/























































































































































Автор: Черных В.П.
Теги: органическая химия химические науки химия строение и реакционные способности углеводородов функциональные производные основные классы органических соединений
ISBN: 5-7768-0137-0
Год: 1993




Текст
В. П. Черных, Б. С. Зименковский, И. С. Гриценко
ОРГАНИЧЕСКАЯ
ЖШЕЯШЯ
В ТРЕХ КНИГАХ
Книга 2 Углеводороды и их функциональные производные
Утверждено Министерством здравоохранения Украины а качестве учебника для студентов фармацевтических вузов н факультетов
1 1' 1 -........
УкраГ'-'^ я * .’н ^и-мччв \ к . д < i я л. -еу в
I & Л I О * Е К А дЦ2>..
, 1111 .„I—„ I» |„ , .
XapKiH «Основа* 1995
ББК 24 2
449
УДК 547
Рецензенты
д-р хим наук проф В Д Орлов (Харьковский государственный университет), д-р фарм наук проф Б А Прийменко (Запорожский медицинский университет)
В учебнике рассмотрены главные аспекты строения и реакционной способности углеводородов и их функциональных производных Материал по основным классам органических соединений изложен с учетом принципа их функциональной принадлежности
Для студентов фармацевтических вузов и факультетов
„ 1705000000-041 _ Ч------— ---------Замовне
226-95
ISBN 5-7768-0137-0 (кн 2)
ISBN 5-7768-0130-3
© В П Черних, Б С Зименковський, I С Гриценко 1995
ОГЛАВЛЕНИЕ
ГЛАВА 1 АЛКАНЫ Г ц
1 1 Строение алканов 1<)
| 2 Номенклатура
1 3 Изомерия jij
I 4 Способы получения
1 4 I Природные источники 1$
1 4 2 Синтетические методы получения 1у
I 5 Физические свойства 1$
I 6 Химические свойства 1g
1 6 1 Реакции радикального замещения ( SR) 2q
1 6 2 Окисление алканов 2j
I б 3 Крекинг алканов 2ц
1 7 Идентификация алканов 2$
1 8 Отдельные представители Применение 2g
Контрольные вопросы и упражнения 2g
ГЛАВА 2 АЛКЕНЫ
2й
2 1 Номенктатура
2 2 Изомерия
2 3 Способы по пучения
2 4 Физические свойства
2 5 Химические свойства
2 5 I Реакции электрофильного присоединения (ЛЕ)
2 52 Реакции восстановления и окисления
2 5 3 Полимеризация алкенов
2 5 4 Аллильное галогенирование алкенов
2 6 Идентификация алкенов
2 7 Отдельные представители Применение Контрольные допросы и упражнения
ГЛАВА 3 АЛКАДИЕНЫ
28
29
3|
38
33
40
43
48
47
48
48
50
3 1 Номенклатура 50
3 2 Строение алкадиенов 51
3 3 Алкадиены с сопряженными связями 52
3 3 I Способы получения 52
332 Химические свойства 53
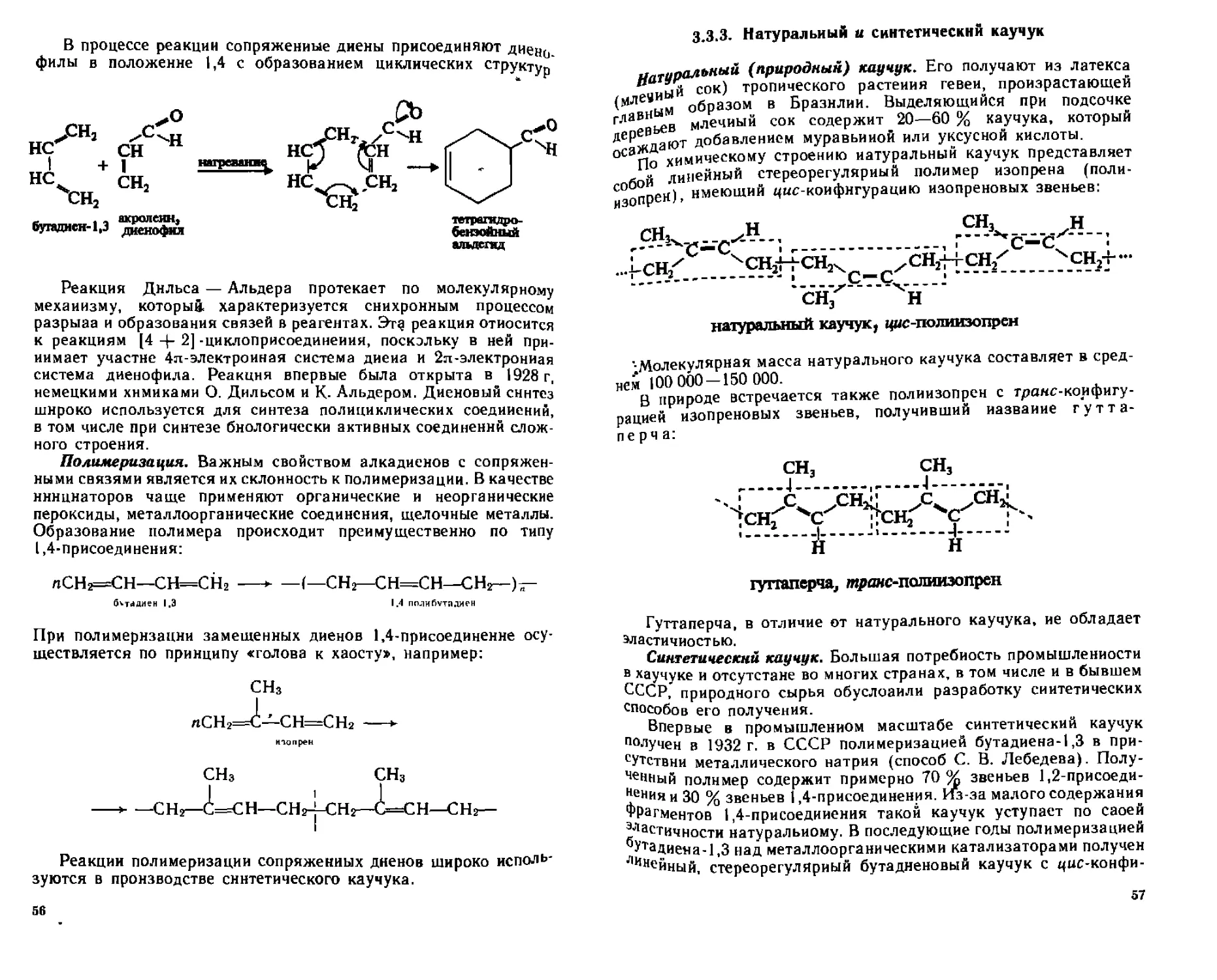
3 3 3 Натуральный и синтетический каучук 57
3 3 4 Идентификация сопряженных диенов 58
33 5 Отдельные представители Применение 59
Контрольные вопросы и упражнения 59
Г Л АВ А 4 АЛКИНЫ
60
4 1 Номенк1атура и изомерия 60
4 2 Способы получения 61
4 3 Физические свойства 62
4 4 Химические свойства 62
4 4 I Реакции электрофильного присоединения (АЕ) 63
4 4 2 Реакции замещения 64
4 4 3 Реакции окисления и восстановления 66
4 4 4 Димеризация, тримернзания и тетрамеризация алкинов 67
4 5 Идентификация алкинов 68
4 6 Отдельные представители Применение 68
Контрольные вопросы и упражнения 64
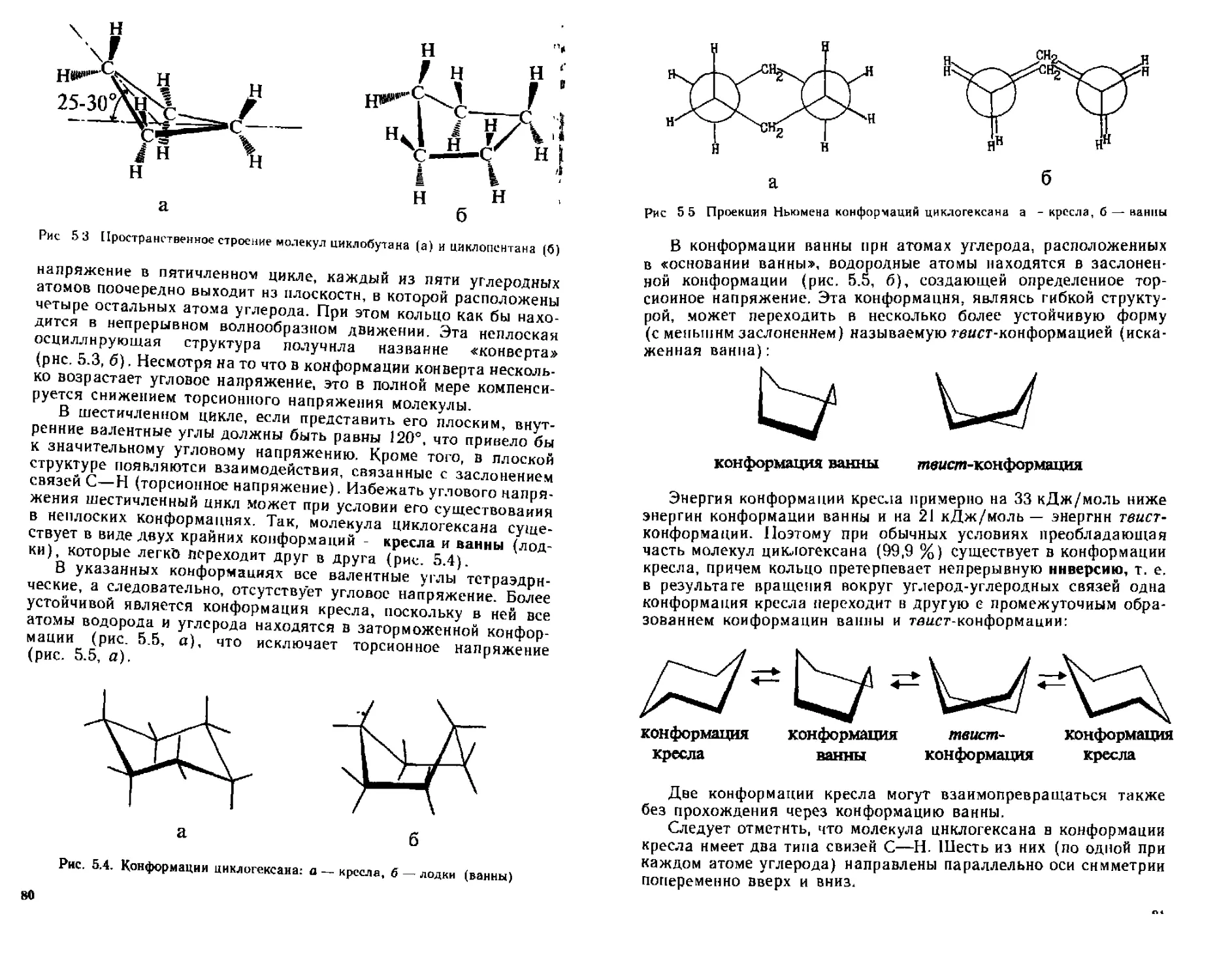
ГЛАВА 5 ЦИКЛОАЛКАНЫ 70
5 1 Классификация и номенклатура 70
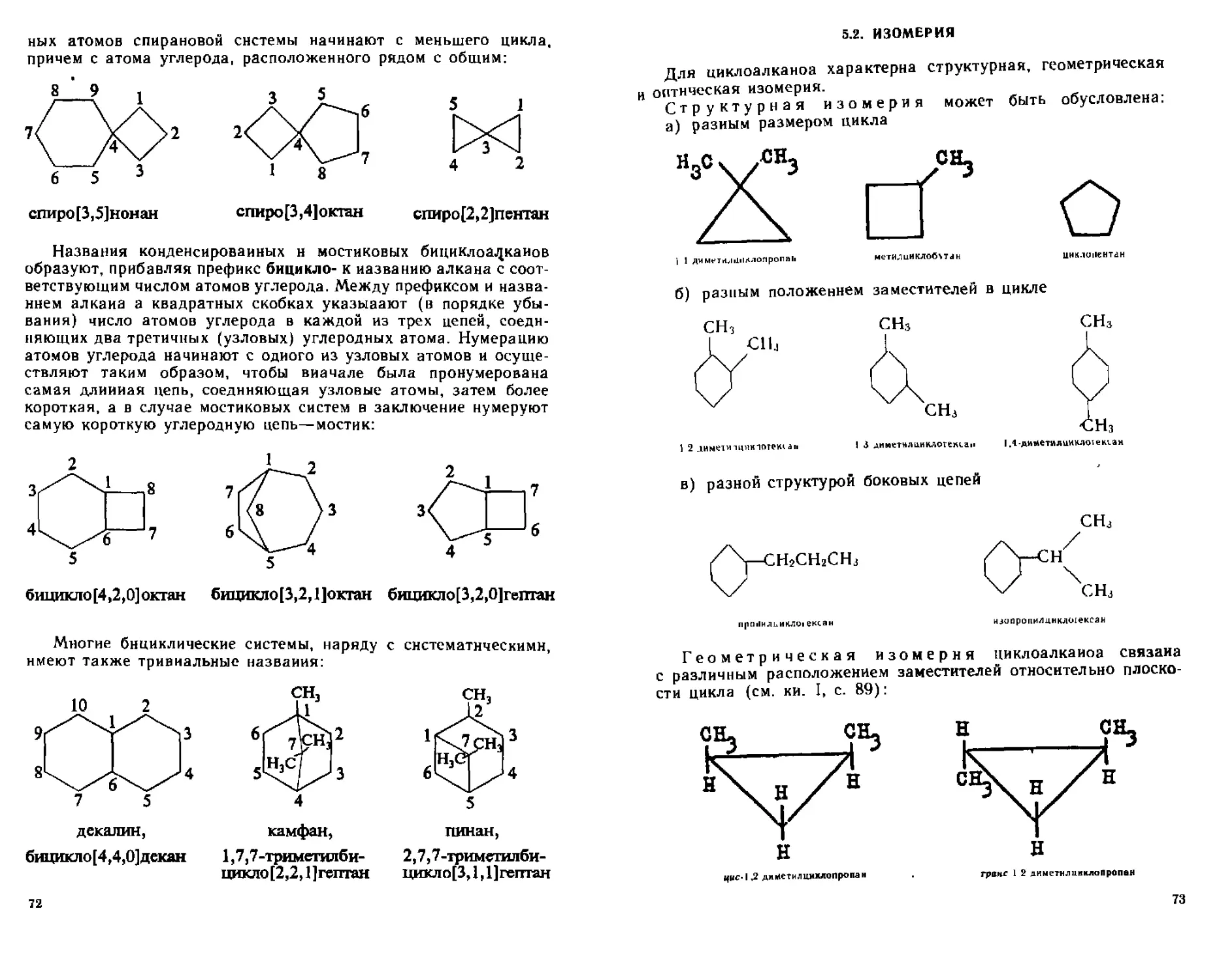
5 2 Изомерия 73
5 3 Способы попучения 74
5 4 Физические свойства 77
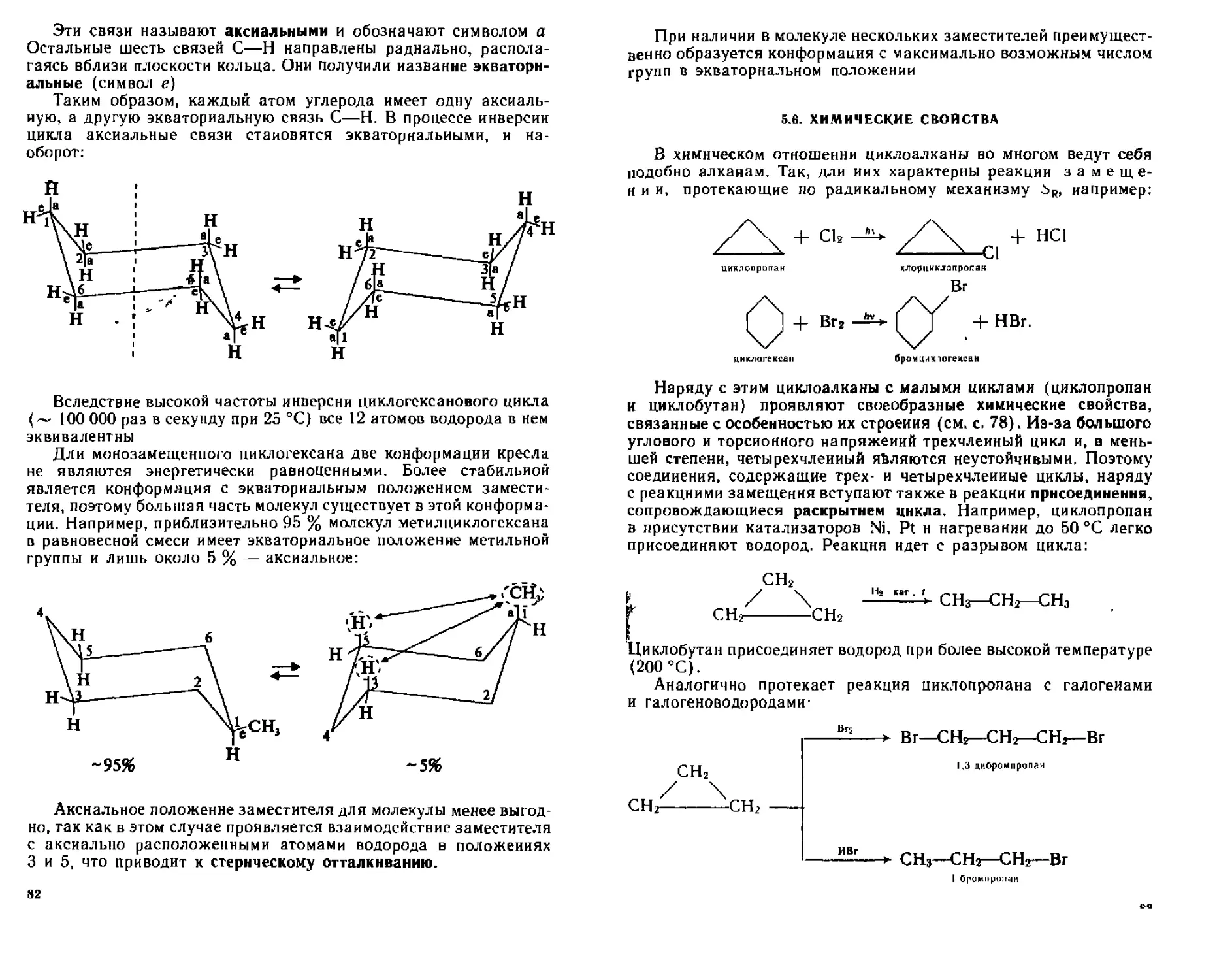
5 5 Строение пик юатканпн 77
5 6 Химические свойства 83
5 7 Идентификация диклоалканов 84
5 8 Отдельные представители Применение 84
Контрольные вопросы и упражнения 86
Г Л А В Л 6 ОДНОЯДЕРНЫЕ АРЕНЫ 88
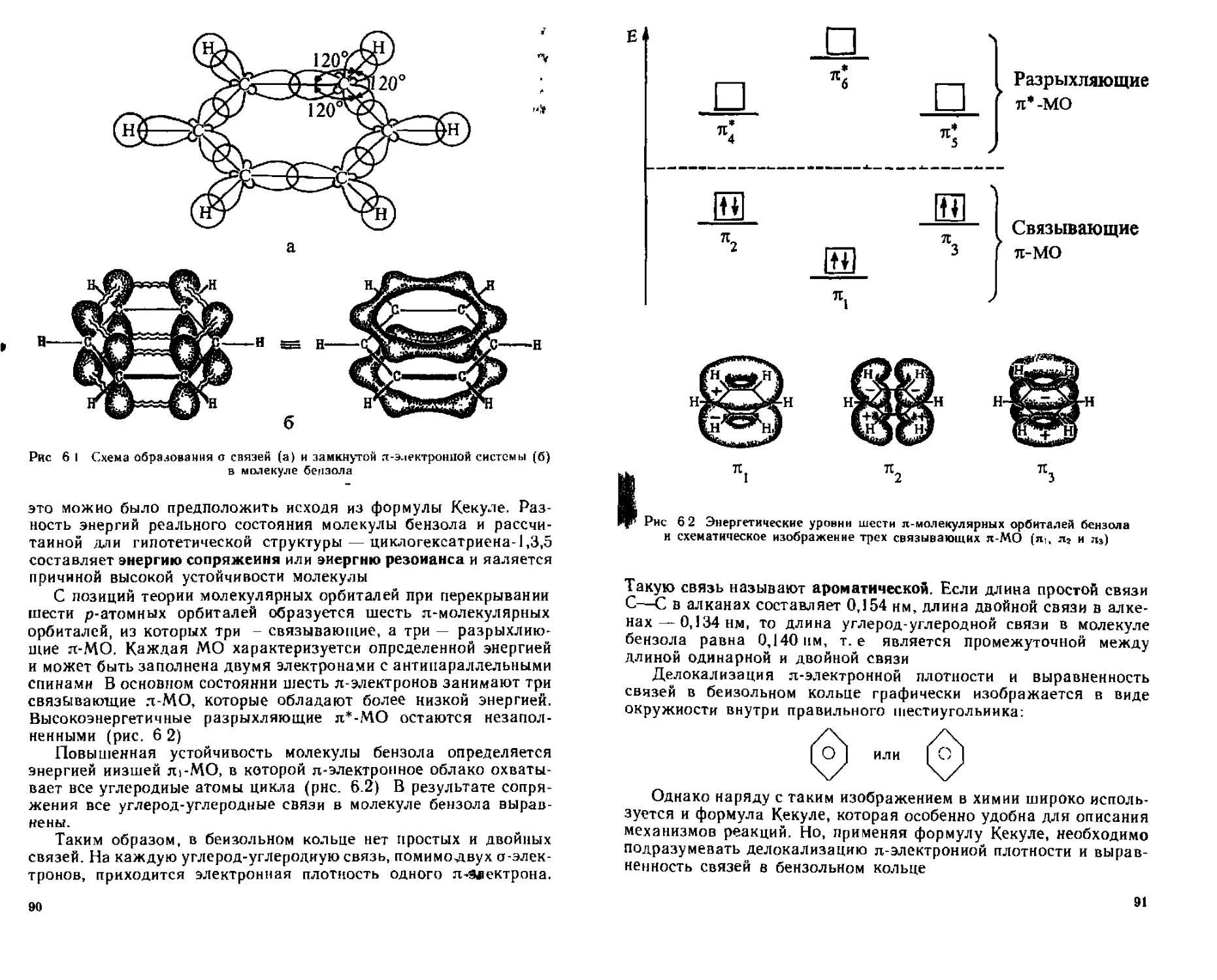
6 1 Строение бензола Ароматичность 88
6 2 Номенклатура и изомерия 92
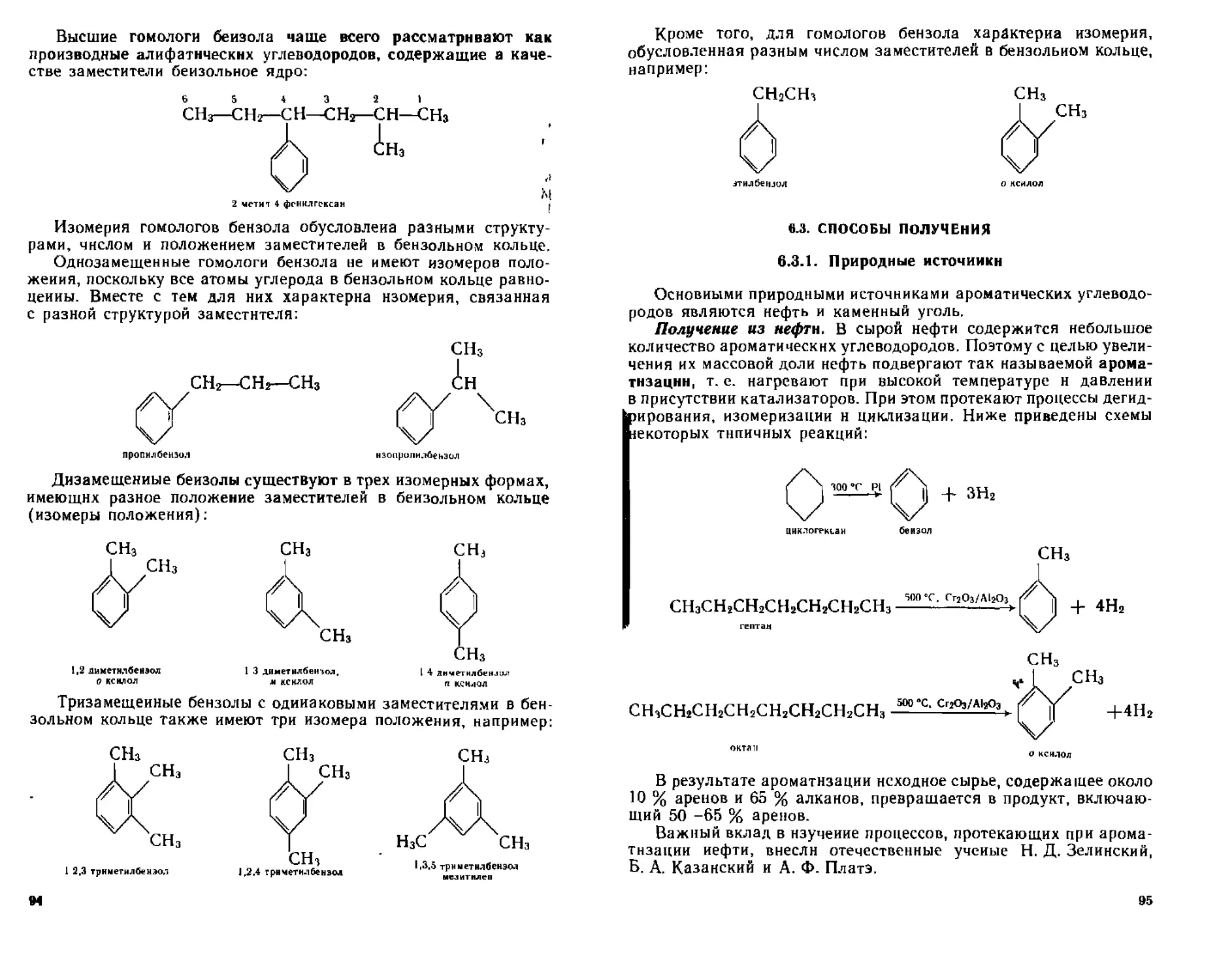
6 3 Способы получения 95
63 1 Природные источники 95
63 2 Синтетические методы получения 9Ь
6 4 Физические свойства 96
6 5 Химические свойства 97
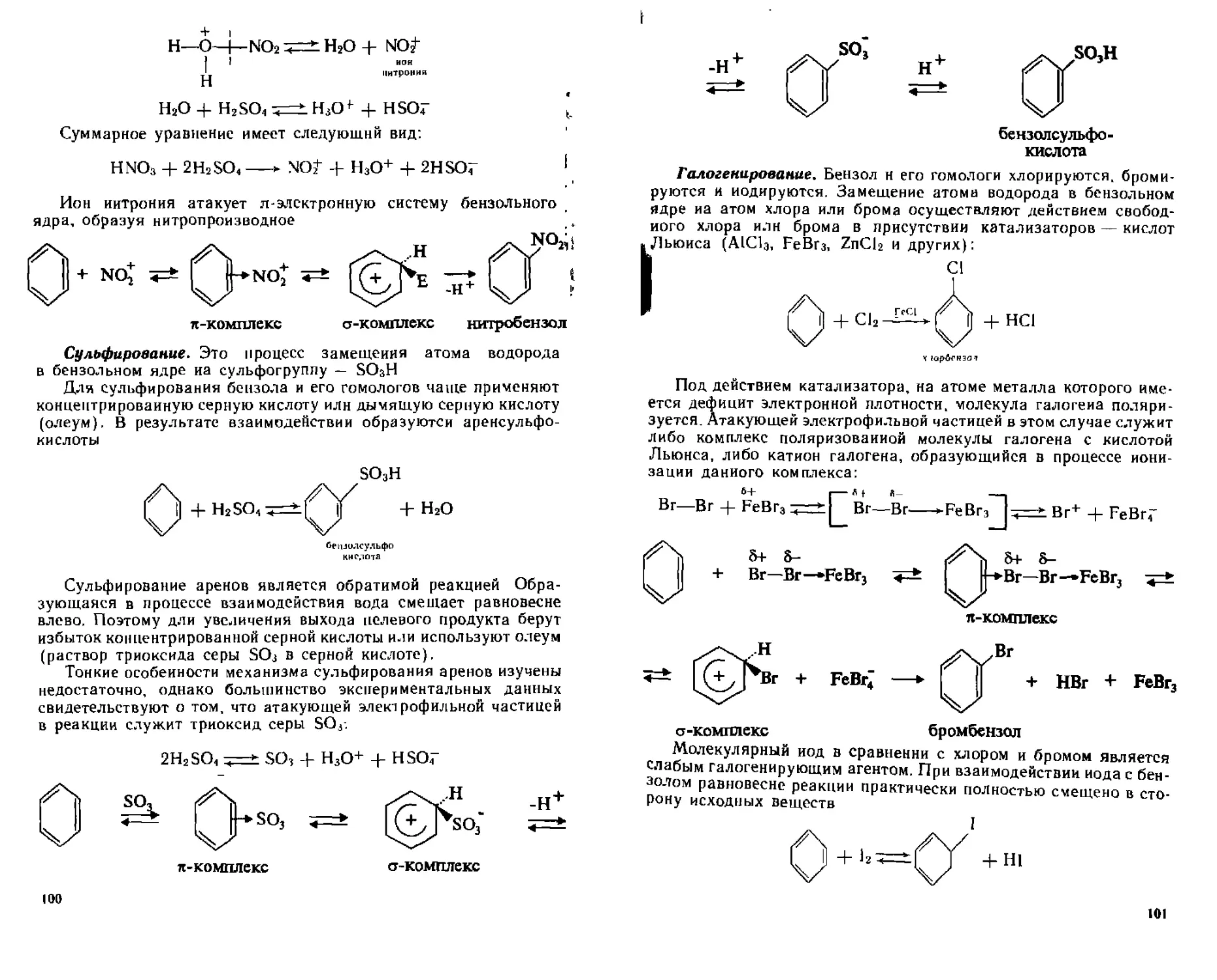
6 5 1 Реакции электрофильного замещения ( SF) 97
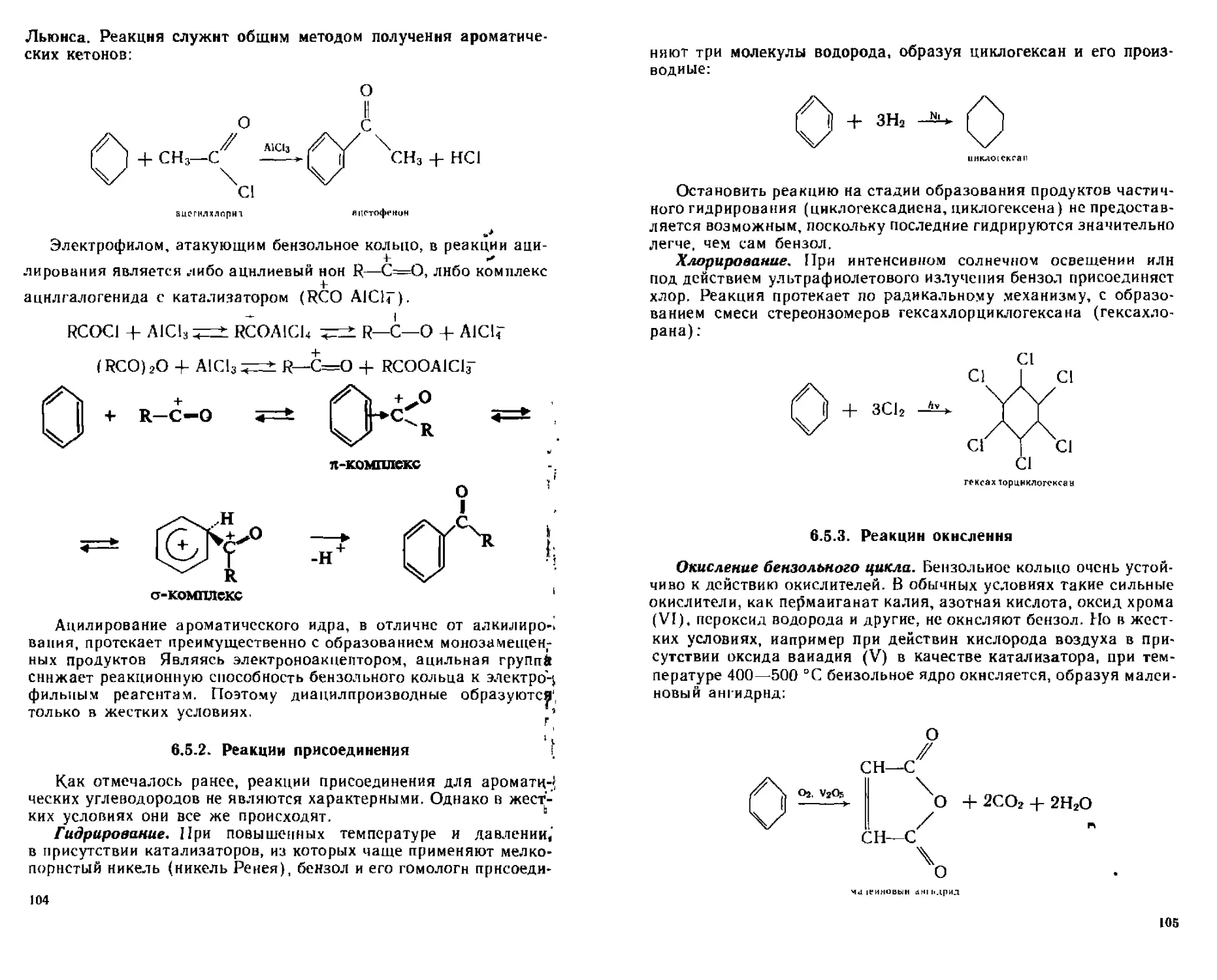
652 Реакции присоединения 104
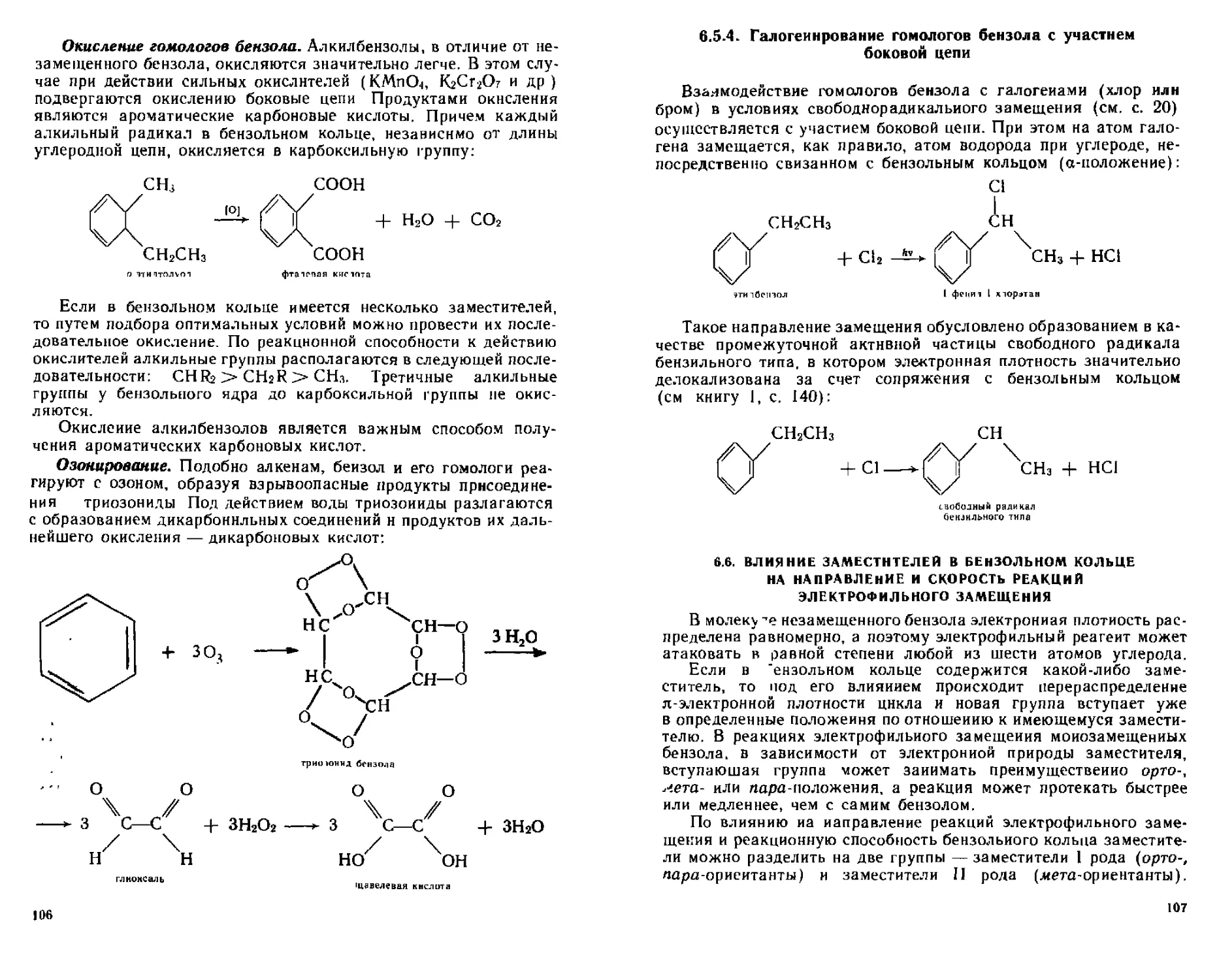
65 3 Реакции окистения 105
654 Галогенирование гомологов бензола с участием боковой цепи 107
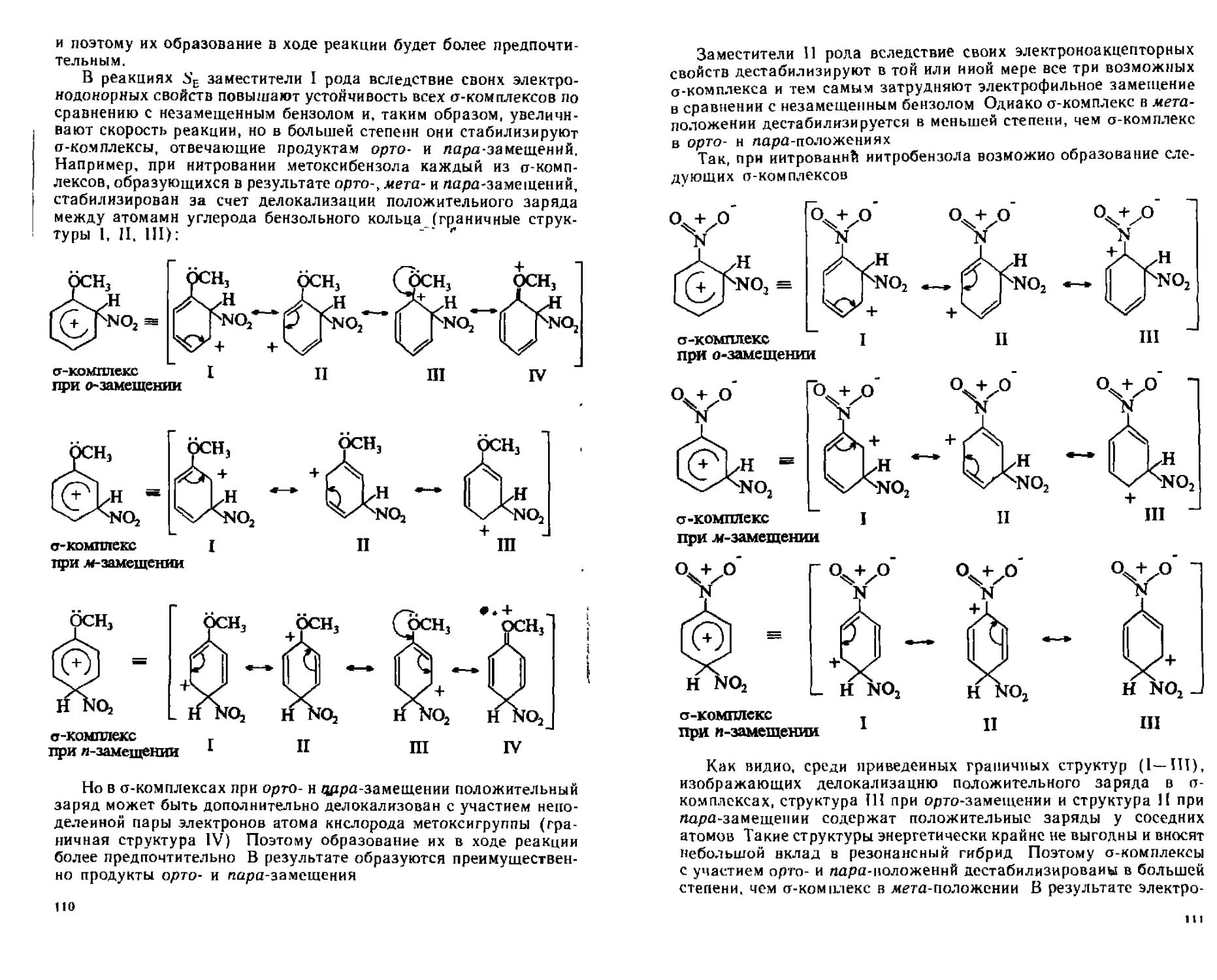
6 6 Влияние заместителей в бензольном кольце на направление и скорость реакций электрофильного замещения 107
6 6 1 Ориентация в дизамещенных бензола 113
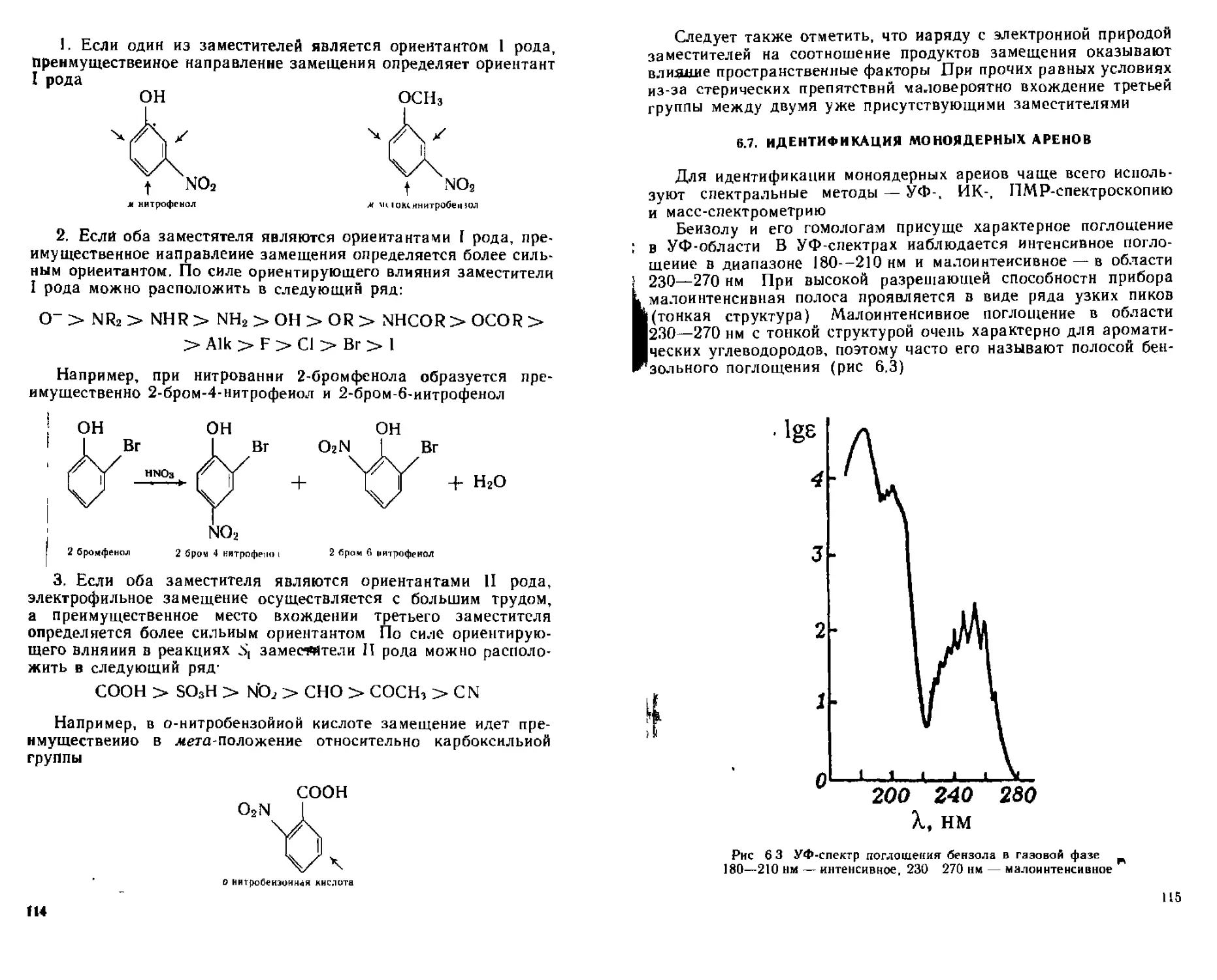
6 7 Идентификация моноядерных аренов 115
6 8 Отдельные представители Применение 116
Контрольные вопросы и упражнения 117
ГЛАВА 7 МНОГОЯДЕРНЫЕ АРЕНЫ С КОНДЕНСИРОВАННЫМИ (АННЕЛИРОВАННЫМИ) БЕНЗОЛЬНЫМИ ЦИКЛАМИ 118
71 Нафтазин 118
7 1 1 Номенклатура и изомерия 118
7 1 2 Способы пшучення 120
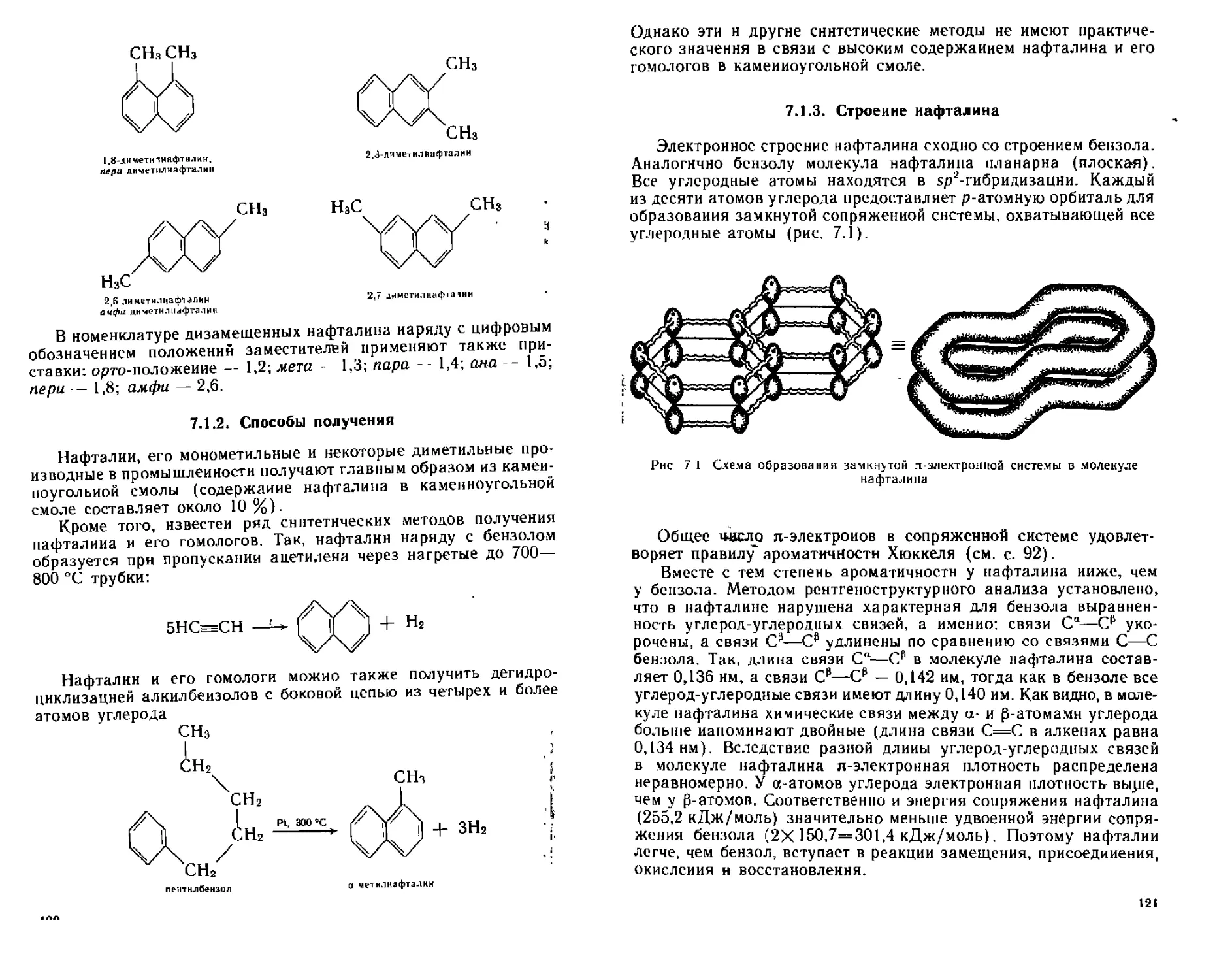
7 1 3 Строение нафта тина 121
7 1 4 Химические свойства 122
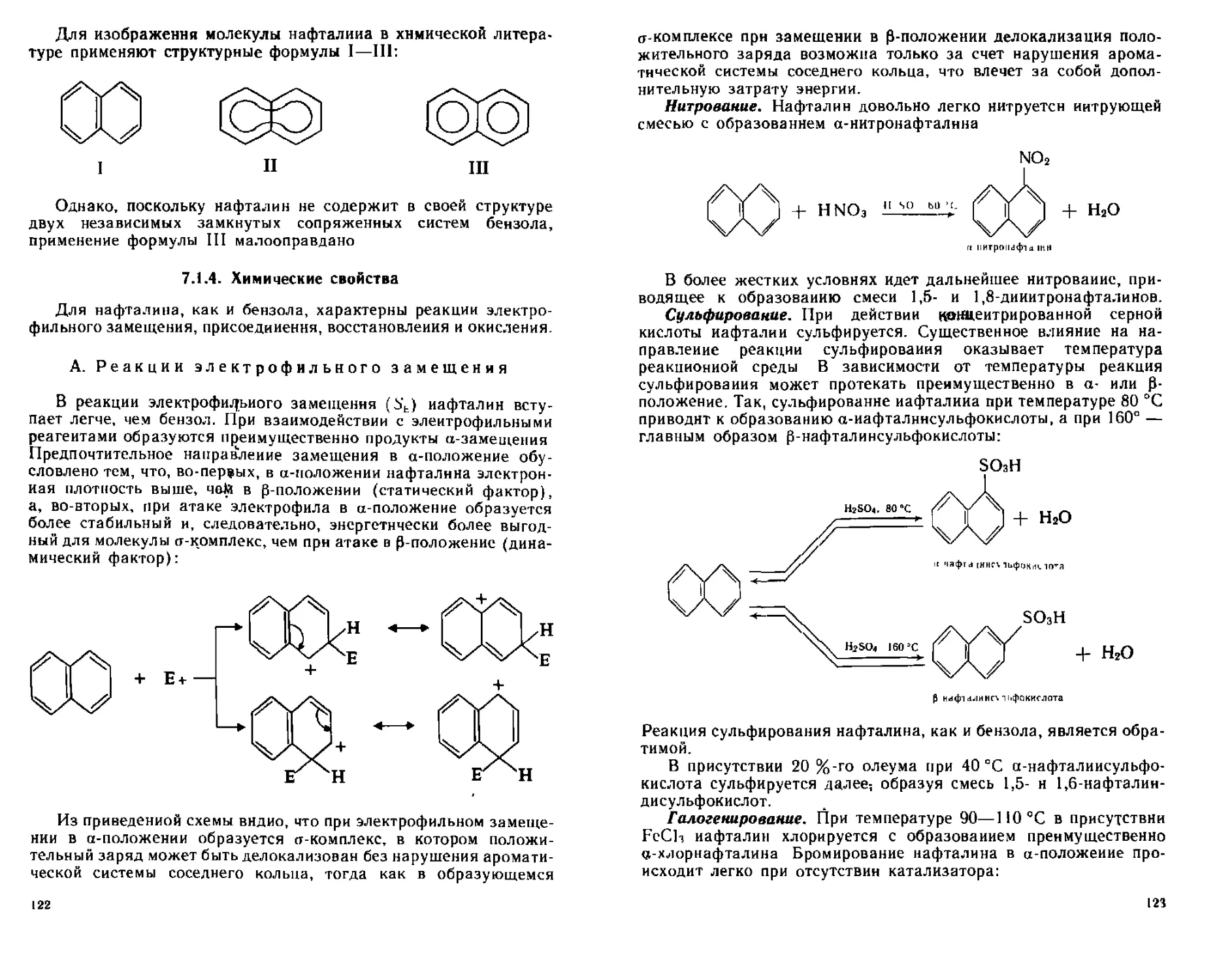
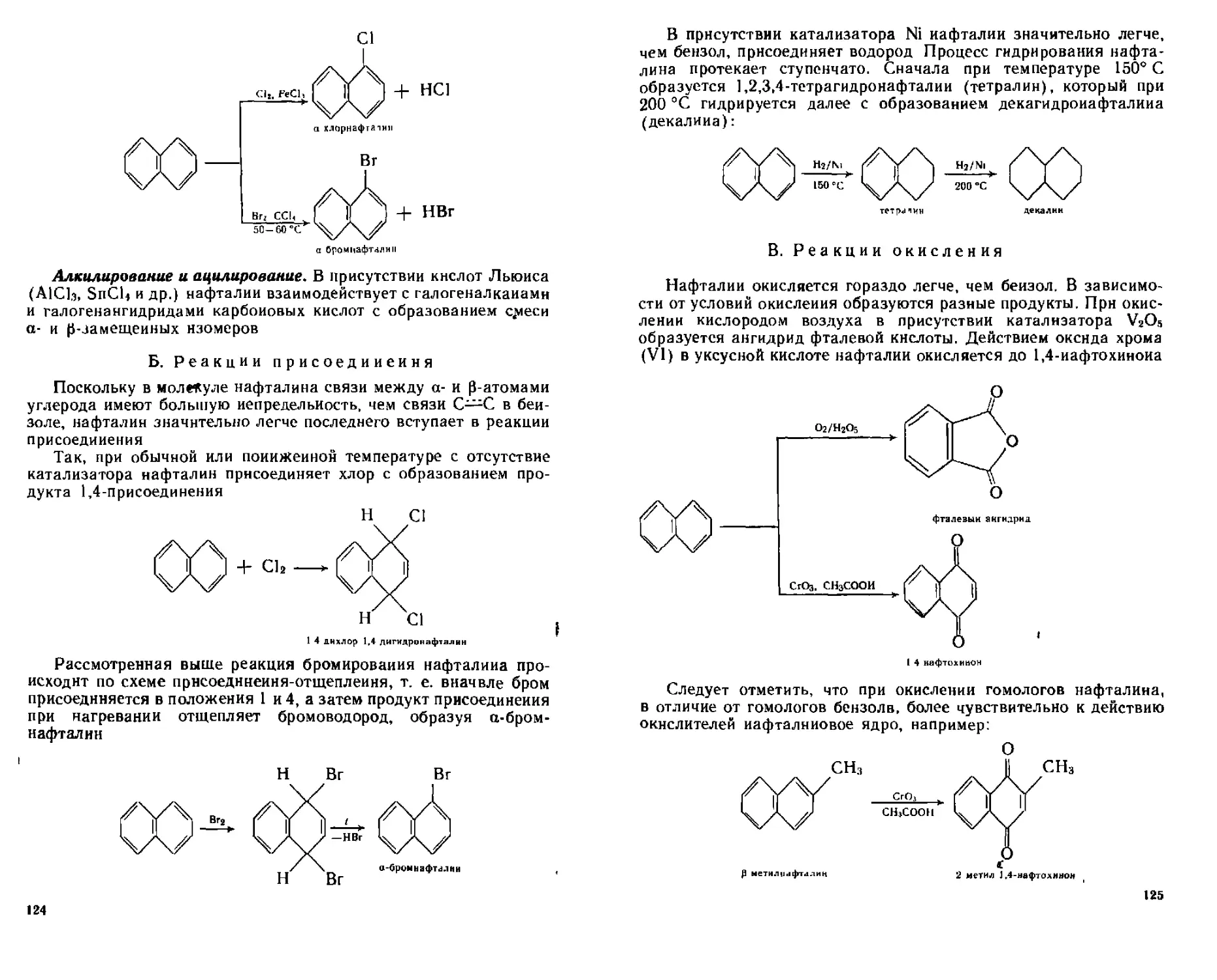
А Реакции электрофильного замещения 122
Б Реакции присоединения 124
В Реакции окисления 125
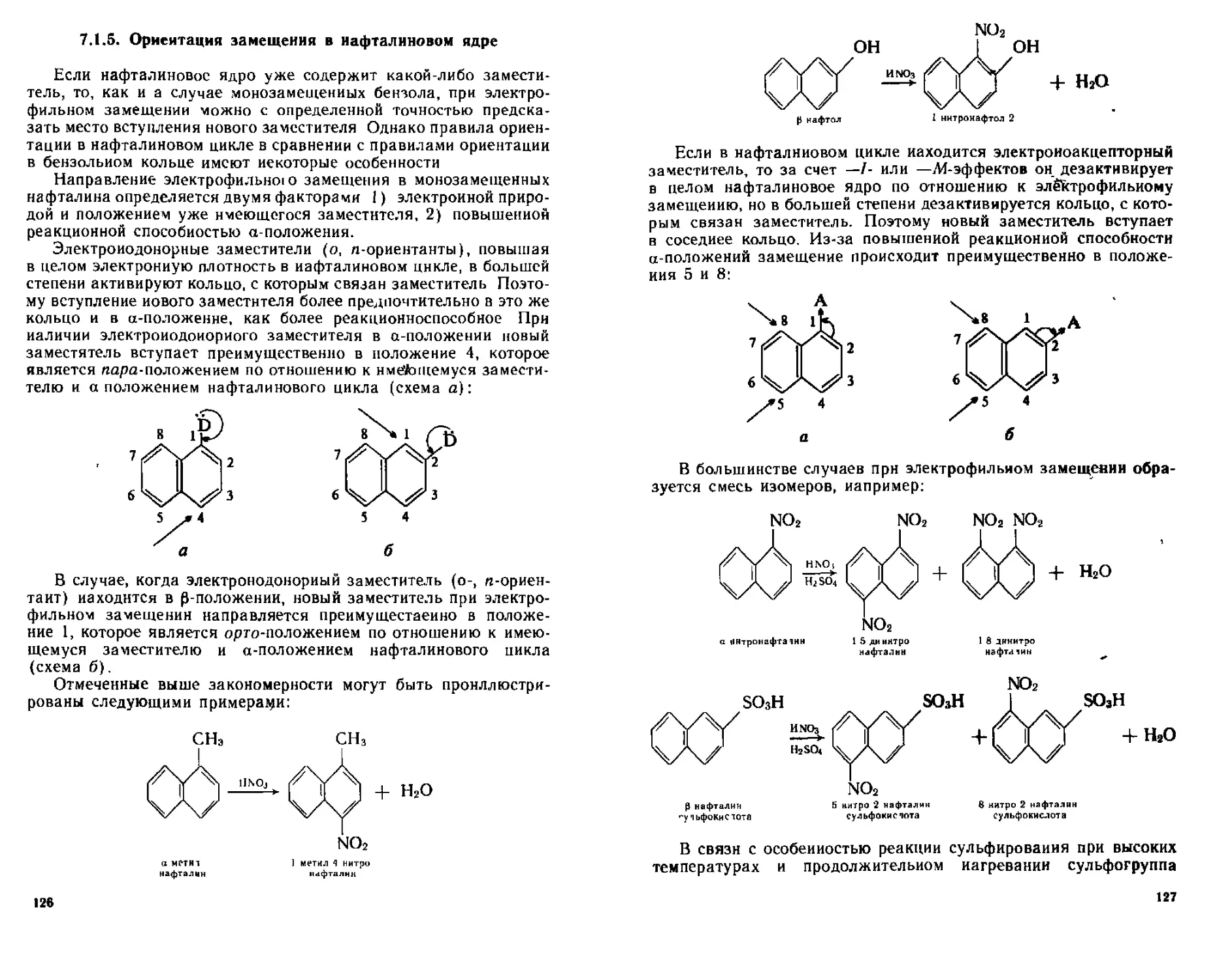
7 1 5 Ориентация замещения в нафталиновом ядре 126
7 1 6 Отдельные представители Применение 128
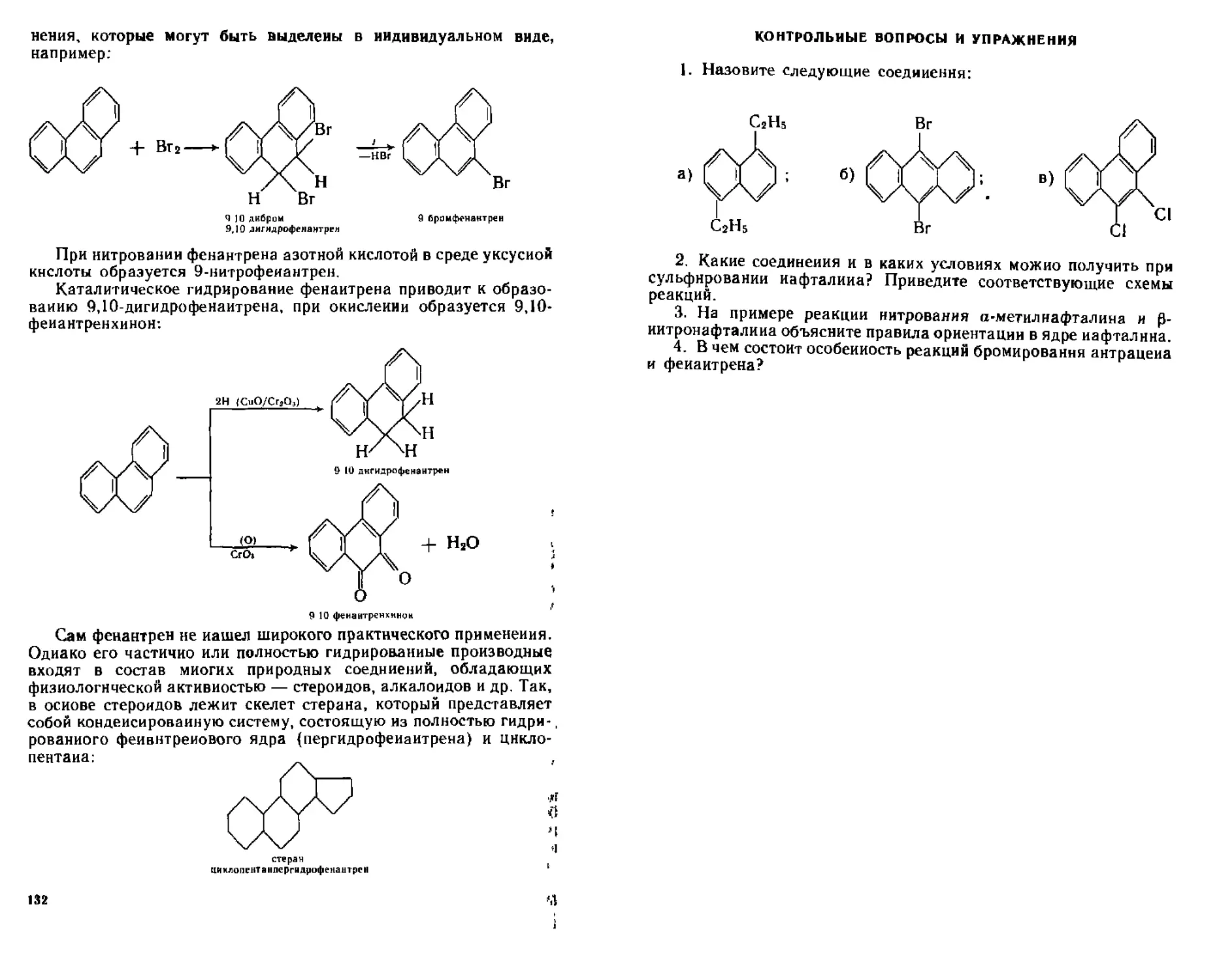
7 2 Антрацен 128
72! Способы получения 124
722 Строение Химические свойства 124
7 3 Фенантрен 131
Контрольные вопросы и упражнения 133
ГЛАВА 8 МНОГОЯДЕРНЫЕ АРЕНЫ С ИЗОЛИРОВАННЫМИ БЕНЗОЛЬНЫМИ ЦИКЛАМИ 134
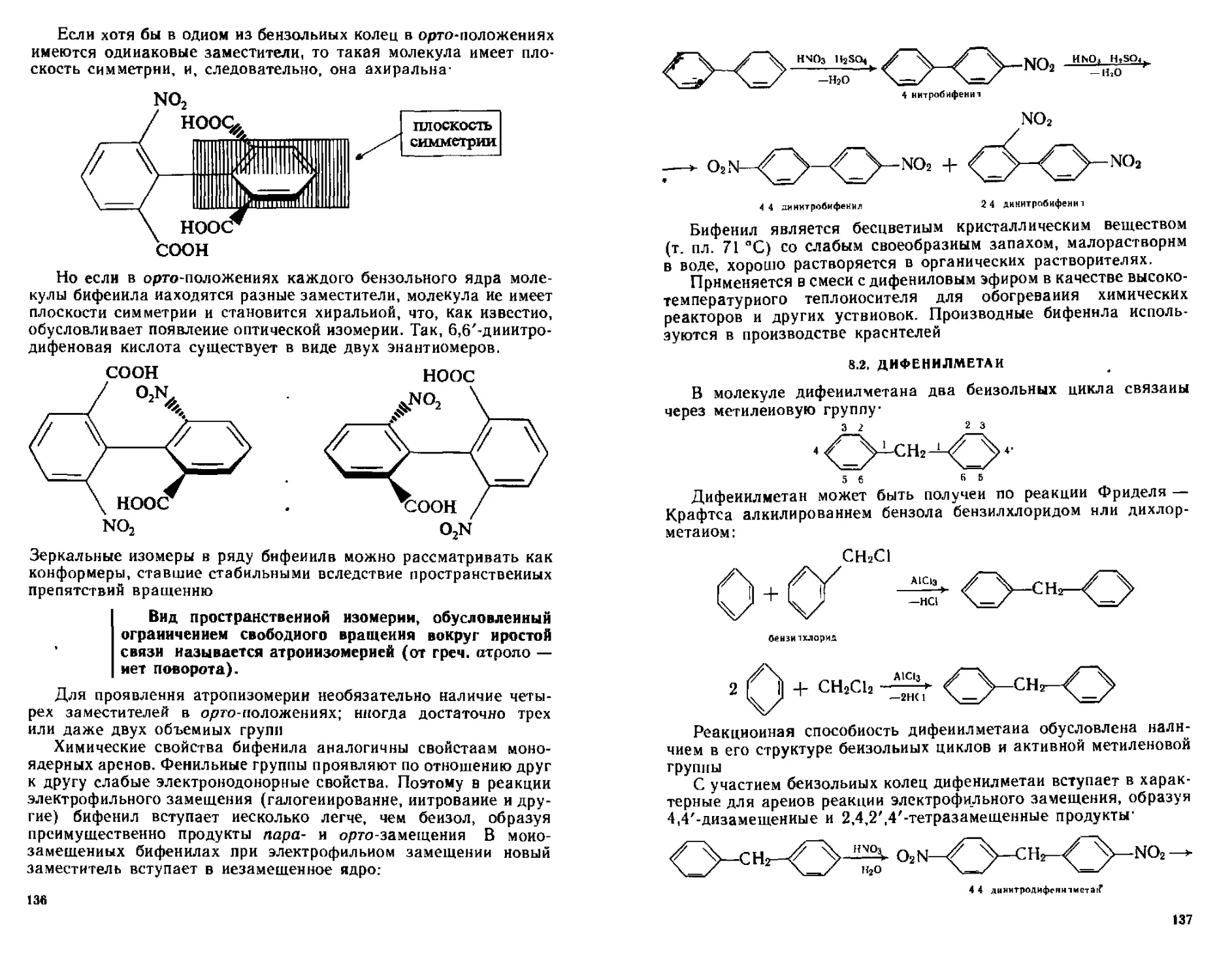
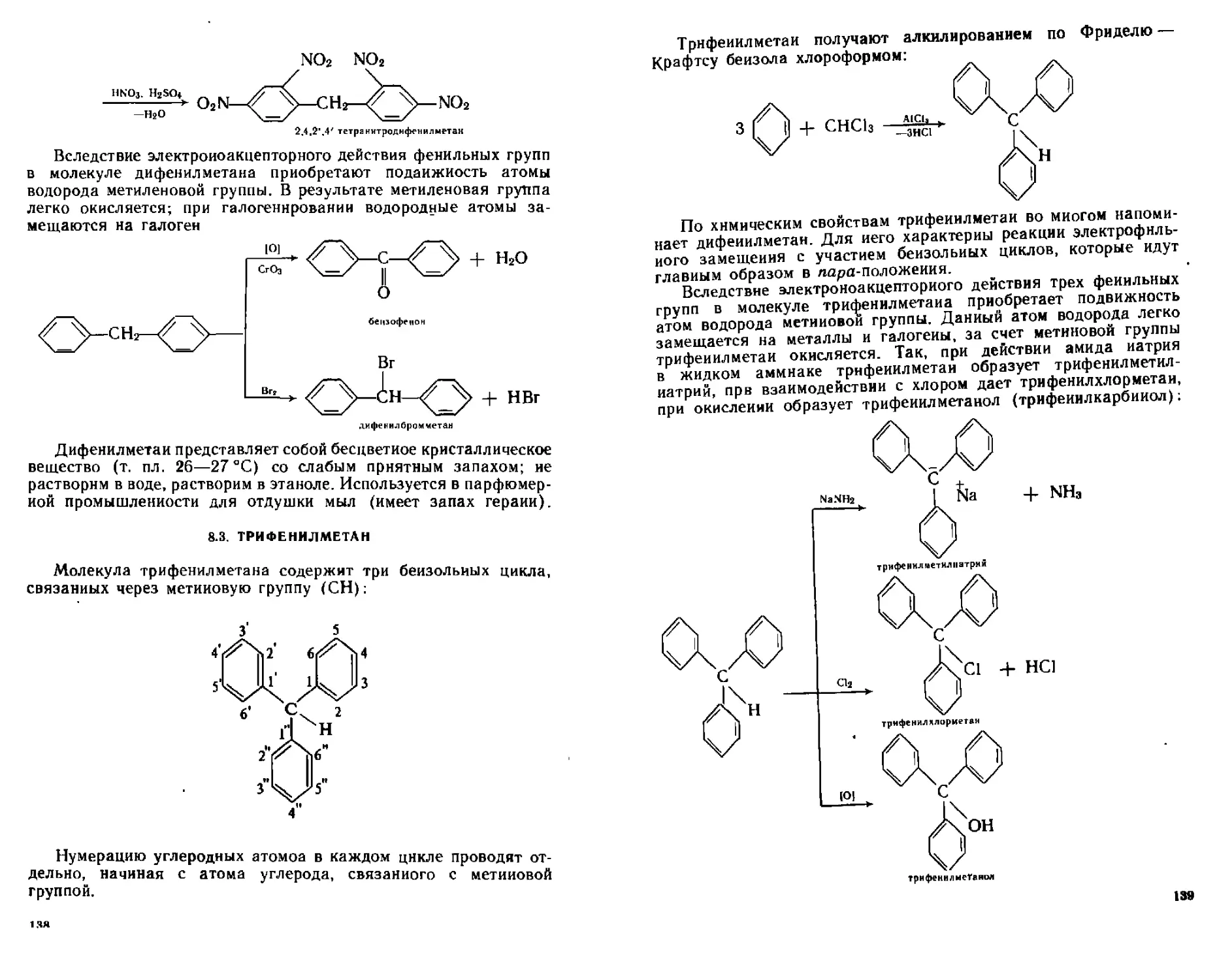
8 ! Бифенил 134
8 1 1 Способы получения 134
812 Строение Химические свойства 135
8 2 Дифенилметан 137
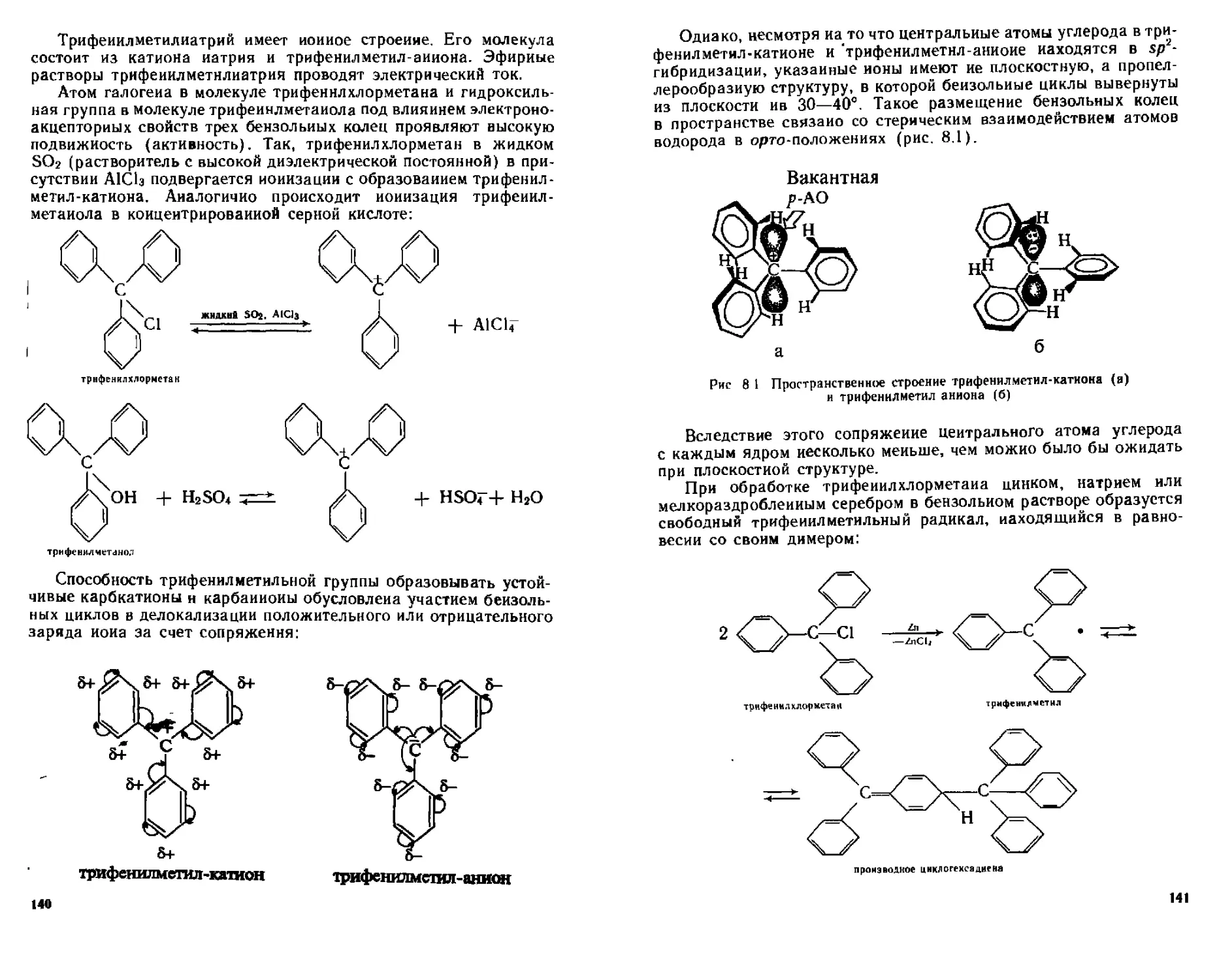
4
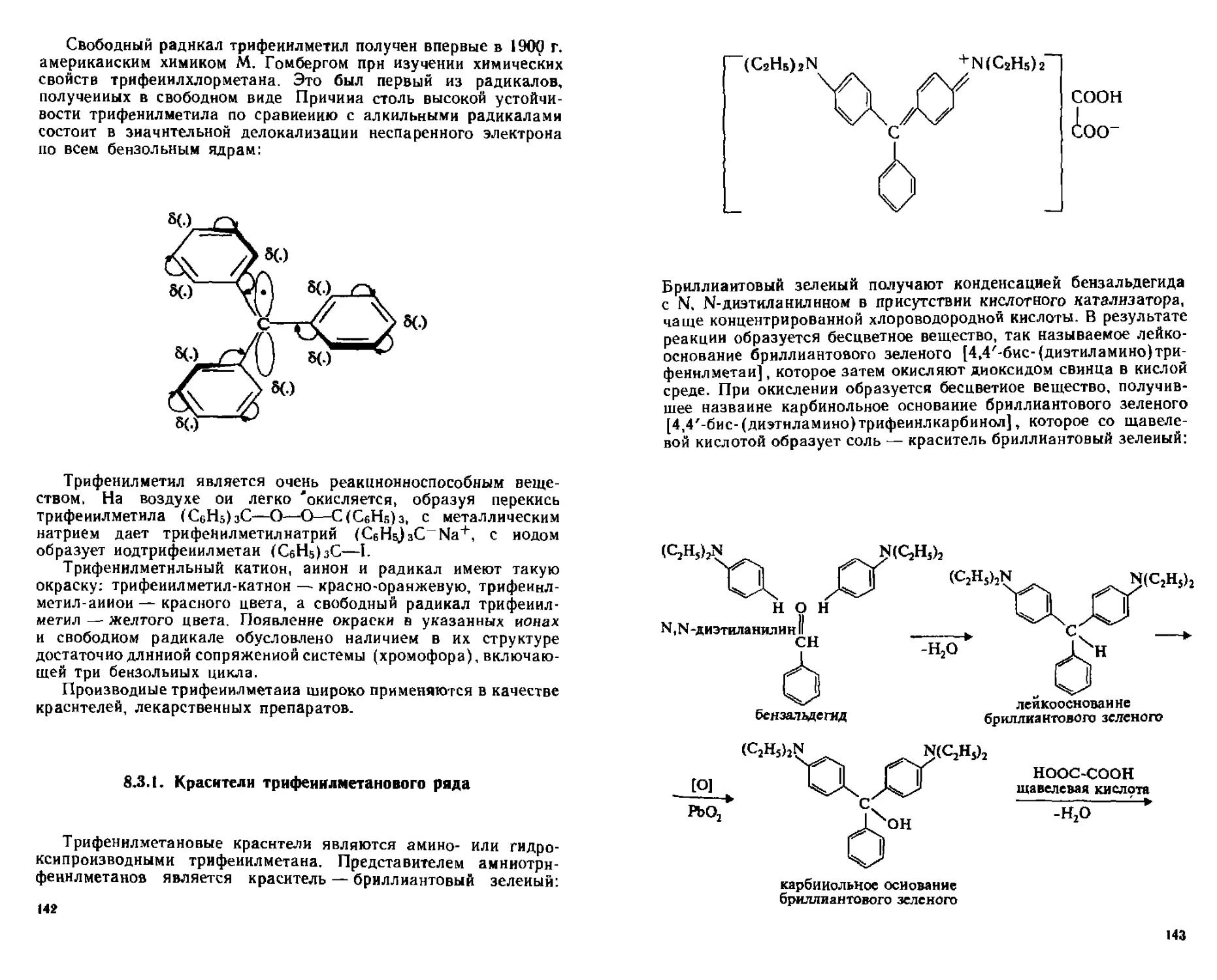
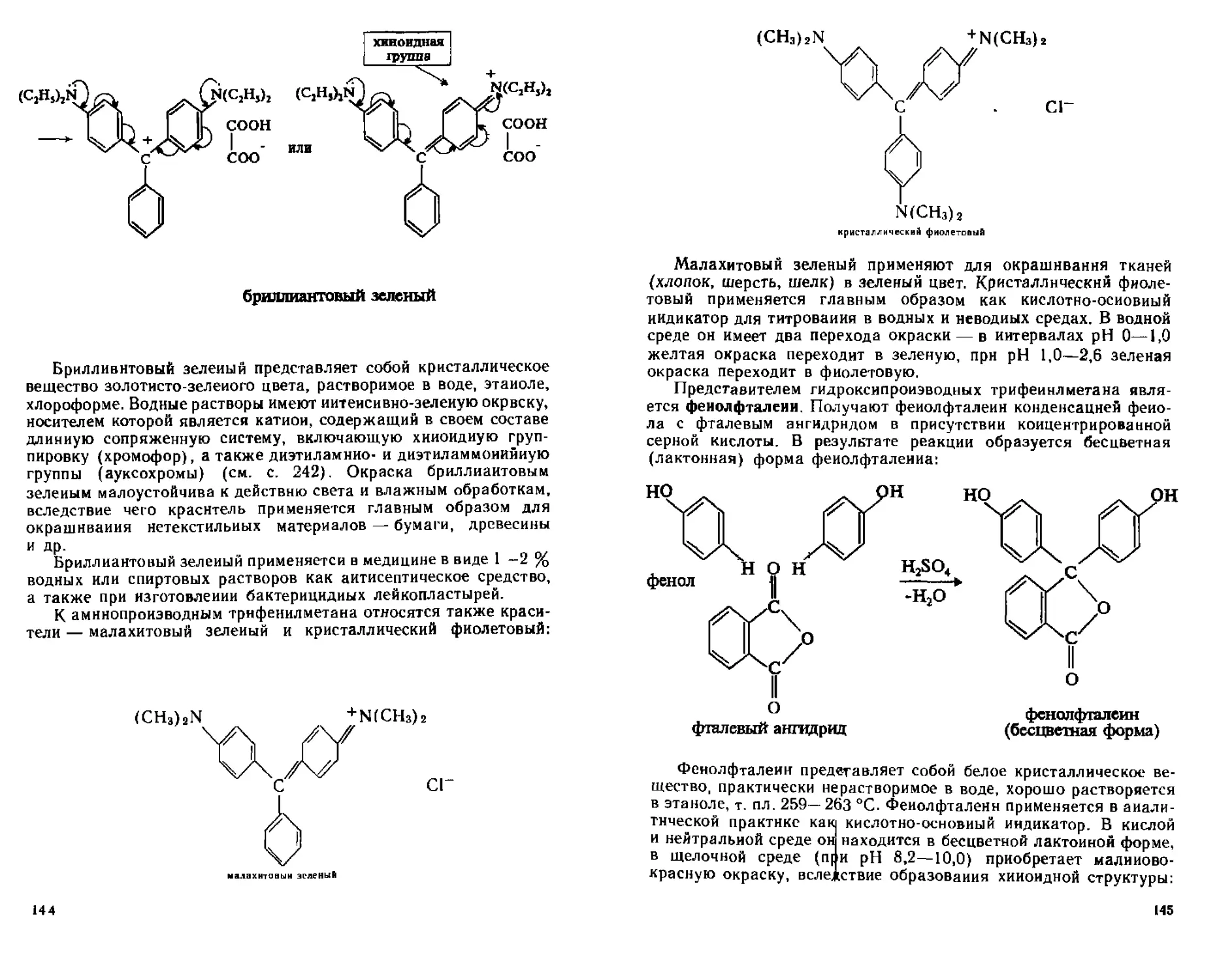
ТРи*е^^ли трифенилмегаиового ряда
Контрольные вопросы и упражнения
I ЛАВ А 9 НЕБЕНЗОИДНЫЕ АРОМАТИЧЕСКИЕ СОЕДИНЕНИЯ
о । Пиклопентадиеннл анион
g 2 Циалогептатриеимл катион
Контрол^ вопросы и упражнения
Г п ДВА Ю ГАЛОГЕНОПРОИЗВОДНЫЕ УГЛЕВОДОРОДОВ
1 т\ р”
10 2 Изомерия
]0 3 Гален еналкапы
10 3 I Способы по |учений
[0 3 2 Физические свойства
10 3 3 Химические свойства
А Реакции нуклеофильного замещения (SN)
Б Реакции элиминирования (Z7)
В Взаимодействие с металлами
Г Восстановление галогеналканов
10 4 Дигалогеналканы
10 4 1 Способы получения
10 4 2 Химические свойства
105 Галогеналкены
10 5 1 Способы получения
(0 5 2 Химические свойства
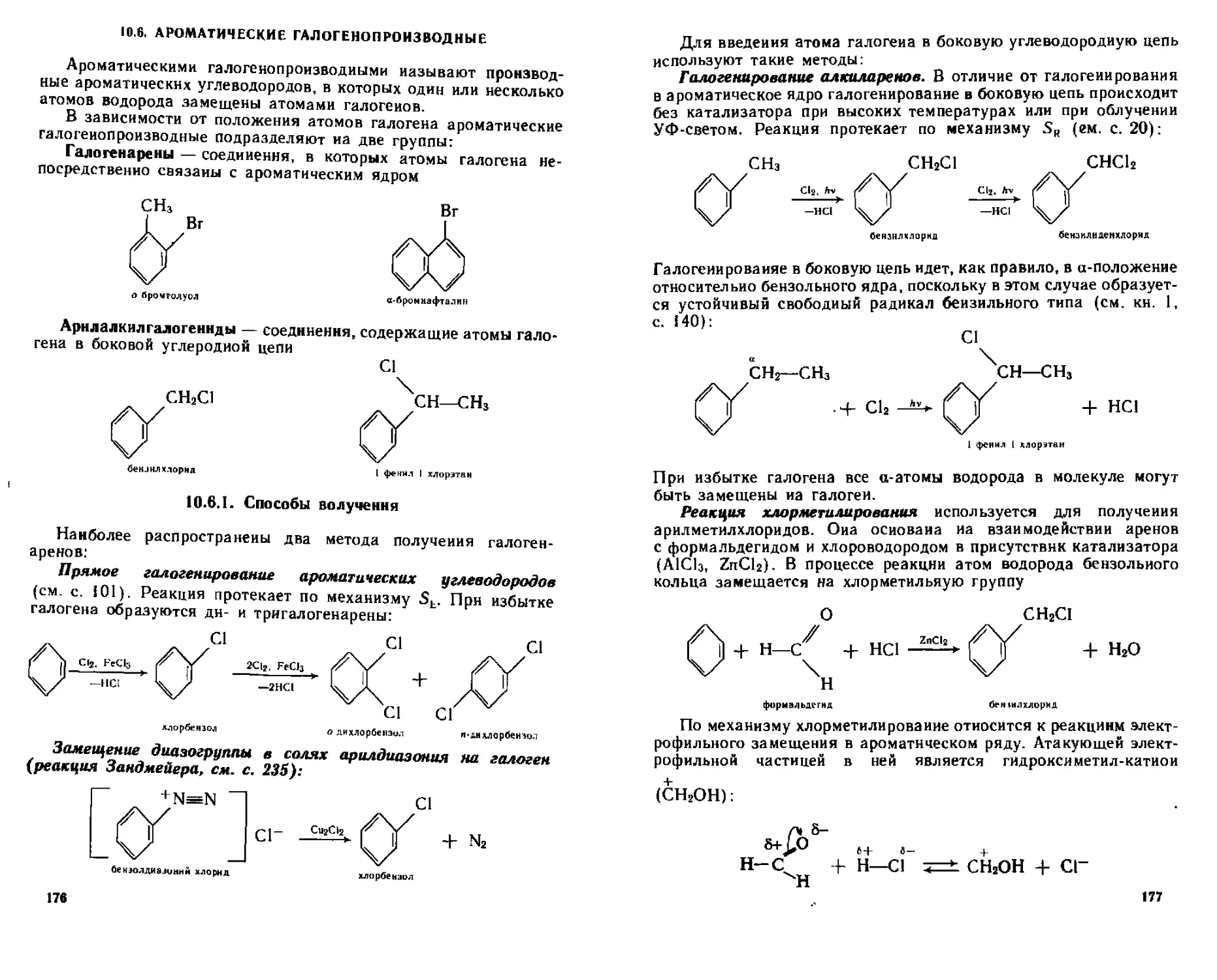
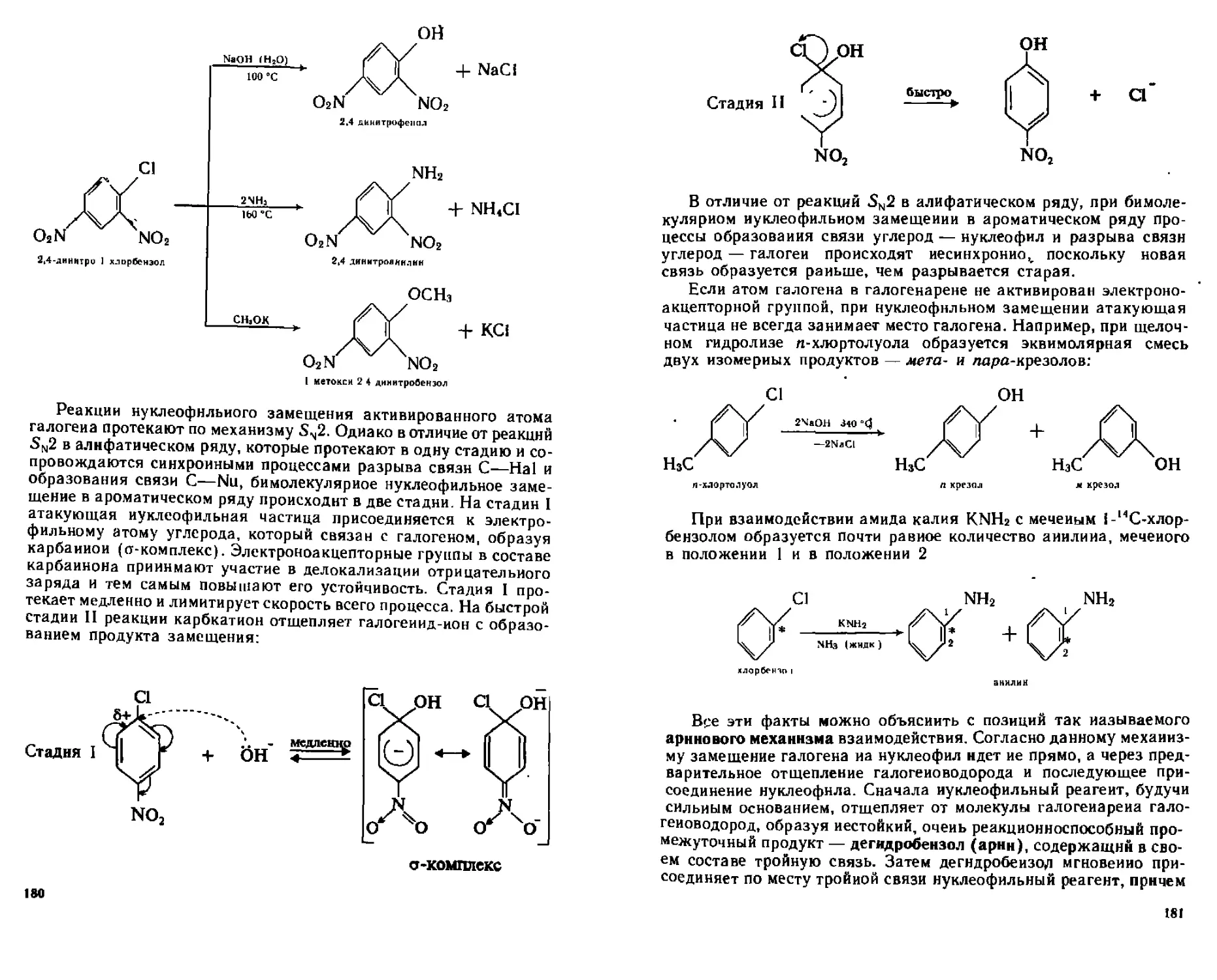
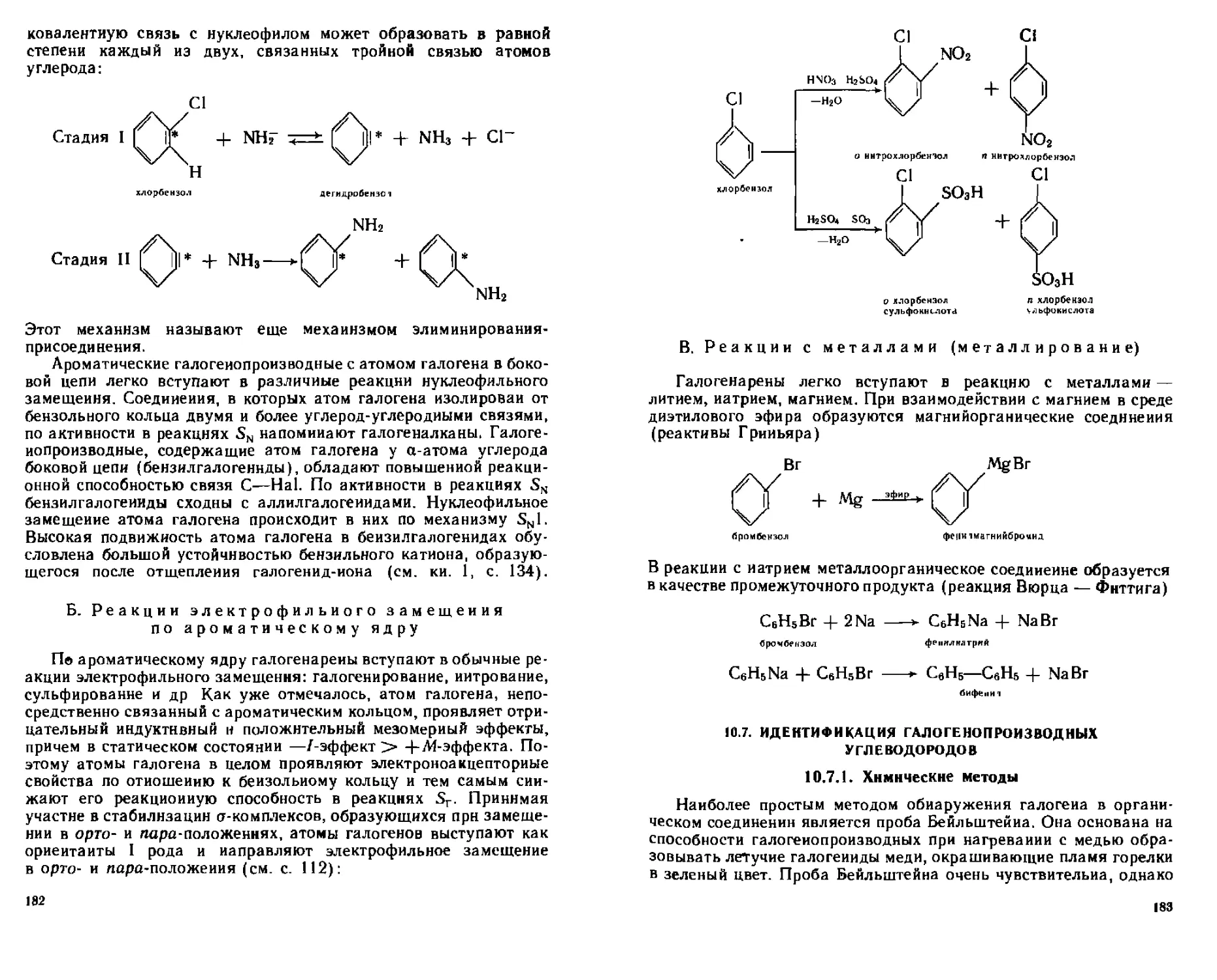
10 6 Ароматические га чогенопроизводные
1Q 6 1 Способы получения
10 6 2 Физические свойства
10 6 3 Химические свойства
А Реакции нуклеофильного замещения
Б Реакции электрофильного замещения по ароматическому ядру
В Реакции с металлами (металлирование)
10 7 Идентификация галогенопроизводных углеводородов
10 7] Химические методы
10 7 2 Инструментальные методы
10 8 Отдельные представители галогенопроизводных углеводородов Контрольные вопросы и упражнения
138
142
147
148
148
150
151
152
153
153
155
156
156
157
158
158
167
170
170
172
173
173
174
176
176
178
178
178
182
183
183
183
184
184
186
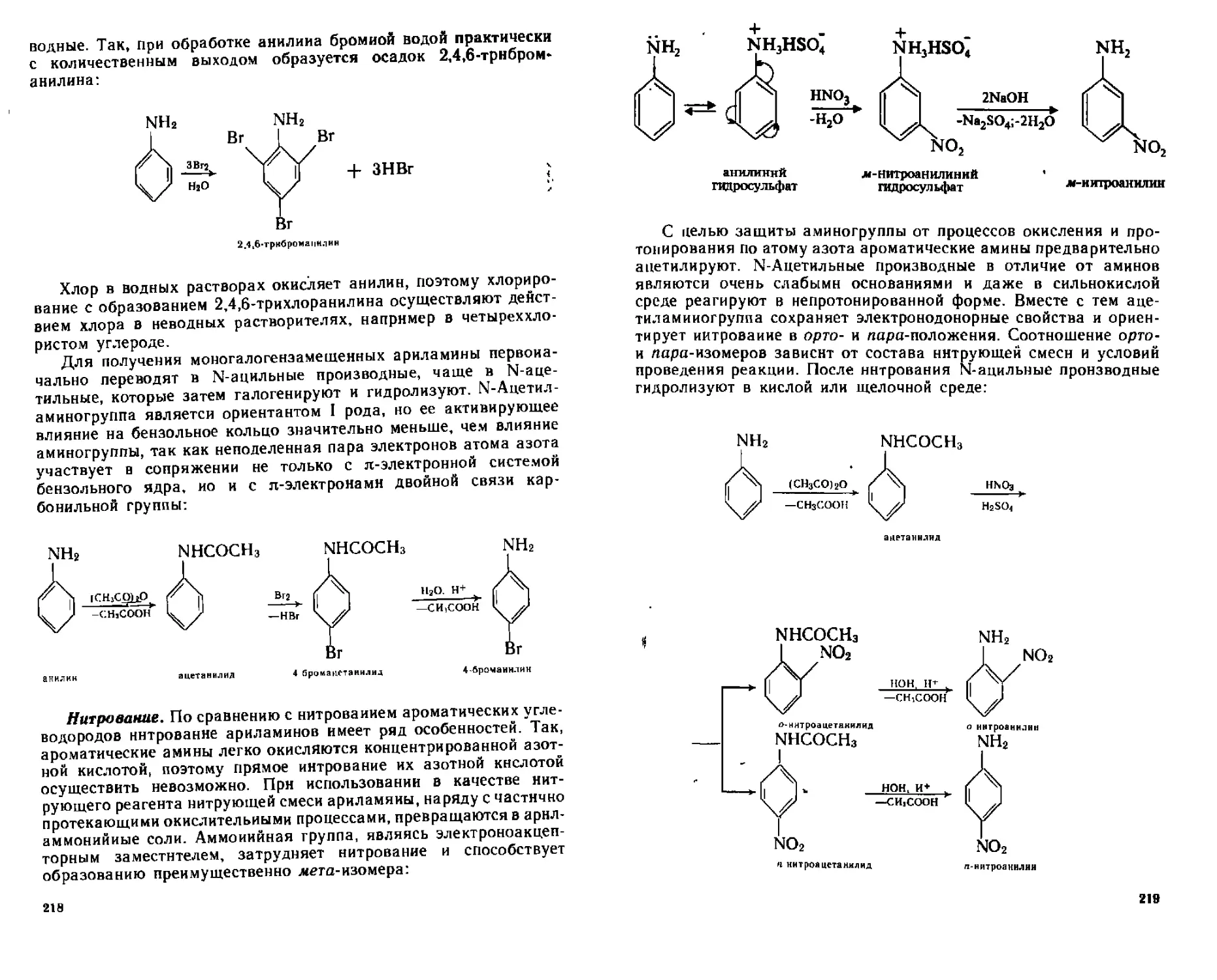
ГЛАВА 11 НИТРОСОЕДИНЕНИЯ 187
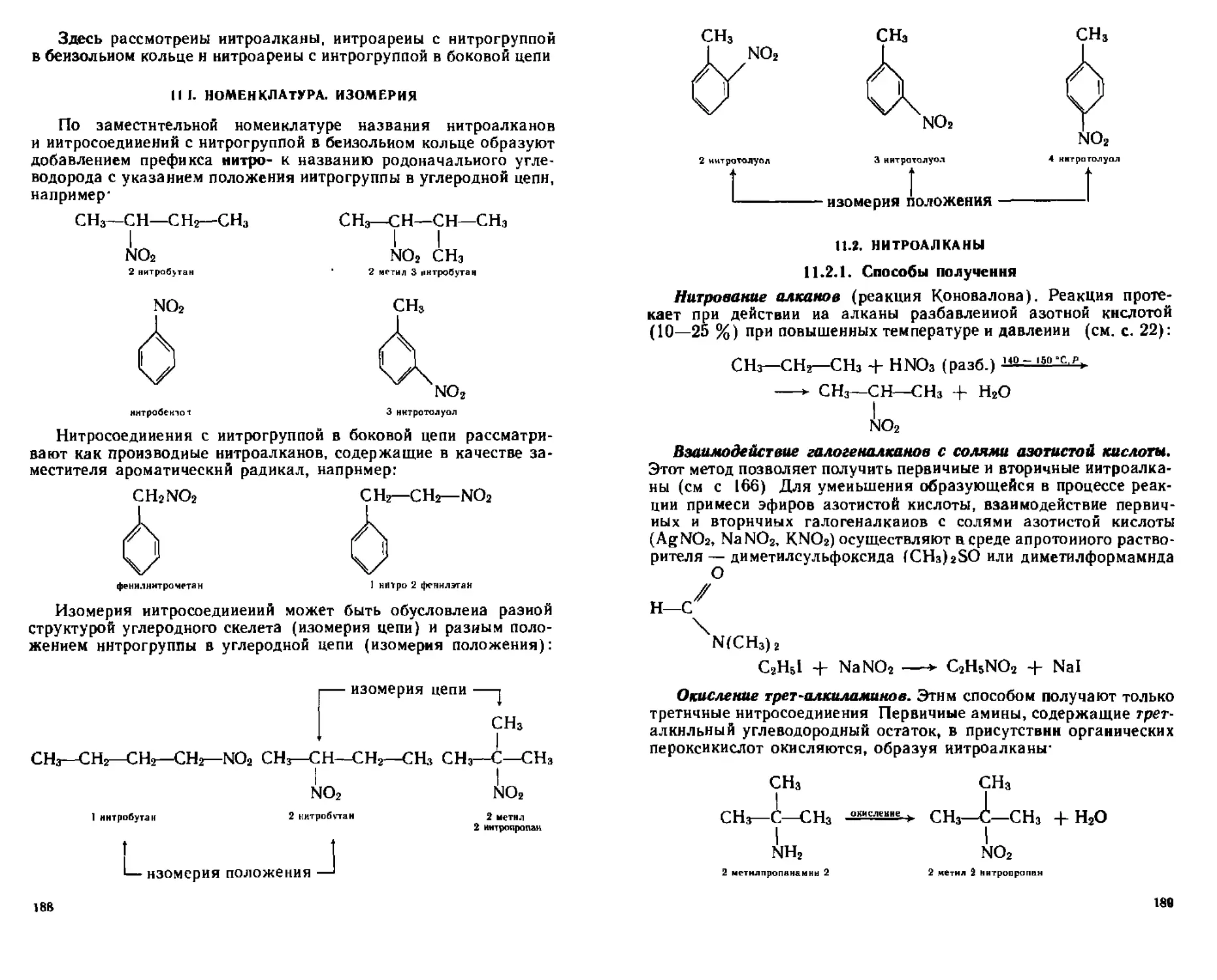
Н 1 Номенклатура Изомерии 188
11 2 Нитроалканы 189
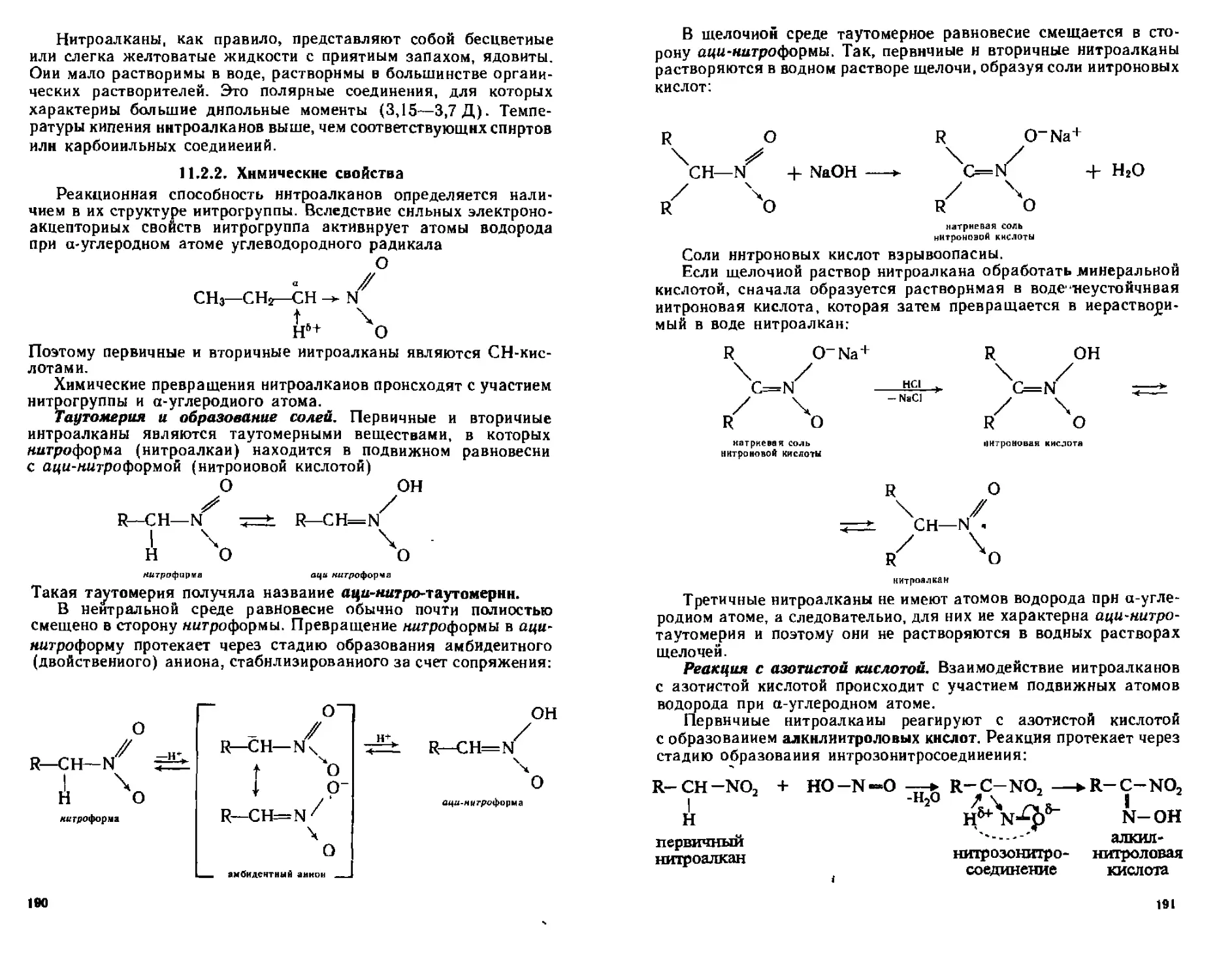
112 1 Способы получения 189
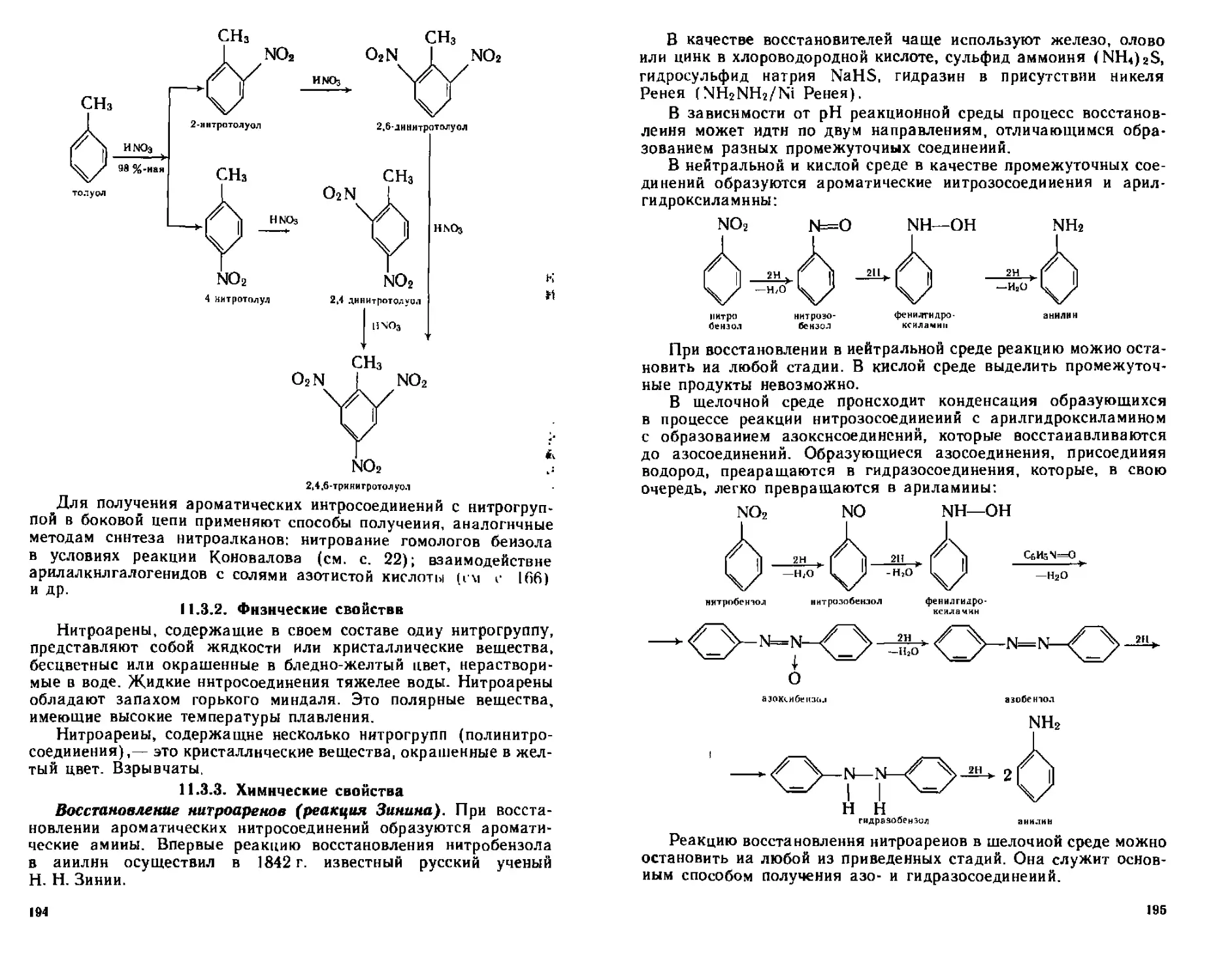
1122 Химические свойства 190
ИЗ Ароматические нитросоединения 193
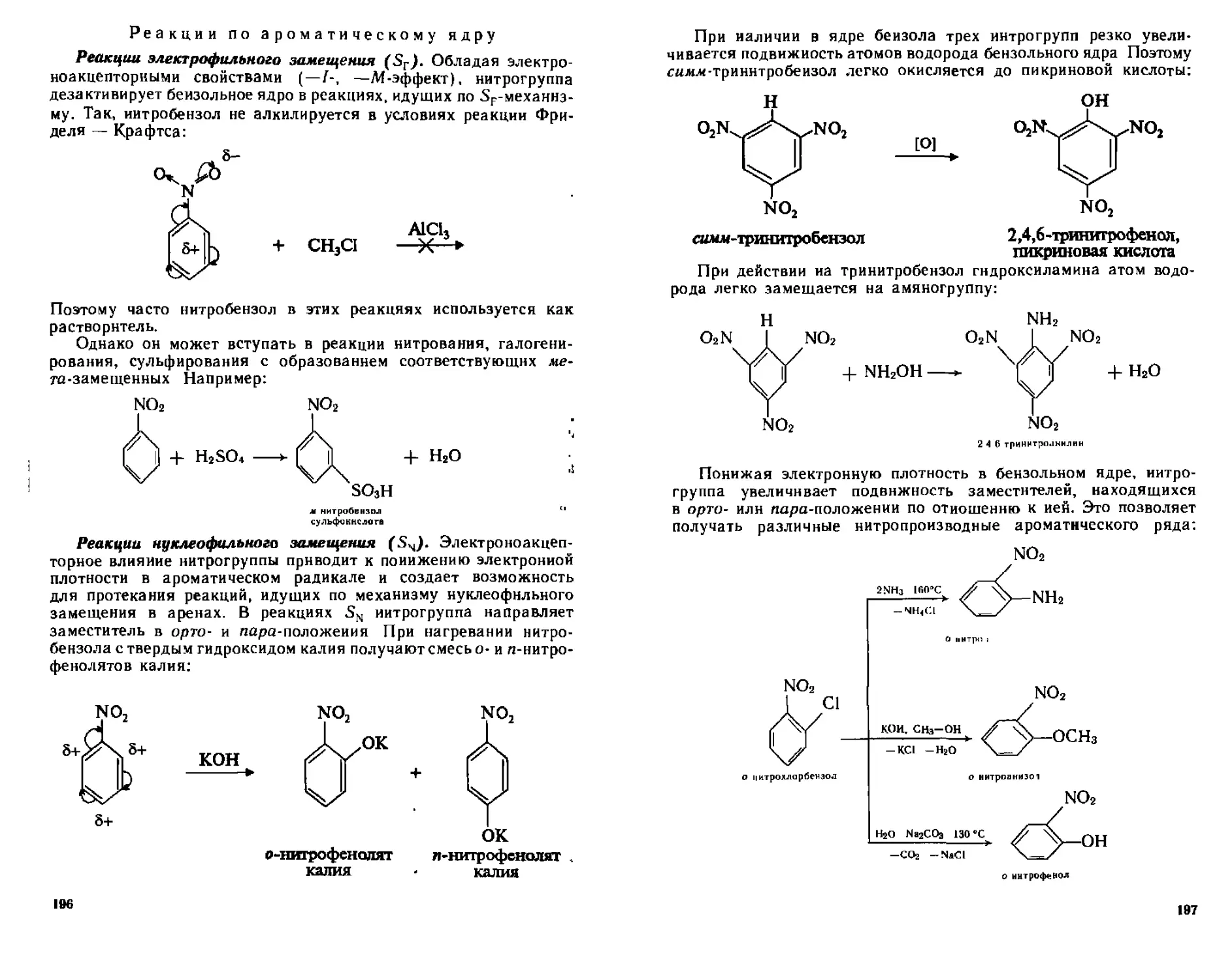
П31 Способы получения 193
1132 Физические свойства 194
11 3 3 Химические свойства 194
А Реакции по ароматическому ядру 196
И 4 Идентификация нитросоединений 198
И 5 Отдельные представители Применение 199
Контрольные вопросы и упражнения 199
ГЛАВА 12 АМИНЫ 200
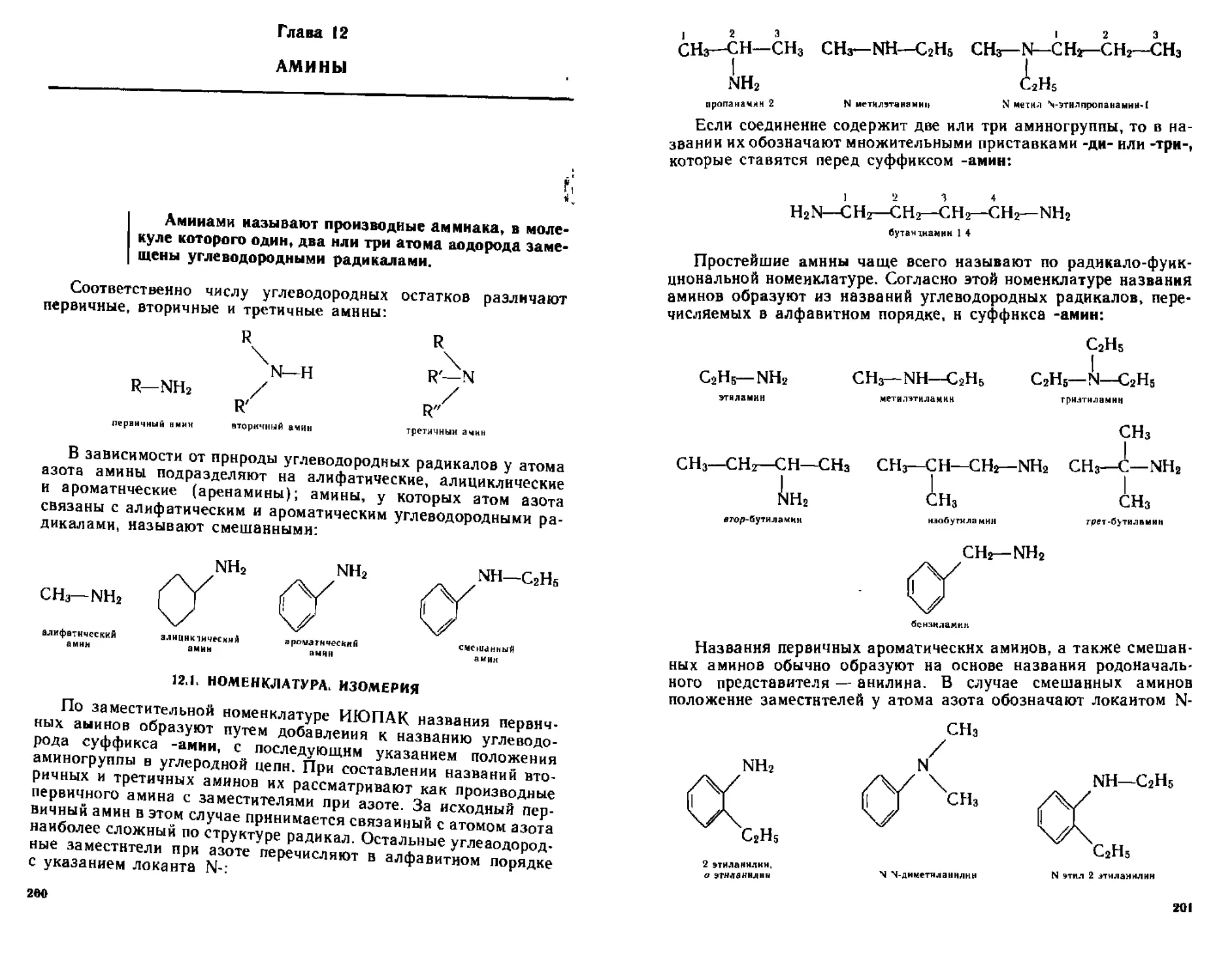
1 Номенклатура Изомерия 200
*2 2 Алкичамнны 202
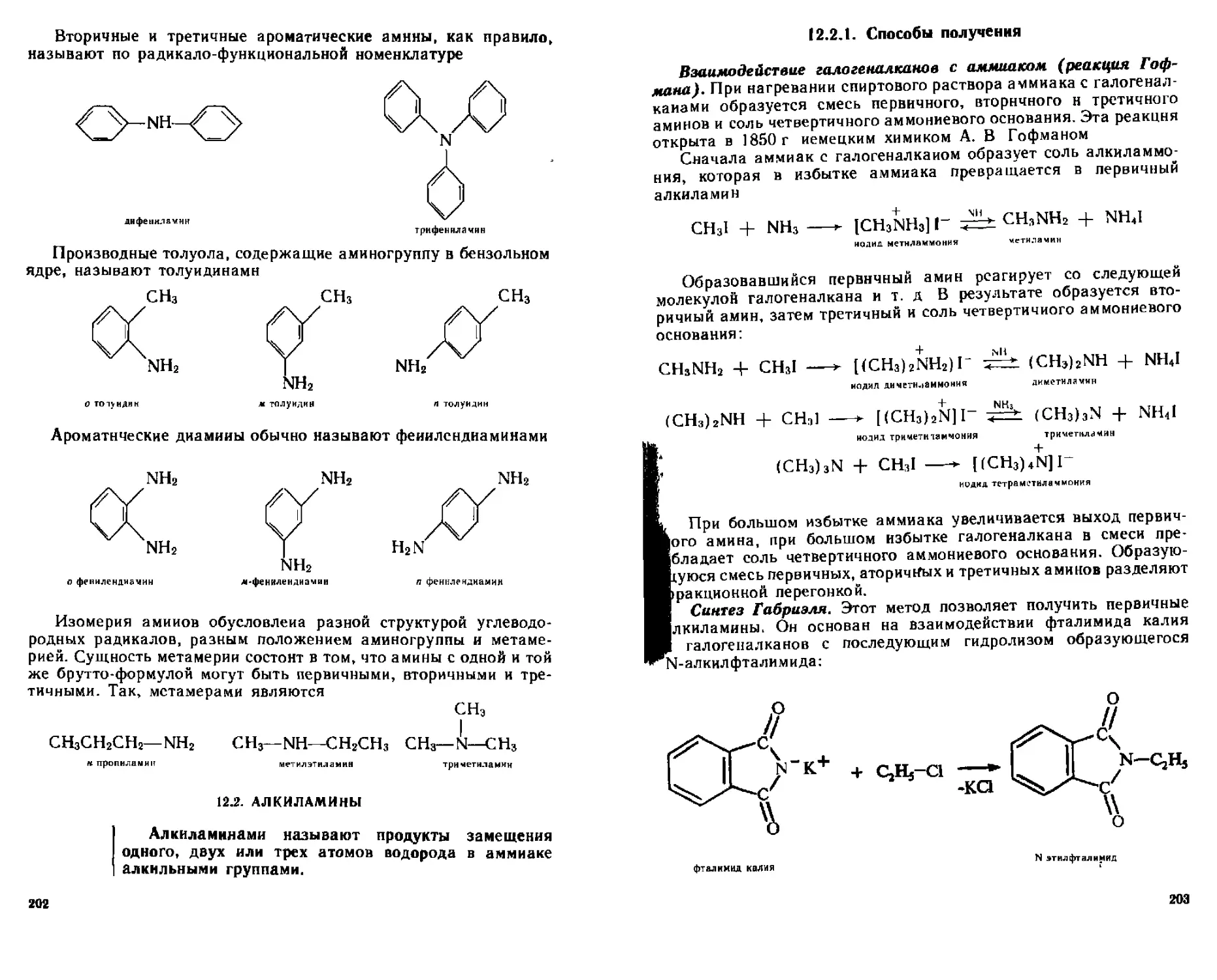
12 2 1 Способы попучения 203
12 2 2 Фшнческие свойства Пространственное строение 204
12.2.3. Химические свойства....................................206
А. Основность...............................................206
Б. Реакции с электрофильными реагентами.................... 208
В. Окисление алкнламинов....................................211
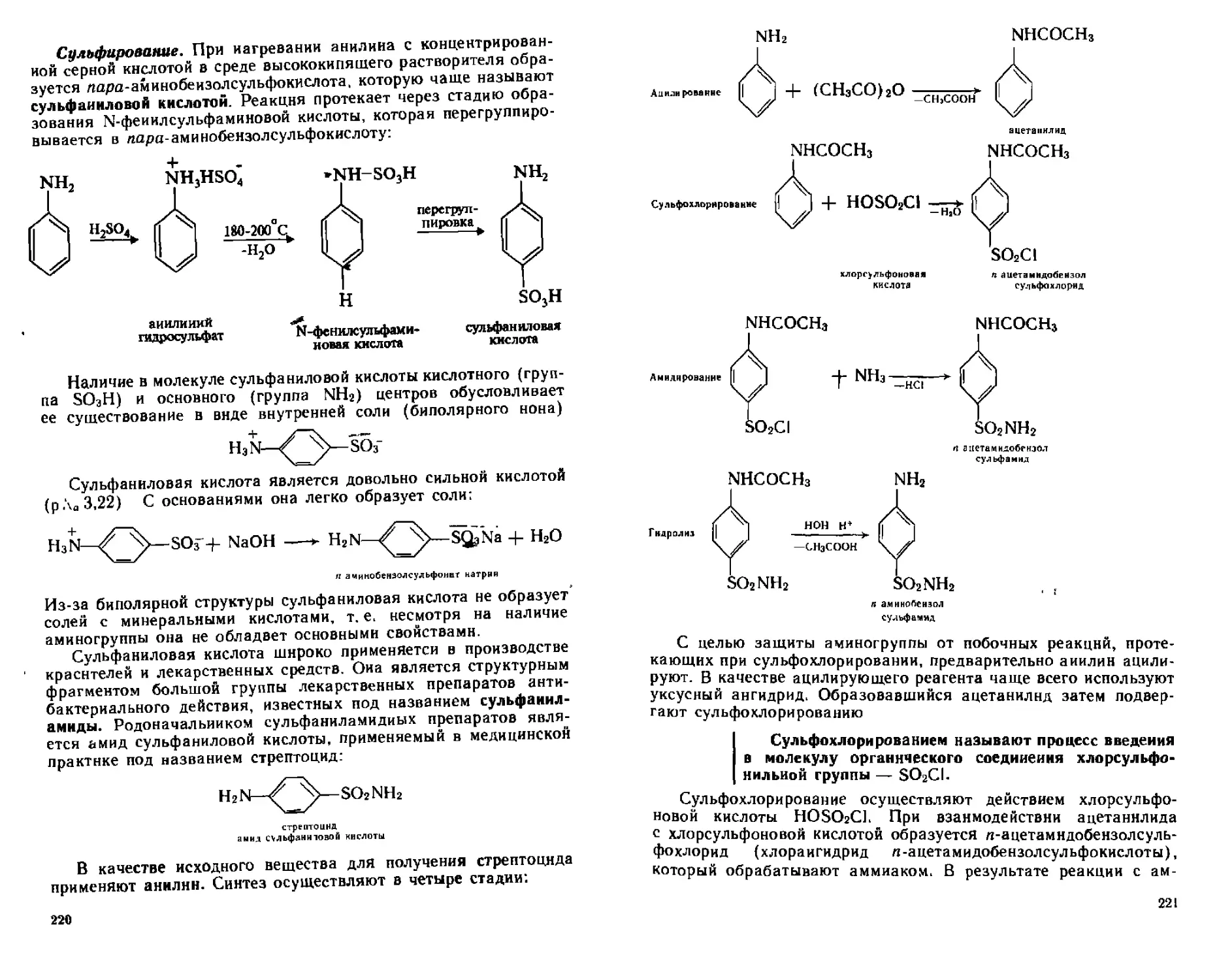
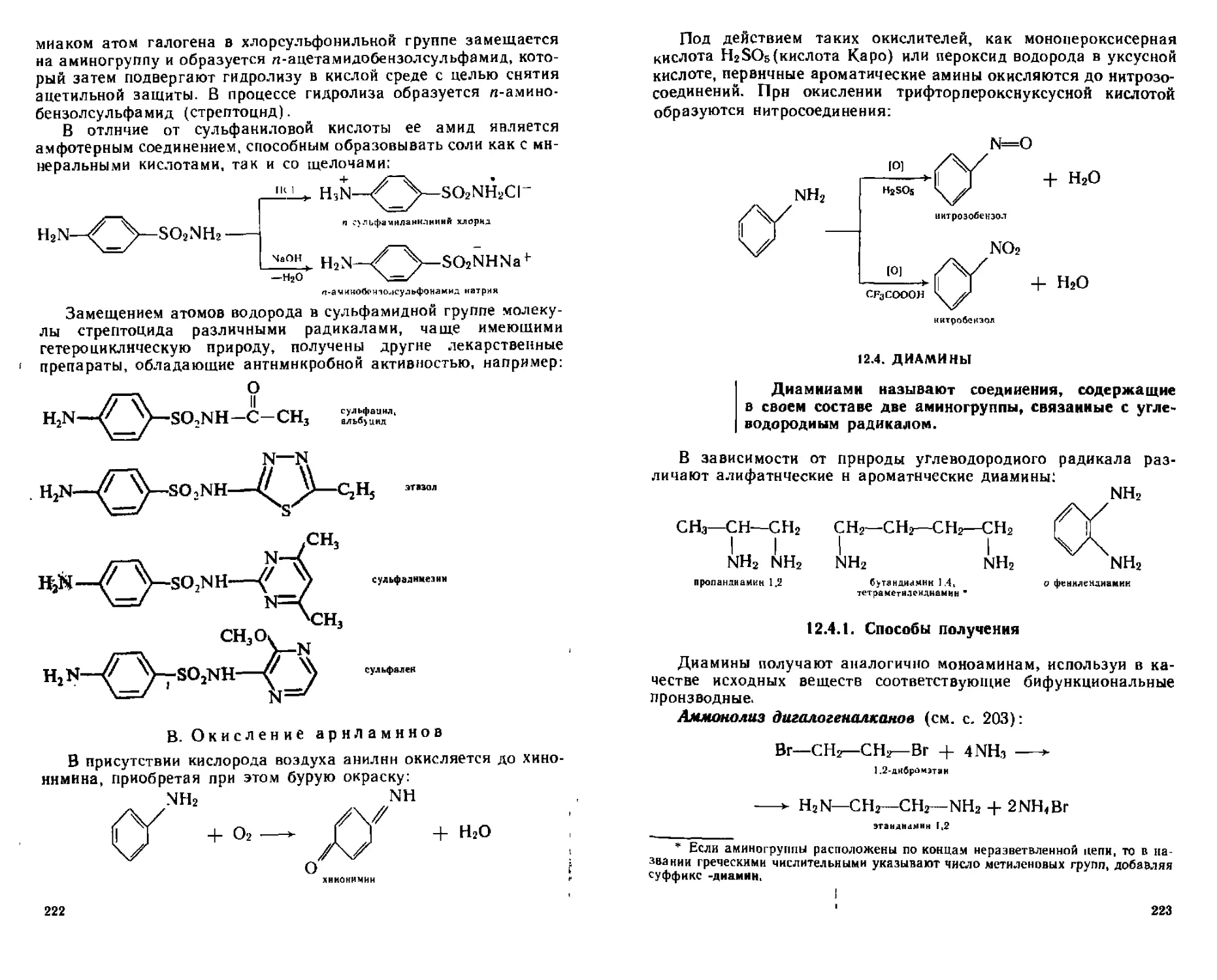
12.3. Ариламины....................................................212
12.3.1. Способы получения......................................212
12.3.2. Физические свойства....................................213
12.3.3. Химические свойства....................................213
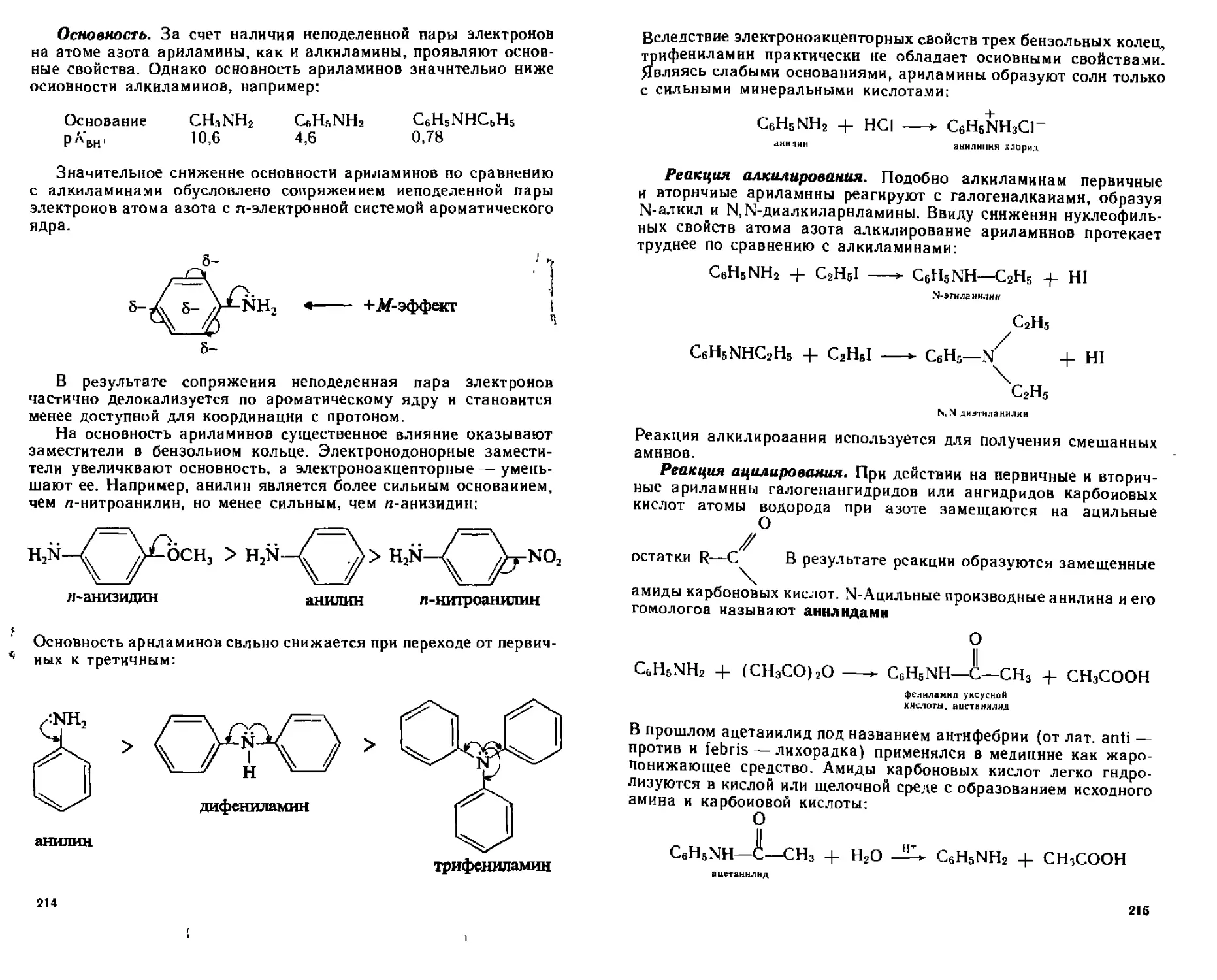
А. Реакции с участием атома азота...........................213
Б. Реакции с участием атомов углерода ароматического ядра .... 217
В, Окисление арилам илов................................... 222
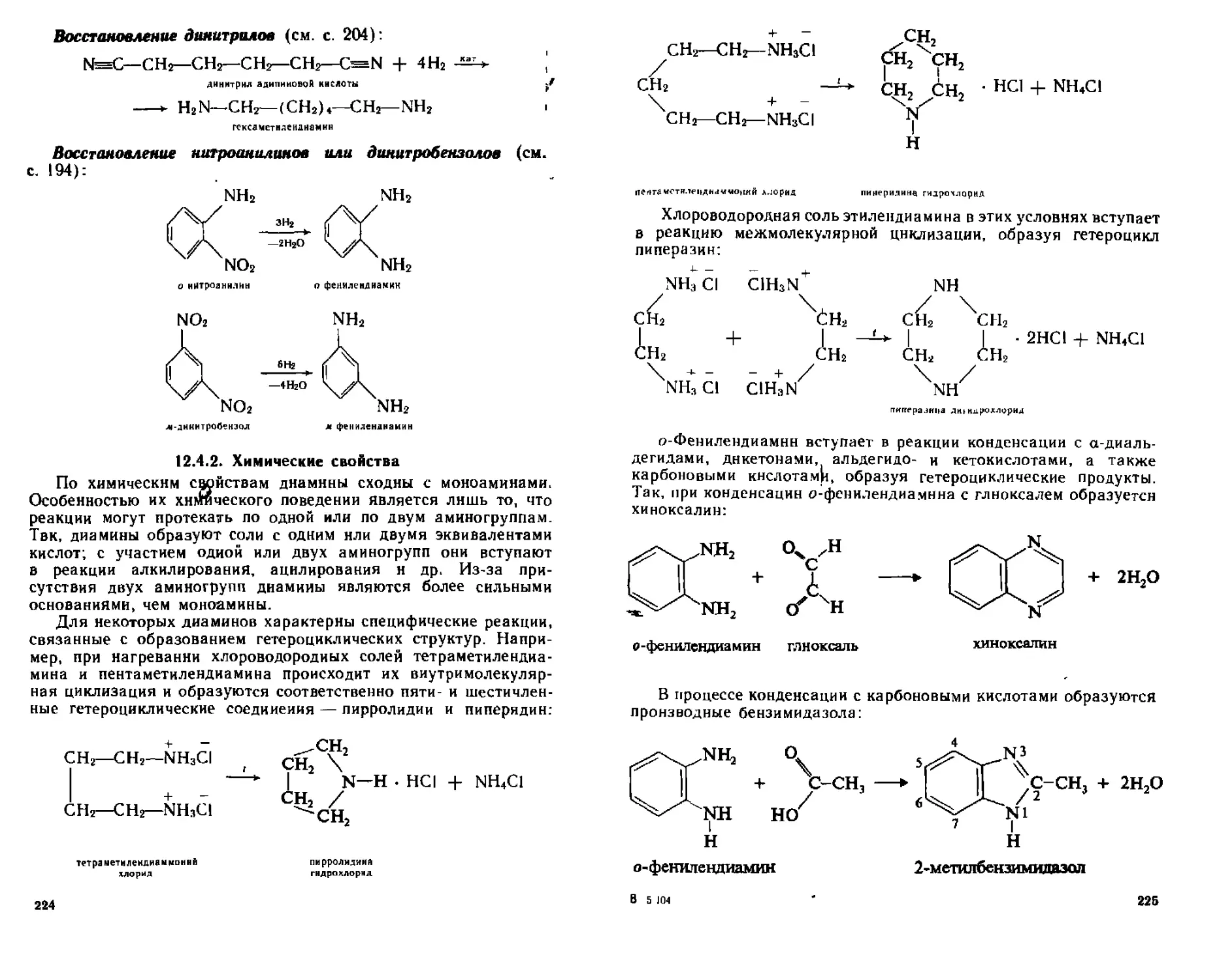
12.4. Днамипы......................................................223
12.4,1. Способы получения......................................223
12.4.2. Химические свойства....................................224
12.5. Идентификация аминов....................................... 226
12.5.1. Химические методы......................................226
12.5.2. Физические .методы.....................................227
12.6. Отдельные представители. Применение........................ 227
Контрольные вопросы и упражнения..................................229
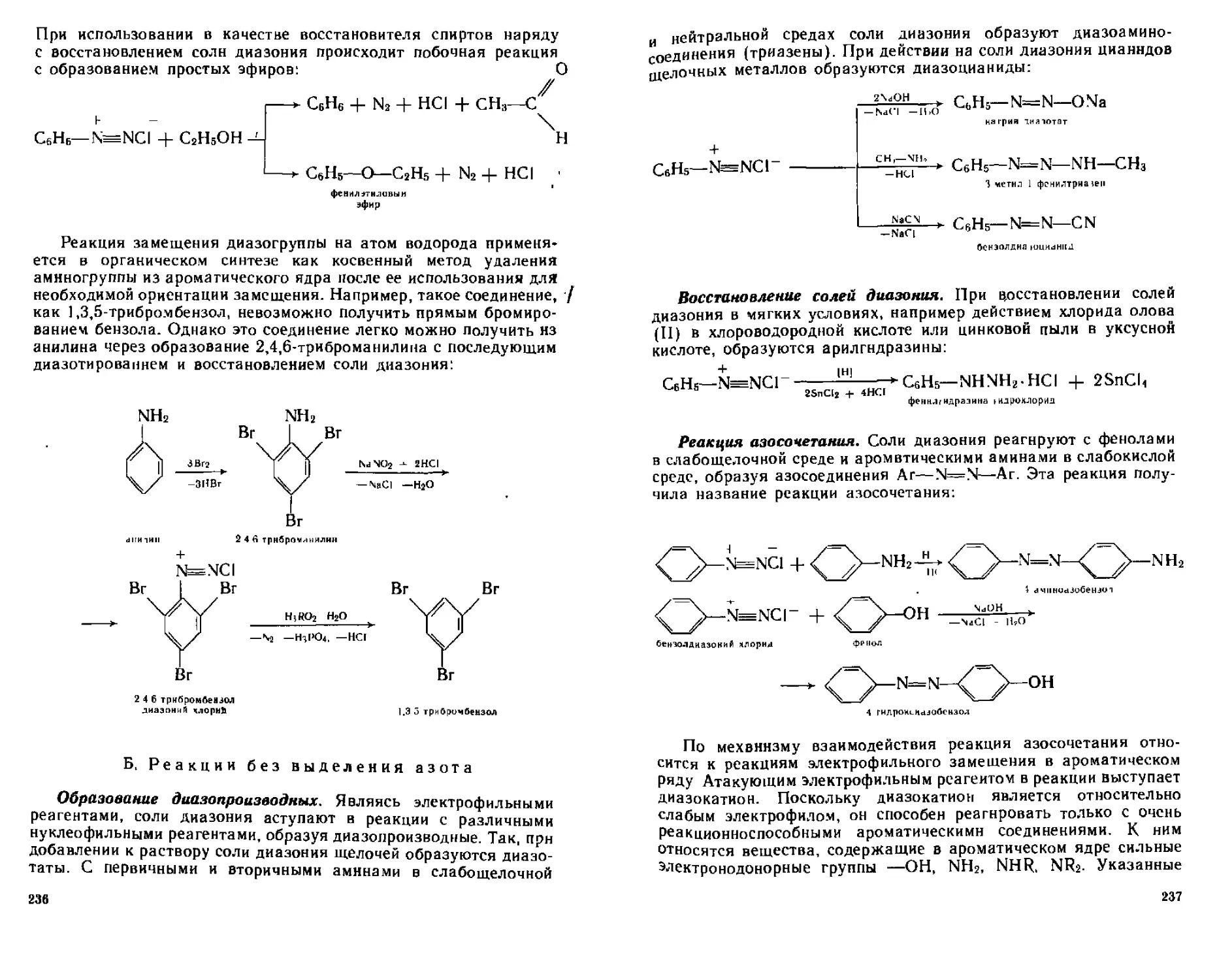
ГЛАВА 13. ДИАЗО- И АЗОСОЕДИНЕНИЯ..............................230
13.1. Диазосоединения..............................................230
13.1.1. Номенклатура. Изомерин.................................231
13.1.2, Способы получения солей арендназония..................231
13.1.3. Физические свойства солей диазония....................233
13.1.4. Химические свойства солей диазония.....................233
А- Реакции с выделением азота................................234
Б. Реакции без выделения азота...............................236
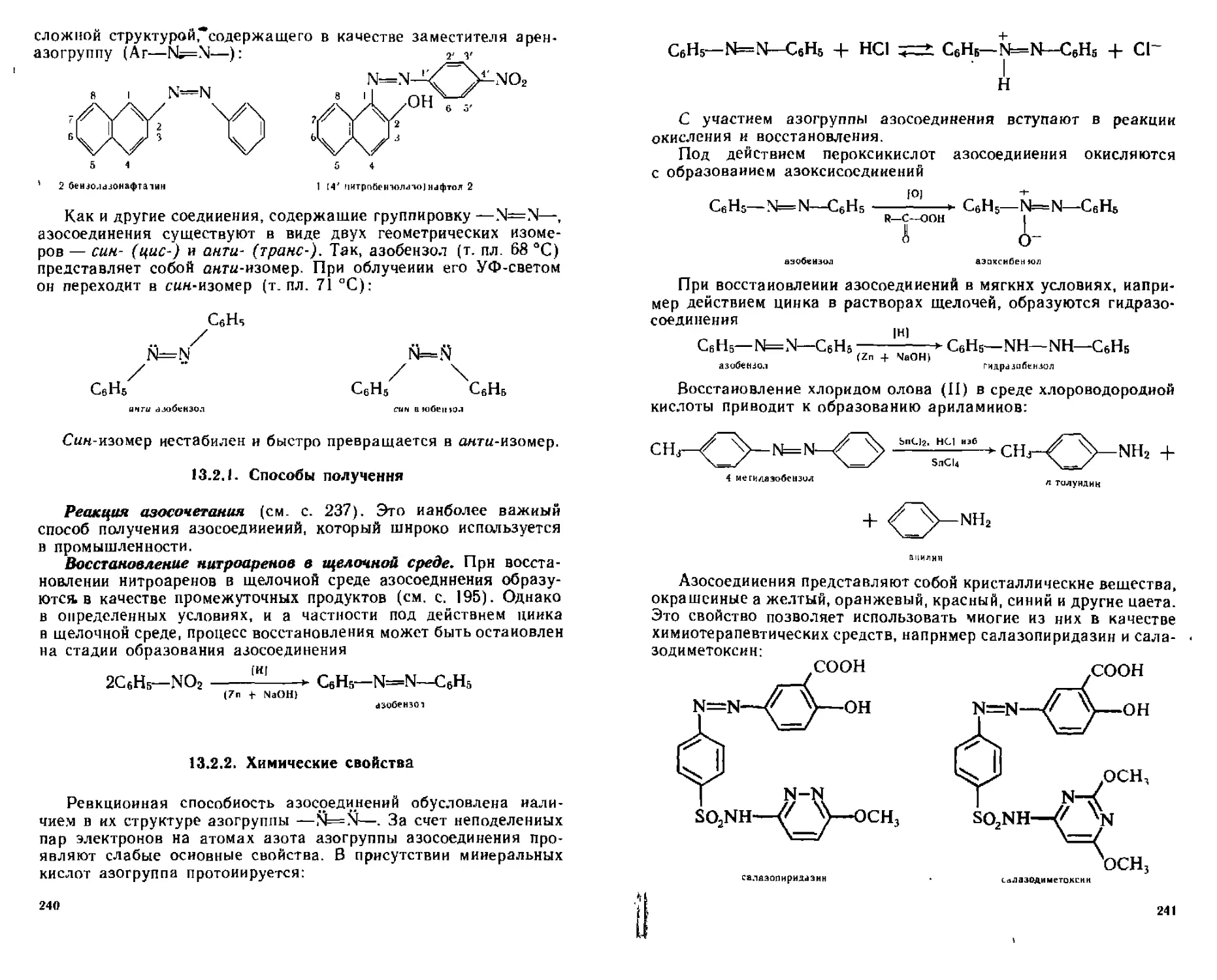
13,2, Азосоединения ..............................................239
*13.2.1. Способы получения......................................240
13.2.2. Химические свойства....................................240
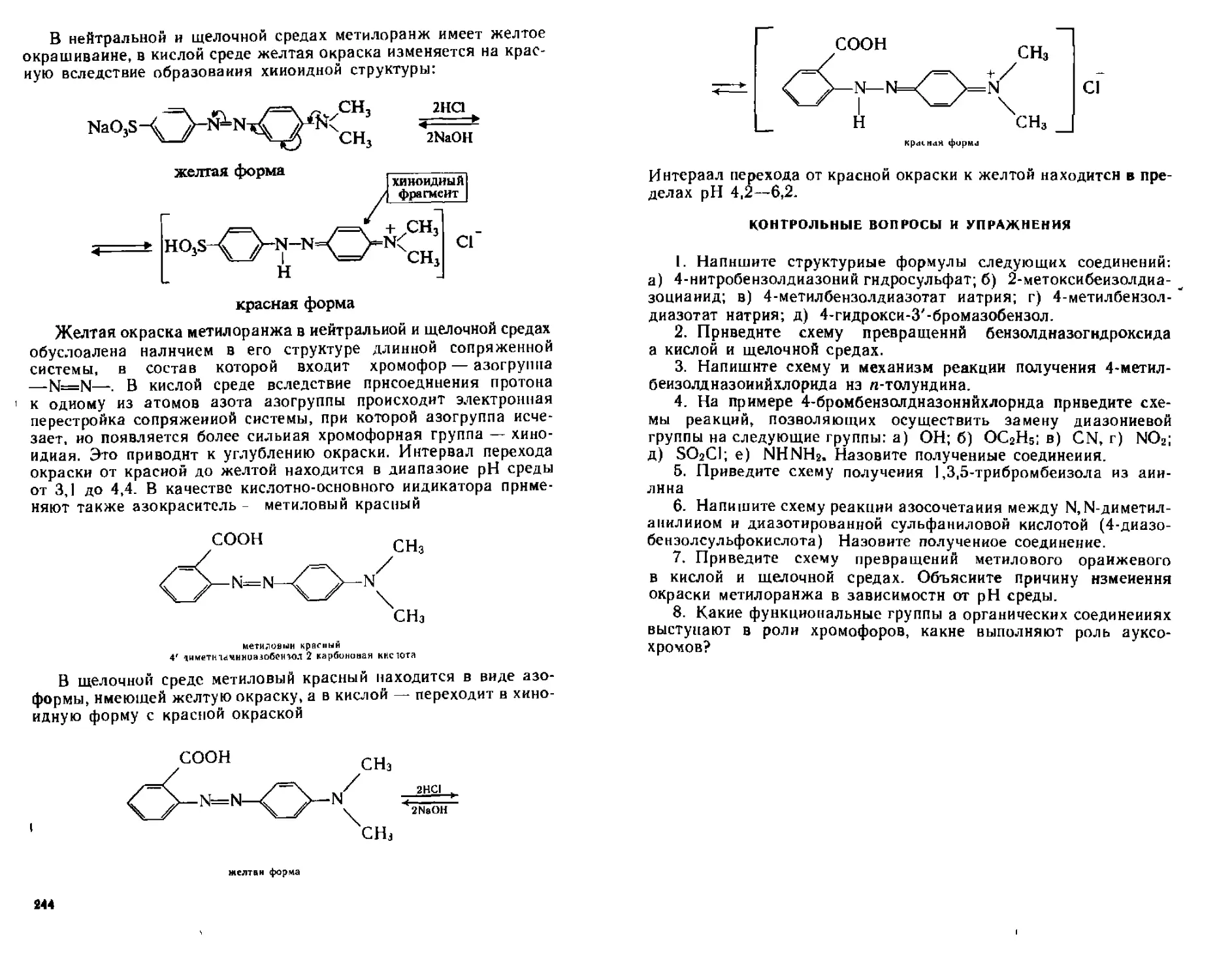
13.2.3. Основные положения теории цветности. Азокрасители......242
Контрольные вопросы и упражнения...................................245
ГЛАВА 14. ГИДРОКСИЛЬНЫЕ ПРОИЗВОДНЫЕ УГЛЕВОДОРОДОВ.
