлическую решетку или преимущественно адсорбироваться на
поверхности частицы. Стабилизатором же может служить всякое
вещество, имеющее такие ионы, независимо от того, является ли
оно одним из исходных веществ, взятым в избытке при образова-
нии дисперсной фазы (например, KI или AgNO3 при образовании
242
золя Agl) или оно специально введено в систему при ее получе-
нии (мыло при образовании эмульсии).
На основании изложенного выше можно уточнить строение ми-
целл различных золей и выразить это строение специальными
формулами.
Обратимся для примера к мицелле золя Agl, находящейся
в межмицеллярной жидкости, представляющей собой слабый рас-
твор KI. Учитывая строение двойного электрического слоя, обра-
зующегося на поверхности дисперсной фазы, и пользуясь прави-
лом Панета — Фаянса, строение этой мицеллы можно представить
схемой, изображенной на рис. VIII, 9.
Внутри мицеллы находится кристалл,
состоящий из молекул Agl. Эту часть
мицеллы, согласно терминологии, пред-
ложенной Н. П. Песковым, будем на-
зывать агрегатом, так как она всегда
состоит из агрегата атомов, молекул
или ионов, образующих дисперсную
фазу. На поверхности кристалла на-
ходятся достраивающие его ионы,
которые придают ему электрический
заряд. Эти ионы, как мы уже знаем,
являются потенциалопределяющими.
Агрегат вместе с адсорбированными
на нем потенциалопределяющими ио-
Рис. VIII, 9. Схема строения
мицеллы золя иодида серебра
в слабом растворе нодида ка-
лия.
нами составляет ядро мицеллы.
В, непосредственной близости от
ядра в адсорбционном слое находится
часть противоионов К+, которые настолько прочно связаны элек-
тростатическими и адсорбционными силами, что в электрическом
поле они движутся совместно с ядром к аноду. Заметим, что эти
противоионы К+ вместе с эквивалентным числом потенциалопреде-
ляющих ионов I-'формально можно рассматривать как недиссо-
циированные молекулы.
Ядро вместе с частью прочно связанных с ним противоионов
будем в дальнейшем называть собственно коллоидной частицей.
В отличие от мицеллы, в целом электронейтральной, частица
всегда имеет заряд (в рассматриваемом случае отрицательный).
Остальные противоионы, находящиеся ближе к периферической
части мицеллы, образуют диффузный слой мицеллы. Благодаря
ослаблению с расстоянием кулоновских и адсорбционных сил эти
ионы относительно свободны и в электрическом поле движутся
уже к катоду. Граница между диффузным слоем и собственно кол-
лоидной частицей, по которой мицелла «разрывается» при элек-
трофорезе, называется плоскостью или границей скольжения. На
рис. VIII, 9 она обозначена пунктирной линией.
Строение мицеллы можно представить также удобной для на-
писания формулой. Например, для золя Agl, межмицеллярной
243
жидкостью которого является слабый раствор KI, можно написать
следующую формулу мицеллы:
мицелла
{т [Agl] пГ • (и — х) К+}хК+
агрегат
—........у
ядро
частица
В этой формуле tn [Agl] соответствует числу молекул Agl, со-
держащихся в агрегате мицеллы, п!~ — числу потенциалопреде-
ляющих ионов, (п — х) К+ — числу противоионов в непосредствен-
ной близости от ядра и хК+ — числу противоионов, находящихся
в диффузном слое. Как правило, т^п. Таким же способом можно
изображать и мицеллу золя с положительно заряжённой частицей,
например мицеллу золя Agl в слабом растворе AgNO3:
{т [Agl] nAg+- (и — х) NO0 xNOJ
Мы рассмотрели строение мицелл, у которых ионогенная часть
образуется в результате адсорбции стабилизующего электролита,
отличающегося по своей химической природе от вещества дисперс-
ной фазы. В других случаях ионогенная часть мицеллы может
образоваться из вещества самого агрегата. Примером такой кол-
лоидной системы может служить достаточно постаревший гидро-
золь двуокиси кремния. Поверхность агрегата, реагируя с окру-
жающей его водой, образует метакремневую кислоту HzSiOg,
которая и будет являться стабилизатором. Строение миЦеллы та-
кого золя, очевидно, следует изображать формулой
{т [SiO2] nSiOr • 2 (n - х) Н*-} 2хН*
Численный коэффициент 2 перед (п — х) и х в этой формуле по-
ставлен ввиду двухосновности метакремневой кислоты.
При пользовании схемами строения мицелл и их формулами
следует помнить, что мицелла лиозоля не является чем-то раз и
навсегда сформированным, а может претерпевать самые различ-
ные изменения. Так, при введении индифферентного электролита
в золь происходит сжатие диффузной части двойного электриче-
ского слоя, а следовательно, радиус мицеллы уменьшается. При
этом противоионы, находящиеся в диффузном слое, проникают за
плоскость скольжения и в результате (п — х) возрастает, а х
уменьшается. При достаточном количестве индифферентного элек-
тролита ионы диффузного слоя могут полностью перейти в адсорб-
ционный слой и частица окажется лишенной заряда. При этом,
например, мицелла золя Agl, для которой стабилизатором яв-
ляется KI, будет иметь вид
m [Agl] «Г • иК+
244
Кроме того, при введении индифферентного электролита с про
тивоионом, отличным от противоиона мицеллы, происходит обмен
противоионов.
При введении в золь электролита, один из ионов которого спо-
собен достраивать кристаллическую решетку, может изменяться
число потенциалопределяющих ионов и даже один потенциалопре-
деляющий электролит может замениться другим, т. е. произойдет
перезарядка, например, при введении избытка AgNO3 к золю Agl,
стабилизованному KI.
3. ПРИМЕРЫ ПОЛУЧЕНИЯ КОЛЛОИДНЫХ СИСТЕМ
Ниже приводятся только наиболее характерные примеры син-
теза коллоидных систем, причем основное внимание обращено на
сущность способа, а не на его технические подробности, которые
можно найти в специальной литературе.
Получение лиозолей методом конденсации. К этой группе син-
тезов относится получение лиозолей прямой конденсацией, заме-
ной растворителя и путем различных химических реакций.
Примером синтеза прямой конденсацией может служить полу-
чение золя ртути. Для этого Нордлунд пропускал пары ртути че-
рез слой воды и получал довольно высокодисперсную эмульсию
ртути в воде. Аналогичным способом могут быть получены золи
серы, селена и теллура. Путем конденсации в жидкости паров
меди, серебра, золота и платины, полученных в вольтовой дуге,
можно получить соответствующие золи в воде, спиртах, глицерине
или бензоле. Строение мицелл этих золей мало изучено. Стабили-
затором при получении всех этих систем служат окислы веществ,
получающиеся при соприкосновении их паров с воздухом при вы-
сокой температуре. Образование в таких условиях окислов, об-
ладающих свойствами электролитов, подтверждается заметным
возрастанием электропроводности системы. Однако более стойкие*
золи получаются в том случае, если в воду, в которой происходит
конденсация паров, вводят стабилизующие электролиты.
Синтез коллоидных систем путем замены растворителя сво-
дится к тому, что вещество, из которого хотят получить золь, рас-
творяют в соответствующем растворителе в присутствии стабили-
затора и затем раствор смешивают с другой жидкостью, в которой
вещество нерастворимо. В результате этого вещество выделяется
из раствора, но ввиду присутствия в системе стабилизатора оно'
не выпадает в виде осадка, а образует золь. Таким образом могут
быть получены гидрозоли канифоли и серы. Этот же метод Перрен
использовал для получения классических дисперсий гуммигута и
мастики. Растворителем для этих веществ служит этанол. Стаби-
лизаторами являются примеси, содержащиеся в ничтожных коли-
чествах в исходных веществах и спирте (продукты окисления).
Строение мицелл полученных таким способом золей еще не изу-
чено, но известно, что во всех случаях коллоидные частицы заря-
жены отрицательно.
245-
/
Коллоидные системы можно получить в результате химических
реакций почти всех известных типов: реакций обмена, восстанов-
ления, окисления, гидролиза и т. д. Следует, однако, заметить, что
коллоидные системы при проведении реакций, способных давать
золи, образуются не. всегда, а лишь при определенных концентра-
’ циях исходных веществ, порядке их смешения, температуре и со-
блюдении некоторых других условий.
В качестве примера получения золя путем реакции двойного
обмена можно привести синтез гидрозоля иодида серебра из раз-
бавленных растворов иодида калия и нитрата серебра. Суще-
ственно, что при смешении растворов, содержащих исходные ве-
щества в количествах, близких к эквивалентным, золя не полу-
чается, а образуется осадок иодида серебра по реакции:
AgNO3 + KI —> AglJ, + KNO3
Если же при смешении одно из исходных веществ взято в избытке,
образуется золь. П. П. Веймарн, указавший на особую положи-
тельную роль избытка одного из веществ, принимающих участие
в образовании коллоидной системы, полагал, что этот избыток не-
обходим для понижения растворимости дисперсной фазы. Однако
гораздо более правильно объяснить получение устойчивого золя
Agl в присутствии избытка AgNO3 или KI тем, что эти электро-
литы являются стабилизаторами частиц иодида серебра, образуя
на них двойной электрический слой.
Интересно, что устойчивость золя иодида серебра с отрица-
тельно заряженными частицами несколько выше, чем с положи-
тельно заряженными. Причина этого заключается в том, что
иодид-ионы адсорбируются на агрегатах иодида серебра сильнее,
чем катионы серебра *.
Аналогичным образом можно получить золи бромида и хлорида
серебра, однако эти золи менее устойчивы. Причина этого заклю-
чается в сравнительно большой растворимости этих соединений.
(Растворимости иодида, бромида и хлорида серебра в воде при
20°C равны соответственно 9,7-10~9, 6,6-10-7 и 1,25-10-5 моль/л).
Чем выше растворимость дисперсной фазы, тем легче происходит
перекристаллизация коллоидных агрегатов и тем быстрее стареет
золь.
Метод синтеза с помощью реакции обмена может быть исполь-
зован не только для получения гидрозолей. Например, золь иодида
серебра в ацетоне можно получить, смешивая растворы нитрата
серебра и иодида калия в ацетоне. Впрочем, этот же золь очень
легко получить путем диализа гидрозоля против ацетона.
Разработанный Зигмонди синтез гидролиза золота путем вос-
становления аурата калия формальдегидом может иллюстриро-
вать получение коллоидной системы при реакции восстановления.
* Вследствие этого изоэлектрическая точка для золя иодида серебра отве-
чает не строгой эквивалентности взятых количеств исходных веществ, а некото-
рому небольшому избытку ионов Ag+ над ионами I*.
246
Исходным веществом в этом случае служит золотохлористоводо-
родная кислота Н [АиСЦ^НгО, из которой при взаимодействии
с карбонатом калия в' водном растворе образуется аурат калия:
2H[AuClJ 4-5К2СО3 —> 2KAuO2 + 5СО2 + 8КС1 + Н2О
Полученный раствор нагревают и к нему по каплям добавляют
слабый раствор формальдегида. Реакция восстановления проте-
кает по следующему уравнению:
2КАиО2 + ЗНСНО + К2СО3 —► 2Аи + ЗНСООК + КНСО3 + Н2О
Получается красный золь Золота. Стабилизатором золя золота
служит аурат калия. Строение мицеллы этого золя можно пред-
ставить следующей формулой:
{т [Au] nAuOJ • (п — х) К+) хК+
При проведении этого синтеза, равно как и всех синтезов золей
благородных металлов, необходимо особенно тщательно следить
за чистотой посуды и реактивов.
Для получения монодиоперсных золей золота этим способом
в раствор перед восстановлением вводят зародышевый золь золота
(т. е. очень высокодисперсный), приготовленный отдельно путем
восстановления хлорида золота фосфором. Золото, выделяющееся
при восстановлении аурата калия в присутствии зародышевого
золя, равномерно распределяется на зародышах, что и обеспечи-
вает монодисперсность конечного золя.,Все выделяющееся золото
отлагается на зародышах, и в полученном золе образуется столько
частиц, сколько было введено зародышей. Размер частиц такого
золя, очевидно, тем больше, чем меньше зародышей было введено
в раствор перед восстановлением. Зародышевый способ получения
монодисперсных коллоидных систем с частицами желаемого раз-
мера широко используется при проведении ряда исследований
в коллоидной химии.
Простейшим примером получения золя путем окислительной
реакции является окисление сероводорода кислородом в водной
среде. Этой реакцией объясняется помутнение сероводородной
воды при стоянии на воздухе. Основная реакция протекает ш>
уравнению:
2H2S + О2 —► 2S4-2H2O
Параллельно протекают более сложные реакции окисления, при-
водящие к образованию политионовых кислот, являющихся стаби-
лизаторами золя серы. Принимая условно, что стабилизующим
электролитом при этом синтезе служит пентатионовая кислота
H2S5O6, строение мицеллы полученного золя серы можно предста-
вить следующей формулой:
{т [S] /iS6Of • 2 (п - х) Н+) 2хН+
Наконец, примером получения коллоидной системы путем реак-
ции гидролиза является синтез золей гидратов окисей тяжелых
247
-металлов нагреванием или диализом растворов их солей. Напри-
мер, золь гидрата окиси железа получается, если в кипящую воду
'добавить небольшое количество хлорида железа FeCU. При этом
образуется красно-бурый золь Ре(ОН)з‘.
FeCl3 + 3H2O —► Fe(OH)3 4-ЗНС1
Кипячение способствует реакции благодаря удалению из системы
хлористого водорода с парами воды. Стабилизатором в этом про-
цессе принято считать хлорокись железа FeOCl, являющуюся про-
дуктом неполного гидролиза хлорида железа:
FeCl3+H2O —► FeOCl+2НС1
Впрочем, согласно другой точке зрения стабилизатором может
быть и,сам хлорид железа. Хлористый водород можно удалять из
системы также диализом. В результате этого золь гидрата окиси
железа может образоваться и при достаточно длительном диализе
раствора хлорида железа. Наконец, согласно третьей точке зре-
ния, стабилизатором может являться хлористый водород. Эта точ-
ка зрения основана на том, что на поверхности частиц гидрата
•окиси железа находится огромное число групп —ОН, способных
адсорбировать водородный ион, всегда присутствующий в системе
в момент образования золя.
Таким образом, мицелла золя Ре(ОН)з в соответствии с тем,
‘что является стабилизатором, может быть выражена формулами:
{т [Fe(OH)3] яРеО+ • (я - х) СГ} хСГ
дли
{т [Fe(OH)3] яРе3+ • 3 (п - х) СГ} ЗхСГ
ДЛИ
{т [Fe(OH)3] яН+ • (я — х) С1“} хСГ
В заключение необходимо указать, что лиозоли получаются
всегда более устойчивыми, если при реакции не образуется ин-
дифферентных электролитов, обусловливающих астабилизацию.
В связи с этим такие реакции, как:
2H2S + SO2 —► 3S + 2Н2О
ИЛИ
As2O3 + 3H2S —► As2S3 + 3H2O
'более благоприятны для получения устойчивых коллоидных си-
стем, чем, например, реакция:
AgNO3 + KI AgI + KNO3
Получение золей методом диспергирования. К этому методу
относится получение коллоидных или микрогетерогенных систем
обычным механическим диспергированием и вибрационным измель-
чением, например с помощью ультразвуковых колебаний. К этому
же методу можно отнести получение золей и с помощью электро-
распыления, хотя по существу электрораспыление является комби*
нацией процессов диспергирования и конденсации.
248
Получение высокодисперсных систем методом механического
диспергирования осуществляют обычно путем дополнительного
дробления частиц сравнительно грубых дисперсий. Для этого гру-
бую дисперсию обрабатывают обычно в жидкой среде в шаровых
мельницах, краскотерках или коллоидных мельницах. В этих аппа-
ратах сравнительно еще большие частицы подвергаются ударам,
раздавливанию или истиранию в зависимости от типа машины, в
на мелкие частицы, которые
результате чего они распадаются
благодаря стабилизатору, вводи-
мому в дисперсионную среду, об-
разуют относительно, высоко дис-
персную устойчивую систему.
Принцип, по которому рабо-
тают шаровые мельницы, понятен
из рис. VIII, 10. Шаровая мель-
ница представляет собой вра-
щающийся полый металлический
цилиндр, который частично за-
полнен тяжелыми металлически-
ми или фарфоровыми шарами.
В этот цилиндр загружают гру-
бую дисперсию -измельчаемого
вещества и дисперсионную среду,
содержащую стабилизатор, после
этого цилиндр приводят в мед-
ленное вращение. Шары под дей-
Рис. VIII, 10. Принципиальная схема
шаровой мельницы.
ствием центробежной силы прижимаются к стенке и вместе с ней
поднимаются на некоторую высоту, затем, оторвавшись, падают на
шары, находящиеся внизу, и дробят попадающие между ними ча-
стицы дисперсной фазы. Понятно, что при перекатывании шаров
во вращающейся шаровой мельнице происходит также и истирание
частиц материала.
Для хорошего диспергирования объем шаров, загружаемых в
мельницу, должен составлять 30—40% от ее общего объема. При
большем числе шаров они падают с меньшей высоты, и, следова-
тельно, измельчение идет хуже; при меньшем числе шаров они
скользят по стенке, что также ухудшает измельчение и, кроме того,
шары быстрее изнашиваются. Объем дисперсии в шаровой мель-
нице не должен превышать 20% от объёма мельницы.
Если измельчение вещества в шаровой мельнице проводить в -
отсутствие дисперсионной среды, то обычно невозможно получить
частицы размером меньше 60 мкм. При мокром помоле в присут-
ствии стабилизатора могут быть получены дисперсии с размерами
частиц, близкими к коллоидным.
Шаровые мельницы имеют ряд преимуществ перед другими
аппаратами для получения высокодисперсных систем, а именно,
измельчение проводится в замкнутом пространстве без потерн рас-
творителя, если он летуч; процесс можно продолжать до тех пор,
249
Рис. VIII, 11. Схема коллоидной мель-
ницы:
/—конический диск ротора; 2—вал ротора;
5 — статор, 4—пришлифованные рабочие по*
верхности.
пока не будет достигнута нужная степень измельчения- обслужи-
вание машины чрезвычайно просто. Недостатком шаровых мель-
ниц является значительное истирание шаров при работе, что
приводит к загрязнению получаемой дисперсии, а также их срав-
нительно малая производительность.
Принцип диспергирования частиц сравнительно грубых диспер-
сий в аппаратах типа краскотерок заключается в раздавливании
частиц между двумя вращающимися валиками или между вра-
щающимся валиком и неподвижной поверхностью. Кроме того, в
-данном случае диспергирование происходит и в результате боль-
ших сдвиговых напряжений, возникающих в тонком слое Жидко-
сти между валиками. К краскотеркам по принципу работы при-
ближаются смесительные валь-
цы, применяющиеся в резиновом
производстве.
Наибольшую степень дисперс-
ности можно получить используя
для диспергирования твердого
вещества в жидкости коллоид-
ные мельницы. Первая коллоид-
ная мельница, сконструированная
Плаузоном, представляла собой
металлический охлаждаемый во-
дой кожух, в котором вращался
эксцентрический вал с частотой
10 000—20 000 об/мин. На валу в
несколько рядов были укреплены
На внутренней стенке к еталличе-
ы металлические зубья, между ко-
торыми при вращении вала проходили била. Суспензию предвари-
тельно грубоизмельченного вещества подавали в кожух сверху.
При вращении вала с большой скоростью жидкость отбрасы-
вается от него, но одновременно засасываются новые ее порции.
Дробление грубых частиц в мельнице Плаузона происходит в ре-
зультате удара частичек о била и зубья и за счет истирания. Жид-
кость, содержащую диспергированные частицы, близкие по разме-
рам к коллоидным, отбирают из кожуха снизу.
Другой тип коллоидной мельницы, широко распространенный
в настоящее время, изображен на рис. VIII, 11. Эта мельница со-
стоит из ротора, представляющего собой конический диск 1, сидя-
щий на валу 2, и статора 3. Ротор приводится во вращение с по-
мощью специального расположенного вертикально мотора, совер-
шающего обычно около 9000 об/мин. Рабочие поверхности ротора
и статора 4 пришлифованы друг к другу и толщина щели между
ними составляет около 0,05 мм. Грубая суспензия подается в мель-
ницу по трубе 5 под вращающийся диск центробежной силой, раз-
вивающейся в результате вращений ротора, проталкивается через
щель и затем удаляется из мельницы через трубу 6. При прохо-
металлические пальцы — била,
ского кожуха были расположе!
250 "
ждении жидкости в виде тонкой пленки через щель взвешенные в
жидкости частицы испытывают значительные сдвиговые усилия и
измельчаются. Степень дисперсности полученной системы зависит
от толщины щели и скорости вращения ротора: чем меньше зазор
и больше скорость, тем больше сдвиговое усилие и следовательно,
выше будет дисперсность.
Разработан ряд конструкций коллоидных мельниц. Однако с
помощью коллоидных мельниц даже самых совершенных Конструк-
ций никогда не удается получить системы такой дисперсности, как
методом конденсации.
Диспергирование е помощью ультразвуковых колебаний, т. е.
колебаний с частотой выше 20 000 в секунду, не улавливаемых
человеческим ухом, является эффективным лишь в том случае, ко-
гда диспергируемое вещество обладает малой прочностью. К та-
ким веществам следует отнести смолы, серу, графит, гипс. При-
меняя ультразвук, можно получать также дисперсии легких метал-
лов и их сплавов в органических жидкостях. Ультразвук может
быть с успехом использован и при пептизации свежеприготовлен-
ных осадков.
Ультразвуковые колебания обычно получают с помощью пьезо-
электрических осцилляторов, принцип действия которых заклю-
чается в превращении электрических колебаний высокой частоты
в механические. В настоящее время с помощью кварцевых осцил-
ляторов получают ультразвуковые волны с частотой до 1 миллио-
на колебаний в секунду. С помощью магнитострикционных осцил-
ляторов, рабочей частью которых является ферромагнитный стер-
жень, получают ультразвуковые волны с меньшей частотой — до
50 тысяч колебаний в секунду.
Диспергирование с применением пьезоэлектрических осцилля-
торов проводят следующим образом. Пьезоэлектрическую пластин-
ку, к которой прилагают разность потенциалов, помещают в жид-
кость с малой диэлектрической проницаемостью, например в транс-
форматорное масло, через которую ультразвуковые колебания
передаются сосуду с системой, подвергающейся диспергированию.
Механизм диспергирования твердых тел ультразвуком еще
сравнительно мало исследован. Под влиянием ультразвуковых ко-
лебаний в системе возникают местные, быстро чередующиеся сжа-
тия и расширения вещества, приводящие к образованию мельчай-
ших полостей — кавитаций, сейчас же исчезающих под влиянием
внешнего давления. Эти сжатия, расширения и кавитации разру-
шают твердую фазу, т. е. диспергируют ее. Следует, впрочем, за-
метить, что ультразвуковые волны в определенных условияхЧюгут
вызывать не только диспергирование, но и коагуляцию, которая
происходит в результате скопления частиц в узлах колебаний и
движения меныпих частиц по направлению к большим. В резуль-
тате такой коагуляции при диспергировании быстро достигается
равновесие, при котором диспергируется столько же вещества,
сколько его выпадает из золя в виде осадка.
2э1
В последнее время для диспергирования стали широко приме-
нять вибрационные измельчители. Работа этих измельчающих ап-
паратов основана на воздействии на материал частопеременных
силовых импульсов, в результате чего материалу сообщается
большое число сравнительно слабых ударов. По этому принципу
действуют вибромельницы, представляющие собой комбинацию ша-
ровой мельницы и специального вибратора Работа вибратора вы-
зывает крлебания корпуса мельницы с частотой, равной числу
оборотов электромотора, приводящего в движение вибратор. Это
вызывает перемещение и соударение шаров, что приводит к дроб-
лению и измельчению частиц материала. Эффективность работы
вибромельииц (т. е. интенсивность действующих измельчающих
усилий и мощность установки) определяется частотой и амплиту-
дой колебаний.
Имеются также вибрационные измельчители без измельчающих тел Работа
этих измельчителей, имеющих весьма различное устройство, основана иа соуда-
рении частиц измельчаемого материала в движущихся с большой скоростью
пересекающихся потоках газа или пара При скоростях движения, превышающих
100 м/с, наступает вибрационный режим разрушения, поэтому в таких измель-
чителях эффективно диспергируются не только хрупкие, но и пластические мате-
риалы Преимущество подобных машин заключается в том, что они дают весьма
равномерный помол и в измельченном материале от9утствуют продукты износа
измельчающих поверхностей Кроме того, такие измельчающие устройства го-
раздо экономичнее обычных мельниц
Дробление частиц дисперсной фазы при получении систем ме-
тодом механического диспергирования, как правило, проводят в
водной среде. Однако водные системы, если их частицы смачи-
ваются органическими жидкостями, легко можно перевести в сус-
пензии с неводной средой. Так, измельчение пигментов обычно
ведут в воде, а затем, не высушивая, влажный пигмент смешивают
с маслом, при этом гидрофобные частицы пигмента переходят в
масло. Интересно, что для высокодисперсных коллоидных систем,
полученных методом конденсации, этот способ замены среды обыч-
но непригоден, так как при смешении гидрозоля с органической
жидкостью частицы коллоидных размеров, как правило, соби-
раются на поверхности раздела жидкостей.
К диспергационным методам получения золей условно можно
отнести и получение золей электрораспылением в вольтовой дуге
металлических электродов, погруженных в дисперсионную среду.
Этот способ можно считать диспергационным потому, что в дан-
ном случае дисперсная фаза образуется путем непосредственного
диспергирования металла, при котором твердые частицы коллоид-
ных размеров отрываются от металлических электродов, посту-
пают в среду и образуют лиозоль. Однако, с другой стороны, этот
метод можно считать, хотя бы отчасти, и конденсационным, так
как при высокой температуре дуги металл электродов превра-
щается в пар, который, соприкасаясь с окружающей средой, ох-
лаждается, конденсируется и образует коллоидные частицы.
252
Метод электрораспыления был предложен Бредигом в 1898 г.
Бредит включал в цепь постоянного тока силой 5—10 А и напря-
жением 30—НОВ амперметр, реостат и два электрода из дис-
пергируемого металла. Электроды он погружал в сосуд с водой,
охлаждаемый снаружи льдом. Схематическое устройство прибора,
которым пользовался Бредит, показано на рис. VIII, 12. При прохо-
ждении тока через электроды между ними под водой возникает
вольтова дуга. При этом у электродов образуется облачко высоко-
дисперсного металла. Для получения более стойких золей в воду,
в которую погружены электроды, целесообразно вводить следы
стабилизующих электролитов, например гидроокисей щелочных
металлов. Интересно, что диспергированию в описанных условиях
подвергается не только катод, но и анод.
Это подтверждает тот факт, что при
электрораспылении существенную роль
играют термические процессы и явления
парообразования.
Метод Бредига из-за высоких темпе-
ратур, создающихся около вольтовой ду-
ги, применим только для получения гид-
розолей. Сведберг усовершенствовал этот
метод, сделав его пригодным для полу-
чения органозолей. Для этого вместо по-
стоянного тока Сведберг применил пере-
Рис. VIII, 12. Получение
золей электрораспылением
по Бредигу.
менный ток высокой частоты, а сам процесс электрораспыления
проводил путем погружения электродов в металлический порошок,
лежащий на дне сосуда в дисперсионной среде. Электрораспыле-
ние в этом случае происходит в результате проскакивания искры
между отдельными частицами порошка. При таком способе сильно
уменьшается термическое разложение окружающей среды и
можно получить золи металлов в различных органических
жидкостях.
М А. Луниной совместно с сотр. усовершенствован метод полу-
чения металлических органозолей. Этот метод сводится к пропу-
сканию электрического тока через металлический порошок в жид-
кой органической среде, при этом переменный ток проходит по
слою металлического порошка, находящегося на дне сосуда с жид-
кой средой, и вызывает в точках неполного касания электрический
разряд. Таким образом путем электрораспыления были получены
органозоли железа, никеля, алюминия, хрома, вольфрама и дру-
гих металлов. Для повышения устойчивости этих золей в систему
добавляют стабилизаторы, обычно нафтенат или стеарат алю-
миния.
Органозоли металлов широко применяются при гидрировании и
восстановлении различных органических соединений, в качестве
катализаторов горения жидкого топлива в ракетах, как наполни-
тели пластических масс, клеев, антикоррозионных лаков и красок,
в медицине для изготовления лекарственных препаратов и т. д.
253
Получение лиозолей методом пептизации. Различают следую-
щие виды пептизации: пептизация промыванием осадка; пеп-
тизация осадка электролитом; пептизация поверхностно-актив-
ными веществами; химическая пептизация.
Пептизация промыванием осадка сводится к удалению из нега
электролита, вызвавшего коагуляцию. В результате этого остав-
шийся двойной электрический слой утолщается, силы отталкива-
ния начинают преобладать над силами притяжения и отделив-
шиеся друг от друга мицеллы в результате броуновского движения
равномерно распределяются в дисперсионной среде, т. е. образуют
коллоидный раствор. Этот вид пептизации, как можно видеть, не
требует введения в систему специального пептизатора, так как ста-
билизатор присутствует в осадке и промывание лишь обеспечивает
увеличение его активности.
Пептизация осадка промыванием хорошо известна из практики
аналитической химии, где она обычно рассматривается как неже-
лательное явление. Очень часто при промывании полученного осад-
ка водой он начинает проходить через фильтр, что указывает на
увеличение его дисперсности и образование гидрозоля.
Ниже в качестве примера приведены данные о поведении при
промывке водой осадка М.О2О5, полученного коагуляцией различ-
ными электролитами; промывку проводили несколько раз, всегда
постоянным объемом воды при 18—20 °C:
Электролит» примененный прн
коагуляции
NaCl..................
NH4CI..................
КС1...................
Морфинхлорид . . . . л .
Sr(NO,)2.................
ВаС12....................
Al2(SOp3.................
Полная пептизация после одной
промывки
То же
То же, после двух промывок
То же, после трех промывок
То же, после шести промывок
Следы пептизации после семи про-
мывок
То же
Как можно видеть, чем больше валентность и радиус коагули-
рующего иона, тем труднее идет пептизация осадка промывкой.
Это объясняется тем, что обусловившие коагуляцию ионы, если они
поливалентны или обладают большим радиусом, прочней удержи-
ваются осадком и этим затрудняют пептизацию.
Пептизация электролитами наблюдается при введении в оса-
док, образовавшийся при коагуляции, электролита, один из ионов
которого может достраивать кристаллическую решетку дисперсной
фазы или, по крайней мере, адсорбироваться на ее поверхности.
Понятно, такой пептизации всегда способствует предварительная
промывка осадка чистым растворителем.
Примером пептизации с помощью электролита является введе-
ние в свежеполученный и промытый водой осадок гидрата окиси
железа раствора хлорида железа. В результате неполного гидро-
лиза из хлорида железа образуется хлорокись железа, которая,
254
как мы видели, способна стабилизовать частицы гидрата окиси
железа. При этом происходит косвенная пептизация, когда вводи-
мое вещество сначала реагирует с водой, а затем уже продукт
этой реакции пептизирует осадок. Существенно, что-на пептизацию
осадка электролитом оказывает влияние не только ион, сообщаю-
щий свой заряд частице, но и противоположно заряженный ион.
Примером пептизации с помощью поверхностно-активных веществ может
служить пептизация высокодисперсиого порошка кровяного угля пикриновой
кислотой и мылами Окись железа также может быть пептизирована мылами, а
окись алюминия — ализарином. Высокодисперсный порошок гидрофильного као-
лина пептизируется гуминовыми кислотами. Хорошим пептизирующим действием
часто обладают высокомолекулярные вещества, макромолекулы которых способ-
ны адсорбироваться на частицах и придавать им заряд или сольватную обо-
лочку. Согласно новым воззрениям Пептизация может обусловливаться и взаим-
ным отталкиванием совершающих тепловое движение гибких цепных молекул,
только частично адсорбировавшихся иа поверхности коллоидной частицы. Более
подробно об этих взглядах сказано в гл. IX.
Химическая пептизация происходит, когда вещество, добавляе-
мое в систему, взаимодействует с веществом осадка, в результате
чего образуется электролит, придающий устойчивость частицам
дисперсной фазы. Примером химической пептизации может слу-
жить пептизация осадка гидрата окиси железа хлористоводородной
кислотой, которая, взаимодействуя с Ре(ОН)з, образует на поверх-
ности частиц осадка стабилизатор — хлорокись железа:
Fe(OH)3 + HCl —> FeOCl+2Н2О
4. ОЧИСТКА КОЛЛОИДНЫХ СИСТЕМ
Очень часто в полученных тем или иным методом лиозолях
помимо мицелл, электролита — стабилизатора и растворителя со-
держатся низкомолекулярные примеси. Например, золь иодида
серебра,# полученный в результате взаимодействия нитрата серебра
и иодида калия, всёгда содержит значительное количество индиф-
ферентного электролита — нитрата калия. В других случаях элек-
тролиты и иные низкомолекулярные примеси могут попадать в
коллоидные системы вследствие загрязненности исходных продук-
тов или по другим причинам.
Так как чужеродные электролиты действуют на коллоидные
системы астабилизирующим образом, полученные золи во многих
случаях приходится очищать. Низкомолекулярные примеси можно
удалять из лиозолей с помощью диализа, электродиализа и ультра-
фильтрации.
Диализ. Этот простейший метод очистки коллоидных систем
мы уже рассматривали в начале курса. Простейший диализатор
представляет собой мешочек из полупроницаемого материала, в
который заливается диализуемая жидкость. Мешочек опускается
в сосуд с водой.
В настоящее время существует много усовершенствованных
конструкций диализаторов, обеспечивающих более быстрый
255
процесс диализа. Интенсификация диализа достигается увеличе-
нием поверхности, через которую идет диализ, уменьшением слоя
диализуемой жидкости, более частой или непрерывной сменой
внешней жидкости и нагреванием, ускоряющим диффузию.
Природа полупроницаемой перегородки в зависимости от си-
стемы, подвергаемой диализу, может быть различной. Ранее в ка-
честве мембраны использовали бычий пузырь или пергамент. В на-
стоящее время чаще всего применяют мембраны, приготовленные
из коллодия — раствора нитрата целлюлозы. Эти перепонки очень
удобны, так как их легко изготовить с порами любого диаметра.
Нужная пористость коллодиевой мембраны обеспечивается путем
подбора растворителя для нитрата целлюлозы и условий сушки
полученной пленки. При очист-
ке органозолей, растворяющих'
обычную коллодиевую мем-
брану, состоящую из нитрата
целлюлозы, для диализа при-
меняют пленку из целлофана
или из денитрированного кол-
Рис. VIII, 13. Схема устройства электро- ЛОДИЯ.
диализатора. В заключение следует на-
помнить, что длительный диа-
лиз обусловливает не только удаление из раствора примесей, но
и вывод из системы стабилизатора, что может привести к астаби-
лизации и коагуляции системы.
Электродиализ. Поскольку низкомолекулярные примеси в золях
обычно являются электролитами, диализ можно ускорить путем
наложения на диализуемую жидкость электрического поля.
Предложено много конструкций электродиализаторов. Схема
относительно простого электродиализатора, применявшегося Пау-
ли, изображена на рис. VIII, 13. Этот диализатор состоит из трех
стеклянных камер, отделенных друг от друга полупроницаемыми
перегородками. В боковых камерах установлены электроды. Кроме
того, в эти камеры по специальным трубкам непрерывно вводится
дистиллированная вода, являющаяся внешней жидкостью, и по
другим трубкам вода отводится после того, как в нее продиффун-
дировали электролиты из средней камеры. В средней камере, в ко-
торую помещается очищаемый золь, находится мешалка, обеспе-
чивающая перемешивание золя при электродиализе. Следует за-
метить, что электродиализ особенно эффективен только после пред-
варительной очистки путем обычного диализа, когда скорость диф-
фузии из-за падения градиента концентрации электролитов между
золем и водой мала и можно применять электрические поля боль-
шого напряжения, не боясь сильного разогревания золя.
Для протекания электродиализа весьма существенно изменение чисел пере-
носа ионов в капиллярах полупроницаемой перегородки по сравнению с теми
же числами, характерными для самого раствора. Явления изменения чисел пере-
носа в капиллярах мембран обнаружены Гитторфом еще в 1902 г. и затем под-
256
том же диаметре капил-
^'7777777777777777^7777777777
тверждены в работах ряда исследователей Этот вопрос особенно подробно был
исследован в работах И. И. Жукова и его школы.
Опыт показывает, что мембраны из целлюлозы и пергамента, а также кера-
мические диафрагмы в растворах электролитов приобретают отрицательный за-
ряд. Некоторые полупроницаемые перегородки, например из дубленой желатины,
наоборот, приобретают в растворах электролитов положительный заряд. Экспе-
риментально установлено, что при отрицательном заряде диафрагмы с умень-
шением диаметра пор перенос электричества анионами уменьшается и в пределе
становится равным нулю. В этих условиях электричество переносится только
с помощью катионов. Если же диафрагма заряжена положительно, иаблюдаегся
обратное явление. Следует отметить, что при одном и
ляров изменение чисел переноса тем больше, чем
выше электрокинетический потенциал стенок капил-
ляров.
Все эти явления можно объяснить, если предпо-
ложить существование двойного электрического слоя
на поверхности капиллиров диафрагмы. В самом
деле, представим себе капиллир с отрицательно за-
ряженной поверхностью, заполненный разбавленным
раствором электролита. Очевидно, в диффузном слое
такого капилляра преобладают катионы. Если диа-
метр капилляра велик по сравнению с толщиной
двойного электрического слоя, разница в соотноше-
нии концентраций катионов и анионов в капилляре
и в растворе вне капилляра невелика и числа пере-
носа в капилляре мало отличаются от чисел переноса
в' растворе. Однако с уменьшением диаметра капил-
ляра, когда он становится соизмеримым с толщиной
двойного электрического слоя, в капилляре увели-
чивается концентрация катионов, что вызывает уве-
личение числа переноса катионов и уменьшение числа
переноса анионов. В пределе, когда диаметр капил-
ляра равен удвоенной толщине двойного электриче-
ского слоя, изменение чисел переноса будет макси-
мальным. Изменение соотношения концентраций катионов и анионов в капил-
ляре с отрицательно заряженной поверхностью при уменьшении его диаметра
можно видеть из схемы, приведенной на рис. VIII, 14. Аналогичным образом
можно объяснить большую проницаемость положительно заряженных мембран
для анионов.
При таком объяснении изменения чисел переноса ионов с уменьшением дна-
метра пор мембраны эти числа должны, очевидно, зависеть от концентрации
электролитов в капиллярах Уменьшение концентрации электролита, приводящее
к увеличению толщины диффузного слоя, должно способствовать большему из-
менению чисел переноса. Обратное отношение должно наблюдаться прн увели-
чении концентрации электролитов Опыт полностью подтвердил правильность
этих выводов.
Возвращаясь к электродиализу, нетрудно видеть, что изменение концентра-
ций электролитов в средней камере электродиализатора может происходить
только в том случае, если по сечению электродиализатора изменяются числа
переноса электролита. Такое изменение чисел переноса, как указывает И И Жу-
ков, может произойти как в результате изменения чисел переноса в порах диа-
фрагмы по сравнению со свободным раствором, так и вследствие изменении
состава электролита в отдельных камерах Последнее явление наблюдается в
работающем электродиализаторе, где в анодной камере образуется кислота и в
катодной — щелочь. Прн этом в средней камере может происходить уменьшение
концентрации электролита, даже если применялись диафрагмы, ие изменяющие
чисел переноса (так называемые электрохимически неактивные диафрагмы с по-
рами достаточно большого размера), так как по мере накопления кислоты и
щелочи в электродных камерах взамен уходящих ионов электролита в среднюю
камеру начнут поступать ионы Нч и ОН*, образующие вбду,
9 Зак. 664 2§7
Рис. VIII, 14. Изменение
соотношения концентра-
ций катионов и анионов
в капилляре с отрица-
тельно заряженной по-
верхностью при умень-
шении его диаметра.
Применяя мембраны, изменяющие числа переноса, т. е. электрохимически
активные, можно значительно ускорить процесс электродиализа. Если поставить
отрицательно заряженную мембрану на катодную сторону трехкамерного диали-
затора, то такая диафрагма будет увеличивать число переноса катионов, а поло-
жительно заряженная мембрана на анодной стороне будет увеличивать число
переноса анионов. Таким образом можно значительно увеличить разницу чисел
переноса ионов между диафрагмами. Такие диафрагмы называют идеально элек-
трохимически активными Разница между числами переноса в этом случае дохо-
дит до единицы, и выход по току достигает 100%.
Электродиализ находит применение не только при лабораторных исследова-
ниях, но и в технике. Например, электродиализ применяется для удаления из
молочной сыворотки солей Очищенная таким образом сыворотка, содержащая
большие количества ценной лактозы и протеинов, используется для получения
продуктов питания. Применение для очистки обычного диализа привело бы
к большим потерям ценной лактозы.
Ультрафильтрация. Ультрафильтрацией называется диализ,
проводимый под давлением. По существу ультрафильтрация яв-
ляется не методом очистки золей, а лишь методом их концентри-
рования. При этом важно, что повышается концентрация только
дисперсной фазы, состав же дисперсионной среды практически
остается постоянным.
Понятно, что если после частичной ультрафильтрации получен-
ный таким образом золь разбавить чистым растворителем до
прежнего содержания дисперсной фазы, он будет содержать мень-
ше низкомолекулярных веществ, но одновременно и стабилизую-
щих электролитов.
Ультрафильтрацией иногда пользуются для получения меж-
мицеллярной жидкости. Однако при этом следует помнить, что
во время ультрафильтрации может происходить адсорбция элек-
тролитов на ультрафильтре и состав полученного ультрафильтра-
та может быть не идентичен составу дисперсионной среды. Кроме
того, следует учитывать, что при этом устанавливается мембран-
ное равновесие, или равновесие Доннана, характеризующееся не-
одинаковым распределением электролитов по обе стороны мембра-
ны (см. гл. XIV).
Применяя ультрафильтры различной пористости, можно ис-
пользовать ультрафильтрацию для разделения коллоидных систем
на более монодисперсные фракции и для приблизительного опре-
деления дисперсности этих фракций. При этом, однако, надо по-
мнить, что размер пор в большинстве мембран колеблется в до-
вольно широких пределах и получить таким способом полностью
монодисперсные системы практически невозможно.
Предложено много приборов для ультрафильтрации. Так как
ультрафильтрация проводится всегда под давлением, то во всех
приборах для ультрафильтрации мембрана либо накладывается
на пластинку с мелкими отверстиями, служащую для нее опорой,
либо непосредственно получается на стенках неглазурованного
фарфорового сосуда. В частности, известные ультрафильтры Бех-.
гольда получают именно путем нанесения на стенки пористого
фарфорового сосуда разбавленного коллодия и последующего его
высушивания.
258
ГЛАВА IX
УСТОЙЧИВОСТЬ И КОАГУЛЯЦИЯ коллоидных СИСТЕМ*
Обычные коллоидные системы в отличие от молекулярных рас-
творов вследствие наличия поверхности раздела частиц с диспер-
сионной средой гетерогенны, большей частью термодинамически
неравновесны и агрегативно неустойчивы. Именно поэтому пробле-
ма устойчивости коллоидных систем является центральной пробле-
мой коллоидной химии, а коагуляция составляет наиболее важный
механизм перехода к более устойчивому состоянию для всех ти-
пичных коллоидных систем.
Устойчивость и коагуляция коллоидных систем имеют огромное
практическое значение в геологии, земледелии, биологии, технике.
Неудивительно, что этому вопросу посвящено огромное число ис-
следований.
Неустойчивость золей может проявляться также в укрупнении
частиц за счет исчезновения или уменьшения размера более мел-
ких. Процесс укрупнения частиц в золях аналогичен изотермиче-
ской перегонке, при которой в замкнутом пространстве крупные
капли или кристаллы растут за счет мелких вследствие большего
давления насыщенного пара малых капель или кристалликов. Та-
кая неустойчивость золей, выражающаяся в появлении крупных
частиц, проявляется тем быстрее, чем больше растворимость дис-
персной фазы. Регулируя растворимость дисперсной фазы путем
изменения состава дисперсионной среды или температуры, можно
влиять на скорость процесса в жидкой среде. Именно на этом ос-
нованы методы укрупнения мелких частиц, проходящих через
фильтр, что особенно важно при проведении анализов в аналити-
ческой химии. Однако в связи с обычно очень малой раствори-
мостью дисперсной фазы разрушение коллоидных систем в резуль-
тате роста больших частиц за счет малых происходит, как правило,
весьма медленно, и с этим видом потери устойчивости исследова-
телю, работающему в области коллоидной химии, приходится
иметь дело сравнительно редко.
Гораздо большее значение для коллоидных систем имеет коагу-
ляция, ведущая к образованию агрегатов.
Коллоидные системы обладают весьма различной агрегативной
устойчивостью. Некоторые системы живут лишь секунды после их
* Глава написана совместно с Б В. Дерягиным.
9*
259
образования, но есть много коллоидных систем, существующих
весьма длительное время. Известно, например, что типичные гид-
рофобные золи иодида серебра и сульфида мышьяка, из которых
удалены чужеродные электролиты, очень устойчивы и могут хра-
ниться годами. Объяснить длительное существование принципиаль-
но неустойчивых коллоидных систем только малой концентрацией
золя и, следовательно, редкостью столкновения частиц, очевидно,
нельзя, так как в хорошо очищенных золях концентрация дисперс-
ной фазы может быть доведена до 10% и более.
Агрегативная устойчивость дисперсных систем в очень сильной
степени зависит от состава дисперсионной среды и может быть
резко изменена введением в нее даже очень малых количеств чу-
жеродных электролитов. По влиянию добавок электролитов на
устойчивость коллоидные системы можно разделить на два класса:
лиофобные и лиофильные системы. В лиофобных системах при до-
бавлении электролитов'резко увеличивается скорость коагуляции.
После перехода через некоторый предел — критическую концентра-
цию — скорость коагуляции достигает предельного значения, ха-
рактеризующего так называемую быструю коагуляцию. Лиофиль-
ные коллоидные системы коагулируют, если концентрация прибав-
ляемого электролита весьма велика — порядка молей на литр.
Критические концентрации электролитов для лиофобных систем
(в противоположность лиофильным) резко уменьшаются с ростом
заряда противоионов — ионов, заряженных разноименно с зарядом
коллоидных частиц.
Эти особенности агрегативной неустойчивости лиофобных си-
стем, например золей металлов, заставили (Гарди, 1901 г.) пред-
положить, что устойчивость лиофобных золей обусловлена элек-
трическим зарядом их частиц, обнаруживающимся в явлениях
электрофореза. После того как эта догадка подтвердилась, стало
ясно, что механизм устойчивости и природа лиофобных дисперс-
ных систем иные, чем лиофильных.
Наиболее основательно и успешно изучены, особенно в теорети-
ческом отношении, лиофобные системы, на устойчивости и коагу-
ляции которых мы в первую очередь и остановимся.
В свое время были сделаны попытки трактовать агрегативную
устойчивость лиофобных коллоидных систем с позиций термодина-
мики. Ряд авторов (например, Марх), учитывая, с одной стороны,
положительную свободную энергию поверхности раздела и, с дру-
гой стороны, понижение свободной энергии в результате образо-
вания на частицах двойного электрического слоя, а также энтро-
пию системы пытались определить условия, при которых фактор,
способствующий коагуляции, уравновешивается противодействую-
щим фактором, и поэтому коллоидная система является агрегатив-
но устойчивой. Однако все~эти попытки, за исключением специаль-
ных случаев (см. гл. VIII), кончились неудачей, так как эти авторы
не учитывали, что при слипании частиц поверхность раздела ча-
стица — дисперсионная среда существенно не меняется (см. гл. I)
260
и изменение энергии необходимо подсчитывать, исходя из дей-
ствия сил притяжения между частицами. Эти силы вместе с сила-
ми отталкивания определяют агрегативное состояние системы.
В связи с этим агрегативная устойчивость системы обычно
означает медленность процесса коагуляции, т. е. носит кинетиче-
ский, а не термодинамический характер. Даже полное агрегатив-
ное равновесие, когда процессы агрегации и распада агрегатов
взаимно уравновешиваются, еще не означает термодинамического
равновесия всей системы в целом.
1. КИНЕТИКА КОАГУЛЯЦИИ
Прежде чем перейти к обсуждению причин устойчивости и коа-
гуляции лиозолей, рассмотрим теорию кинетики коагуляции, кото-
рая, кстати говоря, была разработана гораздо раньше теории
устойчивости коллоидных систем.
Различают быструю и медленную коагуляцию. Под быстрей
коагуляцией подразумевают такую коагуляцию, при которой все
сближения частиц, находящихся в броуновском движении, кон-
чаются их слипанием. При медленной коагуляции вследствие того,
что на поверхности коллоидных частиц частично сохранился двой-
ной электрический слой, сольватная оболочка и т. д., слипание ча-
стицпрдисхбдйт лишь в результате особо удачных сближений.
Таким образом, оба термина являются вполне условными.
Теория кинетики быстрой коагуляции создана польским уче-
ным Смолуховским. Основные положения, из которых исходил
Смолуховский, сводятся к тому, что между частицами золя дей-
ствуют силы притяжения и отталкивания; последние ослабевают
при введении электролита и при концентрации электролита, вы-
зывающей быструю коагуляцию, исчезают вовсе. Дальнейшее при-
бавление электролита не может ускорить коагуляцию. Частицы
такого астабилизованного золя при сближении в процессе броу-
новского движения на достаточно близкое расстояние слипаются
под давлением сил молекулярного притяжения, образуя агрегат,
который совершает в дальнейшем броуновское движение как одно
целое. Природу сил, действующих между частицами, Смолухов-
ский не рассматривал.
Для экспериментального изучения кинетики коагуляции необхо-
димо было определить изменение концентрации частиц в золе по
мере коагуляции. Это можно было бы осуществить путем счета
частиц с помощью ультрамикроскопа. Однако определение числен-
ной концентрации таким методом весьма длительно, а коагуляция
протекает обычно очень быстро, так что к концу счета концентра-
ция частиц в золе оказалась бы совсем иной, чем в его начале.
Выход был найден в том, что в золь, в который был уже введен-
электролит и который таким образом находился в состоянии коа-
гуляции, в определенный момент вводился стабилизатор, «обры-
вающий» коагуляцию. В таком стабилизованном золе численная
261
Рис. IX, 1. Схема, поясняю-
щая сферу действия частиц
при быстрой коагуляции.
концентрация частиц уже не меняется и его можно исследовать
с помощью ультрамикроскопа. В качестве стабилизатора для «об*
рыва» коагуляции обычно применялся желатин. В настоящее вре-
мя метод, основанный на счете частиц в неподвижном объеме, не
применяется.
[/Современный метод изучения процесса коагуляции основан на
счете частиц в потоке. Поточный ультрамикроскоп, разработанный
впервые Б. В. Дерягиным и Г. Я. Власенко, описан в гл. II. Этим
прибором можно определять численную концентрацию, не преры-
вая процесса коагуляции, во много раз быстрее, чем по старому
способу. Одновременно новый способ устраняет многие источник»
ошибок, присущие старому методу изме-
рения численной концентрации золя. Не-
давно этот метод был усовершенствован
Мак Файденом и А. Смитом.
Смолуховский при создании своей
теории принимал, что скорость быстрой
коагуляции, т.е. изменение численной
концентрации частиц в единицу времени,,
зависит от численной концентрации зо-
ля v, от интенсивности броуновского
движения, характеризующейся коэффи-
циентом броуновской диффузии частиц
D, и от критического расстояния р, на
которое должны приблизиться друг к
другу центры двух частиц, чтобы произошло слипание частиц.
Расстояние р может превышать диаметр коллоидных частиц
(рис. IX, 1). Таким образом, если представить себе сферу радиу-
са р, центр которой совпадает с центром одной из частиц, другая
частица прилипнет к ней только тогда, когда центр второй части-
цы коснется поверхности этой сферы, называемой сферой погло-
щения. При расстояниях, больших р, действием молекулярных сил
притяжения на броуновское движение частиц и на процесс их.
сближения Смолуховский полностью пренебрегал.
При быстрой коагуляции агрегация, согласно Смолуховскому»
идет таким образом, что первоначальные одинарные частицы, стал-
киваясь друг с другом, образуют двойные частицы, затем двойные
частицы, сталкиваясь с одинарными, образуют тройные и т. Д-
Возможны столкновения между собой и сложных частиц. Одно-
временное столкновение трех н более простых или сложных частиц
возможно, но вследствие малой вероятности такого события Смо-
луховский для упрощения теории не принимал в расчет подобные
столкновения.
Обозначая через vi, vz, V3 ... численные концентрации частиц,,
состоящих из одной, двух, трех и т. д. первоначальных частиц»
можно написать, что в начале, когда время т = О
Vi = v0 и va = vs== ... v„=0
262
по истечении времени т
v = vt = v, + v, + v3 + ...
где v — конечная численная концентрация.
При этом, очевидно, будет соблюдаться неравенство:
V < Vo
Смолуховский считал, что формально процесс коагуляции можно
приравнять к реакции второго порядка. Тогда скорость коагуля-
ции должна быть прямо пропорциональна квадрату численной кон-
центрации, определяющему вероятность (или частоту) сближения
частиц до расстояния р:
— dvfdr = Kv2 (IX, f)
где к — константа, характеризующая вероятность сближения.
Знак минус перед производной dvfdx стоит потому, что с течением
времени т численная концентрация v падает.
По Смолуховскому, константу к, зависящую от скорости броу-
новской диффузии D и расстояния р, можно выразить так:
к = 4л£>р (IX, 2)
Уравнение (IX, 2) можно вывести следующим образом. Совместим начало
координат с центром какой-либо произвольно выбранной частицы. В этой си-
стеме координаты смещения остальных частиц Д' будут в действительности рав-
няться смещениям относительно выбранной, т. е.
Д' = Д/-Д1
где Д, — действительное смещение какой-либо i-частицы; Д1 — смещение части-
цы, помещенной в начало координат.
Очевидно, что возведя обе части уравнения в квадрат и усредняя по вре-
мени <получим:
(Д7)2 = (Д; - Д,)2 = Д2 - 2Д^] + Д? (IX, 3)
В этом уравнении черта над символом величины означает, что берется среднее
значение этой величины.
При броуновском движении смещения обеих частиц совершенно независимы
друг от друга. Поэтому, как можно строго доказать статистически, порядок t
операций усреднения и умножения можно обратить:
Д^Д] = Д; • Д]
Но Д, = О и Д[ = О, так как все направления смещений равновероятны. По-
этому Д,Д, = 0 и, следовательно
—2 —2 —2
(д')2 = Д1 + д 1 = 2Д7 (IX, 4)
Таким образом, средние квадраты смещений всех частиц относительно первой
удваиваются. Благодаря связи Д2 с коэффициентом диффузии можно сказать,
что броуновское движение остальных частиц относительно первой характери-
зуется вдвое большим коэффициентом диффузии D'. В результате этого время от
времени частицы будут сближаться с первой до критического расстояния их
Центров р. Подсчитаем число таких сближений, не учитывая тех осложнений,
которые возникают при изменении броуновского дви.’кения вследствие слипания
263
частиц. Для этого примем, что после сближения частиц до расстояния центров р
соответствующая частица как бы поглощается центральной. Пусть частицы за-
ключены в очень большой объем. Допустим также, что в рассматриваемый мо-
мент коагуляции численная концентрация v равна vt. Однако в непосредствен-
ной близости от поверхности поглощающей сферы концентрация частиц близка
к нулю, так как ввиду беспорядочного характера броуновского блуждания ве-
роятность попадания частицы, находившейся вблизи поглощающей сферы, в эту
сферу очень велика, а вероятность избежать этого мала. При удалении от
сферы р быстро достигаются значения vT. Совокупное перемещение частиц, со-
вершающих независимо друг от друга беспорядочные блуждания, описывается
уравнением диффузии. Допустим, что концентрация частиц v(J?) зависит только
От расстояния R от начала координат, к которому направлен поток диффузии,
вызванный поглощением частиц сферой радиуса р. В этом случае из законов
диффузии следует:
Q = « D'4nR2 (IX, 5)
aJ\ aJ\
где Q — число частиц, проходящих за единицу времени через поверхность сфе-
ры s радиуса R по направлению к центральной частице.
Число Q, очевидно, равно числу частиц, поглощенных за единицу времен»
центральной частицей при сближении с ней, т. е. Q равно искомой частоте актов
агрегации с участием центральной частицы.
Учитывая граничное условие
v(p) = 0 (IX, 6>
которое следует из того, что частицы, достигнув сферы р, поглощаются централь-
ной частицей, и интегрируя уравнение (IX, 5), получим:
Учитывая другое граничное условие
v(co) = v0 (IX,8)
получим:
Q = 4nD'vop (IX, 9)
Общее число сближений всевозможных частиц в единице объема можно опреде-
лить, умножая Q на численную концентрацию v и деля полученное произведе-
ние на 2, так как иначе каждая пара частиц будет подсчитана дважды. Заменяя
D' на 2D, получим:
- dv/dr = 4nZ>pv2 (IX, 10)
Сравнивая это уравнение с уравнением (IX, 1), мы найдем значение константы к
в уравнении Смолуховского (IX, 2):
к = 4л£>р
Следует заметить, что формула Смолуховского предполагает существование на-
чального момента коагуляции, до которого слипания частиц не происходило.
Поэтому в начальный момент центральная частица окружена частицами, кон-
центрация которых всюду равняется Vo. Вследствие этого в начале процесса
Коагуляции частота сближения и слипаний несравненно больше, чем величина Q
в уравнении (IX, 9). Однако очень скоро именно благодаря высокой начальной
частоте сближений концентрация частиц вблизи центральной уменьшается до
нуля, и за доли секунды установится распределение частиц, подчиняющееся
уравнению (IX, 6). После этого скорость коагуляции будет подчиняться уравне-
нию Смолуховского.
Однако через некоторое время начнутся отклонения от закона Смолухов-
ского, Основная причина заключается в том, что коэффициент к в формуле
(IX, 2), выведенный при рассмотрении процесса сближения двух одинарных ча-
стиц, принимает иное значение при сближении с агрегатом слипшихся частиц
другого агрегата или даже одиночной частицы.
264
Придав уравнению (IX, 1) вид:
— rfv/v2=KrfT (IX, 11)
и проинтегрировав его в пределах от Vo до v и от 0 до т, получим:
1/v — 1/Vo = кт
откуда численная концентрация золя в момент т будет равна:
v = -i-r^-- (IX, 12)
1 + kvot
Вводя время половинной коагуляции 0, можем написать
kvq = 1/е (IX, 13)
Тогда основное уравнение (IX, 12) можно представить так:
v-bft/e (IXJ4)
По уравнению IX, 14, зная vo и v для различных значений т,
можно вычислить 0 и к.
Графически зависимость относитель-
ных изменений общего числа частиц У v
и числа частиц vi, vz, vz ... в единице
объема золя от т/0 показана на рис.
IX, 2. Из уравнения (IX, 14) следует, что
1/v есть линейная функция времени т.
Это следствие теории неоднократно было
подтверждено на опыте.
Теория Смолуховского позволяет так-
же вычислить расстояния р, на которые
должны приблизиться центры двух ча-
стиц для того, чтобы произошло их сли-
пание.
Так как
3
V
I
2
1
ч!/е—~
Рнс. IX, 2. Зависимость от-
носительного числа частиц
различного порядка от т/0
при быстрой коагуляции.
(IX, 15)
е____L_ = _L_
kv0 4n£>pv0
и, согласно Эйнштейну,
D = A.T-J—
6лт)Г
где k — константа Больцмана; г) — Динамическая вязкость дисперсионной среды;
Г — радиус частиц,
то, комбинируя два последних уравнения, получим:
6Т)Г
$pvokT
е
или
Р
яда» <“ ,6>
Согласно эксперименту, р/г = 2,3, следовательно, расстояние р
довольно близко к 2г, т. е. силы аттракции начинают действовать
265
лишь тогда, когда частицы приблизятся на весьма малые расстоя-
ния, на которых энергия 'молекулярного притяжения становится
намного больше энергии теплового, а следовательно, и броуновско-
го движения 3lzkT.
Представления Смолуховского объясняют коагуляцию монодис-
персных золей. Мюллер разработал подобную же теорию для
объяснения коагуляции полидисперсных систем. Он показал, что
частицы различных размеров агрегируются всегда скорее, чем оди-
наковые частицы. При этом большие частицы играют роль как бы
«зародышей» коагуляции; такую же роль могут играть и агрега-
ты, образующиеся в начальной стадии коагуляций приблизительно
монодисперсного золя золота, как об этом свидетельствуют на-
блюдения Б. В. Дерягина и Н. М. Кудрявцевой. Впрочем, положе-
ния Мюллера полностью верны лишь тогда, когда в золе имеются
частицы, существенно превосходящие по размеру малые частицы.
Теория Мюллера объясняет автокаталитический характер коагу-
ляции, скорость которой может постепенно возрастать со временем.
Мюллер также показал, что коагуляция ускоряется, если частицы
имеют удлиненную форму, так как на поступательное броуновское
движение налагается еще вращательное движение, увеличиваю-
щее вероятность столкновения таких частиц.
Теория Смолуховского, как мы неоднократно подчеркивали,
пригодна "Для быстрой коагуляции. Им было сделано предполо-
жение, что разработанная теория может быть приложима и к мед-
ленной коагуляции, когда частицы не полностью астабилизованы.
В этом случае в уравнение IX, 12, описывающее процесс коагуля-
ции, следует ввести эффективность сближения е:
v т“т-г^----- (IX, 17>
Однако, как следует из теории коагуляции Н. А. Фукса, приложи-
мой к частицам, силы взаимодействия между которыми изменяют-
ся с расстоянием по любому закону, параметру е надо придать
другой смысл, так как понятие эффективности сближения по Смо-
луховскому неприменимо к процессу сближения частиц, совер-
шающих броуновское движение.
Теория Смолуховского предполагает, что до сближения частиц на опреде-
ленное расстояние р никаких сил взаимодействия между ними нет. Для учета
сил дальнодействия частиц, как функции от расстояния их центров /?, Н. А. Фукс
дополнил формулу (IX, 5) членом, выражающим дрейф частиц по иаправленик>
к центральной частице под влиянием сил притяжения F:
Q = 4nR2(D' dv/dR + 2$Fv) (IX, 18>
где Р — подвижность частицы в вязкой среде, равная отношению ее скорости
к действующей силе; цифра 2 перед 0 введена потому, что в действительности
обе частицы движутся навстречу друг другу.
Согласно Эйнштейну 0 = D/kT, следовательно, 20 = D’/kT. Далее, заме-
няя силу F на dU/dR (где U — потенциальная энергия молекулярного взаимо-
266
действия двух частиц) и интегрируя уравнение (IX, 18), вместо (IX,9) получим:
Q == «--------------- (IX, 19)
^eu^/kr-dR/R2
р
При £/(/?)= О снова получается формула (IX, 9). Уравнение (IX, 19) было
выведено Н. А. Фуксом первоначально для аэрозолей с целью учета ускорения
коагуляции под влиянием притяжения разноименно заряженных частиц. В этом
случае потенциальная энергия взаимодействия отрицательна и скорость коагуля-
ции возрастает.
В общем случае из (IX, 19) вместо (IX, 10) можно получить уравнение
dv 4nDpv2
"dx** “
J euWkTdRlR2
p
и вместо константы Смолуховского к величину, меньшую в W раз, где
«7= ^euw/kT dR/R2
• р
(IX, 20)
Величину W называют коэффициентом замедления. Легко видеть, что уравнение
медленной коагуляции получится путем замены е на \/W. Таким образом, теория
Фукса дает теоретическое истолкование коэффициента Смолуховского е.
Для коллоидной химии особый интерес представляет случай,
когда между частицами преобладают силы отталкивания. При
этом U(R)> 0, и если для каких-то значений R величина U(R)
много больше, kT, то значение Q становится крайне малым. Это
Значит, Что скорость коагуляции настолько снизится, что можно
говорить о практической агрегативной устойчивости системы.
Для исследования кинетики коагуляции Б. В. Дерягиным и Н. М. Кудрявце-
вой был применен поточный ультрамикроскоп (по схеме, близкой к поточному
ультрамикроскопу для аэрозолей Б. В. Дерягина и Г. Я. Власенко). С помощью
поточного ультрамикроскопа можно определять за 2—3 мин численную концен-
трацию гидрозолей вплоть до 1010—1011 частиц в 1 см3; другие способы счета
частиц не позволяют измерять концентрацию больше 104— 105 частиц в 1 см3.
При применении достаточно концентрированных золей с помощью поточного
микроскопа можно наблюдать не только быструю, но и медленную коагуляцию,
отвечающую малым значениям коэффициента е, не затрачивая для этого чрез-
мерно много времени.
На рис. IX, 3 приведены графики зависимости 1/v от времени т, прошедшего
от начала коагуляционного процесса для сравнительно низкодисперсного голу-
бого гидрозоля золота; отдельные кривые отвечают различным концентрациям
коагулирующего электролита. Можно видеть, что при высокой концентрации
электролита (кривые 3 и 4), при которой потенциальный барьер * исчезает, кине-
тика коагуляции характеризуется линейной зависимостью, вытекающей из теории
* С понятиями потенциальный или
яма учащиеся должны быть знакомы из
но об этих понятиях в приложении их
дующих разделах этой главы.
энергетический барьер и потенциальная
курса физической химии. Более подроб-
к коллоидным системам сказано в сле-
267
Смолуховского. Несколько меньший наклон прямой к оси абсцисс объясняется
согласно В. М. Муллеру тем, что на близких расстояниях вязкое сопротивление
жидкой прослойки сближению сферических частиц возрастает по сравнению с
сопротивлением, рассчитанным по формуле Стокса При малых концентрациях
электролита линейная зависимость ^кривые 1, 2) нарушается. Типичной является
кривая 2. После начального подъема кривой следует участок, почти параллель-
ный оси абсцисс, и в некоторый момент происходит новый подъем кривой, в
дальнейшем не прекращающийся. Согласно Б. В. Дерягину и Н. М. Кудрявцевой
первоначальный подъем кривой и, следовательно, уменьшение численной концен-
Рис. IX, 3. Кинетика коагуляции
голубого гидрозоля золота в при-
сутствии хлорида калии.
Концентрация коагулирующего электро-
лита (в моль/л):
1—5 • 10~3; 2—1 • 10“2; 3 —5 • 10“2;
4 — 1 10“ *.
трации золя означает образование агрега-
тов из двойных частиц. При малых концен-
трациях электролита ближняя потенциаль-
ная яма сравнительно не глубока, энерге-
тические взаимодействия не велики и
потому распады образовавшихся двойных
частиц происходят с достаточной частотой.
Прн достижении определенной концен-
трации двойных частиц их распады урав-
новешивает процесс слипания одиночных
частиц, вследствие чего численная концен-
трация золя становится постоянной. В не-
который момент к одной из двойных частиц
прилипает третья частица, образуя тройную
частицу. Энергия связи каждой из трех
частиц образовавшегося агрегата в два раза
больше, чем у частицы, входящей в двой-
ную частицу. Поэтому такая тройная части-
ца имеет мало шансов распасться. Одновре-
менно происходит дальнейший рост агрега-
тов за счет присоединения новых частиц.
И действительно, визуальные наблюдения
под микроскопом показали, что в некоторый
момент среди сравнительно слабо видимых
частиц (по «вспышкам» в поле зрения по-
точного ультрамикроскопа) появляются все
более яркие и коагуляция все более уско-
ряется. Этим объясняется форма кривых
с перегибом. При более высоких концен-
трациях электролита вследствие снижения
энергетического барьера и углубления по-
тенциальной ямы горизонтальные участки графика укорачиваются и, наконец,
исчезают, ио S-образная форма кривых сохраняется. Таким образом, при изуче-
нии коагуляции необходимо учитывать не только процессы агрегации, но и рас-
пада агрегатов.
Дальнейшее развитие этих положений принадлежит Г. А. Мартынову и
В. М. Муллеру. В определенных условиях может устанавливаться агрегативное
равновесие между одинарными и агрегированными частицами. Хотя вероятность
распада крупных агрегатов меньше, чем парных, все же уменьшение числа оди-
нарных частиц в конечной стадии коагуляции может настолько понизить ско-
рость образования новых агрегатов, что коагуляция будет уравновешена ско-
ростью распада агрегатов. Следовательно, возможно равновесие между коагу-
лятом и оставшимся разбавленным золем. Это явление, однако, не носит общего
характера, так как существуют золи, коагулирующие необратимо, и обнаружен-
ное поведение золей золота в работе Н. М Кудрявцевой, по-видимому, связано
с частичной гидрофилизацией поверхности его частиц за счет адсорбции органи-
ческих компонентов, остающихся в золе после его приготовления.
Следует отметить, что учет образования и распада агрегатов и его ускоре-
ния в градиентном потоке, как было показано уже давно Гудивом, Жнллепси
и др., позволяет объяснить реологическое поведение тиксотропных суспензий,
о чем будет сказано в гл. X.
268
От распада агрегатов в процессе коагуляции следует отличать явление пеп-
тизации — распада агрегатов в результате изменения ионного состава диспер-
сионной среды, о чем уже говорилось в гл. VIII. В этом случае пептизация
происходит благодаря усилению электрической слагающей расклинивающего
давления и уменьшению глубины потенциальной ямы.
2. УСТОЙЧИВОСТЬ тонких СЛОЕВ жидкости И ЭНЕРГИЯ
ВЗАИМОДЕЙСТВИЯ МЕЖДУ ПОВЕРХНОСТЬЮ ДВУХ ТЕЛ
Силы взаимодействия между коллоидными частицами, прояв-
ляющиеся при утоньшении разделяющих их прослоек жидкости,
могут как ускорять коагуляцию, так и сильно ее тормозить. Чтобы
выяснить1 роль таких прослоек и механизм их стабилизующего дей-
ствия, рассмотрим их поведение на примере простой схемы, когда
прослойка жидкости разделяет параллельные поверхности двух
пластинок. В этом случае разделяющая прослойка всюду имеет
одинаковую толщину и по краям граничит с дисперсионной сре-
дой, в которую погружены обе пластинки.
При достаточно малой толщине прослойки (практически мень-
ше 1 мкм) гидростатическое давление в ней отличается от давле-
ния в окружающей жидкости на величину расклинивающего дав-
ления, являющегося функцией толщины прослойки. Поэтому для
сохранения равновесия, при котором толщина прослойки остается
постоянной, к пластинкам необходимо приложить силу, уравнове-
шивающую расклинивающее давление прослойки. Определив зна-
чение уравновешивающего давления, можно тем самым найти и
расклинивающее давление П. Если гидростатическое давление в
прослойке понижено или пластинки притягиваются друг к другу,
то расклинивающее давление имеет отрицательное значение.
Рассмотрим природу сил, определяющих расклинивающее дав-
ление и его зависимость от толщины зазора между пластинками.
Молекулярные силы притяжения. Прежде всего следует рас-
смотреть силы молекулярного притяжения, действующие между
поверхностями любых тел, как одинаковой, так и различной при-
роды.
Закон молекулярного притяжения всего проще проявляется при
взаимодействии пары молекул в отсутствие других молекул, кото-
рые могут изменять силу молекулярного притяжения. Как было
показано впервые с помощью квантовой механики Лондоном, сила
притяжения FM меняется обратно пропорционально седьмой сте-
пени расстояния г между молекулами, а молекулярная энергия
притяжения — обратно пропорционально шестой степени рас-
стояния:
Uu = -a/r* (IX, 21)
Fм = - dU^jdr ~ - 6 а/г7 (IX, 22)
Знак минус в уравнении (IX, 21) указывает на убывание энергии
при сближении молекул; знак минус в уравнении (IX, 22) пока-
зывает, что сила притяжения увеличивается при уменьшении
269
расстояния г. Константа а изменяется в довольно широких преде-
лах в зависимости от природы обеих молекул. Чтобы оценить,
насколько велико влияние потенциальной энергии на движение ча-
стиц, следует разделить величину UM на энергию теплового дви-
жения молекул kT:
kT rs
где b = a/kT.
Существенно, что отношение U^IkT становится весьма малым уже
при расстояниях в несколько молекулярных радиусов.
Формулы (IX, 21) и (IX, 22) применимы и тогда, когда моле-
кул много, но средние расстояния_между ними велики по сравне-
Рис. IX, 4. Схема, поясняющая ра-
счет энергии притяжения к толстой
пластинке.
ния равнодействующей силы,
нию с их радиусами, как это, на-
пример, имеет место в газах.
В таком случае полную энергию
всех молекул можно найти, сум-
мируя выражения (IX, 21) для
всех пар молекул. Кроме того,
зная поправку в уравнении Ван-
дер-Ваальса, учитывающую вза-
имное притяжение молекул, мож-
но найти и значение константы а.
Подобное суммирование мо-
лекулярных взаимодействий в
качестве первого приближения
можно применить и для вычисле-
с которой притягиваются друг
к другу две пластинки, разделенные плоскопараллельным зазором
шириной h *.
Определим сначала энергию взаимодействия 17м одной молекулы со всеми
молекулами пластинки, расположенной на расстоянийг0 от нее (рис. IX, 4).
Предполагается, что толщина пластинки много больше не только h, но и ра-
диуса молекулярного действия г. В этом случае суммирование по всем парам
молекул без заметной ошибки можно заменить интегрированием в пределах от
Го до оо;
оо
и'и = - j qsa dr/r* (IX, 23)
га
тде q — число молекул в единице объема; s — площадь сегмента шара радиуса г,
заключенного внутри объема пластинки, а — константа. Как известно, поверх-
ность шарового сегмента равна:
s — 2лг (г — г0)
Подставляя значение .? в уравнение (IX, 23) и интегрируя, получим
Т1' _ f (г - fo) п<!а
UM-~2rtqa\ ---------dr = -~г
г, г 6r0
(IX, 24)
* Приведенный ниже способ расчета предложен Шелудко.
270
Для перехода от одной молекулы к единице площади слоя толщиной dr0 сле-
дует умножить значение UM на q dr0. Полная энергия притяжения обеих пла-
стинок будет равна:
СО
^м = -$ <^0 = —g--^- (IX,25)
h
Отсюда сила притяжения, приходящаяся на единицу площади, определится
уравнением:
р _ ди* — п
а dh 6
А
6лЛ3
(IX, 26)
где А — константа Гамакера.
Измерив силу притяжения пластинок для малых расстояний,
можно определить константу а, а также константу Гамакера:
A=n2q2a (IX, 27)
Значение Ем (взятое со знаком минус) выражает слагающую
расклинивающего давления Пм. зависящую от молекулярного при-
тяжения.
Впервые измерения силы Ем были проведены Б. В. Дерягиным
и И. И. Абрикосовой в 1953—54 гг. Позднее их измерения были
подтверждены английским исследователем Китченером и голланд-
скими учеными Спарнеем, Овербеком, Де Йонгом и др. Уже на
основании данных первых измерений Б. В. Дерягина и И. И. Абри-
косовой выяснилось, что силы притяжения при расстояниях между
пластинками больших 300 А оказываются меньше, чем можно
было ожидать, исходя из значений а, и убывают с расстоянием
быстрее, чем это подсказывает теория.
Это побудило советского теоретика Е. М. Лифшица разрабо-
тать теорию притяжения тел, состоящих из многих молекул, на
новой основе. Согласно Лифшицу, во всех средах существуют бес-
порядочно флуктурирующие во времени и в пространстве электро-
магнитные-поля. Распространяясь в пространстве в виде волн, в
том числе видимой и ультрафиолетовой области, и проникая в со-
седние тела, они взаимодействуют с их молекулами. Это взаимо-
действие и создает силы притяжения между обоими телами в тех
случаях, когда разделяющий их зазор достаточно узок. Не приводя
сложных расчетов, основанных на квантовой механике, изложим
выводы этой теории.
Теория Е. М. Лифшица показывает, что при компактном распо-
ложении молекул, имеющем место в твердых телах и жидкостях,
энергию взаимодействия нельзя вычислять простым суммирова-
нием энергии взаимодействия отдельных пар молекул вследствие
отсутствия аддитивности молекулярных взаимодействий. Тем не
менее сила притяжения между двумя пластинками зависит от рас-
стояния h так же, как это показано в формуле (IX, 26), однако
константа А получает по Е. М. Лифшицу другое выражение.
Е. М. Лифшиц связал значение этой константы с оптическими
271
свойствами тел. Следует указать, что формула (IX, 26) справедли-
ва только для расстояний между пластинками не больше, чем
200 А. При больших расстояниях надо принимать во внимание так
называемое электромагнитное запаздывание. Оно связано с элек-
тромагнитной природой молекулярных сил, которая была предска-
зана еще русским физиком П. Н. Лебедевым. В силу этой природы
молекулярные силы распространяются не мгновенно, а со ско-
ростью света. Не приводя сложные выводы теории, укажем, что
в результате электромагнитного запаздывания на расстояниях бо-
лее 300 А сила притяжения меняется по закону:
= (IX, 28)
При этом для теоретического определения константы В доста-
точно знать только диэлектрическую проницаемость тел *.
Табором и другими учеными были измерены силы притяжения
твердых тел при значениях ширины зазора, доходивших до 50 А, и
для малой ширины зазора была обнаружена зависимость согласно
формуле (IX, 26), выражающей Молекулярное притяжение при
условии отсутствия электромагнитного запаздывания. Для боль-
шинства твердых и жидких тел значения константы А лежат в ин-
тервале 1014 — 10~12 эрг.
' В коллоидной химии большой интерес представляет взаимодей-
ствие поверхностей, разделенных жидкой прослойкой, или, иначе,
расклинивающее давление жидкой прослойки. Было показано, что
уравнения (IX, 26) и (IX, 28) применимы и в этом, более общем
случае, причем константы А и В зависят от свойств как обоих тел,
так и жидкой прослойки. Последняя может не только уменьшать
притяжение поверхностей, но и превращать притяжение в отталки-
вание. Иными словами, молекулярная или ван-дер-ваальсова сла-
гающая Пм расклинивающего давления жидкой прослойки может
быть как отрицательной, так и положительной. Последний случай
чаще всего наблюдается для смачивающей пленки, отделяющей
пузырек газа от твердой подложки, хотя в этом случае нельзя
говорить о взаимодействии двух тел через прослойку жидкости.
Отсюда, в частности, следует, что понятие расклинивающего дав-
ления шире, чем понятие взаимодействия тел. Уравнение
п = —
м 6лЛ3
хорошо применимо для не слишком толстых пленок.
Силы отталкивания. Кроме молекулярной слагающей раскли-
нивающего давления, следует учитывать и другие его слагающие.
Из них имеет наиболее общее значение и притом наиболее хорошо
изучена сила отталкивания, возникающая при перекрытии диф-
фузных двойных ионных слоев, которые образуются в слоях рас-
* Применительно к металлам эта формула была дана еще раньше Казими-
ром и Польдером.
272
твора электролита на границе с поверхностью любой инородной
фазы.
Метод расчета этой слагающей впервые был разработан и при-
менен Б. В. Дерягиным и далее усовершенствован совместно с
Л. Д. Ландау. Позднее аналогичные расчеты были опубликованы
Ленгмюром, А. Н. Фрумкиным, Бергманом, Лов — Беером и Цохе-
ром. Изложим принципы этих расчетов для более простого случая,
когда потенциал <р0, до которого заряжены поверхности взаимодей-
ствующих фаз, мал.
Рассмотрим распределение электрического потенциала <р между заряжен-
ными до потенциала <₽о поверхностями пластинок, погруженными в раствор
сильного (полностью диссоциированного) бинарного электролита. Поместим на-
чало координат в середине жидкой прослойки толщиной 2хв, направив ось абс-
цисс по нормали к поверхностям пластинок
Если заряженная до потенциала фо поверхность граничит с раствором элек-
тролита, распределение потенциала в образующемся одиночном двойном ионном
слое описывается уравнением:
ф = фое~хг (IX, 29)
где х — величина, обратная эффективной толщине двойного электрического
слоя, I — расстояние от поверхности пластинок. Согласно этому уравнению при
Z = О потенциал <р = фо, а при I -* оо потенциал <р = О
В случае, когда в прослойке раствора электролита происходит перекрытие
двух двойных ионных слоев, интегралом уравнения cPy/dx2 = х2ф будет
Ф — Фо
+ е~х* \
+ е~хх°)
(IX, 30)
7^
где координата х отсчитывается от плоскости, расположенной посередине, между
поверхностями обеих пластинок.
Электрический потенциал согласно уравнению (IX, 30) удовлетворяя соот-
ношению = х2<р, одновременно принимает значение фо при х — ±х0.
Чтобы найти электростатическую силу отталкивания между
пластинками (равную соответствующей слагающей расклиниваю-
щего давления прослойки Пэ), Б. В. Дерягин вычислил разность
сил электрических напряжений и гидростатического давления, при-
ложенных к обеим поверхностям — внутренней и внешней — ка-
ждой из пластинок.
Эта разность равна:
Пэ==8^(Я>-0-(Р1-Ро) (IX, 31)
где е — диэлектрическая проницаемость жидкой прослойки; Н\ и Но — напря-
женность электрического поля- с внутренней и внешней сторон пластинки соот-
ветственно; pi и р0 — соответствующие гидростатические давления.
- Согласно закону Паскаля гидростатическое давление во всех
точках покоящейся жидкости одинаково при отсутствии внешних
сил. В рассматриваемом случае в точках двойных ионных слоев
действуют силы электрического поля, обусловленные зарядами, со-
средоточенными в соответствующих местах. От этого гидростати-
ческое давление изменяется, причем давление в какой-либо точке
жидкой прослойки по сравнению с равномерным давлением в
273
объеме электролита определяется значением электрического потен-
циала в рассматриваемой точке. Поэтому, если потенциал на внут-
ренних поверхностях пластин сохраняет такое же значение, что
и на внешних поверхностях, Р\ = рй.
Может возникнуть мысль, что выражение р, = р0 необходимо дополнить
двумя членами, учитывающими влияние так называемых электрострикционных
членов, дополняющих Максвелловские выражения для напряжений электриче-
ского поля, и влияние аналогичных электрострикционных членов, меняющих
распределение гидростатического давления, в частности давление на пластинки.
Однако, как известно из теории электричества, оба эффекта имеют противопо-
ложные знаки, поэтому компенсируют друг друга
Важно, однако, заметить, что при учете электрострикционных членов избы-
точное гидростатическое давление в медианной плоскости (х = 0) между пла-
стинками, где Н = 0. не равно П8 согласно формуле (IX, 31). Следовательно,
использованный Ленгмюром и вслед за ним другими авторами способ определе-
ния П3 дает совпадающий с (IX, 31) результат только при пренебрежении элек-
трострикционными членами
Полагая, что Н = —d<f/dx и используя формулы (IX, 29) и (IX, 30) при
значениях I = 0 и х = х0, найдем
Я0=хМ (IX, 32)
и
и
« ! — X ф0
(IX, 33)
Наконец, подставляя значения HQ и Нг в уравнение (IX, 31), получим:
тт е 2 2 4
э~ toх Фа (ехх’ +е-х*“)2
(IX, 34)
Возникающая при перекрытии ионных слоев сила отталкива-
ния (электростатическая компонента расклинивающего давления),
пропорциональная квадрату потенциала <р0 поверхностей пласти-
нок, быстро убывает, когда расстояние между пластинками ста-
новится существенно больше, чем эффективная толщина двойных
ионных слоев.
Расчет давления Па сильно усложняется, когда потенциал <ро, возрастай,
нарушает условие малости безразмерного потенциала:
Q = (IX, 35)
где г—валентность иона; е — заряд электрона.
При этом оказывается, что с ростом Q электростатическая слагающая раскли-
нивающего давления, возрастая, стремится к конечному пределу. Это на первый
изгляд мало понятное явление объясняется тем, что вблизи заряженной поверх-
ности с ростом ее потенциала <ро, согласно теореме Больцмана, под влиянием
притяжения противоположных зарядов быстро растет пропорционально экспо-
ненте еа концентрация противоионов В результате действие заряженной поверх-
ности экранируется и потенциал <р с удалением от нее быстро принимает уме-
ренные значения, не увеличивающиеся с дальнейшим возрастанием <р0
Сравнительно легко найти, как меняется давление Па в жидкой прослойке,
в которой происходит резкое падение потенциала с уменьшением ее толщи-
ны 2х0, но значение Q во всех ее точках остается достаточно высоким-
п /- kT\! 1
8 8 \ ze ) х^
(IX, 36)
274
При малых значениях безразмерного потенциала Q = гефо/(А7), получаю-
щихся для больших значений хх0, давление Пэ убывает пропорционально экспо-
ненте е~2хх".
Складывая компоненту расклинивающего давления 1ТМ, завися-
щую от молекулярных сил (имеющую отрицательный знак), со
слагающей, зависящей от перекрытия ионных слоев Пэ (имеющей
положительный знак), получают общее расклинивающее давление
между пластинками с лиофобными поверхностями, у которых от-
сутствует сильное взаимодействие с дисперсионной средой. Если
пластинки имеют лиофильную поверхность, то вследствие сильно-
го взаимодействия с жидкостью может возникнуть третья, струк-
турная слагающая расклинивающего давления слоев жидкости,
структура которых изменена под влиянием лиофильной поверхно-
сти. Эта слагающая расклинивающего давления еще недостаточно
количественно изучена, и мы ее здесь не рассматриваем. _ . ,
До сих пор мы рассматривали взаимодействие одинаковых поверхностей.
Значительно более сложная картина наблюдается, когда жидкая прослойка
разделяет поверхности двух различных тел При этом даже если электрический
потенциал поверхностей одинаков по знаку, отталкивание на достаточно близких
расстояниях переходит в притяжение тем раньше, чем больше различаются
потенциалы Когда толщина прослойки станет много меньше толщины двойного
электрического слоя 1/х, сила притяжения (—Пэ) составит:
-П3 * (^-Ф1 Y (IX, 37)
где ф2 и ф: — потенциалы обеих поверхностей
В случае разноименно заряженных поверхностей притяжение
будет наблюдаться при любых (но не слишком больших) расстоя-
ниях между поверхностями.
Отрицательная молекулярная слагающая расклинивающего
давления (равная ван-дер-ваальсовой силе притяжения) для раз-
нородных поверхностей может стать положительной. Таким обра-
зом, обе слагающие расклинивающего давления могут иметь как
разные, так и одинаковые знаки. Последний случай часто наблю-
дается для смачивающих жидкостей, для которых молекулярная
слагающая положительна так же, как и электростатическая сла-
гающая.
Возвращаясь к рассмотрению тонкого слоя жидкости толщиной
2х0, расположенного между двумя одинаковыми пластинками, изо-
бразим графически зависимость от половинной толщины слоя ха
энергии взаимодействия UM + иэ, вычисленной исходя из стро-
гой теории при условии постоянства потенциала <ро = ЮО мВ
(рис. IX, 5). Показанные кривые относятся к различным значениям
концентрации раствора электролита. При малой концентрации
электролита (кривая 1) почти при всех значениях толщины слоя
превышает отталкивание и только при малых значениях толщины
наблюдается результирующее притяжение, которое достигает ма-
ксимума при некоторой малой толщине. При средней концентра-
ции электролита (кривая 2) потенциальный барьер сдвигается
275
влево, становится круче, уже и выше, при этом при очень большой
толщине прослойки наблюдается неглубокая потенциальная яма.
При достаточно высокой концентрации электролита (кривая 3)
область значений толщины, при которых наблюдается отталкива-
ние, еще больше сдвигается влево и сужается, а правая потен-
циальная яма углубляется. Наконец, при еще большей концентра-
ции (кривая 4) барьер исчезает и обе потенциальные ямы сли-
ваются.
До сих пор речь шла о взаимодействии пластинок. Подобные
случаи реализуются в коллоидных системах, частицы которых
имеют плоскую форму, а также в некоторых глинистых минера-
лах при их внутрикристаллическом набухании. Так, было обнару-
жено, что в кристалле монтмориллонита, насыщенного ионами ли-
тия или натрия, помещенном в слабый раствор хлорида натрия, в
несколько раз меняется расстояние между слоями, составляющими
его слоистую решетку, и соответственно увеличиваются размеры
кристалла. Толщины прослоек раствора NaCl, внедряющихся ме-
жду сетчатыми плоскостями кристалла, могут доходить до 300 А.
Таким образом, совершенно очевидна роль расклинивающего дав-
ления прослоек в процессе набухания кристалликов монтморил-
лонита.
В соответствии с теорией набухание тем больше, чем меньше
концентрация раствора электролита, и процесс прекращается, ко-
гда концентрация одновалентного симметричного электролита до-
стигает некоторого предела — порядка 0,1 М. Однако более рас-
пространены в природе и технике коллоидные системы, частицы
которых имеют более или менее округлую форму. Взаимодействие
(например, сила отталкивания F) двух сферических частиц радиу-
сом г на близком расстоянии h0 может быть вычислено прибли-
женно по формуле:
оо
(IX, 38)
ha
где П — расклинивающее давление между плоскими частицами той же природы,
разделенными прослойкой той же жидкости толщиной h.
Применяя эту формулу и формулы для вычисления Пм и Пэ,
для энергии взаимодействия лиофобных сферических частиц в рас-
творах электролитов можно построить потенциальные кривые, ана-
логичные тем, которые изображены на рис. IX, 5.
Аналогично меняется характер кривых с изменением концен-
трации электролита. Таким образом, применяя указанные выше
диаграммы, исследование агрегативной устойчивости коллоидных
систем сводят к рассмотрению баланса сил молекулярного притя-
жения и электростатического отталкивания.
Выше мы рассматривали взаимодействие между двумя поверхностями, ис-
ходя из представлений о расклинивающем давлении. Однако эти взаимодействия
можно интерпретировать, исходя из энергий (нли сил), действующих между
276
двумя заряженными плоскопараллельными поверхностями, разделенными весьма
малым зазором.
Рассмотрим кратко, не прибегая к использованию довольно сложного мате-
матического аппарата *, окончательные выводы, к которым можно прийти, учи-
тывая энергию взаимодействия Ua между двумя гладкими одноименно заряжен-
ными пластинками, находящимися друг от друга на весьма малом рассстоянии.
Рис. IX, 5. Зависимость энергии взаимодействия пластинок UM + U3
от половинной толщины жидкой прослойки х0.
Л=10~12 эрг; <Рв=100мВ; значения параметра и
I— 105; 2 — 106; 3 —107; 4 —107-5.
Энергию электростатического взаимодействия lfa между этими пластинками,
отнесенную к поверхности в 1 см2, можно выразить следующим уравнением:
,, 64с/? Г 2
=-----—-— уе
. Величина у в этом уравнении имеет следующее значение:
= ехР (zF<pof2RT) — !
Y = ехр (zFtp0/2RT) + 1
Энергия притяжения UK, обусловленная действием молекулярных сил между
пластинками, отнесенная также к поверхности в 1 см2, выражается следующим
уравнением:
“ 12л (2л0)2
где А — константа Гамакера; 2х0— расстояние между пластинками. -
Энергии Ua и Um противоположны по знакам, так как в первом случае
имеет место отталкивание, а во втором случае — притяжение. Таким образом,
общая энергия взаимодействия двух пластинок, отнесенная к 1 см2 поверхности^
в этом случае будет равна:
у — у2е~4хх0 __
х 48лл§
* Соответствующий математический аппарат дан в книге Шелудко, Кол-
лоидная химия. Пер. с болг., под ред. Б. В. Дерягина и С. С. Воюцкого, М.,
Издатннлит, 1960. См. с. 109.
277
В этом уравнении не учитывается компонента, отвечающая структурной сле-
тающей расклинивающего давления слоев жидкости, свойства которых изме-
нены под влиянием поверхностей.
-3. ИЗМЕНЕНИЕ ЭНЕРГИИ ВЗАИМОДЕЙСТВИЯ МЕЖДУ МИЦЕЛЛАМИ
ПРИ ИХ СБЛИЖЕНИИ
На основании материала, изложенного в предыдущем разделе,
можно представить себе, как при сближении двух одноименно за-
ряженных коллоидных частиц будет изменяться энергия их взаи-
модействия, являющаяся результатом сложения молекулярных
сил притяжения и электростатических сил отталкивания. Для это-
го рассмотрим потенциальные кривые (рис. IX, 6 и IX, 7), харак-
теризующие зависимость энергии взаимодействия двух сближаю-
щихся частиц (энергия Отталкивания отложена вверх, а энергия
.притяжения — вниз от нуля) от расстояния Н между частицами.
Рис. IX, 6. Потенциальная кри-
вая для частиц в вакууме, газе
или жидкости, не содержащей
стабилизующих ионов.
Рис. IX, 7. Потенциальные кривые
для частиц в обычном иониоста-
билизованиом лиозоле:
/—изменение энергии молекулярного1'
притяжения; 2—изменение энергии элек-
тростатического отталкивания; 3— ре-
зультирующая потенциальная кривая.
На рис. IX, 6 изображена потенциальная кривая для частиц,
находящихся в вакууме, газе или жидкости, не содержащей стаби-
лизующих ионов и не образующей сольватного слоя. Левая часть
кривой показывает, что при малых значениях Н энергия молеку-
лярного взаимодействия изменяется обратно пропорционально вто-
рой степени расстояния. В правой части кривой при сравнительно
•больших значениях Н энергия молекулярного притяжения из-за
электромагнитного запаздывания изменяется обратно пропорцио-
нально третьей степени расстояния. Расположение всей кривой
ниже оси абсцисс свидетельствует о том, что при отсутствии ста-
билизующего фактора сблизившиеся частицы неизбежно должны
слипнуться. В реальных условиях это отвечает двум частицам
аэрозоля или двум полностью стабилизованным частицам лиозоля.
Скорость коагуляции таких систем определяется только временем,
необходимым для сближения частиц друг с другом в результате
броуновского движения.
278
На рис. IX, 7 изображены кривые, соответствующие сближению
мицелл обычного ионностабилизованного лиозоля, у которого ча-
стицы несут двойной электрический слой. Как можно видеть, кар-
тина здесь гораздо более сложная. Кривая 1 характеризует изме-
нение энергии молекулярного притяжения между частицами, при-
чем она сходна с кривой, изображенной на рис. IX, 6. Кривая 2
характеризует изменение энергии электрического отталкивания
между двойными электрическими слоями частиц и поэтому она
расположена над осью абсцисс.
Кривая 3 является результирующей потенциальной кривой, по-
строенной на основании первых двух путем геометрического сложе-
ния их ординат. При больших расстояниях между частицами ре-
зультирующая кривая лежит под осью абсцисс (вторичная неглу-
бокая потенциальная яма или дальний потенциальный минимум *).
Между частицами наблюдается некоторый перевес сил молекуляр-
ного притяжения, обусловленный тем, что эти силы убывают по-
степенному закону, а силы электростатического отталкивания —
по экспоненциальному. При средних расстояниях, отвечающих тол-
щине эффективных ионных оболочек (порядка 100 нм), кривая
лежит над осью абсцисс, образуя энергетический барьер. Это зна-
чит, что на этом расстоянии превалируют силы электростатиче-
ского отталкивания. Наконец, при более близких расстояниях
опять начинают преобладать силы притяжения, и этот участок кри-
вой снова лежит под осью абсцисс (первичная потенциальная яма
или ближний потенциальный минимум). Для частиц, не обладаю-
щих способностью к коалесценции, первичный минимум обуслов-
лен компенсацией молекулярных сил притяжения борновскими си-
лами отталкивания.
Форма результирующей потенциальной кривой может объяснить-
ряд явлений, наблюдающихся при астабилизации и коагуляции
коллоидных систем.
Отрицательным минимумом,энергии, отвечающим сравнительно
большимД^асст^н^Тм^лежду частицами и представляющим весьма
неглубокую потенциальную яму, объясняется явление тиксотропии,
т. е. превращение при перемешивании гелей в золи с последующим
переходом золей в гели при стоянии системы. Более подробно
о тиксотропии будет сказано в гл. X. Другое явление, которое
* Следует заметить, что взаимодействие частиц на больших расстояниях,
характеризуемое наличием на потенциальной кривой неглубокого отрицательного
минимума, до сих пор не имеет специального названия. Ученые называют это
взаимодействие по разному: дальней коагуляцией, коагуляцией во вторичном
минимуме, дальней агрегацией, флокуляцией. В дальнейшем мы будем поль-
зоваться всеми этими терминами за исключением флокуляции, поскольку термин
«флокуляция» имеет чисто описательный характер (образование хлопьев, фло-
кул) и не зависит от того, происходит ли она в результате истинной коагуляции
или дальней агрегации. Термином коагуляция будем обозначать все виды
агрегации частиц, начиная от коалесценции и непосредственного слипания ча-
стиц и кончая дальней агрегацией. Наконец, под истинной коагуляцией будем
понимать непосредственный физический контакт между частицами.
