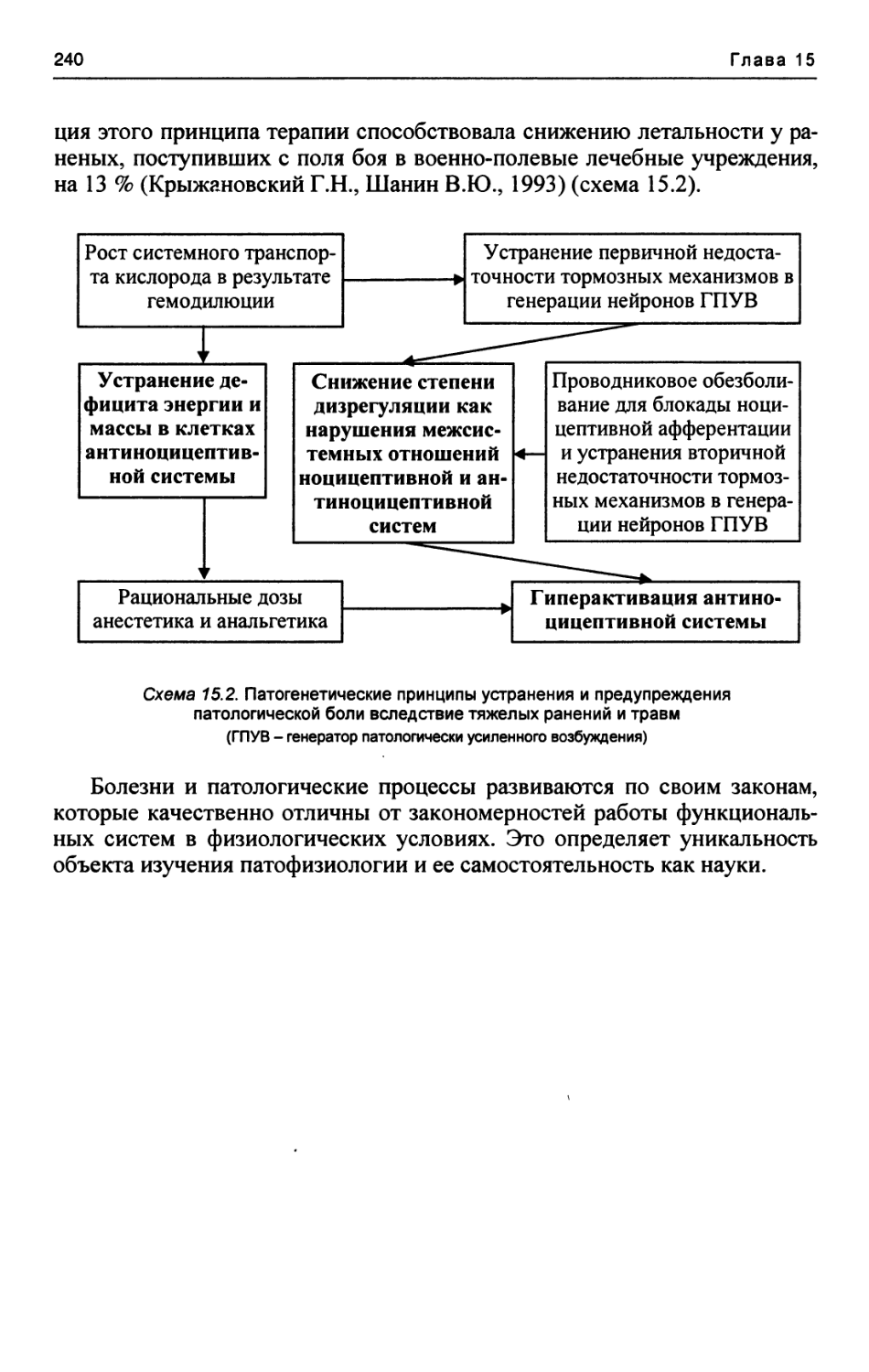
/





























































































































































































































































































































































































































































































































































































Текст
РОССИЙСКАЯ ВОЕННО-МВДИЦИНСКАЯ АКАДЕМИЯ
ФОНД «УЧЕБНАЯ ЛИТЕРАТУРА»
200 ае/н
В. Ю. Шанин
КЛИНИЧЕСКАЯ
ПАТОФИЗИОЛОГИЯ
(Под редакцией Ю. Л. Шевченко)
Учебник для медицинских вузов
Санкт-Петербург
«Специальная Литература»
1998
УДК 616
Ш 22
Учебник одобрен Межвузовским редащионно-издательекнм экспертным советом
по медицинской литературе Санкт-Петербурга и допущен для преподавания
патофизиологии в медицинских высших учебных заведениях
Автор:
В. Ю. Шанин — д. м. н., профессор, начальник кафедры патологической физиологии Российской Военно-
медицинской академии
Редактор:
Ю. Л. Шевченко —r.m.h., профессор, чл.-корр. РАМН, начальник Российской Военно-медицинской
академии
Рецензенты:
Н. А. Гавришева — д. м. н., профессор Санкт-Петербургского медицинского университета;
И. А. Михайлова — д. м. н., профессор Санкт-Петербургского медицинского университета
Шанин В. Ю.
Ш 22 Клиническая патофизиология. Учебник для медицинских вузов.— СПб:
«Специальная Литература», 1998.—569 с.
ISBN 5-86457-063-Х
Учебник клинической патофизиологии — это первое в нашей стране учебное пособие такого
рода. Клиническая патофизиология, взяв на вооружение физиологический метод акад. П. К.
Анохина, исследует общие для разных болезней и патологических состояний закономерности
расстройств взаимосодействия функциональных систем при достижении целей адаптации,
компенсации и поддержании гомеостаза. Такими общими для различных заболеваний механизмами
являются типовые патологические процессы (стереотипные системные патологические реакции),
описание которых вместе с принципами предупреждения и устранения содержит первая часть
учебника. Вторая часть посвящена патофизиологии функциональных систем, то есть описывает
универсальные для различных нозологических форм патогенетические механизмы возникновения
недостаточности конечных полезных приспособительных результатов и трансформации
компенсаторно-защитных реакций в звенья патогенеза. В каждой из глав есть достаточно детальное
описание патогенетических принципов коррекции недостаточности функциональных систем.
Нозологические очерки учебника ограничены в связи с тем, что авторы не ставили себе целью
создать альтернативу современным российским курсам лекций и учебникам патофизиологии, а
лишь дополнить их сведениями, необходимыми для патофизиологического анализа в клинической
практике.
© Шанин В. Ю., J 998
ISBN 5-86457-063-Х © «Специальная Литература», 1998
Профессор Военно-медицинской академии Юрий
Николаевич Шанин - пионер отечественной
анестезиологии, основоположник современной отечественной
послеоперационной интенсивной терапии. В 1983 году
на кафедре патофизиологии ВМедА проф. Ю.Н. Шанин
создал первую в стране лабораторию клинической
патофизиологии. Одновременно начал читать курс
клинической патофизиологии, внедрив метод клинико-пато-
физиологического исследования больного для
преподавания и фундаментальных научных изысканий.
ПРЕДИСЛОВИЕ
Доктор философии и медицины профессор Иоган Петер Франк -
составитель устава Российской Военно-медицинской академии и ее первый
ректор - высказывался о возможности преподавания практическими врачами
в госпиталях следующим образом: «Полагали, будто ординаторы заменят
клинических профессоров и сберегут издержки, употребляемые на
содержание последних. Плохая экономия! Губят научное образование, отнимают
у юношей ежедневно три часа времени, которое ушло бы на усвоение
основ и хотят еще, чтобы выходили хорошие врачи! Дайте ученикам хорошее
клиническое образование и лишь после этого пускайте в госпитали.
Предоставляя практическое образование студентов ординаторам, полагают,
будто последние настолько сведущи, что умеют учить. Так ли бывает на самом
деле? Да и есть ли у них досуг и охота к преподаванию? ... Эмпиризм...,
самое опасное в медицине внушается ученикам в госпиталях».
Эмпиризм и в наши дни представляет определенную опасность, хотя
мы далеки от того, чтобы подозревать наших самоотверженных
клиницистов-преподавателей в невежестве. Дело в том, что степень проникновения
медицинских знаний в патогенез болезней непрерывно возрастает и
является первичной относительно способов лечения. Поэтому объективно
неизбежно отставание теоретического содержания клинической практики от
современных представлений о патогенезе. При этом нельзя игнорировать
того, что в настоящее время основная масса новых фактов патофизиологии
получается через использование новых (в том числе
иммунопатологических и молекулярно-генетических) способов исследования больных. В этой
связи возрастает роль нового исследователя, исследователя-клинициста,
который получает свидетельства эффективности или неэффективности
проводимой терапии, исследуя модуляции патогенеза под влиянием лечения.
В этой связи возникает необходимость клинической патофизиологии,
основоположником которой мы по праву считаем нашего учителя
профессора Юрия Николаевича Шанина.
Представляется возможным выделить следующие основные задачи
клинической патофизиологии:
♦ Исследование общих и частных закономерностей развития болезней
у больного человека. Установление их связи с реактивностью
организма, а также влияния на их реализацию личности врача и
содержания терапии.
♦ Изучение патогенеза конкретной болезни у конкретного больного с
целью повышения эффективности терапии (прикладная задача).
♦ Формирование на определенном этапе развития патофизиологии
патогенетических принципов терапии адекватных современному
уровню представлений о патогенезе.
Инструментом решения этих задач при обучении слушателей Российской
Военно-медицинской академии и является данное руководство. Эту книгу
нельзя считать полным руководством по клинической патофизиологии. Тому
причиной служат исторически сложившиеся в академии традиции
фундаментального преподавания клинической патофизиологии клиницистами-
исследователями в клиниках нервных болезней, психиатрии,
инфекционных болезней, гастроэнтерологии, клинической гематологии. Поэтому
соответствующие разделы в этой книге отсутствуют. Тем не менее, у нас не
было сомнений при направлении рукописи для издательской подготовки.
Мы ориентировались на известное изречение сэра Уильяма Ослера: «Цель
медицинского образования -дать человеку направление, указать путь и
снабдить картой, весьма неполной с точки зрения затеваемого путешествия».
Член-корреспондент РАМН профессор Ю.Л. Шевченко
Профессор В.Ю. Шанин
Часть I
ТИПОВЫЕ
ПАТОЛОГИЧЕСКИЕ
ПРОЦЕССЫ
Глава 1
БОЛЕЗНЬ И ТИПОВОЙ ПАТОЛОГИЧЕСКИЙ ПРОЦЕСС
Болезнь - это ключевое понятие нозологии и общей патологии,
которую в основном составляют патофизиология и патологическая анатомия.
Термин «болезнь» используется для обозначения отдельных заболеваний
(нозологических единиц). Для определения конкретных нозологических
единиц, обладающих спецификой развития при возникновении у разных
больных, необходимо определение болезни как философской категории.
Категория болезни должна вобрать в себя содержание и признаки,
необходимо присущие всем конкретным болезням при их каждый раз новом течении
у разных по реактивности больных.
Болезнь как философская категория, обозначающая определенное
биологическое явление и особый вид жизнедеятельности организма, - это
результат абстрагирования от всей совокупности отдельных нозологических
единиц и всего множества случаев их возникновения и развития у
конкретных больных. В этой связи небесполезно будет привести определение
абстракции, данное К. Юнгом: «Абстракция ... есть извлечение или отвлечение
какого-нибудь содержания (какого-нибудь значения, общего признака и т. д.)
из связного контекста, содержащего еще и другие элементы, комбинация
которых, как нечто целое, является чем-то неповторимым или
индивидуальным и потому не поддающимся сравнению».1
При определении философской категории болезни мы должны извлечь
из всех случаев возникновения и развития конкретных нозологических
единиц те содержание и признаки, которые всегда содержат неповторимые
случаи всех разнообразных болезней у неповторимых по геному и онтогенезу
больных.
Анализ классических и современных определений категории болезни
позволяет выделить основные содержание и признаки, которые всегда есть
при возникновении и течении отдельных заболеваний у каждого больного:
1. Болезнь как объективное явление всегда имеет определенную
первоначальную причину. Причина болезни может быть взаимодействием с чем-
то внешним относительно организма больного (экзогенный
этиологический фактор). Первоначальную причину заболевания может содержать и сам
организм в виде своего взаимодействия с внутренним этиологическим
фактором.
2. Болезнь - всегда системная, то есть всего организма, реакция на
действие этиологического фактора, которую определяют: а) специфика, сила и
длительность действия этиологического фактора; б) реактивность
организма. Реактивность - это генетически детерминированная и находящаяся под
влиянием факторов внешней (физико-химической, биологической и соци-
1 Юнг К. Психологические типы. - СПб.: «Ювента», 1995. - С. 498.
Болезнь и типовой патологический процесс
9
альной) среды совокупность качеств организма, определяющая характер
системной реакции на действие этиологического фактора (Лисицын Ю.П.,
Петленко В.П.)
3. На каждом из этапов своего развития болезнь есть результат
взаимодействия: а) вызванных эффектом этиологического фактора как
раздражителя патологических процессов и патологических реакций, которые могут
привести к нарушениям гомеостазиса или вызывают их; б) защитных
реакций в ответ на угрозу нарушений гомеостазиса или возникшие его
нарушения; в) приспособления к нарушениям гомеостазиса и расстройствам
функциональных систем, связанных с заболеванием; г) компенсации
расстройств функциональных систем, связанных с заболеванием. Защитные,
приспособительные и компенсаторные реакции составляют совокупность
реакций саногенеза, которые через утилизацию резервов самого
организма, восполняемых и повышаемых оптимальной терапией, направлены на
выздоровление и реабилитацию больных.
4. Реакции саногенеза при нормальной реактивности, как правило,
избыточны относительно действия стимулов, их вызвавших, что
обуславливает возможность их превращения в звенья патогенеза патологических
процессов и реакций.
5. На определенном этапе болезнь может потерять связь с
первоначальным этиологическим фактором, и сама через взаимодействие
патологических процессов, реакций и саногенеза может вызывать причины своего
дальнейшего развития (эндогенизация болезни и патологического процесса)
(Крыжановский Г.Н.). При этом развитие болезни начинают
детерминировать патологические системы регуляции, функционирование которых
имеет прямо патогенное значение. Патогенному эффекту таких систем
противостоит функционирование специфических антисистем, которые,
регулируя реакции саногенеза, сами могут стать патологическими системами.
Эндогенизация болезней и патологических процессов, связанная с
взаимодействием патогенных систем регуляции и их антисистем, придает
болезни свойство качественного отличия от физиологических процессов и
состояний организма, при которых функционирование систем регуляции и
функциональных систем направлено на достижение конечного полезного
приспособительного результата.
6. К. Маркс: «Болезнь- есть ограниченная в своей свободе жизнь».1
Н.И. Лосев (1995): «Болезнь - это динамическое состояние организма,
характеризующееся нарушениями нормального течения жизненных
процессов, приводящими к снижению биологических и социальных
возможностей человека».
Типовой патологический процесс - обладающий определенной
последовательностью ряд явлений, который вызывает действие этиологического
фактора. Типовой патологический процесс индуцируется
взаимодействием этиологического фактора и организма. Поэтому его во многом
определяет реактивность.
1 Маркс К., Энгельс Ф. Соч., изд. 2-е. - М, 1955. - Т. 1. - С. 64.
10
Глава 1
На определенном этапе развития патологический процесс может
потерять связь с действием первоначального этиологического фактора, то есть
приобрести свойство эндогенизации. После эндогенизации патологический
процесс содержит внутреннюю причину своего развития в виде
взаимодействия нарушений гомеостазиса и расстройств функциональных систем
и противодействующих им реакций саногенеза. При типовом
патологическом процессе реакции саногенеза часто теряют свое первоначальное
значение и трансформируются в звенья патогенеза типового патологического
процесса и болезни, которую он в данный момент отчасти составляет.
Такая патогенная трансформация реакций саногенеза служит одной из
причин эндогенизации типового патологического процесса. Так при
сепсисе интенсивные межклеточные взаимодействия клеток системы
иммунитета обуславливают гиперцитокинемию, повреждающую органы эффекторы
функций и после того, как патогенные бактерии перестают циркулировать
с кровью. При гипоксии компенсаторная гипервентиляция повышает
потребление кислорода организмом, что обостряет кислородное голодание.
После эндогенизации по ходу смены явлений, в основном
составляющих типовой патологический процесс, происходит и смена его ведущих
звеньев патогенеза. Если сразу после ишемии ведущим звеном ее
патогенеза на уровне клетки можно считать нарушения трансмембранного
переноса ионов, обусловленные гипоэргозом, то после восстановления
артериального кровоснабжения ранее ишемизированных тканей основным
механизмом возникновения клеточных дисфункций и цитолиза является свободно-
радикальное окисление наиболее в функциональном отношении активных
фосфолипидов клеточных мембран. Для типового патологического процесса
после эндогенизации свойственна вариабельность патогенеза на разных
этапах развития.
Типовой патологический процесс могут вызывать различные
этиологические факторы, то есть его характеризует полиэтиологичность.
Патологический процесс - это всегда системная реакция организма. Нельзя
выделить функциональной системы, регуляторные сдвиги и дисфункции в
пределах которой определяют развитие типового патологического процесса на
всех его этапах. Например, гипоксия, ишемия и системная воспалительная
реакция развертываются на разных этапах по-разному через сдвиги супра-
сегментарной и эндокринной регуляции, изменение функционирования
систем внешнего дыхания, транспорта газов с кровью, при
компенсаторных и патологических изменениях периферического кровообращения, а
также вследствие патогенных системных межклеточных взаимодействий.
Поэтому нельзя считать относящимися к типовым патологическим
процессам дыхательные расстройства кислотно-основного состояния, патогенез
которых целиком ограничивается пределами функциональной системы
внешнего дыхания.
В этой связи нельзя признать типовым патологическим процессом и
реакцию повышенной чувствительности первого типа. Ее причина
известна. Это взаимодействие аллергена через антигенную стимуляцию системы
иммунитета с сенсибилизированным организмом. Основное звено патоге-
Болезнь и типовой патологический процесс
11
неза также четко очерчено. Это образование иммунных комплексов на
поверхности тучных клеток как причина их дегрануляции. Известна и
структура комплексов антиген-антитело, состоящих из гомоцитотропных
антител и аллергена. Другие виды реакций повышенной чувствительности
также имеют вполне определенный и специфический круг этиологических
факторов и четко очерченный алгоритм звеньев патогенеза в пределах иммунной
системы. В этой связи не исключено, что аллергические реакции следует
считать не типовым патологическим процессом, а
иммунопатологическими реакциями, которые могут составлять патогенез довольно четко
определенной совокупности болезней.
Типовой патологический процесс сохраняет основную
последовательность составляющего его ряда явлений при развертывании в рамках
различных нозологических единиц. При этом одну нозологическую единицу
могут составлять несколько типовых патологических процессов. При
раневой болезни, сепсисе и системной воспалительной реакции в организме
одновременно происходят воспаление, циркуляторная гипоксия, а также
ускоренное и стрессорное голодание.
Глава 2
НАСЛЕДСТВЕННЫЙ ФАКТОР
В ВОЗНИКНОВЕНИИ И РАЗВИТИИ ЗАБОЛЕВАНИЙ
МУТАЦИИ КАК ПРИЧИНЫ
НАСЛЕДСТВЕННЫХ ЗАБОЛЕВАНИЙ
Основная причина приобретения генотипом свойства вызывать болезни
как дискретные наследственные признаки и обуславливать через экспрессию
в фенотипе предрасположенности к болезни полигенные заболевания - это
мутации (лат. mutatio - изменение, перемена). Мутация - внезапно
возникающее естественное или искусственное изменение генетического материала,
ответственного за хранение генетической информации и ее передачу от клетки
клетке и от предка потомку. Мутации возникают в половых клетках - гаметах
{гаметические мутации) и других клетках организма {соматические
мутации), В зависимости от уровня, на котором происходят мутации, выделяют
геномные, хромосомные и генные мутации. Мутации генов, локализованных вне
цитоплазматического ядра, называют цитоплазматическими.
Если внезапные изменения фенотипа клетки или организма нельзя
связать с обычными генетическими явлениями, а микроскопическое
исследование не выявляет хромосомных аберраций, то причина изменений
фенотипа скрыта на уровне генов. Генная или точечная мутация - результат
изменения нуклеотидной последовательности молекулы дезоксирибонуклеино-
вой кислоты (ДНК) в определенном участке хромосомы. Это изменение
последовательности нуклеотидных оснований в мутантном гене
воспроизводится при транскрипции в структуре информационной
рибонуклеиновой кислоты и меняет последовательность аминокислот в полипептидной
цепи, образующейся в результате трансляции. Существуют различные типы
точечных мутаций, обусловленных добавлением, выпадением или
перестановкой пар нуклеотидов в гене.
В результате мутации появляется новая нуклеотидная
последовательность ДНК, которой не было в генетическом материале гамет родителей
больного. Гаметические мутации аллелей генов переходят в генотип
потомков, в том числе и как причины болезней. Соматические мутации могут
приводить к ряду изменений генома клеток, которые меняют фенотип
клетки таким образом, что она становится злокачественной.
Конформационная мутация - это следствие замены одного нуклеотида
другим в нуклеотидной последовательности ДНК с изменением
пространственного строения ее двойной спирали.
Если мутация меняет генетический материал таким образом, что
меняются молекулярная структура и функции продуктов генов, то она
искажает информацию, хранимую геномом клетки или организма. Такую му-
Наследственный фактор в возникновении и развитии заболеваний
13
тацию называют искажающей биологический смысл наследственной
информации (англ. missense mutation - мутация, искажающая смысл). В этом
контексте под биологическим смыслом понимают функционирование
генетического материала, направленное на поддержание нормальных
функций клеток и всего организма. Искажающую смысл мутацию
составляют изменения кодонов, в результате которых они начинают кодировать
другие аминокислоты. Под не имеющей смысла мутацией (англ. nonsense
mutation) понимают изменение нуклеотидной последовательности ДНК,
нарушающее нормальное транскрибирование кодонов, которые
кодируют информационный сигнал к остановке трансляции с матричной
рибонуклеиновой кислоты (мРНК). Биологического смысла такая мутация не
имеет. Более того, она может изменить экспрессию генов на уровне
трансляции с мРНК.
Точечная мутация представляет собой замену одной пары спаренных
нуклеотидных оснований ДНК на другую. В силу вырожденного кода ДНК
это не всегда искажает информацию, хранимую кодоном, претерпевшим
мутацию. Точечную мутацию, которая вследствие вырожденного кода ДНК
не приводит к изменению информации, хранимой геном, и не меняет
молекулярную структуру его продукта, называют молчащей мутацией. Если
мутация меняет нуклеотидную последовательность ДНК и структуру
продукта гена, то нарушение функции протеина возникает необязательно. Дело в
том, что для модуляции функций белков необходимо изменение структуры
функционально и биохимически активной части белковой молекулы. Если
мутация как изменение структуры кодонов ДНК меняет экспрессию гена
таким образом, что меняется строение активной части молекулы белка
(фермента, элемента рецептора и т. д.), то это вызывает дисфункции
определенного клеточного клона, расстройства функциональных систем и
всего организма.
Точечная мутация может трансформировать кодон, кодирующий
определенную аминокислоту в полипептидной цепи продукта гена, в кодон,
которой при нормальном функционировании генетического материала
кодирует сигнал к прекращению трансляции. В результате мутантный ген
начинает экспрессировать белок с неполноценной абортивной структурой. Тран-
зиционная мутация - это изменение нуклеотидной последовательности
ДНК, состоящее из замены одного пурина на другой (аденина на гуанин, и
наоборот) или одного пиримидина на другой (цитидина на тимидин, и
наоборот). Трансверзионная мутация - это точечная мутация, состоящая в
замене одного пурина на пиримидин, и наоборот.
Мутагены - это воздействия любой природы на генетический
материал, обуславливающие мутации. Выделяют экзогенные и эндогенные
мутагены. К экзогенным мутагенам химической природы (химические
мутагены) относят некоторые пестициды (гербициды, фунгициды, инсектициды),
субстраты промышленного химического синтеза и продукты химической
промышленности (формальдегид, ацетальдегид, уретан, хлорпрен, эпок-
сиды, бензол и др.), а также некоторые лекарственные вещества (ртутные
соединения, коффеин, препараты на основе мышьяка, цитостатики - цик-
14
Глава 2
лофосфамид, митомицин С, 5-фторурацил и др.). Экзогенными
химическими мутагенами могут быть пищевые добавки (цикламаты,
ароматические углеводороды, тартразон). К экзогенным мутагенам физической
природы (физические мутагены) относят все виды ионизирующей радиации и
ультрафиолетовые лучи.
Эндогенные химические мутагены образуются на путях обмена веществ
в организме человека - перекись водорода и липидные перекиси, а также
свободные кислородные радикалы (см. главу, посвященную ишемической
болезни сердца). После фиксации экзогенных радиоактивных элементов в
тканях они становятся источниками физического мутагена,
ионизирующего излучения, вызывающего мутации генома клеток, влияя на него во
внутренней среде.