ПРОСТЫЕ ЭФИРЫ И ИХ ТИОАНАЛОГИ.........................246
14.1. Одноатомные спирты......................................... 246
14.1.1, Номенклатура спиртов...................................247
14.1.2. Изомерия...............................................248
14.1,3. Способы получения......................................249
14.14. Физические свойства....................................252
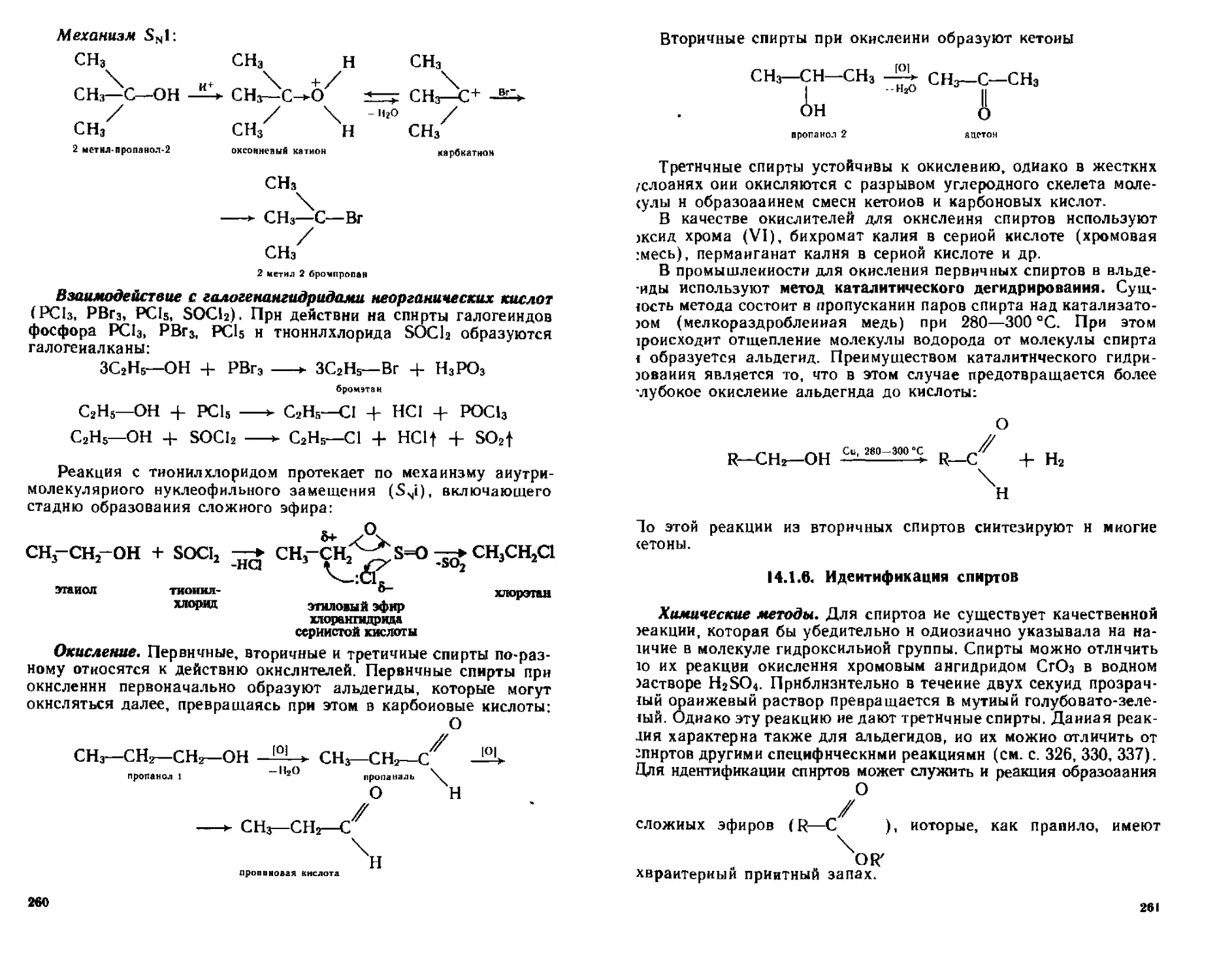
14,1.5. Химические свойства...................................254
14.1.6. Идентификация спиртов . ..............................261
14.1.7. Отдельные нредстанители................................263
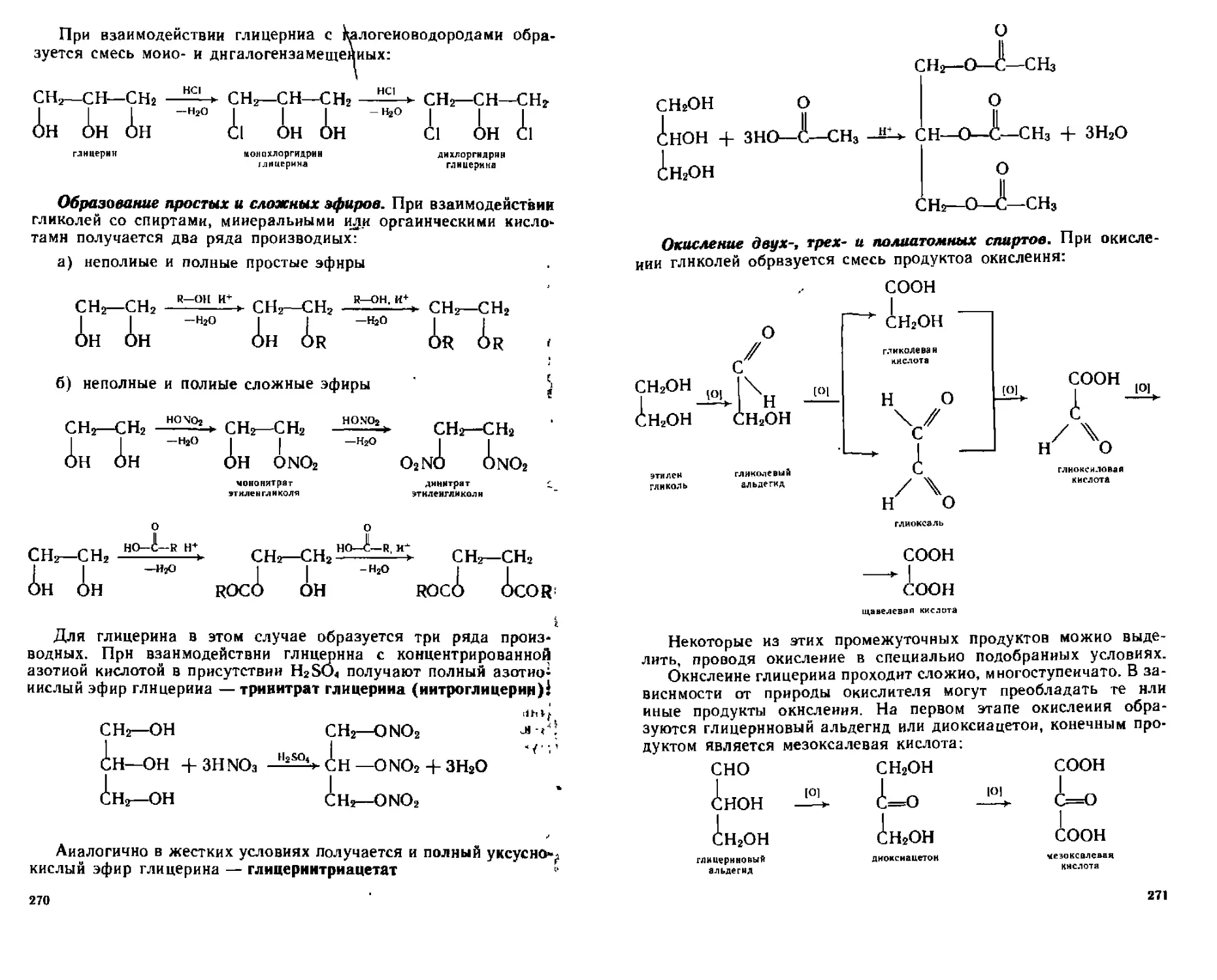
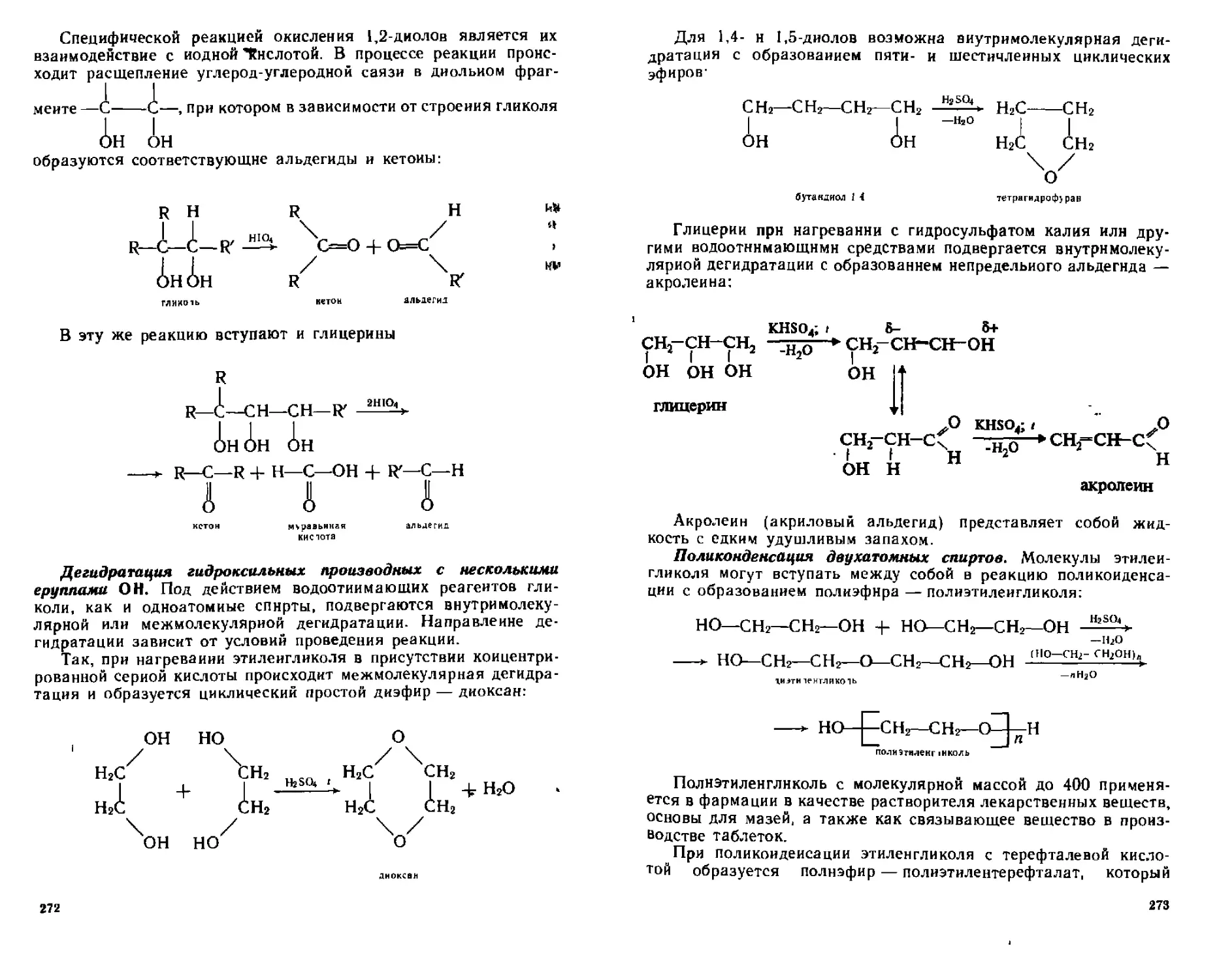
14.2, Двух-, трех- и полиатомные спирты...........................264
14.2.1. Способы получения двух-, трех- и полнатомных спиртов..266
14.2.2. Физические свойства....................................268
14.2.3. Химические свойства.................................. 268
14.2.4. Идентификация диатов и триалов........................274
14.2.5. Отдельные представители...............................274
14.3. Енолы........................................................275
14.4. Аминоспирты..................................................278
14.4.1. Способы получения......................................279
14.4.2. Химические свойства....................................279
14.4.3. Отдельные представители................................280
Контрольные вопросы и упражнения..................................281
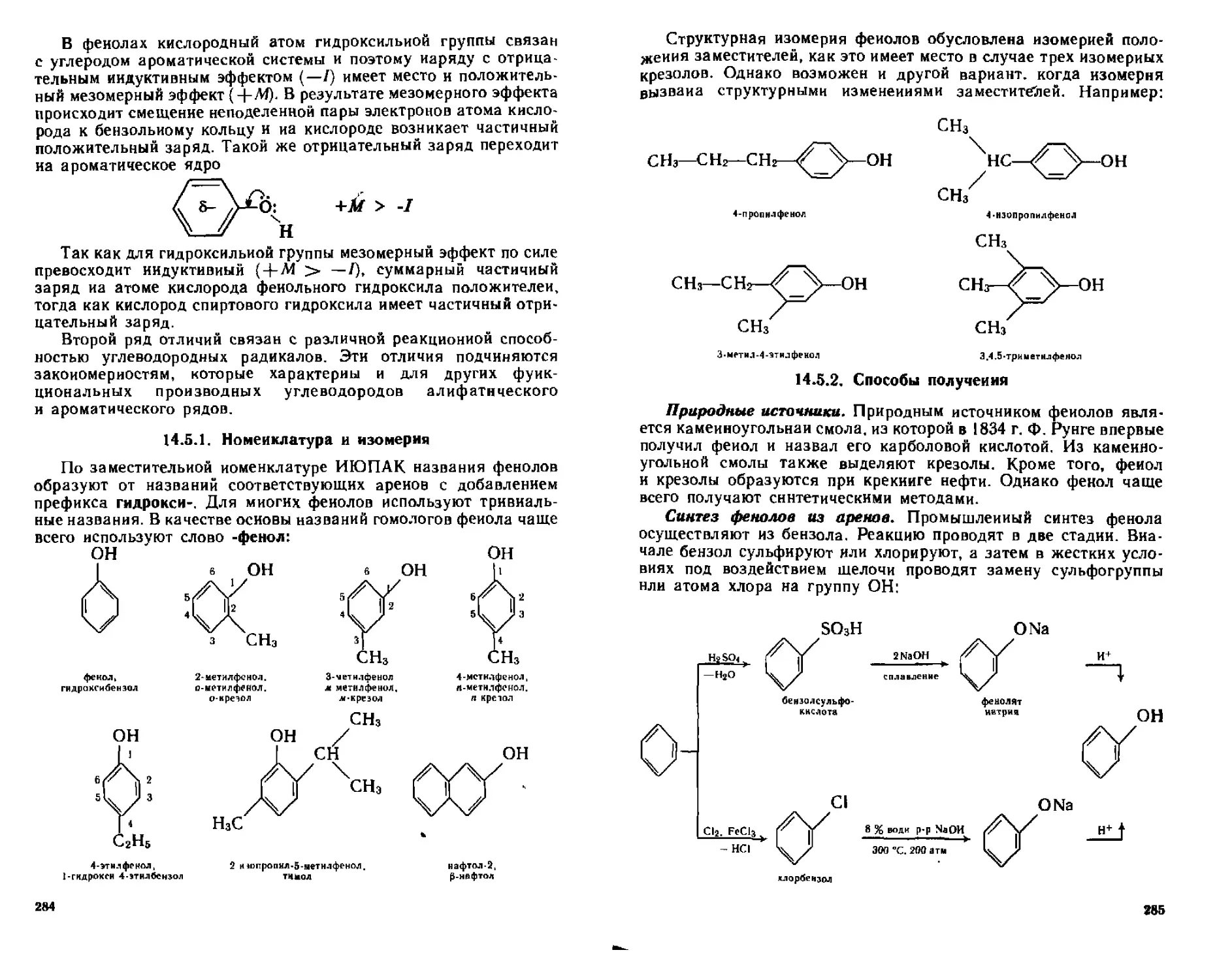
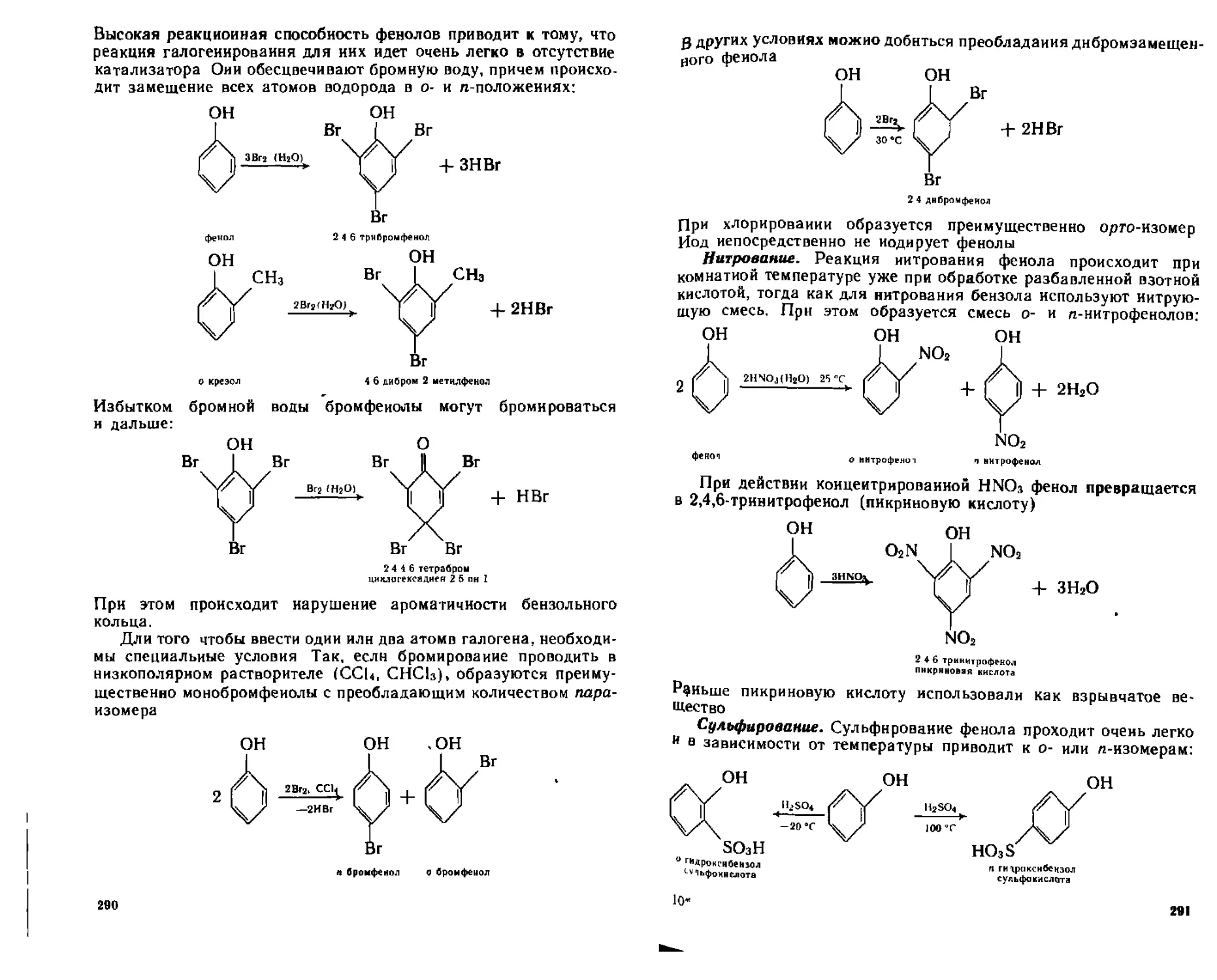
14.5. Одноатомные фенолы...........................................283
14.5.1, Номенклатура и изомерия...............................284
14.5.2. Способы получения.....................................285
14.5.3. Физические свойства...................................287
6
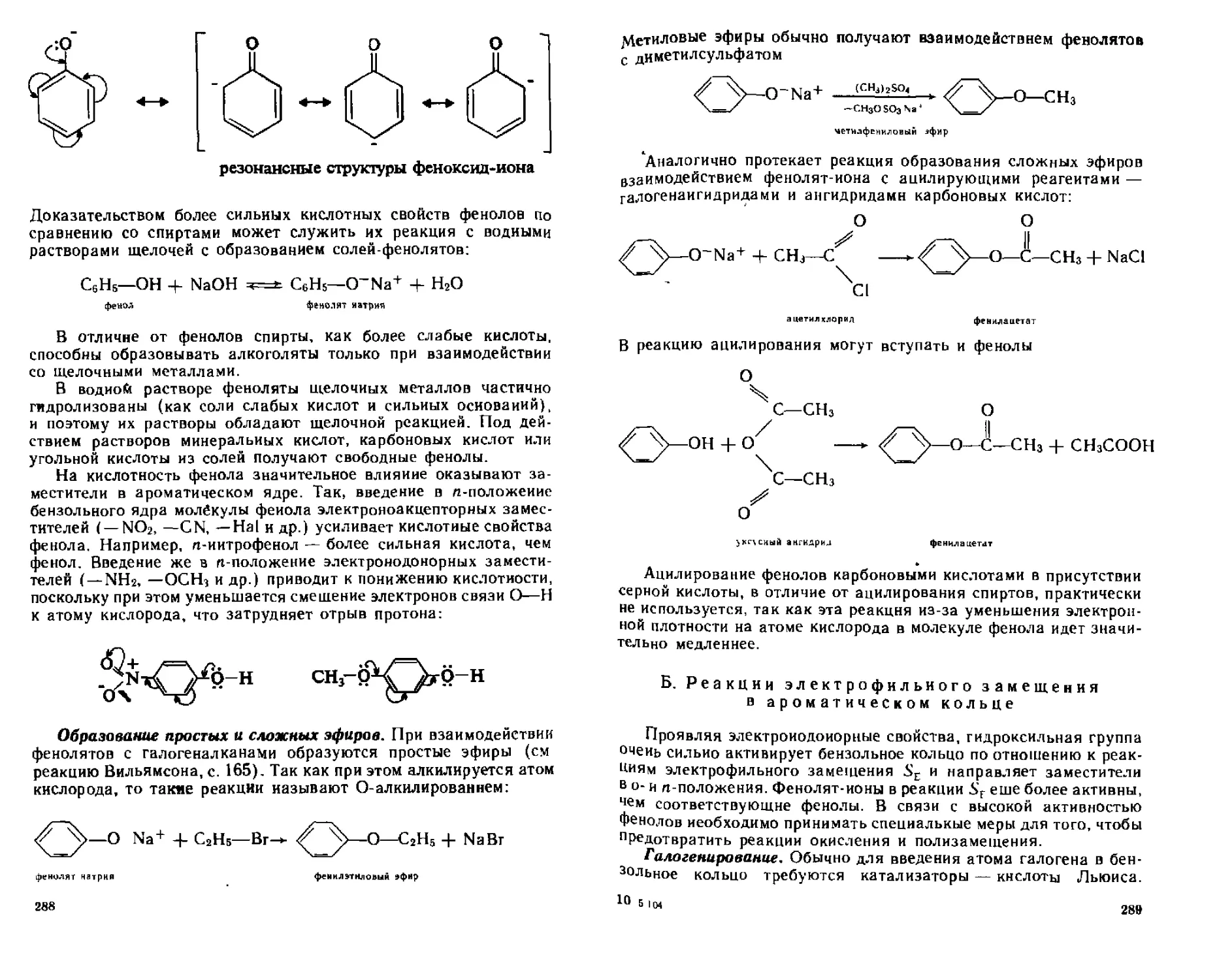
(4 5.4. Химические свойства.....................................287
А. Реакции с участием связи О—Н..............................287
Б. Реакции электрофильного замещения в ароматическом кольце . . 289
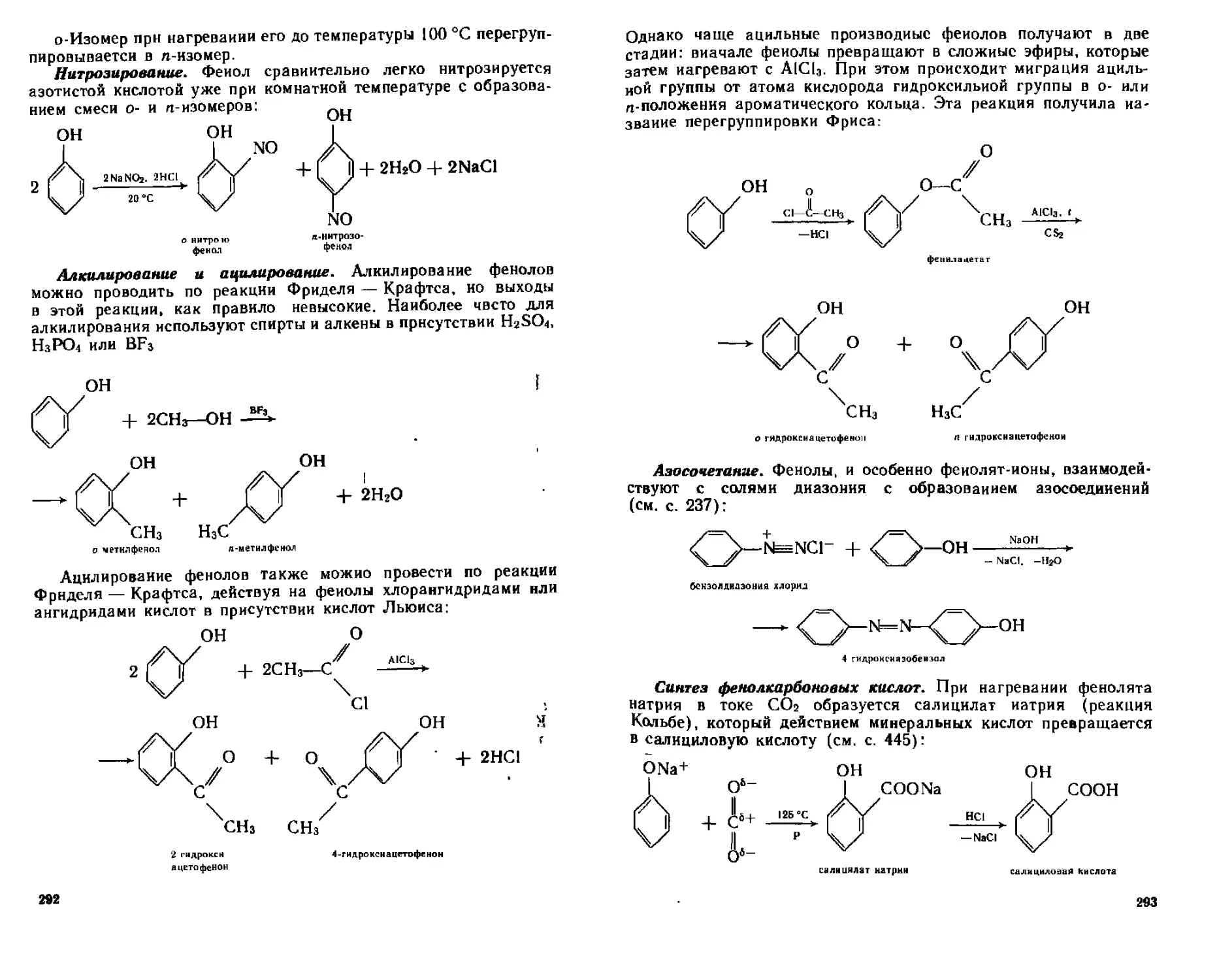
В. реакции восстановления и окисления........................295
14.5.5- Идентификация одноатомных фенолов..................... 296
14.5.6. Важнейшие представители фенолов.........................296
14 6 Двух-, трех- и лолиатомные фенолы...............................298
14.6.1. Способы получения.......................................298
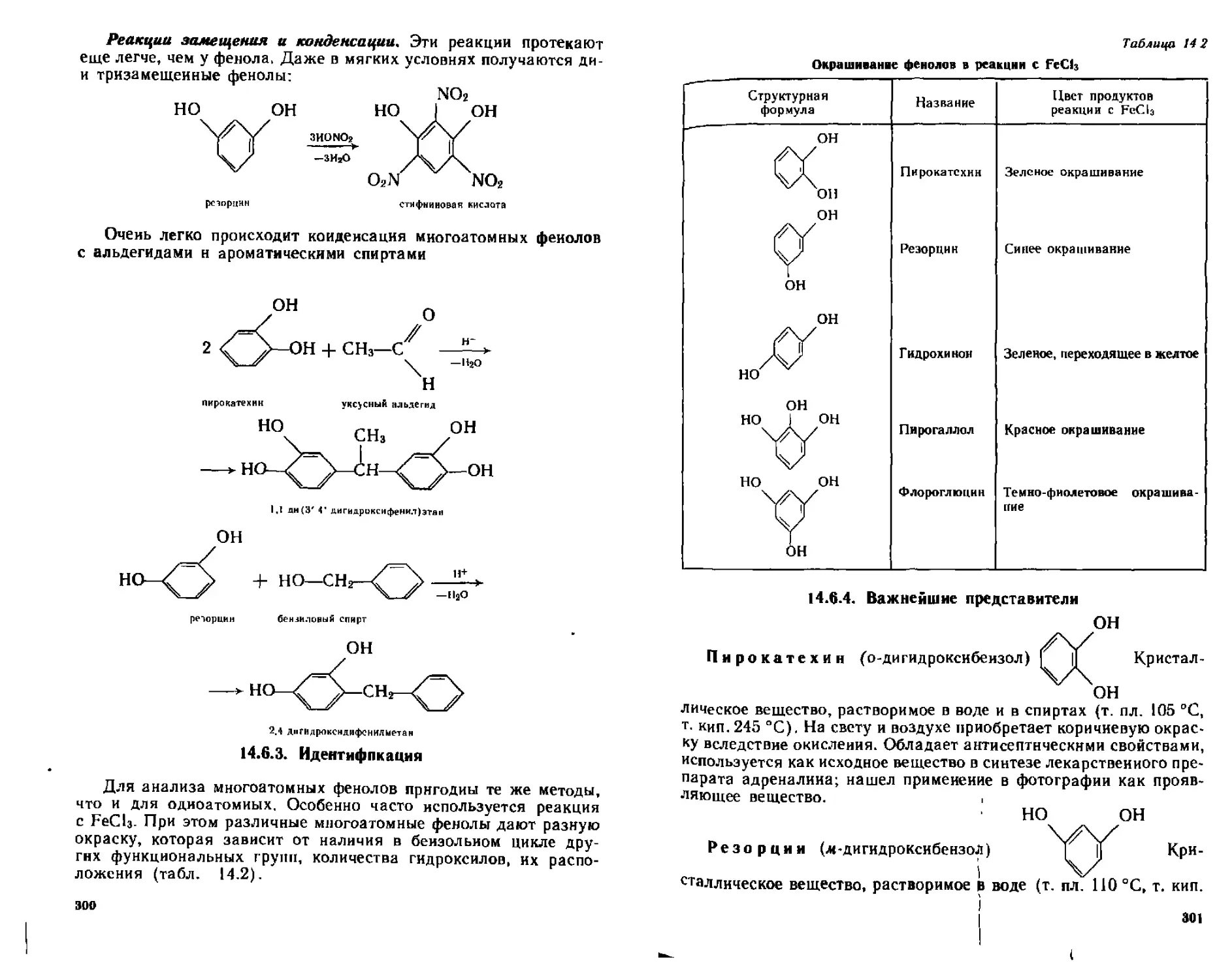
М.6.2. Химические свойства......................................299
14.6.3. Идентификация...........................................300
14.6.4. Важнейшие представители ...............................301
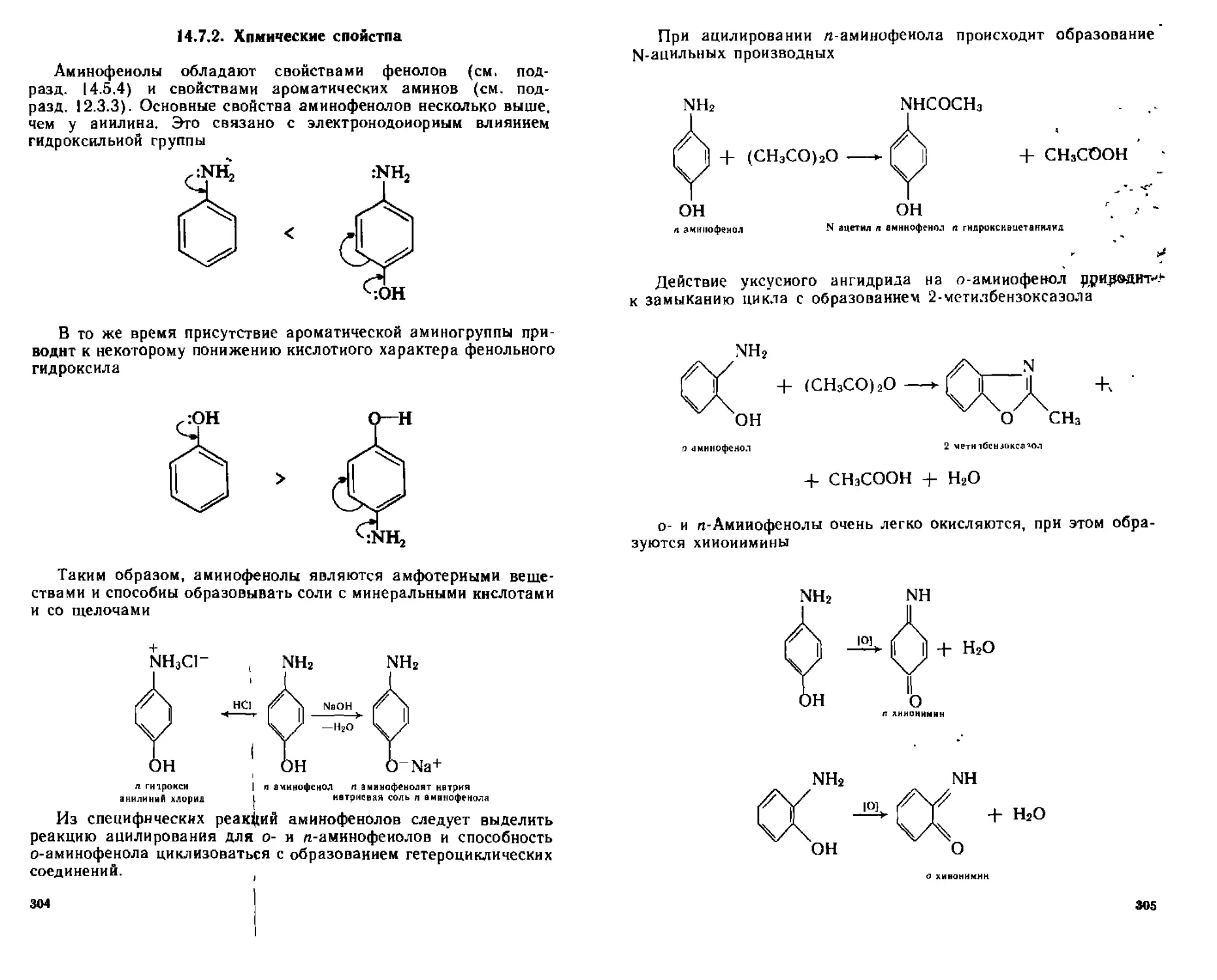
14 7 Аминофенолы....................................................
14.7.1 . Способы получения.....................................
14.7.2 . Химические свойства...................................
14.7.3 . Отдельные представители...............................
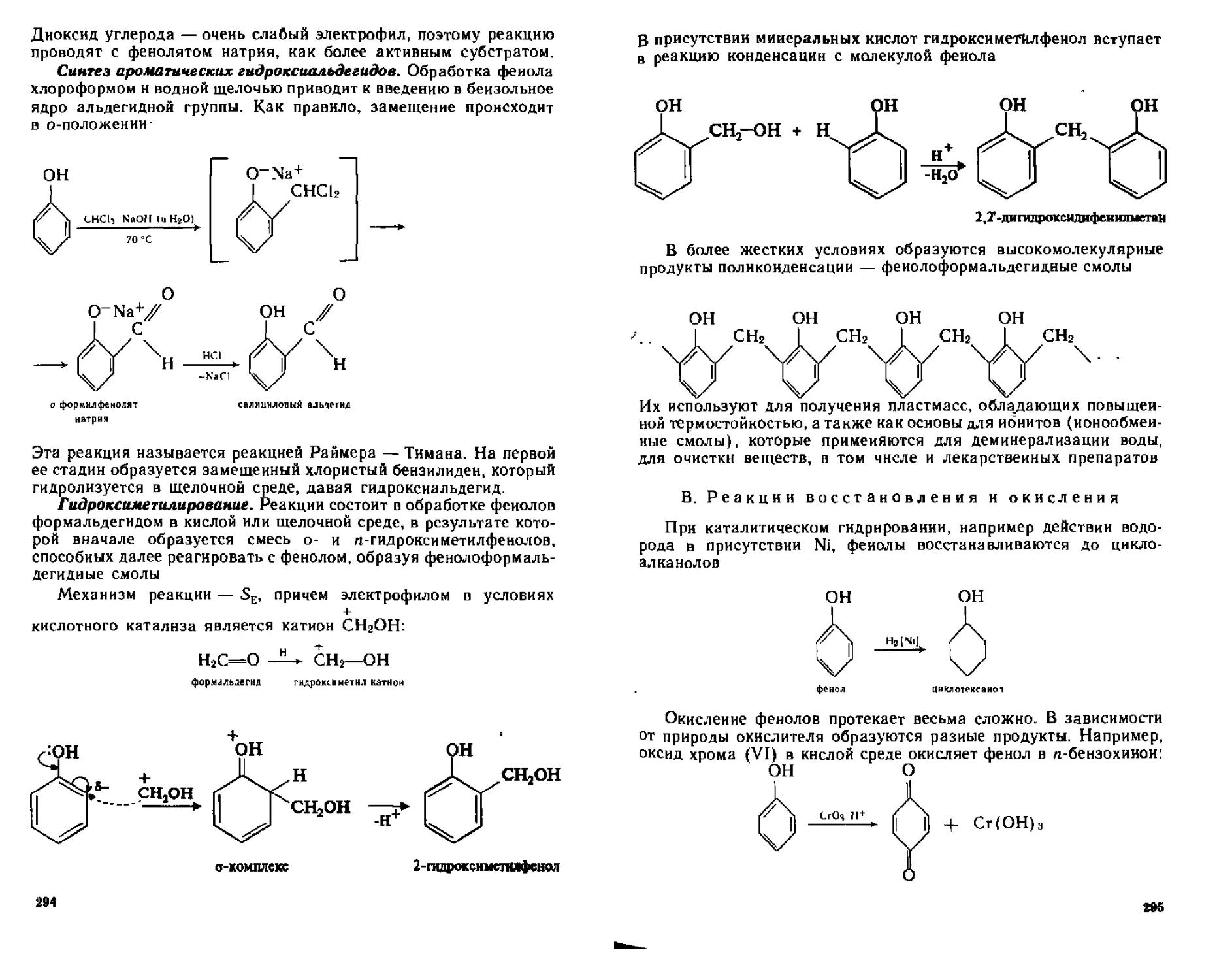
] 4-8. Тиолы........................................................
14.8- 1. Способы получения.....................................
14.8.2. Химические свойства....................................
14.9. Простые эфиры ................................................
14.9.1- Номенклатура и изомерия................................
14.9.2. Способы получения......................................
14.9.3. Физические свойства..........................-.........
14.9.4. Химические свойства....................................
14.9.5. Идентификация простых эфиров...........................
14.9.6. Отдельные представители................................
14.10. Сульфиды.....................................................
14.10.1. Способы получения.....................................
14.10.2. Химические свойства...................................
14.10.3. Отдельные представители. Применение...................
Контрольные вопросы и упражнения....................................
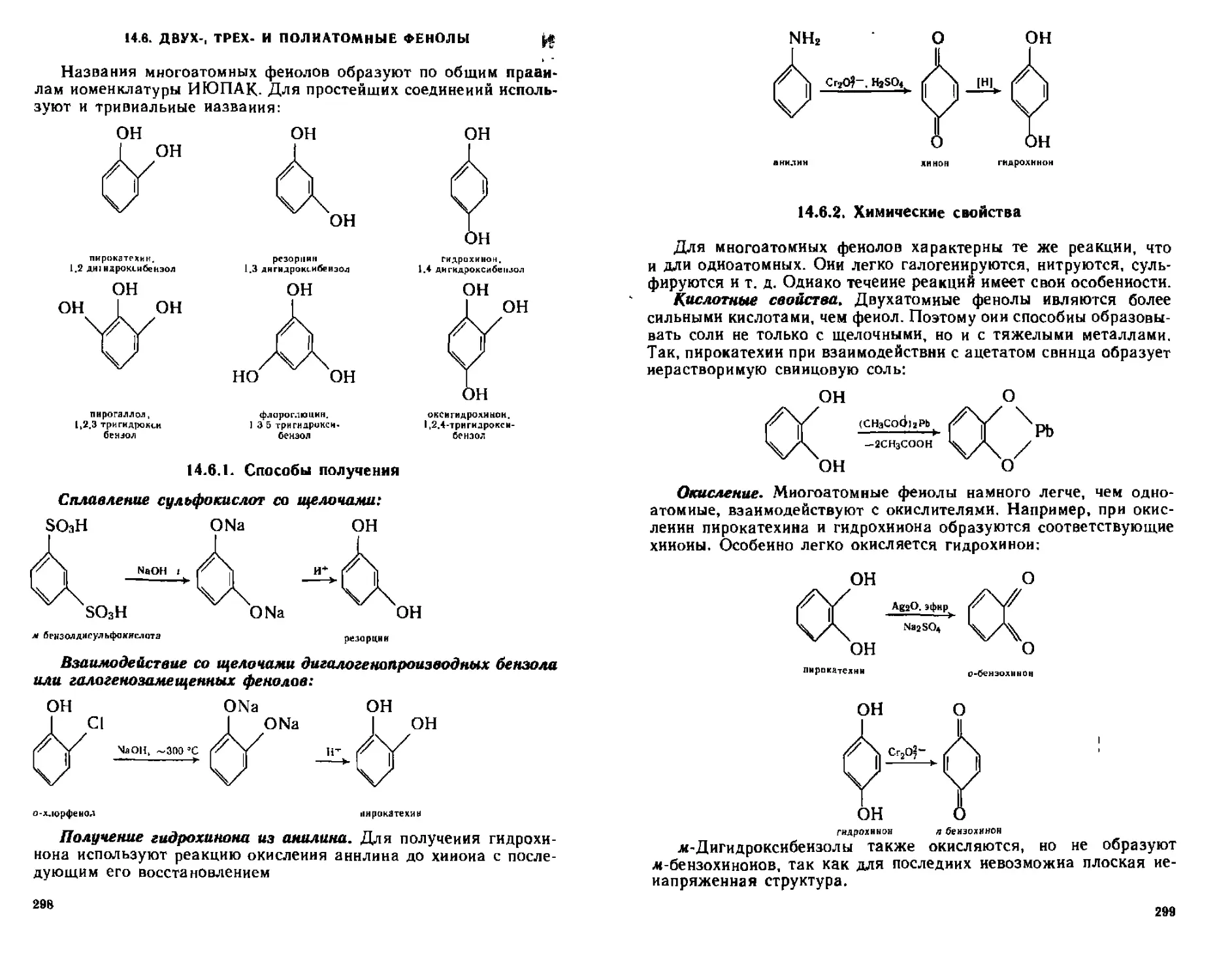
302 303 304
306 307 307
308 310 310
311 311 311
313 313 314
314 315
316 317
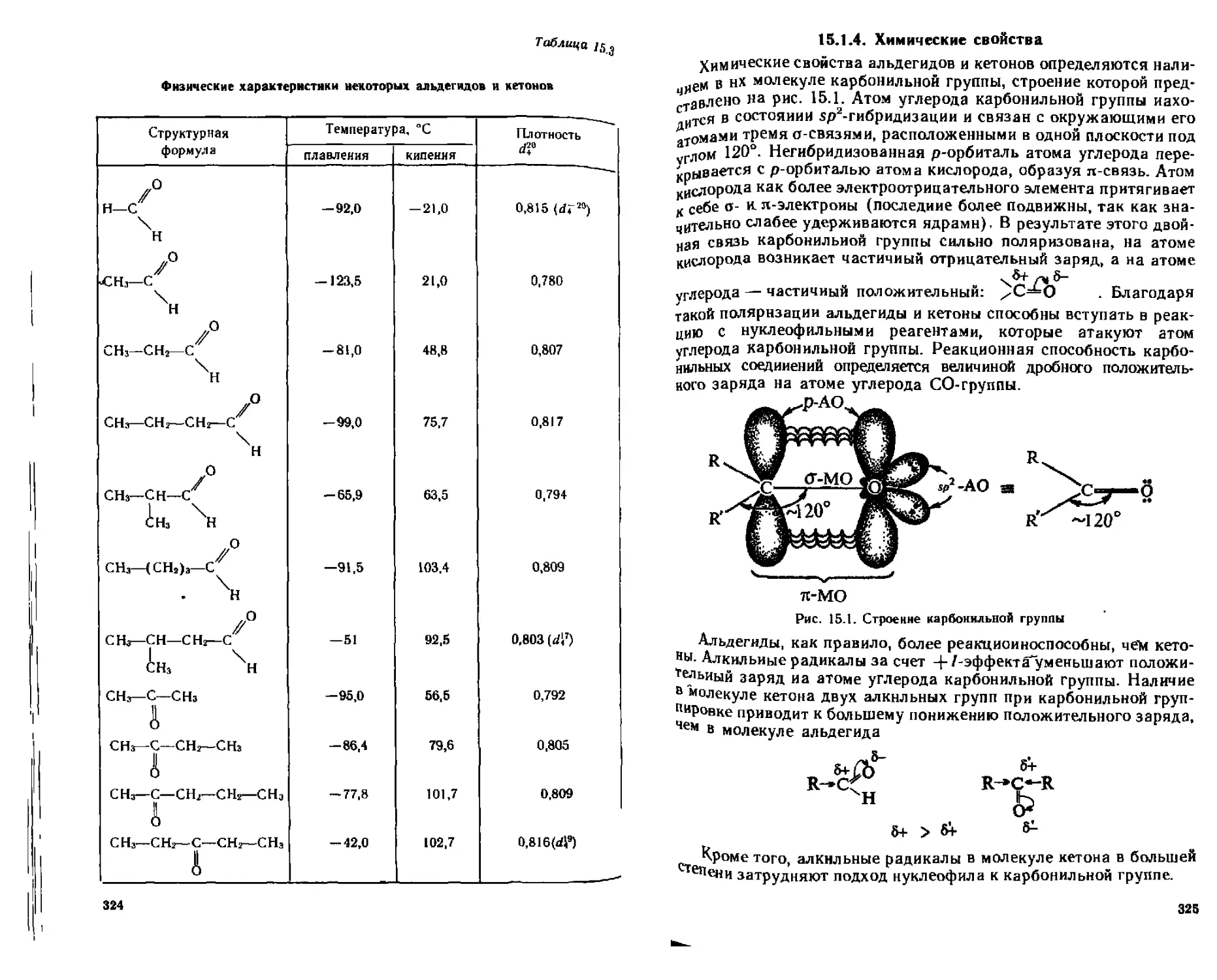
ГЛАВА 15. АЛЬДЕГИДЫ И КЕТОНЫ..................318
15.1. Насыщенные альдегиды и кетоны.................................318
15.1.1. Номенклатура и изомерия.................................318
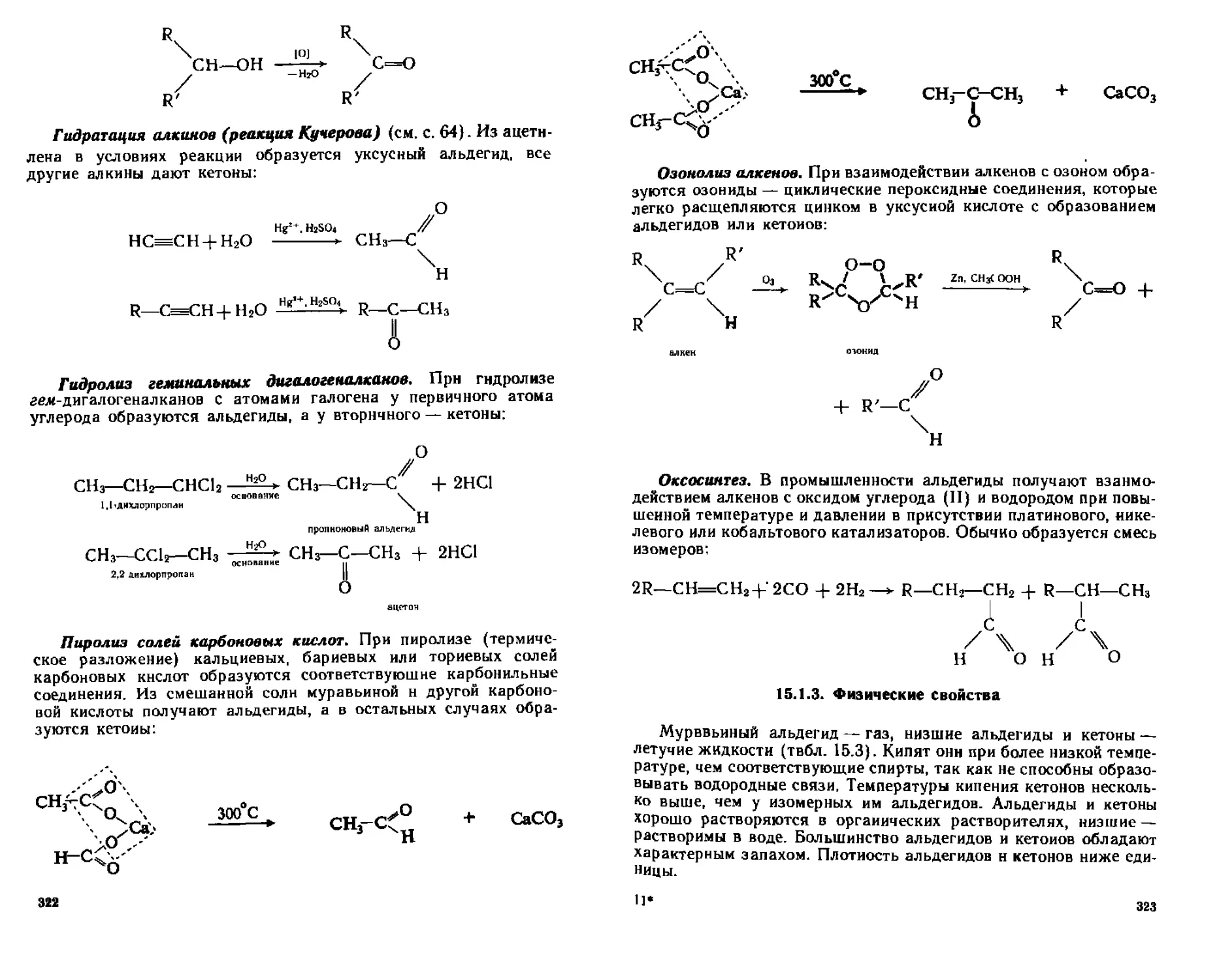
15.1.2. Способы получения.......................................321
15.1.3. Физические свойства.....................................323
15.1.4. Химические свойства.....................................325
А. Реакции нуклеофильного присоединения (As).................326
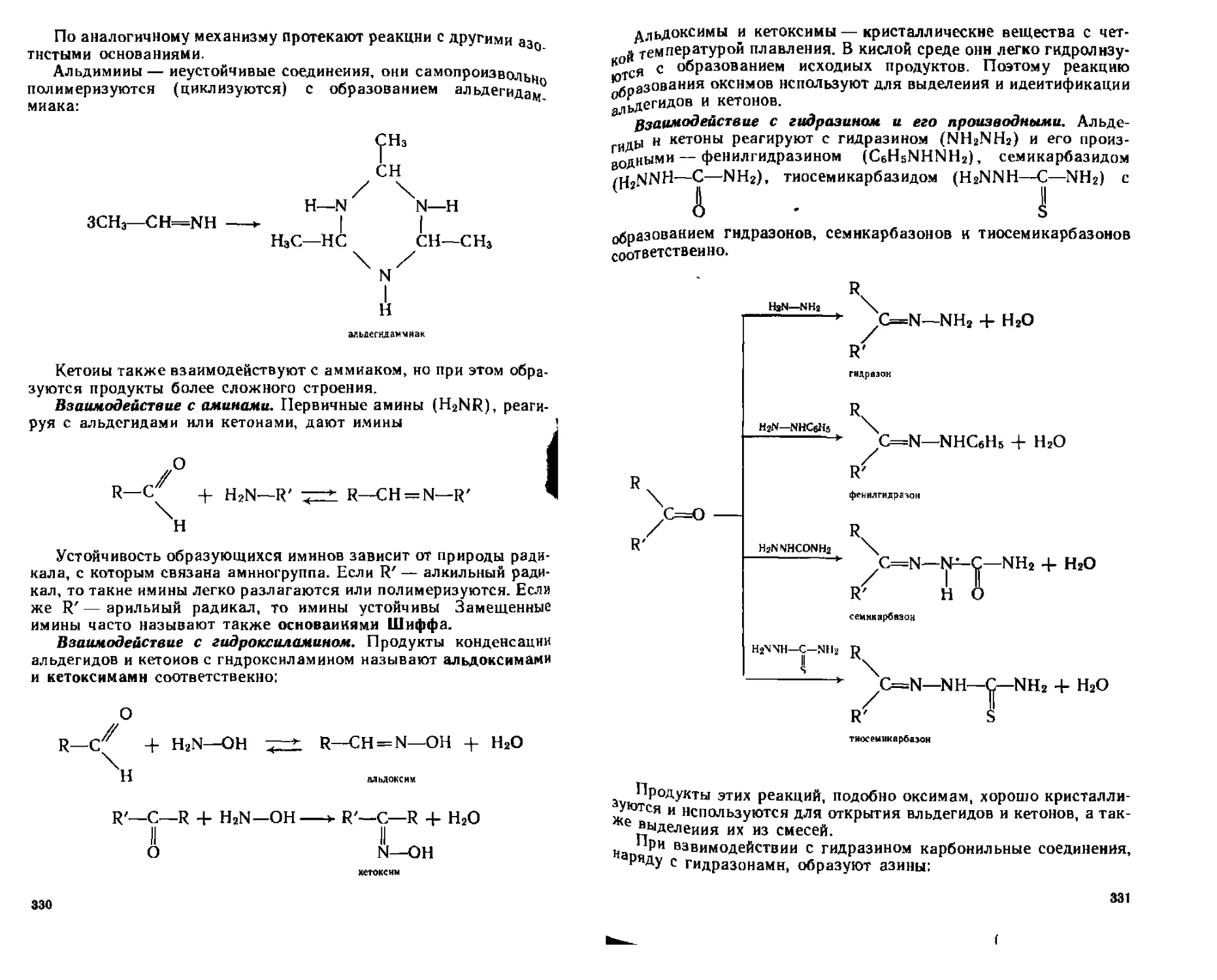
Б. Реакции присоединения-отщепления..........................329
В. Реакции конденсации ..................................... 332
Г. Реакции с участием «-углеродного атома....................334
Д. Реакции полимеризации......................................335
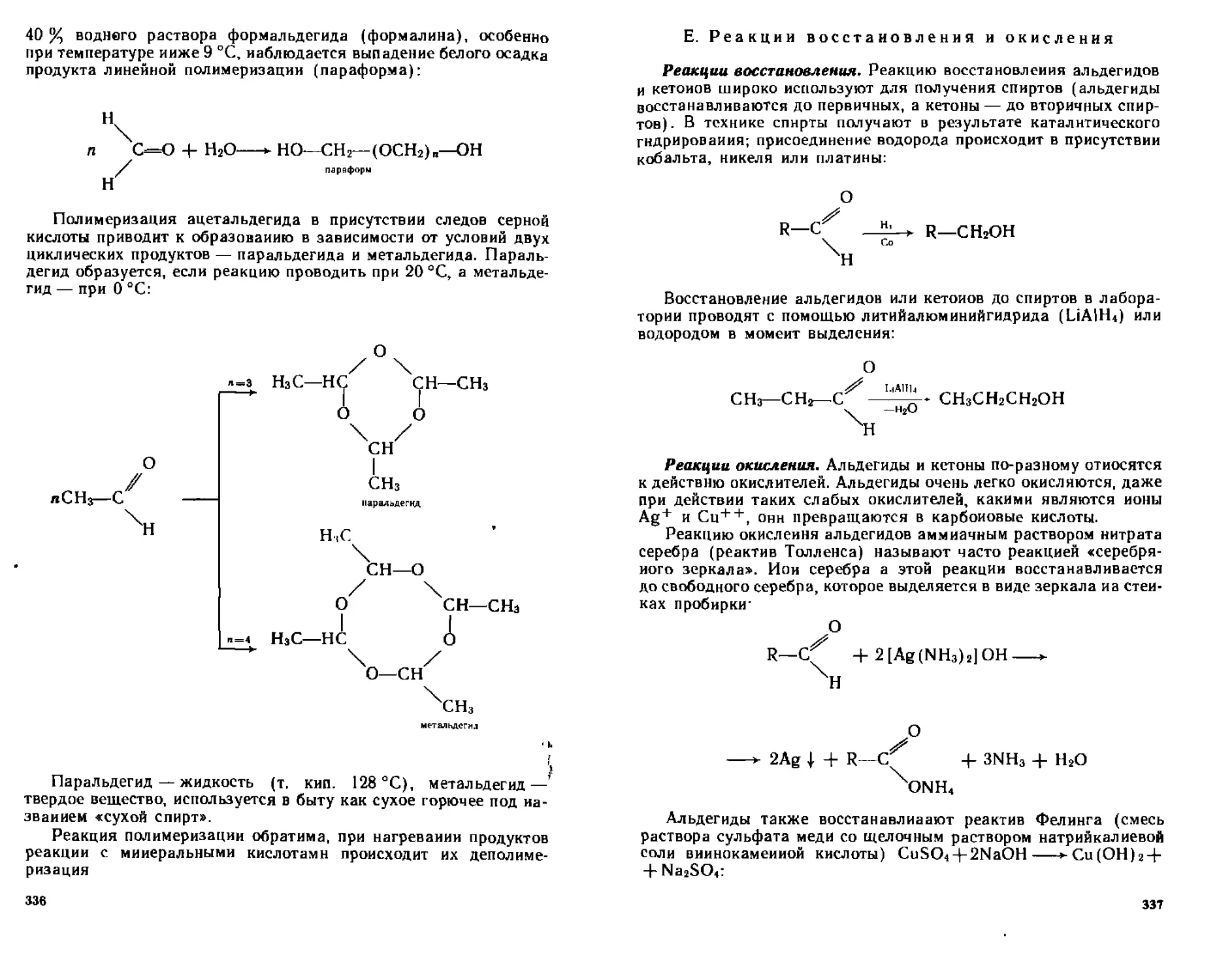
Е. Реакции восстановления и окисления........................337
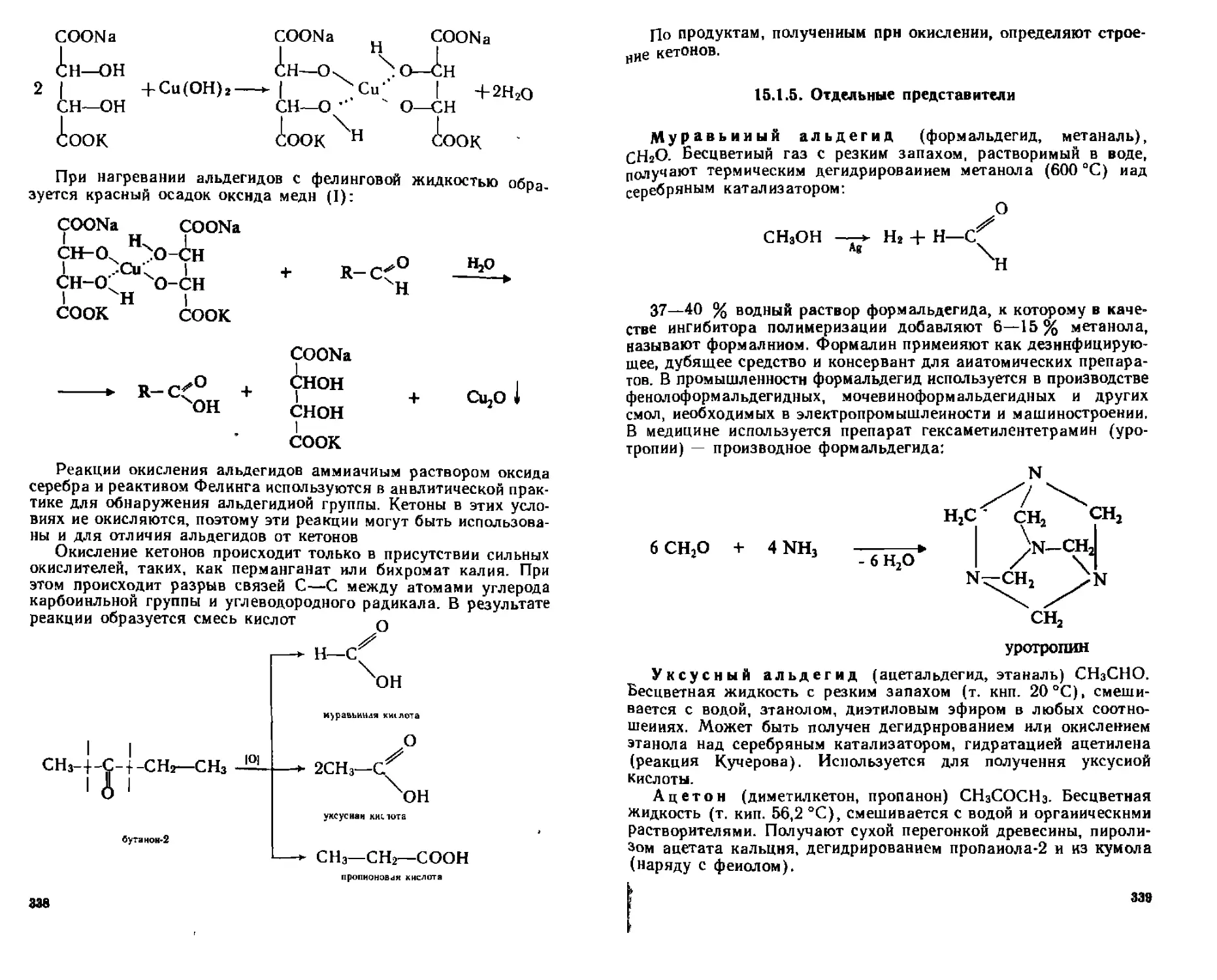
15.1.5. Отдельные представители.................................339
15.2. Непредельные альдегиды........................................340
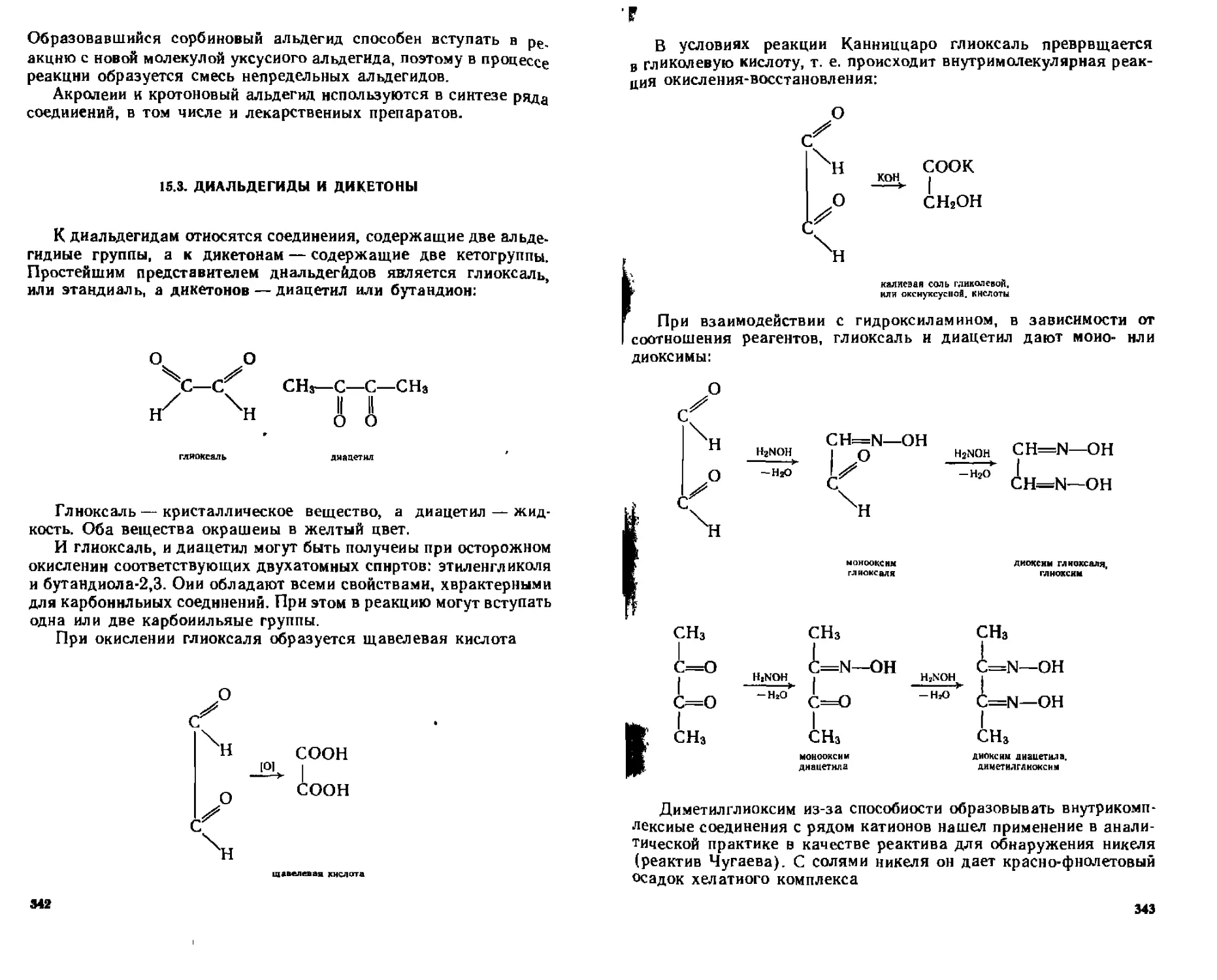
15.3. Диальдегиды и дикетоны . ....................................342
15.4. Ароматические альдегиды и кетоны..............................344
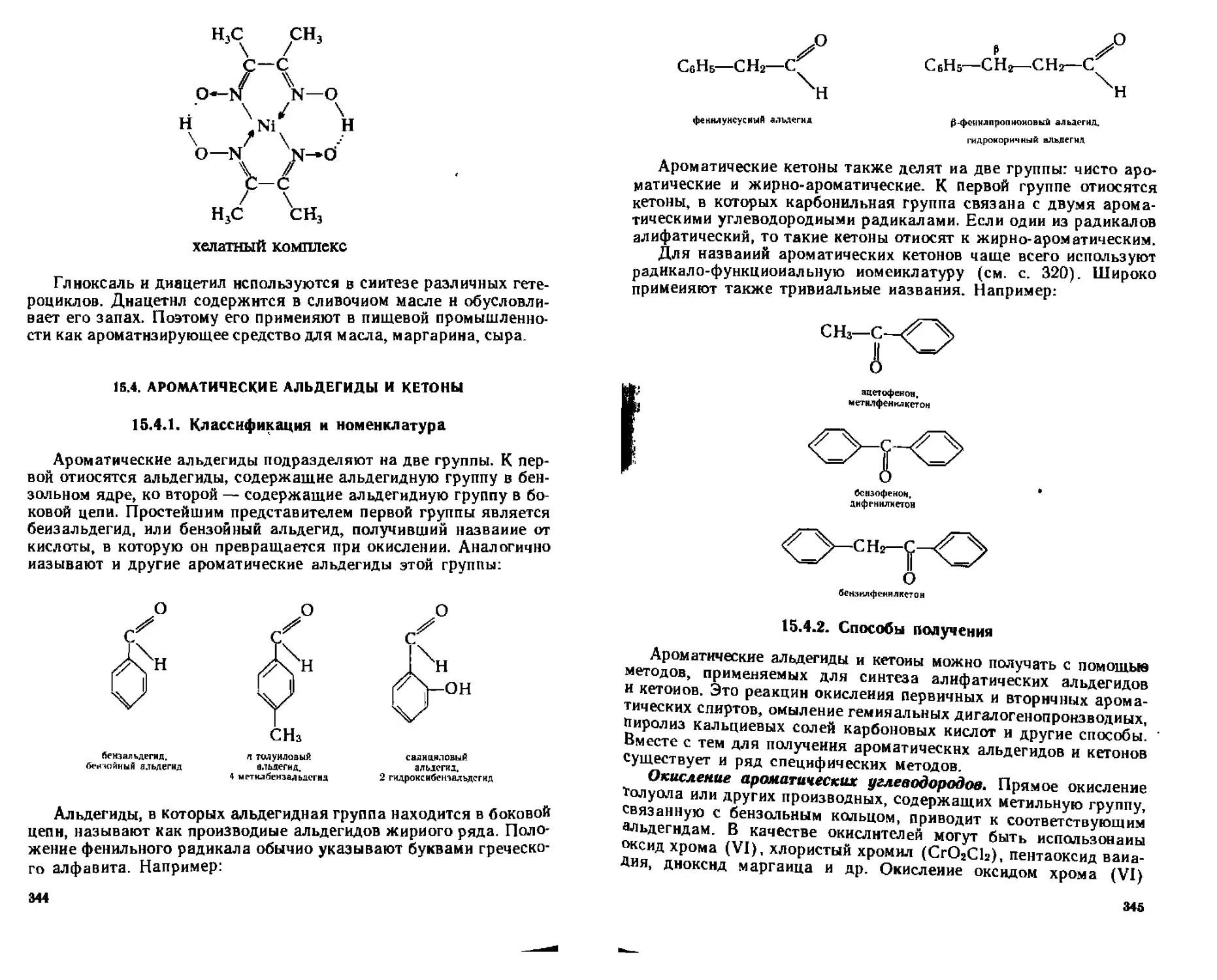
15.4.1. Классификация и номенклатура............................344
15.4.2. Способы получения..................................... 345
15.4.3. Физические свойства ....................................347
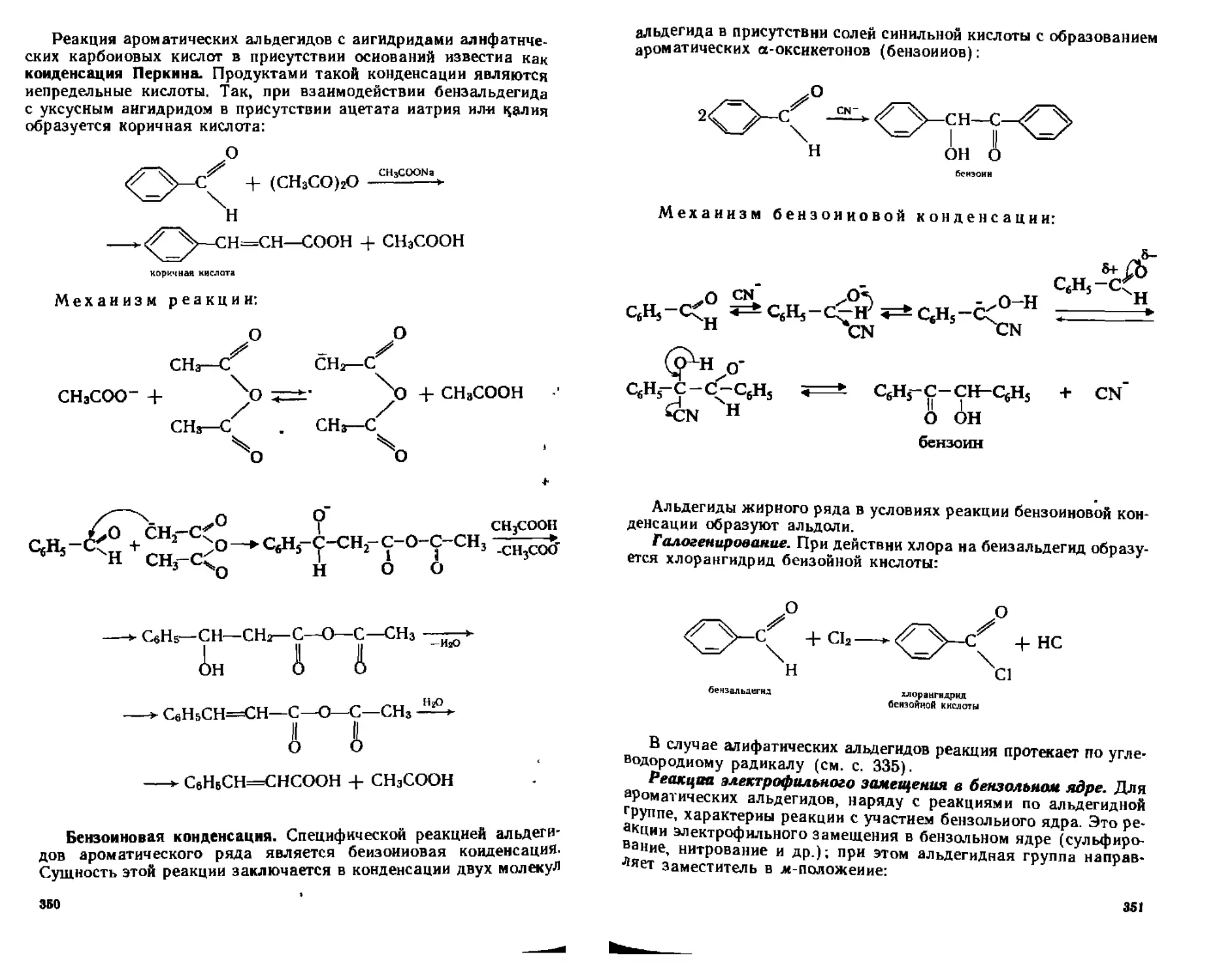
15.4.4. Химические свойства.................................... 348
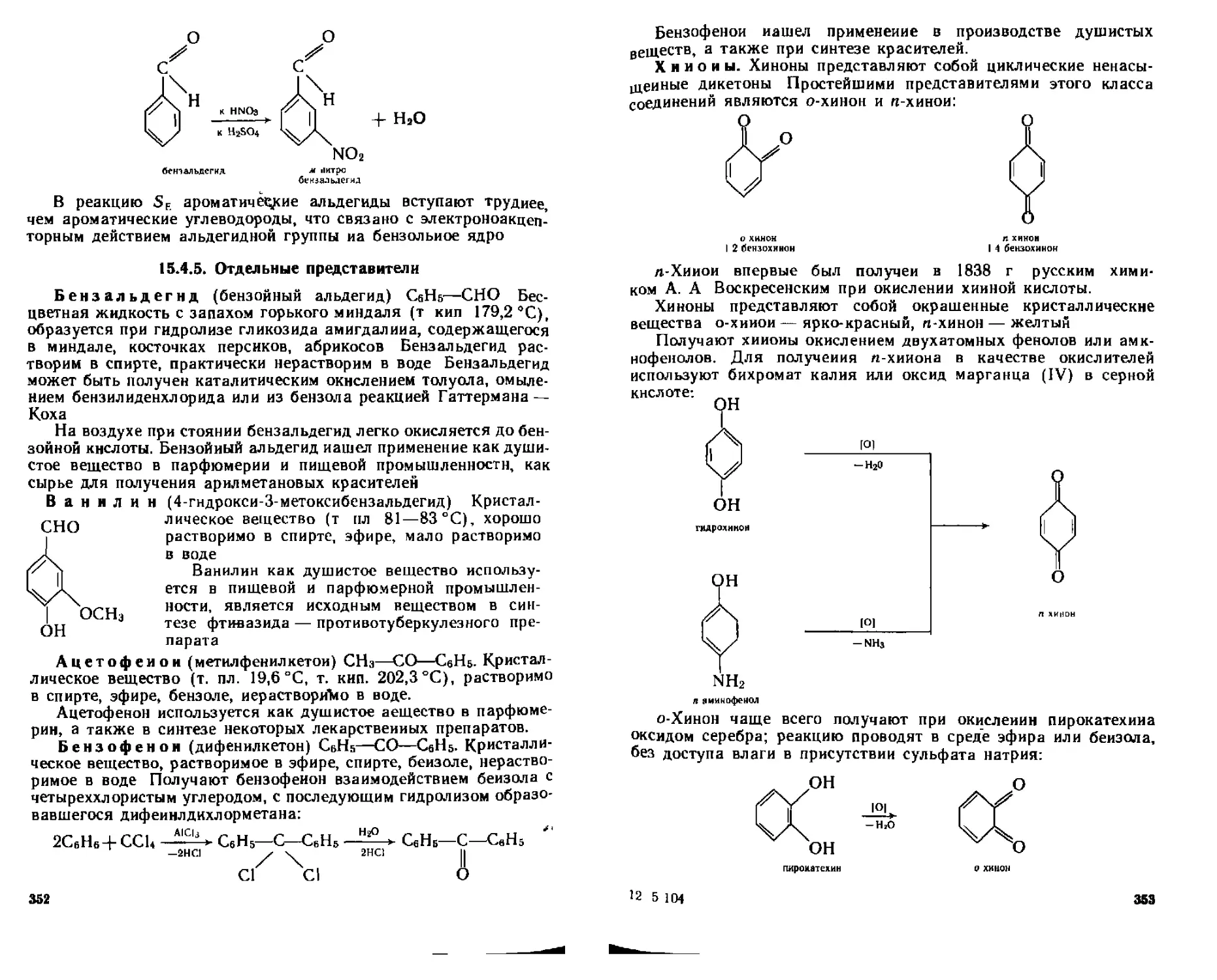
15.4.5. Отдельные представители.................................352
Контрольные вопросы и упражнения....................................355
глава 16. монокарбоновые кислоты.........................358
16.1. Насыщенные монокарбоновые кислоты.............................358
16.1.1. Номенклатура и изомерия................................ 358
16.1,2. Способы получения ......................................361
16.1,3, Физические свойства.....................................363
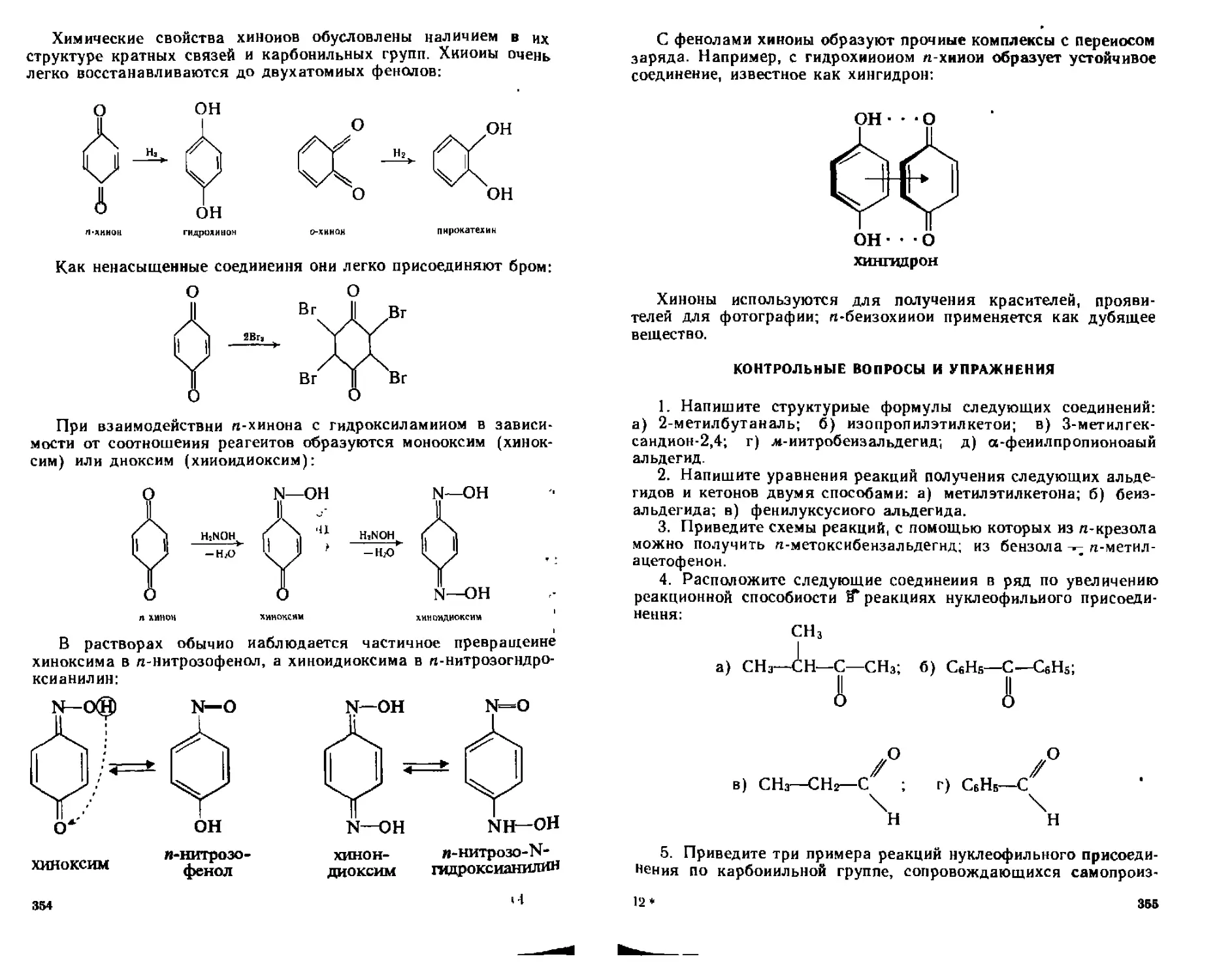
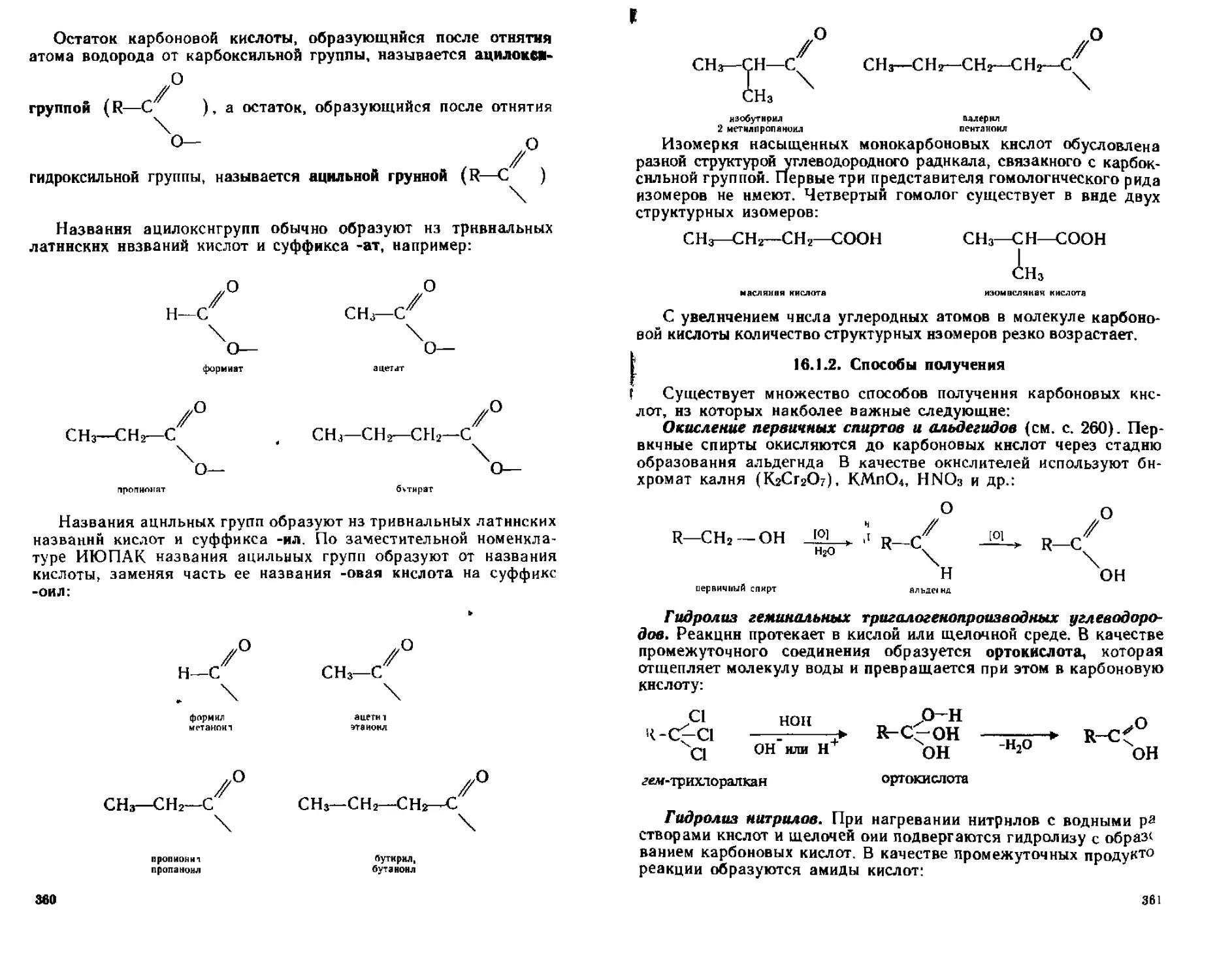
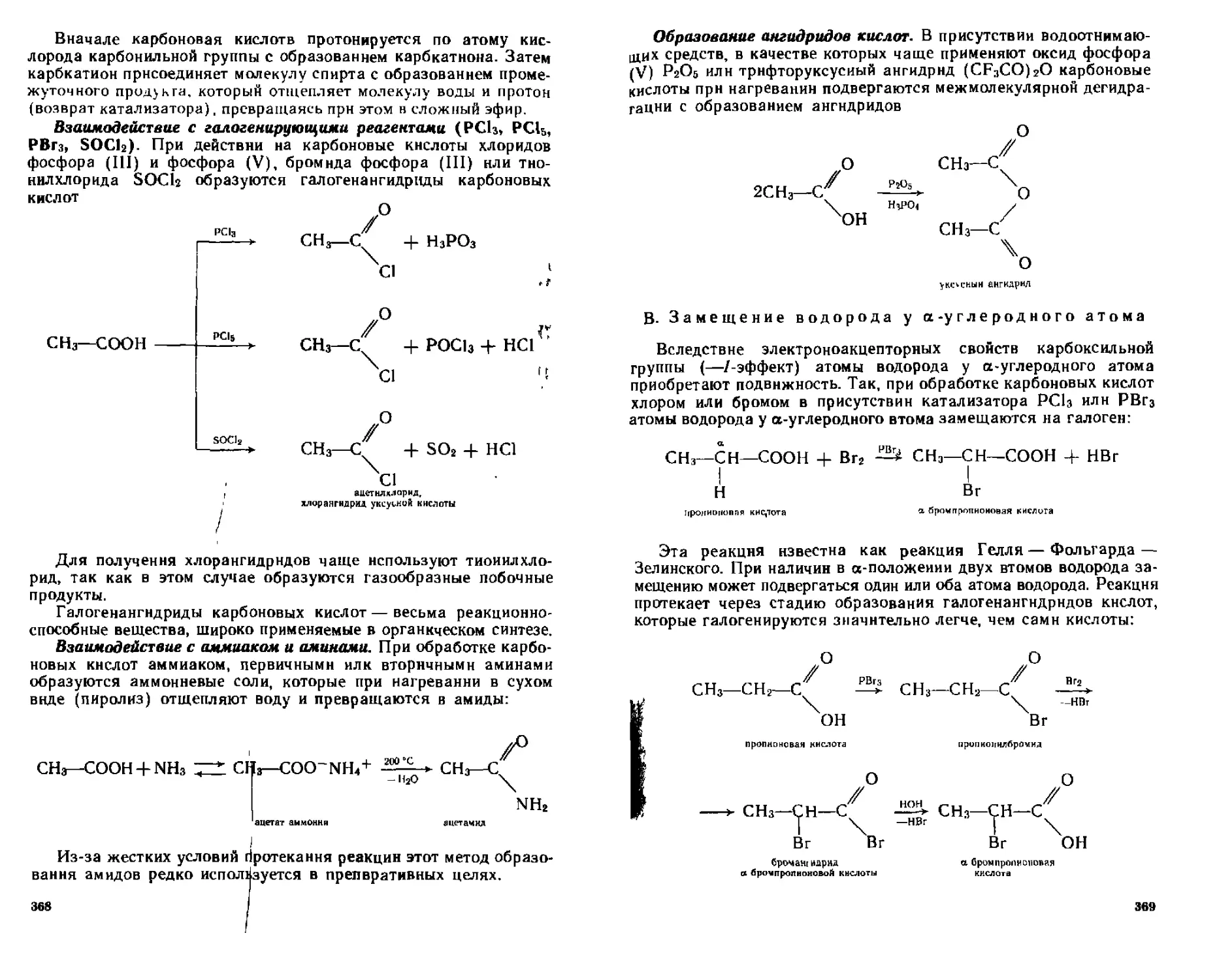
16 I 4 Химические свойства ЗЬ4
А Кислотные свойства 364
Б Реакции нуклеофильного замещении 366
В Замещение у а углеродного атома 369
Г Окисление и восстановление 370
16 1 5 От дельные представители 370
16 2 Ненасыщенные чонокарбоновые кислоты 371
16 2 1 Номенклатура и изомерия 372
16 2 2 Способы потучсния 373
16 2 3 Фи ^ические свойства * 374
16 2 4 Химические свойства 374
16 2 5 Отдельные представители 374
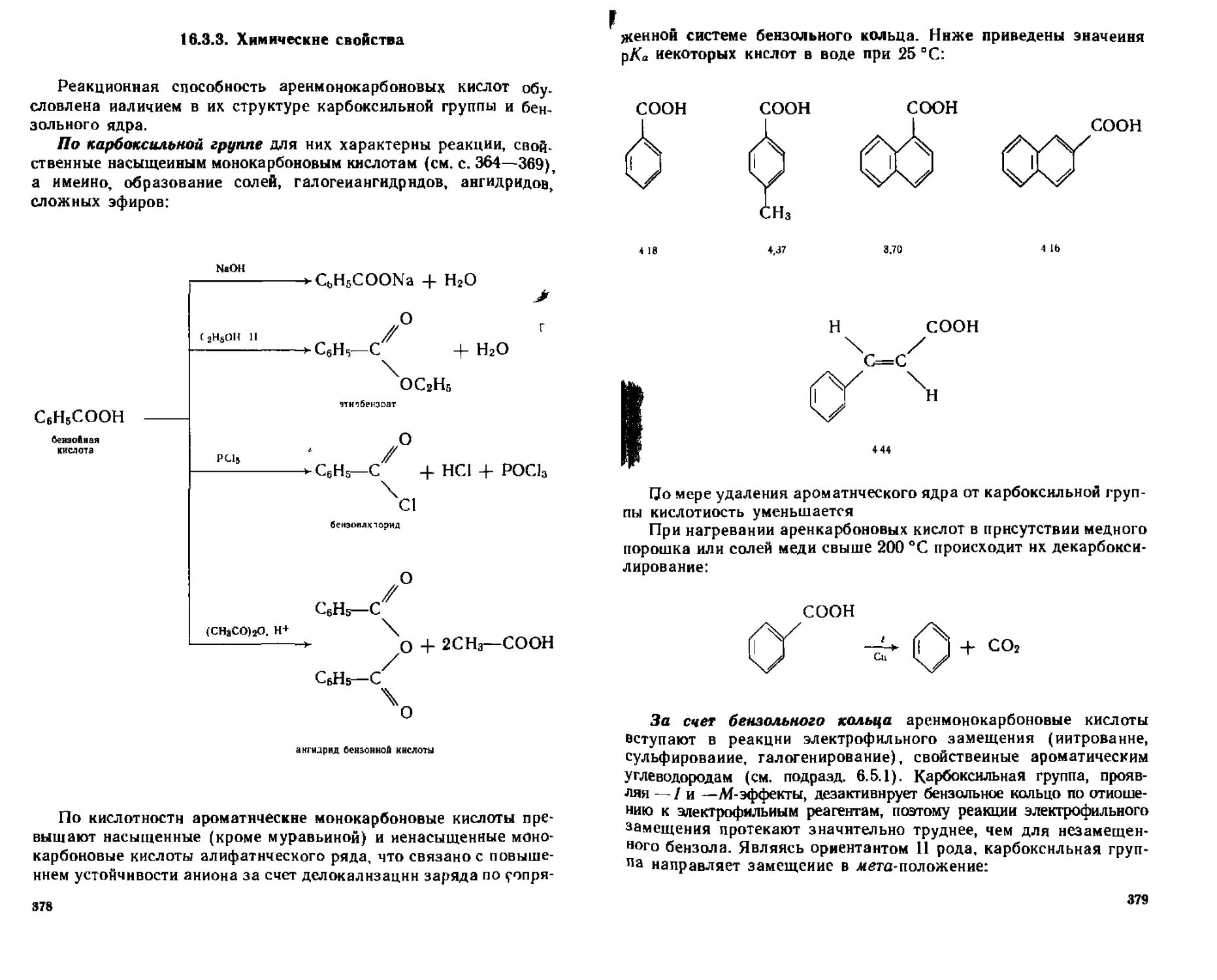
16 3 Ароматические монокарбонавые кислоты 376
16 3 1 Способы получения 377
1632 Физические свойства 377
16 3 3 Химические свойства 378
(6 3 4 Отдельные представитети 380
16 4 Идентификация монокарбоновых кислот 381
Контрольные вопросы и упражнения 382
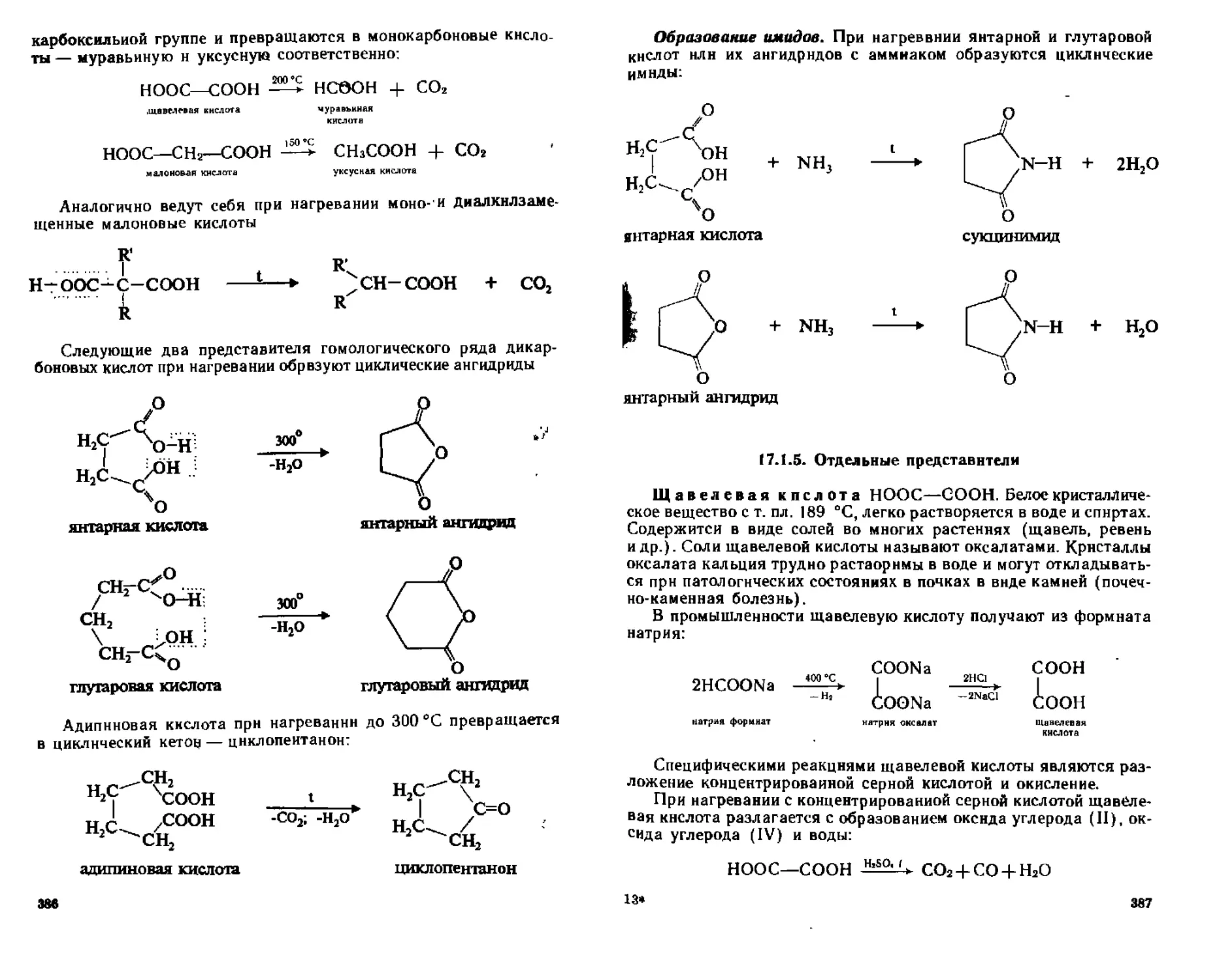
ГЛАВА 17 ДИКАРБОНОВЫЕ КИСЛОТЫ 383
17 1 Насыщенные дикарбононые кислоты 383
17 1 1 Номенклатура и изомерия 383
17 12 Способы получения 384
17 13 Физические свойства 384
17 14 Химические свойства 387
17 15 Отдельные представители 387
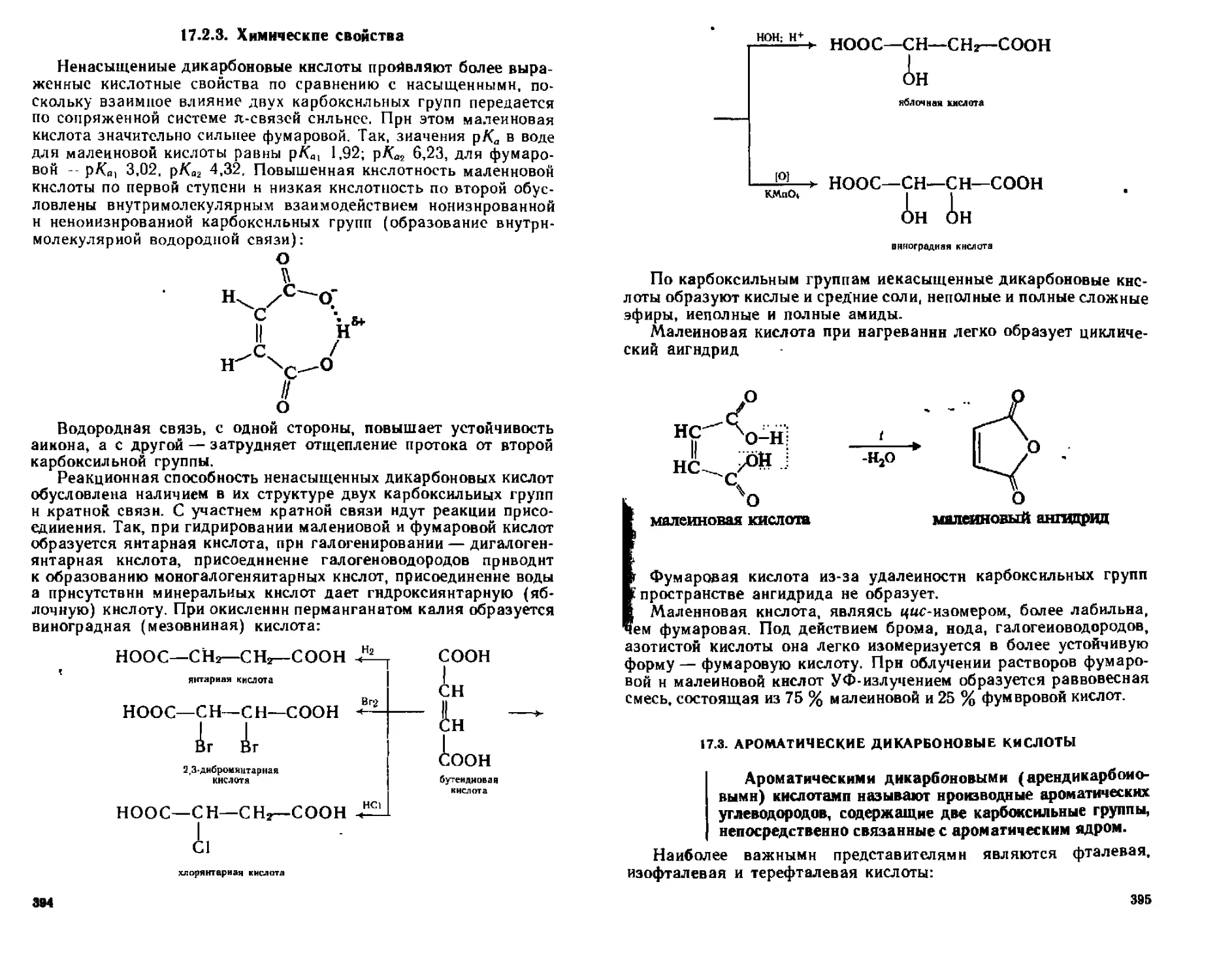
17 2 Ненасыщенные гнкарбоновыс кислоты 392
17 2 1 Способы получения 392
17 2 2 Фи ж>дескис свойства 393
17 2 3 Химические свойства 394
17 3 Ароматические дикарбоновые кислоты 395
17 3 1 Способы получения 396
17 3 2 физические и химические свойства 397
17 4 Идентификация дикарбоновых кислот 398
Контрольные вопросы и упражнения 398
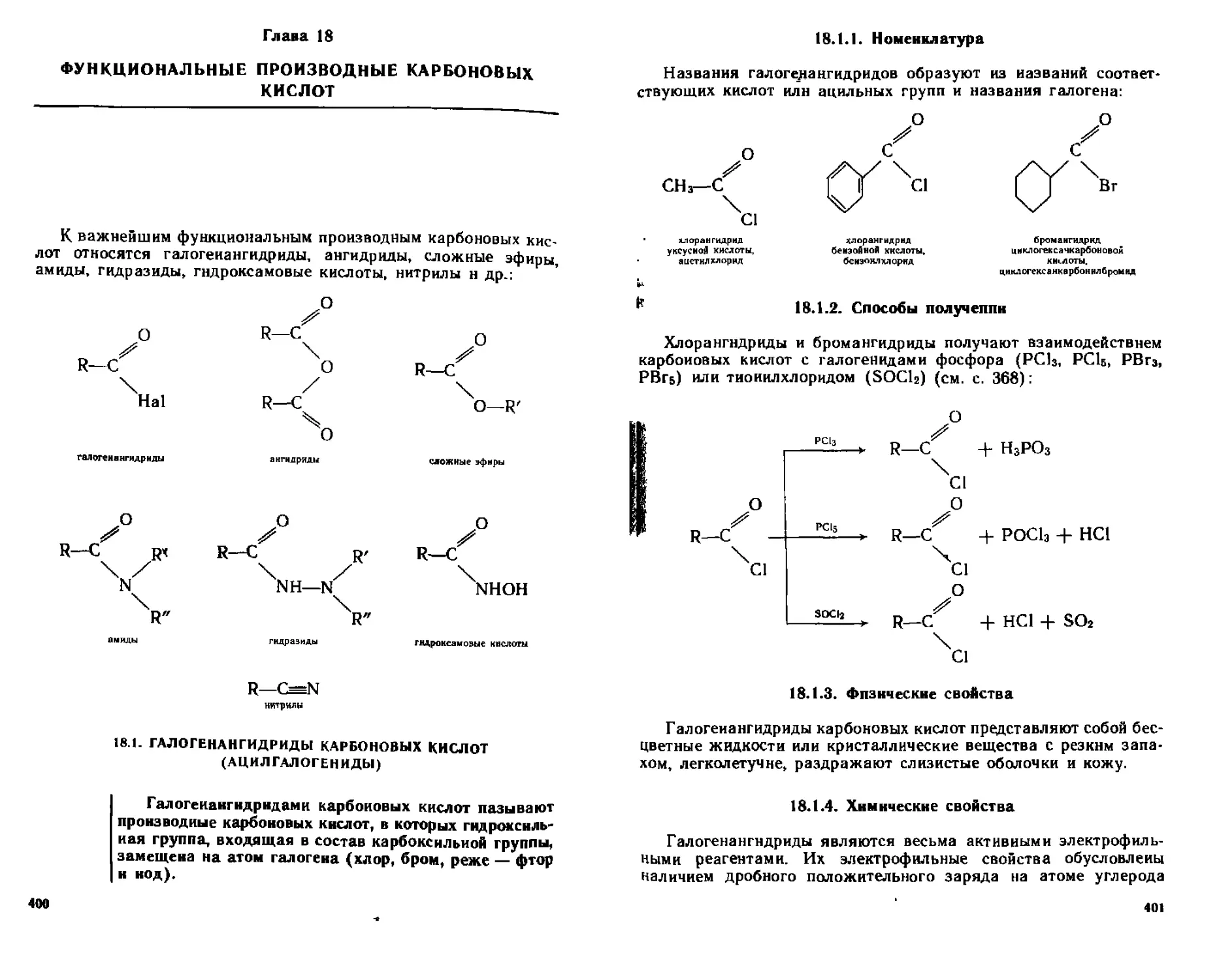
ГЛАВА 18 ФУНКЦИОНАЛЬНЫЕ ПРОИЗВОДНЫЕ КАРБОНОВЫХ
кислот 400
18 1 Галогеналгидриды карбоновых кислот (ацил галоген иды} 400
18 1 1 Номенклатура 401
[8 12 Способы потучсния 401
18 1 3 Фи шческис свойства 401
18 14 Химические свойства 401
18 15 Отдельные представители 403
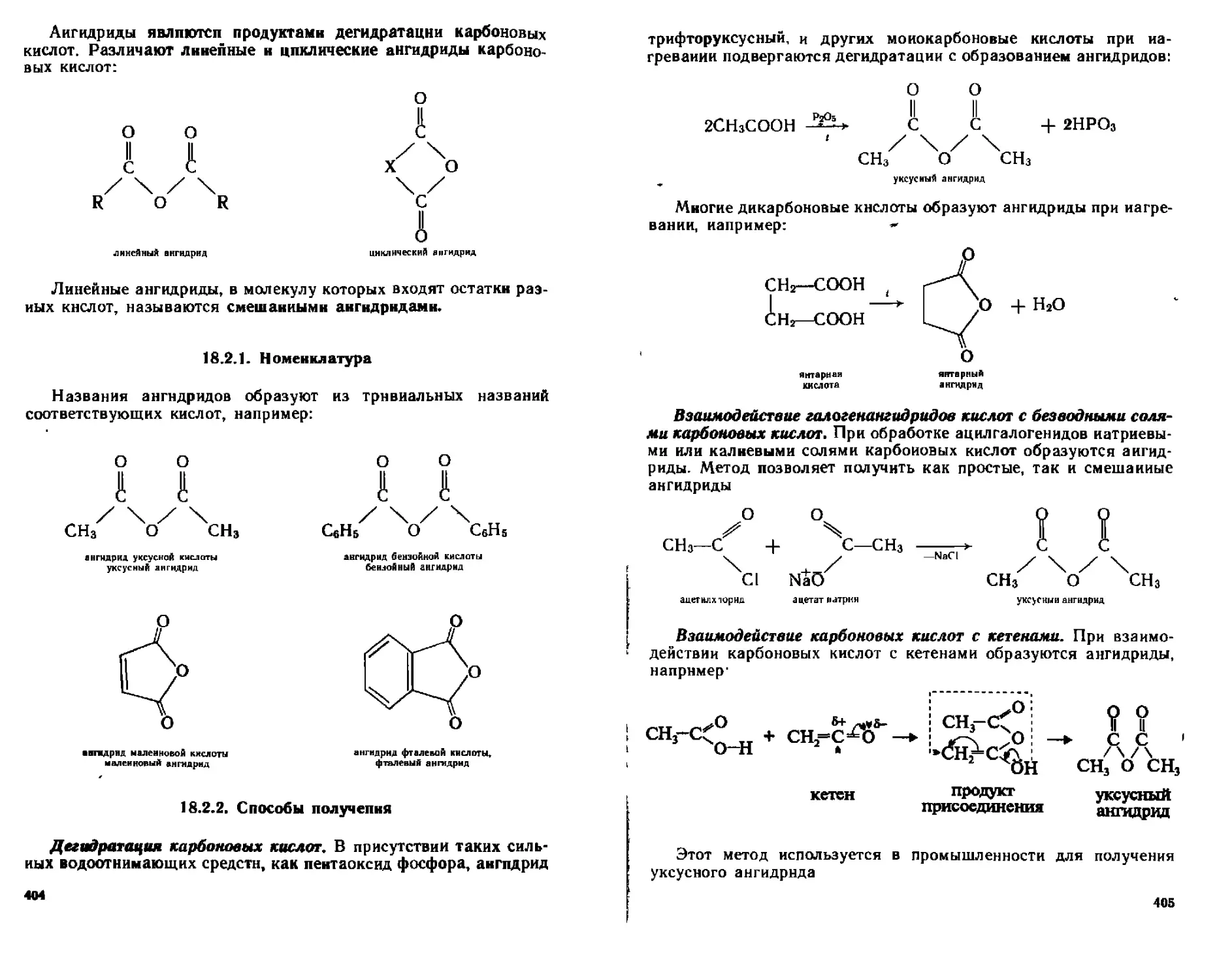
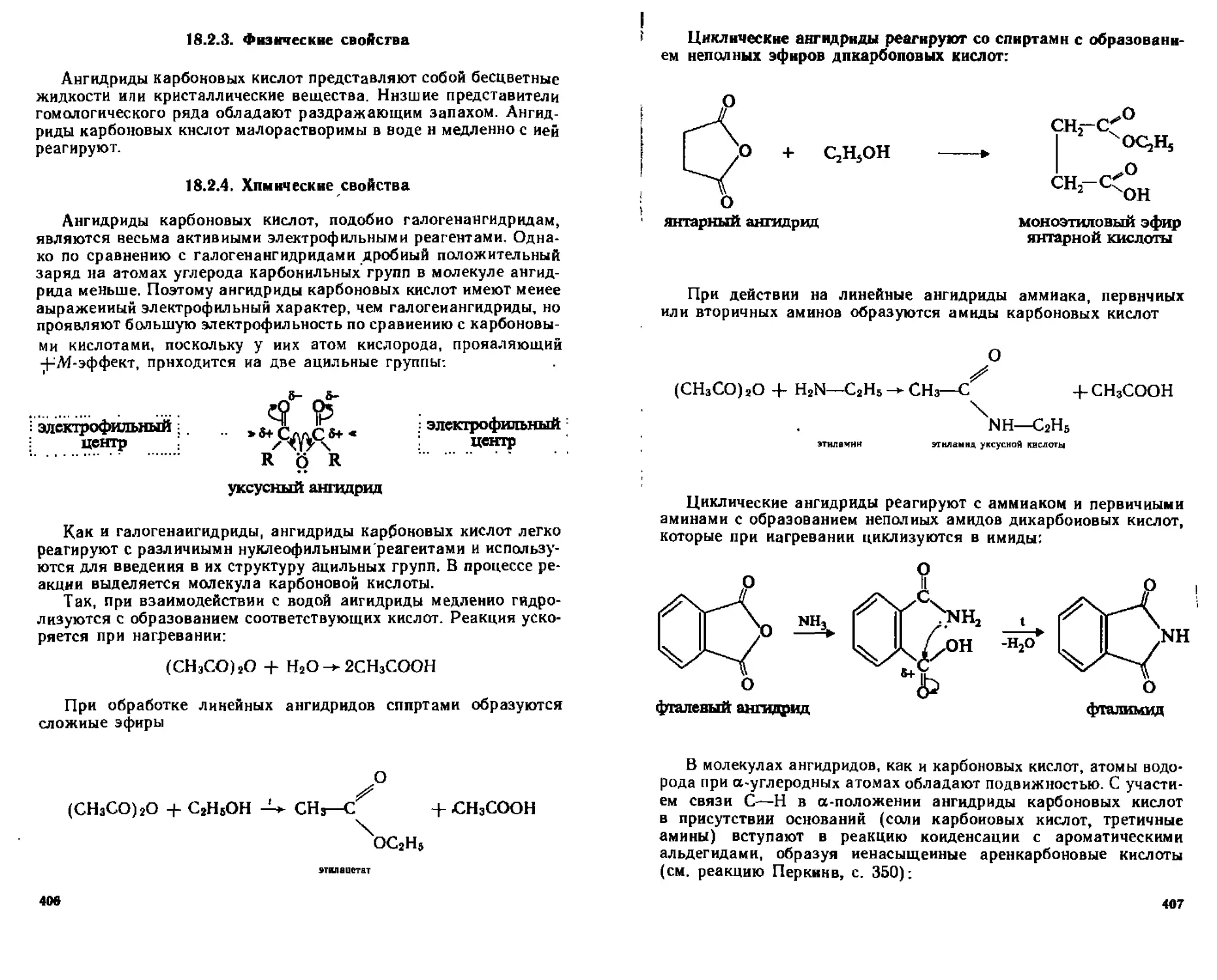
18 2 Ангидриды карбоновых кислот 403
18 2 1 Номенклатура 404
18 2 2 Способы получения 404
18 2 3 Фи жческие свойства 406
18 2 4 Химические свойства 406
18 2 5 Отдетьныс представиюпи 408
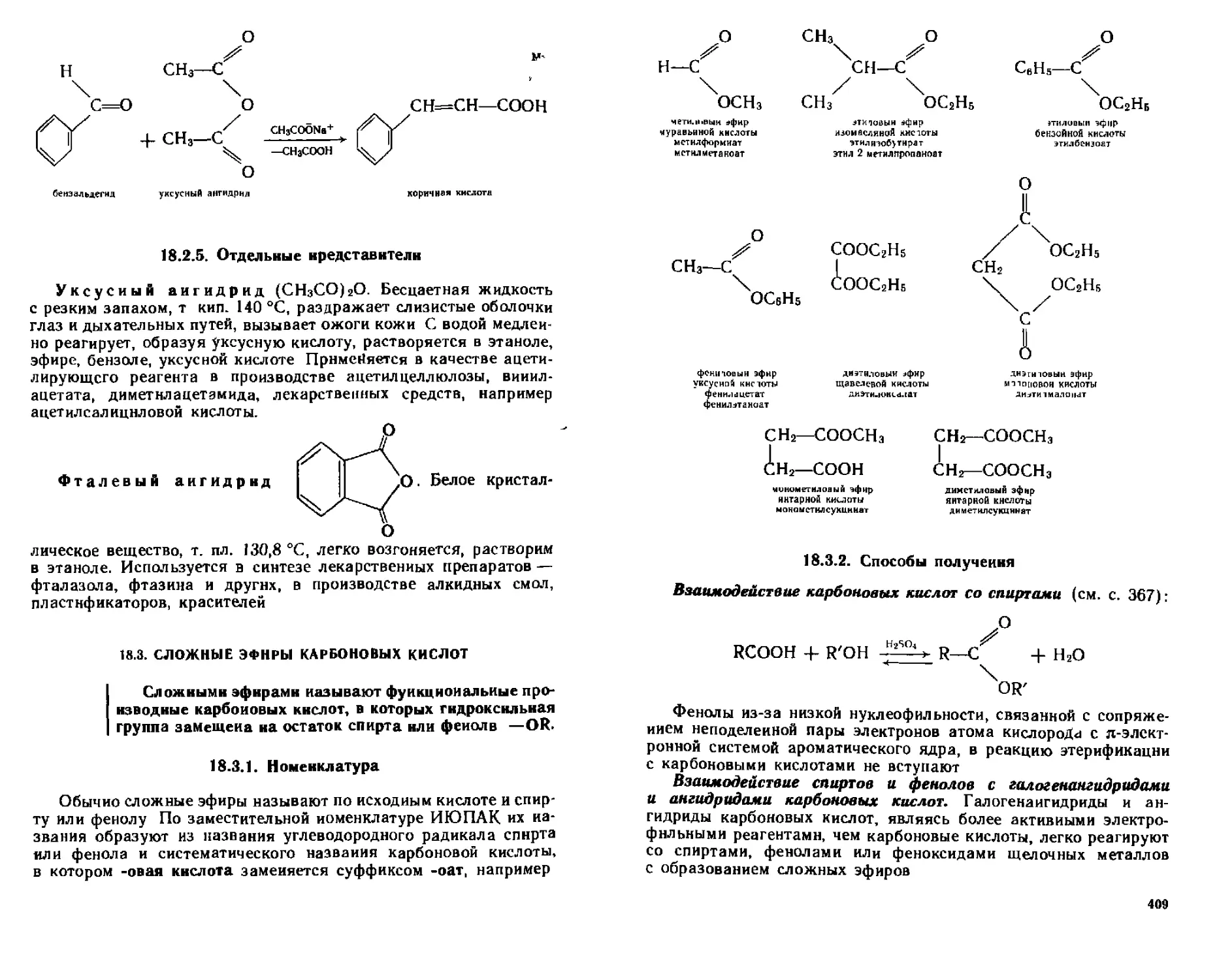
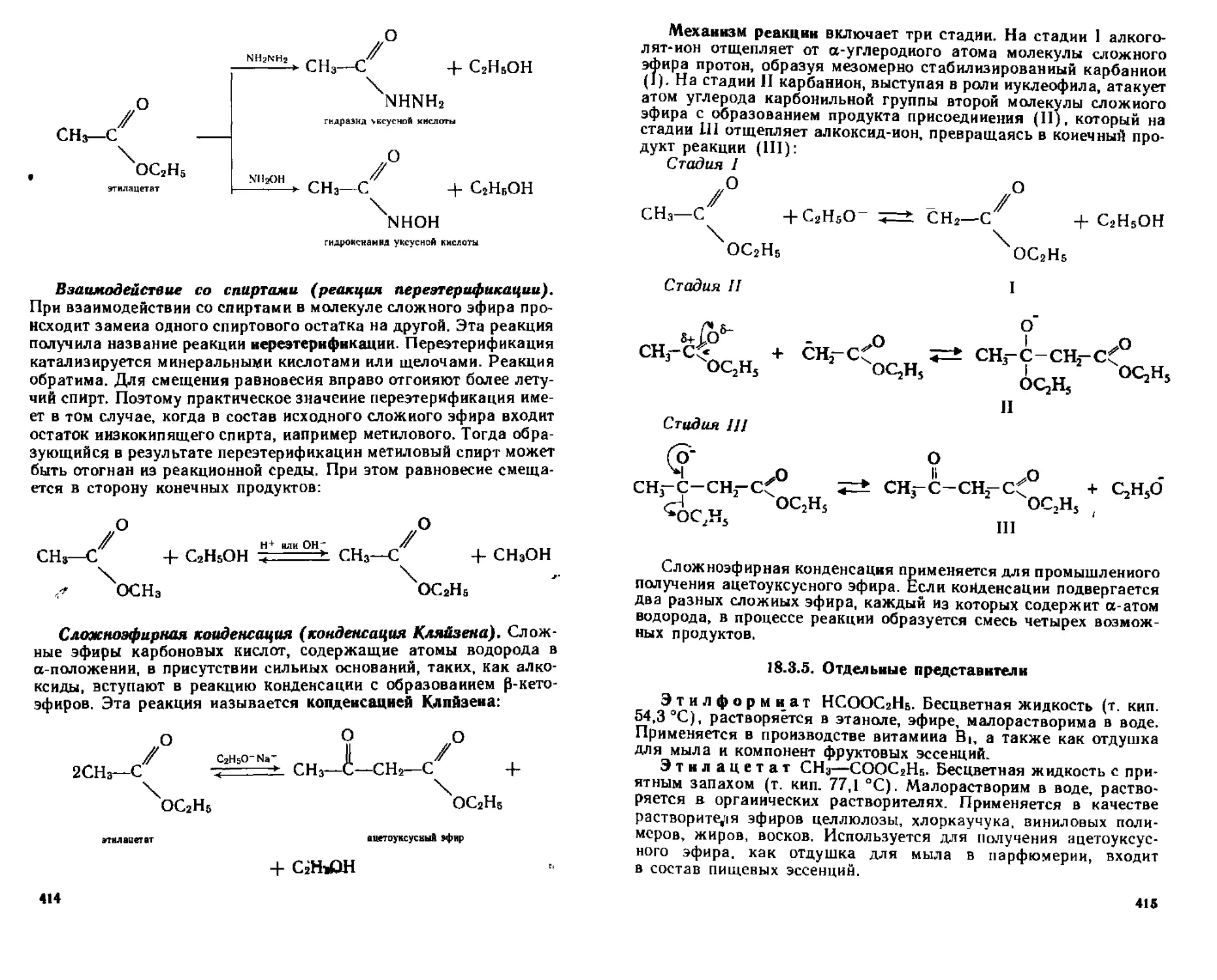
18 3 Сложные эфиры карбоновых кислот 408
183 1 Номенклатура 408
18 3 2 Способы получения 409
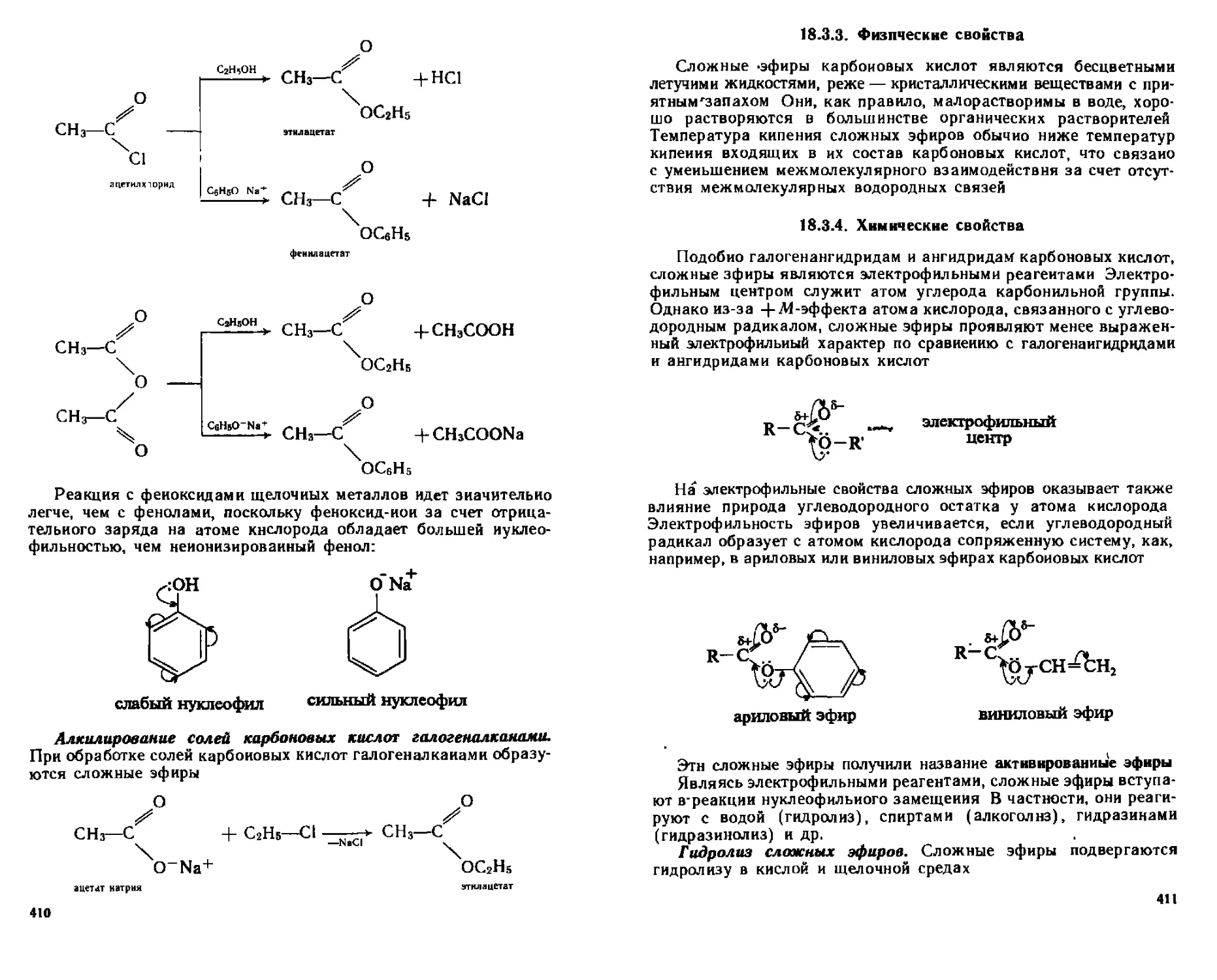
18 3 3 Фи 1 и чес кие свойства 411
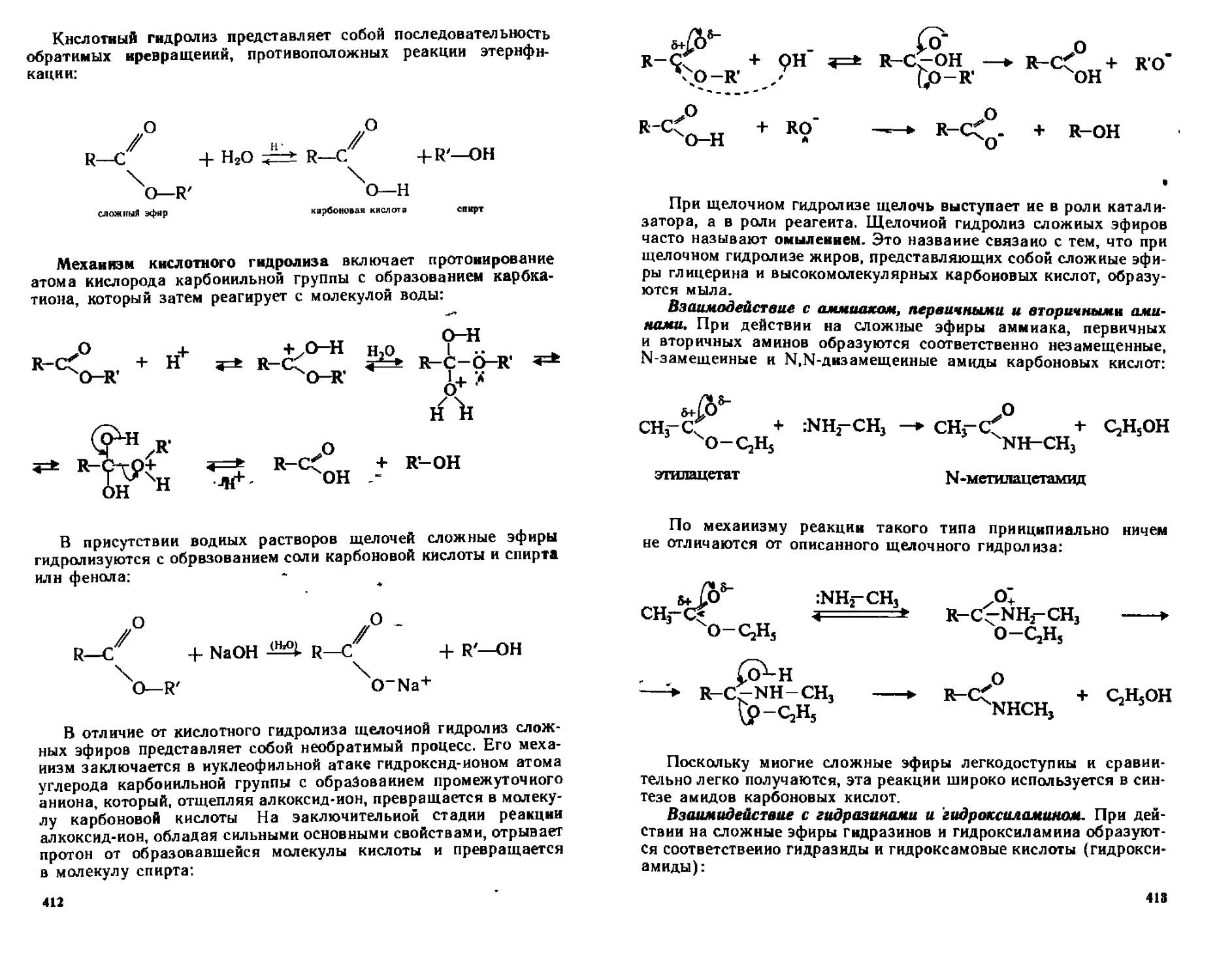
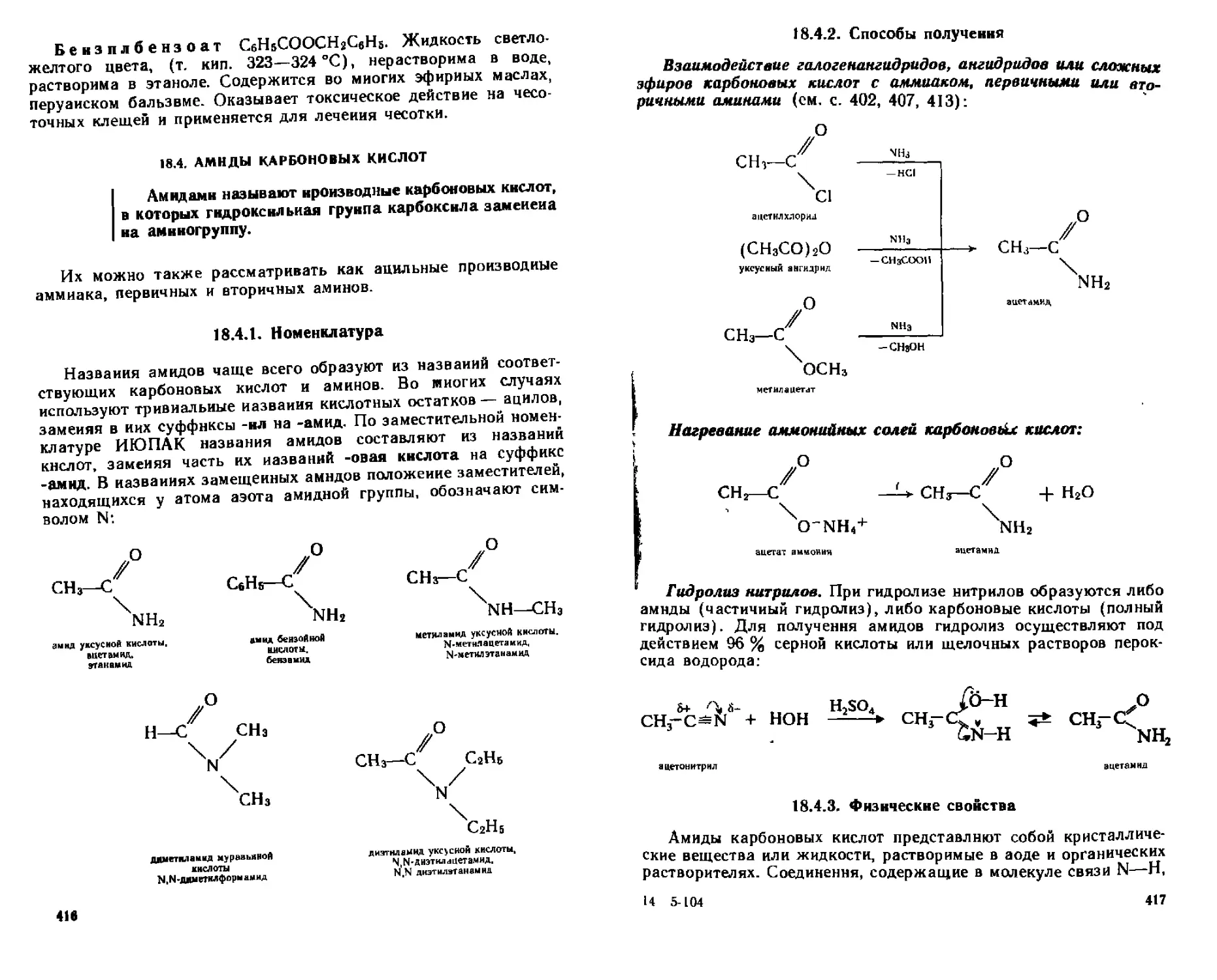
18 3 4 Химические свойства 41 1
18 3 5 Отдельные представители 415
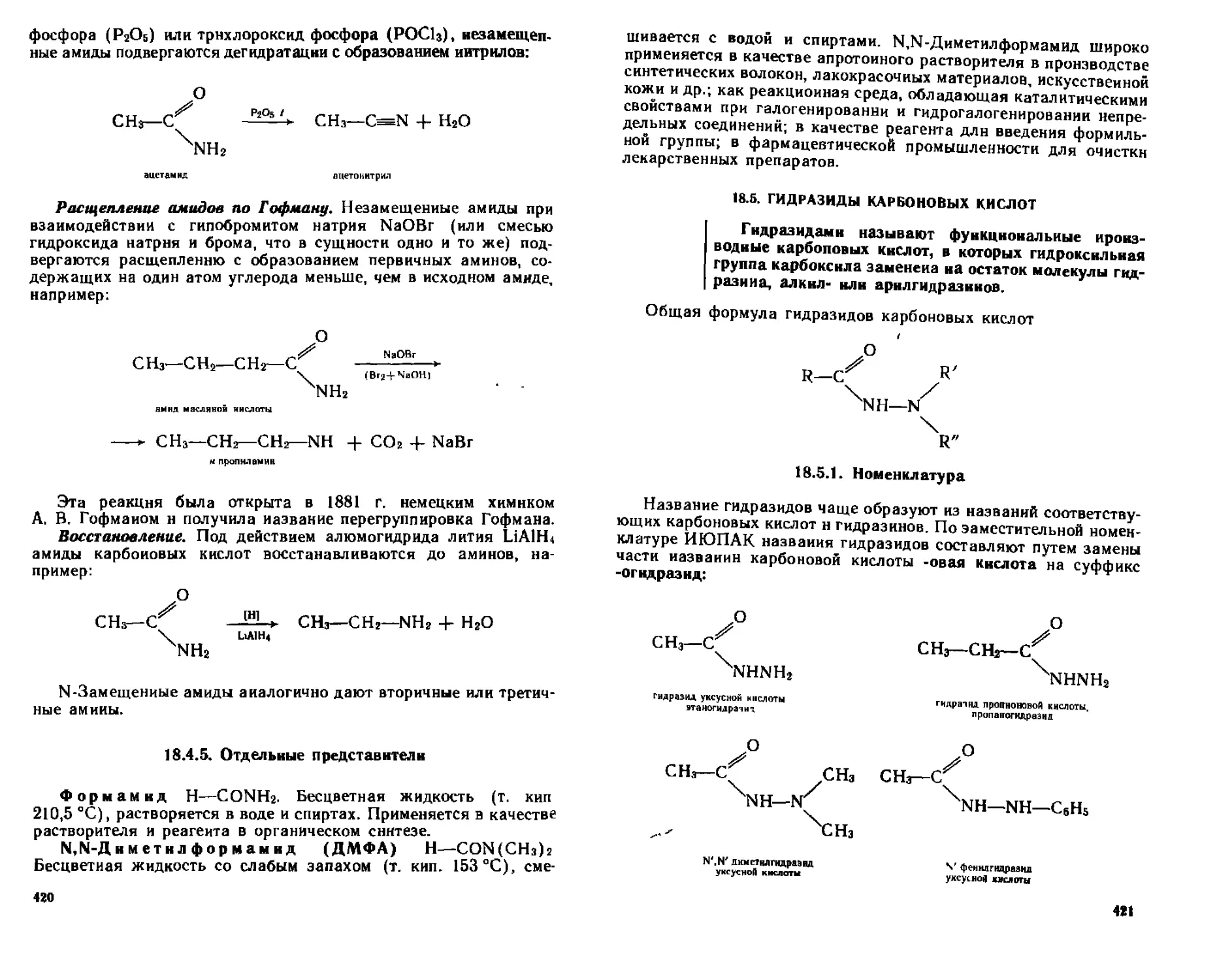
18 4 Амиды карбоновых кислот 416
18 4 1 Номенклатура 416
8
18 4 2 Способы получения 417
[8 4 3 Физические свойства 417
1844 Химические свойства 418
[8 4 5 Отдельные представители 420
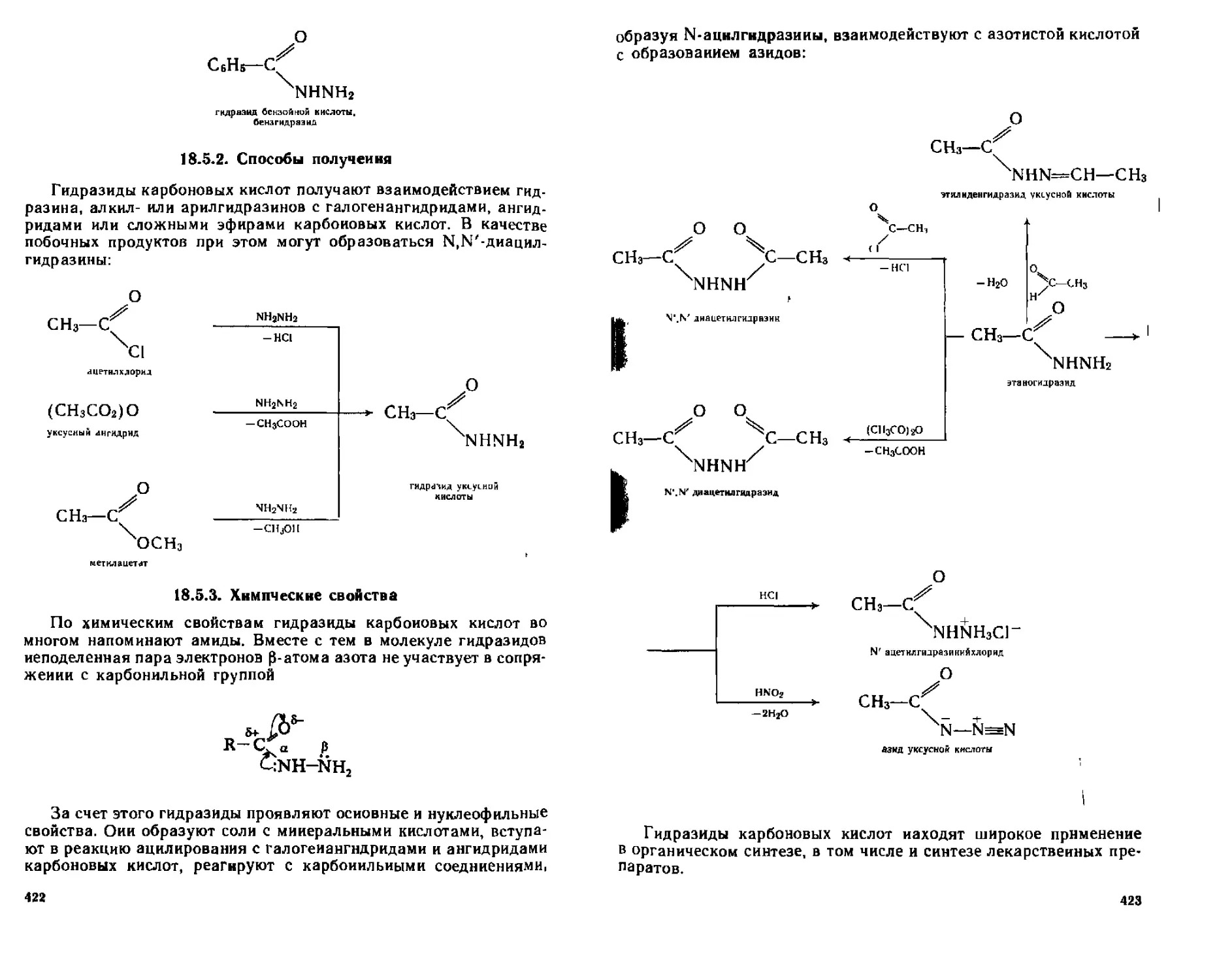
18 5 Гидразиды карбоновых кислот 421
18 5 1 Номенклатура 421
18 5 2 Способы получения 422
18 5 3 Химические свойства 422
18 б Нитрилы (цианиды) 424
(8 6 I Номенклатура 424
18 6 2 Способы получения 424
18 6 3 Химические свойства 425
18 6 4 Отдельные представители 426
18 7 Идентификация функциональных производных карбоновых кислот 426 кислот 426
Контрольные вопросы и упражнения 428
ГЛАВА 19 ГЕТЕРОФУНКЦИОНАЛЬНЫЕ КАРБОНОВЫЕ
КИСЛОТЫ 429
19 1 Г илогенокарбоновые кислоты 429
|9 I 1 Номенклатура 429
|9 1 2 Способы получения 430
19 13 Физические и химические свойства 431
19 1 4 Отдельные представители 434
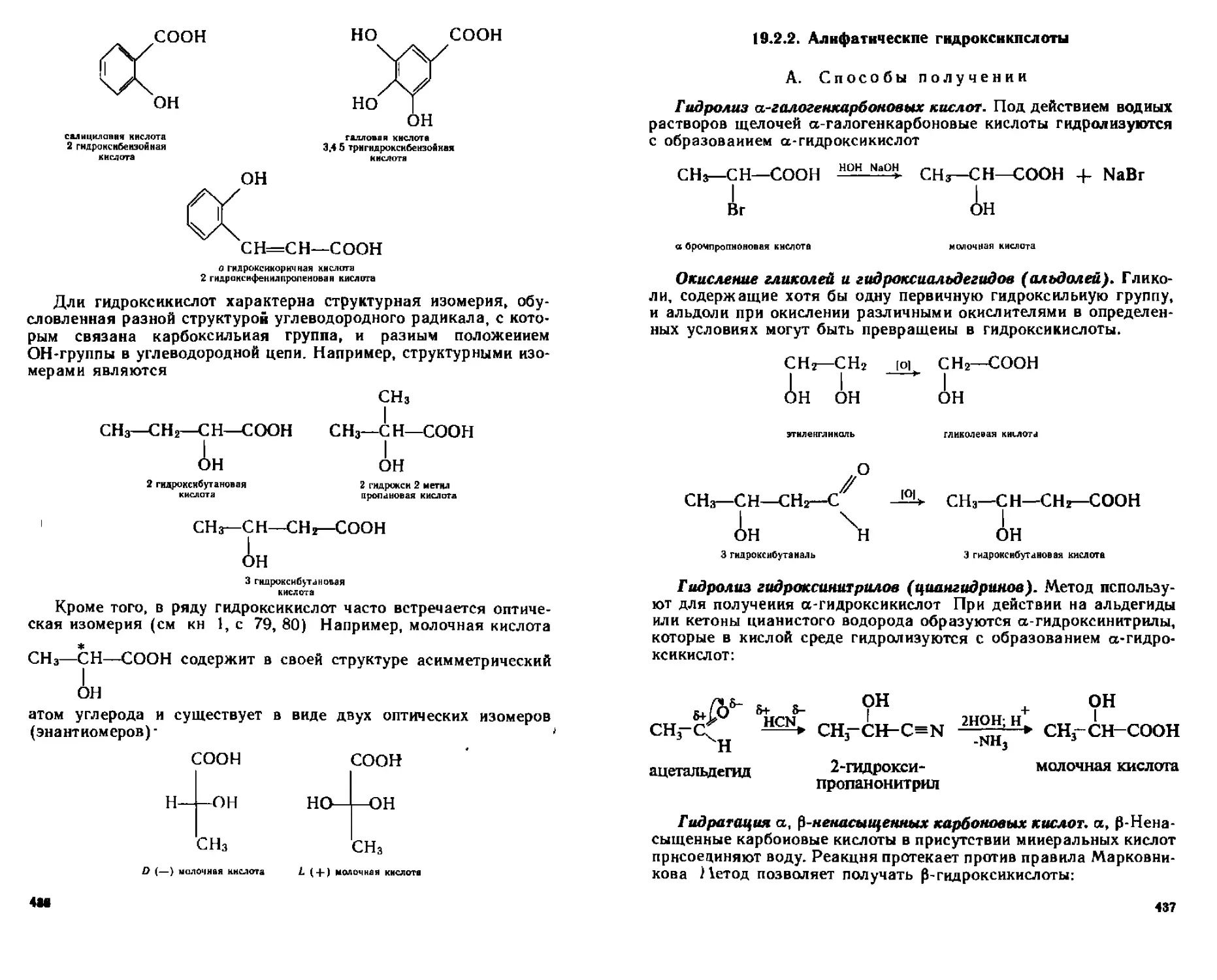
19 2 Гидрокси кислоты 434
19 2 1 Номенклатура изомерия 435
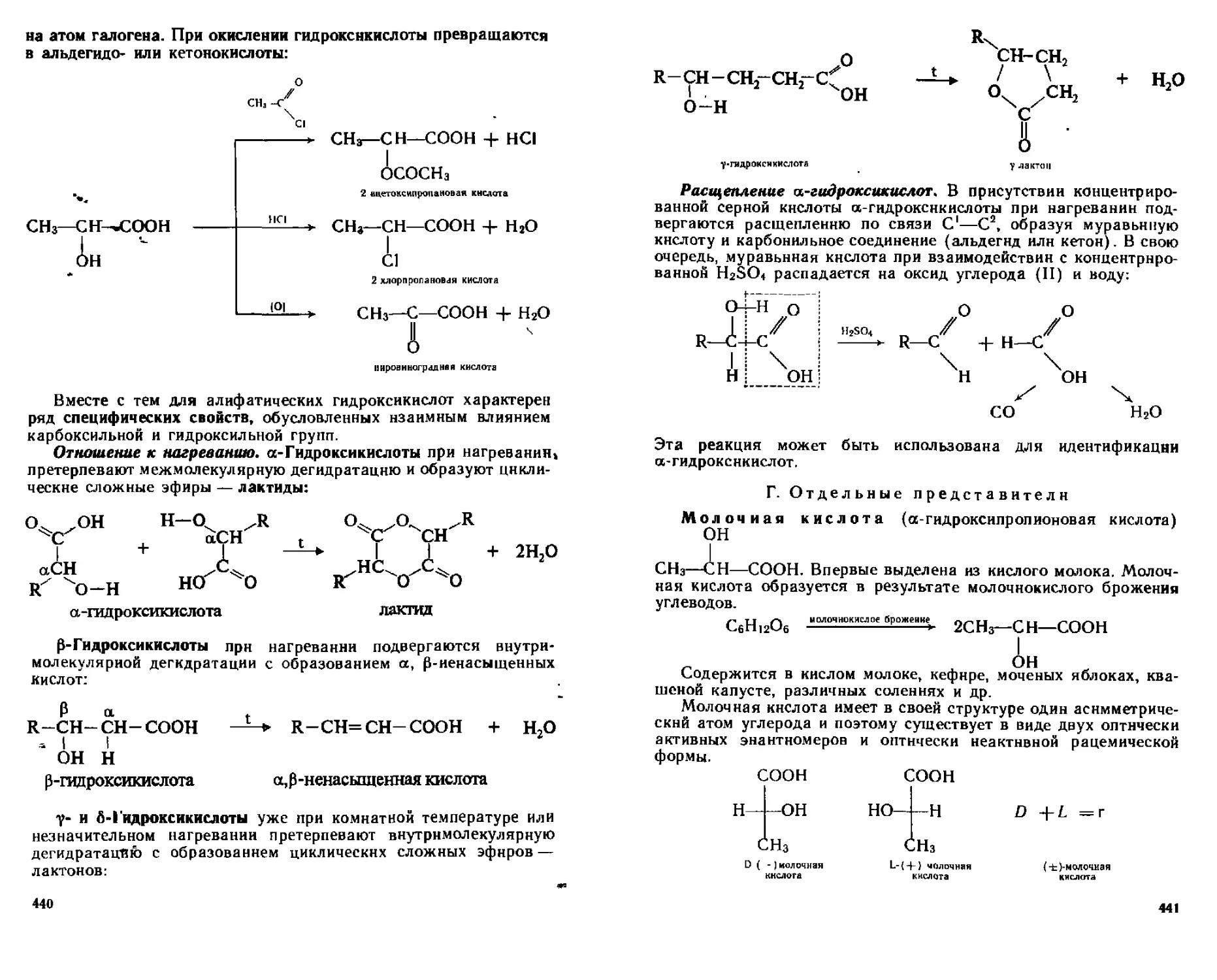
19 2 2 Алифатические гидроксикислоты 437
А Способы получения 437
Б Физические свойства 438
В Химические свойства 438
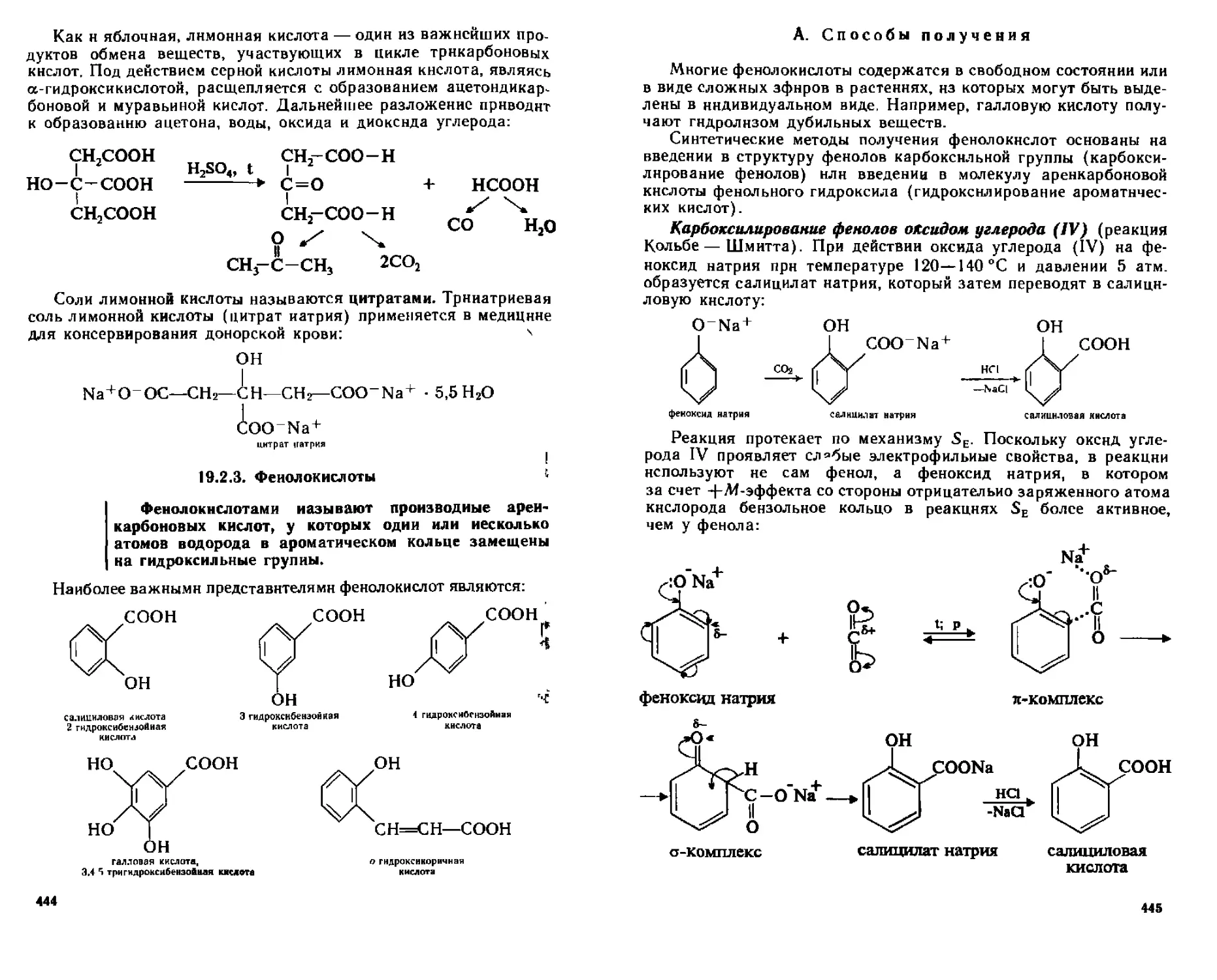
Г Отдельные представители 441
19 2 3 Феполокислоты 444
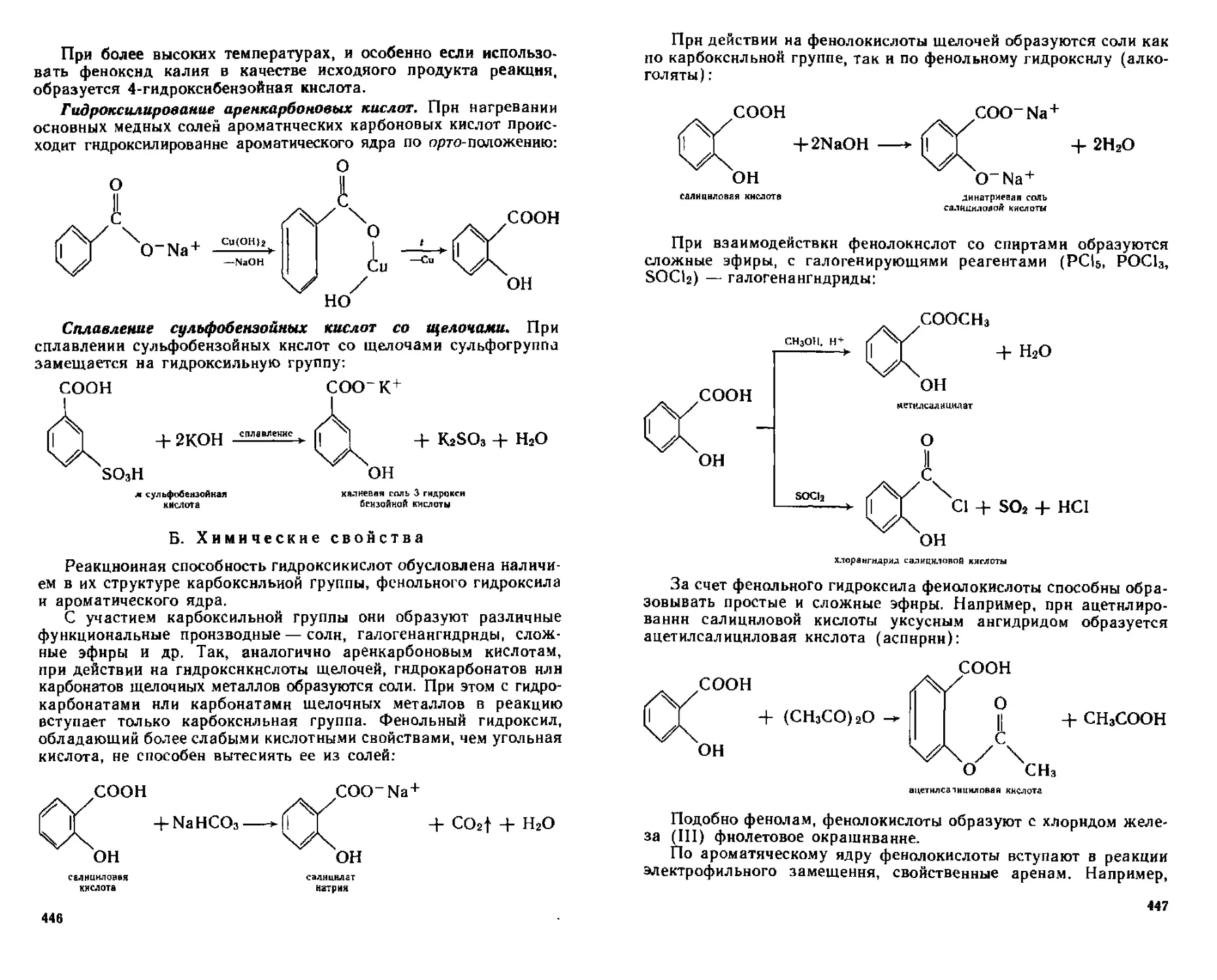
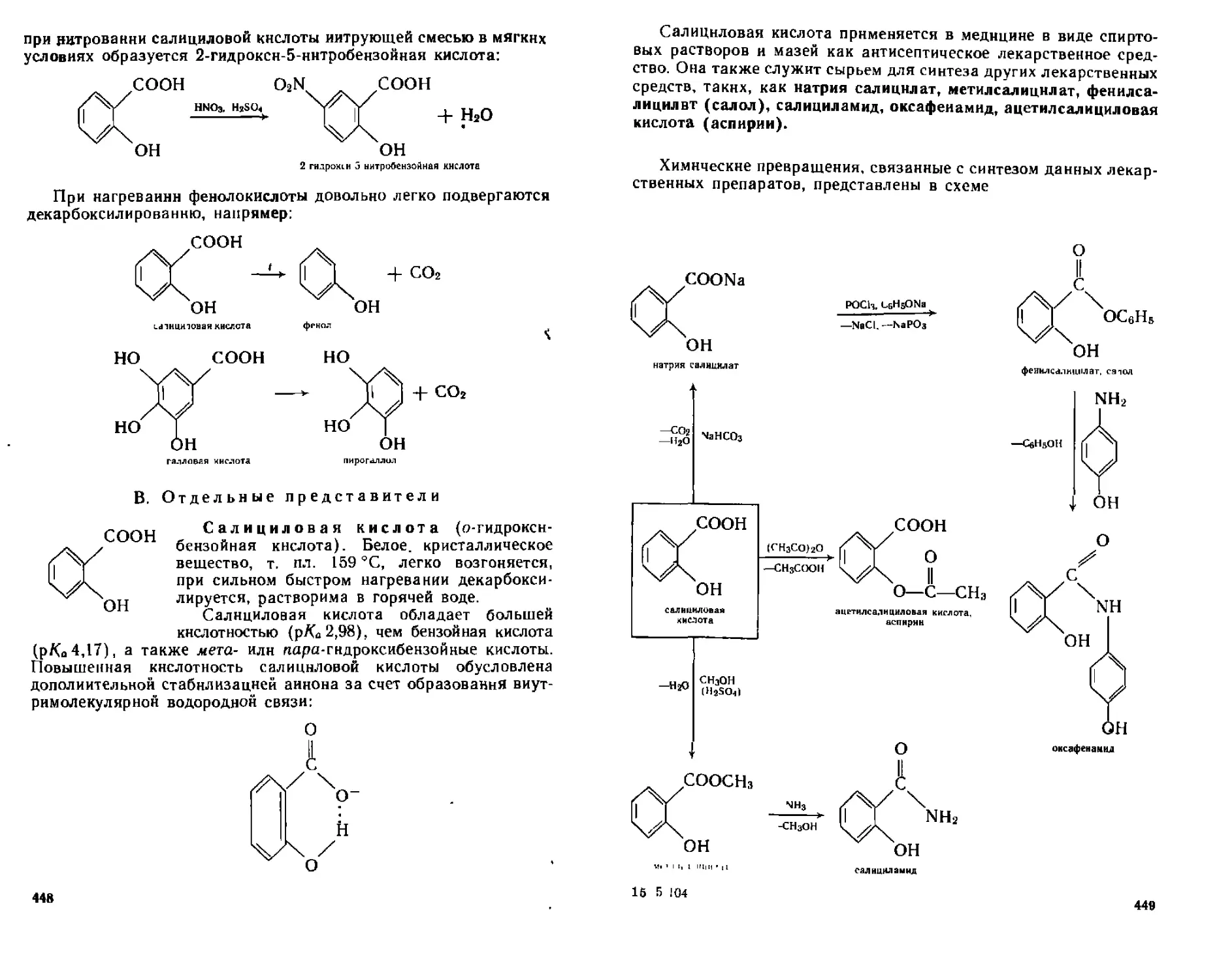
А Способы получения 445
Б Химические свойства 446
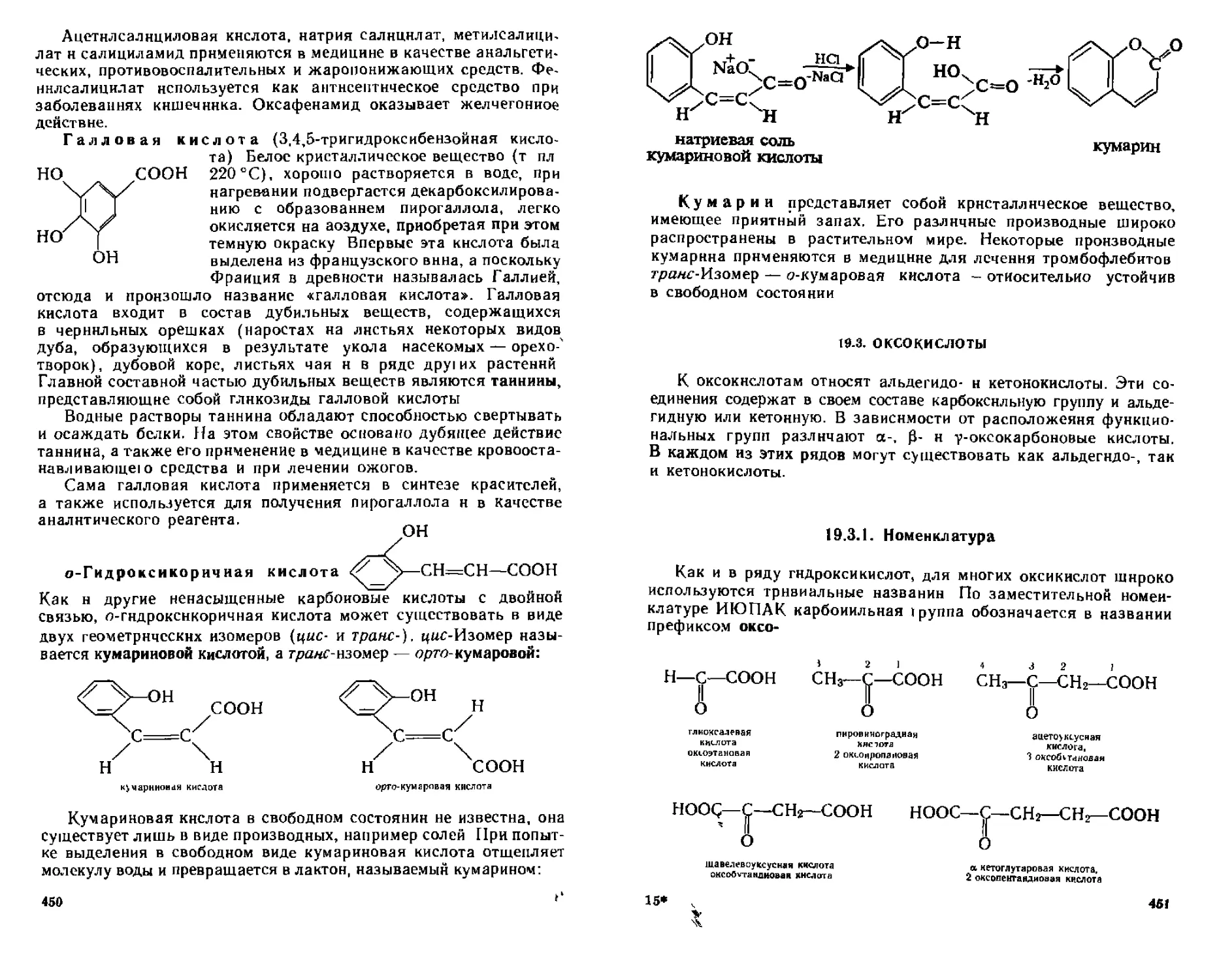
В Отдельные представители 448
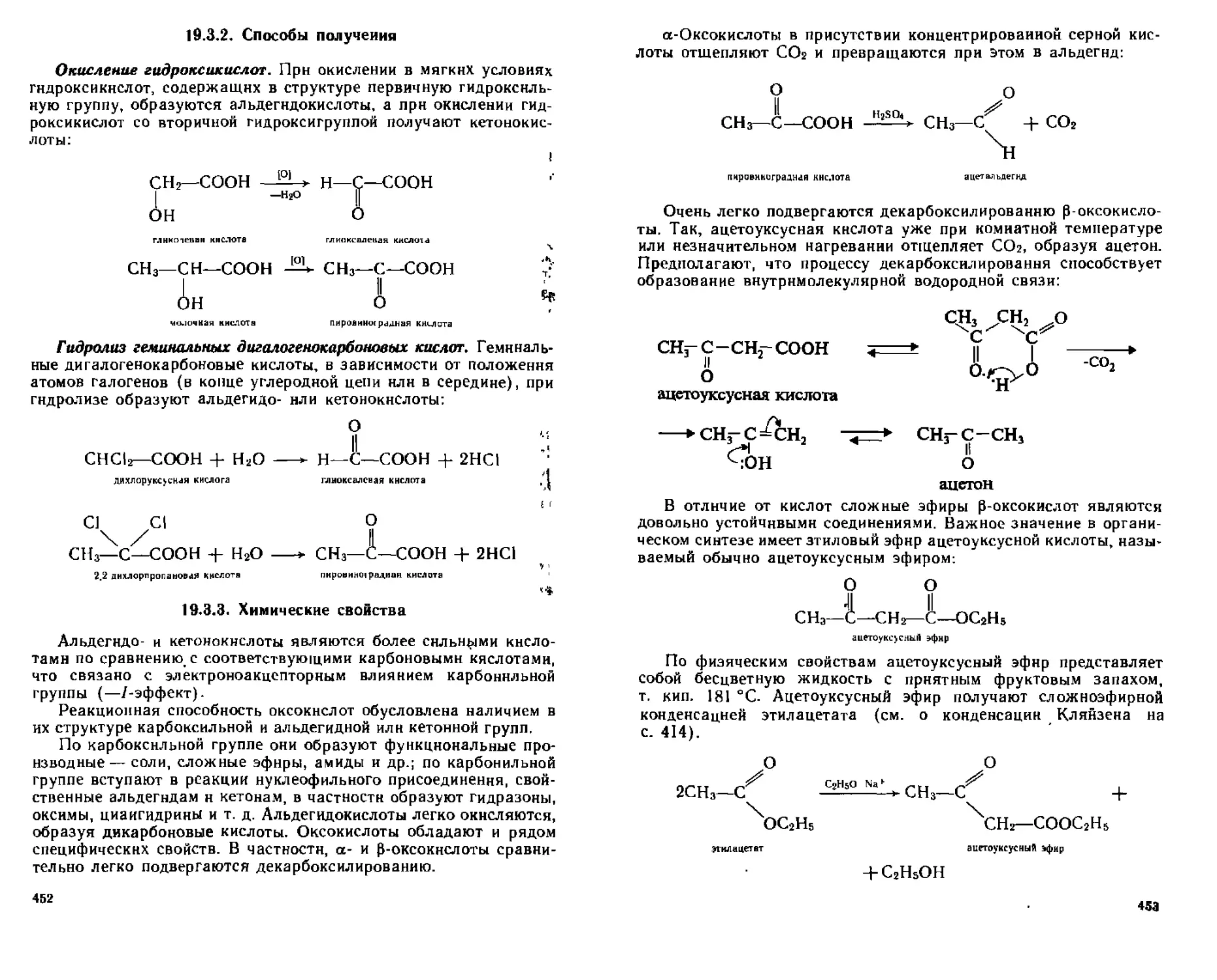
19 3 Оксокислоты 451
19 3 1 Номенклатура 451
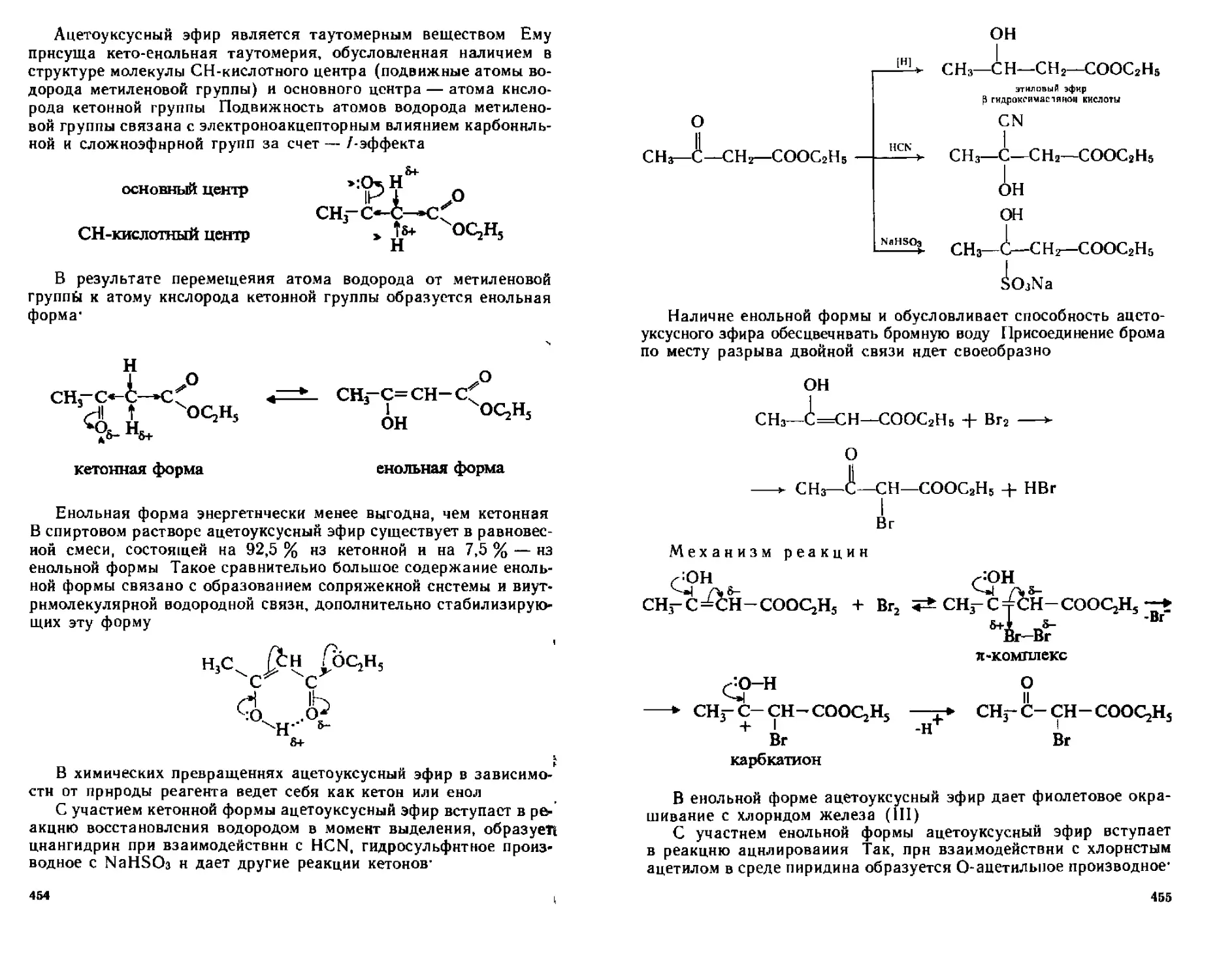
19 3 2 Способы получения 452
19 3 3 Химические свойства 452
19 3 4 Отдельные представители 458
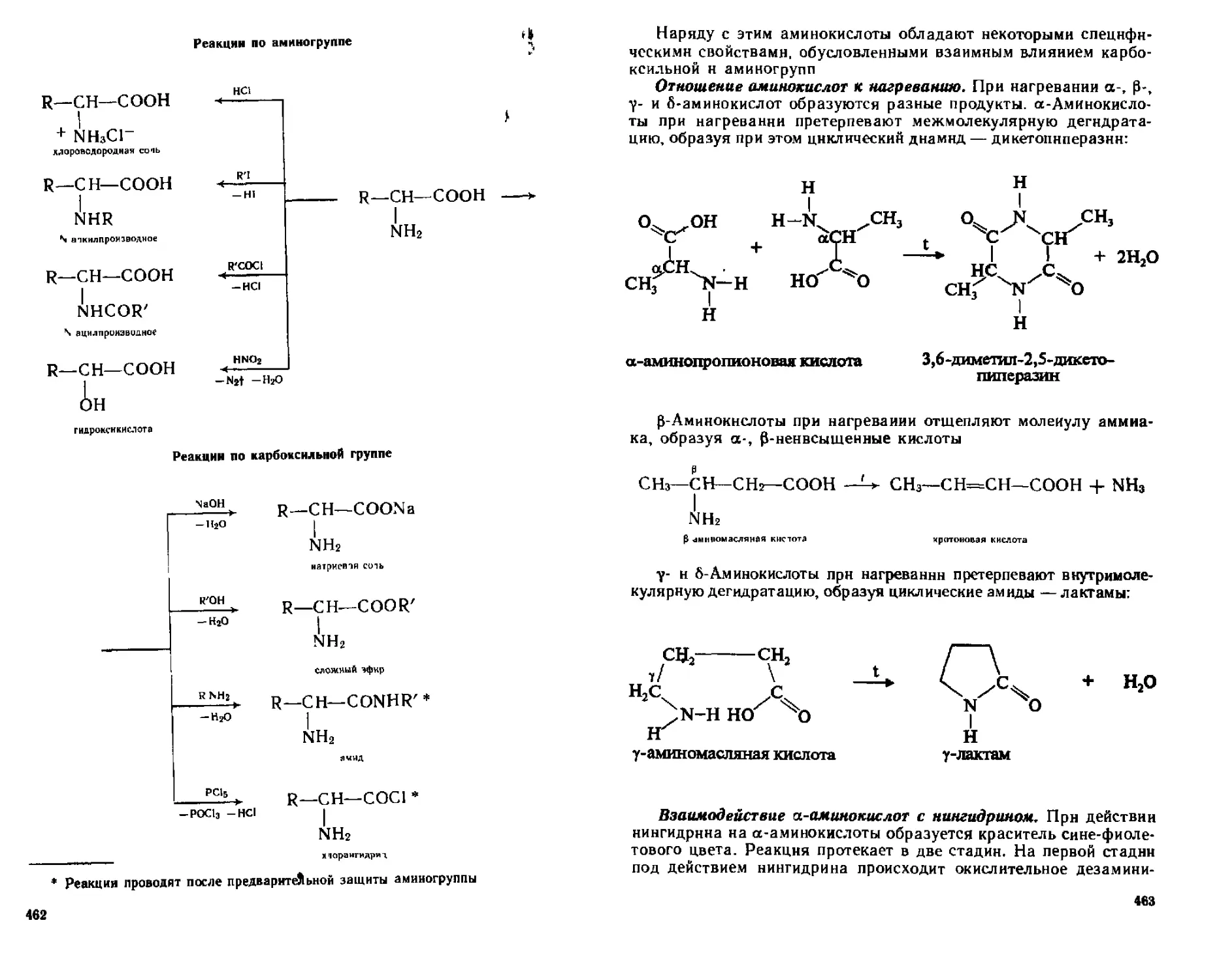
19 4 Аминокислоты 459
19 4 1 Номенклатура Изомерия 459
19 4 2 Способы получения 460
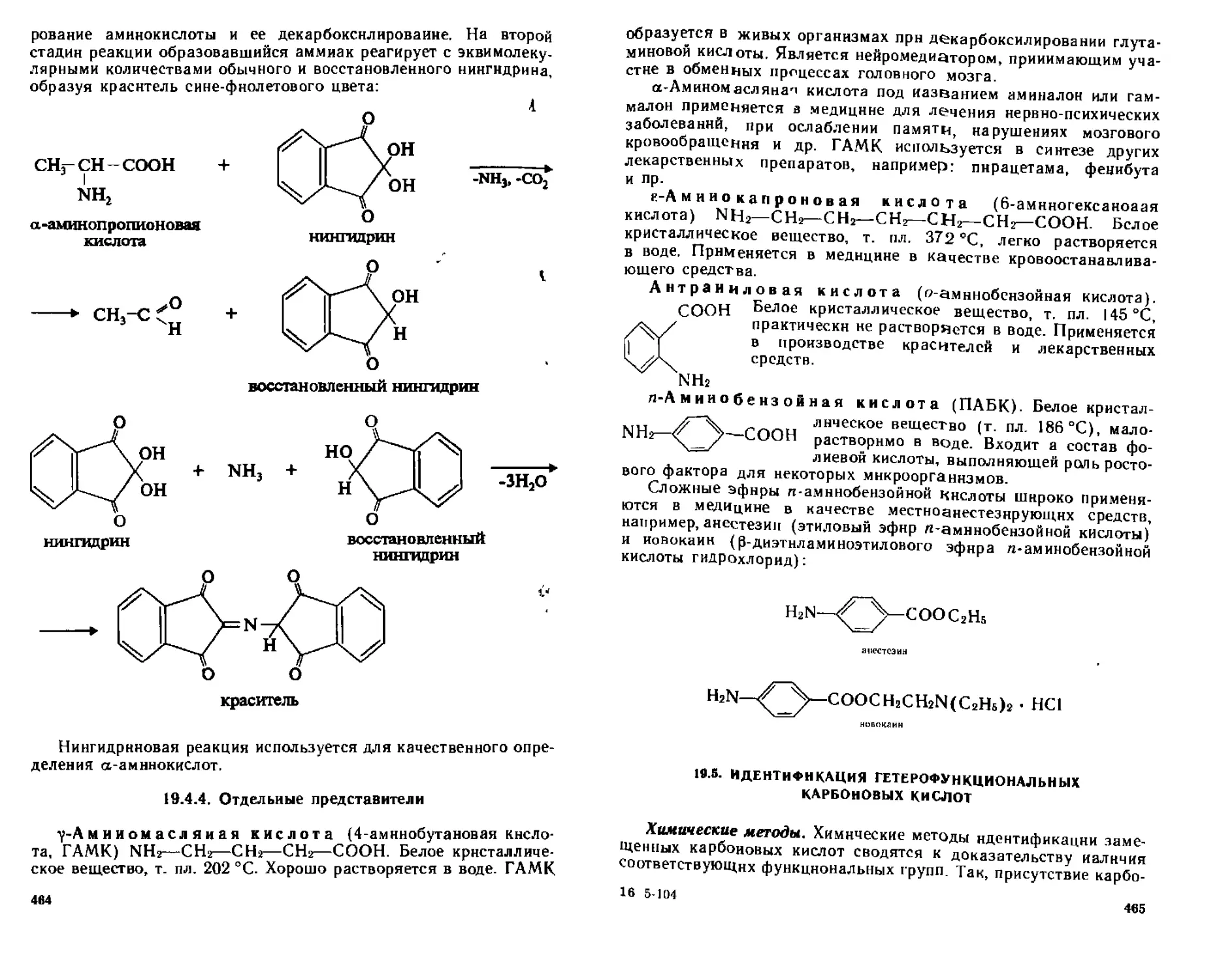
19 4 3 Физические и химические свойства 461
19 4 4 Отдельные представители 464
19 5 Идентификация гетерофуикциональпых. карбоновых кислот 465
Контрольные вопросы и упражнения 466
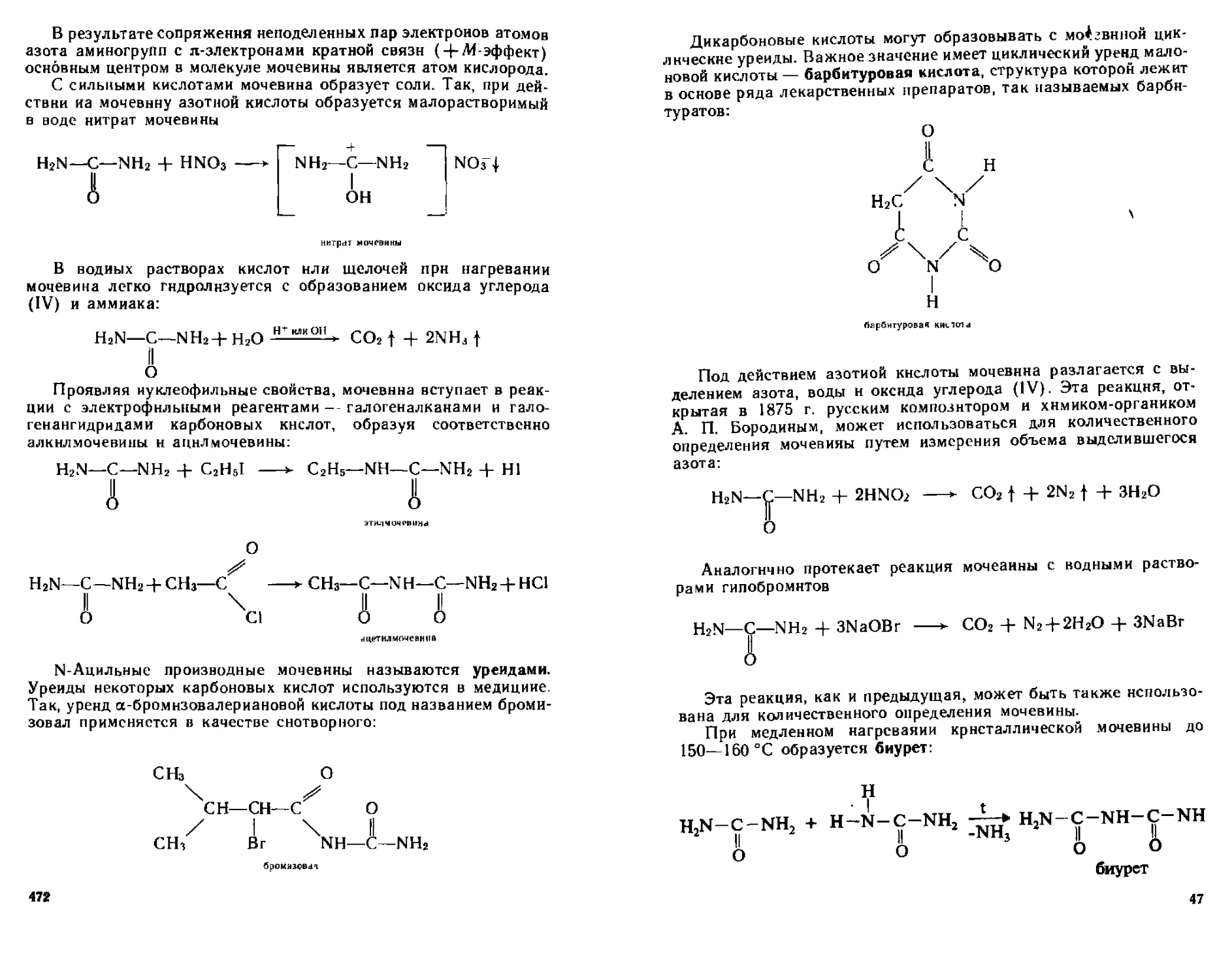
ГЛАВА 20 ПРОИЗВОДНЫЕ УГОЛЬНОЙ КИСЛОТЫ 468
20 1 Хлоран! идриды угольной кислоты 468
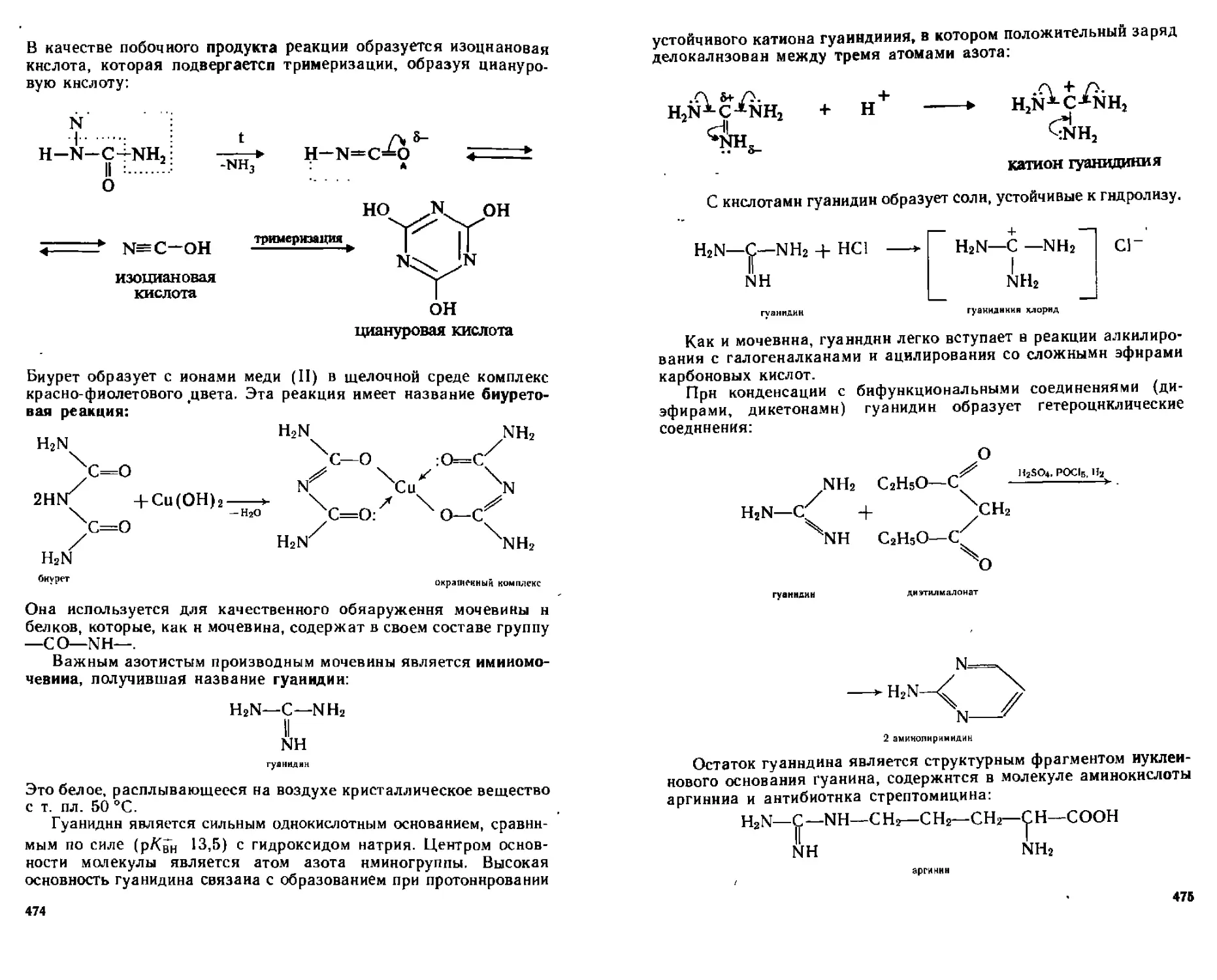
20 2 Амиды угольной кислоты 470
Контрольные вопросы и упражнения 476
предметный указатель 477
9
УГЛЕВОДОРОДЫ
Углеводородами называют органические соединения, молекулы которых состоят только из атомов углерода и водорода.
В зависимости от строения углеродного скелета углеводороды подразделяются на ациклические (алифатические), алициклические и ароматические:
углеводороды
I Г I
АЛИФАТИЧЕСКИЕ АЛИЦИКЛИЧЕСКИЕ АРОМАТИЧЕСКИЕ
I | ( (:-----(
АЛКАНЫ АЛКЕНЫ АЛКАДИЕНЫ АЛКИНЫ ОДНО- МНОГО-
ЯДЕРНЫЕ ЯДЕРНЫЕ
Алифатические углеводороды имеют открытую (незамкнутую) углеродную цепь. По степени насыщенности углерод-углеродных связей их разделяют на алканы (предельные углеводороды), алкены (углеводороды с двойной связью), алкадиены (с двумя двойными связями), алкины (с тройной связью). Ароматические и алициклические углеводороды имеют замкнутую углеродную цепь. К ароматическим относят углеводороды, содержащие одно или несколько бензольных колец. В зависимости от количества бензольных колец их делят на одноядерные и многоядерные. Все другие углеводороды циклического строения относят к алициклическим. Приставка али- в названии указывает на сходство этих углеводородов с алифатическими.
ю
Глава 1 АЛКАНЫ
Алканами называют углеводороды алифатического ряда, в молекулах которых атомы углерода связаны между тобой только простыми ковалентными связями (о -с вязями).
Их еще называют предельными, или насыщенными углеводородами. Раньше эти соединения именовались парафинами (от лат. parum affinitas — мало свойств), что указывает на их сравнительно низкую реакционную способность. Алканы имеют общую формулу СпН2Я+2- Они образуют гомологический ряд, родоначальником которого является метан (табл. 1.1). Каждый член этого ряда отличается от последующего на звено СНз (гомологическая разность).
Таблица 1 1
Первые члены гомологического ряда ал канон
Название Число атомов углерода Молекулярная формула Структурная формула
Метан 1 СН< СН<
Этан 2 СаНд СНэСНз
Пропан 3 С3Нв СН,СН2СНЛ
Бутан 4 С^Ню СН,{СН2)аСНэ
Пентан 5 С5Н|2 СН,(СН2),СНа
Гексан 6 CJM СНзГСНгЬСНэ
Гептан 7 CrHie СН,(СН2)ЧСН4
Октан 8 с8н18 CHs(CHa)sCH,
Нонан 9 СзНм СНЛСН2)7СЖ
Декан 10 СюНза СНДСН2)ВС1Ъ
Ундекан 11 СцНл СНДСНа)вСНа
Додекан 12 СиНгь СНИСН^ЬоСН,
Тридекан 13 CoHsft СНДСНгЬСНз
Тетрадекан 14 СцНзп СН3(СН2)13СН3
Пентадекан 15 CisHaa CHj(CHa) иСНз
Начиная с углеводорода алканы могут иметь как нераз-ветвлениую углеродную цепь, так н разветвленную. Алканы с не-разветвленной цепью атомов углерода называют нормальными, или н-алкаиами.
1.1. СТРОЕНИЕ АЛКАНОВ
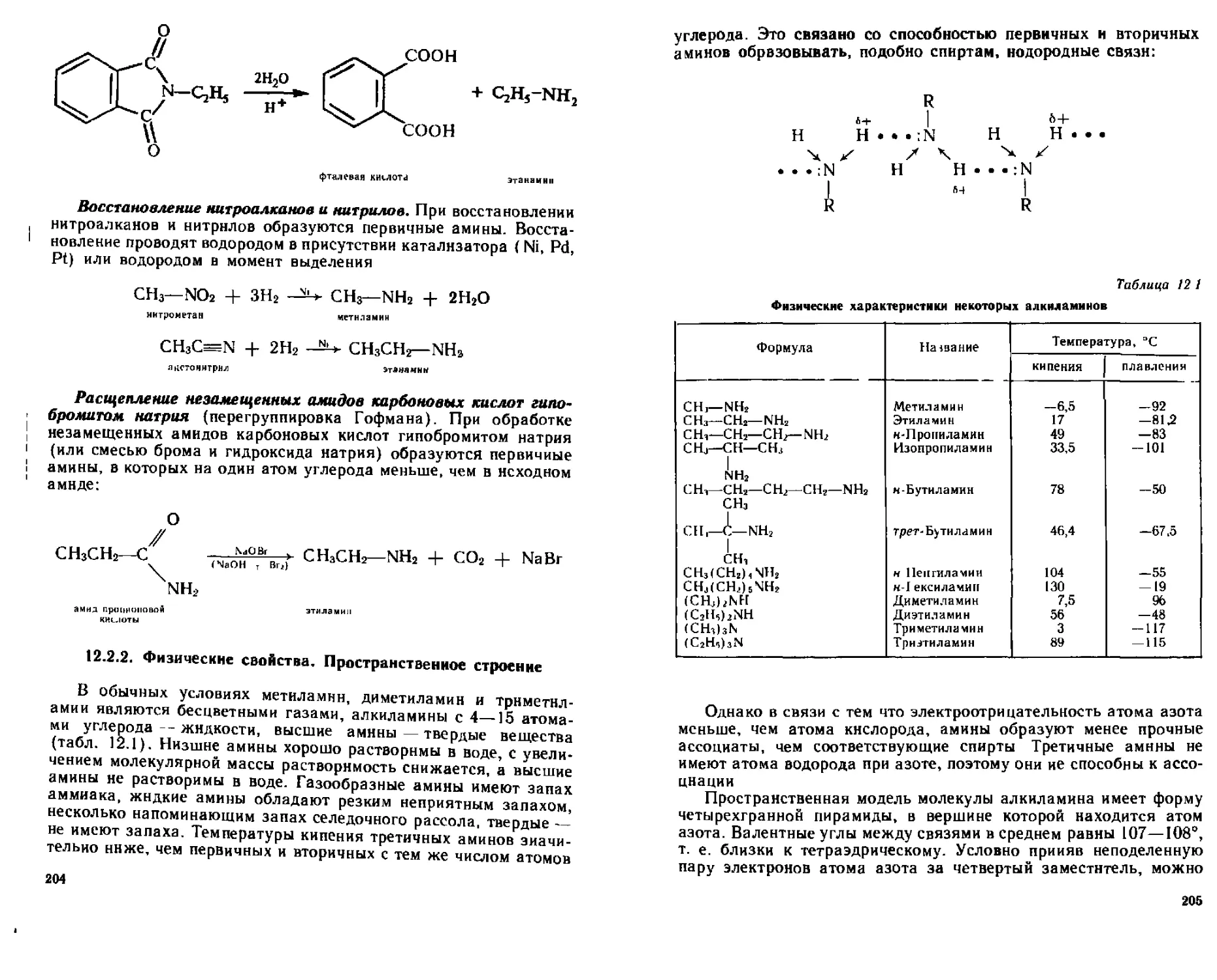
Все атомы углерода в алкаиах находятся в а/?3-гнбрндизации и, следовательно, имеют тетраэдрическую конфигурацию. Валентные углы между связями равны 109°28' (тетраэдрический угол). Если углеродный атом связан с четырьмя разными заместителями, то углы несколько отклоняются от тетраэдрического. Расстояние между атомами углерода в алканах составляет 0,154 нм, а между углеродом и водородом — 0,110 нм. Вокруг связей С—С происхо-дит вращение, в результате которого молекула может принимать разные пространственные формы — конформации (рис. 1.1).
а б в
Рис 1 I Конформации алканов1 а — зигзагообразная, б - нерегулярная;
з - - клешневидная
На основании данных реитгеноструктуриого анализа установлено, что алканы нормального (неразветвленного) строения в кристаллическом состоянии имеют зигзагообразную конформацию. Такое строение для молекулы наиболее выгодно, поскольку в данном случае все атомы углерода по отношению друг к другу находятся в заторможенной анти -бута но вой конформации. При разветвленном строении углеродной цепи конформация может изменяться.
1.2. НОМЕНКЛАТУРА
Первые четыре члена гомологического ряда алканов имеют тривиальные названия — метан, этан, пропан, бутан. Название последующих углеводородов с нормальной углеродной цепью образуется от названия греч. или лат. числительного, указывающего количество атомов углерода в молекуле, с добавлением суффикса -ан, например: пентан, гексаи (см. табл. 1.1). Для обозначения иеразаетвленной углеродной цепи в название алкаиа вводится приставка н-:
СНз—СНг-—СН2-—СНг—-СНз
н «ектан
В названиях алканов с разветвленной цепью, в которых две метильные группы находятся на одном ионце углеродной цели.
12
меЮ1дей других ответвлении, используется приставка изо-; Не и же на конце углеродной цепи имеется трн метильные группы, еСназванне алкана вводится приставка иео-:
СНз снэ
>СН—СНз—CHj—СНз СНз^С—СН^-СНз
f Сн/ СНз
пеогехса н
и ioreKLdH
Названия алканов с разветвленной углеродной цепью образуются согласно заместительной номенклатуре ИЮПАК следующим образом.
j. За основу названия принимают название углеводорода, которому отвечает в рассматриваемом алкане наиболее длинная неразвствленная углеродная цепь (главная углеродная цепь); если в углеводороде имеется несколько цепей одинаковой длины, за главную из них принимается та, которая имеет наибольшее число разветвлений:
главная цепь
цепь не является главной
2. Нумеруют атомы углерода главной цепи с того конца, к которому ближе находится заместитель. Если в молекуле алкана заместители расположены на равном расстоянии от обоих концов, то нумерацию проводят с того конца, к которому ближе расположен заместитель с названием, стоящим в алфавитном порядке раньше;
5 4 3 2 1
С Нэ—СНо—С Hz- CH—С Из
СНз
7 6 5 4 3 2 I
| Скз^Н^Н^СНа—СН—СНг-СНз I
СНз—СНз СНз
Если же на равном расстоянии от обоих концов главной цепи расположены одинаковые заместители, но с одной стороны количество разветвлений больше, чем с другой, нумерацию начинают с того конца, где большее число заместителей:
. CH, 1__2| 4 5 6 ,
|СН3—С—СН2—СН2~(pH—СН3
СН3 СН3
6 5 4 3 2 1
СН3— СН— сн2-сн-сн— СН3
СН3 СН3 СН3
13
3. Составляют название соединения в целом, соблюдая определенные правила:
А. Сначала перечисляют в алфавитном порядке названия заместителей, указывая цифру, соответствующую положению каждого заместителя в главной углеродной цепи. Если углеводород содержит несколько одинаковых заместителей, число их обозначают множительными приставками дн-, три-, тетра- и т. д., а положение в главной цепи, как обычно, цифрами.
Б. Затем называют углеводород, которому отвечает в рассматриваемом соединении главная углеродная цепь.
Заместителями при главной углеродной цепи в алканах являются одновалентные остатки алканов, так называемые алкильные группы, или алкильные радикалы, которые обозначают Aik или R. Названия алкильных групп образуют из названий соответствующих алкаиов, заменяя суффикс -аи на -ил. Наиболее часто встречаются такие алкильные радикалы:
CHd— СН3СН2— СНз—СН-2—СНг—
СН—
пропил
изопропил
СНз-—СН2—СН2——СН2
н бутил
СНз
^>сн—сн сн/
изобутил
СНз—СНз—СН—
СНз
втор бутил
СНз
I СНз—С—
(^Нз грет бутил
В приведенных названиях приставки втор- (вторичный) и трет-(третичный) характеризуют атом углерода со свободной валентностью.
Для названия сложных разветвленных радикалов используют нумерацию углеродной цепи радикала, причем начинают нумерацию всегда с атома углерода, имеющего свободную валентность:
5 4 3 2 1
СНз—СИ—СН—СН?—СИ?—
I I СНз СНз
3.4-диметидиептил
Названия остатков молекул алкаиов, имеющих дае свободные валентности, образуют от названий соответствующих алкапов путем замены суффикса -аи иа -илен (если саободиые аалеитности
14
ходится при разных атомах углерода) или -илиден (если свободные валентности находятся у одного и того же атома углерода), например:
. Сн?- —СН2--СН.,- СНз—СН,—СНа—сн
ыститен этилен эти1нлен пропили
СНз—С—-СНз I изопропилиден
СНз—СН—СН?—
П|ч>цилец
—СН?—СНз—СНа—
тричетнлен
Используя рассмотренные правила составления названий алканов по заместительной номенклатуре ИЮПАК, нижеприведенные углеводороды следует называть таким образом:
СНз
4 3 2| 1
СНз—СНа—С—СНз
СН3
2 2 диметилбутан
НзС' СНз
СНХ СНз
1 2 3[ 4 5| 6 7 8
СНз—СН—СН—СН2—С—СНг—СНг—СНз
(Ll3 СНз—СНз
3 изопропил 2,5 днметнл 5 эти.титан
1.3. ИЗОМЕРИЯ
Для алканов характерна структурная н оптическая изомерия.
Структурная изомерия алканов обусловлена разной последовательностью связывания атомов углерода в молекуле (изомерия Цепи). Она возможна, начиная с бутана, который имеет два структурных изомера: «-бутан и изобутан. Для пентана С5Н12 существует три изомера:
СНз
СНз—СНг—СНз—СНг—СНз СН3—<^Н—СН<г-СН3
н пентан
СНз
С Н з—(Jl—-С Н з !
СН3
неопентан 2 2 диметнипропан
изопентан.
2 мсгилбутак
15
С увеличением числа атомов углерода в молекуле алкана число структурных изомеров быстро возрастает Так, гексан CrH^ имеет 5 изомеров, гептан С?Н|6 — 9, октаи CeHi8 18, декан СюН22 -75, эйкозаи С20Н42 — 366319
Начиная с углеводорода С7Н16, дли алканов возможна оптическая изомерия Например, 3-метилгексан
СНз—СН2— СН—СНг—СНг—СН3 к ГДТз
имеет асимметрический атом углерода и существует в виде двух энантиомеров (зеркальных изомеров):
сн3 СН,
С-Ill Н
СН3-СнГ ^СН2-СН2-СН, СН3-СН2-СН7 сн2-снэ
Я-З-мегилгексан
S- 3-метилгексан
14 СПОСОБЫ ПОЛУЧЕНИЯ
1.4.1, Природные источники
Главными природными источниками алканов являются нефть и природный 1эз Нефть представляет собой сложную смесь органических соединений, основными компонентами которой являются иеразвствленные и разветвленные алканы Природный газ состоит из газообразных алканов, главным образом метана (до 95%), этана, пропана и бутана
Для получения из нефти смеси алканов и других углеводородов ее подвергают фракционной перегонке В результате перегонки получают несколько фракций (петролеипый эфир, бензин, керосин, дизельное топливо, мазут), каждая из которых представляет собой смесь углеводородов, кипящих в определенном интервале температур (табл 1 2)
Таблица 1 2 Фракции, получаемые при перегонке нефти
Фракция Температура кипения, °C Смесь алканов с числом атомов углерода
Петролсйный эфир Бенчин Керосин Дизельное топливо Мазут 20 Ь0 60 180 180 230 230 300 Выше 300 С$. Св Се- Сю Си, С|2 С13 -С17 С|в и выше
Из мазута перегонкой под вакуумом или с водяным паром получают соляровое масло (углеводороды Ci0—С25), смазочные масла (углеводороды Сзв—Сза), вазелин и парафин При
16
дальнейшей перегонке фракций нефти можно получить индивидуальные алканы
Природный газ разделяют на составляющие компоненты путем сжижении с последующей фракционной перегонкой
1.4.2. Синтетические методы получения
Каталитическое гидрирование оксида углерода (II) (синтез Фишера — Тропша) При пропускании смеси оксида углерода (II) и водорода над железиым или кобальтоаым катализатором при температуре 180 -300 °C образуется смесь углеводородов, состоящая главным образом из нормальных алканов, содержащих 6- 10 атомов углерода
СО + 2Нг Не1Сц)> н-алканы + Н2О
Синтез Фишера Тропша применяют в промышленности для получения синтетического бензина и отдельных углеводородов
Каталитическое гидрирование алкенов и алкинов. В качестве катализаторов применяют мелкораздроблеиные металлы — платину, палладий нли никель Реакция протекает при обычных давлении и температуре
СНз—С—С—СНз------------>СН3—СН=СН—СНз H?tPlU
Ovtjjh 2 Pi (Mi Pd) | Nil Pd)
flvreH 2
---> СНз—СНг-СН^—СНз н буттн
Взаимодействие галогеналканов с металлическим натрием (реакция Вюрца) Эта реакция была открьиа в 1854 г французским химиком Ш А Вюрцем
2СЖ—I + 2№ --------> СНз—СП i + 2\lal
{ нодметпи этчи
Реакция протекает через стадию образования металлоорганических соединений-
СНз—1 + 2Na -----> CH3Na + Na!
метил натрин
CH3Na + CHj—I —> СНз—СНз + Nal
Если в качестве исходных веществ используют два разных гало-геналкана, в результате реакции образуется смесь трех углеводородов
t
'.iT—~"wa>’ 4* т3 (Уп п и > к ‘ ( * 1 ?
V.! ь,л нь-р £
СНз—СНз
ГНз—СНг-СНз
этан
______________ пропан ччнвГ____г
+ СНэ—СНа—СНг—СНз бутан
17
Вместо натрия в дайной реакции могут применяться и другие металлы, в частности Zn, Mg, Li. Наиболее легко реакция Вюрца протекает с первичными иодалканами, труднее с бром- и хлор-алканамн. Вторичные н третичные галогенопроизводныс в.условиях реакции Вюрца практически нс образуют алканов. В этом случае образуются преимущественно алкены.
Сплавление солей карбоновых кислот со щелочами. В качестве исходных веществ обычно используют соли карбоновых кислот со щелочными или щелочноземельными металлами и гидроксиды натрия или бария с добавлением натронной извести Са(ОН)2. При сплавлении образуется алкан, имеющий на одни атом углерода меньше, чем в исходной кислоте:
СН3—CH2^CQOn£±51Io]H СНз—СН3 + Na2CO3
пропионат натрия
Взаимодействие металлоорганических соединений с водой. При действии воды иа металлоорганические соединения лития, иатрня, магния, цинка разрывается связь металл—углерод с образованием углеводородов:
СНз—CH2Mgl + НОН --------> СНз—СНз + Mg(OH)I
нодистый этнлмагиии этап
Действием воды на карбид алюминия получают метан:
А14Сз + 12Н2О ----> ЗСН4 + 4А1(ОН)з
Электролиз водных растворов солей карбоновых кислот. Чаще используют натриевые н калиевые соли карбоновых кислот: 2CH3COONa + 2Н2О „ СНз—СН3 _|_ СО2 + 2 NaOH + Нй
ацетат натрин атак
На аноде анион карбоновой кислоты отдает один электрон, образуя неустойчивый свободный радикал, который распадается на СО2 и свободный алкильный радикал. Алкильные радикалы диме-ризуютси с образованием алканов:
2CHjCOO — 2е- -------► 2СН3СОО* ----> 2СН3 + СО2
2СНз ---> СНз—СНз
На катоде образуетси водород и гидроксид соответствующего щелочного металла.
1.5. ФИЗИЧЕСКИЕ СВОЙСТВА
В обычных условиях четыре первых члена гомологического рнда — газообразные вещества; нормальные алканы с числом атомов углерода от 5 до 17 - жидкости; далее следуют твердые вещества (табл. 1.3).
18
Физические характеристики некоторых алканов
Таблица 1.3
Название Структурная формула Температура, °C
плавления кипения
Метан CIL -182,6 — 161,6
Этан СНз—СНз -183,3 -88.5
Пропан СНзСНаСНз -187,1 -42,2
Бутан CH3CHjCHaCH4 — 138,4 —0,50
2-Метилпропан СНаСН—СНз -159,6 — 11,7
(изобутан) 1 СНз
Пентан СН3СН2СН2СНгСНз -129,7 36,1
2-Мети л бутан снзснснгснз -159.9 27.8
(изопентан) Дн Спз
СНз
2,2-л(иметнлпропан , 1
(неопентан) СНз-С-СН, i — 16,6 9.5
СНз
Гексан СНз (СНз) «СНз —94,0 68,7
Гептан СНэ(СНгЛСНз -90.5 98,4
Декан СНэГСНДвСНз —29,7 174.1
Пентадекан СН3(СН2)1зСНа 10 270.7
Эйкозан СН3(СН2)1*СН3 36,4 345,1
По мере увеличения молекулярной массы алканов в гомологическом ряду возрастают температуры плавления и кипения. Температуры кипения изомероа с разветвленной углеродной цепью более низине, чем у нормальных алканов. Газообразные и твердые алкаиы не имеют запаха, жидкие — обладают характерным «бензиновым» запахом. Все алкаиы легче воды и практически не растворяются в ней. Наряду с этим они хорошо растворяются в неполярных растворителях — днэтнлоаом эфире, четыреххлористом углероде, бензоле и др., причем с увеличением молекулярной массы растворимость уменьшается.
1.6. ХИМИЧЕСКИЕ СВОЙСТВА
В обычных условиях алкаиы являются малореакционноспособными соединениями. Они устойчивы к денстаию кислот, щелочей и окислителей. Химическая инертность алканов обусловлена высокой прочностью а-связей С—С и С—Н (см. ки. 1, с. 43). В результате незначительного различия электроотрицательностей $рэ-гиб-ридизованиого атома углерода (2,5) и атомв водорода (2,1) о-связн углерод—углерод и углерод водород в алканах практически
19
не полярны и поэтому не склонны к гетеролитическому разрыву, ио способны расщеплитьсн гомолитически с образованием свободньрс радикалов Химические превращения алканов чаще сопровождаются гомолитическим расщеплением связей С—Н с последующим замещением атома водорода другими атомами или группами, т. е. для иих характерны реакции замещения, происходящие по радикальному механизму (SR). При высоких температурах может наблюдаться гомолитический разрыв связей С—С.
1.6.1. Реакции радикального замещения (SR)
Галогенирование. Алкаиы легко реагируют с галогенами (кроме иода), образуя смесн моно- н полигалогеналканов. По реакционной способности с алканами галогены располагаются в ряд: F- > CI2 > Вг2. Со фтором реакция принимает характер взрыва. Выделяющаяся при замещении атома водорода иа фтор энергия превышает энергию диссоциации связи С—С. Поэтому при фторировании алканов наряду с замещением атомов водорода фтором происходит разрыв угле род-угле родных связей, в результате чего образуется сложная смесь фторпроизводных. По этой причине прямое фторирование алканов имеет ограниченное применение.
Значительно меиее экзотермичиа реакция с хлором. Оиа протекает при освещении УФ-нзлучением или нагревании (300 °C). При взаимодействии метана с хлором атомы водорода постепенно замещаются на атомы хлора:
СН4 4- С1а НС1 4 СН3С1 хлорметан
CH3CI 4 С12—HCI 4- CH2CI2 дихлорметан
СНгСЬ 4 СЬ НС1 4 CHClj трнхлорметан (хлороформ)
CHCI3 4 С12 — ч> НС1 4 СС14 тетрахлорметан (четыреххлористый углерод)
Реакция протекает по цепному свободнорадикальному механизму, который был изучен советским ученым Н. Н. Семеновым. В цепном процессе выделяют три стадии: инициирование; рост цепи; обрыв цепи.
Инициирование. Под действием энергии квантов света (/iv) или нагревания молекула хлора активируется и распадается иа два свободных радикала:
Cl • Cl 2СГ
Рост цепи. Образовавшиеся радикалы хлора атакуют связь С—Н в молекуле метана, отрывая при этом атом водорода с образованием хлороводорода НС1 и свободного метильного радикала СНз:
СН4 4 СГ—> НС1 4 С Ж
20
лл тильный радикал, в свою очередь, атакует молекулу хлора, отрывает атом галогена и образует хлорметан СНзС1 и свободный радикал хлора.
СН5 + С1 . . CI -----------> СНзС1 + СГ
хлорметан
Образовавшийся радикал хлора повторяет цикл указанных превращений, т- е, происходит цепной процесс, в котором атом хлора, израсходованный на предыдущей стадии роста цепи, способствует высвобождению нового радикала хлора на последующей стадии.
Таким образом получается смесь моно-, ди-, трн- и тетрахлор-производных метана:
СГ + СНзС1 н СН2С1 + НС1
СН2С1 ~Г С12 —► СН2С12 4- СГ диклор метен
СГ 4* СН2С12 » СНС12 4- НС1
СНС12 + С12 СНС13 4- СГ трнхлор
и т. д.
Цепной процесс прекращается только после исчезновения всех свободных радикалов, образующихся в ходе реакции.
Обрыв цеп н. Обрыв цепи происходит в результате рекомбинации (димеризации) свободных радикалов:
СНз + CHS---* СНз—СНз
СНз + СГ---->- СНз—С1
СГ + СГ-----^С12
Аналогично протекает реакция бромирования метана, однако она значительно менее экзотермична, чем реакции фторирования и хлорирования, а поэтому происходит не так бурно.
Следует от.метить, что замещение водорода иа атом галогена в алканах происходит в большинстве случаев региосе ле к-т и в н о (избирательно): в первую очередь, как правило, заметается атом водорода при третичном атоме углерода, затем при вторичном и в последнюю очередь при первичном. Например, при бромировании 2-метилпропана образуется продукт замещения у третичного атома углерода:
СНз СНз
СНз—С—СНз 4- Вг2 СНз—С—CH.J + НВг
Н Вг
.................... 2 бром 2-метилиропан
21
Такая последовательность замещения обусловлена устойчивостью образующихся при отрыве атома аодорода свободных радикалов. Чем устойчивее свободный радикал, тем легче он образуется. Поскольку третичные алкильные радикалы более стабильны, чем вторичные и тем более первичные (см. кн. 1, с. 139), реакционная способность связей С—Н при галогенировании алкаиов увеличивается в ряду: первичный < вторичный < третичный атом углерода. Однако эта закономерность строго не выполняется. Региоселективность галогенирования зависит также от активности реагента (атома галогена) и температуры. Чем активнее реагент, тем ниже селективность реакции. Следовательно, у алканов реакция бромирования более селективна, чем хлорирования. Региоселективность галогенирования алканоа возрастает при понижении температуры.
