/












































































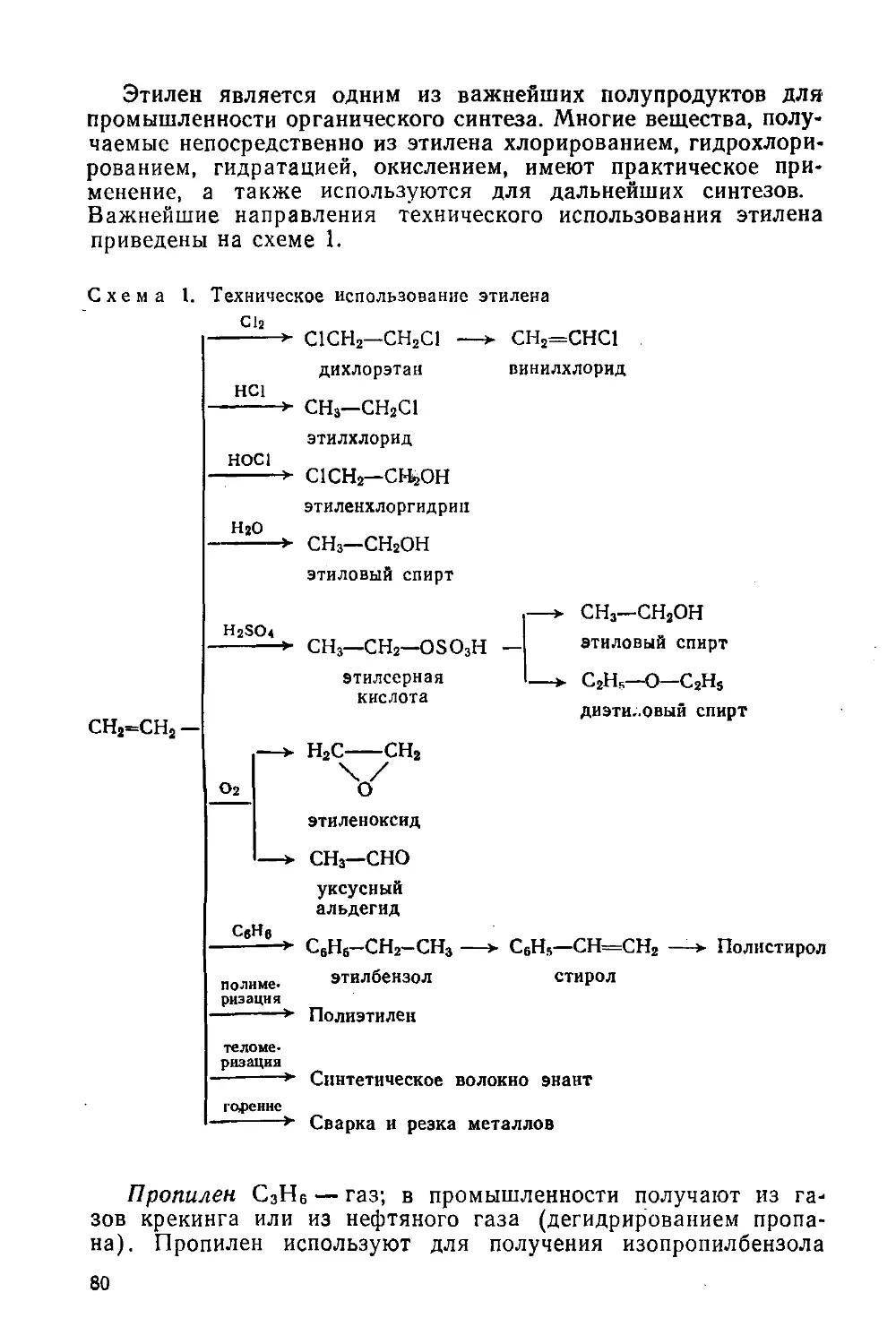







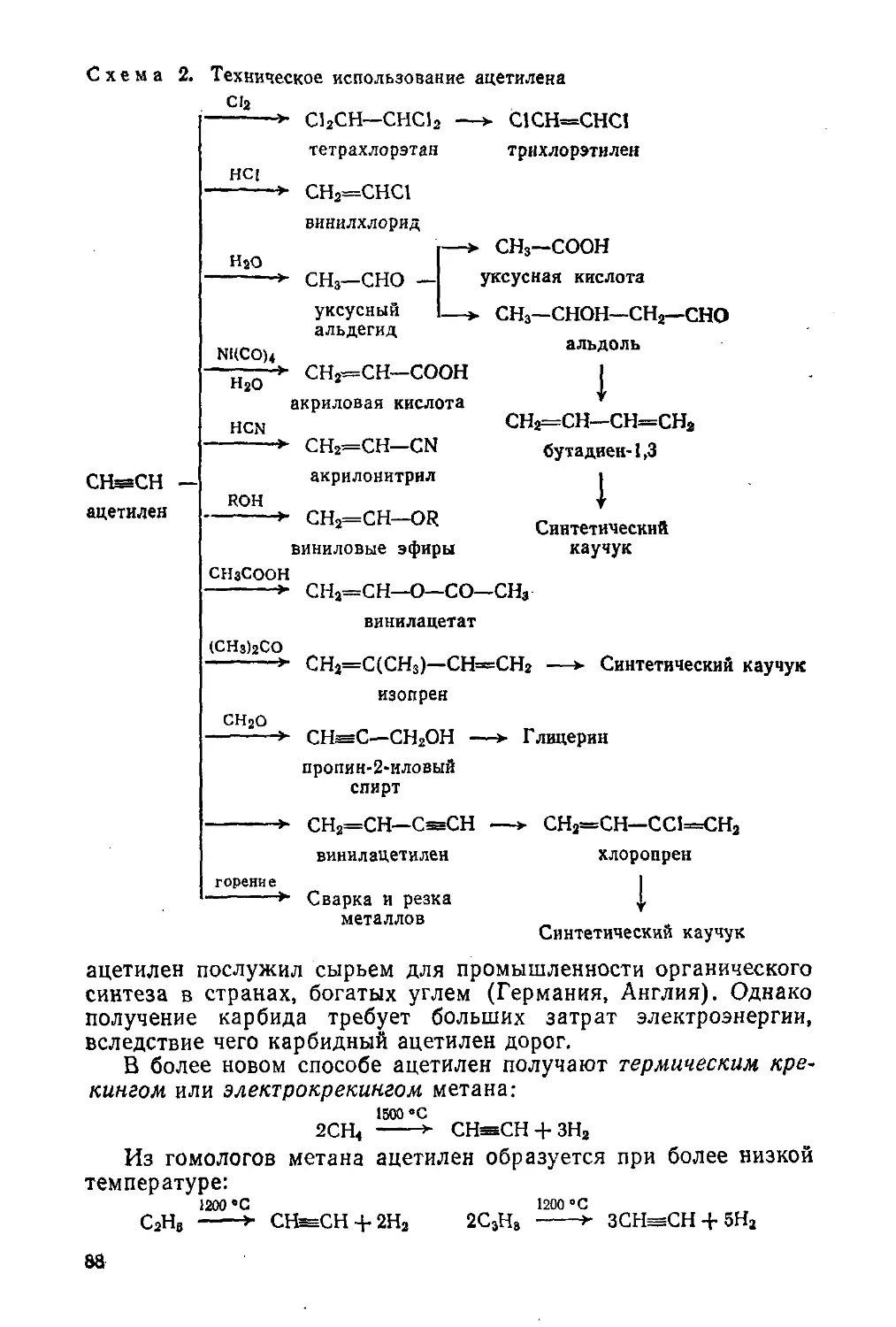




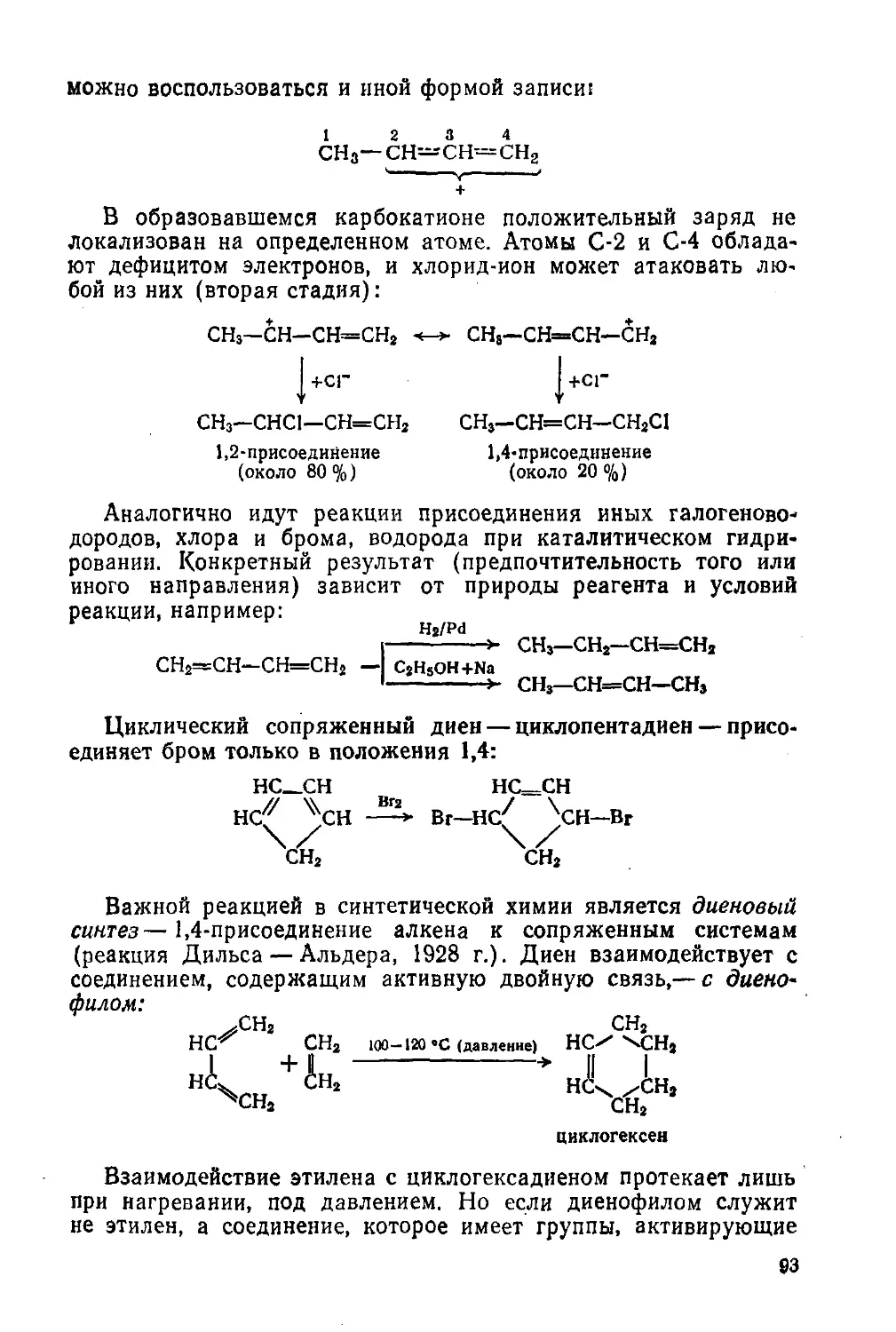







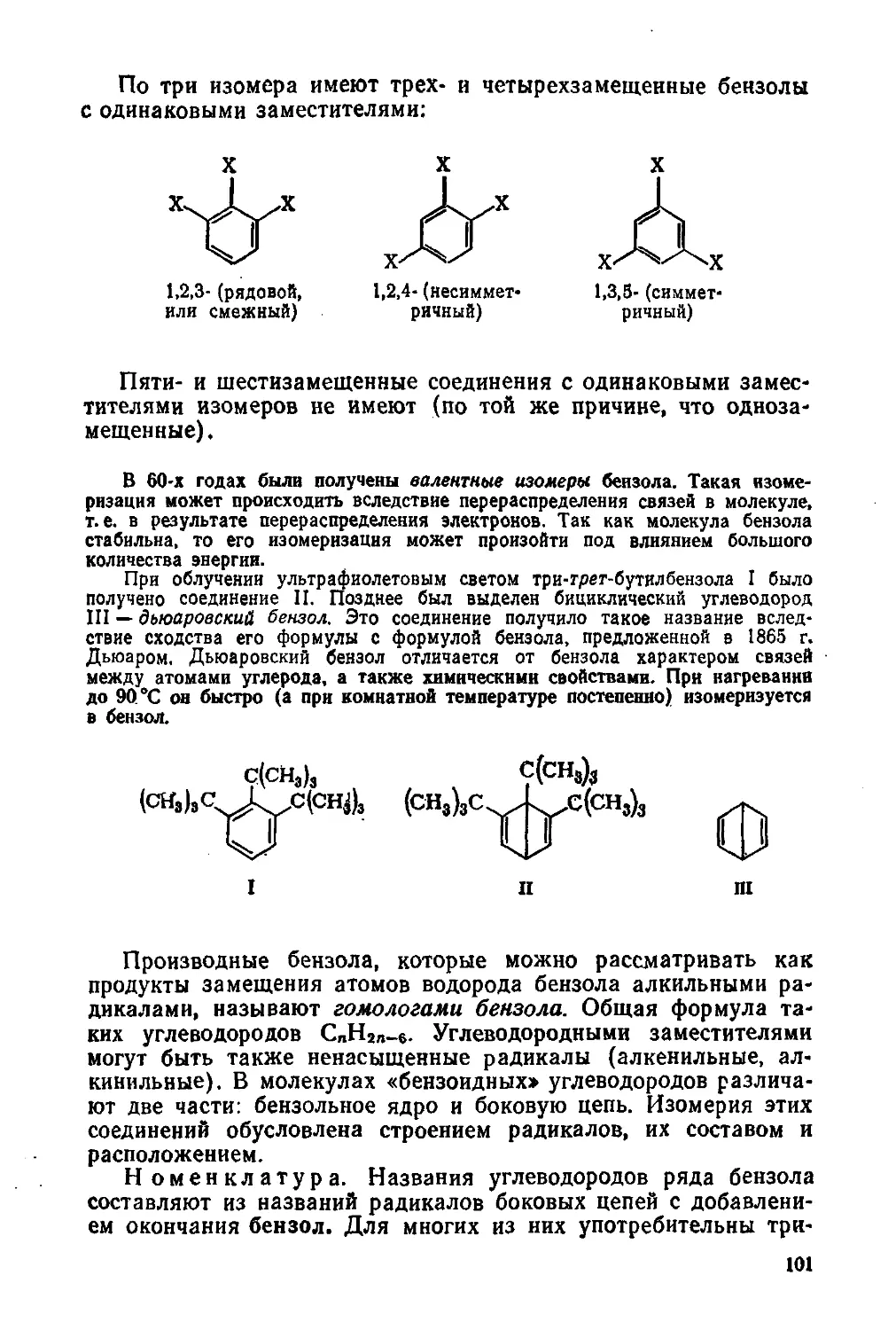





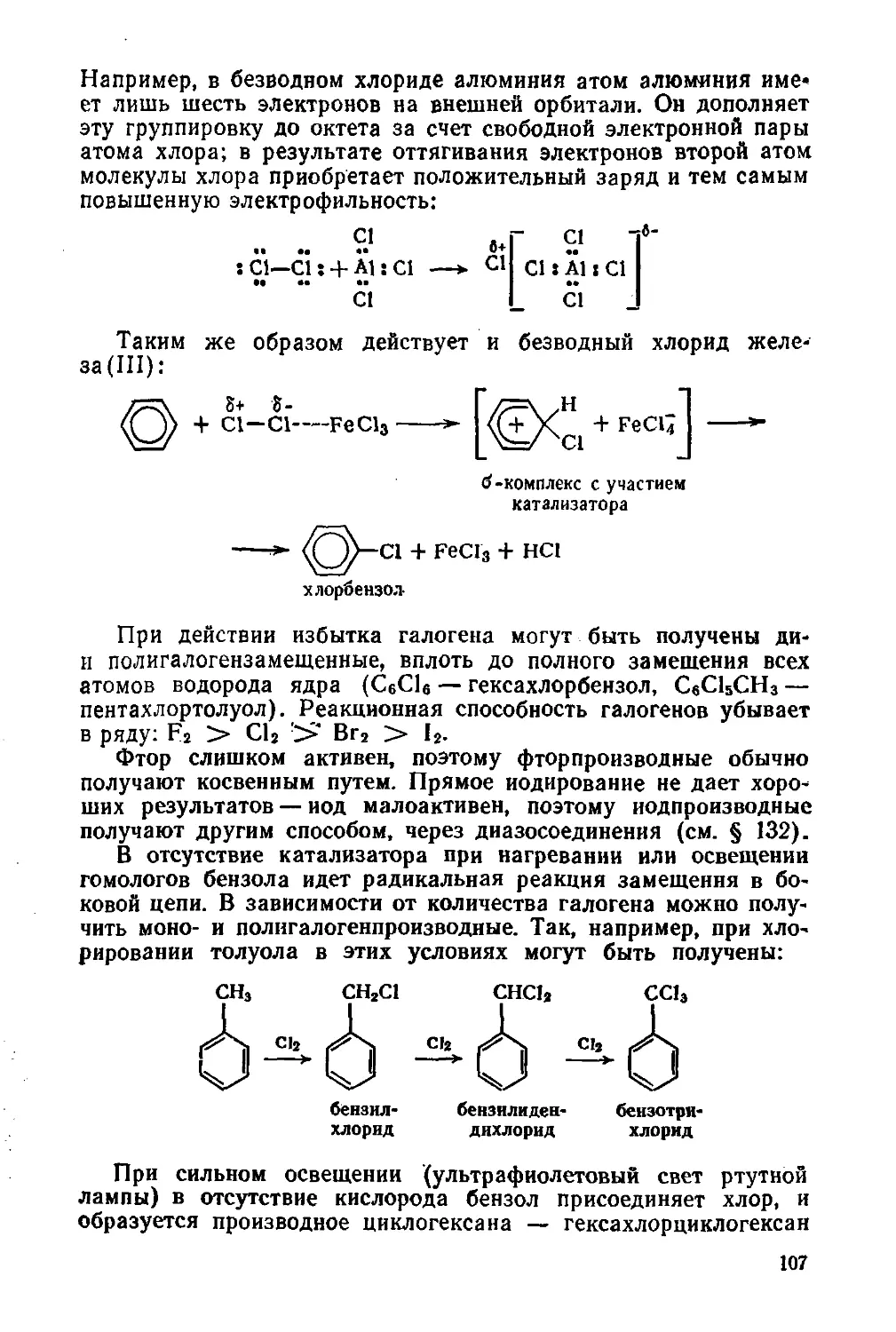











































































































































































































































































Автор: Потапов В.М. Татаринчик С.Н.
Теги: органическая химия здравоохранение медицинские науки химия
ISBN: 5-7245-0256-9
Год: 1989




Текст
В.М. ПОТАПОВС.Н. ТАТАРИНЧИК
ИЗДАНИЕ ЧЕТВЕРТОЕ,
ПЕРЕРАБОТАННОЕ И ДОПОЛНЕННОЕ
Допущен Министерством
химической промышленности СССР
в качестве учебника
для учащихся техникумов
МОСКВА
»химия«
1989
ББК 547
П64
УДК 547(075.3)
Рецензенты:
Голубкова Н. К. (Ленинградский политехникум),
Либерман А. Б. (Московский химико-технологический техникум)
, Татаринчик С. Н.
П64 Органическая химия; Учебник для техникумов,—
4-е изд., перераб. и доп. — М.: Химия, 1989. — 448 с., ил.
ISBN 5—7245—0256—9
Книга является четвертым, переработанным изданием учебника
для учащихся химических и химико-технологических специальностей
техникумов (3-е издание вышло в 1980 г.).
В книге изложен современный курс органической химии, написан¬
ный в соответствии с программой для химических техникумов. В осно¬
ву построения учебника положена классификация органических соедине¬
ний по функциональным группам. На тщательно отобранном фактиче¬
ском и теоретическом материале учащиеся познакомятся с различными
типами реакций и их механизмами, наиболее важными соединениями,
используемыми в различных областях народного хозяйства. Кратко рас¬
смотрены некоторые вопросы биоорганической химии. Рассмотрены ве¬
щества и реакции, составляющие основу важных процессов промышлен¬
ного органического синтеза.
Книга может быть полезной также преподавателям средних школ,
учащимся школ с химическим уклоном, студентам нехимическнх спе¬
циальностей вузов и инженерно-техническим работникам промышленно¬
сти органического синтеза.
И 1705000000—117 сВ- пл. для сред. спец. КБК 547
050(01)—89 учеб, заведений — 112—89
ISBN 5—7245—0256—9 © Издательство «Химия», 1976
© Издательство «Химия», 1989,
с изменениями
Потапов В. М..
СОДЕРЖАНИЕ
От авторов 9
Введение Ю
§ 1. Предмет органической химии и ее практическое значение 10
§ 2. Сырьевая база промышленности органического синтеза П
§ 3. Роль отечественных ученых в развитии органической химии 12
§ 4. Развитие химии в СССР 12
Глава 1. Общие положения органической химии 14
§ 5. Особенности органических соединений 14
§ 6. Выделение и анализ органических веществ 14
§ 7. Явление изомерии 19
§ 8. Первоначальные представления о природе органических соеди¬
нений 19
§ 9. Теория химического строения органических соединений 22
§ 10. Электронная оболочка атома 24
§ 11. Валентные состояния углерода. Гибридизация 27
§ 12. Электронная природа химической связи 28
§ 13. Характеристики ковалентных связей и методы их определения 31
§ 14. Типы органических реакций 34
§ 15. Классификация органических соединений 36
ЧАСТЬ I. УГЛЕВОДОРОДЫ 39
Глава 2. Алканы 39
§ 16. Гомологический ряд, строение, изомерия 39
§ 17. Конформации. Модели молекул 42
§ 18. Номенклатура 45
§ 19. Способы получения 47
§ 20. Общая характеристика физических и химических свойств 49
§ 21. Реакции 52
§ 22. Алканы в природе и в технике 55
Глава 3. Циклоалканы 57
§ 23. Строение, номенклатура, изомерия, нахождение в природе 57
§ 24. Способы получения 58
§ 25. Устойчивость циклов 59
§ 26. Физические и химические свойства 60
§ 27. Пространственная изомерия замещенных циклов 63
§ 28. Отдельные представители 64
Глава 4. Алкены 64
§ 29. Строение 65
§ 30. Изомерия и номенклатура 65
§ 31. Способы получения 68
§ 32. Общая характеристика физических и химических свойств 70
§ 33. Реакции 71
§ 34. Отдельные представители 79
1*
3
Глава 5. Алкины
§ 35. Строение, изомерия и номенклатура
§ 36. Способы получения
§ 37. Общая характеристика физических и химических свойств
§ 38. Реакции
§ 39. Ацетилен в промышленности органического синтеза
Глава 6. Диеновые углеводороды
§ 40. Изомерия, номенклатура и классификация
§ 41. Соединения с сопряженными двойными связями
§ 42. Наиболее важные представители
Глава 7. Ароматические углеводороды
Бензол и его производные
§ 43. Строение бензола. Природа ароматического состояния
§ 44. Изомерия производных бензола. Номенклатура ароматических
углеводородов 100
§ 45. Способы получения гомологов бензола 102
§ 46. Физические свойства 104
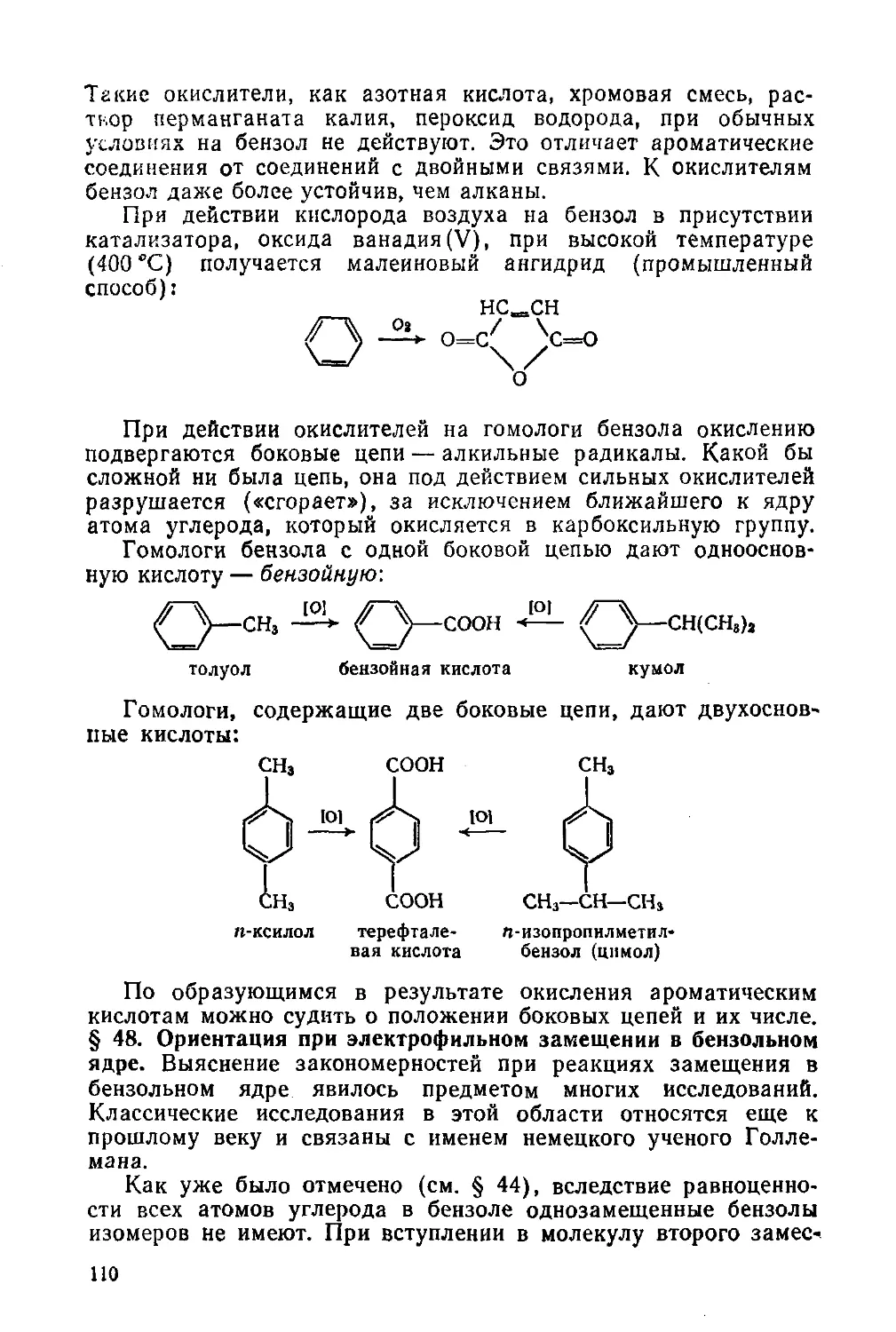
§ 47. Химические свойства бензола и его гомологов 104
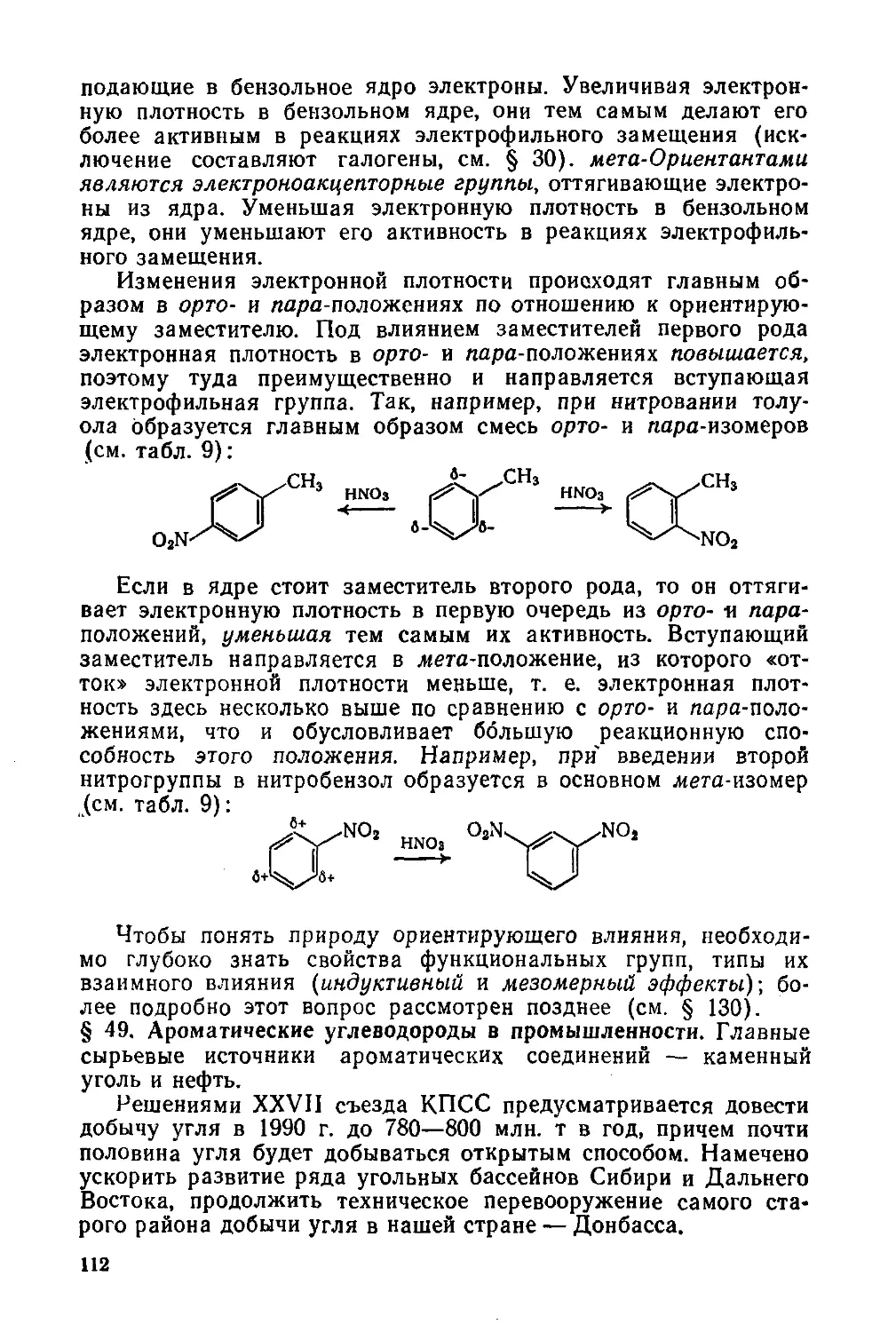
§ 48. Ориентация при электрофильном замещении в бензольном ядре ПО
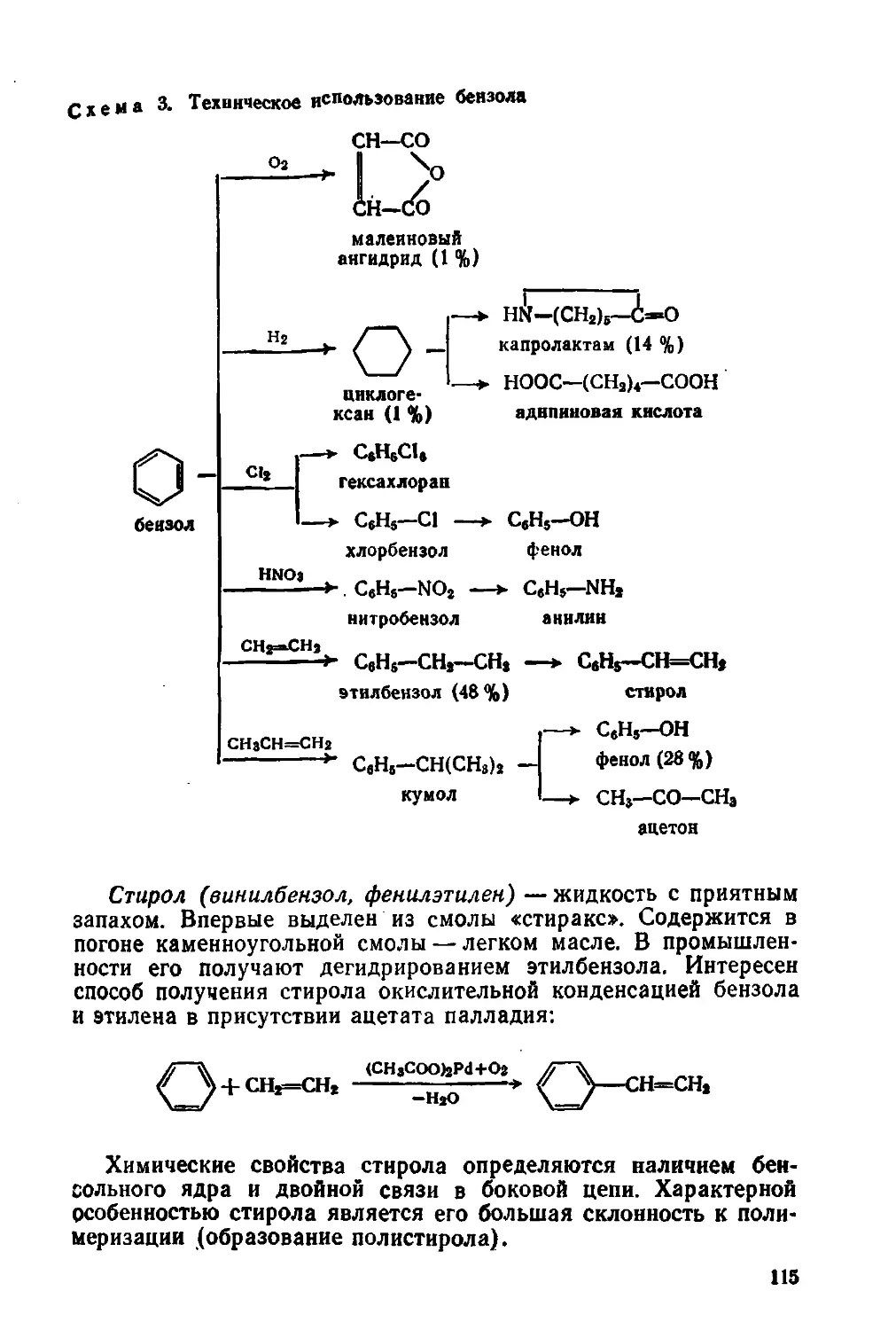
§ 49. Ароматические углеводороды в промышленности 112
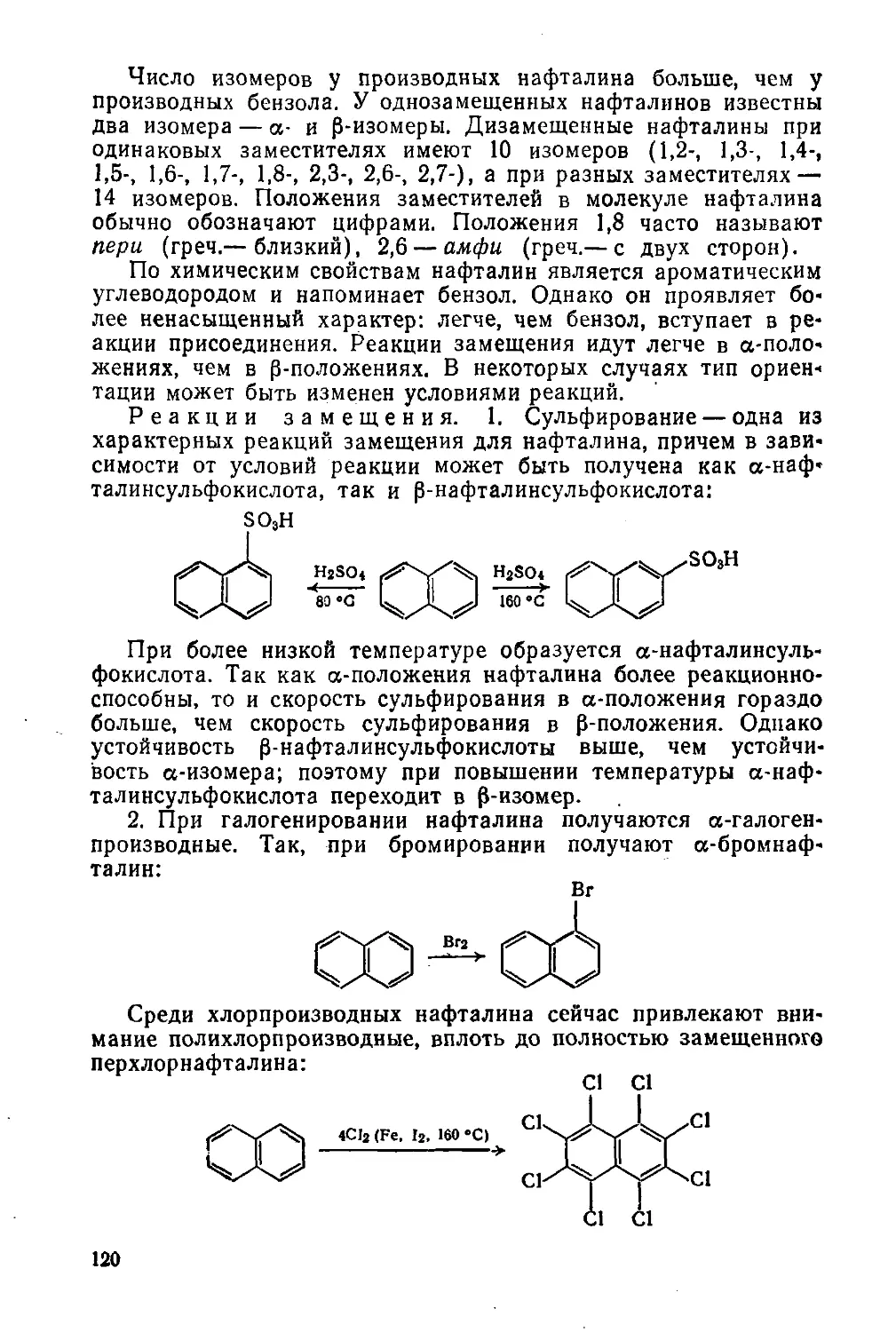
Многоядерные ароматические углеводороды 116
§ 50. Соединения с изолированными бензольными ядрами 116
§ 51. Свободные радикалы 117
§ 52. Соединения с конденсированными бензольными ядрами. Нафта¬
лин 119
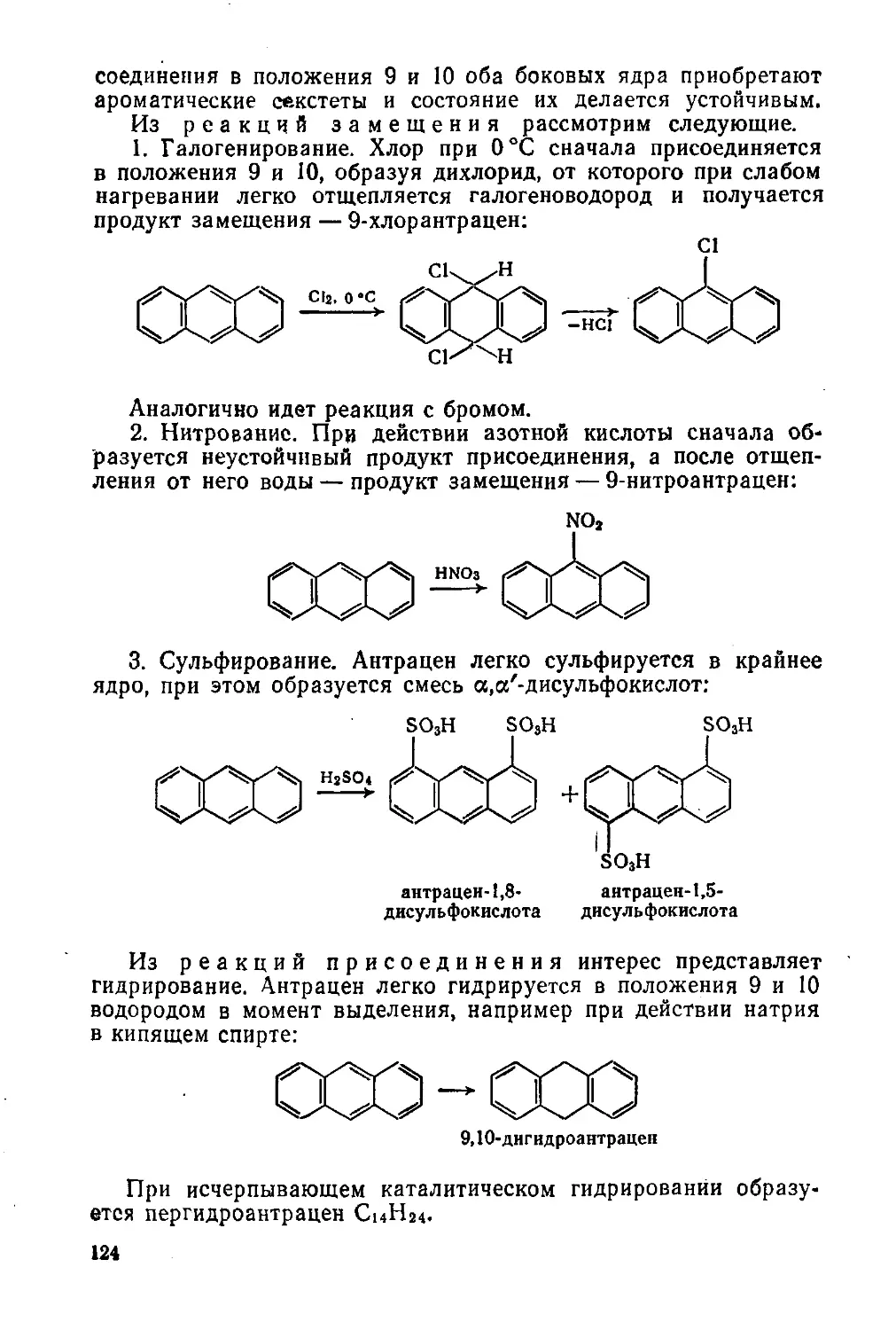
§ 53. Антрацен и фенантрен 123
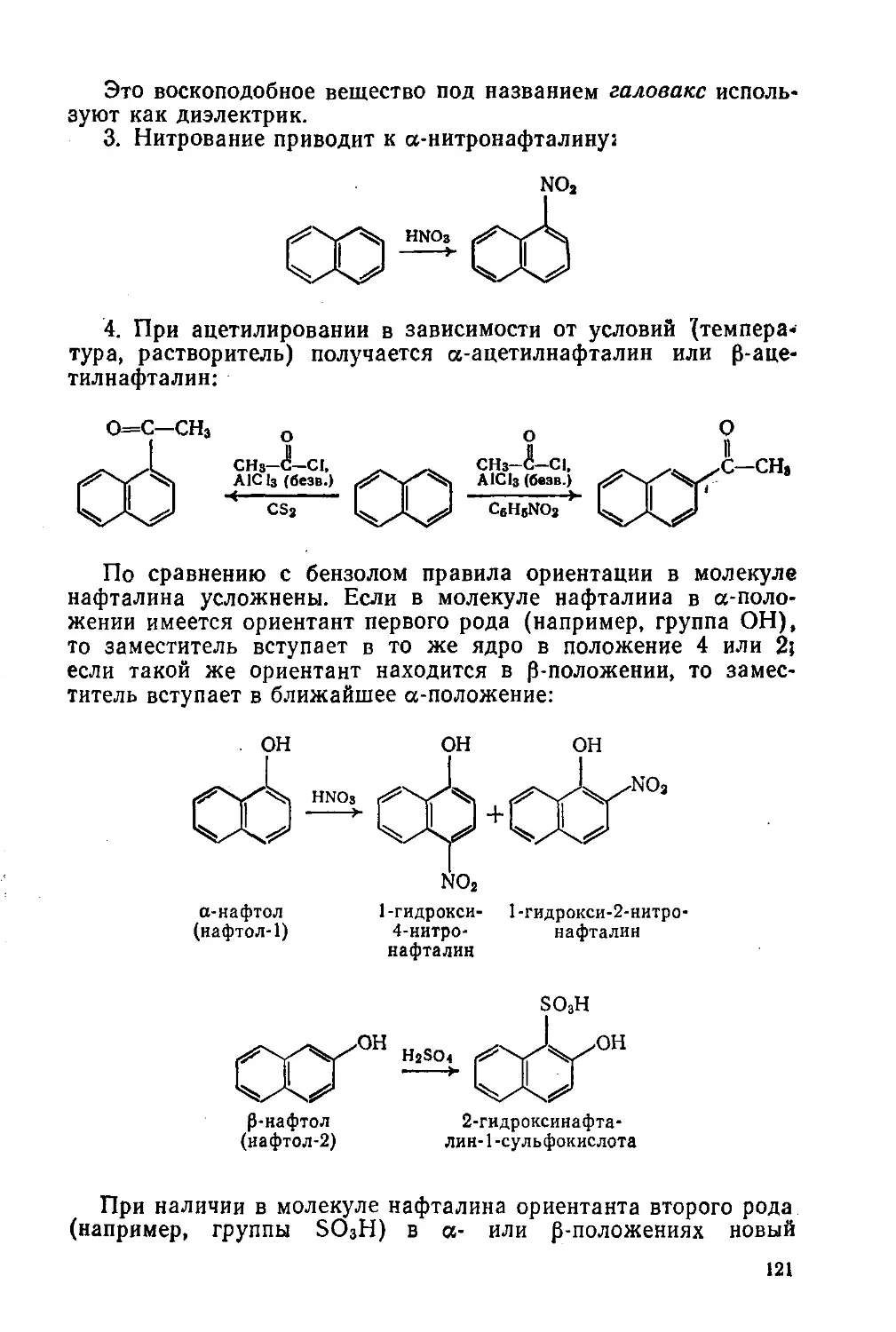
§ 54. Углеводороды со многими конденсированными ядрами 125
Небензоидные ароматические соединения 126
§ 55. Углеводороды — основа органической химии 127
Глава 8. Нефть 129
§ 56. Перегонка нефти 130
§ 57. Нефть как химическое сырье. Выделение углеводородов из нефти 131
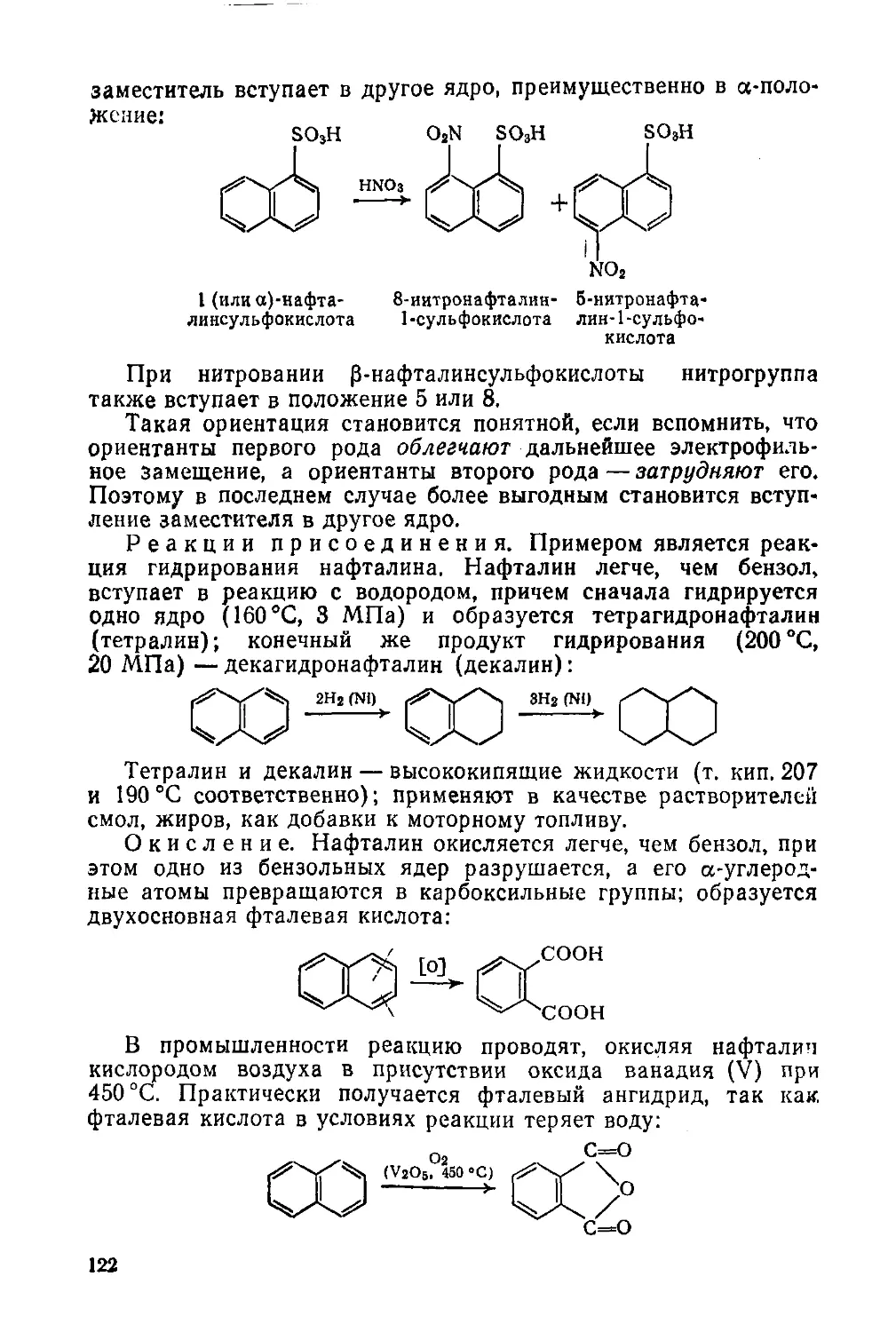
ЧАСТЬ II. СОЕДИНЕНИЯ С ОДНОРОДНЫМИ ФУНКЦИЯМИ 137
Глава 9. Галогенпроизводные 137
§ 58. Изомерия и номенклатура 137
§ 59. Способы получения 138
§ 60. Физические свойства 139
§ 61. Химические свойства 140
§ 62. Хлорорганические продукты в промышленности 145
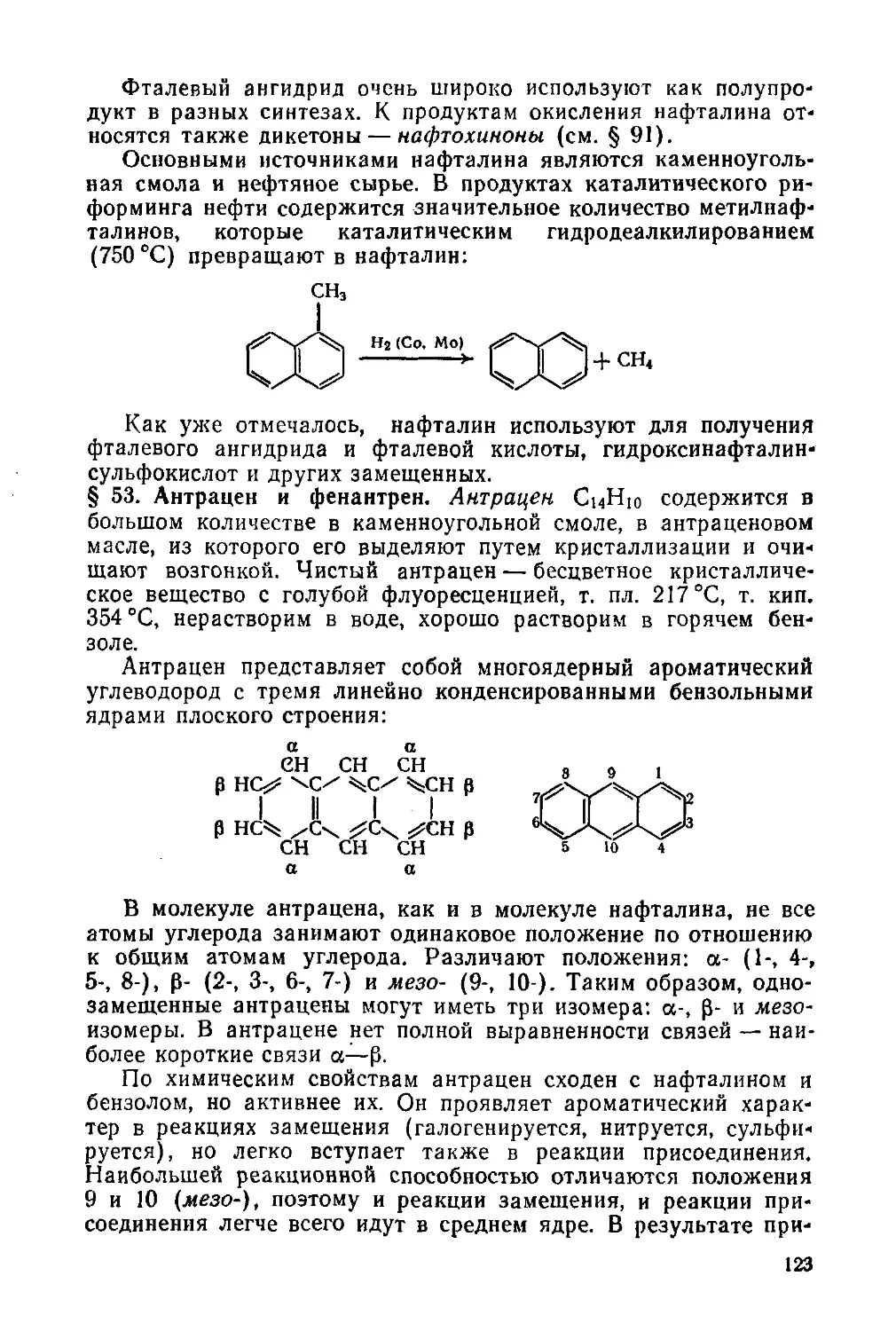
§ 63. Фторпроизводные 147
§ 64. Индуктивный эффект 149
Глава 10. Гидроксильные соединения и их производные 150
Одноатомные спирты 151
§ 65. Изомерия и номенклатура 151
§ 66. Общие способы получения 151
§ 67. Физические свойства. Ассоциация спиртов. Водородная связь 152
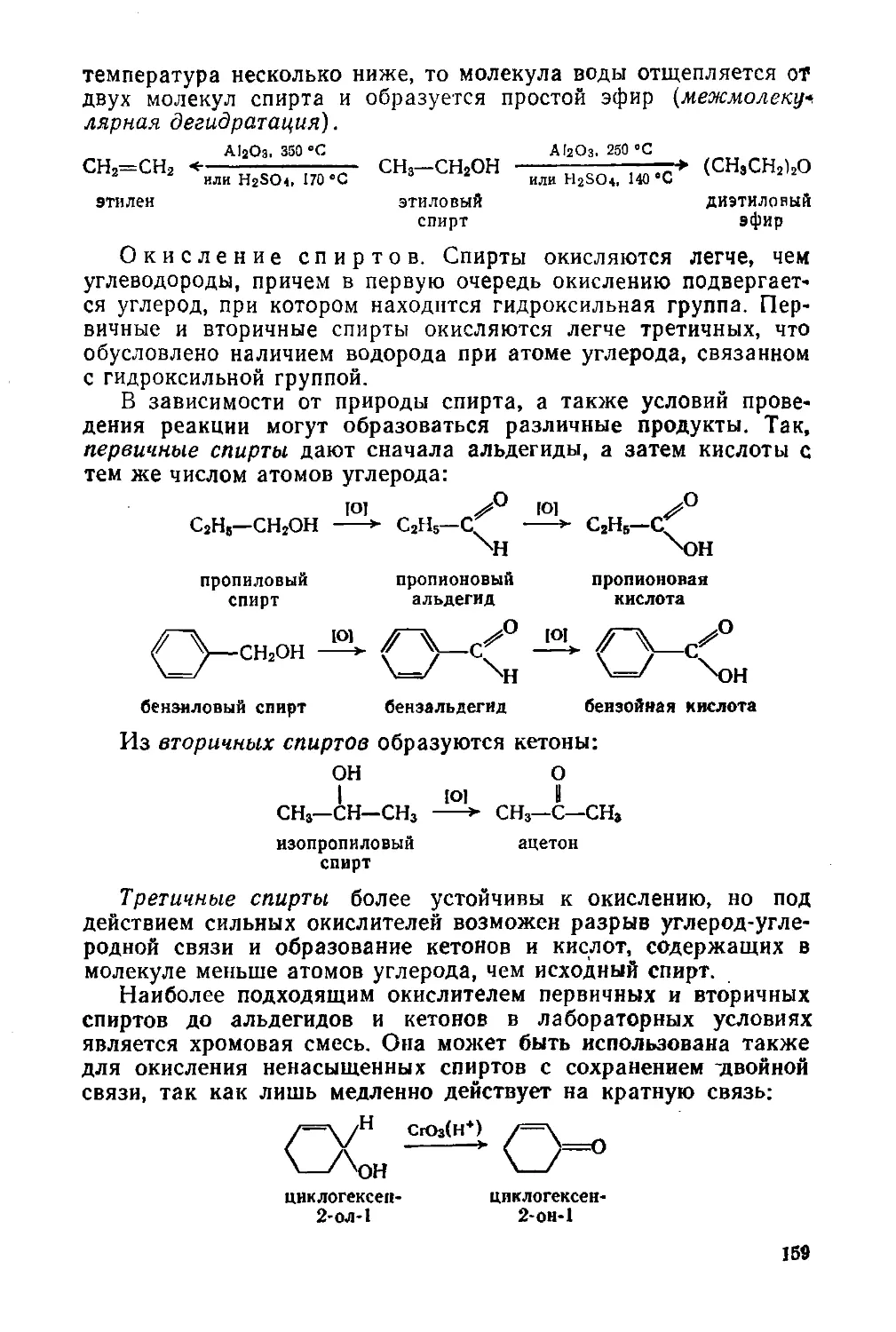
§ 68. Химические свойства 155
§ 69. Важнейшие представители 160
81
81
82
83
84
87
89
89
91
94
97
97
97
4
Многоатомные спирты 165
§ 70. Номенклатура, физические свойства, отдельные представители 165
Фенолы и нафтолы 168
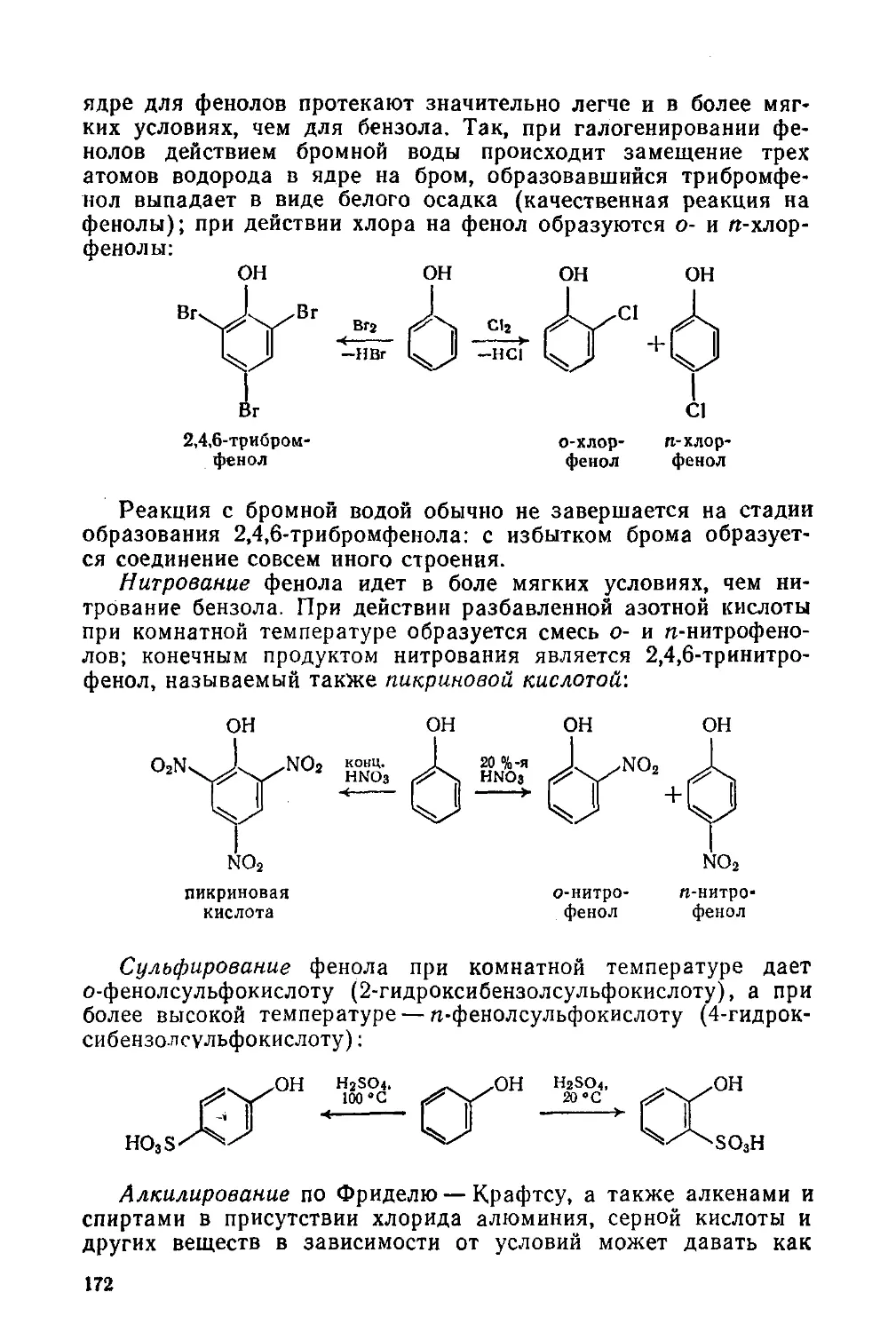
§ 71. Фенолы. Изомерия и физические свойства 168
§ 72. Промышленные способы получения фенолов 169
§ 73. Химические свойства фенолов 170
§ 74. Отдельные представители фенолов 173
§ 75. Нафтолы 176
Простые эфиры. Эпоксиды 176
§ 76. Простые эфиры. Номенклатура, изомерия, физические свойства 176
§ 77. Способы получения простых эфиров 177
§ 78. Химические свойства простых эфиров 178
§ 79. Наиболее важные представители простых эфиров 179
§ 80. Эпоксиды 181
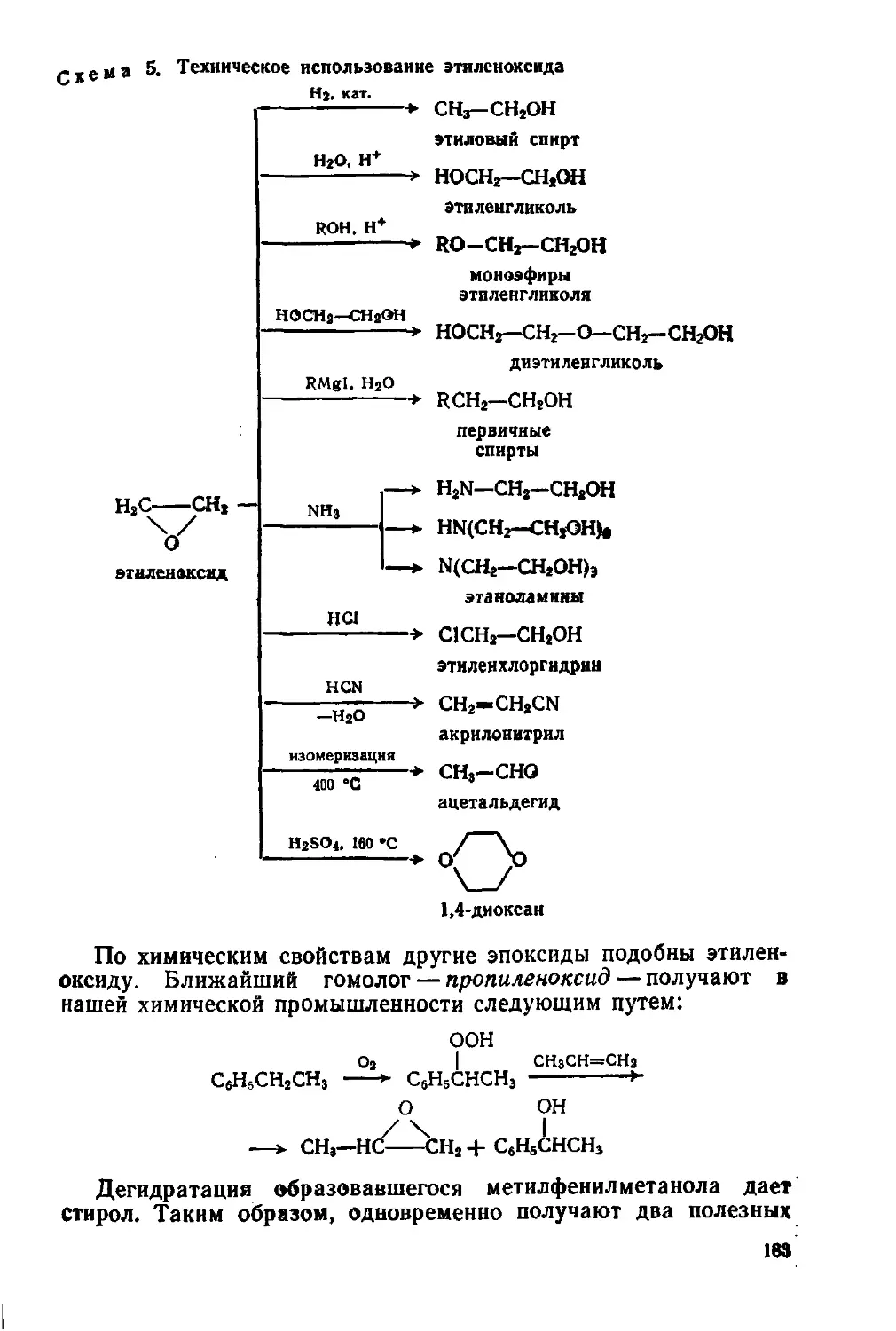
§ 81. Этиленоксид в химической промышленности 181
Органические пероксидные соединения 184
§ 82. Способы получения и химические свойства 184
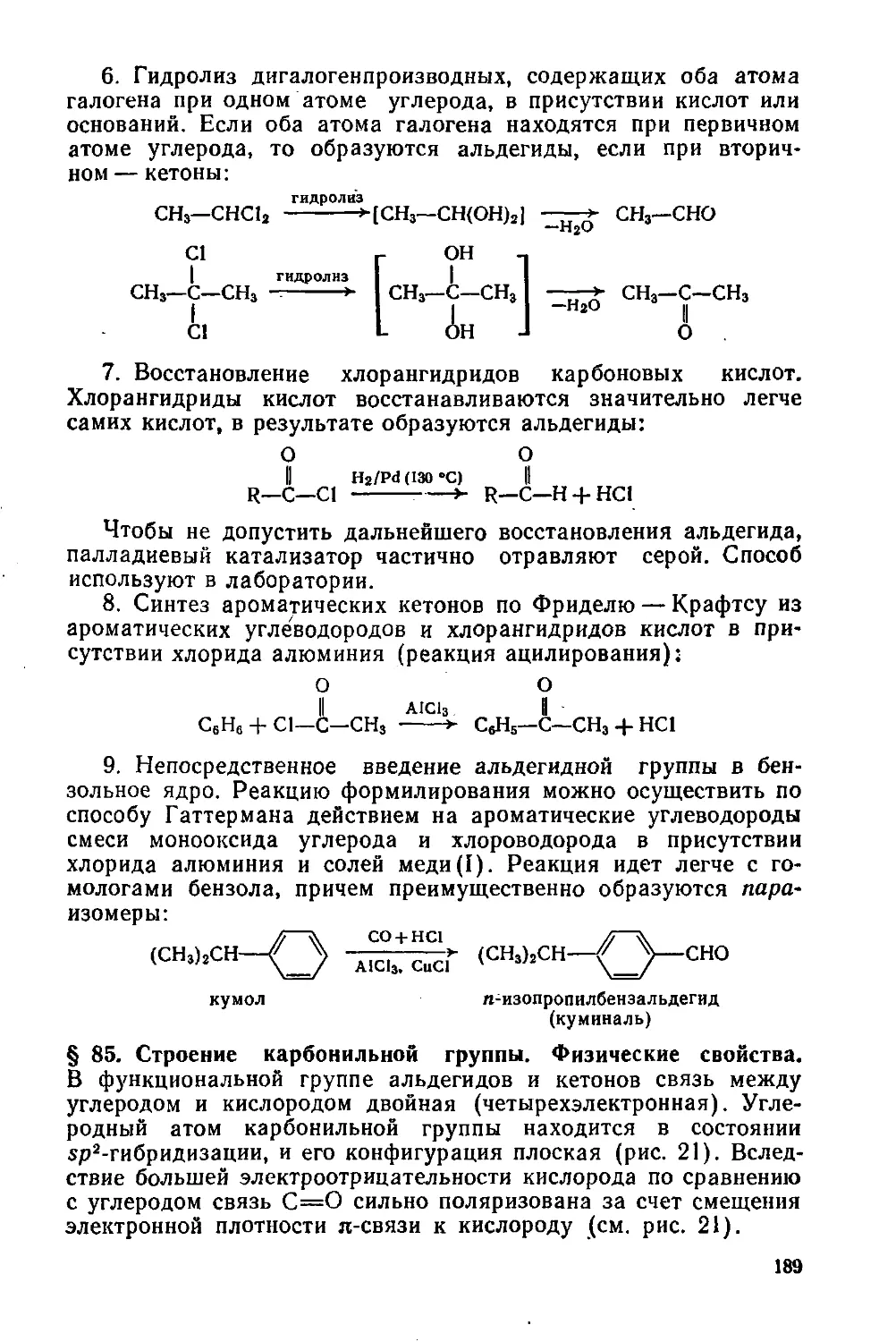
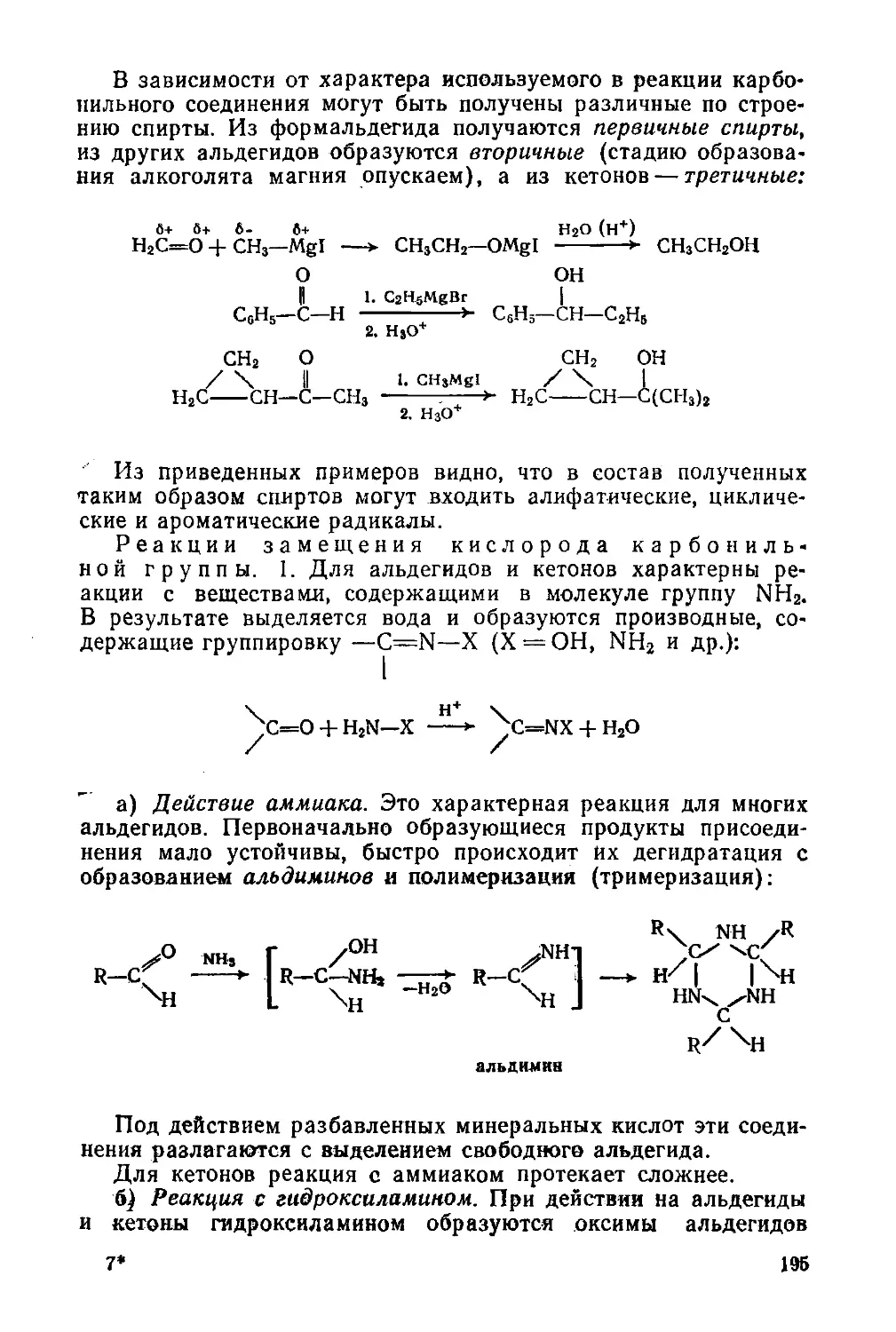
Глава 11. Альдегиды и кетоны 186
§ 83. Изомерия и номенклатура 186
§ 84. Способы получения 187
§ 85. Строение карбонильной группы. Физические свойства 189
§ 86. Химические свойства 190
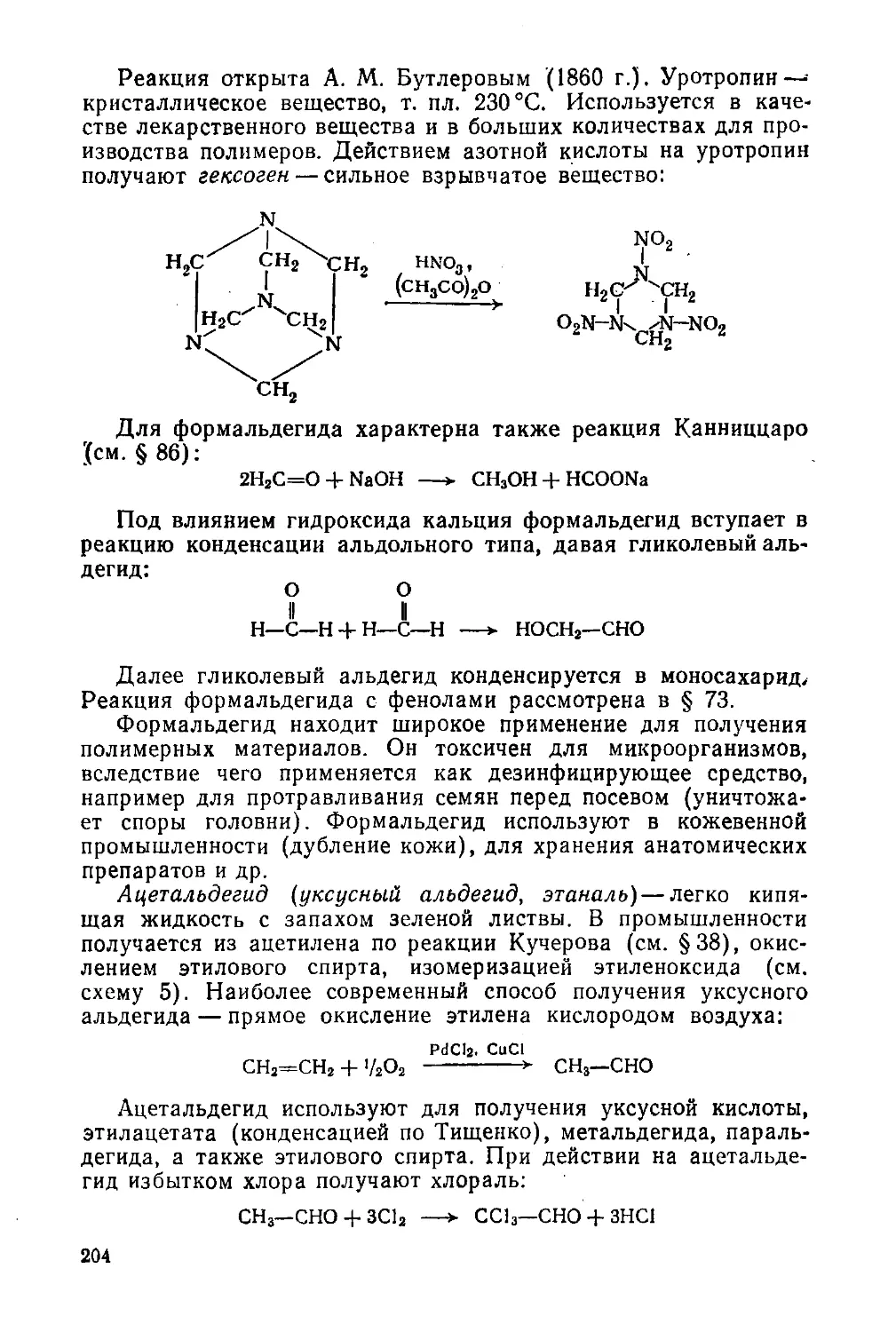
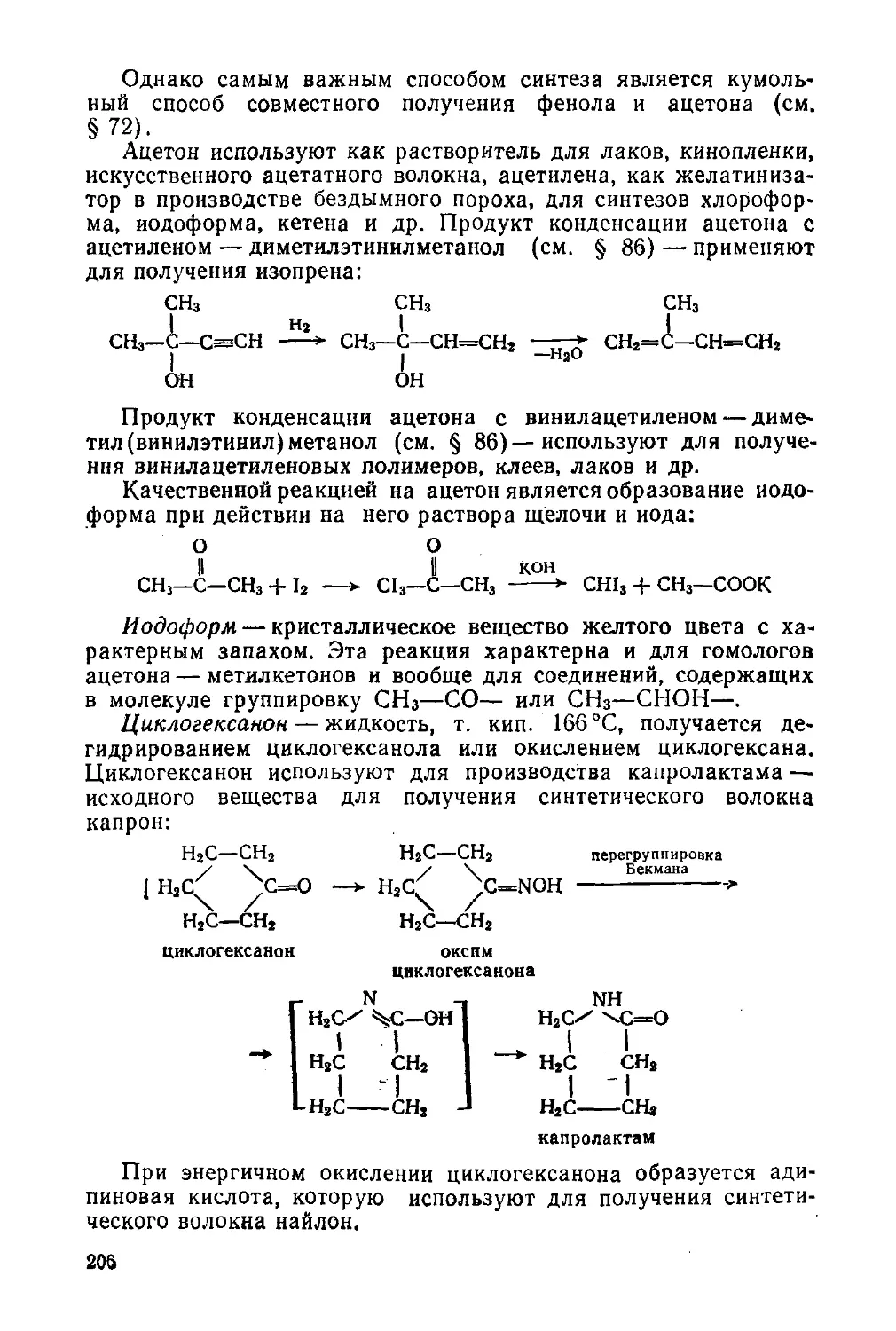
§ 87. Наиболее важные представители 203
Ненасыщенные карбонильные соединения 207
§ 88. Сопряженные или α,β -ненасыщенные альдегиды и кетоны 207
§ 89. Кетены 209
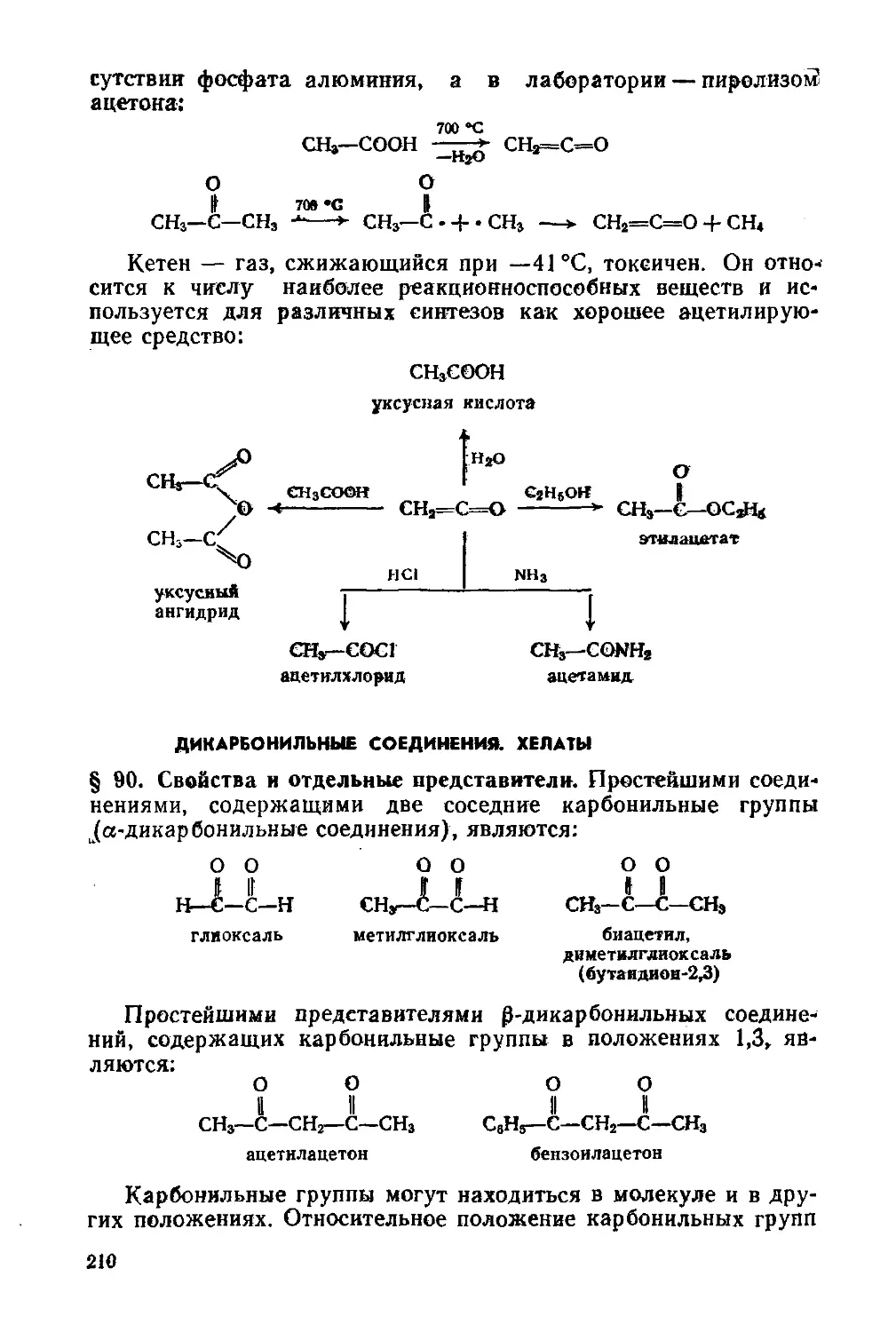
Дикарбонильные соединения. Хелаты 210
§ 90. Свойства и отдельные представители 210
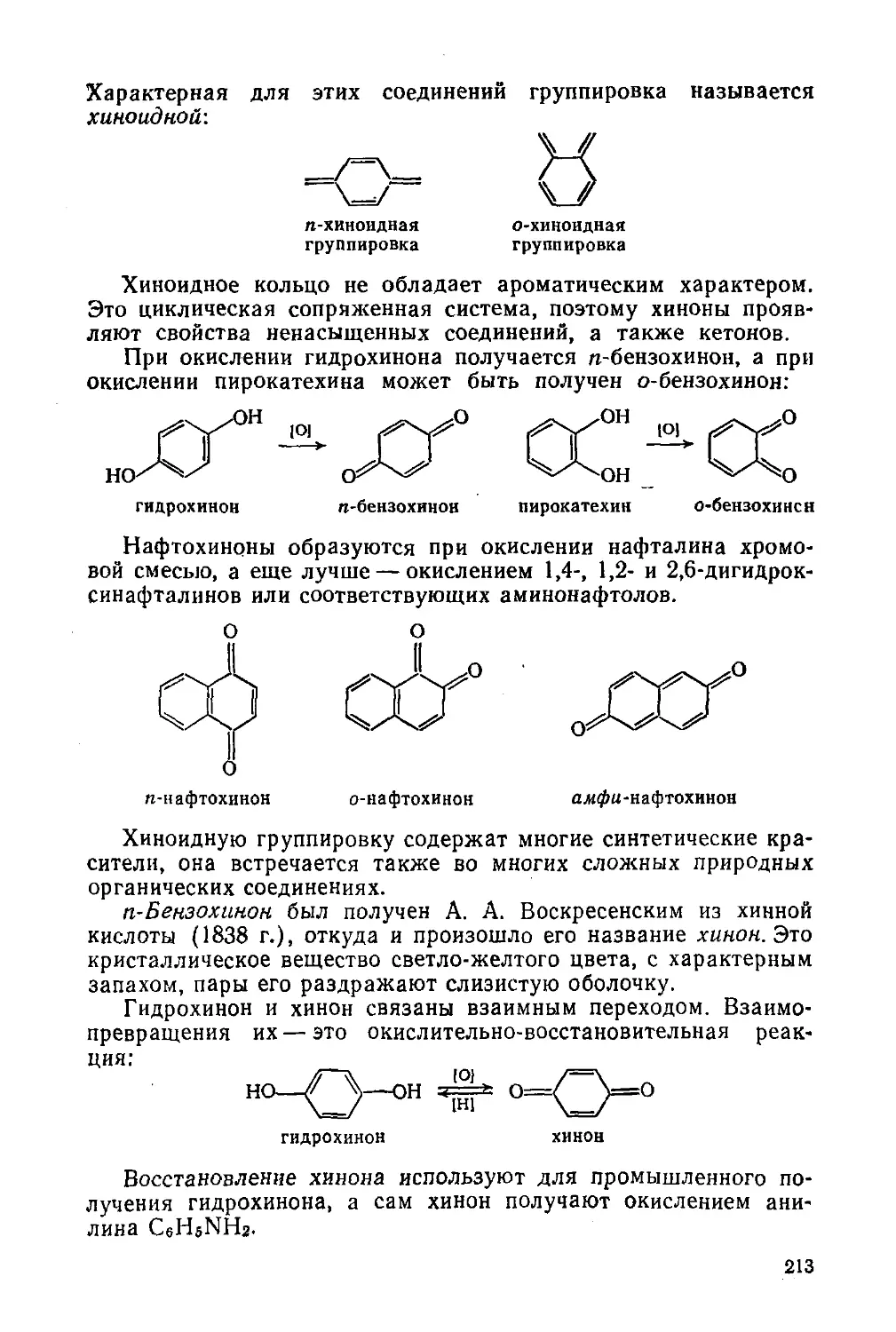
Хиноны 212
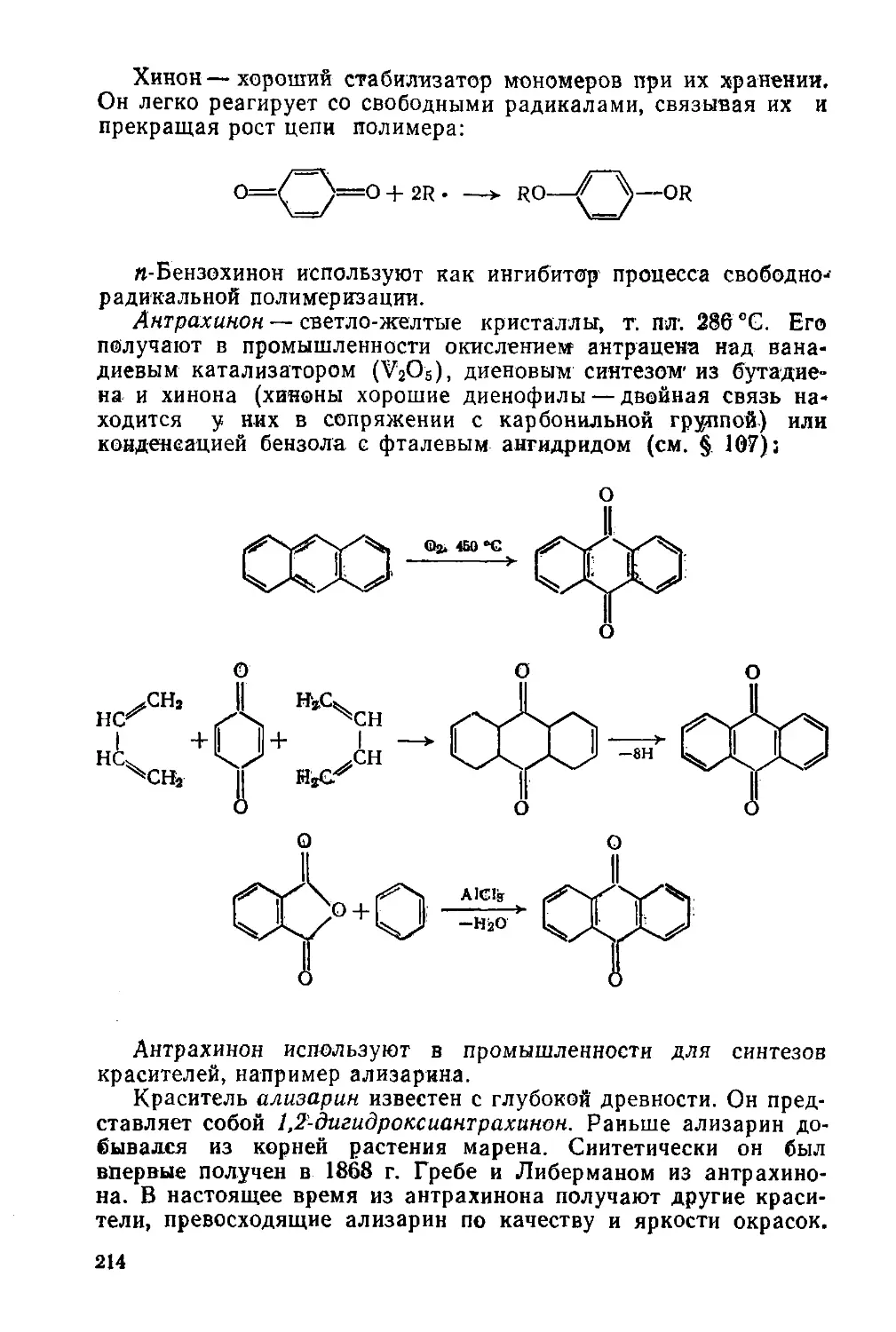
§ 91. Свойства и отдельные представители 212
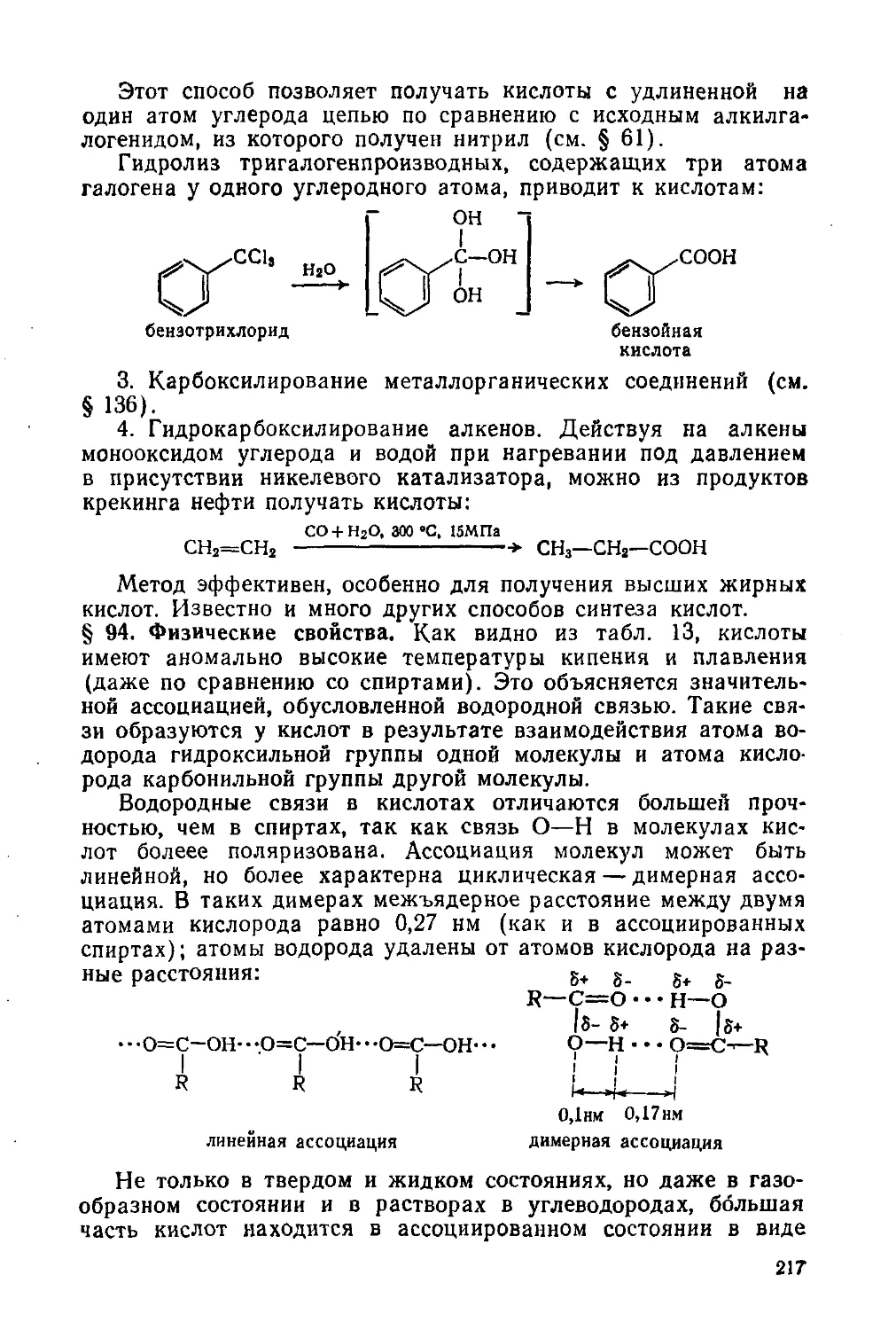
Глава 12. Карбоновые кислоты и их производные 215
Одноосновные кислоты 215
§ 92. Изомерия и номенклатура 215
§ 93. Общие способы получения 216
§ 94.Физические свойства 217
§ 95. Строение карбоксильной группы. Химические свойства 219
§ 96. Наиболее важные представители 223
§ 97. Мыла и моющие средства 226
Непредельные одноосновные кислоты 229
§ 98. Изомерия и способы получения 229
§ 99. Физические и химические свойства 230
§ 100. Отдельные представители 231
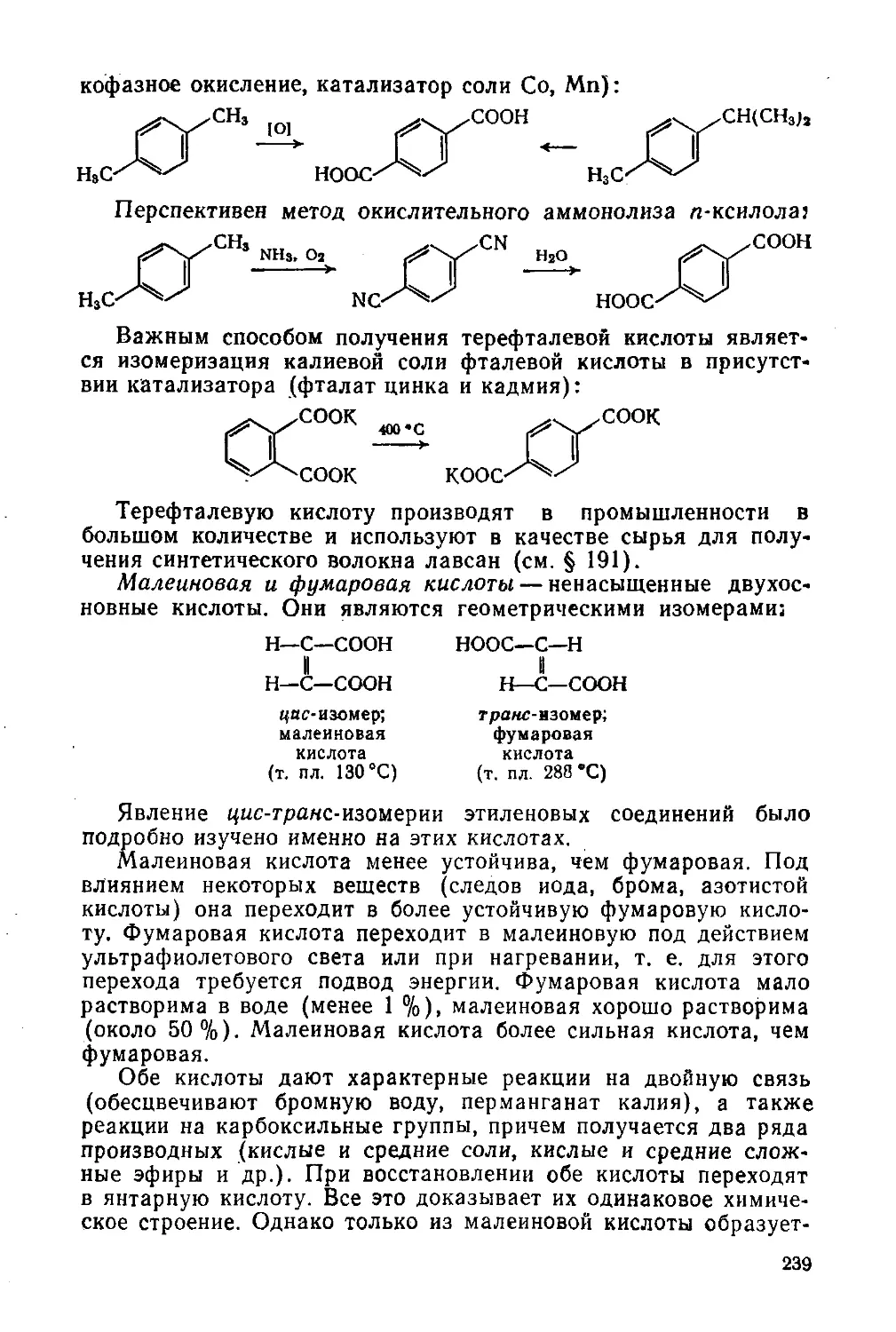
Двухосновные кислоты 233
§ 101. Номенклатура, общие способы получения, физические свойства 233
§ 102. Химические свойства 235
§ 103. Отдельные представители 233
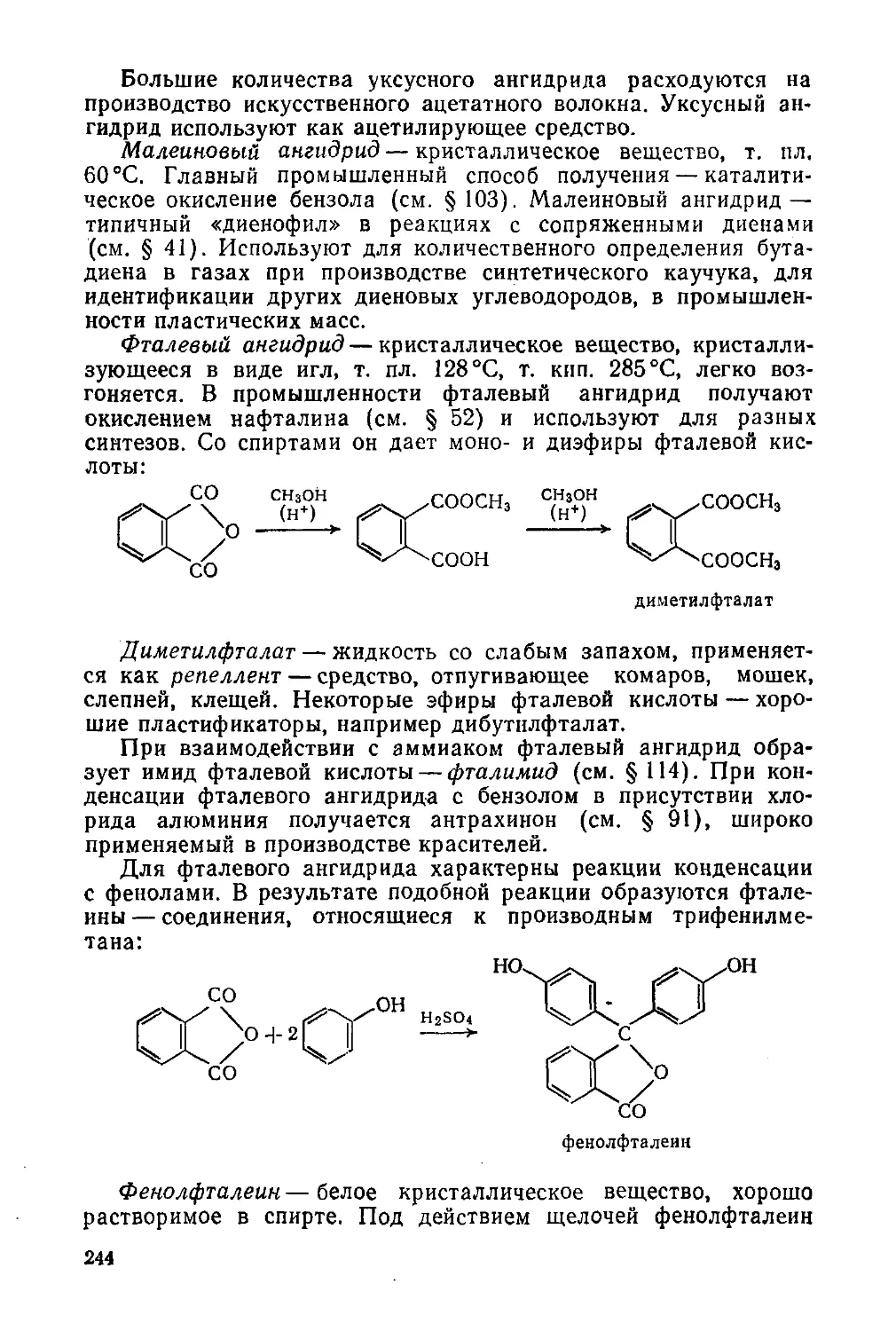
Галогенангидриды карбоновых кислот 240
§ 104. Номенклатура, способы получения и свойства 240
§ 105. Отдельные представители 242
Ангидриды карбоновых кислот 243
§ 106. Способы получения и свойства 243
§ 107. Отдельные представители 243
Пероксиды ацилов. Пероксикислоты 245
§ 108. Способы получения и отдельные представители 245
Сложные эфиры карбоновых кислот 246
§ 109. Изомерия, номенклатура, физические свойства 246
5
§ 110. Способы получения
§ 111. Химические свойства
§ 112. Воска
Жиры и жироподобные вещества
§ 113. Природные жиры и их техническая переработка
Амиды кислот
§ 114. Способы получения и свойства
§ 115. Отдельные представители
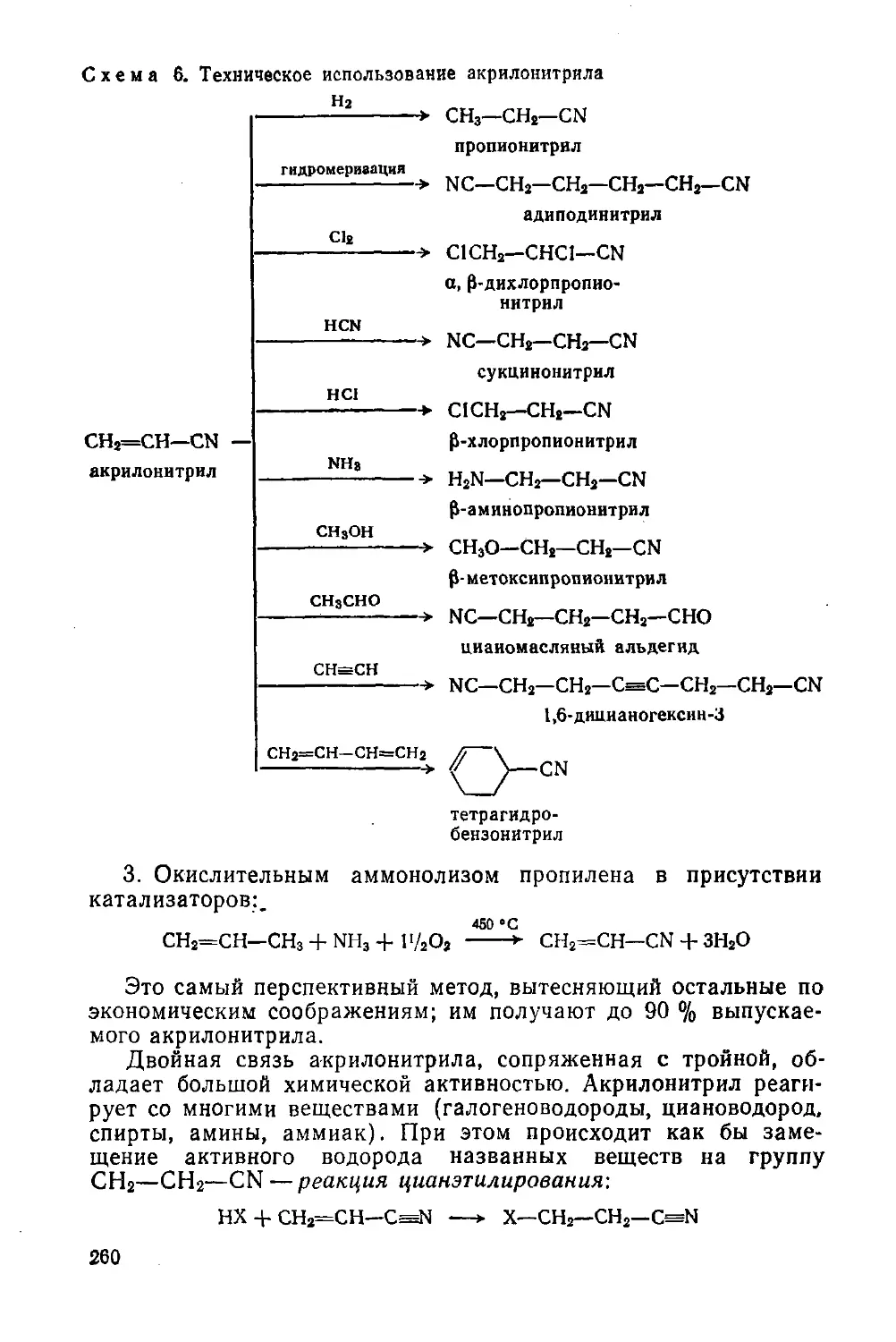
Нитрилы кислот
§ 116. Способы получения и свойства
§ 117. Акрилонитрил в химической промышленности
Глава 13. Органические соединения серы
§ 118. Тиолы и дисульфиды
§ 119. Тиоэфиры, сульфоксиды и сульфоны
§ 120. Сульфоновые кислоты и сульфонилхлориды
Глава 14. Нитросоединения
§ 121. Изомерия, номенклатура и строение
§ 122. Способы получения
§ 123. Физические и химические свойства
§ 124. Важнейшие представители
Глава 15. Амины
§ 125. Строение, изомерия, номенклатура, физические свойства
§ 126. Способы получения
§ 127. Амины — органические основания
§ 128. Химические свойства
§ 129. Важнейшие представители
§ 130. Роль электронных эффектов при электрофильном замещении в
ароматическом ядре
Глава 16. Диазосоединения
§ 131. Реакция диазотирования и строение диазосоединений
§ 132. Реакции диазосоединений с выделением азота
§ 133. Реакции диазосоединений без выделения азота
§ 134. Алифатические диазосоединения
Глава 17. Элементорганические соединения
§ 135. Общая характеристика
§ 136. Магнийорганические соединения
§ 137. Органические соединения щелочных металлов
§ 138. Алюминийорганические соединения
§ 139. Органические соединения переходных металлов
§ 140. Кремнийорганические соединения
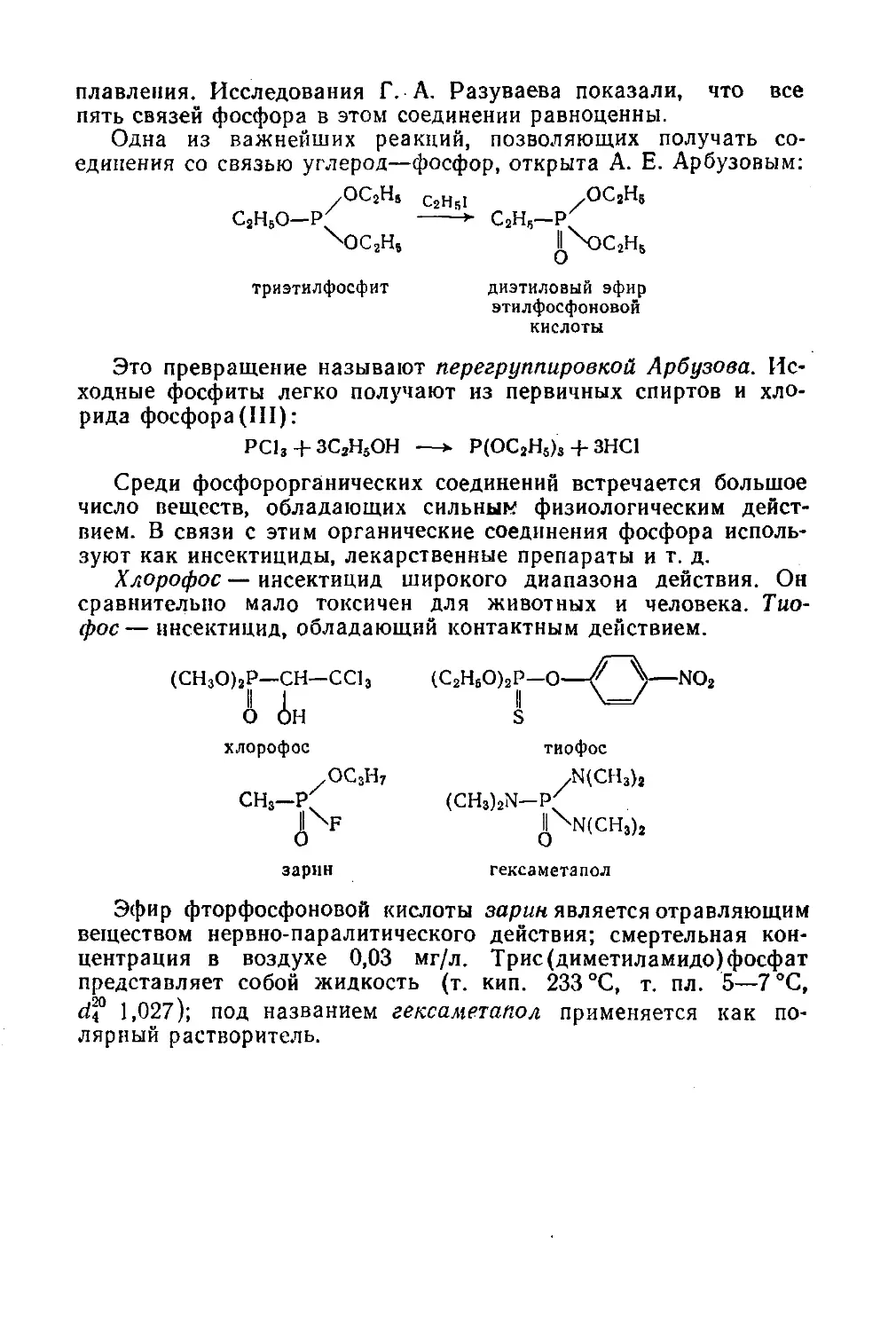
§ 141. Фосфорорганические соединения
ЧАСТЬ III. ГЕТЕРОФУНКЦИОНАЛЬНЫЕ СОЕДИНЕНИЯ
Глава 18. Галогензамещенные кислоты. Гидроксикислоты. Альдегидо- и
кетокислоты
§ 142. Галогензамещенные кислоты
Г идроксикислоты
§ 143. Способы получения, физические и химические свойства
247
248
249
250
250
254
254
256
258
258
259
261
262
263
264
267
267
269
270
275
276
276
278
280
281
284
288
292
292
293
295
298
300
300
300
303
304
305
305
307
310
310
310
313
313
6
§ 144. Оптическая изомерия 316
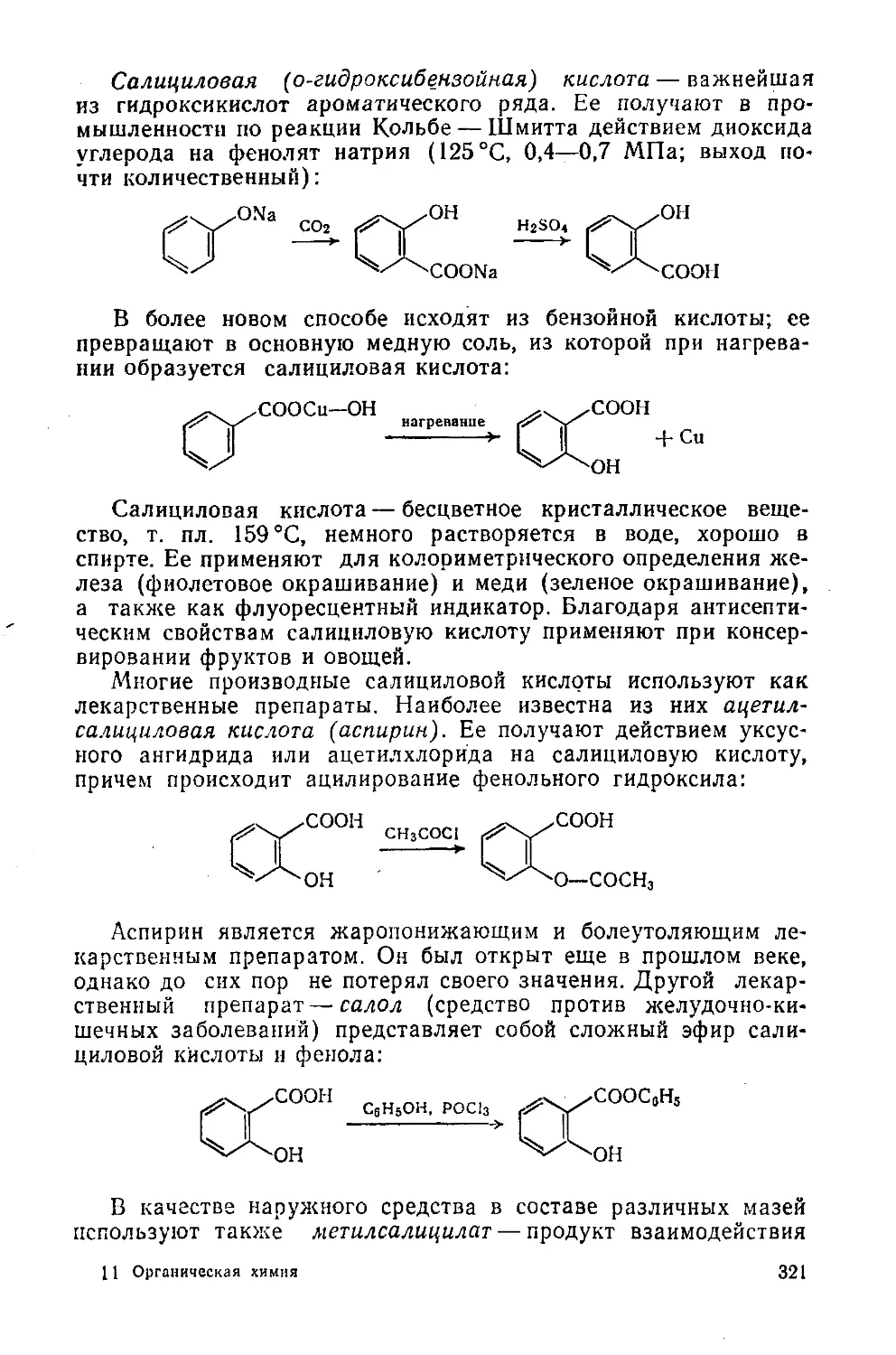
§ 145. Важнейшие представители 320
Полигидроксибензойные кислоты. Дубильные вещества 322
§ 146. Отдельные представители 322
Альдегидо- и кетокислоты 323
§ 147. Номенклатура и способы получения 323
§ 148. Представители кетокислот 324
§ 149. Сложноэфирная конденсация 325
§ 150. Таутомерия ацетоуксусного эфира 326
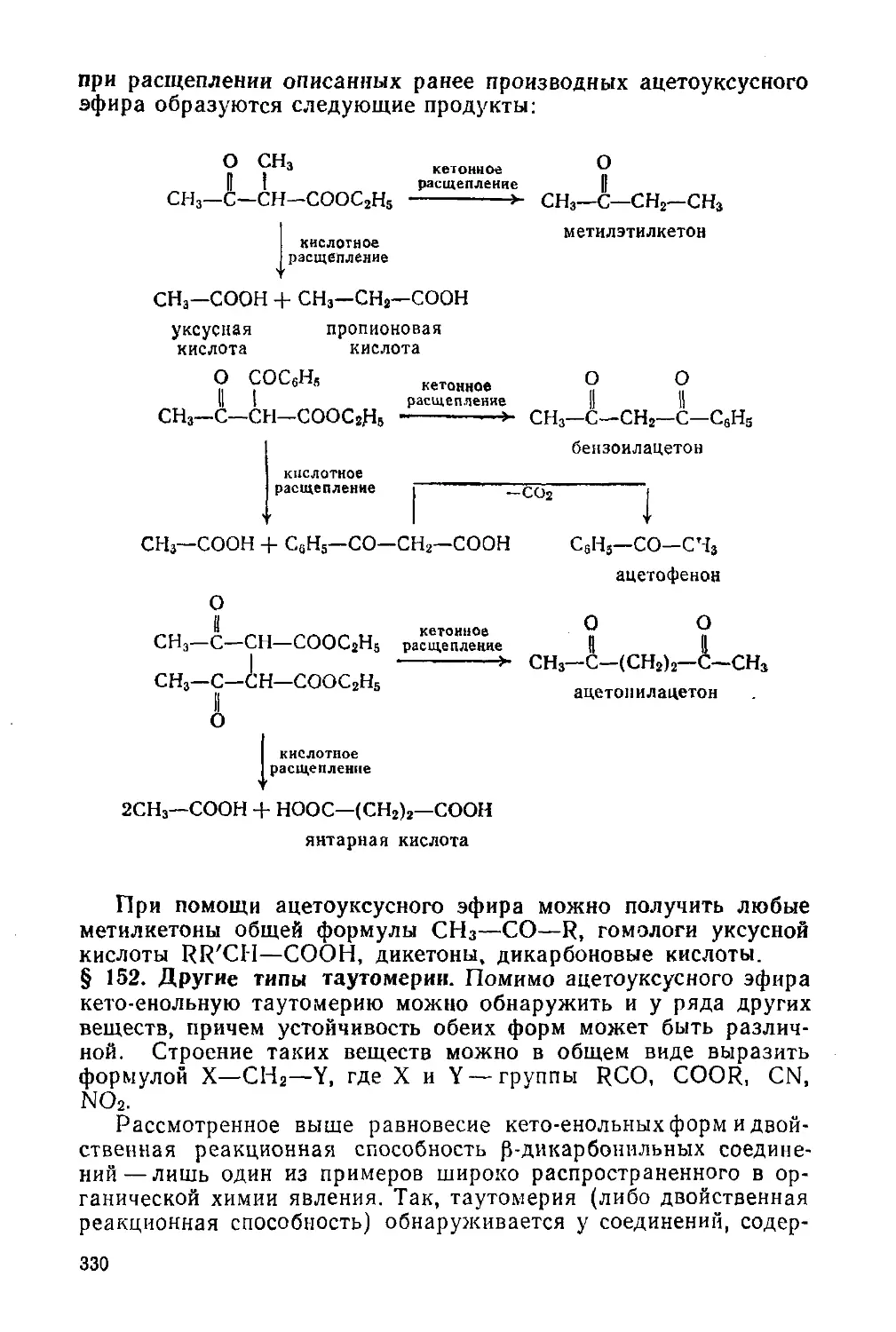
§ 151. Синтезы на основе ацетоуксусного эфира 329
§ 152. Другие типы таутомерии 330
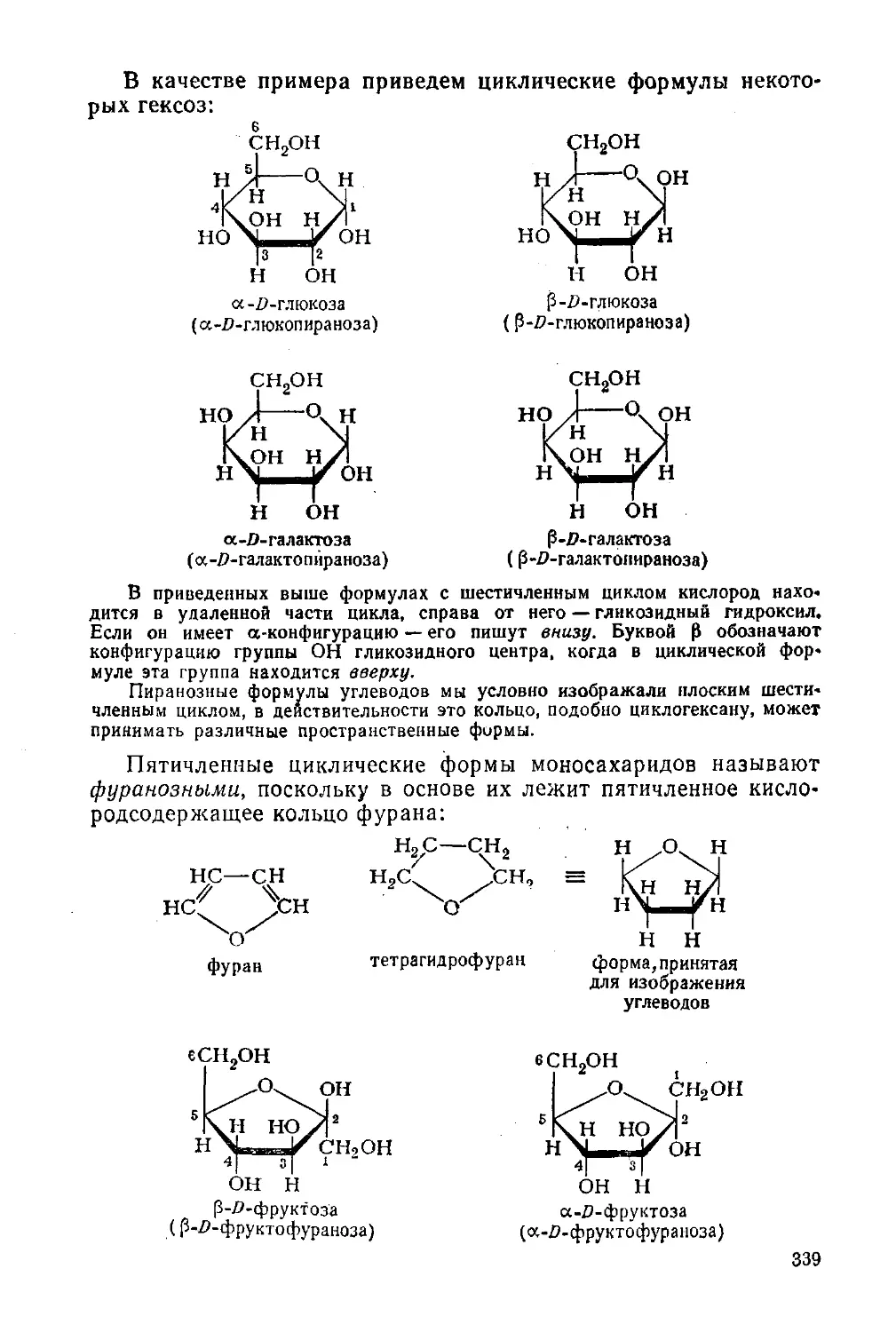
Глава 19. Углеводы 331
§ 153. Классификация, физические свойства, нахождение в природе 331
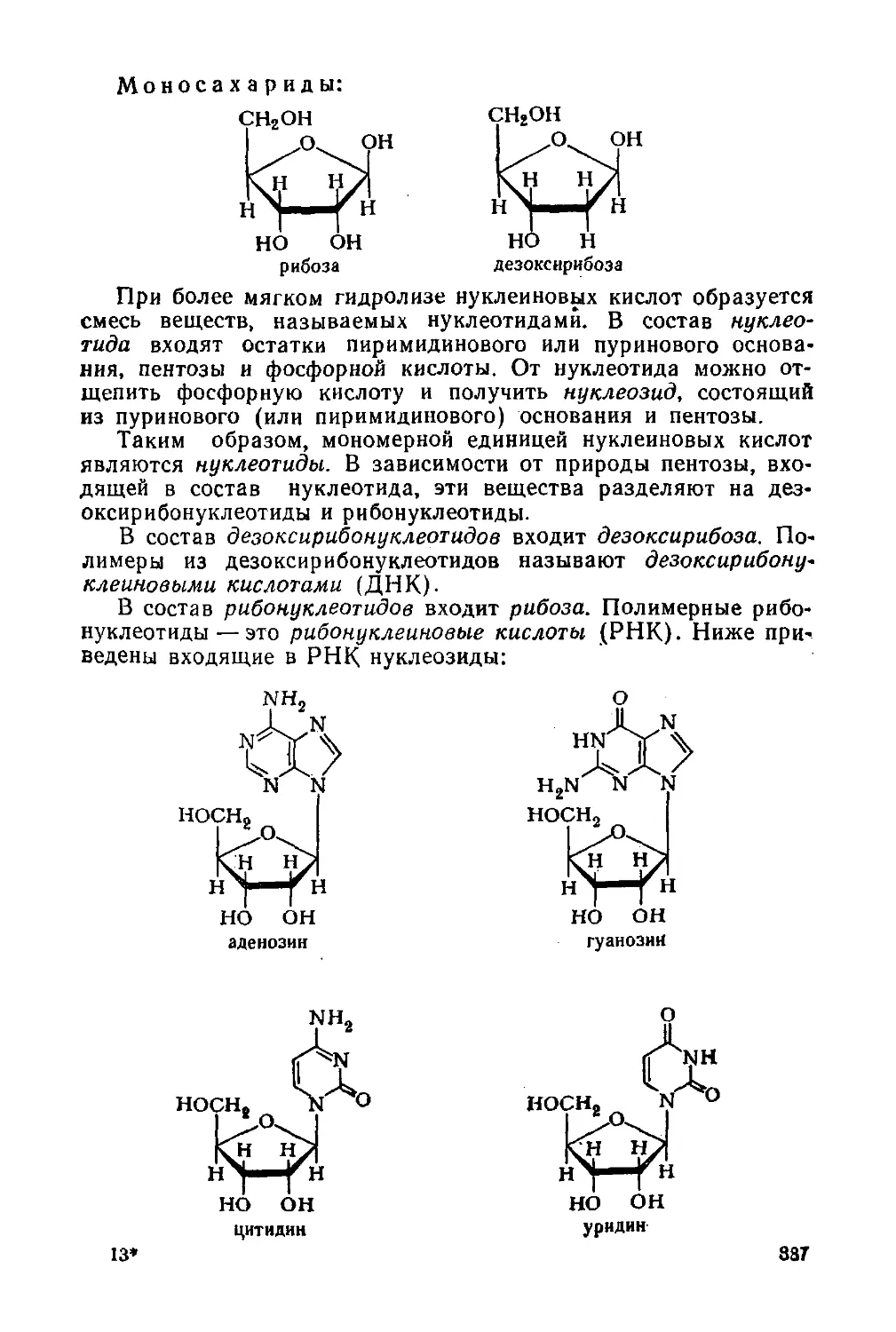
Моносахариды 334
§ 154. Строение 334
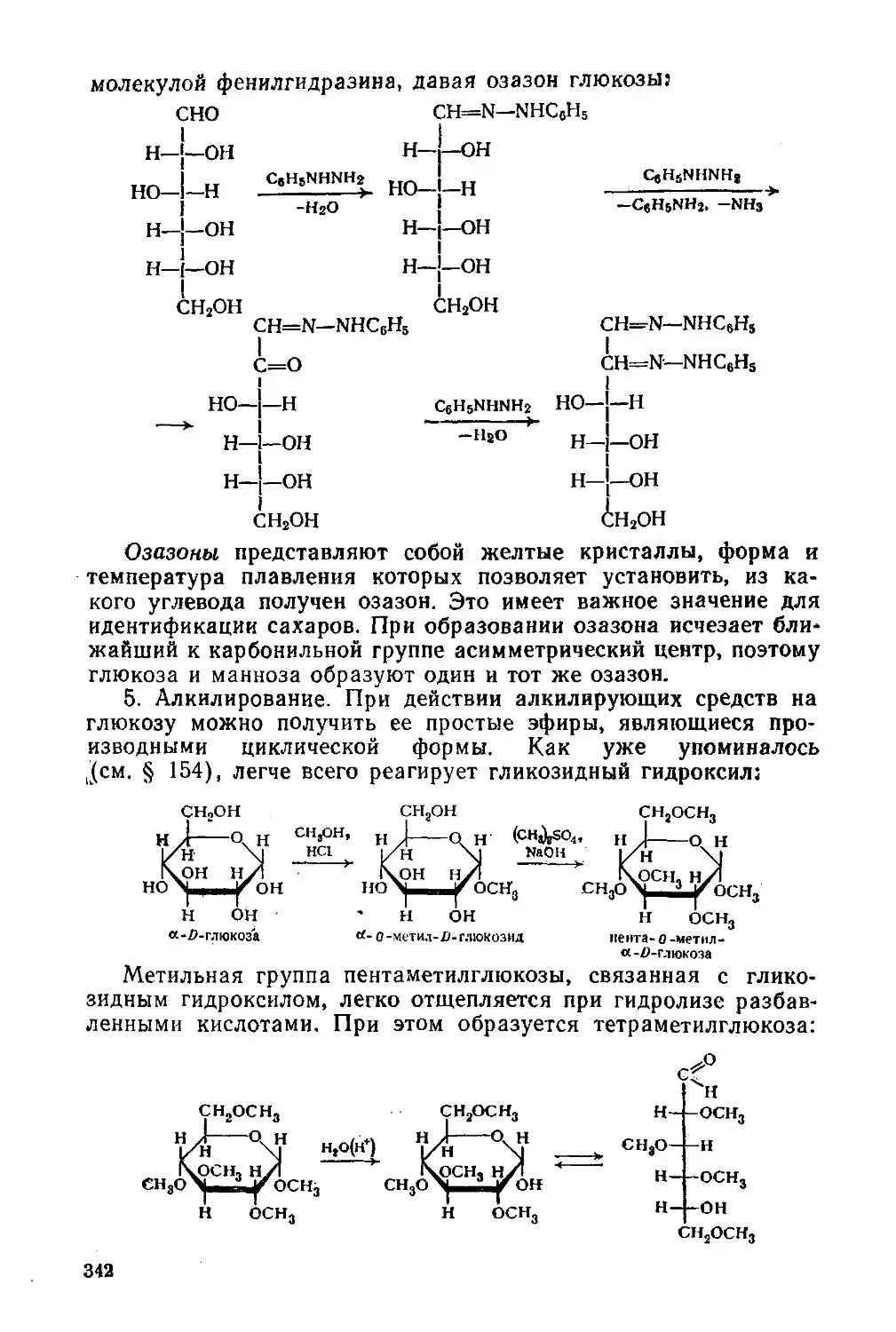
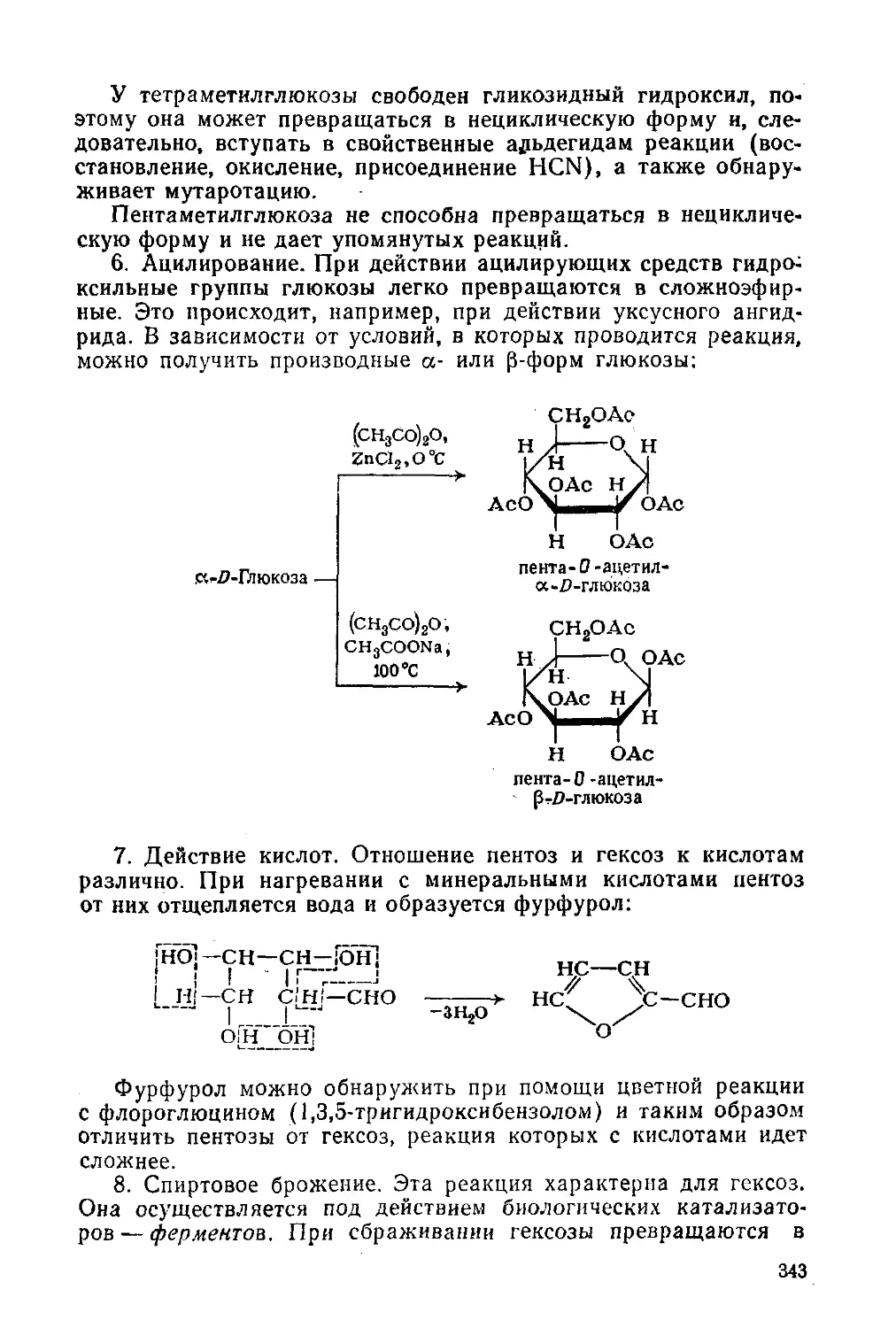
§ 155. Химические свойства 340
§ 156. Стереохимия моносахаридов 344
Олигосахариды 345
§ 157. Строение и свойства 345
Полисахариды 347
§ 158. Крахмал 347
§ 159. Целлюлоза 349
Глава 20. Аминоспирты и аминокислоты 353
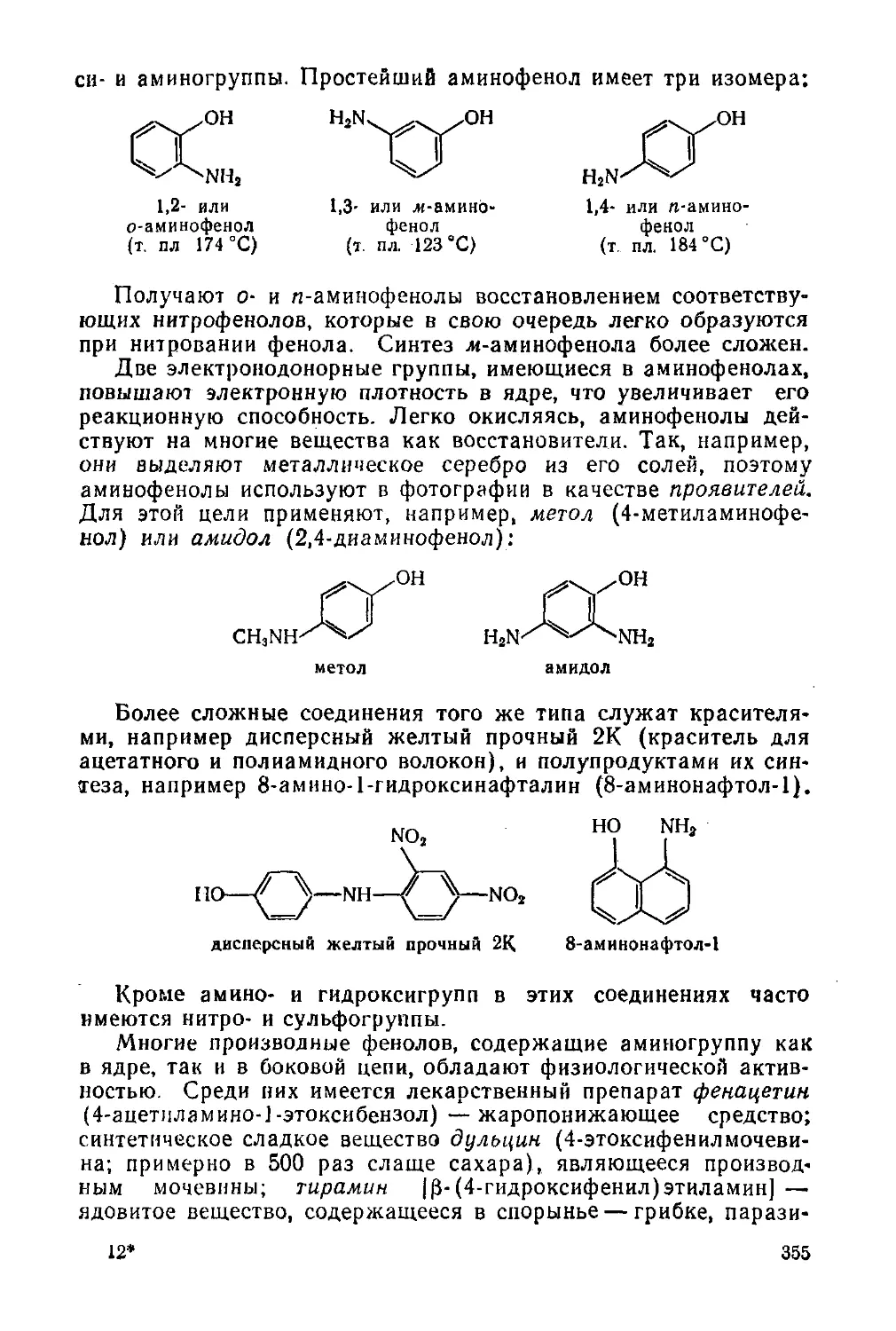
Аминоспирты и аминофенолы 353
§ 160. Аминоспирты 353
§ 161. Аминофенолы 354
Аминокислоты 356
§ 162. Физические свойства и способы получения 356
§ 163. Химические свойства 358
§ 164. Отдельные представители 359
ЧАСТЬ IV. ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ 361
Глава 21. Шестичленные и пятичленные гетероциклы, Алкалоиды 361
§ 165. Классификация и общая характеристика 361
Шестичленные гетероциклы с одним гетероатомом 362
§ 166. Пиридин и его производные 362
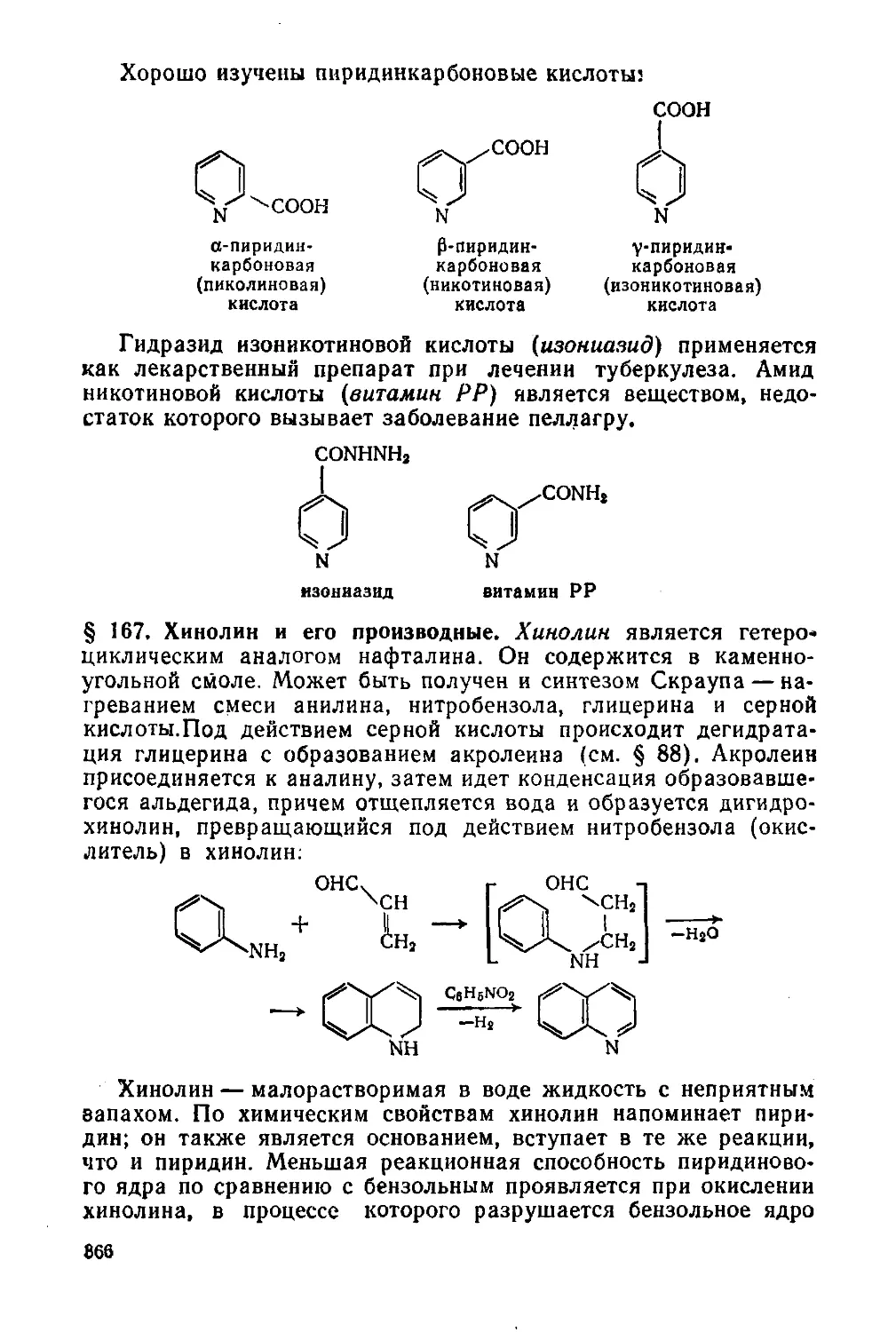
§ 167. Хинолин и его производные 366
Пятичленные гетероциклы с одним гетероатомом 368
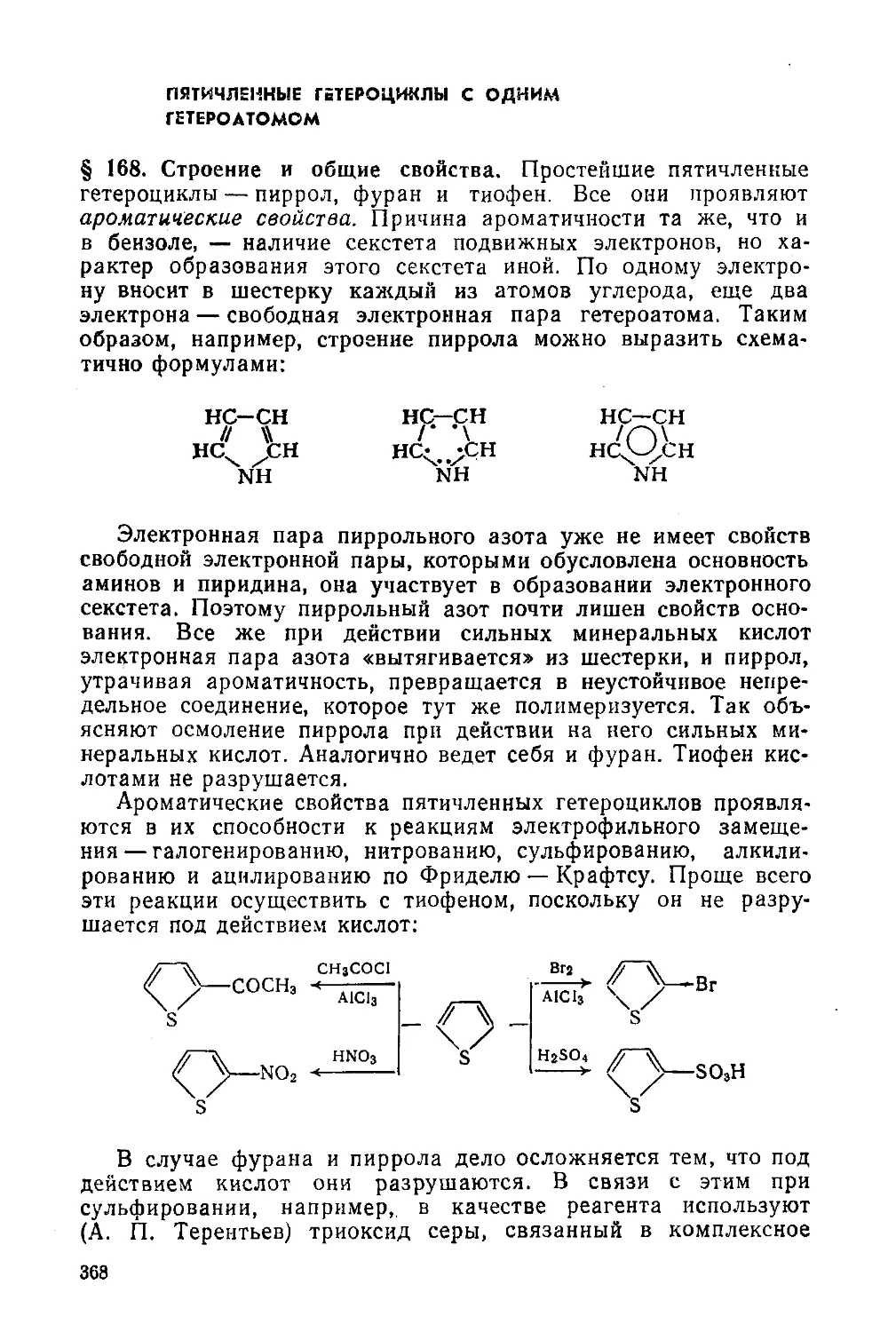
§ 168. Строение и общие свойства 368
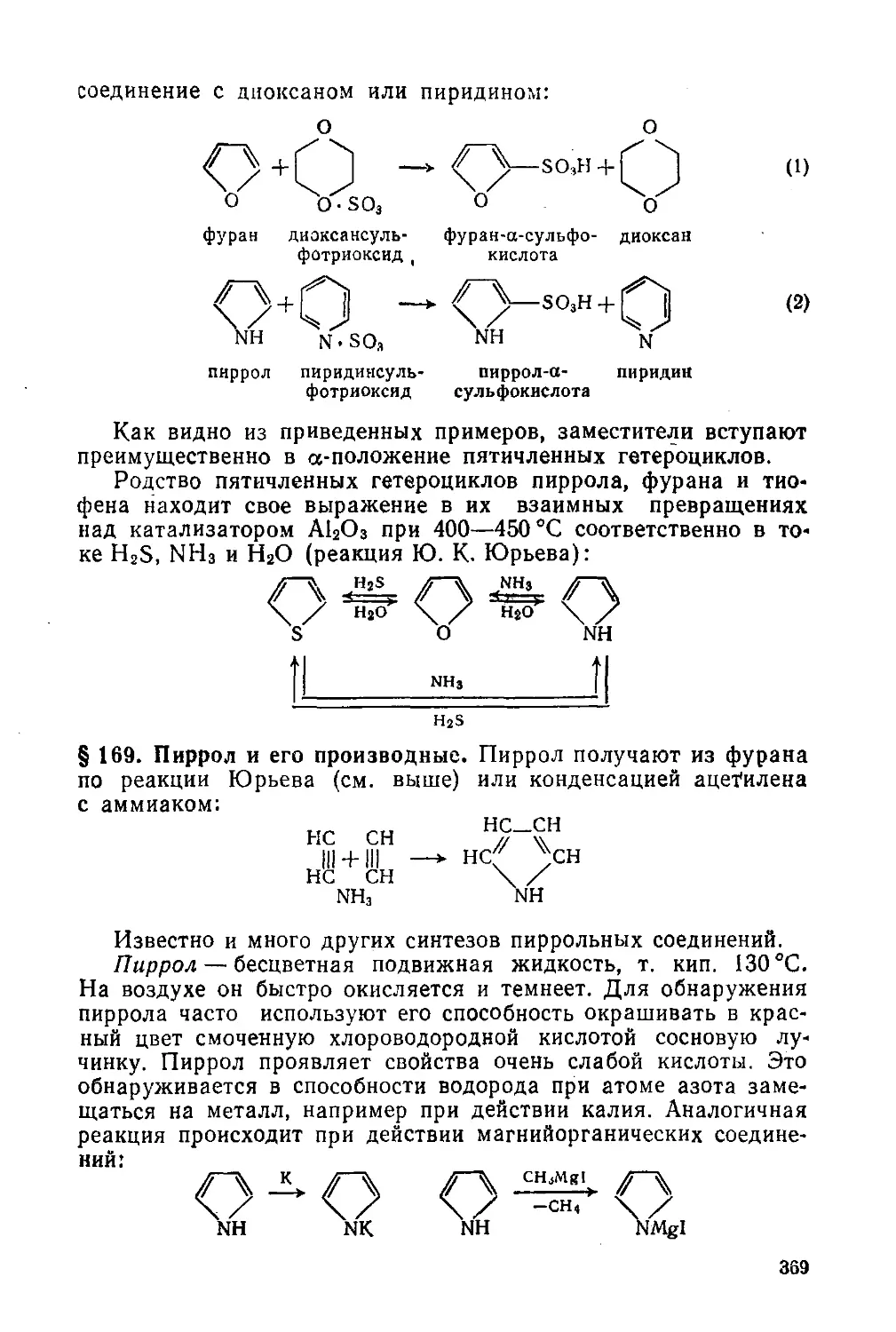
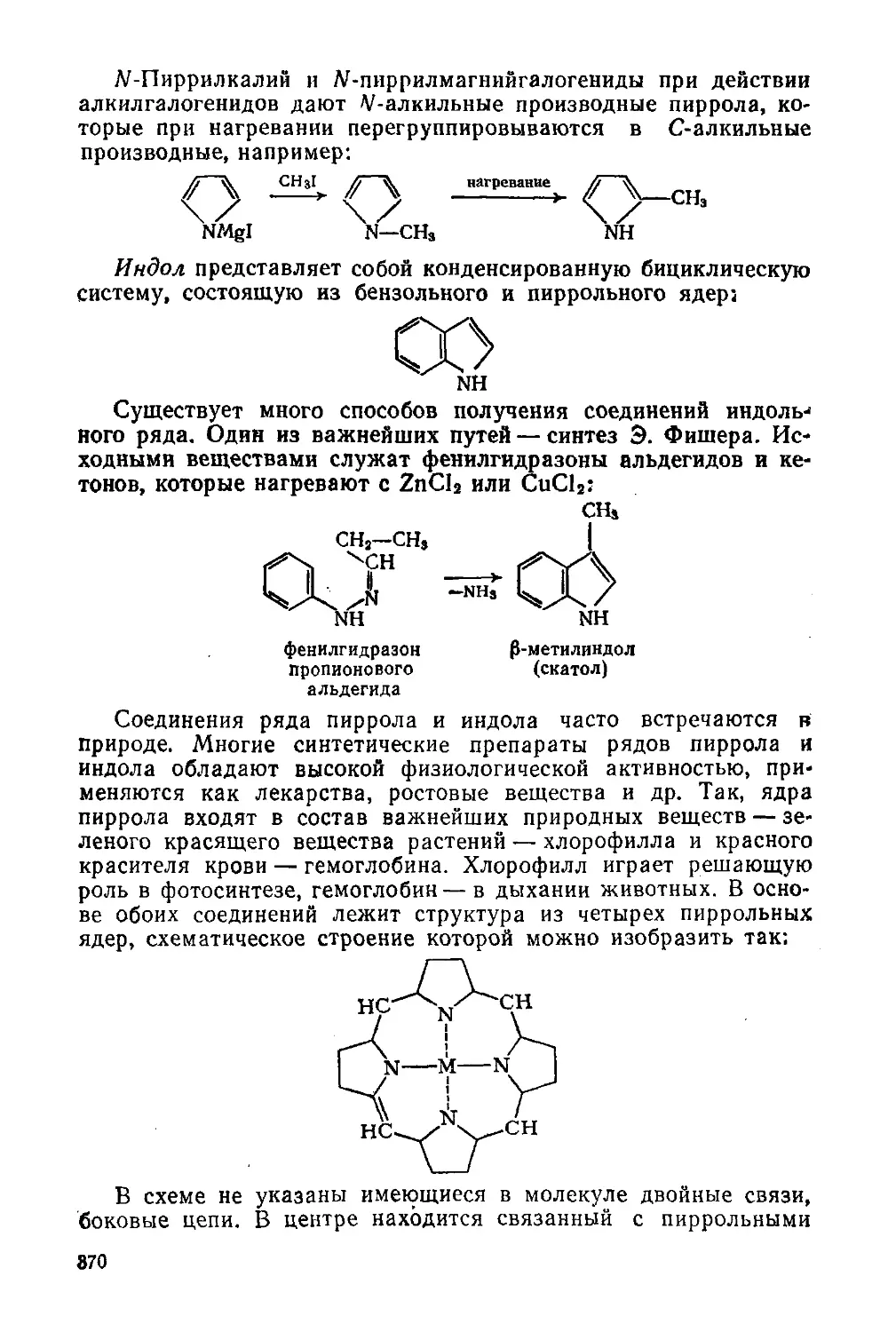
§ 169. Пиррол и его производные 369
§ 170. Фуран и его производные 372
§ 171. Тиофен и его производные 374
Сложные гетероциклы и алкалоиды 374
§ 172. Гетероциклы с несколькими гетероатомами 374
§ 173. Алкалоиды 375
ЧАСТЬ V. СПЕЦИАЛЬНЫЕ ГЛАВЫ ОРГАНИЧЕСКОЙ ХИМИИ 377
Глава 22. Элементы биоорганической химии 377
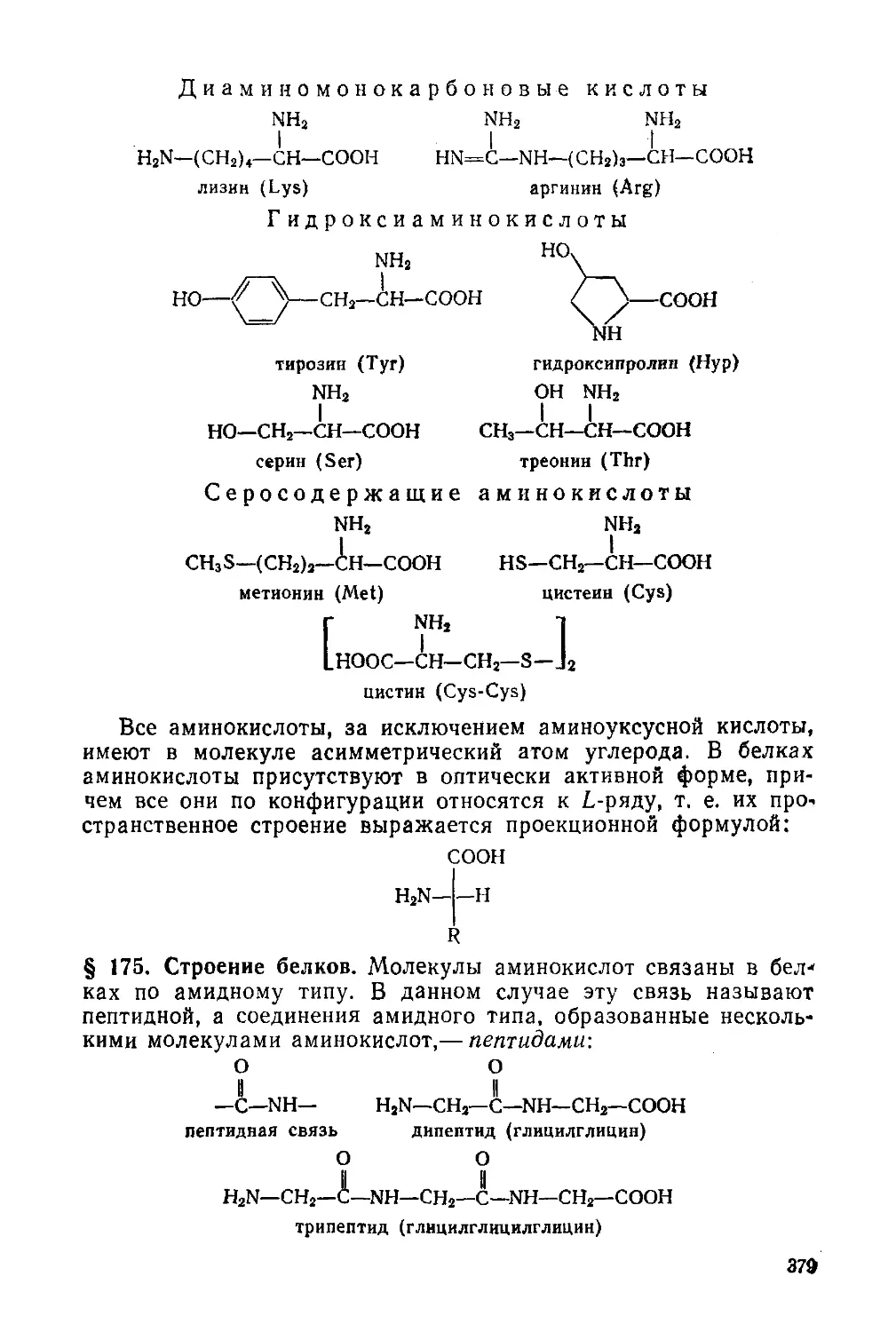
§ 174. Белки. Аминокислотный состав 377
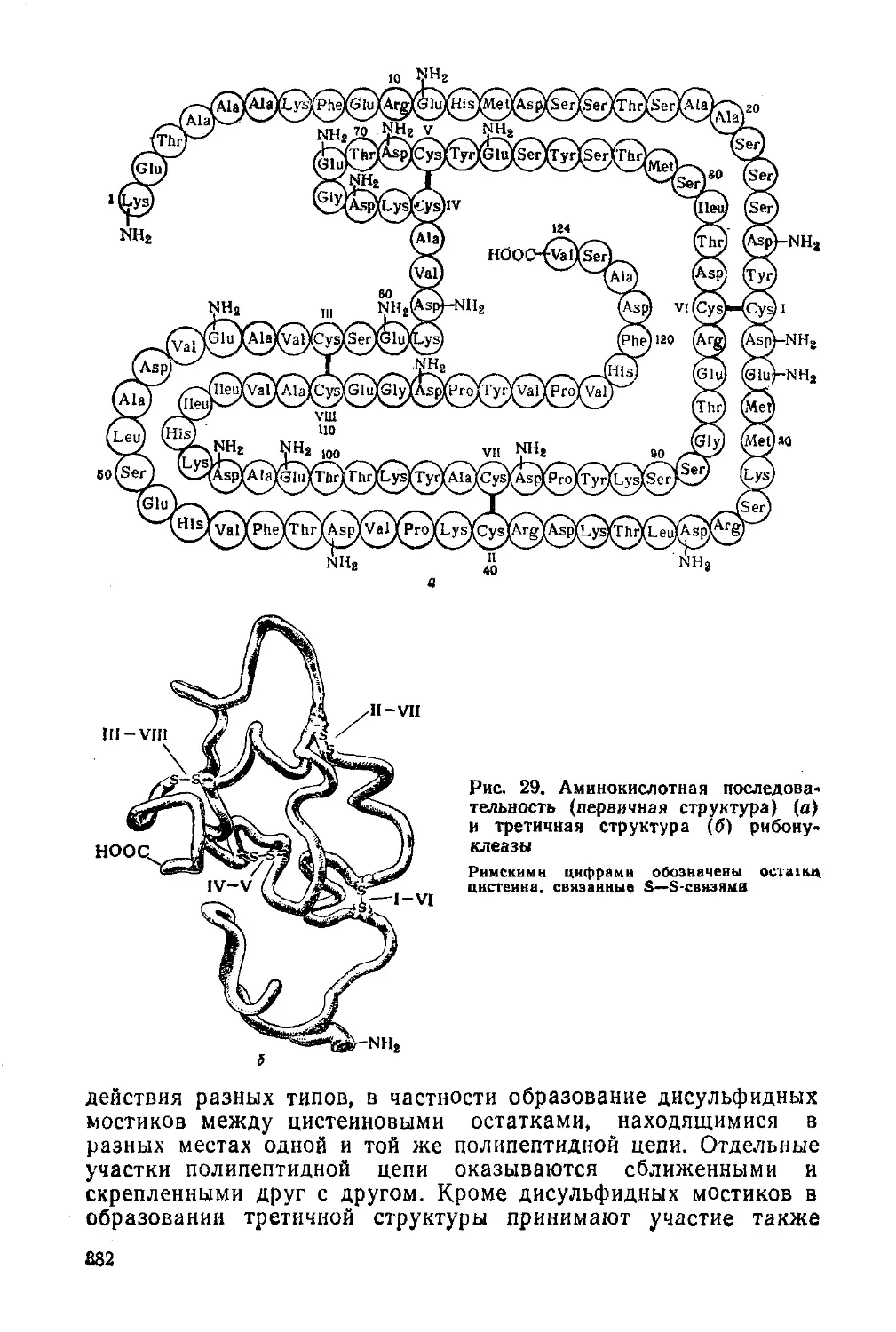
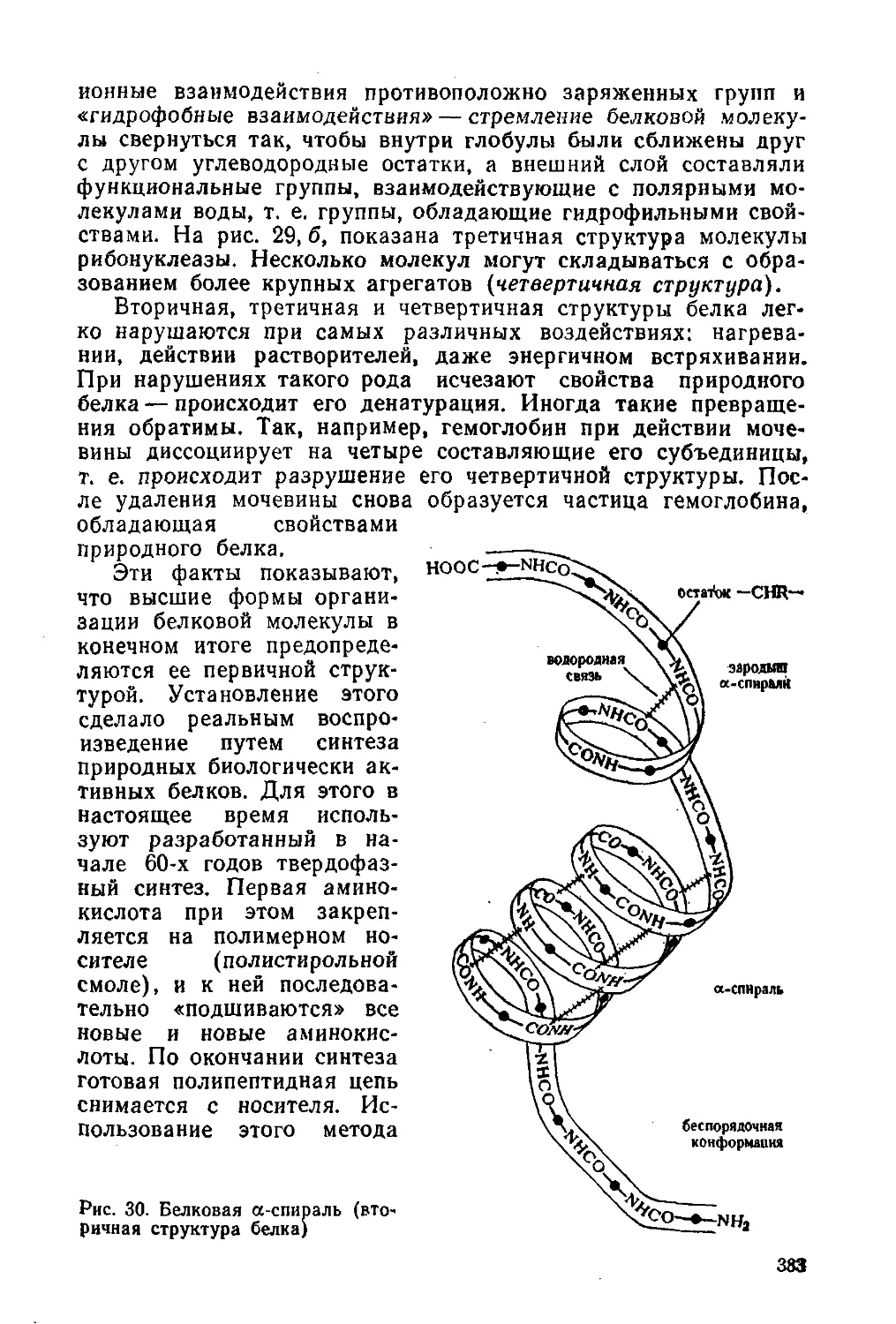
§ 175. Строение белков 379
§ 176. Пищевое и промышленное использование белков 384
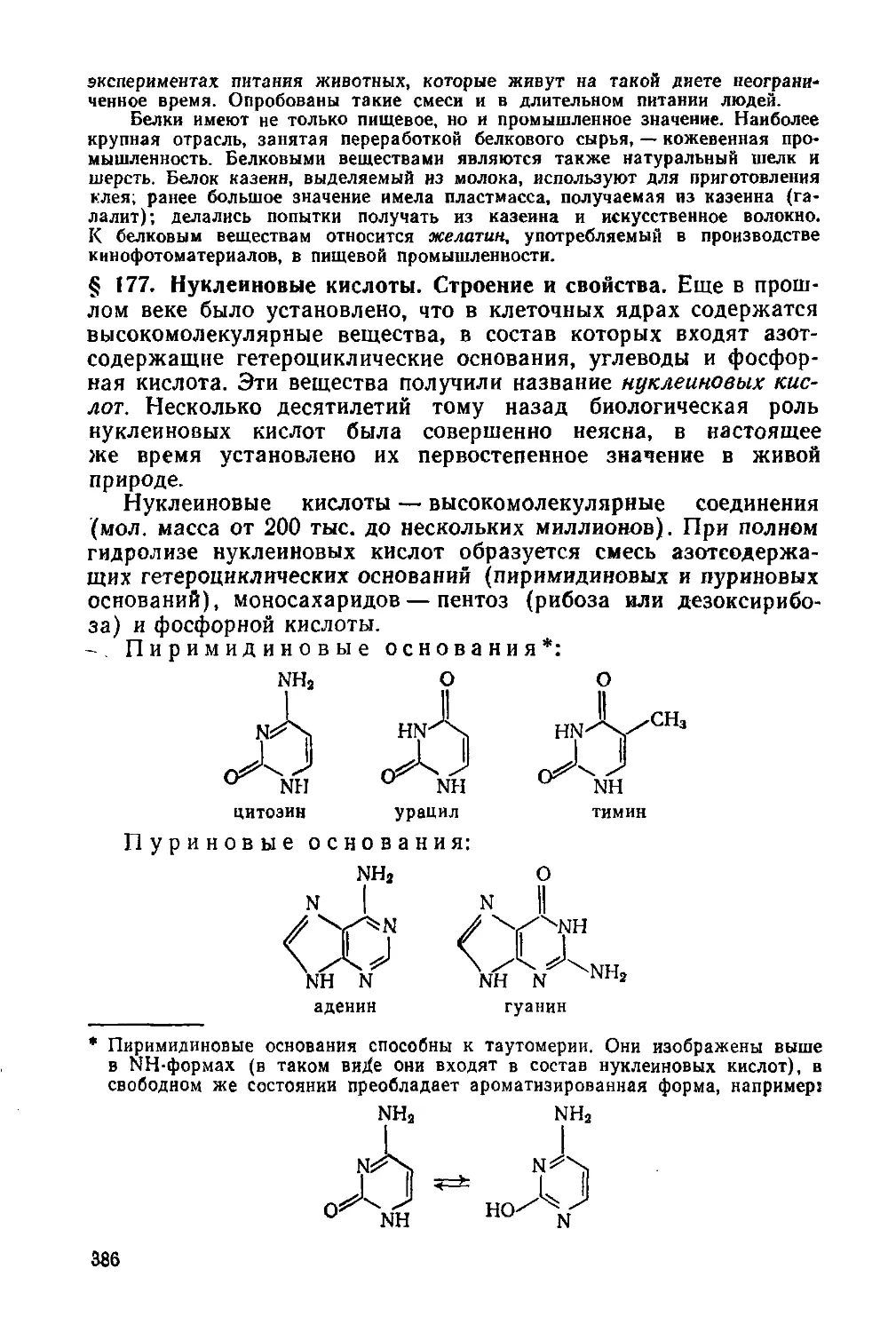
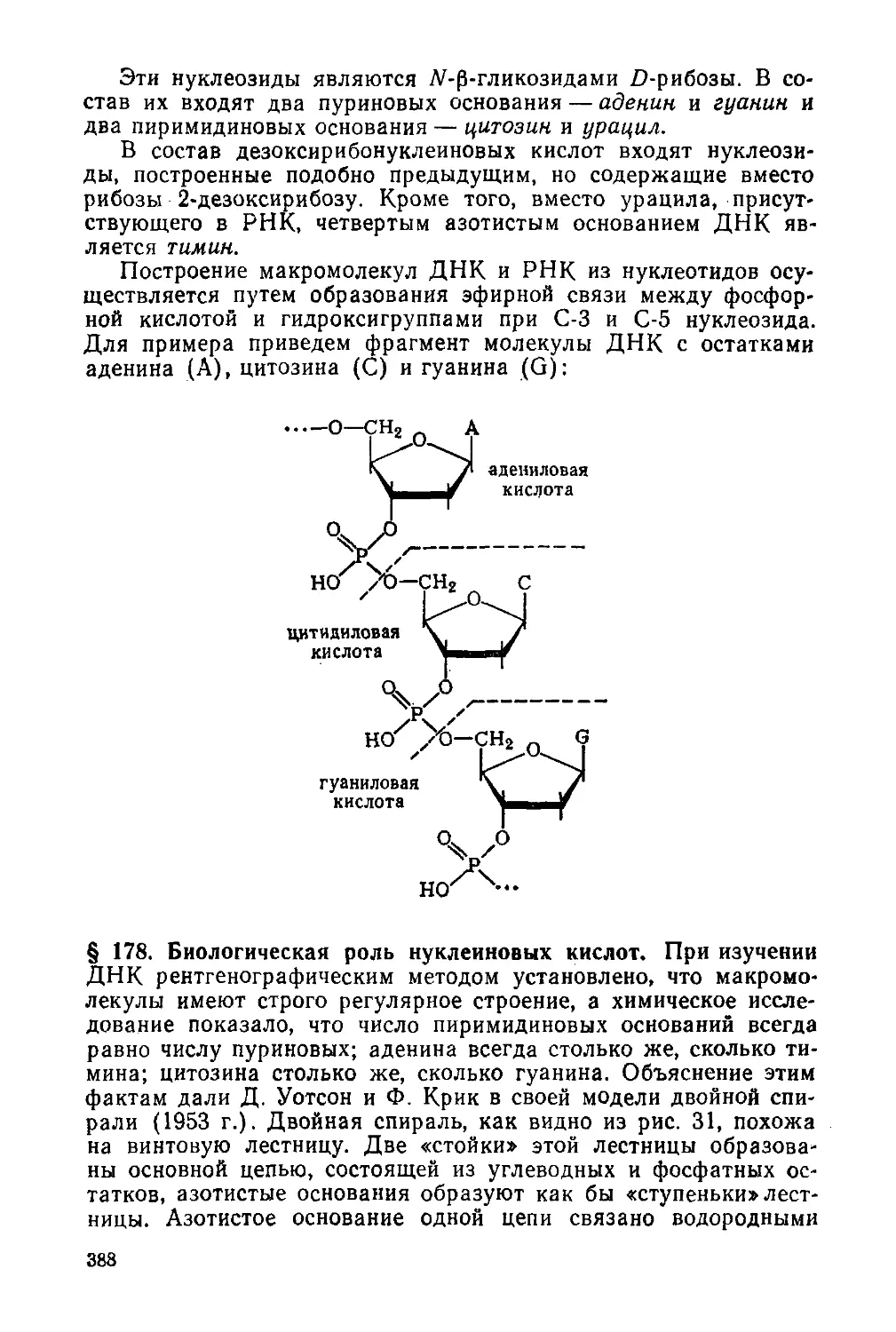
§ 177. Нуклеиновые кислоты. Строение и свойства 386
7
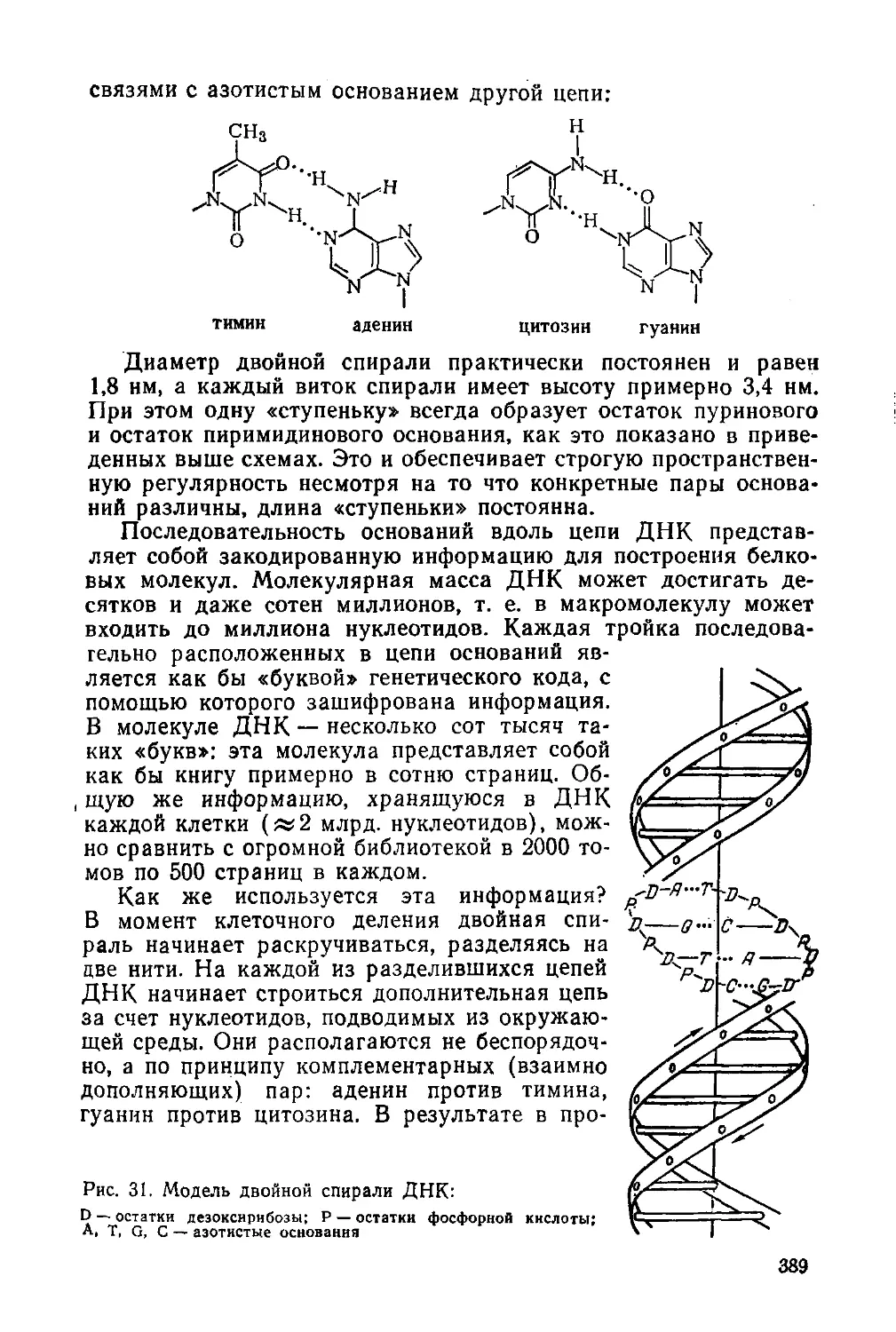
§ 178. Биологическая роль нуклеиновых кислот
§ 179. Ферменты
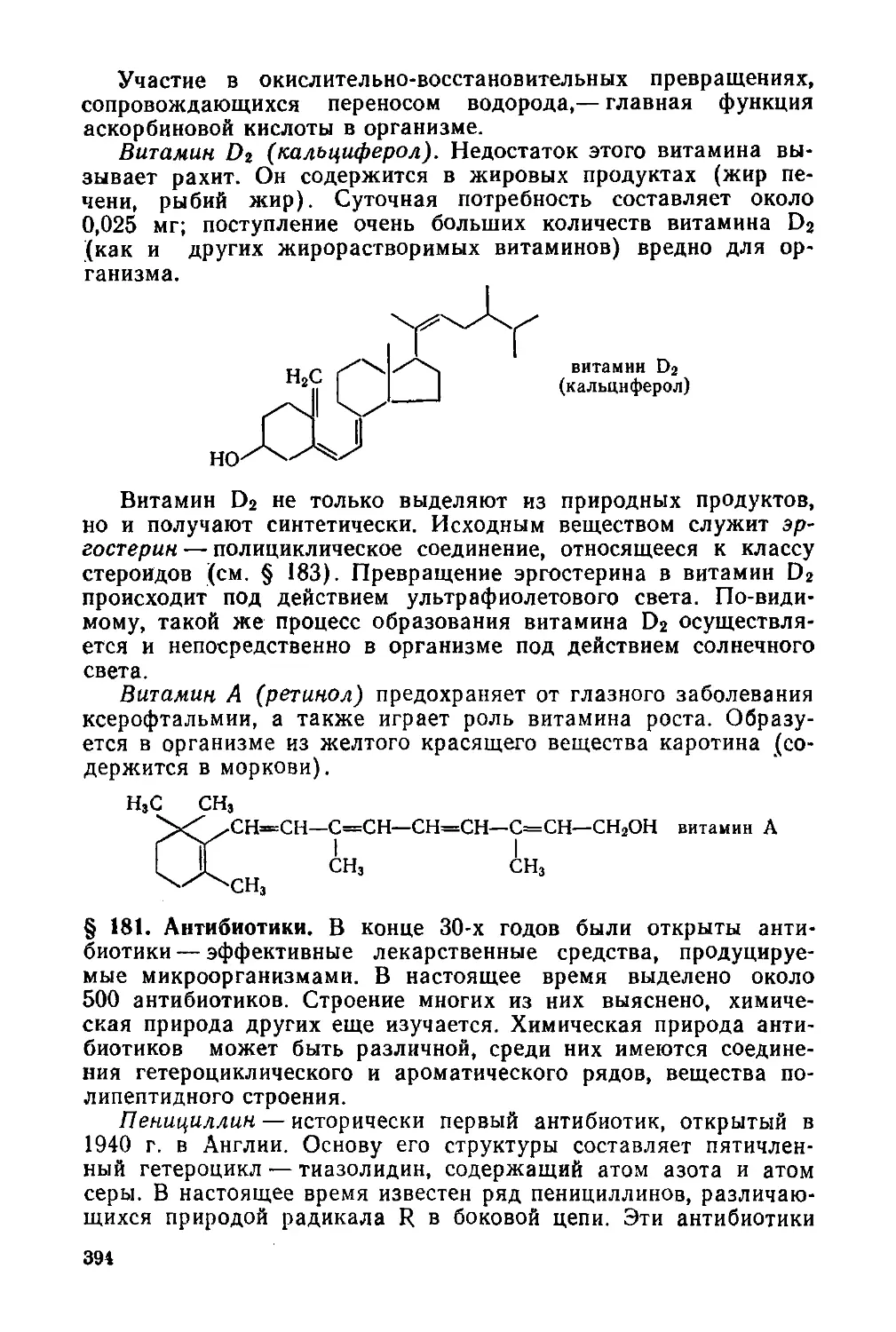
§ 180. Витамины
§ 181. Антибиотики
§ 182. Терпены
§ 183. Стероиды
Глава 23. Высокомолекулярные соединения
§ 184. Классификация. Связь строения со свойствами
Полимеризационные высокомолекулярные соединения
§ 185. Реакции полимеризации
§ 186. Полиалкены
§ 187. Виниловые полимеры
§ 188. Каучук
Поликонденсационные высокомолекулярные соединения
§ 189. Реакции поликонденсации
§ 190. Полиамиды
§ 191. Полиэфиры
§ 192. Фенолоформальдегидные смолы
§ 193. Кремнийорганические полимеры
Глава 24. Методы исследования органических веществ
§ 194. Определение строения
§ 195. Физические методы определения строения
Заключение
Предметный указатель
388
391
393
394
395
399
401
401
403
403
404
407
409
413
413
413
415
416
418
419
419
421
427
428
ОТ АВТОРОВ
В основе органической химии лежит созданная А. М. Бутлеро¬
вым теория химического строения органических соединений.