27$
-можно объяснить, исходя из существования минимума в пра-
вой части потенциальной кривой, заключается в том, что малые
частицы, прилипшие к твердой стенке, способны совершать около
этой стенки довольно интенсивное броуновского движение, что
можно наблюдать под микроскопом (Бузаг, 1929 г). Этим же ми-
нимумом на потенциальной кривой ряд исследователей объясняет
способность многих бактерий и частиц вирусов удерживаться друг
возле друга и образовывать цепочки, явно не соприкасаясь между
собою. Наконец, Лангеланд обнаружил, что в водных дисперсиях
каучука (латексах) сохраняются во времени ансамбли мелких ча-
стиц, движущихся вокруг крупных, что можно объяснить только
силами дальнодействия. Подобным же образом ведут себя микро-
объекты и другой природы.
Щоложите££ьнцйи4акаимум^(энергетический барьер) на кривой,
отвечающий” средним расстояниям, является причиной того, что
при медленной коагуляции не все частицы слипаются друг с дру-
гом при сближении. Если энергия, соответствующая высоте энерге-
тического барьера, меньше или хотя бы одного порядка со средней
кинетической энергией движущихся частиц, то частицы, очевидно,
смогут преодолеть электростатические силы отталкивания, сбли-
зиться на очень малое расстояние, где превалируют молекулярные
силы притяжения, и слипнуться. Если же энергетический барьер
высок, частицы не смогут его преодолеть и образовать агрегаты.
Понятно, что если каким-либо способом, например, прибавляя
электролит в систему, снизить толщину двойного электрического
слоя и тем самым уменьшить силы отталкивания настолько, чтобы
энергетический барьер исчез полностью, частицы при сближении
ддджны обязательно слипнуться.
гНаличие глубокой потенциальной ямы на потенциальной кри-
вой слева от положительного максимума объясняет механическую
'прочность коагулята. Частицы на близких расстояниях прочно свя-
^^ываются друг с другом в результате действия ван-дер-ваальсовых
сил, и образовавшиеся агрегаты приобретают некоторые свойства
твердого тела. Минимум потенциальной кривой, расположенный в
области отрицательных значений энергии взаимодействия, очевид-
но, объясняется уравновешиванием силы молекулярного притяже-
ния силой отталкивания электронных оболочек (силы Борна) и
отвечает физическому контакту обеих частиц. Это наиболее устой-
чивое состояние системы, в котором она обладает наименьшей сво-
•бодной энергией.
Впрочем некоторые исследователи считают, что на поверхности частиц часто
(в основном для лиофильных частиц) существует один или несколько слоев
молекул дисперсионной среды, которые никогда не выдавливаются из зазора,
образующегося между сталкивающимися частицами Это, согласно их мнению,
происходит потому, что энергия десорбции этих молекул больше кинетической
энергии движущихся частиц. Наличие прослоек прочно связанной среды толщи-
ной порядка 6—10 А мешает частицам сближаться на такое расстояние, на кото-
ром энергия молекулярного притяжения становится очень большой Это приво-
дит к качественному изменению вида взаимодействия. Согласно классической
280
теории всегда существует очень глубокая ближняя потенциальная яма, при на-
личии же прослойки среды ближняя яма очень неглубока или вообще отсут-
ствует. В обоих последних случаях коагуляция может происходить лишь за счет
наличия дальней потенциальной ямы.
Следует заметить, что на наличие на поверхности коллоидных частиц прочно
адсорбированных, не выдавливающихся при коагуляции молекул среды указывал
в свое время и П А. Ребиндер.
4. СОЛЬВАТАЦИЯ ЧАСТИЦ, СТРУКТУРНО-МЕХАНИЧЕСКИЙ
И ЭНТРОПИЙНЫЙ ФАКТОРЫ УСТОЙЧИВОСТИ
Представления о молекулярных силах притяжения и электро-
статических силах отталкивания лежат в основе современной тео-
рии устойчивости и коагуляции ионностабилизованных коллоид-
ных систем. Однако существуют и иные причины устойчивости кол-
лоидных систем. Так, устойчивость коллоидных систем с жидкой
дисперсионной средой может быть обусловлена образованием на
поверхности частиц достаточно развитых сольватных слоев из мо-
лекул дисперсионной среды. Способность подобных сольватных
оболочек препятствовать слипанию частиц П. А. Ребиндер объяс-
няет тем, что оболочки обладают сопротивлением сдвигу, мешаю-
щим их выдавливанию из зазора между частицами, а также тем,
что на границе сольватного слоя и свободной среды отсутствует
сколько-нибудь заметное поверхностное натяжение. По Б. В. Деря-
гину, причина нерлипания двух сольватированных частиц при их
сближении заключается в возникновении расклинивающего давле-
ния, обусловленного отличием структуры граничных сольватных
слоев от свойств объемной фазы.
Сольватные оболочки образуются на границе раздела фаз. Ме-
жду сольватными оболочками отсутствует молекулярное притяже-
ние, поскольку сила взаимодействия молекул слоя практически
равна силе взаимодействия молекул среды. При сближении частиц
необходимо совершить работу, расходуемую на удаление сольват-
ного слоя (работа десорбции), что приводит к появлению значи-
тельных сил отталкивания между частицами. Наиболее убедитель-
но подтверждают существование граничных слоев с особой струк-
турой исследования Б. В. Дерягина, Н. В. Чураева, С. В. Нерпина,
Ю. М. Поповского, М. С. Мецика, Г. М. Зорина и др.
Образование достаточно развитых сольватных оболочек мало-
вероятно для коллоидных систем с лиофобной дисперсной фазой
вследствие слабого энергетического взаимодействия среды с дис-
персной фазой. Сольватация может иметь место только в том слу-
чае, когда поверхностные молекулы дисперсной фазы достаточно
сильно взаимодействуют с молекулами дисперсионной среды за
счет химических сил или, по крайней мере, прочных водородных
мостиков.
Примером систем, устойчивость которых можно объяснить соль-
ватацией, являются гидрозоли SiO2 и гидратов окисей некоторых
многовалентных металлов. Эти системы, поскольку они не подчи-
няются обычным закономерностям коагуляции лиофобных золей,
281
в известной мере обратимы и способны существовать без стабили-
затора, можно отнести к лиофильным коллоидным системам. Од-
нако при суждении о причине устойчивости подобных систем надо
быть очень осторожным, так как работами последних лет показа-
но, что во многих случаях эти системы являются не золями, а рас-
творами, содержащими макромолекулы.
Другим примером систем, в которых сольватация, по-видимо-
му, оказывает существенное влияние на устойчивость, могут слу-
жить дисперсные системы с неполярной углеводородной средой,
играющие важную роль при производстве и применении нефтепро-
дуктов. Такие системы, например, растворы поверхностно-актив-
ных веществ и высокодиснерсные взвеси в углеводородах подроб-
но изучены Г. И. Фуксом и его сотр. Оказалось, что устойчивость
этих систем зависит от структуры молекул углеводорода и ее соот-
ветствия структуре молекул частиц дисперсной фазы, а также от
диэлектрической проницаемости среды и от наличия следов ве-
ществ с полярными и дифильными молекулами. Впрочем, для этих
систем, как показал Овербек, нельзя пренебрегать двойным элек-
трическим слоем и электростатическими взаимодействиями.
Были сделаны попытки объяснить устойчивость лиофобных коллоидных си-
стем и с помощью индуцированной сольватации их частиц
Согласно этим представлениям агрегативная устойчивость лиофобных золей,
связанная с возникновением на частицах двойного электрического слоя, объяс-
няется не отталкиванием частиц в результате действия электростатических сил,
а тем, что противоионы двойного слоя сольватируются и таким образом создают
вокруг каждой лиофобной частицы сольватную оболочку
Легко видеть слабые стороны такого объяснения агрегативной устойчивости.
Весьма трудно представить себе возникновение в результате сольватации про-
тивоионов вокруг лиофобных частиц сплошных сольватных оболочек, препят-
ствующих слипанию частиц при их сближении. В самом деле, сольватные
оболочки из полярных молекул среды образуются отдельно вокруг каждого про-
тивоиона двойного слоя. Это должно приводить к тому, что на границе, разде-
ляющей оболочки двух соседних одноименно заряженных противоионов, моле-
кулы среды, представляющие собой диполи, будут обращены друг к другу
-одноименно заряженными концами и, следовательно, будут испытывать взаимное
отталкивание. Кроме того, следует помнить, что микроструктура окружающего
частицы слоя непрерывно меняется в результате теплового движения ионов.
Понятно, что при таких условиях говорить о создании в результате притяже-
ния и ориентации диполей какого-то сплошного слоя из сцепленных друг с дру-
гом ионов и молекул среды, нужного для обеспечения положительного раскли-
нивающего давления или упругости сольватной оболочки, просто невозможно.
Положительное расклинивающее давление, обусловливающее агрегативную
устойчивость лиофобных коллоидов, может возникать лишь в результате дефор-
мации ионных атмосфер, т. е. может определиться только электростатическими
силами
Кроме того, с тем, что устойчивость типичных лиофобных ионностабилизо-
ванных коллоидных систем может определяться сольватацией, не согласуются
способность этих систем коагулировать под влиянием ничтожных количеств
электролитов и вообще все закономерности, наблюдающиеся при электролитной
коа1уляции, о которых мы скажем несколько ниже Далее, при объяснении
устойчивости лиофобных систем сольватацией ионов становится непонятным,
как могут происходить электрокинетические явления.' Для того чтобы воспрепят-
ствовать коагуляции, сольватный слой вокруг частицы должен быть достаточно
толстым и уж во всяком случае простираться за плоскость скольжения мицел-
282
лы Однако в этом случае невозможен разрыв мицеллы по плоскости скольже-
ния прн наложении на нее электрического поля Наконец, объяснение устойчи-
вости коллоидных систем сольватацией противоионов двойного электрического
слоя не позволяет объяснить явление взаимной коагуляции золей с разноименно
заряженными частицами Если же для объяснения устойчивости и коагуляции
коллоидных систем использовать силы электростатического взаимодействия, яв-
ление взаимной коагуляции не требует особых пояснений и может трактоваться
строго количественно
Более приемлемым кажется взгляд, согласно которому устойчивости лио-
фобных коллоидных систем способствует сольватация потенциалопределяющих
ионов Однако и в этом случае возникает ряд вопросов, на которые трудно
ответить
На основании приведенных рассуждений следует сделать за-
ключение, что сольватация может обеспечивать устойчивость лио-
фобных, в частности гидрофобных, коллоидных систем только в
особых случаях или может служить фактором, дополняющим дей-
ствие электрических сил. К этому следует добавить, что даже ко-
гда роль сольватации для устойчивости коллоидной системы не-
сомненна, строгий учет этого фактора весьма затруднен ввиду от-
сутствия количественной теории сольватации.
Наряду с термодинамическими факторами устойчивости, к ко-
торым следует отнести двойной электрический слой и сольватную
оболочку вокруг коллоидной частицы, на устойчивость коллоид-
ных систем может влиять и прочность структурно-механическога
барьера, возникающего по тем или иным причинам на поверхности
частицы. Этот фактор, согласно П. А. Ребиндеру, нельзя назвать
термодинамическим, поскольку при удалении или разрыве обо-
лочки, представляющей структурно-механический барьер, она не
обязательно должна восстанавливаться самопроизвольно. Кроме
того, у этой оболочки отсутствует равновесие с окружающей средой.
Уже давно высказывалось мнение, что устойчивость пен и
эмульсий должна сильно возрастать, когда на поверхности раз-
дела между дисперсной фазой и дисперсионной средой образуется
слой из молекул стабилизатора, обладающий повышенной струк-
турной вязкостью или даже известной механической прочностью и
являющийся как бы структурно-механическим барьером, препят-
ствующим сближению частиц.
Наиболее широко учение о структурно-механическом факторе-
стабилизации развито П. А. Ребиндером. Согласно П. А. Ребин-
деру, стабилизующими свойствами обладают насыщенные или
близкие к насыщению адсорбционные слои ориентированных мо- *
лекул поверхностно-активных веществ, образующие двухмерные '<
структурыАОсобенно сильным стабилизующим действием обла-
дают коллоидные адсорбционные слои, являющиеся своеобразны-
ми пленочными (двухмерными) студнями — лиогелями, сильно-
сольватированными дисперсионной средой и диффузно переходя-
щими в межмицеллярную жидкость. Веществами, способными об-
разовывать такие слои, являются белки и щелочные мыла в гидро-
золях, в олеозолях — смолы, мыла поливалентных металлов и ли-
поиды.
283
По П. А. Ребиндеру, стабилизующее действие гелеобразных ад-
сорбционных слоев стабилизатора обусловливается тем, что высо-
ковязкая прослойка между частицами не успевает выдавиться за
время столкновения частиц дисперсной фазы в результате броу-
новского движения или в потокедВ известных условиях стабилиза-
ция дисперсных систем адсорбционно-сольватными слоями, обла-
дающими упругостью и механической прочностью, может безгра-
нично повышать устойчивость системы вплоть до полной фиксации
ее частиц. Примером этому может служить отвердевание жидких
прослоек между воздушными пузырьками пены в результате геле-
образования или полимеризационных процессов. П. А. Ребиндер
отмечает, что образования структурно-механического барьера до-
статочно для стабилизации только тогда, когда на наружной гра-
нице адсорбционного слоя поверхностная энергия мала и не резко
возрастает на подступах к частице. При наличии хотя и структу-
рированной, но не лиофильной, а лиофобной оболочки все же мо-
жет происходить слипание частиц путем сцепления оболрчек на-
ружными поверхностями. Такого рода явления можно наблюдать
при флотации в результате адсорбции поверхностно-активных ве-
ществ полярными группами на поверхности гидрофильных твердых,
частиц. Направленные в водную среду углеводородные цепи свя-
зываются друг с другом своеобразной местной коалесценцией гид-
рофобных оболочек.
Согласно П. А. Ребиндеру, структурно-механический фактор яв-
ляется наиболее сильным фактором стабилизации и его использо-
вание якобы неизбежно при получении высокоустойчивых, осо-
бенно концентрированных дисперсных систем (например, техниче-
ских пен, эмульсий и суспензий).
Однако о структурно-механических свойствах адсорбционного
слоя можно, очевидно, говорить лишь тогда, когда молекулы ста-
билизатора типа мылообразных веществ или адсорбирующихся по-
лимеров присутствуют в системе и могут образовывать двухмер-
ный гель или студень.
Исходя из этого, можно полагать, что эмульсии могут быть
устойчивыми за счет структурно-механического фактора лишь при
достаточно больших концентрациях стабилизатора, когда на меж-
фазной поверхности образуются компактные, хорошо сольватиро-
ванные гелеобразные слои, обладающие высокой прочностью.
Г. А. Мартынов представляет себе действие адсорбционных полислоев
поверхностно-активных веществ следующим образом. Молекулярные силы при-
тяжения универсальны, т. е. действуют между любыми телами. Представим
теперь себе две одинаковые частицы, дисперсную фазу которых обозначим че-
рез 1, а полимолекулярно адсорбционный слой через 2 (рис. IX, 8), Очевидно,
энергия взаимодействия самих частиц будет равна:
Ui « Kt-ilH* (IX,39)
где Kt_t — константа, характеризующая взаимодействие обеих частиц: // — рас-
стояние между частицами,
284
Энергия взаимодействия адсорбционных слоев (пленок) очевидно составит:
U2 « к2_2/(// - 2Л)2 (IX, 40)
где №-2 — константа, характеризующая взаимодействие адсорбционных слоев;
h—толщина адсорбционного слоя. Как можно видеть, закономерности взаимо-
действия в обоих случаях качественно одинаковы, меняются лишь константы
взаимодействия и знаменатели уравнений.
На малых расстояниях нельзя пренебречь вкладом пленки в энергию взаимо-
действия частиц, и тогда энергия притяжения частиц будет равна:
1/ = к|_1///Ч-К2-2/(^-2Л)2 (IX, 41)
Таким образом, адсорбционный слой, представляющий нечто иное, как струк-
турно-механический барьер, влияет на взаимодействие частиц, не устраняя их
притяжения. Следовательно, адсорбци-
онные пленки не должны были бы при-
вести к повышению устойчивости систе-
мы. Однако если «2-2 = 0, то молеку-
лярным притяжением между пленками
можно пренебречь. В то же время ста-
билизующие пленки могут являться пре-
пятствием, мешающим тесному сближе-
нию частиц. Если частицы не могут при-
близиться друг к другу, то молекулярные
силы притяжения между ними будут
малы, поскольку расстояние велико. Это
и приводит к повышению устойчивости.
В отсутствие двойного электрическо-
го слоя эффективность адсорбционных
стабилизующих пленок определяется сле-
дующими условиями. Во-первых, стаби-
лизующая пленка должна быть доста-
Рис. IX, 8. Схема взаимодействия
частиц, стабилизованных полислоями
поверхностно-активного вещества:
I — частицы; 2 —адсорбционный слой по-
верхностно-активного вещества; Н— рас-
стояние между частицами, Л—толщина
адсорбционного слоя.
точно лиофильной, т. е. к2-2 — 0; во-вторых, чем длиннее молекулы вещества,
образующего защитную пленку, тем больше то расстояние, на которое могут при-
близиться частицы, и тем меньше значение молекулярных сил притяжении между
ними (т. е. тем выше устойчивость системы).
В заключение рассмотрим представления о так называемом
энтропийном факторе устойчивости.
В работах Симха, Эйриха, Фриша и многих других исследова-
телей было показано, что достаточно длинные и гибкие молекулы
поверхностно-активных веществ и полимеров способны адсорбиро-
ваться на поверхности твердого тела своими отдельными звеньями.
При этом большая часть концов цепей или петель, образуемых ад-
сорбированными молекулами, находится в дисперсионной среде и
может совершать микроброуновское движение (рис. IX, 9а), что
обусловливает стабилизацию системы. Согласно Овербеку, стаби-
лизация коллоидных частиц в этом случае происходит в резуль-
тате «отталкивания» друг от друга гибких участков цепных моле-
кул, «торчащих» в среду.
Согласно другому мнению при сближении частиц, на поверхно-
сти которых адсорбированы отдельные звенья макромолекул,
уменьшается число конформаций, осуществляемых гибкими цепями
стабилизатора (рис. IX, 96). Такое уменьшение конформаций
вследствие «стесненности» макромолекул между сблизившимися
частицами приводит к снижению энтропии, а следовательно, и
285
к увеличению свободной энергии системы. Конечно, это будет спо-
собствовать раздельному существованию коллоидных частиц, т. е.
будет являться фактором, обусловливающим агрегативную устой-
чивость системы.
Третье объяснение стабилизующего действия частично адсорби-
рованных макромолекул предложено А. В. Бромбергом еще
в 1946 г. Согласно А. В. Бромбергу, стабилизация в этом случае
определяется осмотическими силами, действующими в адсорбцион-
ных слоях. Неппер также отметил, что перекрытие слоев частично
свободных макромолекул должно приводить к возникновению ос-
мотических снл. Таким образом, изменение энтропии полимерных
а б
Рис. IX, 9. Схематическое изображение коллоидных частиц, адсор-
бировавших отдельные звенья цепных макромолекул, совершающих
микроброуновское движение:
а—отдельная частица; б—две сблизившиеся частицы.
молекул, находящихся в зазоре между сближаемыми поверхно-
стями, должно обусловливать приток жидкости из объема рас-
твора в зазор и возникновение расклинивающего давления, стаби-
лизующего систему.
5. ПРАВИЛА КОАГУЛЯЦИИ ЭЛЕКТРОЛИТАМИ
Коагуляция коллоидных систем может происходить под влия-
нием ряда факторов — старения системы, изменения концентрации
дисперсной фазы, изменения температуры, механических воздей-
ствий, света и т. д. Однако наиболее важное теоретическое и прак-
тическое значение имеет коагуляция при добавлении электролитов.
В нашем курсе мы подробно остановимся только на коагуляции
электролитами и лишь вкратце коснемся других причин коагуля-
ции.
Способность ионностабилизованных коллоидных растворов коа-
гулировать под влиянием электролитов была отмечена еще пер-
выми исследователями коллоидных систем — Сельми, Грэмом,
И. Г. Борщовым. Они обратили внимание на то важное обстоя-
тельство, что коагуляцию способны вызывать все электролиты. Как
показали последующие исследования, в этом отношении не пред-
ставляют исключения и электролиты, являющиеся стабилизато-
рами. Существенно лишь, чтобы концентрация таких электролитов
в системе была достаточно велика для того, чтобы сжать двойной
286
электрический слой и этим понизить энергетический барьер, пре-
пятствующий’слипанию частиц прй йх столкновении.
Гарди в 1900 г. установил, что коагулирующим действием
в электролите обладают не все его ионы, а только те, которые не-
сут здряд, одноименный с зарядом противоиона мицеллы (или, что
то же, заряд, противоположный-по знаку заряду коллоидной ча-
стицы). Таким образом, для золей с отрицательно заряженными
частицами коагулирующими ионами являются катионы, а для зо-
лей с положительными частицами — анионы.
Было также установлено, что для начала коагуляции необхо-
димо превысить некоторую минимальную концентрацию электро-
лита в золе. Эта величина (у), получившая название порогакоа-
гуляции и обычно выражаемая в ммоль/л или” мг-экв/л, очевидно,
отвечает сжатию двойного электрического слоя до той степени,
когда он перестает служить энергетическим барьером, предохра-
няющим частицы от слипания под действием молекулярных сил
притяжения. Ниже в качестве примера приведены пороги коагуля-
ции некоторыми электролитами золя сульфида мышьяка с отри-
цательно заряженными частицами (по Фрейндлиху):
Электролит Y. ммоль/л Электролит Y, ммоль/л
СНзСООК 110,0 ВаС12 0,69
ZnCl2 0,68
L1C1 58,0 CaCl2 0,65
NaCl 51,0 UO2(NO3)2 0,64
KNO3 50,0 ,SrCl2 0,63
КС1 49,5
HCi 31,0 A1(NO3)3 0,095
MgSO4 0,81 AICI3 0,093
MgCl2 0,71 Ce(NO3)3 0,080
Следует заметить, что начало коагуляции может быть опре-
делено по разным признакам — по изменению окраски золя, по-
явлению мути, началу выделения дисперсной фазы в осадок и т. д.
Появление этих признаков не всегда совпадает во времени. Кроме
того, порог коагуляции в известной степени зависит и от концен-
трации золя. Поэтому порог коагуляции является довольно отно-
сительной характеристикой устойчивости золя по отношению
к данному электролиту. Во всяком случае, всегда необходимо точно
указывать условия, при которых проводилось определение порога
коагуляции золя.
Еще в 1882 г. Шульце установил, что коагулирующая сила
иона тем больше, чем больше его валентность. Эта зависимость
была подтверждена Гарди и получила название правила
Шульце — Гарди, или правила значности. Это пра-
вило не предусматривает прямой пропорциональности между ва-
лентностью иона и его коагулирующим действием, коагулирующая
сила возрастает гораздо быстрей валентности. Для золя AS2S3
287
Шульце нашел следующие соотношения коагулирующей силы
одно-, двух- и трехвалентных катионов: 1 : 20 : 350; Фрейндлих для
того же золя и тех же катионов нашел ряд 1:7: 531; Пиктон и
Линдер — ряд 1:20:1500. Расхождение между значениями поро-
гов коагуляции, найденных различными авторами, объясняется
трудностью получения коллоидной системы с одной и той же ха-
рактеристикой и несоблюдением, полностью одинаковых условий
при определении порогов коагуляции.
Дальнейшие опыты показали, что коагулирующая сила ионов
одной и той же валентности возрастает с увеличением радиуса
иона. Иначе говоря, катионы или анионы одной и той же валент-
ности по своему коагулирующему действию располагаются в обыч-
ный лиотропный ряд. Для одновалентных катионов и отрицательно
заряженных частиц золя иодида серебра такая закономерность
видна из следующих данных (по Л. К. Лепинь и А. В. Бромбергу):
лектролит мг-экв/л
LiCl 582
NaCl 300
КС1 255
LiNO3 600
NaNO, 360
KNO3 260
V.
Электролит мг-экв/л
Li2SO4 640
Na2SO4 420
K2SO4 272
Изменение коагулирующего действия в лиотропном ряду ионов
щелочноземельных металлов выражается в более слабой форме,
чем изменение действия в ряду одновалентных катионов. При коа-
гуляции золей с положительно заряженными частицами с помощью
анионов изменение коагулирующего действия анионов в лиотроп-
ном ряду выражено менее заметно.
Весьма своеобразно коагулирующее действие проявляется у ор-
ганических ионов. Положительно заряженные одновалентные ионы
алкалоидов и красителей действуют гораздо сильнее, чем соответ-
ствующие им по валентности ионы неорганических электролитов.
Это объясняется высокой адсорбционной способностью громоздких
органических ионов, обладающих, большей поляризуемостью.
В своих исследованиях Гарди полагал, что коагуляция должна
наступать в изоэлектрической точке, когда ^-потенциал частиц ра-
вен нулю. Однако позднее было установлено, что коагуляция
обычно наступает не в изоэлектрической точке, а при достижении
некоторого критического ^-потенциала. Существенно, что Значение
критического потенциала в общем оказалось мало зависящим от
вида электролита, с помощью которого он был достигнут. Для мно-
гих систем этот потенциал довольно близок к 30 мВ, что можно
видеть из данных табл. IX, 1.
Однако во многих случаях значение критического ^-потенциала,
отвечающее началу коагуляции, сильно отличается от 30 мВ.
Иногда с понижением ^-потенциала золи не только не коагули-
288
Таблица IX, 1. Значения критического потенциала £крит
для золя AS2S3 (по Повису) и золя Fe2O3 (по Гошу)
Золь Коагулирующий электролит Концентрация электролита, мг-экв/л Ькрит» мВ
Золь As2O3 (отрицательно заряжен- КС1 40,0 44
ные частицы) ВаС12 1,0 26
А1С13 0,15 25
Th(NO3)4 0,20 27
Золь Fe2O3 (положительно заряжен- КС1 • 100,0 34
ные частицы) NaOH 7,5 31
K2SO4 6,6 32
К2СгО4 6,5 32
KsFe(CN)e 0,65 30
руют, но увеличивают свою устойчивость и, наоборот, повышение
потенциала подчас сопровождается коагуляцией. Это говорит
о том, что ^-потенциал связан с устойчивостью далеко не так про-
сто, как это думали в свое время некоторые исследователи. Сле-
дует, однако, отметить, что до сих пор большинство определений
электрокинетического потенциала проведено без учета электрофо-
ретического запаздывания, электрической релаксации и других
факторов, и поэтому найденные значения критического потенциала
являются во многих случаях неточными.
Наконец, рядом исследователей было обнаружено, что осадок,
состоящий из скоагулировавшей дисперсной фазы коллоидной си-
стемы, всегда уносит с собой часть коагулирующего иона. Такое
явление становится понятным, если учесть, что при коагуляции
всегда происходит частичный обмен противоионов мицелл золя на
коагулирующие ионы электролита, которым ведется коагуляция.
6. ТЕОРИИ КОАГУЛЯЦИИ ЭЛЕКТРрЛИТАМИ
Приведенные выше правила коагуляции золей охватывают
в основном все характерные ее особенности. Ознакомимся теперь
с тем, как могут быть истолкованы явления, наблюдаемые при
коагуляции электролитами.
Авторами предлагались различные теории коагуляции электро-
литами. Сюда относятся химическая теория коагуляции (Дюкло),
адсорбционная теория (Фрейндлих), электростатическая теория
.(Мюллер, А. И. Рабинович, В. А. Каргин). Однако все они по тем
илн иным причинам утратили свое значение и представляют сейчас
только исторический интерес*. В настоящее время общепризнан-
ной является физическая теория коагуляции электролитами, бази-
рующаяся на общих принципах статистической физики, теории рас-
творов и теории действия молекулярных сил. Физическая теория
* Краткое изложение сущности этих теорий и их критику можно найти в
первом издании этого учебника, с. 304—308,
10 Зак. 664
289
коагуляции электролитами строится на рассмотрении баланса мо-
лекулярных сил притяжения и сил ионно-электростатического от-
талкивания, действующих между частицами, как это было изло-
жено на примере взаимодействия пластинчатых частиц во втором
разделе главы.
Впервые качественный подход к изучению устойчивости золей
наметили Кальман и Вильштеттер в 1932 г. Первые количествен-
ные расчеты были произведены Б. В. Дерягиным в конце 30-х го-
дов и затем завершены в работе Б. В. Дерягина и Л. Д. Ландау
,(1941 г.). Аналогичный подход к изучению устойчивости коллоид-
ных систем в дальнейшем был раз-
вит и в работах голландских иссле-
дователей Фервея и Овербека. По
начальным буквам основных авто-
ров возникшей физической теории
коагуляции эту теорию теперь ча-
сто называют теорией ДЛФО.
При рассмотрении коагуляции
коллоидных систем следует разли-
чать два предельных случая:
1) нейтрализационную коагуля-
цию, когда потеря
происходит в результате
ния коллоидных частиц и
ния их фо-потенциала*;
2) концентрационную
цию, при которой потеря
Фо-потенциала, а вызвана
Рнс. IX, 10. Потенциальные кри-
вые, характеризующие изменение
энергии взаимодействия двух
коллоидных частиц от расстоя-
ния Н между нх поверхностями.
Нумерация кривых возрастает с умень-
шением, фо-потенциала.
устоичивости
разряже-
уменьше-
коагуля-
устойчи-
сжатием
вости связана не с падением
диффузного двойного СЛОЯ.
Рассмотрим эти два типа коагуляции несколько подробнее.
Нейтрализационная коагуляция наблюдается у зо-
лей со слабо заряженными частицами, обладающими сравнительно
низкими значениями фо-потенциала. В этом случае коагуляция
происходит обычно у золей при снижении электрического заряда
частиц из-за уменьшения вследствие тех или иных причин адсорб-
ции потенциалопределяющих ионов. В результате уменьшения за-
ряда электрические силы отталкивания между частицами ослабе-
вают, частицы при сближении слипаются и выпадают в осадок.
Теория, развитая Б. В. Дерягиным, позволяет вычислить, при
каком достаточно низком значении фо-потенциала должен исчез-
нуть энергетический барьер, т. е. когда результирующая потен-
циальная кривая (рис. IX, 10), характеризующая зависимость
энергии взаимодействия частиц от расстояния между ними,
должна только в одной точке коснуться оси абсцисс (пунктирная
кривая). При достаточно малом ф0-потенциале связь между кри-
* Согласно уточненным представлениям область сил отталкивания опреде-
ляется не потенциалом <р0, а потенциалом <ро > отвечающим границе слоя Штерна.
Существенно, что <ро < Фо-
290
тическим потенциалом фо, Крит, при котором энергетический барьер
исчезает, и толщиной 1/х (или h) диффузных слоев, окружающих
обе коллоидные частицы, выражается формулой:
Фо, крит = д/с = aJ С (IX, 42)
где С — константа; А — постоянная притяжения; е — диэлектрическая проницае-
мость раствора.
Связь между фо, крит и х, вытекающая из уравнения (IX, 42),
была эмпирически установлена Эйлерсом и Корфом еще в 1940 г.
Согласно этим исследователям
=,(р2.1/х = сА^ в- (IX,43)
где В — некоторая критическая величина, определяемая из опыта.
Условие ф^/г < В представляет собой условие коагуляции,
а ф^/г > В является условием устойчивости системы.
Для достаточно разбавленных растворов электролитов, когда
толщина диффузного слоя велика, и для малых значений фо-потен-
циала можно принять, что этот потенциал довольно близок к ^-по-
тенциалу, измеренному по подвижности частиц в электрическом
Рис. IX, И. Схема, выражающая зависимость <р0 и ^-потенциалов
для слабо заряженных частиц в разбавленном растворе электро-
лита (а) и для сильно заряженных частиц в достаточно концентри-
рованном растворе электролита (б).
поле (рис. IX, Па). Надо, однако, подчеркнуть, что отождествле-
ние общего скачка потенциала фо или потенциала Штерна ф6, от-
вечающего границе ионный конденсатор — диффузный слой, и £-по-
тенциала, вообще говоря, незаконно и в ряде случаев может при-
водить к неправильным результатам. В самом деле, определение
^-потенциала связано с гидродинамическими особенностями си-
стемы, а именно, с положением границы скольжения у поверхности
раздела частица — раствор или вязкостью прилегающих к ней
слоев жидкости, тогда как потенциалы ф0 и ф6, очевидно, никакого
10*
291
отношения к этим особенностям не имеют. Использование £-потен-
циала как критерия устойчивости коллоидных систем связано в ос-
новном с тем, что ^-потенциал сравнительно легко эксперимен-
тально измерить и в определенных условиях его изменение сходно
с изменением фо-потенциала.
Концентрационная коагуляция наблюдается обыч-
но у золей с сильно заряженными частицами при увеличении кон-
центрации индифферентного электролита в системе. Это обстоя-
тельство позволяет в первом приближении вовсе не учитывать воз-
можное изменение фо-потенциала при различного рода адсорбцион-
ных или десорбционных явлениях. Единственной причиной коагу-
ляции системы в эуом случае является, согласно теории ДЛФО,
чисто электростатический эффект сжатия двойного электрического
слоя.
В предельном случае потенциал поверхности — фо-потенциал —
при коагуляции может сохранять достаточно высокие значения
(более 100 мВ). При этом соответствие между ^-цотенциалом, ко-
торый при увеличении концентрации раствора электролита может
значительно падать, и фо-потенциалом теряется. Это легко понять
из схемы, представленнной на рис. IX, 116. Теряется также связь
между устойчивостью системы и фо- и ^-потенциалами. Таким об-
разом, становится понятным, почему ^-потенциал далеко не всегда
может являться критерием устойчивости золя.
Для того чтобы понять механизм концентрационной коагуля-
ции, необходимо остановиться на весьма важной особенности — за-
висимости сил электростатического взаимодействия от фо-потен-
циала, рассмотренного ранее на примере двух пластинок. Теория '
показывает, что по мере безграничного возрастания фо-пот’енциала
обеих поверхностей сила электростатического отталкивания F3 ме-
жду коллоидными частицами любой формы не возрастает безгра-
нично, а стремится клоненному пределу, подходя к нему уже при
значениях потенциала поверхности, превышающих 100 мВ (рис.
IX, 12). Вследствие этого свойства, как бы насыщения сил, можно
говорить о силе взаимодействия предельно заряженных поверхно-
стей как о величине, не зависящей от точных значений потенциала
поверхности.
Этот на первый взгляд трудно понятный вывод объясняется,
как мы видели, тем, что по мере роста фо-потенциала увеличивается
притяжение противоионов *к поверхности частицы. Таким образом,
параллельно с ростом заряда внутренней обкладки двойного элек-
трического слоя и потенциала поверхности усиливается и экрани-
рование внешнего поля этой обкладки противоионами. Поэтому
дальнейший рост напряженности электрического поля в перифе-
рийных частях ионных атмосфер и сил взаимодействия обеих ча-
стиц прекращается. Таким образом, если коллоидные частицы за-
ряжены достаточно сильно, то их взаимодействие зависит только
от заряда противоионов, экранирующих действие внутренней об-
кладки двойного слоя и обусловливающих его толщину.
292
При прибавлении к системе индифферентного электролита про-
исходит сжатие диффузной части двойного электрического слоя и
толщина ионных атмосфер уменьшается. Одновременно также в ре-
зультате сжатия ионного слоя увеличивается глубина вторичного
потенциального минимума, что приводит к возрастанию вероят-
ности дальней агрегации. Изменение формы потенциальных кри-
вых парного взаимодействия частиц при увеличении содержания
индифферентных электролитов в системе показано на рис. IX, 13.
Теоретические расчеты позволили Б. В. Дерягину и Л. Д. Лан-
дау установить, что энергетический барьер на диаграмме энергия
Рис. IX, 12. Зависимость элек-
тростатической силы отталки-
вания Рэ между двумя кол-
лоидными частицами от потен-
циала <р0 их поверхности.
Рис. IX, 13. Изменение вида резуль-
тирующих кривых, характеризующих
парное взаимодействие частиц, при
увеличении содержания электролита.
Нумерация кривых возрастает с уменьше-
нием Фк-нотеициала.
взаимодействия — расстояние между коллоидными частицами ис-
чезает, когда достигнут порог.коагуляции у, определяемый по фор-
муле:
V С Л2лв~в (IX, 44)
где С — константа, слабо зависящая от асимметрии электролита, т. е. от отноше-
ния числа зарядов катиона и аниона; е — заряд электрона; z— валентность про-
тивоиона.
Порог концентрационной коагуляции, как и следовало ожидать,
не зависит от фо-потенциала, но зависит от значения постоянной/!,
диэлектрической проницаемости г раствора, температуры Т и вау
лентности zпротивоиона. Ион, заряженный одноименно с коллоид-
ной частицей, на коагуляцию оказывает малое влияние, несколько
изменяя лишь значение коэффициента С.
Из уравнения (IX,44) следует также, что значения порога коа-
гуляции для одно-, двух-, трех- и четырехвалентных ионов должны
относиться, как
1: (’/2)в: (’/3)в: (’А)’ или как
1:0,016:0,0013 : 0,00024
293
В некоторых случаях этот ряд в первом приближении согла-
суется с опытными данными, получившими свое выражение в пра-
виле Шульце — Гарди, и как будто дает его теоретическое обосно-
вание.
Ниже приведены собранные Овербеком средние значения порогов коагуля-
ции- (в ммоль/л) некоторых золей, заимствованные из ряда исследований Как
можно видеть, отношения экспериментально определенных порогов коагуляции
ионами различной валентности (I, II, III, IV) довольно хорошо совпадают с тео-
ретически рассчитанными данными (особенно для коагуляции одно- и двух-
валентными ионами).
Золи с отрицательно заряженными Вален! частицами ность иона
I II III IV
Порог коагуляции у золя 55 0,69 0,091 0,090
Соотношение 1 0,013 0,0017 0,0017
Порог коагуляции у золя Au 24 0,38 0,006 0,0009
Coot юшеиие 1 0,016 0,0003 0,00004
Порог коагуляции у золя Agl 142 2,43 0,068 0,013
Соотношение 1 0,017 0,0005 0,0001
Золи с положительно заря ж е н н ы м и частицами
Порог коагуляции у золя Ре20з I 11,8 Валентность иона II III 0,21 - IV
Соотношение 1,0 0,018 — —
Порогу коагуляции у золя 52 0,63 0,080 0,053
Соотношение 1,0 0,012 0,0015 0,0010
Наблюдающиеся отклонения можно объяснить следующими обстоятель-
ствами.
Во-первых, опыты по определению порогов коагуляции проводились в раз-
личных условиях. Между тем иа результаты определения порогов коагуляции
могут влиять малейшие, порой даже трудно учитываемые воздействия Кроме
того, есть все основания полагать, что золи, с которыми проводили экспери-
менты различные авторы, даже если они имели одну и ту же природу, обладали
разной дисперсностью и другими неодинаковыми характеристиками, сказавши-
мися на результатах определения порогов коагуляции.
Во-вторых, при определении порога коагуляции обычно берут то количества
электролита, которое нужно для коагуляции еще не полностью астабилизоваи-
ного золя. Однако остается неизвестным, в каком соотношении находятся най-
денные таким образом значения порога коагуляции к значениям концентрации»
соответствующим коагуляции полностью астабилизоваиного золя, причем для
различных электролитов это соотношение может быть не одинаковым.
В-третьих, фактически найденная концентрация электролита при коагуляции
может значительно отличаться от концентрации, определяемой путем расчета»
вследствие ионного обмена, в особенности когда порог коагуляции низок.
Однако в последнее время получены экспериментальные данные, которые
указывают на иеприложимость в ряде случаев правила Шульце — Гарди в виде
закона Дерягина — Ландау z6. На опыте часто наблюдаются значительные от-
клонения от такой закономерности, а именно, в ряде случаев коагулирующее
действие электролитов пропорционально валентности противоионов в степени
меньше шести. Согласно И. Ф, Ефремову и О. Г. Усьярову, это отклонение от
294
закономерности объясняется взаимодейстием частиц во вторичной потенциальной
яме, что ведет или к образованию сравнительно малопрочных флокул или к же-
латинированию.
Обобщая все сказанное выше о нейтрализационной и концен-
трационной коагуляции, можно прийти к следующим выводам.
Когда потенциал <р0 частиц невысок, устойчивость золя зависит
от значения потенциала поверхности, а адсорбционные явления оп-
ределяют коагуляционный процесс; этим, как мы видели, объяс-
няется правило Эйлерса — Корфа. Для объяснения коагуляции
золя с сильно заряженными частицами теория ДЛФО исходит
уже из представлений о сжатии двойного электрического слоя, со-
гласно которым объясняется правило Шульце — Гарди. Суще-
ственно, что оба правила приложимы к золям одной и той же при-
роды, а иногда и при одинаковом составе электролита.
7. ВЛИЯНИЕ РАЗМЕРА И КОНЦЕНТРАЦИИ ЧАСТИЦ
НА ИХ ВЗАИМОДЕЙСТВИЕ В ДИСПЕРСНЫХ СИСТЕМАХ
Теория ДЛФО ограничивается рассмотрением потенциальных
кривых для двух дисперсных частиц. Это объясняется тем, что
коагуляция, протекающая в~ разбавленных золях, определяется
парным взаимодействием частиц, положенным, как мы видели,
в основу теорий кинетики коагуляции Смолуховского и Н. А. Фукса.
Однако для определения условий устойчивости концентрированных
золей необходимо учитывать коллективные взаимодействия ча-
стиц. Такие золи не только обладают практически достаточной
стабильностью, но часто обнаруживают и периодическое располо-
жение частиц аналогично узлам кристаллической решетки. Подоб-
ные периодические коллоидные структуры образуют, например, не-
которые вирусы и монодисперсные латексы. Условием периодич-
ности, конечно, является прежде всего достаточная монодисперс-
ность системы. Как отметили еще Бернал и Фанкухен, периодиче-
ское расположение свидетельствует о дальнодействующих силах
между коллоидными частицами.
Метод построения кривых взаимодействия трех плоских частиц
разработали И. Ф. Ефремов и С. В.'Нерпин. Из построенных ими
потенциальных кривых следует, что при малой концентрации дис-
персной фазы глубина потенциальных ям уменьшается с увеличе-
нием толщины частиц; одновременно с этим увеличивается размы-
тость ям и крутизна потенциального барьера. При высокой кон-
центрации дисперсной фазы глубина потенциальных ям меньше для
тонких частиц. Наконец, при равных и достаточно больших рас-
стояниях между толстыми и тонкими плоскими частицами энерге-
тический барьер для тонких частиц выше, что ведет к повышенной
устойчивости тонких пластинок. При возрастании концентрации
дисперсной фазы расстояние между поверхностями и, следова-
тельно, устойчивость резко снижаются для мелких частиц, что
является причиной их агрегирования при таких концентрациях,
когда низкодисперсные системы могут быть еще устойчивыми.
295
Такое агрегирование происходит вследствие «тесноты», когда ча-
стицы даже независимо от броуновского движения оказываются
на расстояниях, при которых они принуждены скатываться в пер-
вичную потенциальную яму. В этом случае нарушение агрегатив-
ной устойчивости происходит по механизму, отличному от коагуля-
ции электролитами.
Эти выводы получены в результате рассмотрения модельной
системы, представляющей три плоские частицы, при условии по-
стоянства заряда и потенциала поверхности частиц и при одной и
той же концентрации электролита. Однако, несмотря на допущен-
ные упрощения, эти выводы качественно согласуются и с опытными
данными, полученными при изучении систем, состоящих из мно-
жества частиц, т. е. при изучении реальных дисперсных систем.
Аналогичный метод использован и для изучения влияния кон-
центрации дисперсной фазы лиофобных золей на их устойчивость
при различных концентрациях электролитов. Учет коллективного
взаимодействия коллоидных частиц позволяет объяснить суще-
ственные различия в закономерностях коагуляции электролитами
разбавленных и нарушении устойчивости концентрированных лио-
фобных золей. В частности, было найдено, что при постоянной
объемной концентрации дисперсной фазы устойчивость концентри-
рованных систем с увеличением размера частиц проходит через
максимум. Этот вывод был экспериментально подтвержден Отте-
вилем Шоу. Если же численная концентрация частиц остается не-
изменной, то устойчивость системы с увеличением размера частиц
снижается монотонно. Одновременно для больших сферических
частиц и толстых пластинчатых частиц характерно наличие глубо-
кого вторичного минимума на потенциальных кривых, вследствие
чего процессы дальней агрегации должны быть особенно распро-
странены в низкодисперсных системах.
8. ЗНАЧЕНИЕ АДСОРБЦИОННЫХ ЯВЛЕНИЙ ДЛЯ КОАГУЛЯЦИИ
Как уже было указано, коагуляция коллоидных систем может
осуществляться как по нейтрализационному, так и по концентра-
ционному механизму. Однако практически в большинстве случаев
коагуляция осуществляется по обоим этим механизмам одновре-
менно. Так как нейтрализационный механизм коагуляции лиозолей
весьма существенен для реальных систем и зависит от адсорб-
ционных явлений, на что указывали многие ученые, то именно ко- (
личественный учет влияния адсорбции, на коагуляцию составляет ’
одну из самых актуальных задач дальнейшего развития теории
устойчивости и коагуляции ионностабилизованных систем.
Кратко'рассмотрим роль адсорбционных явлений при коагуля-
ции коллоидных растворов. Имеющиеся литературные данные по
этому вопросу чрезвычайно противоречивы. Это объясняется тем,
что вплоть до последнего времени все исследователи определяли
адсорбцию ионов косвенным методом — по изменению концентра-
296
ции соответствующего иона в растворе до и после коагуляции.
Если принять во внимание сравнительно небольшие величины ад-
сорбции ионов коллоидными частицами и недостаточную точность
применявшихся методов анализа, то становятся понятны причины
противоречивости результатов, полученных в ранних исследова-
ниях. В связи с этим Ю. М. Глазман и Д. Н. Стражеско, а также
югославские исследователи Херак и Мирник применили прямой
метод радиометрического определения адсорбции ионов частицами
лиофобных золей. Это позволило правильно оценить даже самые
малые количества адсорбированных ионов.
Остановимся на работах, проведенных в Советском Союзе.
Опыты проводились в основном с типичными гидрозолями, имею-
щими отрицательно заряженные частицы. В качестве коагулирую-
щих ионов были использованы Na+, Rb+, Ag+, Са2+, Sr2+, Y3+, La3+.
Уже двукратная промывка коагулятов 0,1 М раствором КС1 была
достаточна для практически полной десорбции поглощенных ка-
тионов. Это свидетельствует об и'онообменном характере поглоще-
ния противоионов при коагуляции золей. Отсюда следует, что если
квазимолекулярная адсорбция электролитов и имеет место в ис-
следованных системах, то она должна играть сугубо подчиненную
роль.
Результаты опытов, в которых изучалось влияние избытка элек-
тролита на сорбцию противоионов, показали, что при повышении
концентрации электролита сорбция катионов увеличивается. Это
указывает на то, что при введении электролитов в количестве, пре-
вышающем порог коагуляции, происходит дополнительная адсорб-
ция потенциалопределяющих ионов, всегда присутствующих в ин-
термицеллярной жидкости. В связи с этим возрастает плотность
заряда на поверхности дисперсной фазы и соответственно увели-
чивается ее сорбционная емкость.
Концентрация стабилизующего электролита в интермицеллярном растворе
по-разному влияла на сорбцию противоионов. Так, удаление растворенного се-
роводорода из золя сульфида ртути сопровождалось значительным (в 6—8 раз)
уменьшением сорбции ионов Са2+ и Rb*; одновременно существенно понижалась
устойчввость золя к этим ионам. В то же время, как следует из экспериментов,
а также из литературных данных, сорбционная емкость и устойчивость золей
Agl сравнительно мало зависели от содержания стабилизующих ионов в си-
стеме:
Было установлено, что при коагуляции гидрозолей AS2S3, Sb2S3,
HgS, Agl и MgO происходило эквивалентное поглощение противо-
ионов разной валентности. Таким образом, представление об эк-
вивалентности сорбции ионов-коагуляторов, являвшееся до послед-
него времени дискуссионным, получило для типичных гидрофоб-
ных золей прямое и однозначное подтверждение. Этот факт одно-
временно свидетельствует о том, что теория диффузного слоя
Гуи — Чэпмена не может объяснить наблюдаемых на опыте за-
кономерностей. Эквивалентность сорбции противоионов можно
297
объяснить лишь, исходя из строения двойного электрического слоя,
согласно представлениям Штерна.
Адсорбция побочных ионов электролита, которыми проводили
коагуляцию, т. е. ионов, одноименно заряженных с коллоидной ча-
стицей, согласно радиометрическим исследованиям весьма неве-
лика, за исключением тех случаев, когда ионы обладают сильно
выраженной адсорбционной способностью (например, анион Г и
катион Cs+).
Помимо адсорбции ионов низкомолекулярных электролитов необходимо рас-
смотреть адсорбцию коллоидными частицами поверхностно-активных веществ.
Такая адсорбция представляет большой интерес, так как она вызывает измене-
ние всех свойств коллоидной системы, в частности устойчивости ее к действии*
электролитов, и, следовательно, позволяет расширить наши представления в от-
ношении стабильности и коагуляции коллоидных систем. Кроме того, адсорбция
поверхностно-активных веществ дисперсными системами имеет и большое прак-
тическое значение.
Было изучено влияние на устойчивость и коагуляцию золей гидрата окиси
железа и сульфида мышьяка адсорбции неионогенных поверхностио-активных
веществ, дифильные молекулы которых состоят из неполярного углеводородного
радикала и полярной полиоксиэтиленовой цепи. В зависимости от интенсивности
взаимодействия поверхности коллоидных частиц с дисперсионной средой влия-
ние неионогенных поверхностно-активных веществ на коллоидные системы ока-
залось различным даже в качественном отношении. Поверхностно-активные со-
единения при малых их концентрациях в системе не повышали гидрофильности
частиц гидрата окиси железа и уменьшали устойчивость гидрозоля к действии*
электролитов. Это, очевидно, связано с промежуточным характером золя
Fe(OH)3, имеющего достаточно гидрофильные частицы. При больших концен-
трациях неионогенные поверхностно-активные вещества вызывали коагуляции*
золя Fe(OH)3.
Совершенно иначе вел себя гидрозоль сульфида мышьяка, в котором в ре-
зультате ориентированной адсорбции молекул неионогенных поверхностно-актив-
ных соединений происходила гидрофилизация сильно гидрофобной поверхности
коллоидных частиц. При весьма небольших концентрациях неионогенных поверх-
ностно-активных веществ, когда степень гидрофилизации была мала, имело место
либо повышение чувствительности (сенсибилизация) к действию одновалентных:
ионов, либо совсем незначительное возрастание устойчивости к действию двух-
и трехвалентных противоионов. Сенсибилизацию под действием одновалентных
ионов можно объяснить взаимодействием (притяжением) между собой адсор-
бированных дипольных молекул поверхностно-активных соединений. По мере-
заполнения поверхности дисперсной фазы адсорбированными молекулами поверх-
ностно-активных соединений степень гидрофильности частиц и устойчивости золя
AssS3 все более возрастала и, в конце концов, образовавшийся лиофилизованный
золь сульфида мышьяка коагулировал только при больших концентрациях низ-
комолекулярного электролита.
В целом полученные результаты свидетельствуют о том, что вследствие
ориентированной адсорбпии молекул неионогенных поверхностно-активных ве-
ществ происходит модификация поверхности частиц сульфида мышьяка. Ти-
пично гидрофобный коллоидный раствор AsjSs превращается в золь с лиофиль-
ными свойствами, агрегативная устойчивость которого обусловлена адсорбцион-
ными гидратированными слоями неионогенного стабилизатора, образующимися
вокруг частиц дисперсной фазы. Ориентированная адсорбция молекул неиоио-
генных поверхиостно-активных веществ на поверхности частиц была установлена
экспериментально.
В другой серии работ было исследовано влияние на устойчивость гидрозо-
лей Agl и AgBr различных алкильных эфиров полиэтиленгликоля, самих поли-
этиленгликолей (неионогенные поверхностно-активные вещества), а также нат-
298
риевых солей различных производных сульфоянтарной кислоты и бутилнафта-
линдисульфокислоты (ионогенные поверхностно-активные вещества).
В этих работах в общем были обнаружены те же закономерности, что и в
случае рассмотренного выше действия поверхностно-активных веществ иа свой-
ства золя AS2S3. Особенность заключалась в том, что алкильные эфиры поли-
этиленгликоля вызывали перезарядку положительно заряженных частиц золя
Agl. Это явление было объяснено тем, что на потенциал двойного электрического
слоя налагается адсорбционный скачок потенциала, который возникает в ориен-
тированном у поверхности дисперсной фазы слое полярных молекул алкильных
эфиров полиэтиленгликоля.
Далее полученные данные о зависимости степени стабилизации золей от их
«возраста» и концентрации свидетельствовали о том, что наибольшая устойчи-
вость золя Agl имеет место при достижении определенной (по-видимому, близ-
кой к предельной) плотности покрытия коллоидных частиц монослоем адсорби-
рованных молекул алкильных эфиров полиэтиленгликоля.
Закономерности коагуляции золей Agl, содержащих неионогенные поверх-
ностно-активные вещества, свидетельствуют о том, что высокая стабильность
систем в этом случае в основном обусловлена силами неэлектростатической при-
роды, хогя и в присутствии неионогенного стабилизатора коллоидные частицы
имеют электрический заряд.
При исследовании влияния поверхностно-активных веществ на устойчивость
золей AgBr, было найдено, что метиловый эфир полиэтиленгликоля незначи-
тельно повышает устойчивость коллоидных растворов бромида серебра. Даже
при относительно высоком содержании эфира значения порога коагуляции золей
ионами разной валентности резко отличались между собой. Это означает, что
в присутствии плох® адсорбируемого частицами метилового эфира полиэтилен-
гликоля золи AgBr остаются ионно стабилизованными. При добавлении же к
коллоидным растворам AgBr лучше адсорбирующегося додецилового эфира по-
лиэтиленгликоля свойства золей коренным образом менялись. Золи становились
весьма устойчивыми и коагулировали только в высококонцентрированных рас-
творах электролитов; валентность противоионов переставала влиять на их коа-
гулирующее действие. Следовательно, устойчивость золей AgBr, стабилизованных
этим неионогенным поверхностно-активным веществом, обусловлена неиоиной
слагающей расклинивающего давления. Эта слагающая возникает в результате
перекрытия граничных слоев воды с особой структурой, гидратирующих гидро-
фильные группы поверхностно-активных молекул, адсорбированных на частицах
золя. Таков же механизм стабилизации и в других случаях при адсорбции ана-
логичных веществ на гидрофобных золях.
Стабилизация гидрозолей AgBr самими полиэтиленгликолями имела место
только при оксиэтилеиовой цепи, содержащей более ста звеньев, и выражалась
тем сильнее, чем выше молекулярный вес неионогенного поверхностно-активного
вещества. Закономерности коагуляции таких систем свидетельствуют об образо-
вании в этих случаях гидрофилизованных золей.
Выше речь шла об адсорбции коллоидными частицами противоионов. Суще-
ствуют, однако, данные, что адсорбироваться могут и ионы, одноименно заря-
женные с коллоидной частицей, если они обладают достаточно большим адсорб-
ционным потенциалом (например, I"). Имеются указания, что при этом значи-
тельную роль играет гидратация ионов, причем уменьшение гидратации, как
правило, способствует их адсорбции.
Такого мнения в тридцатых годах этого столетия придерживались Гош,
Дхар и др. В последнее время правильность такой точки зрения подтвердил
“С. Г. Телетов. Исследуя гидрозоли гидрата окиси железа, он нашел, что раз-
личия в коагулирующей способности ацетатов металлов определяются действием
сопутствующих ионов, т. е. ионов, одноименно заряженных с частицей Fe(OH)s.
€ помощью методов полярографии, колориметрии и радиометрии С. Г. Телетов
получил данные, свидетельствующие также об адсорбции катионов меди и цинка
на положительно заряженных частицах гидратов окисей металлов.
Однако способность ионов, одноименно заряженных с дисперсной фазой,
адсорбироваться на ней и тем самым повышать заряд, оспаривается некоторыми
299
исследователями. Паули, например, считает, что ионы, одноименно заряженные
с частицей, понижают в растворе активность противоионов, в результате чего и
происходит повышение £кри,-потенциала при введении индифферентного элек-
тролита. Эта способность понижать активность противоионов тем больше, чем
выше валентность противоположно заряженных ионов индифферентного элек-
тролита. Другое объяснение повышения $крв,-потенциала при введении индиф-
ферентного электролита было выдвинуто Бикермаиом, а также Ратджерсом. Со-
гласно этим авторам, увеличение £Крит-потенцнала в этом случае кажущееся.
Его следует отнести за счет того, что при вычислении потенциала обычно не
учитывалась поверхностная проводимость. При введении соответствующих по-
правок эти исследователи не наблюдали возрастания критического электрокине-
тического потенциала коллоидных частиц при добавлении в систему индиффе-
рентных электролитов.
9. ОСОБЫЕ ЯВЛЕНИЯ, НАБЛЮДАЮЩИЕСЯ
ПРИ КОАГУЛЯЦИИ ЭЛЕКТРОЛИТАМИ
К особым явлениям, наблюдающимся при коагуляции электро-
литами, относятся так называемое явление неправильных рядов,
антагонизм и синергизм ионов при коагуляции, «привыкание» зо-
лей к действию электролитов, коллоидная защита.
Явление неправильных рядов
Это явление наблюдается при введении в коллоидные системы
электролитов, содержащих многовалентные ионы с зарядом, про-
тивоположным заряду частицы. Оно заключается в том, что при
добавлении к отдельным порциям золя различных, все возрастаю-
щих количеств электролитов золь сначала остается устойчивым,
затем в определенном интервале концентраций происходит коагу-
ляция, далее золь снова становится устойчивым и, наконец, при
высоком содержании электролита опять наступает коагуляция, уже
окончательная. Подобное явление могут вызвать и большие орга-
нические ионы красителей или алкалоидов.
Явлейие неправильных рядов объясняется тем, что при весьма
малых количествах введенного электролита многовалентных ионов
недостаточно, чтобы скоагулировать золь. При этой концентрации
электролита t-потенциал частиц выше критического его значения.
При несколько больших количествах электролита его ионы прояв-
ляют уже коагулирующее действие. Этот интервал концентраций
отвечает значениям электрокинетического потенциала частиц от
£крит одного знака до £крит противоположного знака. При еще не-
сколько больших концентрациях многовалентные ионы перезаря-
жают коллоидные частицы и золь становится опять устойчивым.
В этой зоне t-потенциал снова выше критического значения, но
обратен по знаку t-потенциалу частиц исходного золя. Наконец,
при высоком содержании введенного электролита многовалентные
ионы снова и уже окончательно коагулируют золь по механизму
концентрационной коагуляции.
Для золей с отрицательно заряженными частицами и много-
валентных катионов вышеуказанную зависимость хорошо иллю-
стрирует схема, приведенная на рис. IX, 14.
300
Следует заметить, что в последнее время получило распространение мнение,
что перезарядка отрицательно заряженных частиц обусловлена не самими элек-
тролитами с многовалентными катионами, а продуктами их гидролиза, содер-
жащими положительно заряженные ионы окислов или их гидратов. Доказатель-
ством этому служит то, что в кислых растворах перезарядка частиц вообще не
происходит. В слабо щелочной среде изменение знака заряда может быть вы-
звано очень малой концентрацией поливалентного иона, но при еще более высо-
ких значениях pH перезарядка снова становится невозможной из-за того, что
образующиеся в этих условиях иоиы гидратов окисей сами несут отрицательный
заряд.
Правильность подобной точки зрения на перезарядку и явление неправиль-
ных рядов подтверждается тем фактом, что перезарядка имеет место и при вве-
дении электролита с двухвалентными ионами, если только растворимость соот-
ветствующих гидратов окисей достаточно мала. Так, отрицательные частицы
золя Agl могут стать положительно за-
ряженными, если к нему добавить ни-
траты кадмия, цинка или магния и до-
статочное количество NaOH. Такой пере-
зарядки ие происходит, если к золю Agl
добавить Ва(ОН)2, так как раствори-
мость гидрата окиси бария сравнительно
велика.
Явление неправильных рядов,
понятно, может наблюдаться и
при добавлении к золю потен-
циалопределяющих ионов (напри-
мер, при добавлении AgNOa к зо-
лю галогенида серебра с отрица-
тельно заряженными частицами).
Явление неправильных рядов
известно давно и получило свое
название в то время, когда пере-
зарядка частиц золей была мало
исследована. На самом деле чередование зон устойчивости и не-
устойчивости лизолей при добавлении многовалентных ионов
вполне закономерно, и поэтому термин «неправильные ряды» дол-
жен рассматриваться как условный.
Рис. IX, 14. Чередование зон устой-
чивости н неустойчивости при введе-
нии в золь с отрицательно заряжен-
ными частицами электролита с много-
валентными катионами.
Зоны неустойчивости заштрихованы.
Антагонизм и синергизм электролитов
Явления синергизма и антагонизма электролитов можно на-
блюдать при коагуляции золей смесями некоторых электролитов.
Эти явления имеют большое практическое значение, так как даже
при добавлении к коллоидной системе одного коагулятора благо-
даря содержанию в системе стабилизующего электролита коагуля-
ция проходит в действительности под влиянием по крайней мере
двух электролитов. Кроме того, в технике для коагуляции часто
применяют смеси нескольких (обычно двух) электролитов.
При коагуляции золя смесью двух электролитов можно наблю-
дать три предельных случая.
1. Аддитивное действие электролитов. На рис. IX, 15 этот
случай характеризуется прямой /, соединяющей значения порогов
301
способствуют друг
Рис. IX, 15. Антаго-
низм и синергизм при
коагулирующем дей-
ствии электролитов:
У —прямая, характеризую-
щая аддитивное действие
электролитов; 2—кривая,
характеризующая антаго-
низм, 3 —кривая, каракте-
ризующая синергизм.
коагуляции yi и уг каждым электролитом (на оси абсцисс нане-
сены значения концентрации одного электролита, на оси ординат —
значения концентрации другого). Электролиты действуют как бы
независимо друг от друга.
2 Антагонизм электролитов (кривая 2). Электролиты как бы
противодействуют друг другу и для коагуляции золя их нужно до-
бавить больше, чем это требуется по правилу аддитивности.
3. Синергизм электролитов (кривая 3). Электролиты как бы
другу и для коагуляции золя их требуется
меньше, чем это нужно по правилу аддитив-
ности.
Аддитивность обычно наблюдается при
сходстве коагулирующей способности обоих
электролитов (т. е. когда они содержат про-
тивоионы одинаковой валентности), антаго-
низм — при большом различии в коагулирую-
щем действии электролитов. Условия, при ко-
торых наблюдается синергизм, трудно сфор-
мулировать.
Для объяснения антагонистического дей-
ствия электролитов при коагуляции предло-
жен ряд теорий.
Фрейндлих, создатель адсорбционной тео-
рии коагуляции, считал, что причиной анта-
гонизма является способность одного иона
понижать адсорбционную способность, а сле-
довательно, и коагулирующую силу другого
иона.
Согласно Свену Одену, Гошу и Дхару,
а также Кройту, антагонизм ионов объясняет-
ся тем, что при введении смеси электролитов
ионы какого-нибудь одного вида, адсорби-
руясь на одноименно заряженных частицах, могут повысить их
^-потенциал, а следовательно, и устойчивсть системы.
Первая количественная теория коагуляции смесями электроли-
тов была развита Ю. М. Глазманом на основе современной физи-
ческой теории коагуляции лиофобных золей. Согласно этой теории
антагонизм ионов (а также противоположный эффект — синер-
гизм) для сильно заряженных золей есть следствие электростати-
ческих взаимодействий в диффузных атмосферах коллоидных ча-
стиц, а для слабо заряженных золей может обусловливаться кон-
куренцией ионов за места в адсорбционном слое.
В 1961 г. была предложена более сложная теория коагулирую-
щего действия смеси электролитов, которая учитывает адсорбцион-
ные явления и позволяет найти критические концентрации смеси
по критическим потенциалам и определить способность к адсорб-
ции отдельных компонентов. Основные положения этой теории
заключаются в учете соотношения электростатической и ван-дер-
>02
ваальсовой компонент энергии (или силы) взаимодействия ча-
стиц.
В ряде случаев на коагулирующее действие смеси электролитов
может влиять химическое взаимодействие между их ионами, веду-
щее к образованию не обладающих коагулирующим действием
комплексов или нерастворимых соединений. Примером может слу-
жить коагуляция золя Agl смесью электролитов K2SO4 и Th(NO3)4,
образующих комплекс Кг [Th(SO4)3], в котором теряет свою коагу-
лирующую силу.
Привыкание коллоидных систем
Опыт показал, что при постепенном добавлении электролита коллоидные
системы иногда теряют устойчивость при введении большего количества коагу-
лятора, чем при одновременном его добавлении. Коллоидная система как бы
«привыкает» к электролиту, отчего это явление и получило свое название. На-
блюдаются также, и явления отрицательного привыкания, когда прн медленном
добавлении к золю электролита его требуется меньше для коагуляции, чем при
быстром введении.
Существует несколько объяснений положительного и отрицательного привы-
кания коллоидных систем.
Согласно наиболее ранней теории положительное привыкание объясняется
тем, что постепенно выпадающие в осадок агрегированные коллоидные частицы
адсорбируют электролит, в результате чего уменьшается его эффективная кон-
центрация в межмицелляриой жидкости и коагулятора для полной коагуляции
системы требуется добавить больше, чем при одновременном его введеини.
Гош н Дхар считают, что положительное привыкание обусловлено медленно
протекающей адсорбцией коллоидными частицами одноименно заряженных с
этими частицами ионов, что приводит к некоторому увеличению заряда, повы-
шающему устойчивость золя. Одиако было показано, что прн коагуляции ад-
сорбция ионов, заряженных однонмеиио с коллоидными частицами, происходит
только в редких случаях и поэтому точка зрения Гоша н Дхара едва ли имеет
достаточное основание.
Иначе подходит к явлениям привыкания В. Н. Крестинская. Она полагает,
что положительное и отрицательное привыкание является результатом химиче-
ских изменений, происходящих в коллоидной системе при добавлении электро-
литов. Например, явление положительного привыкания при добавлении НС1 в
золь Fe(OH)3 она объясняется тем, что прн быстром введении коагулятора в
золь гидрата окиси железа он коагулирует, как под действием индифферентного
электролита Прн медленном же добавлении коагулятора НС1 реагирует с дис-
персной фазой золя.
Fe(OH)3 + ЗНС1 —> FeCI3 + 3H2O
Тем самым уменьшается количество дисперсной фазы и одновременно увеличи-
вается количество растворимой соли железа, являющейся стабилизатором, вслед-
ствие чего и повышается устойчивость золя.
Отрицательное привыкание, согласно В. Н. Крестинской, сводится к обрат-
ным явлениям — к уменьшению содержания стабилизатора (FeCI3) и увеличению
количества дисперсной фазы [Fe(OH)3J. Отрицательное привыкание у золя гид-
рата окиси железа может быть вызвано, например, добавлением щелочей, умень-
шающих в этом золе количество FeCI3 и увеличивающих количество Fe(OH)3.
Наиболее обоснована точка зрения на явления привыкания, основанная на
физической теории устойчивости. По мере постепенного возрастания концентра-
ции прибавляемого к золю электролита высота энергетического барьера, имеюще-
гося на пути сближения частиц золя, медленно уменьшается. Соответственно
этому увеличивается доля эффективных сближений частиц, что и приводит
к агрегации. В полном согласии с этими представлениями на опыте обычно
303
наблюдается некоторая размытость порога коагуляции. Как следствие этого, в об-
щем случае — при отсутствии побочных явлений — должно иметь место ие поло-
жительное, а отрицательное привыкание. Другими словами, причина отрицатель-
ного привыкания заключается в том, что при постепенном добавлении электро-
лита первые его порции воздействуют иа золь дольше, чем если бы электролит
был добавлен единовременно. Каждая следующая порция электролита действует
уже на изменившийся, несколько астабилизоваииый золь, и поэтому для дости-
жения коагуляции требуется меньше электролита.
Было также показано, что описанные в литературе случаи положительного
привыкания на самом деле являются результатом случайных ошибок или по-
грешностей опыта; при тщательно проведенном эксперименте всегда наблю-
дается отрицательное привыкание, что согласуется с теорией. Положительное
привыкание может иметь место лишь в тех немногих случаях, когда электролит
при очень малых концентрациях вызывает пептизацию коллоидной системы.
Защита коллоидных частиц и сенсибилизация
Как известно, типичные коллоидные системы весьма чувстви-
тельны к действию электролитов. Одиако при введении в золь оп-
ределенных высокомолекулярных веществ и образовании на по-
верхности частиц соответствующего адсорбционного слоя устойчи-
вость системы может быть значительно повышена. Такое явление
получило название коллоидной защиты.
Веществами, способными обусловливать коллоидную защиту,
являются белки, углеводы, пектины, а для систем с неводной дис-
персионной средой — каучук. Часто эти вещества называют защит-
ными коллоидами, хотя такое название является по существу не-
правильным и объясняется лишь исторической традицией.
Защитное вещество как бы придает золю свойства раствора
этого вещества. В присутствии высокомолекулярных защитных ве-
ществ золи, вообще ие поддающиеся концентрированию до высо-
кого содержания дисперсной фазы, можно выпарить досуха и за-
тем полученный сухой остаток можно снова коллоидно растворить.
Электрофоретическая подвижность частиц золей, адсорбировавших
достаточное количество защитного вещества, обычно равна элек-
трофоретической подвижности молекул полимера. Наконец, защи-
щенные золи при добавлении электролитов ие подчиняются пра-
вилу Шульце — Гарди, а ведут себя как растворы защитного вы-
сокомолекулярного вещества, причем для выделения дисперсной
фазы в осадок требуется то же количество электролита, что и для
осаждения высокомолекулярного вещества. Существенно также,
что реагент, способный осаждать защитное вещество, осаждает и
защищенный золь даже в том случае, если исходный золь индиф-
ферентен к этому реагенту. Так, золи, защищенные желатином, те-
ряют устойчивость при добавлении таннидов, образующих с жела-
тином нерастворимое соединение, в то время как незащищенные
золи нечувствительны к действию таинидов.
Для характеристики защитного действия различных высокомо-
лекулярных веществ Зигмонди предложил так называемое «золо-
тое число». Под золотым числом подразумевают число миллиграм-
мов высокомолекулярного вещества, которое необходимо добавить
304
к 10 мл красного золотого золя для того, чтобы предотвратить его
посинение при введении в систему 1 мл 10%-ного раствора хло-
рида натрия. Понятно, что полученные таким образом золотые
числа являются в достаточной мере условными, так как на защит-
ное действие вещества влияет ряд факторов — дисперсность золя,
молекулярный вес защитного вещества, значение pH системы, при
котором проводится испытание, и т. д.
Иногда при определении защитного действия высокомолекуляр-
ного вещества вместо золя золота пользуются коллоидными рас-
творами серебра, красителя конго-рубин, гидрата окиси железа
и др. В этих случаях говорят соответственно о серебряном, руби-
новом, железном и других числах. В табл. IX, 2 приведены значе-
ния этих чисел для некоторых защитных веществ.
Таблица IX, 2. Защитное действие высокомолекулярных веществ
на различные золи
Защитное вещество Число
золотое серебря- ное рубиновое железное сериое ДЛЯ берлин- ской лазурн
Желатин 0,01 0,035 2,5 5 0,00012 0,05
Гемоглобин 0,03-0,07 — 0,8 —
Гуммиарабик 0,5 1,25 — 25 0,024 5
Декстрин 20 100 20 0,125 250
Казеинат натрия 0,01 0,4 — — —
Крахмал картофельный 20 ~20 — —— м
Сапонин 115 35 — 115 0,015 2,5
Яичный альбумин 2,5 1,5 2,0 15 0,025 25
Механизм защитного действия сводится, как мы уже указы-
вали, к образованию вокруг коллоидной частицы адсорбционной
оболочки из высокомолекулярного вещества. Электрониомикроско-
пические снимки непосредственно доказали наличие таких защит-
ных оболочек. Например, адсорбционные слои из метилцеллюлозы
на частицах полистирола имеют толщину 70—100 А. Защитный
слой, если он образован из макромолекул, имеющих полярные или
ионогенные группы, может обеспечивать индуцированную сольва-
тацию частицы и достаточно высокий g-потенциал, что обусловли-
вает повышенную устойчивость системы. Кроме того, согласно но-
вейшим представлениям, стабилизация коллоидных частиц может
происходить вследствие теплового движения и взаимного отталки-
вания гибких макромолекул, только частично связанных с части-
цами золя в результате адсорбции отдельных их участков (энтро-
пийный фактор устойчивости).
Защита коллоидных частиц применяется при изготовлении ле-
карственных препаратов. В организм часто необходимо вводить
лекарственные вещества в мелкораздроблеином, коллоидном
305
состоянии с тем, чтобы они равномерно распределились в нем иг
постепенно растворяясь, оказывали нужное воздействие. Для этого
как раз и пригодны защищенные золи или суспензии лекарствен-
ных веществ. В качестве примера такого препарата можно приве-
сти высокодисперсное серебро, стабилизованное белком, называе-
мое в медицине колларголом.
Иногда введение в коллоидную систему очень малых коли-
честв высокомолекулярного вещества приводит не к защите, а,
наоборот, к сенсибилизации, т. е. к тому, что порог коагуляции
золя, в который введено высокомолекулярное вещество, оказы-
вается меньшим, чем для исходного золя.
Если введенные в систему макромолекулы несут заряд, разно-
именный с зарядом коллоидных частиц, сенсибилизация объяс-
няется как одна из форм взаимной коагуляции, механизм которой
будет рассмотрен ниже. Однако сенсибилизация наблюдается и
тогда, когда частицы золя и молекулы полимера имеют одноимен-
ный заряд. Такая сенсибилизация объясняется тем, что различные
участки одной и той же макромолекулы адсорбируются на поверх-
ности разных коллоидных частиц и таким образом как бы «склеи-
вают» частицы, образуя из них агломераты. При этом адсорбция
происходит обычно уже после добавления коагулирующего элек-
тролита, способствующего адсорбции высокомолекулярного веще-
ства.
10. КОАГУЛЯЦИЯ ЭЛЕКТРОЛИТАМИ ЗОЛЕЙ С НЕВОДНОЙ СРЕДОЙ
Выше мы рассматривали коагуляцию электролитами гидрозолей. Однако
наблюдается коагуляция электролитами и золей с неводной средой. Механизм
и особенности этой коагуляции гораздо менее изучены.
Некоторые исследователе установили, что у органозолей с достаточно боль-
шой диэлектрической проницаемостью среды обнаруживаются явления электро-
фореза и существует известная корреляция между электрофоретической подвиж-
ностью частиц и устойчивостью этих систем. Таким образом, в органозолях,
так же, как и в гидрозолях, коллоидные частицы могут нести двойной элек-
трический слой и обладать ^-потенциалом. Установлено также, что во многих
случаях Для органозолей справедливы закономерности, которым подчиняются
и гидрозоли. К ним приложимо правило Шульце—Гарди, при их коагуляции
наблюдаются явления аддитивности и антагонизма при действии ионов и т. д.
Таким образом, есть все основания считать, что к золям с неводной дисперсион-
ной средой с известными коррективами приложима физическая теория коагу-
ляции.
Понятно, что в связи с меньшей диэлектрической проницаемостью среды и,
следовательно, меньшей диссоциацией молекул стабилизующего электролита за-
ряд частиц органозолей обычно невелик. Однако в связи с малой емкостью
двойного электрического слоя на частицах органозоля этот слой весьма диффу-
зен и обладает большой толщиной, вследствие чего для возникновения электро-
статических сил отталкивания достаточны даже малые заряды. В общем, органо-
золи гораздо менее устойчивы, чем гидрозоли.
Наличие у коллоидных частиц двойного электрического слоя далеко не
всегда является причиной устойчивости органозолей. Устойчивость органозолей
может обусловливаться и сольватацией частиц в результате физического пли
химического взаимодействия среды с дисперсной фазой.
К вопросу об устойчивости коллоидных систем в неводных средах тесно
примыкает вопрос о потере устойчивости гидрозолей при добавлении к ним
306
некоторых неэлектролитов. Известно, что коагуляцию гидрозолей могут вызы-
вать в больших концентрациях такие вещества, как этиловый спирт, ацетон,
сахара. Коагулирующее действие неэлектролитов объясняется тем, что они по-
нижают диссоциацию молекул, содержащихся в системе стабилизующих элек-
тролитов, или вызывают десольватацию коллоидных частиц; это и приводит
в конечном счете к коагуляции золя.
11. ГЕТЕРОКОАГУЛЯЦИЯ И ГЕТЕРОАДАГУЛЯЦИЯ
КОЛЛОИДНЫХ СИСТЕМ
Помимо коагуляции или флокуляции, при которых подобные
друг другу частицы слипаются в крупные агрегаты (гомокоагуля-
ция), в практике ^асто наблюдаются процессы, когда имеет место
слипание разнородных частиц (гетерокоагуляция) или прилипание
частиц дисперсной системы к вводимой в систему чужеродной по-
верхности (гетероадагуляция).
Явление взаимной коагуляции или гетерокоагуляции коллоидов,
наблюдавшееся еще Пиктоном и Линдером в 1897 г., объясняли
действием противоположных зарядов частиц дисперсной фазы
обоих золей, в результате чего происходит слипание частиц. При
этом считали, что для полной коагуляции необходимо, чтобы золи
были взяты в таком количественном соотношении, которое обес-
печивало бы более или менее полную нейтрализацию заряда ча-
стиц. Однако последующие исследования установили, что взаим-
ная коагуляция коллоидных систем может наблюдаться и тогда,
когда частицы обоих золей несут электрический заряд одного и
того же знака.
Гетероадагуляция, наблюдающаяся, например, при отложении
коллоидных или более грубодисперсных частиц на волокне, имеет
большое значение при дублении, крашении, проклеивании бумаж-
ной массы при получении бумаги,- в процессах получения водоот-
талкивающих тканей и т. д.
Согласно взглядам С. И. Соколова и С. С. Воюцкого, основан-
ным на представлениях, развитых ранее Н. П. Песковым, причи-
ной потери устойчивости дисперсных систем в присутствии чу-
жеродной поверхности является адсорбция стабилизатора этой
поверхностью и снижение вследствие этого содержания стабили-
затора в золе *.
Не так давно Б. В. Дерягин предложил общую теорию взаимо-
действия и слипания разнородных частиц, основанную на тех же
положениях, из которых он исходил при рассмотрении гомокоагу-
ляции, и позволяющую объяснить коагуляцию, происходящую как
при взаимодействии двух золей, так и при введении в коллоидную
систему чужеродной поверхности.
* Некоторые сведения о гетероадагуляция и о ее применении в практике
можно найти в монографии С С Воюцкого «Физико-химические основы пропи-
тывания и импрегнирования волокнистых материалов дисперсиями полимеров».
Л., «Химия», 1969. 336 с.
307
Теория гетерокоагуляции Б. В. Дерягина подтверждается неко-
торыми экспериментальными фактами, в частности полученными
Ю. М. Чернобережским. Она объясняет явление так называемой
«групповой коагуляции», когда в смесях некоторых коллоидных
систем и суспензий образуются агрегаты, содержащие частицы
только одного и того же рода.
12. КОАГУЛЯЦИЯ ПОД ДЕЙСТВИЕМ ФИЗИЧЕСКИХ ФАКТОРОВ
Коагуляция под влиянием электролитов является наиболее ти-
пичным случаем коагуляции и обычно применяется в технике,
когда необходимо разрушить коллоидную систему. Однако очень
часто коагуляция обусловливается и другими, чисто физическими
факторами — механическим воздействием на коллоидную систему,
нагреванием или замораживанием золя, разбавлением или концен-
трированием. Коагуляция может также происходить под влиянием
видимого и ультрафиолетового света, рентгеновских лучей, радио-
активного излучения, при действии электрического разряда и уль-
тразвука. Наконец, разрушение системы может наступить спон-
танно при длительном хранении коллоидной системы. К сожале-
нию, особенности и механизм безэлектролитной коагуляции до
настоящего времени изучены недостаточно. Между тем для пони-
мания явления коагуляции во всех его аспектах, для составления
верного представления о его существе подобные исследования
могли бы дать очень много. Несомненно, что правильный взгляд
на явление может быть установлен лишь при всестороннем его
изучении, при подходе к нему с самых различных точек зрения.
Рассмотрим кратко те современные сведения, которые имеются
о коагуляции золей, происходящей под влиянием наиболее важ-
ных физических факторов.
Спонтанная коагуляция. Самопроизвольная коагуляция при
хранении коллоидных систем может происходить либо в резуль-
тате медленно протекающих химических изменений в золе, либо
вследствие того, что всегда имеется некоторая доля эффективных
столкновений частиц, в конце концов приводящих к разрушению
системы.
Самопроизвольное понижение устойчивости золя очень сильно
зависит от условий, в которых хранится золь. Например, стабиль*
ность золя сульфида ртути сравнительно быстро понижается при
хранении в открытом сосуде, что объясняется улетучиванием из
раствора сероводорода, являющегося стабилизатором свежепри-
готовленного золя сульфида ртути. При хранении золя сульфида
ртути в заполненном доверху и герметически закрытом сосуде золь
не только не коагулирует, но становится при хранении даже более
устойчивым к действию электролитов. Это можно объяснить тем,
что при таких условиях в золе в результате хемосорбционного
взаимодействия сероводорода и сульфида ртути вместо мицелл
типа
{т [HgS] nSH~ • (n - х) Н+} хН+
308
постепенно образуются более устойчивые мицеллы типа
{т [HgS] nHgS’“ • 2 (п - х) Н1} 2ХН1-
Коагуляция в результате механического воздействия наблю-
дается при интенсивном перемешивании коллоидных систем, при
перекачке их по трубопроводам и т. п. Причины коагуляции при
механическом воздействии обусловлены, вероятно, временным на-
рушением адсорбционного баланса стабилизатора у поверхности
коллоидных частиц. Такие астабилизованные частицы получают
возможность сближаться на расстояние действия молекулярных
сил и вследствие этого слипаются друг с другом. Доказательством
такого механизма ко'агуляции служит тот факт, что в коагуляте,
полученном в результате механической коагуляции, стабилизатора
содержится всегда меньше, чем в коагуляте, получаемом при коа-
гуляции электролитами.
Интересно, что системы, более стойкие к механическим воздей-
ствиям, обычно более стойки и к действию электролитов. Имеются
данные, что механическая устойчивость коллоидных систем связана
с их устойчивостью при старении. Связь между всеми этими фак-
торами вполне понятна, так как механическая устойчивость, с од-
ной стороны, и устойчивость лиозоля к электролитам и при хране-
нии, с другой стороны, определяется одним и тем же фактором —
свойствами двойного электрического слоя.
Коагуляция коллоидных систем может происходить и в резуль-
тате вибрационных воздействий и влияния ультразвукового поля.
Особенное значение вибрационная коагуляция имеет в технике
при получении различных паст, бетонов и других систем. Напри-
мер, виброобработка бетонной смеси вначале ведет к разрушению
в ней коагуляционной структуры и в результате этого к увеличе-
нию текучести смеси, что облегчает заполнение смесью форм Од-
нако при дальнейшей виброобработке образуется прочная кристал-
лизационная структура.
Коагуляцию коллоидных систем в ультразвуковом поле наблю-
дал еще Дарсинг (1908 г-)- В дальнейшем было установлено, что
в докавитационной области облучение ультразвуком способствуег
коагуляции, однако с увеличением мощности поля начинает уже
преобладать его диспергирующее действие. В ультразвуковых
полях малой мощности малые частицы следуют за средой, в то
Время как крупные, обладающие большой инерцией, почти не увле-
каются жидкостью. Таким образом, малые частицы как бы «про-
шивают» среду и оказываются в поле действия молекулярных сил
больших частиц, что приводит к коагуляции. Д. С. Лычников и
Г. А. Мартынов установили, что преодоление энергетического
барьера и коагуляция возможны лишь, когда амплитуда колеба-
ния частиц соизмерима с расстоянием между частицами. Ультра-
звуковое поле как бы «перебрасывает» мелкие частицы из вторич-
ного потенциального минимума в первичный, Если частицы нахо-
309-
оооо
ссоооо -
оооо
+
Рис. IX, 16. Схема взаимодействия
дипольных частиц и образования це-
почечных агрегатов.
дятся на больших расстояниях друг от друга, то устойчивость не
нарушается и золь остается стабильным.
Не так давно было показано, что в осадках подвергнутых уль-
тразвуковому облучению суспензий содержатся цепочки частиц,
возникающие, по всей вероятности, в результате поляризационного
взаимодействия между частицами. Таким образом, при образова-
нии структур в дисперсных системах под влиянием ультразвуко-
вого поля важное значение имеет деформация двойного электри-
ческого слоя.
Коагуляция под влиянием электрического поля. Уже сравни-
тельно давно обнаружено, что под действием переменного электри-
ческого поля в золях и суспензиях может наблюдаться как разру-
шение структур, так и образование цепочек частиц. Особенно
важно воздействие электрическо-
го поля на эмульсии, поскольку
оно применяется для обезвожива-
ния нефтяных эмульсий и очистки
воды, содержащей примеси ми-
неральных масел.
О. Г. Усьяров, И. С. Лавров
и И. Ф. Ефремов исследовали
влияние переменных и постоян-
ных электрических полей на взаи-
модействие частиц синтетических каучуков, диспергированных
в алифатических спиртах и гексане. Было найдено, что в полярных
средах между частицами, находящимися в электрическом поле,
возникает притяжение, которое заметно проявляется на расстоя-
нии, в 2—3 раза превышающем размер частиц. В результате обра-
зуются линейные агрегаты — цепочки, что не наблюдается в слу-
чае неполярного гексана. Это привело к выводу об ответственности
за процесс ориентированной агрегации неравновесных электропо-
верхностных сил, обусловленных деформацией и поляризацией
двойного электрического слоя. При взаимодействии цепочек друг
< другом по схеме, изображенной на рис. IX, 16, возникают вто-
ричные дендритоподобные структуры. Снятие электрического поля
при фиксации частиц в неглубоком потенциальном минимуме при-
водит к распаду агрегатов или структур; при фиксации частиц
в глубокой потенциальной яме такого явления не происходит.
В более поздней работе И. С. Лавров показал, что наложение
внешнего переменного поля сравнительно невысокой напряжен-
ности на водные суспензии полиакрилонитрила приводит к ориен-
тированному вдоль силовых линий агрегированию частиц.
Коагуляция при разбавлении или концентрировании коллоид-
ной системы. Наблюдающуюся в некоторых случаях коагуляцию
при разбавлении гидрозолей водой можно объяснить десорбцией
стабилизующего электролита с поверхности частиц в дисперсион-
ную среду, что обусловливает падение заряда частицы. При этом,
конечно, может происходить и гидролиз стабилизатора, вслед-
310
ствие которого золь может также терять устойчивость. В практике-
при разбавлении коллоидной системы технической водой, содер-
жащей электролиты, коагуляцию при разбавлении можйо объяс-
нить и действием электролитов.
Гораздо труднее объяснить коагуляцию при концентрировании коллоидных
систем. Правда, при концентрировании коллоидной системы путем выпаривания
в ней повышается концентрация электролитов, всегда содержащихся в гидрозо-
лях, что может действовать на систему астабилизующим образом. Однако опыт
показал, что коагуляция гидрозоля происходит и в том случае, когда концентри-
рование проводится с помощью ультрафильтрации, т. е. когда состав диспер-
сионной среды не меняется.
Некоторые исследователи объясняют коагуляцию золя при концентрировании
увеличением числа столкновений частиц друг с другом. Однако это объяснение
мало соответствует тому факту, что золи проявляют способность к спонтанной
коагуляции только тогда, когда их концентрация превышает определенное кри-
тическое значение. Можно полагать, что неустойчивость коллоидной системы
выше определенной концентрации объясняется увеличением в единице объема
содержания не только чужеродного электролита, но и самих коллоидных частиц,,
которые должны рассматриваться как поливалентные ионы, а также и содержа-
ния соответствующих противоионов. Подобное допущение вполне вероятно.
В самом деле, как показал еще Дюкло, коллоидные частицы вносят свою долю
в электропроводность системы, и поэтому есть все основания думать, что заряд
этих частиц должен учитываться при вычислении ионной силы раствора.
Коагуляция при нагревании или охлаждении. Нагревание даже
до кипения обычно сравнительно мало влияет на устойчивость гид-
розолей. Наблюдающееся в отдельных случаях падение агрегатив-
ной устойчивости при нагревании объясняется, вероятно, десорб-
цией стабилизатора с поверхности частицы и увеличением интен-
сивности броуновского движения. Оба эти фактора способствуют
преодолению энергетического барьера при столкновении частиц.
Охлаждение гидрозолей до температур, выше температуры их
замораживания, обычно также мало сказывается на устойчивости
гидрозолей. Наоборот, охлаждение, сопровождающееся заморажи-
ванием гидрозоля, очень часто приводит к его коагуляции, причем
коагуляция, как правило, бывает тем более полной, чем ниже тем-
пература, до которой охлаждался золь, и чем дольше он пребывал
в замороженном состоянии.
По Лоттермозеру, на коагуляцию при замораживании влияет
не столько температура, сколько степень превращения раствора
в кристаллическую массу. Лоттермозер считает, что при замерза-
нии вода образует между коллоидными частицами микроскопиче-
ские кристаллы. Вследствие увеличения объема в замороженной
системе могут развиваться большие давления. Частицы дисперсной
фазы, спрессованные в результате этого между кристалликами,
могут деформироваться, приходить друг с другом в контакт и сли-
паться. Так, в результате чисто механических воздействий, возни-
кающих при замерзании коллоидной системы, может образовы-
ваться коагулят.
Иной позиции придерживался А. В. Думанский, считавший, что
при замораживании золя постепенно образуются кристаллики чи-
стой воды, в результате чего в оставшейся незамерзшей части
311
системы происходит концентрирование как золя, так и содержа-
щихся в нем электролитов. Вследствие этого может возникнуть
дакая большая концентрация электролитов, что произойдет коагу-
ляция. В доказательство правильности этой точки зрения А. В.Ду-
манский приводил наблюдения Зигмонди, нашедшего, что коллоид-
ная система при замораживании тем более стойка, чем она устой-
чивее к влиянию электролитов или удалению воды при высуши-
вании.
Следует заметить, что в результате замораживания и после-
дующего оттаивания.коагулятов гидратов окисей металлов в зна-
чительной степени меняются многие их свойства: уменьшается
объем осадков,- понижается влажность и улучшаются фильтрую-
щие свойства. Такое изменение свойств коагулятов ценно для ана-
литической химии, прейаративной радиохимии, химической техно-
логии и для очистки вод, в том числе радиоактивных.
ГЛАВА X
СТРУКТУРНО-МЕХАНИЧЕСКИЕ СВОЙСТВА
ДИСПЕРСНЫХ СИСТЕМ
Ознакомившись в предыдущей главе с явлениями коагуляции,
можно перейти к рассмотрению структурно-механических свойств
дисперсных систем. Ранее этого сделать было нельзя, поскольку
образование структур в коллоидных и микрогетерогенных системах
является следствием коагуляции этих систем.
Коллоидные и микрогетерогенные системы с жидкой и твердой
дисперсионной средой, как и все другие конденсированные системы,
обладают определенными механическими свойствами — вязкостью,
во многих случаях пластичностью, упругостью и прочностью. Эти
свойства связаны со структурой подобных систем, поэтому их ча-
сто называют структурно-механическими свойствами.
Эти свойства называют еще реологическими, так как учение
о течении различных тел или, в более общем виде, о процессах
деформации, развивающихся во времени, носит название реоло-
гии.
Все коллоидные и микрогетерогенные дисперсные системы, как
мы уже указывали в гл. I, можно разделить на свободнодисперс-
ные и связнодисперсные системы. Если дисперсионной средой яв-
ляется жидкость, то могут быть и переходные системы, отдельные
частицы которых связаны друг с другом в рыхлые агрегаты, но не
образуют сплошной структуры (структурированные жидкости).
Очевидно, подобные агрегаты можно рассматривать как обрывки
пространственной сетки, которая по тем или иным причинам не
получила полного развития.
На тип системы весьма существенное влияние оказывает кон-
центрация дисперсной фазы. В свободнодисперсных системах кон-
центрация дисперсной фазы не может быть очень большой, так как
в противном случае неизбежно возникал бы контакт между от-
дельными частицами. В результате образовывались бы простран-
ственные сеткн или, по крайней мере, объем системы заполнялся
настолько частицами дисперсной фазы, что свободное перемеще-
ние частиц по отношению друг к другу было бы невозможно. По-
нятно, что при введении в систему стабилизатора, препятствую-
щего сближению частиц, а следовательно, и проявлению молеку-
лярных сил между частицами, можно значительно увеличивать
критическую концентрацию, при которой возникают связи между
313
элементами структурной сетки, т. е. концентрацию, отвечающую
достаточно высокой прочности или структурной вязкости системы,
В связнодисперсных системах концентрация дисперсной фазы
может достигать очень больших значений. Однако следует отме-
тить, что эти системы могут получаться и при очень малых кон-
центрациях дисперсной фазы, если только частицы достаточно
анизодиаметричны, являясь, например, палочками или пластин-
ками. Так, золь пятиокиси ванадия с палочкообразными частицами
образует гель при содержании в нем 0,01—0,001% V2O5.
Понятно, что та или иная структура коллоидной системы при-
дает ей определенные реологические свойства.
Из-за несвязанности друг с другом отдельных частиц в свобод-
нодисперсных системах эти системы проявляют способность к вяз-
кому течению, т. е. к непрерывному изменению своей формы
во времени под влиянием даже очень малых напряжений сдвига.
При этом течение этих систем качественно обычно подчиняется
тем же закономерностям, что и течение чистой дисперсионной
среды. Количественные же отклонения сводятся в основном к тому,
что вязкость такой системы обычно выше вязкости чистой среды.
В противоположность этому связнодисперсные системы, вслед-
ствие наличия сил взаимодействия между их частицами, обладают
в известной степени свойствами твердых тел — способностью со-
хранять форму, некоторой прочностью, упругостью, часто эластич-
ностью. Однако из-за малой прочности связи между отдельными
элементами структуры сетки структуры в связнодисперсных си-
стемах сравнительно легко разрушаются и эти системы приобре-
тают способность течь.
Структурированные жидкости обычно представляют собой си-
стемы с малой концентрацией дисперсной фазы, но с явно выра-
женной тенденцией частиц к слипанию.
Структурированные жидкости, очевидно, должны обладать рео-
логическими свойствами, промежуточными между свойствами сво-
бодно- и связнодисперсных систем. Эти системы способны течь,
но они не подчиняются при этом законам течения обычных не-
структурированных жидкостей.
На реологические свойства коллоидных систем, помимо концен-
трации дисперсной фазы в системе, сильно влияют и такие фак-
торы, как природа дисперсной фазы, дисперсионной среды и при-
сутствующего стабилизатора, поскольку именно от этих факторов
зависит эффективность молекулярных сил, действующих, с одной
стороны, между частицами дисперсной фазы, а с другой — между
частицами и растворителем.
Изучая реологические свойства коллоидных систем, можно оп-
ределить характер образовавшихся в них структур. Значение рео-
логических свойств коллоидных систем важно и с практической
стороны. Такие важные системы, как почва, формовочные глины,
цементный раствор, краски, лаки, пасты, характеризуются рядом
особых структурно-механических свойств.
314
В этой главе рассмотрены реологические свойства свободно-
дисперсных и связнодисперсных систем, а также систем промежу-
точного типа с жидкой дисперсионной средой. Структурно-меха-
нические свойства пен, резко отличающиеся от свойств других
систем с жидкой средой, рассмотрены в гл. XII, а механические
свойства систем с газовой дисперсионной средой — в гл. XI.