Сульфохлорирование. При облучении УФ-излучением алкаиы подвергаются совместному действию SOs н Ch с образованием алкансульфонилхлоридов R—SO2CI:
R—И + SO2 + Ch R—SO2C1 4 HCI алкан аакапсульфонил-
хлорид
Как и в случае галогенирования, реакция сульфохлориро-вання алканов протекает по цепному радикальному механизму ( ^r) '•
Ch СГ 4- СГ
R—Н -J- СГ -----> R* 4 НС1
R* 4 SOa -----> R—SO2
R—SO2 + Ch--------* R— SO2C1 4 СГ н т. д.
В результате реакцнн получается смесь пераичиых и вторичных алкансульфоиилхлоридов. Третичные сульфохлориды не образуются, очевидно, вследствие пространственных препятствий.
Реакция сульфохлорироаания имеет важное значение в производстве синтетических моющих средств.
Нитрование. Реакция основана иа взаимодействии алкаиов с азотной кислотой. При нагреаанин алканов с разбавленной азотной кислотой (температура около 140 °C и небольшое давление) происходит замещение одного из атомов водорода ннтро-группой (М. И. Коновалов, 1888г.):
R—Н 4 HNOg R—NO2 4- Н2О а пиан иитроалкан
Реакция протекает по свободнорадикальному механизму: hono2 -ьл- но- 4 no;
R—Н 4 НО- -------> R* 4 Н2о
R* 4 HONO2 •—> R—NO2 4 ОН*
22
логично реакции галогенирования при нитровании легче замешается водород у третичного атома углерода, затем
ВСвторичиого и, наконец, у первичного. Например:
У СНа СНа
СНз—с—сн2—СН3 + Н2О
I. p
СНз
NO2
2 ие'гил-2-нит|К|б}тан
Н
изопентан
Концентрированная же азотная кислота в обычных условиях ие взаимодействует с алканами; при нагревании она действует главным образом как окислитель.
1.6.2. Окисление алканов
В избытке кислорода или на воздухе алкаиы сгорают с образованием СО2 и Н2О и выделением большого количества теплоты: СН4 + 2 Ог---► СО2 + Н2О + 882 кДж/моль. Окисление алка-
нов кислородом воздуха в присутствии катализатора (солей марганца, хрома, свинца и др.) при температуре 150—200 °C приводит к образованию смеси продуктов, состоящей в основном из карбоновых кислот с различной длиной углеродной цепи, альдегидов, кетонов и спиртов. Реакция протекает по радикальному механизму и сопровождается разрывом углерод-углеродных связей. В качестве промежуточных продуктов окисления образуются органические гидропероксиды. Схема окисления пропана;
СНз—СНа—СНз СНз—СН—СНз
---1- СНз—СН —сиз-—с-сн^
СНз—CH—СНз + СНз—CH—СНз
О—ОН
ГИД рш№ ДОКС Нд
I
о
о
СНзОН + СНз—С
н
уксусник альдегид
СНз—СООН
уксусная кислота
-С—СНз + Н2О
ацетон
в промышленности для по-ацетальдегида и уксусной
мета мол р, нсоон
муравьиная кислота
Реакция окисления используется лучения метанола, формальдегида, кислоты из пропана и бутана, а также высших жирных кислот из алканов с длиной цепи более 25 углеродных атомов.
23
1.6.3. Крекинг алканов
Крекингом называют процесс термического расщепления алканов. Под действием высоких температур алканы разлагаются с разрывом связей С—С и С—Н. При этом одновременно протекают процессы дегидрировании, изомеризации и циклизации. Начальная температура распада алкаиов зависит от нх строения и молекулярной массы. Чем больше молекулярная масса углеводорода, тем легче он расщепляется при нагревании. Различают термический крекинг и каталитический крекинг. Термический крекинг проводят при температурах 800 °C и выше, каталитический - при температурах 450—550 °C в присутствии алюмосиликатных катализаторов (оксид алюминия А12Оз на силикагеле SiO2).
Наиболее устойчив к термическому разложению метан. При температуре 1400 —1500 °C он подвергается распаду с образованием ацетилена:
2СН4 НС^СН + Нг
Этаи разлагается при более низких температурах:
СНз—СНз б-д°^'ч cHjf=ch2 + н2
Высщнс алканы в условиях термического крекинга разлагаются с образованием сложной смеси низших алканов и алкенов. Разрыв углеродной цепи молекулы может произойти в любом месте, например:
СНз—СН2- СНг—СНг—СН2—СНз ——*-
СН4 + СН2=СН—СН2- СН2- -СНз СНз—СНз + СН2=СН—СНг—СНз СН2=СН2 + СНз—СНг—СНг—СНз СНз—СН2—СНз -I- СН2==СН—СНз
Термический крекинг протекает по радикальному механизму.
При каталитическом крекинге расщепление связи С—С сопровождается преимущественно изомеризацией w-алкаиов в алканы с разветвленной цепью
2СНзСН2СН2СН2СН3—СНз
СНз
1
СНз—CH—СН- СНз + СНз—С—СНг—СНз
СНз (^Нз СНз
в присутствии катализатора высшие алканы способны к цикли-иИ с образованней ароматических углеводородов:
СН3
СНлСН2СН2СНгСН2СН2СНз (fY + 4Н2
каталитический крекинг протекает по ионному механизму.
Крекинг-процесс имеет важное промышленное значение и широко используется для получения высокооктановых бензинов, непредельных и ароматических углеводородов.
1.7. ИДЕНТИФИКАЦИЯ АЛКАНОВ
Вследствие аысокой инертности алканов, химические методы лдентификации для них, как правило, не применяют. Чаще всего злканы идентифицируют по физическим константам (температуры <ипении и плавления, показатель преломлении, удельное вращение тля оптически активных веществ и др.) и спектральным характеристикам.
УФ-излучение алканы поглощают а области менее 200 нм, поэтому жндкне алканы используют а качестве растворителей для измерения электронных спектров других веществ.
Для ИК-спектров алканов характерны полосы поглощения в области 2850 —3000 см1, отвечающие валентным колебаниям связей С—Н, н в области 1370— 1470 см характеризующие деформационные колебания связей С—Н.
В ПМР-спектрах алканов разные по расположению протоны имеют близкие значении химических сдвигов (0,5—2м.д.)> что затрудняет интерпретацию ПМР-спектров.
В последние годы достигнуты большие успехи при идентификации алканов методом масс-спектрометрин.
1.8. ОТДЕЛЬНЫЕ ПРЕДСТАВИТЕЛИ- ПРИМЕНЕНИЕ
Метан (СН4). При нормальных условиях бесцветный газ, не имеющий запаха, малорастворимый в воде. Метан является основной составной частью природного газа. Он широко используется в качестве промышленного и бытового топлива, а также служит важным сырьем дли химической промышленности. Из метана получают ацетилен, метиловый спирт, формальдегид, хлороформ, четырёххлористый углерод, газовую сажу н другие вещества.
Этан (С2Н6), пропан (С3На), бутан (С4Н10). При нормальных условиях бесцве^^ые газы, без запаха. Пропан и бутан легко сжижаются, что позволяет использовать их в быту в качест-в* топлива (баллонный сжиженный газ). Как и метан, данные
25
алканы широко используют в химической промышленности в качестве сырья для получения этилена, пропилена, бутадиена и других веществ, имеющих важное народнохозяйственное значение.
Вазелиновое масло — бесцветная маслянистая жидкость без запаха и вкуса, практически нерастворимая в воде. По химической структуре представляет собой смесь алканов с числом углеродных атомов до 15. Применяется в медицине как слабительное средство, в фармации при изготовлении лекарственных форм, а также в парфюмерно-косметической промышленности.
Вазелин — бесцветное илн светло-желтого цвета однородное вещество, практически нерастворимое в воде. В химическом отношении представляет смесь жидких и твердых алканов с числом углеродных атомов от 12 до 25. Вазелин широко используется в фармации в качестве основы для приготовления мазей.
Парафин — белая твердая мелкокристаллическая масса без запаха и вкуса, слегка жирная на ощупь, нерастворимая а воде. Температура плавления 50—57 °C. Парафни состоит из смеси твердых алканов с числом углеродных атомов от 19 до 35. Применяется в фармации в качестве основы для приготовления мазей. В связи с большой теплоемкостью и низкой теплопроводностью парафин используют в медицине для лечения теплом (парафинотерапия) .
Озокерит (горный в о с к) — твердая воскообразная масса черного цвета. Температура плавления около 80 °C. Это ископаемое вещестао иефтниого происхождения. По химической структуре представляет собой смесь высших алкаиов и алкенов, смол и минеральных масел. Как и парафин, озокерит применяют в медицине для лечения теплом при невралгиях, невритах, плекситах и других заболеваниях.
КОНТРОЛЬНЫЕ ВОПРОСЫ И УПРАЖНЕНИЯ
1. Назовите углеводороды по рациональной и заместительной номенклатуре ИЮПАК:
а) СНз—СН—СНз—СН—СН3;
<!:Н2 СНз
С^Г1з
б) СНз—СН—СН—СН2—СНз
СНз СНз—СНз—СНз—СНз к
Ьпз
СН2—СНл—СНз в) СНг—(^Н—СН3
СН—СНз
; СН3
26
2 Назовите алканы, образующиеся в результате следующих
зеакиий:
а) СНз-CH—СНг
СНз Вг
Na
б) СНз—СНг—1 + НзС—СН—СН21 СНз
СНз
a) H.C-C-CHaMgBr-iS-к Спз
г) СНг^СН—СНг—СНа—СНз
д) НзС—СН—COONa
сплавление
е) CH3CH2COONa
электролиз нго
3, Напишите уравнения реакций, с помощью которых можно осуществить химические превращения: а) СНз—СНгСООН
2-нитробутан; б) пропаи -► 2,3-днметилбутан; в) АЬСз-*• ->> м-бутан.
4. Приведите схему реакции Коновалова для следующих углеводородов: а) и-пента на; б) 2-метилбутана, в) метана. В каких условиях протекает взаимодействие? На примере реакции (б) опишите механизм реакции.
5. С какими из приведенных соединений реагирует пропан в заданных условиях? Напишите уравнении реакций. Опишите механизмы взаимодействия с реагентами, отмеченными звездочкамн; а) H2SO4 (конц.), 20 °C; б) Вг21 в темноте, 20 °C; в) Вг2, освещение, 20 °C *; г) 1а, освещение, 20 °C; д) SOa -|- Ch, освещение, 20 °C *; е) HNO3 (разб.), 140 °C, давление.
27
Глава 2 АЛКЕНЫ
Алкенами называют углеводороды алифатического ряда, содержащие одну двойную связь. Общая формула алкенов СпНа«.
Они образуют гомологический ряд, родоначальником которого является этилен (табл. 2.1), что и обусловило еще одно название алкенов — этиленовые углеводороды. Для ния* сохранилось также исторически сложившееся иазаание — олефины (масло-образующне), поскольку низшие гомологи этой группы соединений при взаимодействии с хлором илн бромом образуют маслянистые жидкости.
Таблица 2.1 Первые члены гомологического ряда алкенов
Структурная формула соединения Тривиальное название Название по номенклатуре ИЮПАК
СНз==СНа СН,—СН=СНг СН3—CHs—СН=СНа СНз— СН=СН- CHj СНз—с=сн2 1 Этилен Пропилен а-Бутилен р-Бутилен Изобутилен Этен Пропен Бутен-1 Бутен-2 2-Метилпропен
СНз СНз—CHs—CH^—СН^СНа СНз— СН2—СН=СН—СНз а-Амилен р- Амилен Пентен-1 Пентен-2
2.1. НОМЕНКЛАТУРА
Согласно правилам ИЮПАК названия алкенов образуют от названий соответствующих алканов, заменяя суффикс -ан на -ен с указанием положения двойной связи в цепи углеродных атомов. Например: этен, пропен, бутеи-l и т. д. (см. табл. 2.1).
При построении названий алкенов по заместительной номенклатуре ИЮПАК используют следующие правила:
1. Выбирают самую длинную углеродную цепь, включающую двойную связь (главная углеродная цепь).
2. Углеродные атомы главной цепи нумеруют, причем нумерацию начинают с того конца цепи, к которому ближе расположена двойная саязь.
28
3. Составляют название алкеиа, перечисляя вначале, как и алканах, в алфавитном порядке углеводородные заместители с указанием их положения в главной цепи, затем называют угле-водород, которому соответствует главнаи углеродная цепь и, наконец, после суффикса -ен через дефис ставят цифру, указывающую положение двойной связи (номер первого из двух углеродных атомов, образующих двойную связь). Например:
4 3 2 1
СН3—СН2—СН=СН2
б^теН-1
СНз
1 2 3 |4
СНз—СН=СН—С—СНз 5| 6
СНг—СНз
4-4 диметилгрксен 2
2 3 4 5
СНз—СНз—С—СН,—СН—СНз
I' 1
1 СНз СНз
4 метил-Й этнлпеитен-1
Для низших членов гомологического ряда алкеиов применяют также тривиальные названия — этилен, пропилен, бутилен и т. д. (см. табл. 2.1), причем название первого представителя — этилен — принято правилами ИЮПАК как более предпочтительное, чем этен.
Название одновалентных углеаодородных радикалоа, образованных из алкенов, получают добавлением к назаанию алкена суффикса -ил, например:
3 2 1 3 2 1
СН2=СН— СНа—СН=СН— СН?=СН—СН2—
этеннл пропен-1-ил пролеи-2-ил
Некоторые радикалы имеют также тривиальные названия, например:
СН2=СН—
винил
СНз—С=СН2
и юиротюнил
СН2=СН—СН?—
аллил
2.2. ИЗОМЕРИЯ
Для алкеиов характерна структуриаи и геометрическая изомерия.
Структурная изомерия алкенов обусловлена разной последовательностью связывания атомов углерода в молекуле (изомерия *№
цепи) и разным положением двойной связи при одном и толГ Же углеродном скелете (изомерия положения). Этот вид изомерин возможен, начиная с бутена (СЛв), который может существо^ вать в виде трех структурных изомеров:
изомерия положения
I I
СН^=СН—СНг-СНз СНз—С=СН2 СН3—СН=СН—СНз
СНз
Оугек i 2 мстилпропен бутен 2
t_________________t
изомерии цепи
Кроме того, в ряду алкенов имеет место геометрическая, или так называемая цис-транс-изомерия, что обусловлено различным расположением атомов или групп а пространстве относительно плоскости двойной связи (см ки. 1, с. 86). Например, бутен-2 может существовать в виде двух пространственных изомеров — цис- (два одинаковых заместителя при углеродах с двойной связью расположены по одиу сторону плоскости л-связи) и транс- (два одинаковых заместители расположены по разные стороны плоскости л-саязи):
СНз СНз
цис бутен-2
Если у атомов углерода, связанных двойной связью, имеется три или четыре разных заместителя, используют Е, Z-снстему обозначений конфигурации геометрических изомеров (см. кн. 1, с. 88):
СНз СН2СН3
^с^с/7
СИдСНг^ ^СНзСНгСНз
СНз CHsCHiCHj
СНзСн/7 ЧчСН2СН3
Z Э метил 4 эти.1 гептен 3
Е 3 метил 4 этилгетттен 3
Так, З-метил-4-этилгептеи-З может иметь Z-конфигурацию (старшие заместители при углеродах с двойной связью расположены по одну сторону плоскости л-связи) и £-конфигурацию (старшие заместители расположены по разные стороны плоскости л-связи).
зо
2.3, СПОСОБЫ ПОЛУЧЕНИЯ
ц небольших количествах алкены встречаются в некоторых сторождениях нефти н природного газа, откуда могут быть Ме елены в чистом виде. Как отмечалось ранее, алкены образуют-0 также при термическом крекинге высших алканов.
СЯ Большинство методов синтеза основаны на элиминировании Готшепленин) атомов или атомных групп от молекул алканов, галогеналканов н спиртоа.
Дегидратация предельных спиртов. При нагревании с сильными минеральными кислотами, например серной или фосфорной, предельные спирты отщепляют молекулу воды и образуют соответствующие алкеиы:
алкеиы:
СН2-СН2
;н он;
этанол
H2so4
t
сн2-сн2 + Н2О
этилен
Механизм этой реакции приведен на с. 258.
В промышленности дегидратацию осуществляют пропусканием паров спирта над катализатором — оксидом алюминия AI2O3 при температуре 300—400 °C:
сн,-сн-сн2
;н он;
А1 Л
2 3-> сн3-сн-сн2 + н2о
300-400 С
пропанол-1
пропен
Легче отщепляют воду вторичные и особенно третичные спирты. При этом, если атом углерода связан с неравноценными между собой углеродными атомами, отщепление воды происходит преимущественно таким образом, что вместе с гидроксилом уходит водород от менее гидро генизованпого соседнего атома углерода. Эта закономерность, устаноаленная в 1875 г. русским химнком-оргаии-к°м А. М. Зайцевым, получила название правила Зайцева (см, с 257):
CH,— СН__С__СИ М 'чО 3 2| 1
з С СН3 СИз-СН-С-СН, + Н2О
Грн ’ н ; 1
3-Мегилбутанал-2 2-метилбутеи-2
31
Дегидрогалогенирование моногалогеналканов. При нагревали! моногалогеналканов со спиртовым раствором гидроксида натрщ или калия отщепляется галогеноводород и образуются алкены
СНз—СН—СНз—СН=СН2 + NaBr + Н2О
I । с спирт 1 1
[rtj
I бромпропан пропен
Механизм этой реакции приведен в подразд. 10.3.3.
Как и в случае со спиртами, порядок отщепления галогсно-водорода от вторичных и третичных галогепалканов определяется преимущественно правилом Зайцева, т. е. вместе с галогеном уходит атом водорода, находнщнйся при менее гидрогенизированном соседнем атоме углерода:
СНд
1 2 3| 4 4 3 2 1
СН3-СН-С-СН3 NaOH СНу-С№-С-СН3 + NaBr + Н2(
Где.«: спирт* сн,
2-бром-З-метилбутан 2-метилбутен-2
Дегалогенирование дигалогеналканов. Ди галоген ал кины с атомами галогена у соседних атомов углерода при действии Zn или Mg в водноспиртовом растворе отшеплиют два атома галогена, образуя алкены:
СНз—СН—СН—СН3 + 2п ► СНз—СН=СН—СНз + ZnBr2
I I
Вг Вг
2.3 тбромбутан бутен 2
Дегидрирование алканов (промышленные метод). При температуре 300 500 °C в присутствии катализаторов (мелкораздробленный никель, оксид хрома (III) Сг2Оз и др.) алканы отщепляют водород, образуя алкены:
СНз—СИа—СНз —СНг=СН—СНз + Н2
Пропан пропен
Селективное гидрирование алкинов. В присутствии катализаторов с пониженной активностью (Fe, частично дезактивированные «отравленные» солями тяжелых металлов Pd и Pt) алкины селективно (избирательно) присоединяют водород с образованием алкеиов:
НС=С—СНз------—* НЙС=СН—СНз
Pd/PbCOj
пропин пропей
32
2.4. ФИЗИЧЕСКИЕ СВОЙСТВА
Аналогично алканам четыре первых представителя гомологи-ского ряда алкенов при нормальных условиях — газы, далее
Ч1еду1°т жидкости (С5—Ctr), затем твердые вещества.
с’ Все алкены практически нерастворимы в воде, хорошо раство-яются в органических растворителях.
Р Температуры кипения «-алкенов, как правило, выше, чем нх изомеров с разветвленной цепью углеродных атомов. Z/uc-изомеры имеют, обычно, более высокие температуры кипения и более низкие температуры плавления по сравнению с транс-изомерами.
2.5. ХИМИЧЕСКИЕ СВОЙСТВА
Главной структурной особенностью, определяющей реакционную способность алкенов, является наличие в их молекуле двойной углерод-углеродной связи. Атомы углерода, связанные двойной связью, находятся в состоянии зр2-гибрндизации. Двойная связь (см. кн. 1, с. 40) представляет собой сочетание ковалентных a-связи и л-связп, нз которых л-связь меиее прочная, чем а-связь. Электронная плотность л-связи расположена симметрично выше и ниже плоскости, в которой лежат a-связи 5р2-гибридизоваиных атомов углерода (рис. 2.1).
(У-СВЯЗЬ
электронное облако л-связи
Рис 2 1 Строение двойной связи в молекуле этилена
Благодаря такому расположению электроны л-связи легче П(Мяризуются по сравнению с электронами а-связн. Вследствие высокой поляризуемости и низкой энергии образования л-связи алкены довольно легко вступают в реакции присоединения, протекающие с разрывом л-связи. В большинстве случаев такие реакции иДут по иоииому механизму и иачииаются с атаки электрофильным реагентом (электрофильной частицей) элеитроиов л-саязи, а поэтому их называют реакциями электрофильного присоединения (At).
2 5 НМ ЯЯ
Кроме того, двойной связь влияет па реакционную способность связей С—И у Соседнего с ней $р3-гибрндизова иного атома
углерода
сн-Фн2
н
Благодаря сверхсопряжению (о.л-сопряжеиие) атомы водорода, расположенные у а-углеродного атома по отношению к двойной связи, приобретают подвижность и способность вступать в ре акции замещения (SR), которые протекают значительно легче, чем у алканов
Для алкенов характерны также реакции окисления, восстановления и полимеризации
2.5.1. Реакции электрофильного присоединения (ЛЕ)
За счет наличия а своей структуре п-связи алкены проявляют нуклеофильные (электроиодонориые) свойства и аступают а реакции с электрофильными реагентами, такими, как галогены, галоге-новодороды, сериая кислота, вода в присутствии минеральных кислот и др Эти реакции протекают по механизму электрофильного присоединения (HF)
Механизм включает две последовательные стадии:
Стадия /
л комплекс
На стадии I электрофильная фнльного реагента X6-*- > Уй“ в взаимодействия с электронным
частица или молекула электрорезультате электростатического облаком л-связи образует с
I 34
молекулой алкеиа так называемый л-комплекс Затем образуется ковалеитиая связь между электрофильной частицей Х+ и одним из атомов углерода двойной связи, при этом л-комплекс превращается а карбкатион
На стадии И карбкатион взаимодействует с освободившейся из электрофильного реагента нуклеофильной частицей У~ и образуется конечный продукт присоединения
Присоединение галогенов (галогенирование), Алкены довольно легко присоединяют по двойной связи хлор и бром, труднее — иод, в результате чего образуются днгалогеиопроизводные алканов, содержащие атомы галогена у соседних атомов углерода (вицинальные дигалогеиопроизводиые)
СН2=СН2 4- Вг2 ----► СН2Вг—СН2Вг
Многочисленными исследованиями установлено, что механизм этой реакции аключаст электрофильную атаку молекулы галогена на л-электроны двойной связи
Под влиянием л-электроиного облака двойной связи молекула галогена поляризуется (Вг6+ >Вг{ ) и приобретает способность выступать в качестве электрофильного реагента
На стадии I реакции атом галогена, несущий частичный положительный заряд, вступает во взаимодействие с л-электронами двойной связи, в результате чего образуется л-комплекс.
Br5i->Br6'
1—-Вг^Вг5-
этилен
л-комплекс
Затем в л-комплексе постепенно происходит гетеролитический разрыв связи между атомами галогена с образованием продукта присоединения, строение которого может быть представлено равновесием между ионом карбония и циклическим ионом галогеноиня-
^'Комплекс
ион карбония
циклический ион бромония
2*
35
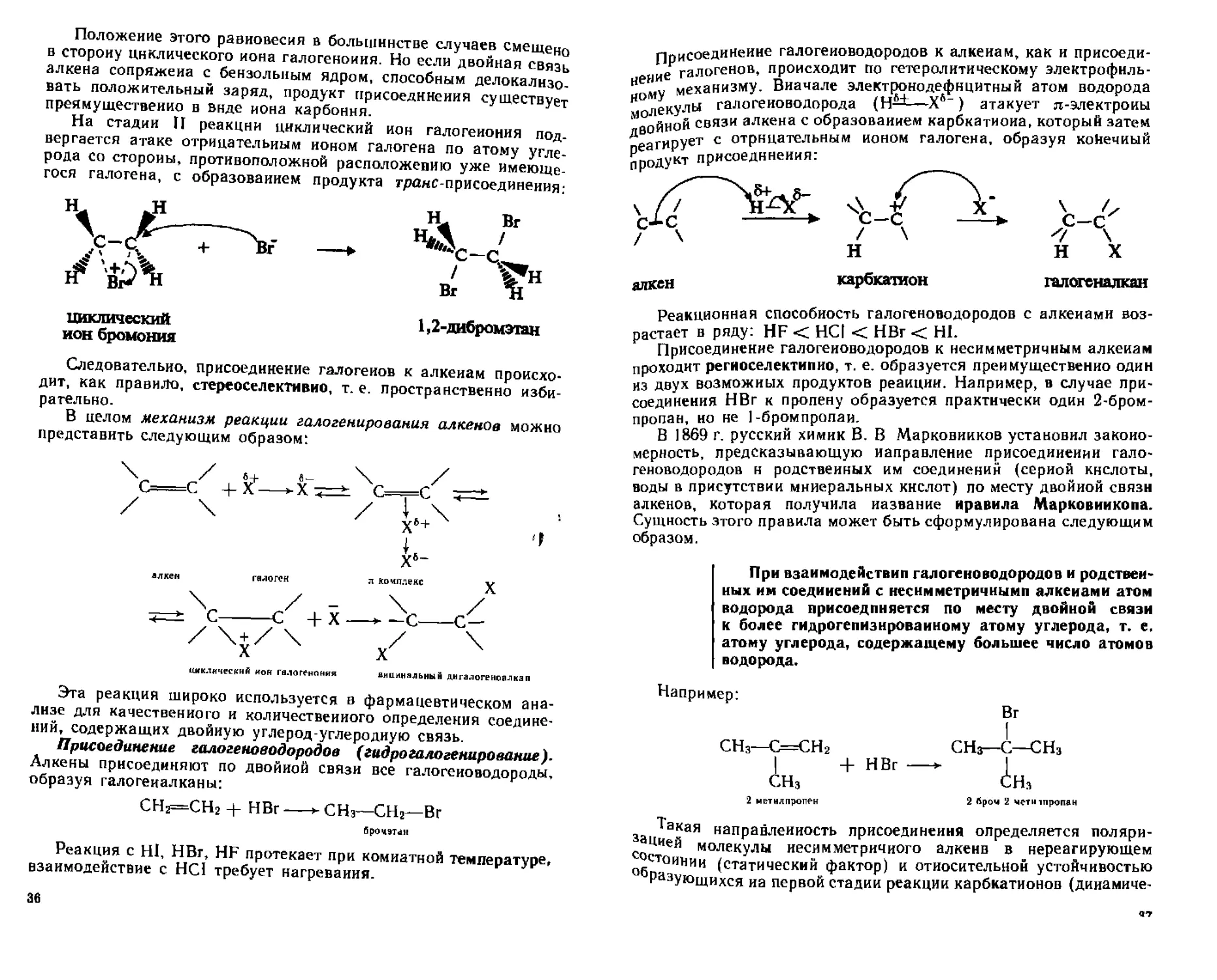
Положение этого равновесия в большинстве случаев смещена в сторону циклического иона галогеноиия. Но если двойная связь алкена сопряжена с бензольным ядром, способным делокализовать положительный заряд, продукт присоединения существует преимущественно в виде иона карбоння.
На стадии II реакции циклический ион галогеиония подвергается атаке отрицательным ионом галогена по атому углерода со стороны, противоположной расположению уже имеющегося галогена, с образованием продукта гранс-присоединеиия:
циклический ион бромония
1,2-дибромэтан
Следовательно, присоединение галогенов к алкенам происходит, как правило, стереоселективио, т. е. пространственно избирательно.
В целом механизм реакции галогенирования алкенов можно представить следующим образом:
циклический ион галогеноиия
вицинальный дигалогеноалкап
Эта реакция широко используется в фармацевтическом анализе для качественного и количественного определения соединений, содержащих двойную углерод-углеродную связь.
Присоединение еалогеноводородов (гидро галогенирование ). Алкены присоединяют по двойной связи все галогеиоводороды, образуя галогеиалканы:
СН2=СН2 + НВг------>- СНз—СНа—Вг
брочзтан
Реакция с HI, НВг, HF протекает при комнатной температуре, взаимодействие с НС1 требует нагревания.
36
присоединение гало ге ио водородов к алкеиам, как и присоеди-е галогенов, происходит по гетеролитическому электрофнль-механизму. Вначале электронодефнцитный атом водорода Н°лекулы галогеиоводорода (Н——X6- ) атакует л-электроиы М°ойной связи алкена с образованием карбкатиона, который затем Драгирует с отрицательным ионом галогена, образуя конечный проДУкт присоединения:
CJ-C
с-с
алкен
Н карбкатион
С-С "7 \
Н X
галогсналкан
Реакционная способность галогеноводородов с алкенами возрастает в ряду: HF < HCI < НВг < Н1.
Присоединение галогеноводородов к несимметричным алкеиам проходит региоселектипио, т. е. образуется преимущественно один из двух возможных продуктов реакции. Например, в случае присоединения НВг к пропену образуется практически один 2-бром-пропан, но не 1-бромпропаи.
В 1869 г. русский химик В. В Марковииков установил закономерность, предсказывающую направление присоедииеиии галогеноводородов н родственных им соединений (серной кислоты, воды в присутствии минеральных кислот) по месту двойной связи алкенов, которая получила название правила Марковиикопа. Сущность этого правила может быть сформулирована следующим образом.
При взаимодействии галогеноводородов и родственных им соединений с несимметричными алкенами атом водорода присоединяется по месту двойной связи к более гидрогенизированному атому углерода, т. е. атому углерода, содержащему большее число атомов водорода.
Например:
СНз—С==СН2
<^Нз
2 метил пропен
+ НВг
Вг
СНз—С—СНз к ЬПЗ
2 бром 2 метитпропан
за ^акая направленность присоединения определяется поляри-Нией молекулы несимметричного алкенв в нереагирующем 0 ~и (Этический фактор) и относительной устойчивостью Разующнхся на первой стадии реакции карбкатионов (дииамиче-
ский фактор). Влияние статического фактора состоит в том, что в иереагирующен молекуле несимметричного алкена вследствие +/-эффекта и а, л-сопряжения со стороны алкильных групп л-электронная плотность двойной связи смещена к более гидрированному ненасыщенному атому углерода. Это определяет наиболее вероитиое место присоединения протона:
СН~ 5-сн/ V
5-Н-Вг i
сн^
СН/| 3
Вг
Влияние динамического фактора обусловлено тем, что нз двух возможных вариантов присоединения протона к несимметричному алкеву преимущественно реализуется тот, при котором в качестве промежуточного продукта присоединении образуется более устойчивый карбкатион. Более устойчивому карбкатиону отвечает переходное состояние с меньшей энергией, а это обеспечивает большую скорость реакции. Делокализация положительного заряда, а следовательно, н устойчивость карбкатиона возрастает с увеличением числа алкильных групп, поэтому третичные карбкатионы устойчивее вторичных, а те, в свою очередь, устойчивее первичных (см. ки. I, с. 134). Поэтому не трудно заметить, что присоединение НВг к 2-метилпропеиу будет протекать преимущественно по направлению а:
СНз
С=СН2 СНз^
а СН3
СН3-^>
СНз^ третичный карбкатион СНз +
СН—СН2-5£1 б /
-^>СН3
СН3
^С—СНз СНз^Вг
СНз i
СН—СН2Вг
2-метил пропен
перпичный карбкатион
Следует отметить, что правило Марковникова соблюдается не всегда. Так, в присутствии пероксидов присоединение бромоводорода к несимметричным алкенам происходит по свободнорадикальному механизму с ориентацией против правила Марковникова:
Н2О2 + 2НВг
2Вг* + 2Н2О
яя
СНз—сн=сн2
СНз—СН—СН2Вг
СНз—СНВг—сн2
- -г- СНз—CHa^-CHsBr + Вг-
НВг— CHj—СНВг—СНз + Вг-
Возникший на первой стадии реакции радикал брома присоединяется по месту двойной связи алкеиа к более гидрированному атому углерода (направление а), так как при этом образуется более устойчивый радикал. Последний атакует новую молекулу НВг с образованием конечного продукта присоединения.
Присоединение концентрированной серной кислоты. Присоединение серной кислоты к алкенам протекает по ионному электрофильному механизму аналогично присоединению галогеиоводородов. Реакция подчиняется правилу Марковинкова и приводит к образованию моиоалкнлсульфатов — кислых эфиров серной кислоты:
OSO3H
сн3—сн=сн2 + h2so4 к. —> СНз—<!:н—СНз
изопропиловый эфир серной кислоты
При нагревании с водой моноалкнлсульфаты гидролизуются, образуя спирты:
СНз—СН—СН3 + Н2О —► СНз—СН—СНз + H2SO4
Aso3h ОН
изопропилсульфат пропанол 2
В промышленности эта реакция используется для получения этилового и изопропилового спиртов.
Присоединение воды (гидратация). В присутствии минеральных кислот — серной, азотной, хлорной и др.— влкены присоединяют по двойной связи воду. Реакция подчиняется правилу Мар-ковникова и приводит к образованию спиртов:
СНз СНз
^С^СНг 4- Н2О СНз—СНз
СНз^ ОН
2-метнлпропен
2 метилипропанол-2
39
Аналогично присоединению галогеноводородов гидратация алкенов протекает по ионному электрофильному механизму с первоначальной атакой протона:
СНз—С—СНз
J-H
СНз
) 'i j
СНз—С—СНз q=zt СНз—С—СН3 + Н+
СНз <^Нз
Реакция применяется для промышленного синтеза спиртов
Присоединение гипогалогенных кислот (гипогалогенирование).
Алкены присоединяют по двойной связи гипогалогениые кислоты (HOCI, HOBr, НО1) с образованием галогенгидринов:
СНг—СН==СН2 + HOCI —СНз—СН—СНг—CI
Н
пропен
1 хлорпропанол 2 припнлепк юргидрнн
Присоединение происходит по электрофильному механизму в соответствии с правилом Марковннкова, т е. положительно заряженный нои галогена направляется к более гидрированному атому углерода при двойной связи.
Гнпогалогеннрованке обычно проводят действием на алкеи водного раствора галогена. Гн по галогенная кислота в этих условиях образуется по реакции
С12 4- Н2О -> HOCI 4- HCI
2.5.2. Реакции восстановления и окисления
Восстановление алкенов (гидрирование), В присутствии катализаторов (тонкоизмельчеииые Pt, Pd нли Ni) алкены присоединяют по даойной саязи водород, образуя алканы
СНз—СН=СН2 4- Н2 СНз—СН?—СНз
пропен
пропан
Каталитическое гидрирование в присутствии палладия и платины протекает в большинстве случаев уже при комнатной темпе-
40
е применение никеля требует нагревания Учитывая боль-стоимость платины и палладия, восстановление алкенов ш^помышлеиности обычно проводят в присутствии то н кои з мель-В₽иносо никеля при температуре 150—300 °C
46 Окисление алкенов’ Окисляются алкеиы довольно легко Ha-явление окисления зависит от используемого окислителя и условий проведения реакции.
Окисление перманганатом калия (реакция Вагиера) Разбавленный раствор КМпО4 в нейтральной нлн гпабошелочной среде окисляет алкены до двухатомных спиртов (гликолей). ~ ~
и выпадает
При этом обесцвечивается розовый раствор КМпО4 коричневый осадок оксида марганца (IV)*
ЗСН2=СН2 + 2КМпЪ4 4Н2О------------->
этилен
---^ЗСН2—СН2 + 2KQH + 2МпО3
in in
этиленгляко ть
Данная реакция была открыта в 1888 г. русским химиком Е Е Вагнером.
Реакция Вагнера широко используется в анализе для обнаружения двойной углерод-углеродной связи.
Концентрированные растворы КМпО4 окисляют алкены с разрывом двойной связи В зависимости от структуры алкена в качестве продуктов окисления образуются кетоны и альдегиды, причем последние окисляются дальше до карбоновых кислот*
СН3
С=СН—СНз
СН3^
2 ыетнлбутен 2
СНз
КМпО+ „
----С=О
СНзСООН
уксусная кислота
Озонирование алкенов. Алкены легко окисляются озоном. Реакция озонирования протекает по сложному механизму с образованием продуктов присоединения озоиа по месту двойной связи, называемых озонидами:
СНз—СН=СН3 4- Оз
пропен
озонид
Многие озониды взрывоопасны. Прн обработке цинком в уксусной кислоте озоннды разлагается, образуя карбонильные соединения (2 моля альдегида или 2 моля кетона илн же 1 моль альдегида и 1 моль кетоиа в зависимости от структуры алкена):
О-^О
/ А \
сн~сн/ \ сн2
"ХО
Zn + CHiCOOH _тт Xм -------2► сн3-< + <
ХН
озонид
уксусный альдегид формы ьдегвд
Полученные карбонильные соединения могут быть идентифицированы, что позволяет использовать реакцию озонирования для определения положения двойной связи.
Поскольку озон реагирует с алкенами количественно (1 моль озона расходуется на 1 моль алкена), эта реакция может также применяться для установления числа двойных углерод-углеродных связей в молекуле.
Окнсленйе алкенов кислородом и. пероксикислотами. Кислород воздуха в присутствии серебряного катализатора окисляет алкеиы при нагреааннн с образованием эпоксидов:
2СНг=СН2 4- 02
зтнленокснд
этилен
Эта реакция применяется в промышленности для получения эти-ленокснда (оксирана).
Аналогично алкены окисляются перокси кислота ми (реакция Прилежаева). Так, прн обработке алкенов пероксибензойной кислотой образуются эпоксиды:
О
СН3—СН=СН2 + СБН5—С ---
^О—ОН
пропен
перокснбензойлая ннслста
--► СНз-СН-СН2 + СбН5СООН
пропиленокснд
бензинная кислота
Реакция изучена в 1909 г. советским хнмиком-оргаинком Н. А. Прилежаевым.
42
2.5.3. Полимеризация алкеиов
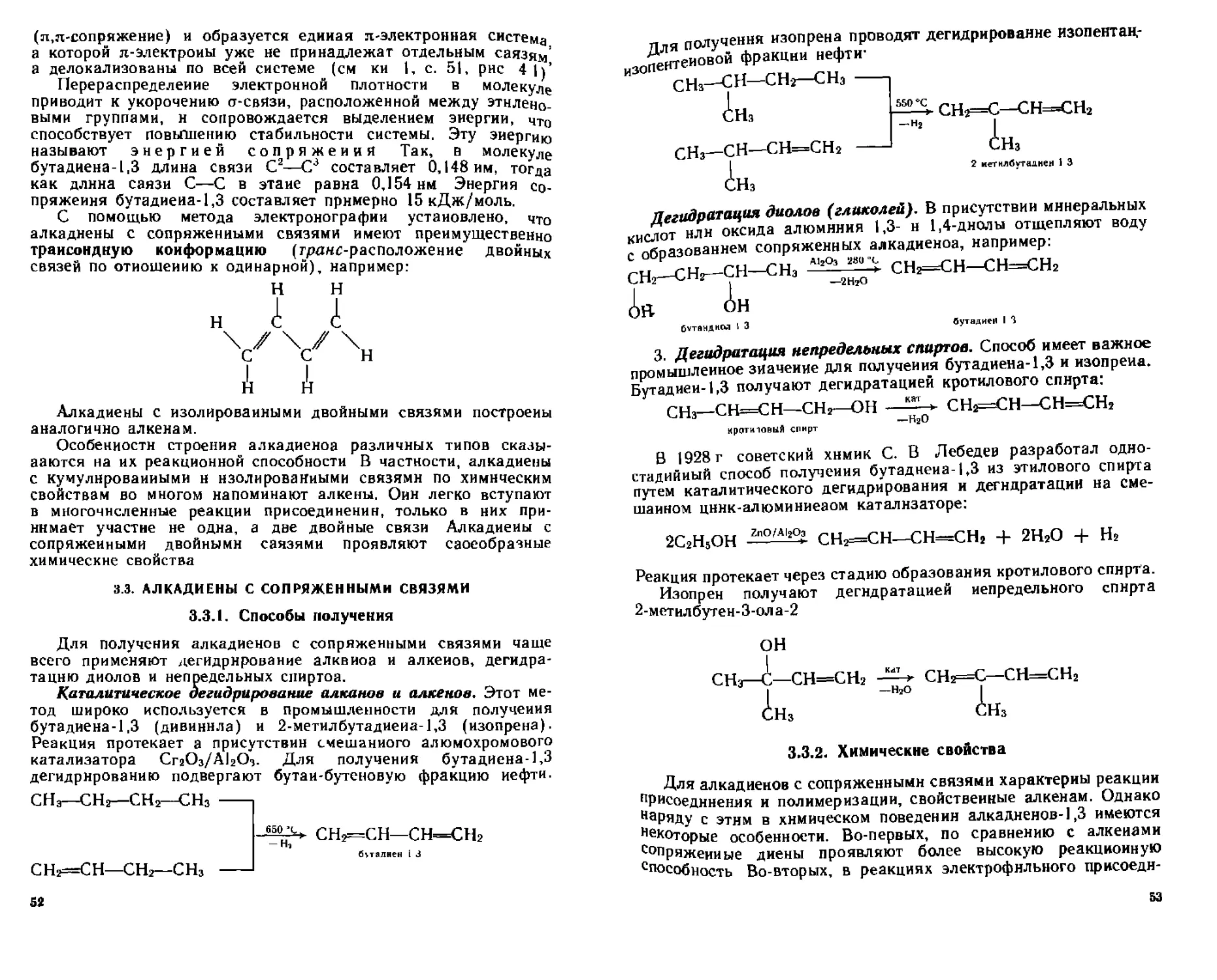
Полимеризацией называют процесс соединения друг с другом молекул низкомолекулярных веществ (мономеров) с образованием высокомолекулярных соединений (полимеров).