В настоящее время она дополнена более глубоким пониманием
природы химической связи, причин протекания реакций. В один
ряд с бутлеровской классификацией по строению молекул вста¬
ла другая классификация — по типам реакций. Именно раскры¬
тие природы органических реакций позволило подходить к
превращениям органических веществ не как к набору разнород¬
ных, трудно запоминаемых частных случаев, а как к стройной
системе.
Насколько возможно в рамках данного курса, авторы стре¬
мились изложить общие закономерности органической химии,
рассматривая изучение теории не как самоцель, а как средство
для лучшего понимания, лучшего запоминания — в конечном
итоге как средство облегчения усвоения материала. В то же
время органическую химию нельзя изучать без фактического
материала — конкретных веществ с их способами получения,
свойствами, областями применения. Авторы стремились в пер¬
вую очередь рассказать о практически важных органических
веществах, получивших применение в промышленности, сель¬
ском хозяйстве, медицине.
В основу построения книги положена классификация органи¬
ческих соединений по функциональным группам, определяющим
химическое поведение органических веществ. Сначала рассмо¬
трены углеводороды разных типов, которые составляют одну
большую семью, связанную многочисленными взаимными пере¬
водами, затем все галогенпроизводные, гидроксильные произ¬
водные и т. д.
При подготовке этого издания авторы внесли в него неко¬
торые дополнения, отражающие новые достижения органической
химии, а также задачи, стоящие перед нашей промышленностью
в свете решений XXVII съезда КПСС.
Со времени выхода предыдущего издания произошли изме¬
нения в номенклатуре органических соединений, что потребова¬
ло пересмотра ряда названий в соответствен с современными
правилами Международного Союза чистой и прикладной химии
(ШРАС).
Авторы будут благодарны читателям за пожелания и крити¬
ческие замечания, которые могут содействовать дальнейшему
улучшению книги.
ВВЕДЕНИЕ
§ 1. Предмет органической химии и ее практическое значение.
Органическую химию обычно определяют как химию соедине¬
ний углерода. Такое определение сразу вызывает вопрос: а как
же обстоит дело с такими хорошо известными неорганическими
веществами, как угольная и синильная кислоты и их соли, —
ведь они тоже содержат углерод? Действительно, абсолютно
челкой грани между органической и неорганической химией
провести невозможно. Однако огромное большинство соедине¬
ний углерода относится к органическим веществам.
Существует и другое определение: органическая химия — хи¬
мия углеводородов и их производных. Глубокий его смысл ста¬
нет понятным позднее. Пока поясним, что углеводородами на¬
зывают простейшие органические вещества, в состав которых
входят атомы только двух элементов — углерода и водорода.
Говоря о производных, имеют в виду более сложные вещества,
которые могут быть получены замещением атомов водорода в
углеводородах на атомы других элементов или сложные груп¬
пировки атомов.
Название органическая химия возникло в начале XIX века,
когда было выяснено, что углеродсодержащие вещества являют¬
ся главной составной частью растительных и животных организ¬
мов. Первоначально задачей органической химии было изучение
веществ, находимых в живой природе. Однако постепенно все
большее значение стали приобретать продукты органического
синтеза — искусственно получаемые вещества, многие из кото¬
рых не встречаются в природе. В изучении же природных ве¬
ществ постепенно переходили от простых к все более сложным,
а затем и к изучению химических процессов, составляющих ос¬
нову жизнедеятельности. Современная органическая химия изу¬
чает как природные, так и синтетические органические веще¬
ства — их строение, пути получения, свойства, возможности
практического использования. При этом помимо чисто химиче¬
ских методов—анализа и синтеза — широко применяют физи¬
ческие методы.
Еще в доисторические времена человек использовал для
своих нужд природные органические вещества (продукты пита¬
ния, дерево, шкуры животных). На протяжении тысячелетий че¬
ловечество постепенно научилось перерабатывать находимые в
природе органические вещества: получать ткани из волокон
хлопка, шерсти, льна и шелка; превращать шкуры животных пу¬
тем дубления в кожу; добывать из растений лекарства, крася¬
щие и душистые вещества; получать из жиров глицерин и жир¬
ные кислоты; выделять сахар из сахарной свеклы; превращать
10
природный каучук в резину; перерабатывать древесину, уголь и
нефть. Ныне переработкой органических веществ заняты многие
отрасли тяжелой и легкой промышленности — нефтяная и газо¬
вая, нефте- и коксохимическая, текстильная, пищевая, фарма¬
цевтическая и др.
Особое значение приобрело производство синтетических вы¬
сокомолекулярных соединений (полимеров), которые исполь¬
зуются в качестве конструкционных материалов, а также для
изготовления различных предметов быта. Сначала эти мате¬
риалы рассматривали лишь как заменители природных материа¬
лов (каучука, дерева, кожи, тканей, смол и др.). Однако теперь
они стали поистине «незаменимыми заменителями»: многие за¬
дачи современной техники могут быть решены лишь при исполь¬
зовании новых синтетических материалов.
§ 2. Сырьевая база промышленности органического синтеза.
В течение многих столетий человечество получало необходи¬
мые ему органические вещества переработкой растительного
и животного сырья. При этом разнообразие получаемых в про¬
мышленности органических веществ было не очень велико,
а масштабы их производства оставались довольно скром¬
ными.
Со второй половины прошлого столетия серьезное значение
в качестве сырья приобрел каменный уголь. Получаемая в виде
побочного продукта коксования каменноугольная смола откры¬
ла путь для промышленного получения бензола, толуола, наф¬
талина и других ароматических углеводородов. Эти вещества в
свою очередь стали сырьем для синтеза красителей, лекарст¬
венных препаратов, взрывчатых веществ. В нашем столетии все
большее значение в качестве сырья стала приобретать нефть,
главной составной частью которой являются парафиновые и
нафтеновые углеводороды. Важным сырьем стал и природный
газ, главная составная часть которого — простейший парафино¬
вый углеводород метан СН4. Органические вещества выделяют
также из сланцев. Сохранила свое значение и древесина, запа¬
сы которой, в отличие от ископаемого сырья, постоянно возоб¬
новляются.
Современная промышленность основана на переработке
углеводородного сырья: простейшие органические соединения —
углеводороды — стали не только формально, но и фактически
основой для получения разнообразных органических веществ.
Доступность и дешевизна углеводородного сырья привели к бы¬
строму росту новой отрасли — промышленности органического
синтеза. Эта отрасль в свою очередь является фундаментом для
производства полимерных материалов (пластических масс, син¬
тетических каучуков, химических волокон), красителей, средств
защиты растений, моющих препаратов, химических реактивов,
лекарственных препаратов, продуктов тонкого органического
синтеза.
11
§ 3. Роль отечественных ученых в развитии органической хи¬
мии. Наша страна имеет все основания гордиться тем большим
вкладом, который внесли отечественные ученые в развитие ор¬
ганической химии. В дореволюционной России работали многие
выдающиеся химики-органики. А. М. Бутлеров (1828—1886 гг.)
в 60-х годах XIX века своей теорией химического строения за¬
ложил фундамент современной органической химии; его ученик
В. В. Марковников (1838—1904 гг.) развил теоретические поло¬
жения своего учителя, раскрыл химическую природу нефти.
Н. Д. Зелинский (1861—1963 гг.) с учениками глубоко изучил
каталитические превращения углеводородов и создал основы
нефтехимии. А. Н. Несмеянов (1899—1980 гг.) стал основате¬
лем новой ветви химической науки — элементорганической хи¬
мии. Н. Н. Зинин (1812—1880 гг.), М. Г. Кучеров (1850—
1911 гг.), М. И. Коновалов (1858—1906 гг.), А. Е. Арбузов
(1877—1968 гг.) открыли важные превращения органических
веществ, послужившие в дальнейшем основой для создания но¬
вых химических производств. Химики всего мира знают имя
Ф. Ф. Бейльштейна (1838—1907 гг.) — основателя фундамен¬
тального справочника, содержащего сведения о всех известных
органических веществах. Перечисляя имена ученых, мы пока
ничего не говорим о сущности их открытий: с ней вы познако¬
митесь позднее.
На Западе открытия русских химиков нередко замалчивают¬
ся. Это, впрочем, не мешало западноевропейским химическим
концернам использовать их на своих заводах. В экономически
отсталой царской России химическая промышленность была
развита слабо и идеи отечественных ученых почти не находили
в ней применения.
§ 4. Развитие химии в СССР. После Великой Октябрьской ре¬
волюции была создана сеть научных учреждений, в которых
ученые-органики старой школы — Н. Д. Зелинский, А. Е. Фа¬
ворский (1860 — 1945 гг.), С. В. Лебедев (1874 — 1934 гг.) и дру¬
гие — получили простор для исследований, для воспитания мо¬
лодых научных кадров.
В годы предвоенных пятилеток была создана химическая
промышленность, обеспечивающая потребности народного хо¬
зяйства и обороны. В послевоенные годы химическая про¬
мышленность нашей страны продолжала развиваться, опережая
по темпам роста другие отрасли.
В двенадцатой пятилетке намечено увеличить выпуск про¬
мышленной продукции на 21—24 %, в химической и нефтехи¬
мической промышленности предусмотрен рост на 30—32%: со¬
поставление этих двух показателей наглядно демонстрирует
опережающий рост химической промышленности.
К 1990 г. годовой выпуск минеральных удобрений возрастет
до 41 — 43 млн. т, химических средств защиты растений — до
440 — 480 тыс. т, синтетических смол и пластических масс — до
12
6,8—7,1 млн. т, химических волокон — до 1,85 млн. т, синтети¬
ческих каучуков — до 2,7—2,9 млн. т. При этом особенно
быстро будет развиваться производство современных конструк¬
ционных пластмасс, заменителей растительных масел и другого
пищевого сырья, расходуемого для технических целей. Значи¬
тельное место продукция химической промышленности займет в
производстве товаров народного потребления — тканей, обуви,
трикотажных изделий.
Важной составной частью химической промышленности яв¬
ляются перечисленные ниже производства, основанные на до¬
стижениях органической химии.
1. Промышленность тяжелого (основного) органического
синтеза, вырабатывающая углеводороды разных типов, кисло¬
родсодержащие органические соединения (спирты, альдегиды,
кетоны, кислоты, эфиры), азотсодержащие органические соеди¬
нения (нитросоединения, амины, нитрилы), галогенсодержащие
органические соединения, вещества, содержащие фосфор, крем¬
ний и другие элементы. Частью промышленности тяжелого
органического синтеза является нефтехимическая промышлен¬
ность.
Химические продукты, вырабатываемые на предприятиях
промышленности основного органического синтеза и нефтехими¬
ческих заводах, частично потребляют непосредственно (моющие
средства, синтетическое топливо, смазочные масла, пластифика¬
торы, растворители), главным же образом используют в каче¬
стве сырья для перечисляемых ниже отраслей.
2. Производство ядохимикатов (для борьбы с вредителями
сельского хозяйства) и органических удобрений (мочевина).
3. Промышленность синтетических смол и пластических
масс.
4. Промышленность синтетического каучука.
5. Промышленность химических волокон.
6. Лакокрасочная промышленность.
7. Анилинокрасочная промышленность.
8. Химико-фармацевтическая промышленность.
9. Производство химических реактивов и высокочистых ве¬
ществ.
Перед химической промышленностью стоит задача не только
развить производство для удовлетворения нужд народного хо¬
зяйства и населения, но и сделать это с наименьшим расходом
природных ресурсов, энергии, трудовых затрат, обеспечить со¬
кращение отходов, устранить вредное влияние химических про¬
изводств на окружающую среду. Для этого необходимо шире
внедрять малоотходные и безотходные технологии, развивать
комбинированные производства, обеспечивающие полное и
комплексное использование природных ресурсов, сырья и мате¬
риалов и исключающие или существенно снижающие вредное
воздействие на окружающую среду.
13
ГЛАВА 1
ОБЩИЕ ПОЛОЖЕНИЯ ОРГАНИЧЕСКОЙ ХИМИИ
§ 5. Особенности органических соединений. В состав органиче¬
ских соединений обязательно входит углерод; почти в каждом
органическом веществе имеется также водород. Многие органи¬
ческие вещества содержат кислород и азот, несколько реже в
них входят галогены, сера, фосфор. Перечисленные элементы
образуют группу органогенов, чаще всего встречающихся в мо¬
лекулах органических веществ. В составе так называемых
элементорганических соединений можно встретить почти любой
элемент (кроме инертных газов).
В настоящее время известно более семи миллионов углерод¬
содержащих соединений, в то время как соединений, не содер¬
жащих углерода, насчитывается лишь несколько сотен тысяч.
Общая причина многообразия органических соединений в том,
что в их молекулы могут входить десятки (а иногда сотни и
тысячи) атомов, располагающихся в разном порядке.
Типичные органические соединения характеризуются опреде¬
ленными свойствами, отличными от свойств неорганических
веществ. В то время как подавляющее большинство неорганиче¬
ских соединений — твердые вещества с высокими температура¬
ми плавления, ббльшая часть органических соединений — низ¬
коплавкие твердые вещества или жидкости. Многие реакции
органических веществ протекают медленнее, чем у веществ не¬
органических, причем превращения нередко идут одновременно
в нескольких направлениях. Органические соединения разруша¬
ются при высоких температурах, многие из них постепенно
окисляются на воздухе. В общем можно сказать, что типичные
органические вещества менее устойчивы, чем типичные неорга¬
нические. В дальнейшем мы узнаем, что эти и другие внешние
отличия имеют глубокую причину, зависят от различий в харак¬
тере химической связи.
§ 6. Выделение и анализ органических веществ. Исследование
любого вещества начинается с получения его в чистом (инди¬
видуальном) виде. Между тем получить чистое органическое
вещество далеко не просто. Во многих случаях легче провести
саму реакцию, чем выделить в чистом виде синтезированный
продукт. Для очистки веществ применяют следующие методы.
Перекристаллизация. Этот метод широко использу¬
ют и в неорганической химии. Простейшим (хотя и не абсолют¬
но надежным) доказательством чистоты кристаллического ве¬
щества может служить постоянство его температуры плавления
при повторных кристаллизациях («перекристаллизация до по¬
стоянной температуры плавления»). Для доказательства иден¬
тичности синтезированного вещества с другим имеющимся в
14
наличии образцом проводят определение температуры плавле¬
ния смеси обоих веществ: отсутствие депрессии температуры
плавления смеси (т. е. понижения ее по сравнению с темпера¬
турой плавления компонентов смеси) укажет на идентичность
смешанных веществ.
Перегонка. Этот метод в различных его видах особенно
часто применяют для очистки органических веществ. Это и про¬
стая перегонка, и перегонка в вакууме, позволяющая снизить
температуру кипения по сравнению с перегонкой при атмосфер¬
ном давлении примерно на 100 °С (обычный вакуум — остаточ¬
ное давление 10 мм рт. ст.)* или 200 °С (высокий вакуум^
остаточное давление 0,1—0,01 мм рт. ст.). Хороший способ
очистки — перегонка с водяным паром. Современные устройства
для перегонки — ректификационные колонки — позволяют раз¬
делять вещества, температуры кипения которых различаются
всего на несколько градусов.
Возгонка — относительно редко применяемый процесс
очистки, при котором используют способность ряда веществ
переходить из твердого состояния в парообразное, минуя жид¬
кое. Возгонка — весьма эффективный способ очистки вещества.
Экстракция основана на разделении веществ путем рас¬
пределения их между несмешивающимися растворителями.
В простейшем случае экстракцию осуществляют встряхиванием
в делительной воронке водного раствора органического веще¬
ства с растворителями, например с эфиром и др. При этом
органическое вещество переходит из водного слоя в слой орга¬
нического растворителя. Таким образом отделяют органические
вещества от неорганических солей, а также разные органиче¬
ские вещества друг от друга. Существуют приборы для автома¬
тического проведения многократной экстракции.
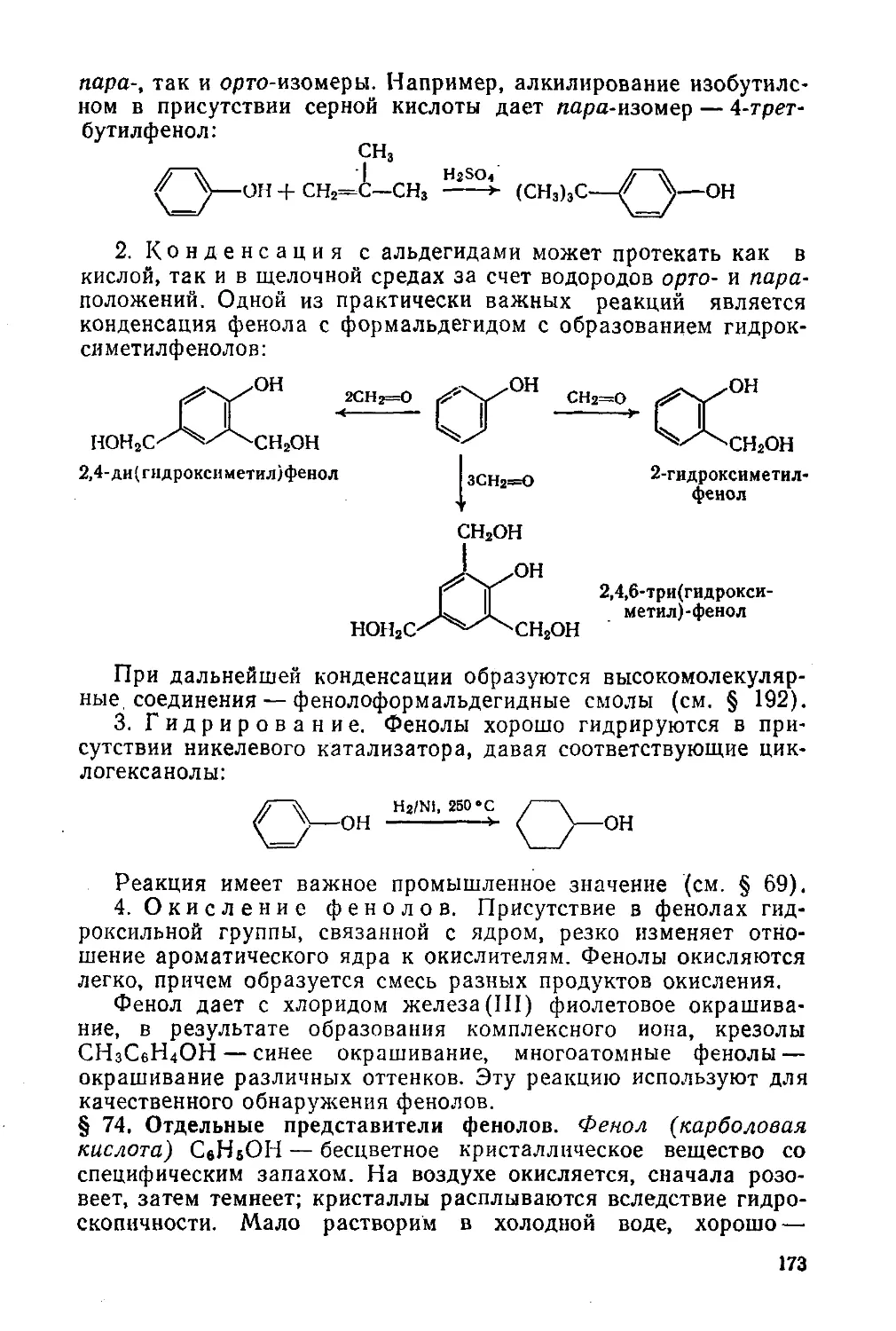
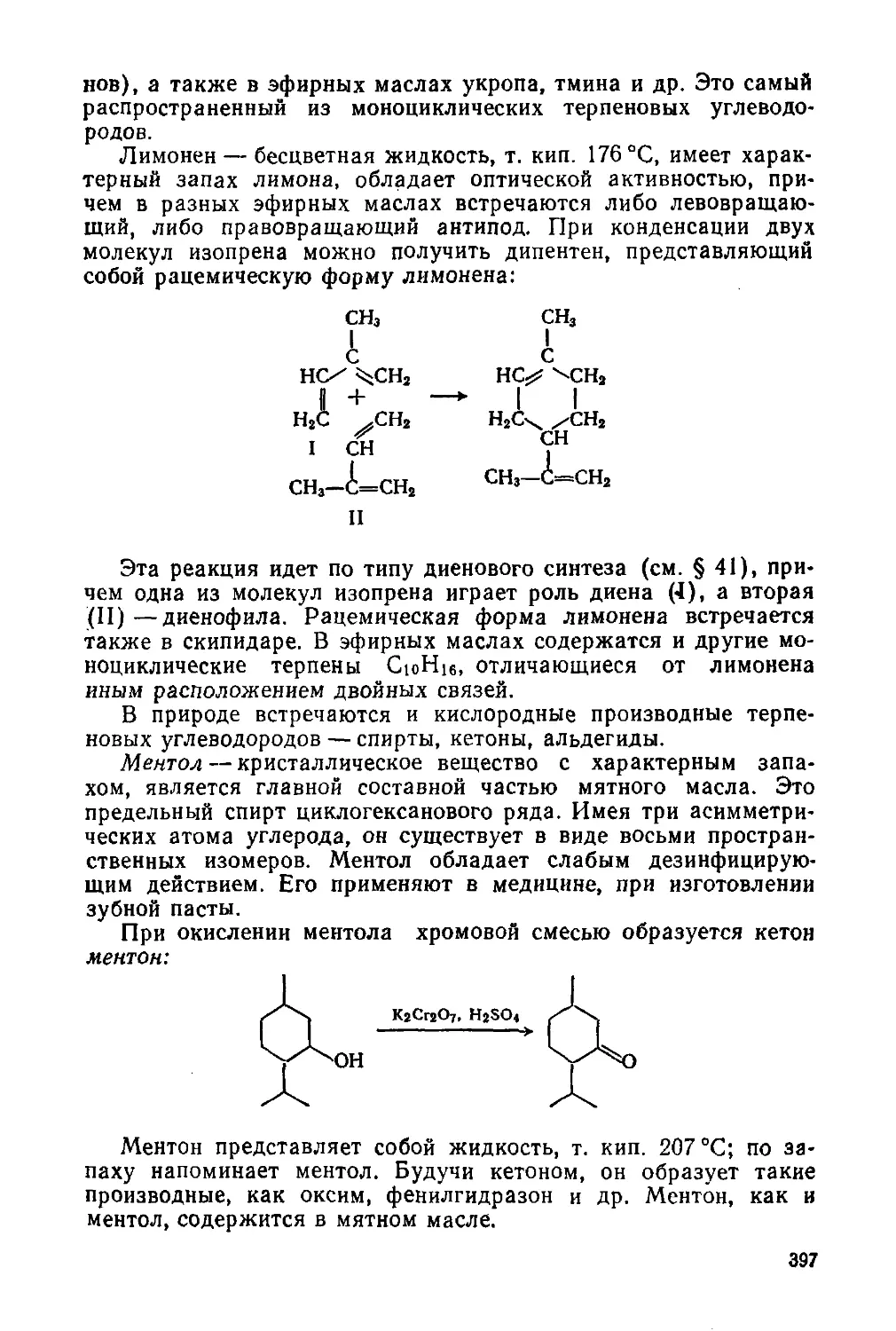
Хроматография основана на различиях в адсорбцион¬
ных свойствах веществ. Основы метода заложил русский ученый
М. С. Цвет в 1903 г. Ныне существует ряд вариантов хромато¬
графии, и этот метод часто применяют для очистки и доказа¬
тельства индивидуальности вещества.
Общий принцип всех хроматографических методов заключа¬
ется в том, что разделяемую смесь пропускают через адсор¬
бент— вещество, способное с разной силой взаимодействовать
с компонентами разделяемой смеси. При этом более сильно
адсорбирующиеся вещества отстают в своем движении от слабо
адсорбирующихся; так происходит их разделение. В наиболее
наглядном виде этот процесс идет в хроматографических ко-
лонках (рис. 1). На таком же принципе построены сложные
автоматические приборы — жидкостные хроматографы.
Малые количества веществ разделяют или идентифицируют
с помощью хроматографии на бумаге или в тонком слое других
* 1 мм рт. ст. = 133,322 Па.
lfl
2
Рис. 1. Хроматографическая
колонка:
1 — растворитель; 2 — адсорбент
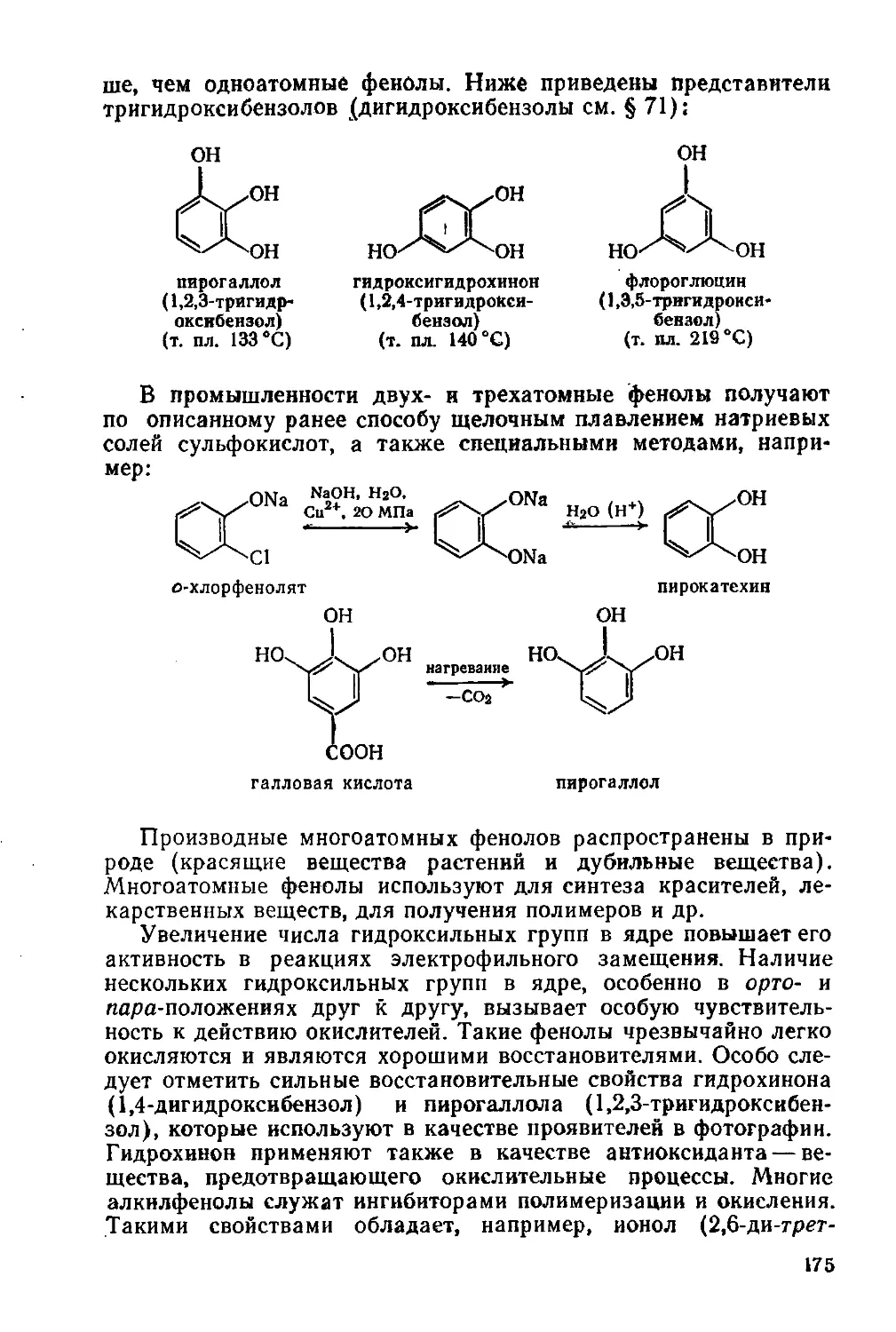
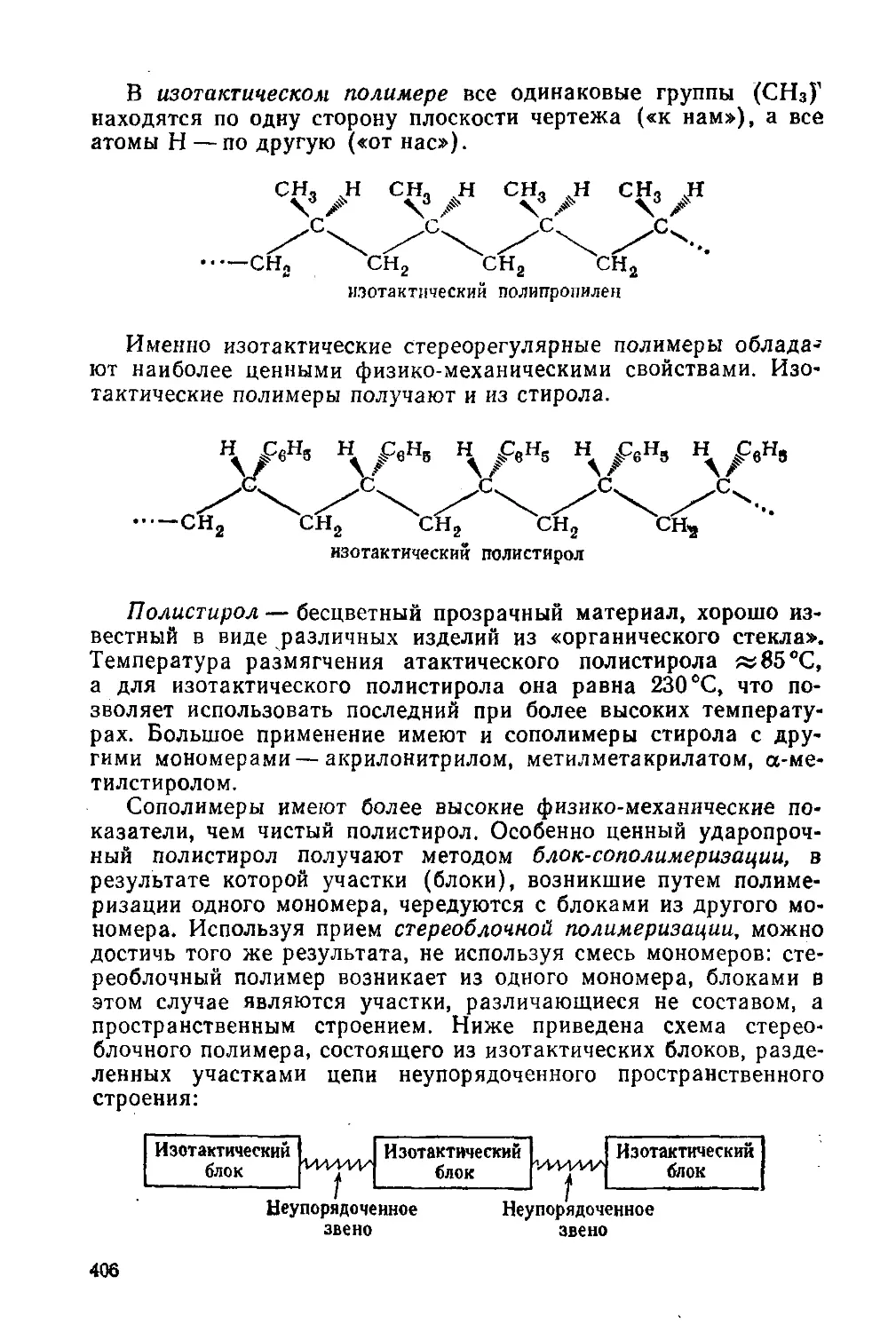
Рис. 2. Обнаружение углерода
и водорода:
1 —безводный CuSO4; 2—смесь орга¬
нического вещества с СuО; 3—ра¬
створ Са(ОН)2
адсорбентов — оксида алюминия, силикагеля и др. (тонкослой¬
ная хроматография).
Особенно эффективна газожидкостная хроматография, при
которой через адсорбент пропускают пары органических ве¬
ществ. Многие вещества, описанные в свое время как индиви¬
дуальные, при исследовании методом газожидкостной хромато¬
графии оказались смесями нескольких компонентов.
Все эти методы очистки используют не только в лаборато¬
риях, но и в промышленности.
Чистое органическое вещество характеризуют обычно сле¬
дующими константами.
Температура плавления. Чем чище вещество, тем уже интер¬
вал плавления. Обычно он составляет 0,6—1,0 °С. Примеси все¬
гда понижают температуру плавления.
Температура кипения. Интервал ее для чистых веществ, пе¬
регнанных на ректификационных колоннах, составляет десятые
доли градуса.
Относительная плотность. Выражается отношением массы
вещества при определенной температуре (обычно 20 °С) к мас¬
се воды, взятой в том же объеме при 4°С.
Показатель преломления. В случае жидких органических
веществ для его определения используют рефрактометр. Пока¬
затель преломления зависит от природы вещества, а также от
его чистоты. Показатель преломления служит основой для рас¬
чета молекулярной рефракции (о которой подробнее рассказа¬
но в гл. 24).
Качественный и количественный элемент¬
ный анализ. Исследование выделенного в чистом виде ор-
16
ганического вещества обычно начинают с выяснения того, какие
элементы входят в его состав, т. е. с качественного элементного
анализа.
Качественное определение углерода и водорода основано на
окислений органических веществ. Вещество смешивают с окси¬
дом меди (II) и- нагревают в пробирке с газоотводной трубкой,
опущенной в известковую воду (рис. 2). Оксид меди окисляют
органическое вещество. Входивший в его состав углерод обра¬
зует СO2, который можно обнаружить по образующемуся осад¬
ку СаСОз. Из водорода получается вода, капельки ее можно
увидеть в верхней части пробирки, а также обнаружить по по¬
синению безводного сульфата меди, помещенного в верхнюю
часть пробирки.
На кислород, часто входящий в состав органических соеди¬
нений, качественных реакций нет, хотя и существуют методы
его количественного определения.
Азот обнаруживают в составе органического вещества,
сплавляя в пробирке небольшое его количество с металличе¬
ским натрием; азот при этом переходит в цианид натрия NaCN.
Последний открывают при помощи известной из неорганической
химии реакции: появление синего окрашивания после добавле¬
ния солей двух- и трехвалентного железа. Химические процес¬
сы, происходящие при обнаружении азота, можно выразить сле¬
дующими уравнениями:
Na + C + N —► NaCN
FeS04 + 6NaCN —► Na4(Fe(CN)6] + Na2S04
4FeCl3 + 3Na4[Fe(CN)6) —► Fe4[Fe(CN)6]3 + 12NaCl
При сплавлении с металлическим натрием переходят в не¬
органическую форму («минерализуются») и другие элементы,
которые могут входить в состав органической молекулы. Так,
сера образует сульфид натрия Na2S, который открывают при
помощи нитрата свинца: выпадающий черный осадок PbS ука¬
зывает на присутствие серы в органическом веществе:
2Na + S → Na2S ↔ 2Na+ + S2- Рb2+ + S2- → PbS↓
Галогены переходят в соответствующие соли, которые дают
осадок с раствором нитрата серебра. Галогены можно обнару¬
жить также при помощи пробы Бейльштейна — по зеленой
окраске пламени, в которое внесена медная проволока с гало¬
генсодержащим веществом.
Вместо сплавления с натрием органическое вещество можно
разлагать действием сильных окислителей (пероксид натрия,
азотная кислота, хромовая смесь). Перешедшие в ионное со¬
стояние элементы открывают после этого обычными реакциями,
известными из неорганической химии.
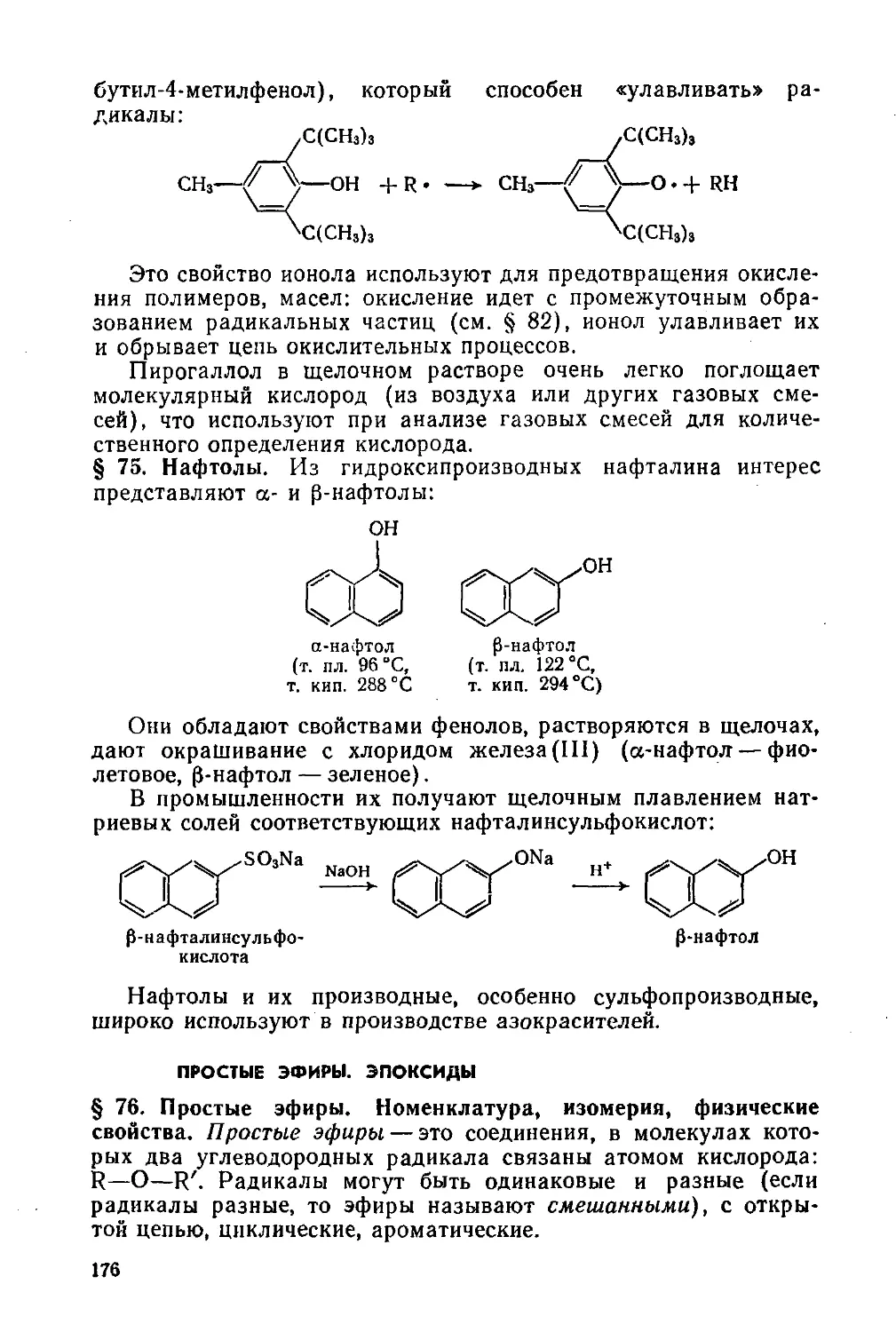
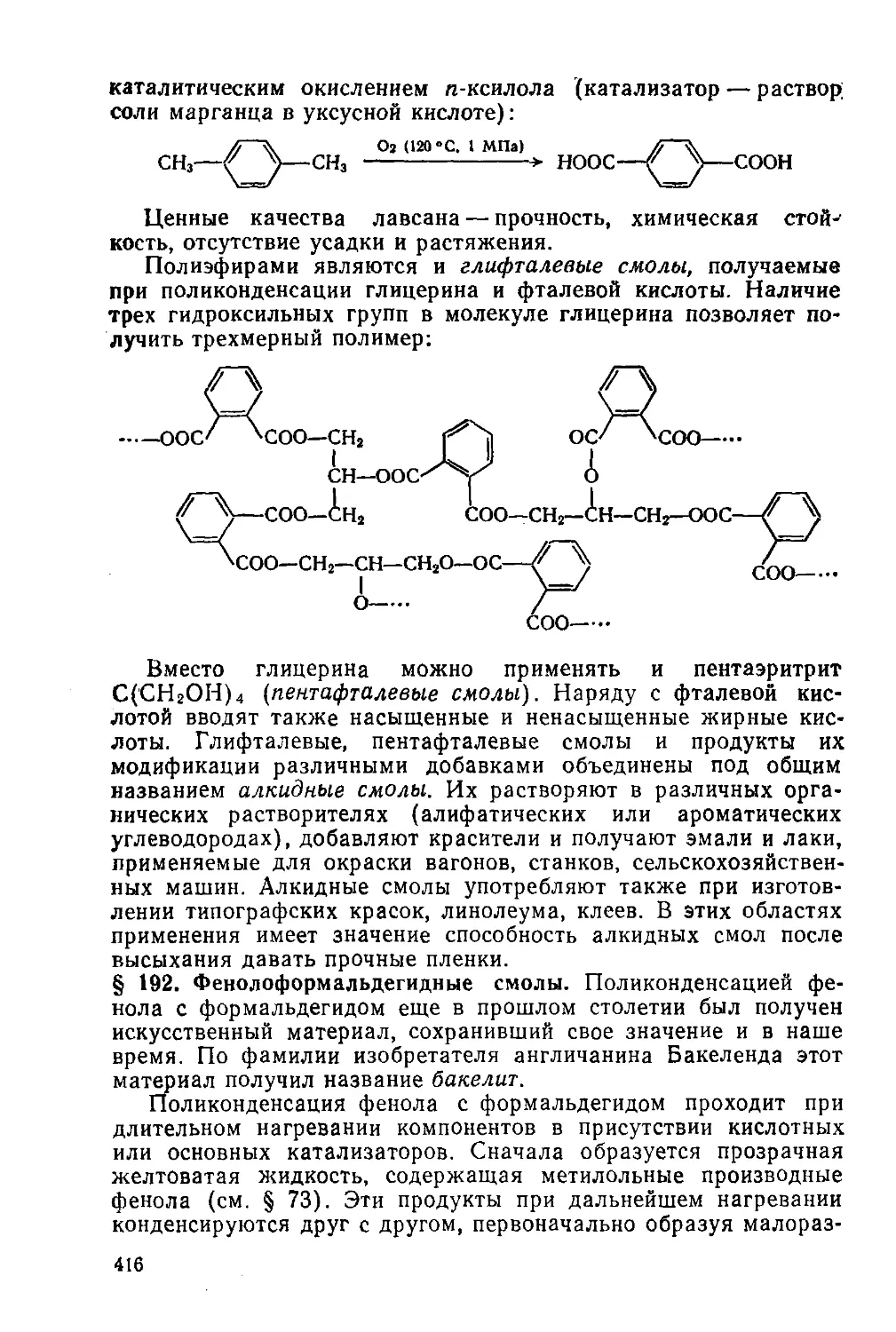
Количественный элементный анализ органических веществ
проводят сожжением их (обычно в токе кислорода) в приборах,
17
Рис. 3. Установка для определения углерода и водорода:
I — газометр; 2 — осушительная склянка; 3 —кран; 4 — прибор для очистки кислорода:
6, 9t 10 — электропечи; б, 7 — аппараты для поглощения Н2O и СО2, образующихся
после очистки кислорода; 8 — трубка для сожжения; // — газовая горелка; 12, 13 —
аппараты для поглощения Н2О и СО2, образующихся после сожжения пробы; 14 —
изолирующая трубка; 15 — аспиратор
позволяющих количественно уловить продукты сгорания. Если
вещество содержит только четыре элемента — углерод, водород,
кислород и азот, то улавливают и взвешивают образовавшиеся
при сожжении диоксид углерода и воду (азот выделяется в
свободном виде). Если имеются, кроме того, сера, галогены
и другие элементы, к прибору присоединяют дополнительные
поглотительные трубки, чтобы уловить соединения этих элемен¬
тов. Разработано много разных вариантов количественного сож¬
жения органических соединений. Эти варианты отличаются
аппаратурой, условиями сожжения и улавливания образующих¬
ся продуктов. На рис. 3 изображена установка для элементного
анализа по методу М. О. Коршун. Существуют и автоматические
приборы для проведения элементного анализа.
Чтобы уменьшить расход вещества и ускорить работу, в на¬
учных лабораториях обычно применяют такие аппараты, кото¬
рые дают возможность использовать для анализа 3—5 мг ве¬
щества.
Допустим, что для проведения элементного микроанализа была взята на¬
веска 4,51 мг органического вещества, состоящего из углерода, водорода и
кислорода, а после сожжения было получено 9,18 мг СO2 и 2,88 мг Н20.
В продуктах сожжения содержатся весь углерод и водород, находившиеся
во взятой навеске анализируемого вещества. По этим данным рассчитывают
массовые количества этих элементов в навеске (2,50 мг углерода и 0,32 мг
водорода), а затем и процентное их содержание. В приведенном примере по¬
лучается 55,4 % С и 7,1 % Н. Остальное (37,5 %) приходится на долю кис¬
лорода. Для перехода от массовых долей элементов к соотношениям их ато¬
мов надо разделить процентные доли на атомные массы соответствующих эле¬
ментов. Таким образом получают: 4,61 С : 7,04 Н : 2,34 О. Проведя деление
этого отношения на наименьшее из этих чисел, получим более удобное соот¬
ношение 1,97 : 3,01 : 1,00 или, округленно, 2:3:1.
18
Найденные числа выражают соотношение между атомами С, Н и О в
молекуле анализировавшегося вещества, т. е. его простейшая формула С2Н3О,
Это не значит, однако, что состав молекулы именно таков, он может быть
удвоенным, утроенным — в общем отвечать формуле (C2H3O)n.
Чтобы решить вопрос о молекулярной формуле, необходимо
определить относительную молекулярную массу (молекулярную
массу) вещества. Для этого пригодны многие методы, например
измерение давления пара или понижения температуры застыва¬
ния. Получив для нашего примера молекулярную массу «90,
мы имели бы право считать молекулярной формулу С4Н6O2.
§ 7. Явление изомерии. При исследовании неорганических со¬
единений молекулярная формула чаще всего служит точной
«визитной карточкой» соединения. Формула H2S04 несомненно
является формулой серной кислоты, NaHC03 — гидрокарбоната
натрия и т. д. Иное дело в органической химии: одну и ту же
молекулярную формулу могут иметь разные вещества. Так, фор¬
мулу С2Н60 имеют два вещества: газ, называемый диметило-
вым эфиром, и жидкость — этиловый спирт. Формулу C4HgO
имеет 21 вещество!
Различные вещества, имеющие одинаковую молекулярную
формулу, называют изомерами, а само явление носит название
изомерии (от греч. «изомерос» — состоящий из равных частей).
В чем же причина различия изомеров? Поскольку состав их
молекул одинаков (это вытекает из самого определения изоме¬
рии), то причину различия можно искать только в разном по¬
рядке связей атомов в молекуле. Тем самым явление изомерии
заставляет, не довольствуясь установлением молекулярной фор¬
мулы, идти дальше, выясняя детали внутреннего строения
молекул органических веществ. Поэтому явление изомерии (от¬
крытое еще в начале прошлого столетия) служило постоянным
стимулом для развития теоретических воззрений органической
химии.
§ 3. Первоначальные представления о природе органических
соединений. Любая наука начинает свой путь с того, что пыта¬
ется упорядочить, классифицировать собранный фактический
материал. В химии, очевидно, речь идет прежде всего о клас¬
сификации химических соединений. Вплоть до XVIII века хими¬
ки не делали различия между минеральными и органическими
веществами. Например, «солями» называли бесцветные кристал¬
лические вещества, растворимые в воде: сюда вместе с неорга¬
нической поваренной (хлорид натрия) и другими солями
попадали также и органические вещества, например янтарная
и щавелевая кислоты. «Маслами» считали все густые жидкости,
к ним причисляли не только растительные масла, но и «купо¬
росное масло» (название, еще и ныне употребляемое в технике
для концентрированной серной кислоты) и «масло винного
камня» (расплывшийся на воздухе гидроксид калия), хлорид
цинка, также легко поглощающий влагу воздуха и превращаю¬
19
щийся в густую жидкость. Спиртовыми веществами считали
летучие жидкости (от лат. spiritus — дух, душа): и винный
спирт, и хлорид олова (IV), и хлороводородную кислоту, и вод¬
ный раствор аммиака. Для последнего название «нашатырный
спирт» сохранилось и поныне.
К середине XVIII века из животных и растительных орга¬
низмов было выделено значительное число веществ, по свой¬
ствам и составу имевших много общего и в то же время замет¬
но отличавшихся от минеральных (неорганических). Вещества
растительного и животного происхождения стали называть ор¬
ганическими. В начале XIX века знаменитый шведский химик
Берцелиус в своем руководстве по химии провел четкую грань
между минеральными и органическими веществами. Принципи¬
альное различие он видел в том, что органические вещества
получаются, якобы, только в живых организмах при участии
таинственной «жизненной силы», в лаборатории же их будто
бы получить нельзя.
Представление о необходимости участия «жизненной силы»
в создании органических соединений получило название вита¬
лизма (от лат. vita — жизнь). Оно было таким же проявлением
идеализма, как и вера в существование «души» в биологии.
Ошибочность виталистических взглядов была доказана еще в
первой половине XIX века синтезом многих органических ве¬
ществ. Важными вехами на этом пути явились получение моче¬
вины из неорганического вещества — цианата аммония (Велер,
1828 г.), синтез жиров (Бертло, 1854 г), синтез сахаристых
веществ (Бутлеров, 1861 г.).
Первые попытки объяснить природу органических соединений
были сделаны в 20—30-х годах прошлого столетия Берце¬
лиусом в его теории радикалов. Берцелиус полагал, что орга¬
нические вещества построены из радикалов. Считалось, что
радикалы — «подлинные элементы органической химии», что
они без изменения могут переходить из одного соединения в
другое. В этих представлениях Берцелиуса правильно отражена
одна из особенностей органических молекул — наличие в них
определенных устойчивых группировок атомов, остающихся не¬
затронутыми при химических превращениях. Берцелиус был, од¬
нако, неправ, когда считал эту устойчивость абсолютной и на¬
стаивал на возможности выделения радикалов в свободном
виде. Наивными были и представления Берцелиуса о роли элек¬
трических зарядов в органических молекулах. Впрочем, твердо
зная теперь, что органические радикалы и ионы являются ре¬
ально существующими промежуточными частицами в ходе пре¬
вращений органических веществ, мы не можем осуждать эти
взгляды Берцелиуса столь решительно, как это делали в конце
XIX — начале XX веков.
В 40—50-х годах XIX века на смену теории радикалов при¬
шла теория типов, связанная главным образом с именем фран¬
20
цузского ученого Жерара. Все Органические соединения рас¬
сматривались им как производные простейших неорганических
веществ: водорода, хлороводорода, воды, аммиака:
Тип
водорода:
Н1
СН3
5 1 С2Н5
) С2н30 }
н }
н
Н
) Н J
водород
метан этан
уксусный
альдегид
Тип
хлороводорода:
Н1
сн8 \
адо 1
Cl J
С1 )
Cl J
хлоро-
метил-
ацетил-
водород
хлорид
хлорид
Тип
воды:
«Ь
сн’)о
ад} о
адо \
адо \0
Н J
н J
с2н6 J
н /
C2HS J
вода
метиловый
диэтиловый
уксусная
этилацетат
спирт
эфир
кислота
При
таком подходе
для многих
органических
соединений
получались формулы, сходные с современными формулами
строения. Однако сторонники теории типов вкладывали в них
совершенно иное содержание: формулы теории типов — это
только формулы превращения. Для одного вещества писали
множество различных формул в зависимости от химических ре¬
акций, которые хотели этими формулами выразить. Внутреннее
строение молекул сторонники теории типов считали принципи¬
ально непознаваемым, становясь тем самым на позиции агно¬
стицизма — ложного философского учения, ставящего границы
человеческому познанию.
Помимо основных теорий существовало немало их разновид¬
ностей. Авторам этих вариантов отличия казались столь суще¬
ственными, что часто они признавали только свое толкование,
отвергая все другие. В результате химики часто просто плохо
понимали друг друга. Яркую картину состояния органической
химии в первой половине XIX века рисует Гьельт в книге
«История органической химии»: «...в 40-х и 50-х годах в этой
области господствовала настоящая анархия, которая приводила
в отчаяние многих химиков и тормозила спокойное развитие
науки. Рядом применялись атомные веса Берцелиуса, Дюма,
Жерара-Лорана, а также эквивалентные веса Гмелина. Поэто¬
му химические формулы органических соединений представляли
собой пеструю, мало привлекательную картину... Если где-либо
встречалась формула Н202, то следовало выбирать, означает ли
21
она воду или перекись водорода. Формула С2Н4 означала —
смотря по тому, к какому лагерю принадлежал автор,— болот»
ный газ (метан) либо этилен».
В 50-х годах прошлого столетия «анархия» в химии посте-
пенно пошла на убыль. В результате работ Франклан-
да и Кекуле в химии утвердилось понятие о валентности.
В частности, Кекуле развил представление о четырехвалентно-
сти углерода. Благодаря трудам Канниццаро была внесена
ясность в вопрос об атомных и молекулярных массах, об экви¬
валентах.
В 1860 г. более 140 ведущих химиков из разных стран Ев¬
ропы собрались на международный конгресс в г. Карлсруэ. На
этом конгрессе было достигнуто единое понимание фундамен¬
тальных понятий химии (атом, молекула, эквивалент), признана
справедливость закона Авогадро, вошли в употребление пра¬
вильные значения атомных масс элементов. Все эти успехи
науки подготовили условия для нового этапа в развитии орга¬
нической химии — появления теории химического строения ор¬
ганических соединений.
§ 9. Теория химического строения органических соединений.
Создателем теории химического строения был великий русский
химик Александр Михайлович Бутлеров (1828—1886 гг.). Ос¬
новные положения своей теории Бутлеров сформулировал в
1858—1861 гг. Некоторая трудность для нас заключается в том,
что создатель теории строения не формулировал пункт за пунк¬
том положения своей теории в одном месте: они пронизывают
все научное творчество А. М. Бутлерова и его учеников.
1. Атомы в органических молекулах связаны друг с другом
в определенном порядке химическими силами. Этот порядок
Бутлеров и называл химическим строением. Во времена Бутле¬
рова лишь в общих словах называли эти силы «силами валент¬
ности»: в наше время выяснено, что силы валентности имеют
йлектронную природу (см. § 12).
2. Строение можно изучить экспериментально, используя
химические методы — анализ и синтез. В наше время при уста¬
новлении строения широко используют также физические мето¬
ды — разные виды оптической спектроскопии, ядерный магнит¬
ный рёзонанс, масс-спектрометрию, рентгенографию, электроно¬
графию, определение Дипольных моментов.
Физические методы, в первую очередь рентгенография и
электронография, позволяют определить реальное положение
каждого атома в молекуле, получить как бы ее фотографию.
Это расположение согласуется с найденным чисто химическим
путем. Поэтому мы чаще говорим просто о строении молекул,
опуская прилагательное «химическое», которое обязательно
употреблял Бутлеров.
3. Формулы строения выражают порядок химической связи
атомов. В формулах строения (структурных формулах) симво-
22
лы элементов соединяют черточками, условно изображающими
химическую связь:
Н
■
Н Н
1 1-
н—С—С—н
U
Н Н Н
1 1 1
н—С—н
А
н—£—£—с—
А <!)—н н
метан СНз
этан С2Нв
изопропиловый
спирт С3Н3О
Чаще используют сокращенные структурные формулы:
он
СНз—СНз СНз—CH— СH3 или СН3—СНОН—СНз
этан изопропиловый спирт
При построении структурных формул органических соедине¬
ний необходимо учитывать найденную опытным путем валент¬
ность элементов-органогенов. Так, валентность углерода 4, во¬
дорода 1, кислорода 2, галогенов 1. Азот, сера и фосфор могут
находиться в разных валентных состояниях.
4. Каждое вещество имеет одну определенную формулу стро¬
ения, отражающую порядок химической связи атомов в реально
существующей молекуле. Это положение глубоко материалис¬
тично, философски правильно: в основе лежит реально суще¬
ствующий в природе объект — молекула, и структурная форму¬
ла должна возможно точнее отразить этот объект. Выполнению
этого требования подчинены все современные «усовершенство¬
вания» в написании формул органических веществ—введение в
них обозначений электронных пар, стрелок, пунктиров, знаков
заряда. Все это улучшает соответствие между формулой и ре¬
альной молекулой, т. е. отвечает принципам теории строения.
5. Связанные в молекуле атомы оказывают друг на друга
взаимное влияние: свойства каждого атома в составе молекулы
зависят не только от его собственной природы, но и от окру¬
жения, в котором этот атом находится. С взаимным влиянием
атомов мы постоянно будем встречаться во всем курсе органи¬
ческой химии. Пока ограничимся простейшим примером: и в
молекуле воды, и в молекуле хлороводорода имеется атом во¬
дорода, но сколь различны его свойства в обоих веществах!
6. Физические и химические свойства органических соеди¬
нений определяются составом и строением их молекул. Во вре¬
мена Бутлерова можно было судить лишь о химическом строе¬
нии — порядке химической связи атомов. В настоящее время
имеется возможность определять также пространственное строе¬
ние; определять распределение электрических зарядов — элек¬
тронное строение. Все три особенности строения важны при
рассмотрении свойств органических соединений.
23
Рис. 4. Формы электронного облака:
а — сферическое s-облако; б — эллипсоидное р-облако
Теория химического строения позволила понять природу изо¬
мерии: молекулы структурных изомеров имеют одинаковый со¬
став, но различаются порядком связи атомов (химическим
строением). Например, упоминавшаяся ранее (см. § 7) форму¬
ла С2Н6О отвечает двум различным веществам:
СН3—О—СН3 СН3—СН2ОН
диметиловый эфир этиловый спирт
А. М. Бутлеров не ограничился лишь теоретическим объясне¬
нием изомерии на основе теории строения. Он провел и ряд
экспериментальных работ, подтвердив предсказания теории по¬
лучением изобутана, третичного бутилового спирта и других
веществ.
§ 10. Электронная оболочка атома. Благодаря успехам химии
и физики в настоящее время установлено, что химические явле¬
ния связаны с процессами, происходящими в электронной обо¬
лочке атомов. Электронное строение атомов должно быть из¬
вестно из курсов физики и неорганической химии; здесь мы
лишь кратко напомним о нем. Атом состоит из положительно
заряженного ядра, окруженного электронной оболочкой с отри¬
цательным зарядом. Первоначально предполагали, что атом
можно представить себе в виде миниатюрной солнечной систе¬
мы, в которой ядро играет роль солнца, а вокруг него движутся
планеты — электроны. Однако вскоре выяснилось, что законы
квантовой механики, действующие в мире элементарных частиц
(протонов, нейтронов, электронов и др.), существенно отличают¬
ся от привычных «обычных» физических законов.
Согласно современным квантово-механическим представле¬
ниям электрон одновременно обладает свойствами частицы и
волны; вместо материальной точки рассматривается электрон¬
ное облако. Местоположение движущегося в атоме электрона
определяется орбиталью — пространством вблизи атомного
ядра, в котором достаточно велика (≈90%) вероятность на¬
хождения электрона.
24
Состояние электрона в атоме описывается с помощью четы¬
рех квантовых чисел.
Главное квантовое число п определяет электронный слой
(К-слой, L-слой, M-слой и т. д.), который занимают электроны
в соответствии со своей энергией, и энергию электронов, т. е.
его энергетический уровень. Главное квантовое число принимает
целые значения: 1, 2, 3, 4, . . . ; значение 1 соответствует энер¬
гетическому уровню с наименьшей энергией электрона, более
высокие значения п отвечают большей энергии электрона.
Форму электронного облака определяет побочное (азиму¬
тальное, орбитальное) квантовое число (l), которое может при¬
нимать значения от 0 до п — 1. Значению l = 0 отвечает сфе¬
рическое электронное облако, в центре которого находится
атомное ядро (рис. 4, а); такие электроны называют s-электро¬
нами. Значению l = 1 отвечает электронное облако в виде двух
эллипсоидов (для наглядности их часто изображают в виде
«гантелей»), между которыми находится атомное ядро
(рис. 4,6); такие электроны называют р-электронами. Два упо¬
мянутых состояния электронов, находясь в пределах одного
энергетического уровня, все же несколько различаются по энер¬
гиям (находятся на разных энергетических подуровнях), причем
энергия р-электронов несколько больше, чем у s-электронов
(рис. 5).
При главном квантовом числе 3 появляются электронные
облака более сложной формы. Для элементов-органогенов такие
облака имеют малое значение, поэтому мы их не рассматри¬
ваем.
Магнитное квантовое число m характеризует у элементов
второго периода ориентирование эллипсоидных облаков в про¬
странстве: магнитные квантовые числа в этом случае обозна¬
чают индексами при букве р (т. е. рх, Ру, Рz), связывая их с
ориентировкой эллипсоидных р-облаков по трем координатным
осям.
Квантовые числа п, l и m определяют, таким образом, обла¬
сти пространства, в которых «разрешено» пребывание электро¬
нов. Такие области пространства вокруг ядра получили назва¬
ние электронных орбиталей. Они мо¬
гут быть занятыми (заполненными б
электронами) или незанятыми (сво¬
бодными).
Четвертое квантовое число — спи¬
новое (s) характеризует вращение
электрона вокруг собственной оси. По¬
добно волчку, электрон может вра¬
щаться по часовой стрелке или про¬
тив нее. Электроны, имеющие одина-
Рис. 5. Энергия электронных состояний
—за
И
li
2s
<s
9 0 А
25
Таблица 1. Электронные состояния атомов
Элемент
Атом¬
ный
номер
Распределение электронов
Электронная
формула
К-слой
(п=1,
тй), (=0)
1-слой (пв 2)
1=0,
т=0
м,
т=-1
1=0,
т=0
1=1.
т=+1
Водород 1
IX
и
Гелий 2 | Т ^ 1 1т2
Литий
Бериллий
Бор
Углерод
Азот
Кислород
Фтор
Неон
1-Т 1.
ш:
5 ГХХ
6 гтт
I—Г.1
ни
хх
10
XX
XX
XX
2ЕХ
XX
XX
XX
тт-
XX
XX
ТХ~)
ХХ~1
XX
lA
lAs2
\s22s22p
ls22s22p2
ls22s22p3
lr22s22p4
ls22s22p6
li22s22p6
ковые квантовые числа n, l и m (занимающие одну электронную
Орбиталь), но различающиеся спином, образуют электронную
пару. Далее мы узнаем, что именно электронные пары ответ¬
ственны за образование связей в молекулах органических ве¬
ществ.
Согласно принципу Паули, в электронной оболочке атома
не может быть двух электронов, у которых все четыре кван¬
товые числа одинаковы.
В табл. 1 приведены электронные состояния для элементов
первого и второго периодов Периодической системы элементов
Д. И. Менделеева. В этой таблице каждый электрон обозначен
стрелкой; противоположные направления стрелок означают про¬
тивоположные спины. Каждая электронная орбиталь условно
изображена в виде прямоугольника или квадрата (квантовая
26
Рис. 6. Электронные орбитали невозбужденного (а) и возбужденного (б)
атомов углерода
ячейка); расположение прямоугольников на разной высоте на¬
поминает о различающихся энергиях соответствующих орбита-
лей. Электронное строение атомов изображают также элек?рон-
ными формулами, которые показывают распределение электро¬
нов по энергетическим уровням (обозначают цифрами 1, %
3, ...) и подуровням (обозначают буквами s, р, d, ...).
Рассмотрение табл. 1 позволяет заметить соответствие меж¬
ду числом неспаренных электронов и валентностью: один — для
водорода, лития, фтора; два — для кислорода; три — для азота.
Есть, однако, и исключения: у бора один валентный электрон,
а обычная валентность — два. У углерода два неспаренных
электрона (рис. 6, а), а его обычная валентность равна че¬
тырем.
§11. Валентные состояния углерода. Гибридизация. Чтобы
согласовать электронное строение атома углерода и валентность
этого элемента, используют представление о возбуждении ато¬
ма углерода:
С → СВОЗБ
В возбужденном состоянии атом углерода имеет четыре не¬
спаренных электрона, следовательно, может проявлять четырех*
валентность. Однако обнаруживается новое противоречие: четы¬
ре валентных электрона не одинаковы по своему состоянию
|[один 2s-, и три 2p-электрона (рис. 6,6)], в то время как дока¬
зано на опыте, что все четыре валентности углерода одинаковы
и направлены друг относительно друга под тетраэдрическими
углами 109°28'. Чтобы согласовать теоретическое рассмотрение
С опытными данными, было введено (Полинг) представление о
гибридизации атомных орбиталей — своеобразном «смешении»
шаровой s- и эллиптических p-орбиталей. Для атома углерода
27
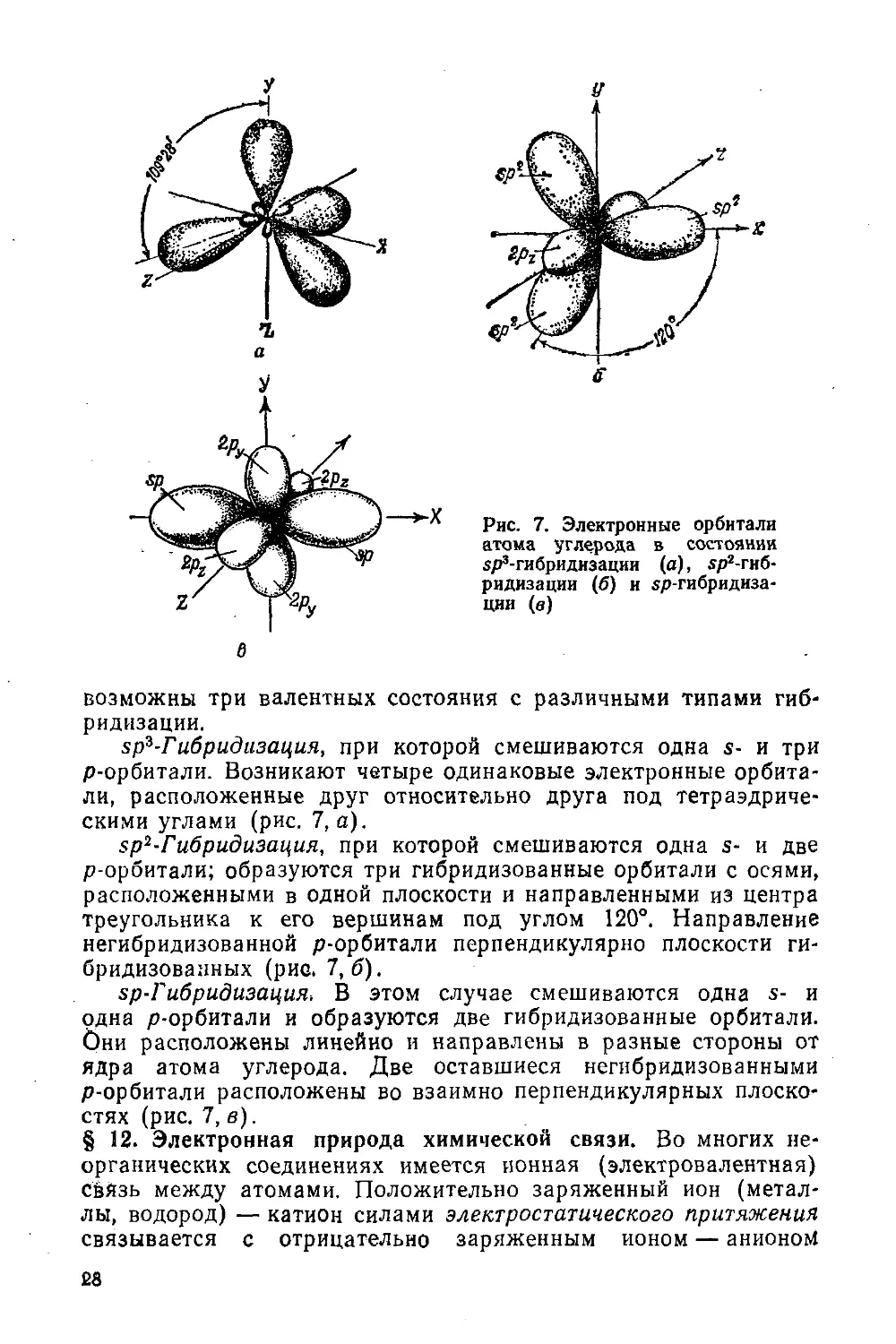
У
V
1.
У
l
X
Рис. 7. Электронные орбитали
атома углерода в состоянии
sр3-гибридизации (а), sp2-гиб¬
ридизации (б) и sp-гибридиза¬
ции (в)
6
возможны три валентных состояния с различными типами гиб¬
ридизации.
sp3 - Гибридизация, при которой смешиваются одна s- и три
р-орбитали. Возникают четыре одинаковые электронные орбита¬
ли, расположенные друг относительно друга под тетраэдриче¬
скими углами (рис. 7, а).
sp2-Гибридизация, при которой смешиваются одна s- и две
р-орбитали; образуются три гибридизованные орбитали с осями,
расположенными в одной плоскости и направленными из центра
треугольника к его вершинам под углом 120°. Направление
негибридизованной р-орбитали перпендикулярно плоскости ги-
бридизованных (рис. 7,6).
sp-Гибридизация. В этом случае смешиваются одна s- и
одна р-орбитали и образуются две гибридизованные орбитали.
Они расположены линейно и направлены в разные стороны от
ядра атома углерода. Две оставшиеся негибридизованными
р-орбитали расположены во взаимно перпендикулярных плоско¬
стях (рис. 7, в).
§ 12. Электронная природа химической связи. Во многих не¬
органических соединениях имеется ионная (электровалентная)
связь между атомами. Положительно заряженный ион (метал¬
лы, водород) — катион силами электростатического притяжения
связывается с отрицательно заряженным ионом — анионом
(гидроксид-ион, кислотные остатки). При этом образуются
устойчивые электронные оболочки, отвечающие электронной
конфигурации инертных газов — гелия (2 электрона на /(-обо¬
лочке), неона (8 электронов на L-оболочке, октет) и др.
Отсутствие диссоциации на ионы у большинства органиче¬
ских соединений и другие их свойства давно уже заставили
предполагать, что тип связи в органических молекулах иной:
эту связь называют ковалентной. Осуществляется ковалентная
связь путем обобщения электронов. Простейший пример кова¬
лентной связи — связь между атомами в молекуле водорода.
Каждый из атомов водорода имеет по одному электрону. Если
сближаются два атома водорода, электроны которых имеют
противоположные спины, то электронная плотность их объеди¬
няется: обобщая свои электроны, оба атома приобретают устой¬
чивую электронную оболочку ближайшего инертного газа —
гелия:
Н• + •Н → Н:Н или H↑+ ↓H → Н↑↓Н
Перейдя от «точечной» модели электрона к представлению
об электронном облаке, получим эллипсоид, в котором элек¬
тронная плотность между ядрами больше, чем в других местах,
что и стягивает положительно заряженные ядра (рис. 8).
Таким же путем образуются ковалентные связи в органиче¬
ских молекулах, например в метане СН4 и этане С2Н6 [для на¬
глядности собственные валентные электроны углерода обозна¬
чены крестиками; электроны, ранее принадлежавшие атомам
водорода,— точками (это сделано только для иллюстрации спо¬
соба образования связи, на самом деле электроны, конечно,
неотличимы)]. В результате обобщения электронов атом угле¬
рода имеет теперь на внешней орбитали восемь электронов, как
у неона, а каждый из четырех атомов водорода — по два, как
у гелия. Каждый из атомов достиг, следовательно, устойчивого,
отвечающего инертному газу электронного состояния:
н н н
н;с;н шсгс;н
'X «х • •
н н н
метан этан
Атомы углерода в метане и этане находятся в состоянии
sp3-гибридизации, имея тетраэдрическую конфигурацию (рис. 9).
Помимо простых (одинарных)
связей, в создании которых уча¬
ствует пара электронов, в орга¬
нических молекулах встречают¬
ся двойные и тройные связи:
Рис. 8. Распределение электронной плот¬
ности в молекуле водорода (цифры
означают доли заряда электрона)
29
Рис. 9. Модель σ-связей в молекулах метана (а) и этана (б)
их образуют соответственно две или три электронные пары, на¬
пример:
н н
с;;с н;с»«с;н
• х • • х
н н
этилен ацетилен
Углерод в этих соединениях находится в состояниях соот¬
ветственно sp2- и sp-гибридизации.
Подробнее с разными типами ковалентных связей в органи¬
ческих соединениях мы будем знакомиться постепенно.
Может возникнуть вопрос: почему углерод образует кова¬
лентные связи путем обобщения электронов, а не склонен к
образованию связей путем отдачи или принятия электронов?
Имея четыре валентных электрона, атом должен был бы при¬
нять еще четыре электрона, чтобы образовать устойчивый октет.
Между тем, после присоединения первого электрона углерод
приобрел бы отрицательный заряд и подход каждого нового
электрона к аниону небольшого размера (радиус атома углеро¬
да мал) требовал бы все большей затраты энергии: поэтому
образование иона С4- крайне невыгодно. Так же труден и от¬
рыв четырех электронов, приводивший бы к образованию иона
С4+, поскольку каждый последующий электрон должен был бы
уходить, преодолевая притяжение остающегося катиона. Гораз¬
до более благоприятны условия для образования ковалентных
связей путем обобщения электронов, при котором не возникают
заряды.
В настоящее время все шире используют иной подход к теоретическому
рассмотрению строения органических соединений: теорию молекулярных ор¬
биталей. Вместо локализованных (закрепленных) электронных пар рассматри¬
вают по существу взаимодействие всех электронов со всеми ядрами. Основой
теории МО является сложный математический аппарат.
30
§ 13. Характеристики ковалентных связей и методы их опре¬
деления. Большим достижением современной науки является
возможность дать каждой конкретной связи точную числен¬
ную характеристику. В нее входят геометрические парамет¬
ры — длины связей и валентные углы, энергия связей, а также
электронные параметры — полярность и поляризуемость (см.
табл. 2).
Длина связи — расстояние между центрами связанных ато¬
мов — зависит от природы атомов и характера связи между
ними (одинарная, двойная или тройная). С повышением крат¬
ности связи становятся короче.
Валентным углом называют угол между направлениями свя¬
зей, образуемых многовалентным атомом. Так, в молекуле воды
две связи О—Н расположены друг относительно друга под
углом около 100°; это и есть валентный угол атома кислорода.
Атом азота в трехвалентном состоянии имеет три связи, направ¬
ленные к углам основания трехгранной пирамиды, в вершине
которой находится азот. В качестве примеров на рис. 10 приве¬
дены длины связей и валентные углы в молекулах воды и
аммиака. Валентный угол атома углерода зависит от типа гиб¬
ридизации.
Геометрические параметры молекул определяют методами
рентгенографии и электронографии. Эти методы основаны на
том, что проходящие через вещество рентгеновские лучи или
поток электронов дают дифракционную картину, на основании
которой можно вычислить расстояния между атомами и валент¬
ные углы. Оба метода хорошо дополняют друг друга: рентгено¬
графический метод обладает большой точностью (расстояния
между атомами измеряются с точностью ±0,0001 нм), но при
его помощи нельзя определить положение атомов водорода.
Метод электронографии менее точен (0,001 нм), но устанавли¬
вает и положение атомов водорода. Рентгенографию применяют
главным образом в случае кристаллических веществ, электроно¬
графию — для газов.
Межатомные расстояния и валентные углы можно опреде¬
лять также методом дифракции нейтронов, вычислять из спек¬
тральных данных. Все эти методы позволили получить надеж¬
ные данные о геометрическом строении молекул органических
Рис. 10. Длины связей и валентные углы а молекулах воды (а) и аммиа
ка (б)
31
соединений. Эти данные подтвердили и уточнили то, что химики
давно вывели своими методами — на основании теории химиче¬
ского строения А. М. Бутлерова и стереохимической гипотезы
Я. Г. Вант-Гоффа.
Энергия связи — величина, характеризующая прочность свя¬
зи. При образовании химической связи всегда выделяется энер¬
гия; ее называют энергией связи и измеряют в килоджоулях на
моль (кДж/моль). Чтобы разорвать связь, надо затратить опре¬
деленную энергию: очевидно, чем больше энергия связи, тем
эта связь прочнее, тем труднее ее разорвать.
Наиболее прямой путь получения сведений об энергиях свя¬
зи — использование термохимических данных, т. е. сведений о
тепловых эффектах реакций. Практически чаще всего эти дан¬
ные получают в виде теплот сгорания, т. е. теплового эффекта,
которым сопровождается полное сгорание органического соеди¬
нения до оксидов составляющих его элементов (С02, Н20, S02).
Азот, бром и иод выделяются в свободном виде, хлор образует
НС1. Сжигание проводят в калориметрах — приборах, состоя¬
щих из прочных металлических сосудов для сожжения веще¬
ства под давлением кислорода, причем по повышению темпера¬
туры в специальной водяной рубашке сосуда учитывают коли¬
чество выделившегося тепла. Полученные данные используют
для расчета теплот образования соединений из атомов состав¬
ляющих их элементов; от теплот образования переходят к энер¬
гиям связей. Так, например, теплота образования метана равна
1660 кДж/моль. Поскольку при образовании метана возникают
четыре С—Н-связи, на долю каждой из них приходится энергия
1660:4 = 415 кДж/моль. Разность между теплотами образова¬
ния двух соседних членов ряда парафинов составляет около
1180 кДж/моль; это значение соответствует теплоте образова¬
ния группы СН2, т. е. созданию дополнительной С—С-связи
и двух С—Н-связей. Вычитая из приведенного выше значения
энергию двух С—Н-связей, можно получить энергию С—С-связи
(≈350 кДж/моль). Аналогично находят и энергии других
связей.
Полярность связи указывает на характер распределения
электронов между связанными атомами. Атомы разных элемен¬
тов обладают различным сродством к электронам: одни из них,
подобно литию и натрию, легко отдают электроны, другие, по¬
добно фтору и хлору, охотно принимают их. Известно, что
стремление к присоединению электронов (так называемая элек¬
троотрицательность атомов) растет в Периодической системе
слева направо и снизу вверх. В соответствии со своей электро¬
отрицательностью атомы ведут себя и в отношении связываю¬
щей их электронной пары: один из партнеров связи может быть
довольно «равнодушен» к электронам и предоставить другому
партнеру возможность завладеть связующей электронной парой
«больше чем наполовину». В результате одна часть молекулы
32
приобретает положительный заряд, другая — отрицательный.
Эти заряды не будут, вообще говоря, равны полному заряду
электрона. Подобные частичные заряды принято обозначать
знаками δ+ и δ-. Простейшим примером может служить мо¬
лекула воды.
. "■ , +е г ~е
8+ О 8+ см ►<>
Иг д=ег
8*
н-
0,127 нм 8-
г-»-С1
Д=1,081Д
Полярность — очень важное свойство связей, так как имен¬
но наличие того или иного заряда на атоме определяет его
реакционные возможности. Полярность отражается и на физи¬
ческих свойствах. В самом наглядном виде она проявляется в
существовании дипольных молекул, полярность которых может
быть экспериментально измерена и выражена в виде дипольно¬
го момента μ — произведения заряда е (в электростатических
единицах) на расстояние между зарядами r (в нм). За едини¬
цу дипольного момента принят дебай (Д).
Полярность ковалентных связей экспериментально определя¬
ется прежде всего из дипольных моментов. Поясним это на
простом примере. Экспериментально определенный дипольный
момент газообразного хлороводорода равен 1,08 Д, а расстоя¬
ние Н—С1 равно 0,127 нм. Используя соотношение между ди¬
польным моментом, зарядами и расстоянием между ними,
можно вычислить значение зарядов на атомах водорода и
хлора:
е = μ/r = 1,08 • 10-18/(0,127 • 10-7) = 0,85 • 10-10 эл.-ст. ед
Это означает, что в газообразном хлороводороде на атомах
находится менее !/б заряда электрона (0,85:4,8); связь Н—С1
в значительной мере ковалентна. Ионной она становится лишь
тогда, когда эта молекула попадает в среду с большой диэлек¬
трической проницаемостью — в воду.
Помимо полярности, присущей связям в статическом состоя¬
нии, каждая связь обладает еще определенной поляризуе¬
мостью — способностью изменять (увеличивать) свою поляр¬
ность под действием внешнего электромагнитного поля. Такое
иоле может создавать и частица, приближающаяся во время
химической реакции, поэтому поляризуемость в значительной
степени определяет химические свойства.
В табл. 2 сопоставлены физико-химические характеристики
разных ковалентных связей. Эти величины могут заметно изме¬
няться под влиянием соседних атомов (соседних связей): в этом
и проявляется, в частности, взаимное влияние атомов. В табли¬
це всюду первым указан атом, образующий положительный
конец диполя.
2 Органическая химия
33
Таблица 2. Длины, энергии, полярности и поляризуемости
некоторых ковалентных связей
Тип
СВЯЗИ
Длина, нм
Энергия,
кДж/моль
Полярность,
Д
Поляризуе¬
мость, см3
С-
-с
0,154
348
0
1,3
С=
=с
0,133
620
0
4,2
с=
еС
0,120
810
0
6,2
с-
-N
0,147
290
0,5
1,6
с=
=N
0,127
615
1,4
3,8
Сз
eN
0,115
880
3,1
4,8
с-
-О
0,143
340
0,7
1,5
с=
о
0,121
710
2,4
3,3
с-
-F
0,140
485
1,4
1,4
с-
-С1
0,176
330
1,5
6,5
с-
-Вт
0,191
280
1,4
9,4
с-
-I
0,212
240
1,3
14,6
н-
-С
0,109
415
0,4
1,7
н-
-О
0,096
465
1,5
1,7
н-
-S
0,134
340
4,8
н-
-N
0,101
390
1,3
1,8
N-
-N
0,148
160
0
2,0
N=
=N
0,124
420
0
4,1
Ns
eN
0,109
950
0
N-
-О
0,137
200
1,0
2,4
N=
=0
0,122
400
3,0
4,0
§ 14. Типы органических реакций. Органические реакции мож¬
но классифицировать, подобно неорганическим, по их результа¬
там, различая
реакции замещения: СН4 + Вr2 → СН3Вr + НВr
реакции присоединения: СН2=СН2 + Вr2 → ВrСН2—СН2Вr
реакции отщепления: ВrСН2—CH2Br+Zn → СН2=СН2 + ZnBr2
Особый, важный вид реакций присоединения составляют
взаимодействия между двумя (несколькими) органическими мо¬
лекулами, ведущие к усложнению углеродного скелета. Такие
превращения называют реакциями полимеризации, конденсации
и поликонденсации (см. §§ 86, 185, 189).
В современной органической химии появилась возможность
классифицировать органические реакции по их механизмам.
При изучении механизмов реакций устанавливают, как именно,
в каком порядке и каким путем разрываются старые и обра¬
зуются новые химические связи в процессе реакции. Классифи¬
цируя реакции по их механизмам, прежде всего обращают вни¬
мание на способ разрыва ковалентной связи в реагирующей
молекуле. Таких способов два — гемолитический и гетеролити-
ческий.
При гомолитическом (радикальном) разрыве ковалентной
связи связующая электронная пара разъединяется, причем каж¬
34
дый из образующихся свободных радикалов сохраняет один не-
спаренный электрон, оставаясь электронейтральной частицей:
Н Н
Н s С:Н —► HsC. + .H
tt ••
н н
ретан радикал атом
метил водорода
Радикальному разрыву подвергаются обычно неполярные
или малополярные связи (С—С, N—N, С—Н) при высокой
температуре, под действием УФ-света или радиоактивного из¬
лучения.
Образовавшиеся при гомолитическом разрыве связи радика¬
лы и свободные атомы неустойчивы и способны существовать
лишь очень непродолжительное время. Эти промежуточные час¬
тицы подвергаются дальнейшим превращениям, переходя в ус¬
тойчивые конечные продукты. Так, например, радикал метил и
атом водорода могут стабилизироваться путем димеризации:
• CH3 + • CH3 → H3C:CH3 Н• + •Н → Н:Н
этан
Если в реакционной смеси присутствуют другие вещества
(примеси или специально добавленные реагенты), то осущест¬
вляются другие пути стабилизации радикалов. С ними мы по¬
знакомимся по мере изучения конкретных реакций.
Гетеролитический (ионный) разрыв ковалентной связи при¬
водит к образованию заряженных осколков — катионов и анио¬
нов. Связующая электронная пара не разъединяется, а целиком
отходит к более электроотрицательному атому, который приоб¬
ретает отрицательный заряд, превращается в анион. Вторая
частица, лишившись электрона, приобретает положительный
заряд, становится катионом. Частица с положительным зарядом
на атоме углерода называется карбокатионом (карбениевым
ионом):
Н
Н
Н: С: I—*- Н;С+ + :Г
н
н
метил-
иодид
метил- иодид-
катион анион
(карбо-
катион)
К гетеролитическому разрыву склонны сильно полярные и
легко поляризуемые связи. Ему способствуют растворители с
высокой диэлектрической проницаемостью и большой поляр¬
ностью.
Карбокатионы неустойчивы, как и свободные радикалы. Они
легко реагируют с частицами, обладающими избытком элек¬
2*
35
тронной плотности в виде полного или частичного отрицатель¬
ного заряда, а также свободными электронными парами:
СН3 + : ОН —► СНз: ОН
метил- гидро- метиловый
катион ксид- спирт
анион
Такие частицы называют нуклеофильными («любящими яд¬
ра»). К нуклеофильным реагентам относятся: вода, щелочи,
аммиак, кислотные остатки (например, -CN, -S03H).
Гетеролитический разрыв ковалентной связи может приво¬
дить и к образованию органических анионов {карбанионов),
например:
СНз : Li → : CH3 + Li+
Стабилизация карбанионов происходит в результате их ре¬
акций с электрофильными («любящими электроны») реагента¬
ми. К их числу относятся: протон, катионы металлов, карбока-
тионы, органические остатки с частичным положительным заря¬
дом. Примером может служить реакция метиллития с водой:
СНз—Li + Н—ОН —► СН4 + Li+ + НО'
Свободные радикалы, карбокатионы и карбанионы играют
роль промежуточных реакционноспособных частиц во многих
органических реакциях.
§ 15. Классификация органических соединений. Основу любого
органического вещества составляет последовательность химиче¬
ски связанных атомов углерода — его углеродный скелет. Раз¬
личают следующие типы углеродного скелета.
а) Ациклический скелет, т. е. углеродные цепи неразветвлен-
ные (нормальные) и разветвленные:
С
I
С—С—С—С—С—С с—с—с—с—с—с
I
с
б) Циклический скелет:
С
<<L>c
С—С
с/ \с
Ч /°
с—с
с—с
с/ \с
сч /с
с=с
скелет
бензола
в) Гетероциклический скелет:
с-с
</ \с
\/
о
с
O'
36
Оч. /С
N
Таблица 3. Функциональные группы и классы органических соединений
Класс соединений
Типичные представители
группы
название
формула
Галогены F, С1,
Вг, I
Г алогенпроизводные
Метилиодид
СНз—I
Г идроксильная
—ОН
Спирты
Метиловый
спирт
СНз—ОН
Фенолы
Фенол
С2Н5—он
Карбонильная
^С=0
Альдегиды
Уксусный аль¬
дегид
CH3—CHO
Кетоны
Ацетон
CH3—CO—CH3
Карбоксильная
<
Карбоновые кислоты
Уксусная кис¬
лота
сн3—соон
Нит^огруппа
Нитросоединения
Нитробензол
C6H5—NO2
Аминогруппа
—NHS
Амины
Этиламин
C2H5—NH2
Диазогруппа
—NjX
Диазосоединения
Бензолдиазо-
нийхлорид
С6Н5—N2Cl
Меркаптогруппа
—SH
Тиолы (тиоспирты)
Метантиол
СНз—SH
Сульфогруппа
—S03H
Сульфокислоты
Бензолсульфо-
кислота
C6H55—SO3H
Металлы (Li, Mg,
Al)
Металлорганические
соединения
Метиллитий
СНз—Li
Все эти схемы показывают порядок химической связи ато¬
мов, но не передают их пространственного расположения. Так,
неразветвленная углеродная цепь на самом деле имеет зигзаго¬
образную форму и может быть изогнута в пространстве самым
причудливым образом.
Органические вещества с нормальными и разветвленными
углеродными цепями называют соединениями жирного ряда,
ациклическими или алифатическими. Соединения с углеродны¬
ми циклами называют алициклическими. В состав гетероцикли¬
ческого кольца входят атомы не только углерода, но и других
элементов — гетероатомы (от греч. «гетерос» — разный); здесь
название «углеродный скелет» условно.
В большинстве органических молекул помимо углерода и во¬
дорода содержатся атомы других элементов. Именно от них в
первую очередь зависят химические свойства органических ве¬
ществ. Эти атомы (кислород, азот, сера и др.) входят в состав
особых группировок, называемых функциональными группами.
Присутствие той или иной функциональной группы определяет
37
принадлежность органических соединений к определенным клас¬
сам. Главные функциональные группы и отвечающие им классы
органических соединений приведены в табл. 3.
«Органическая химия есть химия углеводородов и их произ¬
водных» — говорил К. Шорлеммер еще в прошлом веке; при
этом под производными он имел в виду вещества, получаемые
при введении функциональных групп в молекулы углеводо¬
родов.
Гетероатомы, входящие в состав функциональных групп, по¬
зволяют подразделять их на кислородные функции (ОН, СО,
СООН и др.); азотные функции (NO, NO2, NH2 и др.); серни¬
стые функции (SH, SO, SO2, SO3H и др.); фосфорные функции
[РН2, РО(ОН)2 и др.].
Во многих функциональных группах можно узнать осколки неорганиче¬
ских молекул: ОН — остаток воды, NO2 — остаток азотной кислоты, NH2 —
остаток аммиака, SO3H — остаток серной кислоты. И действительно, соответ¬
ствующие органические соединения в значительной мере сохраняют химиче¬
ские особенности своих неорганических прототипов. Общее число известных
в настоящее время функциональных групп весьма велико и даже в больших
курсах органической химии рассматривается лишь часть их.
В молекуле органического вещества могут находиться и не¬
сколько функциональных групп. Если эти группы одинаковы
(два галогена, два или три гидроксила и т. д.), такое соедине¬
ние называют полифункциональным. Органические соединения,
характеризующиеся присутствием в молекуле нескольких раз¬
ных функциональных групп, называют гетерофункциональными.
ЧАСТЬ I
УГЛЕВОДОРОДЫ
Углеводороды — органические соединения, в молекулы которых
входят атомы лишь двух элементов — углерода и водорода.
Углеводороды могут отличаться друг от друга по строению
углеродного скелета. Благодаря способности атомов углерода
образовывать цепи и циклы различного размера и формы, а
также различные типы связей возможно существование большо¬
го числа углеводородов. Углеводороды разных типов отличают¬
ся друг от друга также степенью насыщения водородом. Число
атомов водорода в молекуле любого углеводорода четное.
ГЛАВА 2
АЛКАНЫ
Алканы являются углеводородами, наиболее богатыми водоро¬
дом, они насыщены им до предела. Отсюда названия — насы¬
щенные (предельные) углеводороды. Их называют также пара¬
финами или углеводородами ряда метана (по первому члену
ряда).
§ 16. Гомологический ряд, строение, изомерия. Г омологиче¬
ски й ряд. Простейший представитель алканов — метан СН*.
Существует ряд углеводородов, подобных метану:
сн4
метан
С9Н20
конан
С2н6
этан
С10Н22
декан
С3Н8
пропан
С11Н24
ундекан
С4Н10
бутан
СцНгв
додекан
С5Н12
пентан
С13Н23
тридекан
С6Н14
гексан
С14Нзо
тетрадекан
С7Н16
с8н18
гептан
октан
с15н32
пентадекан и др.
Рассматривая приведенный ряд соединений, можно заметить,
что по составу каждый последующий член ряда отличается от
предыдущего на одну группу СН2. Можно вывести и общую
формулу ряда СnН2n+2.
Ряд соединений, обладающих сходным химическим строени¬
ем, сходными свойствами и отличающихся друг от друга по
составу на группу СН2, называется гомологическим рядом. Чле¬
ны этого ряда носят название гомологов, разница в составе
двух соседних членов ряда — группа СН2 — называется гомоло¬
гической разностью состава. В дальнейшем мы узнаем, что хи¬
мические свойства всех членов гомологического ряда сходны.
39
Гомология — явление общее для органической химии в целом. Это яркий
пример проявления одного из основных законов природы — закона перехода
количества в качество. Энгельс писал: «...Прибавляя каждый раз группу СН2,
мы получаем тело, качественно отличное от предыдущего тела». «Химию
можно назвать наукой о качественных изменениях тела, происходящих под
влиянием изменения количественного состава» (Ф. Энгельс, Диалектика при¬
роды, 1969, с. 47, 48).
Открытие явления гомологии облегчило изучение органических веществ:
достаточно изучить свойства типичного представителя ряда, чтобы иметь
понятие о свойствах ряда. Однако первый член ряда обычно нетипичен для
ряда в целом.
Строение. Общая черта в строении насыщенных соеди¬
нений — простая или одинарная связь между атомами углерода.
На образование этой связи затрачивается одна пара электро¬
нов, причем максимальное перекрывание электронных орбита-
лей находится на прямой, соединяющей ядра атомов. Такую
связь называют σ (сигма) -связью, а электроны, образующие
σ-связь, — σ-электронами. Распределение электронной плотно¬
сти σ-связи симметрично относительно оси, проходящей через
центры связанных атомов (см. § 12).
В молекулах насыщенных углеводородов атомы углерода
находятся в состоянии sр3-гибридизации и каждый из них об¬
разует четыре σ-связи с углеродом или водородом. Состояние
sр3-гибридизации характеризуется тетраэдрической конфигура¬
цией углеродного атома (см. рис. 10). Данные о длинах и энер¬
гиях связей С—С и С—Н приведены в табл. 2 (см. § 13).
Изомерия. Как уже отмечалось, изомерия проявляется
в существовании различных веществ, имеющих один и тот же
состав молекул. Изомеры чаще всего возникают из-за различий
в химическом строении. Различный порядок связи атомов обус¬
ловливает и некоторое различие в их свойствах, так как наи¬
более сильное влияние друг на друга оказывают атомы, связан¬
ные непосредственно. Изменение свойств отдельных атомов и
связей проявляется в свойствах соединения в целом.
Так, для углеводорода состава С4Н10 известны два изомера:
нормальный бутан — соединение с неразветвленной углеродной
цепью и изобутан, имеющий разветвленную углеродную цепь:
СН3
I
СН3—СН2—CHj—СН3 СН3—СН—СН3
к-бутан изобутан
Оба эти углеводорода — газы, но с различной температурой
кипения: —0,5 °С для бутана и —П,7°С для изобутана. Изо¬
бутан впервые синтезировал А. М. Бутлеров (1867 г.), доказав
тем самым существование изомерии у углеводородов.
Такой вид изомерии, когда вещества различаются порядком
связи атомов в молекуле, называют структурной изомерией.
Изомерия предельных углеводородов обусловлена простейшим
видом структурной изомерии — изомерией углеродного скелета.
40
Соединения, имеющие неразветвленную углеродную цепь, назы¬
вают нормальными.
Первые члены гомологического ряда алканов — метан, этан,
пропан — существуют только в одной форме, они не имеют изо¬
меров. У бутана, как уже отмечалось, существуют два изомера.
Для углеводорода состава C5H12 (пентан) известны три изо¬
мера:
СНз
СНз—СН2—СН2—СН2—СНз СНз—СН—СН2—СНз СНз—С—СНз
I I
СНз СНз
н-пентан изопентан неопентан
С увеличением числа углеродных атомов в молекуле воз¬
растает и число теоретически возможных изомеров. Так, для
гексана оно составляет 5, для гептана 9, для октана 18, для
нонана 35, для пентадекана С15Н32 4347, для триаконтана
С30Н62 4 111 846 673, т. е. более четырех миллиардов. Это не
значит, что все теоретически возможные изомеры действительно
известны. Для углеводородов C1—С9 они получены; для высших
членов ряда известны лишь некоторые.
При выводе формул изомеров рекомендуется предварительно
строить схемы углеродных скелетов, постепенно укорачивая
основную (главную) цепь и располагая изъятые из этой цепи
атомы углерода в виде разветвлений (боковых цепей) во всех
возможных положениях. Рассмотрим, например, схемы углерод¬
ного скелета изомерных гептанов (С7) (полные формулы угле¬
водородов получаются после дополнения схем углеродных ске¬
летов необходимым числом атомов водорода):
С—С—С—С—С—С—С с—с—с—с—с
I
с—с
с—с—с—с—с—с
I
с
А—с—с—с
А
с
с—с—с—с—с—с
I
с
А 1
с—с—с—с—с
I
с
с—с—с—с—с
А А
с—с—с—с—с
А
с
с—А—с—с
I I
с с
41
Как видно из приведенных примеров, атомы углерода в пре¬
дельных углеводородах неравноценны: они могут быть соедине¬
ны с разным числом атомов углерода и водорода.
Если атом углерода затрачивает на углерод-углеродную
связь одну единицу валентности, то его называют первичным,
если две — вторичным, если три — третичным, если все четы¬
ре — четвертичным. В молекулах нормальных алканов бывают
только первичные (на концах цепи) и вторичные (в середине
цепи) атомы углерода.
Одновалентные остатки, получающиеся при отнятии атома
водорода от алканов, называют алкильными радикалами или
алкилами. Названия алкилов производят от названий соответ¬
ствующих углеводородов с заменой окончания ан на ил, напри¬
мер: СНз— метил, С2Н5 — этил, С3Н17 — пропил, С4Н9 — бу¬
тил, С5Н11 — пентил, С6Н13 — гексил. Общая формула алкилов
СnН2n + 1, их часто сокращенно обозначают Alk или R.
Названия алкилов широко ипользуют в номенклатуре орга¬
нических соединений, т. е. при построении названий органиче¬
ских веществ. Позднее мы узнаем, что алкилы могут существо¬
вать короткое время как промежуточные продукты реакций
(см. § 51).
Молекулы метана и этана образуют по одному одновалент¬
ному радикалу каждая (метил, этил). От молекулы пропана
СНз—СН2—СНз можно отщепить атом водорода двумя путями:
от первичного атома углерода с образованием радикала про¬
пила СНз—СН2—СН2— и от вторичного углеродного атома,
тогда образуется радикал вторичный пропил или изопропил
[(СНз)2СН—. Молекулы бутана и изобутана также могут обра¬
зовывать по два одновалентных радикала каждая:
СН3—СН2—СН2—СН2— СНз—СН2—СН—СНз
бутил
СН3
СН3—СН—СН2—
вторичный бутил
(втор-бутил)
СН3
СНз—С—СН3
изобутил третичный бутил
(трет-бутил)
Из этих примеров видно, что алкилы могут быть первичны¬
ми, вторичными и третичными, в зависимости от того, находится
ли свободная валентность у первичного, вторичного или тре¬
тичного атомов углерода.
§17. Конформации. Модели молекул. Структурные формулы
показывают порядок химической связи атомов в молекуле, но
не передают их пространственного расположения.
Рассмотрим модель молекулы этана. Не нарушая ни поряд¬
ка химической связи (химического строения), ни валентных
42
Рис. 11. Перспективные формулы (а, б) и формулы Ньюмена (в, г) двух
конформаций этана (в — заторможенная, г — заслоненная)
углов и длин связей, изменим пространственное положение од¬
ной метальной группы по отношению к другой путем ее враще¬
ния вокруг связи С—С. Молекула этана может принять при
этом различные геометрические формы, отличающиеся взаим¬
ным поворотом метальных групп. Изображения двух из этих
форм в перспективной проекции показаны на рис. 11, а, б. На¬
гляднее эти формы можно изобразить, в виде проекций
(рис. 11, в, г), получающихся при рассмотрении их вдоль связей
С—С. Такой способ изображения называют формулами Ньюме¬
на. Ближайший к наблюдателю атом углерода надо представ¬
лять себе в центре формулы, от него идут три связи к атомам
водорода. Связи второго, удаленного от наблюдателя атома
углерода, как бы «высовываются» из-за круга.
Как показано на рис. 11, в, г, взаимное расположение ме¬
тальных групп в этой модели можно охарактеризовать углом
φ. Если допустить, что каждому значению угла φ отвечает
определенный, отличный от других пространственный изомер, то
возникает противоречие с тем несомненным фактом, что суще¬
ствует только один этан, не имеющий изомеров. Поэтому Вант-
Гофф, выдвигая в 1874 г. гипотезу тетраэдрического атома угле¬
рода, ввел представление о свободном вращении групп вокруг
простых связей, т. е. о том, что не существует никаких особых
форм молекул с определенным значением угла ф. Впоследствии
это представление Вант-Гоффа было уточнено. Оказалось, что
расположение, характеризующееся углом φ° (см. рис. 11, г),
так называемое заслоненное, менее выгодно энергетически, чем
расположение с углом φ1 (рис. 11, в) — заторможенное. Таким
образом, на смену постулату Вант-Гоффа о свободном враще¬
нии групп вокруг простых связей пришло представление о том,
что в результате поворота вокруг простых связей молекула
стремится принять наиболее выгодную, обладающую наимень¬
шей энергией геометрическую форму.
Различные геометрические формы молекул, переходящие
друг в друга путем вращения вокруг простых связей, называют
конформациями или поворотными изомерами (конформерами)
При вращении вокруг σ-связи изменяются расстояния меж¬
ду несвязанными атомами. С уменьшением расстояния растет
43
а 6 в
Рис. 12. Конформации цепи из пяти углеродных атомов:
а — зигзагообразная; б — клешневидная; в — нерегулярная
степень отталкивания между ними, повышается потенциальная
энергия. В заслоненной конформации в молекуле этана расстоя¬
ния между атомами водорода минимальны, а в заторможен¬
ной — максимальны, поэтому в заслоненной конформации по¬
тенциальная энергия больше. Однако разница энергий
составляет всего 12 кДж/моль, поэтому переход из одной кон¬
формации в другую осуществляется легко и выделить их в ка¬
честве устойчивых изомеров нельзя.
Для более сложных молекул возможно множество конфор¬
маций; из них в первую очередь реализуются энергетически
более выгодные. Длинная углеродная цепь может принимать
самые разные геометрические формы. Несколько конформаций
для цепи из пяти атомов углерода показаны на рис. 12. Изо¬
браженные плоские формы [зигзагообразная (а), клешневид¬
ная (б), нерегулярная (в)] представляют собой лишь часть из
общего числа возможных конформаций: атомы углерода отнюдь
не обязаны находиться в одной плоскости. Какую бы конфор¬
мацию ни приняла цепь атомов углерода, связанных простыми
связями, три ее соседних атома никогда не окажутся на одной
прямой.
Органические вещества обычно представляют собой равно¬
весные смеси конформеров, среди которых преобладают наибо¬
лее выгодные, обладающие минимальной внутренней энергией.
Многие физические и химические свойства веществ зависят от
того, какая именно конформация у них преобладает. Как по¬
казывают рентгенографические исследования, алканы нормаль¬
ного строения в твердом состоянии имеют правильную зигзаго¬
образную конформацию.
Простейшие модели, типа изображенных на рис. 10, показы¬
вают порядок связи атомов в молекуле и размер валентного
угла, но не дают верного представления о размерах атомов и
о заполнении пространства внутри молекулы. В действительно¬
сти никаких пустых промежутков в молекулах нет. Правиль¬
ное представление о строении молекулы и о заполнении внутри¬
молекулярного пространства можно получить при помощи полу¬
сферических моделей, называемых также моделями Стюарта —
Бриглеба. При изготовлении моделей учитывают, что каждый
атом характеризуется межмолекулярным или ван-дер-ваальсо-
вым радиусом а и атомным или ковалентным радиусом г. Ван-
дер-ваальсов радиус характеризует размеры несвязанного (сво¬
44
бодного) атома. Размер ковалентного радиуса зависит от при¬
роды элемента, его валентного состояния и кратности связи.
Ковалентный радиус равен половине расстояния между двумя
одинаковыми атомами, связанными ковалентной связью.
Если два атома вступают в химическую связь, то их центры
подходят друг к другу ближе, чем позволяют ван-дер-ваальсо-
вы радиусы; в этом случае центры связанных атомов будут
находиться на расстоянии, равном сумме их ковалентных ради¬
усов. Для передачи этого на модели от центра шара (атома)
откладывают отрезок, равный ковалентному радиусу (в опреде¬
ленном масштабе), и на этом расстоянии отсекают шаровой
сегмент. Атом одновалентного элемента, например водорода,
изображают шаром с одним срезом (рис. 13, а). Сложив сре¬
занными частями две такие модели, получим модель молекулы
водорода (рис. 13,6). В ней правильно передано межатомное
расстояние и ограничена та сфера, внутрь которой не может
проникнуть какой-либо другой атом. Модель атома двухвалент¬
ного кислорода представляет собой шар с двумя срезами, рас¬
положенными под углом около 105°. Из модели атома кисло¬
рода и двух моделей атомов водорода можно построить модель
молекулы воды (рис. 13, в). Модель атома углерода в состоянии
5р3-гибридизации— шар с четырьмя срезами под тетраэдриче¬
скими углами. Приложив к местам среза модели атомов водо¬
рода, получим модели молекулы метана (рис. 13, г), этана
(рис. 13, д).
§ 18. Номенклатура. Номенклатура определяет правила со¬
ставления названий соединений. Название органического соеди¬
нения должно отражать не только состав, но и строение его
молекулы. Иными словами, название должно позволять легко
и точно написать структурную формулу.