Однако прежде чем приступить к рассмотрению реологических
свойств систем с жидкой средой, необходимо хотя бы кратко по-
знакомиться со строением и свойствами структур, образующихся
в таких системах.
1. ВОЗНИКНОВЕНИЕ И ОСОБЕННОСТИ СТРУКТУР
В КОЛЛОИДНЫХ СИСТЕМАХ
Согласно А. И. Рабинерсону и Г. И. Фуксу, структуры, обра-
зующиеся в высокодисперсных системах, можно классифицировать
по их плотности (числу связей в единице объема). По этой клас-
сификации структуры делят на пространственные (рыхлые) и ком-
пактные. Первые структуры характерны для дисперсных систем
с анизодиаметрическими частицами, вторые структуры часто воз-
никают в системах с изодиаметрическими частицами. Первые
структуры при старении и действии коагулирующих факторов мо-
гут переходить во вторые.
В последнее время наибольшее распространение получила
классификация структур, предложенная П. А. Ребиндером.
Согласно П. А. Ребиндеру, структуры в коллоидных и микро-
гетерогенных системах можно разделить на коагуляционные (тик-
сотропно-обратимые) и конденсационно-кристаллизационные (не-
обратимо-разрушающиеся),
Коагуляционные структуры. К ним относятся структуры, обычно
возникающие в результате понижения агрегативной устойчивости
дисперсных систем. При истинной коагуляции, когда частицы пол-
ностью теряют фактор устойчивости (двойной электрический слой,
сольватную оболочку и т. д.), они слипаются друг с другом, об-
разуя компактные агрегаты. Достигнув определенного размера,
эти агрегаты образуют плотный коагулят (или коагулюм). Если
же происходит неполная астабилизация системы, то фактор устой-
чивости будет снят только с некоторых участков поверхности ча-
стиц, да и то не полностью, и в результате этого частицы, слипаясь
по таким местам, образуют пространственную сетку, в петлях ко-
торой находится дисперсионная среда. Происходит, как принято
говорить, гелеобразование * или образование лиогеля. Вид струк-
* Согласно принятой в настоящее время терминологии, гелеобразованием
или желатинированием называют переход коллоидного раствора из свободно-
дисперсного состояния (золя) в связнодисперсное (гель). Термином «застудне-
вание» пользуются для обозначения аналогичного перехода раствора высоко-
молекулярного вещества в студень,
. 31 о
Рис. X, 1. Строение про-
странственных структур,
образующихся в лиозоле
при астабилизации:
1—частицы дисперсной фа-
зы; 2—участки поверхности
частиц, лишившиеся фактора
устойчивости после астаои*
лизации; 3—участки поверх-
ности частиц, сохранившие
фактор устойчивости, 4—пет-
ли структуры, заполненные
дисперсионной средой.
туры, получаемой в результате гелеобразования, схематически изо-
бражен на рис. X, 1.
При достаточно сильной астабилизации прослойки дисперсион-
ной среды, находящиеся между частицами, в местах их соприкос-
новения полностью вытесняются, и благодаря этому осущест-
вляется непосредственный контакт частиц друг с другом. Это
отвечает образованию наиболее прочных, но одновременно и наи-
более хрупких коагуляционных структур. Однако весьма часто при
более слабой астабилизации-в месте контакта между частицами
остаются достаточно толстые слои диспер-
сионной среды. Иначе говоря, между ча-
стицами наблюдается дальнодействие, при-
чины которого рассмотрены в гл. IX. Нали-
чие тонкой жидкостной прослойки между
частицами обусловливает меньшую проч-
ность структуры, но зато придает ей пла-
стичность, а в некоторых случаях и эластич-
ность. Чем толще прослойка среды между
частицами, тем меньше сказывается дей-
ствие молекулярных сил, обусловливающих
сцепление частиц, тем менее прочна струк-
тура и тем жидкообразней система.
На гелеобразование может влиять ряд
факторов. - Концентрация дисперсной фазы
сильно сказывается на скорости образова-
ния геля и его прочности, так как с повы-
шением численной концентрации число
контактов, приходящихся на единицу объе-
ма системы, и скорость установления кон-
тактов возрастают.
Уменьшение размера частиц при по-
стоянной концентрации дисперсной фазы
также способствует гелеобразованию. Очень большое значение для
гелеобразования имеет форма, частиц. Образование лиогеля облег-
чается, если частицы анизодиамётричны и имеют концы, углы и
ребра. В этих местах двойныё электрические слои или сольватные
оболочки наименее развиты, так что слипание астабилизован-
ных частиц происходит именно по этим участкам. Кроме того,
для образования структуры требуется гораздо меньше дисперс-
ной фазы, представляющей собой палочкообразные или пластин-
чатые частицы, чем дисперсной фазы, состоящей из сферических
частиц.
На скорость образования и свойства полученного геля весьма
сильно влияет температура. Время образования геля по тем же
причинам, что и время коагуляции, при повышении температуры
уменьшается. Однако с повышением температуры в результате
увеличения интенсивности броуновского движения лиофобные гели-
могут переходить в структурированную жидкость, а затем, при еще
316
более высоких температурах, становиться даже неструктурирован-
ными жидкостями.
- Механическое воздействие, например перемешивание, обычно
препятствует образованию геля. Однако в некоторых случаях
время образования геля из агрегативно неустойчивых золей с силь-
но анизодиаметрическими частицами (например, золя V2O5) можно
значительно сократить, если сосуд, содержащий золь, медленно
вращать. Это явление, открытое Фрейндлихом, получило назва-
ние реопексии (греч. — образование геля при движении). Причину
реопексии некоторые исследователи видят в том, что параллель-
ная ориентация вытянутых частиц при течении благоприятствует
установлению Между ними контактов и, следовательно, способ-
ствует образованию геля. Другие исследователи считают, что при-
чиной реопексии является возникновение при движении системы
слабой турбулентности, ускоряющей установление контакта ме-
жду частицами.
Специфическим свойством коагуляционных структур является
тиксотропия (от греч. — тиксо — прикосновение, тропе — поворот,
изменение) — способность структур после их разрушения в резуль-
тате какого-нибудь механического воздействия самопроизвольно
восстанавливаться во времени. Иначе говоря, тиксотропия предста-
вляет собой способность к изотермическому обратимому превра-
щению золя в гель. Сущность тиксотропии заключается в том, что
связи, которые были разрушены при механическом воздействии,
восстанавливаются в результате случайных удачных соударений
частиц, находящихся в броуновском движении. Такое постепенное
восстановление структуры и, следовательно, нарастание ее проч-
ности происходит не только, когда система находится в покое,
но и при течении системы со скоростью меньшей той, которая
обусловила данную степень разрушения первоначальной струк-
туры. Существенно, что при переходе от одного режима течения
к другому с большей скоростью обычно, но не всегда, наблюдается
дополнительное разрушение структуры, что понижает эффектив-
ную вязкость и прочность структуры. Наоборот, при переходе от
установившегося режима течения к течению с меньшей скоростью,
как правило, происходит некоторое восстановление структуры и,
соответственно, эффективная вязкость и прочность системы увели-
чиваются.
Явление тиксотропии часто встречается в природе. Тиксотроп-
ные свойства проявляют некоторые грунты («плывуны»). Для про-
топлазмы в клетках живых организмов также характерна тиксо-
тропия. Гели миозина обладают сильно выраженными тиксотроп-
ными свойствами, что свидетельствует о роли тиксотропии в ра-
боте мускулов.
Способность дисперсных систем проявлять тиксотропию часто
используют в технике. Например, при бурении нефтяных скважин
глинистые растворы прокачивают через скважину — промывают ее
для удаления из нее частиц измельченной выбуренной горной
317
породы. Благодаря тиксотропным свойствам этих растворов преду-
преждается оседание частиц породы в скважине и заклинивание
этим самым бурового инструмента при временной остановке бу-
рения.
Другим примером тиксотропных систем, имеющих практическое
применение, могут служить обычные масляные краски, представ-
ляющие собой взвесь минеральных пигментов в олифе. Благодаря
тиксотропным свойствам красок их можно наносить на вертикаль-
ные поверхности в виде жидкости после их механического переме-
шивания, при этом нанесенная краска не стекает в результате
быстро наступающего структурирования. Для повышения тиксо-
тропных свойств в краски иногда вводят специальные добавки,
например полиамиды, бентониты. Характерные реологические свой-
ства, включая тиксотропию таких красок, в том числе и типограф-
ских, исследовали А. А. Трапезников с сотр. с помощью разрабо-
танных ими методов определения предела прочности и вязкости
в широком интервале скоростей деформации. Было показано, что
тиксотропия может выражаться как в разрушении и образовании
сплошной сетки (прочностная тиксотропия), так и в раз-
рушении и восстановлении агрегатов частиц (вязкостная'
тиксотропия).
Явлением, противоположным тиксотропии, является дилатансия, проявляю-
щаяся в небольшом сопротивлении системы при низком напряжении сдвига и
высоком сопротивлении при большом сдвиговом усилии. Дилатансия характерна
для очень концентрированных агрегативно устойчивых суспензий, у которых нет
постоянного контакта между частицами. Рейнольдс, открывший это явление в
1885 г., объяснил его тем, что движение системы возможно только при малых
напряжениях сдвига и малом изменении относительного положения частиц При
больших напряжениях сдвига происходит местное сближение частиц и соответ-
ственно уменьшается свободное пространство для течения, в результате чего
движение жидкости сильно затрудняется или даже приостанавливается.
к тиксотропным системам близко примыкают тактоиды и слои Шиллера,
Под тактоидами подразумевают дисперсии, имеющие участки с хорошо выра-
женной периодичностью в расположении параллельно ориентированных относи-
тельно друг друга анизодиаметрических частиц. Явление анизотропной ориента-
ции частиц было обнаружено на золях Fe(OH)s, на ряде других неорганических
и органических дисперсий, а также на биоколлоидах — колониях вирусов и бак-
терий. Причиной образования анизотропных областей в таких системах яв-
ляется равновесие между молекулярными силами притяжения и электростати-
ческими силами отталкивания, действующими между частицами, являющимися
обычно диполями.
Слои Шиллера представляют собой коллоидные осадки, состоящие из пла-
стинчатых частиц, которые расположены в горизонтальных плоскостях, обычно
отделенных друг от друга расстояниями в несколько тысяч ангстрем. Характер-
ное строение слоев'Шиллера определяется соотношением силы электростатиче-
ского отталкивания заряженных частиц и силы тяжести Структуры, близкие
к слоям Шиллера, могут возникать под действием и других внешних полей —>
центробежных, электрических и магнитных.
Доказательством того, что периодические структуры образуются
в результате электростатического отталкивания при перекрытии
ионных атмосфер, служит закономерное уменьшение расстояний
между соседними частицами при увеличении концентрации элек-
318
тролита. Количественная теория периодических коллоидных струк-
тур была развита И. Ф. Ефремовым и С. В. Нерпиным и в неко-
торых отношениях уточнена С. С. Духиным и др. При этом каж-
дая частица рассматривается как фиксированная в потенциальной
яме, образуемой совокупным действием ее соседей.
Известным аналогом периодических коллоидных структур мо-
жет служить кристалл монтимориллонитовой глины при его вну-
трикристаллическом набухании в водных растворах. При внутри-
кристаллическом набухании кристаллические плоскости толщиной
каждая около 10 А раздвигаются и между ними образуются
жидкие прослойки. Условием набухания является насыщение кри-
сталла ионами Н+, Li+ или Na+. При очень низких концентрациях
внутрикристаллические прослойки достигают толщины в 300 А.
Одинаковость всех прослоек сохраняет периодическую структуру
системы и позволяет по дифракции рентгеновских лучей измерять
толщины прослоек. Полученные данные согласуются с теорией
ДЛФО. Такой набухший кристалл служит хорошей моделью дру-
гих периодических структур. С помощью этой модели можно
также, как показал О. Г. Усьяров, обнаружить существование
ближней и дальней потенциальной ям, энергетического барьера и
влияние валентности ионов на закономерности набухания.
Если концентрация достаточно монодисперсной коллоидной системы, защи-
щенной от коагуляции в ближней потенциальной яме высоким барьером, стано-
вится высокой, то часто в результате сближения частицы образуют квазикри-
сталлическую решетку, в которой они расположены на равных расстояниях
друг от друга. При высокой дисперсности системы можно заметить, как ча-
стицы совершают колебательное броуновское движение около своих положений
равновесия.
Периодические коллоидные структуры образуют многие вирусы, бактерии,
монадисперсные золи металлов, золи пятиокиси ванадия, латексы.
Образование периодических структур означает, что расстояние каждой ча-
стицы от соседних соответствует дальней потенциальной яме в тех случаях,
когда система граничит со свободной дисперсионной средой и, следовательно,
не подвергается внешнему давлению. В этом случае расстояния между части-
цами уменьшаются при добавлении электролита и увеличиваются при разбав-
лении системы.
Чаще встречаются периодические коллоидные структуры, занимающие весь
объем жидкой среды и ограниченные со всех сторон стенками или поверхностью
раздела с воздухом В этом случае потенциальная яма, в которой удерживается
каждая частица, образуется в результате сложения сил отталкивания со сто-
роны соседних частиц. Поэтому соответственные расстояния могут быть меньше,
чем абсциссы дальних потенциальных ям, — система, как принято говорить, на-
ходится в «стенсненном» состоянии. При еще большем «сжатии», когда средние
расстояния между соседними частицами будут меньше абсцисс потенциальных
барьеров, произойдет нарушение равновесия, часть частиц слипнется при попа-
дании в ближние потенциальные ямы, оставшиеся же частицы смогут сохранить
периодическое расположение.
Однако и до такого «сжатия» периодическая коллоидная структура обычно
находится в состоянии только временного равновесия. Время от времени то
одна, то другая частица перескакивает из положения равновесия в узле квази-
кристаллической решетки в ближайшую потенциальную яму. Возникающие де-
фекты квазикристаллической решетки множатся необратимо и не могут «зале-
чиваться». Таким образом, в отличие от кристаллов, периодические коллоидные
структуры находятся часто ие в состоянии термодинамического равновесия, а
319
устойчивы кинетически. Иными словами, их существование обусловливается
медленностью перехода в равновесное состояние
Однако принципиально нельзя исключать возможность равновесных перио-
дических структур, если ближний минимум отсутствует Возможно, что к этой
категории принадлежит ряд вирусов, как, например, вирус табачной мозаики.
Современное состояние проблемы, равно как и собственные исследования
периодических коллоидных структур, изложены в монографии И. Ф. Ефре-
мова *.
Системы с коагуляционными структурами обладают, как пра-
вило, небольшой прочностью, известной пластичностью, а также
некоторой эластичностью. Эластические свойства коагуляционных
структур, согласно П. А. Ребиндеру, можно объяснить изменением
энтропии системы в результате переориентации образующих си-
стему структурных элементов, сопутствующей изменению ее
формы. Такими структурными элементами служат отдельные кол-
лоидные частицы (в отличие от высокомолекулярных соединений,
где эластическая деформация связана с изменением взаимной
ориентации звеньев молекулярных цепей). Системы с коагуля-
ционными структурами проявляют также ползучесть, т. е. способ-
ность при .течении к медленному развитию значительных остаточ-
ных деформаций практически без заметного разрушения простран-
ственной сетки. Ползучесть системы определяется высокой, хотя и
вполне доступной'измерению вязкостью в области весьма малых
скоростей течения. Только при больших скоростях течения в таких
системах происходит значительное разрушение структуры, так как
связи между частицами не успевают восстанавливаться и ско-
рость разрушения становится больше скорости восстановления.
Системам с коагуляционными структурами очень часто при-
суще явление синерезиса. Под синерезисом понимают самопроиз-
вольное уменьшение размеров геля с одновременным выделением
из него дисперсионной среды, содержавшейся в петлях геля. При-
чина синерезиса заключается в том, что при гелеобразовании ме-
жду элементами структуры образуется сравнительно малое число
контактов, не отвечающее предельно уплотненному состоянию
структуры. Затем в результате перегруппировки частиц, обуслов-
ленной их тепловым движением, число этих контактов увеличи-
вается, что неизбежно приводит к сжатию геля и к выпрессовыва-
нию из него дисперсионной среды. Явление синерезиса схематически
представлено на рис. X, 2. Как можно видеть, число палочкообраз-
ных частиц, образующих гель, остается тем же самым, но число
точек соприкосновения их после синерезиса резко увеличивается.
Синерезису благоприятствуют все факторы, способствующие
коагуляции, а именно, увеличение концентрации электролита, по-
вышение температуры, введение в систему десольватирующих аген-
тов и т. д. Синерезису способствует также гибкость и подвижность
элементов коагуляционной структуры. Поэтому особенно значи-
* И. Ф. Ефремов, Периодические коллоидные структуры. Л., « Химия»,
1971, 192 с,
320
тельный синерезис наблюдается у студней высокомолекулярных
веществ, состоящих из гибких макромолекул.
Системы с коагуляционной структурой, из которых высушива-
нием удалена дисперсионная среда, способны в той или иной сте-
пени поглощать эту среду при контакте с ней. Поглощение среды
сухим гелем — ксерогелем (от греч. ксерос — сухой) может
обусловливаться как простым капиллярным всасыванием, так и
раздвижением элементов структуры геля вследствие возникнове-
ния расклинивающего давления и заполнения образовавшихся
промежутков средой. В последнем случае говорят о набухании^
5
Рис. X, 2. Схематическое изображение синерезиса:
в —система до синерезиса, б —система после синерезиса.
ксерогеля. Совершенно очевидно, что набухание является процес-
сом обратным синерезису. Однако вследствие того, что при обра-
зовании пространственной структуры лиофобных систем места кон-
такта закрепляются обычно довольно прочно, лиофобные ксеро-
гели набухают сравнительно незначительно. Наоборот, студни вы-
сокомолекулярных веществ могут очень сильно набухать. Погло-
щение среды сухим гелем или предельно прочной коагуляционной
структурой приводит к пластификации, т. е. к резкому понижению
прочности, сопровождающемуся, однако, возрастанием пластиче-
ских и эластических свойств данного тела.
Более подробно явления синерезиса и набухания рассмотрены
в гл. XIV.
Кондеисациоино-кристаллизациониые структуры. К этому типу
принадлежат структуры, у которых связи между частицами обра-
зованы за счет химических сил. Эти структуры возникают либо
в результате образования прочных химических связей между ча-
стицами (конденсационные структуры), либо вследствие сращива-
ния кристалликов в процессе выкристаллизовывания новой фазы
(кристаллизационные структуры).
Структуры с таким характером связей между частицами не
могут проявлять тиксотропии, пластичности и эластичности,
а должны, наоборот, проявлять упруго-хрупкие свойства. Проч-
ность их обычно значительно выше прочности коагуляционных
структур. Совершенно очевидно, что системы с такого рода
11 Зак. 664
321
структурами не могут в сколько-нибудь заметной степени сине-
резировать или набухать.
Типичный конденсационной структурой является гель кремне-
вой кислоты. Кристаллизационное структурообразование имеет су-
щественное значение для твердения минеральных вяжущих средств
в строительных материалах на основе цементов, гипса или из-
вести.
На образование связей, а следовательно, и на свойствах этих
структур может сильно сказываться присутствие в системе моди-
фицирующих добавок поверхностно-активных веществ, изменяю-
щих форму и размеры образующихся кристаллов, а также условия
их срастания.
Необходимо хотя бы кратко, остановиться на дисперсных системах, в кото-
рых структура в обычном смысле этого слова отсутствует, но у которых наблю-
даются некоторые общие свойства с настоящими структурированными систе-
мами. К таким системам относятся, например, высококонцентрированные стаби-
лизованные суспензии (пасты), а также осадки, образующиеся в результате
седиментации.
В агрегативно устойчивых суспензиях, частицы которых достаточно сольва-
тированы, при предельно высокой концентрации дисперсной фазы (когда частицы
разделены весьма тонкой пленкой жидкости) почти вся дисперсионная среда
может быть сольватно связана с дисперсной фазой. В результате этого вязкость
систем обычно весьма высока
Механическая прочность подобных систем может резко возрастать при
введении в них поверхностно-активных веществ, молекулы которых, адсорби-
руясь и ориентируясь на поверхности частиц, способствуют, согласно Б В. Де-
рягину, развитию и взаимодействию граничных сольватных слоев Механические
свойства концентрированной суспензии можно повысить также введением в нее
высокомолекулярных веществ, обычно адсорбирующихся на поверхности частиц
и вызывающих застудневание жидкой среды При этом застудеванию способ-
ствуют частицы дисперсной фазы, играющие роль «активного наполнителя».
Механические свойства концентрированных систем, в которых частицы дис-
персной фазы имеют сольватные оболочки, все же обычно значительно ниже
механических свойств систем с коагуляционными и конденсационно-кристаллиза-
ционными структурами. Кроме того, благодаря образованию сольватных обо-
лочек у частиц система пластифицируется, понижается ее прочность и у нее
появляются пластично-вязкие свойства, тогда как при возникновении простран-
ственных структур повышаются упруго-хрупкие свойства системы.
Рассмотрим два возможных случая образования осадков в результате осе-
дания частиц седиментационно неустойчивых суспензий.
1. Оседание агрегативно неустойчивых суспензий про-
исходит быстро из-за образования агрегатов; осевший осадок занимает большой
объем; так как частицы сохраняют то случайное -взаимное расположение, в ко-
тором они оказались при соприкосновении. Совершенно очевидно, что подобные
системы, образующиеся при оседании суспензий, довольно близки по строению
и свойствам к рассмотренным выше коагуляционным структурам.
2. Оседание агрегат и.в но устойчивых суспензий, если ча-
стицы достаточно малы, происходит медленно и частицы, осевшие на дно сосуда,
остаются разделенными друг от друга под влиянием тех же сил, которые пре-
пятствуют их агрегации. Вследствие этого частицы, скользя друг по другу, зани-
мают положение, отвечающее минимальной потенциальной энергии н характери-
зующееся максимальной компактностью укладки. Полученный таким образом
осадок, если он достаточно плотен, может обладать всеми механическими свой»
ствами, присущими концентрированным суспензиям.
Из сказанного следует, что объем осадка может служить показателем сте»
пени агрегативной устойчивости седиментирующей суспензии. Это можно видет$
3^2
из следующего примера. Если 40 г порошка кварца с диаметром частиц от 1 до
5 мкм размешать в 25 мл воды, то образуется агрегативно устойчивая суспен-
зия, устойчивость которой обусловлена возникновением вокруг частиц гидратных
оболочек или двойных электрических слоев за счет ионизации кремневой кис-
лоты, образовавшейся прн взаимодействии S1O2 и Н2О. В этой суспензии за
6 ч оседания образуется всего 8,5 мм осадка, содержащего, однако, 54 объемн %
кварца.
Если то же количество порошка кварца распределить в том же объеме
четыреххлорнстого углерода, не способного образовывать на частицах сольват-
ные оболочки н исключающего возможность возникновения двойного электриче-
ского слоя, то получается агрегативно. неустойчивая суспензия В этой суспензии
уже за 15 мин оседания слой осадка достигает предельной толщины в 53 мм,
причем он состоит всего из 7 объемн. % кварца.
Наконец, если 40 г кварцевого порошка поместить в 25 мл четыреххлори-
стого углерода, в который предварительно было введено небольшое количество
олеиновой кислоты, то при оседании суспензии снова образуется малое количе-
ство осадка большой плотности. Это следует объяснить тем, что молекулы олеи-
новой кислоты, адсорбируясь на кварце полярными группами, гндрофобизуют
поверхность частиц, делают их агрегативно устойчивыми в четыреххлористом
углероде и тем самым способствуют компактной укладке частиц в осадке.
2. вязкость ИСТИННЫХ И КОЛЛОИДНЫХ РАСТВОРОВ
Для того чтобы лучше понять особенности вязкости коллоидных систем, на-
помним основные понятия о вязкости и механизме течения обычных низкомоле-
кулярных жидкостей, таких, как вода, спирт,
как это показано на рнс. X. 3, что с помощью
приложенной внешней силы мы привели в не
слишком быстрое движение тонкий слой жид-
кости в направлении, параллельном плоскости
поверхности жидкости, со скоростью U1. Опыт
показывает, что нижележащие слон ие оста-
нутся в покое, а тоже придут в движение.
Верхний слой благодаря внутреннему трению,
возникающему между слоями и являющемуся
следствием хаотического теплового движения
молекул и межмолекулярных сил притяжения,
увлекает нижележащие слон, причем скорость
движения этих слоев уменьшается от верхнего
к нижнему, так как нижние слон тормозят дви-
жение вышележащих. Уменьшение этой ско-
бензол и т. д Представим себе,
Рнс. X, 3. Распределение ско-
ростей движения жидкости
между двумя параллельно дви-
жущимися слоями (/ и 2), на-
ходящимися на расстоянии х.
рости прямо пропорционально расстоянию х
от верхнего слоя до нижнего. Существенно, что такое движение слоев жидкости,
называемое ламинарным течением, может быть вызвано еколь угодно
малой силой, но действующей достаточно длительное время. Внутреннее трение,
как и внешнее, является, конечно, причиной днсснпацни (рассеяния) энер-
гии, т. е. необратимого превращения ее в тепло.
По определению вязкости, данному Ньютоном, сила внутреннего трения F,
равная по значению, но обратная по направлению приложенной извне силе, про-
порциональна площади слоя s, к которому приложена эта сила, и градиенту
скорости движения du/dx между слоями.
Г = r]s (du/dx)
(Х,1)
где г] — коэффициент пропорциональности, называемый коэффициентом вязко-
сти (или вязкостью), зависящей от природы жидкости.
Относя силу F к площади сдвига, уравнение (X. 1) можно переписать в
виде:
Р = F/s = п (du/dx) (X, 2)
где Р — напряжение сдвига, поддерживающее течение жидкости.
И*
323
Из уравнения (X, 1) можно видеть, что единицами измерения вязкости
являются г/(см-с),
В честь французского ученого Пуазейля, впервые изучившего движение
жидкостей в капиллярах, единица вязкости названа пуазом (П); 1 пуаз соот-
ветствует вязкости жидкости, при которой для поддержания градиента скорости
в 1 см/с нужна сила в 1 дин на 1 см2.
Для сравнительно маловязких жидкостей обычно пользуются величиной в
сто раз меньшей — сантипуазом (сП). Следует помнить, что вязкость воды при
20 °C весьма близка к 1 сП.
Вычисленная из уравнения Ньютона вязкость в условиях ламинарного тече-
ния жидкости не зависит ни от способа измерения, ни от типа и размеров
примененного вискозиметра, т. е. является инвариантной характеристикой данной
жидкости.
Величина 1/q, обратная вязкости, называется текучестью. Она характеризует
подвижность жидкости под влиянием внешних воздействий.
Ламинарное течение жидкости по трубкам описывается известным уравне-
нием Пуазейля *:
v = лг4р/(8т]/) (X, 3)
где v — объемная скороеть истечения; г и I — радиус и длина трубки; р — раз-
ность давлений на концах трубки; ri —вязкость жидкости.
Это уравнение найдено Пуазейлем в 1842 г эмпирическим путем
Уравнение Ньютона, а следовательно, и уравнение Паузейля соблюдаются,
если жидкость движется ламинарно, т. е. в виде слоев, имеющих различную
скорость и не смешивающихся друг с другом. Такой режим наблюдается лишь
при сравнительно малых скоростях течения. При больших скоростях •ламинар-
ный характер течения переходит в турбулентный, характеризующийся воз-
никновением в движущейся жидкости завихрений. Если применять к такому
течению уравнения Ньютона и Пуазейля, то коэффициент вязкости теряет свой
обычный смысл, так как его значение при турбулентном течении зависит не
только от природы жидкости, но становится функцией скорости движения жидко-
сти. Очевидно, в этом случае можно говорить лишь об эффективной или кажу-
щейся вязкости, понимая под ней условную величину, вычисленную для данной
скорости течения по уравнениям Ньютона или Пуазейля.
Рейнольдс в 1883 г. показал, что при течении жидкости по трубке с глад-
кими стенками ламинарное движение переходит в турбулентное, когда число
Рейнольдса Re превысит известное значение. Число или критерий Рейнольдса
представляет собой безразмерное отношение:
Re = «rp/n (X, 4)
где р — плотность жидкости.
Как видно из свотношения (X, 4), ламинарное движение переходит в турбу-
лентное при тем меньших скоростях, чем больше радиус трубки и плотность
жидкости и чем меньше ее вязкость. Наличие 'в жидкости взвешенных частиц,
особенно неправильной формы, способствует так называемой ранней турбулент-
ности, т. е. тому, что ламинарное течение переходит в турбулентное при значи-
тельно меньших значениях Re.
Уравнения Ньютона или Пуазейдя количественно описывают течение жидко-
сти, но ничего не говорят о сущности явления. Весьма важно дл.я понимания
процесса течения жидкости разобраться в его молекулярном механизме.
Механизм течения жидкости можно представить себе правильно только
исходя из современных представлений о строении жидкости, развитых
Я. И. Френкелем, а также Эйрингом (см. гл. III).
Как известно, молекулы всякой жидкости находятся в непрерывном тепло-
вом движении, передвигаясь относительно друг-друга путем последовательного
перемещения в дырки, имеющиеся в жидкости. Пока на жидкость не действуют
* Вывод этого уравнения путем интегрирования уравнения Ньютона дается
в учебниках физики; см. также С. С. Воюцкий. Курс коллоидной химии, М.,
«Химия», 1964. 574 с. См. с. 342.
324
а б
какие-нибудь внешние силы, перемещении равновероятны во всех направлениях.
В этом случае зависимость потенциальной энергии от положения молекул вблизи
дырки можно охарактеризовать схемой а иа рис. X, 4. Однако если жидкость
находится под действием внешнего силового поля, которое стремится сместить
молекулы в определенном направлении, то потенциальная энергия после пере-
хода молекул в новое положение Ej будет меньше потенциальной энергии до
перехода £ь При этом потенциальная яма деформируется и примет вид, показан-
ный на схеме б рис. X, 4. Поскольку скачки молекул, приводящие к уменьшению
потенциальной энергии, происходят чаще, чем обратные, хаотическое движение
приобретает направленность и наблюдается перенос вещества, т. е. течение
жидкости.'
Совершенно очевидно, что частота перемещения молекул в жидкости тем
больше, чем выше средняя кинетическая энергия теплового движения молеку-
лы kT, и тем меньше, чем ббльшую работу надо затратить для того, чтобы мо-
лекула могла совершить скачок. Следовательно,
с повышением температуры промежуток времени
между перемещениями молекул в соседние равно-
весные положения становится все короче и короче,
а жидкость при одном и том же напряжении
сдвига будет все более и более подвижной.
Кроме того, при повышении температуры и уве-
личении энергии теплового движения все боль-
шее число молекул обладает энергией, необходи-
мой для совершения скачка. Наконец, с повыше-
нием температуры происходит термическое расши-
рение жидкости, что приводит к возрастанию
числа дырок и к увеличению их размера. Все
это обусловливает значительное снижение внут-
реннего треиия или повышение текучести. Так,
вязкость воды при изменении температуры на
1 °C в интервале не слишком высоких температур
изменяется на 2—3%.
Текучесть и вязкость, как это следует из молекулярной модели вязкого
течения, изменяются с температурой приблизительно по экспоненциальному за-
кону:
1/т) = Л ехр(-£/АТ) (Х,5)
или
П = А' ехр (E/kT) (Х.6)
где А и А' — коэффициенты, сравнительно мало • зависящие от температуры;
Е — энергия активации, которую должна приобрести молекула, чтобы переско-
чить в новое положение равновесия; k — постоянная Больцмана; Т — абсолютная
температура.
При известной температурной зависимости текучести или вязкости, логариф-
мируя эти уравнения, легко найти значение энергии активации, поскольку связь
между In Т) и 1/Т должна выражаться прямой линией. Однако следует помнить,
что эта графическая зависимость для разных жидкостей представляет собой
прямую только в относительно узкой области температур; в более широком
температурном интервале обычно наблюдается отклонение от прямолинейности.
Опыт показывает, что энергия активации вязкого течения жидкости обычно
равна нескольким килокалориям на моль.
Определение вязкости жидкостей
Вязкость жидкостей можно определять с помощью различных методов.
Остановимся кратко лишь на важнейших из них.
Метод падающего шарика сводится к определению скорости свободного па-
дения в жидкости шарика известного объема и массы. Для того чтобы не
возникало турбулентного движения жидкости, скорость падения шарика должна
32S
Рис. X, 4. Зависимость по-
тенциальной энергии моле-
кулы от ее положения
вблизи дырки:
а—в отсутствие внешнего снло-
вого поля; б — прн действии
внешнего силового поля.
быть не слишком большой, а сосуд, в котором падает шарик, достаточно широ-
ким. Коэффициент вязкости вычисляют по уравнению, которое легко выводите®
из уравнения (111,39):
\ , Т) = 2г2(р _ p0)g/(9u) (Х,7>
Нетрудно видеть, что, определяя вязкость по скорости падения шарика,,
экспериментатор решает задачу, обратную той, которую ему приходится решать
при нахождении радиуса частицы суспензии по скорости оседания.
Из уравнения (X, 7) следует, что скорость падения шарика обратно пропор-
циональна вязкости жидкости. Подобная зависимость соблюдается и тогда-
когда шарик падает не в широком сосуде, а движется в наклонной Стеклянной
трубке, заполненной жидкостью, так, как это происходит в известном вискози-
метре Гепплера.
Метод истечения жидкости через капилляр основан на измерении времен»
вытекания определенного объема жидкости через капилляр^ радиус и длина
которого известны. В этом случае вязкость вычисляют по уравнению:
п == лг4рт/(8/7) (X, 8>
где V — объем жидкости, вытекшей за время т; остальные обозначения те же-
что и в уравнении (X, 3).
Определение с помощью ротационных вискозиметров. Приборы, применяе-
мые для определения вязкости по этому методу, представляют собой два коак-
сиальных цилиндра. В кольцевой зазор между цилиндрами заливают исследуе-
мую жидкость. Один из цилиндров (обычно внутренний) приводят во вращение-
например, с помощью груза, блока и шнура. После весьма краткого периода
устанавливается стационарный режим течения жидкости между цилиндрами.
Вязкость находят, определяя число оборотов вращающегося цилиндра в единицу
времени. Ряд конструкций прибора такого типа в Советском Союзе разработай
М. П. Воларовичем.
Другая разновидность ротационного прибора, предложенная еще Ф. Н. Шве-
довым в прошлом столетии, представляет собой также два коаксиальных ци-
линдра, из которых внешний приводится во вращение с постоянной скоростыо
с помощью электромотора, а внутренний подвешен на тонкой упругой нити и
снабжен указателем для отсчитывЗния угла закручивания. Жидкость заливают-
в пространство между цилиндрами. Внешний цилиндр при вращении увлекает-
за собой жидкость, которая в свою очередь приводит во вращение внутренний
цилиндр и закручивает его на некоторый угол до тех пор, пока момент силы
кручения не станет равным моменту сил трения. Так как нить, на которой под-
вешен внутренний цилиндр, упруга, то этот угол всегда пропорционален вяз-
кости.
По уравнению, связывающему скорость вращения и угол закручивания ци-
линдра, зная константы прибора, можно вычислить вязкость.
Очень часто на практике применяют не абсолютные, а относительные методы
определения вязкости, что позволяет исключить из расчета константы приборов..
При этом измеряют время падения шарика, время истечения или другие пара-
метры для стандартной жидкости, а затем определяют ту же величину и для
исследуемой жидкости. Поскольку значения вязкости пропорциональны измерен-
ным величинам, то, зная вязкость стандартной жидкости, можно по полученным
результатам вычислить вязкость исследуемой жидкости. Так как вязкость сильна
зависит от температуры, ее следует измерять всегда при постоянной темпера-
туре, термостатнруя прибор.
Более подробно методы определения вязкости рассматриваются в руковод-
ствах к лабораторным занятиям по коллоидной химии.
Зависимость эффективной вязкости коллоидных систем
от скорости течения
Отлйчие течения золей от течения обычных индивидуальных:
жидкостей или истинных растворов низкомолекулярных вещест»
обуславливается тем, что в первых присутствуют во взвешенном
326
«состоянии коллоидные частицы, размеры которых значительно пре-
вышают размер молекул. Наличие таких частиц изменяет пути
-отдельных молекул текущей жидкости и способствует перемеши-
ванию отдельных слоев. Именно в результате этого у дисперсных
-систем наблюдается ранняя турбулентность, т. е. переход ламинар-
ного течения в турбулентное при меньших числах Рейнольдса; чем
для жидкостей, не содержащих взвешенных частиц. Кроме того,
коллоидные частицы сужают пространство, занятое самой жидко-
стью в потоке, и увеличивают таким образом средний градиент
скорости в направлении, перпендикулярном течению жидкости.
Вследствие этого вязкость золя всегда несколько выше вязкости
дисперсионной среды.'
Наконец, характерной особенностью многих золей является не-
подчинение их зависимостям, выражаемым уравнениями Ньютона
и Пуазейля. Для обычных жидкостей объем жидкости, протекшей
через капилляр в единицу времени, прямо пропорционален раз-
ности давлений р на концах капилляра. Точно так же для обыч-
ных жидкостей наблюдается прямая зависимость между углом
поворота внутреннего цилиндра и скоростью вращения наружного
цилиндра в ротационном приборе типа вискозиметра Ф. Н. Шве-
дова. Для многих же золей, эмульсий и растворов высокомолеку-
лярных веществ такая зависимость отсутствует, а вычисленная по
соответствующему уравнению вязкость имеет переменное значение
и является функцией градиента скорости. Иными словами, вяз-
кость многих дисперсных систем не являемся инвариантной харак-
теристикой системы, а зависит от условии ее определения, напри-
мер от скорости течения жидкости в вискозиметре, от типа и раз-
меров прибора.
Законам Ньютона и Пуазейля не подчиняются коллоидные си-
стемы с удлиненными частицами и частицами, способными дефор-
мироваться, а также структурированные коллоидные системы.
Причина аномалии вязкого течения коллоидных систем с вытяну-
тыми, палочкообразными частицами заключается в том, что по
мере увеличения напряжения сдвига,.обусловливающего течение,
такие частицы ориентируются своей длинной осью в направлении
потока, в результате чего понижается гидродинамическое сопро-
тивление и этим самым убыстряется движение жидкости. Ориен-
тацию вытянутых частиц в направлении потока легко доказать,
измеряя двойное лучепреломление в золе при все возрастающем
градиенте скорости. _
У систем с деформирующимися частицами, например у эмуль-
сий, наблюдается, аналогичная зависимость. Капельки дисперсной
фазы с возрастанием приложенного напряжения сдвига и увели-
чением скорости течения удлиняются, превращаясь из шариков
в эллипсоиды, что, конечно, облегчает течение и понижает вяз-
кость. То же самое наблюдается и при течении растворов высоко-
молекулярных соединений с гибкими, свернутыми в клубок макро-
молекулами. Здесь падение вязкости обусловлено распрямлением
327
молекул и их ориентацией в направлении потока. Конечно, во всех
перечисленных случаях речь идет о кажущейся или эффективной
вязкости г]*, так как истинная вязкость жидкости от скорости
течения зависеть не может.
Законам Ньютона и Пуазейля не подчиняются также структу-
рированные жидкости. В связи с большим практическим значе-
нием и некоторыми принципиальными особенностями вязкость
структурированных жидкостей целесообразно рассмотреть отдельно.
В заключение отметим, что все жидкости, подчиняющиеся за-
кону Ньютона, называются нормальными', системы же, способные
течь, но не подчиняющиеся уравнению Ньютона, принято называть
аномальными. При работе с капиллярным вискозиметром можно
воспользоваться весьма простым приемом для того, чтобы судить,
является ли исследуемая жидкость нормальной или аномальной.
Для этого избыточное давление р, под действием которого исте-
кает жидкость, умножают на соответствующее время истечения
определенного объема жидкости. Так как уравнение Пуазейля
можно представить в виде:
’’“W рТ==КрТ
то совершенно очевидно, если произведение рх не зависит от дав-
ления, под которым происходило истечение, то жидкость является
нормальной, если же зависит, то она аномальна. При работе с ро-
тационным вискозиметром таким же критерием может служить не-
зависимость произведения числа оборотов цилиндра в единицу
времени на величину груза, обеспечивающего вращение цилиндра.
3. СТРУКТУРНАЯ ВЯЗКОСТЬ
Как указывалось выше, структурированные системы не подчи-
няются закону Ньютона. Это может быть обусловлено либо нали-
чием в жидкости несвязанных друг с другом обрывков структуры,
либо малопрочной "сплошной структурной сеткой, способной раз-
рушаться при действии на систему сравнительно малых усилий.
В первом случае система ведет себя при течении как жидкость,
в которой взвешены частицы, способные ориентироваться или де-
формироваться. Обрывки структурной сетки разрушаются в ре-
зультате различной скорости слоев в. потоке, а отдельные элементы
разрушенной сетки, если они имеют вытянутую форму, ориенти-
руются своей длинной осью по направлению течения.
Второй случай более сложен. Сначала Ф. Н. Шведов, затем
Бингам предположили, что течение системы с малопрочной про-
странственной структурой начнется лишь тогда, когда напряжение
сдвига Р превысит какое-то определенное критическое значение 0,
необходимое для разрушения структуры, т. е. когда начнет соблю-.
даться условие Р — 0 > 0. Такое течение Бингам называет пла-
стическим, а критическое (предельное) напряжение сдвига 0 —
пределом текучести.
328
Очевидно, что для систем с пластическим течением уравнение
Ньютона должно быть заменено уравнением Бингама:
р _ 0 = ц' (du/dx)
или
. Р = rf (du/dx) + 0 (Х,9)
где т)' — вязкость, отвечающая пластическому течению системы (пластическая
вязкость).
При отсутствии структурной сетки значение 0, очевидно, равно
нулю и уравнение Бингама переходит в уравнение Ньютона, а пла-
стическая вязкость т/ — в истинную вязкость жидкости. Бингам
принимает, что как только Р превысит 0 и начнется течение, вяз-
кость системы сразу принимает постоянное значение. При таких
Рнс. X, 5. Зависимость между
du/dx и Р для пластической
системы по Бингаму.
Рис. X, 6. Зависимость между du/dx
и Р для реальной пластической си-
стемы.
условиях зависимость du/dx от Р выразится прямой линией
(рис. X, 5). Согласно этому рисунку пластическую вязкость можно
выразить так:
, Р — 6
= <Х’Г0>
где р — угол, образуемый прямой с осью абсцисс.
Примером систем, довольно хорошо подчиняющихся уравне-
нию Бингама, могут служить пасты из глины и консистентные
смазки. Однако для большинства структурированных коллоидных
систем зависимость du/dx от Р выражается не прямой, а кривой
(рис. X, 6). Причина такого явления заключается в том, что при
достижении предела текучести структура разрушается не сразу,
а постепенно по мере увеличения градиента скорости движения
жидкости. Очевидно, можно различать три критических напряже-
ния сдвига: 1) 0/ — первый, или минимальный, предел текучести,
соответствующий началу течения (началу разрушения структуры);
2) 0В — предел текучести по Бингаму, отвечающий отрезку на оси
абсцисс, отсекаемому продолжением прямолинейного участка кри-
вой; 3) 0макс — максимальный предел текучести, соответствующий
значению Р, при котором кривая переходит в прямую линию.
3129
Очевидно, бмакс представляет собой то напряжение, при котором
структура в жидкости разрушается полностью. Все три предела
являются характеристикой механических свойств структуры, суще-
ствующей в системе.
В последние годы структурная вязкость коллоидных систем
была детально изучена П. А. Ребиндером и его школой, а также
Рис. X, 7. Кривые тече-
ния и зависимость- эф-
фективной вязкости 1]*
от напряжения сдвига Р
для структурированных
жидкостей (по П. А. Ре-
бнчдеру):
—напряжение сдвига, от-
вечающее переходу ползу-
чести в течение с измеримой
вязкостью; 0макс—макси-
мальный предел текучести.
А. А. Трапезниковым с сотр. В результате
этих работ было показано, что при любой
скорости течения в коагуляционной струк-
туре протекают два противоположных про-
цесса — разрушение и восстановление. Рав-
новесное состояние между этими процес-
сами в установившемся потоке характери-
зуется эффективной вязкостью.
При малых скоростях течения системе
наносятся незначительные повреждения,,
так как разрушения, неразрывно связан-
ные с течением, успевают тиксотропно вос-
становиться вследствие медленности про-
цесса течения и течение системы происходит’
практически без разрушения структуры, т. е.
наблюдается явление ползучести.
При больших скоростях течения струк-
тура системы значительно разрушается;
при этом разрушенная структура из-за
быстроты процесса восстанавливается не-
значительно.
Для характеристики течения структури-
рованных жидкостей и пластичных тел сле-
дует использовать не пластическую, а эф-
фективную вязкость ц*, которая уменьшает-
ся с ростом действующего напряжения сдви-
га в системе. При малых напряжениях
сдвига эффективная вязкость имеет наи-
большее значение, равное г]' вязкости жид-
кости с практически неразрушенной струк-
турой. При больших напряжениях сдвига
эффективная вязкость уменьшается до предельного значения г]мин —
вязкости, отвечающей полному разрушению структуры (при усло-
вии сохранения ламинарности потока).
Кривые течения (dufdx, Р) и зависимость вязкости от напря-
жения сдвига для структурированных систем, по П. А. Ребиндеру,,
имеют вид, изображенный на рис. X, 7.
Вязкость структурированных коллоидных систем сильно зависит
от условий ее определения, в частности от градиента скорости, при:
котором она измеряется. Поэтому значения вязкости таких си-
стем можно сравнивать только тогда, когда они найдены в таких
состояниях, которые характеризуются одинаковыми значениями
330
числа Рейнольдса. Кроме того, по тем же соображениям вязкость
золей целесообразно определять не при каком-нибудь одном на-
пряжении сдвига, а необходимо получать кривые duldx, Р, харак-
теризующие реологические свойства системы в достаточно боль-
шом интервале значений Р. Однако даже при измерении в совер-
шенно одинаковых условиях с использованием одного и того же
вискозиметра найденные значения вязкости для какой-нибудь оп-
ределенной системы могут существенно различаться в зависимости
ют предыстории системы и от того, когда система была приготов-
лена. Так, при достаточно длительном стоянии коллоидной си-
стемы вязкость ее может постепенно повышаться благодаря про-
цессу структурирования. Такое изменение вязкости можно наблю-
дать на золях гидрата окиси железа или пятиокиси ванадия.
€ другой стороны, в результате механического воздействия, на-
пример при протекании систем через капилляр, структуры, обра-
зовавшиеся в системе, могут разрушаться, вследствие чего вязкость
се уменьшается. Именно этим объясняется то обстоятельство, что
яри следующих непосредственно друг за другом измерениях вяз-
кости с помощью капиллярного вискозиметра очень часто полу-
чают непрерывно уменьшающиеся значения, стремящиеся к опре-
деленному пределу.
4. механические свойства коллоидных систем,
ПРОЯВЛЯЮЩИХ ИСТИННУЮ УПРУГОСТЬ
В предыдущем разделе при рассмотрении структурно-механических особен-
ностей коллоидных систем мы считали эти системы жидкостями. Однако ряд
свойств коллоидных систем можно объяснить, если рассматривать их как твер-
дые тела. Последний подход особенно целесообразен при изучении коллоидных
систем, обладающих в некоторой степени упругостью или эластичностью и ха-
рактеривующихся наличием истинного предела текучести (предела упругости),
т. е. такого предела напряжения сдвига, ниже которого практически никакого
•течения не наблюдается Как правило, таким свойствам отвечают достаточно
концентрированные твердообразные гели и системы с конденсационно-кристалли-
зационной структурой
Однако прежде чем рассматривать механические свойства упругих гелей,
остановимся кратко на таких важных для понимания этих свойств характери-
стиках систем, как модуль сдвига и период релаксации напряжения.
Модуль сдвига Е, как известно, характеризует жесткость всякого тела
« его способность сохранять форму. Эту величину можно вычислить из извест-
ного уравнения Гука: /
г = Р/Е (X, 11)
где е — относительная деформация сдвига в истинно твердых телах; Р — напря-
жение сдвига.
Это уравнение справедливо лишь при малых деформациях, так как при
определенном критическом напряжении, называемом пределом упругости, тело
теряет упругие свойства и сохраняет остаточные деформации. Модуль сдвига Е
при одинаковой скорости приложения нагрузки зависит от природы тела и тем-
пературы Для твердых тел величина Е может достигать весьма больших зна-
чений, для истинных жидкостей Е — 0, так как всякое сколь угодно малое
331
напряжение сдвига приводит к течению жидкости. Ниже в качестве примера
приведены значения модуля сдвига Е (в кгс/см2) для некоторых веществ:
Желатин
0,5%-ный раствор.............. 4-10—’°
10%-ный раствор (студень) . . 5-10~
Каучук............................ 1,7 • 102
Свинец............................. 4,8-10*
Дерево (дуб)...................... 8-10*
Сталь............................. 8 • Ю5
А. А. Трапезников показал, что весьма характерным реологическим пара-
метром системы является предельная обратимая деформация сдвига вмакс, до-
стигаемая в быстро релаксирующих системах (о релаксации см. ниже) при вы-
сокой скорости деформации. В различных коллоидных системах оиа может быть
весьма разной. Например, в пастах она обычно составляет несколько процентов
или десятков процентов, тогда как в эластичных гелях и некоторых растворах
полимеров она может достигать десятка тысяч процентов. Такие значения на-
много превышают привычные значения предельных обратимых деформаций
сдвига каучуков.
Период (или время) релаксации связан с тем, что обычно мо-
лекулы или другие структурные элементы материальной системы обладают не-
которой подвижностью и способны перемещаться относительно друг друга. В ре-
зультате этого напряжение, создавшееся в теле вследствие его деформации, спо-
собно со временем в значительной степени «рассасываться». Подобный процесс
уменьшения напряжения во времени получил название релаксации. Релаксация
является следствием теплового движения и имеет совершенно общий характер.
Предложен ряд уравнений, описывающих деформацию систем, способных
релаксировать Наиболее простым является уравнение Максвелла, вытекающее
из его теории упруго-вязкого тела:
dP/dx = Е (de/dx) - (Р/х*) (X, 12.'
где х* — константа, называемая периодом или временем релаксации.
Легко убедиться, что уравнение Максвелла передает качественно основные
закономерности релаксации при постоянной температуре. Если деформацию тела
поддерживать постоянной (е = const), то de/dx •= 0 и из уравнения Максвелла
следует, что напряжение Р меняется со временем по закону:
(dP/dT) + (P/T*)=0 (X, 13)
Проинтегрировав это уравнение, получим
Р = Рйе~х1х* (X, 14)
Отсюда видно, что с течением времени напряжение в деформированном теле
убывает по экспоненциальному закону, а константа х*, характеризующая ско-
рость релаксации, равна промежутку времени, в течение которого начальное
напряжение тела при постоянной деформации уменьшается в е = 2,72 раза (е —
основание натуральных логарифмов).
Периоды релаксации напряжения низковязких жидкостей весьма малы
вследствие большой подвижности их молекул. С увеличением вязкости периоды
релаксации жидкостей возрастают и приближаются к периодам релаксации на-
пряжения твердых тел. Для кристаллов процесс релаксации протекает беско-
нечно медленно. Ниже в качестве примера приведены значения периода релак-
сации напряжения т* (в с) некоторых веществ:
Вода.......................................3-1°2’
Масло касторовое...........................2-10 3
Лак копаловый............................... 2-10
Канифоль (при 55 °C)...................5-10
Желатин, 0,5%-ный раствор..............8-102
Канифоль (прн 12 °C)....................4-10°
Идеально твердые тела.................... со
332
Рис. X, 8. Зависимость деформации в
от времени т при постоянном напря-
жении для системы, обнаруживающей
мгновенную упругость, запаздываю-
щую упругость и течение.
Обращает внимание сравнительно большой период релаксации для весьма
разбавленного 0,5%-ного водного раствора желатина Период релаксации для
этого раствора ближе к периоду релаксации напряжения твердых тел, чем к пе-
риоду релаксации напряжения жидкостей. Это объясняется наличием в растворе
желатина сравнительно больших структурных элементов (макромолекул жела-
тина), требующих для перегруппировки сравнительно большого времени.
Часто при деформации реальных тел наряду с явлениями релаксации на-
блюдается так называемая запаздывающая упругость. В то время как релакса-
ция приводит к переходу упругой деформации в пластическую, запаздывающая
упругость проявляется в том, что не вся упругая деформация возникает мгно-
венно (как в идеально твердых телах). Часть этой деформации развивается во
времени, так что упругая деформация достигает предельного значения, отве-
чающего заданному напряжению, лишь после определенного промежутка вре-
мени. Как правило, запаздывающая упругость проявляется тем сильнее, чем
неоднороднее структура твердого тела.
Возвратимся к рассмотрению меха-
нических свойств твердообразных микро-
гетерогенных и коллоидных систем, об-
ладающих истинной упругостью. К таким
системам относятся поликристаллические
металлы, самые разнообразные структу-
рированные дисперсные системы, гели,
концентрированные растворы мыл, а
также высокомолекулярные вещества и
их концентрированные растворы, способ-
ные проявлять не только упругость, но
н высокую эластичность.
Поведение всех этих систем при не-
значительных деформациях сходно с по-
ведением идеально упруги^ тел. Однако
при напряжениях, ведущих к разруше-
нию структурной сетки, эти системы
способны течь как вязкие жидкости, причем их эффективная вязкость всегда
падает с увеличением скорости течения или напряжения.
Очень часто при деформации этих систем явления упругой (мгновенной)
деформации, запаздывающей упругости и течения накладываются друг на друга
и дают характерную картину изменения суммарной деформации во времени,
представленную на рис. X, 8 Как можно видеть, под влиянием деформирующей
силы, например напряжения сдвига Р, приложенного к системе в момент Xi,
развивается мгновенная упругая деформация в]. Этой деформации отвечает
мгновенный модуль сдвига Е\ — Р/%\. Затем система под действием силы начи-
нает течь в результате необратимой перегруппировки структурных элементов.
Одновременно в системе развивается запаздывающая упругость, обусловливаю-
щая деформацию е2 вследствие обратимой перегруппировки структурных эле-
ментов Этой замедленно развивающейся упругой деформации отвечает модуль
сдвига Е2 — Р/ъ2. Все это приведет к тому, что кривая на рис. X, 8 будет асимп-
тотически приближаться к некоторой прямой, -соответствующей течению системы.
Если через некоторое время в момент т2 деформирующее усилие будет устра-
нено, упругая деформация 81 исчезнет со скоростью звука. Далее постепенно
исчезнет деформация е2, обусловленная запаздывающей упругостью, а деформа-
ция е3, обусловленная течением (истинной релаксацией), осгаиется как необра-
тимая.
Иногда найденная в этих условиях необратимая деформация представляет
собой не истинно пластическую, а кажущуюся пластическую
деформацию, являющуюся следствием того, что процесс восстановления формы
деформированного тела происходит чрезвычайно медленно. В последнем случае
остаточная деформация частично или полностью исчезает в результате опреде-
ленной обработки тела (например, нагревания или набухания в растворителе),
обусловливающей более быстрое протекание перегруппировки структурных эле-
ментов. Это указывает на то, что деформированное тело после снятия прило-
333
женной извне нагрузки может иметь скрытую упругость. Упругость, т. е. спо-
собность дисперсной системы после прекращения действия деформирующей силы
возвращаться к первоначальной форме, как правило, указывает на особую вну-
треннюю структуру системы, препятствующую необратимому смещению ее эле-
ментов по отношению друг к другу.
Определение механических свойств коллоидных систем
Жесткие системы, обладающие достаточной механической прочностью, мож-
но исследовать обычными методами физико-механического анализа (снятие
кривой, характеризующей зависимость от деформации напряжения, определение
предельного напряжения прн растяжении или сдвиге, определение относительной.
Риг. X, 9. Схема прибора Бейлера и
Ребиидера для определения струк-
турно-механических свойств дисперс-
ных систем:
/ — столик; 2 —кювета; 3 — рифленая пла-
стинка; 4 — жесткая нить; 5 — микроскоп;
6 — динамометр.
Рис. X, 10. Схема крутильного при-
бора для определения структурно-
механических свойств дисперсных
систем:
/ — крутильная головка; 2 —упругая нить;
3 —рифленый цилиндр; 4 — кювета; 5 —осве-
титель; 6—зеркальце; 7 —шкала.
и остаточной деформации и т. д.). При этом необходимо лишь учитывать ско-
рость деформирования, так как последняя сильно влияет на полученные резуль-
таты.
Для определения механических свойств гелей и других структурированных
дисперсных систем, обнаруживающих упругость, предложен ряд специальных
методов, из которых рассмотрим здесь только два.
Метод тангенциального смещения пластинки. Принцип этого метода, пред-
ложенного С. Я. Бейлером и П. А. Ребиндером, заключается в определении
усилия, необходимого для смещения пластинки, погруженной в исследуемую
систему. Устройство прибора, с помощью которого осуществляется измерение,
схематически показано на рис. X, 9.
Прямоугольная рифленая пластинка 3 подвешена с помощью жесткой нити 4
к пружинному динамометру 6. Пластинку полностью погружают в исследуемую
дисперсную систему, помещенную в кювету 2 до начала испытания. Кювету
с дисперсной системой закрепляют на подъемном столике 1. При^ опускании
с постоянной скоростью столика с кюветой пружина растягивается и в системе
возникает напряжение сдвига, которое, Очевидно, пропорционально растяжению
пружины. Последнее может быть измерено с помощью микроскопа 5, снабжен-
ного окулярным микрометром, или с помощью микрошкалы.
334
Напряжение сдвига Р вычисляют по растяжению предварительно прокали-
брованной пружины и соответствующему этому растяжению усилию F по урав-
нению:
Р = F/(2s)
(X, 15)
где s— боковая поверхность пластинки 3.
Предельное напряжение сдвига 0, характеризующее прочность структуры
системы и соответствующее наибольшему усилию (при отсутствии скольжения
системы вдоль поверхности пластинки), вычисляют по уравнению.
6 = -Рмакс/(2s) (X, Тб)
С помощью описанного прибора можно определять ие только предельное
напряжение сдвига, но и модуль упругости, эффективную вязкость, исследовать
процесс релаксации, а также снимать полные деформационные кривые е, Р при
разных скоростях деформации.
Метод закручивания цилиндра. Впервые метод определения упруго-пласти-
ческих свойств структурированных систем по закручиванию цилиндра, подвешен-
ного на упругой инти и погруженного н исследуемую систему, был, как мы уже
указывали, предложен еще Ф. Н. Шведовым н 1889 г. На рис. X, 10 приведена
схема прибора, с помощью которого выполняется определение. Прибор имеет
Крутильную головку 1, в которой закреплена упругая нить 2. На инти поднешеи
рифленый цилиндр 3 с зеркальцем 6. Цилиндр 3 полностью погружают в кю-
вету 4 с исследуемой сцстемой. При повороте крутильной головки на опреде-
ленный угол а крутящий момент передается через нить цилиндру и вызывает
сдвиговые деформации в слое системы, окружающем цилиндр. Цилиндр также
поворачивается иа некоторый угол £ до равновесия между упругим напряже-
нием нити и сопротивлением деформируемой системы. Разность (а — (3) дает
угол закручивания инти ш, соотнетствующий определенному усилию F, задавае-
мому крутильной головкой. Угол поворота цилиндра измеряется по смещению
светового луча, испускаемого осветителем 5 и отражаемого зеркальцем 6 иа
шкалу 7.
Описанный прибор весьма удобен для исследования кинетики развития де-
формации сдвига после приложения заданного постоянного напряжения и кине-
тики спада деформации после разгрузки.
А. А. Трапезниковым сконструирован прибор, иазваиный комплексным эла-
стовискозиметром. Он позноляет применять различные рабочие ячейки (коак-
сиальные цилиндры, конус, диск) и использовать самые разнообразные методы
исследования.
Более подробно методы определения упруго-пластических свойств структу-
рированных коллоидных и микрогетерогенных систем рассматриваются в руко-
водствах к практическим занятиям по коллоидной химии
5. ЗАВИСИМОСТЬ вязкости КОЛЛОИДНЫХ СИСТЕМ
ОТ КОНЦЕНТРАЦИИ ДИСПЕРСНОЙ ФАЗЫ
Вязкость коллоидных систем всегда выше вязкости чистой дис-
персионной среды. Эйнштейн в 1906 г., исходя из чисто гидродина-
мических представлений, вывел уравнение, устанавливающее связь
между вязкостью системы г], и концентрацией дисперсной фазы:
П = no (1 + 2,5<р) (X, 17)
где Цо — вязкость дисперсионной среды; ф— объемная концентрация дисперсной
фазы.
Уравнение (X,17) пригодно только при условии, что взвешен-
ные в жидкости частицы являются твердыми шарообразными те-
лами, концентрация дисперсной. фазы сравнительно невелика и
между частицами отсутствуют взаимодействия. Кроме того, для
335
соблюдения уравнения Эйнштейна необходимо, чтобы система
была несжимаема, течение жидкости носило ламинарный харак-
тер, между частицами и жидкостью отсутствовало скольжение и,
наконец, чтобы частицы были велики по сравнению со свободным
пробегом молекул среды, но малы по сравнению с пространством,
в котором происходит течение.
Значение численного коэффициента в уравнении (X, 17) зави-
сит от формы частиц. Поэтому в общем виде уравнение Эйнштей-
на можно записать так:
Ч = Чо(1 + «ф) (X, 18)
где а — множитель, зависящий от формы частиц.
Уравнение Эйнштейна часто пишут также следующим обра-
зом:
(П/По) — 1 = (Я — Чо)/Чо = «Ф = « (no/V)
где ц/чо — отношение вязкости системы к вязкости среды (относительная вяз-
кость) ; п — общее число частиц в системе; о — объем одной частицы дисперсной
фазы; V — объем системы, мл. '
Величину (г) — г]о)/'По> характеризующую относительное увели-
чение вязкости дисперсионной среды при введении в нее опреде-
ленного количества дисперсной фазы, не совсем правильно назы-
вают удельной вязкостью. Ее обычно обозначают символом т)уд.
Из предыдущего следует, что
ПуД = Потн-1=С1Ф (Х-'Я)
Из всего сказанного следует, что, по Эйнштейну, между вязко-
стью системы и содержанием в ней дисперсной фазы должна су-’
шествовать прямолинейная зависимость. Весьма существенно
также, что, согласно Эйнштейну, вязкость не зависит от дисперс-
ности суспензии.
Экспериментальную проверку уравнения Эйнштейна проводили
Банселен на суспензиях гуммигута, Оден на золях серы и наибо-
лее обстоятельно Эйрих на суспензиях мельчайших стеклянных
шариков, шарообразных спор грибов и дрожжевых клеток. Во
всех этих исследованиях при сферической форме частиц и малых
концентрациях дисперсной фазы численный коэффициент при <р
имел значение, близкое к 2,5. Отклонения наблюдались, когда ча-
стицы не были шарообразны, концентрация дисперсной фазы в сус-
пензии была значительной и между частицами существовали элек-
трические или другие силы взаимодействия.
Влияние анизодиаметричности частиц. При палочкообразной,
эллипсоидной или пластинчатой форме частиц суспензии вязкость
системы всегда больше, чем должна быть согласно уравнению
Эйнштейна. Причина этого заключается в том, что жидкость, по-
падающая в объем (эллипсоид вращения), образующийся вокруг
нешарообразных частиц, находящихся в интенсивном броуновском
движении, становится как бы связанной с частицей. В результате
336
этого возникает кажущееся увеличение объемной доли дириерсной
фазы в системе, что и приводит к повышению вязкости.
Экспериментально показано, что вязкость суспензии С малыми анизодиамет-
рическими частицами, находящимися обычно в интенсивном броуновском дви-
жении, повышается пропорционально квадрату отношения большой и малой осей
эллипсоида вращения, а вязкость суспензии с достаточно большими нешарооб-
разными частицами, совершающими медленное броуновское движение, возрастает
лишь прямо пропорционально отношению осей.
Теоретические вычисления, проведенные Куном, Симха и другими исследо-
вателями с использованием в качестве моделей частиц самой разнообразной
формы, весьма сложны и не всегда убедительны. Поэтому до сих пор еще иет
общей теории зависимости вязкости коллоидных систем от формы частиц.
Вязкость систем, содержащих анизодиаметрические частицы,
как мы видели, зависит от скорости течения. Вытянуты'^ частицы
ориентируются в потоке, вращауельное движение их затрудняется
и в результате этого вязкость системы с увеличением скорости те-
чения снижается. Подобное явление можно наблюдать, например,
при измерении вязкости золя V2O5, частицы которого сильно ани-
зодиаметричны.
Влияние истинной концентрации дисперсной фазы и сольвата-
ции. Отличие вязкости концентрированной дисперсной системы от
значений вязкости, вычисленной по уравнению Эйнштейна, объяс-
няется тем, что в жидкости около частиц возникают взаимовозму-
щающие микропотоки, затрудняющие движение системы. Дебройн
считает, что при этом, помимо гидродинамических взаимодействий,
необходимо учитывать также и механические (столкновения ча-
стиц, образование пар и т. д.).
При очень малых концентрациях суспензии поток, возникаю-
щий вокруг одной частицы, очень мало влияет на потоки, возни-
кающие около других частиц, и на скорость движения всего по-
тока жидкости в целом. Однако с увеличением концентрации дис-
персной фазы это влияние все увеличивается и приводит к откло-
нению от закона Эйнштейна.
Другое объяснение отклрнения вязкости дисперсных систем от
значений, найденных с помощью уравнения Эйнштейна, заклю-
чается в сольватации частиц. Явление сольватации может объяс-
нить и часто наблюдающуюся зависимость вязкости от дисперсно-
сти системы при одинаковой объемной концентрации дисперсной
фазы.
Влияние сольватации можно представить следующим образом. Если к по-
верхности шарообразной частицы радиусом г прилип слой дисперсионной среды
толщиной /г, то влияющий на вязкость эффективный объем частицы (объем са-
мой частицы вместе с объемом сольватного слоя) составляет 4/3л(г-|-/г)3. Для
значений h, малых по сравнению с г, будем иметь 4/зл(г3 + Зг2/г). Соответственно
этому при вычислении эффективной объемной концентрации дисперсной фазы ф
за объем дисперсной фазы следует принять не величину 4/3лг3у, а
4/3л(г3-|-3r2ft)v (где v — численная концентрация). Если принять истинный объем
дисперсной фазы ф0 = 4/3№v, то для ф получаем:
Ф = Фо(1+3/г/г) (X, 20)
337
Следовательно, .величина <р окажется больше объема дисперсной фазы фо,
и эта величина будет тем больше, чем меньше частицы. Иными словами, вяз-
кость возрастает с уменьшением размера частиц золя. Подобное возрастание-
иязкости при повышении степени дисперсности золя серы наблюдал Свен Оден.
Фикенчер и Марк для учета влияния сольватации предложили
модифицировать уравнение Эйнштейна, введя в него соответствую-
щую поправку. Согласно этим авторам, в уравнении Эйнштейна,
так же как и в уравнении Ван-дер-Ваальса, вместо общего объема
системы следует ввести эффективный объем, т. е. объем системы
за вычетом объема частиц. Так как частицы в системе находятся
в сольватированном состоянии и, кроме того, совершают броунов-
ское движение, описывая некие тела вращения, то объем диспер-
сионной среды, энергетически и стерически связанной с частицами,
также следует причислить к объему дисперсной фазы. Тогда урав-
нение (X, 18) примет вид:
I пУд = аис7(^ — nv') (Х,21>
где о'— объем частицы имеете с энергетически и стерически связанной с ней
средой.
Уравнение Фикенчера и Марка хорошо объясняет, почему в не-
которых случаях вязкость возрастает с увеличением концентрации
дисперсной фазы быстрее, чем это должно быть в соответствии
с прямолинейной зависимостью. Действительно, с увеличением
концентрации дисперсной фазы в растворе возрастает пропорцио-
нальная ей' величина п в числителе и одновременно уменьшается
величина (V — nv') в знаменателе, что и приводит к более быст-
рому возрастанию вязкости, чем концентрации.
Влияние взаимодействия между частицами. Причина неприме-
нимости в некоторых случаях уравнения Эйнштейна к дисперсным
системам может заключаться в проявлении сил притяжения ме-
жду коллоидными частицами. При этом в системе образуются бо-
лее или менее рыхлые структуры, которые включают значительные
объемы дисперсионной среды. Подобная иммобилизация, т. е.
уменьшение подвижности растворителя, приводит к тому, что вяз-
кость системы оказывается гораздо больше той, которая может
быть вычислена по уравнению Эйнштейна. Вязкость в таких си-
стемах сильно зависит от скорости течения, так как представляет
собой структурную вязкость, обусловленную наличием в системе
рыхлых пространственных сеток.
. С другой стороны, непрйменимость уравнения Эйнштейна
к коллоидным системам может быть связана и с проявлением сил
отталкивания между частицами, несущими одноименный электри-
ческий заряд. Согласно Смолуховскому, вязкость золей с заряжен-
ными частицами выше вязкости золей с незаряженными частицами.
Повышение вязкости в результате наличия на поверхности частиц
двойного электрического слоя называется электровязкостным эф-
фектом.
338
Смолуховский вывел следующее уравнение, связывающее удель-
ную вязкость золя с электрокинетическим ^-потенциалом двойного
электрического слоя частиц:
<хя>
где Цо — вязкость дисперсионной среды; у — удельная электропроводность; г — ра-
диус частиц; е — диэлектрическая проницаемость
Поправочный член в уравнении (X, 22) может иметь весьма
большое значение. Подсчеты показывают, что для золей, радиус
частиц которых равен 10-6 см, а электропроводность у =
— 10-4 Ом-1-см-1, поправочный член может быть в 10 раз больше
основного.
Согласно уравнению Смолуховского, вязкость коллоидных си-
стем при введении электролитов должна уменьшаться как вслед-
ствие снижения ^-потенциала, так и в результате увеличения элек-
тропроводности межмицеллярной жидкости. В изоэлектрическом
состоянии золя (при £ = 0) уравнение Смолуховского переходит
в уравнение Эйнштейна. Следует, однако, отметить, что при аста-
билизации коллоидной системы введением в нее электролита
(вследствие уменьшения сил отталкивания между частицами
в золе) возможны явления агрегации частиц, приводящие к обра-
зованию структур и появлению структурной вязкости, что не преду-
смотрено уравнением Смолуховского. В результате этого пониже-
ние (^-потенциала частиц золя в определенных условиях может
не только не вызывать понижения вязкости золя, но и обусловить
ее повышение.
Так рак в уравнение Смолуховского входит радиус частиц, то,
очевидно, вязкость золей, частицы которых несут электрический
заряд, в отличие от вязкости золей с незаряженными частицами,
зависит от степени дисперсности.
Буутс в 1948 г. вывел уравиеиие для электровязкостного эффекта, суще-
ственно отличающееся от уравиеиия Смолуховского. Согласно Буутсу, величина
электровязкостцого эффекта значительно меньше, чем об этом можно судить
по уравнению Смолуховского, и независимо от значения ^-потенциала становится
ничтожной, когда толгДииа двойного электрического слоя очень мала по сравне-
нию с радиусом частиц.
ГЛАВА XI
СИСТЕМЫ С ГАЗОВОЙ ДИСПЕРСИОННОЙ СРЕДОЙ
В этой и следующей главах кратко рассмотрены системы с га-
зообразной и твердой дисперсионной средой, а также системы
с жидкой дисперсионной средой, но с газообразной и жидкой дис-
персной фазой. При этом рассмотрены не только системы, частицы
которых отвечают коллоидной степени дисперсности, но и близко
стоящие к ним микрогетерогенные системы, о которых уже говори-
лось во введении.
1. ОБЩАЯ ХАРАКТЕРИСТИКА АЭРОЗОЛЕЙ
Коллоидные системы с газовой дисперсионной средой обычно
называют аэрозолями, хотя большей частью их дисперсность ниже
коллоидной, и поэтому правильнее называть их аэродисперсными
системами.
Отличие аэрозолей от лиозолей обусловлено прежде всего раз-
реженностью и меньшей вязкостью дисперсионной газовой среды.
Поэтому броуновское движение в аэрозолях происходит гораздо
более интенсивно, а седиментация частиц идет значительно быст-
рее, чем в лиозолях. Другое существенное отличие аэрозолей от
лиозолей заключается в том, что в газовой среде не может проис-
ходить электролитическая диссоциация и, следовательно, невоз-
можно образование двойного электрического слоя из ионов вокруг
частиц. В связи с этими особенностями учение об аэрозолях раз-
вивалось в значительной мере самостоятельно, своими собствен-
ными путями.
Классификация аэрозолей. Аэрозоли классифицируют по агре-
гатному состоянию Дисперсной фазы, по дисперсности и по мето-
дам получения.
Исходя из первого принципа, аэрозоли делят на туманы —
системы с жидкой дисперсной фазой и дымы — системы с твер-
дыми частицами. К дымам следует отнести по этой классификации
и пыли — системы с твердыми, но более крупными частицами.
Следует иметь в виду, что часто в практике «дым» означает
аэродисперсную систему, возникающую при сгорании топлива и
содержащую как твердые частицы сажи и золы, так и жидкие
частицы продуктов перегонки топлива и капли воды, образовав-
340
шиеся в результате конденсации водяного пара. Дымы, в которых
частицы дисперсной фазы адсорбировали значительное количество
влаги из атмосферы, очевидно, являются одновременно и дымами,
и туманами. Такие системы, особенно часто образующиеся при
большом содержании влаги в задымленной атмосфере над боль-
шими промышленными городами, называются особым английским
термином «смог» [smog = smoke (дым)+ fog (туман)].
По дисперсности аэрозоли с твердой дисперсной фазой разде-
ляют на дымы с частицами от J0-7 до 10-3 см и на пыли, размер
частиц которых обычно больше 10-3 см. Туманы, как правило,
имеют довольно крупные капельки размером от 10-5 до 10-3 см.
По происхождению системы с газовой дисперсионной средой
разделяют, как и все дисперсные системы,' на диспергацион-
ные и конденсационные аэрозоли. Диспергационные аэро-
золи, образующиеся при измельчении твердых тел или распылении
жидкостей, как и лиозоли, полученные путем диспергирования,
имеют довольно крупные частицы и, как правило, полидисперсны.
Аэрозоли, полученные методом конденсации из пересыщенных па-
ров или в результате химических реакций, наоборот, обычно яв-
ляются высокодисперсными системами с более однородными по-
размеру частицами
Размер и форма частиц. Ниже приведены размеры частиц
(в см) некоторых типичных аэрозолей:
Туман (Н2О)....................
Слоистые облака................
Дождевые облака................
H2SO4 (туман).............. . .
ZnO (дым)......................
Табачный дым...................
Топочный дым...................
Р2О6 (дым).....................
5-1(Г3
1 • 10"4-1. кг3
1.10-3—1 • 10-3
ЫО-4—ЫО
5-10~ .
1 • 10 -1 10-4
1 • 10“ 1 10-’
5.10-4-Ы0“4
Кривая распределения частиц в аэрозоле, т. е. содержание
в нем частиц различных радиусов, зависит от происхождения аэро-
золя и процессов, происходящих в аэрозоле после его получения
(агрегация, коалесценция, изотермическая перегонка).
Форма частиц аэрозолей зависит от агрегатного состояния ве-в
щества дисперсной фазы. В туманах капельки жидкости шарооб-
разны. В дымах они могут иметь самую разнообразную форму,
например, игольчатую, пластинчатую, звездообразную. В дымах
частицы могут представлять собой и сложные агрегаты, тогда как
в туманах столкновение капелек обычно приводит к коалесценции
и образованию капелек большего размера.
В результате рыхлости (пористости) частиц аэрозоля кажу-
щаяся плотность этих частиц, определенная обычно принятыми
способами, часто значительно меньше плотности вещества, из ко-
торого они состоят. Это можно видеть по значениям плотностей
частиц некоторых дымов, полученных различными методами
(табл. XI, 1).
341
Таблица XI, 1. Плотность частиц в дымах
Вещество Плотность» Г/см3 Метод получения дь.ма
истинная кажущаяся
Золото 19,3 0,2—8,0 Испарение в вольтовой дуге
Серебро 10,5 0,64—4,22 То же
Ртуть 13,6 0,07—10,8 Нагревание в лодочке
Окись магния 3,6 0,24—3,48 Сжигание металлического магния
Хлорид ртути 5,4 0,62-4,3 Нагревание в лодочке
Окись кадмия 6,5 0,17—2,7 Испарение в вольтовой дуге
Размер и форму частиц аэрозолей определяют с помощью обыч-
ной микроскопии, ультрамикроскопии и электронной микроскопии.
Для счета частиц в аэрозолях особенно удобен поточный микро-
скоп Б. В. Дерягина и Г. Я. Власенко, о котором уже упомина-
лось в гл. II.
Концентрацию трудно доступных для исследования аэрозолей,
например концентрацию воды в облаке, можно определять с по-
мощью радиолокаторов. «Прощупывающий» пространство направ-
ленный радиолуч испускается источником в виде импульсов через
определенные промежутки времени и регистрируется на экране
осциллографа. С помощью осциллографа регистрируется и излу-
чение, возвратившееся обратно в результате рассеяния объектом
(облаком). По интервалу времени, прошедшему от подачи’радио-
сигнала до приема рассеянного луча, можно определить расстоя-
ние до объекта, а по интенсивности отраженного луча можно су-
дить о концентрации дисперсной фазы в объекте, так как рассея-
ние радиолучей малыми частицами описывается уравнением, в об-
щем аналогичным уравнению Рэлея.
Оптические свойства. Оптические свойства аэрозолей подчи-
няются в общем тем же законам, что и оптические свойства лио-
золей. Следует, однако, помнить, что вследствие большой разницы
в плотностях, а значит, и в показателях преломления дисперсной
и газовой фаз оптические свойства аэрозолей и прежде всего све-
торассеяние проявляются весьма ярко. Благодаря большой спо-
собности рассеивать свет аэрозоли широко применяются для соз-
дания дымовых завес. Из всех дымов наибольшей способностью
рассеивать и отражать свет обладает дым Р2О5; его маскирующая
способность обычно принимается за единицу.
Молекулярно-кинетические свойства. Аэрозоли — сравнительно
сильно разреженные системы, обладающие малым коэффициен-
том внутреннего трения дисперсионной среды. Этим и опреде-
ляются особенности их молекулярно-кинетических свойств.
Движение взвешенных в вязкой жидкости (рассматриваемой как непрерыв-
ная среда) сферических частиц, размеры которых значительно превышают раз-
меры молекул среды, описывается известным уравнением Стокса:
( = 6лтугг (XI, 1)
342
где f — сила трения частицы; г) — вязкость среды, г — радиус частицы, и — ско-
рость движения частицы
Введя коэффициент сопротивления частицы В = f/u, получим
В = 6лти (XI, 2>
Для описания движения частиц, взвешенных в газовой среде, это гидроди-
намическое уравнение пригодно только в том случае, если размер частиц значи-
тельно больше среднего свободного пробега молекул газа Так как при атмо-
сферном давлении эта величина для воздуха составляет приблизительно 10-5см,
то очевидно, уравнение Стокса применимо лишь для грубодисперсных аэрозолей,,
радиус частиц которых превышает 10-4 см При меньших давлениях и, следова-
тельно, при большем свободном пробеге граница применимости уравнения Сток-
са для аэрозолей смещается в сторону еще меньшей дисперсности
Для высоко дисперсных аэрозолей, радиус частиц которых меньше 10-8 см,
движение частиц описывается другим уравнением’
f = 6лг)г2и/(0,3502 • 4,5Л) ' (XI, 3>
или
В = 6лПг2/(0,3502 • 4,5Х) (XI, 4>
где X — средний свободный пробег молекул газа
Это уравнение выводится на основе представлений, аналогичных применяе-
мым в кинетической теории газов, так как для малых частиц (или для низких
давлений), когда отношение Х/г 1, движение частиц аэрозоля происходит по-
добно движению молекул газа
Для описания поведения аэрозолей с промежуточным размером частиц.
(10-6—10~4 см) используются переходные формулы; одной из таких формул
является уравнение Кеннингема.
(х1,5>
или
где А — коэффициент, близкий к единице, значение которого определяется экспе-
риментально
Как можно видеть, для больших значений Х/r (когда А\/г 1) в уравнении
(XI,3) сила сопротивления f пропорциональна квадрату радиуса частицы, а для
малых значений Х/г (когда ДХ/г < 1) уравнение Кеннингема переходит в урав-
нение Стокса
Все сказанное относилось к простейшему случаю — движению сферических,
частиц Зависимость скорости движения частицы от действующей силы для ие-
сферических частиц гораздо сложнее *
Рассмотрим кратко особенности броуновского движения в дисперсных си-
стемах с газовой средой На броуновском движении частиц в аэрозолях весьма
сильно сказывается седиментация вследствие малой вязкости и малой плотности
газовой среды В ранних исследованиях это не было учтено, и поэтому значении
средних смещений в горизонтальном и вертикальном направлениях не совпадали.
Кроме того, благодаря малой вязкости аэрозолей в них легко возникают кон-
векционные токи, что также весьма затрудняло изучение броуновского движения
в этих системах Однако позже благодаря применению усовершенствованных
методов исследования все эти трудности были преодолены и было установлено,
что броуновское движение в аэрозолях подчиняется тем же закономерностям,
что и в лнозолях В настоящее время броуновское движение в аэрозолях изу-
чают путем микроскопического наблюдения за седиментирующими частицами,
которым придают тем или иным способом электрический заряд Благодаря за-
ряду частицы, опустившиеся на некоторое расстояние вследствие седиментации,
можно возвратить в исходное положение при наложении соответственно на-
правленного электрического поля и таким образом проводить множество изме-
343.
-рений с помощью одной и той же частицы, что обеспечивает получение точных
результатов.
Если бы частица аэрозоля не находилась в броуновском движении, то время
ее оседания т на расстояние й было строго постоянным. Однако из-за броунов-
ского движения к ее перемещению добавляется вертикальная составляющая.
Время, необходимое для прохождения частицей расстояния й, может быть
больше (если броуновское смещение за время падения направлено снизу вверх)
или меньше (если броуновское смещение направлено вниз) времени седимента-
ции. Полученное при таких измерениях большое число значений ti, т2, Тз ..«
для продолжительности падения на одно и то же расстояние й можно обрабо-
тать с помощью теории броуновского движения. Не входя в подробности этих
расчетов, укажем, что коэффициент диффузии, вычисленный по полученным та-
ким образом результатам с учетом поправки на седиментацию, для капелек
масляного тумана, как показал Флетчер, прекрасно совпадает с коэффициентом
диффузии, найденным для этой системы другими способами.
Весьма существенно учитывать одновременно идущие диффузию и седи-
ментацию при исследовании поведения аэрозоля, заключенного в небольшое
пространство Это особенно важно для понимания процесса фильтрации.
Допустим для упрощения, что аэрозоль заключен в сферическое простран-
ство с радиусом R. При этом уменьшение численной концентрации аэрозоля мо-
жет идти двумя путями:
1) частицы, находящиеся в броуновском движении, достигают стенок сосуда
и прилипают к ним (вследствие того, что частицы аэрозоля обычно лишены
какого-либо фактора устойчивости, можно считать, что каждая частица, уда-
рившаяся о стенку, остается на ней);
2) частицы седиментируют и оседают на дно сосуда.
Время Тд, необходимое для того, чтобы частица, находящаяся в центре со-
суда и максимально удаленная от стенки, продиффундировала к ней, очевидно,
можно найти, исходя из формулы Эйнштейна — Смолуховского (см. гл. III,
разд 2), путем замены А2 на R2:
Тц = R2/(2D) = R2B/(2kT) (XI, 7)
Время тс, необходимое для седиментации максимально удаленной частицы
на дно, определяется, очевидно, уравнением
тс = 2RB/(mg) (XI,8)
где т — масса частицы; g— ускорение свободного падения (при этом прини-
мается, что седиментация частицы происходит с постоянной скоростью 27?/тс).
Так как для малых частиц коэффициент сопротивления частицы В пропор-
ционален квадрату радиуса, а масса частиц пропорциональна кубу радиуса, то
тд ~ г2, а гс ~ 1/г. Отсюда следует, что средние значения Чд и тс, необходи-
мые для прилипания частиц к стенкам сосуда и для оседания на дно, по-раз-
ному зависят от размера частиц.
Если частицы малы, то диффузия происходит быстрее, чем седиментация,
и разрушение аэрозоля в основном будет вызвано прилипанием частиц к стен-
•кам, а не оседанием на дно Если частицы крупные, наблюдается обратное
явление, т. е. разрушение аэрозолей обусловлено в основном седиментацией.
Из изложенного также следует, что из аэрозоля быстро исчезают как очень
мелкие, так и очень крупные частицы, первые вследствие прилипания к стенкам,
вторые — в результате оседания на дно. Наоборот, частицы промежуточных
размеров дольше пребывают в аэрозоле Частицы, для которых тд = хв, оче-
видно, обладают максимальной устойчивостью. Приравнивая уравнения (XI, 7) и
(XI, 8), легко найти формулу, по которой можно вычислить массу частиц, доль-
ше всего остающихся в аэрозоле:
m0 = 4kT/(gR) (XI, 9)
Все сказанное о разрушении аэрозоля, находящегося в ограниченном объеме,
• относится к разрушению систем с газовой дисперсионной средой при прохожде-
нии нх через пористые фильтрующие материалы. Развитые выше соображения,
844
вероятно, объясняют также и то обстоятельство, что у людей, больных сили-
козом, в легких находят главным образом частицы кварца размером, отвечаю-
щим массе Ото. Можно предполагать, что кристаллы больших и меньщих разме-
ров остаются почти целиком в дыхательных путях человека и не доходят до
легких.
Рассмотрим кратко явления термофореза, фотофореза и термопреципитации,
связанные с кинетическими свойствами и характерные для коллоидных систем
с газовой средой.
Явление термофореза заключается в движении частиц аэрозоля в на-
правлении снижения температуры. При соблюдении условия X/r^> 1, т. е когда
частицы малы, термофорез возникает вследствие того, что на более нагретую
сторону частицы молекулы газа налетают с большей скоростью, чем на менее
нагретую, и, следовательно, сообщают частице импульс в направлении пониже-
ния температуры. Если Х/г 1, причина возникновения термофореза несколько
более сложная. Однако можно показать, что и при K/r «С 1 движение частицы
в поле температурного градиента должно также происходить в сторону пони-
жения температуры.
Строгая количественная теория термофореза для малых и больших частиц
разработана Б. В. Дерягиным, С. П. Бакаевым и Ю. И. Яламовым.
Фотофорез, заключающийся в передвижении частиц аэрозоля при одно-
стороннем их освещении, является частным случаем термофореза. Объяснение
фотофореза более сложно, чем термофореза, поскольку распределение темпера-
туры внутри освещенной частицы зависит от ее размера, формы, прозрачности
и коэффициента преломления и, следовательно, может быть весьма различным.
Для непрозрачных частиц обычно наблюдается положительный фотофорез, т. е.
движение частиц в направлении светового луча. Для прозрачных частиц может
наблюдаться и отрицательный фотофорез в связи с тем, что задняя сторона
частицы может быть нагрета преломившимися в частице лучами сильнее, чем
передняя, обращенная к источнику света. Известны случаи, когда малые ча-
стицы некоторых веществ обнаруживают отрицательный фотофорез, а боль-
шие— положительный. Такое явление можно объяснить тем, что по мере увели-
чения размера частицы свет, прошедший через частицу, ослабляется в большей
степени, а значит, задняя сторона частицы нагревается меньше.
Термофорез и фотофорез имеют большое значение в движении атмосферных
аэрозолей, например при образовании облаков. Термофорез .водяных капелек,
взвешенных в воздухе, возникает при соприкосновении холодных и теплых воз-
душных масс, а фотофорез происходит вследствие освещения облаков солнеч-
ными лучами. Следует вообще заметить, что кинетическая устойчивость атмо-
сферных аэрозолей весьма своеобразна. Благодаря небольшому размеру капелек
и малой скорости оседания (0,05—0,7 см/с) они как бы взвешены в атмосфере,
и поднимающихся от земли сравнительно слабых токов теплого воздуха до-
статочно для того, чтобы облака продолжали свой путь над землей, двигаясь
при этом как одно целое. И только, когда в результате коалесценции или кон-
денсации капельки облаков или туманов становятся больше критического раз-
мера, они выпадают в виде дождя
Под термопреципитацией подразумевают осаждение частиц аэро-
золя иа холодных поверхностях, поскольку при соприкосновении с такими
поверхностями частицы теряют кинетическую энергию. Именно термопрецнпита-
цией объясняется осаждение пыли иа стенах и потолке возле печей, радиаторов,
ламп, а также в трубах.
Электрические свойства. Как уже указывалось, вокруг частиц
в системах с газовой дисперсионной средой не могут возникать
Двойные электрические слои. Тем не менее частицы аэрозолей
в определенных условиях могут быть заряженными, хотя заряд их
обычно невелик. Электрический заряд на частицах в аэрозолях
возникает либо в результате образования и последующего нару-
шения контакта частиц друг с другом или с какой-нибудь поверх-
345
постыо, либо, чаще всего, вследствие адсорбции на поверхности
частиц ионов газов.
В отличие от коллоидных растворов, где величина заряда ча-
стицы обычно обуславливается избирательной адсорбцией ионов
электролита и отвечает равновесию между частицей и окружаю-
щей средой,- у аэрозолей заряд частицы в известной мере случаен
и целиком зависит от причин, его вызывающих. По тем же причи-
нам у аэрозолей не существует строгой зависимости между дис-
персностью и величиной заряда. Однако в общем все же можно
полагать, что заряд частицы аэрозоля тем больше, чем больше ее
размеры. Из практики также установлено, что частицы аэрозолей
металлов и их окислов обычно несут отрицательный заряд, напри-
мер РегОз, MgO, Zn, ZnO, и, наоборот, частицы аэрозолей неме-
таллов и их окислов заряжены, как правило, положительно
(SiOa, Р2О5). Положительно заряжены также частицы NaCl, угля,
крахмала; частицы муки несут отрицательный заряд.
Рассмотрим подробнее причины возникновения электрических зарядов на
частицах аэрозоля.
Допустим, что частица аэрозоля вначале не имеет заряда н адсорбция на
ней ионов, всегда присутствующих в газовой фазе в результате ионизации га-
зов под действием космических или ультрафиолетовых лучей, неспецифична.
Такая частица, сталкиваясь с ионом, адсорбирует его и приобретает заряд. Так
как концентрация ионов в газе невелика, то эти столкновения редки — интервал
времени от одной встречи до другой может измеряться минутами. При новом
столкновении адсорбировавшей частицы с ионом заряд .частицы может увели-
читься или уменьшиться в зависимости от злака заряда и валентности
иоиа, с которым она столкнулась. В результате подобных встреч частица может
даже изменить знак заряда или стать нейтральной. Конечно, одновременно про-
исходит и десорбция ионов, захваченных частицей. Таким образом, частица время
от времени меняет заряд, но колебания заряда в общем должны происходить
около среднего нейтрального состояния. Нетрудно видеть, что колебания заряда
частиц аэрозоля имеют характер флуктуаций и являются отражением молекуляр-
но-кинетического движения ионов и частиц. При таких условиях вероятность W
приобретения частицей какого-либо заряда определяется выражением:
1Г~ехр( — AtkT) (XI, 10)
где А — работа, необходимая для осуществления флуктуации.
Расчеты, подтвержденные опытом, показывают, что заряд частиц аэрозоля
-обычно очень мал и редко превышает элементарный электрический заряд более
чем в 10 раз. Это позволило установить дискретный характер заряда ионов и
измерить абсолютную величину заряда электрона, что и было выполнено Милли-
кеном.
Рассмотренная картина значительно усложняется, когда частицы способны
избирательно адсорбировать ионы какого-нибудь определенного вида, иными сло-
вами, когда проявляется действие адсорбционного потенциала. Кроме того, на
межфазной границе обычно существует скачок потенциала. А. Н. Фрумкин пока-
зал, что на межфазной границе аэрозолей воды или снега благодаря большому
дипольному моменту молекул Н2О и их ориентации существует положительный
электрический потенциал порядка 250 мВ. Скачок потенциала на межфазной гра-
нице может возникать и вследствие так называемой ба ллоэлектризации —
электризации частиц аэрозоля при получении его методом диспергирования.
В результате способности дисперсной фазы к специфической адсорбции ионов
и наличия скачка потенциала у межфазной границы частицы аэрозоля неодина-
ково адсорбируют различные ионы и средний их заряд отличен от нуля.
346
Заряд частиц обусловливает явления, происходящие в больших объемах
аэрозоля, например в облаках. Опытным путем установлено, что заряд капелек
воды в облаках в общем близок к величине, соответствующей потенциалу по-
рядка 250 мВ. В больших объемах атмосферного аэрозоля происходит разделение
частиц по размеру, а следовательно, и по электрическому заряду, вследствие того,
что частицы различных радиусов седиментируют с разной скоростью. В резуль-
тате этого электронейтральность облака нарушается и в нем возникают мощные
электрические поля. При этом иижияя часть облака приобретает обычно отрица-
тельный заряд, а верхняя часть остается положительно заряженной. Расчеты
показывают, что в таких условиях напряженность поля Н в облаке составляет,
в среднем 100 В/см. Однако при значительной полидисперсностн капелек облака,
а также при конвекционных токах, обусловленных ветром, в облаке могут воз-
никать и гораздо большие напряжения, служащие причиной грозовых явлений.
Заряд частиц аэрозолей обычно определяют с помощью приемов, аналогич-
ных методам, используемым для изучения броуновского движения в этих систе-
мах. С большой точностью измеряют скорость свободной седиментации частицы
аэрозоля. После этого определяют скорость падения или поднятия частицы в на-
ложенном на нее электрическом поле н вычисляют заряд частицы Q, пользуясь
уравнением:
и = ис± u3 = ~(mg ± QH) (XI, 11}
D
где ис — скорость свободной седиментации частицы; иа—скорость движения ча-
стицы в электрическом поле; т —масса частицы; g — ускорение свободного па-
дения; Н — напряженность электрического поля.
Ионизируя дисперсионную среду аэрозоля, можно изменять заряд частиц на
величину AQ. Весьма существенно, что изменение AQ всегда оказывается кратным
величине элементарного заряда е = 4,8-10~10 эл.-ст. ед. Заряд частицы можна
найти, не зная коэффициента В, применив поле с напряжением Но, при котором
подъемная сила полностью компенсирует вес частицы. В этом случае, очевидно,
нс = нэ. Заменив ив иа mgIB и иа на QH^jB, получим уравнение:
mg=Qtf0 (XI, 12}
из которого легко найти Q.
Агрегативная устойчивость. Электрический заряд частиц аэро-
золей, возникающий обычно в результате адсорбции ионов, как
правило, весьма невелик, а иногда практически равен нулю. Встает
вопрос, могут ли возникать на поверхности частиц аэрозоля моле-
кулярные адсорбционные слои и способны ли такие слои обуслов-
ливать агрегативную устойчивость аэрозолей.
В ранних работах имеются указания, что при достаточно боль-
ших давлениях на поверхности ч'астиц аэрозоля возникает поли-
молекулярный диффузный слой газа, удерживаемый адсорбцион-
ными силами. Наличием газовой оболочки, в частности, объясняли
неслеживаемость порошков при хранении, способность порошков
течь подобно жидкостям и несмачивание твердых частиц аэрозолей
жидкостью (известно, что дым может проходить через воду, при-
чем частицы дисперсной фазы не остаются в воде). В некоторых
работах приводились даже данные, характеризующие количество
воздуха, адсорбированного аэрозолем, причем объем адсорбиро-
ванного газа обычно во много раз превышал объем адсорбировав-
шей его дисперсной фазы. Однако в последние десятилетия появи-
лись работы, в которых опровергается возможность адсорбции
аэрозолями больших количеств газа, и поэтому считают, что обра-
зование вокруг их частиц диффузных газовых оболочек невозможно.
347
Вне всякого сомнения, что поверхность твердых частиц аэрозо-
лей может быть покрыта пленкой жидкости. Например, у обыч-
ного дыма такая пленка может состоять из жидких продуктов
перегонки топлива и из сконденсированной влаги. Однако стаби-
лизующее действие жидкостных адсорбционных оболочек, на кото-
рое указывалось в некоторых работах, более чем сомнительно. По-
скольку частицы аэрозоля окружены газом, а не жидкостью,
расклинивающее давление не может проявиться. Кроме того, по-
верхностное натяжение на границе раздела жидкость — газ
остается всегда достаточно большим для того, чтобы обеспечить
понижение свободной энергии системы при слипании частиц.