МОНОГЕННЫЕ И ПОЛИГЕННЫЕ БОЛЕЗНИ
Генетические исследования механизмов возникновения болезней
позволили выделить следующие разновидности заболеваний:
1. Заболевания, целиком детерминируемые генетическим фактором, то
есть влиянием патологического гена. Это моногенно наследуемые
болезни. Их этиологический фактор - один ген, определяющий возникновение
болезни как дискретного наследственного признака.
2. Болезни, причина которых - это взаимодействие организма с
экзогенным этиологическим фактором (травма, инфекция и т. д.). В данном
случае наследственность во многом определяет системную реакцию на
действие этиологического фактора, составляя генетическую основу
реактивности организма. Тем самым наследственность определяет особенности
развития (течения) болезни у конкретного больного.
3. Заболевания, при которых наследственность является ведущим
причинным фактором. При таких болезнях ведущий наследственный
этиологический фактор проявляет себя заболеванием в фенотипе только после
аккумуляции в организме последствий влияний этиологических факторов.
Наследование таких болезней не происходит в соответствии с законами
Менделя (неменделирующие заболевания). Они получили название мулътифак-
ториальных или полигенных.
Наследование моногенных болезней происходит в соответствии с
законами Менделя (менделирующие заболевания). Чаще моногенные болезни
наследуются по аутЬсомно-доминантному типу. В последние годы все
больше появляется свидетельств аутосомно-рецессивного наследования менде-
лирующих болезней.
Большинство моногенных болезней почти не зависит от внешних
факторов и определяется генотипом.
Рассмотрим наследование моногенной болезни как дискретного
наследственного признака по аутосомно-доминантному типу.
Наследственный фактор в возникновении и развитии заболеваний
15
Если один из родителей больного моногенным заболеванием
гетерозиготен по гену болезни, то аутосомный ген болезни представлен одним
рецессивным не вызывающим болезнь аллелем и болезнетворным
доминантным аллелем. Предположим, что второй из родителей гомозиготен по гену
болезни, и он представлен у него двумя непатогенными аллелями. Первый
из родителей страдает от моногенного заболевания, а второй здоров.
Сегрегация аллелей гена болезни при образовании гамет у пары таких
родителей приводит к образованию двух типов гамет, один из которых содержит
болезнетворный аллель. Это определяет вероятность заболевания у детей,
составляющую 50 % (табл. 2.1).
Таблица 2.1
Наследование моногенной болезни по аутосомно-доминантному типу
(генотип одного из родителей содержит патогенный доминантный аллель)
Аллели гена болезни в генотипе
родителей
Возможные варианты содержания
аллелей гена болезни в гаметах
Возможные варианты содержания аллелей
гена болезни в генотипе потомства
(вероятность МБ составляет 50 %)
ДР
(МБ есть в фенотипе)
Д Р
РР
Р Р
ДР ДР РР РР
(МБ есть в фенотипе)
Примечание: МБ - моногенная болезнь, Д - патогенный доминантный аллель, Р - рецессивный
непатогенный аллель
Если генотип каждого из родителей содержит гетерозиготный ген
моногенной болезни, содержащий аллели Д и Р, то через разделение аллелей
образуются три типа гамет, два из которых содержат болезнетворный
аллель. В результате доминирования патогенного аллеля любой из детей
может страдать от заболевания с вероятностью в 75 %. Вероятность избежать
болезни составляет 25 %. Не болеют дети, гомозиготный ген болезни
которых не содержит патогенных аллелей. Прогноз особенно неблагоприятен у
потомства с гомозиготным геном заболевания, который представлен двумя
доминантными патогенными аллелями. Это может служить причиной
раннего возникновения моногенного заболевания, его тяжелого течения и
быстрого летального исхода. Вероятность такого характера моногенной
болезни у потомства в данном случае составляет 25 % (табл. 2.2).
Если доминантный патогенный аллель локализован в аутосомах, то
вероятность моногенной болезни, наследуемой по аутосомно-доминантному
механизму, одинакова у потомства как мужского, так и женского пола.
Каждая следующая беременность матери в семье, где один из
родителей болен моногенным заболеванием, определяемым доминантным ауто-
сомным аллелем, может привести к появлению на свет больного ребенка,
несмотря на то, что старшие дети рождались здоровыми. Доминирование
аутосомных патогенных аллелей не всегда приводит к болезни сразу после
рождения, но всегда вызывает заболевание по ходу онтогенеза.
16
Глава 2
Таблица 2.2
Наследование моногенной болезни по аутосомно-доминантному типу
(генотип каждого из родителей содержит патогенный доминантный аллель)
Аллели гена болезни в генотипе
родителей
Возможные варианты содержания
аллелей гена болезни в гаметах
Возможные варианты содержания аллелей
гена болезни в генотипе потомства
(вероятность МБ составляет 50 %)
ДР
(МБ есть в фенотипе)
Д Р
ДР
Д Р
ДД ДР ДР РР
(тяжелое (МБ нет в фе-
течение нотипе, веро-
МБ) ятность 25 %)
Примечание: МБ - моногенная болезнь, Д - патогенный доминантный аллель, Р - рецессивный
непатогенный аллель
Известно уже около 2000 заболеваний, которые наследуются по
аутосомно-доминантному типу (табл. 2.3).
Таблица 2.3
Некоторые заболевания, наследуемые по аутосомно-доминантному типу
Семейный полипоз толстой кишки
Острая перемежающаяся порфирия
Хорея Хантингтона
Геморрагическая телеангиоэктазия
Гипертрофическая обструктивная кардиомиопатия (гипертрофический
субаортальный стеноз)
Поликистозное наследственное перерождение почек
Брахидактилия (короткопалость)
Бугорчатый склероз
Анонихия (недоразвитие ногтей)
Ахондроплазия
Эллиптоцитоз (аномальное строение эритроцитов)
Врожденная устойчивая куриная слепота
Анонихия часто сопровождается деформацией кистей и стоп, а также
отсутствием пальцев (одного и более). Ахондроплазия - форма
карликовости, при которой голова и туловище достигают обычных размеров, а
конечности сильно укорочены вследствие нарушения роста длинных трубчатых
костей. При ахондроплазии деформировано основание черепа, формируемое из
хряща по ходу онтогенеза. Врожденная устойчивая куриная слепота - это
нарушение сумеречного зрения, которое не прогрессирует с возрастом.
Обратимся к рассмотрению аутосомно-рецессивного механизма
наследования заболеваний. При данном механизме наследования болезнь
определяет рецессивный патогенный аллель гена заболевания.
Если один из родителей здоров и гомозиготен, то есть ген болезни у
него представлен двумя доминантными (непатогенными) аллелями, то все
дети в первом поколении будут здоровыми. Дело в том, что в любом случае
Наследственный фактор в возникновении и развитии заболеваний
17
доминирование нормальных аллелей предотвратит экспрессию
рецессивных болезнетворных альтернативных форм гена. Для возникновения
заболевания как дискретного наследственного признака минимально
необходимым условием выступает гетерозиготность отца и матери, ген
болезни у которых представлен доминантным нормальным и рецессивным
патогенным аллелями. Разделение аллелей для образования гамет в данном
случае приводит к образованию трех типов половых клеток, два из
которых содержат рецессивный болезнетворный аллель. В результате
вероятность заболевания составляет 25 % (табл. 2.4). Болеют дети с
монозиготным геном болезни, который содержит два патогенных рецессивных ал-
леля.
Таблица 2.4
Наследование моногенной болезни по аутосомно-рецессивному типу
Аллели гена болезни в генотипе
родителей
Возможные варианты содержания
аллелей гена болезни в гаметах
Возможные варианты содержания
аллелей гена болезни в генотипе
потомства
ДР
Д Р
ДР
Д Р
ДД ДР ДР РР
(МБ в фенотипе,
вероятность 25 %)
Примечание: МБ - моногенная болезнь, Р - патогенный рецессивный аллель, Д - непатогенный
доминантный аллель
Известно более тысячи заболеваний, наследуемых по
аутосомно-рецессивному механизму. По такому механизму наследуется большинство
врожденных ошибок метаболизма.
Еще в 1908 г. английский врач Гаррод высказал предположение, что
некоторые заболевания и патологические состояния представляют собой
следствия неспособности клетки вырабатывать определенные вещества. Эта
неспособность, согласно Гарроду, обусловлена причинами, связанными с
наследственностью. Гаррод назвал такие болезни «врожденными
ошибками метаболизма». Таким образом, Гаррод был первым, кто предположил
наличие связи между генами и ферментами, хотя он и не употреблял слов
«ген» и «фермент».
«Врожденные ошибки метаболизма» -это патологические состояния
и болезни, наследуемые по аутосомно-доминантному и
аутосомно-рецессивному типам, в основе которых лежат ферментопатии и дисфункции
клеток, вызванные нарушениями экспрессии генов, претерпевших
мутации в ряду предшествующих поколений.
Участок дезоксирибонуклеиновой кислоты, несущий информацию о
синтезе одной полипептидной цепи, следует признать тем минимальным
элементом генотипа или генома, мутации которого приводят к
«врожденным ошибкам метаболизма».
18
Глава 2
Недостаточная экспрессия какого-либо гена или образование его
патологических (патогенных) продуктов обуславливают дисфункции на уровне
клеток, которые в свою очередь служат причинами расстройств
функциональных систем и реакции на них всего организма.
Например, наследственная недостаточность активности энзима мет-
гемоглобинредуктазы как «врожденная ошибка метаболизма»
наследуется по аутосомно-рецессивному типу. В результате этой
наследственной ферментопатии метгемоглобин, непрерывно образуемый при
взаимодействии гемоглобина с кислородом, не восстанавливается до
гемоглобина, обладающего нормальными кислородсвязывающими свойствами.
Метгемоглобин накапливается в эритроцитах, что служит причиной ге-
мической гипоксии. Гемическая гипоксия вызывает неспецифическую
стрессорную реакцию, служит стимулом для роста минутного объема
дыхания и минутного объема кровообращения. Это повышает
потребление кислорода всем организмом, что может обострить гемическую
гипоксию.
ЗАКОН ЧАСТОТЫ ГЕНОВ В ПОПУЛЯЦИИ
И НАСЛЕДОВАНИЕ МОНОГЕННЫХ БОЛЕЗНЕЙ
В 1908 г. английский математик Харди и германский физик Вайнберг
независимо друг от друга открыли закон частоты генов в популяции. При
определении количественных характеристик наследования моногенной
болезни по аутосомно-рецессивному типу в популяции, находящейся в
равновесии, в которой незначительна частота мутаций, а скрещивание имеет
случайный характер, уравнение Харди-Вайнберга выглядит следующим
образом:
p2 + 2pq + q2 = (p+q)2 = l,
где р2 - частота гомозиготных генотипов, содержащих два доминантных
непатогенных аллеля р; 2pq - частота гетерозиготных генотипов, которые
содержат аллели р и q; q2 - частота гомозиготных генотипов, содержащих
рецессивные патогенные аллели q; р + q = 1.
Из уравнения частоты генов следует, что генотип представителей
стабильной популяции довольно часто содержит в аутосомах патогенные
рецессивные аллели. Так, фенилкетонурия, вызываемая патогенным
рецессивным аллелем, возникает, у одного из 10000 жителей развитых стран. Так
как патогенный аллель рецессивен, то частота фенилкетонурии
эквивалентна частоте гомозиготного генотипа, содержащего два болезнетворных
аллеля. В данном случае q2 составляет 1/10 000. Определение значения q (1/100)
необходимо для вычисления частоты во всей популяции гетерозиготных
генотипов, содержащих нормальный доминантный аллель р и
рецессивный патогенный аллель q, то есть 2pq. Для расчета частоты гетерозиготных
Наследственный фактор в возникновении и развитии заболеваний 19
генотипов с патогенным аллелем (здоровые носители патогенного
рецессивного аллеля), вызывающим фенилкетонурию, определяют значение р,
составляющее 1-1/100 = 99/100. Теперь есть все необходимое для
вычисления 2pq, то есть частоты в популяции здоровых носителей патогенного
аллеля. Частота в стабильной популяции генотипа здоровых носителей
патогенного рецессивного аллеля фенилкетонурии составляет:
2pq = 2 х 0,99 х 0,01,
то есть приблизительно 0,02 или 2 %.
Как показывают эти вычисления, частота здоровых носителей
рецессивного патогенного аллеля, всегда выше, чем можно ожидать на
основании оценок частоты моногенной болезни, наследуемой по аутосомно-ре-
цессивному типу. Так, частота гомозиготного генотипа, содержащего два
рецессивных аллеля, вызывающих семейную амавротическую идиотию,
которая ведет к слепоте и смерти, составляет 1/40000, а частота здоровых
гетерозиготных носителей патогенного аллеля - 1/100.
СЦЕПЛЕННОЕ С ПОЛОМ НАСЛЕДОВАНИЕ
МОНОГЕННОЙ БОЛЕЗНИ
При аутосомном доминантном и рецессивном путях наследования
моногенной болезни мутантный патогенный ген (аллель) локализован в
одной из двадцати двух аутосом. Ситуация меняется, если болезнетворный
аллель находится в половой Х-хромосоме. Дело в том, что в Х-хромосоме
имеется участок, для которого в Y-хромосоме мужчин нет гомологичного
сегмента. Поэтому у мужчин менделирующие болезни, определяемые
генами этого участка, возникают и в том случае, если один рецессивный
патогенный аллель локализован в части Х-хромосомы, не имеющего
гомологичного участка в Y-хромосоме.
Предположим, что Х-хромосома здоровой женщины содержит
рецессивный патогенный аллель. Представим, что такая женщина выходит
замуж за здорового мужчину, Х-хромосома которого содержит «нормальный»
доминантный аллель, не имеющий альтернативной формы гена в
Y-хромосоме. Мейоз у таких родителей образует четыре гаметы, одна из которых
содержит патогенный рецессивный аллель. Вероятность того, что генотип
дочек этих родителей, будет содержать рецессивный патогенный аллель,
составляет 50 %. Девочки будут здоровыми, так как проявлению в
фенотипе заболевания будет препятствовать доминирование непатогенного
аллеля, локализованного в парной Х-хромосоме. Если родится мальчик, то
вероятность присутствия в его генотипе патогенного аллеля также
составляет 50 %. Мальчик с генотипом, содержащим рецессивный патогенный
аллель, родится больным, или заболевание возникнет у него в последующем.
Причина заболевания - экспрессия патогенного аллеля, несмотря на его
20
Глава 2
рецессивность. Болезнетворный аллель «некому подавлять», так как
другого аллеля просто нет. Вероятность рождения больного мальчика для данной
пары составляет 25 % (табл. 2.5).
Таблица 2.5
Сцепленное с полом наследование моногенной болезни
Мать Отец
Аллели гена болезни в генотипе
родителей
Возможные варианты содержания
аллелей гена болезни в гаметах
Возможные варианты содержания
аллелей гена болезни в генотипе
потомства
ХХ1
(МБ нет)
X X,
Девочки
XXj XX
X
(МБ нет)
X 0
Мальчики
X X,
(МБ есть)
Примечание: X - непатогенный доминантный аллель, локализованный в половой Х-хромосоме;
X, - патогенный рецессивный аллель, локализованный в гомологичном участке второй
Х-хромосомы; МБ - моногенная болезнь
Предположим, что один из родителей, - это мужчина, страдающий
моногенным заболеванием, вызываемым патогенным рецессивным аллелем.
Болезнетворный аллель локализован в сегменте Х-хромосомы, не
имеющем гомологичного участка в Y-хромосоме. Мать - здоровая гомозиготная
по гену болезни женщина. Ее генотип не содержит патогенного
рецессивного аллеля. Мейоз у таких родителей приведет к образованию четырех
типов гамет, одна из которых будет содержать патогенный рецессивный
аллель. В данном случае патогенные аллели могут присутствовать только в
генотипе дочек таких родителей. Девочки будут здоровыми, так как
экспрессию патогенного аллеля заблокирует нормальный доминантный аллель
гомологичной Х-хромосомы. Если родится мальчик, то его генотип не
будет содержать патогенного аллеля, и ребенок будет здоров.
В данном примере связанного с полом наследования моногенная болезнь
выступает как сцепленный с полом рецессивный наследственный признак.
Поэтому гетерозиготные по гену болезни женщины не страдают от
заболевания. Так бывает при наследовании предрасположенности к гемофилии А и
цветовой слепоте. Если же мутантный патогенный аллель обладает свойством
доминантности, то женщины гетерозиготные по гену болезни болеют
наследственным заболеванием. Это в частности относится к наследованию такой
моногенной болезни, как семейный гипофосфатемический рахит.
Если моногенную болезнь вызывает патогенный рецессивный аллель,
который находится в не имеющем гомологичного сегмента Y-хромосомы
участке Х-хромосомы, то отец не может быть источником болезнетворного
аллеля, вызывающего заболевание у его сыновей. Даже, если мужчина
страдает наследственной болезнью, он передает патогенный аллель вместе со
всей Х-хромосомой только дочкам.
Гемофилия А - сцепленный с полом рецессивно наследуемый
дискретный признак, при котором нарушено образование восьмого фактора свер-
Наследственный фактор в возникновении и развитии заболеваний
21
тывания крови. Ген фактора VIII локализован в участке Х-хромосомы, не
имеющем альтернативной формы в Y-хромосоме. Это означает, что в
отсутствии доминирующих влияний второго непатогенного аллеля мутант-
ный болезнетворный и рецессивный аллель всегда вызывает заболевание у
мужчин. В этой связи можно считать, что последний русский император
Николай Александрович не был источником патогенного аллеля,
определившего гемофилию А у наследника престола, цесаревича Алексея, так как
русский царь от этой болезни не страдал.
Если доминантный патогенный аллель локализован в не имеющем
гомологичного сегмента Y-хромосомы участке Х-хромосомы, то вероятность
страдать от редкого менделирующего заболевания у женщин в два раза
выше, чем у мужчин. Причина состоит в том, что вероятность появления
патогенного аллеля в генотипе женщин (две Х-хромосомы) в два раза выше,
чем вероятность присутствия данного аллеля в генотипе мужчин (одна X-
хромосома). Если частота моногенной болезни в популяции достаточно
высока, то в соответствии с законом Харди-Вайнберга (см. выше) отношение
больных женщин к больным мужчинам меньше, чем 2:1.
Для того, что бы выяснить наследуется ли заболевание по сцепленному
с полом доминантному типу, следует подвергнуть генетическому анализу
потомство больного мужчины. Отцовская Х-хромосома, содержащая
болезнетворный доминантный ген, передается через гамету только дочерям,
которые все будут больны в силу доминирования патогенной мутантной
аллели. В генотипе сыновей она не может присутствовать, и они
рождаются здоровыми. Если такого наследования болезни нет, то предположение о
сцепленном с полом механизме передачи болезни от родителей потомству
следует подвергнуть сомнению.
Такой анализ важен при изучении анамнеза у больных, которым
ставят предположительный диагноз семейного гипофосфатемического
рахита, тяжелого заболевания, в основе которого лежит наследственный
дефект тубулярной реабсорбции фосфата. Генетический анализ в данном
случае позволяет поставить окончательный диагноз, и начать лечение,
состоящее в частности из приема внутрь препаратов, содержащих
фосфатный анион.
НЕПРЕРЫВНАЯ ИЗМЕНЧИВОСТЬ
ПРЕДРАСПОЛОЖЕННОСТИ К ПОЛИГЕННОЙ БОЛЕЗНИ
КАК ФЕНОТИПИЧЕСКОГО ПРИЗНАКА В ПОПУЛЯЦИИ
По признаку предрасположенности к мультифакториальной болезни в
популяции людей можно наблюдать полный ряд переходов от одной
крайности к другой:
♦ полное отсутствие предрасположенности к заболеванию, когда
человек не может страдать от мультифакториального заболевания;
22
Глава 2
♦ максимальная предрасположенность к болезни, когда минимальные
по интенсивности влияния этиологических факторов заболевания
приводят к болезни.
Частотное распределение по признаку предрасположенности к мульти-
факториальной болезни соответствует кривой нормального распределения.
При нормальном распределении фенотипического признака
предрасположенности к болезни большинство членов популяции попадает в
среднюю часть кривой, а на ее концах, соответствующих двум крайним
значениям данного признака, находится одинаковое и малое число особей.
Предрасположенность - это результат как взаимодействия многих
составляющих генотипа, так и аккумуляции в организме последствий влияний на него
этиологических факторов.
Уровень предрасположенности к полигенной болезни, выше которого
она всегда проявляется как фенотипический признак, называют пороговой
предрасположенностью (рис. 2.1).
&
О
X
X
I
§
С
8
&
8.
71
Частота предрасположенности определенной
выраженности в основной популяции
Рис. 2.1. Непрерывная изменчивость предрасположенности к полигенной
болезни среди представителей основной популяции
(л - пороговая предрасположенность)
Болезни, генетическая основа которых определяет непрерывную
изменчивость предрасположенности к данным заболеваниям у представителей
людской популяции, представляют собой реализацию предрасположенностит к ним
как результата совместного действия многих различных генов. Поэтому их
называют полигенными или многофакторными (мультифакториальными).
Предрасположенность к развитию полигенных заболеваний в основе
имеет генетически детерминированную реактивность организма, У
некоторой части популяции предрасположенность по ходу онтогенеза
реализует себя болезнью, как фенотипическим признаком.
Передача предрасположенности к полигенным болезням от родителей;
детям не происходит в отличие от моногенных болезней в соответствии с
заранее для данной нозологической формы известной и строго определен-*
ной вероятностью. Можно считать, что вероятность заболеть мультифакто-;
Наследственный фактор в возникновении и развитии заболеваний
23
риальным заболеванием у детей больного выше, чем у других
представителей популяции. Вероятность моногенной болезни у родственников пробан-
да (больной наследственной болезнью, через которого регистрируется вся
семья) второй и третьей степени (бабушки, дедушки, внуки, дяди, тети,
племянники, сводные братья и сестры, двоюродные братья и сестры)
практически не отличается от вероятности болезни у других представителей
популяции. У родственников больного полигенным заболеванием второй и
третьей степени вероятность заболевания выше, чем у других
представителей популяции, и убывает по мере снижения степени родства. Так у
двоюродных братьев больного с язвенной болезнью желудка (пробанд)
вероятность заболеть ею ниже, чем у племянников пробанда, но выше, чем у
других представителей популяции.
При аутосомно-доминантном и аутосомно-рецессивном типах
наследования моногенных болезней как дискретных наследственных признаков оба
монозиготных близнеца страдают от моногенной болезни, так как
патогенная аллель присутствует в генотипе и того и другого близнеца. Если один
из дизиготных близнецов страдает от моногенного заболевания, то
вероятность болезни у второго идентична ее вероятности у родственников
пробанда первой степени родства: родители, дети, другие сибсы (дети от
одной родительской пары) и дизиготные близнецы. Этого нет при
полигенных болезнях. Если один из монозиготных близнецов заболевает
полигенной болезнью, то ее вероятность у другого не составляет 100 %, так как ее
возникновение детерминируется не только генотипом, но и влияниями на
организм экзогенных этиологических факторов, сила которых у близнецов,
живущих раздельно, может быть различной.
ГЕННЫЙ КОМПЛЕКС ЛЕЙКОЦИТАРНЫХ АНТИГЕНОВ
ЧЕЛОВЕКА И СИСТЕМА АНТИГЕНОВ
ГЛАВНОГО КОМПЛЕКСА ТКАНЕВОЙ СОВМЕСТИМОСТИ
КАК МАРКЕРЫ ПРЕДРАСПОЛОЖЕННОСТИ К БОЛЕЗНЯМ
Генный комплекс лейкоцитарных антигенов человека (система HLA,
англ. human leucocyte antigen - лейкоцитарный антиген человека) - это
система генов специфически присущая генотипу больного, которая во
многом определяет индивидуальность его фенотипа, в том числе и
предрасположенность к разнообразным заболеваниям. При этом вероятность
выявления у сибсов одинаковой по составу данной системы генов составляет 25 %.
Впервые существование системы HLA предположили, когда у сыворотки
крови людей, многократно бывших реципиентами при гемотрансфузиях,
обнаружили свойство агглютинировать лейкоциты человека.
Выделяют три класса генов главного комплекса антигенов тканевой
совместимости, представляющих собой три элемента системы HLA. Первый
класс составляют гены групп А, В и С. Второй - DP, DQ и DR. Третий класс
24
Глава 2
состоит из групп генов С2, BF, С4а и С4Ь. Система генов главного
комплекса антигенов тканевой совместимости (ГТС) находится на коротком плече
шестой хромосомы на расстоянии 32 сантиморганов от ее центромеры.
Антигены ГТС впервые идентифицировали по способности вызывать
реакцию отторжения трансплантата. Они есть у всех видов позвоночных. Гены
первого класса кодируют трансмембранные пептиды с молекулярной
массой 44 кДа, которые на поверхности клеток связаны с бета-два-микрогло-
булином. При этом бета-два-микроглобулин кодируется геном,
находящимся в пятнадцатой хромосоме. Антигены, кодируемые генами второго
класса ГТС, - это трансмембранные димеры. Гены третьего класса кодируют
элементы системы комплемента, участвующие в образовании СЗ-конвер-
таз. Все эти гены обладают огромной внутривидовой изменчивостью, что
обуславливает уникальность набора антигенов на поверхности клеток
одного из представителей определенного вида позвоночных.
Антигены, кодируемые генами первого класса (антигены первого
класса ГТС), экспрессируются геномом практически всех клеток организма
человека. У «молчащих» генов из первого класса нет продуктов,
составляющих структуру клеток и обладающих той или иной функцией, которую
можно выявить при «текущей» жизнедеятельности клетки. «Молчащие» гены
способны увеличивать полиморфизм продуктов генного комплекса ГТС
через действие механизма конверсии (лат. conversio - превращение,
изменение) генов. Конверсия генов - изменение одного из аллелей гена под
влиянием другого или других генов, составляющих генотип организма или
геном клетки. Отдельный кластер (совокупность, состоящая из связанных
между собой элементов) генов из систем HLA и ГТС наследуется как
дискретный наследственный признак, а составляющие его гены, выявляются
при кроссинговере, то есть при обмене генетическим материалом между
гомологичными хромосомами.