С середины XIX века началось развитие так называемой
рациональной номенклатуры, согласно которой органические ве¬
щества рассматривали как продукты усложнения простейшего
представителя данного гомологического ряда. Для несложных
соединений такой номенклатурой охотно пользуются и сейчас.
В 1892 г. на международном конгрессе химиков в Женеве были
Рис. 13. Полусферические модели:
а — атом водорода; б — молекула водорода; в — вода; г — метан; д — этан в засло¬
ненное конформации
45
утверждены правила новой научной номенклатуры, получившей
название «женевской». Особенно подробно были разработаны
правила номенклатуры для соединений с открытой цепью.
В основе «женевского» названия любого органического вещест¬
ва лежит название углеводорода, из которого данное соедине¬
ние может быть получено путем замещения. Такую номенкла¬
туру называют заместительной. В 1930 г. в Льеже (Бельгия)
были приняты новые постановления по номенклатуре органиче¬
ских соединений, известные как «льежские правила». В даль¬
нейшем работа над номенклатурой продолжалась, и в настоя¬
щее время "наиболее полными являются правила Комиссии по
номенклатуре органических соединений при Международном
Союзе Чистой и Прикладной химии [кратко называемыми по
первым буквам английского названия этого союза Internatio¬
nal Union of Pure and Applied Chemistry правилами IUPAK
[(ИЮПАК)]. Правила IUPAK обобщают номенклатурный опыт
целого столетия и являются современными международными
правилами номенклатуры органических соединений. По этим
правилам для многих типов соединений можно употреблять как
рациональные (радикально-функциональные), так и замести¬
тельные названия. Знакомиться с современной номенклатурой
мы будем постепенно, по мере надобности. Начнем это знаком¬
ство с названий алканов.
Первые четыре представителя ряда алканов имеют случай¬
ные тривиальные названия (от лат. trivialis — обыкновенный):
метан, этан, пропан, бутан. Названия последующих членов ряда
производят от греческих числительных, указывающих число
углеродных атомов в молекуле; к ним добавляется общее для
всего гомологического ряда алканов окончание ан. Эти назва¬
ния относятся как к неразветвленным, так и к разветвленным
изомерам, однако в случае последних они теряют однозначность:
строения изомера подобные названия не передают.
По рациональной номенклатуре названия всех алканов обра¬
зуются из названия простейшего члена гомологического ряда —
метана, перед которым помещают обозначения углеводородных
радикалов, замещающих в нем атомы водорода; радикалы пере¬
числяют в алфавитном порядке. Если имеется несколько одина¬
ковых радикалов, то число их указывают с помощью приста¬
вок— числительных ди- (два), три-, тетра- (четыре). В приве¬
денных примерах наряду с рациональными названиями в
скобках даны и некоторые тривиальные:
СН3
СНз—СН—СН3 СНз—с—сн.
СНз—СН—СН2-СНз
СНз
СНз
тетраметилметан
(неопентан); II
СНз
триметилметан
(изобутан); I
диметилэтилметан
(изопентан); III
46
СНз
СН3-С-СН2-СН3
cн3
триметилэтилметан
(неогексан); IV
СН3-СН-СН2-СН-СН3
СН3
изобутилдиметилметан; V
При составлении названия по современной заместительной
номенклатуре углеводороды с разветвленной цепью рассматри¬
вают как производные нормальных алканов, в цепи которых
вместо атомов водорода стоят углеводородные радикалы. Глав¬
ную цепь выбирают максимальной длины, углеродные атомы ее
нумеруют, начиная с конца, более близкого к разветвлению.
В названии соединения цифрой указывают номер углеродного
атома, при котором находится замещающий радикал, затем на¬
звание радикала и название главной цепи. Если радикалы по¬
вторяются, то перечисляют цифры, указывающие их положение,
а число одинаковых радикалов указывают приставками ди-,
три-, тетра-. Название алкана имеет, таким образом, следующий
вид:
цифры — алкилы — главная цепь — суффикс ан
Структуры I — V по современной заместительной номенкла¬
туре следует назвать: I — 2-метилпропан, II — 2,2-диметилпро-
пан, III — 2-метилбутан, IV — 2,2-диметилбутан, V — 2,4-диме-
тилпентан.
При выборе начала нумерации углеродных цепей, имеющих
несколько одинаковых заместителей на разных расстояниях от
концов цепи, выбирают тот вариант, при котором заместители
получают меньшие номера (см., например, формулу VI). Если
же разные заместители находятся на равных расстояниях от
концов цепи, то нумерацию начинают с того конца, ближе к
которому расположен радикал, первая буква названия которо¬
го стоит раньше в алфавите (см. формулу VII).
1 2 3 4 5 6
СН3—СН—СН2—СН—СН2-СНЭ
I I
СН3 СНз
2,4-диметилгексан; VI
СНз—СН2—СН2—СН—СНг—СН—СН2—СН2—СНз
СНз С2Н5
4-метил-6-этилнонан; VII
§ 19. Способы получения. Основным промышленным источни¬
ком алканов служат нефть и природный газ. Из нефти и газа
можно выделить индивидуально алканы Ci—С5. Способы по¬
лучения алканов можно разделить на три группы.
47
Реакции, не сопровождающиеся изменением числа углерод¬
ных атомов в молекуле. Примером такой реакции служит гид¬
рирование непредельных углеводородов в присутствии неболь¬
шого количества катализатора — платины, палладия, никеля
(подробнее см. § 33):
H2, катализатор
R—СН=СН—R' > R—СН2—СН2—R'
Реакции, сопровождающиеся уменьшением числа углеродных
атомов в молекуле.
а) Крекинг. При нагревании парафинов выше 400 °С про¬
исходит разрыв углерод-углеродных связей и образуется смесь
более простых насыщенных и ненасыщенных углеводородов (по¬
дробнее см. §§ 21, 31):
>400 °С
C5H12 C3HS + С2Н4
пентан пропан этилен
б) Сплавление солей одноосновных карбоновых кислот с
щелочами:
СН3—COONa + NaOH —► СН4 + Na2C03
Реакции, сопровождающиеся усложнением углеродного ске¬
лета.
а) Реакция Вюрца (1855 г.) —действие металлического нат¬
рия на алкилгалогениды:
2CH3I + 2Na —► СН3—CH3 + 2NaI
Реакция применима в первую очередь для получения угле¬
водородов, молекулы которых состоят из двух тождественных
половин. Для получения углеводородов несимметричного строе¬
ния приходится брать смесь двух алкилгалогенидов, однако в
этом случае получается не один, а три продукта:
NaСН3I + С2Н5I ► СН3-СНз + сн3—С2Н5 + С2Н5-С2Н5
этан пропан бутан
б) Прямой синтез из элементов. Метан образуется в воль¬
товой дуге между угольными электродами в присутствии во¬
дорода:
С + 2Н2 —► СН4
Реакции такого типа, проводимые в иных условиях, получи¬
ли практическое значение в процессе гидрирования угля. Для
этого суспензию каменного угля в тяжелых нефтяных маслах
нагревают с водородом под давлением «30 МПа в присутствии
железных и марганцевых катализаторов при температуре
«400 °С. Получается смесь углеводородов — синтетический
бензин.
48
в) Каталитическое гидрирование монооксида углерода над
железом, кобальтом, никелем при 200—400 °С приводит к сме¬
си жидких углеводородов (синтетический бензин):
пСО + (2/i + 1JI 1г —► CnHjn+г + /1Н2О
Известно, что запасы угля во много раз превосходят запасы
нефти, поэтому способы превращения угля в жидкое горючее
в будущем приобретут важное практическое значение.
§ 20. Общая характеристика физических и химических свойств.
Физические свойства. Как видно из табл. 4, первые
четыре члена гомологического ряда алканов при обычных усло¬
виях— газообразные вещества; соединения от Cs до Cis —
жидкости, от Cie и выше — твердые вещества. В гомологиче¬
ском ряду алканов постепенно повышаются температуры кипе¬
ния, плавления, а также относительные плотности. Это позволя¬
ет предвидеть свойства неизвестного члена ряда, основываясь
на свойствах его соседей. Например, т. кип. гексана 68,6 °С,
гептана 98,4 °С. Разница в составе на одну группу СН2 при¬
водит к повышению температуры кипения на 29,8 °С (гомоло¬
гическая разность температур кипения). На основании этого
можно рассчитать, что температура кипения октана должна
быть равна 98,4 + 29,8 » 128 °С; это примерно на 2°С отли¬
чается от экспериментально найденной. При таком расчете сле¬
дует иметь в виду, что гомологическая разность температур
кипения (как и влияние гомологии на все другие физические
константы) не остается неизменной: у высших членов ряда
изменение состава на группу СН2 относительно меньше влияет
на свойства молекулы. Алканы с разветвленной цепью кипят
Таблица 4. Физические свойства некоторых нормальных алканов
Название
Формула
Т. пл., ®С
T. кип., °С
Относительная
плотность d20
Метан
сн4
— 182,5
-161,5
0,424®
Этан
с2н,
—183,7
-88,6
0,546®
Пропан
с3н8
— 187,6
-42,2
0,585®
Бутан
С4Н,о
— 138,3
-0,5
0,579 8
Пентан
С5н12
— 129,7
+36,1
0,626
Гексан
СвН,4
—95,3
+68,6
0,659
Гептан
с7н,*
—90,6
+98,4
0,684
Декан
С|оНг2
—30,0
+ 173,0
0,730
Тетрадекан
С|4Н3ф
+5,5
+253,0
0,764
Пентадекан
С15Н32
+10,0
+270,5
0,769
Г ексадекан
С,«н34
+18,1
+287,5
0,775 г
Эйкозан
Сг„н„
+36,5
+344,0
0,778 г
Пентаконтан
С$оНмг
+93,0
+421,0“
0,942*
Гектан
СмоНгог
+ 115,4
—
—
Примечания: а—При 15 мм рт. ст. б—При температуре кипения, в—Под давлением.
е—При температуре плавления.
49
при более низкой температуре, чем изомеры с нормальной
цепью. Для примера приводим температуры кипения изомерных
пентанов:
СН3-СН2—СН2—СН2—СН3 (СН3)2СН—СН2—СН3 (СН3)4С
(36,1 °С) (27,8 °С) (9,4 °С)
В неполярных жидкостях между поверхностями молекул действуют ван*
дер-ваальсовы силы притяжения. Чем больше молекула и ее поверхность,
тем сильнее межмолекулярное взаимодействие. С увеличением относительной
молекулярной массы температуры кипения повышаются. Форма разветвлен*
ной молекулы стремится к сферической, площадь ее поверхности уменьшается,
уменьшаются и межмолекулярные силы, которые легче преодолеваются прй
более низких температурах.
Температуры плавления, наоборот, повышаются с ростом
разветвленности углеродной цепи. Плотности всех алканов
меньше единицы. Они практически нерастворимы в воде, однако
растворимы в эфире и других органических растворителях. Ме¬
тан и этан почти лишены запаха, углеводороды Сз—C15 имеют
всем хорошо известный запах бензина или керосина, высшие
члены ряда лишены запаха из-за малой летучести.
Химические свойства. В химическом отношении ал¬
каны мало активны, за что и были названы парафинами (от
лат, parum affinis — лишенные сродства). Известный русский
химик М. И. Коновалов (1858—1906 гг.) образно называл пара¬
фины «химическими мертвецами» за их пассивность.
Химическое поведение насыщенных углеводородов определя¬
ется характером и прочностью имеющихся в их молекулах свя¬
зей. Устойчивость углерод-углеродной (С—С) связи обусловле¬
на малым размером атома углерода и его тетраэдрической
конфигурацией в состоянии $р3-гибридизации, что способствует
максимальной концентрации электронного заряда между ядра¬
ми. Прочность углерод-водородной (С—Н) связи объясняется
тем, что при ее образовании «р3-орбиталь углерода подходит
близко к ядру атома водорода, так как атом водорода в отли¬
чие от атомов других элементов не имеет «внутренних» элек¬
тронов, которые могли бы отталкивать электронный заряд
атома углерода. σ-Связь в молекулах алканов мало поляризо¬
вана вследствие близкой электроотрицательности атомов угле¬
рода и водорода (2,5 и 2,1, соответственно). В силу этого пре¬
дельные углеводороды — вещества мало полярные и трудно по¬
ляризуемые. Они не проявляют склонности к гетеролитическому
разрыву. Атаки нуклеофильных и электрофильных реагентов
затруднены, поэтому к ионным реагентам алканы устойчивы.
На них не действуют при обычных температурах концентриро¬
ванные кислоты (азотная, серная и др.), расплавленные и кон¬
центрированные щелочи, обычные окислители (перманганат ка¬
лия, хромовая смесь). Металлы, даже щелочные, не вытесняют
водород из этих соединений. Эти свойства используют на прак¬
50
тике: например, щелочные металлы хранят в керосине, различ¬
ные металлические изделия с целью предохранения их от кор¬
розии покрывают смазочными маслами, концентрированную
серную кислоту и концентрированные щелочи используют для
очистки нефтепродуктов.
Для насыщенных углеводородов возможен лишь гомолитиче-
ский, радикальный разрыв связей, при этом происходит замеще¬
ние атомов водорода, расщепление углеродного скелета (кре¬
кинг), окисление частичное или полное (сгорание). Все это
определяет круг реакций, к которым способны алканы; это в
первую очередь радикальные реакции замещения, идущие в до¬
вольно жестких условиях (действие света, высокой температу¬
ры и др.). К реакциям присоединения алканы не способны.
В этом их принципиальное отличие от непредельных углеводо¬
родов.
Хотя связь С—Н более прочна, чем связь С—С (ср. энергии связей
в табл. 2), реакции с отщеплением водорода идут чаще, так как С—Н-связи
расположены на поверхности органической молекулы и поэтому более до¬
ступны действию реагентов.
Хорошо изучен механизм замещения атомов водорода в ал¬
канах на атомы хлора и брома. Эти реакции проходят по
радикальному цепному механизму. Сущность цепного механиз¬
ма — в постоянном воссоздании активных частиц, способствую¬
щих превращению все новых и новых молекул реагирующего
вещества. В обычных условиях молекулярные хлор и бром
практически не реагируют с насыщенными углеводородами.
Только в атомарном состоянии они способны вырывать атом
водорода из молекулы алкана. Поэтому предварительно необхо¬
дим разрыв молекулы галогена до свободных атомов, которые
инициируют цепную реакцию. Это может быть осуществлено
действием света или тепла [реакция (I)]. Затем атом хлора
атакует атом водорода (а не атом углерода) в молекуле, на¬
пример, метана. Происходит отрыв водорода и образование
свободного радикала метила [реакция (2)]. Далее метильный
радикал атакует молекулу хлора, в результате чего образуются
метилхлорид и атом хлора [реакция (3)].
С!а —► 2CI • (1) НзС:Н + С1- —► ШС1 + Н3С. (2)
HjC. + CUCl —► HjCsCl + Cl. (3)
Атом хлора атакует следующую молекулу метана и т. д.
Реакции (2) и (3) характеризуют рост цепи, они повторя¬
ются до тех пор, пока не произойдет обрыв цепи — уничтожение
ведущего цепь свободного радикала. В данном случае обрыв
цепи может произойти по реакциям (4), (5) или (6), а также
в результате столкновения радикалов со стенками реакционного
сосуда, с примесями.
2С1 • —► сь (4) 2Н3С • —* СН3—СН3 (5)
Н3С* + С1. —► СН3—С1 (6)
51
Стадии инициирования цепи, ее развития и обрыва харак¬
терны для всех цепных реакций. Интересной особенностью явля¬
ется то, что они не могут начаться без «внешнего толчка» —
появления свободного радикала, инициирующего начало цепной
реакции. При этом достаточно очень небольшого числа радика¬
лов-инициаторов, чтобы осуществить цепное превращение зна¬
чительных количеств вещества. Большие концентрации свобод¬
ных радикалов могут оказаться даже вредными, так как в этих
условиях чаще совершаются акты обрыва цепи по приведенным
выше уравнениям.
Радикальные реакции тормозятся веществами, легко реаги¬
рующими со свободными радикалами. Такие вещества называют
ингибиторами. Большой вклад в изучение цепных реакций внес
акад. Н. Н. Семенов. Его исследования отмечены Нобелевской
премией.
§ 21. Реакции. Галоген и рование. Радикальное галоге-
генирование, механизм которого рассмотрен в предыдущем па¬
раграфе, — важная реакция алканов. В качестве галогенирую-
щих агентов можно применять не только хлор, но и бром. Иод
не способен к реакции прямого замещения водорода, фтор дей¬
ствует слишком энергично и, если не принять особых мер предо¬
сторожности, полностью разрушает органические молекулы.
В промышленности используют термическое хлорирование алка¬
нов при температурах «300°С.
Радикальному замещению на хлор последовательно могут
подвергаться все атомы водорода в метане. При накоплении
первого продукта замещения — метилхлорида СН3С1 — возни¬
кает возможность атаки его атомом хлора и образования сво¬
бодного радикала -СИгСЛ, который при дальнейшей реакции
с радикалом С1 •, дает двузамещенный продукт — дихлорметан
CH2CI2. При избытке хлора произойдет и дальнейшее замеще¬
ние с образованием соединений СН3С1 и СС14. В результате
образуется смесь галогенпроизводных метана:
CI2 С1г ОI2 CI2
сн* -=ЙБГ сн>с| chci> TSET CCI.
метил- дихлор- хлоро- четырех
хлорид метан форм хлористый
углерод
При большом избытке метана получается преимущественно
монохлорзамещенный продукт.
Подобно метану, могут хлорироваться и другие алканы. Пре¬
имущественное направление вступления галогена определяется
закономерностями, впервые изученными В. В. Марковниковым:
легче всего замещение идет у третичного атома углерода, затем
у вторичного, в последнюю очередь — у первичного; при обра¬
зовании полигалогензамещенных соединений атомы галогена
62
преимущественно замещают водород у одного и того же или у
соседних атомов углерода:
СН,
СН,
CI*
CIj
CHs-CHj-CH-СНз СНз—CHj—С—СН3
С1 СНз
I I CI*
СНз—СН—С—СНз
—НС)
<!:i
Cl СНз
СНз—A—i—CHj
I I
Cl Cl
Эти закономерности объясняются тем, что энергия связи
атома водорода с первичным, вторичным и третичным атомами
углерода неодинакова: она составляет соответственно 415, 390
и 376 кДж/моль.
Избирательность (селективность) замещения зависит от при¬
роды галогена и условий, при которых ведется процесс. При
низкой температуре разрывается самая слабая связь С—Н
(у третичного атома углерода). При повышении температуры
селективность снижается. Например, относительная скорость от¬
рыва атома водорода от групп СН3, СН2 и СН при хлориро¬
вании в газовой фазе при 100 °С может быть выражена соот¬
ношением 1 : 4,3 : 7, при хлорировании при освещении— 1 :3,8 :
: 5, а при 300 °С—1 : 3,3 : 4,4, т. е. избирательность уменьша¬
ется. Для брома избирательность выше, чем для хлора. Так,
.при 127 °С относительная скорость бромирования этих же
групп выражается Соотношением 1:82: 1600. При газофазном
бромировании изопентана (СН3)2СНСН2СН3 выход третичного
продукта превышает выход вторичного примерно в 20 раз.
' При расчете выхода различных изомеров надо принимать во
внимание не только относительную реакционную способность,
но и число реагирующих (первичных, вторичных и третичных)
атомов. Так, например, при хлорировании изопентана приведен¬
ную выше относительную скорость замещения водорода надо
умножить на число Н-атомов соответствующего типа, т. е. для
первичных 1-9 = 9; для вторичных 3,8-2 = 7,6; для третичных
5,0-1=5,0. Учитывая это, получаем около 42% первичных
хлорпроизводных (два изомера), 35 % вторичного хлорпроиз-
водного (один изомер) и только 23 % третичного, несмотря на
большую реакционную способность третичного Н-атома.
Нитрование. При обычных температурах алканы ус¬
тойчивы к действию концентрированной азотной кислоты. Од¬
нако при нагревании их с разбавленной азотной кислотой или
с оксидами азота идет реакция нитрования — замещение атома
водорода на нитрогруппу N02. Впервые ее осуществил М. И. Ко¬
новалов (1888 г.) при нагревании алканов до 140°С с 10 %-й
азотной кислотой под давлением. Эта реакция известна ныне
63
как реакция Коновалова (подробнее эта реакция рассмотрена
в § 122):
no2
hono2 I
СНз—сн2—сн3 —
пропан
СНз—СН—СН3
2-нитропропан
Сульфохлорирование. При действии на алканы сме¬
си диоксида серы и хлора идет радикальная реакция сульфо-
хлорирования. Инициируется этот процесс фотохимически —
ультрафиолетовым светом и приводит к сульфонилхлоридам
RS02C1:
УФ-свет
С12 *■ 2С1 • RH + C1. —► R. + HC1
R.+ SOj —► RS02. RS02. + C12 —► RS02C1 + Cl. и т. д.
Сульфонилхлориды (хлорангидриды сульфокислот) легко
гидролизуются до сульфокислот RSO2OH, натриевые соли кото¬
рых RS020Na называют сульфонатами. Практическое значение
имеют сульфонаты с числом атомов углерода Cie—Ci8, они
входят в состав моющих средств (см. § 97).
Сульфоокисление проводят действием на алканы ди¬
оксида серы и кислорода. Оно также приводит к сульфокисло- -
там. Этот путь получения сульфокислот выгоднее, так как не
требует расхода хлора.
RH + S02 + 1/jOi —>• RS020H
Окисление. Как уже отмечалось, алканы при обычной
температуре устойчивы к действию кислорода воздуха и обыч¬
ных окислителей. При поджигании на воздухе алканы воспла¬
меняются и горят, превращаясь в диоксид углерода и воду:
С3Н3 -р 502 —> ЗС02 -{- 4Н20
Алканы — ценное, высококалорийное топливо; в них много
водорода, теплотворная способность которого больше, чем для
углерода. При сгорании 1 кг метана выделяется «55 000 кДж
тепла. Горение — одна из главных реакций алканов, используе¬
мых на практике: природный газ, нефть — важнейшие источни¬
ки тепла и энергии.
СН4 -J- 202 ■ > С02 -р 2Н20 -р 891 кДж/моль
Окисление алканов кислородом воздуха в более мягких ус¬
ловиях приводит к кислородсодержащим органическим вещест¬
вам — спиртам, альдегидам, кетонам, карбоновым кислотам.
Например, при окислении бутана получают в промышленности
уксусную кислоту (см. § 96). Важное промышленное значение
приобрело окисление высших алканов с целью получения выс¬
ших жирных кислот (работы С. С. Наметкина).
Крекинг. Нагревание до 400—600°С сообщает молеку¬
лам алканов достаточно энергии для того, чтобы произошел
64
гемолитический разрыв углерод-углеродных связей. Процесс
расщепления молекул высококипящих углеводородов на моле>
кулы более простых низкокипящих носит название крекинг (от
англ, crack — расщепление).
При крекинге алканов образуются более простые насыщен¬
ные и ненасыщенные углеводороды, например:
СН3—СН2—СН2—СНз —>- СНз—сн8 + сн2=сн2
бутан этан этилен
Эта реакция имеет большое практическое значение. Таким
путем высококипящие фракции нефти (мазут) превращают в
ценные низкокипящие жидкие продукты — бензин, керосин, 9
также в простейшие газообразные углеводороды — сырье для
органического синтеза. О механизме крекинга см. § 31.
На результаты крекинга, т. е. на соотношение жидких и га¬
зообразных продуктов, долю предельных и непредельных угле¬
водородов, «глубину» крекинга (степень распада сложных мо¬
лекул), влияют условия крекинга. При более высоких темпера¬
турах (650—700 °С) идет более глубокий распад — пиролиз,
при котором образуется много водорода, углистого остатка
,(кокса), простейших предельных и непредельных углево¬
дородов.
Наряду с собственно крекингом, т. е. разрывом сложных мо¬
лекул на более простые, при термокаталитической обработке
алканов и их смесей (т. е. нефтяных фракций) идут и другие
процессы, главные из которых: дегидрирование — отнятие водо¬
рода с превращением предельных углеводородов в непредель¬
ные; ароматизация алканов и циклоалканов с превращением
в ароматические углеводороды; изомеризация — перестройка
углеродного скелета, например превращение пентана в изо¬
пентан:
СН3
А1С1з, 100 °с |
СНз—СН2—СН2—СН2—СНз »- СНз—СН—СН2—СНз
Под действием еще более высоких температур (1000 °С и
выше) молекулы алканов распадаются на элементы.
§ 22. Алканы в природе и в технике. В недрах Земли содер¬
жатся большие запасы горючих газов. Они находятся в свобод¬
ном состоянии в виде природного газа (скопления в толще зем¬
ной коры) или в растворенном виде в нефти — попутные нефтя¬
ные газы. Природный газ содержит до 98 % метана, а в
качестве примесей — этан, пропан и др. Попутный нефтяной газ
более беден метаном (30—80 %), но содержит значительные
количества его гомологов: этан (4—20%), пропан (5—22%),
бутаны (5—20 %), а также пентаны и др. Высшие алканы вхо¬
дят в состав нефтей.
65
Простейший парафиновый углеводород — метан СН4 — известен под раз¬
личными названиями, указывающими на его нахождение в природе. Название
болотный газ связано с образованием метана при гниении растительных
веществ на дне болот. Скопления метана нередко можно встретить в зале¬
жах каменного угля, откуда и происходит его название рудничный газ. На¬
капливаясь в шахтах, он может служить причиной опасных взрывов.
Атмосфера больших планет (Сатурн, Юпитер) содержит много метана:
это указывает на то, что метан может возникать в естественных условиях и
в отсутствие органической жизни. Одна из теорий возникновения жизни пред¬
полагает, что жизнь возникла тогда, когда Земля была окружена атмосфе¬
рой, содержащей СН4, NH3, НгО и Н2. Энергия солнца и электрические
разряды способствовали распаду этих молекул до свободных радикалов, ко¬
торые превратились в сложные органические соединения, включая амино¬
кислоты.
По плану 12-й пятилетки в соответствии с решениями
XXVII съезда КПСС добыча газа к 1990 г. должна быть дове¬
дена до 835—850 млрд, м3 в год. Особое развитие получат
газовые промыслы на севере нашей страны, а также в При¬
каспийской низменности.
Добываемый газ используют как топливо для бытовых и
промышленных целей, а также как химическое сырье.
Средняя теплотворная способность 1 м3 природного газа — около
40 000 кДж; энергии, содержащейся в 1 м3 природного газа, достаточно, что¬
бы выплавить 30 кг чугуна. Замена газом других видов топлива, в частно¬
сти угля, дает большую экономию, поскольку добыча угля дороже, чем газа,
а теплотворная способность угля ниже.
Природный газ как химическое сырье используют в следую¬
щих производствах.
1. Получение технического углерода (сажи) и водорода пи¬
ролизом метана:
1000 "С
СН4 ► 2Н2 + С
Сажу применяют как добавку к каучуку в производстве ре¬
зины, для приготовления типографской краски и др., водород —
для различных синтезов.
2. Получение ацетилена (подробнее см. § 39):
1500 «с
2СН4 ► С2Н2 + ЗН2
3. Получение метилового спирта СН3ОН, формальдегида
НСНО, муравьиной кислоты НСООН окислением метана воз¬
духом в присутствии катализаторов.
4. Получение синильной кислоты пиролизом смеси метана и
аммиака в присутствии кислорода:
юоо °с
2СН4 + 2Ш3 + 302 >- 2HCN + 6Н20
5. Получение метилхлорида СН3С1, хлороформа СНС13, че¬
тыреххлористого углерода СС14 хлорированием метана.
56
6. Получение сероуглерода нагреванием метана с серой:
600 °С, катализатор
CH4 + 4S CSj + 2H2S
7. Получение нитрометана парофазным нитрованием метана:
ch4 + hno3 —► ch3no2 + h2o
Нитрометан используют как растворитель и исходное веще¬
ство для различных синтезов.
8. Получение смеси монооксида углерода с водородом (цен¬
ное сырье для органического синтеза) конверсией метана с
помощью воды или диоксида углерода, а также неполным
сжиганием метана:
800 °С, №/А!2Оз
сн4 + н2о * СО + ЗН2
800 -с, ni/ai2o3
СН4 + С02 * 2СО + 2Н2
1200—1500 “С
СН4 + >/202 *■ СО + 2Н2
Из гомологов метана отметим бутан и 2-метилбутан (изо¬
пентан), дегидрированием которых получают бутадиен и изо¬
прен — сырье для производства синтетического каучука (см.
§ 188). Пропан в виде так называемого баллонного газа ис¬
пользуют как горючее при газификации населенных пунктов,
не подключенных к сети газопроводов.
ГЛАВА 3
ЦИКЛОАЛКАНЫ
Циклическими называют соединения, имеющие замкнутые цепи
(от греч. циклос — круг). Молекулы незамещенных циклоалка¬
нов (циклопарафинов) состоят из замкнутых в цикл групп СН2;
по названию этой группы их называют иногда полиметиленовы-
ми углеводородами.
§ 23. Строение, номенклатура, изомерия, нахождение в приро¬
де. Строение и номенклатура. Как и в молекулах
алканов, атомы углерода в молекулах циклоалканов связаны
между собой о-связями (состояние $р3-гибридизации).
Общая формула гомологического ряда циклоалканов (СН2)„ -
или СяН2/). Таким образом, молекула циклоалкана содержит
на два атома водорода меньше, чем алкан с тем же числом
атомов углерода. Названия циклоалканов образуют путем до¬
бавления приставки (префикса) цикло к названию соответ¬
ствующего алкана. Для изображения циклоалканов часто
67
используют условные формулы, в которых опущены символы
элементов, например:
сн2
/Ч
Н2С—сна
/\
ИЛИ а
Н2С—сн2
1 1
Н2С—СН2 или
□
циклопропан
циклобутан
Н2С—СН2
СН3
или <Т>
СН2
Н2С^ ^СН2
1 | или
Н2С\ /СН2
СН2
(3
циклопентан
циклогексан
Изомерия. Структурная изомерия диклоалканов может
быть обусловлена размером цикла, размерами и строением бо¬
ковых цепей и их положением. Это можно проиллюстрировать
на примере пяти изомеров состава СвНю:
циклопентан метилциклобутан этилциклопропан
1,1-диметилцнклопропан 1,2-диметилциклопропан
У циклоалканов встречается также пространственная изоме¬
рия (см. § 27).
Циклоалканы изучали многие русские ученые. Наиболее
известны в этой области работы В. В. Марковникова, Г. Г. Гус-
тавсона, Н. М. Кижнера, Н. Д. Зелинского, С. С. Наметкина,
Б. А, Казанского, А. Ф. Платэ.
Нахождение в природе. Циклоалканы являются
главной составной частью некоторых нефтей. Впервые цикло¬
алканы были обнаружены в бакинской нефти В. В. Марковни-
ковым, который дал им название нафтены. Впоследствии было
установлено, что особенно распространены в природе соедине¬
ния с пяти- и шестичленными циклами, т. е. циклопентан и
циклогексан и их производные. Циклоалканы встречаются так¬
же в эфирных маслах растений. Углеродный скелет гомолога
циклогексана, 1-изопропил-4-метилциклогексана, лежит в осно¬
ве многих терпенов — важных природных соединений (см.
§ 182).
§ 24. Способы получения. Общим способом получения цикло¬
алканов является действие металлов на дигалогенпроизводные
алканов. Так, из 1,3-дибромпропана действием цинка можно
58
получить циклопропан (синтез Г. Г. Густавсона):
усн2вг сн2
Н2с/ + Zn —► Н2с/ | + ZnBr2
'СН2Вг \сн2
Из 1,4-дибромбутана можно таким же путем получить цик¬
лобутан. Для этих реакций применяют также металлический
натрий. Известны и другие реакции замыкания цикла, которые
приводят не к соответствующим циклоалканам, а к их произ¬
водным.
Соединения ряда циклогексана чаще всего получают гидри¬
рованием легкодоступных соединений ряда бензола. Важным
примером такой реакции служит получение в промышленности
чистого циклогексана из бензола:
H2/NI, 150—250 °С, 2,5 МПа
C6H6 ^ с6н12
§ 25. Устойчивость циклов. При изучении циклоалканов было
установлено, что их химические свойства зависят прежде всего
от размера цикла. При этом обычно различают соединения с
малыми циклами (Сз—С4), с обычными циклами (Cs—С7), со
средними циклами (С8—С11) и с большими циклами (от С12
и выше), называемые также макроциклическими. Особенностью
малых циклов является их меньшая устойчивость по сравнению
с другими циклическими соединениями. Повышенное содержа¬
ние энергии в малых циклах отражается, в частности, в тепло-
тах сгорания, которые в расчете на одну группу СН2 для раз¬
ных циклов имеют следующие значения (в кДж/моль):
Циклопропан 695 Циклогексан 657
Циклобутан 686 Макроциклы 657—660
Циклопентан 661
Теплота сгорания ациклических углеводородов в расчете на
одну группу СН2 составляет 660 кДж/моль.
Первая попытка связать устойчивость циклов с особенностя¬
ми их строения принадлежит А. Байеру (1835—1917 гг.).
В основу предложенной им в 1885 г. теории напряжения Байер
положил тетраэдрическую модель атома углерода, предположив,
что всякое отклонение валентностей от их нормального распо¬
ложения (под углом 109°28/) создает в молекуле напряжение,
понижает ее устойчивость. По Байеру, цикл представляется
плоским правильным многоугольником. Простейшее цикличе¬
ское соединение — циклопропан может быть изображен в виде
равностороннего треугольника с углами 60° (рис. 14). Отклоне¬
ние валентностей от их нормального расположения в расчете
на одну связь (введенная Байером мера напряжения а) для
трехчленного цикла равно:
а = (109°28' - 60°)/2 = 24°44'
59
Рис. 14. Циклопропан
Байер предполагал, что циклы имеют плоское строение, т. е.
что все атомы углерода лежат в одной плоскости. В действи¬
тельности это не так. Для плоских циклов С3—С8 вычисленные
углы отклонения составляют: +24°44' (циклопропан),
+9°44' (циклобутан), +0°44' (циклопентан), —5°16' (цик¬
логексан), —9°33' (циклогептан), —12°46' (циклооктан). В этих
цифрах правильно отражается постепенное повышение устойчи¬
вости (т. е. уменьшение напряжения) от трехчленного цикла к
пятичленному. Однако дальше гипотеза Байера уже перестает
соответствовать фактам: шестичленный цикл в действительности
прочнее пятичленного, не наблюдается увеличения напряжения
и в макроциклах.
В настоящее время установлено, что плоским является толь¬
ко трехчленный цикл (три точки всегда лежат в одной плоско¬
сти) и что угловое напряжение — только один из факторов, ко¬
торые надо учитывать,при рассмотрении устойчивости циклов.
Второй существенной причиной напряжения является напряже¬
ние заслонения, связанное с конформационным состоянием мо¬
лекулы.
§ 26. Физические и химические свойства. Физические свойства
циклоалканов приведены в табл. 5. Первые два члена этого
ряда — газы, С5—Сю — жидкости, высшие — твердые вещест¬
ва. Температуры кипения и плавления циклоалканов как и их
плотности, несколько выше, чем у алканов с равным числом
Таблица 5. Физические свойства некоторых циклоалканов
Название
Формула
Т. пл., «С
Т. кип., *С
Относительная
плотность, d
Циклопропан
С3нв
-127,4
-33,3
0,689
(при 40 °С)
Циклобутан
СчН»
-90,2
+ 12,9
0,703
Циклопентан
С5Н10
-93,8
+49,3
0,745
Циклогексан
с6н12
+6,5
+80,7
0,779
Циклононан
CgHig
+ П
+ 178
0,860
Циклотриаконтан
СзоНбо
+56
■“*
0,854
60
атомов углерода. Как и алканы, циклоалканы практически не¬
растворимы в воде.
По химическому характеру малые циклы склонны к реакци¬
ям присоединения, в результате которых происходит разрыв
цикла и образуются алканы и их производные, чем они напо¬
минают ненасыщенные соединения. Циклы с большим числом
звеньев более склонны к реакциям замещения, напоминая этим
алканы.
Действие галогенов. Циклопропан и его производ¬
ные легко присоединяют бром с разрывом цикла, образуя
1,3-дибромпроизводные алканов (реакция идет труднее, чем с
пропиленом):
СН,
Н2С——СН2 + Вг2 —► ВгСН2—СН2—СН2Вг
Большая устойчивость циклобутана по сравнению с цикло¬
пропаном проявляется в том, что он присоединяет бром труд¬
нее— лишь при повышенных температурах, давая 1,4-дибром-
бутан:
н2С—СН2 ви
| I ► ВгСН2—СН2—СН2—СН2Вг
н2с—сн2
У циклопентана, циклогексана и высших циклоалканов при
действии галогенов разрыв кольца не происходит, а идет реак¬
ция замещения атомов водорода на галоген:
н2с—сн2
н2с^ ^сн2
Н2С—сн2
циклогексан
Вг2
— НВг
Н2С—сн2
Н„с/ \н-Вг
н2с—сн2
бромциклогексан
Действие гал огеноводородов. Циклопропан и
его алкилпроизводные легко присоединяют иодоводород с раз¬
мыканием цикла и образованием алкилиодида:
СН2
/ \ + HI —V СНз—СН2—СН21
Н2С—сн2
пропиляодид
С циклобутаном эта реакция идет труднее, при нагревании.
Циклопентан и высшие циклы не присоединяют галогеново-
дородов.
Действие водорода хорошо иллюстрирует постепен¬
ное повышение устойчивости циклов от трехчленного к шести¬
членному: для размыкания таких циклов под действием водо-
61
рода над никелевым катализатором требуется все более высо¬
кая температура:
сн2
н2с^сн2
Ht/Ni, 80-120 "С
СН2—СН2—сн2
Н2С—СН2 180 *С
I I »- СНз—СН2—СН2—СНз
Н2С—СНа
Н2С—СН2
Н2С^^СН2
сн2
Нг/NI, 300 °С
СНз—СН2—СН2—СН2—СНЭ
Циклогексановый цикл в этих условиях не размыкается.
Вместо этого идет дегидрирование (отщепление водорода) с об¬
разованием бензола (см. § 45):
с6н12 —>• СвНз + ЗН2
Окисление. Циклопропан и его производные медленно
окисляются при нормальной температуре раствором перманга¬
ната калия в нейтральной или щелочной средах. При действии
более сильных окислителей или при нагревании окисляются
и другие циклоалканы. При этом происходит разрыв цикла и
образование двухосновных кислот с тем же числом атомов
углерода:
Н2С—сн2
HaG\ /снг
сн2
Ю1
/СН2—соон
Н2с(
ХСН2—соон
>■
циклопентан
глутаровая кислота
Н2С—сн2
н2с^\н2-
[01
сн2—сн2—соон
1
СНг—СН2—СООН
Н2С—СНз
циклогексан
адипиновая кислота
Нитрование. Циклоалканы, имеющие пяти- и шести¬
членные циклы, нитруются в тех же условиях, что и алканы.
Практическое значение имеет нитроциклогексан, из которого
можно получить капролактам, являющийся сырьем для произ¬
водства синтетического волокна капрон.
По современным представлениям, своеобразное химическое поведение
циклопропана и циклобутана в гомологическом ряду циклоалканов объяс¬
няется особым характером связей в молекулах этих соединений. В молекуле
циклопропана углерод-углеродные и углерод-водородные связи отличаются
от обычных or-связей ациклических соединений. Так, длина углерод-углерод-
ной связи равна 0,1526 нм (вместо 0,154 нм в алканах), валентный угол не
62
Рис. 15. «Банановые» связи циклопропана
,НЛ m 1н
отклонен до углов равностороннего тре¬
угольника, т. е. до 60°. Наибольшая элек¬
тронная плотность связи расположена не
по линии, соединяющей центры атомов, а
с внешней стороны треугольника (изогну¬
тая форма — форма «банана», рис. 15).
§ 27. Пространственная изомерия
замещенных циклов. Пространствен¬
ная изомерия (стереоизомерия) про¬
является в существовании веществ,
имеющих одинаковые структурные формулы, но различаю¬
щихся положением атомов в пространстве. Конформационнов
состояние, отмеченное для этана и других парафинов,—
пример пространственной изомерии, однако отдельные кон¬
формации (конформеры) легко превращаются друг в друга.
Существуют и устойчивые стереоизомеры. В частности, они
известны и в ряду циклоалканов. Здесь это явление обус¬
ловлено различным пространственным расположением боковых
цепей относительно кольца. Подобный тип стереоизомерии на¬
зывают геометрической (цис-транс-изомерией). Примером мо¬
жет служить 1,2-диметилциклопропан, метильные группы кото¬
рого могут находиться по одну или по разные стороны от
плоскости кольца:
нэс сн3 Н3С
UN i/\
цис-изомер граяс-изомер
В циклических соединениях свободное вращение вокруг угле-
род-углеродных связей кольца отсутствует, цис-транс-Изомеры
не могут легко переходить друг в друга; для этого необходим
разрыв связей кольца. цис-транс-Изомеры обладают различны¬
ми физическими и химическими свойствами.
Еще сложнее стереоизомерия в неплоских циклах. Так, у циклогексана
внециклические связи его углеродных атомов разделяются на два типа:
аксиальные (а), направленные перпендикулярно к кольцу, и экваториальные
(е), направленные по периферии кольца:
Экваториальное положение заместителя выгоднее аксиального, поэтому
все монозамещенные циклогексаны имеют заместители в экваториальном
63
положении. Аксиальная форма, которую можно представить себе возникаю¬
щей в результате изменения конформации (так называемой инверсии) кольца,
имеет больший запас энергии, поэтому ее существование менее вероятно:
§ 28. Отдельные представители. Циклопентан содержится в
нефти. Его используют как добавку к моторному топливу для
повышения качества последнего и в разных синтезах.
В нефти содержатся также карбоксильные производные цик¬
лопентана— циклопентанкарбоновая кислота и ее гомологи, на¬
зываемые нафтеновыми кислотами. При очистке нефтяных про¬
дуктов щелочью образуются натриевые соли этих кислот, обла¬
дающие моющей способностью {мылонафт).
Циклогексан содержится в нефти, может быть также полу¬
чен гидрированием бензола над никелевыми или платиновыми
катализаторами. Чистота полученного по этому способу цикло¬
гексана достигает 99,99 % и выше. Используют циклогексан
главным образом для синтеза адипиновой кислоты (см. § 103)
и капролактама — полупродуктов для производства синтетиче¬
ских волокон найлон и капрон (см. § 190). Наиболее важными
производными циклогексана являются спирт циклогексано л (см.
§ 69) и кетон циклогексанон (см. § 87).
АЛКЕНЫ
Алкенами называют ненасыщенные углеводороды, содержащие
в молекуле одну двойную связь. Простейшим представителем
таких углеводородов является этилен С2Н4, в связи с чем со¬
единения этого ряда называют также углеводородами ряда
этилена. Часто для них используют название олефины.
Название «олефины» происходит от латинского названия этилена (gaz
olefiant — маслородный газ). Это название было дано этилену в XVIII веке
голландскими химиками из-за его способности образовывать с хлором жид¬
кое маслянистое вещество — дихлорэтан C2H4CI2.
Гомологами этилена являются пропилен С3Нб, бутен C-iHs,
пентен С5Н10, гексен СбН^ и др. Гомологический ряд этилено¬
вых углеводородов имеет общую формулу С„Н2П, т. е. такую
же, как ряд циклоалканов. Таким образом, алканы и циклоал¬
каны — изомеры, относящиеся к различным гомологическим ря¬
дам.
неустойчивая
конформация
устойчивая
конформация
ГЛАВА 4
64
%
Рис. 16. Строение этилена:
а — a-связи н р-электронные облака;
б —а» и л-связи; в — геометрия мо*
лекулы
§ 29. Строение. Характерная особенность строения алкенов—
наличие в молекуле одной двойной углерод-углеродной связи.
Двойная связь образуется при помощи двух пар обобщенных
электронов (четырехэлектронная связь). Углеродные атомы,
связанные двойной связью, находятся в состоянии эр2-гибриди-
зации, каждый из них образует три a-связи, лежащие в одной
плоскости под углом 120°. Негибридизованные орбитали р-элек¬
тронов расположены перпендикулярно к плоскости a-связей и
параллельно друг другу и вследствие «бокового» перекрывания
образуют вторую связь, называемую я-связью. Электронное
облако л-связи частично расположено над плоскостью, а час¬
тично под плоскостью, в которой лежат атомы. Таким образом,
две пары электронов образуют различные по своей природе,
геометрии и прочности связи — а и я. Двойная связь пред¬
ставляет собой сочетание о- и я-связей. Боковое перекрывание
негибридизованных р-орбиталей, образующих я-связь, сближа¬
ет атомы углерода и расстояние между ними становится коро¬
че— длина двойной С=С-связи 0,133 нм (длина простой С—С-
связи 0,154 нм). Схема расположения атомов и связей в моле¬
куле этилена приведена на рис. 16. О том, как влияет природа
двойной связи на свойства алкенов, сказано при рассмотрении
их химических свойств (см. § 32).
Несмотря на хорошее соответствие с опытными данными, представление
о том что двойная связь состоит из двух разных связей (а- и я-) является
лишь гипотезой. Известны и другие гипотезы, которые позволяют создать
другие модели двойной связи (Полинг). Мы рассматривать их не будем.
§ 30. Изомерия и номенклатура. Структурная изоме¬
рия. Структурные изомеры алкенов могут отличаться строе¬
нием углеродной цепи (изомерия углеродного скелета) и поло¬
жением двойной связи (изомерия положения). Первый из эти-
3 Органическая химия
65
леновых углеводородов, для которого возможны изомеры, имеет
четыре атома углерода. Тривиальное название этого углеводо¬
рода бутен С4Н8. Для него известны три изомера:
изомерия углеродного
скелета
СН2—СН—СН2—СН3 3 изомерия положения
СНз—СН=СН—СНз J двойной связи
СН2=С—CHj
I СНз
Углеводород пентен С5Н10 имеет пять изомеров:
изомерия
углеродного
скелета
{
СН2—СН СН2 СН2 СН* "1 изомерия положения
СНз—СН=СН—CHj—СНз J двойной связи
СНз
СН?=С—СН2—СНз
СН;
СНз
СНз—С=СН—СНз
изомерия
> положения
ДВОЙНОЙ связи
СНз—СН—СН=СН2
Эти примеры показывают, что число изомеров у алкенов
выше, чем у алканов.
цис-транс-И з о м е р и я. Для этиленовых соединений возмо¬
жен еще один вид изомерии — пространственная изомерия, ко¬
торая связана с расположением заместителей по отношению к
плоскости двойной связи. Эта изомерия наблюдается в тех слу¬
чаях, когда каждый из атомов углерода, связанных двойной
связью, затрачивает остальные две единицы валентности на
связь с двумя различными заместителями, Расположение пары
Одинаковых заместителей по одну сторону плоскости двойной
связи дает цис-изомер, по разные стороны — транс-изомер, на¬
пример:
транс- бутен-2
(т. пл. -105,8 °с)
(Т. КИП. +0,96 °С)
Превращение этих изомеров друг в друга требует вращения вокруг
двойной углерод-углеродной связи, т. е. необходимо нарушить перекрывание
р-орбиталей и разорвать я-связь. На это требуется затратить свыше
66
250 кДж/моль энергии. Такой энергетический барьер затрудняет вращение и
обусловливает существование цис-транс-изомеров как устойчивых индивиду¬
альных веществ.
Природа этой изомерии такая же, как в замещенных цик¬
лах (см. § 27).
Как правило, транс-изомеры более устойчивы, имеют более
высокую температуру плавления. При нагревании цис-транс-
изомеры могут переходить друг в друга: между ними устанав¬
ливается равновесие. Характерные особенности этого вида про¬
странственной изомерии рассмотрим позднее на примере нена¬
сыщенных карбоновых кислот — фумаровой и малеиновой (см.
§ ЮЗ).
Номенклатура. Тривиальные названия алкенов харак¬
теризуются окончанием илен: этилен, пропилен, бутилен и т. д.
Рациональная номенклатура рассматривает алкены как произ¬
водные этилена, содержащие вместо атомов водорода углеводо¬
родные радикалы; в случае необходимости уточняют положение
радикалов у разных атомов этиленовой группировки, обозначая
их греческими буквами аир, например:
СН2=СН—СН2—СН» СН»—сн=сн—сн»
этилэтилен (бутен-1) cuAt-диметилэтилен,
а, р-диметилэтилен
(бутен-2)
СН»
I
сн3—сн—сн2—сн=сн2
изобутилэтилен
(4-метилпентен-1)
По современным правилам заместительной номенклатуры
'Названия алкенов строят подобно названиям алканов, но окон¬
чание ан заменяют на окончание ен, откуда и происходит об¬
щее название алкены. В качестве главной цепи выбирают цепь,
включающую двойную связь, даже если эта цепь и не является
самой длинной. Нумерацию проводят так, чтобы атом углеро¬
да, от которого начинается двойная связь, получил наименьший
номер; этим номером и указывают положение двойной связи.
В приведенных выше примерах эти названия даны в скобках.
В качестве примеров применения этой номенклатуры можно
привести также названия следующих соединений:
сн» сн» сн2
СН»—СН=СН—СН—СН» СН»—СН—СН2—С—СН2—СН»
4-метилпентеи-2 4-метил-2-этвлпентен-1
Общее название радикалов, образованных из алкенов,—
алкенилы. Приведем для примера названия некоторых наиболее
часто встречающихся радикалов: СН2 — СН — винил (этенил);
сн»
СН»— А=СНа
ссн.к-диметилэтилен,
а,а-диметилэтилен
(2-метилпропен-1)
3*
67
Таблица 6. Состав газообразных продуктов (в %), образующихся
при разных способах переработки нефти
Компоненты
Прямая
перегонка
Пиролиз
Термический
крекинг
Каталитический
крекинг
Водород
1
12
3-9
5-6 .
Метан
11-46
65-57
28—50
10
Этан
3-17
5-7
13-18
3-5
Пропан
3-28
0.5
3-15
18-20
Бутаны
14-34
0,2
1-6
42-46
Этилен
—
16-18
2-23
3
Пропилен
—
7-8
6-18
6-11
Бутены
—
4—5
4—10
5-6
СН2 = С(СНз)— изопропенил; СН3—СН = СН — пропенил;
СН2 = СН—СН2 — аллил.
§31. Способы получения. В противоположность алканам, алке-
ны в природе встречаются редко. Низшие алкены в небольших
количествах могут входить в состав нефтяного газа, высшие —
в состав некоторых нефтей. Важнейшим промышленным постав¬
щиком алкенов является нефтеперерабатывающая промышлен¬
ность. Большие количества алкенов образуются при крекинге и
пиролизе нефти (табл. 6).
Пиролиз — самый эффективный современный промышленный
способ производства низших алкенов. Структура продуктов и
их выходы зависят от используемого сырья и условий процесса
(см. § 57). Газы, получающиеся в количестве до 25 % от сырья,
богаты низшими газообразными алкенами С2—С4. Крекинг-
бензины содержат много жидких алкенов.
Высшие алканы расщепляются под действием высоких тем¬
ператур (400—700 °С). Реакция расщепления протекает с обра¬
зованием свободных радикалов:
СНз—СН3 —► 2-СН3 RCH2—CH2R —► 2-CH2R
Радикал (*СН3, *CH2R), образовавшийся в результате
первичного разрыва связи С—С, атакует молекулу высшего ал¬
кана и отрывает водород от третичного или вторичного атома
углерода, удаленного от конца цепи. В случае нонана процесс
может идти так:
СНз + СНз—СНз—СН2—СН2—СН2—СН2—СН2—СН2—СН3 —>-
—► СН4+ СНз—CHj—СН—СН2—СН2—CHj—СН2—СН2—СНз
Затем идет разрыв молекулы в p-положении к атому угле¬
рода, несущему неспаренный электрон, при этом образуются
алкен и новый радикал:
СНз—СН2—СН—СН2Ч-СН2—СН2—СН2—СН2—СНз —V
—> СНз—СН2—СН=СН2 + • СН2—СН2— СН2— СН2—СНз
68
Многократное p-расщепление приводит к образованию
большого количества алкенов, в частности этилена:
• СН2—СН2-гСН2—СН2—СН3 —► СН2=СН2 + • СН2—СН2—СН3
• СН2—СН2—GH3 —V СН2=СН2 + Н3С .
Одновременно образуются низшие алканы и водород:
сн2=сн2 сн3—сн2—сн3
этилен пропан
|—н- | +н.
СН3—СН2—СН2—СН2—СН3 —v СН3—СН2 + СН2—СН2—СН3
пентан I
димеризация
I радикалов
'1 1
сн3—сн2—сн2—сн3 сн3—сн2—сн2—сн2—сн2—сн3
бутан гексан
Другим важным промышленным способом получения алке¬
нов является дегидрирование алканов. Катализатором служит
оксид хрома(III). Процесс ведут при 450—460 °С:
► СН2=СН—СН2—СН3 + Н2
сн3—сн2—сн2—сн3 —
1—► СН3—СН=СН—СНз + н2
(СН3)2СН—СН3 —►(СН3)2С=СН2 + н2
Лабораторные способы получения алкенов в большинстве
своем являются реакциями отщепления. Важнейший из этих
способов — дегидратация спиртов. При нагревании спиртов с
водоотнимающими веществами (концентрированная серная или
фосфорная кислота) или пропускании паров спиртов над таки¬
ми катализаторами, как каолин, оксид алюминия, оксид тория,
при повышенной температуре идет отщепление воды. Так, из
этилового спирта получается этилен:
сн3—сн2он —► сн2=сн2 + н2о
При дегидратации спиртов водород предпочтительнее отщеп¬
ляется по правилу А. М. Зайцева (1875 г.) —от того из сосед¬
них атомов углерода, который беднее водородом:
СН3—СН2—СН—СНз —► СНз—сн=сн— сн3 + Н20
он
Дегидрогалогенирование. При нагревании алкилгалогенидов
с концентрированными спиртовыми растворами щелочей или
органическими основаниями происходит отщепление галогеново¬
69
дорода (также по правилу Зайцева). Легче всего реакция идет
с третичными галогенпроизводными, труднее с первичными.
КОН(спирт. р-р)
СН3—СН2—СН—CHI—СН3 — > СН3—СН2—С=СН—СН3
I -HI I
СНз СНз
Отщеплением галогена (дегалогенирование) от дигалогенпро-
изводных, у которых галогены находятся при соседних атомах
углерода, также можно получить алкены:
цинковая пыль
СНз—СН—СН—СН—СНз >- СНз—СН=СН—СН—СНэ
I , | ZnBr2 |
Вг Вг СНз СНз
§ 32. Общая характеристика физических и химических свойств.
Физические свойства. Алкены по физическим свойст¬
вам близки к алканам. Однако температуры кипения их не¬
сколько ниже, чем у соответствующих алканов, а плотности
несколько выше. Первые три члена ряда (С2—С4) представляют
собой газы, от С5 до С17 — жидкости, Cie и выше — твердые ве¬
щества. Изомерия углеродной цепи, изомерия положения двой¬
ной связи, а также пространственная конфигурация отражаются
на физических константах изомеров (табл. 7).
Все алкены имеют плотность меньше единицы, обладают
характерным запахом, в воде растворимы плохо, но все же
лучше, чем соответствующие алканы.
Химические свойства. Химическое поведение алке-
нов определяется наличием двойной связи. Электронная плот¬
ность a-связи концентрируется по линии, соединяющей ядра
атомов, а электронная плотность я-связи выходит за эти пре¬
делы, образуя более обширную область отрицательного заряда.
Характерная особенность я-электронов — их подвижность, они
менее прочно удерживаются ядрами атомов, чем о-электроны.
Таблица 7. Физические свойства некоторых алкенов
Соединение
Формула
T. пл„ "С
T. кип., «С
Относительная
плотность
при т. кип.
Этилеп
CHj=CH2
-169
-105
0,570
Пропилен
СН2=СН—СНз
-185,5
-47,8
0,610
Бутен-1
СН2=СН—СН2—СНз
-130,0
-6,3
0,596
Бутен-2
цис-
СНз—СН=СН—СНз
-138,9
+3,5
0,621
транс-
-105,5
+0,9
0,604 *
Изобутен
(СНз)2С=СН2
— 140
-6,9
0,594
* Под давлением.
70
Поэтому я-связь легче поляризуется. Электроны, образующие
я-связь, легко вовлекаются в химические реакции электрофила¬
ми, которые, возбуждая поляризацию я-связи, вызывают ее
гетеролитический разрыв. Энергия образования двойной связи
составляет 620 кДж/моль, а для простой связи она равна
348 кДж/моль. Разница энергии (а + я) -связи и о-связи со¬
ставляет около 270 кДж/моль и характеризует меру прочности
я-связи. При нарушении я-связи происходят реакции присоеди¬
нения с образованием двух новых о-связей. Такие реакции ти¬
пичны для алкенов.
§ 33. Реакции. Реакции присоединения. Большая
часть реакций присоединения к двойной углерод-углеродной
связи происходит по гетеролитическому типу и относится к ре¬
акциям электрофильного присоединения.
Процесс электрофильного присоединения является ступенча¬
тым ионным процессом. Решающей стадией таких реакций яв¬
ляется взаимодействие электрофильного реагента с я-электрон-
ным облаком двойной связи. Часто в роли электрофильного
реагента выступает протон Н+, образующийся при диссоциации
минеральных кислот (НС1, НВг, HI, H2S04). Первоначально
электрофильный реагент за счет своего положительного заряда
взаимодействует с я-электронами двойной связи, образуя
я-комплекс. Затем с одним из ненасыщенных С-атомов с учас¬
тием я-электронов двойной связи образуется обычная а-связь.
Соседний атом углерода — второй партнер имевшейся двойной
связи, лишившись электрона, приобретает положительный за¬
ряд:
^с=с^ + н+
7Г-комплекс карбокатиои
Во второй, завершающей стадии карбокатиои реагирует с
анионом X-, образуя вторую a-связь. В результате получается
конечный продукт присоединения:
Н Н X
—А—с— + х~ —► —А—А—
Обычно первая стадия идет медленнее, чем вторая.
Рассмотрим типичный пример такой реакции — присоедине¬
ние бромоводорода к бутену-2:
НВг
Н+ + Вг~
71
1н+
СН3-СН=СН-СНз+Н+ —[сН3-СН^СН-СНз] —
—► CH3-CH2-CH-CH3+Jl^ CH3—CH2—CHBr—CH3
Таким образом, присоединение бромоводорода протекает как
двухстадийный ионный процесс. Решающая стадия всей реак¬
ции — присоединение протона с образованием карбокатиона.
Это доказывается тем фактом, что присоединяться способны
галогеноводороды, но не их соли.
По электрофильному механизму идет и присоединение гало¬
генов. Под влиянием электронной плотности двойной связи
прежде всего происходит изменение распределения электронной
плотности в молекуле галогена, т. е. поляризация ее. Электро-
фильность симметричной молекулы галогена может проявиться
только после поляризации. Дальше реакция идет по описанному
выше пути:
СН2=СН2 + Вт—Вг >- СН2—СН2Вг - ВГ->- ВгСН2—СН2Вг
~Вг~
Аналогично протекает присоединение и других неорганиче¬
ских веществ (H2SO4, Н20, НОС1 и др.), а также некоторых
органических молекул. После первоначального присоединения
электрофильной частицы во второй стадии реакции может при¬
соединяться любой находящийся в растворе анион. Это было
доказано при реакции этилена с бромом в водном растворе в
присутствии анионов СГ и N03. Кроме ВгСНг—СН2Вг при этом
получаются также ВгСН2—СН2С1 и ВгСН2—CH2ONO2. Струк¬
тура молекулы алкена, а также природа реагента влияют на
скорость реакции. Рассмотрим конкретные примеры реакций
присоединения к алкенам.
1. Присоединение галогена. Алкены при обычных условиях
присоединяют галогены, особенно легко хлор и бром. В резуль¬
тате образуются дигалогенпроизводные алканов, содержащие
галогены у соседних атомов углерода — так называемые вици-
нальные производные (от лат. vicinus — соседний):
СН2=СН2 + С12 —> С1СН2—СН2С1
1,2-дихлорэтан
СН3—СН=СН2 + Вг2 —>• СН3—СНВг—СН2Вг
1,2-дибромпропан
Так, обесцвечивание бромной воды является качественной
реакцией на двойную связь. Реакцию присоединения галогена
используют и для количественного определения степени ненасы-
щенности. Для этого применяют раствор брома в четыреххло¬
ристом углероде или хлороформе, растворы хлорида иода или
72
хлорида брома. Последние особенно легко присоединяются по
двойной связи, так как из-за разницы в электроотрицательности
в- 6+ в- в+
обоих атомов их молекулы поляризованы: С1—I, С1—Вг.
Методы, основанные на присоединении галогенов по двойной
связи, важны для определения степени непредельности жидких
жиров и масел. Реакцию этилена с хлором используют в про¬
мышленности для получения дихлорэтана, применяемого в ка¬
честве растворителя и в производстве винилхлорида.
2. Присоединение галогеноводородов (гидрогалогенирова-
ние). В реакциях присоединения несимметричных реагентов ти¬
па НХ к несимметрично построенным алкенам, например
RCH=CH2, возникает вопрос о порядке присоединения, т. е.
образуется ли соединение RCHX—СН3 или RCH2—СН2Х.
Закономерности подобных реакций были изучены В. В. Мар-
ковниковым (1838—1904 гг.) на примере присоединения к алке¬
нам галогеноводородов. Эти работы привели к формулировке
правила Марковникова (1869 г.): при присоединении галогено¬
водородов к несимметричным алкенам водород присоединяется
к более гидрогенизованному атому углерода, а галоген — к дру¬
гому, связанному с первым двойной связью, т. е. к менее гид¬
рогенизованному:
СНз—СН=СН2 + НВг —> СН3—СНВг—СНз
Правило Марковникова находит теперь объяснение с пози¬
ции электронной теории органических реакций. Вследствие того,
что электроотрицательность углерода больше, чем водорода
атом углерода в метильной группе СН3 несет на себе некото¬
рый отрицательный заряд. Это приводит к сдвигу о-электронов
связи Н3С—СН в направлении центрального атома углерода.
Этот сдвиг в свою очередь влечет за собой гораздо более силь¬
ное смещение подвижных я-электронов двойной связи (в на¬
правлении, указанном изогнутой стрелкой). Таким образом,
электронные сдвиги Н-»-С, Н3С-»-СН вызывают сильный сдвиг
электронов двойной связи, т. е. поляризацию со сдвигом элек¬
тронной плотности к атому углерода, не соединенному с алкиль¬
ной группой:
н
н-*-с->-с!н=0:к2 сн3—сн=сн2
i
Протон присоединяется к тому ненасыщенному атому угле¬
рода, который несет частичный отрицательный заряд. Ясно, что
им будет атом углерода, связанный с меньшим числом алкиль¬
ных групп (более насыщенный водородом). Анионная часть
реагента направляется к атому углерода с наименьшей элек¬
тронной плотностью — к наименее гидрогенизованному.
73
На результат реакции влияет и стабильность промежуточно
образующегося карбокатиона. В принципе на первой стадии ре¬
акции возможно образование двух карбокатионов А и Б, в за¬
висимости от того, к какому из ненасыщенных атомов углерода
направится протон.
СН3-*СН2->СН2 или СН3->СН<-СН3
А Б
Предпочтительным будет направление, требующее меньших
энергетических затрат. В карбокатионе Б положительный заряд
на центральном атоме частично гасится двумя метильными
группами, у которых на атомах углерода имеется частичный
отрицательный заряд. В результате положительный заряд ока¬
зывается «размазанным» по трем атомам углерода и связанным
с ними атомами водорода. Заряд карбокатионного центра в
структуре А будет выше, чем в структуре Б, так как атом
углерода, несущий заряд, связан только с одним радикалом
(этилом). Более устойчивым является карбокатйон с меньшей
энергией, а именно карбокатйон Б, т. е. присоединение водорода
идет к более гидрогенизованному атому углерода.
Правило Марковникова соблюдается только при ионном ме¬
ханизме присоединения. В общем виде его можно сформулиро¬
вать так: электроположительная (катионная) частица реагента
присоединяется к атому углерода, содержащему большее число
атомов водорода, а электроотрицательная (анионная) — к ато¬
му углерода с меньшим числом атомов водорода, например:
СН3—СН=СН2 +Y-C1 —► СН3—СНС1—СН21
хлорид
иода
Ниже приведены катионные и анионные части ряда электро¬
фильных реагентов:
Катион Н Н Н Н Н С! HgOCOCH3 I
Анион Cl Br I OSOjOH ОН ОН СН3СОО CI
Правило Марковникова не имеет абсолютного значения. Хо¬
тя порядок присоединения обусловлен в первую очередь строе¬
нием молекулы алкена, он зависит также от условий реакции.
В частности, в присутствии пероксидов реакция приобретает
радикальный характер и присоединение идет против правила
Марковникова. Это правило неприменимо к некоторым непре¬
дельным альдегидам и кислотам (см. §§ 88, 99).
3. Присоединение серной кислоты. Эта реакция идет анало¬
гично присоединению галогеноводородов. В результате образу¬
ются алкилсерные кислоты (кислые эфиры серной кислоты):
СН2=СН2 + H0S020H —>• сн3—сн2—OS02OH
этилсерная кислота
74
4. Присоединение воды (гидратация алкенов). Прямое при¬
соединение воды к алкенам (прямую гидратацию) проводят в
присутствии фосфатных катализаторов (НЭР04 на алюмосили¬
кате с солями кадмия, меди, кобальта; 300 °С; 8 МПа);
CH2=CH2 + HjO —► СНз—СН2ОН
Подвергая алкилсерные кислоты гидролизу, также можно
получить спирты:
СНз— СН8—0S020H + Н20 —► СНз—СН2ОН + H0S020H
5. Присоединение водорода. Алкены не присоединяют водо¬
род «в момент выделения», поэтому на них не действуют такие
реагенты, как цинк и кислота, натрий и спирт. Однако, действуя
водородом в присутствии катализаторов, можно осуществлять
каталитическое гидрирование:
СНз
3—(!—СН=С—С
СНз—С—СН=С—СНз
<!:Нз СНз
2,4,4-триметилпентен-2
Hj/NI
СНз
I
СНз—С—СН2—СН—СНз
(Ьз СНз
2,2,4-триметилпентан (изооктан)
Наиболее активными катализаторами этой реакции являют¬
ся платина, палладий, но на практике чаще используют мелко¬
раздробленный никель. Особенно активную форму никелевого
катализатора представляет собой «никель Ренея», называемый
также «скелетным» никелем.
Алкилирование. По двойной связи при содействии со¬
ответствующего катализатора могут присоединяться и органиче¬
ские вещества — углеводороды, спирты, карбоновые кислоты
и др.
Очень важной реакцией в современном органическом синтезе
является реакция алкилирования алканов алкенами. Сущность
этой реакции — присоединение алкана (преимущественно с тре¬
тичным атомом углерода) по двойной связи алкена:
(СНз)зС—Н + СНа=С(СН3)2 —► (СН,)зС—СН}—СН(СНзЬ
Катализаторами реакции служат серная и фтороводородная
кислоты, фторид бора, хлорид алюминия. Механизм этой ре¬
акции:
(СНз)2С=СНа + н+ —> (СНз)зС
(СНз)зС + СН2=С(СНз)з —► (СНз)зС—СНз—С(СНзЬ
(СНз)зС—СНз—С(СН3)з + СН(СН3)з —*
—> (СНз)зС—СНз—СН(СН3)з + (СН,)3С*
Получаемые разветвленные алканы — ценные компоненты
горючего для двигателей внутреннего сгорания.
75
Полимеризация. Молекулы алкенов могут вступать в
реакцию между собой. Примером является открытая А. М. Бут¬
леровым реакция димеризации (удвоения) изобутена под дейст¬
вием концентрированной серной кислоты. Сущность этой реак¬
ции в том, что в кислой среде молекула изобутена за счет
электронов двойной связи присоединяет протон. Образовавший¬
ся карбокатион как электрофильная частица присоединяется по
двойной связи второй молекулы алкена. Продукт присоединения
стабилизируется, выбрасывая протон, при этом двойная связь в
образующемся непредельном углеводороде с восемью атомами
углерода может занять два разных положения. Этот синтез те¬
перь осуществляется в промышленном масштабе. Полученную
смесь изомерных углеводородов подвергают далее каталитиче¬
скому гидрированию, получая изооктан:
Н+ СН2=С(СН3)2 +
(СНз)2С=СН2 ► (СН3)зС+ »- (СНз)зС—СН2—С(СНз)8 ^
н2
—► (СНз)зС—СН2— С=СН2 + (СНз)зС—СН=С(СНзЬ >■
СНз
—> (СНз)зС—СН2—СН(СН3)2
Таким путем побочный продукт крекинга нефти — газообраз-1
ный изобутен — превращают в ценное жидкое горючее для дви¬
гателей внутреннего сгорания.
Можно получить и продукты соединения большего числа
молекул алкенов — полимеры — соединения с высокой молеку¬
лярной массой. Полимеризация — одна из наиболее важных в
промышленном отношении реакций алкенов:
сн2=сн2 + сн2=сн2 + ••• —► —сн2—сн2—сн2—сн2
полиэтилен
или х(СН2=СН2) —► (—СН2—СН2—)х
Продукты полимеризации низших алкенов — этилена, пропи¬
лена, изобутена — используют для производства пластических
масс, синтетических волокон и других практически важных ма¬
териалов. Подробнее этот вопрос рассмотрен в специальном
разделе (см. § 186).
Окисление. Алкены легко подвергаются действию раз¬
личных окислителей, резко отличаясь в этом отношении от ал¬
канов и циклоалканов. В зависимости от условий окисления
образуются разные продукты. В самых жестких условиях, при
сжигании на воздухе, алкены превращаются в диоксид углерода
и воду:
С2Н4 + 302 —»- 2С02 + 2Н20
76
В более мягких условиях окисление в первую очередь идет
по месту двойной связи. Осторожное окисление щелочным рас¬
твором перманганата калия приводит к двухатомным спир¬
там — гликолям:
КМпо4 (О + НаО)
СН2=СН2 -» НО—СН2—СН2—ОН
Эту реакцию открыл русский химик Е. Е. Вагнер (1849—
1904 гг.). Реакция протекает быстро, на холоду, причем наблю¬
дается обесцвечивание характерной окраски перманганата. Ре¬
акция Вагнера служит качественной пробой на двойную связь.
При действии более энергичных ‘ окислителей (кислотный
раствор перманганата, хромовая смесь) происходит окислитель¬
ное расщепление молекулы алкена по месту двойной связи. Для
примера приводим реакции окисления трех изомерных бутенов:
10]
СНз—СН2—СН=СН2 *■ СН3—СН2—СООН + С02 (1)
бутен-1 пропионовая
кислота
СНз—СН=СН—СН» 2СНа—СООН (2)
бутен-2 уксусная
кислота
СНз СНз
СНз—i=CH2 101 > СНз—А=0 + С02 (3)
2-метилпропен ацетон
(изобутен)
По продуктам окисления можно сделать вывод о положении
двойной связи в молекуле, о строении углеродного скелета.
Так, если при окислении наряду с карбоновой кислотой обра¬
зуется диоксид углерода, то можно сделать вывод, что двойная
связь была концевой [реакции (1) и (3)]. Если же образуются
только карбоновые кислоты, то очевидно, что алкен имел строе¬
ние R—СН=СН—R' [реакция (2)], так как именно группиров¬
ка —СН= окисляется в карбоксильную группу —СООН. Если
атом углерода, у которого находится двойная связь, не содер¬
жит водорода, то в результате окисления образуются кетоны
[реакция (3)].
Каталитическое окисление кислородом воздуха приводит к
образованию а-оксидов, имеющих большое значение в органи¬
ческом синтезе. Так, например, получают очень важный техни¬
ческий продукт — этиленоксид:
О
Ag. нагревание / \
сн2=сн2 + о2 * Н2С—сн2
77
Озонирование. Алкены легко реагируют с озоном, об¬
разуя продукты присоединения — циклические пероксиды, на¬
зываемые озонидами:
\
/
с=с;
/
\
+ о,
о
/ \ / \
0—0
Озониды — вязкие жидкости или твердые вещества. Они
очень неустойчивы, легко взрываются. Обычно их не выделяют,
а сразу после получения разлагают водой. При этом образуют¬
ся карбонильные соединения (альдегиды и кетоны), строение
которых указывает на строение подвергавшегося озонированию
алкена. В качестве побочного продукта за счет кислорода пер-
оксидного мостика образуется пероксид водорода, который мо¬
жет реагировать с альдегидами. Чтобы не допускать его обра¬
зования, гидролиз проводят в присутствии восстановителя (цин¬
ковой пыли). Так, при озонировании 2-метилбутена-2 сначала
образуется его озонид, затем при разложении водой — кетон и
альдегид:
сн3
I
СНз—С=СН—СНз
Оз
НзС
Н3С
\/\
О
СН—СНз
Н20, Zn
—* (СНз)2С=0 + СНз—с—н + н2о2
ацетон ацеталь¬
дегид
Формальный результат озонирования с последующим дейст¬
вием воды — разрыв молекулы алкена по месту двойной связи и
присоединение атома кислорода к освободившимся в каждом из
осколков валентностям. Для определения строения алкена по
продуктам озонирования надо мысленно удалить из них кисло¬
род и соединить получившиеся осколки двойной связью. Напри¬
мер, если при озонировании были получены метилэтилкетон и
пропионовый альдегид, то исходным алкеном был 3-метилгек-
сен-3:
CHj
CHj—СН*—С=о
метилэтилкетон
Н
I
0=С—СН2—С^з
пропионовый
альдегид
СН*
СНз—СН2—С=СН—СН2—СНз
З-метилгексен-З
Реакции замещения. В некоторых случаях алкены
могут вступать и в реакции замещения. Наиболее легко заме¬
78
щается водород у а-углеродного атома по отношению к двой-
\ I
,С=С—К
/ *
энергия
связи
435 кДж/моль
Так, например, изобутен при действии хлора лишь в незна¬
чительной степени превращается в дихлорзамещенное (10—
15%); главным продуктом оказывается непредельное соедине¬
ние (85—90 %):
СН3 СН, СН3
СНз—с:=снг —► СН2С1—С=СН2 + СН3—(XI—СН2С1
2-метил-З-хлор- 2-метил-1,2-днхлор-
пропен-1 пропан
Ближайший гомолог этилена — пропилен — при хлорирова¬
нии в газовой фазе (500 °С) образует аллилхлорид (см. §59).
Помимо практического значения рассмотренных реакций за¬
мещения, они интересны и в теоретическом отношении, показы¬
вая, что обычное представление о полном сходстве химических
свойств гомологов правильно только в первом приближении.
В начале 60-х годов была открыта своеобразная реакция перераспределе¬
ния остатков RCH= в алкенах, получившая название метатезиса. Она осу¬
ществляется на металлических катализаторах, промежуточно образующих с
алкенами четырехкоординационные комплексы:
N:=c—с—н
/ I °t
энергия
связи
322 кДж/моль
R—СН=ФСН—R
Я—СН—СН—Я
\/
мхя
R—СН
■, 11
R'—СН
СН—R
+ II ,
СН—R
По другим представлениям, в реакции участвуют частицы типа R2C=ме¬
талл. На основе реакции метатеэиса разрабатываются синтезы полимеров,
феромонов и других важных веществ.
§ 34. Отдельные представители. Этилен (этен) С2Н4 — газ;
мало растворим в воде (в 1 объеме воды при 0 °С — 0,25
объема), лучше — в спирте (3,6 объема); горит более ярким пла¬
менем, чем метан, поскольку содержание углерода в нем выше.
Смесь этилена с воздухом взрывчата. Этилен образуется при
сухой перегонке органических веществ, всегда содержится в
светильном газе.
В нашей стране почти весь этилен (более 80 %) получают
пиролизом бензина, около 10 % — дегидрированием этана.
79
Этилен является одним из важнейших полупродуктов для
промышленности органического синтеза. Многие вещества, полу¬
чаемые непосредственно из этилена хлорированием, гидрохлори¬
рованием, гидратацией, окислением, имеют практическое при¬
менение, а также используются для дальнейших синтезов.
Важнейшие направления технического использования этилена
приведены на схеме 1.
Схема 1.
Техническое использование этилена
С12
сн,=сн2 —
НС1
НОС1
н2о
H2SO4
С1СН2—СН2С1 —
дихлорэтан
СНз—СН2С1
этилхлорид
С1СН2—СНвОН
этиленхлоргидриц
СНз—СН2ОН
этиловый спирт
СНз—СНг—OSO3H —
СН2=СНС1
винилхлорид
о2
Свн„
полиме»
ризация
этилсерная
кислота
Н2С—сн2
V
этиленоксид
СНз—СНО
уксусный
альдегид
С6Н6-СН2-СН3
этилбензол
Полиэтилен
СНз—СН2ОН
этиловый спирт
С2Н5—О—С2Н5
ДИЭТИ..ОВЫЙ спирт
С6Н5—СН=СН2
стирол
Полистирол
теломе-
ризация
горение
Синтетическое волокно энант
Сварка и резка металлов
Пропилен С3Н6 — газ; в промышленности получают из га¬
зов крекинга или из нефтяного газа (дегидрированием пропа¬
на). Пропилен используют для получения изопропилбензола
80
(около 40 %), из которого далее получают фенол и ацетон, а
также для получения к-бутанола (20 %), полипропилена (8 %),
акрилонитрила, глицерина, изопропилового спирта, аллилхло-
рида.
АЛКИНЫ
Алкины — ненасыщенные углеводороды, содержащие в молеку¬
ле тройную углерод-углеродную связь —С=С—. Простейшим
представителем алкинов является ацетилен С2Н2, поэтому их
часто называют ацетиленовыми углеводородами. Общая форму¬
ла алкинов СяНгп-2.
§ 35. Строение, изомерия и номенклатура. Строение. В об¬
разовании тройной связи участвуют три пары обобщенных элек¬
тронов (шестиэлектронная связь). Углеродные атомы, образую¬
щие тройную связь, находятся в состоянии sp-гибридизации.
Каждый из них образует две о-связи, направленные под углом
180°. Две негибридизованные р-орбитали каждого углеродного
атома расположены под прямым углом (90°) друг к другу.
Они попарно перекрываются, образуя две я-связи, расположен¬
ные в двух взаимно перпендикулярных плоскостях. Тройная
связь представляет собой сочетание одной о- и двух я-связей.
На рис. 17 изображено расположение связей в молекуле ацети¬
лена. При образовании тройной связи атомы углерода сближа¬
ются еще больше, чем при образовании двойной связи; длина
тройной углерод-углеродной связи 0,120 нм.
Изомерия. Изомерия алкинов обусловлена строением
углеродной цепи и положением тройной связи; в этом у них
сходство с алкенами. Но для ацетиленовых углеводородов не¬
возможно существование цис-транс-томеров, так как две
о-связи, образованные углеродным атомом в состоянии sp-гиб¬
ридизации, направлены под углом 180°, т. е. лежат на одной
прямой. Все четыре атома, составляющие ацетилен, находятся
на одной линии — молекула линейна.
Номенклатура. Принцип построения названий алкинод
тот же, что и алкенов. По рациональной номенклатуре соеди-
ГЛАВА 5
180-
РИС. 17. Строение ацетилена
81
нение рассматривается как производное ацетилена; по совре¬
менной заместительной номенклатуре названия алкинов имеют
окончание ин. При составлении названия выбор главной цепи
и начало нумерации определяет тройная связь, например:
СН3—СН2—С=СН
этилацетилен
(бутнн-1)
•сн3
I
СН3—СН- СН2—С=СН
изобутилацетилен
(4-метилпентин-1)
СН3—СзеС—СН3
диметилацетилен
(бутин-2)
СН3 С2Н5
СН3—(^Н—СН—с=с—снэ
б-метил-4-этилгексин-2
Если в молекуле одновременно имеются тройная и двойная
связи, то начало нумерации определяет двойная связь, напри¬
мер:
СН*=СН—СН2—СН2—СэСН гексен-1-ИН-5
Наиболее часто встречающиеся радикалы алкинов имеют на¬
звания: СН=С — этинил, СН = С—СН2 — пропин-2-ил (про-
паргил).
§ 36. Способы получения. Алкины можно получить следую¬
щими способами.
1. Из дигалогенпроизводных алканов отщеплением галогено-
водорода при действии спиртового раствора гидроксида калия
или амида натрия.
Исходные дигалогенпроизводные могут иметь атомы гало¬
гена у соседних атомов углерода или у одного углеродного
атома:
КОН /спирт, р-р)
С3Н7— СН—СН2Вг — > с3н7—с=сн
Вг
Вг
с2н8—сн2—С2Н5
I
Вг
КОН (спирт, р-р)
-2НВг
С2Н5—СзгС—С2Н5
Дигалогенпроизводные, содержащие атомы галогена при со¬
седних атомах углерода, обычно получают присоединением га¬
логена к алкенам. Таким образом, используя эти реакции, уда¬
ется превращать алкены в алкины:
Вг2 КОН (спирт, р-р)
СН3—СН=СН2 ► СН3—СНВг—СН2Вг > СНз—с=сн
2. Из алкилгалогенидов и ацетиленидов натрия или магния:
СН=С—Na + СН31 —► СН=С—CH3 + NaI
OfeC—MgBr + BrC2H5 •—> CfcC—C2H5 + MgBra
82
Эта реакция позволяет превращать простой алкин в более
сложный.
Соединения, содержащие в молекуле несколько тройных свя¬
зей, найдены в некоторых растениях. Например, из подсолнеч¬
ника выделен углеводород желтого цвета — тридецен-1-пента-
ин-3,5,7,9,11:
СНз—с=с—с=с—с=с—с=с—с=*с—сн=сн2
О получении наиболее важного из алкинов — ацетилена —
см. § 39.
§ 37. Общая характеристика физических и химических свойств.
Физические свойства. По физическим свойствам алки-
ны напоминают алкены и алканы. Низшие алкины С2—С«
представляют собой газы, Се—Cie — жидкости, высшие —
твердые вещества. Температуры кипения и плотности алкинов
несколько выше, чем у соответствующих алкенов. Так, этилен
имеет т. кип. —103 °С, ацетилен кипит при —83,6°С, пропилен
и метилацетилен — соответственно при —47 и —23 °С. Раство¬
римость низших алкинов в воде несколько выше, чем раствори¬
мость алкенов и алканов, однако она все же очень мала.
Химические свойства. Химические свойства ацети¬
леновых углеводородов обусловлены природой тройной связи,
особенностями углеродных атомов в состоянии зр-гибридизации.
Энергия образования тройной (о + 2я) связи 810 кДж/моль.
Разница энергий образования ацетилена и этилена составляет
190 кДж/моль — это меньше, чем разница энергий образования
этилена и этана («270 кДж/моль). Типичными реакциями для
ацетилена и его гомологов как для ненасыщенных соединений
являются реакции присоединения.
Многие из реакций алкинов — это реакции электрофильного
присоединения, протекающие аналогично соответствующим ре¬
акциям алкенов, но в две последовательные стадии. Сначала
идет присоединение по тройной связи с образованием производ¬
ных алкенов, затем присоединение по двойной связи с образо¬
ванием производных алканов. Механизм на примере реакции
ацетилена с бромоводородом можно представить схемой:
Н* ♦ Вг“ н*
СН=СН ► сн*=с—Н ► СН2=СНВг —►
СНа—СНВг - ВГ > CHj—СНВг*
Большая ненасыщенность алкинов по сравнению с алкенами,
казалось бы, должна вызвать ббльшую легкость реакции их
с электрофилами. Но так как я-электроны тройной связи со¬
средоточены у ядер атомов углерода и менее доступны для
атаки реагентов, то типичные электрофильные реакции для аце¬
тиленовых улеводородов протекают медленнее, чем для алке¬
нов.
63
Тройная связь окисляется труднее, чем двойная; это позво¬
ляет избирательно окислять двойную связь в присутствии трой¬
ной; тройная связь окисляется в более жестких условиях. Трой¬
ная связь термически более устойчива по сравнению с простой
связью, что подтверждается получением ацетилена из метана
и его гомологов при термическом крекинге (1200—1500 °С)!
(см. § 39).
Увеличение доли s-компоненты в гибридной sp-орбитали
обусловливает увеличение полярности связи С—Н, так как при
этом увеличивается электроотрицательность углеродного атома:
Доля
дорбитали
Относительная
Гибридизация
алектроотрица-
тельность
Sp3
1/4
2,50
Sp3
1/3
2,62
sp
1/2
- 2,75
Повышенная электроотрицательность углерода в состоянии
sp-гибридизации, вызывает поляризацию связи С—Н в молеку¬
ле ацетилена, что является причиной его кислотных свойств:
СН=С:Н —► СН==С» + Н*
Ацетилен в отличие от этана и этилена способен давать
металлические производные — ацетилениды.
Для ацетилена возможны реакции и с нуклеофильными ре¬
агентами, хотя они протекают медленнее, чем с электрофилами.
Примером может служить реакция с циановодородом:
“CN - Н+
СН=СН ► СН=СН—CN > СН2=СН—CN
акрилонитрил
§38. Реакции. Реакции присоединения. 1. Присоеди¬
нение галогена. Реакция протекает медленнее, чем для алкенов.
Обесцвечивание бромной воды — качественная реакция на крат¬
ную связь:
ВГ2 Вг2
СН=СН ВгСН=СНВг ► Вг2СН—СНВг2
1,2-дибромэтен 1,1,2,2-тетра-
бромэтан
При действии хлора образуются 1,2-дихлорэтен С1СН=СНС1
и затем 1,1,2,2-тетрахлорэтан С12СН—СНС12.
2. Присоединение галогеноводородов. Галогеноводороды
присоединяются к тройной связи труднее, чем к двойной. Реак¬
ция имеет особое значение для получения винилхлорида:
СН=СН + НС1 —► СН2=СНС1
Алкилзамещенные ацетилены R—CssCH присоединяют гало-
геноводород по правилу Марковникова:
НВг НВг
RC=CH >- RBrC=CH2 »- RBr2C—СН3
84
3. Гидрирование. Тройная связь гидрируется легче, чем
двойная:
н2
СН=СН + Н, —>• СН*=СН2 >- СНз—сн,
Реакцию можно остановить на стадии образования алкена.
Для гидрирования тройной связи может быть использован во¬
дород в момент выделения (литий или натрий в жидком ам¬
миаке). Применимо также гидрирование молекулярным водо¬
родом над палладиевым катализатором или скелетным никелем.
4. Гидратация (присоединение воды). Реакция протекает
легче, чем для алкенов. Катализатором служит разбавленная
серная кислота и соли ртути (II). Эта реакция была открыта
М. Г. Кучеровым (1881 г.). Первоначально образуется неустой¬
чивый продукт с гидроксильной группой у атома углерода, свя¬
занного двойной связью,— виниловый спирт, который изомери-
зуется в ацетальдегид:
нон Г ОН "1 О
(Hg2+, H2SO4) I I I
СН==СН * |_СН2=С—ш —V СНз—с—н
Гомологи ацетилена гидратируются легче, чем сам ацетилен.
Реакция идет по правилу Марковникова, в результате образу¬
ются кетоны:
Г ОН 1 о
нон 1| II
СН3—С=СН ► |_СН3—6=CH2J —► СНз—С—СН,
5. Присоединение циановодорода. Этот способ используют в
промышленности для получения важного продукта — акрилони¬
трила (см. § 37).
Большое значение имеют реакции присоединения к ацетиле¬
ну органических соединений — спиртов, карбоновых кислот, аль¬
дегидов, кетонов и др. (см. схему 2 на с. 89).
Окисление. Алкины окисляются легко, причем процесс
часто сопровождается разрывом углеродной цепи по месту трой¬
ной связи. Алкины быстро обесцвечивают раствор перманганата
калия, что можно использовать как качественную реакцию на
тройную связь.
Полимеризация. В присутствии катализаторов алки¬
ны могут реагировать друг с другом, причем в зависимости от
условий образуются различные продукты.
а) Под действием водного раствора хлорида меди (I) и хло¬
рида аммония ацетилен димеризуется, давая винилацетилен:
СНз=СН + СН=СН —► СН2=СН— Cs=CH
Винилацетилен обладает большой реакционной способ¬
ностью; присоединяя хлороводород, он образует хлоролрен, ис¬
85
пользуемый для получения синтетического каучука (см. § 188).
СНг=СН—CssCH + НС1 —► СН^СН—СС1=СН2
б) При пропускании ацетилена над активированным углем
при 600 °С образуется бензол (Н. Д. Зелинский, Б. А. Казан¬
ский) :
НС_СН
зснг=сн —>■ нс^ \:н
нс=сн
В настоящее время сходная реакция с использованием циа¬
нида никеля в качестве катализатора применяется для синтеза
ненасыщенных соединений с восьми- и десятичленными цик¬
лами:
СНз=СН
NI(CN)S, 60 «С, 1,5МПа
— ->
С
циклоокта- циклодека-
тетраен пентаен
Циклооктатетраен — жидкость, т. кип. 143 °С, является ис¬
ходным веществом для многочисленных синтезов.
Реакции замещения. Атом водорода в молекуле аце¬
тилена и его моноалкильных производных RC=CH способен за¬
мещаться металлом. Под действием сильных оснований (ами¬
дов щелочных металлов в жидком аммиаке) идет замещение
водорода щелочным металлом и образуется ацетиленид:
б- б+
R—С=С—Н + NaNHa —v R—С==С—Na + NHj
Алкин выступает здесь как кислота и отдает протон сильно¬
му основанию. Ацетилениды — солеобразные продукты, легко
гидролизующиеся водой. Кислотные свойства в ацетилене выра¬
жены слабее, чем в воде, но значительно сильнее, чем в этиле¬
не и этане. При действии магнийорганических соединений обра¬
зуются магнийацетилениды (впервые получены Иоцичем в
1902 г.) s
R—Сз=С—Н + CH3MgI —► R—Сз=С—Mgl + СН4
Ацетилениды натрия и магния применяют в различных син¬
тезах.
Качественной реакцией на атом водорода при тройной связи
является образование ацетиленида серебра, а также ацетилени-
да меди(1). При пропускании ацетилена (и углеводородов типа
RCs=CH) через аммиачные растворы нитрата серебра или хло¬
рида меди(1) образуются характерные осадки: AgCssCAg бело¬
го цвета и CuC=CCu красно-бурого цвета. В сухом состоянии
они взрывчаты.
86
Аммиачный раствор нитрата серебра содержит комплексное соединение
серебра [Ag(NH3)2]OH, которое реагирует с ацетиленом:
CHsCH + 2[Ag(NH3)2]OH —>- AgC=CAg + 4NH3 + 2H2G
Аналогично аммиачный раствор хлорида меди(1) содержит комплекс ме¬
ди [Cu(NH3)2]OH.
Эта реакция может быть использована для выделения аце¬
тиленовых углеводородов из смесей. После осаждения и отде¬
ления ацетиленидов свободные алкины можно регенерировать
действием минеральных кислот:
AgC=CAg + 2HCl —► CH=CH + 2AgCi
§ 39. Ацетилен в промышленности органического синтеза.
В чистом виде ацетилен не имеет запаха. Неприятный запах
ацетилена, получаемого из карбида кальция, объясняется при¬
месями сероводорода и фосфина. В обычных условиях ацетилен
растворяется в равном объеме воды, с повышением давления
растворимость увеличивается. Смесь ацетилена с воздухом
взрывчата в широких пределах концентраций ацетилена (от 3
до 82 %), поэтому в обращении с ацетиленом требуется боль¬
шая осторожность. Ацетилен в виде раствора в ацетоне (1 объ¬
ем ацетона при нормальном давлении растворяет 25 объемов
ацетилена, а при давлении 1,2—1,5 МПа — 300 объемов) хранят
под давлением в стальных баллонах, содержащих пористый
материал — асбест или кизельгур.
Ацетилен — эндотермическое соединение, для его образова¬
ния требуется затратить большое количество энергии; при сжи¬
гании ацетилена выделяется много тепла (1300 кДж/моль).
Температура кислородно-ацетиленового пламени «3000°С, т. е.
выше температуры горения этилена и этана. При этой темпе¬
ратуре часть ацетилена разлагается на элементы, образуя
мельчайшие ярко светящиеся частицы. В XIX веке карбидные
фонари использовали для освещения улиц, площадей. Кисло¬
родно-ацетиленовые горелки в настоящее время используют для
сварки и резки металлов. При недостатке кислорода пламя аце¬
тилена сильно коптит.
Ацетилен получают в промышленности в огромных количе¬
ствах. Сырьевые источники ацетилена — уголь, природный газ,
нефть.
Прокаливанием угля в смеси с оксидом кальция в электри¬
ческих печах получают карбид кальция, при разложении кото¬
рого водой образуется ацетилен:
2000 »С 2НаО
ЗС + СаО ——► СаС2 ► СН=СН + Са(ОН)2
—СО
Этот способ применяют как в лабораториях, так и в про¬
мышленности, где он освоен еще в прошлом веке. Карбидный
87
Схема 2. Техническое использование ацетилена
С12
СНззСН —
ацетилен
v
НС!
>-
н2о
Ni(CO)4
Н20 >
HCN
ROH
У
СНзСООН
у
(СН3)2СО
сн2о
у
горение
►
СЬСН—СНС12 —V С1СН=СНС1
тетрахлорэтан трихлорэтилен
СН2=СНС1
винилхлорид
СНз—СНО —
уксусный
альдегид
СНз—СООН
уксусная кислота
СНз—СНОН—СН2—СНО
альдоль
СН2=СН—СООН |
акриловая кислота *
СНа=СН—СН=СН2
СН2=СН—CN бутадиен-1,3
акрилонитрил
СН2=СН—OR
виниловые эфиры
С Н2=С Н—О—СО—СНз
1
Синтетический
каучук
винилацетат
СН2=С(СН3)—СН=СН2 —► Синтетический каучук
изопрен
Cite С—СН2ОН —>- Глицерин
пропин-2-иловый
спирт
СН2=СН—С=СН
виниладетилен
Сварка и резка
металлов
СН2=СН—СС1=СН2
хлоропрен
I
Синтетический каучук
ацетилен послужил сырьем для промышленности органического
синтеза в странах, богатых углем (Германия, Англия). Однако
получение карбида требует больших затрат электроэнергии,
вследствие чего карбидный ацетилен дорог.
В более новом способе ацетилен получают термическим кре~
кингом или электрокрекингом метана:
1500 «с
2СН4 >■ СНззСН + ЗН2
Из гомологов метана ацетилен образуется при более низкой
температуре:
1200 'С 1200 °С
С2н„ »- СН==СН + 2Н2 2С3Н3 *■ ЗСН=СН + 5Нг
88
Другой путь — окислительный пиролиз — сожжение метана с
недостаточным количеством воздуха. При этом идет частичное
окисление метана с образованием водорода и монооксида угле¬
рода:
1500 *с
6СН4 + О* 2СН==СН + 2СО + ЮН2
Водород и монооксид углерода используют для получения
спиртов. Сырьем для крекинга и пиролиза служит природный
газ или нефть. Получаемый таким образом ацетилен дешевле
карбидного, однако очистка его — сложная задача.
Ацетилен образуется при сухой перегонке многих органиче¬
ских веществ, всегда содержится в светильном газе каменно¬
угольного происхождения (коксовый газ). На основе ацетилена
развились многие отрасли промышленности органического син¬
теза.
Большое значение в развитии химии ацетилена имеют рабо¬
ты советских ученых. Выдающаяся роль принадлежит здесь
А. Е. Фаворскому (1860—1945 гг.) и его школе: разработаны
безртутный способ получения уксусного альдегида, способы по¬
лучения высокомолекулярных соединений — синтетического кау¬
чука, пластмасс, универсального клея (клей Назарова), лекар¬
ственных веществ (бальзам Шостаковского). Большой вклад в
химию ацетилена внесли также работы Н. Д. Зелинского и
Б. А. Казанского.
Большой интерес представляют реакции ацетилена и ацети¬
леновых углеводородов с кетонами. Например, реакцией ацети¬
лена с ацетоном можно получить изопрен, который является
исходным продуктом для получения синтетического каучука.
На схеме 2 (см. с. 88) приведены важнейшие промышлен¬
ные синтезы на основе ацетилена. Необходимо отметить, что
ряд химических продуктов, получаемых из ацетилена, можно
синтезировать также из этилена, причем часто этот путь более
экономичен.
ГЛАВА 6
ДИЕНОВЫЕ УГЛЕВОДОРОДЫ
Соединения, содержащие в углеродной цепи две двойные связи,
называют диеновыми. Углеводороды такого типа с открытой
цепью называют алкадиенами, а циклические — циклоалкадие¬
нами.
§ 40. Изомерия, номенклатура и классификация. Общая фор¬
мула алкадиенов СпНгл-г (как и ацетиленов). Следовательно,
соединения с двумя двойными связями изомерны соединениям
с одной тройной связью.
Изомерия диеновых соединений обусловлена строением угле¬
родного скелета и относительным расположением двойных свя¬
зей. В названиях положение каждой двойной связи обозначают
цифрами. Перед окончанием ен, символизирующим двойную
связь, ставят греческое числительное ди — так образуется окон¬
чание диен. Многие диены имеют и тривиальные названия. При¬
ведем ряд примеров (в скобках даны тривиальные названия);
СН2=С=СН—CHj
бутадиен-1,2
СНз
I
сн2=сн—сн2—сн2—сн=сн2 _ сн2=с—сн=сн2
гексадиен-1,5 (биаллил) 2-метилбутадиен-1,3
(изопрен)
Относительное расположение двойных связей в молекуле
вследствие их взаимного влияния сказывается на свойствах со¬
единений. Это влияние особенно сильно, когда двойные связи
находятся близко друг к другу. По взаимному расположению
двойных связей различают следующие диеновые углеводороды.
Диены с кумулированными двойными связями содержат не¬
посредственно примыкающие друг к другу двойные связи (по¬
ложения 1,2);
С=С=С СН2=С=СНЬ
кумулированные пропадиен
двойные связи (аллеи)
Диены с сопряженными двойными связями (положения 1,3):
с=с—С=.С СН2=СН—сн=сн2
сопряженные бутадиен-1,3
двойные связи (бивинил)
Диены с изолированными двойными связями — двойные свя¬
зи разделены одним или более атомами углерода:
С=С—(С„)—С=С СН2=СН—СН2—СН=СН2
изолированные пентадиен-1,4
двойные связи
Соединения, в молекулах которых двойные связи удалены
друг от друга, не отличаются по химическим свойствам от ал-
кенов. Реакции присоединения идут у них так же, как у алке-
нов, но в реакцию могут вступать две молекулы реагента.
нс_сн
нс\ /сн
сн2
циклопентадиен-1,3
90
Двойные связи в подобных
соединениях реагируют не¬
зависимо друг от друга, по¬
этому их и называют изоли-
Рис. 18. Строение бутадиена
рованными.
§ 41. Соединения с сопря- тиг
женными двойными связями.
Природа сопряженных двойных с в я з е й. Типичный
пример соединения с сопряженными двойными связями — бута¬
диен-1,3. В молекуле бутадиена все атомы углерода находятся
в состоянии з/Я-гибридизации, они лежат в одной плоскости.
Четыре параллельные друг другу орбитали л-электронов пер¬
пендикулярны плоскости, в которой лежат ядра атомов углеро¬
да (рис. 18). При взаимодействии подвижных л-электронных
облаков двух сопряженных связей возникает единое я-элек-
тронное облако, охватывающее все четыре атома углерода. От¬
дельные я-электроны не закреплены попарно в определенных
связях, а делокализованы, т. е. распределены по всей сопряжен¬
ной системе, по всем находящимся в сопряжении связям, и
простым, и кратным. Максимальная электронная плотность об¬
разуется между атомами углерода 1—2 и 3—4, меньшая — ме¬
жду атомами 2 и 3 (см. рис. 18).
Взаимное влияние сопряженных связей проявляется в неко¬
тором выравнивании межъядерных расстояний. Длина цент¬
ральной связи (между кратными связями) короче обычной
о-связи, а кратные связи несколько длиннее обычной двойной
связи (длина обычной связи С—С равна 0,154 нм, а обычной
двойной связи — 0,133 нм):
: ! 1 I
К—0,136нм —>| Ц—0,166нм—и
Таким образом, три связи, соединяющие четыре атома угле¬
рода, не являются ни истинно простыми, ни истинно двойными.
Сопряжение обусловливает некоторую двоесвязность атомов
углерода, формально соединенных ст-связью (нецелочисленная
кратная связь).
Более точно, чем обычной формулой, строение бутадиена
можно передать, изобразив пунктирными линиями распределе¬
ние я-электронов между всеми С—-С-связями (см. выше).
Содержание энергии в молекуле при образовании сопряжен¬
ных связей снижается, поэтому соединения с сопряженными
двойными связями — наиболее устойчивые из диенов.
X имичес к ие о с о бенно сти сопряженных ди¬
енов. В системе сопряженных связей два ненасыщенных цент¬
С
0,148нм—»-j
91
ра функционируют как единое целое. Реакции присоединения к
системе сопряженных двойных связей могут протекать двояко:
в концевые положения системы — положения 1,4 с образовани¬
ем новой двойной связи между атомами 2 и 3 или по месту
одной из двойных связей, в то время как другая связь остает¬
ся незатронутой. Первый тип называют 1,4-присоединением,
второй—1,2-присоединением. Так, например, при присоедине¬
нии хлора к бутадиену образуются два продукта:
—V С1СН2—СН=СН—СН2С1
1.4- дихлорбутен-2
.—► С1СН2—СНС1—сн=сн2
3.4- дихлорбутен-1
Рассмотрим ход присоединения к сопряженной системе на
примере взаимодействия бутадиена и хлороводорода. Как и все
обычные реакции присоединения к двойной углерод-углеродной
связи, процесс идет по электрофильному механизму и начинает¬
ся с присоединения протона за счет я-электронов двойной свя¬
зи (первая стадия). Присоединение идет по правилу Марков-
никова:
1 2 3 4 Н+ 1 2 3 4
СН2=СН—СН=СН2 > СН3—СН—СН=СН2
с 12
сн2=сн—сн=сн2
В образовавшемся карбокатионе положительный заряд не
сосредоточен на атоме С-2: ведь по соседству с положительным
зарядом находится двойная связь с ее подвижными я-электро-
нами. Притяжение атомом С-2 я-электронов приводит к их
перемещению в центр молекулы, а заряд переносится на атом
С-4, лишившийся электрона при перемещении двойной связи.
+
Таким образом возникает карбокатион СН3—СН=СН—СН2. Од¬
нако в нем взаимодействие между положительным зарядом и
я-электронами двойной связи должно способствовать обратно¬
му превращению в первоначальный катион с зарядом на атоме
С-2. Очевидно, наиболее устойчивым состоянием будет проме¬
жуточное между крайними состояниями, выраженными обеими
формулами. Это условно изображают, соединяя обе формулы
двусторонней стрелкой:
СНз—СН—СН=СН2 СНз—СН=СН—СН2
Структуры, истинное распределение электронов в которых
является промежуточным между описанным обычными форму¬
лами, называют мезомерными, а формулы, ограничивающие со¬
стояние мезомерии,— граничными. Для того чтобы яснее под¬
черкнуть промежуточный характер распределения электронов,
92
можно воспользоваться и иной формой записи!
1 2 3 4
сн3—сн—сн--=сн2
у *
+
В образовавшемся карбокатионе положительный заряд не
локализован на определенном атоме. Атомы С-2 и С-4 облада¬
ют дефицитом электронов, и хлорид-ион может атаковать лю¬
бой из них (вторая стадия):
СНз—сн-сн=сн2
+СГ
• СНз—СН=СН— СН2
|+сг
СНз—СНС1—СН=СН2 СНз—СН=СН—СН2С1
1,2-присоединение
(около 80%)
1,4-присоединение
(около 20%)
Аналогично идут реакции присоединения иных галогеново»
дородов, хлора и брома, водорода при каталитическом гидри¬
ровании. Конкретный результат (предпочтительность того или
иного направления) зависит от природы реагента и условий
реакции, например:
H,/Pd СНз—сн2—сн=сн2
сн2=сн-сн=сн2 —
C2H50H+Na
V
СНз—CH=CH—СНз
Циклический сопряженный диен — циклопеитадиен — присо¬
единяет бром только в положения 1,4:
НС—СН нс^сн
нс^ ^сн —»• вг—нс(^ ^;сн—Вг
сн2 сн2
Важной реакцией в синтетической химии является диеновый
синтез—1,4-присоединение алкена к сопряженным системам
(реакция Дильса — Альдера, 1928 г.). Диен взаимодействует с
соединением, содержащим активную двойную связь,— с диено-
филон:
хи2
Г СН2
сн2
100—120 ®С (давление) Ns>CH*
Vh,+J1h*
НС\ /С)Н2
СН2
циклогексен
Взаимодействие этилена с циклогексадиеном протекает лишь
при нагревании, под давлением. Но если диенофилом служит
не этилен, а соединение, которое имеет группы, активирующие
03
двойную связь (нитрильная, альдегидная, ангидридная и др.),
то реакция протекает быстро и в мягких условиях. Рассматри¬
ваемая реакция имеет значение не только для синтеза шести¬
членных циклов, но и для идентификации диеновых углеводо¬
родов.
Реактивом на сопряженную двойную связь служит малеино¬
вый ангидрид:
НС=СН
о=с^ \z=o
о
Соединения с сопряженными двойными связями склонны к
реакциям полимеризации. Примером может служить полимери¬
зация бутадиена путем 1,4-присоединения:
сн2=сн—сн=сн2 + сн2=сн—сн=сн2 + сн2=сн—сн=сн2 + • • • —►
—► —сн2—сн=сн—сн2—сн2—сн=сн—сн2—сн2—сн=сн-сн2— •
Реакции полимеризации диенов важны в практическом
отношении, с их помощью получают различные синтетические
каучуки. Более подробно о реакции полимеризации бутадиена
см. § 188.
§ 42. Наиболее важные представители. Бутадиен-t,3 — первый
и наиболее важный представитель гомологического ряда со¬
пряженных диенов. Это легко сжижающийся газ (т. кип.
—4,5°С) с характерным неприятным запахом.
В лаборатории бутадиен удобно получать по способу, раз¬
работанному Н. Д. Зелинским,— пропусканием паров циклогек¬
сена над раскаленной проволокой:
СН2
нс/ ^сн2
II I
НС\ ХСН2
сн2
500 °С
НС
I
НС
+ сн2=сн2
чсн2
Это по существу обратный процесс по сравнению с рассмот¬
ренным выше диеновым синтезом. Бутадиен образуется под
действием высоких температур из многих других углеводоро¬
дов. Поэтому он всегда содержится в продуктах, получающихся
при высокотемпературной переработке (пиролизе) нефти.
Бутадиен получают в больших количествах в промышлен¬
ности, так как он является сырьем для производства синтетиче¬
ского каучука. При этом используют следующие пути.
1. Из этилового спирта по С. В. Лебедеву (1874—1934 гг.).
Процесс состоит в одновременном дегидрировании, конденсации
и дегидратации спирта, осуществляемых на смешанном катали¬
заторе (оксид цинка и оксид алюминия с небольшими добав¬
ками других веществ):
ZnO, А|20з (400 «С)
2С2Н5ОН ► СН2=СН— СН=СН2 + 2НаО + Н2
94
По этому способу в начале 30-х годов в нашей стране было
организовано первое в мире крупное производство синтетиче¬
ского каучука. Так как в настоящее время спирт для техниче¬
ских целей получают из непищевого сырья, то промышленный
синтез бутадиена из спирта и сейчас не потерял своего значе¬
ния, несмотря на то, что разработаны другие способы, основан¬
ные на использовании продуктов переработки нефти или угля
(ацетилена).
2. Дегидрированием бутенов (600 °С, катализатор оксид
хрома на оксиде алюминия):
СН3—СН=СН—СНз —7+ СН2=СН—СН=СН2 — СНз-СН,-СН=СНа
“■*п —2Н
Сырьем служит бутан-бутеновая фракция крекинг-газов неф¬
ти, а также попутный нефтяной газ. Содержащийся в этом
сырье бутан также дегидрируется.
3. В промышленности используют и процесс окислительного
дегидрирования бутенов:
к-С4Нв + У202 —>- сн2=сн—сн=сн2 + н2о
Катализаторами служит смесь оксидов железа, висмута и
молибдена. Степень превращения достигает 95% при 90 %-й|
селективности.
4. Из ацетилена (несколькими путями), например через ацет¬
альдегид:
Ацетилен —»■ Ацетальдегид —► З-Гидроксибутаналь —>
—> Бутандиол-1,3 —► Бутадиен
Фракция Са пиролиза легкого бензина содержит до 40 %
бутадиена, который можно извлечь экстракцией специальными
растворителями, в частности диметилформамидом (см. § 115).
Бутадиен используют не только для получения синтетическо¬
го каучука, но и в других промышленных синтезах, например в
синтезе хлоропрена:
СН2=СС1—сн=сн2
| -НС1
—► С1СН2—СНС1—сн=сн2
С12 *
СН2=СН—СН=СН2 изомеризация
—С1СН2-СН=СН-СН2С1
2-Метилбутадиен-1,3 (изопрен)—жидкость, т. кип. 34 °С.
Сам изопрен в природе не встречается, но соединения, углерод¬
ный скелет которых построен из изопреновых звеньев, широко
распространены в растительном и животном мире. Их называют
95
изопреноидными соединениями. К соединениям такого типа от¬
носится натуральный каучук; он является природным полиме¬
ром, молекула которого построена из звеньев изопрена (см. § 188).
Важными изопреноидными соединениями являются терпены
(см. § 182).
Впервые изопрен в чистом виде был получен при нагрева¬
нии без доступа воздуха (сухая перегонка) натурального кау¬
чука. В настоящее время известно более 20 методов его синтеза,
но промышленное значение приобрели лишь немногие.
1. Дегидрирование изопентана (550 °С, пониженное давле¬
ние) :
СН3 сн3
I Сг2Оз/АЬОз |
сн3—сн—сн2—сн3 >- сн2=с—сн=сн2
Изопентан выделяют из нефтяных фракций, а также полу¬
чают каталитической изомеризацией нормального пентана.
2. Димеризация пропилена с последующей изомеризацией и
отщеплением метана:
СНз
(СзН,)3А1 | SIO2/AI2O3. 200 «С
2СН3—СН=СН2 ► СН2=С—СН2—СН2—СНз >
СНз СНз
I НВг, 750 °С |
—► СНз—С=СН—СН2—СНз »- СН2=С—СН=СН2 + СН4
Методы получения изопрена конденсацией ацетилена с аце¬
тоном и изобутена с формальдегидом рассмотрены в § 86.
Циклопентадиен-1,3 — жидкость, т. кип. 42,5 °С. Образуется .
при пиролизе углеводородов нефти, при коксовании каменного
угля. Синтезировать его можно из 1,2-дибромциклопентана от¬
щеплением бромоводорода под действием сильных оснований:
ВгНС СНВг
Н2С^ \н2
сн2
КОН (спирт, р-р)
-2НВг
НС—СН
Н</ \:Н
СН2
Характерным свойством циклопентадиена, отличающим его
от других соединений с двойными связями, является кислотный
характер атомов водорода группы СН2: эти атомы способны
замещаться на металл. При реакции циклопентадиена с порош¬
ком металлического натрия в кипящем ксилоле образуется
циклопентадиенилнатрий CsH5Na. Гидрированием циклопента¬
диена (он содержится в С5-фракциях пиролиза нефти) получа¬
ют циклопентен, используемый для получения высокомолеку¬
лярных соединений.
Циклопентадиен легко полимеризуется и обычно сохраняется
в виде более устойчивого димера. Реакция протекает по схеме
96
диенового синтеза:
НС
НС
^СН
)сн2 +
^СН
нсЛн
II >Н
HC-dH2
н
СН сн
та">
нс^Гс4
н
При нагревании димер циклопентадиена расщепляется на
две молекулы мономера.
Циклопентадиен используют для получения полимерных ма¬
териалов и малотоксичных антидетонаторов (см. § 139).
Аллен. В отличие от диенов, рассмотренных выше, аллен
СН2=С=СН2 относится не к сопряженным, а к кумулирован¬
ным диенам. Он образуется при пиролизе бензина. Может быть
использован в промышленности для получения высокомолеку¬
лярного соединения — полиаллена, имеющего стереорегулярную
структуру, а также для различных синтезов.
ГЛАВА 7
АРОМАТИЧЕСКИЕ УГЛЕВОДОРОДЫ
БЕНЗОЛ И ЕГО ПРОИЗВОДНЫЕ
В начале второй половины XIX века ароматическими называли
вещества, выделенные из природных душистых веществ — рас¬
тительных смол, бальзамов, ладана, эфирных масел. Эти соеди¬
нения противопоставляли алифатическим соединениям, к которым
относили, в частности, жиры. В настоящее время к арома¬
тическим относят вещества, в молекулах которых имеется устой¬
чивая циклическая группировка с особым характером связи.
Химия ароматических соединений — это прежде всего химия
бензола и его производных, а также производных нафталина,
антрацена и др. Особые химические свойства ароматических со¬
единений, а также их многочисленность и значение побудили
многих ученых к изучению этого типа соединений. Раскрытие
природы ароматичности сыграло важную роль в развитии тео¬
ретической и прикладной химии.
§ 43. Строение бензола. Природа ароматического состояния.
Строение бензола. Бензол СвНб — простейший предста¬
витель ароматических углеводородов — был открыт Фарадеем в
1825 г. в светильном газе. В 1845 г. Гофман выделил его из
каменноугольной смолы. Пожалуй, ни одно из соединений не
вызвало такого интереса исследователей, как бензол — это не¬
сложное по составу вещество.
Из многочисленных формул строения, предлагавшихся для
бензола в 60-х годах прошлого века, наиболее удачной оказа¬
4 Органическая химия
97
лась формула Кекуле 865 г.)', согласно которой бензол пред*
ставляет собой циклогексатриен-1,3,5 — шестичленный цикличе¬
ский углеводород с тремя сопряженными двойными связями:
нс_сн
нс/
НС=СН
Такая структура правильно отразила равноценность всех
шести атомов водорода бензола и некоторые другие особенности
этого вещества. Однако формула Кекуле не могла объяснить,
почему бензол не обладает свойствами ненасыщенного углеводо¬
рода: не обесцвечивает бромную воду и щелочной раствор пер¬
манганата калия. Вместо этого при действии брома протекает
реакция замещения водорода, характерная для насыщенных
соединений, и образуется бромбензол СбН5Вг; при действии
азотной кислоты водород замещается на нитрогруппу и образу¬
ется нитробензол C6H5N02.
Все же в определенных условиях возможны и реакции при¬
соединения. При каталитическом гидрировании молекула бен¬
зола присоединяет шесть атомов водорода, образуя циклогек¬
сан:
H2/Pt
СеНв СвН12
При сильном освещении бензол присоединяет шесть атомов
хлора, образуя гексахлорциклогексан С6Н6С16. Бензол реаги¬
рует и с озоном, давая неустойчивый триозонид.
Из приведенных примеров видно, что бензол может прояв¬
лять ненасыщенность, но в обычных условиях это свойство ос¬
лаблено.
Для бензола и его производных характерна термическая
устойчивость, легкость образования; ароматическое ядро оказы¬
вает специфическое влияние на свойства связанных с ним за¬
местителей.
Развитие электронных и квантово-механических представле¬
ний о химических связях позволило объяснить особенности аро¬
матических соединений.
Энергию молекулы бензола можно определить различными
способами. Все они показывают, что реальная молекула бензола
обладает меньшей энергией, чем если бы она представляла
собой циклогексатриен (формула Кекуле), где электроны жест¬
ко закреплены в двойных и простых связях. Гидрирование изо¬
лированной двойной связи алкена сопровождается выделением
энергии порядка 120 кДж/моль. При гидрировании гипотетиче¬
ского циклогексатриена-1,3,5 с его тремя двойными связями
следует ожидать выделения энергии «360 кДж/моль. В дейст¬
вительности же при гидрировании бензола выделяется
«210 кДж/моль. Разность («150 кДж/моль) составляет эне'рг
6
Рис. 19. Модель о- и я-связей в молекуле бензола:
а — связи и отдельные р-электронные облака; б — о-связн (светлые) и делокализован¬
ные я-электронные облака (темные)
гию сопряжения бензола. Она указывает на то, что бензол ус¬
тойчивее, чем гипотетический циклогексатриен-1,3,5.
Природа ароматического состояния. Бен¬
зол — наиболее яркий пример вещества, в котором осуществле¬
на делокализация электронов. В молекуле бензола все углерод-
углеродные связи выравнены, длины всех связей одинаковы
(0,139 нм). Это плоская молекула, где шесть атомов углерода
объединены в правильный шестиугольный цикл. Все атомы
углерода в бензольном цикле находятся в состоянии «^-гибри¬
дизации. Каждый из них образует три обычные о-связи (две
связи С—С и одну связь С—Н) с углом между ними 120°,
затрачивая на это три валентных электрона; четвертый элек¬
трон каждого атома углерода не закреплен. Орбитали всех
шести р-электронов перпендикулярны плоскости кольца и вза¬
имно параллельны (рис. 19,о). Каждый р-электрон данного
углеродного атома взаимодействует с р-электронами смежных
углеродных атомов. Шесть л-электронов не локализованы в
пары, как при образовании обычных двойных связей, а обра¬
зуют общую л-систему — осуществляется круговое сопряжение:
Рентгенографическим методом установлено, что электронная
плотность в бензольном цикле действительно распределена рав¬
номерно, т. е. ни простых, ни двойных связей в бензольном
ядре нет, все связи одинаковы. На рис. 19, б приведена модель
о- и я-связей бензола. Более темным цветом изображена
л-электронная система, образующая как бы внешнюю оболоч¬
ку шестичленного цикла; более светлым цветом переданы «р2-
Н
Н
4*
99
гибридные орбитали, при помощи которых осуществляются
о-связи с атомами углерода и водорода.
Обязательным условием полной делокализации я-электро-
нов, условием полноты сопряжения, является плоское строение
циклической молекулы и определенное число я-электронов: в
бензольном цикле оно равно шести (ароматический секстет,
ароматическая шестерка). Таким образом, молекула бензола —
устойчивый симметричный шестичленный цикл из одинаковых
СН-групп, лежащих в одной плоскости и связанных ароматиче¬
ской системой связей.
Истинное строение бензольного цикла является мезомерным,
промежуточным между изображаемым двумя граничными фор¬
мулами; для обозначения бензола можно использовать также
одну из формул (с кружками), указывающих на промежуточ¬
ный характер связей:
Однако чаще всего для условного изображения молекулы
бензола используют формулу Кекуле, помня о всех ее недо¬
статках.
Существует правило (Хюккель, 1931 г.), по которому арома¬
тической устойчивостью обладают симметричные циклические
полиеновые системы с числом я-электронов Ап + 2, где п = 1,
2, 3 и т. д. Следовательно, ароматичностью обладают систе¬
мы с 6, 10, 14 и т. д. я-электронами. Примером ароматических
систем с 10 и 14 я-электронами являются нафталин и антра¬
цен:
Эти соединения относятся к рассматриваемым далее много¬
ядерным ароматическим углеводородам (см. § 52, 53).
§ 44. Изомерия производных бензола. Номенклатура аромати¬
ческих углеводородов. Изомерия. Однозамещенные бензолы
изомеров не имеют, так как все атомы углерода в бензоле
равноценны. Двузамещенные бензолы существуют в виде трех
изомеров, отличающихся расположением заместителей относи¬
тельно друг друга. Положение заместителей указывают при по¬
мощи приставок орто- (о-), мета- (м-), пара- (п-) или обозна¬
чают цифрами:
ИЛИ
нафталин
антрацен
орто- (1,2-) мета- (1,3-)
пара- (1,4-)
100
По три изомера имеют трех- и четырехзамегценные бензолы
с одинаковыми заместителями:
XXX
1,2,3- (рядовой,
или смежный)
1,2,4- (несиммет¬
ричный)
1,3,5- (симмет¬
ричный)
Пяти- и шестизамещенные соединения с одинаковыми замес¬
тителями изомеров не имеют (по той же причине, что одноза-
мещенные).
В 60-х годах были получены валентные изомеры бензола. Такая изоме¬
ризация может происходить вследствие перераспределения связей в молекуле,
т. е. в результате перераспределения электронов. Так как молекула бензола
стабильна, то его изомеризация может произойти под влиянием большого
количества энергии.
При облучении ультрафиолетовым светом три-грет-бутилбензола I было
получено соединение II. Позднее был выделен бициклический углеводород
III — дьюаровский бензол. Это соединение получило такое название вслед¬
ствие сходства его формулы с формулой бензола, предложенной в 1865 г.
Дьюаром. Дьюаровский бензол отличается от бензола характером связей
между атомами углерода, а также химическими свойствами. При нагревании
до 90 “С он быстро (а при комнатной температуре постепенно) изомеризуется
в бензол.
С(сн3),
(СНз)зСчХ^С(СН^)з
и
Ф
Производные бензола, которые можно рассматривать как
продукты замещения атомов водорода бензола алкильными ра¬
дикалами, называют гомологами бензола. Общая формула та¬
ких углеводородов СпН2п-б. Углеводородными заместителями
могут быть также ненасыщенные радикалы (алкенильные, ал-
кинильные). В молекулах «бензоидных» углеводородов различа¬
ют две части: бензольное ядро и боковую цепь. Изомерия этих
соединений обусловлена строением радикалов, их составом и
расположением.
Номенклатура. Названия углеводородов ряда бензола
составляют из названий радикалов боковых цепей с добавлени¬
ем окончания бензол. Для многих из них употребительны три-
101
виальные названия (приведены в скобках):
СНз С2И, СНэ СНэ—сн-
СНэ—СН—СНз
СНз
СНз
3
метил* этил¬
бензол бензол
(толуол)
о (или 1,2)- изопропилбензол
диметилбен- (кумол)
зол (о-ксилол)
СН3—СН—СНЭ
п( или 1,4)-изо*
пропил-метил-
бензол (цимол)
Можно использовать и сокращенные записи формул. Напри¬
мер, 1,2-диметилбензол СбН4(СНз)2-1,2.
Примеры названий производных бензола с ненасыщенными
боковыми цепями:
Общее название ароматических углеводородов — арены. Об¬
щее название ароматических радикалов — арилы. Примеры на¬
званий радикалов:
СН3СбН4— толил (о-, л-, п-) |
Названия фенил, фенилен происходят от старинного назва¬
ния бензола — фен.
§ 45. Способы получения гомологов бензола. Ароматические
углеводороды можно синтезировать, исходя из ароматических и
неароматических соединений.
Из неароматических соединений арены полу¬
чают несколькими способами.
1. Каталитическое дегидрирование циклогексана и его про¬
изводных. Эта реакция открыта Н. Д. Зелинским в 1911 г,
(катализатор — палладиевая чернь, 300 °С):
В промышленности в качестве катализатора используют пла¬
тину на оксиде алюминия; температура около 450 °С. Эта ре¬
акция обратна реакции гидрирования бензола (см. § 47).
2. Каталитическая дегидроциклизация алканов — отщепле¬
нием водорода с одновременным замыканием цикла (Б. А. Ка¬
занский, А. Ф. Плата, Б. Л. Молдавский). Катализатором мо¬
винилЗензол
(стирол)
этинилбензол
(фенилацетилен)
СбН5— фенил
—С6Н4— фенилен (о-, м-, п-)
С6Н5СН2— бензил
СбН5СН— бензилиден
102
жет служить платинированный уголь при 300 °С, а также
оксиды хрома, молибдена, ванадия. Наиболее часто используют
оксид хрома на оксиде алюминия при 500 °С и 3 МПа:
Н2С_СН3
у \
н2с' хсн2-
н2с-сн3
гептан толуол
СН3 —► —СН3 + 4Н2
Аналогично из октана могут буть получены этилбензол, кси-
лолы.
Обе рассмотренные реакции в настоящее время лежат в ос¬
нове так называемой ароматизации нефти; они позволяют пере-
водить алкановые и циклоалкановые углеводороды нефти в аро¬
матические, что имеет большое практическое значение.
3. Полимеризация ацетилена и его гомологов (см. § 38).
Из ароматических соединений арены получают
следующими способами.
1. Алкилирование ароматических углеводородов галогенпро-
изводными и олефинами (реакция Фриделя— Крафтса; подроб¬
нее см.§ 47):
А1С1з (безв.) // \
+ С1-СН2-СН3 *■ Г у—СН2-СН3 + НС1
этилбензол
2. Восстановление ароматических кетонов:
О
Л I +4H<Zn+HCl) Л-\
f У—С—С2Н8 »- ^ у—CHj—С2Н,
пропилбензол
Исходные кетоны также получают по реакции Фриделя —
Крафтса, действуя на бензол хлорангидридом кислоты (реакция
ацилирования; см. § 47).
3. Синтез Вюрца — Фиттига (1864 г.)—обработка смеси
арилгалогенида и алкилгалогенида металлическим натрием:
С6Н5Вг + BrC2H6 + 2Na ——>■ С$Н$—С2Н6 -f- 2NaBr
Как установил П. П. Шорыгин (1881—1939 гг.), первона¬
чально образуется натрийорганическое соединение — фенилнат-
рий, который затем реагирует с этилбромидом:
2Na С2Н>Вг
c«HsBr ^ с‘НбМа свн6-с2н6
103
Таблица 8. Физические свойства некоторых аренов
Соединение
Т. пл., 'С
Т. кип., °С
Относительная
.20
ПЛОТНОСТЬ
Бензол
+5,5
80,1
0,879
Толуол
-95
110,5
0,866
Этилбензол
Ксилол (диметилбензол)
-94
136,2
0,866
О-
-25
144,4
0,896
-47
139,1
0,881
п-
+ 13
138,4
0,854
Пропилбензол
-99
159,2
0,861
Кумол (изопропилбензол)
-96
152,4
0,862
Стирол (винилбензол)
-31
145,0
0,906
Фенилацетилен
-45
142,0
0,930
4. Сплавление щелочных солей ароматических кислот со
щелочью или натронной известью:
—COONa + NaOH
<^У + NajCOj
соль бензойной
кислоты
5. Декарбоксилирование ненасыщенных ароматических
кислот:
/) 'Х нагревание // \
f У—СН=СН—СООН )- / у—сн=сн2 + со2
коричная кислота стирол
§ 46. Физические свойства. Бензол и его ближайшие гомологи —
бесцветные жидкости с характерным запахом, высшие гомо¬
логи — твердые вещества (табл. 8). Температуры кипения и
плавления зависят от состава и изомерии боковых цепей, от
расположения боковых цепей в цикле. Изомеры с разветвлен¬
ными боковыми цепями обычно кипят при более низких темпе¬
ратурах, чем с нормальными; ляра-изомеры имеют наиболее
высокие температуры плавления. Плотность аренов всегда мень¬
ше единицы. Они мало растворимы в воде, но во всех соотно¬
шениях смешиваются с органическими углеводородами. Жидкие
арены сами являются хорошими растворителями органических
веществ. Они легко воспламеняются и горят ярким, сильно коп¬
тящим пламенем. Пары и жидкости токсичны, некоторые веще¬
ства канцерогенны (являются возбудителями раковых заболе¬
ваний), при работе с ними требуется осторожность,
§ 47. Химические свойства бензола и его гомологов. Реакции
электрофильно го замещения в ароматическом
ряду. При явной ненасыщенности состава (нехватка восьми
атомов водорода по сравнению с гексаном) бензол проявляет
насыщенный характер: склонность к реакциям замещения и
104
устойчивость к действию окислителей. Реакции присоединения
затруднены, для их проведения необходимы особые условия.
Наиболее характерны для ароматических соединений реак¬
ции электрофильного замещения, хотя известны и реакции нук¬
леофильного замещения, и радикальные реакции.
Механизм реакций электрофильного замещения в аромати¬
ческом ряду имеет общие черты с электрофильным присоедине¬
нием к алкенам (см. § 33). Это также двухстадийный ионный
процесс. Ароматическое ядро, обладающее подвижными я-элек-
тронами, является удобным объектом для атаки электрофиль¬
ных реагентов. Процессу замещения предшествует распад мо¬
лекулы реагента XY с образованием электрофильной частицы
Х+ и аниона Y-:
XY =F=fc Х+ + Y”
Электрофильная частица взаимодействует с я-электронным
облаком ароматического ядра. Она притягивается отрицатель¬
ным зарядом ароматического секстета электронов, но пока не
соединена настоящей химической связью, а образует п-комп-
лекс. Затем два электрона из шести я электронов цикла лока¬
лизуются у одного атома углерода и участвуют в образовании
новой ковалентной с-связи с вступающим заместителем, а
остальные четыре электрона распределяются между пятью ато¬
мами углерода цикла. Образуется промежуточный карбокатион,
в котором нарушена ароматичность, так называемый о-комп-
лекс. При образовании такого комплекса один из шести атомов
углерода переходит из состояния sp7- в состояние зр3-гибриди-
зации. Конфигурация его становится тетраэдрической. Наруше¬
ние ароматического состояния невыгодно, поскольку ароматиче¬
ское ядро обладает особой устойчивостью. Поэтому происходит
быстрая потеря протона и ароматичность восстанавливается:-
0"х+н*
Отщепившийся протон образует с анионом Y- побочный про¬
дукт реакции:
Н+ + Y” —* HY
Основное отличие реакций электрофильного замещения в
ароматическом ряду от реакций электрофильного присоединения
в ряду алкенов заключается в быстром отщеплении протона.
Ароматичность воссоздается легче, чем осуществляется реакция
между промежуточным катионом и анионом Y- реагента. В слу¬
чае алкенов образующийся промежуточный катион дает с нук¬
леофильной частицей реагента (анионом) устойчивый продукт
присоединения.
7Г-комплекс <г- комплекс
105
Предпочтительное протекание реакций замещения у бензола
обусловлено тем, что эти реакции не требуют большой затраты
энергии. Присоединяться же к бензолу могут только реагенты,
богатые энергией: свободные атомы хлора, возникшие фото¬
химическим путем из молекул хлора при освещении; атомы водо¬
рода, активированные катализатором; озон.
Реакции электрофильного замещения в ароматическом ряду
имеют большое значение для синтезов, используемых в лабора¬
тории и в промышленности. Ниже приведены наиболее важные
из них.
1. Нитрование — введение нитрогрупы N02. Реакцию про¬
водят обычно смесью концентрированных азотной и серной кис¬
лот. Действующим агентом нитрования является образующийся
в этой смеси нитроний-катион +N02:
HN03 + 2H2S04 5f=fc N02 + 2HSOJ + [Н,0]+
нитро- гидрок-
ний- соний-
катион ион* *
Далее нитроний-катион взаимодействует с ароматическим
углеводородом, атакуя л-облако бензола, в результате чего
сначала образуется л-комплекс, а затем о-комплекс с кова¬
лентной связью между нитрогруппой и углеродным атомом бен-
зольного кольца. В последней стадии ион HS04 отрывает ион
водорода и образуется продукт замещения — нитросоединение:
Суммарную реакцию можно записать так:
АгН + НОЖ>2 —► ArN02 + H20
Присутствие в реакционной смеси воды мешает течению ре¬
акции, так как вода участвует в процессе, обратном образова¬
нию катиона нитрония. Для связывания выделяющейся в реак¬
ции воды берут избыток концентрированной серной кислоты.
2. Галогенирование в ядро проводят при помощи галогенов
в присутствии катализаторов. Чаще всего используют галогени¬
ды алюминия и железа: А1С13, А1Вг3, FeCl3, FeBr3 и т. д.
Катализаторы содействуют созданию активной электрофильной
частицы путем поляризации связи между атомами галогена.
S
* Ион гидроксония присутствует в водных растворах минеральных кислот.
Он образуется из протона Н+ и молекулы воды:
н2о + н+ |”h*oshJ+
106
Например, в безводном хлориде алюминия атом алюминия име-
ет лишь шесть электронов на внешней орбитали. Он дополняет
эту группировку до октета за счет свободной электронной пары
атома хлора; в результате оттягивания электронов второй атом
молекулы хлора приобретает положительный заряд и тем самым
повышенную электрофильность;
С1
! Cl—С1 ! + А1 S С1
м •• ••
С1
в+
С1
С1 -*■
Cl: А1 j С1
••
С1
Таким же образом действует и безводный хлорид желе-
за(Ш):
8+ 8-
+ С1-С1 FeCl3
Н
Сl
+ FeCl-4
d-комплекс с участием
катализатора
®-Cl + FeCl3 + НСl
хлорбензол
При действии избытка галогена могут быть получены ди-
и полигалогензамещенные, вплоть до полного замещения всех
атомов водорода ядра (С6С16 — гексахлорбензол, С6С15СН3 —
пентахлортолуол). Реакционная способность галогенов убывает
в ряду: F2 > С12 > Вr2 > I2.
Фтор слишком активен, поэтому фторпроизводные обычно
получают косвенным путем. Прямое иодирование не дает хоро¬
ших результатов — иод малоактивен, поэтому иодпроизводные
получают другим способом, через диазосоединения (см. § 132).
В отсутствие катализатора при нагревании или освещении
гомологов бензола идет радикальная реакция замещения в бо¬
ковой цепи. В зависимости от количества галогена можно полу¬
чить моно- и полигалогенпроизводные. Так, например, при хло¬
рировании толуола в этих условиях могут быть получены:
СН3 СН2С1 СНС12 СС13
бензил- бензилиден- бензотри-
хлорид дихлорид хлорид
При сильном освещении (ультрафиолетовый свет ртутной
лампы) в отсутствие кислорода бензол присоединяет хлор, и
образуется производное циклогексана — гексахлорциклогексан
107
(гексахлоран), который используют в качестве инсектицида:
СН
СНС1
с\нсу m:hci
ЗС12, УФ-облучение
н _ :н
>
С1НС\ /СНС1
СНС1
СН
Не все методы получения хлорпроизводных углеводородов
экономически выгодны — на производство хлора затрачивается
много энергии, а при обычном хлорировании углеводородов по¬
ловина хлора уходит в виде НС1, который пока используется
не полностью. Наиболее перспективные современные промыш¬
ленные способы получения хлорпроизводных рассмотрены в
§ 62.
Геакции галогенов с ароматическими углеводородами явля¬
ются примером того, как исходя из одних и тех же веществ,
можно получать различные продукты, создавая условия для
протекания реакций либо по электрофильному, либо по ради¬
кальному механизму.
3. Сульфирование — замещение водорода в бензольном ядре
сульфогруппой. Обычно реакцию осуществляют нагреванием
ароматического углеводорода с концентрированной серной кис¬
лотой или олеумом. В результате образуются ароматические
сульфокислоты:
Реакция сульфирования в отличие от реакций нитрования и
галогенирования обратима: при нагревании с перегретым водя¬
ным паром (180 °С) происходит гидролиз сульфокислот. Под¬
робнее о реакции сульфирования, ее механизме см. § 120.
4. Алкилирование — введение в ядро алкильной группы, в
результате чего образуются гомологи бензола. Алкилирование
осуществляется двумя путями.
а) Действие на бензол алкилгалогенидами в присутствии
катализаторов — безводных галогенидов алюминия (алкилиро¬
вание по Фриделю — Крафтсу, 1877 г.):
О
+ HOSOiH
О-
S03H + н20
бензолсульфо-
кислота
О
СНз— CHjCI, А1С13 (безв.)
Аналогично рассмотренному выше галогенированию роль ка¬
тализатора в реакции алкилирования также заключается в уве-
личении полярности реагента:
, Ь* ъ-
+ СН3—СН2—А1С14 -
'Н2—СНз
<0>-СН2-СН3 + А1С13 + НС1
+ А1С14
]
Реакция осложняется дальнейшим алкилированием, так как
гомологи бензола вступают в эту реакцию легче, чем бензол.
В этой области известны работы русского ученого Г. Г. Густав-
сона.
б) Алкилирование бензола алкенами в присутствии хлорида
алюминия или других катализаторов (трифторид, бора, фосфор¬
ная кислота). Эти реакции широко используют в промышлен¬
ности для получения этилбензола и изопропилбензола (кумола)
из бензола и газов крекинга:
+ СН2=СН„ -
этилен
+ СН»=СН—CHj
пропилен
СН2—СНЭ
этилбензол
СН,
Н—СН3
кумол
Механизм реакции сходен с предыдущими: протон кислоты с
молекулой алкена дает карбокатион, например (СН3)2С+Н, ко¬
торый реагирует с бензолом, как рассмотрено выше.
5. Ацилирование — введение в ядро ацильной группы
R—С=0. В результате подобных реакций образуются кетоны
(см. § 84).
Реакции присоединения к ароматическим
углеводородам. 1. Присоединение водорода. Каталитиче¬
ское гидрирование идет гладко в присутствии никелевых
(150 °С) и платиновых (50 °С) катализаторов. Бензол дает
при этом циклогексан, а производные бензола — производные
циклогексана. Это практически важная реакция, так как цикло¬
гексан— хороший растворитель, а также полупродукт для син¬
теза адипиновой кислоты и капролактама.
0 — 0
2. Присоединение хлора на свету (рассмотрено выше).
Окисление. Устойчивость бензольного ядра к окислению
является одним из важнейших свойств ароматических соединений.
109
Такие окислители, как азотная кислота, хромовая смесь, рас¬
твор перманганата калия, пероксид водорода, при обычных
условиях на бензол не действуют. Это отличает ароматические
соединения от соединений с двойными связями. К окислителям
бензол даже более устойчив, чем алканы.
При действии кислорода воздуха на бензол в присутствии
катализатора, оксида ванадия(V), при высокой температуре
(400 °С) получается малеиновый ангидрид (промышленный
способ):
HCUCH
О -21* о=с\ /°-°
о
При действии окислителей на гомологи бензола окислению
подвергаются боковые цепи — алкильные радикалы. Какой бы
сложной ни была цепь, она под действием сильных окислителей
разрушается («сгорает»), за исключением ближайшего к ядру
атома углерода, который окисляется в карбоксильную группу.
Гомологи бензола с одной боковой цепью дают однооснов¬
ную кислоту — бензойную:
а—-
толуол бензойная кислота кумол
СООН
[О]
О
СН(СН3)2
Гомологи, содержащие две боковые цепи, дают двухосное-
пые кислоты:
СН3
I
СООН
1
<*
X
о-
„ Л-
- ril
и
1
у
у
СН3
1
СООН
1
СН3—СН—СНз
«-ксилол
терефтале-
вая кислота
«-изопропилметил-
бензол (цимол)
По образующимся в результате окисления ароматическим
кислотам можно судить о положении боковых цепей и их числе.
§ 48. Ориентация при электрофильном замещении в бензольном
ядре. Выяснение закономерностей при реакциях замещения в
бензольном ядре явилось предметом многих исследований.
Классические исследования в этой области относятся еще к
прошлому веку и связаны с именем немецкого ученого Голле-
мана.
Как уже было отмечено (см. § 44), вследствие равноценно¬
сти всех атомов углерода в бензоле однозамещенные бензолы
изомеров не имеют. При вступлении в молекулу второго замес¬
110
тителя могут образоваться три изомера, различающиеся взаим-
ным положением заместителей,— орто-, мета- или пара-mo-
меры.
Место вступления нового электрофильного заместителя опре¬
деляется природой уже имеющегося в ароматическом ядре за¬
местителя. Иными словами, стоящий в ароматическом ядре
заместитель оказывает при дальнейшем замещении определен'
ное направляющее (ориентирующее) действие. Все заместители
по их ориентирующему действию при реакциях электрофильного
замещения в бензольном ядре можно разделить на две группы.
Заместители первого рода направляют преимущественно з
орто- и пара-положения — это орто-пара-ориентанты. К ним от¬
носятся алкильные радикалы, группы ОН, NH2, NHR, NR2, га¬
логены и др.
Заместители второго рода направляют преимущественно в
лета-положение — это мета-ориентанты. К ним относятся груп¬
пы CF3, N02, S03H, СООН, CN и др.
В табл. 9 приведены результаты нитрования монозамещенных
бензолов. Видно, что не все заместители действуют строго из¬
бирательно. Например, при наличии группы СНС12 образуется
смесь, содержащая значительные количества всех трех изоме¬
ров. Соотношение образующихся изомеров зависит также от при¬
роды реагента, его концентрации, типа растворителя, катализа¬
торов, температуры.
Заместители первого и второго родов по-разному влияют на
реакционную способность бензольного ядра. Заместители пер¬
вого рода (за исключением галогенов) активируют ядро, облег¬
чают введение нового заместителя, вследствие чего реакции
электрофильного замещения протекают значительно легче, чем
для самого бензола. Заместители второго рода затрудняют
дальнейшее электрофильное замещение.
Для истолкования ориентирующего действия заместителей
предлагалось множество теорий. В настоящее время этот во¬
прос полностью выяснен на основе электронных представлений.
орто-пара-0риентантами являются электронодонорные группы,
Таблица 9, Ориентирующее влияние различных групп в реакции
нитрования монозамещенных бензолов
Замести¬
тель
в ядре
Содержание образовавшегося
изомера, %
Замести¬
тель
| в ядре
Содержание образовавшегося
изомера, %
орто-
мета-
пара-
орто-
мета•
пара•
он
40
__
60
CF3
100
СН3
56
4,0
40
no2
6,5
93,2
03
С(СНэ)э
12
8
80
CN
17
81
2
F
12
—
88
СООН
18,5
80
1,5
С1
30
1
69
CHCl,
23
35
42
Вг
36,5
1
62,5
Ш
подающие в бензольное ядро электроны. Увеличивая электрон¬
ную плотность в бензольном ядре, они тем самым делают его
более активным в реакциях электрофильного замещения (иск¬
лючение составляют галогены, см. § 30). мета-0риентантами
являются электроноакцепторные группы, оттягивающие электро¬
ны из ядра. Уменьшая электронную плотность в бензольном
ядре, они уменьшают его активность в реакциях электрофиль¬
ного замещения.
Изменения электронной плотности происходят главным об¬
разом в орто- и пара-положениях по отношению к ориентирую¬
щему заместителю. Под влиянием заместителей первого рода
электронная плотность в орто- и пара-положениях повышается,
поэтому туда преимущественно и направляется вступающая
электрофильная группа. Так, например, при нитровании толу¬
ола образуется главным образом смесь орто- и пара-изомеров
(см. табл. 9):
Если в ядре стоит заместитель второго рода, то он оттяги¬
вает электронную плотность в первую очередь из орто- -и пара¬
положений, уменьшая тем самым их активность. Вступающий
заместитель направляется в лета-положение, из которого «от¬
ток» электронной плотности меньше, т. е. электронная плот¬
ность здесь несколько выше по сравнению с орто- и пара-поло¬
жениями, что и обусловливает большую реакционную спо¬
собность этого положения. Например, при введении второй
нитрогруппы в нитробензол образуется в основном мега-изомер
(см. табл. 9):
Чтобы понять природу ориентирующего влияния, необходи¬
мо глубоко знать свойства функциональных групп, типы их
взаимного влияния (индуктивный и мезомерный эффекты)-, бо¬
лее подробно этот вопрос рассмотрен позднее (см. § 130).
§ 49. Ароматические углеводороды в промышленности. Главные
сырьевые источники ароматических соединений — каменный
уголь и нефть.
Решениями XXVII съезда КПСС предусматривается довести
добычу угля в 1990 г. до 780—800 млн. т в год, причем почти
половина угля будет добываться открытым способом. Намечено
ускорить развитие ряда угольных бассейнов Сибири и Дальнего
Востока, продолжить техническое перевооружение самого ста¬
рого района добычи угля в нашей стране — Донбасса.
сн.
t ^СНз
СН.
112
При коксовании каменного угля при 1000—1200 °С образу¬
ются кокс (75% от массы угля), коксовый газ (300 м3 на 1 т
угля), каменноугольная смола (2—5 % от массы угля) и амми¬
ачная вода. Из продуктов коксования угля получают большое
число ароматических соединений. Коксовый газ содержит около
25—35 г/м3 ароматических углеводородов. После их отделения
промывкой на холоду тяжелыми маслами коксовый газ исполь¬
зуют в качестве топлива. В состав газа входят метан, водород,
монооксид углерода, этилен, ацетилен, азот, синильная кислота,
диоксид углерода и др.
Каменноугольная смола — черно-коричневое масло, тяжелее
воды, с характерным запахом. Состав смолы зависит от темпе¬
ратуры коксования угля. При температуре до 500 °С образует¬
ся первичная (низкотемпературная) смола, содержащая много
алканов и циклоалканов. Смола, полученная при высоких тем¬
пературах (1000°С и выше), содержит в основном ароматиче¬
ские соединения (до 300 различных веществ). Хотя вывод смолы
составляет всего 2—5 % от массы угля, общее количество полу¬
чаемой смолы велико, так как коксованию подвергают сотни
миллионов тонн угля. Каменноугольную смолу перегоняют,
обычно выделяя следующие фракции.
1. Легкое масло (около 2 % от общей массы смолы) отгоня¬
ется до 170 °С, содержит бензол, толуол, ксилолы, стирол,
этилбензол, пиридин и др.
2. Среднее масло (до 12 % от общей массы смолы) отгоня¬
ется в интервале 170—240 °С, содержит нафталин, фенол,
крезолы.
3. Тяжелое масло (до 10 % от общей массы смолы) отгоня¬
ется в интервале 240—270 °С, содержит нафталин и его произ¬
водные.
4. Антраценовое масло (до 25 % от общей массы смолы)
отгоняется в интервале 270—360 °С, содержит антрацен, фенан-
трен и др.
5. Пек (50—60 % от общей массы смолы) — твердый смоли¬
стый остаток после перегонки. Применяется для изготовления
строительных (кровельных) материалов, брикетов топлива и др.