В реакцию полимеризации могут вступать молекулы одного ого же мономера» а также молекулы двух н более разных моио-« . Полимер, состоящий из одинаковых мономеров, назы-
вается гомополимером, а полимер, в состав которого входят двв более разных мономера — сополимером. Реакция получения сополимеров называется реакцией сополимеризации.
Полимеризация алкенов представляет собой последовательное соединение молекул алкена друг с другом вследствие разрыва двойной связи. В общем аиде полимеризацию алкенов можно выразить следующей схемой:
Число повторяющихся мономерных звеньев п называют степенью полимеризации. В зависимости от степени полимеризации из одного и того же мономера можно получить вещества с разными свойствами. Процесс полимеризации осуществляется в присутствии катализаторов (инициаторов) и включает три основные стадии: зарождение цепи (инициирование); рост цепи; обрыв цепи.
В зависимости от структуры исходного мономера, природы инициатора и условий реакции (температуре, давление) полимеризация алкенов может происходить по радикальному и ионному (катионному) механизмам. Особым типом полимеризации является полимеризация в присутствии металлоорганических соединений, получившая название координационная полимеризация.
Свободнорадикальная полимеризация. По радикальному механизму алкены полимеризуются в присутствии пероксндных соеди-О О
нений, таких, как пероксид ацетила — СНз—i—О—О—(!—СНз, О О
ПеРокснд бензоила С6Н5—О—О——СбНь, и других, спо-сй^лЫХ ПРИ высоком давлении и температуре распадаться на °бодные радикалы. Эти свободные радикалы затем присоединяются к алкеиу с образованием новых радикалов. Далее пронс-однт последовательное присоединение других молекул алкена.
СтУш,ая активироааииая цепь при радикальной полимеризации
43
представляет собой свободный радикал. Обрыв цепи осуществ ляется чаще всего путем димеризации свободных радикалов Механизм радикальной полимеризации этилена а присутствии пероксида ацетила можно представить следующим образом:
СНз—С—О—О—С—СНз
2СНз—С—О
сн2=н:н2 4- CHj—С—О* -СН3—С—О—СН2—СН$ (за-
J (| рождение цепи)
О О
СНз—COO—СН2—СН5
сна=<н3
СНз—СОО—СН^—СНг—СНг—Сн2
In -1)CH2=CH2 -------------
—»- СНз—COO—(СНа—СНг)-^СНз—СН2 (рост цепи) СНз—СОО—(СН2—СН2у^-СН2—СН5 +
4- ’СНг—СН2-ЧСН2—CH2)^OOC—СНз *
СНзСОО— (СН<2--СН4^СН2—сн2
СНзСОО—(СНз—СНг)^—СНг—<^Н2 (обрыв цепи)
Так в промышленности получают полиэтилен и полипропилен высокого давления.
Катионная полимеризация. Катионная полимеризация алкенов инициируется протонными кислотами или кислотами Льюиса (AICI3, ВЬ'з и др.). Реакционным центром растущей полимерной цепи является карбкатион. По ионному катиоииому механизму наиболее легко полимеризуются несимметрично построенные
R
алкены обшей формулы С=СНг, из которых образуются
относительно стабильные промежуточные карбкатионы В промышленности этот метод применяют для полимеризации изобутилена:
сн/
изобутилен
+ н+
'с-сн, СН/
зарождение цепи
о
44
СНг^СсНз Сй/
₽ СНз I и Ы2ХС-СНзг
СН, I .
сн3-с—сн2—с I I
сн3
сн3
снэ
VIli 1+ сн-с in vrli Л
рост цепи
СНз
СНз—'
СНз’
С На—С1
СНз—
СНз Г СНзЛ
СНз
-СН^С^
и ХСН3
in? тизобути лен
обрыв цепи
Координационная полимеризация. Координационная полимеризация алкеиов представляет собой довольно сложный процесс, протекающий в присутствии комплексных металлоорганических катализаторов. Эти катализаторы открыты немецким химиком К. Циглером и модифицированы итальянским химиком Дж. Наттом, а поэтому получили название катализаторы Циглера—Натта. Наиболее распространенным из них является комплекс трнэтнл-алюминия с хлоридом титана АНСгНОз* TiCh-
Механизм координационной полимеризации пока точно не известен, однако установлено, что образование полимера про-исходит путем внедрения молекул алкена ио связи металл—углерод растущей полимерной цепи.
Полимеризация алкенов в присутствии катализаторов Циглера — Натта позволяет получать высокомолекулярные полимеры при относительно низких давлении и температуре. Этот метод широко используется в промышленности для производства полиэтилена и полипропилена низкого давления.
Важной особенностью координационной полимеризации является ее стереохимическая направленность При свободнорадикаль-Нон и катионной полимеризации образуется полимер нерегулярного строения, т. е. с произвольной стереохимической конфигурацией. Такой полимер называют атактическим. Полимеризация алкенов с использованием катализатора Циглера — Натта приводит к образованию стереорегулярного полимера, получившего Название изотактический. Пространственное строение атактического и изотактического полипропиленов имеет вид
45
Н СН3СН3 Н СНЭ Н Н сн3
XCH2Z 4CHZ XCH2Z 4CH2Z 4CHZ
атактический полипропилен
CHj Н СН3 Н CHj Н СНЭ Н
С
\ / \ / \ /\ /\ /
СН2 СН2 СН2 СН3 СН3
изотактический полипропилен
Изотактическим полимерам по сравнению с атактическими свойственны большая прочность и более высокие температуры плавления.
2.5.4. Аллильное галогенирование алкенов
При действии на алкеиы таких галогенирующих реагентов, как С1г, N-бромсукцинимнд, н других в присутствии инициаторов процесса образования саободиых радикалов (пероксиды, температура, УФ-свет) происходит не присоединение галогена по месту разрыва двойной связи, а свободнорадикальное замещение на галоген атома водорода, находящегося при атоме углерода в а-положенни к двойной связи (аллильное положение). Так, при температуре 500—600 °C пропен реагирует с хлором, образуя аллилхлорид:
СН2=СН—СНз + С12 СН2=СН-СН2—Cl 4- НС1
пропен а.плилхлорид
Эта реакция используется в промышленности для синтеза глицерина.
Бромирование алкенов по аллильному положению чаще всего осуществляется действием N-бромсукцииимида в присутствии пероксидов.
СНг=СН—СНз—й +
пероксиды
N брочс\,кцниимнд
4А
о
--- СН2=СН—CH—R +1 N— H
Br
О
сукцинимид
Аллильное галогенирование протекает по свободнорадикальному механизму (см. с. 20) и включает стадию образования аллильного радикала. В результате сопряжения неспареиного электрона с л-электронами двойной связи аллильные радикалы являются более устойчивыми, чем обычные алкильные:
сн2=сн-<н2 > снэ—сн=сн2:
Поэтому замещение атома водорода на галоген в аллильном положении алкенов происходит легче, чем в алканах.
2.6. ИДЕНТИФИКАЦИЯ АЛКЕНОВ
В отличие от алканов, алкены идентифицируют с использованием инструментальных и химических методов. Химические методы основаны на наличии в структуре влкенов реакционноспособной двойной связи. Обычно для доказательства наличия двойной связи используют реакцию взаимодействия алкенов с раствором брома в хлороформе или четыреххлористом углероде (наблюдается обесцвечивание растворов брома) и реакцию окисления разбаа-лениым раствором КМпО< в нейтральной или щелочной среде, так называемая проба Байера (наблюдается исчезновение розово-фиолетового окрашивания перманганата). Однако эти реакции не являются строго специфическими иа двойную связь, поэтому Для идентификации алкенов используют комплекс данных, полученных с помощью химических и физических методоа исследования.
Из физических методов широко применяют ИК-, ПМР-спектро-скопию и масс-спектрометрию. В ИК-спектрах алкенов имеется полоса поглощения в области 1640--1670 см-', обусловленная валентными колебаниями связи С=С и полоса в области ЗОЮ— ^690см~1( отвечающая валентным колебаниям связи Cspa—Н. *хроме того, в спектре присутствуют полосы деформационных колебаний связей Сэл—Н, которые для цис-изомера проявляются в области 650—720см-1, для транс-изомера — 960—980см-1.
ПМР-спектры алкенов характеризуются нвличием сигналов алкеновых протонов при 6 = 4,5—6,0 м. д.
Распад алкенов при измерении масс-спектров — достаточно л°Жнын процесс. Вместе с тем легче всего происходит аллильное
47
расщепление молекулярного иона вследствие образования стабц. лизированного за счет полярного резонанса аллил-катиона:
^-^сн2-сн-сн2]+ —►
R*
СН^СН-^ аллил-катиоц
2.7. ОТДЕЛЬНЫЕ ПРЕДСТАВИТЕЛИ. ПРИМЕНЕНИЕ
Этилен (СН2=СНа). Бесцветный газ с очень слабым запахом, мало растворим в воде, горйт ярким- коптящим пламенем, с воздухом, образует взрывчатые смесн. Служит сырьем в промышленности для получения этанола, этиленгликоля, уксусного альдегида, полиэтилена н др.
Пропилеи (СНг=СН—СН3). Бесцветный газ со слабым запахом. Широко используется в промышленном органическом синтезе для получения изопропилового спирта, глицерина, ацетона, полипропилена и других ценных органических соединений.
КОНТРОЛЬНЫЕ ВОПРОСЫ И УПРАЖНЕНИЯ
1 Назовите следующие соединения по заместительной номенклатуре-
СНз
а) СНз—СН—СН=С—СН31 б) СН2=С—^Н—СНз—СН3;
<!н2 СНз <^Н2
СНз СНз
в) СНз—СН—<
СНз
Н3
Н3
2. Какие из приведенных алкенов могут существовать а виде цис- и транс-изомеров: а) пентен-2; б) 2-метилпентен-2; в) гексен-3; г) 3,4-диметилгексен-3; д) 3,4-диэтилгексен-2; е) 3,4-днэтилгексеи-3? Приведите формулы геометрических изомеров н обозначьте конфигурацию по цис, гране- и Е, Z-системе
3 Напишите уравнения реакций и назовите образующиеся а л кеньг
а) СНз—СН—СН—СН3 А~3^; б) СНЭ—СН—СН2—СНз^;
I I 300 °C I 1
СНз ОН ОН
в) СНт—CHg CHBr—CH3 г) СНз-^CHs—C=sC—СНз
кои
- - - > ;
(С2И5ОН) На
Pd. PdO
4 Напишите уравнения последовательных реакций н назовите полученные соединения:
/П № з
CHs-CH—СНз—СНз -S2> № 1 --------——Ь № 2--------№ 4
I ” tc,ifcOH.0 о,
/'ll -> iN? 5
ЬПз H2O1Z11)
5. Приведите механизм реакции пропена с бромом.
6. Напишите схемы взаимодействия бутена-1 с указанными реагентами. Приведите механизмы реакций с реагентами, отмеченными звездочками: а) Вг2 (CCL); б) НВг*; в) HjO (Н+)*; г) H2SO4
7. Какими качественными реакциями можно отличить соединения в следующих парах: а) н-пентан и пентен-2; б) гексен-2 и гексен-3?
Глава 3
АЛКАДИЕНЫ
Алкадиенами называют углеводороды алифатиче< с ко го ряда, содержащие две двойные связи. Общая формула алкадиенов СлН2п-2.
По взаимному расположению двойных связей в молекуле различают три основных типа алкадиенов:
I. С кумулированными двойными связями (двойные саязн
расположены у одного атома углерода): С=С=С . Такие
соединения еще называют алленами, по тривиальному названию простейшего представителя этого ряда — аллеиа СН2=С=СН2. 2. С сопряжёнными двойными саязями (двойные связи разделены одной простой связью); .
3. С изолированными двойными связями (двойные связи разделены двумя или более простыми связями):
3.1. НОМЕНКЛАТУРА
аиалоХано1лкеммНеНя^«^НОМеНКЛаТ-уРе ИЮПАК образуют суффиксом «диен с икячя» Даух двойных связей обозначают Углеродной цепи: УКаЗЗННем “ения каждой нз них в главной
5 4 3 2 1
С Нэ С н=с Н—с Н=С Н 2
пента диен-1,3
СНз—СН=С=СН2
бутадиен 1.2
1 2 3 4
СНу=С—СН=СН2
<?:Нз
2 метилбутадиен-1.3
50
лля некоторых алкадиенов используют тривиальные икрр°ациоиальные названия:
СН2=С'-СН=СНз СН2=СН—сн=сн2
(Ьэ
изопрен дивинил
СН2=СН—СН2—СН--=СН2
днвипн. сметан
3.2. СТРОЕНИЕ АЛКАДИЕНОВ
В молекуле алкаднеиов с кумулированной системой двойных связей атом углерода, образующий дае двойные саязн, находится в состоянии sp-гибриднзацин, а соседние с ним атомы углерода — в sp2-гибридизации (рис. 3.1, а). Эти три атома углерода расположены в пространстве лннейио, а л-саязи находятся в двух взаимно перпендикулярных плоскостях. Четыре заместителя прн углеродных атомах в зр2-гибрндизации тоже расположены в двух взаимно перпендикулярных плоскостях (рис. 3.1, б).
1Г
а б
Рис 3 1 Схема пространственного строения алленов
Вследствие такого пространственного строения молекулы алленов, у которых при каждом из атомов углерода в 5р2-гибридизацни имеются разные заместители, являются хиральиыми, а следовательно, для них возможна оптическая изомерия, например:
С-С
? нк ^СН3
j с-с-с^*
$ сн3х >н
к л
энантцомеры 1,3-димегилаллена
fa ^CJ1H Двойные связи в молекуле разделены одной о-связью
1 лкадиены с сопряженными связями), происходит дополни-льное перекрыааиие р-электрониых облаков соседних л-связей
51
(л,л-£Опряжение) и образуется единая л-электронная система а которой л-электроны уже не принадлежат отдельным связям’ а делокализованы по всей системе (см ки 1, с. 51, рнс 4 И’
Перераспределение электронной плотности в молекуле приводит к укорочению сг-связи, расположенной между этиленовыми группами, н сопровождается выделением энергии, что способствует повышению стабильности системы. Эту энергию называют энергией сопряжения Так, в молекуле бутадиена-1,3 длина связи С2—CJ составляет 0,148 им, тогда как длина саязи С—С в этаие равна 0,154 нм Энергия сопряжения бутадиена-1,3 составляет примерно 15 кДж/моль.
С помощью метода электронографии установлено, что алкадиены с сопряженными связями имеют преимущественно трансоидную конформацию (транс-расположение двойных связей по отношению к одинарной), например:
Н Н
Алкадиены с изолированными двойными связями построены аналогично алкенам.
Особенности строения алкадиеноа различных типов сказываются на их реакционной способности В частности, алкадиены с кумулированными н изолированными связями по химическим свойствам во многом напоминают алкены. Оин легко вступают в многочисленные реакции присоединении, только в них принимает участие не одна, а две двойные связи Алкадиены с сопряженными двойными связями проявляют своеобразные химические свойства
3.3. АЛКАДИЕНЫ С СОПРЯЖЕННЫМИ СВЯЗЯМИ
3.3.1. Способы получения
Для получения алкадиенов с сопряженными связями чаще всего применяют дегидрирование алквиоа и алкенов, дегидратацию диолов и непредельных спиртов.
Каталитическое дегидрирование алканов и алкенов. Этот метод широко используется в промышленности для получения бутадиена-1,3 (дивинила) и 2-метилбутадиеиа-1,3 (изопрена). Реакция протекает а присутствии смешанного алюмохромового катализатора СгзО3/А12Оъ Для получения бутадиена-1,3 дегидрированию подвергают бутаи-бутеновую фракцию нефти.
СНз—С Н г—С Н 2—СНз
СНг^=СН—СНг—СНз
сн^сн—сн=сн2 — П’З
бътаднен I 3
52
Иля получения изопрена проводят дегидрирование изопентац-зопентеиовой фракции нефти-
И СН3-<Н—СН2—СНз -------
<Lh3 СН2=С—сн=<н2
-Н2 |
СНз—СН—СН=СН2 -------- СНз
2 метнлбутаднен 1 3
СНз
Дегидратация диолов (гликолей). В присутствии минеральных кислот НЛН оксида алюминия 1,3- н 1,4-днолы отщепляют воду с образованием сопряженных алкадиеноа, например:
СНг—СНг-СН—СНз СНг=СН—СН=СН2
Ан ОН
б*тандиол 1 3 бутадиен I 3
3. Дегидратация непредельных спиртов. Способ имеет важное промышленное значение для получения бутадиена-1,3 и изопрена. Бутадиен-1,3 получают дегидратацией кротилового спирта:
СНз—СН=СН—СНг—ОН СН2=СН—сн=сн2
. ~Н2О
иротиювый спирт
В 1928 г советский химик С. В Лебедев разработал одностадийный способ получения бутадиена-1,3 из этилового спирта путем каталитического дегидрирования и дегидратации на смешанном цннк-алюминиеаом катализаторе:
2С2Н5ОН СНг=СН—СН=СН2 + 2Н2О + Н2
Реакция протекает через стадию образования кротилового спирта.
Изопрен получают дегидратацией непредельного спирта 2-мет илбутен-З-ола-2
ОН
СНз—С—сн=сн3 сн?=с—СН=СН2
I —НаО I
СНз СНз
3.3.2. Химические свойства
Для алкадиенов с сопряженными связями характерны реакции присоединения и полимеризации, свойственные алкенам. Однако наряду с этим в химическом поведении алкадиенов-1,3 имеются екоторые особенности. Во-первых, по сравнению с алкенами °пряжеииые диены проявляют более высокую реакционную пособность Во-вторых, в реакциях электрофильного присоедн-
53
неиня чаше всего образуется два продукта, из которых один является результатом присоединения по месту двойной связи (1,2-присоединение), а второй — по концам сопряженной системы (1,4-присоедииение). Соотношение этих продуктов зависит от условий проведения реакции (температура, растворитель), а также природы электрофильного реагента.
Гидрирование. Водород в момент выделения образует с алкадиенами- 1,3 обычно продукты 1,4-присоедииеиия, иапрнмер:
СНг=СН—СН=СН2 ~-Н-°Н + а*а> СНз—СН=СН—СН3
В присутствии катализаторов (Ni, Pt) алкадиены-1,3 присоединяют водород а 1,2- и 1,4-положения с образованием соответствующих алкеиов, которые подвергаются дальнейшему гидрированию до алкаиов:
2СНа=СН—СН=СН2 —
СНз—СН2—СН=СН2
--СНз—СН=СН—СНз 1
> 2СН3—СН2—СНг—СНз
Присоединение галогенов. Присоединение галогенов к сопряженным диенам приводит к образованию смеси продуктов 1,2-н 1,4-присоединення, состав которой зависит от природы галогена, структуры диенового углеводорода и условий проведения реакции. Как правило, при повышении температуры и переходе от хлора к ноду возрастает выход продукта 1,4-присоедниеиия. Например, в процессе бромирования бутадиена-1,3 при температуре —80 °C образуется преимущественно продукт 1,2-прнсоединеиия (3,4-ди-бромбутен-1), а при 40 °C — продукт 1,4-присоедннеиня (1,4-дибром бутеи-2):
СНг=СН—СН=СНЙ + Вг2
Вт Вг
1.2 присоединение
СНг—СН—СН=СН2
1 4-ирнсоединенне
СН^СН=СН—сн2
Аналогично алкенам присоединение галогенов к сопряженным алкадиенам происходит по ионному электрофильному механизму-Особенность механизма состоит а том, что электрофильная частица всегда атакует концевой атом углерода сопряженной системы,
54
льку при этом образуется мезомерно стабилизированный п°л«гятион строение которого можно представить граничными структУРами 1 и 11:
СН2=СН—СН=СН2 + Вг2-------> CH2=j=CH—сн=сн2
Вг^^Вг6-
л комплекс
—Вг~
2 СНг—СН=СН—£н2
Вг
И
Последующая атака карбкатиона бромид-иоиом приводит к образованию продуктов 1,2- (111) и 1,4-присоедииеиия (IV):
СН2—Ен—СН—СН2
СНг-СН—сн=сн3
Вг
Вг- / Вг Вг Ш
1
СНг—СН=СН—Ен;
сн^—сн=сн—сн2
Вг
IV
Присоединение гало г еноаодородов. Как и в случае галогенирования, присоединение к сопряженным диенам галогено-водородов происходит с образованием продуктов 1,2- и 1,4-присоединения:
I 2 СНз_СН_сН=СН2
СН2=СН—СН=СН2 -НО-
3 хлорбутен 1
1,4 присоединение ________„„______—
I морбутен 2
Реакция Дильса — Альдера (диеновый синтез). Реакция Дильса — Альдера основана на взаимодействии сопряженных Лиеиов с веществами, имеющими в своем составе двойную илн тройную углерод-углеродную связь и получившими в данном слу-сае Иазвание диенофилы. Особенно легко эта реакция происходит т Диеиофилами, содержащими активированную двойную с а язь, ’ е- когда двойная связь находится в сопряжении с элекгроно-СппПтоРН0^ или электроиодоиорной группой (CN, NO2, СНО,
°’ СООН, COOR, галогены, OR и др.).
55
В процессе реакции сопряженные диены присоединяют диец0 филы в положение 1,4 с образованием циклических структур
бутадиен-1,3
альдегид
Реакция Дильса — Альдера протекает по молекулярному механизму, который характеризуется синхронным процессом разрыва и образования связей в реагентах. Этд реакция относится к реакциям [4 2] -циклоприсоединения, поскольку в ней при-
нимает участие 4л-электроиная система диеиа и 2л-электронная система диенофила. Реакция впервые была открыта в 1928 г, немецкими химиками О. Дильсом и К- Альдером. Диеновый синтез широко используется для синтеза полициклических соединений, в том числе при синтезе биологически активных соединений сложного строения.
Полимеризация. Важным свойством алкадиенов с сопряженными связями является их склонность к полимеризации. В качестве инициаторов чаще применяют органические и неорганические пероксиды, металлоорганические соединения, щелочные металлы. Образование полимера происходит преимущественно по типу 1,4-присоединения:
пСН2=СН—СН=СН2 --------— (—СН2—СН=СН^СНг-)—
диен 1,3 1.4 полиб¥тпдиен
При полимеризации замещенных диенов 1,4-присоединенне осуществляется по принципу «голова к хаосту», например:
СНз пСН2=^—СН—СН2
неопрен
Реакции полимеризации сопряженных дненов широко используются в производстве синтетического каучука.
56
3.3.3. Натуральный и синтетический каучук
(природный) каучук. Его получают из латекса ииый сок) тропического растения гевеи, произрастающей (*ле м образом в Бразилии. Выделяющийся при подсочке гЛа00Ьев млечный сок содержит 20—60 % каучука, который ж да ют добавлением муравьиной или уксусной кислоты.
°СЭПо химическому строению натуральный каучук представляет Лой линейный стереорегуляриый полимер изопрена (поли-изопрен), имеющий ц ис-кои фигурацию изопреновых звеньев:
CHt >н„,
•..............• ^с-с;
...ТСН/ ''CHiH-CHj. /CH2tf_CH2x
ch; h
натуральный каучук, цис -полиизопрен
2 '
’Молекулярная масса натурального каучука составляет в среднем 100 000 — 150 000.
В природе встречается также полиизопрен с транс-кои фигурацией изопреновых звеньев, получивший название гуттаперча:
сн3 сн3
'' J " " "Lc” ’ Jch<[ ' ‘ "с” ’ ’ CH J
>CHf
.....-А-................А"~
гуттаперча, траис-полиизопрен
Гуттаперча, в отличие от натурального каучука, ие обладает эластичностью.
Синтетический каучук. Большая потребность промышленности в каучуке и отсутстане во многих странах, в том числе и в бывшем СССР, природного сырья обусловили разработку синтетических способов его получения.
Впервые в промышленном масштабе синтетический каучук получен в 1932 г. в СССР полимеризацией бутадиена-1,3 в присутствии металлического натрия (способ С. В. Лебедева). Полу-Ченный полимер содержит примерно 70 % звеньев 1,2-присоединения и 30 % звеньев 1,4-присоединения. Из-за малого содержания Фрагментов 1,4-присоедииения такой каучук уступает по саоей ластичности натуральному. В последующие годы полимеризацией ^Та Диен а-1,3 над металлоорганическими катализаторами получен
Нейный, стереорегуляриый бутадиеновый каучук с цис-конфи
57
гурацией мономерных звеньев, не уступающий по свойствам натуральному:
бутадиеновый каучук (цис-полибугаднен)
В настоящее время промышленность производит много видов синтетических каучуков с различными свойствами. Так, полимеризацией изопрена в присутствии комплексных катализаторов Циглера — Натта получают изопреновый каучук стереорегулярио-го строения, полимеризацией 2-хлорбутадиена-1,3 (хлоропрена) СН2=СС1—СН=СН2 — хлоропреновый каучук. Получены также сополимеры бутадиена-1,3 со стиролом СбНэ—СН=СНа (бута-диен-стирольиый каучук), с акрилонитрилом СНа=СН—-CN (бутадиен-нитрнльный каучук) и др.
Вулканизация каучука. Для придания каучуку повышенной прочности, эластичности, устойчивости к изменениям температуры и действию химических реагентов его подвергают вулканизации. Процесс вулканизации состоит в обработке каучука серой при нагревании. В результате вулканизации происходит поперечное сшивание линейных молекул полимера за счет образования сериых мостиков. При этом каучук превращается в эластичную массу, называемую резиной.
3.3.4. Идентификация сопряженных диеиов
Аналогично алкеиам наличие двойных связей в сопряженных диенах может быть доказано обычными реакциями иа кратную связь (реакция с бромом и реакция с КМпОЦ- Если проба на кратную связь положительна, то идентификацию диенов-1,3 проводят по образованию кристаллических продуктов присоединения в реакции диенового синтеза с ангидридом малеиновой кислоты при нагревании:
иаленновЫЙ ангидрид
Для ИК-спектров сопряженных диенов, по сравнению с алкенами, характерно смещение полосы валентных колебаний саязи С=С в длинноволновую область (1600—1620 см’1) и увеличение ее интенсивности. В УФ-спектрах сопряженных диеиов наблюдается интенсивное поглощение в области 200—220 см’1. ПМР-спектры диенов не имеют существенных отличнй от спектров алкеиов.
3.3.5. Отдельные представители. Применение
Бутадиеи-1,3 (дивинил) СН2=СН—СН=СНг. Бесцветный газ с характерным запахом, прн 20 °C существует бреимущественно в виде транс-изомера Хорошо растворяется в эфире и бензоле, нерастворим в воде. При температуре 420 °C самовоспламеняется. Применяется для получения синтетических каучуков, пластиков, в синтезе алициклических соединений
СНз
2-Метнл бутадиен-1,3 (изопрен) СНз=СН—С==СН2. Бесцветная жидкость (т кип. 34,1 °C) Не растаоряется в воде, хорошо растаорим в большинстве углеводородных растворителей. В высоких концентрациях обладает наркотическим действием, в малых — раздражает слизистые оболочки дыхательных путей и глаз. Изопрен является основой многих природных соединений, таких, как натуральный каучук, терпены, стероиды. Широко прнменяетсн в промышленности для получения изопренового каучука, душистых веществ, лекарственных средств.
КОНТРОЛЬНЫЕ ВОПРОСЫ И УПРАЖНЕНИЯ
1. Напишите структурные формулы и укажите, к какому типу алкадиеиоа относятся следующие соединения: а) дивинил; б) пеитадиен-1,4; в) нзопрен; г) 1,2-бутаднен; д) 2,3-диметил-бут адиен-1,3.
2. Объясните, как образуется сопряженная система в молекуле пентадиена-1,3. Имеется ли сопряжение в молекуле пеита-диена-1,4?
3. Приведите два способа получения бутадиена-1,3.
4. Какие продукты образуются при взаимодействии 2-метил-пеитадиена-1,3 с бромоводородом? Приведите механизм реакции.
5. Напишите схему реакции пентадиена-1,3 с акрилонитрилом (СН2=СН—CN).
6. Чем отличается по химическому строению натуральный каучук от гуттаперчи? Приведите фрагмент молекулы стерео-регулярного бутадиенового каучука.
Глава 4 АЛКИНЫ
Алкинами называют углеводороды алифатического ряда, содержащие одну тройную связь. Общая формула алкинов СпН2п__2.
Простейшим представителем этого ряда соединений является ацетилен С2Н2, поэтому алкины называют ещё ацетиленовыми углеводородами.
4.1. НОМЕНКЛАТУРА И ИЗОМЕРИЯ
По номенклатуре ИЮПАК названия алкинов образуют от названий соответствующих алканов, заменяя суффикс -аи на -ин с указанием положения тройной связи в цепи углеродных атомов. Нумерацию главной углеродной цепи начинают с того конца, к которому ближе расположена тройная связь:
5 4 3 2 1
HfeCH СНз—feCH СНз—СН—feC—СНз
<1Н
СНз *тин пропин 4 четилпентнн 2
Наряду с номенклатурой ИЮПАК для простейших углеводородов часто применяют рациональные названия. Согласно рациональной номенклатуре, ацетиленовые углеводороды рассматривают как производные ацетилена, в котором атомы водорода замещены на углеводородные радикалы:
СНз—С=СН СНз—СНг—С=СН СН3—feC—СН3
чети ла нет к лен этнлаиетнлен димети тацетнлен
Для первого представителя гомологического ряда алкинов сохранилось тривиальное название ацетилен.
Названия углеводородных остатков (радикалов) алкинов образуют путем добавления к названию углеводорода суффикса -ил:
3 2 I
нс==с— СНз—С=с— нс^с—CHs—
этннкл пропин-1-ид пропин 2 нл,
пропаргил
00
Для алкиноа харктериа структурная изомерия, которая обусловлена различной структурой углеродной цепи (изомерия цепи) и различным положением тройной связи (изомерия положения):
--СН3—СН—С=СН
З'нетнлбутин I
изомерия положения
I-----------1----------1
--> СНэ—СНз—СНг—С=СН СНа—С=С—СН2—СНз
пентин I пентин 2
4,2. СПОСОБЫ ПОЛУЧЕНИЯ
Длинны чаще всего получают дегидрогалогенированием ди-галогеизамещеииых алканов и алкилированием ацетилена. Для получения ацетилена существует ряд специфических способов.
Дегидрогалогенирование дигалогеналканов и галогеналкенов. Геминальные дигалогензамещеиные алкаиов (атомы галогена расположены у одного и того же атома углерода) и вицинальные дигалогеиалканы (атомы галогена расположены у двух соседних атомов углерода) в присутствии спиртового раствора тидроксида натрия или калия при нагревании отщепляют галогеноводород с образованием алкииоа:
Гн Вг
Ч-’-Н' сн3-с-сн rl—!-.
Н Вг;
2NaOH, t
(С^ОН) * СНз"С=СН + 2NaBr + 2Н2О
IJ днбромлропаи
лропян
Вг
Вг
СНз—СН—СН2 ^‘°н1>СНз—С^СН + 2NaBr + 2Н2О ICiIIbOH)
1.2 либрампрепам пропин
В аналогичных условиях образуют алкины также галоген-алкены, содержащие атом галогена у атома углерода с двойной связью:
СНз -С=СНа --—Л СН3—С=СН + NaCI + Н2О
I (СаН^он)
61
Алкилирование ацетилена. Алкилированием называют ваедение алкильной группы в молекулу органического соединения. Данным способом из ацетилена можно получить его гомологи. С этой целью вначале при действии на ацетилен амидом натрия NaNH2 в жидком аммиаке или алкнлмагнийгалогенидами, например метнлмагнийиодидом CHaMgl а эфире получают ацетиленид натрия или этинилмагиийгалогеннд соответственно, которые затем обрабатывают галогеналкаиом:
НС=СННС—С Na <аЩ~в»г НС==С—С2Н5 '
—ЧНз — МйВг 4,
ацетиленид натрин бутин 1
НС==СН HC==CMgI НС==С—СНЯ
ЭТИ Н И Л Ч А ГН И Й fi ропнн
и одна
Получение ацетилена из метана (промышленный метод). При нагревании метана до 1500 °C образуется ацетилен:
2СН4 СН==СН + ЗН2
Получение ацетилена из карбида кальция (промышленный метод). Прн взаимодействии карбида кальция с водой образуется ацетилен:
СаС2 + 2Н2О ---> НС=СН + Са(ОН)2
Этим способом получают основное количество ацетилена, используемого для сварочных работ,
4.3. ФИЗИЧЕСКИЕ СВОЙСТВА
По физическим свойстаам три первых представителя гомологического ряда алкииоа прн нормальных условиях представляют собой газы, далее следуют жидкости (Сб—С15), а начиная с углеводорода С16Н30 алкииы являются твердыми веществами. Изменения температур плавления и кипения в гомологическом ряду алкинов подчиняются основным закономерностям, характерным для алканов и алкенов.
4.4. ХИМИЧЕСКИЕ СВОЙСТВА
Химические свойства алкинов обусловлены наличием в нх Структуре тройной связи. Атомы углерода, связанные тройной связью, находятся в состоянии sp-гибрндизации. Тройная саязь представляет собой сочетание одной п-связи и двух л-связей, расположенных во взаимно перпендикулярных плоскостях (см. ки.1, с. 42, рис. 3.11). Следовательно, для алкииов, как и для соединений с двойными связями, характерны реакции электрофильного присоединения за счет разрыва л-связей. Эти реакции протекают, как правило, в две стадии. После присоединения
02
одной молекулы реагента алкин превращается в замещенный алкен, который может присоединять по месту разрыва л-связи вторую молекулу реагента. Однако вследствие большей электроотрицательности sp-гибридизоваииых атомов углерода по сравнению с углеродами в $р2-гибриднзацин л-связи алкинов труднее вступают в химическое взаимодействие с электрофильными реагентами, чем л-саязь алкеиоа. Поэтому алкины по сравнению с алкенами несколько менее активны в реакциях электрофильного присоединения. Кроме того, благодаря высокой электроотрицательности атома углерода в ^-гибридизации алкииы с концевой тройной связью R—-С=С—Н обладают слабой СН-кис-лотностью и способны замещать атом водорода на металлы и другие группы. Наконец, аналогично алкенам, алкины вступают также в реакции окисления, восстановления и полимеризации.
4.4.1. Реакции электрофильного присоединения (ДЕ)
Наиболее важными реакциями электрофильного присоединения а ряду алкинов являются галогенирование, гидрогалогеннро-вание и гидратация.
Галогенирование, Алкииы довольно легко присоединяют по месту тройной связи хлор и бром. В реакцию может вступать одна или две молекулы галогена. В результате присоединения одной молекулы галогена образуется преимущественно транс-дигалогеиалкен. Присоединение второй молекулы галогена идет труднее. При этом образуется тетразамещеиный алкай
СН3—С=СН
транс 1.2 дибром 1,1,2 2-тстрабром
пропан
Механизм реакции аналогичен галогенированию алкенов (см. с. 35).
Гидрогалогенирование. Алкииы могут присоединять одну или две молекулы галогеиоаодорода (НС1, НВг) с образованием соответственно моиогалогеиозамещенных алкенов или гемннарь-ных днгалогеиалканов (оба атома галогена находятся при одном атоме углерода). Присоединение идет по правилу Марковникова:
Вг
СНз—С^сн СНз—С=СН2-Щ^ СНз—С—СНз
Вг Вг
। 2 брочнропен 2,2-дибром про пан
63
По механизму реакция аналогична гидрогалогеннроваиню алкеиов.
Гидратация (реакция Кучерова). В присутствии солей ртути (11) в качестве катализатора алкины присоединяют воду. Присоединение происходит а соответствии с правилом Марков-ннкова. При этом из ацетилена образуется уксусный альдегид, другие алкииы образуют кетоны.
Гидратация алкннов протекает через стадию образования непредельных спиртов, содержащих гидроксильную группу у атома углерода с двойной связью. Такие спирты являются неустойчивыми соединениями. В процессе образования они изомеризуются в карбонильные соединения — альдегиды нлн кетоиы. Эта закономерность непредельных спиртов была установлена в 1877 г. русским химиком А. П. Эльтековым и получила название правила Эльтекоаа:
нс=сн + нон
HgSOs
виниловый спирт
сн-с
уксусный алвдегид
HsSO. СНЭ-С=СН + НОН -- »
сн3-с/<5н^
*5+
—► сн3-с-сн
I о
ацетон
Реакция гидратации алкинов открыта в 1881 г. русским химиком М. Г. Кучеровым. Ее используют а промышленности для получения из ацетилена уксусного альдегида.
4.4.2, Реакции замещения
Этн реакции характерны для ацетилена и алкинов с концевой тройной связью R—С=СН.
Образование ацетиленидов. Ацетилен и другие алкины с концевой тройной связью вступают в реакцию с некоторыми осиования-
04
мИ) такими, как амид натрия NaNH2 в жидком аммиаке, соли меди (I) и серебра а водио-аммиачном растворе (Си(МНз)зОН, AgfNHghOH). При этом водород у атома углерода с тройной с0язью замещается на металл, в результате чего образуются соли, называемые ацетиленидами:
НС^СН + 2Ag(NH3)2OH-------► Ag—С=С—Ag + 4NH3 + 2Н3О
яисгнленнд серебра
R--Се=СН + Cu(NH3)aOH-----> R—CesC—Си + 2NH3 + Н2О
замешенным ацетиленид чеди
R—С=СН + NaNH2--------> R—С=С—Na + NH3
намешенный ацетиленид натрия
Ацетилениды меди и серебра в сухом виде взрываются от удара. Аналогично протекает реакция с магнийорганическими соединениями (реактивы Гриньяра):
R—С=СН -4- С2Н6—Mgl ------> R-C=C—Mgl + С2Н6
Металлические производные алкинов являются аесьма реакционноспособными соединениями. Онн легко вступают а реакцию нуклеофильного замещения с галогеналканами (см. с. 62), взаимодействуют с альдегидами и кетонами, образуя продукты присоединения по карбонильной группе:
СНз СН3
R—CsC—Mgl + <Uo R—С=С—С—OMgl -rfe-
СНз СНз
СНз
--► R—CssC—С—ОН к
При действии хлороводородной кислоты ацетилениды разлагаются с выделением исходного алкнна
R—С=С—Na + НС1-------> R—С=С -Н + NaCl
Эту реакцию применяют для выделения алкиноа а чистом аиде из смесей с другими углеводородами.
Замещение водорода при С^р на галоген. Ацетилен и его гомологи с кониеаой тройной связью прн действии галогенов в присутствии щелочей замещают атомы при углероде с тройной связью на атомы галогенов.
3 5 104
65
4.4.3. Реакции окисления и восстановления
Окисление алкинов. Алкины, подобно алкенам, легко окисли, ются. В качестае окислителей используют перманганат кадия в нейтральной и шелочной среде, озон, тетраоксид рутения RuO<, диоксид селена SeOa и др. При окислении перманганатом калия а щелочной среде или озоном происходит расщепление молекулы алкнна по тройной связи и образуются карбоновые кислоты:
CHj—СН2—С=С—СНз КМ"О<'.О1|> пентнн 2
---> СНз—СНг—СООН + СНз—СООН пропановая уксусная
кислота кислота
Алкины с коицеаой тройной связью при окислении в этих условиях образуют карбоновую кислоту и оксид углерода (IV)-
СНз—СН2—С^СН СНз—СН2—СООН + СО2
б*тин I пропановая кипота
Под действием перманганата калия в нейтральной среде, тетраоксида рутения и диоксида селена алкины окисляются до а-дикетонов:
R—С=С—R' КМпО<- Н2^ R— С—С— R'
а дм кетон
Восстановление алкинов. В присутствии катализаторов Pd, Pt или Ni алкины восстанавливаются с образованием алкаиов. Присоединение водорода осуществляется ступенчато:
Н
СНз—С=СН СНз—d=CH2 СНз—СН,—СНз
кат кат
пропни пропел rrpnnaii
Образующийся в качестве промежуточного продукта алкен не удается выделить вследствие его быстрого превращения в алкан Одиако при использовании в качестае катализатора палладия, частично дезактивированного солями свинца, процесс восстановления останавливается на стадии образования алкена, причем водород присоединяется в цис-положе ине*
Н Н
СНз—с—с— СНз —\:=с^
Pd/ РЬСО,
CH3Z СНз
бутин 2 Чис бутен 2
66
восстановлении влкинов с помощью натрия в жидком аммиаке од также присоединяется селективно, но с образованием тр^-алкена:
СН3-С=С-СН3 — ha МН4 бутин 2
4 4.4. Димеризация, тримериэация
транс б>т<н 2
и тетрамеризация алкниов
В присутствии хлорида меди (I) и хлорида аммония ацетилен димеризуется с образованием виннлаиетнлена
н-с»<Цн; + нг=<рн
нс=с-сн=сн2
винилацетилен
Эта реакция имеет важное промышленное значение, так как винилацетилен используют в качестве промежуточного продукта при производстве синтетических каучуков
При нагревании а присутствии комплексных никельоргани-ческих катализаторов, например Ni(CO)2 [ (СеНя) зР] 2, алкины подвергаются циклотримеризацни с образованием бензола и замещенных бензолов:
1,3 5 тричегилбензол
Ацетилен при нагревании в присутствии катализатора цианида никеля подвергается циклотетрамериэации с образованием цикло-°ктатетраена
4НС=эСН
циклею кт атм раен
3*
67
4.5. ИДЕНТИФИКАЦИЯ АЛКИНОВ
Наличие кратной связи в структуре алкинов, как и в алкенах, обнаруживают реакциями с раствором брома или перманганата кал ня (наблюдается исчезновение окраски). Для отличия алкинов с концевой тройной связью от алкеиоа используют реакцию образования ацетиленидов. Выпадение осадка с аммиачным раствором хлорида меди (I) служит доказательством наличия в соединении группы ==С—Н.
В ИК-спектрах алкинов с концевой тройной связью наблюдается полоса поглощения при 3300 см-1, характеризующая валентные колебания группировки =С—Н. Для несимметричных алкинов характерна слабоинтеисивная полоса поглощения в области 2100—2300 см-1, отвечающая валентным колебаниям связи С=С. У симметричных алкинов (R—С=С—R) эта полоса в ИК-спектре определяется с трудом
Аналогично алканам и алкенам алкииы поглощают УФ-излу-ченне в области ниже 200 им.
В ПМР-спектрах алкинов сигналы протона при атоме углерода с тройной связью (=С—Н) проявляются в области 2,3—2,9 м. д.
Расщепление алкинов при получении масс-спектров происходит твким же образом, как и в случае алкенов
4.6. ОТДЕЛЬНЫЕ ПРЕДСТАВИТЕЛИ- ПРИМЕНЕНИЕ
Ацетилен НС=СН. Бесцветный газ, без запаха, плохо растворим в воде, горит яркнм, сильно коптящим пламенем, с воздухом образует взрывчатые смеси. При сгорании ацетилена в кислороде выделяется большое количество тепла (температура пламени достигает 3000 °C). Это позволяет использовать ацетилен для автогенной сварки и резки металлов.
Ацетилен является сырьем для многих химических производств. В промышленности его используют для получения уксусного альдегида н хлорвинила (СН2=СН—С1), из которых затем получают уксусную кислоту и полихлорвинил соответственно.
Большое количество ацетилена используется в технике для получения винилацетилена, который затем действием хлороводорода превращают в хлоропрен (2-хлорбутадиен-1,3), а частичным гидрированием — в бутадиен-1,3:
нс=с—сн=сн2
СН2=СН—сн=сн2
Хлоропрен и бутадиен-1,3 служат ценным исходным сырьем для получения синтетического каучука. Полимеризацией хлоропрена получают хлоропреновый каучук, полимеризацией бутадиена-1,3—бутадиеновый каучук.
КОНТРОЛЬНЫЕ ВОПРОСЫ И УПРАЖНЕНИЯ
1. Напишите структурные формулы следующих алкинов: а) 4,4-диметилпентии-1, б) изопропилацетнлен.
2. Назовите по заместительной номенклатуре ИЮПАК следующие алкины*. СНз—СН—СН—С=СН^ НС^=С~—СНг—CH=CH2i
(^Н3 ^Н3
3 Напишите схему реакции получения бутина-2 из пропина через стадию образования соответствующего магиийорганического соединения.
4 Приведите схемы реакций получения бутина-2 из соответствующих днгалогеиалкаиов.
5. Напишите схемы реакций гидратации бутена-1 н бутина-1. Объясните, почему в случае бутниа-1 конечным продуктом ие является спирт.
6. Приведите схему последовательного взаимодействия 3-ме-тилбутина-1 с двумя молями хлороводорода. Назовите промежуточный и конечный продукты реакций.
7. Напишите схемы реакций окисления бутина-2 перманганатом калия в нейтральной и щелочной средах. Назовите конечные продукты.
8. Напишите схему реакции, с помощью которой можно доказать наличие тройной связи в молекуле бутииа-1.
Глава 5 ЦИКЛОАЛКАНЫ
Циклоалианами называют алициклические углеводороды. в которых все углеродные атомы, образующие цикл, находятся в sp'-гибридизованиом состоянии.
Структурные формулы алициклических соединений чаще записывают в упрощенном виде, изображая только геометрическую форму цикла:
/С\2
Н2С—сн2
5.1. КЛАССИФИКАЦИЯ И НОМЕНКЛАТУРА
Циклоалканы классифицируют по размеру цикла, числу циклов и способу соединения циклов
По размеру цикла различают цнклоалканы с малыми циклами (трех- и четырехчленные), обычными циклами (пяти-, шести-и семичлеииые), средними циклами (восьми- — одиннадцатичленные) и макроциклами (двенадцатичленные и более)
В зааисимости от количества циклов, входящих в состаа молекулы, циклоалкаиы подразделяют иа моиоциилические, бициклические и полициклические.
Бициклические циклоалканы по способу соединения циклов делят иа спираны (два кольца имеют один общий атом углерода), конденсированные (два кольца имеют два общих соседних атома углерода) и мостиковые (два кольца имеют три и более общих атомов углерода):
спирановая конденсированная мостиковая система система система
70
В соответствии с правилами ИЮПАК названия моноцикли-ческих циклоалканов образуют от названий алканов с соответствующим количеством атомов углерода, прибавляя префикс ЦИКЛО-'
циклопропан
циклопентан
циклогексан
циклобутан
Положение заместителей в кольце обозначают цифрами Нумерацию углеродных атомов цикла начинают с атома, имеющего заместитель, и проводят таким образом, чтобы остальные заместители получили возможно меньшие номера:
1 -метил-3-этилциклопентан
1,2-диметилциклогексан
Названия одновалентных радикалов, образованных из цикло-алкапов, получают путем замены в названии соответствующего углеводорода суффикса -ан на -ил:
циклогексил
циклопропил циклопентил
Систематические названия бициклических циклоалканов образуют с учетом общего числа углеродных атомоа, входящих в циклическую систему, н способа соединения циклов между собой
Название спирановой системы состаалиют, прибавляя префикс спиро- к названию алкана с соответствующим числом атомов углерода. Между префиксом и названием алкана в квадратных скобка^ указывают (в порядке возрастания) число атомов углерода, исключая общий, в каждом из циклов. Нумерацию углерод-
71
ных атомов спирановой системы начинают с меньшего цикла, причем с атома углерода, расположенного рядом с общим:
спиро[3,5]нонан спиро [3,4] октан спиро[2,2]пентан
Названия конденсированных н мостиковых бициклоа^каиов образуют, прибавляя префикс бицикло- к названию алкана с соответствующим числом атомов углерода. Между префиксом и названием алкаиа а квадратных скобках указывают (в порядке убывания) число атомов углерода в каждой из трех цепей, соединяющих два третичных (узловых) углеродных атома. Нумерацию атомов углерода начинают с одного из узловых атомов и осуществляют таким образом, чтобы вначале была пронумерована самая длинная цепь, соединяющая узловые атомы, затем более короткая, а в случае мостиковых систем в заключение нумеруют самую короткую углеродную цепь—мостик:
бицикло [4,2,0] октан бицикл о [3,2,1] октан бицикло[3,2,0]гептан
Многие бициклические системы, наряду с имеют также тривиальные названия:
систематическими,
бицикло [4,4,0] декан
камфан, 1,7,7-триметилби-цикло [2,2,1] гептан
пинан, 2,7,7-триметил бицикл о[3,1,1]гептан
72
5.2. ИЗОМЕРИЯ
Для циклоалканоа характерна структурная, геометрическая и оптическая изомерия.
Структурная изомерия может быть обусловлена:
а) разным размером цикла
1 1 ди Mt'THjiiiixjionponah
метилциклоб\тди
б) разным положением заместителей в цикле
I J диметилциклогексан
СНз
I Л-диметилиишилексая
в) разной структурой боковых цепей
Н2СН2СНз
СНз
изопропилцикло1ексйн
пропил циклен ексан
Геометрическая изомерия циклоалканоа связана с различным расположением заместителей относительно плоскости цикла (см. ки. I, с. 89):
73
Оптическая изомерия характерна для циклоалканов, молекулы которых не имеют плоскости снмм<?Трии (см. кн I, с. 90). Она неразрывно связана с геометрической изомерией. Так, транс-\, 2-диметнлциклопропаи существует а виде пары энантиомеров:
энантиомеры транс-1,2-диметилциклопропана
5.3. СПОСОБЫ ПОЛУЧЕНИЯ
Такие цнклоалканы, как циклопропан и циклогексан, а также их гомологи входят в состав некоторых видов нефти, из которых могут быть выделены в чистом виде.
Наряду с этим для получения циклоалканов существует ряд синтетических методов.
Взаимодействие а, ы-дигалогеналканов * с металлическим натрием или цинком. Этот метод, представляющий собой внутримолекулярный вариант реакции Вюрца, позволяет получить трех-, четырех- и пятичленные циклоалканы:
СН2—СН2—Вг CHo-Clb
| 4- Zn ---► | -|- ZnBr2
СНа—СН2—Вг СН2—СН2
1,4 диброчвСтан in1*, ihCa i .111
Пиролиз кальциевых, бариевых или ториевых солей дикарбоно-9ых кислот. Прн пиролизе (сухой перегонке) кальциевых, барне-зых или ториевых солей дикарбоновых кислот образуются цикли-1еские кетоны, которые затем восстанавливают до соответству-ощих циклоалканов:
* Последняя буква греческого алфавита ы (омега) применяется для обоэначе-ия положения заместителя, находящегося в конце углеродной цени
4
сн2- сн/г Сх \ сн - сн2
\ ^Са^- - 5
\ О У -СаСОз /
СН2- СН“С^'' сн2-сн2
сна-сн2
сн2-сн2
ннльцня адипинат
циклопентанаи
циклоиентан
Кальцнеаые и бариевые соли дикарбоновых кйслб*к образуют с хорошими выходами только пяти- и шестичленные цикады. Для получения больших циклов используют солн тория'.
Реакции циклоприсоединения. Циклоприсоединением называют процесс соединенна двух илн нескольких ненасыщенных молекул с образоаанием продукта циклического строения. В зависимости от числа атомов, принимающих участие в образовании цикла, различают [2 + 1] циклоприсоединение, [2 + 2] циклоприсоединение и [4+2] ци клоп ри соединен не.
Реакции циклоприсоединения имеют важное значение для синтеза различных алициклических соединений.
Взаимодействие алкенов с карбенами (циклоприсоединение [2+ 1]). Карбены являются органическими радикалами (см. с. 210), которые образуются в качестве промежуточных продуктов при разложении дназоалканов или дегидрогалогенировании галогеналканов (см. с. 210). Прн взаимодействии карбенов с алкенами образуются производные циклопропана:
CH2N2 + N2;
лнаючетан клрбен
сн2
СНз—СН=СНа + -СНз --------> CHj—cfOcH2
пропен метилциклопропан
Jt Димеризация алкенов (циклоприсоединение [2 + 2]).
* Циклоприсоединение двух молекул алкена происходит только f под действием ультрафиолетового света (фотохимически) и при-1 водит к образованию циклобутаиа и его производных:
СН2 СН2 СНг—сн2
L +ь -MH L
к>Г12 С>Г12 V.IT2 \>Г12
Реакция Д н л ьс а - А л ь д е р а (диеиовый синтез). Эта реакция подробно рассмотрена иа с. 55. В простейшем виде представляет собой присоединение алкеиа к сопряженному
75
диеиу. Дненовый синтез относится к реакциям [4 + 2] циклоприсоединения и находит широкое применение для получения циклогексана и его производных. Реакция протекает легко при комнатной температуре нли незначительном нагревании. Образующийся циклоалкен восстанавливают до цнклоалкана.
Оутаднен 1 3
циклогексен циклогексан
СН3
I хсн СН I
СН хсн
I сн3 гексадиен 2 4
сн2
I 2
сн2
Электроциклические реакции. Электроциклической называют реакцию, в которой происходит образование а-связн между концами открытой сопряженной системы молекулы К электроцик-лическим реакциям относят также обратные процессы, т. е. происходящие с разрывом о-связи и образованием сопряженной системы. С помощью электро циклических реакций замыкания цикла, инициирующихся термически или фотохимически, получают ненасыщенные и алициклические соединения, которые могут быть восстановлены до циклов л канов:
1,3 Д гексйтриен
циклогексаднен 13
циклогексан
76
5.4. ФИЗИЧЕСКИЕ СВОЙСТВА
В обычных условиях циклопропан и циклобутан являются газообразными веществами, циклоалканы с размером цикла от С5 до Си представляют собой жидкости, далее следуют твердые вещества (табл. 5 1) По сравнению с соответствующими алканами (см табл. 13) циклоалканы имеют несколько более высокие температуры кипения и плавления. Все циклоалканы практически нерастворимы в воде.
Таблица 5 I
Температуры плавления н кипения некоторых циклоалканов
Название Брутто-формула Температура, °C
плавления кипения
Циклопропан С3НЬ — 127 -33
Метил ци клопропа н СлНв — 177 0,7
Циклобутан С4На -во 13
Цнклопентан G5H10 —93.8 49,3
Метилциклобутан CSHID -149,3 36,8
Циклогексан С6Н)г 6,5 80,7
Метилциклопентан С6Н|5 -142,2 71,9
Циклогептан С7Н14 -12 118
Циклоокта к СвН16 14 151
5.5. СТРОЕНИЕ ЦИКЛОАЛКАНОВ
Аналогично алканам в молекулах циклоалканов атомы углерода находятся в состоянии 5р3-гнбридизацнн. Но если молекулы алканов обладают значительной гибкостью за счет свободного вращения вокруг углерод-углеродных связей, то молекулы цикло-алканов, несмотря на возможные конформационные повороты, представляют собой в определенной степени жесткие образования
Для молекул циклоалканов, как и для алканов, характерны торсноииое (пнтцеровское) напряжение, связанное с взаимодействием химических связей в заслоненной или частично заслоненной конформациях, и напряжение Ван-дер-Ваальса, обусловленное взаимным отталкиванием заместителей при сближении на расстояние, близкое к сумме их ааи-дер-ваальсовых радиусоа (см. кн. 1, с. 93) Кроме того, для некоторых цнклоалкаиов характерно напряжение, связанное с отклонением валентных углов между углерод-углеродными связями в цикле от нормального (тетраэдрического) значения. Это напряжение получило название угловое напряжение. Его называют еще байеровским напряжением, по имени немецкого химика-оргаиика А. Байера, выдвинувшего в 1885 г. теорию напряжения циклов.
77
Согласно этой теории циклоалканы рассматривались в форме плоских многоугольников и единственным фактором, определяющим прочность никла, считалось напряжение, вызванное отклонением (по сраанению с тетраэдрическим) внутренних аалентныл углоа между атомами углерода в цикле. Чем больше такое отклонение, тем выше напряжение и менее устойчив цикл. В соответ* стаии с этим трехчленный цикл, внутренние валентные углы котог рого составляют 60°, менее устойчив, чем четырехчленный (виут* реинне углы равны 90°), а тот, в свою очередь, менее устойчив, чем пятичленный (углы 108°). Эти представления подтверждались накопленным в то время экспериментальным материалом. Но уже для шестичленного цикла экспериментальные данные вступили в противоречие с теорией. Шестнчлеиные циклы (внутренний угол 120°), имеющие значительное отклонение валентных углоа от тетраэдрического, оказались устойчивее пятичленных, в которых внутренние углы наиболее близки к тетраэдрическим.
Причиной несоответствия теории Байера с экспериментальным материалом явилось ошибочное представление автора о плоском' строении циклов. В действительности единственным цикломJ имеющим плоское строение, является трехчленный. Все остальные' циклы не имеют плоского строения.
Пространственное строение циклоалканов определяется резной" конформационной подвижностью углеродных атомов, зависящей', от числа звеньев в цикле. Молекула любого циклоалкана ст ре-" мится принять в пространстве такую форму (конформацию),-; а которой сумма углового, торсионного и ван-дер-ваальсового'. напряжений была бы минимальной.
Из всех циклоалканоа наиболее жесткую структуру имеют соединения, содержащие трехчленный цикл. Поскольку в соответствии с правилами геометрии три точки всегда лежат в одной плоскости, трехчленный цикл может иметь только плоское строение. Все атомы водорода в .таком цикле находятся в заслонённой конформации (см. рис. 5.1), что создает сильное торсионное напряжение. Поворот вокруг углерод-углеродных свнзсй неаоз-
Рис. 5.1. Модели молекулы циклопропана: а — шаро-стержневая; б - Драйдинга
78
мфкен- Внутренние валентные углы между связями С—С в трех* члённом цикле сильно отклонены от тетраэдрического значения, в результате чего возникает большое угловое напряжение.
Вследствие взаимного отталкивания электронных облаков углерод-углеродных связей максимальная электронная плотность перекрывающихся орбиталей атомов углерода в трехчленном цикле расположена не по прямой, соединяющей центры связываемых атомов, а за пределами треугольника молекулы (рис. 5.2).
Рис. 5.2. Схема образования углерод-углеродных связей в молекуле циклопропана
Образующиеся при этом о-связи отличаются от. обычных ст-связей. Они занимают промежуточное положение между о-» и л-связями и получили название т-(греч, «тау») связен или «банановых» связей. Несмотря на то что перекрывание за пределами треугольника менее эффективно, образование банановых связей для молекулы является аыгодиым процессом, поскольку в результате этого углы между связями, которые теоретически должны быть равными 60°, увеличиваются до 106°, что, следовательно, снижает угловое напряжение молекулы.
Четырехчленный цикл, в отличие от трехчленного, обладает все же незначительной гибкостью. Внутренние валентные углы в нем меньше искажены, чем в трехчленном, а следовательно, и угловое напряжение несколько ниже. Стремясь уменьшить торсионное напряжение в четырехчленном цикле, одни из атомов углерода выходит нз плоскости остальных трех атомов на угол 25 -30°. Поэтому четырехчленный цикл не является плоским, его пространственная форма представлена на рис. 5.3, а.
Еще большая гибкость характерна для пятичлеиного цикла. В отличие от трехчленного и четырехчленного, в пятичлеином цикле практически отсутствует угловое напряжение (отклонение внутренних валентных углов от тетраэдрического составляет менее 1°). Однако в плоском пятичленном цикле связи С—Н находятся в заслоненной конформации, что создает значительное торсионное напряжение в молекуле. Стремясь уменьшить торсионное
7Q
a
Рис 5 3 Пространственное строение молекул циклобутана (а) и никлопентана (6)
напряжение в пятичленном цикле, каждый из пяти углеродных атомов поочередно выходит нз плоскости, в которой расположены четыре остальных атома углерода. При этом кольцо как бы находится в непрерывном волнообразном движении. Эта неплоская осциллирующая структура получила название «конверта» (рнс. 5.3, б), Несмотря на то что в конформации конверта несколько возрастает угловое напряжение, это в полной мере компенсируется снижением торсионного напряжения молекулы.
В шестичленном цикле, если представить его плоским, внутренние валентные углы должны быть равны 120°, что привело бы к значительному угловому напряжению. Кроме того, в плоской структуре появляются взаимодействия, связанные с заслонением связей С—Н (торсионное напряжение). Избежать углового напряжения шестичленный цикл может при условии его существования в неплоских конформациях. Так, молекула циклогексана существует в виде двух крайних конформаций - кресла и ванны (лодки), которые легко переходит друг в друга (рис. 5.4).
В указанных конформациях все валентные углы тетраэдрические, а следовательно, отсутствует угловое напряжение. Более устойчивой является конформация кресла, поскольку в ней все атомы водорода и углерода находятся в заторможенной конформации (рис. 5.5, а), что исключает торсионное напряжение (рис. 5.5, а).
Рис. 5.4. Конформации циклогексана: а — кресла, б — лодки (ванны)
80
Рис 5 5 Проекция Ньюмена конформаций циклогексана а
- кресла, б — ванны
В конформации ванны прн атомах углерода, расположенных в «основании ванны», водородные атомы находятся в заслонен-ной конформации (рис, 5.5, б), создающей определенное торсионное напряжение. Эта конформация, являясь гибкой структурой, может переходить в несколько более устойчивую ф°РмУ (с меныпнм заслонением) называемую твист-конформацией (искаженная ванна):
конформация ванны meucm-конформация
Энергия конформации кресла примерно на 33 кДж/моль ниже энергии конформации ванны и на 21 кДж/моль — энергии твист-конформации. Поэтому при обычных условиях преобладающая часть молекул циклогексана (99,9 %) существует в конформации кресла, причем кольцо претерпевает непрерывную инверсию, т. е, в результате вращения вокруг углерод-углеродных связей одна конформация кресла переходит в другую с промежуточным образованием конформации ванны и таист-конформации:
конформация кресла
конформация твист- конформация ванны конформация кресла
Две конформации кресла могут взаимопревращаться также без прохождения через конформацию ванны.
Следует отметить, что молекула циклогексана в конформации кресла имеет два типа свизей С—Н. Шесть из них (по одной при каждом атоме углерода) направлены параллельно оси симметрии попеременно вверх и вниз.
Эти связи называют аксиальными и обозначают символом а Остальные шесть связей С—Н направлены радиально, располагаясь вблизи плоскости кольца. Они получили название экваториальные (символ е)
Таким образом, каждый атом углерода имеет одну аксиальную, а другую экваториальную связь С—Н, В процессе инверсии цикла аксиальные связи становятся экваториальными, и наоборот:
Вследствие высокой частоты инверсии циклогексанового цикла (—100 000 раз в секунду при 25 °C) все 12 атомов водорода в нем эквивалентны
Дли монозамещснного циклогексана две конформации кресла не являются энергетически равноценными. Более стабильной является конформация с экваториальным положением заместителя, поэтому большая часть молекул существует в этой конформации, Например, приблизительно 95 % молекул метилциклогексана в равновесной смеси имеет экваториальное положение метильной группы и лишь около 5 % — аксиальное:
Аксиальное положение заместителя для молекулы менее выгодно, так как в этом случае проявляется взаимодействие заместителя с аксиально расположенными атомами водорода в положениях 3 и 5, что приводит к стернческому отталкиванию.
82
При наличии в молекуле нескольких заместителей преимущественно образуется конформация с максимально возможным числом групп в экваториальном положении
5.6. ХИМИЧЕСКИЕ СВОЙСТВА
В химическом отношении циклоалканы во многом ведут себя подобно алканам. Так, дли иих характерны реакции замещении, протекающие по радикальному механизму i>R, например:
циклопропан
4- НС1
<1
х/горциклапролан
циклогексан
Вг
+ НВг.
бром цик югексан
Наряду с этим циклоалканы с малыми циклами (циклопропан и циклобутан) проявляют своеобразные химические свойства, связанные с особенностью их строения (см. с. 78). Из-за большого углового и торсионного напряжений трехчленный цикл и, в меньшей степени, четырехчлеииый являются неустойчивыми. Поэтому соединения, содержащие трех- и четырехчлеииые циклы, наряду с реакциями замещения вступают также в реакции присоединения, сопровождающиеся раскрытием цикла. Например, циклопропан в присутствии катализаторов Ni, Pt и нагревании до 50 °C легко присоединяют водород. Реакция идет с разрывом цикла:
СН2
6 / \ На \ СНз—СНз—СНЭ
Г СНз-------СНз
Циклобутан присоединяет водород при более высокой температуре (200 °C).
Аналогично протекает реакция циклопропана с галогенами и галогеноводородами-
Brj
СН2
1,3 дибромпропан
► СН3—СНэ—СНг—Вг
i бромпропан
Присоединение галогеноводородов к алкнлзамешенным циклопропана протекает в соответствии с правилом Марковникова
СН2 Вг
Н3С----CfC-^CH2 + НВг-----^CHj—СНа—СН—СНз
мсти щи к н>пропан 2 бромбутан
Циклобутаи с галогеиоводородами не реагирует, а с галогенами вступает в реакцию замещения SR.
Для циклоалканов и их производных характерны реакции сужения н расширения циклов. Эти реакции протекают в присутствии кислот Льюиса как катализаторов. Так, этилциклобутаи при нагревании с AlClj изомеризуется в метилциклопентаи:
CHg—СН3
А1С1з, ।
не тил циклоне нтан
эти 1циклобутан
5.7. ИДЕНТИФИКАЦИЯ ЦИКЛОАЛКАНОВ
Вследствие высокой инертности циклоалканов (за исключением циклопропана, проявляющего ненасыщенный характер) для идентификации их обычно не применяют химические методы
Как и алканы, циклоалканы поглощают УФ-излучение в области менее 200 нм, что позволяет использовать их в качестве растворителей для измерении электронных спектров других соединений
ИК-слектры циклоалканов, кроме циклопропана и его производных, во многом схожи с ИК-слектрами алканов В ИК-снектрах циклолропанового цикла имеются полосы поглощения в области 1000 и 1050 см L, отвечающие деформационным колебаниям кольца и в области ЗОЮ 3100 см-1, характеризующие валентные колебания связей С—Н
В ПМР-спектрах циклоалканов сигналы протонов цикла проявляются в той же области, что и у алканов. Некоторые особенности имеет опять же трехчленный цикл, протоны которого поглощают излучения в сильном поле (0,1—0,6 м д,),
5-8. ОТДЕЛЬНЫЕ ПРЕДСТАВИТЕЛИ. ПРИМЕНЕНИЕ
Циклопропан. Бесцветный газ (т. кил. —34,5 °C), хорошо растворим в органических растворителях, плохо растворим в воде. Легко воспламеняется, смеси с кислородом или оксидом азота (IV) взрывоопасны. Применяется а медицинской практике как средство для ингаляционного наркоза
Цнклопентаи, Бесцветная жидкость (т. кип. 49,3 °C). Не растворяется в воде, растворим в органических растворителях,
84
является хорошим растворителем для эфиров целлюлозы. Обладает наркотическим действием. Используют в органическом синтезе. Циклопентановое кольцо входит в состав важных биологически активных соединений — простагландинов
Циклогексан. Бесцветная жидкость (т. кип. 80,7 °C), не растворяется в воде, растворяется в органических растворителях Циклогексан широко применяется в качестве растворители, а также как исходное сырье для получения капролактама и адипиновой кислоты
Адамантан
. Бесцветное кристаллическое
вещество со слабым запахом камфоры. Температура плавления адамаитаиа составляет 270 °C, ио уже при комнатной температуре кристаллы возгоняются Впервые получен из нефти в 1933 г.
Молекула адамантана содержит конденсированную систему, состоящую из трех циклогексановых колец, имеющих конформацию кресла Пространственное расположение углеродных атомов соответствует кристаллической решетке алмаза.
Некоторые производные адамантана иашли применение в медицинской практике как лекарственные средства, обладающие сильным противовирусным действием
1 - А мн ноада м анта на гидрохлорид (мидаитаи)
Применяется в медицине как пре-
тарат, обладающий противовирусным действием (против вирусов 'риппа типа Аэ), а также для лечения болезни Паркинсона.
1 - (2-амнноэтнл-) адамантана гидрохлорид
'емантадии)
^рименяетси в медицинской практике как противовирусное средство.
85
КОНТРОЛЬНЫЕ ВОПРОСЫ И УПРАЖНЕНИЯ '•V
о
1, Напишите структурные формулы следующих циклоалканов: а) транс-1,2-диметилциклобутан; б) 1,2-диметил-4-этилциклопентан; в) спиро [2,5] октан; г) бицикло [4,3,0] нонаи; д) 1,8,8-триметнлбицнкло [3,2,1 [октан.
2. Напишите схему реакции получения 1,3-диметнлциклопен-тана из соответствующего дигалогеналкана. Назовите исходное соединение.
3 Приведите схему получения 1-метил-4-этилциклогексана по реакции Дильса —Альдера.
4. Какие виды напряжения существуют в органических молекулах? Какие виды напряжения имеют место в молекуле циклогексана, находящейся в конформации ванны?
5. Что такое инверсия цикла? Приведите схему инверсии метилциклогексаиа с промежуточным образованием конформации ванны н таист-конформации. Обозначьте аксиальные и экваториальные связи в конформациях кресла.
6. На примере циклобутаиа н циклогексана объясните общность и различие в реакционной способности циклоалканов.
АРОМАТИЧЕСКИЕ УГЛЕВОДОРОДЫ (АРЕНЫ)
Термин ароматические первоначально применяли для органических соединений, которые нлн сами имели приятный запах, или же выделялись из природных веществ, обладающих приятным запахом. В дальнейшем это название сохранилось за большой группой органических соединений, проявляющих сходные свойства с бензолом,
К ароматическим углеводородам относят соединения, молекулы которых содержат одно или несколько бензольных колец. Для них применяют также название арены.
В зависимости от числа бензольных циклов, входящих в состав молекулы, различают одноядерные (моноцнклнческне) и многоядерные (полициклические) арены. Многоядерные арены подразделяются на арены с конденсированными циклами (аннелнрован-ные) н изолированными циклами.
87
Глава 6 ОДНОЯДЕРНЫЕ АРЕНЫ
&4S
i
V I
6.1. СТРОЕНИЕ БЕНЗОЛА. АРОМАТИЧНОСТЬ
Простейшим представителем одноядерных ароматических углеводородов является бензол СбШ.
Впервые бензол был получен М. Фарадеем в 1825 г. из конденсированных остатков светильного газа, образующегося в процессе переработки каменного угля. Однако строение его молекулы в течение многих лет оставалось дли химиков загадкой. Несмотри на то что формула С6Нб предполагает достаточно выраженный ненасыщенный характер, бензол, в отличие от непредельных соединений, оказался относительно инертным веществом. Так, он сравнительно устойчив к нагреванию и действию окислителей, практически не вступает в характерные для ненасыщенных соединений реакции присоединения. Наоборот, для бензола более характерными оказались не свойственные непредельным соединениям реакции замещения.
Составу СеНб приписывались разные структурные формулы, но все они не объяснили в полной мере химических свойств бензола.
Важным этапом в установлении строения бензола явилась высказанная немецким химиком А. Кекуле идея о циклическом строении его молекулы. В 1865 I. А. Кекуле предложил формулу в виде цикла из шести атомов углерода с чередующимися простыми и двойными связями. Эта формула вошла в органическую химию как формула Кекуле:
Формула Кекуле предполагает равноценность всех атомов углерода и водорода в молекуле, что позже было подтверждено исследованиями А. Ладенбурга (1874 г.) и Э. Вроблевского (1878 г.).
Вместе с тем в соответствии с формулой Кекуле бензол должен иметь два 1,2-дизамещенных изомера:
Экспериментально же было установлено, что 1,2-днэамешеи-ные бензолы не имеют изомеров положения, т, е, они существуют в виде одного вещества.
Для объяснения этого противоречия в 1872 г. Кекуле выдвинул осцнлляционную гипотезу, согласно которой три двойные связи в молекуле бензола не фиксированы, а непрерывно перемещаются (осциллируют) между двумя возможными положениями:
Правильно отражай некоторые свойства бензола, формула Кекуле тем не менее не согласовывалась с рядом установленных фактов. По-прежнему оставалось неясным, почему при наличии в молекуле трех двойных связей бензол проявляет значительную инертность в реакциях присоединения и гораздо более склонен к реакциям замещении, почему он устойчив к нагреванию и действию окислителен. Все это возвращало химиков к пересмотру структуры бензола.
В соответсгвии с современными представлениями, основанными на данных квантовой химии и фнзико-химичсскнх исследований, молекула бензола представляет собой правильный плоский шестиугольник. Все углеродные атомы находится в состоянии $ра-гибридизации. При этом каждый атом углерода образует три о-связи (одну С—Н и две С С), лежащие в одной плоскости под углом 120° друг к другу (рнс. 6.1, а) и предоставляет одну р-орби-таль для образовании замкнутой сопряженной системы, электронная плотность которой равномерно распределена (делокализована) между углеродными атомами и сконцентрирована в основном над н под плоскостью о-скелета молекулы (рис. 6.1, б).
Образование замкнутой сопряженной системы (ароматиче; скоро секстета) является для молекулы бензола энергетически вьи одним процессом. Экспериментально установлено, что сопряжение в цикле приводит к уменьшению энергии иа 150,7 кДж/моль, т. е реально бензол оказался на 150,7 кДж/моль стабильнее, чем
RQ
Рис 6 I Схема образования о связей (а) и замкнутой л-э.1ектрониой системы (б) в молекуле бензола
это можно было предположить исходя из формулы Кекуле. Разность энергий реального состояния молекулы бензола и рассчитанной дли гипотетической структуры — циклогексатриена-1,3,5 составляет энергию сопряжения или энергию резонанса и является причиной высокой устойчивости молекулы
С позиций теории молекулярных орбиталей при перекрывании шести р-атомных орбиталей образуется шесть л-молекулярных орбиталей, из которых три - связывающие, а три — разрыхляющие л-МО, Каждая МО характеризуется определенной энергией и может быть заполнена двумя электронами с антипараллельными спинами В основном состоянии шесть л-электронов занимают три связывающие л-МО, которые обладают более низкой энергией. Высокоэнергетичные разрыхляющие л*-МО остаются незаполненными (рис, 6 2)
Повышенная устойчивость молекулы бензола определяется энергией низшей л>-МО, в которой л-электроиное облако охватывает все углеродные атомы цикла (рнс. 6.2) В результате сопряжения все углерод-углеродные связи в молекуле бензола выравнены.
Таким образом, в бензольном кольце нет простых и двойных связей. На каждую углерод-углеродиую связь, помимодвух о-элек-тронов, приходится электронная плотность одного л-электрона.
90
Е
Разрыхляющие л*-МО
Рис 6 2 Энергетические уровни шести л-молекулярных орбиталей бензола и схематическое изображение трех связывающих л-МО (п,, лг и л3)
Такую связь называют ароматической. Если длина простой связи С—С в алканах составляет 0,154 нм, длина двойной связи в алкенах—0,134 нм, то длина углерод-углеродной связи в молекуле бензола равна 0,140 им, т. е является промежуточной между длиной одинарной и двойной связи
Делокализация л-электронной плотности и выравненность связей в бензольном кольце графически изображается в виде окружности внутри правильного шестиугольника;
Однако наряду с таким изображением в химии широко используется и формула Кекуле, которая особенно удобна для описания механизмов реакций. Но, применяя формулу Кекуле, необходимо подразумевать делокализацию л-электрониой плотности и выравненность связей в бензольном кольце
91
Совокупность специфических свойств бензола, а нмеиио, высокая стабильность, инертность в реакциях присоединения и склонность к реакциям замещения, получила общее название ароматичность, или ароматические свойства.
В 1931г, немецкий ученый Э, Хюккель иа основе каантово-химических расчетов с помощью метода МО сформулировал правило стабильности циклических сопряженных систем, которое представляет собой теоретически обоснованный метод, позволяющий предсказать, будет ли циклическая сопряженная система ароматической или нет. Согласно правилу Хюккеля критерием ароматичности органического соединения являетси наличие в его
структуре плоского цикла, содержащего замкнутую сопряженную систему, включающую (4п + 2) л-электроиов, где п = 0, 1, 2, 3 н т. д. К наиболее распространенным ароматическим системам, Содержащим бл-электроиов (п = 1) относится бензол и его производные. Правило Хюккеля применимо и к системам с конденсированными ядрами, такими, как нафталин, антрацен и фенантрен:
нафталин антрацен фенантрен
Юл электронов [п=2) 14п-э.юктронов (л—J} 14я электронов (м=Э)
6.2. НОМЕНКЛАТУРА И ИЗОМЕРИЯ
По заместительной номенклатуре ИЮПАК одноядерные арены рассматривают как продукты замещения бензола: метилбензол, этилбензол, винилбеизол и т. д. При наличии в бензольном кольце двух и более заместителей их положение указывают цифрами. Нумерацию атомов углерода бензольного кольца осуществляют таким образом, чтобы заместители имели возможно меньшие номера. В дизамещенных бензолах нариду с цифровым обозначением положений заместителей примениют также приставки: орто-(о-) положение— 1,2; мета- (м-) положение — 1,3 и лара-(л-) положение — 1,4.
Кроме названий по заместительной номенклатуре, в ряду одноядерных аренов сохранились и тривиальные назаания: толуол, ксилол, кумол и другие. Ниже приведены некоторые представители аренов (тривиальные названия даны после заместительных):
изопропилбензол, кумол
нннклбензол, стирол
92
1,4 дммртнл 2 этилбензол
I „3,5 трнметнлбензол. меэнтнлен
В ряде случаев моиоциклические ароматические углеводороды рассматривают как производные другого какого-либо углеводорода, имеющего тривиальное название, иапрнмер толуола, стирола:
З-ЭТИЛТОНУ ол
сн=сн2
СНз
4 метилстирол
Одновалентные радикалы ароматических углеводородов имеют Орщее название арилы (Аг):
Двухвалентный радккал бензола называют фениленом
о фенилен
м фенилен
93
Высшие гомологи бензола чаше всего рассматривают как производные алифатических углеводородов, содержащие а качестве заместители бензольное ядро:
6 5 4 3 2 1
СНз—С Н 2—CH—СН2—СН—СНз
2 метит 4 фенилгексая
Изомерия гомологов бензола обусловлена разными структурами, числом и положением заместителей в бензольном кольце. Однозамещенные гомологи бензола не имеют изомеров положения, поскольку все атомы углерода в бензольном кольце равноценны. Вместе с тем для них характерна изомерия, связанная с разной структурой заместителя:
пропил бензол
СНг—СНг—СНз
изопропилбензол
Дизамещениые бензолы существуют в трех изомерных формах, имеющих разное положение заместителей в бензольном кольце (изомеры положения):
1,2 ди метил бензол о ксилол
Тризамещеиные бензолы с одинаковыми заместителями в бензольном кольце также имеют три изомера положения, например:
Кроме того, для гомологов бензола характерна изомерия, обусловленная разным числом заместителей в бензольном кольце, например:
в.З. СПОСОБЫ ПОЛУЧЕНИЯ
6.3.1. Природные источники
Основными природными источниками ароматических углеводородов являются нефть и каменный уголь.
Получение из нефти. В сырой нефти содержится небольшое количество ароматических углеводородов. Поэтому с целью увеличения их массовой доли нефть подвергают так называемой арома-тнзацнн, т. е. нагревают при высокой температуре н давлении в присутствии катализаторов. При этом протекают процессы дегидрирования, изомеризации н циклизации. Ниже приведены схемы некоторых типичных реакций:
циклогексан бензол
СНэСН2СН2СН2СН2СН2СН3
гептан
сн,сн2сн2сн2сн2сн2сн2сн3
октан
500 °C, CrjOa/AljOj ------------------->.
В результате ароматизации исходное сырье, содержащее около 10 % аренов и 65 % алканов, превращается в продукт, включающий 50 -65 % аренов.
Важный вклад в изучение процессов, протекающих при ароматизации нефти, внесли отечественные ученые Н. Д. Зелинский, Б, А, Казанский и А. Ф. Платэ,
95
Получение из каменного угля. При нагревании каменного угля без доступа воздуха до 1000—1300 °C образуется кокс, коксовый газ и каменноугольная смола, В 1 м3 коксового газа содержится около 30 г бензола н Ю г толуола. Каменноугольная смола представляет собой сложную смесь органических соединений. Подвергая ее фракционной перегонке, получают одноядерные ароматические углеводороды (бензол, толуол, ксилолы), многоядерные арены (нафталин, антрацен), фенолы, гетероциклические соединения н др. Всего из каменноугольной смолы выделено свыше 120 индивидуальных веществ.
6.3.2. Синтетические методы получения
Циклотримериэация алкинов. При нагревании в присутствии комплексных ннкельорганических катализаторов алкяны образуют бензол и его гомологи (см. с. 67).
Взаимодействие смеси алкил- и арилгалогенидов с металлическим натрием (реакция Вюрца — Фиттига). При обработке металлическим натрием смеси галогеналканов и галогенаренов образуются гомологи бензола;
Оринбензол
этилбепзо.1
+ 2NaBr •
Эта реакция является частным случаем реакции Вюрца применительно к аренам (см. с. 17). В качестве побочных продуктов в процессе реакции Вюрца Фиттига образуются алканы, а также дифенил и его гомологи:
2СбН5Вг + 2Na------> СЬН5—С6Н5 + 2NaBr
ди фенил
2C2HsBr + 2Na -----► СНзСНгСНгСНэ + 2NaBr
бу МН
Алкилирование ароматических углеводородов по Фриделю — Крафтсу. Реакция Фриделя — Крафтса (1887 г.) представляет собой общий метод получении гомологов бензола, основанный на взаимодействии ароматических углеводородов с алкилирующими агентами — галогеналканами, алкенами и спиртами (см. алкилирование по Фриделю — Крафтсу, с. 102).
6.4. ФИЗИЧЕСКИЕ СВОЙСТВА
В обычных условиях бензол и низшие члены гомологического ряда являются жидкостями, обладающими сильным специфическим запахом (табл. 6.1).
96
Таблица 6I
Некоторые физические характеристики низших гомологов бензола
На гаание Температура, °C d?
плавления кн ггения
Бензол 5.5 80,1 0,8790
Толуол -95.0 1(0.6 0,8669
о-Кси.юл - 25,0 144,4 0,8802
м- Ксилол 47,9 139,1 0,8641
л-Ксилол 13,3 138,4 0.8610
Кумол —96,0 152,4 0.8618
Mc-jhthjch 44,7 164,7 0,8651
л-Ни мол -67.9 177J 0,8573
Стирол -30.6 145,2 0.9060
Все ароматические углеводороды нерастворимы в воде и хорошо растворяются в органических растворителях. Многие из них сами являются хорошими растворителями для других органических веществ. Из-за высокого содержания углерода ароматические углеводороды горят сильно коптящим- пламенем. Бензол очень ядовит. Продолжительное вдыхание его паров вызывает лейкемию,
6.5. ХИМИЧЕСКИЕ СВОЙСТВА
Реакционная способность бензола н его гомологов определяется главным образом наличием в структуре замкнутой л-элект-ронной системы, которая является областью повышенной электронной плотности молекулы и способна притягивать положительно заряженные частицы — электрофилы. Поэтому ароматические углеводороды, как н алкены, обладают нуклеофильным характером. Однако арены, в отличие от ненасыщенных соединений, при взаимодействии с электрофильными реагентами более склонны к реакциям замещения, а не присоединения, поскольку при этом сохраняется нх ароматическая система. Этн реакции носят название реакций электрофильного замещения 5К.
Реакции присоединения для аренов менее характерны, так как они приводят к нарушению ароматичности, с трудом вступают ароматические углеводороды и в реакции окисления,
6.5.1. Реакции электрофильного замещения (Sh)
Бензол и его гомологи сравнительно легко вступают в реакции электрофильного замещения. Электрофильная частица, атакующая л-электронную систему бензольного кольца, может быть представлена положительно заряженным ионом Е+ нлн частью нейтральной молекулы, имеющей центр с пониженной электронной плотностью Е6+—>-Хв_ (см, кн. 1, с. 129). Образование электро
4 з ни
97
фильных частиц для участия в реакции возможно различными способами—под действием л-электронной системы бензольного кольца, катализатора, растворителя и др.
Несмотря на большое разнообразие электрофильных реагентов и ароматических систем, подавляющее большинство реакций электрофильного замещении в ароматическом ряду описывается в рамках единого общего механизма.
При атаке электрофильной частицей л-электронной системы бензольного кольца сначала в результате электростатического взаимодействия образуется неустойчивый л-комплекс:
Е
л-комплекс
л-Комплекс представляет собой координационное соединение, в котором бензольное кольцо является донором электронов, а электрофил — акцептором. Образование л-комплекса является быстрой и обратимой стадией реакции. Ароматичность бензольного кольца при этом не нарушается. Во многих случаях л-коми-лекс удалось обнаружить при помощи электронных спектров поглощения. Поглощая некоторое количество энергии, л-комплекс превращается затем в о-комплекс (карбкатион). В отличие от л-комнлекса, в п-комплексе электрофильная частица образует ковалентную связь с одним из атомов углерода бензольного кольца за счет двух его л-электронов. При этом происходит нарушение ароматической системы бензольного цикла, так как один из атомов углерода переходит из состояния sp2- в состояние зр3-гибриди-зации. Оставшиеся четыре л-электрона бензольного кольца делокализованы между пятью атомами углерода.
Строение п-комплекса можно представить в виде резонансного гибрида структур 1, 11, III, ио чаще его изображают структурой IV:
СТ-комплекс
IV ст- комплекс
Образование ст-комплекса является наиболее высокоэнергетич-ной стадией реакции, определяющей скорость всего процесса.
Протекание электрофильного замещения через стадию а-комп-лекса подтверждено многочисленными исследованиями, В некоторых случаях a-комплекс удалось обнаружить спектральными методами и даже выделить в кристаллическом виде.
Несмотря на относительную стабильность a-комплекса за счет распределения положительного зарида между литью атомами углерода, он значительно меиее устойчив, чем структуры с ароматическим секстетом электронов. Стремясь к дальнейшей стабилизации, п-комплекс отщепляет протон от атома углерода, связанного с электрофилом, и тем самым восстанавливает ароматичность бензольного кольца:
Восстановление ароматической структуры дает выигрыш энергии, равный 42 кДж/моль,
К наиболее важным реакциям электрофильного замещения в бензольном ядре относится реакции нитрования, галогенирования, сульфирования, алкилирования и ацилирования.
Нитрование. Это процесс замещения атома водорода в бензольном ядре иа нитрогруппу — NO2. В качестве нитрующих агентов в реакции нитровании чаще используют концентрированную азотную кислоту или смесь концентрированных азотной и серной кислот (нитрующая смесь). С концентрированной азотной кислотой бензол и его гомологи реагируют медленно. Поэтому для нитрования аренов наиболее широко используется нитрующая смесь:
Uhno,-1^.^
нитробензол
+ н,о.
Атакующей электрофильной частицей в реакции нитрования является ион иитроиия NO^", который образуется в результате кислотно-основной реакции между азотной и серной кислотами, где азотная кислота играет роль основания:
HONOa + H2SO4 ^=z± Н—О— NO2 + HSO4-
н
протонированнаи «иотная кислота
99
+ I
H—-О—н-NO2 +=* Н2о + NO^
| I ноя
I нитрония
г!
Н2О + H2SO< НзСГ + HSO?
Суммарное уравнение имеет следующий вид:
HNOa + 2H2SO4-----> NOt + Н3О+ + 2HSOF
Ион нитрония атакует л-электронную систему бензольного ядра, образуя нитропроизводное
п-комплекс a-комплекс нитробензол
Сульфирование. Это процесс замещения атома водорода в бензольном ядре иа сульфогруплу - SO3H
Для сульфирования бензола и его гомологов чаще применяют концентрированную серную кислоту илн дымящую серную кислоту (олеум). В результате взаимодействии образуютси аренсульфокислоты
беьиолсулъфп кн с ,ю та
Сульфирование аренов является обратимой реакцией Образующаяся в процессе взаимодействия вода смещает равновесие влево. Поэтому дли увеличения выхода нолевого продукта берут избыток концентрированной серной кислоты или используют олеум (раствор триоксида серы SO3 в серной кислоте).
Тонкие особенности механизма сульфирования аренов изучены недостаточно, однако большинство экспериментальных данных свидетельствуют о том, что атакующей электрофильной частицей в реакции служит триоксид серы SO3-.
2H2SO4 SO, + Н3О+ + HSOT
л-комплекс
ст-комплекс
100
бензол сульфокислота
Галогенирование. Бензол н его гомологи хлорируются, бронируются и иодируются. Замещение атома водорода в бензольном ядре иа атом хлора или брома осуществляют действием свободного хлора илн брома в присутствии катализаторов — кислот Льюиса (А1С1Э, БеВгз, ZnCh и других):
С1
ч юрйгнза1!
Под действием катализатора, на атоме металла которого имеется дефицит электронной плотности, молекула галогена поляризуется. Атакующей электрофильной частицей в этом случае служит либо комплекс поляризованной молекулы галогена с кислотой Льюнса, либо катион галогена, образующийся в процессе ионизации данного комплекса:
6+ I— л f fi— ——।
Вг—Br + FeBr3 Вг—Вг-------------^FeBr , Ь—>► Br+ + FeBrf
+ Вг—Вг —♦Fe Вг3
8-
Вг—Br—FeBr3
л-комплекс
о-комплекс бромбензол
Молекулярный иод в сравнении с хлором и бромом является слабым галогенирующим агентом. При взаимодействии иода с бензолом равновесие реакции практически полностью смещено в сторону исходных веществ
I
101
Поэтому прямое иодирование аренов проводят иодом а присутствии таких окислителей, как HNO3 или HgO. Роль окислителя состоит в связывании образующегося иодоводорода:
2 J + 212 + HgO--------------*-2
+ Hgl2 + Н2О
Алкилирование по Фриделю — Крафтсу. Для введения алкильной группы в молекулу бензола и его гомологов в качестве электрофильных реагентов чаще всего используют галогеналканы Взаимодействие аренов с галогеналканами происходит в присутствии катализаторов — кислот Льюиса (A]Ch, FeClj, ZnC!2, BF31 SnCl2 и др ), из которых наиболее часто применяют хлорид алюминия:
K'lopjTdH
+ НС1
Помимо галогеналкаиов, для алкилирования аренов могут быть использованы спирты и алкеиы. Реакции с участием спиртов протекают в присутствии кислот Льюиса илн минеральных кислот (Н3РО41 H2SO4):
Алкилирование аренов алкенами требует присутствия в качестве катализатора кислоты Льюиса и минеральной кислоты как источника протонов: -
Н3С—С Н—С Нэ
fl + СНг=СН—СНз
н+ Aicia
к\ ча 1
пропилен
Атакующей электрофильной частицей в реакции алкилирова; ния по Фриделю — Крафтсу является карбкатион, который обра-
102
зуется в каждом конкретном случае при взаимодействии алкилирующего агента и катализатора. Например, нз галогеналканов:
С2Н5 ------► Cl + AlCh с2н^ + aici;
KjopiTdH ион карбония
из спиртов:
С2Н50н + AiCU^±C2H5OAiCi2^tC2H^ + OAlClj —nci
этанол
С2Н5ОН + Н+ С2н5—О—Н С2н^ + Н2О
н
из алкенов:
5+ /\ S-
сн3—> снхсн2 + J *
снэ-сн-сн3
н
пропен
По своему механизму реакция алкилирования аналогична рассмотренным выше реакциям нитрования, сульфирования и галогенирования:
х-Комплекс а-комплекс
Введенная в бензольное ядро алкильная группа, являясь элект-ронодонором, повышает реакционную способность ароматического кольца но отношению к электрофильным реагентам. Поэтому образовавшийся в процессе алкилирования продукт более склонен к взаимодействию с электрофилом, чем исходный арен. В результате алкилирование часто не останавливается иа стадии образования монозамещенного продукта, а идет дальше, давая ди-и полиалкиларены.
Ацилирование по Фриделю — Крафтсу. Ацилированием называют процесс введения в молекулу органического соединении ацильной группы (R—С=О) Ацилирование бензола и его гомоло-
I
гов но Фриделю— Крафтсу обычно осуществляют галогенангид-рндами или ангидридами карбоновых кислот в присутствии кислот
103
Льюнса. Реакция служит общим методом получения ароматических кетонов:
ацсгнлхлпрнч
яцгтофенин
Электрофилом, атакующим бензольное кольцо, в реакции аци-Ц- г*
лирования является либо ацнлиевый нон R—С—О, либо комплекс
ацнлгалогенида с катализатором (RCO А1С1Г).
RCOC1 + А1С13 RCOAICk R—С—О + А1С1Г
(RCO)2O + А1С13^: R—С=О + RCOOAlClr
Ацилирование ароматического идра, в отличие от алкилиро-* вапия, протекает преимущественно с образованием монозамещен-ных продуктов Являясь электроноакцептором, ацильная группй снижает реакционную способность бензольного кольца к электро-j фильпым реагентам. Поэтому диацилпроизводные образуются', только в жестких условиях, *
6,5.2, Реакции присоединения i
Как отмечалось ранее, реакции присоединения для ароматц-J ческих углеводородов не являются характерными. Однако в жестких условиях они все же происходят. ь
Гидрирование, При повышенных температуре и давлении* в присутствии катализаторов, из которых чаще применяют мелко-порнстый никель (никель Ренея), бензол и его гомологи присоеди-
ни
няют три молекулы водорода, образуя циклогексан и его производные:
Н + ЗНа
НИКЛОЕ СКГЯ П
Остановить реакцию на стадии образования продуктов частичного гидрирования (цнклогексадиена, циклогексена) не предоставляется возможным, поскольку последние гидрируются значительно легче, чем сам бензол.
Хлорирование. При интенсивном солнечном освещении илн под действием ультрафиолетового излучения бензол присоединяет хлор. Реакция протекает по радикальному механизму, с образованием смеси стереоизомеров гексахлорциклогексана (гексахлорана) :
гсксах тор циклогексан
_|_ зС12
6.5.3. Реакции окисления
Окисление бензольного цикла. Бензольное кольцо очень устойчиво к действию окислителей. В обычных условиях такие сильные окислители, как перманганат калия, азотная кислота, оксид хрома (VI), пероксид водорода и другие, не окисляют бензол. По в жестких условиях, например при действии кислорода воздуха в присутствии оксида ванадия (V) в качестве катализатора, при температуре 400—500 °C бензольное ядро окисляется, образуя малеиновый ангидрид:
02, V2O(5
О
Z сн—с
О + 2СОг + 2Н2О
СН-С
105
Окисление гомологов бензола. Ал кил бензолы, в отличие от незамещенного бензола, окисляются значительно легче, В этом случае яри действии сильных окислителей (КМпО-i, К2СГ2О7 и др ) подвергаются окислению боковые цепи Продуктами окисления являются ароматические карбоновые кислоты. Причем каждый алкильный радикал в бензольном кольце, независимо от длины углеродной цепи, окисляется в карбоксильную группу:
СНз
соон
СН2СН3
соон
+ Н2О + со2
□ эти пто/ivm
фтатрпля кисшта
Если в бензольном кольце имеется несколько заместителей, то путем подбора оптимальных условий можно провести их последовательное окисление. По реакционной способности к действию окислителей алкильные группы располагаются в следующей последовательности: CH Ro >'CHsR >'СНз. Третичные алкильные группы у бензольного ядра до карбоксильной группы не окисляются.
Окисление алкилбензолов является важным способом получения ароматических карбоновых кислот.
Озонирование. Подобно алкенам, бензол и его гомологи реагируют с озоном, образуя взрывоопасные продукты присоединения триозониды Под действием воды триозоииды разлагаются с образованием дикарбоннльных соединений н продуктов их дальнейшего окисления — дикарбоновых кислот:
глиоксаль
щавелевая кислите
106
6.5.4. Галогенирование гомологов бензола с участием боковой цепи
Взаимодействие гомологов бензола с галогенами (хлор илн бром) в условиях свободнорадикального замещения (см. с. 20) осуществляется с участием боковой цепи. При этом на атом галогена замещается, как правило, атом водорода при углероде, непосредственно связанном с бензольным кольцом (а-ноложение):
С1
этитОепэол 1 феинт I кюрыан
Такое направление замещения обусловлено образованием в качестве промежуточной активной частицы свободного радикала бензильного типа, в котором электронная плотность значительно делокализована за счет сопряжения с бензольным кольцом (см книгу 1, с. 140):
СН2СН3
СН
\:н3 + нс1
свободный радикал бензильного типа
6.6. ВЛИЯНИЕ ЗАМЕСТИТЕЛЕЙ В БЕНЗОЛЬНОМ КОЛЬЦЕ НА НАПРАВЛЕНИЕ И СКОРОСТЬ РЕАКЦИЙ ЭЛЕКТРОФИЛЬНОГО ЗАМЕЩЕНИЯ
В молеку незамещенного бензола электронная плотность распределена равномерно, а поэтому электрофильный реагент может атаковать в равной степени любой из шести атомов углерода.
Если в "ензольном кольце содержится какой-либо заместитель, то под его влиянием происходит перераспределение л-электронной плотности цикла и новая группа вступает уже в определенные положения по отношению к имеющемуся заместителю. В реакциях электрофильного замещения моиозамещениых бензола, в зависимости от электронной природы заместителя, вступающая группа может занимать преимущественно орто-, ^.ета- или пара-положения, а реакция может протекать быстрее или медленнее, чем с самим бензолом.
По влиянию иа направление реакций электрофильного замещения и реакционную способность бензольного кольца заместители можно разделить на две группы — заместители 1 рода (орто-, пара-ориеитанты) и заместители II рода (хета-ориентанты).
107
К заместителям I рода относятся атомы н атомные группы, проявляющие положительный индуктивный или положительный мезо-мерный эффекты — О , NRa, NHR, NH2, бн, 6r, NHCOR, 6COR, SR, p, Cl, Sr, Г, Aik, Заместители 1 рода (за исключением галогенов) увеличивают электронную плотность в бензольном кольце и тем самым активируют его в реакциях электрофильного замещения; заместители 1 рода направляют замещение главным образом в орто- и пара- положения:
фенол
о нитрофенол
п нитрофенол
К заместителям II рода относятся группы, проявляющие отрицательный индуктивный илн отрицательный мезомерный эффекты — NR31 NH3, NO2, SO3H, CN, СНО, COR, СООН, COOR, CONH2, CCI3. Заместители II рода уменьшают электронную плотность в бензольном кольце н снижают скорость реакции электрофильного замещении по сравнению с незамещенным бензолом. При этом вновь входящий заместитель направляется преимущественно в жетд-лоложеиие:
NO2 NO2
NO2 нитробензол М дннитробеитол
Следует отметить, что описанная выше ориентация замещения не является абсолютной, а свидетельствует лишь о предпочтительном направлении реакции с преобладающим образованием того или иного изомера. Так, при нитровании нитробензола образуется 93 % мета-, 6 % орто- и 1 % пара-ди нитробензол а.
Механизм влиянии заместителей в бензольном кольце на направление и скорость реакций электрофильного замещении можно объяснить с учетом электронных эффектов, которые играют существенную роль как в распределении электронной плотности в стационарном состоянии молекулы (статический фактор), так н в стабилизации образующихся а процессе реакции о-комплексов (динамический фактор).
108
Заместители 1 рода (кроме галогенов) за счет -(-/-эффекта или + /М-эффекта проявляют электронодонорные свойства Они повышают электронную плотность на всех атомах углерода бензольного кольца, но в большей степени на углеродных атомах в орто- и гаара-лоложеииях (статический фактор):
8-
+7-эффект
Это является причиной облегчения электрофильного замещения в сравнении с реакциями 5Е у незамещенного бензола и преимущественной атаки электрофильной частицей орто- и пара-положений.
Заместители II рода, наоборот, за счет — /- или —М-эффектов проявляют электроноакцелторные свойства, вызывая общее уменьшение электронной плотности в бензольном кольце, но в большей степени это влияние сказывается в орто- и пара-положениях. Поэтому они затрудняют реакции электрофильного замещения вообще, и особенно с участием орто- и napa-положеиий. В результате замещение протекает преимущественно в лета-положении
-Af-эффект
Необходимо отметить, что наряду со статическим фактором существенное, а в некоторых случаях решающее влияние на направление электрофильного замещения оказывает динамический фактор. Его сущность определяется влиянием имеющегося в бензольном кольце заместителя на устойчивость образующегося в момент реакции того или иного о-комплекса. Из всех возможных ^-комплексов для молекулы энергетически более^выгодны те, в которых имеется возможность дополнительной делокализации положительного заряда за счет заместителя. Эти о-комллексы обладают меньшей энергией, а следовательно, они более устойчивы
109
и поэтому их образование в ходе реакции будет более предпочтительным.
В реакциях 5Е заместители I рода вследствие своих электронодонорных свойств повышают устойчивость всех о-комплексов по сравнению с незамещенным бензолом и, таким образом, увеличивают скорость реакции, но в большей степени они стабилизируют сг-комплексы, отвечающие продуктам орто- и лара-замещений. Например, при нитровании метоксибензола каждый из п-комп-лексов, образующихся в результате орто-, мета- и пара-замещений, стабилизирован за счет делокализации положительного заряда между атомами углерода бензольного кольца (граничные структуры 1, II, III): ” ' '
Но в о-комплексах при орто- н qppa-замещении положительный заряд может быть дополнительно делокализован с участием непо-делеиной пары электронов атома кислорода метоксигруппы (граничная структура IV) Поэтому образование их в ходе реакции более предпочтительно В результате образуются преимущественно продукты орто- и napa-замещения
110
Заместители II рода вследствие своих электроноакцепторных свойств дестабилизируют в той или иной мере все три возможных a-комплекса и тем самым затрудняют электрофильное замещение в сравнении с незамещенным бензолом Одиако о-комплекс в мета-лоложении дестабилизируется в меньшей степени, чем о-комллекс в орто- н пара-положениях
Так, прн нитрований нитробензола возможно образование следующих о-комплексов
□-комплекс I
при о-замещении
□-комплекс J П
при .^-замещении
□-комплекс при «-замещении
Как видно, среди приведенных граничных структур (1—III), изображающих делокализацию положительного заряда в п-комплексах, структура III при орто-замещении и структура П при пара-замещении содержат положительные заряды у соседних атомов Такие структуры энергетически крайне не выгодны и вносят небольшой вклад в резонансный гибрид Поэтому о-комллексы с участием орто- и napa-воложеннй дестабилизированы в большей степени, чем о-комилекс в мета-положении В результате электро-
111
фнльное замещение затруднено и протекает преимущественно в л₽та-лоложении.
В большинстве случаев статический и динамический факторы действуют согласованно. Но если их влияние проявляется в противоположных направлениях, то решающее значение на направление электрофильного замещения оказывает динамический фактор. Наглядным примером могут служить галогенбензолы, в которых атомы галогена проявляют электроноакцепторные свойства, но вместе с тем направляют электрофильное замещение в орто-и лара-положения, Причиной такого поведения галогенов в качестве заместителя является несогласованное действие статического и динамического факторов.
Как известно, атом галогена в бензольном ядре проявляет отрицательный индуктивный и положительный мезомерный эффекты, причем в статическом состоянии —/-эффект больше 4*М-эффекта В результате происходит смещение электронной плотности бензольного кольца в сторону атома галогена и, следовательно, реакционная способность цикла по отношению к электрофильным реагентам снижается:
Следовательно, в статическом состоянии галогены, подобно ориентантам II рода, затрудняют электрофильное замещение
Однако в процессе реакции неподеленные пары электронов атома галогена, которые находятся в сопряжении с л-электронной системой бензольного кольца, принимают участие в дополнительной стабилизации a-комплексов, образующихся при орто- и пара-замещениях, но не участвуют в стабилизации лета-о-комплекса. Об этом свидетельствуют граничные структуры:
Поэтому галогены аыступают как ориентанты 1 рода к направляют электрофильное замещение в орто- и пара-положения
Кроме ориентаитов I и II рода имеется небольшое число заместителей, проявляющих смешанное действие К ним относятся CHaNO2, СН2На1, СН2ОН, СННа12 и другие Эти заместители
112
несколько затрудняют электрофильное замещение в бензольном ядре, но в результате реакции, как правило, образуется смесь примерно равных количеств орто-, мета- и пара-изомеров.
6.6.1. Ориентация в днзамещенных бензола
При наличии в бензольном кольце двух заместителей их ориентирующее влияние осуществляется более сложно, но и в этом случае часто можно правильно предсказать предпочтительное место вхождения нового заместителя,
В зависимости от электронной природы заместителей и их взаимного расположения различают согласованную и несогласованную ориентацию.
При согласованной ориентации оба имеющихся заместителя направляют новую группу в одни н те же положения бензольного кольца Согласованная ориентация характерна для дизамещенпых бензола, в которых соблюдается два условия:
1. Заместители находятси в .мета-положении относительно друг друга и принадлежат к ориеитантам одного и того же рода (предпочтительные места вхождения нового заместителя указаны стрелками)
н иптро6енл)л.ульфокислота
2, Заместители находятся в орто- или пара-положении по отношению друг к другу, но один из них является ориентантом I рода, а второй — ориентантом ]1 рода
о ।идрокеибснзойнаи кислота салнцн ювая кислота
При несогласованной ориентации один из заместителей направляет новую группу в одни, а другой — в иные положения бензольного кольца, В результате, как правило, образуется несколько различных изомеров. Вместе с тем предпочтительное направление замещения можно определить, используя следующие правила.
113
1. Если один из заместителей является ориентантом 1 рода,
Преимущественное направление замещения определяет ориентант I рода
л нитрофенол л Mi юК1ннитро6ешол
2, Еслй оба заместителя являются ориеитантами I рода, преимущественное направление замещения определяется более сильным ориентантом. По силе ориентирующего влияния заместители I рода можно расположить в следующий ряд:
О- > NR2 > NHR > NH2 > ОН > OR > NHCOR > OCOR >
> Aik > F > Cl > Br > 1
Например, при нитровании 2-бромфенола образуется преимущественно 2-бром-4-нитрофеиол и 2-бром-6-иитрофенол
3, Если оба заместителя являются ориеитантами II рода, электрофильное замещение осуществляется с большим трудом, а преимущественное место вхождении третьего заместителя определяется более сильным ориентантом По силе ориентирующего влияния в реакциях 5, заместители II рода можно расположить в следующий ряд-
СООН > SOsH > NO2 > СНО > COCH, > CN
Например, в о-нитробензойиой кислоте замещение идет преимущественно в мета-положение относительно карбоксильной группы
1 14
Следует также отметить, что наряду с электронной природой заместителей на соотношение продуктов замещения оказывают влияние пространственные факторы При прочих равных условиях из-за стерических препятствий маловероятно вхождение третьей группы между двумя уже присутствующими заместителями
6.7. ИДЕНТИФИКАЦИЯ МОНОЯДЕРНЫХ АРЕНОВ
Для идентификации моноядерных аренов чаще всего используют спектральные методы — УФ-, ИК-, ПМР-спектроскопию и масс-спектрометрию
Бензолу и его гомологам присуще характерное поглощение ; в УФ-области В УФ-слектрах наблюдается интенсивное поглощение в диапазоне 180--210 нм и малоинтеисивное — в области J 230—270 нм При высокой разрешающей способности прибора L малоинтенсивная полоса проявляется в виде ряда узких пиков (тонкая структура) Малоинтенсивиое поглощение в области ИйЗО—270 нм с тонкой структурой очень характерно для аромати-Ических углеводородов, поэтому часто его называют полосой бен-"зольного поглощения (рис 6.3)
Рис 6 3 УФ-спектр поглоте(£ия бензола в газовой фазе 180—210 нм — интенсивное, 230 270 нм — малоинтенсивное
115
В ИК-спектрах бензола и его гомологов имеютси характерные полосы поглощения в области 3100—3000 см-1, связанные с валентными колебаниями ароматических связей С—Н и полосы в области 1600—1500 см"1, обусловленные валентными колебаниями свизей С—С бензольного ядра. Наличие указанных полос свидетельствует о присутствии в структуре соединения бензольного кольца. По характеру других полос поглощения, особенно в областях 2000 — 1650 см" 1225—950 см \ можно установить число и взаимное расположение заместителей в бензольном ядре.
Сигналы ароматических протонов в спектре ПМР бензола и его гомологов прояаляются в слабом поле (5 = 6,5—8,0 м. д.). Это связано с дезэкранированием протонов за счет образующегося прн действии внешнего магнитного поля кольцевого тока.
В масс-спектрах бензола и его гомологов наблюдается интенсивный пик молекулярного иона.
6.8. ОТДЕЛЬНЫЕ ПРЕДСТАВИТЕЛИ. ПРИМЕНЕНИЕ
Бензол. Бесцветная жидкость с характерным запахом (т. кип. 80,1 °C), нерастворим в воде, горит сильно коптящим пламенем. Бензол сильно ядовит. В химической промышленности широко используется в качестве растворителя и исходного сырья для производства стирола, фенола, циклогексана, анилина, красителей, лекарственных препаратов.
Толуол (метилбеизол). Бесцветная жидкость с характерным запахом (т. кип. 110,6 °C). Толуол практически нерастворим в воде, смешивается с этиловым спиртом и диэтиловым эфиром. Применяется в производстве лаков, типографских красок, бензойной кислоты, взрывчатых веществ, лекарственных препаратов и др. Ислользуетси в качестве добавки к моторному топливу для повышении октанового числа бензинов. Обладает слабым наркотическим действием.
Ксилолы (о-, м- и n-ди метил бензолы). Бесцветные жидкости с запахом бензола. Смешиваются с этанолом, диэтиловым эфиром, ацетоном, хлороформом, бензолом. Смесь изомерных ксилолов, так называемый технический ксилол (т. кип. 138 —144 °C), используется в промышленности в качестве растворителя и высокооктановой добавки к моторным топливам.
Стирол (винилбензол). Бесцветная жидкость со своеобразным запахом (т. кип. 145,2 °C). Используется в производстве полистирола, синтетического каучука.
Кумол (изопропилбензол). Бесцветная жидкость (т. кип. 152,4 °C). Нерастворим в воде, смешивается с этанолом, диэтиловым эфиром, бензолом. Используется в производстве фенола и ацетона.
я-Цнмол (1-изолропил-4-метилбснзол). Бесцветная жидкость с приятным запахом (т. кип. 177,4 °C). Нерастворим в воде, растворим в этаноле, хлороформе, ацетоне, днэтиловом эфире. Входит в состав эфирных масел многих растений.
116
КОНТРОЛЬНЫЕ ВОПРОСЫ И УПРАЖНЕНИЯ
1. Назовите следующие соединения:
2, Сравните химические свойства бензола и, 1,3-циклогекса-днсна. В чем причина имеющихся различий?
3, Исходи из бензола и других необходимых реагентов, предложите схемы синтеза следующих соединений; а) пропнлфеиил-кетона; б) 2,4-дя нитротолуола; в) n-хлорбензойиой кислоты; г) н-бутилбензола.
4, В каких условиях следует проводить реакции бромирования изопропилбензола (кумола) для введения атома галогена в ароматическое ядро и боковую углеводородную цепь? Приведите соответствующие схемы и механизмы реакций.
5. Какие продукты образуются при озоиолнзе: а) бензола; б) толуола? Приведите соответствующие уравнения реакций.
6. Расположите нижеприведенные соединения в порядке увеличении реакционной способности в реакциях электрофильного замещения; а) С6НвОН; б) СбН5\Н2; в) CeHsCl; г) CeHsCOOH; д) C6H5NHCH3. Ответ поясните.
7. Определите положения, по которым преимущественно будет проходить сульфирование следующих дизамещеииых бензола:
117
Глаза 7
МНОГОЯДЕРНЫЕ АРЕНЫ С КОНДЕНСИРОВАННЫМИ (АННЕЛИРОВАННЫМИ) БЕНЗОЛЬНЫМИ ЦИКЛАМИ
Миогоядерные арены с конденсированными циклами содержат в своем составе два или более бензольных ядра, имеющие общие атомы углерода,
В зависимости от способа сочленения циклов различают два основных тина конденсированных систем.
а) конденсированные системы с линейным расположением циклов
антраисн
б) конденсированные системы с ангулярным (угловым) рас-
положением циклов
Наиболее важными представителями конденсированных аренов являются нафталин, антрацен и фенантрен.
7.1. НАФТАЛИН
Впервые нафталин выделен из каменноугольной смолы в 1819 г. Элементный состав его молекулы установлен русским химиком А. А. Воскресенским в 1838 г., а строение доказано немецким химиком К Гребе в 1898 г
7.1.1. Номенклатура и изомерия
Молекула нафталина состоит из двух конденсированных бензольных колец. Нумерацию углеродных атомов нафталинового
пв
ядра в соответствии с правилами ИЮПАК проводят, как показано на формуле
Следует отметить, что в молекуле нафталина, в отличие от бензола, углеродные атомы неравнозначны Атомы углерода в положениях 1, 4, 5, 8 отличаются от углеродных атомов в положениях 2, 3, 6, 7. Учитывая это, четыре равнозначных положения в молекуле нафталина - 1, 4, 5, 8 принято обозначать буквой а н называть а-лоложения, а четыре других равнозначных между собой положения 2, 3, 6, 7 — обозначать буквой 0 и называть соответственно 0-положении,
Вследствие неравнозначности положений одиозамещеиные нафталины существуют в виде двух изомеров — а- и 0-:
р метилнафталнн, 2 мстилнафтатии
Двухзамещенные нафталины с одинаковыми заместителями могут существовать в виде десяти изомеров:
1 3 диметилиафтлни мега димети тнафталин
1 4 днметнл нафта тин пара днметилнафталин
1,6 днметнлнафтатин
1 7 диметилнафтdпни
л
СНз СНз
1,8-ДИчетитняфталнн, пери диметилиафталнн
2,fi лиметнлпафт алии ачфи. ди метил II афта ui mi
В номенклатуре дизамещенных нафталина наряду с цифровым обозначением положений заместителей применяют также приставки: орто-положеиие — 1,2; мета - 1,3; пара -- 1,4; ана -- 1,5; пери — 1,8; амфи — 2,6.
7.1.2. Способы получения
Нафталин, его монометильные и некоторые диметильные производные в промышленности получают главным образом из каменноугольной смолы (содержание нафталина в каменноугольной смоле составляет около 10 %).
Кроме того, известен ряд синтетических методов получения нафталина и его гомологов. Так, нафталин наряду с бензолом образуется при пропускании ацетилена через нагретые до 700— 800 °C трубки:
5НС—СН
Нафталин и его гомологи можно также получить дегидроциклизацией алкилбеизолов с боковой цепью из четырех и более атомов углерода
п^нтилбеизол а чет мл на фтал ни
Однако эти н другие синтетические методы не имеют практического значения в связи с высоким содержанием нафталина и его гомологов в каменноугольной смоле.
7.1.3. Строение нафталина
Электронное строение нафталина сходно со строением бензола. Аналогично бензолу молекула нафталина планарна (плоская). Все углеродные атомы находятся в $р2-гибридизацни. Каждый из десяти атомов углерода предоставляет р-атомную орбиталь для образования замкнутой сопряженной системы, охватывающей все углеродные атомы (рис. 7.1).
Рис 7 1 Схема образования замкнутой л-электрошюй системы в молекуле нафталина
Общее число л-электроиов в сопряженной системе удовлетворяет правилу" ароматичности Хюккеля (см. с. 92).
Вместе с тем степень ароматичности у нафталина ниже, чем у бензола. Методом ронтгеноструктурного анализа установлено, что в нафталине нарушена характерная для бензола выравнен-ность углерод-углеродных связей, а именно: связи С“—Ср укорочены, а связи С3—Се удлинены по сравнению со связями С—С бензола. Так, длина связи Са—С₽ в молекуле нафталина составляет 0,136 нм, а связи Ср—С? — 0,142 им, тогда как в бензоле все углерод-углеродные связи имеют длину 0,140 им. Как видно, в молекуле нафталина химические связи между а- и р-атомамн углерода больше напоминают двойные (длина связи С=С в алкенах равна 0,134 нм). Вследствие разной длины углерод-углеродных связей в молекуле нафталина л-электронная плотность распределена неравномерно. У а-атомов углерода электронная плотность вьцпе, чем у р-атомов. Соответственно и энергия сопряжения нафталина (255,2 кДж/моль) значительно меньше удвоенной энергии сопряжения бензола (2Х 150,7=301,4 кДж/моль). Поэтому нафталин легче, чем бензол, вступает в реакции замещения, присоединения, окисления н восстановлеиня.
121
Для изображения молекулы нафталина в химической литературе применяют структурные формулы I—III:
III
Однако, поскольку нафталин не содержит в своей структуре двух независимых замкнутых сопряженных систем бензола, применение формулы III малооправдано
7.1.4. Химические свойства
Для нафталина, как и бензола, характерны реакции электрофильного замещения, присоединения, восстановления и окисления.
А. Реакции электрофильного замещения
В реакции электрофильного замещения (5fc) нафталин вступает легче, чем бензол. При взаимодействии с электрофильными реагентами образуются преимущественно продукты «-замещения Предпочтительное направление замещения в a-положение обусловлено тем, что, во-первых, в а-положении нафталина электронная плотность выше, чей в p-положении (статический фактор), а, во-вторых, при атаке электрофила в a-положение образуется более стабильный и, следовательно, энергетически более выгодный для молекулы «-комплекс, чем прн атаке в 0-положенис (динамический фактор):
Из приведенной схемы вндио, что при электрофильном замещении в a-положении образуется «-комплекс, в котором положительный заряд может быть делокализован без нарушения ароматической системы соседнего кольца, тогда как в образующемся
122
а-комплексе прн замещении в 0-положении делокализация положительного заряда возможна только за счет нарушения ароматической системы соседнего кольца, что влечет за собой дополнительную затрату энергии.
Нитрование. Нафталин довольно легко нитруетсн нитрующей смесью с образованием а-нитронафталнна
В более жестких условиях идет дальнейшее нитрование, приводящее к образованию смеси 1,5- и 1,8-дииитронафталинов.
Сульфирование. При действии концентрированной серной кислоты нафталин сульфируется. Существенное влияние на направление реакции сульфирования оказывает температура реакционной среды В зависимости от температуры реакция сульфирования может протекать преимущественно в а- или 0-положение. Так, сульфирование нафталина при температуре 80 °C приводит к образованию а-иафталннсульфокислоты, а при 160° — главным образом р-нафталинсульфокислоты:
4- Н2О
р н<<ф1 djiHHCi т ||фпкислота
Реакция сульфирования нафталина, как и бензола, является обратимой.
В присутствии 20 %-го олеума при 40 °C а-нафталиисульфо-кислота сульфируется далее; образуя смесь 1,5- н 1,6-нафталиндисульфокислот.
Галогенирование. При температуре 90—110 °C в присутствии FcCh нафталин хлорируется с образованием преимущественно <л-хлорнафталина Бромирование нафталина в а-положеиие происходит легко при отсутствии катализатора:
123
а бромнафталии
Алкилирование и ацилирование. В присутствии кислот Льюиса (Aids, SnCh и др.) нафталин взаимодействует с галогеналкаиамн и галогенангидридами карбоновых кислот с образованием супеси а- и (З-замещеиных изомеров
Б. Реакции присоедииеиня
Поскольку в молекуле нафталина связи между а- и р-атомами углерода имеют большую иепредельиость, чем связи С—С в бензоле, нафталин значительно легче последнего вступает в реакции присоединения
Так, при обычной или пониженной температуре с отсутствие катализатора нафталин присоединяет хлор с образованием продукта 1,4-присоединения
1 4 днхлор 1,4 д и гидро на фтал ин
Рассмотренная выше реакция бромирования нафталина происходит по схеме прнсоедннеиня-отщеплеиня, т. е. вначвле бром присоединяется в положения 1 и 4, а затем продукт присоединения при нагревании отщепляет бромоводород, образуя а-бром-иафталин
124
В присутствии катализатора Ni нафталин значительно легче, чем бензол, присоединяет водород Процесс гидрирования нафталина протекает ступенчато. Сначала при температуре 150° С образуется 1,2,3,4-тетрагидронафталии (тетралин), который при 200 °C гидрируется далее с образованием декагидроиафталииа (декалина):
тот тычин декалин
В. Реакции окисления
Нафталин окисляется гораздо легче, чем бензол. В зависимости от условий окисления образуются разные продукты. Прн окислении кислородом воздуха в присутствии катализатора V2O5 образуется ангидрид фталевой кислоты. Действием оксида хрома (VI) в уксусной кислоте нафталин окисляется до 1,4-иафтохиноиа
1 4 нафтохинон
Следует отметить, что при окислении гомологов нафталина, в отличие от гомологов бензоле, более чувствительно к действию окислителей нафталиновое ядро, например:
£
р метили а фталин 2 метил J ,4-нафтохинон
125
7.1.5. Ориентация замещения в нафталиновом ядре
Если нафталиновое ядро уже содержит какой-либо заместитель, то, как и а случае монозамещениых бензола, при электрофильном замещении можно с определенной точностью предсказать место вступления нового заместителя Однако правила ориентации в нафталиновом цикле в сравнении с правилами ориентации в бензольном кольце имеют некоторые особенности
Направление электрофильною замещения в монозамещенных нафталина определяется двумя факторами 1) электронной природой и положением уже имеющегося заместителя, 2) повышенной реакционной способностью а-положения.
Электроиодонорные заместители (о, n-ориентанты), повышая в целом электронную плотность в нафталиновом цикле, в большей степени активируют кольцо, с которым связан заместитель Поэтому вступление нового заместителя более предпочтительно в это же кольцо и в а-положенне, как более реакционноспособное Прн наличии электроиодоиориого заместителя в a-положении новый заместитель вступает преимущественно в положение 4, которое является гаара-положением по отношению к нмйЬщемуся заместителю и а положением нафталинового цикла (схема а):
В случае, когда электронодонориый заместитель (о-, га-ориен-таит) находится в 0-положении, новый заместитель при электрофильном замещении направляется преимущественно в положение 1, которое является орто-положением по отношению к имеющемуся заместителю и a-положением нафталинового никла (схема б).
Отмеченные выше закономерности могут быть проиллюстрированы следующими примерами:
a MfTHl нафталин
1 метил -1 нитро нафталин
126
Если в нафталниовом цикле находится электроновкцепторный заместитель, то за счет —или —М~ эффекте в он дезактивирует в целом нафталиновое ядро по отношению к элйстрофильиому замещению, но в большей степени дезактивируется кольцо, с которым связан заместитель. Поэтому новый заместитель вступает в соседнее кольцо. Из-за повышенной реакционной способности a-положений замещение происходит преимущественно в положения 5 и 8:
В большинстве случаев прн электрофильном замещении образуется смесь изомеров, например:
а ННтронафта чин 1 5 ди нитро
нафталин
NOa NO3
1 8 дннитро нафта чин
р нафталин '-учьфокистотй
5 нитро 2 нафталин сульфокис юта
8 нитро 2 нафталин сульфокислота
В связи с особенностью реакции сульфирования при высоких температурах и продолжительном нагревании сульфогруппа
127
направляется преимущественно в 0-положения соседнего кольца (положения 6 и 7), например:
SO3H
р пафтаяшкутьфокнслоти
H2SO* —-----
160 “С
HO3S
нафталин 2 Ь дисульфскислота
SO3H
HO3S SO3H
няфтлтмч 2 7 July (Ьфокислота
+ Н2о
7.1.6. Отдельные представители. Применение
Нафталин. Бесцветное кристаллическое вещество с характерным запахом, обладает высокой летучестью (т, пл 80,3 °C). Хорошо растворяется в органических растворителях, плохо — в воде. Нафталин используется в производстве фталевого ангидрида, тетралина, декалина, красителей, лекарственных средств. Применяется в качестве инсектицида,
2-Метил и афтали и. Бесцветное кристаллическое вещество (т, пл 34,4 °C). Хорошо растворяется в органических растворителях, плохо — в воде. 2-Метилнафталин используют главным образом для получения лекарственного препарата викасола (синтетический витамин К) С этой целью вначале 2-метилнафтални окисляют оксидом хрома (VI), а затем действием NaHSO3 получают водорастворимое производное
2 метилнафта ihh
2 мстил 1 4 нафтохинон
пикаюл 2 3 тигитро 2 мети t
I4 няфтохинон 2 сульфонат натрия
Внкасол увеличивает свертываемость крови.
7.2. АНТРАЦЕН
Антрацен впервые выделен из каменноугольной смолы в 1832 г. французскими химиками Ж. Дюма и О. Лораном,
Молекула антрацена состоит из трех линейно конденсированных бензольных циклов. Нумерацию углеродных атомов проводят,
128
как показано на структурной формуле (нумеруются лишь атомы углерода, несущие водород):
Положения 1, 4, 5, 8 в молекуле антрацена называют а-поло-жеииямн, 2, 3, 6, 7 — р-, а 9, 10 — у- или ц- (лезо-средний) поло-
жениями.
7.2.1. Способы получения
В промышленности антрацен получают из антраценовой фракции каменноугольной смолы.
Синтетически антрацен может быть получен:
Алкилирование бензола по Фриделю — Крафтсу 1,1,2,2-те-трабромэтаном
Восстановлением антрахинона
9 10 антрахинон
7.2.2. Строение. Химические свойства
Молекула антрацена плоская. Все атомы углерода находятся в £рй-гнбридизации. Число л-электронов (14 электронов) подчиняется правилу ароматичности Хюккеля Вместе с тем электронная плотность в молекуле антрацена распределена еще более неравномерно, чем в нафталине Энергия сопряжения антрацена (351,5 кДж/моль) значительно меньше утроенной энергии сопряжения бензола (3X150,5 = 451,5 кДж/моль). Поэтому антрацен
5 5 104 129
обладает меньшей ароматичностью, чем нафталин, и значительно меньшей, чем бензол. В итоге антрацен по сравнению с нафталином в большей степени склонен к реакциям присоединения и окисления. Реакции электрофильного замещения для антрацена протекают легко, ио во многих случаях они сопровождаются образованием промежуточных продуктов присоединения, которые могут быть выделены в индивидуальном виде. Наиболее реакционноспособны в антрацене леэо-положения (положения 9 и 10).
Так, антрацен легко восстанавливается водородом в момент выделения и окисляется концентрированной азотиой кислотой с образованием соответственно 9,10-дигидроантрацена и 9,10-
аитрахинона:
Реакции электрофильного замещения, в частности галогенирование и иитроваиие, протекают по положению 9, При взаимодействии с хлором или бромом сначала образуются продукты присоединения в положениях 9 и 10, которые при нагревании отщепляют галогеноводород, превращаясь в 9-галогеиантрацены:
9 llHTpoaHTpadCH
130
4
В результате сульфирования антрацена серной кислотой при нагревании образуется смесь 1- н 2-аитраценсульфокислот, которые являются более устойчивыми изомерами, чем 9-иафталии-сульфокислота.
SO3H
1 интраценсульфокислота
2 антраиенольфокислота
SO3H
+ н2о
Антрацен применяется главным образом в производстве антра хииона и красителей.
7.3. ФЕНАНТРЕН
Фенантрен является структурным изомером антрацена. Его молекула состоит из трех конденсированных ангулярно бензольных циклов. Структурную формулу фенантрена представляют двояко:
10
Нумерацию углеродных атомов проводят, как показано иа структурной формуле.
Получают фенантрен главным образом из антраценовой фракции каменноугольной смолы Известны также синтетические методы получения фенантрена и его гомологов.
По физическим свойствам фенантрен предстааляет собой бесцветное кристаллическое вещество (т. пл. 101 °C), нерастворимое в воде, легко растворимое в органических растворителях. Бензольные растворы фенантрена обладают голубой флуоресценцией.
По строению и реакционной способности фенаитреи во многом сходен с антраценом. Как и антрацен, он является ароматическим соединением, однако ароматический характер у фенантрена выражен несколько сильнее. Так, энергия сопряжения фенантрена (387 кДж/моль) на 35,5 кДж/моль выше, чем у антрацена
Фенантрен так же, как и антрвцен, легко вступает в реакции электрофильного замещения, восстановления и окисления. Наиболее активными являются положения 9 и 10. Химическая связь между 9-м и 10-м атомами углерода в фенантрене весьма напоминает двойную, поэтому прн электрофильном замещении в отдельных случаях образуются промежуточные продукты присоеди-
5*
131
нения, которые могут быть выделены в индивидуальном виде, например:
9 бри «фенантрен
При нитровании фенантрена азотной кислотой в среде уксусной кислоты образуется 9-нитрофеиантрен,
Каталитическое гидрирование фенантрена приводит к образованию 9,10-дигидрофенаитрена, при окислении образуется 9,10-феиантренхинон:
9 10 феиантренхннчн
Сам фенантрен не иашел широкого практического применения. Однако его частично или полностью гидрированные производные входят в состав многих природных соединений, обладающих физиологической активностью — стероидов, алкалоидов и др. Так, в основе стероидов лежит скелет стерана, который представляет собой конденсированную систему, состоящую из полностью гидрированного феивнтреиового ядра (пергидрофеиаитрена) и цнкло-пентаиа:
стеран
циклопрнтанпергидрофенантрен
я
I
132
КОНТРОЛЬНЫЕ ВОПРОСЫ И УПРАЖНЕНИЯ
1. Назовите следующие соедииення:
2. Какие соединения и в каких условиях можно получить при сульфировании нафталина? Приведите соответствующие схемы реакций.
3. На примере реакции нитрования а-метил нафталина и 0-иитронафталииа объясните правила ориентации в ядре нафталина.
4. В чем состоит особенность реакций бромирования антрацена и феиаитрена?
Глава 8
МНОГОЯДЕРНЫЕ АРЕНЫ С ИЗОЛИРОВАННЫМИ БЕНЗОЛЬНЫМИ ЦИКЛАМИ
К многоядериым аренам с изолированными циклами относят углеводороды, содержащие два или более бензольиых цикла, соединенных между собой либо о-связью, либо через алифати-^ ческую углеродную цепь. Наиболее важными представителями этой группы соединений являются бифенил, дифенилметан и три-» феи ил метан.
i
6.1. БИФЕНИЛ
Молекула бнфеиила содержит два беизольиых кольца, соединенных о-свявью.
Для обозначения положения заместителей в молекуле бифенила углеродные атомы каждого бензольного кольца нумеруют отдельно, начиная с атома углерода, через который осуществляется связь со вторым циклом:
би фс н нл
4.4 ‘-диметилбифен ил, л, пJ-диметилби фенил
Расположение заместителей в положениях 2,6,2',6' часто обозначают в названии приставкой орто-, в положениях 3,5,3\5' — • мета-, в 4,4' — пара-.
8.1.1. Способы получения
В небольшом количестве бифенил содержится в измени оу сольной смоле.
Синтетически его можно получить:
Дегидрированием бензола при 750—800 °C
+ Н2
134
Нагреванием иодбензола до 200—250 °C в присутствии порошка меди (реакция Ульмана)
2
1 2Cj
4- 2CuI
8.1.2. Строение. Химические свойства
Два бензольных кольца в молекуле бнфеиила соединены между собой о-связью, длина которой (0,148 нм) несколько меньше длины углерод-углеродной связи в алканах (0,154 им). Уменьшение длины о-свяэи обусловлено мезомерным взаимодействием л-электроиов бензольных колец, которое, как известно, наиболее эффективно при планарном расположении циклов.
Одиако вследствие взаимного отталкивания атомов водорода в положениях 2 и 2' мезомерное взаимодействие между бензольными циклами в бифениле затруднено:
По данным электроиографических исследований в газовой фазе бензольные кольца бнфеиила расположены под углом 45° друг к другу. Следовательно, вокруг о-связи в молекуле бифенила возможно свободное вращение. При отсутствии в орто-положеииях заместителей барьер вращения невелик. Но если в орто-положе-ииях обоих бензольных колец содержатся объемные заместители, то из-за пространственных препятствий свободное вращение вокруг о-связи, соединяющей два цикла, становится невозможным и бензольные ядра располагаются во взаимно перпендикулярных плоскостях:
136
Если хотя бы в одном из бензольных колец в орто-положениях имеются одинаковые заместители, то такая молекула имеет плоскость симметрии, и, следовательно, она ахиральна-
Но если в орто-положениях каждого бензольного ядра молекулы бифенила находятся разные заместители, молекула ие имеет плоскости симметрии и становится хиральиой, что, как известно, обусловливает появление оптической изомерии. Так, 6,6'-дииитро-дифеновая кислота существует в виде двух энантиомеров.
Зеркальные изомеры в ряду бнфеиилв можно рассматривать как конформеры, ставшие стабильными вследствие пространственных препятствий вращению
Вид пространственной изомерии, обусловленный ограничением свободного вращения вокруг простой ’ связи называется атроиизомерией (от греч. атроло — нет поворота).
Для проявления атропизомерии необязательно наличие четырех заместителей в орто-положениях; иногда достаточно трех или даже двух объемных групп
Химические свойства бифенила аналогичны свойстаам моно-ядерных аренов. Фенильные группы проявляют по отношению друг к другу слабые электронодонорные свойства. Поэтому в реакции электрофильного замещения (галогенирование, нитрование и другие) бифенил вступает несколько легче, чем бензол, образуя преимущественно продукты пара- н орто-замещения В моио-замещениых бифенилах при электрофильном замещении новый заместитель вступает в незамещенное ядро:
136
4 4 диннтробифенил
2 4 днннтробифеннi
Бифенил является бесцветным кристаллическим веществом (т. пл. 71 °C) со слабым своеобразным запахом, малорастворнм в воде, хорошо растворяется в органических растворителях.