Антигены, представляющие собой продукты генов второго класса, -
это протеины (антигены второго класса ГТС), молекула которых состоит
из двух сходных альфа- и бета-цепей с трансмембранными
гидрофильными участками и внеклеточными частями, которые связаны дисульфидной
связью. Эти антигены присутствуют на поверхности смерматозоидов, В-
лимфоцитов, антигенпрезентирующих клеток (моноциты, макрофаги,
клетки Лангерганса и другие клетки системы мононуклеарных
макрофагов), активированных Т-лимфоцитов, эндотелиальных и других клеток;
нормальное функционирование антигенов второго класса - это
необходимое условие эффективности защитной реакции клеточной цитотоксично-
сти. Без нормальной экспрессии антигенов второго класса невозможны
нормальная презентацию антигена иммунокомпетентным клеткам, а
также их ненарушенные взаимодействия, в частности функционирование Т-
хелперов и Т-супрессоров при системной иммунной реакции в ответ на
антигенную стимуляцию.
Существует достоверная связь между разнообразными заболеваниями)
и определенными антигенами, кодируемыми генами из систем HLA и ГТС.
Тесно сцепленные между собой в шестой хромосоме гены HLA распреде-s
Наследственный фактор в возникновении и развитии заболеваний
25
ляются среди всех членов популяции ассоциировано, то есть вместе, а не
случайно, как это бывает при разделении генов в результате рекомбинаций.
Связь между болезнями и генами HLA позволила считать кластеры
аллелей и отдельные аллели генов из ГТС элементами генетического
материала, определяющими предрасположенность к заболеваниям (табл. 2.6).
Таблица 2.6
Связь аллелей генов HLA с некоторыми заболеваниями
Заболевания
Заболевания, связанные с антигенами
второго класса ГТС
Аутоиммунный тиреоидит
Ревматоидный артрит
Герпетиформный дерматит
Хронический аутоиммунный гепатит
Болезнь Аддисона
Инсулин зависимый сахарный диабет
Рассеянный склероз
Болезни, связанные с геном HLA-B27
из первого класса ГТС
Анкилозирующий спондилоартрит (болезнь Бехтерева)
Гонококковый артрит
Болезни, связанные с другими генами
из первого класса ГТС
Псориаз
Миастения
Аллель
гена HLA
DR5
DR4
DR3
DR3
DR3
DR3
DR4
DR2
В27
В27
Cw6
В8
Вероятность
болезни (%)
3,2
5,8
56,4
13,9
6,3
5,0
6,8
4,8
87,4
14,0
13,3
4,4
Существует несколько теорий связи между антигенами HLA и болезнями:
♦ Рецепторная теория, согласно которой сходство молекул антигенов
HLA с рецепторами некоторых эндогенных лигандов обуславливает
конкурентные процессы между ними как причины нарушений
регуляции и дисфункций.
♦ Гипотеза молекулярной мимикрии, то есть наличия антигенов на
поверхности некоторых из возбудителей бактериальных инфекций
сходных по структуре с антигенами HLA. В результате стимуляция
системы иммунитета бактериальными антигенами приводит к
образованию аутоантител и аутоиммунным заболеваниям.
♦ Гипотеза модификации HLA-антигенов, согласно которой
измененные патогенными воздействиями антигены из системы HLA
вызывают аутоиммунную реакцию.
Обычно заболевания, связанные (ассоциированные) с генами HLA,
представляют собой результат иммунопатологических состояний и процессов.
Так, заболевания, ассоциированные с аллелями HLA-D, относятся к
аутоиммунным. При этом с антигеном (и соответственно геном) DR3 связаны
26
Глава 2
органоспецифические аутоиммунные заболевания, патогенез которых во
многом составляет аутоиммунное поражение рецепторов наружной
клеточной поверхности. Предположительно продукты генов HLA, соединяясь с
вирусами, обуславливают проникновение последних в клетки, что через
образование соединений вирусов с аутоантигенами индуцирует
аутоиммунные реакции.
Аутоиммунные заболевания в основном детерминированы генами из
второго класса ГТС. Их связывают с генами иммунореактивности,
определяющими реакцию системы иммунитета на аутоантигены. Например, гены
DR3 и DR4 считаются определяющими аутоиммунные реакции,
приводящие к инсулинзависимому сахарному диабету. Кроме того, полагают, что
аллели генов из группы DR детерминируют системную регуляцию
иммунных реакций, а не системную иммунную реакцию на конкретные
антигены. Схожесть продукта гена В27 с некоторыми бактериальными
антигенами послужила основой для отчасти подтвердившихся предположений о том,
что аутоиммунные заболевания, связанные с В27, обусловлены
перекрестным реагированием антигена В27 с бактериальными антигенами.
ХРОМОСОМНЫЕ МУТАЦИИ
(ИЗМЕНЕНИЯ ЧИСЛА И МАКРОСТРУКТУРЫ ХРОМОСОМ)
Изменения числа и макроструктуры хромосом как причины удаления
из генома клетки, генотипа организма или добавления к ним определенной
совокупности генов могут вызывать патологические состояния клетки
(малигнизация и др.), всего организма, синдромы и болезни.
Полиплоидия - увеличение числа хромосом до величины кратной их
нормальному гаплоидному набору (23 хромосомы). Нормальное
диплоидное число хромосом соматической клетки составляет 46. Триплоидия -
это полиплоидия, при которой общее число хромосом в организме
равняется 69. При этом каждая хромосома представлена в генотипе организма
и геноме клеток своими тремя копиями. Как правило, триплоидия
приводит к спонтанной гибели плода во чреве матери. В противном случае
триплоидия обуславливает тяжелые генетические дефекты анатомического
строения и работы функциональных систем у новорожденных. Тетрапло-
идия - патологическое состояние генотипа организма и (или) генома
клеток, при котором каждая хромосома представлена своими четырьмя
копиями, а общее число хромосом составляет 92. Тетраплоидия в геноме
определенной совокупности клеток плода сочетается с нормальным
диплоидным набором хромосом в другой части и всегда приводит к гибели
плода до рождения ребенка.
Анэуплоидия - рост или снижение общего числа хромосом в генотипе;
организма (геноме клетки) от его нормальной величины, которые не
захватывают каждую хромосому в нормальном гаплоидном наборе хромосом.
Наследственный фактор в возникновении и развитии заболеваний
27
При анэуплоидии может меняться как число аутосом, так и количество
половых хромосом. Трисомия - анэуплоидия, при которой общее число
хромосом в соматических клетках больше их числа в нормальном диплоидном
наборе, так как одна или несколько хромосом представлена своими тремя
копиями. Трисомии во всем генотипе организма могут быть причинами ряда
синдромов, для которых характерны тяжелые дефекты анатомического
строения костей, всего тела и умственного развития. Например, трисомия, при
которой восьмая хромосома представлена своими тремя копиями,
проявляет себя нарушениями развития костей, аномалиями строения позвонков и
костей таза, низкой подвижностью суставов, аномально большой головой
(макроцефалия), задержкой умственного развития и другими дефектами.
Трисомия, при которой генотип содержит три двадцать первые хромосомы
и две половые X хромосомы (общее число хромосом в диплоидном наборе
составляет 48) приводит к появлению к фенотипе как синдрома Дауна, так
и синдрома Кляйнфельтера.
При моносомии одна из хромосом нормального гаплоидного набора
представлена лишь одной копией, что снижает общее число хромосом в
нормальном диплоидном наборе от 46 до 45. Это относительно редкая ци-
тогенетическая аномалия обычно представляет собой снижение общего
числа хромосом группы G (хромосомы 21 и 22). Анэуплоидия может
возникнуть в результате нерасхождения в анафазе мейоза I одной или
нескольких пар гомологичных хромосом. При нерасхождении обе гомологичные
хромосомы одной пары смещаются к одному полюсу клетки. В результате
мейоз приводит к образованию гамет, содержащих на одну или несколько
хромосом больше или меньше, чем нормальные гаметы. Когда гамета с
недостающей или лишней хромосомой сливается с гаметой с нормальным
гаплоидным набором хромосом, то образуется зигота с нечетным числом
хромосом. В такой зиготе нет нормального диплоидного набора
гомологичных хромосом. Она вместо каких-либо двух гомологов содержит три
или только одну хромосому.
Зигота, в которой число хромосом меньше диплоидного, обычно не
развивается. Зиготы с лишними хромосомами часто способны к развитию. Из
таких зигот развиваются больные с тяжелыми наследственными
аномалиями. Одна из наиболее часто встречающихся хромосомных мутаций,
возникающих в результате нерасхождения, - это трисомия, при которой зигота,
из которой развивается организм, содержит три двадцать первые
хромосомы, две из которых не разошлись в анафазе мейоза I (трисомия-21). Данная
трисомия обуславливает синдром Дауна. При синдроме Дауна в части
случаев (около 4 %) выявляют транслокацию (см. главу, посвященную
канцерогенезу) лишней хромосомы двадцать первой пары в четырнадцатую или
двадцать вторую. Симптомами и патологическими состояниями,
связанными с синдромом Дауна, являются задержка умственного развития,
сниженная резистентность по отношению к инфекциям, сердечная недостаточность,
связанная с врожденными сердечными пороками, короткое коренастое
туловище и толстая шея, характерные складки кожи над внутренними углами
глаз («монголоидный разрез глаз»). Синдром Дауна и хромосомные мута-
28
Глава 2
ции сходные с трисомией-21 чаще встречаются у детей, рожденных
немолодыми женщинами.
Если нерасхождение при мейозе двух половых X хромосом приводит к
образованию в организме матери гаметы, содержащей две X хромосомы,
которую оплодотворяет спермий, содержащий Y-хромосому, то генотип
ребенка содержит две X хромосомы и одну Y хромосому. Такой генотип (XXY)
в фенотипе проявляет себя синдромом Кляйнфельтера. Мужчина с
синдромом Кляйнфельтера бесплоден, его яички развиты слабо, волос на лице у
пациента мало, а молочные железы развиты избыточно. При этом
умственное развитие больного замедлено.
Синдром Тернера обусловлен нерасхождением половых хромосом X и
Y при мейозе с образованием гамет отца больного. В результате
оплодотворения материнской гаметы, содержащей одну Х-хромосому, спермием, не
содержащим половых хромосом, образуется генотип больного с одной X-
хромосомой. У больных женщин с синдромом Тернера отсутствуют
обычные половые признаки, длина тела снижена, а соски сближены.
Если при мейозе у матери происходит нерасхождение половых
хромосом, то оплодотворение материнской гаметы, содержащей две половые
хромосомы спермием, гаплотип которого содержит одну Х-хромосому,
образует генотип ребенка XXX (синдром трисомии X). Больные женщины с
таким синдромом внешне нормальны, плодовиты, но отсталы в
умственном отношении.
Глава 3
ГИПОКСИЯ
Гипоксия (hypoxia; гип- + лат. oxygenium, кислород) - типовой
патологический процесс, который вызывают недостаточное поступление
кислорода в ткани и клетки организма или нарушения его использования при
биологическом окислении. Гипоксия всегда приводит к недостатку
свободной энергии, гипоэргозу.
Различают экзогенный и эндогенный виды гипоксии (табл. 3.1).
Экзогенная гипоксия связана с изменением парциального давления кислорода
во вдыхаемой газовой смеси. Эндогенную гипоксию вызывают
расстройства внешнего дыхания, транспорта кислорода с кровью и нарушения
тканевого дыхания.
Таблица 3.1
Этиопатогенетическая классификация гипоксии
Экзогенная гипоксия
Гип оксиче екая
Гипероксическая
Эндогенная гипоксия
Респираторная
Гемическая
Циркуляторная
Тканевая
Гипероксическая гипоксия возникает вследствие патогенно высокого
парциального давления кислорода во вдыхаемой смеси газов, которое
обуславливают: а) рост содержания кислорода во вдыхаемой газовой смеси;
б) увеличение давления (барометрического, атмосферного) смеси газов.
Гипероксическая гипоксия - это следствие токсичного действия
кислорода при его аномально высоких парциальном давлении в альвеолярной
газовой смеси и напряжении в артериальной крови и тканях. Под
токсичным действием кислорода понимают повреждения тканей, клеток и
межклеточного матрикса, обусловленные свободнорадикальным окислением.
Токсическому действию кислорода особенно подвержены старики, у
которых со старением падает активность антиоксидантных систем, в частности
ферментов супероксиддисмутазы, каталаз и пероксидазы. Недостаточный
транспорт кислорода с кровью при травматическом шоке предрасполагает
к токсическому эффекту кислорода, вызывая через свободнорадикальное
окисление и дефицит свободной энергии недостаточность
антиоксидантных систем.
При гипероксической гипоксии высокое напряжение кислорода в
тканях ведет к окислительной деструкции митохондриальных структур, что
снижает эффективность улавливания клеткой свободной энергии при
биологическом окислении. У гипероксической гипоксии весьма сложный па-
30
Глава 3
тогенез, так как она обуславливает разнообразные сдвиги обмена веществ
вследствие высокого напряжения кислорода в тканях. Прежде всего
происходит инактивация многих энзимов, особенно тех, что содержат сульф-
гидрильные группы. Одним из следствий системной ферментопатии при
гипероксической гипоксии выступает падение содержания в мозге гамма-
аминобутирата, главного тормозного медиатора серого вещества, что
обуславливает судорожный синдром кортикального генеза. Высокое
напряжение кислорода в тканях приводит к усиленному образованию свободных
кислородных радикалов, нарушающих образование дезоксирибонуклеино-
вой кислоты, и тем самым извращающих внутриклеточный синтез белка.
Кроме того, свободнорадикальное окисление фосфолипидов клеточных
мембран как фактор первичной альтерации тканей служит инициирующим
моментом воспаления. При увеличении парциального давления кислорода во
вдыхаемой газовой смеси в первую очередь патологические изменения
возникают в легочной паренхиме, в которой в наибольшей степени возрастают
напряжение кислорода и образование свободных кислородных радикалов.
Токсический эффект кислорода может клинически значимо проявить
себя уже при возрастании парциального давления кислорода во вдыхаемой
газовой смеси до 200 мм рт. ст., если больной непрерывно дышит такой
смесью в течение нескольких часов. При парциальных давлениях
кислорода во вдыхаемой смеси газов меньших, чем 736 мм рт. ст., гистотокси-
ческий эффект высокого напряжения кислорода в тканях приводит в
основном к воспалительным изменениям легочной ткани, а у некоторых
больных вызывает некардиогенный отек легких. Кроме того, у больных
выявляют диффузное микроателектазирование легких из-за разрушения
свободнорадикальным окислением системы сурфактанта. Дыхание газовой
смесью, парциальное давление кислорода в которой выше, чем
4416 мм рт. ст., приводит к тонико-клоническим судорогам и потере
сознания в течение нескольких минут. Резистентность по отношению к
гистотоксическому эффекту высокого напряжения кислорода в тканях
снижает наследственная недостаточность антиоксидантных систем клетки,
в частности низкая активность ферментов гйютатионпероксидазы и
супероксиддисмутазы.
Высокие парциальное давление кислорода в альвеолах и его
напряжение в артериальной крови и тканях являются патогенными
раздражителями, которые вызывают дисфункции на уровне респиронов. В основном эти
расстройства складываются из нарушений легочной микроциркуляции,
обусловленных спазмом микрососудов в ответ на избыточные нервные регуля-
торные влияния. Это индуцирует патологическую вариабельность венти-
ляционно-перфузионных отношений структурно-функциональных
элементов легких, а через активацию эндотелия легочных микрососудов предрас-1
полагает к воспалению, лишенному биологической цели, вторичная
альтерация при котором разрушает респироны.
Гипоксическую гипоксию вызывает ограничение транспорта кислорода
с кровью от легких на периферию из-за артериальной гипоксемии вследствие;
низкого парциального давления кислорода во вдыхаемой газовой смеси.
Гипоксия
31
Содержание кислорода в атмосферном воздухе остается неизменным и
составляет 20,9 % при подъеме от уровня моря до высоты в 100000 м над
ним. Атмосферное давление при подъеме падает, что обуславливает
неуклонное снижение парциального давления кислорода во вдыхаемом
воздухе. Это приводит к падению напряжения кислорода в артериальной крови.
Артериальная гипоксемия ограничивает транспорт кислорода с кровью
через снижение насыщения кислородом гемоглобина. Гипервентиляция,
стимулом для которой служит возбуждение периферических хеморецепторов,
не может восстановить кислородной емкости крови на высотах,
превышающих 3500 м над уровнем моря. Более того, интенсификация энерготрат в
результате гипервентиляции повышает потребление кислорода всем
организмом. Это ведет к дальнейшему снижению парциального давления
кислорода в альвеолярной газовой смеси и его напряжения в артериальной
крови. На таких высотах напряжение кислорода в артериальной крови как
правило ниже 50 мм рт. ст., если только не обогащать вдыхаемую газовую смесь
кислородом.
Компенсаторная гипервентиляция при адаптации к гипоксической
гипоксии на больших высотах вызывает респираторный алкалоз. Для
компенсации респираторного алкалоза растет экскреция бикарбонатного
аниона почками. Для поддержания электронейтральности мочи вслед за би-
карбонатным анионом в ее состав устремляется катион натрия. Организм
теряет натрий. Снижение содержания натрия в организме влечет за собой
снижение объема внеклеточной жидкости вплоть до гиповолемии
(недостаточного объема циркулирующей плазмы).
Интенсификация физической нагрузки обостряет артериальную гипок-
семию при гипоксической гипоксии, так как увеличивает потребление
кислорода организмом.
Гипоксическая гипоксия может быть следствием низкого содержания
кислорода во вдыхаемой газовой смеси. У больных концентрация
кислорода во вдыхаемой смеси газов чаще всего падает в результате ятрогенных
дефектов искусственной вентиляции легких.
Респираторная гипоксия возникает тогда, когда транспорт
кислорода с кровью падает и не соответствует потребностям клеток и тканей в
кислороде вследствие артериальной гипоксемии, обусловленной расстрой-
ствами внешнего дыхания. Респираторную гипоксию чаще всего
вызывают гиповентиляция альвеол, нарушения диффузии свободных молекул
кислорода через легочную мембрану, патологическая вариабельность венти-
ляционно-перфузионных отношений структурно-функциональных единиц
легкого (респиронов) и отделов легких (самая частая причина
респираторной гипоксии у больных), а также патологическое шунтирование
смешанной венозной крови в легких.
Гемическая (кровяная) гипоксия выступает результатом уменьшения
кислородной емкости крови в результате: а) дефицита объема циркулирующих
эритроцитов и низкой концентрации гемоглобина в крови; б) снижения кис-
лородсвязывающих свойств гемоглобина. Кровяную гипоксию, которая
развивается вследствие дефицита объема циркулирующих эритроцитов, назы-
32
Глава 3
вают анемической. Гемическая гипоксия развивается не только в результате
блокады кислородсвязывающих свойств гемоглобина, но может быть
следствием избыточного сродства гемоглобина к кислороду, снижающего
восстановление гемоглобина и транспорт кислорода в клетку на периферии.
Врожденные гемоглобинопатии, которые служат причиной гемической
гипоксии подразделяют на два вида:
♦ гемоглобинопатии, вызывающие цианоз;
♦ гемоглобинопатии с изменением сродства гемоглобина к кислороду.
Если кислород на периферии высвобождается в виде супероксидного
аниона, не возвращая электрон атому железа, то образуется метгемогло-
бин, неспособный к обратимому связыванию кислорода. Образование
метгемоглобина идет непрерывно, и у здорового человека в метгемоглобин
превращается 1 % всего гемоглобина. Большему образованию
метгемоглобина противостоят антиоксидантные системы клеток, в частности каталазы и
глютатиона. Рост содержания метгемоглобина до 15 г/л и более вызывает
гемическую гипоксию и цианоз. Патологически высокая концентрация
метгемоглобина в крови может быть следствием врожденных нарушений
реакций деструкции метгемоглобина, содержания в крови патологических
форм гемоглобина, устойчивых по отношению к физиологическим
механизмам их разрушения и элиминации, а также токсического окисления
двухвалентного иона железа молекулы восстановленного гемоглобина.
При врожденных гемоглобинопатиях, сопровождающихся цианозом,
вследствие изменения аминокислотного состава молекулы гемоглобина в
участке, прилегающим к гему (образование гемоглобина М), окисление
железистой группы гемоглобина приводит к образованию метгемоглобина,
чаще, чем при окислении нормального гемоглобина. В результате
замещения в крови гемоглобина способного к переносу кислорода метгемоглоби-
ном падает кислородная емкость крови.
Побочные эффекты ряда лекарств и токсинов могут привести к
патологическому росту содержания в крови метгемоглобина (нитриты и нитраты,
анилиновые красители и сульфаниламиды). К образованию
метгемоглобина в результате побочного действия лекарственных веществ и токсических
эффектов химических соединений существует наследственная
предрасположенность.
Известно более чем 20 видов врожденных дефектов гемоглобина, при
которых кривая диссоциации гемоглобина патологически сдвинута влево,
что приводит к недостаточному высвобождению кислорода из соединения
с гемоглобином на периферии и гипоксии.
Угарный газ (окись углерода, СО) обладает сродством к гемоглобину в
240 раз большим сродства к нему кислорода. При парциальном давлении
СО во вдыхаемой газовой смеси, составляющем 1/240 парциального
давления в ней кислорода, окись углерода блокирует временное соединение
кислорода с гемоглобином, образуя с ним относительно стойкое соединение,
карбоксигемоглобин. В результате кровь содержит оксигемоглобин в
количестве недостаточном для адекватного транспорта кислорода. Токсический
эффект СО вызывает патологический сдвиг кривой диссоциации оксиге-
Гипоксия
33
моглобина влево, что обостряет гипоксию вследствие недостаточного
содержания оксигемоглобина в крови.
Тканевой тип гипоксии характеризует снижение способности клеток
использовать кислород для биологического окисления. Кроме того, под
тканевой гипоксией традиционно понимают системный гипоэргоз,
обусловленный падением эффективности улавливания клеткой свободной энергии,
высвобождаемой при биологическом окислении, что обычно связано с
разобщением окисления и фосфорилирования.
Использование клетками кислорода для биологического окисления
нарушают: а) снижение активности энзимов цепи дыхательных ферментов
митохондрий; б) нарушения гомеостазиса, а также изменения
внутриклеточной среды, блокирующие аэробное окисление; в) нарушения синтеза
ферментов, участвующих в аэробном биологическом окислении, и
разрушение наружной клеточной и цитоплазматических, в том числе и митохон-
дриальных, мембран.
Следует заметить; что патологические изменения среды обитания
клеток и внутриклеточной среды, обусловленные гипоэргозом вследствие
гипоксии, (лактатный метаболический ацидоз, рост содержания в клетке и в
интерстиции аденозинмонофосфата и др.) сами по себе тормозят
активность ферментов аэробного биологического окисления и растормаживают
анаэробный гликолиз. Когда локальная гипоксия обусловлена ишемией, то
из тканей и клеток в состоянии гипоксического гипоэргоза не удаляются с
кровью как протоны, так и аденозинмонофосфат (АМФ). В результате
концентрации в клетке свободных ионов водорода и АМФ растут в такой
степени, что это тормозит уже последний для клетки источник свободной
энергии, анаэробный гликолиз.
Нарушение способности клеток утилизировать кислород для
биологического окисления и улавливать при нем свободную энергию в виде
макроэргов, связанное с разрушением аппарата тканевого дыхания в
результате гипоксии-ишемии, называют вторичной тканевой гипоксией.
Во многом вторичная гипоксия связана со свободнорадикальным
окуклением наиболее в функциональном отношении активных фосфолипидов
клеточных мембран, в том числе и митохондриальных. Свободнорадикаль-
ное окисление мембранных фосфолипидов предельно интенсивно после
восстановления кровотока в ранее гипоксичных или ишемизированных
тканях. При этом в тканях высока концентрация таких субстратов синтеза
свободных кислородных радикалов как протоны, и есть второй его субстрат,
кислород. В результате угнетения антиоксидантных систем гипоэргозом
образование свободных кислородных радикалов при восстановлении
притока артериальной крови после ишемии почти не ограничено.
Кроме свободнорадикального окисления фосфолипидов клеточных
мембран к их деструкции может приводить широкий спектр патогенных
воздействий, повреждающих клетку: гипо- и гипертермия, многочисленные
экзогенные яды, проникающая радиация и т. д.
Согласно Н.И. Лосеву, ингибирование ферментов биологического
окисления как причина тканевой гипоксии происходит по трем основным пу-
34
Глава 3
тям. Первый состоит в специфическом связывании активных центров
дыхательных ферментов токсичными соединениями со способностью
стойко связывать активные центры энзимов через специфическую реакцию,
субстратами которой являются активный центр молекулы фермента и
токсичный агент. Такими токсичными соединениями являются цианиды,
содержащие токсичный ион циана, а также соединения, диссоциирующие с
высвобождением токсичного сульфидного иона, некоторые антибиотики и
др. Второй - это неспецифическое связывание функциональных групп
белковой части молекулы фермента ионами тяжелых металлов и в
результате реакции с алкилирующими агентами. Третий заключается в
конкурентном торможении вследствие блокады активного центра ферментом
«псевдосубстратом». «Псевдосубстрат» - это соединение, которое
образуется на естественных путях метаболизма в норме или при их расстройствах,
и, не являясь субстратом определенного энзима, блокирует его активный
центр. Примером тут может служить ингибирование сукцинатдегидроге-
назы малоновой и другими дикарбоновыми кислотами.
Нарушения синтеза ферментов, участвующих в биологическом
окислении, могут быть следствием качественного витаминного голодания,
которое обуславливает недостаток в организме специфических нутриентов,
необходимых для образования этих энзимов: витаминов группы В, тиамина,
никотиновой кислоты и др. Алиментарная дистрофия как состояние
системной недостаточности экспрессии генома клетки также нарушает синтез
дыхательных ферментов.
Снижение эффективности улавливания клеткой свободной энергии при
аэробном биологическом окислении часто происходит через снижение
сопряженности окисления и фосфорилирования на дыхательной цепи
ферментов в митохондриях. При этом потребление кислорода растет, но
аккумуляция свободной энергии в виде макроэргов падает, и ее значительная
часть рассеивается в виде тепла. Разобщают аэробное биологическое
окисление и фосфорилирование внутриклеточный ацидоз, избыток в клетке
ионизированного кальция, неэстерифицированных жирных кислот, а
также избыточное влияние на клетку адреналина и гормонов щитовидной
железы. Таким свойством обладают и микробные токсины. Разобщение
окисления и фосфорилирования может быть побочным эффектом
лекарственных веществ (антибиотики и др.).