Рассматривая влияние влажности на коагуляцию дымов, необ-
ходимо упомянуть о наблюдениях Далавала и Орра. ЭтгГисследо-
ватели нашли, что скорость седиментации аэрозолей MgO и, осо-
бенно, NH4CI, значительно повышается во влажной атмосфере.
Микроскопическое исследование показало, что агрегаты частиц
становятся при этом более компактными. По всей вероятности это
вызвано стягиванием агрегатов конденсированной водной пленкой-.-
Таким образом, аэрозоли, обладая при высокой дисперсности
достаточной седиментационной устойчивостью, обычно являются
весьма агрегативно неустойчивымц_системами и в них всегда идет
процесс коагуляции. Этим объясняется сравнитёльнсг-небольшой'
срок жизни любого аэрозоля. Существенно, что максимальную не-
устойчивость проявляют аэрозоли с наиболее крупными и наибо-
лее мелкими частицами. Первые системы неустойчивы из-за боль-
шой скорости оседания их частиц, вторые не могут долго суще-
ствовать вследствие интенсивного броуновского движения, приво-
дящего к столкновению частиц и образованию агрегатов.
Коагуляция аэрозолей, являющаяся, как правило, процессом
быстрой коагуляции, обычно протекает значительно быстрее, чем
коагуляция лиозолей, из-за болееинтенсивного броуновского дви-
женияидц»£1£мах_£_1^30ййй_,Дисперсионной средой? Расчеты пока-*
зывают, что скорость коагуляций~чретвычайно сильно возрастает
с увеличениемчислениой каццентрации аэрозоля.. Ниже приведены
данные, характеризующие скорость коагуляции аэрозолей в зави-
симости от концентрации:
Начальная численная
концентрация в 1 см3 1012 1О10 10е 10е
Время, требующееся
для уменьшения чи-
сленной концентрации
на два порядка . . . Доля 15—30 с 30 мин Несколь-
секунды ко суток
Из этих данных следует, что независимо от начальной концентра-
ции аэрозоля через несколько минут после его получения численная
концентрация в 1 см3 не может быть выше 108— 106. Это примерио!
в 108 раз меньше численной концентрации лиозолей (например,
обычный золь золота содержит около 1O1S частиц в 1 см3). Таким
348
•образом, как в природе, так и в производственных условиях мы
почти всегда имеем дело с весьма сильно разбавленными аэрозо-
лями.
Приведенные данные характеризуют скорость коагуляции аэро-
золей только в первом приближении. На скорость разрушения си-
стем с газовой дисперсионной средой, помимо частоты столкнове-
ния частиц, влияют и другие факторы Так, .коагуляции аэрозолей
способствует полидисперсность и анизодиаметрическая форма ча-
стиц. Разрушение аэрозолей ускоряется' при наличии в них проти-
воположно заряженных частиц. Наоборот, если частицы аэрозоля
обладают одинаковым по знаку и
достаточно большим по величине
зарядом, то наблюдается рассея-
ние частиц.
Весьма интересно поведение аэрозо-
лей, содержащих частицы жидкости с вы-
соким давлением пара. Частицы таких
аэрозолей могут упруго отскакивать друг
от друга при столкновениях. Причина
этого, кдк установили Б В. Дерягин и
П С. Прохоров, заключается в испаре-
нии жидкости с поверхности капелек и
образовании вследствие этого диффузно-
конвекционного газового потока, препят-
ствующего коалесценции капель Расчеты
подтвердили, что давление пара, возни-
кающее в результате такого испарения,
вполне достаточно, чтобы неограниченно
долго препятствовать слиянию двух ка-
Сопротивление среды \
Скорость испарения I
Скорость охлаждения J
Константа коагуляции
Рассеяние сдета
Область микроскопичес-
кой Видимости
Область упьтрамикроско
пическод Видимости
Преобладание дшррузии
или оседания
Давление пара
(Водяных капелек)
пелек жидкости, находящихся в непо-
средственной близости (при условии по-
полнения испаряющейся жидкости). Ин-
тересно, что если предотвратить испаре-
Рнс. XI, 1. Зависимость основных
свойств аэрозолей от их дисперс-
ности (по Н. А. Фуксу).
ние, например путем насыщения окру-
жающего воздуха парами той же жидкости, то капли тотчас коалесцируют
Повышения агрегативной устойчивости эмульсий и суспензий вследствие раство-
рения дисперсной фазы в дисперсионной среде никогда не наблюдается; оче-
видно, это можно объяснить тем, что диффузия в жидкой среде протекает с
очень малой скоростью.
На скорость коагуляции аэрозоля, конечно, влияют конвек-
ционные потоки, механическое перемешивание, ультразвуковые ко-
лебания, поскольку все эти воздействия увеличивают вероятности
столкновения частиц друг с другом.
В заключение отметим, что в аэрозолях, как и лиозолях, могут
изменяться размеры частиц не только за счет явления коалесцен-
ции и агрегации, но и вследствие изотермической перегонки дис-
персной фазы, что приводит к укрупнению больших частиц за счет
испарения более мелких. Испарение капелек туманов может при*
водить в соответствующих условиях и к Переходу аэрозоля в гомо*
генную систему подобно тому, как растворение дисперсной фазы
лиозоля приводит к образованию истинного раствора,
349
Зависимость свойств аэрозолей от радиуса их частиц. На
рис. XI, 1 наглядно представлена зависимость основных свойств
аэрозолей от их дисперсности. Нетрудно видеть, что зависимость
многих свойств и особенно молекулярно-кинетических от радиуса
частицу аэрозолей выражена ярче, чем у лиозолей.
2. ПОРОШКИ И ИХ СВОЙСТВА
К аэрозолям по свойствам близко примыкают порошки, которые можно рас-
сматривать как аэрозоли с твердой дисперсной фазой, скоагулировавшие и обра-
зовавшие осадок (аэрогель). К порошкам следует отнести также и грубодис-
персные системы, которые вследствие большого размера частиц седиментацион-
ио неустойчивы.
Размеры первичных частиц порошков колеблются в весьма широких пре-
делах. Ниже в качестве иллюстрации приведены значения диаметров (в мкм)
частиц некоторых порошков, широко применяемых й технике и пищевой промыш-
ленности:
Сажа
газовая канальная......................... 0,03—0,09
газовая печная........................ 0,10—0,30
ламповая.............................. 0,30—0,60
Окнсь магния........................... 0,2—0,5
Титановые белила....................... 0,2—0,7
Окнсь цнака................... . . . . 0,2—0,8
Литопон (пигмент)...................... 0,3—0,8
Окись железа (пигмент) . . . .,........ 0,3—1,5
Сульфат бария................................ 1—3
Мел
осажденный.............................. 1—5
молотый................................. 5—50
Каолин...................................... 2—20
Крахмал
рисовый................................ 6—10
кукурузный......................... 15—25
картофельный....................... 100—150
Мука пшеничная
высшего сорта............................. 50—200
3-го сорта......................... до 800
Какао.................................. 100—200
Как можно видеть из этих данных, только некоторые сорта сажи имеют
частицы, отвечающие коллоидным размерам; все остальные порошки являются
микрогетерогенными системами
Размеры частиц порошков, а следовательно, и их удельная поверхность
имеют огромное значение для практического применения порошков. Так, яркость
окраски й кроющая способность пигментов (титановые белила, литопон, окнсь
железа), усиливающее действие наполнителей (сажа, окись цинка, окись маг-
ния), вкусовые свойства порошков, применяемых в пищевой промышленности
(какао, мука), сильно зависят от их дисперсности.
Размер частиц порошков можно определять микроскопически, методом седи-
ментации и с помощью ситового анализа. Удельную поверхность порошков опре-
деляют либо по адсорбции азота на частицах, либо путем фильтрации жидкости
через порошок, либо, наконец, путем просасывания через него разреженного или
неразрежениого воздуха.
Рассмотрим кратко наиболее характерные свойства порошков — способность
к течению и распылению, флуидизацию и гранулирование. При этом будем в
основном придерживаться изложения, принятого в монографии Н. А. Фукса
«Механика аэрозолей», М., изд-во АН СССР, 1955.
350
Рассмотрим движение малых частиц на поверхности слоя, состоящего из
тех же частиц Такое движение лежит в основе переноса песка и почвы ветром,
пневматического транспорта сыпучих материалов и т. д. Это движение может
осуществляться тремя способами: 1) частицы перекатываются по поверхности;
2) частицы отрываются от поверхности и сейчас же падают обратно, т. е. пере-
двигаются «прыжками»;-3) частицы переносятся в состоянии аэрозоля Эти виды
движения частиц можно наблюдать в аэродинамической трубе, на дне которой
насыпан толстый слой песка. При определенной скорости воздуха частицы, вы-
ступающие из слоя песка, начинают перекатываться. Однако эти песчинки скоро
останавливаются, попав, например, в небольшие углубления. Если несколько
увеличить скорость воздуха, снова некоторое число песчинок перекатится и
остановится и т. д. Движущиеся таким образом песчинки, сталкиваясь с дру-
гими более крупными частицами, выступающими над поверхностью, подскаки-
вают. При некоторой скорости воздуха, называемой критической, большая часть
песчинок начинает передвигаться путем прыжков; траектории таких прыгающих
песчинок представлены на рис. XI, 2.
Механизм отрыва песчинок от поверхности воздушным потоком при совер-
шении ими прыжка еще не вполне ясен. Если частица выпрыгивает из потока
воздуха, двигающегося ламинарно у самой поверхности, то ее подхватывают
турбулентные вертикальные пульсирующие потоки воздуха. Также причиной
Рис. XI, 2. Траектории прыгающих песчинок.
отрыва частиц может быть местный отрыв вихрей песка от поверхности (напри-
мер, у возвышений на песке или почве). Песчинки иногда отскакивают от "по-
верхности рикошетом, повторяя свой прыжок; иногда, падая, зарываются в пе-
сок и передают свой импульс другим песчинкам, которые начинают перекаты-
ваться или в свою очередь подскакивают. Таким образом, процесс переноса пе-
ска и любого порошка имеет характер цепной реакции.
Крупные песчинки радиусом большим 0,2—0,3 мм при скорости ветра, не
превышающей 10 м/с, не могут прыгать, а лишь перекатываются, а песчинки
радиусом большим 1 мм вовсе остаются неподвижными. Поэтому из полидис-
персного песка ветер выдувает более мелкую фракцию. Самая тонкая фракция
под действием воздушного потока переходит в состояние аэрозоля и в таком
виде передвигается над поверхностью песка.
Раз начавшись, унос песка или почвы ветром распространяется на большое
расстояние. Начинается же этот процесс на небольших возвышениях, под
действием вихрей и случайных причин. Эффективность даже невысоких препят-
ствий в борьбе против ветровой эрозии' объясняется тем, что частицы почвы не
могут перепрыгнуть через препятствие и процесс приостанавливается.
Опыты показали, что для песчинок радиусом большим 50 мкм критическая
скорость «течения» по поверхности песка пропорциональна квадратному корню
из радиуса песчинок. Для песчинок радиусом меньшим 50 мкм критическая
скорость возрастает при переходе к более мелким частицам благодаря молеку-
лярным силам, действующим между частицами.
Ниже приведены значения скорости ветра (по Гензелю), при которых на-
чинается передвижение песков различной дисперсности:
Радиус песчинок, мм.................. 0,087—0,12 0,12—0,25 0,25—0,5 0,5—1,0
Критическая скорость ветра, см/с
для сухого песка.................... 380 480 600 900
для песка с 2% влаги 600 750 950 1200
351
Заметный перенос снега начинается, по Л. Дановскому, при скорости ветра
4—5 м/с (на льду уже при 2 м/с) и быстро растет по мере усиления ветра.
Быстрым ростом переноса песка и снега при увеличении силы ветра объясняется
катастрофический характер этих явлений во время сильных бурь.
Способность частиц прилипать к различным поверхностям имеет большое
значение при нанесении инсектофунгицидных порошков на растения. В этом
случае частицы отрываются от поверхностей (листьев и т. д), расположенных
под различными углами к горизонтальной плоскости. Такой отрыв облегчается
с увеличением массы частиц, т. е. прилипаемость усиливается с уменьшением
размера частиц. Помимо размера частиц имеет значение их форма и природа, ,а
также материал стенки, от которой зависит поиерхностное натяжение. Большую
роль играет также и пластичность частицы и стенки. Пластическая деформация
соприкасающихся неровных поверхностей, обусловливающая увеличение пло-
щади контакта, приводит к значительному возрастанию силы, необходимой для
отрыва частицы от стенки. Поэтому частицы мягких веществ прилипают лучше,
чем твердых.
Большое значение для прилипания имеет и влажность: образующийся в
месте соприкосновения стенки и шарообразной смачиваемой частицы радиусом г
водный мениск в результате действия поверхностного натяжения притягивает
частицу к стенке с силой, пропорциональной го (где о — поверхностное натя-
жение воды). Так как радиус частицы значительно больше радиусов кривизны
тех субмикроскопических выступов, по которым происходит фактическое сопри-
косновение, прилипаемость весьма сильно повышается с влажностью. С другой
стороны, при прилипании частиц к стенкам нередко возникают большие элек-
тростатические силы, н в этом случае влажность, способствующая нейтрализа-
ции электрических зарядов, может отрицательно сказываться на прилипаемости.
Перейдем теперь к рассмотрению явлений распыления порошков и их флуи-
дизации (перевод в состояние, подобное жидкому состоянию). Эти явления
происходят при распылении угольной пыли в топках с помощью форсунок, при
воздушной сепарации порошкообразных материалов, распылении инсектофунги-
цидов, проведении химических реакций в «кипящем слое» и т. д.
Если через слой порошка, находящийся в цилиндрическом сосуде с пори-
стым дном, пропускать снизу с постепенно возрастающей скоростью какой-ни-
будь газ, то наблюдаются следующие яиления. При малых скоростях течения
частицы порошка остаются неподвижными, а высота слоя и коэффициент за-
полнения пространства Ф постоянны. Когда градиент давления газа сравнивается
с градиентом гидростатического давления порошка, равнодейстиующая всех
действующих на частицу сил станет равной нулю и при дальнейшем повышении
скорости течения среды слой начнет расширяться. Слой порошка с достаточно
крупными частицами в этих условиях расширяется равномерно; контакт между
соседними частицами сохраняется, но структура порошка становится более рых-
лой. В более дисперсных порошках, в которых заметную роль играют силы
сцепления между частицами, при достаточно больших скоростях течения среды
наблюдается уже не равномерное расширение слоя порошка, а распадение его
на отдельные агрегаты, -между которыми образуются каналы, по которым и про-
ходит значительная часть газа.
Следует заметить, что влияние сил сцепления на свойства порошка сказы-
вается и в статических условиях, при отсутствии движения воздуха. Так назы-
ваемая насыпная плотность порошка, равная Фр, в грубых порошках почти не
зависит от размера частиц, так как определяется отношением силы тяжести
частиц к пропорциональной ей силе трения между частицами. Однако по мере
увеличения дисперсности порошка начинают сказываться межмолекулярные силы,
увеличивающие силу трения между частицами и способствующие образованию
более рыхлой структуры; поэтому насыпная масса начинает уменьшаться.
Угол естественного откоса порошкои также не зависит от размера частиц
в очень грубых порошках и возрастает при переходе к порошкам с мелкими ча-
стицами. По мере расширения слоя его текучесть (измеряемая, например, по
сопротивлению, оказываемому слоем вращающейся в нем мешалке) возрастает
в несколько десятков раз, а угол откоса значительно уменьшается, т. е, порошок
приближается по свойствам к жидкости.
352
После расширения слоя порошка до определенного размера (5—20%’от на-
чального объема в зависимости от свойств порошка) частицы начинают дви-
гаться, а сопротивление слоя протекающей среде несколько уменьшается, ве-
роятно, благодаря уменьшению трения между частицами.
При известной скорости течения среды через равномерно расширившийся
слой грубых порошков газ начинает барботировать, как через жидкость В ре-
зультате этого высота слоя продолжает расти, но сильно колеблется. В этом
состоянии порошок весьма напоминает кипящую жидкость, отчего такой слой и
получил название «кипящего слоя». По мере увеличения скорости течения ча-
стицы, скорость оседания которых меньше скорости течения среды над слоем,
увлекаются из «жидкой фазы» т. е. кипящего слоя, в «газообразную» (аэрозоль-
ную) фазу. Концентрация частиц в аэрозольной фазе все возрастает, наконец,
граница между обеими фазами исчезает, и порошок целиком выдувается газом.
У тонких порошков в результате 'действия сил сцепления наблюдается так
называемая агрегативная флуидизация. При небольших скоростях
течения в слое образуются каналы, через которые и проходит основная масса
газа. При увеличении скорости течения каналы разрушаются, в слое начинается
интенсивное перемешивание н непрерывное образование н распад агрегатов,
сопровождающееся уносом отдельных частиц в аэрозольную фазу. Так как с
увеличением размера частиц гидродинамические силы возрастают, а действие
молекулярных сил ослабевает, то следует ожидать, что при некоторой средней
степени дисперсности порошка условия для флуидизации порошка будут опти-
мальными. И действительно, наиболее равномерная и полная флуидизация на-
блюдается для порошков с частицами, радиус которых близок к 20—25 мкм.
Так как трение порошков о стенки трубки убывает по мере увеличения раз-
мера частиц (уменьшается эффект молекулярных сил), то в грубых порошках
прн малом диаметре трубки наблюдается явление «захлебыв-ания» в слое: он
целиком поднимается газом, затем газ пробивается через слой, н слой снова
спадает. Если после достижения полной флуидизации порошка постепенно умень-
шать скорость течения, то при полной остановке тока газа слой порошка оста-
нется в расширенном состоянии; для возвращения в первоначальное нерасширен-
ное состояние его надо утрясти. Отсюда следует, что в расширенном слое кон-
такт между частицами сохраняется и встречающееся в литературе утверждение,
что в расширенном слое частицы окружены со всех сторон «газовой пленкой»
и, следовательно, не соприкасаются друг с другом, необоснованно.
Указанным способом удобно определять значения коэффициента заполнения
Фмин при расширенном состоянии порошка. Эти значения, соответствующие
максимально возможной пористости или максимально возможному объему дан-
ного порошка при сохранении контакта между частицами, являются одной из
важных характеристик порошкообразных материалов. В табл. XI, 2 приведены
значения ФМин для некоторых пррошков и Фо, соответствующие покоющемуся
нерасширенному слою порошка.
Как видно из приведенных в табл. XI, 2 данных, степень заполнения про-
странства уменьшается вместе с размером частиц не только в покоящемся, но и
в расширенном слое, что также указывает на действие молекулярных сил, а
следовательно, и на контакт между частицами при расширении слоя.
Некоторые порошки можно перевести в расширенное состояние не только
пропуская через них газ, но и просто осторожным пересыпанием. В таком со-
стоянии многие порошки также обладают большой текучестью и напоминают
по свойствам жидкость. Очевидно, находящийся между частицами воздух тор-
мозит их падение и способствует образованию рыхлой структуры. Так как в
Данном случае сила тяжести не уравновешивается, текучесть некоторых грубых
порошков, вероятно, объясняется сравнительно малыми молекулярными силами,
а следовательно, и малыми силами трения между частицами.
Известно, что некоторые порошки сильно «пылят» при пересыпании, а дру-
гие при той же дисперсности не пылят. Распыляемость порошков имеет боль-
шое значение в технике. Распыляемость в первую очередь определяется силами
сцепления между частицами, она увеличивается при возрастании размеров ча-
стиц до известного предела и уменьшается с увеличением влажности порошка.
Поэтому неувлажняющиеся гидрофобные порошки (например, тальк) распыляются
12 Зак. 664 353
лучше гидрофильных (кварца, известняка). Порошки из мягких, пластичных ма-
териалов распыляются хуже, чем из твердых; моноднсперсные порошки распы-
ляются лучше полидисперсных, так как в последних степень заполнения
пространства, а следовательно, и число точек соприкосновения между частицами
больше, чем в первых
Таблица XI, 2. Значения коэффициентов заполнения пространства
для некоторых порошков (по Н. А. Фуксу)
Порошок Средний радиус частиц, мкм Фо ^МИН
Стеклянные шарики 285 0,67 0,55
115 0,66 0,54
Песок с округленными частицами 185 .— 0,58
80 — 0,56
42 — 0,53
25 — 0,46
Песок с угловатыми частицами 160 .— 0,50
100 —— 0,47
42 0,43
27 — 0,42
Окись железа 187 0,50 0,48
100 0,48 0,44
35 0,42 0,39
В заключение рассмотрим процесс сухого гранулирования, по своей природе
как бы обратный таким явлениям, как флуидизация и распыление Давно из-
вестно, что в результате не слишком сильных механических воздействий многие
порошки могут быть переведены в гранулы — крупные агрегаты обычно сфери-
ческой формы, распыляющиеся в порошок при сравнительно легком надавлива-
нии. Этот процесс детально изучен на примере сажи Гранулированная сажа
имеет меныпнй объем, ие пылит и обладает повышенной текучестью, что удобно
для ее перевозки и переработки
Сажу можно гранулировать встряхиванием ее на вибрирующих плоскостях,
а также под действием ультразвука. Однако наиболее эффективное гранулирова-
ние достигается при обкатывании сажи в специальных вращающихся бараба-
нах. С С. Воюцкий с сотр. показали, что гранулирование сажи прн обкатыва-
нии идет эффективно лишь при введении в порошкообразную сажу «зародышей».
Такне зародыши можно получить из сажи в виде мелких плотрых комочков при
пропускании ее через узкий зазор вальцов, применяемых в резиновом производ-
стве, илн каким-нибудь иным способом. В качестве зародышей при гранулирова-
нии можно использовать н сажевые гранулы малого диаметра. Гранулирование
вызывают также любые мелкие инородные частицы, например зерна растений,
кристаллы сахара и т. д. Однако слишком тяжелые чужеродные тела, как
свинцовая дробь, не могут служить зародышами, очевидно вследствие того, что
образующийся на поверхности сажевый слой разрушается под тяжестью дро-
бинки, когда последняя при обкатывании сталкивается со стенкой барабана или
другой дробинкой.
При обкатывании порошкообразная сажа как бы налипает на зародыши, и
при этом образуются сферические гранулы. Число гранул обычно равно числу
зародышей, н это позволяет, меняя соотношение между количествами порошко-
образной сажи н зародышей, получать гранулы любого размера, вплоть до
1—2 см в поперечнике. В этом отношении имеется формальная аналогия с об-
разованием коллоидных систем методом конденсации с введением в систему
354
зародышей. Важно отметить, что с увеличением соотношения порошкообразная
сажа — зародыши снижается прочность гранул.
Опыт показал, что гранулирование идет наиболее эффективно при средней
скорости обкатывания. При очень больших скоростях центробежная сила при-
жимает всю сажу к стенкам барабана и сажа в барабане не обкатывается. При
очень малых скоростях не достигается вращательного движения сажи, при ко-
тором один слой, накатываясь -на другой, обуславливает трение частиц друг
о друга, необходимое для гранулирования.
Поскольку гранулы образуются при обкатывании самых разнообразных по-
рошков, следует полагать, что механизм гранулирования должен иметь вполне
общий характер. Вероятно, отдельные частицы того или иного порошка под
действием молекулярных сил вступают в энергетическое взаимодействие, обра-
зуя агрегаты. Причиной, определяющей достаточно прочную связь между ча-
стицами, может быть либо случайный контакт частиц в особо активных участ-
ках, либо соприкосновение частиц плоскими гранями, в результате чего
межмолекулярные силы могут действовать иа сравнительно большой пло-
щади.
Если порошок перемешивать или пересыпать не со слишком большой ско-
ростью, ведущей к разрушению агрегатов, то совершенно очевидно, что в
порошке постепенно будут накапливаться агрегаты частиц. Вследствие меньшей
кривизны поверхности этих агрегатов при столкновении их с отдельными ча-
стицами, последние прилипают к ним особенно прочно. Все это приведет к тому,
что постепенно весь порошок превратится в агрегаты — гранулы. Форма этих
агрегатов должна быть близкой к сферической, так как все выступы на поверх-
ности таких гранул сглаживаются при перемешивании или пересыпании си-
стемы.
Причинам возникновения прочной связи между частицами при перемешива-
нии или пересыпании порошка можно дать и несколько другое толкование. Для
образования контакта и прочной связи частицам необходимо преодолеть некий
энергетический барьер, например преодолеть силы адсорбции молекул газов
частицами пыли. Очевидно, образование агрегатов в этом случае вызовут только
особо удачные столкновения частиц, обладающих кинетической энергией, доста-
точной для преодоления этого энергетического барьера. При такой точке зрения
факторы, обуславливающие удаление адсорбированных молекул, должны спо-
собствовать образованию контактов и, следовательно, действию молекулярных
сил. Это и наблюдается в действительности: как показали опыты, при об-
катывании под вакуумом и при повышенной температуре гранулирование улуч-
шается.
Положительную роль зародышей • при гранулировании можно объяснить
большей массой и меньшей кривизной поверхности зародыша по сравнению с
частицей порошка. Вследствие большой массы зародыша происходят более силь-
ные удары, что может способствовать преодолению энергетического барьера и,
следовательно, обусловливать слипание. Меньшая кривизна поверхности заро-
дыша также должна благоприятствовать взаимодействию между зародышем и
элементарной частицей сажи, т. е. налипанию отдельных частиц на зародыш. По-
ложительное действие зародышей может объясняться также и тем, что зародыш
при обкатывании катится между элементарными частицами и вызывает извест-
ную их ориентацию, что тоже способствует налипанию их на зародыш.
Основная причина связи между гранулированными частицами (контакт
между элементарными частицами в отдельных местах) также хорошо согла-
суется с наблюдающимся возрастанием плотности и прочности с увеличением
длительности обкатывания. Вследствие толчков при перекатывании и легких
ударов гранулы о гранулу возникает большее число контактов, в результате
чего вея система становится более плотной и прочной.
Рассмотренный выше метод сухого гранулирования порошков с применением
только одних механических и притом не интенсивных механических воздействий
не следует смешивать с другими методами уплотнения порошков и переведения
их в непылящее состояние — брикетированием с применением связующей среды,
получением гранул из порошка, смоченного небольшим количеством жидкости,
и т. д.
12*
355
3. МЕТОДЫ ПОЛУЧЕНИЯ АЭРОЗОЛЕЙ
Аэрозоли, равно как и другие коллоидные системы, можно по-
лучать с помощью методов конденсации и диспергирования.
Методы конденсации. В основе всех конденсационных методов
образования аэрозолей лежит конденсация пересыщенных паров.
Пересыщение паров может быть достигнуто либо за счет охлаж-
дения системы, либо при образовании пара в результате химиче-
ской реакции.
Охлаждение, пересыщение и конденсация паров может проис-
ходить различными путями, например при адиабатном расширении
газа, содержащего пары какой-либо жидкости. Именно так обра-
зуются обычные кучевые облака, когда теплые массы влажного
воздуха поднимаются в более высокие слои атмосферы. Перистые
облака, возникающие на больших высотах, также являются ре-
зультатом конденсации водяных «паров, однако в этом случае при
конденсации в верхних слоях атмосферы вследствие низкой тем-
пературы образуются не жидкие капельки, а твердые кристаллики
льда. Таким образом, перистые облака следует отнести к системам
с твердой дисперсной фазой.
Охлаждение, пересыщение и конденсация паров может также
происходить при их соприкосновении с холодной поверхностью или
при смешении с холодным воздухом. Так образуются в природе
туманы. Чаще всего туман появляется при ясной погоде ночью,
при сильном охлаждении поверхности земли в результате тепло-
вого излучения. Влажный воздух вторгается в зону с более низкой
температурой или соприкасается с охладившейся землей, вслед-
ствие чего в нем и образуются капельки тумана.
Химические реакции, при которых возможно образование аэро-
золей, могут иметь самый различный характер. Так, в результате
окисления при сгорании топлива образуются дымовые газы, содер-
жащие продукты с весьма малым давлением пара. Смешиваясь
с более холодным воздухом, эти продукты конденсируются и об-
разуют топочный дым. Дымы получаются также при сгорании фос-
фора на воздухе (возникают частицы Р2О5), при взаимодействии
газообразного аммиака и хлористого водорода (образуются ча-
стицы NH4CI), в результате, фотохимических реакций, например
при освещении влажного хлора (возникает туман хлористоводо-
родной кислоты), и т. д. Окисление металлов на воздухе, проис-
ходящее при различных металлургических и химических процес-
сах, очень часто сопровождается образованием дымов, состоящих
из частиц окислов металла, например окиси цинка, окиси магния
и т. д. Стойкие туманы могут давать в смеси с воздухом такие ве-
щества, как SO3 и НС1. Наконец, дым образуется при соприкосно-
вении с влажным воздухом хлорида алюминия. Последний дымит
на воздухе потому, что между А1С13 и водяным паром происходит
химическая реакция с образованием высокодисперсных частиц
А1(ОН)3.
356
При получении аэрозолей путем конденсации из пересыщенного
пара должна возникнуть новая фаза. Выделение новой фазы прн
образовании аэрозоля, равно как н при образовании любой другой
системы, связано с большими энергетическими препятствиями. Это
можно понять, если учесть огромную удельную поверхность, ко-
торая соответствует началу возникновения гетерогенной системы,
когда ее дисперсность чрезвычайно высока. Поэтому при образо-
вании аэрозолей, как и лиозолей, либо должно быть очень большое
пересыщение, либо в системе должны присутствовать или возни-
кать в результате какого-нибудь особого явления зародыши или
ядра конденсации, вокруг которых может уже отлагаться веще-
ство дисперсной фазы. Такими зародышами в атмосфере могут
служить мельчайшие кристаллики соли, ультрамикроскопические
пылинки и другие образования.
Рассмотрим сначала механизм образования новой фазы в отсутствие посто-
ронних зародышей, пользуясь представлениями, развитыми Фольмером. Для
простоты возьмем случай, когда образуется дисперсная система с жидкой дис-
персной фазой. Если только система не близка к критическому состоянию, воз-
никновение новой (жидкой) фазы без сильного пересыщения невозможно. При-
чина этого заключается в том, что первоначально образующиеся мельчайшие
капельки, необходимые для получения тумана со сравнительно большими ча-
стицами, обладают очень малым радиусом кривизны, вследствие чего давление
пара у поверхности таких капелек весьма велико и они легко испаряются. Это
становится более понятным из следующих рассуждений.
Как известно, давление пара рг на поверхности капелек с радиусом г может
быть выражено с помощью уравнения В. Томсона
Рг = Рэехр(^^) (XI, 13)
где ps — давление пара над плоской поверхностью жидкости; Умол— мольный
объем жидкости.
, Согласно уравнению (XI, 13) капелька жидкости, находящаяся в атмосфере
пара с давлением ра, обладает более высоким давлением рг, чем давление пара
над плоской поверхностью, и потому она начнет испаряться. Причем она испа-
ряется так, что с уменьшением ее радиуса испарение ускоряется, поскольку
разность между рг и р3 непрерывно возрастает. Очевидно, ’капельки могут на-
ходиться в равновесии только с пересыщенными (по отношению к плоской по-
верхности) парами, причем любой степени пересыщения должны отвечать ка-
пельки определенного размера. Однако для того, чтобы в пересыщенном паре
возникли такие равновесные капельки, они должны вырасти из более мелких,
неустойчивых капелек. Равновесные капельки могут возникать лишь в результате
флуктуации их размера Этот процесс может идти сравнительно легко при боль-
ших пересыщениях.
Из сказанного следует, что в отсутствие посторонних зародышей образова-
ние новой фазы может быть только флуктуационным процессом и число спон-
танно возникающих зародышей, так же как и скорость образования новой
фазы /о, пропорциональны числу флуктуаций. Вероятность флуктуаций пропор-
циональна ехр(—AF/kT) (где ДЕ—изменение свободной энергии системы при
данной флуктуации). Величина ДЕ равна работе А, нужной для изотермического
и обратимого образования зародыша. Таким образом:
/0 = кехр(-4/АТ) (XI, 14)
Приведенное уравнение показывает, что скорость образования новой фазы в
отсутствие чужеродных зародышей определяется в основном работой, необходи-
мой для спонтанного образования скопления молекул пара с давлением, равным
Давлению пара пересыщенной системы.
357
a
Правильность теории Фольмера подтверждена экспериментально рядом ис-
следователей. В частности, такая проверка проводилась путчем адиабатического
расширения воздуха насыщенного парами данной жидкости, в камере Вильсона.
В результате расширения в камере происходит охлаждение, а следовательно, и
пересыщение паров до вполне определенного значения. Применяя камеру Виль-
сона, можно визуально устанавливать начало конденсации, т. е. пересыщение,
отвечающее образованию тумана. Чтобы исключить возможность образования
капелек на чужеродных зародышах, система должна быть предварительно очи-
щена путем многократной конденсации, при которой посторонние ядра конден-
сации удаляются из газовой фазы. При этом критическое пересыщение, отвечаю-
щее началу образования новой фазы, непрерывно возрастает до определенного
предела.
Расчеты, основанные на полученных таким образом данных, показывают, что
относительные пересыщения, отвечающие заметному спонтанному образованию
зародышей, очень велики и достигают 200—300% и выше. Столь большие сте-
пени пересыщения наблюдаются далеко не всегда.
Таким образом, следует прийти к заключению, что образование капелек про-
исходит, как правило, в результате наличия в газовой фазе чужеродных актив-
ных центров, понижающих работу А. Такими центрами могут быть либо ионы,
либо нейтральные жидкие и твердые частицы, присутствующие в системе.
Значение ионов при образовании новой фазы в газовой среде легко дока-
зать с помощью камеры Вильсона. Для этого камеру следует заполнить возду-
хом и паром'исследуемой жидкости, пересыщение которого недостаточно для
образования тумана в неионизированном газе, и вызвать в камере ионизацию
газа, например, путем облучения частицами высоких энергий (продуктами рас-
пада радиоактивных элементов, космическими лучами). В таких условиях в ка-
мере можно наблюдать дорожки из тумана, соответствующие пути частиц. При-
чиной образования таких дорожек является образование ионов в результате
столкновения частиц высоких энергий с молекулами газа и конденсация на этих
ионах паров.
Основные положения теории образования новой фазы иа ионах, находя-
щихся в парах, следующие. При столкновении с капелькой жидкости, являю-
щейся проведником, ионы как бы равномерно распределяются по поверхности
капельки. В результате этого на поверхности капельки возникают силы, стре-
мящиеся увеличить ее поверхность, и, следовательно, действующие против по-
верхностного натяжения. Это приводит к снижению поверхностного натяжения,
а следовательно, и работы,,необходимой для образования зародыша. Значение
поверхностного натяжения заряженной капельки можно найти по уравнению:
О2
где Оо — поверхностное натяжение незаряженной капельки; г и Q — радиус и
заряд капельки.
Давление насыщенного пара над заряженной капелькой можно найти из
уравнения В. Томсона, подставив в него значение а:
График этого уравнения, выражающего связь между давлением пара заря-
женной иапельки и ее радиусом, изображен на рис. XI, 3. В отсутствие ионов
в системе невозможно образование капельки без флуктуаций размера (давление
окружающего пара всегда ниже давления пара над незаряженной капелькой).
В присутствии ионов при некотором давлении пара для образования капелек
флуитуацйи не нужны. В самом деле, заряженная капелька имеет равновесное
Давление пара всегда меньшее, чем давление пара окружающей среды. Таким
образом, капельки будут легко возникать в результате конденсации. Подобный
вывод находится в полном согласии с наблюдениями, которце были сделаны
с помощью камеры Вильсона.
358
Прн некотором давлении ра < рмакс (этот случай изображен на рис. XI, 3)
в системе, в которой присутствуют ионы, самопроизвольно образуются капельки
радиусом га. Даже при р = ps, т. е. в отсутствие пересыщения, образуются ка-
пельки весьма малым радиусом г0 При этом характер кривой / на рис. XI, 3
показывает, что капельки с радиусом, отвечающим уравнению (XI, 16), обра-
зуются в результате конденсации без флуктуаций, поскольку любая заряженная
капелька с меньшим радиусом имеет меньшее давление пара, чем давление окру-
жающего пара, и должна расти в результате конденсации до тех пор, пока дав-
ление ее пара не станет равным давлению окружающего пара. Очевидно, в
результате этого в системе возникнет столько капелек, сколько в ней имелось
ионов.
Однако возникшие в таких условиях мельчайшие заряженные капельки не
способны служить зародышами для образования сравнительно больших капелек
тумана, так как они не могут беспре-
пятственно расти дальше. Действи-
тельно, из кривой 1 рис. XI, 3 видно,
что при увеличении радиуса от га до
гк капельки должны были бы посте-
пенно расти в таких условиях, когда
давление 'их пара выше давления
пара окружающей среды, что, конеч-
но, невозможно. Поэтому образова-
ние капелек радиусом гк может осу-
ществляться только в результате
флуктуаций. Понятно, что когда зна-
чение гк уже достигнуто, дальнейший
рост капелек может идти уже беспре-
пятственно и каждая такая капелька
будет' являться теперь настоящим за-
родышем новой фазы *.
Несмотря на возможность опи-
санного выше механизма образования
новой фазы в парах на ионах, этот
механизм, вероятно, не имеет суще-
ственного значения при возникнове-
нии атмосферных аэрозолей. Дело в
Рис. XI, 3. Зависимость давления пара
капельки от ее радиуса:
1—для заряженных капелек; 2—для незаря-
женных капелек.
том, что в атмосфере всегда присут-
ствуют ядра конденсации, обеспечивающие конденсацию при сравнительно малых
пересыщениях. Такими ядрами могут служить мельчайшие кристаллики соли,
содержащиеся в атмосфере над морями и океанами, ультрамикроскопические
пылинки и другие образования. Теория конденсации на ядрах еще мало разрабо-
тана, так как структура и свойства ядер весьма сложны и разнообразны. Отме-
тим лишь, что скорость возникновения зародышей должна быть пропорциональна
численной концентрации ядер.
Методы диспергирования. К методам получения аэрозолей пу-
тем диспергирования относятся измельчение и истирание твердых
тел, распыление жидкостей, а также получение аэрозолей в резуль-
тате взрыва. Как правило, методами диспергирования получаются
гораздо более низкодисперсные и более полидисперсные аэрозоли,
чем методами конденсации.
Распылением жидкости путем продавливания через специаль-
ные пульверизаторы и форсунки пользуются для равномерного
нанесения красок и лаков на окрашиваемые поверхности, для
* Более подробно о возникновении капелек на ионах см. А. Шелудцо.
Коллоидная химия. Пер. с англ, под ред. Б. В. Дерягина и С. С. Воюцкого. М„
Издатинлит, 1960, с. 299—307.
359
распыления жидкого топлива и других целей. Интересно, что если
при таком распылении струю жидкости зарядить до электриче-
ского потенциала в несколько тысяч вольт, то распыление жидко-
сти идет гораздо легче.
В природе аэрозоли, содержащие капельки жидкой дисперсной
фазы, получаются путем диспергирования в результате падения
больших масс воды в водопадах.
4. МЕТОДЫ РАЗРУШЕНИЯ АЭРОЗОЛЕЙ
К разрушению аэрозолей приходится прибегать, если из аэро-
золя нужно выделить дисперсную фазу, например при улавлива-
нии из дыма металлургических печей содержащихся в нем ценных
Рис. XI, 4. Движение
газа в циклоне.
продуктов, либо при очистке газов
или воздуха. В последнее время ме-
тоды разрушения (коагуляции) ат-
мосферных аэрозолей применяют
при искусственном дождевании или
рассеивании облаков и тумана.
Рис. XI, 5. Схема филь-
тра с развернутой по-
верхностью.
На практике частицы дисперсной фазы выделяют из газовой
среды путем изменения скорости и направления потока аэрозоля
(инерционное осаждение) фильтрацией, действием ультразвука или
электрического поля, введением зародышей и коагуляцией.
Выделение дисперсной фазы из аэрозоля путем изменения ско-
рости и направления потока аэрозоля осуществляют обычно с по-
мощью центробежных отделителей, называемых циклонами. Цик-
лоны представляют собой металлические цилиндры, в которых’
аэрозоль движется по спирали сверху вниз. При этом частицы осе-
дают на стенках цилиндра, а освобожденный от них газ подни-
мается по специальной трубе и выводится из циклона. Движение
газа в циклоне схематически показано на рис. XI, 4. Этот способ
применяется лишь для разрушения сравнительно грубых аэрозо-
лей, содержащих частицы диаметром более 3 мкм.
С помощью фильтрации от газовой фазы можно отделить го-
раздо более мелкие частицы. Фильтры применяют в противогазах
'360
для задержания частиц ядовитых дымов, для получения стериль-
ного воздуха и в ряде других случаев. Существуют сетчатые и во-
локнистые фильтры.
Сетчатые фильтры служат для задержания сравнительно гру-
бых частиц аэрозолей. Их изготовляют из одного или нескольких
слоев ткани или металлической сетки. Действие этих фильтров
основано на механическом задерживании больших частиц, не про-
ходящих через ячейки сетки, а также на инерционном осаждении
частиц. Эффективность сетчатых фильтров заметно увеличивается
по мере забивания их отфильтрованной дисперсной фазой, по-
скольку в результате образования на поверхности фильтра слоя
пыли уменьшается диаметр отверстий, через которые протекает
аэрозоль. Поэтому иногда на тканевые фильтры перед их исполь-
зованием наносят асбестовую пыль, особенно эффективную при
фильтрации, или при очистке тканевых фильтров на их поверхности
целесообразно оставлять часть пылевого слоя.
Волокнистые фильтры делают из фильтровальной бумаги, спе-
циального картона и некоторых других волокнистых материалов.
Вследствие значительного гидравлического сопротивления эти
фильтры применяют лишь при небольших скоростях течения аэро-
золя. С целью повышения производительности волокнистых филь-
тров их часто изготовляют с «развернутой» (увеличенной) поверх-
ностью. Схема устройства такого фильтра показана на рис. XI, 5.
Характер течения аэрозоля в волокнистом фильтре очень сло-
жен, поскольку поток, огибая отдельные, беспорядочно располо-
женные волокна, все время изменяет свое направление. Действие
волокнистых фильтров сводится к инерционному осаждению, при-
липанию движущейся частицы к какому-нибудь выступу на по-
верхности волокна (эффект зацепления), седиментации и, нако-
нец, к диффузии частицы к поверхности волокна с последующей
фиксацией. Различные факторы действуют неодинаково на разные
явления, на которых основано выделение дисперсной фазы при
фильтрации аэрозоля. Инерционное осаждение и седиментация
увеличиваются при возрастании размера и плотности частиц,
а также скорости течения, диффузионному осаждению способ-
ствует уменьшение размера частиц, но оно не зависит от плотно-
сти частиц.
Конечно, эффективность фильтрации, независимо от преобла-
дающего механизма осаждения, возрастает с уменьшением рас-
стояния между волокнами, однако при этом увеличивается сопро-
тивление фильтра. В связи с тем, что очень часто, в особенности
при фильтрации с большой скоростью, основную роль играют
инерционное осаждение, эффективность волокнистого фильтра оп-
ределяется не столько размерами пор, сколько их извилистостью
и радиусом волокон.
Разрушение аэрозоля под действием ультразвука известно
давно, но только сейчас оно начинает получать практическое при-
менение. Согласно одной из теорий, действие ультразвука на
361
аэрозоли так же, как и на лиозоли, объяснялось тем, что во всех
реальных полидисперсных системах разные по размеру частицы
в различной степени увлекаются колебаниями среды. В результате
этого мелкие частицы, обладающие большой амплитудой колеба-
ний, как бы «прочесывают» аэрозоль. Это способствует тому, что
они скорее сталкиваются с более крупными, почти неподвижными
частицами. Однако против этой теории говорит то обстоятельство,
что самые мелкие, наиболее' энергично колеблющиеся частицы
остаются в звуковом поле нескоагулированными.
Согласно другой теории ультразвуковая коагуляция обусловли-
вается притяжением между частицами, движущимися в ультра-
звуковом поле. Такое притяжение может возникнуть между части-
цами аэрозоля, если они совершают быстрое, параллельное и
одинаково направленное движение. Нужны всего секунды для того,
чтобы туман, Движущийся в ультразвуковом поле, скоагулировал
на 90%. Полученные в результате коагуляции крупные капли легко
отделяются от газа в обычных циклонах.
Ультразвук применяют для разрушения сернокислотных и дру-
гих производственных туманов. В настоящее время для осаждения
аэрозолей ультразвуком разработаны промышленные установки
производительностью до 1000 м3/мин. К сожалению, в ультразву-
ковом поле остается нескоагулировавшей обычно самая высоко-
дисперсная часть тумана. Другой недостаток коагуляции аэрозо-
лей с помощью ультразвука заключается в том, что ультразвук
малоэффективен при разрушении сильно разбавленных систем.
Ряд методов разрушения атмосферных аэрозолей основан на их
коагуляции. Практическое значение таких методов очень велико
для сельского хозяйства, так как процесс коагуляции обычно со-
провождается отделением дисперсной фазы атмосферных аэрозо-
лей в виде дождя или снега. Большое значение методы коагуля-
ции имеют и в авиации для искусственного рассеивания облакоз
над аэродромами.
В литературе имеются указания, что "коагуляция атмосферных
аэрозолей может быть вызвана разбрасыванием с самолета высо-
кодисперсного песка, частицы которого несут электрический заряд,
по знаку обратный заряду частиц аэрозолей. Другой метод искус-
ственного рассеивания облаков и туманов с помощью коагуляции
заключается в распылении в аэрозоль растворов гигроскопических
веществ, например, концентрированных растворов хлорида каль-
ция (В. А. Федосеев, 1933 г.). Капельки этой жидкости захваты-
вают капельки воды, укрупняются и выпадают в виде дождя. Для
разрушения переохлажденных атмосферных аэрозолей можно при-
менять также дымы иодида серебра или иодида свинца, частицы
которых являются зародышами и вызывают в облаках образова-
ние крисФалликов льда.
Эффективным методом искусственного рассеивания облаков и
туманов, вполне оправдавшим себя на практике, является метод
с использованием твердой двуокиси углерода. Метод этот также
362
применим при температуре воздуха ниже О °C и при капельной
структуре облаков или тумана, т. е. для переохлажденного аэро-
золя. Разбрасываемая в таком аэрозоле твердая двуокись угле-
рода с температурой —79,8 °C вызывает быстрое охлаждение при-
лежащего к ней слоя воздуха и в нем образуется огромное число
ледяных кристалликов, служащих далее центрами кристаллиза-
ции. Кристаллики быстро растут, продолжая рост и после выхода
из зоны искусственного охлаждения, поскольку давление насыщен-
Лзрозоль
Пыль
Рис. XI, 6. Устройство эле-
мента электрофильтра:
1, 4 — патрубки; 2—труба;
3 —электрод; 5—бункер.
ных паров воды над льдом меньше, чем над
водой. Достигнув критического размера,
кристаллики выпадают из аэрозоля в виде
снежинок. Опыт показал, что образование
снега при таком методе рассеивания обла-
ков происходит через 5—7 мин, а через
15—30 мин зона, в которую была введена
двуокись углерода, полностью освобо-
ждается от тумана. Следует заметить, что
при слишком больших количествах двуоки-
си углерода, а также малом содержании
воды в аэрозоле, или малом переохлажде-
нии, процесс рассеивания не идет или идет
очень медленно.
Наконец, аэрозоли можно разрушать
действием электрического поля высокого
напряжения. Этот метод, разработанный
Коттрелем, используется в промышленности
для очистки газов от пыли, разрушения
дыма перед его выбросом в атмосферу и
других целей. Поскольку частицы аэрозоля
обычно слабо заряжены или практически
электронейтральны, им придают достаточно
ский заряд. Для этого дым или туман пропускают между электро-
дами, создающими поле весьма высокого напряжения. В таких
условиях происходит так называемый коронный разряд, при ко-
тором катод (имеющий форму проволоки для того, чтобы заряд
обладал наибольшей плотностью) светится и выделяет огромное
количество электронов. Электроны ионизируют молекулы газа
(воздуха), находящегося в пространстве между электродами. Об-
разовавшиеся отрицательные ионы адсорбируются частицами
аэрозоля и придают им достаточно большой здряд, что обеспечи-
вает частицам передвижение в электрическом поле и осаждение на
аноде. Передвижению частиц к аноду способствует также «элек-
трический ветер», возникающий между электродами.
Схематическое устройство одного из элементов батареи обыч-
ного электрофильтра показано на рис. XI, 6. Электрическое поле
возникает между отрицательным коронирующим электродом 3 и'
положительным электродом, которым служит металлическая тру-
ба 2. На электроды подается постоянный ток высокого напряжения
большой электриче-
363
(70—100 кВ). Аэрозоль поступает в элемент через патрубок /.
Под действием электрического поля и ионного ветра получившие
отрицательный электрический заряд частицы аэрозоля направ-
ляются к аноду 2, ударяются о него и оседают. Осевшие и поте-
рявшие заряд частицы осыпаются в низ трубы и удаляются из нее
через бункер 5. Если частицы аэрозоля жидкие, то после коалес-
ценции капелек жидкость непрерывно стекает по стенкам трубы
и удаляется через тот же бункер 5. Очищенный газ уходит из эле-
мента через патрубок 4.
Имеются электрофильтры, в которых процессы заряжения и
осаждения частиц пространственно разделены. В таких электро-
фильтрах образуется значительно меньше продуктов окисления
воздуха (окислов азота и т. д.), и поэтому их применяют для кон-
диционирования воздуха.
5. ПРАКТИЧЕСКОЕ ЗНАЧЕНИЕ АЭРОЗОЛЕЙ
Аэрозоли благодаря широкому распространению в природе и
технике играют важную роль в жизни и деятельности человека.
Природные аэрозоли — облака и туманы — имеют огромное
значение для метеорологии и сельского хозяйства, поскольку они
определяют выпадение осадков и в значительной степени обусло-
вливают климат того или иного района. Такие природные явления,
как дождь или снег, гроза, радуга, целиком определяются нали-
чием в атмосфере аэрозолей. Известную роль играют аэрозоли и
в биологии — пыльца растений, споры бактерий и плесени, а также
легкие семена переносятся в природе в форме аэрозоля.
Не меньшее значение имеют аэрозоли, получающиеся в резуль-
тате практической деятельности человека. В металлургических и
химических производствах в воздух выбрасываются огромные ко-
личества дыма. Например, по данным Н. П. Пескова, с дымом на
медеплавильных предприятиях при переработке 10 000 т руды
количества различных веществ
в воздух уносятся следующие
(в кг):
A12S3 .............. 26 900
Sb2S3............... 1906
Си ................ 1 907
Pb.........." . . . . 2 170
Zn........................... 2 785
Fe + Al...................... 8 100
Bi............................ 400
Мп........................... .г80
Помимо того, что с дымом уносятся ценные вещества, которые
могли бы быть использованы в производстве, дым загрязняет
окрестности, уничтожает растительность и вредно влияет на
здоровье людей. Наличие в атмосфере промышленных городов
большого количества дыма при достаточной влажности воздуха
способствует образованию тумана. Именно поэтому, например, в
Лондоне наблюдается особенно много туманных дней в году.
В производстве аэрозоли образуются при работе разного рода
машин — дробилок, мельниц, вальцов, просеивающих приспособ-
364
лений и т. д. Выделяющаяся пыль загрязняет производство, попа-
дая между трущимися частями машин, ускоряет их износ, создает
антисанитарные условия для работы человека. Особенно вредна
пыль, содержащая мельчайшие, острые осколки кварца. Продол-
жительное вдыхание такой пыли вызывает тяжелое, часто со смер-
тельным исходом заболевание — силикоз. Кварц, попадая в лег-
кие в виде микроскопических острых осколков, разрушает ткань
легких и способствует проникновению в организм разнообразных
инфекций и, в частности, способствует заражению туберкулезом.
Сходные заболевания вызывают аэрозоли окислов некоторых ме-
таллов, например окиси цинка.
В угольных шахтах образование пыли может служить причи-
ной сильных взрывов. Опасность взрыва возможна на всех пред-
приятиях, перерабатывающих в порошкообразном состоянии мате-
риалы, способные гореть, но в обычном виде вполне безопасные
(мука, сахар, сера). Это объясняется тем, что благодаря огромной
удельной поверхности дисперсной фазы, а значит, огромной пло-
щади ее соприкосновения с воздухом и малой теплопроводности
аэрозоля, способствующей местному разогреванию, реакция окис-
ления при сгорании дисперсной фазы аэрозоля идет с колоссаль-
ной скоростью, что приводит к взрыву. Взрывы аэрозолей опаснее,
чем взрывы газов, так как переход от твердого или жидкого со-
стояния вещества к газообразному сопровождается гораздо боль-
шим увеличением объема системы, чем при газовых реакциях.
Из всего сказанного выше понятно, почему в СССР такое боль-
шое внимание уделяется очистке выхлопных газов и улавливанию
содержащихся в дыме ценных веществ, а также борьбе с пылью
на производстве.
Выше мы касались главным образом отрицательного значения
аэрозолей, образующихся в производственных условиях. Однако
в некоторых случаях аэрозоли играют положительную роль и их
приходится специально получать особыми методами. Например,
распыление до состояния аэрозоля или микрогетерогенной системы
применяют при подаче твердого или жидкого топлива в топки. По-
лучение аэрозоля краски или лака путем пневматического распы-
ления с помощью специальных пульверизаторов широко исполь-
зуется для окрашивания различных поверхностей и предметов. По-
добный же прием применяют и при металлизации поверхностей.
Огромное значение имеет распыление инсектицидов, фунгицидов и
гербицидов в сельском хозяйстве при борьбе с вредными насеко-
мыми, грибками и сорняками. В медицине аэрозоли применяют
для введения лекарственных веществ в организм путем ингаляции.
Особое значение аэрозоли имеют в военном деле при свето-
маскировке.
ГЛАВА XII
СИСТЕМЫ С ЖИДКОЙ И ТВЕРДОЙ ДИСПЕРСНОЙ ФАЗОЙ
Излагая материал, касающийся аэрозолей, мы не принимали
во внимание различия между аэрозолями с жидкой и твердой дис-
персной фазой, так как по свойствам они сравнительно мало отли-
чаются друг от друга. При ознакомлении с лиозолями рассмотрим
отдельно в соответствии с принятой во введении классификацией
системы с газовой, жидкой и твердой дисперсной фазой, поскольку
их свойства весьма различны.
Прежде -всего остановимся на золях с твердой дисперсной фа-
зой, поскольку эти системы нам уже хорошо известно. В предыду-
щих главах мы рассматривали свойства коллоидных систем на
примере именно этих лиозолей и поэтому в данной главе приведем
только несколько дополнительных замечаний в отношении суспен-
зий, т. е. систем с твердой дисперсной фазой и жидкой дисперсион-
ной средой, размеры частиц которых превышают коллоидные раз-
меры Далее рассмотрим эмульсии, поскольку они по свойствам
более близки к суспензиям, и, наконец, пены — системы, весьма
своеобразные и по ряду свойств резко отличные от суспензий и
эмульсий. В конце главы кратко остановимся на системах с твер-
дой дисперсионной средой.
1. СУСПЕНЗИИ
Суспензии или взвеси"порошков в жидкости имеют исключи-
тельно большое значение в природе и технике, далеко превосходя-
щее значение типичных золей с твердой дисперсной фазой. К сус-
пензиям при достаточном содержании влаги относятся почвы и
грунты, глиняное тесто, используемое в керамическом производ-
стве, цементные и известковые растворы, применяемые в строи-
тельном деле. Суспензиями являются взвеси пигментов в органи-
ческих средах, применяющихся в качестве масляных красок и
цветных лаков, взвеси графита и угля, используемые для созда-
ния центров кристаллизации с целью предотвращения образования
накипи в котлах и т. д. В химических производствах технологу
приходится встречаться с суспензиями при получении солей в мел-
кокристаллическом состоянии.
Суспензии одновременно поглощают и рассеивают свет, хотя
их Цастицы по размеру больше коллоидных частиц. При этом в от-
366
личие от опалесцирующих золей суспензии проявляют мутность
не только при боковом освещении, но и в проходящем свете. Од-
нако суспензии, так же как и золи, способны проявлять двойное
лучепреломление в потоке.
Поскольку частицы суспензий обладают сравнительно боль-
шими размерами (они видимы в микроскоп), суспензии седимен-
тационно неустойчивы, если плотность дисперсной фазы не очень
близка к плотности дисперсионной среды н вязкость этой среды не
очень Йелика. По той же причине суспензии не обнаруживают ос-
мотического давления и броуновского движения и не способны
к диффузии.
Если по оптическим и молекулярно-кинетическим свойствам
суспензии и золи с твердой дисперсной фазой резко различны, то
по агрегативной устойчивости они имеют много общего. Как пра-
вило. частицы суспензий, равно как и частицы лиофобных коллои-
дов, имеют на поверхности двойной электрический слой или
сольватную оболочку. Электрокинетический потенциал частиц сус-
пензий можно определить с помощью макро- или микроэлектро-
фореза, причем он имеет величину того же порядка, что и ((-потен-
циал частиц типичных золей. Под влиянием электролитов суспензии
коагулируют, т. е. их частицы слипаются, образуя агрегаты.
В определенных условиях в суспензиях, так же как и в золях, об-
разуются пространственные коагуляционные структуры, способные
к синерезису. Явления тиксотропии и реопексии при соблюдении
соответствующих условий, проявляются у суспензий почти всегда
в большей степени, чем у лиофобных коллоидных систем.
Для суспензий характерен ряд процессов, не свойственных кол-
лоидным системам или протекающих у последних иначе, чем у сус-
пензий. К таким процессам относятся седиментация и флотация,
которые рассмотрены в предыдущих главах, а также фильтрация
и кольматация.
Фильтрация суспензий определяется дисперсностью и степенью
агрегации частиц, а также образованием коагуляционной струк-
туры и способностью ее к самоуплотнению в фильтрующем осадке.
Поэтому фильтрация является сложным физико-химическим про-
цессом, на который влияют все факторы, управляющие агрегиро-
ванием частиц и развитием коагуляционных структур. Не менее
сложен и процесс кольматации — «в мы в» мельчайших гли-
нистых или илистых частиц в поры грунта для уменьшения водо-
проницаемости различных гидротехнических сооружений — дамб,
плотин и т. д.
2. ЭМУЛЬСИИ
Гетерогенные системы, называемые эмульсиями, широко рас-
пространены в природе (молоко, млечный сок растений и т. д),
их легко изготовить также искусственным путем (пропиточные со-
ставы для придания тканям водонепроницаемости, смазки, марга-
рин, косметические кремы и т. д.).
367
Условия, необходимые для образования эмульсий, сходны
с теми условиями, которые нужны для получения коллоидных си-
стем с твердой дисперсной фазой и жидкой дисперсионной средой.
Обе жидкости, образующие эмульсию, должны быть нерастворимы
или мало растворимы друг в друге, и в системе должен присут-
ствовать стабилизатор, который в этом случае называют эмульга-
тором. Эмульсии тем седиментационно устойчивее, чем ближе
плотности обеих фаз.
Отличительной особенностью не слишком концентрированных
эмульсий является сферическая форма частиц (капелек). Как пра-
вило, дисперсность лиофобных эмульсий значительно ниже дис-
персности золей с твердой дисперсной фазой.
От типичных лиофобных эмульсий следует отличать так назы-
ваемые критические — лиофильные эмульсии. Критические эмуль-
сии— это системы, образующиеся обычно из двух ограниченно
смешивающихся жидкостей (например, анилина и воды, изоами-
лового спирта и воды) при температурах, весьма близких к крити-
ческой температуре смешения, когда поверхностное натяжение на
границе фаз становится весьма малым (порядка 0,01 эрг/см2) и
теплового движения молекул уже достаточно для диспергирования
одной жидкости в другой. В результате такого самопроизвольного
диспергирования образуется тончайшая эмульсия, в которой коа-
лесценция отдельных капелек уравновешивается стремлением
обеих жидкостей равномерно распределиться в объеме (см.
гл VIII, разд. 1).
Всякая критическая эмульсия является термодинамически
устойчивой равновесной системой, для существования которой не
требуется эмульгатор. Другими отличительными свойствами кри-
тической эмульсии являются возможность существования ее лишь
в очень узком интервале температур и непостоянство частиц дис-
персной фазы: капельки критической эмульсии все время обра-
зуются в системе и тотчас же исчезают, напоминая в этом отноше-
нии ассоциаты, образующиеся в жидкости в результате флуктуации
ее плотности.
Классификация эмульсий
Обычные лиофобные эмульсии классифицируют либо по поляр-
ности дисперсной фазы и дисперсионной среды, либо по концен-
трации дисперсной фазы в системе.
Согласно первой классификации различают эмульсии неполяр-
ной или слабополярной жидкости в полярной (например, эмульсия
масла в воде)—эмульсии первого рода, или прямые,
и эмульсии полярной жидкости в неполярной (например, вода
в масле)—эмульсии второго рода, или обратные.
Эмульсии первого рода очень часто обозначают через м/в, где
под буквой «м» подразумевается масло или иная неполярная
жидкость, а под буквой «в» — вода или другая полярная жидкость.
368
Эмульсии второго рода обозначают соответственно через в/м.
В особый класс выделяют эмульсии жидких металлов (ртути, гал-
лия) в воде, поскольку в этом случае и дисперсная фаза, и дис-
персионная среда ведут себя как полярные жидкости.
Тип эмульсий устанавливается очень легко путем определения
свойств ее дисперсионной среды. Для этого либо определяют спо-
собность эмульсии смачивать гидрофобную поверхность, либо про-
веряют возможность эмульсии разбавляться водой, либо испыты-
вают способность эмульсии окрашиваться при введении в нее кра-
сителя, растворяющегося в дисперсионной среде, либо, наконец,
определяют электропроводность эмульсии. Если эмульсия не сма-
чивает гидрофобную поверхность, разбавляется водой, окраши-
вается при введении водорастворимого красителя (например,
метиленового голубого) и обнаруживает сравнительно высокую
электропроводность, то это эмульсия типа м/в. Наоборот, если
эмульсия смачивает гидрофобную поверхность, не окрашивается
водорастворимым красителем (или окрашивается при введении
маслорастворимого красителя, например Судана III) и не обнару-
живает заметной электропроводности, то это эмульсия типа в/м.
Согласно второй классификации, эмульсии делят на разбавлен-
ные, концентрированные и высококонцентрированные, или жела-
тинированные.
К разбавленным эмульсиям относятся системы жид-
кость— жидкость, содержащие до 0,1 объемн.% дисперсной фазы.
Типичным примером таких систем может служить эмульсия ма-
шинного масла в конденсате, образующемся при работе паровых
машин. Следует, впрочем, отметить, что термин «разбавленные
эмульсии» весьма условен, так как к разбавленным эмульсиям от-
носят не просто эмульсии с малой концентрацией дисперсной фазы,
полученные, например, разбавлением концентрированных эмуль-
сий, но системы, обладающие характерными свойствами.
Прежде всего, разбавленные эмульсии по размеру частиц резко
отличаются от концентрированных и высококонцентрированных
эмульсий, являясь наиболее высокодисперсными. Диаметр капелек
в разбавленных эмульсиях составляет, как правило, порядка
Ю-s см, т е близок к размеру коллоидных частиц. Далее, разбав-
ленные эмульсии обычно образуются без введения в систему спе-
циальных эмульгаторов. Тем не менее, как показал опыт, частицы
этих эмульсий обнаруживают электрофоретическую подвижность
и, следовательно, несут электрический заряд. Заряд возникает на
частицах дисперсной фазы таких эмульсий в результате адсорб-
ции ионов неорганических электролитов, которые могут присут-
ствовать в среде в ничтожных количествах. Некоторые исследова-
тели полагают, что в отсутствие чужеродных электролитов на
поверхности капелек таких эмульсий могут адсорбироваться гидр-
оксильные или водородные ионы, всегда присутствующие в воде
в результате диссоциации ее молекул. Наконец, разбавленные
эмульсии по свойствам более, чем все остальные эмульсии, сходны
369
а
5
с лиофобными золями. Именно с разбавленными эмульсиями про-
водил свои работы Повис, который установил подчинение эмуль-
сий правилу Шульце — Гарди и существование у их частиц кри-
тического электрокинетического потенциала. Помимо заряда агре-
гативной устойчивости разбавленных эмульсий способствует еще
чрезвычайно малая численная концетрация этих
систем, обусловливающая очень редкие столк-
новения капелек.
К концентрированным эмульсиям
относятся системы жидкость — жидкость со срав-
нительно значительным содержанием дисперс-
ной фазы, вплоть до 74 объемн.% (рис. XII, 1а).
Эту концентрацию часто указывают как макси-
мальную для эмульсий этого класса потому,
что она в случае монодисперсной эмульсии со-
ответствует максимально возможному объем-
ному содержанию недеформированных сфериче-
ских капель независимо от их размера. Для
полидисперсных эмульсий указанный предел,
конечно, является условным, так как в таких
эмульсиях маленькие капельки могут разме-
щаться между большими, как это показано на
рис. XII, 16.
Так как концентрированные эмульсии полу-
чаются обычно методом диспергирования, то
размер их капелек относительно велик и состав-
ляет 0,1—1 мкм и больше. Такие капельки хо-
рошо видны под обычным микроскопом, и кон-
центрированные эмульсии должны быть отнесе-
ны к микрогетерогенным системам. Капельки
концентрированных эмульсий также совершают
броуновское движение тем более интенсивное,
чем меньше их размер. Концентрированные
эмульсии легко седиментируют, причем седимен-
тация происходит тем быстрее, чем больше раз-
ница между плотностями дисперсной фазы и
дисперсионной среды. Если дисперсная фаза обладает меньшей
плотностью, чем среда, то наблюдается всплывание капель дис-
персной фазы.
Агрегативная устойчивость концентрированных эмульсий мо-
жет быть обусловлена различными причинами в зависимости от
природы эмульгатора. Поэтому этот вопрос целесообразно обсу-
дить при рассмотрении типов эмульгаторов, применяемых для
получения эмульсий.
К высокою концентрированным, или желатиниро-
ванным, эмульсиям обычно относят системы жидкость — жид-
кость с содержанием дисперсной фазы выше чем 74 объемн.%.
Отличительной особенностью таких эмульсий является взаимное
6
Рис. XII, 1. Упа-
ковка капелек в
эмульсиях с боль-
шим содержанием
дисперсной фазы:
а—моно дисперсная
эмульсия, содержащая
<74 объемы. % диспер-
сной фазы, б —поли-
дисперсная эмульсия;
в —желатинированная
эмульсия.
370
деформирование капелек дисперсной фазы, в результате чего они
приобретают форму многогранников (полиэдров), разделенных
тонкими пленками — прослойками дисперсионной среды. Такая
эмульсия при рассматривании в микроскоп, как это видно из
рис. XII, 1в, напоминает соты. Вследствие плотной упаковки капе
лек высококонцентрированные эмульсии не способны к седимен-
тации и обладают механическими свойствами, сходными со свой-
ствами гелей. Последняя особенность и привела к тому, что
высококонцентрированные эмульсии иногда называют желатини-
рованными.
Высококонцентрированные эмульсии в некоторых условиях
можно приготовить с очень большим содержанием дисперсной
фазы и соответственно с ничтожным содержанием дисперсионной
среды. Например, эмульгируя бензол в 1%-ном растворе олеата
натрия, удается получить эмульсию, содержащую выше
99 объемн.% диспёрсной фазы. В таких предельно концентриро-
ванных эмульсиях раствор эмульгатора находится между части-
цами дисперсной фазы в виде тончайших пленок. По данным
Л. Я. Кремнева, толщина таких пленок может достигать 100 А и
даже меньше в зависимости от природы эмульгатора.
Естественно, что особые механические свойства высококонцен-
трированных эмульсий проявляются в тем большей степени, чем
выше их концентрация. Так, подвижность эмульсий м/в с содер-
жанием дисперсной фазы, немного превышающим 74 объемн.%,
еще достаточно высока. Эмульсии же, содержащие 95% углеводо-
рода, обладают уже свойствами, подобными свойствам геля, на-
пример их можно резать ножом.
Агрегативная устойчивость эмульсий и природа эмульгатора
Эмульсии, как и все коллоидные и микрогетерогенные системы,
агрегативно неустойчивы из-за избытка свободной энергии на
межфазной поверхности. Агрегативная неустойчивость эмульсий
проявляется в самопроизвольном образовании агрегатов капелек
с последующим слиянием (коалесценцией) отдельных капелек
друг с другом. В пределе это может приводить к полному разру-
шению эмульсии и разделению ее на два слоя, из которых один
соответствует жидкости, образующей в эмульсии дисперсную фазу,
а другой — жидкости, являющейся дисперсионной средой.
Агрегативную устойчивость эмульсий характеризуют либо ско-
ростью расслаивания эмульсии, либо продолжительностью суще-
ствования (временем живни) отдельных капелек в контакте друг
с другом или с межфазной поверхностью.
По первому методу находят объем дисперсной фазы (или дис-
персионной среды), отслоившейся за определенное время с мо-
мента приготовления эмульсии. Откладывая значения объема, вы-
раженного в процентах от объема всей эмульсии, по оси орди-
нат, а соответствующее время по оси абсцисс, можно получить
371
кинетическую кривую, характеризующую устойчивость эмульсии..
Так как при разрушении эмульсии в цилиндрическом сосуде объем
выделившейся фазы пропорционален высоте соответствующего"
слоя жидкости, то вместо объемов можно пользоваться высотой
образовавшегося слоя той или иной фазы.
Если количество выделившейся фазы пропорционально вре-
мени, то устойчивость эмульсии- -можно также характеризовать-
временем ее существования т. Это время можно найти по урав-
нению:
х = Н!и (XII, 1>
где Н — высота столба эмульсии; и — скорость выделения фазы.
Время жизни капельки эмульсии определяют по времени суще-
ствования капли у межфазной поверхности обеих жидкостей. На-
пример, если хотят определить время жизни капли бензола в воде
в присутствии эмульгатора, то<
о слой бензола наслаивают на слой
воды и с помощью пипетки осто-
рожно наносят каплю воды на
межфазную поверхность со сто-
роны бензола, как это показано
на рис. XII, 2а. Подобным же об-
разом наносят каплю бензола на
межфазную поверхность со сто-
роны водного слоя с помощью пи-
петки с загнутым концом снизу
(рис. XII, 26). В обоих случаях
Рис. XII, 2. Определение времени
жизни капель эмульсии.
регистрируют время, которое про-
ходит с момента нанесения капли
до момента ее коалесценции с
соответствующей фазой. Это вре-
мя обычно связано с устойчивостью эмульсии. Если при получении
эмульсии применяли водорастворимый стабилизатор, устойчивость
капли бензола у меж"фазной границы в водном растворе стабили-
затора всегда выше устойчивости капли раствора стабилизатора
в бензоле. Это показывает, что при применении водорастворимого
эмульгатора образуется эмульсия типа м/в. Если применяли эмуль-
гатор, растворимый в бензоле, результаты обратные, т. е. при та-
ком стабилизаторе образуется эмульсия типа в/м. О причинах об-
разования в первом случае эмульсии типа м/в, а во втором — типа
в/м будет сказано ниже.
На агрегативную устойчивость эмульсий сильнее всего влияют
природа и содержание в системе эмульгатора. С термодинамиче-
ской точки зрения эмульгатор, адсорбируясь на межфазной гра-
нице, понижает межфазное поверхностное натяжение и в отдель-
ных случаях может приводить даже к образованию равновесных
коллоидных систем (эмульсии, получаемые из эмульсолов). Другое
объяснение заключается в том, что при наличии стабилизатора на
372
границе раздела фаз между капельками возникают силы отталки-
вания (энергетический барьер). Повышение в известных пределах
концентрации эмульгатора в системе способствует устойчивости,
эмульсии.
Природа эмульгатора определяет не только устойчивость, но и
тип эмульсии. Опыт показывает, что гидрофильные эмульгаторы,
лучше растворимые в воде, чем в углеводородах, способствуют об-
разованию эмульсии типа м/в, а гидрофобные (или олеофильные)
эмульгаторы, лучше растворимые в углеводородах, — эмульсии-
типа в/м (правило Банкрофта). Это вполне понятно, так как
эмульгатор препятствует слипанию, или коалесценции, капелек,
только тогда, когда он находится у поверхности с наружной сто-
роны капельки, т. е. лучше растворяется в дисперсионной среде.
В качестве эмульгаторов могут применяться самые различные
по природе вещества: поверхностно-активные вещества, молекулы-
которых содержат ионогенные полярные группы (мыла в широком
смысле слова), неионогенные поверхностно-активные вещества,
высокомолекулярные соединения. Эмульгирующей способностью,
обладают даже порошки. Стабилизация более или менее концен-
трированных эмульсий с помощью обычных неорганических элек-
тролитов невозможна вследствие недостаточной адсорбции их
ионов на межфазной границе неполярный углеводород — вода.
Эффективность эмульгатора характеризуют специальным чис-
лом— гидрофил ьно-.липофил ьным балансом (ГЛБ).
Если число ГЛБ лежит в пределах 3—6, образуется эмульсия в/м.
Эмульгаторы с числом ГЛБ 8—13 дают эмульсию м/в. Изменяя-
природу эмульгатора и его концентрацию, можно добиться обра-
щения фаз эмульсии. Более подробно о ГЛБ сказано в гл. XIII.
Стабилизующее действие мыл и мылоподобных веществ на-
эмульсии типа м/в в настоящее время объясняется несколькими 1
факторами устойчивости. /
Первый фактор — электрический заряд, возникший на поверх-
ности капелек эмульсий, стабилизованных ионогенными мылами
при адсорбции органических ионов мыла. В результате образуется
двойной электрический слой, аналогичный тому, который суще-
ствует на поверхности частиц типичных гидрофобных золей. Этот-
двойной слой и обуславливает устойчивость эмульсий. Поэтому
прямые эмульсии, стабилизованные ионогенными мылами, харак-
теризуются всеми свойствами, присущими типичным гидрозолям,,
т. е. для них соблюдается правило Шульце — Гарди, возможность,
перезарядки частиц эмульсий с помощью поливалентных ионов-
и т. д.
Для того чтобы происходила адсорбция органического иона,
он должен хорошо адсорбироваться дисперсной фазой, т. е. иметь,
достаточно длинную углеводородную цепь. Поэтому эмульсии типа
м/в могут быть стабилизованы только сравнительно высокомоле-
кулярными мылами (щелочными солями лауриновой и более вы-
сокомолекулярных жирных кислот). Отсутствие эмульгирующей
37а
•способности у щелочных солей жирных' кислот, являющихся низ-
шими членами гомологического ряда, объясняется, вероятно, также
и тем, что при высоких концентрациях, необходимых для адсорб-
ции таких мыл, начинает уже преобладать коагулирующее дей-
\ ствие иона щелочного металла.
Второй фактор устойчивости концентрированных эмульсий типа
'м/в заключается в образовании на поверхности их капелек струк-
турированных гелеобразных слоев эмульгатора, обладающих вы-
сокой структурной вязкостью и прочностью при одновременной гид-
ратированности. Этот фактор, особенно подробно рассмотренный
в работах П. А. Ребиндера и А. Б. Таубмана, приобретает еще
большее значение для высококонцентрированных эмульсий.
А. Б. Таубманом с сотр. показано, что устойчивые эмульсии
могут образовываться также в результате возникновения на по-
поверхности капелек основной эмульсии нескольких слоев ми-
крокапелек, служащих структурно-механическим барьером. Такие
микрокапельки возникают вследствие явлений турбулентности у
поверхности капелек основной эмульсии, обладающей малым меж-
фазным натяжением.
Устойчивость эмульсий типа в/м, стабилизованных мылами с
поливалентным катионом, ранее объяснялась главным образом на-
личием на поверхности капелек эмульсии структурно-механического
барьера. Объяснение же устойчивости эмульсий типа в/м суще-
ствованием на межфазной поверхности двойного электрического
слоя на первый взгляд кажется невозможным вследствие малой
диэлектрической проницаемости дисперсионной среды. Однако, как
уже указывалось (гл. IX, разд. И), в последние годы было пока-
зано, что даже в неполярных средах может происходить некоторая
диссоциация молекул эмульгатора. Соли поливалентных металлов
и органических кислот в углеводородных средах обычно имеют
константы диссоциации порядка 10~8. Следовательно, если, на-
пример, концентрация такой соли в бензоле равна 10 ммоль/л, то
концентрация ионов в растворе будет иметь значение порядка
10'10 н. При таких условиях двойной электрический слой будет,
конечно, очень диффузным: расчеты показывают, что его толщина
должна составлять несколько микрометров. Отсюда емкость двой-
ного слоя в неполярной жидкости должна быть весьма невелика
и нужен очень небольшой заряд для того, чтобы обусловить зна-
чительный поверхностный потенциал. Таким образом, электроста-
тические силы отталкивания могут играть существенную роль и
в устойчивости обратных эмульсий, особенно не очень концентри-
рованных.
Третий фактор, который может обусловливать лишь устойчи-
вость эмульсий второго рода, стабилизованных мылами с полива-
лентным катионом, сводится к адсорбции на поверхности капелек
воды полярных концов достаточно длинных и гибких углеводород-
ных участков молекул мыла, растворенных во внешней неполярной
фазе эмульсии и способных совершать микроброуновское движе-
374
ние. В этом случае устойчивость, очевидно, определяется тепловым
движением и взаимным отталкиванием углеводородных радикалов,
т. е. фактором, носящим энтропийный характер.
Наконец, иногда причина устойчивости обратных эмульсий мо-
жет заключаться в стабилизации капелек полярной дисперсной
фазы «броней» из крупинок, образовавшихся из нерастворимых
солей щелочноземельных металлов или продуктов гидролиза.
В этом случае механизм стабилизации полностью аналогичен меха-
низму стабилизации эмульсий с помощью
порошкообразных эмульгаторов, который
рассмотрен ниже.
В последнее время для стабилизации
прямых эмульсий широко применяют не-
ионогенные эмульгаторы, дифильные мо-
лекулы которых состоят из углеводород-
ного радикала и углеводородной цепи с
расположенными по всей ее длине по-
лярными, но не способными к ионизации
группами, чаще всего гидроксильными
(более подробные сведения о неионо-
генных эмульгаторах даны в гл. XIII).
Устойчивость эмульсий типа м/в, стаби-
лизованных неионогенными эмульгатора-
ми, естественнее всего объяснить следую-
щим образом. Дифильные молекулы
эмульгатора ориентированы на межфаз-
ной границе так, что углеводородные
участки направлены в дисперсную фазу, а
Рис. XII, 3. Поведение раз-
лично сбалансированных ди-
фильных молекул эмуль-
гатора:
а—молекулы с преобладающей
неполяоной частью; б—хорошо
с''а таьс 1рованные молекулы;
в — молекулы с преобладающей
полярной частью.
полярные сильно гидра-
тированные группы в воду, при этом онц либо образуют доста-
точно толстый гидратный слой, обусловливающий расклинивающее
давление, либо совершают микроброуновское движение (энтропий-
ный фактор устойчивости).
Эмульгирующее действие как ионогенных, так и неионогенных
поверхностно-активных веществ тем эффективнее, чем лучше сба-
лансированы полярные и неполярные части молекулы эмульгатора
между обеими фазами эмульсии. Это значит, что дифильная мо-
лекула хорошего эмульгатора должна обладать сродством как
к полярным, так и к неполярным средам. Только при этом усло-
вии молекулы эмульгатора не будут растворяться преимуществен-
но в какой-нибудь одной из фаз и будут находиться на межфаз-
ной поверхности. Сбалансированность молекул эмульгатора в
простейшем случае определяется, с одной стороны, длиной углево-
дородной цепи и с другой — сродством ионогенной или полярной
группы к воде.
Поведение различно сбалансированных дифильных молекул
можно пояснить.схемой, изображенной на рис. XII, 3.
Именно хорошей сбалансированностью объясняется наилуч-
шее стабилизующее действие мыл, содержащих в углеводородной
375-
цепочке от 12 до 18 атомов углерода. Сбалансированностью моле-
кул эмульгатора объясняется и род эмульсий, которые получаются
с применением этого эмульгатора. Так, эмульгаторы, в молекуле
которых действие полярной части превалирует над действием не-
полярной и которые лучше растворяются в воде, образуют эмуль-
сии первого рода. Наоборот, эмульгаторы, у которых действие не-
полярной группы молекулы преобладает над действием полярной
группы и которые лучше растворяются в углеводородах, способ-
ствуют образованию эмульсии
второго рода.
Эмульгирующее действие вы-
сокомолекулярных веществ, та-
ких, как желатин, казеин, поли-
метакриловая кислота, Метилцел-
люлоза, поливиниловый спирт, а
также их действие как защитных
коллоидов, вероятно, можно объ-
яснить энтропийным фактором.
Впрочем, можно также допу-
стить, что прямые эмульсии, ста-
билизованные защитными кол-
лоидами, молекулы которых со-
держат ионогенные группы,
устойчивы благодаря образова-
нию на поверхности капелек
двойного электрического слоя в
результате ионизации этих групп.
В заключение рассмотрим так
называемые твердые эмульгато-
ры и причины их эмульгирующе-
го действия. Твердыми эмульга-
Рис. XII, 4. Расположение частичек
твердого эмульгатора у межфазной
поверхности при образовании капе-
лек эмульсии:
/—гидрофильный эмульгатор (каолин),
II — гидрофобный эмульгатор (сажа).
торами могут служить достаточно высокодисперсные порошки, спо-
собные смачиваться как полярной, так и неполярной жидкостями,
•образующими эмульсию. К таким порошкам относятся глины, гипс,
гидрат окиси железа, сажа.
При встряхивании полярной жидкости с неполярной в присут-
ствии твердого .эмульгатора его крупинки прилипают к межфаз-
ной поверхности, причем большая часть поверхности частиц
эмульгатора находится в той жидкости, которая их лучше смачи-
вает. Таким образом, на капельках образуется как бы «броня»,’
предотвращающая их коалесценцию. Понятно, что если твердый
эмульгатор лучше смачивается водой (например, каолин), такая
броня возникает со стороны водной фазы; при этом образуется
эмульсия типа м/в. Если же твердый эмульгатор лучше смачивается
неполярным углеводородом (например, сажа), то образуется
эмульсия типа в/м. Сказанное иллюстрируется схемой, изображен-
ной на рис. XII, 4. В случаях I а и II б крупинки твердого эмульга-
тора находятся с наружной стороны капелек, и поэтому соответ-
376
ствующие эмульсии оказываются устойчивыми. В случаях I б и II а
крупинки твердого эмульгатора находились бы у межфазной по-
верхности с внутренней стороны капелек, в результате чего обра-
зование таких эмульсий невозможно.
Понятно, что наличие на поверхности капелек одного только
слоя твердого эмульгатора еще недостаточно для агрегативной
устойчивости системы. Капельки такой эмульсии при столкнове-
нии должны были бы слипаться, поскольку всегда существует меж-
фазное натяжение на границе твердый эмульгатор — дисперсион-
ная среда. Очевидно, устойчивость эмульсии в этом случае опре-
деляется возникновением на частицах твердого эмульгатора либо
двойного электрического слоя, либо достаточно толстой сольват-
ной оболочки.
Рассмотренные выше классические представления о роли «бро-
нирования» в устойчивости эмульсий, стабилизованных твердыми
эмульгаторами, значительно расширены А. Б. Таубманом. В его
работах показано, что в реальных условиях высокая устойчивость
эмульсий, стабилизованных твердыми эмульгаторами, определяется
обычно совместным действием твердого высокодисперсного эмуль-
гатора и поверхностно-активного компонента и это стабилизующее
действие обусловлено образованием весьма прочной стабилизи-
рующей оболочки. В этих случаях структурно-механический барьер
непосредственно измерен и сопоставлен с устойчивостью.
Стабилизация эмульсии твердым эмульгатором возможна
только при условии, что размер частиц порошка меньше размера
капелек эмульсии. В то же время слишком мелкие частицы по-
рошка, способные совершать интенсивное броуновское движение,
не прилипают к поверхности капелек и не образуют защитного
слоя. В доказательство этого можно привести известный факт, что
эмульсии типа м/в получают только с помощью золя AS2S3 с до-
статочно крупными частицами. Высокодисперсные золи AS2S3,
равно как и грубый осадок AS2S3, не способны стабилизовать
эмульсии.
Методы получения и разрушения эмульсий
Эмульсии получают механическим диспергированием дисперс-
ной фазы в дисперсионной среде в присутствии соответствую-
щего эмульгатора. Для диспергирования эмульгируемые жидкости
сильно перемешивают, встряхивают или подвергают вибрацион-
ному воздействию. Для этого используют специальные эмульса-
торы, мешалки, коллоидные мельницы. В последнее время для
эмульгирования начинают применять ультразвук. Иногда получен-
ные грубые эмульсии подвергают дополнительной гомогенизации
в специальных гомогенизаторах разнообразных конструкций. Наи-
более часто в качестве гомогенизаторов применяют устройства,
в которых дополнительное диспергирование капелек грубой эмуль-
сии достигается продавливанием ее через малые отверстия под
377
высоким давлением. При обработке в таких гомогенизаторах, на-
пример молока, диаметр жировых капелек понижается с 3 до
0,2 мкм. В результате значительно увеличевшейся седиментацион-
ной устойчивости такое молоко расслаивается значительно мед-
леннее.
Процесс эмульгирования состоит из собственно диспергирова-
ния, т. е. образования капелек дисперсной фазы в дисперсионной
среде и их стабилизации в результате адсорбции на поверхности
эмульгатора. Процесс гомогенизации всегда заключается в обра-
зовании из дисперсной фазы тонких цилиндриков, которые весьма
неустойчивы и легко распадаются на ряд капелек. Как известно
из молекулярной физики, цилиндрик жидкости начинает распа-
даться на капельки, когда его длина становится больше окружно-
сти его сечения.
Следует учесть, что при эмульгировании наряду с диспергиро-
ванием всегда в той или иной степени происходит коалесценция
возникших капелек, так как эмульгатор не успевает полностью
адсорбироваться на поверхности капелек и они еще не обладают
той устойчивостью, которая соответствует устойчивости капелек
в готовой эмульсии. П. А. Ребиндер показал, что при эмульгирова-
нии всегда образуется два типа эмульсии — м/в и в/м и только
вследствие большей устойчивости «выживает» та эмульсия, кото-
рая соответствует природе примененного эмульгатора.
Влияние эмульгатора на образование эмульсии того или иного
рода становится более сложным, когда эмульгатор способен да-
вать как эмульсию м/в, так и в/м. В этом случае на род образую-
щейся эмульсии может влиять природа стенок сосуда и мешалки,
равно как и другие предметы, с которыми соприкасается эмульсия.
Например, если стенки сосуда смачиваются только какой-нибудь
одной жидкостью, то соприкосновение эмульсии с этой стенцой
может приводить к обращению типа эмульсии, причем жидкость,
смачивающая стенки сосуда, становится дисперсионной средой.
На результат эмульгирования влияет не только природа при-
мененного эмульгатора и вид механического воздействия, но и ряд
других условий — температура, количественное соотношение фаз
и т. д. При всех прочих равных условиях более низкоконцентриро-
ванная эмульсия получается более устойчивой, так как вероятность
столкновения двух ее частиц меньше.
Таким образом, эмульгирование представляет собой весьма
сложный процесс и для приготовления стойких высокодисперсных
эмульсий от технолога требуется много знаний и опыта.
Помимо механического диспергирования эмульсии могут быть
получены путем самопроизвольного диспергирования, механизм ко-
торого был рассмотрен в гл. VIII. Однако при самодиспергирова-
нии полученные весьма высокодисперсные равновесные системы
резко отличаются по термодинамической устойчивости от обычных
эмульсий, агрегативная устойчивость которых является временной.
Поверхностно-активные вещества, применяемые при самопроиз-
378
вольном диспергировавши, также не могут рассматриваться как
типичные эмульгаторы, поскольку их берут в таких количествах,
что происходит изменение объемных свойств фазы.
Самопроизвольное эмульгирование играет существенную роль
в процессах, связанных с перевариванием и усвоением пищи орга-
низмом. При попадании, например, в кишечник жира сначала про-
исходит самодиспергирование жира под влиянием поверхностно-
активных веществ (холевых кислот), содержащихся в желчи, а за-
тем полученная таким образом высокодисперсная эмульсия вса-
сывается через стенку кишечника в организм.
Часто перед технологом стоит задача не получить эмульсию,
а наоборот, предупредить ее возникновение или разрушить (д е-
эмульгировать) уже образовавшуюся систему. Эмульсии типа
м/в, полученные с применением ионогенных эмульгаторов, обычно
разрушают с помощью коагуляции электролитами с поливалент-
ными ионами. Так как такие электролиты, взаимодействуя с ионо-
генной группой эмульгатора, обычно дают соединения, нераство-
римые Ё воде, то введение их в систему равнозначно переводу
эмульгатрра в неактивную форму. Иногда для деэмульгирования
эмульсий, полученных с применением ионогенных эмульгаторов,
вводят в систему эмульгатор, способствующий образованию эмуль-
сии обратного типа и таким образом как бы нейтрализующий дей-
ствие первоначального эмульгатора. Следует заметить, что такой
эмульгатор практически всегда образуется при введении электро-
литов с поливалентным катионом в эмульсии типа м/в, стабилизо-
ванные щелочными мылами, так как образующиеся при этом мыла
с поливалентными катионами способствуют образованию эмульсий
типа в/м.
Эмульсии, стабилизованные неионогенными стабилизаторами,
разрушаются гораздо труднее. Электролиты разрушают такие
эмульсии только при больших концентрациях, когда происходит
уже не коагуляция, а высаливание. Более эффективным способом
разрушения таких эмульсий является нагревание, вызывающее
десорбцию молекул неионогенных стабилизаторов с капелек эмуль-
сии или дегидратацию полярной части молекулы неионогенного
стабилизатора.
Разрушение всех эмульсий можно достичь введением в систему
поверхностно-активного вещества, вытесняющего из адсорбцион-
ного слоя эмульгатор, но неспособного стабилизовать эмульсию.
Именно на этом основана возможность разрушения некоторых
эмульсий первого рода введением в них амилового спирта. Эмуль-
сии можно также разрушить путем центрифугирования, фильтро-
вания, электрофореза. При центрифугировании и фильтровании
происходит собственно концентрирование эмульсии. Однако в
эмульсиях с очень высокой концентрацией дисперсной фазы и не-
достаточным содержанием эмульгатора, как правило, происходит
коалесценция капелек, и таким образом система разрушается.
С. С. Воюцкйм с сотр. разработан метод непрерывного разрушения
379
эмульсий воды в углеводородах путем пропускания эмульсий через
специальный фильтр. Капельки дисперсной фазы (воды) адсорби-
руются на фильтрующем материале, коалесцируют на его поверх-
ности и стекают с фильтра (самоочищение фильтра). Разрушение
эмульсий при повышении температуры обусловливается уходом
эмульгатора с поверхности капелек в результате его десорбции
или растворения в дисперсной фазе.
Обращение фаз эмульсий
Рассмотрим так'называемое явление обращения фаз, весьма
характерное для эмульсий. При введении в эмульсию в условиях
интенсивного перемешивания большого количества поверхностно-
активного вещества, являющегося стабилизатором эмульсий про-
тивоположного типа, первоначальная эмульсия может обра-
щаться, т. е. дисперсная фаза становится в ней дисперсионной
Рис. XII, 5. Схема обращения эмульсии типа м/в в эмульсию
типа в/м.
средой, а дисперсионная среда — дисперсной фазой. Так, эмульсия
типа м/в, стабилизованная олеатом натрия, может быть превра-
щена в эмульсию типа в/м путем введения в систему при сильном
перемешивании олеата кальция. Такое же явление наблюдается и
при введении в эмульсию веществ, способных изменять природу
эмульгатора, например, при введении хлорида кальция в эмульсию
первого рода, стабилизованную олеатом натрия.
Интересно, что обращение эмульсий в определенных условиях
может быть вызвано и длительным механическим воздействием.
Так, сбивание сливок ведет к получению масла. При этом эмуль-
сия типа м/в (сливки) переходит в эмульсию типа в/м (масло) со
сравнительно весьма малым содержанием воды в виде дисперсной
фазы.
Наблюдения под микроскопом показали, что при обращении
фаз капельки дисперсной фазы сначала растягиваются, превра-.;
щаются в пленки и затем образовавшиеся пленки охватывают дис-
380
Рис. ХП, 6.' Микрофотография мно-
жественной эмульсии.
Природной эмульсией является
к эмульсиям помимо молочных
-персионную среду первоначальной эмульсии, которая в результате
этого становится дисперсной фазой. На рис. XII, 5 наглядно можно
видеть эти этапы при переходе слева направо. Интересно, что
в результате неравномерного распределения эмульгатора в раз-
личных микроучастках системы, при обращении фаз могут воз-
никать так называемые «множественные> эмульсии, в которых,
например, капельки масла эмуль-
сии первого рода содержат в себе
мельчайшие капельки воды. Ми-
крофотография такой множе-
ственной эмульсии представлена
на рис. XII, 6.
Практическое значение
эмульсий и эмульгирования
К природным эмульсиям отно-
сится ряд ценнейших раститель-
ных и животных продуктов. Так,
эмульсией является молоко —
стабилизованная животными бел-
ками эмульсия жиров в воде. Мо-
локо является сырьем молочной
промышленности и служит для
получения множества молочных
продуктов — сливок, простоква-
ши, кефира, масла, сыра и т. д.
также яичный желток.
В пищевой промышленности
продуктов принадлежат такие продукты, как маргарин, майонез,
различные соусы. В фармацевтической промышленности многие
лекарства применяются в виде эмульсий, причем, как правило,
для приема лекарств внутрь применяются эмульсии первого рода,
а эмульсии второго рода используются для наружного примене-
ния.
3. ЛАТЕКСЫ
По своим свойствам к эмульсиям с водной дисперсионной средой прибли-
жаются натуральные и синтетические латексы, очень широко применяемые в на-
родном хозяйстве для получения эластичных пленок, шаров-пилотов, для изго-
товления эластичных пористых материалов, заменителей кожи, для придания
водонепроницаемости тканям, для пропитки корда в шинной промышленности
и т. д. Натуральные латексы представляют собой млечный сок бразильской
гевеи, синтетические латексы получают путем полимеризации в водной среде не-
предельных углеводородов — бутадиена, хлоропрена и других мономеров. Часто
латексы изготовляют путем сополимеризации двух или даже нескольких мономе-
ров (бутадиен-стирольные латексы, бутадиен-акрилонитрильные латексы и т. д.).
Латексы, как и эмульсии, содержат микроскопические или ультрамикроско-
пические частицы (глобулы), приближающиеся по форме к сферическим, на
поверхности которых адсорбирован стабилизатор — соединения типа белков для
381
натурального латекса и мыла или другие поверхностно-активные вещества у син-
тетических латексов. Вещество дисперсной фазы латексов состоит из каучука,
макромолекулы которого представляют собой гибкие углеводородные цепи, не
содержащие или содержащие полярные группы В этих цепях имеется всегда
некоторое количество непредельных связей, обусловливающих возможность вул-
канизации каучуков, т. е. сшивания макромолекул по месту ненасыщенных свя-
зей с помощью серы или других вулканизующих агентов.
Здесь уместно указать, что стабилизованные мылами или мылоподобными
веществами синтетические латексы являются хорошими моделями ионностабили-
зованных коллоидных систем. Это объясняется следующими свойствами латек-
сов.
Химический состав водной фазы (дисперсионной среды} синтетических ла-
тексов сравнительно прост, а дисперсная фаза обычно состоит из достаточно
инертного в химическом отношении и в большинстве случаев гидрофобного
вещества. Поэтому едва ли можно ожидать, что при астабилизации этих систем
на поверхности частиц-могут происходить какие-нибудь реакции, за исключением
тех хорошо изученных 'реакций, в которых участвует стабилизатор. У латексов
с гидрофобным полимером сольватация дисперсной фазы, которая может влиять
на устойчивость коллоидной системы, безусловно, отсутствует. Сферическая или
близкая к сферической форма частиц устраняет влияние на их взаимодействие
неровностей поверхности и позволяет считать, что при столкновении двух глобул
они ведут себя как два идеальных шарика. Дисперсная фаза латексов, как
правило, является диэлектриком, и при электрофорезе можно не учитывать по-
правку на проводимость частиц. Большая вязкость полимеров позволяет рас-
сматривать латексные глобулы как твердые частицы. Это значительно упрощает
трактовку экспериментальных результатов, так как такие частицы не могут де-
формироваться под влиянием движения окружающей жидкости. Наконец, весьма
существенно, что синтетические латексы можно получать с применением почти
любого эмульгатора. Это представляет огромное удобство для экспериментатора,
изучающего влияние на свойства латекса природы стабилизующих веществ.