Применяется в смеси с дифениловым эфиром в качестве высокотемпературного теплоносителя для обогревания химических реакторов и других уствиовок. Производные бифенила используются в производстве красителей
8.2, ДИФЕИИЛМЕТАН
В молекуле дифеиилметана два бензольных цикла связаны через метиленовую группу
Дифеиилметан может быть получен по реакции Фриделя — Крафтса алкилированием бензола бензилхлоридом нли дихлор-метаиом:
СН2С1
Реакционная способность дифеиилметана обусловлена наличием в его структуре беизольиых циклов и активной метиленовой группы
С участием беизольиых колец дифеиилметан вступает в характерные для аренов реакции электрофильного замещения, образуя 4,4'-дизамещениые и 2,4,2',4'-тетразамещенные продукты’
Si- O2N< V-СНг--Z ►
Н2О \ Z \— Z
4 4 дияитродифени1мета[Г
137
no2 no2
hno3. H2SOt
-нго
N02
2,4,2‘,4' тетранитродифенилметаи
Вследствие электроиоакцепторного действия фенильных групп в молекуле дифенил метана приобретают подаижиость атомы водорода метиленовой группы. В результате метиленовая группа легко окисляется; при галогенировании водородные атомы замещаются на галоген
дифенил бромиетаи
Дифенилметаи представляет собой бесцветное кристаллическое вещество (т. пл. 26—27 °C) со слабым приятным запахом; не растворим в воде, растворим в этаноле. Используется в парфюмерной промышленности для отдушки мыл (имеет запах герани).
8.3, ТРИ ФЕ НИЛ МЕТАН
Молекула трифенил метана содержит три беизольиых цикла, связанных через метииовую группу (СН):
Нумерацию углеродных атомоа в каждом цикле проводят отдельно, начиная с атома углерода, связанного с метииовой группой.
1.ЧЯ
Трнфеиилметаи получают алкилированием по Фриделю — Крафтсу бензола хлороформом:
По химическим свойствам трнфеиилметаи во многом напоминает дифенилметаи. Для него характерны реакции электрофильного замещения с участием бензольных циклов, которые идут главным образом в лара-положеиия.
Вследствие электроиоакцепторного действия трех фенильных групп в молекуле трифен ил метан а приобретает подвижность атом водорода метииовои группы. Данный атом водорода легко замещается на металлы и галогены, за счет метнновой группы трнфеиилметаи окисляется. Так, при действии амида натрия в жидком аммиаке трнфеиилметаи образует трифенилметил-иатрий, прв взаимодействии с хлором дает трифенилхлорметаи, при окислении образует трифеиилметаиол (трифеиил карбинол}:
Ма.ЧН2 - — >
(О|
139
Трифеиилметилиатрий имеет иоииое строение. Его молекула состоит из катиона натрия и трифенилметил-аииона. Эфирные растворы трифеиилметнлиатрия проводят электрический ток.
Атом галогена в молекуле трифеннлхлорметана и гидроксильная группа в молекуле трифеинлметаиола под влиянием электроно-акцепторных свойств трех бензольных колец проявляют высокую подвижность (активность). Так, трифенилхлорметан в жидком SO2 (растворитель с высокой диэлектрической постоянной) в присутствии AlCh подвергается ионизации с образованием трифенил-метил-катиона. Аналогично происходит ионизация трифеиил-метаиола в концентрированной серной кислоте:
трифенилхлорметан
Способность трифеннлметильной группы образовывать устойчивые карбкатионы н карбаииоиы обусловлена участием бензольных циклов в делокализации положительного или отрицательного заряда иоиа за счет сопряжения:
трифенилметил-катион
трифенилмстал-аниои
140
Одна ко, несмотря иа то что центральные атомы углерода в трифенил метил-катионе и "трифенилметнл-анионе находятся в sp'2-гибридизации, указанные ионы имеют ие плоскостную, а пропел-лерообразиую структуру, в которой беизольиые циклы вывернуты из плоскости ив 30—40°. Такое размещение бензольных колец в пространстве связано со стерическим взаимодействием атомов водорода в орто-положениях (рис, 8.1).
Вакантная
а
Рис 8 1 Пространственное строение трифенилметил-катиона (я) и трифенилметил аниона (б)
Вследствие этого сопряжение центрального атома углерода с каждым ядром несколько меньше, чем можно было бы ожидать при плоскостной структуре.
При обработке трифеиилхлорметаиа цинком, натрием или мелкораздроблеииым серебром в бензольном растворе образуется свободный трифеиил метильный радикал, находящийся в равновесии со своим димером:
141
Свободный радикал трифеиилметил получен впервые в 1900 г. америкаиским химиком М. Гомбергом прн изучении химических свойств трифеиилхлорметана. Это был первый из радикалов, полученных в свободном виде Причина столь высокой устойчивости три фен ил метил а по сравнению с алкильными радикалами состоит в значительной делокализации неспаренного электрона по всем бензольным ядрам:
Трифеиилметил является очень реакционноспособным веществом, На воздухе ои легко 'окисляется, образуя перекись трифеиилметила (СбНз^зС—О—О—С(СбН5)з, с металлическим натрием дает трифеиилметилнатрий (С6Н5)эС-На+, с иодом образует иодтрифеиил метай —1.
Трифенилметнльный катион, аинон и радикал имеют такую окраску: трифеиилметил-катнон — красно-оранжевую, трифеинл-метил-аииои — красного цвета, а свободный радикал трифеиилметил - желтого цвета. Появление окраски в указанных ионах и свободном радикале обусловлено наличием в их структуре достаточно длинной сопряженной системы (хромофора), включающей три бензольных цикла.
Производные трифеиилметаиа широко применяются в качестве красителей, лекарственных препаратов.
8.3.1. Красители трифеиилметанового ряда
Трифенилметановые красители являются амино- или гидроксипроизводными трифеиилметаиа. Представителем амниотрн-фецнлметанов является краситель — бриллиантовый зеленый:
142
Бриллиантовый зеленый получают конденсацией бензальдегида с N, N-диэтиланилином в присутствии кислотного катализатора, чаще концентрированной хлороводородной кислоты. В результате реакции образуется бесцветное вещество, так называемое лейко-основание бриллиантового зеленого [4,4'-бис-(диэтиламино)три-феннлметаи], которое затем окисляют диоксидом свинца в кислой среде. При окислении образуется бесцветное вещество, получившее назваине карбинольное основание бриллиантового зеленого [4,4'-бис-(диэтнламино)трифеинл карбинол], которое со щавелевой кислотой образует соль — краситель бриллиантовый зеленый:
ноос-соон щавелевая кислота
-Н1О *
карбинольное основание бриллиантового зеленого
143
бриллиантовый зеленый
Бриллиантовый зеленый представляет собой кристаллическое вещество золотисто-зеленого цвета, растворимое в воде, этаноле, хлороформе. Водные растворы имеют иитеисивно-зелеиую окрвску, носителем которой является катион, содержащий в своем составе длинную сопряженную систему, включающую хиноидную группировку (хромофор), а также диэтиламнио- и диэтиламмоиийиую группы (ауксохромы) (см. с. 242). Окраска бриллиантовым зеленым малоустойчива к действию света и влажным обработкам, вследствие чего краситель применяется главным образом для окрашивания нетекстильиых материалов — бумаги, древесины и др.
Бриллиантовый зеленый применяетси в медицине в виде 1 -2 % водных или спиртовых растворов как антисептическое средство, а также при изготовлении бактерицидных лейкопластырей.
К амннопроиэводным трнфенилметана относятся также красители — малахитовый зеленый и кристаллический фиолетовый:
(CH3)2N +N(CH3)2
малахитовым зеленый
144
кристаллический фиолетовый
Малахитовый зеленый применяют для окрашивания тканей (хлопок, шерсть, шелк) в зеленый цвет. Кристаллический фиолетовый применяется главным образом как кислотно-осиовиый индикатор для титрования в водных и неводных средах. В водной среде он имеет два перехода окраски — в интервалах pH 0—1,0 желтая окраска переходит в зеленую, прн pH 1,0—2,6 зеленая окраска переходит в фиолетовую.
Представителем гидроксипроиэводных трифеинлметана является фенолфталеин. Получают фенолфталеин конденсацией фенола с фталевым ангидридом в присутствии концентрированной серной кислоты. В результате реакции образуется бесцветная (лактонная) форма фенолфталеина!
фталевый ангидрид
фенолфталеин (бесцветная форма)
Фенолфталеин представляет собой белое кристаллическое вещество, практически нерастворимое в воде, хорошо растворяется в этаноле, т. пл. 259—263 °C. Фенолфталеин применяется в аналитической практике каю кислотно-основный индикатор. В кислой и нейтральной среде он находится в бесцветной лактонной форме, в щелочной среде (при pH 8,2—10,0) приобретает малиново-красную окраску, вследствие образования хиноидной структуры;
145
фенолфталеин (бесцветная форма)
фенолфталеин (форма с малиново-красной окраской)
В сильно щелочи ой среде (pH 3>12) малииово-красиая окраска фенолфталеина исчезает в результате образования соли бензоидной структуры:
форма с малиново красной окраской
бесцветная форма
+ Н2О
Фенолфталеин применяется в медицине как слабительное средство при хронических запорах. Препарат выпускается в таблетках под названием пурген.
146
КОНТРОЛЬНЫЕ ВОПРОСЫ и УПРАЖНЕНИЯ
1. Назовите приведенные соединения:
2. Предложите способы получения следующих соединений, исходя из беизолв; а) 4,4'-диметилднфенила; б) 4,4',4"-трибром-дифенилметаиа; в) 4-иитротрнфеиилкарбннола.
3. Почему 6,6'-дииитродифеновая кислота, несмотря на отсутствие центров хиральности, обладает оптической активностью и существует в виде пары оптических антиподов?
4. Напишите схемы получения промежуточных активных частиц трифенилметаиового ряда. Объясните причину их высокой устойчивости. Как влияют электронодоиорные и электроноакцепторные заместители на их стабильность?
Главе 9 НЕБЕНЗОИДНЫЕ АРОМАТИЧЕСКИЕ СОЕДИНЕНИЯ
Как уже? отмечалось, критерием ароматичности соединения является иалйчЛе ^._его структуре плоского цикла, имеющего замкнутую сопряженную систему, содержащую (4п -f- 2)л-элек-тронов. Этим требования# удовлетворяет ряд соединений, не содержащих в своем составе бензольных циклов. Наиболее важными представителями небЬнзоидных ароматических систем являются циклопеитадиенил-аииои, циклогептатриеиил-катиои (тропилий-катиои) и бициклический углеводород азулеи:
нс—сн
// \\
нс. « .сн хс^
1 н
цикло пента диски л аннон 6л ллектронов irt = 1)
нс=сн
сн
цнкпогептатрненил катион
(тропилиft ион) бл-электронов {а — I)
нс
НС—СН
.сн
сн
азулеи
10л электронов (и = 2}
9.1. ЦИКЛОПЕНТАДИЕНИЛ-АННОН
При взаимодействии циклопеитадиеиа-1,3 с металлическим натрием в кипящем ксилоле активная метиленовая группа отщепляет протоиш образуя цнклопентадиеиилиатрий, содержащий ароматический циклопентадиеиил-аиион
НС—СН
Н Н
нс—сн
Na
цнклопемтадиен 1 3
циклопентадиенилнатрнй
В циклoneитадиеиил-аииоие все пять атомов углерода находятся в «р2-гнбридиэацни. На пяти р-орбиталях размещается шесть
148
тр2-гибридиэоваииый атом углерода
Рис 9 | Схема образования ароматической системы циклопентадненил-аннона
р-электроиов (4 электрона двух л-связей и 2 электрона аиноииого центра) (рнс. 9.1). В результате перекрывания р-орбиталей збразуется замкнутое л-электроиное облако, имеющее ароматический секстет электронов При этом отрицательный заряд анионного центра равномерно распределяется между пятью атомами углерода.
Ароматические свойства циклопеитадиеиил-аииона подтверждаются его способностью вступать в реакции электрофильного замещения (сульфирования, азосочетания), а тацрте образовывать устойчивые л-комплексы с катионами двухвалентных металлов группы железа (железо, кобальт, никель). Например, при взаимодействии циклопентадиенилнатрия с солями железа (II) образуется ферроцен (дициклопентадиеиилжелезо):
Ферроцен является представителем группы металлоорганических соединений, получивших общее название — металлоцены.
Методом рентгеиоструктуриого анализа установлено, что циклопеитадиенильиые анионы в ферроцене размещены друг над другом в двух параллельных плоскостях, между которыми расположен атом железа. Такое строение, напоминающее расположение масла между двумя кусками хлеба в бутерброде, получило название «сэндвичевой структуры» (от англ, sandwich — бутерброд).
Молекула ферроцена представляет собой л-комплекс, который образуется в результате перекрывания связывающих л-МО двух цнклопентадиеиил-аиионов с вакантными АО катиона железа
149
Ферроцен является типичным ароматическим соединением. Он устойчив к нагреванию и каталитическому гидрированию, вступает в реакции электрофильного замещения — алкилирования и ацилирования по Фриделю — Крафтсу, сульфирования.
В технике ферроцеи применяется в качестве антидетонатора и термически стойкого теплоносителя. Производное ферроцена — ферроцерон применяется в медицине для лечения железодефицит-иых анемий
ферроцерон
9.2. циклогептатриеннл-катион
Циклогептатриеиил-катион (тропилий-ион) образуется прн от- , щеплеиии гидрид-иоиа (атома водорода с двумя электронами) ; от метиленовой группы циклогептатриена-1,3,5 (тропилидеиа). Так, при взаимодействии циклогептатриена-1,3,5 с бромом обра- t зуется тропилийбромнд, который содержит циклогептатриеиил-катион:
циклогептатриен-1,3,5
тропилий бромид
Тропилий-ион обладает ароматичностью. В семичленной циклической структуре тропилиевого иона все атомы углерода находятся в зра-гибрндизацнн. В результате перекрывания шести одно-электронных р-орбиталей двойных связей и одной вакантной р-орбнталн атома углерода катионного центра образуется замкнутое л-электрониое облако (ароматический секстет), охватывающее все углеродные атомы цикла. При этом положительный заряд
150
_v2-гибридизоваиный атом углерода
Рис. 9 2. Схема образования ароматической системы циклогептатриенил-катнона
катионного центра равномерно распределяется между семью атомами углерода (рис. 9.2).
Вследствие ароматичности тропнлий-катион довольно устойчив. Имея дефицит электронной плотности, тропнлий-катион проявляет электрофильные свойства и легко вступает в реакции с нуклеофильными реагентами, образуя продукты присоединения,
9.3. АЗУЛЕН
Азулен представляет собой бициклическую конденсированную систему, состоящую из цнклопеитаднеиового и циклогептатриено-вого колец: 7 R
азулен, бицикл о (5,3.0] деклке нтаен
Молекула азулена имеет биполярное строение — атомы углерода пятичлениого цикла несут частичный отрицательный, а семичленного — частичный положительный заряд. Образование биполярной структуры обусловлено стремлением каждого кольца Иметь ароматический секстет р-электроиов. Такая возможность появляется при переходе одного электрона из семичлеиного цикла
151
в пятичлеиный, вследствие чего пятичлеиное кольцо приобретает отрицательный заряд, а семичлеиное — положительный:
Поэтому азулеи можно рассматривать как конденсированную систему, состоящую из циклопентадиеиил-аниоиа и тропилий-катиоиа. Азулен обладает ароматичностью. Ароматический характер азулеиа проявляется в склонности к реакциям электрофильного замещения (галогенирование, нитрование, сульфирование, ацилирование), которые протекают по пятичлениому кольцу в положениях 1 или 1 и 3.
При нагревании азулен изомеризуется в нафталин
атутен нафталин
Алкилпроизводиые азулена содержатся в эфирных маслах ряда лекарственных растений, например, ромашки, тысячелистника, полыни, эвкалипта и других С их присутстаием связывают противовоспалительное действие этих растений.
КОНТРОЛЬНЫЕ ВОПРОСЫ И УПРАЖНЕНИЯ
1. Почему ферроцен вступает в реакцию ацилирования легче, чем бензол, а тропнлийбромид не подвергается ацилированию?
2. Напишите схемы реакций тропилийбромида со следующими реагентами: a) NaNH2; б) СН3О№. Обладают ли ароматичностью полученные продукты?
3. Напишите схемы реакций азулеиа со следующими реагентами: а) Вг2 (А1Вг3); б) HNO3 (коиц.); в) СН3СОС1 (А1С13).
Глава 10 ГАЛОГЕНОПРОИЗВОДНЫЕ УГЛЕВОДОРОДОВ
Галогеиоироиз водны ми углеводородов называют продукты замещения в углеводородах одного или нескольких атомов водорода атомамя галогенов.
В зависимости от природы углеводородного радикала галоге-иопроизводные подразделяют иа алифатические, алициклические н ароматические. Среди алифатических различают насыщенные (галогеналкаиы) и ненасыщенные (галогеиалкены, галогеиалки-иы). Ароматические галогеиопроизводиые делят на соединения, в которых атом галогена непосредствеиио связан с ароматическим ядром (галогеиареиы) и вещества, содержащие атом галогена в боковой цепи (арилалкилгалогеииды). В соответствии с природой атома галогена галогеиопроизводиые подразделяют на фтор-, хлор-, бром- и иодпроизводные. По числу атомов галогена в молекуле различают моно-, ди-, три- и полигалогенопроизводные углеводородов.
10.1. НОМЕНКЛАТУРА
По заместительной номенклатуре ИЮПАК названия галогено-
производных углеводородов составляют аналогично названиям соответствующих углеводородов. К названию атомов галогенов прибавляют название родоиачальной структуры. За родоначаль-иую структуру в алифатических галогеиопроизводиых принима-
ется главная углеродная скнх — цикл:
цепь, в алициклических и ароматиче-
4 3 2 1
СНз—СНз—С Н—СНз
I Вг
2 бром бутам
бромбензол
хлор пик юге ксвн
Если при родоначальной структуре имеется несколько заместителей, которыми кроме атомов галогенов могут быть и углеводородные радикалы, то в названии перечисляют их в алфавитном порядке. Атомы углерода родоначальной структуры нумеруют
153
в этом случае таким образом, чтобы заместитель, который обозначен в названии первым, получил возможно меньший иомер, например-
1 2 3 4 5
СНз—СН-СНз—СН—СНз
2 бром 4 мстн,1пента||
5 4 3 2 1
СНз—С Н—СНз—СН—СНз
2 метил 4 клорпеитан
СНз—CH—СН—СНз—СНз
3 бром б метилпен-ган
1 бром 3 изопропилбензол
I изопропил 3 хлорбензол
В галогеналкеиах и галогеналкииах нумерацию атомов углерода главной цепи проводят так, чтобы возможно меньшие номера получили атомы углерода кратной связи:
5 4 3 2 1
СНг-СНг—СН—СН=СН2
Вг С^Н3
5 бром 3 метилпентен 1
3 2 1
СНз—feCH
3 хлорпропии
Для простейших галогенопроизводиых продолжают использо- , вать радикало-фуикциоиальиую номенклатуру, согласно которой название составляют из названия углеводородного радикала, связанного с галогеном, и суффикса -фторид, -хлорид, -бромид или -иодид: (
СН3С1 метнлхлорид
С2Н5Вг
ЬТИ 1броыид
С6Н5СН2С1
бенлюхлорид
СбН5СНС12
СНа=СН—С1
СН2=СН—СНз—I
бензнлидеихлорид
винилхлорид
а 11илиодиД
i
154
Для галогенопроизводных, у которых все имеющиеся в молекуле атомы водорода замещены иа галЪгеи, в названиях используют префикс пер-:
CF3—CF3 CF2=CF2
перфторэтан перфторзтнлен
За некоторыми галогеиопроизводиыми сохранились тривиаль-
иые названия- CI
СН3С1 СНС1з СНВгз - CH1S СНа=С—СН=СН2
хлористый метил '-лороформ бромоформ йодоформ хлоропрен
10.2 ИЗОМЕРИЯ
Для галогенопроизводных углеводородов характерна структурная, геометрическая и оптическая изомерия.
Структурная изомерия обусловлена разной структурой углеродного скелета молекулы и разным положением атомов галогенов в цепи. Так, хлорбутан C+HgCl существует в виде четырех структурных изомеров:
ch3ch2ch2—сн2 сн3—сн2—сн—сн3
Cl (L
1 хлорбутан 2 хлорбутан
СНз С1
2 метил I хлорпропан
СН3
2 метил 2 хлорпропан
Днгалогенопроизводиые могут быть представлены несколькими структурными изомерами, отличающимися взаимным расположением атомов галогенов, например-
СН2- СН—СНз СН2— СНг—СН2
<ii h ci (ii
1 2 дихлорпропан 13 ди хлорпропан
С1 СНз—(!: —СНз
I
С1
2 2 дихлорпропа»
1 1 дихлорпропан
156
Дигалогеиопроизводные с атомами галогенов у одного и того же атома углерода называют геминальными (сокр. гем), у соседних атомов углерода — вицинальными (сокр. виц).
Для галогеналкаиов, имеющих разные заместители при атомах углерода, образующих двойную связь, наряду со структурной изомерией возможна геометрическая изомерия, иапрнмер:
цис 1 хлорпропен
транс 1 хлорпропек
Для галогеналкаиов, содержащих в своей структуре асимметрический атом углерода, характерна оптическая изомерия (см. кн. 1, разд. 5.2.2.). Так, 2-хлорбутаи СНэСН2СНС1СН3 существует в виде двух зеркальных изомеров (энантиомеров):
СНз СНз
Н---------С1 С1--------Н
С2Н6
D 2 хлор бутан S 2-*лорбутап
С2Н5
L 2 хлорбутан Я 2-хлорбутан
10.3. ГАЛОГЕНАЛКАНЫ
10.3.1. Способы получения
Галогенирование алканов. Взаимодействие алкаиов с галогенами протекает при УФ-облучении по свободнорадикальному механизму SR (см. с. 20). Метод позволяет получить хлор- и бром-алкаиы. Недостатком этого способа является образование смеси моио-, ди- и полигалогеиалкаиов:
СН< С1а,-% СНэС! СН2С12 и т. д.
-НС) - HC1
хлориетан дихлорметан
Присоединение галогеноводородов к алкенам (см. с. 36).
Вт
СН3—*СН^Н2 + НВг ------------► СН3-СН-СН3
пропен 2-бромпропан
С помощью этого метода можно получить фтор-, хлор-, бром-и иодалкаиы.
156
Получение из спиртов. Для получения галогеналкаиов нз спиртов в качестве галогенирующих реагентов используют галогеио-водороды (НС1, НВг, HI), галогениды фосфора (РС13, РС1Б, РВг3, РВгБ) и тиоиилхлорнд SOC12.
Взаимодействие спиртов с галогеиоводородами протекает по следующей схеме:
С2Н5ОН 4- НВг С2Н5Вг 4- Н2О этанол бромэтан
Обычно галогеиоводороды выделяют непосредственно в процессе реакции взаимодействия солей галогеиоводородных кислот с концентрироааиной H2SO*:
С2Н5ОН 4- КВг 4- На SO* ---->- C2HsBr 4- КН SO* 4- Н2О
С помощью этой реакции можно получить хлор-, бром- и нодал-каиы. Особенности протекания реакции в зависимости от строения спирта и природы галогеиоводорода, а также механизм взаимодействия рассмотрены на с. 259>
С более высокими выходами галогеиалкаиы образуются при взаимодействии спиртов с галогенидами фосфора (III) или фосфора (V), а также тионилхлоридом SOC12:
ЗС2Н5ОН 4- РС1з ----► ЗС2Н5С1 4- Н3РО3
С2Н5ОН 4- PCU ------>- С2Н5С1 4- НС! 4- РОС1Э
С2Н5ОН 4- SOC12----->- C2H5CI + НС1| + SOat
Реакция с тионилхлоридом очень удобна тем, что все побочные продукты являются газообразными веществами и легко уходят из реакционной среды.
Взаимодействие галогеналкаиов с солями галогеноводородных кислот (реакция Финкельштейна). При действии на хлор- или бром алканы иодида натрия в среде ацетона происходит замещение атома хлора или брома иа иод
С2Н5Вг 4- Nal зцетон > С2Н51 4- NaBrj
Реакцию используют для получения иодалкаиов из более доступных хлор- илн'бромпроиэводных.
10.3.2. Физические свойства
При обычных условиях низшие галогеналкаиы — бесцветные газы или жидкости со своеобразным сладковатым запахом, средине-жидкости, высшие — твердые вещества (табл. 10.1.).
Температура кипения возрастает с увеличением атомной массы галогена, числа атомов галогена (за исключением фторпроизвод-ных) и длины углеродной цепи молекулы. Низшие галогеиалкаиы
157
Таблица 10 ]
Физические характер нети ин некоторых галогеналканов
Галогеналкан Температура, °C
плавления кипения
СН3—F -141,8 -78,6
СНз—CI -96,7 -23,8
СНз—Вг —93,6 —3,6
СН3—I -66,1 42,5
СНэСНг— F — 143,2 —37,7
СНэСНг— CI -138,7 12,2
СНэСНг— Вг -119 38,2
СНзСНг—1 -108,5 72,2
СНаСНгСНг—С1 -122,8 47.2
CHaCh -96,7 40,1
CHCh -63.5 61.3
СНВгз 8 150
снь 119 210 (разл )
СС14 -22.8 76,8
практически не растворимы в воде, ио легко растворяются в ор- ' гаиических растворителях. Некоторые из них сами являются хорошими растворителями. Многие галогеиалквны обладают наркотическим действием
10.3.3. Химические свойства
Галогеиалканы являются весьма реакционноспособными веществами. Наиболее характерны для иих реакции нуклеофильного замещения (SN) и отщепления (£). Оии также-способны к взаимодействию с металлами и восстановлению.
А. Реакции нуклеофильного замещения (SN)
Галогеиалканы являются электрофильными реагентами. Их электрофильные свойства обусловлены полярностью связи С—Hal. Поскольку атом галогена проявляет большую электроотрицательность, чем атом углерода, электронная плотность связи С—Hal в галогеиалкаиах смещена к атому галогена. В результате атом галогена приобретает частичный отрицательный (6—), а атом углерода — частичный положительный (б-|-) заряды «4- t>-(С-»-На1), Электронодефицитиый атом углерода становится электрофильным центром молекулы галогеиалкаиа и может быть атакован нуклеофильным реагентом. В процессе атаки нуклеофил предоставляет пару электронов для образования химической связи с электронодефнцитным атомом углерода, а атом галогена отщепляется от молекулы галогеиалкаиа с электронной парой связи С—Hal:
6- .. .
галогеналкан нуклеофил
-JC-Nu продукт замещения
4- НаГ
уходящая Группа
= 158
в
i акую реакцию называют реакцией нуклеофильного замещения и обозначают символом Нуклеофильными реагентами могут быть вещества, содержащие в молекулах атомы с неподелениыми парами электронов (NH3, R—NH21 RsNH, НбН и др ), либо вещества, образующие при диссоциации аяиоиы (нуклеофильные частицы): №ОН (ОН-), СгНбСЖа (С2Н5О-), KCN(CN“), NaSH(SH“), NaNO2(NOr), CH3COONa(CH3COO-), КВг(Вг"), KI (1“) и др.
В результате нуклеофильного замещения в структуру нуклеофила вводится алкильный заместитель, т. е. происходит алкилирование нуклеофила. Поэтому галогеиалканы являются алкилирующими реагентами.
Склонность галогеиалканов к реакциям определяется полярностью связи С—На! (статический фактор). В зависимости от электроотрицательности атомов галогенов (см. кн. 1, стр 44) полярность связи С—На! увеличивается в ряду: С—1 < С—Вг < < С—Cl с С—F. Следовательно, можно предположить, что фторпроизводиые галогеиалканов более реакционноспособны, чем хлорпроизводиые, а последние более активны, чем бром- и иод-производиые.
Реально же реакционная способность галогеиалканов в реакциях уменьшается в ряду: R—1 > R—Вг > R—Cl >>R—F Наблюдаемая закономерность объясняется поляризуемостью связи С—Hal, т. е. способностью связи увеличивать полярность при подходе нуклеофильного реагента (динамический фактор). Как известно, поляризуемость связи тем больше, чем более объемиста и подвижна электронная оболочка атомов, образующих связь. Среди галогенов наиболее высокой поляризуемостью обладает атом иода, наименьшей — атом фтора, а поэтому связь С—I более склонна к гетеролитическому разрыву, чем связь С—F. Из-за низкой поляризуемости связи С—F фторалканы практически не вступают в реакцию нуклеофильного замещения. Активность остальных галогеиалканов по отношению к нуклеофилам падает в ряду R—I > R—Вг 2> R—С1.
В зависимости от строения галогеиалкаиа, природы нуклеофила и растворителя реакции нуклеофильного замещения протекают по двум основным механизмам: S42 и Shl.
Механизм 5Ч2 (бимолекулярное нуклеофильное замещение). По механизму SN2 реакции происходит в одну стадию через образование переходного состояния, в построении которого принимает участие как молекула галогеиалкаиа, так и нуклеофильный реагент
В механизме S42 нуклеофил атакует электрофильный центр молекулы галогеиалкаиа (атом углерода, связаиный с галогеном) со стороны, противоположной связи С—На! (атака с тыла). При сближении нуклеофила с галогеналканом в их молекулах происходит перераспределение электронной плотности химических связей. В результате образуется переходное состояние, представляющее собой предельно неустойчивое сочетание двух реагентов,
159
в котором связь С—Hal ослабляется и начинает формироваться связь С—Nu. Переходное состояние находится в равновесии с исходными реагентами. Ему соответствует максимум на энергетической диаграмме реакции. По мере дальнейшего сближения реагентов в переходном состоянии происходит синхронный процесс разрыва связи С—Hal и образование связи С—Nu:
tin' + ^С—Hal
/ 5-
Nu—C — Hal I
Nu-C-
+ Hal
переходное состояние
С позиций теории МО при образовании переходного состояния происходит перегибридизация центрального атома углерода, а именно: исходная $р3-гибридизация изменяется иа 5р2-гибриди-зацию. В результате три нереагирующие связи атома углерода располагаются в одной плоскости, перпендикулярно к которой находится негнбридизированная р-орбиталь Одна доля этой р-орбитали частично перекрывается с орбиталью атакующего нуклеофила, а вторая — с орбиталью атома галогена (рис 10.1).
Рис 10 1 Схема перекрывания р орбитали атома углерода в переходном состоянии с орбиталью атакующего нуклеофила и атома гадснена
Если электрофильным центром в молекуле галогеналкана является асимметрический атом углерода, то нуклеофильное замещение атома галогена сопровождается обращением конфигурации. Это служит подтверждением того, что в механизме нуклеофильная атака электрофильного центра галогеналкана осуществляется с тыла:
% >СНз
С- Вг
I
С2Н5
А-изомер
Х>сн;
Nu—C — Br I С2Н5
% >СНз
Nu-C
I C2HS 5-изомер
Вг
160
По предложению английского химика К- Иигольда, описанный механизм получил обозначение S42. Буква S указывает на замещение, N — на нуклеофильный тип реакции, а цифра 2 обозначает, что реакция является бимолекулярной, т. е в стадии, определяющей скорость реакции в целом (в данном случае образование переходного состояния) участвует два реагента (галоген-алкан и нуклеофил). Скорость реакций, протекающих по механизму S*j2, зависит от концентрации обоих реагентов и описывается кинетическим уравнением второго порядка и = A[R—Hal] [Nu].
Факторы, способствующие протеканию реакций по механизму S^2. Реакционная способность галогеналкаиов по механизму SN2 тем выше, чем больше частичный положительный заряд на атоме углерода, связанном с галогеном- Поэтому среди первичных галогеналкаиов активность уменьшается в ряду
5+ 6+ 6<-"
СНз^Вг > СН^СНг^Вг > СНз *СН2^СН2—Вг а-1- > й-r > й+
Одиако наряду с электронными факторами значительно большее влияние на реакционную способность галогеналкаиов по механизму SN2 оказывают пространственные факторы. Чем больше количество и объем заместителей у атома углерода, связанного с галогеном, тем труднее образуется переходное состояние из-за пространственных препятствий. В связи с этим по механизму 5^2 легко реагируют первичные и несколько труднее — вторичные га-логеналканы. Третичные галогеналканы из-за пространственных препятствий в реакции нуклеофильного замещения по механизму Sn2 не вступают.
Для реакций, протекающих по механизму S42, важное значение имеет природа нуклеофила, так как нуклеофил принимает участие в образовании переходного состояния Чем выше реакционная способность нуклеофила, тем легче протекает реакция по механизму Sn2-
Под нуклеофильностью (нуклеофильной силой) реагента понимают его способность образовывать за счет неподелеииой пары электронов ковалентную связь с электронодефицитным атомом углерода.
Нуклеофильность реагента зависит от ряда факторов электроотрицательности и поляризуемости нуклеофильного центра (атома с иеподелеиной парой электронов или несущего отрицательный заряд), характера заместителей, связанных с нуклеофильным центром, и природы растворителя. При равных других факторах по мере увеличения электроотрицательности атома нуклеофильного центра в пределах одного и того же периода Периодической системы элементов Д. И. Менделеева неподелениые пары электронов удерживаются прочнее и, следовательно, нуклеофильность реагента снижается. Так, нуклеофильность следующих соедине
6 > ни
161
ний и соответствующих им анионов снижается в рядах ЙНз > Н2О > HF; NHf > ОН” > F”
Следует отметить, что нуклеофильность иона значительно выше нуклеофильности соответствующей нейтральной молекулы: NH2” > N'H3; ОН > Н2б; SH > H2S.
В пределах группы Периодической системы нуклеофильность увеличивается сверху вниз в соответствии с возрастанием поляризуемости нуклеофильного центра. Так, у галогеиид-ионов нуклеофильность увеличивается в ряду F“ < CI” < Вг” < I”.
Аналогично нуклеофильность серосодержащих нуклеофилов выше, чем у кислородсодержащих аналогов: RS” > RO”; SH”> ОН”
На нуклеофильность реагента оказывают влияние заместители, связанные с нуклеофильным центром. Электроиодонорные заме--стители повышают электронную плотность в нуклеофильном центре и приводят к увеличению нуклеофильности, а электроноакцепторные, наоборот, снижают нуклеофильность, например: CHj—NH2 > NH3 > СеНз— NH2.
Влияние растворителя иа нуклеофильность определяется эффектом сольватации. Протонные полярные растворители — вода, спирты, муравьиная кислота, уксусная кислота и другие — хорошо сольватируют как катионы, так и анионы. Сольватируя нуклеофил, они снижают его реакционную способность и, следовательно, замедляют реакции, идущие по механизму Хл2. Апротонные полярные растворители (днметнлформамид HCON(CH3)2, ди-метилсульфоксид (CH3)2SO, ацетон СН3СОСН3, ацетонитрил СНз-*-С =N и др.) сольватируют только катионы, поэтому скорость реакций S42 в них существенно повышается.
Механизм S^l (мономолекулярное нуклеофильное замещение).
В отлнчие от механизма SN2, где разрыв связи С—На] и образование связи С—Nu происходят синхронно, в механизме SN1 разрыв саязи С—Hal предшествует образованию связи С—Nu.
По механизму SN1 реакция протекает в две стадии:
Стадия 1 —С—На! + НаГ
карбкатион
Стадия II
быстро
На стадии I происходит ионизация молекулы галогеналкана с образованием карбкатиона н галогенид-иона. Процесс ионизации протекает медленно, а поэтому ои определяет скорость всей реакции. В ионизации галогеналкана оказывает содействие растворитель.
На стадии II образовавшийся катион быстро взаимодействует с нуклеофильным реагентом, образуя конечный продукт реакции.
162
Механизм нуклеофильного замещения, протекающий по рассмотренной схеме, называется моно молекулярным, так как на стадии, определяющей скорость всего процесса (стадия I), принимает участие молекула только одного реагента — галогеналкана. Такой механизм обозначают S4I.
Скорость реакций, протекающих по механизму Shl, зависит только от концентрации галогеналкана н описывается кинетическим уравнением первого порядка: v = fe[R—Hal].
Оптически активные третичные галогеиалканы, в которых атом галогена связан с асимметрическим атомом углерода, в реакциях как правило, образуют рацемические формы. Это связано с тем, что образующийся на стадии 1 реакции карбкатион имеет плоскостную конфигурацию (катионный центр находится в $р2-гибридизацни) и может быть атакован нуклеофил’ьным реагентом с равной вероятностью с обеих сторон плоскости. А это приводит к образованию равных количеств двух стереоизомеров, т. е. к рацемизации:
Указанные экспериментальные данные подтверждают механизм SN1.
Факторы, способствующие протеканию реакций по механизму 5^1.
1. Пространственные препятствия для атаки нуклеофилом электроиодефицитиого атома углерода галогеналкана.
2. Способность соединений образовывать весьма устойчивые карбкатионы.
3. Высокая ионизирующая и сольватирующая способность растворителя По механизму 5Ч1 происходит нуклеофильное замещение в третичных и прн определенных условиях — во вторичных галогеналканах.
В молекуле третичного галогеналкана, например грег-бутил-бромида (СНэ)эС—Вг, три метильные группы создают пространственное препятствие для подхода нуклеофила к электрофильному
6*
163
центру, и, следовательно, его атака с тыла станоантся невозможной. Вместе с тем третичные галогеналканы в снльнополярных средах способны к ионизации:
СНз СНз
СНз—С—Вг СНз—С+ + Вг-
СНз^ СНз^
Как известно, ионизация соединения тем легче, чем стабильнее образующиеся яоиы. Устойчивость алкильных карбкатионов обусловлена делокализацией положительного заряда за счет +/-эффекта алкильных групп и возрастает в ряду:
СНз СНз
СНз < СНз-*СН2 < ^СН < С^-СН3 СНэ^ СНз^
Поэтому образование третичного карбкатиона для молекулы энергетически выгоднее, чем вторичного, а тем более — первичного. Следовательно, третичные галогеналканы легче подвергаются ионизации, чем вторичные и первичные, и тем самым наиболее реакционноспособны в реакциях, идущих по механизму SNI.
Влияние растворителя на протекание реакций по механизму SNI обусловлено эффектом сольватации. Протонные полярные растворители, такие, как вода, спирты, муравьиная кислота, уксусная кислота, способствуют ионизации галогеиалкаиа и хорошо сольватируют образующиеся карбкатионы и анионы, повышая нх устойчивость. Этн растворители ускоряют реакции, идущие по механизму SNI.
Таким образом, иа основании вышеизложенного можно сделать вывод, что первичные галогеналканы обычно реагируют по механизму SN2, третичные — по механизму S^l. Вторичные гало-геналкаиы, в зависимости от природы нуклеофила и растворителя, могут реагировать как по механизму SN2, так и по механизму SN1. Сильные нуклеофильные реагенты я апротоииые полярные растворители (нитробензол, диметилформамид, ацетонитрил и др.) благоприятствуют протеканию реакции по механизму SN2, протонные полярные растворители (вода, метиловый спирт, этиловый спирт, муравьиная кислота, уксусная кислота и др.), обладающие высокой сольватирующей способностью, благоприятствуют взаимодействию по механизму SN1.
Некоторые нуклеофильные реагенты содержат анионы, имеющие в своем составе два и более нуклеофильных центра, например, ннтрит-ион (:б—N=Oj цнанид-ирн [:C=J4]~ Та.кие иоиы
IH4
обладают двойственной реакционной способностью н называются амбидентными.
В реакциях, протекающих по механизму S^2, амбидентные ионы атакуют электрофильный реагент реакционным центром с большей нуклеофильностью, т. е. атомом азота в нитрнт-иоие и атомом углерода в цианид-ионе. В реакциях, идущих по механизму SN1, промежуточно образующийся карбкатион активнее взаимодействует с реакционным центром с большей электронной плотностью, а именно, атомом кислорода в нитрит-ионе и атомом азота в цианид-ионе. Довольно часто замещение в галогеналкаиах осуществляется параллельно по механизмам S42 и SN1.
Галогеналканы вступают в многочисленные реакции нуклеофильного замещения. Важное синтетическое значение имеют:
Гидролиз галогеналкаков. При гидролизе галогеналканы образуют спирты. Реакция с водой протекает медленно и является обратимой:
С2Н5Вг + НОН C2HSOH + НВг бромэтан этанол
Поэтому обычно гидролиз проводят в присутствии водных растворов щелочей или карбонатов щелочных металлов:
СгНвВг + NaOH-----► С2Н5ОН + NaBr
Взаимодействие с алкоголятами н фенолятами (реакция Вильямсона). При действии на галогеналканы алкоголитов и фенолятов образуются простые эфиры. Третичные галогенал-каиы образуют в качестве побочных продуктов алкены:
4- -
С2Н5Вг NaOCaHs -------► С2Н5—О—С2Нб -Г NaBr
этипат Ацетиловый
натрии эфНр
С2Н5Вг + ----►СгНй—NaBr
фенолит натрин фенилэтиловый эфир
Реакция открыта в 1851 г. английским химиком А. У. Вильямсоном и до сих пор используется в качестве одного из лучших методов получения простых эфиров.
Взаимодействие с солями карбоновых кислот. При действии на галогеналканы солей карбоновых кислот в среде апротонного полярного растворителя (диметилформамид, диметнлсульфоксид) с высокими выходами образуются сложные эфиры. Реакция идет по механизму SN2:
С2Н5Вг + NaO—С—СНз--------С2НЬ—О—С—СНЯ + NaBr
А А
ацетат натрнч этила пета т
165
Взаимодействие с аммиаком, алкил- и ариламинами. При взаимодействии галогеиалканов с избытком аммиака образуется смесь первичных, вторичных и третичных аминов, а также соли четвертичных аммониевых оснований. Подробное описание реакции см. на с. 203. Образование первичного амина происходит по схеме
С2Н5ВГ+ NH3------► [C2H5NH3]+Вг~ —^>C2H5NH2
этнламчоння бромид »тилачнн
Аналогично реагируют галогеиалканы с алкил- и ариламинами (см. с. 208).
Взаимодействие с солями циановодородной кислоты. Поскольку циаиид-иои является амбидентным нуклеофилом, при взаимодействии галогеиалканов с солями цн а и оводородной кислоты, в зависимости от условий проведения реакции, образуются нитрилы или изоцианиды (нзоинтрилы).
Первичные и вторичные галогеиалканы с солями щелочных металлов циаиоводородиой кислоты (KCN, NaCN) в среде апротонного полярного растворителя с хорошими выходами образуют нитрилы (механизм Sh2)
C2H5Br + NaCN--------> С2Н5—C=N + NaBr
пропанонмтрил
Основными продуктами реакции вторичных и третичных галогеиалканов с цианидом серебра в среде протонного полярного растворителя являются изоцианиды (нзонитрилы) (механизм SN1).
СНз
^СН—Вг + AgCN
СН,
нзопрппнлизоцнаннд
Взаимодействие с солями азотистой кислоты. Взаи моде й-ствие гвлогеналкаиов с солями азотистой кислоты, содержащими амбидентный нитрит-ион, протекает с образованием нитросоединений или эфиров азотистой кислоты в зависимости от условий проведения реакции.
Первичные и вторичные галогеиалканы с нитритом натрия в условиях реакции SN2 образуют преимущественно ннтросо-единения
С2Н5—Вг + NaNO2
С2Н5—NO2 + NaBr ннтроэтан
166
Вторичные н третичные галогеиалканы с AgNOa в условиях реакции SNI образуют с хорошими выходами эфиры азотистой кислоты:
СНз
СН—Вг + AgNO2
СНз
СНз
СН—О—N=O + AgBrl СНз^
II |ОПрО]|МЛННТрНГ
Взаимодействие с салями галогеноводородных кислот (реакция Финкельштейна). С помощью этой реакции в молекуле галогеналкана можно заменить один галоген другим. Взаимодействие галогеиалканов с солями галогеиоводородиых кислот является обратимым процессом. Для смещения равновесия впраао используют разную растворимость исходных веществ и продуктов реакции. Реакция имеет практическое значение для получения первичных фтор- и иодалканов иэ более доступных хлор- и бром-производиых. Для получения иодидов реакцию проводят в ацетоне, так как иодид натрия растворим в ацетоне, а образующиеся в процессе взаимодействия NaCI или NaBr выпадают в осадок:
СаНБС1 4- Nal -a-ueTOi^ C2H5I + NaClj
Взаимодействие с гидросульфидами и сульфидами щелочных металлов. При действии на первичные и вторичные галогенал-каны гидросульфидов щелочных металлов образуются тиоспирты (меркаптаны), при действии сульфидов—тноэфиры:
С2Н51 4- NaSH ----и C2H5SH + Nal
2С2Нб1 + Na2S-----► С2Н5—S—С2Н5 + 2Nai
ли этил сульфид
Б. Реакции элиминирования (£)
Каждый реагент, содержащий атом с неподеленной парой электронов или несущий отрицательный заряд, наряду с нуклеофильными свойствами проявляет также основные свойства (см. ки. 1, с. 101). Следовательно, он способен не только предоставлять лару электронов дйя образования связи с электрофильным атомом углерода, но и отщеплять от молекулы, имеющей подвижные атомы водорода, протон. В молекуле галогеналкана вследствие — /-эффекта атома галогена атомы водорода у 0-углеродиого атома приобретают подвижность:
«+• о-
R—СН---->СН2--► На 1
Нб+-'
167
Поэтому в большинстве случаев параллельно с реакцией нуклеофильного замещения атома галогена протекает реакция отщепления галогеиоводорода (^-элиминирование) с образованием алкеиа, например:
нуклеофильное замещение
СНз—СН2—i + NaOH
СНЭ—СНг—ОН
CH2=CHs
— Nal
fl элиминирование
—Nal —Н3О
Реакции нуклеофильного замещения и элиминирования конкурируют друг с другом и в определенных условиях каждая из них может стать доминирующей. Отщепление галогеиоводорода от галогеналкана становится основным процессом в присутствии нуклеофильных реагентов, обладающих высокой основностью. К ним относятся спиртовые растворы гидроксидов щелочных металлов (например, спиртовый раствор NaOH, КОН) или алкого-ляты щелочных металлов (например, CaHsONa). Элиминированию способствуют также повышение температуры реакционной смеси и коицентрацнн реагентов. Благоприятное влияние оказывает увеличение числа заместителей у электрофильного атома углерода Поэтому особенно легко элимиинроваиие протекает у третичных галогеналкаиов.
Если в молекуле галогеналкана имеется несколько альтернативных путей отщепления галогеиоводорода, то нре имуществен ио реализуется тот нз них, при котором двойная связь образуетсн у наиболее замещенного атома углерода; т. е. вместе с галогеном уходит водород от наименее гидрогенизированного соседнего атома углерода.
Эта закономерность получила название правила Зайцева:
СНз
СНз—СН—СНз
Вг
NaOH ILjHsOH)
—NaBr —НаО
2 Ороч 3 четнлбута и
СНз
СНз—С=СН—СНз
(ОСНОВНОЙ продукт реакции)
СНз
СНз—СН—СН=СН2
3 метилбутси-| (побочный продукт реакции)
1ВД
Диалогично нуклеофильному замещению элиминирование гало* геналканов может протекать по бимолекулярному (£2) и моно-молекулярному (£1) механизмам.
Механизм Е2 (бимолекулярное элиминирование). Механизмы бимолекулярного элиминирования £2 н бимолекулярного нуклеофильного замещения SN2 сходны. Механизм £2 представлен следующей схемой;
В“ + основание
S+ IP la H-C-C-Hal
галогеналкан
В-Н +
в Д..Н.Л..С^-С..(.. Нд!
переходное состояние
C-Cf + Hal
Реакция по механизму £2, как н по SN2, идет в одну стадию с образованием переходного состояния, в формировании которого принимают участие молекулы двух реагентов. А поэтому скорость такой реакции зависит от концентрации обоих реагентов и описывается кинетическим уравнением второго порядка. Процессы разрыва и образования связей в переходном состоянии происходят синхронно.
Различие же между механизмами SN2 и £2 состоит в том, что в механизме SN2 частица с неподелеииой парой электронов или несущая отрицательный заряд атакует электрофильный атом углерода молекулы галогеналкана, действуя при этом как нуклеофил, а в механизме £2 она атакует атом водорода при р-угле-родном атоме, действуя как основание. Поэтому процессы SN2 и £2 являются конкурирующими. Наиболее легко по механизму £2 происходит элиминирование у первичных галогеналкаиов.
Механизм £1 (мономолекулярное элиминирование). Как механизм £2 сходен с механизмом SN2, так и механизм £1 имеет большое сходство с SN1 и конкурирует с ним.
Реакция, идущая по механизму £1, представляет собой двухстадийный процесс. На стадии I, как и в реакциях по механизму SaJ, под влиянием растворителя происходит ионизация молекулы галогеналкана с образованием карбкатиона. Процесс ионизации идет медленно и определяет скорость реакции в целом. На стадии II образовавшийся карбкатион стабилизируется, отщепляя протон от (J-углеродного атома с образованием алкена:
Стадия I —С—С—Hal < —С—С+ 4- Hal-
11 ' Н '
галоген алкан
инрбкатнон
169
Стадия И —С-----*С+ 6ь)стр^ С=С + Н+
Н ' марбкатипн алкен
Акцептором протона часто служит сам растворитель, например, вода, поэтому протекающая по механизму £1 реакция обычно не требует присутствия основания как реагента. В реакции элиминирования по механизму £1 наиболее легко вступают третичные галогеналканы.
В. Взаимодействие с металлами
Галогеналканы реагируют с некоторыми металлами, образуя металлоорганические соединения. Чаще асего в качестве металла используют магний. При взаимодействии галогеналкаков с металлическим магнием в среде безводного днзтилового эфира образуются магнийорганические соединения, известные под названием реактивов Гриньяра:
C2H5CI + Mg C2H5MgCl этилма! ннйхлорид Реактивы Гриньяра ивляются весьма реакционноспособными веществами. Их активность обусловлена полярностью связи углерод—магний. Поскольку атом углерода имеет большую электроотрицательность, чем магний, связь С—Mg поляризована таким образом, что на атоме углерода появляется частичный отрица-6+
тельиый заряд (СНз—CHs-^-MgCl) Вследствие этого магнийорганические соединения являются сильными нуклеофильными реагентами и сильными основаниями.
При взаимодействии галогеналканов с металлическим натрием металлоорганические соединения образуются в качестве промежуточного продукта (см. реакцию Вюрца на с. 17).
Г. Восстановление галогеналканов
При восстановлении галогеналканов образуются алканы. В качестве восстановителей используют водород в присутствии катализаторов гидрирования или иодоводородную кислоту:
С2Н5С1 —С2Н6 + НС1
C2H5Ci + 2HI —!+ С2Н6 + НС1 + Ь
170
10.4. ДИГАЛОГЕНАЛКАНЫ
Дигалогеналканы содержат в своем составе два атома галогена. Атомы галогенов могут находиться у одного и того же атома углерода (геминальные дигалогеналканы), у соседних атомов углерода (вицинальные дигалогеиалкаиы) или разделены несколькими углерод-углеродными связями.
10.4.1. Способы получения
Присоединение галогеноводородов к алкинам (см. с. 63). В результате реакции образуются геминальные дигалогеналканы
НС^СН СН2=СН—С1 СНз—СНС12
1,1 дихлорэтан
Взаимодействие альдегидов и кетонов с пентагалогенидами фосфора (РСЬ, РВг5). В процессе реакции образуются геминальные дигалогеналканы
О
СНз—С + PCls -J— CHjCHCIs + POCI3
н
уксусный альдегид 1 I дихлорэтан
Вг
СН3С—СНз + РВгБ —U- СНз—А—СНз + РОВгз
А А.
ацетон 2,2 дибром пропан
Присоединение галогенов к алкенам (см. с. 35). В результате реакции образуются вицинальные дигалогеналканы:
СН3—СН==СН2 + Вг2
СНз—СН—СН2
1.2 днбромпрппан
171
10.4,2. Химические свойства
Химические свойства дигалогеиалканов аналогичны свойствам моногалогеиалкаиов. Они вступают в различные реакции нуклеофильного замещения и отщепления, свойственные галоген-алканам.
Так, при щелочном гидролизе ди га логе нал каков, в которых атомы галогена находятся при разных атомах углерода, образуются двухатомные спирты — гликоли:
СНз—СН—СН2 JfsdO-> СНз—СН—СН2 + 2NaCi + 2Н2О
I I ШгО) II’
Cl CI он он
1,2 днхлорпропдн ролиленгликоль
В присутствии спиртовых растворов щелочей при нагревании геминальные и вицинальные дигалогеиалканы подвергаются элиминированию с образованием алкинов:
С1
СНз—СН^ —CbteCH + 2NaCl + 2Н2О (CjHsOH)
С1
1 1 дихлорэтан
ацетилен
СНз—СН—СН2—2—н > СНз—С=СН ч- 2NaCl + 2Н2О Г I (С,Н>ОП) 1
С1 С1
I 2 дихлорпропам пропин
Наряду с этим геминальные дигалогеиалканы в химическом поведении имеют некоторые особенности.
При щелочном гидролизе они образуют альдегиды нлн кетоны:
xci
СН3-С1<С1 +
Н2О
-НС1
.о-н
СНз-СНС
ЛТсг
1Л-дихлорэтан
уксусный альдегид
СНт. ,С1 +
СН3 С1
Н2О
-HQ
СН^ ,0-Н сн3х '"Cl
ацетон
2,2-дихлор пропаи
172
10.5. ГАЛОГЕНАЛКЕНЫ
Гало генал йенам и называют производные алкенов, в которых одни или несколько атомов водорода замещены атомами галогенов.
По взаимному расположению двойной связи и атома галогена галогеналкены можно условно разделить на три группы:
1. Внннлгалогениды — соединения, содержащие атом галогена у углерода, образующего двойную связь:
С1
I
СНг=СН—CI СНз—СН=СН— Вг СН2=С—СНз
хлористый винн7 1 бромпролен 2 хлорпропен
«
2. Аллилгалогеннды — соединения, в которых атом галогена находится в a-положении к углероду, образующему двойную связь:
СН2=СН—СН2—С1
СНз—СН—СН^=СН2
С1
хлористый аллнл
j х.торбутен I
3- Соединения, в которых атом галогена н атом углерода, образующий двойную связь, разделены двумя и более простыми углерод-углеродными связями:
СНг=СН—СНа—СНа—Вг СНз—СН=СН—СНг—С Нт—CI
4 бромбутвн 1 S хюриентен 2
10.5.1. Способы получения
Основными способами получения галогеиалкеиов являются:
Гидрогалогенирование алкинов (см. с. 63):
СН=СН + НС1 --------* СНа=СН—CI
хлористый винил
Галогенирование алкенов в аллильное положение (см. с 46):
СНа=СН—СНз + С12 —СН2=СН—СН2С1 + НС1 хлористый аллил
Действие галогенирующих реагентов (PCI3, PCI&, SOCk) на непредельные спирты (см. с. 260):
СНа=СН—СНг—ОН + РС15 --------
а мн.товый спирт
---► СН2==СН—CH2Ci + РОС1з + НС1
хлористый аллил
173
10.5.2. Химические свойства
•Химические свойства галогена л кеиов обусловлены наличием в их структуре двух групп: С=С и —С—Hal. Являясь бифункциональными соединениями, галогеиалкеиы по двойной связи способны вступать в реакции, характерные для алкенов (присоединение, полимеризация н др., см. с. 34), а по связи углерод—галоген — в реакции замещения и отщепления, свойственные галогеналканам. Однако реакционная способность галогенал-кенов во многом зависит от взаимного расположения данных групп в молекуле. Если в соединении атом галогена и диойиая связь достаточно удалены друг от друга (разделены двумя и более простыми углерод-углеродными связями), то каждая из этих групп ведет себя независимо от другой.
Вместе с тем галогеиалкеиы, содержащие атом галогена у углерода, образующего двойную связь (виннлгалогениды), вследствие взаимного влияния указанных групп характеризуются низкой реакционной способностью связи С—Hal и двойной связи. Атом галогена в этих соединениях малоподвижен н с большим трудом замещается на другие атомы н группы. Реакции присоединения по двойной связи также идут труднее, чем, например, в этилене
Причиной низкой реакционной способности связи С—Hal является сопряжение неподеленной пары электронов атома галогена с л-электронами двойной связи (атом галогена проявляет +Л1-эффект):
<!н^-сн-£с1
В результате сопряжения связь С—На! укорачивается и становится значительно прочнее, чем в галогеналканах. Поэтому реакции нуклеофильного замещения для винилгалогеиидов в большинстве случаев осуществить не удается. При обычной температуре они практически не идут, а при нагревании протекают побочные процессы — отщепление галогеиоводорода, присоединение по двойной связи, полимеризация.
В присутствии концентрированных растворов щелочей винил-галогеииды, содержащие атом водорода у углерода, образующего двойную связь, отщепляют галогеноводород и превращаются в алкины:
СНг=С—СНз + NaOH ----------> C№sC—СН3 + NaCl + Н2О
(L
2 клорпропен прОпнн
174
Наряду с этим в молекуле вниилгалогенида атом галогена за счет сильного отрицательного индуктивного эффекта уменьшает электронную плотность двойной связи и тем самым снижает ее реакционную способность в реакциях электрофильного присоединения. Поэтому винилгалогеииды присоединяют галогены, галогеноводороды и другие электрофильные реагенты труднее, чем соответствующие алкены. Присоединение галогеноводородов идет по правилу Марковникова:
СН2=СН—Cl + НС1 -------► СН3—СНС12
хлористый винил I 1-дихлорэтан
В присутствии катализаторов виннлгалогеннды легко вступают в реакции полимеризации, которые широко применяются в производстве полимерных материалов:
п сн,-сн а —► +сн2-сн+ к CI ]а
ПОЛИНИНИЛхлорид
В отличие от вниилгалогенидов, в молекулах аллилгалоге-иидов атом галогена обладает повышенной подвижностью. Аллил-галогсниды вступают в реакции нуклеофильного замещения легче, чем галогеналканы. Замещение, как правило, происходит по механизму SN1.
Высокая активность аллилгалогеиидов в реакциях нуклеофильного замещения объясняется нх склонностью к ионизации, поскольку при этом образуется весьма устойчивый аллильный катион
СН2=СН—СН2Вг СН2=СН—СН2 + Вт
Стабильность аллильного катиона обусловлена делокализацией положительного заряда по сопряженной системе
сн2^сн^сн2 ---------* снХсн^сн2
Вследствие электроноакцепторного влияния атома галогена реакционная способность двойной связи в аллилгалогенидах несколько ниже, чем в соответствующих алкенах, однако присоединение галогеноводородов идет по правилу Маркоаникова:
СН2=СН^СН2-С1 + НС1 ---------СНз—СН—СН2
di
хлористый аллил 1.3 дихлорпропам
175
10.6. АРОМАТИЧЕСКИЕ ГАЛОГЕНОПРОИЗВОДНЫЕ
Ароматическими галогенопроизводиыми называют производные ароматических углеводородов, в которых один или несколько атомов водорода замещены атомами галогенов,
В зависимости от положения атомов галогена ароматические галогеиопроизводные подразделяют иа две группы:
Гало ген арены — соединения, в которых атомы галогена непосредственно связаны с ароматическим ядром
Арнлалкилгалогеннды — соединения, содержащие атомы галогена в боковой углеродной цепи
С1
бензил хлорид
I фенил I хлората и
10.6.1. Способы во лучения
Наиболее распространены два метода получения галоген-аренов:
Прямое галогенирование ароматических углеводородов (см. с. 101). Реакция протекает по механизму St. Прн избытке галогена образуются дн- и тригалогенарены:
С1 С1 С1
Замещение диазогруппы в солях арилдиазоння на галоген (реакция Зандмейера, см. с. 235):
бензолдиааиний хлорид
СГ
CugCia
+ Na
176
Для введения атома галогена в боковую углеводородную цепь используют такие методы:
Галогенирование алкиларенов. В отличие от галогенирования в ароматическое ядро галогенирование в боковую цепь происходит без катализатора при высоких температурах или при облучении УФ-светом. Реакция протекает по механизму SR (ем. с. 20):
С1а, Л*
—НС1
СН2С1
Clj, hy ------►
—НО
CHCla
бензнлхлорид
бенз или денхлорид
Галогеиироваияе в боковую цепь идет, как правило, в а-положение относительно бензольного ядра, поскольку в этом случае образуется устойчивый свободный радикал бензильного типа (см. кн. 1, с. 140):
С1
1 фенил I хлор этан
При избытке галогена все а-атомы водорода в молекуле могут быть замещены иа галоген.
Реакция хлорметилирования используется для получения арилметилхлоридов. Оиа осиоваиа иа взаимодействии аренов с формальдегидом и хлороводородом в присутствнк катализатора (А1С1з, ZnCl2). В процессе реакции атом водорода бензольного кольца замещается на хлорметильяую группу
+ НС1
СН2С1
+ Н2О
формальдегид бен >и л хлорид
По механизму хлорметилироваиие относится к реакциям электрофильного замещения в ароматическом ряду. Атакующей электрофильной частицей в ней является гидроксиметил-катиои
(СН2ОН):
г* 5-
S+jr® 6+ в- +
Н-С + Н—Cl сн2он + С|-
177
+ СН2ОН —г —н
СН2ОН
HCI, ZnCle
—Н20
СН2С1
Кроме того, для получения ароматических галогенопроизводных с атомами галогена в боковой цепи пригодны все способы получения галогенопроизводных алифатического ряда.
10.6.2. Физические свойства
Галогенопроизводные бензола и его гомологов являются жидкостями илн кристаллическими веществами. Температуры кипения галогеиареиов возрастают в ряду: фтор-, хлор-, бром-, иодпроизводные. Все соединения этого ряда не растворимы в воде, но легко растворимы в органических растворителях. Галогено-производные с атомами галогена в a-положении боковой цепи обладают раздражающим действием на слизистые оболочки, вызывая слезотечение.
10.6.3. Химические свойства
Для ароматических галогенопроизводных характерны реакции нуклеофильного замещения с участием связи углерод — галоген, реакции электрофильного замещения по ароматическому идру и реакции с металлами.
А. Реакции нуклеофильного замещения
Галогеиопроизводные, у которых атом галогена непосредственно связан с бензольным кольцом, характеризуются низкой реакционной способностью связи углерод — галоген. Атом галогена в этих соединениях малоподвижен и замещается с большим трудом. В данном отношении галогенареиы сходны с вннилгалоге-нндами.
Низкая активность связи С—Hal в галогенареиах, как и в ви-иилгалогеиидах, обусловлена сопряжением иеподеленной пары электронов атома галогена с л-электроииой системой бензольного кольца:
17S
В результате сопряжения происходит укорочение и уменьшение полярности связи углерод — галоген, что приводит к ее упрочнению. Поэтому нуклеофильное замещение атома галогена в галоген-аренах происходит лишь в очень жестких условиях.
Так, в молекуле хлорбензола атом хлора замещается иа гидроксильную группу под дейстаием концентрированного раствора NaOH при температуре свыше 300 °C н давлении 150 атм.:
хлорбензол
+ NbCI
Реакция с аммиаком происходит при температуре 200 °C в присутствии катализатора — порошка медн илн солей одновалентной меди (реакция Ульмана)
+ 2NH3
200 “Ct Си
анилин
Вместе с тем подвижность атома галогена в галогеиаренах резко возрастает при наличии в орто- иля napa-положеииях по отношению к галогену сильных электроноакцепторных заместителей, например: NO?, NO, CN, СООН, SO3H. Такне соединения называют активированными галогенаренами. Повышение реакционной способности связи углерод—галоген в активированных галогенареиах связано с увеличением дробного положительного заряда на атакуемом атоме углерода под влиянием электроноакцепторного заместителя:
Так, в молекуле 2,4-днннтро-1-хлорбензола атом хлора довольно легко замещается на группы ОН, NH2, ОСНз и др.:
179
ort
NaOH (Н2О)
100 °C
2.4 диннтрофеиил
+ NaCi
2,4-днннтри 1 хлорбензол
aNHa
1Ы °C
+ NH4CI
2.4 лийитрояиилин
СН,ОК
I метокси 2 4 димнтроОензол
+ KCi
Реакции нуклеофильного замещения активированного атома галогена протекают по механизму SN2. Однако в отличие от реакций SN2 в алифатическом ряду, которые протекают в одну стадию и сопровождаются синхронными процессами разрыва связи С—Hal и образования связи С—Nu, бимолекулярное нуклеофильное замещение в ароматическом ряду происходит в две стадии. На стадии I атакующая нуклеофильная частица присоединяется к электрофильному атому углерода, который связан с галогеном, образуя карбаииои (о-комплекс). Электроноакцепторные группы в составе карбаниона принимают участие в делокализации отрицательного заряда и тем самым повышают его устойчивость. Стадия 1 протекает медленно и лимитирует скорость всего процесса. На быстрой стадии 11 реакции карбкатион отщепляет галогеиид-ион с образованием продукта замещения:
с-комплекс
180
быстро
а
В отличие от реакций 5N2 в алифатическом ряду, при бимолекулярном нуклеофильном замещении в ароматическом ряду процессы образования связи углерод — нуклеофил и разрыва связи углерод — галоген происходят несинхронноv поскольку новая связь образуется раньше, чем разрывается старая.
Если атом галогена в галогенарене не активирован электроноакцепторной группой, при нуклеофильном замещении атакующая частица не всегда занимает место галогена. Например, при щелочном гидролизе n-хлортолуола образуется эквимолярная смесь двух изомерных продуктов — мета- и пара-крезолов:
2МаОН 340 °<5
—2NaCl
л-хлортолуол
л крезол
jm крезол
При взаимодействии амида калия KNHa с меченым 1-иС-хлор-бензолом образуется почти равное количество аиилииа, меченого в положении 1 и в положении 2
хлор бен зп ।
анилин
Все эти факты можно объяснить с позиций так называемого аринового механизма взаимодействия. Согласно данному механизму замещение галогена иа нуклеофил идет ие прямо, а через предварительное отщепление галогеиоводорода и последующее присоединение нуклеофила. Сначала нуклеофильный реагент, будучи сильным основанием, отщепляет от молекулы галогеиареиа гало-геиоводород, образуя нестойкий, очень реакционноспособный промежуточный продукт — дегидробензол (арин), содержащий в своем составе тройную связь. Затем дегндробеизод мгновенно присоединяет по месту тройной связи нуклеофильный реагент, причем
181
ковалентную связь с нуклеофилом может образовать в равной степени каждый из двух, связанных тройной связью атомов углерода:
Стадия
+ NH3 + С1
хлорбензол
дегидробензо i
Этот механизм называют еще механизмом элиминирования-присоединения.
Ароматические галогеиопроизводиые с атомом галогена в боковой цепи легко вступают в различные реакции нуклеофильного замещения. Соединения, в которых атом галогена изолирован от бензольного кольца двумя и более углерод-углеродными связями, по активности в реакциях SN напоминают галогеиалканы, Галоге-иопроизводные, содержащие атом галогена у а-атома углерода боковой цепи (бензилгалогеннды), обладают повышенной реакционной способностью связи С—Hal. По активности в реакциях SN бензилгалогеииды сходны с аллилгалогенидами. Нуклеофильное замещение атома галогена происходит в них по механизму SN1. Высокая подвижность атома галогена в беизилгалогенидах обусловлена большой устойчивостью бензильного катиона, образующегося после отщепления галогенид-иона (см. ки. 1, с. 134).
Б. Реакции электрофильного замещения по ароматическому ядру
По ароматическому ядру галогенареиы вступают в обычные реакции электрофильного замещения: галогенирование, нитрование, сульфирование и др Как уже отмечалось, атом галогена, непосредственно связанный с ароматическим кольцом, проявляет отрицательный индуктивный и положительный мезо мерный эффекты, причем в статическом состоянии —/-эффекту -|-Л1-эффекта. Поэтому атомы галогена в целом проявляют электроноакцепториые свойства по отношению к бензольному кольцу и тем самым снижают его реакционную способность в реакциях Sr. Принимая участие в стабилизации о-комплексов, образующихся при замещении в орто- и napa-положеннях, атомы галогенов выступают как ориеитаиты I рода и направляют электрофильное замещение в орто- и пара-положения (см. с. 112):
182
Cl
6-хлорбензол
о хлорбензол п хлорбензол
сульфокислота ул ьфоки слота
В. Реакции с металлами (металлирование)
Галогенарены легко вступают в реакцию с металлами — литием, натрием, магнием. При взаимодействии с магнием в среде диэтилового эфира образуются магнийорганические соединения (реактивы Гриньяра)
MgBr
феНитмагнийброчид
В реакции с натрием металлоорганическое соедииеине образуется в качестве промежуточного продукта (реакция Вюрца — Фиттига)
С6Н5Вг + 2 Na брочбенэол
CeH5Na -|- СбН5Вг
C6H5Na + NaBr фенил натрий
СвН5—СвН5 -|- NaBr
бифени1
10.7. ИДЕНТИФИКАЦИЯ ГАЛОГЕНОПРОИЗВОДНЫХ УГЛЕВОДОРОДОВ
10.7.1. Химические методы
Наиболее простым методом обнаружения галогена в органическом соединении является проба Бейльштейиа. Она основана на способности галогеиопроизводных при нагревании с медью образовывать летучие галогениды меди, окрашивающие пламя горелки в зеленый цвет. Проба Бейльштейиа очень чувствительна, однако
183
оиа не позволяет определить природу галогена. Нельзя также с ее помощью открыть фтор, поскольку фториды меди нелетучи.
Для определения природы галогена в органическом веществе применяют метод сплавления с металлическим натрием. При сплавлении галогеиопроизводных с натрием образуются галогениды натрия, в которых галогеннд-иои определяют реакцией с нитратом серебра в азотнокислой среде.
10.7.2. Инструментальные методы
В ИК-спектрах галогенопроизводных углеводородов наблюдаются полосы поглощения, обусловленные валентными колебаниями связи С—Hal Связь С—F проявляется в области 1350— 1000 см-1, связь С—С1 в области 800—600 см-1. Валентные колебания связей С—Вг и С—1 находятся в дивпазоие ниже 600 см~\ который не доступен для измерения большинством существующих ИК-спектрометров.
Химические сдвиги протонов в ПМР-спектрах галогеналканов зависят от природы галогена и разветвления углеродной цепи у атома углерода, с которым связан галоген. Вследствие дезэкранирующего эффекта атомов галогена сигналы протонов, находящиеся у того же атома углерода, что и галоген, сдвинуты в слабое поле и характеризуются химическим сдвигом 2,8—4,2 м. д.
Для идентификации галогеиаренов широко применяют метод масс-спектрометрии. Ароматический характер этих соединений способствует образованию довольно стабильных молекулярных нонов, интенсивность пяков которых достаточна для качественного и количественного определения галогена-
10.8. ОТДЕЛЬНЫЕ ПРЕДСТАВИТЕЛИ ГАЛОГЕНОПРОИЗВОДНЫХ УГЛЕВОДОРОДОВ
Хлората и (этилхлорид) C2H5CI. В обычных условиях хлор-этан является газообразным веществом, прн температуре ниже 12 °C представляет собой бесцветную жидкость. Смешивается с этанолом и диэтиловым эфиром. Огнеопасен. Обладает сильным наркотическим действием. При попадании иа кожу хлорэтан вследствие быстрого испарения вызывает сильное охлаждение участка кожи и потерю болевой чувствительности, что позволяет использовать его в медицине для местного обезболивания при невралгиях, иейромиозитах, небольших поверхностных порезах.
Хлороформ (трихлорметаи) CHClg — бесцветная жидкость с характерным сладковатым запахом (т. кнп. 6] ,2 °C). Хлороформ мало растворим в воде, хорошо растворим в органических растворителях. Он растворяет многие органические вещества и широко применяется как растворитель в химических производствах, а также для извлечения многих веществ из растительного сырья, в частности жиров, алкалоидов, смол и др. На свету хлоро
184
форм медленно окисляется кислородом воздуха с образованием высокотоксичного вещества — фосгена и хлороводорода. Фосген может окисляться далее, образуя молекулярный хлор и оксид углерода IV:
О снсь ci—ci—ci + нс1
фосген
О
С1—<!—Cl J2U С12 + СО2
Хлороформ обладает сильным наркотическим действием. Ранее он применялся в медицине для усиления действия закиси азотв при комбинированном ингаляционном наркозе, но в настоящее время из-за высокой токсичности не используется.
Йодоформ (трииодметаи) СН13. Твердое кристаллическое вещество лнмоиио-желтого цвета с резким характерным устойчивым запахом (т. пл. 116 °C). Практически нерастворим в воде, хорошо растворим в хлороформе и диэтиловом эфире. Йодоформ с давних времен применяется в качестве антисептического средства в стоматологии, а также в форме присыпок н мазей для лечения инфицированных рай н язв.
F Вг Фторотан (2-бром-1,1,1-трифтор-2-хлорэтан) F—С—<^Н.
I I
F С1 Бесцветная подвижная жидкость с запахом, напоминающим запах хлороформа (т. кип. 49-51 °C). Мало растворим в воде, хорошо растворяется в этаноле, диэтяловом эфире, хлороформе. Фторэтаи обладает сильным наркотическим действием и низкой токсичностью. Он широко применяется в медицинской практике в качестве средства для комбинированного ингаляционного наркоза.
Дифтордихлорметан (фреон-12) CF2C12- В обычных условиях дифтордихлорметан является газообразным веществом без запаха. Ои не горюч, не взрывоопасен, не токсичен и ие вызывает коррозии металлов. Указанные свойства позволяют использовать фреон-12 в качестве хладоагента в холодильных установках, а также в качестве пропеллента в производстве аэрозольных лекарственных препарвтов.
Хлорбензол СбНйСГ Бесцветная жидкость со своеобразным запахом (т. кнп. I32°C). С водой образует азеотропную смесь, содержащую 71,6 % хлорбензола. Растворяется в бензоле, этаноле, хлороформе и других органических растворителях. Хлорбензол применяется в производстве фенола, анилина, лекарственных средств.
184
КОНТРОЛЬНЫЕ ВОПРОСЫ И УПРАЖНЕНИЯ
1. Назовите по заместительной номенклатуре ИЮПАК следующие соединения: а) СНз—СН—СН2—СН—СНз;
ir (^Нз
б) СНз—СН3—СН—CHz—СН—СН3;
Вг СНз
в) СНз—СН—СН=СН—СН3;
I С1
д) СН=С—СН—СН2—С1;
I
СНз
Г) СНг—СН— СН=СН2;
I I
С1 Вг
С1 '
I п
е) СНг—СН=СН—СНа <,
С1
ж) CeHs—СН2—СН—СН3—С1
2. Расположите следующие соединения в порядке увеличения подвижности атома галогена в их молекулах: а) хлорэтан; б) бромэтан; в) нодэтан. Ответ поясните.
3. Напишите схемы реакций 2-хлорбутана с такими реагентами: a) NaOH (Н2О); б) NaOH (в С2Н5ОН); в) C2H5O-Na + ; г) AgNO2; д) KCN;e) Mg. В схемах «а»— «д» обозначьте механизмы реакций.
4. В каждой из приведенных пар выберите соединения с более подвижным атомом галогена: а) бензнлхлорид н хлорбензол; б) аллилхлорид и винилхлорид; в) аллилхлорид и ] -хлорпропан.
5. Какие из перечисленных веществ реагируют с водным раствором NaOH и аммиака в обычных условиях: а) хлорбензол; б) хлорэтан; в) n-интробензол; г) n-хлортолуол; д) винилхлорид; е) аллилхлорид?
6. Напишите схемы реакций аллилхлорида, винилхлорида и 3,3,3-трихлорпропеиа с хлороводородом. Объясните направления реакций.
7. Приведите схемы реакций, с помощью которых можно получить геминальные и вицинальные дигалогеиопроизводные алканов.
8. Напишите уравнения реакций 2,2-дибромбутана и 1,1-ди-бромбутана с водным и спиртовым растворами NaOH.
9. Приведите механизмы реакций 1-хлорпропаиа и 2-метил-2-хлорпропана с водным и спиртовым растворами NaOH.
10. Объясните с позиции теории МО, как образуется переходное состояние в реакциях, протекающих по механизму SN2.
II. Назовите факторы, способствующие протеканию реакций по механизмам SN2 н S^l.
Глава 11 НИТРОСОЕДИНЕНИЯ
Нитросоедннениямн называют производные углеводородов, содержащие в своем составе одну или несколько ннтрогрунн — NO2.
Нитрогруппа имеет планарное (плоское) строение; атомы азота и кислорода находятся в состоянии 5/?2-гибридизации. Электронное строение нитрогруппы можно представить с помощью двух граничных структур, в которых один из атомов кислорода образует с атомом азота двойную, а другой — семиполярную связь:
б
\):
О
R— N
Однако в реальной молекуле химические связи обоих атомов кислорода с атомом азота равноценны, что изображается формулой с дробными зарядами
+ R-N
о1/2
В зависимости от природы углеводородного радикала, с которым связана нитрогруппа, различают алифвтнческие и ароматические нитросоедииения. Алифатические могут быть насыщенными (нитроалканы) и ненасыщенными (нитроалкены, иитро-алкины). Ароматические нитросоединеиия могут содержать иитрогруппу, непосредственно связанную с бензольным ядром (нятроарены), н нитрогруппу в боковой цепи (ннтроалкилареиы). По расположению иитрогруппы в углеродной цепи нитроалканы и нитроарены с ннтрогруппой в боковой цепи подразделяют на первичные, вторичные н третичные:
R— СНг— NO2
первичное нитросоедннекие
R ^СН—NO2
вторичное ннтросоеднненне
R—С—NO2
третичное ннтросоедн ненне
187
Здесь рассмотрены иитроалканы. иитроареиы с нитрогруппой в бензольном кольце н нитроареиы с интрогруппой в боковой цепи
II I. НОМЕНКЛАТУРА. ИЗОМЕРИЯ
По заместительной номенклатуре названия нитроалканов и иитросоедииений с нитрогруппой в бензольном кольце образуют добавлением префикса нитро- к названию родоначальиого углеводорода с указанием положения иитрогруппы в углеродной цепи, например'
СНз—СН—СНг— СНз
no2
2 нитрабутан
СНэ—СН—СН—СНз
I I
NO2 СНэ
2 мстил 3 ннтробутан
Нитросоедииения с интрогруппой в боковой цепи рассматривают как производные нитроалканов, содержащие в качестве заместителя ароматический радикал, например:
С Н2 NO2 С Нг—С Ha— NO2
фенил нитрометан
1 нитро 2 фенил этан
Изомерия иитросоедииений может быть обусловлена разной структурой углеродного скелета (изомерия цепи) и разным положением ннтрогруппы в углеродной цепи (изомерия положения):
— изомерия цепи
СНз
СНэ—С Н2—С Н2—СН:
no2
no2
1 нитробутан
2 нитробута и
2 метил
2 нитропропан
— изомерия положения
18В
3 нитротолуол 4 нитротолуол
изомерия положения------------------
11.2. НИТРОАЛКАНЫ
11.2.1. Способы получения
Нитрование алканов (реакция Коновалова). Реакция протекает при действии иа алканы разбавленной азотной кислотой (10—25 %) при повышенных температуре и давлении (см. с. 22):
СНз—СНа—СНз + НЫОз (разб.) l5f'А
---► СНз—СН—СНз Н2О
11о2
Взаимодействие галогеиалканов с солями азотистой кислоты. Этот метод позволяет получить первичные и вторичные иитроалка-ны (см с 166) Для уменьшения образующейся в процессе реакции примеси эфиров азотистой кислоты, взаимодействие первичных и вторичных галогеиалканов с солями азотистой кислоты (AgNCb, NaNO2, KNO2) осуществляют н среде апротонного растворителя— диметилсульфоксида (СНз)а5О или диметилформамнда О
н—С
^NfCHj)!!
СаН51 + NaNO2 -----> C2H5NO2 + Nal
Окисление трет-алкиламинов. Этим способом получают только третичные нитросоедниения Первичные амины, содержащие трет-алкильный углеводородный остаток, в присутствии органических пероксикислот окисляются, образуя иитроалканы
СНз СНз
СНз—С—СН3 окнслеыне-> СНз—(t—СНз + Н2О
NH2 no2
2 метилпропанамни 2 2 метил 2 ннтропропан
180
Нитроалканы, как правило, представляют собой бесцветные или слегка желтоватые жидкости с приятным запахом, ядовиты. Они мало растворимы в воде, растворимы в большинстве органических растворителей. Это полярные соединения, для которых характерны большие дипольные моменты (3,15—3,7Д). Температуры кипения ннтроалканов выше, чем соответствующих спиртов или карбонильных соединений.
11.2.2. Химические свойства
Реакционная способность ннтроалканов определяется наличием в их структуре иитрогруппы. Вследствие сильных электроно-акцепторных свойств иитрогруппа активирует атомы водорода при а-углеродном атоме углеводородного радикала
О
СНз—СН^СН N t \ н6+ о Поэтому первичные и вторичные иитроалканы являются СН-кис-лотами.
Химические превращения нитроалкаиов происходят с участием нитрогруппы и а-углеродиого атома.
Таутомерия и образование солей. Первичные и иитроалканы являются таутомерными веществами, нитроформа (нитроалкаи) находится в подвижном с дци-яитроформой (нитроновой кислотой) О ОН
R—CH—N
R—CH=N
вторичные в которых равновесии
О
Н О О
«итрофнрми аци нитроформа
Такая таутомерия получила название аци-нитро-таутомернн.
В нейтральной среде равновесие обычно почти полностью смещено в сторону нитроформы. Превращение нитроформы в аци-нитроформу протекает через стадию образования амбидентного (двойственного) аниона, стабилизированного за счет сопряжения:
on
О
/ и
R—СН— N 5=
н о
хигроформа
R—CH=N
H+,
R—CH=N
OH
О р-
О
аци - н и графа р м а
t
О амбидентный аннон ___
190
В щелочной среде таутомерное равновесие смещается в сторону аци-нитроформы. Так, первичные н вторичные нитроалканы растворяются в водном растворе щелочи, образуя соли нитроновых кислот:
R О
\зН—-Ь NaOH
RZ \>
+ Н,0
натриевая соль
нитронпвой кислоты
Соли нитроновых кислот взрывоопасны.
Если щелочной раствор нитроалкана обработать минеральной кислотой, сначала образуется растворимая в воде “неустойчивая нитроновая кислота, которая затем превращается в нерастворимый в воде нитроалкан:
НС1 э — NaCl
натриевая соль нитроновой кислоты
ННтроновая кислота
нитроалкаи
Третичные нитроалканы не имеют атомов водорода прн а-угле-родиом атоме, а следовательно, для них ие характерна аци-нитро-таутомерия и поэтому они не растворяются в водных растворах щелочей.
Реакция с азотистой кислотой. Взаимодействие ннтроалканов с азотистой кислотой происходит с участием подвижных атомов водорода при а-углеродном атоме.
Первичные иитроалканы реагируют с азотистой кислотой с образованием алкнлиитроловых кислот. Реакция протекает через стадию образования интрозонитросоедииеиия:
r-ch-no2 I н
первичный нитроалкан
+ НО—N“O —►
-Н2О
r-c-no2 —►r-c-no2
N—OH
'.... алкил-
нитрозонитро- нигрол овая соединение кислота
191
Алкилиитроловые кислоты представляют собой бесцветные кристаллические вещества. Соли нитроловых кислот со щелочными металлами в растворе окрашены в красный цвет.
Вторичные иитроалкаиы дают с азотистой кислотой псевдо» нитролы;
СН—NO2 + НО—N==O
R
\—NO2 + Н2О
вторичный нитроалкан
псевдоннтрол
Растворы и расплавы псевдоиитролов окрашены в синий цвет.
Третичные нитросоединения с азотистой кислотой не реагируют.
Реакцию с азотистой кислотой используют для отличия первичных, вторичных и третичных иитросоедииений друг от друга.
Реакция с альдегидами и кетонами. Первичные и вторичные ннтроалканы в слабощелочной среде вступают в реакцию конденсации с альдегидами и кетонами, образуя нитроалканолы. Эта реакция протекает по типу альдольной конденсации (см. с. 352),
NaOH
£^с-^н-снэ NO, ОН
При конденсации первичных нитроалканов с карбонильными соединениями образующиеся нитроалканолы в большинстве случаев отщепляют молекулу воды и реакция завершается образованием непредельных нитросоединеннй ।
О
R—СН2 + С—СНз » R—СН—СН—СНз -птТ J
I / II
NO2 н NO2 он
—г R—С=СН—СНз
| Я
no2
С формальдегидом первичные иитроалкаиы часто образуют продукты конденсации с двумя, а нитрометан — с тремя молекул ла ми альдегида:
192
R—СН2
no2
о
CH2OH СН2ОН
I \ I
R— CH -------* R—C—CH2OH
NO2 no2
Восстановление митроалканов. При восстановления нитроалканов образуются алканамины
C2H5NO2 + 6Н-----н C2H5NH2 + 2Н2О
В качестве восстановителей применяют хлорид олова (II), железо в присутствии хлороводородной кислоты, сульфиды щелочных металлов и др.
11.3. АРОМАТИЧЕСКИЕ НИТРОСОЕДИИЕНИЯ
11.3.1. Способы получении
Нитрование аренов. Нитросоедннеиия, содержащие нитро-группу, связанную с ароматическим радикалом, получают нитрованием ареиов смесью концентрированных азотной и серной кислот (см. с. 99):
Н + HONO2 --25°*
Для введения второй нитрогруппы требуются более жесткие условия: высокая температура, концентрированные кислоты, длительное нагревание. Введение третьей нитрогруппы в молекулу бензола происходит с большим трудом при избытке дымящей азотной и серной кислот:
HNOgfHjSCM). t
—НаО
HN03(H2S04>, \
—НзО
При наличии в ядре электронодонорных заместителей реакция нитрования значительно облегчается. Так, при нитровании толуола можно в обычных условиях ввести три иитрогруппы по схеме
7 5 КМ
193
NOa NO2 k
4 нитротолуд 2,4 ди нитротолуол
ц\Оэ
NO2
2,4,6-тринигротолуол
Для получения ароматических иитросоедииений с нитрогруп-пой в боковой цепи применяют способы получения, аналогичные методам синтеза нитроалканов: нитрование гомологов бензола в условиях реакции Коновалова (см. с. 22); взаимодействие арилалкнлгалогенидов с салями азотистой кислоты (гм с 166) и др.
11.3.2. Физические свойства
Нитроарены, содержащие в своем составе одну нитрогруппу, представляют собой жидкости или кристаллические вещества, бесцветные или окрашенные в бледно-желтый цвет, нерастворимые в воде. Жидкие ннтросоединения тяжелее воды. Нитроарены обладают запахом горького миндаля. Это полярные вещества, имеющие высокие температуры плавления.
Нитроареиы, содержащие несколько нитрогрупп (полинитро-соедииения),— это кристаллические вещества, окрашенные в желтый цвет. Взрывчаты.
11.3.3. Химические свойства
Восстановление нитроаренов (реакция Зинина). При восстановлении ароматических иитросоедииений образуются ароматические амииы. Впервые реакцию восстановления нитробензола в аиилнн осуществил в 1842 г. известный русский ученый Н. Н. Зинии.
194
В качестве восстановителей чаще используют железо, олово или цинк в хлороводородной кислоте, сульфид аммоиня (NH^S, гидросульфид натрия NaHS, гидразин в присутствии никеля Ренея (NHaNHj/Ni Ренея).
В зависимости от pH реакционной среды процесс восстановления может идти по двум направлениям, отличающимся образованием разных промежуточных соединений.
В нейтральной и кислой среде в качестве промежуточных соединений образуются ароматические иитрозосоедииения и арил-гидроксиламнны:
нитро нитрозо- фенидггидро-
бензол бензол ксиламнп
2Н
—ИаО
При восстановлении в нейтральной среде реакцию можно остановить иа любой стадии. В кислой среде выделить промежуточные продукты невозможно.
В щелочной среде происходит конденсация образующихся в процессе реакции нитрозосоедииеиий с арилгидроксиламином с образованием азокснсоединений, которые восстанавливаются до азосоединений. Образующиеся азосоединения, присоединяя водород, превращаются в гидразосоединения, которые, в свою очередь, легко превращаются в ариламииы:
no2 no nh—он
нитробензол нитрозобензол фенилгидро-
не ила чин
азокеибепзо.1
н н
гидразобензол
азобензол
Реакцию восстановления нитроареиов в щелочной среде можно остановить иа любой из приведенных стадий. Она служит основным способом получения азо- и гидразосоединеиий.
195
Реакции по ароматическому ядру
Реакции электрофильного замещения (Sr). Обладая электроноакцепторными свойствами (——Af-эффект), нитрогруппа дезактивирует бензольное ядро в реакциях, идущих по 5Р-механнз-му. Так, нитробензол не алкилируется в условиях реакции Фриделя — Крафтса:
+ СН3С1
A1CL
Поэтому часто нитробензол в этих реакциях используется как растворитель.
Однако он может вступать в реакции нитрования, галогенирования, сульфирования с образованием соответствующих же-та-замещенных Например:
+ Н2О
м нитробензол (|
сульфокислота
Реакции нуклеофильного замещения Электроноакцепторное влияние нитрогруппы приводит к понижению электронной плотности в ароматическом радикале и создает возможность для протекания реакций, идущих по механизму нуклеофильного замещения в аренах. В реакциях SN иитрогруппа направляет заместитель в орто- и пара-положеиия При нагревании нитробензола с твердым гидроксидом калия получают смесь о- и п-нитро-фенолятов калия:
84-
КОН ----►
о-нигрофенодят л-нитрофенолят . калия • калия
106
При наличии в ядре бензола трех интрогрупп резко увеличивается подвижность атомов водорода бензольного ядра Поэтому гижл-триннтробеизол легко окисляется до пикриновой кислоты:
симм-тринитробснзол 2,4,6 -тринитрофенол,
пикриновая кислота
При действии иа тринитробензол гндроксиламина атом водорода легко замещается на аминогруппу:
Понижая электронную плотность в бензольном ядре, иитро-группа увеличивает подвижность заместителей, находящихся в орто- илн пара-положении по отношению к иен. Это позволяет получать различные нитропроизводные ароматического ряда:
197
В данном случае нитрогруппа принимает участие в делокализации отрицательного заряда образующегося о-комплекса, облег- . чая тем самым замещение галогена
н-нитрохлорбензол
л-нитрофенол
Реакция замещения галогена в галогеннитроаренах также протекает по механизму SN.
Ароматические нитросоединения, содержащие нитрогруппу в боковой цепи, в реакциях, идущих по группе NO2, напоминают нитроалканы. Оин легко восстанавливаются до аминов, растворяются в щелочах с образованием солей a^w-нитроформы (см. с. 191).
Наличие ароматического радикала позволяет нм вступать и различные реакции замещения по ароматическому ядру.
11.4. идентификация нитросоединении
Для идентификация ннтросоеднненнй используют инструментальные (ИК-, УФ-спектроскопия, масс-спектрометрия) и химические методы анализа.
ИК-спектры нитросоединений характеризуются наличием интенсивных полос поглощения, отвечающих валентным колебаниям нитрогруппы. Для иитроалканов отмечается полоса в области 1375 и 1580 см-1. В ИК-спектрах нитроаренов присутствуют две сильные полосы поглощения в области 1490—1560 н 1320 - 1380 см *.
В УФ-спектрах нитросоединеиий присутствуют полосы поглощения, соответствующие л->л* переходу. В нитроалканах такой переход наблюдается в области 210 нм (е як 1500). В УФ-спектрах нитроаренов присутствует полоса поглощения в области 250—300 им (е як 6000).
£ качестве химических методов ндеитнфикации обычно используют химические реакции, которые позволяют отличить одно соединение от другого или различить группы соединений,
Чтобы отличить первичные, вторичные и третичные нитроалка-ны друг от друга, используют реакции со щелочью н азотистой кислотой (см. с. 191). Эти же реакции позволяют отличить нитро-алкаиы от нитроареиов, а также нитроарены от ароматических иитросоедииений с нитрогруппой в боковой цепи.
198
Для идентификации ароматических интросоединеннй, содержащих нитрогруппу в ядре, применяется реакция восстановления до аминов с последующим диазотированием и азосочетанием (см. с. 226).
11.5. ОТДЕЛЬНЫЕ ПРЕДСТАВИТЕЛИ. ПРИМЕНЕНИЕ
Нитрометан СНэ—NOa— простейший представитель иитроалканов. Это бесцветная жидкость (т. кнп. 101,2 °C). Применяется как растворитель эфирцеллюлозных лаков, различных виниловых полимеров, взрывчатых веществ, хлорпикрина СС13—NO2.
Нитробензол CeHgNOz — жидкость (т. кип. 210 °C), обладающая запахом горького миндаля. Применяется для получения анилина, бензидина, в производстве красителей, как мягкий окислитель в химических реакциях, в качестве растворителя.
Ннтротолуолы CH3C6H4NO2. п-Нитротолуол — это кристаллическое вещество (т. пл, 52 °C), о- и ж-нитротолуолы — жидкие вещества (т. кип. 221,7 и 232,6 °C соответственно). Применяются нитротолуолы для синтеза красителей и других ароматических соединений.
КОНТРОЛЬНЫЕ ВОПРОСЫ И УПРАЖНЕНИЯ
1. Напишите структурные формулы следующих иитросо-единений: а) 2-метил-4-нитропеитаи; б) 1-нитро-2-феиилпропан; в) З-метил-1-иитробутан. Укажите среди изображенных структур первичные, вторичные и третичные нитросоединения.
2. На примере 2-метил-1-иитропропана изобразите схему ацц-нитро-таутомерии.
3. Какие из приведенных иитросоединекий реагируют с азотистой кислотой: а) I-иитропропаи; б) 2-метил-2-интропропан; в) 2-нитропропан; г) нитробензол? Напишите схемы н назовите продукты реакций.
4. Какие продукты образуются при взаимодействии этилбензола: а) с коиц. HNO3; б) с разб. HNOs, р, /? Напишите схемы реакций.
5. Напишите схемы последовательных реакций, протекающих при восстановлении 4-иитротолуола: а) в кислой среде; б) в щелочной среде. Назовите промежуточные и конечные продукты.
6. Приведите схемы реакций, позволяющих различить феинл-интрометаи и нитробензол.
Глава 12
АМИНЫ
Аминами называют производные аммиака, в моле* куле которого один, два или три атома аодорода заме* щены углеводородными радикалами.
Соответственно числу углеводородных остатков различают первичные, вторичные и третичные амнны:
R—NH2
первичный вммн
R
^N—Н R//Z вторичный амин
третичный амин
В зависимости от природы углеводородных радикалов у атома азота амины подразделяют на алифатические, алициклические и ароматические (аренамины); амины, у которых атом азота связаны с алифатическим и ароматическим углеводородными радикалами, называют смешанными:
СНз— NH2
алифатический амии
алиинктнчесний амии
ароматический амин
NH—С2Н5
смешанный амин
12.1. НОМЕНКЛАТУРА. ИЗОМЕРИЯ
По заместительной номенклатуре ИЮПАК названия первичных аыинов образуют путем добавления к названию углеводорода суффикса -амии, с последующим указанием положения аминогруппы в углеродной цепн. При составлении названий вторичных и третичных аминов их рассматривают как производные первичного амина с заместителями при азоте. За исходный первичный амин в этом случае принимается связанный с атомом азота наиболее сложный по структуре радикал. Остальные углеводородные заместители при азоте перечисляют в алфавитном порядке с указанием локанта N-:
200
12 3 12 3
CHj—СН—СИ3 CHj—NH—C2Hs CHs—N—CH»—CHj—СНз
NH2 (Lh5
пропаначнн 2 N метилзтаиамии N метил \-этн л проча намин- (
Если соединение содержит две или три аминогруппы, то в названии их обозначают множительными приставками -дн- или -три-, которые ставятся перед суффиксом -амин:
12 3 4
H2N—СНг—СНз—СНз—СНг—NH2
вутан тиамин 1 4
Простейшие амнны чаще всего называют по радикало-фуик-цнональной номенклатуре. Согласно этой номенклатуре названия аминов образуют из названий углеводородных радикалов, перечисляемых в алфавитном порядке, н суффикса -амин:
С2Н5—NH2
этиламин
СН3—NH—С2Н5
мети.иткламкн
с2н5
С2Н5—N—С2Н5
тризтиламнн
СНз—СНг—СН—СНз
лгод-бутилаиин
СНз—СН—СНа—NHa
СНз
нзобутила мин
СНз I СНз—С—NH3
СНз rpfj-бутилвынц
бензиламин
СНг— NH2
Названия первичных ароматических аминов, а также смешанных аминов обычно образуют на основе названия родоначального представителя — анилина. В случае смешанных аминов положение заместителей у атома азота обозначают локантом N-
2 этиланилин, о этиланнлнн
N N-диыетнланилнн
NH—С2Н5
С2Нб
N этил 2 этиланнлнн
201
Вторичные и третичные ароматические амины, как правило, называют по радикало-функциональной номенклатуре
дифенилачин
Производные толуола, содержащие аминогруппу в бензольном ядре, называют толуидинами
п толуидин
Ароматические диамины обычно называют феиилсндиаминами
о фенилендиачии
Изомерия аминов обусловлена разной структурой углеводородных радикалов, разным положением аминогруппы и метамерией. Сущность метамерии состоит в том, что амины с одной и той же брутто-формулой могут быть первичными, вторичными и третичными. Так, метамерами являются
СНз
СНзСН2СНа—NH2 СН3—NH—СН2СН3 СН3—N—СН3
к пропилами к
мет ил этил а мин
три метиламин
12.2. АЛКИЛАМИНЫ
Алкнламннами называют продукты замещения одного, двух или трех атомов водорода в аммиаке алкильными группами.
202
12.2.1. Способы получения
Взаимодействие галогеналкаиов с аммиаком (реакция Гофмана). При нагревании спиртового раствора аммиака с галогенал-каиами образуется смесь первичного, вторичного н третичного аминов и соль четвертичного аммониевого основания. Эта реакция открыта в 1850 г немецким химиком А. В Гофманом
Сначала аммиак с галогеналкаиом образует соль алкиламмо-ния, которая в избытке аммиака превращается в первичный алкиламин
СН31 + NH3 -------[CH3NH3] Г CH3NH2 + NH4I
иодил метнламмония «етилачин
Образовавшийся первичный амин реагирует со следующей молекулой галогеналкана и т. д В результате образуется вторичный амин, затем третичный и соль четвертичного аммониевого основания:
CHsNH2 + СН31 --------> (!СН3)2Ш2)Г 5^ (СНЭ)2МН 4- NH4I
ипдил дичетн.(аммония диметнлячин
(CH3)2NH + СНз1 --------> [(CH3)aN]I- (CH3)3N + NH41
иоднд три чети 1аи чо мня триметнла^ин
(CH3)3N + СН3! -------► [ГСН3)Л]1-
иодкд тетрамгтилвммония
При большом избытке аммиака увеличивается выход первич-|ого амина, при большом избытке галогеналкана в смеси пре-бладает соль четвертичного аммониевого основания. Образующуюся смесь первичных, аторичНых и третичных аминов разделяют шакционной перегонкой.
Синтез Габриэля. Этот метод позволяет получить первичные лкиламины, Он основан на взаимодействии фталимида калия : галогеналкаиов с последующим гидролизом образующегося N-алкил фталимида:
Г |Т N“K+ + CJij-Cl - фталимид калия О —* [ 11 \\ О N этилфталимид
203
фталевая KHUiOTd
этананнн
Восстановление нитроалканов и нитрилов. При восстановлении нитроалканов и нитрилов образуются первичные амины. Восстановление проводят водородом в присутствии катализатора (Ni, Pd, Pt) или водородом в момент выделения
СНз—NO2 + ЗН2 СНз—NH2 + 2Н2О
нитрометан метиламин
СН3С=\’ + 2Н2 СН3СН2—NHS
я^стоннтрил этянамнн
Расщепление незамещенных амидов карбоновых кислот гипо-броматом, натрия (перегруппировка Гофмана). При обработке незамещенных амидов карбоновых кислот гипобромитом натрия (или смесью брома и гидроксида натрия) образуются первичные амины, в которых на один атом углерода меньше, чем в исходном амиде:
О
СНзСН2- -с/ СНзСН=~NHs + с°2 + №Вг
'nh2
амид пропионовой этиламил
кислоты
12.2.2. Физические свойства. Пространственное строение
В обычных условиях метиламин, диметиламин и трнметнл-амии являются бесцветными газами, алкиламины с 4—15 атомами углерода — жидкости, высшие амнны — твердые вещества (табл. 12.1). Низшие амины хорошо растворимы в воде, с увеличением молекулярной массы растворимость снижается, а высшие амины не растворимы в воде. Газообразные амины имеют запах аммиака, жидкие амины обладают резким неприятным запахом, несколько напоминающим запах селедочного рассола, твердые — не имеют запаха. Температуры кипения третичных аминов значительно ниже, чем первичных и вторичных с тем же числом атомов
204
углерода. Это связано со способностью первичных и вторичных аминов образовывать, подобно спиртам, недородные связи:
R
6+ | й +
Н Н • • .;N н Н • • •
\ / / \
. . . :N Н Н • • • :N
| м I
R R
Таблица 12 1
Физические характеристики некоторых алкиламинов
Формула Название Температура, ЭС
кипения плавления
CHj—nh2 Метиламин -6,5 -92
СНз—СНа—NH2 Этиламин 17 —81,2
сн,—сн2—СН/—nh2 н-11ропиламин 49 —83
CHj—СН—С Нл Изопропиламин 33,5 -101
। NH3 С H, СНа—С Н2—СН2—NHa н-Бутнламин 78 -50
CHj СН,—(L- nh2 трет'Б^тиламин 46,4 —67.5
1 СН, CH3(CH2),NH2 н Пепгиламии 104 -55
CHj(CH2)sNH2 н-I ексиламип 130 -19
(CHj)2NH Диметиламин 7,5 96
(CjIM/NH Диэтил а мин 56 -48
(С Н,) з N Триметиламин 3 -117
(C2H-,)3N Трнэтиламнн 89 — 115
Однако в связи с тем что электроотрицательность атома азота меньше, чем атома кислорода, амины образуют менее прочные ассоциаты, чем соответствующие спирты Третичные амнны не имеют атома водорода при азоте, поэтому они ие способны к ассоциации
Пространственная модель молекулы алкиламина имеет форму четырехгранной пирамиды, в вершине которой находится атом азота. Валентные углы между связями в среднем равны 107—108°, т. е. близки к тетраэдрическому. Условно приняв неподеленную пару электронов атома азота за четвертый заместитель, можно
205
представить конфигурацию атома азота в аминах аналогично тетраэдрической конфигурации атома углерода:
Исходя из этого атом азота в аминах находится в 5р3-гибри-дизации, а неподеленная пара электронов занимает sp -гибридную орбиталь
Такое строение предполагает существование оптической изомерии у соединений, имеющих три разных заместителя у атома азота (роль четвертого заместителя выполняет неподеленная пара электронов). Однако вследствие инверсии, т. е. быстрого взаимного превращения одной тетраэдрической конфигурации в другую, разделить оптические изомеры не представляется возможным:
Поэтому алкиламины не проявляют оптической активности!
12.2.3. Химические свойства
Реакционная способность алкиламинов определяется главным образом наличием у атома азота неподеленной пары электронов'. За счет пары электронов атома азота амины, с одной стороны, способны присоединять протон от кислоты, проявляя прн этом основные свойства, а с другой — могут атаковать в молекуле реагента электрофильный центр (чаще атом углерода, несущий частичный или полный положительный заряд) и образовывать химическую связь с ним, проявляя нуклеофильные свойства.
Ввиду сравнительно низкой электроотрицательности атома азота и -(-/-эффекта со стороны алкильных групп связь N—Н в алкиламннах поляризована незначительно. Поэтому первичные и вторичные алкиламииы являются очень слабыми NH-кислотами.
А. Основность
Являясь производными аммиака, алкиламины, подобно аммиаку, проявляют ярко выраженные основные свойства, которые обусловлены наличием на атоме азота неподеленной пары электро
206
нов. При этом алкиламины являются более сильными основаниями, чем аммиак.
Как известно, сила основания определяется устойчивостью катиона, образующегося в результате присоединения протона. Чем устойчивее катион, тем сильнее основание. На устойчивость катиона оказывает влияние электронная природа заместителей при катионном центре и природа растворителя. Алкильные заместители, обладая -(-/-эффектом, способствуют делокализации положительного заряда катиона и тем самым повышают его устойчивость. Поэтому можно предположить, что третичные алкиламины, имеющие три донорных заместителя, будут более сильными основаниями, чем вторичные, а вторичные, в свою очередь, более сильными, чем первичные и аммиак:
СНз СНз
CH3-*-N > N— Н > СН3 > NH2 > NH3
/ /
СН3 СНз
Такая закономерность изменения основности алкиламинов наблюдается в газовой фазе и в неводных растворах. Однако в водных растворах на устойчивость катиона влияет наряду с электронными эффектами заместителей сольватационный эффект растворителя, Степень сольватации катиона тем выше, чем меньше его объем и степень делокализации в нем заряда.
Способность к сольватации алкиламмониевых катионов аозрас-тает в ряду
207
Как видно, электронные эффекты заместителей и сольватационный эффект растворителя оказывают на основность противоположное воздействие Поэтому в результате совместного проявления этих двух факторов основность третичных аминов в водных растворах, как правило, ниже основности первичных и вторичных: t
R' R' и
/ \ 1
R— N < R— NH2 < NH t,
R" R w
третичный первичный вторичный f
Водные растворы алкиламинов имеют щелочную среду.
+
R—NH2 + НОН q=± RNH3 + ОН"
С кислотами алкнламины образуют соли алкиламмония:
C2H5NH2 + НС1 ------► [С2Н5ЙНЭ] С1-
х торил этила чмония
При действии на аммониевую соль сильного основания, напрна! мер гидроксида натрия, высвобождается исходный амин;
[C2H5NH3]C1- + NaOH -----C2H5NHa + NaCI + Н2О
J
Реакцию солеобразования с последующим выделением свободного амина часто используют для очистки аминоа.
Б. Реакции с электрофильными реагентами
Взаимодействие с галогеналканами. С помощью этой реакции^ в структуру амина вводят алкильный заместитель и поэтому она^ получила название реакции алкилирования. При взаимодейст-1 вии с галогеналканами первичные амины превращаются во вто- ? ричиые, вторичные — в третичные, а третичные — образуют чет-л вертичные аммониевые соли, например: Ц
3+ 3- + $
C2H5NH2 + СН3-1 -► [C2H5NH2CH3]I C2H5-NH-CH3 + HI?
иодид ‘
метилэтиламмония метилэтиламин |
>
Аналогично вторичный амин алкилируется с образованием третичного, а третичный — с образованием четвертичной аммониевой солн (см. о реакции Гофмаиа на с. 203).
В зависимости от природы галогеналкана и алкиламина реакция может протекать по механизму SN1 нлн SN2 (см. с. 159).
208
Ацилирование. Первичные и вторичные алкилвмины вступают в реакцию с функциональными производными карбоновых кислот — галогенангидридами, ангидридами или сложными эфирами, образуя амнды (см. с. 417):
О О
C2H5NH2 + СНз——* C2H5NH-JL-СНз + НС1 \:1
этнламин
хлорангидрид уксусной кислоты
этиламнд уксусной кислоты
c2h5nh2 +
о / СНз—С
C2H5NH
о
о
СНз—С
\
уксусный ангидрид
О о I
C2H5NH2 4- СНз——u C2H5NH—C—СНз + С2НЯОН ^ОСгНв
В процессе реакции атом водорода при азоте в молекуле амина О //
замещается иа остаток карбоноаой кислоты R—С , называемый ацильной группой Реакции, с помощью которых в молекулу органического вещества вводится ацильная группа, называют реакциями ацилирования.
Третичные амнны не содержат при атоме азота атома водорода и поэтому в реакцию ацилирования не вступают.
Взаимодействие с азотистой кислотой. С азотистой кислотой реагируют первичные н вторичные алканамины. Так как азотистая кислота очень неустойчива, обычно ее получают непосредственно в процессе реакции взаимодействием солей азотистой кислоты (NaNOs, KNO2) с какой-либо сильной минеральной кислотой, чаще хлороводородной илн серной.
При действии азотистой кислоты на первичные алканамины выделяется свободный азот и образуются спирты. Реакция протекает через стадию образования неустойчивых солей алкилдиазо-
200
ния, которые в водной среде разлагаются с выделением азота и образованием спиртов;
C2H5NH2 + НО—N=O —[C2H5N=N]Cr
—2Н2О этиламин этаидиаэонии хлорид
---> С2НБОН + N2 + HCI
этанол
Механизм образования соли дназоння изложен на с. 231,
В качестве побочных продуктов реакции часто образуются алкены и галогеналканы.
Вторичные алканамины в реакции с азотистой кислотой образуют N-иитрозоамины:
(CH3)2NH + НО—N=O ---------> (CH3)2N— N=O 4- Н2О
\ нигрозодиметиламнн
Ннтрозоамины представляют собой желтые или оранжевые маслянистые жидкости. При обработке концентрированными минеральными кислотами оии расщепляются с образованием исходного амина и азотистой кислоты:
'(£H3)2N—N=O + HCI 2^ (CH3)2NH-HCI + HNO2
Третичные алканамины в обычных условиях с азотистой кислотой не реагируют. Реакция взаимодействия алкиламинон с азотистой кислотой может быть использована для отличия первичных, вторичных и третичных аминов друг от друга.
Изонитрилъная реакция. Эта реакция характерна только для первичных аминов. При нагревании первичных алкиламинов с хлороформом в присутствии щелочей (NaOH, КОН) в спиртовой среде образуются изоцианиды (изонитрилы):
c2h5nh2 + снсь + зкон СэН5°н>
jth ламин хлороформ
--->-С2Н5—N=C“ + 3KCI + ЗН2О этн чмзоцнаннд
Изоцнаниды обладают очень сильным неприятным запахом. На этом свойстве основано использование изонитрнльной реакции для качественного обнаружения первичных амниов.
Реакция протекает по механизму нуклеофильного замещения.
При взаимодействии хлороформа со щелочью образуется в качестве промежуточного продукта дихлоркарбен:
□
СНС1з + NaOH -----------► ;СС12 + NaCI + Н2О
дихлоркарбен
210
Дихлоркарбен представляет собой двухвалентный радикал, имеющий вакантную орбиталь, которая подвергается атаке неподеленной парой электронов атома азота. Образующийся продукт присоединения отщепляет в присутствии спиртового раствора щелочи две молекулы хлороводорода и превращается в нзоцианид;
C2H5NH2 + :СС12 -----► C2H5-N-C<°
И
2NaOH + “
———► CsHsfeC + 2NaCI + 2Н2О
I 2115мИ )
лилигоцианид
N-галоге пирование. При взаимодействии первичных и вторичных алкиламинов с галогенами (С12, Вг2) или гипогалогеиитамн (NaOCI, NaOBr) атомы водорода при азоте замещаются на атомы галогена и образуются N-галогено- или N, N-дигалогеноамииы:
Н С1
C2H6NH2 -2!— С2Н5—С2Н5—N"/ НС1 -НО
Cl CI
N х юрэтиламин
N.N дил.юрэтиламнн
N-Галогеналкиламнны являются неустойчивыми соединениями.
В> Окисление алкиламинов
Первичные алкиламины при окислении озоном образуют с хорошим выходом нитроалканы:
C2H5NH2 -%* C2H5NO2 + Н2О
этиламин нитроэтан
Третичные алкиламины при окислении пероксидом водорода О
или пероксикислотами R—С образуют N-окснды аминов:
\)ОН
(C2Hs)3N (C2H5)3N^O
триэтнламкн N оксид триэтила мина
При окислении в более жестких условиях первичные, вторичные и третичные алкиламины образуют альдегиды, кетоны или карбоновые кислоты.
211
12.3. АРИЛАМННЫ
Арнламинами называют производные аммиака, в молекуле которого один, два или три атома водорода замещены остатками ароматических углеводородов.
12.3.1. Способы получения
Восстановление нитроаренов (реакция Зинина): C6HS—NO2 -!!!!► CoHuNHs + Н50 нитробензол зиизии
В качестве восстановителей используют металлы (Fc, Zn, Sn) в среде хлороводородной кислоты, NaaS, SnC^ водород в присутствии катализатора и др. В зависимости от pH реакционной среды процесс восстановления сопровождается образованием разных промежуточных продуктов (см, с. 195).
Взаимодействие галогенаренов с аммиаком и аминами. Га-логенарены реагируют с аммиаком, первичными и вторичными аминами в жестких условиях (высокое давление и температура, присутствие в качестве катализатора меди или се солей). При взаимодействии с аммиаком образуются первичные ариламнны:
С6Н3—Cl + 2NFh 20 —С6Н5—NH2 + NH4C1 Си клорбензол
Прн взаимодействии галогенаренов с арнламинами образуются вторичные и третичные ариламнны'
СеН5—CI + С6Н5—NH2 с6Н5—NH—CfaH5 + НС1
р f и дифенн танин
“С
С6Н5—I + (C6H5)aNH (C6H5)3N + HCI
р, Си
иодбе!под дифениламин тркф< ни i 1 мi, 11
Если в орто- илн пара-положении молекулы галогенарена находится электроноакцепторный заместитель (NO2, CN, COR и др.), то замещение атома галогена осуществляется значительно лете (см. с. 179).
Алкилирование первичных ариламинов. Этот метод позволяет получить смешанные N-алкил- и N,N-диалкилариламины. В качестве алкилирующих реагентов чаще всего используют галоген-алканы или спирты в присутствии кислот, В ’процессе реакции образуется смесь вторичного и третичного аминов:
СН3
N-метиланилин
N.N дннетиланнлии
212
12.3.2. Физические свойства
В обычных условиях ариламнны представляют собой бесцветные высококипящие жидкости или твердые кристаллические ве- 1 щества со слабым неприятным запахом (табл. 12.2). Они мало-растворимы в воде, сильно токсичны, окисляются кислородом" воздуха, из-за чего при хранении приобретают желтоватую окраску.
Таблица 12.2
Физические характеристики некоторых ариламинов, N-алкил-и К^диалкилариламинов
Формула Название Температура, °C
кипения плавления
CoH8NH2 анилин 184 -6
CoHsNHCHj N-метиланилин 196 -57
С6Н5М(СНэ)г М.М-диметиланилин 194 3
C»H?N(CaHB)a N. N-диэтиланилин 217 —21,3
(C6HS)2NH дифениламин 302 53
трифепиламик 365 127
NU Са о-толуидин 200,2 -24.5
\нэ МНг 6. и- толундин 204 -30
х/\ СНз NHa п-толундин 200,6 45
н,с
12.3.3. Химические свойства
Для ариламннов характерны реакции с участием атома азота и реакции с участием атомов углерода ароматического ядра.
А. Реакции с участием атома азота
По атому азота ариламнны вступают практически во все реакции, рассмотренные ранее для алкиламинов, однако протекание некоторых из них имеет свои особенности.
213
Основность. За счет наличия нелоделенной пары электронов на атоме азота ариламины, как и алкиламины, проявляют основные свойства. Однако основность ариламинов значительно ниже основности алкиламииов, например:
Основание СНэМН2 CeHaNHa СбН5МНСьН5
рА' , 10,6 4,6 0,78
“ on 7 7
Значительное снижение основности ариламинов по сравнению с алкиламинами обусловлено сопряжением иеподеленной пары электронов атома азота с л-электронной системой ароматического ядра.
+ЛГ-эффект
В результате сопряжения неподеленная пара электронов частично делокализуется по ароматическому ядру и становится менее доступной для координации с протоном.
На основность ариламинов существенное влияние оказывают заместители в бензольном кольце. Электронодонорные заместители увеличивают основность, а электроноакцепторные — уменьшают ее. Например, анилин является более сильным основанием, чем л-нитроанилин, но менее сильным, чем л-анизидин:
л-анизидин
анилин л-нитроанилин
Основность ариламинов свльно снижается при переходе от первичных к третичным:
анилин
трифенил амин
214
Вследствие электроноакцепторных свойств трех бензольных колец, трифениламнн практически не обладает основными свойствами. Двляясь слабыми основаниями, ариламины образуют солн только с сильными минеральными кислотами;
C6H5NH2 + HCI -------* C6HeNH3Cr
анилин знилипия хлорид
Реакция алкилирования. Подобно алкиламинам первичные и вторичные ариламнны реагируют с галогеналканами, образуя N-алкил и N,N-диалкиларнламины. Ввиду снижении нуклеофильных свойств атома азота алкилирование ариламинов протекает труднее по сравнению с алкиламинами:
C6H5NH2 C2H5I -------»- C6H5NH—С2Н5 -|- HI
.N-этила лилия
С2Н5
C6H5NHC2H& + C2HBI ------С6Н5—+ HI
С2Нз
Ь, N ДИЛТН.ЧЯ НИЛИН
Реакция алкилирования используется для получения смешанных аминов.
Реакция ацилирования. При действии на первичные и вторичные ариламнны галогенангидридов или ангидридов карбоновых кислот атомы водорода при азоте замещаются на ацильные О
J о
остатки R—С В результате реакции образуются замещенные
амиды карбоновых кислот. N-Ацильные производные анилина и его гомологоа называют анилидами
О
CeHsNHa + (СН3СО)2О---------- C6H5NH—С—СН3 + СН3СООН
фенилаиид уксусной кислоты, ацетанилид
В прошлом ацетанилид под названием антифебрин (от лат. anti — против и febris — лихорадка) применялся в медицине как жаропонижающее средство. Амиды карбоновых кислот легко гидролизуются в кислой или щелочной среде с образованием исходного амина и карбоновой кислоты:
О
C6H5NH СНз + Н2О C6H5NH2 + СНзСООН
ацетанилид
2IS
Ни этом свойстве амидов карбоновых кислот основано применение реакции ацилирования аминов для временной защиты аминогруппы от окисления и протекания по ней других реакций, если они нежелательны.
В N-ацильиых производных ароматических аминов неподеленная лара электронов атома азота находится в сопряжении не только с л-электронной системой бензольного ядра, но и с л-элек-тронами двойной связи карбонильной группы С=О, что приводит к значительному снижению в сравнении с аминами ее электронодонорных свойств по отношению н бензольному кольцу:
Образование изоцианидов. Аналогично алкилами нам первичные ароматические амины при нагревании с хлороформом и щелочью в спиртовой среде образуют изоцианиды — вещества с неприятным, тошнотворным запахом:
C6H5NH2 + CHCh + 3NaOH-^^ C6H5N^C 4-
фенилнзонианид
+ 3NaCl 4- 3H2O
О механизме реакции см. на с. 210.
Взаимодействие с азотистой кислотой. Первичные, вторичные и третичные ароматические амины при взаимодействии с азотистой кислотой образуют разные продукты. Прн действии азотистой кислоты на первичные ароматические амины в присутствии сильной минеральной кислоты образуются соли диазония. Эта реакции получила название реакции диазотирования:
C6H5NH2 + НО—NO —С-н- > [CeH5feN]Cl- + 2НаО
Об условиях протекания и механизме реакции диазотирования см. на с. 231.
Вторичные арнламины и N-алкилариламины при взаимодействии с азотистой кислотой, подобно алкиламинам, образуют N-нитрозоамнны;
СсНъ— NH—С6Н5 + НО—NO ----------С6Н5—N—С6Н5 + Н2О
NO
N нитрсэадифсни-ляннн
С6Н5—NH—СНз 4- НО—NO ----------► С6Н5— N—СН3 + Н2О
I
NO
51 метил N нитразааннлнн
216
Третичные 1Ч,М-диалкнлариламины под действием азотистой кислоты подвергаются нитрозированию в пара-положение бензольного кольца, а если оно занято — в орто-положение:
СНз
СНз
СНз + НО—N==O
СНЭ
N N днмстнланилин
O=N
4 нитрозо N N димстнланилнн
Взаимодействие с ароматическими альдегидами. В отличие от алкиламинов первичные ароматические амины реагируют при нагревании с ароматическими альдегидами, образуя азометнны*:
C6H,-NCJ + 'C-C6HS —► С6Н —N=CH—С6Н; +‘Н20 Н Н
бензальдегид
N-бензилиденанилин
Эта реакция была открыта а 1864 г. итальянским химиком И. X. Шиффом, поэтому продукты реакции называют основаниями Шиффа. Под действием разбавленных кислот основания Шиффа гидролизуются с образованием исходного амина и альдегида.
Б. Реакции с участием атомов углерода ароматического ядра
Для ариламинов характерны реакции электрофильного замещения по ароматическому ядру, свойственные ароматическим углеводородам. Аминогруппа в молекуле арнламина в результате проявления -|-Al-эффекта аыстулает в качестве сильного электро-нодонора по отношению к бензольному кольцу н тем самым активизирует его реакционную способность в реакциях электрофильного замещения. Поэтому арнламины вступают в реакции электрофильного замещения значительно легче, чем бензол. Являнсь орнентантом 1 рода, аминогруппа направляет электрофильное замещение в орто- и пар®-положении.
Галогенирование. Анилин легко реагирует с галогенами (Ch, ВГ2) в отсутствие катализатора, образуя 2,4,6-тригалогенопроиз-
* К азо мети нам относят органические вещества общей формулы N=N—R".
где R и R'=H, Aik, Аг, R"---Aik, Аг
217
водные. Так, при обработке анилина бромной водой практически с количественным выходом образуется осадок 2,4,6-трнбром-анилина:
2.4,6-трнброманнлнн
Хлор в водных растворах окисляет анилин, поэтому хлорирование с образованием 2,4,6-трихлоранилина осуществляют действием хлора в неводных растворителях, например в четыреххлористом углероде.
Для получения моногалогензамещенных ариламнны первоначально переводят в N-ацильные производные, чаще в N-аце-тильные, которые затем галогенируют и гидролизуют. N-Ацетил-аминогруппа является ориентантом I рода, но ее активирующее влияние на бензольное кольцо значительно меньше, чем влияние аминогруппы, так как неподеленная пара электронов атома азота участвует в сопряжении не только с л-электронной системой бензольного ядра, ио и с л-электронамн двойной связи карбонильной группы:
Нитрование. По сравнению с нитрованием ароматических углеводородов ннтрованне ариламннов имеет ряд особенностей. Так, ароматические амины легко окисляются концентрированной азотной кислотой, поэтому прямое нитрование их азотной кислотой осуществить невозможно. Прн использовании в качестве нитрующего реагента нитрующей смеси ариламяиы, наряду с частично протекающими окислительными процессами, превращаются в арнл-аммонийиые соли. Аммонийная группа, являясь электроноакцепторным заместителем, затрудняет нитрование и способствует образованию преимущественно лета-изомера:
218
анилиннй jw-нитроанилиннй
гидросульфат гидросульфат
С целью защиты аминогруппы от процессов окисления и протонирования по атому азота ароматические амины предварительно ацетилируют. N-Ацетильные производные в отличие от аминов являются очень слабыми основаниями и даже в сильнокислой среде реагируют в непротонированной форме. Вместе с тем аце-тиламииогруппа сохраняет электронодонорные свойства и ориентирует нитрование в орто- и пара-положения. Соотношение орто-и пара-изомеров зависит от состава нитрующей смесн и условий проведения реакции. После нитрования N-ацильные производные гидролизуют в кислой или щелочной среде:
ЫНСОСНз
НЬОз
H2SO4
анетаннлид
NHCOCHa
о-ннтроац.етаиилид
NHCOCH3
NO2
»i нмтроацетанилнд
нон, 1Г
—сн^соон
НОН, и* —CHjCOOH
219
Сульфирование. При нагревании анилина с концентрированной серной кислотой в среде высококипящего растворителя образуется пара-аминобеизолсульфокислота, которую чаще называют сульфаниловой кислотой. Реакция протекает через стадию образования N-феиилсульфаминовой кислоты, которая перегруппировывается в пара-аминобензолсульфокислоту:
аиилииий гипрскутгифат N-фснилсульфами- сульфаниловая
новая кислота кислота
Наличие в молекуле сульфаниловой кислоты кислотного (группа SOgH) и основного (группа NH2) центров обусловливает ее существование в виде внутренней соли (биполярного нона)
H3N—SO,’
Сульфаниловая кислота является довольно сильной кислотой (рля3,22) С основаниями она легко образует соли;
SOf + NaOH —> S^Na + H2O
<1 аминобензолсульфоннт натрин
Из-за биполярной структуры сульфаниловая кислота не образует солей с минеральными кислотами, т. е, несмотря на наличие аминогруппы она не обладает основными свойствами.
Сульфаниловая кислота широко применяется в производстве красителей и лекарственных средств. Она является структурным фрагментом большой группы лекарственных препаратов антибактериального действия, известных под названием сульфаниламиды. Родоначальником сульфаниламидных препаратов является амид сульфаниловой кислоты, применяемый в медицинской практике под названием стрептоцид:
стрептоцид амид Сульфзииюэой кислоты
В качестве исходного вещества для получения стрептоцида применяют аннлнн. Синтез осуществляют в четыре стадии:
220
nh2
NHCOCH3
Адили ром пне
+ (СНзСО)2О
-------->
-СНэСООН
ацетанилид
Су льфохл&рнрова нме
NHCOCH3 NHCOCH3
SO2C1
хлорсульфоновая п аиетамндобензол
кислота сульфахлорид
Амидирование
NHCOCH3
SO2NH2
NHCOCHj
п анетамндобензол сульфамид
NHCOCH3 nh2
Гидролиз
нон н* —СНэСООН
SO2NH2 SO2NH2
п амннобензол сульфамид
С целью защиты аминогруппы от побочных реакций, протекающих при сульфохлорировании, предварительно аиилин ацилируют. В качестве ацилирующего реагента чаще всего используют уксусный ангидрид. Образовавшийся ацетанилид затем подвергают сульфохлорированию
Сульфохлорированием называют процесс введения в молекулу органического соединения хлорсульфо-нильиой группы — SO2CI-
Сульфохлорирование осуществляют действием хлорсульфоновой кислоты HOSO2CL При взаимодействии ацетанилида с хлорсульфоновой кислотой образуется л-ацетамндобензолсуль-фохлорид (хлораигидрид л-ацетамидобензолсульфокислоты), который обрабатывают аммиаком. В результате реакции с ам
221
миаком атом галогена в хлорсульфонильной группе замещается на аминогруппу и образуется ц-ацетамидобензолсульфамид, который затем подвергают гидролизу в кислой среде с целью снятия ацетильной защиты. В процессе гидролиза образуется л-амино-бензолсульфамид (стрептоцид).
В отлнчие от сульфаниловой кислоты ее амид является амфотерным соединением, способным образовывать соли как с минеральными кислотами, так и со щелочами;
п-ачинобгн'юлсульфон.амид натрия
Замещением атомов водорода в сульфамидной группе молекулы стрептоцида различными радикалами, чаще имеющими гетероциклическую природу, получены другие лекарственные препараты, обладающие антимикробной активностью, например:
В. Окисление ариламннов
В присутствии кислорода воздуха анилин окисляется до хино-ннмина, приобретая при этом бурую окраску:
NH2
+ о2
+ н2о
222
Под действием таких окислителей, как монопероксисерная кислота H2SO5(кислота Каро) или пероксид водорода в уксусной кислоте, первичные ароматические амины окисляются до нитрозосоединений. Прн окислении трифтор л ер окси уксусной кислотой образуются нитросоединения:
нитробензол
12.4. ДИАМИНЫ
Диамниамн называют соединения, содержащие в своем составе две аминогруппы, связанные с углеводородным радикалом.
В зависимости от природы углеводородного радикала различают алифатические и ароматические диамины:
NH2
СНэ—СН—СН2
nh2 nh2
пропанздаиин 1 .‘2
СН2—CHj—CHj—CHj
I I
nh2 nh2
о феннленаианкн
б^танднгмнн 1,4, тетра мети.теиднам ин *
12.4.1 . Способы получения
Диамины получают аналогично моноаминам, используя в качестве исходных веществ соответствующие бифункциональные производные.
Аммонолиз дигалогеналканов (см. с. 203):
Вг—СН^СНз—Вг + 4NH.3 ->
1.2-днбрймэтаи
> H2N—CHj—CHj— NH2 + 2NH,Br этанднамнн 1,2
* Если аминогруппы расположены по концам неразветвленной цепи, то в названии греческими числительными указывают число метиленовых групп, добавляя суффикс -диамин,
I
223
Восстановление динитрилов (см. с. 204):
N=C—СН?—СН2—СН?—СН?—C=N + 4Н2 динитрил адипиновой кислоты
---► H2N—СН?—(СН2)4- -СН2—nh2
гексаметнленднамкн
Восстановление нитроанилинов с. 194):
или динитробензолов (см.
знг
—2Н2О
о нНТроаннлин
о фенилендиамин
л-дикитробензол
м фениленднамин
12.4.2 . Химические свойства
По химическим свойствам днамнны сходны с моноаминами. Особенностью их химического поведении является лишь то, что реакции могут протекать по одной или по двум аминогруппам. Твк, диамины образуют соли с одним нли двумя эквивалентами кислот; с участием одной или двух аминогрупп они вступают в реакции алкилирования, ацилирования н др. Из-за присутствия двух аминогрупп диамииы являются более сильными основаниями, чем моноамины.
Для некоторых диаминов характерны специфические реакции, связанные с образованием гетероциклических структур. Например, при нагревании хлороводородных солей тетраметилендиамина и пентаметилендиамина происходит их внутримолекулярная циклизация и образуются соответственно пяти- и шестичленные гетероциклические соединения — пирролидин и пиперидин:
СНа—СН2—NH3CI
СН2—СН2—NH3C1
^сн2 сн, \ I * сн2 /
"'СН,
тетра мети лендиаммоннй
хлорид
ПИррОЛНДИНА
гидрохлорид
224
СНг-СНг— NHaCl cfh + —
CHa—CHs— NH3CI
^CH2
ch2vch2
сн2/ён2 -hci + nh4ci
I H
nsMTa метиле одним ионий хлорид
пиперидина гидрохлорид
Хлороводородная соль этилендиамина в этих условиях вступает в реакцию межмолекулярной циклизации, образуя гетероцикл пиперазин:
NH3 Cl
СН2
I
СН2
Cl
C1H3N + \:н2
I
CH2
CIH3N
NH
cfC \:h2
I I - 2HC1 4- NH4CI
CH2 CH2
гтиггеразнпа ди» ндрохлорнд
о-Фенилендиамнн вступает в реакции конденсации с а-диаль-дегидами, днкетонами, альдегидо- и кетокислотами, а также карбоновыми кислотами, образуя гетероциклические продукты. Так, при конденсации о-фенилендиамнна с глиоксалем образуется хиноксалин:
е-фениленднамин глиоксаль
хиноксалин
В процессе конденсации с карбоновыми кислотами образуются производные бензимидазола:
о- фенилендиамин
2-метилбензимидазол
В 5 104
225
12.5. ИДЕНТИФИКАЦИЯ АМИНОВ
12.5.1. Химические методы
Первичные алифатические н ароматические амины можно обнаружить с помощью изонитрильной реакции (см. с. 210) по характерному неприятному запаху образующихся изонитрилов
R -NH2 + СНСЬ + 3NaOH > R— N=C + 3NaCl + 3H2O
нзонитрил
Для обнаружения первичных ароматических аминов используют реакцию диазотирования (см. с, 231) с последующей конденсацией образовавшейся солн диазония с р-нафтолом (см. реакцию азосочетання, с. 237). При этом образуется азокраситель красного цвета
NaMO2 4- HCI (изб }
\=NC1 + NaCl 4- Н2О
бсггтолдназоний клорит
р нафто
азикрасител!
Первичные, вторичные и третичные амины алифатического н ароматического ряда можно отличить друг от друга реакцией с азотистой кислотой (см. с. 209).
Идентификацию первичных н вторичных аминов часто осуществляют через образование N-ацильных производных. Для характеристики алифатических аминов обычно получают N-бен-зоильные производные, для ароматических - N-ацетнльные. Бензоильные производные легко образуются при обработке аминов бензоилхлоридом, а ацетильные — действием уксусного ангидрида:
СаН5—NH2 + С6Н5СОС1 -> С2Н5—NH—С—С6Н5 + НС1
бензоил хлорид
этиламид йевзоннон кислоты
226
о
C6H5—NH2 + (СН3СО)аО ----к С6Н5—NH—(Lch, +
ацет а ннлид.
4- СНзСООН
N-Ацильные производные аминов представляют собой кристаллические вещества с четкими температурами плавления.
12.5.2. Физические методы
В ИК-спектрах аминов наблюдаются полосы поглощения, обусловленные валентными колебаниями связи С—N. В алифатических аминах vc_ ч проявляются в области 1030- 1230 см-1, в ароматических - 1250—1300 см-1. ИК-спектры первичных и вторичных аминов имеют полосы поглощения в области 3320— 3550 см-1, отвечающие валентным колебаниям связи N—Н. У первичных аминов в этой области имеется две полосы поглощения, а у вторичных — одна. Образование межмолекулярных водородных связен приводит к смещению полос поглощения связи N—Н в область 3000—3330 см-1.
Метод УФ-спектроскопии применяется в основном для идентификации ариламинов Алифатические амнны поглощают УФ-излучение в дальней ультрафиолетовой области, которая малодоступна для измерения. Наличие в молекуле ариламина аминогруппы, связанной с бензольным ядром, приводит к значительному смещению полосы бензольного поглощения в длинноволновую область и увеличению ее интенсивности.
Гак, в УФ-спектре анилина бензольная полоса проявляется прн Хмаке 280 нм (е 1430).
В ПМР-слектрах первичных и вторичных аминов наблюдается широкий нерасщеплеиный сигнал в области 0,5—4,7 м. д,, отвечающий протонам групп NH2 и NH
12.6. ОТДЕЛЬНЫЕ ПРЕДСТАВИТЕЛИ. ПРИМЕНЕНИЕ
Метиламин CH3NH2. Газ с острым запахом, напоминающим запах аммиака, хорошо растворяется в воде. Насыщенный водный раствор содержит 35—40 % метиламина. Метиламин применяется в производстве лекарственных средств, красителей, инсектицидов, фунгицидов и др.
Тетраметилендиамин (путресцин)
HsN—СНйСНаСНзСНа—NH2 Кристаллическое вещество (т. пл. 27—28 °C), легко растворяется в воде. Образуется в процессе гниения белков прн ферментативном декарбоксилировании аминокислоты орнитина:
СН2-СН2-СН2-СН-СООН - » сн,-сн2~сн2-сн2
I I -СО2 I 2 2 2 I 2
nh2 nh2 nh2 nh2
орнитин путресцин
8’
227
Путресцин обладает неприятным запахом. Он также образуется при гнилостном разложении трупов, из-за чего его относят к группе птомаинов (трупных ядов). В организме человека путресцин используется для синтеза биологически активных полнаминов — спермидина и спермина, принимающих участие в регуляции биосинтеза нуклеиновых кислот и белков:
NHz— (СН2) ч—NH—(СН2) з— NH2
спермидин
NH2— (СН2)3—NH— (СН2)4—NH—(СНй)з— NH2
спермин
П еитаметилеиди а мин (кадаверин)
NH2—СН2(СН2)3СН2—NH2. Жидкость (т. кип. 178 °C), легко растворяется в воде. Образуется в процессе гниения белков при ферментативном декарбоксилировании аминокислоты лизииа
СН2—СН?—СНа—СНг—СН—СООН
nh2 nh2
СН2—С Hz— СН2—СН;>—сн2
nh2 nh2
калыверин
Кадаверин, как и путресцин, образуется при гнилостном разложении трупов и принадлежит к группе птомаинов.