Индивидуальные соединения выделяют из фракций путем
повторной перегонки и кристаллизации твердых продуктов. Ве¬
щества с кислотными свойствами (фенолы) выделяют из смесей
обработкой водными растворами щелочей, -а вещества с основ¬
ными свойствами (пиридин) — извлечением разбавленной сер¬
ной кислотой. Из 1 т каменноугольной смолы получается около
16 кг бензола, 2,5 кг толуола, 0,3 кг ксилолов, 40—60 кг
нафталина, 5—20 кг антрацена, 20 кг веществ фенольного ха¬
рактера.
Коксохимическая промышленность — важный сырьевой ис¬
точник для производства продуктов органического синтеза. Од¬
нако вследствие быстрого роста химической промышленности
ИЗ
коксохимия не может удовлетворить потребность в ароматиче¬
ском сырье. Кроме того, продукты коксохимического происхож¬
дения трудно получать достаточно чистыми.
Основным поставщиком ароматического сырья является неф¬
теперерабатывающая промышленность. Нефть некоторых место¬
рождений содержит значительное количество ароматических
углеводородов, но основное количество ароматических соедине¬
ний образуется ори ароматизации нефти, т. е. при каталитиче¬
ской дегидроциклизации алканов и каталитическом дегидриро¬
вании циклоалканов (см. § 45). В промышленности этот про¬
цесс называют каталитическим риформингом. В качестве ката¬
лизатора используют платину на оксиде алюминия (500 °С,
3—4 МПа). Сырьем служит нефтяная фракция, выкипающая
до 180°С. В продуктах риформинга соотношение бензола, то¬
луола и ксилолов составляет 1:4:5. Более подробно нефть и
продукты ее переработки рассмотрены в гл. 8.
Ароматические углеводороды — важное сырье для изготовле¬
ния разнообразных синтетических материалов, полупродуктов и
красителей, физиологически активных веществ и др. В промыш¬
ленном отношении наиболее важен бензол: по значимости он
уступает лишь этилену. В начале 80-х годов в нашей стране
40 % бензола получали из продуктов сухой перегонки угля,
24 % — из продуктов каталитического риформинга, 21 % —де¬
алкилированием толуола, 17%—при пиролизе нефтяных
фракций.
Некоторые примеры использования бензола и его гомологов
приведены на схеме 3 (в скобках вслед за названиями важней¬
ших продуктов указана доля бензола, расходуемого для данно¬
го синтеза).
Важными гомологами бензола являются следующие соеди¬
нения.
Толуол в большом количестве используют как добавку к
моторному топливу, как растворитель и для синтезов взрывча¬
тых веществ, полупродуктов анилинокрасочной и фармацевти¬
ческой промышленности. Больше половины производимого толу¬
ола переводят в бензол термическим деалкилированием в при¬
сутствии катализатора:
Кумол (изопропилбензол) используют в промышленности
для синтеза фенола и ацетона (см. § 72).
о-Ксилол используют для получения фталевого ангидрида
(см. § 107) —очень важного исходного вещества для производ¬
ства полиэфиров и синтетических смол.
п-Ксилол применяют для получения терефталевой кислоты
СеН4(СООН) 2-1,4, являющейся важным сырьем для изготовле¬
ния синтетического волокна лавсан.
114
Схема 3. Техническое использование бензола
СН—СО
о-
бензол
^12
н2
малеиновый
ангидрид (1 %)
'О-
циклоге¬
ксан (1 %)
Cl*
НЫОз
-» С*НвС1»
гексахлоран
-» С*Н5—С1 -
хлорбензол
С*Н6—N0»
нитробензол
HN—(СН2)6—i*o
капролактам (14 %)
НООС—(СН*)Ч—СООН
адипиновая кислота
С6Н$-ОН
фенол
► С«Н5—NH*
анилин
сн*=>»сн2
*■ CeHs—CHj—CHj
этилбензол (48%)
СНзСН=СН2
СвН8—СН(СН3)г —
кумол
С*Н*~СН=СН»
стирол
> С*Н5—ОН
фенол (28 %)
► CHj—СО—СНЭ
ацетон
Стирол (винилбензол, фенилэтилен) — жидкость с приятным
запахом. Впервые выделен из смолы «стиракс». Содержится в
погоне каменноугольной смолы — легком масле. В промышлен¬
ности его получают дегидрированием этилбензола. Интересен
способ получения стирола окислительной конденсацией бензола
и этилена в присутствии ацетата палладия:
Q+сц-сн. ^-сн-сн,
Химические свойства стирола определяются наличием бен¬
зольного ядра и двойной связи в боковой цепи. Характерной
особенностью стирола является его большая склонность к поли¬
меризации (образование полистирола).
115
МНОГОЯДЕРНЫЕ АРОМАТИЧЕСКИЕ УГЛЕВОДОРОДЫ
Многоядерными ароматическими соединениями называют веще¬
ства, содержащие в молекуле два или несколько бензольных
ядер, связанных между собой углерод-углеродной связью. Раз¬
личают соединения с изолированными бензольными ядрами, в
которых бензольные ядра соединены непосредственно (напри¬
мер, бифенил) или алифатической цепью (например, дифенил-
метан), и соединения с конденсированными бензольными ядра¬
ми. В конденсированных системах, например в нафталине и
антрацене, бензольные циклы имеют общие орго-углеродные
атомы (см. § 52).
бифенил дифенилметан
§ 50. Соединения с изолированными бензольными ядрами. Про¬
стейшим и наиболее важным представителем соединений с
изолированными бензольными циклами является бифенил. Это
кристаллическое вещество (т. пл. 70 °С, т. кип. 254 °С), рас¬
творяющееся в спирте, эфире и других органических раствори¬
телях. Небольшое количество бифенила содержится в каменно¬
угольной смоле. В промышленности бифенил получают пропус¬
канием паров бензола через расплавленный свинец:
600-700 'С
2СвНа >- С6Н6—С6Н5 + Н2
В лаборатории бифенил получают по реакции Вюрца —
Фиттига действием металлического натрия на бромбензол или
при нагревании иодбензола в присутствии мелкораздробленной
меди:
2Na 2Cu
2С0Н6Вг
—2NaBr
С6Н5-СвН6 2С6Н51
Бифенил — вещество ароматического характера. В реакциях
электрофильного замещения из него образуются главным обра¬
зом /гара-изомеры и небольшие количества орго-изомеров; сле¬
довательно, фенильный радикал проявляет свойства орто-пара-
ориентанта:
HNOa
NO*
Бифенил термостоек, поэтому его применяют в химической
промышленности в качестве теплоносителя для обогревания раз¬
личных аппаратов.
Трифенилметан представляет собой кристаллическое веще¬
ство с т. пл. 92,5 °С. Наиболее удобно синтезировать его по
116
реакции Фриделя — Крафтса:
Трифенилметан обладает значительной реакционной способ¬
ностью из-за особой подвижности атома водорода при третич¬
ном атоме углерода:
С12 ГО!
(СвН5)3С-С1 ч (СвН6)3СН >- (С8Н5)зС-ОН
трифенилхлор- трифенилмета-
метан нол
Амино- и гидроксипроизводные трифенилметана являются
красителями.
§51. Свободные радикалы. Влияние фенильных групп на тре¬
тичный углеродный атом в молекуле трифенилметана проявля¬
ется в способности этого соединения образовывать свободный
радикал трифенилметил.
Открытие трифенилметила связано с работами Гомберга
(1900 г.) по синтезу полифенильных производных этана. Так,
при действии металлического серебра на трифенилбромметан в
отсутствие кислорода Гомберг рассчитывал получить гексафе-
нилэтан:
2(С,Н5)3СВг + 2Ag —(CeH8)3C-C(C,H5), + 2AgBr
гексафенилэтан
Однако у полученного соединения обнаружились некоторые
неожиданные свойства. При нагревании его бесцветного раство¬
ра в бензоле появлялось желтое окрашивание, исчезавшее при
охлаждении. Как выяснилось, в действительности образовался
свободный радикал трифенилметил, находящийся в равновесии
со своим димером. Этот димер первоначально считали гексафе-
нилэтаном; теперь установлено, что он имеет более сложное
строение:
трифенилмешл димер трифенилметила
117
Повышение температуры способствует диссоциации димера,
т. е. увеличению содержания трифенилметила, имеющего жел¬
тую окраску. С кислородом воздуха трифенилметил мгновенно
реагирует, образуя бесцветный бис (трифенилметил) пероксид
(СбНе)зС—О—О—С(СбН5)3. Реакцию с кислородом исполь¬
зуют для открытия и количественного определения триарильных
радикалов.
Позднее Панету (1929 г.) при термическом разложении ал¬
кильных соединений свинца в газовой фазе удалось получить
короткоживущие радикалы — метил, этил и др.:
РЬ(СН3)4 —> РЬ + 4Н3С •
Для этого он пропускал через кварцевую трубку ток водо¬
рода, увлекавший с собой пары тетраалкилсвинца. В опреде¬
ленном месте трубку сильно нагревали, и здесь происходило
разложение свинецорганического соединения, о чем свидетель¬
ствовало образование на стенке трубки налета металлического
свинца (свинцовое зеркало) по приведенной выше реакции. До¬
казательством наличия свободных радикалов в продуктах раз¬
ложения служило исчезновение другого зеркала, заранее нане¬
сенного на стенку трубки на некотором удалении от места
нагревания. Свободные радикалы, реагируя с металлом зерка¬
ла, давали летучие соединения (R2Hg, R3Sb, R2Zn и др.), кото¬
рые уносились током водорода; после улавливания в охлаждае¬
мой ловушке их анализировали.
Короткоживущие свободные радикалы (полупериод сущест¬
вования Н3С«0,005 с) могут образовываться в качестве про¬
межуточных продуктов реакций и благодаря высокой реакцион¬
ной способности влиять на течение реакции. Многие практиче¬
ски важные реакции идут с участием свободных радикалов,
например реакции галогенирования алканов, термического раз¬
ложения углеводородов, реакции окисления, полимеризации
и др.
Свободный радикал — это нейтральная частица с нечетным
числом электронов. Причиной неустойчивости свободных ради¬
калов является наличие неспаренного электрона. Сравнительная
устойчивость трифенилметильного радикала (СеН5)зС« обус¬
ловлена тем, что неспаренный электрон не локализован у цент¬
рального атома углерода, его плотность в результате сопряже¬
ния распределена между центральным атомом углерода и всеми
атомами углерода трех бензольных ядер (т. е. между 19 атома¬
ми углерода). Свободный радикал может стать устойчивым и
в том случае, если молекулам реагентов затруднен доступ к
тому участку, где локализован неспаренный электрон («экрани¬
рование»). Если оба стабилизирующих фактора — сопряжение и
экранирование — действуют одновременно, то свободные ра¬
дикалы могут стать очень устойчивыми.
118
Советским ученым (Институт химической физики АН СССР) удалось
синтезировать и выделить в кристаллическом виде «сверхстабильные» ради*
калы, например иминоксильные (радикалы Розанцева):
В иминоксильных радикалах неспаренный электрон «размазан» по связи
N—О, что приводит к выигрышу энергии; кроме того, объемистые соседние
группы затрудняют доступ к участку, на котором локализован неспаренный
электрон. Иминоксильные радикалы вступают в обычные химические реакции,
протекающие и без затрагивания неспаренного электрона. Эти радикалы ока*
зались сильными ингибиторами реакций окисления и полимеризации. Они
обладают рядом интересных свойств и находят разнообразное применение.
Известен также бирадикал карбен • СН2 — важная промежуточная ча*
стица в ряде реакций органического синтеза.
§ 52. Соединения с конденсированными бензольными ядрами.
Нафталин. Наиболее важные представители многоядерных аро¬
матических соединений такого типа — нафталин, антрацен, фе-
нантрен.
Нафталин СюН8 открыт в 1819 г. в каменноугольной смоле.
Состав его установлен А. А. Воскресенским (1858 г.). Это бес¬
цветное кристаллическое вещество (т. пл. 80 °С, т. кип. 218 °С)
с характерным запахом, легко возгоняется. Он испаряется даже
при обычной температуре, о чем свидетельствует характерный
запах нафталина. Нерастворим в воде, легко растворим в орга¬
нических растворителях.
Углеродный скелет молекулы нафталина состоит из двух
бензольных ядер, сконденсированных при помощи двух общих
соседних углеродных атомов. В молекуле нафталина, так же
как и в молекуле бензола, нет ни двойных, ни простых связей,-
хотя в приведенных ниже формулах условно показаны пять
двойных связей, чередующихся с простыми. Ароматичность
здесь обусловлена сопряжением 10 я-электронов (см. § 43,
правило Хюккеля).
Рентгенографические исследования кристаллов нафталина
показали, что молекула нафталина имеет плоское строение,
длина всех углерод-углеродных связей занимает промежуточное
положение между длинами простых и двойных связей. В моле¬
куле нафталина не все атомы углерода равноценны. Различают
а-углеродные (положения 1, 4, 5, 8) и p-углеродные (положе¬
ния 2, 3, 6, 7) атомы.
(СНз)зС—N—С(СНа)}
а
СН СН
а
119
Число изомеров у производных нафталина больше, чем у
производных бензола. У однозамещенных нафталинов известны
Два изомера — а- и (3-изомеры. Дизамещенные нафталины при
одинаковых заместителях имеют 10 изомеров (1,2-, 1,3-, 1,4-,
1,5-, 1,6-, 1,7-, 1,8-, 2,3-, 2,6-, 2,7-), а при разных заместителях —
14 изомеров. Положения заместителей в молекуле нафталина
обычно обозначают цифрами. Положения 1,8 часто называют
пери (греч.— близкий), 2,6 — амфи (греч.— с двух сторон).
По химическим свойствам нафталин является ароматическим
углеводородом и напоминает бензол. Однако он проявляет бо¬
лее ненасыщенный характер: легче, чем бензол, вступает в ре¬
акции присоединения. Реакции замещения идут легче в а-поло-
жениях, чем в p-положениях. В некоторых случаях тип ориен¬
тации может быть изменен условиями реакций.
Реакции замещения. 1. Сульфирование — одна из
характерных реакций замещения для нафталина, причем в зави¬
симости от условий реакции может быть получена как а-наф*
талинсульфокислота, так и р-нафталинсульфокислота:
S03H
При более низкой температуре образуется а-нафталинсуль-
фокислота. Так как a-положения нафталина более реакционно-
способны, то и скорость сульфирования в a-положения гораздо
больше, чем скорость сульфирования в p-положения. Однако
устойчивость р-нафталинсульфокислоты выше, чем устойчи¬
вость о-изомера; поэтому при повышении температуры а-наф-
талинсульфокислота переходит в р-изомер.
2. При галогенировании нафталина получаются а-галоген-
производные. Так, при бромировании получают а-бромнаф-
талин:
Вг
Среди хлорпроизводных нафталина сейчас привлекают вни¬
мание полихлорпроизводные, вплоть до полностью замещенного
перхлорнафталина:
120
Это воскоподобное вещество под названием галовакс исполь-
зуют как диэлектрик.
3. Нитрование приводит к а-нитронафталинуг
NOj
4. При ацетилировании в зависимости от условий ’(темпера*
тура, растворитель) получается а-ацетилнафталин или р-аце-
тилнафталин:
По сравнению с бензолом правила ориентации в молекуле
нафталина усложнены. Если в молекуле нафталина в a-поло¬
жении имеется ориентант первого рода (например, группа ОН),
то заместитель вступает в то же ядро в положение 4 или 2}
если такой же ориентант находится в p-положении, то замес¬
титель вступает в ближайшее а-положение:
ОН ОН он
а-нафтол 1-гидрокси- 1-гидрокси-2-нитро-
(нафтол-1) 4-нитро- нафталин
нафталин
S03H
[3-нафтол 2-гидроксинафта-
(нафтол-2) лин-1-сульфокислота
При наличии в молекуле нафталина ориентанта второго рода
(например, группы S03H) в а- или p-положениях новый
121
заместитель вступает в другое ядро, преимущественно в а-поло'
Жение:
SOjH
hno3
OsN S03H
+
no2
S03H
1 (или а)-нафта- 8-нитронафталин- 5-нитронафта*
линсульфокислота 1-сульфокислота лин-1-сульфо-
кислота
При нитровании р-нафталинсульфокислоты нитрогруппа
также вступает в положение 5 или 8.
Такая ориентация становится понятной, если вспомнить, что
ориентанты первого рода облегчают дальнейшее электрофиль¬
ное замещение, а ориентанты второго рода — затрудняют его.
Поэтому в последнем случае более выгодным становится вступ¬
ление заместителя в другое ядро.
Реакции присоединения. Примером является реак¬
ция гидрирования нафталина. Нафталин легче, чем бензол,
вступает в реакцию с водородом, причем сначала гидрируется
одно ядро (160°С, 3 МПа) и образуется тетрагидронафталин
(тетралин); конечный же продукт гидрирования (200°С,
20 МПа) — декагидронафталин (декалин):
2Н2 fNI) ЗН2 (N1)
—*оо—
Тетралин и декалин — высококипящие жидкости (т. кип. 207
и 190 °С соответственно); применяют в качестве растворителей
смол, жиров, как добавки к моторному топливу.
Окисление. Нафталин окисляется легче, чем бензол, при
этом одно из бензольных ядер разрушается, а его а-углерод-
ные атомы превращаются в карбоксильные группы; образуется
двухосновная фталевая кислота:
.соон
соон
В промышленности реакцию проводят, окисляя нафталин
кислородом воздуха в присутствии оксида ванадия (V) при
450 °С. Практически получается фталевый ангидрид, так как,
фталевая кислота в условиях реакции теряет воду:
122
Фталевый ангидрид очень широко используют как полупро¬
дукт в разных синтезах. К продуктам окисления нафталина от¬
носятся также дикетоны — нафтохиноны (см. § 91).
Основными источниками нафталина являются каменноуголь¬
ная смола и нефтяное сырье. В продуктах каталитического ри¬
форминга нефти содержится значительное количество метилнаф-
талинов, которые каталитическим гидродеалкилированием
(750 °С) превращают в нафталин:
СНз
Н2 (Со, Мо)
+ СН4
Как уже отмечалось, нафталин используют для получения
фталевого ангидрида и фталевой кислоты, гидроксинафталин*
сульфокислот и других замещенных.
§ 53. Антрацен и фенантрен. Антрацен СиНю содержится в
большом количестве в каменноугольной смоле, в антраценовом
масле, из которого его выделяют путем кристаллизации и очи¬
щают возгонкой. Чистый антрацен — бесцветное кристалличе¬
ское вещество с голубой флуоресценцией, т. пл. 217 °С, т. кип.
354 °С, нерастворим в воде, хорошо растворим в горячем бен¬
золе.
Антрацен представляет собой многоядерный ароматический
углеводород с тремя линейно конденсированными бензольными
ядрами плоского строения:
а а
СН СН СН
Р НС^ ^СН р
I II I I
р НС^ ^сн р
СН СН СН
а а
В молекуле антрацена, как и в молекуле нафталина, не все
атомы углерода занимают одинаковое положение по отношению
к общим атомам углерода. Различают положения: а- (1-, 4-,
5-, 8 ), р- (2-, 3-, 6-, 7-) и мезо- (9-, 10 ). Таким образом, одно-
замещенные антрацены могут иметь три изомера: а-, р- и мезо-
изомеры. В антрацене нет полной выравненности связей — наи¬
более короткие связи а—р.
По химическим свойствам антрацен сходен с нафталином и
бензолом, но активнее их. Он проявляет ароматический харак¬
тер в реакциях замещения (галогенируется, нитруется, сульфи¬
руется), но легко вступает также в реакции присоединения.
Наибольшей реакционной способностью отличаются положения
9 и 10 (мезо-), поэтому и реакции замещения, и реакции при¬
соединения легче всего идут в среднем ядре. В результате при¬
123
соединения в положения 9 и 10 оба боковых ядра приобретают
ароматические секстеты и состояние их делается устойчивым.
Из реакций замещения рассмотрим следующие.
1. Галогенирование. Хлор при 0°С сначала присоединяется
в положения 9 и 10, образуя дихлорид, от которого при слабом
нагревании легко отщепляется галогеноводород и получается
продукт замещения — 9-хлорантрацен:
Аналогично идет реакция с бромом.
2. Нитрование. При действии азотной кислоты сначала об¬
разуется неустойчивый продукт присоединения, а после отщеп¬
ления от него воды — продукт замещения — 9-нитроантрацен:
Ш2
HNO3
3. Сульфирование. Антрацен легко сульфируется в крайнее
ядро, при этом образуется смесь а.а'-дисульфокислот:
антрацен-1,8-
дисульфокислота
антрацен-1,5-
дисульфокислота
Из реакций присоединения интерес представляет
гидрирование. Антрацен легко гидрируется в положения 9 и 10
водородом в момент выделения, например при действии натрия
в кипящем спирте:
9,10-дигидроантрацен
При исчерпывающем каталитическом гидрировании образу¬
ется пергидроантрацен С14Н24.
124
Окисление антрацена хромовой смесью и другими окис-
лителями приводит к антрахинону (см. § 91):
Большие количества антрацена расходуются для производ¬
ства антрахинона и различных красителей.
Фенантрен СыНю изомерен антрацену, но отличается от
него расположением бензольных ядер:
з
или
Это бесцветное кристаллическое вещество (т. пл. 100 °С;
т. кип. 340 °С), содержится в антраценовой фракции каменно¬
угольной смолы.
Фенантрен обладает ароматическим характером. По химиче¬
ским свойствам он напоминает нафталин, но отличается боль¬
шей ненасыщенностью. Это сказывается в легкости протекания
реакций присоединения. Особенной активностью отличаются по¬
ложения 9 и 10. Эта связь в молекуле фенантрена напоминает
обычную двойную связь.
Сам фенантрен не представляет практического интереса, но
многие сложные природные соединения, имеющие физиологиче¬
ское значение, содержат фенантреновое кольцо. Это стероиды,
половые гормоны, витамин D, алкалоиды и другие вещества,
которые будут рассмотрены позднее.
§ 54. Углеводороды со многими конденсированными ядрами.
Высшие фракции каменноугольной смолы содержат'значитель¬
ное количество сложных соединений со многими конденсирован¬
ными ядрами. Они могут служить исходным материалом для
синтеза различных полупродуктов, красителей и лекарственных
веществ. По строению (по расположению циклов) одни поли¬
циклические ароматические соединения имеют сходство с антра¬
ценом, другие — с фенантреном.
В молекуле антрацена бензольные ядра сконденсированы
линейно (центры ядер лежат на одной прямой). Общее назва¬
ние таких соединений — ацены.
антрацен нафтацен
125
Ацены — окрашенные вещества. С увеличением числа циклов
уменьшаются ароматические свойства (размазывание одного
секстета электронов). Эти соединения напоминают полиеновые
углеводороды с сопряженными связями (легко окисляются).
Частично гидрированные производные тетрацена лежат в ос¬
нове важных антибиотиков тетрациклинового ряда.
Многоядерные системы, построенные по типу фенантрена,
где центры не всех бензольных ядер лежат на одной прямой,
носят название ангулярных систем. Они обладают более выра¬
женным ароматическим характером, чем ацены. Примером мо¬
гут служить пирен, хризен и бензопирен:
Установлено, что некоторые многоядерные циклические со¬
единения вызывают экзему и образование злокачественных опу¬
холей, т. е. обладают канцерогенными свойствами. Примером
может служить бензопирен С20Н12 — один из наиболее опасных
канцерогенов. Бензопирен образуется при пиролизе углеводо¬
родных топлив при недостатке окислителя. В продуктах коксо¬
вания каменных углей, в табачном дыме и выхлопных газах
автотранспорта содержится много веществ, обладающих канце¬
рогенными свойствами.
НЕБЕНЗОИДНЫЕ АРОМАТИЧЕСКИЕ СОЕДИНЕНИЯ
Существуют соединения, в молекулах которых я-электроны находятся в кру¬
говом сопряжении, хотя в этих молекулах нет бензольных циклов. При этом
ароматический характер проявляется наиболее ярко в тех соединениях, где
в цикле имеется шесть л-электронов, т. е. образуется ароматический элек¬
тронный секстет. Такие соединения называют небензоидными ароматическими
соединениями.
Ароматическим характером могут обладать не только карбоциклические
соединения, скелет которых построен из атомов углерода, но и гетероцикли¬
ческие, имеющие в цикле и другие атомы — кислород, азот, серу. Кроме ней¬
тральных молекул ароматические свойства могут проявлять некоторые орга¬
нические ионы (анионы и катионы). Характерными примерами являются
циклопентадиенилий-анион и тропилий-катион.
Циклопентадиен-1,3 и циклопентадиенилий-анион. Углеводород С5Н6 яв¬
ляется циклическим диеновым углеводородом, обладающим ненасыщенным
характером. В отличие от других подобных соединений он обладает кислот¬
ными свойствами, даже более сильными, чем алкины-1. Водород группы СН2
способен замещаться металлом. При действии на циклопентадиен-1,3 щелоч¬
ных металлов в органических растворителях отщепляется протон и обра-
пирен
хризен
бензопирен
120
зуется соединение,
анион:
несущее отрицательный заряд, — циклопентадиенилий-
НС—СН
н</ Vh
ОН,
Na
—Н+
нс_сн
нс\ /сн
хн
В этом анионе два неподеленных электрона не локализуются у одного
атома углерода, а вместе с четырьмя я-электронами двух двойных связей
образуют ароматический секстет я-электронов. В анионе все атомы углерода
равноценны:
НС—СН
нс(*
чсн
.он
или
Тропилий-катион. В результате отщепления гидрид-иона Н~ от молекулы
циклогептатриена в кольце остается шесть я-электронов, которые делокали¬
зуются, образуя устойчивый электронный секстет. Тропилий-катион (СтН7) +
весьма реакционноспособен.
тропилий-катион
Азулен. Представителем бициклических соединений, не содержащих бен¬
зольных циклов, но обладающих ароматическим характером, является азулен
СюН8. Азулен — кристаллическое вещество темно-синего цвета. Он изомерен
нафталину, но в его молекуле, в отличие от нафталина, сконденсированы
циклы различной величины — пяти- и семичленный:
4
Азулен менее устойчив, чем нафталин; при нагревании выше 350 ®С в
отсутствие воздуха он изомеризуется в нафталин. Молекула азулена поляриа
(р, = 1,0 Д). В пяти- и семичленных циклах содержится по шести делокали¬
зованных электронов, и таким образом молекула азулена представляет собой
сочетание Двух ионов: аниона циклопентадиенилия и катиона тропилия. Од¬
нако, как установлено, здесь нет полной ионизации, 10 я-электронов участ¬
вуют в образовании единого я-электронного облака.
§ 55. Углеводороды — основа органической химии. В последние
десятилетия углеводороды стали основным видом сырья для
промышленности органического синтеза. Промышленность орга¬
нического синтеза перерабатывает более доступное углеводо¬
родное сырье в углеводороды требуемого состава и строения.
Главные методы переработки: расщепление (крекинг, пиролиз),
дегидрирование, алкилирование (изоалканов и ароматических
углеводородов алкенами), изомеризация, полимеризация. О вза¬
имопревращениях углеводородов дает представление схема 4.
127
Низшие алканы — этан, пропан, изобутан — при термиче¬
ском дегидрировании образуют этилен, пропилен и изобутилен,
используемые в промышленности для различных синтезов. Ка¬
талитическое дегидрирование высших нормальных алканов яв¬
ляется эффективным способом промышленного производства
высших (линейных) алкенов (Си—Cig), что имеет большое
значение для получения биоразлагаемых синтетических моющих
средств.
Замещая атомы водорода в углеводородах на другие атомы
или группы, можно переходить к новым классам органических
соединений. Для этого используют реакции хлорирования, суль-
фохлорирования, сульфирования, нитрования, окисления, окис¬
лительного аммонолиза, оксосинтеза. Важно превращение аро¬
матических углеводородов (бензола и его производных) в
ценные алифатические соединения (капролактам, адипиновую
кислоту и др.). Многочисленные полупродукты, получаемые в
промышленности из углеводородов, используют для синтеза ка¬
учука, пластмасс, синтетических волокон, моющих средств, кра¬
сителей, фармацевтических препаратов.
При разработке и освоении новых технологических процес¬
сов стремятся использовать более дешевое сырье. Например,
Схема 4. Взаимопревращения углеводородов
128
если сравнить стоимость алканов, алкенов, ароматических угле¬
водородов и ацетилена, то наиболее дорогим является ацетилен,
а наиболее дешевыми — алканы. Поэтому постепенно заменяют
ацетилен на этилен, а алкены на алканы. Самыми перспектив¬
ными являются методы прямого синтеза (одностадийные) или
совмещенные (когда в одном аппарате осуществляют несколько
реакций).
ГЛАВА 8
НЕФТЬ
Нефть — одно из важнейших природных ископаемых и один из
основных источников получения технического топлива. Главные
кладовые нефти и газа в СССР находятся в Западной Сибири.
О происхождении нефти существуют разные гипотезы. Сто¬
ронники органического происхождения нефти считают, что
нефть образовалась из продуктов разложения органических
веществ. В качестве доказательства приводится наличие в неф¬
ти веществ, характерных для живых организмов,— жирных кис¬
лот, стероидов, гормонов, хлорофилла. Сторонники неорганиче¬
ского происхождения нефти полагают, что углеводороды могли
получаться из элементов в начальной стадии образования пла¬
нет. Одним из доводов является обнаружение. значительных
количеств метана в атмосферах больших планет. В космиче¬
ских телах обнаружены (методами газовой хроматографии и
спектроскопическими) углеводороды, аминокислоты и другие
соединения, встречающиеся в живых организмах и продуктах
их распада.
По предположениям сторонников карбидной теории (впер¬
вые ее высказал Д. И. Менделеев), нефть образовалась при дей¬
ствии воды на карбиды металлов, находящиеся в глубинах
земли. Эта гипотеза объясняет нахождение богатых месторож¬
дений нефти на глубине 2—3 км. На такой глубине нефть не
могла возникнуть из живых организмов.
Нефть известна человеку с глубокой древности (несколькр веков до на¬
шей эры). Образующийся из выступающей на поверхность нефти смолообраз¬
ный битум использовали как вяжущий строительный материал, а также для
лекарственных целей. Древние греки, римляне и китайцы использовали нефть
для военных целей. Арабы в I веке до нашей эры имели установки для
перегонки нефти.
До середины XIX века нефть добывали примитивным способом — вруч¬
ную, ее черпали из неглубоких колодцев. В начале 60-х годов XIX века от
ручного способа бурения перешли к механическому; добываемую нефть начали
подвергать разгонке. Первая буровая скважина в России начала работать
в Ухте (1855 г.).
В начальный период развития нефтяной промышленности главным про¬
дуктом переработки нефти был керосин, который применяли для освещения.
Бензин из-за опасности в обращении выбрасывали (сливали в овраги, в
5 Органическая химия
129
море). Оставшуюся после отгонки керосина часть нефти сжигали в топках.
В 80-х годах было установлено, что из нефти можно получать ароматические
углеводороды. Первая в мире опытная крекинг-установка была предложена
инженером-конструктором В. Г. Шуховым (1890 г.).
Сырая нефть представляет собой маслянистую жидкость от
светло-коричневого до черного цвета (в зависимости от место¬
рождения). У разных нефтей различен не только цвет, но и
запах, а также вязкость. Плотность нефти 0,7—0,9 г/см3.
По своему составу нефть — сложная смесь, главной частью
которой являются алканы, циклоалканы (нафтены) и аромати¬
ческие углеводороды. Низшие газообразные алканы сопутству¬
ют нефти (попутный нефтяной газ), частично растворены в ней,
и если их накапливается много, нефть под их давлением бьет
из земли фонтаном. В жидких углеводородах растворены также
высшие твердые углеводороды.
Нефти, содержащие большое количество алканов, называют парафиновы¬
ми, а нефти, богатые циклоалканами — нафтеновыми. Есть нефти, богатые
ароматическими углеводородами.
Кроме углеводородов в состав нефтей в малом количестве
входят соединения, содержащие кислород (нафтеновые кислоты,
фенол), серу (тиофен и его производные), азот (разные гетеро¬
циклические соединения).
Нефть — очень ценное горючее; она, как и газ, обладает вы¬
сокой теплотворной способностью. При сгорании 1 кг нефти
выделяется 46 000 кДж тепла.
§ 56, Перегонка нефти. Первичная переработка нефти заклю¬
чается в ее перегонке, т. е. разделении на погоны (фракции),
различающиеся по температуре кипения. Это физический метод
переработки нефти. Наиболее важны следующие фракции.
1. Фракция до 40 °С — газообразные продукты, преимуще¬
ственно алканы Ci—Cs нормального и изостроения. Из нефтя¬
ного газа получают низкокипящий газовый бензин.
2. Бензиновая фракция (40—180 °С) содержит углеводоро¬
ды С5 — Сю: алканы нормальные и разветвленные, циклоалка¬
ны, алкилбензолы; всего свыше 100 различных соединений.
3. Керосиновая фракция (180—270 °С) содержит углеводо¬
роды Сю — Сю, используется для крекинга, а также как топли¬
во для реактивных двигателей.
4. Газойлевые фракции (соляровые масла, 270—360 °С) содер¬
жат углеводороды Сю—С2о, используются в качестве сырья для
крекинга, как дизельное топливо, а также для получения сма¬
зочных масел.
5. Нефтяные остатки (>360°С), обычно называемые мазу¬
том (40—50% от исходной нефти), используют как сырье для
крекинга, а также для получения тяжелых смазочных масел,
вазелина, парафина и др.
Твердый остаток после отгонки всех фракций — гудрон, со¬
держащий высшие алканы до С5о, окисляют кислородом возду¬
130
ха; образующийся битум используют для строительства дорог.
Из гудрона может быть получен также кокс.
Вторичной разгонкой легкие фракции могут быть разделены
на фракции, кипящие в более узких пределах. Например, из
бензиновой фракции выделяют петролейный эфир (40—70 °С),
авиационный бензин (70—100 °С), автомобильный бензин
(100—120 °С) и др. Так же могут быть получены более узкие
керосиновые фракции. Из высококипящих бензиновых фракций
выделяют индивидуальные углеводороды: пентаны, гексаны,
циклопентан, циклогексан, их метальные производные, аромати¬
ческие углеводороды — бензол, толуол, ксилолы.
Из нефти выделено и идентифицировано около 150 индиви¬
дуальных углеводородов с т. кип. до 250 °С. Продукты пере¬
гонки подвергают очистке от сернистых и других примесей.
§ 57. Нефть как химическое сырье. Выделение углеводородов
из нефти. Химическая переработка нефти. Класси¬
ческими работами В. В. Марковникова и его учеников
(М. И. Коновалов, Н. М. Кижнер и др.) было положено начало
подробному исследованию и изучению химии нефти. Большое
значение для изучения превращений углеводородов, создания
современных путей переработки нефти имеют работы Н. Д. Зе¬
линского и его учеников (Б. А. Казанский, А. Ф. Платэ и др.).
Важнейшие химические реакции, используемые при перера¬
ботке нефти, были описаны в 'предыдущих главах. Рассмотрим
применение этих реакций в процессах переработки нефти.
Один из основных процессов химической переработки неф¬
ти — крекинг, в результате которого возрастает выход моторных
топлив, а также простейших газообразных алкенов и алканов —
ценного сырья для дальнейшей химической переработки. Суще¬
ствует несколько вариантов крекинга.
Термический крекинг проводят при температурах «500°С.
Это радикальный процесс. Цепь зарождается при распаде ал¬
кана с разрывом С—С-связи и образованием свободных ра¬
дикалов. Например, для нонана CgH2o процесс может идти так:
СН3—(СН2)4—СН2—СН2—СН2-СН3 —► СН3-(СН2)4-СН2-СН2-СН2+ .сн3
Образовавшийся октальный радикал подвергается распаду
по p-углеродной связи (считая от радикального С-атома):
Р а
СН3—(СН2)4—сн2—сн2—сн2. —> СН3—(СН2)4—сн2 + сн2=сн2
А
Возникший при зарождении цепи метильный радикал может
оторвать водород от другой молекулы нонана и в свою очередь
превратить ее в радикал:
СН3—(СН2)4—сна—сн2—сн2—сн3 + • сн3 —►
—*■ СН9—(СН,)4—СН*—СНа—СН—CHj + СН4
Б
5*
131
Образовавшиеся в этих реакциях радикалы А и Б могут
подвергаться дальнейшим процессам p-распада или захваты*
вать имеющиеся в реакционной смеси простые радикалы и ато*
мы водорода, например:
• • СНв
СН3—(СН2)2—СН2 + СН2=СН2 ч— Радикал А ► СН3—(СН2)5—СНЭ
СН3—(СН2>4—СН2 + СН2=СН—СНз ч— Радикал Б СН3-(СН2)7СНЭ
Р-распад захват радикалов
Таким образом, в результате крекинга углеводороды с боль*
шим числом С-атомов, входящие в состав высококипящих
фракций нефти, расщепляются на жидкие низкокипящие и газо*
образные алканы и алкены. Так, например, при крекинге пара*
фина образуется ж 20% газообразных и 80 % жидких про¬
дуктов.
Каталитический крекинг на алюмосиликатных катализато¬
рах идет по ионному механизму. Распад углеводородов на более
мелкие осколки сопровождается изомеризацией нормальных це¬
пей в разветвленные, что повышает качество бенвина (см. ниже
об октановом числе).
При действии на нефть (или отдельные нефтяные фракции)
более высоких температур («700—900 °С), происходит пиро¬
лиз—более глубокий распад, при котором образуется много
простейших алкенов, а жидкая фракция обогащается аромати¬
ческими углеводородами. Пиролиз полученной прямой перегон¬
кой бензиновой фракции служит в нашей стране главным ис¬
точником этилена (около 70 %). Нафтены в процессе пиролиза
превращаются в арены: из циклогексана образуется бензол, из
метилциклогексана — толуол.
Для перехода от алканов к аренам в промышленности ис¬
пользуют и другой процесс — ароматизацию:
СНз
НгС-^ СН2— СНз
Н2С\ /СН2
СН2
■Pt/AI203
>
500 -С. 4—5 МПа
СНз
Бензол — более ценное химическое сырье, чем толуол, поэто¬
му в промышленности проводят деалкилирование толуола или
его диспропорционирование:
700 “С, 5 МПа, Н2
сен5сн3 * С6Н6 + СН4
350- 500 °С, 1 МПа
2С6Н5СН3 -> СвНв + СвН4(СН3)2
При диспропорционировании толуола кроме бензола образу¬
ются также изомерные ксилолы — ценное химическое сырье.
132
Реакции изомеризации с образованием изоалканов, реакции
дегидрогенизации нафтенов, ароматизации алканов, деалкили¬
рования аренов под действием соответствующих катализаторов
могут протекать одновременно, приводя к повышению качества
нефтепродуктов — росту октанового числа. Такой процесс назы¬
вают риформингом (от англ, reform — преобразование). В про¬
цессе риформинга происходит существенное изменение углеводо¬
родного состава, что приводит к повышению октанового числа
с 45—50 до 70—80. Так, например, если риформингу подвер¬
гнуть фракцию нефти, в которой содержится 58 % алканов,
29 % нафтенов, 12 % аренов и 1 % алкенов, то после риформинга
в ней оказывается 30 % алканов, 4 % нафтенов, 66 % аренов и
1 % алкенов.
Выделение углеводородов из нефти и их
переработка. Смеси веществ (Ci2—Cie), которые могут
быть использованы в промышленности без разделения (напри¬
мер, в производстве синтетических моющих средств), получают
методом ректификации. Для выделения индивидуальных низших
парафинов (С3 — С5) из попутных газов проводят ректифика¬
цию при повышенном давлении (0,9—1,8 МПа). Для выделения
индивидуальных углеводородов широко используют также азео¬
тропную перегонку. К перегоняемой смеси добавляют жидкость,
изменяющую летучесть компонентов смеси,— азеотропный
агент, образующий с одним из .компонентов постояннокипящую,
или азеотропную, смесь, имеющую минимальную температуру
кипения. Так выделяют, например, бутадиен из смеси углеводо¬
родов С4, используя в качестве азеотропного агента аммиак.
Неразветвленные парафины, содержащие в молекуле не
меньше семи атомов углерода, могут быть выделены из раз¬
личных нефтяных фракций (бензина, керосина, газойля) мето¬
дом карбамидной депарафинизации. Насыщенный водный рас¬
твор карбамида [мочевины CO(NH2)2] смешивают с нефтяной
фракцией при температуре не выше 40 °С. (Кристаллы мочеви¬
ны способны -«включать» в себя соединения с нормальной
цепью.) После фильтрования осадок разлагают горячей водой.
Соединения извстроения не связываются мочевиной.
до 40 *с
nCO(NH2)2 + RH "( RH • rtCO(NHj)2
70— loo °С
Смесь высших н-алканов (твердый парафин) выделяют из
смазочных масел и газойлевой фракции методом экстракции,
основанным на различной растворимости углеводородов. Опре¬
деленные растворители обладают избирательной растворяющей
способностью (селективностью), хорошо растворяют только
часть компонентов смеси и не смешиваются с остальной частью.
Образуются две жидкие фазы с различной плотностью, которые
133
можно разделить. Экстрактивной перегонкой разделяют также
смесь низкокипящих алканов и алкенов (Сг — С$). Алкены об¬
ладают большей растворимостью в полярных жидкостях, поэто¬
му их летучесть по сравнению с алканами уменьшается. Смесь
высших алкенов (Сб—С18) может быть разделена ректифи¬
кацией.
Изоалканы — изобутан и изопентан — получают изомериза¬
цией н-алканов под влиянием А1С1з или металлов платиновой
группы на оксиде алюминия. Прямым окислением выделенных
низших алканов (пропана, бутана) получают соответствующие
кислоты, при их хлорировании — ценные полупродукты. Бутан
и бутены — сырье для получения бутадиена-1,3—мономера для
производства синтетического каучука. Низшие алкены и арома¬
тические углеводороды являются очень важными полупродукта¬
ми. Синтез высокомолекулярных соединений немыслим без до¬
ступного и дешевого нефтехимического сырья — мономеров.
Многие микроорганизмы могут развиваться на средах, где
единственным источником углерода являются углеводороды
нефти. Наиболее предпочтительны для них нормальные алканы,
начиная с додекана Q2H26. Освоен микробиологический метод
получения белково-витаминного концентрата из углеводородов
нефти. Из нефтехимического сырья производят биологически
активные и лекарственные вещества, душистые вещества, краси¬
тели, растворители, пластификаторы, лаки, мастики, эмали, об¬
лицовочные материалы, инициаторы полимеризации, ингибиторы
коррозии.
Нефть многих месторождений содержит значительное коли¬
чество органических соединений серы. Эти соединения затрудня¬
ют процессы химической переработки нефти; недопустимо их
присутствие и в моторном топливе (усиленная коррозия двига¬
телей, загрязнение атмосферы). Поэтому одним из важных
процессов является обессеривание нефти. Его осуществляют
действием водорода на молибденовых катализаторах. При этом
выделяется сероводород, который сжигают и используют обра¬
зовавшийся диоксид серы для производства серной кислоты.
Вместе с тем ведутся работы по выделению из нефти органи¬
ческих соединений серы, для того чтобы непосредственно ис¬
пользовать их в синтетических целях.
Значение нефти для народного хозяйства непрерывно воз¬
растает. Для транспорта, сельскохозяйственной техники нефте¬
продукты используют в качестве топлива. Нефтяные масла при¬
меняют для трансф'орматоров электрических подстанций, для
двигателей, различных станков и др. В последнее время все
чаще вспоминают о том, что запасы нефти на Земле не так уж
велики и задумываются над необходимостью сократить расход
нефти для энергетических целей. Об этом говорил еще в свое
время Д. И. Менделеев: «Нефть не топливо — топить можно и
ассигнациями».
134
Все же и в настоящее время нефть главным образом исполь¬
зуют как топливо. В мировой выработке энергии и тепла нефть
в 1985 г. занимала первое место (более 4 млрд, т условного
топлива), далее шли природный газ (около 2 млрд, т), твердое
топливо (1,6 млрд, т), ядерная энергия (около 1 млрд, т), гид¬
ростанции (0,5 млрд, т). Менее 10 % добываемой нефти исполь¬
зуют как химическое сырье.
По плану 12-й пятилетки в нашей стране намечено повысить
эффективность добычи нефти в результате применения рацио¬
нальных систем разработки месторождений; продолжить разви¬
тие нефтедобычи в Западной Сибири, на севере европейской
части страны, в Казахской ССР, приступить к разработке глу-
бокозалегающих месторождений в Прикаспии. К 1990 г. наме¬
чено достичь ежегодной добычи 625—640 млн. т. В нефтепере¬
рабатывающей промышленности предусматривается обеспечить
дальнейшее углубление переработки нефти и существенное уве¬
личение выработки моторных топлив, а также сырья для хими¬
ческой, нефтехимической и микробиологической промышленно¬
сти, расширить выпуск смазочных масел, довести использование
попутного нефтяного газа не менее чем до 90 %.
В настоящее время нефть — один из главных источников хи¬
мического сырья; 1 т нефти может дать столько сырья для
химической промышленности, сколько 15 т бурого угля. Нефте¬
химическая промышленность — основа, на которой развивается
и будет развиваться в дальнейшем промышленность органиче¬
ского синтеза.
Детонация топлива и октановое число. Коэффициент по¬
лезного действия двигателя зависит от степени сжатия горючей смеси. Сте¬
пень сжатия — отношение первоначального объема бензино-воздушной смеси,
которая засасывается в цилиндр, к конечному объему после сжатия. Повы¬
шение степени сжатия дает возможность экономить топливо и увеличивать
мощность двигателя. Увеличение же мощности двигателя, например автомо¬
биля, означает увеличение скорости и грузоподъемности, уменьшение расхода
топлива. При нормальном сгорании топлива давление внутри цилиндра повы¬
шается непрерывно, скорость сгорания 20—25 м/с. При неправильном сгора¬
нии происходит детонация — смесь бензина с воздухом вспыхивает мгновенно
со взрывом, скорость сгорания 1500—2000 м/с. При этом быстро выделяется
огромное количество газов, что приводит к резкому повышению давления
внутри цилиндра. Удар детонационной волны о стенки цилиндра и поршень
создает «стук» мотора. Следствие детонации — неправильная работа мотора,
снижение мощности двигателя, повышение расхода горючего, прогар и разру¬
шение отдельных частей мотора.
За образец топлива с высокими антидетонационными свойствами был
выбран изооктан (2,2,4-триметнлпентан). Его октановое число (ОЧ) принято
равным 100. За образец топлива с низкими антидетонационными свойствами
взят «-гептан, октановое число которого считается равным 0,
(СН3)3С—СН2—СН(СНзЬ
изооктан
СНз—СН2—СН2-СН2—СН2—CHj—CHj
к-гептан
135
Испытуемый бензин сравнивают с эталонными смесями я-гептана и изо¬
октана. Например, бензин с ОЧ 80 обладает такими антидетонационными
свойствами, как смесь 80 % изооктана и 20 % и-гептана. Чем выше октано¬
вое число, тем выше качество горючего. Изучение многих углеводородов по¬
казало, что в ряду алканов октановое число уменьшается по мере удлинения
нормальной цепи углеродных атомов и увеличивается с ее разветвлением.
Алкены имеют более высокие октановые числа, увеличивающиеся по мере
смещения двойной связи к центру молекулы. Октановые числа циклоалканов
еще выше, а ароматические углеводороды наиболее устойчивы к детонации.
Антидетонационные свойства горючего могут быть повышены прибавле¬
нием к топливу некоторых веществ. Наиболее важной добавкой является тет¬
раэтилсвинец РЬ(С2Н5)4 (ТЭС). В технике состав, содержащий эту добавку,
называют этиловой жидкостью, а бензин, к которому добавлена эта жид¬
кость, — этилированным бензином. В состав этиловой жидкости входят 63 %
РЬ(С2Н5)4. 26 % дибромэтана СН2Вг—СН2Вг, 9 % дихлорэтана СН2С1—СН2С1,
краситель. Прибавление 1—2 мл ТЭС на 1 л бензина повышает октановое
число на 8—10 единиц, но большие количества этой жидкости добавлять не
рекомендуется. При сгорании топлива свинец выделяется в виде оксида, от¬
ложение которого на стенках двигателя нарушает его нормальную работу.
Добавленные к тетраэтилсвинцу дибромэтан и дихлорэтан превращают оксид
свинца в летучие галогениды свинца, выбрасываемые из цилиндра вместе с
выхлопными газами. Двигатели, работающие на этилированном бензине, соз¬
дают, следовательно, ядовитый выхлоп. В современных двигателях с высокой
степенью сжатия чаще применяют тетраметилсвинец РЬ(СНз)4 и другие ме-
таллорганические соединения.
Дизельное топливо — это горючее, содержащее неразветвленные и мало-
разветвленные высококипящие углеводороды (керосино-газойлевая фракция).
Качество дизельного топлива определяют по цетановой шкале и характери- .
зуют цетановым числом. За 100 принята способность к воспламенению цетана
(н-гексадекана С]бНз4), за 0 — способность к воспламенению а-метилнафта-
лина. Обычно используют дизельное топливо с цетановым числом 50—60.
Для реактивных двигателей пригодно самое разнообразное горючее,
обычно применяют к-алканы с температурой кипения до 315 °С.
ЧАСТЬ II
СОЕДИНЕНИЯ С ОДНОРОДНЫМИ
ФУНКЦИЯМИ
К соединениям с однородными функциями относят органические
соединения, содержащие в молекуле одну или несколько одина¬
ковых функциональных групп.
ГЛАВА 9
ГАЛОГЕНПРОИЗВОДНЫЕ
Производные углеводородов, в молекулах которых один или не¬
сколько атомов водорода замещены галогеном, называются га-
логенпроиззодными. Известны фтор-, хлор-, бром- и иодпроиз-
водные. По числу атомов газогена различают моно-, ди- и по-
лигалогенпроизводные.
Производные алканов носят название алкилгалогениды или
галогеналканы, производные циклоалканов — циклоалкилгало-
гениды, производные ароматических соединений, содержащие
галоген, связанный с атомом углерода ядра, — арилгалогениды.
§ 58. Изомерия и номенклатура. Алкилгалогениды имеют об¬
щую формулу CreH2n+iX (где X=F, Cl, Вг, I). Изомерия га-
логенпроизводных обусловлена изомерией углеродного скелета
и положением галогена.
Рациональные названия галогенпроизводных обычно состав¬
ляют из названия углеводородного радикала и соответствующе¬
го галогена. При этом название галогена может быть поставле¬
но либо в форме прилагательного перед названием углеводо¬
родного радикала, либо после названия радикала. Например:
СН3С1 — хлористый метил или метилхлорид; С2Н5Вг — бро¬
мистый этил или этилбромид; СбНбСН2С1 — хлористый бензил
или бензилхлорид. Для более сложных соединений к названию
прибавляют обозначения: первичный, вторичный или третичный,
в зависимости от того, у какого углеродного атома находится
галоген, например:
СНз—СН—СН2С1
первичный хлористый
изобутил, первичный
изобутилхлорид
(СНз)зСВг
третичный броми¬
стый бутил, трет-
бутилбромид
По современной заместительной номенклатуре название га¬
логена и цифру, указывающую его положение, помещают перед
137
названием главной цепи в общем алфавитном порядке с други¬
ми фрагментами, помещаемыми в префиксной части названия.
Углеродный атом, несущий галоген, должен входить в состав
главной цепи, а нумерацию цепи проводят с того конца, к кото¬
рому ближе любой заместитель — алкил или галоген. Если раз¬
личные заместители одинаково удалены от концов цепи, то их
нумеруют, начиная с того конца цепи, к которому ближе замес¬
титель, стоящий первым в алфавите. Перечисление всех замес¬
тителей также ведут по алфавиту.
12345 54321
СН3—СН—СН2—СН—СН3 СН3—СН—СН2—СН—СН3
II II
СНз С1 СНз Вг
2-метил-4-хлорпентан 2-бром-4-метилпентан
4 3 2 1
СНз—СН—СН—СНз
С1 Вг
2-бром-З-хлорбутан
В ненасыщенных галогенпроизводных начало нумерации
определяет кратная связь (двойная, тройная): она получает
наименьший возможный номер:
СНз—СН=С—СН—СН2—СНз
Hji ^Н2С1
СН,—СНз—СН—СН=СН—СН—СНз
I I
Вг С1
3 • метил-5-хлор-4-эти лпентен-2
5-бром-2-хлоргептен-3
§ 59. Способы получения. Одним из наиболее важных агентов
промышленного галогенирования является хлор. Хлорорганиче-
ский синтез играет важную роль для получения мономеров
'(например, винилхлорида), растворителей (например, четырех¬
хлористого углерода), ядохимикатов, а также промежуточных
продуктов синтеза.
Получение насыщенных галогенпроизвод¬
ных. 1. Прямое галогенирование алканов (хлорирование или
бромирование) рассмотрено ранее как типичный пример ради¬
кальной цепной реакции (см. § 20). Практическое использова¬
ние этой реакции затруднено тем, что при ее проведении всегда
образуются смеси изомеров положения, моно- и полизамещен-
ных. Несмотря на это реакция имеет промышленное значение,
так как смеси часто используют без разделения или выделяют
отдельные вещества фракционной перегонкой. Аналогично про¬
текает реакция замещения в циклоалканах.
2. Галогенирование и гидрохлорирование алкенов (см.
§ 33).
3. Действие галогеноводородов или галогенпроизводных фос¬
фора на спирты:
RBr + Н20
ROH + НВг
ROH + РС16
RC1 + НС1 + РОС13
Рис. 20. Зависимость от температуры выходов
продуктов замещения (/) и присоединения (2)
при хлорировании пропилена
too
Для получения иодпроизводных берут
спирт, свободный иод й красный фосфор:
31а + 2Р—*• 2PIj
3ROH + Р13 —*• 3RI + Н3РО3
в 25-
125 250 375 500
Температура,"О
Из-за доступности спиртов эту реакцию часто используют для
получения алкилгалогенидов.
Получение ненасыщенных галогенпроиз-
водных. Способы получения ненасыщенных галогенпроизвод-
ных рассмотрены при изучении алкинов (см. § 38) и диеновых
углеводородов (см. § 41).
Термическое хлорирование этилена (300—600 °С) приводит
к винилхлориду:
300-6ОЭ “С
СН2=СН2 + С12 *- СН2=СНС1+НС1
При термическом галогёнировании гомологов этилена идет
замещение водорода в аллильном положении. Так, пропилен
при хлорировании по Львову — Шешукову (при »500°С в га¬
зовой фазе) дает аллилхлорид (З-хлорпропен-1):
С12, 500 °С
СН2=СН—СНз »- СН2=СН—СНгС1
Эта реакция составляет часть важного для современной про¬
мышленности процесса — синтеза глицерина из нефтяного
сырья (см. § 70). Интересно это превращение и как яркий при¬
мер зависимости направления реакции от внешних условий
(рис. 20). Познавая подобные зависимости, химики получают
возможность лучше управлять химическими реакциями.
Получение ароматических га л о геипро из¬
вод ных. При действии галогена на ароматические соедине¬
ния в зависимости от условий реакции образуются различные
продукты (см.§ 47).
§ 60. Физические свойства (табл. 10). Как видно из таблицы,
первые члены ряда алкилгалогенидов и алкенилгалогенидов
имеют низкие температуры кипения. По мере увеличения ради¬
кала в гомологическом ряду и атомного номера галогена тем¬
пература кипения повышается. Наиболее высокие температуры
кипения характерны для иодпроизводных. Низшие члены гомо¬
логического ряда обладают более высокой плотностью, из них
самая большая плотность у иодпроизводных. Алифатические
хлориды легче воды, бромиды и иодиды — тяжелее. С увеличе¬
нием углеводородного радикала плотность уменьшается.
Большинство галогенпроизводных — бесцветные жидкости с
характерным запахом; полииодпроизводные — твердые вещест¬
ва желтого цвета. Все галогенпроизводные практически нерас-
139
Таблица 10. Физические свойства некоторых галогенпроизводных
Радикал
Хлорид
Бромид
Иодид
т. кип.,
,20
г. кип.,
А20
т. кип.,
W20
название
формула
°с
di
°с
°С
Метил
СНз
—24,2
0,92
4,3
1,7
42,3
2,27
Этил
с2н5
13
0,9
38,4
1,43
72,3
1,93
Пропил
CSH7
46,4
0,89
70,9
1,35
102
1,74
Изопропил
(СНзЬСН
36,5
0,86
59,5
1,32
89,4
1,70
Винил
сн2=сн
— 14
—
15,8
—
56
—
Аллил
сн2=снсн2
45,7
0,93
70
—
102
1,84
Фенил
С.Н.
132
1,10
155
1,49
189
1,83
Бензил
свн5сн2
179
1,09
198
1,43
96
1,73
Бензилиден
С6Н5СН
205
1,25
—
—
—
—
творимы в воде, растворимы в органических растворителях —
углеводородах, спиртах, эфире; нерастворимы в концентриро¬
ванной серной кислоте, поэтому их можно очищать от примесей
спиртов и эфиров встряхиванием с серной кислотой, в которой
спирты и эфиры растворяются.
Некоторые алкилгалогениды обладают анестезирующим
(обезболивающим) действием. Так, например, этилхлорид ис¬
пользуют для местной анестезии, хлороформ — для общей ане¬
стезин. Йодоформ СШз обладает антисептическими свойствами.
Производные ароматических углеводородов, которые содер¬
жат галоген в боковой цепи у атома углерода, связанного с
ароматическим ядром, имеют раздражающий запах, токсичны.
Некоторые из них, например бензилбромид, применяли в первую
мировую войну в качестве слезоточивых отравляющих веществ,
так называемых лакриматоров (лат. lakrima — слеза).
§ 61. Химические свойства. Реакции нуклеофильного
замещения. В молекулах галогенпроизводных атом га¬
логена более электроотрицателен, чем атом углерода, связь
углерод —галоген поляризована:
ч е+ е-
,С—X Х = С1, Вг, I
/|
Нуклеофильный реагент, атакуя молекулу галогенпроизвод-
ного, образует связь с атомом углерода, обладающим понижен¬
ной электронной плотностью, и вытесняют из молекулы атом
галогена. В результате реакции галоген замещается нуклео¬
фильной группой. Такие реакции называют реакциями нуклео¬
фильного замещения и условно обозначают Sn (англ. Substitu¬
tion —■ замещение, Nucleophylic — нуклеофильное). При нук¬
леофильном замещении реакции могут протекать по двум раз¬
ным механизмам, условно обозначаемым 5ы2 и S\l. Цифры
указывают молекулярность реакции, т. е. число частиц, которые
участвуют в реакции на стадии, определяющей ее скорость.
НО
Бимолекулярная реакция нуклеофильного замещения в об*
щем виде может быть записана так:
• б+ б- б" б+ 7
Y: + R * X Y * R + : X
В решающей стадии реакции учдствук>т оба реагента, поэто¬
му процесс условно обозначается Sn2. Если они содержатся в
соизмеримых концентрациях (без брльшого избытка одного из
них), то скорость реакции Sn2 пропорциональна концентрации
обоих реагентов.
При гидролизе алкилгалогенидов бимолекулярная реакция
представляет собой одностадийный процесс с Образованием пе¬
реходного комплекса или, иначе, переходного состояния. Типич¬
ным примером такой реакции является гидролиз метилбромида»
б+ в-
НО ; + Н3С: Вг
' б- Н\б+
НО—с—
н/\н
б- 6+
НО: СНз + : Вг
Подход гидроксид-иона к атому углерода возможен только
со стороны, противоположной той, где находится атом брома.
Приближение гидроксид-иона к атому углерода, удаление бро¬
ма и превращение его в ион брома происходят одновременно.
Отрицательный заряд в переходном комплексе распределяется
между вступающей и уходящей нуклеофильными группами.
Для осуществления реакции по механизму SN2 имеет значе¬
ние легкость подхода нуклеофила к молекуле. Реакция проте¬
кает легче для первичных алкилгалогенидов, имеющих нормаль¬
ную цепь углеродных атомов; разветвленный углеродный скелет
тормозит подход нуклеофила.
Мономолекулярная реакция нуклеофильного замещения
представляет собой двухстадийный ионный процесс. Первая ста¬
дия — ионизация реагента и образование карбокатиона (карбе-
ниевого иона) [реакция (1)], вторая стадия — взаимодействие
карбокатиона с нуклеофильной частицей [реакция (2) ]:
6+ 6“ + » + — б+ б*
RjX ;<=* R + sX (1) R + :Y —► R: Y (2)
Скорость реакции зависит от концентрации только одного из
реагирующих веществ. Она определяется диссоциацией, образо¬
ванием карбокатиона. Весь процесс условно обозначается SN1.
Рассмотрим этот процесс на примере трет-бутилхлорида. Пер¬
вая стадия — ионизация трет-бутилхлорида и образование кар¬
бокатиона трег-бутила:
«-
Н3с/ \сн3
медленно
■ — ■ >
(СН3)зС+ +: с г
141
Вторая стадия — реакция карбокатиона с гидроксид-иономг
+ - быстро
(СН3)3С + : ОН »- (СН3)3СОН
По общему закону скорость многостадийного процесса опре¬
деляется самой медленной стадией, в данном случае первой.
Накопление алкильных групп у атома углерода стабилизи-
рует карбокатион и тем самым способствует протеканию реак¬
ции по механизму SN1; такие реакции характерны для третич¬
ных алкилгалогенидов. Вторичные алкилгалогениды могут ре¬
агировать в зависимости от условий реакции по механизмам
SnI или 5N2. В приведенном ниже ряду алкилбромидов показа¬
ны направления изменения скоростей реакций SN2 и Sn1:
Скорость реакции 5^2 возрастает
<•" 1 — 1
СНзВг, CHjCH2Br, (СН3)*СНВг, (СНз)зСВг
Скорость реакции возрастает
Во многих реакциях нуклеофильного замещения реагентом
является растворитель: при гидролизе — вода, при алкоголи¬
зе — спирт, при ацетолизе — уксусная кислота, при аммоноли-
зе — аммиак.
Влияние углеводородного радикала на
свойства галоген производных. Механизм реакции
нуклеофильного замещения и реакционная способность галоген-
производных зависят от природы галогена и углеводородного
радикала, связанного с галогеном, от характера реагента, от
растворителя и условий реакции — температуры, катализато¬
ра и др.
Реакционная способность различных типов алкилгалогени¬
дов рассмотрена выше. Галогенциклоалканы напоминают по
своему поведению вторичные алкилгалогениды. Чаще всего они
реагируют по механизму S^l, но скорость реакции зависит так¬
же от размера цикла. Хорошо идет реакция для циклов с пятью
и более атомами углерода, в то время как производные цикло¬
пропана напоминают винилгалогенид.
Свойства галогена в ненасыщенных галогенпроизводных за¬
висят от взаимного расположения атома галогена и двойной
связи. Галоген в аллильном положении по отношению к двой¬
ной связи, как в аллилхлориде СНг=СН—СН2С1, более акти¬
вен, чем в насыщенных соединениях. Это объясняется большой
устойчивостью карбениевого иона вследствие сопряжения:
СН2=СН— СН2:С1:^ГСНг=СН—СН8 + :С1:~
t
+
сн2=сн^:н
142
Поэтому реакция по механизму SN1 для аллилхлорида про¬
текает легче, чем для алкилгалогенидов.
Соединения типа бензилхлорида СбН5СН2С1 и трифенил-
хлорметана (CsHsbCCl также легщэ образуют карбокатионы,
стабилизованные сопряжением, поэтому реакции замещения по
механизму SN1 протекают у них легче, чем у других насыщен¬
ных галогенпроизводных. Если галоген удален от двойной связи
или от бензольного кольца на два и более атома углерода, то
в таких соединениях активность галогена близка к активности
его в алкилгалогенидах. Соединения, содержащие галоген при
двойной связи (типа винилгалогенида), мало активны. Связь
углерод—галоген здесь короче обычной; то же относится к аро¬
матическим соединениям, замещенным галогеном в ядре:
Атом галогена в этих соединениях за счет своих свободных
электронов сопряжен с л-электронами двойной связи или с
ароматическим секстетом. В результате возникает частичная
двоесвязность связи С—X, которая приводит к уменьшению
длины связи и возрастанию ее энергии. Например, строение ви¬
нилхлорида можно точнее Бсего передать двумя граничными
формулами или одной мезомерной:
— +
СН2=СН—CI ■< *- сн2—СН=С1 или СН2=СН^С1:
Сопряжение уменьшает вызванную индуктивным эффектом
хлора полярность связи С—С1 и тем самым ухудшает условия
протекания реакций нуклеофильного замещения.
Подвижность атома галогена в различных галогенпроизвод¬
ных хорошо видна при рассмотрении условий их гидролиза.
Так, алкил- и циклоалкилхлориды гидролизуются водными рас¬
творами щелочей при нагревании, аллил- и бензилхлориды —
избытком воды при кипячении, трифенилхлорметан — водой на
холоду. Соединения типа винилхлорида СН2 = СНС1 и хлорбен¬
зол С6НбС1 в этих условиях не гидролизуются; они содержат
«неомыляемый» галоген. В более жестких условиях и они всту¬
пают в реакции нуклеофильного замещения.
В соединениях, содержащих одинаковые радикалы, реакци¬
онная способность увеличивается в ряду: RC1 < RBr < RI.
Реакции нуклеофильного замещения галогена часто сопро¬
вождаются реакциями отщепления галогеноводорода с образо¬
ванием алкенов. Промежуточно образующийся карбокатион
н
0,177 нм
0,173 нм
0,173 нм
143
превращается в устойчивый продукт не вследствие присоеди¬
нения аниона, а путем отщепления протона, например:
(CHshCCl —> (СНз)зС+ + СГ (СНз)зС+ +■ (СН3)2С=СН2
-нт
Реакции отщепления ускоряются в сильноосновной среде,
при нагревании.
Важнейшие реакции галогенпроизводных.
1. Гидролиз (в щелочной или кислой среде). Моногалоген-
производные в результате гидролиза дают спирты, например:
RX + HOH —► ROH + HX
С6Н5—СН2С1 + НОН —V СбН5—СН2ОН + НС1
бензиловый
спирт
При гидролизе полигалогенпроизводных, содержащих два
атома галогена у одного атома углерода, образуются альдеги¬
ды или кетоны (см. § 84), а при гидролизе аналогичных трига-
логенпроизводных — кислоты (см. § 93). Гидролиз полигалоген¬
производных, у которых атомы галогена находятся у разных
атомов углерода, приводит к многоатомным спиртам, например:
С1СН2—СН2С1 + 2НОН —V НОСН2—СН2ОН + 2НС1
этиленгликоль
2. Обмен галогена на алкоксигруппу OR в реакции с алко-
голятами щелочных металлов. При этом образуются простые
эфиры:
СНз—CH2Br + NaOCH3 —* CHj—СН2—О—СН3 + NaBr
3. Обмен галогена на нитрильную группу — образование ни¬
трилов при реакции с цианидом калия. При этом идет наращи¬
вание углеродной цепи (см. § 116):
СНз—CH2I + KCN —► СНз—СН2—CN + KI
пропионитрил
При помощи реакций нуклеофильного замещения из галоген-
производных можно получать также нитросоединения RN02
(см. § 122), амины RNH2 (см. § 126), тиолы RSH (см. § 118),
и др. Из других реакций галогенпроизводных большое значение
имеют отщепление от них галогеноводородов с образованием
алкенов и алкинов, синтез гомологов бензола при алкилирова¬
нии по реакции Фриделя — Крафтса, синтез алканов по реакции
Вюрца.
Арилгалогениды подвергаются реакциям электрофильного
замещения подобно ароматическим углеводородам. Галоген,
хотя и является орго-пара-ориентантом, дезактивирует ядро
(см. § 130).
144
Особое место занимает реакция образования магнийоргани-
ческих соединений (см. § 136). Она идет практически с любыми
галогенпроизводными независимо от природы радикала R:
эфир
RI+Mg RMgl
К действию окислителей галогенпроизводные довольно ус¬
тойчивы. Восстановители превращают их в углеводороды, на¬
пример:
СвНп1+Н1 - > C8H18+l2
§ 62. Хлорорганические продукты в промышленности. Галоген¬
производные отличаются большой реакционной способностью.
Они расположены как бы на «оживленном перекрестке» орга¬
нической химии: к ним и от них ведет много путей. Для синте¬
за галогенпроизводных в промышленности практически исполь¬
зуют только относительно дешевый реагент — хлор (отдельную
область, которую мы сейчас не обсуждаем, образуют фторорга-
нические соединения). В мире для производства хлорорганиче-
ских продуктов расходуется несколько десятков миллионов
тонн хлора. В промышленности получают следующие важней¬
шие хлорорганические продукты:
Исходный углеводород
Метан СН4
Этан С2Нв
Бутан C^Hij
Алканы С9—С25
Этилен СН2=СН2
Пропилен СН2=СН—СНэ
Бутадиен СН2=СН—СН=СН2
Ацетилен СН==СН
Бензол CSH6
Толуол СвН6—СН3
Главные продукты
Метилхлорид, дихлорметан, хлороформ, че¬
тыреххлористый углерод
Этилхлорид
Г ексахлорбутадиен
Хлорсодержащие компоненты смазок и ох¬
лаждающих жидкостей, пластификаторы
Дихлорэтан, винилхлорид
Аллилхлорид
Изомерные дихлорбутены
Тетрахлорэтан, трихлорэтилен, винилхлорид, '
хлоропрен
Моно- и дихлорбензолы, гексахлорцикло-
гексан
Бензилхлорид, бензилидендихлорид, бензо-
трихлорид
Винилхлорид. В наибольших количествах производят винил¬
хлорид (бесцветный газ, т. кип. —13,9°С) и из него — поливи¬
нилхлорид. На примере получения винилхлорида познакомимся
с некоторыми проблемами промышленности хлорорганического
синтеза и путями их решения.
Исходным веществом для синтеза винилхлорида может слу¬
жить ацетилен или этилен:
CffeCH+HCI —> СН2=СНС1
Cl2 Ca(OHfe
СН2=СН2 ► С1СН2—СН2С1 ——СН2=СНС1
Первый метод одностадийный и безотходный — оба исход¬
ных вещества входят в конечный продукт. Это важное
145
преимущество метода, особенно сейчас, когда обращается серь*
езное внимание на охрану окружающей среды. Однако эконо¬
мически метод не очень выгоден, так как ацетилен — сырье до¬
рогое. Второй метод базируется на более дешевом сырье — эти¬
лене, однако хлор (реагент не слишком дешевый!) используется
только наполовину: другая половина переходит в бесполезные
отходы. Важным усовершенствованием явилась разработка бес-
щелочного процесса превращения дихлорэтана в винилхлорид.
Реакцию проводят под действием высоких температур (пиро¬
лиз):
400-500 “С
С1СН2—СН2С1 >• СН2=СНС1 + НС1
Еще одно усовершенствование — разработка метода окисли¬
тельного хлорирования, при котором реагентами служат хлоро-
водород и кислород воздуха. В качестве катализатора при этом
используют хлорид меди(II), который по существу служит
«переносчиком» хлора: хлорирует этилен и вновь превращается
в дихлорид под влиянием кислорода и хлороводорода:
СН2=СН2 + 2СиС12 —► С1СН2—СН2С1+ 2СиС1
2CuCI + '/202 + 2НС1 —► 2CuC12 + H20
Комбинируя описанные процессы, удалось реализовать в про¬
мышленности такой путь получения винилхлорида:
СН2=СН2 + С12 —► С1СН2—СН2С1
СН2=СН2 + 2НС1 + Vj02 —► С1СН2—СН2С1 + Н20
2С1СН2—СН2С1 —v 2СН2=СНС1 + 2НС1
Суммарное уравнение:
2СН2=СН2 + С12 + 7202 —V 2СН2=СНС1 + Н20
Таким образом, новый способ получения винилхлорида заключается в
комбинировании процессов обычного и окислительного хлорирования и терми¬
ческого дегидрохлорирования дихлорэтана. Здесь используется дешевое
сырье — этилен, весь хлор входит в состав конечного продукта, в принципе
отсутствуют отходы производства (оговорка «в принципе» существенна: от¬
ходы все же могут появляться за счет побочных процессов). В то же время
есть и существенный недостаток: многостадийность процесса. В связи с этим
представляет интерес новый процесс получения винилхлорида, где сырьем
служит этан (сырье еще более дешевое, чем этилен), а процессы хлорирова¬
ния, окислительного хлорирования и дегиДрохлорирования совмещены в од¬
ном реакторе.
При окислительном хлорировании метана получают четырех¬
хлористый углерод:
СН4 + 4С12 —>• СС14 + 4НС1
СН4 + 4НС1 + 02 —> СС14 + 2НгО
2СН4 + 4С12 + 02 —». 2СС14 + 2НгО
Четыреххлористый углерод — бесцветная жидкость, т. кип.
76,5 °С, df 1,59. Хорошо растворяет каучук, жиры, масла, смо¬
146
лы и др. Негорюч, применяется в огнетушителях для тушения
пожаров в лабораториях, книгохранилищах, однако образую¬
щийся при работе этих огнетушителей фосген делает опасным
их применение в закрытых помещениях.
Полихлорпроизводные используют в качестве инсектицидов
для борьбы с вредителями сельскохозяйственных растений. На¬
пример, гептахлор применяется для борьбы со свекловичным
долгоносиком, гексахлорбутадиен — для борьбы с вредителями
винограда.
CI
Cl CI
гептахлор
С12С=СС1—СС1=СС12
гексахлорбутадиен
§ 63. Фторпроизводные. Фторпроизводные углеводородов по
свойствам и способам получения отличаются от других галоген-
производных.
Получение. Прямое фторирование углеводородов невоз¬
можно, так как в атмосфере фтора они воспламеняются. Ино¬
гда процесс идет со взрывом, реакция сильно экзотермична. Вы¬
деляется много энергии, часть ее расходуется на разрыв угле¬
родных связей, поэтому органическое вещество разрушается.
Несмотря на трудности получения фторпроизводных, разрабо¬
тано много методов их синтеза, так как эти вещества вследствие
их ценных свойств приобрели большое значение в современной
технике. Наиболее важными промышленными способами полу¬
чения фторорганических соединений являются следующие.
1. Каталитическое фторирование углеводородов. Пропускают
смесь паров углеводородов с азотом через слой нагретого фто¬
рида кобальта (III):
300 °с
R—Н + 2CoF3 ► R—F + 2CoF, + HF
Фторид кобальта (III) регенерируют фторированием образо¬
вавшегося фторида кобальта(II):
F2. 250 -с
2CoF2 *■ 2CoFj
Таким путем можно заменить все водородные атомы в угле¬
водородах и получить перфторуглеводороды (или фторуглеро¬
ды). Приставка пер означает, что замещены все водородные ато¬
мы, например CF4, C2F6 и др. (перфторметан, перфторэтан и др.),
2. Усовершенствованным методом фторирования является
«струйное» фторирование. В специальную горелку, имеющую
два отверстия для впуска газов, подают через одно отверстие
фтор, а через другое — органическое соединение, разбавленное
147
инертным газом. Происходит «мягкое» горение, причем образу¬
ются фторзамещенные с тем же углеродным скелетом.
3. Действие фтороводорода на алкены:
CH2=CH2 + HF —► СНз—ch2f
4. Широко применяется метод получения смешанных фтор,
хлоралканов из полихлорпроизводных действием фтороводорода
или фторида сурьмы (III) в присутствии SbCls (реакция
Свартса):
SbCl5
CCI4 + 2HF > CF2C12 + 2НС1
SbCl5
ЗССЬ + 2SbF3 »- 3CF2C12 + 2SbCU
Из образующегося хлорида сурьмы (III) действием фторово¬
дорода вновь регенерируется SbF3.
Если 50 лет тому назад были известны только немногие
фторуглероды, то сейчас фторорганические соединения исчисля¬
ются тысячами и эта область химии быстро развивается.
Свойства фторорганических соединений.
Фторуглероды имеют более низкие температуры кипения и бо¬
лее летучи, чем соответствующие алканы. Например, н-гептан
имеет т. кип. 98 °С, перфторгептан кипит при 82 °С. Атомы
фтора действуют стабилизирующе на другие атомы галогена в
молекуле. Например, в дифтордихлорметане хлор не гидроли¬
зуется. Фторуглероды негорючи, отличаются термостойкостью
(до 400 °С) и высокой химической инертностью к действию
кислот, щелочей. Атомы фтора в перфторсоединениях создают
вокруг атома углерода плотный барьер, защищающий углерод¬
ный скелет от воздействия других реагентов.
Фтор более электроотрицателен, чем кислород, и поэтому
фторуглероды устойчивы к окислителям. Расплавленный метал¬
лический натрий разлагает их только при 400 °С. По химической
устойчивости высокомолекулярные перфторуглероды напоми¬
нают благородные металлы и даже превосходят их.
Отдельные представители и их использо¬
вание. Дифтордихлорметан (фреон-12) CF2CI2 — бесцвет¬
ный газ, т. кип. —30 °С. Широко используется как хладоагент
в холодильных установках глубокого охлаждения и бытовых
холодильниках, в установках для кондиционирования воздуха;
применяется в качестве пропеллента (легко испаряющегося рас¬
творителя) для получения аэрозолей, используемых для распы¬
ления ядохимикатов, лаков, красок и др.
Бромтрифторметан, дибромдифторметан, 1,2-дибромтетра-
фторэтан употребляются как пламягасящие жидкости для за¬
полнения огнетушителей в реактивных самолетах, для тушения
жидких ракетных горючих. Они менее токсичны, чем СС14.
Тетрафторэтилен CF2 = CF2 — бесцветный газ, т. кип.
—78 °С, т. пл. —142 °С. В промышленности его получают из
148
хлороформа и фтороводорода с последующим пиролизом (700—
1000°С) промежуточного дифтор хлор метана:
СНС13 + 2HF —► CHF2C1+2HC1 2CHF2C1 —► CF2=CF2 + 2НС1
Тетрафторэтилен легко полимеризуется в присутствии пер¬
оксидов в политетрафторэтилен (фторопласт-4, тефлон).
Трифторхлорэтилен CF2 = CFC1 — бесцветный газ, т. кип.
—27 °С. Получают дехлорированием трифтортрихлорэтана:
Zn
CF2CI—CFCI2 ► CF2=CFCI + ZnCl2
На его основе готовят полимер — фторопласт-3.
§ 64. Индуктивный эффект. При изучении строения и свойств
органических соединений и отдельных функциональных групп
очень важно учитывать взаимное влияние атомов в молекуле.
Идеи о взаимном влиянии атомов, не только непосредствен¬
но связанных, но и не находящихся в связи, впервые были вы¬
сказаны А. М. Бутлеровым (1861 г.) и развиты- его учеником
В. В. Марковниковым ('1869 г.). Марковниковым был сделан
также вывод, что влияние какого-нибудь элемента на другие
ослабляется по мере удаления элементов друг от друга в общей
цепи связанных атомов. Эти идеи получили свое дальнейшее
развитие в электронной теории (Ингольд и др.).
При образовании о-связи между атомами с различной элек¬
троотрицательностью электронная плотность в молекуле смеща¬
ется в сторону более электроотрицательного атома. Это вызыва¬
ет поляризацию молекулы и образование частичных зарядов
(см. § 12). Так, например, в молекуле метилхлорида электрон¬
ная плотность смещена в сторону хлора, что условно может
быть выражено формулами
б+ в-
НзС->С1 или СН3—С1
Если атом хлора связан с углеродом насыщенной цепи, на¬
пример в пропилхлориде, то происходит дальнейшее смещение
электронной плотности по a-связям углеродных атомов:
Н3с->СН2->.СН2->С1
Атом С-1, имея недостаток электронов, оттягивает их от С-2,
а С-2 от С-3. Однако влияние атома хлора на С-1 больше, чем
влияние С-1 на С-2, а последнее, в свою очередь, больше, чем
влияние С-2 на С-3, и доля частичного заряда (6+), харак¬
теризующая электронную недостаточность, уменьшается:
в"'+ «"+ в'+
CHj—СН2—СН2—С1 б' > 6" > б"'
Смещение электронной плотности по о-связям в молекуле
называют индукционной поляризацией, индуктивным влиянием
или индуктивным эффектом (/-эффект). Индуктивный эффект —
149
эффект, обусловленный стремлением атома или группы атомов
подавать или отнимать электроны. Индуктивный сдвиг элек¬
тронной плотности осуществляется в одном направлении по на¬
сыщенной цепи (смещение а-электронов) и не сопровождается
переходом электронов из оболочки одного атома в оболочку
другого. Индуктивный эффект — влияние быстро затухающее.
Если смещение электронной плотности идет в сторону заме¬
стителя (например, хлора), то такой заместитель называют
электроноакцепторным, а вызываемый им эффект — отрицатель¬
ным индуктивным эффектом (—/-эффект). Если же влияние
группы приводит к повышению электронной плотности у сосед¬
него углеродного атома, т. е. если группа отталкивает электро¬
ны (например, метальный радикал),— такой заместитель назы¬
вают электронодонорным, а эффект положительным индуктив¬
ным эффектом (+/-эффект) (подробнее см. § 130).
Характерным примером проявления —/-эффекта является
влияние галогена в галогензамещенных карбоновых кислотах на
их кислотность (см. § 142).
ГЛАВА 10
ГИДРОКСИЛЬНЫЕ СОЕДИНЕНИЯ
И ИХ ПРОИЗВОДНЫЕ
Продукты замещения водорода в углеводородах гидроксильной
группой (гидроксигруппой) называют спиртами. Общая форму¬
ла спиртов R—ОН.
Гидроксильные производные ароматических углеводородов
называют ароматическими спиртами, если гидроксильная груп¬
па находится в боковой цепи, и фенолами — когда гидроксиль¬
ная группа связана с углеродом ядра.
По характеру углеводородного радикала алифатические
спирты делят на насыщенные и ненасыщенные. В зависимости
от числа гидроксильных групп в молекуле различают:
одноатомные спирты.
СНз—СНгОН
этиловый
спирт
двухатомные спирты:
НОСН2—СН2ОН СНз—снон—СН2ОН
этиленгликоль пропиленгликоль
трехатомные спирты.
НОСН2—СНОН—СН2ОН глицерин
СН2=СН—СН2ОН
аллиловый
спирт
С6Н5—СН2ОН
бензиловый
спирт
150
Углеродный атом способен прочно удерживать только одну
гидроксильную группу; не может гидроксил стоять и при крат¬
ной углеродной связи, хотя из этих правил есть исключения.
ОДНОАТОМНЫЕ СПИРТЫ
§ 65. Изомерия и номенклатура. Изомерия спиртов обу¬
словлена строением радикала (изомерия углеродного скеле¬
та) и положением гидроксигруппы (изомерия положения). По
положению гидроксигруппы в молекуле, в зависимости от того,
с каким атомом углерода она связана (с первичным, вторичным
или третичным), различают первичные RCH2OH, вторичные
R2CHOH и третичные R3COH спирты.
Номенклатура. Широко используется рациональная
номенклатура спиртов. Название производится от соответствую¬
щего углеводородного радикала в сочетании со словом «спирт».
Это удобно в случае несложных радикалов: метиловый спирт
СНзОН, изопропиловый спирт СНз—СНОН—СНз- Более слож¬
ный по строению спирт можно рассматривать как производное
метилового спирта.
По правилам современной заместительной номенклатуры к
названию углеводорода добавляют окончание ол и цифру, обо¬
значающую номер атома углерода, у которого стоит гидроксил.
Углеродный атом, связанный с гидроксилом, обязательно вклю¬
чается в главную цепь и определяет начало нумерации. Приве¬
дем ряд примеров:
СН3 ОН
СНз—СН—СН2ОН (СНз)зСОН свн5—сн—сн2—сн=сн2
изобутиловый спирт грег-бутиловый аллилфенилметанол
(2-метилпропанол-1) спирт, триметил- (1-фенилбутен-3-ол-1)
метанол (2-метил-
пропанол-2)
СН2ОН СНз ОН
СНз—СН2—£н—СН2— СНз СНз—in—сн2—СН—СН2—СНз
(2-этилбутанол-1) изобутилэтилметанол
(5-метилгексанол-З)
§ 66. Общие способы получения. 1. Гидратация алкенов в кис¬
лой среде. Это самый распространенный способ, имеющий
большое промышленное значение, так как позволяет получать
спирты из газов крекинга. Из этилена получают первичный
спирт — этиловый, из пропилена — изопропиловый, из изобути¬
лена — трет-бутиловый спирт. Присоединение идет по правилу
Марковникова:
СН2=СН2 + НОН —► СНз—СН2ОН
СНз—СН=СН2 + НОН —► (СНз)2СН—ОН
(СНз)2С=СН2 + НОН —► (СНз)зС—ОН
151
В промышленности используют способ прямой гидратации,
заключающийся в пропускании смеси алкенов с водяным паром
над фосфатным катализатором (см. § 33).
2. Другим важным промышленным способом является фер¬
ментативное превращение углеводов (см. § 69), где попутно с
главным продуктом — этиловым спиртом — получаются первич¬
ные спирты Сз—Се и другие соединения.
3. Гидролиз галогенпроизводных (подробнее см. § 61):
Способ имеет значение для получения спиртов из углеводо¬
родов. При гидролизе смеси изомерных хлорпентанов (получен¬
ных хлорированием изопентана) образуется смесь изомерных
пентиловых спиртов, из которых может быть выделен перегон¬
кой наиболее высококипящий пентанол-1. Смесь пентиловых
спиртов используют в качестве растворителя.
4. Восстановление карбонильных соединений. Восстановле¬
ние альдегидов приводит к первичным спиртам, а восстановле¬
ние кетонов — к вторичным спиртам:
Синтезы спиртов из альдегидов и кетонов при помощи маг-
нийорганических соединений рассмотрены в § 86.
§ 67. Физические свойства. Ассоциация спиртов. Водородная
связь. Физические свойства. Низшие и средние члены
ряда предельных'одноатомных спиртов (Ci—'Сц) — жидкости,
температуры кипения которых возрастают по мере усложнения
состава. Высшие спирты- от С12 и выше—твердые вещества.
Спирты изостроения кипят при более низкой температуре, чем
спирты нормального строения. Низшие представители имеют ха¬
рактерный алкогольный запах и жгучий вкус, средние С4— Се —
неприятный запах, высшие — лишены запаха. Плотность спир¬
тов меньше единицы, лишь у некоторых ароматических спиртов
она больше единицы. Первые три члена ряда предельных спир¬
тов смешиваются с водой во всех отношениях, по мере услож¬
нения радикала растворимость уменьшается. Высшие спирты,
так же как и углеводороды, практически нерастворимы в воде.
Физические константы некоторых спиртов приведены в табл. 11.
RX + HOH —► ROH + HX
О
уксусный
альдегид
О
этиловый
спирт
ОН
152
Таблица 11. Физические свойства некоторых спиртов
Спирт
Формула
Т. пл.,
•с
Т. кип.,
вС
Относи¬
тельная
плотность,
*5°
Метиловый
сн3он
-98
64,7
0,79
Этиловый
СН3—СН2ОН
-117
78,3
0,79
Пропиловый
сн3—сн,—СН2ОН
-127
97,2
0,80
Изопропиловый
СНз—СИОН— сн3
-88,5
82,2
0,78
Аллиловый
сн2=сн—СН2ОН
96,7
0,85
ДеЦиловый
СНз— (СН2)8—СН2ОН
+6
228
0,80
Циклогексиловый
с6н„он
+25,5
161,5
0,96
Бензиловый
С6Н5-СН2ОН
—
205
1,04
Р-Фенилэтиловый
С6Н5—сн2—СН2ОН
220
1,02
Температуры кипения спиртов выше, чем у других соедине-
ний с близкой молекулярной массой:
Мол. масса
Т. кип., »С
СН3ОН
32
+64,7
СН3-СН3
30
-88,6
CH3NHj
31
-6,7
CH3SH
48
+7,6
CH3CI
50,5
-24,2
В этом проявляется сходство спиртов с водой, температура
кипения которой значительно выше, чем это соответствует ее
молекулярной массе (аналог воды—сероводород H2S — кипит
при —60 °С).
Ассоциация спиртов, водородная связь. Мо¬
лекулы спиртов в твердом и жидком состояниях, как и молеку¬
лы воды, ассоциированы (объединены друг с другом); при этом
существенно увеличивается молекулярная масса и, следователь¬
но, уменьшается летучесть вещества. При переходе спиртов в
парообразное состояние ассоциация нарушается. Явление ассо¬
циации объясняют возникновением между молекулами так на¬
зываемых водородных связей.
Водородная связь — особый вид связи, возникающий за счет
атома водорода таких функциональных групп, как ОН, СООН,
NH2, и электроотрицательных атомов (О, N, F и др.), обладаю¬
щих свободными электронными парами. Происходит электроста¬
тическое взаимодействие частично положительно заряженного
водорода одной функциональной группы и частично отрицатель¬
но заряженного атома X другой функциональной группы. Чем
больше дробный положительный заряд атома водорода, тем
легче образуется водородная связь, «водородный мостик». Обыч¬
но водородную связь обозначают тремя точками:
- Н-Х- Н—Х - Н—X—
163
Для образования водородной связи кроме электроотрица¬
тельности имеет значение размер атома. Так, например, элек¬
троотрицательности атомов азота и хлора близки («3), но
малый размер атома азота и большая концентрация заряда по¬
зволяют ему подойти ближе к ядру атома водорода по сравне¬
нию с атомом хлора, атомный радиус которого больше, чем у
азота. Поэтому хлор проявляет меньшую склонность к образо¬
ванию водородной связи, чем азот. В неполярных молекулах и
углеводородных радикалах атомы водорода не образуют водо¬
родную связь.
В гидроксильной группе ковалентная связь кислород—водо¬
род поляризована, что обусловливает межмолекулярное взаимо¬
действие водорода, несущего частичный положительный заряд,
с кислородом, несущим частичный отрицательный заряд:
б+ б- 6+- б- 5+ б-
О-Н—О"
н
н
н
6+ 6- 6+ б- 6+ 6-
•Н—О-Н—О-Н—о-
I к I
R R R
ассоциация молекул воды ассоциация молекул спирта
Энергия образования водородной связи обычно не превышает
20—30 кДж/моль, в то время как средняя энергия ковалент¬
ной связи колеблется в пределах 330—460 кДж/моль. Расстоя¬
ние между двумя атомами кислорода в двух ассоциированных
молекулах спирта (или воды) равно 0,27 нм, среднее расстоя¬
ние между молекулами неассоциированных веществ в жидком
состоянии равно 0,34 нм. Атом водорода находится ближе к
тому из атомов кислорода, с которым он связан ковалентной
связью:
0,27 им
И
1-0, ГО пи
н—о • • • н—о • • • н—о • • •.
СНз
сн.
сн.
При растворении спиртов в воде у них возникают новые
водородные связи с молекулами воды.
Чем меньше разветвлена углеродная цепь спирта и чем
меньше алкильных групп связано с атомом углерода, при ко¬
тором стоит гидроксильная группа, тем легче осуществляется
ассоциация, поэтому первичные спирты и спирты нормального
строения имеют более высокие температуры кипения. Так, на¬
пример, бутиловые спирты С4Н9ОН имеют следующие темпера¬
туры кипения (в °С):
СН3(СН2)2СН2ОН (СНз)2СНСН2ОН СН3СН2СН.(ОН;СН3 (СНз)зСОН
117,7 107,3 99,5 82,8
При замещении атома водорода гидроксильной группы в
спиртах другими атомами (или группами) водородная связь
исчезает, и это сразу сказывается на температуре кипения
(в °С):
СН3СН2ОН СН3СН2—О—СН2СН3
78,3 34,6
Рассмотренные выше водородные связи относятся к межмо¬
лекулярным связям. Водородная связь может возникать и вну¬
три одной и той же молекулы.
Прочные водородные связи образуют карбоновые кислоты (см. § 94).
Признаки слабых водородных связей из-за недостаточной полярности связи
Н—С обнаружены в хлороформе Н—СС15, синильной кислоте H-CsN, Боль¬
шое значение водородные связк имеют в таких биологически важных при¬
родных веществах, как белки, ферменты, целлюлоза, а также в синтетических
полиамидах.
§ 68. Химические свойства. Функциональная группа спиртов —
гидроксильная группа—обусловливает главные химические
свойства этих соединений. Спирты отличаются большой химиче¬
ской активностью. Вследствие многообразия реакций спиртов и
доступности сырья для их получения спирты являются ценным
исходным материалом для синтеза многих соединений.
6+ й— 6+
Связи углерод—кислород и кислород—водород С—О—Н по¬
ляризованы, причем отрицательным концом диполя является
кислород, как наиболее электроотрицательный элемент. На ато¬
мах углерода и водорода имеются частичные положительные
заряды. [Характеристики связей (длина, энергия, полярность,
поляризуемость) см. табл. 2.] Такой электронный характер гид¬
роксильной группы предопределяет ее склонность к реакциям
гетеролитического типа, в ходе которых может разрываться
связь С—О или О—Н.
Кислотные и основные свойства. Образо¬
вание алкоголятов. Спирты—практически нейтраль¬
ные вещества: они не изменяют окраски индикаторов, не всту¬
пают в реакцию ни с водными растворами щелочей, ни с раз- -
бавленными кислотами. Однако в определенных реакциях спир¬
ты все же проявляют свойства очень слабых кислот и основа¬
ний, т. е. являются амфотерными, подобно воде.
Кислотные свойства у спиртов выражены несколько слабее,
чем у воды. Алкильные группы отталкивают от себя электроны,
снижая тем самым легкость отщепления протона по сравнению
с водой. У третичных спиртов кислотные свойства выражены
наиболее слабо, примером может служить грег-бутиловый
спирт. Если же в радикал спирта ввести электроноакцепторный
атом, то кислотные свойства усиливаются. Примером может
служить перфторпроизводное грег-бутилового спирта, кислотные
155
свойства его заметно выражены (разлагает соли угольной кис¬
лоты) .
н
4 в- б+
Н->С->Оч-Н
4
н
СНз
4 в- б+
СНз">С->0<-Н
t
СН3
CF3
t б- б»
CF3<-C<-0<-H
■I
CF3
метиловый
спирт
грег-бутиловый
спирт
перфтор-трет-
бутиловый спирт
Влияние метильных групп и атомов фтора на кислотность
атома водорода гидроксильной группы является примером ин¬
дуктивного влияния (см. § 64).
При действии на спирты щелочных металлов в безводной
среде вытесняется атом водорода гидроксильной группы и об¬
разуются алкоголяты (лат. alkohol — спирт)
2C2H6OH + 2Na —v 2C2H5ONa+H2
Реакция щелочных металлов со спиртами протекает медлен¬
нее, чем с водой. Спирты могут образовать алкоголяты и дру¬
гих активных металлов (Са, Mg, А1).
Алкоголяты имеют характер солей очень слабой кислоты
(спирта) с основанием. В присутствии воды они гидролизуются:-
C2H50Na + H20 —> С2Н5ОН + NaOH
Алкоголяты щелочных металлов имеют ионный характер:
катионом является щелочной металл, а анионом — алкоксигруп-
па RO- (алкоксид-ион, или, иначе, алкоголят-ион):
СНзО" С2Н5СГ (СНзЪСНСГ (СНз)зССГ
метилат- этилат- изопропилат- трег-бутилат
Спиртовые растворы алкоголятов обладают сильноосновны¬
ми свойствами, поэтому их часто используют в реакциях для
создания сильноосновной среды.
Основные свойства спиртов проявляются в их взаимодейст¬
вии с сильными кислотами. При этом спирты подобно воде да¬
ют соли оксония:
Н—О—Н + НВг —Н30* Вг"
бромид
гидроксония
СНз—О—Н +НВг —> £сн3—О—Н^+ Вг"
бромид метилоксония
Образование простых эфиров. Простые эфиры
R—О—R' получают при взаимодействии алкоголятов (вернее,
156
алкоксид-ионов) с алкилгалогенидами:
R—О—Na + R'I —► R—О—R' + Nal
Это один из важнейших способов получения простых эфиров
и типичная реакция нуклеофильного замещения у галогенпро-
изводных. Более подробно простые эфиры рассмотрены в § 77.
Замещение гидроксильной группы галоге¬
ном. Действие галогеноводородов на спирты — одна из важ¬
нейших реакций нуклеофильного замещения:
ROH + HBr RBr+HaO
Реакция обратима: при действии воды на алкилгалогениды
идет гидролиз с образованием спирта. Для смещения реакции
вправо, в сторону образования галогенпроизводного, нужно
брать избыток галогеноводорода или удалять воду.
Как уже отмечалось, при взаимодействии спирта с сильной
кислотой образуется ион алкоксония, от которого легко отрыва¬
ется молекула воды (см. ниже). Таким образом, роль минераль¬
ной кислоты состоит в передаче протона атому кислорода
спирта:
НВГ :?=*= Н+ + Вг“ С2Н5ОН + Н+ ?=>: [с2Н6<!>—н]
катион
этилоксония
Далее реакция идет как обычное нуклеофильное замещение
по механизму SN2 (преимущественно для первичных спиртов):
СН3—СНг—ОН2 + Вг" <=* СНз—СН2—Вг + Н20
Для третичных спиртов реакция идет по механизму SN1:
первая стадия
+ медленно
(СНз)зС—ОН2 * (СН,)зС+ + Н20
вторая стадия
быстро
(СН3)зС++Вг_ *- (СНз)зС—Вг
Образование сложных эфиров. Для спиртов ха¬
рактерна реакция с кислотами, приводящая к образованию
сложных эфиров (реакция этерификации). Она обратима, так
как сложные эфиры под действием выделяющейся при их об¬
разовании воды гидролизуются.
Сложные эфиры минеральных кислот. При действии на спир¬
ты концентрированной серной кислоты образуются как кислые,
так и средние эфиры серной кислоты. Кислые алкилсульфаты,
или алкилсерные кислоты, получаются при действии H2SO4 на
157
спирт (механизм реакции сходен с получением алкилгалоге-
Нидов):
СН3СН2ОН + HOSO3H [СН3СН2—ОН2 +'OSO3HI
СН3СН2—OSO3H + Н20
этилсерная кислота
Средние эфиры можно готовить разными путями. Так, дей¬
ствие триоксида серы на метанол дает диметилсульфат:
2СН3ОН + SOj —> (CH30)2S02 + н2о
Диметилсульфат — важный реагент, с помощью которого
проводят реакции метилирования, например получают простые
метиловые эфиры (см. § 77).
Действием на спирты концентрированной азотной кислоты
можно получить ее сложные эфиры — нитраты-.
C2H50H + H0N02 с2н5оыо2 + н2о
этилнитрат
Практическое осуществление реакции сильно осложняется
окислительным действием азотной кислоты. Сами нитраты
взрывчаты.
Сложные эфиры карбоновых кислот. Взаимодействие спир¬
тов с карбоновыми кислотами ускоряется присутствием неболь¬
шого количества минеральной кислоты (подробнее см. § 110):
X)
СНз—+HOC2Hs СНз—+н2о
хон Х)С2Н5
этилацетат
Дегидратация спиртов. В определенных условиях
спирты способны терять воду. В качестве водоотнимающих
средств можно применять концентрированную серную или фос¬
форную кислоты. Легче всего дегидратируются третичные спир¬
ты, труднее — первичные. Иллюстрацией к сказанному могут
служить условия дегидратации бутиловых спиртов:
Концентрация Температура,
H2SO4, % °С
«-Бутиловый спирт 75 170
егор-Бутиловый спирт 60 100
гряг-Бутиловый спирт 20 90
Спирты дегидратируются также при пропускании их паров
над нагретым твердым катализатором (силикагель, каолин, ок¬
сид алюминия, оксид тория и др.).
В зависимости от условий реакция может идти по одному
из двух направлений. В жестких условиях (при высокой тем¬
пературе) каждая молекула спирта теряет молекулу воды с об¬
разованием алкена (внутримолекулярная дегидратация)-, если
158
температура несколько ниже, то молекула воды отщепляется от
двух молекул спирта и образуется простой эфир (межмолеку«
лярная дегидратация).
СН2=СН2
этилен
А120з. 350 °С
или H2SO4. 170 °С
сн3—СН2ОН
этиловый
спирт
А120з. 250 °С
или H2S04, 140 'С
•>
(СНЭСН2)20
ДИЭТИЛОРЫЙ
эфир
Окисление спиртов. Спирты окисляются легче, чем
углеводороды, причем в первую очередь окислению подвергает¬
ся углерод, при котором находится гидроксильная группа. Пер¬
вичные и вторичные спирты окисляются легче третичных, что
обусловлено наличием водорода при атоме углерода, связанном
с гидроксильной группой.
В зависимости от природы спирта, а также условий прове¬
дения реакции могут образоваться различные продукты. Так,
первичные спирты дают сначала альдегиды, а затем кислоты с
тем же числом атомов углерода:
Ю1 Ю1
с2н5—сн2он —► с2и5—сг >- с2н5—с;'
Ч)Н
пропиловыи
спирт
СН2ОН
[О]
пропионовый
альдегид
ГЛ^ с^°
W и
пропионовая
кислота
_!£!>. с^°
\=/ \)Н
бензиловый спирт бензальдегид бензойная кислота
Из вторичных спиртов образуются кетоны:
ОН о
СНа—СН—СН3 *■ СНз—С—СН*
изопропиловый ацетон
спирт
Третичные спирты более устойчивы к окислению, но под
действием сильных окислителей возможен разрыв углерод-угле-
родной связи и образование кетонов и кислот, содержащих в
молекуле меньше атомов углерода, чем исходный спирт.
Наиболее подходящим окислителем первичных и вторичных
спиртов до альдегидов и кетонов в лабораторных условиях
является хромовая смесь. Она может быть использована также
для окисления ненасыщенных спиртов с сохранением -двойной
связи, так как лишь медленно действует на кратную связь:
оС—о°
циклогексеп-
2-ол-1
циклогексен-
2-ОН-1
159
При сгорании спирты образуют диоксид углерода и воду:
2СН3ОН + 302 —V 2С02 + 4Н20
Каталитическое дегидрирование спиртов.
Превращение спиртов в альдегиды и кетоны можно осущест¬
вить также дегидрированием — пропусканием паров спиртов
над нагретым металлическим катализатором, например медью
или серебром, при 300 °С без добавления окислителя. Типичная
реакция дегидрирования — превращение этилового спирта в
ацетальдегид:
Си. 300 “С
сн3—сн2он ► снз—с; +н2
Эта реакция эндотермична. Оптимальную температуру мож¬
но поддерживать за счет сожжения части выделяющегося водо¬
рода. В промышленных условиях для этого применяют принцип
непрерывного потока, используя дешевый окислитель — воздух
(при тщательном контроле, чтобы не произошло более глубоко¬
го окисления). Так, окисление метилового спирта дает форм¬
альдегид:
'/г02, о
катализатор ^
СН3—ОН *■ н—с' +Н20
чн
Кислород играет роль акцептора водорода, отщепляющегося
под действием катализатора.
Реакция Чугаева — Церевитинова. Эта реакция
положена в основу аналитического метода определения в спир¬
тах «активного водорода» (водорода гидроксильной группы).
Взаимодействие спиртов с метилмагнийиодидом идет по урав¬
нению:
R—ОН + CH3MgI —► R—OMgl + СН4
Зная навеску исследуемого вещества и измерив объем выде¬
лившегося метана, можно определить число гидроксильных
групп в молекуле спирта.
§ 69. Важнейшие представители. Метиловый, спирт (метанол)
получают в больших количествах в промышленности из моно¬
оксида углерода и водорода:
СО + 2Н2 —► СН3ОН
Эту реакцию долгое время проводили над катализаторами
(оксиды цинка и хрома) при 350—400 °С и давлении до
30 МПа. В настоящее время разработан новый катализатор
(смесь оксидов цинка и меди), который позволяет осуществлять
160
синтез в более мягких условиях (250 °С, 7 МПа). Это дает
большой экономический эффект, позволяет упростить аппара¬
туру.
При сухой перегонке дерева среди других веществ в каче¬
стве побочного продукта образуется и метиловый спирт (на¬
званный из-за этого «древесным»), В настоящее время этот
способ сохранил значение лишь при переработке отходов дре¬
весины.
Главное промышленное применение метилового спирта — по¬
лучение формальдегида. Он используется также для синтеза
метилирующих средств (метилхлорид, диметилсульфат), диме-
тиланилина, для получения уксусной кислоты (см. § 96). При
реакции метанола с изобутиленом получают грег-бутилметило-
вый эфир — антидетонатор, который в отличие от тетраэтил¬
свинца не токсичен.
Синтезы на основе метанола относятся к перспективной об¬
ласти «Ci-химии» — построению сложных органических веществ
из простейших соединений с одним атомом углерода. Сырьем в
производстве метанола служит монооксид углерода, который
в настоящее время получают сжиганием угля, в перспективе же
существует возможность использования диоксида углерода, по¬
лучаемого термическим разложением неорганического сырья —
карбонатных горных пород (известняк, мрамор).
Метиловый спирт — растворитель органических веществ. Ток¬
сичен в любых дозах, в малых количествах вызывает слепоту,
в больших — смерть.
Этиловый спирт (этанол, винный спирт) имеет характерный
алкогольный запах, жгучий вкус. Обычный спирт содержит
около 4,5 % воды. Это нераздельно перегоняющаяся, так назы¬
ваемая азеотропная смесь кипит при 78,15°С.
Для получения абсолютного (безводного) спирта воду мож¬
но удалить химическим путем — кипячением спирта с оксидом
кальция или безводным сульфатом меди. В промышленности
для производства абсолютного спирта к очищенному спирту
(ректификату) добавляют бензол и смесь перегоняют. Сначала
отгоняется тройная азеотропная смесь спирт — вода — бензол
(т. кип. 64,9 °С). При этом полностью удаляется вода. Затем
при 68,3 °С полностью удаляется бензол в виде двойной азео¬
тропной смеси спирт—бензол и после этого при 78,3 °С отго¬
няется чистый спирт.
Абсолютный спирт смешивается во всех отношениях с бен¬
зином (в отличие от 95%-го), что дает возможность использо¬
вать его в составе смесей топлива для двигателей внутреннего
сгорания. Спирт является хорошим растворителем многих орга¬
нических веществ, обладает антисептическими свойствами, кон¬
сервирует белок.
В промышленности этанол получают синтезом или из
природных сложных углеводов (крахмал, целлюлоза). Из
6 Органическая химия
161
синтетических способов в настоящее время наибольшее значение
имеет гидратация этилена (см. § 33).
Сущность самого старого способа производства спирта —
спиртового брожения сахаристых веществ — заключается в том,
что углеводы (в первую очередь, глюкоза С*НцОв) в присут¬
ствии особых микроорганизмов — дрожжей — подвергаются
расщеплению (брожению). Механизм реакции брожения сло¬
жен, суммарно процесс молено выразить уравнением:
С6Н,2Ов —>■ 2СОа 4* 2С2Н5ОН
Сырьем могут служить ягоды, виноград, свекловичная пато¬
ка, которые содержат сахаристые вещества в свободном состоя¬
нии, а также крахмалсодержащие вещества — зерна хлебных
злаков, кукуруза, картофель. Крахмал (СвНщОб)* сначала
(Подвергают «осахариванию» — гидролизу до глюкозы, а затем
брожению. Для получения спирта, применяемого для техниче¬
ских целей, в промышленности используют также целлюлозу
(СбНюОб)*. Для этого отходы лесотехнической промышлен¬
ности (опилки, щепа и др.) гидролизуют 5 %-й серной кислотой
под давлением 1 МПа. В продуктах гидролиза содержится глав¬
ным образом глюкоза. Гидролизат нейтрализуют известью
(серная кислота осаждается в виде CaS04), фильтруют и затем
сбраживают, получая гидролизный спирт:
гидролиз брожение
(СвН10О5)* ► С6Н120„ V С2Н5ОН
Из 1 т сухих опилок можно получить столько же спирта,
сколько из 1 т картофеля или 300 кг зерна.
Этиловый спирт относится к числу наиболее важных техни¬
ческих продуктов. С начала 30-х годов спирт в нашей стране
использовали для производства синтетического каучука по мето¬
ду С. В. Лебедева (см. § 42). В настоящее время производство
синтетического каучука переводится на нефтяное сырье. Спирт
используют как растворитель для экстракции и кристаллизации,
для изготовления лаков и красок, для консервирования анато¬
мических препаратов, как дезинфицирующее средство в медици¬
не, для изготовления лекарственных веществ, в парфюмерной
промышленности для изготовления одеколона и духов. Большие
количества спирта расходуются для синтеза диэтилового эфира,
этилхлорида, сложных эфиров органических кислот, хлорофор¬
ма, йодоформа и др. Как уже отмечалось, спирт можно приме¬
нять и в качестве топлива для двигателей.
Этиловый спирт входит в состав алкогольных напитков. Их
употребление — большое социальное зло. Особенно опасны
спиртные напитки для молодежи. Хотя этиловый спирт менее
ядовит, чем метиловый, но острое отравление этанолом может
вызвать смерть, а постоянное употребление спиртных напитков
разрушает физическое и психическое здоровье.
162
Пропиловые, бутиловые и пентиловые спирты используют
как в свободном виде, так и в виде эфиров уксусной кислоты
в качестве растворителей. Бутиловые эфиры двухосновных и
высших одноосновных кислот используют как пластификаторы.
Сырье для получения этих спиртов поставляет нефтеперераба¬
тывающая промышленность.
Высшие жирные спирты (ВЖС). В начале XIX века фран¬
цузский химик Шеврель впервые выделил высший жирный
спирт С16Н33ОН из жироподобного вещества, имеющегося в
голове кашалота, и назвал его цетиловым (лат. cetus — кит).
Это вещество внешне напоминает жир; т. пл. 50 ®С. В зависи¬
мости от длины радикала и от консистенции различают спир¬
ты: Се — Ci2 — сиропообразные жидкости, т. кип. 157—263 °С;
С13 — С20 — мазеобразные вещества, напоминающие застывшее
сало, т. пл. 31—66 °С, по свойствам они более близки к угле¬
водородам; и С21—С40 — твердые вещества, т. пл. 69—95вС.
До конца 40-х годов основным источником получения ВЖС
оставался китовый жир. В настоящее время ВЖС производят
в большом количестве синтетически. Мировое производство этих
спиртов составляет около 1 млн. т в год. Сырье для их полу¬
чения поставляет нефтеперерабатывающая промышленность.
Один из способов синтеза — восстановление высших жирных
кислот, получаемых окислением парафина. Вторичные ВЖС
можно приготовлять прямым окислением нормальных алканов
кислородом в присутствии борной кислоты. Первичные нормаль¬
ные спирты С6—С20 получают полимеризацией этилена по спо¬
собу Циглера с последующим окислением и гидролизом (см.
§ 138).
Применяют ВЖС в качестве поверхностно-активных ве¬
ществ— антииспарителей (защита водоемов), пенообразовате¬
лей (флотация руд) и др. Они являются хорошими растворите¬
лями. Диэфиры на основе ВЖС — хорошие пластификаторы,
смазочные масла для авиационных двигателей, работающие при
температурах от —50 до +200 °С. В большом количестве ВЖС
используют для получения синтетических моющих средств, ко¬
торые полностью разрушаются при биохимической очистке сточ¬
ных вод и потому не загрязняют водоемы. На основе ВЖС
получают четвертичные аммониевые соли, микродобавки кото¬
рых в воду обеззараживают (стерилизуют) ее, что имеет боль¬
шое значение в пищевой промышленности при переработке мяс¬
ных продуктов, рыбы, фруктов, овощей, промывке аппаратуры
и др.
Циклогексанол СвНцОН — при обычной температуре кри¬
сталлическое вещество (т. пл. 25,5°С) с характерным камфар¬
ным запахом. В промышленности получают гидрированием фе¬
нола (см. § 73).
При окислении циклогексанона в зависимости от условий
получаются кетон циклогексанон или адипиновая кислота,
6*
163
которые используют в промышленности для получения синтети¬
ческих волокон капрон и найлон (см. §§ 87, 190).
окисление
энергичное
окисление
НООС—(СН2),—соон
циклогексанон
адипиновая кислота
Аллиловый спирт СН2 = СН — СН2ОН — представитель нена¬
сыщенных спиртов. В промышленности его получают гидроли¬
зом аллилхлорида. Более новым промышленным способом
получения аллилового спирта является каталитическое окисле¬
ние пропилена в акролеин и восстановление последнего:
02 (Сц, 270 °С) |Н|
СН2=СН—СНз >- сн2=сн—сно —>- сн2=сн—СН2ОН
Аллиловый спирт — промежуточный продукт синтеза глице¬
рина (см. § 70).
Ненасыщенные спирты типа винилового спирта СНг=СНС)Н,
которые содержат гидроксильную группу у атома углерода,
связанного двойной связью, так называемые енолы, в свободном
состоянии неустойчивы (правило Эльтекова). Неустойчивость
винилового спирта связана с тем, что свободные электронные
пары кислорода вступают в сопряжение с л-электронами двой¬
ной связи:
Ь- О. 8+
сн2==сн—о-^-н
Атом водорода гидроксильной группы, приобретая повышен¬
ный положительный заряд, легко отрывается в виде протона,
который затем присоединяется к имеющему избыток электрон¬
ной плотности атому углерода СН2-группы.
В момент образования такие спирты изомеризуются в кар¬
бонильные соединения — альдегиды и кетоны:
Н Н
R—СН=С—ОН —► R—СН2—С=0
ОН О
I II
R—С=СН—R —» R—С—СН2—R'
Простые и сложные эфиры этих спиртов устойчивы. Их ис¬
пользуют в промышленности. Простые эфиры получают при
реакции спиртов с ацетиленом (см. § 77), сложные эфиры —
при реакции карбоновых кислот с ацетиленом (см. § 110).
Бензиловый спирт СбН5—СН2ОН — простейший ароматиче¬
ский спирт — в свободном состоянии и в виде сложных эфиров
содержится в эфирных маслах жасмина и других цветов. При¬
меняется в парфюмерии. Получают его гидролизом бензилхло-
рида, а также восстановлением бензальдегида.
164
$-Фенилэтиловый спирт СеН5—СН2—СН2ОН является глав¬
ной составной частью розового масла. Используется в парфю¬
мерии. В промышленности его синтезируют из этиленоксида и
бензола:
О
/ \ А1С13
С6Нв + Н2С СН2 >- С„Н6—СН2—СН2ОН
МНОГОАТОМНЫЕ СПИРТЫ
§ 70. Номенклатура, физические свойства, отдельные предста¬
вители. Двухатомные спирты называют гликолями или диолами,
трехатомные—глицеринами или триолами. Положение гидрок¬
сильных групп указывают цифрами:
ОН он
НОСН2—СН2ОН СНз—сн—СН2ОН носн2—сн—СН2ОН
этиленгликоль
(этандиол-1,2)
пропиленгликоль
(пропандиол-1,2)
глицерин
(пропантриол-1,2,3)
НОСН2—СН2—СН2ОН НОСН2—СН2—СН2—СН2ОН
триметиленгликоль тетраметиленгликоль
(пропандиол-1,3) (бутандиол-1,4)
Многоатомные спирты хорошо растворимы в воде, плохо —
в органических растворителях, имеют высокие температуры
кипения. Могут быть получены теми же способами, что и-одно-
атомные спирты.
Этиленгликоль. Наиболее важным из диолов является эти¬
ленгликоль. Это бесцветная жидкость, т. кип. 197 °C,df 1,11
смешивается с водой во всех отношениях. Его производят в
промышленности из этилена разными путями:
02» Н2О
сн2=сн2 —
НОСН2—СН2ОН
С12 н2о
► С1СН2—СИ2С1 >- НОСН2—СН2ОН
о
нею 02(Ag) / \
С1СН2—сн2он •* сн2=сн2 ■—*■ Н2С—сн2
I н2о н2о
I v НОСН2—СН2ОН -е
Перспективным является способ получения этиленгликоля
из синтез-газа (250 °С, 200 МПа, катализатор):
2СО + ЗН2 —► НОСН2—СН2ОН
Химические свойства этиленгликоля и вообще многоатомных
спиртов напоминают свойства одноатомных спиртов. В зависи¬
мости от того, участвует ли в реакции одна гидроксильная
группа или обе, получаются два ряда соединений.
163
Гликоля так же, как и одноатомные спирты, не изменяют
окраски индикаторов, но этиленгликоль обладает несколько бо¬
лее кислотным характером, чем этиловый спирт. Увеличение
числа гидроксильных групп усиливает кислотный характер.
Гликоляты образуются под действием не только щелочных ме¬
таллов, но и гидроксидов тяжелых металлов. Например, при
реакции этиленгликоля со свежеосажденным гидроксидом меди
получается гликолят меди (синего цвета). Причиной легкого
образования гликолятов тяжелых металлов является не только
повышенная кислотность этиленгликоля, но и повышенная ус¬
тойчивость этих гликолятов, имеющих характер внутрикомп-
лексных соединений.
СН2—ОН
21 + Си(ОН)2
сн2—ОН
н
св2—о А-сн2
,Си^ | + 2НаО
СН2—О О—CIIj
н
Гидроксильные группы в этиленгликоле могут замещаться
на атомы галогена. При действии хлоро- или бромоводорода
замещается одна гидроксильная группа и получается галоген-
гидрин:
HCI
НОСН2—СН2ОН ► С1СН2—СН2ОН
этиленхлоргидрин
Вторая гидроксильная группа замещается труднее — под
действием РСЦ или SOCb-
Этиленгликоль образует простые и сложные эфиры. Некото¬
рые из них нашли промышленное применение как растворители,
например метилцеллозольв СН3ОСН2СН2ОН (жидкость с т. кип.
125 °С).
Водный 50 %-й раствор этиленгликоля используют в качестве
незамерзающей жидкости (антифриз) для охлаждения автомо¬
бильных двигателей в зимних условиях (температура застыва¬
ния —34 °С). Этиленгликоль применяют для синтеза полимер¬
ных материалов и для других синтезов.
Глицерин (пропантриол-1,2,3) — важнейший представитель
многоатомных спиртов. В обычных условиях это вязкая гигро¬
скопическая жидкость сладкого вкуса (т. пл. 17 °С, т. кип.
290 °С, df 1,26). Смешивается с водой во всех отношениях.
Глицерин входит в состав жиров и других веществ, образующих
животные ткани.
Наиболее старый способ производства глицерина в промыш¬
ленности— гидролиз жиров и масел (см. § 113). В более но¬
вом и важном промышленном способе исходят из пропилена,
166
являющегося продуктом нефтепереработки. Пути превращения
пропилена в глицерин различны.
При термическом хлорировании пропилена получают аллил-
хлорид, при гидролизе которого образуется аллиловый спирт.
Аллиловый спирт под действием хлорноватистой кислоты
(СЬ + HjO) переводят в З-хлорпропандиол-1,2, а последний
гидролизуют в глицерин:
сн, С1г, СН2С1 „г0, СН2ОН сн*он СН.ОН
500 °С I NaOH I НОС1 I известь \
сн *• сн *• сн ► снон *- снон
II II II I I
СН2 СН2 СН* CHjCl CHjOH
Более перспективный способ—окисление пропилена кисло¬
родом воздуха (катализатор Си, 3700С) до акролеина, восста¬
новлением которого получают аллиловый спирт. Под действием
пероксида водорода аллиловый спирт превращается в глицид-
ный спирт (глицидол), а последний гидролизуется в глицерин:
СН3
СНО
СН2ОН
СН2ОН
СН2ОН
1
о2 I
[HI 1 Н202
1
н2о I
СН
1
—► сн
-II
*■ СН >
II
НСЧ
[О
► СНОН
1 л
СН2
СН2
СН2
н*6/
СН2ОН
При действии пероксида водорода непосредственно на акро¬
леин получается глицериновый альдегид, который каталитиче¬
ским гидрированием переводят в глицерин:
Н202 Hi/Nl
сн2=сн—сно >- носн2—снон—сно ►
—». носн2—снон—сн2он
Перспективен способ получения глицерина из синтез-газа
при нагревании под давлением в присутствии катализатора:
ЗСО + 4Н2 —► носн2—снон—СН2ОН
Получепие глицерина гидролизом пищевых жиров постепен¬
но теряет значение.
По свойствам глицерин подобен этиленгликолю: легко дает
с гидроксидами тяжелых металлов глицераты, гидроксильные
группы обмениваются на галоген, образует простые и сложные
эфиры, например эфир глицерина и азотной кислоты — трини-
трат глицерина, неточно называемый «нитроглицерином»:
ОН ono2
| hono2. h2so4 I
HOCH2—СН—СН2ОН ■> o2no—СН2—СН—СН2—ono2
При обычной температуре это жидкость (т. пл. 13 °С), чув¬
ствительная к удару, является взрывчатым веществом.
167
Разложение нитроглицерина — сильно экзотермическая ре¬
акция (3000 °С), при этом выделяется большое количество
газов:
Для повышения стабильности нитроглицерина и обеспечения
безопасности в обращении нитроглицерином пропитывают по¬
ристую массу (инфузорную землю, древесную муку и др.) и
готовят так называемые динамиты. Динамиты используют в во¬
енном и горном деле.
При конденсации глицерина и двухосновной фталевой кис¬
лоты образуются высокомолекулярные соединения — глифтале-
вые смолы (см. § 191).
Глицерин гигроскопичен, поэтому его используют в качестве
увлажняющего средства при изготовлении фармацевтических
препаратов и косметических средств, а также в кожевенной и
текстильной промышленности. В пищевой промышленности его
применяют для подслащивания ликеров и др.
ФЕНОЛЫ И НАФТОЛЫ
§ 71. Фенолы. Изомерия и физические свойства. Фенолы —
гидроксилсодержащие ароматические соединения, в молекулах
которых гидроксил связан непосредственно с углеродным ато¬
мом бензольного ядра (в отличие от ароматических спиртов).
Общая формула фенолов Аг—ОН, где Аг — ароматический ра¬
дикал. По числу гидроксильных групп различают одно-, двух- и
многоатомные фенолы:
ОН ОН ОН
Изомерия фенолов обусловлена положением гидрок¬
сильной группы. Для двухатомных фенолов, как и для других
двузамещенных ароматических соединений, известны три изоме¬
ра — орто-, мета- и пара-:
фенол о-крезол
(гидрокси- (2-гидрокси-
бензол) толуол)
гидрохинон
(1,4-дигидрокси-
бензол)
пирогаллол
(1,2,3-три-
гидроксибензол)
пирокатехин
(1.2-дигидроксИ'
бензол)
(т. пл. 105 °С)
резорцин
(1,3-дигидрокси-
бензол)
(т. пл 110°С)
гидрохинон
(1,4-дигидрокси-
бензол)
(т. пл. 171 °С)
168
Физические свойства. Фенолы — кристаллические
вещества или высококипящие жидкости, обладающие сильным
характерным запахом. Плотность фенолов около единицы. Боль¬
шая кислотность атома водорода гидроксильной группы по срав¬
нению со спиртами отражается на физических свойствах фено¬
лов. Они образуют более прочные водородные связи, имеют
более высокие температуры кипения, большую растворимость в
воде по сравнению с соответствующими циклическими спирта¬
ми, например:
Фенол
Циклогексанол
Т. пл„ «С
41
25,5
181
161
Растворимость
(в Н*0, 20 °С), г/100 г
9,0
3,6
§ 72. Промышленные способы получения фенолов. Большие ко¬
личества фенола и крезолов (метилфенолов) содержатся в
среднем масле каменноугольной смолы и в древесном дегте. Из
смолы их извлекают обработкой раствором гидроксида натрия
в виде фенолятов, затем осаждают кислотами (см. § 73).
В синтетических методах для получения самого фенола исход¬
ным сырьем является главным образом бензол или кумол.
Один из наиболее старых методов получения фенолов за¬
ключается в сплавлении натриевых солей бензолсульфокислот
со щелочами. Образовавшиеся феноляты разлагают минераль¬
ными кислотами. В настоящее время метод используют главным
образом для получения многоатомных фенолов:
S03Na ONa ОН
-S03Na
NaOH,
300 °с
►
—Na2S03
о
ONa
H2So4
/ \
\=/
ОН
резорцин
Наиболее важным промышленным способом получения про¬
стейшего из фенолов является кумольный способ. Исходным
веществом служит кумол (изопропилбензол), получаемый алки¬
лированием бензола пропиленом (см. § 47). Окисление кумо-
ла проводят кислородом воздуха, а образовавшийся гидропер¬
оксид кумола разлагают серной кислотой. Способ экономически
выгоден, так как одновременно с фенолом получается и другой
важный продукт—ацетон:
СУ
СН(СНзЬ
о2
С(СНзЬ
ООН
H2S04
У
с
он
+ СН;
о
.—А—'
сн3
Этот метод был впервые освоен в СССР (1949 г.). Из ди¬
изопропилбензолов аналогично могут быть получены резорцин
и гидрохинон.
Стадия разложения пероксида интересна тем, что при этом
происходит перегруппировка, во время которой группа ОН пе¬
реходит к бензольному ядру.
169
Применяют в промышленности и старый способ Рашита —
окислительное хлорирование бензола смесью хлороводорода и
воздуха с последующим гидролизом образующегося хлорбен¬
зола:
СвН, + НС1 + У,О, f С6Н5С1 ——С»Н5ОН
Метод выгоден тем, что хлороводород постоянно регенериру¬
ется, гидролиз не требует расхода щелочи.
В лаборатории фенолы можно получать гидролизом солей
диазония (см. § 132).
§ 73. Химические свойства фенолов. По строению фенолы ана¬
логичны спиртам. Однако гидроксильная группа и бензольное
ядро оказывают сильное влияние друг на друга, что обусловли¬
вает специфичность свойств фенолов, отличающих их от арома¬
тических спиртов.
Для характеристики важнейших свойств фенолов рассмот¬
рим отдельно реакции фенольной гидроксильной группы и бен¬
зольного ядра.
Для фенольной гидроксильной группы характерны следую¬
щие реакции.
1. Кислотные свойства. Образование фено¬
лятов. Фенолы, как и спирты, не изменяют окраски индика¬
торов, но кислотные свойства у них выражены хотя и слабо, но
сильнее, чем у спиртов и воды, например: С6Н5ОН > НгО >
> С2Н5ОН.
Более сильный кислотный характер фенолов по сравнению
со спиртами объясняется влиянием бензольного ядра. Неподе-
ленная пара электронов атома кислорода вступает в сопряже¬
ние с я-электронами бензольного ядра. В результате электрон¬
ная плотность кислородного атома перемещается частично на
связь кислород—углерод, увеличивая при этом электронную
плотность в орто- и пара-положениях в бензольном ядре. Элек¬
тронная пара связи кислород—водород сильнее притягивается
к атому кислорода, способствуя тем самым созданию большего
положительного заряда на атоме водорода гидроксильной груп¬
пы и, следовательно, отщеплению этого водорода в виде про¬
тона:
В отличие от спиртов фенолы реагируют с водными раство¬
рами щелочей, образуя феноляты (поэтому простейший фенол
был назван «карболовой кислотой»). Феноляты подобно всем
солям слабых кислот и сильных оснований легко гидролизуют¬
200 -с
Н20, 500 -С,
соли меди
170
ся, водные растворы их имеют еильнощелочную реакцию:
Аг—OH + NaOH Аг— ONa + Н20
фенолят
натрия
Под действием сильных кислот гидролиз протекает необра¬
тимо:
C6H6ONa + H2SO4 - * С6Н5ОН + NaHS04
По сравнению с кислотами кислотные свойства у фенолов
выражены чрезвычайно слабо. Так, фенол значительно слабее
угольной кислоты (примерно в 3000 раз), поэтому фенолы не¬
растворимы в водных растворах карбонатов щелочных металлов
и не разлагают солей угольной кислоты. Наоборот, при дейст¬
вии на фенолят угольной кислотой образуется свободный фенол:
C6H5QNa + С02 + Н20 —► СсН5ОН + NaHCOj
Фенолят-ион СбН50~—более слабое основание, чем
алкоксид-ион, так как в нем ионный заряд распределяется на
всю сопряженную систему.
Кислотные свойства фенола усиливаются под влиянием свя¬
занных с ядром электроноакцепторных групп, например нитро¬
группы, галогенов и др. (см. § 123).
2. Образование простых эфиров. Простые эфи¬
ры фенолов образуются аналогично простым эфирам спиртов:
Аг—ONa + RX —► Аг—О—R-f NaX
3. Образование сложных эфиров. В отличие от
спиртов фенолы не образуют сложных эфиров ври непосред¬
ственном действии кислот. Обычно их получают при действии на
фенолы хлорангидридами или ангидридами кислот:
О О
II II
С.Н5ОВ+Cl—С—СН3 —V С6Н5—О—С—СНа + НС1
хлорангидрид фенилацетат
уксусной кислоты
4. Восстановление. При перегонке фенолов с цинко¬
вой пылью гидроксильная группа восстанавливается и образу¬
ется соответствующий углеводород:
С6Н6ОН + Zn —>- С6Н6 + ZnO
5. Обмен гидроксильной группы на галоген.
Эта реакция практически не идет, так как связь углерод—кис¬
лород в фенолах прочнее, чем в спиртах.
Реакции бензольного ядра. 1. Реакции электро¬
фильного замещения. Гидроксильная группа относится
к числу наиболее сильных орто-гаарв-ориентантов (см. § 48).
Реакции электрофильного замещения водорода в бензольном
171
ядре для фенолов протекают значительно легче и в более мяг¬
ких условиях, чем для бензола. Так, при галогенировании фе¬
нолов действием бромной воды происходит замещение трех
атомов водорода в ядре на бром, образовавшийся трибромфе-
нол выпадает в виде белого осадка (качественная реакция на
фенолы); при действии хлора на фенол образуются о- и п-хлор-
фенолы:
ОН ОН ОН ОН
2,4,6-трибром-
фенол
о-хлор- п-хлор-
фенол фенол
Реакция с бромной водой обычно не завершается на стадии
образования 2,4,6-трибромфенола: с избытком брома образует¬
ся соединение совсем иного строения.
Нитрование фенола идет в боле мягких условиях, чем ни¬
трование бензола. При действии разбавленной азотной кислоты
при комнатной температуре образуется смесь о- и п-нитрофено-
лов; конечным продуктом нитрования является 2,4,6-тринитро-
фенол, называемый также пикриновой кислотой:
ОН
V
no2
пикриновая о-нитро- и-нитро-
кислота фенол фенол
Сульфирование фенола при комнатной температуре дает
о-фенолсульфокислоту (2-гидроксибензолсульфокислоту), а при
более высокой температуре — n-фенолсульфокислоту (4-гидрок-
сибензолсульфокислоту):
H03S
H2SO4.
100 “С
.ОН H2SO4,
20 «С
ОН
•S03H
Алкилирование по Фриделю — Крафтсу, а также алкенами и
спиртами в присутствии хлорида алюминия, серной кислоты и
других веществ в зависимости от условий может давать как
172
пара-, так и орто-изомеры. Например, алкилирование изобутиле¬
ном в присутствии серной кислоты дает /гора-изомер — 4-трет-
бутилфенол:
СН3
—ОН + сн2=с-
-сн3
H2SO<
►
(СНз)зС
ОН
2. Конденсация с альдегидами может протекать как в
кислой, так и в щелочной средах за счет водородов орто- и пара-
положений. Одной из практически важных реакций является
конденсация фенола с формальдегидом с образованием гидрок-
симетилфенолов:
Н0Н2О
он
2СН2=0
СН2ОН
2,4-ди(гидроксиметил)фенол
НОН2С-
СН2=о
*-
ОН
СН2ОН
2-гидроксиметил-
фенол
СН2ОН
1 ,0„
СН2ОН
2,4,6-три(гидрокси-
метил)-фенол
При дальнейшей конденсации образуются высокомолекуляр¬
ные, соединения— фенолоформальдегидные смолы (см. § 192).
3. Гидрирование. Фенолы хорошо гидрируются в при¬
сутствии никелевого катализатора, давая соответствующие цик-
логексанолы:
Н2/Ы1, 250 «с
ОН *■
Реакция имеет важное промышленное значение (см. § 69).
4. Окисление фенолов. Присутствие в фенолах гид¬
роксильной группы, связанной с ядром, резко изменяет отно¬
шение ароматического ядра к окислителям. Фенолы окисляются
легко, причем образуется смесь разных продуктов окисления.
Фенол дает с хлоридом железа(III) фиолетовое окрашива¬
ние, в результате образования комплексного иона, крезолы
СНзСбН4ОН — синее окрашивание, многоатомные фенолы —
окрашивание различных оттенков. Эту реакцию используют для
качественного обнаружения фенолов.
§ 74. Отдельные представители фенолов. Фенол (карболовая
кислота) СвНбОН — бесцветное кристаллическое вещество со
специфическим запахом. На воздухе окисляется, сначала розо¬
веет, затем темнеет; кристаллы расплываются вследствие гидро-
скопичности. Мало растворим в холодной воде, хорошо —
173
в горячей. Смешивается во всех отношениях со спиртом, эфиром,
бензолом. Обладает сильными антисептическими свойствами.
Его водный раствор применяется для дезинфекции. Токсичен,
вызывает ожоги кожи.
Самый распространенный и экономически выгодный способ
получения фенола — кумольный; так получают около 85 % об¬
щего количества фенола. Более новые способы получения фено¬
ла в промышленности, например так называемый циклогексано¬
вый метод, связаны с использованием бензола:
О <5) СУ~он О~он
Другой перспективный способ — окислительная конденсация
бензола и уксусной кислоты в присутствии хлорида палладия:
О
+HOOCCHs
02, PdCl2
-н2о
ОСОСНа
н2о
ОН
Фенол — один из важнейших полупродуктов современной
промышленности органического синтеза; его используют для по¬
лучения фенолоформальдегидных смол, циклогексанола и син¬
тетических волокон на его основе {капрон, найлон), в синтезе
красителей, лекарственных веществ, салициловой кислоты, гер¬
бицидов, анилина, термостойких смол, поликарбонатов, эпоксид¬
ных смол и др.
Продукт исчерпывающего хлорирования фенола — пента-
хлорфенол CeCUOH, обладает сильным бактерицидным дейст¬
вием, применяется для консервации древесины; пентахлорфено-
лят натрия используют как гербицид.
При конденсации фенола с ацетоном в присутствии кислот¬
ных катализаторов образуется дифенилолпропан [2,2-ди(4-гидр-
оксифенил)пропан], используемый для синтеза полимеров, анти¬
оксидантов, гербицидов:
СН3
н+
2С6Н8ОН + СНзСОСН, ——►
—НдС)
но—о—
сн,
Крезолы (о-, м-, п-метилфенолы) СНзСбН4ОН получают из
каменноугольной смолы в виде смеси, носящей название «сырой
крезол». Используют их для получения синтетических смол,
красителей, ядохимикатов. Водные эмульсии крезолов с раство¬
ром мыла (лизол, креолин) — антисептики, применяемые в вете¬
ринарии.
Двух- и трехатомные фенолы (дигидрокси- и тригидрокси-
бензолы) — кристаллические вещества, растворимые в воде луч-
174
ше, чем одноатомные фенолы. Ниже приведены представители
тригидроксибензолов (дигидроксибензолы см.§ 71):
он
он
Ч
он
но
■он
он
пирогаллол
(1,2,3-тригидр-
оксибензол)
(т. пл. 133 °С)
гидроксигидрохинон
(1,2,4-триги дрокси-
бензол)
(т. пл. 140 °С)
флороглюцин
(1,3,5-тригидрокси*
бевзол)
(т. пл. 219 °С)
В промышленности двух- н трехатомные фенолы получают
по описанному ранее способу щелочным плавлением натриевых
солей сульфокислот, а также специальными методами, напри¬
мер:
галловая кислота пирогаллол
Производные многоатомных фенолов распространены в при¬
роде (красящие вещества растений и дубильные вещества).
Многоатомные фенолы используют для синтеза красителей, ле¬
карственных веществ, для получения полимеров и др.
Увеличение числа гидроксильных групп в ядре повышает его
активность в реакциях электрофильного замещения. Наличие
нескольких гидроксильных групп в ядре, особенно в орто- и
пара-положениях друг к другу, вызывает особую чувствитель¬
ность к действию окислителей. Такие фенолы чрезвычайно легко
окисляются и являются хорошими восстановителями. Особо сле¬
дует отметить сильные восстановительные свойства гидрохинона
(1,4-дигидроксибензол) и пирогаллола (1,2,3-тригидроксибен-
зол), которые используют в качестве проявителей в фотографии.
Гидрохинон применяют также в качестве антиоксиданта — ве¬
щества, предотвращающего окислительные процессы. Многие
алкилфенолы служат ингибиторами полимеризации и окисления.
Такими свойствами обладает, например, ионол (2,6-ди-трет-
175
бутил-4-метилфенол), который
дикалы:
/С(СНз)з
СНз—/ \—ОН +R.
способен «улавливать» ра-
СНз
,С(СНз),
./ \—о • + RH
'С(СН3)з хС(СН3)з
Это свойство ионола используют для предотвращения окисле¬
ния полимеров, масел: окисление идет с промежуточным обра¬
зованием радикальных частиц (см. § 82), ионол улавливает их
и обрывает цепь окислительных процессов.
Пирогаллол в щелочном растворе очень легко поглощает
молекулярный кислород (из воздуха или других газовых сме¬
сей), что используют при анализе газовых смесей для количе¬
ственного определения кислорода.
§ 75. Нафтолы. Из гидроксипроизводных нафталина интерес
представляют а- и р-нафтолы:
ОН
а-нафтол р-нафтол
(т. пл. 96 “С, (т. пл. 122 °С,
т. кип. 288 °С т. кип. 294 °С)
Они обладают свойствами фенолов, растворяются в щелочах,
дают окрашивание с хлоридом железа(III) (а-нафтол — фио¬
летовое, р-нафтол — зеленое).
В промышленности их получают щелочным плавлением нат¬
риевых солей соответствующих нафталинсульфокислот:
fS-нафта ли нсульфо- (1-нафтол
кислота
Нафтолы и их производные, особенно сульфопроизводные,
широко используют в производстве азокрасителей.
ПРОСТЫЕ ЭФИРЫ. ЭПОКСИДЫ
§ 76. Простые эфиры. Номенклатура, изомерия, физические
свойства. Простые эфиры — это соединения, в молекулах кото¬
рых два углеводородных радикала связаны атомом кислорода:
R—О—R'. Радикалы могут быть одинаковые и разные (если
радикалы разные, то эфиры называют смешанными), с откры¬
той цепью, циклические, ароматические.
176
Названия простых эфиров обычно составляют по назва¬
ниям радикалов. Так, СН3—О—С2Н5 — метплэтиловый эфир,
СН2=СН—О—С2Н5 — винилэтиловый эфир. Если радикалы
одинаковые, то перед названием ставится приставка ди, напри¬
мер: СеН5—О—СбНв — дифениловый эфир (иногда приставку
опускают). Употребляют также названия, производимые от угле¬
водородов, в которых один атом водорода заменен на алкокси-
группу:
СНз—О—СН2—СНз С6Н5—О—СНз
метоксиэтан метоксибензол
Используют и тривиальные названия: метиловый эфир фено¬
ла СвН5ОСНз носит название анизол, а его этиловый эфир
СвН5ОСгН5 — фенетол.
Изомерия простых эфиров обусловлена строением ради¬
калов и их составом:
СНз
СНз—О—СН2—СН2—СНз СНз—о—СН—СНз
метилпропиловый
эфир
СНз—СН2—О—СН2—СНз
изопропилметиловый
эфир
СН,—О—СН2—СН2—СН»
диэтиловый эфир
метилпропиловый эфир
Простые насыщенные эфиры изомерны насыщенным одно¬
атомным спиртам, например СН3—О—СН3 и СН3—СН2—ОН.
Они имеют общую формулу СлНгл+гО.
Физические свойства. Простые эфиры имеют более
низкие температуры кипения, чем изомерные им спирты или
фенолы, например:
Т. кип., «С Т. кип., °С
СНз—О—СНз —23,7 СН3СН2ОН 78,3
С6Н5—О—СНз 155 СН3С6Н4ОН 191
Объясняется это тем, что простые эфиры не образуют водо¬
родные связи и не ассоциируют, как молекулы спирта. В отли¬
чие от низших спиртов эфиры не смешиваются с водой во всех
отношениях, но первые представители ряда частично раствори¬
мы в воде (например, растворимость в воде диэтилового эфира
7%). Эфиры—хорошие растворители органических веществ.
Плотность эфиров меньше единицы, они имеют приятный запах.
§ 77. Способы получения простых эфиров. 1. Дегидратация
спиртов — наиболее важный промышленный способ получения
эфиров (см. § 68):
H2SO4, нагревание
2R—ОН -> R—О—R + Н20
Реакция ограничена получением симметричных эфиров, так
как если взять два различных спирта, то получится трудно раз¬
деляемая смесь трех эфиров.
177
2. Наиболее важным лабораторным способом является син¬
тез Вильямсона — действие алкоголятов на алкилгалогениды.
Способ можно использовать для получения как симметричных,
так и несимметричных простых эфиров:
C2H5ONa + CH3I —V С2Н5—О—СН3 + Nal
C6HsONa + C2H5Br —у С«Н6—О—C2H5 + NaBr
Вместо алкилгалогенидов можно применять диалкилсульфа-
ты, особенно для получения эфиров фенолов:
С„Н5(Жа + (CH30)2S02 —у С*Н5ОСН3 + CH30S03Na
3. Присоединение спиртов к алкенам. Реакция идет в кислой
среде в присутствии трифторида бора. Направление присоеди¬
нения соответствует правилу Марковникова:
BF3
СН3—СН=СН2 + НОС2Н5 *■ (СН3)2СН—о—С2Н5
нзопропилэтиловый
эфир
4. Простые алкилвиниловые эфиры получают при нагрева¬
нии (150 °С) ацетилена со спиртами и твердой щелочью под
давлением 1,5—1,6 МПа (реакция А. Е. Фаворского и М. Ф. Шо-
стаковского, 1943 г.):
СН=СН + НОСН2-СН, —У сн2=сн—о—С2Н5
винилэтиловый
эфир
Реакцию можно проводить и в кислой среде в присутствии
солей ртути(II) (А. Е. Фаворский, 1887 г.) аналогично присо¬
единению воды.
§ 78. Химические свойства простых эфиров. Простые эфиры
значительно уступают спиртам по реакционной способности.
Связь кислород — углерод весьма прочна и расщепляется с
трудом.
1. Характерной реакцией для простых эфиров является раз¬
ложение их при нагревании с иодоводородом (иод связывается
с низшим радикалом):
СвН30—СН3 + Ш —у СвН6ОН + СН31
Ни водные растворы щелочей, ни разбавленные кислоты
разложения не вызывают.
2. Сильные кислоты дают с эфирами продукты присоедине¬
ния:
(С2Н5)20 + НС1 —-У (С2Н5)2О.НС1
Этим соединениям обычно придают строение солей оксония,
хотя отсутствие электропроводности противоречит такой фор¬
178
муле и заставляет считать, что присоединение идет за счет во¬
дородной связи:
[(С,Н1),0 : Н]+ СГ (C,H*)S0-H-C1
3. Многие эфиры легко подвергаются самоокислению (ауто-
окисление) при хранении в контакте с воздухом, особенно на
свету. Процесс идет медленно при участии молекулярного кис¬
лорода. При этом образуются гидропероксиды, например:
ООН
02 I
СНз—СН2—О—СН2—СНа >- СН3—СН—О—СН2—СН3
Пероксиды взрывоопасны, особенно при нагревании; они
имеют свойства сильных окислителей (окрашивают иодокрах-
мальную бумажку). При перегонке эфира нелетучий пероксид
накапливается в остатке и может быть причиной взрывов. По¬
этому перед перегонкой для удаления пероксидных соединений
эфир промывают гидроксидом натрия или обрабатывают восста¬
новителями (Na2S03, FeS04 и др.). f
4. Простые виниловые эфиры могут полимеризоваться и яв¬
ляются важными полупродуктами для производства высокомо¬
лекулярных соединений.
5. В отличие от насыщенных простых эфиров виниловые
эфиры легко гидролизуются в кислой среде, причем вместо не¬
устойчивого винилового спирта получается ацетальдегид (см.
§69):
н2о (н+)
R—О—СН=СН2 *- R—ОН + СН3—СНО
Так как эфиры являются хорошими растворителями органи¬
ческих веществ и химически стойки, их часто используют в ка¬
честве среды при проведении различных реакций; особенно ши¬
роко применяют диэтиловый эфир и циклические эфиры — те-
трагидрофуран, диоксан. Высококипящие моноалкиловые эфиры
этиленгликоля — целлозольвы и диэтиленгликоля — карбитолы
используют для растворения эфиров целлюлозы:
СН3ОСН2—сн2он С4Н,—ОСН2—СН20—СН2—СН2ОН
метилцеллозольв бутилкарбитол
(т. кип. 125 °С) (т. кип. 231 °С)
§ 79. Наиболее важные представители простых эфиров. Диэти¬
ловый эфир (этиловый эфир, просто «эфир», серный эфир) —
жидкость с характерным запахом; т. кип. 34,6 °С, т. пл. —
116,3 °С, df 0,714. Легко воспламеняется, его пары в смеси с
воздухом взрывоопасны, способен к аутоокислению. Требует
тщательного хранения и осторожного обращения. Используется
как растворитель многих органических веществ. Эфир обладает
179
анестезирующим действием, его применяли в медицине при хи¬
рургических операциях; в настоящее время вытеснен другими
препаратами.
Простые эфиры фенолов и нафтолов имеют своеобразный
запах, благодаря чему используются в парфюмерии. Наиболее
важными являются метиловый (анизол) и этиловый (фенетол)
эфиры фенола, которые применяют в качестве растворителей и
в синтезе красителей, лекарственных веществ и др. Эфиры фе¬
нолов, содержащие ненасыщенную боковую цепь, распростране¬
ны в природе. Например, анетол содержится в анисовом масле.