Если нет грубых нарушений легочного газообмена и транспорта газов с
кровью, напряжение кислорода в артериальной крови у больных с
тканевой гипоксией находится в нормальных пределах при почти максимальном
насыщении кислородом в ней гемоглобина. При этом растет напряжение
02 в смешанной венозной крови, и падает различие артериальной и
смешанной венозной крови по содержанию в них кислорода.
Выделяют три уровня адаптации к гипоксии (табл. 3.2).
Если доставка кислорода тканям и клеткам становится неадекватной
их потребностям вследствие снижения минутного объема кровообращения,
концентрации гемоглобина в крови или расстройств легочного газообмена,
то растет экстракция тканями кислорода из капиллярной крови. В резуль-
Гипоксия
35
Таблица 3.2
Уровни адаптации к гипоксии
Уровень
Увеличение поглощения
кислорода легкими, несмотря на низкое
парциальное давление кислорода
во вдыхаемой газовой смеси
Увеличение транспорта
кислорода в клетки, несмотря на
артериальную гипоксемию
Увеличение способности клеток
утилизировать кислород для био-
логич еского о кисления и улавли-
вать свободную энергию при
биологическом окислении, несмотря
на низкое напряжение кислорода
в их цитозоле и митохондриях
Механизмы адаптации
Гипервентиляция и адекватный ей рост
минутного объема кровообращения. Рост содержания
эритроцитов в крови. Повышение кислородной
емкости крови, в частности связанное с изменением
содержания в эритроцитах 2,3-дифосфоглицерата
Расширение артериол и раскрытие прекапилляр-
ных сфинктеров в ответ на рост концентрации
протонов, увеличение содержания в клетке адено-
зин монофосфат а и действие других местных
факторов расширения микрососудов, обусловленное
гипоэргозом клеток. Мобилизация резерва
капилляров вследствие увеличения объемной скорости
кровотока на уровне микрососудов. Снижение
диффузионного расстояния для кислорода между
просветом микрососудов и митохондриями за счет
мобилизации резерва капилляров, увеличения
количества митохондрий и изменений структуры
клеточных мембран. Увеличение градиента
напряжения кислорода между просветом микрососудов и
клетками за счет роста содержания в клетках
миоглобина, обратимо связывающего кислород
Рост сродства конечного фермента цепи
дыхательных энзимов митохондрий цитохромоксидазы
к кислороду. Увеличение количества митохондрий
в клетках. Повышение эффективности улавливания
клеткой свободной энергии при анаэробном
гликолизе
тате снижается напряжение кислорода в смешанной венозной крови. При
падении напряжения кислорода в смешанной венозной крови до 30 мм рт. ст.
и ниже клетки начинают улавливать свободную энергию в ходе
анаэробного биологического окисления, что приводит к аккумуляции в них и во
внеклеточной жидкости молочной кислоты.
При полном угнетении транспорта кислорода на уровне всего организма
при условии достаточного содержания в клетках глюкозы при анаэробном
гликолизе образуются 3600 ммоль лактата в час. Внеклеточная жидкость
содержит всего 375 ммоль бикарбонатного аниона, которые не могут связать
протоны, высвобождаемые при диссоциации молочной кислоты. Это ведет
к стремительному прогрессированию лактатного метаболического
ацидоза, который сам по себе обуславливает цитолиз (гибель клеток) еще
до того, как причинами гибели клеток становятся другие следствия
гипоксии.
При хронических циркуляторной и гемической гипоксии
компенсаторный сдвиг вправо кривой диссоциации оксигемоглобина приводит к
большему высвобождению кислорода на периферии при восстановлении окси-
36
Глава 3
гемоглобина. Сдвиг кривой диссоциации оксигемоглобина вправо
вызывают: а) рост содержания в клетках 2,3-Дифосфоглицерата; б) ацидоз.
Достижение максимального уровня транспорта кислорода клеткам и
тканям возможно при значениях гематокрита от 40 до 50 %. При
компенсаторном возрастании содержания в крови эритроцитов в ответ на
хроническую гипоксию увеличение гематокрита до 55 % приводит к росту
транспорта кислорода от легких на периферию. Дальнейшее увеличение
содержания эритроцитов и гематокрита приводит к падению минутного объема
кровообращения из-за роста общего периферического сосудистого
сопротивления и расстройств микроциркуляции в результате увеличения
вязкости крови.
У больных частой причиной возникновения и обострения гипоксии
служит усиление несоответствия между доставкой кислорода в клетки, ткани
и резко возросшей в нем потребностью всего организма вследствие
интенсификации аэробного обмена на системном уровне. Потребности
организма в кислороде меняются в зависимости от потребления кислорода на уровне
всего организма. У больных острое повышение потребности в кислороде в
частности вызывают судорожный синдром, мышечная дрожь и лихорадка.
Особенно потребность в кислороде возрастает при диэнцефально-катабо-
лическом синдроме у молодых больных после черепно-мозговых ранений
и травм, при котором возникают одновременные лихорадка и судороги.
Примером хронически повышенной потребности тканей в кислороде
служит состояние аэробного обмена у больных с гипертиреозом.
Наиболее ранний и эффективный механизм аварийной компенсации
остро возросшей потребности клеток в кислороде - это возрастание
минутного объема кровообращения. В условиях относительного покоя
минутный объем кровообращения может возрастать в три-четыре раза, что в 3-4
раза повышает системный транспорт кислорода. При компенсации
гипоксии, связанной с высокой потребностью в кислороде при физической
нагрузке, именно рост минутного объема кровообращения служит детерми-
нантой как максимального поглощения кислорода легкими, так и
максимального потребления кислорода на периферии.
Гипервентиляция (рост минутного объема дыхания) так же быстро
развивается в ответ на развитие гипоксического состояния, как и возрастание
минутного объема кровообращения. Если только у больного нет
критических обструктивно-рестриктивных нарушений альвеолярной вентиляции, то
снижение напряжения углекислого газа в артериальной крови выступает
нормальным признаком срочной «аварийной» компенсации гипоксии. В этой
связи следует считать возвращение значений напряжения углекислого газа
в пределы средне-статистической «нормы» у больных с респираторной
гипоксией признаком прогрессирования недостаточности внешнего дыхания,
что у больных в астматическом статусе служит показанием для
искусственной вентиляции легких.
Напряжение кислорода в артериальной крови прямо пропорционально
его поглощению легкими и обратно пропорционально потреблению
кислорода организмом. Если интенсификация внешнего дыхания не приводит к
Гипоксия
37
росту поглощения кислорода легкими, которое соответствует возрастанию
потребления кислорода организмом, то напряжение кислорода в
артериальной крови падает. При сниженных функциональных резервах внешнего
дыхания и кровообращения быстрое и значительное возрастание
потребления кислорода организмом приводит к респираторной гипоксии.
Одновременно с ростом потребления кислорода растет образование
углекислого газа. При ограничении роста выделения углекислого газа
вследствие обструктивно-рестриктивных расстройств альвеолярной вентиляции
повышение его парциального давления в альвеолярной газовой смеси
снижает в ней парциальное давление кислорода, и артериальная гипоксемия
прогрессирует.
При хронически и патологически высоком потреблении кислорода
организмом (тиреотоксикоз, акромегалия) устойчивый компенсаторный сдвиг
кривой диссоциации оксигемоглобина вправо обусловлен ростом
содержания в эритроцитах 2,3-дифосфоглицерата (ДФГ). Этот неорганический
фосфат образуется в эритроцитах при реализации там цикла биохимических
реакций Раппопорта-Люберинга. ДФГ связывается с бета-цепью молекулы
восстановленного гемоглобина, снижая его сродство к кислороду, что
служит причиной большего восстановления гемоглобина на периферии с
увеличением доставки кислорода непосредственно в клетку. Кроме того, ДФГ
сдвигает кривую диссоциациии оксигемоглобина вправо через снижение
рН в эритроцитах.
Основной этиологический фактор горной болезни - это снижение
парциального давления кислорода в альвеолярной газовой смеси,
обусловленное низким парциальным давлением кислорода во вдыхаемой газовой
смеси. Предрасполагают к развитию острой горной болезни большие дозы
ультрафиолетового облучения и сверхинтенсивные физические нагрузки при
восхождении. От горной болезни страдают 30 % неадаптированных к
высотной гипоксемии людей после быстрого подъема на высоту большую,
чем 3000 м над уровнем моря. У 75 % неприспособленных субъектов
симптомы острой горной болезни выявляют после быстрого подъема на
высоту, превышающую 4500 м над уровнем моря. Головная боль как первый
признак начала развития горной болезни связана со спазмом сосудов
головного мозга в ответ на падение напряжения углекислого газа в
артериальной крови в результате компенсаторной гипервентиляции,
обуславливающей гипокапнию, но не устраняющей артериальной гипоксемии. Когда
напряжение кислорода в артериальной крови не больше, чем 60 мм рт. ст.,
то значительный гипоэргоз церебральных нейронов, несмотря на
противодействие системы ауторегуляции локальной скорости мозгового
кровотока, обуславливает расширение артериол и раскрытие прекапиллярных
сфинктеров в системе микроциркуляции мозга. В результате увеличивается
кровоснабжение головного мозга, что повышает внутричерепное давление и
проявляет себя головной болью..
Компенсаторная гипервентиляция у страдающих горной болезнью на
высотах в диапазоне 3000-4500 м над уровнем моря вызывает
респираторный алкалоз и бикарбонатурию как компенсаторную реакцию на снижение
38
Глава 3
содержания протонов и рост концентрации бикарбонатного аниона во
внеклеточной жидкости и клетках. Бикарбонатурия усиливает натрийурез и,
снижая содержание в организме натрия, уменьшает объем внеклеточной
жидкости и даже обуславливает гиповолемию. При подъеме на высоты, на
которых компенсаторные реакции в ответ на гипоксическую гипоксию не в
состоянии предотвратить связанного с ней гипоэргоза клеток,
гипервентиляция через повышение потребления кислорода организмом обостряет
системный гипоэргоз. Усиление системного гипоэргоза на уровне всего
организма повышает интенсивность анаэробного гликолиза, что вызывает
метаболический лактатный ацидоз типа А.
Патологически низкое парциальное давление кислорода во вдыхаемой
газовой смеси служит стимулом для «альвеоло-капиллярного рефлекса» с
еще не выявленным центральным звеном. В эфферентном звене, на уровне
эффектора, рефлекс сужает легочные венулы и артериолы, что
обуславливает легочную первичную как венозную, так и артериальную гипертензию.
Легочная артериальная гипертензия может приводить к острой правожелу-
дочковой недостаточности в результате патогенно высокой постнагрузки
правого желудочка.
Полагают, что легочная венозная гипертензия и отрицательные нервные
влияния на легочную паренхиму в ответ на гипоксическую гипоксию
обуславливают некардиогенный отек легких как осложнение горной болезни.
При этом отрицательные нервные влияния на легкие в частности приводят
к росту экспрессии флогогенного потенциала клеточных эффекторов
воспаления, локализованных в легких, то есть эндотелиоцитов, нейтрофилов,
тучных клеток и клеток системы мононуклеарных фагоцитов {не имеющее
биологического смысла нейрогенное воспаление).
Глава 4
НАРУШЕНИЯ ОБМЕНА ВОДЫ И НАТРИЯ
Организм здорового человека за сутки теряет минимум 0,8 л воды. Для
восполнения этих потерь и предупреждения задержки воды в организме
необходимо ненарушенное действие ряда физиологических механизмов.
♦ Нормальное функционирование центра жажды. При оптимальном
функционировании центра рост осмоляльности жидкой части
плазмы крови на 1-2 % выше нормального уровня вызывает сильную
жажду.
♦ Оптимальная секреция антидиуретического гормона (АДГ).
Высвобождение АДГ задней долей гипофиза растет в ответ на увеличение
осмоляльности внеклеточной жидкости, снижение объема
циркулирующей крови. Рост секреции АДГ может быть элементом защитно-
патогенной стрессорной реакции.
♦ Достаточная способность почек разводить или концентрировать
мочу. О нормальном функциональном состоянии почек
свидетельствуют выделение минимального количества максимально
концентрированной мочи при секреции АДГ, а также усиление диуреза и
падение осмоляльности мочи в ответ на падение концентрации АДГ
в плазме крови.
Вода составляет 60 % массы тела (МТ) здорового и не страдающего
от оэюирения муэючины. Две трети общего содерэюания воды в организме
приходятся на внутриклеточный жидкостной сектор, а одна - на
внеклеточный (табл. 4.1). В зависимости от того, какое вещество, не
мигрирующее во внутриклеточный сектор, разводят во внеклеточной жидкости для
расчета ее объема (ОВнЖ), величина ОВнЖ варьирует от 0,27 до 0,45
общего содержания воды в организме. Вода почти незатрудненно
перемещается из внутриклеточного во внеклеточный жидкостной сектор и обратно.
Осмоляльность внутриклеточной и внеклеточной жидкости одинакова, так
как вода быстро мигрирует из клеток в интерстиций и обратно под
влиянием градиента осмотических концентраций. Вследствие быстрого
перемещения Н20 через наружные клеточные мембраны сдвиги содержания в
организме молекул мочевины, этилового спирта и других веществ, легко
проникающих из интерстиция в клетку и свободно выходящих из нее, не
влияют на распределение воды между жидкостными секторами организма.
Так как вода свободно проходит через плазматическую клеточную
мембрану, то выделение клеточной и внеклеточной дегидратации как видов
обезвоживания всего организма представляется не совсем верным. Если
организм теряет свободную воду из интерстиция, то потери Н20 без натрия
происходят одновременно из внеклеточного жидкостного сектора и
клеток. При этом быстрая миграция воды в сторону большей осмотической
концентрации в интерстиций, повышенной потерями воды без потерь на-
40
Глава 4
Таблица 4.1
Жидкостные секторы организма взрослых
Жидкостной сектор
Внекчеточная жидкость
Вода плазмы крови
Вода в интерстиции
Внутриклеточная жидкость
Общее содержание воды в организме
Мужчина (МТ - 70 кг)
Женщина (МТ - 70 кг)
% от массы тела
20
4
16
40
60
50
Объем, л/70 кг
14
2,8
НД
28
42
35
трия во внешнюю среду, уменьшает содержание свободной Н20 в клетках
одновременно с обезвоживанием внеклеточного пространства.
Если организм из внутренней среды теряет жидкость с осмоляльнос-
тью более низкой, чем осмоляльность плазмы и внеклеточной жидкости
(повышенное потоотделение, вследствие спонтанной и искусственной
гипервентиляции легких), то осмоляльность внеклеточной жидкости и
содержание в ней натрия растут. Рост содержания натрия ведет к перемещению
воды из клеток во внеклеточный сектор, что предотвращает дефицит ОВнЖ.
Поэтому до появления симптомов гиповолемии как следствия дефицита
ОВнЖ (ортостатическая артериальная гипотензия, запустевание
периферических вен и др.), организм теряет значительную массу свободной воды.
В результате клетки страдают от обезвоживания, находясь в среде
обитания с патологически низким содержанием свободной воды. О потерях воды
из внутренней среды и клеток, преобладающих над потерями из организма
натрия, у таких больных свидетельствует сильная жажда, которая должна
служить показанием к экстренной коррекции патогенно высокой осмоляль-
ности жидкости в клетках и во внеклеточном пространстве.
Основная функция главного внеклеточного катиона Na+ и
сопутствующих ему во внеклеточной жидкости хлоридного и бикарбонатного
анионов - это удержание воды во внеклеточном жидкостном секторе. Внутри
клеток воду удерживают главный внутриклеточный катион К+ и макромо-
лекулярные органические анионы, покидающие клетки только после
цитолиза.
Концентрация натрия во внеклеточной жидкости ([Na*]) - это
критерий объема внутриклеточного жидкостного сектора (рис. 4.1). Ее рост
говорит о сужении данного сектора, а падение свидетельствует о
наводнении клеток. Рост содержания во внеклеточной жидкости глюкозы, приводя
к миграции воды из клеток во внеклеточное пространство, может снижать
[Na+] без роста содержания Н20 в клетках.
Детерминантой ОВнЖ выступает общее содержание натрия в
организме, а не [Na+]. Так, у больных с застойной сердечной недостаточностью
содержание натрия во внеклеточной жидкости и жидкой части плазмы
крови часто снижено в результате падения водного диуреза. Одновременно с
падением ренальнои экскреции воды у таких пациентов растет задержка
Нарушения обмена воды и натрия
41
Снижение массы доступной
для функциональных систем
внеклеточной жидкости
Сужение жидкостных
секторов организма и
гипернатриемия
Г о 1!Го'if о 1
У. У.Ч ■ А \ J.
( €Э );[ © 1Г@1
........ ..V111 ■ 11*1 Г?11111 г.
|. ....... 71111Г. ГТТТГГ .J
\
Потери во внешнюю среду
или секвестрация в
организме натрия и свободной воды
из внеклеточной жидкости
Потери свободной воды
из внеклеточного сектора
и клеток
\
%ш
нею
GO
1 V 1
Го):
т
к Л.1
Падение водного диуреза,
преобладающее над
снижением натрийуреза
\
Рост содержания в организме
натрия при неограниченном
поступлении во внутреннюю
среду свободной воды
N.
1,'У* "Ч
?:Г © J
; О 1'
:[ О }
v s
:fo1;
' ' ' ' ' ^
:[ © ]:
-Na+
Of
©
lf©l
о
Г©
Расширение жидкостных
секторов, отек клеток и
гипонатриемия
Увеличение объема
внеклеточной жидкости
Рис. 4.1. Общее содержание натрия в организме как детерминанта объема
внеклеточной жидкости и [Na+] как детерминанта содержания воды в клетках
почками в организме натрия, что повышает общее содержание катиона в
организме и объем внеклеточной жидкости.
Потери натрия вместе с водой во внешнюю среду (наружное
кровоизлияние) и (или) его секвестрация в организме (в просвете кишки при
острой кишечной непроходимости, в некробиотически измененных клетках
после минно-взрывных ранений, вследствие внутреннего кровоизлияния и
т. д.) снижают массу натриевых ионов как детерминанту ОВнЖ. В
результате возникает дефицит объема внеклеточной жидкости. Дефицит ОВнЖ у
42
Глава 4
таких пациентов устраняют внутривенным вливанием изоосмоляльных по
отношению ко внеклеточной жидкости (ВнЖ) растворов.
У тяжелых хирургических больных потери натрия вместе с водой
часто возникают одновременно с потерями воды без потерь натрия.
Например, у больного с перитонитом возможны одновременные
патологическая секвестрация части ВнЖ в просвете кишечника и потери гипоосмо-
ляльной жидкости в результате интенсивного потоотделения, связанного
с лихорадкой. Поэтому у пациентов в тяжелом состоянии часто выявляют
дефицит ОВнЖ при высоком содержании натрия во внеклеточной
жидкости. Это может служить показанием к одновременной внутривенной ин-
фузии изо- и гипоосмоляльных относительно нормальной осмоляльнос-
ти плазмы растворов.
ГИПОНАТРИЕМИЯ
Гипонатриемия - это патологическое состояние, которое
характеризует снижение содержания натрия в сыворотке крови до уровня меньше-
го, чем 135 ммоль/л, как критерия его концентрации во внеклеточной
эюидкости. Это наиболее частое из расстройств водно-солевого обмена, которое
выявляют у 1 % больных, находящихся в стационаре.
Гипонатриемию вызывают:
♦ избыточное поступление воды в организм при сохранной
экскреторной функции почек (редкая причина);
♦ снижение способности почек к водному диурезу, то есть к
выделению свободной воды (частая причина).
Общую массу выделяемой почками воды составляют осмоляльный
клиренс и клиренс свободной воды.
Осмоляльный клиренс - это объем воды, разведение в котором всех
осмотически активных молекул, выделяемых с конечной мочой за единицу
времени, дает жидкость с осмоляльностью ВнЖ. Осмоляльный клиренс
представляет собой выделяемую почками во внешнюю среду часть
внеклеточной эюидкости, которая содержит все подлежащие ренальной экскреции
растворенные и находящиеся в конечной моче вещества.
Клиренс свободной воды - это объем конечной мочи, из которого не-
фроны удалили все растворенные в нем вещества для генерации гипото-
ничной относительно плазмы крови и ВнЖ эюидкости (мочи). Если из
общего объема конечной мочи вычитают ее осмоляльный клиренс, то получают
клиренс свободной воды. Рост осмоляльного клиренса и падение клиренса
свободной воды ведут к гипонатриемии.
Есть три необходимых условия нормального водного диуреза.
1. Нормальная скорость клубочковой фильтрации без избыточной ре-
абсорбции воды в проксимальных отделах нефрона как необходимое
условие достаточного для разведения мочи (водного диуреза) поступления жид-
Нарушения обмена воды и натрия
43
Таблица 4.2
Виды гипонатриемии
Вид
гипонатриемии
Гиповоле-
мический
Эуволеми-
ческий
Гиперво-
леми чес-
кий
Симптомы
- снижение т>ргора кожи
- сухая кожа и высохшие
слизистые оболочки
- ортостатическая артериальная
гипертензия
- нет клинических признаков
обезвоживания и гиповолемии
- набухание подкожных шейных
вен
- периферические отеки
(в области нижних третей голени,
крестца и в др. местах)
Данные специальных
исследований
- рост гематокрита и концентрации
белка в сыворотке крови
- увеличение отношения содержания
в сыворотке крови азота мочевины
крови (АМК) к концентрации в ней
креатинина (КК)
- содержание натрия в моче меньше,
чем 20 ммоль/л
- кроме низкой концентрации натрия в
сыворотке крови, других признаков нет
- снижение гематокрита, содержания
белка в сыворотке крови и АМК/КК
кости по почечным канальцам в отделы нефрона, осуществляющие водный
диурез. Водный диурез осуществляют восходящее колено петли Генле и
проксимальный отдел дистального извитого канальца нефрона.
2. Нормальное функционирование сегментов нефрона, разводящих
жидкость в просвете канальцев.
3. Прекращение или низкий уровень секреции АДГ, действие которого
вызывает реабсорбцию воды из просвета собирательных канальцев
нефрона, концентрирует конечную мочу и препятствует ренальной экскреции
свободной воды.
Выделяют три вида гипонатриемии: гиповолемический (с
дефицитами объемов циркулирующей крови и ее плазмы); эуволемический, при кото-
ром объемы циркулирующей крови и ее плазмы находятся в нормальных
пределах, и гиперволемический. При гиперволемической гипонатриемии она
развивается одновременно с патологическим возрастанием объемов
внеклеточной жидкости и циркулирующей крови (табл. 4.2).
Под изоосмолярной гипонатриемией понимает псевдогипонатриемию,
связанную с увеличением твердого остатка плазмы крови в результате
роста в ней содержания липидов или белка. Строго говоря, при
определении содержания натрия в сыворотке крови, мы определяем не его
содержание во внеклеточной жидкости, находящейся в сосудистом секторе, а
концентрацию катиона в единице объема плазмы. Чем больше будет
твердый остаток, тем больше будет превалировать концентрация натрия во
внеклеточной жидкости над его содержанием в единице объема плазмы
крови, то есть над концентрацией натрия в сыворотке. Поэтому у
больных с гиперлипидемией и гиперпротеинемией (макроглобулинемия Валь-
дёнстрёма, множественная миелома и другие патологические состояния
и болезни), которые существенно снижают содержание внеклеточной
44
Глава 4
жидкости и воды в плазме крови, концентрация натрия в ее сыворотке
перестает быть критерием его содержания во внеклеточной жидкости и
всегда ниже [Na+]. Изоосмоляльную гипонатриемию вследствие гиперли-
пидемии выявляют тогда, когда концентрация триглицеридов в плазме
крови больше, чем 1,5 г/л.
Внутривенные инфузии гиперосмоляльных по отношению к
внеклеточной жидкости растворов, которые содержат молекулы, не проникающие в
клетки (маннитол и др.), через увеличение осмоляльности внеклеточной
жидкости вызывают перемещение в нее воды из клеток, разводя в ней
натрий и приводя к гиперосмоляльной гипонатриемии.
У большинства больных с патологическим снижением концентрации
натрия в сыворотке крови осмоляльность внеклеточной жидкости
снижена, то есть гипонатриемия является гипоосмолялъной.
Гиповолемическую гипонатриемию обуславливают почечные и/или вне-
почечные потери натрия как ведущего элемента ионного скелета
внеклеточной жидкости. Наиболее частая причина гиповолемической
гипонатриемии у больных - это побочное действие мочегонных.
Мочегонные, увеличивая экскрецию из организма натрия, могут
снизить ОВнЖ, что у части пациентов приводит к падению скорости клубочко-
вой фильтрации и усиленной реабсорбции натрия в проксимальных
отделах нефрона. В результате падает доставка натрия и воды в сегменты
нефрона, осуществляющие водный диурез, а значит и выделение почками
Н20. Задержка воды в организме ведет к гипонатриемии.
Фуросемид (лазикс), этакриновая кислота и диуретики тиазидовой
группы угнетают реабсорбцию натрия в сегментах нефрона, ответственных за
разведение конечной мочи водой. Это увеличивает осмоляльный клиренс и
снижает клиренс свободной воды. Увеличение осмоляльного клиренса без
восполнения потерь натрия и воды может служить причиной дефицита
ОВнЖ без изменения осмоляльности внеклеточной жидкости. Снижение
клиренса свободной воды, одновременное с ростом осмоляльного
клиренса, вызывает гипонатриемию, которая при определенной степени
дефицита ОВнЖ может стать гиповолемической.
Чаще всех других мочегонных гипонатриемию вызывают диуретики
тиазидовой группы. Тиазиды, угнетая реабсорбцию натрия в дистальном
извитом канальце, повышают осмоляльность конечной мочи, осмоляльный
клиренс и снижают экскрецию свободной воды. При этом в отличие от
мочегонных, действующих на петлю нефрона, они не ограничивают
активный транспорт ионов через стенку канальцев на уровне петли, то есть не
препятствуют водному диурезу.
Гиповолемическую гипонатриемию может обусловить снижение
разведения мочи и ограничение потребления натрия хлорида с пищей у
больных с гипертонической болезнью. Поражение почек вследствие
хронической артериальной гипертензии снижает водный диурез. Снижение водного
диуреза повышает осмоляльный клиренс и выделение натрия с мочой. Рост
потерь натрия с мочой при ограничении его поступления во внутреннюю
среду с пищей служит причиной гиповолемии.