Беизогексо н и й, [1,6-бис- (N-триметиламмоний)-гексана дибензолсульфонат] или N,N^i cKcaметил-1,6-гекса метилендиаммония дибензолсульфонат:
“НэС СН3~
НзС— N— (СН2) 6—fc^CHa \:н3
2CfiH5SOr
Белое кристаллическое вещество (т. пл. 196—202 °C), легко растворяется в воде. Применяется в медицине как ганглиоблокирующее и гипотензивное средство.
А нил и в СбН5\Н2. Бесцветная жидкость со своеобразным запахом (т. кип. 184,4 °C), легко окисляется кислородом воздуха, приобретая красно-бурую окраску. Анилин ядовит. Производится в больших количествах и широко применяется в синтезе красителей, пластмасс, лекарственных препаратов, фотоматериалов и др.
228
Фенамин, 1-фенилпропанамнн-2 —СН2—СН -СН3.
NH2
Белое кристаллическое вещество, малорастворнмое в воде. В виде сернокислой соли применяется в медицине как стимулятор центральной нервной системы.
NH2
орто- Феи и лен ди ам нн Бесцветное кри-
NH2
сталлическое вещество, темнеющее на воздухе (т. пл. 102—104 °C), хорошо растворим в воде. Широко применяется в производстве лекарственных препаратов (дибазол и др.), красителей, пестицидов.
КОНТРОЛЬНЫЕ ВОПРОСЫ И УПРАЖНЕНИЯ
1. Напишите структурные формулы следующих аминов: a) N, №-диэтилбутанамин-2; б) N, №-диэтил-4-этнланилин; в) а-нафтиламин; г) Ь1-этнлпропанамин-2; д) вгор-бутилметил-амин. Укажите среди них первичные, вторичные и третичные амины
2. Назовите по заместительной и радикало-функциональной номенклатуре следующие амины:
a) C2H5NHC2H5, б) C6H5NHC2H5; в) С6Н5— N--С2Н5
С>г1з
3. Напишите схемы реакций последовательного превращения метиламина в тетраметиламмоний бромид
4. Предложите четыре способа получения этиламина.
5. Расположите в порядке уменьшения основных свойств следующие амины: а) этилами н; б) диэтила мин; в) анилин; г) л-нитроанилин.
6. Приведите схемы реакций алкилирования и ацилирования н-пропиламина. На примере л-толуидина охарактеризуйте реакционную способность ароматических аминов с участием аминогруппы н бензольного кольца.
7. Напишите схемы качественных реакций, позволяющих отличить этиламин, анилин и N-этиланилин друг от друга.
8. Предложите способ получения л-нитроанилина из анилина,
9. Особенности реакционной способности диаминов.
Глава 13
ДИАЗО- И АЗОСОЕДИНЕНИЯ
13.1. ДИАЗОСОЕДИНЕННЯ
Диазосоединевиями называют органические вещества, содержащие в своей структуре группировку из двух атомов азота, связанную с углеводородным радикалом и остатком минеральной кислоты.
Общая формула диазосоединений RN2X, где R—углеводородный радикал, X — остаток минеральной кислоты (CI -, Вг , NOT, SO4H“, ОН ", С.\Г, SO3H ", SH"). В зависимости от природы углеводородного радикала различают алифатические и ароматические дназосоединения. В главе рассмотрены ароматические диазосоединения, имеющие большое значение в синтезе красителей, лекарственных препаратов, в фармацевтическом анализе н др.
Общая формула ароматических диазосоединсний ArN2X. В зависимости от природы кислотного остатка (аниона) связь между ArNa и X может быть ионной или ковалентной. Если X — остаток сильной минеральной кислоты (СР, Вг-, HSO4“, NOT), то диазосоединения имеют ионное строение и их называют соли диазо ни я — +
Аг—N=NX-. Если же X — остаток слабой минеральной кислоты (CN-, HSOr, ОН-, SH-) диазосоединення имеют ковалентное строение — Аг—N=N—X. Диазосоединення обшей формулы Аг—N=N—О‘М+, где М- металл, получили название диазота-ты. В растворе указанные формы диазосоединений в зависимости от pH среды могут превращаться друг в друга. В кислой среде диазосоединення существуют в форме солей диазония, в среде, близкой к нейтральной, имеют ковалентное строение, а в щелочной среде находятся в форме диазотатов. Так, при действии на соль диазония аодного раствора NaOH в нейтральной среде образуется диазосоединение ковалентного строения (диазогидроксид), которое в щелочной среде превращается в диазотат. И наоборот, под действием хлороводородной кислоты диазотат в нейтральной среде переходит в диазогидрокенд, а в кислой образует соль диазония;
кислая среда нейтральная среда
+ NaOH + — NaOH
Ar—N=NCI- Ф--------> Аг—N=NOH 7—Аг—N=N—ОН *
саль диазония НС) арнлдназогидроксид HCI
щелочная среда
< Аг-—N=N—О Na+
« 4 арилдназотат натрия
230
13.1.1. Номенклатура. Изомерия
Согласно номенклатурным правилам ИЮПАК названия ароматических диазосоединений образуют путем добавления к названию исходного углеводорода суффикса -диазо-, а названия солей диазония—добавлением окончания -диазоний с последующим указанием аниона:
СбН5— N-=N-- ОН
бензолдна I «гидрокеиД (диазогидрат)
Сбн5— N=N—O_Nar
бензолдиазотат натрия
4 метидбензолдназпний бромид
Ковалентнопостроенные диазосоединення могут существовать в виде двух геометрических изомеров — син-(цис-) и анти-(транс-), из которых более устойчивой является анти-форма (см. кн. I, с. 89):
сан-диазосоединение анти-диазосоединение
13.1.2. Способы получения солей арендиазония
Реакция диазотирования. Эта реакция, открытая в 1858 г. немецким химиком П. Гриссом, основана иа взаимодействии первичных ароматических аминов с азотистой кислотой в среде сильной минеральной кислоты, чаще хлороводородной или серной. Поскольку сама азотистая кислота очень неустойчива, на практике обычно ароматический амин обрабатывают раствором соли азотистой кислоты (NaNChi KNOa) в присутствии сильной минеральной кислоты, причем на один моль амина используют не менее 2,5 моля минеральной кислоты. Один моль минеральной кислоты расходуется на получение азотистой кислоты, один — на образование соли диазония, а избыток кислоты необходим для создания кислой среды. Минеральная кислота принимает также участие в растворении ароматического амииа, однако в реакции диазотирования вступает не соль амина, а находящийся в равновесии с ней в небольшом количестве свободный амин:
+
CeH5NH3Cl - C6H5NH2 + HCI
231
Реакция диазотирования экзотермична, н поскольку соли диазония при нагревании легко разлагаются, диазотирование проводят при температуре 0—5 °C:
NaNO;. H^t (изб ) 0—5 “С
—N=NCI + NaCl + Н2О
бенлоииазоний хлорит.
Механизм реакции. При взаимодействии NaNO2 с минеральной кислотой образуется азотистая кислота, которая в кислой среде образует несколько диазотирующих агентов — + ' протонированную азотистую кислоту Н—О—N=O( ннтрозоний-
+ Н ‘
катион N=O и оксид азота (III) N2O3
Н—б— N=O + Н+ нДэ—N=O NO + Н2О
протонированная нитроэонин- 7
азотистая кислота катион
I + + ?' NO -Н :О— N=O О—N—О—N=O ------------------> N2O3
I I
H H
171 ° оксид азота (III)
Наиболее активным среди них является нитрозоиий-катион. Механизм реакции, в которой он выступает в качестве диазотирующего агента, можно представить следующим образом.
Вначале нитрозоний-катион атакует атом азота ароматического амина с образованием N-нитрозоамина, который в кислой среде легко переходит в свою таутомерную форму — диазогндроксид. Затем диазогидроксид в кислой среде превращается в соль диазония:
+ | 1
С6н5—NH2 4- NO С6Н5— N—N=O CgH^-N^N^
N-нитрозоамин
^±С6Н5—N=N—ОН C6HS—N=NCT -f- Н2О
1на шгидроксид соль диазония
В результате реакции получают водные растворы солей диазоиия, которые непосредственно используют для дальнейших реакций.
232
Взаимодействие первичных ароматических аминов с алкил-нитритами. При действии на первичные ароматические амины эфиров азотистой кислоты в присутствии минеральной кислоты в среде этанола образуются соли диазония в кристаллическом виде:
+
С6Н5— NH2 + СаНб—ONO + НС1 --------------- СЬН5— N=ssNCI“ +
этн тннтрит бенли.инадэний
хлорид
+ С2НБОН + Н2О Применение солей диазония в кристаллической форме ограничено из-за их взрывоопасности.
13.1.3. Физические свойства солей диазония
Соли диазония — бесцветные кристаллические вещества, легко растворимые в воде. Они нестабильны, прн нагревании и механических воздействиях разлагаются со взрывом. Поэтому в реакциях обычно используют их свежеполученные водные растаоры.
13.1.4. Химические свойства солей диазония
Реакционная способность солей диазония обусловлена наличием в их структуре диазокатиона. Методом рентгеноструктурного анализа установлено, что атомы азота в диазокатионе расположены линейно в плоскости бензольного кольца. Связь между атомами азота по длине соответствует тройной (0,11 нм). Эт© свидетельствует о том, что оба атома азота находятся в состоянии sp-гибридизацнн. При этом один нз них содержит положительный заряд, а второй — имеет нсподеленную пару электронов (формула I). Положительный заряд катиона делокализован в основном между атомами азота и лишь частично за счет л-электроиной системы бензольного кольца (формула II):
I II
В результате делокализации каждый из атомов азота приобретает частичный положительный заряд, что можно изобразить с помощью резонансных структур
Реакции с участием солей диазония можно условно разделить иа две группы: с выделением азота и без выделения азота.
233
А. Реакции с выделением азота
Реакции солей диазония с выделением азота сопровождаются разрывом связи С—N в диазокатионе и замещением диазогруппы на другие атомы или группы атомов. В зависимости от способа разрыва связи С—N замещение диазогруппы может происходить по механизму 8Ч1 или SR.
Механизм SN1 представляет собой двухстадийиый процесс. На первой, медленной, стадии происходит гетеролитический разрыв связи С—N в диазокатионе с образованием очень неустойчивого арил-катнона и молекулы азота. На второй стадии арил-катион быстро взаимодействует с нуклеофилом, образуя продукт замещения +
Аг— Nh=NX мелленн°> Аг+ + N2 + Х“
арил-катион
Ar+ -J- Nu- —Ar—Nu
Протеканию реакций по механизму SN1 способствуют нагревание и растворители, обладающие высокой сольватирующей способностью.
Замещение диазогруппы по радикальному механизму происходит в присутствии катализатора — солей одновалентной меди или порошкообразной меди. Как металлическая, так и одновалентная медь способны выступать в качестве донора электронов.
На первой стадии реакции ион меди (I) отдает один электрон дназокатнону, превращаясь в ион меди (II), При этом в диазокатионе происходит гомолитический разрыв связи С—N с образованием арильного радикала и молекулы азота. На второй стадии, арильный радикал отрывает атом галогена от галогенида меди (11), регенерируя тем самым галогенид меди (I)
+
С6Н6— NasNCr + CuCI ------СеНв + N2 + CuCI2
C6Hs + Cl • : • Cu—Cl ----> С6НЯ—Cl + CuCI
Реакции солей диазония с выделением азота позволяют ввести в ароматическое ядро различные заместители, такие, как ОН, F, CI, Вг, I, CN, NO2, OR и др.
Замещение диазогруппы на гидроксильную группу. Для замещения диазогруппы на гидроксильную группу подкисленные водные растворы солей диазония подвергают кипячению. При этом выделяется азот и образуются фенолы. Реакцию можно проводить с растворами любых солей, но лучше с этой целью использовать гидросульфаты. Взаимодействие происходит по механизму <S\1
+
C6H5N=NHSOr + Н2О н1>'>С5НбОН + N2 + H2SO4 бенэолдназоний гидросульфат фенол
234
Чтобы предотвратить реакцию азосочетания (см. с. 237) между солью диазония и образующимся фенолом, обычно раствор солн диазония прибавляют по частям к кипящей разбавленной H2SO4.
Замещение диазогруппы на атом иода. При нагревании растворов солей диазония с раствором иодида натрия или калия, диазогру ппа замещается на атом иода. Реакция является одним из наиболее удобных методов введения иода в ароматическое ядро
С6Н5— №NCI“ + KI —C6H5I + N2 + КС1 бенчолдиаэоний иодбснюл
хлорид
Замещение диазогруппы, катализируемое солями одновалентной меди (реакция Зандмейера). При каталитическом действии солей одновалентной меди диазогруппа может быть замещена на атом хлора, брома, нитрогруппу, цнаногруппу, хлорсульфонильную группу SO2CI и др. Реакции названа в честь швейцарского химика Т. Зандмейера, который в 1884 г. нз солей диазоння в присутствии солей одновалентной меди получил хлор- и бромареиы. Замещение протекает по механизму SR
— ‘ > C6H5C1 + n2 хлорбензол
kb., cusr, , >CaH5Br + n2 _|_ KCI бромбснэол
+
СвН5— N=NC1“
бензолдняjoний хлорид
KCN ^VCsHsCN + Ns + KCI бензонитрил
Na-°2- Cu-Qa- >C6H5NO2 + N2 + NaCl нитробензол
-' C“C1,1 ► C8H8SO2C1 + Ns
бен юл vульфохлорид
Замещение диазогруппы на атом водорода. При нагревании солей диазония с восстанавливающими агентами, такими, как фосфорноватистая кислота НэРОа, формальдегид, спирты и др., диазогруппа замещается на атом водорода. В результате соль диазония превращается в ароматический углеводород. Реакция идет по механизму SH
ь
С6Н5—N=NCr 4- Н3РО2 4- Н2О—
—►- СбНе 4" N2 4~ НС! 4~ Н3РО3
236
При использовании в качестве восстановителя спиртов наряду
с восстановлением солн диазония происходит побочная реакция с образованием простых эфиров: О
---► С6Н6 + N2 + HCI + СН:1—
h - \
С6Н6— N=NCI + С2Н5ОН -L Н
---С6Н&—О—С2Н5 + N2 + HCI . фенил этиловым
эфир
Реакция замещения диазогруппы на атом водорода применяется в органическом синтезе как косвенный метод удаления аминогруппы из ароматического ядра после ее использования для необходимой ориентации замещения. Например, такое соединение, / как 1,3,5-трибромбензол, невозможно получить прямым бромированием бензола. Однако это соединение легко можно получить нз анилина через образование 2,4,6-триброманилина с последующим диазотированием и восстановлением соли диазония:
ЗВгг ----- ►
-ЗНВг
2 4 fl трнброчмнллнн
ЩйОз НгО
_\j -ЩР04. —НСГ
2 4 6 трибромбеизол диазиний члорий
1.3 5 трибрючбеизол
Б. Реакции без выделения азота
Образование диазопроизводных. Являясь электрофильными реагентами, соли диазония аступают в реакции с различными нуклеофильными реагентами, образуя диазопроизводные. Так, прн добавлении к раствору соли диазония щелочей образуются диаэо-таты. С первичными и вторичными аминами в слабощелочной
236
и нейтральной средах соли диазония образуют диазоаминосоединения (триазены). При действии на соли диазония цианидов щелочных металлов образуются диазоцианиды:
+
CeHs—N==NC1-
СЬН5—N=N—ONa нагрнн iHaiOTar
-СН,~7'' > С6Н5—N=N—NH—СНз
3 «етнл 1 фенилтрна ieu
> СвН5—N=N—CN
ОСНЭПЛДМа jOUHdHI[±l
Восстановление солей диазония. При восстановлении солей диазония в мягких условиях, например действием хлорида олова (II) в хлороводородной кислоте или цинковой пыли в уксусной кислоте, образуются арилгндразины:
+ |Н]
СбН5—N=NCr------------L-L—-*С6Н5— NHNHa.HCI + 2SnCI<
2SnCIj + 4НСГ 1
феннлгидрадина ,илрохлорид
Реакция азосочетания. Соли диазония реагируют с фенолами в слабощелочной среде и ароматическими аминами в слабокислой среде, образуя азосоединения Аг—N=N—Аг. Эта реакция получила название реакции азосочетания:
бензолдиазоний хлорид
фр НОЛ
4 гидроксиазабензол
По механизму взаимодействия реакция азосочетания относится к реакциям электрофильного замещения в ароматическом Ряду Атакующим электрофильным реагентом в реакции выступает диазокатион. Поскольку диазокатион является относительно слабым электрофилом, он способен реагировать только с очень реакционноспособными ароматическими соединениями. К ним относятся вещества, содержащие в ароматическом ядре сильные электронодонорные группы —ОН, NH?, NHR, NRa- Указанные
237
заместители за счет проявления 4-/И-эффекта увеличивают электронную плотность в бензольном ядре и тем самым активируют его в реакциях SE Замещение идет главным образом в пара-положение по отношению к активирующей группе, если же это положение занято, происходит орто-замещение Соль диазония в реакции азосочетания называют диазосоставляющей (диазокомпонента), а фенол или ароматический амин — азосоставляющей (азокомпонента) Активность диазосоставляющей возрастает при наличии в орто- или паря-положениях бензольного кольца электроноакцепторных заместителей, например NO?, SO3H, СООН
Большое влияние на протекание реакции азосочетания оказывает pH реакционной среды, т е ее кислотность или щелочность
Реакцию с фенолами проводят в слабощелочной среде, где они превращаются в более реакционноспособные феноляты, поскольку группа —О' обладает ббльшнми электронодонорнымн свойствами, чем группа —ОН'
Сильно щелочная среда недопустима, так как в этих условиях соль диазония превращается в диазотат, который не аступает в реакцию азосочетания
С ароматическими аминами реакцию азосочетания проводят в слабокислой среде (pH 5—7) В сильнокнслой среде реакция не идет, поскольку в этом случае аминогруппа практически полностью превращается в аммонийную, которая дезактивирует бензольное ядро в реакциях SE
Катион диазония, являясь электрофильным реагентом, атакует в бензольном кольце ароматического амина или фенола атом угле-^1 рода с наибольшей электронной плотностью, образуя n-комплекс^ который отщепляет протон и превращается в азосоединение:4
tUn + <T=z>-N(CH3)2 -----►
о-комплекс
В случае первичных и вторичных ароматических аминов, наряду с атакой бензольного кольца диазокатион конкурентно } атакует атом азота аминогруппы В результате этой атаки обра-'>
238
зуются диазоаминосоединеиия (трназены), которые при нагревании в кислой среде подвергаются перегруппировке с образованием азосоединений
диазоаминобензол
перегруп-
пировка
4-амииоазобензол
Используя в реакции азосочетания различные диазо- и азосоставляющие, можно получить большое число азосоединений Ввиду того, что все азосоединения являются окрашенными веществами, реакция азосочетания широко применяется в фармацевтическом анализе для подтверждения подлинности лекарственных препаратов, содержащих в своем составе первичную ароматическую аминогруппу
132 АЗОСОЕДИНЕНИЯ
Азосоедннеинямн называют органические вещества, содержащие в своем составе группировку —№=№— (азогрувпу), связанную с двумя углеводородными радикалами.
В зависимости от природы углеводородного радикала, различают алифатические и ароматические азосоединения Наиболее важное значение имеют ароматические азосоединения
Ароматические азосоединения имеют общую формулу Ar— N=N—Аг'
Названия азосоединений с одинаковыми углеводородными радикалами составляют из префикса азо- и названия углеводорода Положение заместителей в углеводородных радикалах обозначают цифрами или локантами орто-, мета- и пара
НО
азобепки
2 гидрокси 4 метила юбеизол о гидрокси и метилааобензоп
Азосоедииения с разными углеводородными радикалами при азогруппе рассматривают как производные углеводорода с более
239
сложной структурой,^содержащего в качестве заместителя ареназогруппу
(Ar—N^N—):
Б
5
2 бензолазонафтатин
N=N
1 !4' пнтрпбенчолло] нафтол 2
Как и другие соединения, содержащие группировку —N=N—, азосоединения существуют в виде двух геометрических изомеров — сил- (цис-) и анти- (транс-). Так, азобензол (т. пл. 68 °C) представляет собой анти-изомер. При облучении его УФ-светом он переходит в син-изомер (т. пл. 71 °C):
untu азобензол
СбН5
сын в юбеи юл
Син-изомер нестабилен и быстро превращается в анти-изомер.
13.2.1. Способы получения
Реакция азосочетания (см. с. 237). Это наиболее важный способ получения азосоедииеиий, который широко используется в промышленности.
Восстановление нитроаренов в щелочной среде. При восстановлении нитроаренов в щелочной среде азосоедннения образуются, в качестве промежуточных продуктов (см. с. 195). Однако в определенных условиях, и а частности под действием цинка в щелочной среде, процесс восстановления может быть остановлен на стадии образования азосоединения
(И|
2С6Н5—NO2-------------ь С6Н5—N=N—С6Н5
(7п f NaOHJ
азобензси
13.2.2. Химические свойства
Ревкциоиная способность азосрединений обусловлена наличием в их структуре азогруппы —N=N—. За счет неподелениых пар электронов на атомах азота азогруппы азосоединения проявляют слабые основные свойства. В присутствии минеральных кислот азогруппа протоиируется:
240
Сбн5—N==N—<бН5 + НС1 С6НБ—N=N—С6Н5 + СГ
н
С участием азогруппы азосоединения вступают в реакции окисления и восстановления.
Под действием пероксикислот азосоедииения окисляются с образованием азоксисосдииений
foj -+
СбНз—N=N—CgHs-------------*- CeHs—N=N—СеН&
R—С- ООН |
А 6-
азобензол аэпксибен юл
При восстановлении азосоедииений в мягких условиях, например действием цинка в растворах щелочей, образуются гидразо-соединения
|н)
СбН5— N=N—С6Н5---------------> С6Н5—NH—NH—С6Н5
(Zn + ЧаОН}
азобензол гидразобензол
Восстановление хлоридом олова (II) в среде хлороводородной кислоты приводит к образованию ариламннов:
Азосоедииения представляют собой кристаллические вещества, окрашенные а желтый, оранжевый, красный, синий и другие цаета. Это свойство позволяет использовать многие из них в качестве химиотерапевтических средств, например салазопиридазин и сала- <
салааопиридаэнн
241
13.2.3. Основные положения теории цветности. Азокрасители
Теория цветности рассматривает зависимость окраски органических соединений от строения их молекул.
Окраска любого вещества обусловлена его способностью поглощать электромагнитное излучение в видимой области спектра (400—760 нм). При этом человеческий глаз воспринимает предмет окрашенным в дополнительный цвет к поглощаемому (табл. ) 3.1).
Таблица 13 1
Спектральные и дополнительные цвета
Длина волны, нм Спектральный цвет (поглощенное излучение) Дополнительный цвет (цвет вещества)
400—435 435 —480 480- 490 490 -500 500—560 560—580 580—595 595 605 605- 730 730-760 Фиолетовый Синий Зеленовата-скний Синевато-зеленый Зеленый Желтовато зеленый Желтый Оранжевый Красный Пурпурный Зеленовато-желтый Желтый Оранжевый Красный Пурпурный фиолетовый Синий Зслсновато-синий Синевато-зеленый Зеленый Углубление окраски (батохромный сдвиг) Повышение окраски (гипсохромяый сдвигу
I loi лощение веществом света в узком диапазоне длин волн приводит к появлению ярких окрасок, например красной, синей, зеленой и др. Если вещество поглощает свет в широком диапазоне андимой области, появляются неяркие окраски (коричневая, бордо, хаки и др.). При поглощении веществом практически всей видимой области спектра появляется черная или серая окраска. Вещества, не поглощающие излучение в видимой области, являются бесцветными.
Поглощаемая молекулой энергия в видимой области расходуется на возбуждение образующих л-связи электронов (п-элект-роиов) и неподелениых электронов (п-электронов), которые при этом переходят иа более высокоэнергетичные разрыхляющие л*-М0 (см. кн. 1, с. 109-111). Таким образом, поглощение в видимой области и связанное с ним появление окраски обусловлено л -► л*- и п л*-переходамн. Особенно легко осуществляются л -* л*-переходы в сопряженной системе кратных связей.
Структурные фрагменты молекулы, поглощающие излучение а видимой области, т. е. являющиеся согласно хромофорной теории О. Витта (1876 г.) ответственными за окраску, получили название хромофоры (от греч. «хромое» — цвет и «форос» — носитель). Основными хромофорами являются: достаточно длинная сопряженная система кратных связей, азогруппа —N=N—, хиноидная группа ==<\^/>==’ литрогруппа —NO2, иитрозогруппа —N=O 242
и др. Для появления окраски часто достаточно одного такого хромофора в структуре молекулы. Если молекула содержит несколько хромофоров, включенных в единую цепь сопряжения, интенсивность окраски увеличивается.
Одиако для того чтобы вещество стало красителем, в его структуре должны присутствовать группировки, которые без хромофоров не способны вызвать окраску, но находясь с ними в единой сопряженной системе, усиливают окраску. Такие группы получили название ауксохромы (от греч. «ауксео» — увеличиваю и «хромое»— цает). К ним относятся группы ОН, NHa, NHR, NRs, OR, SH и др- Ауксохромами обычно являются заместители, обладающие -j-/И-эффектом. Ауксохромные группы не только усиливают окраску красителя, но н способствуют его взаимодействию с окрашиваемым материалом, что повышает устойчивость окраски к действию моющих средств.
По химическому строению различают иитро-, иитрозо- азокрасителя, трифенилметановые, антрахиноновые, индигоидные и др. Наиболее распространенным классом красителей являются азокрасители. Их применяют для крашения шерстяных и искусственных тканей, кожи, бумаги и т. д. Окраска некоторых азокрасителей изменяется в зависимости от pH среды, что позволяет использовать их в качестве индикаторов. Примером такого азокрасителя является метиловый оранжевый (метилоранж, гелнаитни}
NaO3S-^^^^N=N-^^>—N(CH3) 2
метиловый оранжевый
Метилоранж получают азосочетанием диазотированной сульфаниловой кислоты с ЧМ-диметиланнлнном.
цетиловый оранжепый (красная форма)
NaO3
—N(CH3)S
метиловый оранжевый (желтая форма)
243
В нейтральной и щелочной средах метилоранж имеет желтое окрашивание, в кислой среде желтая окраска изменяется на красную вследствие образования хиноидной структуры:
красная форма
Желтая окраска метилоранжа в нейтральной и щелочной средах обусловлена наличием в его структуре длинной сопряженной системы, в состав которой входит хромофор — азогруппа —N=N—. В кислой среде вследствие присоединения протона к одному из атомов азота азогруппы происходит электронная перестройка сопряженной системы, при которой азогруппа исчезает, ио появляется более сильная хромофорная группа — хиноидная. Это приводит к углублению окраски. Интервал перехода окраски от красной до желтой находится в диапазоне pH среды от 3,1 до 4,4. В качестве кислотно-основного индикатора применяют также азокраситель - метиловый красный
тли
СНэ
мети^овин красным
4Г аниетнчачиниэзобомзол 2 карбиноаая кис юга
В щелочной среде метиловый красный находится в виде азоформы, имеющей желтую окраску, а в кислой — переходит в хино-
идную форму с красной окраской СООН ( желтан форма СНэ ЭНС1 2NaOH CHj
244
Интераал перехода от красной окраски к желтой находится в пределах pH 4,2—6,2.
КОНТРОЛЬНЫЕ ВОПРОСЫ И УПРАЖНЕНИЯ
I. Напишите структурные формулы следующих соединений; а) 4-нитробензолдиазоний гндросульфат; б) 2-метоксибеизолдиа-_ зоциаиид; в) 4-метилбензолдиазотат натрия; г) 4-метилбензол-диазотат натрия; д) 4-гидрокси-3'-бромазобензол.
2. Приведите схему превращений бензолдназогндроксида а кислой и щелочной средах.
3. Напишите схему и механизм реакции получения 4-метил-беизоддназоиийхлорида нэ и-толундина.
4. На примере 4-бромбензолдназоннйхлорнда приведите схемы реакций» позволяющих осуществить замену диазониевой группы на следующие группы: а) ОН; б) ОС2Н5; в) CN, г) NO2; д) SO2CI; е) NHNHg, Назовите полученные соединения.
5. Приведите схему получения 1,3,5-трибромбеизола из анилина
6. Напишите схему реакции азосочетаиия между N,N-диметил-анилииом и диазотированной сульфаниловой кислотой (4-диазобензолсульфокислота) Назовите полученное соединение.
7. Приведите схему превращений метилового оранжевого в кислой и щелочной средах. Объясните причину изменения окраски метилоранжа в зависимости от pH среды.
8. Какие функциональные группы а органических соединениях выступают в роли хромофоров, какие выполняют роль ауксохромов?
Глава 14 ГИДРОКСИЛЬНЫЕ ПРОИЗВОДНЫЕ УГЛЕВОДОРОДОВ. ПРОСТЫЕ ЭФИРЫ И ИХ ТИОАНАЛОГИ
Гидроксильными производными называют производные углеводородов, в которых один или несколько атомов водорода замещены на гидроксильную группу.
В зависимости от типа гибридизации углеродного атома, непосредственно связанного с гидроксильной группой, гидроксильные производные делят иа спирты (гидроксильная группа находится при атоме углерода в $рэ-гнбрндизации) и фенолы (гидроксильная группа расположена при sp2-гнбридизованиом атоме углерода, аходящем в ароматическую систему) Соединения с гидроксильной группой при $р2-гибридизованиом атоме углерода, ие входящем в ароматическую систему, получили название енолы Такие вещества, как правило, неустойчивы Гидроксильные производные с группой —ОН при атоме углерода в sp гибридизации неизвестны
В зависимости от числв гидроксильных групп в молекуле различают одно-, двух-, трех- и полиатомиые спирты и фенолы
По расположению гидроксильной группы в углеродной цепи спирты класси фи цирмот на первичные (группа —ОН расположена при первичном атоме углерода), вторичные (гидроксильная группа расположена при вторичном атоме углерода) и третичные (группа —ОН находится при третичном атоме углерода)
СНз—СН2—ОН
Н3С
^СН—ОН НзС^
н3с
Н3С—с—он
НзС^
1ервнчным ctiHpT
вторичный спирт
третичный спирт
Прн замене кислородного атома гидроксильных производных углеводородов иа атом серы образуются серные аналоги гидро ксильных производных, которые называют тиолами (тиоспирты и тиофеиолы) Иногда для этих соединений используется название — меркаптаны Функциональная iруппа —SH, которая входит в состав тиолов, называется тиольной или меркаптогруппой.
246
14.1. ОДНОАТОМНЫЕ СПИРТЫ
Одноатомными спиртами * называют гидроксильные производные углеводородов, содержащие одну гидроксильную группу, связанную с атомом углерода в sp^-гибриднэации.
В соответстаии с природой углеводородного радикала спирты подразделяют на три группы
насыщенные (алканолы и циклоалканолы) —гидроксильные производные алкаиоа и циклоалканов,
ненасыщенные (алкенолы, алкинолы, цнклоалкеиолы и др ) — гидроксипронзводные ненасыщенных углеводородов, у которых гидроксильнвя группа не находится при кратной связи, ароматические (арилалкаиолы) — гидроксильные производные ароматических углеводородов с группой —ОН в боковой цепи
14.1.1. Номенклатура спиртов
Для названия спиртов чаще всего применяют заместительную и радикало-функциональную номенклатуру ИЮПАК
По заместительной номенклатуре название спирта образуют из названия углеводорода, соответствующего главной углеводородной цепи, к которому прибавляют суффикс -ол с указанием положения гидроксильной группы в цепи углеродных атомов Нумерацию главной углеродной цепи начинают с того конца, к которому ближе расположена гидроксильная группа
Например
2 1 3 2 1 4 3 2 1
СНз—СН2—ОН СНз—СН—СН3 СН2=СН—СН—СНз—ОН
ОН
СНз
этанон
проламп 1 2
2 чети твитен 3 ол 1
СНз—с=с- СН2—С Hz—ОН < С Hz—С Hz— он
пентин 3 ол I
2 фенил этанол
циклогексанот
Очень часто одноатомные спирты называют просто «спиртами»
247
По радикало-функциональной номенклатуре название спиртов образуют из названия углеводородного радикала, связанного с гидроксильной группой, к которому добавляют -овый и спирт,
например:
С2Н5—ОН
Н3С
НзС—С—ОН
ИзС^
СНз—ОН
бензиловый спирт
этиловый спирт трет бутиловый спирт
СН2=СН—сн2—ОН
аллиловый спирт
Иногда для названия спиртов используют рациональную номенклатуру, согласно которой спирты рассматривают как производные метилового спирта (СН3ОН), получившего название карбинол, например:
СН3-СН-ОН
метилкарбинол
трифенилкарбинол
CH3-CH2-CH-OHJ сн3 метилэтилкарбинсф г
Для некоторых спиртов применяют тривиальные названия:
СНзОН С2Н5ОН
древесный винный ментол
спирт
спирт
бензгидрол
14.1.2. Изомерия
Для спиртов характерна структурная, геометрическая н оптическая изомерия. Структурная изомерия спиртов обусловлена разным строением углеродного скелета, а также различным положением гидроксильной группы в углеродной цепи:
248
С Нз—С Н2—С Н2—С Ня—-ОН
СН3—СНг—СН—СНэ
бутанол I
он
бутанол 2
СНз—С Н—СН 2—о н
L113
2 мети 1пропанол 1
ОН
СНз—с—СНз
<^Нз
2 метилпропанол 2
Для ненасыщенных спиртов структурная изомерия может обусловливаться н положением кратной связи
4 3 2 1
СН2=СН—СНг—СНа—ОН
бутен 3 ол I
4 3 2 1
СНз—СН=СН—СНа—ОН
бутен S ол 1
Геометрическая изомерия характерна для непредельных спиртов и определяется различным расположением заместителей относительно двойной связи:
Н3С СН2—ОН
^С^С^
Н3С Н
^С^^
Н^ ^СНг—ОН
чме бутен 2 ол I
транс бутен 2 wi I
Оптическая изомерии возможна для спиртов, имеющих в сво- " ей структуре асимметрический атом углерода:
СН2-СНЭ
HO'C>W,
▼CHj Я-бутанол-2
CH2-CH3 нкЛ
ОН н3с< 5-бутанол-2
14.1.3. Способы получения
Гидролиз галогенопроизводных углеводородов. Галогенопроизводные углеводородов, атом галогена которых связан с углеродным атомом в $р3-гибридизаиии, в присутствии водных растворов
249
щелочей при нагревании подвергаются гидролизу с образованием спиртов (см. с. 165):
СНз—-CHr—Cf СНз—СН2—ОН + NaCl
\ 10рэтан этанол
Гидратация алкенов. Присоединение воды к алкенам приводит к образованию предельных спиртов (см. с. 39).
ОН
СН3—СН=СН2 Нг0 -г» СНз—СН—СНз
пропен пропанол 2
Поскольку присоединение воды к алкенам происходит по правилу Марковникова, в зависимости от строения углеводорода по этой реакции образуются вторичные и третичные спирты. Из первичных спиртов этим способом можно получить только этанол.
Восстановление карбонильных соединений — альдегидов, кетонов, карбоновых кислот и сложных эфиров. Восстановление
карбонильной группы С=О до гидроксильной является доволь-
но распространенным методом получения спиртов. В качестве восстановителей используют различные реагенты. Наиболее часто употребляемый метод гидрирования карбонильных соединений заключается в обработке нх натрием в этаноле. Очень часто используют каталитическое гидрирование в присутствии никеля Ренея, платины, палладия и др. Кроме того, для восстановления карбонильных соединений используют комплексные гидриды металлов, такие, как борогидрид натрия Na^AlHi-, алюмогидрид лития U+AlHr и др.
При восстаноаленин альдегндоа, карбоновых кислот и сложных эфиров образуются первичные, а при восстановлении кетонов — вторичные спирты:
О
СНз—СНз—СН2—он
уксусный эльлсгяд этанол
о
СНз—L1—и\ СНз—СНз—ОН
\ эфир
ОН
уксусная кислота
250
о
СНз—С \)С2Н6
этилацетвт
СНз
ацетон
СНз \:Н—ОН
пропанол Э
Для восстановления ненасыщенных карбонильных соединений используют мягкий селектнанын восстановитель — борогид-рид натрия Na+BHf, ие затрагивающий при восстановлении кратные углерод-углеродиые связи-.
О
СНг=СН—СНг—СН2———-й
печтеп 4 аль
--> СНг=СН—СНг—СНг—СНг—ОН
пентен 4 ол 1
Этот метод используется для получения непредельных спиртов из альдегидов и кетонов,
Взаимодействие карбонильных соединений с магнийорганиче* скими соединениями (реактивами Гриньяра). Для получения спиртов используют реакцию магнийоргаиических соединений RMgX с альдегидами, кетонами и сложными эфирами. Синтез осуществляют в две стадии. На первой стадии молекула магний-органического соединения присоединяется к молекуле карбонильного соединения по месту разрыва л-связи карбонильной грчпгты Направление присоединения обусловлено полярностью карбо-
в—
мильной группы С=О и полярностью связи углерод - магний
L в+
в реактиве Гриньяра (R—С-<- MgX)
Н-С% + СН3—СН2~MgBr н ----------
формальдегид этилмагнийбромид
Н
* CH3-CH2-C-OMgBr
Н броммагнийалкоголя т
251
На второй ступени образовавшийся алкоголят подвергают гидролизу, в результате которого образуется спирт
СНз—СНг—СНг—OMgBr + НОН------->
---► СН3—СН2—СН2—ОН ч- Mg(OH) Вг
пропанол I
Прн действии магинйоргаиических соединений на формальдегид получают первичные спирты, с другими альдегидами образуются вторичные спирты:
уксусный альдегид
CaFfe—МйВг ----------у-
OMgBr
СН,—С—С2Н, I н
брони агнийал ко гол нт
НОН
—Mg(OH)Br
он
СНз—СН—СНг—СНз
При действии магнийорганнческих соединений на сложные эфиры образуются третичные спирты. В этой реакции с одной молекулой эфира реагируют дае молекулы реактива Гриньяра:
6
СНз^-С^ \>С2Н5
этилацетат
С2Н5
Ci-HsMg->r СНз—С—OMgBr
ОС2Н5
CjHsMgBr
—CaHjOMgBr
С2Н5
СНз—(L-OMgBr
1
С2Н5
С2Н5
—сн,—с—он
— Mg(OH)Hr |
С2н5
бром магний а эко гот пт
3 иетилпентанол 3
14.1.4. Физические свойстве
Насыщенные спирты — это, как правило, бесцветные жидкости или кристаллические вещества со специфическим запахом (табл. 14.1). Низшие члены гомологического ряда имеют характерный «спиртовый» запах; для бутанолов и пентанолов присущ неприятный «сивушный» запах; высшие алканолы обладают приятным запахом. Циклоалкаиолы, непредельные и ароматические спирты в большинстве случаев представляют собой жидкие или твердые вещества, имеющие приятный ароматный запах. Так,
252
Таблица 14 1
Физические характеристики некоторых спиртов
Название Структурная формула Температура, °C
плавления кипения
Метанол Эта иол Пропзнол-1 Пропанол-2 СНзОН СН,СН2ОН СНэСНгСН2ОН С ж—сн - СНз 1 -97,8 117,3 -127.0 -88,5 64.7 78.4 97.2 82,3
Бутапол-1 Бутанол 2 он С Из—CHz—СН2- СН2ОН СНз—СНг—СН—СНз 1 —89.5 — 114.7 117,7 100
2-Метил пропанол-1 ОН СНз—сн—снг—он -108,0 108,4
СНз СНз
2 - Мети л п ро па нол -2 CHj—с—он 1 25.5 83,0
Пропен-2-ол-1, аллиловый спирт Пропин-2-ол-1, пропаргиловый спирт СНз СН^--С11—СН, ОН СНзг-C-ClL- он -129,0 -48 96,9 115
Цнклогексанол он 25 161
Фенилмета нол, бензиловый спирт СНг—ОН -15 205,8
цнклогексанол имеет запах иамфоры, пропаргиловый спирт (НС=С—СНз—ОН) обладает запахом герани, а 2-феиилэтанол (СвН5—СН2—С На—-ОН) — запахом роз.
Спирты имеют более высокие температуры плавления и кипения, большую растворимость в воде, чем соответствующие углеводороды.
Такое резкое различие между физическими свойствами спиртов и алканов обусловлено в первую очередь тем, что спирты являются полярными соединениями. Онн имеют две полярные связи С—О н О—Н. Существование на атомах гидроксильной группы частичных зарядов разного знака приводит к межмолеку-ляриому взаимодействию гидроксильных групп и образованию водородных связей
Л— 6—- б-п й— й-|-
.... о^-н -О^Н.- О^Н...
RZ RZ RZ
253
В результате такого взаимодействия происходит ассоциация молекул спирта. Водородные связи значительно слабее ковалентных, однако их образование существенно уменьшает летучесть, повышает температуру кнпеиия, так как образующиеся агрегаты имеют большую молекулярную массу. Например, этан кипит прн —89 °C. тогда как этанол —при 78,5 °C.
Спирты с небольшой молекулярной массой хорошо растворимы в воде. А такие спирты, как метанол, этанол, пропанолы, алли-лоаый и йропаргнловый спирты, смешнааются с водой во всех соотношениях.
В водных растворах спиртов образуются водородные связи между молекулами воды и спирта (
t
а- а+ а- 6+ ь- а+ а- в+
-О^Н- -О^Н - О^Н.
/ Z Z Z
R Н R Н Р
I
Образующиеся водородные связи более прочны, чем связи между молекулами спирта, что приводит к уменьшению суммарного объема воды и спирта при смешивании (явление контракции спирта).
14.1.5. Химические свойства
Для спиртов характерны ревкции с участием саязей О—Н, С—О и окислительные реакции. Присутствие в молекуле спирта кратных связей или ароматического радикала не изменяет принципиально химические свойства гидроксильной группы, а придает спиртам свойства, характерные для ненасыщенных или ароматических соединений (см. подразд. 2.5, 6.5).
Кислотно-основные свойства. Спирты проявляют слабые кислотные и слабые основные свойства, т. е. они являются амфотерными веществами. Их кислотные свойства обусловлены подвижностью атома водорода гидроксильной группы. Атом кислорода, как более электроотрицательный элемент, смещает электронную
6- С+
плотность связи О-*— Н на себя, образуя при этом на атоме водорода частичный положительный заряд. Под действием сильных оснований спирты отщепляют от гидроксильной группы протон, т. е. проявляют свойстаа ОН-кислот. Однако спирты являются более слабыми ОН-кислотами, чем вода. Это обусловлено положительным индуктивным эффектом углеводородного радикала, связанного с гидроксильной группой. Дополнительное увеличение электронной плотности на атоме кислорода за счет -(-/-эффекта углеводородного радикала приводит к уменьшеивю полярности связи О—Н и соответственно подвижности атома водорода.
254
Поэтому при переходе от первичных спиртов к третичным кислотные свойства снижаются:
Н^О^Н6+ > СНз-»-О-«-Н6+' > вода чет а дол
СНа СНз
> СН3-»-О^Нв+* > СН3^С-^О^-Нв+"
СНз СН,
пропанол 2 2 метилпропанол 2
6+ > 6+' > 6+" > 6-Н"
Спирты как кислоты реагируют со щелочными металлами, образуя алкоголяты (алкоксиды)
2С2Н5—ОН + 2 Na -------► 2С2Н5—O“Na+ + H2f
Зтднол этилат натрия,
этикснд натрия
В спнртоаой среде алкоголяты подвергаются ионизации с образованием алкоксид-аииона, проявляющего сильные нуклеофильные и сильные основные свойства:
R— 0“ Na+ R—О” + Na+
алкоксид натрия алкокснд-днион
В связи с этим алкоголяты широко используются в органическом синтезе в качестве сильных оснований и сильных нуклеофильных реагентов. Алкоголяты легко разлагаются под действием воды до исходных спиртов, что подтверждает более низкую кислотность спиртоа по сравнению с водой:
С2Н5—O~Na+ 4- НОН С2Н5—ОН + NaOH
Из-за низкой кислотности спирты почти ие вступают а реакцию со щелочами.
Основные свойства спиртов обусловлены наличием иа атоме кислорода гидроксильной группы неподелеиной пары электронов, способной присоединять протон. Так, с сильными кислотами первичные спирты образуют на холоде неустойчивые соли алкилоксоиия
С2Н5—ОН + НВг
С2НБ—О
Вг~
этнлоксонкя бромид
2Б5
Во многих реакциях с участием спиртов алкоксоииевые соли образуются в качестве промежуточных продуктов.
По сравнению с кислотными основные свойства спиртов изменяются в противоположном порядке, т. е. при переходе от первичных к третичным спиртам основные свойства возрастают:
СНЭ СНз
СНз-*-О^-Н < \н-*О—Н < СН3-*-С-*-О-<-Н
CH3Z СНЭЛ
Взаимодействие с минеральными и органическими кислотами. Спирты реагируют с минеральными кислотами (серная, азотная, азотистая и др.) и органическими кислотами с образованием сложных эфиров. Эта реакция получила название реакции этерификации (от лат. aether — эфир):
t
с2н5-о-н + HO-SO3H «=* c.Hs OSO3H + н2о
моноэтилсульфат
СЗз-О-Н + ho-no2 г► c2h5-ono2 + Н2О
этилнитрат
0 Н ЧП 0
I H2SO4 1
C)HS O-H + но-с-сн3 С2Н5-О-С-СН3 + Н2о
этилацетат
Реакция этерификации обратима. Для смещения равновесия вправо либо берут избыток одного из реагентов (обычно спирта), либо удаляют одни из продуктов реакции Взаимодействие спиртов с карбоновыми кислотами протекает в присутствии катализатора, чаще всего с концентрированной H2SO4. Скорость реакции этерификации зависит от строения спирта и карбоновой кислоты. Русским химиком Н. А. Меишуткииым установлено, что при одной и той же кислоте скорость образования сложных эфиров уменьшается в ряду спиртов: первичный, вторичный, третичный. Реакции спиртов с карбоновыми кислотами происходят по механизму нуклеофильного замещения (см. с. 367). Выделяющаяся в процессе взаимодействия молекула воды образуется за счет атома водорода группы —ОН спирта и гидроксильной группы карбоновой кислоты.
Дегидратация спиртов. При нагревании спиртоа в присутствии концентрированной серной кислоты, безводной фосфорной кислоты нли при пропускании паров спирта над катализатором—-оксидом алюминия А(2ОЭ, спирты отщепляют воду, т. е. подвергаются дегидратации. В зависимости от природы спирта н уело-
256
вий проведении реакции дегидратация может протекать межмолекулярно н внутримолекулярно
При межмолекулярной дегидратации спиртов образуются простые эфиры
+ Н : I
С^НгО-Н + НСНС,Н5 -------------► С,Н§- О-С3Н5 + Н2О
ДИЭТИЛОВЫЙ эфир
В результате внутримолекулярной дегидратации образуются алкеиы
СНз—СНз—ОН СН2=СН4 4- Н2О
этилен
При этом, если с атомом углерода, несущим гидроксильную группу, непосредственно саязаны неравноценные атомы углерода, отщепление аоды происходит по правилу Зайцева, т. е. водород уходит от соседнего, менее гидрогенизированного атома углерода:
СНз—СН2—СН—СН3 СНз—СН=СН—CHjf + Н2О
Межмолекуляриая н внутримолекулярная дегидратации спиртов представляют собой два конкурирующих процесса, из которых каждый в определенных условиях может стать доминирующим. Отщепление воды от двух молекул спирта с образованием простых эфиров — межмолекулярная дегидратация — становится преобладающим процессом при нагревании спиртов в присутствии каталитических количеств минеральной кислоты (спирт в избытке) при температуре 140—160 °C.
Внутримолекулярная дегидратация, т. е. превращение спирта в алкен, становится доминирующей при нагревании спиртов с избытком минеральной кислоты при температуре выше 170 °C. Особенно легко она протекает у третичных спиртов.
В случае пропускания паров спирта иад А12Оз при температуре 200—250 °C происходит межмолекуляриая дегидратация с образованием простых эфиров, а при более высокой температуре (300—400 °C) прбтекает внутримолекулярная дегидратация, т. е. образуются алкены.
Межмолекулярная дегидратация спиртов протекает по механизму 5^2 или S41. При этом вначале молекула спирта под действием минеральной кислоты протоннруется с образованием оксо-ииевого катиона, а затем происходит замещение группы —ОН:
I Л-----k I +ХН
R-C-OH + Н R-C-Of
I I Н
спирт оксониевый катион
9 5 104
257
Механизм SN2 включает образование переходного состояния, которое формируется в процессе нуклеофильной атаки электрофильного атома углерода оксониевого катиона второй молекулой спирта:
нуклеофильный электрофильный
центр центр
I I,,н R-C-OH +
спирт оксоннсвый катион
переходное состояние
Механизм SN1 протекает через стадию образования карбо-ниевого катиона;
+ Н2О
оксонненый карбон невый
катион
катион
R-C+ + Нб-C-R +=* I I
I + I II
C-O-C-R —> R-C-O-C-R
III -Н+ I I
спирт
простой эфир
Подобно галогеиалкаиам первичные спирты вступают в реакцию межмолекулярной дегидратации, как правило, по механизму Sn2, третичные- по механизму SNI, вторичные могут реагировать как по SN2, так и по SN1 -механизму.
Внутримолекулярная дегидратация спиртов в зависимости от их строения может проходить по механизму Е\ и Е2. Прн этом первичные спирты реагируют в большинстве случаеа по механизму Е2, а вторичные, и особенно третичные — по механизму EI. Как и в случае нуклеофильного замещения, элиминирование спиртов протекает через образование оксониевого катноиа. Ниже приведена схема внутримолекулярной дегидратации по механизму Е\:
258
нярбониепый катион
алкен
Взаимодействие с галогеноводородными кислотами. При взаимодействии спиртов с галогеноводородны мн кислотами (НС1, НВг, HI) гидроксильная группа замещается на атом галогена и образуются галогеналканы. Реакция является обратимой. Для смещения равновесия вправо обычно нз реакционной среды отгоняют воду или галогеиалкаи
С2Н5—ОН + HI <=+ С2Н5—I + Н2О
По реакционной способности со спиртами галогеноводородные кислоты располагаются в ряд НС1 < НВг < HI. С иодово-дородиой н бромоводородной кислотами реакция протекает легко, с хлороводородной — значительно труднее. Первичные и вторичные спирты реагируют с хлороводородной кислотой только в присутствии хлорида циииа (кислота Льюисв).
Реакционная способность спиртов по отношению к галогеноводородным кислотам убывает в ряду бензиловым спирт > аллиловый спирт > третичные > вторичные > первичные. С первичными спиртами реакция протекает, как правило, по механизму с третичными — по SN1. Вторвчиые спирты реагируют н по механизму Shl, и по SN2. Ниже приведены схемы взаимодействия спиртов с бромоводородной кислотой по механизму SN2 и SN1.
Механизм SN2:
Н
СНз—сНг—он СНз—сн^с/7
этанол окспннрйый катион
переходное состояние
бромэтзн
259
Механизм SN1:
СН3 СН3 Н СНэ
СНз^С—ОН СНг^С—OZ СНз—1+ -S£»-
СН,/ СНз^ ’ СНз^
2 метил-пропанол-2 оксонневый катион карбкатион
СНз
---> СНз—С—Вг СНэ^
2 метил 2 бронирован
Взаимодействие с галогенангидридами неорганических кислот (РС1з, РВгз, PCI5, SOCh). Прн действии на спирты галогенидов фосфора PCh, РВг3, PCU и тноннлхлорида SOCh образуются галогеиалканы:
ЗС2Н5—ОН 4- РВгэ -------* ЗС2Н5—Вг 4- НзРОз
броматан
С2Н5—ОН 4- РС15 —► С2Н5—С1 4" HCI 4- РОС13
С2Н5—ОН 4- SOC12 —► С2Н5—С1 4- HClf 4- SO2f
Реакция с тионилхлоридом протекает по механизму внутримолекулярного нуклеофильного замещения (S^i), включающего стадию образования сложного эфира:
8+ 'Оу
CHj-CHj-OH + SOCI1 CH3-CH/^S=O ^CHjCHjCl этанол тионил- б— хлорэтан
хлорвд этиловый эфир
хлорангндрида сернистой кислоты Окисление. Первичные, вторичные и третичные спирты по-разному относятся к действию окислителей. Первичные спирты при окислении первоначально образуют альдегиды, которые могут окисляться далее, превращаясь при этом в карбоновые кислоты: О
СНз—СНз—СНз—ОН -'0| ► СНз—СНз— пропанол 1 —HjO пропаналь
О Н
> СНз—СН2—С \l пропановая кислота
260
Вторичные спирты при окислении образуют кетоны
СНз—СН—СНз СНз—С—СНз
i-HaO я
Н О
пропанол 2 ацетон
Третичные спирты устойчивы к окислению, одиако в жестких /слоанях они окисляются с разрывом углеродного скелета моле-сулы и образоааинем смеси кетоиов и карбоновых кислот.
В качестве окислителей для окисления спиртов используют жсид хрома (VI), бихромат калия в серной кислоте (хромовая :месь), перманганат калия в серной кислоте и др.
В промышленности для окисления первичных спиртов в вльде-'иды используют метод каталитического дегидрирования. Сущность метода состоит в пропускании паров спирта над катализато-юм (мелкораздроблеииая медь) при 280—300 °C. При этом 1роисходит отщепление молекулы водорода от молекулы спирта < образуется альдегид. Преимуществом каталитического гидрирования является то, что в этом случае предотвращается более 'лубокое окисление альдегида до кислоты:
О
r—сиг—он 9l_28Q3Q0^. r—-|- н2
1о этой реакции из вторичных спиртов синтезируют и многие сетоны.
14.1.6. Идентификация спиртов
Химические методы. Для спиртоа ие существует качественной реакции, которая бы убедительно и однозначно указывала на на-1ичие в молекуле гидроксильной группы. Спирты можно отличить ю их реакции окисления хромовым ангидридом СгОэ в водном растворе H2SO4. Приблизительно в течение двух секунд прозрачней оранжевый раствор превращается в мутный голубо вато-зеле-1ый. Одиако эту реакцию ие дают третичные спирты. Данная реакция характерна также для альдегидов, ио их можно отличить от гпнртов другими специфическими реакциями (см. с. 326, 330, 337). Для идентификации спиртов может служить и реакция образования
О
/
сложных эфиров (R—С ), иоторые, как прапило, имеют
\>R'
характерный приятный запах.
261
Для определения того, является лк спирт первичным, вторичным или третичным, используется проба Лукаса. Она основана иа различной скорости взаимодействия первичных, вторичных и третичных спиртов с раствором хлористого цннка в коицеитрн-роваииой хлороводородной кислоте (реактив Лукаса). В результате образуется галогеиопронзводное углеводорода, которое выделяется в виде мелкодисперсного осадка. Взаимодействуя с реактивом Лукаса, третичные спирты обнаруживаются почти сразу же, аторичные — приблизительно через 5 мин, а первичные спирты при комнатной температуре практически не реагируют.
Н
Спирты, имеющие в молекуле фрагмент ——СН3, дают положи-
ОН
тельиую йодоформную пробу. Оиа заключается в обработке сйирта иодом и гидроксидом натрия или гипоиодитом натрия NaOI:
Н
R—С—СНз + Nal 4- Н2О (окисление)
R—С—СНЭ 4~ 3 Nal R—С—С1з 4- 3NaOH (галогенирование)
R—С—Ch 4- NaOH --------------->- CHI3| 4- R—COO Na+ (расщепление)
|| желтый
Л ОСАДОК
В результате реакции образуется желтый осадок йодоформа СН1Э, имеющий характерный запах. Йодоформная проба ие является строго специфической реакцией на спирты. Положительную йодоформную пробу также дают другие соединения, содержащие
в своих молекулах фрагмент —С—СНз (см. с. 335).
Физические методы. Спирты, в молекуле которых отсутствуют хромофоры, не поглощают ультрафиолетового саета с длиной волны более 200 нм. Электронные спектры ненасыщенных и ароматических спиртов характеризуются поглощением за счет углеводородного радикала. Поэтому для анализа спиртов электронная спектроскопия используется очень редко.
В инфракрасных спектрах наиболее характерной является интенсивная широкая полоса в области 3200—3600 см-1, обусловленная валентными колебаниями группы О—Н. Уширение этой полосы происходит а результате образования межмолекулярных
262
водорОДиых связей. В отсутствие водородных связей (разбавленный раствор спирта а неполярном растворителе, нвпрнмер СС14) в ИК-спектрах спиртов наблюдается узкая полоса в области 3610—3640 см-1. Другая интенсивная широкая полоса, вызванная валентными колебаниями связи С—О, расположена в области 1000— 1200 см-1.
В ПМР-спектрах спиртов сигнал протона гидроксильной группы обычно обнаруживается в области 2,0—5,0 м. д. Химический сдвиг зависит от степени ассоциации и прочности водородных связей. При уменьшении степени ассоциации, например при разбавлении спирта четыреххлористым углеродом, сигнал протона ОН-группы смещается в более сильное поле. В соответствии с этим положение сигнала гидроксильной группы сильно зависит от температуры, концентрации и природы растворителя. Если в ИК-спектрах наблюдаются полосы, обусловленные как колебаниями водородносаязаиных, так и свободных гидроксильных групп, то в ПМР-спектрах спиртов наблюдается лишь одни сигнал. Это связано с тем, что для регистрации ПМР-спектра необходимо больше времени, чем для ИК-спектра. Поэтому фиксируется лишь один усредненный сигнал протонов ОН-группы. При снятии ПМР-спектров в диметнлсульфоксиде обмен протонами замедляется за счет возникновения сильных водородных связей между растворителем и спиртом. Это позволяет наблюдать расщепление сигнала гидроксильного протона зв счет спин-спииового взаимодействия с соседними протонами и, таким образом, по мульти-плетности сигивла устанавливать принадлежность к первичным (триплет), вторичным (дублет) или третичным (синглет) спиртам.
14.1.7. Отдельные представители
Метанол СНзОН. Бесцветная горючвя жидкость.с запахом, напоминающим запах этилового спирта, смешивается с водой во всех отношениях (т. кип. 64,7 °C). Ядовит, смертельная доза внутрь — 25 г, меньшие количества приводят к слепоте. При окислении в организме превращается в формальдегид и муравьиную кислоту. Имеет значение как исходное соединение для синтеза органических веществ как растворитель. До 1925 г. метанол получали сухой перегонкой древесины. В настоящее время его получают каталитическим гидрированием оксида углерода (II). Реакция протекает при температуре 400 °C, давлении 250—400 атм.:
2Н2 + СО ' “ , СНзОН
Этанол СйНбОН. Бесцветная горючая жидкость с обжигающим вкусом и характерным запахом, смешивается с водой а любых соотношениях (т. кип. 78,3 °C). В небольших количествах действует опьяняюще, большие дозы приводят к наркотическому состоянию. В организме окисляется до уксусного альдегида, а затем до углекислого гвза и воды. Широко используется как сырье и рвство-
263
ритель в органическом синтезе, в фармации и медицине (изготовление настоек, экстрактов, растворов), как обеззараживающее средство, для консервации различных анатомических препаратов, как горючее и др. Входит в состав алкогольных напитков. Для получения этанола с древнейших времен широко используется ферментативный гидролиз углеводов. В настоящее время этанол получают также гидратацией этилена:
СН2=СН2 + НОН нЛЗМГ<? СНз—СНг—ОН 60 атм
С аодой этанол образует азеотропную смесь (т. кип. 78,15 °C), состоящую нз 95,572 % спирта. Поэтому выпускаемый в промышленности спирт-ректификат содержит около 96,6 % этанола. Для получения безводного (абсолютного) этанола применяют специальные методы обезвоживания, такие как длительное нагревание с оксидом кальция, безводным сульфатом меди или другими веществами, способными саязывать воду химическим путем:
(С2Н5ОН + Н2О) + СаО ------► С2Н5ОН + Са(ОН)2
азеотропная смесь
Аллиловый спирт СН2=С И СН>—ОН. Бесцветная жидкость с резким запахом (т. кнп. 96,9 °C). Смешивается с водой. Используют в производстве глицерина. В промышленности получают из 3-хлорпропеиа:
СНа=СН—СНз—CI КиОН‘ Н2^> СН2=СН—СН^—ОН 250 “С
Пропаргиловый спирт СН=С—СН2ОН. Бесцветная жидкость с запахом герани (т. кип. 115 °C). С водой смешивается во всех соотношениях. Образует азеотропную смесь, кипящую при 97 °C и содержащую 45 % пропаргилового спирта. Получают из ацетилена и формальдегида. Используют в органическом синтезе.
Бензиловый спирт. Бесцветная жидкость, плохо растворимая в воде, лучше — в оргаии-Оческих растворителях (т. пл. 15 °C, —СНг—ОН т. кип. 205 °C). Содержится в эфир-
ных мвслвх и бальзамах. Относится к душистым веществам и используется в парфюмерии как фиксатор запаха, а также как растворитель для красителей, чернильных паст, казеина, восков и др.
14.2. ДВУХ-, ТРЕХ- И ПОЛНАТОМНЫЕ СПИРТЫ
Двухатомные свирты (содержат две гидроксильные группы) называют диолами или гликолями. В завнснмости от положения гидроксильных групп в углеродной цепи гликоли делят на а-гликоли (гидроксильные группы находятся у соседних
264
углеродных атомов, т.е. в положении 1,2); р-гликоли (ОН-группы в положении 1,3); у-гликоли (ОН-группы в положении 1,4) и т. д.:
R—СН—СН—R'
а-гл и ко ль
R—СН—СН2—СН— R' in он
р-ГЛИКОЛЬ
R—СН—СНа—СНг—С Н— R' он Ан
у-гликоль
а-Глнколи еще называют вицинальными гликолями. Гликоли с гидроксильными группами при одном углеродном атоме (геми-вальные гликоли) —очень неустойчивые соединения. В момент образования они отщепляют молекулу воды и превращаются в альдегид нли кетон
НО R R
геминальный ли ил
иксосоединение
Твкие диолы могут существовать только при наличии в молекуле сильных электроноакцепториых заместителей. Так, хлоралгидрат является устойчивым соединением
С1 ОН
ci^4 С—С—Н Cl^ \>Н
По систематической номенклатуре ИЮПАК. названия гликолей образуют от названия соответствующего углеводорода, добавляя суффикс -диол и указывая положение гидроксильных групп в углеродной цепи. По радикало-фуикцноиальной номенклатуре названия а-гликолей производят от названия соответствующего двухвалентного радикала, к которому добавляют суффикс -гликоль:
СН^-СН2
Ан Ан
этандиол-1,2, этиленгликоль
СНй—СН—сн3
пропэндиол-1.2, пропиленгликоль
сн2—сн=сн—сн2
Ан Ан
бутен-2-лнол-1.4
265
Трехатомные спирты (содержат три гидроксильные группы) называют триолами нлн глицеринами. По заместительной номенклатуре названия трехатомных спиртов образуют добавлением к названию соответствующего углеводорода суффикса -триол
СНг—СН—СН2
ОН (!>Н он
пропант рно 112 3 глиисрнн
Спирты, содержащие более трех гидроксильных групп, называют полиолами или просто многоатомными спиртами. Четырехатомные спирты имеют общее название — эритриты, пятиатомные — пеититы, шестнатомиые — гекситы и т д
Структурная изомерия двух-, трех- и полиатомиых спиртов обусловлена разным строением углеродного скелета, разным расположением гидроксильных групп
ОН ОН
СН;1^Н-^Н2
пропандиол 1 2
ОН он
пропаиднол I 3
Для полиатомиых спиртов характерна оптическая изомерна, обусловленная появлением в их структуре асимметрических атомов углерода Так, эритрит (бутантетраол) содержит в своем составе два асимметрических атома углерода и существует в виде трех пространственных изомеров (см кн 1, с. 75):
сн2он СН2ОН СН2ОН
но тт н он н он
н —Т1 он но н п ы он
( ял-(- ^jn :н2он ( 5 5—(4- :н2он эрятри-? п СН2ОН
14.2.1. Способы получения двух-, трех- и полиатомиых спиртов
Гидроксильные производные углеводородов с несколькими гидроксильными группами можно получить теми же способами, что и одноатомные спирты, используя а качестве исходных веществ днфункцнональные производные (см подразд. 14.1,3) Например, гидролизом дигалогеипроизводных, содержащих атомы галогенов прн разных атомах углерода, получают гликоли
260
СН г—СНг—СН2 СНз—СНз—СН2
I | —2НВг | I
Вг Вг ОН ОН
1 3 аибромпропаи
пропандиол I 3
Кроме того, существует ряд специфических способов получения а-гликолей.
Гидроксилирование алкенов. Эту реакцию проводят, действуя на алкены водным раствором перманганата калия иа холоду (реакция Вагнера):
ЗСН-г—СН=СН2 + 2КМпО4 + 4Н2О
пропен
ЗСНз—СН—СН2 4-он Ан
проканднол I 2
+ 2 кон 4- 2МпО2
Гидратация оксиранов. Оке и рай и его производные присоединяют молекулу воды, образуя а-гликоли. Реакция в кислой среде сопровождается раскрытием цикла
Н2С--—СНа 4- Н3О-^СНа0Н—СН2ОН
оксиран
этакдиол 1 2
Этот способ используется а промышленности для получения этиленгликоля
Наиболее важный представитель триолов— глицерин (пропаи-триол-1,2,3) может быть получен кислотным или щелочным гидролизом жиров (сложных эфиров глицерина и высших гомологов карбоновых кислот) :
СНг—О—СО—R ^н2—о—со—r -25%
CHj—О—СО—R
Триглицерид (жир)
СН2ОН
Анон 4- 3R—СООН
I
СН2ОН
глицерид карбоновая кислота
В промышленности синтез глицерина осуществляют из пропилена по схеме
СНз СН2С1 1 1 СН2ОН СНзОН сн2он
in Ди. (Lh -2225. Ан - НОС1-> Анон - -^Uchoh
11 НС 1 II — NaU II (Clj + HjjO) I — NaCJ |
СН2 СН2 СН2 СН2С1 СН2ОН
Арипнлен яплрнстый аллиловый 3 кяорпропан глинеряд
аллил спирт диол I 2
267
14.2.2. Физические свойства
Низшие члены гомологического ряда днолов представляют собой вязкие жидкости, высшне — кристаллические вещества. Жидкие глнколн имеют большую плотность и более высокие температуры плавления и кипения, чем одноатомные спирты, хорошо растворяются в воде. Трехатомиые спирты — вязкие жидкости или трудно кристаллизующиеся твердые веществе. Вязкость, растворимость в воде, температуры плавления и кипения гидроксильных производных алифатических углеводородов увеличиваются в ряду одноатомные спирты < гликоли < < глицерины. Это является следствием усиления ассоциации молекул за счет образования межмолекуляриых водородных связей.
Отличительной особенностью гидроксильных производных углеводородов с несколькими ОН-группамн является их сладковатый вкус, усиливающийся, как правило, с увеличением числа гидроксильных групп в молекуле.
14.2.3. Химические свойства
Двухатомные спирты вступают в те же реакции, что и одноатомные, с той лишь разницей, что они могут протекать с участием одной или двух гидроксильных групп. Еще большее разнообразие продутктов возможно для реакций с участием трех- и полиатомных спиртов. Например, глицерин образует три ряда производных: моно-, ди- и тризамещеииые. При этом для моио-и дизамешенных возможны структурные изомеры, обусловленные различным положением заместителей.
Образование алкоголятов. Гликоли являются более сильными ОН-кислотамн, чем одноатомные спирты, поэтому оии образуют алкоголяты (гликоляты) не только со щелочными металлами, ио и с другими активными металлами, такими, как Al, Mg и др., а также со щелочами и гидроксидами тяжелых металлов. Повышение кислотных свойств гликолей связано с элек-троноаицепторным влиянием одной гидроксильной группы на другую (—/-эффект). При взаимодействии с активными металлами безводные гликоли образуют неполные и полные гликоляты:
СН2ОН CH2ONa
'I 21 -iX
СН2ОН -* СН2ОН 1,1
лилен чоно! ликолйт
гликоль натрия
CH2ONa ibbONa
ДМ глмколят натрия
288
С гидроксидом меди (II) гликоли образуют глнколят меди — комплексное соединение синего цвета:
н
СНа—ОН
2| +Си(ОН)2
СНз—ОН
—2НаО
Н2С—о
Н2С—о
этилеигликоль
гли колят меди
С увеличением числа гидроксильных групп в молекуле кислотные свойства соединения усиливвются. Так, глицерин, по сравнению с этиленгликолем, имеет более выраженные кислотные свойства. В водном растворе щелочи глицерин легко образует моноглицераты [НОСНг—CH(ONa)—CHZOH]. При взаимодействии с гидроксидом меди (II) глицерин образует глицерат меди (раствор синего цвета). Хорошая растворимость глицератов в воде объясняется их комплексным строением:
СНаОН
СНг—ОН
2(^Н—ОН + Си (ОН) 2 ™-°и,
Г —4НаО
СНг—ОН
СНг—О
_Na
О—СН
СН—о
I
СН2ОН
Na
О~СН2
Взаимодействие с галогеноводородами. При взаимодействии гликолей с галогеиоводородамн (НС1 или НВг) образуются хлорил и бромгндрнны
СНу—СН2 — Ня-^ СНй—сн2
1 I "Н2° 1 I ОН ОН ОН Hal
Вторая гидроксильная группа замещается труднее (лучше использовать РС15 или SOCI2)
СН,—сн2 ----—► CHz-сн2
il —2SOj, —SHCI I I
H OH Cl Cl
200
При взаимодействии глицерина с калогсио водорода ми образуется смесь моио- и днгалогензамещениых:
СНг—СН—СНа - НС1 > СНг—СН—СНз НС1 > СН2—СН—СНг
||Т I 11-^1 II
ОН ОН ОН С1 ОН ОН С1 ОН С1
глицерин
МОШ) хлоргидрин /лицермна
дихлоргндрин глицерина
Образование простых и сложных зфиров. При взаимодействии гликолей со спиртами, минеральными или органическими кислотами получается два ряда производных:
а) неполные и полные простые эфиры
СН2—СН2
r—он и~
—нго
СНг—СНг
R—ОН. И+
—Н2О
СН?—СН2
б) неполные и полные сложные эфиры
СН2—СН2
HONQ2 —н2о
СНг—СН2
Ан ONO2
ЧОНОНИТрИт этиленгликоля
HONOa
—Н2О
СН2—С Н2
диннтрат этиленгликоли
CHz—сн2
А>н он
о
НО-Д—R н+
—ИаО
CHa—CH2 и; с Hz— сн2
1 I -На° 1 1
ROCO OH ROCO ОСО It
Для глицерина в этом случае образуется три ряда производных. Прн взаимодействии глицерина с концентрированной азотиой кислотой в присутствии H2SO4 получают полный азотио-иислый эфир глицерина — тринитрат глицерина (нитроглицерин)}
СН2—ОН CH2-4)NO2 л -Л
Ан—ОН +3HNO3 Ан —ONO2 + ЗНаО
A Hz—ОН А На—О NO2
Аналогично в жестких условиях получается и полный уксусно*^ кислый эфир глицерина — глицериитриацетат '
270
о
СН2—О—С—СНз
СН2ОН О
(^НОН + ЗНО—(!—СНз
(!:н2он
о
сн—О—Л—СНз + ЗН2О
О
Окисление двух-, трех- и полиатомных спиртов. При окислении гликолей образуется смесь продуктов окисления:
О
СН2ОН (^Н2ОН
СН2ОН
этилен гликолевый
гликоль альдегид
соон
СН2ОН
гликоле вея кислота
глиоксаль
СООН
глиоксиловая кислота
10]
СООН
—I
СООН
щавелевая кислота
Некоторые из этих промежуточных продуктов можно выделить, проводя окисление в специально подобранных условиях.
Окисление глицерина проходит сложно, многоступенчато. В зависимости от природы окислителя могут преобладать те или иные продукты окисления. На первом этапе окисления образуются глицериновый альдегид или диоксиацетои, конечным продуктом является мезоксалевая кислота:
СНО (^НОН (^Н2ОН глицериновый альдегид
[О]
СН2ОН
Н2ОН
диоксиацетон
Ю|
СООН
СООН
мезоксалевая кислота
271
Специфической реакцией окисления 1,2-диолов является их взаимодействие с иодной Тнслотой. В процессе реакции происходит расщепление угле род-угле родной саязи в диольиом фраг-
менте —С----С—, при котором в зависимости от строения гликоля
Ан ОН
образуются соответствующие альдегиды и кетоиы:
ГЛИИО1Ь
кетон альдегид
В эту же реакцию вступают и глицерины
2НЮ4
R—С—R 4- Н—С—ОН + R'—С—Н
А А А
кетон муравьиная альдегид
кисюта
Дегидратация гидроксильных производных с несколькими группами ОН. Под действием водоотиимающих реагентов гликоли, как и одноатомные спирты, подвергаются внутримолекулярной или межмолекуляриой дегидратации. Направление дегидратации зависит от условий проведения реакции.
Так, при нагревании этиленгликоля в присутствии концентрированной серной кислоты происходит межмолекулярная дегидратация и образуется циклический простой диэфир — диоксан:
ОН НО
НгС^ \н2
ОН НО
о
НзС^ ^СНз H2SO4 t । ।
Н2С СНа
ХХ
4г Н2О
дноксан
272
Для 1,4- н 1,5-диолов возможна внутримолекулярная дегидратация с образованием пяти- и шестичлеиных циклических эфиров’
СН2—СН,- —СН2 сн2 Hg5Q4* Н2С—сн2 1 —на0 | |
он он н2с сн2
О
бута «диол 1 4
тетрягидроф! ран
Глицерин прн нагревании с гидросульфатом калия илн другими водоотннмающнмн средствами подвергается внутримолекулярной дегидратации с образованием непредельного альдегида — акролеина:
KHSO4; t 6- 6+
СН2-СН-СН2 _Нз0 ► CH2CH-Clf~OH он он ОН ОН '
глицерин
СН2-СН-С I 2 I
он н
с°
KHSOz t £>
»CH2=ClRCf
-н2о \
акролеин
Акролеин (акриловый альдегид) представляет собой жидкость с едким удушливым запахом.
Поликонденсация двухатомных спиртов. Молекулы этиленгликоля могут вступать между собой в реакцию поликонденсации с образованием полиэфира — полиэтилеигликоля:
НО—СН2—СН2—ОН 4- НО—СН2—СН,—ОН H*S^ -то
-> НО—СН2—СНг—О—СН2—СН2—ОН сн^онч
IMJTH 1?НТЛИКО1Ь
-пН2О
но
-СНг—СНг—О^-Н
полиэтилене школь
Полнэтиленглнколь с молекулярной массой до 400 применяется в фармации в качестве растворителя лекарственных веществ, основы для мазей, а также как связывающее вещество в производстве таблеток.
При поликонденсации этиленгликоля с терефталевой кислотой образуется полиэфир — полиэтилентерефталат, который
273
используется для изготовления синтетического волокна —-лавсана:
СН?—ОН
СН2—ОН
-пИгО
пол нянчен терефталат
14.2.4. Идентификация диолов и триодов
/ Обнаружить гидроксильные группы в диолах и триолах можно по образованию окрашенного в сиинй цвет раствора комплексной соли медн (см. с. 269).
Для обнаружения глицерина может быть использована реакция дегидратации с образованием акролеина — вещества с резким раздражающим запахом (см. с. 273).
Длн идентификации а-гликолей используют реакцию окисления с иодной кислотой. При окислении происходит разрыв химической связи между углеродными атомами, связанными с группами —ОН, н образуются соответствующие карбонильные соединения, по которым можно установить место расположении диольного фрагмента молекулы:
СНа—ОН
I
СН—он + НЮ4 -------» ’Н
I
СНЯ
пропилен гликоль >
О о
н-J—н + Н—ci—СНз + НГОз + н2о
метаналь этаналь
14.2.5. Отдельные представители
Этиленгликоль (этандиол-1,2) НО—СН2—СН2—ОН. Бесцветная вязкая жидкость (т. кип. 197,6 °C, т. пл. —11,5 °C), имеет плотность бл0 == 1,113. Гигроскопичен, смешивается с водой н этанолом Сильно понижает температуру замерзания воды и используется для приготовления антифриза. Очень токсичен. Широко используется для получения синтетических волокон.
274
он он он
Глицерин (пропаитриол-1,2,3) (^На—(Lh—(^Н». Бесцвет-
ная сиропообразная жидкость без запаха, со сладким вкусом (т. пл. 18 °C, т. кип. 290 °C с разложением). Гигроскопичен, смешивается с водой и этанолом в любых соотношениях. Применяется в качестве основы для мазей и паст, добввкв к мылам. В больших количествах глицерин используется для получения нитроглицерина.
ono2 ono2 ono2
Нитроглицерин СН2-----СН---СН2. Тяжелая маслянис-
тая жидкость со сладковатым жгучим вкусом, при нагревании или ударе взрывается, используется для изготовления динамита. В виде разбавленных спиртовых растворов нитроглицерин оказывает сосудорасширяющее действие и применяется в медицине при стенокардии. Выпускается также в таблетках с содержанием 0,0005 г вещества.
М.З. ЕНОЛЫ
Гидроксильные производные углеводородов с ОН-группой прп двойной углерод-углеродной связи называют енолами (суффикс
-ей обозначает двойную связь С=С , а -ол — гидроксильную
группу).
Формально енолы можно отнести к ненасыщенным спиртам, у которых ОН-группа связана с углеродным атомом в $р2-гибри-дизацни. Одиако по своим свойствам эти гидроксипрорзводные настолько отличаются от ненасыщенных спиртов с ОН-группой прн $р3-гнбридизоваииом атоме углерода, что целесообразно про-, вести их отдельное рассмотрение.
Названия енолов по систематической номенклатуре ИЮПАК образуют из названия алкеиа, к которому добввляют суффикс -ол:
3 4 5
СН2=СН—ОН СН2=С—СНз СНз—СНз—С=С—СНт—СНз
этенол, виниловый спирт
нропенол 2
4 метил гексен з ол 3
Рассмотрим два изомерных непредельных гидроксильных производных
СН2=СН—СНг—ОН
про пен о Л 2 (енол)
пропенол I (непредельный спирт)
275
Эти два изомера очень существенно отличаются по своим физическим и химическим свойствам. Первое вещество относится к енолам (гидроксильная группа при связи С=С), а второе — к непредельным спиртам (алкенолвм), у которых гидроксильная группа находится при углероде в ярэ-гнбридизации.
Енолы, в отличие от непредельных спиртов, являются неустойчивыми соединениями Попытки получить соединения с енольной структурой С=С—ОН приводят к соединениям, содержащим
карбонильную группу С=О
Так, при гидратации ацетилена в качестве промежуточного вещества образуется виниловый спирт (енол), который изомеризуется в уксусный альдегид
£н^-СН^-Н
ацетилен
виниловый спирт
уксусный альдегид
Это свойство еиолов отражено в правиле Эльтекова — Эрлен-мейера:
Соединения, у которых гидроксильная группа находится при углеродном атоме, образующем кратную углерод-углеродиую связь, неустойчивы и изомеризуются в карбонильные соедииеиия — альдегиды или кетоны.
Причина неустойчивости еиолов вызвана миграцией протона от атома кислорода ОН-группы к соседнему атому углерода, образующему двойную связь Такое явление называется прототропной изомерией или таутомерией. При этом происходит изомеризация енольной формы в кетонную.
8+ . U
♦ Н-^С-С'
енольная форма
кетонная форма
Приведенная прототропная изомерия называется кето-еноль-иой таутомерией.
270
В случае кето-еиольной таутомерии равновесие сильно сдвинуто в сторону образования кетониой формы Это объясняется тем, что в равновесной смеси таутомеров всегда больше той формы, в которой атом водорода прочнее связан, т е равновесие смещено в сторону более слабой кислоты Естественно, что в кетоиной структуре (СН-кислота) атом водорода удерживается сильнее, чем в енольной структуре (ОН-кислота), и поэтому равновесие сильно сдвинуто в сторону образования кетониой формы В связи с этим становятся понятными причины неудач в синтезах енолов
В отличие от енолов их простые и сложные эфиры весьма устойчивы Они не содержат подвижного атома водорода и в обычных условиях не перегруппировываются в карбонильные соединения Так, изаестны сложные эфиры винилового спирта, например винилацетат, который получают реакцией присоединения уксусной кислоты к ацетилену
О
СНз—СООН + СН=СН-----------СНз-J—о—сн=сн2
винилацетат
уксусновмнитпвый эфир
Винилацетат способен полимеризоваться с образованием поли-винилацетвта (ПВА):
пСН=СН2 АсОСНз
СН—СН2---
ОСОСНз л
поливинил аиетлт (ПВА)
Молекулярная масса ПВА может колебаться от 500 до 1600 тыс. Он широко применяется в производстве лаков, красок, как основа для клеев
При гидролизе ПВА получают поливиниловый спирт (ПВС), представляющий собой белое аморфное вещество, которое не растворяется в органических растворителях, ио растворяется в горячей воде:
СН2—СН----
ЬсОСНз
+ пСНзСООН
поливиниловый спирт (ПВС)
Поливиниловый спирт применяется в производстве искусственных волокон, лекарственных средств В частности, он используется при изготовлении саморассасывающнхся хирургических ниток, крове- и плазмозаменителей.
377
14.4. АМИНОСПИРТЫ
’Аминоспиртами называют производные углеводородов, содержащие аминогруппу (1У-алкил- или Ч М-диалкиламиногруппу) и спиртовый гидроксил.
Устойчивыми являются амииоспирты, у которых аминогруппа и спиртовый гидроксил расположены у разных атомов углерода. Для названия аминоспиртов чаще всего применяют заместительную номенклатуру, согласно которой нх называют как производные спиртоа, содержащие в качестве заместителя аминогруппу. Положение аминогруппы указывают цифрами или буквами греческого алфавита:
СН,—CHz— ОН I
NH,
2-аниноэт.а ноя.
Р-аминоэтнлопыЙ спирт
СН2—СН2—ОН
I
NHCH3
2-N-мети.та мн но этанол
СНз—СН—СНз—ОН AlHa
2 ачннопротганол 1, fj-аминопрогтиловый спирт
СНг—СНг—ОН
N(C3H5)2
2 N, N дизтиламнноэтаиол
Если молекула амииоспирта содержит в своем составе две или три гидроксиалкильные группы, связанные через атом азота, в этом случае за основу берется название амииа
Н I НО—СН,—СН,— N—СН,—СН,—ОН
. оксиэтил) амин.
ди (2 гидроксиэтил) амии
СНг—СНг—ОН
НО--СН2—СН2— N—СН2—сн2—он
три (0-оисизтил )амин.
три (2 гидроксиэтнл) амин
Изомерия спиртов обусловлена теми же причинами, что и у ди-замещенных углеводородов.
Аминоспирты представляют собой ассоциированные соединения, в которых меж молекулярные водородные связи образованы с участием аминогрупп и спиртовых гидроксилов:
...NHz—СНг—СНг—OH--NH2—СНг—СНг—ОН--
278
14.4.1. Способы получения
Присоединение аммиака или аминов к а-окисям. Одним нз самых распространенных способов получения амииоспиртов служит реакция раскрытия а-окнсного цикла аммиаком или аминами
СН2—-СН2—он
Н2С----СН2 + NH3---> I
\ / nh2
о
оксиран, ЭПОКСИЭтВН
2 аминолзнол
Восстановление нитроспиртов. При восстановлении соответствующих нитроспиртов образуются аминоспирты
СНз—СН—СНзОН СНз—СН—СНаОН 4- 2Н2О
no2 nh2
Взаимодействие галогенсодержащих спиртов с аммиаком или аминами:
СНг-СН2ОН
I
С1
2-хлор этанол
+ (CHahNH -------► СН^СНг—ОН + НС1
N(CH3)2
диметиламин 2 N.N диметилачнноэтанол
14.4.2. Химические свойства
Амниоспирты относятся к бифункциональным соединениям, проявляющим свойства спиртов (см. подразд. 14.1.5) н аминов (см. подразд. 12.2.3).
Как основания аминоспирты образуют соли с минеральными кислотами
НО—СНз—сн,—NH2 + НС1 НО—СНг—СН,—NH3CI
2 амино-этанол 2 гндрокеиэтилачмпинй хлорид
гидрохлорид 2 амииоэтанола
Наличие гидроксильной группы, проявляющей электроноакцеп-торные свойства, приводит к некоторому понижению основности аминоспиртов. С увеличением числа метиленовых групп между
спиртоаым гидроксилом и аминогруппой это влияние ослабевает.
Амниоспирты, содержащие аминогруппу и спиртовый гидроксил у соседних атомов углерода, прн нагревании с серной
кислотой отщепляют молекулу воды, образуя гетероциклические
Структуры: н2С------СН2
СН2—СН2 „ ilipOj
н
+ Н2О
азкрндин. зтмлеиимин
279
Обладая нуклеофильными свойствами, амииоспирты взаимодействуют с эпокисями с образованием соответствующих дн-и тризамещеиных производных
* + СНг—СНг—NH2 4- Н2С--СН2
---НО—СНл—СН2—N—СНз—СН2—ОН -------------е—-
н
ди (2 гндроксиэтил)амии1 диэтаноламин
СНг—СНз—ОН
---> НО—СНг-CHz—N—СНз—СНг-ОН
трн(2-гидроксиэтил}вмии, триэтаноламин
При обработке аминоспиртов тионилхлоридом SOCla образуются галогеиосодержащне амины
СН2—СНз—ОН
Н— 4- 2SOCI2 -->
^СНз—СНз—ОН
дн (2 гидро кснэтн.1) а м и и
! |
ГВ
I
А г
СН^СНз—С1
Н—
^СНз—СН?—С1
ан (2 кюрэтнл (амин
+ 2SOB + 2НС1
Из три (2-гидроксиэтил)амина при взаимодействии с SOCI2 получается Х(СН2СН2С1)з — три (2-хлорэтил) амии — азотистый иприт, соединение, обладающее кожионарывиым действием, боевое отравляющее вещество.
14,4.3. Отдельные представители
Колам ии (2-аминоэтанол) H2N—СНз—СНз—ОН — вязкая гигроскопичная жидкость (т. кип. 171 °C), хорошо растворимая в воде. Водный раствор коламина имеет сильно щелочную реакцию. Коламин входит в состав сложных липидов. Образуется при расщеплении фосфатидов.
280
Холин [2-(гидроксиэтил)-триметиламмоиия гидроксид] +
[НО—СН2—СНа—1Ч(СН3)з]ОН_ представляет собой бесцветное кристаллическое вещество (т. пл. 180 °C), Холин содержится в животных тканях, растениях и микроорганизмах.
Синтетически холии можно получить действием трнметилами-на на оксиран в присутствии воды
Н2С---+ N(CH3)3 [НО—СН2—СН2— N(CH3)3]OH“
Холии необходим для жизнедеятельности человека и животных. Недостаток холииа может вызвать жировое перерождение печени и ее цирроз. Он относится к витаминам группы В, участвует в транспорте жиров и метаболизме белков и углеводов. Из продуктов литания наиболее богаты холином мясо, рыба, яичный желток и др.
В процессе гниения или при кипячении с баритовой водой холин подвергается дегидратации с образованием нейрина
+
[НО—СН2—СН2—N(CH3)3]OH~ --------«-
—н2о
4-
---- [CH2=CH--N(CHj)3]OH
Нейрин относится к токсичным веществам.
КОНТРОЛЬНЫЕ ВОПРОСЫ И УПРАЖНЕНИЯ
1. Назовите по рациональной, радикало-функциональной и заместительной номенклатуре ИЮПАК следующие соединения:
СН3
а) СНз—СН—СНз—СНз; б) СН3—С—СН3;
ОН ОН
в) СН3—CHs—СНз—СН2—ОН; г) СНг=СН—С На—ОН;
Какие из приведенных соединений являются изомерами?
281
2. Приведите четыре способв получения пропаиола-2.
3. Напишите схемы реакций изобутилового спиртв со следующими реагентами: a) Na; б) НВг; в) HNO3; г) H2SO4; д) СНэ—СООН; е) C2HSOH; ж) РС15; з) КзСг2О7.
4. Охарактеризуйте кислотио-основиые свойства спиртов. Расположите в ряд по уменьшению кислотных свойств следующие соединения:
а) этанол, пропаиол-1, пропаиол-2, трет-бутанол, метанол;
б) этанол, этиленгликоль, глицерин.
5. Приведите схемы реакций, которыми можно различить соединения в следующих парах: а) метаиол и этанол; б) этанол и этиленгликоль; в) этиленгликоль и глицерин; г) этанол и бензиловый спирт.
6. На примере вгор-бутилового спирта напишите механизм внутримолекулярной дегидратации спиртов, (
ФЕНОЛЫ
Гидроксильные производные углеводородов, у которых гидроксильная группа находится при углероде в $р2-гибридизации, входящем а цикл ароматического углеводорода, называют фенолами (ареноламн).
По числу гидроксильных групп фенолы делят иа одноатомные (ареиолы); двухатомные (арендиолы), трех-и полиатомные (ареи-триолы, аренполиолы).
14.5. ОДНОАТОМНЫЕ ФЕНОЛЫ
Фенолы, как и енолы, имеют гидроксильную группу при sp'2-гибридизованном атоме углерода. Одиако, если енолы неустойчивы и почти полностью изомеризуются в карбонильные соединения, то фенолы — устойчивые соединения и существуют исключительно в енольной форме:
Большая стабильность еиольиой формы обусловливается ароматическим строением углеводородного радикала н высокой степенью сопряжения ОН-группы с бензольным ядром.
Фенолы существенно отличаются по своим физическим и химическим свойствам и от спиртоа Глазной причиной этих отличий явлиется различный характер электронных взаимодействий гидроксильной группы с углеводородным радикалом, В спиртах гидроксильная группа связана с атомом углерода в 5рэ-гибридизации. За счет —/-эффекта гидроксильной группы происходит смещение электронной плотности по о-связи и образование на атоме кислорода частичного отрицательного заряда fi —, а иа атоме углерода — частичного положительного заряда 6-|-
(-0
2 83
В фенолах кислородный атом гидроксильной группы связан с углеродом ароматической системы и поэтому наряду с отрицательным индуктивным эффектом (—/) имеет место и положительный мезомерный эффект (Ч-Л1). В результате мезомерного эффекта происходит смещение неподеленной пары электронов атома кислорода к бензольному кольцу и на кислороде возникает частичный положительный заряд. Такой же отрицательный заряд переходит иа ароматическое ядро
Так как для гидроксильной группы мезомерный эффект по силе превосходит индуктивный (Ч-А1 > — /), суммарный частичный заряд иа атоме кислорода фенольного гидроксила положителен, тогда как кислород спиртового гидроксила имеет частичный отрицательный заряд.
Второй ряд отличий связан с различной реакционной способностью углеводородных радикалов. Эти отличия подчиняются закономерностям, которые характерны и для других функциональных производных углеводородов алифатического и ароматического рядов.
14.5.1. Номенклатура и изомерия
По заместительной номенклатуре ИЮПАК названия фенолов образуют от названий соответствующих ареиов с добавлением префикса гидрокси-. Для многих фенолов используют тривиальные названия. В качестве основы названий гомологов фенола чаще
всего используют ОН
слово -фенол:
2-иетилфенол. о-метилфенол. о-крезол
3-четилфенол м метил фенол, м-к ре зол
фенол, гидрокембензол
2 топропил-5-метнлфенол. тимол
4-этилфенол, I-гидрокси 4-этилбензол
нафтол-2, fj-нафтол
2Н4
Структурная изомерия фенолов обусловлена изомерией положения заместителей, как это имеет место в случае трех изомерных крезолов. Однако возможен и другой вариант, когда изомерия вызвана структурными изменениями заместителей. Например:
С Нэ—С Н2—С Нг—Ч—ОН
4-пропнл фенол
3- метил-4-этил фенол
14.5.2 . Способы получения
Природные источники. Природным источником фенолов является каменноугольная смола, из которой в 1834 г. ф. Рунге впервые получил феиол и назвал его карболовой кислотой. Из каменноугольной смолы также выделяют крезолы. Кроме того, феиол и крезолы образуются при крекинге нефти. Одиако фенол чаще всего получают синтетическими методами.
Синтез фенолов из аренов. Промышленный синтез фенола осуществляют из бензола. Реакцию проводят в две стадии. Вначале бензол сульфируют или хлорируют, а затем в жестких условиях под воздействием щелочи проводят замену сульфогруппы нли атома хлора на группу ОН:
SO3H
хлорбензол
2NaOH сплавление
фенолят натрии
8 % води р-р NaOH ---- --------------*-
300 ’С. 200 а тм
285
Эти реакции используют для получения и других ареиолов. Так, * для синтеза Р-нвфтола вначале сульфируют нафталин концентрированной H2SO4, а затем полученную р-нафталинсульфокислоту сплавляют со щелочью при 300 °C (реакция «щелочного плавления»). После прибавлении кислоты получают 0-нафтол:
H2SO4.
160 "С
Получение из кумола (изопропилбензола). Еще одни промышленный способ получения фенола основан иа окислении кумола. В качестве исходных продуктов используют бензол и пропилен
С||г--С.н-С1^
А1С13
Окисление кумола — экономически выгодный метод, так как позволяет получить наряду с фенолом другой важный продукт — ацетон. В настоящее время этот метод наиболее широко используется в промышленности.
Окислительное декарбоксилирование ароматических карбоно* вых кислот. При нагревании ареикарбоиовых кислот до температуры 200—300 °C в присутствии солей меди (II) происходит образование фенолов:
СООН
Оз, HjO
Cua+
феяол
бензойная кислота
286
Получение из солей арилдиазония. Фенолы можно получать при нагрепании водных растворов солей арилдиазония (см. с. 234):
^NCl + НОН —ОН + N2 + ЙС1
бензолдказоннн хлорил
фенол
14.5.3 . Физические свойства
Простейшие фенолы представляют собой вязкие жидкости или яизкоплавкие твердые вещества с очень специфическим устойчивым запахом («карболовый 'запах»). фенол растворим в воде (9 частей на 100 чвстей воды), другие фенолы в воде малорастворимы. Большинство фенолов — бесцветные вещества, однако при хранении под действием кислорода воздуха они могут окисляться и приобретать темную окраску за счет примеси продуктов окисления.
14.5.4 . Химические свойства
Все возможные реакции фенолов можно подразделить иа реакции с участием связи О—Н, связи С—О, арильного радикала, а также реакции восстановления и окисления. Одиако вследствие р, л-сопрйження неподеленной пары электронов атома кислорода с л-электроиной системой ароматического ядра прочность связи С О у фенолов значительно больше, чем у спиртов. Поэтому реакции фенолов с разрывом связи С—О протекают редко.
А. Реакции с участием связи О—Н
Кислотные свойства. Фенолы — более сильные ОН-кислоты, чем спирты. Это вызвано тем, что иеподелеиная пара электронов атома кислорода в молекуле фенола смещена к ядру (-|-Л4-эффект ОН-группы), что приводит к образованию частичного положительного заряда 64- на атоме кислорода н увеличению поляризации связи О—Н по сравнению со спиртами. Кроме того, образующийся феяолят-ион (фенокснд-ион) имеет повышенную стабильность за счет делокализации отрицательного заряда по ароматическому радикалу:
основание
сопряженная кислота
фсноксиц-иои (сопряжен кое основание)
287
Доказательством более сильных кислотных свойств фенолов по сравнению со спиртами может служить их реакция с водными растворами щелочей с образованием солей-фенолятов:
С6Н5—ОН + NaOH =₽=t С6Н5—O’Na+ + Н2О фенол фенолят натрия
В отличие от фенолов спирты, как более слабые кислоты, способны образовывать алкоголяты только при взаимодействии со щелочными металлами.
В водной растворе феноляты щелочных металлов частично гидролизованы (как соли слабых кислот и сильных оснований), и поэтому их растворы обладают щелочной реакцией. Под действием растворов минеральных кислот, карбоновых кислот или угольной кислоты из солей получают свободные фенолы.
На кислотность фенола значительное влияние оказывают заместители в ароматическом ядре. Так, введение в п-положеиие бензольного ядра молёкулы фенола электроноакцепторных заместителей (— NO2, —CN, —Hal н др.) усиливает кислотные свойства фенола. Например, п-иитрофенол — более сильная кислота, чем фенол. Введение же в п-положение электронодонорных заместителей (— NH2, —ОСНз и др.) приводит к понижению кислотности, поскольку при этом уменьшается смещение электронов связи О—Н к атому кислорода, что затрудняет отрыв протона:
Образование простых и сложных эфиров. При взаимодействии фенолятов с галогеналканами образуются простые эфиры (см реакцию Вильямсона, с. 165). Так как при этом алкилируется атом кислорода, то такие реакции называют О-алкилированием:
фенолят натрия
Na+ + С2Н5—Вг->-
О—С2Н5 -|- NaBr
фен и л этиловый эфир
288
фетиловые эфиры обычно получают взаимодействием фенолятов с диметилсульфатом
O'Na+
(CHjbSOj
-СН30 so3\a'
О—СН
з
чети лфенил оный дфир
Аналогично протекает реакция образования сложных эфиров взаимодействием фенолят-иона с ацилирующими реагентами — галогенаигидридами и ангидридами карбоновых кислот:
О
О
O'Na+ + СНэ
—С—СНз + NaCl
С1
ацетилклорид фенилаиетат
В реакцию ацилирования могут вступать и фенолы
С—СН3
jнеясный ангидрид
О - С- СНз + СНзСООН
фенилацетат
О
Ацилирование фенолов карбоновыми кислотами в присутствии серной кислоты, в отличие от ацилирования спиртов, практически не используется, так как эта реакция из-за уменьшения электронной плотности на атоме кислорода в молекуле фенола идет значительно медленнее.
Б. Реакции электрофильного замещения в ароматическом кольце
Проявляя электроиодоиорные свойства, гидроксильная группа °чеиь сильно активирует бензольное кольцо по отношению к реакциям электрофильного замещения SE и направляет заместители в о- и п-положения. фенолят-ионы в реакции \ еше более активны, чем соответствующие фенолы. В связи с высокой активностью фенолов необходимо принимать специальные меры для того, чтобы предотвратить реакции окисления и полизамещения.
Галогенирование, Обычно для введения атома галогена в бензольное кольцо требуются катализаторы — кислоты Льюиса.
Ю 5 Км
28»
Высокая реакционная способность фенолов приводит к тому, что реакция галогенирования для них идет очень легко в отсутствие катализатора Оии обесцвечивают бромную воду, причем происходит замещение всех атомов водорода в о- и м-положеииях:
ЗВгз (НгО)
2 4 6 трнбромфенол
фенол
о крезол 4 6 дибром 2 метндфенол
Избытком бромной
воды бромфеиолы могут броми роваться
и дальше:
2 4 4 6 тетрабром цикло гекса диен 2 5 пн 1
При этом происходит нарушение ароматичности бензольного кольца.
Дли того чтобы ввести одни илн два атомв галогена, необходимы специальные условия Так, если бромирование проводить в низкополяриом растворителе (CCL, СНС1з), образуются преимущественно монобромфеиолы с преобладающим количеством лара-изомера
290
0 других Усл°виях можно добиться преобладания днбромэамещен-ного фенола
ОН ОН
[J) 4- 2НВг
4^ / зо ‘с < /
Вг
2 4 дийромфенол
При хлорировании образуется преимущественно орто-изомер Иод непосредственно не иодирует фенолы
Нитрование. Реакция нитрования фенола происходит при комнатной температуре уже при обработке разбавленной взотной кислотой, тогда как для нитрования бензола используют нитрующую смесь. Прн этом образуется смесь о- и п-нитрофенолов;
При действии концентрированной HNOa фенол превращается в 2,4,6-тринитрофеиол (пикриновую кислоту)
+ ЗН2О
2 4 6 тринитрофенол пикриновая кислота
Раньше пикриновую кислоту использовали как взрывчатое вещество
Сульфирование. Сульфирование фенола проходит очень легко и в зависимости от температуры приводит к о- или п-изомерам:
° гилрокгнбенюл ^Чьфохнслота
HtSQj
-20 4С
H2SO4
100°Г
HO3S
п гигракснбензол сульфо кислота
ОН
JO-
291
о-Изомер прн нагревании его до температуры 100 °C перегруп-
пировываетси в п-изомер.
Нитрозирование. Фенол сравнительно легко нитрозируется азотистой кислотой уже при комнатной температуре с образова-
о нитрою фенол
д-нитрозо-фенол
+ 2HSO + 2NaCl
Алкилирование и ацилирование. Алкилирование фенолов можно проводить по реакции Фриделя — Крафтса, ио выходы в этой реакции, как правило невысокие. Наиболее чвсто для алкилирования используют спирты и алкены в присутствии H2SO4, Н3РО4 или BFs
Ацилирование фенолов также можно провести по реакции Фриделя — Крафтса, действуя на фенолы хлорангидридами пли ангидридами кислот в присутствии кислот Льюиса:
2 гидрокси 4-гидрокснацетофенон
ацетофенон
292
Однако чаще ацильные производные фенолов получают в две стадии: вначале фенолы превращают в сложные эфиры, которые затем нагревают с А1С1Э. При этом происходит миграция ацильной группы от атома кислорода гидроксильной группы в о- или п-положения ароматического кольца. Эта реакция получила название перегруппировки Фриса:
фе II илацетат
о гидрокси ацетофенон
и гидрокси ацетофено и
Азосочетание. Фенолы, и особенно феиолят-ионы, взаимодействуют с солями диазония с образованием азосоедииений (см. с. 237):
—N=NC1- +
- NaU. -НЙО
NaOH
бензолднаэония хлорид
4 гидроксиазобензол
Синтез фенолкарбоновых кислот. При нагревании фенолята натрия в токе СОз образуется салицилат натрия (реакция Кольбе), который действием минеральных кислот превращается в салициловую кислоту (см. с. 445):
салниялат натрин салициловая Кислота
293
Диоксид углерода — очень слабый электрофил, поэтому реакцию проводят с фенолятом натрия, как более активным субстратом.
Синтез ароматических гидроксиальдегидов. Обработка фенола хлороформом н водной щелочью приводит к введению в бензольное ядро альдегидной группы. Как правило, замещение происходит в о-положении-
HCI
— NaCI
салициловый альдегид
Эта реакция называется реакцией Раймера —Тимана. На первой ее стадии образуется замещенный хлористый бензилиден, который гидролизуется в щелочной среде, давая гидроксиальдегид.
Гидроксиметилирование. Реакции состоит в обработке фенолов формальдегидом в кислой или щелочной среде, в результате которой вначале образуется смесь о- и л-гидроксиметилфенолов, способных далее реагировать с фенолом, образуя фенолоформаль-дегидиые смолы
Механизм реакции — SE, причем электрофилом в условиях +
кислотного катализа является катион СН2ОН:
н2с=о сн2—он
формальдегид гндрокенметнл катион
294
В присутствии минеральных кислот гидроксиметилфеиол вступает в реакцию конденсации с молекулой фенола
З.Т'Дигидроксидифенилметам
В более жестких условиях образуются высокомолекулярные продукты поликонденсации — феиолоформальдегидные смолы
ОН ОН он он
Их используют для получения пластмасс, обладающих повышенной термостойкостью, а также как основы для ионитов (ионообменные смолы), которые применяются для деминерализации воды, для очистки веществ, в том числе и лекарственных препаратов
В. Реакции восстановления и окисления
При каталитическом гидрировании, например действии водорода в присутствии Ni, фенолы восстанавливаются до циклоалканолов
Окисление фенолов протекает весьма сложно. В зависимости от природы окислителя образуются разные продукты. Например, оксид хрома (VI) в кислой среде окисляет фенол в п-бензохииои: ОН О
+ Сг(ОН)з
29S
При окислении фенола персульфатом калия KsSaOa в щелочной среде образуется гидрохинон (ревкция Эльбса)
ОН ОН
он
гидрохинон
14.5.5 . Идентификация одноатомных фенолов
Химические методы. Для большинства фенолов характерна цветная реакция с хлоридом железа (111). В результате реакции образуются комплексные соединения, имеющие интенсивную окраску. Например, при взаимодействии фенолв с FeCh образуется смесь комплексных соединений СбН5ОРеС12, (СвН5О) aFeCl, (СбНзО)эРе, окрашенных в фиолетовый цвет. Крезолы с FeCh дают голубую окраску, фенолы со свободными орто- и параположениями обесцвечивают бромиую воду и образуют при этом продукты замещения, которые обычно выпадают а осадок (см. с. 290).
Для идентификации фенолов может быть использована реакция азосочетания с солями диазония (см. с. 237). Обычно с этой целью применяется -диазотированная сульфаниловая кислота. В процессе взаимодействия с фенолами образуются азокрасители (окрашенные продукты).
Спектральные методы. Для фенолов в ИК-спектрах характерна сильнан широкая полоса валентных колебаний связи О—Н в той же области, что и для спиртов: 3200—3600 см-1. Положение полосы ввлентных колебаний связи С—О (~1230cm j) отличается от положения этой полосы в спектрах спиртов (1050— 1200 см"1). Для фенолов в ИК-спектре наблюдается и поглощение ароматического кольца В спектрах ПМР положение сигнала протона гидроксильной группы, как и у спиртов, очень зависит от того, насколько сильна водородная связь, а следовательно, и от температуры, концентрации и природы растворителя. Сигнал может проявляться в виде уширенной полосы в области 6 4—7 м. д. и при наличии водородных связей смещается в еще более слабое поле (fi 6—12 м. д.).
В УФ-спектрах фенолов наблюдается поглощение в областях 210—230 нм н 270—280 нм. *
14.5.6 . Важнейшие представители фенолов >,
Фенол СбНэОН. Представляет собой бесцветные, розовеющие на воздухе вследствие окисления кристаллы (т. пл. 43 °C, т. кип. 182 °C), растворяется в воде (при 15 °C — около 8 %).
296
Обладает антисептическими свойствами. В виде 5 % водного раствора (карболовая кислота) используется в качестве дезинфицирующего средства. Широко применяется в производстве пластмасс, красителей, взрывчатых веществ, лекарственных средств. Фенол токсичен, может вызвать ожоги кожи.
о~, м-, н-Крезолы —ОН. Применяются для получения
Н3С ”
пластмасс, красителей и др. Смесь изомерных крезолов с мылами используется как дезинфицирующее средство в ветеринарной практике (лизол, креолин).
Тимол (2-изопропил-5-метилфеиол) НзС—р—СН(СН3)2-
Кристаллическое вещество (т. пл. 50 °C, т. кип. 232,9 °C), хорошо рвстворимое в этаноле, диэтиловом эфире, бензоле. Применяется в медицине как антисептическое и противоглистное средство. Используется в производстве ментола.
Пикриновая кислота (2,4,6-тринитрофеиол)
азокрасителей, анализе.
Представляет собой желтые кристаллы, мало растворимые в воде, лучше - - в органических растворителях (т. пл. 122 °C). Взрывается при трении, нагревании до 300 °C, ударе, контакте с металлами и их оксидами Пикриновая кислота применяется в производстве
широко используется в фармацевтическом
а - Нафтол
(1 -гидроксииафталии)
ОН
. Желтоватые
кристаллы, плохо растворимые в воде, растворимые в спирте, диэтиловом эфире, хлороформе, бензоле (т. пл. 122 °C, т. кип. 286 °C). Применяется в производстве иафтолсульфокислот, галогене- и иитронафтолов, азокрасителей.
ОН
Р -Нафтол (2-гидроксинафталин) Белый
кристаллический порошок, растворимый в органических растворителях и нерастворимый в воде (т. пл. 93 °C, т. кип. 351 °C). Используется в производстве нафтолсульфокислот и антиоксидантов для каучуков, а также .как реагент в фармацевтическом анализе,
297
14.6. ДВУХ-, ТРЕХ- И ПОЛ И АТОМНЫЕ ФЕНОЛЫ
Названия многоатомных фенолов образуют по общим правилам номенклатуры ИЮПАК- Для простейших соединений используют и тривиальные названия:
гидрохинои.
1.4 ди г и дрок с и бе и зо л
пирокатехин.
1.3 ДН1 н дроке и бе изол
резорцин
1.3 днгидроксибеизол
пирогаллол,
1,2.3 тригидрокси бензол
флороглюцин.
1 3 5 три гидрокси-бензол
оксигидрохинон.
1,2.4-трнгидрокси-бенэол
14.6.1. Способы получения
Сплавление сульфокислот со щелочами:
SOaH
л/ бензол ди сульфокислота
NaOH 1 -------►
резорцин
Взаимодействие со щелочами дигалогенопроизводных бензола или галогена замещенных фенолов:
о-х.юрфеиол
NaOH, -300 ’С —----------->-
пирокатехин
Получение гидрохинона из анилина. Для получения гидрохинона используют реакцию окисления анилина до хииоиа с последующим его восстановлением
296
14.6.2. Химические свойства
Для многоатомных фенолов характерны те же реакции, что и дли одноатомных. Они легко галогенируются, нитруются, сульфируются и т. д. Однако течение реакций имеет свои особенности.
Кислотные свойства. Двухатомные фенолы ивляются более сильными кислотами, чем феиол. Поэтому оии способны образовывать соли не только с щелочными, но и с тяжелыми металлами. Так, пирокатехин при взаимодействии с ацетатом свинца образует нерастворимую свинцовую соль:
(CHaCodhPb —
-асн3соон
Окисление. Многоатомные фенолы намного легче, чем одноатомные, взаимодействуют с окислителями. Например, при окислении пирокатехина и гидрохинона образуются соответствующие хииоиы. Особенно легко окисляется гидрохинон:
ОН
пирокатехин
ОН
AgaO. эфнр^ Na2SO4
о-бензохннон
гидрохинон л бензохинон
м-Дигидрокси бензолы также окисляются, но не образуют м-бензохиноиов, так как для последних невозможна плоская ненапряженная структура.
299
Реакции замещения и конденсации. Эти реакции протекают еще легче, чем у фенола. Даже в мягких условиях получаются ди-и тризамещеиные фенолы:
NO2
o2n no2
резорцин
стифниновая кислота
Очень легко происходит конденсация многоатомных фенолов с альдегидами н ароматическими спиртами
1,1 дн(3' 4' дигидриксифенил)этян
2.4 Дигидрокендифенилметан
14.6.3. Идентификация
Для анализа многоатомных фенолов пригодны те же методы, что и для одноатомных. Особенно часто используется реакция с FeCh- При этом различные многоатомные фенолы дают разную окраску, которая зависит от наличия в бензольном цикле других функциональных групп, количества гидроксилов, их расположения (табл. 14,2).
300
Таблица 142
Окрашивание фенолов в реакции с FeCh
Структурная формула Название Цвет продуктов реакции с FeCla
он
а он он Пирокатехин Резорцин Зеленое окрашивание Синее окрашивание
он он
но7 0х Гидрохинон Зеленое, переходящее в желтое
Н°\ он он Пирогаллол Красное окрашивание
НО\ он Флороглюцин Темно-фиолетовое окрашивание
14.6.4. Важнейшие представители
ОН
Пирокатехин (о-дигидроксибеизол) Кристал-
ОН
лическое вещество, растворимое в воде и в спиртах (т. пл. 105 °C, т. кип. 245 °C), На свету и воздухе приобретает коричневую окраску вследствие окисления. Обладает антисептическими свойствами, используется как исходное вещество в синтезе лекарственного препарата адреналина; нашел применение в фотографии как проявляющее вещество. ,
НО ОН
Резорцин (лс-дигндроксибензол) Кри-
сталлическое вещество, растворимое в воде (т. пл. ПО °C, т. кип.
)
I 301
I
178 °C). Применяется в производстве красителей, резорцииоформ-альдегидных смол. Хороший антисептик при лечении кожиых заболеваний (в составе примочек и мазей). Резорцин раздражает кожу и слизистые оболочки.
Адреналин (а-3,4-дигидроксифенил-р-метиламиноэтаиол)
НО—)>—СН—СН2 . Кристаллическое, вещество, хорошо
in NHCH3 nU
растворимое в горячей воде. Является гормоном группы катехоламинов, вырабатываемым внутренней мозговой частью надпочечников. Принимает участие в регуляции углеводного и жирового обменов Нашел применение в медицинской практике в виде гидрохлорида или гидротартрата в связи со способностью вызывать сужение мелких кровеносных сосудов, повышать артериальное давление, стимулировать деятельность сердца и др.
НО
Пирогаллол (1,2,3-тр11гидроксибензол) НО
hoz
Белое кристаллическое вещество, растворимое в воде, спиртах (т. пл. 134 °C, т. кип. 309 °C). На свету темнеет. Сильный восстановитель. Чрезвычайно быстро реагирует с кислородом в щелочном растворе, в связи с чем используется для поглощения О2 в газоанализаторах. Используется в производстве красителей, как восстановитель при органическом синтезе, проявитель в фотографии.
НО
Флороглюцин (1,3,5-тригидроксибеизол) —ОН.
hoz
Кристаллическое вещество, растворимое в спиртах и плохо — в воде (т. пл. 223 °C, воэгоияетси). Применяется как азосоставляющий компонент светочувствительных бумаг и пленок. Используется также в анализе как аналитический реагент, в частности, для обнаружения пентоз.
14.7. АМИНОФЕИОЛЫ
Амипофенолами называют производные ароматических углеводородов, содержащие в своем составе фенольный гидроксил и аминогруппу.
По взаимному расположению аминогруппы и фенольного гидроксилв различают орто-, мета- w пара-аминофенблы:
302
о амннофснол
м аминофенол
П яминофенпл
14.7.1. Способы получения
Восстановление нитрофенолов. Это один из наиболее распространенных способов получения амииофенолов. Восстановление обычно проводят цинковой пылью в щелочном растворе:
ОН
+ 2Н2О
я нитрофенол
я аминофенил
Взаимодействие двухатомных фенолов с аммиаком. При нагревании двухатомных фенолов с аммиаком происходит замещение одного из фенольных гидроксилов иа аминогруппу:
+ Н2О
Восстановление нитробензола. Этот метод используется для промышленного получения п-аминофенола. Он оГнован на перегруппировке образующегося феиилгидроксиламина под действием концентрированной серной кислоты:
нитрозобензол
N=O
фенил гидроксила мин
п аминоферол )
30
14.7,2. Химические свойства
Аминофеиолы обладают свойствами фенолов (см. подразд. 14.5.4) и свойствами ароматических аминов (см. под-разд, 12.3.3). Основные свойства аминофенолов несколько выше, чем у анилина. Это связано с электронодоиорным влиянием гидроксильной группы
В то же время присутствие ароматической аминогруппы приводит к некоторому понижению кислотного характера фенольного гидроксила
Таким образом, амииофенолы являются амфотерными веществами и способны образовывать соли с минеральными кислотами и со щелочами
л гитрокси | п ачннофенол л амииофенолят натрия
анилиннй хлорид | натриевая соль л амннофенолл
Из специфических реакций аминофенолов следует выделить реакцию ацилирования для о- и n-аминофеиолов и способность о-аминофенола циклизоваться с образованием гетероциклических соединений.
304
При ацилировании /г-аминофеиола происходит образование N-ацильных производных
nh2 7 NHCOCHa А
(A + (CH3CO)2O > - f J + СНзСООН
1 он 1 он
п амнпофеиол N ацетил п а мни оф енол п гидрокси ацетанилид
Действие уксусного ангидрида на о-амииофенол к замыканию цикла с образованием 2-метилбензоксаэола
о аминофенол
2 мети1бензокса'ол
+ СНэСООН + Н2О
о- и ц-Амииофенолы очень легко окисляются, при этом образуются хииоиимины
305
14.7.3. Отдельные, представители
NH2
орго-Амииофеиол Кристаллическое ве-
ОН
щество, быстро темнеющее на воздухе (т. пл. 174 °C). Применяется в производстве некоторых азокрасителей, лекарственных препаратов (нитроксолин и др,), входит в состав красителя для меха,
NH2
ме та - А м и но ф е но л Бесцветное кристалли-
ОН
ческое вещество (т. пл. 123 °C), Является важным исходным продуктом для получения роданиновых и розамииовых красителей. Особое значение в производстве красителей имеет его N.N-диме-тильное производное.
я а ра - А м и н о фе н о л НО——NH2 . Кристалличе-
ское вещество (т, пл. 184 °C), быстро темнеющее на воздухе вследствие образования продуктов окисления. Применяется как проявляющее вещество в фотографии, широко используется в качестве промежуточного продукта в производстве красителей. Его ацетильное производное — n-ацетиламинофенол (парацетамол) применяется в медицине как жаропонижающее и болеутоляющее средство
НО—£>-ИНСОСН.
п ацетдмндофено! 1ыраиет<шол
Большое значение в синтезе лекарственных препаратов и красителей имеют эфиры аминофенолов. Метиловые эфиры аминофенолов называются аиизидинами, а этиловые - фенетидииами:
°СНз
метиловый эфир я аиинофеноid, этиновый эфир п амннофенола
п анмзндин и фенэтидин
Ацетильное производное п-фенетндина - фенацетин применяется в качестве жаропонижающего и антиневралгического средства:
C.H.O^-.H- |РСНз
фенацетин О
306
14,8. ТИОЛЫ
Тиолами называют производные углеводородов, в молекулах которых один или несколько атомоа водорода замещены мериаптогруппой —SH.
Меркаптогруппу еще называют тиольной или сульфгидрильной группой Тиолы можно также рассматривать как тиоаиалоги гидроксильных производных углеводородов, в молекулах которых атом кислорода группы -ОН заменен на серу. Для соединения этого класса используют еще название меркаптаны. Тиолы, в которых меркаптогруппа связана с алифатическим радикалом, называют тиоспиртами, а с ароматическим радикалом -- тпофенолами:
С2Н5—SH
тиоспнрт
тнофенол
Номенклатура тиолов аналогична номенклатуре гидроксильных производных с той лишь разницей, что вместо суффикса -ол используется суффикс -тиол или вместо префикса гидрокси- применяется префикс меркапто-, например:
СНз—SH СНз—СН 2— S”Na+ HS—СН2—СНг—SH
метан тиол, четилмеркантан
этантиолят натрия
Этилмеркаптид натрия
этандитиол 1,2
SH
SH
SH
СНз
SH
меркаптобенаол, тиофенол
2 метнлтиафсноя
1,2 -дн м е р ка птобенэол
Тиолы, за исключением метантиола,— жидкие или твердые вещества. Как правило, их температуры плавления и кипения ниже гидроксианалогов, что связано с меиьшей электроотрицательностью атома серы по сравнению с атомом кислорода и соответственно меиьшей склонностью к образованию водородных связей. По этой же причине тиолы хуже растворяются в воде, чем спирты и фенолы. Как правило, меркаптаны ядовиты и обладают чрезвычайно неприятным запахом.
14.8.1. Способы получении
Взаимодействие галогеналканов с гидросульфидами щелочных металлов. При действии иа первичные и вторичные галогеналканы гиДросульфнда калия или натрия образуются тиоспирты. Реакция Протекает по механизму (см. с. 158).
307
С2Н5С1 + NaSH -* C2H5SH + NaCl
хлорэтан этантии 1
С третичными галогеиалканами эта реакция ие идет, поскольку в данном случае преобладает процесс элиминирования.
Взаимодействие спиртов с сероводородом. При обработке спиртов сероводородом в присутствии катализатора AI2O3 при температуре 350 400 °C образуются тиоспирты-
с2н5он + H2S -3^. C2H5SH + Н2О
эта hoi этантнол
В реакцию вступают только первичные спирты.
Восстановление аренсульфокислот и аренсульфонилхлоридов. При восстановлении аренсульфокислот и аренсулъфопилхлоридов образуются тиофенолы В качестве восстановителей используют цинк в растворе серной кислоты или алюмогидрид лития LiAlH*:
бензоле у ть фок и слота тиофено i
SO2CI
+ Н2О + НС1
бензо icy тьфохлорид
14.8.2. Химические свойства
По химическим свойствам тиолы во многом сходны с гидроксильными производными углеводородов. Особенности их химического поведения обусловлены уменьшением прочности связи S—Н по сравнению со связью О—Н в спиртах и фенолах, поэтому тиолы обладают более выраженными кислотными свойствами, чем соответствующие им гидроксианалог и (см кн 1,с. 100), Реакции тиолов в основном обусловлены ионизацией связи S—Н и нуклеофильными свойствами атома серы.
Образование гиолятов (меркаптидов). Тиолы, как более сильные кислоты по сравнению с аналогичными О—Н кислотами, легко образуют соли — тиоляты (меркаптиды) ие только со щелочными металлами, ио и с ионами тяжелых металлов:
С2Н5—SH + NaOH -> С2Н5—SNa + + Н2О
этантио л этаитнотнт натрия
2С2Н5—SH + -Ь (C2H5S)2Hg + 2Н +
этил меркаптид ртути
308
На этом свойстве тиолов основано их применение в медицине в качестве антидотов при отравлении тяжелыми металлами.
Взаимодействие тиолов с алкенами. В присутствии пероксидов или под действием УФ-излучения тиолы присоединяются к алкенам по месту разрыва двойной связи с ориентацией против правила Марковиикова. Реакция протекает по механизму
о
I-------1 сн‘-с(
СНз—S—Н + СНг=СН—СНз---------------
t__________t
истантиол iiponrw
+ СНЭ—S—СН2—СН2—СНа
гегил н пропил сульфид
Ацилирование тиолов. Реакция тиолов с карбоновыми кисло* тами катализируется сильными кислотами и приводит к образованию тноэфиров карбоновых кислот:
О О
СНз—4- HS—С2Н5 сн3—+ н2о
^ОН S—С2Н5
уксусная кислота этантнол этиловый лфир
тиоуксусной кислоты
Эта реакция аналогична реакции этерификации карбоновых кислот и спиртов (см. разд. 16.1.4).
Окисление тиолов. В отличие от спиртов, тиолы окисляются не по атому углерода, а по атому серы. Продукты реакции зависят от условий окисления. При окислении в мягких условиях (действие таких окислителей, как Н2О2, СиС12 и др.) образуются диалкилдисульфиды:
R-S-H + О + H-S-R —► R-S-SR + Н2О диалкил-дисульфид
При окислении сильными окислителями (KMnCh, HNO3 или HOI) образуются сульфокислоты:
О
R— S—Н -IHU. R—S—ОН А тиот су1ьфоновая кислота,
сульфокислота
ЗОВ
14.0. ПРОСТЫЕ ЭФИРЫ
Простые эфиры можно рассматривать как производные спнр. то в, еиолов и фенолов, образующиеся в результате замещения атома водорода гидроксильной группы углеводородным остатком:
R—О— R' простой эфир
Радикалы в простых эфирах могут быть одинаковыми (симметричные эфиры) или разными (несимметричные или смешанные эфиры):
С2Н5—О—С2Н5;
симметричные эфиры
СНз—
аНэ
несимметричные эфиры
-ц-
14.9.1. Номенклатура и изомерия
Названия простых эфиров по радикало-функциональной номенклатуре обычно образуют из названий углеводородных радикалов, связанных с кислородом, и суффикса -овый
СНз—О—СНз дн мет иловый эфир
С2Н5—О—C2H5
диэтиловый эфир
СНз—О—С2Н5
метнлэтил пвый эфир
О—СНз
метилфен иловый
эфир
В заместительной номенклатуре ИЮПАК простые эфиры рассматривают как производные углеводородов, в которых один из атомов водорода замещен алкоксигруппой (RO—). За родоначальную структуру принимается более сложный по структуре радикал:
2 1 12 3 4
С 2 Hs—О—С Н—С Нз С Нэ—О—С На—С Н 2—С Н—С Нэ
3СНз СНз
2 этоксипропаи
1-метоксн 3 мети л бутан
Изомерии простых эфиров обусловлена изомерией радикалов, связанных с кислородом
310
СНз—О—-СНа—СНг—СНз
метили ро а иловый »фнр
1 метокси пропан
СНз—СНг—О—СНг-СНз
диэтиловый эфир, этоксизтан
СНз
СНз—О—СН
СНэ
метил и зопроп иловый эфир. 2 метоксипропяи
14.9,2. Способы получении
Взаимодействие алкоголятов и фенолятов с еалогекаяканами (см. реакция Вильямсона, с. 165):
С2Н5—O“Na+ + СаН5—I ------> С2Н5—О—С2Н5 + Nal
диэтнловын эфир
СбНз—O“Na+ -|- С2Н5—I ----► СбНь—О—CaHs -f- Nal
феннлэтнловый эфир
Межмолекулярная дегидратация спиртов, При нагревании спиртов в присутствии концентрированной H2SO4 или других водоотнимающих средств образуются простые эфиры (см. с. 257)
СНэ—СНг-ОН- На~*> СИз—СНг—О—СНа—СНз + Н2О 140 °C
14.9.3. Физические свойства
Диалкиловые эфиры — это бесцветные жидкости со своеобразным «эфирным» запахом и относительно низкими по сравнению с соответствующими спиртами температурами кипения. Алкилариловые и диариловые эфиры являются бесцветными жидкостями или кристаллическими веществами с приятным запахом (табл. 14.3). Молекулы простых эфиров не образуют межмолекулярных водородных связей,
14.9.4. Химические свойства
В химическом отношении простые эфиры являются весьма инертными веществами. Разведенные минеральные кислоты и щелочи иа холоду с ними ие реагируют. За счет наличия на атоме кислорода иеподелеииых пар электронов простые эфиры проявляют слабые основные свойства. Под действием иодоводородной и концентрированной серной кислот простые эфиры подвергаются расщеплению. Реакционная способность виниловых и ариловых эфиров обусловлена наличием углеводородного радикала.
311
Таблица 14з
Физические характеристики некоторых простых эфиров
Соединение Температура. “С
плавления кипения
СНз—О—СНа СН-)—СНг— О—СНг—СНз СНз- СН2 -О—СН=СН2 -138,5 -116,3 — 23,7 34.6 36
СНз — 155
26 259
Образование оксониевых солей. Концентрированные минеральные кислоты (НС1, H2SO4, HNO3 и др.) образуют с простыми эфирами оксониевые соли Протон кислоты присоединяется к атому кислорода эфира за счет неподелеиной пары электронов атома кислорода:
СНз—СН?—О—СН3 + HClz^tl-СНз—СН2—О—CH3"VC1-
мети л этиловый эфир
окссниевая соль
Реакция расщепления простых эфиров (ацидолиз). Реакция с иодоводородной кислотой. Простые эфиры разлагаются концентрированной HI уже иа холоду с образованием 'алкилиодида и спирта
СНз—О—СНз + Н1 --> СНзОН + СН31
ля мсти юпый эфир
метиловый метил
спирт иоднд
Реакция расщепления диметилового эфира HI проходит по механизму Sn2 и начинается с образования оксониевого иона, который затем подвергается атаке нуклеофилом:
Н1^±Н4 + Г
v + + - Н
снэ-о-сн3 + н <±сн3-о-с-н + I СН3ОН + СН31 tr н диметиловый п _ меткл-
эфир оксомиевый ион метанол • ИОдЦд
Если реакция ведется при нагревании, то образуется вторая молекула метилиодида
312
СНЭ-ОН + н+ —► f + *сн3-о-н —► CHJ + Н2О н
реакция с концентрированной серной кислотой. Взаимодействие с концентрированной H2SO4 приводит к расщеплению простых эфиров- Реакция проходит или по механизму 5n2 (эфиры первичных спиртов), или по механизму SN1 (эфиры третичных спиртов) и приводит к образованию сложных эфиров
серной кислоты
СгНе—О—С2Н5-
2Н;5О<
2С2Н5—О—SO3H + Н2О
этил сульфат
днэтиловый эфир
кснц c2Hs—O--SO2—О—С2Н5 _|_ Н2о
ди эти п сульфат
Окисление простых эфиров. При длительном пребывании иа воздухе простые эфиры окисляются с образованием взрывоопасных гидропероксидов R—О—ОН и пероксидов R—О—О—R. Поэтому перегонку эфиров нельзя вести досуха из-за опасности взрыва. Для разрушения пероксидов эфир обрабатывают восстановителем, разрушающим пероксид. Хранят свободные от пероксидов эфиры иад металлическим натрием или гидридом кальция.
14.9.5. Идентификации простых эфиров
Ввиду низкой реакционной способности простых эфиров идентификацию их с помощью химических методов осуществляют редко,
Обычно строение простых эфиров устанавливают анализом продуктов их расщепления иодоводородной кислотой.
В ИК-спектрах простых дналкиловых эфиров наблюдаются полосы валентных колебаний связи С—О (1060—1150см-1), а для ариловых и виниловых эфиров — в области 1200—1275 см-1. В спиртах также наблюдается полоса vc 0 в области 1100 см-1, но для простых эфиров в ИК-спектрах нет характеристической полосы О~-Н-группы.
В ПМР-спектрах строение простых эфиров может анализироваться лишь по влиянию атома кислорода иа сигналы соседних связей С—Н.
14.9.6. Отдельные представители
Диэтиловый эфир* (этоксиэтан) С2Н5—О—С2Н5. Бесцветная, летучая, легковоспламеняющаяся жидкость со специфическим запахом (т. кип. 36,5 °C), образующая с воздухом взрыв-
* Ди-этиловый эфир обычно называют эфиром или этиловым эфиром
313
чатую смесь.‘Смешивается с большинством органических растворителей. Не смешивается с водой, значительно легче последней. Широко применяется как растворитель, а в медицине в качестве препарата для общего наркоза. При действии солнечного света иа воздухе ои легко окисляется с образованием уксусного альдегиде, гидропероксида (СН3—СНОН—ООН), пероксида (СН2—СНОН—О—О—СНОН—СН3) и других весьма ядовитых веществ, присутствие которых недопустимо в эфире для наркоза.
ДиоксаиО О. Прозрачная бесцветная жидкость
(т. пл. 11,8 °C, т. кип. 101,5 °C), смешивается с водой, горит голубым пламенем. Эффективный растворитель органических веществ. Ядовит.
Анизол (метоксибензсл, метилфеииловый эфир) CgHg—О—СНз. Жидкость, смешивается с органическими растворителими, не смешивается с водой (т. пл. —37,5 °C, т. кип. 153,8 °C). Раздражает слизистые оболочки. Применяется в производстве душистых веществ, дисперсных красителей.
Фенетол (этоксибензол, фенилэтиловый эфир) С6Н5—О—С2Н5. Бесцветней жидкость, смешивающаяся с этанолом, эфиром и другими органическими растворителями, практически не смешивается с водой (т. пл. —29,5 °C, т. кип. 170 °C). Используется как растворитель и в производстве душистых веществ.
м.10. СУЛЬФИДЫ
К сульфидам (тиозфирам) относят тиоаналоги простых эфиров общей формулы R—S—R'. Например:
СНз—СН2—S—СН2—СНз
фенн л эт ил сульф и д, этилтиобенлол
днэтилсульфид. зтнлтиоэтаи
14.10.1. Способы получения
Алкилирование тиолятов галогеналкйнами. Этот метод аналогичен синтезу простых эфиров по Вильямсону:
C2HS—Вг + NaSCH3——С2Н5—S—СН3
—NaBr
четилэтидсульфид
Симметричные сульфиды получают действием сульфида нат,-рия иа галогеиалканы
2СаН5—Вг + Na2S • - в > С2НВ—S—С2Н5
—JNa вг дм эти л сульфид
Взаимодействие еалогенидов серы с аренами. Для получения ди а рил сульфидов используют реакцию электрофильного заме-
314
щеиия в аренах. В качестве электрофильного реагента используют галогениды серы в присутствии кислот Льюиса:
4- 2НС1 4- S
дифен ил сульфид
Сульфиды представляют собой бесцветные вещества с неприятным запахом. Они практически нерастворимы в воде.
14.10.2. Химические свойства
Сульфиды по своим химическим свойствам во многом напоминают эфиры, однако для них характерны и особенности, связанные с большей поляризуемостью атома серы по сравнению с атомом кислорода.
Образование солей сульфония. Сульфиды, как и простые эфиры, являются слабыми основаниями. Поэтому они способны про-тонироваться в кислой среде. Например, в концентрированной серной кислоте образуются соли сульфоиип:
R
R—S—R Н HSO?
R^
сульфид соль сульфония
Алкилирование сульфидов. Сульфиды сравнительно легко реагируют с галогеналканами с образованием достаточно стабильных галогенидов триалкилсульфоиия:
Н3С
СНз—S—СНз > НзС—S+ Г
НзС^
дн метил сульфид и од ид триметиясульфония
Окисление сульфидов. Продукты окисления сульфидов могут быть различными в зависимости от использующихся окислителей и условий проведения реакции. Так, при окислении сульфидов иодо водородной кислотой при О °C образуются сульфоксиды:
О
R—S—R -12U R— S— R
сул ьфокснд
315
Дальнейшее окисление сульфоксидов приводит к сульфоШиц h
О
R— S— R -SU R—1—R
4 А
сульфо и
Реакция с солями тяжелых металлов. Сульфиды легко реагируют с солями тяжелых металлов с образованием комплексов, например:
RaS -|- HgCla —-> RzS’HgClg диалкнлсу-и.фи! сулема комплексное соединение
Эта реакция применяется в анализе для идентификации сульфидов. Кроме того, на этой реакции основано использование некоторых сульфидов в качестве антидотов при отравлении солями тяжелых металлов.
14.10.3. Отдельные представители. Применение
Диметилсульфид СНз—S—СН3. Представляет собой бесцветную летучую жидкость с иеприитиым запахом. Получают в присутствии катализатора из метанола и сероводорода. Используют для производства диметилсульфоксида.
Иприт (фф'-ди хлордиэтил сульфид) CICH2CH2—S—СНгСН2С1 Бесцветное кристаллическое вещество, плохо растворимое в воде, хорошо — в органических растворителях (т. кип. 157 °C). Технический иприт представляет собой буроватую жидкость с запахом горчицы. Чрезвычайно ядовит. Это боевое отравляющее вещество (БОВ) кожио-нарывного и общетоксического действия, противоплазматический яд. Как БОВ был впервые применен Германией в 1917 году у бельгийского города Ипр (откуда и название - иприт) против англо-французских войск. Смертельная концентрация иприта при попадании на кожу 70 мг/кг. В качестве защиты используют противогаз и спецодежду. Для дегазации используют хлорирующие и окисляющие агенты.