°СзН6
анизол
(т. кип. 165 °С)
фенетол
(т. кип. 172 °С)
дифениловый эфир
(т. кип. 259 °С;
запах герани)
ОСН,
неролин
(т. пл. 72 °С,
т. кип. 275 °С;
запах апельсина)
OCjH,
новый неролин
(т. пл. 37 °С,
т. кип. 275 °С;
запах акации)
СН30—<
СН=СН—СНЭ
анетол
Тетрагидрофуран (тетраметиленоксид, оксолан)—цикличе¬
ский простой эфир, представляет собой жидкость с т. кип.
66 °С, смешивается с водой. Получают его гидрированием пя¬
тичленного гетероцикла фурана или дегидратацией бутанди-
ола-1,4:
НОСН2—СН2—СН2—СН2ОН
<о е>
фуран тетра* бутандиол-1,4
гидро-
Тетрагидрофуран часто используют как растворитель при
проведении органических реакций, например при восстановле¬
нии алюмогидридом лития, в реакциях Гриньяра вместо диэти-
лового эфира.
1,4-Диоксан — жидкость, т. кип. 101,5 °С, df 1,03, смеши¬
вается с водой, эфиром, бензолом. Хороший растворитель орга¬
нических и многих неорганических веществ. Ядовит. При хра¬
нении образует взрывоопасные пероксиды. Получается из эти¬
ленгликоля под действием серной кислоты:
h2so4 / \
2НО—сн2—сн2—он —°у_у°
180
§ 80. Эпоксиды. Циклические простые эфиры с трехчленным
(а-оксидным) кольцом называют эпоксидами (приставку «эпи»
применяют для обозначения соседних положений), а также алкен-
оксидами. Формула этих соединений в общем виде может быть
записана так [(где R, R', R" и R"' — водород или радикалы
(одинаковые или разные)]:
R>—<
R,/ V
R"
R'"
В качестве примеров эпоксидов можно привести следующие
соединения:
О
/\
Н,С CHS
о
/\
СНз—НС—сн2
о
/ \
свн5—НС—сн2
этиленоксид пропиленоксид
(оксиран) (1,2-эпоксипропан)
стиролоксид
(1-фенилоксиран)
Наиболее важный метод получения эпоксисоединений — ре¬
акция Прилежаева — действие пероксикарбоновых кислот (на¬
пример, пербензойной) на алкены:
О О
^С=С^ + С6Н5—С—ООН —»• ^С С^ + С„Н5СООН
В отличие от простых эфиров с открытой цепью и цикличе¬
ских эфиров с большими циклами, эпоксиды — химически ак¬
тивные вещества. Их трехчленный цикл, подобно циклопропану,
неустойчив и легко размыкается под влиянием разных реаген¬
тов. Характерны реакции присоединения нуклеофильных реаген¬
тов, обычно проводимые в кислой среде. .Наиболее важный
представитель эпоксисоединений —этиленоксид.
§ 81. Этиленоксид в химической промышленности. Этилен¬
оксид— газообразное, легко сжижающееся вещество, т. кип.
10,7 °С, плотность 0,89 г/см3, хорошо растворим в воде, обра¬
зует гидрат С2Н40-7Нг0 — твердое вещество с т. пл. 12,8°С.
С воздухом образует взрывоопасные смеси, ядовит.
Главный промышленный способ получения этиленоксида —
окисление этилена кислородом воздуха в присутствии серебря¬
ного катализатора:
О
02 . 200- 300 «С / \
СНг=СН2 -V Н2С СН2
Этиленоксид — весьма реакционноспособное вещество, легко
вступающее в реакции с размыканием эпоксидного цикла. Ти¬
пичны реакции присоединения веществ, имеющих подвижный
атом водорода (вода, хлороводород, синильная кислота, спирты,
181
карбоновые кислоты). Общий механизм реакции сводится к
активированию электрофилом этвлевокенда (присоединение по
кислородному атому), чем облегчается последующая атака ато¬
ма углерода нуклеофилом.
О НО* сг
/\ / \
Н2С СН2 + НС1 —> Н2С CHj —> НОСН2—СН2С1
этиленхлоргидрин
В присутствии фосфорной кислоты или при нагревании до
180—200 °С этиленоксод присоединяет воду, давая этиленгли¬
коль:
О
/\
н2с—сн2 + н2о —► носн2—СН2ОН
Образующийся этиленгликоль содержит активный водород,
способный к дальнейшей реакции с этиленокеидом, поэтому в
результате получается смесь продуктов:
НОСН2—СН2ОН
н2с—сн2
о
НОСН2—СН2—О—СН2—СН2ОН
диэтиленгликоль
Н2С СН2
О
—► носн2—снц—о—снг—енг-о—cHr-CHjOH
триэтиленгликоль
Аналогично идет реакция со спиртами, приводящая к эфи¬
рам гликолей:
о
/ \
ROH + Н2С СНа
ROCHj—СН2ОН
При реакции зтиленоксида с высшими спиртами (Cj2—Сю)
и алкилфенолами (АГк = С8—Сю) получаются вещества с
большой молекулярной массой, обладающие поверхностной ак¬
тивностью. Важной реакцией зтиленоксида является реакция с
аммиаком, в результате которой образуются этаноламины (см.
§ 160).
Этиленоксид — один из важнейших полупродуктов органиче¬
ского синтеза. Его используют в основном для производства
гликолей и их эфиров, этаноламинов, поверхностно-активных
веществ (ПАВ). Исходя из гликолей, синтезируют эфиры, поли¬
эфиры, являющиеся растворителями, пластификаторами и сма¬
зочными маслами. Полигликоли применяют как высокотемпера¬
турные теплоносители, смазочные материалы, неподвижные
фазы в газожидкостной хроматографии. Наиболее важные на¬
правления использования зтиленоксида приведены на схеме 5.
Этиленоксид используют также для дезинфенции транспортных
средств и жилых помещений.
182
Схема 5. Техническое использование этиленоксида
СНз—СН2ОН
этиловый спирт
НОСН2—СН,ОН
этиленгликоль
Н2С СН,
V
этиленоксид
Н2. кат.
НгО, Н+
ROH, Н+
НОСНг—СНгОН
RMgl. НгО
NH3
-*
НС1
HCN
-НгО
изомеризация
О
О
о
H2SO4. 160
•с
—►
RO-CHj—СНгОН
моноэфиры
этиленгликоля
диэтилеигликоль
RCH2—СНгОН
первичные
спирты
HjN—CHj-CHsC»I
HN(CH2-CH,OH)b
N(CH2—СНгОН)э
этаноламины
С1СН2—СНаОН
этилеихлоргидрин
CH2=CH*CN
акрилонитрил
CHj-CHO
ацетальдегид
О
1,4-диоксан
По химическим свойствам другие эпоксиды подобны этилен-
оксиду. Ближайший гомолог — пропиленоксид — получают в
нашей химической промышленности следующим путем:
ООН
o2 I сн3сн=сна
С6Н5СН2СН3 ► С6Н5СНСН, *-
О ОН
—V СН,—НС^СНг + С6Н5СНСН3
Дегидратация образовавшегося метилфенилметанола дает
стирол. Таким образом, одновременно получают два полезных
183
продукта. Подобные процессы называют совмещенными: они
представляют особый интерес из-за высокой экономичности и от¬
сутствия отходов производства. Получать пропиленоксид непо¬
средственным действием кислорода на пропилен нельзя, так как
в этих условиях образуются продукты окисления по а-углерод-
ному атому — аллиловый спирт, акролеин (см. § 69).
ОРГАНИЧЕСКИЕ ПЕРОКСИДНЫЕ СОЕДИНЕНИЯ
§ 82. Способы получения и химические свойства. Органические
пероксиды можно рассматривать как органические производные
пероксида водорода НО—ОН, у которого один атом водорода
(гидропероксиды) или оба атома водорода (пероксиды) заме¬
щены на алкильные радикалы:
RO—ОН RO—OR
гидропероксиды пероксиды
Способы получения. 1. Окисление кислородом воз¬
духа. В конце прошлого века А Н. Бах установил, что неко¬
торые органические вещества в контакте с воздухом окисляются.
Механизм реакций аутоокисления раскрыт Н. Н. Семеновым
(1927 г.)—это свободнорадикальные цепные реакции. Реакция
может быть инициирована парами ртути, ультрафиолетовым
светом и др.
R—Н + X • —► R • + НХ (инициирование цепи)
R • + О2 —R—О—О •
R—О—О • + Н—R —► R—О—О—Н + R •
R • + R • —> R-R ) , * ,
}• (обрыв цепи)
R—О—О • + R • —► R—О—О—R J
Свободный радикал, инициирующий реакционную цепь, от¬
рывает атом водорода наиболее реакционноспособной группы
(СН или СН2) и образует свободный радикал, который связы¬
вает молекулярный кислород.
Алканы и циклоалканы окисляются сравнительно легко,
предпочтительно затрагивается третичный атом углерода:
(СНз)зСН (СНз)зС—ООН
изобутан грег-бутилгидро-
пероксид
1 (развитие цепи)
В алкенах легко окисляется атом углерода в аллильном по¬
ложении:
сн3—сн,—сн=сн:
ООН
02 I
»- СН3—СН—сн=сн.
бутен-1
бутен-1-илгидро-
пероксид-3
184
В ароматических углеводородах легче всего окисляется бли¬
жайший к бензольному ядру атом углерода боковой цепи (на¬
пример, образование гидропероксида кумола, см. § 72).
2. Присоединение пероксида водорода к алкенам:
ООН
<СН3)2С=СН2 + НО—ОН —> (СН3)2С—снэ
3. Алкилирование пероксида водорода алкилсульфатами в
присутствии щелочей:
кон
(CHsO)2S02 + НО—ОН >- СН3—ООН + СНзО—OSOjK + н20
метилгидро-
пероксид
кон
СНз—ООН + (CH30)2S02 ► СНзО—ОСНз + CH30—0S02K + н2о
диметилпероксид
4. При реакции грег-бутилового спирта с пероксидом водо¬
рода в кислой среде можно получить грег-бутилгидропероксид
и ди-грег-бутилпероксид:
н+
(СНз)зС—ОН + НО—ОН ► (СНз)зС—ООН
(СНз)зС—ООН + (СНз)зС—ОН —* (СН3)3С—О—О—С(СН3)з + Н20
Химические свойства. Гидропероксиды обладают
более сильными кислотными свойствами, чем спирты. Так, с
концентрированными растворами щелочей они образуют соли.
Гидропероксиды являются окислителями, например легко реаги¬
руют с иодоводородом, выделяя при этом иод. Эту реакцию ис¬
пользуют для количественного иодометрического определения
гидропероксидов:
СНз—OOH + 2HI —*. СН3ОН + 12 + н20
Пероксиды реагируют с иодоводородом труднее, чем гидро¬
пероксиды. Они легко распадаются на свободные алкоксильные
радикалы:
(СНа)зС—О—О—С(СНз)з —► 2(СН3)3СО •
Низшие гидропероксиды — неустойчивые взрывоопасные
жидкости. Так, трег-бутилгидропероксид (т. кип. 33 °С при
17 мм рт. ст.) прй нагревании выше 100 °С разлагается со
взрывом; при осторожном нагревании образуется свободный
алкоксильный радикал:
(СНз)зС—ООН —► (СНз)зСО - + • ОН
Свободные алкоксильные радикалы используют как инициа¬
торы цепных реакций полимеризации.
Пероксидные соединения кислот и их производных — перокси*
кислоты и пероксиды ацилов описаны ниже (см. § 108).
185
ГЛАВА 11
АЛЬДЕГИДЫ И КЕТОНЫ
Функциональной группой этого типа соединений является кар¬
бонильная группа С=0. Соединения, в молекулах которых кар¬
бонильная группа связана с одним органическим радикалом и
с атомом водорода, называют альдегидами, а соединения, в ко¬
торых карбонильная группа связана с двумя углеводородными
радикалами,—
кетонами:
0
О
о
О
II
ц
II
1
R—С—H
—С—н
R-C-R'
—С—
альдегид
альдегидная
группа
кетон
кетогруппа
(оксогруппа)
Карбонильную группу называют также оксогруппой, а аль¬
дегиды и кетоны — оксосоединениями. В молекуле оксосоедине-
ций может содержаться одна или несколько карбонильных
ррупп. В зависимости от радикала, связанного с карбонильной
Группой, карбонильные соединения могут быть насыщенными,
ненасыщенными (если в радикале содержится кратная связь),
ароматическими и гетероциклическими.
§ 83. Изомерия и номенклатура. Изомерия и номенкла¬
тура альдегидов. Изомерия альдегидов обусловлена
строением углеводородного радикала.
Наиболее употребительны названия альдегидов, произведен¬
ные от соответствующих карбоновых кислот: муравьиный аль¬
дегид (формальдегид), уксусный альдегид (ацетальдегид),
масляный альдегид. По рациональной номенклатуре альдегиды
с разветвленной цепью рассматривают как производные уксус¬
ного альдегида. По заместительной номенклатуре названия аль¬
дегидов производят от соответствующих углеводородов, наличие
альдегидной группы обозначают окончанием аль. Атом углерода
альдегидной группы определяет начало нумерации. Названия
некоторых альдегидов приведены ниже:
н—сно
сн,—сно
(СН,)*СН—сн2—сно
(CHj)jC—сно
СН*=С(СНз)—сно
С6Н5—сн2—сно
Тривиальное и рациональное
названия
Муравьиный, формаль¬
дегид
Уксусный, ацетальдегид
Изовалериановый, изо-
пропилуксусный
Т риметилуксусный
Метакриловый
Фенилуксусный
Название по современной
заместительной номенклатуре
Метаиаль
Этаналь
З-Метилбутаналь
2,2-Диметилпропаналь
2-Метилпропеналь
2-Фенилэтаналь
Изомерия и номенклатура кетонов. Изомерия
кетонов обусловлена строением радикалов и положением оксо-
группы в углеродной цепи.
185
По рациональной номенклатуре название кетона строится из
названий радикалов, связанных с карбонильной группой, и
окончания кетон. По современной заместительной номенклатуре
кетогруппу обозначают окончанием он и цифрой, указывающей
номер атома углерода карбонильной группы (нумерацию начи¬
нают от ближайшего к кетогруппе конца цепи), а также с по¬
мощью префикса оксо.
Тривиальное и рациональное
названия
Название'; но современной
заместительной
номенклатуре
СН3—СО—СН3 Диметилкетон, ацетон
СНз—СО—CHj—СН2—СН3 Метилпропилкетон
СН3—СО—СН(СН3)2 Изопропилметилкетон
СН3—СН2—СО—СН2—СН3 Диэтилкетон
СН3—СО—СН2—СН=СН2 Аллилметилкетон
Пропанон
Пентанон-2
З-Метилбутанон-2
Пентанон-3
Пентен-4-он-2
§ 84. Способы получения. I. Получение из спиртов. При окис<
лении первичные спирты дают альдегиды, вторичные—кетоны
(см. § 68). Метод используют в лаборатории (окислитель—*
хромовая смесь) для получения легколетучих альдегидов и ке¬
тонов (формальдегида, ацетальдегида, ацетона, циклогексано¬
на), которые по мере образования отгоняют. В промышленных
способах в качестве окислителя используют кислород воздуха в
присутствии катализатора или проводят каталитическое дегид¬
рирование спиртов (см. § 68).
2. Декарбоксилирование солей карбоновых кислот и самих
кислот. «Сухая перегонкад кальциевых (бариевых) солей карбо¬
новых кислот является общим методом получения любых кето¬
нов. Соли одноосновных кислот дают кетоны с открытой цепью,
а соли двухосновных кислот — циклические кетоны. Молекула
образующегося кетона содержит на один атом углерода меньше,
чем молекула исходной соли:
О -| о
СНз—С—oJ2Ca —> СНз—С—СНз + СаСОз
ацетат кальция
О
II
СНг—СН*—С—“Оч
I /
сн2—сн2—с—ск
II
о
адипинат кальция
ацетон
СН2—СН2
Са —► ^)С=0 + СаСОэ
!-ОТ2
СН2
циклопентанон
Из смеси солей двух разных кислот получаются несиммет¬
ричные кетоны:
° 1 Г ° 1 °
И II II
СвН5—С—О J2Ca + L СНз—С—О J2Ca —► 2С6Н5—С—СН3 + 2CaCOs
ацетофенон
187
Попутно в этой реакции образуются ацетон (С113)2С=0 и
бензофенон (СбН5)2С=0.
Если один из компонентов — соль муравьиной кислоты, то
образуются альдегиды:
Г ° 1 Г ? 1
LCH3—С—0/2Са + LH—С—OjjCa —V 2СНз—СНО + 2СаС03
ацетат кальция формиат кальция
Более новым методом получения кетонов является пропуска¬
ние паров кислот над катализатором. Хорошими катализатора¬
ми реакции являются оксиды марганца (II) и тория:
СН2—СН2—СООН
Ан2—СН2—СООН
МпО, 300 °с
>■
СН2—СН2
^С=0 + СО, + Н20
СН2—СН2
Используя смесь муравьиной кислоты с какой-либо другой
кислотой, получают альдегиды:
RCOOH + НСООН —У RCHO + С02 + Н20
3. Гидроформилирование алкенов (оксосинтез, карбонилиро-
вание). По этому способу альдегиды получают присоединением
к олефинам смеси монооксида углерода и водорода (синтез-
газ). Реакцию проводят в присутствии дикобальтоктакарбо-
нила [Со (СО) 4] 2 при 140 °С и 20 МПа:
со+н2
СН2=СН2 ► СНз—СН2—СНО
Из гомологов этилена образуются альдегиды с нормальной
и разветвленной цепями:
СНз
СО + Н2 |
СНз—СН=СН2 ► СНз—СН—СНО + СНз—СН2—СН2—СНО
На основе использования монооксида углерода в присутствии карбонилов
металлов в промышленности получают альдегиды, кислоты, эфиры, спирты.
4. Окисление углеводородов кислородом воздуха в присут¬
ствии катализаторов. Способ используется в промышленности и
приобретает все большее значение. Так получают уксусный аль¬
дегид из этилена (см. § 87), ацетон — из кумола (см. § 72).
5. Из ацетиленовых углеводородов гидратацией по Кучерову
(см. § 38). Другой путь получения альдегидов и кетонов из
ацетиленовых углеводородов — через виниловые эфиры:
СНзОН, кон
НС=СН >- Н2С=СН—ОСНз
давление
HjO. Н+
-СН3ОН
СНз—СНО
СН3ОН. КОН н2о. Н+
R—С=СН V R—С=СН2 ———R-C-CH»
■ —ензон у
ОСНз О
188
6. Гидролиз дигалогенпроизводных, содержащих оба атома
галогена при одном атоме углерода, в присутствии кислот или
оснований. Если оба атома галогена находятся при первичном
атоме углерода, то образуются альдегиды, если при вторич¬
ном — кетоны:
гидролиз
СНа—СНСЬ :
>-[СНз—СН(ОН)2]
—н2о
СНз—сно
С1
Г ?Н 1
| гидролиз
1 1 1
СНз—С—СНз ►
I
СНз—С—СНз
—н2о
СНз—С—СНз
С1
L (!)Н J
О
7. Восстановление хлорангидридов карбоновых кислот.
Хлорангидриды кислот восстанавливаются значительно легче
самих кислот, в результате образуются альдегиды:
О О
II H2/Pd (130 »С) ||
R—С—С1 : »- R—С—Н + НС1
Чтобы не допустить дальнейшего восстановления альдегида,
палладиевый катализатор частично отравляют серой. Способ
используют в лаборатории.
8. Синтез ароматических кетонов по Фриделю — Крафтсу из
ароматических углеводородов и хлорангидридов кислот в при¬
сутствии хлорида алюминия (реакция ацилирования):
О о
II AICI3 II
С6Н6 + Cl—С—СНз >- С6Н5—С—СН3 + НС1
9. Непосредственное введение альдегидной группы в бен¬
зольное ядро. Реакцию формилирования можно осуществить по
способу Гаттермана действием на ароматические углеводороды
смеси монооксида углерода и хлороводорода в присутствии
хлорида алюминия и солей меди(1). Реакция идет легче с го¬
мологами бензола, причем преимущественно образуются пара-
изомеры:
(СНзЬСН—(3 -Л^Г (CH3)2CH-<Qb-CHO
кумол п-изопропилбензальдегид
(куминаль)
§ 85. Строение карбонильной группы. Физические свойства.
В функциональной группе альдегидов и кетонов связь между
углеродом и кислородом двойная (четырехэлектронная). Угле¬
родный атом карбонильной группы находится в состоянии
«р2-гибридизации, и его конфигурация плоская (рис. 21). Вслед¬
ствие большей электроотрицательности кислорода по сравнению
с углеродом связь С=0 сильно поляризована за счет смещения
электронной плотности я-связи к кислороду (см. рис. 21).
189
Рис. 21. Строение карбониль¬
ной группы
Дипольные моменты
альдегидов и кетонов
близки к 2,4 Д, т. е. мно¬
го больше, чем у спиртов,
имеющих дипольные мо¬
менты порядка 1,7 Д. Полярность карбонильной группы сказы¬
вается на физических свойствах. Многие альдегиды и кетоны хо¬
рошо растворимы в воде. Низшие (ацетальдегид, ацетон) сме¬
шиваются с водой во всех отношениях. Температуры кипения
низших членов ряда альдегидов и кетонов выше, чем у соответ¬
ствующих углеводородов, и ниже, чем у соответствующих спир¬
тов; последнее указывает на отсутствие у оксосоединений суще¬
ственной молекулярной ассоциации. Низшие альдегиды обла¬
дают резким запахом, многие высшие альдегиды и кетоны имеют
приятный запах, напоминающий запах цветов, поэтому приме¬
няются в парфюмерии. Физические свойства некоторых альдеги¬
дов и кетонов приведены в табл. 12.
§ 86. Химические свойства. Полярность карбонильной группы
определяет и ее реакции. Эта группа — одна из наиболее реак¬
ционноспособных функций. Характерные реакции: 1) присоеди¬
нение по двойной связи карбонильной группы; 2) замещение
атома кислорода карбонильной группы азотсодержащими груп¬
пировками; 3) конденсация.
Реакции присоединения. Эти реакции являются
процессами нуклеофильного присоединения. Они начинаются с
взаимодействия положительно заряженного атома углерода кар¬
бонильной группы со свободной электронной парой нуклеофиль¬
ного реагента (протекает медленно), после чего к образовавше-
Таблица 12. Физические свойства некоторых альдегидов и кетонов
Соединение
т. пл., °с
Т. кип., °С
Относительная
.,20
плотность d4
Формальдегид
-92
-20
0,81
(при -20 °С)
Ацетальдегид
-121
21
0,80
Пропионовый альдегид.
-81
49
0,807
Акролеин
-87
52
0,84
Бензальдегид
—26
180
1,05
Ацетон
-94
56
0,792
Метилэтилкетон
-86
80
0,805
Циклопентанон
—58
130
0,94
Циклогексанон
-40,5
156
0,94
Ацетофенон
+20
202
1,02
190
муся аниону присоединяется (эта стадия протекает быстро) про'
тон (или другой катион):
X X
Ч б+ б- медленно Ч I быстро Ч
V=C + : X" ► С—0~ - *- ,С—ОН
/ / н+ /
Активность карбонильных соединений и скорость протекания
реакций зависят от положительного заряда на атоме углерода
карбонильной группы. Чем больше этот заряд, тем больше ско¬
рость присоединения нуклеофила. Электроноакцепторные замес¬
тители, оттягивая электронную плотность, увеличивают положи¬
тельный заряд этого атома углерода, вследствие чего реакция
присоединения нуклеофильных реагентов облегчается, скорость
ее увеличивается. Так, например, положительный заряд углеро¬
да карбонильной группы в трихлоруксусном альдегиде (хлора¬
ле) больше, чем в ацетальдегиде, поэтому альдегидная группа
в хлорзамещенном альдегиде более активна!
8+ Н‘
«
Электроноотталкивающие '(электронодонорные) заместите-'
ли, наоборот, замедляют реакцию присоединения, так как они
увеличивают электронную плотность на карбонильном атоме
углерода, снижают его положительный заряд. Алкилы относятся
к электронодонорным группам, поэтому положительный заряд
карбонильного атома углерода в ацетальдегиде меньше, чем в
формальдегиде. Арилы обладают еще более выраженными элек¬
тронодонорными свойствами, поэтому ароматические альдегиды
и кетоны менее активны, чем алифатические. Таким образом,
карбонильные соединения можно расположить в следующий ряд
по убывающей активности:
Н—СНО > СН3—СНО > С6Н5—СНО > СНз—со—СНэ >
> СНз—со—С6Н6 > С6Н6—со—С6Н6
На реакционную способность карбонильной группы влияет и
размер радикалов, связанных с ней: при большом их объеме
затрудняется доступ реагента к атому углерода карбонильной
группы.
Ациклические кетоны реагируют медленнее, чем их цикличе¬
ские аналоги, из-за пространственных препятствий, которые мо¬
гут создавать алкильные группы для подхода реагента к карбо¬
нильной группе:
(СН3)2СН—СО—СН(СН3)2 < СН3СН2—СО—СН2СН3 <
О <
< СНа—СНО < Н—СНО
191
Для многих реакций альдегидов и кетонов требуется кислая
среда (кислотный катализ). Присоединение протона приводит к
возрастанию положительного заряда на карбонильном атоме
углерода:
б + б- +
R—С=0 + Н+ —► R—С—ОН
Различные полярные реагенты присоединяются к карбониль¬
ной группе по общей схеме, приведенной на с. 191. Приведем
следующие важнейшие реакции такого типа.
1. Присоединение водорода с образованием спиртов. Из аль¬
дегидов получаются первичные спирты, а из кетонов — вторич¬
ные:
R ч /ОН R4 Rv /ОН
r—+ 2Н —► ;с( ;с=о + 2Н —► ;с(
\н н/ Ni w r/ хн
Эффективными восстановителями карбонильных групп явля¬
ются алюмогидрид лития LiAlH4 и борогидрид натрия ЫаВН4.
При восстановлении ненасыщенных альдегидов эти восстанови¬
тели не затрагивают двойной углерод-углеродной связи. Алюмо¬
гидрид лития чувствителен к действию влаги, кислот. Применя¬
ется в виде раствора в абсолютном эфире, тетрагидрофуране
или пиридине.
В комплексных гидридах атом водорода несет частичный отрицательный
варяд и вместе со связующей электронной парой переходит к положительно
заряженному карбонильному атому углерода. Требующийся для завершения
восстановления второй атом водорода приходит не из восстановителя, а из
кислоты (или протонного растворителя) при разложении реакционной смеси:
0+ 8-
^С = 0
\L)''
Н—Al—Н
—С—О
I
н
н
н
I
-> —с—он
I
н
Li+
Н
Остальные три гидридных атома водорода используются для восстанов¬
ления других молекул карбонильного соединения. Таким образом, 1 моль
алюмогидрида лития восстанавливает 4 моль карбонильного соединения.
В условиях каталитического гидрирования (катализаторы
Ni, Pt, Pd, хромит меди) восстанавливается и двойная углерод-
углеродная связь ненасыщенных альдегидов и кетонов.
2. Присоединение циановодорода. В результате этой реакции
образуются циангидрины (гидроксинитрилы или нитрилы а-
гидроксикислот). Цианид-ион атакует карбонильный углерод,
192
образующйся в результате этого анион связывает протон:
н+ 1
CN
А-
в+
—С—
! CN |
*- — С—CN
О'
,в-
—С—CN
Ан
Практически в реакцию вводят смесь карбонильного соеди¬
нения, цианида натрия и небольшое количество сильной мине¬
ральной кислоты, так как синильная кислота не реагирует
вследствие ее малой диссоциации. Циангидрины используют в
качестве промежуточных продуктов синтеза гидроксикислот,
аминокислот и др.
3. Присоединение гидросульфита натрия к альдегидам, ме-
тилкетонам и несложным циклическим кетонам идет легко в
водном растворе, причем образуются так называемые гидро¬
сульфитные соединения:
CHj О Н»С О
I 1 + I II
R—Ce+ + S S—О" Na —>. R—С—S— О" Na+
а-
он
I I
но о
Эти вещества хорошо кристаллизуются, нерастворимы в
избытке гидросульфита натрия. Гидролизуются как в кислой,
так и в щелочной средах, образуя исходные карбонильные со¬
единения. Эти свойства используют для очистки альдегидов и
кетонов и выделения их из смесей.
4. Присоединение воды. Формальдегид легко присоединяет
воду при комнатной температуре (без катализатора), образуя
гидрат формальдегида:
H2C=0 + H20 Н2С(ОН)2
Гидраты альдегидов не удалось получить в свободном со¬
стоянии. О течении реакции свидетельствует тепло, выделяю¬
щееся при растворении формальдегида и ацетальдегида в воде.
Спектральными методами не обнаружена карбонильная группа
в водных растворах формальдегида: спектр раствора сходен со
спектром гликоля.
У галогензамещенных альдегидов и кетонов, например у хло-
раля, трихлорацетона и гексафторацетона, склонность к присо¬
единению воды больше, чем у незамещенных, и их гидраты
можно выделить в свободном состоянии.
ССЦ—СНО ССЦ—СО—СН3 CF3—СО—CF,
хлораль трихлорацетон гексафторацетон
/ОН
ССЦ—СН0 + Н20 —► ССЦ—НС^
ЧЖ
хлораль хлоральгидрат
(т. кип. 98 °С) (т. пл. 57 °С)
7 Органическая химия
193
5. Присоединение спиртов. Альдегиды могут присоединять
спирты. Первоначально образуются полуацетали — неполные
простые эфиры гем-лиолов *. При нагревании с избытком спир¬
та в присутствии хлороводорода образуются полные простые
эфиры гидратных форм — ацетали:
/ОН r'oh, hci /OR'
R— СГ + R'OH =?=fc R—HG^ - - ">■ R—HC; + H20
XH 4)R' X)R'
полуацеталь ацеталь
Ацетали устойчивы и могут быть выделены в свободном
виде. В отличие от обычных простых эфиров ацетали гидроли¬
зуются под действием кислот, образуя спирт и альдегид. В ще¬
лочной среде они устойчивы.
Ацетализацию применяют для «защиты» альдегидной груп¬
пы. Наиболее важны в этом смысле циклические ацетали эти¬
ленгликоля:
но—сн2
R—СГ + I
\Н НО—сн2
При проведении некоторых реакций возникает необходимость защитить
от действия реагента одну из активных групп, содержащихся в молекуле
реагирующего вещества. Для этого ее предварительно превращают в- группу,
устойчивую в данной реакции, но легко регенерируемую после реакции.
Для кетонов не характерно непосредственное взаимодействие
со спиртами с образованием ацеталей. В случае кетонов ацета¬
ли легко образуются по этой реакции только с метиловым
спиртом и этиленгликолем. Другие ацетали получают по реак¬
ции с триэтиловым эфиром ортомуравьиной кислоты:
В\ R\
;с=0 + НС(ОС2Н»)» —>■ Х(ОС2Н5)2 + нсоос2н5
R'/ R'/
6. Присоединение магнийорганических соединений (реакти¬
вов Гриньяра) — одна из типичных реакций альдегидов и кето¬
нов. Углеводородный радикал R, несущий в магнийорганическом
соединении частичный отрицательный заряд, выступает в этой
реакции в роли нуклеофильного реагента, присоединяясь к кар¬
бонильному углероду, а остаток MgX соединяется с кислоро¬
дом:
\в+ в- в- I нон (н+) |
\c=o + R-MgX -> R-C-OMgX _Me(QH^>' R-C-OH
/О—сн2
—»- R—нс; | +н2о
хо—сн,
* Приставка гем- происходит от слова «геминальный», означающего, что две
функциональные группы находятся у одного атома углерода.
194
В зависимости от характера используемого в реакции карбо¬
нильного соединения могут быть получены различные по строе¬
нию спирты. Из формальдегида получаются первичные спирты,
из других альдегидов образуются вторичные (стадию образова¬
ния алкоголята магния опускаем), а из кетонов — третичные:
8+ 6+ 6- в+ н2о (н+)
Н2С=0 + CHj—Mgl —* СН3СН2—OMgl ► СН3СН2ОЦ
0 он
1 1. c2H5MgBr |
с0н5-с-н —»- С6Н5-СН-С2Н5
2. н8о+
сн2 о сн2 он
/ \ I 1. CH»Mgi / \ I
Н2С СН—С—СНз : »- Н2С СН—С(СН3)2
2. Н30+
Из приведенных примеров видно, что в состав полученных
таким образом спиртов могут входить алифатические, цикличе¬
ские и ароматические радикалы.
Реакции замещения кислорода карбониль¬
ной группы. 1. Для альдегидов и кетонов характерны ре¬
акции с веществами, содержащими в молекуле группу NH2.
В результате выделяется вода и образуются производные, со¬
держащие группировку —C=N—X (X = ОН, NH2 и др.):
\
/
С=0 + H2N—X
н+
\
/
C=NX + Н20
а) Действие аммиака. Это характерная реакция для многих
альдегидов. Первоначально образующиеся продукты присоеди¬
нения мало устойчивы, быстро происходит Их дегидратация с
образованием альдиминов и полимеризация (тримеризация):
R-C
NHs
\н
,он
С—NHj
\н
—н2о
-сз
альдимин
К\ NH /R
С/ МУ
y/ | | \н
HN\ /NH
С
y/ Ni
Под действием разбавленных минеральных кислот эти соеди¬
нения разлагаются с выделением свободного альдегида.
Для кетонов реакция с аммиаком протекает сложнее,
б) Реакция с гидроксиламином. При действии на альдегиды
и кетоны гидроксиламином образуются оксимы альдегидов
7*
195
(iальдоксимы) и кетонов (кетоксимы):
СН3—СН=0 + Н2ЫОН —► СН3—CH=NOH + Н20
оксим
ацетальдегида
\=0 + H2NOH
NOH + Н20
оксим
циклогексанона
Оксимы кетонов способны к реакции с изменением химиче¬
ского строения, к так называемой перегруппировке Бекмана
(1886 г.). Эта реакция проходит в кислой среде. Формально
перегруппировка заключается в том, что один из радикалов
оксима перемещается от карбонильного атома углерода к азоту,
а гидроксильная группа переходит к карбонильному атому угле¬
рода. Так как гидроксильная группа у двойной связи неустой¬
чива (правило Эльтекова), водород гидроксильной группы пе¬
ремещается к атому азота и образуется амид кислоты:
"1
rzb
СН3—C=N ОН!
I * 1 J
[н_з_с}-]
оксим ацетона
ОН
СН3—C=N—СН3
О
II
- СН3— с—NHCH3
метиламид
уксусной кислоты
в) Реакция с гидразином приводит к гидразонам:
Ч R\
v=o + h2n—nh2 —>• ;c=n—nh2 + h2o
w w
гидразон
альдегида
При нагревании гидразонов с гидроксидом калия в присут¬
ствии платины образуются углеводороды и выделяется свобод¬
ный азот. Эту реакцию почти одновременно открыли русский
ученый Н. М. Кижнер (1911 г.) и немецкий ученый Л. Вольф
(1912 г.). Называют это превращение реакцией Кижнера —
Вольфа:
N—NH2
II КОН (Pt). 300 “С
СН3—С—СН3 > СНз— СНу-СНз + Ns
г) Реакция с фенилгидразином приводит к фенилгидразонам:
С6Н6—СНО + H2N—NH—С„Н5 —>- CeH5—CH=N—NHC6HS + Н20
Оксимы, гидразоны, фенилгидразоны — твердые кристалли¬
ческие вещества с характерными температурами плавления. Это
196
свойство используется для идентификации альдегидов и ке¬
тонов.
2. Замещение карбонильного атома кислорода хлором. Реак¬
ция осуществляется при действии хлорида фосфора (V):
циклопен-
танон
РС1$
/—\/а
< У + рось
чм
1,1-дихлор-
циклопентан
Реакции полимеризации. При полимеризации аль¬
дегидов происходит разрыв двойной связи карбонильной груп¬
пы, и атом кислорода одной молекулы соединяется с атомом
карбонильного углерода другой. В результате могут образовать¬
ся линейные и циклические продукты. Так, при стоянии водного
раствора формальдегида постепенно выделяется белый осадок.
Это линейный полимер — параформ, или полиоксиметилен:
пСН2=0 + Н20 —>. НОСН2[—ОСН2—]„_2ОСН2ОН (/* —10—50)
При нагревании параформа до 150 °С выделяется мономер¬
ный формальдегид. В настоящее время полимеризацией форм¬
альдегида получают ценный высокомолекулярный материал —
полиформальдегид. При нагревании с кислотой формальдегид
полимеризуется с образованием циклического триоксиметилена
(триоксана).
Уксусный альдегид под влиянием кислот (H2SO4, НС1) об¬
разует жидкий циклический тример — паральдегид и твердый
тетрамер — метальдегид («сухой спирт»). Последний образует¬
ся при проведении полимеризации уксусного альдегида ниже
0°С.
сн2
ОУ мэ
H2d:\ /сн2
о
сн—сн*
о/ м>
СН3
г-Н Он-
сн*—нс—о
о/ \СН—сн3
СН*
СНз—НС\
О—СН—СН*
триоксиметилен
паральдегид
(т. пл. 10 °С,
т. кип. 124 °С)
метальдегид
(т. возг. 112 "С)
При нагревании этих полимеров в присутствии разбавленных
минеральных кислот они деполимеризуются, образуя исходный
альдегид.
Ароматические альдегиды с альдегидной группой, связанной
непосредственно с бензольным ядром, не полимеризуются. Со¬
единения, содержащие удаленную от бензольного ядра альде¬
гидную группу, ведут себя аналогично алифатическим альдеги¬
дам.
Реакции конденсации. В молекулах альдегидов и
кетонов водородные атомы, стоящие у соседнего с карбонильной
197
группой атома углерода (a-положение), под влиянием карбо¬
нильной группы приобретают частичный положительный заряд
и тем самым способность отщепляться в виде протона. Альдегид
или кетон превращается при этом в карбанион. Вследствие это¬
го альдегиды и кетоны способны к реакциям конденсации, про¬
текающим по механизму нуклеофильного присоединения. В ре¬
зультате реакции конденсации образуются новые углерод-угле-
родные связи.
1. Конденсация альдегидов с альдегидами. В реакции уча¬
ствует одна молекула своей карбонильной группой (карбониль¬
ный компонент), а другая молекула — сс-водородом (метилено¬
вый компонент). Под действием оснований молекула карбониль¬
ного соединения превращается в карбанион (гидроксид'-ион от¬
рывает протон от а-углеродтюго атома):
Н
н->с—сн=о + “он =?=* н2о + г сн2—ен=0
Образующийся карбанион является нуклеофилом. Он взаи¬
модействует с карбонильной группой другой молекулы оксосо-
единения по общей схеме реакции с нуклеофильными реаген¬
тами:
8+
СН3—СН + : СН2-СНО —► СН3—СН—СН2—СНО
1®- СГ
Образовавшийся анион отрывает протон от воды; образуя
р-гидроксиальдегид:
О" ОН
I И* •
сн3—сн—сн2—сно > сн3—сн—сн2—сно
альдоль
(3-гидрокеибутаналь)
Продукт конденсации двух молекул уксусного альдегида на¬
зывается альдоль (сокращение от слова «альдегидо-алкоголь»),
что указывает на присутствие в молекуле двух функциональных
групп—альдегидной и спиртовой. Описанная реакция носит
название альдольной конденсации.
Если альдегид не содержит а-водорода, например
(СНз)зС—СНО, Аг—СНО и др., то альдольная конденсация не
происходит.
При нагревании альдоля отщепляется вода и образуется
кротоновый альдегид (бутен-2-аль):
СН3—СН—CHj—СНО ——у СНз—СН=СН—СНО
| —н2 о
ОН
198
Реакция конденсации с отщеплением воды и образованием
непредельного карбонильного соединения получила название
кротоновой конденсации.
Подобно уксусному альдегиду конденсируются и его гомоло¬
ги, всегда за счет а-водородных атомов:
0 СН, О ОН СНз о
1 I II III
СН3—СН2—С—Н + СН2—С—Н —► СНз—СН2—СН—СН—С—Н —-£■
—н2о
З-гидрокси-2-метилпентаналь
Н3С О
I II
—► сн3—сн2—сн=с—с—н
2-метилпентен-2-аль
Образующиеся соединения, содержащие двойную углерод-
углеродную связь в сопряжении с двойной углерод-кислородной
связью, энергетически особенно выгодны. Этим объясняется
легкость протекания такой реакции.
В реакцию могут вступать и молекулы разных альдегидов.
Так, для формальдегида характерна конденсация с другими
альдегидами с образованием многоатомных альдегидоспиртов:
СН2ОН
зн2с=о + сн3—сно —> носн2—с—сно
СН2ОН
Эта реакция протекает под влиянием карбонатов щелочных
металлов ■(■КвСО». Na2C03).
Ароматические альдегиды, содержащие альдегидную группу
у ядра, не могут конденсироваться между собой из-за отсутст¬
вия а-водородов. С алифатическими альдегидами, содержащи¬
ми а-водород, они вступают в кротоновую конденсацию:
СбНз—СН=0 + СНз—СНО —► С6Н5—СН=СН—СНО + Н20
коричный альдегид
2. Конденсация альдегидов с кетонами. В этой реакции ке¬
тоны выступают в качестве метиленового компонента, так как
оксогруппа кетонов менее активна, чем у альдегидов:
НО О
I II II
СвН5—С=0 + СНз—С—СНз —► СвН5—СН=СН—С—СНз
бензилиденацетои
3. Конденсация кетонов с кетонами. Наиболее активные ке¬
тоны способны конденсироваться аналогично альдегидам:
О
II
сн3—с + н-
<!:Нз
-сн2-
-С—СН3
II
о
он
сн3—с—сн2—с—сн3
I II
СНз о
диацетоновый спирт
(4-гидрокси-4-метил-
пентанон-2)
—* СНз—С=СН-С—СНз
СН3 О
окись мезитила
(4-метилпентен-3-он-2)
4. Конденсация альдегидов и кетонов с углеводородами,
а) Конденсация с алкенами. При взаимодействии избытка
формальдегида с изобутиленом в присутствии серной кислоты
образуется 4,4-диметил-1,3-Диоксан. Это вещество интересно
тем, что из него под действием фосфорной кислоты (200 °С)
можно получить важный технический продукт изопрен:
СН3
СН3—С=СН2
2CH20(H2S04)
НзСч сн2
^сн2
н3с/| I
с.п2
НзРО,
»-
20Э "С
сн2
СНз
=с—сн=сн2 + сн2о + н2о
изопрен
В настоящее время разработан одностадийный промышлен¬
ный процесс, в ходе которого из изобутена и метанола (проме¬
жуточно окисляемого в формальдегид) получают изопрен:
СНз
I
СНз—С=СН2 + СНзОН
Оа, Н3РО4 + М0О3
на силикагеле
-2НгО
СНз
СН2=С—СН=СН2
б) Конденсация с ацетиленом и алкинами-1. Реакция проте¬
кает в присутствии порошкообразных щелочей или алкоголятов
щелочных металлов (реакция Фаворского):
2СН2=0 + СН=СН —* НОСН2—С=С—СН2ОН
бутиндиол-1,4
ОН
(СН3)2С=0 + CHssCH —(СН3)2С-С=СН
200
диметилэтинил-
метанол
он
I
(СН3)2С=0 + СН=С—СН=СН2 —► (СН3)2С—с=с—сн=сн2
винилацетилен диметил(винилэтинил)метанол
Получаемые по этим реакциям ацетиленовые спирты служат
исходными веществами в разных синтезах.
в) Конденсация с ароматическими углеводородами. Арома¬
тические углеводороды при конденсации с альдегидами дают со¬
единения ряда дифенил- и трифенилметанов:
h2so4
с6нв + сн2=о + с6нв ► с6н5—сн2—с6н6 + н2о
дифенилметан
О С8Н5
II H2so4 |
CeH5—С-Н + 2С6Н„ ► С6Н5—СН—С6Н5
трифенилметан
Окисление альдегидов и кетонов. Альдегиды и
кетоны по-разному относятся к действию окислителей. Наличие
в альдегидной группе атома водорода обусловливает легкость
окисления этой группы до карбоксильной, в результате получа¬
ются кислоты с тем же числом углеродных атомов в молекуле:
О О
II [О] II
R—С—Н ► R—С—ОН
Альдегиды окисляются слабыми окислителями, например
аммиачным раствором нитрата серебра, содержащим комплекс¬
ное соединение [Ag(NH3)2] ОН. При этом на поверхности
стеклянного сосуда образуется слой металлического серебра —
«серебряное зеркало».
О о
II II •
R—С—H + 2[Ag(NH3)2]OH —v R—С—ONH, + 2Ag + 3NH3 + Н20
Эту реакцию, а также реакции восстановления фелинговой
жидкости * или свежеосажденного гидроксида меди использу¬
ют для идентификации альдегидов (кетоны этих реакций не
дают):
Н ОН
R— С=0 + 2Си(ОН)2 —>■ R—С=0 + Си20 + 2Н20
(голубой (красный
цвет) осадок)
* Фелингову жидкость готовят смешением водного раствора сульфата ме-
ди(П) с щелочным раствором сегнетовой соли (тартрат калийнатрия). При
этом образуется темно-синий раствор комплексной медной соли винной кис¬
лоты.
201
Многие альдегиды легко окисляются кислородом воздуха.
В качестве промежуточных продуктов образуются пероксидные
соединения — лероксикислоты. Наиболее хорошо изучено окис¬
ление бензальдегида:
О о
II о2 II
с6н5—С—Н ► С6Н5—С—ООН
пербензойная
кислота
(гидропероксид
бензоила)
Кетоны окисляются значительно труднее, они устойчивы к
слабым окислителям и к кислороду воздуха. Действие на ке¬
тоны сильных окислителей в жестких условиях приводит к раз¬
рыву углерод-углеродной связи, в результате образуется смесь
разных продуктов (кислот и кетонов) с меньшим числом атомов
углерода, чем исходный кетон. По продуктам окисления можно
судить о строении исходного кетона.
О
II Ю1 ,—► СНзСООН + СНзСООН
СНз—С—СН2—СН3
1—► НСООН + СН3—СН2—СООН
(НСООН С02 + Н20)
о
II [О]
(CHa)jCH—С—СН(СНз)2 ► (СН3)2СН—СООН + (СН3)2С=0
Для альдегидов, не имеющих в молекуле а-водорода, ха¬
рактерна реакция Канниццаро (1853 г.) — окисление одной мо¬
лекулы за счет другой при действии концентрированных щело¬
чей (одна молекула альдегида окисляется до кислоты, а другая
восстанавливается до спирта):
О о
II II
2С6Н5—С—Н + NaOH —v С6Н5—С—ONa + CeH5—СН2ОН
В изученной В. М. Родионовым «перекрестной» реакции Кан¬
ниццаро участвует смесь высшего альдегида с формальдегидом.
В этой реакции ароматический альдегид восстанавливается, а
формальдегид окисляется:
С6Н5—СНО + НСНО + NaOH —> СВН5—СН2ОН + HCOONa
Алифатические альдегиды, содержащие а-водород, под дей¬
ствием щелочей ведут себя иначе: они конденсируются сначала
в альдоли, из которых затем образуются высокомолекулярные
соединения. Однако под действием этилата алюминия окисли¬
тельно-восстановительное превращение, подобное рассмотренно¬
о
с$н5сно ||
► 2С6Н6—С—ОН
бензойная
кислота
202
му выше, возможно (реакция Тищенко, 1906 г.). При этом об¬
разуются сложные эфиры:
о
II
2СН3—С—Н
А1(ОС2Н5)3
О
II
СНз—C^OCjHs
Для бензойного альдегида характерна бензоиновая конден¬
сация (Н. Н. Зинин, 1839 г.) под действием цианида калия:
О О ОН О
II II кем I II
с6н5—с—н + нс—свн5 »- С6Н5—сн—с—с„н5
бензоин
Другие реакции. Как уже отмечалось при рассмотре¬
нии реакций конденсации, карбонильная группа увеличивает
подвижность сс-водородов в молекуле. Так, а-водород легко
замещается на галоген (хлор, бром, иод):
О О
СНз— С—СНз + С12 —► С1СН2—С—СНэ + нет
хлорацетон
В полученных соединениях галоген в a-положении также
отличается большой активностью.
При характеристике химических свойств альдегидов и кето¬
нов отмечалось их сходство, обусловленное наличием в молеку¬
лах этих соединений карбонильной группы. Однако между ними
существуют и различия, прежде всего по отношению к окисли¬
телям. Цветную реакцию с фуксинсернистой кислотой дают
только альдегиды. Для альдегидов характерна реакция полиме¬
ризации.
§ 87. Наиболее важные представители. Формальдегид (муравьи¬
ный альдегид, метаналь) — газ с резким раздражающим
запахом. Образуется при неполном сгорании многих органиче¬
ских веществ. Следы формальдегида всегда содержатся в дыме,
этим объясняется его консервирующее действие при копчении
мясных и рыбных продуктов.
В промышленности формальдегид получают из метанола —
пропусканием паров спирта вместе с воздухом над нагретым
медным (или серебряным) катализатором. Формальдегид сохра¬
няют в виде 40%-го водного раствора, называемого формали-
ном, или в виде твердых соединений — триоксана (СН20) 3 и
параформальдегида (СН20)л.
Отсутствие органического радикала в молекуле формальде¬
гида сказывается на его химических свойствах. Так, в отличие
от других альдегидов формальдегид дает с аммиаком цикличе¬
ское соединение гексаметилентетрамин (уротропин):
6CH20 + 4NH3 —»- C16H12N4 + 6H20
203
Реакция открыта А. М. Бутлеровым (I860 г.). Уротропин—-
кристаллическое вещество, т. пл. 230 °С. Используется в каче¬
стве лекарственного вещества и в больших количествах для про¬
изводства полимеров. Действием азотной кислоты на уротропин
получают гексоген — сильное взрывчатое вещество:
NO,
I 2 •
Н2С"^''СН2
02N-Nx /N-N02
CHg
Для формальдегида характерна также реакция Канниццаро
(см. § 86):
2Н2С=0 + NaOH —► СН3ОН + HCOONa
Под влиянием гидроксида кальция формальдегид вступает в
реакцию конденсации альдольного типа, давая гликолевый аль¬
дегид:
О О
II II
н—с—н + н—с—н —» носн2—сно
Далее гликолевый альдегид конденсируется в моносахарид.
Реакция формальдегида с фенолами рассмотрена в § 73.
Формальдегид находит широкое применение для получения
полимерных материалов. Он токсичен для микроорганизмов,
вследствие чего применяется как дезинфицирующее средство,
например для протравливания семян перед посевом (уничтожа¬
ет споры головни). Формальдегид используют в кожевенной
промышленности (дубление кожи), для хранения анатомических
препаратов и др.
Ацетальдегид (уксусный альдегид, этаналь) — легко кипя¬
щая жидкость с запахом зеленой листвы. В промышленности
получается из ацетилена по реакции Кучерова (см. §38), окис¬
лением этилового спирта, изомеризацией этиленоксида (см.
схему 5). Наиболее современный способ получения уксусного
альдегида — прямое окисление этилена кислородом воздуха:
PdCI2, CuCl
СН2=СН2 + '/202 * СН3—СНО
Ацетальдегид используют для получения уксусной кислоты,
этилацетата (конденсацией по Тищенко), метальдегида, параль-
дегида, а также этилового спирта. При действии на ацетальде¬
гид избытком хлора получают хлораль:
СН3—СНО + ЗС12 —► СС13—СНО + 3HCI
N
(сн3со)2о
204
Хлораль — жидкость, т. кип. 98ЛС, с водой образует устой¬
чивый гидрат — хлоральгидрат СС13СН(ОН)2 (см. § 86). Это
один из редких примеров устойчивости гел-диолов. При дейст¬
вии на хлораль водного раствора щелочи происходит расщепле¬
ние углерод-углеродной связи и образуется хлороформ:
ССЦ—СНО + NaOH —► СНСЦ + HCOONa
Эту реакцию используют в промышленности для получения
хлороформа из ацетальдегида или из этилового спирта. Снача¬
ла спирт действием гипохлорита кальция (хлорной извести)
окисляют в ацетальдегид, который затем хлорируют до хлораля,
а щелочная среда способствует его расщеплению и образованию
хлороформа.
Бензальдегид — бесцветная маслянистая жидкость с запахом
горького миндаля. Бензальдегид встречается в природе в виде
гликозида амигдалина (см. § 154). В промышленности бенз¬
альдегид получают хлорированием толуола до бензилиденхло-
рида с последующим гидролизом или каталитическим окислени¬
ем толуола:
ci2 н2о
С6Н6—СН3 ► С6Н,—CHClj >- с6н5—сно
окислитель
с6н5—сн3 >- С6Н5—сно
Особенности химических свойств бензальдегида, связанные с
отсутствием в его молекуле а-водорода, уже отмечались при
рассмотрении химических свойств альдегидов. Реакция бензаль¬
дегида с хлором также протекает иначе, чем в случае алифати¬
ческих альдегидов — идет замещение атома водорода альдегид¬
ной группы. Эту реакцию используют для промышленного про¬
изводства бензоилхлорида:
О О
II Clj II
СвН6-С-Н С6Н5-С-С1
Бензальдегид как ароматическое соединение дает и харак¬
терные реакции бензольного ядра, например нитруется. Альде¬
гидная группа относится к -мега-ориентантам. Бензальдегид
используют для синтезов красителей, душистых веществ и др.
Ацетон — жидкость с характерным запахом, смешивается с
водой во всех отношениях, хороший растворитель органических
веществ. В промышленности ацетон раньше получали сухой
перегонкой ацетата кальция (см. § 84). Сейчас существуют
другие способы — каталитическая кетонизация уксусной кисло¬
ты, ацетонобутиловое брожение сахаров, дегидрирование изо¬
пропилового спирта, полученного из пропилена, или прямое
окисление пропилена:
О
Ог (PdC)2, CuCJ) II
CHs=CH—СН3 * СНЭ—С—СН3
205
Однако самым важным способом синтеза является кумоль-
ный способ совместного получения фенола и ацетона (см.
§ 72).
Ацетон используют как растворитель для лаков, кинопленки,
искусственного ацетатного волокна, ацетилена, как желатиниза-
тор в производстве бездымного пороха, для синтезов хлорофор¬
ма, йодоформа, кетена и др. Продукт конденсации ацетона с
ацетиленом — диметилэтинилметанол (см. § 86) — применяют
для получения изопрена:
СНз
СНз—С—С==
СН,
;СН
н*
СНз—с—сн=сн2
—н2о
сн2
СНз
.=(!:—сн=
сн2
он
он
Продукт конденсации ацетона с винилацетиленом — диме¬
тил (винилэтинил) метанол (см. § 86)—используют для получе¬
ния винилацетиленовых полимеров, клеев, лаков и др.
Качественной реакцией на ацетон является образование йодо¬
форма при действии на него раствора щелочи и иода:
о о
II 1 КОН
СНз—С—СНз + Ь —► С1з—с—СНз *- CHIj + СНз—COOK
Йодоформ — кристаллическое вещество желтого цвета с ха¬
рактерным запахом. Эта реакция характерна и для гомологов
ацетона — метилкетонов и вообще для соединений, содержащих
в молекуле группировку СН3—СО— или СНз—СНОН—.
Циклогексанон — жидкость, т. кип. 166 °С, получается де¬
гидрированием циклогексанола или окислением циклогексана.
Циклогексанон используют для производства капролактама —■
исходного вещества для получения синтетического волокна
капрон:
н2с—СН2
1 Н2С^ ^с=0
н2с—сн»
Н2С-СН2
Н2С^ ^С=МОН
н2с—сн»
перегруппировка
Бекмана
циклогексанон
окспм
циклогексанона
г N
н2с^ чс—он
1 1
н2с сн2
1 “I
Lh2C—сн2
NH
н2с/ \с=о
i I
н2с сн2
I 'I
Н2С СН»
капролактам
При энергичном окислении циклогексанона образуется ади¬
пиновая кислота, которую используют для получения синтети¬
ческого волокна найлон.
20S
Ацетофенон (метилфенилкетон) — вещество с приятным
цветочным запахом, вследствие чего его применяют в производ¬
стве туалетного мыла. Синтезируют по реакции Фриделя —
Крафтса или окислением этилбензола кислородом воздуха в
присутствии катализаторов;
0 °
/~\+ ci-i-сн, ^VJUh»
О
о2 II
СвН5—СН2—СНз > С6Н,—С—СНз + н2о
Ацетофенон используют в различных синтезах. При действии ,
хлора на ацетофенон образуется хлорацетофенон:
О О
I) сн II
CeH5—С—СНз —СвН5—С—СН2С1
Это кристаллическое вещество, т. пл. 52 °С, один из силь¬
ных лакриматоров, в первую мировую войну его использовали
как боевое отравляющее вещество.
НЕНАСЫЩЕННЫЕ КАРБОНИЛЬНЫЕ СОЕДИНЕНИЯ
Наличие в молекуле вещества карбонильной группы и близко
расположенной к ней двойной связи придает особые свойства
соединению вследствие взаимного влияния этих групп. Наибо¬
лее важными являются сопряженные системы С—С—С=0 и ку¬
мулированные системы С=С=0. Если карбонильная группа
удалена от двойной связи, то каждая из групп сохраняет инди¬
видуальность и проявляет присущие ей свойства.
§ 88. Сопряженные или а,р-ненасыщенные альдегиды и кетоны.
Общим способом их получения является кротонова» кон¬
денсация альдегидов и кетонов (см. § 86). Таким путем были
получены, например, следующие оксосоединения;
О О СНз о
В а || В а II f II
СН2=СН—С—Н СНз—СН=СН—С—Н СНз—С=СН—С—СНз
В а
акролеин кротоновый альдегид окись мезитила
При образовании а,р-ненасьиценных карбонильных соеди¬
нений в молекуле возникает система сопряженных двойных
связей:
4 3 2 1
С=С—С=Ю
В реакциях присоединения такие системы в зависимости от
условий и применяемого реагента могут реагировать в положе¬
ния 1,2- 3,4- и 1,4-, т. е. в реакции участвует либо только
207
карбонильная группа, либо только двойная углерод-углеродная
связь, либо та и другая одновременно.
Двойная углерод-углеродная связь а,р-ненасыщенных со¬
единений в отличие от изолированных двойных связей обладает
повышенной активностью: ее можно гидрировать не только
водородом в присутствии катализатора, но и водородом в мо¬
мент выделения (амальгамой натрия в водно-спиртовом рас¬
творе, цинком в уксусной кислоте). При гидрировании а,р-
ненасыщенных альдегидов образуются насыщенные альдегиды и
ненасыщенные спирты; конечным продуктом гидрирования
являются насыщенные спирты:
С i3—СН=СН—СНО
h2/ni
*■ СН3—СН2—СН2—СНО —
H2/Pt
>- СНз—сн=сн—СН2ОН —
—► сн3—сн2—сн2—СН2ОН
Гидрирование а,р-ненасьпценных кетонов идет по двойной
углерод-углеродной связи:
О о
II H2/Pd II
(СН3)2С=СН—С—СНз * (СН3)2СН—СН2—С—СНз
а,р-Ненасыщенные карбонильные соединения дают типич¬
ные реакции на карбонильную группу — образуют циангидрины,
оксимы, гидразоны и др. Окислители атакуют и карбонильную
группу, и двойную связь, но при действии влажного оксида
серебра окисляется только карбонильная группа:
О о
II Ag20 II
СН2=СН—С—Н -9- СН2=СН—С—ОН + 2Ag
Галоген присоединяется по месту С=С-связи, а присоедине¬
ние галогеноводорода идет под влиянием карбонильной группы
против правила Марковникова:
О о
СН2=СН—С—Н + Вг2 —► СН2Вг—СНВг—С—н
8+ z' -v 5- sQ
сн2=сн-с=о + НВс ► СН2Вг-СН2-С^
н
а,р-Ненасыщенные карбонильные соединения могут приме¬
няться в качестве диенофилов в реакции Дильса — Альдера:
СН.
Повышенная активность ос,р-двойной углерод-углеродной
связи объясняется ее сопряжением с карбонильной группой:
С=С—С=0. Передача влияния при сопряжении может распро¬
страняться и на несколько сопряженных связей. Например, сор-
биновый альдегид вступает в реакцию конденсации за счет
водорода метильной группы (как и уксусный альдегид):
Н
I
R—с=о + н2сн—сн=сн—сн=сн—сно —►
—> R—СН=СН—сн=сн—сн=сн—сыо + н2о
Сходные по химическим свойствам вещества, отличающиеся
друг от друга на группу —СН=СН—, называют винилогами.
Из отдельных представителей а,|3-ненасыщенных альдеги¬
дов интерес представляют следующие.
Акролеин СН2=СН—СНО — жидкость с резким раздражаю¬
щим запахом, т. кип. 52 °С. Его получают в промышленности
конденсацией формальдегида с уксусным альдегидом или ката¬
литическим окислением пропилена:
02. Си
н2с=о + н2сн—сно ——сн2=сн—сно < ■ — СН2=СН—CHj
—Н2О 300 °С.
В лаборатории акролеин готовят нагреванием глицерина с
обезвоживающими веществами (например, с гидросульфатом
калия KHSO4):
СН2-ОН
СН—ОН
£н2—он
-н2о
[СН—он
fH
СН2—ОН-1
СНО сно
сн2
I
сн2он
-н2о
СН
II
сн2
Акролеин используют для различных синтезов и для получе¬
ния пластмасс, отличающихся прозрачностью и твердостью.
Кротоновый альдегид — жидкость с резким раздражающим
запахом, т. кип. 102°С; получают кротоновой конденсацией
уксусного альдегида.
При окислении кротонового альдегида легко образуется кро¬
тоновая кислота СН3—СН=СН—СООН, эфиры которой ис¬
пользуют для производства полимерных материалов. При гид¬
рировании альдегида получается масляный альдегид и бута¬
нол- 1.
§ 89. Кетены. Кетены— соединения с кумулированными карбо¬
нильной и двойной углерод-углеродной связями. Простейшим
и важнейшим их представителем является соединение
СНг=С=О, носящее собственно название кетен. В промыш¬
ленности его получают дегидратацией уксусной кислоты в при¬
209
сутствии фосфата алюминия, а в лаборатории — пиролизом
ацетона:
700 “С
СН»—СООН ——► CHj=C=0
—Н30
о о
If 70в *G |
СНг—С—СН3 - ► СНз—С • + • CHj —* СН2=С=0 + СН«
Кетен — газ, сжижающийся при —41°С, токсичен. Он отно-*
сится к числу наиболее реакционноспособных веществ и ис¬
пользуется для различных синтезов как хорошее ацетилирую-
щее средство:
СНзСООН
сн*—с;
СНз—с:
уксусная кислота
[н*о
О ■*-
СНзСООН
о
е2н6ок |
CHj=C=0 ► СН*—G—OCjH*
N>
уксусный
ангидрид
этидацатат
НС1
NH3
СИ»—СОС1
адетилхлорид
СНз—СОКН2
ацетамид
ДИКАРБОНИЛЬНЫЕ СОЕДИНЕНИЯ. ХЕЛАТЫ
§ 90. Свойства и отдельные представители. Простейшими соеди¬
нениями, содержащими две соседние карбонильные группы
(а-дикарбонильные соединения), являются:
О О
О О
о о
Н-1-С-Н
СНу-1—с—н
I 0
СНз—С—С—СН9
глиоксаль
метилглиокеаль
биацетил,
диметилгдиоксаль
(бутандион-2,3)
Простейшими представителями р-дикарбонильных соедине¬
ний, содержащих карбонильные группы в положениях 1,3, яв¬
ляются:
О О О О
II II II II
СНз—С—СНз—С—СНз CeHs—С—СН2—С—СНз
ацетилацетон бензоилацетон
Карбонильные группы могут находиться в молекуле и в дру¬
гих положениях. Относительное положение карбонильных групп
210
влияет на свойства соединений. Соединения, содержащие карбо¬
нильные группы в a-положении, вследствие сопряжения я-свя-
зей окрашены в желтый цвет. Они обладают повышенной хими¬
ческой активностью, в щелочном растворе подвергаются вну¬
тримолекулярной реакции Канниццаро:
0 О ОН О
1 ! I II
СН,—С—С—Н 4- NaOH —► СН,—СН—С—ONa
метилглиоксаль натриевая соль
молочной кислоты
Из отдельных представителей дикарбонильных соединений
рассмотрим следующие:
Биацетил СН3—СО—СО—СН3—жидкость желтого цвета,
т. кип. 88 °С. Получают из метилэтилкетона действием азотис¬
той кислоты на холоду (в реакцию В9тупает а-водород метиле¬
новой группы) с последующим разложением монооксима:
О HON О 0 0
II ногга || || н2о(н+) II ||
СНз—СН2—С—СНз СН3—С—С—СН, ► СН3—С—С—СН3
Диоксим биацетила (диметилглиоксим; реактив Чугаева)
дает с ионами никеля характерный осадок розового цвета. Эта
реакция открыта Л. А. Чугаевым в 1906 г. Ее используют в
аналитической химии для качественного и количественного
определения никеля.
HON NOH
«А
СНз—С—С—CHj диметилглиоксим
Соединения, содержащие карбонильные группы в р-положе-
нии, проявляют особые свойства. Выше было отмечено, что
карбонильная группа активирует а-водородные атомы, делает
их более подвижными. В p-дикарбонильных соединениях а-во-
дороды находятся между двумя карбонильными группами и по¬
движность их еще больше.
Представителем p-дикарбонильных соединений является
ацетилацетон СНз—СО—СН2—СО—СНз. Это бесцветная жид¬
кость, т. кип. 139 °С, df 0,98; получают конденсацией этилаце-
тата с ацетоном:
С2Н5ОЫа
СНз—СО—ОС2Н5 + СНз—СО—СНз —„ - - >• СНз—СО—СН*—СО—СНз
—С.2Н5ОН
Ацетилацетон представляет собой равновесную смесь моле¬
кул двух форм — ацетил ацетона и изомерного ему ненасыщен¬
211
ного оксоспирта с гидроксильной группой при двойной связи
(енола):
0 0 он о
СИ*— С—СН2—I—СНЭ СН3—С=СН—С—СН3
кетонная форма (15%) енольная форма (85%)
Это явление — кето-енольная таутомерия — более подробно
рассмотрено в § 150.
Енольная форма ацетилацетона обладает более выраженны¬
ми кислотными свойствами, чем спирты, поэтому ацетилацетон
легко образует с основаниями металлические производные —
еноляты. Катионы поливалентных металлов (меди, бериллия,
алюминия, хрома, железа и др.) образуют с ацетиланетоном
стабильные еноляты, содержащие металл в цикле:
/'
\
НаС/
с=о
НС
С-0
/ \
о—с
о=с
/
\
/
\
СН3
СН
СНз
Эти соединения известны под названием хелатов (от греч.
«хела» — клешня), или внутрикомплексных соединений. Более
подробно о характере связей, обозначенных стрелкой, см. § 121.
В настоящее время доказано, что хелатное кольцо в ацетилаце-
тонах проявляет ароматические свойства, все связи в нем вы¬
равнены.
Характерным хелатообразующим агентом является ион двух¬
валентной меди. При смешении ацетилацетона со свежеосаж-
денным гидроксидом меди последний сначала переходит в рас¬
твор, а затем выделяется трудно растворимый темно-синий
ацетилацетонат меди (структура аналогична приведенной вы¬
ше). Хелаты почти нерастворимы в воде, хорошо растворяются
в органических растворителях. Многие современные органиче¬
ские реактивы, используемые в аналитической химии для обна¬
ружения и количественного определения ионов металлов,— ве¬
щества, образующие внутрикомплексные соединения, например
диметилглиоксим (см. выше).
Хелатные соединения приобрели значение и для получения
особо чистых металлов.
хиноны
§91. Свойства и отдельные представители. Двухатомные фено¬
лы с гидроксильными группами в орто и пара-положениях
легко окисляются, образуя циклические дикетоны — хиноны.
212
Характерная для этих соединений группировка называется
хиноидной:
я-хиноидная о-хиноидная
группировка группировка
Хиноидное кольцо не обладает ароматическим характером.
Это циклическая сопряженная система, поэтому хиноны прояв¬
ляют свойства ненасыщенных соединений, а также кетонов.
При окислении гидрохинона получается n-бензохинон, а при
окислении пирокатехина может быть получен о-бензохинон:
гидрохинон л-бензохинон пирокатехин о-бензохинсн
Нафтохиирны образуются при окислении нафталина хромо¬
вой смесью, а еще лучше — окислением 1,4-, 1,2- и 2,6-дигидрок-
синафталинов или соответствующих аминонафтолов.
О О
л-нафтохинон о-нафтохинон ал<ф«-нафтохинон
Хиноидную группировку содержат многие синтетические кра¬
сители, она встречается также во многих сложных природных
органических соединениях.
п-Бензохинон был получен А. А. Воскресенским из хинной
кислоты (1838 г.), откуда и произошло его название хинон. Это
кристаллическое вещество светло-желтого цвета, с характерным
запахом, пары его раздражают слизистую оболочку.
Гидрохинон и хинон связаны взаимным переходом. Взаимо¬
превращения их — это окислительно-восстановительная реак¬
ция:
но—CZ)—°н °=(^у=о
гидрохинон хинон
Восстановление хинона используют для промышленного по¬
лучения гидрохинона, а сам хинон получают окислением ани¬
лина C6H5NH2.
213
Хинон—хороший стабилизатор мономеров при их хранении.
Он легко реагирует со свободными радикалами, связывая их и
прекращая рост цепи полимера:
0=О=°
+ 2R.
RO
OR
«-Бензохинон используют как ингибитор процесса свободно^
радикальной полимеризации.
Антрахинон — светло-желтые кристаллы, т. гот. 286 °G. Его
получают в промышленности окислением антрацена над вана¬
диевым катализатором (V205), диеновым синтезом1 из бутадие¬
на и хинона (хиноны хорошие диенофилы — двойная связь на¬
ходится у них в сопряжении с карбонильной группой) или
конденсацией бензола с фталевым ангидридом (см. § 107);
Антрахинон используют в промышленности для синтезов
красителей, например ализарина.
Краситель ализарин известен с глубокой древности. Он пред¬
ставляет собой 1,2'-дигидроксиантрахинон. Раньше ализарин до¬
бывался из корней растения марена. Синтетически он был
впервые получен в 1868 г. Гребе и Либерманом из антрахино-
на. В настоящее время из антрахинона получают другие краси¬
тели, превосходящие ализарин по качеству и яркости окрасок.
214
ГЛАВА 12
КАРБОНОВЫЕ КИСЛОТЫ И ИХ ПРОИЗВОДНЫЕ
Карбоновыми кислотами называют соединения, содержащие в
0
1
молекуле функциональную группу —С—ОН. Эта группа носит
название карбоксильной, происходящее от названий составляю¬
щих ее групп — карбонильной и гидроксильной.
Карбоновые кислоты можно рассматривать как производные
углеводородов, в молекулах которых атом водорода замещен
на карбоксильную группу.
Водород карбоксильной группы обладает кислотным харак¬
тером. По числу карбоксильных групп различают одноосновные
(монокарбоновые), двухосновные (дикарбоновые) и многоос¬
новные (поликарбоновые) кислоты. По характеру радикала,
связанного с карбоксильной группой, кислоты могут быть пре¬
дельными, непредельными, ароматическими и др.
ОДНООСНОВНЫЕ кислоты
§ 92. Изомерия и номенклатура. Общая формула гомологиче¬
ского ряда дредельных одноосновных кислот C„H2n+jCOQH,
или СяНгпОг.
Одноосновные кислоты с алкильными или алкенильным ра¬
дикалами называют жирными кислотами, так как некоторые из
них в виде сложных эфиров входят в состав растительных и
животных жиров и масел.
Изомерия одноосновных кислот, как и альдегидов, обус¬
ловлена строением радикала.
Номенклатура. Для низших членов ряда карбоновых
кислот наиболее употребительны тривиальные названия: му¬
равьиная, уксусная, масляная и т. д.
Приставку изо используют в названии тех разветвленных
кислот, которые содержат в разветвлении метальную группу на
удаленном от карбоксильной группы конце цепи, у предпослед¬
него углеродного атома, т. е. имеют группировку (СНз)2СН—.
По правилам заместительной номенклатуры название карбо¬
новой кислоты составляется добавлением к названию углеводо¬
рода окончания овая и слова кислота. Углеродный атом карбок¬
сильной группы определяет начало нумерации и входит в счет
атомов главной углеродной цепи.
Во многих случаях кислоту рассматривают как продукт за¬
мещения атома водорода в углеводороде на карбоксильную
группу. Например, уксусную кислоту можно было бы назвать
метанкарбоновая, пропионовую — этанкарбоновая, а для кислоты
с пятичленным циклическим радикалом самое употребительное
название — циклопентанкарбоновая. Названия некоторых кар¬
боновых кислот приведены ниже:
Тривиальное
и рациональное
названия
Название по современное!
заместительной
номенклатуре
н—соон
СНз—соон
(СНа)2СН—СООН
(CHS)8CH—СН2—СООН
(СНз)2СН—СН=СН— СН2 —СООН
Муравьиная
Уксусная
Изомасляная,
диметилуксусиая
Изовалериановая,
изопропилуксусная
Метановая
Этановая
2- Метилпропановая
3- Метилбутановая
б-Метилгексен-З-овая
Кислотным радикалом или ацилом называют одновалентный
остаток R—СО, образующийся из молекулы кислоты при удале¬
нии гидроксильной группы. Названия ацилов произведены от
латинских названий кислот:
0 0 о о
II И I I
н—С— СНз—С— СНз—СНг—С— CeH5—С—
формил ацетил пропионил бензоил
Названия солей производят от названий ацилов с заменой
окончания ил на ат. Например, соли кислот, соответствующих
указанным ацилам, называют: формиат, ацетат, пропионат,
бензоат, добавляя название соответствующего металла, напри¬
мер CH3COONa— ацетат натрия.
§ 93. Общие способы получения. 1. Способы, основанные на
окислении. Со многими реакциями этого типа мы уже знакомы.
При окислении алкенов энергичными окислителями (КМп04,
Сг03) происходит разрыв молекулы по месту двойной связи.
Окисление первичных спиртов и альдегидов приводит к кисло¬
там с тем же числом атомов углерода, а при окислении кетонов
получаются кислоты с меньшим числом атомов углерода в мо¬
лекуле, чем исходный кетон (см. §§ 68 и 86).
Кислоты ароматического ряда получают окислением гомоло¬
гов бензола; при этом атом углерода боковой цепи, связанный
с бензольным ядром, окисляется до карбоксильной группы (см.
§47).
Для получения высших жирных кислот большое практиче¬
ское значение имеет окисление высших алканов нефти.
2. Способы, основанные на гидролизе. Так, гидролиз нитри¬
лов в кислой или щелочной средах сначала дает амиды кислот,
гидролизующиеся далее до кислот:
,ch2c^n Н20
ч
бензилцианид
СН2С—NH2
II
О
амид фенилук-
сусной кислоты
Н2о
>
СН2СООН
фенилуксусная
кислота
216
Этот способ позволяет получать кислоты с удлиненной на
один атом углерода цепью по сравнению с исходным алкилга-
логенидом, из которого получен нитрил (см. § 61).
Гидролиз тригалогенпроизводных, содержащих три атома
галогена у одного углеродного атома, приводит к кислотам:
сусс" ^
бензотрихлорид
Ч
ОН ‘
I
С—ОН
ь
соон
бензойная
кислота
3. Карбоксилирование металлорганических соединений (см.
§ 136).
4. Гидрокарбоксилирование алкенов. Действуя на алкены
монооксидом углерода и водой при нагревании под давлением
в присутствии никелевого катализатора, можно из продуктов
крекинга нефти получать кислоты:
С0 + Н20, 300 -с. 15МПа
СН2=СН2 -> СНз—сн2—соон
Метод эффективен, особенно для получения высших жирных
кислот. Известно и много других способов синтеза кислот.
§ 94. Физические свойства. Как видно из табл. 13, кислоты
имеют аномально высокие температуры кипения и плавления
(даже по сравнению со спиртами). Это объясняется значитель¬
ной ассоциацией, обусловленной водородной связью. Такие свя¬
зи образуются у кислот в результате взаимодействия атома во¬
дорода гидроксильной группы одной молекулы и атома кисло¬
рода карбонильной группы другой молекулы.
Водородные связи в кислотах отличаются большей проч¬
ностью, чем в спиртах, так как связь О—Н в молекулах кис¬
лот болеее поляризована. Ассоциация молекул может быть
линейной, но более характерна циклическая — димерная ассо¬
циация. В таких димерах межъядерное расстояние между двумя
атомами кислорода равно 0,27 нм (как и в ассоциированных
спиртах); атомы водорода удалены от атомов кислорода на раз-
8+ 8- 8+ 8-
R—с=о • • • н—о
Is- 8+ 8- |s+
о—н • • • о=с—Я
0,1нм 0,17нм
димерная ассоциация
ные расстояния:
* • -о=с-он-• -о=с—ОН- • -0=С—ОН-
I ' I I
R R R
линейная ассоциация
Не только в твердом и жидком состояниях, но даже в газо¬
образном состоянии и в растворах в углеводородах, большая
часть кислот находится в ассоциированном состоянии в виде
217
димеров. Только при высоких температурах димеры распада¬
ются на мономеры.
Из табл. 13 видно, что с увеличением углеводородного ра¬
дикала температуры кипения плавно возрастают. Температуры
плавления в общем также возрастают, но наблюдается своеоб¬
разное чередование: каждая кислота с четным числом углерод¬
ных атомов в молекуле плавится при более высокой темпера¬
туре, чем два ее соседа с нечетным числом атомов углерода
(рис. 22). Кислоты с нормальной цепью имеют более высокие
температуры кипения, чем их изомеры с разветвленной цепью.
С увеличением углеводородного радикала плотность кислот
уменьшается. Плотность циклических кислот больше, чем аци¬
клических.
Низшие кислоты обладают острым кислым запахом, средние
(С4—Се) имеют неприятный прогорклый запах. Высшие жир¬
ные (от С8) и ароматические кислоты запаха не имеют.
Низшие кислоты хорошо растворяются в воде. С увеличени¬
ем углеводородного остатка растворимость в воде уменьшается.
Углеводородный остаток гидрофобен (отталкивает воду, как и
углеводороды). Высшие жирные кислоты, начиная от С9, в воде
практически нерастворимы. Однако все кислоты растворяются в
водных растворах щелочей, образуя соли. Высшие кислоты, как
и углеводороды, хорошо растворимы в эфире и бензоле.
Таблица 13. Физические свойства некоторых одноосновных
карбоновых кислот
Кислота
Формула
Т. пл.,
•с
Т. кнп.,
Относи¬
тельная
плотность
d4
Константа
диссо¬
циация
в воде
при 25 °С»
*а-ю-5
Муравьиная
Н—СООН
+8,4
100,7
1,22
17,7
Уксусная
СН3-СООН
+ 16,6
118,1
1,049
1,75
Пропишювая
CjHs-COQH
-22,0
141.1
0,99
1-3
Масляная
С3Н7—СООН
-7,0
163,5
(при 15 °С)
0,96
1,5
Изомасляная
(СН3)2СН—СООН
-47,0
154,5
0,95
1,4
Валериановая
С4Н9-СООН
-34,5
187,0
0,94
1,6
Гексановая
С5НИ—СООН
-3,9
205,8
0,92
1,32
Энантовая
С6Н13—СООН
-7,5
223,5
0,918
1,28
Пальмитиновая
С15н31—СООН
+64,0
390
0,84
Стеариновая
Ci7H35-COOH
69,4
(разя.)
360
(при 80 вС)
0,84
_
Циклопента нкар-
с5н8-соон
-4
(разл.)
215
(при 80°С)
1,05
—
Циклогексанкар-
С6Н,,—СООН
+31
232
1,03
боновая
Бензойная
С6Н5—СООН
122,0
249,0
(ири 34°С)
6,5
Фенилуксусная
С6н5сн2—СООН
76,7
265
—
5,6
218
ГШШШ/
•////////////////////>
//////////////////////
so,
?! I— ! 1 I 1
0 3 S 9 12 15 Ш
Число атомоВ углерода 6 цела
Рис. 22. Температуры плавления карбоновых кислот
Рис. 23. Ориентация молекул в кристаллах жирных кислот
Кристаллы высших жирных кислот (например, стеариновой}'
обладают скользкостью (жирные на ощупь). Это явление объ¬
ясняют следующим. Молекулы в кристаллах расположены па¬
раллельными рядами: карбоксильные группы повернуты друг к
другу (рис. 23). Плоскость, в которой расположены метальные
группы, называют плоскостью спайности кристаллов. Связь ме«
жду метальными группами менее прочна, чем связь между
карбоксильными группами, и по этой плоскости происходит
скольжение.
§ 95. Строение карбоксильной группы. Химические свойства.
Карбоксильная группа формально представляет собой сочетание
двух групп — карбонильной С=0 и гидроксильной ОН. Вслед¬
ствие близкого расположения этих групп они оказывают друг
на друга сильное влияние; свойства карбоксильной группы не
являются простой суммой свойств карбонильной и гидроксиль¬
ной групп. Карбоксильная группа — это новая функциональная
группа с присущими ей свойствами. Карбоновые кислоты отли¬
чаются по свойствам как от альдегидов и кетонов, так и от
спиртов.
Гидроксильная группа кислот легче отщепляет протон, чем
гидроксильная группа спиртов. Причина усиления кислотно¬
сти — влияние полярной карбонильной группы. Карбонильный
атом углерода, несущий положительный заряд, стремится пога¬
сить дефицит электронов, притягивая электронные пары не
только связей R—С и С=0, но и свободные электронные пары
кислорода гидроксильной группы. Поэтому гидроксильный кис¬
лород сильнее оттягивает электронную пару связи О—Н, усили¬
вая положительный заряд на водороде:
(о-н
8- 8+
-о*-н
Карбоновые кислоты диссоциируют с образованием карбо-<
219
ксилат-аниона и протона; в водном растворе существует равно¬
весие:
О о
II II
R-C—ОН ч=* R—С—O' + Н RCOOH + HjO з=±= RCOO'+ Н30+
Константа равновесия является константой кислотности К*
’(«а» —от англ, acid — кислота); служит для сравнения силы
кислот (см. табл. 13).
/Са = [RCOCT] [H3O+]/[RC00H]
Водные растворы кислот изменяют окраску индикаторов и
являются электролитами. Хотя кислотные свойства у карбоно¬
вых кислот выражены сильнее, чем у спиртов и воды, по срав¬
нению с сильными минеральными кислотами — это слабые
кислоты (исключением является муравьиная кислота — кислота
средней силы).
На силу кислот, т. е. на легкость отщепления протона, вли¬
яет радикал, связанный с карбоксильной группой. Из значений
констант диссоциации (см. табл. 13) видно, что наиболее силь¬
ной является муравьиная кислота. Это можно понять, приняв
во внимание, что алкильные группы, обладая электроноотталки¬
вающими свойствами, уменьшают заряд на атоме углерода
карбоксильной группы и тем самым — его влияние на группу
ОН. Введение в радикал, особенно в a-положение, электроно¬
притягивающих групп усиливает кислотные свойства (см.
§ 142). Наличие двойной связи в а,р-положении значительно
повышает силу карбоновых кислот, облегчает образование кар-
боксилат-аниона. Ароматические кислоты сильнее алифатиче¬
ских; так, бензойная кислота СбН5СООН сильнее уксусной.
Как показали рентгеноструктурные исследования, в карбок-
силат-анионе оба атома кислорода находятся на одинаковых
расстояниях от атома углерода — при образовании аниона про¬
исходит выравнивание связей. Карбоксилат-анион — типичный
пример мезомерии. Его строение можно выразить рядом фор¬
мул: I — с указанием электронных сдвигов, II — набором гра¬
ничных формул и III—формулой с выравненными связями:
8+ 5-
—с=о
о~
I
С—о
I
L от
-> — с-о~
II
о
п
/OV.
-с< ц.
О 2
ш
Делокализация электронов, как мы уже знаем, снижает
энергию. Следовательно, образование карбоксилат-аниона энер¬
гетически выгодно и является второй причиной повышенной
кислотности карбоновых кислот по сравнению со спиртами.
Формула III особенно наглядно показывает, что связи между
220
углеродом и кислородом не являются ни простыми, ни двойны¬
ми— они выравнены.
Таким образом, ни одна из связей углерод — кислород не
имеет карбонильного характера.
Активность карбонильной группы определяется величиной
положительного заряда на атоме углерода, а в карбоксильной
группе положительный заряд атома углерода в значительной
мере погашен вследствие сопряжения со свободными электрон¬
ными парами гидроксильного кислорода. Под влиянием гидрок¬
сильной группы карбонильная группа теряет способность реаги¬
ровать с нуклеофильными реагентами.
В функциональных производных кислот нет такой выравнен-
ности углерод-кислородных связей, как в карбоксилат-анионе.
Поэтому, например, такие производные кислот, как сложные
эфиры RCOOR', вступают в некоторые реакции, характерные
для соединений, содержащих карбонильную группу.
Для кислот наиболее типичны реакции, связанные с кислот¬
ными свойствами, т. е. реакции, которые сопровождаются раз¬
рывом связи кислород—водород, а также реакции замещения
гидроксильной группы. Для остальной части молекулы возмож¬
ны реакции, обусловленные ее структурой — наличием кратной
связи, ароматического кольца, других функциональных групп.
Рассмотрим важнейшие реакции кислот.
1. Образование солей — разрыв О—Н-связи. Карбоновые кис¬
лоты легко реагируют с основаниями, основными оксидами,
активными металлами. При этом атом водорода карбоксильной
группы замещается металлом и образуются соли:
RCOOH + NaOH ——> RCOONa 2RCOOH -f Mg ——*■ (RCOO)jMg
— H2O —H2
Соли карбоновых кислот легко гидролизуются, как и вообще
соли слабых кислот. Поэтому водные растворы солей щелочных
металлов имеют щелочную реакцию:
[СН3СООГ Na+ + НОН СН3СООН + Na+ + "ОН
Минеральные кислоты, как более сильные, вытесняют карбо¬
новые кислоты из их солей:
ICH3COO)' Na+ + H+ + Cl' —► СН3СООН + Na++ СГ
Карбоновые кислоты, за исключением муравьиной кислоты
(см. § 96), устойчивы к действию концентрированных минераль¬
ных кислот.
Свойства солей карбоновых кислот—образование углеводо¬
родов при сплавлении со щелочами; сухая перегонка, приводя¬
щая к образованию кетонов и альдегидов,— уже рассматрива¬
лись при изучении других классов органических соединений.
Сами кислоты устойчивы к нагреванию, но если у а-угле-
родного атома стоит сильная электроноакцепторная группа, то
декарбоксилирование идет уже при нагревании до 100—150°С.
221
Например, из трихлоруксусной кислоты образуется хлороформ
и отщепляется С02:
СС13—СООН —► CHCI3 + CO2
2. Образование функциональных производных — разрыв
С—ОН-связи. При замещении гидроксильной группы различны¬
ми группами можно получать функциональные производные
кислот:
*->
R—с;
N>
R—сС
ХС1
*-сС
*-<°
XNH*
ангидриды
хлорангидриды
сложные
эфиры
амиды
Для этих соединений характерно, что при гидролизе из них
вновь образуются кислоты, Реакции получения этих производ¬
ных, а также их свойства рассмотрены отдельно (см. §§ 104,
106,110,111,114).
3. Галогенирование (реакция Зелинского — Гелля — Фоль-
гарда). В присутствии брома и небольших количеств красного
фосфора образуются а-бромзамещенные кислоты:
вrg (Р)
СН,—СН2—СООН ► СНзСНВг—СООН + НВг
пропионовая а-бромпропионовая
кислота кислота
4. Восстановление. Кислоты восстанавливаются с трудом.
Под действием алюмогидрида лития или диборана В2Нб они
восстанавливаются до первичных спиртов; иодоводород восста¬
навливает их в углеводороды:
4Н
RCOOH >- RCHjOH + Н20
г СН3—СООН + 6HI —► СН3—СН3 + 2НгО + 312
Легче восстанавливаются производные кислот—хлорангид-
риды, сложные эфиры.
5. Окисление. Насыщенные кислоты с нормальной углерод¬
ной цепью окисляются с трудом. Так, уксусную кислоту приме¬
няют даже в качестве растворителя в реакциях окисления хро¬
мовой смесью. В отличие от других кислот муравьиная кислота
легко окисляется, причем образуются вода и С02:
[01 гноч л
Н— ► ;С=о1 —* н2о + со*
Х)Н \ж>/ J
222
Кислоты с третичным атомом углерода дают при окислений
гидроксикислоты с тем же числом атомов углерода в молекуле!
т
(СНзЬСН—СООН >- (СНзЪСОН—соон
а-гидроксиизомасляная
кислота
При действии пероксида водорода получаются р-гидрокси-<
кислоты:
н2о3
СНз—CHj—СН2—СООН »- СНЭ—СНОН— СН*—СООН
р-гидрокеимасляная кислота
В животных организмах жирные кислоты также окисляются
до р-гидроксикислот.
Специфические свойства непредельных кислот подробно рас¬
смотрены в §§ 98—100.
§ 96. Наиболее важные представители. Муравьиная кислота
НСООН. Впервые была выделена в XVII веке из красных лес¬
ных муравьев. Содержится также в соке жгучей крапивы. Без¬
водная муравьиная кислота — бесцветная жидкость с острым
запахом и жгучим вкусом, вызывающая ожоги на коже. В про¬
мышленности ее получают нагреванием монооксида углерода С
порошкообразным гидроксидом натрия е последующей обработ¬
кой образовавшегося формиата натрия разбавленной серной
кислотой:
200 *С, 0.7 МПа H2SO4
NaOH + CO -> HCOONA - - —НСООН
Технический продукт после перегонки представляет собой
85 %-ую муравьиную кислоту (азеотропная смесь, т. кип.
107 °С).
Из спиртов и монооксида углерода можно под давлением
получать эфиры муравьиной кислоты:
ROH + CO —► HCOOR
Как уже отмечалось, муравьиная кислота отличается рядом
особенностей. Под влиянием водоотнимающих веществ (концен¬
трированная H2SO4) муравьиная кислота разлагается. Эту ре¬
акцию используют для получения чистого монооксида углерода!'
H2SO4
НСООН ► со + н2о
В присутствии некоторых мелкораздробленных металлов,
главным образом группы платины, муравьиная кислота разла*
гается с выделением диоксида углерода:
НСООН —► COj + H*
Наличие в молекуле муравьиной кислоты альдегидной груп¬
пы обусловливает ее восстанавливающие свойства. Она восстав
£23
навливает соли некоторых тяжелых металлов: HgCl2 до Hg2Cl2,
соли серебра и палладия — до свободных металлов (дает реак¬
цию серебряного зеркала). Формиат никеля восстанавливается
до свободного никеля:
190 «С
(HCOO)2Ni >- Ni + 2C02 + H2
Образовавшийся в виде тонкого порошка никель используют
для каталитического гидрирования.
Применяют муравьиную кислоту в текстильной промышлен¬
ности в качестве протравы при крашении тканей, в кожевенной
промышленности (дубление кож). Большие количества муравь¬
иной кислоты в настоящее время используют для консервирова¬
ния кормов.
При быстром нагревании формиата натрия до 400°С обра¬
зуется соль щавелевой кислоты — оксалат натрия. Эту реакцию
используют для промышленного получения щавелевой кислоты.
400 °С
2HCOONa NaOOC—COONa + Н2
Уксусная кислота СН3СООН широко распространена в при¬
роде — содержится в выделениях животных (моче, желчи, ис¬
пражнениях), в растениях (в зеленых листьях), образуется при
брожении, гниении, скисании вина, пива, содержится в кислом
молоке и сыре. Образуется при окислении многих органических
веществ. Безводная уксусная кислота плавится при +16,6°С,
кристаллы ее прозрачны как лед, поэтому ее назвали ледяной
уксусной кислотой. Впервые была получена в таком виде в
конце XVIII века русским ученым Т. Е. Ловицем. Обычная тех¬
ническая уксусная кислота имеет концентрацию 70—80 %.
Уксусная кислота может быть получена любым из общих
способов получения кислот. В промышленности ее производят
окислением ацетальдегида кислородом воздуха в присутствии
марганцевых катализаторов, уксуснокислым брожением жидко¬
стей, содержащих этиловый спирт. Последний из упомянутых
способов относится . к биохимическим (микробиологическим)
процессам. Под влиянием «уксусного грибка», зародыши кото¬
рого всегда присутствуют в воздухе, содержащие спирт жид¬
кости «скисают», образуя натуральный уксус. Процесс сложен,
но суммарно уравнение реакции можно записать так:
СН3СН2ОН + о2 —► СН3С00Н + Н20
Натуральный уксус содержит около 5 % уксусной кислоты.
Из него путем фракционной перегонки приготовляют уксусную
эссенцию (70—80 %-я кислота), используемую в пищевой про¬
мышленности для консервирования овощей, грибов, рыбы.
Уксусная кислота получается также при сухой перегонке
дерева; этот способ сейчас имеет значение лишь для утилизации
отходов лесотехнической промышленности. В последние годы
224
приобрели большое значение методы промышленного получения
уксусной кислоты из углеводородов нефти. По одному из спо¬
собов кислоту производят прямым окислением бутана:
02. 200 *С
СН3—СН2—СН2—СН3 ► 2СН3—СООН
Как и во многих других современных процессах, успех це¬
ликом зависит от чистоты сырья, в данном случае от чистоты
бутана.
Уксусную кислоту получают также из метилового спирта
оксосинтезом в присутствии тетракарбонилникеля Ni(CO)4:
№(СОЬ, давление
СНзОН + СО ■+ СН3—СООН
Разработан способ, в котором исходным веществом служит
нитроэтан (см. § 124).
Уксусная кислота устойчива к окислителям и к концентри¬
рованным минеральным кислотам. Смешивается во всех отно¬
шениях с водой, спиртом, эфиром, бензолом. Ледяная уксусная
кислота — хороший растворитель многих органических веществ.
Уксусную кислоту широко используют в химической про¬
мышленности для различных синтезов. Большие количества
кислоты расходуют для получения эфиров — этилацетата, про-
пилацетата, ацетата целлюлозы, для получения уксусного анги¬
дрида, монохлоруксусной кислоты. Ацетаты алюминия, хрома,
железа применяют при крашении тканей в качестве протрав,
соли меди—для борьбы с огородными, полевыми и лесными
вредителями (парижская зелень), соли свинца—свинцовый са¬
хар (СН3С00)2РЬ-ЗН20 и свинцовый уксус (СНзСОО)гРЬ'
•РЬ(ОН)2 для изготовления свинцовых белил, свинцовой при¬
мочки в медицине.
Кислоты с нечетным числом углеродных атомов и изострое¬
ния встречаются в природе реже, главным образом в эфирных
маслах; например, изовалериановая кислота—в эфирном масле
корней лекарственного растения валерианы. Производные гек¬
сановой и энантовой СН3(СН2)1бСООН кислот используют для
получения синтетических полиамидных волокон.
Особое место среди жирных кислот занимают высокомолеку¬
лярные кислоты — пальмитиновая CH3(CH2)i4COOH и стеари¬
новая СН3(СН2)1бСООН. Глицериды этих кислот являются
главной составной частью природных жиров и масел.
В настоящее время большие количества высших жирных
кислот получают окислением парафина. Окисление проводят,
продувая воздух через расплавленный парафин в присутствии
оксидов марганца при 100 °С. После промывки низшие кисло¬
ты (Ci—С4) переходят в раствор, откуда их выделяют отгон¬
кой. Жирные кислоты С5—С22 нейтрализуют, после чего выде¬
ляют действием серной кислоты и затем разгоняют. Из 1000 кг
парафина получается 50—60 кг низших и 600—700 кг высших
$ Органическая химия
225
кислот. Получаемые из нефти жирные кислоты используют в
технике. Из них изготовляют мыла, смазочные материалы для
защиты металлов, применяют в горно-рудной и металлообраба¬
тывающей промышленности, при изготовлении резины, линоле¬
ума, лакокрасочных изделий.
Нафтеновые кислоты, кислоты нефти (открыты М. В. Мар-
ковниковым, 1892 г.). Природная нефть содержит небольшое
количество (до 1 %) этих кислот. Главным образом это алицик-
лические кислоты с пятичленным циклом — циклопентанкарбо-
новая кислота и ее гомологи, например:
от
С ООН \^^СООН
При очистке нефтяных продуктов щелочью образуются нат¬
риевые соли этих кислот, обладающие моющей способностью,
так называемый мылонафт. Нафтенаты кобальта, марганца,
свинца и других металлов — сиккативы — используют как ката¬
лизаторы отверждения олиф (см. § 113).
Бензойная кислота СбН5СООН — наиболее важный предста¬
витель ароматических кислот. Распространена в природе в рас¬
тительном мире: в бальзамах, ладане, эфирных маслах. В жи¬
вотных организмах она содержится в продуктах распада белко¬
вых веществ. Это кристаллическое вещество, т. кип. 122 °С,
легко возгоняется. В холодной воде растворяется плохо, с по¬
вышением температуры растворимость в воде увеличивается;
хорошо растворима в спирте, эфире. Получается в промышлен¬
ности окислением толуола или других гомологов бензола с од¬
ной боковой цепью в молекуле, гидролизом бензотрихлорида
С6Н5СС13.
Бензойная кислота, подобно жирным кислотам, образует
обычные для кислот функциональные производные: бензоилхло-
рид, амид бензойной кислоты (бензамид):
О О о
fl II II
С„Н8-С—О—О—С—С„Н, СвН5—С—NH,
дибензоилпероксид бензамид
Кроме того, для бензойной кислоты возможны реакции за¬
мещения в ядро — галогенирование, нитрование, сульфирование.
Карбоксильная группа относится к мега-ориентантам, поэтому
при реакциях электрофильного замещения образуются лета-
производные: л-бромбензойная кислота, ж-нитробензойная кис¬
лота и др.
§ 97. Мыла и моющие средства. Соли высших жирных кислот
называют мылами. Обычное твердое мыло представляет собой
смесь натриевых солей главным образом пальмитиновой и сте¬
ариновой кислот. Смесь солей калия дает жидкое зеленое мыло.
О
свн5—cl;—ci
бензоилхлорид
226
Получают мыла из растительных и животных жиров гидроли¬
зом (омылением) их в щелочной среде или из синтетических
жирных кислот:
н2о, "ОН
Жир *• Глицерин + Соли жирных кислот (мыло)
Твердые жиры (природные и полученные гидрогенизацией
жидких жиров) дают твердые мыла. Жидкие масла (содержа¬
щие много ненасыщенных жидких кислот) дают более мягкие
мыла.
Моющая способность мыла связана с его поверхностной ак¬
тивностью, т. е. со способностью резко снижать поверхностное
натяжение воды, вызывая смачивание поверхностей (или час¬
тиц) и способствуя образованию эмульсий, суспензий и др. Это
сложный физико-химический процесс.
Все загрязнения гидрофобны (отталкивают воду), вода их
не смачивает, а скатывается с них в виде капель. Если в воду
добавить мыльный раствор или другое поверхностно-активное
вещество, смачивающая способность воды возрастает.
В поверхностно-активных веществах (ПАВ). достаточно
длинная углеводородная цепь, обладающая гидрофобными (во¬
доотталкивающими) свойствами, связана с гидрофильной (лю¬
бящей воду) группой. Такая молекула способна взаимодейство¬
вать с органической и водной фазами системы. По такому же
принципу построены молекулы мыла RCOONa. Гидрофильная
группа погружена в воду, а гидрофобная выталкивается из
воды (рис. 24). Вследствие такой ориентации молекул мыла
на границе двух фаз поверхностное натяжение снижается, а
смачивающая и пенообразующая способности мыльного раство¬
ра и, следовательно, его моющее действие возрастают. В жест¬
кой воде (богатой солями кальция, магния, железа) образуются
хлопья нерастворимых солей, и моющее действие мыла снижа¬
ется:
2Cl7H35COONa + Са>+ —>• (С17Н35СОО)2Са + 2Na+
В морской воде обычным мылом стирать нельзя. Осадок
солей кальция и магния делает ткань жесткой. Недостатком
мыла является и то, что при действии на него воды оно час¬
тично гидролизуется как соль слабой кислоты и создается ще¬
лочная среда, действующая от¬
рицательно на некоторые типы
ткани.
В настоящее время для стир¬
ки в быту и для промывки шер¬
сти и тканей в промышленности
Рис. 24. Расположение молекул в рас¬
творах мыла
8*
227
используют синтетические моющие средства (детергенты), кото¬
рые обладают более сильным моющим действием по сравнению
с мылом, не портят тканей и применимы в жесткой и морской
воде. Молекулы синтетических моющих веществ, так же, как и
мыла, содержат полярную гидрофильную группу (но другого
характера), связанную с углеводородным радикалом (С12—Qg,
как и в обычном мыле). Сырьем для получения синтетических
моющих веществ являются не пищевые жиры, а продукты пе¬
реработки нефти. При окислении парафина воздухом кроме выс¬
ших жирных кислот в качестве побочных продуктов образуются
высшие жирные спирты. Высшие жирные спирты можно полу¬
чить также каталитическим гидрированием высших жирных
кислот:
Н2
RCOOH ► RCH2OH
Под действием концентрированной серной кислоты высшие
жирные спирты образуют кислые эфиры — алкилсерные кисло¬
ты, обладающие сильными кислотными свойствами и дающие со
щелочами соли — алкилсулыраты:
H2SOi NaOH
RCH2OH ► RCH2—OSO3H ► RCH2—0S03Na
Эти соли обладают прекрасной моющей способностью.
При фотохимическом сульфохлорировании парафинов (см.
§ 21) образуются сульфонилхлориды, из которых при действии
щелочи получаются соли сульфокислот — алкилсульфонаты
RS03Na, также являющиеся хорошими моющими средствами.
Алкиларилсульфонаты получают из ароматических угеводо-
родов с длинной боковой цепью, подвергая их сульфированию
и последующей обработке щелочью:
H2S04 NaOH
С8Н,7—С6Н5 ► С8Н,7—СбН4—S020H ► С»Н17—СВН„—SOaONa
Алкилсульфаты, алкилсульфонаты и алкиларилсульфонаты
можно применять в жесткой воде (их кальциевые соли раство¬
римы). Они, являясь солями сильных кислот, не гидролизуются,
и, таким образом, не создается щелочная среда. В качестве
катиона в состав таких поверхностно-активных веществ может
входить не только натрий, но и калий, аммоний, органические
основания, например амины, и др.
Перечисленные типы соединений составляют группу анионо¬
активных моющих средств. Существуют и другие моющие веще¬
ства — катионоактивные, амфотерные, неионогенные. Поверхно¬
стно-активные вещества, получающиеся при реакции этиленок-
сида с высшими спиртами и алкилфенолами, обладают неионо¬
генным характером и используются для получения синтетиче¬
ских моющих средств, которые не боятся жесткой воды. Их
применяют в текстильной промышленности, для мойки и обра¬
ботки шерсти, в кожевенной промышленности и др.
228
Амфотерными свойствами обладают вещества, в которых од¬
новременно содержатся аминогруппы и кислотные остатки,
например С!2Н25—N+(CH3)2—СН2—СОО~; по химической при¬
роде это внутренние соли.
Современные синтетические моющие средства (СМС) пред¬
ставляют собой сложные композиции, в состав которых входят
поверхностно-активные вещества рассмотренных выше типов,
силикаты, фосфаты, душистые вещества, оптические отбеливате¬
ли для повышения белизны тканей. Моющая способность син¬
тетических моющих средств примерно в 10 раз выше моющей
способности обычного мыла. При всех своих преимуществах
синтетические моющие средства имеют, однако, и недостатки:
попадая в водоемы, они вызывают образование обильной пены;
входящие в их состав фосфаты «удобряют» водоемы и вызыва¬
ют бурный рост водных растений. Выходом из положения явля¬
ется использование биоразлагаемых СМС: органический остаток
у них должен представлять собой неразветвленную алифатиче¬
скую цепь. Это показано выше на примере амфотерного детер¬
гента.
НЕПРЕДЕЛЬНЫЕ ОДНООСНОВНЫЕ КИСЛОТЫ
§ 98. Изомерия и способы получения. В молекулах непредель¬
ных кислот в радикале, связанном с карбоксильной группой,
имеются кратные связи. Ненасыщенные кислоты с одной двой¬
ной связью в молекуле имеют общую формулу C„H2n_iCOOH.
Простейшей является кислота СН2 = СН—СООН — акриловая
кислота. Изомерия ее гомологов обусловлена изомерией угле¬
родного скелета и положением двойной связи по отношению к
карбоксильной группе. Так, для кислот состава С3Н5СООН
известны следующие изомеры:
СН3—СН=СН—СООН
кротоновая
(бутеи-2-овая)
кислота
СН3
СН2=С—СООН
метакриловая
(2-метилпропен-
2-овая) кислота
сн2=сн-сн2-соон
винилуксусная
(бутен-3-овая)
кислота
Кроме того, как и для других этиленовых соединений, для
ненасыщенных кислот возможна цис-транс-изомерия:
СНз—С—Н
СНз—С-Н
Н—С—СООН ноос—с—н
грамс-изомер;
кротоновая
кислота
(т. пл. 72 °С,
т. кип. 189 °С)
цис-изомер;
изокротоновая
кислота
(т. пл. 15,5 °С,
т. кип. 172 °С)
229
Непредельные кислоты могут быть получены следующими
способами.
1. Из замещенных насыщенных кислот путем образования в
радикале двойной связи.
а) Дегидрогалогенирование галогензамещенных кислот:
СНз—СН2—СНС1—соон —
КОН, спирт
СНз—сн=сн—соон
кротоновая кислота
-нс:
а-хлормасляная кислота
СНз—СНС1—СНз—СООН —
Р-хлормасляная кислота
б) Дегидратация гидроксикислот:
НОСНз—СНз—СООН
СНз=СН—соон
—н3о
Р-гидроксипропиоповая акриловая кислота
Особенно легко протекают реакции для соединений, содер¬
жащих отщепляемые группы (С1, ОН и др.) в р-положении.
2. Из галогенсодержащих непредельных соединений синте¬
зом через нитрилы:
KCN HgO (н+)
СН2=СН—СН2С1 ► СН2=СН—СН2—CN *■
—>■ СН2=СН—сн2—СООН
§ 99. Физические и химические свойства. Кислотность непре¬
дельных кислот выше, чем соответствующих предельных. Так,
акриловая кислота примерно в четыре раза сильнее пропйоно-
вой. Влияние кратной связи особенно сказывается, если она
находится в а,p-положении к карбоксильной группе. В этом
случае протон отщепляется легче, так как образуется устойчи¬
вый вследствие сопряжения карбоксилат-анион СН2=СНСОО~.
По мере удаления кратной связи от карбоксильной группы ее
влияние уменьшается.
Ряд свойств непредельных кислот обусловлен свойствами
карбоксильной группы (образование солей, сложных эфиров
и др.) и двойной связи (различные реакции присоединения).
Специфическими свойствами обладают кислоты с близко распо¬
ложенной от карбоксила двойной связью — а,р-непредельные
кислоты. Так, присоединение галогеноводородов и гидратация
идут для них против правила Марковникова по типу 1,4-присо¬
единения. На первой стадии протон присоединяется к кислоро¬
ду карбоксильной группы, нуклеофил атакует p-углерод, а
двойная связь переходит в положение 2,3. Далее промежуточ¬
ный продукт изомеризуется:
б+ fl- fl+ б-
СН2=СН—С=0 + Н+ + Вг~ —> ГСН2Вг—СН=С—ОНт —►
он L он J
—► CHjBr—СНз—СООН
230
Для непредельных кислот характерна реакция присоедине¬
ния брома (или иода). По количеству присоединившегося брома
(или иода) можно определить число кратных связей.
Аналогично идут и другие реакции присоединения.
В отличие от трудно окисляемых предельных кислот непре¬
дельные кислоты, как и алкены, окисляются легко. При осто¬
рожном окислении щелочным раствором перманганата калия
образуются дигидроксикислоты:
[OJ. н2о
сн2=сн—соон ► СН2ОН—снон—соон
акриловая кислота дигндроксипропионовая
кислота
При энергичном окислении происходит разрыв молекулы по
месту двойной связи и образуется смесь продуктов, по которым
можно установить место двойной связи. Таким образом было
установлено положение двойной связи в олеиновой кислоте:
СН3—(СН2)7—СН=СН—(СН2)7—соон
олеиновая кислота
—► СН3—(СН2)г—СООН + НООС—(СН2)7—СООН
пеларгоновая кислота азелаииовая кислота
§ 100. Отдельные представители. Акриловая (пропеновая) кис¬
лота представляет собой жидкость с резким запахом, т. кип.
140 °С, т. пл. -)-130С, df 1,06. С водой смешивается во всех
отношениях. В промышленности ее получают гидролизом акри¬
лонитрила или карбонилированием ацетилена (реакция Реппе):
н2о
СН2=СН—CN *- СН2=СН—СООН
Ni(CO)4. CuCl
сн=сн + со + н2о ► СН2=СН—СООН
Разработан также двухстадийный процесс получения акри¬
ловой кислоты из пропилена. Катализаторами для обеих стадий
служит смесь В1гОз и МоОз, выходы на каждой из стадий
«90%.
СН2=СН—СН3 СН2=СН—СНО —СН2=СН—СООН
Сама кислота и ее производные (акрилонитрил, эфиры) лег¬
ко полимеризуются и служат сырьем для получения разнооб¬
разных высокомолекулярных соединений..
Широко используется в промышленности метиловый эфир
акриловой кислоты — метилакрилат, который легко полимеризу-
ется с образованием прозрачных продуктов. Его получают из
акрилонитрила и метилового спирта в присутствии серной кис¬
лоты:
СНзОН. h2so4, н2о
СН2=СН—CN * СН2=СН—COOSH,
231
Метакриловая кислота — жидкость, т. кип. 160°С, т. пл.
15°С, df 1,05. Получают в промышленности из ацетона и си¬
нильной кислоты:
9^ гидролиз, 9Нз
HCN | дегидратация (H2SO4) |
(СН3)2С=0 ► (СН3)2С—CN -> СН2=С—СООН
Другой путь основан на окислении изобутилена (аналогич¬
но синтезу акриловой кислоты из пропилена).
Производные метакриловой кислоты также служат мономе¬
рами в реакциях полимеризации. Наиболее важным производ¬
ным является метилметакрилат, который получают из ацетон¬
циангидрина при нагревании с метиловым спиртом в присутст¬
вии H2SO4:
ОН СН3
I СНзОН, H2SO4. н2о |
(СН3)2С—CN > СН2=С—СООСНз
При полимеризации метилметакрилата получают полиметил¬
метакрилат— органическое стекло (см. § 187).
Сорбиновая кислота— представитель кислот с двумя двой¬
ными связями в молекуле СН3СН=СНСН=СНСООН. Это кри¬
сталлическое вещество, т. пл. 134 °С. Встречается в природе,
а также получается синтетически (см. § 103). Сорбиновая
кислота — ценное консервирующее средство для пищевых про¬
дуктов. В концентрации около 0,1 % она предохраняет от порчи
мясные и рыбные продукты, готовые кулинарные изделия, сыр,
предотвращает скисание плодово-ягодных соков, сиропов, вин
и др.
Олеиновая кислота С17Н33СООН — одна из важнейших выс¬
ших непредельных кислот. Весьма распространена в природе: в
виде эфиров с глицерином входит в состав всех жиров (в не¬
которых жирах до 50 % от общей массы кислот). Она представ¬
ляет собой бесцветную жидкость, затвердевающую на холоду
(т. пл. 14°С).
Общей формуле СНз(СН2)7СН==СН(СН2)7СООН соответст¬
вуют два геометрических изомера: цис- (олеиновая кислота) и
транс- (элаидиновая кислота):
СНз—(СН2)7—С—Н СНз—(СН2)7—с—н
НООС—(СН2)7—с—н
Н—С—(СН2)7—СООН
олеиновая кислота
элаидиновая кислота
Элаидиновая кислота в природе не встречается. Ее можно
получить из олеиновой кислоты каталитическим действием азо¬
тистой кислоты. В отличие от олеиновой кислоты она представ¬
ляет собой твердое вещество, т. пл. 51—52 °С.
Линолевая С17Н31СООН и линоленовая Ci7H29COOH кисло¬
ты входят в состав растительных масел. Например, льняное
232
масло содержит около 25 % линолевой кислоты и до 58 % ли-
ноленовой. Обе кислоты при гидрировании дают стеариновую
кислоту, что подтверждает нормальное строение их углеродного
скелета:
СН3—(CHS)4—СН=СН—СНа—СН=СН—(СН2)7—СООН
линолевая кислота (т. пл. —5 °С)
СН3— СН2—СН=СН— СН2—СН=СН— СН2—СН=СН-(СН2)7—СООН
линоленовая кислота (т. пл. 11 °С)
Обе кислоты легко окисляются кислородом воздуха и поли-
меризуются (см. § 113).
Коричная кислота С6Н5СН=СНСООН известна в виде двух
геометрических изомеров, обладающих разными физическими
константами:
С6Н6-С—Н С6Н5-С—Н
ноос—С—н
Ч«с-коричная
(т. пл. 57 °С)
Н—С—СООН
траяс-коричная
(т. пл. 133 °С)
Одним из важных способов получения коричной кислоты яв¬
ляется конденсация бензальдегида с уксусным ангидридом в
присутствии ацетата натрия (реакция Перкина)-.
С6Н8—СНО + (СН3С0)20 —>• СбН6—СН=СН—СООН + СНзСООН
Коричная кислота входит в состав душистой бальзамной
смолы стиракс (ладан). При нагревании коричная кислота де-
карбоксилируется и образуется стирол (см. § 49):
С6Н5—СН=СН—СООН —> С6Н5—сн=сн2 + со2
ДВУХОСНОВНЫ^ кислоты
Простейшим представителем этого ряда кислот является щаве¬
левая кислота НООС—СООН; ближайший ее гомолог — малоно¬
вая кислота НООС—СН2—СООН, в которой карбоксильные
группы разделены группой СН2. Удлиняя последовательно цепь
на группу СН2, можно получить ряд двухосновных кислот.
§ 101. Номенклатура, общие способы получения, физические
свойства. Многие кислоты носят тривиальные названия (см.
табл. 14). В названиях по современной номенклатуре двух¬
основные кислоты получают окончание диовая или дикарбоно-
вая кислота. Например, щавелевая кислота — этандиовая кис¬
лота, малоновая — пропандиовая или метандикарбоновая и т. д.
Наиболее употребительны тривиальные названия.
От каждого из членов ряда двухосновных кислот (кроме
хцавелевой) можно вывести свой гомологический ряд с одина-
233
новым для всех членов ряда взаимным расположением карб¬
оксильных групп:
/СООН
Н2<
хсоон
/СООН
сн3—н</
ХЮОН
СНз^ /СООН.
С2Н5/ ^соон
малоновая
кислота
метилмалоновая
кислота
метилэтилмалоновая
кислота
Упомянем следующие общие способы получения двухоснов¬
ных кислот. 1. Окисление двупервичных гликолей. Реакция идет
через промежуточное образование диальдегида:
Pi Р1
НОСН2—СН2ОН ► ОНС—СНО >- НООС—СООН
этиленгликоль глиоксаль щавелевая кислота
2. Действие цианида калия на дигалогенпроизводные с по¬
следующим гидролизом динитрилов:
KCN Н20
С1СН2—СН2С1 ► ncch2—ch2cn ► НООССН2—CHiCOOH
1,2-дихлорэтан динитрил янтарная кислота
янтарной
кислоты
Двухосновные кислоты — кристаллические вещества. Низ¬
шие члены ряда значительно растворимы в воде. С увеличени¬
ем числа атомов углерода в молекуле растворимость уменьша¬
ется, но у соединений с нечетным числом атомов углерода рас¬
творимость выше. Температуры плавления у соединений с чет¬
ным числом атомов углерода выше, чем у соединений с нечет¬
ным числом углеродов (см. § 94).
Таблица 14. Физические свойства некоторых двухосновных кислот
Кислота
Формула
T. пл..
Раство*
римость
Константы
диссоциации
в воде при 25 °С
•с
при 20 вС,
в г на 100 г
воды
«аГ1»"5
«а*'10-5
Щавелевая
НООС—СООН
189,5
8,6
5400
5,2
Малоновая
НООС-СН2—СООН
135,6
73,5
140
0,22
Янтарная
НООС—(СН2)2—СООН
185,0
5,8
6,6
0,25
Г лутаровая
НООС—(СН2)з—СООН
97,5
63,9
4,7
0,29
Адипиновая
НООС—(СН2>4—СООН
152
1,5
3,7
0,39
Пимелино-
вая
НООС—(СН2)6—СООН
105
5,0
3,1
0,37
Малеиновая
НООС—СН=СН—СООН
130
79
1000
0,55
Фумаровая
НООС—СН=СН—СООН
288
0,7
86
4,1
Фталевая
о-СвН4(СООН)2
206-208
(разл.)
0,8
110
0,4 .
234
Дикарбоновые кислоты в водном растворе диссоциируют
последовательно:
НООС—СООН *=>: (НООС—СООГ + н+
(НООС—COO)' (ООС—COO)s_ + н+
В табл. 14 приведены обе константы диссоциации. Двухос¬
новные кислоты по силе превосходят одноосновные. Влияние
одной карбоксильной группы на кислотность другой тем боль¬
ше, чем ближе друг к другу они находятся в молекуле. Наи¬
более сильной является щавелевая кислота (сильнее уксусной
примерно в 2000 раз). Вторая константа диссоциации у кислот
значительно ниже первой (отрыв второго протона более тру¬
ден).
§ 102. Химические свойства. По общим химическим свойствам
дикарбоновые кислоты напоминают монокарбоновые: образуют
соли, сложные эфиры, хлорангидриды и др. В зависимости от
того, вступает ли в реакцию одна карбоксильная группа или
две, получаются кислые и средние соли, кислые и средние слож¬
ные эфиры, полные и неполные хлорангидриды и др.
Взаимное расположение карбоксильных групп в молекуле
оказывается на свойствах соединений. При близком расположе¬
нии карбоксильных групп возможность их взаимодействия уве¬
личивается. Если карбоксильные группы разделены цепью из
шести и более атомов углерода, их взаимное влияние сказыва¬
ется мало.
Из специфических свойств следует отметить отношение к
нагреванию; характер превращений зависит от расстояния меж¬
ду карбоксильными группами. Щавелевая кислота, у которой
карбоксильные группы находятся в положениях 1,2, при быст¬
ром нагревании декарбоксилируется, образуя муравьиную
кислоту и диоксид углерода. Если нагревание вести в присут¬
ствии концентрированной серной кислоты, то разлагается и му¬
равьиная кислота:
H2SO4 200 °с
CO, + CO + HjO ■* НСХ)С—СООН ► СОа + НСООН
Малоновая кислота и ее гомологи, т. е. кислоты, в молеку¬
лах которых карбоксильные группы находятся в положениях
1,3 (обе карбоксильные группы связаны с одним атомом угле¬
рода), легко декарбоксилируются с образованием одноосновной
уксусной кислоты и ее гомологов:
НООС—СНа—СООН ——* СН3—СООН
— COj
R—СН(СООН)2 -3^- R—CHj—СООН
Близость карбоксильных групп обусловливает термическую
неустойчивость соединений. При нагревании янтарной и глута-
ровой кислот или их гомологов, т. е. соединений, в которых
235
карбоксильные группы находятся в положениях 1,4 и 1,5, от¬
щепляется вода и образуются внутренние циклические ангидри¬
ды с пяти- и шестичленными циклами:
СН2-СО
\
/
СН2—со
о
сн2—со
Н2С^
сн2—со
янтарный глутаровый
ангидрид ,ангидрид
Так же ведет себя и ароматическая о-дикарбоновая кисло¬
та— фталевая кислота, превращаясь во фталевый ангидрид
(см. § 52).
Адипиновая кислота декарбоксилируется и циклизуется в
циклопентанон:
300 °С, МпО
НООС—(СН2)4—СООН *-
=Ю f С02 + н2о
§ 103, Отдельные представители. Щавелевая кислота сущест¬
вует обычно в виде кристаллогидрата НООС—С00Н-2Н20, т. пл.
101°С; безводная кислота плавится при 189,5°С. Распро¬
странена в природе в виде оксалатов и в свободном виде. Ок¬
салат кальция содержится во всех растениях, калиевая кислая
соль КООС—СООН — в щавеле, кислице. Оксалаты щелочных
металлов растворимы в воде, оксалат кальция практически не¬
растворим в холодной воде. При некоторых нарушениях обмена
веществ в организме человека оксалат кальция накапливается,
вызывая образование камней в печени, почках, мочевыводящих
путях. Малая растворимость оксалата кальция используется в
аналитической химии для качественного и количественного
определения кальция.
Характерными свойствами щавелевой кислоты являются
описанная выше способность декарбоксилироваться при нагре¬
вании и легкая окисляемость. Являясь продуктом окисления
многих органических веществ, щавелевая кислота сама легко
окисляется, например перманганатом калия:
[01
НООС—СООН —► со2 + н2о
Это используют, например, в аналитической химии для уста¬
новления титра раствора перманганата калия. В промышлен¬
ности щавелевую кислоту применяют при крашении тканей, как
отбеливающее средство, для удаления с тканей ржавчины, чер¬
нил.
Промышленный способ получения щавелевой кислоты — на¬
гревание формиата натрия (см. § 96).
Малоновая кислота. В промышленности малоновую кислоту
получают из монохлоруксусной кислоты через мононитрил с
236
последующим гидролизом:
kcn н2о
CJ—СН2—СООН 5» NC—сн2—соон *• ноос—сн2—соон
Карбоксильные группы активируют водородные атомы стоя¬
щей между ними группы СНг. Повышенная реакционная способ¬
ность водородных атомов сказывается как на свойствах самой
кислоты, так и на свойствах ее эфиров, что используется в
различных синтезах. Так, например, при конденсации с альде¬
гидами и кетонами малоновая кислота дает а,р-непредельные
кислоты:
CHS—СНО + СН2(СООН}2 СН3—СН=С(СООН)2
этилиденмалоновая
кислота
—> СН3—СН=СН—СООН
кротоновая кислота
Из непредельных альдегидов по этой реакции получают по-
лненовые кислоты:
СНз—СН=СН— СНО + СН2(СООН)2 —>. СН3—СН=СН—СН=С(СООН)2 —►
—► СНз—СН=СН—СН=СН—СООН + С02
сорбиновая кислота
Наиболее важным производным малоновой кислоты являет¬
ся ее диэтиловый эфир, так называемый малоновый эфир. Это
жидкость с приятным запахом (т. кип. 199 °С), широко исполь¬
зуется для различных лабораторных синтезов. При действии на
малоновый эфир металлического натрия или этилата натрия
получается натриймалоновый эфир, который легко алкилирует¬
ся галогенпроизводными, образуя замещенные у углерода про¬
изводные малонового эфира:
/СООС2Н5
Н2С(
хСООС2Н5
Na
-'ЛНа
[/СООСгНз
не'
'СООС2Н5,
СНз!
—Nal
/СООСцНз
СН3-НС(
ХСООС2Н5
натриймалоновый
эфир
метилмалоновый
эфир
В полученном эфире атом водорода группы СН также может
быть замещен натрием и затем опять углеводородным радика¬
лом, в результате чего образуются эфиры диалкилмалоновых
кислот:
/СООС2Н5 г.- с2н5вг снз\ /СООС2Н5
СНз—нс( ► ;с(
^СООС2Н6 С2Н5/ ^COOCjHs
эфир метилэтилмалоновой
кислоты
237
При гидролизе алкил- и диалкилмалоновых эфиров образу¬
ются соответствующие замещенные малоновые кислоты. При де-
карбоксилировании этих кислот получают одноосновные кис¬
лоты:
СН3 СН3
С2Н5-С(СООН)2 ——> С2Н5—СН—СООН
2-метилмасляная
кислота
Адипиновая кислота НООС—(СН2)4—СООН легко раство¬
рима в спирте, трудно — в холодной воде, в эфире. В промыш¬
ленности ее получают окислением циклогексанона или циклоге¬
ксана (см. §26). Интересным и перспективным является синтез
адипиновой кислоты гидрокарбоксилированием бутадиена
j(200°C, 20—50 МПа, катализатор дикобальтоктакарбонил):
СН2=СН—СН=СН2 + 2СО + 2Н20 —► НООС—(СН2)4—СООН
Большие количества адипиновой кислоты используют для
получения синтетического волокна найлон. В пищевой промыш¬
ленности адипиновая кислота может заменять лимонную и вин¬
ную кислоты. Эфиры адипиновой кислоты — бутил- и октилади-
пинаты — применяют в производстве пластических масс в каче¬
стве пластификаторов.
Фталевые кислоты. При окислении ароматических углеводо¬
родов, содержащих в молекуле две боковые цепи, в зависимо¬
сти от расположения боковых цепей могут быть получены три
изомерные дикарбоновые кислоты:
фталевая изофталевая терефталевая
кислота кислота кислота
Фталевая кислота — кристаллическое вещество, плавится
при 206—208°С (разл.), растворима в горячей воде. Ее полу¬
чают окислением нафталина (см. § 52). Широкое применение
нашли диэфиры фталевой кислоты — дибутил- и диоктнлфтала-
ты, которые имеют очень малые давления паров, высокие тем¬
пературы кипения. Их используют вместо ртути в вакуумных
и диффузионных насосах, в качестве пластификаторов для пла¬
стических масс.
Терефталевая кислота при нагревании до 300°С возгоняет¬
ся, не плавясь, трудно растворима в воде и органических рас¬
творителях. В промышленности ее получают окислением «-кси¬
лола или цимола — вещества, выделяемого из скипидара (жид-
233
кофазное окисление, катализатор соли Со, Мп):
Важным способом получения терефталевой кислоты являет¬
ся изомеризация калиевой соли фталевой кислоты в присутст¬
вии катализатора (фталат цинка и кадмия):
COOK
COOK
400-С
КООС-
COOK
Терефталевую кислоту производят в промышленности в
большом количестве и используют в качестве сырья для полу¬
чения синтетического волокна лавсан (см. § 191).
Малеиновая и фумаровая кислоты — ненасыщенные двухос¬
новные кислоты. Они являются геометрическими изомерами:
Н—С—СООН НООС—С—Н
Н—С—СООН
Ч«с-изомер;
малеиновая
кислота
(т. пл. 130 °С)
Н—С—СООН
транс-изомер;
фумаровая
кислота
(т. пл. 288 *С)
Явление цис-транс-тоыерш этиленовых соединений было
подробно изучено именно на этих кислотах.
Малеиновая кислота менее устойчива, чем фумаровая. Под
влиянием некоторых веществ (следов иода, брома, азотистой
кислоты) она переходит в более устойчивую фумаровую кисло¬
ту. Фумаровая кислота переходит в малеиновую под действием
ультрафиолетового света или при нагревании, т. е. для этого
перехода требуется подвод энергии. Фумаровая кислота мало
растворима в воде (менее 1 %), малеиновая хорошо растворима
(около 50%). Малеиновая кислота более сильная кислота, чем
фумаровая.
Обе кислоты дают характерные реакции на двойную связь
(обесцвечивают бромную воду, перманганат калия), а также
реакции на карбоксильные группы, причем получается два ряда
производных (кислые и средние соли, кислые и средние слож¬
ные эфиры и др.). При восстановлении обе кислоты переходят
в янтарную кислоту. Все это доказывает их одинаковое химиче¬
ское строение. Однако только из малеиновой кислоты образует¬
239
ся циклический ангидрид, что подтверждает ее цис-конфигура-
цию:
СН—СООН
II
СН—СООН
СН—со
/О + п2о
СН—со
При транс-расположении карбоксильные группы слишком
далеки друг от друга, чтобы замкнуться в циклический ангид¬
рид.
Фумаровая кислота распространена в природе, встречается
в грибах, лишайниках, в незначительном количестве — в живот¬
ных клетках, участвует в обмене веществ. Малеиновая кислота
в природе не найдена; ядовита.
Малеиновую кислоту получают в промышленности в виде
малеинового ангидрида при окислении ряда веществ кислородом
воздуха в присутствии соединений ванадия (V):
о2
СН—СО
\
СН-
:—do
о
— СНа—СН=СН— СНО
— СНз—СН=СН—СН3
ГАЛОГЕНАНГИДРИДЫ КАРБОНОВЫХ КИСЛОТ
Галогенангидридами кислот называют такие производные кис¬
лот, у которых гидроксил карбоксильной группы замещен на
галоген; эти соединения называют также ацилгалогенидами.
§ 104. Номенклатура, способы получения и свойства. Названия
галогенангидридов строят аналогично названиям алкилга-
логенидов, но исходят в этом случае из названия ацильной
группы, например:
О
II
R—С—X
ацилгало-
генид
О
hJ-p
фтористый
формил
(формил-
фторид)
о
СНз—С—С1
хлористый
ацетил
(ацетил-
хлорид)
О
II
С6Н6—С—С1
хлористый
бензоил
(бензоил-
хлорид)
Их получают разными путями. Выбор реагента связан с ме¬
тодом разделения получаемых продуктов — температура кипе¬
ния галогенангидрида должна отличаться от температуры кипе¬
ния побочных продуктов. Практически используют следующие
способы.
1. Действие на кислоты галогенидов фосфора:
ЗСН:
О
ОН + РВгз
. О
II
ЗСНз—С—Br + Н3РО3
о
о
С6Н6—С—ОН + РС18 —> С6Н5—с—С1 + РОСЦ + НС1
240
2. Действие на кислоты или ангидриды кислот тионилхло-
рида S0C12 в присутствии пиридина как катализатора:
soci2
СН3—СН2—СН2—СООН ► СН3—СИ2—СН2—СОС1 + S02 + НС1
масляная кислота бутирилхлорид
Этот способ удобен тем, что в качестве побочных продуктов
образуются газообразные вещества, поэтому продукт реакции
легко получить в чистом виде.
Фторангидриды готовят реакцией хлорангидридов с KHF2:
R—СОС1 + KHF2 —► R—COF + KC1+IIF
Иодангидриды мало устойчивы. Наиболее важны хлорангид-
риды.
Хлорангидриды низших кислот — жидкости с резким запа¬
хом, хлорангидриды высших кислот — твердые вещества. Они
не растворяются в воде, но гидролизуются. Так, многие жид¬
кие хлорангидриды дымят на воздухе:
R—СОС1 + Н20 —> RCOOH + НС1
Это весьма реакционноспособные вещества. Реакционным
центром является карбонильный углерод, несущий значительный
положительный заряд. Этот заряд возникает под действием
двух сильноэлектроотрицательных атомов — кислорода и хлора.
В связи с этим карбонильный углерод легко реагирует с нук¬
леофильными реагентами. На первой стадии происходит присо¬
единение нуклеофильного реагента (воды, спирта, амина) по
двойной связи С=0. Затем от промежуточного продукта отщеп¬
ляется хлороводород. Так, в реакциях со спиртом образуется
сложный эфир:
г ОСН3
6+ Л- ••
R—С—С1 + Н—О—СНз — ►
Ан
R—С—О СН3 + НС1
II
О
В результате реакции в молекулу органического соединения
вводится ацильная группа. Эти реакции носят название реакций
ацилирования. К ним относятся следующие реакции.
1. Ацилирование воды (гидролиз) — образование кислот:
СНз—СОС1 + н20 —► СНз—СООН + НС1
ацетилхлорид уксусная
кислота
2. Ацилирование спирта (алкоголиз)—образование слож¬
ных эфиров:
СНз—С ОС1 + НОС2Н5 —► СНз—СООС2Н6 + НС1
241
3. Ацилирование аммиака (аммонолиз)—образование ами¬
дов кислот:
4. Ацилирование ароматических углеводородов (синтез кето¬
нов по методу Фриделя — Крафтса) уже рассматривалось ранее
(см.§ 84).
§ 105. Отдельные представители. Простейшим представителем
хлор ангидридов является формилхлорид НСОС1. Этот ангидрид
не выделен в свободном виде (распадается на СО и НС1), од¬
нако возможность его существования подтверждается реакция¬
ми, например реакцией Гаттермана (см. § 84).
Формилфторид HCOF — газ с сильным запахом, сжижаю¬
щийся при —26 °С. При комнатной температуре разлагается на
СО и HF. Получен впервые А. Н. Несмеяновым из бензоилфто-
рида и муравьиной кислоты.
Ацетилхлорид СН3СОС1 — жидкость, кипящая при 51 °С.
На воздухе дымит, с водой реагирует бурно. Широко использу¬
ется как ацетилирующее средство. В промышленности его полу¬
чают из безводного ацетата натрия и сульфурилхлорида (диок¬
сид-дихлорид серы):
Бензоилхлорид С6Н6СОС1 — жидкость, т. кип. 197 °С, об¬
ладает неприятным раздражающим запахом. Получают дейст¬
вием РСЬ на бензойную кислоту (см. § 104) или действием
хлора на бензальдегид (см. § 87). Используют для разных
синтезов как бензоилирующее средство.
Фосген СОС12 — полный хлорангидрид угольной кислоты,
т. кип. 8,3 °С, имеет запах свежего сена. В промышленности
его получают из монооксида углерода и хлора в присутствии
активированного угля:
Фосген использовали в первую мировую войну в качестве
боевого отравляющего вещества удушающего и общеядовитого
действия. Имеет скрытый период действия — в течение первых
двух часов не дает симптомов отравления, токсичнее хлора при¬
мерно в 10 раз. Несмотря на крайнюю опасность работы с фос¬
геном, его довольно часто используют в разных лабораторных
синтезах и в промышленности.
CeHs—СОС1 + NH3 —>
бензоилхлорид
CeH6—CONH2 + НС1
бензамид
О
2СН3— С—ONa + SOsCI2 —►
242
АНГИДРИДЫ КАРБОНОВЫХ КИСЛОТ
Важными функциональными производными кислот являются их
ангидриды, которые можно рассматривать как продукты деги¬
дратации кислот или как. оксиды ацилов:
о о о о
И г I! 1 II
R—С—О! Н + НО —С—R ——> R—С—О—С—R
! 1 — rljO
В молекулах ангидридов два кислотных радикала соединены
атомом кислорода. Названия их строят, исходя из названий
соответствующих кислот.
§ 106. Способы получения и свойства. Ангидриды можно полу¬
чить реакцией хлорангидридов с солями карбоновых кислот.
Если исходить из производных разных кислот, то образуются
смешанные ангидриды:
О о 0 0
II II II II
R—С—Cl + NaO—С—R' —► R—С—О—С—R' -f NaCl
Ангидриды низших кислот — жидкости с резким запахом,
нерастворимы в воде. Ангидриды высших кислот — кристалли¬
ческие вещества, без запаха. Температуры кипения ангидридов
выше, чем температуры кипения соответствующих кислот.
По химическим свойствам ангидриды напоминают хлоранги-
дриды. Они также являются ацилирующими средствами, но ре¬
акции с ними протекают более мягко, чем с хлорангидридами.
Действием ангидридов на спирты получают сложные эфиры,
действием на аммиак — амиды. Реакции эти аналогичны реак¬
циям хлорангидридов, но в результате образуется смесь основ¬
ного продукта и органической кислоты. Например, со спиртами
реакция идет по схеме:
О О о
II 1 II
СНз—С—О—С—СНз + НОСгН, —► СН3—С—ОС,Н5 + CHjCOOH
Ангидриды гидролизуются водой до кислот, но медленнее,
чем хлорангидриды.
§ 107. Отдельные представители. Уксусный ангидрид
[(СНзСО)гО — жидкость с т. кип. 140 °С, не смешивающаяся
с водой и на холоду лишь медленно с ней реагирующая. Полу¬
чают в промышленности в больших количествах из кетена и
уксусной кислоты, а также термокаталитической дегидратацией
уксусной кислоты:
0 О О О
1 1 II 1
СН*=С=0 + СНз—С—ОН —► СНз—С—О—С—СНз 2СНз—С—ОН
—HgO
243
Большие количества уксусного ангидрида расходуются на
производство искусственного ацетатного волокна. Уксусный ан¬
гидрид используют как ацетилирующее средство.
Малеиновый ангидрид — кристаллическое вещество, т. пл.
60°С. Главный промышленный способ получения — каталити¬
ческое окисление бензола (см. § 103). Малеиновый ангидрид—
типичный «диенофил» в реакциях с сопряженными диенами
(см. § 41). Используют для количественного определения бута¬
диена в газах при производстве синтетического каучука, для
идентификации других диеновых углеводородов, в промышлен¬
ности пластических масс.
Фталевый ангидрид — кристаллическое вещество, кристалли¬
зующееся в виде игл, т. пл. 128 °С, т. кип. 285 °С, легко воз¬
гоняется. В промышленности фталевый ангидрид получают
окислением нафталина (см. § 52) и используют для разных
синтезов. Со спиртами он дает моно- и диэфиры фталевой кис¬
лоты:
СО
СНзОН
(н+)
СООСНз с(нн3°н
■СООН
СООСНз
СООСНз
диметилфталат
Диметилфталат — жидкость со слабым запахом, применяет¬
ся как репеллент — средство, отпугивающее комаров, мошек,
слепней, клещей. Некоторые эфиры фталевой кислоты — хоро¬
шие пластификаторы, например дибутилфталат.
При взаимодействии с аммиаком фталевый ангидрид обра¬
зует имид фталевой кислоты — фталимид (см. § 114). При кон¬
денсации фталевого ангидрида с бензолом в присутствии хло¬
рида алюминия получается антрахинон (см. § 91), широко
применяемый в производстве красителей.
Для фталевого ангидрида характерны реакции конденсации
с фенолами. В результате подобной реакции образуются фтале-
ины — соединения, относящиеся к производным трифенилме-
тана:
Фенолфталеин — белое кристаллическое вещество, хорошо
растворимое в спирте. Под действием щелочей фенолфталеин
244
переходит в соль ярко-малинового цвета:
Его используют как индикатор, а в медицине под названием
пурген — как легкое слабительное. При конденсации фталевого
ангидрида с резорцином образуется краситель флуоресцеин.
Фталевый ангидрид вступает в реакцию поликонденсации с
многоатомными спиртами, образуя алкидные смолы (см.
§ 192).
ПЕРОКСИДЫ АЦИЛОВ. ПЕРОКСИКИСЛОТЫ
§ 108. Способы получения и отдельные представители. При
взаимодействии ангидридов или хлорангидридов кислот с пер¬
оксидами металлов получаются пероксиды ацилов:
о О О
II ИагОг II II
2С6Н5—С—С1 ,1М С6Н5—С—О—О—С—C6HS
—zNaL.1
дибензоилпероксид
При взаимодействии пероксидов ацилов с алкоголятом нат¬
рия и последующим действием минеральной кислоты получают¬
ся гидропероксиды ацилов или пероксикислоты:
о о о о
II II СНзОЫа II 1
С0Н5—С—О—О—С—СбН5 ► СсН5—С—OONa + С6Н5—С—ОСНэ
О О
II Н- ||
С6Н5—С—OONa ► С6Н6—С—ООН
пербензойная
кислота
Пероксиды ацилов и пероксикислоты — сильные окислители.
На холоду пероксикислоты медленно разлагаются, выделяя
кислород, а при нагревании взрываются, поэтому перегонять их
нельзя. Действуя как окислители, они выделяют галоген из га-
логеноводородов, окисляют алкены до оксидов. Обычно ими
пользуются в виде растворов в хлороформе или четыреххло¬
ристом углероде.
Пероксид бензоила (дибензоилпероксид)—кристаллическое
вещество, т. пл. 108 °С. Более устойчив, чем другие пероксиды
ацилов. Его можно хранить и транспортировать. При нагрева¬
нии или под действием ультрафиолетовых лучей он разлагается
245
со взрывом. При нагревании раствора пероксида в органическом
растворител’е идет медленное разложение и образуются свобод¬
ные радикалы:
0 0 о
II I ' I
СвН,—С—О—О—С—С5Н5 —> 2СвН,—с—о.
Пероксид бензоила находит промышленное применение как
инициатор полимеризации.
Гидропероксид бензоила (пербензойная кислота) — кристал¬
лическое вещество, т. пл. 43 °С, имеет запах гипохлорита. По¬
лучают его из пероксида бензоила и алкоголята натрия с по¬
следующим подкислением. Гидропероксид бензоила применяют
для синтеза и как реактив для определения положения двойной
связи (реакция Прилежаева). При действии его на алкены об¬
разуются а-оксиды:
+ CbUs~cf —► N;—с( + с6н5—соон
/ \ Чюн /\/\
о
Вместо пербензойной кислоты удобнее использовать ее м-
хлорзамещенный аналог, обладающий более высокой устойчи¬
востью.
СЛОЖНЫЕ ЭФИРЫ КАРБОНОВЫХ КИСЛОТ
Сложные эфиры — наиболее важные функциональные производ¬
ные кислот. Их можно рассматривать как производные кислот,
у которых гидроксильная группа замещена алкоксильной
группой.
О О
II И
R—С—OR R—С—О—
сложный аци латная
эфир группа
§ 109. Изомерия, номенклатура, физические свойства. Изоме¬
рия сложных эфиров обусловлена строением радикалов, связан¬
ных с ацилатной группой. Сложные эфиры изомерны не только
внутри класса (из-за различного состава и строения радика¬
лов); они изомерны и с кислотами. Например:
О О О
СН„—h—OCsH, CjH5—С—ОСНз CjH7—он
Название сложных эфиров обычно производят от углеводо¬
родного радикала спирта и корня латинского названия кисло¬
ты с добавлением окончания ат (или оат) вместо окончания
овая в кислоте; используют также названия, образуемые из на-
246
званий спиртов и кислот., дающих эфир:
О
н—осн,
метиловый эфир
муравьиной
кислоты
(метилформиат)
О
ц
СН3—С—ОС,Н5
этиловый эфир
уксусной кислоты
(этилацетат)
CI О
СН,=СН—СН—С—OCjfH,
этил-2-хлорбу тен -3-оат
Сложные эфиры широко распространены в природе. Аромат
многих цветов, плодов, ягод обусловлен присутствием в них
сложных эфиров. Жиры, чрезвычайно распространенные в рас¬
тительном и животном мире, также относятся к сложным
эфирам.
Эфиры низших одноатомных спиртов и низших кислот — ле¬
тучие жидкости с приятным запахом цветов, фруктов. Этилфор-
миат имеет запах рома, изопентил ацетат — запах груш, этил-
бутират— запах абрикосов, изопентил бутират — запах анана¬
сов, изопентиловый эфир изовалериановой кислоты — запах
яблок, бензйлацетат — запах жасмина и др. Эти эфиры исполь¬
зуют в парфюмерной и пищевой промышленности как «фрукто¬
вые эссенции». Они мало растворимы в воде, хорошо растворимы
в органических растворителях, сами являются растворителями.
Температуры кипения сложных эфиров ниже, чем у соот¬
ветствующих кислот, так как им не присуща ассоциация. Так,
температура кипения уксусной кислоты 118 °С, а этилацетата
78 °С.
§ 110. Способы получения. Важнейший способ получения слож¬
ных эфиров — взаимодействие карбоновых кислот со спир¬
тами (реакция этерификации):
0 О
1 н+ *
R—С—ОН + R'—ОН =S=t R—С—OR' + Н20
В качестве катализаторов применяют концентрированную
H2SO4, газообразный НС1, BF3 и др. Влияние кислотного ка¬
тализатора заключается в протонировании атома кислорода кар¬
бонильной группы и, следовательно, в увеличении положитель¬
ного заряда на атоме углерода карбоксильной группы, что
делает его более способным к присоединению нуклеофильного
реагента — спирта:
247
Атом кислорода спирта остается в составе сложного эфира;
это было доказано при использовании спирта, меченного изото¬
пом 180.
Реакция этерификации обратима. Избыток спирта или кисло¬
ты благоприятствует сдвигу равновесия вправо. Увеличить вы¬
ход сложного эфира можно также постоянным выведением од¬
ного из образующихся веществ (эфира или воды) из сферы
реакции.
Скорость реакции этерификации зависит от природы кисло¬
ты и спирта: от того, с каким атомом углерода связана гидро¬
ксильная группа (первичным, вторичным или третичным) и
каково строение углеродной цепи, связанной с карбоксильной
группой. Объемистые радикалы создают пространственные за¬
труднения и мешают образованию промежуточных продуктов
присоединения. Особенно плохо этерифицируются третичные
спирты (Н. А. Меншуткин, 1887 г.).
Из других способов получения сложных эфиров упомянем
об ацилировании спиртов хлорангидридами или ангидридами
кислот (так, в частности, получают сложные эфиры третичных
спиртов и фенолов), а также об имеющем промышленное зна¬
чение присоединении карбоновых кислот к алкенам:
О О
II BF3 ||
(СН3)2С=СН2 + НО—С—СН3 >- (СНз)3С—О—С—СЧз
т рет-бутилацетат
В промышленности используют также присоединение карбо¬
новых кислот к ацетилену. Так получают сложные виниловые
эфиры (200 °С, в присутствии солей кадмия или цинка):
О о
II II
СНз—С—ОН + СН==СН —У СНз—с—о—сн=сн2
винилацетат
Винилацетат получают в промышленности также окислитель¬
ной конденсацией этилена и уксусной кислоты на металлорга-
нических катализаторах: •
02 (кат.)
СНз—СООН + СН2=СН2 —Г*- СНз—соо—сн=сн2
—Н20
Винилацетат — важный полупродукт для синтеза полимеров.
§ 111. Химические свойства. Для сложных эфиров характерны
следующие химические реакции.
1. Действие воды (гидролиз). Кислая или основная среда
способствует этой реакции. В кислой среде реакция обратима,
в результате образуются'кислота и спирт:
о О
II (Н2о> н+ ||
R—С—OR' ~ * R—С—ОН + R'OH
248
В щелочной среде реакция необратима протекает быстрее,
в результате образуются спирт и соль кислоты.