Нарушения обмена воды и натрия
45
Гиповолемическую гипонатриемию иногда вызывают поражения
почечного интерстиция при поликистозном перерождении почек, кистозных
изменениях их мозгового слоя, а также в результате хронического
пиелонефрита. У таких больных поражение почек приводит к одновременным
патологическим потерям с мочой натрия и снижению водного диуреза, что
снижает объем внеклеточной жидкости и вызывает гипонатриемию.
При низкой секреции альдостерона и обладающего свойствами ми-
нералкортикоида кортизола вследствие снижения реабсорбции Na+B не-
фронах растет экскреция натрия почками, увеличивается осмоляльный
клиренс и падает водный диурез. Это приводит к снижению содержания
натрия в организме, тем самым вызывая дефицит объемов
внеклеточной жидкости и плазмы крови. Одновременно падение водного диуреза
вызывает гипонатриемию. Гиповолемия снижает скорость клубочковой
фильтрации (СКФ). Падение СКФ также ведет к гипонатриемии,
которую усиливает защитно-патогенная реакция роста секреции
антидиуретического гормона (АДГ) в ответ на гиповолемию и падение минутного
объема кровообращения.
У больных с осмотическим диурезом осмоляльность конечной мочи
повышают молекулы, которые, находясь во внеклеточном пространстве, не
мигрируют в клетки, но попадают в состав ультрафильтрата (глюкоза и др.).
Это повышает осмоляльный клиренс и ограничивает водный диурез, что
служит причиной гиповолемической гипонатриемии.
При эуволемической гипонатриемии у больных нет как признаков
дефицитов ОВнЖ и объема циркулирующей крови, так и периферических
отеков, то есть задержки воды в интерстициальном жидкостном секторе. У
больных с эуволемической гипонатриемией общее содержание воды в
организме обычно повышено на 3-5 л. Причинами гипонатриемии данного
вида выступают:
♦ избыточная секреция АДГ,
♦ аномально сильное действие аргинин-вазопрессина как гормона,
♦ потенцирование эффекта антидиуретического гормона.
Одну из этих эндокринопатий всегда следует считать причиной
гипонатриемии, при которой нет:
♦ гиповолемии;
♦ наводнения интерстициального жидкостного сектора как причины
периферических отеков;
♦ сниженного уровня секреции гормонов щитовидной железы;
♦ почечной недостаточности;
♦ гипонатриемии как следствия побочного эффекта лекарственных
средств;
♦ болезни Аддисона.
При эуволемической гипонатриемии в результате действия АДГ на эпи-
телиоциты собирательных канальцев растет осмоляльность конечной мочи
и концентрация в ней натрия становится большей, чем 20 ммоль/л.
Часто к эуволемической гипонатриемии у больных ведет синдром
неадекватной секреции антидиуретического гормона.
46
Глава 4
Синдром неадекватной секреции АДГ характеризуют угнетение
водного диуреза, снижение осмоляльности внеклеточной жидкости и рост
почечного осмоляльного клиренса. Эти нарушения водно-солевого обмена
возникают в результате а) превращения защитной неспецифической реакции
роста секреции АДГ в звено патогенеза нарушений водно-солевого обмена
или б) (предположительно) патогенной секреции подобных АДГ пептидов
клетками злокачественных новообразований или тканями, в норме не
высвобождающими АДГ.
Синдром неадекватной секреции АДГ выявляют у больных с
пневмониями, абсцессами и туберкулезом легких, бронхиальной астмой, при
тяжелой артериальной гипоксемии и аспергиллезе легких. Не исключено, что
эти опасные заболевания и патологические состояния обуславливают
длительное действие спектра сильных и неотвратимых раздражителей как
стимулов хронического патогенного стресса с неадекватно высоким уровнем
секреции АДГ.
Синдром неадекватной секреции АДГ выявляют у больных с
мелкоклеточным (овсяноклеточным) раком легкого, ходжкинской и неходжкинской
лимфомой, при раке двенадцатиперстной кишки и поджелудочной железы,
а также при тимоме.
Заболевания и травмы центральной нервной системы, нарушая внутри-
центральные отношения и выступая индукторами системной патогенной
стрессорной реакции, ведут к синдрому неадекватной секреции аргинин-
вазопрессина. Синдром вызывают менингит, энцефалит, абсцесс
головного мозга, черепно-мозговая травма, опухоли головного мозга, субарахнои-
дальные и субдуральные кровоизлияния, острая перемежающаяся
порфирия, острые психозы и белая горячка, нарушения мозгового
кровообращения, множественный склероз и др.
У небольшой части больных эуволемическая гипонатриемия возникает
и без нарушений водного диуреза. Такую гипонатриемию определяют как
эссенциальную или «синдром слабой клетки». Полагают, что в основе
синдрома лежит «перенастройка установочной точки» осморецепторов
гипоталамуса, в результате которой конечным результатом функциональной
системы поддержания осмоляльности жидких сред организма выступает ее
новый уровень, более низкий, чем обычный. В результате такой
перестройки рост содержания натрия во внеклеточной жидкости относительно вновь
установленного уровня конечного приспособительного результата служит
стимулом для секреции АДГ и ведет к выделению концентрированной мочи.
Обильное питье или внутривенное введение гипоосмоляльных
относительно внеклеточной жидкости растворов не меняют [Na+], которую
удерживает на прежнем низком уровне усиление водного диуреза через
прекращение секреции АДГ. Патогенез эссенциальной эуволемической гипонатрие-
мии остается не вполне ясным.
У части больных с выраженной эмоциональной лабильностью
эуволемическая гипонатриемия выступает следствием полидипсии как
результата нарушения внутрицентральных отношений, в том числе связанного и с
острым психозом.
Нарушения обмена воды и натрия
47
Таблица 4.3
Лекарственные средства, вызывающие гипонатриемию
Механизм индукции гипонатриемии
Потенцирование действия АДГ
Прямая стимуляция секреции АДГ
Прямая и непрямая стимуляция
секреции АДГ и потенцирование
действия аргинин-вазопрессина
Лекарственные средства
ацетаминофен (парацетамол)
нестероидные противовоспалительные
средства
аналоги АДГ
винкристин
карбамазепин (тегретол) ,/*t>&M*z£eAug'&s*
наркотические анальгетики
барбитураты
хлорпропамид
циклофосфамид (циклофосфан)
диуретики из группы тиазидов
Некоторые из лекарственных средств, чье действие меняет регуляцию
водно-солевого обмена через модуляцию секреции АДГ и его эффекта на
соответствующий сегмент нефрона, вызывают эуволемическую
гипонатриемию (табл. 4.3).
Если у больных возникает патологическое наводнение интерстициаль-
ного жидкостного сектора, которое обуславливают застойная сердечная
недостаточность, нефротический синдром, цирроз печени как причина
портальной гипертензии и асцита, другие болезни и патологические состояния,
то общее содержание в организме воды растет в большей степени, чем
содержание в нем натрия. В результате развивается гиперволемическая гипо-
натриемия, которую можно определить как гипонатриемию вследствие
снижения объема циркулирующей плазмы крови.
Все болезни и патологические состояния, выступающие причинами так
называемой гиперволемической гипонатриемии, ведут к сужению внутри-
сосудистого жидкостного сектора. При застойной сердечной
недостаточности миграция жидкости из сосудов в интерстиций в частности
вызывается ростом гидростатического давления в капиллярах. Снижение
коллоидно-осмотического давления плазмы крови у больных с нефроти-
ческим синдромом ведет к перемещению воды из сосудистого сектора в
межклеточные пространства. Результатом такого перераспределения воды
между жидкостными секторами выступает падение объема
циркулирующей плазмы крови, которое снижает минутный объем кровообращения
через падение преднагрузки сердца. Снижение объема циркулирующей
плазмы и минутного объема кровообращения уменьшает СКФ и служит
стимулом защитно-патогенной реакции усиления реабсорбции воды в
проксимальных отделах нефрона. Все это обуславливает снижение клиренса
свободной воды как причину гипонатриемии. Часто сопутствующая
застойной сердечной недостаточности хроническая почечная недостаточность
усиливает гипонатриемию, снижая доставку воды и натрия в сегменты
нефрона, осуществляющие водный диурез. При гипонатриемии вследствие
снижения минутного объема кровообращения и объема циркулирующей
48
Глава 4
плазмы в результате активации ренин-ангиотензин-альдостеронового
механизма концентрация натрия в моче обычно ниже 15 ммоль/л. Несмотря
на снижение концентрации натрия в конечной моче, ее осмоляльность
вследствие падения клиренса свободной воды растет выше 350 мосм/кг Н20. По
мере прогрессировать хронической почечной недостаточности вследствие
застойной сердечной недостаточности почки менее интенсивно реабсор-
бируют натрий в ответ на действие альдостерона. В результате в моче
растет содержание натрия.
При быстром снижении содержания натрия во внеклеточной жидкости
до 120 ммоль/л и ниже (острая тяжелая гипонатриемия), вероятность
летального исхода составляет 50 %. Причина смерти у больных с острой
тяжелой гипонатриемией - отек нейронов головного мозга. Его основная
причина - это падение осмоляльности внеклеточной жидкости. Наводнение
нейронов быстро приводит к опасному подъему в них давления, которое
может быть индуктором процесса цитолиза. Рост внутриклеточного
давления в нейронах вследствие гипергидратации особенно значителен, так как
клеточные элементы головного мозга находятся в замкнутом пространстве
полости черепа. При хронической гипонатриемии риск отека нейронов ниже,
благодаря тому, что защитная реакция роста выведения из клеток натрия
успевает снизить степень гипергидратации церебральных нейронов.
Первыми из симптомов гипонатриемии о ее развитии свидетельствуют
сонливость, тошнота, рвота, психомоторное возбуждение, слабость и
головные боли. По мере прогрессирования гипонатриемии к ним
присоединяются галлюцинации. Острая тяжелая гипонатриемия вызывает судороги,
кому, патологические рефлексы, псевдобульбарный паралич и дыхание
Чейн-Стокса, представляющие собой следствия отека головного мозга.
Следует заметить, что до падения концентрации натрия в сыворотке
крови на уровень более низкий, чем 120 ммоль/л, гипонатриемия обычно
не проявляет себя перечисленными симптомами.
Для эффективной коррекции гипонатриемии в ходе интенсивной
терапии необходима идентификация ее причин в соответствии с алгоритмом
дифференциальной диагностики (схема 4.1).
Если гипонатриемия приводит к параличам и коме, то она служит
показанием к неотложной интенсивной терапии, патогенетически
ориентированной на подъем концентрации натрия во внеклеточной жидкости,
снижающий отек церебральных нейронов. При этом ближайшей целью
терапии посредством внутривенной инфузии гипертонического раствора
натрия хлорида служит подъем концентрации катиона в сыворотке крови
до уровня, который выше, чем 120 ммоль/л. Несмотря на свой
неотложный характер, такая коррекция гипонатриемии должна быть очень
осторожной, так как может вызвать летальное осложнение, центральный ми-
елинолиз в области моста. Центральный миелинолиз в области моста -
это деструкция мозговых оболочек в центре основания моста.
Клинические проявления центрального миелинолиза в области моста варьируют от
минимальных дисфункций нервной системы до вялой тетраплегии. Риск
центрального миелинолиза в области моста особенно велик, если при кор-
Нарушения обмена воды и натрия
49
Определение осмоляльности сыворотки крови (ОСК)
Изоосмоляльная
гипонатриемия
(ОСК - 280-295 мосм/кг Н,0)
Псевдогипонатриемия
Внутривенная инфузия изо-
осмоляльных по отношению
к ВнЖ растворов веществ,
не проникающих в клетки
(глюкоза, маннитол и др.)
Гипоосмоляльная
гипонатриемия
(ОСК< 280 мосм/кг Н20)
Оценка ОВнЖ и
состояния жидкостных
секторов организма в
соответствии с табл. 4.1.
Гиперосмоляльная
гипонатриемия
(ОСК>295 мосм/кг КЛ)
Внутривенная инфузия ги-
перосмоляльных
относительно ВнЖ растворов
веществ, не мигрирующих
через наружные клеточные
мембраны
Гиповолемическая
гипонатриемия
т
Определение
концентрации в моче натрия
(КНМ) и
осмоляльности мочи (ОМ)
Эуволемическая гипонатриемия
Гипотиреоз
Побочное действие лекарств
Синдром неадекватной секреции АДГ
Гипокалиемия
Эссенциальная гипонатриемия
Психогенная полидипсия
Гиперволемическая
гипонатриемия
Определение
концентрации в моче натрия
(КНМ) и
осмоляльности мочи (ОМ)
КНМ<20 ммоль/л
ОМ>400 мосм/кг Ц,0
Внепочечные потери
натрия в результате:
♦патогенно
усиленного потоотделения;
♦секвестрации
внеклеточной
жидкости в просвете
кишечника и
некробиотически
измененных тканях
КНМ>20 ммоль/л
ОМ<400 мосм/кг Hfl
Рост ренальной
экскреции натрия
вследствие:
♦ интерстициального
нефрита;
♦низкого уровня
секреции альдостерона
и глюкокортикоидов;
♦осмотического
диуреза
КНМ<20 ммоль/л,
ОМ>350 мосм/кг Н,0
Гипонатриемия
вследствие снижения
объема циркулирующей
плазмы крови, которое
вызывают:
♦ нефротический
синдром;
♦цирроз печени и асцит,
♦застойная сердечная
недостаточность
КНМ>20 ммоль/л,
ОМ<350 мосм/кг Н,0
Хроническая
почечная недостаточность
Схема 4.1. Алгоритм патогенетически обоснованной
дифференциальной диагностики гипонатриемии
рекции гипонатриемии содержание натрия в сыворотке возрастает на
25 ммоль/л в течение 48 ч после начала коррекции гипонатриемии в ходе
интенсивной терапии.
Высока вероятность центрального миелинолиза при коррекции
гипонатриемии у больных, перенесших острую гипоксию того или иного вида.
Гипоксия как причина нарушений трансмембранного транспорта ионов
предрасполагает к отеку нейронов головного мозга вследствие
гипонатриемии, снижая эффективность защитных изменений переноса ионов через
наружную клеточную мембрану.
50
Глава 4
Для предотвращения острой ятрогенной гипернатриемии скорость
возрастания содержания натрия в сыворотке крови следует ограничить
верхним пределом в 2,0 ммоль/л/ч.
При интенсивной терапии больного с гипонатриемией прежде всего
необходимо ограничить объемы выпиваемой воды и внутривенные инфу-
зии гипоосмоляльных по отношению к плазме крови растворов. Цель
интенсивной терапии у больных с гипонатриемией - это снижение уровня
секреции антидиуретического гормона. При гиповолемической гипонатри-
емии внутривенной инфузией изоосмоляльных относительно внеклеточной
жидкости растворов устраняют дефицит ОВнЖ как причину усиления
секреции АДГ. Внутривенное вливание кристаллоидных растворов сочетают
с инфузией в вену растворов альбумина, декстранов и гидроксиэтилкрах-
мала, которые не мигрируют в интерстиций и задерживают жидкость в
сосудистом секторе.
У больных с сердечной недостаточностью стимулом для секреции АДГ
является патологически низкий минутный объем кровообращения. В этой
связи фармакотерапию, ориентированную на улучшение насосной
функции сердца (средства с положительным инотропным действием,
препараты, снижающие постнагрузку, антиаритмические средства), следует
считать патогенетически обоснованным способом коррекции гипонатриемии.
Если сердечную недостаточность, вызывающую гипонатриемию,
обуславливает низкий уровень секреции гормонов коры надпочечников или
щитовидной железы, то коррекция эндокринопатии как причины низкой
насосной функции сердца введением соответствующих гормональных препаратов
устраняет гипонатриемию.
ГИПЕРНАТРИЕМИЯ
Гипернатриемия - это патологическое состояние, обусловленное
ростом [Na+] в результате потерь из организма свободной воды или
избыточного поступления в него натрия. Гипернатриемия вызывает
обезвоживание клеток вследствие роста осмоляльности внеклеточной
жидкости.
В подавляющем большинстве случаев гипернатриемия представляет
собой следствие потерь организмом свободной воды, которые преобладают
над потерями натрия. Свободную воду организм может терять при
избыточном водном диурезе или по внепочечным путям.
Можно выделить два основных варианта этиологии и патогенеза
избыточного водного диуреза как причины гипернатриемии. Если после
приема больным или введения ему средств, обладающих свойствами АДГ,
снижается объем выделяемой мочи и растет ее осмоляльность, то причина
гипернатриемии - центральный несахарный диабет (несахарное
мочеизнурение). Когда после введения таких препаратов моча остается гипо- или
\ Нарушения обмена воды и натрия
51
изосмоляльной по отношению ко внеклеточной жидкости, причина
избыточной ренальной экскреции воды и гипернатриемии - нефрогенный
несахарный диабет.
Рост содержания натрия во внеклеточной жидкости и жидкой части
плазмы крови служит стимулом для усиления синтеза антидиуретического
гормона (АДГ) в паравентрикулярном и супраоптическом ядрах
гипоталамуса. Транспорт АДГ в заднюю долю гипофиза происходит посредством
аксонального тока. Повреждения церебральных нейронов на одном из этих
трех уровней (гипоталамус, аксоны, по которым происходит транспорт
гормона, задняя доля гипофиза) обуславливают центральный несахарный
диабет. Наиболее частые причины этих повреждений - это черепно-мозговая
травма (особенно переломы основания черепа), инфекции,
злокачественный клеточный рост и доброкачественные опухоли (краниофарингиома,
опухоли гипофиза, киста шишковидной железы, метастазы в мозг опухолей
другого происхождения), нейрохирургические вмешательства.
Удаление хирургическим путем задней доли гипофиза приводит лишь
к временному несахарному центральному диабету. Прекращение
несахарного мочеизнурения после удаления задней доли связывают со
способностью клеток, образующих гормон, секретировать его в кровь. Основные
симптомы несахарного диабета, полиурия и полидипсия, при его центральном
происхождении развиваются остро. Выделяют три фазы центрального
несахарного диабета как осложнения нейрохирургических вмешательств:
1) полиурия в течение первых двух суток после возникновения
несахарного мочеизнурения как результат прекращения секреции АДГ;
2) фаза снижения диуреза, который падает вследствие высвобождения
аргинин-вазопрессина некробиотически измененными клеточными
элементами головного мозга;
3) фаза постоянного несахарного мочеизнурения, в которую объем
конечной мочи, выделяемый за единицу времени, меньше, чем в первую
фазу. Снижение диуреза относительно его уровня в первую фазу связывают
с восстановлением секреции АДГ интактными нейронами, синтезирующими
гормон.
В острую фазу несахарного мочеизнурения больной за сутки выделяет
до 20 л мочи с патологически низкой осмоляльностью. Лекарственные
средства со свойствами АДГ не сразу повышают осмоляльность до уровня,
соответствующего максимальной концентрационной способности почек, так
как необходимо некоторое время для возрастания осмоляльности
внеклеточной жидкости в интерстиции мозгового слоя почек.
Патологические состояния, которые снижают доставку воды и натрия
в сегменты нефрона, осуществляющие водный диурез, уменьшают степень
обезвоживания и гипернатриемии вследствие центрального несахарного
диабета. Если нейрохирургическое вмешательство затрагивает и переднюю
долю гипофиза (резекция передней доли), то его осложнением может быть
угнетение секреции кортизола и гормонов щитовидной железы. Эндокри-
нопатия такого рода приводит к недостаточности кровообращения и
снижает СКФ. Это ограничивает ренальную экскрецию свободной воды и
52
Глава 4
снижает степень гипернатриемии вследствие центрального несахарного
мочеизнурения.
У 50 % больных с центральным несахарным мочеизнурением выявить
его этиологию не представляется возможным. В таких случаях говорят об
идиопатическом несахарном центральном диабете.
Патогенетическая классификация нефрогенного несахарного
мочеизнурения (диабета) выделяет два его основных вида. При первом рост
клиренса свободной воды связан с тем, что нет роста проницаемости для воды
обращенной в просвет собирательных канальцев части наружной
клеточной мембраны эпителиоцитов под влиянием АДГ. Второй вид усиления
водного диуреза характеризует снижение осмоляльности жидкости в интер-
стиции мозгового слоя почек вследствие его патологических изменений.
Отравления литием, диметилхлортетракциклином ведут к
приобретенному нефрогенному несахарному диабету через угнетение роста
содержания в эпителиоцитах собирательных канальцев циклического аденозинмо-
нофосфата под влиянием АДГ. У больных с наследственным нефрогенным
несахарным диабетом этот рост значительно меньше, чем у людей, не
страдающих данным заболеванием.
Снижение осмоляльности интерстициальной жидкости в мозговом слое
почек обуславливают его поражения вследствие амилоидоза, инфекций
(пиелонефрит), побочного действия лекарств, гипоксии (в том числе и геми-
ческой, связанной с серповидноклеточной анемией), обструктивной уропа-
тии, поликистозного перерождения почек. Поддержанию высокой
осмоляльности в интерстиции мозгового слоя почек противодействует
эффект диуретиков, воздействующих на петлю нефрона.
Причиной нефрогенного несахарного диабета могут быть гипокалие-
мия и гипокальциемия.
Объем мочи, выделяемой за сутки больным с нефрогенным сахарным
диабетом, колеблется от 4 до 5 л. При этом ее осмоляльность обычно равна
осмоляльности жидкой части плазмы крови.
Полиурию вследствие несахарного мочеизнурения любого происхождения
всегда характеризует изо- или гипоосмоляльная относительно плазмы крови
конечная моча. Если у больного выявляют полиурию и гиперосмоляльность
мочи относительно плазмы крови (гиперосмоляльная полиурия), то это
свидетельствует о повышенном высвобождении осмолей во внеклеточную
жидкость как о причине высокой осмоляльности мочи. Когда при
гиперосмоляльной полиурии общее содержание натрия и калия в моче меньше,
чем 150 ммоль/л, то следует заподозрить осмотический диурез. При
осмотическом диурезе высокое содержание в просвете канальцев осмолей, не реаб-
сорбируемых нефроном, повышает осмоляльный клиренс в такой степени, что
возникает полиурия. Такими осмолями чаще всего являются молекулы
глюкозы и мочевины. Высокое содержание глюкозы в моче - это следствие сахарного
диабета. Повышенную ренальную экскрецию мочевины обуславливают:
♦ диета с высоким содержанием белка, при которой организм
вынужден подвергать часть белковых нутриентов распаду до азотистых
шлаков без какой-либо утилизации;
Нарушения обмена воды и натрия
53
♦ патологические состояния, при которых на системном уровне
катаболизм преобладает над анаболизмом: травматическая, раневая
болезнь, системные патологические сдвиги при злокачественном
клеточном росте, сепсис и др.;
♦ высокое содержание мочевины в циркулирующей крови после
устранения обструктивной уропатии.
Некоторые больные с полиурией вынуждены мочиться редко, но
помногу, одномоментно выделяя в среднем 1 л конечной мочи. Это ведет к
растяжению мочевого пузыря, мочеточников и даже служит причиной
гидронефроза.
Здоровый человек при внешнем дыхании и в ходе неощутимых потерь с
поверхности кожи (перспирация) за сутки в среднем теряет немногим менее
чем 1 л свободной воды. Перспирация возрастает во много раз при высокой
температуре окружающей среды и физической нагрузке. При этом
одновременно с ростом перспирации растут ощутимые потери воды с поверхности
кожи, то есть заметное испарение обильно отделяемого пота. Потери воды
по внепочечным путям повышают лихорадка и гипервентиляция, особенно
при искусственной вентиляции легких. Если повреждение центральной
нервной системы на супрасегментарном уровне препятствует возникновению
чувства жажды в ответ на рост осмоляльности внеклеточной жидкости, то
риск гипернатриемии особенно велик. Часто гипернатриемия развивается у
детей, которые не могут сказать, что они испытывают жажду.
Наиболее частый и нередко единственный симптом гипернатриемии -
это угнетение сознания той или иной степени в первую очередь вследствие
обезвоживания нейронов головного мозга. Когда концентрация натрия в
сыворотке крови становится большей, чем 160 ммоль/л, следует ожидать
субарахноидальные и интрацеребральные кровоизлияния и кому.
Если у больного с гипернатриемией выявляют снижение общего
содержания натрия в организме, которое проявляет себя симптомами гиповоле-
мии (табл. 4.2), то гипернатриемию устраняют одновременным
внутривенным вливанием 5 %-го раствора глюкозы и 0,45 %-го раствора натрия хлорида,
увеличивая объем внеклеточной жидкости и снижая ее осмоляльность.
Если у пациента с гипернатриемией нет дефицита объема
внеклеточной жидкости и гиповолемии, то дефицит свободной воды в организме
восполняют внутривенным вливанием 5 %-го раствора глюкозы. Для
определения объема 5 %-го раствора глюкозы, который необходимо инфузировать
внутривенно для коррекции гипернатриемии, производят расчеты, ход
которых представляется целесообразным проиллюстрировать на конкретном
примере.
У больного с несколько угнетенным, но сохраненным сознанием, без
явных симптомов снижения объема плазмы крови, масса тела которого до
дегидратации составляла 73 кг, концентрация натрия в сыворотке крови -
162 ммоль/л. Расчет объемов жидкостных секторов будем производить в
соответствии с данными табл. 4.1. Концентрацию натрия в сыворотке
крови признаем эквивалентной его содержанию во ВнЖ. За нормальный
уровень [Na+] принимаем ее величину, составляющую 140 ммоль/л. Для у про-
54
Глава 4
щения расчетов считаем, что весь натрий, который содержит организм
находится во внеклеточном пространстве. Изучение анамнеза позволяет
исключить кровопотерю и патологическую секвестрацию ВнЖ.