Всеми этими свойствами и объясняется широкое применение латексов в по-
следнее время в исследовательских лабораториях.
Ниже мы остановимся только на работах, проведенных с использованием
латексов, стабилизованных мылами и неионогеиными поверхностно-активными
веществами, представляющих лишь общий интерес для коллоидной химии.
Несмотря на то, что вопрос о связи между ^-потенциалом и агрегативной
устойчивостью лиофобных коллоидных систем чрезвычайно сложен, нельзя от-
рицать мнение ряда ведущих ученых о том, что электрокинетический потенциал
до сих пор остается одной из важнейших характеристик устойчивости лиофоб-
ных коллоидов. На значение ^-потенциала для устойчивости коллоидных систем,
и в частности латексов, указывали Кройт, Овербек, Гаузер и др.
С. С. Воюцкий и Р. М. Панич еще в начале пятидесятых годов исследовали
влияние на устойчивость и одновременно на ^-потенциал латексов таких факто-
ров, как концентрация латексов, pH среды и валентность коагулирующего иона.
Исследования проводили как с иедиализованными, так и диализованными син-
тетический латексами, стабилизованными олеатом и нафтенатом аммония.
Электрофоретическую подвижность определяли с помощью макроэлектрофореза,
поскольку в задачу работы входило определение зависимости ^-потенциала от
концентрации дисперсной фазы. Электрокинетический потенциал вычисляли по
формуле Генри, причем численный коэффициент подбирали в соответствии с
известным критерием ха, пользуясь графиками Овербека.
Опыты показали, что при разбавлении латексов вероналовым буфером с
pH = 7,7, одинаковым с pH исходных латексов, абсолютные значения Е-потенциа-
ла сначала возрастали, достигали 75 4- 85 мВ, а затем при содержании сухого
остатка в латексах менее 0,02—0,1 % падали. Однако даже при весьма больших
разбавлениях, когда уже трудно вести наблюдения за электрофорезом, значения
электрокииетического потенциала не были меньше —50 мВ.
Начальное возрастание отрицательного электрокииетического,потенцияла при
разбавлении буферной смесью е постоянной ионной силой С. С. Воюцкий объ-
ясняет только уменьшением ионной силы системы вследствие снижения содержа-
382
ния в единице ее объема числа глобул, которые следует рассматривать в этом
случае как поливалентные ионы. Падение абсолютной величины ^-потенциала
при сильном разбавлении латексов, отмечавшееся и другими авторами (Боулер,
Мунро, Сексмис), следует объяснить десорбцией стабилизатора с поверхности
глобул. Правильность подобного объяснения была подтверждена установлением
перехода молекул мыла с поверхности дисперсной фазы в межглобулярную
жидкость. Однако было найдено, что заметная десорбция мыла с поверхности
глобул происходит только лишь при содержании в латексе сухого остатка менее
0,02%. В достаточно концентрированных латексах как недиализованных, так и
диализованных почти все мыло (свыше 99%) находится в адсорбированном
состоянии, причем слой мыла на глобулах ненасыщен.
Латексы не флокулировали и не коагулировали при любых, сколь угодно
больших разбавлениях. Флокуляция и коагуляция не наблюдались и при стоянии
разбавленных латексов в течение
двух суток. Однако прн введении „г
даже в сильно разбавленные ла-
тексы хлорида кальция тотчас на-
ступала коагуляция. Это говорит
о том, что устойчивость сильно
разбавленных латексов опреде-
ляется не частотой столкновения
частиц друг с другом, а достаточ-
ной их защищенностью стабилиза- °
тором.
При постепенном добавлении
0,1 и. НС1 в стабилизованные мы- но
лами концентрированные латексы _ „
происходила их полная коагуля- Рис. XII, 7. Зависимость ^-потенциала ла-
цня. Быстрое же введение доста- тексных глобул (кривая 1) и удельной элек-
точно большого количества кисло- тропроводности у латекса АПД-2 (кривая 2)
ты в латексы, разбавленные до от среды (по С. С. Воюцкому).
3—4%-ного содержания в них су-
хого остатка, не вызывало коагуляции или вызывало ее лишь частично. Это
явление оказалось совершенно неожиданным, так как шло вразрез с обычными
представлениями о том, что латексы, стабилизованные мылами (не амфотерными
веществами), не могут быть устойчивыми прн низких pH. Определение знака
заряда частиц в подкисленных латексах показало, что глобулы в этом случае
претерпевают перезарядку и становятся положительно заряженными.
На рис. XII, 7 представлено изменение электрокинетического потенциала
глобул диализованного синтетического латекса, содержащего 2% сухого остатка,
в зависимости от pH среды при ионной силе, равной 0,01. Электрокннетнческнй
потенциал прн повышении активной кислотности системы в некотором диапа-
зоне pH не изменяется, затем начинает уменьшаться, очевидно, в результате
перехода мыла с поверхности глобул в слабо ионизированную кислоту, дости-
гает нуля при pH = 3,9 и, наконец, принимает положительное значение в сильно
кислых средах, вероятно, за счет адсорбции ионов водорода. Существенно, что
положительный потенциал глобул в кислой среде по абсолютному значению
намного меньше, чем в щелочной, что, безусловно, связано с различной приро-
дой и концентрацией ионов, стабилизующих частицы в кислой и щелочной
средах.
Как показал эксперимент, с ростом абсолютного значения ^-потенциала
растет и агрегативная устойчивость латексов Повышение устойчивости латек-
сов с увеличением отрицательного ^-потенциала общеизвестно. Но подобная же
зависимость наблюдается и для перезаряженных латексов с положительно заря-
женными частицами При pH = 3,9 в изоэлектрическом состоянии стабилизован-
ные латексы тотчас коагулируют. При pH — 3,1 идет процесс скрытой коагуля-
ции, переходящий за сутки в явную. При pH = 2,7 явной коагуляции не про-
исходит в течение 25 дней.
В заключение отметим, что перезаряженные глобулы латексов обладают по-
вышенной способностью адсорбироваться отрицательно заряженными поверхио-
383
стями, например хлопчатобумажным волокном, и, следовательно, могут быть с
успехом использованы для проклеивания бумаги и картона н пропитывания тка-
ней. Эти выводы полностью совпадают с литературными указаниями *.
При изучении влияния катионов различной валентности на электрокинети-
ческнй потенциал латексных глобул было установлено, что для латексов с от-
рицательно заряженными частицами соблюдается правило Шульце—Гарди,
которому подчиняются лиофобные коллоидные системы. На рис. XII. 8 приведены
результаты электрофоретических исследований диализованного синтетического
латекса, содержащего 1 % сухого остатка. Последние точки на кривой, отмечен-
ные стрелками, соответствуют предельной концентрации электролита, при кото-
рой еще можно провести электрофорез.
Некоторое повышение потенциала при введении небольших количеств хло-
рида натрия наблюдали и другие исследователи (Гаузер и Бендер, Марон с
сотр). Причина подобного явления заключается, возможно, в адсорбции глобу-
Рис. XII, 8. Влияние ионной силы / на
g-потенцнал глобул латекса В1Д1 при
введении в него различных электроли-
тов:
1 — NaCl; 2—СаС12; 3 —А1С13.
ненасыщенности адсорбционного слоя на
чни вокруг частиц двухмерных студней
ламн анионов, присутствующих в си-
стеме. Падение потенциала при повы-
шении концентрации хлорида натрия
объясняется, конечно, сжатием двой-
ного электрического слоя. Этим же
объясняется и резкое снижение по-
тенциала прн введении хлоридов каль-
ция и алюминия. Прибавление боль-
ших количеств хлорида алюминия,
как можно видеть на рисунке, ведет
к перезарядке глобул.
В последние годы довольно ши-
рокое распространение получило мне-
ние, что основную роль в агрегатив-
ной устойчивости обычных латексов
играет структурно-механический фак-
тор. Однако эту точку зрения приме-
нительно к латексам, стабилизован-
ным мылами, нельзя считать правиль-
ной. Было показано, что поверхность
глобул стабилизованных латексов
обычно покрыта слоем эмульгатора
лишь на 30—40%. При значительной
поверхности глобул говорить о нали-
и о их структурно-механических свой-
ствах едва ли возможно. Устойчивость латексов, стабилизованных мылами, опре-
деляется, в основном, действием отталкивающих сил между двойными электриче-
скими слоями, возникающих при перекрытии ионных атмосфер. При этом
собственно стабилизующей частью молекулы стабилизатора является ее гидрати-
рованные ионизированные группы, а роль углеводородного радикала сводится
к фиксации молекулы стабилизатора иа межфазной поверхности полимер —
вода. ।
Другим доказательством двойного электрического слоя как основной при-
чины устойчивости водных дисперсий полимеров, стабилизованных мылами, яв-
ляется тот факт, что стабильные латексы можно получить с помощью эмульга-
торов, не способных давать механически прочные адсорбционные пленки (напри-
мер, с помощью некалей).
Все сказанное выше ни в коем случае не снижает значения структурно-меха-
нических свойств адсорбционного слоя как причины агрегативной устойчивости
вообще и латексов в частности. В определенных условиях при образовании на
* Некоторые сведения о взаимодействии частиц латексов с волокном можно
найти В монографии: С. С. Воюцкий. Физико-химические основы пропитыва-
ния и импрегнирования волокнистых материалов дисперсиями полимеров. Л.,
«Химия», 1969. 336 с,
384
поверхности частиц достаточно прочного и мощного слоя гидратированного ста-
билизатора структурно-механические свойства этого слоя могут иметь решающее
значение для стабильности системы.
Согласно Р. Э. Нейману, с увеличением плотности адсорбционных слоев
происходит все большая замена двойного электрического слоя сильно развитыми
гидратными оболочками на поверхности частиц. Таким образом, имеет место
переход от систем, стабилизованных двойным электрическим слоем, к системам,
стабильность которых обусловлена структурно-механическим барьером. Иначе
говоря, при увеличении адсорбции поверхностью латексных глобул происходит не
только количественное, но и качественное изменение механизма стабилизации.
Возникает новый по своей природе энергетический барьер, препятствующий коа-
гуляции, близкий к представлениям П. А. Ребиндера об образовании структури-
рованных гелеобразных слоев эмульгатора. Электрический заряд двойного элек-
трического слоя при этом уменьшается или исчезает совсем благодаря тесному
контакту ионогенных групп и возрастанию ионной силы. На неэлектростатиче-
скую природу стабилизующего барьера в этом случае, согласно Р. Э Нейману,
указывает и то, что коагуляция адсорбционно насыщенных латексов не подчи-
няется закономерностям, характерным для латексов, частицы которых несут
двойной электрический слой. Очевидно, существует иной, неэлектростатический
механизм стабилизации, связанный со структурой и гидратацией плотно упако-
ванных насыщенных слоев эмульгатора.
Как было указано, сравнительно недавно широкое распространение у нас
и за рубежом получили неионогенные поверхностно-активные вещества, моле-
кулы которых имеют несколько неионогенных полярных групп, например —ОН.
Гидрофобными частями молекул этих веществ являются углеводородные ради-
калы алифатических спиртов или алкилфенолов.
В СССР первые работы по исследованию свойств латексов, стабилизован-
ных неионогенными поверхностно-активными веществами, выполнены Р. М. Па-
нич и С. С. Воюцким с сотрудниками еще в 1961 г. В этих исследованиях ими
было установлено, что латексы, полученные с применением неионогенных по-
верхностно-активных веществ, представляющих собой продукты сополимеризаций
моно- и диалкилфенолов с достаточными количествами окиси этилена, вполне
устойчивы к действию электролитов, что имеет немаловажное практическое зна-
чение. Латексы с более гидрофильными стабилизаторами, имеющими длинную
оксиэтиленовую цепь, оказались устойчивыми к интенсивному перемешиванию,
тогда как в латексе с более гидрофобным стабилизатором при перемешивании
образуется коагулят. Разбавленные латексы с неионогенными эмульгаторами
обладают небольшим отрицательным электрокинетическим потенциалом. При-
чина этого явления, по мнению авторов, заключается в адсорбции латексными
глобулами посторонних ионов, присутствующих в системе. Абсолютное значение
отрицательного электрокинетического потенциала латексных глобул с неионоген-
ными стабилизаторами возрастает с увеличением pH среды Это указывает на
то, что адсорбирующимися ионами, обусловливающими заряд, могут являться
гидроксильные ионы.
Введением А1С13 были получены латексы в изоэлектрическом состоянии, при-
чем в них не происходило явной коагуляции. Это указывает на то, что их
устойчивость обусловлена не электростатическими силами, а в основном гидра-
тацией полярных участков цепей стабилизатора. Однако агрегативная устойчив
вость латексов, содержащих неионогенный стабилизатор, в изоэлектрическом со-
стоянии ниже, чем агрегативная устойчивость исходных латексов. Таким обра-
зом, заряд латексных глобул, обусловленный адсорбцией ионов, все же способ-
ствует повышению устойчивости латексов.
Стабилизирующая роль гидратации, вероятно, заключается в том, чтобы
обеспечить эффективное отталкивание при взаимодействии таких гидратирован-
ных слоев (расклинивающее давление).
Процесс скрытой коагуляции и образования геля под действием электроли-
тов у латексов, стабилизованных неионогенными поверхностно-активными веще-
ствами, наступает только при повышенной температуре. Скорость коагуляции
тем выше, чем интенсивнее дегидратирующие факторы: температура, концентра-
ция электролита, тип коагулирующего иона.
13 Зак. 664
385
4. ПЕНЫ
Типичные пены представляют собой сравнительно весьма гру-
бые высококонцентрированные дисперсии газа (обычно воздуха)
в жидкости. Пузырьки газа в таких системах имеют размер по-
рядка несколько миллиметров, а в отдельных случаях и санти-
метров. Благодаря избытку газовой фазы и взаимному сдавлива-
нию пузырьки пены имеют не сферическую форму, а представляют
собой полиэдрические ячейки, стенки которых состоят из весьма
тонких пленок жидкой дисперсионной среды. Пленки пены часто
обнаруживают интерференцию; это свидетельствует о том, что их-
толщина соизмерима с длиной световых волн.
В результате того, что пена состоит из таких полиэдрических
ячеек, она имеет сотообразную структуру. Плато установил, что
в соответствии с требованием минимума свободной поверхностной
энергии на одном'жидком ребре ячейки всегда сходятся три
пленки, образующие между собой равные углы в 120°, и что в од-
ной точке могут сходиться лишь четыре ребра. Большой размер
отдельных газовых пузырьков и тесное расположение их в пене
исключают возможность броуновского движения. Кроме того, в ре-
зультате особой структуры устойчивые пены обладают некоторой
жесткостью или механической прочностью. Вообще, по строению
обычные пены весьма напоминают высококонцентрированные
эмульсии.
От типичных пен, представляющих высококонцентрированные
дисперсии газа в жидкости, следует отличать низкоконцентриро-
ванные системы Г/Ж, в которых газовые пузырьки находятся на
сравнительно большом расстоянии друг от друга. Примером такой
дисперсной системы могут служить газированная вода, пиво или
шипучее вино, содержащие пузырьки двуокиси углерода. Эти си-
стемы по свойствам ближе к разбавленным эмульсиям. Однако
благодаря большой -разнице в плотностях жидкой и газовой фазы
такие системы обладают очень малой седиментационной устойчи-
востью и существуют непродолжительное время.
Пены образуются при диспергировании газа в жидкости в при-
сутствии стабилизаторов или, как их целесообразно называть
в этом случае, пенообразователей. Жидкости без пенообразовате-
лей сколько-нибудь устойчивой пены не дают.
Прочность и продолжительность существования (время жизни)
пены зависят от свойств пленочного каркаса, в свою очередь оп-
ределяющихся природой и содержанием в системе пенообразова-
теля, адсорбированного на межфазной поверхности. К типичным
пенообразователям в случае водных пен принадлежат такие по-
верхностно-активные вещества, как спирты, жирные кислоты, мыла
и мылоподобные вещества, белки, сапонин (экстрагируемый из
растений глюкозид, обладающий поверхностно-активными свой-
ствами). Существенно, что эти вещества обусловливают и устой-
чивость эмульсий углеводородов в воде.
386
Устойчивость пей
Устойчивость пен зависит от природы и концентрации пенооб-
разователя. Со временем пленки между пузырьками пены стано-
вятся тоньше вследствие стекания жидкости, пузырьки лопаются,
пена разрушается и, наконец, вместо пены остается одна жидкая
фаза — раствор пенообразователя в воде или другой жидкости.
Устойчивость пен можно характеризовать’ временем существо-
вания пены, т. е. временем, прошедшим с момента образования
пены до момента полного ее разрушения. Другой способ оценки
устойчивости пены заключается в \1ропускании с заданной ско-
ростью через вспениваемую жидкость пузырьков воздуха и опре-
деления равновесной высоты образующегося при этом столба
пены. Постоянная высота столба пены устанавливается в тот мо-
мент, когда скорость разрушения пены равна скорости ценообра-
зования и, очевидно, может служить мерой устойчивости пены.
Устойчивость пены удобно изучать также по рремени жизни от-
дельного газового пузырька на поверхности жидкости, граничащей
с воздухом. С этой целью пузырек воздуха выдавливают в жид-
кость с помощью капилляра с загнутым концом. Пузырек всплы-
вает и, достигнув поверхности, задерживается там на некоторое
время, прежде чем лопнет. Это время жизни пузырька обычно про-
порционально времени существования столба пены в целом.
Существенно, что во время пребывания пузырька воздуха на
поверхности жидкости пленка, покрывающая пузырек, становится
все тоньше, о чем иногда можно судить по изменению интерферен-
ционных цветов пленки. Когда пленка достигает толщины меньше
0,01 мкм, интерференция становится уже почти незаметной, пленка
темнеет, так как почти не отражает света, и затем через некоторое
время разрушается. Однако в особых условиях, когда исключены
испарение жидкой среды, сотрясения и другие внешние воздей-
ствия, пены могут существовать неограниченно долго. Например,
Дьюару удалось обеспечить существование мыльного пузыря в те-
чение трех лет.
По наблюдению Б. В. Дерягина и А. С. Титиевской мыльная
пленка, достигшая наименьшей толщины, состоит из двух моно-
слоев молекул пенообразователя, разделенных полимолекулярным
слоем воды.
Сравнительно малое время существования пены и тот факт, что
разрушению ее пузырька всегда предшествует стекание жидкости
в пленке пены, приводит к выводу, что устойчивость пены в обыч-
ных условиях носит кинетический характер, а роль пенообразо-
вателя сводится в значительной степени к замедлению стекания
жидкости.
Как видно из данных, приведеииых в табл. XII, 1, водные растворы спиртов
и жирных кислот образуют малоустойчивые пены с продолжительностью суще-
ствования, не нревышающей 20 е. Максимальная продолжительность существова-
ния пены приходится иа средние члены гомологических рядов. Низшие члены
13*
387
обоих рядов, очевидно, слишком мало поверхностно-активны для того, чтобы
образовывать устойчивые пены; высшие же члены ряда обладают недостаточной
для этого растворимостью.
Таблица XII, 1. Оптимальная концентрация водного раствора
пенообразователя и максимальная продолжительность существования пены
Пенообразователь Опти- мальная концен- трация, ммоль/л Макси- мальная продолжи- тельность существо- вания пены, с Пенообра зователь Опти- мальная концен- трация, ммоль/л Макси- мальная продолжи- тельность существо- вания пены с
Спирты Этиловый 280 5 Кислоты Муравьиная 450 4
Пропиловый 340 11 Уксусная 200 8
Язо-бутиловый 90 12 Пропионовая Масляная 250 1000 11 18
Язо-амиловый 36 17 Валерьяновая 15 9
Третичный амило- 34 10 Капроновая 7,5 13
вый Гептиловый 0,7 8 Гептиловая Каприловая 1,5 0,25 16 12
Октиловый 0,3 5 Нониловая 0,07 5
Каждому спирту или кислоте отвечает оптимальная концентрация, при ко-
торой пенообразователь наиболее эффективен. Обычно наиболее устойчивые пены
образуются при некоторой средней, но в общем небольшой концентрации спирта
или кислоты. На рис. XII, 9 приведена изотерма,
Рис. XII, 9. Зависимость
времени жизни пены т от
концентрации азо-амилового
спирта с.
вания пен в этом случае
характеризующая зависимость продолжительности
существования пены от концентрации изо-амило-
вого спирта.
Мыла дают гораздо более устойчивые
пены, чем спирты и кислоты, очевидно
благодаря наличию в их молекулах ионо-
генной группы. Так же, как для спиртов
и кислот, максимальная устойчивость
пены отвечает мылам со средней длиной
углеводородного радикала и их раство-
рам средней концентрации.
Иначе ведут себя высокомолекуляр-
ные пенообразователи. Время существо-
очень велико и может составлять в обыч-
ных условиях сотни и даже тысячи секунд. При этом время суще-
ствования пен всегда тем больше, чем выше концентрация высоко-
молекулярного пенообразователя.
Помимо природы и концентрации пенообразователя на устой-
чивость пены влияют температура, вязкость дисперсионной среды,
введение в жидкую фазу электролитов и pH среды. К сожалению,
точных данных о влиянии этих факторов на устойчивость пен
очень мало. Повышение температуры обычно неблагоприятно ска-
зывается на устойчивости пены. Действие повышения температуры
. 388
можно объяснить десорбцией пенообразователя с межфазной по-
верхности и понижением вязкости дисперсионной среды, что спо-
собствует более быстрому стеканию жидкости в пленке. Повыше-
ние температуры, очевидно, вызывает более быстрое разрушение
пены и вследствие того, что ускоряется испарение дисперсионной
среды и пленка обезвоживается. Введение в жидкую фазу не раз-
рушающих пену электролитов уменьшает устойчивость пен, обра-
зованных низкомолекулярными пенообразователями. Повышение
вязкости среды всегда повышает устойчивость пен.
Весьма интересны исследования над «черными» пленками, на-
чатые еще Перреном. Если наблюдать отдельную свободную
пленку (или пузырек), образованную из достаточно концентриро-
ванного раствора мыла (например, олеата натрия), то легко за-
метить, что пленка постепенно становится все более тонкой, меняя
цвета интерференции. После достижения толщины в 1000 А пленка
становится белой. При дальнейшем утоньшении пленки количество
отраженного света уменьшается, пленка становится серой, а от-
дельные ее участки приобретают неодинаковую толщину.
Существенно, что неодинаковое утоньшение пленки, образован-
ной из концентрированных мыльных растворов, происходит ступе-
необразно. В наиболее тонких участках пленка приобретает со-
вершенно черный цвет.
Веллс провел ряд измерений толщины мыльных пленок с по-
мощью интерферометрических и колориметрических методов и
нашел, что толщина черной пленки в несколько раз меньше тол-
щины наиболее тонкой части обычной пленки, что согласуется
с опытами Перрена. Минимальная толщина черной пленки состав-
ляла 42—45 А, ч~то соответствует примерно удвоенной длине моле-
кулы олеата натрия.
Шелудко и Эксерова (1961 г.) и Дюйвис (1962 г.) провели точ-
ные исследования толщины черной пленки, полученной из раство-
ров поверхностно-активных веществ, и подтвердили результаты
предыдущих исследований. Было найдено, что тончайшая пленка,
полученная из раствора олеата натрия, имела толщину 40 А, в то
время как толщина пленки, полученной из растворов смачивателей
ОП-7 и ОП-20, составляла 85 и 100 А соответственно. Толщина
этих пленок примерно в два раза больше длины молекулы поверх-
ностно-активного вещества. Следует заметить, что такая малая
толщина получается только при достаточной концентрации элек-
тролита в растворе. Если содержание электролита слишком мало,
то образуются более толстые пленки, причем их равновесная тол-
щина уменьшается постепенно с увеличением содержания элек-
тролита в полном соответствии с теорией ДЛФО.
Выше мы принимали, что при хранении пены отдельные ее пу-
зырьки лопаются и в результате этого пена полностью разру-
шается. Однако, как показывает опыт, при хранении пены может
происходить не только разрушение пузырьков, но и изменение
их размеров. При этом размеры мелких пузырьков всегда
389
уменьшаются, а крупных — увеличиваются. Причина этого явления
заключается в том, что по законам капиллярности газ, находя-
щийся в мелких пузырьках пены, испытывает большее давление,
чем газ, заполняющий крупные пузырьки. Давление стремится вы-
равняться путем диффузии газа через жидкую пленку, что приво-
дит к уменьшению размеров мелких пузырьков и увеличению раз-
меров больших пузырьков (Де Фриз, 1957 г.). Это явление напоми-
нает изотермическую перегонку, когда более крупные капельки
жидкости растут за счет более мелких вследствие того, что в по-
следних жидкость обладает большим давлением пара. Подобное
старение пены играет особенно большую роль, когда пленка пены
весьма устойчива и разрушение пены обычным образом не проис-
ходит (например, для пен, полученных из каучукового латекса).
Старение пены сопровождается также и уменьшением общего
ее объема. Причина этого вполне понятна, если учесть, что газо-
вую фазу над пеной надо рассматривать как один бесконечно
большой пузырек, в который путем диффузии газ будет переходить
из отдельных пузырьков пены. Де Фриз установил, что в старею-
щей пене квадрат радиуса малых пузырьков линейно уменьшается
со временем. Такой вывод полностью отвечает теории, развитой на
основе указанных выше представлений.
Причины устойчивости пен. Устойчивость пен можно объяснять
разными факторами, а именно действ,ием так называемого эффекта
Гиббса, наличием у пленки сравнительно высокой поверхностной
вязкости или особых механических свойств (структурно-механиче-
ский фактор устойчивости) и существованием в приповерхностном
слое пленки гидратных или двойных электрических слоев, препят-
ствующих ее утоньшению (термодинамический фактор устойчиво-
сти). Рассмотрим последовательно эти три фактора устойчивости
пены.
Пленки пены при ее получении путем пропускания через жид-
кость пузырьков воздуха, а также при медленном уменьшении
объема пены в результате сжатия отдельных ее пузырьков или их
разрушения испытывают локальные деформации и поэтому должны
хорошо переносить как сжатия, так и растяжения. Можно было
бы считать, что легкой деформируемости пены и ее прочности
должно способствовать малое поверхностное натяжение на границе
пенообразующая жидкость—воздух. Однако это не так. Опыт по-
казал, что для устойчивости пены имеет значение не столько малое
поверхностное натяжение, сколько способность жидкой пленки
легко и быстро изменять его значение. Чтобы выдержать локаль-
ные деформации без разрыва, пленка должна обладать способ-
ностью повышать поверхностное натяжение при локальных рас-
тяжениях и уменьшать его при локальных сжатиях. Этими изме-
нениями компенсируются локальные деформации и разности в на-
пряжениях, возникающих в разных участках пленки, и обеспечи-
вается ее прочность. Гиббс называл эту способность эффективной
упругостью пленки. Ее причина заключается в том, что если один
390
участок пленки подвергается, например, растяжению, то его по-
верхность увеличится и вследствие этого концентрация поверх-
ностно-активного вещества на межфазной границе уменьшится.
Уменьшение поверхностной концентрации обусловит повышение
поверхностного натяжения в растянутом участке, вследствие чего
участок стремится сжаться в большей степени, чем все соседние
нерастянутые участки. Обратное явление наблюдается при дефор-
мации, вызывающей сжатие пленки.
Совершенно очевидно, что «упругостью» в том смысле, в каком
понимал ее Гиббс, могут обладать только пленки, полученные из
растворов поверхностно-активных веществ. Пленки из индивиду-
альных жидкостей, обладающих постоянным поверхностным натя-
жением, не изменяющимся при их растяжении или сжатии, лишены
подобной упругости, и поэтому получить из таких жидкостей
устойчивые пены невозможно. Существенно также, что наиболее
устойчивые пены обычно получаются из растворов поверхностно-
активных веществ, обладающих не минимальным поверхностным
натяжением, а способных наиболее резко изменять поверхностное
натяжение с концентрацией.
При объяснении устойчивости реальной пены с точки зрения
Гиббса следует иметь в виду особое строение этой системы.
Именно благодаря своеобразной структуре пены эффект Гиббса
вызывает значительные затруднения в стекании жидкости в плен-
ках пены, что очень сильно сказывается на устойчивости всей си-
стемы. Каркас пены, как было показано, состоит из приблизи-
тельно плоских жидких пленок, являющихся стенками отдельных
ячеек. Там, где сходятся три пленки, образуются ребра пузырька,
в которых жидкость имеет сильно вогнутую поверхность. По зако-
нам капиллярности в этих местах жидкость имеет пониженное
давление, что вызывает отсасывание ее из плоских частей каркаса
пены в вогнутые. В результате этого в пленках пены возникает те-
чение жидкости к ребрам. Это течение способствует самопроиз-
вольному утоньшению пленок пены. Однако такое течение жидко-
сти может происходить лишь внутри пленки, на поверхности оно
невозможно из-за эффекта Гиббса. В самом деле, при течении
жидкости от центральной части пленки к ребрам должно было бы
увеличиться поверхностное натяжение в центральных частях
пленки и в результате этого на поверхности ее тотчас возник бы
противоток жидкости, направленный от ребер к центру, из-за чего
течение прекратилось бы. Таким образом, стекание жидкости про-
исходит так, как если бы поверхность пленки была неподвижной,
т. е. жидкость как бы протекает по плоскому капилляру. Очевидно,
стекание по такому капилляру происходит тем медленнее, чем
тоньше пленка.
Рассмотренные выше представления не могут все же полностью
объяснить устойчивость пены. Во-первых, с этой точки зрения
трудно понять зависимость устойчивости пены от природы и кон-
центрации пенообразователя. При наличии в жидкости достаточно
391
активного пенообразователя режим стекания жидкости в пленках,
казалось бы, не должен зависеть от этих обоих факторов. Однако
опыт показывает, что это не так. Во-вторых, согласно этой точки
зрения упругость пленки должна возрастать по мере ее утоньше-
ния в результате отсасывания жидкости к ребрам пены. Это обус-
ловлено тем, что запас поверхностно-активного вещества, содер-
жащегося в тонких пленках, меньше, а следовательно, и уменьше-
ние их поверхностной концентрации при растяжении будет больше.
Однако, если с уменьшением толщины пленки упругость ее возра-
стает, то непоятно, почему же пленка рвется.
Рассмотренный фактор устойчивости является, по-видимому,
определяющим для малоустойчивых пен, стабилизованных срав-
нительно низкомолекулярными пенообразователями.
Устойчивость высокоустойчивых пен объясняется существова-
нием в пленках высоковязкого или механически прочного адсорб-
ционного слоя- из молекул пенообразователя. Такое объяснение
было предложено впервые еще в прошлом столетии Плато, а затем
особенно широко было развито в работах П. А. Ребиндера и его
школы. П. А. Ребиндер считает, что на поверхности растворов мыл
или мылоподобных веществ образуются высоковязкие адсорбцион-
ные слои с гелеобразным строением, диффузно распространяю-
, щиеся в глубь раствора. Эти слои, с одной стороны, замедляют
стекание жидкости в пленке, с другой — придают пленке пены
высокую структурную вязкость и механическую прочность. Од-
нако исследования А. А. Трапезникова, Лоуренса и других иссле-
дователей показали, что стойкие пены могут получаться и тогда,
когда не обнаруживается заметная поверхностная вязкость или
структурно-механические свойства на границе раствор — воздух.
К. В. Зотовой и А. А. Трапезниковым обнаружен интересный факт, позво-
ляющий в некоторых случаях по-новому объяснить устойчивость пленок пены.
Эти авторы установили, что поверхностно-активные коллоидные компоненты мо-
гут переходить в пленку в большем количестве, чем в адсорбционный слой на
поверхности исходного раствора. Это обусловлено особыми условиями образова-
ния пленки, способствующими непрерывному обновлению поверхности и обмену
поверхностно-активными компонентами. В результате перехода в пленку не-
прочных коллоидных агрегатов, возникших по тем или иным причинам в рас-
творе, в глубине пленки между адсорбционными слоями может образоваться
тиксотропная структура, сильно повышающая вязкость этой части пленки. Сами
же адсорбционные слои остаются при этом маловязкими. Понятно, что благо-
даря такой структуре сильно замедляется процесс стекания и повышается устой-
чивость пен. С таким объяснением устойчивости пены хорошо согласуется исклю-
чительная длительность существования пен, стабилизованных высокомолекуляр-
ными соединениями. В этом случае образование высоковязкой тиксотропной
структуры в глубине.пленки пены почти не вызывает сомнений.
Перейдем теперь к объяснению стабильности высокоустойчивых
пен с помощью термодинамического фактора устойчивости.
Б. В. Дерягин, первый показавший значение этого фактора,
объясняет возможность существования пен, исходя из разработан-
ных им представлений о расклинивающем давлении. Причиной
расклинивающего давления в пленках пены, стабилизованной ионо-
392
генными веществами, является отталкивание двойных электриче-
ских слоев, образованных ионами пенообразователя в растворе
около обеих поверхностей пленки. Наличие такого отталкивания
доказано Б. В. Дерягиным и А. С. Титиевской при исследовании
сжатия двухсторонних пленок, образованных в месте соприкосно1
вения двух пузырьков пены, с помощью очень тонкой методики и
специально сконструированного прибора.
Эти исследования показали, что пленки, полученные из водных
растворов олеата натрия, при утоньшении в результате наложения
давления достигали некоторой постоянной толщины, которая
дальше уже не изменялась. Равновесная толщина таких пленок
составляла сотни ангстрем при малом содержании в пенообразую-
щей жидкости электролитов. Наоборот, в достаточно концентри-
рованных растворах электролитов равновесная толщина пленок
была значительно меньше по сравнению с теоретически вычислен-
ной. Последнее обстоятельство явилось прекрасным доказатель-
ством электростатической природы расклинивающего давления
в этом случае. При сравнительно высоких концентрациях электро-
лита (порядка 0,1 н. и более), когда диффузные ионные слои
сжаты до предела, зависимость толщины пленки от концентрации
не соблюдалась. Однако пленки при этом оставались устойчивыми.
Это указывает на то, что при таких условиях в действие вступают
уже силы отталкивания неэлектростатической природы, вероятно,
связанные с гидратацией монослоев пенообразователя. Установ-
ленное Б. В. Дерягиным и А. С. Титиевской положение, что рав-
новесные толщины адсорбированных пленок как ионогенных, так
и неионогенных пенообразователей не зависят от высоких концен-
траций электролитов, а также от температуры, указывает на спе-
цифическую структуру этих слоев, придающих им свойства особой
граничной фазы.
На значение гидратации для устойчивости пен указывал А. А. Трапезников,
а еще раньше Д. А. Талмуд. С точки зрения А. А. Трапезникова стабильность
пены обусловливается гидратацией полярных групп молекул пенообразователя,
что тормозит стекание жидкости в пленке пены. Сцепление концов углеводо-
родных цепей, расположенных на межфазной поверхности со стороны газовой
фазы, нужно лишь для обеспечения связности (цельности) адсорбционного слоя.
При этом адсорбционный слой должен быть достаточно легкоподвижным и, сле-
довательно, разреженным для того, чтобы разрывы, образующиеся в результате
стекания жидкости в пленке, успевали своевременно «залечиваться». Причиной
разрушения пены А. А. Трапезников считает дегидратацию полярных групп
адсорбционного слоя, наступающую вследствие непрерывного отсоса дисперсион-
ной среды. В результате возникают сначала поверхностные, а затем и трехмер-
ные агрегаты из молекул пенообразователя, не обладающие стабилизующим
действием, и пленка в конце концов разрывается.
Таким образом, существует несколько факторов, объясняющих
устойчивость пен. В настоящее время все больше исследователей
приходит к выводу, что вообще не может быть единой теории
устойчивости пен и что причины существования пен зависят от
пенообразователей и условий получения пен.
393
Методы получения и разрушения пен.
Практическое значение пен
Пены получают путем пропускания пузырьков соответствую-
щего газа (обычно воздуха) через раствор пенообразователя или
путем интенсивного механического перемешивания раствора пено-
образователя. Пузырьки газа в жидкости окружаются адсорбцион-
ным слоем пенообразователя, всплывают к поверхности, подходят
к имеющемуся на ней адсорбционному слою, растягивают его и
таким образом образуют двухсторонние пленки. Если эти пленки
достаточно прочны, возникшие пузырьки образуют пену.
О роли природы стабилизатора при ценообразовании было ска-
зано в предыдущем разделе этой главы.
Очень часто технологу приходится разрушать образовавшуюся
пену или принимать меры для предупреждения ее образования.
С этой целью в систему обычно вводят такие вещества, которые,
обладая высокой поверхностной активностью, не дают стойкой
пены. Эти вещества вытесняют пенообразователь с поверхности
жидкости и этим делают невозможным существование пены. В ка-
честве подобных противопенных, или пеногасящих, веществ при-
меняют различные вещества, например, спирты, сложные эфиры.
Из спиртов для гашения пены чаще всего применяют циклогекса-
нол, амиловый и октиловый спирты, а также смеси высших спир-
тов, получающихся как побочные продукты при синтезе метилового
спирта. Другой метод пеногашения заключается в «пережигании»
пленки пены путем воздействия на нее высоких температур. Для
разрушения стойких пен могут быть использованы и различные
механические воздействия.
Пенообразование и пены имеют большое практическое значе-
ние. Роль пены при флотации уже отмечалась в гл. VI, разд. 5.
’Образование пены является положительным фактором при стирке.
С помощью вспенивания и последующего удаления пены можно
очищать некоторые жидкости от содержащихся в них поверхност-
но-активных примесей, переходящих при вспенивании в пену.
И наоборот, пользуясь тем же приемом, из раствора можно из-
влекать содержащиеся в нем ценные поверхностно-активные ве-
щества. Исключительно значение пен в противопожарном деле.
Поскольку применяемые при тушении пожаров пены содержат
в виде дисперсной фазы обычно двуокись углерода, такая пена
при нанесении на горящие предметы препятствует доступу к ним
воздуха и способствует затуханию огня.
В ряде случаев пенообразование может иметь отрицательное
значение. Например, благодаря легкому образованию пены возни-
кают трудности при перемешивании некоторых растворов. Обра-
зование обильной пены мешает выпариванию растворов в выпар-
ных аппаратах и приводит к потерям ценной жидкости при пере-
бросах пены. В этих случаях применяют средства, предупреждаю-
щие пенообразование, о которых уже было сказано выше.
394
5. СИСТЕМЫ С ТВЕРДОЙ ДИСПЕРСИОННОЙ СРЕДОЙ
Коллоидные системы с твердой дисперсионной средой, или, как
их часто называют, твердые золи, несмотря на огромное практиче-
ское значение, до сих пор привлекали в коллоидной химии срав-
нительно мало внимания. Причиной этого являются их необычные
по сравнению с остальными коллоидными системами свойства.
Понятно, что для систем с твердой дисперсионной средой бессмыс-
ленно говорить о таких важных для других коллоидных систем
особенностях, как молекулярно-кинетические свойства и агрега-
тивная устойчивость. Рассмотрим некоторые из этих систем и их
специфичные свойства.
Коллоидные и микрогетерогенные системы с твердой диспер-
сионной средой, так же как и лиозоли, можно разделить на си-
стемы с газовой, жидкой и твердой дисперсной фазой.
Системы с твердой дисперсионной средой и газовой дисперсной
фазой — Г/Т часто называют твердыми пенами. Твердые пены,
так же как и жидкие пены, вследствие большого размера пузырь-
ков газовой фазы обычно относят к микрогетерогенным или даже
грубодисперсным системам. Примером природной твердой пены
может служить пемза — пористая губчато-ноздреватая очень лег-
кая горная порода вулканического происхождения, применяемая
как абразив для полировки и шлифования, а также в строитель-
ном деле для изготовления пемзобетона. Из искусственных твер-
дых пен можно указать пеностекла и пенобетоны,-широко приме-
няемые в качестве строительных и изоляционных материалов.
Достоинствами этих материалов являются малая плотность, малая
теплопроводность и довольно большая прочность, обусловленная
их ячеистой структурой и прочностью дисперсионной среды. Сюда
же надо отнести искусственные губчатые материалы, изготовлен-
ные на основе полимеров (микропористая резина, различные пено-
пласты).
Системы с твердой дисперсионной средой и жидкой дисперсной
фазой — Ж/Т, которые по аналогии можно считать твердыми
эмульсиями, встречаются довольно редко. Примером таких систем
может служить так называемый черный фосфор, получающийся
путем диспергирования металлической ртути в расплавленном
фосфоре.
Наибольшее значение имеют системы Т/Т с твердой диспер-
сионной средой и твердой дисперсной фазой. К этому классу кол-
лоидных и микрогетерогенных систем относятся некоторые окра-
шенные драгоценные и полудрагоценные камни, цветные стекла,
эмали, многие минералы, горные породы, некоторые сплавы.
О том, что драгоценные и полудрагоценные камни являются,
как правило, глиноземом или кварцем с диспергированными в них
окислами различных металлов, мы уже говорили в начале курса.
Цветные стекла представляют собой обычные силикатные стекла,
содержащие ничтожные примеси коллоидно диспергированного
395
вещества, придающего стеклу окраску. Например^ рубиновое
стекло, которое получал еще М. В. Ломоносов, содержит в виде
дисперсной фазы высокодиспергированное золото. Содержание зо-
лота в рубиновом стекле колеблется от 0,01% (розовое стекло) до
0,1% (ярко-красное стекло), а размер частиц составляет 4—30 нм,
редко превышая 150 нм. Менее дорогие стекла с той же рубиновой
окраской могут быть получены диспергированием (вместо золота)
таких металлов, как серебро, селен, медь. Стекла других цветов
можно получить при сплавлении стекла с другими веществами.
Эмали, стеклообразные непрозрачные вещества, наносят в рас-
плавленное состоянии на металлические или керамические поверх-
ности, чтобы придать им красивый вид или большую устойчивость
к действию химических факторов. Они представляют собой также
силикатные стекла, в которых содержатся частицы пигментов, при-
дающие эмалям непрозрачность и окраску. В качестве таких пиг-
ментов могут использоваться окислы и соли, например ЗЮг, ТЮг,
фосфат кальция.
Весьма близки к цветным силикатным стеклам стекла, полу-
чаемые сплавлением окислов металлов с бурой. Как известно, та-
кие стекла («перлы») имеют большое значение в аналитической
химии, поскольку их цвет может указывать на химическую при-
роду сплавленного с бурой вещества.
Очень интересным представителем рассматриваемых коллоидных систем яв-
ляется встречающаяся в природе голубая каменная соль. Причиной голубой
окраски каменной соли является присутствие в кристаллах хлорида натрия ни-
чтожного количества (0,0001%) коллоидно диспергированного металлического
натрия. Зидентопф еще в 1905 г. получил голубую каменную соль искусственно,
нагревая кристаллы хлорида натрия в парах натрия. Сначала соль приобретала
желтую окраску, соответствующую высокой степени дисперсности частиц натрия.
Однако при дальнейших последовательных нагреваниях и охлаждениях проис-
ходила постепенная агрегация частиц натрия и окраска кристаллов становилась
голубою Опыты, проведенные позднее, показали, что искусственная голубая соль
может быть получена и при действии на кристаллы хлорида натрия рентгенов-
ских лучей и радиоактивного излучения.
Наличие в голубой каменной соли частиц металлического' натрия подтвер-
ждается тем, что растворение такой соли в воде сопровождается заметным вы-
делением водорода, освобождающегося из разлагаемой натрием воды. Образова-
ние металлического натрия в каменной соли в природных условиях объясняется
тем, что ионы натрия могут восстанавливаться до металла за счет присоедине-
ния электронов под действием [3-лучей. Источником радиоактивного излучения в
горных породах может служить радиоактивный изотоп калия 40К, всегда при-
сутствующий в небольших количествах в природной каменной соли. Вычисления
показывают, что содержащегося в каменной соли калия 40К вполне достаточно,
чтобы вызвать появление голубой окраски в течение геологических периодов.
Весьма важными представителями систем типа Т/Т являются
горные породы. Коллоидная или микрогетерогенная структура
многих вулканических пород объясняется тем, что различные ком-
поненты магмы при ее застывании выделяются в виде кристалли-
ков и образуют таким образом дисперсную фазу. Осадочные по-
роды имеют коллоидную структуру вследствие того, что образуются
при оседании в континентальных или морских водах мельчайших
396
частиц кремнезема, глинистых минералов, гумусовых веществ, гид-
роокиси железа, панцырей диатомовых водорослей. Следует заме-
тить, что в свою очередь горные породы являются материалом, ид
которого в результате выветривания и размывания в природе об-
разуются аэрозоли и лиозоли *.
Наконец, весьма важными системами Т/Т являются гетероген-
ные сплавы, к которым принадлежит большинство технических
металлов. В зависимости от условий получения сплав может иметь
строение, соответствующее молекулярному раствору, коллоидной
системе и грубой дисперсной системе. Например, в стали мы встре-
чаемся со всеми переходами от истинного раствора (аустенит)
через коллоидные растворы (мартенсит) вплоть до микрогетеро-
генных систем (перлит). В чугуне дисперсной фазой являются ча-
стицы углерода, размеры которых близки к коллоидным.
Из типичных коллоидных свойств у систем с твердой диспер-
сионной средой, пожалуй, лучше всего выражена способность
к светорассеянию. Как было указано еще в начале курса, термин
«опалесценция» произошел от минерала опала, обладающего весь-
ма сильно выраженной способностью рассеивать свет. Интересно,
что рубиновое стекло послужило Зидентопфу и Зигмонди объек-
том для первых ультрамикроскопических наблюдений. Понятно,
что светорассеяние у систем с твердой дисперсионной средой
можно наблюдать только тогда, когда дисперсионная среда про-
зрачна.
Выше указывалось, что обычная коагуляция в системах с твер-
дой дисперсионной средой невозможна из-за огромной вязкости
среды, препятствующей столкновению частиц между собой. Од-
нако все же некоторое укрупнение частиц в таких системах воз-
можно за счет изотермической перегонки вещества дисперсной
фазы. Такое укрупнение частиц наблюдается, например, при дли-
тельном нагревании рубинового стекла при температуре, когда
давление пара металла уже достаточно высоко. При очень высо-
ких температурах, когда происходит плавление дисперсионной
среды, в подобных системах может наблюдаться и истинная коа-
гуляция. При этом, если среда прозрачна, меняется и цвет си-
стемы. Например, при высокой температуре красный цвет рубино-
вого стекла переходит в фиолетовый, а затем 1? синий вследствие
агрегации частиц. Интересно, что двуокись олова, присутствующая
в стекле, оказывает защитное действие и препятствует образова-
нию агрегатов.
Коллоидные и микрогетерогенные системы с твердой диспер-
сионной средой обычно образуются методом конденсации из рас-
плава. При охлаждении расплава, представляющего собой гомо-
генную систему, выделяется дисперсная фаза, которая и остается
* Много сведений о коллоидном строении горных пород и о значении
коллоидных процессов при образовании земной коры можно найти в кн.:
Ф. В. Чухров. Коллоиды в земной коре. М., изд-во АН СССР, 1955, 672 с.
397
распределенной в виде частиц того или иного размера в затвердев-
шей системе. Таким образом получаются горные породы с кол-
лоидной или микрогетерогенной структурой при застывании магмы.
Пенопласты в промышленности получают несколько иным спосо-
бом. В полимер вводят порообразователь, который при нагревании
или каком-нибудь ином воздействии выделяет газ. Последний мо-
жет оставаться в полимере в виде мельчайших замкнутых пузырь-
ков или образовывать в нем извилистые незамкнутые поры.
При получении коллоидных и микрогетерргенных систем
с твердой дисперсионной средой методом диспергирования в рас-
плавленной среде диспергируется газ, жидкость или. твердое ве-
щество. Такой расплав, обладающий еще свойствами жидкости,
называется пирозолем. При охлаждении пирозоля он затвердевает
и образует коллоидную илй микрогетерогенную систему с твердой
дисперсионной средой. Как мы видели на примере рубинового
стекла, устойчивость пирозоля, а следовательно, и дисперсность си-
стемы с твердой средой можно повысить введением соответствую-
щего стабилизатора.
ГЛАВА XIII
КОЛЛОИДНЫЕ ПОВЕРХНОСТНО-АКТИВНЫЕ ВЕЩЕСТВА
Подавляющее большинство коллоидных растворов являются
гетерогенными и термодинамически неравновесными системами.
Однако существуют системы, которые в одних условиях могут
представлять собой истинные растворы, а в других становятся зо-
лями, структурированными жидкостями или даже гелями. Такие
системы обратимы и термодинамически равновесны
истинный раствор золь <=£ гель
Для того чтобы произошел переход из одного состояния в дру-
гое, необходимо лишь изменить концентрацию раствора, темпера-
туру, pH или ввести в систему электролит. Изменяя условия суще-
ствования системы, можно получать либо истинные (гомогенные)
растворы с молекулярной степенью дисперсности, либо гетероген-
ные системы, частицы которых представляют собой агрегаты, со-
стоящие из множества молекул. Такие частицы, подобно электро-
нейтральным частицам в лиофобных коллоидных системах, назы-
вают мицеллами. Однйко в отличие от мицелл коллоидных систем
они термодинамически стабильны и не изменяются до тех пор,
пока под действием внешних факторов не сместится равновесие,
в котором находилась система. Устойчивость мицелл характери-
зуется скоростью диссоциации, т. е. средним временем пребывания
молекулы в мицелле.
К системам, в которых наблюдаются обратимые переходы по-
добного рода, относятся водные растворы многих поверхностно-
активных веществ, например, мыл и мылоподобных веществ,
а также растворы таннидов (дубильных веществ) и некоторых
красителей. Эти растворы, если в них содержатся частицы, со-
стоящие из большого числа молекул, с полным правом можно от-
нести к лиофильным коллоидным системам, так как они обладают
признаками коллоидных систем — гетерогенностью и высокой дис-
персностью, но в отличие от лиофобных коллоидных систем тер-
модинамически равновесны и агрегативно устойчивы.
Ранее вещества, дающие такие растворы, называли полукол-
лоидами (или семиколлоидами). Однако этот термин нельзя
* Глава написана совместно с Р. М. Панич.
399
признать удачным. В дальнейшем все вещества поверхностно-
активные, способные переходить в растворах из молекулярного
раздробления в коллоидное, мы будем называть принятым теперь
термином «коллоидные поверхностно-активные вещества» или со-
кращенно коллоидными ПАВ.
Поскольку мыла являются наиболее типичными коллоидными
поверхностно-активными веществами и широко применяются в са-
мых различных отраслях народного хозяйства, свойства коллоид-
ных ПАВ в этой главе в основном рассмотрены на примере мыл.
1. ОСНОВНЫЕ ПОНЯТИЯ И КЛАССИФИКАЦИЯ КОЛЛОИДНЫХ ПАВ
Коллоидные поверхностно-активные вещества характеризуются,
подобно всем поверхностно-активным веществам, малой истинной
। растворимостью и способностью снижать поверхностное и межфаз-
ное натяжение в разбавленных растворах вследствие адсорбции и
ориентации молекул на поверхности раздела. Однако наряду
с этим при некоторой концентрации — критической концен-
трации мицеллообразования (ККМ) — в растворе на-
чинают образовываться агрегаты молекул — мицеллы, вследствие
чего общая растворимость ПАВ, обусловленная образованием на-
ряду с истинным также и коллоидного раствора, резко увеличи-
вается, тогда как молекулярная растворимость остается неизмен-
ной и равной ККМ.
\Для того чтобы ПАВ было способно образовывать мицеллы,
оно должно удовлетворять двум требованиям — с одной стороны,
иметь достаточно большой углеводородный радикал, снижающий
растворимость в воде, а, с другой — обладать достаточно сильной
полярной группой, способствующей его растворимости. Этому тре-
бованию удовлетворяют не все поверхностно-активные вещества.
Например, для гомологического ряда алифатических спиртов ми-
целлообразование вовсе не характерно. При этом для соединений
с числом углеродных атомов меньше 7 мицеллообразованию ме-
шает малая длина углеводородного радикала, а для более высоких
гомологов — сравнительно низкая гидрофильность полярной груп-
пы. Известно также, что для многих коллоидных ПАВ, например,
для оксиэтилированных спиртов, независимо от числа оксиэтиле-
новых групп, т. е. от полярности гидрофильной части молекулы,
мицеллообразование становится возможным лишь при длине
углеводородного радикала, превышающей 7—8 углеродных
атомов.
Изотерма поверхностного натяжения, характеризующая зави-
симость этой величины от концентрации коллоидного ПАВ в рас-
творе, состоит из почти прямолинейного участка падения поверх-
ностного натяжения, криволинейного участка, описываемого урав-
нением Шишковского, и почти горизонтального участка в области
концентраций выше ККМ. На этом последнем участке поверхност-
нее натяжение почти перестает изменяться, так как вновь вводи-
400
свойства
мое ПАВ не адсорбируется на границе раствор — воздух, а обра-
зует мицеллы в растворе. Для ПАВ, не обладающих коллоидной
растворимостью, горизонтальный участок на изотерме поверхност-
ного натяжения сдвинут в область более высоких концентраций.
Коллоидные ПАВ в соответствии с особенностями строения их
молекул подразделяются на три основные группы: анионные, ка-
тионные и неионогенные.
Анионные ПАВ диссоциируют в воде, образуя отрицательно
заряженные поверхностно-активные ионы. При адсорбции таких
ПАВ из раствора на поверхности адсорбируются анионы, в резуль-
тате чего поверхность приобретает отрицательный заряд. Важней-
шими представителями этой группы коллоидных ПАВ являются
обычные мыла и соли сульфокислот.
Обычные мыла представляют собой соли предельных и неко-
торых непредельных карбоновых кислот,
проявляются только у солей тех жирных
кислот, у которых число атомов углерода в
цепи не меньше 10 и не больше 22. Соли
низкомолекулярных жирных кислот, как
отмечалось в гл. V, слишком хорошо рас-
творимы в воде, чтобы проявлять указан-
ные свойства в заметной степени. Соли
жирных кислот, углеводородная цепь кото-
рых содержит больше 22 атомов углерода,
практически нерастворимы в воде.
Для технических целей особое значение
имеют натриевые мыла пальмитиновой, сте-
ариновой и ненасыщенной олеиновой кис-
лот, получаемых в больших количествах из животных жиров. Это —
пальмитат натрия Ci5H3ICOONa, стеарат натрия CnHasCOONa
и олеат натрия Ci7H33COONa. Меньшее значение имеют калие-
вые и аммонийные мыла этих же кислот, жидкие в обычных усло-
виях.
Мыла с двух- и трехвалентным катионом (кальциевые, магние-
вые, алюминиевые и т. п.) нерастворимы в воде, но образуют кол-
лоидные системы в углеводородных средах. Они используются
в консистентных смазках на минеральном масле, а также для ста-
билизации эмульсий второго рода (в/м).
Обычные продажные мыла содержат большое количество воды,
а часто и примеси различных веществ, особенно электролитов,
о чем нельзя забывать при их использовании. Чистые мыла можно
выделить из растворов в кристаллическом виде. Кристаллы мыл,
как показал рентгенографический анализ, обладают слоистым
строением, причем молекулы в бимолекулярных слоях обращены
друг к другу неполярными, т. е. углеводородными концами, так,
как это показано схематически на рис. XIII, 1. При этом обычно
молекулы расположены под определенным углом к плоскости,
в которой лежат полярные группы.
шиши
rrrffrrrr?
гттгг
Рис. XIII, 1. Схемати-
ческое строение кристал-
ла олеата натрия:
1 — плоскость, отвечающая
группам —COONa; 2—пло-
скость; отвечающая груп-
пам —СН4.
401
Моющим действием обладают не только соли жирных кислот»
но и соли нафтеновых кислот, представляющие собой алицикли-
ческие соединения. Эти соли содержатся в мылонафте — продукте»
получаемом при очистке керосина и солярового масла.
К коллоидным ПАВ, содержащим в качестве активной группы
сульфо-группу, следует в первую очередь отнести соли высокомо-
лекулярных сульфокислот общего строения СпН2п+150зМ— ал-
килсульфонаты или СпНгп-нСвНчЗОзМ — алкиларилсульфонаты
(где М обозначает одновалентный катион натрия, калия или ам-
мония) . Сюда же относятся соли алкилзамещенных нафталинсуль-
фокислот, например, натриевая соль нзо-бутилнафталинсульфокис-
лоты:
SO3Na
/СН3
СН2—СН^
ХСН3
Соединения последнего типа в технике называют некалями.
В качестве ПАВ в настоящее время широко применяют и ал-
килсульфаты — сульфоэфиры высших спиртов, а также их соли
СпН2п+1—О—SO3M.
Поскольку сульфокислоты являются сильными кислотами, не
только их соли с одновалентными катионами, но и соли с много-
валентными катионами, а также и сами сульфокислоты достаточно
хорошо растворимы в воде и образуют водные растворы со всеми
характерными свойствами «мыльных» растворов. Это является
важным преимуществом сульфомыл перед обычными мылами, так
как позволяет использовать их в жесткой воде и даже в кислой
среде.
Катионные ПАВ, диссоциируя в воде, образуют положительно
заряженные поверхностно-активные ионы. Из растворов таких ПАВ
поверхностью адсорбируются катионы, вследствие чего она стано-
вится положительно заряженной. Во многих случаях это пред-
ставляет практический интерес. Так, положительно заряженные
частицы дисперсной фазы, стабилизованные такими ПАВ, избира-
тельно поглощаются (связываются) из пропиточной ванны отри-
цательно заряженным растительным волокном гораздо более ин-
тенсивно и полно, чем отрицательно заряженные частицы, стаби-
лизованные анионными ПАВ.
Примером катионных ПАВ может служить октадециламмоний-
хлорид:
C18H37NH3C1
Сюда же относятся соли четырехзамещенных аммониевых основа-
ний, например, цетилтриметиламмонийхлорид:
C16H33(CH3)3NC1
402
Другим примером таких коллоидных ПАВ являются пиридиновые
соединения, замещенные у атома азота, например, цетилпириди-
нийхлорид:
С1в1
Все эти вещества в воде распадаются на анионы С1~ и катионы
соответственно
[C18H37NH3]+, [C16H33(CH3)N3]+, [cI6h33n
Одновременное присутствие в водном растворе анионных и ка-
тионных ПАВ обычно невозможно, так как в таком растворе из
большого катиона и большого аниона образуется весьма слабо
диссоциирующая соль с большим молекулярным весом, практи-
чески нерастворимая в воде.
Неиоиогенные ПАВ (НПАВ). Это — вещества, молекулы кото-
рых не способны к диссоциации. Дифильные молекулы таких ПАВ
обычно состоят из длинной углеводородной цепочки с несколькими
полярными; но неионогенными группами на конце, обуславливаю-
щими растворимость этих веществ. Такими группами обычно яв-
ляются гидроксильные или эфирные группы.
Примером неионогенных ПАВ являются соединения, получае-
мые при взаимодействии одной молекулы высокомолекулярного
спирта (или другого органического соединения—кислоты, фено-
лов) с несколькими молекулами окиси этилена:
С„Н2„+|ОН + тСН2—СН2 —► C„H2n+1(OCH2CH2)mOH
Схематически молекулы НПАВ обозначают следующим обра-
зом:
углеводородный радикал оксиэтиленовая цепочка
Оксиэтиленовая цепь обладает некоторой гидрофильностью
вследствие взаимодействия эфирного атома кислорода с молеку-
лами воды. Наряду с гидратносвязанной водой оксиэтиленовая
цепь, которая имеет достаточную длину и образует в растворе
клубки, связывает воду также и за счет энтропийного эффекта.
Поэтому продукты присоединения, начиная с некоторого опреде-
ленного числа оксиэтиленовых групп, которое зависит от молеку-
лярного веса и строения гидрофобной части молекулы, приобре-
тают растворимость в воде.
Существенным преимуществом оксиэтилированных ПАВ яв-
ляется возможность при их синтезе регулировать гидрофильность
путем изменения не только числа атомов углерода в гидрофобной
цепи, но и числа оксиэтиленовых групп. Благодаря этому можно
403
получать вещества с заранее выбранными свойствами, рассчитан-
ными на конкретную область применения. Другой особенностью
этих веществ является то, что они не образуют соли и поэтому хо-
рошо растворимы в жесткой воде. Кроме того, НПАВ могут быть
использованы в сочетании с ионогенными ПАВ. Адсорбция неионо-
генных ПАВ из водных растворов превращает гидрофобные по-
верхности в гидрофильные.
Мы остановились здесь только на типичных представителях
коллоидных ПАВ и мылоподобных веществ. За последние 40—
50 лет химия и технология производства ПАВ очень быстро раз-
вивались, и в настоящее время известны уже многие сотни синте-
тических поверхностно-активных веществ.
Важной характеристикой молекул коллоидных ПАВ, имеющей
решающее значение для их поверхностных и объемных свойств,
а значит, и для их применения, является соотношение двух проти-
воположных групп молекулы •— гидрофильной и гидрофобной (ли-
пофильной), так называемый гидрофильно-липофильный баланс
(ГЛБ). В настоящее время не существует строгой теории, позво-
ляющей определить значение ГЛБ, исходя из строения молекулы
или из физико-химических свойств вещества. В качестве первого
приближения пользуются предложенной Гриффином и развитой
Дэвисом полуэмпирической системой ГЛБ, позволяющей с энерге-
тических позиций количественно оценить и выразить в виде услов-
ных групповых чисел степень взаимодействия с водой отдельных
групп, из которых состоит молекула ПАВ. Групповые числа гидро-
фильных групп положительны, а липофильных —- отрицательны.
Числа ГЛБ различных ПАВ могут быть вычислены по спе-
циальным формулам как сумма групповых чисел или определены
экспериментально. Чем больше баланс сдвинут в сторону гидро-
фильности, тем выше число ГЛБ. Обычно число ГЛБ неионоген-
ных ПАВ не превышает 20, для ионогенных ПАВ, например олеата
калия, оно равно 20, а для лаурилсульфата калия оно составляет
около 40. Числа ГЛБ определяют области применения ПАВ. Для
эмульсий типа в/м значение ГЛБ должно изменяться в интервале
от 3 до 6, для смачивателей — от 7 до 9, для моющих веществ оно
обычно лежит между 13 и 15 и для эмульсий типа м/в в зависи-
мости от природы масла изменяется от 8 до 18.
Система ГЛБ является в известной степени формальной, так
как она исходит из стехиометрического состава соединения и не
учитывает геометрических особенностей строения его молекул, на-
пример, изомерии. Она позволяет определить области применения
ПАВ, не характеризуя достаточно полно его эффективность.
2. СОСТОЯНИЕ ПАВ В РАСТВОРЕ
Истинные растворы ПАВ. Максимально возможная концентра-
ция, при которой коллоидные ПАВ еще находятся в водном рас-
творе в молекулярной (ионной) форме, т. е. критическая концен-
404
трация мицеллообразования, не велика и изменяется в пределах
10-5— 10-3 моль/л. Мыла, которые являются слабыми электроли-
тами, могут существовать в растворе не только в виде ионов
(RCOO-, М+), но и в виде недиссоциированных молекул (RCOOM)
и продуктов их гидролиза — молекул жирной кислоты (RCOOH),
Соли сильных кислот, например сульфокислот, находятся в рас-
творе в виде ионов, а неионогенные ПАВ — в виде недиссоцииро-
ванных молекул.
Причины мицеллообразования. Выше критической концентра-
ции мицеллообразования ПАВ в водных растворах начинается
агрегация молекул и образуются мицеллы.
Состояние раствора при мицеллообразовании можно рассма-
тривать как обратимое равновесие, подчиняющееся закону дей-
ствия масс. Это равновесие для простейшего случая недиссоцииро-
ванных молекул, например молекул НПАВ, может быть описан»
уравнением*
тх (Х)щ
Другой подход к этому явлению основан на предположении, что-
мицеллообразование заключается в возникновении новой фазы
в системе вода —ПАВ. При этом мицеллы рассматривают каю
своеобразную фазу с предельной дисперсностью (псевдофазу), по-
скольку в отличие от истинного фазового разделения мицеллооб-
разование не приводит к бесконечно большому числу молекул
в агрегате (число молекул в агрегате сокращенно называют чис-
лом агрегации). Подтверждением фазовой теории мицеллообразо-
вания, которой придерживается большинство ученых, являются
по крайней мере, два обстоятельства: во-первых, постоянство
концентрации молекулярно растворенного ПАВ выше ККМ и, во-
вторых, наличие в области ККМ резкого излома на кривых зави-
симости физико-химическое свойство — концентрация. Термодина-
мическое рассмотрение показывает, что при больших числах агре-
гации (25 и больше) оба подхода равноценны.
Агрегат из молекул ПАВ образуется из углеводородных цепей,
а гидрофильные части молекул располагаются на внешней поверх-
ности мицеллы. Основной причиной возникновения в водных рас-
творах агрегатов из углеводородных цепей являются когезионные
силы между молекулами воды, которые больше, чем взаимное при-
тяжение молекул воды и углеводородных цепей. Молекулы воды
как бы «вытесняют» углеводородные цепи из раствора, что сопро-
вождается уменьшением энтальпии системы. Образованию мицелл
препятствуют силы взаимного отталкивания гидрофильных частей
молекул ПАВ. Для ионных ПАВ это отталкивание обусловлено
электростатическим взаимодействием одноименно заряженных
ионогенных групп, для НПАВ — осмотическими силами, которые
возникают из-за повышения концентрации оксиэтиленовых цепей
в периферической части мицеллы. Кроме того, образованию мицелл
препятствует падение энтропии из-за возрастания при агрегации
405
упорядоченности системы. Возрастание энергии отгалкирания по-
лярных групп и падение энтропии компенсируют убыль энергии
системы при агрегации углеводородных цепей и приводят к уста-
новлению равновесных значений К.КМ и размера мицелл.
Согласно этим традиционным представлениям, уменьшение свободной энер-
гии системы при мицеллообразоваиии связано с падением энтальпии системы и,
следовательно, мицеллообразоваиие должно сопровождаться выделением тепла.
•Однако при экспериментальном определении теплот мицеллообразоваиии было
установлено, что эти теплоты малы, а в некоторых случаях даже имеют отрица-
тельное значение, т е мицеллообразоваиие сопровождается поглощением тепла.
Этот результат можно объяснить особыми свойствами воды, обусловленными
существованием в воде структур ближнего порядка
Предложено много различных моделей состояния воды, но во всех этих
моделях признается образование льдоподобной ажурной тетраэдрической струк-
туры — каркаса, в котором молекулы воды соединены друг с другом водород-
ными связями В такой структуре каждая молекула воды в среднем окружена
четырьмя другими молекулами воды Наличие тетраэдрической структуры воды
было впервые предсказано в классической работе Бернала и Фаулера и под-
тверждено позже рентгенографическими исследованиями Наряду с молекулами,
входящими в каркас, существуют свободные молекулы воды, ие связанные водо-
родными мостиками Эти молекулы частично заполняют области неплотной
упаковки внутри структуры воды, перемещаясь в иих В результате теплового
движения между молекулами каркаса и свободными молекулами происходит
постоянный тепловой обмен Понижение температуры приводит к уменьшению
числа свободных молекул, тек упрочнению, или иначе, к стабилизации струк-
туры воды Повышение температуры дает обратный эффект — уменьшается чис-
ло молекул, входящих в каркас, и тетраэдрическая структура воды ослабляется.
Области неплотной упаковки внутри льдоподобиой структуры воды можно
приближенно рассматривать как пустоты, которые играют существенную роль
при растворении в воде неполярных органических веществ При растворении
молекулы этих веществ внедряются в структуру воды, что приводит к стабили-
зации структуры и к уменьшению внутренней энергии системы Одно из воз-
можных объяснений этого явления, предложенное О Я Самойловым, состоит
в том, что молекулы неполярных органических веществ, заполняя пустоты, огра-
ничивают перемещение свободных молекул воды и их тепловой обмен с моле-
кулами воды каркаса Это способствует стабилизации структуры воды Ввиду
того чю ПАВ содержат неполярную, углеводородную цепочку, их растворение
также приводит к стабилизации структуры воды
Одновременно с растворением ПАВ происходит объединение углеводород-
ных частей молекулы ПАВ в водной среде — так называемое гидрофобное вза-
имодействие Причиной гидрофобного взаимодействия является ослабление струк-
туры воды при переходе углеводородных цепочек из водных растворов ПАВ в
ядро мицеллы, где между ними возникают ван-дер-ваальсовы связи Разрушение
структуры воды, а также увеличение конформационной энтропии углеводородных
цепочек в ядре мицеллы по сравнению с их энтропией в водной фазе приводят
к повышению энтропии системы В работах Немети и Шерага, а также в иссле-
дованиях П А Ребиидера и 3 Н Маркиной показано, что процесс образования
мицелл является типичным случаем гидрофобного взаимодействия Это озна-
чает, что в энергетическом балансе мицеллообразовання появляется новая со-
ставляющая — увеличение энтропии системы, что и определяет во многих слу-
чаях энтропийный характер мицеллообразоваиии Конечно, ие следует забывать,
что при образовании мицелл идет и противоположный процесс — падение энтро-
пии В результате в качестве компромисса возникают малые мицеллы, так как
при образовании крупных мицелл происходила бы значительная убыль энтропии.
Образование мицеллярных растворов ионных ПАВ возможно лишь выше
некоторого критического значения температуры, так называемой точки Крафта
(рис XIII, 2) Прн низких температурах гидратированное твердое ПАВ образует
набухший гель, равновесный с истинным раствором (участок 1) В точке Крафта
406
иеионогенных ПАВ являются
Рис XIII,2. Фазовая диа-
грамма растворов ПАВ
вблизи точки Крафта (с —
концентрация ПАВ в рас-
творе, t — температура).
1 — кривая молекулярной раство-
римости ПАВ ниже точки Крафта;
2—кривая растворимости (моле-
кулярной и мицеллярной) выше-
точки Крафта, 3— кривая моле-
кулярной растворимости (ККМ>
выше точки Крафта, 4—точка^
Крафта.
углеводородные цепи ПАВ переходят как бы в жидкое состояние При stow
в результате теплового движения углеводородных цепей ориентированные слои
молекул ПАВ в геле распадаются на частицы коллоидных размеров — мицеллы.
Образование мицелл из геля сопровождается возрастанием энтропии, способствую-
щим протеканию этого процесса Общая растворимость ПАВ резко возрастает
(участок 2), тогда как истинная растворимость, характеризуемая критической-
концентрацией мицеллообразования, существенно не изменяется (участок 3).
Точка Крафта не совпадает с температурой плавления твердого ПАВ, а лежит
ниже, так как в набухшем геле ПАВ гидратировано и это облегчает плавление.
То, что мицеллы образуются лишь выше точки Крафта, позволяет рассматривать,
их как жидкую фазу Жидкое состояние углеводородного ядра мицелл подтвер-
ждается также и тем, что они могут образовывать смешанные мицеллы с раз-
личными добавками Большинство гидратированных ~
жидкими при достаточно низких температурах, и
поэтому для них точка Крафта отсутствует
Строение мицелл. При критической
концентрации мицеллообразования в си-
стеме из отдельных молекул ПАВ обра-
зуются так называемые мицеллы Гарт-
ли. Эти мицеллы представляют собой
сферические агрегаты, в которых-углево-
дородные цепи молекулы переплелись,
а полярные группы обращены наружу
(в воду). Число молекул ПАВ в одном
таком агрегате обычно находится в пре-
делах 50—100, но может доходить и до
1000. Диаметр подобного сферического
агрегата, по Гартли, примерно вдвое
больше длины молекулы ПАВ, из кото-
рого он образован.
Нетрудно видеть, что для ионогенных
ПАВ строение такого сферического агре-
гата полностью сходно со строением типичной коллоидной ми-
целлы. В самом деле, агрегированные углеводородные цепи, кото-
рые образуют как бы капельку жидкого углеводорода, играют
в мицелле роль агрегата обычной мицеллы, а частично диссоции-
рованные ионогенные группы, находящиеся в воде, образуют двой-
ной электрический слой.
При более высоких концентрациях ПАВ в растворах обра-
зуются мицеллы уже иного строения По мере повышения концен-
трации раствора размер мицелл увеличивается и углеводородные
цепи располагаются в них все более и более параллельно друг
другу. В результате образуются пластинчатые мицеллы, состоящие
из двух слоев молекул ПАВ, обращенных друг к другу углеводо-
родными цепями, а ионогенными группами наружу Эти мицеллы
напоминают по своему строению двухмерный кристалл и могут
иметь неограниченно большие размеры в двух направлениях. Пла-
стинчатые мицеллы ионогенных ПАВ заряжены гораздо более
слабо, чем сферические, поскольку при сравнительно высокой
концентрации ПАВ, при которой они образуются, уменьшается
407
диссоциация ионогенных групп. Благодаря этому пластинчатые
мицеллы располагаются параллельно друг другу, причем соседние
мицеллы обращены одна к другой своими поверхностями, на ко-
торых находятся гидратированные группы молекул ПАВ. Вслед-
ствие образования пластинчатых мицелл и их характерного рас-
положения достаточно концентрированные растворы способны пе-
реходить в гель, показывающий под микроскопом своеобразную
структуру, состоящую как бы из лент и пленок.
Существование пластинчатых мицелл в достаточно концентри-
рованных растворах мыла впервые предположил Мак-Бен, поэтому
их часто называют мицеллами Мак-Бена. Впоследствии существо-
вание пластинчатых мицелл мыла было доказано рентгенографии
чески.
Образование различных мицелл в растворах ПАВ по мере по-
вышения их концентрации изображено схематически на рис. XIII, 3.
Определение критической концентрации мицеллообразования
основано на том, что при образовании мицелл, как уже отмечалось
выше, происходит изменение всех свойств раствора ПАВ, тем более
резкое, чем больше число агрегации молекул. Нефелометрическим
методом установлено, что образование мицелл приводит к резкому
увеличению светорассеяния раствором ПАВ вследствие того, что
он становится гетерогенным.
Мак-Бен показал, что концентрационная зависимость осмоти-
ческого коэффициента * для растворов мыл выражается кривой,
схематически изображенной на рис. XIII, 4. Эта кривая в отличие
ют монотонной кривой, характерной для слабых электролитов, не
-образующих мицелл, состоит из трех участков, соответствующих
предкритической области, области, в которой осмотический коэф-
фициент резко изменяется с концентрацией, и послекритической
области, где кривая идет почти параллельно оси концентраций.
Характер первого участка кривой изменяется в той точке, которая
соответствует уменьшению осмотической активности раствора в ре-
зультате образования сферических мицелл, т. е. отвечает ККМ.
Аналогично этой кривой изменяется с концентрацией и эквива-
лентная электропроводность, раствора ПАВ. На рис. XIII, 5 пока-
заны типичные концентрационные кривые эквивалентной электро-
проводности для растворов обычных мыл. Выбранный масштаб
графика не позволяет изобразить на нем начальные участки кри-
вых, отвечающие предкритической области. Однако по форме вто-
рого и третьего участков можно видеть, что эквивалентная элек-
тропроводность растворов мыл, содержащих 12 и более атомов
углерода в цепи, резко падает в области небольших концентраций
и достигает минимального значения в точке, отвечающей образо-
ванию мицелл, а затем несколько увеличивается. При этом с уве-
* Осмотический коэффициент Бьеррума равен отношению фактического чис-
ла частиц в растворе к тому, которое имело бы место при полной диссоциации
электролита.
408
личением молекулярного веса ПАВ этот минимум сдвигается
в сторону меньших концентраций и становится более выраженным.
Понижение эквивалецтной электропроводности с повышением
р \ <
г
Рис. XIII, 4. Зависимость осмотиче-
ского коэффициента G от концентра-
ции раствора мыла с.
Рис. XIII, 5. Зависимость эквивалент-
ной электропроводности Л растворов
мыл от концентрации с (при 18°С):
1 — каприлат калия; 2—лаурат калия;
3— олеат калия.
Рис. XIII, 3. Схема образова-
ния мицелл в растворах мыла:
а—разбавленный раствор, содержа-
щий отдельные молекулы мыла и
ионы; б—более концентрированный
раствор, содержащий сферические
мицеллы; в, г—концентрированные
растворы, содержащие пластинчатые
мицеллы.
концентрации мыла в растворе, согласно Гартли, обусловливается
частичным присоединением противоионов к ионизированным ми-
целлам. Это присоединение происходит под действием значитель-
ных электрических сил, вызванных присутствием образовавшихся
в системе высокозаряженных ионных мицелл. Причины повыше-
ния эквивалентной электропроводности после достижения мини-
мума не вполне ясны,
409
. Наконец, ККМ можно определять по изменению поверхностного
натяжения коллоидного раствора ПАВ при повышении его кон-
центрации. С увеличением содержания ПАВ поверхностное натя-
жение раствора всегда падает, достигая в точке, отвечающей ККМ,
обычно предельного постоянного значения.
На ККМ в растворах ПАВ может влиять ряд факторов. Так,
ККМ снижается с увеличением молекулярного веса углеводород-
ной цепи ПАВ. Такая зависимость вполне понятна, потому что
-с увеличением длины углеводородной цепи уменьшается истинная
растворимость и возрастает склонность молекул ПАВ к ассоциа-
ции. Влияние температуры на ККМ различно для ионогенных и
неионогенных ПАВ. Для ионогенных ПАВ ККМ обычно повы-
шается с увеличением температуры вследствие дезагрегирующего
.действия теплового движения молекул. Однако этот эффект неве-
лик, так как он ослаблен гидрофобными взаимодействиями, сопро-
вождающимися увеличением энтропии системы. Поэтому влияние
температуры на ККМ проявляется тем слабее, чем'больше выра-
жены гидрофобные свойства мыл. Для неионогенных ПАВ ККМ
всегда уменьшается при повышении температуры. Это вызвано
тем, что при повышении температуры водородные связи между
эфирным атомом кислорода и молекулами воды разрушаются,
оксиэтиленовые цепи дегидратируются и уменьшается их взаимное
отталкивание, препятствующее агрегации.
Заканчивая рассмотрение процесса мицеллообразования, от-
метим, что мицеллы могут образоваться не только в водных рас-
творах коллоидных ПАВ, но и в растворах ПАВ в углеводородах.
При этом молекулы ПАВ в мицелле ориентированы полярными
группами внутрь мицеллы, а углеводородными концами обращены
к растворителю. Вместе с тем необходимо указать, что в спирте
ПАВ обычно дают молекулярные растворы. Это объясняется тем,
что спирт по своей полярности стоит между водой и углеводоро-
дами и, следовательно, является растворителем как для полярной,
так и для неполярной части молекул ПАВ.
3. СТАБИЛИЗУЮЩЕЕ ДЕЙСТВИЕ ПАВ
Стабилизующее действие ПАВ определяется их способностью
адсорбироваться на межфазной поверхности. Вследствие высокой
поверхностной активности концентрация ПАВ в поверхностном
слое в десятки тысяч раз превышает объемную концентрацию.
В адсорбционных пленках, так же как и в мицеллах ПАВ, проис-
ходит ассоциация неполярных групп. Строение адсорбционного
слоя зависит от природы ПАВ и межфазной поверхности, степенй
заполнения поверхности, введения в среду различных добавок. Из-
менение строения адсорбционного слоя отражается на его защит-
ных свойствах.
Адсорбционные слои ПАВ лиофилизируют, а в водных раство-
рах гидрофилизируют поверхность, вследствие чего при сближе-
410
нии частиц развиваются силы отталкивания. Для ионогеннык
ПАВ — это электростатическое отталкивание двойных электриче-
ских слоев, которое описывается рассмотренной в гл. IX теорией
ДЛФО. Для неионогеняых ПАВ электростатическое отталкивание,
как правило, не играет роли; вопрос о стабилизации дисперсных
систем этими веществами является одной из нерешенных проблем
коллоидной химии.
Наиболее перспективными кажутся следующие представления, развиваемые
как для неионогенных ПАВ, так и для адсорбционных слоев полимеров
При сближении частиц на расстояние меньшее, чем удвоенная толщина ад-
сорбционного слоя, происходит перекрытие (взаимопроникновение) адсорбцион-
ных слоев, и концентрация НПАВ в области перекрытия увеличивается по
сравнению с ее значением в адсорбционном
слое При этом, если среда представляет собой
хороший растворитель для вещества, образую-
щего адсорбционный слой, возникает осмотиче-
ское давление, подобное давлению набухания
(рис XIII, 6). Это обуславливает приток
жидкости из объема раствора в область
перекрытия адсорбционных слоев и возникно-
вение расклинивающего давления. Осмотиче-
ское давление, в зависимости от природы взаи-
модействия НПАВ и растворителя, может быть
функцией изменения энтропии или изменения
энтальрии системы в области перекрытия. В
первом случае падение энтропии определяется
тем, что в области перекрытия уменьшается
число конформаций гибких цепей стабилиза-
тора, что в конечном счете вызывает повыше-
ние агрегативной устойчивости. Во втором слу-
чае в области перекрытия некоторые контакты
Рис. XIII, 6. Схема, иллюстри-
рующая взаимодействие адсорб-
ционных слоев неионогенных
ПАВ.
г —радиус частиц; б—толщина ад-
сорбционного слоя, Н — расстояние-
между частицами.
между молекулами воды и полярными груп-
пами НПАВ заменяются контактами между
молекулами НПАВ, т е происходит дегидратация адсорбционного слоя Это
приводит к возрастанию энтальпии системы, вызывает отталкивание, т. е также
повышает агрегативную устойчивость системы
На основании представлений о роли осмотических сил в стабилизации дис-
персных систем немецким ученым Фишером было предложено уравнение, позво-
ляющее рассчитать энергию отталкивания UacM при сближении частиц на рас-
стояние меньше чем 26:
^осм=2ВУА"2^ (ХП U >
где В — коэффициент, характеризующий взаимодействие молекул растворяемого
вещества с растворителем, NА — число Авогадро, с — концентрация НПАВ в
межфазном слое; Гд — объем области перекрытия, который можно рассчитать,,
если известен радиус частиц, толщина адсорбционного слоя 6 НПАВ и расстоя-
ние между частицами
Как видно из уравнения (XIII, 1), энергия отталкивания тем
больше, чем выше концентрация адсорбтива в межфазном слое.
Она возрастает также с увеличением объема области перекрытия.
Кроме того, энергия отталкивания зависит от знака и значения ко-
эффициента В. В хороших растворителях, когда В > 0, возникает
положительное расклинивающее давление, стабилизующее си-
стему. Если среда является плохим растворителем для вещества,
образующего адсорбционный слой, то В < 0 и расклинивающее
411
давление отрицательно, т, е. среда вытекает из области перекры-
тия. В этом случае осмотический эффект, наряду с силами межмо-
лекулярного притяжения, способствует агрегации частиц. Нако-
нец, при В = 0 энергия отталкивания равна нулю.
Следует иметь в виду, что адсорбционные слои, даже при от-
сутствии взаимодействия с растворителем при В 0, представ-
ляют собой стерический барьер, препятствующий сближению ча-
стиц на достаточно малые расстояния, при которых существенную
роль начинают играть силы межмолекулярного притяжения. Ко-
нечно, эффективная стерическая стабилизация осуществляется
лишь тогда, когда адсорбционные слои насыщены, а образующие
их молекулы не способны десорбироваться при соударениях ча-
стиц. Для таких стерически стабилизованных систем невозможна
коагуляция с непосредственным контактом частиц, а возможна
лишь дальняя агрегация.
4. СОЛЮБИЛИЗАЦИЯ В РАСТВОРАХ ПАВ
Солюбилизация является важным свойством растворов ПАВ,
которое связано с их мицеллярной структурой.
При введении в достаточно концентрированные растворы ПАВ
практически нерастворимых в воде органических веществ (алифа-
тические и ароматические углеводороды, маслорастворимые кра-
сители и т. д.) последние способны коллоидно растворяться, или
солюбилизироваться. В результате такой солюбилизации обра-
зуются почти прозрачные термодинамически равновесные растворы.
Вещество, которое растворяется в растворах ПАВ, принято назы-
вать солюбилизатом.
Солюбилизирующая способность разных ПАВ весьма различна;
количество коллоидно растворенного органического вещества в го-
мологических рядах ПАВ возрастает с увеличением длины углево-
дородного радикала.
Количество солюбилизированного вещества увеличивается про-
порционально концентрации раствора ПАВ в области существова-
ния сферических мицелл и резко возрастает при образовании пла-
стинчатых мицелл. Солюбилизация зависит также от молекуляр-
ной структуры солюбилизата. Так, если в качестве солюбилизата
применяют углеводород, то при уменьшении его молекулярного
веса коллоидная растворимость повышается. Введение в солюби-
лизат полярных групп обычно также увеличивает солюбилизацию.
Согласно современным взглядам солюбилизация представляет
собой растворение органических веществ в мицеллах ПАВ. Воз-
можны различные механизмы солюбилизации. Неполярные угле-
водороды растворяются в ядре мицеллы (рис. XIII, 7 а), а поляр-
ные органические вещества (спирты, амины) располагаются в
мицеллах так, что их углеводородные цепи направлены внутрь
мицелл, а полярные группы — в водную фазу (рис. XIII, 76). Для
неионогенных ПАВ, содержащих полиоксиэтиленовые группы, из-
вестен еще один особый способ солюбилизации. Молекулы солю-
412
по-видимому, водородную СВЯЗЬ
Солюбилизат
Подерхностнмк/нибное
Вещество
Рис. XIII, 7. Схематическое изображение
солюбилизации углеводородов (а) и по-
лярных органических веществ (б) в ми-
целлах ПАВ.
билизата (например, фенола) не проникают в глубь мицелл, а рас-
полагаются в их периферической части, между изогнутыми окси-
этиленовыми цепями, образуя,
с эфирным атомом кислорода.
При солюбилизации мицел-
лярный вес ПАВ возрастает
не только за счет включения
молекул солюбилизата, но и
из-за увеличения числа моле-
кул ПАВ в мицелле. Подоб-
ная перестройка мицелл вы-
звана тем, что при солюбили-
зации углеводородов увеличи-
вается гидрофобность ядра ми-
целлы, а поэтому для сохра-
нения равновесия должно уве-
личиться и число молекул
ПАВ, образующих мицеллу.
При солюбилизации в пластинчатых мицеллах органическое
вещество входит внутрь мицеллы,
располагаясь между углеводо-
1Щ1ШЩГ
>_ 2_—_бензол —-
яшшпш
— — Вода -
Nrniimffi
s
Рис. XIH, 8. Солюбилизация бензола в мицелле олеата натрия:
а—мицеллярный раствор до солюбилизации; б —то же после солюбилизации.
родными концами молекул мыла и тем самым раздвигая слои мо-
лекулярных цепей. Правильность такой точки зрения подтверж-
дается рентгенографическими исследованиями, показавшими, что
при солюбилизации расстояние между молекулами мыла в ми-
целле увеличивается. Изменение строения пластинчатых мицелл
при солюбилизации изображено на рис. ХШ.8,
413
Явление солюбилизации чрезвычайно важно для проведения
полимеризации непредельных углеводородов в эмульсиях при син-
тезе латексов. Как показали многочисленные исследования, про-
цесс полимеризации идет в основном не в капелькаж непредель-
ного углеводорода, а внутри или на поверхности мицелл мыла,,
в которых солюбилизован этот углеводород.
Обратная солюбилизация — коллоидное растворение
воды в маслах в присутствии соответствующих коллоидно раство-
римых в масле поверхностно-активных веществ имеет большое
значение в пищевой промышленности, в частности, в маргариновом
производстве.
5. ПРАКТИЧЕСКОЕ ЗНАЧЕНИЕ РАСТВОРОВ КОЛЛОИДНЫХ ПАВ
Коллоидные поверхностно-активные вещества имеют огромное
практическое значение. Нет почти ни одной отрасли народного хо-
зяйства, где бы не использовали мыла или мылоподобные веще-
ства. Ценные технические свойства ПАВ обусловлены образованием
в растворах мицелл либо высокой поверхностной активностью,,
т. е. способностью их молекул образовывать поверхностные ад-
сорбционные слои.
О значении ПАВ для улучшения смачивания различных поверх-
ностей водой, для получения стойких эмульсий и пен, а также для
процессов флотации указано в гл. VI и XII. Остановимся теперь
на некоторых других применениях коллоидных ПАВ в технике.
Из процессов, при которых основную роль играет способность
коллоидных ПАВ образовывать адсорбционные слои, рассмотрим
несколько более подробно (см. гл. VI) моющее действие мыл.
Как известно, твердые или жидкие загрязнения удаляются
с поверхности волокон тканей чистой водой с большим трудом
даже при повышенной температуре и интенсивном механическом
воздействии. Однако процесс этот идет сравнительно легко, если
для стирки применяют раствор ПАВ. Моющее действие ПАВ свя-
зано с рядом различных эффектов.
1. В присутствии в воде ПАВ понижается поверхностное натя-
жение раствора, тем самым улучшается смачивание ткани моющей
жидкостью. Это способствует проникновению жидкости в, тонкие
капилляры загрязненной ткани, в которые чистая вода проникнуть
не может.
2. Молекулы мыла, адсорбируясь на поверхности волокна и
частицах твердых или жидких загрязнений, создают хорошо гид-
ратированный адсорбционный слой, что обусловливает возникно-
вение расклинивающего давления. Это способствует отрыву частиц
загрязнений от поверхности волокна и переходу их в моющую
жидкость.
3. Адсорбционные пленки на поверхности частиц загрязнений
придают этим частицам высокую агрегативную устойчивость и
предупреждают их прилипание к поверхности волокна в другом
месте.
414
4. В присутствии ПАВ в моющей жидкости образуется пена,
содействующая механическому уносу загрязнений или флотации
тех загрязнений, частицы которых вследствие пониженной способ-
ности к смачиванию прилипают к пузырькам воздуха.
5. Наконец, моющее действие ПАВ в известной степени свя-
зано со способностью Частиц загрязнений, особенно если они
имеют маслянистый характер, солюбилизироваться в их раство-
рах. Это подтверждается тем, что моющее действие часто обна-
руживается лишь при концентрациях, превышающих ККМ.
6. ТАН НИДЫ И КРАСИТЕЛИ
Свойства коллоидных ПАВ проявляют почти все дубящие ве-
щества, являющиеся, как известно; производными многоатомных
фенолов, в которых полярными и ионогенными группами являются
фенольные и карбоксильные группы. Имеются указания, что обра-
зование мицелл в водных растворах таннидов может обусловли-
ваться не только агрегацией молекул по гидрофобным участкам, но
и возникновением водородных связей. Согласно А. Н. Михайлову,
мицеллярный вес продуктов ассоциации в растворах дубящих
веществ составляет примерно 20 000, в то время как молекулярный
вес таннидов колеблется в пределах 1000—2000. Таким образом,
мицелла в этом случае состоит из 10—20 молекул.
К красителям, проявляющим в растворах все особенности,
свойственные растворам коллоидных ПАВ, относится ряд синтети-
ческих красителей, например, бензопурпурин, ночной голубой
и т. д. Ионогенными группами у коллоидных красителей служат
карбоксильные группы, фенольные группы, сульфо-группы, амино-
группы и т. д. Растворы этих красителей сходны с растворами вы-
сокомолекулярных соединений — они обладают сравнительно вы-
сокой агрегативной устойчивостью, а образующийся при введении
электролитов осадок способен диспергироваться в чистой воде.
Растворы этих красителей проявляют такие же аномалии в отно-
шении электропроводности и осмотического давления, как и рас-
творы мыл и таннидов. С. М. Липатов показал, что благодаря
большому размеру молекул красителей ассоциация в растворах
протекает значительно в большей степени, чем в растворах мыл, и
весьма сильно зависит от концентрации, температуры, pH системы,
присутствия электролитов и других факторов. Как и мыла, многие
красители, дающие коллоидные растворы в воде, в спирте обра-
зуют молекулярные растворы.
ГЛАВА XIV
ПРИРОДА И НЕКОТОРЫЕ СВОЙСТВА РАСТВОРОВ
ВЫСОКОМОЛЕКУЛЯРНЫХ ВЕЩЕСТВ
Растворы высокомолекулярных веществ представляют собой
истинные растворы, термодинамически устойчивые и обратимые,
не нуждающиеся в стабилизаторе. Частицы, содержащиеся в та-
ких растворах, состоят не из множества малых молекул, как это
имеет место у коллоидов, а представляют отдельные молекулы,
правда, относительно очень больших размеров. В этом собственно
и заключается отличие растворов высокомолекулярных соедине-
ний от растворов низкомолекулярных веществ. Тем не менее ряд
ученых (Кройт, Бунгенберг де Йонг, И. И. Жуков, Эдельман и др.)
относят растворы высокомолекулярных веществ к коллоидным
растворам, причем некоторые называют растворенные в них веще-
ства обратимыми коллоидами (Кройт, Бунгенберг де Йонг), а не-
которые— молекулярными коллоидами (И. И. Жуков, Эдельман).
Нетрудно видеть, что основные особенности, присущие лиозолям,
объясняются в случае растворов высокомолекулярных веществ
просто большим размером молекул, приближающимся, а в неко-
торых случаях даже превосходящим размер коллоидных частиц.
Поэтому растворы высокомолекулярных веществ едва ли правиль-
но относить к коллоидным растворам.
Однако существуют некоторые причины, действительно сбли-
жающие растворы высокомолекулярных веществ с коллоидными
системами. Так, растворы высокомолекулярных соединений в пло-
хих растворителях содержат молекулы (или агрегаты молекул),
свернутые в компактный клубок с явно выраженной межфазной
поверхностью. По существу, они представляют отдельную фазу.
Такие растворы высокомолекулярных соединений действительно
можно отнести к коллоидным системам. Далее, в концентрирован-
ных растворах высокомолекулярных веществ обычно возникают
достаточно большие ассоциаты макромолекул, существующие не-
определенно долго. Эти частицы также можно рассматривать как
вторую фазу или, по крайней мере, как зародыши этой фазы. На-
конец, растворы высокомолекулярных веществ благодаря большим
размерам их молекул обладают, как будет показано ниже, рядом
свойств лиозолей, что позволяет рассматривать многие проблемы
одновременно и для коллоидных растворов, и для растворов вы-
сокомолекулярных веществ.
416
Все это приводит к выводу о необходимости хотя бы весьма
кратко рассмотреть в нашем курсе природу и свойства растворов
высокомолекулярных веществ, обратив главное внимание на те
системы и те явления, которые ближе всего к коллоидным. Однако
прежде чем говорить о растворах высокомолекулярных веществ,
кратко остановимся на самих высокомолекулярных веществах и
рассмотрим особенности строения их молекул.
1. ОБЩИЕ СВЕДЕНИЯ О ВЫСОКОМОЛЕКУЛЯРНЫХ ВЕЩЕСТВАХ
К высокомолекулярным веществам * относятся вещества, со-
стоящие из больших молекул (макромолекул) с молекулярным
весом не менее 10 000—15 000. Нередко молекулярный вес природ-
ных высокомолекулярных соединений достигает нескольких мил-
лионов. Размер макромолекул весьма велик по сравнению с раз-
мерами обычных молекул. Например, если длина молекулы этана
равна всего нескольким ангстремам, то длина линейных молекул
каучука и целлюлозы достигает 4000—8000 А (при поперечном
размере 3—7,5 А). Моделью подобных молекул могут служить
нити длиной от 800 до 1600 мм и диаметром 1 мм.
Высокомолекулярные вещества вследствие их большого моле-
кулярного веса нелетучи и не способны перегоняться. По той же
причине высокомолекулярные вещества весьма чувствительны
к воздействию различных внешних факторов. Макромолекулы
легко распадаются под действием самых незначительных количеств
кислорода и других деструктирующих агентов. Причина различия
в чувствительности к химическим воздействиям низко- и высоко-
молекулярных веществ становится понятной из следующего при-
мера. Допустим, что для расщепления одной молекулы вещества
на две достаточно одной молекулы кислорода. Тогда для низко-
молекулярного вещества с молекулярным весом, равным, напри-
мер 100, количество кислорода, необходимое для такого расщепле-
ния, должно составлять 32% от его массы. Если же молекулярный
вес окисляемого вещества 100 000, то кислорода для той же цели
потребуется лишь 0,032% от его массы, что на 1 г вещества со-
ставит всего 32-10~5 г кислорода, т. е. количество, которое с тру-
дом может быть обнаружено даже с помощью самых точных ана-
литических весов. Большинство высокомолекулярных веществ при
повышении температуры размягчается постепенно и не имеет опре-
деленной температуры плавления. Температура разложения этих
веществ ниже температуры кипения. Следовательно, высокомоле-
кулярные вещества могут находиться только в конденсированном
состоянии.