Способность сложных эфиров гидролизоваться в щелочной
среде принципиально отличает их от простых эфиров и аце¬
талей. Как известно, ацетали могут гидролизоваться только в
кислой среде, а простые эфиры расщепляются только под дей¬
ствием сильных галогеноводородных кислот.
2. Реакция переэтерификации. При нагревании сложных
эфиров со спиртами в присутствии минеральной кислоты или
алкоголята (щелочная среда) происходит обмен алкоксигрупп.
Реакция обратима, степень превращения зависит от количества
взятого спирта:
О О
R—С—OR' + R"OH R—С—OR" + R'OH
Реакцию используют для получения различных сложных
эфиров.
3. Действие аммиака (аммонолиз) приводит к амидам кис¬
лот:
о О
II II
сн3—С—ОС2Н5 + NH3 —> СНз—С—NH2 + С2Н5ОН
этилацетат ацетамид
4. Восстановление сложных эфиров протекает легче, чем са¬
мих кислот. В результате получается первичный спирт, соот¬
ветствующий кислоте сложного эфира. В качестве восстанови¬
теля используют алюмогидрид лития, натрий в кипящем спирте,
водород в присутствии медно-хромового катализатора:
О
II восстановление
R—С—OR' »• R—CII2OH + R'OH
§ 112. Воска. Природные воска представляют собой смесь
сложных эфиров высших жирных кислот и высших спиртов,
содержащую также свободные высшие спирты, свободные кис¬
лоты и алканы с нормальной цепью углеродных атомов. Пче¬
линый воск (т. пл. 60—62 °С) состоит из эфиров первичных
спиртов и кислот с четным числом углеродных атомов (С24—
С34) и свободных алканов с нечетным числом углеродных ато¬
мов (С25—Сл).
Некоторые растения выделяют на листьях и плодах защит¬
ную пленку воска. Состав хлопкового воска близок к составу
пчелиного. Воск на листьях табака почти исключительно состо¬
ит из алканов С25—Сзз, а воск на листьях капусты — на 95 %
из алкана С2дН6о.
Из головы морского млекопитающего кашалота выделен
твердый воск — спермацет (т. пл. 41—48°С) и жидкий воск —
249
спермацетовое масло. Спермацет состоит в основном из гекса-
децилпальмитата (гексадециловый эфир пальмитиновой кисло¬
ты) С15Н3|СООС|6Изз. Главной составной частью спермацето¬
вого масла являются эфиры непредельных спиртов (олеинового
спирта С17Н33СН2ОН) и высших непредельных кислот С12—Ci6
с четным числом углеродных атомов.
Ланолин — жиропот овечьей шерсти — представляет собой
смесь эфиров кислот Сю—С2о и спиртов Сю—С2о (в его состав
входят также гидроксикислоты).
Консистенция воска, температура его застывания обусловле¬
ны составом (длиной цепи радикалов, непредельностью). Чем
длиннее цепь углеродных атомов кислоты или спирта, тем выше
температура плавления. Наличие в молекуле двойных связей
снижает температуру плавления. Спермацет и ланолин исполь¬
зуют в медицине и парфюмерии для приготовления различных
мазей и кремов.
ЖИРЫ И ЖИРОПОДОБНЫЕ ВЕЩЕСТВА
§ 113. Природные жиры и их техническая переработка. В состав
растительных и животных организмов наряду с белковыми
веществами и углеводами входят жиры и жироподобные веще¬
ства, которые объединяют под общим названием липиды.
Липиды находятся в организме либо в виде протоплазмати-
ческого жира (входят в состав протоплазмы клеток), либо в
виде запасного, резервного жира (входят в состав жировой
ткани). Протоплазматический жир как составная часть клетки
находится в организме в виде сложных соединений с белком и
количество его не меняется при истощении организма от голода
или при ожирении. В этих случаях изменяется количество ре¬
зервного жира.
Липиды играют важную биологическую роль: они являются
источником энергии для животного организма; при окислении
в организме 1 г жира выделяется 39 кДж энергии. Они хоро¬
шие растворители биологически активных веществ (например,
витаминов),- необходимы для осуществления нормальных функ¬
ций животного организма. Жировая ткань образует мягкую
изолирующую прослойку, защищая внутренние органы и все
тело от толчков, ударов и переохлаждения.
Количество накапливаемого жира в животном организме зависит от ре¬
жима питания, возраста и других условий. Обычно количество жира в чело¬
веческом организме составляет 10—20 % от общей массы организма (при
патологических изменениях в организме его может быть больше). У живот¬
ных, специально откармливаемых «на сало», количество жира достигает 50 %
от общей массы. У растений растительные масла накапливаются обычно в
семенах, плодах (льняное семя, конопляное, семена подсолнуха, плоды оливы
и др.) тоже до 50 % от общей массы.
Растения синтезируют жиры из крахмала, который является
продуктом ассимиляции угольной кислоты. В животном орга¬
250
низме жиры образуются из жиров пищи, а также из угле¬
водов.
Главной составной частью растительных и животных жиров
являются сложные эфиры глицерина и высших жирных кислот.
Химическая природа жиров была установлена в первой четвер¬
ти XIX века Шеврелем. Первый синтез жиров осуществил
Бертло (1854 г.) нагреванием глицерина со стеариновой кис*
лотой:
СНг—ОН О СН2—О—СОС17Н35
| И 200 *С I
СН—ОН + ЗНО—С—С17Н35 1 ^ СН—О—COC|7H3j зн2о
СН2—ОН СН2—О^СОСпНз,
тристеарин
Во всех жирах остаток спирта один и тот же (остаток гли*
церина); разница между жирами обусловлена остатками кислот»
Наиболее важными кислотами, входящими в состав жиров Я
жироподобных веществ, являются монокар боновые кислоты, со*
держащие четное число углеродных атомов и неразветвленнуЮ
углеродную цепь. Из предельных кислот в жиры входят}
масляная (С4), гексановая (С6), октановая (С8), декановая
(Сю), лауриновая (Си), пальмитиновая (Си), стеариновая
[(Сю) и арахиновая (С20). Из непредельных — пальмитоолеи*
новая, олеиновая, линолевая, линоленовая.
Масляная кислота содержится в количестве 2,4 % в виде
эфира с глицерином в коровьем масле-, откуда она была впер¬
вые выделена (Шеврель, 1814). При прогоркании масла (при
длительном его хранении) наряду с другими продуктами разло¬
жения образуется масляная кислота, придающая маслу непри¬
ятный запах и вкус. Свободная масляная кислота содержится в
потовых выделениях (неприятный запах пота — запах масляной
кислоты). Гексановая кислота СН3(СН2)4СООН в виде эфира о
глицерином содержится в козьем молоке.
Жиры масличных плодов содержат много непредельной оле¬
иновой кислоты (оливковое масло до 80%, подсолнечное до
36 %). Жир человека содержит до 70 % олеиновой кислоты, до
25 % пальмитиновой. В жирах жвачных животных много стеа¬
риновой кислоты. Жиры молока содержат до 40 % олеиновой
кислоты, до 25 % пальмитиновой; характерно также присутст¬
вие в них низших предельных кислот С4—Сю. Жиры морских
животных содержат много непредельных кислот Сю и Сю-
Различный кислотный состав обусловливает различные фи¬
зико-химические и химические свойства жиров. Так как природ¬
ные жиры представляют собой сложные смеси глицеридов, они
не имеют резкой температуры плавления, а, предварительно
размягчаясь, плавятся в определенном интервале температур.
Температура затвердевания жира тем выше, чем больше в нем
содержится предельных кислот (пальмитиновой, стеариновой).
2S1
Жиры легко растворяются в органических растворителях:
эфире, хлороформе, четыреххлористом углерода, бензоле, толу¬
оле, бензине. Они практически нерастворимы в воде, но в при¬
сутствии поверхностно-активных веществ могут образовывать с
водой стойкие эмульсии. В природных условиях в животном
организме жиры эмульгируются белками, солями желчных кис¬
лот и др.
Примером водной эмульсии жира, стабилизованной белком,
является молоко. Эмульгирование необходимо для быстрого
переваривания жиров пищи пищеварительными органами. Во
время пищеварения жиры под действием фермента липазы
(находится в слюне, печени, желудочном и кишечном соках),
гидролизуются до свободных кислот и глицерина, которые про¬
ходят через стенки кишечника и затем снова связываются в
различных комбинациях, отлагаясь в виде резервного жира.
Техническая переработка жиров. В промышлен¬
ности реакцию гидролитического расщепления жиров широко
используют для получения глицерина и жирных кислот или их
солей, т. е. мыла. В кислой среде образуются глицерин и смесь
свободных кислот — стеарин:
СН2—ОСОС,7Н35
I н20 (н+)
СН—ОСОС15Н31 ►
Глицерин
Стеариновая
кислота
СН2-ОСОС16Н31
Пальмитиновая
кислота
Стеарин
В щелочной среде наряду с глицерином получаются соли
этих кислот — мыла (см. § 97).
Очень важной реакцией, имеющей промышленное значение,
является реакция гидрогенизации (гидрирования) жиров — пре¬
вращение жидких жиров в твердые. Сущность этой реакции
состоит в присоединении водорода к непредельным жирным
кислотам и превращении их в предельную кислоту (Ni, 200°С,
1,5 МПа):
СН2—ОСОС17Н33 СН2—ОСОС17Н35
I +6Н2 |
СН—ОСОС,7Н31 >- СН—ОСОС,7Н35
СН2—ОСОС17Н29 СН2—ОСОС,7Н36
жидкий жир твердый жир
Гидрогенизации подвергают растительные масла, рыбий жир,
жиры морских млекопитающих животных. В результате исчеза¬
ют темный цвет и неприятный запах. Твердые жиры удобнее
транспортировать, из них можно получать стеарин для свечей,
твердое мыло и др. Лучшие сорта гидрированного жира исполь¬
зуют для получения маргарина.
2Ь2
Маргарин представляет собой эмульсию гидрированного жи¬
ра, животного жира (легкоплавкого говяжьего жира) или рас¬
тительного масла в молоке. По виду и запаху напоминает сли¬
вочное масло; для придания продукту желтого цвета в него
добавляют яичный желток, а для запаха—дикетон биацетил
СН3СОСОСН3—главное душистое вещество сливочного масла.
Для растительных масел характерны процессы аутоокисле¬
ния и полимеризации. Эти свойства используют в промышлен¬
ности для приготовления олифы, лаков и красок. Олифу изго¬
товляют из масел, содержащих большое количество эфиров
линолевой и линоленовой кислот и носящих название высыхаю¬
щих. К ним относятся, льняное, конопляное, подсолнечное и дру¬
гие масла. Олифа — густая темная жидкость, на воздухе «вы¬
сыхает», образуя пленку. В процессе высыхания идет окисление
масел кислородом воздуха. Для ускорения этого процесса к
олифе добавляют сиккативы (см. § 96). Окисленный полимер
льняного масла — густая эластичная масса линоксин — приме¬
няется для изготовления линолеума и клеенок.
Для приготовления некоторых смазочных масел употребляют
смеси растительных масел с минеральными. Густые смазки го¬
товят на животных жирах. Жиры используют также для фар¬
мацевтических целей и в парфюмерии (кремы, помады и др.).
Важной группой липидов, входящих в состав мозга и нерв¬
ных тканей, являются фосфатиды. Фосфатиды при гидролизе
дают глицерин, жирные кислоты, фосфорную кислоту и амино-
спирт. В зависимости от того, остаток какого спирта входит в
состав фосфатидов, различают лецитины, содержащие остаток
аминоспирта холина, и кефалины, содержащие остаток этанол-
амина:
СН2—О—COR СН2—О—COR
СН-О—COR' СН—О—COR'
I ♦ I
СН2—О—Р—О—СН2—СН2—N(CH3)3 СН2- О—Р—О—СН2—СН2—NH8
лецитин кефалин
В отличие от жиров в молекуле фосфатидов одна из гидрок-
сигрупп глицерина связана не с жирной кислотой, а с фосфор¬
ной (в виде ее эфира с аминоспиртом).
Лецитины — белые воскоподобные вещества, растворимы в
эфире, спирте и других органических растворителях. В воде
набухают. Входят в состав мозговых и нервных тканей, яично¬
го желтка. В растениях они содержатся в семенах и ростках.
При гидролизе лецитинов образуются свободные карбоновые
кислоты, холин и глицерофосфорная кислота.
О
[НОСН2—СН2—N(CH3)3]+ 'ОН НОСН2—СНОН—СН2—О—Р(ОН)2
холин глицерофосфорная кислота
253
При гидролизе кефалины дают карбоновые кислоты, глице-
рофосфорную кислоту и эганоламин (НОСНг—СН2ЫНг). Осо¬
бая роль принадлежит лецитинам и кефалинам в клеточных
мембранах — «перегородках» внутри клеток, очень важных для
их жизнедеятельности. Лецитины принимают участие в обмене,
всасывании и транспортировке жиров, холестерина. При недо¬
статке фосфолипидов в организме создаются условия для раз¬
вития атеросклероза.
АМИДЫ КИСЛОТ
§ 114. Способы получения и свойства. Амиды можно рассматри¬
вать как продукты замещения гидроксильной группы в карб¬
оксильной группе остатком аммиака — группой NH2. В молеку¬
лах амидов группа NH2 связана с ацилом; общая формула
амидов R— CONH2.
Примерами амидов и их названий могут служить следую¬
щие:
О О О О
sunn
Н—С—NHj СН3—С—NH2 С„Н5—с—NH2 СНз—С—NH—СвНз
формамид ацетамид бензамид ацетанилид
Соединения, содержащие в молекуле группу —NH—, связан¬
ную с двумя ацильными радикалами, называют имидами.
Некоторые амиды представляют собой жидкости, большая
же часть — твердые вещества. Низшие амиды растворимы в
воде.
При изучении межатомных расстояний установлено, что в амидах длины
связей С = 0 и С—N соответственно равны 0,124 и 0,132 нм, т. е. отличаются
от длины аналогичных связей в других соединениях (см. табл. 2). Эти и
другие особенности строения амидов указывают на то, что их истинное
строение мезомерное н может быть выражено граничными формулами:
Важным промышленным способом получения амидов являет¬
ся нагревание (сухая перегонка) аммониевых солей карбоновых
кислот;
Амиды получают действием аммиака на хлорангидриды, ан¬
гидриды и эфиры кислот (см. §§ 104 и 111). Кроме того, они
образуются при неполном гидролизе нитрилов. Гидролиз до
О
О'
О
О
R—С—ONH« —► R—С—NHj + НгО
£54
амидов можно проводить, например, действием пероксида водо¬
рода в слабощелочной среде:
R—C=N
HjO, Н4О2
*
R—С—NHS
Амиды в отличие от аммиака почти лишены основных
свойств, они дают соли только с сильными кислотами, напри¬
мер RCONH2-HC1, которые легко разлагаются водой с выде¬
лением исходного амида. При действии на амиды металличе¬
ского натрия в жидком аммиаке образуются металлические
производные амидов типа RCONHNa. В этом проявляются кис¬
лотные свойства амидов. Еще более кислотным характером об¬
ладают циклические амиды (имиды). Так, фталимид при обра¬
ботке его раствором гидроксида калия образует калиевое про¬
изводное— фталимид калия:
СО
J^JJ^ ^NH + КОН
СО
;NK + Н20
Как и другие функциональные производные кислот, амиды
при гидролизе в кислой или щелочной средах образуют кисло¬
ты (или их соли):
О
NaOH И НС1
R-COONa + NH3 ч—- R—С—NH, »■ R—COOH + NH«Cl
HjO П2“
Превратить амиды в кислоты можно также действием азо¬
тистой кислоты:
HONO
R—CONHj ► R—СООН + + HjO
При нагревании амидов с сильными водоотнимающими сред¬
ствами (например, Р2О5) образуются нитрилы:
СН3—СО—NH2 CH3-C^N
ацетамид ацетонитрил
При восстановлении амидов (алюмогидрид лития или ката¬
литическое гидрирование) образуются первичные амины с тем
же числом углеродных атомов в молекуле:
восстановитель
R—СО—NH2 ■» R—СН2—NH2
Своеобразная реакция — перегруппировка Гофмана — про¬
исходит при действии на амиды гипохлоритом или гипоброми*
том натрия в водном щелочном растворе. Эта реакция приво¬
дит к аминам, содержащим в молекуле на один атом углерода
меньше, чем исходные амиды. В ходе перегруппировки группа
NH2 перемещается от группы СО к углеводородному радикалу:
R—СО№2 + NaOCl + 2NaOH —► R—NH2 + Na2C03 + NaCl + HaO
§ 115. Отдельные представители. Формамид HCONH2 — жид¬
кость, т. кип. 193 °С, т. пл. 2,5 °С, df 1,13. Его получают при
реакции аммиака с монооксидом углерода под давлением.
Ацетамид CH3CONH2, т. пл. 82 °С, т. кип. 222 °С, получа¬
ют сухой перегонкой аммониевой соли уксусной кислоты.
Диметилформамид— жидкость, т. кип. 150 °С, смешивается
с водой, эфиром, бензолом. Получают при реакции диметил¬
амина и монооксида углерода под давлением в присутствии
метилата натрия:
О
CH3ONa II
(CH3)2NH + CO ► Н—С—N(CH3)2
Формамид и диметилформамид применяют в качестве рас¬
творителей в лаборатории, а также в производстве синтетиче¬
ских волокон при прядении. Диметилформамид—селективный
растворитель некоторых газов; его используют для выделения
ацетилена из газовых смесей (1 объем диметилформамида рас¬
творяет 31 объем ацетилена).
Карбамид (диамид угольной кислоты, мочевина)—кристал¬
лическое вещество, т. пл. 133 °С, легко растворяется в воде,
труднее в спирте, не растворяется в эфире, углеводородах.
Впервые мочевину синтезировал Велер (1828 г.) путем нагрева-
'ния цианата калия и сульфата аммония:
KOCN
(NH4)2SQ4
-k2so4
О
100 °с II
NH4OCN >- H2N—С—nh2
Мочевина является продуктом обмена веществ в организмах
млекопитающих. При распаде белка около 80 % азота выделя¬
ется в виде мочевины. Моча человека содержит около 2 % мо¬
чевины. Мочевина — важный технический продукт, который в
промышленности получают следующими путями.
1. Из диоксида углерода и аммиака при нагревании под
давлением:
200 »С, 20 МПа
С02 + 2NH3 -> H2N—COONH4 H2N—CO—NH2
—rl2*J
2. Из фосгена и аммиака:
Cl—СО—Cl + 4NH3 —> H2N—CO—NH2 + 2NH4C1
3. Из цианамида:
h2n—cn + h2o —► H2N—CO—nh2
256
Рис. 25. Соединения включения
с мочевиной
Мочевина обладает
слабоосновными свойст¬
вами, образует соли с кис¬
лотами. Характерной со¬
лью является ее мононит¬
рат C0(NH2)2-HN03,
трудно растворимый в во¬
де; используется для вы¬
деления мочевины из рас¬
твора. Мочевина имеет
обычные свойства ами¬
да— гидролизуется при
нагревании как в кислой,
так и в щелочной средах, а также под действием азотистой кис¬
лоты:
2HN02 н2о
СО» + 2NS + ЗНцО *« HjN—СО—NH* ► СО, + 2NH,
Для мочевины характерна реакция конденсации с формаль¬
дегидом. В слабощелочном растворе образуются моно- и ди-
(гидроксиметил)мочевины (метилол- и диметилолмочевины):
HjN—СО—NH2
сн2о t H2N—СО—NHCH2OH
гидроксиметилмочевнна
2СН20
► HOCHjNH—СО—NHCH2OH
ди(гидроксиметил)мочевина
В сильнощелочной или кислой среде эти продукты дегидра¬
тируются и полимеризуются в высокомолекулярные смолы —
аминопласты.
Мочевину используют в промышленности для получения по¬
лимеров, лекарственных веществ, гербицидов, для стабилизации
взрывчатых веществ и др. В сельском хозяйстве ее применяют
в качестве богатого азотом удобрения и как добавку в корм
жвачным животным.
Соединения включения (клатраты). Кристаллы мочевины, смоченные ме¬
тиловым спиртом, способны поглощать органические соединения (углеводо¬
роды, спирты, алкилгалогениды, амины и др.) только с нормальной цепью
углеродных атомов, образуя устойчивые кристаллические продукты.
Установлено, что молекулы мочевины ориентированы в кристаллической
решетке, так, что между ними образуются шестигранные каналы, напоминаю¬
щие пчелиные соты. В этих каналах могут размещаться молекулы неразвет-
вленных соединений (рис. 25), разветвленные молекулы не проходят. Таким
способом можно разделить соединения нормального и изостроения.
В образующихся соединениях включения молекулы «гостя» не связаны
с молекулами «хозяина» химической связью, а лишь заключены в каналы,
образованные кристаллической решеткой. При растворении кристаллов погло¬
щенные вещества выделяются. Подобные соединения получили название со-
9 Органическая химия
257
единений включения или клатратов '(от лат. clat — замок, ratus — загоро¬
женный).
Свойство мочевины давать соединения включения используется в нефте¬
перерабатывающей промышленности для повышения качества топлива. Из
бензина удаляют таким путем нормальные углеводороды, которые снижают
октановое число горючего (являются причиной детонации топлива). При уда¬
лении к-алканов из топлива для реактивных двигателей снижается его тем¬
пература застывания, так как углеводороды с разветвленной цепью плавятся
при значительно более низких температурах, чем нормальные углеводороды.
НИТРИЛЫ кислот
§ 116. Способы получения и свойства. Нитрилы (цианиды)'
R—Cs=N являются производными карбоновых кислот, хотя и
не содержат ацильной группы. При гидролизе нитрилов группа
C = N превращается в карбоксильную группу. Таким образом,
нитрилы ведут себя как и другие функциональные производные
кислот. Связь нитрилов с амидами отмечалась уже ранее. Ни¬
трилы молено рассматривать и как производные синильной кис¬
лоты Н—C=N, в которой атом водорода замещен на углево¬
дородный радикал.
Названия нитрилов производят от названий кислот с тем
же числом углеродных атомов; их называют также алкилци-
анидами:
СНз—C=sN
ацетонитрил,
нитрил уксус¬
ной кислоты
(метилцианид)
СН2=СН—C=N
акрилонитрил,
нитрил акрило¬
вой кислоты
(винилиианид)
С6Н5—C=N
бензонитрил,
нитрил бен¬
зойной кислоты
(фенилцианид)
С6Н5—CHj—G=N
фенилацетонитрил,
нитрил фенилук-
сусной кислоты
(бенэилцианид)
Нитрилы можно получать несколькими способами. 1. Дей¬
ствием цианида калия на галогенпроизводные. Метод интересен
тем, что приводит к наращиванию цепи углеродных атомов:
RBr + KCN —RCN + KBr
2. Нагреванием амидов кислот (см. § 114) или оксимов
альдегидов с водоотнимающими средствами:
(CHjCOhO
CeH5CH=N—ОН —- >■ С6Н5—Cs=N
““П20
бензальдегидоксим бензонитрил
3. Ароматические нитрилы получают при сплавлении солей
сульфокислот с цианидом калия (см. § 120).
Основным промышленным способом получения является
окислительный аммонолиз алкенов. С ним мы познакомимся в
следующем параграфе на примере получения акрилонитрила.
Для нитрилов характерна реакция гидролиза, которая может
протекать как в кислой, так и в щелочной средах:
HjO, NaOH H2O. Н+
RCOONa + NH3 -« R—C=N »- RCOCT + NHj
258
При осторожном гидролизе сначала образуются амиды, а
затем кислоты (см. § 114).
Восстановление нитрилов приводит к первичным аминам.
В качестве восстановителя можно применять натрий в спирте
или алюмогидрид лития, можно проводить каталитическое гид¬
рирование:
4Н
R—C=sN ► R-CH8-NH2
Водородные атомы при а-углероде в молекуле нитрила
подвижны, так как группа C==N является сильной электроноак¬
цепторной группой. В этом отношении она напоминает карбо¬
нильную группу. Сходное влияние обеих групп на а-водород-
ные атомы имеет одинаковые причины: и карбонил, и нитриль-
ная группа несут значительный положительный заряд на атоме
углерода:
а* 6-
—С=зЫ
Особой подвижностью обладают водородные атомы при угле¬
роде, связанном с двумя нитрильными группами, как, напри¬
мер, в динитриле малоновой кислоты NC—СН2—CN, или с од¬
ной нитрильной и одной фенильной группой, как, например,
в фенилацетонитриле C6Hs—СН2—CN. Такие соединения обра¬
зуют натриевые производные и затем алкилируются:
CN CN CN
I C8H5ON3 1 CjH5I 1
С6Н5—СН2 ► С6Н5—CHiVa CSH5—СН—С2Н5
За счет а-водорода эти соединения легко вступают в реак¬
цию конденсации с альдегидами. Эти свойства используются
для различных синтезов.
§ 117. Акрилонитрил в химической промышленности. Акрилони¬
трил CH2=CHCN — жидкость, т. кип. 78 °С, растворим в воде
(около 7%), хорошо растворим в органических растворителях;
с воздухом образует взрывоопасные смеси [3—17 % (объемн.)],
ядовит.
В промышленности акрилонитрил получают несколькими
способами. 1. Из этиленоксида и синильной кислоты под дав¬
лением 0,25 МПа:
О
/ \
Н2С—сн2
он см
HCN, во "С I I 170 "С
*■ СИг-СНг CH3=CH-CN
2. Из ацетилена и синильной кислоты в присутствии хлори¬
да меди(1) (процесс осложняется побочными реакциями):
СНззСН + HCN —> СН,=СН—CN
9*
2S9
Схема 6. Техническое использование акрилонитрила
— ► СНз—СН2—CN
гидромериаация
>
пропионитрил
NC—СН2—СНа—СНа—СН2—CN
СН2=СН—CN
акрилонитрил
С12
HCN
■>
HCI
NHa
>
СН3ОН
>
СН3СНО
— >
адиподинитрил
ClCH2—СНСl—CN
α, β-дихлорпропио-
нитрил
NC—СН2—СН2—CN
сукцинонитрил
ClCH2—CH2—CN
β-хлорпропионитрил
H2N—СН2—СН2—CN
β-аминопропионитрнл
СН3О—СН2—СН2—CN
β-метоксипропионитрил
NC—СН2—СН2—СН2—СНО
сн=сн
цианомасляный альдегид
> NC—СН2—СН2—С≡С—СН2—СН2—CN
1,6-дицианогексин-З
СНз=СН-СН=СНз
>.
CN
тетрагидро-
бензонитрил
3. Окислительным аммонолизом пропилена в присутствии
катализаторов:.
450º ССН2=СН—СНз + NH3 +1*1/2 О2 ► СН2=СН—CN + ЗН2O
Это самый перспективный метод, вытесняющий остальные по
экономическим соображениям; им получают до 90 % выпускае¬
мого акрилонитрила.
Двойная связь акрилонитрила, сопряженная с тройной, об¬
ладает большой химической активностью. Акрилонитрил реаги¬
рует со многими веществами (галогеноводороды, циановодород,
спирты, амины, аммиак). При этом происходит как бы заме¬
щение активного водорода названных веществ на группу
СН2—СН2—CN — реакция цианэтилирования:
НХ + СН2=СН—C=N —► X—СН2—СН2—C≡N
260
Кажущееся нарушение правил Марковникова объясняется
сопряжением, как и у а,р-непредельных кислот (см. § 99):
8+ 6- 8+ 6-
СН2=СН—C==N
В реакцию цианэтилирования вступают, также вещества,
содержащие подвижный атом водорода при атоме углерода —
альдегиды и кетоны, нитросоединения, малоновый эфир. В свя¬
зи с разнообразными возможностями, открываемыми реакцией
цианэтилирования, акрилонитрил широко используется в лабо¬
раторных и промышленных синтезах (см. схему 6).
Акрилонитрил легко полимеризуется под влиянием неболь¬
шого количества пероксидов (пероксид бензоила и др.), вслед¬
ствие чего является одним из важнейших мономеров в произ¬
водстве синтетических каучуков, синтетических волокон, плас¬
тических масс и др. Важной реакцией, имеющей перспективное
значение, является реакция гидроформилирования акрилони¬
трила:
со. н2
NC—СН=СН2 ► N С—СН2—СН2—СНО
Р-цианопропионовый
альдегид
На основе (3-цианопропионового альдегида синтезируют
важные аминокислоты — глутаминовую и триптофан (см.
§ 174).
В промышленных масштабах производится также ацетони¬
трил CH3CN. Это жидкость с т. кип 82 °С, смешивается с во¬
дой во всех отношениях, хороший растворитель многих неорга¬
нических солей. Получают ацетонитрил при действии на
ацетамид водоотнимающими средствами (см. § 114). В послед¬
нее время ацетонитрил широко применяют в промышленности
как растворитель, например для удаления смол и фенолов из
углеводородов нефти. Как и другие нитрилы, ацетонитрил до¬
вольно токсичен, хотя конечно не столь опасен, как свободная
синильная кислота. Значительные количества ацетонитрила об¬
разуются как побочный продукт при окислительном аммонолизе
пропилена. Это и является главным промышленным источником
ацетонитрила.
глава 13
ОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ СЕРЫ
Сера и кислород, являясь элементами одной и той же группы
Периодической системы элементов Д. И. Менделеева, обнару¬
живают сходство в образовании не только неорганических, но
и органических соединений. Однако в отличие от кислорода
261
сера в органических соединениях может проявлять различную
валентность.
Типы органических соединений серы приведены ниже:
S
RSH
R—S—R' R—S—
•S—R'
R—С—R'
R—S—ОН
тиолы
сульфиды дисульфиды
тиокетоны
сульфеновые
(тиоэфиры)
кислоты
О
О
О
О
II
II
II
1
R—S—ОН
X
0
1
С/Э
1
0£
R-S-R
R-S-R
R3S+ Х~
II
О
II
О
сульфиновые сульфоновые
сульфо¬
сульфо-
соли
КИСЛОТЫ
кислоты
ксиды
ны
сульфо-
(сульфокислоты)
пия
§ 118. Тиолы и дисульфиды. Тиолы (тиоспирты) R—SH — одно-
замещенные органические производные сероводорода. Их
можно также рассматривать как сернистые аналоги спиртов,
Названия образуют добавлением окончания тиол к названию
углеводорода: например, метантиол.
Тиолы обладают характерным, очень навязчивым запахом,
более сильным и неприятным, чем запах сероводорода. Для
низших тиолов запах ощущается при концентрации 1 ч. на
400 млн. ч. воздуха. Добавкой к природному газу ничтожных
количеств изопентантиола пользуются для лучшего обнаруже¬
ния по запаху утечки газа в жилых помещениях. Тиолы и их
производные встречаются в растительном и животном мире,
например, пропантиол C3H7SH — в свеженарезанном луке, бу-
тантиол C4H9SH — в выделениях скунса. Сера входит в состав
некоторых белковых аминокислот (см. § 174) и ферментов (см.
§ 179).
В отличие от спиртов и воды тиолы и сероводород не ассо¬
циированы, так как сера не образует водородных связей. По
этой причине тиолы значительно хуже растворимы в воде, чем
спирты, и кипят при значительно более низких температурах:
Т. кип., °С T. кип., °С
Н20 100 H2S —61
СН3ОН 64.7 CH3SH 7,6
CsH5OH 78,3 C2H5SH 34,7
Тиолы можно получать следующими способами.
1. Реакция алкилгалогенидов с гидросульфидом натрия:
C2H6Br -+■ NaSH —V C2H6SH + NaBr
2. Пропускание паров спиртов и сероводорода над нагретым
катализатором (оксид тория, 450°С):
ROH + H2S —► rsh + h2o
262
3. Взаимодействие алкенов с сероводородом в кислой среде
H2SO«
(СН3)2С=СН2 + H2S ► (СНз)зС—SH
По химическим свойствам тиолы сходны с сероводородом.
Они обладают более выраженными кислотными свойствами, чем
соответствующие спирты: растворяются в водных растворах ще¬
лочей, образуя соли — тиолаты R—SNa. С ртутью образуют ха¬
рактерные нерастворимые тиолаты:
2CH3SH + HgO —> (CH3S)2Hg + H20
Аналогично спиртам тиолы реагируют с хлорангидридами,
образуя тиоаналоги сложных эфиров:
О О
II И
СНз—С—Cl + C2H5SH —> СНз—С—SC2H6 + HC1
этилтиоацетат
Тиолы и спирты по-разному относятся к окислению. При
окислении спиртов увеличивается степень окисления углерода, а
не кислорода. При окислении тиолов в первую очередь окисля¬
ется сера. В мягких условиях образуются дисульфиды:
С2Н5—SH + 0 + HS—С2Н5 ——> С2Нд—S—s—с2н5 + н2о
диэтилдисульфид
Диаллилдисульфид СН2=СН—СН2—S—S—СН2—СН=СН2,
т. кип. 117°С (16 мм рт. ст.). Содержится в чесноке. Отсюда
и произошло название аллильного радикала (лат. allium sati¬
vum — чеснок). Из чеснока получают антибиотик аллицин —
аллилпропен-2-тиосульфинат:
О
II
СН2=СН—СН2—S—S —сн2—сн=сн2
§ 119. Тиоэфиры, сульфоксиды и сульфоиы. Тиоэфиры (диал-
килсульфиды) R—S—R — дизамещенные органические произ¬
водные сероводорода. Их можно получать реакцией алкилгало-
генидов с сульфидами или тиолатами щелочных металлов:
2CH3I + Na2S —► CHr-S—СНз + 2NaI
C2H5Br + C2H6SNa —C2H5—S—C2H5 + NaBr
Тиоэфиры — нейтральные жидкости, кипящие при более вы¬
соких температурах, чем соответствующие тиолы.
Представителем диалкилсульфидов является $,$'-дихлор~
диэтилсульфид (иприт) С1СН2—СН2—S—СН2—СН2С1, приме¬
нявшийся в первую мировую войну в качестве отравляющего
вещества кожно-нарывного действия.
263
Тиоэфиры легко окисляются (Н202, СНзСОООН) с образо¬
ванием сульфоксидов и сульфонов:
. о о
[О], 25 »С II (О] ||
СН3—S—СН3 >- СНз—S—СНз >- СН3—S—СНЭ
диметил- диметилсульфоп
сульфоксид (т. кип. 238 °С,
(т. кип. 86 °С) т. пл. 110°С)
Диметилсульфоксид— важный растворитель.
§ 120. Сульфоновые кислоты и сульфонилхлориды. Сульфоно¬
выми кислотами, или сульфокислотами, называют органические
соединения, содержащие в молекуле сульфогруппу SO3H (оста¬
ток серной кислоты), связанную через атом серы с углеводо¬
родным радикалом.
Алифатические сульфокислоты можно получить энергичным
окислением тиолов, а также реакцией галогенпроизводных с
сульфитом натрия:
lO]
CH3SH ► СНз—S02OH
метансульфо-
кислота
С2Н61-f Na2S03 —► C2H5S03Na + Nal
этансульфо-
нат натрия
Под действием минеральных кислот из солей могут быть
выделены свободные сульфокислоты.
Промышленное значение имеет фотохимическое сульфохло-
рирование алканов или циклоалканов (это цепная свободнора¬
дикальная реакция; см. § 21):
RH 4- S02 + С12 —► RSOsCl+HCl
Полученные сульфонилхлориды при гидролизе образуют
сульфокислоты:
rso2ci + h2o —► rso2oh + hci
При действии щелочей на сульфонилхлориды или свободные
сульфокислоты легко образуются соли — алкилсульфонаты:
2NaOH NaOH
RSOsC1 RS0’0Nat^T RS0’0H
Алкилсульфонаты — хорошие моющие вещества (см. § 97).
Ароматические сульфокислоты получают прямым сульфиро¬
ванием ароматических углеводородов серной кислотой. Реакция
сульфирования — одна из характерных реакций электрофильно¬
го замещения у ароматических соединений. Для сульфирования
используют концентрированную серную кислоту или олеум. Ди-
£64
и трисульфокислоты получают в более жестких условиях
(300°С, повышенное содержание S03 в олеуме).
Относительно механизма реакции сульфирования единого
мнения нет. Полагают, что в условиях реакции сульфирования
(отсутствие воды или малое ее содержание) молекулы серной
кислоты, взаимодействуя друг с другом, образуют активные
электрофильные частицы—оксид серы(У1) и ион HS03:
2H2SO« S03 + Н30+ + HSO;
3H2so4 hsoj + н3о+ + 2hso;
Электронная плотность на атоме серы в этих частицах силь¬
но уменьшена под влиянием атомов кислорода, поэтому атом
серы обладает повышенной электрофильностью.
Первая стадия реакции сульфирования, протекающая мед¬
ленно, приводит к образованию карбениевого иона (а-комп-
лекс):
+ S03 ^
,Н
SO3
Вторая стадия протекает быстро:
+ HS04
SOj + H2S04
£>so; + н3о+ ^=t (>so3h + н2о
Сульфокислоты в водных растворах находятся в ионизиро¬
ванном состоянии. Это сильные кислоты, по силе сравнимые с
минеральными кислотами.
После окончания реакции смесь сульфокислот и серной кис¬
лоты разбавляют большим количеством воды (до полного рас¬
творения) и нейтрализуют карбонатом кальция или бария.
Сульфаты этих металлов осаждаются, а в растворе остаются
кальциевые или бариевые соли сульфокислот, из которых могут
быть выделены свободные сульфокислоты.
В качестве сульфирующего агента может быть использована
хлорсульфоновая кислота H0S02C1 (в эквимольных количе¬
ствах):
C6He + H0S02Cl —V C6HS—SO3H + НС1
Избыток хлорсульфоновой кислоты реагирует с образовав¬
шейся сульфокислотой, давая сульфонилхлориды (см. ниже).
Ароматические сульфокислоты — кристаллические, гигроско¬
пичные, легко растворимые в воде вещества. Часто сульфо-
группу вводят в молекулу органического соединения для повы¬
шения его растворимости в воде. Это имеет большое практиче¬
265
ское значение в производстве красителей. Большое применение
в производстве органических красителей находят сульфокисло¬
ты нафтолов.
Сульфогруппа легко замещается другими группами, поэтому
сульфокислоты являются важными промежуточными продукта¬
ми для синтезов. Приведем наиболее важные их превращения.
1. Сульфокислоты гидролизуются перегретым водяным па¬
ром в кислой среде; при этом регенерируется ароматическое со¬
единение и выделяется серная кислота (реакция десульфиро¬
вания) :
НгО (пар), 180 вС
CeH5—S03H + CeHe + H2S04
2. При замещении сульфогруппы гидроксильной группой об¬
разуются фенолы (щелочное плавление солей сульфокислот; см.
§72).
3. При сплавлении солей сульфокислот с цианидом калия
образуются нитрилы:
ocrso,K
Р-нафтонитрил
4. При обработке солей сульфокислот хлоридом фосфора (VJ
образуются сульфонилхлориды:
СбН5—SOjONa + РС15 —► С6Н5—S02C1 + POCU + NaCl
Сульфонилхлориды ароматических кислот можно получить
непосредственным сульфохлорированием ароматических углево¬
дородов хлорсульфоновой кислотой (избыток):
hoso2ci
СНз—CeHs ► СНз—С6Н4—S02C1
толуолсульфонилхлорид
Это весьма реакционноспособные вещества. Так, они легко
реагируют со спиртами и фенолами, образуя эфиры сульфокис¬
лот, являющиеся хорошими алкилирующими средствами:
CgHs—S02C1 + НОСН3 —► С6Н5—S02—ОСНз + HCI
метиловый эфир
бензолсульфокислоты
С6Н3—S02C1 + НОС6Н5 —► С6Н5— S02—ОС5Н5 + HCI
фениловый эфир
бензолсульфокислоты
С аммиаком они легко образуют амиды сульфокислот —
сульфонамиды:
CeHs— SO2CI + 2NH3 —> с6н5—so2—nh2 + nh4ci
Многие производные сульфонамидов являются лекарствен¬
ными и дезинфицирующими средствами (см. § 129).
266
ГЛАВА 14
НИТРОСОЕДИНЕНИЯ
Нитросоединениями называют органические вещества, содержа¬
щие в качестве функциональной группы нитрогруппу NO2. Атом
азота нитрогруппы непосредственно связан с атомом углерода
в отличие от эфиров азотистой кислоты, содержащих функцио¬
нальную группу ONO, где связь осуществляется через атом кис¬
лорода, Эфиры азотистой кислоты изомерны нитросоединениям:
R—N02 R—О—NO
нитросо- эфир азотистой
единение кислоты
§ 121. Изомерия, номенклатура и строение. В зависимости от
природы органического радикала, с которым соединена нитро¬
группа, различают нитросоединения алифатические (предельные
и непредельные), алициклнческие, ароматические и гетероцик¬
лические. По характеру углеродного атома, связанного с нитро¬
группой, нитросоединения подразделяют на первичные, вторич-
ные и третичные (подобно галогенпроизводным и спиртам).
В состав молекулы может входить одна или несколько нитро¬
групп. В названиях нитросоединений употребляют префикс нит¬
ро. Ниже приводятся формулы и названия нитросоединений
разных типов:
СНа NO*
СНа—N0» СНа—i—СНа СН3—СН=С—СНа
IsFOa
нитрометан 2-метил*2-нитро-
пропан
aN°2
СХ
нитроцикло- нитробензол
гексан
2-нитробутен-2
N
3-нитро¬
пиридин
Строение. Электронная конфигурация атома азота выра¬
жается схемой ls22s22p3, т. е. атом азота имеет во внешнем слое
пять валентных электронов. Использовав все пять валентных
электронов для образования ковалентных связей, мы полу¬
чили бы вокруг атома азота десятиэлектронную группировку,
что противоречит правилу октета. Поэтому нельзя изображать
нитрогруппу формулой 0=N=0. Для подхода к вопросу о ее
подлинном строении рассмотрим сначала строение нитрозосо-
••
единений R—N=0.
267
За счет свободной электронной пары азот группы N0 спо¬
собен присоединять различные атомы и группы. Присоединяя
кислород, нитрозосоединения превращаются в нитросоединения:
R— n^o + 05
•• XX
R—N==0
* >{*
Присоединение кислорода произошло здесь «за чужой
счет» — связующая электронная пара принадлежала атому азо¬
та. В результате у всех входящих в состав нитрогруппы атомов
образовались устойчивые октеты электронов. Атом азота, пре¬
доставивший свободную пару электронов для образования свя¬
зи с атомом кислорода, как бы «потерял» электрон (принадле¬
жавшая ранее азоту свободная пара теперь поделена между
азотом и кислородом), а кислород получил электрон (один
электрон из пары, которой кислород владеет вместе с азотом).
В результате азот приобрел положительный заряд, равный за¬
ряду электрона, а кислород—соответствующий отрицательный
заряд. Кроме обычной ковалентной связи с помощью пары
электронов (лишь происхождение этой пары необычно!) азот и
кислород связаны еще и электрическим притяжением разно¬
именных зарядов.
Такая связь весьма распространенная в соединениях азота,
носит название координационной или семиполярной (т. е. полу-
полярной от английского semi — наполовину), или донорно-ак¬
цепторной. Атом, представивший электроны для образования
такой связи, называется донором (он заряжается положитель¬
но); атом, принявший электроны для пополнения своего октета
и зарядившийся отрицательно, называется акцептором. Для
обозначения такого типа связи в нитросоединениях используют
различные способы:
R—N=0 или R—N=0
В первой формуле указаны возникшие на атомах заряды,
во второй — направление передачи электронов.
Приведенные выше формулы, отвечающие правилу октета,
однако, тоже не вполне точно выражают истинное строение
нитрогруппы. В ней проявляется мезомерия; связи имеют про¬
межуточный характер, распределение 'электронной плотности
выравнивается. Поэтому более точно строение нитрогруппы пе¬
редают следующие формулы:
Мезомерная структура нитрогруппы наглядно подтверждает¬
ся рентгенографическими определениями: обе связи N—О име¬
ют одинаковую длину, промежуточную между длинами простой
N—О и двойной N=0 связей:
R—О N =='0
0,137 НМ 0,122 нм
По строению нитрогрупца похожа на рассмотренный ранее
карбокснлат-анион (см. § 95), для которого характерна такая
же мезомерия.
§ 122. Способы получения. 1. Нитросоединения могут быть по¬
лучены в результате обменной реакции между галогенпроизвод-
ными и нитритом серебра. В качестве побочных продуктов все¬
гда образуются изомерные эфиры азотистой кислоты:
AgNOj
С2Н5Вг С2Н5—N02 + С2Н5—О—NO
нитроэтан этилнитрит
2. Главный способ получения нитросоединений—реакция
нитрования — замена водорода на нитрогруппу:
R—Н —► R—NOj
Применяют различные нитрующие агенты — смесь азотной
кислоты с серной (нитрующая смесь), соли азотной кислоты в
смеси с серной кислотой, оксиды азота и др.
Нитрованию могут подвергаться самые разнообразные орга¬
нические соединения — алканы, циклоалканы, ароматические
углеводороды, гетероциклы, а также многие вещества (особенно
в ароматическом ряду), уже содержащие в молекуле другие
заместители. Широкая применимость реакции нитрования, а
также важность получающихся при этом продуктов делают ни¬
трование одним из важнейших превращений в органической хи¬
мии.
Нитрование алканов и циклоалканов проводят разбавленной
азотной кислотой (реакция Коновалова; см. § 21) или оксида¬
ми азота при нагревании в жидкой или паровой фазе. Эта ре¬
акция идет по радикальному механизму. В условиях высокой
температуры азотная кислота распадается по радикальному
типу:
НО—no2 —*- НО • + • N02
Образовавшиеся радикальные частицы стабилизируются, от¬
рывая водород от молекул углеводорода и превращая их в свою
очередь в углеводородный радикал R-, который с радикалом
•N02 дает нитросоединение:
• no2
HO- + RH R- >- RN02
269
В этой последовательности реакций не происходит регенера¬
ции радикальных частиц. Реакция не имеет цепного характера
(точнее, цепи здесь очень короткие) и активные частицы надо
постоянно воссоздавать нагреванием.
Процесс нитрования всегда сопровождается частичной дест¬
рукцией органического вещества — окислением, разрывом угле-
род-углеродных связей. Так, например, при нитровании пропана
лишь около 40 % его превращается в нитросоединения. Образо¬
вавшаяся смесь нитросоединений имеет следующий состав:
HNO*. 420 *С
СН3—СН2—СНз *- CH3NO2 + CjH5N02 + c3h7no2
(9 %) (26 %) 1-нитропро-
пан (32 %)
2-нитропро¬
пан (33 %)
Совершенно по иному механизму протекает замещение на
нитрогруппу атома водорода ароматического ядра. Это реакция
электрофильного замещения; механизм ее уже подробно рас¬
сматривался в § 47. В этом случае нитрование проводят нитру¬
ющей смесью при комнатной или несколько более высокой тем¬
пературе.
В бензольное ядро можно непосредственно ввести не более
трех нитрогрупп, причем введение каждой последующей группы
проводится во все более жестких условиях. Как заместитель
второго рода нитрогруппа уменьшает способность ароматическо¬
го ядра к реакциям электрофильного замещения. Это можно
иллюстрировать на примере нитрования бензола:
Для получения ди- и тринитросоединений требуются не толь¬
ко более высокие температуры, но и применение более концент¬
рированной азотной кислоты (дымящая азотная кислота), а
также олеума вместо серной кислоты.
Нитроваться могут не только различные ароматические угле¬
водороды (в том числе многоядерные), но и соединения, уже
имеющие в ядре другие группы — галоген, гидроксильную, суль-
фогруппу, аминогруппу и др. Получающиеся соединения имеют
большое значение как полупродукты для синтеза красителей,
фармацевтических препаратов и др.
§ 123. Физические и химические свойства. Физические
свойства нитросоединений тесно связаны с особенностями
270
строения нитрогруппы. Семиполярная связь создает значитель¬
ный дипольный момент. Это ясно видно при сопоставлении ди¬
польных моментов различных производных бензола. Особенно
наглядно сильное возрастание дипольного момента при перехо¬
де от нитрозосоединений (обычная двойная связь N=0) к ни¬
тросоединениям (семиполярная связь N+—О-):
и. Д и. д
С6Н81 1,30 CeH5NO 3,14
СвН3ОН 1,45 CeH5NO, 4,01
Большой дипольный момент нитросоединений служит под¬
тверждением приписываемого им электронного строения.
Соединения с сильнополярными молекулами отличаются от
неполярных и другими физическими свойствами. Разноименные
заряды полярных молекул притягиваются; такие молекулы
труднее оторвать друг от друга, чем неполярные. Поэтому по¬
лярные вещества имеют повышенные температуры плавления и
кипения. Это видно при сравнении температуры кипения нитро¬
этана с температурами кипения других производных этана
.(в °С):
С2Н8Вг 38 C2H5NOa 114
CjH5OH 78
Физические свойства некоторых нитросоединений приведены
в табл. 15. Большая часть нитросоединений — жидкости, имею¬
щие довольно высокие температуры кипения. Ди- и полинитро¬
соединения — твердые вещества. В совершенно чистом виде
нитросоединения почти бесцветны, но обычно они содержат
примеси, придающие им желтоватую окраску. Плотность первых
представителей гомологического ряда больше единицы, по мере
роста углеводородного радикала она уменьшается. С ростом
углеводородного радикала уменьшается растворимость в воде,
как это всегда наблюдается в гомологических рядах. С органи¬
ческими растворителями нитросоединения смешиваются во всех
соотношениях и сами являются хорошими растворителями для
многих органических веществ.
Таблица 15. Физические свойства некоторых нитросоединений
Соединение
Т. пл.,
°С
Т. кип.,
•С
Относи¬
тельная
ПЛОТНОСТЬ
d20
4
Диполь¬
ный
момент,
д
Растворимость
в воде при 20 °С,
г/100 мл
Нитрометан
—28,в
101
1,138
3,17
9,5
Нитроэтан
—89,5
114
1,051
3,19
4,5
Ннтропропан
—104
131
1,001
3,57
1.4
Нитробензол
+5,8
211
1,223
4.01
0,2
а-Нитронафталин
+57,8
304
3,98
Практически
нерастворим
271
Химические свойства. Восстановление. По химиче¬
скому поведению нитросоединения обнаруживают определенное
сходство с азотной кислотой. Это сходство проявляется при
окислительно-восстановительных реакциях. Наиболее характер¬
ное свойство азотной кислоты, отличающее ее от большинства
других кислот,— окислительное действие. Нитросоединения,
действуя, подобно азотной кислоте, как окислители, при этом
сами восстанавливаются. В зависимости от применяемых вос¬
становителей и условий реакции из нитросоединений можно
получить различные вещества. Конечными и наиболее важными
продуктами являются амины:
R—NO„ + 6H —>- R—NHj + 2Н20
Эта реакция аналогична электрохимическому восстановле¬
нию азотной кислоты до гидроксиламина:
НО—NO2 + 6H —>. НО—NH2 + 2H20
Электроноакцвпторные свойства нитрогруппы. Реакции кон¬
денсации. Сильным положительным зарядом на атоме азота
нитрогруппа напоминает карбонильную группу с ее положитель¬
но заряженным атомом углерода. Вспомним, что карбонильная
группа, оттягивая на себя электроны, увеличивает подвижность
атомов водорода у соседнего атома углерода — их способность
отщепляться в виде протона (см. § 86). Так же действует на
а-водородные атомы и нитрогруппа.
Подвижность ос-водородных атомов проявляется в способ¬
ности первичных и вторичных нитросоединений играть роль
метиленового компонента в реакциях конденсации с карбониль¬
ными соединениями. Эти конденсации идут по альдольно-крото-
новому типу:
О ОН
с6н5—С—Н + CH3NOj —► свн5—сн—сн2—no2 ——+■
“Н20
Р-нитро-а-фенилэтиловый
спирт
—► С6Н5—СН=СН—N02
Р-нитростирол
Действие щелочей. Таутомерия нитросоединений. Подвиж¬
ность а-водородных атомов первичных и вторичных нитросо¬
единений проявляется и в их способности реагировать со щело¬
чами. Рассмотрим эту реакцию на примере фенилнитрометана
C6H5CH2NO2. Это вещество представляет собой жидкость с
т кип. 226 °С, нерастворимую в воде. Однако в водных рас¬
творах щелочей фенилнитрометан растворяется, образуя соль, в
которой органический остаток играет роль аниона. При осто¬
рожном действии кислоты на эту соль выделяется твердое ве¬
щество— аци-форма фенилнитрометана (т. пл. 84 °С). Посте-
272
пенно эта форма превращается в обычный жидкий фенилнитро-
метан:
о
О
NaOH |
С6Н5—СН2—N=0
>• С„Н5—CH=N—ONa
Jhci
о
t
CeH6—CH=N—ОН
Способность существовать в нескольких взаимопревращаю-
щихся формах (таутомерия) встречается не только у нитросо¬
единений, но и у ряда других органических веществ. Подробнее
с явлением таутомерии мы познакомимся позднее (см. § 150).
Влияние нитрогруппы на ароматическое ядро. Нитрогруппа
относится к заместителям второго рода: оттягивая электроны из
ароматического ядра, она уменьшает его активность в реакциях
электрофильного замещения. Вступающий заместитель направ¬
ляется в лета-положение:
Электронная плотность особенно сильно уменьшается в ор¬
то- и пара-положениях к нитрогруппе. Обедненные электронами
орто- и пара-положения ядра приобретают частичный поло¬
жительный заряд, а вместе с тем и способность к необычным
для ароматических соединений реакциям нуклеофильного заме¬
щения, например:
1,3-динитробензол 2,4-динитроанилин
Под влиянием нитрогрупп увеличивается реакционная спо-*
собность галогена, стоящего в орто-пара-положениях:
02N.
273
Влияние нитрогрупп распространяется и на атомы водорода
метильной группы, связанной с бензольным ядром: она приоб¬
ретает способность играть роль метиленового компонента в ре¬
акциях конденсации с карбонильными соединениями в присут¬
ствии оснований:
N02
Ш2
02№
•ОН.
3 о=сн-с6н5
•NO*
2,4,6-тринитротолуол
СН=СН—С6Н5
2,4,6-тринитростильбен
Оттягивая электроны из ядра, нитрогруппа также повышает
кислотность стоящих в орто- и /гара-положениях гидроксильных
групп.
Особенно сильным оказывается согласованное действие не¬
скольких нитрогрупп. Влияние нитрогрупп на кислотность фе¬
нольного гидроксила наглядно показывают значения констант
кислотной диссоциации Ка следующих соединений:
Последнее из приведенных в этом ряду соединений—<
2,4,6-тринитрофенол называют пикриновой кислотой. Действи¬
тельно, это вещество ведет себя как настоящая кислота (сила
ее превышает силу угольной кислоты) — образует соли — пи-
краты:
ОН
OAg
ОСНз
OjNs
•no2
no2
пикриновая
кислота
снз. °’NN^
*-
N02
no2
метиловый эфир
пикриновой кислоты
Ее метиловый эфир имеет свойства сложного эфира: гидро-
274
лизуется при кипячении со щелочами, с аммиаком образует
пикрамид:
§ 124. Важнейшие представители. Алифатическиенитро-
соединения. В промышленном масштабе производятся
первые представители гомологического ряда с числом углерод¬
ных атомов Ci—С3. Эти нитросоединения применяют как рас¬
творители (в частности, для синтетических смол, каучуков), а
также для различных синтезов — получения нитроспиртов, нит-
роалкенов и других соединений. Промышленное значение имеет
производство пропионовой кислоты и гидроксиламина из нитро¬
пропана при действии серной кислоты:
н2о. h,so4. 13о-с
СН3СН4СНг—NO, * СН,СН,СООН + NH,OH - H,SO«
Все нитросоединения — довольно сильные яды для централь¬
ной нервной системы. Некоторые из них обладают раздражаю¬
щим действием. Таков, например нитротрихлорметан CCI3NO2,
который под названием хлорпикрин применяют в качестве
инсектицида. Хлорпикрин обладает сильным слезоточивым дей¬
ствием, в первой мировой войне его применяли как боевое от¬
равляющее вещество.
Тетранитрометан C(N02)4 — жидкость, т. кип. 126°С; ис¬
пользуют как окислитель для ракетного топлива. Его получают
из уксусного ангидрида или из ацетилена:
4(СН»С0)20 + 4HNO, —> C(NO,)« + 7СН,СООН + СО,
HNO3 HNO,. H2so4
СН=СН ► HC(NO,)3 *- C(NO*),
нитроформ
Нитроциклогексан — жидкость, т. кип. 205 °С. Получают в
промышленном масштабе нитрованием циклогексана азотной
кислотой в жидкой фазе при 150°С и повышенном давлении.
Нитроциклогексан превращают в циклогексаноноксим — сырье
для получения капролактама — исходного вещества в синтезе
капрона.
Нитробензол — бесцветная жидкость (обычно он окрашен
примесями в желтый цвет), т. кип. 211 °С, df 1,203. Получают
275
в больших количествах в промышленности и используют глав¬
ным образом для восстановления в анилин С6НдЫН2.
Нитротолуолы существуют в виде трех изомеров, из которых
наибольшее значение имеют орто- и пара-изомеры, образующие¬
ся при нитровании толуола. Эти изомеры можно разделить вы¬
мораживанием пара-изомера.
сн3
НзСЧ*^/к°2
•NO.
о-нитротолуол
(т. пл. —3°С,
т. кип. 222 °С)
.«-нитротолуол
(т. пл. 16 °С,
т. кип. 233 °С)
Нитротолуолы восстанавливают в соответствующие амины,
которые используют далее в синтезе красителей.
Тринитротолуол (тротил, тол) — продукт исчерпывающего
нитрования толуола:
N02
СН3
HNO3, HsS04
w
СНэ
■NOj
Это твердое вещество (т. пл. 80 °С) — одно из самых рас¬
пространенных взрывчатых веществ. Ценным свойством тола яв¬
ляется сравнительно малая чувствительность к удару, трению,
вследствие чего он относительно безопасен. При зажигании на
воздухе спокойно горит, взрыв может наступить лишь при сго¬
рании больших количеств или под действием детонаторов.
а-Нитронафталин — твердое вещество, т. пл. 61 °С. Легко
получается при нитровании нафталина. Восстановление его дает
а-нафтиламин — исходное вещество для синтеза других а-за-
мещенных нафталинов, красителей.
ГЛАВА 15
АМИНЫ
Амины — органические производные аммиака, которые можно
рассматривать как продукты замены атомов водорода аммиака
на углеводородные радикалы.
§ 125. Строение, изомерия, номенклатура, физические свойства.
В зависимости от природы радикала амины могут быть
алифатическими (предельными и непредельными), алицикличе-
скими, ароматическими и гетероциклическими. В зависимости
от числа замещенных на радикалы атомов водорода аммиака
различают амины первичные RNH2, вторичные R2NH и третич¬
276
ные R3N; органическими производными солей аммония являют¬
ся четвертичные аммониевые соединения [R4N] + О-.
Обратим внимание на то, что классификация спиртов и га-
логенпроизводных на первичные, вторичные или третичные за¬
висит от положения функциональной группы, у аминов же эта
классификация отражает степень замещения атомов водородов
в остатке аммиака.
Существование аминов разной степени замещения создает
дополнительные возможности для изомерии, увеличивает число
возможных соединений. Вспомним, что углеводород бутан имеет
два изомера, производящиеся от него спирты — четыре, аминов
же с формулой C4H11N имеется восемь.
Названия первичных аминов строят из названий углеводоро¬
дов или радикалов, добавляя к ним окончание амин. Названия
вторичных и третичных аминов чаще всего образуют по принци¬
пам рациональной номенклатуры, перечисляя имеющиеся в со¬
единении радикалы. Для примера приведем названия упоминав¬
шихся выше изомерных аминов C4HuN.
Первичные амины:
NHj
СН,—CHs—CH—CHj
erop-бутиламин,
бутана мнн-2
СН,
СН3—С—NH2
I
СН3
грег-бутиламин,
2-метилпропанамин-2
СН,
I
СН,—СН—NH—СН,
Л^-метилпропанамин-2,
изопропилметиламин
СН3—СН2—NH—СНз—СНз
ЛГ-этиламиноэтан,
диэтиламин
Третичный амин:
СНз—СН2—N(CHj)2
Nt Л/-диметилэтанамин,
диметилэтиламин
Названия, в которых присутствие аминогруппы отражено в
префиксной части, не предусмотрены правилами ШРАС, одна¬
ко употребляются достаточно широко.
СНз—СН2—СНз—СН,—NH2
н-бутиламин,
бутанамин-1
СН,—СН—CHS—NH2
Ан3
изобутиламин,
2-метилпропанамин-1
Вторичные амины:
СНз—СНг—CH2—NH—СНз
Лг-метилпропанамин-1,
метилпропиламин
277
Таблица 16. Физические свойства некоторых аминов
Амив
Формула
T. пл.. °C
T. кип., *C
Относительная
плотность
Метиламин
ch3nh2
■
-7,6
0,769 (при -79 °C)
Диметиламин
(CH3)2NH
—
+7
0,680 (при 0°C)
Т риметиламин
(CH3)3N
-117
+3,5
0,671 (при 0°C)
Этиламин
c2h6nh2
—
16,6
0,706 (при 0eC)
Диэтиламин
(CsH5)2NH
-50
56
0,711
Пропиламин
C3H7NH2
—
49
0,714
Бутиламин
c4h9nh2
—
78
0,742
Гексиламин
CeH,3NH2
— 19
130
0,763
Анилин
CeH5NH2
—6
184
1,027
Метила нилин
CeH6NHCH3
_
196
0,989
Диметиланилин
C6H5N(CH3)2
+2,5
194
0,956
Дифениламин
(CeH5)2NH
+54
302
1,158
Простейшие алифатические амины (метиламин, диметил¬
амин, триметиламин и этиламин)—газообразные вещества,
амины с большим числом атомов углерода — жидкости, а начи¬
ная с C12H25NH2 — твердые вещества. Запах низших аминов
напоминает запах аммиака. Первые представители ряда хорошо
растворимы в воде, по мере роста углеродного скелета раство¬
римость в воде уменьшается. Физические свойства некоторых
аминов приведены в табл. 16.
§ 126. Способы получения. Способы получения аминов весьма
разнообразны, но главными являются алкилирование аммиака
(реакция Гофмана) и восстановление нитросоединений. Первая
из этих реакций применяется главным образом для получения
аминов жирного и жирноароматического рядов, вторая служит
основным путем синтеза ароматических аминов.
1. Алкилирование аммиака. При этом происходит постепен¬
ное замещение атомов водорода аммиака на углеводородные
радикалы с образованием смеси аминов всех степеней замеще¬
ния (в виде солей), например:
+ NH3
NHa + СгН6Вг —>• C2H5NH3 Вт" ► C2H5NH2 + NH4Br
С2Н5ВГ + nh8
C2H5NH2 ► (C2H5)2NH2 Вг- ► (C2H5)2NH + NH4Br
C2H5Br + NH3
(C2H5)2NH >- (C2H5)3NH Br- *■ (C2H6)3N + NH4Br
С2Н5ВГ +
(CSH6)3N »■ (C2He)4N Br"
Эта реакция — один из классических примеров нуклеофиль¬
ного замещения: вследствие поляризации связи С—Вг на атоме
углерода возникает положительный заряд, на который и на¬
правляется атака аммиака, проявляющего нуклеофильные свой¬
ства вследствие наличия свободной электронной пары у азота.
278
При алкилировании аммиака по Гофману образуется смесь
солей первичных, вторичных, третичных аминов и четвертичных
аммониевых оснований. Поэтому реакция Гофмана не очень
удобна для получения первичных аминов. Чаще ее применяют
для получения вторичных и третичных аминов, например моно-
метил- и диметиланилинов из анилина. В качестве более деше¬
вого алкилирующего средства при промышленном проведении
этой реакции вместо галогенпроизводных используют метиловый
спирт. Реакция идет при нагревании под давлением:
СНзОН
CeH5NH2 + СНзОН C6H5NHCH3 ■ _н^>- CeH5N (СН3)2
Для получения первичных аминов алкилируют не сам амми¬
ак, а некоторые его производные. Например, в одном из вари¬
антов исходят из фталимида калия (синтез Габриэля):
со СО
2. Восстановление ароматических нитросоединений. Эта ре¬
акция впервые осуществлена Н. Н. Зининым (1842 г.). Действуя
на нитробензол сульфидом аммония, он получил анилин:
CeH5N02 + 3(NH4)2S —► C6H5NH2 + 6NH3 + 3S+2НгО
Открытие Зинина послужило исходной точкой развития ани¬
линокрасочной промышленности. Хорошо известно высказыва¬
ние А. Гофмана, что за одно это открытие имя Зинина заслу¬
живает быть записанным золотыми буквами в истории химии.
Для превращения нитросоединений в амины могут быть ис¬
пользованы и другие восстановители — олово, цинк, железо в
кислой среде, хлорид олова (II), водород в присутствии катали¬
заторов. Восстановление можно осуществить и электрохими¬
чески.
Наибольшее значение в технике получило восстановление
нитробензола в анилин действием железа в присутствии хлоро¬
водородной кислоты. При техническом осуществлении процесса
нет необходимости брать большие количества кислоты. После
образования следов FeCl2 восстановление идет дальше за счет
воды и металлического железа по суммарному уравнению:
4C8H5N02 + 9Fe + 4Н20 —► 4C6H6NH2 + 3FesO,
В лабораторных условиях восстановление нитросоединений
до аминов часто проводят оловом и хлороводородной кислотой:
CeH6N02 + 3Sn + 7НС1 —► C6H6NH2-HCI + 3StiCI2 + 2H20
279
Удобный современный лабораторный метод восстановления
нитросоединений основан на использовании гидразина в при¬
сутствии катализатора — скелетного никеля:
N1
2C6H5N02 + 3H2N—NHa ► 2CeH5NHs + 4НгО + 3Na
Способ этот привлекает тем, что в числе продуктов реакции
нет веществ, мешающих выделению полученного амина простой
перегонкой реакционной смеси.
Восстановление нитросоединений в амины проходит через
несколько промежуточных стадий. Строение промежуточных
продуктов зависит от типа примененного восстановителя, от ус¬
ловий проведения реакции (в первую очередь от кислотности
среды). Разнообразие промежуточных продуктов восстановле¬
ния иллюстрирует следующая схема:
Нейтральная или
кислая среда
CeHe—N0*
нитробензол
I
С„Н6—N0 —
нитрозобензол
I
С„Н5—NHOH
фенилгидроксил
амин
1
CeH6—NH* ч— СвН6—NH—NH—СвН6 ч— CeH6—N=N—С6Н5
анилин гидразобензол азобензол
Все промежуточные продукты при дальнейшем восстановле¬
нии превращаются в амины.
3. Можно восстановить до аминов и другие азотсодержащие
соединения, например оксимы, нитрилы, амиды, действием ме¬
таллического натрия в спирте или LiAIH4.
4. Своеобразный способ получения аминов представляет со¬
бой перегруппировка Гофмана (см. § 114).
§ 127. Амины — органические основания. Являясь органиче¬
скими производными аммиака, амины сохраняют главные его
химические особенности. В частности, подобно аммиаку, амины
проявляют свойства оснований. Это обнаруживается по щелоч¬
ной реакции водных растворов аминов:
Шз + НлО NHJ + HO' RNH2 + H20 ч=* RNHJ + HCT
конденсация
в щелочной среде
Щелочная среда
С6Н5—N=N—С6Н5
4
о
азоксибензол
280
По мере роста углеводородного остатка растворимость ами¬
нов в воде уменьшается. Поэтому высшие амины уже не дают
щелочной реакции. Однако и они сохраняют свойства осно¬
ваний: способны образовывать с кислотами соли. Причина
основных свойств — свободная электронная пара атома азота.
В аммиаке или аминах атом азота из пяти валентных элек¬
тронов затрачивает на образование ковалентных связей только
три электрона. Два электрона остаются свободными — за счет
этой свободной электронной пары может присоединяться протон
с образованием солей аммония:
Н
Н8С% + Н+ + СГ
Н
или иначе CH3NH2*HC1
В четвертичных аммониевых соединениях азот образует че¬
тыре совершенно одинаковые ковалентные связи, является че¬
тырехковалентным; пятая связь — ионная; положительно заря¬
женная аммониевая группировка удерживает анион путем
обычного электростатического притяжения.
Природа радикала, с которым связана аминогруппа, влияет
на основные свойства аминов. В частности, ароматические
амины — гораздо более слабые основания, чем амины жирного
ряда. Это объясняется тем, что свободная электронная пара
аминного азота вступает в сопряжение с подвижными электро¬
нами ароматического ядра. Распределение электронной плотно¬
сти в анилине можно условно выразить следующими форму¬
лами:
Такое распределение приводит к уменьшению электронной
плотности на атоме азота и, тем самым, к ослаблению его спо¬
собности присоединять протон. Введение в ядро анилина нитро¬
группы, обладающей электроноакцепторным действием, еще бо¬
лее понижает основность аминогруппы. Это следует из сравне¬
ния констант основности Кь анилина и л-нитроанилина:
3800-10 13 1,3 -кг13
§ 128. Химические свойства. Свободная электронная пара атома
азота не только обусловливает основность аминов, но и опре¬
деляет многие другие их свойства. Реакции аминов чаще всего
начинаются со взаимодействия свободной электронной пары с
электрофильными реагентами, т. е. веществами, реакционный
281
центр которых имеет положительный заряд (недостаток элек¬
тронов). Некоторые реакции на этом и заканчиваются, в других
вслед за присоединением следует стадия отщепления. Важней¬
шие из реакций аминов следующие.
Присоединение алкилгалогенидов. Образую¬
щиеся соли аммония устойчивы, и реакция на этом заверша¬
ется:
•• в* в- Г С,Н5Ч п*
свн5ын2 + c2h5ci —> ;nh2 с г
1с2н/ J
хлорид
этилфениламмония
(CH3)3Ns + CH3I —у [(CH3)4N]+ Г
иод ид
тетраметиламмония
Действуя на образовавшиеся соли основаниями, можно
выделить в свободном виде соответствующие амины и четырех-
замещенные аммониевые основания:
[С6Н5—NH2—С2Н5]+ СГ + КОН —у С9Н5—NH—С2Н5 + Н20 + КС1
этиланилин
I(CH3)<N]+ Г + AgOH —у I(CH3)4NJ+ 'OH + Agl
гидроксид тетра¬
метиламмония
Проведя последовательно обе реакции, получают из первич¬
ного амина — вторичный, из вторичного — третичный, из тре¬
тичного— четвертичное аммониевое основание. Результатом яв¬
ляется введение к атому азота нового углеводородного остатка
вместо атома водорода, т. е. алкилирование амина.
Ацилирование. При взаимодействии аминов с хлоран-
гидридами, ангидридами или сложными эфирами образуют¬
ся амиды кислот. Процесс начинается так же, как и алкилиро¬
вание, со взаимодействия свободной электронной пары азота с
электрофильным центром реагента:
н С1
| 8+1 I
с2н5—N: + с—сн3 —;
! + '
I
н о
8-
Н С1
С2Н5—N—i-CH3
Н—(!)
Б
Н С1
1 I
С2Н5—NH—С—СН3
. + А- .
А
Н
з I
—HCI
f С2Н5—N-C-CH3
О
этиламид уксусной
кислоты
Продукт присоединения образуется в результате взаимодей¬
ствия свободной электронной пары азота с карбонильным угле¬
232
родом, на котором вследствие поляризации связи С=0 имеется
частичный положительный заряд. При этом электронная пара
азота переходит в совместное владение с карбонильным углеро*
дом. Но последний уже до реакции имел заполненный октет на
внешней электронной оболочке и принять еще пару электронов
не может. Поэтому одновременно с установлением связи между
азотом и карбонильным углеродом подвижная электронная
пара п-связи С=0 оттесняется на атом кислорода, который
приобретает таким образом отрицательный заряд (стадия /)<
В создавшейся структуре (промежуточный продукт А) на близ¬
ком расстоянии друг от друга находятся положительно заря¬
женный центр (азот) и отрицательный (кислород). Естествен¬
ное стремление к нейтрализации осуществляется путем пере¬
хода протона от азота к кислороду, обладающему к тому же
большей электроотрицательностью (стадия 2). Однако соеди¬
нения, подобные промежуточному продукту Б, которые имеют
у одного атома углерода гидроксильную группу и галоген, не¬
устойчивы. Стабилизация происходит в результате отщепления
хлороводорода (стадия 3). Окончательным результатом реак¬
ции является введение к азоту ацильной группы вместо атома
водорода, т. е. ацилирование амина.
Ре акции с альдегидами и кетонами. Эти реакции
тоже идут через промежуточные продукты присоединения (по¬
казаны в квадратных скобках). Стабилизация осуществляется
разными путями для первичных и вторичных аминов (третич¬
ные амины в реакцию не вступают).
Из первичных аминов и альдегидов (кетоны вступают в эту
реакцию труднее) с отщеплением воды образуются так называе¬
мые основания Шиффа (имины, азометины):
С«Н9—NH, + СН—СН3 —». Г С4Н9—NH2—СН—СН3
А- I А-
С4Н9—NH—СН—СНл
Ан J
^ С4Н9-М= сн-сн3
основание Шиффа
Аналогичные соединения, образующиеся в реакции анилина
с альдегидами, носят название анилы.
Из вторичных аминов и кетонов образуются енамины, например
.. ХН2— СН2 г XHj—снл
(CH3)2N-H + 0=cr J —> (СН3)2Ш-< —>
хсн2-сн2 L _^>ch,-chJ
XHs-CHjI
(ch3)2n-c;
hANch’
2-CHj-I
2-CH2J
-H20
/CH2-CH2
(CH3)2N-C' I
^CH—CH2
283
Продукт присоединения содержит здесь только один атом
водорода у азота; при стабилизации промежуточного продукта
йтот атом переходит к кислороду, а в образовании воды участ¬
вует подвижный водород, стоящий у а-углеродного атома.
Реакции с азотистой кислотой. Практически
для проведения этих реакций берут соль азотистой кислоты и
сильную минеральную кислоту. Считают, что при этом образу¬
ется нитрозоний-катион. +N=0, который и играет роль электро¬
фильной частицы (подобно нитроний-катиону +N02 в реакциях
нитрования):
HCl НС1 +
NaN02 -гг^Г Н—О—N==0 ——> N=0 + СГ
Подобно рассмотренным ранее реакциям, прежде всего сво¬
бодная электронная пара аминного азота взаимодействует с
нитрозоний-катионом. Получающиеся промежуточные продукты
присоединения стабилизируются разными путями, и поэтому,
несмотря на общность механизма для первичных, вторичных,
третичных, жирных и ароматических аминов, конечные продук¬
ты оказываются разными.
Первичные ароматические амины превращаются при дейст¬
вии азотистой кислоты в соли диазония (см. гл. 16). Первичные
алифатические амины дезаминируются с выделением азота и
образованием спиртов или непредельных углеводородов. Вто¬
ричные амины (алифатические и ароматические) при действии
азотистой кислоты дают N-нитрозамины:
RjHN : + N=0 —► [R2HN—N=0] R2N—N=0
-н+
Третичные амины жирного ряда с азотистой кислотой обра¬
зуют лишь непрочные соли, а третичные амины ароматического
ряда -п-нитрозосоединения, например:
(CH3)2N-
в- + N=0
-н+
(СНзЫМ—
N=0
диметиланилин
га-нитрозодиметиланилин
Эта реакция идет как обычное электрофильное замещение в
ароматическом ядре, сильно активированном аминогруппой.
§ 129. Важнейшие представители. Простейшие алифатические
амины — метиламин, диметиламин, диэтиламин — применяют
при синтезе лекарственных веществ, ускорителей вулканизации
и других продуктов органического синтеза.
Гексаметилендиамин H2N—(СН2)6—NH2 является одним
из исходных веществ для получения важного полимерного ма¬
териала— найлона (см. § 190). Производится в промышлен¬
284
ности гидрированием амида или нитрила адипиновой кислоты:
Н2-^М H2/NI
HsNCO(CH2)4CONH2 -> H2N(CH2)eNHs -• NC(CH2)«CN
Анилин C6HsNH2 — важнейший из ароматических аминов.
Получается в промышленности в больших количествах восста¬
новлением нитробензола чугунными стружками в присутствии
воды и небольших количеств кислоты. Все большее значение
приобретает также каталитическое восстановление нитробензо¬
ла водородом в газовой фазе. Хлорбензол можно превратить в
анилин, пропуская его смесь с аммиаком при 400°С над ката¬
лизатором — солью меди(1). Новым промышленным способом
получения анилина является каталитический аммонолиз фе¬
нола:
Анилин применяют для получения красителей, лекарствен¬
ных препаратов, вулканизаторов и стабилизаторов для резины,
пластических масс, фотографических проявителей.
Основные свойства анилина ослаблены влиянием ароматиче¬
ского ядра на аминогруппу (константа основности анилина рав¬
на 3,8-10—,0, в то время как для метиламина она равна
5'10-4). Аминогруппа в свою очередь оказывает влияние на
ароматическое ядро: как электронодонорный заместитель она
повышает электронную плотность в ядре, облегчая тем самым
реакции электрофильного замещения. Анилин легко бронирует¬
ся, образуя уже при действии бромнрй воды триброманилин:
При нагревании с концентрированной серной кислотой он
сульфируется с образованием сульфаниловой кислоты — важно¬
го промежуточного продукта в синтезе красителей и лекарствен¬
ных препаратов:
Анилин легко окисляется. Поэтому при хранении он обычно
темнеет, хотя в совершенно чистом виде бесцветен. При дейст¬
вии хлорной извести CaCI(OCl) на водный раствор анилина
(растворимость его в воде 3,6 %) в результате окисления появ¬
ляется интенсивное фиолетовое окрашивание. Эта реакция слу-
S03H
285
Схема 7. Продукты, получаемые на основе анилина
диазотирование
С*Н„—Ш2
анилин
-> CeHs—NjCl
фенилдиазо-
нийхлорид
Красители
гидрирование
CICHj-COOH
CHjO
* С5Нц—nh2
циклогексиламин
CeHs—NH—СН2—СООН
фениламиноуксусная
кислота
сульфирование
ацилирование
алкилирование
арилировапие
Индиго
Анилиноформальдегидные смолы
Красители
/i-H2N—CeHe—S03H —j
сульфаниловая
кислота
-> С6Н5—NH—СО—СН3
C,H8-N(CH9)2 -
диметиланилип
С6Н5—NH—CeHs
дифениламин
-► Лекарственные
препараты
n-OsN—С6Н4—NH2
л-нитроанилии
1
—*• Красители
t
жит качественной пробой на анилин. Азотная кислота также
действует на анилин как окислитель. Поэтому перед нитровани¬
ем защищают аминогруппу, превращая анилин в М-фениламид
уксусной кислоты — ацетанилид:
NH2 + (СНзС0)20
NH—СОСНз + СНзСООН
Ацетанилид нитруется, образуя в основном я-нитроацетани-
лид и небольшое количество орто-изомера:
При нагревании нитроацетанилидов с разбавленными кисло¬
тами они гидролизуются до нитроанилинов:
o2n
-О
NHCOCH3
н2о (н+)
►
o2n
-NH2 + СН3СООН
286
Нитроанилины — твердые вещества желтого цвета, приме*
няются в качестве промежуточных продуктов синтеза краси¬
телей.
О разнообразии продуктов, которые получают в промышлен¬
ности из анилина, дает представление схема 7.
Сульфаниловая кислота H03S—С6Н4—NH2 — твердое веще¬
ство, выпадающее при кристаллизации из горячей воды в виде
блестящих чешуек, т. пл. «290°С (разл.). Как уже упомина¬
лось, она получается при сульфировании анилина. Обладая
свойствами как основания (аминогруппа), так и кислоты (суль-
фогруппа) сульфаниловая кислота представляет собой внутрен-
нюю соль +NH3—С6Н4—S03.
Важнейшее производное сульфаниловой кислоты — ее амид
(сульфаниламид) H2N—С6Н4—S02—NH2. Это бесцветное кри¬
сталлическое вещество, мало растворимое в холодной воде и
хорошо — в горячей, является основой важного класса лекарст¬
венных веществ — сульфамидных препаратов. Простейший из
них — сам амид сульфаниловой кислоты — стрептоцид. Изве¬
стны и многочисленные его производные — продукты замещения
атома водорода амидной группы на различные органические
остатки:
л—N
# ^—NHSO2C0H4NH2
S
норсульфазол
с2н5—^
‘I
-nhso2c6h4nh2
этазол
СНз
I
НзС^ N nHS02C6H4NH2
сульфадимезин
НООС,
( \—NHS02CeH4—NHCO—\~^У
г/
фталазол
Лекарственное действие- сульфамидных препаратов открыто в начале
30-х годов. С тех пор синтезировано более 6000 соединений этой группы,
однако практическое применение получили лишь около 20 препаратов. В йа*
стоящее время сульфамидные препараты широко применяют при лечений
различных инфекционных заболеваний. Например, норсульфазол, сульфадиме¬
зин, этазол применяют для лечения заболеваний дыхательных путей, фтал£*
зол — при лечении желудочно-кишечных инфекций.
Нафтиламины CioH7NH2, как и другие монозамещенные
нафталины, существуют в виде двух изомеров (а- и р-нафтил?»
амины). Эти соединения, и в особенности их сульфо- или гидр-
оксипроизводные, имеют большое значение как полупродукты
887
в синтезе красителей. Большая их часть имеет тривиальные
названия, например:
ын2 но nh2
so3h
нафтионовая
кислота
(1-аминонафталин-
4-сульфокислота)
SOaH
Аш-кислота
(1-амино-8-гидроксинафталин-
3,6-дисульфокислота)
а-Нафтиламин и Л^-фенил-р-нафтиламин применяют как
антиоксиданты для стабилизации крекинг-бензинов, смазочных
масел. р-Нафтиламин канцерогенен, в связи с чем в нашей
стране в промышленности не применяется.
Дифениламин (CeHs^NH — бесцветные кристаллы, практи¬
чески нерастворимые в холодной воде. Получается при нагрева¬
нии под давлением смеси анилина с гидрохлоридом анилина:
C6H5NH2 + C6H6NHj • НС1 —► С6Н6—NH—CeH5 + NH4C1
Основные свойства выражены у дифениламина еще слабее,
чем у анилина. Дифениламин образует соли только при раство¬
рении в концентрированных кислотах Эти соли при разбавлении
водой гидролизуются, и выпадает свободное основание. Дифе¬
ниламин применяют для получения красителей и в других син¬
тезах. При окислении дифениламина появляется синяя окраска;
реакцию используют для открытия азотной кислоты и других
окислителей.
Диметиланилин СвН5М(СНз)2 — жидкость, т. кип. 194 °С,
важный промежуточный продукт при синтезе красителей, взрыв¬
чатых веществ, проявителей для цветной фотографии. Получа¬
ют алкилированием анилина метиловым спиртом (см. § 126).
§ 130. Роль электронных эффектов при электрофильном заме¬
щении в ароматическом ядре. Познакомившись со строением
и свойствами главных функциональных групп, можно продол¬
жить начатое ранее (см. § 48) рассмотрение ориентации при
электрофильном замещении в ароматическом ядре.
Стоящие у ароматического ядра заместители могут либо
увеличивать электронную плотность в ядре, либо уменьшать ее.
Влияние на ядро может осуществляться двумя путями — с по¬
мощью индуктивного и мезомерного (эффект сопряжения) эф¬
фектов.
Индуктивный эффект обусловливает сдвиг электронной плот¬
ности простых связей (т. е. их поляризацию; см. § 64). По силе
288
и направлению индуктивного эффекта заместители можно рас¬
положить в ряд, где водород считается условным нулем шкалы,
т. е. заместителем, с которым сравнивается влияние всех дру¬
гих:
-/-Эффект: (CH3)3N > N02 > CN > СО > COOR > F >
> Cl > Br > I > ОН > NH2 > H
4- /-Эффект: H < CH3 < С2Н5 <Z изо-С3Н7 м-С3Н7 < С(СН3)3
Мезомерный эффект проявляется в сопряженных системах.
Он является отрицательным (—М), если заместитель оттягивает
электроны из сопряженной системы, и положительным (+М),
когда заместитель отдает свободную электронную пару для
участия в общем сопряжении. По знаку и силе мезомерного
эффекта заместители располагаются в следующие ряды:
Ш2 > CN > СО > СООН > Н
— М (акцепторы электронов)
Н < Вг < С! < ОСН3 < ОН < NH* < О'
+ М (доноры электронов)
Сравнивая приведенные выше ряды для мезомерного и ин¬
дуктивного эффектов, видно, что знаки обоих эффектов для
конкретной группы могут быть и одинаковыми, и разными.
Характер ориентирующего действия заместителя определяет¬
ся знаком проявляемого им мезомерного и индуктивного эффек¬
тов. Заместители, у которых преобладает +Л1 или +/-эффект,
относятся к орто-пара-ориентантам, заместители с преоблада¬
нием —М- или -/-эффекта, являются лшга-ориентантами.
Рассмотрим на примере анилина, каким образом электрон¬
ные влияния в ароматическом ядре передаются преимуществен¬
но в орто- и пера-положения. Аминогруппа проявляет в основ¬
ном -{-/W-эффект. Это приводит к тому, что электронная плот¬
ность свободной электронной пары атома азота частично
смещается к ароматическому ядру. Полное смещение означало
бы образование двойной связи N = C. Углеродный атом .С-1 при¬
обрел бы в таком случае заполненный октет за счет имеющихся
четырех связей; тем самым оказалось бы нарушенным круговое
сопряжение в ароматическом ядре. Шестерку л-электронов приня¬
лось бы размещать в локализованных, закрепленных двойных
связях. Однако, не нарушая правил валентности, можно пред¬
ставить себе создание в кольце только двух двойных связей, при
этом одна электронная пара останется свободной.
Напишем обычную формулу анилина и три получающиеся
после указанного сдвига структуры, отличающиеся расположе¬
289
Ю Органнчсскал химия
нием двойных связей и свободной электронной пары *:
6
шн2
Эти формулы представляют собой граничные структуры, ко¬
торыми изображается мезомерное состояние молекулы анилина.
Его можно изобразить и одной формулой (см. выше формулу
справа). Оба способа изображения распределения электрон¬
ной плотности в молекуле анилина показывают, что отрицатель¬
ный заряд сосредоточивается в орто- и пара-положениях, т. е.
именно там создаются благоприятные условия для электрофиль¬
ного замещения.
Положительным мезомерным эффектом обладает и гидрок¬
сильная группа. Свободная электронная пара атома кислорода
ОН-группы фенола вступает в сопряжение с ароматическим
ядром и в орто-пара-положениях создается повышенная элек¬
тронная плотность, подобно тому, как это происходит в анилине.
Иная природа орго-пара-ориентирующего действия алкиль¬
ных групп: здесь проявляется их -(-/-эффект. Как известно, в
связях Н—С частичный отрицательный заряд несет атом угле¬
рода: часть накопленной на нем электронной плотности переда¬
ется в ядро и сосредоточивается преимущественно в орто-пара-
положениях:
Увеличивая электронную плотность в ядре, орто-пара-орк-
ентанты повышают активность ароматического ядра в реакциях
электрофильного замещения. Исключением являются лишь га¬
логены, которые, будучи ориентантами первого рода, тем не
менее уменьшают реакционную способность ароматического
ядра. Это объясняется своеобразным соотношением между ха¬
рактером индуктивного и мезомерного эффектов галогенов.
Галогены, обладая сильным —/-эффектом, оттягивают электро¬
ны из ядра, уменьшают его электронную плотность, что приво¬
дит к понижению его реакционной способности. В то же время
вследствие присущего им слабого +.М-эффекта происходит час¬
тичная передача свободной электронной пары атома галогена в
* В мега-положении свободную электронную пару поместить нельзя: при
этом не удается разместить в кольце двойные связи.
6+
290
орто- и «ара-положения ароматического ядра, чем обеспечива¬
ется принадлежность галогенов к ориентантам первого рода.
Типичным л1ега-ориентантом является нитрогруппа, оттяги¬
вающая электроны из ядра как за счет —М-,, так и за счет
■—/-эффектов. Отрицательный мезомерный эффект приводит
здесь к вытягиванию электронной плотности из ядра и появле¬
нию в орто- и пара-положениях положительного заряда:
+
В том же направлении действует и индуктивный эффект ни¬
трогруппы. Другой сильный мета ориентант — аммониевая груп¬
па — из-за своего положительного заряда вытягивает электрон¬
ную плотность из ядра по индуктивному механизму.
Под действием лага-ориентантов, следовательно, появляется
положительный заряд в орто- и «ара-положениях. В мета¬
положения х электронная плотность не столь сильно уменьша¬
ется, поэтому действие электрофильных реагентов направляется
в эти места. Поскольку общая электронная плотность ядра по¬
нижена, оно менее реакционноспособно при электрофильном
замещении.
Электронные эффекты, ориентирующее действие заместите¬
лей и их влияние на ядро приведены ниже:
Заместитель Влияние на ядро
Заместители I рода (орто-пара-ориентанты)
Алкил +/ Активация
Аминогруппа +Л1»—1 »
Гидроксигруппа +М » — / »
Галоген -\-М < —1 Пассивирование
Заместители
Нитрогруппа
Карбоксигруппа
Аммониевая группа
И рода (.иега-ориентанты)
—М, —1 Пассивирование
—М, —I »
-I »
Ю*
ГЛАВА 16
ДИАЗОСОЕДИНЕНИЯ
Диазосоединения Ar—N2X — это вещества с функциональной
группой, составленной из двух атомов азота и соединенной с
ароматическим радикалом и неорганическим остатком X.
§ 131. Реакция диазотирования и строение диазосоединений.
Общий и важнейший способ получения диазосоединений заклю¬
чается в действии азотистой кислоты на соли первичных аро¬
матических аминов (реакция диазотирования). Практически
вместо неустойчивой в свободном состоянии азотистой кислоты
берут ее соль и сильную минеральную кислоту (хлороводород¬
ную, серную) в количестве, достаточном для образования соли
амина, выделения азотистой кислоты из ее соли и сохранения
кислой среды после окончания процесса (на 1 моль амина 2,5—
3 моль кислоты):
ArNH2 + NaNO* + 2НС1 ArN2Cl + NaCt + 2Н20
Диазосоединения почти никогда не выделяют в твердом ви¬
де (это опасно из-за взрывчатости сухих диазосоединений), а
используют непосредственно раствор для дальнейших превраще¬
ний. Если же требуется выделить твердое диазосоединение, то
диазотирование ведут в органическом растворителе, например
спирте, применяя в качестве источника азотистой кислоты пен-
тилнитрит C5H11ONO и используя для подкисления уксусную
кислоту. Затем диазосоединение осаждают эфиром. В твердом
виде диазосоединения выделяют также в виде тетрафторборатов
ArN2BF4 или комплексов с солями тяжелых металлов.
Диазосоединения — весьма реакционноспособные вещества.
На возможность существования диазосоединений в нескольких
взаимопревращающихся формах указывают следующие их свой¬
ства. Диазосоединения легко растворяются в воде. Эти раство¬
ры проводят электрический ток, т. е. в водном растворе проис¬
ходит диссоциация на ионы — анион кислоты, взятой при про¬
ведении диазотирования (чаще всего, С1~), и катион диазония
АгN2. Таким образом, в водном растворе диазосоединения суще¬
ствуют в виде солей диазония. Поскольку раствор имеет нейт¬
ральную реакцию, необходимо допустить, что содержащееся в
соли основание диазония обладает ярко выраженными основ¬
ными свойствами *. Это подтверждается следующим опытом.
Если на раствор соли диазония подействовать свежеприготов¬
ленным оксидом серебра, содержащим следы гидроксида сереб¬
ра, то образуется гидроксид диазония:
ArN2Cl + AgOH —»■ ArN2OH + AgCl
* Если бы основание диазония было слабым, то его соль с сильной кислотой
(например, хлороводородной) в результате гидролиза создавала бы в вод¬
ном растворе кислую реакцию.
292
Раствор приобретает сильнощелочную реакцию, причиной
которой не может быть AgOH — основание очень слабое: сле¬
довательно, сильным основанием является образовавшийся в
этой реакции гидроксид диазония ArN2OH. Через некоторое
время щелочная реакция сама собой исчезает. При этом проис¬
ходит не разложение диазосоединения, как можно было бы по¬
думать, а его изомеризация — превращение в диазогидрат —
форму, обладающую свойствами слабой кислоты. При действии
щелочей на диазогидрат образуется соль, диазотат ArN2ONa,
в которой остаток ArN20~ играет роль аниона. Если на эту
соль подействовать хлороводородной кислотой, то можно вер¬
нуться к исходной соли диазония ArN2Cl.
Все эти факты не оставляют- сомнения в том, что диазосо¬
единения существуют в нескольких изомерных формах, способ¬
ных переходить друг в друга. Явления сосуществования находят
щихся в равновесии изомерных форм называют таутомерией.
Как именно построены таутомерные формы диазосоединений,
представляли себе по-разному. Современную точку зрения на
этот вопрос выражает следующая схема:
НС! + AgOH +
► [Ar—N=N] СГ ► [Аг—N=N] 'ОН -
соль диазония гидроксид диазония
NaOH
Ar—N=N—ONa •* Аг—N=N—ОН «—
изомеризация
диазотат диазогидрат
Диазотаты могут существовать в виде двух пространствен¬
ных изомеров — син- и ангы-диазотатов:
Ar-N
II
NaO—N
сик-диазотат
Ar—N
II
N-ONa
анги-диазотат
сик-Диазотаты гораздо более реакционноспособные соедине¬
ния, чем анги-диазотаты.
Реакции диазосоединений отличаются большим разнообрази¬
ем. Их разделяют обычно на две группы — реакции с выделе¬
нием азота и реакции без выделения азота. Первые имеют
большое значение для синтеза разнообразных ароматических
соединений, вторые служат основой целой отрасли промышлен¬
ности — получения азокрасителей.
§ 132. Реакции диазосоединений с выделением азота. Важней¬
шими реакциями диазосоединений с выделением азота являются
следующие реакции замещения.
При стоянии водных растворов диазосоединений, быстрее
при их нагревании, диазогруппа замещается на гидроксил:
C„H8N2C1 + НгО —С,Н6ОН + Ns + НС1
фенилдиазо- фенол
нийхлорид
293
При действий иодида калия диазогруппа замещается на иодг.
СНз——N2C1 + KI —> СН3——I + N2 + KC1
ft-толилдиаэонийхлорид п-иодтолуол
Замещение диазогруппы на бром или хлор, а также на груп¬
пу CN идет при действии солей меди(1) (реакция Зандмейера):
Р-нафтилдиазоний- Р-бромнафталии
хлорид
/СНз /СНз
СНз——NaCl + CuCN —V СНз——CN + N2+CuC1
2,4-диметилфенил- 1,3-диметил-4-циано-
диазонийхлорид бензол
При добавлении к водным растворам диазосоединений кис¬
лоты HBF* выпадают нерастворимые в воде тетрафторбораты
диазония:
C6H5N2C1 + HBF4 —> C6H6N2BF, + НС1
Эту реакцию используют для выделения солей диазоння из
растворов. Кроме того, тетрафторбораты служат промежуточны¬
ми продуктами при замене диазогруппы на фтор. Реакция идет
при осторожном нагревании:
CeH6N2BF4 —*• C6H5F + Ns + BF3
Поскольку фтор нельзя ввести в ароматическое ядро пря¬
мым галогенированием, эта реакция имеет большое значение
для получения ароматических фторпроизводных.
При нагревании спиртовых растворов диазосоедин.ений одно¬
временно идут две реакции — замещение диазогруппы на оста¬
ток спирта (образование эфиров фенолов) и замещение на во¬
дород (образование углеводородов), причем в последней из
реакции спирт окисляется в альдегид:
,—► С6Н6ОСНз + N2 + НС1
CeH6N2CI + СН3ОН -
1—* С6Нб + N2 + НС1 + нсно
В зависимости от природы спирта и условий реакция на¬
правляется преимущественно по одному или другому пути.
Заместить диазогруппу на водород можно также действием
некоторых слабых восстановителей (муравьиная кислота, соли
фосфоноватистой кислоты). ,
294
При действии хлорида ртути (II) и медного порошка образу¬
ются ртутьорганические соединения:
C6H6N2C1 + HgCl2 + 2Cu —> C6H5HgCl + N2 + 2CuCl
При помощи сходных реакций диазогруппа замещается на
сурьму, висмут и олово. Эти реакции были открыты академи¬
ком А. Н. Несмеяновым и широко используются при синтезе
разнообразных металлорганических соединений.
При обработке диазосоединений гидросульфидом калия об¬
разуются тиофенолы:
CSH5*N2C1 + KSH —-* C6H5SH + КС1 + n2
Диазогруппу можно заменить и на другие группы — на ази-
догруппу (действие азида натрия), нитрогруппу (действие солей
азотистой кислоты в присутствии солей одновалентной меди,
т. е. в условиях реакции Зандмейера), альдегидную группу СНО,
фосфор- и мышьяксодержащие группы.
Соли диазония реагируют также с веществами, содержащи¬
ми сопряженные кратные связи (реакция Меервейна):
С1
I
C6H5N2C1 + сн2=сн—соон —>. с6н5—сн2—сн—соон + n2
Все это делает диазосоединения важными промежуточными
продуктами для синтеза самых разных ароматических соеди¬
нений.
§ 133. Реакции диазосоединений без выделения азота. Одна из
реакций без выделения азота — образование гидразинов при
восстановлении диазосоединений. Эта реакция положена в осно¬
ву промышленного производства фенилгидразина:
SnCI2 или NaHS03+Zn
C6H5N2C1 ■» c0h5nh—nh2
Производные гидразина используют как реагенты на карбо¬
нильную группу (см. § 86).
Наиболее важной реакцией диазосоединений без выделения
азота является 'реакция азосочетания, при которой группа
ArN=N замещает атом водорода ароматического ядра фенола
или амина, образуя гидроксиазо- и аминоазосоединения:
C„HsN2C1 + С6Н5ОН + NaOH —>■ С&Н5—N=N—С6Н4ОН + Н20 + NaCl
я-гидроксиазобензол
СНзСООЫа
CeH5-N2Cl + C6H6-N(CH3)2 ——C8Hs-N=N—C6H4-N(CH3)2
—Н LI
я-диметиламинобензол
По своему механизму азосочетание относится к реакциям
электрофильного замещения в ароматическом ядре: благодаря
положительному заряду катион диазония ArNa играет роль
электрофильной частицы. Однако, поскольку электрофильные
295
свойства катиона диазония весьма слабы, он реагирует лишь с
теми ароматическими соединениями, ядра которых активирова¬
ны электронодонорными группами — гидрокси- или аминогруп¬
пой. Участвующие в реакции ароматические амины или фенолы
называют азокомпонентами или азосоставляющими, а введен¬
ные в реакцию диазосоединения — диазокомпонентами или
диазосоставляющими. При проведении реакции большое значе¬
ние имеет создание необходимой среды — слабощелочной при
сочетании с фенолами и слабокислой при сочетании с аминами.
Это требуется для того, чтобы создать нужные условия для су¬
ществования азосоставляющих в наиболее активной форме:
фенолов в виде фенолят-ионов Аг—0~, аминов — в виде солей,
легко гидролизующихся с образованием свободных оснований,
которые и вступают в реакцию.
Реакции азосочетания идут в пара- или орто-положении к
гидроксильной или аминогруппе. Активность азокомпонентов
тем больше, чем выше электронная плотность в их ядре: по¬
этому особенно легко реагируют многоатомные фенолы (резор¬
цин, флороглюцин), полиамины, аминофенолы. Активность ди¬
азокомпонентов повышается при наличии в ароматическом ядре
электроноакцепторных заместителей, способствующих повыше¬
нию положительного заряда катиона диазония. Поэтому, напри¬
мер, диазотированный пикрамид может сочетаться даже с
углеводородом — мезитиленом:
Вещества, образующиеся при азосочетании, относятся к
классу ароматических азосоединений. Для них характерно при- .
сутствие группы —N=N—. Обе свободные валентности этой
группы связаны с ароматическими радикалами. Все азосоёдине-
ния окрашены. В зависимости от строения исходных азо- и
диазосоставляющих можно получать азосоединения самой раз¬
личной окраски. Многие из этих веществ способны закрепляться
на различных материалах, сообщая им окраску. Они составляют
очень обширный и разнообразный по оттенкам, хотя и не самый
важный (по качеству окраски) класс синтетических красите¬
лей — азокрасителей.
Еще в прошлом веке было замечено, что в молекулах окра¬
шенных органических веществ присутствуют определенные груп¬
296
пировки атомов, получившие название хромофоров, например:
—N=N—
—С—С—
II II
О о
азогруппа хиноидная
группировка
а-дикарбонильная
группировка
Однако окрашенное вещество будет красителем лишь в том
случае, если оно способно закрепляться на волокне. Такую спо¬
собность органическим веществам придают ауксохромы — силь¬
нополярные группы, имеющие, как правило, кислотный или ос¬
новной характер. К ауксохромам относятся- амино-, карбокси-,
сульфо- и гидроксигруппы.
Примером азокрасителей может служить краситель нафто¬
ловый сине-черный В, применяемый для окраски шерстяных
тканей:
ОН NH2
Хромофорами здесь являются азогруппы, ауксохромами —
амино-, гидрокси- и сульфогруппы. Этот азокраситель может
быть получен азосочетанием Аш-кислоты (см. § 129) с диазоти-
рованными анилином и я-нитроанилином.
Многие азокрасители способны менять окраску в зависимо¬
сти от pH среды, т. е. служить индикаторами, например гелиан-
тин (метиловый оранжевый, диметиламиноазобензолсульфонат
натрия), получаемый из диазосульфаниловой кислоты и диме-
ткланилина:
+
N(CH3)2
CH3COONa
►
NaOs
S —ft \—N=N—ft \—
NtCHab
Метиловый оранжевый — ярко окрашенное вещество, до¬
вольно хорошо растворимое в воде. Окраска его меняется в за¬
висимости от среды: в щелочной и нейтральной средах — жел¬
тая, в кислой — красная, поэтому его широко применяют в ана¬
литической химии как кислотно-основной индикатор. Изменение
окраски метилового оранжевого под действием кислот связано
с переходом одного из бензольных ядер в хиноидную форму:
NaQ3S
N(CH3)3
HCl
Na03S
NH—N
=Q==N(CH3b СГ
297
Азокрасители (азосоединения вообще) способны при дейст¬
вии сильных восстановителей (хлорид олова, гидросульфит нат¬
рия) расщепляться по связи N==N, причем по месту разрыва
образуются аминогруппы:
/Л
НО;
S^0-N“N-0
[HI
но'
Р-нафтоловын оранжевый
NHj
—> ho3s—
сульфанилоаая 1-аминонафтол-2
,кислота
Реакцию используют для получения некоторых аминосоеди-
нений, а также для установления строения азокрасителей.
§ 134. Алифатические диазосоединения. Простейшим и самым
важным представителем алифатических диазосоединений явля¬
ется диазометан CH2N2. Алифатические диазосоединения имеют
совсем иные свойства и иное строение, чем ароматические.
; Прежде им приписывали формулу с трехчленным циклом. Од¬
нако электронографические исследования показали, что молеку¬
ла диазометана имеет линейное строение (формула I):
I
CH2=N=N : СН3—N—NO
• • ••
I II
Диазометан получают разложением ряда соединений, содер¬
жащих группировку II, например действием щелочи на N-ue-
тил-М-нитрозоацета мид:
О CHj О
« I II
CHj—С—N—NO + NaOH —> СН3—С—ONa + CH2N2 + Н20
Диазометан — газообразное вещество желтого цвета, т. кип.
—24 °С, крайне опасен из-за токсичности и взрывчатости. Его
применяют обычно в виде раствора в диэтиловом эфире. Ди¬
азометан обладает высокой реакционной способностью и в мяг¬
ких условиях реагирует с разнообразными соединениями. Это
объясняется образованием очень активного промежуточного
продукта с двухвалентным атомом углерода — карбена:
CIV=N=N —»- :CH2 + N2
298
Карбен способен внедряться по О—Н- и С—С-связям. Вне¬
дрение по О—Н-связям наблюдается в реакциях с веществами
кислотного характера. Так, на карбоновые кислоты и фенолы
карбен действует как метилирующее средство;
СНэСООН + CHjN2 —V СН3СООСН3 + n2
CfiH6OH + CHjNj —V CeH6—OCH3 + N2
Конечно, ни уксусную кислоту, ни фенол нет необходимости
метилировать диазометаном — это можно осуществить другими,
более простыми способами. Однако для метилирования разла¬
гающихся при нагревании или действии кислот соединений диа¬
зометан является идеальным средством.
С кетонами реакция идет с расширением цикла в резуль¬
тате внедрения по С—С-связи:
^ \=о + ch2n2 —0>=о + n2
циклогекса- циклогептанон
нон (суберон)
Диазометан способен также реагировать с соединениями, со¬
держащими кратные связи. Так, из диазометана и этилена об¬
разуется циклическое азотсодержащее соединение пиразолин,
которое после разложения дает циклопропан:
н2с=сн2 + ch2n2
н2с.
,/
-СН2
н<\ >н
N
НО"
-N2
При действии диазометана на хлорангидриды кислот образу¬
ются диазокетоны (замещение хлора на остаток диазометана);
О о
II I * -
R—С—Cl + CH2N2 —»■ R—С—CH=N=N + НС1
При нагревании со спиртом диазокетон в результате пере¬
группировки образует сложный эфир, имеющий в кислотном
радикале на один атом углерода больше, чем в исходном хлор-
ангидриде:
О о
и + - с2н5он II
R—С—CH=N=N *- R—СН2—С—ОС2Н5 + N2
Таким образом, при помощи диазометана можно удлинять
углеродную цепь карбоновых кислот (синтез Арндта — Эйстер-
та).
299
ГЛАВА 17
ЭЛЕМЕНТОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ
Органические соединения, в которых имеется непосредственная
связь элемент—углерод, принято объединять под общим назва¬
нием элементорганических соединений.
§ 135. Общая характеристика. Помимо элементов-органогенов
(С, Н, О, N, S, Cl, Br, I, Р) в состав органических молекул мо¬
гут входить и многие другие химические элементы. Классифи¬
цируют эти соединения прежде всего по содержащемуся в
них элементу. Органические производные элементов, принадле¬
жащих к одной группе Периодической системы элементов
Д. И Менделеева, имеют сходные свойства, поэтому их можно
объединить и рассматривать совместно. Так возникает естест¬
венная классификация элементорганических соединений по
группам Периодической системы.
В зависимости от строения различают полные элементорга-
нические соединения (у них все валентности элемента затраче¬
ны на связь с углеродными атомами) и смешанные (только
часть валентностей элемента связана с углеродом, остальные
же затрачены на связь с кислородом, серой, азотом, галогена¬
ми). Примерами могут служить следующие соединения:
Полные элемент-
органические соединения
Смешанные элемент
органические соединения
C^jLi бутиллитий
(CH3)2Zn диметилцинк
(СН3)3Р триметилфосфин
(СвН3)5Р пентафенилфосфоран
CH3Mgl
СН3РО(ОН)2
CH3AsC1j
метилмагнийиодид
метилфосфоновая
кислота
метилдихлорарсин
Среди элементорганических соединений можно выделить бо¬
лее узкую область металлорганических соединений, имеющих
одну объединяющую их особенность — в связи С — металл уг¬
лерод является отрицательным концом диполя в отличие от
встречавшихся нам ранее связей С—X (X = галоген, О, N, S
и др.), где направление поляризации обратное. Именно наличие
частичного отрицательного заряда на атоме углерода Св_—Мей+
и определяет свойства металлорганических соединений — их
способность реагировать с электрофильными, а не нуклеофиль¬
ными реагентами, что характерно для связей с поляризацией
Сй+—X6-. Это различие наглядно проявляется, например, при
сравнении действия воды на метнлиодид и метилнатрий:
6+ в- б+ ft- ft*
СН3—I + H—О-Н —► СНз—ОН + III
ft- 6+ ft* ft- ft*
CH3—Na + H—О—H —► СН4 -}- NaOH
§ 136. Магнийорганические соединения. По имени впервые ис¬
следовавшего их французского ученого Гриньяра магнийорганн-
300
ческие соединения, часто называют соединениями Гриньяра, а
синтезы с их участием — реакциями Гриньяра.
Для получения магнийорганических соединений стружки ме¬
таллического магния покрывают безводным (абсолютным) ди-
этиловым эфиром и к этой смеси постепенно прибавляют гало-
генпроизводное, например метилиодид. В ходе реакции магний
постепенно переходит в раствор. Процесс протекает по схеме:
R-X + Mg —* R-Mg-X
Эфир не является в этой реакции простым растворителем.
Он образует с магнийорганическими соединениями комплексы
состава RMgX-2(C2Hs)20, называемые эфиратами. Вопрос об
истинном состоянии магнийорганических соединений в эфирном
растворе до конца не решен, но их реакции вполне удовлетво¬
рительно выражаются обычной формулой смешанных магнийор¬
ганических соединений (алкилмагнийгалогенидов) RMgX.
Магнийорганические соединения не выделяют из растворов,
в которых они получены; синтезы проводят, добавляя к этим
растворам второй компонент реакции. Кислород воздуха легко
окисляет магнийорганические соединения; защитой от окисления
служат пары эфира, вытесняющие воздух.
Являясь типичными представителями металлорганических
веществ, магнийорганические соединения обладают соответству¬
ющей поляризацией связи С6-—Mg6+. Это находит свое выра¬
жение в способности вступать в реакции замещения (1) и при¬
соединения (2):
б- б+ б+ б-
R—Mg—Х + А—В —► R—А + В—Mg—X (1)
б- б+ 6+ б-
R—Mg—X + А=В —R—А—В— Mg—X (2)
Важнейшие реакции магнийорганических соединений -приве¬
дены ниже.
1. С водой магнийорганические соединения образуют углево¬
дороды, например:
б- 6+ б+ б- 6+
СН3—Mg—I + Н—О—Н —V CH4 + Mg(OH)I
Это пример реакции замещения, в которой атом углерода,
несущий частичный отрицательный заряд, соединяется с ато¬
мом водорода — положительным концом диполя, имеющегося в
воде.
Аналогично идут реакции и с другими соединениями, содер¬
жащими подвижный водород,— спиртами, кислотами, аминами:
CH3MgI + CH3OH —* CH4 + CH3OMg[
CeH5MgBr + СН3СООН —V С6Н6 + CH3COOMgBr
C6H6CH2MgCl + C3H7NH2 —► C6H5CH3 + C3H7NHMgCl
301
Во всех этих реакциях наряду с углеводородом образуется
не имеющее практического значения солеобразное соединение
магния.
Эту реакцию применяют в аналитической химии, на ней ос¬
нован метод определения активного водорода по Чугаеву — Це-
ревитинову. В качестве реагента используют раствор метилмаг-
нийиодида CH3Mgl. При реакции с органическими соединения¬
ми, содержащими подвижный водород (спирты ROH, амины
RNH2 и RNHR), выделяется 1 моль метана на 1 моль экв ак¬
тивного водорода. Измерив объем метана, выделившегося при
действии избытка CHsMgl, и зная навеску гидроксил- или ами¬
носодержащего вещества, можно рассчитать число гидроксиль¬
ных или аминогрупп, содержащихся в данном веществе.
2. Реакции с подвижным водородом, стоящим у атома угле¬
рода, идут по той же общей схеме, что и рассмотренная выше
реакция. В этом случае, однако, интерес представляют продук¬
ты, содержащие магний, например:
2CHjMgI + СН=СН —► 2СН4 + IMg—С==С—Mgl
ацетилен реактив Иоцича
CH3MgI + /^ _* сн4 + <Г)
СН2 СИ—Mgl
цикло- циклопентадие-
печтадиен нилмагнийиодид
Получающиеся в таких реакциях соединения содержат маг¬
ний, связанный с углеродом. Они являются настоящими магний-
органическими соединениями, вступающими во все характерные
для них реакции.
3. Присоединение магнийорганических соединений к поляри¬
зованным кратным связям. Наибольший интерес представляет
присоединение к карбонильным соединениям с образованием
спиртов (см. § 86). В роли карбонильного соединения может
выступать и диоксид углерода 0=С=0. Реагируя с ним, маг-
нийорганические соединения образуют соли карбоновых кислот,
которые затем можно превратить в соответствующие кислотьи
н2о (н+)
R—MgX+0=C=0 —*• R—COOMgX ► R—СООН
По типу реакций с карбонильными соединениями взаимодей¬
ствуют и вещества, содержащие группировки C=N и C = N; при
этом в первом случае получаются амины, во втором — кетоны.
4. С галогенпроизводными магнийорганические соединения
реагируют по типу реакции Вюрца, образуя углеводороды:
СН2=СН—CH2I + CH3Mgl —» СН2=СН—СН2—СНз + Mgl2
Получаемые по реакции Гриньяра спирты, кетоны, кислоты
могут служить исходными веществами для синтеза многих дру¬
гих органических соединений.
302
При действии серы магнийорганические соединения превра¬
щаются в тиолаты, гидролиз которых дает тиолы:
Магнийорганические соединения могут также служить для
синтеза других металлорганических (и вообще элементоргани-
ческих) соединений.
До открытия магнийорганических соединений для синтеза спиртов ши¬
роко использовали цинкорГаничеСкие соединения. Синтезы с Помощью цинк-
органических соединений были разработаны А. М. Бутлеровым и его учени¬
ками. В этих синтезах применяли полные цинкорганические соединения соста¬
ва R2Zn. Используя реакцию между диметилцинком и ацетилхлоридом, Бут¬
леров в 1864 г. синтезировал первый представитель ряда третичных спир¬
тов — триметилметанол. Другая реакция цинкорганических соединений, ис¬
пользовавшаяся школой Бутлерова, приводит к углеводородам:
§ 137. Органические соединения щелочных металлов. Соедине¬
ния лития, натрия, калия вступают в те же реакции, что и
магнийорганические соединения. Однако из-за крайней чувстви¬
тельности этих соединений к кислороду воздуха (самовоспламе¬
нение!) и влаге с органическими соединениями щелочных ме¬
таллов работать менее удобно, чем с магнийорганическими.
Поэтому их применяют только в тех случаях, когда требуются
соединения с повышенной реакционной способностью.
Большая химическая активность органических соединений
щелочных металлов является следствием того, что связь угле¬
род — металл имеет у них ионный характер. В реакциях этих
соединений участвуют карбанионы типа ~СН3, -C6Hs и т. п.
Активность органических соединений щелочных металлов возра¬
стает в следующем ряду: Li < Na < К < Rb < Cs.
Литийорганические соединения используют для синтеза
сильно разветвленных спиртов, которые нельзя получать маг-
нийорганическим синтезом, например:
S
Н20
C6H5SH
С6Н5—MgBr —> СбН„—SMgBr
(СН3)3СС1 + (C2H5)2Zn —► (СНз)зС—СН2-СН, + C2H5ZnI
2,2-диметилбутан
(неогексан)
1. (СН3)2СНЫ
2. Н20
СН(СН3)2
(СИзЬСН—С—СН(СН3)а
I
он
триизопропилметанол
Повышенная реакционная способность органических соеди¬
нений щелочных металлов проявляется также в их способности
присоединяться по двойным углерод-углеродным связям:
сн3 сн3
I I н2о
С„Н5—CH2Na + СН=СН2 ■—► С,Н5—СН2—СН—CH2Na "lNaC;^
—v С6Н5—СН2—СН(СН3)2
§ 138. Алюминийорганические соединения. Триалкильные соеди¬
нения алюминия R3AI были известны еще в прошлом веке.
Однако они привлекли к себе внимание лишь после того, как
немецкий химик Циглер открыл простой прямой способ их по¬
лучения из металлического алюминия, водорода и этиленовых
углеводородов (1955 г.):
100 «С, 8 МПа
2А1 + ЗН2 + 6C2H4 *- 2(С2Н5)зА1
Простейшие алюминийтриалкилы — жидкости, самовоспла¬
меняющиеся на воздухе. Работать с ними можно только в ат¬
мосфере инертного газа (азота).
Алюминийорганические соединения за последние десятиле¬
тия превратились из весьма редких «экзотических» веществ в
важнейшие продукты современной техники. Эта область про¬
должает быстро развиваться. Для промышленного синтеза алю-
минийтриалкилов используют реакцию Циглера.
Соединения типа RA1C12 и R2A1C1 образуются при дей¬
ствии алкилгалогенидов на металлический алюминий в авто¬
клаве:
3RC1 + 2A1 —► RA1C12 + R2A1C1
Алюминийтриалкилы приобрели важное значение в ряде об¬
ластей. Их используют как катализаторы процессов полимери¬
зации. С их помощью получают полиэтилен низкого давления
и другие полиалкены, бутадиеновый и изопреновый каучуки.
Для синтеза высших спиртов из этилена и триэтилалюминия
получают высшие алюминийтриалкилы, которые окислением
кислородом воздуха превращают в алкоголяты алюминия, раз¬
лагаемые водой на гидроксид алюминия и высший спирт:
о2
(С2Н5)зА1 + 12СН2=СН2 — (С10Н2,)зА1 *-
н2о
—>• (СюН210)3А1 СюН21ОН
Высшие спирты служат для получения флотореагентов, упо¬
требляемых при обогащении руд цветных металлов; для произ¬
водства синтетических моющих веществ и для других целей.
Из алюминийтриалкилов получают высшие жирные кислоты:
со2 н2о
(СюН2|)зА1 _ (С,0Н21СОО)зА1 ^ С|эН2|СООН
Эти кислоты также используют для производства моющих
веществ. В отличие от кислот, получающихся при окислении
304
алканов, кислоты, синтезированные при помощи алюминийорга-
нических соединений, имеют неразветвленную цепь— как и те
кислоты, которые входят в состав жиров. В будущем это может
оказаться важным для синтеза пищевых жиров.
§ 139. Органические соединения переходных металлов. Пере¬
ходными называют металлы, у которых происходит достройка
внутренних rf-уровней после того, как уже произошло заполне¬
ние s-уровней с большим квантовым числом. К переходным от¬
носятся металлы с порядковыми номерами 21—30 (от скандия
до цинка), 39—47 (от иттрия до серебра), 71—80 (от лютеция
до ртути).
Многие из этих элементов образуют органические соедине¬
ния необычного строения. Примером такого рода соединений
может служить дициклопентадиенилжелезо (ферроцен). Он
имеет состав CioHioFe и представляет собой кристаллы цвета
ржавчины; т. пл. 174 °С. Ферроцен кипит без разложения при
250°С, растворяется в органических растворителях. Ферроцен
можно получать пропусканием паров циклопентадиена над по¬
рошком железа при 300 °С:
С8Н„ (C5H5)2Fe
Ферроцен обладает свойствами ароматического соединения:
ацилируется по Фриделю — Крафтсу, сульфируется, не разла¬
гается при нагревании с концентрированной НС1 или с 10 %-м
раствором NaOH. Необычные для металлорганического соедине¬
ния свойства ферроцена являются следствием его своеобразного
строения (формула 1). Структуры подобного типа получили на¬
звание «сэндвичеобразных» (англ, «сэндвич» — бутерброд): в
данном случае атом железа прикрыт сверху и снизу циклопен-
тадиенильными циклами. Такое строение ферроцена подтверж¬
дено рентгеноструктурным анализом, на него указывают и
спектральные данные. В изучение ферроцена и его производных
большой вклад внесен А. Н. Несмеяновым и его учениками.
,Мп
/| \
СО СО
I II
Трикарбонилциклопентадиенилмарганец (формула II) ис¬
пользуют как антидетонационную добавку для повышения окта¬
нового числа горючего. Он менее ядовит, чем тетраэтилсвинец.
§ 140. Кремнийорганические соединения. В Периодической си¬
стеме элементов Д. И. Менделеева кремний, как и углерод,
находится в четвертой группе и расположен непосредственно
305
под углеродом. Между соединениями обоих элементов имеется,
однако, гораздо меньше сходства, чем следовало бы ожидать
по их близкому соседству в Периодической системе элементов.
Органические соединения кремния по строению сходны с соеди¬
нениями углерода (оба элемента четырехвалентны), но сущест¬
венно отличаются от них по свойствам.
Первые два члена гомологического ряда кремневодородов
(силанов) SiH4 и Si2He — газы, последующие — жидкости. Все
они имеют неприятный запах, ядовиты, смешиваются с органи¬
ческими растворителями. Один из способов получения сила¬
нов — действие кислот или щелочей на силициды металлов, на¬
пример:
Mg2Sl + 4HCl —>■ SiH4 + 2MgCI2
В отличие от углеводородов силаны — мало устойчивые со¬
единения: на воздухе загораются, легко гидролизуются водой:
SiH4 + 202 —► Si02-f2H20 Si2He + 4H20 —> 2SiOa + 7H3
Более устойчивы соединения, в которых вместо водорода с
атомами кремния связаны углеводородные радикалы.
Известны кремниевые аналоги галогенпроизводных, спиртов,
углеводородов, простых эфиров и др. Алкил- и арилсиланы или
их галогенпроизводные получают при помощи магнийорганиче-
ских соединений:
CHaMgl CHsMgl CHjMgl
SiCl4 »- CH3SiCl3 ► (CH3)2SiCl2 ►
CH3MgI
—► (CH3)3SiCl *■ (CH3)4Si
В промышленности для синтеза таких соединений использу¬
ют другой способ — пропускают галогенпроизводные, например
метилхлорид, при 300—350 °С над смесью кремния и меди
'(медь служит катализатором);
CHjSlCl;
(CH3)3SiCl
метилтри-
хлорсилан
(т. кип. 66 °С)
СН3С1 + Si
триметилхлор-
силан
(т. кип. 58 °С)
(CH3)2SiCl2
(CH3)4Si
диметилдихлор-
силан (т. кип. 70 °С)
тетраметилсилан
(т. кип. 26 °С)
Тетраметилсилан — представитель полностью алкилирован¬
ных соединений кремния. Это легкокипящая жидкость, по хими¬
ческой инертности напоминающая алканы.
Алкилхлорсиланы легко гидролизуются, напоминая в этом
отношении тетрахлорид кремния:
н2о
(CH3)3SiCl ► (CH3)3SiOH + НС1
зов
Образующиеся при гидролизе аналоги спиртов — сила-
нолы — неустойчивы и легко переходят с отщеплением воды в
силоксаны — кремнийорганические аналоги простых эфиров:
2(CH3)3SiOH —-*• (CH3)3Si—О—Si(CHa)3 + Н20
триметилсиланол гексаметилдисилоксан
Аналогично идет и гидролиз диалкилдихлорсиланов, но при
этом образуются соединения полимерного характера. Диалкил-
дихлорсиланы используют для получения кремнийорганических
полимеров (см. § 193).
Подобно углероду, кремний способен образовывать линейные
и циклические соединения. Так, известен гексаметилдисилан
(CH3bSi—ЭЦСНзЬ {жидкость с т. кип. 112—114 °С), а также
его высшие гомологи, содержащие в цепи до девяти атомов
кремния.
Эти вещества довольно легко окисляются и гидролизуются с
разрывом связей Si—Si.
§ 141, Фосфорорганические соединения. Фосфор — элемент пя¬
той группы Периодической системы, аналог азота. Между орга¬
ническими производными этих элементов наблюдается опреде¬
ленное сходство, например: R—NHa—первичные амины,
R—РН2 — первичные фосфины. Подобно соответствующим со¬
единениям азота, известны вторичные и третичные фосфины, а
также аналоги четвертичных аммониевых соединений — соли
фосфония. Так же как амины, фосфины являются основаниями,
способными давать с кислотами соли:
СН3РН2 + НС1 —► [СН3РН3Г С Г
метилфосфин метилфосфонийхлорид
Однако основные свойства фосфинов значительно слабее,
чем у аминов.
Отличия соединений фосфора от соединений азота связаны
главным образом со способностью фосфинов легко реагировать
с элементами, обладающими большим сродством к электронам
(галогены, кислород). Объясняется это тем, что свободная
электронная пара фосфинов и других соединений фосфора(III),
находится дальше от ядра, чем в азоте. Чем дальше располо¬
жен электрон от ядра, тем меньше он притягивается ядром и
тем легче может перейти к другому атому. В частности, фос¬
фины легко окисляются с образованием соответствующих окси¬
дов (на воздухе простейшие фосфины самовоспламеняются).
При окислении первичных и вторичных фосфинов образуются
соответственно алкилфосфоновые и диалкилфосфиновые кисло¬
ты, из третичных фосфинов — оксиды. Эти же соединения об¬
разуются из хлорфосфинов; в результате присоединения к хлор-
307
фосфинам хлора получаются хлорфосфораны, гидролизующиеся
при действии воды:
о
[О] I н2о С12
RPH2 >- R—Р(ОН)2 -< RPCU -« RPCI2
алкилфосфо-
новая кислота
О
Ю1 II Н20 С12
R2PH >- RaP—он -< r2pci3 •«—- R2PC1
диалкилфосфи-
новая кислота
ЕО]
R3P ► R3P=0
оксид
третичного
фосфина
Формулы алкилфосфоновых и диалкилфосфиновых кислот, а
также оксидов фосфинов, написанные выше, содержат пятико¬
валентный фосфор. Не нарушает ли это правила октета? Мо¬
жет быть, эти соединения, подобно оксидам аминов и их
аналогам, содержат четырехвалентный фосфор, а кислород свя¬
зан семиполярной связью? Этот вопрос был тщательно изучен.
Измерения длин связей и дипольных моментов показали, что
связи фосфора с кислородом в алкилфосфоновых и диалкил¬
фосфиновых кислотах имеют промежуточный характер между
простыми и двойными, т. е. их истинное состояние является
мезомерным между граничными структурами:
/ОН +/он
R—р( R-P; (CH3)3P=0 (СНз)зР—O'
^ Ч)Н о- он
Таким образом, формулы с пятиковалентным фосфором
лишь приближенно передают строение этих соединений.
Известны соединения, в которых фосфор несомненно образу¬
ет пять совершенно одинаковых ковалентных связей, например
пентафенилфосфоран, впервые полученный Виттигом (1949 г.):
С6НЗЧ /CJHt
СвН5'
/Т\
1хсвн5
СцН6
Пентафенилфосфоран обладает характерными свойствами
ковалентного соединения: растворим в органических растворите¬
лях и нерастворим в воде, имеет довольно низкую температуру
308
плавления. Исследования Г. А. Разуваева показали, что все
пять связей фосфора в этом соединении равноценны.
Одна из важнейших реакций, позволяющих получать со¬
единения со связью углерод—фосфор, открыта А. Е. Арбузовым:
уОСгН5 C2H5I .ОС2Н5
с2н6о-р( + С2Н5-Р(
ЧОС2Н5 ^\ОСгН5
триэтилфосфит диэтиловый эфир
этилфосфоновой
кислоты
Это превращение называют перегруппировкой Арбузова. Ис¬
ходные фосфиты легко получают из первичных спиртов и хло¬
рида фосфора (III):
РСЫ-ЗС2Н5ОН —► Р(ОС2Н5)3 -f ЗНС1
Среди фосфорорганических соединений встречается большое
число веществ, обладающих сильным физиологическим дейст¬
вием. В связи с этим органические соединения фосфора исполь¬
зуют как инсектициды, лекарственные препараты и т. д.
Хлорофос — инсектицид широкого диапазона действия. Он
сравнительно мало токсичен для животных и человека. Тио-
фос— инсектицид, обладающий контактным действием.
(СН30)2Р—СН—СС13
II 1
о он
хлорофос
СН:
/ОС3Н7
<
IIN*
о
(С2Н60)2Р-0
II
S
тиофос
/Н(СН3)2
(CH3)2N-P(
llxN(CH3)2
зарин
гексаметапол
Эфир фторфосфоновой кислоты зарин является отравляющим
веществом нервно-паралитического действия; смертельная кон¬
центрация в воздухе 0,03 мг/л. Трис(диметиламидо) фосфат
представляет собой жидкость (т. кип. 233 °С, т. пл. 5—7°С,
df 1,027); под названием гексаметапол применяется как по¬
лярный растворитель.
ЧАСТЬ 111
ГЕТЕРОФУНКЦИОНАЛЬНЫЕ
СОЕДИНЕНИЯ
Вещества, в молекуле которых содержится несколько разных
функциональных групп (функций), принято называть соедине¬
ниями с разнородными (смешанными) функциями, или гетеро-
функциональными. Такие вещества заслуживают особого внима¬
ния по многим причинам. Именно эти соединения являются
наиболее удобными моделями для рассмотрения ряда общетео¬
ретических вопросов органической химии — проблемы взаимно¬
го влияния функциональных групп, таутомерии, пространствен¬
ной изомерии. Среди гетерофункциональных соединений встре¬
чается много практически важных веществ.
ГЛАВА 13
ГАЛОГЕНЗАМЕЩЕННЫЕ КИСЛОТЫ.
ГИДРОКСИКИСЛОТЫ. АЛЬДЕГИДО- И КЕТОКИСЛОТЫ
§ 142. Галогензамещенные кислоты. К числу галогензамещен-
ных кислот относят соединения, содержащие одновременно атом
галогена и карбоксильную группу. Обе функции могут быть
представлены в молекуле один или несколько раз. У галоген-
замещенных кислот, как и у других поли- и гетерофункциональ¬
ных соединений, приходится учитывать возможность проявления
трех типов структурной изомерии. Помимо изомерии углеродно¬
го скелета и изомерии положения, у них появляется еще изо¬
мерия взаимного положения функциональных групп. Это созда¬
ет возможности для существования большого числа изомеров.
Так, кислот состава С4Н802 имеются две — масляная и изомас-
ляная, в случае же хлорзамещенных кислот C4H7O2CI число
изомеров возрастает до пяти.
Названия всех гетерофункциональных соединений в совре¬
менной международной номенклатуре строят на основе назва¬
ний монофункциональных соединений с добавлением приставки,
характеризующей дополнительную функцию, и греческой буквы
или цифры, указывающей положение этой функции. Познако¬
мимся с такими названиями на примере хлорбутановых кислот;
СН3—СН2—СНС1-СООН
а-хлормасляная
(2-хлорбутановая)
кислота
СН3-СНС1-СН2-СООН
р-хлормасляная
(3-хлорбутановая)
кислота
31С
Cl—CHj—CHjj—CH2—COOH
Y-хлормасляная
(4-хлорбутановая) кислота
СН3 СН3
СНз—С—COOH Cl—СН2—СН—СООН
С1
а-хлоризомасляная
(2-метил-2-хлор-
пропановая) кислота
р-хлоризомасляная
(2-метил-З-хлорпропа-
новая) кислота
Получать галогензамещенные кислоты можно разными спо¬
собами: введением в галогенсодержащее соединение карбоксиль-
ной группы и, наоборот, введением в карбоновую кислоту га¬
логена. Для введения галогена наибольшее значение имеют
следующие два способа.
1. Замещение водорода в кислотах жирного ряда на галоген,,
Например, при хлорировании уксусной кислоты водородные
атомы группы СН3 постепенно замещаются на атомы хлора:
С1г С1? С^2
СНз—СООН —► С1СН2—соон —► сьсн—СООН —► СС13—СООН
уксусная монохлоруксусная дихлоруксусная трихлоруксусная
кислота кислота кислота кислота
Однако свободные кислоты трудно поддаются галогенирова-
нию. Гораздо легче идут подобные реакции с производными
кислот, например со сложными эфирами и галогенанГидридами.
Одним из удобных вариантов этой реакции является бромирова-
ние кислот по методу Н. Д. Зелинского. На кислоту действуют
бромом в присутствии бромида фосфора(Ш), при этом обра«.
зуется бромангидрид, подвергающийся далее бромированию:
РВгз Bf2
СНз—СН2—СООН *- СНз—СН2—СОВг > СНз—СНВг—СОВг
Галоген вступает в a-положение к карбоксильной группе.
2. Присоединение галогенов или галогеноводородов к непре¬
дельным кислотам. В первом случае получаются дигалоген-, во
втором — моногалогензамещенные кислоты.
Вг2
СНз—СН=СН—СООН *- СНз—СНВг—СНВг—СООН
Галогеноводород присоединяется к акриловой кислоте и ее
аналогам против правила Марковникова с образованием (3-гало-
гензамещенных кислот (см. § 99).
Атомы галогена и карбоксильная группа в галогензамещен-
ных кислотах обладают в основном обычными, характерными
для этих функций свойствами. Так, галоген способен к реакци¬
ям нуклеофильного замещения (см. § 61), карбоксильная груп¬
311
па проявляет кислотные свойства, а также дает обычные для
кислот функциональные производные.
Реакции карбоксильной группы:
PC15 NaOH
С1СН2СН2СОС1 ■< С1СН2СН2СООН *■ ClCH2CH2COONa
NH3
ClCH2CH2CONH2
с2я5он
(н+)
С1СН2СН2СООС2Н6
Реакции галогена:
NH3
h2nch2ch2coonh, •*-
CH3COONa
СН3СООСН2СН2СООН <
СН2С1 CH3ONa
| ► CH3OCH2CH2COONa
СН2 — кон
I (спирт.)
соон ► сн2=снсоок
Вместе с тем обе функциональные группы оказывают опре¬
деленное взаимное влияние, особенно значительное тогда, когда
они расположены близко друг от друга. Так, в эфирах а-гало-
гензамещенных кислот атомы галогена обладают повышенной
подвижностью, т. е. их реакционная способность возрастает под
влиянием карбоксила. Это происходит вследствие того, что со¬
вместное действие галогена и группы С=0, оттягивающих элек¬
троны, создает на стоящем между ними атоме углерода значи¬
тельный положительный заряд, облегчающий атаку нуклеофиль¬
ного реагента. В более отдаленных положениях (р-, у-
и т. д.) взаимное влияние групп ослабевает.
В свою очередь галоген, оттягивая электроны, увеличивает
кислотные свойства карбоксильной группы. Это — проявление
отрицательного индуктивного эффекта галогена (-/-эффект):
6- 6+ Й+
Х<-СН2-^С<-0<-Н
Сила, индуктивного эффекта может быть оценена количест¬
венно по константе диссоциации соответствующей кислоты.
Индуктивный эффект возрастает при переходе от иода к фтору,
а также по мере увеличения числа атомов галогена и быстро
убывает с увеличением расстояния между галогеном и карбок-
сигруппой. Эти закономерности видны при сопоставлении при¬
веденных ниже констант диссоциации кислот:
Ка.10-5
Ка-ю~5
Уксусная
1,75
Дихлоруксусная
3 320
Иодуксусная
75
Трихлоруксусная
20 000
Бромуксусная
138
р-Хлорпропионовая
8,5
Хлоруксусная
140
а-Хлорпропноновая
140
312
Другие заместители, обладающие индуктивным эффектом и
способные вызывать поляризацию связей, также влияют на
кислотные свойства карбоксила.
Из галогензамещенных кислот наибольшее практическое зна¬
чение имеет хлоруксусная кислота С1СН2СООН. Ее получают в
больших количествах хлорированием уксусной кислоты и приме¬
няют в органическом синтезе, в частности при получении инди¬
го, а также гербицидов—веществ, при помощи которых прово¬
дят «химическую прополку» полей — уничтожают сорняки.
Один из распространенных гербицидов — 2,4-дихлорфеноксиук-
сусную кислоту — получают из дихлорфенола и хлоруксусной
кислоты:
С1СН2СООН
>-
ГИДРОКСИКИСЛОТЫ
§ 143. Способы получения, физические и химические свойства.
Гидроксикислотами называют органические вещества, в молеку¬
лах которых присутствуют две функции — гидроксильная и
карбоксильная. Возможности для изомерии в этих соединениях
совершенно такие же, как и у галогензамещенных кислот.
В зависимости от числа карбоксильных групп гидроксикис-
лоты бывают одноосновными, двухосновными и т. д., в зависи¬
мости от числа гидроксильных групп — двухатомными, трех¬
атомными и т. д. (учитывается и гидроксил, входящий в состав
карбоксильной группы). Таким образом, простейшая гидрокси-
кислота — гликолевая НО—СН2—СООН является одноосновной
двухатомной кислотой.
Гидроксильная группа в молекулах гидроксикислот может
находиться на разном расстоянии от карбоксильной: в зависи¬
мости от этого различают а-, р-, у- и другие гидроксикис-
лоты:
У Р О
С—С—С—СООН
Названия гидроксикислот строят по тому же принципу, что
и названия галогензамещенных кислот: основой служит назва¬
ние соответствующей карбоновой кислоты, присутствие гидрок¬
сила указывается приставкой гидроксц с буквой или цифрой,
определяющей положение гидроксигруппы. Так, изомерные
813
гидроксимасляные кислоты имеют названия:
ОН ОН
сн3—сн2—сн—соон сн3—сн—сн2—соон
а-гидроксимасляная
(2-гидроксибутановая)
кислота
Р-гидроксимасляная
(3-гидроксибутановая)
кислота
НО—СН2—СН2—СН2—СООН
у-гидроксимасляная
(4-гидроксибутановая)
кислота
СНз
СН,
СН,—С—СООН
А„
а-гидроксиизомасляная
(2-гидрокси-2-метил-
пропановая) кислота
НО—СН2—СН—СООН
Р-гидроксиизомасляная
(З-гидрокси-2-метил-
пропановая) кислота
Вторая функция обязательно должна входить в главную
цепь, которая при этом может оказаться и не самой длинной,
Яапример
С2Нь
НО—CHg—СН—СООН З-гидрокси-2-этилпропионовая кислота
Из весьма разнообразных способов получения гидроксикис-
лот приведем несколько, имеющих наибольшее значение.
1. Гидролиз галогензамещенных кислот, например:
НгО (Н* или но-)
С1СН2СООН * НО—СН2—СООН
гликолевая кислота
2. Получение НЗ альдегидов и кетонов через циангидрины
[(циангидринный синтез):
ОН ОН
HCN I Н20 |
СНз—СНО ^ СН,—СН—CN »- СНз—СН—СООН
молочная
(а-гидроксипропио-
новая) кислота
Первый из этих способов пригоден для получения гидрокси-
кислот с любым взаимным расположением функций, второй —
только для а-гидроксцкислот.
По физическим свойствам гидроксикислоты — высококипя-
щие жидкости или кристаллические вещества. Как правило,
они хорошо растворимы в воде и плохо — в малополярных ор¬
ганических растворителях .(эфир, бензол). Это часто затрудняет
314
отделение гидроксикислот от неорганических продуктов, обра¬
зующихся в ходе синтезов.
Как и в других гетерофункциональных соединениях, обе
функции в общих чертах сохраняют свои обычные свойства.
Так, например, гидроксильная группа может подвергаться ал¬
килированию [реакция (1); образование простых эфиров], аци¬
лированию [реакция (2); образование сложных эфиров], заме¬
щаться на галоген [реакция (3); получение галогензамещенных
кислот], отщепляться в виде воды [реакция (4); образование
непредельных кислот]. Карбоксильная группа способна образо¬
вывать соли, сложные эфиры [реакция (5)], ангидриды и гало-
генангидриды, амиды [реакция (6)] и другие функциональные
производные. Проиллюстрируем некоторые из этих реакций
на примере молочной кислоты:
(CH3)2S04
СН3—СНОН—СООН > СНз—СН(ОСНз)—СООН (1)
CH3COC1
СНз—СНОН—СООН ► СНз—СН(ОСОСН3)СООН (2)
РС1з н20
СНз—СНОН—СООН ► СНз—СНС1—СОС1 ► СНз—СНС1—СООН
(3)
СНз—СНОН—СООН ► СН2=СН—СООН (4)
-н2о
СНзОН, н+
СНз—СНОН-СООН ► СНз—СНОН—СООСНз (5)
NH3
СНз—СНОН—СООСНз ► СНз—СНОН—CONH2 (6)
Вместе с тем в свойствах гидроксикислот появляется и нечто
новое по сравнению со свойствами спиртов и кислот: это реак¬
ции, в которых гидрокси- и карбоксигруппы участвуют одно¬
временно. Одна из наиболее характерных реакций такого типа
происходит при нагревании гидроксикислот, причем превраще¬
ние идет по-разному в зависимости от взаимного расположения
гидрокси- и карбоксигрупп.
При нагревании ос-гидроксикислот R—СНОН—СООН отщеп¬
ляется вода, причем в реакции участвуют две молекулы кисло¬
ты. Например, молочная (а-гидроксипропионовая) кислота
реагирует с образованием циклического соединения — лактидаъ
/СООН НО/ °~р—°ч
снз-нс^ + )сн-сн3 ——> сиз-нс; ;сн-снэ
N)H ноос/ ~2Н2° V/n
Лактиды являются циклическими сложными эфирами, при
образовании которых каждая молекула а-гидроксикислоты ре¬
агирует и как кислота, и как спирт.
315
Р-Гидроксикислоты R—СНОН—СН2—СООН при нагревании
легко теряют воду, переходя в непредельные кислоты:
носн2—сн2—соон ■ > СН2=СН—СООН
—•Н2О
p-гидроксипропионовая акриловая кислота
кислота
При нагревании у-гидроксикислот отщепляется вода и об¬
разуются внутренние сложные эфиры — лактоны:
СНз—сн—сн2—сн2—соон ——*■
I —н2о
он
СНз
-нс_сн2
о^ ^сн2
с==о
у-гидроксивалериановая кислота
у-валеролактон
§ 144. Оптическая изомерия. Многие гидроксикислоты содержат
асимметрический атом углерода. Так называют атом угле¬
рода, связанный с четырьмя разными заместителями. Тетраэдр,
в вершинах которого находятся разные заместители, не имеет
ни одного элемента симметрии. Это приводит к тому, что суще¬
ствуют две несовместимые одна с другой пространственные
формы. Отличаются они друг от друга как несимметричный
предмет от своего изображения в зеркале (рис. 26). Вещества
с асимметрическим атомом углерода существуют в виде двух
пространственных изомеров. Такие изомеры называют зеркаль¬
ными или оптическими. Второе название связано с тем, что
наглядное различие, которое имеется в этом случае между изо¬
мерами,— их отношение к поляризованному свету. В то время
как один из зеркальных изомеров вращает плоскость поляри¬
зации света влево, другой вращает ее точно на такой же угол
вправо. В связи с этим пару зеркальных изомеров называют
еще оптическими антиподами. Все другие физические свойства
у оптических антиподов одинаковы.
Способность веществ вращать плоскость поляризации света
называется оптической активностью. Для ее измерения пользу¬
ются прибором, именуемым поляриметром. Устройство поляри¬
метра схематически изображено на рис. 27.
Свет источника / проходит через поляризационное устройство 2 (призма
Николя или поляроид). При этом он становится поляризованным, т. е его
колебания совершаются теперь в одной плоскости — плоскости поляризации.
Если на пути поляризованного
света поставить анализатор 4—
вторую призму Николя или
поляроид, то сила света, по¬
падающего в глаз наблюдате¬
ля, будет зависеть от взаимной
ориентации призм, т. е. поляри¬
затора 2 и анализатора 4.
Рис. 26. Модели антиподных
(зеркальных) тетраэдров
316
При одинаковой ориентации поляризующих плоскостей обеих призм
(«параллельные николи») свет будет проходить без ослабления, в положении
же «скрещенных николей» свет полностью гасится. Если в поляриметр, уста¬
новленный на полное гашение, внести трубку 3 с оптически активным веще¬
ством, плоскость поляризации света изменится, полного гашения уже не
будет. Для того чтобы снова погасить проходящий через прибор луч, надо
повернуть анализатор 4 на угол, соответствующий вращению находящегося
в трубке вещества. Этот угол отсчитывают с помощью шкалы 5. Наблюдае¬
мое вращение а зависит от природы вещества, длины слоя /, а для растворов
также от концентрации. Измеренное вращение принято пересчитывать в
удельное вращение, V. е. вращение плоскости поляризации света (в градусах)
1 г вещества, содержащегося в 1 мл раствора, при длине слоя 1 дм (10 см).
Для расчета используют формулу
[а] = aVl(pt),
где [а] — удельное вращение; а — наблюдаемое вращение; V —объем рас¬
твора, мл; р — количество растворенного вещества, г; /— длина поляримет¬
рической трубки, дм.
Удельное вращение — чисто расчетная величина, поэтому не надо удив¬
ляться, что иногда его значение превышает 360°. Оно зависит от длины вол¬
ны; чаще всего его определяют для желтой линии натрия при 20 °С и обозна¬
чают тогда [а]д.
Одно из простейших оптически активных веществ — молоч¬
ная кислота СН3СНОНСООН существует в трех формах: право¬
вращающая [обозначается знаком (+)], левовращающая
[(-)] и оптически неактивная [(±)].
(+)-Молочная кислота имеет [а]^ + 3,82° (10%-й водный ра¬
створ). Это кристаллическое вещество, т. пл. 25—26 °С. Имен¬
но эта правовращающая форма содержится в мышечном соке,
почему ее и назвали мясомолочная кислота.
Левовращающая (—)-молочная кислота совершенно неотли¬
чима по свойствам от правовращающей формы, за исключением
знака вращения, [а]^ = —3,82°.
Оптически неактивная (±) -молочная кислота, так называе¬
мая молочная кислота брожения,— кристаллическое вещество,
т. пл. 18 °С. В твердом виде получить ее очень трудно, обычно
она существует в виде густого сиропа, смешивающегося с водой
во всех отношениях. Исследования показали, что отсутствие
317
оптического вращения у (±)-молочной кислоты — результат
того, что она представляет собой смесь одинаковых количеств
право- и левовращающей молочной кислот. Такие смеси назы¬
вают рацематами.