Сознание пациента несколько угнетено, но клинических признаков ги-
поволемии нет. Это позволяет сделать вывод, что общее содержание
натрия в организме после развития гипернатриемии не претерпело
изменений, и гипернатриемия обусловлена исключительно потерями свободной
воды. До дегидратации ОВнЖ составлял 0,2x73=14,6 (л). Общее
содержание в организме катиона натрия равнялось до обезвоживания 140х 14,6=2044
(ммоль). После развития гипернатриемии ОВнЖ снизился от исходного
уровня до 2044:162=12,6 (л). Из внеклеточного жидкостного сектора
организм потерял 14,6-12,6=2 (л) свободной воды. Потеря свободной воды из
организма всегда происходит одновременно из клеточного и
внеклеточного секторов в соответствии с соотношением их объемов 2:1. Значит
клеточный жидкостной сектор потерял 4 л Н20. Общий объем свободной воды,
потерянной во внешнюю среду, составляет 6 л. Такой дефицит воды в
организме восполняют медленно, в течение двух суток, вливая внутривенно 5 %
раствор глюкозы таким образом, чтобы скорость снижения осмоляльности
внеклеточной жидкости находилась на уровне в 1-2 мосм/кг Н20 в час.
У небольшой части больных гипернатриемия выступает следствием
избыточного поступления натрия в организм. К гипернатриемии такого гене-
за приводят:
♦ внутривенные инфузии гиперосмоляльного раствора хлорида натрия;
♦ попадание морской воды в желудок и кишечник;
♦ случайная замена сахара солью в детском питании;
♦ восполнение потерь свободной воды внутривенными инфузиями
только изоосмоляльных относительно плазмы крови растворов (раствор
Рингера и др.).
Глава 5
НАРУШЕНИЯ ОБМЕНА КАЛИЯ
Гипокалиемия - это патологическое состояние организма вследствие
падения содержания калия во внеклеточной эюидкости и эюидкой части
плазмы крови ({K+J) до уровня меньшего, чем 5 ммолъ/л.
Гипокалиемия вызывает опасные дисфункции и проявляет себя
соответствующими симптомами, когда [К+] меньше, чем 3,0 ммоль/л.
Внеклеточная жидкость содержит всего 60 ммоль калия, что составляет лишь 2 %
его общего содержания в организме. Поэтому изменения [К+] не всегда
отражают патологические сдвиги общего содержания катиона в клетках и во
внеклеточной жидкости. [JC] меньше 3,0 ммоль/л свидетельствует о потере
организмом не менее, чем. 200 ммоль калия.
У большинства больных с хронической гипокалиемией ее обуславливают
потери калия во внешнюю среду. Их вызывают рвота, потери содержимого
желудка по гастральному зонду, понос или действие мочегонных средств.
Обычно при этом выявляют симптомы и получают данные специальных
исследований, которые свидетельствуют о развитии дефицита внеклеточной
жидкости. Если признаков дефицита ОВнЖ нет, то причиной гипокалиемии
следует считать избыточную секрецию альдостерона (вторичный или
первичный альдостеронизм). Так, избыточная секреция минералкортикоидов при
вторичном альдостеронизме как следствии и (или) причине артериальной
гипертензии ведет к гипокалиемии, которую может опасно обострить
действие мочегонных^ повышающих экскрецию калия. Перемещение калия в
клетки из внеклеточного сектора редко вызывает хроническую опасную ги-
покалиемию. Калий перемещается в клетки из внеклеточного пространства
под действием гормонов (инсулин, эндогенные катехоламины), в результате
метаболического алкалоза, вследствие преобладания на системном уровне
анаболических процессов над катаболическими.
Одна из наиболее частых причин гипокалиемии у тяжелых больных в
условиях стационара - это усиленная экскреция калия с мочой.
Почти весь калий, который попадает в состав ультрафильтрата в
клубочках нефронов, реабсорбируется до того, как жидкость в просвете
канальцев достигнет дистальных сегментов нефрона, где происходит секреция
калия в просвет канальцев, вторичная по отношению к активной реабсорб-
ции натрия. Ключевой гормон в регуляции экскреции калия почками - это
усиливающий ее альдостерон. Стимулом для секреции альдостерона
служат гиперкалиемия, рост содержания в циркулирующей крови ангиотензи-
на II и усиление высвобождения передней долей гипоталамуса альдосте-
рон-рилизинг-фактора, которое происходит вне зависимости от секреции
адренокортикотропного гормона. Содержание ангиотензина II в плазме крови
возрастает в результате увеличения в ней активности ренина.
Высвобождение ренина клетками юкстагломерулярных аппаратов нефронов растет в
56
Глава 5
Рис. 5.1. Активная секреции натрия в дистальных сегментах нефрона
ответ на развитие дефицита ОВнЖ при стенозе почечной артерии (реновас-
кулярная артериальная гипертензия), а также в результате возбуждения бета-
один-адренорецепторов. Увеличение объема внеклеточной жидкости и
действие бета-один-адреноблокаторов угнетают секрецию ренина, уменьшают
высвобождение альдостерона надпочечниками и секрецию калия в просвет
дистальных сегментов нефрона. Деструкция юкстагломерулярных
аппаратов нефронов при заболеваниях, поражающих почечный интерстиции,
особенно характерная для диабетической нефропатии, резко снижает
активность ренина в плазме. Ингибиторы ангиотензинпревращающего фермента
снижают концентрации в крови ангиотензина II и альдостерона, несмотря
на высокую активность ренина в плазме крови. Предсердный натрийурети-
ческий пептид угнетает секрецию альдостерона клетками клубочковой зоны
коры надпочечников.
Альдостерон открывает натриевый канал обращенной в просвет канальца
части наружной клеточной мембраны эпителиоцита, одновременно снижая
ее проницаемость для хлоридного аниона (рис. 5.1). Кроме того, гормон
повышает активность натрий-калий-АТФазы базолатеральной мембраны
эпителиоцита. Натрий из просвета канальцев устремляется в сторону своей
меньшей концентрации в эпителиоцит, из которого его выводит механизм
активного трансмембранного переноса (натрий-калий-АТФаза).
Одновременно действие этого механизма перемещает в обратном направлении
через базолатеральную мембрану калий. Рост содержания калия в клетке
ведет к его выходу в интерстиции и в просвет канальца. В просвете калиевые
катионы удерживаются анионами, в том числе и хлоридным, которые не
реабсорбируются из просвета канальцев.
Все катионы калия, которые в ходе активной секреции попадают в
просвет дистальных извитого и кортикального собирательных канальцев,
выделяются в составе конечной мочи. В собирательных канальцах мозгового
слоя реабсорбция воды не меняет общего содержания калия в просвете
канальцев, а только повышает концентрацию К+ в конечной моче.
В этой связи выделяют две детерминанты экскреции почками калия:
♦ общее количество натрия, которое по просвету канальцев достигает
дистальных сегментов нефрона, где происходит активная секреция калия;
♦ действие на эпителиоциты этих сегментов альдостерона.
Нарушения обмена калия
57
Наиболее частая причина повышенной секреции альдостерона,
приводящей к гипокалиемии, - это увеличение секреции ренина клетками юк-
стагломерулярного аппарата в ответ на падение объема внеклеточной
жидкости. Дефицит объема внеклеточной жидкости ведет к увеличению
секреции ренина не только через гиповолемию, снижающую объемную
скорость кровотока через клубочки нефронов. Кроме того, он вызывает
неспецифическую стрессорную реакцию, при которой системная адренергичес-
кая стимуляция повышает секрецию ренина через возбуждение бета-один-
адренорецепторов . Если объем внеклеточной жидкости падает в такой
степени, что это снижает скорость клубочковой фильтрации, то вторичный аль-
достеронизм как реакция на дефицит ОВнЖ может не вызывать
гипокалиемии из-за низкого уровня доставки натрия в сегменты нефрона, где
происходят сопряженные активная реабсорбция натрия и секреция калия.
Вторичный альдостеронизм как звено патогенеза реноваскулярной
артериальной гипертензии также может быть причиной гипокалиемии.
Дефицит в организме магния повышает секрецию альдостерона, что у части
больных обуславливает гипокалиемию.
Вторичный альдостеронизм как причина гипокалиемии у части больных
связан с ренин-продуцирующей опухолью почек или гипертрофией
образующих и высвобождающих ренин клеток юкстагломерулярного аппарата.
Наконец, вторичный альдостеронизм характеризует такое редкое
системное расстройство как синдром Бартера (см. главу, посвященную
метаболическому алкалозу). При всех этих видах вторичного альдостеронизма в
плазме крови высока активность ренина.
Первичный альдостеронизм, обусловленный двусторонней
гиперплазией коры надпочечников или их аденомой, также приводит к гипокалиемии.
При этом активность ренина в плазме крови находится на низком уровне.
В главе, посвященной артериальной гипертензии, описан патогенез
гипокалиемии, а) связанной с высокой концентрацией кортизола как мине-
ралкортикоида в плазме крови, б) вызванной ферментопатиями,
нарушающими синтез гормонов коры надпочечников, в) обусловленной экзогенными
веществами со свойствами минералкортикоидов или усиливающих синтез
кортизола в организме, и г) как следствия мутаций генов,
детерминирующих синтез гормонов коры надпочечников.
Действие мочегонных средств, которые не относятся к группе
препаратов, обладающих свойством не вызывать потери калия (спиронолактон, ами-
лорид и др.), приводит к росту экскреции калия с конечной мочой. Диуретики
блокируют реабсорбцию натрия в почечных канальцах таким образом, что
повышают поступление натрия хлорида в сегменты нефрона, где под
влиянием альдостерона происходят активные реабсорбция натрия и сопряженная с
ней секреция калия. Таким образом действие диуретиков усиливает влияние
на экскрецию калия с мочой одной из ее детерминант. Одновременно через
усиление натрийуреза мочегонные средства снижают объем внеклеточной
жидкости, что служит стимулом для усиления действия второй детерминанты ре-
нальной экскреции калия, эффекта альдостерона на эпителиоциты сегментов
нефрона, осуществляющие секрецию калия в просвет канальцев (схема 5.1)
58
Глава 5
Действие мочегонного средства на почки
^
т
Снижение реабсорбции натрия в
дистальных сегментах нефрона
т
V
Усиление натрийуреза как
причина дефицита ОВнЖ
Рост количества катионов натрия, поступающих
в сегменты нефрона, где под влиянием
альдостерона происходит секреция калия
^
г
Вторичный
альдостеронизм
у
Рост содержания калия в конечной моче
Схема 5.1. Патогенез гипокалиемии в результате побочного действия диуретиков
Патогенетически обоснованная коррекция гипокалиемии как следствия
побочного действия диуретиков должна быть ориентирована на снижение
двух детерминант величины выделения калия с мочой. Количество
катионов натрия, поступающих в сегменты нефрона, в которых под влиянием
альдостерона происходит секреция калия в просвет канальцев, снижают,
ограничивая суточное потребления натрия с пищей и напитками 100 ммо-
лями. Вторичный альдостеронизм стремятся устранить через действие
ингибиторов ангиотензин-превращающего фермента. Одновременно повышают
суточное потребление с пищей и напитками калия, снижают дозу
диуретика или применяют мочегонные средства, в меньшей степени повышающие
выделение калия почками.
Содержание калия в желудочном содержимом относительно невелико и
обычно не больше, чем 15 ммоль/л. Обычно потери содержимого желудка
при частой рвоте и по зонду вызывают гипокалиемию не через потери
катиона во внешнюю среду. Ведущее звено патогенеза гипокалиемии вследствие
потерь содержимого желудка - это вторичный альдостеронизм, связанный
с дефицитом ОВнЖ. Понос может быть непосредственной причиной
гипокалиемии, так как содержание калия в кишечнике выше, чем в просвете
более проксимальных отделов желудочно-кишечного канала. При холерном
поносе за несколько часов больной может потерять 6 л воды, содержащей
750 ммоль натрия и 100 ммоль калия. Когда диарея связана с дисфункцией
более каудальных отделов кишечника, в частности обусловленных
ворсинчатой аденомой прямой кишки, потери калия больше, так как в просвете
этих отделов концентрация К+особенно велика.
Вне зависимости от причины, вызвавшей перемещение калия из
внеклеточного пространства в клетки, такая миграция катиона сама по себе не
может быть причиной клинически значимой гипокалиемии, так как
снижает концентрацию калия не более, чем на 1 ммоль/л.
Нарушения обмена калия
59
сопряженный трансмембранный
перенос натрия и протонов
падение [Н*]
натрий-калий-АТФаза
""•• рост [НС03']
Рис. 5.2. Патогенез гипокалиемии вследствие метаболического алкалоза
(см. объяснения в тексте, пунктиром обозначены причинно-следственные связи)
Перемещение калия в клетки может обострить гипокалиемию, которую
в основном вызывает какогой-либо другой патологический процесс. Так,
если гипокалиемию у больного выявляют одновременно с метаболическим
алкалозом (ростом содержания бикарбонатного аниона во внеклеточной
жидкости и в жидкой части плазмы крови), то она часто обусловлена
вторичным альдостеронизмом вследствие дефицита ОВнЖ, связанного с
потерями желудочного содержимого или ростом натрийуреза в результате
действия мочегонных.
Наиболее клинически значимое перемещение калия в клетки из всех
расстройств кислотно-основного состояния вызывает метаболический
алкалоз (рис. 5.2).
Рост при метаболическом алкалозе содержания во внеклеточной
жидкости бикарбонатного аниона ([НС03*]) снижает концентрацию в ней
протонов ([Н+]). Это служит причиной выхода из клеток свободных ионов
водорода, сопряженного с перемещением натрия в клетку. Натрий, поступивший
в клетку в обмен на протоны, выводится обратно во внеклеточное
пространство через действие механизма активного трансмембранного переноса с
участием натрий-калий-АТФазы. Работа этого механизма одновременно с
выводом натрия из клетки перемещает в нее калий.
Действие инсулина перемещает калий в клетки из внеклеточного
пространства через увеличение трансмембранного клеточного потенциала
покоя и содержания в клетке органических анионов. Если больному сахарным
диабетом и гипокалиемией ввести инсулин, то действие гормона может
опасно снизить содержание калия в плазме крови.
Возбуждение бета-два-адренорецепторов вызывает перемещение калия
в клетки. Особенно велик уровень возбуждения бета-два-адренорецепторов
вследствие системной адренергической стимуляции, обусловленной
патогенным стрессом тяжелого ранения, гиповолемией, хирургической
операцией в условиях неэффективной анальгезии, а также в результате расстройств
внутрицентральных отношений у больных белой горячкой. Передозировка
бета-два-адреномиметиков может снизить концентрацию калия в плазме
крови на 1,5-2,0 ммоль/л.
60
Глава 5
Таблица 5.1
Связь патогенеза и симптомов гипокалиемии
Эффектор
функциональной системы, состояние
которого патологически
изменяет гипокалиемия
Скелетные мышцы
Гладкие мышцы
стенок желуцка и
кишечника
Сердце
Почки
Звено патогенеза гипокалиемии
Увеличение трансмембранного
потенциала скелетных мышц.
Падение возбуцимиости миоцитов
Рост наружного клеточного
потенциала покоя миоцитов гладких
мышц и клеток дуоденального
водителя ритма. Расстройства
генерации и распространения по
желудочно-кишечному каналу
мигрирующего миоэлектрического
комплекса
Увеличение трансмембранного
потенциала покоя клеток
водителей ритма проводящей системы
сердца и рабочих кардиомиоцитов
Падение концентрационной
способности почек
Клинические признаки
Мышечная слабость,
параличи
Угнетение кишечной
моторики вплоть до
развития острой кишечной
непроходимости
Сердечные аритмии,
патологические изменения
электрокардиограммы:
уплотнение и инверсия зубца Т,
депрессия сегмента S-T,
появление волны U
Полиурия, полидипсия
Без интенсивной коррекции тяжелая гипокалиемия может вызвать
опасные нарушения сердечного ритма и обусловить прогрессирование
нарушений водно-солевого обмена, в частности через развитие паралитической
кишечной непроходимости (табл. 5.1).
Так как способность почек ограничивать выделение калия с мочой
исключительно велика, то гипокалиемия у больных почти не бывает следствием
низкого поступления калия в организм. Низкая величина потребления
калия с пищей и напитками приводит к снижению его общего содержания в
организме на 100 ммоль от нормальной величины в 3000—4000 ммоль за
недели или месяцы.
При патологических потерях калия из внеклеточной жидкости, в
частности вследствие побочного эффекта некоторых мочегонных средств,
низкое потребления калия с пищей и напитками ускоряет развитие
гипокалиемии и гипокалии (низкого общего содержания калия в организме). Особенно
часто гипокалиемия такого происхождения развивается у пожилых
больных, страдающих гипертонической болезнью, которым назначают
диуретики, обладающие свойством повышать ренальную экскрецию калия.
Гиперкалиемия - это патологическое состояние вследствие роста
содержания во внеклеточной жидкости и жидкой части плазмы крови калия.
Гиперкалиемия приводит к опасным дисфункциям, когда концентрация
калия в плазме становится выше, чем 6,0 ммоль/л. При остром развитии
гиперкалиемии она вызывает сердечные аритмии при меньших значениях
[К+], чем при хронической гиперкалиемии.
Нарушения обмена калия
61
Хроническая гиперкалиемия — это всегда результат сниженной
экскреции калия.
Если гиперкалиемия не представляет собой следствия почечной
недостаточности, то ее чаще всего обуславливают низкая активность альдостерона в
циркулирующей крови или сниженная объемная скорость тока жидкости в
просвете канальцев дистальных сегментов нефрона, в которых происходят
сопряженные активные реабсорбция натрия и секреция калия. Эту скорость
снижает защитно-патогенная реакция спазма приводящих артериол нефро-
нов в ответ на гиповолемию, циркуляторную гипоксию и дефицит объема
внеклеточной жидкости. Системная патогенная болевая реакция может
вызывать и обострять спазм приводящих артериол как причину гиперкалиемии.
Если при гиперкалиемии снижена активность ренина в плазме, то это
свидетельствует о недостаточности секреции ренина и вторичному по
отношению к ней падению секреции альдостерона, вызывающему гиперка-
лиемию. Это может быть следствием потери почками значительного числа
нормально функционирующих юкстагломерулярных аппаратов нефронов,
которую обуславливают:
♦ интерстициальный нефрит как результат инфекций;
♦ нефротоксическое действие нестероидных противоспалительных
средств и некоторых антибиотиков (метициллин и др.);
♦ амилоидоз почек или отложения в них уратов;
♦ диабетическая нефропатия.
Побочное действие бета-адренолитиков через снижение уровня
возбуждения бета-один-адренорецепторов ведет к снижению активности в плазме
крови ренина, альдостерона и гиперкалиемии.
У части больных гиперкалиемия - это элемент синдрома хлоридного
шунта, который, кроме нее, характеризуют:
♦ умеренный рост ОВнЖ;
♦ незначительная артериальная гипертензия;
♦ метаболический ацидоз при нормальном АПП;
♦ низкая концентрация калия в конечной моче.
У больных с синдромом хлоридного шунта действие альдостерона на
тубулярные эпителиоциты, секретирующие калий, не приводит к снижению
проницаемости стенки канальцев для хлоридного аниона, то есть не
вызывает деполяризации обращенной в просвет канальцев части наружной
клеточной мембраны клетки тубулярного эпителия. Без деполяризации нет
секреции калия и протонов в просвет канальцев. Сниженная секреция протонов
служит причиной метаболического ацидоза. Рост концентрации протонов
во внеклеточной жидкости при метаболическом ацидозе у больных с
синдромом хлоридного шунта - это результат не только сниженной секреции
протонов в просвет канальцев, но и торможения вследствие гиперкалиемии
образования аммиака эпителиоцитами. Предположительно гиперкалиемия
служит стимулом усиленной секреции альдостерона, которая повышает ре-
абсорбцию натрия. Усиленная реабсорбция натрия служит причиной роста
объема внеклеточной жидкости и артериальной гипертензии у пациентов с
синдромом хлоридного шунта.
62
Глава 5
Если у больного выявляют гиперкалиемию и высокую активность
ренина в плазме крови, то патологический рост содержания калия в плазме
обусловлен или низкой активностью в крови альдостерона или угнетением
физиологической реакции почек на действие минералкортикоида.
Низкую активность альдостерона в циркулирующей крови чаще всего
обуславливает первичная недостаточность надпочечников (см. главу,
посвященную эндокринопатиям). Вероятность недостаточности надпочечников
как причины гиперкалиемии велика при синдроме приобретенного
иммунодефицита (результат обширного некроза надпочечников), саркоме
Калоши, цитгломегаловирусной инфекции, туберкулезе, инфекциях, вызванных
атипичными микобактериями и Cryptococcus.
Недостаточная активность альдостерона в плазме крови может быть
следствием действия ингибиторов ангиотензинйревращающего фермента,
антибиотиков из класса циклоспоринов и диуретика спиронолактона.
В основе синдрома тубулярной гиперкалиемии без дефицита
альдостерона лежит угнетение физиологической реакции клеток тубулярного
эпителия, осуществляющих секрецию калия в просвет канальцев, на действие
гормона. Синдром вызывают:
♦ обструктивная уропатия;
♦ постишемические изменения эпителиоцитов в пересаженной почке;
♦ системные красная волчанка и амилоидоз, поражающие паренхиму
почек;
♦ нефропатия вследствие серповидно-клеточной анемии;
♦ избыточное действие диуретиков, не обладающих свойством
повышать выделение калия с мочой.
Молекулы этих лекарственных средств (спиронолактон и др.),
связываясь с альдостероновыми рецепторами, обуславливают ареактивность
эпителиоцитов к действию минералкортикоида.
Гиперкалиемию может вызывать выход калия из клеток во
внеклеточное пространство вследствие эндокринопатий, ацидоза и цитолиза.
Недостаточная секреция инсулина, инактивация гормона на пререцеп-
торном уровне, угнетение физиологической реакции клеток на действие
инсулина на рецепторном и пострецепторном уровнях, которые лежат в
основе развития сахарного диабета, ограничивают вход калия в клетку с
глюкозой и аминокислотами в качестве субстратов анаболических процессов.
Резистентность к инсулину (см. главу, посвященную сахарному диабету)
обуславливает гиперкалиемию. Выход калия из клеток у больных
сахарным диабетом не связан с диабетическим кетоацидозом. Это следствие
резистентности к инсулину.
Бета-адренолитики Могут вызвать гиперкалиемию, так как возбуждение
соответствующих адренорецепторов перемещает калий в клетки.
Бета-адренолитики, не обладающие свойством селективных бета-адреноблокато-
ров, одновременно с бета-два-адренорецепторами блокируют бета-один-
адренорецепторы. Это снижает синтез ренина и секрецию альдостерона,
вызывая гиперкалиемию. Вот почему действие бета-адреноблокаторов
может приводить к клинически значимой гиперкалиемии. Альфа-адреноми-
Нарушения обмена калия
63
метики усиливают выход калия из клеток. Их действие может усилить ги-
перкалиемию, в основном обусловленную другими причинами.
Особенно выражена гиперкалиемия когда ее обуславливают
резистентность к инсулину и снижение секреции альдостерона при низкой
активности ренина в плазме крови у больного с инсулинзависимым сахарным
диабетом и диабетической нефропатией, разрушающей клетки юкстагломе-
рулярных аппаратов, секретирующие ренин.
Из расстройств кислотно-основного состояния острую гиперкалиемию
может вызвать острый метаболический ацидоз при нормальной величине
анионного пробела плазмы (АПП), обусловленный потерями бикарбонат-
ного аниона во внешнюю среду, которые в частности происходят при
поносе и бикарбонатурия. Падение [НС03] вызывает рост содержания во
внеклеточной жидкости протонов. В результате роста концентрации протонов
во внеклеточной жидкости они начинают мигрировать в клетки без обычно
присутствующих во внеклеточной жидкости анионов (хлоридного, бикар-
бонатного и других). В клетках протоны связываются внутриклеточными
буферными системами, на что расходуются внутриклеточные анионы.
Нарушение электронейтральности внутри клеток ведет к выходу из клетки
калия и гиперкалиемии. Если рост концентрации протонов во
внеклеточной жидкости происходит одновременно с появлением в ней органических
анионов, которое увеличивает АПП (лактатный ацидоз, диабетический ке-
тоацидоз), то он не вызывает выхода калия из клеток. Дело в том, что эти
органические анионы свободно мигрируют в клетки вместе с протонами
(рис. 5.3). В результате электронейтральность внутриклеточной среды
изменений не претерпевает, так как расходуемые на связывание протонов
внутриклеточные анионы замещаются анионами из внеклеточной жидкости.
У наружная клеточная
у Н v / мембрана
/ \ I Л лаюгатныи
U —>\ v+ I I к I анион
Рис. 5.3. Схема патогенеза гиперкалиемии, вызванной метаболическим ацидозом
Рост концентрации калия во внеклеточном секторе снижает
трансмембранный потенциал покоя миоцитов скелетных мышц и сердца, а также
нейронов. В результате возникают расстройства возбудимости данных клеток,
которые проявляют себя парестезиями, слабостью мышц вплоть до
паралича, высокими и острыми зубцами Т электрокардиограммы, а также
депрессией ее сегмента S-T. В тяжелых случаях гиперкалиемии, когда концентрация
калия в циркулирующей крови превышает 7 ммоль/д, возникают
фибрилляция желудочков и асистолия.
Глава 6
НАРУШЕНИЯ КИСЛОТНО-ОСНОВНОГО СОСТОЯНИЯ
Концентрацию протонов во внеклеточной жидкости и в жидкой части
плазмы крови ([Н+]) физиологические системы удерживают на уровне,
который можно признать исключительно низким (40 нмоль/л) относительно
массивного высвобождения ионов водорода в клетки и во внутреннюю
среду (7x107 наномолей1 каждый день). При обычном полноценном питании
высвобождение протонов в клетки и во внутреннюю среду организма
составляет 1 ммоль/кг массы тела в день, что в 106 раза превышает общее
содержание протонов во внеклеточной жидкости.
Свободные ионы водорода связываются буферными системами: 40 %
всех высвобождаемых ионов водорода нейтрализует главная внеклеточная
буферная система угольной кислоты и гидрокарбоната натрия, а 60 % -
внутриклеточные буферные системы. После взаимодействия протона с
буферной системой он переходит во временное состояние связывания и
нейтрализации. Буферные системы -это первая защитная линия в противодействии
постоянной тенденции к закислению клеток и внутренней среды. Для
поддержания нормального кислотно-основного состояния необходима
элиминация протонов из форм их временного связывания во внешнюю среду.