* К высокомолекулярным веществам относятся и высокополимеры или, про-
сто, полимеры, т. е. высокомолекулярные вещества, получаемые в результате
полимеризации или конденсации и обладающие обычно линейным строением
молекул и часто высокой эластичностью.
14 Зак, 664
417
Свойства высокомолекулярных веществ зависят не только от
размера, но и от формы молекул.
Высокомолекулярные соединения с изодиаметрическими моле-
кулами (например, гемоглобин, печеночный крахмал — гликоген)
обычно представляют собой порошкообразные вещества. При рас-
творении они почти не набухают, а растворы этих веществ не
обладают высокой вязкостью даже при сравнительно больших кон-
центрациях и подчиняются закону вязкости Пуазейля, закону диф-
фузии Эйнштейна и закону осмотического давления Вант-Гоффа.
Высокомолекулярные соединения с сильно асимметрическими
вытянутыми молекулами (например, желатин, целлюлоза и ее про-
изводные, натуральный и синтетические каучуки) при растворе-
нии очень сильно набухают и образуют высоковязкие растворы,
не подчиняющиеся закономерностям, приложимым к растворам
низкомолекулярных веществ.
Характерной особенностью высокомолекулярных веществ с ли-
нейными молекулами является волокнистая структура, обусловли-
вающая анизотропию свойств и высокую механическую прочность.
Поэтому такие вещества обладают способностью образовывать во-
локна и пленки. Некоторые полимеры обладают ценнейшим свой-
ством — высокой эластичностью.
Природные высокомолекулярные вещества
К природным высокомолекулярным веществам относятся такие
важнейшие для биологии и техники органические вещества, как
белки, высшие полисахариды, натуральный каучук.
Белки (протеины) представляют собой сложнейшие высокомолекулярные со-
единения. Это основное вещество, которое входит в состав протоплазмы клеток
мышц, хрящей, сухожилий и кожи животных и человека. Они содержатся также
в шелке, молоке (казеин) и растениях, особенно в зернах пшеницы, семенах
бобовых (растительные белки). Все известные энзимы, многие гормоны и ви-
русы также состоят из белков. К белкам, применяемым в технике, следует отне-
сти желатин, казеин, яичный альбумин.
Молекулы белков состоят из аминокислот, содержат ионогенные группы
(—СООН, —NH3OH) и обладают амфотерными свойствами. Белки растворимы
в растворах щелочей, некоторые из них растворимы в воде и разбавленных рас-
творах солей и кислот. Растворы белков очень нестойки к действию температур:
при нагревании происходит денатурация многих белков и переход их в нерас-
творимую форму. Белки осаждаются из растворов электролитами, спиртом и
ацетоном. До сих пор многие белки из-за сложности строения не получены син-
тетическим путем.
Высшие полисахариды. К этим веществам относятся крахмал и целлюлоза.
Крахмал, макромолекула которого состоит из звеньев глюкозы, представляет
собой не индивидуальное вещество, а смесь полисахаридов, отличающихся не
только размером макромолекул, но И строением. Крахмал является одним из
важнейших продуктов фотосинтеза, образующихся в зеленых частях растений,
и составляет основную часть питательного вещества хлеба, картофеля и различ-
ных круп. В воде при определенной температуре крахмал набухает и клейсте-’
ризуется, образуя внешне однородную густую жидкость — крахмальный клей-
стер, который широко применяют в технике в качестве клея, для шлихтования
и отделки тканей, для проклеивания бумаги и т. д. Путем гидролиза из крахмала
получают декстрин, патоку и глюкозу.
418
Целлюлоза является главной составной частью организма растений, она
придает ему прочность и эластичность. Целлюлоза также состоит из длинных
цепочек, составленных из остатков глюкозы, но соединенных друг с другом не-
сколько иначе, чем в молекуле крахмала. Попытки синтезировать целлюлозу
еще не привели к положительным результатам, и поэтому ее получают из древе-
сины, соломы и других растительных материалов путем горячей обработки рас-
творами веществ, растворяющих содержащиеся в этих материалах лигнин и
другие примеси. Целлюлозу широко используют для получения бумаги. Хлопок
и другие виды растительного волокна, представляющие собой почти чистую цел-
люлозу, применяют в текстильном производстве для получения тканей. Производ-
ные целлюлозы — нитрат целлюлозы, ацетат целлюлозы и другие простые и слож-
ные эфиры целлюлозы — применяют для получения кинофотопленок и искусствен-
ного волокна
Натуральный каучук — чрезвычайно ценный материал, обладающий высокой
эластичностью Его добывают из млечного сока (латекса) некоторых растений
(каучуконосов). По своей природе — это углеводород, причем его макромолекулы
состоят из изопентеновых (изопреновых) остатков. Растворим в углеводородах,
обладает пластичностью, особенно заметно проявляющейся при повышении тем-
пературы При нагревании с небольшим количеством серы каучук вулкани-
зуется — молекулы его химически связываются друг с другом посредством мо-
стиков из серы. Вулканизованный каучук (резина) теряет способность раство-
ряться и размягчаться при нагревании, но сохраняет при этом эластические-
свойства. При нагревании с большим количеством серы в результате образова-
ния большого числа поперечных связей между его молекулами каучук теряет
эластичность и образует твердый вулканизат, называемый эбонитом.
Синтетические высокомолекулярные вещества
Помимо природных высокомолекулярных веществ в настоящее
время в технике и быту применяют ряд синтетических высокомоле-
кулярных продуктов. Сюда следует отнести синтетические каучуки
и различные синтетические полимеры. Эти продукты, чрезвычайно
разнообразные по химическому строению и свойствам, не только
являются полноценными заменителями природных высокомолеку-
лярных веществ, но и получают часто совершенно новое примене-
ние. Так, их используют для получения разнообразных пластмасс,
в виде органического стекла, в качестве ионообменных материалов
(ионитов) для очистки воды и выделения индивидуальных веществ,
из смесей, для изготовления деталей самолетов и автомобилей и
даже корпусов малотоннажных судов. Показательно, что произ-
водство синтетических высокомолекулярных веществ значительно
превысило производство не только традиционных конструктивных
материалов, но и таких сравнительно новых материалов, как алю-
миниевые и магниевые сплавы.
К достоинствам синтетических высокомолекулярных веществ,
относится то, что их можно получать с заданными свойствами —
прочностью, эластичность^), химической стойкостью, диэлектриче-
ской проницаемостью, подбирая подходящие исходные материалы
и регулируя технологический процесс. Кроме того, из синтетиче-
ских высокомолекулярных веществ можно получать изделия ме-
тодом непосредственного формования без потери материала в виде
стружки и без необходимости последующей обработки изделий.
• Широкое применение синтетических высокомолекулярных ве-
ществ объясняется также и неограниченными ресурсами сырья,
14:
419
необходимого для' их получения. Для синтеза высокомолекуляр-
ных веществ используются простейшие продукты переработки
нефти и угля, ацетилен, а также отходы ряда отраслей промыш-
ленности.
Синтетические каучуки. К синтетическим каучукам относятся изготовляемые
в промышленных масштабах полибутадиен, полихлоропрен, бутадиен-стирольные
и бутадиен-нитрильные сополимеры и ряд других продуктов. Строение молекул
этих веществ рассматривается во всех курсах органической химии.
Помимо собственно синтетических каучуков, которые благодаря наличию
в их макромолекулах двойной связи способны вулканизоваться так же, как и
натуральный каучук, известен ряд синтетических полимеров, лишенных этой спо-
собности, но обладающих высокой эластичностью. Сюда бтносится, например,
продукт низкотемпературной полимеризации изобутилена — полиизобутилен.
Пленки из полиизобутилена газонепроницаемы и не изменяются под действием
воздуха и озона, вследствие чего полиизобутилеи широко применяют для изго-
товления оболочек аэростатов, шаров-пилотов и т. д. При сополимеризации
изобутилена с небольшим количеством (2—3%) диена, например изопрена, полу-
чаются продукты, уже содержащие в молекуле небольшое число двойных связей.
Эти продукты, способные вулканизоваться, получили название бутилкаучуков.
Полисилоксаны. Это весьма интересные новые высокомолекулярные продук-
ты, применяемые в качестве материала для термостойкой электроизоляции. По-
лисилоксаны, как и каучуки, обладают высокой эластичностью, но благодаря
тому, что основная цепь полисилоксана состоит из атомов кремния и кислорода,
эти соединения весьма прочны и более стойки к температурным воздействиям.
Синтетические полимеры. К синтетическим полимерам, в обычных условиях
не обладающим высокой эластичностью, относятся полиэтилен, поливинилхлорид,
поливинилиденхлорид, поливинилацетат, полиметилакрилат, полиметилметакри-
лат, полистирол и ряд других широко известных Продуктов, идущих для изго-
товления изделий из пластмасс, пленок и т. д. Этн вещества являются термо-
пластичными, поскольку они могут размягчаться и формоваться при нагрева-
нии. К синтетическим полимерам относятся также термореактивиые смолы, теку-
чие в исходном состоянии и способные при нагревании в результате химических
реакций необратимо отвердевать. К таким смолам следует отнести феноло-форм-
альдегидные и мочевино-формальдегидные смолы, применяемые в технике уже
несколько десятилетий.
Из сравнительно новых полимеров назовем лишь полиамиды и политетра-
фторэтилен.
Полиамиды получают при поликонденсации диаминов с дикарбоновыми кис-
лотами, например при конденсации гексаметилендиамина и адипиновой кислоты,
полимеризацией со-аминокислот и другими методами, В результате этих реакций
получается полигексаметиленадипамид. Из полигексаметиленадипамида в США
изготовляют искусственное волокно найлон. Это волокно по свойствам близко
к шерстяному и шелковому волокнам, а по некоторым свойствам даже превос-
ходит их. Исключительно высокое сопротивление разрыву пайлонового волокна,
достигающее 4000—4500 кгс/см2, объясняется полярностью молекулы полигекса-
метиленадипамида, возможностью образования водородной связи между отдель-
ными молекулярными цепочками и тем, что в вытянутом волокне полиамид на-
ходится главным образом в ориентированном, кристаллическом состоянии. Близ-
ко по свойствам к найлону полиамидное волокно капрон, получаемое в Совет-
ском Союзе путем полимеризации капролактама.
Быстрому развитию химии соединений фтора обязано производство поли-
тетрафторэтилена, Молекулы этого полимера вследствие того, что сильно элек-
троотрицательные атомы фтора расположены в них симметрично, неполярны.
Благодаря правильной линейной структуре молекул политетрафторэтилен сравни-
тельно легко кристаллизуется.
Политетрафторэтилен является одним из наиболее высокоплавких кристал^
лических полимеров. Вместе с тем температура, при которой он становится хруп-
ким, очень низка. В результате этого изделия из политетрафторэтилена могут
420
работать в очень широком интервале температур. По термостабильности, хими-
ческой стойкости и диэлектрическим свойствам политетрафторэтилен превосходит
все известные до сих пор полимеры.
Неорганические высокомолекулярные вещества
Помимо органических, хорошо изученных высокомолекулярных соединений
существуют также и неорганические высокомолекулярные вещества. К сожале-
нию, строение их молекул, равно как и свойства их растворов, еще недостаточно
изучены.
К высокомолекулярным неорганическим веществам с цепйым строением мо-
лекулы можно отнести, например, одну из модификаций серы («пластическая»
сера), получаемую быстрым охлаждением расплава серы, нагретого выше 300 °C.
Благодаря цепному строению молекулы высокомолекулярная сера обладает
каучукоподобной эластичностью.
Рис. XIV, 1. Строение графита и алмаза:
а — плоскостные решетки графита; б — трехмерная структура алмаза.
Неорганическим высокомолекулярным веществом является также полифос-
фонитрилхлорид.
Примером двухмерных высокомолекулярных неорганических веществ служит
обыкновенная слюда, молекулы которой имеют форму пластинок. Сюда же
следует отнести и алюмосиликаты, из которых состоят глины, и графит, кри-
сталлы которого построены из углеродных шестиугольников, расположенных друг
под другом в виде отдельных плоскостей. Благодаря сравнительно большому
расстоянию между отдельными слоями и непрочности связи между ними можно
считать, что в графите молекулой является каждая отдельная плоскость.
Примером трехмерного неорганического высокомолекулярного соединения
может служить алмаз, состоящий из атомов углерода. В кристалле алмаза
каждый атом углерода связан с четырьмя другими ближайшими атомами угле-
рода. Кристалл алмаза можно считать одной гигантской молекулой, лишенной
ряда свойств, типичных для обычных молекул. Вследствие такого строения алмаз
не способен набухать, не растворяется ни в одном из растворителей и обладает
очень большой твердостью. Структура графита и алмаза показана на рис. XIV, 1.
Промежуточные системы
Особо следует отметить вещества промежуточного типа, рас-
творы которых, в зависимости от условий их получения, могут
проявлять свойства как типичных золей, так и растворов высоко-
молекулярных соединений. К таким веществам относятся окиси
и гидроокиси элементов, образующих слабокислые или амфотер-
ные соединения, например гидроокись кремния,
421
Свежеприготовленные растворы этих окисей и гидроокисей во
многом сходны с растворами высокомолекулярных соединений.
Эти вещества осаждаются из растворов при введении электролита,
но осадок легко вновь переходит в коллоидный раствор, если коа-
гулятор удалить. Такое осаждение и диспергирование, напри-мер
двуокиси олова, может быть произведено сколь угодно большое
число раз. Действие электролитов на растворы таких веществ и
влияние валентности иона, вызывающего понижение ^-потенциала
частиц, далеко не столь значительны, как для типичных коллоид-
ных систем, например металлических золей и золей сульфидов ме-
таллов. Относительная вязкость растворов подобных веществ зна-
чительно выше, чем обычных золей. Наконец, растворы их обла-
дают способностью давать студни, очень сходные по свойствам со
студнями высокомолекулярных веществ.
Все приведенное выше позволяет считать, что такие растворы
содержат макромолекулы. Например, можно предположить, что
в свежеприготовленном растворе ортокремневой кислоты ее мо-
лекула имеет следующее строение:
(HO)3SiO— [Si(OH)2—OJn—Si(OH)3
Вероятно, таким же образом можно представить и строение моле-
кул в проявляющих коллоидные свойства растворах гидроокиси
железа, гидроокиси алюминия и др. Однако известно, что подобные
растворы при стоянии или при добавлении электролитов могут
приобретать типичные свойства обычных коллоидных систем. Для
жидкого стекла это явление можно объяснить наличием у молекул
ортокремневой кислоты гидроксильных групп, благодаря чему при
добавлении, например, кислоты происходит сшивание молекул по-
перечными химическими связями. Если растворы достаточно раз-
бавлены, то вследствие сшивания участков одной и той же гибкой
макромолекулы могут образоваться отдельные мицеллы, причем
роль стабилизатора играет сама кремневая кислота.
Если растворы достаточно концентрированы, то вследствие по-
перечного сшивания макромолекул образуется трехмерная сетка
и раствор переводится в необратимый студень. Такого рода явле-
ние очень близко к тем процессам конденсации, которые происхо-
дят при отвердении феноло-формальдегидных смол. Отличие за-
ключается в том, что студни кремневой кислоты после высушива-
ния и прокаливания способны обнаруживать кристаллическую
структуру (в результате образования кристалликов).
Промежуточные системы, стоящие между растворами высоко-
молекулярных соединений и типичными коллоидными растворами
или же способные переходить из одного класса растворов в дру-
гой, не следует смешивать с растворами коллоидных поверхностно-
активных веществ, отличающимися тем, что в одних условиях они
проявляют свойства истинных растворов, а в других — типичные
свойства золей.
422
2. ПОЛИДИСПЕРСНОСТЬ И МОЛЕКУЛЯРНЫЙ ВЕС
ВЫСОКОМОЛЕКУЛЯРНЫХ ВЕЩЕСТВ
Полидисперсность полимеров
При полимеризации или поликонденсации, вследствие неодина-
ковости технологических условий, обычно получаются макромоле-
кулы различных размеров, т. е. образуется полимергомологический
ряд. Поэтому принято говорить о полидисперсности высокомоле-
кулярного вещества, которую удобно характеризовать кривыми
распределения, аналогичными тем, которые рассматривались
в гл. III. На рис. XIV, 2 изображены типичные кривые распреде-
ления для одного и того же полимера. Одна кривая характеризует
распределение по числу (а), другая — по массе макромолекул- (б).
Рис. XIV, 2. Кривые распределения фракций высокомолекулярных
соединений:
а—по числу частиц; б—по массе частиц.
Существенная разница в форме обеих кривых объясняется тем,
что малые макромолекулы, обычно присутствующие в продукте
реакции в большом количестве, составляют сравнительно неболь-
шую весовую фракцию.
Следует заметить, что набухание и растворение высокомолеку-
лярных соединений, свойства их растворов, а также физико-меха-
нические свойства самих высокомолекулярных веществ существен-
ным образом зависят от их полидисперсности. Поэтому определе-
ние и регулирование степени полидисперсности высокомолекуляр-
ных веществ играют в технике большую роль.
Для получения более однородных по степени полимеризации
или по молекулярному весу высокомолекулярных продуктов обыч-
но проводят их фракционирование методами растворения или оса-
ждения.
Фракционирование растворением основано на зависимости растворимости
полимергомологов от размера молекул: чем меньше молекула, тем лучше раство-
ряется гомолог. Практически для разделения на фракции высокомолекулярное
вещество обрабатывают сначала жидкостью, растворяющей низкомолекулярные
члены полимергомологического ряда, затем оставшийся продукт обрабатывают
жидкостью, растворяющей уже более высокомолекулярные фракции, и т. д. Часто
вместо того, чтобы пользоваться различными жидкостями для экстрагирования,
применяют смеси двух жидкостей, _из которых одна служит хорошим раствори-
телем для всех фракций, а другая является для них нерастворителем. Изменяя
в смесях, которыми последовательно проводится экстрагирование, соотношение
423
растворителя и нерастворителя, продукт можно разделить на сколь угодно
большое число фракций различного молекулярного веса. Из растворов, получен-
ных в результате фракционированного растворения, экстрагированную фракцию
осаждают, прибавляя большое количество нерастворителя. Например, для фрак-
ционирования нитрата целлюлозы можно использовать смеси воды с ацетоном,
для фракционирования каучука — смеси этилового спирта с бензолом.
Фракционирование осаждением проводят из раствора высокомолекулярного
вещества, в котором содержатся все фракции. Приливая к этому раствору все
возрастающие количества нерастворителя, в осадке последовательно получают
ряд фракций. Естественно, что молекулярный вес этих фракций будет тем. меньше,
чем больше было прилито нерастворителя, так как наиболее низкомолекулярные
фракции всегда обладают наибольшей растворимостью.
Следует отметить, что при фракционировании как растворением, так и оса-
ждением выделение совершенно однородных фракций, в которых все молекулы
имели бы одну и ту же длину, практически невозможно, поскольку с уменьше-
нием разницы в длине цепи различие в растворимости ' (равно как и во всех
других физических свойствах) становится весьма незначительным.
Очень часто высокомолекулярные вещества содержат примеси — электро-
литы, низкомолекулярные органические вещества. Для очистки высокомолекуляр-
ных веществ применяют диализ. Техника диализа растворов высокомолекуляр-
ных веществ ничем не отличается от диализа типичных коллоидных систем. Если
в водном растворе очищаемого продукта присутствуют только электролиты, для
удаления их с успехом можно применять электродиализ. Если продукт нерас-
творим в жидкости, выбранной для диализа, а способен только набухать в ней,
то диализ можно заменить простым вымачиванием высокомолекулярного веще-
ства при периодической смене жидкости.
Методы определения молекулярного веса
высокомолекулярных веществ
От молекулярного веса (как и от полидисперсности) высоко-
молекулярных соединений зависят такие их свойства, как проч-
ность и эластичность, способность к набуханию и растворению.
Поэтому химику, имеющему дело с высокомолекулярными соеди-
нениями, важно уметь определять их молекулярный вес.
Обычные методы определения молекулярного веса органиче-
ских соединений, мало пригодны для определения молекулярного
веса, высокомолекулярных соединений. Поэтому разработаны со-
вершенно новые методы определения молекулярного веса этих ве-
ществ, многие из которых близки к методам определения числен-
ного веса, применяемым в коллоидной химии. Здесь мы лишь
кратко укажем принципы, на которых основаны эти методы.
Все методы определения молекулярного веса высокомолекулярных соедине-
ний могут быть разделены на четыре группы.
1. Химические методы. Если в молекуле высокомолекулярных соедине-
ний имеется известное, строго определенное число реакционноспособных групп
(например, групп, расположенных на конце молекулы), то количественное опре-
деление этих групп может служить методом определения молекулярного веса.
Эти методы, в связи с рядом экспериментальных трудностей, сравнительно мало
применяются на практике. Они подробно рассматриваются в курсах химии вы-
сокомолекулярных веществ.
2. Термодинамические методы определения молекулярных весов
основаны иа термодинамических закономерностях, характерных для разбавлен-
ных растворов, и сводятся к определению мольной доли вещества в растворе
известной концентрации.
424
Молекулярный вес в этом случае определяют по осмотическому давлению,
по понижению температуры замерзания раствора (криоскопический метод) и по
повышению температуры кипения раствора полимеров (эбулиоскопический ме-
тод).
3. Молекулярно-кинетические методы основаны на перемеще-
нии макромолекул относительно растворителя и сводятся, в конечном счете, к
определению соответствующей силы трения. К этой группе методов относятся
определение молекулярного веса по скорости диффузии, с помощью ультрацен-
трифуги и по вязкости растворов.
4. Оптический ме.тод, получивший в последнее время широкое рас-
пространение, основан на измерении интенсивности рассеянного света раство-
рами высокомолекулярных соединений.
Опыт показал, что из термодинамических методов единственно приемлемым
является метод определения молекулярного веса по осмотическому давлению.
В этом методе не приходится встречаться с теми трудностями, которые препят-
ствуют применению этого метода для определения численного веса коллоидных
систем, так как растворы высокомолекулярных соединений хорошо выдерживают
очистку.
Криоскопический и эбулиоскопический методы пригодны для определения
молекулярного веса полимеров с сравнительно малыми молекулами, так как в
случае очень большого молекулярного веса растворенного вещества в разбавлен-
ных растворах (к которым только и приложимы эти методы) не удается устано-
вить ощутимой разности температур замерзания и кипения.
Из молекулярно-кинетических методов определений молекулярного веса наи-
более проверенным и теоретически обоснованным является метод ультрацентри-
фугирования.
Особого внимания заслуживает метод определения молекулярного веса по
седиментационному равновесию, так как в этом случае в расчетную формулу
не входит время анализа и сила трения и, следовательно, исключаются те про-
извольные допущения в отношении формы частиц, к которым приходится прибе-
гать при использовании других молекулярно-кинетических методов.
Из молекулярно-кинетических методов определения молекулярного веса наи-
более простым, хотя и не очень точным, является вискозиметрический метод.
Размеры молекул высокомолекулярных веществ, имеющие очень большой
молекулярный вес порядка 10б—107 (вирусы, гемоцианин и т. д.), можно опре-
делить и непосредственно с помощью электронной микроскопии.
Средний молекулярный вес
Выше уже указывалось, что высокомолекулярные вещества состоят из смеси
молекул различного размера, но построенных по одному и тому же принципу из
одних и тех же мономерных остатков. Это понятие об индивидуальном высоко-
молекулярном веществе существенно отличается от понятия об индивидуальном
низкомолекулярном соединении, молекулы которого имеют один и тот же строго
определенный размер.
Поскольку высокомолекулярное вещество всегда представляет собой смесь
полимергомологов, т. е. молекул различного молекулярного веса, то приходится
говорить лишь о его среднем молекулярном весе. Существенно, что это среднее
значение зависит от того, какой экспериментальный метод был применен для его
нахождения.
Обычно молекулярный вес полимеров выражают через так называемые чис-
ловые и весовые средние значения Мп и Ма. Среднее числовое значение моле-
кулярного веса определяется по уравнению:
где П{ — число молекул, имеющих молекулярный вес Mt. Средний числовой мо-
лекулярный вес находят с помощью всех тех методов, которые позволяют опре-
делять число молекул в исследуемом растворе высокомолекулярного вещества.
Сюда относятся осмотический, криоскопический и эбулиоскопический методы.
425
Среднее весовое значение молекулярного веса находят по уравнению:
MG = ^GzAfI./^Gi = ^niM?/£nlAf; (XIV, 2)
где G{ — масса фракции с молекулярным весом М,. Средний весовой молекуляр-
ный вес определяют с помощью методов, позволяющих устанавливать средний
размер молекулы в растворе. К таким мотодам относятся, например, определе-
ние молекулярного веса по скорости диффузии, седиментации в ультрацентри-
фуге, а также по светорассеянию. _
Совершенно очевидно, что для монодисперсного продукта Мп = Ма- Для
полидисперсного вещества Afn < Мо, поскольку Ма растет с увеличением поли-
дисперсности. Очевидно также, что чем сильнее различаются молекулярные веса
фракций высокомолекулярного вещества, тем больше должно быть отношение
Мс/Мп. Величина Ма/Мп, характеризующая полидисперсность высокомолеку-
лярного вещества, называется показателей, или коэффициентом,
полидисперсности.
При малом содержании в высокомолекулярном веществе низкомолекулярной
фракции значения Мп и Ма незначительно отличаются друг от друга, и отноше-
ние Ма[Мп приближается к единице. При большом содержании низкомолеку-
лярной фракции расхождения между значениями Мп и Ма резко возрастают
и отношение Ма/Мп становится весьма большим. Ниже приводятся вычислен-
ные значения Мп и Ма для смеси, состоящей из молекул с молекулярным весом
100 000 и 1000:
Содержание фракции с молекулярным весом
100 ООЭ, %.................................... 99 50
Содержание фракции с молекулярным весом 1000, % 1 50
Мо ............................................ 99 000 50 500
Мп............................................. 50 000 1 980
MJMn......................................... 1,98 2550
Сг' п
Определять Мп и Ма полезно и тогда, когда известно, что высокомолеку-
лярное вещество более или менее монодисперсно. Если в этом случае наблю-
дается значительное расхождение между Мп и Mg, то это свидетельствует
о разветвленном строении молекул.
3. СТРОЕНИЕ МАКРОМОЛЕКУЛ И СТРУКТУРА
ВЫСОКОМОЛЕКУЛЯРНЫХ ВЕЩЕСТВ
Молекулы высокомолекулярных веществ могут быть линейными
и разветвленными, причем длина молекулярных цепей может быть
сравнительно большой — превышать 1 мкм. Именно линейной фор-
мой макромолекул определяются типичные свойства полимеров:
каучукоподобная эластичность, способность образовывать проч-
ные пленки и нити, набухать, давать при растворении вязкие рас-
творы и т. д.
Кроме цепных молекул, в определенных технологических усло-
виях или в результате специальной обработки могут быть
получены полимеры с пространственной структурой. Конечно, по-
нятие молекулы у трехмерных полимеров теряет смысл, так как
пространственные сетки могут достигать весьма больших разме-
ров. Типичным примером полимера с пространственной структурой
может служить вулканизованный каучук, состоящий из каучуко-
вых молекул, «сшитых» друг с другом серными мостиками,
426
Ниже рассмотрены свойства цепных макромолекул, так как
растворы, содержащие такие. молекулы, представляют наиболь-
ший интерес для теории и практики. Полимеры с пространственной
структурой не способны растворяться, поэтому свойства молеку-
лярных сеток в нашем курсе не рассматриваются.
Гибкость линейных макромолекул
Еще не так давно некоторые ученые считали линейные макро-
молекулы жесткими. Однако опыт последних десятилетий показал,
что в большинстве случаев линейные макромолекулы обладают
гибкостью.
Чтобы понять причину гибкости линейных макромолекул, рас-
смотрим строение молекулы этана. Ее можно представить как две
группы —СН3, соединенные одинарной связью. Из органической
химии известно, что группы —СН3 в молекуле этана способны
вращаться вокруг одинарной связи С—С. Это хорошо согласуется
с тем, что, например, ^симметричный дихлорэтан не имеет изоме-
ров. Однако при низких температурах свободное вращение вокруг
связи С—С затруднено, так как не все возможные положения
групп —СН3 относительно друг друга равноценны в энергетиче-
ском отношении. Такая неравноценность обусловлена тем, что при
повороте одной группы —СНз по отношению к другой изменяются
расстояния между атомами водорода обеих групп. Это ведет к из-
менению энергии взаимодействия между группами —СН3. По-
этому при низких температурах —СН3-группы не вращаются во-
круг оси С—С, а лишь вращательно колеблются на сравнительно
небольшой угол. Только при достаточно высокой температуре бла-
годаря увеличению кинетической энергии может быть преодолен
энергетический барьер и группы — СН3 будут свободно вращаться
вокруг соединяющей их связи.
Высота потенциального барьера вращения, естественно, тем
больше, чем больше разность максимального и минимального зна-
чений энергии взаимодействия атомов или групп атомов при их
вращении относительно друг друга. Понятно также, что свободное
вращение затруднено тем больше, чем больше по размеру атомы,
связанные с углеродными атомами. Опыт показал, что даже когда
углеродные атомы связаны со сравнительно малыми атомами во-
дорода, свободное вращение вокруг связи С—С требует энергии
активации порядка 3000 кал/моль.
Следует заметить, что вращение возможно, если атомы угле-
рода соединены одинарной связью. Вращение групп = СН2 или
=СН (например, в молекуле этилена или ацетилена), т. е. вра-
щение вокруг двойной или тройной связи, невозможно. Вместе
с тем наличие двойной связи в углеводородной цепи благоприятно
влияет на подвижность смежных с этой связью атомов или групп,
соединенных одинарной связью. Так, в группе —СН2—СН=СН—
потенциальный барьер, отвечающий вращению вокруг связи
427
СН2—СН, на 500 кал/моль ниже энергетического барьера враще-
ния вокруг связи СН2—СН2.
Перейдем теперь к рассмотрению какой-нибудь линейной мак-
ромолекулы, например цепочки насыщенного углеводорода. Хо-
рошо известно, что такая цепочка, состоящая из связанных оди-
нарной валентностью атомов углерода и присоединенных к каж-
дому из них двух атомов водорода, расположена не в плоскости,
а в пространстве и имеет в идеальном случае зигзагообразную
форму. При этом угол между каждыми тремя соседними атомами
углерода имеет постоянное значение, равное 109°28', и для изме-
нения его требуется значительная затрата энергии. Строение це-
почки насыщенного углево-
дорода схематически изо-
бражено на рис. XIV, 3.
Гибкость углеводородной
цепочки, как показали Кун,
Марк и Гут, обусловливает-
ся в основном вращением
одних участков цепи относи-
тельно других вокруг оди-
Рис. XIV, 3. Строение линейной углеводо-• парной валентной связи, со-
роднои цепи. единяющей соседние атомы
углерода. Так как таких от-
дельных связей в макромолекуле множество, то становится по-
нятной та исключительная гибкость, которой обладают углеводо-
родные цепочки.
Кун, Марк и Гут в первом приближении полагали, что враще-
ние вокруг связей С—С в макромолекуле может происходить со-
вершенно свободно, и считали углеводородные молекулы весьма
гибкими нитями, которые могут принимать любую форму, совме-
стимую с постоянством валентного угла. Однако Я. И. Френкель
и С. Я. Бреслер показали, что и в этом случае, как и для моле-
кулы этана, необходима некоторая энергия активации, т. е. необхо-
димо преодолеть энергетический барьер, зависящий от природы
атомов, из которых построена цепь. Иными словами, вращение во-
круг связи С—С возможно только при достаточно высоких тем-
пературах. Такая зависимость свободы вращения от температуры
становится понятной из следующих рассуждений.
С повышением температуры возрастает энергия теплового дви-
жения, равная, как известно, для каждой частицы в среднем про-
изведению kT (yjifi k — постоянная Больцмана, Т — абсолютная
температура). Пока величина kT значительно меньше энергетиче-
ского барьера, группы атомов только вращательно колеблются
около положения равновесия. С возрастанием температуры ам-
плитуда этих колебаний увеличивается и, когда значение kT ста-
новится соизмеримым со значением энергетического барьера, на-
чинается вращение и молекулы приобретают гибкость. Естественно,
что при понижении ’температуры наблюдается обратное явление—-
428
свободное вращение вокруг одинарной связи становится невозмож-
ным и молекула теряет гибкость.
Если при вращении вокруг связи С—С молекула может при-
нимать не одно, а несколько положений равновесия, то полимер
в каждом из таких положений по существу представляет собой
отдельный изомер. Такие изомеры М. В. Волькенштейн назвал по-
воротными изомерами. Очевидно, обычные полимеры являются
смесями поворотных изомеров. Поворотные изомеры отличаются от
цис- и транс-изомеров тем, что энергетический барьер у них срав-
нительно невысок и они легко переходят друг в друга.
Мы рассматривали линейную макромолекулу как растянутую
зигзагообразную цепочку. Такая форма отвечает наименьшей по-
тенциальной энергии молекулы, и при очень низких температурах
линейные макромолекулы стремятся принять эту форму. Вытяну-
тая форма молекулярных цепочек способствует их ориентации,
обеспечивает возможность компактной упаковки; именно этим и
объясняется способность полимеров, состоящих из линейных мак-
ромолекул, кристаллизоваться при растяжении. С повышением
температуры, вследствие увеличения гибкости макромолекул, воз-
растания интенсивности теплового движения отдельных звеньев И
благодаря толчкам, получаемым от соседних молекул, линейные
макромолекулы могут свертываться и образовывать клубки. По-
скольку предельно вытянутое состояние линейной макромолекулы
может быть лишь одним, а конформаций *, которые может прини-
мать макромолекула при свертывании, очень много, то естествен-
но, что при достаточно высоких температурах все гибкие молекулы
полимера будут представлять собой клубки. При этом состоянии
энтропия полимерной системы максимальна, благодаря чему клуб-
кообразное состояние гибких макромолекул при достаточно высо-
ких температурах отвечает и минимуму свободной энергии.
Следует отметить, что цепные макромолекулы с ограниченным
вращением звеньев часто представляют как цепи, состоящие из
жестких участков, способных совершать тепловое движение как
бы независимо друг от друга. Такое движение принято называть
микроброуновски м, а принимающие в нем участие отрезки
цепи — сегментами. Под длиной сегмента, согласно В. А. Кар-
гину и Г. Л. Слонимскому, следует понимать значение молекуляр-
ного веса, которое должны были бы иметь молекулы полимера
для того, чтобы полимер или его раствор подчинялись обычным
для низкомолекулярных веществ закономерностям. Таким обра-
зом, сегмент является всего лишь эквивалентной величиной, по-
зволяющей описывать поведение реальных полимерных систем при
помощи хорошо известных закономерностей, свойственных идеаль-
ным системам.
Длиной сегмента часто приходится пользоваться при изучении
физических свойств полимера или его раствора. Если эти свойства
* Конформациями называются формы молекул, переходящие друг в друга
за счет свободного внутримолекулярного вращения,
429
независимы друг от друга, то и сегменты могут не совпадать друг
с другом. Так, длина сегмента, вычисленная по данным вискози-
метрии, обычно не совпадает с длиной сегмента, найденной на ос-
новании осмометрических измерений. Поэтому, говоря о длине сег-
мента, всегда следует указывать, к какому свойству относится эта
величина.
Понятно, что чем гибче молекула, тем меньше должен быть ее
сегмент. Как правило, сегмент гибких молекул представляет собой
отрезок цепи, состоящий из 20—30 атомов элемента, образующего
главную цепь. У макромолекул, содержащих полярные группы,
сегмент значительно больше.' У весьма жестких молекул размер
молекулы и длина сегмента совпадают. Поскольку гибкость моле-
кул зависит от температуры, то, понятно, что с увеличением тем-
пературы длина сегмента должна уменьшаться. Длина сегмента
зависит также от времени воздействия внешней силы. Так как
релаксационный переход молекулы от одной конформации к дру-
гой требует определенного времени, то при быстрых внешних воз-
действиях на молекулу часть конформаций не может осуществиться
и конформационный набор обедняется. Это приводит к увеличению
жесткости макромолекулы и возрастанию длины сегмента.
Из сказанного следует, что сегмент, несмотря на условность,
является весьма показательной характеристикой гибкости цепи
в данных конкретных условиях.
Выше мы рассматривали линейную макромолекулу независимо
от остальных молекул вещества. Однако реальные макромолекулы
всегда находятся в конденсированной фазе, т. е. окружены дру-
гими макромолекулами. Поэтому при рассмотрении поведения мо-
лекулы необходимо учитывать как взаимодействие между входя-
щими в молекулу группами атомов, так и взаимодействие между
этими группами и атомными группами соседних молекул. Таким
образом, свобода вращения вокруг связи С—С зависит не только
от расположения атомных групп в молекуле, но определяется и
близлежащими атомными группами других молекул. Иначе, вслед-
ствие межмолекулярных взаимодействий возможность изменения
формы цепных макромолекул определяется не только самой моле-
кулой, но и соседними молекулами.
Рассмотрим теперь связь гибкости макромолекул с их химиче-
ской природой. Наиболее гибкими линейными макромолекулами
являются цепочки углеводородов, так как из-за незначительного
взаимодействия —СНг— или —СН3-групп между собой энергети-
ческий барьер свободного вращения невелик. К таким высокомо-
лекулярным углеводородам принадлежат натуральный каучук, по-
либутадиен (бутадиеновый синтетический каучук), полиизобути-
лен *.
* На эластические свойства высокомолекулярных углеводородов в сильной
степени может влиять их способность кристаллизоваться. Естественно, что кри-
сталлизующиеся высокомолекулярные углеводороды будут проявлять малую эла-
стичность и обладать достаточно высокой жесткостью,
430
Наличие в макромолекуле полярных заместителей, например
—С1, —ОН, —CN, —СООН, делает молекулу менее гибкой, так
как взаимодействие между этими заместителями повышает энер-
гетический барьер. Кроме того, полярные заместители обусловли-
вают увеличение взаимодействия с полярными группами соседних
молекул. Между этими группами, являющимися диполями, могут
возникать как значительные межмолекулярные силы (например,
в поливинилхлориде между атомами хлора), так и водородная
связь, если имеются соответствующие условия (например, в поли-
мерах акриловой кислоты между карбоксильными группами). Все
это приводит к уменьшению гибкости цепи и повышает жесткость
полимеров. К полимерам с цепями ограниченной гибкости (из-за
содержания в них полярных групп) можно отнести целлюлозу,
поливинилхлорид, полиакрилонитрил и т. д.
Разветвление у макромолекул также сильно влияет на их гиб-
кость. Короткие и часто расположенные боковые цепи увеличивают
жесткость макромолекул вследствие возрастания энергетического
барьера вращения отдельных звеньев. Более редкие, но более
длинные разветвления способствуют уменьшению межмолекуляр-
ных взаимодействий и обусловливают «разрыхление» высокомо-
лекулярного вещества. Очевидно, при увеличении длины боковой
углеводородной цепи выше оптимальной стерические затруднения
начинают перекрывать эффект снижения молекулярных сил при-
тяжения.
Существенно, что на гибкость макромолекулы могут влиять не
только соседние молекулы того же полимера, но и молекулы рас-
творителя или пластификатора (например, в растворах или пла-
стифицированных полимерах). При этом наличие растворителя
между молекулами полимера может приводить, не только к умень-
шению энергетического барьера из-за понижения вязкости системы
и экранирования полярных групп макромолекул, но и к увеличе-
нию энергетического барьера в результате своеобразной «адсорб-
ции» молекул растворителя на макромолекулах, что, конечно, пре-
пятствует свободе вращения вокруг связи С—С. Короче говоря,
следует помнить, что при растворении в зависимости от условий
цепи полимера могут становиться как более гибкими, так и более
жесткими.
Фазовые состояния высокомолекулярных веществ
Как известно, вещества могут находиться в трех фазовых состояниях —
газообразном, жидком и кристаллическом. Критерием того, что данное вещество
находится в том или иной фазовом состоянии, служат не внешние признаки,
а степень упорядоченности ионов или молекул, из которых состоит вещество.
Газообразное состояние является наиболее беспорядочным фазовым состоянием.
В кристаллическом состоянии, наоборот, вещество имеет наиболее упорядочен-
ную структуру; в этом случае наблюдается дальний порядок, т. е. в кристалле на
всем его протяжении повторяется в определенном порядке один и тот же
структурный элемент. Жидкое состояние занимает среднее положение, в нем
отсутствует дальний порядок, но наблюдается ближний порядок, т. е. упорядо-
чение структурных элементов в отдельных участках жидкости.
431
В результате многочисленных исследований структуры высокомолекулярных
веществ установлено, что твердые высокомолекулярные вещества могут нахо-
диться как в жидком, так и в кристаллическом фазовом состоянии. К числу
высокомолекулярных веществ, имеющих аморфную жидкую структуру при обыч-
ной (комнатной) температуре, следует отнести большинство высокомолекулярных
веществ, в частности эфиры целлюлозы, натуральный и большинство синтетиче-
ских каучуков, поли-изо-бутилен и другие продукты. Кристаллическими поли-
мерами в этих же условиях являются полиамиды, полиэтилен.
Еще не так давно принимали, что в аморфном состоянии полимеры пред-
ставляют собой систему хаотически перепутанных макромолекул. Однако работы
последних десятилетий показали, что на самом деле уже в аморфном состоянии
в полимерах наблюдается некоторая структурная упорядоченность. Эта упоря-
доченность, естественно, резко возрастает при кристаллизации. Согласно
В. А. Каргину и Г. Л. Слонимскому, в аморфных прлимерных веществах точно
так же, как и в обычных жидкостях, имеются области ближнего порядка, в ко-
торых молекулы ориентированы параллельно друг другу, образуя достаточной
длины пучки, или пачки. Существование таких пачек ни в коей мере не про-
тиворечит высокой эластичности полимеров, так как макромолекулы могут при-
нимать различные конформации и тогда, когда они образуют пачки. Молекулы
могут различно располагаться в пачках, да и сами пачки могут принимать
самую разнообразную форму.
В свою очередь пачки в полимерах могут образовывать более сложные
структуры, вплоть до хорошо видимых в микроскоп кристаллов и сферолитов.
Таким образом, очень часто полимеры, особенно с жесткими и стереоспецифиче-
скими молекулами, не имеют той простой структуры, которую им приписывали
ранее, а представляют собой весьма сложные системы, содержащие как отдель-
ные молекулы, так и надмолекулярные структуры различной степени сложности.
Понятно, что при растворении полимеров в зависимости от природы раствори-
теля и условий растворения в раствор могут переходить как отдельные моле-
кулы, так и целые фрагменты надмолекулярной структуры.
4. ТЕОРИИ РАСТВОРОВ ВЫСОКОМОЛЕКУЛЯРНЫХ ВЕЩЕСТВ
Еще 20—40 лет тому назад существовали две теории растворов
полимеров. Согласно одной из них (мицеллярная теория), разви-
той Майером и Марком, макромолекулы находятся в растворе в
виде мицелл, согласно второй — достаточно разбавленные растворы
высокомолекулярных веществ содержат отдельные, друг с другом
не связанные макромолекулы (молекулярная теория).
В соответствии с мицеЛлярной теорией кинетическими единицами в растворе
высокомолекулярных веществ являются пачки макромолекул, удерживаемые
вместе межмолекулярными силами. Основанием для создания этой теории по-
служил взгляд на полимеры как иа кристаллические вещества, состоящие из
кристаллитов (мицелл) — пучков из 40—60 ориентированных макромолекул. Пред-
полагалось, что при растворении эти кристаллиты не распадаются, а продолжают
существовать в растворе. Представления, положенные в основу мицеллярной
теории, по существу являлись переносом на раство'ры высокомолекулярных ве-
ществ представлений о природе типичных коллоидных систем — лиозолей/ И те
и другие системы признавались гетерогенными, термодинамически неравновес-
ными и неустойчивыми. Различие между этими двумя типами систем видели
в том, что для обеспечения агрегативной устойчивости типичных лиозолей в них
должен быть стабилизатор, в то время как растворы высокомолекулярных ве-
ществ достаточно устойчивы и без стабилизатора из-за большого сродства дис-
персной фазы к дисперсионной среде. Поэтому растворы высокомолекулярных
веществ раньше обычно и назывались «лиофилными коллоидными системами»,
432
Весьма сильной сольватацией объясняли и отличия других свойств раство-
ров полимеров и типичных гидрофобных золей. Высокую вязкость растворов
полимеров объясняли либо энергетическим связыванием растворителя, либо его
иммобилизацией в мицеллах полимера. Более быстрое возрастание осмотиче-
ского давления в растворах полимера с увеличением концентрации вещества,
чем это требуется по линейному закону, также интерпретировали, исходя из того,
что в этих растворах растворитель частично связывается полимером и в резуль-
тате этого увеличивается отношение числа частиц дисперсной фазы к числу
молекул свободного растворителя.
Однако представление о каком-то особом сродстве полимеров к раствори-
телям не имеет достаточных оснований. Еще в 1932 г. Маринеско, определяя
количество воды, энергетически связываемой крахмалом, путем сравнения зна-
чений диэлектрической проницаемости раствора со значениями диэлектрических
проницаемостей его компонентов получил данные, указывающие, что это количе-
ство воды незначительно и приблизительно соответствует образованию моно-
молекулярного слоя. А. В. Думанский, а также С. М. Липатов в результате
калориметрических исследований пришли к такому же выводу. Наконец, к ана-
логичным выводам пришел и А. Г. Пасынский, определявший сольватацию по
сжимаемой части растворителя. Этот метод основан на том, что в сольватной
оболочке растворитель находится под большим внутренним давлением; сжимае-
мость он определял по скорости распространения ультразвука в растворах.
Ниже приведены обобщенные результаты исследований А. Г. Пасынского по
гидратации различных полярных групп ряда органических соединений;
Группа................ —ОН —СООН >СО —СНО
Число молекул воды, свя-
зываемых группой ... 3 4 2 2
Еще более убедительны результаты исследований растворов полимеров в их
собственных гидрированных мономерах. В этом случае говорить о сольватации
вообще бессмысленно, поскольку силы взаимодействия между большими и ма-
лыми молекулами полимера и мономера имеют одну и ту же природу и должны
быть примерно одинаковыми. Однако и здесь наблюдались все явления, харак-
терные для раствора# полимеров. Все это опровергает представление о каком-то
особом значении сольватации для устойчивости растворов полимеров и указы-
вает на несостоятельность исходных положений мицеллярной теории.
В настоящее время мицеллярная теория потеряла свое значе-
ние. Как показали тщательные исследования, высокомолекулярные
соединения в подходящих растворителях самопроизвольно диспер-
гируются до отдельных молекул. Сходство типичных золей и рас-
творов полимеров основано не на том, что и в этих системах су-
ществуют мицеллы, а на том, что в обоих случаях в растворе со-
держатся частицы сравнительно большого размера (в первом слу-
чае — мицеллы, во втором — макромолекулы).
Почти одновременно с мицеллярной возникла молекулярная
теория. Один из авторов молекулярной теории Штаудингер пред-
ставлял себе эти макромолекулы в растворе в виде жестких пало-
чек. Однако впоследствии было показано, что поведение этих мо-
лекул в растворе более сходно с поведением свернутых в клубок
гибких нитей.
Следует указать, что конформации свернутой нити в растворе
в результате теплового движения все время меняются. В целом,
однако, форма клубка всегда остается близкой к форме вытяну-
того эллипсоида вращения, Это подтверждается тем, что в то
433
время как длина линейных макромолекул превосходит их попереч-
ный размер в сотни и тысячи раз, экспериментально определенная
степень асимметрии этих молекул в растворе достигает всего де-
сяти.
Молекулярный клубок при не слишком большой длине цепи
еще не плотен, и сквозь просветы может протекать растворитель.
Однако по мере увеличения длины макромолекулы изогнутые
участки цепи все больше закрывают просветы и в конце концов
такой клубок почти полностью теряет способность пропускать рас-
творитель, если, конечно, градиент скорости потока растворителя
не достаточно велик для того, чтобы развернуть молекулярный
клубок в вытянутую цепочку. Понятно, что чем жестче молекула,
тем менее компактен клубок и тем сильнее он вытянут.
Макромолекулы в растворе, конечно, могут находиться в соль-
ватированном состоянии. Однако, как уже указано, сольватация
макромолекул в общем невелика, если под сольватно связанной
средой понимать растворитель, энергетически взаимодействующий
с полимером.
Молекулярная теория находит подтверждение в ряде фактов
и наблюдений. Во-первых, определение молекулярных весов в раз-
бавленных растворах полимеров методами, прямо указывающими
молекулярный вес частиц (например, методом светорассеяния),
однозначно показало отсутствие в таких растворах мицелл, т. е.
частиц, состоящих из агрегатов молекул. Во-вторых, растворение
высокомолекулярного вещества, как и растворение низкомолеку-
лярных соединений, идет самопроизвольно, часто с выделением
тепла. Например, достаточно желатин внести в воду, а' каучук
в бензол, чтобы через некоторое время без какогб-либо вмешатель-
ства извне образовался раствор полимера в растворителе. При
диспергировании же вещества до коллоидного состояния, как из-
вестно, требуется затрата энергии на преодоление межмолекуляр-
ных сил. В-третьих, растворы полимеров термодинамически устой-
чивы и при соответствующих предосторожностях могут храниться
сколь угодно долго. Коллоидные растворы, наоборот, термодина-
мически неустойчивы и способны стареть. Это объясняется тем, что
при растворении полимеров всегда образуется гомогенная система
и свободная энергия уменьшается, как и при получении растворов
низкомолекулярных • веществ, либо за счет выделения тепла в ре-
зультате взаимодействия полимера с растворителем, либо за счет
увеличения энтропии. При получении же гетерогенной коллоидной
системы ее свободная энергия всегда возрастает в результате уве-
личения поверхности дисперсной фазы. В-четвертых, растворение
высокомолекулярных соединений не требует присутствия в системе
специального стабилизатора. Лиофобные же золи не могут быть
получены без специального стабилизатора, придающего системе
агрегативную устойчивость. Наконец, растворы полимеров нахо-
дятся в термодинамическом равновесии и являются обратимыми
системами: к ним приложимо известное правило фаз Гиббса.
434
Отдельные указания на применимость правила фаз к раство-
рам высокомолекулярных веществ имелись еще в начале XX сто-
летия. В. А. Каргин с сотр. подробно исследовал подобные си-
стемы и установил связь между применимостью правила фаз
к растворению высокомолекулярных соединений и термодинами-
ческой устойчивостью и обратимостью растворов. Наиболее важ-
ной в этой области является работа В. А. Каргина, 3. А. Роговина
и С. П. Папкова по исследованию растворов ацетата целлюлозы
в различных растворителях — хлороформе, дихлорэтане, метило-
вом спирте, нитробензоле, метилэтилкетоне,
бензоле, толуоле, этилацетате. Авторы
установили, что при ограниченной рас-
творимоСти ацетата целлюлозы после во
расслаивания системы и достижения рав- 50
новесия как в верхнем, так и в нижнем
слое раствора устанавливается опреде-
ленная концентрация ацетата целлюлозы 30
в зависимости от температуры. Процесс 20
растворения оказался строго обратимым jq
и термодинамически равновесным, кон-
центрации были одними и теми же при о
подходе к заданной температуре как пу-
метилпропилкетоне,
1,0 2,0 3,0 4,0 5,0 бр 7,0
Ацетилцеппюлом, %
тем нагревания, так и путем охлаждения.
На рис. XIV, 4 в качестве примера приве-
дена диаграмма состояния раствора аце-
тата целлюлозы в хлороформе. Эта диа-
Рис. XIV, 4. Диаграмма со-
стояния системы ацетил-
целлюлозы — хлороформ.
грамма чрезвычайно сходна с диаграммой состояния двух ограни-
ченно смешивающихся жидкостей с верхней критической темпера-
турой растворения. На основании этих данных авторы пришли к
однозначному выводу, что растворы ацетата целлюлозы подчи-
няются правилу фаз и являются обратимыми системами.
Равновесность и обратимость растворов полимеров были также
доказаны В. А. Каргиным, 3. А. Роговиным, А. А. Тагер и другими
исследователями на опытах с бензилцеллюлозой, нитратом целлю-
лозы, поливинилхлоридом, желатином и другими высокомолеку-
лярными веществами. Эти исседователи получали насыщенные
растворы полимеров в плохих растворителях различными путями.
Опыты показали, что если равновесие (расслоение раствора на
две фазы или достижение предела растворения) достигалось при
одной и той же температуре ..и давлении, то всегда получались
растворы одинаковой концентрации. Для перечисленных высоко-
молекулярных веществ были получены диаграммы состояния
с верхней критической температурой растворения. Однако имеются
данные, что для растворов метилцеллюлозы и этилцеллюлозы
в воде получаются диаграммы с нижней критической температу-
рой.
Все это свидетельствует о том, что при растворении высокомо-
лекулярных веществ, как и при растворении низкомолекулярных
435
соединений, соблюдается правило фаз, не приложимое к коллоид-
ным системам. Правда, установление равновесия в растворах поли-
меров вследствие их весьма большой вязкости может длиться
весьма долго (например, несколько месяцев), и химику, получаю-
щему подобные системы, практически очень часто приходится
иметь дело с неравновесными системами. Однако это не меняет
существа вопроса *.
Все сказанное выше о том, что молекулы полимеров не связаны
друг с другом и ведут себя вполне самостоятельно, верно лишь
в том случае, когда они находятся в относительно разбавленных
растворах. В концентрированных растворах, когда вероятность
столкновения молекул растворенного вещества достаточно велика,
макромолекулы могут взаимодействовать и образовывать так на-
зываемые рои, или асссоциаты. Эти ассоциаты, состоящие из
сравнительно малого числа молекул и представляющие собой про-
образ пачек, о которых мы уже говорили при рассмотрении струк-
туры полимеров, обычно не обладают достаточной протяженно-
стью и поэтому не могут считаться фазой. Кроме того, ассоциаты,
в отличие от мицелл, существуют не постоянно, они возникают
в одном месте, затем распадаются и снова возникают в другом.
Таким образом, ассоциаты в разбавленных растворах полимеров
не являются постоянно существующими образованиями и не имеют
определенного состава.
Следует отметить, что образование ассоциатов характерно не
только для растворов высокомолекулярных веществ. Ассоциаты
образуются и в растворах низкомолекулярных веществ, а также
и в индивидуальных жидкостях за счет столкновения и сцепления
двух, трех, четырех и более молекул. Таким образом, явление
ассоциации близко к явлению флуктуации плотностей в растворах
и индивидуальных жидкостях. Особенностью образования ассоциа-
тов в растворах высокомолекулярных веществ является то, что
длинные и гибкие макромолекулы могут входить отдельными
своими участками в состав различных ассоциатов.
Связи, которые образуются между макромолекулами, входя-
щими в ассоциат, имеют ту же природу, что и связи между моле-
кулами в ассоциатах, возникающих в низкомолекулярных систе-
мах. Эти связи могут образовываться за счет межмолекулярных
взаимодействий, возникновения водородной связи, а в водных рас-
творах и за счет электростатических сил. Какие силы превалируют
.при образовании тех или иных ассоциатов, необходимо решать
в каждом отдельном случае. При этом обязательно следует учиты-
вать природу растворителя. Понятно, например, что в воде взаи-
модействие должно происходить, в основном, по углеводородным
частям молекул, а в углеводородной среде по полярным группам.
* Более подробно о применимости правила фаз к растворам высокомолеку-
лярных веществ см. Воюцкий С. С. Растворы высокомолекулярных соединений.
Изд 2-е, М., «Химия», 1960. См. с. 46.
436
На ассоциацию сильно влияет температура. С повышением
температуры увеличивается тепловое движение ассоциатов, и это»
ведет к их разрушению. Наоборот, понижение температуры спо-
собствует ассоциации.
Средний период жизни ассоциатов в растворах низкомолеку-
лярных веществ очень мал и составляет около 10-10 с. В растворах,
высокомолекулярных веществ средний период жизни ассоциатов,
значительно больше, так как для того, чтобы громоздкие макромо-
лекулы оторвались друг от друга, требуется гораздо больше вре-
мени. Процессы образования и разрушения ассоциатов при изме-
нении температур-ы или концентрации обратимы, т. е. при заданных
условиях среднестатистический размер ассоциатов в растворе вы-
сокомолекулярных веществ является вполне определенным. Одна-
ко поскольку периоды жизни ассоциатов, состоящих из макромоле-
кул, более длительны, то, естественно, что равновесное значение
степени ассоциации в растворах высокомолекулярных веществ
устанавливается обыкновенно не сразу.
С увеличением концентрации растворов высокомолекулярных
веществ или с понижением их температуры размер и длительность
существования ассоциатов макромолекул увеличиваются. Это мо-
жет привести к тому, что при известных условиях ассоциаты сде-
лаются настолько большими и прочными, что их можно будет
рассматривать как новую фазу. При образовании этой второй
фазы система приобретает способность к расслаиванию, которое
может проявиться, например, в коацервации — выделении ново-
образовавшейся фазы в виде мельчайших капель.
Благодаря тому, что макромолекулы имеют значительную
длину и гибкость, а также могут входить в состав различных ас-
социатов, явление ассоциации в растворах в итоге может привести
и к образованию в системе пространственной сетки, что прояв-
ляется в застудневании раствора. Наличие таких сеток, обеспечи-
вающих эластические свойства даже у сравнительно разбавленных
растворов полимеров, было доказано Фрейндлихом и Зейфрицем
\же в начале XX столетия. В результате наблюдений под микро-
скопом эти исследователи установили, что если на мельчайшую
крупинку никеля в даже очень вязкой жидкости действует магнит-
ное поле, то эта крупинка может перемещаться в жидкости на
сколь угодно большое расстояние. В растворах же высокомолеку-
лярных веществ крупинка передвигается в магнитном поле на
очень небольшое расстояние и затем останавливается, а после пре-
кращения действия поля возвращается в первоначальное положе-
ние под влиянием эластических сил, обусловленных существова-
нием в растворе сетки из макромолекул. Застудневший раствор
обычно со временем претерпевает синерезис, разделяясь на две
фазы, — раствор высокомолекулярного вещества в растворителе и
раствор растворителя в высокомолекулярном компоненте. Из ска-
занного следует, что ассоциаты — это не что иное, как зародыши
новой фазы.
437
Наряду с растворами полимеров могут существовать и их дис-
персии, обладающие всеми свойствами коллоидных систем (см.
гл. XII, разд. 8). Частицы таких дисперсий представляют*собой
большие агрегаты макромолекул.
5. ТЕРМОДИНАМИКА РАСТВОРЕНИЯ ВЫСОКОМОЛЕКУЛЯРНЫХ
ВЕЩЕСТВ
Растворение высокомолекулярных веществ принято рассматри-
вать как процесс смешения двух жидкостей. Эта точка зрения при-
нимает во внимание как энергетическое взаимодействие между мо-
лекулами растворяемого вещества и растворителя, так и действие
энтропийного фактора, обусловливающего равномерное распреде-
ление молекул растворенного вещества в растворе. Аналогия ме-
жду растворением высокомолекулярного вещества и смешением
двух жидкостей не является формальной, а отвечает самому суще-
ству явления. Так, ограниченное набухание высокомолекулярного
вещества соответствует процессу ограниченного смешения, а неог-
раниченное набухание, переходящее в растворение, — процессу не-
ограниченного смешения.
Самопроизвольное растворение высокомолекулярного соедине-
ния, равно как и всякого другого вещества, при постоянном дав-
лении должно сопровождаться уменьшением изобарно-изотерми-
ческого потенциала (свободной энергии при постоянном давле-
нии) .
Согласно второму закону термодинамики изменение изобарно-
изотермического потенциала системы составляет:
&G = &Н — Т &S (XIV, 3)
где Н — энтальпия; Т — абсолютная температура; S— энтропия.
Когда система не изменяет своего объема, что с известным
приближением можно принять для процесса растворения, уравне-
ние (XIV, 3) можно заменить следующим:
&F — &U — Т AS (XIV, 4)
где F — изохорно-изотермический потенциал (свободная энергия); U — внутрен-
няя энергия
Очевидно, для того чтобы произошло • самопроизвольное рас-
творение полимера, AG или AS должны иметь отрицательное зна-
чение. Это может быть в двух случаях.
1. При условии АЯ < 0 (или At7<ZO), которое соблюдается,
если при растворении выделяется тепло, так как изменение эн-
тальпии (или внутренней энергии) равно интегральной теплоте
растворения с обратным знаком. Такое условие часто соблюдается
на практике, например при растворении полярных полимеров в по-
лярных растворителях. Положительный тепловой эффект при рас-
творении объясняется тем, что теплота сольватации макромоле-
кул больше теплоты собственно растворения, а, как извстпо, об-
438
щий тепловой эффект растворения равен алгебраической сумме
теплот сольватации и собственно растворения.
2. При условии AS > 0, которое всегда осуществляется на
практике при растворении, так как энтропия смешения всегда по-
ложительна. Энтропия смешения высокомолекулярного вещества
с растворителем, рассчитанная на весовую долю вещества, лежит
между значениями энтропии растворения низкомолекулярных ве-
ществ и типичных коллоидных систем. Поэтому относительная
роль энтропийного фактора при растворении высокомолекулярных
соединений меньше, чем при растворении низкомолекулярных, и
энергетический фактор (сольватация) имеет относительно большое
значение. Вместе с тем, поскольку в рассматриваемом случае эн-
тропийный член не равен нулю, а может иметь сравнительно боль-
шие значения, некоторые полимеры способны растворяться с по-
глощением, а не с выделением тепла, т. е. при А// > 0 (или
At/>0). Это обусловлено тем, что в таких случаях
(или TAS > At/) и, следовательно, AG (или AF) меньше 0.
Как известно, мольная энтропия смешения двух веществ для идеальней си-
стемы или изменение энтропии системы при растворении некоторого количества
вещества в данном объеме растворителя находится из следующего термодина-
мического уравнения-
Д5ид = — /? (щ In + гс2 1п У2) (XIV, 5)
где R— универсальная газовая постоянная; ni и п2— число молей смешиваемых
компонентов; и N2— мольные доли обоих компонентов *.
Парциальные же мольные энтропии смешения можно вычислить по уравне-
ниям: _
ДЗ,,Ид=<?д5Ж1 = —fllntf! (XIV, 6>
И _
Д32, Ид = д ,\S/dK2 = - R In N2 (XIV, 7)
Если исходить из представлений, используемых при вычислении
идеальной энтропии смешения изодиаметрических молекул, зави-
сящей только от числа смешивающихся молекул, то повышение
молекулярного веса одного компонента должно приводить к умень-
шению числа молекул этого компонента в 1 г системы и, следова-
тельно, к уменьшению энтропии смешения данных весовых коли-
честв обоих веществ. Энтальпия (или внутренняя энергия) не
должна изменяться, поскольку весовые количества смешиваю-
щихся компонентов, а следовательно, и число взаимодействующих
атомных групп не изменяются. Отсюда следует, что по мере по-
вышения молекулярного веса полимера значение энтропийного
члена TAS в уравнениях (XIV, 3) и (XIV, 4) будет неограниченно
уменьшаться и определяющее значение должен приобрести энер-
гетический член А// или At/. Однако при исследовании растворе-
ния высокомолекулярных веществ с цепными молекулами было
* В этом уравнении, равно как и во всех уравнениях термодинамики, ин-
дексами 1 и 2 обозначены величины, относящиеся к растворителю и растворен-
ному веществу соответственно,
439>
установлено, что подобная зависимость соблюдается в той или
иной степени только для полимеров с жесткими молекулами *.
При растворении же гибких цепных молекул в низкомолекулярных
жидкостях, как показал ряд тщательных исследований, энтропия
«мешения обычно значительно превышает энтропию смешения
идеальной системы, причем тем больше, чем больше молекуляр-
ный вес полимера. Аномально большая энтропия смешения при
растворении полимеров, превышающая в сотни и тысяч раз идеаль-
ную, объясняется тем, что при этом сильно возрастает возможность
движения в растворе отдельных участков (сегментов) гибких цеп-
ных молекул. Можно считать, что в растворе сегменты макромо-
лекул играют, по существу, роль отдельных кинетических единиц.
Иначе это можно сформулировать таким образом, что полимеры
с гибкими молекулами ведут себя так, как если бы они обладали
идеальной энтропией смешения, но зато меньшим молекулярным
весом (при том же весовом количестве).
Согласно статистической физике увеличение энтропии при рас-
творении высокомолекулярных веществ объясняется тем, что мак-
ромолекулы в растворе могут быть расположены различным обра-,
зом, причем каждая макромолекула может осуществлять большое
число конформаций. В нерастворенном полимере цепные моле-
кулы мешают друг другу принимать любые конформации. При
наличии же между макромолекулами обычных малых молекул
растворителя стесняющее действие молекулярных цепочек друг на
друга становится все меньше, и, наконец, в предельно разбавлен-
ном растворе, когда макромолекулы очень далеки друг от друга,
•они могут принимать практически любые конформации. Иначе
говоря, термодинамическая вероятность состояния макромолекул
в маловязком растворе больше, чем в исходном полимере.
Обозначим через IFH начальную термодинамическую вероят-
ность состояния макромолекул (до растворения) и через ГГК — ко-
нечную (после растворения). Как известно, вероятность W связана
< энтропией S уравнением Больцмана:
S = AlnF (XIV,8)
где k — постоянная Больцмана.
Тогда изменение энтропии при переходе вещества в раствор
•будет:
AS = SK - SH = k in FK - k In FH = k In (rK/FH) (XIV, 9)
Так как вероятность IFK всегда больше WH, то приращение эн-
тропии растворения всегда положительно. Из приведенного урав-
нения видно, что с увеличением вероятности в результате раство-
рения энтропия системы всегда возрастает.
* Даже при растворении веществ с жесткими, сильно вытянутыми моле-
кулами в низкомолекулярной жидкости с изодиаметрическими молекулами эн-
тропия смешения будет больше идеальной, поскольку число размещений таких
вытянутых молекул намного превысит чйсло размещений того же числа изо-
диаметрических молекул двух сортов.
440
Из сказанного можно сделать важный вывод, что высокомоле-
кулярные вещества с гибкими макромолекулами должны всегда
лучше растворяться, чем с жесткими, поскольку первые могут
располагаться в растворе значительно большим числом Способов.
Кроме того, следует помнить, что у жестких макромолекул, обычно
ориентированных более или менее параллельно, энергия взаимо-
действия между отдельными молекулярными цепочками очень
велика, и такие цепи трудно оторвать друг от друга. Этими обстоя-
тельствами и можно объяснить обычно весьма ограниченное число
растворителей для высокомолекулярных веществ с жесткими це-
пями (целлюлоза, поливинилхлорид, полиамиды).
Как уже было указано, для растворения существенно соблю-
дение одного условия — уменьшение изобарно-изотермического по-
тенциала системы. Поэтому растворение может осуществляться и
тогда, когда тепловой эффект отрицателен, но энтропия системы
при растворении возрастает настолько, что в итоге обуславливает
уменьшение изобарно-изотермического потенциала.
При повышении температуры значение энтропийного фактора
становится все больше, и, таким образом, для всякого высокомо-
лекулярного вещества и растворителя должна существовать кри-
тическая температура растворения Ткрит, выше которой наблю-
дается их смешение во всех отношениях. Теоретически такая кри-
тическая температура должна существовать для любой комбина-
ции высокомолекулярного вещества и растворителя. Практически
же она во многих случаях не может быть достигнута из-за низкой
температуры кипения растворителя и низкой температуры деструк-
ции высокомолекулярного вещества. Критическую температуру
смешения легко найти из условия ДН — ГкритДЗ = 0 или \Н —
== Т'критДЗ.
Отсюда
7’крит = ЛЯ/AS (XIV, 10>
где Д/f и AS — соответственно изменение энтальпии и энтропии при растворении.
Опыт последних лет показал, что при растворении полимеров
энтропийный фактор благодаря аномально большой энтропии сме-
шения в некоторых случаях играет основную роль. Например, он
почти целиком определяет растворимость неполярных полимеров
в углеводородах. Поэтому-то оказались неправы те исследователи
[(Кац, Бренстед), которые пытались объяснить растворимость по-
лимеров только влиянием энергетического фактора. Однако, когда
имеют дело с полярными полимерами, например, когда растворяют
желатин в воде или нитрат целлюлозы в ацетоне, энергетический
фактор может играть существенную роль и его влиянием нельзя
пренебрегать.
Рассмотрим причины растворения полимеров на некоторых типичных приме-
рах. Для этого воспользуемся табл. XIV, I, заимствованной у А. Г. Пасынского.
Как видно из данных, приведенных в табл. XIV,!, растворение полярного
нитрата целлюлозы в полярном циклогексаноне идет как вследствие уменьшения
441
Таблица XIV, 1. Соотношение между термодинамическими величинами
при растворении некоторых высокомолекулярных соединений
Система Характер растворения ЛЯ AS AG
Нитрат целлюлозы в цик- Экзотермический <0 >0 <0
логексаноне
Яичный альбумин в воде » <0 <0 <0 (при Atf>TAS)
Поли-изо-бутилеи в изо- Изотермический 0 >0 <0
октане
Каучук в толуоле Э гдотермический >0 >0 <0 (при ЬН < Т AS)
энтальпии (ЛЯ < 0), так и в результате увеличения энтропии (AS > 0). Благо-
даря тому, что смешению способствуют оба фактора, циклогексанон является
хорошим растворителем нитрата целлюлозы.
При растворении полярного яичного альбумина в воде (полярный раствори-
тель) процесс вначале идет только вследствие уменьшения энтальпии. Энтропия
ъ начальной стадии растворения имеет даже отрицательные значения из-за силь-
ной гидратации полимера. (Молекулы воды в гидратной оболочке переходят в
более упорядоченное состояние. Кроме того, не исключено уменьшение гибкости
молекул полимера при гидратации его молекул.) Но, конечно, для того, чтобы
полимер мог растворяться, абсолютное значение А// должно быть больше ГAS.
Однако по мере гидратации гидратированные молекулы полимера растворяются
в избытке воды со все меньшим выделением тепла. Растворение в этой конеч-
ной стадии идет уже главным образом за счет возможности распределения мо-
лекул по всему объему системы, т. е. уже за счет энтропийного фактора.
Растворение неподярного поли-изо-бутилена в неполярном же изо-октане
идет только вследствие повышения энтропии без выделения тепла, т. е. смешение
имеет здесь изотермический характер. Существенно, что цри растворении поли-
лзо-бутилена в изо-октане, гидрированном димере изо-бутилена, энергетический
барьер при вращении отдельных звеньев цепи молекулы не изменяется, так как
действие межмолекулярных сил в растворе такое же, что и в самом поли-изо-
бутилене. Иными словами, растворение в этом случае происходит без изменения
гибкости макромолекул.
Наконец, растворение каучука в толуоле идет тоже за счет увеличения эн-
тропии смешения. Отличие этого случая от предыдущего заключается в том, что
здесь наблюдается небольшой отрицательный тепловой эффект Одиако этот
эффект с избытком перекрывается увеличением энтропии, поскольку молекулы
каучука весьма гибки 5
Представленные в табл XIV, 1 комбинации изменения энтальпии и энтропии
полностью охватывают все типичные случаи растворения полимеров При осталь-
ных комбинациях этих величин растворение не происходит, так как в этих слу-
чаях AG > 0.
Пути определения величин ДО, А// и AS описаны в первом издании этого
учебника на с 481.
6. НАБУХАНИЕ ВЫСОКОМОЛЕКУЛЯРНЫХ ВЕЩЕСТВ
Растворение высокомод§куля_рщдх—веществ с линейными гиб-
кими молекулами в отличие от растворения низкомолекулярных
соединений сопровождается набуханием, или, вернее, набухание
таких веществ является первым этапом их растворения. При на-
бухании высокомолекулярное вещество поглощает низкомолеку-
442
лярный растворитель, значительно увеличивается в массе, при
этом изменяет механические свойства без потери однородности.
Объем высокомолекулярного вещества при набухании может уве-
личиваться до 1000—1500% *•
Набухание имеет' большое значение в природе и технике. На-
бухание лежит в основе таких процессов, как клейстеризддцц крах-
мала, мерсеризация в текстильной технологии, придание нажора
обезволошенной шкуре в кожевенном производстве. Ряд явлений
в организмах животных и растений также можно объяснить набу-
ханием.
Причиной набухания является то, что при растворении проис-
ходит не только диффузия молекул растворяемого вещества в рас-
творитель, как это- ямеё*: место при растворении низкомолекуляр-
ных веществ, но, главным образом, диффузия молекул раствори-
теля в высокомолекулярное вещество. Последнее связано с тем, что
макромолекулы в обычных аморфных высокомолекулярных ве-
ществах упакованы сравнительно неплотно и вLрезультате тепло-
вого движения гибких цепей между пй\Гй"'перйиДйчески образуются
весьма малые пространства, в которые могут проникать молекулы
растворителя. Так как подвижность маленьких молекул раствори-
теля во много раз больше подвижности макромолекул, сначала,
главным образом, происходит диффузия_зтлекул растворителя
в полимер, что сопровождается увеличением объема последнего, и
только уже затем макромолекулы, связь между которыми сильна
ослабилась, отрываются от основной массы вещества и диффун-
дируют в среду, образуя однородный истинный раствор.
Заметим, что поскольку растворимость связана с движением
в растворе не всей макромолекулы, а. ее сегментов, то она не
должна зависеть от молекулярного веса полимера. Однако он
весьма значительно сказывается на скорости растворения. Чем
меньше молекулярный вес, тем больше растворение высокополи-
мера похоже на растворение низкомолекулярного вещества. Из-
вестно, например, что деструктиронанный каучук растворяется без
набухания. Наоборот, с увеличением молекулярного веса раство-
рение полимеров замедляется. При весьма малых скоростях рас-
творения, что наблюдается, когда молекулы полимера очень боль-
шие, может даже создаться неправильное представление о нерас-
творимости вещества. Из сказанного также понятно, что если мо-
лекулы полимера жесткие, т. е. если длина сегмента практически
равна длине всей цепи, растворимость всегда должна зависеть от
степени полимеризации.
Существенно, ~что—высокомолекулярные вещества со сфериче-
скими^лолекулами при растворении не набухают или набухают
очейь слабо. В качестве типичного примера можно привести гли-
• С набуханием нельзя смешивать процесс капиллярного впитывания, при
котором жидкость заполняет микроскопические пустоты, имеющиеся в твердом
теле Размеры твердого тела при этом обычно не увеличиваются.
443
коген, состоящий из глобулярных молекул. Образцы гликогена,
обладающие весьма большим молекулярным весом 800 000, при
растворении не набухают. Это указывает, что диффузия не может
рассматриваться как единственный фактор, управляющий набуха-
нием. Отсутствие способности набухать у веществ со сфериче-
скими макромолекулами объясняется тем, что когезионные силы,
приходящиеся на сферическую молекулу, благодаря ее сравни-
тельно небольшой поверхности всегда меньше, чем те же силы,
приходящиеся на линейную макромолекулу. Благодаря малой ко-
гезионной энергии высокомолекулярных веществ со сферическими
молекулами их растворение облегчается.
Описанная картина набухания высокомолекулярных соедине-
ний с линейными макромолекулами наблюдается в простейшем
случае, когда набухание и растворение носят почти чисто энтро-
пийный характер, Например при растворении каучуков в углеводо-
родах. Когда молекулы растворителя энергетически взаимодей-
ствуют с молекулами высокомолекулярного вещества, как, напри-
мер, при растворении желатина в воде, механизм растворения
усложняется. В этом случае можно считать, что растворение про-
текает в две стадии, хотя, конечно, вторая стадия практически на-
чинается раньше окончания первой.
На первой стадии набухания происходит сольватация макромо-
лекул в результате диффузии растворителя в высокомолекулярное
вещество. Эта стадия характеризуется выделением тепла и упоря-
дочением расположения молекул растворителя около макромоле-
кул, в результате чего энтропщя-системы в первой стадии раство-
рения обычно даже понижается. Основное значение этой стадии^
при растворении сводится"'!?-разрушению связей -между отдель-
ными макромолекулами, вследствие чего цепи становятся свобод-
ными и способцы^^срверщать тепловое движение в целом.
Второй стадией является набухание или растворение, обуслов-
ленное чисто энтропийными причинами. В этой стадии, поскольку
сольватация уже завершилась, тепловой эффект равен нулю или
даже имеет отрицательное значение, а энтропия резко возрастает
вследствие смешения громоздких и гибких макромолекул с ма-
ленькими молекулами растворителя. Вторую стадию растворения
можно рассматривать как чисто осмотический процесс.
На набухании и растворении полимеров сказывается их физи-
ческое состояние. Конечно, легче всего набухают и растворяются
полимеры в вязкотекучем и высокоэластическом состоянии, моле-
кулы которых связаны друг с другом наименее прочно. Значительно
труднее растворяются полимеры, находящиеся в застеклованном;
состоянии. В этом случае сначала при контакте полимера с раство-
рителем молекулы растворителя прбникают в поверхностный слой
полимера, что вызывает поверхностное набухание его. Далее на-
бухший полимер растворяется таким же образом, как и высоко-
эластичный полимер. Граница раздела между твердым полиме-
ром, в который еще не проник растворитель, и набухшим его слоеЦ
444
постепенно продвигается внутрь вещества со скоростью диффузии
растворителя в стеклообразный полимер.
Наиболее трудно растворимы кристаллические полимеры.
С растворителем в первую очередь будут взаимодействовать неупо-
рядоченные области таких полимеров. В кристаллические области
растворитель сможет проникать, если он экзотермически взаимо-
действует с полимером, так как разрушение кристаллических об-
ластей всегда сопровождается значительным поглощением тепла.
В противном случае для растворения кристаллической части си-
стему необходимо нагревать.
Набухание далеко не всегда кончается растворением. Очень
часто после достижения известной степени набухания процесс пре-
кращается. Одна из причин такого явления может заключаться
в том, что высокомолекулярное вещество и растворитель способны
смешиваться ограниченно. Поэтому в результате набухания в си-
стеме образуются две фазы — насыщенный раствор полимера
в растворителе (собственно раствор) и насыщенный раствор рас-
творителя в полимере (гель, студень). Такое ограниченное набу-
хание носит равновесный характер, т. е. объем набухшего до пре-
дела высокомолекулярного вещества неограниченно долго остается
неизменным, если только в системе не произойдут химические из-
менения. Примерами набухания, обусловленного ограниченным
растворением, являются набухание поливинилхлорида в ацетоне и
полихлоропрена в бензоле. Следует отметить, что ограниченное на-
бухание, причина которого кроется в ограниченном растворении,
очень часто при изменении условий опыта переходит в неограни-
ченное. Так, желатин и агар, набухающие ограниченно в холодной
воде, в теплой воде набухают неограниченно.
В настоящее время еще невозможно точно установить связь
между природой растворителя и его способностью растворять дан-
ное высокомолекулярное вещество. Обычно ограничиваются эм-
пирическим правилом — подобное-растворяется в подобном. Иными
словами, неполярные полимеры растворяются в неполярных рас-
творителях, а полярные — в полярных. Джи установил связь ме-
жду способностью растворителей вызывать набухание и растворе-
ние полимера и значениями плотностей когезионных энергий этих
растворителей. Удельная плотность когезионной энергии £7[7МОЛ
(где Е — когезионная энергия или скрытая теплота испарения,
[7МОЛ — мольный объем) представляет собой энергию, которую не-
обходимо затратить для того, чтобы раздвинуть молекулы, содер-
жащиеся в 1 см3 полимера, на расстояние, превышающее сферу
их действия. На ряде примеров было показано, что максимальное
набухание наблюдается, когда удельные плотности когезионной
энергии растворителя и полимера равны или близки.
Другая причина ограницеяНйгю._набухания высокомолекуляр-
ного вещества заключается в том, что между молекулами поли-
мера могут существовать поперечные химические связи (так назы-
ваемые мостики), и все вещество по существу представляет собой
445
пространственную сетку. Это препятствует отрыву макромолекул
друг от друга и переходу их в раствор. Кроме того, если даже не
все молекулы полимера связаны в пространственную сетку, то
такая сетка может играть роль мембраны, проницаемой для ма-
лых молекул растворителя и препятствующей диффузии макромо-
лекул из объема набухшего полимера. В результате увеличения
объема высокомолекулярного вещества при набухании в простран-
ственной сетке появляются напряжения, что и приводит к прекра-
щению набухания. Примером ограниченного набухания, обуслов-
ленного наличием прочной пространственной сетки, является на-
бухание вулканизованного каучука в бензоле.
Ограниченное растворение полимера вследствие наличия в нем
пространственной молекулярной'сётки можно трактовать и с тер-
модинамической точки зрения. Действительно, при набухании та-
кого полимера гибкие участки макромолекул, лежащие между
узлами сетки, растягиваются и распрямляются и, следовательно,
«энтропийные пружины» переходят в менее вероятное состояние.
В результате энтропия системы уменьшается, причем это умень-
шение может стать равным увеличению энтропии в результате
смешения. В этот момент набухание прекратится, т. е. система при-
дет в равновесное состояние. Правильность приведенных рассуж-
дений подтверждается наличием связи между модулем упругости
полимеров и их способностью к набуханию (Флори).
Следует заметить, что при большом числе поперечных химиче-
ских связей между макромолекулами высокомолекулярное веще-
ство перестает не только растворяться, но и набухать. Примером
может служить эбонит — вулканизованный каучук с 25—30%-ным
содержанием серы. Вследствие частоты и жесткости простран-
ственной сетки молекулы растворителя уже не могут проникать
в полимер и раздвигать его цепи. Поэтому набухание эбонита не
происходит. При малом числе связей между макромолекулами ог-
раниченно набухающий полимер, в результате его механической
обработки, например вальцевания, можно превратить в неограни-
ченно набухающий. При вальцевании редкие поперечные связи
разрываются, и полимер приобретает способность растворяться.
Между обоими возможными случаями ограниченного набуха-
ния, один из которых обусловлен ограниченностью смешения,
а другой — пространственной структурой полимера, нет принци-
пиального различия. Можно принять, что и в первом случае вы-,
сокополимер представляет собой пространственную сетку, но
только связи здесь обусловлены не химическими, а межмолекуляр-
ными силами. Тогда ограниченное растворение такого полимера
можно объяснить тем, что плохой растворитель просто не может
разрушить эти связи или разрушает их частично, что и находит
свое выражение в ограниченном набухании.
Возможно н третье объяснение ограниченного набухания высокомолекуляр-
ных веществ, предложенное В А Каргиным Представим себе, что в результате
сольватации при набухании и растворении гибкость макромолекул уменьшается.
446
Это может быть обусловлено увеличением потенциального барьера вращения
участков молекулы в результате взаимодействия с растворителем. Тогда энтро-
пия полимера при набухании будет убывать, поскольку макромолекулы стано-
вятся все более и более жесткими и не могут осуществлять тех конформаций,
которые были им доступны в отсутствие растворителя. Как только понижение
энтропии из-за потери гибкости макромолекул станет больше уменьшения вну-
тренней энергии в результате энер! етического взаимодействия макромолекул с
молекулами растворителя, т. е. как только изобарно-изотермический потенциал
системы перестанет уменьшаться, набухание прекратится Подобный случай имеет
место, например, при набухании ацетата целлюлозы в тетрахлорэтане. Аналогич-
ные явления часто наблюдаются также при набухании полярных высокомолеку-
лярных веществ в воде В табл XIV, 2 приведены парциальные величины АН/,
AGi и TAS] воды в набухших до различных степеней агаре, казеине, кератине
и целлюлозе. По мнению В. А. Каргина и А. А Тагер, при набухании всех при-
веденных веществ в результате взаимодействия молекул растворителя с макро-
молекулами резко увеличивается потенциальный барьер вращения, что приводит
к уменьшению энтропии и возрастанию свободной энергии системы. Вследствие
этого набухание прекращается на определенной степени и растворения не про-
исходит.
Таблица XIV, 2. Парциальные термодинамические характеристики воды
(в кал/моль) в некоторых набухших высокомолекулирных веществах
Высокомолеку- лярное вещество t, °C ДН1 AGt Т ASi ДЩ ДО] Т ASj
(5,7% ДС Н2О, 0,943 лей вещест мол. ва) (10,89 ДС Н2О, 0,89. лей вещест мол. ва)
Агар 5 -3200 -1850 — 1350 -1850 -1000 -850
Казеин 5 —2050 -900 -1150 —1000 -350 —650
Кератин 5 -2250 -1200 -1050 -1150 -500 -950
» 20 —2800 -1100 — 1400 —1€50 —450 -1200
Целлюлоза 50 -1000 -230 -770 -200 -30 -175
Набухание полимера в жидкости характеризуется степенью на-
бухания а, вычисляемой по уравнению:
а = (т — т0)/т0 (XIV, 10)
где т0 и т — навеска полимера до и после набухания.
Очевидно, степень набухания численно равна массе (в г) жидко-
сти, поглощенной 1 г высокомолекулярного вещества. При опреде-
лении а следует учитывать, что в ходе набухания из полимера вы-
мываются низкомолекулярные фракции, и поэтому вычисленная
по вышеприведенной формуле степень набухания может быть не-
сколько ниже той, которая соответствовала бы набуханию в парах
растворителя.
Определяя степень набухания через заданные промежутки,
можно получить кривые, характеризующие кинетику набухания.
Часто о набухании судят не по привесу, а по увеличению объема
образца. На рис. XIV, 5 изображены типичные кинетические кри-
вые набухания. Кривые 1 и 2 характеризуют ограниченное набу-
хание, причем кривая 1 соответствует сравнительно быстро набу-
хающему полимеру, Но с малым значением предельного набухания,
447
а кривая 2 — медленно набухающему полимеру с большим значе-;
кием предельного набухания. Как видно из рис. XIV, 5, обе кривые/
вначале круто поднимаются, а затем плавно переходят в прямую,
параллельную оси абсцисс. Аналитически эти кривые можно пред-
ставить дифференциальным уравнением:
daldx = k (амакс — ат) (XIV, 11)
где амакс — степень предельного набухания; at — степень набухания к моменту
времени т; k — константа скорости набухания, зависящая от природы полимера,
природы растворителя и температуры
В результате интегрирования уравнение принимает вид:
й=—1п—амакс .
®макс *
Скорости набухания различных полимеров надо сравнивать по
наклону ( касательных, проведенных к кривым из начала коорди-
(XIV, 12)
Рис. XIV, 5. Типы кинетических
кривых набухания:
/—неограниченное набухание; 2—бы-
строе набухание с малым значением
предельного набухания, 3 — мед пенное
набухание с большим значением пре-
дельного набухания; 4 — ограниченное
набухание с экстрагированием низко-
молекулярных фракций.
нат, а способность полимеров к на-
буханию следует характеризовать
по предельной степени набухания.
Кривая 1 (рис. XIV, 5) характе-
ризует неограниченное набухание,
когда полимер растворяется в рас-
творителе. В этом случае о предель-
ной степени набухания говорить
нельзя, хотя на кривой и имеется
максимум. Кривая 4 характеризует
ограниченное набухание, когда из
набухающего вещества экстраги-
руется значительное количество
низкомолекулярных фракций, что
уменьшает предельную степень на-
бухания.
На набухание д сильной степени
может влиять форма образца. По-
этому опыты, в которых хотят срав-
нить набухание различных полимеров, надо проводить всегда с об-
разцами одинаковой формы.
Набухание высокомолекулярного вещества может привести
к возникновению значительного давления, если что-нибудь препят-
ствует увеличению объема образца Например, при набухании дре-
весины в воде может развиваться давление в десятки атмосфер.
Это давление настолько велико, что в древние времена это явление
использовали для дробления скал' в трещины скалы забивали.де-
ревянные клинья и затем заливали воду, клинья набухали в воде,
стремились увеличить свои размеры и тем самым создавали дав-
ление, разрушающее скалу.
Давление р, которое развивается при набуханий, можно вычислить по урав-
нению Позняка:
p = pocfc (XIV, 13)
448
где ро — константа, зависящая от природы высокомолекулярного вещества, рас-
творителя и температуры; с — содержание сухого полимера в набухающем студ-
не; k — константа, значение которой обычно близко к 3. Очевидно, что при
с = 1 значение р должно равняться р0. При логарифмировании уравнение Поз-
няка принимает вид:
1п р = 1п Ро + k In с (XIV, 14)
Рис. XIV, 6. Зависимость 1п р
от 1п с при набухании нату-
рального каучука в различных
растворителях:
1—четыреххлористый углерод:
2—хлороформ; 3 —тетрахлорэтаи;
4—толуол; 5—бензол; 6—диэтило»
вый эфир; 7— дихлорэтан.
Это уравнение прямой позволяет графически легко определить величины р0
и k, а прн известных константах и при заданном значении с можно определить
н р.
Для примера на рис. XIV, 6 представлена логарифмическая зависимость дав-
ления от концентрации для каучука, набухающего в различных растворителях.
Как можно видеть, для всех испытанных растворителей зависимость между
1п р и 1п с носит линейный характер и констан-
та не меняется при переходе от одной жидко-
сти к другой. Интересно, что если жидкости,
в которых набухал каучук, расположить в по-
рядке увеличения константы рв, то они обра-
зуют ряд, совпадающий с рядом, в котором эти
же жидкости стоят в порядке возрастания
предельной степени набухания в них. Это ука-
зывает на связь между давлением набухания
и предельной степенью набухания.
Помимо увеличения объема высоко-
молекулярного вещества, в начале на-
бухания часто наблюдаются уменьше-
ние объема всей системы и тепловой
эффект набухания. Эти явления весь-
ма существенны для понимания меха-
низма набухания.
В то время как при набухании объ-
ем полимера всегда увеличивается,
объем всей системы (полимер + рас-
творитель) обычно уменьшается. Это
особенно заметно при набухании по-
лярных полимеров в полярных растворителях. Уменьшение объема
системы при набухании, называемое контракцией, в большин-
стве случаев хорошо описывается следующим эмпирическим урав-
нением с двумя константами:
V = <хт/(0 + т)
(XIV, 15)
где V — контракция; т — масса жидкости, поглощенной при набухании 1 г поли-
мера (степень оводнения); а и р — константы.
Контракция системы при набухании полимера объясняется
ориентацией молекул растворителя в результате их «адсорбции»
макромолекулами, что способствует увеличению плотности си-
стемы. Кроме того, частично контракция происходит за счет чисто
стерического фактора — при набухании малые молекулы раство-
рителя проникают в пространства между громоздкими макромоле-
кулами, вследствие чего компактность упаковки молекул возрас-
тает.
Тепловой эффект при набухании высокомолекулярного веще-
ства обычно положительный, например, при набухании полярных
15 Зак. 664
449
полимеров в полярных же растворителях. Это и понятно, так как
выделение тепла при набухании связано с взаимодействием моле-
кул полимера и растворителя. Различают интегральную теплоту
набухания <гИнт, т. е. общее количество тепла, выделившееся при
набухании 1 г сухого полимера, и дифференциальную теплоту на-
бухания <7диф, представляющую собой количество тепла, выделив-
шегося при поглощении 1 г жидкости сухим или уже набухшим
высокомолекулярным веществом.
В качестве примера в табл. XIV, 3 приведены интегральные и дифференци-
альные теплоты набухания некоторых высокомолекулярных веществ (по данным
Каца н Марка).
Табл 1ца XIV. 3. Интегральные и дифференциальные теплоты набухания
(в кал/г) некоторых высокомолекулярных веществ
Высокомолекулярное вещество Растворитель ^ИНТ ^диф (для сухого вещества)
Казеин Вода 25,4 265
Нуклеин » 23,4 310
Индулин » 21,8 420
Древесное волокно > 16,9
Целлюлоза » 10,7 390
Ацетат целлюлозы Трихлорэтилен 11,4 108
То же Бензилхлорид 8,1 76
» Бензиловый спирт 8,2 69
Нитрат целлюлозы Муравьиная кислота 6,8 60
То же Этиловый/ спирт - 5,5 87
Интегральная теплота набухания дИнт складывается из трех величин:
?инт — теплоты, соответствующей работе, необходимой дли разъединения макро-
молекул, 9„нТ—теплоты, соответствующей работе разъединения молекул раство-
рителя, и 9'нт — теплоты, выделяющейся в результате взаимодействия раство-
рителя с высокомолекулярным веществом. Первые две величины имеют всегда
отрицательное значение, третья — положительное:
?инт = 9иит ?нит 9инт (XIV, 16)
При набухании неполярного высокомолекулярного вещества в неполярной
жидкости абсолютные значения величии 9ИНТ, 9ИНТ и qHHT близки и невелики;
поэтому тепловой эффект близок к нулю. При набухании полярного высоко-
молекулярного вещества в полярном растворителе величина 9ИНТ, как правило,
большая и растворение сопровождается выделением тепла. Дли кристаллических
высокомолекулярных веществ величина днит обычно высока. В этом случае
теплота набухания может быть отрицательной, вследствие чего набухание идет
с трудом.
Дифференциальная теплота набухания тем меньше, чем больше степень на-
пухания высокомолекулярного вещества В качестве примера приведем найден-
ные интерполяцией дифференциальные теплоты набухания'для желатина:
Масса воды, поглощенная 1 г же-
латина, г............................ 0,000 0,005 0,041 0,103 0,242
^диф’ кал/г............................. 228 222 186 142 86
450
Как можно видеть, дифференциальная теплота набухания быстро падает
с увеличением оводненности высокомолекулярного вещества. При значительном
оводнении тепловой эффект набухания может равняться нулю или даже прини-
мать отрицательное значение.
Интегральная теплота набухания ^Инт может быть представ-
лена уравнением, аналогичным уравнению контракции:
?иит = ш7(6 + 1) (XIV, 17)
где (— степень оводиення; а и Ь — константы.
Существенно, что отношение контракции к интегральной теп-
лоте набухания является, как правило, величиной постоянной, т. е.
И/<7инт = const (XIV, 18)
Это указывает на то, что контракция и выделение тепла при
набухании обычно являются процессами взаимно связанными, ко-
торые в основном протекают в результате взаимодействия моле-
кул высокомолекулярного вещества с диффундирующими в него
молекулами растворителя, т. е. в результате сольватации.
Обращает на себя внимание, что уравнения (XIV, 15) и
(XIV, 17) сходны с уравнением адсорбции Ленгмюра (IV, 10). Это
является еще одним доказательством того, что сольватация имеет
характер адсорбционного явления.
В заключение рассмотрим влияние температуры на ограниченное набухание.
Влияние температуры на набухание легко определить исходя нз термодина-
мического рассмотрения процесса. Если набухание экзотермический процесс, что
характерно для первой стадии набухания, то равновесная степень набухания
понижается с повышением температуры. В соответствии с этим ограниченное
набухание, например целлюлозы в воде или растворе едкого натра, представ-
ляющее типичный экзотермический процесс, сильно уменьшается при повышении
тмпературы. Однако, как мы видели, во второй стадии набухание может стать
эндотермическим процессом. В этом случае набухание, следовательно, должно
увеличиваться с возрастанием температуры. Опыты показали, что набухание
желатина в общем с повышением температуры увеличивается Скорость набу-
хания с повышением температуры, конечно, всегда должна возрастать, так как
повышение температуры прн всех обстоятельствах способствует ускорению уста-
новления равновесного состояния системы.
Влияние электролитов на набухание полимеров рассмотрено ниже в раэд. 8
при описании свойств растворов высокомолекулярных электролитов, так как
у полиэлектролитов это влияние проявляется наиболее сильно.
7. НЕКОТОРЫЕ СВОЙСТВА РАСТВОРОВ
ВЫСОКОМОЛЕКУЛЯРНЫХ ВЕЩЕСТВ
Закон Рауля и кажущийся молекулярный вес
высокомолекулярных веществ
Как известно, идеальнее растворы, т. е. растворы, образую-
щиеся без изменения энтальпии, энтропия которых равна энтро-
пии смешения идеальных растворов, подчиняются закону Рауля,
определяющему ряд важных свойств растворов в зависимости от
содержания в них растворенного вещества. Если растворенное ве-
щество нелетуче, т.е. если давление его паров практически равно
15*
451
нулю, закон Рауля может быть выражен следующим образом:
P1/po = JVi = l-^
(XIV, 19)
где pi — давление паров растворителя над раствором; р® — давление паров над
чистым растворителем; и — соответственно мольные доли растворителя и
растворенного вещества в растворе.