Существование пары оптических антиподов—проявление
зеркальной изомерии молочной кислоты (рис. 28). При помощи
проекционных формул Фишера строение оптических антиподов
.молочной кислоты изображается так:
СООН
1
СООН
]
но—J—н
1
1
н—}—ОН
СНз
1
СН3
£-(+)-молочная
кислота
зеркало
£>-(—)-молочная
кислота
В этих формулах верхний и нижний заместители (СООН,
СН3) надо представлять себе лежащими за плоскостью чертежа
^для напоминания об этом вертикальную линию мы будем в
проекционных формулах изображать пунктиром), боковые заме¬
стители Н, ОН —лежащими перед плоскостью чертежа. В цент¬
ре формулы в точке пересечения обеих линий находится не
изображенный в проекционной формуле асимметрический атом
углерода, лежащий в плоскости чертежа. Оптические антиподы
нельзя совместить никаким вращением, они отличаются друг от
друга как правая рука от левой, как предмет от своего изо¬
бражения в зеркале.
I Обозначения D и L в названиях гидроксикислот указывают
условно их конфигурацию, т. е. пространственное расположение
заместителей вокруг асимметрического атома углерода. Обозна¬
чение D получают те гидроксикислоты, у которых гидроксиль¬
ная группа в проекционной формуле Фишера стоит справа;
обозначение L ставится в тех случаях, если гидроксильная
группа в проекционной формуле оказывается слева.
Рис. 28. Оптические антиподы молочной кислоты
818
Оптическая изомерия наблюдается и у яблочной кислоты
НООС—СНОН—СН2—СООН:
соон
I
н—|—он
CHjCOOH
0-(+)-яблочная
кислота
СООН
I
НО—J—Н
CHjCOOH
£-(—)-яблочная
кислота
В природе встречается (—)-яблочная кислота.
В молекуле винной кислоты НООС—СНОН—СНОН—СООН
имеются два одинаковых асимметрических атома. Винная кис¬
лота была первым оптически активным веществом, для которого
в 50-х годах нашего столетия при помощи специального рентге¬
нографического метода была определена абсолютная конфигу¬
рация, т. е. установлено соответствие между знаком оптическо¬
го вращения и пространственной моделью и условно выражаю¬
щей эту модель проекционной формулой;
СООН
СООН
СООН
но-
-н
н—
—он
н—
н—
—он
но—
—н
н—
СООН
соон
-он
он
соон
(—)-винная
кислота
(+)-винная
кислота
ыезовинная
кислота
Равномолекулярная смесь право- и левовращающего антипо¬
дов образует рацемическую винную кислоту, называемую также
виноградной; мезовинная кислота оптически недеятельна вслед¬
ствие внутримолекулярной компенсации вращения асимметриче¬
ских центров. Легко увидеть, что в формуле мезовинной кисло¬
ты «верхний» асимметрический центр имеет конфигурацию
,'(+)-винной кислоты, а «нижний»— (—)-винной. Обе оптически
активные винные кислоты образуют пару оптических антиподов,
все их физические свойства одинаковы, за исключением знака
оптического вращения. По отношению к мезовинной кислоте
(—)-винная и (+)-винная кислоты являются диастереомерами.
Так называют группы стереоизомеров с несколькими асим¬
метрическими центрами, конфигурация части которых совпа¬
дает, а части — различается. Главная особенность диастереоме-
ров в том, что физические свойства у них в каждой паре
различны.
В природе широко распространена (+)-винная кислота.
Еще более сложный случай пространственной изомерии воз¬
никает, когда в соединении имеются два разных асимметриче¬
ских атома. В общем случае молекула, имеющая п неодинако-
319
вых асимметрических атомов, существует в виде 2" индивиду¬
альных пространственных форм (рацематы не учитываются).
§ 145. Важнейшие представители. Среди гидроксикислот име¬
ются соединения, играющие важную роль в живой природе, а
также используемые в промышленности.
Молочная кислота была впервые обнаружена в прокисшем
молоке. Источником молочной кислоты служит молочный сахар
(лактоза), подвергающийся брожению под действием особых
микроорганизмов — молочнокислых бактерий. Молочнокислому
брожению могут подвергаться и другие сахара. Такие процессы
идут при приготовлении сыра, квашении капусты, силосовании
кормов.
С6Н,20« —► 2СН3—СНОН—СООН
В промышленности молочную кислоту до последнего време¬
ни получали исключительно путем молочнокислого брожения.
В настоящее время освоен синтез ее из уксусного альдегида:
О ОН ОН
| HCN | Н20 |
СНз—С—Н >- СНз—СН—CN ► СНз—СН—СООН
Молочную кислоту используют как консервирующее сред¬
ство в пищевой промышленности, кроме того, ее применяют при
обработке кож, в текстильной промышленности, как исходное
вещество во многих синтезах.
(—)-Яблочная кислота часто встречается в различных фрук¬
тах и плодах (особенно незрелых) —яблоках, винограде, ряби¬
не. Это кристаллическое вещество, т. пл. 100°С, легко раство¬
римое в воде.
(-\-)-Винная кислота — кристаллическое вещество, содер¬
жится во многих растениях, особенно в винограде, рябине. Кис¬
лая калиевая соль винной кислоты трудно растворима в воде
(осаждается в винных бочках в виде так называемого винного
камня). Реакцию ее образования используют для обнаружения
калия. При нейтрализации «винного камня» гидроксидом натрия
- образуется смешанная калиево-натриевая соль винной кисло¬
ты — сегнетова соль.
Лимонная (2-гидроксипропантрикарбоновая-1,2,3) кислота
СООН
I
НООС—СНг—СОН—СН2—СООН
содержится в плодах цитрусовых растений (лимоны, апель¬
сины), винограде, крыжовнике, малине, свекле; в виде соли с
никотином содержится в табаке; кристаллическое вещество,
т. пл. 153 °С, легко растворима в воде. Лимонную кислоту
применяют при кращении, в медицине, в кондитерской и пище¬
вой промышленност и
320
Салициловая (о-гидроксибензойная) кислота — важнейшая
из гидроксикислот ароматического ряда. Ее получают в про¬
мышленности по реакции Кольбе — Шмитта действием диоксида
углерода на фенолят натрия (125°С, 0,4—0,7 МПа; выход по¬
чти количественный):
ONa
со2
ОН
H2SO,
*-
ОН
^-COONa
СООН
В более новом способе исходят из бензойной кислоты; ее
превращают в основную медную соль, из которой при нагрева¬
нии образуется салициловая кислота:
СООСи—ОН
нагревание
СООН
+ Си
ОН
Салициловая кислота — бесцветное кристаллическое веще¬
ство, т. пл. 159 °С, немного растворяется в воде, хорошо в
спирте. Ее применяют для колориметрического определения же¬
леза (фиолетовое окрашивание) и меди (зеленое окрашивание),
а также как флуоресцентный индикатор. Благодаря антисепти¬
ческим свойствам салициловую кислоту применяют при консер¬
вировании фруктов и овощей.
Многие производные салициловой кислоты используют как
лекарственные препараты. Наиболее известна из них ацетил¬
салициловая кислота (аспирин). Ее получают действием уксус¬
ного ангидрида или ацетилхлорида на салициловую кислоту,
причем происходит ацилирование фенольного гидроксила:
СООН
CH3COC1
ОН
-СООН
О—СОСНз
Аспирин является жаропонижающим и болеутоляющим ле¬
карственным препаратом. Он был открыт еще в прошлом веке,
однако до сих пор не потерял своего значения. Другой лекар¬
ственный препарат — салол (средство против желудочно-ки¬
шечных заболеваний) представляет собой сложный эфир сали¬
циловой кислоты и фенола:
•СООН
ОН
СбН5ОН, РОС13
>
соос„н5
он
В качестве наружного средства в составе различных мазей
используют также метилсалицилат — продукт взаимодействия
11 Органическая химия
321
карбоксильной группы салициловой кислоты с метиловым спир-1
ПОЛИГИДРОКСИБЕНЗОЙНЫЕ КИСЛОТЫ.
ДУБИЛЬНЫЕ ВЕЩЕСТВА
§ 146. Отдельные представители. Галловая кислота по своему
строению является 3,4,5-тригидроксибензойной кислотой. Она
встречается в природе как составная часть чернильных орешков
(см. ниже), дубовой коры и т. д., из которых ее впервые
получил Шееле еще в конце XVIII века. Это кристаллическое
вещество, т. пл. 293 °С, мало растворимое в холодной воде и
легко — в горячей. На воздухе окисляется и темнеет. При на¬
гревании легко теряет диоксид углерода и превращается в пи¬
рогаллол (см. § 74).
Другие фенолокислоты, карбоксильная группа которых на¬
ходится в орто- или пара-положениях к гидроксильной, также
сравнительно легко декарбоксилируются (это относится и к са¬
лициловой кислоте).
Дубильные вещества. В процессе выделки кожи шку¬
ры животных подвергают дублению, в ходе которого увеличи¬
вается прочность кожи, уменьшается набухание под действием
воды, придается необходимая эластичность. Вещества, вызы¬
вающие дубление, называют дубильными веществами. Это
название напоминает о том, что в свое время главным источ¬
ником дубильных веществ была кора дуба. В настоящее время
для дубления используют неорганические вещества (соли трех¬
валентного хрома, алюминия и др.), органические вещества
растительного происхождения или полученные синтетически.
Одним из важнейших дубильных веществ являются танины,
добываемые из наростов на дубовых листьях (чернильных ореш¬
ков*). Известно несколько типов танинов. Многие из них
представляют собой сложные эфиры, построенные из несколь¬
ких молекул галловой кислоты,— депсиды.
* Это название связано с тем, что чернила ранее готовили, смешивая танин
с солями двухвалентного железа; такой раствор почти бесцветен, при окис¬
лении же железа на воздухе и взаимодействии с ним танина происходило
постепенное «проявление» написанного — образование прочной черной
краски.
том;
СООСНз
ОН
галловая кислота
дидепсид (л-дигалловая кислота)
322
о
/СООН
Lo_/4
НО7 Ч)Н
НО' хон
тридепсид
Другие танины представляют собой эфиры галловой кислоты
с углеводами, чаще всего с глюкозой (см. § 154).
Синтетические дубильные вещества чаще всего представляют
собой сульфопроизводные многоядерных фенолов, например:
ОН ОН
АЛЬДЕГИДО- И КЕТОКИСЛОТЫ
§ 147. Номенклатура и способы получения. Альдегидо- и кето-
кислоты — это гетерофункциональные соединения, в молеку¬
лах которых содержатся одновременно оксо- (альдегидная или
кетонная) и карбоксильная группы. Названия альдегидо- и ке-
токислот строят на основе названий соответствующих карбоно¬
вых кислот с указанием положения карбонильной группы; часто
используют и тривиальные названия:
Н—С—СООН СНз—С—СООН
О
о
глиоксиловая
кислота
Э
СН3—С—СН2—СООН
А
пировиноградная
(2-оксопропановая)
кислота
СНз—С—СН2—СН2—СООН
О
ацетоуксусная 4-оксопентановая кислота
(3-оксобутановая)
кислота
Для получения альдегидо- и кетокислот могут быть исполь¬
зованы многие реакции, при помощи которых либо вводится
карбонильная группа в карбоксильное соединение, либо, наобо¬
рот, карбоксильная группа вводится в соединение, уже содер¬
жащее альдегидную или кетонную группы.
11*
323
1. Окисление гликолей:
но—сн2—сн2—он —>- н—с—со
2. Гидролиз дигалогензамещенных кислот:
Г 0Н 1
н2о I
С12СН—СООН ——f н—с—соон ——»■
НС1 | “-Н2О
- он
о
101 II
н—с—соон
о
соон
3. Окисление гидроксикислот:
ОН
СН3—CH—СНг—CHji—СООН
СНз—С—СН2—СН2—СООН
О
4'Гидроксипентановая
4-оксопентановая
кислота
кислота
§ 148. Представители кетокислот. Простейшей по строению
является а-оксопропионовая кислота, называемая обычно пиро-
виноградной. В названии отражается способ ее получения —
нагреванием виноградной (винной) кислоты (греч. пирос—
огонь). В процессе этой реакции молекула винной кислоты те¬
ряет диоксид углерода и воду:
НООС—СН—СН—СООН > ГСН2=С—СООНт —► СНз—с—соон
I 1 1 II
он он 2 L он J о
Пировиноградная кислота — жидкость с характерным запа¬
хом. При дальнейшем нагревании она теряет еще одну молеку¬
лу диоксида углерода и превращается в уксусный альдегид:
Подобная реакция декарбоксилирования при нагревании ха¬
рактерна и для других а-кетокислот.
Карбоксильная группа пировиноградной кислоты проявляет
присущие этой группе свойства: известны соли, эфиры, амиды
и другие производные пировиноградной кислоты по карбоксиль¬
ной группе. Кетонная группа, в свою очередь, вступает в ха¬
рактерные для нее реакции: образует оксим, гидразон и фснил-
гидразон, присоединяет гидросульфит натрия, синильную
кислоту. Все реакции карбонильной группы протекают у пиро¬
виноградной кислоты легче, чем у обычных кетонов. Это —
результат усиления положительного заряда на атоме углерода
карбонильной группы под влиянием карбоксильной группы:
СНз—СО—СООН —* СНз—сно + со2
6+ 6+
СНз—с—с—о—н
324
В свою очередь карбонильная группа, оказывая влияние на
карбоксильную, увеличивает ее кислотность: поэтому пировино-
градная кислота почти в 500 раз сильнее пропионовой.
Простейшая по строению 0-кетокислота — ацетоуксусная
СН3—СО—СН2—СООН. Подобно всем другим (3-кетокислотам,
в свободном состоянии она неустойчива, распадается с выделе¬
нием диоксида углерода:
СНз—СО—СН2—СООН —► СНз—со—сн3 + со2
Соли, эфиры и другие функциональные производные ацето-
уксусной кислоты вполне устойчивы. Среди производных ацето-
уксусной кислоты особое значение имеет ее этиловый эфир,
обычно называемый ацетоуксусным эфиром. Его получают при
сложноэфирной конденсации, как й другие эфиры р-кетокислот.
§ 149. Сложноэфирная конденсация. Сложноэфирная конденса¬
ция— взаимодействие двух молекул сложного эфира алифа¬
тической карбоновой кислоты с образованием сложного эфира
р-кетокислоты. Эта реакция идет под влиянием основных
катализаторов. Ее простейший и в то же время важнейший
пример — конденсация двух молекул этилацетата с образова¬
нием ацетоуксусного эфира:
0 0 о о
СН3—(!—OC2Hs -f СНз—С—ОС2Н5 СНз—с—сн2—с—ос2н5
-с2н5он
Конденсирующими агентами кроме металлического натрия
могут служить алкоголяты, амид и гидрид натрия.
Механизм этой реакции напоминает изученную ранее аль-
дольную конденсацию (см. § 86). Прежде всего под действием
сильного основания (этилата натрия) отщепляется протон из
СНз-группы уксусноэтилового эфира и образуется карбанион:
СН3СООС2Н5 + С2Н5СГ =«=± : СН2СООС2Н5 + С2Н5ОН
Этот карбанион играет далее роль метиленового компонента
и присоединяется к другой молекуле этилацетата, являющейся
карбонильным компонентом:
ОС2Н5
6* I
СН3—С—ОС2Н5 + : СН2—СООС2Н5 —► СН3—с—СН2—СООС2Н,
После отщепления молекулы спирта образуется новый анион,
который при разложении реакционной смеси разбавленной
325
кислотой превращается в ацетоуксусный эфир;
ос,н6
СНЭ—С—СНа—СООС2Н5
I
О'
—CjH5OK
СН3—С=СН—СООС2Н5
О"
СНз—С—СН2—СООС2Н5
н+
О
Подобно этилацетату, в сложноэфирную конденсацию всту¬
пают и другие эфиры общей формулы RCH2COOR'. Эфиры типа
R2CHCOOR' к конденсации под действием натрия не способны.
§ 150. Таутомерия ацетоуксусного эфира. Ацетоуксусный эфир
явился первым из органических соединений, на примере кото¬
рого было открыто и подробно изучено явление таутомерии —
сосуществование изомерных форм, находящихся в равновесии и
способных легко переходить друг в друга (динамическая изоме¬
рия). Для ацетоуксусного эфира речь идет о следующих струк¬
турах:
СИз—С—СН2—СООСаНз
СНз—С=СН—COOCjHs
ОН
кетонная форма
енольная форма
Превращение обеих форм друг в друга заключается в пере¬
мещении двойной связи (т. е. подвижных л-электронов) и во¬
дорода:
—С—СНз— ?=ь —СН=СН—
Такой тип таутомерии называют кето-енольной. Он является
частным случаем таутомерии с переходом водорода — прото-
тропии. В данном случае водород приобретает подвижность
под влиянием групп СО и COOR.
Вывод о существовании кетанной и енольной форм ацето¬
уксусного эфира первоначально был сделан при изучении хи¬
мических свойств этого соединения (см. ниже), позднее обе
формы удалось выделить в индивидуальном состоянии. Если
обычный ацетоуксусный эфир (он содержит «93 % кетонной и
«7 % енольной форм) растворить в петролейном эфире и силь¬
но охладить, например до —78 °С сухим льдом в ацетоне,
кетонная форма выделится в виде кристаллов, плавящихся при
—30 °С (плотность чистой кетонной формы df 1,0368, показа¬
тель преломления «о 1,4425). Постепенно она превращается в
326
равновесную смесь кетонной и енольной форм. Чистую енольную
форму проще всего получить перегонкой ацетоуксусного эфира;
температура кипения енольной формы ниже, чем у кетонной;
т. пл.— 44 °С, 1,0119, по 1,4480. При стоянии енольная фор¬
ма также превращается в равновесную смесь.
Взаимопревращение таутомерных форм ацетоуксусного эфи¬
ра ускоряется кислотами и щелочами. Даже ничтожные следы
щелочи, попадающие в жидкость из стекла лабораторной посу¬
ды, из табачного дыма, оказывают сильное каталитическое дей¬
ствие. Содержание кетонной и енольной форм в растворах
зависит от природы растворителя и от температуры. Например,
раствор ацетоуксусного эфира в воде содержит 0,4 % енольной
формы, в метаноле 6,87 %, в бензоле 16,2 %, в гексане 46,4 %.
Обычный ацетоуксусный эфир дает реакции как кетонной,
так и енольной формы. При этом, несмотря на малое содержа¬
ние енольной формы в равновесном ацетоуксусном эфире, при
соответствующих реакциях весь ацетоуксусный эфир может
перейти в производное енольной формы, так как по мере рас¬
ходования этой формы запас ее вновь пополняется вследствие
енолизации кетонной формы.
Реакции, характерные для кетонной фор-
па ы. а) Присоединение гидросульфита натрия с образованием
характерного для карбонильных соединений гидросульфитного
производного;
S03Na
ИаНвОз
CHj—С—СН-—COOCjHs »-
О ОН
СН,
,-С-
СН«—СООС2Н5
б) Присоединение синильной кислоты с образованием циан¬
гидрина;
CN
HCN I
СНз—С—СН2—СООС2Н5 »- СНз—С—СН2—СООС2Н5
о
он
Реакции, характерные для енольной формы.
а) Красно-фиолетовое окрашивание с раствором хлорида
железа (111); при этом происходит образование сложных внутри-
комплексных соединений железа.
б) Реакция с хлоридом фосфора (V), приводящая к р-хлор-
кротоновому эфиру:
ОН С1
I РС15 I
СНз—С=СН—СООС2Н5 ► СНз—с=сн—СООС2Н5
327
в) Быстрое присоединение на холоду брома по двойной свя¬
зи. Первоначально образовавшийся дибромид легко отщепляет
бромоводород и превращается в производное кетонной фор¬
мы — а-бромацетоуксусный эфир:
ОН НО Вг
I Вг2 [|
СН-,—с=сн—СООС2Н5 —► СНЭ—С—СН—COOC2HS ——>
| ~НВг
Вг
О Вг
П I
—»• СНз—С—СН—СООС2Нз
г) Ацилирование по ОН-группе под действием ацетилхлорида
в присутствии пиридина:
ОН О—СОСНз
I снзсоа |
СНз—С=СН—СООС2П6 СН3—С=СН—СООС2Н8
д) Алкилирование по ОН-группе диазометаном:
ОН ОСНз
I ch2n2 |
СНз—С=СН—СООС2Н5 ——»- СНз—С=СН—СООС2Н5
—N2
е) Реакции с металлическим натрием, алкоголятом натрия
или щелочью, приводящие к натрийацетоуксусному гфиру (ал-
коголят (3-гидроксикротонового эфира):
ОН ONa
I C2H5ONa I
СНз—С=СН—СООС2Н5 —► СНз—С=СН—СООС2Н6
— С2Н50Н
Натрийацетоуксусный эфир имеет большое значение для
синтеза разнообразных органических соединений. Многие синте¬
зы осуществляют действием на него алкилирующих и ацнлирую-
щих средств. Интересная и важная особенность этих реакций —
перенос реакционного центра.
Реакция алкилирования протекает следующим образом:
ONa О СНз
I сн31 II I
СНз—С=СН—СООС2Н5 —ТГТ СНз—С—СН—СООС2Н5
—Nal
В этой реакции радикал алкилгалогенида становится не на
место своего предшественника — атома натрия, связанного с
кислородом, а оказывается у «--углерода.
328
Аналогично идет и ацилирование натрнйацетоуксусного
эфира:
ONa
СНз—С=СН—СООС2Н5
С6Н5СОС1
>.
О СОС6Н5
II I
СНз—С—СН—СООС2Н5
Действием иода можно осуществить «сшивание» двух моле¬
кул ацетоуксусного эфира, протекающее по такой же схеме:
2СН;
ONa
i=CH—СООСоН.
la
—2NaI
СНз—со—CH—COOC2Hs
СНз—со—Ан—СООС2Н5
§ 151. Синтезы на основе ацетоуксусного эфира. Полученные
путем алкилирования или ацилирования замещенные ацетоук-
сусные эфиры представляют интерес не сами по себе, а как
исходные вещества для дальнейших превращений. Большое зна¬
чение имеют реакции кетонного и кислотного расщепления
этих соединений.
Процесс кислотного расщепления, протекающий под дейст¬
вием концентрированного спиртового раствора щелочи, фор¬
мально сводится к разрыву связи между а-углеродом и кето-
группой и присоединению воды (щелочи) при одновременном
гидролизе сложноэфирной группировки, причем образуются две
молекулы соли уксусной кислоты:
О
СНз—<LСН2—СООС2Н5 + 2К.ОН —> 2СН3СООК + С2Н5ОН
В действительности этот процесс более сложен, поэтому его
механизм здесь рассматриваться не будет.
Под действием слабых щелочей или разбавленных минераль¬
ных кислот идет омыление сложноэфирной группы и выделяет¬
ся свободная ацетоуксусная кислота. Подобно всем р-кетокис-
лотам, она неустойчива: происходит кетонное расщепление —
отщепляется диоксид углерода и образуется кетон, в данном
случае — ацетон:
О О
II н2о |
СНз—С—СН2—СООС2Н5 *- СНз—с—сн2—соон —>
о
—> СНз—I—СНз + со2
Аналогично могут подвергаться кетонному и кислотному рас¬
щеплению замещенные ацетоуксусные эфиры; при этом могут
быть получены более сложные кетоны, гомологи уксусной кис¬
лоты, дикетоны, дикарбоновые кислоты и др. Так, например,
329
при расщеплении описанных ранее производных ацетоуксусного
эфира образуются следующие продукты:
О СН3
II I
СН3—С—СН—СООС2Н5
кетонное
расщепление
У-
кислотное
расщепление
О
СНз—С—СН2—СН3
метилэтилкетон
СН3—СООН + СН3—СН2—СООН
уксусная
кислота
пропионовая
кислота
О СОСвН,
II I
СН3—С—СН—СООСлНз
кетонное
расщепление
—>.
О
О
СНз—С—СН»—С—CeHs
бензоилацетон
кислотное
расщепление
-со2
СНз—СООН + CSH5—СО—СН2—СООН
о
СН3—С—СН—СООС2Н5 расщепление
С6Н(—СО—СНз
ацетофенон
О
СНз—С—СН—СООС2Н5
II
О
кислотное
расщепление
0
1
СНз—с—(СН2)2—С—СНз
ацетонилацетон
2СН3—СООН + НООС—(СН2)2—СООН
янтарная кислота
При помощи ацетоуксусного эфира можно получить любые
метилкетоны общей формулы СН3—СО—R, гомологи уксусной
кислоты RR'CIT—СООН, дикетоны, дикарбоновые кислоты.
§ 152. Другие типы таутомерии. Помимо ацетоуксусного эфира
кето-енольную таутомерию можно обнаружить и у ряда других
веществ, причем устойчивость обеих форм может быть различ¬
ной. Строение таких веществ можно в общем виде выразить
формулой X—СН2—Y, где X и Y — группы RCO, COOR, CN,
N Оа.
Рассмотренное выше равновесие кето-енольных форм и двой¬
ственная реакционная способность p-дикарбонильных соедине¬
ний— лишь один из примеров широко распространенного в ор¬
ганической химии явления. Так, таутомерия (либо двойственная
реакционная способность) обнаруживается у соединений, содер¬
330
жащих одновременно гидроксильную и карбонильную группы,
например:
СН2—СО—СН3 Н2С_СН2СН3
I *=± н,с' X
СН2-СН2^-ОН \ / \он
о
у-ацетопропиловый а-гидрокси-«-метил-
спирт (оксоформа) тетрагидрофуран
(циклическая форма)
Наибольшее значение этот вид таутомерии имеет в ряду
углеводов, где мы с ним и познакомимся подробнее.
В настоящее время таутомерию понимают как динамическую
изомерию — равновесие форм, способных легко переходить друг
в друга. При внимательном рассмотрении можно установить,
что резкой границы между таутомерией и изомерией в сущности
нет. Так, кетонная и енольная формы ацетоуксусного эфира,
находящиеся в таутомерном равновесии при комнатной темпе¬
ратуре, при охлаждении до —70 °С становятся устойчивыми,,
раздельно существующими изомерами. В условиях антарктиче¬
ской зимы для этих соединений пришлось бы говорить уже не
о таутомерии, а об изомерии!
Пропил- и изопропилбромиды при обычных условиях — ус¬
тойчивые, раздельно существующие изомеры. При нагревании
до 250 °С между обоими веществами устанавливается таутомер¬
ное равновесие:
250 °С
СН3—СН2—СН2Вг *=* СНз—СНВг—сн3
ГЛАВА 19
УГЛЕВОДЫ
Углеводы — вещества состава СпН2пОп, имеющие первостепен¬
ное биохимическое значение, широко распространены в живой
природе. К ним относятся различные сахаристые вещества,
крахмал, целлюлоза (клетчатка). Название углеводы сохрани¬
лось за ними с тех времен, когда строение этих соединений еще
не было известно, но установлен был их состав, отвечающий
общей формуле С„(Н20)т. По этой формуле углеводы рассма¬
тривали как гидраты углерода, т. е. соединения углерода с во¬
дой — «угле-воды».
§ 153. Классификация, физические свойства, нахождение в при¬
роде. Класс углеводов объединяет:
моносахариды — соединения, имеющие химическую природу
гидроксиальдегидов или гидроксикетонов, но существующие
преимущественно в таутомерных циклических формах;
331
олигогахариды (греч. олнгос — мало) — продукты конденса¬
ции нескольких молекул моносахаридов друг с другом с выде¬
лением воды (по типу простых эфиров); важнейшими олигоса¬
харидами являются дисахариды, молекулы которых построены
из двух остатков моносахаридов;
полисахариды — высокомолекулярные вещества, продукты
конденсации большого числа молекул моносахаридов.
Моносахариды в зависимости от числа входящих в их со¬
став атомов кислорода (обычно это число равно числу атомов
углерода) разделяют на группы тетроз, пентоз, гексоз и т. д.
В зависимости от того, имеется ли в молекуле моносахаридов
альдегидная или кетонная группа, их разделяют на альдозы и
кетозы. Классификацию углеводов можно изобразить следую¬
щей схемой:
Моносахариды
Дисахариды С,2Н220и
Тетрозы С4Н804
эритроза
треоза
Пентозы С5Н,0С>5
арабиноза
ксилоза
рибоза
Гексозы С6Н12О0
глюкоза
манноза
галактоза
фруктоза
сахароза
лактоза
мальтоза
целлобиоза
Полисаха риды
(С*Нв04)„
пентозаны
(C0HioOs)n
целлюлоза
крахмал
гликоген
Из всего многообразия моносахаридов наиболее распро¬
странены альдопентозы и альдогексозы. Все приведенные
выше моносахариды за исключением фруктозы относятся
к альдозам. Самой распространенной альдогексозой является
глюкоза.
Глюкозу называют также виноградным сахаром, поскольку
она содержится в виноградном соке. Кроме винограда глюкоза
находится и в других сладких плодах и вообще в разных час¬
тях растений. Не менее распространена глюкоза и в животном
мире: около 0,1 % ее содержится в крови. Глюкоза разносится
кровью по всему телу и служит источником энергии для орга¬
низма. Она входит в состав важнейших природных дисахари¬
дов— сахарозы, мальтозы, лактозы и полисахаридов — целлю¬
лозы, крахмала.
Другой моносахарид, широко распространенный в раститель¬
ном мире, — это фруктоза (кетогексоза), или плодовый сахар.
Наряду с глюкозой фруктоза содержится в сладких плодах,
входит в состав дисахарида сахарозы, полисахарида инулина
(гидролизом последнего обычно и получают фруктозу). Изв.тс-
332
кая из цветов сладкие соки, пчелы приготовляют мед, который
по химическому составу представляет собой в основном смесь
глюкозы и фруктозы. Эта смесь образуется при ферментативном
гидролизе сахарозы, содержащейся в собираемых пчелами со¬
ках.
Другие моносахариды почти не встречаются в свободном
виде в природе, но входят в состав важных олиго- и полиса¬
харидов. Так, например, ксилоза (древесный сахар) является
составной частью полисахарида ксилана, сопровождающего
целлюлозу в соломе, кукурузных стеблях, хлопке; арабиноза
встречается в растениях в виде полисахарида арабана, входя¬
щего в состав вишневого клея, аравийской камеди (отсюда и
название арабиноза); рибоза имеет очень большое биологиче¬
ское значение из-за своей связи с нуклеиновыми кислотами (см.
§ 177); манноза — составная часть полисахаридов маннанов;
галактоза входит в состав дисахарида лактозы—молочного са¬
хара.
В классе углеводов приходится встречаться с самыми слож¬
ными особенностями, которые могут быть присущи органиче¬
ским соединениям. Моносахариды способны существовать в ряде
таутомерных форм. Присутствие в их молекулах нескольких
асимметрических атомов углерода приводит к появлению боль¬
шого числа оптически активных стереоизомеров. Так, вещество
состава СбН^Ов может образовать около 100 структурно и
пространственно изомерных форм или таутомерных модифи¬
каций.
Моно- и дисахариды представляют собой бесцветные крис¬
таллические вещества, очень хорошо растворймые в воде, но
нерастворимые в неполярных органических растворителях. При
нагревании до температур порядка 120—150 °С они разлагаются,
не плавясь (образование карамели). Все они оптически активны,
обладают сладким вкусом. Известный всем обычный тростнико¬
вый (или свекловичный) сахар — типичный представитель диса¬
харидов — сахароза.
Полисахариды — соединения высокомолекулярные. Они об¬
ладают свойствами, присущими веществам такого типа: не пла¬
вятся, образуют коллоидные растворы. Главная химическая осо¬
бенность олиго- и полисахаридов — их способность к гидролизу
с образованием моносахаридов.
В природе углеводы образуются в процессе фотосинтеза: так
называют превращение диоксида углерода воздуха в сахара
за счет солнечной энергии. Этот процесс идет в зеленых листь¬
ях растений на солнечном свету. Катализатором служит хлоро¬
филл — зеленый пигмент дистьев. Синтетические способы полу¬
чения углеводов не имеют практического значения, хотя в
принципе известны. Первый синтез сахаристого вещества был
осуществлен в 1861 г. А. М. Бутлеровым действием известковой
воды на формальдегид. Реакция заключается в альдольной кон-
ззз
денсации нескольких молекул формальдегида:
Л—с—н + н-
6, i
-с—н + н—с—н + н-
II I II I
о*- 1 ск '
-С—Н + Н—С—Н + Н—С—Н
I I II ! А
СИ- ' 0<- 1 о
сн2—сн—сн—сн—сн—с—н
Ан Ан он Ан Ан о
МОНОСАХАРИДЫ
§ 154. Строение. Альдегидные формулы. Строение мо¬
носахаридов мы рассмотрим на примере глюкозы. Впервые она
была получена в 1811 г. русским , химиком Г. 3. Кирхгофом при
гидролизе крахмала. В конце 60-х годов прошлого столетия
работавший в Московском университете А. А. Колли доказал,
что в молекуле глюкозы имеется пять гидроксильных групп.
Шестой кислородный атом, очевидно, должен входить в состав
альдегидной группы, поскольку глюкоза дает многие качествен¬
ные реакции на альдегидную группу — окисляется оксидом се¬
ребра (реакция /), присоединяет синильную кислоту (реак¬
ция 4):
снэ
1
о
II
с—н
CHI
HI
Ag2o СООН
I 1 —Г* I
(CH*)s 2 (СНОН)4 1 (СНОН)4
JL I I ....
СНз
СН2ОН
СН2ОН
2-иод¬
глюкоза
глюконовая
гексан
кислота
ацилирование
(CH3CO)jO
HCN
«)
1
(3)
CN
алкилирование
(CH»)2S04
(5)
1
Пента-О-ацетилглюкоза
(Пентаацетат глюкозы)
(СНОН)б Пентаметилглюкоза
(]Н2ОН
нитрил
глюкогептановой
кислоты
Наличие неразветвленной цепи углеродных атомов в глюкозе
доказано ее превращением под действием иодоводорода в
2-иодгексан с нормальной цепью углеродных атомов (реакция
2). Образование в результате ацилирования (реакция 3) и ал¬
килирования (реакция 5) соответственно пента-О-ацетил- и пен-
тз-0-метилглкжозы также доказывает существование в глюкозе
ш}ти гидроксильных групп. Однако построены эти производные
н$ столь просто, как можно было бы думать, принимая альде¬
гидную формулу глюкозы. С такими производными мы позна¬
комимся позже,
834
В молекулах альдогексоз четыре асимметрических атома
углерода; вещества с таким строением могут иметь 24, т. е.
16 стереоизомеров. В конце прошлого столетия в классических
работах Э. Фишера была установлена конфигурация (простран¬
ственное расположение атомов) природной правовращающей
глюкозы, выражаемая следующей проекционной формулой:
СНО
н—С—он
•Н
или в упрошенной
•ОН условной записи
I
н—с—он
I
СН2ОН
•0-(+)-глюкоза
НО—С
Н_А
СНО
он
но
—он
—он
сн2он
Знак (+) показывает направление оптического вращения,
обозначение D указывает конфигурацию. Условное обозначение
D получают те сахара, в проекционной формуле которых гидр-
оксигруппа у самого нижнего асимметрического атома углеро¬
да стоит справа.
Другие моносахариды могут отличаться от глюкозы числом
углеродных атомов, иметь кетонную группу вместо альдегидной
(например, фруктоза), а также иную конфигурацию асимметри¬
ческих центров. Приводим формулы важнейших моносахаридов.
Альдо- и кетогексозы:
СНО
СН20Н
1
СНО
СНО
\
Н—с—он
1
j
н—с—он
с=о
1
1
но—с—н
1
но-с—н
НО—С—Н
1
но—с—н
но—с—н
1
н—с—он
1
Н-С—он
1
Н— С—ОН
1
НО—С—Н
1
1
н—с—он
1
Н—С—ОН
j
Н—С—ОН
н—с—он
1
сн.
1
СНаОН
1
СН2ОН
1
СН2ОН
£)-(+)-глюкоза
Альдопентозы:
D-(—)-фруктоза
£>-( -(-)'Манноза
0-(+)-галакто
СНО
СНО
СНО
1
1
н—с—он
1
1
НО—С—Н
н-с-он
1
н—с—он
1
Н—С—он
но—с—н
1
н—с—он
1
1
Н—С—он
1
н—с—он
1
СНаОН
j
сн2он:
сн2он
/).(—).рибоза D-(—)-арабиноза £)-(+)-ксилоза
335
Циклические формулы. Как уже упоминалось, не¬
которые свойства .моносахаридов не могут быть объяснены фор¬
мулой альдегидоспирта. Так, глюкоза вступает не во все
характерные для альдегидов реакции, например, она не образует
гидросульфитного соединения, не дает окрашивания с фуксин-
сернистой кислотой. Оказалось также, что не все гидроксильные
группы глюкозы имеют одинаковые химические свойства. Это
проявляется в том, что при действии метилового спирта и хло-
роводорода на глюкозу метилированию подвергается одна гидр-
оксигруппа остальные гидроксильные группы алкилируются лишь
в более жестких условиях (метилирование метилиодидом и
оксидом серебра, диметилсульфатом и щелочью). В получен¬
ной таким путем пентаметилглюкозе одна из метильных групп
отщепляется при гидролизе легко, остальные удерживаются
прочно.
При растворении глюкозы в воде наблюдается постепенное
изменение значения удельного вращения — так называемая му-
таротация. Свежеприготовленный водный раствор глюкозы име¬
ет удельное оптическое вращение [а]о+112°С; при стоянии
раствора вращение постепенно снижается, достигая значения
+52,5°.
Как было установлено позже, это конечное вращение создает¬
ся равновесной смесью двух форм: р-глюкозы с [a] D +112° и
0-глюкозы с [а]д+ 18,7°. Чистую а-форму можно выделить пу¬
тем кристаллизации из воды, чистую p-форму — кристаллизацией
из пиридина. (О сущности различия между а- и p-формами см.
ниже.)
Свойства глюкозы, которые не могут быть объяснены при
рассмотрении ее альдегидной формулы, находят объяснение,
если придать ей циклическую формулу, которую называют так¬
же оксидной или полуацетальной. Это таутомерная модифика¬
ция, находящаяся в равновесии с альдегидной формой:
6 5 4 3 2 1
НОСН2—СНОН—СНОН—СНОН—СНОН—СНО ^=fc
НОСН2—СН—СНОН—СНОН—СНОН—СНОН
Сущность таутомерного перехода альдегидной формы в цик¬
лическую заключается в том, что атом водорода гидроксигруп-
пы при пятом атоме углерода перемещается к атому кислорода
карбонильной группы. Углерод С-1 становится асимметриче¬
ским, а между атомами С-1 и С-5 устанавливается связь через
атом кислорода (отсюда термин окисная форма) с образова¬
нием шестичленного цикла. Вновь возникший асимметрический
атом С-1 может иметь две антиподные конфигурации, т. е. воз¬
336
можно существование двух циклических изомеров, называемых
а- и р-формами:
и
н—с—он
1
но—с—н
н—с—он
1 £
н—с—он
U о
U °
но—с—н
1
но—с—н
н—С—он
1
н—с—он
1 jt;
Н—С- ;
1
н—с-
сн2он
а-форма
Д-глюкозы
сн2он
(3-форма
/7-глюкозы
В этих двух диастереомерах конфигурации «старых» асим¬
метрических атомов углерода одинаковы, а конфигурации у
вновь образовавшегося асимметрического атома углерода —
противоположны. В этом и заключается различие между ос-
и р-глюкозой.
Буквой а принято обозначать конфигурацию гликозидного асимметри¬
ческого центра сахаров D-ряда, в которой ОН-группа при С-1 (глнкозидный
гидроксил) стоят в проекционной формуле справа.
Гидроксигруппа у С-1, возникшая при переходе в цикличе¬
скую форму, существенно отличается от остальных гидроксиль¬
ных групп. В то время как при атомах С-2—С-6 стоят обычные
спиртовые гидроксилы, при С-1 гидроксильная группа входит в
состав так называемой полуацетальной группировки (см. §86).
Структура этой группировки аналогична структуре полуацета-
лей, возникающих при присоединении молекулы спирта к аль¬
дегиду, например:
н3а
>=о + сн3он
н
Н,с.
3^
н"
„он
"о-
сн,
1 он
<
о—
Полуацетали легко образуются и столь же легко гидролизу¬
ются. В случае глюкозы образование полуацеталя — реакция
внутримолекулярная. В растворах глюкозы устанавливается
равновесие между альдегидной и полуацетальными а- и f$-
формами.
В присутствии кислотных катализаторов полуацетали пре¬
вращаются в ацетали:
R. .ОН н+ Rv /ОСНз
;с( + снзон 4=*s V; + н2о
W ХОСН3 W ХОСН3
Аналогичное превращение происходит и с моносахаридами.
При действии на них метилового спирта и хлороводорода обра¬
337
зуются два диастереомерных ацеталя — производные цикли-»
ческой формы глюкозы:
Г
V*-
(
н—С—ОСИ-,
1 6
С
СНдО—с—Н
н—с—он
сн3он.
н—с—он
сн3он,
Н—С—ОН
1
НС1
1
на
1
но—с—н с
1
3
■ч ■ ■
но—с—н
]
у
но—С—Н (
5
н—с—он
1
н—с—он
1
н—с—он
н—с J
1
н—с—он
X
1
-о-
сн2он
1
СН2ОН
сн2он
ос-метилглюкозид
глюкоза
|? -метилглюкозид
Такие соединения называют гликозидами, а
в случае глюко-
зы — глюкозидами.
В гликозидах гидроксильная группа заменена на алкокси-
группу OR и, следовательно, исключена возможность таутомер¬
ного превращения в альдегидную форму. И действительно —
гликозиды не обнаруживают мутаротации.
Гликозиды встречаются в природе, Роль спиртового компонента (агли-
кона) в них могут играть такие соединения, как фенолы, циангидрины и др.
К гликозидам относятся, в частности, красящие вещества растений; сердечные
гликозиды, обладающие сильным физиологическим действием; дубильные ве¬
щества. Примером может служить гликозид амигдалин C20H27O11N, содержа¬
щийся в зернах горького миндаля и косточках других плодов. Он является
гликозидом дисахарида генциобиозы и циангидрина бензальдегида. При гид¬
ролизе кислотами амигдалин распадается на углевод, бензальдегид и синиль¬
ную кислоту:
гн2о (н+)
CsoHsA.N >- 2C6HISOe + С6Н6СНО + HCN
Ядовитое действие ядер плодовых косточек при употреблении их в боль¬
ших количествах объясняется выделением синильной кислоты.
Циклические формулы в том написании, которое мы исполь¬
зовали до сих пор, недостаточно наглядно передают простран¬
ственное строение молекулы. Гораздо нагляднее иной способ
написания с привычным изображением пяти- или шестичленного
кольца. Шестичленные циклические формы моносахаридов на¬
зывают пиранозными, поскольку основу их структуры составля¬
ет шестичленное кольцо пирана — гетероциклического соедине¬
ния, содержащего в цикле атом кислорода:
- £Н2
СН9
* /• < л
f
НС. >сн
о
н2сг хн.
H2cv ^сн.
пиран
тетрагидро-
пиран
н Н
форма, принятая
для изображения
углеводов
338
В качестве примера приведем циклические формулы некото¬
рых гексоз:
сс-5-глгокоза
(а-5-глюкопираноза)
[В-5-глюкоза
(p-5-глюкопираноза)
а-О-галактоза
(ос-5-галактопйраноза)
Р-5-галактоза
((3-5-галактонираноза)
В прицеленных выше формулах с шестичленным циклом кислород нахо¬
дится в удаленной части цикла, справа от него — глнкозидный гидроксил.
Если он имеет а-конфигурацию — его пишут внизу. Буквой Р обозначают
конфигурацию группы ОН гликозидного центра, когда в циклической фор¬
муле эта группа находится вверху.
Пиранозные формулы углеводов мы условно изображали плоским шести¬
членным циклом, в действительности это кольцо, подобно циклогексану, может
принимать различные пространственные формы.
Пятичлениые циклические формы моносахаридов называют
фуранозными, поскольку в основе их лежит пятичленное кисло¬
родсодержащее кольцо фурана:
нс—сн
ж/ \:н
ту
фуран
н2/с—сн2
н2а. Ън,
ТУ
тетрагидрофуран
Н Н
форма, принятая
для изображения
углеводов
ОН Н
Р-5-фруктоза
(Р-5-фруктофураноза)
ОН н
а-5-фруктоза
(а-5-фруктофураноза)
339
Р-/7-рибоза
(p-Д-рибофураноза)
а-/?-рибоза
(а-Д-рибофураноза)
§ 155. Химические свойства. Зная строение моносахаридов, лег¬
че понять их химические свойства. Мы рассмотрим эти свой¬
ства в основном на примере глюкозы. При этом глюкозу мы
будем изображать то в альдегидной, то в циклической формах.
Поскольку обе формы находятся в равновесии друг с другом,
расходование одной из них в ходе реакции сдвигает таутомер¬
ное равновесие в сторону реагирующей формы. Таким образом,
несмотря на малое содержание альдегидной формы, глюкоза
может быть полностью превращена в производные этой формы.
В иных реакциях образуются лишь производные другой фор¬
мы — циклической.
1. Восстановление. При действии восстановителей (амальга¬
ма натрия, борогидрид натрия) глюкоза превращается в шести¬
атомный спирт сорбит:
СНО
СН2ОН
н—
но—
н—
н—
он
н
он
—он
сн2он
Na/Hg или NaBH4
н—
но—
н—
н—
—он
—н
—он
—он
сн2он
2. Окисление. Хлорная (или бромная) вода окисляет прежде
всего альдегидную группу моносахарида до карбоксильной.
Получающиеся соединения называют альдоиовыми кислотами,
например:
СНО
соон
н—j—ОН
Н—1—он
1
НО—1-Н
Ю1 НО—!—H
н—!—ОН
н—j—он
н—]—он
н—J—он
сн2он
сн2он
глюкоза
глюконовая
340
Более сильные окислители (например, концентрированная
HNO3) превращают в карбоксильную не только альдегидную,
но и первичную спиртовую группу, причем образуются двухос¬
новные кислоты. По номенклатуре IUPAC название этих кис¬
лот строится из названия соответствующего моносахарида пу¬
тем замены окончания оза окончанием аровая кислота:
СНО
н ОН
но—н
н—он
н—он
сн2он
соон
1
Н—j—он
[о] но—!—н
н—j—он
1
н—j—он
соон
глюкоза глюкаровая
(сахарная)
кислота
Для качественного обнаружения углеводов используют реак¬
цию серебряного зеркала или действие фелинговой жидкости
(см. § 86). При этом также происходит окисление, но процесс
протекает сложно с образованием разнородных продуктов. Ке¬
тозы в отличие от обычных кетонов также окисляются фелин¬
говой жидкостью.
3. Присоединение синильной кислоты к альдегидной группе
глюкозы. При этом углеродная цепь удлиняется на один атом
и возникает новый асимметрический центр. Эту реакцию ис¬
пользуют для перехода от низших сахаров к высшим:
CN
СНО
СНОН
н—
но—
н—
н
—он
—н
—он
он
сн2он
н—
HCN НО—
>
н—
н—
—он
—н
—он
—он
сн2он
4. Реакция с фенилгидразином. При нагревании глюкозы с
избытком фенилгидразина сначала реагирует ее альдегидная
группа и образуется продукт замещения атома кислорода кар¬
бонильной группы на остаток фенилгидразина (см. § 86). Вто¬
рая молекула фенилгидразина действует как окислитель, пре¬
вращая соседний с бывшей альдегидной группой гидроксил в
группу С=0, которая реагирует обычным образом с третьей
341
молекулой фенилгидразина, давая озазон глюкозы:
сно
(
;H=N—NHC0H5
н—!—он
н—
—он
но 1 н
1
CeH6NHNH2 нп
CeH5NHNHs
-н2о
—CeH6NH2. -NHs
н—J—он
н—
-он
н—1—он
н—
—ОН
СН2ОН
(
(
:н2он
:h=n—nhc6h5
CH=N—NHCeH5
(
:=о
CH=N-NHCeHs
но—
—Н c6h5nhnh2 НО—J—Н
н—
—он
-HaO H—1—ОН
н—
—он
н—[—он
(
:н2он
£н2он
Озазоны представляют собой желтые кристаллы, форма и
температура плавления которых позволяет установить, из ка¬
кого углевода получен озазон. Это имеет важное значение для
идентификации сахаров. При образовании озазона исчезает бли¬
жайший к карбонильной группе асимметрический центр, поэтому
глюкоза и манноза образуют один и тот же озазон.
5. Алкилирование. При действии алкилирующих средств на
глюкозу можно получить ее простые эфиры, являющиеся про¬
изводными циклической формы. Как уже упоминалось
к(см. § 154), легче всего реагирует гликозидный гидроксил:
ct-0-глкжоза *-о-метил-0-глюкозид пента* о-метил-
«-0-глюкоза
Метильная группа пентаметилглюкозы, связанная с глико-
зидным гидроксилом, легко отщепляется при гидролизе разбав¬
ленными кислотами. При этом образуется тетраметилглюкоза:
сн2осн3
342
У тетра метил глюкозы свободен гликозидный гидроксил, по¬
этому она может превращаться в нециклическую форму и, сле¬
довательно, вступать в свойственные альдегидам реакции (вос¬
становление, окисление, присоединение HCN), а также обнару¬
живает мутаротацию.
Пентаметилглюкоза не способна превращаться в нецикличе¬
скую форму и не дает упомянутых реакций.
6. Ацилирование. При действии ацилирующих средств гидрой
ксильные группы глюкозы легко превращаются в сложноэфир¬
ные. Это происходит, например, при действии уксусного ангид¬
рида. В зависимости от условий, в которых проводится реакция,
можно получить производные а- или p-форм глюкозы:
пента-0-ацетил-
' р-Д-глюкоза
7. Действие кислот. Отношение пентоз и гексоз к кислотам
различно. При нагревании с минеральными кислотами пентоз
от них отщепляется вода и образуется фурфурол:
оГн ~ он]
-зн2о >
НС—сн
// \
^с-сно
Фурфурол можно обнаружить при помощи цветной реакции
с флороглюцином (1,3,5-тригидроксибензолом) и таким образом
отличить пентозы от гексоз, реакция которых с кислотами идет
сложнее.
8. Спиртовое брожение. Эта реакция характерна для гексоз.
Она осуществляется под действием биологических катализато¬
ров — ферментов. При сбраживании гексозы превращаются в
343
этиловый спирт (см. § 69). Для пентоз брожение не харак¬
терно.
§ 156. Стереохимия моносахаридов. Присутствие нескольких асимметрических
атомов углерода создает условия для существования большого числа стерео-
изомерных моносахаридов. Так, глюкоза — один из членов обширного семей¬
ства шестнадцати альдогексоз C6Hi206. Арабиноза — одна из восьми стерео-
изомерных альдопентоз.
Обозначение, конфигурации буквами D и L нам уже знакомо. Однако
в сложных веществах с несколькими асимметрическими атомами обозначение
L или D относится только к одному «ключевому» асимметрическому атому.
. Так, например, название «О-глюкоза» показывает лишь, что гидроксил у ниж¬
него асимметрического атома углерода находится в проекционной формуле
справа, о конфигурации же остальных трех асимметрических атомов в на¬
звании никаких указаний нет.
Иная система обозначения конфигурации асимметрического атома пред¬
ложена в начале 50-х годов А. П. Терентьевым с сотр.; сходные предложения
сделали за рубежом Кан, Ингольд, Прелог. Обе системы описывают не про¬
екционную формулу, а непосредственно пространственную структуру, т. е.
объемную модель молекулы. Этот принцип положен в основу стереохимиче-
ской номенклатуры IUPAC.
Прежде всего определяют старшинство заместителей у асимметрического
центра, руководствуясь правилом: старшими являются заместители с боль¬
шим атомным номером. Как пример рассмотрим глицериновый альдегид, за¬
писав его формулу следующим образом:
Согласно атомным номерам атомов, непосредственно связанных с асим¬
метрическим центром, видно, что ОН-группа, содержащая атом кислорода с
атомным номером кислорода 8, — самая старшая, атом водорода—самый
младший. Группы , же СНО и СН2ОН связаны с асимметрическим центром
атомами углерода: для этого, чтобы расставить их по старшинству, обра¬
щаются к атомам «второго слоя». При этом у каждого из заместителей в
скобках записывают тройки связанных с ним атомов «второго слоя». Рас¬
полагают их по уменьшающемуся старшинству, причем связанный двойной
связью атом, в данном случае кислород альдегидной группы, записывается
дважды:
Таким образом видно, что группа СНО имеет во втором слое атомы с
большими атомными номерами и, следовательно, она старше, чем группа
СН2ОН.
По старшинству атомных номеров заместители у асимметрического атома
глицеринового альдегида располагаются в ряд: ОН > СНО > СН2ОН > Н.
Для определения стереохимического обозначения модель располагают так,
чтобы младший заместитель у асимметрического атома был наиболее удален
от наблюдателя, остальные же три заместителя находились в обращенном к
наблюдателю основании тетраэдра: при убывании старшинства групп против
часовой стрелки конфигурацию обозначают знаком 'S, при убывании стар-
/ ' \ \
! н—с—О-гН
V I / I
группа СН2ОН: С(0, Н, Н) группа СНО: С (О, О, Н)
344
шинства групп по часовой стрелке — знаком R, например:
НОН2С^
ОНС
но—'с—н
но—с—н
ОНС
(+)-глицериновый
альдегид
^-конфигурация;
^'-конфигурация;
(—)-глицериновый
альдегид
сно
сно
I
н—он
но
н
СН2ОН
СН2ОН
ОЛИГОСАХАРИДЫ
§ 157. Строение и свойства. Соединения, построенные из не¬
большого числа остатков моносахаридов (2—6), называют оли¬
госахаридами. Важнейшими из олигосахаридов являются диса¬
хариды. Они могут быть весьма разнообразными по строению:
в них могут входить остатки разных моносахаридов; в создании
связи между этими остатками могут принимать участие разные
гидроксильные группы. В молекулы дисахаридов входят цик¬
лические формы моносахаридов: они могут быть пиранозными
и фуранозными, с а- или p-конфигурацией гликозидного гид¬
роксила.
Известны два типа дисахаридов, различие между которыми
зависит от того, участвуют ли в создании связи оба гликозид-
ных гидроксила или один гликозидный и один спиртовой.
Примером дисахаридов первого типа может служить саха¬
роза (обычный сахар), состоящая из остатков a-D-глюкозы и
Р-7)-фруктозы. В дисахаридах такого типа нет свободных гли-
Н
ОН Н
сахароза
целлобиоза
345
козидных гидроксилов. Они не способны переходить в таутомер¬
ные альдегидные формы, а следовательно, не могут вступать в
свойственные этой форме реакции. В частности, дисахариды
гликозидо-гликозидного типа не дают столь характерной для
моносахаридов реакции серебряного зеркала, не восстанавли¬
вают фелингову жидкость. Поэтому их называют невосстанав¬
ливающими дисахаридами. Они способны вступать лишь в ре¬
акции, обусловленные присутствием гидроксильных групп (ал¬
килирование, ацилирование).
Второй тип дисахаридов — это дисахариды гликозидо-гли-
козного типа (восстанавливающие дисахариды). К ним отно¬
сятся важнейшие природные продукты — целлобиоза, мальтоза,
лактоза. Целлобиоза (формулу см. выше) и мальтоза состоят
из остатков глюкозы и отличаются друг от друга тем, что в
молекуле целлобиозы остатки моносахаридов связаны р-глико-
зидными связями, а в молекуле мальтозы — а-гликозидными.
В состав дисахарида лактозы (молочный сахар) входят остатки
глюкозы и галактозы.
остаток остаток
ос-0-глюкозы a-ZJ-глюкозы
остаток
|3-/?-галактозы
мальтоза
лактоза
У всех дисахаридов этого типа имеется свободный гликозид-
ный гидроксил (в формулах он отмечен ззездочкой), следова¬
тельно, они могут переходить в альдегидную форму, отсюда и
восстановительные свойства этих дисахаридов. Восстанавливаю¬
щие дисахариды вступают в те же химические реакции, что и
моносахариды, они способны образовывать алкильные и ациль¬
ные производные, окисляться до монокарбоновых кислот (типа
глюконовой), давать озазоны, реакцию серебряного зеркала,
выделять оксид меди(1) из фелинговой жидкости.
Характерным для всех дисахаридов (как и олигосахаридов
и полисахаридов вообще) является способность при кислотном
или ферментативном гидролизе превращаться в моносахариды.
846
Гидролиз сахарозы сопровождается изменением знака оптиче¬
ского вращения: правовращающая сахароза превращается в ле¬
вовращающую смесь глюкозы и фруктозы. Этот процесс часто
называют инверсией.
Важнейший из дисахаридов — сахароза — очень распростра¬
нен в природе. Это химическое название обычного сахара, на¬
зываемого также тростниковым, или свекловичным. Получают
сахарозу из сахарного тростника, произрастающего в тропиках;
(на о-ве Куба и в других странах Центральной Америки) или
из сахарной свеклы (содержит 12—15 % сахарозы).
Сахарную свеклу измельчают и извлекают из нее сахарозу горячей водой
в специальных аппаратах — диффузорах. Полученный раствор обрабатывают
известью для осаждения примесей, а перешедший частично в раствор избы¬
точный гидроксид кальция осаждают пропусканием диоксида углерода. Да¬
лее после отделения осадка раствор упаривают в вакуум-аппаратах. Так по¬
лучается мелкокристаллический песок — сырец. После его дополнительной
очистки получают рафинированный (очищенный) сахар. В зависимости от
условий кристаллизации он выделяется либо в виде мелких кристаллов, либо
в виде компактных «сахарных голов», которые раскалывают или распиливают
на куски. Быстрорастворимый кусковой сахар готовят прессованием мелкоиз-
мельченного сахарного песка.
ПОЛИСАХАРИДЫ
Молекулы полисахаридов можно рассматривать как продукты
конденсации большого числа молекул моносахаридов друг с
другом. Состав их выражается формулой (C6Hio05)n; молеку¬
лярные массы природных полисахаридов составляют от не¬
скольких тысяч до нескольких миллионов. Важнейшие из поли¬
сахаридов — крахмал и целлюлоза.
§ 158. Крахмал. Крцхмал образуется в растениях при фотосин¬
тезе и откладывается в корнях, клубнях и семенах. Зерна рис-а,
пшеницы, ржи и других злаков содержат 60—80 % крах¬
мала, клубни картофеля 15—20%. В животном мире роль
«запасного углевода» играет родственный крахмалу полисахарид
гликоген, откладывающийся в основном в печени. Внешний вид
крахмала хорошо известен: это белое вещество, состоящее
из мелких зерен. Крахмал нерастворим в холодной воде, набу¬
хает и постепенно растворяется в горячей. Образующиеся вязкие
растворы при понижении температуры превращаются в студне¬
видную массу — клейстер.
Крахмал представляет собой смесь полисахаридов — амило-
пектина и амилозы. Применяя особую обработку растворителя¬
ми, из крахмала можно выделить чистую, кристаллическую
амилозу. Она не образует при набухании клейстера, с иодом
дает характерное темно-синее окрашивание; амилопектин от¬
ветственен за образование клейстера, а с' иодом дает лишь
слабое фиолетовое окрашивание. И амилоза, и амилопектин
состоят из остатков глюкозы, связанных сс-гликозидными свя¬
зями, однако они различаются формой молекул. Амилоза пред¬
347
ставляет собой линейный полисахарид, построенный из несколь¬
ких тысяч остатков глюкозы, соединенных а-гликозидной
связью. Строение амилозы схематически выражается следую¬
щей формулой:
По данным рентгеноструктурного анализа, молекула амило¬
зы свернута в спираль. Внутри спиралевидной молекулы оста¬
ется канал диаметром около 0,5 нм, в котором могут распола¬
гаться подходящие по размеру молекулы, образуя особого типа
комплексы — так называемые соединения включения (см.
§ 115). Одним из них является упомянутое ранее соединение
амилозы с иодом.
Молекула амилопектина, в отличие от амилозы, имеет раз¬
ветвленное строение, приближающееся к шарообразной форме.
Определенная физическими методами молекулярная масса ами¬
лопектина составляет ж 10®, следовательно,, степень полимери¬
зации равна примерно 6000.
Применение крахмала. Ферментативный гидролиз
крахмала имеет промышленное значение в производстве этило¬
вого спирта из зерна или картофеля (см. § 69). Процесс начи¬
нается с превращения крахмала в глюкозу, которую затем
сбраживают. Используя специальные культуры дрожжей и из¬
меняя условия, можно направить брожение и в сторону полу¬
чения бутилового спирта, ацетона, молочной, лимонной и
глюконовой кислот и др.
Подвергая крахмал гидролизу кислотами, можно получить
глюкозу в виде чистого кристаллического препарата или в виде
патоки — окрашенного некристаллизующегося сиропа.
Наибольшее значение крахмал имеет в качестве пищевого
продукта': в виде хлеба, картофеля, круп, являясь главным ис¬
точником углеводов в нашем рационе питания. Кроме того,
чистый крахмал применяют в пищевой промышленности в про¬
изводстве кондитерских и кулинарных изделий, колбас. Значи¬
тельные количества крахмала употребляют для проклеивания
(шлихтования) тканей, склеивания бумаги и картона, производ¬
ства канцелярского декстринового клея.
В аналитической химии крахмал служит индикатором в
иодометрическом методе титрования. Для этих целей лучше
применять очищенную амилозу; ее растворы не загустевают, а
образуемая с иодом окраска более интенсивна.
348
§ 159. Целлюлоза. Целлюлоза (клетчатка) представляет собой
полисахарид, построенный из глкжозных звеньев. Ее строение
напоминает строение амилозы, т. е. она тоже имеет линейное
строение, но остатки глюкозы связаны между собой р-глико-
зидными связями, а не а-гликозидными, как в амилозе.
Молекулярная масса целлюлозы очень велика — порядка
500 тыс., она может достигать и нескольких миллионов, т. е. в
приведенной формуле п равно десяткам тысяч.
Целлюлоза — главный «строительный материал» растений,
из которого состоят стенки растительных клеток. Например,
волокна хлопчатника, хорошо известные всем в форме ваты,
на 98 % состоят из целлюлозы. Целлюлоза не плавится и не
переходит в парообразное состояние: при нагревании примерно
до 350 °С она разлагается — обугливается. Она нерастворима
в воде и большинстве других неорганических и органических
растворителей.
Неспособность целлюлозы растворяться в воде—неожидан¬
ное свойство для вещества, содержащего по три гидроксильные
группы на каждые шесть атомов углерода.. Хорошо известно,
что полигидроксильные соединения легко растворяются в воде.
Нерастворимость целлюлозы объясняется тем, что ее волокна
представляют собой как бы «пучки» расположенных парал¬
лельно нитевидных молекул, связанных множеством водородных
связей, которые образуются в результате взаимодействия гид¬
роксильных групп. Внутрь подобного «пучка» растворитель про¬
никнуть не может, а следовательно, не происходит и отрыва
молекул друг от друга.
Целлюлоза растворима в растворе гидроксида меди в кон¬
центрированном водном аммиаке (реактив Швейцера). Кон¬
центрированные кислоты (серная, фосфорная) и концентриро¬
ванный раствор хлорида цинка также растворяют целлюлозу, но
при этом происходит ее частичный распад (гидролиз), сопро¬
вождающийся уменьшением молекулярной массы.
Химические свойства целлюлозы определяются прежде
всего присутствием гидроксильных групп. Действуя метал¬
лическим натрием, можно получить алкоголят целлюлозы
[СбН702 (ONa) 3J п. Под действием концентрированных водных
растворов щелочен происходит так называемая мерсеризация —
частичное образование алкоголятов целлюлозы, приводящая к
349
набуханию волокна и повышению его восприимчивости к краси¬
телям. В результате окисления в макромолекуле целлюлозы
появляется некоторое число карбонильных и карбоксильных
групп. Под влиянием сильных окислителей происходит распад
макромолекулы. Гидроксильные группы целлюлозы способны
алкилироваться и ацилироваться, давая простые и сложные
эфиры.
Применение целлюлозы. Целлюлоза используется
человеком с очень давних времен. Сначала применяли древе¬
сину как горючий и строительный материал; затем хлопковые,
льняные и другие волокна стали использовать как текстильное
сырье. Первые промышленные способы химической переработки
древесины возникли в связи с развитием бумажной промыш¬
ленности.
Бумага — это тонкий слой волокон клетчатки, спрессованных
и проклеенных для создания механической прочности, гладкой
поверхности, для предотвращения растекания чернил. Первона¬
чально для изготовления бумаги употребляли растительное
сырье, из которого чисто механически можно было получить
необходимые волокна, стебли риса (так называемая рисовая
бумага), хлопка, использовали также изношенные ткани. Одна¬
ко по мере развития книгопечатания перечисленных источников
сырья стало не хватать для удовлетворения растущей потреб¬
ности в бумаге. Особенно много бумаги расходуется для печа¬
тания газет, причем вопрос о качестве (белизне, прочности,
долговечности) для газетной бумаги значения не имеет. Зная,
что древесина примерно на 50 % состоит из клетчатки, к бу¬
мажной массе стали добавлять размолотую древесину. Такая
бумага непрочна и быстро желтеет (особенно на свету).
Для улучшения качества древесных добавок к бумажной
массе были предложены различные способы химической обра¬
ботки древесины, позволяющие получить из нее более или ме¬
нее чистую целлюлозу, освобожденную от сопутствующих ве¬
ществ— лигнина, смол и др. Для выделения целлюлозы было
предложено несколько способов, из которых мы рассмотрим
сульфитный.
По сульфитному способу измельченную древесину «варят»
под давлением с гидросульфитом кальция. При этом сопутст¬
вующие вещества растворяются, и освобожденную от примесей
целлюлозу отделяют фильтрованием. Образующиеся сульфит-
ные щелока являются в бумажном производстве отходами. Од¬
нако вследствие того, что они содержат наряду с другими
веществами способные к брожению моносахариды, их использу¬
ют как сырье для получения этилового спирта (так называемый
гидролизный спирт, см. § 69).
Целлюлоза применяется не только как сырье в бумажном
производстве, но идет еще и на дальнейшую химическую пере¬
работку. Наибольшее значение имеют простые и сложные
S50
эфиры целлюлозы. Так, при действии на целлюлозу смесью
азотной и серной кислот получают нитраты целлюлозы. Все они
горючи и взрывчаты. Максимальное число остатков азотной
кислоты, которые можно ввести в целлюлозу, равно трем на
каждое звено глюкозы:
НЫОэ
[С6Н,02(0Н)3]ч ► [C6H702(0N02).,]„
Продукт полной этерификации — тринитрат целлюлозы (три-
нитроцеллюлоза) — должен содержать в соответствии с форму¬
лой 14,1 % азота. На практике получают продукт с несколько
меньшим содержанием азота (12,5—13,5%), известный в тех¬
нике под названием пироксилин. При обработке эфиром
пироксилин желатинизируется; после испарения растворителя
остается компактная масса. Мелконарезанные кусочки этой
массы — бездымный порох.
Продукт нитрования, содержащий около 10 % азота, отве¬
чает по составу динитрату целлюлозы: в технике такой продукт
известен под названием коллоксилин. При действии на него сме¬
си спирта и эфира образуется вязкий раствор, так называемый
коллодий, применяемый в медицине. Если к такому раствору
добавить камфору (0,4 ч. камфоры на 1 ч. коллоксилина) и
испарить растворитель, то останется прозрачная гибкая плен¬
ка — целлулоид. Исторически — это первый известный тип пла¬
стмассы. Еще с прошлого века целлулоид получил широкое
применение как удобный термопластичный материал для произ¬
водства многих изделий (игрушки, галантерея и т. д.). В осо¬
бенности важно использование целлулоида в производстве кино¬
пленки и нитролаков. Серьезным недостатком этого материала
является его горючесть, поэтому в настоящее время целлулоид
все чаще заменяют другими материалами, в частности ацета¬
тами целлюлозы.
При действии на целлюлозу смеси уксусного ангидрида, ук¬
сусной кислоты и серной кислоты или хлорида цинка (послед¬
ние играют роль катализатора) образуется триацетат целлю¬
лозы:
(СН3С0)20
[С6Н702(0Н)з)„ ь [СвН702(ОСОСН3)з]*
Неполное ацетилирование целлюлозы или частичный гидро¬
лиз триацетата приводят к вторичному ацетату (2,4—2,7 остат¬
ков уксусной кислоты на элементарное звено), условно называе¬
мому диацетатом. Из ацетатов целлюлозы готовят лаки, негорю¬
чую кинопленку, а также ацетатное волокно.
Если посмотреть под микроскопом волокна главных природ¬
ных текстильных материалов — хлопка, шерсти и натурального
шелка, то обращает на себя внимание различие между первы¬
ми двумя и шелком. Волокна хлопка и шерсти имеют «мохна¬
тую» поверхность. Волокна шелка — более гладкие, отсюда
блеск и плотность шелковых тканей. Подметив это, уже давно
351
пытались создать искусственный шелк, изменяя характер по¬
верхности целлюлозных волокон.
Для получения волокна триацетат целлюлозы растворяют в
смеси дихлорметана и этилового спирта, а диацетат — в смеси
ацетона с водой и затем продавливают этот раствор (формова¬
ние волокна) через сосуд с тонкими отверстиями — фильеру.
Вытекающие тончайшие струйки при испарении растворителя
(сухое прядение) превращаются в очень тонкие нити, которые
далее скручивают в более толстую, уже пригодную для ткаче¬
ства нить ацетатного шелка. Этот вид искусственного волокна
обладает рядом преимуществ по сравнению с другими искус¬
ственными волокнами, например с вискозным (см. ниже). По¬
этому его производство в последние годы успешно развивается.
Очевидно, что общий принцип, лежащий в основе получения
ацетатного волокна (растворение, затем формование ни1-ей),
можно осуществлять и по-иному. Большое техническое значение
имеет вискозный способ. Сущность этого способа — образование
растворимого в воде соединения целлюлозы при действии на
нее сероуглерода и щелочи:
Образовавшееся соединение представляет собой натриевую
соль сложного эфира дитиоугольной (ксантогеиовой) кислоты
и целлюлозы. Это соединение называют ксантогенатом целлю¬
лозы. Водный (точнее, щелочной) раствор ксантогената целлю¬
лозы (вискоза) продавливают через фильеры в прядильную
ванну, содержащую серную кислоту, сульфаты натрия и цинка
и воду (так называемое мокрое прядение). Под действием кис:
лоты ксантогенатные группы отщепляются и регенерируется
целлюлоза.
Такая же нить, но несколько более толстая и «нарубленная»
на мелкие куски представляет собой штапельное волокно, из
которого получают ткани, заменяющие хлопчатобумажные. Если
вискозу вместо фильер продавливать через узкие щели, полу¬
чается прозрачная пленка — целлофан.
Было предложено получать искусственное волокно и из рас¬
творов целлюлозы в аммиачном растворе гидроксида меди.
Получаемое таким способом медноаммиачное волокно обладает
хорошим качеством, однако стоимость его высока.
Нашли техническое применение и простые эфиры целлю¬
лозы. Так, обрабатывая целлюлозу сначала щелочью, а затем
метилхлоридом (под давлением), получают метилцеллюлозу:
Метилцеллюлоза обладает некоторой растворимостью в во¬
де; применяется главным образом как загуститель (вместо
NaOH, СН3С1
[СвН,0,(0Н),]п
+■ [С6Н702(0Н) (ОСНзЫ*
352
крахмала) в текстильной, косметической и пищевой промыш¬
ленности. Аналогично получают этилцеллюлозу, которую ис¬
пользуют для производства прочных морозостойких пленок.
Искусственные волокна на основе целлюлозы ныне занимают
видное место в общем балансе текстильного сырья.
ГЛАВА 20
АМИНОСПИРТЫ И АМИНОКИСЛОТЫ
Аминоспиртами называют соединения, в молекуле которых од¬
новременно содержатся амино- и гидроксильная группы, ами¬
нокислотами— соединения с аминной и карбоксильной функ¬
циями.
АМИНОСПИРТЫ И АМИНОФЕНОШ
§ 160. Аминоспирты. Важнейшие представители этого, класса —
этаноламины (гидроксиэтиламины) — получают в промышлен¬
ности из аммиака и этиленоксида:
О °
/ \ н2с^Ьн2
Н2С СН2 + Ш3 —► HOCHs—CHjNHj *
этаноламин
о
/ \
Н2С—сн2
—► (HOCH2CH2)2NH >- (HOCH2CH2)3N
диэтаноламин триэтаноламин
Нитроспирты, получаемые конденсацией нитросоединений с
альдегидами и кетонами (см. § 123), при восстановлении обра¬
зуют аминоспирты:
CH3N02 Hj/NI
н2с=о ► носн2—ch2no2 ► носн2—ch2nh2
Этаноламины — высококипящие жидкости, смешивающиеся с
водой во всех отношениях. Их применяют в качестве раствори¬
телей. Присутствие аминогруппы придает всем этим соедине¬
ниям свойства оснований. Триэтаноламин образует с кислотами
соли, например:
(HOCH2CH2)3N + H2S —► (HOCH2CH2)3N-H2S
При нагревании эта соль разлагается. Этот способ исполь¬
зуют для очистки различных промышленных газов от сероводо¬
рода. Образующийся сероводород можно использовать, например,
как сырье для производства серной кислоты, а регенерирован¬
ный триэтаноламин вновь применить для очистки газа. Анало-
12 Органическая химия
353
гично триэтаноламин связывает диоксид углерода и циановодо-
род, что также применяется в газоочистке.
Конденсацией этаноламина с высшими карбоновыми кисло¬
тами получают iV-гидроксиэтиламиды, которые являются компо¬
нентами неионогениых моющих средств:
С,7Н16СООН + H2NCH2CH2OH ——*- C,7H3sCO—NHCH2CH2OH
При замене в диэтаноламине гидроксильных групп на гало¬
ген образуется азотистый аналог иприта (C1CH2CH2)2NH. Со¬
единения такого типа привлекли к себе внимание исследовате¬
лей как препараты, перспективные для лечения злокачествен¬
ных опухолей.
К числу важных физиологически активных веществ отно¬
сится аминоспнрт холин. По своему строению это четвертичное
аммониевое основание [НОСН2СН2—N(CH3)3]+ [ОН]-. Холин
входит в состав фосфатидов (см. § 113). Исходя из способности
холнна участвовать в регулировании обмена веществ, синтетиче¬
ский холин стали использовать в животноводстве и птицевод¬
стве как добавку к кормам. При дегидратации холина об¬
разуется весьма ядовитый непредельный амин — нейрин
[СН2=СН—N(CH3)3]+ [ОН]-. Процесс протекает, в част¬
ности, при гниении белков.
Некоторые аминоспирты — производные бензола, у которых
аминогруппа и гидроксигруппа находятся в боковой цепи, от¬
носятся также к числу важных физиологически активных ве¬
ществ. Например, эфедрин, содержащийся в некоторых видах
растений, применяют как лекарственное средство, возбуждаю¬
щее центральную нервную систему и сужающее сосуды. В его
молекуле имеются два разных асимметрических атома углерода
и поэтому он существует в виде четырех (22) пространственных
изомеров. В природе встречается левовращающий изомер.
Адреналин играет в организме животных и человека роль
гормона, регулирующего важные жизненные процессы (обмен
углеводов, сердечную деятельность). По строению он сходен с
эфедрином, но у него имеются гидроксильные группы и в ядре.
Природный адреналин представляет собой левовращающий изо¬
мер. Интересно, что его правовращающий антипод в 15 раз
слабее по физиологическому действию.
§ 161. Аминофенолы. Аминофенолами называют ароматические
соединения — производные бензола, содержащие в ядре гидрок-
НСК
эфедрин
адреналин
854
си- и аминогруппы. Простейший аминофенол имеет три изомера:
1,2- или
о-аминофенол
(т. пл 174 °С)
1,3- или ж-амино-
фенол
(т. пл. 123 °С)
1,4- или ге-амино'
фенол
(т. пл. 184 °С)
Получают о- и n-аминофенолы восстановлением соответству¬
ющих нитрофенолов, которые в свою очередь легко образуются
при нитровании фенола. Синтез ж-аминофенола более сложен.
Две электронодонорные группы, имеющиеся в аминофенолах,
повышают электронную плотность в ядре, что увеличивает его
реакционную способность. Легко окисляясь, аминофенолы дей¬
ствуют на многие вещества как восстановители. Так, например,
они выделяют металлическое серебро из его солей, поэтому
аминофенолы используют в фотографии в качестве проявителей.
Для этой цели применяют, например, метол (4-метиламинофе-
нол) или амидол (2,4-диаминофенол):
метол амидол
Более сложные соединения того же типа служат красителя¬
ми, например дисперсный желтый прочный 2К (краситель для
ацетатного и полиамидного волокон), и полупродуктами их син¬
теза, например 8-амино- 1-гидроксинафталин (8-аминонафтол-1).
дисперсный желтый прочный 2К
НО NH4
8-аминонафтол-1
Кроме амино- и гидроксигрупп в этих соединениях часто
имеются нитро- и сульфогруппы.
Многие производные фенолов, содержащие аминогруппу как
в ядре, так и в боковой цепи, обладают физиологической актив¬
ностью. Среди них имеется лекарственный препарат фенацетин
(4-ацетиламино-] -этокси бензол) —жаропонижающее средство;
синтетическое сладкое вещество дульцин (4-этоксифенилмочеви¬
на; примерно в 500 раз слаще сахара), являющееся производ¬
ным мочевины; тирамин ||J-(4-гидроксифенил)этиламин]—
ядовитое вещество, содержащееся в спорынье — грибке, парази-
12*
355
тирующем на злаках (рожь, пшеница)
ОС*Н» ОС2Н5
ОН
NHCOCH3
фенацетин
NHCONHj
CH2CH2NH,
дульцин
тирамин
АМИНОКИСЛОТЫ
§ 162. Физические свойства и способы получения. В зависи¬
мости от взаимного положения обеих функциональных групп
различают а-, (3- и у-аминокислоты. Простейшие аминокислоты
содержат одну амино- и одну карбоксильную группы; такие со¬
единения называют моноаминомонокарбоновыми кислотами. Из¬
вестны, однако, аминокислоты, содержащие в молекуле две
аминогруппы и одну карбоксильную группу,— диамино кар боно¬
вые кислоты, одну аминогруппу и две карбоксильные группы —
аминодикарбоновые кислоты и т. д.
Названия аминокислот строят по уже известному нам прин¬
ципу: к названию соответствующей карбоновой кислоты добав¬
ляют приставку амино. Широко распространены и тривиаль¬
ные названия важнейших аминокислот. Приведем несколько
примеров:
Одновременное присутствие в молекуле аминокислоты кис¬
лотной и основной групп приводит к внутримолекулярной ней¬
трализации. Учитывая это, свободные аминокислоты правильнее
представлять себе в виде внутренних солей типа
Это сказывается и на физических свойствах аминокислот;
подобно обычным неорганическим солям, аминокислоты пред¬
ставляют собой кристаллические вещества, растворимые в воде
и мало растворимые в органических растворителях. Они плавят¬
ся при высоких температурах и обычно при этом разлагаются.
NH2
I
h2n—сн2—сн2—соон
Р-аминопропионовая
(2-аминопропановая)
кислота (Р-аланин)
а-аминО'Р-фенилпропионовая
С6Н5—СН2—СН—СООН
кислота (фенилаланин)
NH2
СНз NH2
I I
сн3—сн—сн—СООН
I
НООС—СН2—СН—соон
a-а миноянтарная
а-аминоизовалериановая
кислота (валин)
(аспарагиновая) кислота
356
Переходить в парообразное состояние аминокислоты не спо¬
собны.
Разработано много разнообразных способов синтеза амино¬
кислот.
1. Замена галогена в галогензамещенных кислотах на ами¬
ногруппу при действии избытка аммиака (способ разработан на
рубеже XIX и XX веков Э. Фишером):
С1СН2—СООН + 3NHS —*• HSN—СНа—COONH4 + NH4CI
Из соответствующих галогензамещенных кислот по этому
способу можно получать аминокислоты любого строения. Основ¬
ная трудность процесса — не проведение самой реакции, а от¬
деление образовавшейся аминокислоты от побочного продук¬
та — хлорида аммония. В настоящее время очистку чаще всего
производят с помощью ионообменных смол.
2. Действие цианида аммония (он ведет себя как смесь
синильной кислоты и аммиака NH4CN NH3 + HCN) на аль¬
дегиды и кетоны дает возможность получать а-аминокислоты
(синтез Н. Д. Зелинского). Процесс идет через промежуточное
образование циангидрина, который затем превращается в ами-
нонитрил, дающий при гидролизе а-аминокислоту:
О ОН NH2
]| HCN I NH3 I Н20 (н+)
R—С—Н ► R—СН—CN ► R—СН—CN ►
NH,
—► R—£н—СООН
3. Присоединение аммиака к а,р-непредельным кислотам;
при этом образуются р-аминокислоты:
NH3
СН4=СН—СООН H2N—СН2—СН2—СООН
Такой порядок присоединения (против правила Марковнико-
ва) предопределяется поляризацией двойной связи под влияни¬
ем карбоксильной группы и сопряжением (см. § 99).
4. Действие альдегидов и аммиака на малоновую кислоту
(синтез р-аминокислот по В. М. Родионову). В первой стадии
этой реакции идет конденсация типа альдольной, в которой
роль метиленового компонента играет малоновая кислота с ее
подвижными водородами СН2-группы. В продукте конденсации
действием аммиака гидроксигруппа заменяется на аминогруппу
(см. способ 2, синтез а-аминокислот по Зелинскому), образо¬
вавшийся промежуточный продукт декарбоксилируется.
О ОН СООН
л I II NHj
СвН„—С—Н + НООС—СН2—СООН —>• СвИ5-СН—СН—СООН ►
NHj СООН NH2
—»- cgh5—<!;н—сн—соон с0н5—сн—сн2—соон
—С02
357
5. Восстановление нитрозамещенных кислот. Этот путь имеет
наибольшее значение для синтеза ароматических аминокислот:
СООН
hno3
- >-
СООН
[Н]
h2n.
Г
СООН
Приведенные способы — лишь небольшая часть из числа
разнообразных путей, предложенных для синтеза аминокислот.
§ 163. Химические свойства. Как и у других гетерофункциональ-
ных соединений, свойства аминокислот в первом приближении
являются суммой свойств имеющихся в их составе функций.
Своеобразие аминокислот определяется прежде всего тем,
что в них имеются две функции противоположного химического
характера — аминогруппа со свойствами основания и карбок¬
сильная группа с кислотными свойствами. Подобно таким неор¬
ганическим веществам, как гидроксиды алюминия или цинка,
аминокислоты являются соединениями амфотерными, сочетаю¬
щими в себе свойства и кислот, и оснований. Амфотерность
аминокислот позволяет им образовывать соли как с кислотами,
так и с основаниями:
Я R
H2N—Ан—СООН + NaOH —> H2N—СН—COONa + Н20
Я Я
I ♦ I
HjN—СН—СООН + НС1 —> СГ H3N—СН—СООН
В других реакциях аминокислот также может участвовать
либо одна, либо другая их функциональная группа. Эти реакции
подобны реакциям аминов и кислот, поэтому их можно подроб¬
но не рассматривать, а ограничиться лишь рядом типичных
реакций, проведенных с аминоуксусной кислотой (глицином).
Реакции по карбоксильной группе:
СН3ОН, Н+ NH3
H2N—СН2-СООН — *■ H2N—СН2—СООСНз ►
—► H2N—сн2—conh2
нагревание
H2N—СН2—СООН *- H2N—сн3 + со2
Реакции по аминогруппе:
СН3СОС1
Н2Ы—СН2—СООН ► CH3CO—NH—СН2—СООН
сн31
H2N—СН2—СООН -—>- (CH3)2N—СН2—СООН
с6н5сно
H2N—СН2—СООН *■ CeH6CH=N—СН2—СООН
hno2
h2n—СН2—СООН “*■ НО—СН2—СООН
358
Как и у других гетерофункциональных соединений, свойства
аминокислот зависят от взаимного расположения амино- и
карбоксигрупп. Например, различно их отношение к нагре¬
ванию.
а-Аминокислоты образуют циклические амиды, построенные
из двух молекул а-аминокислот,— диоксопиперазины:
H2N
НО
\
;сн—
/ Х)Н
+
N:—сн^
NH,
-2Н2С>
\сн3
н3сх
^СН— с
HISr
Nl—НС
\
/
\
О
NH
СН3
а-аминопропионовая
кислота
3,6'Диметил-2,5-диоксо-
пинеразин
р-Аминокислоты легче других аминокислот теряют молекулу'
аммиака и превращаются в непредельные кислоты:
СНз—СН—СН2—СООН ——> СН3—СН=СН—СООН
j •—NH3
nh2
Р-аминомасляная кротоновая кислота
кислота
у-Аминокислоты образуют внутримолекулярные циклические
амиды — лактамы:
h2n—сн2—сн2—сн2—СООН
—н2о
н2с_сн2
в2с/ ^;с=о
NH
у-аминомасляная кислота лактам у-амино-
масляной кислоты
В отношении а-, р- и у-аминокислот к нагреванию мож¬
но заметить определенное сходство с соответствующими гидр-
оксикислотами (см. § 143).
§ 164. Отдельные представители. Важнейшими из аминокислот
являются а-аминокислоты. Они образуют мономерные звенья
белковых молекул (см. §175). Подробнее познакомимся с ними
в § 174, здесь упомянем лишь о простейшей из а-аминокислот
а-аминоуксусной кислоте (гликол, глицин) H2N—СН2—СООН.
Как и все аминокислоты, глицин представляет собой бесцветное
кристаллическое вещество, растворимое в воде. Его получают
из хлоруксусной кислоты и аммиака.
Аминокислоты, содержащие функции в бензольном или наф¬
талиновом ядре, используют как полупродукты при синтезе кра¬
сителей и лекарственных препаратов.
359
Аминобензойные кислоты. Из трех изомерных кислот наи¬
большее значение имеют о-аминобензойная (антраниловая) и
я-аминобензойная кислоты. Исходным веществом для их синте¬
за служит толуол, который нитрованием превращают в смесь
о- и /г-нитротолуолов, затем окислением — в о- и я-нитробензой-
ные кислоты, восстановление которых дает о- и я-аминобензой-
ные кислоты:
СНз СООН соон
no2 no2 nh2
пара
Из антраниловой кислоты получают азокраситель метило¬
вый красный, который используют как индикатор: в кислой
среде он имеет красный цвет, в нейтральной и щелочной —
желтый.
НООСч
(CH3)2N—^ ^—N=N—метиловый красный
Среди производных я-аминобензойной кислоты имеются
важные лекарственные препараты:
H2N——СО—ОС2Н5 н.м—
СО—OCH2CH2N(C2Hs)2
анестезин (этиловый эфир новокаин (эфир л-аминобензойной кислоты
я-аминобензойной кислоты) и диэтиламиноэтилового спирта)
Оба вещества обладают местным анестезирующим (обезбо¬
ливающим) действием. Они заменяют природный алкалоид кока¬
ин, использование которого опасно из-за развивающегося болез¬
ненного привыкания к его употреблению.
я-Аминосалициловая кислота — противотуберкулезный ле¬
карственный препарат. Ее получают из л-аминофенола и диок¬
сида углерода (реакция аналогична синтезу салициловой кис¬
лоты; см. § 145):
HjN.
ОН
С02, КНСОз
»-
H2N.
ОН
360
соон
ЧАСТЬ IV
ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ
ГЛАВА 21
ШЕСТИЧЛЕННЫЕ И ПЯТИЧЛЕННЫЕ
ГЕТЕРОЦИКЛЫ. АЛКАЛОИДЫ
Гетероциклами называют циклические соединения, в состав ко¬
лец которых кроме атомов углерода входят и атомы других
элементов — гетероатомы.
§ 165. Классификация и общая характеристика. Эти соединения
классифицируют по природе гетероатома, затем по числу гетеро-
атомов, числу звеньев в цикле и другим признакам.
Наибольшее значение имеют гетероциклические соединения,
в которых атомы N, О или S входят в состав шести- и пяти¬
членных колец. Эти соединения по строению могут быть анало¬
гами циклоалканов, но важнее формально непредельные гетеро¬
циклы, являющиеся аналогами ароматических соединений.
Ниже приведены формулы и названия важнейших гетероцик¬
лических ядер. Как показано на формулах, нумерация всегда
начинается с гетероатома. Для простых соединений используют
и другой способ: атомы обозначают греческими буквами — со¬
седние с гетероатомами — а, более удаленные — р и у:
4 V
5 СН
3 НС/ ЧСН р
II I3
а НС\ ^СН а
6 N 2
СН2
Н2С/ \сн2
Н2(к. /СН2
NH
5 4 V
6 СН СН
НС^ \С/ чсн 3
I II I3
HC'nn /С\ j^CH а
7 СН N 2
8 1
пиридин
Р НС_СН р
а НС^ ^СН а
5\ /2
NH
пиперидин
И2С_СН2
н2с(^ ^сн2
NH
5 сн сн р
НС^ \С/ \
I II /СН а
НС^ /С/ / 2
6 СН NH
7 1
пиррол
СН2
р НС/ Ч-СН р
а НС\ /СН а
О
пирролидии
индол
р НС_СН р
НС^ ^СН а
О
Н2С_СН2
н2с(^ ^СН2
О
пиран
фуран тетрагидрофуран
361
тиофен
тиофан
Гетероциклические ядра обычно изображают не полными
формулами, а условными схемами, подобно тому, как это
принято для ароматических соединений:
Подобно углеводородам, гетероциклические соединения ис¬
пользуют для получения разнообразных производных, имеющих
в молекулах различные заместители. Так, существуют гетеро¬
циклические галоген- и гидроксипроизводные, альдегиды и кето¬
ны, карбоновые и сульфокислоты, нитросоединения, амины
и т. д. Свойства гетероциклических соединений в общем сходны
со свойствами аналогичных нециклических или карбоцикличе-
ских соединений. Это существенно облегчает изучение гетеро¬
циклических соединений, превращая его во многом в повторение
уже пройденного. При изложении материала мы будем больше
обращать внимание на различия в свойствах, чем на сходство.
Своеобразие гетероциклическим соединениям придает тот
факт, что их ядро вносит свои особенности, которые нередко в
значительной степени и определяют свойства соединения в це¬
лом.
Химия гетероциклических соединений развивается в послед¬
нее время довольно быстро. Это объясняется тем, что к данно¬
му классу соединений относятся важные природные вещества,
ряд ценных лекарственных препаратов, красители и другие
практически важные вещества. Описанию гетероциклов посвя¬
щены многоатомные издания; в рамках данной книги мы можем
лишь кратко познакомиться с основами этой обширной области.
ШЕСТИЧЛЕННЫЕ ГЕТЕРОЦИКЛЫ С ОДНИМ ГЕТЕРОАТОМОМ
§ 166. Пиридин и его производные. Пиридин можно рассматри¬
вать как аналог бензола, в котором одна группа СН заменена
атомом азота:
О
NH
N
фуран
индол
хинолин
СН
НС^ ^СН
II
N
862
В написанной выше формуле пиридина мы изобразили три
двойные связи. Однако в действительности шесть л-электронов
этих двойных связей образуют ароматический секстет. Поэтому
пиридин, подобно бензолу, проявляет ароматические свойства;
он устойчив к окислению, вступает в реакции электрофильного
замещения.
Пиридин — жидкость с т. кип. 115 °С, обладающая харак¬
терным неприятным запахом, смешивается с водой в любых
отношениях. Пиридин и его ближайшие гомологи содержатся в
каменноугольной смоле, которая и служит их промышленным
источником. Их можно получить и синтетически.
Строение пиридина наиболее наглядно следует из синтеза,,
в котором исходными веществами служат ацетилен и синиль¬
ная кислота:
хн сн
нс^ сн нс^ \сн
hc%j + «ЦДн
По А. Е. Чичибабину, гомологи пиридина можно получать
конденсацией альдегидов с аммиаком (синтез имеет промыш¬
ленное значение). Так, из ацетальдегида был получен а-метил-
пиридин:
СН
\сн3
СНз О
СН + СН-СНз
1
О NH3
у
~зн2о, -н2
СН
НС^ "СН
НС^ Jc—СНз
N
а-метилпиридин
(а-пиколин)
В промышленности таким путем получают 2-метил-5-этил-
пиридин, а из него дегидрированием — 5-винил-2-метилпиридин,
который при совместной полимеризации с бутадиеном дает цен¬
ный синтетический каучук.
4СН3СНО
NH3
-н2о
НзО"'
С2Н5
-н2
N
НзС-^
СН=СН3
N
Как и амины, пиридин обладает свойствами основания, что
связано со свободной электронной парой, имеющейся у атома
азота. Однако пиридин—более слабое основание, чем алифати¬
ческие амины.
Химические свойства. Из-за наличия ароматического
секстета свойства пиридинового ядра во многом сходны со
363
свойствами бензола. Для пиридина, как и для бензола, харак¬
терны реакции электрофильного замещения. Он подвергается
галогенированию, нитрованию, сульфированию. При этом атом
азота пиридинового ядра играет роль заместителя второго рода,
направляющего вступающие группы в жега-положение и затруд¬
няющего (по сравнению с бензолом) реакции электрофильного
замещения:
.NOj
hno3
ci2
Cl
N
-Н20 У -НС1
N N
H2SO4
-н2о
N
S03H
Атомы С-2, С-4 и С-6 (а- и у-положения) обеднены электро¬
нами и способны вступать в реакции нуклеофильного замеще¬
ния. Одним из таких превращений является прямое аминирова-
ние пиридина (реакция Чичибабина), осуществляемое амидом
натрия или калия при нагревании в инертном углеводородном
растворителе (керосин, ксилол):
N N
у-аминопиридин
Однозамещенные гомологи пиридина могут существовать
в виде трех изомеров, аналогичных орто-, мета- и пара-азоме-
рам в ряду бензола, например:
СН3
а-метилпиридии p-метилпиридин у-метилпиридин
(а-пиколнн) (р-пиколин) (у-пиколин)
364
При действии окислителей гомологи пиридина ведут себя
аналогично углеводородам ряда бензола: затрагивается только
боковая цепь соединения, превращаясь в карбоксильную группу
(см. ниже).
В ядро пиридина можно вводить различные заместители:
галогены, гидрокси-, нитро-, сульфо- и аминогруппы. При этом
свойства соответствующих соединений сильно зависят от поло¬
жения заместителя в пиридиновом ядре. Как правило, (5-заме-
щенные пиридины наиболее похожи на соответствующие арома¬
тические соединения. Так, например, в p-хлорпиридине атом
галогена отличается малой реакционной способностью, что на¬
блюдается и у хлорбензола. Иные свойства присущи а- и
у-производным пиридина. В а- и у-хлорпиридинах атом гало¬
гена более подвижен, подобно тому, как это наблюдается у
о-и л-нитрохлорбензолов. Это становится понятным, если вспом¬
нить, что мезомерный эффект передает влияние заместителя в
орто- и лара-положения ароматического ядра. У пиридина этому
соответствуют а- и у-положения; они находятся под влиянием
стоящего в цикле атома азота, который ведет себя как замес¬
титель второго рода.
При каталитическом гидрировании пиридин превращается в
пиперидин, имеющий строение и свойства вторичного амина:
0-Q
N NH
Своеобразными свойствами обладают а- и у-амино- или
гидроксипиридины. Они ведут себя как вещества таутомерные,
например:
а-гидроксипиридин а-пиридон
Это доказывается тем, что в химических превращениях полу¬
чаются производные как гидроксипиридиновой, так и пиридоно-
вой форм:
Л^-метилпиридон-2 2-гидрокси- 2-метокси-
пиридин пиридин
Таутомерия подобного типа существует также у пиримиди¬
новых оснований, входящих в состав нуклеозидов (компонентов
нуклеиновых кислот).
365
Хорошо изучены пиридинкарбоновые кислоты:
соон
I
^/СООН
1
L JJ
^соон
N
и
N
сс-пиридин-
карбоновая
(пиколиновая)
кислота
р-пиридин-
карбоновая
(никотиновая)
кислота
у-пиридин-
карбоновая
(изоникотиновая)
кислота
Гидразид изоникотиновой кислоты (изониазид) применяется
как лекарственный препарат при лечении туберкулеза. Амид
никотиновой кислоты (витамин РР) является веществом, недо¬
статок которого вызывает заболевание пеллагру.
CONHNHj
6 crC0NH‘
N N
изониазид витамин РР
§ 167. Хинолин и его производные. Хинолин является гетеро*
циклическим аналогом нафталина. Он содержится в каменно¬
угольной смоле. Может быть получен и синтезом Скраупа — на¬
греванием смеси анилина, нитробензола, глицерина и серной
кислоты.Под действием серной кислоты происходит дегидрата¬
ция глицерина с образованием акролеина (см. § 88). Акролеин
присоединяется к аналину, затем идет конденсация образовавше¬
гося альдегида, причем отщепляется вода и образуется дигидро¬
хинолин, превращающийся под действием нитробензола (окис¬
литель) в хинолин:
Хинолин — малорастворимая в воде жидкость с неприятным
эапахом. По химическим свойствам хинолин напоминает пири¬
дин; он также является основанием, вступает в те же реакции,
что и пиридин. Меньшая реакционная способность пиридиново¬
го ядра по сравнению с бензольным проявляется при окислении
хинолина, в процессе которого разрушается бензольное ядро
866
^образуется хинолиновая кислота) г
ноос.
ноос
я
В реакциях нуклеофильного замещения заместитель всту¬
пает в пиридиновое кольцо, в реакциях электрофильного заме¬
щения — в бензольное:
При гидрировании хинолин, как и пиридин, превращается в
соответствующие насыщенные соединения, по химическим свой¬
ствам не отличающиеся от обычных алифатических аминов:
тетрагидро- декагидро¬
хинолин хинолин
8-Гидроксихинолин (кристаллическое вещество, т. пл. 75 °С)’
широко применяют как реактив на ионы многих металлов, о
которыми он образует внутрикомплексные соединения:
Производными хинолина являются цианиновые красители,
которые употребляют в качестве сенсибилизаторов — веществ,
придающих фотоматериалам чувствительность к длинноволно¬
вым излучениям (желтая, красная, инфракрасная части спек¬
тра). Представителем такого типа красителей является ксено-
цианин:
сн—сн=сн—сн=сн—сн=сн
367
ПЯТИЧЛЕННЫЕ ГЕТЕРОЦИКЛЫ С ОДНИМ
ГЕТЕРОАТОМОМ
§ 168. Строение и общие свойства. Простейшие пятичленные
гетероциклы — пиррол, фуран и тиофен. Все они проявляют
ароматические свойства. Причина ароматичности та же, что и
в бензоле, — наличие секстета подвижных электронов, но ха¬
рактер образования этого секстета иной. По одному электро¬
ну вносит в шестерку каждый из атомов углерода, еще два
электрона — свободная электронная пара гетероатома. Таким
образом, например, строение пиррола можно выразить схема¬
тично формулами:
Электронная пара пиррольного азота уже не имеет свойств
свободной электронной пары, которыми обусловлена основность
аминов и пиридина, она участвует в образовании электронного
секстета. Поэтому пиррольный азот почти лишен свойств осно¬
вания. Все же при действии сильных минеральных кислот
электронная пара азота «вытягивается» из шестерки, и пиррол,
утрачивая ароматичность, превращается в неустойчивое непре¬
дельное соединение, которое тут же полимеризуется. Так объ¬
ясняют осмоление пиррола при действии на него сильных ми¬
неральных кислот. Аналогично ведет себя и фуран. Тиофен кис¬
лотами не разрушается.
Ароматические свойства пятичленных гетероциклов проявля¬
ются в их способности к реакциям электрофильного замеще¬
ния— галогенированию, нитрованию, сульфированию, алкили¬
рованию и ацилированию по Фриделю — Крафтсу. Проще всего
эти реакции осуществить с тиофеном, поскольку он не разру¬
шается под действием кислот:
В случае фурана и пиррола дело осложняется тем, что под
действием кислот они разрушаются. В связи с этим при
сульфировании, например,, в качестве реагента используют
(А. П. Терентьев) триоксид серы, связанный в комплексное
НС—сн
NH
368
соединение с дноксаном или пиридином:
О
SO,H +
о
N - SO,
пиридинсуль-
фотриоксид
фуран-а-сульфо- диоксан
кислота
^~y~so3H +
NH
пиррол-а-
сульфокислота
N
пиридин
(о
(2)
Как видно из приведенных примеров, заместители вступают
преимущественно в «-положение пятичленных гетероциклов.
Родство пятичленных гетероциклов пиррола, фурана и тио¬
фена находит свое выражение в их взаимных превращениях
над катализатором АЬОз при 400—450 °С соответственно в то¬
ке H2S, NH3 и Н20 (реакция Ю. К. Юрьева):
0
*-HsS Г~\
н2о* \ / н2о’
0
5
0
NH
|
NH3
t|
H2S
§ 169. Пиррол и его производные. Пиррол получают из фурана
по реакции Юрьева (см. выше) или конденсацией ацетилена
с аммиаком:
НС сн
III+ 111
НС сн
NH,
нс_сн
нс\ >
NH
Известно и много других синтезов пиррольных соединений.
Пиррол — бесцветная подвижная жидкость, т. кип. 130 °С.
На воздухе он быстро окисляется и темнеет. Для обнаружения
пиррола часто используют его способность окрашивать в крас¬
ный цвет смоченную хлороводородной кислотой сосновую лу¬
чинку. Пиррол проявляет свойства очень слабой кислоты. Это
обнаруживается в способности водорода при атоме азота заме¬
щаться на металл, например при действии калия. Аналогичная
реакция происходит при действии магнийорганических соедине¬
ний:
0-^0 о^го
NH NK NH NMgl
359
iV-Пиррилкалий и Д^-пиррилмагнийгалогениды при действии
алкилгалогенидов дают ty-алкильные производные пиррола, ко¬
торые при нагревании перегруппировываются в С-алкильные
производные, например:
NMgl N—СН3
нагревание
►
СНэ
Индол представляет собой конденсированную бициклическую
систему, состоящую из бензольного и пиррольного ядер:
С
>
NH
Существует много способов получения соединений индоль-
ного ряда. Один из важнейших путей — синтез Э. Фишера. Ис¬
ходными веществами служат фенилгидразоны альдегидов и ке¬
тонов, которые нагревают с ZnCl2 или СиС12:
СН2
сн2—сн,
фенилгидразон Р-метилиндол
пропионового (скатол)
альдегида
Соединения ряда пиррола и индола часто встречаются в
природе. Многие синтетические препараты рядов пиррола и
индола обладают высокой физиологической активностью, при¬
меняются как лекарства, ростовые вещества и др. Так, ядра
пиррола входят в состав важнейших природных веществ — зе¬
леного красящего вещества растений — хлорофилла и красного
красителя крови — гемоглобина. Хлорофилл играет решающую
роль в фотосинтезе, гемоглобин—в дыхании животных. В осно¬
ве обоих соединений лежит структура из четырех пиррольных
ядер, схематическое строение которой можно изобразить так:
В схеме не указаны имеющиеся в молекуле двойные связи,
боковые цепи. В центре находится связанный с пиррольными
870
атомами азота металл: магний в хлорофилле, железо в гемо¬
глобине. Вся структура соединена еще с белковой частью, без
которой ни хлорофилл, ни гемоглобин не способны осуществ¬
лять свои биохимические функции.
Индолилуксусная кислота (гетероауксин) обладает способ¬
ностью ускорять рост растений, способствует черенкованию.
Она имеет следующее строение:
j^j—-j—сн2-соон
NH
Производным индола является одна из важных белковых
аминокислот — триптофан (см. § 174).
Краситель индиго также является производным индола. В те¬
чение многих веков его добывали из растений. В прошлом сто¬
летии Байеру удалось установить формулу индиго, после чего
был освоен и его промышленный синтез. Один из способов син¬
теза основан на использовании анилина и хлоруксусной кис¬
лоты:
Непосредственно окрашивать ткань с помощью индиго нельзя,
так как этот краситель нерастворим в воде. Поэтому действием
слабых восстановителей в щелочной среде индиго переводят
в раствор в виде бесцветного белого индиго:
С
С=0 NH
w ^
NH 0=С
4?
+211
—-2Н
С—ОН NH
. / %
NH НО—С
Л»
Этим раствором пропитывают ткань, на которой затем в ре¬
зультате окисления кислородом воздуха образуется индиго. Та¬
кой способ крашения называют кубовым крашением.
Производство индиго явилось одним из первых достижений
промышленного органического синтеза в прошлом веке. Инди¬
го выпускается и в наше время, но сам краситель потерял свой
титул «короля красок», так как получены другие красители,
дающие прочные красивые окраски. При крашении современны¬
371
ми кубовыми красителями используется принцип, применяемый
при крашении с помощью индиго.
§ 170. Фуран и его производные. Фуран получают декарбокси-
лированием пирослизевой кислоты:
^ у—соон v Р у + со2
о о
Он представляет собой бесцветную, подвижную, легкокипя-
щую жидкость (т. кип. 31 °С), которая на воздухе окисляется и
темнеет. Фуран легко разрушается кислотами. Поэтому реак¬
ции электрофильного замещения проводят так, чтобы предо¬
хранить его от разрушительного действия кислот: при сульфи¬
ровании используют диоксансульфотриоксид, при нитровании —
ацетилнртрат, при ацилировании в качестве катализатора при¬
меняют трифторид бора вместо более активного хлорида алю¬
миния:
Л—\ СНзСОО— N02
у—ш2
диоксан^ЭОз
->
\/
о
(CH3CO)2O.BF3
>
о
СОСН3
Реакции электрофильного замещения проходят в ядре фура-
на очень легко; по реакционной способности фуран можно срав¬
нить с фенолом. Азотная кислота окисляет фуран с разруше¬
нием цикла. При каталитическом гидрировании получается тет-
рагидрофуран, представляющий собой внутримолекулярный
простой эфир бутандиола-1,4. Как и все простые эфиры, это до¬
вольно инертное в химическом отношении соединение. Исполь¬
зуется как хороший растворитель.
9
Н2С_СН2
н2/ш / ч
>- н2с^ усн2
о
Наиболее доступное соединение фуранового ряда — альде¬
гид фурфурол образуется при нагревании пентоз с разбавлен¬
ными кислотами (см. § 155). Источником пентоз служат поли¬
сахариды пентозаны, содержащиеся в больших количествах в
различных продуктах растительного происхождения — отрубях,
шелухе подсолнечника, кукурузных початках. Переработкой
этих и других отходов сельского хозяйства в промышленности
получают большие количества фурфурола. Его используют в
производстве полимерных материалов, как растворитель в не¬
которых процессах переработки нефти, а также как исходное
вещество для синтеза других соединений ряда фурана.
372
Фурфурол — бесцветная жидкость, т. кип. 161 °С. Запах хле¬
ба обусловлен следами фурфурола, который при выпечке обра¬
зуется из содержащихся в муке пентоз. На воздухе фурфурол
легко окисляется, превращаясь сначала в бурую жидкость, а
затем в черную смолу. Он имеет все свойства альдегида: можно
получить его оксим, фенилгидразон и другие производные:
NH2OH // \
*■ у—CH=NOH
О
оксим фурфурола
CeH5NHNH2
■CH=N—NHCeHs
О
фенилгидразон фурфурола
Фурфурол вступает в реакцию Канниццаро:
—сно —СН2ОН + <^^
О О о
COONa
фуриловый натриевая соль
спирт пирослизевой кислоты
При восстановлении фурфурола образуется фуриловый
спирт, а при окислении — пирослизевая кислота.
Ароматические свойства фуранового ядра в фурфуроле про¬
являются в его способности вступать в реакции электрофильно¬
го замещения. Так, нитрованием фурфурола был получен 5-нит¬
рофурфурол и далее его семикарбазон:
ГУ-
'СНО
hno3
-*■ o2n
_/ л
H2N—NH—CONH2
семикарбаэид
сно >
о
о
—► 02N——CH=N—NHCONH2
О
Семикарбазон 5-нитрофурфурола под названием фурацилин
'(желтые кристаллы, т. пл. 227—232 °С) применяют как наруж¬
ное дезинфицирующее средство и при лечении дизентерии.
Реакциями электрофильного замещения являются также хло¬
рирование и меркурирование фурфурола:
о—Гу—сн° ч~ —сно HgCI*~ сш8—\/—сно
С> С> О
5-хлорфурфурол б-хлормеркурфурфурол
373
§ 171. Тиофен и его производные. Тиофен — самый устойчивый
из рассматриваемых пятичленных гетероциклов. Он не разру¬
шается кислотами. Это типичное ароматическое соединение: его
можно хлорировать, нитровать, получать кетоны по реакции
Фридела — Крафтса.
Тиофен имеет очень близкую к бензолу температуру кипения
(соответственно 84 и 80°С), поэтому эти вещества трудно отде¬
лить друг от друга перегонкой. Очистить бензол от тиофена
можно лишь химическим путем, воспользовавшись тем, что ти¬
офен сульфируется и меркурируется легче, чем бензол:
Интересна история открытия тиофена. Известный немецкий химик
В. Мейер демонстрировал на лекции цветную реакцию, которая считалась
характерной для бензола. Однажды эта реакция не удалась. Расследуя при¬
чины неудавшегося опыта, Мейер установил, что цветную реакцию дает лишь
бензол, выделенный из каменноугольной смолы, в то время как бензол, по¬
лученный декарбоксилированием бензойной кислоты, цветной реахции не дает.
На этом основании Мейер сделал вывод, что в бензоле каменноугольного
происхождения содержится какая-то примесь, которая и дает окрашивание.
В дальнейшем Мейеру удалось выделить содержащее серу вещество, кото¬
рое он назвал тиофеном.
1 Соединения ряда тиофена содержатся в значительных коли¬
чествах в некоторых нефтях. Присутствие сернистых соединений
в нефти весьма нежелательно; если их не удалить при перера¬
ботке, то при сгорании топлива будет образовываться диоксид
серы, который вызывает сильную коррозию. Поэтому нефть
подвергают обессериванию. Один из способов основан на рабо¬
тах Н. Д. Зелинского и заключается в каталитическом гидриро¬
вании (250 °С, 5 МПа) тиофеновых соединений с выделением
серы в виде сероводорода:
Содержание сернистых соединений в нефти может быть зна¬
чительным, поэтому большой интерес представляло бы не раз¬
рушение, а полезное их использование. Такие способы разраба¬
тываются.
СЛОЖНЫЕ ГЕТЕРОЦИКЛЫ И АЛКАЛОИДЫ
§ 172. Гетероциклы с несколькими гетероатомами. Среди гете¬
роциклов с несколькими гетероатомами встречаются важные
природные вещества; на основе этих гетероциклов получают ле¬
карственные препараты, красители и другие продукты. Важ¬
нейшие из гетероциклов этого типа содержат несколько атомов
азота или азот в сочетании с другими гетероатомами.
S
s
s
//—Л, H2/NI
р у ► СН,СН2СН2СН3 + H2S
374
Пиримидин — шестичленный гетероцикл с двумя атомами
азота.
Тиазол — жидкость с т. кип. 117°С, растворим в воде, обла¬
дает свойствами слабого основания. Ядро тиазола содержится
в молекуле витамина Вь антибиотика пенициллина, сульфамид¬
ного препарата норсульфазола.
сн
NCH
N СН
нс_сн
N СН
1 II
нс/ \сп
Nf \;Н
нс/ \:н
НС^-СН
V
NH
\/
NH
пиримидин
тиазол
пиразол
имидазол
Пиразол и имидазол — пятичленные гетероциклы с двумя
атомами азота. Это изомеры, различающиеся расположением
гетероатомов. Пиразол и имидазол — кристаллические вещест¬
ва, имеющие свойства слабых оснований и обладающие
ароматичностью. Так, пиразол очень устойчив к действию окис¬
лителей, вступает в реакции электрофильного замещения
(нитрование, сульфирование, бромирование). К числу важных
производных этих гетероциклов относится жаропонижающее,
противовоспалительное средство — антипирин и одна из белко¬
вых аминокислот — гистидин:
антипирин гистидин пурин
Из числа более сложных гетероциклов упомянем пурин, со¬
стоящий из конденсированных друг с другом ядер имидазола и
пиримидина.
§ 173. Алкалоиды. Алкалоидами называют органические осно¬
вания, встречающиеся в растениях. Большая часть алкалоидов
относится к азотистым гетероциклическим соединениям. Для
животных и человека алкалоиды, как правило, являются силь¬
ными ядами, но в малых дозах многие из них применяют как
лекарства.
Для обнаружения алкалоидов разработано много групповых
качественных реакций осаждения или цветных реакций. Так,
многие алкалоиды осаждаются фосфорномолибденовой кисло¬
той, танином, пикриновой кислотой, тетракалийгексацианофер-
ратом K4[Fe(CN)6].
Один из простейших по структуре алкалоидов — кониин —
является а-пропилпиперидином. Имея асимметрической атом уг¬
лерода (С-2), кониин, как и все другие алкалоиды, существует
в природе в оптически активной форме. Это жидкость, т. кип.
(166 °С. Представляет собой сильное основание, образует соли
375
с кислотами. Сильный яд, вызывает паралич дыхательных нер¬
вов.
Производным пиридина является и алкалоид анабазин.
Этот алкалоид был впервые выделен А. П. Ореховым (1928 г.)
из среднеазиатского растения Anabasis aphylla. В настоящее
время анабазин получают в больших количествах промышлен¬
ным путем и используют для борьбы с вредителями сельского
хозяйства.
Никотин является изомером анабазина. Это главный алка¬
лоид табака, в котором он находится в виде солей лимонной и
яблочной кислот. В природе встречается левовращающий анти¬
под никотина, в несколько раз более ядовитый, чем правовра¬
щающий. Такое явление — один из примеров того, как прост¬
ранственное строение органических веществ влияет на их фи¬
зиологическое действие. Извлеченный из табака никотин (жид¬
кость т. кип. 246 °С) служит для борьбы с вредителями сельского
хозяйства. Никотин — сильный яд и для человека, смертельная
доза — около 40 мг.
Общее число известных алкалоидов составляет более 1000.
И в настоящее время из растений выделяют все новые алкало¬
иды, устанавливают их строение. Несмотря на это, до сих пор
еще не пришли к единому мнению о роли алкалоидов в расте¬
ниях.
кониин
анабазин
никотин
ЧАСТЬ V
СПЕЦИАЛЬНЫЕ ГЛАВЫ
ОРГАНИЧЕСКОЙ ХИМИИ
ГЛАВА 22
ЭЛЕМЕНТЫ БИООРГАНИЧЕСКОЙ ХИМИИ
В начале прошлого столетия произошло разделение химии на
неорганическую и органическую химию — науку о веществах
растительного и животного происхождения. Хотя в дальней¬
шем большое место стало занимать исследование синтетиче¬
ских, не встречающихся в природе веществ, химики-органики
никогда не забывали об исследовании природных веществ. При
этом по мере развития науки ученые могли успешно решать
все более сложные задачи. В XIX и начале XX веков это было
исследование химического строения веществ растительного и
животного происхождения, сначала простых, затем все более и
более сложных. Постепенно стали развиваться и исследования
химических процессов, происходящих в живых организмах. На
стыке химии и биологии возникли новые науки — биохимия и
биоорганическая химия. Провести четкую грань между ними
трудно. Можно сказать, что биохимия в основе своей является
все же областью биологической науки, использующей химиче¬
ские методы для изучения явлений в живых организмах. Био¬
органическая химия — это область химии, изучающая химиче¬
ские основы жизненных процессов. Здесь приходится встре¬
чаться с наиболее сложными органическими веществами, ис¬
пользовать самые тонкие методы исследования.
§ 174. Белки. Аминокислотный состав. Знакомясь с аминокис¬
лотами, мы уже упоминали о том, что высокомолекулярные со¬
единения, построенные из остатков аминокислот, называют
белковыми веществами — белками. Нет ни одного живого ор¬
ганизма, растительного или животного, в котором белки не вы¬
полняли бы жизненно важных функций. В прошлом веке
Ф. Энгельс дал известное определение, что жизнь есть способ
существования белковых тел. Несмотря на то, что с тех пор
наука несравненно глубже познала сущность жизни, это опре¬
деление сохранило свою силу. Действительно, всюду, где есть
жизнь, встречают и белковые вещества.
Физическое состояние белков может быть столь же разно¬
образным, как и функции, которые они выполняют в организ¬
ме. Белок куриного яйца, мышцы, части скелета и суставов,
кожа, волосяной покров, рога, копыта — это все различные
377
виды белков. В крови в растворенном виде содержится группа
белков, в том числе гемоглобин, обеспечивающий перенос кис¬
лорода. В молоке содержится белок казеин и большая группа
других белков. Многочисленные ферменты — катализаторы об¬
мена веществ в живых организмах — относятся к белковым ве¬
ществам.
У растений белки не выполняют «структурных» функций;
остов растительной клетки образует полисахарид целлюлоза,
но тем не менее и в растениях белки выполняют жизненно важ¬
ные функции, сосредоточиваясь в основном в семенах.
Несмотря на внешнее несходство, различные представители
белков обладают некоторыми общими свойствами. Так, все
белки образуют коллоидные растворы. При повышении темпе¬
ратуры, действии УФ-излучения или радиации, под влиянием
кислот, щелочей и других реагентов происходят изменения фи¬
зико-химических свойств белков, называемые денатурацией.
При этом теряется биологическая активность. Денатурацию в
настоящее время связывают с изменениями конформации бел¬
ковых молекул.
Белковые вещества осаждаются фосфорновольфрамовой,
фосфорномолибденовой, пикриновой, трихлоруксусной, сали¬
циловой и другими кислотами, а также солями многих тяжелых
металлов. Белки дают и многие цветные реакции. Например,
специфические окраски возникают при взаимодействии их с
щелочными растворами солей меди (биуретовая реакция).
Главной составной частью белковых веществ являются при¬
веденные ниже а-аминокислоты.
-Моноаминомонокарбоновые кислоты
h2n—сн*—соон
глицин (Gly)
NHj
I
СН3—СН—СООН
аланин (Ala)
Н3С NH2
NHj
NH2
(СН3)2СН—СН—СООН
валин (Val)
NH2
С2Н5—СН—СН—СООН С6Н5—СН2—СН—СООН
изолейцин (Не) фенилаланин (Phe)
NH,
лейцин (Leu)
СООН
NH
вролин (Pro) гистидин (His) триптофан (1
Моно аминодикарб оно вые кислоты
триптофан (Try)
NH2
NH2
НООС—СН2—СН—СООН НООС—СН2— СН2—СН— СООН
аспарагиновая кислота (Asp) глутаминовая кислота (Glu)
878
Диамин омонокарбоновьге кислоты
NH2 NH2 NH2
H2N—(CH2)4—СН—СООН HN=C—NH—(СН2)э—сн—СООН
лизин (Lys) аргинин (Arg)
Г идроксиаминокислоты
NH2 но\
НО——СН2—СН—СООН —СООН
NH
тирозин (Туг)
NH2
I
НО—СН2—СН—СООН
серин (Ser)
гидроксипролин (Нур)
ОН NH2
СНз—СН—СН—СООН
треонин (Thr)
Серосодержащие аминокислоты
NHj NHa
CH3S—(СН2)2—Ан—СООН HS—СН2—СН—СООН
метионин (Met) цистеин (Cys)
Г NHa I
Lhooc—сн-сн2—s—J2
цистин (Cys-Cys)
Все аминокислоты, за исключением аминоуксусной кислоты,
имеют в молекуле асимметрический атом углерода. В белках
аминокислоты присутствуют в оптически активной форме, при¬
чем все они по конфигурации относятся к L-ряду, т. е. их про¬
странственное строение выражается проекционной формулой:
соон
h2n—
-н
§ 175. Строение белков. Молекулы аминокислот связаны в бел-*
ках по амидному типу. В данном случае эту связь называют
пептидной, а соединения амидного типа, образованные несколь¬
кими молекулами аминокислот,— пептидами:
О о
II II
—с—NH— H2N—СН2—C—NH—СН2—СООН
пептидная связь дипептид (глицилглицин)
О О
h2n—сн2—<?:—NH—СН2—С—NH—СН2—СООН
трипептид (глицилглицилглицин)
379
Белки представляют собой высокомолекулярные вещества.
Важной характеристикой является размер их молекул — моле¬
кулярная масса. Для ее определения используют различные фи¬
зические и химические методы. Один из простейших химических
методов заключается в определении содержания какого-либо
характерного химического элемента. Так, например, анализ ге¬
моглобина показывает, что он содержит около 0,08 % железа.
Поскольку менее одного атома железа в молекуле содержать¬
ся не может, из этого следует, что минимальная молекулярная
масса гемоглобина около 70 тыс. Молекулярные массы белков
колеблются от нескольких тысяч до нескольких миллионов, т. е.
число аминокислотных остатков в макромолекуле белка состав¬
ляет от нескольких десятков до сотен тысяч. Например, природ¬
ный полипептид окситоцин — гормон, выделяемый задней до¬
лей гипофиза, состоит из 9 аминокислот, его молекулярная мас¬
са 1007; молекулярная масса адренокортикотропного гормона
(23 аминокислоты) 3200, фермента рибонуклеазы (124 амино¬
кислоты)— около 15 тыс., а молекулярные массы белков виру¬
сов могут достигать 50 млн. Уже эти вариации молекулярной
массы создают большое разнообразие белков. Но гораздо бо¬
лее существенная причина разнообразия белков по сравнению,
например, с полисахаридами заключается в том, что в состав
макромолекул белка могут входить около 20 различных амино¬
кислот, в то время как обычные полисахариды (целлюлоза,
крахмал) построены нз одного моносахарида — глюкозы.
После установления размера молекулы дальнейшим шагом
в изучении белка является определение его аминокислотного
состава. Основы для этого были заложены на рубеже XIX—
XX веков работами известного немецкого ученого Э. Фишера.
Сущность разработанного им метода заключалась в кислотном
гидролизе изучаемого белка; образующиеся аминокислоты пре¬
вращали в этиловые эфиры:
В отличие от самих аминокислот их эфиры — вещества ле¬
тучие; их разделяли перегонкой в вакууме и другими методами.
Все это требовало больших количеств исходного белка (сотни
граммов), анализ продолжался много месяцев. Теперь созданы
автоматические аминокислотные анализаторы, которые позво¬
ляют, имея всего несколько миллиграммов белка, через не¬
сколько часов определить, какие аминокислоты и в каких со¬
отношениях входят в состав белка.
н2о, н+
R
H2N—СН—СООН
с2н6он, н+
R
—► H2N—СН—COOC2Hs
880
Однако с определения аминокислотного состава изучение
белка только начинается. Можно подсчитать, что сравнительно
простой белок, построенный из 20 остатков разных аминокис¬
лот, может з0 счет различного порядка их чередования иметь
2-101а изомер00- Для белка, состоящего из 1000 аминокислот,
могло бы существовать 201000 изомеров: чтобы синтезировать
хотя бы по одной молекуле каждого изомера, не хватило бы
всего вещества солнечной системы.
Определение последовательности аминокислотных остат¬
ков — первичной структуры белка, т. е. его химического строе¬
ния,— еще более сложная задача. Например, на выяснение
первичной структуры гормона инсулина (это белок с относи¬
тельно небольшой молекулярной массой, участвующий в регу¬
лировании сахарного обмена в организме) английскому биохи¬
мику Ф. СэИДжеру потребовалось 10 лет. В основе работы
Сэнджера лежало гидролитическое расщепление белка на не¬
большие фрагменты и определение аминокислотной последова¬
тельности в них. Для гидролиза был использован набор специ¬
фических ферментов, каждый из которых был способен расщеп¬
лять полипептидную цепь в определенном месте. Сэнджер уста¬
новил, что молекулу инсулина образуют две полипептидные цепи
(21 и 30 аминокислотных остатков), связанные друг с другом
дисульфидными связями (—S—S—), которые образуются меж¬
ду остатками содержащей серу аминокислоты — цистеина.
Вслед за инсулином была расшифрована структура рибо-
нуклеазы — фермента, в молекуле которого 124 аминокислот¬
ных остатка (рис. 29, а).
Созданы приборы, которые автоматизируют эту трудоемкую работу.
К настоящему времени установлена первичная структура многих белков.
Изучение строения белков, выполняющих важные физиологические функ¬
ции, позволило 0 ряде случаев вскрыть химическую первопричину некоторых
болезней. Так, при тяжелой наследственной болезни, так называемой серпо¬
видной анемии, аномальным оказывается белок крови — гемоглобин: в слож¬
ной цепи этого белка всего одна аминокислота заменена другой. Подобных
«молекулярных болезней» (термин, предложенный известным американским
ученым Полинг0м) 0 настоящее время известно уже несколько.
Однако свойства белковой молекулы зависят не только от
первичной структуры, но и от конформации полипептидной це¬
пи (вторичной структуры). Одной из моделей вторичной струк¬
туры белка является а-спираль, в которой полипептидную цепь
надо представлять себе в виде нити, как бы обвивающей по¬
верхность цилиндра. Устойчивость а-спирали обеспечивается
водородными связями между группами NH и СО (рис. 30). Дру¬
гая структура —p-форма, представляет собой «жгут» связанных
водородными связями вытянутых полипептидных цепей.
ПолипептНДная цепь, имеющая ту или иную вторичную
структуру (т. е. конформацию а-спирали, p-формы или неупоря¬
доченную), может приобретать еще одну форму упорядоче¬
ния— третичную структуру. При этом могут возникать взаимо-
381
20
Рис. 29. Аминокислотная последова¬
тельность (первичная структура) (а)
и третичная структура (б) рибону-
клеазы
Римскими цифрами обозначены ociaiiut
цистеина, связанные S—S-связяма
действия разных типов, в частности образование дисульфидных
мостиков между цистеиновыми остатками, находящимися в
разных местах одной и той же полипептидной цепи. Отдельные
участки полипептидной цепи оказываются сближенными и
скрепленными друг с другом. Кроме дисульфидных мостиков в
образовании третичной структуры принимают участие также
£82
ионные взаимодействия противоположно заряженных групп и
«гидрофобные взаимодействия» — стремление белковой молеку¬
лы свернуться так, чтобы внутри глобулы были сближены друг
с другом углеводородные остатки, а внешний слой составляли
функциональные группы, взаимодействующие с полярными мо¬
лекулами воды, т. е. группы, обладающие гидрофильными свой¬
ствами. На рис. 29, б, показана третичная структура молекулы
рибонуклеазы. Несколько молекул могут складываться с обра¬
зованием более крупных агрегатов (четвертичная структура).
Вторичная, третичная и четвертичная структуры белка лег¬
ко нарушаются при самых различных воздействиях: нагрева¬
нии, действии растворителей, даже энергичном встряхивании.
При нарушениях такого рода исчезают свойства природного
белка — происходит его денатурация. Иногда такие превраще¬
ния обратимы. Так, например, гемоглобин при действии моче¬
вины диссоциирует на четыре составляющие его субъединицы,
т. е. происходит разрушение его четвертичной структуры. Пос¬
ле удаления мочевины снова образуется частица гемоглобина,
обладающая свойствами
природного белка.
Эти факты показывают,
что высшие формы органи¬
зации белковой молекулы в
конечном итоге предопреде¬
ляются ее первичной струк¬
турой. Установление этого
сделало реальным воспро¬
изведение путем синтеза
природных биологически ак¬
тивных белков. Для этого в
настоящее время исполь¬
зуют разработанный в на¬
чале 60-х годов твердофаз¬
ный синтез. Первая амино¬
кислота при этом закреп¬
ляется на полимерном но¬
сителе (полистирольной
смоле), и к ней последова¬
тельно «подшиваются» все
новые и новые аминокис¬
лоты. По окончании синтеза
готовая полипептидная цепь
снимается с носителя. Ис¬
пользование этого метода
Рис. 30. Белковая а-спираль (вто¬
ричная структура белка)
383
позволило синтезировать, например, инсулин и рибонуклеазу,
причем оба полученных белка обладали биологической актив¬
ностью. При синтезе рибонуклеазы необходимо было осущест¬
вить более 10 тыс. отдельных операций. В настоящее время
синтезированы и другие природные белки.
Белки, дающие при гидролизе исключительно аминокисло¬
ты, называют протеинами В зависимости от их свойств и био¬
логических функций белки подразделяют в биохимии на мно¬
жество групп (например, альбумины, глобулины, протамины,
глутелины).
Сложные белки (протеиды) являются соединениями белков
с небелковой частью В зависимости от природы небелковой
части различают следующие группы протеидов.
1. Фосфопротеиды содержат фосфор. Наиболее важный
представитель этой группы — содержащийся в молоке казеин.
2. Липопротеиды — соединения белков с веществами, род¬
ственными жирам,— фосфатидами, сфингомиелинами, а также
полиеновыми пигментами типа каротина. К белкам этого типа
относится, например, зрительный пурпур сетчатки глаза.
3. Гликопротеиды и мукопротеиды — соединения белков с
углеводами; к протеидам этого типа относятся альбумины и
глобулины сыворотки, гиалуроновая кислота стекловидной жид¬
кости глаза.
4. Металлопротеиды — сложные белки, содержащие ком¬
плексно связанный металл. Белками такого типа являются: ге¬
моглобин — содержащий железо дыхательный пигмент крови
млекопитающих; гемоцианины — содержащие комплексно свя¬
занную медь дыхательные пигменты крови моллюсков, улиток,
крабов, и другие.
5. Нуклеопротеиды — соединения белка с нуклеиновыми
кислотами. Ввиду большой важности этих веществ им посвящен
отдельный параграф (см. § 177).
§ 176. Пищевое и промышленное использование белков. Растения способны,
синтезировать аминокислоты и белки, используя в качестве источника азота
неорганические соединения. Животные же для нормального существования
должны получать белки с пищей. В процессе пищеварения белки расщеп¬
ляются на низкомолекулярные пептиды или аминокислоты, которые всасы¬
ваются кишечником и разносятся током крови. Они и служат строительным
материалом, из которого организм создает белки своего тела. Таким образом,
белки в питательном рационе вполне могут быть заменены аминокислотами.
Некоторые необходимые для жизни аминокислоты организм может вырабаты¬
вать сам из других азотсодержащих соединений, поступающих с пищей..
Другие же аминокислоты организм синтезировать не в состоянии, и их надо
вводить в готовом виде, с белковой пищей. Такие аминокислоты получили
название незаменимых. К их числу относятся: лизин, триптофан, фенилала¬
нин, валин, метионин, треонин, лейцин, изолейцин, гистидин, аргинин.
Белковая пища должна покрывать не только общую потребность в ами¬
нокислотах, как главном «строительном материале» организма, но и обяза¬
тельно содержать требуемые количества незаменимых аминокислот. При не¬
достатке незаменимых аминокислот нарушается нормальное существование
организма. Так, например, белок кукурузы зеин не содержит лизина и почти
884
не содержит триптофана. В опытах над животными, которым вводили с пи¬
щей только один этот белок, наблюдалась, несмотря на обильное кормление,
потеря в массе. При отсутствии в пище триптофана развивается заболевание
глаз — катаракта.
Потребность в белках человек удовлетворяет за счет продуктов сельскохо¬
зяйственного производства, животноводства, растениеводства. Однако сель¬
ское хозяйство с его зависимостью от природных условий, необходимостью
использования огромных земельных массивов, большими затратами челове¬
ческого труда — далеко не идеальный источник пищи. В связи с этим ученые
давно уже задумываются над проблемами синтеза продуктов питания и в
первую очередь наиболее ценной части пищи — белков. В настоящее время
наиболее перспективным представляется микробиологический синтез белков
из углеводородов нефти. В конце 50-х годов были найдены микроорганизмы,
которые могут питаться алканами. При этом из 1 т углеводородов полу¬
чается 0,7 т полноценных белковых веществ. В образовавшейся массе содер¬
жатся также витамины группы В. Метод получения белково-витаминных
концентратов из нефти освоен у нас в стране в промышленном масштабе.
Сырьем служат тяжелые фракции нефти с добавками обычных калийных,
азотных и фосфорных удобрений, а также микроэлементов. Эту смесь микро¬
организмы перерабатывают в белковую массу, которую используют в живот¬
новодстве.
В настоящее время для повышения питательной ценности рациона сель¬
скохозяйственных животных к их пище добавляют наиболее «дефицитные»
аминокислоты. Для этой цели организовано крупное промышленное производ¬
ство лизина, глутаминовой кислоты, метионина. Для синтеза лизина исполь¬
зуют капролактам — то же сырье, что и для производства капрона. В него
вводят нитрогруппу (процесс идет в несколько стадий), а затем восстанавли¬
вают ее с одновременным размыканием лактамного цикла:
Недостаток лизина в пище вызывает анемию, головную боль, повышенную
раздражительность. Синтетический лизин вводят в пищу детей для повыше¬
ния аппетита, при лечении тяжелых отравлений.
При промышленном производстве глутаминовой кислоты в качестве сырья
используют акрилонитрил:
Глутаминовую кислоту добавляют для улучшения вкуса в консервы и
пищевые концентраты.
Другие составные части пищи в принципе также могут быть получены
синтетическим путем. Оба компонента жиров — глицерин и жирные кисло¬
ты — производят в промышленных масштабах из нефтяного сырья. Никаких
принципиальных трудностей не представляет и получение жиров из этих ком¬
понентов. Витамины — необходимую составную часть полноценной пищи —
получают в настоящее время синтетически в промышленном масштабе. Наи¬
менее ясны перспективы синтетического получения пищевых углеводов: труд¬
ности здесь связаны с необходимостью получения определенного простран¬
ственного изомера — глюкозы. Однако уже сейчас глюкозу получают из не¬
пищевого сырья — древесины — путем гидролиза.
Все компоненты, которые должны содержаться в полноценной пище, в
настоящее время известны. Приготовленные из них смеси использовали в
со+Н2
NH<CN
СН2=СН—CN ►
ЫН2
—» N С—СН—СН2—СН2—CN
ОНС—СН2—СН2—CN
NaOH. ^Н2
NaOH.
н2о
НООС—СН—сн2—сн2—соон
385
13 Органическая химия
экспериментах питания животных, которые живут на такой диете неограни¬
ченное время. Опробованы такие смеси и в длительном питании людей.
Белки имеют не только пищевое, но и промышленное значение. Наиболее
крупная отрасль, занятая переработкой белкового сырья, — кожевенная про¬
мышленность. Белковыми веществами являются также натуральный шелк и
шерсть. Белок казеин, выделяемый из молока, используют для приготовления
клея; ранее большое значение имела пластмасса, получаемая из казеина (га¬
лалит); делались попытки получать из казеина и искусственное волокно.
К белковым веществам относится желатин, употребляемый в производстве
кинофотоматериалов, в пищевой промышленности.
§ 177. Нуклеиновые кислоты. Строение и свойства. Еще в прош¬
лом веке было установлено, что в клеточных ядрах содержатся
высокомолекулярные вещества, в состав которых входят азот¬
содержащие гетероциклические основания, углеводы и фосфор¬
ная кислота. Эти вещества получили название нуклеиновых кис¬
лот, Несколько десятилетий тому назад биологическая роль
нуклеиновых кислот была совершенно неясна, в настоящее
же время установлено их первостепенное значение в живой
природе.
Нуклеиновые кислоты — высокомолекулярные соединения
(мол. масса от 200 тыс. до нескольких миллионов). При полном
гидролизе нуклеиновых кислот образуется смесь азотсодержа¬
щих гетероциклических оснований (пиримидиновых и пуриновых
оснований), моносахаридов — пентоз (рибоза или дезоксирибо-
за) и фосфорной кислоты.
- Пиримидиновые основания*:
NH, О
цитозин урацил
О
-СНз
NH
тимин
Пуриновые основания:
NH2
N
О
/
IJ
NH N
XNH
''"'NH,
аденин
гуанин
* Пиримидиновые основания способны к таутомерии. Они изображены выше
в NH-формах (в таком вш(е ови входят в состав нуклеиновых кислот), в
свободном же состоянии преобладает ароматизированная форма, например!
NH3 NH2
О
N
НО-"'
N
386
Моносахариды:
сн2он
о он
снгон
о ОН
но
рибоза
он
но н
дезоксирибоза
При более мягком гидролизе нуклеиновых кислот образуется
смесь веществ, называемых нуклеотидами. В состав нуклео¬
тида входят остатки пиримидинового или пуринового основа¬
ния, пентозы и фосфорной кислоты. От нуклеотида можно от¬
щепить фосфорную кислоту и получить нуклеозид, состоящий
из пуринового (или пиримидинового) основания и пентозы.
Таким образом, мономерной единицей нуклеиновых кислот
являются нуклеотиды. В зависимости от природы пентозы, вхо¬
дящей в состав нуклеотида, эти вещества разделяют на дез-
оксирибонуклеотиды и рибонуклеотиды.
В состав дезоксирибонуклеотидов входит дезоксирибоза. По¬
лимеры из дезоксирибонуклеотидов называют дезоксирибону¬
клеиновыми кислотами (ДНК).
В состав рибонуклеотидов входит рибоза. Полимерные рибо¬
нуклеотиды— это рибонуклеиновые кислоты (РНК). Ниже при¬
ведены входящие в РНК нуклеозиды:
NH,
О
но он
аденозин
НО ОН
гуанозий
НО ОН
уридин
НО ОН
цитидин
13*
337
Эти нуклеозиды являются А-р-гликозидами £>-рибозы. В со¬
став их входят два пуриновых основания — аденин и гуанин и
два пиримидиновых основания — цитозин и урацил.
В состав дезоксирибонуклеиновых кислот входят нуклеози¬
ды, построенные подобно предыдущим, но содержащие вместо
рибозы 2-дезоксирибозу. Кроме того, вместо урацила, присут¬
ствующего в РНК, четвертым азотистым основанием ДНК яв¬
ляется тимин.
Построение макромолекул ДНК и РНК из нуклеотидов осу¬
ществляется путем образования эфирной связи между фосфор¬
ной кислотой и гидроксигруппами при С-3 и С-5 нуклеозида.
Для примера приведем фрагмент молекулы ДНК с остатками
аденина (А), цитозина (С) и гуанина (G):
§ 178. Биологическая роль нуклеиновых кислот. При изучении
ДНК рентгенографическим методом установлено, что макромо¬
лекулы имеют строго регулярное строение, а химическое иссле¬
дование показало, что число пиримидиновых оснований всегда
равно числу пуриновых; аденина всегда столько же, сколько ти¬
мина; цитозина столько же, сколько гуанина. Объяснение этим
фактам дали Д. Уотсон и Ф. Крик в своей модели двойной спи¬
рали (1953 г.). Двойная спираль, как видно из рис. 31, похожа
на винтовую лестницу. Две «стойки» этой лестницы образова¬
ны основной цепью, состоящей из углеводных и фосфатных ос¬
татков, азотистые основания образуют как бы «ступеньки» лест¬
ницы. Азотистое основание одной цепи связано водородными
388
связями с азотистым основанием другой цепи:
Н
цитозин гуанин
Диаметр двойной спирали практически постоянен и равен
1,8 нм, а каждый виток спирали имеет высоту примерно 3,4 нм.
При этом одну «ступеньку» всегда образует остаток пуринового
и остаток пиримидинового основания, как это показано в приве¬
денных выше схемах. Это и обеспечивает строгую пространствен¬
ную регулярность несмотря на то что конкретные пары основа¬
ний различны, длина «ступеньки» постоянна.
Последовательность оснований вдоль цепи ДНК представ¬
ляет собой закодированную информацию для построения белко¬
вых молекул. Молекулярная масса ДНК может достигать де¬
сятков и даже сотен миллионов, т. е. в макромолекулу может
входить до миллиона нуклеотидов. Каждая тройка последова¬
тельно расположенных в цепи оснований яв¬
ляется как бы «буквой» генетического кода, с
помощью которого зашифрована информация.
В молекуле ДНК — несколько сот тысяч та¬
ких «букв»: эта молекула представляет собой
как бы книгу примерно в сотню страниц. Об-
, щую же информацию, хранящуюся в ДНК
каждой клетки («2 млрд, нуклеотидов), мож¬
но сравнить с огромной библиотекой в 2000 то¬
мов по 500 страниц в каждом.
Как же используется эта информация?
В момент клеточного деления двойная спи¬
раль начинает раскручиваться, разделяясь на
цве нити. На каждой из разделившихся цепей
ДНК начинает строиться дополнительная цепь
за счет нуклеотидов, подводимых из окружаю¬
щей среды. Они располагаются не беспорядоч¬
но, а по принципу комплементарных (взаимно
дополняющих) пар: аденин против тимина,
гуанин против цитозина. В результате в про-
Рис. 31. Модель двойной спирали ДНК:
D — остатки дезоксирибозы; Р — остатки фосфорной кислоты;
А, Т, G, С — азотистые основания
389
цессе клеточного деления возникают две новые двойные спира¬
ли, являющиеся точными копиями исходной: каждая половинка
разделившейся клетки получает свой набор ДНК, идентичный
набору ДНК материнской клетки.
Молекулы ДНК хранят в клеточных ядрах наследственную
информацию, «записанную» в виде различной последовательно¬
сти нуклеотидов. Молекулы ДНК играют роль «матрицы», с ко¬
торой «отпечатываются копии» молекул РНК, непосредственно
участвующих в синтезе белка. Таким образом, молекулы РНК
служат передатчиками информации от ДНК к местам клетки,
где непосредственно осуществляется синтез белка. Эти моле¬
кулы РНК называют информационными. Наряду с ними в про¬
цессе синтеза белка участвуют транспортные РНК, каждая из
которых специфично связывается с определенной аминокисло¬
той и доставляет ее к нужному месту информационной РНК,
после чего остатки аминокислот соединяются пептидной связью,
образуя макромолекулу белка. Расшифрована последователь¬
ность оснований примерно в 80 транспортных РНК.
Роль РНК в процессе синтеза белка была подтверждена
опытами, выполненными в начале 60-х годов. Из клеток бакте¬
рии Echerichia coli после полного разрушения была получена
бесклеточная жидкость, содержащая все необходимые для
синтеза белка ферменты, ранее находившиеся в клетке. Эта
система способна некоторое время осуществлять синтез белка,
затем он замедляется. Если в этот момент добавить РНК, то
синтез белка возобновляется. Из этого непосредственно следует,
что РНК участвует в процессе синтеза белка. Установив это,
стали добавлять не природную, а синтетическую РНК. Синтез
белка продолжался и в этом случае. Когда добавляли синтети¬
ческую РНК, содержащую только один нуклеотид, а именно
урацил, образовывался пептид, состоящий почти целиком из
одной аминокислоты — фенилаланина.
Дальнейшее развитие подобных опытов позволило расшиф¬
ровать «генетический код»: определить, как именно в молекуле
РНК записан «приказ» включать в молекуле белка определен¬
ные аминокислоты. Каждая аминокислота имеет свой «шифр»,
записанный в виде последовательности трех нуклеотидов из
числа четырех, встречающихся в молекулах РНК: аденозина
(А), гуанозина (G), цитидина (С) и уридина (U). Так, вклю¬
чение аланина задается в виде одного из трех кодов CCG,
UCG и ACG, триптофана — кодом GGU (латинскими буквами
обозначены не азотистые основания, а нуклеотид в целом).
За последние десятилетия наука глубоко проникла в самые
сокровенные процессы жизни. Однако по мере выяснения одних
вопросов возникают другие, ответ на которые еще более углуб¬
ляет наши знания. Молекулярная биология — быстро разви¬
вающаяся область науки, в которой огромное значение имеет
органическая химия.
3S0
Большое биологическое значение имеют не только полину¬
клеотиды, но и соответствующие мономеры. Они входят в со¬
став ряда ферментов. В качестве примера рассмотрим аденозин-
трифосфат (АТФ). Накопленная в нем энергия освобождается,
когда она нужна клеткам для выполнения определенных
функций.
О .
ОН
О
II
-Р—I
I
ОН
о
он
Энергия освобождается при экзотермической реакции гидро¬
лиза АТФ с отщеплением одной фосфатной группы и образова¬
нием аденозиндифосфата: энергетический эффект составляет
35—50 кДж/моль.
§ 179. Ферменты. Ферменты играют роль катализаторов хими¬
ческих превращений в животных и растительных организмах.
Характерные черты каталитического действия ферментов —
высокая эффективность и строгая направленность. Это обеспе¬
чивает осуществление сложных превращений органических
веществ с большими скоростями и в мягких условиях—при
низкой температуре, нормальном давлении, в разбавленных рас¬
творах, при реакции среды, близкой к нейтральной. Поэтому
изучение ферментов важно не только с биохимической точки
зрения; оно может указать пути создания высокоэффективных
катализаторов для промышленности. В настоящее время изве¬
стно более тысячи ферментов; каждый из них является катали¬
затором определенной реакции.
По химической природе ферменты относятся к белкам. В их
молекулах белковая часть соединена с небелковым компонен¬
том, который называют коферментом. Химическое строение мно¬
гих коферментов известно. Изучены и механизмы их пре¬
вращений в процессе осуществления каталитических функций.
Однако это еще не значит, что полностью понята каталитиче¬
ская роль фермента. Дело в том, что сам по себе кофермент
обычно либо вовсе не обладает каталитической активностью,
либо проявляет лишь слабое каталитическое действие. Актив¬
ность кофермент приобретает лишь в комбинации с соответ¬
ствующим белком.
Носителем биологической активности в ряде случаев оказа¬
лась не вся белковая молекула в целом, а определенная ее
часть. Так, в растительном ферменте папаине, построенном из
391
180 аминокислотных остатков, можно «отстричь» до 2/з его по-
липептидной цепи, не оказав заметного влияния на его биологи¬
ческую активность. Факты подобного рода позволяют глубже
понять природу каталитического действия ферментов, а также
вселяют надежду на возможность создания синтетических фер¬
ментов, при помощи которых можно будет упростить получение
многих нужных человеку органических веществ. Важно и то, что,
освоив синтезы, исследователи смогут наряду с воспроизведением
природных продуктов синтезировать сходные с природными, но
определенным образом отличные от них вещества. Имея серию
подобных аналогов, можно проследить за изменениями их фи¬
зиологической активности в зависимости от изменений химиче¬
ского строения, что важно как с теоретической, так и с практи¬
ческой точек зрения.