Легкие выступают эффектором системы главного буфера внеклеточной
жидкости, осуществляя экскрецию углекислого газа и вызывая сдвиг
реакции системы бикарбонатного буфера вправо:
н+ + нсо3 = н2со3 = н2о + со2
Почки восполняют потери бикарбонатного аниона, метаболизируя глю-
тамин, синтезируемый печенью, с образованием бикарбонатного аниона
и одновременной экскрецией NH4+. Физиологическая система,
поддерживающая на нормальном уровне концентрацию протонов во внеклеточной
жидкости, для достижения своего конечного полезного
приспособительного результата объединяет в качестве эффекторов легкие, почки и печень
(схема 6.1).
Для удержания [Н+] в нормальных пределах необходима
соответствующая нейтрализации свободных ионов водорода регенерация бикарбонатных
анионов почками.
Кислотно-основное состояние больного характеризуют величины трех
его параметров:
♦ концентрация протонов во внеклеточной жидкости ([Н+]);
♦ содержание в ней бикарбонатного аниона([НС03~]);
♦ напряжение углекислого газа в артериальной крови (РаС02).
1 Наномоль- Ю9моля(нмоль).
Нарушения кислотно-основного состояния
65
Протоны
▼
^ ™"|
Система бикарбо-
натного буфера
4
Углекислый газ
*
Легкие
+
<
1 Белок 1
j
Печень
Генерация
НСОз'
Экскреция С02
►
«
Глутамин
▼
Почки
i
г
Экскреция NH4+
Схема 6.1. Взаимосодействие эффекторов при удержании [Н+]
во внеклеточной жидкости в нормальных пределах
Функциональную связь между ними отражает уравнение Гендерсона-
Гассельбаха:
[Н+](нмоль/л)=23,9хРаСЮ2(мм рт. ст.)/[НС03](ммоль/л).
В состав желудочного содержимого каждые сутки попадают 150 ммоль
протонов. Секреция протонов в просвет желудка сопровождается
эквивалентной генерацией гидрокарбонатного аниона и его поступлением во
внеклеточную жидкость. Тонкая кишка и поджелудочная железа секретируют
бикарбонатные анионы, тем самым нейтрализуя протоны в просвете
желудочно-кишечного канала. В физиологических условиях те бикарбонатные
анионы, которые не нейтрализуют протоны, поступившие в кишечник из
желудка, реабсорбируются в кровь.
Потери кислого содержимого усиливают образование бикарбонатного
аниона в эпителиоцитах желудка. Это повышает [НС03]. Потери
содержимого кишечника (диарея, кишечные свищи и др.) уменьшают реабсорбцию
бикарбонатного аниона из просвета желудочно-кишечного канала. Это
снижает [НС03].
Микроорганизмы в просвете кишечника могут перерабатывать
питательные вещества таким образом, что вызывают патологические
изменения [НС03] и [Н+]. Образование ими не усваиваемых организмом
органических кислот служит причиной метаболического ацидоза. Полное
окисление в ходе жизнедеятельности микроорганизмов органических
анионов из состава пищи до углекислого газа и воды ведет к аккумуляции в
просвете кишечника бикарбонатного аниона, что вызывает
метаболический алкалоз.
Для оценки кислотно-основного состояния необходимо определение
четырех его параметров (табл. 6.1).
66
Глава 6
Таблица 6.1
Нормальные величины параметров кислотно-основного
состояния организма
Параметр кислотно-основного
состояния
[Н1 (рН)
РаС02
[НС03]
Анионный пробел плазмы
Пределы нормальных
колебаний
40±2 нмоль/л (7,40±0,02)
4Ш:3 мм. рт. ст.
24±2 ммоль/л
12±2 мэкв/л
Таблица 6.2
Связь между рН и [Н+/
рН 7,0 7,1 7,2 7,3 7,4 7,5 7,6 7,7
[Н+], ммоль/л 100 79 63 50 40 32 25 20
Таблица 6.3
Компенсаторные реакции в ответ на нарушения
кислотно-основного состояния
Расстройство кислотно-
основного обмена
Метаболический ацидоз
Метаболический алкалоз
Острый респираторный
ацидоз
Хронический
респираторный ацидоз
Острый респираторный
алкалоз
Хронический
респираторный алкалоз
Ненарушенная компенсаторная реакция
физиологичеких систем
Снижение [НС03] на 1 ммоль/л от уровня в 25 ммоль/л
приводит к падению РаС02 на 1 мм рт. ст. от 40 мм рт. ст.
Рост [НС03] на 1 ммоль/л от уровня в 25 ммоль/л
приводит к подъему РаС02 на 1 мм рт. ст. от 40 мм рт. ст.
Возрастание РаС02 на 1мм рт. ст. повышает [Н~] на
0,77 нмоль/л. Для возвращения [Н ~] к исходному
нормальному уровню [НС03] возрастает на 1 ммоль/л от 25 ммоль/л
Рост РаС02 на 1мм рт. ст. увеличивает [Н+] на 0,32 нмоль/л.
Для востановления нормальной [Н "] [НС03] растет на
03 ммоль/л от 25 ммоль/л
Уменьшение РаС02 в два раза ведет к снижению
[НС03] на 2,5 ммоль/л
Снижение РаС02 на 10 мм рт. ст. от уровня в 40 мм рт. ст.
ведет к снижению [НС03] на 5 ммоль/л
Клинический опыт исследований кислотно-основного состояния (КОС)
и коррекции его нарушений позволил считать, что концентрация
водородных ионов во внеклеточной жидкости и жидкой части плазмы - это более
информативный показатель, чем производная [Н+], отрицательный
десятичный логарифм концентрации водородных ионов (рН). Например, при
снижении рН на три десятых от 7,4 до 7,1 [Н+] возрастает почти в два раза
(табл. 6.2). Кроме того, определение концентрации протонов, а не рН, по-
Нарушения кислотно-основного состояния
67
зволяет более точно дозировать инфузию растворов, содержащих протоны
или бикарбонатные анионы, которую производят для возвращения [Н+] в
нормальные пределы.
Выделяют четыре основных вида нарушений кислотно-основного
состояния: метаболический ацидоз, метаболический алкалоз, респираторный
ацидоз, респираторный алкалоз.
Метаболический ацидоз -это подъем [Н+] и снижение [НС03].
Метаболический алкалоз- падение [Н+] и рост [НС03]. Респираторный ацидоз-
это подъем [ЬГ] вследствие роста РаС02, а респираторный алкалоз -
падение [Н+] вследствие снижения РаС02.
Патологические сдвиги концентрации протонов во внеклеточной
жидкости и плазме крови вызывают реакции физиологических систем (табл. 6.3),
которые направлены на возвращение [Н+] в нормальные пределы, что
достижимо только при хроническом респираторном алкалозе.
МЕТАБОЛИЧЕСКИЙ АЦИДОЗ
Метаболический ацидоз - это патологическое состояние, которое
характеризуют рост во внеклеточной жидкости и жидкой части плазмы
крови концентрации протонов ([Н+]) и снижение в них содержания бикар-
бонатного аниона ([НС05]). К метаболическому ацидозу приводят
задержка в организме сильно диссоциирующих кислот и (или) потеря им бикар-
бонатного аниона. Для идентификации основного звена патогенеза
метаболического ацидоза необходимо определить анионный пробел плазмы.
Число анионов всегда равно числу катионов как в клеточной, так и во
внеклеточной жидкости. Если из величины содержания во внеклеточной
жидкости и жидкой части плазмы их главного одновалентного катиона
натрия ([Na+]) вычесть общее в них содержание главных одновалентных
анионов, хлоридного ([О"])и бикарбонатного ([НС03]), то мы получим
значение анионного пробела плазмы (АПП):
АПП = [Na+] -([С1] + [НСОЗ]).
АПП - это различие между общим содержанием катионов во
внеклеточной жидкости и жидкой части плазмы с вычетом из нее [Na+] и
суммарной концентрацией в них анионов без [НСОЗ] и концентрации во ВнЖ и
плазме хлоридного аниона ([О"]). АПП можно определить как различие
между общим содержанием во внеклеточной жидкости и жидкой части
плазмы катионов кальция, калия и магния, и суммарной концентрацией в них
сульфатного, фосфатных, альбуминового и органических анионов.
Во внеклеточной жидкости в силу внутренних или внешних причин в
результате диссоциации кислот одновременно могут расти содержание
протонов и концентрация анионов. При этом 1 ммоль эндо- или экзогенной
органической кислоты диссоциирует с высвобождением 1 ммоль протонов и
68
Глава 6
Таблица 6.4
Этиопатогенетическая классификация метаболического ацидоза
С ростом АПП
Лактатный ацидоз
Кетоацидоз
Вследствие почечной
недостаточности
В результате интоксикации
Как следствие абсорбции
органических кислот из просвета
кишечника
Без роста АПП
Вследствие потерь натрия бикарбоната:
- прямых, из просвета желудочно-кишечного канала
или с мочой;
- непрямых, в результате низкого содержания в моче
NH4+ или повышенной ренальной экскреции
органических анионов
Как результат разведения натрия бикарбоната во
внеклеточной жидкости
1 ммоль аниона. На нейтрализацию 1 ммоль протонов уходит 1 ммоль би-
карбонатного аниона. На место этого ммоль бикарбонатного аниона во
внеклеточной жидкости приходит ммоль органического аниона
диссоциирующей кислоты. В результате, несмотря на то, что общее содержание анионов
во внеклеточной жидкости остается неизменным, АПП становится на
1 ммоль больше. Поэтому рост АПП выше верхнего предела нормальных
колебаний (10-14 ммолъ/л) свидетельствует о повышенном
высвобождении или поступлении извне во внутреннюю среду эндо- или экзогенных
диссоциирующих кислот как о причине метаболического ацидоза. Если АПП
при метаболическом ацидозе не растет, то его причина - потеря
организмом бикарбонатного аниона.
Содержание альбуминовых анионов во внеклеточной жидкости и
плазме при нормальной концентрации белка в крови составляет 12 ммоль/л.
Гипоальбуминемия уменьшает АПП без нарушений кислотно-основного
состояния. Поэтому у больных с гипоальбуминемией диссоциация
органических кислот в клетках и во внеклеточной жидкости ведет к
метаболическому ацидозу при АПП в «нормальных» пределах.
Этиопатогенетическая классификация метаболического ацидоза
выделяет два его вида: метаболический ацидоз с увеличенным АПП и
метаболический ацидоз с нормальным АПП (табл. 6.4). Наиболее частая причина лак-
татного метаболического ацидоза - это недостаточный транспорт в клетку
кислорода вследствие артериальной гипоксемии, нарушений системного и
периферического кровообращения. Накопление лактата в цитозоле клеток всех
тканей происходит только при замедлении трансформации лактата в пируват,
которая невозможна без сопряженного с ней окисления восстановленной
формы никотинамидадениндинуклеотида (НАДН). Гипоксия через падение
напряжения кислорода в митохондриях ведет к накоплению в них НАДН, что
блокирует передачу протона от восстановленной формы НАДН в цитозоле к
ее окисленной форме в митохондриях. Это повышает содержание НАДН в
цитозоле и ведет к накоплению в клетке молочной кислоты. Концентрация
молочной кислоты в клетке растет вследствие блокады ее трансформации в
пируват, обусловленной угнетением или прекращением окисления НАДН.
Второй непосредственной причиной роста содержания лактата в цитозоле
клеток может быть аккумуляция в них пировиноградной кислоты.
Нарушения кислотно-основного состояния
69
Лактатный ацидоз, связанный с гипоксией, называют лактатным
ацидозом типа А. Острая респираторно-циркуляторная гипоксия может
приводить к высвобождению во внутреннюю среду протонов со скоростью
60 ммоль/мин, чему не в состоянии противостоять система бикарбонатного
буфера внеклеточной жидкости. В этой связи острый лактатный ацидоз
можно считать одним из основных механизмов необратимости тяжелого
шока и клинической смерти. Локальная ишемия не приводит к лактатному
ацидозу типа А, так как одновременно с прекращением доставки в клетки
кислорода в них перестает поступать глюкоза как предшественник
молочной кислоты на пути ее синтеза. Кроме того, печень трансформирует
молочную кислоту, высвобождаемую локальным очагом ишемии, в глюкозу.
Лактатный ацидоз типа Б - это следствие повышенного высвобождения
молочной кислоты печенью. Его может вызвать угнетение трансформации
лактата в глюкозу гепатоцитами на уровне всей печени при печеночной
недостаточности. Лактатный ацидоз может быть следствием недостатка в
организме тиамина, обусловленного витаминным голоданием или алкоголизмом.
Дефицит тиамина приводит к лактатному ацидозу через угнетение
утилизации пирувата на путях обмена веществ. Аналогичным образом ведут к
лактатному ацидозу типа Б наследственные нарушения обмена веществ:
непереносимость фруктозы и недостаточная активность некоторых ферментов,
участвующих в глюконеогенезе. Отравление этанолом через его
интенсивную переработку в печени ведет к накоплению в цитозоле гепатоцитов
НАДН, что увеличивает образование лактата.
Опухоли печени при определенной распространенности поражения ее
паренхимы уменьшают очищение плазмы крови от лактата печенью,
вызывая лактатный ацидоз типа Б. Злокачественные новообразования другой
локализации ведут к лактатному ацидозу типа Б, высвобождая метаболиты,
которые угнетают глюконеогенез в печени. К ним в частности относят
метаболит триптофана, угнетающий активность ключевого фермента глюко-
неогенеза, фосфоенолпируваткарбоксикиназы. Предположительно,
злокачественные опухоли большой массы образуют и высвобождают молочную
кислоту столь интенсивно, что это превышает способность печени очищать
плазму крови от лактата и ведет к метаболическому ацидозу.
Некоторые из бактерий в просвете желудочно-кишечного тракта могут
трансформировать клетчатку, которую содержит пища, в органические
кислоты. Образование органических кислот бактериями из клетчатки
усиливают замедление прохождения пищевых масс по желудочно-кишечному
каналу (слепые петли кишки, обструкция, непроходимость) и изменение состава
кишечной флоры под влиянием терапии антибиотиками. В основном
бактерии образуют из клетчатки D-изомер молочной кислоты. Так как организм
человека метаболизирует этот изомер лактата медленнее, чем эндогенный
L-изомер, то абсорбция D-изомера ведет к метаболическому ацидозу с
увеличенным АПП. Существующие способы определения концентрации
лактата во внеклеточной жидкости и жидкой части плазмы крови
ориентированы на L-изомер, что затрудняет идентификацию этиологии метаболического
ацидоза данного генеза. Одновременно с развитием метаболического аци-
70
Глава 6
доза бактериальный рост в просвете кишечника ведет к усиленному
образованию бактериями в ее просвете аминов, попадание которых в
значительных количествах в кровь вызывает угнетение сознания. Часть D-изомера
лактата выводится из крови почками. Это обуславливает несоответствие
между увеличением АПП и снижением концентрации бикарбонатного
аниона в плазме крови.
Выделяют два вида кетоацидоза:
1. При сохраненной способности бета-клеток поджелудочной железы
образовывать и секретировать инсулин. При первом виде кетоацидоза он
представляет собой следствие торможения или недостаточной стимуляции
бета-клеток.
2. Вследствие патологических изменений бета-клеток (диабетический
кетоацидоз).
К кетоацидозу первого вида приводит гипогликемия как причина
угнетения секреции инсулина. Одна из частых причин гипогликемии у
больных - это голодание, которое редко снижает содержание глюкозы в плазме
крови до уровня более низкого, чем 3 ммоль/л. При метаболическом кетоа-
цидозе как следствии голодания концентрация бикарбонатного аниона в
плазме не падает ниже 18 ммоль/л, а ее анионный пробел не становится
большим, чем 19 ммоль/л. При этом умеренно выраженном
метаболическом ацидозе внутривенное вливание глюкозы прекращает кетогенез и
подвергает обратному развитию метаболический ацидоз.
Если гипогликемия как причина кетоацидоза связана с нарушениями
накопления и хранения гликогена в гепатоцитах, то она часто сочетается с
низким уровнем глюконеогенеза в печени. При этом снижено образование
глюкозы печенью из лактата. В результате метаболический ацидоз
обусловлен как кетогенезом, так и диссоциацией молочной кислоты во
внеклеточной жидкости (лактатный метаболический ацидоз типа Б). Поэтому при
таком патогенезе метаболического ацидоза, к развитию которого приводит
действие двух механизмов, в отличие от кетоацидоза, вызванного
голоданием, [НС03"] часто ниже, чем 18 ммоль/л. Этиопатогенетическая терапия
основного заболевания, и внутривенное вливание растворов глюкозы в
частности, устраняет кетоацидоз данного генеза.
Торможение секреции инсулина через возбуждение альфа-адреноре-
цепторов бета-клеток поджелудочной железы обуславливают действие
этанола как адреномиметика и побочный эффект лекарственных адрено-
миметических средств. Кроме того, оно может быть следствием
неспецифической защитно-патогенной стрессорной реакции в ответ на дефициты
ОВнЖ и объема циркулирующей крови. Поэтому, если при отравлении
этанолом частая и обильная рвота ведет к дефициту объема внеклеточной
жидкости, то кетоацидоз вследствие отравления этиловым спиртом
развивается быстрее. При такой этиологии кетоацидоза он может быть
тяжелым метаболическим ацидозом. У таких больных вследствие усиления
гликогенолиза и глюконеогенеза в результате падения секреции инсулина
часто выявляют гипергликемию. Внутривенное вливание им раствора
глюкозы устраняет кетоацидоз, только если его причина - это гипогликемия.
Нарушения кислотно-основного состояния
71
Интенсивная терапия в основном сводится к внутривенному вливанию
растворов натрия и калия хлорида, направленной на коррекцию
дефицитов ОВнЖ и калия в организме.
МЕТАБОЛИЧЕСКИЙ АЦИДОЗ, СВЯЗАННЫЙ С ДИСФУНКЦИЯМИ
НА УРОВНЕ СЕГМЕНТОВ НЕФРОНА
Почечная недостаточность может приводить к метаболическому
ацидозу с увеличением АПП. У здоровых людей и у пациентов с хронической
почечной недостаточностью нормальное высвобождение протонов при
утилизации аминокислот, поступивших в организм с пищей, составляет около
1 ммоль/кг массы тела за сутки. Это количество протонов нейтрализует
эквивалентная масса гидрокарбонатного аниона. Потери гидрокарбонатного
аниона из внеклеточной жидкости восполняет генерация новых анионов
почками, сопряженная с образованием и экскрецией NH4+. По мере прогрес-
сирования хронической почечной недостаточности падают экскреция NH4+
и образование почками бикарбонатных анионов. Снижение концентрации
бикарбонатного аниона во внеклеточной жидкости и жидкой части плазмы
ведет к метаболическому ацидозу. Одновременное снижение скорости клу-
бочковой фильтрации служит причиной падения экскреции органических
анионов, что повышает АПП. При этом рост АПП и снижение
концентрации бикарбонатного аниона во внеклеточной жидкости относительно
независимы друг от друга и не находятся в прямой связи.
Почечный тубулярный ацидоз - это метаболический ацидоз при
нормальном анионном пробеле плазмы и повышенной концентрации хлоридно-
го аниона во внеклеточной жидкости и жидкой части плазмы крови, ко-
торый представляет собой следствие несостоятельности почек как
эффектора систем удержания концентрации протонов во ВнЖ в нормаль-
ных пределах.
Если почки выделяют протоны с мочой недостаточно относительно их
высвобождения в клетки и во внутреннюю среду, то развивается почечный
тубулярный ацидоз.
В основе функции почек как эффектора систем удержания [Н+] в
нормальных пределах лежит активная секреция ионов водорода клетками тубулярно-
го эпителия в просвет канальцев. Нормальная секреция протонов эпителио-
цитами проксимальных сегментов нефрона - это необходимое условие
ненарушенных реабсорбции бикарбонатного аниона, экскреции протонов в
виде одноосновного фосфатного аниона (Н2Р04", «титруемая кислотность
мочи») и в виде NH4+ с конечной мочой. При обратном поступлении
бикарбонатных анионов из просвета канальцев нефрона в плазму крови, ни один
новый свободный ион водорода не высвобождается в клетки и во внеклеточную
жидкость. Если секреция протонов эпителиоцитами нормальна, то в
конечной моче практически нет бикарбонатного аниона. В этой связи появление
72
Глава 6
НС03" в конечной моче в значительных количествах (бикарбонатурию)
следует рассматривать как признак недостаточной секреции протонов в просвет
почечных канальцев или результат компенсаторного усиления ренальной
экскреции бикарбонатного аниона в ответ на развитие алкалоза.
Общее количество бикарбонатных анионов, которое из почек поступает
в плазму крови и во внеклеточную жидкость, составляют:
♦ число бикарбонатных анионов, реабсорбируемых в проксимальных
сегментах нефрона из ультрафильтрата в плазму, которое определяет
величина секреции протонов клетками тубулярного эпителия;
♦ количество бикарбонатных анионов, которое образуется в почках при
синтезе и экскреции NH4+.
Основная масса протонов, которые поступают в просвет канальцев в ходе
их активной секреции эпителиоцитами, попадает туда в проксимальных
сегментах нефрона. В просвете канальцев проксимальных сегментов бикарбо-
натные анионы из состава ультрафильтрата и активно секретируемые
протоны образуют угольную кислоту. Угольная кислота в просвете канальцев служит
субстратом карбоангидразы, которая ведет к распаду Н2С03 на воду и
углекислый газ. В результате возникает градиент напряжений углекислого газа
между просветом канальцев и цитозолем их эпителиоцитов, и углекислый газ
диффундирует в клетки тубулярного эпителия. В этих клетках карбоангидра-
за образует угольную кислоту из воды и углекислого газа. Диссоциация
образованной в эпителиоцитах угольной кислоты ведет к высвобождению в них
протонов и бикарбонатных анионов в количествах равных тем, которые
затрачиваются на синтез угольной кислоты в просвете канальцев
проксимальных сегментов. Все это заставляет определить поступление бикарбонатных
анионов из просвета канальцев проксимальных сегментов в плазму крови как
рециркуляцию, а не как реабсорбцию.
В проксимальных сегментах протоны секретируются в просвет
канальцев в обмен на натриевые катионы. Катионы натрия мигрируют в эпителио-
циты в силу градиента концентраций натрия между цитозолем
эпителиоцитов и просветом канальцев, в ультрафильтрате которых концентрация натрия
выше. В этой связи ключевыми ферментами активной секреции протонов и
рециркуляции бикарбонатного аниона в проксимальных сегментах нефрона
следует признать натрий-калий-АТФазу базолатеральных мембран
эпителиоцитов, которая перекачивает натрий в интерстиций почек, и карбоангидразу в
просвете канальцев (рис. 6.1).
Главная биологическая цель секреции протонов в проксимальных
отделах нефрона - это обеспечение рециркуляции бикарбонатных анионов,
которая предотвращает их потерю с конечной мочой. В этих сегментах
нефрона эпителиоциты секретируют в сутки в просвет канальцев 4000 ммоль
протонов, что позволяет возвратить в плазму эквивалентное количество
бикарбонатных анионов. При нормальных содержании бикарбонатного
аниона в плазме (25 ммоль/л) и скорости клубочковой фильтрации,
составляющей 180 л в день, за сутки в состав ультрафильтрата попадают 4500 ммоль
бикарбонатного аниона, из которых 4000 возвращаются в плазму крови в
ходе рециркуляции в проксимальных сегментах.
Нарушения кислотно-основного состояния
73
NaHCO
Рис. 6.1. Активная реабсорбция натрия и секреция протонов
в проксимальных сегментах нефрона
(ПК- просвет канальца, БЛМ - базолатералльная мембрана эпителиоцита)
Если на организм больного оказывают действие лекарственные
средства, которые снижают активность карбоангидразы в просвете канальцев,
то это тормозит реакцию распада угольной кислоты на углекислый газ и
воду в просвете канальцев. Одновременно замедляется реакция
образования угольной кислоты из протонов и бикарбонатных анионов в просвете
канальцев. В результате в просвете канальцев резко возрастает содержание
протонов, что тормозит их секрецию.
Гипокалия (патологическое снижение общего содержания калия в
организме) и рост напряжения углекислого газа в интерстиции и клетках
организма повышают содержание протонов в эпителиоцитах канальцев. Это
повышает трансмембранный перенос в просвет канальцев свободных ионов
водорода в обмен на натрий. Метаболический алкалоз, снижая содержание
протонов в эпителиоцитах, уменьшает их секрецию в просвет канальцев
проксимальных сегментов.
Рост общего содержания натрия в организме через рост ОВнЖ может
вызвать бикарбонатурию. Число бикарбонатных анионов, которые рецир-
кулируют в проксимальных сегментах, при этом не снижается. Увеличение
ОВнЖ повышает скорость клубочковой фильтрации (СКФ). Рост СКФ
увеличивает общее содержание бикарбонатных анионов в просвете канальцев
проксимальных сегментов. Это обуславливает преобладание общего
содержания НС03" в проксимальных канальцах над количеством протонов,
которые эпителиоциты секретируют в их просвет. Преобладание служит
причиной бикарбонатурии. Таков один из механизмов бикарбонатурии у больных,
которым вливают растворы гидрокарбоната натрия, тем самым увеличивая
общее содержание натрия в организме.
74
Глава 6
Гиперкальциемия и патологически низкий уровень секреции парат-гор-
мона увеличивают секрецию протонов в проксимальных отделах нефронов.
Недостаточность секреции протонов в проксимальных сегментах как
причина бикарбонатурии приводит к сдвигам кислотно-основного состояния,
которые проявляют себя:
♦ падением содержания бикарбонатного аниона во внеклеточной
жидкости;
♦ отсутствием роста [НС03] вследствие внутривенной инфузии
растворов, содержащих бикарбонатные анионы;
♦ рН мочи менее 7,0, несмотря на метаболический ацидоз.
Под проксимальным тубулярным (каналъцевым) метаболическим
ацидозом (почечный канальцевый ацидоз II типа) понимают патологическое
состояние вследствие недостаточности секреции протонов эпителиоцита-
ми канальцев проксимальных сегментов. При этом недостаточность
секреции протонов может быть одним из звеньев патогенеза синдрома Фанкони.
При цистинозе ацидоз данного генеза наследуется как фенотипический
признак по аутосомальному рецессивному типу. К нему могут приводить
хроническая гипокальциемия и вторичный гиперпаратиреоз. Почечный
канальцевый ацидоз может быть осложнением синдрома недостаточного
всасывания из просвета кишечника. Повреждения паренхимы почек под
влиянием лекарств и токсинов (свинец и др.), при поликистозном
перерождении мозгового слоя, вследствие множественной миеломы и при нефроти-
ческом синдроме также служат причиной почечного канальцевого ацидоза
второго типа.