К реальным растворам закон Рауля часто неприменим, причем
наблюдаются два типа отклонений: отрицательные отклонения,
когда отношение pjp° меньше мольной доли летучего компонента
в растворе (т. е. < AQ, и положительные отклонения, когда
Рнс. XIV, 7. Зависимость
Pi/Pi от ^2 для идеального
раствора (/)н реальных рас-
творов с отрицательным
отклонением от закона Рау-
ля (2) и с положительным
отклонением от закона
Рауля (3).
отношение больше мольной доли
растворителя в растворе (т. е. р,/р? > AQ.
На рнс. XIV, 7 показана зависимость Р\[Р\
от мольной доли растворенного вещества Ы2 для
идеального раствора н для реальных растворов
с отрицательным и положительным отклонением
от закона Рауля.
Долгое время причину отклонения в поведе-
нии реальных растворов от закона Рауля объяс-
няли только энергетическим взаимодействием
между молекулами растворителя н растворенного
вещества. Отрицательные отклонения объясняли
тем, что молекулы растворителя прочнее удержи-
ваются нелетучими молекулами растворенного
вещества, чем себе подобными молекулами. Такое
сильное взаимодействие указывает на сродство
между растворителем н растворенным веществом,
н поэтому отрицательные отклонения должны
всегда наблюдаться прн хорошей совместимости
обоих компонентов, а также прн сольватации
нлн образовании аддитивных соединений. Понят-
но также, что при отрицательных отклонениях,
вызванных этими причинами, наблюдается конт-
ракция системы и выделение тепла. Положительные отклонения объясняли тем,
что взаимодействие молекул одного рода друг с другом сильнее, чем взаимодей-
ствие молекул различной природы. Это приводит к вытеснению молекул раство-
рителя из раствора, и следовательно, давление его пара над раствором будет
выше, чем должно быть по закону Рауля. Естественно, что положительные
отклонения указывают на плохую совместимость обоих компонентов. Отсюда
понятно, что прн положительных отклонениях всегда происходит поглощение
тепла и увеличение объема системы.
Однако, как показали дальнейшие исследования, отклонения в поведении
реальных растворов от закона Рауля могут объясняться не только энергетиче-
ским взаимодействием обоих компонентов, но н тем, что в реальных растворах
энтропия смешения может быть н не равна энтропии смешения для идеальной
системы. Если молекулы одного компонента могут располагаться средн молекул
другого компонента большим числом способов, чем средн себе подобных моле-
кул, то энтропия раствора будет больше значения идеальной энтропии смеше-
ния. Понятно, что это приведет к отрицательным отклонениям от закона Рауля.
Особенно важную роль подобное явление играет в случае растворов высоко-
молекулярных веществ, в которых молекулы растворенного вещества обладают
аннзоднаметрней и гибкостью, что способствует увеличению энтропии смешения.
Наоборот, если молекулы одного компонента располагаются среди молекул дру-
гого меньшим числом способов, чем средн себе подобных, то энтропия смешения
452
будет меньше энтропии смешения Для идеальной системы. Такой случай наблю-
дается, когда растворенное вещество по тем или иным причинам лишает воз-
можности молекулы растворителя свободно менять свои места в растворе.
Закон Рауля, связывающий давление пара растворителя над раствором и
концентрацию растворенного вещества, может быть использован для определе-
ния молекулярного веса растворенного вещества. Сначала опытным путем мож-
но найтн отношение давлений паров Pi/p?, равное Aft. Затем, исходя нз извест-
ного уравнения, можно вычислить молекулярный вес растворенного вещества:
N — _21—_= mifMi
1 «1 + п2 mtlMi+mz/Mz
(XIV, 20)
где ni н п2 — соответственно число молей растворителя и растворенного веще-
ства в идеальном растворе; mt и т2 — соответственно масса растворителя и рас-
творенного вещества; Aft и М2 — молекулярные веса растворителя и растворен-
ного вещества.
Поскольку для растоворов высокомолекулярных веществ значение Р\ р\ не
равно Ni, подобный метод определения молекулярного веса для них непригоден.
Однако приняв, что растворы высокомолекулярных веществ ведут себя как
идеальные растворы и определив рирр можно вычислить кажущийся, нлн экви-
валентный, молекулярный вес высокомолекулярного вещества, т. е. величину,
эквивалентную молекулярному весу вещества, которое давало бы в данном
растворителе идеальный раствор.
Значение кажущегося молекулярного веса представляет собой не что иное,
как молекулярный вес отрезка цепи (сегмента), являющегося в растворе кине-
тической единицей. Понятно, что сегмент достаточно длинных молекул не дол-
жен зависеть от молекулярного веса и что значения кажущегося молекулярного
веса всегда меньше значений истинного молекулярного веса. Однако чем более
разбавлены растворы, взятые для определения, тем ближе значения кажущегося
молекулярного веса к значениям истинного. В пределе, при бесконечном раз-
бавлении раствора, кажущийся молекулярный вес может стать равным истин-
ному, так как в таком растворе кинетической единицей является уже вся макро-
молекула в целом. Этим можно воспользоваться для нахождения истинного
молекулярного веса.
х Здесь надо отметить, что длина сегмента макромолекулы в растворе опре-
деляется не только гибкостью цепи, но и ассоциацией макромолекул или их
участков. Как мы видели, ассоциация зависит как от природы растворителя, так
и от природы макромолекул, т. е. от содержания в ннх полярных или ноноген-
ных групп, по которым может устанавливаться связь. Таким образом, кажу-
щийся молекулярный вес является величиной весьма условной, о чем никогда
не следует забывать.
Поскольку в растворе мольная доли высокомолекулярных веществ, вслед-
стие их огромного молекулярного веса, обычно очень невелика, то относительное
понижение давления пара растворителя даже над концентрированными раство-
рами высокомолекулярных соединений весьма мало и экспериментально его
определить крайне трудно. Поэтому на практике для нахождения молекулярного
веса определяют не давление пара растворителя над раствором, а находят
мольную долю растворенного вещества по осмотическому давлению, как из-
вестно, также зависящему от концентрации раствора.
j , , • .
Осмотическое давление
Осмотическое давление л достаточно разбавленных растворов
низкомолекулярных веществ подчиняется, как известно, закону
Бант-Гоффа;
n = {c/M)RT (XIV, 21)
453
Рис. XIV, 8. Зависимость осмо-
тического давления от концен-
трации раствора:
/—высокомолекулярное вещество,
2—низкомолекулярное вещество
(сахар); 3—слабый электролит.
где с — весовая концентрация; М — масса 1 моль; R — универсальная газовая
постоянная; Т — абсолютная температура
Уравнение Вант-Гоффа по внешнему виду сходно с уравнением
состояния идеального газа. Однако это сходство чисто формаль-
ное. Осмотическое давление не имеет ничего общего с давлением
газа, поэтому его нельзя представлять как результат механических
ударов молекул о стенки сосуда. Осмотическое давление обуслов-
ливается тем, что растворитель переходит в раствор через мем-
брану до тех пор, пока этот поток растворителя не будет компен-
сирован направленным навстречу потоком растворителя из осмо-
тической ячейки, возникшим вследствие повышения уровня жидко-
сти в капилляре осмометра. Давлением со стррон-ы-раствора, т. е.
гидростатическим давлением, в мо-
мент равновесия системы и измеряет-
ся осмотическое давление.
К растворам полимеров закон
Вант^Соффа в приведенном виде не-
приложим. Опыт показал, что осмоти-
ческое давление растврров полимеров;
значительно рыше. чем это требует за-
кон Вант-Гоффа. Объясняется это тем»
что макромолекула благодаря гибко-
сти ведет себя в растворе как несколь-
ко более коротких молекул, т. е. что
роль_ кинетического элемента играет
уже не макромолекула, а ее сегмент.
Понятно, что чем более, гибка молеку-
ла, тем при прочих равных условиях
осмотическое давление выше и тем больше оно отклоняется от
значения, вычисленнопШо уравнению Вант-Гоффа. Кроме того»
с повышением концентрации осмотическое давление растворов по-
лимеров возрастает не._прямолинейно, в то время как согласна
закону~Вапт-£офФа осмотические ,дявл§ние увеличивается пряма
пропорционально концентрации.
На рис. XIV, & схематически показано изменение с повышением
концентрации осмотического давления раствора низкомолекуляр-
ного вещества (сахара), слабого электролита и высокомолекуляр-
ного соединения. Как видно из рисунка, осмотическое давление
раствораХ/низкомолекулярного вещества возрастает прямо пропор-
ционально концентрации. В случя^Ослабого электролита. когда
осмотическое давление раствора обуславливается не только чис-
лом молекул; но и ионов, осмотическое давление’возрастает более
медленно, чем концентрация, о 'чём"свидетельствует выпуклость
кривой, обращенная в сторону ординаты. Такое явление объяс-
няется умрнмпрнием степени ионизации с довышением концентра-
ции. Наконец, осмоТйчеекее-давление'раствораХввысокомолекуляр-
•юго вещества возрастает быстрее, чем увеличивается концентра-
ция. Согласно Галлеру, это объясняется тем, что при повышении
154
концентрации возрастает число сегментов, на которые условно
можно"разделить гибкую молекулу. На основании представлений
Галлера для описания зависимости осмотического давления от
концентрации полимеров было предложено уравнение:
(XIV, 22)
где Ь — второй вириальный коэффициент, с — константа, характеризующая от-
клонение от уравнения Вант-Гоффа, зависящая от природы растворителя и рас-
творенного вещества, но не зависящая от молекулярного веса растворенного
вещества.
U— cRT/M + be2
При малых значениях с величина второго члена правой части
этого уравнения приближается к нулю и выражение принимает вид
уравнения Вант-Гоффа. Уравнение (XIV, 22)
можно переписать так:
да/с = RT/M + be (XIV, 23)
f
В таком виде это уравнение представляет
собой уравнение прямой, по которой графиче-
ски легко найти -молекулярный вес полимера.
Для этого определяют осмотическое давление
при разных концентрациях, строят график в
координатах л/с, с и экстраполируют прямую
до пересечения с осью ординат. Отрезок, отсе-
ченный прямой на оси ординат, равен RT/M,
а b определится как тангенс угла, образуе-
мого прямой и осью абсцисс.
На рис. XIV, 9 в качестве иллюстрации по-
казана зависимость л/с от с для раствора ни-
трата целлюлозы в циклогексаноне и гемогло-
бина в воде, причем для удобства сравнения
Рис. XIV, 9. Зависи-
мость л/с от концен-
трации с растворов:
выбраны высокомолекулярные вещества с
примерно одинаковым молекулярным весом
(70 000). Как видно из рисунка, для нитрата
1—нитрата целлюлозы в
циклогексаноне; 2—гемо-
глобина в воде.
целлюлозы (молекулы которого имеют линейное строение) значе-
ние л/с с увеличением с возрастает, что, конечно, обусловлено
увеличением числа сегментов с повышением концентрации; для
гемоглобина (белка со сферическими молекулами} значение л/с,
«естественно, не зависит от концентрации. __
Определение молекулярного веса по. осмотическому давлению
разбавленных растворов является д „настоящее время одним из
наиболее распространенных 'методов нахождения молекулярного
веса. Благодаря тому, что растворы полимеров легко очистить от
лримёсей, полученные этим методом средние молекулярные веса
являются гораздо более достоверными, чем численные веса, опре-
деленные тем же методом для коллоидных систем. Неточности при
определении молекулярного веса осмотическим методом возможны
чаще всего из-за тенденции молекул к ассоциации. Поэтому во
избежание ошибок молекулярный вес высокомолекулярного веще-
455-
ства целесообразно определять путем нахождения осмотического
давления его растворов в нескольких разных растворителях.
Молекулярный вес, определенный с помощью осмометрии, как
мы уже указывали, дает усредненный по числу частиц, или сред-
нечисденный, молекулярный вес Мп, поскольку осмотическое дав-
ление пропорционально числу молекул растворенного вещества.
На осмотическое давление в достаточно концентрированных
растворах полимеров может существенным образом влиять способ
приготовления раствора. Предварительное нагревание и перемеши-
вание способствуют повышению осмотйчеёкбРЭ^давления, а охлаж-
дение или длительное выдерживание раствора, наоборот, пониже-
нию его. Причина этих явлений заключается в образовании ассо-
циатов или даже пространственных сеток макромолекул в доста-
точно концентрированных растворах полимеров и их разрушении
при перемешивании или нагревании раствора.
Диффузия и седиментация
Вследствие большого размера макромолекул растворы высоко-
молекулярных веществ по своей малой диффузионной способности
близки к типичным коллоидным системам. Тем не менее опреде-
ление коэффициента диффузии широко используется для установ-
ления молекулярного веса высокомолекулярных соединений, на-
пример белков.
При определении молекулярного веса полимеров со сфериче-
скими молекулами диффузионным методом поступают так же, как
и при нахождении этим же способом численного веса коллоидных
систем. Сначала экспериментально определяют коэффициент диф-
фузии, затем, пользуясь известным уравнением Эйнштейна, вычис-
ляют радиус молекулы и, наконец, зная радиус молекулы и плот-
ность растворенного вещества, находят массу 1 моля вещества.
Определение молекулярного веса нитеобразных гибких молекул диффузион-
ным методом гораздо более сложно, так как такие молекулы диффундируют
иначе, чем сферические частицы, к которым только и приложимо уравнение
Эйнштейна в его обычном виде. Поэтому при расчетах необходимо учитывать
тдк называемый коэффициент днснмметрни (подробно об этрм см. первое изда-
ние этого учебника, с. 498).
Практически наиболее распространенным методом определения
коэффициента диффузии высокомолекулярных веществ является
метод Ламма, основанный иа фотографировании шкалы через
столб жидкости, в которой происходит диффузия. Из-за различия
коэффициентов преломления, обусловленных градиентом концен-
трации в жидкости, расстояния между делениями шкалы на фото-
снимке будут иными по сравнению с подобными расстояниями на
фотоснимке, снятом через чистый растворитель. Измеряя отклоне-
ние в расстояниях через определенные промежутки времени, можно
получить распределение градиента концентраций по всему столбу
жидкости при различной продолжительности диффузии. По этим
данным можно вычислить коэффициент диффузии,
456
Несмотря на малый коэффициент диффузии, растворы высоко-
молекулярных соединений обладают, как правило, высокой седи-
ментационной устойчивостью, чему значительно способствует
обычно малая плотность растворенного вещества. Поэтому молеку-
лярный вес высокомолекулярных веществ можно определить мето-
дом седиментации только с помощью достаточно мощной ультра-
центрифуги.
Как н прн определении численного веса коллоидных систем, для определе-
ния молекулярного веса полимеров применяются два метода: по скорости седи-
ментации н по седиментационному равновесию. Второй метод обладает тем
преимуществом, что полученные с его помощью результаты не зависят от
формы частиц; недостатком же его является длительность установления седи-
ментационного равновесия.
Интерпретация результатов, полученных этими методами, сильно ослож-
няется, когда в растворе находятся не компактные частицы, а рыхлые клубки
гибких молекулярных цепей. Если молекулы компактны, нх взаимодействие не-
велико н влияние его становится заметным лишь прн высоких концентрациях.
Прн исследовании компактных структур результаты легко экстраполировать до
нулевой концентрации. При исследоваиин рыхлых клубкообразных структур воз-
никают осложнения, вызываемые взаимодействием макромолекул н гидродина-
микой нх оседания. В этом случае скорость седиментации так быстро изме-
няется с повышением концентрации, что экстраполяция может повести к суще-
ственным ошибкам.
Оптические свойства
Цепные молекулы полимеров нельзя обнаружить в растворах
при ультрамикроскопических наблюдениях. Это объясняется тем,
что растворы полимеров гомогенны и линейные макромолекулы
приближаются к коллоидным частицам только по длине, а в двух
других направлениях соответствуют размерам обычных молекул
Кроме того, линейные макромолекулы нельзя обнаружить под
ультрамикроскопом из-за сольватации макромолекул (если она
имеет место) и еще потому, что коэффициент преломления поли-
меров, как правило, сравнительно близок к коэффициенту прелом-
ления среды.
Растворы высокомолекулярных веществ способны рассеивать
свет, хотя и в меньшей степени, чем типичные коллоидные си-
стемы. Дебаем предложен даже оптический метод определения мо-
лекулярного веса полимеров, основанный на измерении мутности
их разбавленных растворов (величины, представляющей собой
коэффициент ослабления света в результате светррассеяния при
прохождении луча через слой раствора определенной толщины).
Светорассеяние концентрированных растворов полимеров обусловлено их
неоднородностью, возникающей вследствие непрерывных небольших отклонении*
концентрации, которые вызывают в свою очередь отклонения (флуктуации)
показателя преломления от его среднего значения.
Согласно Эйнштейну:
З2п3 nl (dnldcfc Нс
3 ’ ’ d[nl(RT)]ldc ~ d[n/(RT)]fdc ' ’ ’
437
где т— мутность раствора; По н п — соответственно показатель преломления рас-
творителя и раствора; дп/дс— концентрационный градиент показателя прелом-
ления раствора {величина постоянная, которая может быть заменена на
(п— п0)/с, так как в области используемых при определении концентраций раз-
ность п — п0 пропорциональна с]; X — длина волны падающего света;
д [n/(RT)]/dc— концентрационный градиент осмотического давления; Н — по-
стоянная величина для данной системы, равная 32л3По (дя/дс)2ДЗА?АХ4); Na—
число Авогадро.
Обратная зависимость т от концентрационного градиента осмотического дав-
ления отвечает положению Эйнштейна, что флуктуации плотности в растворе
тем больше, чем меньше осмотическое давление.
Уравнение (XIV, 24) можно переписать в виде:
Нс d[n/(RT)]
х дс
(XIV, 25>
Осмотическое давление растворов полимеров с линейными молекулами, как
мы видели выше, может быть выражено уравнением (XIV, 22):
Рис. XIV, 10. Зависимость
Helt от концентрации с
растворов высокомолеку-
лярных соединений.
я = cRT/М + Ьс2
где константа Ь учитывает отклонение от идеаль-
ного раствора.
Подставляя уравнение (XIV, 22) в уравнение
(XIV, 25), получим:
я с __. <? [гс/(/?Г)] _
т дс
__ д [RTcl(MRT) + bc2/(RT)]
дс =
~ dc/M+bdc2KRT) 1+2Вс щу,26>
дс М 1 Л
где В = b/(RT).
Множитель В характеризует взаимодействие
между молекулами растворенного вещества. При
В = 0 уравнение (XIV, 26) принимает вид:
Hcjx = 1/М
Из уравнения (XIV, 26) следует, что величина Нс/х линейно зависит от с.
Такая зависимость представлена на рис. XIV, 10. Очевидно, отрезок, отсекаемый
прямой на оси ординат, численно равен величине 1/М.
Для определения молекулярного веса по описанному методу работу ведут
с растворами, содержащими около 0,1 % полимера. Совершенно очевидно, что
для получения правильных результатов исследуемые растворы должны быть,
полностью свободны от посторонних частиц, способных рассеивать свет. Порядок
работы следующий:
1) измеряют п0 и п для какой-нибудь одной концентрации раствора. Так как
разность (п — п0) в уравнении (XIV, 24) возведена в квадрат, ее необходим»
измерить с весьма высокой точностью;
2) с помощью фотометра или нефелометра измеряют т для нескольких зна-
чений с;
3) строят график в координатах Нс/х— с, находят значение Нс/х при-
с->0, равное 1/М, и вычисляют М. Найденное значение для полндисперсных.
высокомолекулярных иеществ представляет средневесовое значение молекуляр-
ного веса Мт- '
Следует иметь в виду, что оптический меюд определения молекулярног»
веса пригоден только в том случае, если частицы меньше 0,1 длины волны рас-
сеянного света. Это условие обычно соблюдается только для растворов полиме-
ров, в которых макромолекулы свернуты в клубок.
458
Описанный метод позволяет определять также степень взаимодействия по-
лимера с растворителем, характеризуемую отклонением системы от поведения
идеального цаствора
Растворы полимеров помимо светорассеяния обнаруживают
способность избирательно поглощать световые лучи. По ультра-
фиолетовым и инфракрасным спектрам поглощения можно судить
с строении полимера — наличии в его молекулах определенных
атомных групп, сопряженных двойных связей и т. д. Однако по-
скольку эти методы применяются для исследования растворов не
только полимеров, но и органических веществ вообще, мы здесь
останавливаться на них не будем.
Вязкость
Вязкость растворов, содержащих макромолекулы, обычно выше
вязкости растворов низкомолекулярных соединений и коллоидных
растворов тех же концентраций. Например, у растворов каучука
аномально высокая вязкость наблюдается уже при концентрациях
порядка 0,05%. Только очень разбавленные растворы высокомоле-
кулярных соединений можно считать подчиняющимися законам
Ньютона и Пуазейля. Вязкость растворов высокомолекулярных ве-
ществ не подчиняется также закону Эйнштейна и возрастает
с увеличением концентрации. Графически эта зависимость изобра-
жается кривой, обращенной выпуклостью к оси концентраций.
Долгое время высокую вязкость растворов высокомолекуляр-
ных соединений объясняли большой сольватацией макромолекул.
Однако впоследствии, в связи с обнаружением сравнительно не-
значительной^ сольватации макромолекул, пришли к убеждению,
что отклонения вязкости растворов высокомолекулярных соедине-
ний от законов, которым подчиняются растворы низкомолекуляр-
ных веществ, следует объяснить особенностями гидродинамики си-
стем, содержащих вытянутые и гибкие макромолекулы и нали-
чием в них ассоциа!ов и легко разрушаемых структур.
Рассмотрим сначала сильно разбавленные растворы полиме-
ров, в которых макромолекулы не взаимодействуют друг с другом.
Штаудингер, представлявший себе макромолекулы в растворе
в виде жестких палочек, предложил следующую зависимость
удельной вязкости т)уд от концентрации растворов полимеров:
ЧУД=М« (XIV, 27)
где Км —константа для каждого полимергомологического ряда; М — молекуляр-
ный вес растворенного полимера; с — концентрация раствора, выраженная в
«основных молях» на литр. («Основной моль» — число граммов, равное молеку-
лярному весу мономера, из которого получена молекула полимера; например,
у полнбутадиена «основной» одномолярнын раствор должен содержать 54 г по-
лимера в 1 л).
По уравнению (XIV, 27), представленному в виде:
ЛТуд = п/(Км«) (XIV, 28)
459
можно вычислить молекулярный вес полимера, если определена
удельная вязкость его раствора, известна его концентрация с и
константа /См-
Значение константы Кл, необходимое для определения молеку-
лярного веса, Штаудингер находил путем измерения молекулярных
весов низших членов данного полимергомологического ряда крио-
скопическим методом и определения удельной вязкости их рас-
творов. Подставляя найденные величины в уравнение:
Ям = т)уд/(Мс) (XIV, 29)
Штаудингер вычислял значения Kw. данного полимергомологиче-
ского ряда.
Уравнение (XIV, 27) можно представить так:
Т1ул/г = Хмм (XIV, 30)
В таком виде уравнение указывает на линейную зависимость
между величиной т)уд/с, называемой приведенной (к единице
Рис. XIV, 11. Зависимость
приведенной вязкости от
концентрации раствора:
/— раствор, подчиняющийся
уравнению (XIV, 30); 2—раствор
полимера*
концентрации) вязкостью, и молекуляр-
ным весом полимера. Уравнение XIV, 30
показывает также, что для одного и того
же полимера (при постоянном молеку-
лярном весе) приведенная вязкость не
должна зависеть от концентрации, как
это и изображено на рис. XIV, 11 (пря-
мая /).
Первые исследования Штаудингера и
его сотр. как будто подтвердили пра-
вильность предложенного им уравнения.
Однако дальнейшие работы показали,
что приведенная вязкость растворов од-
ного и того же полимера обычно воз-
растает с повышением концентрации,
причем это возрастание в интервале не-
больших концентраций происходит по прямой, как это показано
на рис. XIV, 11 (прямая 2). Отрезок, отсекаемый этой прямой на
оси т)уД/с, отвечает величине так называемой характеристи-
ческой вязкости [т)], отражающей гидродинамическое сопротив-
ление потоку молекул данного полимера. Характеристическая вяз-
кость представляет собой приведенную вязкость при бесконечно
большом разбавлении раствора.
Очевидно
[q] = lim (пуд/с)
с-»0
(XIV, 31)
а уравнение прямой в этом случае может быть представлено как
Луд/с = [т)1 + “с (XIV, 32)
где а — тангенс угла, образуемого прямой с осью концентраций.
460
Зависимость значения т]уд/с от концентрации следует отнести за
счет взаимодействия макромолекул.
Далее исследования показали, что во многих случаях постоян-
ная Лм в уравнении Штаудингера зависит от молекулярного веса
полимера, как правило, уменьшаясь с его увеличением. Штаудин-
гер пытался объяснить это тем, что на величину Лм влияет раз-
ветвленность макромолекул, обычно возрастающая по мере увели-
чения степени полимеризации. Однако было установлено, что зна-
чение Км снижается по мере увеличения молекулярного веса и для
строго линейных молекул. Хаггинс показал, что изменение /См
с увеличением молекулярного веса обусловлено не увеличением
разветвленности макромолекул, а тем, что более длинные моле-
кулы способны сильнее изгибаться и тем самым оказывают отно-
сительно меньшее сопротивление потоку жидкости.
В настоящее время для определения молекулярного веса пред-
ложено уравнение, учитывающее взаимодействие макромолекул
даже в разбавленных растворах и изменение константы /См с дли-
ной молекулы:
[T)] = feMa (XIV, 33)
где k — коэффициент, постоянный для растворов соединений полимергомологиче-
ского ряда в данном растворителе; а — величина, характеризующая форму ма-
кромолекул в растворе и связанная с гибкостью цепей.
Если а = 1, то приведенное уравнение переходит в уравнение,
аналогичное уравнению Штаудингера. Если же a = 0, то уравне-
ние (XIV, 33) переходит в уравнение Эйнштейна, выведенное из
предположения о сферичности частиц, согласно которому вязкость
раствора не зависит от размера частиц.
На форму макромолекулы в растворе, как это показали экспе-
риментальные исследования, существенным образом влияет при-
рода растворителя. В одном растворителе молекулы полимеров
могут быть более вытянуты, в другом — более свернуты в клубок.
Как правило, чем-лучше полимер растворяется в данной жидко-
сти, чем более он сольватирован, тем меньше участки молекуляр-
ной цепи взаимодействуют друг с другом, тем более вытянуты
макромолекулы и тем выше вязкость раствора. В плохом раство-
рителе макромолекулы мало сольватированы и поэтому образуют
более компактные клубки. Поэтому введение в раствор полимера
нерастворителя обычно значительно снижает вязкость раствора.
Таким образом, для одного и того же полимера, растворенного
в различных растворителях, будет меняться гидродинамическое со-
противление макромолекул потоку жидкости, а следовательно, и
характеристическая вязкость. Экспериментально было показано,
что на зависимость т]уд/с от концентрации с влияет природа рас-
творителя, т. е. меняется наклон прямых т[уя/с, с. На основании не-
которых соображений считают, что коэффициент, определяющий
наклон прямой, пропорционален квадрату [tj]. Тогда уравнение
(XIV, 32) можно написать в виде
= + (XIV, 34)
461
Хаггинс считает, что коэффициент k в этом уравнении может
служить характеристикой интенсивности взаимодействия молекул
полимера и растворителя.
Для вычисления молекулярного веса по уравнению (XIV, 33)
должны быть известны величины k к а. Эти величины вычисляют
по экспериментально найденным значениям вязкости растворов и
молекулярному весу какого-нибудь члена данного полимергомоло-
гического ряда, определенного другим более точным методом.
В табл. XIV,4 приведены значения величин k и а для некоторых
Таблица XIV, 4. Значения k и а для вискозиметрического
определения молекулярных весов некоторых полимеров
по уравнению (jn]=feAf“
Полимер Растворитель Темпера- тура, °C й-104 а Область молеку- лярных весов, лью-5
Целлюлоза Медио-аммиачный 25 0,85 0,81 *—
Полиизобутилен Диизобутилен 20 3,6 0,64 0,05—13,0
Натуральный каучук Толуол 20 5,02 0,67 0,4-15,0
Полистирол Толуол 30 3,7 0,62 2,0-18,0
Полиметилметакрилат Хлороформ 20 0,49 0,82 0,56-9,8
Поливинилацетат Ацетон 50 2,8 0,67 0,77-8,5
полимеров. Для более низких молекулярных весов, чем указанные
в последней колонке таблицы, значения а для всех полимеров по-
степенно приближаются к единице в соответствии с первоначаль-
ным уравнением Штаудингера.
Перейдем теперь к рассмотрению вязкости растворов средней
концентрации. Как уже указывалось, эти растворы не подчиняются
законам Ньютона и Пуазейля. Коэффициент вязкости для этих
растворов не является постоянным, а зависит от градиента ско-
рости или давления, если определение ведут с помощью капилляр-
ного вискозиметра. При этом, так же как и для структурирован-
ных коллоидных систем, с возрастанием градиента скорости вяз-
кость раствора падает, постепенно приближаясь к некоторому
пределу.
Зависимость вязкости от градиента скорости для растворов по-
лимеров средней концентрации обусловлена двумя причинами.
Во-первых, при течении раствора длинноцепные молекулы, нахо-
дящиеся в растворе в виде клубков, распрямляются и ориенти-
руются по направлению течения, что, конечно, уменьшает гидро-
динамическое сопротивление потоку. Это объяснение аналогично
объяснению зависимости коэффициента вязкости от градиента ско-
рости для коллоидных систем, содержащих жесткие удлиненные
частицы. Понятно, что ориентация макромолекул происходит и при
течении разбавленных растворов полимеров. Однако в этом слу-
462
чае, вследствие малой концентрации дисперсной фазы в растворе,
отклонения от закона Пуазейля проявляются значительно слабее.
При течении с весьма большими градиентами скорости растяже-
ние молекулярных цепей может быть настолько сильным, что мо-
жет приводить к разрыву макромолекул. Этим, вероятно, и объ-
ясняется некоторая деполимеризация полимеров в растворах при
очень большой скорости их истечения сквозь тонкие капилляры.
Во-вторых, в достаточно концентрированных растворах макромо-
лекулы могут взаимодействовать друг с другом и образовывать
ассоциаты или даже обрывки пространственной сетки, сильно ме-
шающие течению. По мере ускорения течения эти структуры по-
степенно разрушаются, что также должно понижать сопротивление
молекул потоку, а следовательно, и вязкость растворов. Такое объ-
яснение вполне аналогично объяснению подобного же явления для
структурированных коллоидных систем.
А. А. Трапезниковым с сотр. с помощью новых методов измерения и при-
боров проведены многочисленные исследования реологических свойств концен-
трированных растворов полимеров преимущественно в неполярных растворите-
лях. При этом определяли не только напряжение сдвига, но и обратимую де-
формацию и исследования проводили не только в стационарном потоке, но и в
предстационарной стадии деформации. Эти исследования показали, что для мно-
гих систем можно наблюдать свойства, присущие как типичным пластическим
системам, так и жидкостям, не подчиняющимся закону Ньютона и вязкость
которых при истечении определяется ориентацией молекул. Для объяснения
сложного комплекса свойств подобных систем необходимо отказаться от при-
вычного представления о том, что ниже предела текучести невозможно течение.
Совершенно очевидно, что если в принципе необратимая релаксация возможна
при любых малых напряжениях сдвига, то и течение возможно при таких же
малых напряжениях. Вопрос заключается только в продолжительности измере-
ния и чувствительности регистрирующих приборов. В связи с этим было пред-
ложено новое понятие о пределе текучести как отражающем не появление те-
чения, а изменение скорости течения, связанное со структурными изменениями
в системе.
При повышении температуры увеличивается интенсивность дви-
жения сегментов, что препятствует образованию структур, и вслед-
ствие этого отклонение от законов Ньютона и Пуазейля при повы-
шенных температурах наблюдается' в меньшей степени. Кроме
того, при повышении температуры понижается истинный коэффи-
циент внутреннего трения, что также обуславливает понижение
вязкости раствора. Здесь, однако, уместно отметить, что повыше-
ние температуры не всегда ведет к понижению вязкости раствора
высокомолекулярного вещества. Такое понижение характерно для
растворов, содержащих сйльно разветвленные макромолекулы,
у которых сегментарный тип движения мало выражен. Вязкость
растворов, содержащих длинные неразветвленные молекулярные
цепи, с повышением температуры может даже повышаться из-за
увеличения интенсивности движения сегментов, прейятствующего
ориентации макромолекулы в потоке.
Помимо зависимости коэффициента вязкости от градиента ско-
рости для растворов средней концентрации наблюдаются и другие
463
Рис. XIV, 12. Зависимость
удельной вязкости от концен-
трации водных растворов са-
хара (I) и казениата натрия (2).
аномалии, например увеличение вязкости при стоянии раствора,
уменьшение вязкости после нагревания и последующего охлажде*
ния раствора, после перемешивания и т. д. Все эти аномалии яв-
ляются следствием неравновесности раствора, с которым имеет
дело экспериментатор, и происходят из-за склонности системы
к структурированию.
Весьма характерно изменение вязкости растворов полимеров
с увеличением их концентрации. На рис. XIV, 12 показана схема-
тически зависимость т]УД от с для водных растворов сахара и ка-
зеината натрия. Как можно видеть, для раствора сахара эта за-
висимость выражается прямой линией. Совершенно иначе ведут
себя растворы полимеров, например водный раствор казеината
натрия. С повышением концентрации
вязкость этого раствора возрастает
очень резко по кривой, обращенной
выпуклостью к оси концентрации. При-
чину такой аномалии чисто качествен-
но можно объяснить тем, что в урав-
нении Эйнштейна (так же, как и в
уравнении Ван-дер-Ваальса) должен
фигурировать не общий объем систе-
мы, а эффективный объем, т. е. объем
системы за вычетом объема макромо-
лекул. Так как макромолекулы в рас-
творе находятся в виде клубков, вклю-
чающих большое количество раство-
рителя, то объем этого растворителя, геометрически связанного
с полимером, также следует отнести к объему дисперсной фазы
(более подробно об этом см. в гл. X).
Следует заметить, что помимо указанной причины, резкое уве-
личение вязкости с повышением концентрации может происходить
и в результате образования в системе структур. Понятно, что при
этом раствор будет обладать уже структурной вязкостью, в то
время как при первом объяснении концентрационной аномалии
допущение о взаимодействии макромолекул не обязательно.
На вязкость так же, как и на осмотическое давление доста-
точно концентрированных растворов высокомолекулярных веществ,
может влиять способ приготовления раствора. И здесь такое влия-
ние объясняется медленным установлением равновесия в системе.
Наконец, на вязкость растворов высокомолекулярных веществ
может влиять введение в раствор небольших количеств некоторых
веществ. Из практики, например, известно, что вязкость растворов
эфиров целлюлозы при введении все возрастающих количеств
спирта сначала падает, а затем возрастает. Подобные же явления
наблюдаются и при введении в эти же растворы воды. При до-
бавлении в растворы эфиров целлюлозы солей алюминия, железа,
свинца, кальция, магния и цинка, как правило, вязкость повы*
шается; при введении некоторых мыл она уменьшается.
464
Повышение вязкости растворов высокомолекулярных веществ
при введении в них различных добавок объясняется либо увели-
чением взаимодействия макромолекул друг с другом в результате,
освобождения под влиянием примесей активных мест на молеку-
лярных цепях, либо образованием химических связей между моле-
кулами полимера и примесей (действие окислов металлов, альде-
гидов). Понижение вязкости также можно объяснить двумя при-
чинами: либо деструкцией макромолекул под влиянием примесей
(действие аммиака, альдегидов, кислот и т. д.), либо уменьшением
взаимодействия цепей друг с другом в результате взаимодействия
примесей с активными группами макромолекулы. „
В заключение следует отметить, что существенной особенностью
растворов высокомолекулярных веществ является неподчинение их
известному правилу Вальдена. Вальден установил, что эквивалент-
ная электропроводность X обычных вязких растворов, содержащих
низкомолекулярные электролиты, обратно пропорциональна вяз-
кости, т. е.
A = fe(l/t]) (XIV, 35)
или
АЛ — const (XIV, 36)
В растворах высокомолекулярных веществ, вязкость которых
обусловлена присутствием свернутых в клубок длинных цепных
молекул, ионы низкомолекулярных электролитов способны совер-
шенно свободно продвигаться в водной среде между этими макро-
молекулами. В результате электропроводность вязких растворов
высокомолекулярных веществ, содержащих низкомолекулярные
электролиты, практически равна электропроводности растворов
этих электролитов. Отсюда понятно, почему электропроводность
растворов высокомолекулярных веществ не зависит от их вязко-
сти. Это наблюдается даже тогда, когда раствор высокомолеку-
лярного вещества перейдет в состояние студня.
Агрегативная устойчивость
Растворы высокомолекулярных веществ, если они находятся
в термодинамически равновесном состоянии, агрегативно устой-
чивы, как и истинные растворы. Поэтому специальные теории
о устойчивости лиофильных коллоидных систем, например теория
Кройта и Бунгенберг де Йонга, согласно которой агрегативную
устойчивость растворов желатина, агара и других высокомолеку-
лярных соединений авторы пытались объяснить либо электриче-
ским зарядом частиц, либо сольватацией, либо, наконец, действием
обоих этих факторов одновременно, представляют теперь только
исторический интерес.
При введении больших количеств электролитов наблюдается
выделение высокомолекулярных веществ из раствора. Однако это
явление не следует отождествлять с коагуляцией типичных кол-
лоидных систем. Коагуляция золей происходит при введении срав-
465
нительно небольших количеств электролита и представляет собой'
обычно необратимое явление. Выделение же из раствора высоко-
молекулярного вещества происходит при добавлении относительна
больших количеств электролита, не подчиняется правилу Шульце—
Гарди и является обычно обратимым процессом — после удаления
из осадка электролита промыванием или диализом высокомолеку-
лярное вещество снова способно к растворению.
Различен и механизм обоих явлений. Коагуляция золей проис-
ходит обычно в результате сжатия двойного электрического слоя
и уменьшения или полного исчезновения электрического заряда на
поверхности частицы, являющегося в этом случае основным фак-
тором устойчивости. Выделение же из раствора полимера при до-
бавлении электролита объясняется уменьшением растворимости
высокомолекулярного вещества в концентрированном растворе
электролита. По аналогии с подобными явлениями в растворах
низкомолекулярных веществ такое' выделение высокомолекуляр*
ного вещества из раствора можно называть высаливанием. Дебай
считает, что при высаливании молекулы растворенного вещества
вытесняются из электрического поля введенных ионов, которые
связываются с полярными молекулами растворителя. Таким обра-
зом, высаливание принципиально не отличается от выделения вы-
сокомолекулярного вещества из раствора при добавлении к по-
следнему нерастворителя. Как правило, высаливающее действие
ионов изменяется соответственно тому порядку, в каком они стоят
в лиотропном ряду. Так, катионы по мере уменьшения их высали-
вающего действия могут бы'ть расположены в ряд:
Li+>Na+>K+>Rb+>Cs+
Подобный же ряд анионов имеет вид
SO*’ > СГ > NO’ > Вг' > Г> NCS-
Взаимосвязь между высаливающим действием и местом иона
в лиотропном ряду вполне понятна: чем больше ион способен свя-
зывать растворитель, тем больше он будет уменьшать способность
среды растворять высокомолекулярное вещество.
Высаливание лежит в основе одного из методов фракциониро-
вания высокомолекулярных веществ, поскольку способность этих
соединений выделяться из раствора весьма сильно зависит от их
химической природы и резко возрастает С увеличением молекуляр-
ного веса. Особенно широко применяется фракционирование с по-
мощью высаливания для разделения белков. При этом высалива-
ние часто сочетают с введением в систему нерастворителя (напри-
мер, спирта) и охлаждением раствора. Высаливание белков целе-
сообразно проводить при значении pH, близком к изоэлектриче-
ской точке, так как при значении pH, большем или меньшем изо-
электрической точки, возрастает заряд и гидратация белковых мо-
лекул и увеличивается их растворимость.
466
В растворах высокомолекулярных соединений при изменении
температуры или pH или при введении низкомолекулярных ве-
ществ иногда наблюдается явление коацервации, о котором мы
уже указывали. Это явление, свойственное только растворам, не
находящимся в термодинамическом равновесии, заключается
в разделении системы на две фазы, из которых одна представляет
собой раствор высокомолекулярного вещества в растворителе,
а другая — раствор растворителя в высокомолекулярном веще-
стве. Раствор, более богатый высокомолекулярным веществом,
обычно выделяется в виде мельчайших капелек. Если раствори-
телем является полярная жидкость, например вода, мельчайшие
капельки в определенных условиях могут приобретать электриче-
ский заряд, что доказывается их способностью к электрофорезу.
Понятно, что электрический заряд капелек придает этим системам,
вообще термодинамически неравновесным, некоторую устойчивость.
Такие системы, называемые коацерватами, по существу близки
к типичным эмульсиям. Высказывались предположения, что они
играют важную роль в живых организмах.
Если мельчайшие капельки коацерватов не обладают доста-
точной агрегативной устойчивостью и в то же время не способны
и коалесценции (слиянию), то они могут соединяться друг с дру-
гом, образуя флокулы, которые всплывают или опускаются на дно
сосуда в виде рыхлого осадка. Такая флокуляция происходит
-обычно, когда фаза с большим содержанием высокомолекуляр-
ного компонента обладает достаточной вязкостью. Если же вяз-
кость фазы небольшая, то происходит обычно коалесценция от-
дельных мельчайших капелек и постепенное образование более
крупных капелек. Обычно при длительном стоянии системы, в ко-
торой произошла коацервация, образуются два гомогенных жидких
слоя, состоящих из фаз с различным содержанием высокомолеку-
лярного вещества. Наконец, в достаточно концентрированных рас-
творах высокомолекулярных соединений за счет сцепления макро-
молекул в отдельных местах могут образовываться постоянные
пространственные сетки, благодаря чему раствор превращается
в студень.
В заключение необходимо хотя бы кратко остановиться на яв-
лениях старения растворов высокомолекулярных веществ. Принято
считать, что старение наглядней всего проявляется в спонтанном
(самопроизвольном) изменении вязкости равновесных растворов.
Ранее, когда к растворам высокомолекулярных веществ подходили
с тех же позиций, что и к коллоидным системам, эти изменения
вязкости объясняли медленно протекающими явлениями пептиза-
ции или, наоборот, агрегирования. В настоящее время, когда уста-
новлена гомогенность не слишком концентрированых растворов
высокомолекулярных веществ, такое объяснение не может быть
признано удовлетворительным.
Сейчас следует считать, что изменение вязкости растворов вы-
сокомолекулярных веществ при стоянии происходит в результате
467
воздействия на молекулярные цепи присутствующего в системе
кислорода или других примесей. Кислород может вызывать либо-
деструкцию макромолекул, либо связывание отдельных нитевид-
ных молекул в большие образования. В первом случае вязкость
уменьшается, во втором случае — увеличивается. Аналогично на
вязкость растворов высокомолекулярных соединений способны
действовать и некоторые другие примеси. Таким образом, в на-
стоящее время можно считать установленным, что растворы высо-
комолекулярных соединений в отличие от типичных коллоидных
систем не подвержены процессу собственно старения, а изменение
некоторых их свойств при длительном стоянии обусловлено мед-
ленным действием присутствующих в них посторонних веществ.
8. ПОЛИЭЛЕКТРОЛИТЫ
В предыдущих разделах рассмотрены - свойства растворов по-
лимеров, макромолекулы которых не содержат ионогенных групп.
К таким полимерам относятся натуральный и синтетический кау-
чуки, полиизобутилен, нитрат целлюлозы, ацетат целлюлозы и
многие другие полимеры. Однако молекулы ряда высокомолеку-
лярных веществ содержат ионогенные группы и в растворах спо-
собны распадаться на ионы. Такие высокомолекулярные электро-
литы, или полиэлектролиты, по природе содержащихся
в них ионогенных групп можно разделить на три категории:
1. Полиэлектролиты, содержащие кислотную группу, например
—СОО~ или —OSO3. Группу —СОО" содержат гуммиарабик, аль-
гинаты, растворимый крахмал, а группу —OSO3 — агар.
2. Полиэлектролиты, содержащие основную группу, например
—NH3. Такие вещества в природе не встречаются, но могут быть
синтезированы.
3. Полиэлектролиты, содержащие одновременно как кислотную,
так и основную группы (полиамфолиты). Сюда следует отнести
белки, содержащие Труппы —СОО~ и —NH3. В последнее время
получены синтетические полиамфолиты, например сополимеры
акриловой кислоты и винилпиридина, глютаминовой кислоты и ли-
зина.
Свойства растворов полиэлектролитов
Полиэлектролиты, за исключением белков, характеризуются
высокой плотностью расположения ионогенных групп — обычно на
одно звено цепи приходится по одной ионогенной группе. У белков
одна карбоксильная группа или амино-группа приходится на 6—
8 остатков аминокислот. Вследствие этого молекулы полиэлектро-
литов могут испытывать в растворах значительные электростати-
ческие взаимодействия, что приводит к сильной деформации цепей
гибких молекул. Такая деформация, естественно, зависит от сте-
пени ионизации групп, которая в свою очередь является следствием
468
присутствия в системе низкомолекулярных электролитов и pH рас-
твора. Канальский показал, что при изменении pH раствора цепи
полиакриловой кислоты могут самопроизвольно растягиваться и
сокращаться в несколько раз.
Все высокомолекулярные электролиты растворяются в поляр-
ных растворителях, так как макромолекулы с ионогенными груп-
пами взаимодействуют с полярными жидкостями сильнее, чем с не-
полярными. Именно вследствие значительного взаимодействия вы-
сокомолекулярных электролитов (белков) со средой (водой) вы-
сокомолекулярные вещества одно время называли лиофильными
коллоидами.
Рассмотрим важнейшие представители полиэлектролитов,
а именно, белки, так как они ведут себя в растворах наиболее
сложно. У других высокомолекулярных электролитов, содержащих
либо одни кислотные, либо одни основные группы, исчезают только
свойства, присущие отсутствующей ионогенной группе.
Молекулы белков, построенные из аминокислот, в водной среде
содержат основные группы HONH3— и кислотные группы —СООН
и поэтому являются амфотерными соединениями. Схематически
белковую молекулу можно изобразить так: HONH3—R—СООН,
где R— достаточно длинная углеводородная цепочка, обычно со-
держащая группы —CONH—. Однако следует помнить, что ионо-
генные группы OHNH3— и —СООН могут располагаться не
только на концах молекулы, но и в виде коротких боковых цепей,
распределенных по всей длине основной цепи.
В водном растворе при определенной концентрации водород-
ных ионов, отвечающей изоэлектрической точке, у всякого амфо-
лита (амфотерного электролита) число ионизированных основных
групп равно числу ионизированных кислотных групп. При этом
число как тех, так и других групп минимально. Молекулу белка
в изоэлектрическом состоянии следует считать в целом нейтраль-
ной, хотя она и имеет еще ионизированные группы. Условно ее
можно изобразить в этом состоянии следующим образом:
ОН- 4- NH+—R—COO- + Н+
Поскольку белок обычно является более сильной кислотой, чем
основанием, то изоэлектрическая точка его лежит при pH ниже 7.
Иначе говоря, для достижения изоэлектрической точки в растворе
белка должно содержаться некоторое количество кислоты, подав-
ляющее избыточную ионизацию кислотных групп. Так как в изо-
электрической точке число взаимодействующих ионизированных
основных и кислотных групп в молекуле одинаково, то гибкая
макромолекула в этом состоянии свернется в клубок. Плотность
клубка вследствие сил притяжения между разноименно заряжен-
ными группами будет больше той плотности, которая отвечает наи-
более статистически вероятной форме макромолекулы или макси-
мальной ее энтропии.
469
В кислой среде, например в присутствии НС1, когда в резуль-
тате избытка водородных ионов подавлена ионизация карбоксиль-
ных групп, происходит следующая реакция:
ОН- + NHj— R—СООН + Н+ + С1" —>- СГ + NHJ—R—СООН + Н2О
Молекула белка, ведущая себя в этом случае как основание, при-
обретает положительный заряд и при электрофорезе движется
к катоду. Поскольку между одноименно заряженными группами,
разбросанными по всей длине молекулы, действуют электрические
•силы отталкивания, свернутая в клубок цепная молекула белка
в кислой среде будет стремиться распрямиться. Плотность моле-
кулярного клубка в результате этого понизится и может стать
даже ниже той плотности, которая соответствует наиболее стати-
стически вероятной форме гибкой макромолекулы. Однако при
большом избытке НС1 из-за н-аличия большого количества хлорид-
ионов степень ионизации соединения CINH3—R—СООН, являюще-
гося солью сильной кислоты и слабого основания, будет пони-
жаться и молекула снова свернется в более плотный клубок.
В щелочной среде, например в присутствии NaOH, из-за боль-
шого количества находящихся в растворе гидроксильных ионов
ионизация групп HONH3— подавлена и в растворе протекает сле-
дующая реакция:
HONH3—R—СОО" + Н4 + Na+ + ОН- —>- HONH3—R—СОО" + Na+ + Н2О
Молекула белка ведет себя здесь как кислота, она приобретает от-
рицательный заряд и при электрофорезе передвигается к аноду.
В этом случае также в результате взаимодействия одноименно за-
ряженных групп —СОО- цепная молекула стремится распрямиться
и плотность молекулярного клубка уменьшается. Однако при из-
бытке NaOH, из-за наличия большого количества ионов Na+ и сни-
жения ионизации соли HONH3—R—COONa, заряд будет умень-
шаться и макромолекула снова свернется в более плотный клубок.
На форму макромолекул действует не только изменение pH
•среды, но и введение в раствор индифферентного электролита. Не
«лишком большое количество электролита в растворе полиэлектро-
лита подавляет ионизацию ионогенных групп и, очевидно, приводит
к приближению формы макромолекул к наиболее статистически ве-
роятным конформациям. При больших концентрациях электролитов
начинает сказываться уже их высаливающее действие, что снижает
растворимость высокомолекулярных веществ и приводит к образо-
ванию более плотных молекулярных клубков. При этом действие
ионов низкомолекулярных электролитов, конечно, будет соответ-
ствовать тому порядку, в каком они стоят в лиотропном ряду.
Так как молекулы высокомолекулярного электролита обладают
все^и свойствами обычных электролитов и в том числе способ-
ностью подавлять ионизацию других электролитов, то увеличение
концентрации самого высокомолекулярного электролита в рас-
470
творе будет действовать так же, как если бы в систему вводили
индифферентный электролит.
Из сказанного видно, что pH и введение электролитов влияют
на заряд и форму молекул высокомолекулярных электролитов.
Очевидно, эти факторы также должны влиять и на те свойства
раствора, которые зависят от формы растворенных макромолекул.
К таким свойствам относятся вязкость, осмотическое давление й
объем студня набухшего высокомолекулярного вещества, если он
не растворяется в данной среде.
Рассмотрим, как влияет pH -на свойства растворов белков.
На рис. XIV, 13 изображена экспериментально найденная зави-
симость относительной вязкости 0,67%-ного раствора желатина от
Рис. XIV, 13. Зависимость отно-
сительной вязкости 0,67%-иого
раствора желатина от pH среды.
Рис. XIV, 14. Зависимость на-
бухания желатина от pH среды.
pH этого раствора. Можно видеть, что наименьшая вязкость отве-
чает изоэлектрической точке (pH = 4,8), при которой молекула
свернута в наиболее плотный клубок и благодаря малому объему
этого клубка менее всего препятствует течению жидкости. Как
с уменьшением, так и с увеличением pH вязкость раствора воз-
растает, так как в результате распрямления молекул объем клубка
увеличивается. Однако при очень больших и очень малых значе-
ниях pH, вследствие уплотнения молекулярного клубка в резуль-
тате подавления ионизации одноименно заряженных ионогенных
групп,- вязкость раствора снова уменьшается.
Аналогичную форму должна иметь и кривая, выражающая за-
висимость объема студня желатина от pH жидкости, с которой
студень находится в состоянии равновесия. Действительно, опыты
Лёба показали, что степень набухания желатина в воде в зависи-
мости от pH может быть представлена седлообразной кривой с ми-
нимумом, отвечающим изоэлектрической точке, и с двумя макси-
мумами, лежащими по правую и левую сторону от минимума. Эта
кривая изображена на рис. XIV, 14.
В связи с вопросом о влиянии ионизации ионогенных групп
макромолекул на объем студня уместно хотя бы кратко остано-
виться на исследованиях,, приведших к созданию общей теории на-
471
бухания и «отбухания» студней высокомолекулярных электроли-
тов при изменении степени ионизации.
Если к студню высокомолекулярного электролита приложить
какую-нибудь нагрузку, то при поочередной ионизации и нейтрали-
зации (моляризации) его мол'екул студень способен производить
механическую работу. Таким образом, студень может служить не
чем иным, как механо-химической машиной. Сетка из молекул вы-
сокомолекулярного электролита, подобно хорошо известным теп-
ловым машинам, может производить работу, превращая химиче-
скую энергию в механическую, причем это может происходить при
постоянной температуре. Работа мускулов живого организма тесно
связана с механо-химическими процессами; именно ими объяс-
няется способность человека производить работу.
Нетрудно представить, что кривая, выражающая зависимость
осмотического давления раствора желатина от pH, также должна
иметь седлообразную форму. В изоэлектрическом состоянии свер-
нутая в плотный клубок, макромолекула обладает очень малой
гибкостью и число сегментов, играющих роль кинетических единиц,
минимально. При значениях pH выше и ниже изоэлектрической
точки макромолекула желатина, распрямляясь, становится все
болеё гибкой, что и обуславливает увеличение числа движущихся
сегментов, а следовательно, и рост осмотического давления. При
добавлении в раствор избытка кислоты или щелочи, как было по-
казано выше, гибкость молекулярной цепочки начнет опять умень-
шаться, уменьшится и число движущихся сегментов, в результате
понизится также и осмотическое давление раствора.
На основании подобных же рассуждений нетрудно представить,
как будет изменяться вязкость раствора высокомолекулярного
электролита, его осмотическое давление или объем набухающего
студня при введении в систему низкомолекулярного индифферент-
ного электролита.
Мембранное равновесие Доннана. Зависимость свойств растворов высоко-
молекулярных электролитов от pH и содержания низкомолекулярных электро-
литов можно объяснить не только изменением формы макромолекулы в рас-
творе, но и мембранным равновесием Д о н н а н а.'Правда, равно-
весие Доннана является формальным термодинамическим истолкованием про-
цесса и с его помощью нельзя сделать вывода о молекулярном механизме явле-
ния. Кроме того, следует подчеркнуть, что равновесие Доннана, как и всякая
термодинамическаи интерпретация процесса, пригодно в том случае если имеет
место равновесное состояние системы.
Рассмотрим зависимость осмотического давления растворов полиэлектроли-
тов от pH и присутствия электролитов так, как ее представлял себе Дониан,
а затем уже объясним, исходя из этой концепции, влияние pH и низкомолеку-
лярных электролитов на объем набухшего студня и вязкость растворов поли-
меров
Как мы знаем, осмотическое давление растворов высокомолекулярных ве-
ществ можно определять гораздо более точно, чем осмотическое давление типич-
ных лиозолей. Это обусловлено тем, что растворы высокомолекулярных веществ
можно очищать с помощью длительного диализа или переосаждения, не опа-
саясь коагуляции. Однако практически обычно трудно удалить из раствора высо-
комолекулярного электролита все чужеродные электролиты, и осмотическое
472
давление приходится определять в их присутствии. Чтобы исключить влияние
низкомолекулярных электролитов на результаты определения, Дюкло и Маль-
фитано пытались измерить осмотическое давление растворов высокомолекуляр-
ных электролитов, помещая диализуемый раствор в осмометре в ультрафильтрат.
Однако, как показал Доннан, результаты этих исследований нельзя считать пра-
вильными.
Чтобы понять причину этого, представим себе, что при определении осмо-
тического давления с помощью обычного осмометра с полупроницаемой мем-
браной в начале опыта в осмотическую ячейку налит раствор высокомолекуляр-
ного электролита, полностью распадающегося на не способные к диализу высоко-
молекулярные иоиы Rz+ н на малые ионы, например С1“, проникающие сквозь
мембрану. Примем, что концентрация ионов Rz+ во внутреннем растворе рав-
на Ci, тогда концентрация ионов в том же растворе будет zct. Пусть во внешней
жидкости осмометра содержится низкомолекулярный электролит, например хло-
рид натрия, оба иона которого (Na+ и С1“) способны проходить через мембрану.
Обозначим концентрацию NaCl во внешнем растворе через с2. Наконец, для
упрощения допустим, что объемы внутреннего и внешнего раствора равны.
Тогда распределение ионов в начале опыта будет следующим:
Внутренняя жидкость (/) Внешняя жидкость (е)
[Rz+]i = Ci [Na+]e = с2
(СГ], = гС1 [СГ], = с2
Можно было бы предположить, как принимали Дюкло и Мальфитано, что
хлорид натрия в результате осмоса распределится равномерно по обе стороны
полупроницаемой мембраны. Однако опыт показал, что это не так. Доннаном
было установлено, что в рассматриваемом случае достигается равновесие об-
щего вида:
Внутренняя жидкость (i) Внешняя жидкость (е)
[Rz+L = Ci [Na+]e = с2 - х
[CfJj-zCj+x [СГ]е = с2 — х
[№+].= х
При установлении равновесия иоиы низкомолекулярного электролита не мо-
гут проникать через мембрану порознь, так как в противном случае возникли
бы электрические поля, препятствующие диффузии. Число ударов, производимых
ионами С1" и Na+ одновременно на единицу площади внутренней стороны пере-
городки, зависит от произведения концентраций ионов, т. е. [Na+Ji-ICl-],. Для.
наружной стороны перегородки число одновременных ударов ионов будет про-
порционально произведению [Na+]e-[Cl"]e. Очевидно, в системе установится раи-
новесие, когда число пар ионов, проходящих в единицу времени через перего-
родку справа налево, будет равно числу пар ионов, проходящих слева направо.
Иначе говоря, условием равновесия является равенство произведений концентра-
ций ионов, находящихся внутри н снаружи мембраны и свободно проникающих
сквозь нее:
[Na+]f - [СГ]( = [Na+]e • [СГ]е (XIV, 37)
Подставляя в уравнение (XIV, 37) найденные значения для [Na+],-, [Cl-],, [Na+]»
и [С1"]е, получаем:
x(zC] +х)=>(с2 — x)s (XIV, 38)
или
zcxx = с2 — 2с2х (XIV, 39)
Откуда
х = ^/(жС1+2Сз) (XIV, 40)
473
При условии Ci Сг, т, е. когда в системе индифферентного электролиту
мало
х=су(гС1) (XZV.41J
Из этого уравнения легко получить пропорцию:
х/с2 =» c2/(zcj) (XIV, 42)
Эта пропорция показывает, что низкомолекулярный электролит, когда его мало,
будет преимущественно находиться во внешней жидкости.
При условии Ci С с2, т. е когда в системе индифферентного электролита
много
х = сЦ(2с2) = с2/2 (XIV, 43)
Иными словами, электролит распределится равномерно по обеим сторонам
мембраны.
Рассмотренная схема относится к случаю, когда в начале опыта низкомоле-
кулярный электролит находился только во внешней жидкости. Однако картина
существенно не меняется, если низкомолекулярный электролит присутствует в
начале опыта и в осмотической ячейке. Не меняется положение и в том случае,
если у высокомолекулярного и низкомолекулярного электролитов нет общего
иона, например, когда во внутренней жидкости осмометра находятся ионы Rx+
и С1", а во внешней — Na+ и Вг".
Нетрудно определить, из каких слагаемых состоит осмотическое давление
в рассмотренном выше случае.
Если не считаться с приведенными выше соображениями и принять, что
-присутствие низкомолекулярного электролита не оказывает влияния, поскольку
юн свободно проходит через мембрану, то наблюдаемое осмотическое давление
можно было бы выразить следующим уравнением:
« = RTc1(z +I) (XIV, 44)
На самом деле в результате установления равновесия Доннана осмотическое
давление раствора будет равно:
л = RTcy + RT (zci + х) + RTx — 2RT (c2 + x) (XIV, 45)
В правой части этого уравнения член RTct отвечает осмотическому давле-
нию, обусловленному высокомолекулярными нонами, член RTXzcj-f-x)—осмоти-
ческому давлению хлорнд-ионов; член RTx — осмотическому давлению нонов
натрия инутрн Ячейки осмометра, а член 2RT (с2 + х) — осмотическому давлению
низкомолекулярного электролита в наружной жидкости Подставляя в приведен-
ное уравнение значения х нз уравнения (XIV, 40) и проведя некоторые преоб-
разования, получим:
л = -Ге| Jj -1 + 2сг (XIV, 46)
zcj + 2с2 ' 1
Это уравнение при условии с, с2, т. е. когда в системе почти нет низко-
молекулярного электролита, превращается в следующее:
п = ^с,(г+1) (XIV, 47)
Иными словами, осмотическое давление рредставляет в этом случае давление
высокомолекулярного электролита.
При условии Ci с с2, т. е. когда в системе много низкомолекулярного элек-
тролита, прииеденное выше уравнение принимает вид:
n=>RTct (XIV, 48)
Это значит, что найденное осмотическое давление является давлением, обуслов-
ленным одними высокомолекулярными ионами.
474
При всех промежуточных соотношениях содержаний высокомолекулярного
и низкомолекулярного электролитов осмотическое давление будет меняться в
пределах от /?Tci(z-|- 1) до RTct.
Из изложенного совершенно ясно, что при исследовании осмотического дав-
ления растворов высокомолекулярных электролитов всегда необходимо учиты-
вать эффект Доннана. Практически для получения правильных результатов экс-
периментатор либо определяет концентрацию электролитов, находящихся в си-
стеме, и затем вводит в расчеты соответствующую поправку, либо измеряет
осмотическое давление в присутствии избытка низкомолекулярного электролита.
В последнем случае найденное осмотическое давление отвечает осмотическому
давлению одних высокомолекулярных иоиов.
Поскольку осмотические равновесие устанав-
ливается при неравномерном распределении ионов
по обе стороны мембраны, в системе должна
возникать разность электрических потенциалов
между жидкостью внутри и снаружи осмотиче-
ской ячейки (так называемый «мембранный по-
тенциал»). Эту разность потенциалов можно об-
наружить, вводя, например, во внутреннюю и
внешнюю жидкости осмометра одинаковые кало-
мельные электроды. Зная распределение электро-
литов в системе, по уравнению Нернста можно
вычислить разность потенциалов. Лёб показал, что
значения разности потенциалов, вычисленные и
найденные экспериментально, довольно хорошо
совпадают.
Теория мембранного равновесия Доннана по-
зволяет объяснить седлообразную форму кривой,
характеризующей зависимость осмотического дав-
ления раствора белка от pH среды. В самом деле,
в изоэлектрической точке амфолита число ионизи-
рованных ионогеиных групп минимально. Это и
обусловит минимальное давление. При добавле-
нии кислоты, например НС1, содержание анионов
(хлорид-ионов) в растворе сначала растет бы-
Рис. XIV, 15. Вычисленные
и найденные значения осмо-
тического давления подкис-
ленных растворов желатина:
1 — НС1, найдено; 2—НС1, вы-
числено; 3 — H2SOi, найдено;
4—НгЗОд, вычислено.
стрее, чем содержание ионов водорода (последние
связываются с гидроксилами гидратированных амино-групп белка, образуя
воду). Так будет продолжаться до тех пор, пока все гидроксильные группы
не свяжутся с ионами водорода и группы OHNH3— не перейдут в группы
CINH3—. В • этот момент осмотическое давление будет максимально, во-
первых, потому, что группы CINH3— ионизируют значительно сильнее, чем груп-
пы OHNH3—, и, во-вторых, потому, что в системе не содержится еще чужерод-
ного низкомолекулярного электролита (НС1), способного проникать через мем-
брану и, как уже было показано, понижающего осмотическое давление самого
высокомолекулярного электролита Прн дальнейшем добавлении кислоты число
хлорид-ионов вследствие подавления диссоциации молекул белка будет стано-
виться все меньше по сравнению с числом хлорид-ионов и водородных ионов,,
образовавшихся в результате диссоциации молекул кислоты Благодаря избытку
низкомолекулярного электролита, распределяющегося равномерно во внутренней
и внешней жидкостях осмометра, осмотическое давление будет обусловлено од-
ними высокомолекулярными ионами Кроме того, благодаря наличию в системе
избытка хлорид-ионов диссоциация группы CINH3 — уменьшается. Все это при-
ведет к тому, что осмотическое давление при добавлении избытка кислоты
будет падать
Следует заметить, что теория Доннана не только объясняет форму кривой,
характеризующей изменение осмотического давления при изменении pH, но и
позволяет предсказать, что при введении в систему кислоты с двухвалентным
анионом кривая должна лежать ниже кривой для кислоты с одновалентным
анионом. На рис XIV, 15 изображены подобные кривые, полученные Лёбом При
добавлении к раствору желатина НС1 и Нг5О«. Сплошными линиями показаны
475
экспериментально найденные кривые, пунктирными — теоретически вычисленные
(по Доннану). Некоторое расхождение экспериментальных и теоретических кри-
вых объясняется, очевидно, изменением степени ионизации групп C1NH3— Прй
добавлении кислоты.
Аналогичная картина наблюдается и при повышении pH: осмотическое дав-
ление раствора белка при добавлении щелочи сначала возрастает, а затем, д0.
стигнув максимума, начинает уменьшаться.
Теория Доннана удовлетворительно объясняет форму кривых, характеризую-
щих изменение осмотического давления не только от pH раствора, но и от
введения нейтральных электролитов. Однако рассмотрение этого случая выходит
за пределы настоящего курса.
Очень просто объясняется с позиций теории мембранного равновесия и из-
вестная зависимость объема набухшего белка (например, студня желатина) от
pH среды. Минимальная степень набухания студня должна соответствовать изо-
электрической точке белка, так как при этом минимально и осмотическое давле-
ние, являющееся причиной набухания. По обе стороны от этого минимума кривая
зависимости объема от pH поднимается и, достигнув максимума, спускается,
поскольку таким же образом от pH зависит и осмотическое давление. При трак-
товке набухания с точки зрения Доннана совершенно все равно, являются ли
макромолекулы белка кинетическими отдельностями или образуют трехмерную
сетку. Иначе говоря, безразлично, какими причинами удерживаются вместе
поливалентные ионы в системе — в результате ли наличия полупроницаемой пере-
городки или тем, что эти поливалентные ионы связаны друг с другом прочными
связями и образуют трехмерную сетку.
Аналогичным образом можно объяснить влияние нейтральных электролитов
и на набухание белков. В кислой и щелочной средах все нейтральные соли умень-
шают набухание белка, что вполне согласуется с теорией. Однако вблизи изо-
электрической точки соли могут как понижать, так и увеличивать набухание.
При этом решающую роль играют анионы, которые по влиянию на набухание
можно расположить в следующие ряды:
Анионы, увеличивающие набухание:
NCS" > Г > Вг” > NO- > С1О-
Анионы, уменьшающие набухание:
СГ < СН3СОО" < —ООС—СН2—С(ОН)(СОО-)—СН2—СОО" <
цитрат-иои
< “ООС—СН(ОН)—СОО- < SO42-
тартрат-иои
Различное влияние анионов на набухание объясняется тем, что они обла-
дают различным растворяющим или, наоборот, высаливающим действием на
белки. Особая роль аниона по сравнению с катионом объясняется тем, что
обратимый характер имеет лишь действие катионов щелочных металлов. Все
поливалентные катионы дают при взаимодействии с RCOO- (обычным анионом
высокомолекулярных электролитов) нерастворимые соединения, т. е. вызывают
образование необратимого осадка.
В заключение рассмотрим, как можно объяснить мембранным равновесием
влияние pH и нейтральных электролитов на вязкость белков. Седлообразный
характер кривой, характеризующей изменение вязкости раствора белка в зави-
симости от pH, объясняется способностью кинетических отдельностей макромо-
лекул, находящихся в растворе, изменять свой объем в результате набухания
или «отбухания». Изменение объема частиц в растворе должно сопровождаться
и соответствующим изменением вязкости. Лёб, впервые предложивший такое
объяснение, считал мицеллу кинетической отдельностью в растворе белка. Од-
нако в настоящее время, когда доказано, что высокомолекулярное вещество в
растворе раздроблено до молекул, следует говорить о набухании не мицелл, а
476
отдельных молекулярных клубков. Такой взгляд полностью согласуется с изло-
женными выше представлениями об изменении объема молекулярных клубков.
Исходя из аналогичных представлений, можно объяснить влияние на вяз-
кость раствора полиэлектролитов и нейтральных электролитов. Интересно, что
изменение вязкости в сильной степени зависит от валентности противоиона и
лишь незначительно зависит от его положения в лиотропном ряду. Такое расхо-
ждение в действии нейтральных электролитов на набухание студия и вязкость
раствора до известной степени понятно: в растворах взаимодействие между
отдельными макромолекулами ие играет существенной роли, и лиотропное дей-
ствие, приводящее к упрочнению или ослаблению связи между отдельными
макромолекулами, здесь не может сказаться в той степени, в какой оно сказы-
вается при набухании.
Эффект Доннана обусловливает распределение электролитов в тканях орга-
низма и является причиной возникновения биопотенциалов. Для лиофобных си-
стем, как мы указывали в гл. III, эффект Доннана также имеет большое значе-
ние. Здесь роль мембраны или геля играют сами коллоидные частицы, на кото-
рых адсорбированы недиффундирующие ионы, что приводит к неравномерному
распределению электролита в растворе. Особенно такое неравномерное распреде-
ление сказывается при центрифугировании золей (золь-концентрационный эф-
фект) или при оседании суспензии (суспензионный эффект Пальмана — Вигнера).
При ультрафильтрации доннановскин эффект может приводить к неравномер-
ному распределению электролитов в ультрафильтрате и в межмицеллярной
жидкости.
Электрические свойства растворов полиэлектролитов. Электро-
кинетический потенциал, как известно, с большей или меньшей
точностью может быть подсчитан по уравнениям Гельмгольца —
Смолуховского или Генри только для коллоидных частиц, размер
которых значительно превосходит толщину двойного электриче-
ского слоя. Для частиц же, диаметр которых мал по сравнению
с толщиной двойного электрического слоя, при расчете электроки-
нетического потенциала следует вводить ряд поправок и в первую
очередь поправку на электрическую релаксацию. Кроме того, если
макромолекулы находятся в растворе в виде рыхлого клубка, то
должно быть принято во внимание движение среды через петли
свернутой цепи. К сожалению, до сих пор теория электрофореза
для,свернутых в клубок макромолекул отсутствует. Поэтому в на-
стоящее время распространено определение электрофоретической
подвижности не отдельных макромолекул, а макромолекул, адсор-
бированных на достаточно крупных частицах кварца или угля или
на капельках масла. В этом случае электрокинетический потенциал
легко определить с помощью микроэлектрофоретических методов.
Как показали многочисленные исследования, при достаточной тол-
щине слдя полимера, покрывающего частицу, подобный прием
дает вполне воспроизводимые результаты.
Однако следует указать, что значения электрокинетического по-
тенциала, найденные для молекул высокомолекулярного электро-
лита в растворе, весьма условны. Надо помнить, что ионогенные
группы макромолекул, а следовательно, и заряды расположены
дискретно по всей длине макромолекулы, и о двойном электриче-
ском слое в обычном смысле слова здесь говорить трудно. Скорее,
уместно проводить аналогию с отдельными обычными ионами, ок-
руженными дебаевской ионной атмосферой. Поэтому найденные
477
значения электрокинетического потенциала для растворов высоко-
молекулярных электролитов только качественно характеризуют
электрические свойства макромолекул в растворе.
Применение полиэлектролитов
Полиэлектролиты широко применяются в народном хозяйстве.
Здесь мы только рассмотрим их применение в качестве флокулян-
тов суспензий с полярной (водной) средой и в качестве ионооб-
менных смол.
Полиэлектролиты как флокулянты. Наиболее эффективным»
флокулирующими агентами, как показал Ла Мер, являются поли-
электролиты с достаточно высоким молекулярным весом. При этом
полиэлектролиты следует применять в очень малых количествах,
гак как их избыток стабилизует суспензии.
Флокуляция может происходить за счет того, что длинные цеп»
молекулы полиэлектролита адсорбируются одним концом на одной
гастице суспензии, а другим — на другой частице, образуя между
гастицами достаточно прочнцй мостик. Конечно, практически об-
разуются флокулы, состоящие не из двух, а из большего числа
гастиц.
Другой механизм флокуляции заключается в образовании связи
лежду активными группами отдельных молекул полиэлектролита,
«угорые в свою очередь связаны с различными частицами суспен-
1ий. Такие флокулы, естественно, могут быть легко отделены от
юдной среды благодаря большому размеру.
Отделенные флокулы образуют рыхлый осадок, обладающий
гекоторой прочностью. Если раствор полиэлектролита, способный
«ызывать флокуляцию, вводить в суспензию порошка или волокна,
о полиэлектролит связывает частицы порошка или отдельные во-
юкна, в результате чего можно получать после высушивания связ-
ью систему, представляющую практический интерес. Наиболее
ффективно действие полиэлектролита совместно с обычными
лектролитами. При этом полиэлектролит следует вводить в сус-
ензию раньше низкомолекулярного электролита. В противном
лучае. агрегация частиц суспензии протекает плохо и осадок легко
ептизируется.
Весьма важное значение для флокуляции имеет строение мо-
екулы полиэлектролита и природа активных групп, а также спо-
обность молекул полиэлектролита пребывать в водной среде в бо-
ее или менее распрямленном состоянии. Молекулы полиэлектро-
ита, находящиеся в среде в виде свернутых клубков с малым ко-
ффициентом асимметрии, обладают плохими флокулирующими
войствами, так как часть звеньев цепи, экранируемая соседними
ктивными группами, образует внутримолекулярные связи, и, та-
им образом, эта часть цепи не может быть адсорбирована части-
ами суспензии.
В настоящее время в Советском Союзе и за рубежом в про-
мышленном масштабе выпускаются различные типы флокулянтов.
Одним из наиболее применяемых флокулянтов является частично
гидролизованный полиакриламид, который содержит группы —NH2
и —СООН (гидролиз, ведущий к образованию групп —СООН, сле-
дует проводить так, чтобы гидролизовалось всего Чз —NHa групп.
При большей степени гидролиза молекула полиакриламида приоб-
ретает настолько большой отрицательный заряд, что перестает
адсорбироваться на обычно отрицательно заряженных частицах
суспензий).
Полиэлектролиты в качестве флокулянтов прйменяются при
коагуляции оборотной воды в угольной промышленности, для из-
влечения золота из промывных и сточных вод в золотообрабаты-
вающей промышленности, что снижает потери золота на 99,9%;
в бумажной промышленности для удержания наполнителя в бу-
маге и снижения потерь волокна; для очистки сточных вод. Од-
нако, пожалуй, наиболее важно применение флокулянтов в сель-
ском хозяйстве для придания нужных свойств почве. Введение
в почву даже очень малых количеств флокулянтов (0,02—0,05%
от слоя почвы глубиной 15 см) уменьшает эрозию, структурирует
почву, что улучшает ее обрабатываемость, увеличивает влагоудер-
живающую способность и водопрочность почвы. Введенный в почву
полиэлектролит обычно сохраняет свое действие в течение 3 лет.
Особенно эффективно введение флокулянтов в мелкозернистые
глинистые почвы наших среднеазиатских республик. Поэтому цен-
тром синтеза и изучения применения новых флокулянтов является
Ташкентский государственный университет (школа академика
УзССР К. С. Ахмедова).
Полиэлектролиты как иониты. Ионитами, или ионообменными
материалами, называются нерастворимые вещества, способные об-
менивать содержащиеся в них ионы на другие ионы того же знака,
присутствующие в среде, с которой соприкасается ионит. В гл. VI
мы познакомились с типичным представителем ионитов — перму-
титом, применяющимся для извлечения из воды нежелательных
поливалентных катионов (Са2+, Mg2+ и т. д.).
В последнее время в качестве ионитов стали применять синте-
тические смолы, причем существуют смолы, способные обменивать
как катионы (катиониты), так и анионы (аниониты). Преимуще-
ство ионообменных смол перед ионитами других типов заклю-
чается в их высокой механической прочности, химической стойко-
сти и большой сорбционной (обменной) емкости. Обмен ионов
с помощью синтетических смол может происходить во всем объеме
смолы, так как растворенные ионы обычно свободно проникают
сквозь структурную решетку смолы.
Ионообменные смолы получают либо конденсацией или поли-
меризацией мономеров, уже содержащих активные группы, либо
вводят эти группы в уже готовые смолы. При увеличении числа
активных групп в ионообменной смоле возрастает ее обменная
479
способность, но одновременно возрастает способность набухать и>
растворяться в воде. С увеличением числа поперечных связей
в структуре смолы ее способность набухать и растворяться падает.
Размер зерен ионитов обычно лежит в пределах от 0,25 до
2,0 мм. Естественно, что чем меньше зерна ионита, тем быстрее
идет обмен, но, с другой стороны, тем больше возрастает гидро-
динамическое сопротивление слоя.
Рис. XIV, 16. Структура полисульфостирола (знаком +обозначены обме-
ниваемые катионы).
Обменная емкость ионитов обычно выражается числом грамм-
эквивалентов, извлекаемых с помощью 1 г сухого ионита. Обмен-
ная емкость зависит от характера содержащихся в ионите актив-
ных групп, от структуры ионита, а также От природы обмениваю-
щихся ионов, концентрации раствора и его pH. С увеличением pH
емкость катионитов увеличивается, а анионитов падает. С умень-
шением pH происходит обратное явление. Обменная емкость совре-
менных ионитов составляет 3—10 мг-экв/г. После насыщения
ионита обычно его регенерируют. Для этого катиониты обрабаты-
вают кислотой, а аниониты — растворами щелочей. После про-
мывки регенерированного ионита он снова становится пригодным
для использования,
480
Катиониты могут содержать в качестве активных групп —SO3H,
—СООН, —ОН (фенольные). Катионитами в сущности могут яв-
ляться уже обычные феноло-формальдегидные смолы. Однако ак-
тивная группа, содержащаяся в таких смолах (гидроксил), яв-
ляется слабой, поэтому, как правило, применяются катиониты, со-
держащие группу —SO3H или комбинацию групп —SO3H и —ОН.
На рис. XIV, 16 изображена в схематическом виде структура про-
стейшего катионита — полисульфостирола.
Аниониты могут содержать в качестве активной группы —NH2,
= NH и eN. Анионактивные смолы получили широкое распро-
странение лишь сравнительно недавно. Достижением в области,
анионактивных смол явился синтез сильноосновных анионитов.
С их помощью удается удалять из воды опасную для котлов сла-
бодиссоциирующую кремневую кислоту. Раньше это было невоз-
можно, так как известные в то время аниониты имели слабооснов-
ные свойства.
В последнее время были синтезированы синтетические смолы
селективного действия, избирательно сорбирующие отдельные
ионы, а также амфотерные иониты, пригодные для разделения
аминокислот и амфотерных элементов. Начинают также приме-
няться иониты с оптически активйыми группировками, с помощью
которых можно разделить оптические изомеры.
Благодаря большим достижениям в синтезе ионообменных смол
их стали применять далеко за пределами первоначальной области
их использования — в водоочистке. Иониты применяются всюду,
где требуется удаление, выделение и концентрирование ионов в
растворах. Иониты используются в энергетической, химической, пи-
щевой, фармацевтической, металлургической и в ряде других от-
раслей промышленности. Ионообменные смолы применяются для
разделения ионов, которые до настоящего времени не могли быть
разделены с помощью других методов. В частности, их применяют
для разделения редкоземельных элементов, продуктов распада ра-
диоактивных веществ и т. д. Широкое применение иониты находят
при изготовлении чистых реагентов.
9. СТУДНИ
Растворы высокомолекулярных веществ, равно как и лиозоли,
в известных условиях теряют свою текучесть, т. е. переходят
в студни. Застудневание может происходить спонтанно (самопро-
извольно), в результате изменения температуры, при концентриро-
вании раствора или при добавлении к нему не слишком больших
количеств электролита. Как правило, под действием этих факторов
структурная вязкость системы возрастает, что приводит к превра-
щению жидкости в студень — систему, проявляющую ряд свойств
твердого тела.
Студни и процесс застудневания имеют большое значение в ме-
дицине и биологии, так как из студней в основном состоит орга-
16 Зак. 664
481
низм животных и растений. Большое значение имеет застудневание
растворов высокомолекулярных веществ и в технике. Образование
клеевого слоя при склеивании, желатинирование пироксилина, по-
лучение искусственного волокна представляют собой не что иное,
как процессы застудневания. С застудневанием связан также ряд
процессов в пищевой и хлебопекарной промышленности. Студнем
является голье, лишенная волосяного покрова и специально под-
готовленная животная шкура, из которой получается кожа при
дублении.
Застудневание
Причина застудневания состоит в возникновении связей между
молекулами высокомолекулярного вещества, которые в растворе
представляли собою кинетические отдельности. Между молеку-
лами полимера в растворе могут образовываться кратковременные
связи, приводящие к возникновению ассоциатов. Однако если сред-
ний период существования связей между макромолекулами ста-
новится очень большим (практически бесконечным), то ассоциаты
не будут распадаться и возникшие образования проявляют в неко-
торой степени свойства твердой фазы. Постоянные связи между
молекулами в растворах высокомолекулярных веществ могут об-
разовываться в результате взаимодействия полярных групп мак-
ромолекул или ионизированных ионогенных групп, несущих элек-
трический заряд различного знака, и, наконец, между макромоле-
кулами могут возникать химические связи (например, при вулка-
низации каучука в растворе). Таким образом, застудневание есть
не что иное, Жак процесс появления и постепенного упрочнения
в застудневающей системе пространственной сетки. При этом для
застудневания растворов высокомблекулярных веществ характерно,
что связи образуются не по концам кинетических отдельностей,
как это происходит при переходе в гель лиозолей с удлиненными
жесткими частицами, а могут возникать между любыми участ-
ками гибких макромолекул, лишь бы на них имелись группы, ко-
торые могут взаимодействовать друг с другом.
Очень часто при образовании студня наблюдается упорядоче-
ние отдельных участков молекул, взаимодействующих друг с дру-
гом. Эти участки обычно ориентируются параллельно друг другу,
так как такая'ориентировка способствует уменьшению свободной
энергии системы. При этом степень ориентации зависит как от
природы высокомолекулярного вещества, так и от условий студне-
образования. На рис. XIV, 17 схематически показаны различные
типы ориентации участков макромолекул в студне, расположен-
ные в порядке увеличения их прочности.
Существует непрерывный переход от простейших точечных кон-
тактов макромолекул друг с другом к кристаллитам, т. е. обла-
стям с упорядоченной кристаллической структурой. Поскольку
в одном и том же студне контакты могут иметь самый различный
характер, целесообразно пользоваться понятием о спектре моле-
482
кулярных контактов для данного студня. Этот спектр может быть
представлен кривой распределения, характеризующей фракцион-
ный состав контактов либо по их прочности, либо по их размеру.
Весьма вероятно, что структурными единицами пространствен-
ной сетки студней могут являться не только отдельные молекулы,
но и их пачки. К сожалению, этот вопрос остается до сих пор со-
вершенно не исследованным.
Если образовавшиеся между макромолекулами связи не слиш-^
ком прочны, то механическое воздействие (перемешивание или
встряхивание) может разрушить структуру и студень (так же, как
и коллоидный гель) превращается в жидкость. При устранении
внешнего воздействия полученные растворы обычно снова само-
произвольно застудневают. Однако когда соединение молекул
Рнс. XIV, 17. Типы контактов макромолекул в студне (расположен-
ные в порядке увеличения их прочности).
в одну сплошную пространственную сетку обусловлено возникно-
вением химических связей, сильные механические воздействия вы-
зывают необратимое разрушение студня.
Повышение температуры, если только при этом в системе не
происходит необратимых химических изменений, обычно препят-
ствует застудневанию из-за возрастания интенсивности микроброу-
новского движения сегментов и уменьшения вследствие этого числа
и длительности существования связей, возникающих между макро-
молекулами. Наоборот, понижение температуры, как правило, спо-
собствует застудневанию, так как при этом спектр контактов ме-
жду макромолекулами расширяется и сдвигается в сторону боль-
шей прочности. Следует заметить, что переход раствора в студень,
равно как и студня в раствор, с изменением температуры совер-
шается непрерывно, т. е. в этом случае не существует температур,
подобных температурам кристаллизации или плавления.
Иногда нагревание способствует застудневанию и, наоборот,
охлаждение приводит к разжижению системы. Подобное явление,
наблюдающееся, например, для метилцеллюлозы в воде и нитрата
целлюлозы в спирте, связано с отрицательным температурным ко-
эффициентом растворимости высокомолекулярного вещества в дан-
ном растворителе.
Застудневанию растворов высокомолекулярных веществ всегда
способствует повышение концентрации, так как при этом возрас-
тает частота столкновений между макромолекулами или их участ-
16*
483
ками и увеличивается число связей, образующихся в единица
объема студня. Однако благодаря вытянутой форме макромоле-
кул застудневание может происходить и в очень разбавленных
растворах. Так, при обычной температуре раствор агара застуд-
невает уже рри содержании в нем 0,2% сухого вещества.
Чем выше концентрация, тем выше температура, при которой
растворы высокомолекулярных веществ переходят в студни. На-
пример, достаточно концентрированные (30—45%-ные) растворы
желатина способны застудневать уже при температуре около 30 °C,
более разбавленные (10%-ный) растворы переходят в студень при
температуре около 22 °C. Растворы агара застудневают при еще
более высоких температурах, и студни при этом получаются более
прочными, чем студни желатина. Наоборот, растворы каучука за-
студневают только при температурах, лежащих значительно ниже
нуля. Так, 3%-ный раствор натурального каучука переходит в сту-
день при температуре —41 °C. Плохое застудневание растворов
каучука объясняется, конечно, отсутствием в его молекулах поляр-
ных групп, способных, вступая друг с другом в контакт, образовы-
' вать достаточно прочную связь.
Объем системы при застудневании обычно уменьшается. Впро-
чем, при застудневании раствора кремневой кислоты наблюдается
увеличение объема системы. Это увеличение, протекающее парал-
лельно с полимеризацией кремневой кислоты, объясняется выделе-
нием воды, которая ранее была химически связана с SiOj.
В отдельных случаях застудневание сопровождается выделе-
нием тепла. Это может быть объяснено образованием кристалли-
тов и выделением теплоты кристаллизации. Однако при застудне-
вании растворов высокомолекулярных веществ кривые охлажде-
ния не претерпевают резкого скачка, так как процесс кристалли-
зации как бы размыт из-за неодновременного образования кристал-
литов.
На способность к застудневанию водных растворов амфотерных
высокомолекулярных электролитов, например белков, весьма
сильно влияет pH раствора. Застудневание лучше всего идет при
значении pH, отвечающем изоэлектрической точке, так как при
этом по всей длине молекулярной цепи расположено одинаковое
число противоположно заряженных ионизированных групп, что спо-
собствует установлению связи между отдельными макромолеку-
лами. С изменением pH (в обе стороны от изоэлектрической точки)
макромолекулы приобретают одноименный заряд, что препятствует
образованию между ними связи. При добавлении больших коли-
честв кислоты или щелочи степень ионизации ионогенных групп
уменьшается и тенденция к застудневанию снова увеличивается.
Короче говоря, 'способность к застудневанию у растворов белков
при изменении pH изменяется по седлообразной кривой, как и дру-
гие свойства.
Действие низкомолекулярных нейтральных электролитов на за-
студневание растворов белков прямо противоположно действию
484
этих электролитов на набухание. Ионы, увеличивающие набуха-
ние, замедляют застудневание или делают его вообще невозмож-
ным. Наоборот, ионы, уменьшающие объем набухшего студня, спо-
собствуют застудневанию. Как и на набухание, на застудневание
в основном влияют анионы.
Помимо образования связей между молекулами в известных
условиях могут возникать связи и между участками одной и той
же макромолекулы, если она имеет несколько групп, способных
взаимодействовать, и молекулярная цепочка настолько гибка, что
отдельные части ее в результате теплового движения могут всту-
пать в контакт. При этом образуются так называемые глобуляр-
ные или корпускулярные студни (В. А. Каргин и П. И. Зу-
бов).
Образование глобулярных студней, у которых каждая глобула
состоит только из одной макромолекулы, является предельным
случаем. Глобулярные студни могут образовываться в определен-
ных условиях и в результате ассоциации двух и более молекул
полимера.
Следует отметить, что глобулярные студни можно называть
студнями только условно, так как они способны течь. Благодаря
тому, что макромолекулы в этом случае свернуты в плотный клу-
бок и участки их не могут совершать микроброуновского движе-
ния, вязкость таких систем обычно даже меньше, чем растворов
соответствующей концентрации, в которых макромолекулы обра-
зуют рыхлые клубки.
Примером глобулярных студней могут служить системы, кото-
рые получаются из 0,1%-ных растворов нитрата целлюлозы в аце-
тоне при добавлении к ним воды. При выпаривании таких раство-
ров под вакуумом могут быть получены устойчивые студни, содер-
жащие до 2% сухого остатка. Эти системы обладают рядом кол-
лоидных свойств: обнаруживают способность к опалесценции, с по-
мощью ультрамикроскопа в них можно определить численную кон-
центрацию, при добавлении к ним электролитов они подчиняются
правилу Шульце — Гарди, при достаточном разбавлении системы
водой резко возрастает электрокинетический потенциал их частиц
и резко падает вязкость системы.
Другим примером глобулярных студней может служить рас-
твор желатина, приготовленный в строго определенных условиях.
При охлаждении достаточно концентрированного раствора жела-
тина ниже температурного интервала «плавления» студня в ре-
зультате образования межмолекулярных связей получается обыч-
ный, нетекучий студень. Однако если охлаждать сильно разбав-
ленный раствор желатина, в котором молекулы удалены друг от
друга и вследствие этого возникают, главным образом, внутримо-
лекулярные связи, то раствор остается текучим. Если осторожно
сконцентрировать такой раствор, не нагревая его выше темпера-
турного интервала «плавления» студня, то получается глобу-
лярный студень, остающийся текучим при тех концентрациях и
485
температурах, которые отвечают существованию обычного студня.
Если эту систему затем нагреть, то в результате увеличения интен-
сивности микроброуновского движения связи, возникшие между
отдельными участками молекулы, разрушаются и глобулярный
студень переходит в обычный вязкий раствор желатина, который
при охлаждении дает уже нормальный нетекучий студень. Таким
образом, переход глобулярного студня в обычный является обра-
тимым процессом.
Аналогичные системы, вероятно, образуются при охлаждении
раствора желатина при интенсивном перемешивании. Бунгенберг
де Йонг еще в 1932 г. показал, что в этих условиях образуется не
студень, а раствор, содержащий мельчайшие частицы желйтина,
способные медленно седиментировать. Возможно, что эти частицы
являются не чем иным, как свернутыми в плотный клубок молеку-
лами желатина.
Глобулярные студни могут существовать весьма долго, т. е. оци
являются практически устойчивыми системами. Таким образом,
возможно существование и неравновесных растворов высокомоле-
кулярных веществ. Последнее объясняет тот известный факт, что
различно приготовленные растворы высокомолекулярных соеди-
нений одной и той же концентрации часто обладают разными
свойствами — вязкостью, осмотическим давлением и т. д.
Из всего сказанного о глобулярных студнях ясно, что оии по
своим свойствам являются промежуточными системами между
растворами полимеров и коллоидными растворами.
Свойства студией
Наиболее интересными особенностями обычных студней яв-
ляются их механические свойства, в частности эластичность.
Растворы высокомолекулярных веществ, в которых межмоле-
кулярные связи чрезвычайно непрочны, способны течь, т. е. реаги-
руют даже на очень слабые сдвиговые усилия. Однако если время
существования хотя бы части контактов между макромолекулами
становится очень большим, образовавшийся студень уже способен
противостоять течению вплоть до какого-то определенного значе-
ния напряжения сдвига и ведет себя при сдвиговых усилиях ниже
этого критического значения, как эластическое твердое тело. Зна-
чение критического напряжения сдвига зависит от числа и проч-
ности молекулярных контактов.
На механические свойства студней сильно влияет их концен-
трация. Студни, содержащие в единице объема малое число по-
стоянных межмолекулярных связей, обычно весьма эластичны,
Наоборот, студни с большим числом связей между макромолекул
лами сравнительно мало эластичны, так как чем больше свя-
зей между цепями полимера, тем меньше возможность изме-
нения формы макромолекулы, тем более жестка образовавшаяся
сетка.
486
Ниже приведены значения критического напряжения сдвига
раствора различных концентраций типичного высокомолекуляр-
ного вещества — агара:
Концентрация агара, %...... 0,05 0,06 0,08 0,10 0,20 0,40
Критическое напряжение сдвига,
гс/см2 .................. 0,004 0,036 0,32 1,23 27 525
Можно видеть, что критические напряжения весьма быстро уве-
личиваются с концентрацией, и уже при очень малом содержании
агара в растворе система проявляет свойства студня, т. е. система
нетекуча.
При температуре выше тех температур, при которых происхо-
дит «плавление» студня, ни разбавленные, ни концентрированные
студни не обнаруживают критического напряжения сдвига. Неза-
студневающие растворы, например растворы нитрата целлюлозы
в ацетоне, не обнаруживают критического напряжения сдвига ни
при каких концентрациях. Они текут при самых малых напряже-
ниях сдвига, хотя это течение вследствие высокой вязкости раство-
ров и происходит весьма медленно.
Релаксационные процессы в студнях обычно протекают с боль-
шей скоростью, чем в растворах полимеров, так как в студнях мо-
гут перемещаться только отдельные, сравнительно короткие от-
резки цепей. Поэтому студни обладают всегда более ярко выра-
женной эластичностью, чем структурированные растворы.
Некоторые студни полимеров обладают явно выраженными
тиксотропными свойствами. У таких студней прочность связей ме-
жду макромолекулами должна быть достаточно малая, чтобы они
могли легко разрушаться под действием приложенного усилия
сдвига. Кроме того, у подобных студней должен быть достаточно
узкий спектр молекулярных контактов. Студни, у которых этот
спектр размыт, обычно не проявляют тиксотропии. В самом деле,
когда связность структуры нарушается путем механического воз-
действия, при узком спектре молекулярных контактов большинство
связей разрушается и затем восстанавливается при стоянии си-
стемы. Это и составляет сущность явления тиксотропии. Если же
спектр контактов широкий, разрушается только небольшое-число
связей, обладающих наименьшей прочностью. Система распадается
на большие куски, которые не могут соединиться и образовать
структуру с первоначальным значением критического напряжения
сдвига.
Поскольку макромолекулярные системы обычно обладают ши-
рокими спектрами молекулярных контактов, то тиксотропия про-
является только при особых условиях, способствующих сужению
спектра, например, при значительном разбавлении или в началь-
ной стадии студнеобразования, когда не наступило еще образова-
ния структур из ориентированных участков макромолекул.
К своеобразным свойствам студней следует отнести их «память»
к тому,, как они были получены. Например, если высушить при
487
низкой температуре до одного и того же содержания влаги два
студня, из которых один получен из разбавленного, а другой из
концентрированного раствора желатина, и затем дать этим под-
сушенным студням снова набухнуть в воде, то первый студень на-
бухает гораздо больше, чем второй. Причина этого заключается
в том, что при высушивании до известной степени в студнях сохра-
няется внутренняя структура, возникшая при их образовании.
Во многих случаях студни обладают большой способностью
рассеивать свет. Это можно объяснить образованием в студне кри-
сталлитов или межмолекулярных контактов с большой площадью.
Наличие в студнях высокомолекулярных веществ кристаллитов
можно также доказать тем, что рентгенограммы таких студней ука-
зывают на присутствие в них кристаллов.