Кофермент вместе с определенными частями белковой моле¬
кулы образует активный центр. Все составные части этого
центра находятся на строго определенных расстояниях, зани¬
мают определенное место в пространстве. Строение активного
центра согласовано со структурой той молекулы (субстрата),
превращение которой катализирует данный фермент. Это со¬
здает благоприятные условия для образования фермент-суб-
стратного комплекса, затем одновременно осуществляется раз¬
рыв связей и образование новых.
Ферменты в живых организмах частично находятся в рас¬
творенном состоянии, но во многих случаях они образуют стро¬
го организованные системы, как бы конвейерные линии, на
которых осуществляется цепь химических превращений, обеспе¬
чивающих, например, процессы.дыхания, усвоения пищи, по¬
строение необходимых организму белков и других веществ.
Число известных коферментов значительно меньше общего
числа известных ферментов: один и тот же кофермент в комби¬
нации с разными белками может образовать разные ферменты.
В состав ряда ферментов, катализирующих различные превра¬
щения аминокислот, например их синтез, переаминирование,
декарбоксилирование, входит кофермент пиридоксаль-5-фосфат:
В процессе всех превращений альдегидная группа кофермен-
та прежде всего реагирует с аминогруппой аминокислоты:
СНО
НО^ уСНз
на ,сн3
R—СН—NH2 + 0=СН
соон
:н2-оро(он)2
СН2—ОРО(ОН)2
392
Характер дальнейшего превращения зависит от конкретной
природы фермента, т. е. от строения его белковой части.
Ферментные препараты имеют большое значение и для про¬
мышленности. Ферментативными процессами являются при¬
готовление сыра, чая, табака, обработка кож, виноделие. Фер¬
ментативные превращения протекают при комнатной темпера¬
туре, не требуют повышенных давлений, энергичных реагентов;
таков, например, процесс фиксации атмосферного азота клу¬
беньковыми бактериями. Раскрытие механизма действия фер¬
ментов и воспроизведение его в промышленных условиях по¬
зволило бы решить многие технологические задачи. Одним из
путей этого является использование иммобилизованных фер¬
ментов-. так называют ферменты, закрепленные на полимерных
носителях. При этом повышается устойчивость ферментов.
§ 180. Витамины. Витаминами (лат. vita — жизнь) были назва¬
ны составные части пищи, содержащиеся в ней в ничтожных ко¬
личествах, но совершенно необходимые для нормальной жизни
человека и животных. Когда химическая природа витаминов
была еще неизвестна, их условно обозначили буквами латин¬
ского алфавита. Хотя теперь строение витаминов установлено,
до сих пор вместо громоздких химических названий употребляют
привычные буквенные обозначения.
В настоящее время известно более 20 витаминов. Выяснены
их строение и роль, которую они играют в организме. Оказа¬
лось, что многие витамины используются для синтеза фермен¬
тов. Витамины разделяют на водорастворимые (витамин С,
группа витаминов В, витамин РР и некоторые другие) и жиро¬
растворимые— витамины групп A, D и К.
Витамин С (аскорбиновая кислота)— вещество, родственное
по химической природе сахарам. Содержится в свежих овощах,
фруктах. Получается также синтетически из шестиатомного
спирта сорбита СН2ОН—(СНОН)4—СН2ОН. Недостаток этого
витамина вызывает заболевание цингой. Главная химическая
особенность аскорбиновой кислоты — способность легко окис¬
ляться с образованием дегидроаскорбиновой кислоты; послед¬
няя в свою очередь может легко восстанавливаться в аскорби¬
новую кислоту:
о=с
о=с
с—он
с=о
о
+2Н н—с
-2Н
с=о
о
но—с—н
но—с—н
СН2ОН
аскорбиновая
кислота
СН2ОН
дегидроаскорбиновая
кислота
393
Участие в окислительно-восстановительных превращениях,
сопровождающихся переносом водорода,— главная функция
аскорбиновой кислоты в организме.
Витамин £>2 (кальциферол). Недостаток этого витамина вы¬
зывает рахит. Он содержится в жировых продуктах (жир пе¬
чени, рыбий жир). Суточная потребность составляет около
0,025 мг; поступление очень больших количеств витамина D2
(как и других жирорастворимых витаминов) вредно для ор¬
ганизма.
Витамин D2 не только выделяют из природных продуктов,
но и получают синтетически. Исходным веществом служит эр-
гостерин — полициклическое соединение, относящееся к классу
стероидов (см. § 183). Превращение эргостерина в витамин D2
происходит под действием ультрафиолетового света. По-види¬
мому, такой же процесс образования витамина D2 осуществля¬
ется и непосредственно в организме под действием солнечного
света.
Витамин А (ретинол) предохраняет от глазного заболевания
ксерофтальмии, а также играет роль витамина роста. Образу¬
ется в организме из желтого красящего вещества каротина (со¬
держится в моркови).
Н3С СНз
/СН=СН—С=СН—СН=СН—С=СН— СН2ОН витамин А
Г 1 I
СНз
СИ,
§ 181. Антибиотики. В конце 30-х годов были открыты анти¬
биотики — эффективные лекарственные средства, продуцируе¬
мые микроорганизмами. В настоящее время выделено около
500 антибиотиков. Строение многих из них выяснено, химиче¬
ская природа других еще изучается. Химическая природа анти¬
биотиков может быть различной, среди них имеются соедине¬
ния гетероциклического и ароматического рядов, вещества по-
липептидного строения.
Пенициллин — исторически первый антибиотик, открытый в
1940 г. в Англии. Основу его структуры составляет пятичлен¬
ный гетероцикл — тиазолидин, содержащий атом азота и атом
серы. В настоящее время известен ряд пенициллинов, различаю¬
щихся природой радикала R в боковой цепи. Эти антибиотики
394
обладают сравнительно узким спектром действия, т. е. препят¬
ствуют развитию только определенных микроорганизмов.
В 1942 г. советские ученые Ф. Гаузе и М. Бражникова вы¬
делили из почвенных микроорганизмов грамицидин С («грами¬
цидин советский»). Этот антибиотик также имеет узкий спектр
действия. Грамицидин имеет полипептидную структуру, т. е.
построен из соединенных амидными связями остатков амино¬
кислот:
В состав грамицидина входит D-антипод фенилаланина, т. е.
аминокислота необычной конфигурации. Это наблюдается и у
других антибиотиков. Возможно, что именно это и придает ан¬
тибиотикам своеобразные свойства антагонистов микроорга¬
низмов.
Важную группу антибиотиков образуют тетрациклины. На¬
звание указывает на их строение: в молекуле имеются четыре
сконденсированных шестичленных цикла:
Отличительная особенность этой группы антибиотиков—-
широкий спектр действия. Они находят применение для борьбы
с самыми различными микроорганизмами.
В практике лечения различных заболеваний обнаружилось явление при¬
выкания микроорганизмов к антибиотикам: постепенно появляются новые
расы микроорганизмов, на которые определенный, привычный антибиотик уже
не действует. Это может создать затруднения при лечении и требует, чтобы
«соревнование» между создателями новых антибиотиков и защищающимися
микроорганизмами продолжалось.
§ 182. Терпены. К терпенам относят содержащиеся в эфирных
маслах растений вещества углеводородной, спиртовой или ке-
тонной природы. Большинство терпенов являются производными
алициклических углеводородов, хотя к числу терпенов причис¬
ляют и некоторые соединения с открытой цепью.
Терпены, как правило, представляют собой жидкости с при¬
ятным запахом, практически нерастворимые в воде. Их способ¬
НООС—НС N—С=о О
(CHsbcL, /СН—СН—NH—i—R
S
(СНзЬСх, /СН—СН—NH—1
пенициллин
н3с. .он n(ch3)2
^ /*С /\ JL ,ОН
ОН О НО о.
он
395
ность перегоняться с водяным паром используют при выделении
эфирных масел из растений.
Смесью терпенов является скипидар, получаемый перегонкой
смолы хвойных деревьев — живицы. Эту смолу собирают, делая
надрезы на стволах ели, сосны и кедра. Скипидар используют
как сырье для получения индивидуальных терпеновых углеводо¬
родов, главным образом а-пинена. Кроме того, скипидар при¬
меняют как растворитель для лаков, красок, эмалей, для полу¬
чения смазочных и флотационных масел, ядохимикатов,' инсек¬
тицидов, лекарств.
Терпеновые углеводороды относятся к числу широко рас¬
пространенных в природе изопреноидных соединений — веществ,
в основе структуры которых лежит звено изопрена. В эту груп¬
пу соединений входит и натуральный каучук (см. § 188).
В основе большинства терпенов лежит циклогексановый уг¬
леводород п-ментан, как бы построенный из двух остатков изо¬
прена. Формулу ментана часто изображают условно (формула
справа):
сн*
1
сн
Н2С^ СН2 1
ЩС ^СЩ А|
' си V
СН Дч
н3су чсн3
ментан
Терпены, в основе которых непосредственно лежит углеводо¬
род ментан, называют монотерпенами; в их составе 10 атомов
углерода. Терпены Си называют сесквитерпенами («полутор¬
ные терпены»), С20 — дитерпенами, Сзо — тритерпенами. В за¬
висимости от характера углеродного скелета терпены разделяют
на ациклические, моноциклические, бициклические и полицик¬
лические.
Кроме углеводородов в природе встречаются относящиеся к
классу терпенов спирты, кетоны, кислоты. В настоящем разделе
рассмотрены лишь простейшие представители моно- и дитер¬
пенов.
Терпены, как правило, содержат несколько асимметрических
атомов углерода и встречаются в природе в оптически актив¬
ном виде.
Моноциклические терпены — углеводороды состава
CioHu — являются двунепредельными производными ментана.
Представитель этой группы терпенов лимонен получил такое
название потому, что входит в состав лимонного масла. Содер¬
жится он и в маслах других цитрусовых (апельсинов, мандари¬
396
нов), а также в эфирных маслах укропа, тмина и др. Это самый
распространенный из моноциклических терпеновых углеводо¬
родов.
Лимонен — бесцветная жидкость, т. кип. 176 °С, имеет харак¬
терный запах лимона, обладает оптической активностью, при¬
чем в разных эфирных маслах встречаются либо левовращаю¬
щий, либо правовращающий антипод. При конденсации двух
молекул изопрена можно получить дипентен, представляющий
собой рацемическую форму лимонена:
СН3
А
нс/ чсн2
1 +
Н2С ^сн2
I сн
сн3—£=сн2
II
Эта реакция идет по типу диенового синтеза (см. § 41), при¬
чем одна из молекул изопрена играет роль диена (4), а вторая
(II) —диенофила. Рацемическая форма лимонена встречается
также в скипидаре. В эфирных маслах содержатся и другие мо-
ноциклические терпены СюНи, отличающиеся от лимонена
иным расположением двойных связей.
В природе встречаются и кислородные производные терпе¬
новых углеводородов — спирты, кетоны, альдегиды.
Ментол — кристаллическое вещество с характерным запа¬
хом, является главной составной частью мятного масла. Это
предельный спирт циклогексанового ряда. Имея три асимметри¬
ческих атома углерода, он существует в виде восьми простран¬
ственных изомеров. Ментол обладает слабым дезинфицирую¬
щим действием. Его применяют в медицине, при изготовлении
зубной пасты.
При окислении ментола хромовой смесью образуется кетон
ментон:
КгСггОу, H2SO4
ОН
Ментон представляет собой жидкость, т. кип. 207 °С; по за¬
паху напоминает ментол. Будучи кетоном, он образует такие
производные, как оксим, фенилгидразон и др. Ментон, как и
ментол, содержится в мятном масле.
сн3
I
С
НС^ \СН2
I I
Н2Сч. /СН2
сн
СНз—<U=CH2
397
Бициклические терпены являются производными
трех изомерных углеводородов состава C)0Hig:
карам
пинан
борнан
Наиболее распространенный в природе бициклический тер-
пеновый углеводород группы пинана— а-пинен представляет
собой жидкость, т. кип. 156 °С. Он является одной из основных
составных частей скипидара, имеет приятный запах хвои. Ши¬
роко применяется как растворитель для приготовления лаков и
красок. Из пинена получают синтетическую камфору и другие
соединения, а также инсектициды (полихлорпинен), масла для
флотации руд. При гидрировании а-пинен дает пинан, который
в природе не встречается.
а-пинен
борнеол
*
камфора
Борнеол (производное борнана)—распространенный био-
циклический терпеновый спирт. Встречается во многих эфир¬
ных маслах: правовращающий изомер — в камфарном, лавандо¬
вом и розмариновом маслах, левовращающий — в пихтовом
масле. Борнеол—твердое кристаллическое вещество, мало рас¬
творим в воде, легко — в органических растворителях. Любо¬
пытно необычайно малое различие между температурами плав¬
ления и кипения этого спирта (они равны соответственно 204
и 210°С), поэтому борнеол легко возгоняется.
Камфора — кристаллическое вещество, т. пл. 179 °С, обла¬
дает характерным запахом. Наличие в молекуле двух асиммет¬
рических атомов углерода (узловые атомы, отмеченные в фор¬
муле звездочками) должно было бы, по общему правилу, вы¬
зывать существование четырех оптических изомеров. В дей¬
ствительности же известна лишь пара оптических антиподов
камфоры: (+)- и (—)-камфора и оптически неактивная (ра¬
цемическая) камфора. Меньшее число стереоизомерных форм
камфоры объясняется тем, что в жесткой бициклической систе¬
ме, которую представляет собой камфора, конфигурации обоих
асимметрических атомов взаимосвязаны. Эти конфигурации
можно изменить лишь одновременно.
В природе камфора чаще всего встречается в листьях, кор¬
нях и древесине камфорного лавра в виде (-f-)-камфоры. Мас¬
ло камфорного лавра и служит источником природной камфо-
398
ры. В меньших количествах она содержится в других эфирных
маслах (базиликовом, полынном, шалфейном). Камфору ис¬
пользуют в медицине как средство, возбуждающее сердечную
деятельность. Большие количества камфоры расходуются в ка¬
честве пластификатора при получении целлулоида (см. § 159).
Поскольку природные источники не могут полностью удовле¬
творить потребность в камфоре, ее производят и_синтетическим
путем. В СССР камфору получают дегидрированием борнеола,
извлекаемого из эфирного масла сибирской пихты, в котором
он содержится в количестве около 40 % (свободный и в виде
ацетата):
В другом способе в качестве исходного вещества использу¬
ют а-пинен, являющийся главной составной частью скипидара.
Пары пинена пропускают над катализатором (ТЮа), при этом
идет перегруппировка его углеродного скелета с образованием
камфена. К камфену присоединяют уксусную кислоту, эта реак¬
ция также сопровождается перегруппировкой и приводит к бор-
нилацетату:
тю2
CHgCOOH
(h2so4)
у
OCOCHg
Далее борнилацетат омыляют в борнеол, из которого окис¬
лением (дегидрированием) получают камфору.
Перегруппировки углеродного скелета, подобные приведен¬
ным выше,— весьма частое явление при химических превраще¬
ниях бициклических терпенов. Именно они придают большое
своеобразие этому классу соединений; это свойство сильно ос¬
ложняло установление строения терпенов. Русский ученый
Е. Е. Вагнер, который внес большой вклад в изучение терпенов,
образно называл их «химическими хамелеонами».
§ 183. Стероиды. В основе стероидных соединений лежит тетра¬
циклическая структура циклопентанопергидрофенантрена. Схе¬
ма ее с принятой нумерацией приводится ниже (ядра принято
обозначать латинскими буквами):
399
В стероидах содержатся обычно метильные группы в поло¬
жениях 10 и 13, боковые цепи и заместители (чаще всего в
положениях 3 и 17) и кратные связи. Все узловые атомы цикли¬
ческой структуры (5, 8, 9, 10, 13, 14) являются асимметриче¬
скими (кроме того, асимметрия появляется и при монозамеще¬
нии других атомов), вследствие чего стероиды имеют большое
число пространственных изомеров. В сочетании со структурной
изомерией это создает большое разнообразие стероидных со¬
единений.
Соединения стероидного типа широко распространены в при¬
роде. К стероидам относится холестерин — жироподобное ве¬
щество, содержащееся в каждой клетке животного организма.
В растительных клетках содержатся иные стероиды. Стероид-
-ный скелет имеют желчные кислоты, яды жаб, некоторые ядо¬
витые растительные вещества (сапонины). Стероидную природу
имеют и гормоны, вырабатываемые поджелудочной железой и
половыми железами.
Холестерин представляет собой жирные на ощупь пластин¬
ки, т. пл. 149 °С. Нерастворим в воде, растворим в органических
растворителях. Он содержится во всех тканях животных и че¬
ловека, особенно много его в мозге, печени, почках, а также в
крови. Нарушение нормального содержания холестерина в
крови у человека (160—220 мг в 100 мл) наблюдается при не¬
которых заболеваниях. Часть холестерина поступает с пищей
(особенно много его в жирах, яичном желтке), однако «80%
синтезируется, как теперь установлено, в самом организме. По¬
этому отложение холестерина на стенках кровеносных сосудов
при атеросклерозе является в первую очередь следствием на¬
рушения холестеринового обмена в организме, а не избыточно¬
го поступления холестерина с пищей.
Гормоны представляют собой вещества, вырабатываемые
железами внутренней секреции и являющиеся регуляторами
важнейших жизненных процессов. Мы рассмотрим" здесь неко¬
торые стероидные гормоны, хотя известны гормоны полипептид-
ной природы (инсулин) и ароматического ряда (адреналин).
Стероидные гормоны вырабатываются корой надпочечников и
половыми железами. Они регулируют рост организма и процес¬
сы его старения, влияют на функции размножения и многие
другие стороны жизнедеятельности. Ниже приведены формулы
мужского (тестостерон) и женского (эстрон) половых гормо¬
нов:
ОН
О
тестостерон
эстрон
400
Кортизон — один из 20 гормонов, вырабатываемых корой
надпочечников. Регулирует обмен углеводов, применяется при
лечении многих тяжелых болезней (ревматизм, бронхиальная
астма, воспалительные процессы, аллергические заболевания).
Кортизон получают синтетически, исходя из более доступных
стероидных соединений, например холестерина или соласодина
(последний выделяют из растения «паслен птичий»). Получают
синтетически и аналоги кортизона, например преднизолон.
ГЛАВА 23
ВЫСОКОМОЛЕКУЛЯРНЫЕ СОЕДИНЕНИЯ
§ 184. Классификация. Связь строения со свойствами. В пре¬
дыдущих главах мы неоднократно упоминали о различных
органических соединениях, отличающихся большим размером
молекул; к ним относятся каучуки, белки, полисахариды. По¬
добные соединения с молекулярными массами от нескольких
тысяч до миллионов получили название высокомолекулярных
(полимерных). Некоторые из них выполняют важные функции
в живых организмах. В настоящее время научились синтезиро¬
вать много разных высокомолекулярных соединений, нашедших
применение для изготовления различных материалов: пласт¬
масс, волокон, эластомеров. Для этих материалов очень важны
физико-механические свойства — прочность, эластичность, термо¬
стойкость. Установлено, что физико-механические свойства вы¬
сокомолекулярных соединений зависят прежде всего от формы
молекул.
По форме молекул различают два основных типа полимеров!
линейные (нитевидные) и трехмерные (объемные, глобуляр¬
ные). Существует много полимеров, занимающих промежуточ¬
ные положения между этими крайними типами.
Характерные особенности линейных полимеров — это способ¬
ность образовывать прочные волокна и пленки, значительная
эластичность, способность растворяться, а при повышении тем¬
пературы— плавиться. Типичные представители — каучук и его
синтетические аналоги, полиамидные волокна.
Трехмерные полимеры не плавятся, нерастворимы, значи¬
тельно менее эластичны, чем линейные полимеры, часто даже
401
хрупки. В таких полимерах, собственно говоря, утрачивают свой
смысл понятия—молекула и молекулярная масса: каждый
предмет из трехмерного полимера — это одна гигантская моле¬
кула. Употребляемое иногда для них название «сшитые» напо¬
минает о том, что линейные полимеры можно превратить в
трехмерные, «сшивая» цепные макромолекулы в пространствен¬
ную сетку. Именно такой процесс происходит при вулканизации
каучука. Другие типичные представители трехмерных полиме¬
ров— это фенолоформальдегидные и глифталевые смолы.
Конечно, не только форма, но и химическая природа макро¬
молекулы влияет на физико-механические свойства полимера.
Если между макромолекулами линейного полимера не возни¬
кает значительного взаимодействия (а это значит, что в макро¬
молекуле нет сильно взаимодействующих друг с другом поляр¬
ных групп), то макромолекулы могут легко передвигаться друг
относительно друга, и материал оказывается тягучим. Таковы
невулканизованный каучук, полиэтилен (особенно при нагрева¬
нии). Эластичность таких материалов ограничена. По мере того
как возрастает взаимодействие между макромолекулами линей¬
ного полимера (т. е. по мере накопления в полимере полярных,
взаимодействующих друг с другом групп), его свойства посте¬
пенно приближаются к свойствам трехмерного полимера. Имен¬
но так обстоит дело у целлюлозы, линейные макромолекулы
которой сильно связаны друг с другом водородными связями.
Того же результата можно достигнуть, химически «сшивая» ли¬
нейные макромолекулы.
В зависимости от состава основной цепи различают поли¬
меры карбоцепные (полимерная цепь состоит только из атомов
углерода) и гетероцепные (в состав полимерной цепи входят
атомы разных элементов). Важный класс образуют элементор-
ганические полимеры, в которых помимо обычных элементов-
органогенов входят атомы других элементов — кремния, фос¬
фора, бора, титана и др.
Высокомолекулярные соединения возникают в результате со¬
единения множества молекул низкомолекулярных веществ —
мономеров. Важнейшими мономерами являются соединения сле¬
дующих классов: алкены (этилен, пропилен, изобутен); диены
(бутадиен, изопрен); виниловые мономеры (виниловые простые
эфиры, винилацетат, винилхлорид, винилиденхлорид); много¬
атомные спирты (этиленгликоль, глицерин, пентаэритрит); фенол
и его гомологи; альдегиды (формальдегид, в меньшей степени
ацетальдегид); производные ненасыщенных кислот (эфиры и
нитрилы акриловой, метакриловой, малеиновой кислот); дикар-
боновые кислоты (адипиновая, малеиновая, терефталевая, фта-
левая); полиамины (гексаметилендиамин); соединения амидно¬
го типа (мочевина, капролактам).
Мономеры могут соединяться друг с другом в результате
реакций полимеризации и поликонденсации.
402
ПОЛИМЕРИЗАЦИОННЫЕ ВЫСОКОМОЛЕКУЛЯРНЫЕ
СОЕДИНЕНИЯ
§ 185. Реакции полимеризации. Реакции полимеризации заклю-
чаются в присоединении друг к другу большого числа моле»
кул мономеров:
пМ —>• М „
где М — молекула мономера, М„ — молекула полимера (макромолекула),
в —число мономерных звеньев в макромолекуле (степень полимеризации).
Реакции полимеризации идут в результате присоединения к
кратным связям или за счет раскрытия циклов. Для этого до¬
статочно, чтобы в мономере была одна реакционноспособная
группировка.
Реакции полимеризации обычно имеют цепной характер.
Цепной реакции дает толчок какой-то инициатор. Инициатором
часто служат вещества, легко распадающиеся на свободные ра¬
дикалы. Такой радикал переводит молекулу мономера в ради¬
кальную форму и тем самым вызывает начало цепной реакции
полимеризации (радикальная полимеризация). Так, например,
стирол под действием пероксида бензоила превращается в поли¬
стирол.
Образование инициатора:
[С6Н5СОО], —► с„н6 - + СО»
пероксид
бензоила
Начало цепи:
СвН5—СН=СН, + С6Н5.
стирол
- СН—CHS—С„Н4
с6н5
Процесс полимеризации:
CeH5CH=CHs
СН=СН, + • СН—СН2—СвН5 —► • СН—сн2— СН—сн,—с„н5 *-
i6H, C„HS ieH5 СвН5
—► • СН—СН2—СН—CHj—СН—СН2—СвН5 и т. д.
ieHs СвНв CqHs
Цепная полимеризация идет с большой скоростью даже при
низких температурах. Иногда скорость полимеризации так вы¬
сока, что реакция проходит взрывоподобно. Промежуточные
Продукты полимеризации — неустойчивые частицы. В нашем
примере это радикалы, но могут быть и ионы. Рост цепи после
первоначального инициирования реакции продолжается до тех
пор, пока не исчерпается весь мономер или не произойдет об¬
рыв цепи.
403
Кроме радикальных известны реакции полимеризации, иду¬
щие по ионному механизму: активными частицами их могут
быть положительные ионы (катионная полимеризация) или от¬
рицательные (анионная полимеризация).
Мономеры, полимеризующиеся по одному из механизмов,
могут оказаться инертными в условиях проведения полимери¬
зации по другому механизму. Так, например, стирол подверга¬
ется катионной полимеризации, но не анионной. Метилметакри¬
лат, наоборот, полимеризуется по анионному механизму, но не
по катионному. В условиях радикальной полимеризации оба
мономера обладают примерно одинаковой активностью.
Известна не только цепная, но и ступенчатая полимеризация,
на каждой стадии которой возникают устойчивые частицы.
Примером ее может служить полимеризация изобутена в при¬
сутствии серной кислоты (см. § 33).
В технике используют ряд вариантов проведения полимери¬
зации. Блочная полимеризация проводится с жидкими мономе¬
рами и приводит к образованию монолитных блоков полимера.
Полимеризацию можно проводить и в растворителе, способном
растворять мономер; полимер при этом может либо остаться в
растворе, либо выпасть из него. Эмульсионная полимеризация
проводится с мономером, взвешенным в виде капелек в нерас¬
творяющей его жидкости: в результате такой полимеризации
возникает латекс — суспензия мельчайших частиц полимера, по¬
добная молоку (в котором частицы жира суспендированы
в водной среде). Большое значение имеет совместная полимери¬
зация смеси мономеров: такой процесс называется сополимери-
зацией. Меняя состав смеси мономеров, можно тонко регули¬
ровать свойства получаемых материалов.
§ 186. Полиалкены. При полимеризации алкенов образуются
цепные высокомолекулярные материалы.
Первые наблюдения относительно «уплотнения» этиленовых
углеводородов принадлежат еще А. М. Бутлерову. Он, в частно¬
сти, описал получение димера изобутена и отметил, что этот
процесс может идти дальше, с присоединением третьей и по¬
следующих молекул. Осуществить полимеризацию алкенов с
образованием твердых полимерных продуктов удалось значи¬
тельно позднее, лишь в 40—50-х годах нашего столетия.
Простейший из алкенов — этилен — можно полимеризовать
различными способами: под давлением 120—150 МПа, иниции¬
руя полимеризацию кислородом или органическими пероксида¬
ми (полиэтилен высокого давления); при низком давлении на
металлорганических катализаторах — смесях алюминийалкилов
с солями титана (полиэтилен низкого давления).
СН2=СН2 —► СН2—СН2—СН2—СН2—СН2—СНг—СНг—СН2
Полиэтилен — это по существу насыщенный углеводород с
молекулярной массой от 20 тыс. до 1 млн. Он представляет
404
собой прозрачный материал, обладающий высокой химической
стойкостью; температура размягчения 100—130 °С; разрушаю¬
щее напряжение при растяжении 12—34 МПа, имеет низкую
тепло- и электропроводности. Полиэтилен применяют для изо¬
ляции электрических проводов, изготовления прозрачных пле¬
нок, которые используют в качестве упаковочного материала,
вместо стекол для укрытия растений в парниках. Из полиэти¬
лена производят также посуду и другие изделия.
Полипропилен получают из пропилена аналогично полиэти¬
лену. Долгое время считали, что при полимеризации пропилена
можно получать лишь маслообразные продукты. Когда же на¬
учились проводить стереоспецифическую полимеризацию (см.
ниже) пропилена, оказалось, что при этом получается прозрач¬
ный материал с температурой размягчения 160—170°С (раз¬
рушающее напряжение при растяжении 26—35 МПа), обладаю¬
щий хорошими электроизоляционными' свойствами. Продавли¬
вая расплав полипропилена через фильеры, получают нити
полипропиленового волокна. Это волокно обладает большой
прочностью, химической стойкостью. Из него изготовляют кана¬
ты, рыболовные сети, фильтровальные ткани. Применение по¬
липропиленового волокна в текстильной промышленности не¬
сколько ограничено из-за его невосприимчивости к обычным
красителям. Однако имеются красители, окрашивающие этот
полимер в массе.
Сущность стереоспецифической полимеризации пропилена
заключается в создании строго упорядоченной пространствен¬
ной структуры линейной цепи полимера. При полимеризации в
цепи полимера возникают асимметрические атомы углерода. Их
конфигурация может чередоваться беспорядочно; это характер¬
но для атактического полипропилена.
Условно изображают, что главная цепь макромолекулы по¬
липропилена лежит в плоскости бумаги *, а атомы водорода и
группы СН3 «торчат» по обе стороны от этой плоскости.
В атактическом полипропилене группы СН3 и атомы водорода
располагаются то «под плоскостью» (пунктирные клинья), то
«над плоскостью» (жирные клинья), без какой-либо регуляр¬
ности;
Н СН3 СН3 Н Н «СН3Н чЧСН, СН, Л
v# \V \ 3 \ /• 3 V/
/-*4.
СЩ -СНз СНг CHj CHj
атактический полипропилен
При стереоспецифической полимеризации возникают про¬
странственно упорядоченные структуры.
* Это не вполне точно, так как в действительности главная цепь образует
пространственную спираль.
405
В изотактическом полимере все одинаковые группы (СН3)’
находятся по одну сторону плоскости чертежа («к нам»), а все
атомы Н — по другую («от нас»).
н сн, н
сн, чн
V >
сн, чн
А \3 А
\* А
\ /с\.
/с\
сн2 сн2
сн2
изотактический полипропилен
Именно изотактические стереорегулярные полимеры облада¬
ют наиболее ценными физико-механическими свойствами. Изо¬
тактические полимеры получают и из стирола.
Н
к
£бН3
Н £еНк
'с.
Н
5
н сйн,,
\/в 9
н
У
С,Щ
—сн
2
сн
сн.
сн.
2 V-J12
изотактический полистирол
СН»
Полистирол — бесцветный прозрачный материал, хорошо из¬
вестный в виде различных изделий из «органического стекла».
Температура размягчения атактического полистирола «85°С,
а для изотактического полистирола она равна 230 °С, что по¬
зволяет использовать последний при более высоких температу¬
рах. Большое применение имеют и сополимеры стирола с дру¬
гими мономерами — акрилонитрилом, метилметакрилатом, а-ме-
тилстиролом.
Сополимеры имеют более высокие физико-механические по¬
казатели, чем чистый полистирол. Особенно ценный ударопроч¬
ный полистирол получают методом блок-сополимеризации, в
результате которой участки (блоки), возникшие путем полиме¬
ризации одного мономера, чередуются с блоками из другого мо¬
номера. Используя прием стереоблочной полимеризации, можно
достичь того же результата, не используя смесь мономеров: сте-
реоблочный полимер возникает из одного мономера, блоками в
этом случае являются участки, различающиеся не составом, а
пространственным строением. Ниже приведена схема стерео-
блочного полимера, состоящего из изотактических блоков, разде¬
ленных участками цепи неупорядоченного пространственного
строения:
Изотактический
блок
VWVTVH
f
Изотактический
блок
Неупорядоченное
WWV
I
Изотактический
блок
звено
Неупорядоченное
звено
406
В зависимости от протяженности блоков, частоты и разме¬
ров неупорядоченных участков могут меняться свойства поли¬
мера. Известно, в частности, что упорядоченные, кристалличе¬
ские участки полимера обусловливают его прочность, а неупо¬
рядоченные, аморфные — эластичность. Для получения матери¬
ала с заданными свойствами необходимо создать соответствую¬
щее соотношение между кристаллической и аморфной частями.
Интересно, что аналогично обстоит дело и в металлах: их
прочность и упругость также зависят от степени кристаллич¬
ности.
§ 187. Виниловые полимеры. Кроме углеводородов способны
полимеризоваться и многие другие соединения с двойной связью,
носящие название виниловых мономеров. Их общая формула
СН2=СН—X (где Х = хлор, CN, СООСН3 и др.). Формула по¬
лучаемых полимеров следующая:
СН2—СН—СН2—СН—СН2—СН—СН2—СН
Из полимеров этого типа особое значение имеет поливинил¬
хлорид (Х = С1).
Поливинилхлорид — прочный термопластичный материал,
молекулярная масса 300—400 тыс. При обычной температуре —
это твердый материал, однако его можно сделать мягким и
гибким, смешивая с труднолетучими растворителями, так назы¬
ваемыми пластификаторами — эфирами фталевой или фосфор¬
ной кислот, например дибутил- и диоктилфталатами, трикрезил-
фосфатом и др. Из пластифицированного поливинилхлорида из¬
готовляют гибкие листы (для покрытия полов, отделки стен),
пленки, формуют под давлением разные изделия, употребляют
для производства искусственной кожи, защитных перчаток. Из
жесткого непластифицированного поливинилхлорида изготов¬
ляют трубы (они не подвергаются коррозии и заменяют свинцо¬
вые при изготовлении химической аппаратуры), детали дверей и
окон. В электротехнике поливинилхлорид служит для изоляции
проводов и изготовления деталей аппаратуры. Производят из
него и игрушки, спортивные и канцелярские товары, скатерти,
занавески. Из поливинилхлорида можно получать и волокна.
Это один из самых дешевых видов синтетического волокна. Его
применяют для изготовления фильтровальных технических тка¬
ней, рыболовных сетей, трикотажа и медицинского белья. При¬
меняя особую обработку, поливинилхлорид можно получить в
виде пористого, напоминающего губку материала — пенополи-
винилхлорида. Из него готовят искусственную кожу, подложки
для ковров, покрытия для пола.
Большое значение приобрели благодаря термостойкости и
химической устойчивости полимеры из фторированных этиле¬
401
нов — фторопласт-3 из трифторхлорэтилена и фторопласт-4 из
тетр афторэтил ена:
«CFj=CClF —► CF2—CCIF—CF2—CC1F
фторопласт-3
rtCFs=CFj ■ v •••—CF2—CF2—-CF2—CF2—CF2—CF2—• ••
фторопласт-4
Фторопласты по химической инертности приближаются к
благородным металлам: на них не действуют концентрирован¬
ные кислоты, окислители, водные растворы щелочей, никакие
обычные органические растворители. Их можно использовать
при температурах 250—300 °С. Эти полимеры имеют важное
значение во многих областях техники.
Широко используют полиметилакрилат и полиметилметакри-
лат, получаемые из эфиров ненасыщенных кислот — акриловой
и метакриловой. Эти мономеры также относятся к виниловым.
Из них получается родственное полистиролу «органическое
стекло»:
пСН^СН —► —сн2—сн—сн2—сн—сн2—сн
Ценные материалы получают при полимеризации нитрила
акриловой кислоты (акрилонитрила):
яСН2=СН —*■ —сн2—СН—сн2—СН—сн2—СН
Из полиакрилонитрила изготовляют волокно нитрон. Эти
волокна можно получать сухим прядением — продавливанием
через фильеры нагретого до 120—130 °С раствора полиакри¬
лонитрила в диметилформамиде в шахту с горячим (180—
250°С) воздухом. В ней испаряется растворитель и образуется
нить. Более распространен способ мокрого прядения, заключаю¬
щийся в том, что раствор полимера после фильеры поступает в
осадительную ванну (смесь растворителя с водой). Получаемую
нить используют для изготовления трикотажных изделий, кос¬
тюмных и технических тканей.
При нагревании на воздухе до температур порядка
200—250 °С полиакрилонитрил циклизуется и дегидрируется,
превращаясь в полимерный материал, отличающийся
СООСНз
СООСНз СООСНз СООСНз
метилакрилат
СНз
СН3 СНз СНз
полиметилакрилат
СООСНз
СООСНз СООСНз
СООСНз
метилметакрилат
полиметилметакрилат
CN
CN
CN
CN
408
термостойкостью и имеющий полупроводниковые свойства!
СН2 СНа
/\/\
-НС сн сн-
^ L i
Ч|
N
N N
Мономерами служат также простые и сложные эфиры вини¬
лового спирта (см. §§ 79 и 110), например винилацетат. По¬
ливинилацетат применяют для приготовления лаков, клеев
(канцелярский клей ПВА), пропитывают им ткань для прида¬
ния ей несминаемости. При гидролизе в присутствии небольшо¬
го количества щелочи из поливинилацетата получают поливини¬
ловый спирт\
[—СН2—СН— 1 н20. "ОН Г—СН2—СН—П
0-CO-CH3J„ * L ОН
В отличие от других полимеров поливиниловый спирт вслед¬
ствие наличия гидроксигрупп легко растворяется в воде, но не¬
растворим в углеводородных органических растворителях. По¬
ливиниловый спирт используют для получения синтетического
волокна «винол». Благодаря наличию свободных гидроксильных
групп в это волокно можно вводить остатки различных карбо¬
новых кислот придавая получающемуся модифицированному
полимеру ионообменные, бактерицидные и другие необходимые
свойства. Ценным свойством винилового волокна является его
гигроскопичность; пленки из поливинилового спирта отличаются
малой газопроницаемостью.
§ 188. Каучук. Натуральный каучук представляет собой упру¬
гую аморфную массу, получаемую из млечного сока (латекса)
растений-каучуконосов. Наиболее известный каучуконос — дере¬
во гевея, родина которого — тропические леса Бразилии. Мень¬
шие количества содержатся в ряде других растений, преимуще¬
ственно тропических.
В XVIII веке образцы каучука были привезены в Европу, но особенного
применения этот материал не находил. Лишь в 1823 г. ирландец Мак-Интош
открыл способ пропитки тканей каучуком и стал изготовлять из такой ткани
непромокаемые плащи. Качество таких плащей было невысоким: в теплую
погоду пропитывавший их каучук делался липким, зимой же становился
твердым и хрупким. История промышленного применения каучука началась
в 1839 г., когда путем обработки серой (вулканизация) сырой каучук научи¬
лись превращать в резину — материал, в котором особенно ценным свойством
является эластичность (упругость). С этого времени начался быстрый рост
промышленного применения каучука. Наибольшие его количества стала
вскоре потреблять автомобильная промышленность, на втором месте стоит
409
электротехническая промышленность и производство различных резинотехни¬
ческих изделий.
При изучении химических свойств каучука было установле¬
но, что это типичное непредельное соединение: присоединяет
бром, бромоводород, а также подвергается каталитическому
гидрированию. При сухой перегонке, т. е. при нагревании без
доступа воздуха, каучук распадается с образованием диено¬
вого углеводорода — изопрена (2-метилбутадиен-1,3).
Основу современных представлений о строении каучука за¬
ложил Гарриес в 1909—1912 гг., применивший для этих целей
реакцию озонирования. С тех пор эту реакцию часто исполь¬
зуют для исследования непредельных соединений (см. § 33).
При озонировании каучука Гарриес получил левулиновый
альдегид СН3—СО—СН2—СН2—СНО. Это доказывает, что ка¬
учук — линейный полимер, в котором молекулы изопрена со¬
единены друг с другом по схеме 1,4-присоединения:
СН3 СНз
I I Оз
—сн2—сн=с—сна—сн2—сн=с—сн2— —►
О СНз о СНз
/ \ 1 /41 н2о
—► —сн2—нс' х:—сн2—сн2—нс' ^с—сн2— >-
0—0 0—0
/Н Н3СЧ /Н н,сч
—„ сн2—с' + хс—сн2—сн2—о( + ХС—сн2
Чо 4)0^
Натуральный каучук является смесью полимергомологов —
'молекул с разными молекулярными массами — от 50 тыс. до
3 млн. Основную массу составляют фракции с молекулярной
массой более одного миллиона.
Способность изопрена полимеризоваться под действием УФ-
света и химических реагентов была известна еще в конце про¬
шлого столетия. Естественно, что не- было недостатка в попыт¬
ках воспроизвести натуральный каучук полимеризацией изопре¬
на. Однако долго эти попытки не приводили к успеху; причина
выяснилась лишь после установления всех деталей строения
каучука.
Натуральный каучук — линейный полимер изопрена, обла¬
дающий не только строго регулярным строением, но и опреде¬
ленной конфигурацией у двойной связи. Каучук имеет строение
1,4-полиизопрена с грш-конфигурацией полимерной цепи:
Н/ /СНз Н/ /СНз
с=с сн2 сн, с=с
/ \/\ / \ / \
—сн2 сн2 с=с сн2 сн2
y/ \сн3
41»
Трудность воспроизведения натурального каучука заключа¬
ется в том, что надо синтетически построить эту строго регу¬
лярную (без разветвлений) цепь цыс-конфигурации.
В СССР первые исследования по изучению полимеризации
диеновых углеводородов связаны с именем С. В. Лебедева.
Предложенный им метод было решено положить в основу про¬
ектирования первого в мире крупного производства синтетиче¬
ского каучука. Уже в 1931 г. на опытном заводе были получе¬
ны первые 260 кг синтетического каучука, а через год вступили
в строй два завода синтетического каучука.
Сырьем для получения синтетического каучука по Лебедеву
служит этиловый спирт. Под действием разработанного Лебеде¬
вым катализатора (смесь оксидов алюминия, цинка и некоторых
других металлов) происходят одновременно дегидратация и де¬
гидрирование этилового спирта с образованием бутадиена:
Бутадиен очищают от непрореагировавшего этилового спир¬
та и многочисленных побочных продуктов и подвергают полиме¬
ризации под действием металлического натрия. Полимеризация
идет не только в положения 1,4, но и в положения 1,2, поэтому
получается не строго линейная, а разветвленная макромолекула
(пунктиром отделены друг от друга мономерные звенья):
Лишь в 50-е годы научились проводить стереоспецифическую
г(т. е. приводящую к созданию строго регулярной пространствен¬
ной формы) 1,4-полимеризацию бутадиена под действием ме-
таллорганических катализаторов (алюминийалкилы в смеси с
солями титана, циркония и другими добавками). Это позволило
существенно улучшить качество бутадиенового синтетического
каучука. Стереорегулярный бутадиеновый каучук имеет, подоб¬
но натуральному изопреновому, полимерную цепь цис-конфигу¬
рации:
Изменилась в настоящее время и сырьевая база промыШ'
ленности синтетического каучука. Исходный этиловый спирт
теперь получают не из пищевого сырья, а из этилена или иро-
2С2Н5ОН —► СНг==СН—СН=СН2 + 2Н20 + Н2
н/ \н
411
дуктов гидролиза древесины. Все шире используют и непосред¬
ственный переход от бутана и бутена (газы крекинга нефти)
к бутадиену путем дегидрирования. В странах, не располагаю¬
щих нефтью, например в Германии, производство бутадиена
было организовано на базе угля через уксусный альдегид или
бутиндиол-1,4.
Бутадиеновый синтетический каучук явился первым каучу¬
ком, производство которого было освоено в крупных промыш¬
ленных масштабах во многих странах. Первые заводы были
построены в СССР (1932 г.), а в 1937—1940 гг. производство
бутадиенового синтетического каучука было организовано в
Германии и в США. Этот вид синтетического каучука не поте¬
рял значения и сейчас (особенно в виде стереорегулярного по¬
лимера), но наряду с ним производят и другие виды синтетиче¬
ского каучука: сополимеры бутадиена с другими мономерами
(стиролом, акрилонитрилом, винилметилпиридином). Быстро
развивается производство стереорегулярного изопренового кау-.
чука, по строению и свойствам полностью воспроизводящего на¬
туральный. Необходимый для этого изопрен получают катали¬
тическим дегидрированием изопентана и другими способами.
Промышленное значение имеет и хлоропреновый синтетиче¬
ский каучук, сырьем для которого служит ацетилен (см. § 39).
Полимеризация хлоропрена в положения 1,4 дает полимер:
—сн2—с=сн—сн2—сн2—с=сн—сн5—сн2—с=сн—сн2
I I I
а а а
Молекулярная масса хлоропренового каучука более 100 тыс.
Изделия из этого материала превосходят натуральный каучук
по свето- и термостойкости, по сопротивлению действию нефтя¬
ных продуктов. Кроме того, хлоропреновый каучук негорюч
1,(он содержит около 40 % хлора).
В настоящее время производят очень много разных видов
синтетического каучука, каждый из которых имеет свои преиму¬
щества в определенной области применения. Так, бутадиен-сти-
рольный каучук из-за высокой износостойкости (прочности при
истирании) особенно пригоден для изготовления автомобильных
шин; малая газопроницаемость бутилкаучука (полимер изобуте¬
на) делает его особо пригодным для изготовления автомобиль¬
ных камер. Хлоропреновый каучук, как мы уже говорили, стоек
к маслам, растворителям; этилен-пропиленовый каучук устой¬
чив к действию озона. Силоксановые каучуки можно использо¬
вать при высоких температурах, они сохраняют эластичность
и при низких температурах (см. § 193).
В 1980 г. мировое производство синтетического каучука со¬
ставляло более 7 млн. т в год, примерно в 2 раза превышая
производство натурального каучука.
412
Для изготовления резины каучук подвергают вулканизации,
предварительно смешивая с разными добавками, улучшающими
качество,— наполнителями, противостарителями, красителями
и др.
В первые десятилетия XX века каучук являлся единствен¬
ным в своем роде незаменимым материалом, без которого не¬
возможно было представить себе автомобильный транспорт,
авиацию, электро- и радиотехнику, изготовление многочислен¬
ных бытовых изделий. В настоящее время каучук утратил свое
монопольное положение; появились другие синтетические поли¬
меры, успешно заменившие каучук во многих областях.
ПОЛИКОНДЕНСАЦИОННЫЕ ВЫСОКОМОЛЕКУЛЯРНЫЕ
СОЕДИНЕНИЯ
§ 189. Реакции поликонденсации. В реакции поликонденсации
участвуют полифункциональные мономеры, молекулы которых
присоединяются друг к другу с отщеплением какой-либо прос¬
той молекулы (обычно воды). Классический пример — поликон¬
денсация адипиновой кислоты с гексаметилендиамином, приво¬
дящая к получению полиамида:
СООН NH» СООН
| | HOOC—<СН2)4-СООН
(СН2>6 -тгтГ (сн,)4 : *
I | -н2о ,
СООН NH2 СО—NH—(CH2)e—nh2
(СН2)4—СООН
I H2N-(CH2)u-NH2
—► СО—NH—(CH2)e—NH—СО—(СН2)4—СООН •>
(СН2)4—СООН
—► СО—NH—(СН2)6—NH—СО—(СН2)4—СО—NH—(CH2)e—NH2 и т. д.
Процесс поликонденсации идет ступенчато: одна молекула
присоединяется за другой. Все промежуточные продукты — ус¬
тойчивые соединения, которые нужно каждый раз активировать
для осуществления дальнейшего присоединения. Активируют
нагреванием. В процессе поликонденсации молекулярная масса
полимера постепенно растет.
Кроме двух главных путей синтеза высокомолекулярных соединений —
поликонденсапии и полимеризации — имеет значение еще один способ — по-
липрисоединение. В частности, именно этим путем получают ценный мате¬
риал — полиуретановый каучук. Мономерами служат диизоцианаты и гли-
коли, реагирующие по схеме:
0=C=N—(СН2)„—N=C=0 + НО—(CH2)m—ОН —->
—* О—(СН2)т—О—СО—NH—(СН2)„—NH—СО—О—(СН2)т—О
§ 190. Полиамиды. Важную группу синтетических материалов
образуют полиамиды — высокомолекулярные соединения, в ко¬
торых мономерные звенья соединены группами СО—NH. По
(СН2>4 +
413
строению полиамиды родственны белковым веществам (см.
§ 175). Сырье для получения полиамидов менее доступно, чем
простые виниловые мономеры; это делает полиамиды более доро¬
гими материалами. Несмотря на это, из-за исключительно ценных
физико-механических свойств полиамиды производят в больших
количествах. Их главная область применения — изготовление
синтетических волокон. Эти полимеры получают путем поли¬
конденсации. Из адипиновой кислоты и гексаметилендиамина по¬
лучают полимер СО—(СН2)4—СО—NH—(СН2)б—NH ,
Продавливая расплав этого полимера при 280 °С через фильеры,
получают волокно анид (СССР), найлон 66 (США), перлон Т
(ФРГ).лурон (Англия).
Исходным веществом для получения полиамидного волокна
капрон (в США его называют найлон-6, в ФРГ — перлон, в
ГДР — дедерон, в Чехословакии — силон, в Швейцарии — гри-
лон) служит капролактам:
Н2С—сн2—СО
^NH —► NH—(СН2)5—СО—NH—(СН2)е—СО
Н2С—СН2—Oi2
Родственное волокно — энант — получают при поликонденса¬
ции ш-аминоэнантовой кислоты. Оно имеет строение:
NH—(СН2)5—СО—NH—(СН2)в—СО—NH—(СН2)в—СО
Исходную мономерную кислоту синтезируют из этилена и
четыреххлористого углерода (реакция теломеризации):
NH3
ЗСН2=СН2 + ССЦ —► С1—сн2—сн2—сн2—сн2—сн2—сн2—ссц —►
н2о
—► H2N—(СН2)в—ССЦ *- H2N—(СН2)6—соон
Получают полимеры и из аминокарбоновых кислот, содер¬
жащих 8—12 атомов углерода. -
Особо ценные свойства имеют полиамиды, получаемые на
основе ароматических соединений. Так, при поликонденсации
м-фенилендиамина и хлорангидрида м-фталевой (изофталевой)
кислоты образуется полиамид, известный в нашей стране под
названием фенилон (в США — номекс). Это волокно можно
использовать при температуре до 300 °С. Еще более высокой
термостойкостью обладают так называемые «лестничные» поли¬
амиды, получающиеся при поликонденсации ароматических те¬
трааминов с диангидридами ароматических тетракарбоновых
кислот, например:
[
414
Как легко понять, особая устойчивость обеспечивается здесь'
двойной полимерной цепью. Рекордную прочность и термостой¬
кость (до 600—700 °С) имеет полимер из дифенилового эфира
изофталевой кислоты и 3,3',4,4'-тетрааминобифенила. В костю¬
мах из такой ткани американские астронавты высаживались на
Луну.
Полиамидные волокна отличаются высокой прочностью (раз¬
рушающее напряжение при растяжении до 400 МПа), которая
почти не изменяется и во влажном состоянии, хорошей упру¬
гостью, устойчивостью к действию многих химических реаген¬
тов, микроорганизмов. Волокна эти хорошо окрашиваются.
Полиамидные волокна используют для изготовления бытовых
тканей, кроме того, они имеют и важное техническое примене¬
ние: из них изготовляют парашюты, каркасы высокопрочных
шин для автомашин и самолетов, транспортерные ленты, рыбо¬
ловные сети, фильтровальные ткани.
Для большей устойчивости к действию света и кислорода
воздуха полиамидные волокна стабилизируют добавлением со¬
лей некоторых металлов, ароматических аминов. Для повыше¬
ния белизны вводят оптические отбеливатели — вещества, по¬
глощающие ультрафиолетовый свет и преобразующие его в
голубой.
§ 191. Полиэфиры. Путем поликонденсации дикарбоновых кис¬
лот с многоатомными спиртами получают высокомолекулярные
материалы полиэфирного типа. Примером может служить поли-
этилентерефталат — высокомолекулярный сложный эфир эти¬
ленгликоля и терефталевой кислоты:
Вместо терефталевой кислоты можно исходить из ее диме-
тилового эфира.
Полиэтилентерефталат используют для изготовления пленок
и волокон. Его торговые названия: лавсан (СССР), терилен
(Англия), дакрон (США). В смеси с хлопком, шерстью и дру¬
гими волокнами лавсан используют для изготовления тканей,
трикотажных изделий.
Полиэфирные волокна в настоящее время занимают почти
половину в общем производстве синтетических волокон, на до¬
лю полиамидов приходится менее 30 %, на долю полиакрило¬
нитрильных волокон — около 20%. Экономичность производ¬
ства лавсана целиком определяется стоимостью терефталевой
кислоты. Сейчас ее научились получать с высоким выходом
£97—98 %) и нужной степени чистоты (до 99,9999 %!),
ноос
СООН + НОСН2—СН2ОН —>
—> О—СНа—сн2—о—ос
415
каталитическим окислением п-ксилола
соли марганца в уксусной кислоте):
02 (120» С, I МПа)
(катализатор — раствор;
СН:
'~0“сНз
-> ноос
СООН
Ценные качества лавсана — прочность, химическая стой^
кость, отсутствие усадки и растяжения.
Полиэфирами являются и глифталевые смолы, получаемые
при поликонденсации глицерина и фталевой кислоты. Наличие
трех гидроксильных групп в молекуле глицерина позволяет по¬
лучить трехмерный полимер:
/~\ /Л
—оос/ 'СОО—СН2
I
СН—оос
Ч—соо—Ан,
соо—сн2—сн—сн2о—ос
I
о
ос/ 'СОО
А
СОО—СН2—Ан—СНг—ООС \
■ vr\ г*
соо
соо—
Вместо глицерина можно применять и пентаэритрит
С(СН2ОН)4 (пентафталевые смолы). Наряду с фталевой кис¬
лотой вводят также насыщенные и ненасыщенные жирные кис¬
лоты. Глифталевые, пентафталевые смолы и продукты их
модификации различными добавками объединены под общим
названием алкидные смолы. Их растворяют в различных орга¬
нических растворителях (алифатических или ароматических
углеводородах), добавляют красители и получают эмали и лаки,
применяемые для окраски вагонов, станков, сельскохозяйствен¬
ных машин. Алкидные смолы употребляют также при изготов¬
лении типографских красок, линолеума, клеев. В этих областях
применения имеет значение способность алкидных смол после
высыхания давать прочные пленки.
§ 192. Фенолоформальдегидные смолы. Поликонденсацией фе¬
нола с формальдегидом еще в прошлом столетии был получен
искусственный материал, сохранивший свое значение и в наше
время. По фамилии изобретателя англичанина Бакеленда этот
материал получил название бакелит.
Поликонденсация фенола с формальдегидом проходит при
длительном нагревании компонентов в присутствии кислотных
или основных катализаторов. Сначала образуется прозрачная
желтоватая жидкость, содержащая метилольные производные
фенола (см. § 73). Эти продукты при дальнейшем нагревании
конденсируются друг с другом, первоначально образуя малораз-
416
ветвленный полимер типа:
Молекулярная масса нарастает постепенно. Если поликон¬
денсацию остановить при достижении молекулярной массы
700—1000, то получается резол — твердая, очень хрупкая про¬
зрачная масса, напоминающая янтарь. Этот материал легко
растворяется в органических растворителях (спирт, ацетон).
Такие растворы используют в качестве лаков.
При повышении температуры до 60—90 °С резол плавится,
его применяют для изготовления пресс-порошков. Для этого
смолу смешивают с наполнителем (древесная мука, каолин,
молотая слюда, кварц, графит, волокнистые материалы, в том
числе стеклянное волокно), красителями и другими добавками.
В горячей пресс-форме из этой смеси формуют различные изде¬
лия. Такие изделия непосредственно после прессования приоб¬
ретают красивую гладкую поверхность (не требуют дополни¬
тельной окраски, полировки и т. д.), имеют высокую механиче¬
скую и химическую прочность. В процессе прессования идет даль¬
нейшая поликонденсация резольной смолы с образованием трех¬
мерного полимера — резита:
Резит не плавится и не растворяется. Таким образом, резол
обладает термореактивностью, т. е. необратимо изменяет свое
строение и свойства при нагревании.
14 Органическая химия
417
В зависимости от типа наполнителя материалы, полученные
из фенолоформальдегидных полимеров, известны в технике под
разными названиями: фаолит (на основе асбеста), стекловолок-
нит (на основе стеклянного волокна), арзамит (на основе гра¬
фита). Литые фенолоформальдегидные смолы (без наполните¬
ля) известны под названием карболит. Можно получать из смол
и легкие, пористые материалы — пенопласты.
По такому же типу построены продукты поликонденсации
фенола и его аналогов с другими альдегидами или с а-окси-
дами, продукты поликонденсации альдегидов с анилином, моче¬
виной и другими азотсодержащими соединениями.
§ 193. Кремнийорганические полимеры. Высокомолекулярные
вещества, в состав которых наряду с органическими радикала¬
ми входит кремний, приобрели большое техническое значение.
Разработка этой области химии высокомолекулярных соедине¬
ний связана главным образом с именем акад. К. А. Андрианова.
Наибольшее распространение получили кремнийорганические
полимеры с цепями, состоящими из атомов кремния и кислоро¬
да, — полисилоксаны. Их получают гидролизом диалкилдихлор-
силанов. Первоначальные продукты гидролиза — силандиолы —
конденсируются с отщеплением воды, превращаясь в полимер¬
ные силоксаны:
СНз
I
C1-S1—С1
I
СНз
н2о
г СНз п
НО—Si—ОН
I
L СНз -I
конденсация
>.
-НгО
п
По такой схеме получают метилсилоксановый каучук, осо¬
бенно ценным свойством которого является термостойкость. Ре¬
зина, полученная на основе кремнийорганического каучука, со¬
храняет эластичность в интервале температур —90—|-300 °С.
Она обладает высокими электроизоляционными свойствами, хо¬
рошей устойчивостью к действию химических реагентов. Жидкие
полисилоксаны применяют в качестве теплостойких смазок.
Пропитывая полисилоксанами ткань, бумагу или дерево, им
придают водоотталкивающие свойства.
Ценные свойства имеют полимеры, в состав которых наряду
с кремнием входят алюминий, титан, бор и другие элементы.
* *
*
Разнообразие производимых ныне синтетических материалов
очень велико. Их используют в самых различных областях:
заменяют ответственные металлические части в самолетах, ра¬
кетах, автомашинах, употребляют как конструкционные мате¬
риалы в машиностроении, изоляционные материалы в электро¬
технической промышленности, используют для получения
тканей, предметов потребления, для структурирования почвы,
418
изготовления всевозможных труб и пленок, заменяют керамиче¬
ские изделия (ванны, плитки) в строительстве.
Полимерные материалы широко используют в медицине —
В зубопротезной практике, для изготовления искусственных кро¬
веносных сосудов, клапанов сердца, аппаратов «искусственная
цочка» и другой медицинской техники. В сельском хозяйстве
полимерные пленки заменяют стекло в парниках, пластмассы
служат для изготовления разнообразной тары.
Важное значение приобрели ионообменные смолы (иониты),
Это высокомолекулярные вещества, содержащие ионогенные
группы (S03H, СООН, NH2 и др.). С помощью ионитов извле¬
кают ценные компоненты из морской воды, очищают сточные
воды, обессоливают воду, очищают от неорганических ионов са¬
харные сиропы, растворы лекарственных веществ. Иониты ис¬
пользуют также для разделения сложных смесей электролитов
{ионообменная хроматография), служат катализаторами реак¬
ций этерификации, гидратации и др. Используют полимеры и в
качестве адсорбентов при разделении органических веществ.
ГЛАВА 24
МЕТОДЫ ИССЛЕДОВАНИЯ ОРГАНИЧЕСКИХ
ВЕЩЕСТВ
§ 194. Определение строения. Читая работы классиков органи¬
ческой химии, невольно обращаешь внимание на то, с какой
тщательностью они описывают полученные ими органические
вещества, сколько внимания уделяют их очистке и характери¬
стике. В современных публикациях эта стадия работы химика-
органика выглядит гораздо более лаконичной: даются брутто-
формула, температура плавления или кипения, а далее следуют
различные современные характеристики, полученные с помощью
физико-химических методов исследования,— данные оптических
спектров, ядерного магнитного резонанса (ЯМР), масс-спектро-
метрии и др. Такое описание зачастую создает у начинающего
химика-органика ложное впечатление, что современные методы
исследования избавляют его от необходимости тщательной очи¬
стки вещества, что эти методы «сами по себе» способны дать
правильный ртвет. Нет ничего опаснее и вреднее этого заблуж¬
дения! Правильный'аналлз, точная температура плавления, пра¬
вильная спектральная или иная характеристика возможны
только при работе с идеально чястыда веществом. Данные ис¬
следования загрязненного вещества мргут явиться причиной
серьезных ошибок. Поэтому проблема очистки веществ остается,
как и раньше, весьма актуальной.
С методами очистки органических веществ мы знакомились
в § 6. Чистое органическое вещество подвергают качественному
14»
419
и количественному анализу, определяя суммарную (эмпириче¬
скую) формулу. Однако это составляет лишь начало его иссле¬
дования. Следующий этап заключается в установлении струк¬
турной формулы. Большую помощь в этом оказывают современ¬
ному исследователю различные физико-химические методы (см.
ниже). Однако в принципе задачу установления строения можно
решить и чисто химическим путем. Покажем это на конкретном
примере вещества, для которого была установлена эмпириче¬
ская формула С4Нб02.
Исследуемое вещество представляет собой кристаллы
с т. пл. 71 °С, несколько растворимые в воде, причем водный
раствор показывает кислую реакцию. Последнее свойство наво¬
дит на мысль, что речь может идти о карбоновой кислоте.
Йредположение о наличии карбоксильной группы можно под¬
твердить получением ряда функциональных производных — со¬
лей, эфиров, хлорангидрида, амида. Убедившись, что в веществе
присутствует карбоксильная группа, мы получаем право уточ¬
нить формулу, записать ее в виде С3Н5—СООН.
В радикале С3Н5 недостает двух атомов водорода до пол¬
ного насыщения: следовательно, он должен содержать двойную
связь или цикл. Проделав реакции на двойную связь (обесцве¬
чивание бромной воды или щелочного раствора перманганата
калия) и убедившись в том, что вещество ненасыщенное, при¬
ходим к трем структурным формулам, которые может иметь
исследуемое вещество:
СН3—СН=СН—СООН СН2=С—СООН СН2=СН—СН2—СООН
СНз
I II III
Чтобы сделать выбор между этими структурами, необходимо
исследовать продукты деструктивного окисления. Установив в
них присутствие уксусной и щавелевой кислот, можно считать
Доказанной формулу I:
[О]
СНз—СН=СН—СООН —»- СНз—СООН + НООС—СООН
Окончательным химическим доказательством структуры ор¬
ганического вещества служит его синтез. В данном случае та¬
кой синтез можно было бы осуществить осторожным окислением
кротонового альдегида:
[О]
СНз—СН=СН— СНО —► СН3—СН=СН—СООН
Таким образом, вещество состава С4Н602 в рассмотренном
примере — это кротоновая кислота.
Чаще всего химическим путем пользуются для установления
природы имеющихся в исследуемом веществе функциональных
групп. Присутствие кратных связей обнаруживают реакциями с
420
бромом или перманганатом. Спирты переводят действием хлор-
ангидрида динитробензойной кислоты в соответствующие эфиры
или действием фенилизоцианата в фенилуретаны. Альдегиды и
кетоны обнаруживают по образованию оксимов, фенилгидразо-
иов, кислоты — в виде амидов и других производных, из ами¬
нов получают ацетильные или бензоильные производные и т. д.
Все эти и многие другие производные являются кристалличе¬
скими веществами с определенными температурами плавления.
Они ценны тем, что не только позволяют обнаружить присут¬
ствие той или иной функциональной группы, но и распознать
конкретное вещество. Это делают, сравнивая имеющиеся в спе¬
циальных таблицах температуры плавления соответствующих
производных с находимыми опытным путем.
Так, например, сделав вывод, что упоминавшееся ранее ве¬
щество состава С^НбОг — кислота, можно было бы дальнейшее
исследование построить так: подействовать на это вещество
(для получения хлорангидрида) тионилхлоридом (оксид-дихло¬
рид серы), а затем анилином. Определив температуру плавле¬
ния полученного анилида (т. пл. 118°С), по таблицам можно
было бы установить, что такой температурой плавления обла¬
дает анилид кротоновой кислоты. Тем самым задача идентифи¬
кации исследуемого вещества С4НбОг была бы решена без
дополнительного исследования природы радикала, только на
основании установления природы функциональной группы и по¬
лучения кристаллического производного (анилида). Надежность
результата целиком зависит от умения выделить производное в
чистом виде. Так, например, недостаточно очистив анилид и
установив, что полученное вещество имеет т: пл. 104 °С, можно
было бы сделать ошибочный вывод, что исследуемая кислота —
акриловая.
§ 195. Физические методы определения строения. Рефракто¬
метрия. Определение показателей преломления с помо¬
щью оптических приборов, называемых рефрактометрами,—
рефрактометрия— один из самых старых методов физического
исследования органических соединений. Этот метод вследствие
его простоты и чувствительности охотно используют для иден¬
тификации жидких органических веществ, для оценки их чис¬
тоты.
Чистоту оценивают сравнением найденного показателя пре¬
ломления со взятой из таблиц константой для чистого вещества.
Вещество можно считать чистым, если его показатель преломле¬
ния отличается от указываемого в литературе не более чем на
единицу в третьем знаке после запятой (0,001).
При проведении идентификации вещества из измеренных по¬
казателей преломления и плотности вещества вычисляют моле¬
кулярную рефракцию (формула Лорентц—Лоренца):
MR
М п*- I
d «* + 2
421
Молекулярная рефракция органического вещества — вели¬
чина аддитивная: ее можно вычислить по структурной формуле
вещества как сумму атомных рефракций и инкрементов для
кратных связей.
Ниже приведены значения атомных рефракций некоторых
элементов и инкрементов кратных связей, вычисленных по
Зйзенлору (для D-линии натрия, 589 нм):
Углерод
Атомная
рефракция
2,418
Бром
Атомная
рефракция
8.865
Водород
1,100
Иод
13,900
Кислород
Азот (в первичных али¬
2,322
в гидроксила
1,525
фатических аминах)
эфирный
1,643
Инкременты
карбонильный
2,2Ц
С=*С
1,733
Хлор
5,967
CsC
2,389
Совпадение вычисленной таким образом молекулярной ре¬
фракции с найденной из экспериментальных данных служит
Подтверждением структуры вещества.
Присутствие в молекуле вещества сопряженных двойных
связей вызывает увеличение найденной из опытных данных мо¬
лекулярной рефракции по сравнению с вычисленной. Это явле¬
ние, называемое экзальтацией молекулярной рефракции, ис¬
пользуют для обнаружения сопряженных двойных связей.
Спектроскопия. Среди физических методов исследова¬
ния органических соединений особенно важное место заняли
методы, основанные на изучении спектров поглощения. Общий
принцип всех этих методов сводится к следующему: когда свет
(вернее говоря, любое электромагнитное излучение, см. ниже)
проходит через вещество, он может поглощаться. Энергия света
при этом частично превращается во внутреннюю энергию веще¬
ства — энергию его молекул, атомов, электронов, ядер.
Поглощением управляют квантовые законы: поглощается не
любой свет, а только тот, энергия квантов (фотонов) которого
соответствует разностям энергий двух состояний молекулы —
основного и возбужденного (т. е. создающегося после поглоще¬
ния соответствующего кванта). Математически это выражается
в условии, сформулированном Бором:
Б' — Е0 =* hv
где Е' — анергия возбужденного состояния; Е0 — энергия основного состоя¬
ния; h — постоянная Планка; V —частота Излучения (при измерении частоты
6 «обратных сантиметрах», бм-1, она равна 1Д, т. е. является величиной, об¬
ратной длине волны).
Если через вещество проходит «белый» свет (т. е. излучение,
содержащее кванты света самой различной величины, другими
словами, лучи разных длин волн), то поглощается только та
его часть, которая отвечает приведенному выше условию. Ос¬
тальная же часть проходит без ослабления, и этот прошедший
422
Рис. 32. Спектр поглощения перманганата калия
через вещество свет приобретает окраску, до¬
полнительную к поглощенному. Так, например, z
перманганат калия интенсивно поглощает кван¬
ты сине-зеленой части спектра (480—560 нм); в
результате прошедший свет окрашен в фиолето-;
вый цвет — всем хорошо известный цвет раство¬
ров перманганата. Зависимость поглощения от soo $оо too
длины волны — спектр поглощения пермангана- длина Волны, нм
та — имеет вид, приведенный на рис. 32. Погло¬
щение может наблюдаться не только в видимой области спектра
(как у перманганата), но также и в его невидимых частях —
ультрафиолетовой, инфракрасной.
Чтобы понять, как связан характер поглощения со строени¬
ем органического вещества, вернемся к условию Бора: Е' —
—- Ео = hv, Чем ближе друг к другу находятся оба энергети¬
ческих уровня, тем меньше затрата энергии на возбуждение,
тем меньшей энергией может обладать квант света, т. е. меньше
частота v (и, наоборот, тем больше соответствующая этой час¬
тоте длина волны). Разность энергии Е' — Е0 определяется в
конечном итоге подвижностью электронов. Так, электроны о-
СВязей связаны весьма прочно: для возбуждения их нужны
кванты с большой энергией. Поэтому алканы, спирты, простые
Эфиры поглощают лишь в очень далекой ультрафиолетовой об¬
ласти. Этилен, имеющий подвижные я-электроны, поглощает
При 193 нм. Сопряженные двойные связи в бутадиене
СН2=СН—СН=СН2, обладая еще большей подвижностью я-
эдектронов, поглощают уже при 217 нм. В бензоле я-электрон-
ная система имеет несколько полос поглощения, наиболее длин¬
новолновые из которых расположены в области 260—270 нм.
Нафталин поглощает уже при 314 нм, антрацен — при 380 нм.
На этих примерах видно, что с ростом сопряжения (ростом по¬
движности электронов) наблюдается постепенный сдвиг погло¬
щения в длинноволновую область. Однако все упоминавшиеся
до сих пор соединения бесцветны, так как их избирательное
поглощение лежит в ультрафиолетовой области спектра. Види¬
мая желтая окраска появляется лишь у нафтацена (Я,Макс =
«= 480 нм):
Таким образом, избирательным поглощением в видимой об¬
ласти спектра и, следовательно, окраской обладают соединения
с особенно подвижными, легко возбуждаемыми электронами.
Эта подвижность обеспечивается созданием сопряженных сис¬
тем, включением в сопряжение свободных электронных пар или
423
е
Рис. 33. УФ-Спектры бензола (1), толуола (2) и бензальдегида (3)
неспаренных электронов, создающих анионный заряд. Так нахо¬
дят свое физическое обоснование закономерности, ранее уста¬
новленные чисто эмпирически.
Например, бензол (рис. 33) имеет в области 240—260 нм
характерную полосу с несколькими максимумами (тонкая
структура); введение алкильных групп или других заместите¬
лей, не имеющих кратных связей или свободных электронных
пар, не изменяет характера спектра, лишь немного сдвигает
поглощение в сторону длинных волн. Если же бензольное коль¬
цо вступает в сопряжение с кратными связями или свободными
электронными парами заместителя, то характер спектра изме¬
няется, как это видно на примере спектра поглощения бенз¬
альдегида. При этом не только исчезает тонкая структура, но
ив 100 раз возрастает поглощение (на рисунке по вертикали
шкала логарифмическая!).
Таким образом, ультрафиолетовые спектры (диапазон волн
200—400 нм) дают ценную информацию о строении органиче¬
ских молекул. Поглощение в этой области связано с возбужде¬
нием валентных электронов, поэтому УФ-спектры особенно хо¬
рошо отражают строение систем с подвижными электронами, в
частности ароматических соединений.
Кванты длинноволнового излучения (инфракрасные лучи)
несут относительно небольшую энергию, она может возбудить
колебания атомов в молекулах. Энергия же колебаний атомов
зависит от их собственной природы и от характера связи в
молекуле (т. е. с какими атомами и каким путем осуществля¬
ется эта связь). Поэтому инфракрасные спектры поглощения
(диапазон волн 1—10 мкм, т. е. частоты порядка 500—
5000 см-1) дают особенно ценную информацию о строении мо¬
лекул органических соединений. В качестве примера на рис. 34
приведен ИК-спектр ацетофенона СбН5—СО—СН3.
424
Полоса 1680 см-1 вызывается колебаниями карбонильной
группы: эта характерная полоса имеется вблизи 1700 см-1 у
всех карбонильных соединений. Полосы 1600, 1580, 1450, 765
и 647 см-1 характеризуют монозамещенное бензольное кольцо,
полосы 1430 и 1360 см-1 относятся к метальной группе.
Очень большое значение имеют спектры ядерного магнитного
резонанса (ЯМР). Не вдаваясь в подробности, отметим, что в
этом случае измеряется поглощение электромагнитных излуче¬
ний очень высоких частот (т. е. длинных волн). ЯМР имеет
дело с частотами 0,1—0,01 см-1, т. е. с областью сантиметро¬
вых радиоволн; в связи с этим метод ЯМР называют также
радиоспектроскопией. Наиболее часто этот метод применяется в
форме протонного магнитного резонанса (ПМР), позволяя по¬
лучить точную характеристику атомов водорода, имеющихся в
исследуемом соединении.
Спектр ПМР диацетонового спирта (СН3)2СОНСН2СОСНз
(рис. 35, а) совсем прост: каждому типу водородных атомов со¬
ответствует свой сигнал в
виде одиночной линии. Та¬
кой спектр свидетельствует
о том, что в этом соединении
между протонами разных ти¬
пов нет никакого взаимодей¬
ствия; происходит это пото¬
му, что все содержащие во¬
дород группировки изолиро¬
ваны друг от друга: между
ними находятся атом угле¬
рода и группа С = 0, не
имеющие протонов. Хотя
этиловый спирт — соедине-
Рис. 35. ПМР-Спектры диацртрно-
вого спирта (а) и этанбла (в)
SU*
Рис. 36. Масс-спектры бутана (а) и изобутана (б)
уие боле§ проетое, чем диацетонорый спирт, его спектр ПМВ
Сложнее (рис. 35,6), чем спектр диацетонового спирта, У этило¬
вого спирта проявляется взаимодействие протонов, стоящих у
(юседних атомов. В результате вместо одиночных линий возни¬
кают группы их (расщепление линий в результате спин-спино-
рого взаимодействия). Внимательный анализ характера расщеп¬
ления дает дополнительные данные о структуре молекулы. Та¬
ким образом, в спектрах ЯМР используют два вида информации:
химический сдвиг (положение сигнала) и константы спин-спино-
вого расщепления.
Метод ЯМР — наиболее мощный из применяемых в настоя¬
щее время методов исследования органических соединений. Он
дает информацию не только о химическом и пространственном
Строении вещества, но позволяет экспериментально оценить элек-
Троотрицательность отдельных групп, направление и силу индук¬
тивных и мезомерных электронных эффектов.
Масс-спектрометрией называется метод исследования органи¬
ческих веществ, основанный на изучении «осколочных ионов»,
образующихся под действием «электронного удара» пучка элек¬
тронов с энергией в несколько десятков электронвольт. С по¬
мощью масс-спектрометрии определяют строение органических
соединений и их молекулярную массу. Уже небольшие различия
р строении отражаются в масс-спектрах, как это видно на при¬
мере масс-спектров бутана и изобутана (рис. 36).
Помимо научных лабораторий масс-спектрометрия находит
применение и как метод контроля в нефтехимической и других
отраслях промышленности.
Электронный парамагнитный резонанс (ЭПР) основан на
явлении поглощения электромагнитных волн парамагнитными
веществами в постоянном магнитном поле. Парамагнитными
свойствами обладают, в частности, свободные радикалы, для -
обнаружения и исследования которых и применяется метод
ЭПР в органической химии.
426
ЗАКЛЮЧЕНИЕ
Зародившись как наука о веществах живой природы, органическая хи¬
мия затем в значительной степени обратилась к изучению синтетических сЙ»
единений. Успехи синтетической органической химии послужили еще в про¬
шлом веке основой для создания целых отраслей промышленности — произ¬
водств синтетических красителей, лекарственных препаратов, взрывчатйй)
веществ. Уже в нашем столетии на основе использования нефтяного сырья
развилась промышленность тяжелого органического синтеза—многотоннажный
производства чистых углеводородов, спиртов, кетонов, кислот и их производ¬
ных, являющихся в свою очередь сырьем для получения разнообразных проч
дуктов. Большое значение приобрело получение синтетических высокомоле¬
кулярных соединений. Если бы вдруг по какому-то злому волшебству исчезло
все, изготовленное при содействии синтетической органической химий, челове¬
чество лишилось бы одежды и обуви, большинства предметов домашнего
обихода, лекарств и многого другого. Получение все новых и новых полез¬
ных веществ и материалов является и в настоящее время важной задачей
органической химии.
В принятой ЦК КПСС и Советом Министров СССР Комплексной про¬
грамме химизации народного хозяйства СССР значительное место уделяется
продуктам органической химии. Предусматривается наращивать производство
углеводородного сырья и нефтехимических продуктов за счет углубления пе¬
реработки нефти, широкого использования газового конденсата, комплексного
использования ценных углеводородов, природного и попутного нефтяного
газа, вовлечения в производство ненефтяных видов сырья (монооксида и ди¬
оксида углерода, метанола, продуктов переработки угля, сланцев). Сырье
должно использоваться более эффективно за счет применения высокоселектив¬
ных и ресурсосберегающих технологических процессов.
Выполнение Комплексной программы невозможно без широкого использо¬
вания достижений науки, в частности применения катализаторов с попышеп-
Иой активностью, селективностью, надежностью, с большим сроком службы.
Поэтому в «Основных направлениях экономического и социального развития
СССР на 1986—1990 годы и на период до 2000 года» имеется специальный
пункт, где говорится о необходимости развивать «научные основы катализа,
химической технологии, биотехнологии, а также создавать новые конструкци¬
онные материалы».
В Комплексной программе ставится задача усовершенствовать выпуск
хлорорганических продуктов путем внедрения безотходных, сбалансированных
по хлору технологических процессов, в том числе получение винилхлорида из
этилена. В промышленности синтетических смол и пластических масс наме¬
чается развитие производства полиэтилена, полипропилена, полистирола, по¬
ливинилхлорида с повышенными прочностными характеристиками. Подсчита¬
но, что применение 1 млн. т полимерных материалов вместо традиционных
(металл, дерево) дает возможность высвободить 300 тыс. человек, а исполь¬
зование 1 млн. т химических волокон вместо натуральных (хлопок, шерсть,
лен) высвобождает 1,4 млн. человек. Кроме того, энергетические затраты на
производство химических материалов в 2—3 раза меньше, чем на получение
естественных.
Химизация внесет большой вклад и в выполнение Продовольственной
программы: увеличатся поставки минеральных удобрений, в том числе к
эффективного азотного удобрения — карбамида (мочевины); должен быть
увеличен выпуск средств защиты растений с высокой избирательностью дей¬
ствия, быстро разлагающихся в окружающей среде, позволяющих резко сни¬
зить погектарные нормы расхода. В то же время достижения органической
химии должны быть направлены на сокращение расхода пищевого сырья
для технических целен: например, замена растительных масел в лакокрасоч¬
ном производстве полиэфирныЫй, глифталевыми, эпоксидными смолами, за¬
мена жиров и масел в производстве мыла синтетическими жирными кисло¬
тами.
427
Значительное место в Комплексной программе уделено развитию мало¬
тоннажных химических производств; такая продукция важна для ускорения
научно-технического прогресса, повышения качества продукции. Сюда входят
синтетические красители, стабилизаторы полимеров, фото- и киноматериалы,
биохимические препараты, лекарственные вещества, реактивы для лабора¬
торий.
Задача глубокого познания связи между строением и физическими, хи¬
мическими, физиологическими свойствами веществ остается на ближайшее
будущее одной из важнейших научных задач в области органической химии.
Успех в решении практических и теоретических проблем определяется даль¬
нейшим развитием методов синтеза, анализа, разнообразных физико-химиче¬
ских методов исследования.
Химики-органики всегда занимались исследованием природных веществ,
Ныне, когда в союзе с биологией органическая химия вплотную подошла К
пониманию химических основ жизни, это направление приобретает особую
важность, не только теоретическую, но и практическую. Оно создает научный
фундамент для борьбы с самыми тяжелыми заболеваниями, для поднятия
продуктивности сельского хозяйства.
Соприкасаясь буквально со всеми сторонам и научной и технической дея¬
тельности человека, органическая химия составляет одну из основ научно-
технического прогресса.
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Адениловая кислота 388
Аденин 386, 388 сл.
Аденозин 387, 390
Аде'нбзинтрифосфат (АТФ) 391 сл.
АдиПицрвйя кислота 109, 206, 236,
238, 402, 413 сл.
получение 62, 115, 164, 238
Адиподинитрил 260
Адреналин 354, 400
Адренокортикотропный гормон 380
Азелаиновая кислота 231
Азокомпоненты 296
Азокрасители 296 сл.
Аз оксибензол 280
Азометины 283
Азосоединения 280, 296 сл.
Азосочетание 295 сл.
Азот, обнаружение 17
Азулен 127
Акриловая кислота 88, 230, 231 сл.,
316
Акрилонитрил 258, 259 сл.
получение 84, 88, 183, 259 сл.
реакции 231, 260 сл., 385, 408
Акролеин 190, 207, 209
получение 164, 167, 209, 366
реакции 164, 167
Акцепторы электронов 268
Р-Аланин 360, 378
Ализарин 214
Алкалоиды 375 сл.
Алканы 39 сл.
гаЛогенирование 52 сл., 138,
145 сл.
дегидрирование 55, 57, 69
изомеризация 40 сл., 134
окисление 54, 216
Получение 48 сл., 75
реакции 50—57, 68 сл., 75, 102 сл.
сульфохлорирование 54, 228, 2б4
Алкбноксиды (Эпоксиды) 77, 181
Алкены (Олефины) 64 сл., 70 сл.,
402
алкилирование 75
аммонолиз 258, 260
галогенирование 73, 138 сл., 145
гидратация 74, 151 сл.
гидрирование 48, 75
гидрогалогенирование 72, 138, 148
гндрокарбоксилирование 217
гидроформилирование 188, 227
конденсация 202 сл.
метатезис 79
нитрование 53 сл.
номенклатура 67 сл.
озонирование 77
окисление 54, 76 сл., 185, 216
полимеризация 76 сл., 413 сл.
получение 47 сл., 68 сл.
реакции 71, 178, 181, 185, 200, 248,
263
Алкидные смолы 416
428
Алкиларилсульфонаты 228
Алкилгалогениды 137, 139 сл., 145,
157
реакции 69, 82, 140, 144, 178, 262,
282, 304
Алкилирование
алканов 75
алкенов 75
аминов 282
аммиака 278
ароматических соединений 103,
108 сл.
ацетоуксусного эфира 328
гетероциклов 368
моносахаридов 342
пероксида водорода 185
фенолов 172 сл., 299
Алкилмалоновые эфиры 237
Алкилсерные кислоты (Алкилсульфа-
ты кислые) 74 сл., 157 сл.;
185, 228
Алкилсульфонаты 228 сл., 264
Алкилфосфоновые кислоты 307 сл.
Алкилхлорсиланы 306
Алкины 81 сл., 188, 205, 200 сл.
Алкоголиз 241
Алкоголяты 156, 178
Аллен (Пропадиен) 90, 97
Аллиловый спирт 150, 153, 164, 167
Аллилхлорид 79, 139 сл., 142, 164,
167
Аллицин 263
Альбумины 384
Альдегидокислоты 323 сл.
Альдегиды 186 сл., 190, 402
ароматические 199
восстановление 152, 192
гидразоны 196 сл.
гидратация 193
конденсация 197 сл., 200, 207, 209,
237
ненасыщенные 207 сл.
окисление 201 сл., 216
оксимы 195 сл.
получение 159, 164, 187 сл.
реакции 173, 190 сл., 283, 361, 363
Альдимины 195
Альдозы 332, 335
Альдоль 88, 198
Альдоновые кислоты 340
Алюминийорганические соединения
304 сл.
Амигдалин 205, 338
Амидол (2,4-Диаминофенол) 355
Амиды 222, 254 сл., 258
аминокислот 359
получение 196, 243, 254 сл.
реакции 216, 255 сл., 258
сульфокислот 266
циклические 255
Амилоза 347 сл.
Амилопектин 347 сл.
Аминоазосоединение 295
Аминобензойные кислоты 360
Аминогруппа 289, 291
Аминокислоты 356 сл., 378 сл.
Аминонафтолы 298, 355
Аминопиридины 364
Аминопласты 257
Р-Аминопропионитрил 261
я-Аминосалициловая кислота 360
Аминоспирты 353 сл.
Аминофенолы 354 сл.,
ы-Аминоэнантовая кислота 414
Амины 255, 272, 276 сл., 292
Аммония соли 281 сл.
Аммонолиз 242
алкенов 258, 260
и-ксилола 239
сложных эфиров 249
фенола 285
Анабазин 376
Анализ элементный 16 сл.
Ангулярные системы 126
Анестезин 360
Анетол 180
Анид 414
Анизол (Метоксибензол) 177, 180
Анилин 278, 281, 285, 290
получение 115, 279, 285
реакции 213, 285 сл., 366, 371
Анилы 283
Антибиотики 394 сл.
Антиоксиданты 175
Антипирин 375
Антифриз 166
Антраниловая кислота 360
Антрахинон 125, 214
Антрацен 100, 123 сл., 125, 214, 423
Арабан 333
Арабиноза 332 сл., 335
Арахиновая кислота 251
Арбузова перегруппировка 309
Аргинин 379, 384
Арилгалогениды 137, 144 ел.
Арндта — Эйстерга синтез 299
Ароматизация
алканов 55
нефти 103, 114, 132
Ароматические углеводороды 97 сл.
алкилирование 103, 108 сл„ 173
гидрирование 59, 64, 109, 122, 124
конденсированные 119 сл., 125 сл.
многоядерные 116 сл.
небензоидные 126 сл.
номенклатура 101 сл.
реакции 104 сл., 108 сл., 189, 201,
216, 270
Асимметрический атом углерода 316
429
Аскорбиновая кислота 393
Аспарагиновая кислота 356, 378
Аспирин 321
Атомная рефракция 422
АТФ (Аденозинтрифосфат) 391 сл.
Ауксохромы 297
Ацены 125 сл.
Ацетали 194, 337
Ацетальдегид (Уксусный альдегид)
186, 190, 204, 368
эдсим 196
получение 73, 95, 160, 179, 183
реакции 95, 152, 197 сл., 209, 363
Ацетамид 210, 249, 254, 255 сл.
Йетанилид 254, 285
.цетиламино-1-этоксибензол 355
Ацетилацетон 210 сл.
Ацетилен 81, 87 сл., 145
карбонилирование 231
конденсация 200 сл., 206
нитрование 275
получение 56, 87 сл., 204
реакции 85, 86, 88, 95, 145, 164,
178, 248, 259, 275, 302, 363,
369
тримеризация 86
Ацетилениды 84, 86
Ацетнлнафгалины 121
Ацетилсалициловая кислота 321
Аиетилхлорид 210, 240, 242, 321
Ацетон 187, 190, 196, 205
получение 77, 78, 115, 159, 169,
187, 205
реакции 152, 174, 206, 210, 232
Ацетонилацетон 330
Ацетонитрил 255, 258, 261
Ацетонциангидрин 232
v-Ацетопропиловый спирт 331
Ацетоуксусная кислота 323, 325
цетоуксусный эфир 325—330
цетофенон 187, 190, 207, 330, 424
Ацилгалогениды см. Карбоновых кис¬
лот галогенангидриды
Ацилирование 241
аминов 282
ароматических углеводородов 109,
121, 189
ацетоуксусных эфиров 328
гетероциклов 368, 372
моносахаридов 343
спиртов 248
Байера теория напряжения 69
Бакелит 41о
Бейлыитейна проба 17
Бекмана перегруппировка 196
Белки 377—386
Бензальдегид 159, 164, 190, 202, 205,
233 242
Бензамид 226, 242, 254
430
Бензилгалогениды 107, 140, 143, 164
Бензилиденацетон 199
Бензилиденгалогениды 107, 140
Бензиловый спирт 144, 150, 153, 159,
164
Бензилцианид 216, 258
Бензины 48 сл., 55, 130, 135, 258
Бензоилацетон 210, 330
Бензоилхлорид 205, 226, 240, 242
Бензоин 203
Бензоиновая конденсация 203
Бензойная кислота 218, 226, 242, 321
получение ПО, 159, 202, 217, 226
производные 104, 226, 258
Бензол 97 сл., 104, 115, 145
валентные изомеры 101
гомологи 101 сл., 114
озонид 98
получение 62, 86, 106, 114, 132
реакции 59, 64, 98, 109, 115, 145,
165, 169, 174, 270
спектры 423 сл.
Бензолсульфокислота(ы) 108, 169
эфиры 266
Бензонитрил (Фенилцианид) 258
Бензопирен 126
Бензотрихлорид 107, 217, 226
Бензофенон 188
Ееизохиноны 213
Берцелиуса теория радикалов 20
Биацетил (Бутандион-2,3, диметилгли-
оксаль) 210 сл., 253
Бивинил см. Бутадиен-1,3
Битум 131
Бифенил 116
Бора условие 422
Борнан 398
Борнеол 398 сл.
Борнилацетат 399
Брожение 348
ацетонобутиловое 205
молочнокислое 319
спиртовое 162, 343
уксуснокислое 224
Бромбензол 116
Бромирование
анилина 285
ацетоуксусного эфира 328
кислот 222, 311
циклопентаднена 93
Бромнафталины 120, 294
а-Бромпропионовая кислота 222
Бромтрифторметан 148
Бромциклогексан 61
Бутадиен-1,2 90
Бутадиен-1,3 (Бивинил) 88, 90, 94,
145
получение 57, 89, 94 сл., 411
реакции 92, 93, 94 сл„ 145, 214,
238, 411 сл.
Бутан 40, 42, 48, 49, 55, 57, 69
Бутанамины 277
Бутандиол-1,4 165, 180
Бутандион-2,3 см. Биацетил
Бутанол-1 209
Бутантиол 262
Бутены (Бутилены) 64, 66, 67, 70 ел.,
77, 95, 184
Бутиламины 277 сл.
Трет-Бутилацетат 248
грет-Бутилгидропероксид 184, 185
Бутилкаучук 422
грет-Бутилметиловый эфир 161
Бутиловые спирты 151, 156, 163, 185,
308
4-грет-Бутилфенол 173
Трет-Бутилхлорид 141
Бутиндиол-1,4 200
Бутлерова теория строения 22 сл.
Вагнера реакции 77
Валентность 22
Валериановая кислота 218
-Валеролактон 316
алии 356, 378, 384
Ван-дер-ваалъсов радиус 44 сл>
Вант-Гоффа постулат 43
Вильямсона синтез 178
Винилацетат 88, 248, 402, 409
Винилацетилен 85, 88, 201, 206
Винилбензол см. Стирол
5-Винил-2-метилпиридин 363
Виниловые мономеры 402
Виниловые эфиры 88, 179, 188, 402
Виниловый спирт 85, 164, 409
Винилоги 209
Винилхлорид 80, 88, 140, 143, 145 сл,,
402
Винилэтиловый эфир 178
Винные кислоты 319 сл.
Виноградная кислота 319
Винол 409
Вискоза 352
Витамины 366, 385, 393 сл.
Вициналъные производные 72
Влияние
атомов 149 сл.
заместителей 150, 156, 273, 285,
289, 291
Внутрикомплексные соединения 212
Водород 17, 45
активный, определение 160, 302
Возгонка 15
Воска 249 сл.
Восстановление
азосоединений 298
амидов 255
карбонильных соединений 152, 192
карбоновых кислот 222
Восстановление
моносахаридов 340 сл.
нитрилов 259
нитросоединений 272, 278 сл.,
358
оксимов 280
Сложных эфиров 249
фенолов 171
хлорангидридов 189
Высокомолекулярные соединения (По¬
лимеры) 76, 401 сл.
атактические 405
виниловые 407 сл.
гетероцепные 402
изотактические 405 сл.
карбоцепные 402
линейные 401
Ноликонденсационные 413 сл,
полимеризационные 403 сл:
«сшитые» (трехмерные) 401 сл.
элементорганические 402, 418 сл.
Вюрца реакция 48
Вюрца— Фиттига синтез 103
Габриэля синтез 279
Галактоза 332 сл., 335, 339
Галалит 386
Галловая кислота 175, 322
Галовакс см. Перхлорнафталин
Галогензамещенные кислоты 222, 230,
310 сл., 314, 357
Г алогенирование
алканов 52 сл., 138, 145
алкенов 72, 138
алкинов 84
ароматических углеводородов
106 сл., 124
гетероциклов 368
диенов 92
карбоновых кислот 222, 311
циклоалканов 61
Галогенпроизводные 137 сл., 140 сл,,
142
гидролиз 138, 144, 152, 189, 217
реакции 140 сл., 144, 258, 264,
269, 301 сл., 306
Галогенуксусные кислоты 312
Галогены, обнаружение 17
Гаттермана реакция 189
Гексадецилпальмитат 250
Гексаметапол 309
Гексаметилендиамин 284, 402, 413 сл,
Гексаметилентетрамин 203 сл.
Гексан 49, 69
Гексановая кислота 218, 225, 251
ГексаЛенилэтан 117
Гексафторацетон 193
Гексахлоран (Гексахлорциклогексан}'
98, 107 сл, 115
431
Гексахлорбензол 107
Гексахлорбутадиен 146
Гексен 64
Гексиламин 278
Гексоген 204
Гексозы 332
Гектан 49
Гелиантин 298
Гемоглобин 370, 378, 380, 383 сл.
Гемоцианины 384
Генетический код 389 сл.
Генциобиоза 338
Гептан 49, 102, 135
Гептахлор 147
Гербициды 313
Гстероауксин 371
Г стеролйтический (ионный) разрыв
связей 35 сл.
Гетерофункциональные соединения 38,
310 сл.
Гетероциклические соединения 37,
361 сл.
Гиалуроновая кислота 384
Гибридизация 27 сл.
Гидразины 196, 295
Гидразобензол 280
Гидразоны 196 сл.
Гидратация
алкенов 151 сл.
алкинов 85, 188
карбонильных соединений 193
непредельных кислот 230
Гидрирование
алкенов 49, 75
алкинов 85
ароматических углеводородов 59,
64, 98, 109, 122, 124
гетероциклических соединений 365,
367, 372, 374
диенов 93, 96
жиров 252 сл.
карбонильных соединений 207
карбоновых кислот 228
монооксида углерода 48
угля 48
фенолов 173
циклоалканов 61 сл.
Г идрогалогенирование
алкенов 72, 138, 148
алкинов 84
альдегидов непредельных 208
диенов 92 сл.
карбоновых кислот 230, 311
спиртов 138
циклоалканов 81
Гидрогенизация 252 сл.
Гпдрокарбоксилирование 217, 238
Гндроксиазосоединения 295
Гидроксиаминокислоты 379
Гидроксибензолсульфокислоты (Фе-
нолсульфокислоты) 172
Гидроксигидрохинон 175
Гидроксикислоты 223, 230, 313 сл.,
316, 324
Гидроксиламин 195, 275
Гидроксильная группа 290 сл.
Гидроксиметилмочевина 257
З-Гидрокси-2-метилпентаналь 199
Г идроксиметилфенол ы 173
Гидроксинитрилы (Циангидрины)
193 сл., 327
Гидроксипиридины 365
Гидроксипролин 379
а-Гидроксипропионовая кислота
314 сл.
8-Гидроксихинолин 367
Гидроксиэтиламиды 354
Гидроксиэтиламины 353
Гидроксоний-ион 106
Гидроксония бромид 156
Г идролиз
акрилонитрила 231
алкилгалогенидов 141, 144
алкилхлорсиланов 306 сл.
алкоголятов 156
аллилхлорида 164, 167
амидов 216, 255
АТФ 391
белков 380
бензотрихлорида 226
виниловых эфиров 179
галогенангидридов 241
галогензамещенных кислот 314
галогенпроизводных 138, 144, 152,
189, 217
жиров 166, 227, 252
крахмала 348
малоновых эфиров 238
мочевины 257
нитрилов 216, 236, 254 сл., 258
нитроацетанилидов 286
нуклеиновых кислот 386 сл.
сложных эфиров 248 сл.
солей карбоновых кислот 221
сульфокислот 266
сульфонилхлоридов 264
фенолятов 170
углеводов 346 сл.
фосфатидов 253
Гидролизный спирт 162, 350
Гидропероксид(ы) 184 сл.
ацилов (Пероксикислоты) 181,
202, 245
кумола 169
Гидросульфитные соединения 193,327
Г идроформилироваиие
акрилонитрила 260
алкенов 188
Гидрохинон 168, 169, 175, 213
432
Гистидин 375, 378, 384
Гликоген 322, 347
Гликозиды 337
Гликокол 363
Гликоли СДиолы) 77, 165, 234, 324,
413
эфиры 180 182
Гликопротеиды 384
Глиоксаль 210, 234
Глиоксиловая кислота 323
Глифталевые смолы 169, 401, 416
Глицерин 150, 165, 402
получение 88, 139, 164, 166 сл., 252
реакции 209, 366, 416
сложные эфиры 167, 251
Глицериновый альдегид 167, 345
Глицерофосфорная кислота 253
Глицидный спирт (Глицидол) 167
Глицин 359, 363, 378
Глобулины 384
Глутаминовая кислота 378, 385
Глутаровая кислота 62, 234 сл., 236
Глутелины 384
Глюкаровая (сахарная) кислота 341
Глюкогептоновая кислота, нитрил
334
Глюкоза 332 сл., 334 сл., 345, 348
озазон 342
реакции 162, 338, 340 сл.
а- и (3-формы 337, 339
Глюкозиды 338
Глюконовая кислота 340
Гомолитический (радикальный) раз¬
рыв связей 34 сл.
Гомологи 39
Гормоны 400 сл.
Гофмана
перегруппировка 255 сл.
реакция 278
Грамицидин С 395
Граничные структуры 290
Г риньяра
реактивы 144, 194, 300 сл.
реакция 301
Гуаниловая кислота 388
Гуанин 386, 388 сл.
Гуанозин 387, 390
Гудрон 130
Густавсона синтез 59
Дегалогенирование 70
Дегидратация
альдоля 198
гидроксикислот 230, 315
пентоз 343 сл.
спиртов 69, 158 сл., 177, 366
уксусной кислоты 209 сл.
Дегидрирование
алканов 55, 57, 69, 79 сл., 96
Дегидрирование
борнеола 399
бутана 57
бутенов 95
спиртов 160, 205
циклоалканов 62, 102, 114
этилбензола 115
Дегидроаскорбиновая кислота 393
Дегидрогалогеиирозание
алкилгалогенидов 69, 82, 143
галогензамещенных кислот 230
1,2-дибромциклопентана 96
Дегидроциклизация алканов 102 сл.,
114
Дезаминирование аминов 284, 363
Дезоксирибоза 387
Дезоксирибонуклеиновые кислоты
387 сл.
Дезоксирибонуклеотиды 387
Декагидронафталин (Декалин) 122
Декагидрохинолин 367
Декановая кислота 251
Декарбоксилирование 104, 187,
235 сл, 324
Денатурация белков 383
Депарафинизация карбамидная 133
Депсиды 322
Десульфирование 266
Детергенты (Моющие средства) 228
Детонация топлива 134
Дециловый спирт 153
Дяазокетоны 299
Диазокомпоненты 296
Диазометан 298 сл.
Диазония производные 292 сл, 294
Диазосоединения 292 сл,
Диазотаты 293
Диазотирование 292
Дналкилдихлорсиланы 418
Дналкилсульфаты 178
Диалкилфосфиновые кислоты 307 сл,
Диаллилдисульфид 263
Диаминокарбоновые кислоты 356
2.4- Диаминофенол (Амидол) 355
Диастереомеры 319, 337
Диацетоновый спирт 200, 425
Дибензоилпероксид 226, 245
1.4- Дибромбутан 61
Дибромдифторметан 148
Дибромпропаны 58 сл, 72
1.2- Дибромтетрафторэтан 148
1.2- Дибромэтен 48
2,6-Ди-трег-бутил-4-метилфенол (Ио-
нол) 175 сл.
Ди-грег-бутилпероксид 185
м-Дигалловая кислота 322
9,10-Дигидроантрацен 124
1.2- Дигидроксиантрахинон 214
Дигидроксибензолы 168, 175, 213
Дигидроксиметилмочевина 257
433
Дидепсид 322
Диеновый синтез (Реакция Дильса—
Альдера) 93 сл., 208, 214
Диенофилы 93
Диеновые углеводороды 89 сл.
Дизельное топливо 136
Диизоцианаты 413
Дикарбонильные соединения 210 сл.
Дикарбоновые кислоты 233 сл., 402
Дильса—Альдера реакция см. Дие«
новый синтез
Димеризация
ацетилена 85
изобутена 76
пропилена 96
радикалов 35
Циклопентадиена 96 сл.
Диметиламин 278, 284
п-Диметиламиноазобензол 295
Диметиламинобензолсульфонат нат«
рия 298
Диметиланилин 278, 284, 288, 297
Диметилбенэолы см. Ксилолы
2.2- Диметилбутан 47, 303
Диметил (винилэтинил) метанол 201,
206
2.4- Диметилгексан 47
Диметилглиоксаль см. Биацетил
Диметилглиоксим 211
4.4- Диметил-1,3-диоксан 200
Диметилдихлорсилан 300
Диметилолмочевина 257
Диметилпероксид 185
2.2- Диметилпропаналъ 186
Диметилсульфат 158
Диметилсульфоксид 264
Диметилсульфон 264
Диметилуксусная кислота 216, 218
Диметилформамил 256
Диметилфталат 244
Диметилциклопропаны 58, 63
Динамиты 168
2.4- Динитроанилин 273
Диоксан 179, 180, 183, 368
Диоксансульфотриоксид 368, 372
Диоксопиперазины 359
Диолы 194. См. также Гликоли
Дипольные моменты 33
Дисахариды 332, 345 сл.
Дисульфиды 262
Дитерпены 396
Дифениламин 278, 288
Дифенилметан 116, 201
Дифениловый эфир 177, 180
Дифенилолпропан 174
Дифтордихлорметан (Фреон-12) 148
Дифторхлорметан 199
Дифракция нейтронов, метод 31
р.р'-Дихлордиэтилсульфид 263
Дихлорметан 52, 145
Дихлоруксусная кислота 312
2,4-Дихлорфеноксиуксусная кислота
313
1,2-Дихлорэтан 72, 80, 146, 234
1,6-Дицианогексин-З 261
Дициклопентадиенилжелезо 305
Диэтаноламин 353
Диэтиламин 278, 284
Диэтиленгликоль 182 сл.
эфиры 180
Диэтиловый эфир 80, 159, 179 сл.
ДНК 387 сл.
Доноры электронов 269
Древесный сахар 333
Дубильные вещества 322 сл.
Дульцин 355 сл.
Дыоаровский бензол 101
Енамины 283
Енолы 164, 212
Желатин 385
Жиры 166, 227, 247, 250 сл., 385
Зайцева правило 69
Заномейера реакция 294
Зарин 309
Заряды частичные 294
Зеин 384
Зеленое мыло 226
Зелинского
метод 311
реакция 102
Синтез 357
Зелинского — Гелля — Фолыарда
реакция 222
Зинина реакция 279
Зрительный пурпур 384
Изобутан 40, 42, 46, 47, 134, 426
Изобутен (2-Метилпропен) 67, 70, 77,
79, 402, 404
окисление 77, 232
реакции 76, 79, 151, 161, 173, 200
Изобутиловый спирт 151, 154, 205
Изобутилэтилметанол (5-Мети лгекса»
нол-3) 151
Изовалериановая кислота 216, 225,
247
Изовалериановый альдегид 186
Изолейцин 378, 384
Изомасляная кислота 216, 218
Изомеризация
алканов 55, 134
диазосоединений 293
спиртов 164
фталата калия 239
434
Изомерия 19, 24
алканов 40 сл.
алкенов 65 сл.
алкинов 81
аминов 276
бензолов замещенных 100 сл. ■
галогенпроизводных 137
геометрическая (цис-транс) 63,
66 сл., 239
гидроксикислот 316 сл,
диенов 90
динамическая 331
карбонильных соединений 186 сл.
карбоновых кислот 215, 229, 310
нитросоединений 267
оптическая (зеркальная) 316 сл.
пространственная 66
скелета (структурная) 40, 66
спиртов 151
фенолов 168
диклоалканов 58, 63
эфиров 177, 246
Изомеры
валентные бензола 101
оптические (зеркальные) 316
поворотные 43 сл.
зониазид 366
зоникотиновая кислота 366
Изооктан 75 ел., 135
Изопентан (2-Метилбутан) 41, 46,
47, 57, 96, 134
Изопрен (2-Метилбутадиен-1,3) 57,
90, 95 сл.. 397, 410
получение 5/, 88 сл., 96, 200, 206,
412
Изопреноидные соединения 896
«-Изопропилбензальдегид 189
Изопропилбензол см. Кумол
п-Изопропилметилбензол 102, 110
Изопропиловый спирт 151, 153, 159
Изопропилуксусная кислота 216
Изопропилуксусный альдегид 186
Изофталевая кислота 238
ИК-спектры 423 сл.
Имидазол 375
Имиды 255
Имины 283
Инверсия 64, 347
Ингибиторы 52
Индиго 371
Индоксил 371
Индол 361 сл., 370 сл.
Индолилуксусная кислота 371
Индуктивный эффект 149, 288, 812
Инициаторы полимеризации 403
Инкременты кратных связей 422
Инсулин 381, 384, 400
Иодбензол 116
Йодоформ 140, 206
я-Иодтолуол 294
Иониты 419
Ионный (гетеролитический) разрыв
связей 35 сл.
Ионол (2,6-Ди-грет-бутил-4-метилфе'
нол) 175 сл.
Ионообменные смолы 419
Иоцича реактив 302
Иприт 263, 354
Казеин 378, 384, 386
Кальциферол 394
Каменноугольная смола 113, 366
Камфен 399
Камфора 398 сл.
Кана—И нгольда—Прелога система
349
Канниццаро реакция 202, 204, 211,
373
Капролактам 62, 64, 109, 115, 206,
385, 402, 424
Капрон 206, 414
Каран 398
Карбамид (Мочевина) 133, 256 сл.
Карбанионы 36, 325
Карбен 119, 298 сл.
Карбениевые ионы см. Карбокатио-
ны
Карбид кальция 87
Карбитолы 180
Карбокатионы (Карбениевые ионы)
35 сл., 71, 141
Карбоксилат-анион 219 сл.
Карбоксильная группа 215, 219, 312,
315
Карболит 418
Карболовая кислота см. Феиол
Карбонилярозание алкенов 188, 225
Карбонильная группа 189 сл.
Карбонильные соединения (Оксосое-
динения) 186 сл.
восстановление 152, 192
конденсация 198 сл., 237
ненасыщенные 207 сл.
реакции 190 сл., 207 сл., 216, 283,
302
Карбонильный компонент 198
Карбоновые кислоты 215 сл.
алициклические 226
амиды см. Амиды
ангидриды 222, 243 сл.
ароматические 104
высшие 163 сл., 226, 228, 250 сл.,
304, 309
галогена цгидрид ы (Аци лга логени -
ды) 189, 222, 240 сл., 299
галогензамещенные 222,230,310 сл.,
314, 357
Гидрирование 228
гидрогалогенирование 311
435
Карбоновые кислоты
двухосновные 233 сл.
декарбоксилирование 104, 187,
235 сл., 324
непредельные 104, 229 сл., 311,
316, 357
нитрилы см. Нитрилу
нитрозамещенные 358
одноосновные 215 сл., 229 сл.
полиеновые 237
получение 201, 216 сл., 229 сл.,
234 сл., 255, 302
реакции 187, 220 сл., 230 сл., 248,
299, 301, 311 сл.
сложные эфиры 222, 246 сл.
соли 48, 104, 187, 220, 226 сл.,
254, 307
Каротин 394
Каучук(и) 95, 401 сл.
вулканизация 409, 413
бутадиеновые 411 сл.
бутилкаучук 412
натуральный 409
полиуретановый 413
силоксановые 412, 418
хлоропреновый 412
этилен-пропиленовый 412
Квантовые числа 25 сл.
Кекуле формула бензола 98, 100
Керосин 55, 130
Кетены 209 сл.
Кетогексозы 335
ртокислоты 327 сл.
ётоны 186 сл.
ароматические 103
восстановление 103, 152, 192
изомерия 186
конденсация 200 сл., 206, 237
ненасыщенные 207 сл.
окисление 201 сл., 206, 216
оксимы 195 сл.
получение 159, 164, 187 сл.
реакции 190 сл., 195 сл., 283, 299
фенилгидразоны 370
Кефалины 253 сл.
Кижнера — Вольфа реакция 196
Клатраты 257 сл.
Ковалентный радиус 44 сл.
Коллодий 351
Коллоксилин 351
Кольбе — Шмитта реакция 321
Комплекс
переходный 141
О- 107
я- 71, 105, 106
Конденсация 34
алкенов 200, 369
алкинов 200 сл., 206
альдегидов 197, 200 сл., 207, 209,
233, 333 сл., 353
Конденсация
альдольная 198, 204
ароматических углеводородов 201,
214
бензоиновая 203
изопрена 397
кетонов 200 сл., 206, 237, 353
кротоновая 199, 207
нитросоединений 272, 353
сложноэфирная 325 сл.
фенолов 173, 244
фталевого ангидрида 214, 244
Кониин (а-Пропилпиридин) 375 сл.
Коновалова реакция 54, 269 сл.
Константа кислотности 220
Конфигурация
абсолютная 319
а-аминокислот 379
гидроксикислот 318
моносахаридов 344
Конформации 42 сл.
Конформеры 43 сл.
Коричная кислота 104, 233
Коричный альдегид 199
Кортизон 401
Коферменты 391 сл.
Крахмал 162, 332, 347 сл.
Крезолы 168, 174
Крекинг 48, 54 сл., 68, 88, 131 сл.
Кремнийорганические соединения
305 сл., 418 сл.
Кротоновая кислота 229
получение 209,. 230, 237
Кротоновый альдегид 198, 207, 209
Ксеноцианин 367
Ксил'ан 333
Ксилоза 332 сл., 335
Ксилолы (Диметилбензолы) 102, 104,
114, 132
окисление 110, 238 сл., 416
Куминаль 189
Кумол (Изопропилбензол) 102, 104,
114 сл., 189
гидропероксид 169
окисление 169
получение 80, 109, 110, 115
Кумольный способ 169, 204
Кучерова реакция 85, 188, 204
Лавсан 415
Лаки 416
Лакриматоры 140
Лактамы 359
Лактиды 315
Лактоза 319, 332 сл., 345 сл.
Лактоны 316
Ланолин 250
Латекс 404, 409
Лауриновая кислота 251
436
Левулиновый альдегид 410
Лейцин 378, 384
Лецитины 253 сл.
Лизин 379, 384 сл,
Лизол 174
Лимонен 396 сл.
Лимонная кислота 320
Линоксин 253
Линолевая кислота 232 сл., 251
Линоленовая кислота 232 сл., 251
Липиды 250
Липопротеиды 384
Литийорганические соединения 303
Лорентц—Лоренца формула 421
Львова—Шешукова хлорирование 139
Магнийорганические соединения 86,
144, 194, 300 сл.
Мазут 55, 130
Макроциклические соединения 59
Малеиновая кислота 234, 239 сл., 402
Малеиновый ангидрид 94, 109, 115,
240, 243
Малоновая кислота 233 сл., 235 сл.,
357
динитрил 259
Малоновый эфир 237, 260
Мальтоза 332 сл., 345 сл.
Маннаны 333
Манноза 332 сл., 335, 342
Маргарин 253
Марковникова правило 52 сл., 72 сл.,
84, 208, 230, 311
Масло (а)
антраценовое 113
высыхающие 253
каменноугольной смолы 113
смазочные 163
Масляная кислота 218, 241, 253
Масляный альдегид 209
Масс-спектрометрия 426
Меервейна реакция 295
Мезитилен 396
Мезитила окись 200, 207
Мезовинная кислота 319
Мезомерия 92
карбоксилат-аниона 220
нитросоединений 268 сл.
Мезомерный эффект 289
ц-Ментан 396
Ментол 397
Ментон 397
Меркурирование гетероциклов 373 сл.
Мерсеризация 349 сл.
Метакриловая кислота 229, 232
Металлопротеиды 384
Металлорганическне соединения 295,
300
Метальдегид (Сухой спирт) 197
Метан 145, 42, 49, 52
получение 301 сл.
реакции 56 сл., 89, 145
Метанол см. Метиловый спирт
Метансульфокислота 264
Метатезис 79
Метилакрилат 231, 408
Метиламин 278, 284
4-Метиламинофенол (Метол) 355
Метиланилин 278
Метилбензол см. Толуол
2-Метилбутадиен-1,3 см. Изопрен
2- Метилбутан см. Изопентан
3- Метилбутаналь 186
З-Метилбутановая кислота 216
2- Метилбутен-2 78
Метилгалогениды 52, 56, 141, 300
3- Метилгексен-З 78
Метилгидропероксид 185
Метиленовый компонент 198
Метилглиоксаль 210 сл.
Метилглюкозиды 337, 342
Д-Метилиндол (Скатол) 370
Метилмагнийиодид 302
Метилмалоновая кислота 234
эфир 237
Метилметакрилат 232, 408
Метилнатрий 300
Метиловый красный 360
Метиловый оранжевый 298
Метиловый спирт (Метанол) 56, 151,
153, 156, 160 сл.
реакции 158, 160, 200, 225, 232,
279
МетилокСония бромид 156
Метилолмочевина 257
4- Метилпентен-3-он-2 200, 207
Метилпиридины (Пиколины) 363 сл.
М-Метилпиридон-2 365
2-Метилпропановая кислота 216, 218
Метилпропанолы 151
2-Метилпропен см. Изобутен
Метилсалицилат 321
Метилтрихлорсилан 306
Метилфенилметанол 183
Метилфенолы 168, 174
Метилфосфин 307
Метилфосфонийхлорид 307
Метилцеллозольв 166, 179
Метилцеллюлоза 352
Метилциклобутан 58
Метилэтилкетон 78, 190, 211, 330
Метилэтилмалоновая кислота 234
эфир 237
Метилэтиловый эфир 177
4-Метил-6-этилнонан 47
2-Метил-5-этилпирндин 363
Метионин 379, 384
Метоксибензол (Анизол) 177, 180
437
2-Метоксипиридии 365
Р-Метоксипропионитрил 261
Метоксиэтан 177
Метол (4-Метиламинофенол) 355
Модели молекул 42 сл., 45
Молекулярная рефракция 421 сл.
Молочная кислота 314 сл., 317, 319
соль 211
Молочный сахар 319, 346 сл.
Мономеры 402
Монооксид углерода 57
Моносахариды 331 сл., 334 сл,
реакции 340 сл.
стереохимия 344
Монотерпены 396
Монохлоруксусная кислота 311
Мочевина (Карбамид) 133, 256 сл.,
402
Моющие средства (Детергенты) 228,
354
Мукопротеиды 384
Муравьиная кислота 216, 218, 222 сл.,
235
эфиры 56, 188, 223 сл., 247
Муравьиный альдегид см. Формальде¬
гид
Мутаротация 336
Мыла 226 сл., 252
Мылонафт 64, 226
Мясомолочная кислота 317
Найлон 64, 206, 238
Напряжение циклов 59
Натрийацетоуксусный эфир 328
Натриймалоновый эфир 237
Нафталин 100, 119, 123, 423
реакции 120 сл., 122 сл., 213, 238
Нафтацен 125
Нафтеновые кислоты 64, 226
Нафтены 58
Нафтиламины 276, 287 сл.
Нафтионовая кислота 288
Нафтолы 121, 176, 180
Нафтохиноны 213
Нейрин 354
Неогексан 47, 303
Неопентан 41, 46, 47
Неролины 180
Нефть (и) 129 сл.
ароматизация 103, 114
крекинг 68, 131 сл.
нафтеновые 130
обессеривание 134, 374
парафиновые 130
перегонка 68, 130 сл., 133
пиролиз 68, 132
риформинг 114, 123, 133
фракции 130
Никотин 376
Никотиновая кислота 366
Нитраты 158. 351
Нитрилы (Цианиды) 258 сл., 402
ароматические 258, 266
восстановление 259
а-гидрокспкислог 193 сл.
гидролиз 216, 254 сл., 258
получение 144, 255, 258 сл., 266
реакции 258 сл.
Нитроанилины 281, 286
я-Нитроацетанилид 286
Нитробензойные кислоты 360
Нитробензол 98, 115, 267. 270 сл.,
275
реакции 279, 280, 285, 366
Нитрование
алканов 53 сл., 57, 269 сл.
ароматических соединений 106,
112, 121, 124, 269 сл., 360
гетероциклов 368, 372 сл.
фенола 172, 355
циклоалканов 62, 269
Нитроглицерин 168
Нитрогруппа 272, 289, 291
W-Нитрозамины 284
Нитрозобензол 280
п-Нитрозодиметиланилин 284
Нитрозосоединения 267 сл., 284
Нитрозоний-катион 284
Нитрометан 57, 267, 271
а-Нитронафталин 121, 271, 276
Нитрон 408
Нитроний-катион 106, 284
Нитросоединения 106, 267 сл.
алифатические 275
ароматические 279
восстановление 272, 278 сл., 362
atju-формы 272 сл.
Нитроспирты 353
Нитротолуолы 276
Нитрофенолы 172, 355
Нитроформ 275
5-Нитрофурфурол 373
Нитроциклогексан 62, 2С7, 275
Нитроэтан 271
Новокаин 360
Новый неролин 180
Номенклатура
алканов 45 сл.
алкенов 67 сл.
алкилгалогенидов 137 сл.
алкинов 81 сл.
альдегидов 186
альдегидокислот 323
аминов 277
ароматических углеводородов
101 сл.
галогенангидридов карбоновых
кислот 240
галогензамещенных кислот 310
гидроксикислот 313 сл,
438
Номенклатура
диенов 90
карбоновых кислот 215 сл., 233 сл.
кетокислот 323
кетонов 187
Нитросоединений 267
Простых эфиров 177
^Ложных эфиров 246
<;пмртов 151
циклоалканов 57 сл.
Нонан 41, 68 сд,
Норсульфазол 287
Нуклеиновые кислоты 386 сл,
Нуклеозиды 387 СЛ.
НУклеопротеиды 384
Нуклеотиды 887 сл.
Нуклеофильное замещение 140 сл.,
273, 364, 367
Ну{$деофидь«ре присоединение 190 сл-
Нуклеофильные реагенты 36
Рзаэоны 342
рзониды 77
Озонирование 77, 410
Окисление
Влканов 54, 216, 225
алкенов 54, 76, 184, 216
ддкинов 84, 85
альдегидов 201 сл„ 208, 216
аминонафтолов 213
анилина 213, 285
антрацена 125, 214
ароматических соединений 109 сл„
216
гидроксикислот 324
гидрохинона 213
гдиколей 234, 324
изобутена 77, 232
карбоновых кислот 222, 231, 236
кетонов 201 сл., 206, 216
ксилола 238, 416
кумола 169, 188
метана 56
моносахаридов 340
нафталинов 122 сл., 213, 238
парафина 225, 228
пиридинов 365
пирокатехина 213
пропилена 164, 167, 205, 209, 231
простых эфиров 179
спиртов 159, 187, 216
тиолов 263 сл.
толуола ПО, 226
углеводородов 188
фенолов 173
фурана 372
циклоалканов 62
’ цимола 238
Окисление
этилбензола 207
этилена 181, 188, 204
Оксимы 195 сл., 258
Оксиран см. Этиленоксид
Окситоцин 380
Оксония соли 156, 178
Оксосинтез 188, 225
Оксосоединения см. Карбонильные
соединения
Октан 41, ЮЗ
Октановая кислота 25.1
Октановое число 133, 135 сд.
Олеиновая кислота 231 сл., 251
Олефины см. Алкены
Олигосахариды 332, 345 сл.
Олифа 253
Оптическая активность 316
Оптические антиподы 316
Оптические отоеливатели 415
Орбитали электронные 24 сл,
Органические соединения серы
261 сл.
Органическое стекло 232, 408
Ориентация заместителей 110 сл,,
121 сл., 205, 226, 290, 364
Ортомуравьиная кислота, эфир 194
Основания Шиффа 283
Пальмитиновая кислота 218, 225, 250,
251 сд.
Пальмитоолеиновая кислота 251
Папаин 391
Паральдегид 197
Парафин 133, 225, 228
Параформ (Полиоксиметилен) 197
Параформальдегид 203
Паули принцип 26
Пек 113
Пеларгоновая кислота 231
Пенициллин 394 сл.
Пенополивинилхлорид 407
Пентаацетилглюкоза 334, 343
Пентадекан 49
Пентаконтан 49
Пента-О-метил-а-ТЗ-глюкоза 334, 342
Пентан 41, 48, 49 сл.
Пентафенилфосфоран 308
Пентафталевые смолы 416
Пентахлортолуол 107
Пентахлорфенол 174
Пентаэритрит 402, 416
Пентиловые спирты 163
Пентозаны 332, 372
Пентозы 332, 343, 372
Пептиды 379
Пербензойная кислота 202, 245 сл.
Пергидроантрацен 124
Пербензойная кислота 202, 245 сл,
43»
Пергидроантрацен 124
Перегонка (Ректификация) 15
азеотропная 133
сухая 88, 161, 187, 224, 254, 410
Перегруппировка
Арбузова 309
Бекмана 1S6
Гофмана 255 сл.
Перекристаллизация 14
Переходное состояние 141
Переэтерификация 249
Перкина реакция 233
Пероксид(ы) 184 сл., 245 сл.
Пероксикислоты (Гидропероксиды
ацилов) 181, 202, 245
Перфторуглеводороды 147 сл.
Перхлорнафталин (Галовакс) 120 сл.
Петролейный эфир 131
Пиколиновая кислота 366
Пиколины (Метилпиридины) 363 сл,
Пикрамид 275, 296
Пикриновая кислота 172, 273 сл.
Пимелиновая кислота 234
Пинан 398
а-Пинен 396, 398 сл.
Пиперидин 361, 365
Пиразол 375
Пиразолин 299
Пиран 338, 361
Пиранозы 338
Пирен 126
Пиридин 113, 361, 362 сл.
Пиридицкарбоновые кислоты 366
Пириди'нсульфотриоксид 369
Пиридоксаль-5-фосфат 392
а-Пиридон 365
Пиримидин 375
Пиримидиновые основания 386
Пировиноградная кислота 323 сл.
Пирогаллол 168, 175 сл.
Пирокатехин 168, 175, 213
Пироксилин 351
Пиролиз 55
алканов 56, 68 сл., 89
нефти 68, 132
Пирослизевая кислота 373
Пиррол 361, 368, 369 сл.
Пирролидин 361
Пластификаторы 163, 407
Плодовый сахар 332
Плотность относительная 16
ПМР-спектры 425
Поверхностно-активные вещества 163,
182, 227 сл.
Показатель преломления 16
Полиакрилонитрил 408 сл.
Полиалкены 404 сл.
Волиаллен 97
олиамиды 401, 4J3 сл.
Поливинилацетат 409
Поливиниловый спирт 409
Поливинилхлорид 407
Полигидроксибензойные кислоты
322 сл.
1,4-Полиизопрен 410
Поликонденсация 34, 413 сл.
Полимергомологи 410
Полимеризация 34, 403 сл.
акрилонитрила 260, 408
алкенов 76 сл., 404 сл., 412
алкинов 85 сл., 103
альдегидов 197
блочная 404, 406
блок-сополимеризация 406
виниловых мономеров 407
диенов 94, 411
ионная 404
радикальная 403
стереоспецифическая 405
стирола 403
ступенчатая 404
фторэтиленов 407
эмульсионная 404
эфиров винилового спирта 409
— ненасыщенных кислот 408
Полимеры см. Высокомолекулярные
соединения
Полиметилакрилат 408
Полиметилметакрилат 232, 408
Полиоксиметилен см. Параформ
Полиприсоединение 413
Полипропилен 405 сл.
Полисахариды 332 сл., 347 сл.
Полисилоксаны 418
Полистирол 80, 115, 403, 406
Политетрафторэтилен 149
Полиформальдегид 197
Полифункциональные соединения 38
Полиэтилен 76, 80, 402, 404
Полиэтилентерефталат 415
олиэфиры 415 сл.
олуацетали 194, 337
Поляризация связи 71, 73, 84, 149
Поляризуемость связи 33
Полярность связи 32 сл.
Поляриметр 316 сл.
Порох бездымный 351
Правило
Зайцева 69
Марковникова 52 сл., 72 сл., 208,
230, 311
Хюккеля 100
Эльтекова 164, 196
Яреднизолон 401
ресс-порошки 417
Прилежаева реакция 181, 246
Пролин 378 сл.
Йропадиен см. Аллен
ропан 48, 49, 54, 57, 80, 270
Пропандиолы 165
440
Пропантиол 262
Пропилбензол 103, 104
Пропилгалогениды 61, 140, 149, 331
Пропилен 64, 70, 80, 145, 402
окисление 164, 167, 205, 209, 231
полимеризация 405
реакции 96, 109, 151, 260
хлорирование 79, 139, 145, 167 сл.
Пропиленгликоль 150, 165
Пропиленоксид (1,2-Эпоксипропан)
181, 183 сл.
Пропиловый спирт 153, 159, 163
а-Пропилпиридин (Кониин) 375 сл.
Пропин-2-иловый спирт 88
Пропионовая кислота 77, 159, 218,
222, 330
Пропионовый альдегид 78, 159, 190
Протеиды 384
Протеины 384
Протонный магнитный резонанс
(ПМР) 425
Прототропия 326
Пурген (Фенолфталеин) 244 сл.
Пурин 375
Пуриновые основания 386
Радикальный (гемолитический) раз¬
рыв связей 34 сл.
Радикалы
иминоксильные 119
свободные 35, 117 сл.
Радиоспектроскопия 425
Рацематы 318
Рашига способ 170
Реактив(ы)
Гриньяра 144, 194, 300 сл.
Иоцича 302
Чугаева 211
Швейцера 349
Реакция (и)
бимолекулярные 141
биуретовая 378
Вагнера 77
Вюрца 48
Гаттермана 189
Гофмана 278
Гриньяра 301
Дильса—Альдера 93 сл., 208, 214
замещения 34, 140 сл.
Зандмейера 294
Зелинского 102
Зелинского — Гелля — Фольгарда
222
Зинина 279
ионные 71 сл.
Канниццаро 202, 204, 211, 373
качественная на двойную связь 72
— на Н-атом при связи CsC 86
Кпжнера — Вольфа 196
Кольбе—Шмитта 321
Коновалова 54, 269 сл.
Кучерова 85, 188, 204
Меервейна 295
мономолекулярные 141
Перкина 233
Прилежаева 181, 246
радикальные 184
Реппе 231
Свартса 148
серебряного зеркала 201, 341
Тищенко 203
Фаворского 200
Фаворского — Шостаковского 178
Фриделя — Крафтса 103, 108 сл.,
172, 189, 207
Циглера 304
Чичибабина 363 сл.
Чугаева — Церевитинова 160
Юрьева 369
Ректификация см. Перегонка
Резины 409
Резит 417
Резол 417
Резорцин 168, 169
Рентгенография 31
Реппе реакция 231
Ретинол 394
Рефрактометрия 421
Рибоза 332 сл., 335, 340, 387
Рибонуклеаза 380, 381 сл., 384
Рибонуклеиновые кислоты (PHKJ
387 сл.
Рибонуклеотиды 387
Риформинг 114, 123, 133
Родионова синтез 357
Розанцева радикалы 119
Ртутьорганические соединения 295
Сажа 56
Салициловая кислота 321
Салол 321
Сахарная (глюкаровая) кислота 341
Сахароза 332 сл., 345 сл., 34?
Свартса реакция 148
Связь(и) 29 сл.
аксиальные 63
«банановые» 63
водородные 153 сл., 217
двойные 65, 90, 91
длина 31
донорно-акцепторная 268
изолированные
ковалентные 29 сл., 31, 34 сл., 50
координационная 268
кратные, качественная реакция 84
кумулированные 90
пептидная 379
поляризуемость 33
441
Связь (и)
полярность 32 сл.
семиполярная 268
сопряженные 90, 207
тройные 29, 81
экваториальные 63
электронная природа 28 сл.
энергия 32
Сегнетова соль 320
Сенсибилизаторы 367
Сера, обнаружение 17
Серин 379
Сероуглерод 57
Сиккативы 226, 253
Силандиолы 418
Силанолы 307
Силаны 306 сл.
Силоксаны 307
Синильная кислота 56
Синтез
Арндта — Эйстерта 299
Вильямсона 17§
Вюрца — Фиттига 103
Габриеля 279
Густавсоиа 59
Зелинского 8й7
Родионова 357
Скраупа 386
Твердофадщлй 383
фишерэ 370
циангидринный 314
Синтез-газ 165, 167
Скатол (Р-Метилиндол) 370
Скипидар 396, 398
Скраупа синтез 866
Соединения включения 257 сл,, 348
Сополимеризация 404
Сорбиновая кислота 232, 237
Сорбиновый альдегид 209
Сорбит 340, 393
Спектроскопия 422 сл,
Спермацет 249
Спермацетовое масло 250
Спии-cnniiQBoe взаимодействие 423
Спираль 381, 388 сл.
Спирты 150 сл.
ароматические 150
ассоциация 1оЗ сл.
высшие 163 сл., 304
двухатомные 150, 165
дегидратация 69, 158, 177
дегидрирование 160
многоатомные 165 сл.
насыщенные 150
ненасыщенные 150, 200
одноатомные 151 сл.
окисление 159, 187, 216
получение 151 сл„ 195, 303
реакции 138, 155 сл., 177, 194,
262, 294, 299, 301, 309
Стеарин 252
Стеариновая кислота 218, 225, 251 сл,
Стереоизомерия 66
Стероиды 399 сл.
Стирол (Винилбензол) 102, 104, 115,
403
получение 80, 104, 115, 183,
233
сополимеры 406
Стиролоксид 181
Стрептоцид 287
Суберон (Циклогептанон) 299
Сукцинонитрил 261
Сульфадимезин 287
Сульфамидные препарату 287
Сульфаниловая кислота 285, 287, 298
амид 287
Сульфеновые кислоты 262
Сульфиды 262 сл.
Сульфиновые кислоты 262
Сульфирование 264 сл,
едилнна 285
витрацена 124
ароматических соединений 108,
264 сл.
Гетероциклов 368 сл., 372, 374
нафталина 120
-фенола 172
Сульфокислоты (Сульфоновые кис*
лоты) 262, 264 сл.
алифатические 264
ароматические 204 сл.,
соли 54, 258, 264
хдорангидриды 54, 228, 266
эфиры 266
Сульфоксиды 262, 264
Сульфонамиды 266
Сульфонаты 54, 258, 264
Сульфония соли 262
Сульфоновые кислоты см, Сульфокис¬
лоты
Сульфоны 262, 264
Сульфоокисление алканов 54
Сульфонилхлориды 54, 230, 266
Сульфохлорирование алканов 54,228,
264
Сухой спирт см. Метальдегид
Сфингомиелины 384
Сэндвичеобоазные соединения 805
Танины 322
Таутомерия 330 сл.
ацетоуксусного эфира 326
диазосоединений 293
кето-енольная 212, 326
моносахаридов 336
нитросоединений 272 сл.
пиримидиновых оснований 386
Теломеризация 414
442
Теория
молекулярных орбиталей 30
напряжения Байера 59
радикалов Берцелиуса 20
типов 20 сл.
химического строения Бутлерова
22 сл.
Тепловые эффекты реакции 32
Терефталевая кислота 238 сл., 402
получение ПО, 238 сл., 415 сл.
Терпены 58, 395 сл.
Тестостерон 400
Тетраалкилсвинец 118
1,1,2,2-Тетрабромэтан 84
Тетрагидробензонитрил 261
Тетрагидронафталин (Тетралин) 122
Тетрагидропиран 338
Тетрагидрофуран 179, 180, 339, 361,
372
Тетрагидрохинолин 367
Тетралин (Тетрагидронафталин) 122
Тетраметилглюкоза 342
Тетраметиленгликоль (Бутандиол-1,4)
165, 180
Тетраметилсилан 306
Тетранитрометан 275
Тетрафторэтилен 148, 408
Тетрахлорэтан 88
Тетрациклины 395
Тетрозы 332
Тефлон 149
Тиазол 375
Тиазолидин 402
Тимин 386, 388 сл,
Тиокетоны 262
Тиолы 262 сл., 303
Тиофан 362
Тиофен 362, 368, 374 сл.
Тиофос 309
Тиоэфиры 262 сл.
Тира мин 355 сл.
Тирозин 379
Тищенко реакция 203
Тол см. Тринитротолуол
Толуол (Метилбензол) 102, 104, 114,
114, 145, 424
окисление 110, 226
получение 103, 113
реакции 107, 110, 112, 114, 132,
145, 205. 276, 360
Треоза 332
Треонин 379, 384
Триаконтан 41
Триброманилин 285
2.4.6- Трибромфенол 172
Три-грег-бутил бензол 101
Тригидроксибензолы 168, 175, 343
2.4.6- Три (гидрокси метил) фенол 173
Трядепсид 323
Триизопропилметанол 303
Трикарбонилциклопентадиенилмарга«
нец 305
Триметиламин 278
Триметиленгликоль 165
Триметилсиланол 307
Триметилуксусный альдегид 186
Триметилхлорсилан 306
Триметилэтилметан 47
1.3.5- Тринитробензол 273
Тринитротолуол (Тротил, Тол) 274,
276
2.4.6- Тринитрофенол (Пикриновая кио»
лота) 172, 273 сл.
Триоксан (Триоксиметилен) 197, 203
Триптофан 371, 378, 384
Трис(диметиламидо)фосфат 309
Тристеарин 251
Тритерпены 396
Трифенилбромметан 117
Трифенилметан 116 сл,, 201
Трифенилметанол 117
Трифенилметил 117 сл.
Трифенилхлорметан 117, 143
Трифтортрихлорэтан 149
Трифторхлорэтилен 149, 407
Трихлорацетон 193
Трихлоруксусная кислота 222, 312
Трихлоруксусный альдегид 193,204 сл,
Трихлорэтилен 88
Триэтаноламин 353
Трнэтиленгликоль 182
Триэтилфосфит 309
Тропилии-катион 127
Тротйл см. Тринитротолуол
Углеводы 152, 161 сл., 331 сл.
Углерод 17
валентные состояния 27 сл.
диоксид, реакции 256, 302, 321,
360
■ монооксид 49, 160, 188, 223, 242,
256
технический 56
Углеродный скелет 36 сл., 40
Удельное вращение 317
Уксус 224
свинцовый 225
Уксусная кислота 209 сл., 216, 218,
224 сл., 312
амиды 196, 282
нитрил 255, 258, 260
получение 77, 88, 210, 225, 241, 330
реакции 205, 209 сл., 248, 311, 321
У-фениламид см. Ацетанилид
этиловый эфир см, Этилацетат
эфиры 187, 225, 351
Уксусный альдегид 80, 88, 152, 186.
197 сл., 204, 209, 319. См. так*
же Ацетальдегид
446
Уксусный ангидрид 210, 243, 275, 343
Урацил 386, 388
Уридин 387, 390
Уротропин 203 сл.
УФ-спектры 423
Фаворского реакция 200
Фаворского — Шостаковского реак¬
ция 178
Фелингова жидкость 201, 341
Фенантрен 125
Фенацетин 355 сл.
Фенетол 177, 180
Фенилаланин 356, 378, 384
Фениламиноуксусная кислота 286
Фенилацетат 171
Фенилацетилен 102, 104
Феиилаиетоиитрил 258
Фенилгалогениды 139
Фенилгидразин 196, 295, 341 сл.
Фенилгидразоны 196, 370
Фенилгидроксиламин 280
Фенилдиазонийхлорид 293
Фенилнатрий 103
N-Фенил-Р-нафтиламин 288
Фенилнитрометан 272 сл.
I -Фенилоксиран 181
Фенилуксусная кислота 216, 218
Фенилуксусный альдегид 186
Фенилцианид (Бензонитрил) 258
р-Фенилэтиловый спирт 153, 165
Фенол (Карболовая кислота) 168, 169,
173 сл., 290, 402
получение ИЗ, 115, 174, 293
реакции 170 сл., 172 сл., 285, 416
Фенолоформальдегидные смоль/ 173,
401, 416 сл.
Фенолсульфокислоты (Гидроксибен-
золсульфокислоты) 172
Фенолфталеин (Пурген) 244 сл.
Фенолы 168 сл., 173 сл.
многоатомные 174 сл.
получение 115, 169 сл., 293
реакции 170 сл., 244, 299
эфиры 171, 180, 294
Феноляты 170, 321
Ферменты 343, 378, 391 сл.
Ферроцен 305
Фишера
проекционные формулы 318
синтез 370
Флороглюцин 175, 343
Флуоресцеин 245
Формалин 203
Формальдегид (Муравьиный альде¬
гид) 186, 190, 203, 402
конденсация 199, 200, 209, 334
получение 160
реакции 173, 193, 195, 197, 202 сл.,
257, 416
Формамид 254, 256
Формиаты 188, 223 сл., 247
Формилгалогениды 240, 242
Формула (ы)
Кекуле 98, 100
Лорентц — Лоренца 421
моносахаридов 334 сл., 338 сл.
Ньюмена 43
перспективные 43
проекционные Фишера 318
структурные 22 сл., 420
Фосген 242
Фосфатиды 253, 384
Фосфины 307
Фосфопротеиды 384
Фосфорорганические соединения
307 сл.
Фотосинтез 333
Фреон-12 см. Дифтордихлор метан
Фриделя — Крафтса реакция 103,
108 сл., 172 сл., 189, 207
Фруктоза 332 сл., 335, 339, 345
Фталазол 287
Фталевая кислота 122, 234, 238, 402,
416
эфиры 238, 244, 407
имид см. Фталимид
Фталевый ангидрид 122 сл., 214,
244
Фталеины 244
Фталимид 244, 255
калия 255, 279
Фторопласты 149, 407 сл.
Фторуглероды 147
Фторхлоралканы 148
Фторэтилены 407 сл.
Фуксинсернистая кислота 203
Фумаровая кислота 234, 239 сл.
Функциональные группы 37 сл.
Фуран 339, 361 сл., 368, 372 сл.
реакции 180, 369, 372
Фуранозы 339
Фурациллин 373
Фу()иловый спирт 373
Фурфурол 343, 372 сл.
Хелаты 212
Химический сдвиг 425
Хиноидная группировка 212 сл., 297
Хинолин 361 сл., 366 сл.
Хинолиновая кислота 367
Хиноны 212 сл.
Хлораль J93, 204 сл.
Хлоральгидрат 193, 205
Хлорацетон 203
Хлорацетофенон 207
Хлорбензол 107, 115, 140, 143, 285
Хлорирование
алканов 56, 138, 145 сл.
444
Хлорирование
алкенов 79, 139, 145
алкинов 84
антрацена 124
ацетофенона 207
бензальдегида 205
бензола 98, 145, 170
бутадиена 92, 145
нафталина 120
окислительное 146
толуола 107, 145, 205
уксусной кислоты 311
фурфурола 373
Хлоропрен 85, 88, 95, 412
Хлорофилл 370
Хлороформ 52, 56, 140, 149, 205, 222
Хлорофос 309
Хлорпикрин (Нитротрихлорметан) 275
Хлорсульфоновая кислота 265
Хлоруксусная кислота 312 сл., 371
Хлорфенолы 122
Хлорфосфины 307
Хлорфосфораны 307 сл.
5-Хлорфурфурол 373
Холестерин 399 сл., 401
Холин 253, 354
Хризен 126
Хроматография 15 сл.
Хромофоры 297
Хюккеля правило 100
Циклогексатриен 98
Циклогексен 93, 94
Циклогексен-2-ол 159
Циклогексен-2-он-1 159
Циклогексиламин 286
Циклогептанон (Суберон) 299
Циклогептатриен 127
Циклодекапентаен 86
Циклононан 60
Циклооктатетраен 86
Циклопентадиен-1,3 90, 93, 66 сл.,
126, 302
Циклопеитадиенилий-анион 126 сл.
Циклопентадиенилмагнийиодид 302
Циклопентадиенилнатрий 96
Циклопентан 58 сл., 60 сл., 62, 64
Циклопентанкарбоновая кислота 64,
218 226
Циклопентанон 187, 190, 197, 236
Циклопентанопергидрофенантрен 399
Циклопентен 96
Циклопропан 58 сл., 60 сл., 62, 299
Циклотриаконтан 60
Цимол 102, 110, 238 сл.
Цинкорганические соединения 303
Цистеин 379
Цистин 379
Цитидиловая кислота 388
Цитидин 387, 390
Цитозин 386, 388 сл.
Целлобиоза 332, 345
Целлозольвы 166, 180
Целлофан 352
Целлулоид 351
Целлюлоза 162, 332, 349 сл.
эфиры 350 сл.
Цетановое число 136
Цианамид 256
Циангидрины (Гидроксинитрилы)
193 сл., 327
Цианиды см. Нитрилы
Цианиновые красители 367
Цианэтилирование 260
Циглера реакция 304
Циклоалканы 57 сл.
реакции 55, 61 сл., 114, 264
Циклоалкилхлориды 143
Циклобутан 58 сл., 60 сл.
Циклогексан 58 сл., 60
получение 59, 109, 115
реакции 61 сл., 102
стереоизомеры 63 сл.
Циклогексанкарбоновая кислота 218
Циклогексанол 64, 153, 163, 173
Циклогексанон 64, 164, 169, 190, 206,
299
оксим 196, 206
получение 163, 206
Четыреххлористый углерод 52, 56,
146 сл., 414
Чичибабина реакция 363 сл.
Чугаева реактив 211
Чугаева — Церевитинова
метод 302
реакция 160
Швейцера реактив 349
Шиффа основания 283
Щавелевая (этандиовая) кислота 224,
233 сл., 235 сл.
Эйкозан 49
Экзальтация молекулярной рефрак-
ции 422
Экстракция 15, 133
Элаидиновая кислота 232
Электронное облако 24 сл.
Электронный парамагнитный резонано
426
Электроноакцепторные заместители
112, 150
Электроотрицательность 32
Электрофильное замещение 105 сл.,
110 сл., 171 сл., 288, 295 сл.,
364, 367
443
Электрофильное присоединение 71, 83
Электрофильные реагенты 36
Элементорганические соединения
300 сл.
Эльтекова правило 164, 196
Эмали 416
Энант 80, 414
Энантовая кислота 218, 225
Эпоксиды 17, 181
1,2-Эпоксипропан см. Пропиленоксид
Эргостерин 394
Эритроза 332
Эстрон 400
Этазол 297
Этан 35, 42, 48, 49, 55, 79, 145
Этандиол-1,2 см. Этиленгликоль
Этандиовая кислота см. Щавелевая
кислота
Этаноламины 183, 254, 353 сл.
Этансульфонат натрия 264
Этен см. Этилен
Этерификация 247 сл.
Этилацетат 158, 210, 211, 247, 249
Этилбензол 80, 102, 103, 104, 109,
115
реакции 115, 207
Этилбутират 247
Этилен (Этен) 48, 55, 64, 69, 70,
79 сл„ 145, 402, 423
гидратация 151
окисление 181, 204
полимеризация 40*
реакции 109, 115, 165, 204, 248,
299, 414
хлорирование 139, 145
Этилгалогеннды 103, 137, 140
Этиленгликоль (Этандиол-1,2) 150,
165, 402
ацетали 194
получение 144, 165, 182 сл.
Этиленгликоль
реакции 166, 180, 234
эфиры 183, 415
Этиленоксид (Оксиран) 77, 80, 165,
181 сл.
Этиленхлоргидрин 80, 16$, 182 сл.
Этилиденмалоновая кислота 237
Этилнитрат 158
Этиловая жидкость 136
Этиловый спирт 151, 153, 161 сл.,
425
абсолютный 161
дегидратация 69, 159
получение 80, 151, 152, 183
реакции 94, 160, 411
Этилсерная кислота 74, 158
Этилформиат 247
Этилцеллюлоза 353
Этилэтилен 67
Этинилбензол 102, 104
4-Этоксифенилмочевина 355
Эфедрин 354
Эфиры
виниловые 89, 179, 188, 402, 409
гликолей 179, 182
минеральных кислот 157
простые 144, 156 сл., 171, 176 сл.,
402
сложные 157 сл., 171, 222, 243,
246 сл., 299
сульфокислот 266
фенолов 171, 299
циклические 180, 181
Юрьева реакция 369
Яблочная кислота 319 сл.
Ядерный магнитный резонанс 425
Янтарная кислота 234, 235, 330
производные 231, 236
Учебник
ПОТАПОВ Виктор Михайлович
ТАТАРИНЧИК Софья Наумовна
ОРГАНИЧЕСКАЯ
ХИМИЯ
Редактор М. Н. ПАСТУШЕНКО
Художник Н. М. БИКСЕНТЕЕВ
Художественный редактор Н. В. НОСОВ
Технический редактор С. Ю. ТИТОВА
Корректор Т. С. ВАСИНА
ИБ № 2369
Сдано е набор 20.10.8d. Подписано в печать 03.04.89. Формат
60Х901/!* Бумага тип, JSfe 2. Гарнитура литературная.
Печать высокая. Уел. печ. л. 28,0. Уел. кр.-отт. 28,0. Уч.-изд.
л. 31,07. Тираж 50 000 экэ. Заказ 808. Цена 1 р. 30 к.
Ордена «Знак Почета» издательство «Химия»,
107076. Москва, Стромынка, 21, корп. 2.
Ленинградская типография № 2 головное предприятие
ордена Трудового Красного Знамени Ленинградского объеди¬
нения «Техническая книга» им. Евгении Соколовой Союз-
полиграфпрома при Государственном комитете СССР поделай
издательств, полиграфии и книжной торговли
198052, г, Ленинград, Л-52, Измайловский проспект, 29
Отпечатано с матриц в Ленинградской типографии № 6
издательства «Машиностроение» при Государственном ко?
митете СССР по делам издательств, полиграфии и книжной
торговли. 193144, г. Ленинград, ул. Моисеенко, 10.