Диафеиилсульфон (4,4'-днаминодифенилсульфои). При-
>—ч х меняется в медицине как ле-
H2N—Г у—SO2—у—NH2 карствеииый препарат для ле-
' \=/ чеиия лепры (проказы).
Димексид (ди метил сульфоксид, ДМСО) (СН3) 2^—О. Бесцветная жидкость (т. кип. 189 °C). Хороший органический, растворитель, широко использующийся в органическом синтезе, промышленности. Применяется как лекарственный препарат с противовоспалительным, обезболивающим и антимикробным действием. В основном используется для наружного применения (примочки, мази, растворы и др.).
316
Дпгидрохлорид цистамина [дигидрохлорид бис-й-амнноэтил дисульфида]
н H2N—CHi-CHa—S—S—CH2--CH2—NH2-2HCI.
Нашел применение как лекарственный препарат для профилактики лучевой болезни.
КОНТРОЛЬНЫЕ ВОПРОСЫ И УПРАЖНЕНИЯ
Какие из приведенных соединений являются изомерами?
2. Сравните кислотно-основные свойства одноатомных спиртов и фенолов. Расположите в ряд по уменьшению кислотных свойств соединения: а) этанол и фенол; б) фенол, д-нитрофеиол, п-ами-нофенол.
3. Напишите схемы реакций фенола и резорцина со следующими реагентами: a) NaOH; б) (СН3СО)2О; в) Вгг(НзО); г) Вг2(СС14); д) HNO3KOHU.;e) HNO3 разб.; ж) H2SO4 (100 °C).
4. Какие качественные реакции можно использовать для обнаружения: а) фенола; б) резорцина; в) пирокатехина; г) гидрохинона?
5. Почему простые эфиры имеют более низкую температуру кипения по сравнению с соответствующими спиртами?
6. Напишите уравнения реакций диметилового эфира со следующими реагентами: а) НС1; б) HI; в) H2SO4 коиц.; г) О2.
7 Сравните кислотно-основные свойства тиолов и их гидроксильных аналогов. Ответ поясните.
8. Приведите уравнения реакций этантиола со следующими соединениями:
a) NaOH; б) СН-^СН—СН—СН3; в) С2Н5—СООН; г) О2.
СНз
Глава 15 АЛЬДЕГИДЫ И КЕТОНЫ
Альдегидами н кетонами называют производные углеводородов, содержащие в своем составе карбонильную группу ( С=О).
Поэтому их еще называют карбонильными соединениями.
В альдегидах карбонильная группа связана с углеводородным радикалом и атомом водорода. Общая формула альдегидов о О
Z //
R—С Группировка —С получила название альде-
Хн \
гидная группа, В кетонах карбонильная группа связана с двумя углеводородными радикалами. Общая формула кетонов R—С—R Карбонильную группу в кетонах часто называют
кетогруппой.
В зависимости от строения углеводородного радикала альдегиды н кетоны подразделяют иа алифатические, алициклические и ароматические. Среди алифатических альдегидов и кетонов различают насыщенные и ненасыщенные.
16.1. НАСЫЩЕННЫЕ АЛЬДЕГИДЫ И КЕТОНЫ
15.1.1. Номенклатура и изомерия
В номенклатуре альдегидов н кетоиов используют тривиальные и систематические названия. Тривиальные названия альдегидов происходят от названия кислот, в которые они превращаются при окислении. Например, альдегид, при окислении которого получают муравьиную кислоту, имеет название муравьиный альдегид, или формальдегид (от лат. acidum formicum); альдегид, при окислении которого образуется уксусная кислота — уксусный альдегид, или ацетальдегид (от лат, acidum aceticum) и т. д.
По заместительной номенклатуре ИЮПАК названия альдегидов образуют от названия углеводорода с тем же числом атомов углерода (включая углерод альдегидной группы), прибавляя суф-
31B
фикс -аль. Нумерацию главной углеродной цепи начинают с атома углерода альдегид ион группы. Примеры названий альдегидов по тривиальной и заместительной номенклатуре приведены в табл. 15.1.
Таблица 15 1
Номенклатура некоторых представителей альдегидов алифатического ряда
1— Название
Структурная
формула 4 тривиальное по заместительной номенклатуре
О J н—с н /°
Муравьиный альдегид, формальдегид Метаналь
СНз—с н О я Уксусный альдегид, ацетальдегид Этаналь
СНз—СН1—С О Пропионовый альдегид Прол ан аль
СНз—СНа—СНг— О Масляный альдегид, бутиральдегид Бутаналь
СНз—с н—с/ Ан3 О Изомасляный альдегид 2-Металпропаналъ
СНз—С Нв—СНа—С На—С О * S 2 \ # Валериановый альдегид Пентаналь
СНз— СН—СНв—с in. \i Изовалериановый альдегид 3 - Метил бутаналь
Нередко в названиях альдегидов положение заместителей указывают греческими буквами а, р, у н др. При этой буквой а обозначают атом углерода, соседний с альдегидной группой, например:
319
6 Y - Р « X СНз—cm—сн2—сн—с
СНз
<х четилвалсриановый альдегид
? Р а #
СНг—СНа—СНа—С ,.
I \ь
Вг н
7 бромыасляиый альдегид
Для названий кетонов широко используют радикало-функциональную номенклатуру, согласно которой к названиям в алфавитном порядке углеводородных радикалов при карбонильной группе прибавляют суффикс -кетон, например:
С Нз—С—С Нз С На—С Нг—С—СН3
£ &
диметмл кетон «етнлэтнлкетон
При составлении названия кетонов по заместительной номенклатуре выбирается самая длинная углеродная цепь, в состав t которой входит кетогруппа. Нумерацию проводят таким образом, чтобы атом углерода карбонильной группы получил возможно меньший номер. Затем к названию предельного углеводорода, содержащего такое же количество атомов углерода, прибавляют суффикс -он и цифрой обозначают атом углерода, входящий в кетогруппу, например:
СНа—СН— СН,—С—СНз
<Lhs А
4 метил пентанон-!
1 2 3 4 5
CHj—СН—С—СНа—СНз
2 метил пента нон 3
Для некоторых кетонов сохранились тривиальные названия, например, диметилкетон чаще называют ацетоном. Примеры названий кетонов по радикало-функциональной и заместительной номенклатуре приведены в табл. 15.2.
Для карбонильных соединений характерна структурная изомерия. Альдегиды и кетоны, содержащие одинаковое количество атомов углерода, изомерны между собой. Так, пропанон
СНз—С—СНз в пропаиаль СН3СН2С являются структурными
А \
изомерами.
Изомерия альдегидов и кетонов может быть связана с различной структурой углеродной цепи:
320
о / СНз—СН2—СН2—с
масляный альдегид
СНз о
1 //
СНз—с н—с
изомасляный альдегид
СНз—СНз—СНз—С—СНз
А
пеНтанон-2
СНз—СН—С—СНз
<1н3 I)
3 мети л бут а нон 2
Номенклатура некоторых представителей кетонов алифатического ряда
Таблица 15.2
Структурная формула Название
по радикало-функциональной номенклатуре по заместительной номенклатуре
СНз—С—СНз д Диметил кетон Пропанон
о СН3—С—СНг—СНЭ Метнлэтилкетоп Бутанон
СНз—С—СНг—СНг—СНз Метилпропилкетон Пентанон-2
О СНз—СНг—С—СНэ—СНз Диэтил кетон Пентанон-3
Для кетонов характерна также изомерия, обусловленная положением карбонильной группы, например:
5 4 3 2 1
С Нз—С Н2—С Нг—С—С Нз
I 2 3 4 5
СНз—СНа—С—СНз—С Нз
пентанон 2
пентанон 3
15.1.2. Способы получения
Окисление спиртов. Первичные спирты окисляются до альдегидов, а вторичные — до кетонов (см. с. 260, 261).
/>
R—СНг—ОН —R—С
лдО
н
11 5-104
321
СН—он
|П]
-НгО
Гидратация алкинов (реакция Кучерова) (см. с. 64). Из ацетилена в условиях реакции образуется уксусный альдегид, все другие алкины дают кетоны:
НС=СН + Н2О
Hg‘ '. H2SO4
R—С=СН+Н2О
Hg'+. H2SO4
R—С—СНз
Гидролиз геминальных дигалогеналканов. Прн гидролизе геи-дигалогеналканов с атомами галогена у первичного атома углерода образуются альдегиды, а у вторичного — кетоны:
СНз—СНг—СНС12 Н2°> СНз—СНг—+ 2НС1
основание \
1.1 ’ДИХЛ орпрьпан н пропионовый альдегид
СНз—CCla—СН3 120 > СНз—С—СН3 + 2НС1
* м основание ц 1
2,2 дихлорпропа н
ацетон
Пиролиз солей карбоновых кислот. При пиролизе (термическое разложение) кальциевых, бариевых или ториевых солей карбоновых кислот образуются соответствующие карбонильные соединения. Из смешанной солн муравьиной н другой карбоновой кислоты получают альдегиды, а в остальных случаях образуются кетоиы:
300°с г
.О
снз-сСн
+ СаСО3
322
\
СН3-~С^у
300°C
— »
CH3—c—CH3
CaCO3
Озонолиз алкенов. При взаимодействии алкенов с озоном образуются озониды — циклические пероксидные соединения, которые легко расщепляются цинком в уксусной кислоте с образованием альдегидов или кетоиов:
алкен
Оз —>-
озонид
R'—С
Z
о
Оксосинтез, В промышленности альдегиды получают взаимодействием алкенов с оксидом углерода (II) и водородом при повышенной температуре и давлении в присутствии платинового, никелевого или кобальтового катализаторов. Обычно образуется смесь изомеров:
2R—СН=СН2-|-'2СО + 2Н2-* R—СН2—СН2 + R—СН—СН3 с с
/ X / X
15.1.3. Физические свойства
Мурввьиный альдегид — газ, низшие альдегиды и кетоны — летучие жидкости (твбл. 15.3). Кипят они при более низкой температуре, чем соответствующие спирты, так как не способны образовывать водородные связи, Температуры кипения кетонов несколько выше, чем у изомерных им альдегидов. Альдегиды и кетоны хорошо растворяются в органических растворителях, низшие — растворимы в воде. Большинство альдегидов и кетоиов обладают характерным запахом. Плотность альдегидов н кетонов ниже единицы.
11*
323
Таблица is 3
Физические характеристики некоторых альдегидов и кетонов
1 Структурная формула Температура, °C Плотность 4е
плавления кипения
/° н—с — 92,0 -21,0 0,815 (dr20)
^н
/° <н3—с -123,5 21,0 0,780
^н
/° СНз—с На—С — 81,0 48,8 0,807
о СНз—СНг—СНг— -99,0 75,7 0,817
^Н
/> СНэ—С Н—С -65,9 63,5 0,794
। х. СНз н
о СНз—{СНя)э—с^ —91,5 103,4 0,809
СНз—СН—СН2—С^ I < —51 92,5 0,803 (di7)
СНз н
СНз—с—СНэ д -95,0 56,5 0,792
О СНз - с—СНг— СНз -86,4 79,6 0,805
СНз—С—CHj—СНг—СНэ — 77,8 101,7 0,809
О С Нз—СНг—С—СНг—СНз А -42,0 102,7 0,816 (di9)
324
15.1.4. Химические свойства
Химические свойства альдегидов и кетонов определяются нали-е(Л в нх молекуле карбонильной группы, строение которой пред-4 явлено на рис. 15.1. Атом углерода карбонильной группы иахо-сиТся в состоянии зр2-гибридизации и связан с окружающими его \омами тремя о-связями, расположенными в одной плоскости под гЛом 120°. Негибридизованная р-орбиталь атома углерода пере-кпываетсЯ с р-орбиталью атома кислорода, образуя л-связь. Атом кислор°да как более электроотрицательного элемента притягивает себе о- к л-электроиы (последние более подвижны, так как значительно слабее удерживаются ядрами), В результате этого двойная связь карбонильной группы сильно поляризована, на атоме кислорода возникает частичный отрицательный заряд, а на атоме л* б-
углерода — частичный положительный: ^С—О . Благодаря
такой поляризации альдегиды и кетоны способны вступать в реакцию с нуклеофильными реагентами, которые атакуют атом углерода карбонильной группы. Реакционная способность карбонильных соединений определяется величиной дробного положительного заряда на атоме углерода СО-группы.
Л-МО
Рис. 15.1. Строение карбонильной группы
Альдегиды, как правило, более реакциоиноспособны, чем кетоны. Алкильные радикалы за счет 4-Лэффектауменьшают положительный заряд иа атоме углерода карбонильной группы. Наличие в молекуле кетона двух алкильных групп при карбонильной группировке приводит к большему понижению положительного заряда, Чем в молекуле альдегида
5+ > 5+
б+ R-*C*-R &
Кроме того, алкильные радикалы в молекуле кетона в большей Зелени затрудняют подход нуклеофила к карбонильной группе.
325
Наряду с реакциями, протекающими с участием карбониль ной группы, для альдегидов и кетоиов характерны также превращения по а-атому углерода. Все реакции альдегидов и кетонов условно можно разделить на следующие группы:
• нуклеофильного присоединения;
• присоединения - отщепления;
• конденсации;
с участием а-углеродного атома;
полимеризации; окисления и восстановления.
А. Реакции нуклеофильного присоединения (AN)
К реакциям AN альдегидов и кетонов относятся: присоединение синильной кислоты, гидросульфита натрия, воды, спиртов, взаимодействие с магнийорганическими соединениями. Нуклеофильное присоединение начинается с атаки нуклеофилом электронодефн-цитного атома углерода карбонильной группы. В качестве нуклеофила могут быть ионы или нейтральные частицы.
Присоединение синильной кислоты. Синильная (цианоаодород-ная) кислота присоединяется к карбонильным соединениям, образуя циангндрииы, или а-гидроксинитрилы:
СН3-СС + H-CN ------------------► CH.-CH-CN
Ан
шприл а-окснпропяоновой кислота
сн3
♦ CH-CH-C-CN 1 ОН
нитрил сх-метил-а-окси-масляной кислоты
Реакция протекает является цианид-ион:
в присутствии основания, нуклеофилом
С=О -|- CN"
циангидрин
Образующиеся циангидрины можно легко гидролизовать до соответствующих а-гидроксикислот, что используется в синтезе этих кислот, а также для удлинения углеродной цепи сахаров.
326
Присоединение гидросульфита натрия. Альдегиды и содержащие группировку СНэСО кетоны реагируют с гидросульфитом (бисульфитом) натрия, образуя «бисульфитные соединения». Кетозу более сложного строения этой реакция не дают. Реакция протекает без катализатора, • ------ ж— ---
сильный нуклеофил:
так как гидросульфит-ион — достаточно
О
R- " :S-ОН
Н т +
он
♦ R-C-SOjNa Н
бисульфита ое соединение
Т" t
R—С—SOfNa + I СНз
бисульфит ное соединение
Бисульфитные соединения плохо растворимы в воде и выделяются в виде кристаллического осадка. Нагревание с водным раствором минеральной кислоты или карбоната натрия приводит к их разрушению с выделением свободного альдегида или кетоиа.
Реакция с бисульфитом натрия является качественной на карбонильную группу, а также используется для выделения и очистки альдегидов и кетоиов.
Присоединение воды. Растворение альдегидов в воде сопровождается образованием гидратов, которые представляют собой продукты присоединения молекулы воды по карбонильной группе. Как правило, гидраты альдегидов неустойчивы. В водных растворах они находятся в динамическом равновесии с альдегидом:
+ нон <
н
хо-н R-C-O-H '"н
гидрат альдегида, гем-диол
Положение равновесия определяется строением карбонильных соединений. Формальдегид в воде практически полностью гидратирован, ацетальдегид — наполовину, а ацетон почти не взаимодействует с водой. Гидраты альдегидов существуют только в растворе
327
и выделить их невозможно; при перегонке онн разлагаются. Суще ствование гидратов доказывают с помощью физических методов исследования.
В некоторых случаях, когда карбоиильиая группа связана с сильным электроиоакцепторным заместителем, образовавшийся гидрат может быть выделен в свободнЪм виде. Например, трихлор.
ацетальдегид или хлораль, присоединяя молекулу воды, превра. щается в хлоралгидрат, представляющий собой устойчивое крис,
таллическое вещество:
С13с—сс н
8+ 8—
+ НОН
хлораль
хон
* CLC-C
хлоралгидрат
Отнять воду от хлоралгидрата удается только при действии серной кислоты. Хлоралгидрат применяется в медицинской практике как успокаивающее и противосудорожное средство.
Присоединение спиртов. При взаимодействии альдегидов со спиртами образуются полуацетали, а в присутствии следов минеральных кислот — ацетали:
R'—ОИ,|Н‘|
OR'
R—С — OR' + Н2О
Полуацетали, как правило, малоустойчивы. Ацеталн устойчивы в щелочной среде, но легко гидролизуются до свободного альдегида в разбавленных кислотах. Такое свойство ацеталей используется в органическом синтезе для защиты альдегидной группы.
Кетоны из-за низкой реакционной способности и пространственных препятствий со спиртами не взаимодействуют, поэтому кетали получают, используя другие синтетические методы.
Взаимодействие с магнийорганическими соединениями. Альдегиды и кетоиы реагируют с алкил- и арилмагиийгалогеиидами (реактивы Гриньяра) с образованием продуктов присоединения по карбонильной группе, которые гидролизуются в присутствии разбавленных минеральных кислот до спиртов:
328
^етальдегиц
5- OMgBr
C2H?-MgBr -► сн3-с-с2н5
н
этилмагний-
бромид броммагний-
алкоголят
ОН
сНз-6-сл
-Mg(OH)Br 3 5
Н
бутаиол-2, emop-бут кловый спирт
снз-с-сн.
ацетон
е =. OMgBr
—------ I НОН, [Н I
+ С2НГМ6Вг^СН3-С^СгН&1—;;
сн3
он
СН3—С—С2Н5 сн3
2-мста лбуганол-2, mpem-амиловый спирт
Из формальдегида образуется первичный спирт, все остальные альдегиды дают вторичные спирты, а кетоны — третичные. Реакция присоединения магнийорганических соединений к альдегидам и кетонам является одним из важных способов получения спиртов.
Б. Реакции присоедииения-отщеплеиия
Альдегиды и кетоны взаимодействуют с азотистыми основаниями (H2N—X, где Х = Н, Aik, Аг, ОН, NH2 и др.) с образованием неустойчивых продуктов нуклеофильного присоединения, которые стабилизируются благодаря отщеплению молекулы воды. Эта группа реакций получила название реакций прнсоединения-от-щепления.
Взаимодействие с аммиаком. Альдегиды, присоединяя молекулу аммиака, образуют альдимииы. В процессе реакции вначале образуется неустойчивый амниоспирт, от которого затем отщепляется молекула воды.
Механизм реакции:
сНз-сС; + nh3
хн 3
о он
I +• I
СН—ONH3 CH3-C-NH2 4^ н н
н ch3-c^n'h I н
CHr CH=NH + H2O
ацетальдимин, этанимии
329
По аналогичному механизму протекают реакции с другими азп тнстыми основаниями.
Альдимииы — неустойчивые соединения, они самопроизвольц полимеризуются (циклизуются) с образованием альдегида^0 миака:
ЗСНэ—CH=NH
Н—N
НЭС—НС
<рН3
СН
^N—Н
I
СН—СНз
I
Н
альдегидам мнак
Кетоиы также взаимодействуют с аммиаком, но при этом образуются продукты более сложного строения.
Взаимодействие с аминами. Первичные амины (H2NR), реагируя с альдегидами или кетонами, дают имины
О
D г/
R—С 4- H2N—R'
R—СН —N—R'
Н
Устойчивость образующихся иминов зависит от природы радикала, с которым связана аминогруппа. Если R'— алкильный радикал, то такие имины легко разлагаются или полимеризуются. Если же R' — арильный радикал, то имины устойчивы Замещенные имины часто называют также основаниями Шиффа.
Взаимодействие с гидроксиламином. Продукты конденсации альдегидов и кетоиов с гидроксил амином называют альдоксимами и кетоксимамн соответственно;
О
R—С^ + H2N—ОН
R—CH = N—ОН -|- Н2О
альдоксим
R'—С—R 4- HaN— ОН--------> R'—С—R 4- Н20
II II
О N—ОН
кетокснм
330
дльдоксимы и кет о кси мы — кристаллические вещества с чет-я температурой плавления. В кислой среде они легко гидролнзу-я с образованием исходных продуктов. Поэтому реакцию ^пазования оксимов используют для выделения и идентификации Альдегидов и кетонов.
взаимодействие с гидразином и его производными. Альде-иды н кетОНЫ реагируют с гидразином (NH2NH2) и его производными— фенилгидразином (CeHsNHNbb), семикарбазидом (p[2NNH—С—NH2), тиосеми карб аз и дом (H2NNH—С—NH2) с
образованием гидразонов, семнкарбазонов и тиосемикарбазонов соответственно.
HaN—N На )
C=N—NH2 4- H2O
h2n—NHCgHis
гидразон
C=N—NHCeH5 4- H2O
фенилгидразон
HaNNHCONHa
C=N—N—(jj—NH2 4- H2O H О
семик арб аз он
h2nnh—с—nh2
C=N—NH—
nh2 + H2O
тиосемикарбазон
Продукты этих реакций, подобно оксимам, хорошо кристалли-У’отся и используются для открытия вльдегидов и кетонов, а так-е выделения их из смесей.
При взаимодействии с гидразином карбонильные соединения, Ряду с гидразонами, образуют азины:
331
R\ R
2 C=0 + NH2NH2 -> C=N—N=C^ + 2H2O ;
R^ R^ R'
азин
В. Реакции конденсации
Альдольная конденсация. Альдегиды, содержащие атомы водорода у а-углеродного атома, в присутствии каталитических количеств основания способны вступать в реакцию альдольной конденсации. При этом молекулы альдегидов соединяются друг с другом посредством связей С—С и образуют альдоль — соединение со спиртовой и альдегидной группами:
О О
2СНз—С СНз—CjLH—СНг-С 7
\1 ОН
Р □кснмасляныЙ альдегид
В слабощелочной среде гидроксид-ион отщепляет протон ОТ а-углеродного атома альдегида с образованием сопряженного еиолят-аниона
ОН
-Н2О
.О сн2-сС„
/О СНу=С<
2 хн
енолят-анион
Енолят-аннон обладает сильными нуклеофильными свойствами н атакует атом углерода карбонильной группы второй молекулы альдегида с образованием альдоля:
&+• 4о - ж
снг сСн + сн2-сСн
О' снгс-сн2-сб° 1 н
нон
он I .о сн3-с-сн2-с^ +
но"
332
Продукты альдольной конденсации — 0-окснальдегиды при [агреванин легко теряют воду, превращаясь в а, р-ненасыщенные альдегиды
О / СН3—(^Н—CHr—С он н
о
/
СНз—СН=СН—с Ч-Н2О
кротоновый альдегид
Реакция отщепления молекулы воды от альдоля с образованием «насыщенного альдегида известна в органической химии под названием кротоновая конденсации.
В реакцию альдольной конденсации вступают и кетоиы, однако лз-за более низкой реакционной способности кетогруплы реакция протекает в более жестких условиях:
Нз
СНз—С—СН?—С—СНз
О
ОН О
дна цето новый спирт
s В сильиокислой среде кетоиы вступают в реакцию кротоновой конденсации с образованием непредельных кетоиов
2СНз—С—СНз Н‘ > СНз—С=СН—С—СНз II нзО I II
СНз О
окись чезитша
Сложноафарная конденсация (реакция Тищенко). В 1906 г. русский химик В. Е. Тищенко обнаружил, что при нагревании альдегидов с этилатом алюминия образуются сложные эфиры карбоновых кислот, например:
2СНЭ—С^ ,,AI<QC^); СНз—
н ОСНаСНз
этилацетат
В этой реакции одна молекула альдегида восстанавливается до спирта, а вторая — окисляется до кислоты. Реакции, в процессе которых происходит самоокисление-самовосстановление соединения, получили название диспропорционирования, или дисмутации.
333
г
Реакция начинается с присоединения этилата алюминия к карбонильной группе альдегида. Образовавшийся продукт присоединения (А) вступает во взаимодействие с другой молекулой альдегида, образуя неустойчивое промежуточное соединение (Б), в котором происходит перенос гидридного иона (Н~), отщепление этилата алюминия и в результате образуется сложный эфир:
О
✓ +
СНг—С + А1(ОСгНб)3 СН,—С—О—А1(ОС2Н5)э
А
А
d / +
CHj—С + СНз—С—О—Al (ОС2Н5) 3
н А
А
* -Н'
снэ- С-О-сААлЦОСзНОэ н сн3
Г, Реакции с участием а-углеродного атома
Карбонильная группа оказывает активирующее влияние на углеводородный радикал. Как электроноакцепторный заместитель она увеличивает подвижность атомов водорода при а-углеродиом атоме (СН-кислотиость):
К числу реакций, протекающих с участием а-углеродного это-' ма, относятся галогенирование и рассмотренная выше альдольная^1 конденсация. 1
334
Реакция галогенирования. Альдегиды и кетоны как СП-кислоты легко вступают в реакции с галогенами с образованием а-гало-генопроизводных, например:
О
О
СНз—СН?—С
Вга
НВг
СНз—СН—Сч
'Н
Вг
а бромпропио и оный альдегид
С12
НС!
СНз—(jf—СН2С1
О
хлорзцетон
а-Галогенопроизводиые альдегидов и кетонов проявляют слезоточивое действие и называются лакриматорами (от лат. lacri-гпа— слеза). ос-Моногалогенопроизводные получают в присутствии минеральной кислоты. В щелочной среде происходит дальнейшее замещение атомов водорода у а-углеродного атома и можно получить ди- и тригалогензамещенные альдегиды и кетоны. Тригалогенкарбонильные соединения в щелочной среде расщепляются с образованием кислоты и тригалогеиметана (СННа1з). Например, в случае иодирования в щелочной среде реакция идет с выделением желтых кристаллов йодоформа:
О
R—С—СНз
RCOCT + CHhi
Реакция образования йодоформа используется в аналитической практике под названием «йодоформная проба». Оиа примени-
г о он
ется для обнаружения групп СНз—С
. Спирты, Н
содержащие СНз—СН(ОН)-группировку, в условиях реакции сначала окисляются до соответствующего альдегида или кетоиа и лишь затем подвергаются иодированию.
Д. Реакции полимеризации
Альдегиды, в отличие от кетонов, способны полимеризоваться. Реакция полимеризации протекает в обычных условиях и ускоряется в присутствии минеральных кислот. Так, уже при стоянии
335
40 % водного раствора формальдегида (формалина), особенно при температуре ниже 9 °C, наблюдается выпадение белого осадка продукта линейной полимеризации (параформа):
п С=О 4- НзО-
НО- СН2— (ОСН2)„—он
параформ
Полимеризация ацетальдегида в присутствии следов серной кислоты приводит к образованию в зависимости от условий двух циклических продуктов — паральдегида и метальдегида. Паральдегид образуется, если реакцию проводить при 20 °C, а метальдегид — при 0 °C:
О
л=3
НзС—Н
НзС—Н
СН—СНз
СН
СНз
паральдегид
н<с
СН—о
СН—СНз
О
о
О
О—СН
СНз
метальдегид
1 h f
Паральдегид — жидкость (т. кип. 128 °C), метальдегид — твердое вещество, используется в быту как сухое горючее под названием «сухой спирт».
Реакция полимеризации обратима, при нагревании продуктов реакции с минеральными кислотами происходит их деполимеризация
ззв
Е. Реакции восстановления и окисления
Реакции восстановления. Реакцию восстановления альдегидов и кетоиов широко используют для получения спиртов (альдегиды восстанавливаются до первичных, а кетоны — до вторичных спиртов). В технике спирты получают в результате каталитического гидрирования; присоединение водорода происходит в присутствии кобальта, никеля или платины:
—R—СН2ОН
Восстановление альдегидов или кетоиов до спиртов в лаборатории проводят с помощью литийалюминийгидрида (LiAIH4) или водородом в момент выделения:
□
СНз—СНз—1<А1^ - СН3СН2СН2ОН
Реакции окисления. Альдегиды и кетоны по-разному относятся к действию окислителей. Альдегиды очень легко окисляются, даже при действии таких слабых окислителей, какими являются ионы Ag+ и Си+ + , они превращаются в карбоновые кислоты.
Реакцию окисления альдегидов аммиачным раствором нитрата серебра (реактив Толленса) называют часто реакцией «серебряного зеркала». Иои серебра а этой реакции восстанавливается до свободного серебра, которое выделяется в виде зеркала иа стейках пробирки-
R—+ 2[Ag(NH3)s]OH
2Ag -j- R—С -р 3NH3 -|- НзО
\)NH4
Альдегиды также восстанавлиаают реактив Фелинга (смесь раствора сульфата меди со щелочным раствором натрийкалиевой соли виннокаменной кислоты) CuSO4-|-2NaOH------*-Си(ОН)зЧ-
+ Na2SO4:
337
COONa
in—ОН
I + Си (ОН) 2
СН—ОН
COON a
COONa
>о-сн
I ,Cu I 4-2Н2О сн—О ’* о—СН
ioOK н
:оок
При нагревании альдегидов с фелинговой жидкостью образуется красный осадок оксида медн (I):
COONa COONa I Нх I
С1Ь0. осн
I ^cu; i
“ O-CH
СН-О' I Н
COOK
R-C^° Н
HjO
COOK
R- СГ°
ХОН
COONa СНОН
CHOH
COOK
Реакции окисления альдегидов аммиачным раствором оксида серебра и реактивом Фелинга используются в анвлитической практике для обнаружения альдегидной группы. Кетоны в этих условиях ие окисляются, поэтому эти реакции могут быть использованы и для отличия альдегидов от кетонов
Окисление кетонов происходит только в присутствии сильных окислителей, таких, как перманганат или бихромат калия. При этом происходит разрыв связей С—С между атомами углерода карбонильной группы и углеводородного радикала. В результате реакции образуется смесь кислот О
H—c
OH
муравьиная кислота
о
СНЗ-+-С- + -СН2—СНз —
бута нон-2
* 2СН3—С
\)Н
уксус нам кипота
СНз— сн2—соон
npOflHQHOBdH кислота
338
По продуктам, полученным прн окислении, определяют строение кетонов.
15.1.5 . Отдельные представители
Муравьиный альдегид (формальдегид, метаналь), СН2О. Бесцветный газ с резким запахом, растворимый в воде, получают термическим дегидрированием метанола (600 °C) над серебряным катализатором:
О
СНзОН —Н2 + Н—cf*
Ав \
н
37—40 % водный раствор формальдегида, к которому в качестве ингибитора полимеризации добавляют 6—15% метанола, называют формалином. Формалин применяют как дезинфицирующее, дубящее средство и консервант для анатомических препаратов. В промышленности формальдегид используется в производстве фенолоформальдегидных, мочевиноформальдегидных и других смол, необходимых в электропромышленности и машиностроении. В медицине используется препарат гексаметилентетрамин (уротропин) — производное формальдегида:
6 СНаО + 4 NH3
- 6H2Ok
уротропин
Уксусный альдегид (ацетальдегид, этаналь) СНзСНО. Бесцветная жидкость с резким запахом (т. кнп. 20 °C), смешивается с водой, этанолом, диэтиловым эфиром в любых соотношениях. Может быть получен дегидрированием или окислением этанола над серебряным катализатором, гидратацией ацетилена (реакция Кучерова). Используется для получения уксусной Кислоты.
Ацетон (диметилкетон, пропанон) СНзСОСНэ. Бесцветная жидкость (т. кип. 56,2 °C), смешивается с водой и органическими растворителями. Получают сухой перегонкой древесины, пиролизом ацетата кальция, дегидрированием пропаиола-2 и из кумола (наряду с фенолом).
339
Ацетон нашел применение как растворитель органических веществ (лаки, нитроцеллюлоза) и как исходное вещество в синтезе некоторых лекарственных препаратов, например йодоформа:
СН3—jp—СНз О
12. Off
CHI3 + СНз—С
Этой реакцией часто пользуются для открытия ацетона, однако в этих условиях йодоформ образуется и из других веществ. Ацетон дает интенсивное виино-красное окрашивание с нитропруссидом натрия. Обе реакции используются для обнаружения ацетона в моче при сахарном диабете.
15.2. НЕПРЕДЕЛЬНЫЕ АЛЬДЕГИДЫ
В молекуле непредельных альдегидов карбонильная группа связана с углеводородным радикалом, содержащим кратную связь. Положение двойной связи в углеводородном радикале обозначают греческими буквами а, 0, у и т. д. В соответствии с этим различают а, 0-, 0, у- и т. д. непредельные альдегиды. Например:
°
СНз—С Нг—СН=СН—С^
Y 0 “
С Н3—С Н=С Н— СНг—
а. 0-непредельный альдегид
0 Y непредельный альдегнд
Наибольший интерес представляют а, 0-непредельные альдегиды, у которых двойная углерод-углеродная связь сопряжена с карбонильной группой. Простейшими представителями этой группы соединений являются акролеин и кротоновый альдегид
° °
С Н2=С Н—сХ С Нз—С Н=С Н—С^
акролеин кротоновый альдегид
Акролеин получают дегидратацией глицерина (см. с. 273) при нагревании с KHSO*, а кротоновый альдегид — из уксусного, а результате кротоновой конденсации (см. с. 333).
Для непредельных альдегидов характерны реакции карбонильных соединений, а также ненасыщенных углеводородов, а, 0-Не-предельные альдегиды, содержащие в своей структуре сопряженную систему, имеют ряд особенностей, отличающих их от осталь-
340
Нь1Х альдегидов. В частности, присоединение к ним галогеио-водородов протекает против правила Марковиикова. Вначале происходит присоединение бромистого водорода по концам сопря-деииой системы с образованием неустойчивого еиола, который сразу же изомеризуется в альдегид (правило Эльтекова—Эрлеи-мейера):
6+.
СН2=СН—с
е О
НВг
он
СНз—СН=С
сн;
н
акролеин
г
енол
Диалогично присоединяется молекула в кислой среде:
Br Н
— 3 бром пропа нал ь.
р брочпропииновый альдегид
воды. Реакция протекает
,0
СН2=СН—С + н3о —
СНг—СН2—С
-О
н
н
о
3-гид роке нпроп а нал ь, p оксипроп нон оный альдегид
F Как карбонильные соединения непредельные альдегиды взаимодействуют с синильной кислотой, образуя циангидрины:
о
СН2=СН—с^ -h HCN -Ж
он
CN
Наличие сопряженной системы в молекуле кротонового альдегида приводит к повышенной подвижности атомов водорода метильной группы, благодаря чему он способен вступать в реакции кротоновой конденсации. Например:
о
о
СНз—С + СНз— СН=СН—С
-Н?О
Н
ацетальдегид
О
С Нз—с н=с н—с н=с н—с
н
сербнловыв альдегид
841
Образовавшийся сорбиновый альдегид способен вступать в ре, акцию с новой молекулой уксусного альдегида, поэтому в процессе реакции образуется смесь непредельных альдегидов.
Акролеии и кротоновый альдегид используются в синтезе ряда соединений, в том числе и лекарственных препаратов.
15.3. ДИАЛЬДЕГИДЫ И ДИКЕТОНЫ
К диальдегидам относятся соединения, содержащие две альдегидные группы, а к дикетонам — содержащие две кетогруппы. Простейшим представителем днальдегйдов является глиоксаль, или этандиаль, а дикетонов — диацетил или бутандион:
CHs—С—С—СН3
глиоксаль дна цетил
Глиоксаль — кристаллическое вещество, а ди ацетил — жидкость. Оба вещества окрашены в желтый цвет.
И глиоксаль, и диацетил могут быть получены при осторожном окислении соответствующих двухатомных спиртов: этиленгликоля и бутандиола-2,3. Они обладают всеми свойствами, характерными для карбонильных соединений. При этом в реакцию могут вступать одна или две карбоиильяые группы.
При окислении глиоксаля образуется щавелевая кислота
О с\
СООН
[01 > | о СООН
с
щавелевая кислота
М2
г
В условиях реакции Канниццаро глиоксаль преврвщается в гликолевую кислоту, т. е. происходит внутримолекулярная реакция окисления-восстановления:
О
С
Н
COOK
кон ।
,0
СН2ОН
С
Н
калиевая соль гликолевой, или оксиуксус нов. КИСЛОТЫ
При взаимодействии с соотношения диоксимы:
гидроксиламином, в зависимости от реагентов, глиоксаль и
диацетил дают моио- нли
HaNOH -Н20
CH=N—ОН
Hjnoh С H=N—ОН
-нао 1
CH=N—ОН
ыоноокснм диоксим глиоксаля,
глиоксаля глиоксим
СНз
с=о I с=о
СНз
HiNOH
-НЮ
СНз
C=N—ОН
I
С=О
I
СНз
монооксии ди ацетил а
HjNOH
-ню ъ
СНз (L=N—он
C=N—ОН
СНз
диоксим ди ацетил а. диметилглноксим
Диметилглиоксим из-за способности образовывать внутрикомп-лексиые соединения с рядом катионов нашел применение в аналитической практике в качестве реактива для обнаружения никеля (реактив Чугаева). С солями никеля он дает красно-фнолетовый осадок хелатного комплекса
343
СН3 /
—с
N—О
хелатный комплекс
Глиоксаль и диацетил используются в синтезе различных гетероциклов. Днацетнл содержится в сливочном масле н обусловливает его запах. Поэтому его применяют в пищевой промышленности как ароматизирующее средство для масла, маргарина, сыра.
15.4. АРОМАТИЧЕСКИЕ АЛЬДЕГИДЫ И КЕТОНЫ
15.4.1. Классификация и номенклатура
Ароматические альдегиды подразделяют на две группы. К первой относятся альдегиды, содержащие альдегидную группу в бензольном ядре, ко второй — содержащие альдегидную группу в боковой цепи. Простейшим представителем первой группы является бензальдегид, или бензойный альдегид, получивший название от кислоты, в которую он превращается при окислении. Аналогично называют и другие ароматические альдегиды этой группы:
СНз
бензальдегнд.
бензойный альдегид
п толуиловый альдегид, 4 мстил бензальдегид
салициловый альдегид, 2 гидроксибензальдегид
Альдегиды, в которых альдегидная группа находится в боковой цепи, называют как производные альдегидов жирного ряда. Положение фенильного радикала обычно указывают буквами греческого алфавита. Например:
344
СбНБ—сн2—с
О
СбНй—СНг—СНг—
фенил уксусный альдегид
Р-фенилпропиоиовый альдегид, гидрокоричнай альдегид
Ароматические кетоны также делят иа две группы: чисто ароматические и жирно-ароматические. К первой группе относятся кетоны, в которых карбонильная группа связана с двумя ароматическими углеводородными радикалами. Если одни из радикалов алифатический, то такие кетоны относят к жирно-аром этическим.
Для названий ароматических кетонов чаще всего используют радикало-функциоиальную номенклатуру (см. с. 320). Широко применяют также тривиальные названия. Например:
СНз—С
ацетофенон, м етнл фен ил кетон
бензофенон, дифенилкетон
бе нзилф ени л кет о н
15.4.2. Способы получения
Ароматические альдегиды и кетоны можно получать с помощью методов, применяемых для синтеза алифатических альдегидов и кетоиов. Это реакции окисления первичных и вторичных ароматических спиртов, омыление геминальных дигалогенопронзводиых, пиролиз кальциевых солей карбоновых кислот и другие способы. Вместе с тем для получения ароматических альдегидов и кетонов существует и ряд специфических методов.
Окисление ароматических углеводородов. Прямое окисление Толуола или других производных, содержащих метильную группу, связанную с бензольным кольцом, приводит к соответствующим альдегидам. В качестве окислителей могут быть использонаиы оксид хрома (VI), хлористый хромил (CrOjCh), пентаоксид ваиа-Дия, диоксид марганца и др. Окисление оксидом хрома (VI)
345
проводят в среде уксусного ангидрида. Образующийся в процессе окисления еем-диацетат не подвергается дальнейшему окислению, а при его гидролизе с хорошим выходом образуется альдегид:
гем дмвпетят
бензальдегид
Окисление ароматических углеводородов с целью получения альдегидов используется не только в химической лаборатории, но и в промышленности.
Реакция Гаттермана — Коха. Метод основан иа прямом введении альдегидной группы в бензольное ядро, поэтому данную реакцию называют еще реакцией формилнроваиия. Форматирование ароматических углеводородов проводят смесью оксида углерода (II) и хлороводорода в присутствии хлорида алюминия и хлорида меди (I)-
СО+НС1 ------------*-CujCIj, AICI3
толуол
О
п толуиловый альдегид
Реакция используется в основном для получения алкил- и гало-генозамещеииых ароматических альдегидов. Бензальдегид образуется с небольшим выходом.
Реакция Фриделя — Крафтса. Основным методом получения ароматических кетонов является ацилирование ароматических углеводородов по Фриделю - Крафтсу. В качестве ацилирующих веществ используют чаще всего галогеиангидриды или ангидриды карбоновых кислот, а также сами кислоты; реакцию проводят в присутствии кислот Льюиса:
СОСНз
Oft CH3COCI ZZ \
Ц + НС|
ацетофенон
Реакция протекает по механизму (см. с. 103).
346
15.4.3. Физические свойства
Ароматические альдегиды и кетоны — жидкости или твердые вещества (табл. 15 4), нерастворимые в воде. Ароматические альдегиды обладают запахом горького миндаля, с удалением альдегидной группы от бензольного ядра запах становится резче. Ароматические кетоны также обладают запахом, например ацето-фенои имеет запах черемухи.
Таблица 15 4
Физические характеристики некоторых ароматических альдегидов н кетонов
Структурная формула Название Температура “С Плотность d^. г/см3
плавления кипения
^0 СбНв—С Бензальдегид —26 179,5 1,0447(4°)
СбНв—С—СНз Ацетофенон 20 202,3 1,0281 (d3°)
0 СвНз—С—СеНз Бензофенон 48 306 1,0976(4°)
II 0 0 cf НзС |\ ч/К н о-Толуиловыи 195,5 1,0386(4°)
и 0 с 6: альдегид м -Толуиловый 199 1,0189(4')
СНз ^0 Ан альдегид п-Тол у иловый 205 1,0194 (di7)
I, альдегид
347
15.4.4, Химические свойства
По химическим свойствам ароматические альдегиды и кетоиы во многом сходны с альдегидами и кетонами жирного ряда. Так, ароматические альдегиды дают реакцию «серебряного зеркала», образуют ацетали, циангидрины, гндросульфитные соединения, альдоксимы, гидразоны, азометины. Одиако ароматические альдегиды ие вступают в реакцию вльдольной конденсации, очень трудно полимеризуются, в других соотношениях взаимодействуют с аммиаком.
Ароматические кетоны обладают меиьшей реакционной способностью, чем кетоиы жирного ряда. Поэтому они не образуют гид-росульфитиых соединений, бензофенон не реагирует с синильной кислотой.
Кроме того, ароматические альдегиды и кетоны Дают ряд специфических реакций.
Взаимодействие с аммиаком. Ароматические альдегиды вступают во взаимодействие с аммиаком в соотношении 3:2 с образованием гидробензамида
о С6Н5—CH=N
ЗСбН6—+2NH3 -----------> . СН—С6Н5 + ЗН2О
С6Н5—CH=N'Z
гндробентамид
Образующийся в процессе реакции имии бензальдегида не способен полимеризоваться, две его молекулы вступают в реакцию с новой молекулой бензальдегида.
Реакция Канниццаро. Реакция открыта в 1853 г. итальянским ученым С. Канниццаро. В присутствии сильного основания или концентрированной щелочи ароматические альдегиды вступают-в реакцию диспропорционирования. При этом одна итдвух молекул альдегида окисляется до соответствующей кислоты, а другая восстанавливается до спирта:
О
2С6Н5-С^ С6Н5СОО-К+ + с8н5сн2он
кал ней ия соль бензиловый
гг бензойной кислоты спирт
Механизм реакции:
С6Н3-С^°
6 з -хн
он"
О".'' С6Н5-С$:Н ОН
*8+.О
СбН5-С(н
348
н
СвН5—С—О' -|- СбН8СООН—> с6нБснгон+ С«Н5СОО-
н
Реакция протекает с переносом гидридного иона (Н“).
В реакцию Канниццаро вступают также некоторые альдегиды жирного ряда, в частности формальдегид и альдегиды, не содержащие атомов водорода при а-углеродном атоме:
О
2Н—НСООК +СНзОН о о
2(СНз)зС—(СНз)зС—+ (СНэ)зС—СН2ОН * ОК
Альдегиды, содержащие атомы водорода при а-углеродиом атоме, в условиях реакции Канниццаро осмоляются.
Реакции конденсации, В присутствии оснований ароматические альдегиды способны вступать в реакции конденсации с альдегидами, кетонами, ангидридами карбоновых кислот, содержащими подвижные атомы водорода при а-углеродном атоме. Например, бензальдегид вступает в реакцию альдольной конденсации с уксусным альдегидом. Альдольная конденсация двух разных карбонильных соединений получила название перекрёстной альдольной конденсации:
О
О
С6Н5С 4-СНз—С'
С6Н5— СН—С Hz—с
О
-НгО
2-гидрокси 2 феннд-пропаналь
коричный альдегид
н
н
н
Образовавшийся 0-окси-0-феинлпропионовый альдегид легко теряет воду, превращаясь в коричный альдегид.
349
Реакция ароматических альдегидов с ангидридами алифатических карбоновых кислот в присутствии оснований известна как конденсация Перкина. Продуктами такой конденсации являются непредельные кислоты. Так, при взаимодействии бензальдегида с уксусным ангидридом в присутствии ацетата натрия ил-и ^алия образуется коричная кислота:
коричная кислота
Механизм реакции:
СНг—С
СНзСОО" 4- + СНзСООН
СНз—С . СНз—С
Л С'‘Н — I CHiCOOH
ОД-ёСн + ро—сл-с-снгС-о-^-сн, ^35
н сн3-сЧо йод
--С6Н5—СН—СНг—С—О—С—СНз
СбН&СН==СН—С—О—С—СНз
СеНБСН=СНСООН + СНзСООН
Бензоиновая конденсация. Специфической реакцией альдеги-дов ароматического ряда является бензоиновая конденсация. Сущность этой реакции заключается в конденсации двух молекул
ЗБО
альдегида в присутствии солей синильной кислоты с образованием ароматических а-оксикетонов (бензоинов):
бензоин
Механизм бензоиновой конденсации:
(^НХ с6н5—с—с—с6н3
<CN Н
5=е с6н5—с—сн-с6н3 о он
+ CN'
бензоин
Альдегиды жирного ряда в условиях реакции бензоиновой конденсации образуют альдали.
Галогенирование. При действии хлора на беизальдегид образуется хлорангидрид бензойной кислоты:
В случае алифатических альдегидов реакция протекает по углеводородному радикалу (см. с. 335).
Реакция электрофильного замещения в бензольном ядре. Для ароматических альдегидов, наряду с реакциями по альдегидной группе, характерны реакции с участием бензольного ядра. Это реакции электрофильного Замещения в бензольном ядре (сульфирование, нитрование и др.); при этом альдегидная группа направляет заместитель в ле-положение:
351
о
о
бензальдегид
м Питре бензальдегид
В реакцию SF ароматические альдегиды вступают труднее, чем ароматические углеводороды, что связано с электроноакцепторным действием альдегидной группы иа бензольное ядро
15.4.5. Отдельные представители
Бензальдегид (бензойный альдегид) СаНь—СНО Бесцветная жидкость с запахом горького миндаля (т кип 179,2 °C), образуется при гидролизе гликозида амигдалина, содержащегося в миндале, косточках персиков, абрикосов Бензальдегид растворим в спирте, практически нерастворим в воде Бензальдегид может быть получен каталитическим окислением толуола, омылением бензилиденхлорида или из бензола реакцией Гаттермана — Коха
На воздухе при стоянии бензальдегид легко окисляется до бензойной кислоты. Бензойный альдегид иашел применение как душистое вещество в парфюмерии и пищевой промышленности, как сырье для получения арилметановых красителей
Ванилин (4-гндрокси-З-метоксибензальдегид) Кристал-
CHQ лическое вещество (т пл 81—83 °C), хорошо
। растворимо в спирте, эфире, мало растворимо
J. в воде
(/ |Ч Ванилин как душистое вещество использу-
L ется в пищевой и парфюмерной промышлен-
(ЭСН ности, является исходным веществом в син-
3 тезе фтивазида — противотуберкулезного пре-
парата
Ацетофенон (метилфенилкетои) СНз—СО—СвНь. Кристаллическое вещество (т. пл. 19,6 °C, т. кип. 202,3 °C), растворимо в спирте, эфире, бензоле, иераствориТио в воде.
Ацетофенон используется как душистое вещество в парфюмерии, а также в синтезе некоторых лекарственных препаратов.
Бензофенон (дифенилкетон) СбНб—СО—CeHs. Кристаллическое вещество, растворимое в эфире, спирте, бензоле, нерастворимое в воде Получают бензофенон взаимодействием бензола с четыреххлористым углеродом, с последующим гидролизом образовавшегося дифеинлдихлорметана:
2С6н6+ecu *'C|J > С6Н5—С—С6Н& —Нг° > С6Нб—С—СвН5
—2НС1 у 2НС1 ||
Cl CI о
352
Бензофенон иашел применение в производстве душистых веществ, а также при синтезе красителей.
Хниоиы. Хиноны представляют собой циклические ненасыщенные дикетоны Простейшими представителями этого класса соединений являются о-хинон и п-хинои:
о хиной п хинон
I 2 бензохинон I 4 бензохинон
л-Хииои впервые был получен в 1838 г русским химиком А. А Воскресенским при окислении хниной кислоты.
Хиноны представляют собой окрашенные кристаллические вещества о-хииои — ярко-красный, и-хинон — желтый
Получают хииоиы окислением двухатомных фенолов или амк-нофенолов. Для получения л-хииона в качестве окислителей используют бихромат калия или оксид марганца (IV) в серной кислоте*.
NH2
я аминофеиол
о-Хинон чаще всего получают при окислении пирокатехина оксидом серебра; реакцию проводят в среде эфира или беизола, без доступа влаги в присутствии сульфата натрия:
пирокатехин о хинон
12 5 104
353
Химические свойства хиноиов обусловлены наличием в их структуре кратных связей и карбонильных групп. Хниоиы очень легко восстанавливаются до двухатомных фенолов:
пирокатехин
Как ненасыщенные соедииеиня они легко присоединяют бром:
При взаимодействни n-хинона с гидроксил амином в зависимости от соотношения реагентов образуются монооксим (хинок-сим) или дноксим (хииоидиоксим):
О N—ОН N'-OH
п хинон ХИНОКС.НМ хиноиднокснчл
В растворах обычно наблюдается частичное превращение хиноксима в л-нитрозофенол, а хиноидиоксима в л-нитрозогндро-
кси анилин:
ХИНОКСИМ
и-нитрозо-фенол
хинон- я-нитрозо-N-
диоксим гидроксианилин
354
С фенолами хиноны образуют прочные комплексы с переносом заряда. Например, с гидрохиноном n-хииои образует устойчивое соединение, известное как хингидрон:
хингвдрон
Хиноны используются для получения красителей, проявителей для фотографии; п-беизохииои применяется как дубящее вещество.
КОНТРОЛЬНЫЕ ВОПРОСЫ И УПРАЖНЕНИЯ
1. Напишите структурные формулы следующих соединений: а) 2-метилбутаналь; б) иэопропилэтилкетои; в) 3-метилгек-сандион-2,4; г) м-иитробеизальдегид; д) а-феиилпропионоаый альдегид.
2. Напишите уравнения реакций получения следующих альдегидов и кетонов двумя способами: а) метил эти л кетон а; б) бензальдегида; в) фенилуксусиого альдегида.
3. Приведите схемы реакций, с помощью которых из л-крезола можно получить гс-метоксибензальдегнд; из бензола гс-метил-ацетофенон.
4. Расположите следующие соединения в ряд по увеличению реакционной способности реакциях нуклеофильного присоединения:
СН3
а) СНз—СН—С—СН3; б) С6Н5—С—СбН5;
в) СНз—СН
г) С6Н5—С
5. Приведите три примера реакций нуклеофильного присоединения по карбоиильной группе, сопровождающихся самопроиз-
12 *
355
вольной дегидратацией. На одном из примеров рассмотрите механизм взаимодействия.
6. Чем обусловлена активность атомов водорода при а-угле-родном атоме в молекуле ацетона? Напишите схему реакции бромирования ацетона.
7. Какие из альдегидов — муравьиный, пропионовый, три метил, уксусный, фенил уксусный, «-толуиловый — вступают в реакцию альдольной конденсации и в реакцию Канниццаро? Приведите схемы реакций.
8. Приведите механизм альдольной и кротоновой конденсации пропионового альдегида. Назовите полученные продукты.
9. Предложите схемы качественных реакций, позволяющих отличить: а) масляиый альдегид от метилэтилкетона; б) п-метил-ацетофеиои от п-гидроксиацетофеиона; в) ацетофенон от бензофенона.
КАРБОНОВЫЕ КИСЛОТЫ
Карбоновыми кислотами называют производные углеводородов, содержащие в своем составе карбоксильную группу —СООН.
В зависимости от природы углеводородного радикала, с которым связана карбоксильная группа, различают алифатические, алициклические и ароматические карбоновые кислоты. В соответствии с числом карбоксильных групп кислоты подразделяют иа монокарбоновые (содержат одну группу —СООН), дикарбоновые (две), трикарбоновые (три) и поликарбоновые (более трех). Алифатические карбоновые кислоты классифицируют по степени насыщеиности углеводородного радикала на насыщенные и ненасыщенные.
Глава 16 монокдрбоновые КИСЛОТЫ
1в.|, НАСЫЩЕННЫЕ МОНОКАРБОНОВЫЕ КИСЛОТЫ
16.1.1. Номенклатура и изомерия
В названиях карбоновых кислот очень широко используют тривиальную номенклатуру. Например, НСООН — муравьиная кислота, СНз—СООН — уксусная кислота, СНз—СНг—СООН — пропионовая и т. д. Положение заместителей по отношению к карбоксильной группе в тривиальных названиях обозначают греческими буквами а, 0, у и др , например: *
7 р «
СНз—СН2—СН—соон
СНз
₽ «
СНэ—СН2—СООН
Вг
р бромпропионовая кислота
а метил масляная кислота
По заместительной номенклатуре ИЮПАК названия карбоновых кислот образуют из названий исходных углеводородов с тем же числом атомов углерода, включая и атом углерода карбоксильной группы, к которым прибавляют -овая и кислота. Нумерацию главной углеродной цепи начинают с атома углерода карбоксильной группы:
Н—СООН
СНз—СНг—СООН
метановая кислота
пропановая кислота
3 3 1
СНз—СН—СООН
<^н
V.H3
2 метилпропановая кислота
Иногда названия карбоновых кислот образуют от названия углеводорода, содержащего в качестве заместителя карбоксильную группу, и словосочетания карбоновая кислота, например:
СНз—СООН СНз—СНз—СН?—СООН СН3—СН—СООН
<1Н
С rig
метаикярбоиовая 1-лропанкарбоновви 3 пропапиарбоновля кислота
кислота кислота
356
Этот способ чаще применяют в тех случаях, когда карбоксильная группа связана с циклической структурой.
По рацвональиой номенклатуре насыщенные монокарбоновые кислоты рассматривают как производные уксусной кислоты, например:
Спз
СНз—СНг—СООН ^СН—СООН
C2hZ
метилуксуеная кислота
метилэтилуксусная кислота
Названия некоторых м оиок арб оновых кислот приведены
в табл. 16.1,
Таблица 16.1
Названия некоторых монокарбоновых кислот
Формула Название по номенклатуре
тривиальной заместительной рациональной
нсоон Муравьиная кислота Метановая кислота -
СНз—соон Уксусная кислота Этановая кислота Уксусная кислота
СНзСНэСООН Пропионовая кислота Пропановая кислота Мет ил уксусная кислота
СНзСНаСНгСООН Масляная кислота Бутановая кислота Этилуксусная кислота
СНэСНСООН 1 Изомасляная кислота 2-Метили ропа новая кислота Ди мет ил уксусная кислота
СНз
СНзСНзСНгСНгСООН Валериановая кислота Пентановая кислота Пропилуксусная кислота
СН3СНСН2СООН 1 Изовал ер на новая кислота З-Метилбутаноная кислота Изопропилуксусная кислота
СНз
СНзСНаСНСООН 1 а-Метилмасля-ная кислота 2-Метилбутановая кислота Мет ил этил уксусная кислота
СНз
СНз(СНг)+СООН СНз(СН2)10СООН СНз(СН2)12СООИ СНэ(СН2)нСООН СНЭ(СН9) 1вСООН Капроновая кислота Лауриновая кислота Миристиновая кислота Пальмитиновая кислота Стеариновая кислота Гексановая кислота Додекановая кислота Тетрадекановая кислота Гексадекановая кислота Октадекановая кислота я- Бутнлуксусная кислота
309
Остаток карбоновой кислоты, образующийся после отнятия атома водорода от карбоксильной группы, называется ацилоксм-
группой (R—С ), а остаток, образующийся после отнятия
гидроксильной группы, называется ацильной грунной (R—С )
Названия ацилокснгрупп обычно образуют нз тривиальных латинских нвзваний кислот и суффикса -ат, например:
формиат
СНз—
^О—
ацетат
/°
СНз—-СНг—С
^О—
пропионат
/°
СНз—СНз—СН2—С
бчтират
Названия ацильных групп образуют нз тривиальных латинских названии кислот и суффикса -ил. По заместительной номенклатуре ИЮПАК названия ацильных групп образуют от названия кислоты, заменяя часть ее названия -овая кислота на суффикс -оил:
формил метанонп
а нети 1
ЭТ8НОНЛ
-г //О
СНз—сн2—с
/°
СНз— СНэ—СНг—С
ПрОПИОНН! пропаноил
бутирил, бутаноил
360
CH
н—с
Нз
СНз—СН
изобутнрил валерил
2 метилиропаниил пентаноил
Изомерия насыщенных монокарбоновых кислот обусловлена разной структурой углеводородного радикала, связанного с карбоксильной группой. Первые три представителя гомологического рида изомеров не имеют. Четвертый гомолог существует в виде двух структурных изомеров:
СНз—СНг—сн2—СООН
масляная кислота
СНз—С Н—СООН
<1Н иэомвсляная кислота
С увеличением числа углеродных атомов в молекуле карбоновой кислоты количество структурных изомеров резко возрастает.
I 16.1.2. Способы получения
t Существует множество способов получения карбоновых кислот, нз которых наиболее важные следующие:
Окисление первичных спиртов и альдегидов (см. с. 260). Первичные спирты окисляются до карбоновых кислот через стадию образования альдегида В качестве окислителей используют бн-хромат калня (К2СГ2О7), КМпО4, HNO3 и др.:
R—СН2 —ОН
первичный спирт
10]
нд
[01
R—С
Гидролиз геминальных тригалогенопроизводных углеводородов. Реакции протекает в кислой или щелочной среде. В качестве промежуточного соединения образуется ортокислота, которая отщепляет молекулу воды и превращается при этом в карбоновую кислоту:
С1
НОН
он’или Н+
о-н R-C-0H он
-н2о
R-CC ОН
гем-трихлоралкан ортокислота
Гидролиз нитрилов. При нагревании нитрилов с водными ра створами кислот и щелочей оии подвергаются гидролизу с образа ванием карбоновых кислот. В качестве промежуточных продукте реакции образуются амиды кислот:
361
нитрил
нон
H“ нлк OH
hoh
H 1 или OH
nh2
R—-I- NHa
\)H
амид карбоновой кислоты
Карбиновая
КИСЛОТА
R— feN
Взаимодействие магнийорганических соединений с СО2. При действии магнийорганических соединений (реактивов Гриньяра) на оксид углерода (IV) образуются соли карбоновых кислот, из которых в кислой среде выделяют соответствующие кислоты:
О
С2Н5—MgCl + СО2 —с2н5—с
\)MgCl
этил магния хлормагния
хлорид пропионат
> С2Н;—СООН + MgCl2 пропионовая кислота
Окисление алканов. Этот метод используется в промышленности для получения многих карбоновых кислот. При окислении алканов кислородом воздуха в присутствий катализатора образуется смесь карбоновых кислот, которую затем разделяют. Реакция сопровождается разрывом углерод-углеродных связей (см. с. 23).
Гидрокарбоксилирование алкенов. Алкены с оксидом углерода (И) и водой в присутствии кислотного катализатора при нагревании и давлении образуют карбоновые кислоты:
СН2=СН2 + СО + Н2О СНз—СНу—СООН
лролионавая кислота
Механизм реакции:
СН2=СН2 СНг—СН2 СНз—СИ2—С=О
—н+
—к СНз—СНз—с
\)Н
Метод используется в промышленности для получения ряда кислот.
362
16.1.3. Физические свойства
Низшие карбоновые кислоты (с числом атомов углерода до трех) в обычных условиях представляют собой легко подвижные жидкости с острым запахом. Кислоты с —Сз — маслянистые жидкости с неприятным запахом, напоминающим запах пота. Кар-боноаые кислоты с числом атомов углерода Сю н выше являются твердыми веществами. Муравьинвя, уксусная и пропионовая кислоты смешиваются с водой в любых соотношениях. С увеличением молекулярной массы кислоты растворимость в воде значительно уменьшается. Внешне карбоновые кислоты нерастворимы в воде. Температуры кипения кислот значительно выше температур кипения спиртов с тем же числом атомов углерода (табл. 16.2). Это свидетельствует о том, что кислоты более ассоциированы, чем спирты. В отличие от спиртов, для которых хврактерны только линейные ассоциаты, карбоновые кислоты вследствие образования межмолекулярных водородных связей образуют как линейные, так и циклические ассоциаты в виде димеров:
R R
-сХ н ?
Таблица 16.2
Физические характеристики некоторых насыщенных монокарбоновых кислот
Формула Па знание Температура, ;С
плааления кипения
нсоон СНзСООН CFbCHjCOOH СНЭСН3СН2СООН СНз—СН—соон к СНзСНиСН2СН2СООН CITj—СН—СН2СООН Ъг1з СНз(СН2)4СООН CHj(CHa)uCOOH СИ1(СН2)16СООН Муравьиная кислота Уксусная кислота Пропионовая кислота Масляная кислота Изомасляная кислота Валериановая кислота Изовалериановая кислота Капроновая кислота Пальмитиновая кислота Стеариновая кислота 8.4 16,7 —22,4 — 7,9 —47.0 -34,5 —37,6 — 1.5 64,0 70.0 100,7 118.L 141.1 163.5 154,4 187,0 176,7 205,3 271,5(100 мм) 291 (НО мм)
ЗАЗ
16.1 Л. Химические свойства
Реакционная способность карбоновых кислот определяется в основном наличием в их структуре карбоксильной группы. Карбоксильная группа представляет собой сопряженную систему, в которой неподеленная пара электронов атома кислорода гидроксильной группы вступает в сопряжение с n-электронами карбонильной группы (р, л-сопряжение). Вследствие +Л4-эффекта со стороны группы —ОН электронная плотность в сопряженной системе смещена в сторону атома кислорода карбонильной группы 'ЧчС = О, неподеленные пары электронов которого не участвуют в сопряжении. В результате смещения электронной плотности связь О—Н оказывается сильно поляризованной, что приводит к появлению в карбоксильной группе ОН-кислотного центра. Но в то же время за счет -|-Af-эффекта со стороны группы—ОН в молекулах карбоновых кислот в некоторой степени уменьшается частичный положительный заряд на атоме углерода карбонильной группы по сравнению с альдегидами и кетонами. Кроме того, вследствие —/-эффекта карбоксильной группы в молекуле карбоновой кислоты происходит смещение электронной плотности с углеводородного остатка, что приводит к появлению С—Н-кнслотного центра у а-углеродного втома:
электрофильный центр
СН-кислотный центр
ОН-кисдотный центр
Исходя из приведенного строения карбоновых кислот, основ? ные их реакции можно условно разделить ка четыре группы:
• с участием связи О—Н (кислотные свойства);
• нуклеофильное замещение с участием атома углерода карбо нилыгой группы,
• замещение атомов водорода прн а-углеродном атоме;
• окисление и восстановление.
А. Кислотные свойства
Кислотные свойства карбоновых кислот обусловлены их способностью отщеплять атом водорода карбоксильной группы в виде протона.
304
В водных по схеме
растворах карбоновые кислоты диссоциируют
карбиновая кислота
+ н2о
+ нао+
О"
карбоксилат кон
нон гндроксоння
В процессе диссоциации образуется карбокснлат-нон, в котором оба атома кислорода равноценны, а отрицательный заряд равномерно делокализован между ними. Делокализацию заряда в карбоксил ат-ионе можно представить в виде двух граничных структур илн структуры с равными дробными зарядами на атомах кислорода:
Го Го’ .о,д-
R-C<b. <----------► R-C<& ИЛИ R-C<q1/2
В результате делокализации отрицательного заряда карбокси-лат-ион обладает высокой устойчивостью. А поскольку, как известно, сила кислоты определяется устойчивостью образующегося аннона, то карбоновые кислоты превосходят по кислотным свойствам спирты и фенолы, где возможность делокализации заряда в анионе меиьшая.
На силу карбоновых кислот оказывает также влияние структура углеводородного радикала, связанного с карбоксильной группой, н заместители в нем. Электронодонорные заместители увеличивают электронную плотность в кислотном центре и тем самым дестабилизируют карбоксил ат-ион, что в конечном итоге приводит к ослаблению кислотных свойств. Электроноакцепторные заместители, наоборот, смещая электронную плотность на себя, повы-щают устойчивость карбокснлат-иона, что приводит к усилению кислотности. В табл. 16 3 приведены константы диссоциации некоторых карбоновых кислот в единицах рКа
По мере удаления заместителя от карбоксильной группы ого влияние на кислотные свойства ослабевает, иапрнмер
СНз—СН2—СН—СООН СНз—СН—СНг—соон
С1 1 С1
а хлормаслиная I 3 хлормаслнная
кислота (рК«2 84) j кислота (рК„4 06)
СН2—СНг—CH-rJ-COOH
Y ай ори асл я на и кислота (рК* 4,52) ]
38S
Таблица 16 3
Константы диссоциации некоторых карбоновых кислот в воде при 25° С
Название Формула
Муравьиная кислота НСООН 3.75
Уксусная кислота СНэСООН 4,76
Пропионовая кислота СНэСНгСООН 4,87
Масляная кислота СНаСНаСНгСООН 4,82
Валериановая кислота СНзСНаСНгСНгСООН 4,86
Фторуксусная кислота CHaFCOOH 2,57
Хлоруксусная кислота CHaCICOOH 2.85
Бром уксусная кислота СНаВгСООН 2.90
Иодуксусная кислота CHalCOOH 3,[6
Дихлоруксусная кислота CHChCOOH 1,25
Трихлоруксусная кислота CChCOOH 0,66
Трифторуксусная кислота CFaCOOH 0.23
Образование солей. Карбоновые кислоты при взаимодействии с активными металлами, основными оксидами, гидроксидами и карбонатами щелочных металлов образуют соли:
2СНз—COOH-pZn >- (СНз—COO)2Zn + Н2
2СНа—СООН + MgO —> ‘(CHi^COOJiMg + н2о •
ацетат магния &
СНз—СООН + NaOH ► СН3—COONa + Н2О
СНз—СООН + NaHCO3 > СН3—COONa + СО2 + Н2О
В названиях солей карбоновых кислот чаще применяют тривиальные латинские названия кислот. Соли муравьиной кислоты имеют общее название — формиаты, уксусной — ацетаты, пропионовой— пропионаты, масляной — бутираты, нзомасляной — нзо-бутираты. По заместительной номенклатуре название аннона образуется путем замены части названия кислоты -овая кислота на суффикс -оат, например:
СНз—С Н г—С Hr—COONa
б}тират натрия бутаноат натрия
СНз—СНг—СНг—СНг—СНг—COONa
гексаноат натрия
Б. Реакции нуклеофильного замещения
В результате электроноакцепторных свойств атома кислорода карбонильной группы атом углерода карбоксильной группы приобретает частичный положительный заряд н становится электро-
366
фильным центром, который может быть атакован нуклеофильным реагентом;
электрофильный центр
Nu’
В процессе атаки происходит замещение гидроксильной группы на нуклеофильную частицу.
Взаимодействие со спиртами (реакция этерификации). Карбоновые кислоты при нагревании в присутствии кислотного катализатора реагируют со спиртами, образуя сложные эфиры. Эта реакция получила название реакции этерификации:
снэ-с<° + он
уксусная
кислота
H2SO4 q
н-оед сн3-<~ + Н1О
этанол этилацетат
Реакция этерификации обратима. Образовавшийся сложный эфир в кислой среде подвергается гидролизу до исходных кислоты и спирта. Для смещения равновесия в сторону образования сложного эфирв либо используют избыток одного из реагентов (обычно спирта), либо удаляют из реакционной среды воду. Легче всего сложные эфиры образуются из первичных спиртов н низших карбоновых кислот Вторичные спирты и высшие кислоты реагируют медленнее. Третичные спирты из-за пространственных препятствий вступают в реакцию этерификации с большим трудом Кроме того, под действием минеральных кислот третичные спирты легко подвергаются внутримолекулярной дегидратации с образованием алкенов (см. с. 257). Каталитическое действие серной кислоты состоит в активировании молекулы карбоновой кислоты. Механизм этерификации может быть представлен следующей схемой:
И
сн3-сС°:< <=
ОН
+ ОН
•Зп5,
' он карбкатион
ОН
СНз-С-О-ед
:ОН Н А
сн3—с—ос2н5
сн3-с
-Н2О; -Н
ОС2Н5
н н
307
Вначале карбоновая кислоте протонируется по атому кислорода карбонильной группы с образованием карбкатиона. Затем карбкатион присоединяет молекулу спирта с образованием промежуточного продукта, который отщепляет молекулу воды и протон (возврат катализатора), превращаясь прн этом в сложный эфир.
Взаимодействие с галогенирующими реагентами (РС1з, РС15, РВгз, SOCh)- При действии на карбоновые кислоты хлоридов фосфора (III) и фосфора (V), бромида фосфора (III) или тно-нилхлорнда SOCh образуются галогенангидриды карбоновых
кислот
РС1Э
СН
-|- Н3РО3
СНз—СООН
PCI&
СНз—С
С
+ РОС13 + НС1
ч
sock
СНз—С + SO2 + НС1
\:i аяетнлморнд, хлор ангидрид уксусной кислоты
Для получения хлорангидрндов чаще используют тиоиилхло-рид, так как в этом случае образуются газообразные побочные продукты.
Галогенангндриды карбоновых кислот — весьма реакционноспособные вещества, широко применяемые в органическом синтезе.
Взаимодействие с аммиаком и аминами. При обработке карбоновых кислот аммиаком, первичными илк вторичными аминами образуются аммониевые соли, которые при нагревании в сухом виде (пиролиз) отщепляют воду и превращаются в амиды:
СНз—COOH + NH3 ZZT СНз—COO~NH4+
ацетамид
nh2
ацетат аммония
Из-за жестких условий йротекання реакции этот метод образования амидов редко используется в препаративных целях.
368
Образование ангидридов кислот. В присутствии водоотнимающих средств, в качестве которых чаще применяют оксид фосфора (V) Р2О5 или трнфторуксусиый ангидрид (CF3CO)2O карбоновые кислоты прн нагревании подвергаются межмолекулярнон дегидра-гацни с образованием ангидридов
2СН3—о/ \)Н
РгО5
НгРО<
СНа--С
СНз—С
укосныи ангидрид
В- Замещение водорода у а-углеродного атома
Вследствие электроноакцепторных свойств карбоксильной группы (—/-эффект) атомы водорода у а-углеродного атома приобретают подвижность. Так, при обработке карбоновых кислот хлором или бромом в присутствии катализатора РС1з или РВг3 атомы водорода у а-углеродного втома замещаются на галоген:
СНз—СН—СООН + Вг2 СНз—СН—СООН 4- НВг
Н Вг
продиопопая кнс/тота а бром пропионовая кислота
Эта реакция известна как реакция Гелл я — Фольгарда — Зелинского. При наличии в а-положеиии двух втомов водорода замещению может подвергаться один или оба атома водорода. Реакция протекает через стадию образования галогенангндрндов кислот, которые галогенируются значительно легче, чем сами кислоты:
О СН3—СН2—сУ
ОН
пропионовая кислота
о / СНз—(pH—С Вг Вг
/
СНз—СН2—С^
Вг
[фОП иоиилбромид
НОН^
—НВг
СНз—С^
Вг ОН
броман[ и др нд а брочпропноновоЙ кислоты
а бромпропионовая кислота
369
Г. Окисление и восстановление
Монокарбоновые кислоты, за исключением муравьиной, довольно устойчивы к действию окислителей. Муравьиная кислота легко окисляется КМпО* и другими окислителями с образованием угольной кислоты, которая разлагается на окснд углерода (IV) н воду:
НСООН СО2| + Н2О
При восстановлении монокарбоновые кислоты образуют, в за* виснмости от условий, альдегиды нли первичные спирты.
16.1.5 . Отдельные представители
Муравьиная кислота (метановая кислота) НСООН. Бесцветная жидкость с резким запахом (т. пл. 8,4 °C, т. кип. 100,8 °C); растворима в воде, этаноле, эфире. В свободном состоянии содержится в выделениях желез муравьев, в крапиве. Промышленный способ получения основан на взаимодействии оксида углерода (II) с натронной известью:
j-
СО + NaOH Н—COONa Н—СООН
формил натрия
В связи с особенностью строения (наличием альдегидной группы) муравьннвя кислота дает реакцию «серебряного зеркала»:
альдегидная группа
+ 24820 (NH4OII*
+ 2Ag|
СО2 + Н2О
(Три нагревании с коицеитрироваиной серной кислотой муравьиная'кислота разлагается с образованием оксида углерода (П) и воды:
Н—СООН СО + Н2О
Реакции с оксидом серебра н концентрированной серной кисло-той-являются качественными на муравьиную кислоту.
Муравьиная кислота широко используется в органическом синтезе как протрава при крашении текстиля, в пчеловодстве против варроатоза, для получения пестицидов н др. В медицине муравьиная кислота применяется в виде 1 % спиртового раствора (муравьиный спирт) как растирка при невралгиях, миозитах н др.
370
Уксусная кислота (этановая кислота) СНз—СООН. Бесцветная жидкость с резким запахом, смешивается с водой, этанолом, эфиром. Безводная (ледяная) уксусная кислота имеет т. пл. 16 °C, т, кип. 118 °C.
Уксусная кислота широко применяется как реагент и растворитель в органическом синтезе, 3—6 % растворы используют в качестве вкусовой приправы и консерванта, В больших количествах уксусная кислота используется в производстве искусственных волокон на основе целлюлозы, а также в синтезе лекарственных препаратов.
Пропионовая кислота (пропановая кислота) СНз—СНг—СООН. Бесцветная жидкость (т. кип. 141,1 °C), смешивающаяся с водой и органическими растворителями. Получается окислением пропионового альдегида. Применяется в производстве витаминов, душистых веществ, гербицидов и др.
«-Масляная кислота (бутановая кислота) СНэСНзСНзСООН. Бесцветная вязкая жидкость с неприятным запахом (т. кип. 163,5 °C), растворимая в воде и спирте. В свободном виде содержится в животных жирах, прогоркшем масле, в поте. Получают окислением н-бутанола нли н-мвслиного альдегида. Применяется в синтезе душистых веществ, лекарственных средств, эмульгаторов и др, В виде сложного эфира с глицерином входит в состав коровьего масла.
Валериановая кислота (пентановая кислота) СН3СН2СН2СН2СООН. Жидкость (т. кип. 185,4 °C), растворима в воде. Содержится в корне валерианы. Получают окислением н-амнлового спирта. Применяют для получения ароматизирующих веществ, в производстве лекарственных средств.
н-Валернановой кислоте изомерна изовалериановая кислота, содержащаяся в свободном состоянии и в виде эфира в корнях валерианы:
СНз
^СН—СНг—СООН
СНз^
3-четнлбутановая кислота нзовалериагюпаА к чел от а
Применяется для получения лекарственных препаратов бромизо-вала (см, с. 434) и валидола.
16.2. НЕНАСЫЩЕННЫЕ МО НО КАРБО НОВЫЕ КИСЛОТЫ
К ненасыщенным карбоновым кислотам относят карбоновые кислоты, содержащие в углеводородном радикале кратную связь.
371
16.2.1. Номенклатура н изомерии
В номенклатуре ненасыщенных кислот широко применяются тривиальные названия. По заместительной номенклатуре ИЮПАК названия ненасыщенных кислот образуют аналогично насыщенным, используя суффикс -ен для обозначения двойной связи н суффикс -ин для обозначения тройной, с указанием положения кратной связи в углеродной цепи. Названия некоторых ненасыщенных монокарбоновых кислот приведены в табл. 16.4.
Таблица 16.4
Названия некоторых ненасыщенных монокарбоновых кислот
Формула Название по номенклатуре
тривиальной заместительной
СН^СН—СООН СНа=СН—СООН С —С На—СООН Н СООН ^о-с^ сн^ ^н н н сн^ ^соон CHrf^COOH СН,—С^С—СООН СН—(СН2)7—СНз II СН— (СН2)г—СООН СН3—(СН2)4—СН НС—CHs-Jh Н<!— (СНг)т—СООН НС—СНа—СНз H<L СН2—СН нс—сн2- 4н hL (СНа)7—СООН Акриловая кислота Метакриловая кислота Винилуксуснаи кислота Кротоновая кислота Иэокротоновая кислота Нропиоловая кислота Тстроловая кислота Олеиновая кислота Линолевая кислота Линоленовая кислота Пропеновая кислота 2-Мети.1пропеновая к и мота 3-Б утеновая кислота транс-2- Бутеновая кислота цмс-2-Бутеновая кислота Пропиновая кислота 2-Бутиновая кислота цпс-9-Октадеиеновая кислота цис-9-цис-12-Окта-дскадиеповая кислота Чис-9-ци£>12-ци£-]5-Октадекатриеновая кислота X
372
В зависимости от положения кратной связи по отношению к карбоксильной группе различают а, 0-; 0, у-; у» 6- и другие ненасыщенные^ислоты, например:
р а у 0 а
СНз=СН—СООН СН?=СН—СН2СООН
а р-неиасыщеинаи кислота 0 у ненасыщенная кислота
Для ненасыщенных монокарбоновых кислот характерна структурная изомерия, обусловленная разной структурой углеводородного радикала н положением кратной связи, а также геометрическая изомерия, связанная с разным расположением заместителей относительно плоскости двойной связи.
16.2.2. Способы получения
Для получения ненасыщенных монокарбоновых кислот могут использоваться многие из методов синтеза насыщенных карбоновых кислот. В качестве исходных Веществ применяются ненасыщенные соединения, например окисление ненасыщенных первичных спиртов и альдегидов в мягких условиях, гидролиз нитрилов и др. Кроме того, существуют специфические методы получения;
Гидрокарбоксилирование алкинов (реакция Реппе). В присутствии карбонилов металлов алкины взаимодействуют с оксидом углерода (II) в водной среде с образованием а, 0-ненасыщеиных монокарбоновых кислот:
НС=СН + СО + Н2О
ацетилен
СНг=СН—СООН
акрнчовая кислота
Элиминирование ft-галогене- и fi-гидроксшсарбоновых кислот. 0-Галогенокарбоиовые кислоты при нагревании с водой отщепляют галогеноводород и превращаются в а, 0-ненасыщенные кислоты:
р « ,
СНз—СН?—СООН —СН2—СН—СООН + НС1
С1
Р Х.’.орпрооиомовая кислота акриловая кислота
0-Гидроксикарбоновые кислоты при нагревании отщепляют молекулу воды и превращаются в а, 0-ненасыщенные кислоты:
CHs—СН?—СООН —СН2==СН—СООН + Н2О
Ан
р гидрокснпропноновая кислота акриловая кислота
373
16.2.3. Физические свойства
В обычных условиях ненасыщенные монокарбоновые кислоты являются бесцветными жидкостями нли кристаллическими веществами. Низшие представители хорошо растворимы в воде, обладают резким раздражающим запахом. С увеличением молекулярной массы кислоты растворимость в воде снижается Высшие ненасыщенные кислоты не растворимы в воде и хорошо растворяются в органических растворителях.
16.2.4. Химические свойства
Реакционная способность ненасыщенных монокарбоновых кислот обусловлена наличием в их структуре карбоксильной группы н кратной связи.
За счет карбоксильной группы ненасыщенные кислоты вступают в реакции, характерные для насыщенных кислот, в частности они образуют солн, галогеиангидриды, ангидриды, сложные эфиры, амнды (см. с. 364—369). По кратной связи в углеводородном радикале ненасыщенные инслоты проявляют свойства влкенов (алкинов). Так, для инх характерны реакции присоединения, окисления н полимеризации (см. подразд. 2.5, 4.4).
Однако присоединение галогеноводородов к а, 0-иенасыщен-ным кислотам протекает против правила Марковникова. Это объясняется электроиоакцепторным влиянием карбоксильной группы за счет —М н —/-эффектов;
\ 'О* сн/С1Дс'он+ Н—Вг
Т е~ ОН
Вг
trHrCHi’c''on Вг
акриловая кислота Р-бромпропионовая
кислота
а, р-Ненасыщенные кислоты, особенно с тройной связью, являются более сильными кислотами по сравнению с соответствующими насыщенными. Это объясняется повышением устойчивости аниона за счет делокализации заряда по сопряженной системе. Ниже приведены значения р/<д в воде (при 25 °C) некоторых ненасыщенных карбоновых кислот и пропионовой кислоты:
СН2=СН—СООН СНз—С=С—СООН НС=С--СООН
4 26 2 60 1 84
СНз—СНз—СООН
4 87 .
16.2.5. Отдельные представители
Акриловая кислота СН2—СН—СООН. Бесцветная жидкость с резким запахом (т. пл. 13 °C, т. кип 141 °C), хорошо
374
растворяется в воде. Акриловая кислота легко полимеризуется с образованием полиакриловой кислоты. Важное практическое значение имеют полимеры на основе сложных эфиров акриловой кислоты, так называемые полиакрилаты
,О
«сн2=сн-сС „
2 OR
сн2-сн—
I
orJ
и
полиакрилат
В медицине полиакрилаты применяются в изготовлении зубных протезов,
СН3
Метакриловая кислота СН2==^—СООН. Бесцветная жидкость (т. кип. 160,5 °C). Метакриловая кислота легко полимеризуется. Важное значение имеет метиловый эфир метакриловой кислоты, полимеризацией которого получают полиметилметакрилат — органическое стекло (плексиглас):
п СН2=С~СООСН СН3
метилметакрилат
СНз снгс-
COOCHJ „
полиметилметакрилат
Олеиновая кислота (цис-9-окт а де ценова я кислота) СНз—(СНг)?—СН—СН—(СНа)?—СООН. Бесцветная маслянистая жидкость без вкуса и запаха. В виде сложных эфиров глицерина входит в состав растительных масел. Особенно много содержится в оливковом, миндальном и подсолнечном маслах. Олеиновая кислота является цис-изомером. Под действием азотистой кислоты или прн УФ-облученни олеиновая кислота изомеризуется в элаидиновую кислоту, являющуюся Грайс-изомером:
СНз—(СН2)7—СН 1ЛОг СНз—(СН2)Г—СН
НООС— (СН2)7-—hL(CH2)7-COOH
олеиноаан кислота элаидиновая кислота
Смесь этиловых эфиров олеиновой (около 15%), линолевой (около 15 %) и линоленовой (около 57 %) кислот входит в состав лекарственного препарата линетол, который применяется в медицине для профилактики и лечения гипертонии н атеросклероза, а Также при ожогах и лучевой болезни.
375
16.3. АРОМАТИЧЕСКИЕ МОНОКАРБОНОВЫЕ КИСЛОТЫ
Ароматическими карбоновыми (аренкарбоиовыми) кислотами называют органические кислоты, в которых карбоксильная группа непосредственно связана с ароматическим ядром.
Простейшим представителем аренкарбоновых кислот бензольного ряда является бензойная кислота. В соответствии с номенклатурными правилами ИЮПАК другие гомологи этого ряда рассматриваются как производные бензойной кислоты, например:
бензойная кислоте
CsHs
3 этидбензойная Кислота
2 метил 5 этилбензойидя кислота
Метилбеизойные кислоты имеют тривиальное название — толуиловые кислоты;
о толуиловая кислота
At толуиловая ккслотя
СНз
СООН
л-тол у иловая кислота
Названия карбоновых кислот нафталинового, антраценового, фенантренового и других рядов образуются из названия соответствующего углеводорода добавлением слов — карбоновая и кислота:
2 и а фтал мн кар боновая
кислота
СООН
3 метил-Ьнафталинкарбоиовая
кислота
376
Карбоновые кислоты, в которых карбоксильная группа расположена в боковой углеродной цепи ароматического углеводорода, рассматривают как производные кислот алифатического ряда:
Сн 2—СООН
фенил у кем: мая кислота, фенилэтановая кислота
транс 3 фенилпропеновая кислота, коричная кислота
16.3.1. Способы получения
Ароматические карбоновые кислоты могут быть получены теми же способами, что и алифатические.
Окисление алкиларенов. Этот метод является одним из наиболее часто применяемых для получения аренкарбоновых кислот. Окислению подвергают главным образом метил арены, В качестве окислителей обычно используют КМпО-*. СгОз нлн кислород в присутствии солей кобальта и марганца:
СеН5—СНз С6Н5— СООН
—НэО
толуол бензойная кислота
Гидролиз тригалогенопроизводных ароматических углеводородов. Прн нагревании с водным раствором щелочи ароматических трнгалогенонроизводных, содержащих атомы галогена у одного и того же атома углерода, последние подвергаются гидролизу, образуя аренкарбоновые кислоты:
т р и хл о рм стнлбе нзол
3NaOH
СООН
+ 3NaCl 4- Н2О
бензойная кислота
Гидролиз нитрилов. В водных растворах кислот и щелочей нитрилы гидролизуются с образованием карбоновых кислот
С6Н5—C=N -22-* CsHsCOOH + (NH.)2SO<
H2SO4 бензонитрил бензойная кислота
16.3.2. Физические свойства
Ароматические монокарбоновые кислоты — бесцветные кристаллические вещества, некоторые нз них имеют слабый приятный запах. Низшие гомологи малорастворимы в воде и перегоняются водяным паром. Аренкарбоновые кислоты хорошо растворяются в этаноле и эфире,
377
16.3.3. Химические свойства
Реакционная способность аренмонокарбоновых кислот обусловлена наличием в их структуре карбоксильной группы и бензольного ядра.
По карбоксильной группе для них характерны реакции, свойственные насыщенным монокарбоновым кислотам (см. с. 364—369), а именно, образование солей, галоген ан гидридов, ангидридов, сложных эфиров:
С6Н5СООН
бензойная кислота
NaOH
С6Нв— С
ангидрид бензойной кислоты
По кислотности ароматические монокарбоновые кислоты превышают насыщенные (кроме муравьиной) и ненасыщенные моно-карбоновые кислоты алифатического ряда, что связано с повышением устойчивости аниона за счет делокализации заряда по <опря-
378
ценной системе бензольного кольца. Ниже приведены значения рКа некоторых кислот в воде при 25 °C:
Цо мере удаления ароматического ядра от карбоксильной группы кислотность уменьшается
При нагревании аренкарбоновых кислот в присутствии медного порошка или солей меди свыше 200 °C происходит нх декарбоксилирование:
СООН
+ СО2
За счет бензольного кольца аренмонокарбоновые кислоты вступают в реакции электрофильного замещения (нитрование, сульфирование, галогенирование), свойственные ароматическим углеводородам (см. подразд. 6.5.1). Карбоксильная группа, проявляя — / и —Л4-эффекты, дезактивирует бензольное кольцо по отношению к электрофильным реагентам, поэтому реакции электрофильного замещения протекают значительно труднее, чем для незамещенного бензола. Являясь ориентантом 11 рода, карбоксильная группа направляет замещение в мета-положение:
379
соон
HNOj, На5О<
+ H2O
NO-2
М нитробентойнгн кис юта
соон
бензойная кислота
Brj, FeBrj
соон
HaSC>4
+ H2O
SO3H
м сульф обе той па я кислота
СООН
+ НВг
Вг
л бромбензойна я кислота
16.3.4. Отдельные представители
Бензойная кислота СбН5—СООН. Белое кристаллическое вещество (т. пл. 122 °C), легко возгоняется, плохо растворяется в воде, хорошо - в этаноле н бензоле. В виде сложных эфиров содержится в некоторых природных маслах, например, в гвоздичном; ее бензиловый эфир входит в состав перуанского бальзама. Бензойная кислота используется квк исходное вещество в производстве красителей, душистых веществ, лекарственных средств, а также как антисептическое н противогрибковое средство в медицине. Натриевая соль бензойной кислоты (бензоат иатркя) применяется как отхаркивающее средство при бронхитах.
Фенил уксусная кислота СЙН5—СНа—СООН. Белое кристаллическое душистое вещество (т. пл. 77 °C). Благодаря наличию активированной метиленовой группы легко вступает в реакции конденсации с альдегидами, кетонами, ангидридами кислот н др. Используется в органическом синтезе для получения душистых веществ и лекарственных средств.
Коричная кислота (транс-3-фенилпропеновая кислота) С6Н3—СН=СН—СООН. Белое кристаллическое вещество (т. пл. 133 °C). Содержится в виде сложных эфиров в эфирных маслах, смолах, бальзамах. Применяется в синтезе душистых веществ и лекарственных средств.
380
16.4. ИДЕНТИФИКАЦИЯ МОНОКАРБОНОВЫХ КИСЛОТ
Идентификация карбоновых кислот с помощью химических методов основана на особенностях химического поведения карбоксильной группы, в частности иа способности к образованию труднорастворимых солей с тяжелыми металлами (Fe, Ag, Си н др.) н достаточно легко идентифицируемых производных (амидов, сложных эфнров). Прн действии карбоновых кислот на гидрокарбонаты щелочных металлов выделяется окснд углерода (IV), образующий с баритовой водой белый осадок ВаСОз- Для идентификации ненасыщенных карбоновых кислот используют также реакционную способность кратной связи (обесцвечивание бромной воды или раствора перманганата калия). Ароматические карбоновые кислоты могут быть обнаружены по образованию окрашенных продуктов замещения с участием бензольного ядра.
УФ-спектры для алифатических монокарбоновых кнслот малопоказательны, так как они поглощают ниже 220 нм. Для ароматических кнслот характерно смещение полосы «бензольного поглощения» (около 260 нм) в сторону более длинных волн (за счет удлинения цепи сопряжения).
ИК-спектры карбоновых кислот. Для карбоксильной группы характерны две полосы поглощения: в диапазоне 2500—3300 см-1 (vQH для ассоциированных молекул) и 1700—1720 см-1 (у ). Области валентных колебаний ОН- н СН-групп совпадают (около 3000 см-1). Для ароматических кнслот характерно поглощение ароматического ядра.
Идентификацию карбоновых кислот с помощью масс-спектрометрии используют редко. Для этой цели их переводят в летучие сложные эфиры, которые дают достаточно отчетливый пнк молекулярного ионв:
+ I--- R +
О: О:
D / //
R—С —^R—С------------------> RC=O+
OR'
-->- [COOR'] +
ПМР-сиектры карбоновых кислот характеризуются сигналом протона карбоксильной группы, который расположен в слабом поле (6=10—13 м.д.).
КОНТРОЛЬНЫЕ ВОПРОСЫ И УПРАЖНЕНИЯ
I. Нвпишите структурные формулы следующих карбоновых Кнслот: а) изовалериановая кислота; б) а-метил масляная кисло-Та; в) 3-метил пента новая кислота; г) виннлуксусиая кислота; Д) кротоновая кислота; е) транс-2-бутеиовая кислота; ж) коричная кислота. Укажите, по какой номенклатуре приведены названия.
381
2. Приведите схемы реакций синтеза нзомасляной кислоты тремя способами.
3. Исходя из соответствующего галогенопроизводного, через стадию образования нитрила приведите схему получения изовале-рнвновой кислоты.
4. Расположите следующие кислоты в порядке увеличения кислотности и объясните различие кислотных свойств, исходя из стро-ения молекул: а) пропионовая кислота; б) акриловая кислота, в) бензойная кислота; г) 4-нитробеизойная кислота.
5. Напишите схему реакции получения сложного эфира из этанола и пропионовой кислоты. Опишите механизм реакции,
6. Приведите схемы реакций бензойной кислоты с приведенными ниже соединениями: а) РС15; 6)NHa, i\ в) (СНзСО)2О, Н + ; г) СНаОН, Н+; д) №НСОз. Назовите продукты реакций.
7. Напишите схемы реакций, с помощью которых можно отличить муравьиную, уксусную и метакриловую кислоты друг от друга.
8. Напишите схемы реакций: а) акриловой кислоты с хлороводородом; б) бензойной кислоты с концентрированно^ серной кислотой. Назовите продукты реакций.
Глава 17 ДИКАРБОНОВЫЕ КИСЛОТЫ
Дикарбоновыми кислотами называют производные углеводородов, содержащие в своем составе две карбоксильные группы.
17.1. НАСЫЩЕННЫЕ ДИКАРБОНОВЫЕ КИСЛОТЫ
17.1.1. Номенклатура и изомерия
В номенклатуре дикарбоновых кислот широко применяют тривиальные названия. По заместительной номенклатуре ИЮПАК названия дикарбоновых кислот образуют из названий соответствующих по количеству атомов в главной цепи монокврбоно-вых кнслот с добавлением множительной приставки -ди-. Более упо-требнмы тривиальные названия.
В табл. 17.1 приведены названия важнейших представителей дикарбоновых кислот.
Таблица 17,1
Названия некоторых дикарбоиовых кнслот
Формула Название по номенклатуре
тривиальной заместительной
НООС—СООН НООС—СНг—СООН НООС—СН—СООН 1 CHj НООС—СНг—СНг—СООН НООС—СНг—СНг—СНг—СООН НООС—СНг—С Нг—СНг—СНг—СООН НООС—(СНг)Б—СООН НООС— (СНг)б—СООН Щавелевая кислота Малоновая кислота Метил малоновая кислота Янтарная кислота Глутаровая кислота Адипиновая кислота Пимелиноная кислота Пробковая кислота Этандиовая кислота Пропандиован кислота Метилпропа ндно-вая кислота Бутандиовая кислота Пентандновая кислота Гександиовая кислота Гептандиовая кислота Октандновая кислота
383
Изомерия дикарбоиовых кислот обусловлена разной структурой углеродного скелета молекулы.
17.1.2. Способы получения
Днкарбоновые кислоты получают теми же методами, что н монокарбоновые кислоты. В качестве исходных веществ используют соответствующие бифункциональные соединения.
Окисление двупервичных гликолей, диальдегидов и гидрокси-кислот:
СН-2—СН-2—СНУ
он он
припанднот 1 3
НООС--СНг— СООН
малоновая инслотз
НООС--СООН
[Лиоксаль
щавечеван кислота
СНг—СНг—СООН
1°1
-!12О
НООС—СНг—СООН
Р гндроксипронионппая кислота
ма чиновая кисчота
Гидролиз динитрилов:
N = С—С Нг—СНг—С = N
- > НООС—СНг—СНг—СООН
- NIL,UH
янтарная кислота
17.1.3. Физические свойства
Дикарбоновые кислоты — белые кристаллические вещества, хорошо растворимые в воде. Температуры плавления кислот с чет ным числом атомов углерода выше температур плавления соседних кислот с нечетным числом атомов углерода.
17.1.4. Химические свойства
Днкарбоновые кислоты, имея в молекуле две карбоксильные группы, диссоциируют ступенчато, образуя аиион (pKaJ н дианион (рКа2):
НООС—СООН НООС—COO ^zi ООС—СОО~
— Н анион — Н+ дианиои
384
По первой ступени дикарбоновые кислоты обладают более сильными кислотными свойствами, чем монокарбоновые кислоты с тем же числом атомов углерода. Это обусловлено взаимным влиянием карбоксильных групп (индуктивный эффект). Вторая карбоксильная группа, обладая электроноакцепторнымн свойствами, способствует делокализации заряда аниона и тем самым повышает его устойчивость. По мере удаления карбоксильных групп друг от друга их взаимное влияние ослабевает, вследствие чего кислотность по первой ступени снижается.
Отрыв протона от второй карбоксильной группы происходит значительно труднее из-за низкой стабильности дианнона Поэтому по второй ступени кислотность дикарбоиовых кислот значительно ниже, чем по первой, особенно для щавелевой н малоновой кислот. Значения рЛа (в Н2О) для некоторых дикарбоиовых кислот:
СООН СН<2—СООН
СООН / СНг—СООН /
iooH Н2С ^соон <^Нг—СООН Н2С ^СН2—СООН
pKai 1,27 2,86 4,21 4,34
p/G2 4,27 5,70 5,64 СНг—СНз—СООН СНг— СНг— СООН 5,2? ‘
РКЙ1 4,41
рКа? 5,28
По химическим свойствам днкарбоновые кислоты во многом сходны с монокарбоновыми. Они образуют одни н те же функциональные производные, с тем лишь отличием, что реакции могут идти с участием как одной, так и двух карбоксильных групп. Например, они образуют нейтральные н кислые соли [NaOOC—(СНг)л—COONa, НООС—(СН2)„—COONa], полные н неполные сложные эфиры [ROOC—(СН2)П—COOR, НООС—(СНа)п—COOR], полные и неполные галогенан-гндрнды [HalOC—(CH2)n—COHal, НООС—(СН2)Я—COHal], полные и неполные амиды [H2NCO—(СН2)П—CONH2, НООС—(СН2)п—CONH2].
Наряду с этим дикарбоновые кислоты проявляют н ряд специфических свойств, в частности по-разному относятся к нагреванию.
Отношение дикарбоновых кислот к нагреванию. Щавелевая и малоновая кислоты при нагревании выше температур плавления подвергаются декарбоксилирован ню (отщепляют СОа) по одной
13 5 104
385
карбоксильной группе и превращаются в монокарбоновые кислоты — муравьиную н уксусную соответственно:
5П0 ФГ
ноос—соон —t нсаон + со2
.щавелевая кислота муравьиная
кислота
НООС—СНа—СООН
малоновая кислота
СНзСООН + СО2 уксусная кислота
Аналогично ведут себя при нагревании моно-'и Диалкнлзаме-щенные малоновые кислоты
R*
..... I
Н-ГООС-С-СООН
к
он-соон + со, R
Следующие два представителя гомологического ряда дикарбоновых кислот при нагревании образуют циклические ангидриды
янтарная кислота
Li2
\ >ОН :
сн2-сС^
2
300°
-Н2О
глутаровая кислота
Адипиновая кислота прн нагревании в циклический кетоц — цнклопеитанон:
до 300 °C превращается
, ^СН2
Н2? чсоон н>с-с£ООН
t
-СО2; -Н2О*
н2с-
Н2С^_
сн2 \=о
сн2
адипиновая кислота
циклопентанон
386
Образование имидов. При нагревании янтарной и глутаровой кнслот нлн их ангидридов с аммиаком образуются циклические Ц.МНДЫ1
2Н2О
О янтарный ангидрид
17.1.5. Отдельные представители
Щавеле вая к пел от а НООС—СООН. Белое кристаллическое вещество с т. пл. 189 °C, легко растворяется в воде и спиртах. Содержится в виде солей во многих растениях (щавель, ревень и др.). Соли щавелевой кислоты называют оксалатами. Кристаллы оксалата кальция трудно растаорнмы в воде и могут откладываться при патологических состояниях в почках в виде камней (почечно-каменная болезнь).
В промышленности щавелевую кислоту получают из формиата натрия:
2HCOONa
натрия формиат
COON а
400 °C । 2НС1
COONa ”2NaC1
нятрня оксалат
СООН ioOH
щавелевая кислота
Специфическими реакциями щавелевой кислоты являются разложение концентрированной серной кислотой и окисление.
При нагревании с концентрированной серной кислотой щавелевая кислота разлагается с образованием оксида углерода (II), оксида углерода (IV) и воды:
НООС—СООН H’s0,f> СОз + СО + Н2О
13*
387
При окислении они образует оксид углерода (IV) и воду
НООС—СООН 2СО2+Н2О
Эта реакция используется в аналитической практике для установления титра КМпО4, применяемого в количественном анализе:
5НООС—СООН + 2КМпО4 + 3H2SO4->
1ОСО2 + IGSO, + 2MnSO4 4- ВН2О
Качественной реакцией для обнаружения щавелевой кислоты и ее растворимых солей является образование нерастворимой соли океалвта кальция
NaOOC-COONa + СаСЦ
2NaCl
В аналитической практике этой реакцией обнаруживают также иокы Са2"^
Щавелевая кислота применяется для очистки металлов от ржавчины и накипи, а также в качестве протравы в кожевенном производстве.
Малоиовап кислота НООС—СНг—СООН. Белое кристаллическое вещество с т. пл. 135 °C, растворимое в воде, этаноле, эфире Содержится в соке сахарной свеклы. Впервые получена прн окислительном декарбоксилировании яблочной кислоты:
НООС—СН—СНа—СООН
Ан
яблочная кислота
--► НООС—СНг—СООН + СО2 + Н2О мал о новая кислота
В настоящее время ее полуъают гидролизом цивнуксусной кислоты:
2N=C— СНг— СООН —нн^ > 2НООС—СН2—COOH-h (NH4)2SO4 цнануксусная кислота
Солн малоновой кислоты называют малонатамн В результате электроноакцепторного влияния карбоксильных групп в молекуле
388
малоновой кислоты атомы водорода метиленовой группы приобретают подвижность (СН-кислотность)
Н6+
I
НООС^-С-»СООН
Н®+
Поэтому в присутствии оснований малоновая кислота способна образовывать не только дианион с участием двух карбоксильных групп, но и частично— трианнои за счет ионизации активной метиленовой группы С участием метиленовой группы малоновая кислота в присутствии оснований вступает в реакцию конденсации с альдегидами и кетонами, не содержащими атома водорода прн углероде в a-положении (конденсация Кневенагеля) В результате реакции образуются ненасыщенные монокарбоновые кислоты, например:
с6н5-
бензалъдегид
Нх ,СООН ЬГ ^СООН малоновая кислота
он k -Н2о
с4н5-сн-^°°н
5 СООН со2
бснзилмдснмалоновая кислота
СбНг-СН- сн- соон
коричная кислота
Важным производным малоновой кислоты является ее диэтп-ловый эфир, получивший название малоновый эфир
Q
C2HsO'
OC2Hfi
В молекуле малонового эфира, как и в молекуле соответствующей кислоты, имеется активная метиленовая группа, за счет которой он вступает в различные реакции замещения Малоновый эфир используется в органическом синтезе для получения моно-и днкарбоновых кислот Так, при действии на малоноаый эфир ал-коголята натрия атом водорода метиленовой группы замещается на натрий и образуется натрнймалоновый эфир, в котором аннон Весьма устойчив вследствие делокализации заряда за счет карбоксильных групп.
389
диэтил малонат
+ CjHjO'Na*
5 OC2H5
>czOC2Hs
Na + CjHjOH
натрнймалоновый эфир
Натриймалоновый эфир легко алкилируется, ацилируется и реагирует с другими электрофильными реагентвми. При алкилировании образуется алкилмалоновый эфир, который в процессе гидролизе превращается в алкилмалоновую кислоту. Алкилмалоновые кислоты легко декарбоксилируются с образованием карбоновых кислот:
СООС2Н6
~СН ^СООСгНй
СООС2Н5
Na+ - СНз1 > СНз—Сн"
-NaI
СООС2Н5
натриймилоно вы в эфир
метили ал о новый эфир
СООН
---СНз—СН ^соон
метил мал снова я кислота
—U СНз—СНг—СООН
пропионовая кислота
По аналогичной схеме можно и второй атом водорода метиленовой группы в малоновом эфире последовательно заменить алкильной группой, например:
СООС2Н5
-сн ^СООСгНв натрий малоновый эфир
СООС2Н5
—-а1-> СНз— СН
-Nal
СООС2Н5
метил малоновый эфир
CjHgO Nn + - CjHsOH
СООС2Н5
\зоос2н5
натрнйметилмалоновый эфир
СНз СООС2Н5
CH3I ) ''S'''c?// НдО н*
—aCjHjOH
ООС2Н5
— Nal у СНз
дичстилмалоновый эфир
С
3»0
СООН
дниетилчалонов-ая кислота
СНз—С Н—СООН к ^гПЗ
изомасляная кислота
При действии на натрнймалоновый эфир эфиров сс-галогено-карбоновых кислот образуются днкарбоновые кислоты, например-
СООС2Н5
-ctf \:оос2н5
натрнймалонопый эфир
Na+ + СНз—СН—COOC2H5
ЭТИЛОВЫЙ эфир <Х хлор пропионовой кислоты
СООС2Н5
--* С2Н5ООС—сн—сн Н1° >
I -зсан5он
СН3 <ООС2Н5
хсоо-н ,
-^ноос-сн-сн НООС-СН-СН^-СООН
1Н ^соон 02 I
СНз
четидянтарнэя кислота
Малоновый эфир широко применяется в синтезе гетероциклических соединений и лекарственных препаратов.
Янтарная кислота НООС—СНг—СНэ—СООН. Белое кристаллическое вещество (т. пл. 185 °C), растворимое в воде и этаноле. Впервые выделена из продуктов перегонки янтаря, откуда и получила свое название. В промышленности янтарную кислоту получают каталитическим гидрированием малеиновой кислоты:
НС
II
НС-^
/ОН
N1
малеиновая кислота
Н2^ \)Н
Н2С _ /ОН с
чо янтарная кислота
391
Соли янтарной кислоты называют сукцинатамп. Янтарная кислота, ее сложные эфиры н имид широко используются В органическом синтезе.
Адипиновая кислота НООС—(СН2>4—СООН. Белое кристаллическое вещество (т. пл. 152 °C), малорастворимое в воде. Соли аднпиноаой кислоты называют адипинатами. Применяется главным образом в производстве синтетического волокна —-найлона, а также в пищевой промышленности и органическом синтезе.
17.2. НЕНАСЫЩЕННЫЕ ДИКАРБОНОВЫЕ КИСЛОТЫ
Ненасыщенные днкарбоновые кислоты содержат в своем составе две карбоксильные группы и кратную углерод-углеродную связь. Простейшими представителями ненасыщенных дикарбоновых кислот являются малеиновая н фумаровая кислоты, которые представляют собой геометрические изомеры бутендновой кислоты НООС—СН=СН—СООН. Малеиновая кислота является цис-, а фумаровая — транс-изомером:
НООС н
\z
II с
HZ ^СООН
малеиновая кислота
цисбутендиовая кислоте
фумаровая кислота транс буте ил иов а я кислота
17.2.1. Способы получения
Малеиновую кислоту можно получить дегидратацией яблочной кислоты прн температуре 250 °C. Первоначально в этих условиях образуется малеиновый ангидрид, который реакцией гидратации превращается в малеиновую кислоту*
НО—СН—СООН t
сн2—соон -2Н2°
яблочная кислота
Н2о
малеиновый ангидрид
сн—СООН сн—соон
малеиновая кислота
392
В промышленности малеиновую кислоту получают гидратацией малеинового ангидрида — продукта каталитического окисления бензола или бутена-2 кислородом воздуха:
VjOs, 400-450 С
-2СО2. -2Н2О
„с''° nv \
н2о
V,O<. 400 С
сн-сн=сн-сн3 \ЗИ;0
НС. / о
СН—СООН II сн—соон
о
малеиновый ангидрид
Фумаровая кислота встречается во многих растениях. Синтетически может быть получена конденсацией глиоксиловой кислоты с малоновой кислотой по Кневенагелю:
ноос-с^° + н
НХс/СООН _он^
Нх СООН -Н2о
соо-н t ноос-сн=сС
СООН со2
глиоксиловая малоновая
кислота
кислота
ноосх н
* н/СС^соон
фумаровая кислота
И
И В промышленности фумаровую кислоту получают термической ризомернзацней малеиновой кислоты
НООС СООН НООС Н
\;=с// \:=с//
Н^ ^СООН
17.2.2. Физические свойства
Малеиновая кислота представляет собой белое кристаллическое вещество (т. пл, 131 °C), легко растворяется в воде и спиртах. Фумаровая кислота — белое кристаллическое вещество, обычно возгоняется без плавления, в запаянном капилляре (т. пл. 287 °C). Фумаровая кислота в отличие от малеиновой труднорастворима в воде.
393
17.2.3. Химические свойства
Ненасыщенные дикарбоновые кислоты проявляют более выраженные кислотные свойства по сравнению с насыщенными, поскольку взаимное влияние двух карбоксильных групп передается по сопряженной системе д-связей сильнее. Прн этом малеиновая кислота значительно сильнее фумаровой. Так, значения рКд в воде для малеиновой кислоты равны рКЯ1 1,92; р/С, 6,23, для фумаровой - рКй1 3,02. рК02 4,32, Повышенная кислотность малеиновой кислоты по первой ступени н низкая кислотность по второй обусловлены внутримолекулярным взаимодействием ионизированной н неноиизнрованиой карбоксильных групп (образование внутримолекулярной водородной связи):
О
Н. /С~~О" С в,
II н .С / н \С ^О
Водородная связь, с одной стороны, повышает устойчивость аикона, а с другой — затрудняет отщепление протока от второй карбоксильной группы.
Реакционная способность ненасыщенных дикарбоновых кислот обусловлена наличием в их структуре двух карбоксильных групп н кратной связи. С участием кратной связи идут реакции присоединения. Так, при гидрировании малениовой и фумаровой кислот образуется янтарная кислота, прн галогенировании — дигалогенянтарная кислота, присоединение галогеноводородов приводит к образованию моногалогеняитарных кнслот, присоединение воды а присутствии минеральных кнслот дает гндроксиянтарную (яблочную) кислоту. При окислении перманганатом калия образуется виноградная (мезовниная) кислота:
НООС—СН2—СНг—СООН янтарная кислота
НООС—СН—СН—СООН
2,3-днброиянтарная кислота
НООС—СН—СНг—СООН
СООН
СН
ioOH
бутендцован кислота
НС1
хлорянтарная кислота
зм
НОН: н+ > НООС—СН—СНг-соон
яблочная кислота
[О* > НООС—сн—сн—соон КМлО< I <
н
виноградная кислота
По карбоксильным группам ненасыщенные дикарбоновые кислоты образуют кислые и средние соли, неполные и полные сложные эфиры, неполные и полные амиды.
Малеиновая кислота при нагревании легко образует циклический ангидрид
малеиновая кислота малеиновый ангидрид
F Фумаровая кислота из-за удаленности карбоксильных групп f пространстве ангидрида не образует.
j Малеиновая кислота, являясь цис-изомером, более лабильна, :ем фумаровая. Под действием брома, иода, галогеноводородов,
азотистой кислоты она легко изомеризуется в более устойчивую форму — фумаровую кислоту. Прн облучении растворов фумаровой н малеиновой кнслот УФ-излучением образуется равновесная смесь, состоящая из 75 % малеиновой и 25 % фумвровой кислот.
17.3. АРОМАТИЧЕСКИЕ ДИКАРБОНОВЫЕ КИСЛОТЫ
Ароматическими дикарбоновыми (аренд и карбоновыми) кислотами называют производные ароматических углеводородов, содержащие две карбоксильные группы, непосредственно связанные с ароматическим ядром.
Наиболее важными представителями являются фталевая, иэофталевая и терефталевая кислоты:
395
соон
соон
соон
терефталевая кислота, I 4 бензолднкарбоновяя кислота
фталевая кислота, изофталевая кислота
I 2 бе изолдикарвоновая I 3 бснзолдикарбоиопая
кислота
кислота
17.3J. Способы получения
Главным способом получения арендикарбоновых кнслот является каталитическое окисление ксилолов кислородом воздуха, например прн окислении «-ксилола образуется терефталевая кислота
Q КСИЛОЛ
СНз
Од, £ квт
ноос
соон
+ н2о
терефталевая кислота
При окислении о-ксилол а сначала образуется фталевый ангидрид, который затем в присутствии воды превращается во фталевую кислоту:
В промышленности фталевую кислоту получают окислением нафталина кислородом воздуха в присутствии катализатора. Образующийся в процессе окисления фталевый ангидрид гидролизуют:
396
17.3.2. Фпэпческие и химические свойства
Фталевые кислоты являются белыми кристаллическими веществами с высокими температурами плавления. Фталевая и терефталевая кислоты малорастворнмы в воде, изофталевая кислота легкорастворима в воде.
Фталевые кислоты проявляют большую кислотность, чем бензойная кислота. Значения рКа в воде такие:
По химическим свойствам арендикарбоновые кислоты в целом не отличаются от ароматических монокарбоновых кнслот. С участием одной или двух карбоксильных групп они образуют соли, сложные эфиры и амиды. Галогенангидрнды образуются только по двум карбоксильным группвм. Моногалогенангидриды фталевых кислот в момент образования внутримолекулярно реагируют с карбоксильной группой, образуя ангидриды или полимерные вещества.
Фталевая кислота отличается от других изомеров способностью легко превращаться при нагревании в ангидрид:
фталевая кислота
t
-н2о
фталевый ангидрид
Фталевый ангидрид используется в больших количествах для
получения эфиров — диметил- и диэтнлфталата, применяемых в качестве пластификаторов; дибутилфталат используют как отпугивающее средство (репеллент) против нвсекомых. При нагревании фталевого ангидрида с аммиаком образуется фталимид, находящий широкое применение в органическом синтезе:
397
р
фталевый ангидрид
фталимид
н20
При конденсации фталевого ангидрида с фенолом образуется фенолфталеин (см. с. ). Реакция идет прн нагревании в присутствии водоотннмающих средств (H2SO4, ZnCh):
фталевый ангидрид
фенолфталеин (бесцветная форма)
Фенолфталеин применяется в аналитической практике в качестве кислотно-основного индикатора (см. с. 145).
17.4. ИДЕНТИФИКАЦИЯ ДИЦАРБОИОВЫХ КИСЛОТ
При идентификации дикарбоновых кислот принимаются во внимание нх физнко-химнческне свойства. В основном это твердые кристаллические вещества с определенными температурами плавления, растворимые в воде. Как и монокарбоновые кислоты, они образуют соли, сложные эфиры, амнды. Вместе с тем некоторые из ннх образуют циклические ангидриды н имнды, способные декарбоксилироваться.
Для определения некоторых дикарбоновых кислот существует и ряд специфических методов. Например, сплав щавелевой кислоты и дифениламина окрашивает каплю спирта в синий цвет за счет образования красителя — анилинового синего.
УФ-спектры дикарбоновых кислот, как и монокарбоновых, малопоказательны. *
Некоторые дикарбоновые кислоты в ИК-спектре имеют две полосы поглощения, соответствующие валентным колебаниям карбонильной группы, например: щавелевая кислота — прн 1710 и 1650 см-1, малоновая — при 1740 н 1710 см1, янтарная— при 1780 и 1700 см-1. Высшие члены гомологического ряда имеют
398
лишь одну полосу поглощения карбонильной группы около 1700 см1.
ЯМР-Спектры дикарбоновых и монокарбоиовых кнслот аналогичны.
КОНТРОЛЬНЫЕ ВОПРОСЫ и УПРАЖНЕНИЯ
1. Напишите структурные формулы следующих карбоновых кислот' а) малоновая кислота; б) бутандновая кислота; в) малеиновая кислота; г) транс-бутендиовая кислота; д) изофталевая кислота. Укажите, по какой номенклатуре даны названия.
2. Приведите схемы реакций получения янтарной кислоты: а) из соответствующего динитрила; б) из соответствующей гидроксикислоты.
3. Напишите схемы взаимодействия щавелевой кислоты со следующими реагентами: а) КОН (1 моль); б) Са(ОН)?; в) SOCh (2 моля); г) нзб. С2Н5ОН, Н + . Назовите продукты реакций.
4. Напишите схемы реакций, протекающих при нагревании: а) щавелевой кислоты; б) малоновой кислоты; в) янтарной Кислоты; г) малеиновой кислоты; д) фталевой кислоты. Назовите конечные продукты.
5. Напишите схему реакции получения 2-метилпропаиовон кислоты, используя в качестве исходного вещества малоновый эфир.
6. Напишите схему реакции конденсации фталевого ангидрида с фенолом. Назовите продукт реакции.
Глава 18
ФУНКЦИОНАЛЬНЫЕ ПРОИЗВОДНЫЕ КАРБОНОВЫХ КИСЛОТ
К важнейшим функциональным производным карбоновых кислот относятся галогена н гидр иды, ангидриды, сложные эфиры, амиды, гидразиды, гидроксамовые кислоты, нитрилы и др.:
ангидриды
галогена нгид р иды
«ложные эфиры
амиды гидразиды гидроксамовые кислоты
R—feN нитрилы
18.1. ГАЛОГЕНАНГИДРИДЫ КАРБОНОВЫХ КИСЛОТ (АЦИЛ ГАЛОГЕНИДЫ)
Галогеиангндрндами карбоновых кислот называют производные карбоновых кнслот, в которых гидроксильная группа, входящая в состав карбоксильной группы, замещена на атом галогена (хлор, бром, реже — фтор н иод).
400
18.1.1. Номенклатура
Названия галогедангидридов образуют из названий соответ* ствующих кислот илн ацильных групп и названия галогена:
СНз—С
С1
ил орангидрид уксусной кислоты, ацетилхлорид
хлорангидрид бензойной кислоты, бензоилхлорид
б ром ангидрид цикл огексачкарбоновой кислоты, диклогексанкирбонилбраннд
О
18.1.2. Способы получеппн
Хлорангндриды и бромангидриды получают взаимодействием карбоновых кислот с галогенидами фосфора (РС1з, PCI5, РВгз, РВгб) или тионилхлоридом (SOC12) (см. с. 368):
I
I
РС13 —--------
PCi5
SOCI2
О
R—С + НзРОз \:i о
R—С + РОС1Э + НС1
^С!
О
R—С + HCI + SO2
^Cl
18.1.3. Фпзнческне свойства
Галогеиангидриды карбоновых кислот представляют собой бесцветные жидкости или кристаллические вещества с резким запахом, легколетучне, раздражают слизистые оболочки и кожу.
18.1.4. Химические свойства
Галогенангидриды являются весьма активными электрофильными реагентами. Их электрофильные свойства обусловлены наличием дробного положительного заряда на атоме углерода
401
карбонильной группы. В результате электроноакцепторных свойств атома галогена (—/-эффект) иа атоме углерода карбо-нильной группы электронная плотность значительно снижается поэтому галогенангидриды являются более сильными электрофильными реагентами, чем карбоновые кислоты:
электрофильный центр
Ацил галоген иды легко вступают в различные реакции нуклеофильного замещения. Так, под действием воды онн гидролизуются до карбоновых кислот; со спиртами, алкоголятами и фенолятами образуют сложные эфиры; с аммиаком, первичными и вторичными аминами дают амиды; с гидразином, алкил- и арилгндразинами образуют гидразиды кислот, с гидроксиламином — гидроксамовые кислоты, с солями карбоновых кнслот образуют ангидриды кислот:
2NH3
—1ЧИ4С1
амид кислоты
о
R—
\lHOH
гидроксамовая кислота
R—С
\1HNH2
2NHaNHg
—NHaNH2 HCI
—на
nh2oh
-ч-----
—НС1
гидразид кислоты
НОН
карбоновая
кислота
—NaCI
ангидрид кислоты
402
В процессе реакций нуклеофильного замещения в молекулу нуклеофильного реагента вводится ацильная группа, следовательно, галогенаигидриды являются ацилирующими реагентами, а реакции называют реакциями ацилирования.
Со слабыми нуклеофильными реагентами, такими, как арены, галоген ангидриды реагируют в присутствии кислот Льюиса (А1С1з, РеВгз, SnCl2 н др.). В этом случае кислоты Льюнса активируют молекулу ацилгалогеннда за счет образования донорно-акцепториого комплекса (н-комплекса) или иона ацилия
R-Cf + A1CL а
Cl:—А1С13
О_ + 'C+[AJC141
IX
донорно-акцепторный комплекс (&+' > 8+)
катион ацилия
Ацилирование ароматических углеводородов по Фрнделю — Крафтсу описано иа с. 102.
Ацилгалогениды, содержащие атомы водорода при а-углеро-де, в присутствии сильных оснований, например третичных аминов, отщепляют молекулу галогеиоводорода, образуя кетеиы:
[ 1 н а
NRa
-на
СН,. л
3 С-С-О
СН;
изобутирилхлорид
диметилкетеи
18,1.5. Отдельные представители
Ацетнлхлорнд СНзСОС!. Бесцветная жидкость с острым запахом, т. кип. 51,8 °C, быстро гидролизуется водой, растворяется в большинстве органических растворителей. Ацетилхлорид используется как ацилирующий реагент в производстве красителей н лекарственных средств.
Бензонлхлорнд CeHsCOCl. Бесцветная жидкость с острым запахом, т. кип. 197,2 °C, раздражает слизистые оболочки дыхательных путей и глаз. Беизоилхлорид хорошо растворим в эфире, бензоле, сероуглероде, гидролизуется водой. Применяется для введения бензоильной группы в синтезе ннднгоидных красителей н лекарственных средств.
18.2. АНГИДРИДЫ КАРБОНОВЫХ КИСЛОТ
Ангидридами называют производные карбоновых кнслот, у которых атом водорода карбоксильной группы замещен на ацильную группу.
403
Ангидриды я вл п юте п продуктами дегидратации карбоновых кислот. Различают линейные и циклические ангидриды карбоновых кислот:
линейный ангидрид
циклический ангидрид
Линейные ангидриды, в молекулу которых входят остатки разных кислот, называются смешанными ангидридами.
18.2.1. Номенклатура
Названия ангидридов образуют из тривиальных названий соответствующих кислот, например:
ангидрид уксусной кислоты уксусный ангидрид
ангидрид бензойной кислоты бензойный ангидрид
ангидрид фталевой кислоты, фталевый ангидрид
ангидрид малеиновой кислоты малеиновый ангидрид
18.2.2. Способы получения
Дегидратация карбоновых кислот, В присутствии таких сильных водоотнимающих средстн, как пентаоксид фосфора, ангпдрид
404
трифторуксусный, и других моиокарбоновые кислоты при нагревании подвергаются дегидратации с образованием ангидридов:
2СНзСООН
i
+ 2НРО3
уксусный ангидрид
Многие дикарбоновые кислоты образуют ангидриды при нагревании, например: *
+ н2о
янтарная кислота
СНг—СООН ; (^Н г—СООН
Взаимодействие галогенангидридов кислот с безводными солями карбоновых кислот. При обработке ацил галогенидов натриевыми или калиевыми солями карбоновых кислот образуются ангидриды. Метод позволяет получить как простые, так и смешанные ангидриды
О О
СНз—С + С—СНз ------->
у — NaCl
Cl NaOZ
yxc^CHirfH ангидрид
ацетила iop ид ацетат натрин
Взаимодействие карбоновых кислот с кетенами. При взаимодействии карбоновых кислот с кетенами образуются ангидриды, например1
сн3-сС° о-н
/4*3-+ СН2=СХО
АУ
СН3-<
С с
/X /X сн3 о сн3
кетен
продукт присоединения
уксусный ангидрид
Этот метод используется в промышленности для получения уксусного ангидрида
405
18.2.3. Физические свойства
Ангидриды карбоновых кислот представляют собой бесцветные жидкости или кристаллические вещества. Низшие представители гомологического ряда обладают раздражающим запахом. Ангидриды карбоновых кнслот малорастворимы в воде н медленно с иен реагируют.
18.2.4. Химические свойства
Ангидриды карбоновых кислот, подобно галогенангидридам, являются весьма активными электрофильными реагентами. Однако по сравнению с галогенангидридами дробный положительный заряд на атомах углерода карбонильных групп в молекуле ангидрида меньше. Поэтому ангидриды карбоновых кислот имеют менее аыражеииый электрофильный характер, чем галогеиангидриды, но проявляют большую электрофильность по сравнению с карбоновыми кислотами, поскольку у них атом кислорода, проявляющий ^М-эффект, приходится иа две ацильные группы-.
электрофильный центр
; электрофильный: центр
уксусный ангидрид
Как и галогенаигидриды, ангидриды карбоновых кислот легко реагируют с различными нуклеофильными реагеитами и используются для введения в их структуру ацильных групп. В процессе реакции выделяется молекула карбоновой кислоты.
Так, при взаимодействии с водой ангидриды медленно гидролизуются с образованием соответствующих кислот. Реакция ускоряется при нагревании:
(СН3СО)2О + Н2О2СН3СООН
При обработке линейных ангидридов спиртами образуются сложные эфиры
О (СН3СО)2О + С2Н5ОН СНз—+ СНзСООН \)С2Н5
этил ацетат
406
Циклические ангидриды реагируют со спиртами с образованием неполных эфиров дпкарбоповых кислот:
О
)о + С2Н5ОН
О янтарный ангидрид
СН2-<
сн2-с
о
OCjH,
он
моноэтиловый эфир янтарной кислоты
При действии на линейные ангидриды аммиака, первичных или вторичных аминов образуются амиды карбоновых кислот
О
(СН3СО)2О 4- H2N—С2Н5 —СНз—4-СН3СООН ^NH—С2Н5
этил а ми и этилам нд уксусной кислоты
Циклические ангидриды реагируют с аммиаком и первичными аминами с образованием неполных амидов дикарбоиовых кислот, которые при нагревании циклизуются в имиды:
фталевый ангидрид
В молекулах ангидридов, как и карбоновых кислот, атомы водорода при а-углеродных атомах обладают подвижностью. С участием связи С—Н в a-положении ангидриды карбоновых кислот в присутствии оснований (соли карбоновых кислот, третичные амины) вступают в реакцию конденсации с ароматическими альдегидами, образуя ненасыщенные аренкарбоновые кислоты (см. реакцию Перкннв, с. 350);
407
о
бензальдегид уксусный ангидрид
CH3COONa+ --1------->-
—CH3COOH
коричная кислота
18.2.5. Отдельные представители
Уксусный ангидрид (СНзСО)2О. Бесцветная жидкость с резким запахом, т кип. 140 °C, раздражает слизистые оболочки глаз и дыхательных путей, вызывает ожоги кожи С водой медленно реагирует, образуя уксусную кислоту, растворяется в этаноле, эфире, бензоле, уксусной кислоте Применяется в качестве ацетилирующего реагента в производстве ацетилцеллюлозы, винилацетата, диметнлацетзмида, лекарственных средств, например ацетилсалициловой кислоты.
О
Фталевый ангидрид I | О. Белое Кристал-
О
лическое вещество, т. пл. 130,8 °C, легко возгоняется, растворим в этаноле. Используется в синтезе лекарственных препаратов — фталазола, фтазина и других, в производстве алкидных смол, пластификаторов, красителей
18.3. СЛОЖНЫЕ ЭФИРЫ КАРБОНОВЫХ КИСЛОТ
Сложными эфирами называют функциональные производные карбоновых кислот, в которых гидроксильная группа замещена на остаток спирта или феиолв —OR.
18.3.1. Номенклатура
Обычно сложные эфиры называют по исходным кислоте и спирту или фенолу По заместительной номенклатуре ИЮПАК их названия образуют из названия углеводородного радикала спирта или фенола и систематического названия карбоновой кислоты, в котором -овал кислота заменяется суффиксом -оат, например
о
н—
СНз О
^сн—сХ
О сен5—с^
\юн3
мети, и .в ын эфир муравьиной кислоты
мет и л формиат мстил мета ноат
OCaHs
эти швин эфир иэоиаслиной кислоты этилнчоб)тират этил 2 метилпропаноат
О
СНз— ^OCeHs
СООС2Н5
(!:оос2Н5
ОС2НБ
этиловып эфир бензойной кислоты
этилбсизоат
ДНЭГН1ОВЫН эфир MTIOtiOBOH кислоты ди эти! мало пат
фснитовын Эфир уксусной КНСЮТЫ фенил ацетат фенилэтаноат
диэтнловын эфир щавелевой кислоты
ди эти-юк салат
СН2—СООСНз
<^Н2—СООН
ми комет иловый эфир янтарной кислоты мономстнлсукцинат
СНг—СООСНз I
СНг—СООСНз
дим ст иловый эфир янтарной кислоты ди метнлсукцинат
18.3.2. Способы получения
Взаимодействие карбоновых кислот со спиртами (см. с. 367):
О
RCOOH 4- R'OH > R—С + Н2О
\>R'
Фенолы из-за низкой нуклеофильности, связанной с сопряжением неподелеиной пары электронов атома кислороДа с л-элскт-ронной системой ароматического ядра, в реакцию этерификации с карбоновыми кислотами не вступают
Взаимодействие спиртов и фенолов с галогенангидридами и ангидридами карбоновых кислот. Галогенаигидриды и ангидриды карбоновых кислот, являясь более активными электрофильными реагентами, чем карбоновые кислоты, легко реагируют со спиртами, фенолами или феноксидами щелочных металлов с образованием сложных эфиров
409
о
СНз—+НС1 \)С2Н5
ацетил и порид CgHjO Na ------------------------------►
этил ацетат
о
сн3—с ^OCeHs
+ NaCI
фен ил ацет ат
CsHsOH
О
СНз—С
CeHsO“Na+ ---------и
О
СНз—С 4-СНэСООН
\эс2н5
СНз—С 4-CHaCOONa
\)CGH5
Реакция с фенокс идам и щелочных металлов идет значительно легче, чем с фенолами, поскольку феноксид-иои за счет отрицательного заряда на атоме кислорода обладает большей нуклеофильностью, чем ненонизироваиный фенол:
О Na"
слабый нуклеофил
сильный нуклеофил
Алкилирование солей карбоновых кислот галогеналканами. При обработке солей карбоновых кислот галогеналканами образуются сложные эфиры
° Z
СНз—С + С’Н»~С1^Л С|1’ с O_Na+ OC2HS
ацетат натрия этилацетат
410
18.3.3. Физпческне свойства
Сложные -эфиры карбоновых кислот являются бесцветными летучими жидкостями, реже — кристаллическими веществами с при-ятным'запахом Они, как правило, малорастворимы в воде, хорошо растворяются в большинстве органических растворителей Температура кипения сложных эфиров обычно ниже температур кипения входящих в их состав карбоновых кислот, что связано с уменьшением межмолекулярного взаимодействия за счет отсутствия межмолекулярных водородных связей
18.3.4. Химические свойства
Подобно галогенангидридам и ангидридам карбоновых кислот, сложные эфиры являются электрофильными реагентами Электрофильным центром служит атом углерода карбонильной группы. Однако из-за -|-Л1-эффекта атома кислорода, связанного с углеводородным радикалом, сложные эфиры проявляют менее выраженный электрофильный характер по сравнению с галогенангидридами и ангидридами карбоновых кислот
электрофильный центр
На электрофильные свойства сложных эфиров оказывает также влияние природа углеводородного остатка у атома кислорода Электрофильность эфиров увеличивается, если углеводородный радикал образует с атомом кислорода сопряженную систему, как, например, в ариловых или виниловых эфирах карбоновых кислот
R C\Oj-CH=^CH2
ариловый эфир
виниловый эфир
Эти сложные эфиры получили название активированные эфиры
Являясь электрофильными реагентами, сложные эфиры вступают в'реакции нуклеофильного замещения В частности, они реагируют с водой (гидролиз), спиртами (алкоголиз), гидразинами (гидразинолиз) и др.
Гидролиз сложных эфиров. Сложные эфиры подвергаются гидролизу в кислой и щелочной средах
411
Кислотный гидролиз представляет собой последовательность обратимых превращений, противоположных реакции этерификации:
R—С + Н2О R—С 4-R'—ОН
R' ^О—Н
сложный эфир карбоновая кислота спирт
Механизм кислотного гидролиза включает протонирование атома кислорода карбонильной группы с образованием карбкатиона, который затем реагирует с молекулой воды:
В присутствии водных растворов щелочей сложные эфиры гидролизуются с обрвзованием соли карбоновой кислоты и спирта илн фенола;
О _
+ NaOH R—С
O"Na+
В отличие от кислотного гидролиза щелочной гидролиз сложных эфиров представляет собой необратимый процесс. Его механизм заключается в нуклеофильной атаке гидроксид-ион ом атома углерода карбонильной группы с образованием промежуточного аниона, который, отщепляя алкоксид-ион, превращается в молекулу карбоновой кислоты На заключительной стадии реакции алкоксид-ион, обладая сильными основными свойствами, отрывает протон от образовавшейся молекулы кислоты и превращается в молекулу спирта:
412
r-C* + <?н \.O-R' J
JO .
R—C—OH —► R-Cf + RO
fr>-R' OH
JO
—R-cC - + R—OH
При щелочном гидролизе щелочь выступает ие в роли катализатора, а в роли реагента. Щелочной гидролиз сложных эфиров часто называют омылением. Это название связано с тем, что при щелочном гидролизе жиров, представляющих собой сложные эфиры глицерина и высокомолекулярных карбоновых кислот, образуются мыла.
взоилеодейегвяе с аммиаком, первичными и вторичными аминами. При действии на сложные эфиры аммиака, первичных и вторичных аминов образуются соответственно незамещенные, N-эамещеиные и N.N-днзамещеиные амиды карбоновых кислот:
CH,-cf + :NH2—СН3 -t- СН,-< + C2HSOH
5 ХО-С,Н, NH—СН3
этилацетат
N-метилацетамид
По механизму реакции такого типа принципиально ничем не отличаются от описанного щелочного гидролиза:
S+LW
СН3~С<
О—СзН5
:NH2-CH3
ч -
r-c<nh2-ch3 ------►
О-С2Н5
^^NHCHj +
С,Н5ОН
Поскольку многие сложные эфиры легкодоступны и сравнительно легко получаются, эта реакции широко используется в синтезе амидов карбоновых кислот.