Интересные особенности наблюдаются при диффузии в студни
низкомолекулярных веществ и частиц коллоидной степени дис-
персности. Так как не слишком концентрированные студни пред-
ставляют собой трехмерные сетки со сравнительно большими пет-
лями, диффузия в них малых молекул или ионов идет практически
с той же скоростью, что и в чистой среде, если только диффунди-
рующее вещество не способно взаимодействовать с высокомолеку-
лярным веществом студня. Диффузией в студень, а не в жидкость
иногда пользуются при определении коэффициента диффузии для
того, чтобы исключить влияние конвективных потоков или сотря-
сений.
Низкодисперсные системы не обладают способностью диффун-
дировать в студни, так как частицы не могут проникнуть в петли
молекулярной сетки, если размер их больше размера петель.
В этом случае складываются такие же условия, как и при ультра-
фильтрации. Сходство еще более увеличивается благодаря тому,
что полупроницаемые мембраны обычно являются типичными
студнями.
Высокодисперсные коллоидные системы, а также растворы вы-
сокомолекулярных веществ с неслишком большими молекулами
диффундируют в студни с различной скоростью, зависящей от раз-
мера диффундирующих кинетических отдельностей и частоты мо-
лекулярной сетки студня.
Диффузия в студни имеет место при крашении и дублении, так
как набухшее волокно и голье являются типичными студнями.
Весьма своеобразные явления наблюдаются, если в студень,
содержащий какое-нибудь низкомолекулярное вещество, диффун-
дирует другое вещество, способное с низкомолекулярным веще-
ством образовывать нерастворимое соединение. В этом случае, как
установил Лизеганг еще в прошлом столетии, реакция осаждения
часто идет только в определенных зонах системы, чередующихся с
зонами, в которых осадка не образуется вовсе (образование «рит-
мических осадков»).В результате получаются слои,если диффузия
идет в глубь студня, или кольца, если диффузия начинается в центре
поверхности студня и продолжается параллельно последней.
488
Роль самого студня при образовании таких «ритмических осад-
ков», очевидно, сводится к предупреждению образования в системе
конвективных потоков. Это подтверждается тем, что «ритмиче-
ские осадки» могут возникать и при диффузии низкомолекуляр-
ного вещества в порошки, пропитанные раствором другого низко-
молекулярного вещества, способного давать осадок с первым.
Несмотря на множество работ, посвященных изучению слоев и
колец Лизеганга, причина их образования еще не совсем ясна.
Следует отметить, что слои и кольца Лизенганга очень часто встре-
чаются в природе. Так, полосатость или слоистость описанного
выше характера можно наблюдать у некоторых минералов (агаты),
у мускульной ткани (поперечно-полосатые мышцы).
Электропроводность студней близка к электропроводности рас-
творов, из которых эти студни были получены, несмотря на то, что
вязкость системы резко возрастает при застудневании. Это объяс-
няется тем, что трехмерная сетка, образующаяся в студне, не ме-
шает движению сравнительно малых ионов в растворителе; иначе
говоря, «микровязкость» системы при ее застудневании остается
неизменной.
Специфичным свойством некоторых студней является возмож-
ность вытеснения жидкости, находящейся в петлях молекулярной
сетки, какой-нибудь другой жидкостью. Если вторая жидкость сме-
шивается с первой, то процесс можно осуществить простым погру-
жением студня во вторую жидкость. Если жидкости не смеши-
ваются, то необходимо использовать какую-нибудь третью жид-
кость, смешивающуюся с обеими. Так, если нужно заменить воду
в водном студне углеводородом, то сначала воду вытесняют спир-
том, который затем можно вытеснить смешивающимся с ним угле-
водородом.
Примечательно, что студень, в котором жидкая среда заменена
чужеродной жидкостью, часто обладает повышенной жесткостью и
не претерпевает усадки при высушивании. Молекулярная сетка та-
кого модифицированного студня как бы теряет эластичность и
способность сокращаться при удалении из цее среды. Кроме того,
в таких студнях, как показал Германе, даже после их тщательного
высушивания остается некоторое количество жидкости, что, ве-
роятно, обусловлено чисто стерическими причинами. Ниже опре-
деленной степени набухания молекулярные цепи в студне весьма
плотно упакованы и молекулы среды остаются как бы окклюдиро-
ванными между ними. Продвигаться между тесно сблизившимися
макромолекулами могут только молекулы жидкости, растворяю-
щейся в высокомолекулярном соединении. Все вышеуказанное в не-
которой степени объясняет тот общеизвестный факт, что остатки
(следы) жидкостей, находящихся в пленках высокомолекулярных
веществ, удаляются из них с большим трудом.
Большинство студней, встречающихся в растительном и живот-
ном мире, обладают анизотропией, обусловленной условиями об-
разования этих студней. Причиной анизотропии искусственно по-
489
лученных студней обычно является неравномерная деформация их
при образовании или неравномерная усадка при высушивании.
Примером может служить студень, полученный высушиванием
рцствора желатина на стеклянной пластинке. Очевидно, усадка
в таких условиях может происходить только по толщине пленки
студня, в результате чего макромолекулы желатина будут ориен-
тированы преимущественно параллельно плоскости пластинки.
Естественно, что у анизотропных студней такие свойства, как из-
менение линейных размеров образца студня при набухании или
усадке, преломление и поглощение света, двулучепреломление,
а также механические свойства разные в различных направлениях.
В заключение необходимо указать, что студни высокомолеку-
лярных веществ при малом содержании в них растворителя очень
близки по свойствам к самим высокомолекулярным веществам. Ко-
нечно, высокомолекулярное вещество, не содержащее раствори-
теля, не является студнем или гелем, но оно может быть получено
из студня путем высушивания и легко превращено в студень в ре-
зультате набухания. Поэтому высокомолекулярные вещества, спо-
собные набухать, часто рассматривают как предельное состояние
геля при удалении из него растворителя и называют ксерогеля-
ми, т. е. «сухими» гелями (ксёрос — по-гречески сухой). Высоко-
молекулярные вещества, не способные к набуханию, обычно назы-
вают смолами.
В качестве примеров ксерогелей можно назвать сухой желатин,
полистирол, каучук, целлюлозное волокно, примером смол яв-
ляется бакелит.
Синерезис
Свежеприготовленные студни высокомолекулярных веществ при
стоянии часто претерпевают синерезис: их объем уменьшается и
среда спонтанно выпрессовывается из эластичного студня. В ре-
зультате образуются две макрофазы — жидкая и студнеобразная,
причем последняя сохраняет форму сосуда, в котором находился
первоначальный студень. Синерезис очень распространенное явле-
ние и может наблюдаться не только в студнях с сеткой из макро-
молекул, но, как мы видели в гл. X, и в лиогелях, сетка которых
состоит из кристаллических частиц. Многие явления синерезиса
детально исследованы С. М. Липатовым.
Общей причиной синерезиса является то, что при образовании
студня не достигается еще равновесия системы и в ней продол-
жаются процессы, связанные с его установлением. У лиогелей эти
процессы могут сводиться к рекристаллизации. У высокомолеку-
лярных студней эти процессы сводятся к установлению связей ме-
жду макромолекулами, что способствует дальнейшему уменьшению
объема студня. Синерезис может являться также следствием до-
стижения равновесия в студне при его охлаждении, если высоко-
молекулярное вещество не полностью растворимо в среде. Нако-
нец, в некоторых случаях непосредственной причиной синерезиса
490
являются химические процессы, протекающие в веществе студня.
Например, водные студни ксантогената целлюлозы претерпевают
синерезис в результате разложения ксантогената.
Синерезис 'свойствен не только водным студням (например,
студню желатина и крахмала), но и студням, где средой являются
органические жидкости (например, каучуковому студню).
Предел, к которому стремится объем студня при синерезисе,
зависит от концентрации студня. Синерезис обычно тем больше,
чем выше концентрация растворителя в исходном студне. Опре-
деленной зависимости скорости синерезиса от концентрации исход-
ного студня нет. Например, при высоких концентрациях синерезис
каучукового студня ускоряется, а студня крахмала и агара замед-
ляется. Предельным объемом студня при синерезисе, по С. М. Ли-
патову, является сумма объемов самих макромолекул и объема
растворителя, сольватно связанного с высокомолекулярным веще-
ством. Незначительное повышение температуры, как правило, спо,-
собствует синерезису, облегчая перемещение молекул, необходимое
для усадки студня. Однако при значительном повышении темпера-
туры может произойти переход студня в раствор. Внешнее давле-
ние на студень, конечно, всегда способствует синерезису.
Для студней амфотерных белков максимальный синерезис на-
блюдается в изоэлектрической точке, так как в таком состоянии
молекулы несут равное число разноименных зарядов, что способ-
ствует сжатию молекулярной сетки студня. С изменением pH
среды (относительно изоэлектрической точки) синерезис умень-
шается, так как молекулярные^цепочки приобретают одноименный
заряд, обусловливающий их распрямление и отталкивание друг от
Друга.
Влияние на синерезис студня низкомолекулярных электролитов
весьма сложно. Однако, как правило, электролиты, способствую-
щие набуханию, уменьшают синерезис и наоборот.
Найдено, что синерезис зависит от размеров образца студня, и
причем синерезис тем меньше, чем больше образец. Это вполне
понятно, так как жидкость, которая должна быть выпрессована
при синерезисе из образцов студня большего размера, должна пре-
одолеть большее сопротивление.
Явления синерезиса имеют очень большое значение в биологии,
медицине и технике. Его можно рассматривать как положительное
явление при получении некоторых синтетических смол, поскольку
при этом происходит самопроизвольное отделение смолы от рас-
творителя. Положительное значение имеет синерезис и в молочной
промышленности, так как на нем основано получение творога.
В крахмало-паточной, мармеладной и некоторых других отраслях
пищевой промышленности это явление имеет отрицательное зна-
чение, и усилия технолога направлены на его предупреждение.
В производстве взрывчатых веществ синерезис (например, выделе-
ние из студня, которым является бездымный порох, самовзрываю-
щегося нитроглицерина) чрезвычайно опасен.
491
РЕКОМЕНДУЕМАЯ ЛИТЕРАТУРА ПО КОЛЛОИДНОЙ ХИМИИ
А. УЧЕБНИКИ И РУКОВОДСТВА
Ребиндер П. А. Конспект общего курса коллоидной химии. Изд. 2-е. М.,
Изд-во МГУ, 1950. 112 с.
Кройт Г. Р. Наука о коллоидах. Пер. с англ., под ред. В. П. Мишина. М.,
Издатинлит, 1955. 538 с.
Шелудко А. Коллоидная химия. Пер. с болг., под ред. Б. В. Дерягина и
С. С. Воюцкого. М., Издатинлит, 1960. 332 с.
Пасынский А. Г. Коллоидная химия. Изд. 2-е. М„ «Высшая школа», 1963.
Успехи коллоидной химии. (Сборник) Под ред. П, А. Ребиндера и Г. И. Фукса,
М., «Наука», 1973. 267 с.
Фридрихсберг Д. А. Курс коллоидной химии. Л., «Химия». 1974. 352 с.
Б. СПЕЦИАЛЬНЫЕ МОНОГРАФИИ И ВАЖНЕЙШИЕ ОБЗОРНЫЕ СТАТЬИ
К ГЛАВЕ I
Ребиндер П. А. Новые проблемы коллоидной химии. Вестник АН СССР,
1955, т. 2, с. 8.
Воюцкий С. С., Ребиндер П. А. О предмете и принципах построения курса
коллоидной химии. Коллоидн. ж., 1956, т. 18, с. 253.
Ребиндер П. А. Развитие коллоидной химии р СССР за 40 лет. Усп. хим.,
1957, т. 26, с. 320.
К ГЛАВЕ II
Шифрин К. С. Рассеяние света в мутной среде. М. — Л., Гостехтеоретиздат,
1951. 288 с.
Лукьянович В. М. Электронная микроскопия в физико-химических исследо-
ваниях. М., Изд-во АН СССР, 1960. 273 с.
К ГЛАВЕ III
Ф и г у р о в с к и й'Н. А. Седиментометрический анализ. М., Изд-во АН СССР,
1948. 415 с.
К ГЛАВАМ IV—VI
Де Бур, Я. Динамический характер адсорбции. Пер. с англ., под ред.
В. М. Грязнова. М., Издатинлит, 1962, 290 с.
Курс физической химии. Т. I. Под ред Я. И. Герасимова. М., «Химия», 1970.
592 с. См. с. 412—557.
Липатов Ю. С., Сергеева Л. М. Адсорбция полимеров. Киев, «Наукова
думка», 1972. 233 с.
К ГЛАВЕ VII
Электрические свойства капиллярных систем. (Сборник) Под ред. П. А. Ре-
биндера. М. — Л., Изд-во АН СССР, 1956. 352 с.
Электроповерхностные явления в дисперсных системах. (Сборник) Под ред.
О. Н. Григорова и Д. А. Фридрихсберга. М., «Наука», 1972. 192 с.
Григоров О. Н. Электрокинетические явления. Изд-во ЛГУ, 1973. 168 с.
К ГЛАВЕ VIII
Берестнева 3. Я., Каргин В. А. О механизме образования коллоидных
частиц. Усп. хим'., 1955, т. 24, с. 249.
Ребиндер П. А. Современные проблемы коллоидной химии. Коллоидн ж.
1958, т. 20, с. 527.
Ребиндер П. А. и др. О термодинамически равновесных двухфазных дисперс-
ных системах. Коллоидн. ж., 1970, т. 32, с. 480.
К ГЛАВЕ IX
Дерягин Б. В. Современная теория устойчивости лиофобных суспензий и
золей. Труды 3-й Всесоюзной конференции по коллоидной химии, М., Изд-во
АН СССР, 1956, с. 235.
492
В о ю цк н й С. С., П а н н ч Р. М. Агрегативная устойчивость дисперсий полиме-
ров и дзета-потенциал. Усп. хим., 1956, т. 25, с. 157. -
Дерягин Б. В., А б р и к о с о в а И. И., Л и ф ш и ц Е. И. Молекулярное при-
тяжение конденсированных тел. Усп. физ. наук, 1958, т. 64, с. 493.
Зонтаг Г., Штренге К Коагуляция и устойчивость дисперсных систем. Пер.
с нем., под ред. О. Г. Усьярова. Л., «Химия», 1973. 152 с.
Исследования в области поверхностных снл. (Сборник) Под ред. Б. В. Деря-
гина. В 5 т. Т. 1—5. М., «Наука», 1961—1974.
К ГЛАВЕ X
Воларовнч М. П. Исследование реологических свойств дисперсных систем.
Коллоидн. ж., 1954, т. 16, с. 227.
Михайлов Н. В., Ребнндер П. А. О структурно-механических свойствах
дисперсных н высокомолекулярных систем. Коллондн. ж, 1955, т. 17,
с. 107.
Структурообразованне в дисперсных системах в присутствии полнэлектролитов.
(Сборник). Под ред. К. С. Ахмедова. Ташкент, Изд-во ФАН Узбекской ССР,
1970. 174 с.
Ефремов И. Ф. Периодические коллоидные структуры. Л, «Химия», 1971.
Исследование по физико-химни контактных взаимодействий. (Сборник) Под ред.
Г. И. Фукса. Уфа, Башкирское книжное нзд-во, 1971. 228 с.
К ГЛАВЕ XI
Амелин А. Г. Теоретические основы образования тумана при конденсации
пара. Изд. 3-е. М., «Химия», 1972. 304 с.
Фукс Н. А. Механика аэрозолей. М„ Изд-во АН СССР, 1955. 352 с.
Дерягнн Б. В. Аэрозоли (дымы н туманы). М., «Знание», 1961. 32 с.
Фукс Н. А. Успехи механики аэрозолей. М., Изд-во АН СССР, 1961. 159 с.
К ГЛАВЕ хп
Клейтон В. Эмульсин. Пер. с англ., под ред. П. А. Ребиндера. М., Издат-
ннлит, 1950. 680 с.
Чухров Ф. В. Коллоиды в земной коре. М., Изд-во АН СССР, 1955. 671 с.
Воюцкнй С. С. О причинах агрегативной устойчивости эмульсий. Усп. хим.,
1961, т. 30, с. 1237.
Шерман Ф. Эмульсин. Пер. с англ., под ред. А. А. Абрамзона. Л., «Химия»,
1972. 448 с.
К ГЛАВЕ XIII
Виноградов Г. В. Мыла, растворы н гели мыл. Усп. хнм., 1961, т. 20.
Шварц А., Перри Дж., Берч Дж. Поверхностно-активные вещества и
моющие средства. Пер. с англ., под ред. А. Б. Таубмана. М, Издатинлит,
1960. 555 с.
Ребнндер П. А. Поверхностно-активные вещества и нх применение. Хим.
наука н пром., 1959, т. 5, с. 554.
Ш т ю п е л ь Г. Сннтетнческне моющие и очищающие средства. Пер. с ием., под
ред. А. И. Гершеновича. М., Госхимнздат, 1960. 672 с.
Шенфельд. Неионогенные моющие средства. Пер. с ием., под ред. А. И. Гер-
шеновнча. М, «Химия», 1965. 487 с.
Ш н н о д а К. Коллоидные поверхностно-активные вещества. Пер. с англ., под
ред. А. Б. Таубмана и 3. Н. Маркиной. М., «Мир», 1966. 320 с.
К ГЛАВЕ XIV
Воюцкнй С. С. Растворы высокомолекулярных соединений. Изд. 2-е. М., Гос-
хнмиздат, 1960. 131 с.
Тагер А. А. Физнко-хнмня полимеров. Изд. 2-е. М„ «Хнмня», 1968. 536 с.
Моравец Г. Макромолекулы в растворе. Пер. с англ., под ред. В. А. Кар-
гина и И. А. Туторского. М., «Мнр», 1967. 398 с.
493
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Абрамсон, прибор для электрофореза
211 сл.
Абсорбция света 39—42
и светорассеяние 40
фиктивная 40 сл.
Аеогадро, число 64 сл., 72 сл.
Агар
застудневание раствора 484
как полиэлектролит 468
набухание, функции 447
раствор, критическое напряжение
сдвига 48/
Агрегат(ы) (мицеллы) 243 сл.
молекул поверхностио-активиых ве-
ществ 405
Агрегативная
неустойчивость 11, 18 сл.
— лиофобных систем 260
устойчивость аэрозолей 347 сл.
— золей 282
— коллоидов 259 сл.
— латексов 383 сл.
— лиофобных систем 260 сл.
— растворов полимеров 465 сл.
— суспензий 367
— эмульсий 371 сл.
флуидизация 353
Агрегатное состояние дисперсной фазы
и дисперсной среды 24 сл.
Агрегация
в* лиозолях 68
дальняя 279
частиц при коагуляции 262, 268
число 405
Адгезия 167 сл.
Адсорбат 81
Адсорбеит(ы)" 81, 109 сл.
активность динамическая 112
— статическая 112
аморфные 149
кислотные 149
неполярные 139, 141
иепористые 109
основные 149
поверхность удельная 99, 135
полярные 139, 141
пористость 139
пористые 109
свойства 109, 139
характеристическая кривая 95
--------коэффициент афинности 96
Адсорбтив 81
моиомолекуляриый слой 90
свойства, влияние иа адсорбцию 111 сл.
Адсорбционные
азеотропия 143
колонка 144
объем 93 сл.
понижение твердости 233
потенциал 94 сл., 187, 189,
— общий 187
— электрический 187
равновесие 107, 142
силы 85 сл., 89
— потенциал 86
слой 128 сл., 185
— высоковязкий 392
— заряд 187
— мицеллы 244
ориентация молекул 129, 141
— ПАВ 410 сл
— полимолекуляриый 284 сл
— стабилизующее действие 283 сл.
— строение 97, 128 сл.
— Штерна 198
теория коагуляции 289
494
Адсорбционные
теория кристаллизации 226
Адсорбция 81 сл.
активированная 103
ван-дер-ваальсова 81
влияние иа коагуляцию 296
ПАВ 126 сл
— пористости адсорбента 139 сл.
в статических условиях 112
газов из смеси 112 сл.
— иа твердом теле 88
иа угле 111
гидролитическая 153
динамическая 112
зависимость от времени 141 сл.
— от давления 83
• — от концентрации раствора 141 сл.
«— от природы адсорбента 146 сл.
— от растворителя 138 сл.
— от свойств адсорбента 109 сл.
— —. — адсорбтива 111 сл.
— от температуры 83, 141 сл.
и водородная связь 87 сл.
и достройка кристаллов 147 сл,
избирательная 172
изопикны 83
изостеры 83
изотермы 83 сл., 91 сл., 96, 98, 123,
142 сл.
из растворов, молекулярная 137 сл.
---в природе и технике 143 сл.
из смеси 137
ионная 146 сл»
кинетические кривые 107 сл.
кислорода иа угле 104
количественная характеристика 83
кристаллами 147 сл.
молекулярная 137 сл.
— влияние адсорбента и адсорбтива
139 сл.
— — времени 141 сл.
— — концентрации раствора 141 сл.
---среды 138 сл.
— — температуры 141 сл.
— из растворов 137 ел.
моиомолекуляриая 88 сл.
на границе раствор — газ 114 сл«
— — твердое тело — газ 81 сл.
— — твердое тело — раствор 137 сл,
иа угле 111, 151 сл.
иа флоридине 139
неэлектролитов 137
обменная 148 сл.
отрицательная 83, 118, 123
— изотерма 142 сл.
полимолекуляриая 93 сл.
положительная 117, 123, 143
полярная 146
предел 84, 92
работа, максимальная 127
роль в гетерогенном катализе 105
скорость 107 сл , Г42
— влияние температуры 108
— температурный коэффициент 108 сл,
ступенчатая 92
теория БЭТ 93, 97 сл.
. — Ленгмюра 88 сл.
— потенциальная, Поляни 93 сл.
теплота 98, 105 сл.
физическая 81 сл., 103
— локализованная 82, 90
— иелокализоваииая 82
— скорость 107
химическая 81, 103 си
электролитов 137, 146 сл
Азеотропия адсорбционная 143
Активность
адсорбента 112
капиллярная 125
поверхностная 120» 123, 125, 126
Активные центры 89 сл , 92
Активный уголь 109 сл.
Алкозоли 25 '
Алмаз, строение 421
Альбумин
защитное действие на золи 305
растворение в воде, термодинамиче-
ские функции 440
Алюминия гидроокись, золь
как промежуточная система 422
кристаллизация частиц 229
перезарядка 195
порог коагуляции 294
Алюмогель 109
Алюмосиликагель 109
Алюмосиликаты как двухмерные высоко-
молекулярные вещества 421
Амикроны 24
Аморфное состояние полимеров 432
Анализ
дисперсионный 77 сл.
седиментационный 73, 76
Анизодиаметричиость 47
частиц, влияние иа вязкость 336 сл»
Анизотропия студней 489 сл.
Аиионактивные смолы 479
Аниониты 479, 481
Антагонизм
ионов 300, 302
электролитов 301 сл.
Антонов, правило 116
Ассоциаты 436 сл.
образование и осмотическое давление
455 сл.
период жизни 437
Ассоциация макромолекул 436 сл.
Аустеинт как истинный раствор 397
Ацетат целлюлозы
раствор, диаграмма состояния 435
теплота набухания 450
Аэрогель 350
Аэродинамическая труба 351
Аэрозоли 25, 28, 340 сл.
атмосферные 345, 362
взрывы 365
в сельском хозяйстве 352, 365
днспергациоииые 341
дисперсность и свойства 349 сл.
заряд частиц 345 сл.
инерционное осаждение частиц 360 сл.
классификация 340 сл.
коагуляция 348 сл.
конденсационные 341
концентрация 342
получение 356 сл.
— диспергированием 359
— иоидеисацией 356 сл.
практическое значение 364 сл.
природные 364
размер и форма частиц 341 сл.
разрушение 344, 349, 360 сл,
— под действием электрического поля
363 сл
— ультразвуком 361 сл.
свойства молекулярио-книетическне
342 сл.
— оптические 342
— электрические 345 сл,
седиментация 344
устойчивость агрегативная 347 сл.
» фильтрация 360 сл.
Аэросилы 109
Бакелит 490
Баллоэлектрнздция 346
Банкрофт, правило 373
Бедекер — Фрейндлих, уравнение 84 сл«
Белки 14, 418, 469
высаливание 466
вязкость 476
денатурация 32
изоэлектрическая точка 469
исследование методом Тизелиуса 209-
как защитные коллоиды 304
— пенообразователи 386
— полиэлектролиты 468 сл.
набухание 476
разделение 466
раствор, застудневание 484 сл.
— осмотическое давление 476
электрофорез 470
Бензозоли 25
Берлинская лазурь, золь
окраска 43
получение 227
Бехгольд, ультрафильтры 258
Бингам, уравнение 329
Биопотенциалы и эффект Доннана 41?
«Боковая жидкость» 207 сл.
Больцман
закон распределения 178
уравнение 178, 440
Брединг, получение гидрозолей электро*
распылением 253
Брикетироваине порошков 355
Броуновское движение 55—58
молекулярно-кинетическая природа 54
теории 65
частиц аэрозолей 343 сл.
Брунауэр, Эммет, Теллер
теория адсорбции 97
уравнение изотермы адсорбции 98
Бугер — Ламберт, закон 39
Бугер — Ламберт — Бэр, закон 39
Бутилкаучуки 420
Бьеррум, осмотический коэффициент 408
Вальден, правило 465
Ванадия пятиокись, золь
гелеобразование 314
кристаллизация частиц 229
Ван-дер-Ваальс, уравнение 130
В ант-Гофф
закон 453 сл
уравнение 67, 454 сл
Вейлер — Ребиндер, прибор 334
Векторная диаграмма Ми 34 сл.
Весы
Ленгмюра 132 Ъл.
Мак-Вена 108
седиментационные 76
Вещество, фазовые состояния 431
Взвеси 366
Вибрационное нзмельченне 248, 252
Внбромельиицы 252
Вигнер, седиментометр 76 сл.
Вильсон, камера 358
Внскознметр ротационный 326
Вода
модели состояния 406
растворение неполярных веществ 406
«Водоотталкивающие» вещества 163
Водоочистка 32
Водородная связь и адсорбция 87 сл.
Волокно целлюлозное как ксерогель 490
Время
жизни капли эмульсин 371 сл.
— пей 386 сл
— пузырька пеиы 387
половинной коагуляции 265
релаксации 205, 332
существования эмульсий 371 сл.
495
Вулканизация 419
Высаливание 466
Высаливающее действие ионов 466
Высокомолекулярные вещества (соедине-
ния) 14, 417 сл., см. также Поли-
меры
аморфное состояние 432
ассоциация 436 сл.
вымачивание 424
дисперсии 438
защитное действие иа золи 304 сл.
как эмульгаторы 373, 376
кривые распределения фракций 423
кристаллические 432
— растворение 445
молекулярный вес 417
-------кажущийся (эквивалентный) 451,
453
—— определение 455—457, 462
набухание 442 сл.
— кинетические кривые 447 сл.
— ограниченное 445 сл.
— поверхностное 444
— скорость 448
— степень 447
— теплота 499 сл.
— термодинамические функции 447
неорганические 421
Очистка 424
пептизирующее действие 255
подвижность молекул 212
полидисперсиость 423
— показатель (коэффициент) 426
природные 418 сл.
разделение смеси 209 сл.
размер молекул 417
растворение 438 сл.
— ограниченное 445 сл.
— самопроизвольное 26, 438
— термодинамика 438 сл.
растворы 416 сл.
— ассоциаты 455
— высаливание 466
— вязкость 459 сл., 467 сл-
и молекулярный вес 461
— диффузионная способность 456
— застудневание 437, 482 сл.
— и правило фаз Гиббса 435 сл.
— и уравнение Больцмана 440
— коацервация 437
— конформации молекул 440
— критическое напряжение сдвига
486 сл.
— осмотическое давление 453 сл., 458
— равновесность и обратимость 435
— свойства оптические 451 сл.
реологические 463
— седиментация 458
— синерезис 437
—---старение 467
— теория мицеллярная 432 сл.
---молекулярная 432, 433 сл,
— устойчивость агрегативная 465 сл.
--- седиментационная 457
синтетические 419 сл.
фазовые состояния 431 сл.
фракционирование высаливанием 466
— методом Тизелиуса 209
— осаждением 424
— растворением 423 сл.
В ысокомолекуляриые
пенообразователи 388
соединения см. Высокомолекулярные
веществз и Полимеры
электролиты см. Полиэлектролиты
Вязкость
жидкостей 323 сл.
— в двойном электрическом слое
201 сл.
- определение 325 сл,
496
Вязкость
— структурированных 328 сл.
зависимость от концентрации днсперс*
ной фазы 335 сл., 337 сл.
— от сольватации 337 сл.
— от температуры 325
золя 327 сл.
и аиизодиаметричиость частиц 336
и взаимодействие частиц 338 сл.
и определение молекулярного веса
460 сл.
изменение при введении электролитов
338
кажущаяся 324, 328
коэффициент 323
определение методом истечения 326
-----падающего шарика 325 сл.
— относительное 326
— ротационным вискозиметром 326
относительная, зависимость от pH 471
пластическая 329
приведенная 460
растворов 323 сл.
— полимеров 459 сл., 462 сл., 467 сл,
— самопроизвольное изменение 467
структурная 328» 330
увеличение относительное 336
удельная 336
— зависимость от концентрации 459
— и электрокинетический потенциал 339
уравнение Фикеичера и Марка 338
уравнение Эйнштейна 335 сл.
характеристическая 460 сл.
эффективная 324, 326, 326, 330
— зависимость от напряжения сдвига
330
* — *— от скорости течения 326
Газ(ы) 55
адсорбция из смесей 112 сл.
двухмерный 128 сл.
— аналогия с трехмерным газом 129
— уравнение состояния 130
очистка от пыли 363
реальный, уравнение Ваи-дер-Ваальса
130
Газоочистка 32, 363
Гамакер, константа 271
Гартли, мицеллы 407
Гелеобразование 28, 315
влияние концентрации дисперсной фа-
зы 316
и форма частиц 316
Гель 28, 315, 445
образование, влияние механических
воздействий 317
-----температуры 316 сл.
переход в золь 399
Гельмгольц, двойной электрический слой
(гельмгольцевскнй слой) 185
Гельмгольц — Перрен
двойной электрический слой, строение
175 сл., 198
теория 175 сл.
Гельмгольц — Смолуховский, уравнение 200
Гемоглобин, защитное действие иа золи
305
Генри, уравнение 91, 203 сл.
Гетероадагуляцня 307
Гетерокоагуляцня 307 сл.
Гиббс 123, 127
Г иббс
правило фаз 436
уравнение 120 сл.
эффект 390
Гидратация
значение для устойчивости пен 393
ионов, зависимость от радиуса 186
Гидратация
— и сжатие двойного электрического
слоя 186
Гидрозоли 25
коагулирующее действие неэлектроли-
тов 307
получение методом Брединга 253
электропроводность, влияние напряже-
ния и частоты электрического поля
221 сл.
Гидрофилизация поверхности 163
Гидрофильно-липофильный баланс (ГЛБ)
373, 403
Гидрофильность, поверхностная и объем-
ная 160
Гидрофобизация поверхности 163
Гидрофобизироваииая ткань, схема иесма-
чивания водой 164
Гидрофобное взаимодействие 406
Гидрофобность, поверхностная и объемная
160
Гистерезис 100
при капиллярной конденсации 100 сл«
смачивания 159
Гитторф
метод определения скорости электро*
фореза 207
о числах переноса в капиллярах мемб-
ран 256
Глаукониты 150
Глина, суспензия
электрокинетический потенциал частиц
186 сл.
Гомогенизаторы 377 сл.
Гомокоагуляция 307 сл.
Горные породы как коллоидные системы
типа Т/Т 395, 396 сл.
Гофмейстер, ряды 147
Градиент концентрации при диффузии 59
Граница (плоскость) скольжения 176, 178,
185, 189 сл., 243
Гранулирование 354 сл.
Гранулы 354, 355
Графит, Строение 421
Грозовые явления 345
Гуи
двойной электрический слой 185
диффузный слой 198
Гуи — Чэпмен, теория 176 сл.
Гук, уравнение 331
Гуммиарабик
защитное действие на аоли 305
как полиэлектролит 468
Гуммигут
дисперсия 245
суспензия, концентрация, снижение с
высотой 71
Гюккелъ, уравнение 203
Давление
внутреннее 114
осмотическое см. Осмотическое давле-
ине
поверхностное 132 сл.
расклинивающее см. Расклнннвающее
давление
электроосмотическое 216
Движение
броуновское 55 сл.
микроброуновское 285, 429
тепловое 55 сл.
Двойной электрический слой 146
деформация 197
Гельмгольца 185
Гельмгольца — Перрена 176, 198
диффузная часть 177
емкость 183, 188
и электрофорез 171
Двойной электрический слой
образование 172 сл.
падение потенциала 175, 177 сл., 189
поведение при электрических явлениях
198 сл.
распределение ионов 186
сжатие 179
— н гидратация ионов 186
строение 174 сл., 198, 201
— схема 175, 185
— схема по Ефремову 190
— теория 184 с л.
— — Гельмгольца — Перрена 175 сл.
— — Гуи — Чэпмена 176 сл.
— — Штерна 184 сл.
— у частиц сферических 190
— с постоянным диполем 190 сл.
структура, изменение с концентрацией
188
толщина 176, 179, 183
— зависимость от валентности ионов
186
— — от поляризуемости ионов 186
Дебаевский радиус 183
Дебай — Шеррер, диаграммы 53 сл.
Дезагрегация 68
Декстрин, защитное действие на золи 305
Денатурация белков 32
Дерягин
теория гетерокоагуляции 307 сл.
— коагуляции, физическая 290
о расклинивающем давлении 21
о термодинамическом факторе устой-
чивости 392
Десорбция 82
работа 281
при хемосорбции 103
Деформация
зависимость от времени 333
кажущаяся пластическая 333
необратимая 333
сдвига, предельная обратимая 332
Дефекты кристаллической решетки 232 сл.
Деэмульгирование 379
Диаграммы Дебая — Шеррера 53 сл.
Диализ 10, 255 сл.
очистка высокомолекулярных веществ
424
Диализатор 255
Диафрагмы см. Мембраны
Дилатансия 318
Диспергирование 232 сл., 248 сл.
механизм 232 СЛ1.
механическое 248, 252
получение аэрозолей 359 сл.
— эмульсии 3/7
практическое значение 232
самопроизвольное 223, 236 сл., 378 сл.
— и образование эмульсии 368
с помощью пьезоэлектрических осцил-
ляторов 251
— — ультразвуковых колебаний 248, 251
Дисперсии 12
высокомолекулярных веществ 438
газа в жидкости 386
Дисперсионная среда 13
агрегатное состояние 24
влияние на электрокинетический по-
тенциал 194 сл.
Дисперсная фаза 13
агрегатное состояние 24 сл.
---------и скорость электрофореза 206
концентрация эффективная объемная
337
определение концентрации 52
теория образования 225 сл.
Дисперсность 15
катализаторов и их активность 23
мера 15
пигментов 350
497
Дисперсные системы 16
двухфазные, термодинамическая устой-
чивость 238 сл.
концентрация, снижение с высотой 71
кривые распределения 75
поверхность удельная 16 сл.
свободнодисперсные 27 сл.
свойства, влияние дисперсности 22 сл.
— реологические 313 сл.
— структурно механические 313 сл.
--------- определение 334 сл.
связнодисперсные 27 сл.
стабилизация 284
явления, наблюдаемые при падении
светового луча 33 сл.
«Днсперсоидология» 23
Диссипация энергии 323
Дифильиость поверхностно-активных ве-
ществ 117 сл , 126
Дифильные молекулы, ориентация при
адсорбции 141
Диффузионный поток 59
Диффузиофорез 197, 219
Диффузия 58 сл.
в коллоидных растворах 10
в растворах высокомолекулярных ве-
ществ 456
в студне 488
градиент коицеитрацнн 59
и дисперсность 22
и набухание высокомолекулярных ве-
ществ 443
и скорость роста кристаллов 226
растворенных веществ 56
скорость, эффективная 60
удельный поток 59
энергия активации 56
Диффузный слой
Гун 198
заряд 187 сл.
мицеллы 243
Диэлектрическая проницаемость
жидкости в двойном электрическом
слое 201 сл
и заряд коллоидной частнцы 171
изменение с концентрацией 202
лиозолей 222
Дождевание искусственное 360
Доннан, мембранное равновесие 68, 258,
472 сл,
Дорн
потенциал 199
эффект 170
Драгоценные (н полудрагоценные) камни,
окраска 43
Дымовые завесы 342
Дымы 24, 340
плотность частиц 342
разрушение 363
топочные 356
Дырочная теория жидкостей 55 сл.
Дюкло
строение коллоидных частиц 241
химическая теория коагуляции 289
Дюкло — Траубе
правило 126 сл.
— обращение 139 сл.
Дюпре, уравнение 168
Емкость
двойного электрического слоя 183, 188
молекулярного конденсатора 188
Желатин
вязкость, относительная 471
защитное действие на золи 305
Желатин
как ксерогель 490
— стабилизатор для «обрыва» коагу*
ляции 262
период релаксации 332
раствор, застудневание 484
— как глобулярный студень 485
— модуль сдвига 332
— осмотическое давление 472, 475
Желатинирование 315
Железа гидрат окисн
золь, получение 248, 254 сл.
—• стабилизаторы 248
— строение мицеллы 248
— устойчивость и коагуляция 298
раствор как промежуточная система
422
Железа окись, золь
порог коагуляции 294
электрокннетический потенциал, кри-
тический 289
Жндкость(н) 55
* аномальное 328
вязкость в двойном электрическом
слое 201 сл.
диэлектрическая проницаемость в двой-
ном электрическом слое 201 сл.
межмицелляриая 243 сл , 258
нормальное 328
перенос электроосмотический 196 сл.
поверхностное натяжение 115 сл.
распыление как метод получения аэро-
золей 359 сл.
структура 55 сл.
структурированные 313 сл.
ь- вязкость 328 сл.
---эффективная, зависимость от на-
пряжения сдвига 330
— кривые течения 329 сл.
— определение упруго-пластииеских
свойств 336
течение, вязкое 56, 314
— механизм 324 сл.
— стационарное в капилляре 213
тонкие слои (прослойки) 20 сл.» 269 сл.
влияние иа прочность структур
316
---расклинивающее давление 21, 272
----устойчивость 269 сл
Жирные кислоты как пенообразователя
386
Закон
Больцмана 178
Бугера — Ламберта 39
Бугера — Ламберта — Бэра 39 сл.
Вант-Гоффа 453 сл
гипсометрический 70
Рауля 451 сл.
Томсона 116 сл., 225
Фика 59
Зародыши и образование новой фазы 357
Заряд электрический
объемная плотность 181
поверхностный 175
частнцы 171, 242
— аэрозоля 345 сл.
Застудневание 315, 437, 481 сл.
зависимость от концентрации 483 сл,
— от температуры 483
растворов полимеров 437, 482 сл
спонтанное (самопроизвольное) 481
Защитные коллоиды 304
Зидентопф н Зигмонди, ультрамикроскоп
44
Зигмонди и Фрейндлих, классификация
коллоидных систем 26 сл.
Зигмонди, синтез гидрозоля золота 246
498
Золь(и) 9, 24, 315
вязкость 327
— удельная 339
гидроокисей металлов 27, 248
действие электролитов 300 сл.
защищенные 304
воны устойчивости и неустойчивости
301
концентрированные, устойчивые 295
лиофильные 27
лиофобные, устойчивость 296
.металлов, абсорбция света 41 сл.
— окраска 43
— опалесценция 38 сл.
— уравнение Светопоглощеиия 42
неметаллические (белые), окраска 43
неустойчивость 259
окраска 43 сл.
опалесценция 37 сл.
оптические свойства 33
переход в гель 317
получение методом диспергирования
248 сл.
— — конденсации 245 сл.
— электрораспылеиием 252 сл.
привыкание к электролитам 303 сл.
с неводной средой, коагуляция элект-
ролитами 306 сл.
твердые 24, 395
течение 326 сл.
устойчивость агрегативная 282
— зоны 301
— понижение устойчивости 308
частицы, укрупнение 259
электронные микрофотографии 230
электронограммы 236
Золота золь
абсорбция света 41
коагуляция 267 сл.
концентрация, снижение с высотой 71
кристаллизация частиц 229
окраска 43
получение 246 сл.
порог коагуляции 294
размер частиц 16
среднеквадратичное смещение частиц
64 /
стабилизаторы 247
строение 247
флуктуации числа частиц 66
«Золотое число» 304 сл.
Золь-концеитрационный эффект 477
Измельчение
вибрационное 248, 252
как метод получения аэрозолей 359
ультразвуковым колебанием 248
Изобары адсорбцнн 83
окиси углерода и а палладии 104
Изодиаметричиость 47
Изомеры поворотные 429
Изопикны адсорбции 83
Изостеры адсорбции 83
Изотермы
адсорбции 83, 84, 123, 142 сл.
— в логарифмических координатах 85
— гистерезис 102
— и характеристическая кривая 98
— отрицательной 142 сл.
— пзров (по теории БЭТ) 98
— полимолекулярной 93
— положительной 143
— построение по изотерме поверхност-
ного натяжении 123 сл.
— уравнение 91
Ленгмюра, уравнение 91 сл.
поверхностного давления 132 сл«
Изотермы
— иатяжеиия 119
-------и изотерма адсорбции 123
-------коллоидных поверхностно-актив-
ных веществ 400 сл. '
Изоэлектрическая точка
белков 469
золя иодида серебра 246
и коагуляция 288
Изоэлектрическое состояние 191
Изумруд 43
Иммобилизация растворителя 338
Индулии, теплота набухания 450
Ионизация и образование двойного элект-
рического слоя 172
Иониты 479
амфотерные 481
обменная емкость 480
регенерация 480
Иоииая атмосфера
радиус 133
толщина 179 сл.
«Иоииое отложение» 218
Ионный обмен 191 сл.
Ионообменные смолы 151
получение 479
применение 481
селективное действие 481
Ионы
антагонизм 300, 302
гидратация 186
— и сжатие двойного электрического
слоя 186
гидратированные, радиусы 146
лиотропный ряд по коагулирующему
действию 288
объемный обмен 148
потенциалопределяющие 171, 174, 198.
243
— концентрация 177
синергизм 300
Истечение, объемная скорость 324
Кавитация 251
Казеин
набухание, теплота 450
— термодинамические функции 447
Казеинат натрия, 'вязкость раствора 464
Какао, дисперсность 350
Камера Вильсона 358
Камии драгоценные и полудрагоценные
как коллоидные системы типа Т/Т
395
Канифоль
гидрозоль 245
период релаксации 332
Каолин
дисперсность 350
пептизация 255
Капиллярная
активность 125
конденсация 93, 99 сл.
— в порах различной формы 101 сл.
— гистерезис 100 сл.
«сверхпроводимость» 215
Капиллярное впитывание 443
Капля
давление пара и радиус 357 сл.
давление насыщенного пара 358
поверхностное натяжение 358
Капрон 420
Касторовое масло, период релаксации 332
Катализ гетерогенный, роль адсорбции 105
Катализаторы, активность, зависимость от
дисперсности 23
Катафорез 169
Катиониты 479, 481
499
Каучук(и) 14
вулканизованный 426
вязкость растворов, аномалии 459
дисперсия 310
застудневание раствора 484
защитное действие иа золь 304
как защитные коллоиды 304
— ксерогель 490
модуль сдвига 332
натуральный 419 •
— набухание 449
растворение в толуоле, термодинами-
ческие функции 440
синтетические 420
Квазикристалличиость 55
Кварца суспензия, скорость оседания ча-
стиц 77
Кен
правило 171
прибор для электрофореза 206 сл.
Кеннингем; уравнение 343
Кератин, набухание, термодинамические
функции 447
Кинетические кривые
адсорбции 107 сл.
устойчивости эмульсий 371 сл.
«Кипящий слой» 353
Клейнманн, нефелометр 51
Коагели 28
Коагулирующее действие
ионов 288
неэлектролитов 307
Коагулюм 11, 315
Коагулят 11, 315
гидратов окисей металлов 312
механическая прочность 280
Коагуляторы 11
Коагуляция 11, 13, 19, 32, 191, 259 слм
279 сл.
аэрозолей 348 сл.
безэлектролнтная 308
быстрая 260 сл.
влияние адсорбции 296 сл.
во вторичном минимуме 279
время половинной коагуляции 265
групповая 308
дальняя 279
изучение кинетики 261 сл.
истинная 279, 315
коллоидов, взаимная 307
концентрационная 290, 292
латексов 383, 385
медленная 261
— уравнение 266
начало 287 сл.
нейтрализационная 290 сл.
«обрыв» 261 сл.
под действием ультразвуковых коле-
баний 251
— — физических факторов 308
-------электрического поля 310
полидисперсных систем 266
-------теория Мюллера 266
порог 287, 293 сл.
при замораживании 308, 311 сл.
— охлаждении 308, 311 сл.
— концентрировании 308, 310 сл.
— механических воздействий 308 сл.
— нагревании 308, 311 сл.
— разбавлении 308, 310 сл. ,
скорость 262 сл.
—* и численная концентрация 263
спонтанная 308
суспензий 367
теория 289 сл.
— адсорбционная 289
— Дерягина 290
<— Дюкло 289
— Мюллера 289
— Смолуховского 261 сл.
Коагуляция
— физическая 289 сл.
-------приложение к органозолям 306
— Фрейндлиха 289
— Фукса 266
— химическая 269
электролитами 286 сл.
— антагонизм и синергизм 301 сл,
—• золей с неводной средой 306 сл.
— и явления неправильных рядов
300 сл.
•— по Мюллеру 289
— по Фрейндлиху 289
— теория 289 сл.
-------физическая, Дерягина 290
-- электростатическая 289
Коалесценция 19, 371, 467
Коацерваты 467
Коацервация 437, 467
Колларгол 31, 306
Коллоидная
защита 304 сл.
растворимость 412
химия, определение 13
Коллоидное состояние вещества 12
Коллоидно-химические методы исследова-
ния 9
Коллоидные поверхностно-активные ве-
щества 399 сл.
анионные 401 сл.
катионные 402 сл.
классификация 400 сл,
неиоиогеиные 403 сл.
растворы, практическое значение 414
Коллоидные растворы
агрегативная неустойчивость 11
диффузия частиц 10
коагуляция, роль адсорбции 296 сл.
осмотическое давление 10
особенности 10
размер частиц 10
электропроводность 219
Коллоидные системы
бесцветные, опалесценция 37
вязкость 335 сл.
газообразные 11
гетерогенность 18 сл.
гидрофильные 26
гидрофобные 26
дисперсность 15, 17
жидкие 11
защита 304 сл.
значение в природе н технике 28 сл,
классификация 23—28
Зигмонди и Фрейндлиха 26 сл,
— по агрегатному состоянию 24 сл.
— по взаимодействию дисперсной фазы
и дисперсионной среды 25 сл,
( — *---между частицами 27 сл.
по дисперсности 24
коагуляция взаимная 307
лиофильные 26 сл., 236 сл., 260
лиофобные 26, 237, 260
механизм образования 230 сл.
необратимые 25 сл.
неустойчивость 259
— агрегативная 18 сл.
обладающие истинной упругости 333
обратимые 25 сл.
окраска 43 сл.
определение численной концентрации 47
оптические методы исследования
44 сл.
осмотическое давление 66 сл.
отличие от истинных растворов 12
очистка 223 сл., 255 сл.
поверхность удельная 15—18
получение 223 сл., 239 сл.
— ‘ диспергационйым методом 223,
232 сл., 248 сл.
500
Коллоидные системы
получение диспергационным методом,
термодинамические условия 239
— конденсационным методом 223 сл.
вз--------термодинамические условия 240
— методом пептизации 234 сл.
— > — термодинамические условия 240
— путем аамены растворителя 245
® — химических реакций 245 сл.
— самопроизвольным диспергированием,
условия 240
понятие 9 сл.
привыкание к электролитам 303 сл«
примеры 11 сл.
свободнодисперсное состояние (золь)
315
свойства, молекулярно-кинетические
55 сл.
— - механические 331 сл.
— оптические 33 сл.
еэ фнзико-мехаНическне 334
«— электрические 169 сл., 219 сл.
связнодисперсное состояние (гель) 315
спонтанное образование 15
старение 230
структура, возникновение 315 сл.
твердые 11, 13
течение вязкое 327
устойчивость агрегативная 231 сл.,
259 сл.
— и коагуляция 259 сл.
— седиментацноиная 68 сл,
« факторы 281 сл.
Коллоидные частнцы
адсорбция поверхностно-активных ве-
ществ 298
аморфные 228 сл.
— продолжительность существования
229 сл.
анизоднаметричность 47
взаимодействие н вязкость 338 сл,
— потенциальные кривые 293
диаметр эквивалентный 16
заряд 171, 242
защита 304 сл.
нзодиаметрнчность 47
направление и скорость движения
57 ел.
объем средний, определение 47
перезарядка 189» 192, 194 сл«
радиус, определение 77
— эквивалентный 47, 74
м эффективный 77
размеры 15, 16, 44, 77
— определение 46 сл., 52, 68, 74 сл., 77
распределение по высоте 70 сл.
с дипольной структурой 190
скорость седиментации 74
смещение 68
— среднеквадратичное значение 58, 63 сл,
схема передвижения при электрофорезе
173
тепловое движение 57
укрупнение 259
форма 47
— и гелеобразование 316
электрофоретическая подвижность 304
Коллоиды 9, 12 сл. см. также Коллоидные
системы
защитные 304
молекулярные 416
обратимые 416
Кольматацня суспензий 367
Кольца (слои) Лизеганга 488 сл.
Кометы 29
Конденсация
капиллярная 93, 99 сл.
— в порах различной формы 101 сл*
получение аэрозолей 356
Конденсатор темного поля 46
Кондуктометрическое титрование, измере*
ние электропроводности гидрозолей-
220 сл.
Константа
Гамакера 271
седиментации 79
Смолуховского 263, 267
уравнения Фрейндлиха 85
Контракция при набухании 449
Конус Тиндаля 10
Конформации 429
Концентрация
мццелообразовання, критическая 400,
404 сл. _
— определение 408 сл,
частичная 35
численная 35
— определение 47 сл., 262, 267
Копаловый лак, период релаксации 332
Коэффициент (ы)
абсорбции, фиктивной 41
афинности 96
вязкости 323, 462
— растворов средней концентрации 462=
днффузнн 59, 61
— высокомолекулярных веществ 456
« методы определения 62, 456
замедления 267
заполнения пространства порошкам»
352, 353 сл.
осмотический, Бьеррума 408
поглощения, молярный 40
поли дисперсности высокомолекулярных
веществ 426 трения 60
Краевой угол 153 сл.
и поверхностное натяжение 155 сл,
определение 158
равновесный 155
Красители
как коллоидные поверхностно-активные
вещества 415
растворы 399
Краскотерки 249 сл.
Крахмал 418
дисперсность 350
ващитное действие на золи 305
как полнэлектролнт 468
Крафт» точка 406 с л.
Кремневая кислота
гель 322
застудневание раствора 484
золь, кристаллизация частиц 230
как промежуточная система 422
Кремния гидроокись как промежуточная
система 420
Кремния двуокись, гидрозоль
двойной электрический слой на части-
цах 172 сл.
строение частиц 244
Кривая (ые)
взаимодействия частиц 295
зависимости эффективной вязкости от
напряжения сдвига 330
потенциальные взаимодействия мицелл
278 сл.
— энергии взаимодействия коллоидных
частиц 293 распределения, нормаль-
ные 238
— фракций высокомолекулярных ве-
ществ 423
« — частиц в аэрозоле 341
седиментации 75
течения 329 сл.
характеристическая 95
— коэффициент афниности 96
Кристаллизация
при адсорбции 147
теория, адсорбционная 226
центры (зародыши) 223 сл.
Кристаллоиды 12
501
Кристаллы
адсорбции иоиов 147 сл.
достройка при адсорбции 147 сл
поверхностное натяжение 116 сл
скорость роста» зависимость от диффу-
зии 226
Критерий Рейнольдса 324
Критическая концентрация мицеллообразо»
вания (ККМ) 400, 404 сл.
определение 408 сл.
Ксерогели 321? 490
набухание 321
Ламинарное течение 323 сл.
переход в турбулентное 324, 327
Ламм, метод определения коэффициента
диффузии 456
Лаплас, оператор 181
Латекс (ы) 14 сл., 381 сл., 419
коагуляция 383, 385
натуральный 381
образование плеиок 218
применение правила Шульце—Гарди
384
синтетические 31, 232, 381 сл.
стабилизация поверхиостио-активными
веществами 385
мылами 384
.устойчивость 382 сл.
— агрегативная 383 сл.
электрокинетический потенциал 382 сл,
Ленгмюр
весы 132 сл.
теория моиомолекуляриой адсорбции
88 сл.
уравнение 91 сл., 124 сл.
— изотермы адсорбции 91 сл.
Леннард — Джонс, уравнение 86
Лизеганг, слои (кольца) 488 сл.
Л иогели 26
образование 315
строение пространственных структур
316
Лнозоли 25, 26
астабилизации 316
диэлектрическая проницаемость 222
ионностабилизоваииые, потенциальные
кривые 278 сл.
осмотическое давление 67 сл.
получение методом конденсации 245 сл<
---пептизации 254 сл.
электропроводность 219 сл.
Лиотропный ряд 147
иоиов по высаливающему действию 466
— по коагулирующему действию 288
Лиофильные системы 26 сл , 236 сл , 260
Лиофобные системы 26, 237, 260
устойчивость агрегативная 260 сл.
«=е и индуцированная сольватация ча-
стиц 282
Литопон (пигмент), дисперсность 350
Лифшиц, теория притяжения тел 271
Лондон, уравнение 269
Лучепреломление двойное в потоке 44
Магния окись, дисперсность порошка 350
Мак-Бен
весы 108
мицеллы 408
опыты по проверке уравнения Гиббса
123
-Макромолекулы
ассоциация 436 сл.
гибкость, влияние растворителя и пла-
стификатора 431
— и химическая природа 430 сл*
Макромолекулы
линейные, гибкость 427
ориентация в студие 482 сл.
пачки (пучки) 432
строение 426 сл.
термодинамическая вероятность состоя**
иия 440
форма, влияние индифферентного
электролита 470
в растворах 461
электрофоретическая подвижность 477
Максвелл, теория упруго-вязкого тела к
уравнение 332
Мартенсит как коллоидная система 397
Масляные краски как тиксотропные си-
стемы 317
Мастика, дисперсия 245
Медь, золь 245
Межзвездная материя 29
Межмицелляриая жидкость, получение
ультрафильтрацией 258
Межфазная граница (поверхность) 237
ааряд 183
потенциал 181 сл.
Мел, дисперсность 350
Мельницы коллоидные 249 сл.
Мембраны
активные и неактивные электрохими-
чески 257 сл.
для диализа 256
заряд при электродиализе 257
полупроницаемые 488
Менделеев, о роли примесей в коллоид*
иых растворах 10 сл.
Менделеев — Клапейрон, уравнение 130
Метастабильиое состояние 225
Метод
Вейлера и Ребиндера 334
Гитторфа 207
закручивание цилиндра (определение
вязкости) 336
истечение жидкости через капилляр
(определение вязкости) 326
падающего шарика (определение вяз-
кости) 325 сл.
подвижной границы (определение fc-
потенциада) 207 сл.
Тизелиуса 208 сл.
Ми. векторная диаграмма 34 сл.
Микроброуновское движение 285, 429
Микрон 24
Микроскоп 44 сл.
разрешающая способность 44, 49
электронный 48 сл.
Микроскопия электронная 44, 48 сл.
Микрофотографии электронные 50, 229
Микрощели, схема развития 233
Микроэлектрофорез 211
прибор Абрамсона 211 сл.
Минералы
гидрофильность 158
как коллоидные системы типа Т/Т 395
Миозин, гель 317
Мицелла (ы) 172, 399
Гартли 407
изменение структуры при введении
электролита 244 сл.
Мак-Бена 408
мыла 409
пластинчатые 407, 413
потенциальные кривые взаимодействия
278 сл
строение 240 сл., 407 сл.
— физические, или адсорбционныеа
теории 241 сл.
₽- химические гипотезы 240 сл.
энергия взаимодействия 278 сл.
Мицеллообразоваиие 400, 405 сл., 410
теплота 406
Многофазная система 12
«502
Модуль сдвига 331 сл.
Молекулярное притяжение 269 сл,
Молекулярные снта 111
Молекулярный вес
высокомолекулярных веществ 424
--------кажущийся (эквивалентный) 451,
453
-------- средний 425
определение 424 сл , 455 сл.
— вискозиметрическое 462
— диффузионным методом 456
— оптическим методом 457 сл.
осмотическим методом 68, 455
— по коэффициенту диффузии 61 сл.
— по мутности раствора 457
fe- по седиментационному равновесию
457
по скорости седиментации 457
— прн седиментации 79 сл.
растворенного вещества, определение
453
Моль основной 459
Моющее действие
мыл 414 сл.
поверхностно-активных веществ 414 сл,
Моющие вещества 163
Мука, дисперсность 350
Мутность
раствора 457
— и определение молекулярного веса
457
эмульсий 37
Мыл А
аммониевые 401
и стирка 162
как анионные поверхностно-активные
вещества 401
— пенообразователи 386
смачиватели 162
калиевые 401
кристаллы 401
мицеллы, образование 409
моющее действие 414
натриевые 401
растворы 399
— концентрационная зависимость осмо-
тического коэффициента 408 сл.
*— — *-* эквивалентной электропровод-
ности 408 сл.
С двух- и трехвалентными катионами
401
стабилизация латексов 384
стабилизирующее действие 373 сл.
Мылонафт 402
Мышьяка сульфид, золь
кондуктометрическое титрование 220 сл«
окраска 43
порог коагуляции 287, 294
строение частиц 241
устойчивость 260
— и коагуляция, влияние ПАВ 298
электрокинетический потенциал, крити-
ческий 289
Мюллер
теория коагуляции полндисперсиых си-
стем 266
« — электростатическая 289
Набухание
влияние анионов 476
— pH 471
в природе н технике 32, 443
высокомолекулярных веществ 442 сл
— термодинамические функции 447
давление 448
— расклинивающее 276
кинетические кривые 447 сл,
контракция 449
Набухаине
ксерогеля 321
иеограиичеииое 448
ограниченное 445 сл.
— влияние температуры 451
скорость 448
степень 447
теплота 449 сл.
Найлон 420
Наполнители 23, 350
Напряжение сдвига и эффективная вяз-
кость 330
предельное 336
Насыпная плотность 352
Натрия олеат
кристалл, строение 401
мицеллы, солюбилизация бензола 413-
Натяжение поверхностное см. Поверхност-
ное натяжение
Нафтеновых кислот соли, моющее дейст-
вие 402
Некали 402
Нефелометр 51
Клейнманна 51
фотоэлектрический 52
Нефелометрия 44, 49 сл.
Неэлектролиты
адсорбция 137
коагулирующее действие 307
Никель восстановленный, поверхность (по
Тейлору) 89
Никольский, уравнение 149
Нитрат целлюлозы
раствор, образование глобулярного
студня 485
растворение в циклогексане, термоди-
намические функции 440
теплота набухания 450
Нуклеин, теплота набухания 450
Ньютон
определение вязкости 323
уравнение 323 сл.
Обезвоживание, применение электроосмоса^
219
Облака 29 сл., 364
заряд частиц 347
искусственное рассеяние 360, 362 сл.
напряженность электрического поля 345-
образование 345, 356
размер частиц 341
Обогащение руд» флотационное 165
Обращение фаз 380 сл
Объем эффективный 464
Окись углерода, адсорбция иа палладий
104
Окраска коллоидных систем 43 сл.
Опалесценция 10, 13 34, 397
бесцветных коллоидных систем 37
золей 37 сл.
истинных растворов 38
Оператор Лапласа 181
Оптическая плотность 40
Опыты
Мак-Бена по проверке уравнения Гиб«-
бса 123
Рейса, по электрофорезу 169 сл.
— по электроосмосу 169 сл
Органозоли 25, 306
металлов 41 сл., 253
очистка 256
получение 253
устойчивость 306
Осмометрия 68, 456
Осмотическое давление 58 сл., 453 сл,
дисперсных систем 22
зависимость от pH 472
изменение с концентрацией 454
503
Осмотическое давление
коллоидных растворов 10
•— систем 66 сл.
концентрационный градиент 458
раствора высокомолекулярных веществ
453 сл., 458
—» по Доинаиу 472 сл.
Оствальд
вывод уравнения Гиббса 121
классификация коллоидных систем 24
•Осцилляторы пьезоэлектрические и диспер-
гирование 251
Пальмер — Вигнер, суспензионный эффект
477 5
Палладий, адсорбция окиси углерода 104
Пасты 28, 322, 329
Паули, строение коллоидных частиц 241
Пектины как защитные коллоиды 304
Пемза как твердая пена 395
Пенобетоны 395
Пеногасящие вещества 394
Пеногашение 394
Пенообразоваине
прн стирке 163, 394
прн флотации 165, 167
Пенообразователи 386
концентрация и время жизни пен 388
— оптимальная 388
при флотации 167
Пенопласты 395, 398
Пеностекла 395
Пены 24, 28, 386 сл.
время жизни 386 сл,
м — и концентрация пенообразователя
388
высокоустойчивые 392
методы получения 394
минерализованные 165, 167
образование 386
— при стирке 394
— прн флотации 165, 167
практическое значение 394
прочность 386
разрушение 394
старение 390
структура 386, 391
твердые 24, 395
устойчивость 387 сл.
— мера 387
причины 390 сл.
— структурно-механический фактор 390,
392
—• термодинамический фактор 390, 392
Пептнзаторы 234
Пептнзацня 223, 234 сл., 269
гидроокиси железа 254 сл.
значение в технике 236
и получение лиозолей 254 сл.
коалнна 255
кровяного угля поверхностно-активны-
ми веществами 255
окиси алюминия 255
— железа 255
поверхностно-активными веществами
254 сл.
промыванием осадка 254
скорость 235
химическая 254 сл.
электролитами 254 сл.
Перезарядка 184, 301
н явление неправильных рядов 301
коллоидных частиц 189, 192, 194 сл.
Период релаксации 332
Перлит как микрогетерогеииая система 397
Перлы 396
Пермутит 150, 479
Перрен
определение числа Авогадро 65, 72
опыты по изучению распределения ча-
стиц по высоте 69
прибор для электроосмоса 215 -сл.
Песок
коэффициент заполнения пространства
354
критическая скорость «течения» 351
перенос в аэродинамической трубе 35t
Пигмент (ы)
кроющая способность, зависимость от
дисперсности 22 сл.
литопон, дисперсность 350
окнсь железа, дисперсность 350
свойства 350
Пнрозоли 398
Пластификаторы
влияние на гибкость макромолекул 431
Пластификация 321
Платина, золь 245
Пленки
агрегатное состояние 131
конденсированные 128 сл., 130 сл.
моиомолекуляриые 128
пены 386 с л.
— «пережигание» 394
— толщина 389
S4 — равновесная 393
— эффективная упругость 390 сл
поверхностные, газообразные 128 сл,
*=» и площадь, занимаемая молекулой
134 сл.
— растворимых веществ 134
растянутые 131
«черные» 389
Плоскость (граница) скольжения 176, 178,
185, 189 сл., 243
Плотность
поверхностного заряда и потенциал
поверхности 183
свободной энергии у межфазиой гр а*
ницы 237
электричества, поверхностная 199
Плывуны, тиксотропные свойства 317
Поверхностная(ые)
активность 120, 123
гомологов жирных кислот 126
связь с капиллярной активностью 125
проводимость 214
реакции, теплота 105
соединения 105
Поверхностно-актнвиые вещества 117 сл,
адсорбция коллоидными частицами
126 сл., 298
анионные 401 сл.
днфнльность 117 сл., 126
ноногенные 299
н пептизация 254
н разрушение эмульсий 379
как пенообразователи 386
— эмульгаторы 373 '
катионные 402 сл.
коллоидные 399 сл.
мнцелообразование 400
моно- н полнмолекулярные слон 161
моющее действие 414 сл.
!№ноногенные 298, 403 сл.
ориентация молекул в адсорбционном
слое 129, 141
— в поверхностном слое 127, 141
относительно воды 117
поверхностное иатяжеиие 117
полнмолекулярный слой 134, 284 сл.
размер молекул, определение 132 сл.
растворимость 117
растворы истинные 404 сл.
смачиватели 160
солюбилизирующая способность 412
стабилизация латексов 385
504
Поверхностно-активные вещества
стабилизирующее действие 410 сл,
строение 117 сл.
эмульгирующее действие 375
Поверхностное натяжение 114 сл.
влияние различных веществ 118
водных растворов гомологов 126, 127
жидкостей 115 сл.
— методы определения 115
зависимость от концентрации раствора
119, 124
₽— от температуры 119 сл.
заряженной капли 358
и краевой угол 155 сл.
изотермы 119
кристаллов 116 сл.
критическое значение 238
на границе жидкость — газ 115
— жидкость — жидкость 115 сл.
поверхиостио-актнвного вещества 117
при критической температуре раство-
римости 115
равновесное значение 120
твердых тел 116 сл.
ПоверхнЬстиО'Ниактивиые вещества 117 сл.
относительно воды 118
Поверхность
гндрофилнзация 163
гидрофильная 157
гндрофобнзация 163
гидрофобная 157
никеля (восстановленного) по Тейлору
89
олефнльиая 157
работа создания 114 сл.
удельная 15, 16
— адсорбента 99, 135
— активного угля 110
«— зависимость от дисперсности 17, 18
ея порошков 350
— снлнкагеля НО
энергия 114 сл.
— изменение при смачивании 156
— полная 138
Поглощающий комплекс 150
Поглощение света раствором, относнтель
ное 40
Позняк, уравнение 448 сл.
Ползучесть 320, 330
коагуляционных структур 320
Полиакриламид как флокулянт 479
Полиамиды 420
Полиамфолиты 468
Полибутаднеи 420
Полннзобутилен 420
раствсфенне в нзооктане, термодина-
мические функции 442
Полимергомологнческнй ряд 423
Полимеры 417, см. также Высокомолеку*
ляриые вещества
полнднсперсиость 423
растворы 14
*— обратимость 435
ь- применимость правила фаз Гиббса
434 сл.
— равновесность 435
синтетические 420 сл,
Полисахариды 418 сл.
Полнсилоксаны 420
Полистирол как ксерогель 490
Полнсульфестирол как ионит 480
Политетрафторэтилен 420 сл.
Полнхлоропреи 420
Полиэлектролиты 468 сл.
вязкость, влияние электролитов 477
застудиеваине 483 сл.
как иониты 479 сл.
— флокулянты 478 сл
набухание и отбухаиие 471 сл,
применение 478 сл.
Полиэлектролиты
растворы, зависимость свойств от pH*
471 сл.
вя осмотическое давление, зависимость
от pH 472
* — свойства 468 сл.
— — электрические 477 сл.
Полуколлоиды 399 сл.
Поляни, теория полимолекулярной адсорб-
ции 93 сл.
Поляризуемость иоиов 186
Понизители твердости 233
Порог коагуляции 287, 293 сл.
Порошки 28, 350 сл.
брикетирование 355
взвеси в жидкости 366
движение в аэродинамической трубе-
351
как твердые эмульгаторы 376
коэффициент заполнения пространства*
353 сл.
насыпная плотиость 352
обкатывание 354 сл.
поверхность удельная 350
прилнпаемость 352
размеры частиц 350
— — определение 350
распыление 352
распыляемость 353 сл.
расширенное состояние 353
свойства 347, 350 сл.
слежнваемость прн хранении 347
угол естественного откоса 352
эмульгирующая способность 373
Постоянная капиллярная, удельная 124
связь с поверхностной активностью 125
Потенциал
адсорбционный 94 сл.
адсорбционных сил 86
двойного электрического слоя 175
----------изменение при перезарядке 18!>
Дориа 199
изобарно-изотермический, уменьшений
при растворении
высокомолекулярных веществ 438, 441
иоиов, адсорбционный 187
мембранный 475
на межфазной границе 181 сл , 291 сл.
падение по Штерну 185
поверхности 163
— и плотность поверхностного заряда
183
протекания 170
седиментации 170
течения 170, 199
химический, связь с изобарно-изотер-
мическим 121 сл.
Штерна н общий скачок потенциала
на межфазной границе 291 сл.
электрический между заряженными
пластиками 273
электрокинетический (^-потенциал) см>
Электрокинетический потенциал
Поток удельный
диффузии 59
диффузионный 69
седиментационный 69
Почва
известкование 30
как коллоидная система 30
Правило
Антонова 116
Банкрофта 373
Вальдена 465
Дюкло — Траубе (26 сл.
— обращение 127 сл., 139 сл.
аначиости (Шульце — Гарди) 287
Кеиа 171
осадка 235
Траубе 126
ТТравило
уравнивания полярности, Ребиндера
140 сл
фаз Гиббса н растворы полимеров
434 сл.
Шульце — Гарди 287
»— — применимость к латексам 384
------к органозолям 306
Предел
адсорбции 92
текучести 328 сл., 331, 463
~ максимальный 329
— по Бингаму 328 сл.
упругости 331
Предельное напряжение сдвига 338
Прибор
Бейлера — Ребнидера 334
Кена 206 сл.
Перрена для электроосмоса 215 сл.
Тизелиуса 208 сл.
Уметсу для электроосмоса 216
Прозрачность раствора, относительная 40
Промежуточные системы 27
Пропитка
пористая, водоотталкивающая 163
тканей 163
Протеины 418
Противоионы 172, 175, 198, 243
диффузный слой 177
и понижение электрокинетического по-
тенциала 180, 182
концентрация 177
определение подвижности 207
сорбция, влияние электролита 297
схема передвижения прн электроосмосе
174
— _ прн электрофорезе 173
Противопениые вещества 394
Протоплазма клеток, тиксотропные свой-
ства 317
«Псевдорастворы» 9
Пуаз 324
Пуазейль, уравнение 216, 324, 328
Пуассон, уравнение 180 сл.
Пузырек, время жизни 387
Пульпа флотационная 165
Пыль 24, 340
Работа
адгезии 168
адсорбции 127
десорбции 281
диспергирования 232
Равновесие
адсорбционное 107, 142
Доннана 68, 258, 472
мембранное 68, 258, 472
— теория Доннана 472 сл.
Радиус эффективный или эквивалентный
77
Расклинивающее давление 20 сл., 269 сл«
в пленках пеиы 392 сл.
и набухание 276
и равновесная толщина пленок 393
и устойчивость коллоидов 281
природа сил 269 сл.
слагающая ионно-электростатическая
21 сл., 272 сл.
— молекулярная 21, 271 сл., 275
«структурная» 22, 275
Распыление 352
как метод получения аэрозолей 359 сл.
распыляемость порошков 353 сл
Рассеяние света 34 сл.
Расслаивание эмульсий 371
Растворение высокомолекулярных веществ
438 сл.
изменение свободной энергии 434
Растворимость ПАВ 117
Растворитель
влияние иа гибкость макромолекул 43Т
иммобилизация 338
Растворы
высокомолекулярных веществ, см. Вы-
сокомолекулярные вещества, рас-
творы
вязкость, самопроизвольное изменение
467
идеальные 451
истинные, опалесценция 38
коллоидные, см. Коллоидные растворы
мыл, эквивалентная электропровод-
ность 408 сл.
оптическая плотность 40
относительное поглощение 'света 40
прозрачность относительная 40
светопропускаине 40
Рауль, закон 451 сл.
Ребиндер
классификация структур 315
понятие о лиофильных золях 27
правило уравнивания полярности 140 сл.
о самопроизвольном возникновении
двухфазных систем 236 сл.
о структурно-механическом факторе
стабилизации 283 сл.
эффект (диспергирование под влиянием
адсорбции) 233
Резина 419
Рейнольдс, критерий (или число) 324
Рейсс, опыты по электрофорезу и электро-
осмосу 169 сл.
Рекристаллизация
высокомолекулярных студней 490
и пептизация 234
Рекуперация 102
растаорителя 102 сл.
схема установки 103
Релаксация 332
время 205, 332
для кристаллов 332
электрическая 204
Рентгенография 44, 53 сл.
Реология 313
Реопексия 317
у суспензий 367
Реплнкн 49
Рои 436
Ртути золь 245
Ртуть сульфид, золь, понижение устойчи-
вости 308
Рубии 43
Рэлей, уравнение 35 сл., 50
Ряды Гофмейстера 147
Сажа(и)
белые 109
гранулированная 354
размер частиц 350
черные 109
Самоднффузнн 56
Сапонин
защитное действие на золн 305
как пенообразователь 386
Сахара, вязкость растворов 464
Свет
абсорбция 39 сл.
дифракция 34
рассеяние 34 сл.
— интенсивность 36
Светопропускаине 40
Светорассеяние
воды 37
воздуха 37
дисперсных систем 22
506
Светорассеяние
растворов полимеров 457
студне* 488
Сегменты макромолекулы 429 сл.
длина 249 сл.
— и гибкость цепи 430
молекулярный вес 453
Седиментационная устойчивость 68 сл.
Седиментационный анализ 74
методы 76
Седиментационный поток 69
Седиментация 73 сл.
аэрозолей 344
влияние сольватации частиц 77
дисперсных систем 22
и определение молекулярного веса 457
константа 79
кривая распределения 75
обратная 73
потенциал 170
растворов высокомолекулярных веществ
458
скорость 73 сл.
суспензий 367
Седимеитометр
Вигиера 76 сл.
Фигуровского 76
Селей, золь 245
Семнколлоиды 399 сл.
Сенсибилизация 298, 304, 306
Сера, золь 245
получение 247
строение мицеллы 247
Сера
«пластическая» 421
суспензия 234
Серебра бромид, золь 246
устойчивость, влияние ПАВ 299
Серебра гидрозоль 71, 245
скорость движения частиц 71
Серебра иодид, золь 246
изоэлектрическая точка 246
мицелла 243 сл.
перезарядка частиц 194, 245, 299, 301
порог коагуляции 294
строение частиц 241 сл.
устойчивость 260
— влияние ПАВ 298 сл.
элеитрокинетический потенциал и об-
щий сиачок потенциала межфазной
поверхности 193 сл.
Серебра нодид, кристаллы
двойной электрический слой 171 сл.
достройка кристаллической решетки
147 сл., 171 сл., 242
Серебра хлорид, золь 246
Силикагель 109 сл.
адсорбция воды 100 сл.
образование водородной связи 88
удельная поверхность ПО
Силикоз 345, 365
Силы
адсорбционные 85 сл., SS
аттракционные 20
аттракции 265 сл.
Борна (борновские отталкивания) 86,
280
внутреннего треиия 323
дисперсионные 86 сл.
индуициоииые 86 сл.
молекулярного притяжения 269, 272,
278 сл.
неравновесные электроповерхностные 197
ориентационные 86
осмотические 286
отталкивания, электростатические 272,
278 сл, 290, 292
отталкивательные 20
поверхностного натяжения 114
сцепления 20
Синергизм
иоиов 300, 302
электролитов 301 сл.
Синерезис 32, 320, 437, 490 сл.
и растворы высокомолекулярных ве-
ществ 437
практическое значение 491
у суспензий 357
Системы
азродисперсиые 340
гетерогенные 12, 13
гидрофильные 26
гидрофобные 26
гомогенные 12
грубодисперсные 18
двухфазные, самопроизвольное возник-
новение по Ребиидеру 236 сл.
дисперсные см. Дисперсные системы
«изоколлоидные» 13
изоэлектрическое состояние 191
коллоидные см. Коллоидные системы
лиофильные 28 сл.
лиофобные 28
микрогетерогеииые 15 сл., 18
— классификация 23—28
многофазные 12
молекулярно-дисперсные 18
монодисперсные 16
необратимые 25 сл.
обратимые 25 сл.
объем эффективный 338
переходные 313
полуднсперсиые 16
промежуточные 27, 421
разбавленные 35
свободиодисперсиые 27 сл., 313
связиодисверсные 27 сл., 314
с тв^дой^ дисперсной средой (типа
тиксотропные 318
ультрамикрогетерогеииые 15 сл.
устойчивость кинетическая 69
— седиментационная 68 сл.
Скорость
адсорбции 107 сл., 142
коагуляции 262 сл.
Слипание частиц, критическое расстояние
282, 265
Слой (кольца) Лиаегаига 488 сл.
Слой(и)
адсорбционный см. Адсорбционный
слой
гельмгольцевский 185
Гуи 185
двойной электрический см. Двойной
электрический слой
диффузный, см. Диффузный слой
поверхиостно-актнвиых иещеетв 134, 161
лолнмолекуляриые граничные 143
противоионов, диффузный 177
Шиллера 318
штерновский 185
Слюда как двухмерное высокомолекуляр-
ное вещество 421
Смачивание 153 сл.
гистерезис 159
избирательное 157
и изменение поверхностной энергии 156
неполное 154 сл.
полное 154, 156
практическое значение 161 сл.
теплота 138 сл., 158
Смачиватели 160
«Смог» 341
Смолуховский
константа 263, 267
теория кииетини быстрой иоагуляци»
261 сл.
уравнение 263 сл„ 339
Смолы 420, 490
507
Сольватация
и вязкость 337
как первая стадия набухания 444
потенциалопределяющнх нонов н устой*
чивость коллоидов 283
частиц 281 сл.
Сольватная оболочка 20
и устойчивость коллоидов 281
Солюбилизат 412
Солюбилизация 412
обратная 414
Софндофильные группы 166
Сплавы
гетерогенные как системы типа Т/Т
397
как коллоидные системы типа Т/Т 24,
395 сл
твердость и дисперсность 23
Стабилизаторы 13
для «обрыва» коагуляции 262
коллоцдных растворов 11
— систем 242
Старение
и пептизация 234
растворов полимеров 467
Стекло(а)
рубиновое 43, 396, 397
цветные как коллоидные системы типа
Т/Т 395 сл.
Стирка 162
и ценообразование 394
схема отмывания углеводорода 162
Стокс, уравнение 342 сл.
Структурно-механические свойства
определение методом закручивания
цилиндра 335
тангенциального смещения пла*
стинкн 334 сл«
Структурно-механический фактор устой*
чивостн 281, 283 сл., 390, 392
латексов* 384
Структуры
возникновения в коллоидах 315 сл,
классификация Ребнндера 315
коагуляционные 315 сл.
— эластические свойства 320
компактные 315
конденсационно-кристаллизационные
315, 321
— — необратимо разрушающиеся 315
периодические 318 сл.
пространственные 315
рыхлые 315
тиксотропно-обратимые 315
Студни 445, 467, 481 сл.
анизотропия 489 сл.
глобулярные 485
— устойчивость 486
диффузия в них низкомолекулярных
веществ 488
как механо-химнческая машина 472
корпускулярные 485
критическое напряжение сдвига 486 сл,
механическое разрушение 483
переход в раствор 483
пространственные сеткн 482
свойства 486 сл.
— механические 486
тиксотропные 487
электропроводность 489
Субмнкроны 24
Сульфат бария, суспензия
рассеяние света 36
Сульфокислот соли, как анионные ПАВ
401
Сульфомыла 402
Сурьмы сульфид, золь, окраска 43
Суспензии 24, 25, 366 сл.
агрегативно неустойчивые, оседание 322
— устойчивые, оседание 322 i
508
Суспензии
в природе и технике 366
высококонцентрированные 322
вязкость, зависимость от анизоднамет*
ричности частиц 336 сл.
коагуляция 367
концентрированные 28
полидисперсиые, кривая распределения
75
— преимущественная фракция 75
радиус частиц 74
свойства молекулярно-кинетические 367
— оптические 367
седиментация 73 сл
устойчивость агрегативная 367
—е — и объем осадка 322
Суспензоиды 26
Тактоиды 318
Таиннды
растворы 399
как коллоидные ПАВ 415
Твердость, понизители 233
Текучесть 324
изменение С температурой 325
предел 463
Тела
пористые 24
твердые 55
— поверхностное натяжение 116 сл.
упруго вязкие 332
Теллура, золь 245
Температура растворения (смешения), крн*
тнческая 441
Температурный коэффициент скорости
адсорбции 108
хемосорбции 108 сл.
Теория
адгезии 168
ядсорбцнн потенциальная 93 сл.
броуновского движения 65
БЭТ 93, 97 сл.
Гельмгольца — Перрена 175 сл.
гетерокоагуляции Дерягина 307 сл,
Гун — Чэпмена 176 сл.
----и теория электролитов Дебая —•
Гюккеля 179
двойного электрического слоя с диф-
фузным слоем противоионов 176 сл»
ДЛФО 290
жидкостей 55
кинетики быстрой коагуляции 261 сл,
коагулирующего действия смесей элект-
ролитов 302 сл
коагуляция полнднсперсных систем 266-
в® химическая 289
— электростатическая 289
мономолекулярной адсорбции 88 сл.
----Ленгмюра, применимость 96 сл/
Мюллера 289
обменной адсорбции на угле 152 сл.
образование новой фазы, Фольмера
357 сл.
планет 29
периодических коллоидных структур
319
полимолекулярной адсорбции Поляни
93 сл.
притяжения тел 271
роста кристаллов, адсорбционная 226
флуктуаций 65 сл.
Фукса коагуляции 266
Фрейндлиха 289
Штерна 184 сл.
Тепловое движение 55 сл.
Теплота
адсорбции 105 сл
— дифференциальная 106 сл.
Теплота
— интегральная 106
— физический 103
химический 103
— чистая 98
мицелообразовання 406
набухания 449 сл.
поверхностных реакций 105
смачивания 138 сл , 158
— как критерий пригодности раствор»*
теля при адсорбции 138
Термодинамика растворения высокомоле-
кулярных веществ 438 сл.
Термодинамические факторы устойчивости
293, 390, 392
Термопреципитация 345
Термофорез 345
Течение
вязкое 58, 314
— аномалии 327
жидкостей, механизм 324 сл.
золей 326 сл.
кривая 329 сл.
ламинарное 323 сл.
объемная скорость истечения 324
пластическое 328 сл.
порошков 347
турбулентное 324
Тизелиус
метод определения скорости электро-
фореза 208 сл.
прибор 208 сл
Тиксотропия 279, 317 сл<
вязкостная 318
в природе 317
использование в технике 317 сл<
прочностная 318
студней 487
суспензий 367
Тиндаль, конус 10
Тнндальметр 51
Титана двуокись, золь
кристаллизация частиц 229
электронограммы частиц 230
электронные микрофотографии частиц
229
Томсон
закон 116 сл», 225
уравнение 99 сл, 357
Точка Крафта 406 сл.
Траубе, правило 126
Туман 24, 30, 340, 364
искусственное рассеяние 360, 362 сл.
образование 356
размер частиц 341
разрушение ультразвуком 362
Трение внутреннее, сила 323
Турбидиметрия 44, 53
Турбулентность ранняя 324, 327
Углеводородная цепь, строение 428
Угол
естественного откоса 352
краевой см Краевой угол
смачивания 154
Уголь 109 сл
адсорбция кислорода 104 сл.
— обменная 151 сл.
активирование 110
кровяной, пептизация 255
обеззоленный 153
пептизация поверхностно-активными
веществами 245
применение в качестве адсорбента НО
Удельный поток
диффузии 59
диффузионный 69
седиментационный 69
Ультразвук
применение для эмульгирования 377
разрушение аэрозолей 361 сл.
Ультразвуковые колебания
и диспергирование 248, 251
и коагуляция 251
Ультрамнкроны 24
Ультрамикроскоп 44 сл
Зидентопфа и Зигмонди 44
поточный 47 сл., 262, 267
щелевой 45
Ультрамнкроскопия 44 сл
Ультрафильтрация 255, 258
Ультрафильтры Бехгольда 258
Ультрацентрифуга 77 сл., 80
воздушная 80
применение 80
Уметсу, прибор для электроосмоса 216
Умягчение воды 150 сл.
Упругость 333 сл.
запаздывающая 333
коллоидных систем 332
пленки пен, эффективная 390 сл.
Уравнение
Бедекера — Фрейндлиха 84 сл.
Бингама 329
Больцмана 178, 440
Ван-дер-Ваальса 130
Вант-Гоффа 67. 454
Гельмгольца — Смолуховского 200
Генри 91, 203 сл.
— кривая 205
Гиббса 120 сл.
— и уравнение Ленгмюра 124 сл,
Гука 331
Гюккеля 203
Дюпре 168 z
изотермы адсорбции 91 сл.
----паров (по теории БЭТ) 98
« Ленгмюра 91 сл,
Кеиииигема 343
Леинарда — Джойса 86
Лондона 269
Максвелла 332
Менделеева — Клапейрона 130
Никольского 149
Ньютона 323 сл.
Позняка 448 сл«
Пуазейля 216, 324, 328
Пуассона 180 сл.
Рэлея 36 сл.» 50
седиментации 73
скорости адсорбции 107 сл.
Смолуховского 263 сл., 338
состояния двухмерного газа 130
Стокса 342 сл.
Томсона (Кельвина) 99 сл., 357
Фнкенчера и Марка 338
Фишера 411
Шишковского 124 сл.
Штаудингера 459 сл.
Эйнштейна 59—61, 336 сл., 457 сл.
Эйнштейна Смолуховского 64
Юнга 155
Устойчивость
глобулярных студией 486
золей, теория ДЛФО 290
концентрированных золей 295
пен 387 сл.
Фазы
образование 357
обращение 380 сл.
Фактор шероховатости 160
Фаянс, строение коллоидных частиц 241
Фигуровский, седиментометр 70
Фик, закон диффузии 59
Фикенчер и MapKt уравнение 338
509-
Фильтрация
аэрозолей 360 сл.
как метод разрушения эмульсии 379
суспензий 367
Фильтрпресс электроосмотнчесний 219
Фильтры 361
самоочищение 360
Фишер, уравнение 411
Флокулы 467
Флокулянты 476 сл.
Флокуляция 279, 307, 467, 478
Флоридии, адсорбция бензойной кислоты
139
Флотация 161, 164 сл.
вероятность 166
пеииая 165
— изменение свободной энергии свете*
мы 165 сл.
свиицово-цинковых руд 167
селективная 167
суспензий 367
Флуидизация 352
агрегативная 353
Флуктуации 38, 65 сл.
Флуоресценция 39
Фольмер
адсорбционная теория роста кристал-
лов 226
теория образования новой фазы 357 сл.
Фосфор черный как твердая эмульсия 396
Фотофорез 345
Фракционирование полимеров 423
Фрейндлих» теория коагуляции электроли-
тами 289
Фрумкин
определение электрофоретической по-
движности капель ртути 206
теория обменной адсорбции на угле
152 сл.
уравнение состояния двухмерного газа
130
Фукс, теория коагуляции 266
«Хвосты» при флотации 165
Хемосорбция 81, 103 сл.
температурный коэффициент 108 сл.
Хлорофилл, хроматограмма 144
Хроматограмма
аминокислот 145
«проявление» 144 сл,
хлорофилла 144
Хроматография 144 сл.
Целлюлоза 14, 419
иабухаиие, теплота 450
— термодинамические функции 447
Центрифугирование как метод разрушения
эмульсий 379
Центры (зародыши) кристаллизации 223 сл.
влияние веществ на рост зародышей
228
Цеолитй 111
Циклон 360
Цинка окись, дясиерсность порошка 350
Частицы
агрегаты, образование и распад 262, 268
агрегация 252, 268
анизотропная ориентация 318
аэрозолей, броуновское движение
343 СЛ.
— кривые распределения фракций 341
Частицы
взаимодействие 278 сл.
гидрофильные 184
гидрофобные 164
изменение структуры под действием
электрического поля 310
инерционное оеаждеиие 360 сл.
коллоидные см. Коллоидные частицы
кривые взаимодействия 295
объем эффективный 337
размер, определение 79 сл.
распределение по высоте 69
силы отталкивания, зависимость от по-
тенциала поверхности 292 сл.
слипание, критическое расстояние 26%
265
смещение, среднеквадратичное значе-
ние 263
сольватация 28J сл.
— индуцированная 282
с постоянным диполем, строение двой-
ного электрического слоя 190 сл.
стабилизованные ПАВ 285
сферические, строение двойного элект-
рического слоя 190
счет в потоке 252
электрокииетнческий потенциал н общий
скачок потенциала поверхности 291
электрофоретическая подвижность 200,
304
энергия отталкивания 411
Частичная концентрация 35 ~
Числа переноса в капиллярах мембран
прн электродиализе 256 сл.
Численная концентрация 35
и скорость коагуляции 263
определение поточным ультрамикро-
скопом 252, 257
Число
Авогадро, определение 64 сл., 82 сл,
агрегации 405
железное 305
золотое 304 сл.
Рейнольдса 324
рубиновое 306
серебряное 305
сериое 305
Шаровые мельницы 248 сл.
Шилов
поверхностные соединения 105
теория обменной адсорбция на угле
152 сл.
Шишковский, уравнение 124 сл.
Шмидт, теория образования планет 29
Штаудингер, уравнение 459 сл.
Штерн
адсорбционный слой 198
теория 184 сл.
Шульце — Гарди, правило 287
приложение к органозолям 306
Эбонит 419, 446
Эйнштейн — Смолуховский, уравнение 64
Эйнштейн, уравнение 59—61, 335 сл., 457 сл.
Экстинкция 40
Эластичность х
коагуляционных структур 320
коллоидных систем 332
студней 486
Эластовискозиметр 336
Электродиализ 255 сл. _
очистка высокомолекулярных веществ
424
применение в технике 258
Электродиализатор 256
510
Электрокинетический потенциал 176, 178,
474
влияние индифферентных электролитов
191 сл.
— концентрации коллоидной системы
195 сл., 202
₽= неиндифферентных электролитов
193 сл.
г* неравновесных электроповерхност-
ных сил 197
“ природы дисперсионной среды 196
— pH среды 194 сл.
=- температуры 196
вычисление при электрофорезе и элек*
троосмосе 200 сл.
для полиэлектролита в растворе 477
зависимость от радиуса ионов 186 сл.
--- капилляра 214
значения, найденные разными спосо-
бами 217 сл.
изменение знака 184, 188
и общий скачок потенциала иа меж-
фазной поверхности 292 и толщина
ионной атмосферы 180
критический 288
— влияние электролитов 300
— и коагуляция 288 сл.
латексов 382 сл.
межфазной поверхности и влияние
электролитов 192 сл.
определение 202 сл.
— по объему электроосмотически пере-
несенной жидкости 212 сл.
— по электроосмотическому давлению
216
— электрофоретические методы 202 сл.,
212 сл.
органозолей 306
падение при изменении концентрации
противоионов 180, 182
связь со скоростью электрофореза 198,
200
Электролиз 11
Электролиты
аддитивное действие при коагуляции
301 сл.
адсорбция 137, 146 сл.
антагонизм в коагулирующем действии
301 сл.
влияние на вязкость 338, 476 сл.
на критический электрокинетический
потенциал 300
, — иа синерезис 320
Электролиты
высокомолекулярные см. Полиэлектро-
литы
индифферентные 191
— влияние на форму макромолекул
иеиндифферентиые 193
— влияние на электрокинетический по-
тенциал 193 сл.
синергизм в коагулирующем действии
301 сл.
смеси, теория коагулирующего дейст-
вия 302 сл.
Электромагнитное запаздывание 272
Электронограммы частиц золя 229 сл.
Электронография 44, 53 сл.
Электроосмос 170, 197 сл.
объем перенесенной жидкости 212 сл.
прибор Перрена 215 сл.
— Уметсу 216
схема передвижения жидкости и про-
тивоионов 174
Электроосмотический перенос жидкостей
зависимость от дипольного момента
молекул 196
от диэлектрической проницаемости
Электропроводность
лиозолей 219 сл.
студией 489
Электрораспыление 248, 252 сл.
Электрофильтры 363 сл.
Электрофорез 11, 169 сл., 197 сл.
белков 470
и двойной электрический слой 171
на бумаге 210
скорость 203 сл.
— влияние агрегатного состояния дно
персной фазы 206
— — электрической релаксации 205
!-= — электролитов 205
— и электрокинетический потенциал
198,' 200
>— определение 206 сл.
---методом Гитторфа 207
—------микроскопическим 210 сл.
---подвижной границы 207 сл.
----- — Тизелиуса 208 сл.
------ультрамикроскопическим
210 сл.
схема передвижения частиц 173
Электрофоретические /
запаздывание (торможение) 203
перенос 200, 211
подвижность 200
изменение с концентрацией 202
Электроэндоосмос 170
Эмали как коллоидные системы типа
Т/Т 396
Эмульгаторы 368
гидрофильные и гидрофобные (олео-
фильные) 373
иеионогенные 372
природа н агрегативная устойчивость
эмульсий 372 сл.
твердые 376 сл.
Эмульгирование 377 сл.
практическое значение 32, 381
самопроизвольное 379
Эмульгирующее действие
высокомолекулярных веществ 376
мыл 373 сл.
твердых эмульгаторов 376
Эмульсии 24, 28, 367 сл.
время жизни капли 371 сл.
второго рода, обратные 386
высококонцентрированные 370 сл.
— механические свойства 371
действие электрического поля 310
деэмульгирование 379
желатинированные 371
жидких металлов 369
кинетическая кривая устойчивости
371 сл.
классификация 368 сл.
концентрированные 370
критические 15, 236, 239, 368
лиофильные 368
лиофобные 368
«множественные» 381
мутность 37
неустойчивость агрегативная 371
образование при самопроизвольном
диспергировании 368
обращение фаз 380 сл.
первого рода, прямые 368
получение 377 сл.
практическое значение 381
природные 381
прямые, стабилизованные 373 сл.
разбавленные 369
разрушение 377, 379 сл.
— непрерывное 379 сл.
— фильтрованием 379
— центрифугированием 379
расслаивание, скорость 371
стабилизация 373 сл.
511
Эмульсии
тип. 369. 373
твердые 24, 395
устойчивость 368
— агрегативная 371 сл.
и природа эмульгатора 372 сл.
— равновесная 239
— факторы 373 сл.
Эмульсоиды 26
Эмульсолы 236, 372
растворение 239
Энергия *
активации
f— диффузии 56
— свободного вращения вокруг связи
С-С 427
взаимодействия дисперсионного 87
— индукционного 87
между поверхностью двух тел
270 сл., 277
— молекул 270
ориентационного 87
изменение при самопроизвольном дне*
пергироваиии системы 238
когезионная, удельная плотность 445
коллоидной системы, свободная 434
отталкивания частиц 411
поверхности, удельная свободная 114 сл.
— полная 138
поверхностная, изменение при смачива-
ини 156
притяжения пластинок 270 сл.
системы, свободная 237
---изменение прн флотации 165 сл.
— — при мицелообразоваиин 406
— уменьшение при растворении по-
лимеров 434
Энтальпия системы при растворении поля^
мера 439, 442
Энтропийный фактор устойчивости 281, 2%
Энтропия
изменение при смешении или раство-
рении 439 сл.
растворения полимеров 440
смешения 452
— полимера с растворителем 439 сл.
увеличение при дроблении веществ 239
Эрозия ветровая 351
Этерозоли 25
Эффект
Гиббса 390
Доннана 472 сл.
— и возиикиовеиие биопотенциалов 477
Дориа 170
зацепления 361
Пальмаиа — Вигнера, суспензионный
Ребиидера 233
электровязкостиый 338 сл.
Юнг, уравнение 155
Явление (ня)
неправильных рядов 300
— и перезарядка 301
— при коагуляции электролитами
300 сл.
электрокииетические 170 сл., 197 сл.»
218 сл.
Ядро мицеллы 243 сл.
Сергей Сергеевич Воюцкий
Курс коллоидной химии
Редактор Ларичева Л. Н.
Технический редактор Косачева Г. И.
Художник Бекетов Е. В.
Корректоры Сидорова Г. Н, Саликов А. И.
Т-15407 Сдано в иаб. 21,3.1975 г. Подп, к печ. 21,8.1975 г. Формат бумаги
60X90V1S Бумага тип. .У, 2 Усл. печ. л. 32,0 Уч.-изд. л. 39,7 Тираж
64 000 эка. (1-ый завод—30 000 эка.) Зак. 664 Изд. № 422 Цена 1 р. 60 к.
Издательство „Химия” 107076, Москва, Стромынка, 13, корп. 2.
Ордена Трудового Красного Знамени
Ленинградская типография Ws 2 имени Еигеиии Соколовой
Союзполиграфпрома при Государственном комитете Совета Министров СССР
по делам издательств, полиграфии и книжной торговли.
198052, Ленинград, Л-52, Измайловский проспект, 29.
Отсканировал Семенюченко Владимир
chem_vova@mail.univ.kiev.ua; vova2002@mail.ru
с.с.$оюцкии
г*-#:.. . _ .t.'/.as
КУРС
коллоидной
химии