Взаимодействие с гидразинами и гидроксиламином. При действии на сложные эфиры гидразинов и гидроксиламииа образуются соответственно гидразиды и гидроксамовые кислоты (гидроксиамиды):
413
этнлацетат
NHaNHi> СНз—С + СзНзОН
nhnh2
гидразид уксусной кислоты
О
ЧНоОН $
* > СНз—С + С2НбОН
\чнон
гидрокснамнд уксусной кислоты
Взаимодействие со спиртами (реакция переэтерификации). При взаимодействии со спиртами в молекуле сложного эфира происходит замена одного спиртового остатка на другой. Эта реакция получила название реакции переэтерификации. Переэтерификация катализируется минеральными кислотами или щелочами. Реакция обратима. Для смещения равновесия вправо отгоняют более летучий спирт. Поэтому практическое значение переэтерификация имеет в том случае, когда в состав исходного сложного эфира входит остаток иизкокипящего спирта, например метилового. Тогда образующийся в результате переэтерификации метиловый спирт может быть отогнан из реакционной среды. При этом равновесие смещается в сторону конечных продуктов:
СНз—4- С2Н5ОН СНз—С^ 4- СНзОН
^ОСНз ^ОСаНй
Сложноэфирная конденсация (конденсация Кляйзена). Сложные эфиры карбоновых кислот, содержащие атомы водорода в a-положении, в присутствии сильных оснований, таких, как алкоксиды, вступают в реакцию конденсации с образованием 0-кето-эфиров. Эта реакция называется конденсацией Клпйзена:
/ снД-сн^с7 +
\)С2Н5 ос2н5
амилацетат
ацетоуксусный эфир
+ C2HW3H
414
Механизм реакции включает три стадии. На стадии 1 алкого-лят-ион отщепляет от а-углеродного атома молекулы сложного эфира протон, образуя мезомерно стабилизированный карбанион (I). На стадии II карбанион, выступая в роли нуклеофила, атакует атом углерода карбонильной группы второй молекулы сложного эфира с образованием продукта присоединения (II), который на стадии Ш отщепляет алкоксид-ион, превращаясь в конечный продукт реакции (III):
Стадия I
СНз—С +С2Н5О- Zi СН2-С + С2Н5ОН
^ОСгНй ^ОСзНб
Стадия II I
снэ-с$ + сн2-с ОС2Н5
.О
ос2н5
о
сн3-с-сн2-сС I (
OCjHj
II
>0
ос,н5
Стадия III
О
СН3-С-СН2-СС г-| ос2н5
<*ос,н5
.О
-cf ОС2Н5
III
Сложноэфирная конденсация применяется для промышленного получения ацетоуксусного эфира. Если конденсации подвергается два разных сложных эфира, каждый из которых содержит а-атом водорода, в процессе реакции образуется смесь четырех возможных продуктов.
18.3.5 . Отдельные представители
Этилформнат НСООС2Н5. Бесцветная жидкость (т. кип. 54,3 °C), растворяется в этаноле, эфире, малорастворима в воде. Применяется в производстве витамина Bi, а также как отдушка для мыла и компонент фруктовых эссенций.
Этнлацетат СН3—СООС2Н5. Бесцветная жидкость с приятным запахом (т. кип. 77,1 °C). Малорастворим в воде, растворяется в органических растворителях. Применяется в качестве растворителя эфиров целлюлозы, хлоркаучука, виниловых полимеров, жиров, восков. Используется для получения ацетоуксусного эфира, как отдушка для мыла в парфюмерии, входит в состав пищевых эссенций.
415
Бензплбензоат CeHsCOOCHaCeHg. Жидкость светло-желтого цвета, (т. кип. 323—324 °C), нерастворима в воде, растворима в этаноле. Содержится во многих эфирных маслах, перуанском бальзаме. Оказывает токсическое действие на чесоточных клещей и применяется для лечения чесотки.
18.4, АМИДЫ КАРБОНОВЫХ КИСЛОТ
Амидами называют производные карбоновых кислот, в которых гидроксильная группа карбоксила заменена на аминогруппу.
Их можно также рассматривать как ацильные производные аммиака, первичных и вторичных аминов.
18.4.1. Номенклатура
Названия амидов чаще всего образуют из названий соответствующих карбоновых кислот и аминов. Во многих случаях используют тривиальные названия кислотных остатков — ацилов, заменяя в них суффиксы -ил на -амид. По заместительной номенклатуре ИЮПАК названия амидов составляют из названий кислот, заменяя часть их названий -овал кислота на суффикс -амид. В названиях замещенных амидов положение заместителей, находящихся у атома аэота амидной группы, обозначают символом N‘.
// СНз—с
^МНя
амид уксусной кислоты, вистам ад этанам ад
СбНв—С
\jh2
амид бензойной
ад слот ы. бензамид
/
СНз—С
^NH—СНэ
мстил амид уксусной КИСЛОТЫ.
N-метнДацетаиид, N-мстил этаиамад
димсткламид муравьиной кислоты
N.N-димстилформ амид
СНз—С С2Н6
С2Н5 диэтнламид уксусной кислоты, N, N-днэтнл ацетамид, N.N дизтнлзтанамнд
416
18.4.2. Способы получения
Взаимодействие галогенангидридов, ангидридов или сложных эфиров карбоновых кислот с аммиаком, первичными или вторичными аминами (ем. с. 402, 407, 413):
СНэ—С
ацетил хлорид
(СНзСО)2О
уксусный ангидрид
УНз
— HCI
кн3
— СНзСООИ
Z
СНз—с
\ih2
аист анид
/ СНз—С
\)СН3
NH3
-СНвОН
метил ацетат
Нагревание аммонийных солей карбонов&с кислот:
СНг—С
ONH4
/
СНз—с + н2о
^NHa
ацетат аммония
ацетамид
Гидролиз нитрилов. При гидролизе нитрилов образуются либо амнды (частичный гидролиз), либо карбоновые кислоты (полный гидролиз). Для получения амидов гидролиз осуществляют под действием 96 % серной кислоты или щелочных растворов пероксида водорода:
S+ й-
CHf-C-N + НОН
H2SO4
—— — >
сн3-
ацетамид
ацетонитрил
18.4.3. Физические свойства
Амиды карбоновых кислот представляют собой кристаллические вещества или жидкости, растворимые в аоде и органических растворителях. Соединения, содержащие в молекуле связи N—Н,
14 5-104 417
О
образуют прочные межмолекулярные водородные связи и имеют более высокие по сравнению с карбоновыми кислотами температуры плавления и кипения.
18.4.4. Химические свойства
Реакционная способность амидов обусловлена наличием в их J /
структуре амидной группировки —С—N По электронному
строению амидная группа сходна с карбоксильной. Неподеленная пара электронов атома азота находится в сопряжении с л-электро-иами карбонильной группы (р, л-сопряжение). В результате сопряжения связь С—N становится короче, а связь С = О — несколько длиннее по сравнению с иесопряженными соединениями. За счет сильного смещения неподеленной пары электронов атома азота ( 4-Л1-эффект) к группе С=О частичный положительный заряд на атоме углерода карбонильной группы в амидах меньше, чем у галогенангидридов, ангидридов и сложных эфиров:
R-Cf
СО-ОД
S+ > 8+' > 8+” > б+’”
Вследствие такого электронного строения амиды карбоновых кислот практически не вступают в реакции с нуклеофильными реагентами и в отличие от аминов являются очень слабыми основаниями. Амиды, имеющие в своей структуре связь N—Н, являются слабыми NH-кислотами.
Кислотно-основные реакции. Как слабые основания амиды образуют соли лишь с сильными минеральными кислотами, причем протоиированию подвергается атом кислорода карбонильной группы, поскольку образующийся прн этом катион стабилизируется за счет сопряжения:
R-C^°
nh2
/о—н
-1- 2
н
Протонированная форма амидов является сильной ОН-кислотой, например рКвн ацетамида равен 0,1. В результате сопряжения неподеленной пары электронов атома аэота с л-электронамн кар-416
бон ильной группы в молекулах незамещенных и N-замещенных амидов атом водорода связи N—Н приобретает подвижность. Такие амиды проявляют свойства слабых NH-кислот. При взаимодействии с металлическим натрием или амидом натрия NaNHs они образуют соли:
О О
R—-R NaNH2 R—С -R NH3
^NHa ^NHNa
Гидролиз амидов. Амиды гидролизуются намного труднее, чем другие функциональные производные карбоноаых кислот. В нейтральной среде гидролиз вдет очень медленно. В присутствии минеральных кислот или щелочей амиды гидролизуются довольно легко В щелочной среде амиды превращаются в соль карбоновой кислоты н аммиак или амины:
«О <Н2°> zS-J1’4 Л
R-< + NaOH R-c£o^H —► R-cf + NH,
Г NH2 iNH; ONa
' В процессе гидролиза в кислой среде амиды образуют карбо-иовые кислоты и аммониевые соли. Под действием минеральной (Кислоты сначала образуется протонированная форма амида, которая затем вступает в реакцию с водой:
Н-/О-Н
С>2
+ Н
{ Cnh2
амид протонированная'
форма амида
Дегидратация незамещенных амидов. При нагревании с сильными водоотнимающими средствами, такими, как пентаоксид
14*
419
фосфора (РаО5) или трнхлороксид фосфора (POCI3), незамещенные амиды подвергаются дегидратации с образованием нитрилов:
О CHj—С^ \мН2
СНз—C^N 4- Н2О
ацетамид
ацетонитрил
Расщепление амидов по Гофману, Незамещенные амиды при взаимодействии с гипобромитом натрия NaOBr (или смесью гидроксида натрня и брома, что в сущности одно и то же) подвергаются расщеплению с образованием первичных аминов, содержащих на один атом углерода меньше, чем в исходном амиде, например:
О
СНз—СН2—СНг—С^
\ш2
амил масляной кислоты
NaOBr (Вгя+NaOH)
СНз—СН2—СН2—NH + СО3 4- NaBr н пропил донн
Эта реакция была открыта в 1881 г. немецким химиком А. В. Гофманом н получила название перегруппировка Гофмана.
Восстановление. Под действием алюмогидрнда лития LiAlHi амиды карбоновых кислот восстанавливаются до аминов, например:
О
NHa
СНз—СНг—NHj -Ь Н2О
L1AIH4
N-Замещенные амиды аналогично дают вторичные или третичные амины.
18.4.5. Отдельные представители
Формамид Н—CONH2. Бесцветная жидкость (т. кип 210,5 °C), растворяется в воде и спиртах. Применяется в качестве растворителя и реагента в органическом синтезе.
N,N-Aи м етнл фор мам ид (ДМФА) Н—CON(CI-h)s
Бесцветная жидкость со слабым запахом (т. кип. 153 °C), сме
420
шивается с водой и спиртами. N,N-Диметил формамид широко применяется в качестве апротонного растворителя в производстве синтетических волокон, лакокрасочных материалов, искусственной кожи и др.; как реакционная среда, обладающая каталитическими свойствами при галогенировании и гидрогалогенировании непредельных соединений; в качестве реагента дли введения формильной группы; в фармацевтической промышленности для очистки лекарственных препаратов.
18.5. ГИДРАЗИДЫ КАРБОНОВЫХ КИСЛОТ
Гидразидами называют функциональные производные карболовых кислот, в которых гидроксильная группа карбоксила заменена на остаток молекулы гвд-разииа, алкил- или арнлгидразннов.
Общая формула гидразидов карбоновых кислот о '
R—СГ
^NH—N
^R"
18.5.1. Номенклатура
Название гидразидов чаще образуют из названий соответствующих карбоновых кислот н гидразинов. По заместительной номенклатуре ИЮПАК названия гидразидов составляют путем замены части иазваиин карбоновой кислоты -овая кислота на суффикс -Огндразнд:
О
СН3—С
гидразид уксусной кислоты этаногидрачн*:
о
CHj—СНз—С
\qHNH2
гидрат ид пропионовой кислоты, пропаногцдраэнд
О
CHj—С^ СНз
^NH— К
, ^СНэ
N'.N' диметил гидраз ид уксусной кислоты
О
NH—NH—СбН5
V феннлгвдразид уксусной кислоты
421
о
СбНб—с
nhnh2
гидрнаид бензойной кислоты, бензгидразид
18.5.2. Способы получения
Гидразиды карбоновых кислот получают взаимодействием гидразина, алкил- или арилгидраэинов с галогенангидридами, ангидридами или сложными эфирами карбоновых кислот. В качестве побочных продуктов при этом могут образоваться М,1Ч'-диацил-гидразины:
аи^тнлхлорид
(СН3СО2)О
уксусный ангидрид
NH3NHa -НС1
NH2NH2 — СН3СООН
о
СНэ—С
\mHNH2
СНэ—С
осн
ЧН2ЧНа
-CHjOH
гидр «п ид уксусной кислоты
мет кладет ат
18.5.3. Хнмпческне свойства
По химическим свойствам гидразиды карбоновых кислот во многом напоминают амиды. Вместе с тем в молекуле гидразидов иеподеленная пара электронов р-атом а азота не участвует в сопряжении с карбонильной группой
R С, а р
Gnh-nh2
За счет этого гидразиды проявляют основные и нуклеофильные свойства. Оии образуют соли с минеральными кислотами, вступают в реакцию ацилирования с галогенангидридами и ангидридами карбоновых кислот, реагируют с карбонильными соединениями, 422
образуя N-ацнлгндразииы, взаимодействуют с азотистой кислотой с образованием азидов:
О
СНз—С
о
ч с—сн.
NHN—СН—СНз
этил идеи гидр азид уксусной кислоты
О О
СНэ—С
С—СНз
- НС1
NHNH
-Н2О
V.N' днацетнл гидр азин
% уС—СНз ,нх
О
— СНз—С
NHNH2
этаногидраэнд
о О
СНз—С
С—СНз
NHNH
(СИзСОэО
-СНзСООН
N'.N' ди ацетил гидр азид
HCI
снэ—с
NHNH3C1“
N' ацетнлгндразннийхлорид
О
HNO2 —гнгО
СНз—С
N—N=N
азид уксуснок кислоты
Гидразиды карбоновых
в органическом синтезе, в том числе и синтезе лекарственных препаратов.
кислот находят широкое применение
О
423
18.6. НИТРИЛЫ (ЦИАНИДЫ)
Нптрнламн называют органические соединения, содержащие одну нлн несколько циаиогрупп —feN, связанных с углеводородным радикалом.
От других функциональных производных карбоновых кислот , нитрилы отличаются отсутствием карбонильной группы.
Общая формула нитрилов R—C=N.
18.6.1. Номенклатура
Названия нитрилов образуют из тривиальных названий ацильных остатков карбоновых кнслот или систематических названий карбоновых кислот с тем же числом атомов углерода, включая атом углерода группы —C=N, к которым добавляется суффикс -нитрил:
СНз—teN
ацетонитрил, этаионитрил
С6Н5—C=N бензонитрил
СН2=СН—C==N
акрилонитрил пр опекой нт рнл
С6Н5—СН2—C=N
фен ил ацето нитрил, фенилэтаноннтрил
18.6.2. Способы получения
Дегидратация амидов (см. с. 419):
О
СН3-С^ \чн2
СНз—C=N 4- Н2О
ацетамид
д нет о нитрил
Взаимодействие галогеналкаиов с цианидами щелочных металлов (см. с. 166):
СНз—СНг—Вг + KCN --------► СН3—СНг—C===N +КВг
бромзтан проианонитрил
Так как Цнанид-иои является амбидентным ионом, в качестве побочного продукта реакции образуются язонитрилы, которые удаляют из реакционной среды добавлением разведенной хлороводородной кислоты.
Дегидратация альдоксимов. При взаимодействии альдегидов с гидроксиламином образуются альдоксимы, которые в присут
424
ствии минеральных кислот подвергаются дегидратации, образуя нитрилы:
О N—ОН
-h NH?—ОН ---> R—
Ч — HgO ч
хн н
-HjO
R—C=N
альдегид
альдоксим нитрил
18.6.3. Химические свойства
। Реакционная способность нитрилов обусловлена наличием в их структуре цнаногруппы —C^N. Цианогруппа имеет линейное строение, атомы углерода и азота находятся в sp-гнбриднзацни и образуют между собой полярную тронную связь, электронная .плотность которой смещена к атому азота. Проявляя отрицательней индуктивный эффект, цианогруппа смещает на себя электрон-
<ую плотность с углеводородного радикала и тем самым увеличивает подвижность атомов водорода прн ct-углеродном атоме (СН-кислотность). По месту разрыва тройной связи нитрилы вступают в реакции нуклеофильного присоединения, за счет а-углерод-яых атомов для них характерны реакции конденсации:
реакции конденсации
a* &~
R-CH—C=N
&
нуклеофильное присоединение
Гидролиз нитрилов. При нагревании с водными растворами кислот или щелочей нитрилы подвергаются гидролизу с образованием карбоновых кислот. Гидролиз идет в две стадии. Вначале по месту разрыва кратной связи присоединяется одна молекула воды, в результате чего образуются амиды, которые могут быть выделены в индивидуальном состоянии. Дальнейший гидролиз амидов приводит к карбоновым кислотам:
II- "сЛЛ -НОНЧ кЛН ч=* R-cC° HoH|It R-cr°
\N—Н NH2 NH3 он
нитрил амид карбоновая
кислота
Восстановление. При восстановлении нитрилов алюмогидри-дом лития или водородом в присутствии катализатора образуются первичные амины:
R—C=N R—СНг-NH2
кат
425
Конденсация нитрилов (реакция Торпа). В присутствии оснований, таких, как алкоксиды или амиды металлов (CaHsONa, NaNHa и др ), нитрилы подвергаются конденсации с образованием 0-имиионитрилов. Реакция аналогична альдольной конденсации (см. с. 332), т. е. происходит присоединение а-атома углерода одной молекулы нитрила к атому углерода группы —C^N другой молекулы:
р-иминонитрил
18.6.4. Отдельные представители
Ацетонитрил СНз—C=N Бесцветная жидкость (т. кип. 81,6 °C), хорошо смешивается с водой н многими органическими растворителями. Ацетонитрил применяется в качестве растворителя для выделения жирных кислот из растительных и животных жиров, используется в производстве витамина Вь
Акрилонитрил СНг=СН—feN. Бесцветная жидкость (т кип. 77,3 °C), растворяется в воде и многих органических растворителях. Применяется в производстве синтетического волокна (нитрон), бутадиеи-иитрильного каучука и др. Широко используется в органическом синтезе.
Малонодннптрнл N=C—СН?—C=N. Белое кристаллическое вещество (т. пл. 32 °C), малорастворим в воде, растворяется в органических растворителях. Малонодннитрил вступает в различные реакции конденсации с участием как цианогрупп, так и активной метиленовой группы. Широко используется в органическом синтезе для получения гетероциклических соединений. Применяется в производстве витаминов Bi и Вб, пестицидов, красителей.
18.7. ИДЕНТИФИКАЦИЯ ФУНКЦИОНАЛЬНЫХ ПРОИЗВОДНЫХ КАРБОНОВЫХ КИСЛОТ
Галогенангидриды и ангидриды. Химические методы определения этих соединений основаны на реакциях гидролиза, а также образования амидов и гидразидов — кристаллических веществ с четкими температурами плавления. В галогеиангидридах атом галогена переводят в ионное состояние и определяют известными методами:
RCOHal + C2H5ONa --► RCOOC2H5 + NaHal
Галогенангидриды и ангидриды поглощают в УФ-областн менее 220 нм, поэтому их УФ-спектры малопоказательиы.
426
В ИК-спектрах ангидридов наблюдаются две полосы поглощения, связанные с валентными колебаниями карбонильных групп при 1820 (асимметричные) н 1760см-1 (симметричные). Валентные колебания карбонильной группы хлораигндридов проявляются при более высоких частотах в области 1815—1790см-1, что связано с сильным — /-эффектом атома клора и увеличением частичного положительного заряда на карбонильном атоме углерода.
Галогенангидриды и ангидриды ие содержат протонов, непосредственно связаннык с карбонильной группой, и поэтому функциональные группы этих соединений ие могут быть определены с помощью ПМР-спектров, однако эти группы вызывают сдвиг сигнала протонов, находящихся в a-положении по отношению к карбонильной группе.
Сложные эфиры. Для идентификации сложных эфиров используют реакции гидролиза либо образование амидов или гидразидов— кристаллических аеществ с четкими температурами плавления.
УФ-спектры не дают большой информации, так как слабое поглощение, связанное с п->-л*-переходом, наблюдается около 210 им.
В ИК-спектрах проявляются валентные колебания карбонильной группы в области 1750—1735 см-1 и две интенсивные полосы поглощения при 1275—1185 см-1 и 1160—1050 см-', соответствующие vc _о
Амиды. Идентификацию амидов чаще осуществляют по продуктам гидролиза — карбоновым кцслстгам и аммиаку или аминам.
Амиды поглощают в УФ-области спектра при 210—220 нм* (п-*-л*-переход).
В ИК-спектре амидов проявляется интеисивиап полоса поглощения в области 1700—1680 см-1, соответствующая vc-о- Кроме того, для первичных и вторичных амидов наблюдается полоса валентных колебаний N—Н в области 3500—3000 см-1.
Амндиая группа первичных и вторичных амидов содержит прогой, сигнал которого расположен при 5—6,5 и 6—9,4 м. д. соответственно.
Нитрилы. Характерной реакцией нитрилов является их гидролиз в присутствии щелочи до карбоновых кислот с выделением аммиака. Используется также реакция нитрилов с гидроксиламииом. Образующиеся амидоксимы дают устойчивые соли с кислотами:
NH NH2
R—C.sN "*N~°> R—R— ^NHOH XN—ОН
аиндоксим
* Не наблюдается в спектрах третичных амидов в связи с наложением полос других поглощений.
427
ч
В УФ-диапазоне алифатические нитрилы поглощают в вакуумной области.
В ИК-спектре проявляется полоса средней интенсивности в области 2275—2215 см-1, связанная с vc_N.
КОНТРОЛЬНЫЕ ВОПРОСЫ И УПРАЖНЕНИЯ
1. Напишите структурные формулы следующих соединений: а) пропионилхлорид; б) беизоил бромид; в) ангидрид бензойной и и ело ты; г) этил формиат; д) фенилбензоат; е) N-этил ацетамид; ж) гидразид бензойной кислоты; з) пропаиоиитрил.
2. Напишите схемы получения этилпропаиоата, используя три разных ацилирующих реагента.
3. Приведите схемы получения ацетамида с применением четырех различных ацилирующих реагентов.
4. Напишите схемы получения фенил гидразида уксусной кислоты, используя три разных ацилирующих реагента.
5. На примере этилпропионата приведите механизмы кислотного и щелочного гидролиза сложных эфиров карбоновых кислот.
6. На примере ацетвмида напишите механизмы кислотного и щелочного гидролиза амидов карбоновых кислот.
7. Исходя из электронного строения карбоновых кислот, галогеном гид ридов, ангидридов, сложных эфиров н амидов, охарактеризуйте в сравнительном аспекте их активность во взаимодействии с нуклеофильными реагентами.
8. Напишите схему реакции получения этиламина из: а) соответствующего амида карбоновой кислоты; б) соответствующего нитрила.
Глава 19
ГЕТЕРОФУНКЦИОНАЛЬНЫЕ КАРБОНОВЫЕ КИСЛОТЫ
г
I
К гетерофуикцнональиым карбоновым кислотам относятся производные карбоновых кислот, в углеводородном радикале которых Одни или несколько атомов водорода замещены иа другие атомы или группы атомов — галоген, гндрокснгруппу, аминогруппу, карбонильную группу и др.
Наиболее важными гетерофункцнональиыми карбоновыми кислотами являются галогенокарбоновые кислоты (га логе иокислоты), гидроксикарбоновые кислоты (гидроксикислоты), оксокарбоновые кислоты (альдегидо- и кетонокислоты) и аминокарбоновые кислоты (аминокислоты).
19.1. галогенокарбоновые кислоты
Галогеиокарбоновымн кислотами называют производные карбоновых кислот, в углеводородном радикале которых одни или несколько атомов водорода замещены атомами галогенов.
1 По природе углеводородного радикала различают алифатические, алициклические и ароматические галогенокарбоновые кислоты. Алифатические галогеиокислоты по взаимному расположению атома галогена и карбоксильной группы подразделяют иа а-, р-, у- и т. д.: а р а
R—СН—СООН R—СН—С Нг—СООН
!Hal Hal
а-галогенокарбонован 0 галогенакарбо новая
кислота кислота
СНг—СНг—СООН
Hal
у галогенокарбоновая кислота
19.1.1. Номенклатура
Номенклатура галогеиокарбоновых кислот аналогична номенклатуре карбоновых кислот. По заместительной номенклатуре ИЮПАК название обычно образуют из тривиальных названий
429
карбоновых кислот. Положение атома галогена обозначают греческими буквами а, 0, у и т. д. По систематической номенклатуре ИЮПАК название составляют из систематических названий карбоновых кислот, указывая положение атома галогена цифрами:
СНг—СООН
хлоруксусная кислота, хлорэтановая кислота
СНз—С Н—СООН
а бромпропионовая кислота 2-броипропанояая кислота
СООН
СНг— СН?— СН2—СООН
<L
у хлормвсляная кислота. 4 хяорбутановея кислота
С На—С Н—СООН
С1
о хлорбензойная кислота, 2 хлорбензойная кислота
CF3—СООН
а р днбромпрапиоиовая кислота. 2,3 дибромпропановая кислота
Трифтор уксусная кислота, трифторэтаиовая кислота
19.1.2. Способы получения }
Галогенирование карбоновых кислот (реакция Гелля — Фольгарда — Зелинского), При действии на карбоновые кислоты хлора или брома в присутствии красного фосфора или РС1э (РВгз), что одно и то же, образуются а-хлор и а-бромкарбоиовые кислоты. Реакция протекает через стадию образования галогеиангидрида кислоты, который вследствие —/-эффекта атома галогена галогенируется значительно легче, чем сама кислота:
R—СНг—СООН
Р+С12(РС13) ------------>
-Н-)РОэ
С12 ----->
-НС)
HjO
-HCI
Присоединение галогеноводородов к а, ^-ненасыщенным кислотам. При взаимодействии а, 0-ненасыщенных карбоновых кислот с галогеноводородами образуются 0-галогенокарбоиовые
430
кислоты. Присоединение галогеноводородов протекает с ориентацией против правила Марковиикова (см. с. 374);
\ LO &+ 8-сн/снксС + НС1 он
акриловая кислота
-* СН2-СН2-СООН а p-хлорпропионовая кислота
Галогенирование аренкарбоновых кислот. Бензойная кислота галогенируется в условиях электрофильного замещения с образованием главным образом ж-галогенобензойных кислот:
СООН
4- CI2
м члорбензонпля кисло [а
19.1.3. Физические и химические свойства
По физическим свойствам галогенокарбоновые кислоты представляют собой бесцветные жидкости или кристаллические вещества, растворимые в воде.
В химическом отношении для иих характерны реакции с участием карбоксильной группы и реакции с участием атома галогена.
Реакции по карбоксильной группе. Введение атома галогена в углеводородный радикал карбоновой кислоты приводит к повышению кислотных свойств. Это обусловлено электроноакцепторным влиянием атома галогена, который, обладая отрицательным индуктивным эффектом, смещает электронную плотность с атома углерода карбоксильной группы н тем самым повышает устойчивость аниона. Например, коистаита кислотности рКа уксусной кислоты в водном растворе составляет 4,76, а для хлоруксусной — 2,86. По мере удаления атома галогена от карбоксильной группы индуктивный эффект затухает и поэтому кислотность уменьшается, т. е сила кислот снижается в ряду и т. д., например:
« р
СН3—СНг—СН—СООН СНз—СН—СНаСООН
СНг—СНг—СНзСООН
(L
рХа=4.60
431
При переходе от моно- к ди- и полнгалогенокарбоиовым кислотам кислотность увеличивается. Наиболее сильной карбоновой кислотой является трифторуксусная — CFgCOOH (рХи 0,23).
Галогенокнслоты образуют все описанные в гл. 18 функциональные производные — соли, галогенангидриды, сложные эфиры, амиды и др. Например: СНг—COONa NaOH ।
хлпращггат натрия О
СН2—СООН--------------------сн2—с^
I -SOj -HCI I х
61 Al ci
хлоруксуснвя кислота хлорангнтрид хлоруксусной
кислоты
СгН5ОН. н+ СН2 СООС2Н5 I
-нао Q |
ЭТШОВЫЙ Эфир ХЛОРУКСУСНОЙ кислоты I
Реакции с участием атома галогена. Алифатические галогеио-карбоновые кислоты с участием атома галогена вступают в различные реакции нуклеофильного замещения, свойственные галогенопроизводным углеводородов. При этом а-галогеиокарбоиовые кислоты значительно более реакционноспособны, чем галогеиалка-иы. Это связано с электроноакцепторным влиянием карбоксильной группы, которая, проявляя — /-эффект, увеличивает подвижность атома галогена. Замещение протекает, как правило, по механизму Sn2. Так, при взаимодействии Галогеиокарбоновых кислот с аммиаком, первичными или вторичными аминами образуются аминокислоты, при действии цианида калия или натрия -цианокар-, боновые кислоты:
2NH3
-НВг
СНз—СН—COO“NH4+
nh2
аммонийная соль а ачннопропиоиовой кислоты
СНз— сн— СООН I Вг
а бромпропионовая кислота
KCN ------->-— КВг
) 1 И
СНз—сн—соон
а цианопропионовая кислота
432
Отношение алифатических галогеиокислот к водным растворам щелочей зависит от взаимного расположения атома галогена и карбоксильной группы.
ct-Галогеиокарбоновые кислоты при действии водных растворов щелочей образуют а-гидроксикислоты:
СНз—сн—соон нон tNa0H) — NaCl
а хлорпропионован кислота
СНз—сн—соон
молочная кислота
0-Галогенокарбоиовые кислоты в присутствии водных щелочей образуют 0-гндроксикислоты, которые при повышенных температурах отщепляют воду и превращаются в а, 0-иенасыщениые кислоты:
нон
СН2-СН2-СООН СН2 СН СООН н^СН2-СН-СООН
а он н 2
В-хлорпропионовая р-гидрокси пропионовая акриловая
кислота кислота кислота
у- и 6-Г ал о генок исл оты в этих условиях сначала образуют у-или б-гидроксикислоты, которые при нагревании отщепляют воду, образуя у- илн соответственно 6-лактоиы — внутримолекулярные (циклические) сложные эфиры:
О сн2-сн2-сн2-сС i 2 2 2 хон
С1
нон (Na ОН)
-Nad
сн2-сн2-сн2-с^
I I
о-н он
о
—► сн2-сн3-сн2-с^ 1___---------о—_J
у-бутиролактон
В ряду ароматических галогеиокарбоновых кислот нуклеофильное замещение атома галогена протекает в более жестких условиях. Более реакционноспособными являются атомы галогенов, расположенные в орто- или пара-положении по отношению к карбоксильной группе (активированные атомы галогенов рассмотрены иа с. 179). Так, о-хлорбеизойиая кислота при нагревании с анилином в присутствии СиО и КаСОэ образует N-феиилантраниловую кислоту:
433
СООН
N фенил антраниловая кислота
19.1.4. Отдельные представители
Хлоруксусные кислоты: монохлоруксусная кислота С1—СН2—СООН, дихлоруксусная CI2CH—СООН и трихлоруксусная CI3C—СООН.
Монохлоруксусиая кислота — кристаллическое вещество, т. пл. 62 °C. Используется в производстве карбоксиметиленцеллюлозы, гербицидов, комплексонов.
Дихлоруксусная кислота—иизкоплавкое вещество (т. пл. 13 °C, т. кип. 194 °C), используется в органическом синтезе.
Трихлоруксусная кислота — гигроскопическое вещество (т. пл. 58 °C). Применяется в органическом синтезе; ее натриевая соль используется в качестве гербицида.
а-Бромнзовалериановая кпслота (2-бром-З-метилбутановая кислота) СНз—СН—СН—СООН. Кристаллическое вещество
СНз
(т. пл. 44 °C).
У рейд а-бромизовалериановой кислоты под названием бром и-зов ал используется в медицинской практике в качестве снотворного средства:
О
СНз—СН—СН—С
СНз Вг
NH—С-- NH2
уреид а-бромнзойалЕрмаковой кислоты, бромизсвал
19.2. ГИДРОКСИ КИСЛОТЫ
Гидрокси кн слотам и называют производные карбоновых кислот, содержащие в углеводородном радикале одну или несколько гидроксильных групп.
434
В зависимости от природы углеводородного радикала различают алифатические гидроксикислоты (спиртокислоты) и ароматические (фенолокислоты). Алифатические гидроксикислоты по взаимному расположению карбоксильной н гидроксильной групп подразделяют на а-, 0-, у- и т. д.
Число карбоксильных групп в молекуле гидроксикислоты определяет основность, а число гидроксильных групп, включая и гидроксилы, входящие в состав карбоксильных групп, характеризуют атомность. Так, гликолевая кислота НО—СНг—СООН является одноосновной двухатомной кислотой, а яблочная кислота НООС—СНг—СН—СООН — двухосновной трехатомиой Ан кислотой.
19.2.1. Номенклатура, изомерия
В номенклатуре гидроксикислот широко представлены тривиальные иазваиия.
По заместительной номенклатуре ИЮПАК за родоначальное название принимается тривиальное нли систематическое название карбоновой кислоты. Гидроксильная группа обозначается префиксом гидрокси- (реже окси-). При использовании тривиального ивзвания родоначальиой структуры положение гидроксильной группы в углеводородной цепи обозначается буквами греческого алфавита а-, 0-, у- и т. д., а при употреблении систематического иазваиия родоначальной структуры положение ОН-группы указывается цифровыми локантами:
СНг—СООН
Ан
гликолевая кислота, гидрокси уксусная кислота, гидрокси этановая кислота
СНг—СНг—СНг—СООН
Ан
Т гидро кс им асл иная кислота, 4-гидроксибутановая кислота
НООС—СН—СН—СООН
винная кислота, а.а'-дигидрокснннтарная кислота. 2,3 днгидрокеибутандиовая кислота
СНз—СН—СООН Ан
молочная кислота, а гндроксипропноновая кислота. 2-гидрок.снпропановая кислота
НООС—С Н—С Нг—СООН I он
яблочная кислота, гидрокси янтарная кислота, гидроксибут а ид новая кислота
ОН
НООС—СНг—с—СНг—СООН
Аоон
ди ионная кислота, 2-гндроксн 1,2,3-пролантрикарбеновая кислота
435
салициловая кислота 2 гидрокснбензонная кислота
галловая кислота
3,4 5 трнгндроксибензойная кислота
ОН
сн=сн—соон
о гилрслксикорнчная кислота
2 гидрокснфенилпроленован кислота
Дли гидроксикислот характерна структурная изомерия» обусловленная разной структурой углеводородного радикала, с которым связана карбоксильная группа, и разным положением ОН-группы в углеводородной цепи. Например, структурными изомерами являются
СНз—СН2—СН—СООН
2 гидроксибутановая кислота
СНз
СНз—С Н—СООН I он
2 гидрокси 2 метил пропановая кислота
1 СНз—СН—СНа—СООН
Ан
3 гидроксибут ановая кислота
Кроме того, в ряду гидроксикислот часто встречается оптическая изомерия (см кн 1, с 79, 80) Например, молочная кислота
*
СНз—СН—СООН содержит в своей структуре асимметрический
ОН
атом углерода и существует в (энантиомеров)-
СООН
н-----он
СНз
D (—) молочная кислота
виде двух оптических изомеров
СООН
НО-----ОН
СНз
L ( + ) молочная кислота
4М
19.2.2. Алнфатнческпе гидрокси кп слоты
А. Способы получении
Гидролиз а-галогенкарбоновых кислот. Под действием водных растворов щелочей tz-галогенкарбоновые кислоты гидролизуются с образованием а-гидроксикислот
СНз—СН—СООН H-H-Na-°“ СНз—СН—СООН + NaBr
Вг Ан
а брочпропионовая кислота
молочная кислота
Окисление гликолей и гидроксиальдегидов (альдолей). Гликоли, содержащие хотя бы одну первичную гидроксильную группу, и альдоли при окислении различными окислителями в определенных условиях могут быть превращены в гидроксикислоты.
[о] С Н 2—СООН
Ан
этиленгликоль
СНз—С Н—СНг—С
3 гидроксибутаналь
гликолевая киилота
СНз—СН—СНг—СООН
I
ОН
3 гидроксибутановая кислота
Гидролиз гидроксинитрилов (циангидринов). Метод попользуют для получения а-гидроксикислот При действии на альдегиды или кетоны цианистого водорода образуются а-гидроксинитрилы, которые в кислой среде гидролизуются с образованием ct-гидро-ксикислот:
ацетальдегид
ОН
СН3—СН—C=N
2-гидрокси-пропанонитрил
+ он
н» сн3-сн-соон
-NH3 J
молочная кислота
Гидратация а, ^-ненасыщенных карбоновых кислот, а, р-Ненасыщенные карбоновые кислоты в присутствии минеральных кислот присоединяют воду. Реакция протекает против правила Марковни-кова Метод позволяет получать р-гидроксикислоты:
437
сн2=От^с^
* A он
&+ 8-
+ и-ОН
акриловая Гислота
* СН-сн2-СООН
ОН
р-гидроксипропиононая кислота
Взаимодействие сложных эфиров а-галогенкарбоновых кислот с карбонильными соединениями (реакция Реформатского). При взаимодействии сложных эфиров ct-галогенкарбоновых кислот с карбонильными соединениями в присутствии цинка образуются fj-гидроксикислоты. Реакция протекает через стадию образования цинкорганического соединения:
СН2 СООС2Н5 -h Zn
СН2—СООС2Н5
1пВг
этиловый эфир брому юсу с ной кислоты
(cSr ?пВг
8+-LO* Т
СН3-С<н + CH^COOCjHj -* он
2НОН; н» сн3—сн—сн2—соон
цннкорганическое соединение
OZnBr
СНз-С1^СН-СООС2Н5 -►
+ Zn(OH)Br + С2Н5ОН
р-гидрокси масляная кислота
Метод разработай отечественным хпмиком-органиком С. Н. Реформатским в 1887 году.
Б. Физические свойства
По физическим свойствам гидроксикислоты представляют собой бесцветные жидкости или кристаллические вещества, растворимые в воде. Многие из представителей гидроксикислот обладают оптической активностью.
В. Химические свойства
Реакционная способность гидроксикнслот обусловлена наличием в их структуре двух функциональных групп — карбоксильной и гидроксильной.
По карбоксильной группе гидроксикислоты дают все реакции, свойственные карбоновым кислотам. В частности, они образуют соли, сложные эфиры, галогенангидриды, амиды. В некоторые из этих реакций одновременно может вступать и ОН-группа. Так, при действии на гидррксикислоты галогенирующих реагентов (РСЬ,
438
SOCk и др.) образуются галогепапгидриды галогенкарбоновых кислот:
CHj—СНг—С 4- 2РС16 -----» СНг—CHj—С +
Ан \)Н А1
0 гидроксил ропн снова я хлор ангидрид
кислота 0-хлор пропио новой кислоты
4- 2РОС13 + 2НС1
При взаимодействии со спиртами в присутствии концентрированной H2SO4 гндроксикислоты сначала превращаются в сложные эфиры, а затем могут образоваться и простые эфиры по гидроксильной группе:
СН2—CHj—СООН
Ан
СНзОН |р
—HjO
0 гидрокси пропионовая кислота
СН2—CHj—СООСН3
Ан
метиловым эфир 0 гидроксипропионовой кислоты
сизоин* ; CHj—СНг—СООСНз
-Н..0 |
ОСНз
метиловый эфир 3 метоксипрогановой кислоты
По сравнению с соответствующими незамещенными кислотами гндроксикислоты проявляют более выраженные кислотные свойства. Это обусловлено электроноакцепторным влиянием ОН-группы, которая, проявляя отрицательный индуктивный эффект, дополнительно стабилизирует образующийся в процессе ионизации кар-боксилат-ион. Так, если коистаита ионизации пропионовой кислоты СН3СН2СООН в водном растворе составляет 4,87 единицы рКа, то для молочной кислоты СН3—СН—СООН она равна 3,06 единицы фКа. I
ОН
По мере удаления ОН-группы от карбоксильной сила гидрокси-кислот уменьшается (а>р>-у и т. д.).
С участием гидр^чсильной группы алифатические гидроксикнс-лоты вступают в реакции, характерные для спиртов. Так, они образуют простые и сложные эфиры. Для получения сложных эфиров в качестве ацилирующих реагентов используют галогенангидриды или ангидриды кислот. При взаимодействии с галогеноводородами (НС1, НВг) происходит нуклеофильное замещение ОН-группы
439
на атом галогена. При окислении гидрокснкислоты превращаются в альдегиде- или кетонокислоты:
о
J
СНз “С
-------СНз—С Н—СООН + HCI
ОСОСНэ
. 2 ицетокс«пропановая кислота
СНз—СН—СООН-----------------——* СНз—СН—СООН + НаО
(кн ci
2 хлорпролановая кислота
---—► СНз—С—СООН + Н2О
А
пировиноградная кислота
Вместе с тем для алифатических гидроксикислот характерен ряд специфических свойств, обусловленных взаимным влиянием карбоксильной и гидроксильной групп.
Отношение к нагреванию. а-Гидроксикислоты при нагревания* претерпевают межмолекулярную дегидратацию и образуют циклические сложные эфиры — лактиды:
О. ЮН
I аСН
RX ^О-Н
Н-О^ ,R аСН
НО О
лактид
о.-гидроксикислота
Гидрокси кислоты при нагревании подвергаются внутримолекулярной дегидратации с образованием а, р-иенасыщенных кислот:
Р а R—СН—СН—СООН I I ОН н
р-гидроксикислота
R—СН=СН—СООН + Н2О
а, р-ненасыщенная кислота
у* и 6-Гид роке и кислоты уже при комнатной температуре или незначительном нагревании претерпевают внутримолекулярную дегидратацию с образованием циклических сложных эфиров — лактонов:
«9
440
.0
R-CH-CH2-CH,-C^
i-н 0H
Y - гидр ок сн кислота
Rx
CH-CH, / \
° CH2 c
+ H2O
у лактон
Расщепление а-гидроксикислот\ В присутствии концентрированной серной кислоты а-гидрокснкислоты при нагревании подвергаются расщеплению по связи С1—С2, образуя муравьиную кислоту и карбонильное соединение (альдегид или кетон). В свою очередь, муравьиная кислота при взаимодействии с концентрированной H2SO4 распадается на оксид углерода (II) и воду:
n2so4
СО
Эта реакция может быть использована для идентификации а-гидрокснкислот.
Г. Отдельные представители
Молочная кислота (а-гидрокснпропионовая кислота) ОН
СНз——СООН. Впервые выделена нз кислого молока. Молочная кислота образуется в результате молочнокислого брожения углеводов.
СбН1гОб ^нокиелое брожение 2СН3—СН—СООН I
ОН
Содержится в кислом молоке, кефире, моченых яблоках, квашеной капусте, различных солениях и др.
Молочная кислота имеет в своей структуре один асимметрический атом углерода и поэтому существует в виде двух оптически активных энантиомеров и оптически неактивной рацемической формы.
С .ООН СООН
н— -он но- -н D +£ =г
с D ( :нз -)молочная кислота с L-N ;н3 >-) мелочная кислота (±)-молочная кислота
441
L- (-}-)-молочная кислота образуется в живых организмах в процессе расщепления углеводов. При интенсивной физической работе она накапливается в мышцах, вызывая характерную боль. Соли и сложные эфиры молочной кислоты называют лактатами, Лактаты кальция н железа (II) применяются в медицине в качестве лекарственных препаратов. Лактат кальция [СНзСН (ОН)СОО] гСа- 5НгО используют пря заболеваниях, связанных с недостатком кальция в организме. Лактат железа (II) [СНзСН (ОН)COOJjFe применяется при хронических анемиях.
у-Г и д р о к с и м а с л я и а я кислота (4-гидрокснбутановая кислота) НО—СНг—СНг—СНг—СООН. В свободном состоянии неустойчива, легко образует лактон. Натриевая соль у-гидроксн-масляной кислоты (натрия окснбутират) применяется в медицине в качестве лекарственного средства для неннгаляцнонного наркоза.
Яблочная кислота (гидрокснянтарная кислота) *
НООССН(ОН)СНзСООН. Бесцветное кристаллическое вещество, кислое на вкус, хорошо растворимое в воде. Яблочная кислота соа держит один асимметрический втом углерода, поэтому она существует в виде двух оптически активных энантиомеров и оптически неактивной рацемической формы:
СООН
н----ОН
СНзСООН
D ( + ) яблочная кислота
СООН
СНаСООН
-яблочная кислота
-яблочная кислота
н
В природе встречается L-(—)-яблочная кислота. Она содержится в незрелых яблоках, ягодах рябины, клюквы, малины, барбариса и др.
Яблочная кислота образуется в живых организмах в процессе обмена углеводов, принимает участие в цикле трикарбоновых кислот (цикл Кребса).
В промышленности широко применяется L-(—)-яблочная кислота в производстве вина, фруктовых вод и кондитерских изделий, а также в синтезе лекарственных средств.
Винная кислота (а, а'-дигндроксиянтарная кислота) НО—СН—СООН
НО—СН—СООН. Молекула винной кислоты содержит два аснм-метрических атома углерода с одинаковым набором заместителей. Поэтому для иее известны трв стереоизомера (см. кн. I, с. 75) — два оптически активных (£)-( -|-)-винная и Д-(—)-винная кислоты) и одни оптически неактианый (лсезо-виииая кислота):
442
н-un С :оон —он н с но :оон н н С :оон он
ц —он юон ная кислота Г1 н он
( D ( + ) ВИН! :оон ая кислота ( (— )-вин с 9 :оон ннная кислота
Рацемическая форма D- и А-винных кнслот называется виноградной кислотой.
В природе встречается только О-(4-)-винная кислота. Особенно много ее содержится в винограде, который является исходным сырьем для ее получения. Винная кислота образует кислые и средние солн. Кислые соли называются гндротартратами, средние — тартратами. Калнево-натрневая соль винной кислоты называется сегкетовой солью:
К+СГОС—СН—СН—COONa+ 4Н2О
il I
ОН он
ссгнстова соль
Прн взаимодействии с гидроксидом меди (II) в щелочной среде сегнетова соль образует комплекс ярко-сниего цвета, получивший название реактив Фелиига, применяемый для качественного обнаружения альдегидной группы.
При нагревании в присутствии гидросульфата калия винная кяслота отщепляет диоксид углерода и воду, превращаясь в пировиноградную кислоту:
н81 /Соон
40—СН—СООН KHSO4, t С
I --------► II —►
Ю-СН-СООН -H2o с
н ^соон
винная кислота гидроксималеиновая кислота
О=С-ООН i . снгс-соон
---► I " -СО, II сн2-соо-н v 3 о
щавелевоуксусная кислота
пировиноградная кислота
Лимонная кислота (2-гидрокси-1,2,3-пропантрикарбоно-
СН СООН I
40—С—СООН
I
СНаСООН
вая кислота). Бесцветное кристаллическое вещество, т. пл 153 °C. В больших количествах содержится в цитрусовых (лимоны, апельсины), а также в смородине, малине и др.
443
Как н яблочная, лимонная кислота — один из важнейших продуктов обмена веществ, участвующих в цикле трнкарбоновых кислот. Под действием серной кислоты лимонная кислота, являясь а-гидроксикислотой, расщепляется с образованием ацетондикарбоновой и муравьиной кислот. Дальнейшее разложение приводит к образованию ацетона, воды, оксида и диоксида углерода:
СН2СООН но-с-соон I СН3СООН
h2so4, t ---------►
сн2-соо-н
с=о
I
сн2—соо—н
о s II СНз-С-СН3
2СО2
нсоон
СО Н2о
Соли лимонной кислоты называются цитратами. Трниатриевая соль лимонной кислоты (цитрат натрия) применяется в медицине для консервирования донорской крови: '
ОН
Na^O ОС—CHv— CH GHo—COO“Nar - 5,5 Н2О
(toO“Na+
цитрат натрия
1
19.2.3. Феиолокислоты 1
Фенол окн слотам и называют производные арен-карбоновых кислот, у которых одни или несколько атомов водорода в ароматическом кольце замещены на гидроксильные группы.
Наиболее важными представителями фенолокислот являются:
салициловая кислота 2 гидроке«бензойная кислота
3 гидрокенбензониая кислота
4 гидроксибснзойиая кислота
галловая кислота, 3,4 l тригидроксибенаойная кислота
444
А. Способы получения
Многие феиолокислоты содержатся в свободном состоянии или в виде сложных эфиров в растениях, нз которых могут быть выделены в индивидуальном виде, Например, галловую кислоту получают гидролизом дубильных веществ.
Синтетические методы получения фенолокнслот основаны на введении в структуру фенолов карбоксильной группы (карбоксилирование фенолов) нлн введении в молекулу аренкарбоновой кислоты фенольного гидроксила (гидроксилирование ароматических кислот).
Карбоксилирование фенолов оксидом углерода (IV) (реакция Кольбе — Шмитта). При действии оксида углерода (IV) на феноксид натрия при температуре 120—140 °C и давлении 5 атм. образуется салицилат натрия, который затем переводят в салициловую кислоту:
ONa+ ОН ОН
феноксид натрия салицилат натрия салициловая кислота
Реакция протекает по механизму SE. Поскольку оксид углерода IV проявляет слабые электрофильные свойства, в реакции используют не сам фенол, а феноксид натрия, в котором за счет -|-Л4-эффекта со стороны отрицательно заряженного атома кислорода бензольное кольцо в реакциях SE более активное, чем у фенола:
a-комплекс салицилат натрия салициловая
кислота
445
При более высоких температурах, и особенно если использовать фенокснд калия в качестве исходного продукта реакция, образуется 4-гидроксибенэойная кислота.
Гидроксилирование аренкарбоновых кислот. Прн нагревании основных медных солен ароматических карбоновых кислот происходит гидроксилирование ароматического ядра по орто-положению:
Сплавление сульфобензойных кислот со щелочами. При сплавлении сульфобензойных кнслот со щелочами сульфогруппа замещается на гидроксильную группу:
СООН
+ 2КОН
сплавление
-ф- K2SO3 -ф- НаО
м сульфовензойная кислота
калиевая соль 3 гидрокси бензойной кислоты
Б. Химические свойства
Реакционная способность гидроксикислот обусловлена наличием в их структуре карбоксильной группы, фенольного гидроксила и ароматического ядра,
С участием карбоксильной группы они образуют различные функциональные производные — солн, галогенангидриды, сложные эфиры и др. Так, аналогично аренкарбоновым кислотам, при действии на гндроксикислоты щелочей, гндрокарбонатов нлн карбонатов щелочных металлов образуются соли. При этом с гидрокарбонатами нли карбонатами щелочных металлов в реакцию вступает только карбоксильная группа. Фенольный гидроксил, обладающий более слабыми кислотными свойствами, чем угольная кислота, не способен вытеснять ее из солей:
СООН
салициловая кислота
+ NaHCO3
-ф- COsf -ф- Н2О
446
Прн действии на фенолокнслоты щелочей образуются соли как по карбоксильной группе, так и по фенольному гидроксилу (алко-гол яты):
СООН
ОН
+ 2NaOH
COO”Na+
салициловая кислота
динатриеван соль
СЗЛНЦИЛОДОЙ КИСЛОТЫ
При взаимодействкн фенолокнслот со спиртами образуются сложные эфиры, с галогенирующими реагентами (PCU, РОС13, SOCh) — галогенангидриды:
хлорангидрид салициловой кислоты
За счет фенольного гидроксила феиолокислоты способны образовывать простые и сложные эфиры. Например, прн ацетилировании салициловой кислоты уксусным ангидридом образуется ацетилсалициловая кислота (аспирин):
СООН
он
+ (СН3СО)2О
+ СНзСООН
ацетилсатнцилпвай кислота
Подобно фенолам, феиолокислоты образуют с хлоридом железа (Ill) фиолетовое окрашивание.
По ароматическому ядру феиолокислоты вступают в реакции электрофильного замещения, свойственные аренам. Например,
447
при нитровании салициловой кислоты нитрующей смесью в мягких условиях образуется 2-гидроксн-5-ннтробензойная кислота:
СООН
ОН
2 гндронт 3 нитробензойная кислота
+ Н2О
При нагревании феиолокислоты довольно легко подвергаются декарбоксилированию, напрямер:
СООН
ОН
141нци1оная кислота
В. Отдельные представители
СООН Салициловая кислота (о-гидроксн-
✓v > бензойная кнслота). Белое, кристаллическое
(l вещество, т. пл. 159 QC, легко возгоняется,
U при сильном быстром нагревании декарбокси-
лируется, растворима в горячей воде.
Салициловая кислота обладает большей кислотностью (р/С2,98). чем бензойная кислота (рКа 4,17), а также мета- илн пара-гндроксибензойные кислоты. Повышенная кислотность салициловой кислоты обусловлена дополнительной стабилизацией аннона за счет образования внутримолекулярной водородной связи:
О
448
Салициловая кислота применяется в медицине в виде спиртовых растворов и мазей как антисептическое лекарственное средство. Она также служит сырьем для синтеза других лекарственных средств, таких, как натрия салицилат, метилсалицнлат, фенилса-лицилвт (салол), салициламид, оксафенамнд, ацетилсалициловая кислота (аспирин).
Химические превращения, связанные с синтезом данных лекарственных препаратов, представлены в схеме
РОС1з, СбНвОМа
—NaCl, — КаРОз
15 5 104
449
Ацетилсалициловая кислота, натрия салицилат, метиле алии, и-лат и салициламид применяются в медицине в качестве анальгетических, противовоспалительных и жаропонижающих средств. Фе-ннлсалицилат используется как антисептическое сродство при заболеваниях кишечника. Оксафенамид оказывает желчегонное действие.
Галловая кислота (3,4,5-тригидроксибензойная кислота) Белое кристаллическое вещество (т пл НО СООН 220 °C), хорошо растворяется в воде, при
\аК/ нагревании подвергается декарбоксилирова-
II \ нию с образованием пирогаллола, легко
/х/ окисляется на аоздухе, приобретая при этом
"° I темную окраску Впервые эта кислота была
ОН выделена нз французского вина, а поскольку
Фракция в древности называлась Галлией, отсюда и произошло название «галловая кислота». Галловая кислота входит в состав дубильных веществ, содержащихся в чернильных орешках (наростах на листьях некоторых видов дуба, образующихся в результате укола насекомых — орехо-' творок), дубовой коре, листьях чая и в ряде дру| их растений Главной составной частью дубильных веществ являются таинииы, представляющие собой гликозиды галловой кислоты
Водные растворы таннина обладают Способностью свертывать и осаждать белки. На этом свойстве основано дубящее действие таннина, а также его применение в медицине в качестве кровоостанавливающею средства и при лечении ожогов.
Сама галловая кислота применяется в синтезе красителей, а также используется для получения пирогаллола и в Качестве аналитического реагента.
о-Гидроксикоричиая кислота —СН=СН—СООН
Как н другие ненасыщенные карбоновые кислоты с двойной связью, о-гндрокснкоричная кислота может существовать в виде двух геометрических изомеров (цис- и транс-). цас-Изомер называется кумариновой кислотой, а гралс-нзомер - орто-кумаровой:
орто-кум яровая кислота
Кумариновая кислота в свободном состоянии не известна, она существует лишь в виде производных, например солей При попытке выделения в свободном виде кумариновая кислота отщепляет молекулу воды и превращается в лактон, называемый кумарином:
Р
450
кумарин
натриевая соль кумариновой кислоты
Кумарин представляет собой кристаллическое вещество, имеющее приятный запах. Его различные производные широко распространены в растительном мире. Некоторые производные кумарина применяются в медицине для лечения тромбофлебитов тралг-Изомер — о-кумаровая кислота - относительно устойчив в свободном состоянии
19-3. оксокислоты
К оксокнслотам относят альдегидо- и кетонокислоты. Эти соединения содержат в своем составе карбоксильную группу и альдегидную или кетонную. В зависимости от расположения функциональных групп различают а-, и у-оксокарбоновые кислоты, В каждом из этих рядов могут существовать как альдегидо-, так и кетонокислоты.
19.3.1 . Номенклатура
Как и в ряду гндроксикислот, для многих оксикислот широко используются тривиальные названии По заместительной номенклатуре ИЮПАК карбонильная j руппа обозначается в названии префиксом оксо-
Н—С—СООН
СНз—(j:—соон о
4 3 2 J
СНз—С—сн2—соон
о
глиоксалевая кислота оксоэтановая кислота
пировиноградная кнетота
2 оксопропановая кислота
ацетоуксусная кислота, 3 оксобстановая кислота
HOOQ—^—СНг—СООН О
НООС—(р—СНг—СНг—СООН О
щавелев о уксус на я кислота оксоб¥танднован кислота
а кетоглутаровая кислота, 2 океoneига идиоаая кислота
15*
45 f
19.3.2 . Способы получения
Окисление гидроксикислот. Прн окислении в мягких условиях гидр оке и кислот, содержащих в структуре первичную гидроксильную группу, образуются альдегндокислоты, а прн окислении гидроксикислот со вторичной гидроксигруппой получают кетонокис-лоты:
I
СНг—СООН [ - нао
ОН
Н—(J—СООН о
глнкспенан кнслота глисксаленая кислота
СНз—СН—СООН СНз—с—соон
чо-ючкая кислота пировиноградная кнелита
Гидролиз геминальных дигалогенокарбоновых кислот. Геминальные ди галогенокарбоновые кислоты, в зависимости от положения атомов галогенов (в конце углеродной цепи или в середине), при гидролизе образуют альдегиде- или кетонокнелоты:
л
CHCI2—СООН + Н2О ---------► Н—С—СООН + 2HCI
дихлорукс^сная кислота глиоксалевая кислота /|
Cl CI о
СНз—С—СООН + Н2о -----------> СНз—С—СООН + 2НС1
к ’
2.2 дихлорпропановая кислота пировино|радиан кислота 1
19.3.3 . Химические свойства
Альдегиде- и кетонокнелоты являются более сильными кислотами по сравнению с соответствующими карбоновыми кислотами, что связано с электроноакцепторным влиянием карбонильной группы (—/-эффект).
Реакционная способность оксокнслот обусловлена наличием в их структуре карбоксильной и альдегидной или кетонной групп.
По карбоксильной группе они образуют функциональные производные— соли, сложные эфиры, амиды и др.; по карбонильной группе вступают в реакции нуклеофильного присоединения, свойственные альдегидам и кетонам, в частности образуют гидразоны, оксимы, циангидрины и т. д. Альдегндокислоты легко окисляются, образуя дикарбоновые кислоты. Оксокислоты обладают и рядом специфических свойств. В частности, а- и р-оксокнслоты сравнительно легко подвергаются декарбоксилированию.
452
а-Оксокислоты в присутствии концентрированной серной кислоты отщепляют СОг и превращаются при этом в альдегид:
СНз—С—СООН СНз—С^ + со2
пировиноградная кислота
ацетальдегид
Очень легко подвергаются декарбоксилированию р-оксокисло-ты. Так, ацетоуксусная кислота уже при комнатной температуре или незначительном нагревании отщепляет СОг, образуя ацетон. Предполагают, что процессу декарбоксилирования способствует образование внутримолекулярной водородной связи:
СН3 СН2 О
снгс-сн2-соон
II 2 о ацетоуксусная кислота
сн3-с^сн2
СНз-С-СН3 II о
ацетон
В отлнчие от кислот сложные эфиры р-оксокислот являются довольно устойчивыми соединениями. Важное значение в органическом синтезе имеет этиловый эфир ацетоуксусной кислоты, называемый обычно ацетоуксусным эфиром:
О О
С Нэ—(!—С Н 2—С—ОС2Н5
ацетоуксусный эфир
По физическим свойствам ацетоуксусный эфир представляет собой бесцветную жидкость с приятным фруктовым запахом, т. кип. 181 °C. Ацетоуксусный эфир получают сложноэфирной конденсацией этилацетата (см. о конденсации Клянзена на с. 414).
О О
2СН3—СгН5° Na" > СН3—+
\)С2Н5 ^СНг—СООС2Н5
этил а нет нт ацетоуксусный эфир
С2Н5ОН
453
Ацетоуксусный эфир является таутомерным веществом Ему присуща кето-енольная таутомерия, обусловленная наличием в структуре молекулы СН-кислотного центра (подвижные атомы водорода метиленовой группы) и основного центра — атома кислорода кетонной группы Подвижность атомов водорода метиленовой группы связана с электроноакцепторным влиянием карбонильной и сложноэфнрной групп за счет — /-эффекта
основный центр
СН-кислотный центр
В результате перемещения атома водорода от метиленовой группа к атому кислорода кетонной группы образуется енольная форма*
сн3-с=сн-с 1 он
хОСгН3
кетонная форма
енольная форма
Енольная форма энергетически менее выгодна, чем кетонная В спиртовом растворе ацетоуксусный эфир существует в равновесной смеси, состоящей на 92,5 % из кетонной и на 7,5 % — из енольной формы Такое сравнительно большое содержание енольной формы связано с образованием сопряженной системы и внутримолекулярной водородной связи, дополнительно стабилизирующих эту форму
Н,с^ £н TdCjHj
6+
В химических превращениях ацетоуксусный эфир в зависимости от природы реагента ведет себя как кетон или енол
С участием кетонной формы ацетоуксусный эфир вступает в реакцию восстановления водородом в момент выделения, образует! циангидрин при взаимодействии с HCN, гидросульфнтное производное с NaHSOa н дает другие реакции кетонов*
454
[hi
он
СНз—СН—СН2—СООС2Н5
этиловый эфир ft гидроксимастипом кислоты
СНз—С—СНг—СООС2Н5
HCN
CN
СНз—С—СН2—СООС2Н5
он
он
СНз—i—CHz-COOC2H5
^O3Na
Наличие енольной формы и обусловливает способность ацетоуксусного эфира обесцвечивать бромную воду Присоединение брома по месту разрыва двойной связи идет своеобразно
ОН
СНз—(t=CH—СООС2Н5 + Вг2 ----►
О
О
Механизм реакции
qOH g_ qOH 5_
СН—С ^СН—СООС,Н5 + Вг2 ^СНгС^СН СООСН,—►
Вг-В?7
л-комплекс
qo-н
+ СНз-С-СН-СООСзЩ
+ Вг
О
СНГ с- сн—СООСЕН 5
Вг
карбкатион
В енольной форме ацетоуксусный эфир дает фиолетовое окрашивание с хлоридом железа (III)
С участием енольной формы ацетоуксусный эфир вступает в реакцию ацилирования Так, прн взаимодействии с хлористым ацетилом в среде пиридина образуется О-ацетилыюе производное*
455
ОИ О
CHj—С=СН—СООС2Н5 + СНз—'"'"''•“V
— HCI
ХС1
ацетоуксусный эфир ацстнлхлорид
ОСОСНз
-----> СНз—i=CH—СООС2Н5
этиловый эфир 3 ацртпксикротоновой кислоты
По енольной форме протекает реакция ацетоуксусного эфира с металлическим натрием или гидроксидом натрия. При этом обра-; зуется натрнйацетоуксуснын эфир:
ОН
2СНз—С=СН—СООС2Н5 J- 2Na -------------►
ацетоуксусный эфир
O“Na +
-----► 2СНз—СН—СООС2Н5 + Н2 патрийацетоуксусный эфир
Натрийацетоуксусный эфир широко применяется в органическом синтезе. Он содержит в своем составе амбидентный аннон (аннон с двойственной реакционной способностью). С точки зрения теории резонанса строение аниона можно изобразить в виде двух граничных структур:
qo
снэ-с СООС2Н5
о
«---► CHj-с- сн соос^н5
Натрийацетоуксусный эфир легко реагирует с галогеналкана-мн. Прн этом алкилирование протекает не по атому кислорода, как можно было ожидать, а по атому углерода (С-алкилирование).
г~ о- ~
L снз-
-Nal
натрийацетоуксусный эфир
О
СНз—*
с2н5
этилацетоуксусный эфир
456
Характерным свойством ацетоуксусного эфира является способность к кето иному и кислотному расщевлеиию.
Кетон иое расщепление происходит при нагревании в присутствии разбавленных растворов кислот илн щелочей. В этих условиях ацетоуксусный эфир гидролизуется по сложиоэфирной группе с образованием этилового спирта н ацетоуксусной кислоты, которая легко декарбоксилируется, образуя ацетон (кетон);
|| Н- или он*
С—СНг—СООС2Н5 —СНз—С— СНг—СООН
-CaHsOH —СО,
ацетоуксусный эфир ацетоуксусная кислота
---СНз—С—СНа
ацетон
Под действием концентрированных растворов щелочей гидролитическому расщеплению подвергается сложноэфирная группа и углерод-углеродная связь С2—С\ В результате реакции образуется молекула этилового спирта н две молекулы соли уксусной кислоты. Эта реакция называется кислотным расщеплением:
СООС2Н5 Na0H (KOH,V 2CH3COONa-|-C2H5OH
Механизм реакции:
ч* он
СН-С - СНг COOCjH, <=
+ сн3-сС° о-н
qo'
► снГ с - <5н2- соосен.
он
СН2СООС2Н5
-сн3соо“
-----> СНз—СООС2Н5 пон он» СНзСОО" + СгНвОН
Ацетоуксусный эфир используется в органическом синтезе для получения различных кетонов н карбоновых кислот. Рассмотрим, например, как из ацетоуксусного эфира можно получить бутанон или пропионовую кислоту С этой целью под действием этокснда натрия ацетоуксусный эфир сначала превращают в натрийацетоуксусный эфир, который затем алкилируют и подвергают кетонно-му или кислотному расщеплению;
СНз—С—СНг—СООС2Н5 —
А
457
СНз—с==сн—СООС3Н5
О~
Na + -^1
-Nal
СН3—С—СН—СООС2Н5
о
натрий ацетоуксусный эфир
СНз
СНз—С—СН—СООС2Н5
четнлацето7КС¥<.ный эфир
н- или ОН-(Н^ СНз—с—СН2—СНз + С2Н5ОН + СО2
кето иное ц
расщепление ||
о
б)таион NaOH
L |К-Н^ > CH3COONa + СН3—СН2—COONa + С2Н5ОН кислотное растепление натрия натрия
ацетат пропионат
Ацетоуксусный эфир широко применяется в синтезе лекарст венных веществ, красителей и производных пиридина.
19,3.4 . Отдельные представители
Пировиноградная кислота (2-оксопропаиовая кис-О
лота) СНз——СООН, Бесцветная жидкость (т. кип. 165 °C); растворима в воде, спирте, эфире. Впервые получена пиролизом виноградной кислоты, откуда в произошло название. В настоящее время получают перегонкой винной кислоты в присутствии гидро-сульфатв калия (см. с. 443).
Пировиноградная кислота — промежуточный продукт обмена углеводов и белков в живых организмах, одни нз основных метаболитов в цикле трикарбоновых кнслот. Образуется также при кислотном и спиртовом брожении углеводов. Пировиноградная кислота применяется в производстве лекарственных веществ, например атофана (цинхофена).
Щав е л е в оу к с у с и а я кислота (2-оксобутандиовая кислота) НООС—С—СНг—СООН. Эта кислота является одновре-
А
меино а- н 0-кетокислотой. В живых организмах щавелевоуксус-ная кислота участвует в цикле трикарбоновых кислот, где образуется прн окислении яблочной кислоты, а в дальнейшем превращается в лимонную кислоту.
458
а-Кет о гл у тарой ая кислота (2-оксопентандиовая кислота) НООС—С—СНг—СНг—СООН. Белое кристаллическое
вещество (т. пл. 115—116°C), растворяется в воде и этаноле. Имеет важное биохимическое значение, принимает участие в цикле трикарбоновых кислот.
19.4. АМИНОКИСЛОТЫ
Аминокислотами называют производные карбоновых кислот, в углеводородном радикале которых один или несколько атомов водорода заменены иа аминогруппу.
В зависимости от природы углеводородного радикала, с которым связана карбоксильная группа, аминокислоты подразделяют на алифатические и ароматические. Алифатические аминокислоты по взаимному расположению аминогруппы и карбоксильной группы делятся на а-, 0-, у-амннокислоты и т. д. Наиболее распространенными в природе являются сс-аминокнслоты, входящие в состав белков.
19.4.1. Номенклатура. Изомерия
По заместительной номенклатуре ИЮПАК названия аминокислот образуют из тривиальных или систематических казааннй соответствующих карбоновых кислот и префикса амино-. При использовании тривиальных названий карбоновых кислот положение аминогруппы обозначают буквами греческого алфавита се, 0, у и т. д.; для образования систематического названия кислот — цифровыми локантами 2, 3, 4 н т. д. Для аминокислот, входящих в состав белков, чаще всего применяют тривиальные названия. Ароматические аминокислоты бензольного ряда рассматривают как производные бензойной кислоты:
СНг—СООН
Анг
глииин пмнноуксуснан кислота
СНз—сн—соон
nh2
а а-танми, а нминппролионовая кислота,
2 аминояренанпвяя кислота
СНз—СН—СНг—СООН
i!jh2
Р амииомаслянан кислота
3-амннобутя новая кислота
СООН
СН2—СНг—СНг-СООН
i!ih2
СООН
актраннлопйя кислота, л ачннобьи юйняя Кислота, 2 аминобен юйная кислота
Т-амняоиасляявя кислота. 4-аымнобута новая кислота
п аминобензоПная кислота, Ч-аяннобензойная кислота
459
Йзомерня аминокислот аналогична изомерии гидроксикислот. Она может быть обусловлена разной структурой углеводородного радикала, с которым связана карбоксильная группа и разным положением аминогруппы в углеродной цепи (структурная изомерия); для аминокислот, содержащих асимметрический ат)м углероде, изомерия связана с разным расположением заместителей в пространстве (оптическая изомерия).
19.4.2. Способы получения
Среди множества способов получения аминокислот наиболее важными являются:
Действие аммиака на галогенокарбоновые кислоты. Прн взаимодействии галогеиокарбоновых кнслот с аммиаком атом галогена замещается на аминогруппу. Ввиду доступности а-галогенокарбо-новых кнслот этот метод в основном применяется для получения сс-аминокислот:
СНз—СН—СООН + 2NH3 ---------► СНз—СН—COOHH-NH4CI »
I I *
Cl nh2
а-хлорпролпоновей кислота ot-вминопропионовая кислота
Действие аммиака и циановодородной кислоты на альдегиды (синтез Штреккера). Этот способ применяется для синтеза «-аминокислот. При взаимодействии альдегидов с аммиаком вначале образуется альдимин, который в присутствии циановодородной кислоты превращается в а-аминонитрил. Образовавшийся нитрил легко гидролизуется до кислоты:
ацетальдегид
СНЭ—С^ ^н3
\и Н2О
СНз—СН—C=N
nh2
2-а м инопропанонитрнл
ЗНэО, н + — NHj
-----СНз—СИ—СООН
I NH2 а-аминопропнонован кислота
Присоединение аммиака к а, ^-ненасыщенным кислотам. При действии аммиака на а,0-ненасыщенные кислоты образуются 0-аминокислоты. Присоединение аммиака протекает против правила Марков никова:
460
CHj^H -СООН +• акриловая кислота
:NH3
-* СН2-СН2СООН nh2 p-аминопропионовая кислота
Восстановление нитробензойных кислот. При восстановлении нитробензойных кнслот по реакции Зимина образуются соответствующие аминобензонные кислоты:
п-нитробенлойнйн кислота я-аминобекэойная кислота
19.4.3. Физические и химические свойства
. Аминокислоты представляют собой белые кристаллические вещества с высокими температурами плавления, хорошо растворимые в воде. Вследствие наличия в структуре кислотного центра (группа—СООН) и основного центра (группа—NH2) аминокислоты кристаллизуются из нейтральных водных растворов в виде внутренних солей, получивших название биполярные ноны, или цвиттер-ионы:
R—СН—СОО~
+
ивнттер нои
В химическом отношении аминокислоты проявляют свойства первичных аминов и карбоновых кислот. По карбоксильной группе они образуют функциональные производные карбоновых кислот — солн, сложные эфиры, амнды, галогенангидриды.
С участием аминогруппы аминокислоты образуют соли с минеральными кислотами, вступают в реакции алкилирования, ацилирования, реагируют с азотистой кислотой, а также дают другие реакции, свойственные первичным аминам. Аминокислоты обладают амфотерными свойствами, поскольку образуют солн с минеральными кислотами и с осноааниямн.
Некоторые химические превращения аминокислот представлены на схеме. *
461
Реакции по аминогруппе
R—СН—СООН
+ NH3C1~
хлоройодородная сочь
R—СН—СООН
A HR
% азкилпроизводное
R—СН—СООН
NHCOR'
N ацилпроизволнос
R—СН—СООН
он
гидрокси кислот а
Реакции по карбоксильной группе
—R—CH—COONa
— HjO |
NH2
нйтриспзя соть
---R—СН—COOR7
-н2о |
______ nh2
сложный эфир
—R—CH—CONHR'* *
-НгО |
NH2
АМИД
---^21^ R—CH—COCI *
-POCI3 -HCl I
NH2
, X юраигидрид
* Реакции проводят после предварительной защиты аминогруппы
462
Наряду с этим аминокислоты обладают некоторыми специфическими свойствами, обусловленными взаимным влиянием карбоксильной н аминогрупп
Отношение аминокислот к нагреванию. При нагревании а-, 0-, у- и 6-аминокислот образуются разные продукты. «-Аминокислоты при нагревании претерпевают межмолекулярную дегидратацию, образуя при этом циклический днамнд — дикетопнперазнн:
а-аминопропионовая кислота 3,6-диметил-2,5-диксто-
пиперазин
р-Аминокнслоты при нагревании отщепляют молекулу аммиака, образуя а-, (J-ненвсышенные кислоты
₽
СНз—СН—СНг—СООН —СН3—СН=СН—СООН + NH3
NH2 р dM и ««масляная кистота ир«тоновая кислота
у- и 6-Аминокислоты прн нагревании претерпевают внутримолекулярную дегидратацию, образуя циклические амиды —лактамы:
си,-------сн,
Y/ \
N-Н НО'
+ Н2О
у-аминомасляная кислота
у-лактам
Взаимодействие а-аминокислот с нингидрином, Прн действии нингидрина на ot-аминокислоты образуется краситель сине-фиолетового цвета. Реакция протекает в две стадии. На первой стадии под действием нингидрина происходит окислительное дезамини
463
рование аминокислоты и ее декарбоксилирование. На второй стадии реакции образовавшийся аммиак реагирует с эквимолекулярными количествами обычного и восстановленного нингидрина, образуя краситель сине-фиолетового цвета:
CHj-CH COOH +
I
nh2
а-аминопропионовая кислота
-NH3, -СО*
|,-С С
3 И
восстановленный нингидрин
нингидрин
NH3 +
восстановленный
нингидрин
-ЗН2О*
Нингидриновая реакция используется для качественного определения а-амннокислот.
19.4.4. Отдельные представители
у-А м и и ом асл я и а я кислота (4-амннобутановая кислота, ГАМК) NHa—СН2—СН2—СН2—СООН. Белое кристаллическое вещество, т. пл. 202 °C. Хорошо растворяется в воде. ГАМК
464
образуется в живых организмах прн декарбоксилировании глутаминовой кислоты. Является нейромедиатором, принимающим участие в обменных процессах головного мозга.
ot-Аминомаслянао кислота под названием аминалон или гам-малон применяется в медицине для лечения нервно-психических заболеваний, при ослаблении памяти, нарушениях мозгового кровообращения и др. ГАМК используется в синтезе других лекарственных препаратов, например: пирацетама, фецибута и пр.
г-Амиио капроновая кислота (6-амнногексаноаая кислота) NHa—СНз—СНг—СНг—СНг—СНг—СООН. Белое кристаллическое вещество, т. пл. 372 °C, легко растворяется в воде. Применяется в медицине в качестве кровоостанавливающего средства.
Антраниловая кислота (о-амннобензойная кислота).
СООН Белое кристаллическое вещество, т. пл. 145 °C, а практически не растворяется в воде. Применяется
в производстве красителей и лекарственных средств.
NHs
л-А м и и о б е нз о й н а я кислота (ПАБК). Белое кристал-Олнческое вещество (т. пл. 186 °C), мало-СООН растворимо в воде. Входит а состав фолиевой кислоты, выполняющей роль ростового фактора для некоторых микроорганизмов.
Сложные эфиры н-амннобензойной кислоты широко применяются в медицине в качестве местноацестезнрующих средств, например, анестезии (этиловый эфнр rt-амннобензойной кислоты) и новокаин (0-диэтнл аминоэтил оного эфира п-аминобензойной кислоты гидрохлорид):
СООС2Н5
анестезин
H2N—COOCH2CH2N(C2H5)2 • НС1
новокаин
19.5. ИДЕНТИФИКАЦИЯ ГЕТЕРОФУНКЦИОНАЛЬНЫХ КАРБОНОВЫХ кислот
Химические методы. Химические методы идентификации замещенных карбоновых кислот сводятся к доказательству наличия соответствующих функциональных групп. Так, присутствие карбо-
16 5-104
465
ксильиой группы может быть доказано по образованию солей, сложных эфиров, амидов.
Атом галогена в галогеиокарбоновых кислотах определяют реакцией с AgNOa после сплавления вещества с натрием.
Для определения спиртовой группы в гидроксикислотах используется реакция этерификации илн дегидратации прн нагреваний. а-Гидрокснкислоты при нагревании образуют труднорастворимые в воде лактиды. Р-Гндрокснкислоты при кипячении с 10 % раствором гидроксида натрия образуют непредельные кислоты, в которых наличие двойной связи определяют известными методами. у-Гидроксикислоты легко образуют лактоны, которые восстанавливают алюмогидридом лития LiAIH4 до двухатомных спиртов, легко поддающихся идентификации,
Подобно фенолам, фенолокислоты дают с хлоридом железа (III) фиолетовое окрашивание,
Карбонильную группу в оксокислотах определяют по се способности образовывать нерастворимые кристаллические осадки с гидрокенламином, гидразинами, семикарбазидом и тносемикар-базндом.
С целью идентификации а-амннокнелот используют нингидриновую реакцию.
Физические методы. Введение галогена в «-положение карбоновой кислоты смещает полосу валентных колебаний карбонильной группы в ИК-спектрс в сторону более высоких частот иа 10— 20 см-1.
Из-за образования внутримолекулярной водородной связи поглощение карбонильной группы в а-гидрокснкислотах смещено в сторону низких частот.
Поскольку а-амннокислоты существуют в виде внутренних солей, для них в ИК-спектрах характерна широкая сильная полоса v“sHa+B области 3100—2600 см-1, а также слабая полоса v^|lf при 1550—1485 см-1. Карбоксилат-анион имеет сильную полосу поглощения в области 1590см-1 и слабую — при 1400см-1. Эти полосы связаны с асимметричными и симметричными валентными колебаниями связи С—О,
КОНТРОЛЬНЫЕ ВОПРОСЫ И УПРАЖНЕНИЯ
1. Напишите структурные формулы следующих соединений: а) 0-броммасляная кислота; б) лимонная кислота; в) салициловая кислота; г) яблочная кислота; д) щавелевоуксусная кислота; е) пировиноградная кислота; ж) вцетоуксусиая кислота; з) антраниловая кислота.
2. Расположите нижеприведенные соединения в порядке возрастания их кислотности: а) хлоруксусиая кислота; б) трихлоруксусная кислота; в) p-хлорпропионовая кислота; г) дихлоруксусная кислота.
466
3, Изобразите проекционные формулы Фишера энвнтиомеров молочной кислоты и винной кислоты. Обозначьте их конфигурации no D, /.-системе.
4, Приведите уравнение реакции получения салициловой кислоты. Приведите схемы качественных реакций, позволяющих различить салициловую кислоту, метилсалицнлат н ацетилсалициловую кислоту.
5. Какие продукты образуются при нагревании a-, (J- и у-гид-рокснкнслот и соответствующих им аминокислот? Приведите схемы реакций,
6, Приведите схему динамического равновесия между таутомерными формами ацетоуксусного эфира.
7. Напишите схему кислотного и кетоиного расщепления ацетоуксусного эфира.
8. Объясните понятия «атомность» и «основность» на примере лимонной кислоты. Напишите схему реакции разложения лимонной кислоты при нагревании.
16»
Глава 20
ПРОИЗВОДНЫЕ УГОЛЬНОЙ кислоты
Угольная кислота Н2СО3 является неустойчивым соединением, которое легко разлагается на оксид углерода (IV) я воду, формально угольную кислоту можно рассматривать как двухосновную карбоновую кислоту
но-с
он
о
Угольная кислота как двухосновная образует два ряда функциональных производных — неполные и полные галогенангид-рнды, сложные эфиры, амиды, а также смешанные функциональные производные — эфиры хлоругольной кислоты, эфиры карбаминовой кислоты (уретаны) н др.
—ОН
Cl—С1
о
моноэтнловый эфир угольной кислоты, моноэтил карбонат
h2n—nh2
о
монохл прангндрид угольной кислоты хлоругольная кислота
С2Н5О—С—ОС2Нб
дихлорангидрид угольной кислоты фосген
О
моноамнд угольной кислоты, карбаминовая кислота
ДНЭТИЛОИЫЙ эфир угольной кислоты днэтилкарбонат
днамнд угольной кислоты, карбамид мочевина
Cl—ОС2Н5
о
С2Н5О—С—NH2
этиловый эфир хлоругольной кислоты
этиловый эфир карбаминовой кислоты, уретан
20.1. ХЛОРАНГИДРИДЫ угольной кислоты
Теоретически для угольной как двухосновной кислоты можно предположить существование двух типов хлорангндрндов — неполного (монохлорангндрида) и полного (днхлораигидрида)
468
Монохл op ангидрид получил название хлоругольная (хлормуравьиная) кислота, а днхлорангидрид — фосген:
хлорусильная кислота
фосген
Хлоругольная кислота в свободном состоянии не существует. В момент образования она распадается на СОг н HCI. Однако эфиры хлоругольной кислоты являются довольно устойчивыми соединениями.
Дихлорангидрнд угольной кислоты (фосген) в обычных условиях представляет собой газообразное вещество с запахом прелого сена, в 3—4 раза тяжелее воздуха, малорастворим в воде. Фосген очень ядовит. Прн вдыхании вызывает отек легких. В первую мировую воину применялся как боевое отравляющее вещество.
Получают фосген взаимодействием оксида углерода (IJ) с хлором на свету:
СО + С12 Cl—Cf—С1
о
фосген
В химическом отношении фосген проявляет свойства галоген-аигидридов кврбоновых кнслот. Так, в присутствии воды фосген медленно гидролизуется с образованием оксида углерода (IV) и хлорводорода:
Cl—ft—Cl + Н2О ------>- СО2 + 2НС1
О
С эквимолекулярными количествами спиртов фосген образует эфиры хлоругольной кислоты:
Cl—ft—С1 + СНзОН О
фосген
Cl—ft—ОСН3 + HCI о
метиловый эфир хлоругольной кислоты
Эфиры хлоругольной кислоты представляют собой жидкости с удушливым запахом, вызывают слезотечение. Они содержат в своей структуре подвижный атом хлора, что позволяет широко использовать их в органическом синтезе. В химическом отношении онн проявляют свойства галогенангндрндов и сложных эфиров.
46S
В присутствии избытка спирта или алкоголята фосген образует
полный эфнр угольной кислоты
Cl—С—С1 + 2СН3ОН ------> СНзО— С— ОСНз + 2НС1
диметиюиый эфир угольной кислоты
При действии иа фосген аммиака атомы хлора замещаются на аминогруппы При этом образуется диамид угольной кислоты, получивший название карбамцд, или мочевина
Cl—С—Cl + 2NH3
NHs— ft—NH2 + 2HC1 О
мочевина
В промышленности фосген используется в синтезе лекарственных средств, а также для получения красителей, пластмасс н др
20.2 АМИДЫ УГОЛЬНОЙ КИСЛОТЫ
Угольной кислоте соответствует два амнда — неполный (моно-амнд) и полный (диамид) Неполный амид имеет название карбаминовая кислота, а полный — мочевина, или карбамид:
но—с—nh2
h2n—с—nh2
карб аминов вя кислота
мочевина карбамид
Карбаминовая кислота в свободном состоянии не известна, однако ее соли и эфиры являются довольно устойчивыми соединениями Эфиры карбаминовой кислоты называются уретанами
Уретаны получают действием аммиака на эфиры хлоругольной кислоты или днэфнры угольной кислоты
Cl—ft—ОСНз + NH3 О
NH2—С—ОСНз + НС1
метиловый эфир хлоругольной кислоты
метиловый эфнр карбаминовой кислоты
СйНбО—С—OC2HS + NH3 > NHz—С—ОС2Н6 + С2Н5ОН
диэтиювый эфнр угольной кислоты
этиловый эфнр карбаминовой кислоты
470
Уретаны — бесцветные кристаллические вещества, проявляющие свойства амидов и сложных эфиров Некоторые из них, например мепротан (мспроб/мат), находят применение в медицинской практике в качестве лекарственных препаратов, обладающих снотворным н транквилизирующим действием
О СНз О
H2N—Lo—СНз—С— СН2—О—LnH2
<^Н2—СН2—СНз
мепротан (4v poOdMdT) днкярбячат 2 четнп 2 пропилпропакдиоза I 3
Особый интерес представляет диамнд угольной кислоты — мочевина Мочевина является конечным продуктом распада белков, содержится в моче человека (около 2 %) Впервые была выделена из мочн
Синтетически впервые получена в 1828 г немецким химиком Ф Велером при упаривании водного раствора цианата аммония, который в этих условиях подвергается изомеризации
ч—
h2no—c=n —+ h2n—с—nh2
В промышленности мочевину получают в больших количествах взаимодействием аммиака с оксидом углерода (IV)-
2NH3 + СО2 NHr-С—NH2 + Н2О
Это белое кристаллическое вещество с т пл 133 °C, хорошо растворяется в воде
В химическом отношении мочевина проявляет свойства амидов Однако в отличие от амидов алифатических и ароматических карбоновых кислот у мочевины более выражены нуклеофильные и основные свойства Это связано с тем, что в ее молекуле при карбонильной группе расположены две аминогруппы
H2NiCJtNH2
С11
471
В результате сопряжения неподеленных пар электронов атомов азота аминогрупп с п-злектронами кратной связи (4-А4-эффект) основным центром в молекуле мочевины является атом кислорода.
С сильными кислотами мочевина образует соли. Так, при действии иа мочевину азотной кислоты образуется малорастворимый в воде нитрат мочевины
С—NH2 + HNO3
А
nh2- с—nh2 I он
NOr|
нитрат мочевины
В водных растворах кислот нли щелочей прн нагревании мочевина легко гидролизуется с образованием оксида углерода (IV) и аммиака:
HaN—С—NH2+ НзО Н+илиОИ> СО2 | + 2NH3 J
(I О
Проявляя нуклеофильные свойства, мочевина вступает в реакции с электрофильными реагентами -- галогеналканами и гало-генангидридами карбоновых кислот, образуя соответственно алкнлмочевины и ацнлмочевины:
H2N—С— NH2 + С2Н51 С2Н5—NH—С—NH2 + Hl
3T№14O4?IHI№
О
H2N- - С—NH2 + CH3—С^ ► СНз—С—NH—С—NH2 + HC1
II \ Д II
О CI о о
ацетил мочевина
N-Ацильные производные мочевины называются ур ей дам и. Уреиды некоторых карбоновых кислот используются в медицине. Так, уренд а-бромнзовалериановой кислоты под названием броми-зовал применяется в качестве снотворного:
СНз о
^СН—сн-с^ о
СНз'/ Вг ^NH—С- NH2
брОМИЗОВач
472
Дикарбоновые кислоты могут образовывать с мо^звнной циклические уреиды. Важное значение имеет циклический уренд малоновой кислоты — барбитуровая кислота, структура которой лежит в основе ряда лекарственных препаратов, так называемых барбитуратов:
барбитуровая киислд
х
Под действием азотиой кислоты мочевина разлагается с выделением азота, воды н оксида углерода (IV). Эта реакция, открытая в 1875 г. русским композитором и химиком-органиком А. П, Бородиным, может использоваться для количественного определения мочевины путем измерения объема выделившегося азота:
HjN-j-NHj + 2HNO; О
СОз | -f- 2N21 4- ЗН2О
Аналогично протекает реакция мочеаины с водными растворами гилобромнтов
H2N—С—NH2 4- 3NaOBr -СО2 + N2 + 2H2O 4- 3NaBr
Эта реакция, как и предыдущая, может быть также использована для количественного определения мочевины.
При медленном нагревании кристаллической мочевины до 150—160 °C образуется биурет:
H2N-C-NH2 +
II О
н
I t
H-N-C-NH, —+ H.N-C-NH-C-NH
II -NH3 2 || ||
о о о
биурет
47
В качестве побочного продукта реакции образуется изоциановая кислота, которая подвергается тримеризации, образуя циануровую кислоту:
N
I....:
h-n-c-nh2
II :.
о
4— * N=C-OH
изоциановая кислота
t -NH3*
циануровая кислота
Биурет образует с ионами меди (II) в щелочной среде комплекс красно-фиолетового .цвета. Эта реакция имеет название биурето-
вая реакция:
H2N
^С=О
2НЬГ +Си(ОН)2-------»-
\ - Н2О
с=о
h2n
с—о
окрашенный комплекс
биурет
Она используется для качественного обнаружения мочевины н белков, которые, как н мочевина, содержат в своем составе группу —СО—NH—.
Важным азотистым производным мочевины является имииомо-чевииа, получившая название гуаицдии:
h2n—с—nh2
гуанидин
Это белое, расплывающееся на воздухе кристаллическое вещество с т. пл. 50 °C.
Гуанидин является сильным однокислотным основанием, сравнимым по силе (р/Свн 13,5) с гидроксидом натрия. Центром основности молекулы является атом азота нминогруппы. Высокая основность гуанидина связана с образованием при протоннровании
474
устойчивого катиона гуаиндииия, в котором положительный заряд делокализован между тремя атомами азота:
Н21Дс-^Н2 + н
с1’ ЧГТЕ
• • о—
О +
-----► H2N-*-C-*-NHj Cnh,
катион гуанидиния
С кислотами гуанидин образует соли, устойчивые к гидролизу.
H2N—NH2 + HCI
NH
+ ““
H2N—с —nh2
nh2
Cl-
гуанидин
гуанидиния хлорид
Как и мочевина, гуанидин легко вступает в реакции алкилирования с галогеналканами и ацилирования со сложными эфирами карбоновых кислот.
Прн конденсации с бифункциональными соединениями (диэфирами, дикетонамн) гуанидин образует гетероциклические соединения:
H2N—С
С2Н5О—С
С2Н5О—С
H2SO4, РОС1Б, Ни
гуа нидии ди этил калоиат
2 аминолнримидин
Остаток гуанидина является структурным фрагментом нуклеинового основания гуанина, содержится в молекуле аминокислоты аргинина и антибиотика стрептомицина:
H2N——NH—С Нг—С Н2—СН2—(j: Н—СООН
NH NH2
аргинин
476
он н
гуанин
стрептомицин
КОНТРОЛЬНЫЕ ВОПРОСЫ И УПРАЖНЕНИЯ
1. /Напишите структурные формулы следующих соединений: а) мочевина; б) этиловый эфир карбаминовой кислоты; в) фосген; г) днэтнлкарбонат.
2, Напишите схему образования биурета.
3. Напишите схемы реакций мочевины с: а) азотной кислотой; б) азотистой кислотой.
4. Приведите схему взаимодействия фосгена с этанолом,
5. Напишите схему реакции гидролиза мочевины.
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Адамантан 85
Адреналин 302
Азиды 423
Азины 332
Азиридин 279
Азобензол 195, 239 -241
Азокрасители 226, 243—245, 296
Азоксибензол 195, 241
Азо метины 217, 330
Азосоединения 195, 226, 237 сл., 293
Азотистый иприт 280
Азулен 148, 151, 152
Акрилонитрил 58, 424, 426
Акролеин 55, 273, 340, 341
а-Аланин 459, 460, 463, 464
Аллены 50, 51
Аллнлгалогениды 154, 173, 175, 182
Алкадиены 10, 50 и ел.
Алканы 10, 11 сл.. 31, 32, 52, 77, 135, 156, 253
Алкансульфонклхлориды 22
Алкены 10, 18, 24, 28 и сл., 52, 58, 63, 75, 96, 102, 156, 168—171, 210, 250, 257, 267,
309. 323, 362
Алкиламины 166, 189, 202 сл.
Алкилмагнингалогениды 62, 328
Алкилнитриты 233
Алкилсульфаты 39, 256, 289, 313
Алкины 10, 32, 60 сл., 171 — 174, 373
Альдегид
— акриловый, см. Акролеин
— бензойный, см. Бензальдегид
— валериановый 319, 324
— гликолевый 271
- глицериновый 271
— изовалериановый 319, 324
— нзомасляны А 319, 324
— коричный 349
- - кротоновый 333, 340
- масляный 319, 324
— муравьиный, см. Формальдегид
— пропионовый 260, 319, 324
— салициловый 294, 344
— сорбиновый 341
— л-толуилоиый 347
- о-толу иловый 347
— п-толуиловый 344, 347
— уксусный 23. 41, 48, 64, 68, 171, J72, 192, 236, 250. 263, 274. 276, 300, 305. 314
319, 324. 329, 336, 339
Альдегидаммиаки 330
Альдегиды 23. 42. 64, 172, 192, 217, 260. 318 сл., 361
Альди мины 329, 460
Альдоксимы 330, 424
Альдоли 332
Амид
— бензойной кислоты (бензамид) 416
— масляной кислоты 420
— пропионовой кислоты 204
— уксусной кислоты, см. Ацагадид Амндоксим 427
47
Амиды
— карбоновых кислот 204, 209, 215, 226, 361
— я-амивобензолсульфокислоты, см. Сульфанил амиды
а-Амилен, см. Пентен-1
0-Амилен, см. Пентен-2
Амины 166, 199, 200 сл., 236, 425
Аминокислоты 228, 459 сл.
а-Аминоннтрил 460
2-Аминопирнмидин 475
Аминоспирты 278 сл.
Аминофенолы 302 сл., 353
Аминоэтанал 278--280
Ангидрид
— бензойный 378, 404
— глутаровый 386
— малеиновый 58, 105, 392, 393, 395, 404
- уксусный 209, 221, 227, 289, 369, 404—408, 410. 422
— фталевый 125. 128, 145, 396—398, 404, 407, 408
— янтарный 386, 387, 405, 407
Анестезин 465
Анизндины 214, 306
Анилиды 215
Анилин 116, 181, 195. 201, 212 сл., 227. 228, 236, 241. 298
Антрахинон 129, 131
Антрацен 92, 96, 118. 128 сл.
Антраценсульфокислоты 131
Аргинин 475
Аренам ины, см. Ариламины
Аренолы, см. Фенолы
Арены, см. Углеводороды ароматические
Арил алкил галогениды 153, 176 сл.. 377
Ариламины 166, 195, 212 сл., 231 -233, 237, 238, 241
Ароматичность 92, 97- 99
Аспирин, см. Кислота ацетилсалициловая
Атом углерода
— асимметрический 16
— электрофильный (электронодефицитный) 158, 163, 167—169, 258, 367, 402 иерегибридизация 160
Атропизомерия 136
Ауксохромы 144, 243
’’ Ацетали 328
Ацетальдегид, см. Альдегид уксусный
Ацетамид 368, 416, 417. 420
Ацетанилид (антифебрин) 215, 218- 221, 227
Ацетилен 24. 25, 60 и сл., 120. 172, 275, 373
Ацетилхлорид 104, 209. 289, 368, 401. 403, 405, 410, 422. 456
Ацетон 23, 41. 48, 64. 116. 162, 171, 172, 252, 261, 286, 320 и сл., 453, 457
Ацетонитрил 162. 164, 204, 417. 420, 424, 426
Ацетофенон 104, 345, 347
Бензальдегид 143, 217, 344 . 346 и сл., 389
Бензгидразид 422
Бензгидрол 248
Бензилбензоат 416
Бензилиден хлор ид 154, 177
Бенз ил фенил кетон 345
Бензилхлорид 137, 154, 176, 177
Бензимидазол 225
Бензин 16, 25
Бенэогексоний 228
Бензоилхлорид 226, 351, 378, 401, 403
478
Бензоин ЗБ1
Бензол 19, 67. 87 и сл., 119. 121, 130, 134, 139, 193, 217, 285, 286, 352
БензолдиазонийхлорИД 176. 226. 232—239, 287. 293
Бензол Сульфокислота 100, 101, 285, 308
Бен зол сульфохлорид 235, 308
Бензонитрил 235, 377. 424
Бензофенон 138, 345, 347, 352
Бензохиноны 295, 299, 353—355
Бисульфитные соединения 327
Биурет 473
Бифенил 96, 134, 183
Бриллиантовый зеленый 142—144
Брожение молочнокислое 441
Бромбензол 96, 10|, 153. 183, 235
Бромиэовал 434, 472
Броммагнийалкоголят 251, 252, 329
Бромметагг 158
N-Бромсукцинимид 46
Бромэтан 158, 165, 259, 260, 424
Бутадиен-1,3 26. 51 и сл., 68, 76
Бутан И, 15, 17, 19, 23. 25, 96
Бутаналь (бутиральдегид), см. Альдегид масляный
Бутанон. см. Метилэтилкетон
Бутен-1 28—30,,52
Бутен-2 17—28, 30, 32, 66, 67. 257
Бутиламииы 20|, 205
<х-Бутилен, см. Бутен-1
8-Бутилен, см. Бутен-2
Бутин-1 62, 66
Бутин-2 17, 66, 67
у-Бутиролактон 433, 442
Вазелин 16, 26
Ванилин 352
Внкасол 128
Винилацетат 277
Винилацетилен 67. 68
Ви и ил галоген иды 154. 173- 175. 178
Газ
коксовый 96
— природный 16, 31
светильный 88
Галогеиалканы 17, 31. 32, 37. 75. 96, 102, 103, 124, 153. 156 сл., 189, 203, 208. 210, 212,
215, 258—260, 314, 361, 410
Галогеналкены 61, 63, 153, 173 сл.
Галогеналкины 153, 154
Гал огенан гидриды
— карбоновых кислот 103, 124, 209, 215, 289, 368, 369, 378, 400 сл.
Сульфокислот 221
Галогенарены 96, 153, 176 сл.. 212
Гексаметилсндиамин 224
Гексаметилентетрамин 339
Гексан 11. 16, 19
Гексахлоран 105
Гексиламин 205
Гекситы 226
Гелиантин. см. Метиловый оранжевый
Гептан 11.16, 19. 95
Гидразид
— бензойной кислоты, ем. Бензгидразид
уксусной кислоты (этаногидразид) 414, 421—423
— пропионовой кислоты (пропаногидразид) 421
47
Гидразобензол 195, 241
Гидразоны 330
Гидраты альдегидов 327
Гидробензамид 348
Гндроксикислоты 373, 433, 435 сл, а-Гндроксинитрилы, см. Циангидрины Гидропероксиды 23, 286, 313, 314 Гидрохинон 296, 298, 299, 301, 353 355 Гликоли, см. Спирты двухатомные Глиоксаль 106, 225, 271, 342 344, 384 Глиоксим 343
Глицерин 48, 266 и сл.
Глицеринтри ацетат 270
Глицин, см. Кислота аминоуксусная Гомополимеры 43 Горный воск, см Озокерит Гуанидин 474, 475 Гуанин 476 Гуттаперча 57
Декалин 72, 125, 128
Декан 11, 16, 19
Диазоа м н нобензол
Диазогидрокенды (диаэогидраты) 230, 232
Диазометан 75
Диазосоединения 75, 176, 226, 230 сл,
Диазотаты 230, 236, 238
Диальдегиды 225, 342 ел , 384
Диамины 202, 223 сл.
Диафенил сульфон 316
Диацетил (бутандион) 342- 344
Дибутил фтал ат 397
Дивинил, см Бутадиен-], 3
Ди галогенопроизводные углеводородов 32, 74, 155, 223, 298 - вицинальные 32, 35, 36, 61, 156, 171, 172 — геминальные 61, 63, 156, 171, 172
Диенофилы 55
Дизельное топливо 16 Дикетоны 66, 342 сл Дикетопиперазин 463 Димеризация 21, 44, 67, 75 Диметиламиь 203 205, 279 Диоксан 272, 314 М,М-Диметиланнлин 212, 213, 217, 243 Ди метил гл и оксим 343 Диметил кетен 403 Диметилкетон, см. Ацетон Диметилсукцинат 409 Диметнлсульфид 315, 316
Ди мет ил сульфоксид (димекемд) 162, 165, 189, 316 Ы,Ы-Диметилформамид (ДМФА) 162, 164, 165, 189, 416, 420 Дифенил, см. Бифенил Дифениламин 202, 205, 212 -214, 398 Ди фенил кетон, см. Бензофенон Дифеиилметан 137 сл.
Дифепилсульфид 315
Дихлоркарбен 210
Диклорметан 20, 21, 137, 156, 158 Диэтаноламин 280 Диэтнламип 205
N.N-Диэтиланилнн 143, 213, 215 №,М-Днэтилацетамид 416 Диэтилкетон 321, 324
480
Диэтнлоксалат 409
Диэтнлсульфид 167, 314
Додекан 11 > '
Енолы 246, 275 сл., 283, 341
Енолят-анион 332
Жиры 267
Д Д ит^л и
электроноакцепторные 104, 109, 127, 162, 179, 212, 214, 238, 265, 288, 365 — электронодонорные 103, 109, 126, 162, 193, 214, 288, 365
Изобутан 19
Изобутилен 28, 44
Иэобутнрилхлорид 403
Изомеры
оптические (см. также Энантиомеры) 16, 160, 206, 436
- структурные 16, 155, 320, 361
— сим-, анти- 231, 240
- цис-, транс- 30, 33, 47, 59, 249. 450
Изомерия
— углеродной цепи 15, 29, 30, 61, 188, 321
— геометрическая (цис-, транс-изомерия) 29, 30, 73, 74, 155, 156, 248
- оптическая 15, 16, 51, 74, 136, 155, 156, 206, 248, 249, 266, 436
положения 30, 61, 94, 188, 285, 321, 436
прототропная, см. Таутомерия
— структурная 15, 29, 61, 73, 155, 248, 266, 285, 320, 361, 460
Изонитрилы, см. Изоцианиды
Изопентан 15, 19, 23, 53
Изопрен 15 -53, 56 - 59
Изопропнлнитрит 167
Изоцианиды (изонитрилы) 166, 210, 216, 226
Имины 329, 348
Иминомочевина, см. Гуанидин
Инверсия
— триалкиламннов 206
- циклоалканов 81, 82
Индикаторы 145, 243—245
Иодбензол 212, 235
Иодмстан 17, 158, 312
Йодоформ 155, 158, 185
Иодэтап 158
Ионы
— алкоксид- 255, 412, 415
— амбидентные 165, 166, 190, 424, 456
ацилия 104, 403
биполярные 220, 461
— галогенония 35, 36
— карбония, см. Карбкатионы
— карбоксилат- 365
— нитрозония 232
нитрония 99
- трифенил метильные 140, 141
— фепоксид- 287— 289, 293, 410
— цвиттер, см. Ионы биполярные
Иприт 316
Кадаверин, см. Пентаметилендиамин
Калия
— бензоат 348
- фталимид 203
481
Кальция
— адипинат 75
— ацетат 323
— карбид (ацетиленид) 62
— лактат 442
- оксалат 388
Камфан 72
Карбамид, см. Мочевина
Карбены 75, 210
Карбинол, см. Метанол
Карбкатионы (ионы карбония) 34—38, 44, 55, 98, 102, 103, 162—164, 169, 258
260, 367, 455
Катализатор Циглера-Натта 45, 58
Каучук
— вулканизации 58
— натуральный 57
- синтетический 56 -59, 67, 68
— бутадиеновый 57, 58, 68
- бутадиен-нитрильный 58
— бутадиен-стирольный 58
— изопреновый 58
— хлоропреновый 58, 68
Керосин 16
Кетали 328
Кетокснмы 330
405
Кетоны 41, 64. 74, 104, 171, 172, 192, 211, 250. 261, 318 ел.
Кислота
— адипиновая 85, 383, 386, 392
- акриловая 372—375, 431, 433, 461
— н-аминобензойная (ПАЕК) 459, 461, 465
— е-аминокйпроновая 465
— у-аминомасляная (аминалон, или гаммалон) 459, 463, 464
- а-аминопропионовая, см, а-Аланин
— амнноуксусная 459
— антраниловая 459, 465
— ащчн кллицилован (<пнирнн) 447, 449
- - ацетонднкарбоновая 444
- - ацетоуксусная 451, 453, 457
— барбитуровая 473
— бензойная 42, 286, 348, 376 -380
— а-бромнзовалериановая 434
— бромуксусная 366
— бутандновая, см. Кислота янтарная
— бутановая, см. Кислота масляная
— валериановая 359, 363, 366, 371
— винная 435, 442, 443
— виноградная (мезовинная) 395, 443
— винилуксусная 372
— галловая 436, 444, 448, 450
— гексадекановая, см. Кислота пальмитиновая
— гександиовая, см. Кислота адипиновая
— гексановая, см. Кислота капроновая
— гептандиовая, см. Кислота ш/мелиновая
— у-гидрокснмасляная 442
— гликолевая 271, 343, 435, 437, 452
— глиоксиловая (глиоксалевая) 271, 393, 451
— глутаровая 383, 385, 386
— дихлоруксусная 366, 434
— додекановая, см. Кислота лауриновая
— изовалериановая 359, 363, 371
— иэомасляная 359, 361, 363, 391
— изофталевая 396
— иодуксусная 366
482
— капроновая 359, 363
— карбаминовая 468
— карболовая, см. Фенол
— а-кетоглутароная 451, 459
- коричная 377, 379, 380, 389, 408
— кротоновая 372, 463
— кумариновая ,450
— о-кумаровая 450
— лауриновая 359
— лимонная 435, 443
— линолевая 372
— линоленовая 372
— малеиновая 391—395
— малоновая 383—386, 388, 389, 393
— масляная 359, 361, 363, 366, 371
— мезоксалевая 271
— метакриловая 372, 375
— метановая, см. Кислота муравьиная
— миристиновая 359
- молочная 433, 435—437, 439, 441, 452
- муравьиная 23, 57, 162, 164, 263, 272, 338, 358, 359, 363, 366, 370, 386, 44)
— октадека новая, см. Кислота стеариновая
— октандиовая, см. Кислота пробковая
— олеиновая 372, 375
— пальмитиновая 359, 363
— пентандиовая, см. Кислота глутаровая
— пентановая, см, Кислота.валериановая
— пикриновая 197 , 291, 297
— пимелиновая 383
— пировиноградная 440, 443, 451—453, 458
— пробковая 383
— пропандиовая, см. Кислота малоновая
— пропановая, см. Кислота пропионовая
— пропеновая, см, Кислота акриловая
— пропиоловая (пропиновая) 372
— пропионовая 66, 260, 338, 358, 359, 362, 363, 366, 369, 371, 390
— салициловая 113, 293, 436, 444-449
— стеариновая 359, 363
— стифннновая 300
— сульфаниловая 220, 243, 296
— терефталевая 273, 396
— тетра декановая, см. Кислота миристиновая
— тетроловая 372
— трифторпероксиуксусная 223
— трифторуксусная 366, 430
— трихлоруксусная 366, 434
— уксусная 23, 41, 57, 66, 68, 162, 164, 250, 309, 338, 359, 363, 366—369, 371, 386, 405, 431
— — «ледяная» 371
— Nl-фенилантраниловая 433
— фенилуксусная 377, 380
— фталевая 106, 396, 397
— фторуксусная 366
— фумаровая 392—395
— хлоругольная 468, 469
— хлоруксусная (монохлоруксусная) 430—432, 434
— циануксусная 388
- - циануровая 474
— щавелевая 106, 143, 271, 342, 383—388, 398
— щавеле в оуксу сна я 443, 451, 458
— элаидиновая 375
— этандиован, см. Кислота щавелевая
— этановая, см. Кислота уксусная
483
й
— яблочная 388, 392, 395, 435, 442
— янтарная 383—387, 391, 394, 405
Кислоты
— алкнлнитроловые 191
— ароматические 106, 286, 376 сл., 395 сл.
— галогенокарбоновые 373, 420 сл,
— гидроксамовые (гидроксиамиды) 402, 413 ,
— о-гидрокснкоричные 436, 444, 450
— дикарбоновые 74, 106, 383 сл.
- Льюиса 44, 101-104, 124, 259, 289, 291, 315, 403
- монокарбоновые 23, 358 сл.
— насыщенные 358 сл , 383 сл.
— нафталинкарбоновые 376, 379
— NH- 206, 418
— ненасыщенные 371 сл., 392 сл.
— нитроновые 190. 191
— толуиловые 376, 379
— трикарбоновые 357, 442—444
— хлормасляные 365, 430, 431
— СН- 63, 190, 277, 335, 364, 389. 425, 454
Коламин, см. Аминоэтанол
л-Комплексы 34—36. 55, 98. 100, 101, ЮЗ. 104, 149, 445, 455
о-Комплексы 98—101, 103, 104, 108—112, 122, 170, 182, 198, 238, 294, 445
Конденсация
— альдольная 332
— перекрестная 349
— бензоиновая 350
— кротоновая 333
— Кляйзена 414
— Кневенагеля 393
— Перкина 350, 407
— сложноэфирная, см. Конденсация Кляйзена
— сложноэфирная альдегидов, см. Реакция Тищенко
Константм диссоциации (p/G, рК±) 214, 220, 365, 366, 374, 379, 385, 394, 397, 418, 431, 439, 448, 474 н
Контракция спирта 254
Конфигурация 45
— Z- и £- 30
— обращение 160
— тетраэдрическая 12, 206
— цис- и транс- 30, 57
Конформация 12
— ванны (лодки) 80, 81
— заторможенная 12, 80
--- анты-бутановая 12
— заслоненная 77, 78
— зигзагообразная 12
— клешневидная 12
— конверта 80
— кресла 80, 81
— нерегулярная 12
— твист (искаженная ванна) 81
— трансоидная 52
Конформеры 136
Красители 142, 243—245, 398, 464
Крекинг 24, 25, 31
Крезолы 181, 284, 285. 290, 296, 297
Кристаллический фиолетовый 145
Ксилолы 92, 93, 95 — 97, 396
Кумарин 451
Кумол 92, 97, 102, 116, 286
Лавсан, см. Полиэтилентерефталат
Лактамы 463
Лактиды 440, 466
484
Лактоны 433, 440, 466
Лакриматоры 335
Лизин 228
Линетол 375
Мазут 16
Малахитовый зеленый 144, 145
Малонодинитрил 426
Масло
- вазелиновое 26
— соляровое 16
Меди
— гликолят 269
— глицерат 269
Мезитила окись 333
Мезитиле» 93, 94, 97
Ментол 248
Мспротан (мепробамат) 471
Меркаптаны, см. Тиоспирты
Меркаптобензол 307
Металлоорганические соединения 17, 43, 45, 57, 14S
Металлоцены 149
Метальдегид 336
Метан 11, 18—20, 24, 25, 62
Метаналь, см. Формальдегид
Метанол 23, 25, 164, 248, 253—256, 260, 312
Метантиол 307, 309
Метиламин 203 —205, 227
N-Метиланилин 212, 213
2-Мет ил бензоксазол 305
Метилмагннйиодид 62
Метил меркаптан, см. Метантиол
Метилметакрилат 375
Метилнатрий 17
Метил на фтал ины 119, 120, 126, 128
Метиловый красный 244
Метиловый оранжевый (метилоранж) 243, 244
Метил пропил кетон 321, 324
Метилсалицнлат 447, 449
Метилфенилкетов, см. Ацетофенон
Метил фор ми ат 409
Метил циклобутан 73, 77
Метил циклопентан 77, 84
Метил циклопропан 75, 77
Метил этил кетон 320, 321, 324, 338, 458
Метод
— Лебедева 57
— Фишера-Тропша 17
Механизм реакций
— азосочетания 237
- гидрокарбоксилирования 362
— гидроксиметилирования 294
- - гидролиза амидов 419
— дегидратации спиртов 257—259
— диазотирования 232
— Зандмейера 234
— иэонитрильной 210
— Канниццаро 348
- карбоксилировании 445
— кислотного гидролиза сложных эфиров 419
— конденсации
-- альдольной 332
-- бензоиновой 351
---Кляйзена (сложноэфирная конденсация) 415
---Перкина 350
— нитрования по Коновалову 22
— нуклеофильного замещения ($ч) 158 сл., 210, 367, 412, 413
---ариновый (отщепления-присоединения) 181
---бимолекулярный (SN2) 159, 180, 257—259, 312
---внутримолекулярный (SNJ) 260
— мономолекулярный (SJ) 162, 234, 257— 260
— нуклеофильного присоединения (Д^) 326
— полимер»сшил 44
— присоединения-отщепления 329
— радикального замещения (5 ) 20, 22, 234, 235
— Тищенко 334
— щелочного гидролиза сложных эфиров (омыления) 413
— электрофильного замещения (S_) 98, 100, 104, 445
— электрофильного присоединения* (Др) 34
— элиминирования (Е) 169,257 259'
---бимолекулярный (Е2) 169, 258
---мономолекулярный (Е1) 169, 258, 259
— этерификации 367
Мкдантан 85
Мономеры 43
Монометилсукцинат 409
Мочевина (карбамид) 468. 470 сл.
Найлон 392
Напряжение циклов
— Ван-дер-Ваальса 77, 78
— торсионное (пнтцеровское) 77—80
— угловое (байеровское) 77—79
Натрия
— н-амииобензолсульфонамид 222
— п-аминобензол сульфонат 220, 243
— ацетат 18, 165, 405, 410, 458
— ацетиленид 62, 65
— бензолдназотат 231
— бутират 366
— днглнколят 268
— моногликолят 268
— нитропруссид 340
— оксалат 387
— оксибутират 442
— пропионат 18, 458
— салицилат 293, 445, 446, 449
— фенолят 165, 285, 288, 293, 445
- формиат 370, 387
— хлорацетат 432
- цитрат 444
— этантнолят (этилмеркаптид) 307, 308
— этилат (этоксид) 165, 255
Нафталин 92, 96, 118 сл., 396
Нафталинсульфокислоты 123, 127, 128, 131
Нафтолы 127, 226. 284, 286, 297
Нафтохиноны 125
Нейрин 281
Неогексан 13
Неопейтан 15, 19
Нефть 16, 31, 74, 85
— ароматизация 95
— крекинг 285
— фракции 16, 52, 53
Нингидрин 463, 464
Нитрилы 166, 204, 224, 326, 361, 377, 384, 417, 424 сл.
Нитроалканы 187, 189 сл., 204
486
Нитроанилины 197, 214, 219, 224
Нитробензол 99, 100, 108, 164, 188, 195, 199, 212, 223, 235, 303
Нитроглицерин 270, 275
N-Нитрозоамины 210, 216, 232
Нитрозобензол 195, 223, 303
Нитрозосоединения 195, 217, 223, 354
Нитрометан 199, 204
Нитрон 426
Нитро нафталины 123, 127
Нитросоединения 166, 187 сл„ 223
Ннтротолуолы 199
Нитрофенолы 108, 113, 197, 198, 291, 303
Ннтроэтан 166, 211
Нитрующая смесь 99, 123, 218
Новокаин 465
Нонан 11
Озокерит 26
Озониды 41, 106
Оксафенамид 449
N-Оксиды аминов 211
Оксиран, см. Этиленаксид
Оксокислоты 451 сл.
Оксосинтез 323
Октан 16, 95
Олеум 100
Олефины, см. Алкены
орто- и лара-Орнентанты (заместители I в II рода) 107 сл., 126,182, 2
Орнитин 227
Ортокислота 361
Основания Шиффа, см. Азометины
Паральдегид 336
Парафин 16, 26
Парафины, см. Алканы
Параформ 336
Парацетамол 306
Пентадекан 11, 19
Пентаметилендиамин 224, 228
Пентан 11, 15, 19
Пентанон-2, см, Метилпропиякепш
Пентанон-3, см. Диэтилкетон
Пентен-1 28
Пентен-2 28
Пентиламин 205
Пентнты 266
Перегруппировка
Гофмана (расщепление амидов по Гофману) 204, 420
- - Фриса 293
Пероксиды 43, 46, 309, 313, 314
Пероксикислоты 42, 189, 211, 241
Пинан 72
Пиперазин 225
Пиперидин 224, 225
Пирогаллол 298, 301, 302, 448
Пирокатехин 298 -30], 353, 354
Пирролидин 224
Плексиглас, см, Полиметилметилметакрилат
Полиакрнлат 375
Поливинилацетат (ПВА) 277
Полимеры 43
— атактические (нерегулярные) 45, 46
— изотактические (стереорегул ярные) 45, 46
Поли метил метакрил ат 375
Полиолы, см. Спирты многоатомные
Полипропилен 44 46, 48
Полихлорвинил (поливинилхлорид) 68, 175
Полиэтилен 44, 48
Полиэтнленгликоль 273
Полиэтилентерефталат 273, 274
Полуацетали 328
Правила
— Зайцева 31, 32, 168, 257 '
- Марковникова 37 -40, 63, 64, 84, 250, 309, 341, 374
- ориентации (в реакциях SF) 107 — 115, 126—128
— Кюнкеля 92, 121, 129
— Эльтекова (Элътекова-Эрленмейера) 64, 276, 341
Проба
Байера 47
Бейлыитейна 183
— йодоформная 262, 335
— Лукаса 262
Пропан 11, 17, 19. 23, 25, 32, 40. 66
Пропаналь. см. Альдегид пропионовый
Пропанон, см Ацетон
Пропанонитрил 166, 424
Пропелленты 185
Пропен 26, 28, 31. 32, 40, 41, 46, 48, 66, 75, 102, 103, 156, 250, 267, 309
Пропила мини 202, 205, 420
Пропилен, см Пропен
Пропиленхлоргидрин 40
Пропин 32, 60 и сл., 172
Пропионилбромид 369
Псевдонитролы 192
Птомаины 228
Пурген, см. Фенолфталеин
Путресцин, см. Тетраметилендиамин
Радикалы 14, 15. 29, 60, 71
— алкильные 14, 18, 47
аллильные 29, 47
— ароматических углеводородов 93, 234
свободные 18, 20—22, 43, 46, 234
— бензильного типа 107, 177
— трифенилметильные 141, 142
рекомбинация 21
Рацемизация 163
Рацемические формы 163, 441, 443
Реагенты
- алкилирующие 96, 159, 212
— ацилирующие 221. 403
галогенирующие 46, 157, 173
— нитрующие 99
— нуклеофильные 158 н сл., 181, 236
— нуклеофильность 34, 97, 161, 410
— электрофильные 33—35, 54, 97, 102—104, 107, 122, 158, 236 -238, 315, 401, 411 Реактивы
— Гриньяра (магннйоргзнические соединения) 65, 170, 183, 251, 328, 362
Лукаса 262
— Толленса 337
— Фелинга 337
Чугаева, см. Диметилглиоксим
Реакции
азосочетания 199, 237, 293, 296
— алкилирования 62, 96, 102, ЮЗ, 124, 129, 150, 159, 196, 208 212, 215, 288,
291, 314, 410
алкоголиза 411
488
— амидирования 221, 413
— аммонолиза 223
— ацидолиза 312
- ацилирования 103, 104, 124. 130, 152, 209, 215, 221, 289, 291, 304, 309, 403, 455
— восстановления 40. 66, 129, 130, 170, 193. 194, 204. 212, 236, 241, 250, 303. 337, 420
— галогенирования 20, 35, 36, 46, 54, 63, 101, 105, 107, 123, 124, 130, 136, 138, 152, 156, 176, 196, 21 I. 217, 236, 289, 308, 351
— регноселектнвность 21, 37
— стереоселективность 36
— гидралннолиэа 411, 413
— гидратации 39, 64, 250, 264, 267, 272, 438
гидрирования (см, также Реакции восстановления) 17, 32, 40, 54, 104, 125, 150, 263, 295
гидрогалогенирования 36, 55, 63, 173, 374, 430
— гидрокарбоксилировання 362, 373
— гидроксилирования 267. 446
— гидроксиметилирования 294
- гидролиза 165, 203, 215, 217, 221, 249, 252. 264, 267, 361, 377, 406, 411, 417, 419, 425, 437
— гипогалагенировапия 40
— дегалогенирования 32
— дегидратации 31, 256- 259, 311, 369, 392, 404, 419, 424
— дегидрирования 24, 32, 52, 134, 261
— дегидрогалогенирования 32, 61, 75
дегидроциклизации 25, 95, 120
— декарбоксилирования 228, 286, 379. 385, 390, 448, 453
— деполимеризации 336
— диазотирования 199, 216, 226, 231, 236
— диспропорционирования (дисмутации) 333, 348
— карбоксилирования 445
— конденсации (см. ещё Конденсация) 225, 226, 295, 300, 425, 426
— металлировання 183
нитрования 22, 99, 123, 130, 136, 137, 152, 189, 193, 196, 218, 291
— нитрозирования 292
— нуклеофильного замещения (SN) 158 сл,, 175, 178 182, 196, 367 сл.
— нуклеофильного присоединения (Д^) 326
— озонирования 41, 323
- окисления 23, 41, 66, 97, Юб, 125, 130, 138, 189, 211, 241, 260, 271, 274, 286, 298, 309, 313, 337, 345, 353. 370, 388. 396
— отщепления, см. Реакции элиминирования
— переэтерификации 414
— пиролиза 74, 322, 368
- поликонденсации 273, 295
— полимеризации 43—45, 56, 57, 335
присоединения'отщеплени» 124, 329
— радикального замещения (5Н) 20—22, 34, 46, 83, 84, 156
— радикального присоединения 38, 39, 105, 309
— расширения циклов 84
— скорость 38, 99, 107, НО, 163, 169, 180
— сополимеризации 43
— сужения циклов 84
- сульфирования 100, 123, 150, 152, 196, 29]
— сульфохлорирования 22, 221 *
— формилирования 346
— хлорметилирования 177
— циклоприсоединения 56, 75
- электрофильного замещения (5 ) 97 сл., 122 и сл,, 130, 136, 137, 139, 150, 152, 196. 217, 237, 289, 294, 35!
— электрофильного присоединения (Ль) 33, 34, 53, 62
489
— электроциклические 76
- элиминирования (Е) 31, 168 и сл., 172, 373
— этерификации 256, 367
Реакция
— биуретовая 474
- Вагнера 41, 267
— Вильямсона 165, 288, 311
— Вюрца (Вюрца-Фиттига) 17, 74, 96, 183
— Габриэля, см, Синтез Габриэля
— Гаттермана-Коха 346
— Гелля-Фольгарда-Зелинского 369, 430
— Гофмана 203
— Дильса-Альдера (диеновый синтез) 55, 77
— Зандмейера 176, 235
— Зинина 194, 211
— изонитрильная 210. 226
— Канниццаро 343, 348
— Кольбе (Кольбе-Шмитта) 293
— Коновалова 22, 189
— Кучерова 64
— нингидриновая 464
— Прилежаева 42
— Раймера-Тимана 294
— Реппе 373
— Реформатского 438
— «серебряного зеркала» 337, 370
— Тищенко 333
— Торпа 426
— Ульмана 135, 179
— Финкельштейна 157, 167
— Фриделя-Крафтса 96, 102 -104, 129, 137, 139, 150, 196, 291, 346
— «щелочного плавления» 286, 298, 446
— Эльбса 296
Резина 58
Резорцин 298, 300 303
Ремантадин 85
Сала зоди метокси и 241
Салазолиридазин 241
Салицнламид 449
Салол, см. Фенилсалицилат
Сверхсопряжение 34, 38
Связи
— аксиальные 82
— ароматические 91, 116
- водородные 205, 227,253, 254, 263, 268, 278,296, 307, 363, 394,448, 453. 454
— двойные 28, 30, 33, 35 сл., 55, 61, 62, 90, 91. 121, 173, 174, 216, 218, 275
— длина 12, 52, 91, 121, ]35, 174
— л- 30, 33, 34, 51, 62, 79, 325
— семиполярные 187
— п- 19, 33, 51, 62, 76, 79, 89, 134, 135, 283, 325
— тройные 55, 60 и сл., 181, 233
- т-(«банановые») 79
— экваториальные 82
Сдвиг
- батохромный 242
- гипсохромнын 242
— химический 25, 184, 263
Сегнетова соль 443
Семикарбазоны 331
Синтез
— Габриэля 203
— Фишера-Тропша 17
— Штреккера 460
490
Смолы
— ионообменные (иониты) 295
— каменноугольная 96, 118, 120, 128, 129, 131, 134, 285
— мочевиноформ альдегидные 339
— фенолоформальдегидные 294, 339
Соли диазония 176, 209, 216, 226, 230 сл., 287, 293, 296
Сополимеры 43
Сопряжение 47, 90, 140, 174, 179, ] 90, 216, 218, 243, 364, 422
— р, п- 287, 364, 418
— л, л- 52
— а, л-; см. Сверхсопряжение
Спектры
- инфракрасные (ИК) 25, 47, 59, 68, 84, 116, 184. 198, 227, 262, 296, 313, 381, 398, 427, 466
— масс- 25, 47, 68, 116, 184, 381
— протонного магнитного резонанса (ПМР) 25, 47, 59, 68, 84, 116, 184, 227, 263,
296, 313, 381, 427
ультрафиолетовые (УФ) 25, 59, 68, 84, 115, 198, 227, 262, 296, 381, 427
Спермидин 228
Спермин 228
Спираны 70, 71
Спирт
аллиловый 173, 248, 253, 254, 259, 264, 267
— бензиловый 248, 253, 259, 264, 300, 348
— бутиловый (бутанол-1) 249, 253
— агор-бут иловый (бутанол-2) 249, 253, 257, 329
— трет-бутнловый (2-метилпроланол-2) 248, 249, 253, 260
— виниловый 64, 275 277
— винный, см. Этанол
— диацетоновый 333
- древесный, см. Метанол
— изобутиловый (2-метилпропанол-1) 249, 253
— изопропиловый (пропанол-2) 39, 48, 247, 250, 252—256, 261
— кротиловый 53
— метиловый, см. Метанол
— муравьиный 370
— поливиниловый (ПВС) 277
— пропаргиловый 253, 254, 264
— пропиловый (пропанол-1) 31, 252, 253, 260
цнклогексиловый (циклогексанол) 247, 253, 295
этиловмй, или винный, см. Этанол
Спирты 96, 102, 103, 157, 162, 212, 246 сл., 288, 291,
— ароматические (арилалканолы) 247, 252, 262, 300
- двухатомные (диолы) 41. 53, 172, 246, 264 сл., 384
— многоатомные (полиолы) 246, 266
— насыщенные (предельные) 31, 247, 252
— ненасыщенные (непредельные) 53. 64, 173, 247, 251, 252, 262, 275
— одноатомные 246 сл.
— трехатомные (триолы) 246, 266
Степень полимеризации 43
Стеран 132
Стирол 58, 92, 97, 116
Стрептомицин 476
Стрептоцид 220—222
Сукцинимид 47, 387
Сульфадимезин 222
Сульфален 222
Сульфаниламиды 220—222
Сульфацил (альбуцид) 222
Сульфиды (тиоэфиры) 167, 314 сл.
Сульфоксиды 315
491
Сульфоны 316 Сульфохлориды 22, 221, 235 сСухой спирт», см. Метальдегид Таутомерия 276 — аци-нитро- 190 кето-енольная 276, 454
Теория цветности 242 244 ~ молекулярных орбиталей 90, 160 — напряжения циклов 77 сл. Тетрагидрофуран 273 Тетрадекан 11 Тетралин 125, 128 Тетрамеризация 67 Тетраметилсндиамнн 223, 224, 227 Тетр а хлор метан 19, 20, 25, 47, 158, 352 Тетрацен 118 Тимол 284, 297 Тиолы 246, 307 сл. Тносемикарбазоны 331 Тиоспирты 167, 246, 307 сл. Тиофенол, см. Меркаптобензол Тиофенолы 246, 307 сл. Толуидины 202, 205, 213 Толуол 92, 95-97, 116, 194. 377 Триазены 237, 239 2,4,6-Триброманилин 218, 236 1,2,3-Трибромбензол 236 Трибромметан 158 2,4,6-Трибромфенол 290 Тридекан 11 Тримеризация 67 Триметиламин 202, 204, 205, 281 Трифениламип 202, 205, 212—215 Три фенил карби иол 139, 248 Трнфенилметан 138 сл, Трихлорметан, см. Хлороформ Триэтаноламин 280 Трнэтнламин 205, 211 Тропилиден 150 Тропнлий-катион, см Циклогептатриенил-катион
Углеводороды 10 и сл.
— алифатические, см. Углеводороды ациклические алициклические 10, 70
— ароматические 10, 25, 87 и сл., 177, 235, 285, 314, 345 - ацетиленовые, см, .Алкины — ациклические 10 — насыщенные, см, Длконы — этиленовые, см. Алкены
Ундекан 11
Уреиды кислот 434, 472
Уретаны 468, 471
Уротропин, см. Гексаметилентетрамин
Фенамин 229
Фенантрен 92, 118, 131 сл.
Фенантренхинон 132 Фенацетин 306 Фенетидины 306 Фенетол, см. Эфир фенилэтиловый Фенилацетат 289, 293, 409, 410 Фенилгндразин 237, 331
492
Фенилгидразоны 331
Фенил гидроксил амин 195, 303
Фенилендиамины 202, 223—225, 229
Фенилнитрометан 188
Фенилсалицнлат 449
Фенол 108, 116, 145, 179, 234, 237, 238, 283 сл„ 448
Фенолы 96, 234, 237, 238, 246, 283 сл.
— двухатомные 246, 283, 298 сл„ 353, 354
— одноатомные 246, 283 сл.
— трехатомные 246, 283, 298 сл.
Фенолфталеин 145
Ферроцен 149, 150
Ферроцерон ]50
Флороглюцин 298, 301, 302
Формалин 336, 339
Формальдегид 23, 25, 177, 192, 235, 251, 263. 274, 294, 319, 32
Формамид 420
Фосген 185, 468—470
Фреон-12 185
Фталимид 398, 407
Фторметан 158
Фторотан 185
Фторэтан 158
Хингидрон 355
Хиноксалин 225
Хнноксим 354
Хинондноксим 354
Хиноны 298, 353
Хинонимины 222, 305
Хлоралгидрат 265, 328
Хлораль 328
Хлорангндриды
— бензойной кислоты, см. Бекзоилхлорид
— карбонавык кислот 292, 401 сл.
- салициловой кислоты 447
— сульфокислот (сульфохлорнды) 22!
- -- уксусной кислоты, см, Ацетилхлорид
Хлорбензол 176, 179, 181 -183, 185, 212, 235, 285
Хлорвинил (винилхлорид) 68, 155, 173
Хлоргидрины 40, 269, 270
Хлористый метилен, см, Дихлорметан
Хлормагнийпропионат 362
Хлорметан 20, 21, 156, 158
Хлоропрен 68, 155
Хлороформ 20, 21, 25, 47, 139. 155, 158. 184. 210, 216, 294
Хлорпикрин 199
Хлорпропан 158
Хлорэтан (этилхлорид) 102, 103, 158, 184, 250, 260, 308
Холин 281
Хризен 118
Хромофоры 142, 144, 242, 244
Циангидрины 326, 341, 437
Цинлоалканы 70 и сл.
Циклобутан 74, 77, 80, 83
Циклогексан 71, 75, 77, 80- 83, 85, 95
Циклогептан 77
Циклогептатрненил-катион (тролилий-катион) 148, 150—152
Циклооктан 77
Цнклооктатетраен 67
Циклопентадиенил-аннон 148—150, 152
Циклопентан 73, 75, 77, 80, 84
Циклопентанон 75, 386
Циклопентан пергидрофен антрен, см. Стеран
Циклопропан 7|, 75, 77, 79, 83, 84
Циклотримеризация, см, Тримеризация
п-Цимол 93, 116
Цистамина дигидрохлорид 317
Четыреххлористый углерод, см. Тетрахлорметан
Эйкозан 16, 19
Энантиомеры (зеркальные изомеры) 16, 51, 74. 136, 156, 436, 443
Энергия
— делокализации 89
— конформаций 81
- сопряжения (энергия резонанса) 52, 90, 91, 121, 129, 131
— химической связи 33
Эпоксиды 42
Эритриты 266
Этазол 222
Этан И, 17—19, 24, 25, 254
Этаналь, см. Лльдегид уксусный
Этандиаль, см. Глиоксаль
Этандиамин (этилендиамин) 223, 225
Этанол 31. 39, 48, 102, 103, 116, 157, 164, 165, 210, 247, 248, 250,253 сл., 260, 263, 308. 367
— абсолютный 264
Этантиол 167, 308, 309
Этен, см. Этилен
Этиламин 166, 201, 204, 205, 209—21 I, 226, 407, 420
Этилацетат 165, 209, 251, 252, 256, 333, 367, 406, 410, 413, 415
Этнлбенэоат 378, 409
Этилен 24, 26, 28, 31, 35, 41, 42, 48, 174, 257, 264
Этиленгликоль 41, 48, 265 и сл., 437
Этиленимин, см. Азиридин
Этиленокснд 42, 267, 279, 281
Этилизобутират 409
Этилмагннйгалогениды 18, 170, 251, 329, 362
Этил формиат 415
Этин, см. Ацетилен
Эфир
— ацетоуксусный 414, 453—458
— винилэтиловый 312
— диметнловый 310, 312
— дифениловый 312
— диэтиловый 19, 165, 257, 310—313
— малоновый (диэтилмалонат) 389—391, 409
— метил фен иловый (анизол) 310, 312, 314
— петролейный 16
— уксусновиниловый, см. Винилацетат
— фенилэтиловый (фенетол) 165, 236, 288, 311, 314
Эфиры
— активированные 411
— ариловые 41L
— виниловые 411
— карбаминовой кислоты, см. Уретаны
— простые 165, 236, 257, 258. 270, 272, 277, 288, 310 сл.
— сложные 165, 209, 250, 252, 256, 26], 270, 277, 288, 333, 367, 408 сл.
Эффект
— индуктивный (/) 38, 108, 109, 164, 167, 182, 196, 206, 207, 254, 268, 283, 325, 364, 369, 373, 402, 430, 432
— мезомерный (М) 108, 109, 174, 178, 182, 196, 214, 217, 237, 243, 284, 287, 364, 373, 406, 411, 418
— сольватации 162, 207
Наумове видання
ЧЕРНИХ ВАЛЕНТИН ПЕТРОВИЧ ЗИМЕНКОВСЬКИЙ БОРИС СЕМЕНОВИЧ ГРИЦЕНКО IBAH СЕМЕНОВИЧ
ОРГАН1ЧНА Х1М1Я
У трьох книгах
Книга 2
ВУГЛЕВОДН1 ТА IX ФУНКЦЮНАЛЬН! ПОХ1ДН1 (РосШською мовою)
Редактор А, Л, Алиева
Художник оправн О. Г. Круглое
Худож1ПЙ редактор В. С. Петренко Виконавець рисунюв €, Л. Сштковський ТехшчниЙ редактор /. А. Омельченко Ко ректор Л. П. Сич
Здано до склалання 24 03,95. ГПдписано до друку 10.08,95. Формат 60X90/16, Ilanip друк, № 2. Гарнггура л!тературна. Друк офсетний Умов. друк. арк. 31, Умов. фарбо-в!дб, 31,25 Обл.-вид, арк. 33, Вид, № 2355. Тираж 10 000 прим. Зам. 5-104, Замовне,
Держание спец!ал i зон а не видавннптво «Основа», УкраТна, 310005 Харк1в, майдан Повстання, 17,
Харшвська книжкова фабрика im. М, В, Фрунзе.
УкраТна, 310057 Харк1в, вул. Донець-Захаржевського, 6/8,