Если причиной недостаточности секреции свободных ионов водорода
выступает низкий уровень активного перемещения натрия через базолате-
ральную мембрану эпителиоцитов, то в просвете канальцев
проксимальных сегментов возрастает содержание Na\ Увеличение концентрации
катиона натрия в просвете проксимальных канальцев происходит одновременно
с ростом содержания в них бикарбонатного аниона. При этом концентрация
бикарбонатного аниона в проксимальных канальцах растет вследствие
угнетения рециркуляции НСОэ", обусловленного низким уровнем секреции
протонов. В результате сумма положительных зарядов катиона натрия в
просвете проксимальных канальцев начинает в некоторой степени
уравновешиваться суммой зарядов бикарбонатных анионов. Это ведет к
перемещению хлоридных анионов из просвета канальцев в почечный ин-
терстиций данного уровня и далее в плазму крови. Таков механизм
возникновения патологически высокого содержания аниона С1" во
внеклеточной жидкости и жидкой части плазмы крови, гиперхлоремии, часто
сопутствующей почечному канальцевому ацидозу. Усиление натрийуреза,
которое у больных с почечным канальцевым ацидозом второго типа во
многом обусловлено тем, что бикарбонатные анионы задерживают в
просвете канальцев катионы натрия, вызывает дефицит ОВнЖ и через поли-
урию приводит к гипокалиемии.
При почечном канальцевом ацидозе II типа бикарбонатурия ведет к
снижению концентрации бикарбонатного аниона в плазме крови. По мере сни-
Нарушения кислотно-основного состояния
75
жения [НС03] падает концентрация бикарбонатного аниона в
ультрафильтрате клубочков нефронов, и низкое содержание протонов в просвете
проксимальных канальцев становится адекватным содержанию в нем НС03\
В результате падает число бикарбонатных анионов, поступающих в дис-
тальные сегменты нефрона, и бикарбонатурия временно исчезает.
Кислотно-основное состояние переходит на новый уровень, при котором нет
потерь бикарбонатного аниона, но концентрация протонов во внеклеточной
жидкости остается патологически высокой.
Рост объема внеклеточной жидкости, который увеличивает преднагруз-
ку сердца и минутный объем кровообращения (гиперволемическая гемоди-
люция у больного с нормальной насосной функцией сердца), приводит к
падению реабсорбции натрия в проксимальных сегментах, что может
служить причиной почечного тубулярного ацидоза и гиперхлоремии.
В дистальных отделах нефрона секреция протонов происходит в
основном в собирательных канальцах коркового и мозгового слоя. В этих
сегментах нефрона секреция свободных ионов водорода происходит активно с
участием Н+-АТФазы, локализованной в мембране эпителиоцита, обращенной
в просвет канальца.
Эпителиоциты дистальных сегментов образуют из глутамина,
синтезированного в печени, NH3 (аммиак), который, претерпев ряд
последовательных превращений, оказывается в просвете канальцев дистальных
сегментов. Там NH3 функционирует как акцептор активно секретируемых протонов.
Свободные ионы водорода и NH3 в просвете канальцев дистальных
сегментов образуют аммониевый катион (NH4+). Если образование аммиака в
почках недостаточно, то концентрация протонов в просвете канальцев
дистальных сегментов растет, что ведет к их обратной диффузии в эпителиоциты.
Образование одной молекулы аммиака и экскреция одного аммониевого
катиона с конечной мочой сопровождаются сопряженным с ними
образованием из глутамина одного бикарбонатного аниона.
Дистальный почечный канальцевый ацидоз - это патологическое
состояние вследствие недостаточного образования в клетках тубулярного
эпителия дистальных сегментов нефрона бикарбонатных анионов. Все виды ди-
стального почечного канальцевого ацидоза характеризует низкий уровень
экскреции аммониевого катиона с мочой.
Почечный канальцевый ацидоз I типа (классический дистальный
почечный канальцевый ацидоз) впервые описал Альбрихт в 1946 году. Его
характеризуют патологически высокий рН конечной мочи, бикарбонатурия, ги-
похлоремия и гипокалиемия. В основе развития ацидоза этого типа лежит
недостаточность активной секреции протонов в просвет канальцев
дистальных сегментов нефрона (рис. 6.2).
Непосредственными причинами падения секреции протонов в дисталь-
ные канальцы являются:
♦ недостаточная активность Н+-АТФазы;
♦ изменения трансмембранного потенциала эпителиоцитов вследствие
повышенного поступления в них хлоридного аниона, снижения
доставки натрия в дистальные сегменты при низкой скорости клубочко-
76
Глава 6
НСОз'
NH.
Конечная моча
Рис. 6.2. Основное звено патогенеза почечного тубулярного ацидоза первого типа:
падение активной секреции протонов в просвет канальцев дистальных сегментов нефрона
ведет к бикарбонатурии, росту рН конечной мочи (>6,0) и низкому содержанию в ней
аммониевого катиона
вой фильтрации, а также в результате действия средств, тормозящих
реабсорбцию Na+;
♦ снижение содержания аммиака как акцептора протонов в просвете
канальцев, которое через рост концентрации в нем Н+ тормозит
активную секрецию свободных ионов водорода;
♦ патологическое обратное поступление протонов в секретирующий
их эпителиоцит.
Первичный канальцевый ацидоз первого типа как моногенное
заболевание наследуется по аутосомальному доминантному типу. Вторичный
почечный канальцевый ацидоз первого типа выявляют у больных с системной
красной волчанкой и серповидноклеточной анемией. К нему также может
приводить кальциноз почек вследствие идиопатической гиперкальциурии
и первичного гиперпаратиреоза. Нефропатия, связанная с экзогенными
интоксикациями литием и другими токсичными веществами, также служит
одной из причин классического дистального канальцевого ацидоза.
Несмотря на низкую активность карбоангидразы в просвете
собирательных канальцев коркового и мозгового слоев, в этих отделах нефрона, как и в
его проксимальных сегментах, происходит рециркуляция бикарбонатного
аниона. При недостаточной активной секреции протонов в просвет
канальцев этих сегментов часть анионов НС03" не соединяется с ионами водорода
для образования угольной кислоты, и бикарбонатные анионы, оставаясь в
просвете канальцев, попадают в состав конечной мочи. Бикарбонатурия
(патологическое содержание бикарбонатного аниона в конечной моче),
вытесняя хлоридные анионы из состава мочи, приводит к гиперхлоремии. Кроме
того, бикарбонатурия усиливает ренальную экскрецию натрия, так как
натриевые катионы устремляются за бикарбонатными анионами в состав
конечной мочи. Усиление экскреции почками натрия снижает ОВнЖ.
Дефицит ОВнЖ запускает ренин-ангиотензин-альдостероновый механизм,
Нарушения кислотно-основного состояния
77
активация которого вызывает гипокалиемию через усиление ренальной
экскреции калия. Другая причина гипокалиемии - это полиурия вследствие
натрийуреза, усиленного бикарбонатурией.
Под почечным канальцевым ацидозом III типа в настоящее время
понимают почечный канальцевый ацидоз первого типа у детей.
Образование почками новых бикарбонатных анионов снижают:
♦ падение содержания аммиака как акцептора протонов в просвете
канальцев дистальных сегментов нефрона;
♦ снижение секреции свободных ионов водорода в просвет
собирательных канальцев.
Выделяют кортикальный и медулярный механизмы падения
содержания аммиака в просвете собирательных канальцев как акцептора
свободных ионов водорода. При действии первого из двух механизмов синтез
аммиака в почках снижен вследствие низкой скорости клубочковой фильтрации
игиперкалиемии у больных с почечной недостаточностью. Деструкция
клеток мозгового слоя паренхимы почек, обусловленная пиелонефритом или в
результате токсического действия лекарственных средств (ненаркотические
анальгетики и др.), приводит к снижению числа тубулярных эпителиоци-
тов, в которых не нарушено образование аммиака (медулярный механизм).
В результате возникают метаболический ацидоз с нормальным АПП,
снижение содержания в конечной моче аммониевого катиона и падение рН мочи
до уровня более низкого, чем 5,3. Снижение рН связано с избирательным
расстройством функций эпителиоцитов дистальных сегментов, когда они
теряют способность образовывать аммиак, но продолжают секретировать
протоны в просвет канальцев. В результате часть протонов не встречает своих
акцепторов в просвете собирательных канальцев, что снижает рН конечной
мочи. Внутривенное вливание растворов гидрокарбоната натрия у таких
больных ведет к росту [НС03] и увеличению содержания бикарбонатного
аниона в ультрафильтрате. Повышенное содержание бикарбонатного
аниона в ультрафильтрате приводит через соединение этих анионов с протонами
к образованию угольной кислоты в собирательных канальцах и ее
диссоциации, что повышает напряжение углекислого газа в конечной моче. Таким
образом, рост напряжения углекислого газа в ответ на внутривенное
вливание растворов, содержащих гидрокарбонат натрия, свидетельствует о
сохраненной способности эпителиоцитов канальцев дистальных отделов
нефрона активно секретировать протоны.
Если развитие метаболического ацидоза обуславливает низкий уровень
экскреции аммониевого катиона, связанный с гиперкалиемией, то такой
ацидоз называют почечным тубулярным ацидозом IV типа (гиперкалиеми-
ческий почечный тубулярный ацидоз). Гиперкалиемия представляет собой
звено патогенеза метаболического ацидоза, снижая образование аммиака в
почках. У части больных такой вид метаболического ацидоза вызывают
недостаточная концентрация в циркулирующей крови минералкортикоидов
и действие лекарственных средств антагонистов альдостерона как причины
снижения реабсорбции натрия из просвета канальцев нефрона, секреции в
канальцы протонов и выведения в них калия. Несмотря на то, что у многих
78
Глава 6
больных с почечным канальцевым ацидозом четвертого типа выявляют
почечную недостаточность в результате диабетической нефропатии или
интерстициального нефрита, падение скорости клубочковой фильтрации
нельзя признать единственной причиной гиперкалиемии у больных с данным
расстройством кислотно-основного состояния. Гиперкалиемия у таких
больных гораздо больше той, которую может вызвать снижение скорости
клубочковой фильтрации у пациентов с почечной недостаточностью и
почечным канальцевым ацидозом четвертого типа.
МЕТАБОЛИЧЕСКИЙ АЛКАЛОЗ
Метаболический алкалоз вызывают рост содержания во внеклеточной
жидкости и в эюидкой части плазмы крови бикарбонатных анионов и его
следствие, одновременное падение в них концентрации протонов. Рост
[НСОэ] обуславливают:
♦ усиленное образование бикарбонатного аниона в организме;
♦ снижение объема внеклеточной жидкости без уменьшения общего
содержания в организме бикарбонатного аниона.
Необходимое условие сохранения общего содержания бикарбонатного
аниона на неизменном уровне - это ненарушенное образование
бикарбонатных анионов почками.
Клинико-патофизиологическая классификация метаболического
алкалоза выделяет два его вида:
♦ метаболический алкалоз, который устраняет внутривенное вливание
растворов, содержащих хлорид натрия и хлорид калия;
♦ метаболический алкалоз, не устраняемый такой инфузией.
Метаболический алкалоз первого вида чаще всего выступает
следствием обильной рвоты, потерь желудочного содержимого по гастральному зонду
или побочного эффекта диуретиков (рис. 6.3). Его характеризует низкая
экскреция хлоридного аниона, если только на почки не действуют
мочегонные средства.
Метаболический алкалоз второго вида встречается реже. К нему ведут:
♦ высокая активность минералокортикоидов в циркулирующей крови;
♦ синдром Бартера;
♦ почечная недостаточность при повышенном эндо- или экзогенном
поступлении бикарбонатных анионов во внеклеточную жидкость.
При первом виде метаболического алкалоза у больных развивается
дефицит ОВнЖ. Одновременно в моче падает содержание О" до уровня более
низкого, чем 20 ммоль/л.
Потеря с содержимым желудка одного протона ведет к высвобождению
одного бикарбонатного аниона в париетальных клетках желудочного
эпителия. Для поддержания электронейтральности внутри эпителиоцита би-
карбонатный анион уходит из него во внеклеточную жидкость и в жидкую
Нарушения кислотно-основного состояния
79
Потери протонов и
хлоридных анионов с
желудочным содержимым
Просвет
желуцка
Активная секреция
протонов в просвет желудка
париетальными клетками
Поступление хлоридного
аниона из внутренней среды
в обкладочную клетку и в
просвет желудка
Поступление
бикарбонатного аниона
во внутреннюю среду
Рис. 6.3. Ведущее звено патогенеза метаболического алкалоза первого вида
часть плазмы крови, что повышает [НС03]. Для поддержания
электронейтральности внеклеточной жидкости «в обмен» на гидрокарбонатный анион
в просвет желудка уходит один анион С1\ Это снижает содержание С1" во
внеклеточной жидкости и плазме крови (гипохлоремический алкалоз).
Потери содержимого желудка при частой рвоте и по гастральному
зонду ведут к снижению содержания во внеклеточной жидкости хлоридного
аниона, который в ней замещает бикарбонатный анион. Если при этом не
падают объем внеклеточной жидкости и скорость клубочковой фильтрации,
то рост [НС03] обуславливает повышение содержания бикарбонатного
аниона в ультрафильтрате почечных клубочков. При этом секреция протонов в
просвет канальцев проксимальных сегментов нефрона не возрастает, так
как еще не действуют ее стимулы - дефицит ОВнЖ и снижение во
внеклеточной жидкости содержания калия и хлоридного аниона. В результате
низкой относительно содержания НС03" в ультрафильтрате секреции протонов
в проксимальных сегментах нефрона часть бикарбонатных анионов
остается в просвете канальцев и увлекает за собой в состав конечной мочи
катионы натрия. Поэтому у больных с потерями содержимого желудка еще до
появления признаков дефицита ОВнЖ и гиповолемии фиксируют высокое
содержание натрия в конечной моче.
По мере увеличения потерь кислого содержимого из желудка растут
[НС03] и ренальная экскреция натрия. К потерям натрия присоединяются
потери калия как следствие роста секреции альдостерона в ответ уже на
80
Глава 6
развившиеся дефицит ОВнЖ (следствие снижения содержания в организме
натрия) и падение объемной скорости кровотока в почках.
При таком происхождении метаболического алкалоза общее
содержание натрия в организме снижают как повышенная ренальная экскреция Na+,
так и потери натрия вместе с содержимым желудка. Возрастающие потери
натрия постепенно приводят к значительному дефициту ОВнЖ, который
ограничивает выделение бикарбонатного аниона с мочой. Во-первых,
дефицит ОВнЖ уменьшает скорость клубочковой фильтрации, снижая общее
число бикарбонатных анионов, которые попадают в состав
ультрафильтрата. Во-вторых, дефицит ОВнЖ как причина гиповолемии вызывает
усиленную секрецию минералкортикоидов. Повышенная концентрация минерало-
кортикоидов в циркулирующей крови увеличивает секрецию протонов в
собирательных канальцах, что усиливает реабсорбцию бикарбонатного
аниона и экскрецию калия. Поэтому тяжелый метаболический алкалоз первого
вида характеризуют гипокалиемия и клинические признаки дефицита ОВнЖ.
Мочегонные средства вызывают метаболический алкалоз прежде всего
через сужение внеклеточного жидкостного сектора. Дело в том, что
диуретики вызывают потери катиона натрия и хлоридного аниона, не увеличивая
экскрецию НС03". Потери натрия снижают ОВнЖ при неизменном общем
содержании в нём бикарбонатного аниона. В результате растет [НС03] и
развивается метаболический алкалоз. Кроме того, дефицит ОВнЖ через
снижение объема плазмы крови уменьшает скорость клубочковой фильтрации.
Падение скорости клубочковой фильтрации обуславливает неэффективность
защитной реакции выделения НС03' с мочой в ответ на рост [НС03~].
Гипокалиемия как побочный эффект диуретиков усиливает
образование бикарбонатного аниона почками.
Повышенный диурез как побочный эффект диуретиков вызывает
дефицит объемов внеклеточной жидкости и плазмы крови. Это служит
причиной усиленной секреции минералкортикоидов. Рост содержания
минералкортикоидов в крови увеличивает секрецию протонов в просвет канальцев
нефрона. Это способствует росту во внутренней среде бикарбонатных
анионов, образованных почками.
Если концентрации в моче Na+ и О" определяют во время
продолжающегося действия диуретиков, то они больше, чем 20 ммоль/л. В другой
момент они ниже этого уровня, как и при гипохлоремическом алкалозе
вследствие потерь содержимого желудка.
У некоторых больных внеклеточная жидкость может содержать
экзогенные анионы (в результате приема или введения антибиотиков и т.д.),
которые попадают в состав ультрафильтрата, но не реабсорбируются из
просвета канальцев. Эти анионы, находясь в просвете канальцев,
препятствуют реабсорбции натрия в проксимальных сегментах нефрона.
Растет число катионов натрия, поступающих в дистальные сегменты
нефрона. Это повышает активную реабсорбцию натрия, сопряженную с
секрецией в просвет канальцев протонов, калия и образованием НС03\
У таких больных повышенный уровень активной реабсорбции натрия в
дистальных сегментах нефрона не предотвращает потерь катиона с ко-
Нарушения кислотно-основного состояния
81
нечной мочой. Это снижает ОВнЖ, и его дефицит обостряет
метаболический алкалоз. Повышенные потери калия с мочой служат причиной
снижения его концентрации во внеклеточной жидкости. Гипокалиемия
(патологически низкая концентрация калия в плазме крови) еще в
большей степени усиливает образование почками бикарбонатного аниона и
метаболический алкалоз.
При хронической дыхательной недостаточности задержка во внутренней
среде бикарбонатного аниона почками компенсирует респираторный ацидоз.
При этом бикарбонатный анион в составе конечной мочи замещается на хло-
ридный. Представим, что у пациента после устранения гипокапнии есть
дефицит ОВнЖ. Мы уже знаем, что дефицит ОВнЖ сам по себе служит
причиной метаболического алкалоза. Кроме того, в данной ситуации он препятствует
выведению из внеклеточной жидкости с мочой бикарбонатных анионов,
которые были задержаны во внутренней среде во время гиперкапнии. Все это
приводит к постгиперкапническому метаболическому алкалозу. Следует
заметить, что длительной гиперкапнии часто сопутствуют критические
состояния, вызывающие обезвоживание и дефицит ОВнЖ.
Метаболический алкалоз, который не устраняет внутривенная инфу-
зия изоосмоляльных относительно внеклеточной жидкости растворов,
характеризуют :
♦ снижение концентрации хлоридного аниона в моче до уровня
меньшего, чем 20 ммоль/л;
♦ повышенный или нормальный ОВнЖ у большинства больных;
♦ обычно сопутствующая метаболическому алкалозу данного генеза
артериальная гипертензия.
Необходимое условие развития метаболического алкалоза второго вида
при любой его этиологии - это поступление катиона натрия и хлоридного
аниона по канальцам в собирательные протоки коркового слоя почки при
действии на эти сегменты нефрона повышенной концентрации минерал-
кортикоидов в циркулирующей крови. Это приводит к росту реабсорбции
натрия и усилению экскреции протонов и калия. В результате роста
выделения калия развивается гипокалиемия. Гипокалиемия как стимул
образования аммиака в эпителиоцитах канальцев увеличивает генерацию
бикарбонатных анионов почками. Увеличение образования аммиака ведет к росту
выделения протонов в виде аммониевых катионов и метаболическому
алкалозу. Одновременно гипокалиемия снижает скорость клубочковой
фильтрации, что уменьшает эффективность бикарбонатурии как компенсаторной
реакции на метаболический алкалоз.
Рост активности минералкортикоидов в циркулирующей крови
обуславливают:
♦ активация ренин-ангиотензин-альдостеронового механизма в ответ на
дефицит ОВнЖ;
♦ избыточная секреция альдостерона при гиперплазии или аденоме
надпочечников;
♦ опухоль гипофиза как причина патологически интенсивной секреции
адренокортикотропного гормона, которая увеличивает содержание в
82
Глава 6
циркулирующей крови кортизола, обладающего свойствами минерал-
кортикоида.
При реноваскулярной артериальной гипертензии и ренинпродуцирую-
щих опухолях почек рост концентрации альдостерона в крови вторичен по
отношению к высокой активности в ней ренина.
Ятрогенный метаболический алкалоз второго вида обусловлен
действием лекарств, обладающих свойствами минералкортикоидов.
До сих пор неясным остается патогенез метаболического алкалоза
второго вида, связанного с дефицитом в организме магния.
При синдроме Бартера, этиология и патогенез которого также остаются
неясными, артериальной гипертензии нет, а ОВнЖ снижен, что проявляет
себя ортостатической артериальной гипотензией и другими признаками
гиповолемии. Кроме того, у больных с этим синдромом выявляют рост
экскреции натрия с мочой, метаболический алкалоз и гипокалиемию.
Исторический интерес представляет метаболический алкалоз,
вызванный обильным употреблением в пищу молочных продуктов как причина
роста содержания во внеклеточной жидкости бикарбонатного аниона у
пациентов с низкой скоростью клубочковой фильтрации. Низкая скорость клу-
бочковой фильтрации снижает эффективность бикарбонатурии как
компенсаторной реакции.
У больных с метаболическим алкалозом первого вида его устраняет
восстановление объема внеклеточной жидкости внутривенным вливанием изо-
осмоляльных растворов. Во-первых, такая инфузия восстанавливает массу
внеклеточной жидкости, дефицит которой служит стимулом для активации
ренин-ангиотензин-альдостеронового механизма как причины
метаболического алкалоза. Устранение дефицита ОВнЖ увеличивает скорость
клубочковой фильтрации, что усиливает бикарбонатурию, компенсирующую
метаболический алкалоз. Коррекция дефицита в организме калия нормализует
кислотно-основное состояние только в тех редких случаях, когда гипокалие-
мия представляет собой ведущую причину метаболического алкалоза. При
такой гипокалиемии содержание К+ в плазме крови меньше чем 2,4 ммоль/л.
Если причиной вторичного альдостеронизма и низкой скорости
клубочковой фильтрации, вызывающих метаболический алкалоз, является
застойная сердечная недостаточность, инфузию изоосмоляльных растворов нельзя
признать патогенетически обоснованным способом коррекции
метаболического алкалоза. У таких больных, кроме кардиотропной фармакотерапии и
других лечебных воздействий, направленных на усиление насосной функции
сердца, при гипокалиемии метаболический алкалоз может устранить
внутривенное вливание растворов калия хлорида. Инфузия растворов калия
хлорида устраняет гипокалиемию, которая снижает скорость клубочковой
фильтрации и увеличивает образование почками бикарбонатных анионов.
Если метаболический алкалоз второго вида развивается у больных с
почечной недостаточностью, как это бывает при реноваскулярной
артериальной гипертензии, то концентрация протонов во внеклеточной жидкости и
жидкой части плазмы крови падает до уровня более низкого чем 20 нмоль/л
при рН больше 7,7. Такое значительное возрастание рН связано с неэффек-
Нарушения кислотно-основного состояния
83
тивностью при низкой скорости клубочковой фильтрации бикарбонатурии
как компенсаторной реакции в ответ на метаболический алкалоз.
Метаболический алкалоз такой степени служит показанием к внутривенному
вливанию растворов соляной кислоты или хлорида аммония. Если таким
больным проводят гемодиализ, то в растворе для гемодиализа снижают
содержание бикарбонатных и ацетатных анионов для уменьшения [НС03].
Если у больного выявляют избыточную секрецию альдостерона как
причину метаболического алкалоза второго вида, то применение лекарственных
средств, блокирующих реабсорбцию натрия в собирательных канальцах
(амилорид и др.) или антагонистов альдостерона выступает
патогенетическим средством коррекции нарушений кислотно-основного состояния.
Следует заметить, что все перечисленные способы коррекции
метаболического алкалоза не ориентированы на этиологический фактор болезней
и патологических состояний, вызывающих метаболический алкалоз. Так,
при внутрибрюшном абсцессе как причине высокой кишечной
непроходимости, частой рвоты и метаболического алкалоза, нормального кислотно-
основного состояния часто не восстановить без лапаротомии с целью
радикальной санации очага гнойной инфекции.
Глава 7
НАРУШЕНИЯ ПЕРИФЕРИЧЕСКОГО КРОВО-
И ЛИМФООБРАЩЕНИЯ
Нормальная жизнедеятельность организма во многом обусловлена
адекватной работой органов крово- и лимфообращения, которые находятся в
морфофункциональном единстве. При этом сердце - источник кровотока,
сосуды - магистрали кровераспределения и лимфосбора, микроциркуля-
торное русло - плацдарм транскапиллярного обмена и тканевого
метаболизма.
Значение нарушений нормального крово- и лимфообращения состоит
в том, что они ведут к ухудшению тканевого (клеточного) метаболизма с
последующим повреждением структур ткани (клетки) через развитие того
или иного вида дистрофии или некроза. При возникновении
патологического процесса в сердце расстройства его деятельности приводят к общим,
а расстройства функционирования сосудистого русла на том или ином
участке - к местным нарушениям крово- и лимфообращения. Местные
нарушения кровообращения могут стать причиной общих (например,
кровоизлияние в головной мозг). Общие и местные нарушения крово- и
лимфообращения наблюдаются при различных болезнях (травмах), могут
осложнять их течение и приводить к опасным последствиям. Различают
следующие виды нарушений кровообращения: полнокровие (артериальное и
венозное), малокровие, тромбоз, эмболия, нарушения микроциркуляции,
лимфообращения и др., причём многие из нарушений кровообращения
патогенетически связаны и могут находиться в причинно-следственной
взаимосвязи.
ПОЛНОКРОВИЕ
Полнокровие (гиперемия) -это повышение содержания циркулирующей
крови в том или ином участке сосудистой сети. Различают артериальное и
венозное полнокровие. Каждая из указанных разновидностей может носить
общий и местный характер.
Артериальное полнокровие - это увеличенное кровенаполнение органов,
тканей вследствие повышенного притока артериальной крови. Общее
артериальное полнокровие отражает увеличение сердечного выброса и
объёма циркулирующей крови, что соответствует понятию «плетора» и
необязательно сопровождается увеличением общего количества эритроцитов
(эритремией). Клинически это проявляется повышением систолического
артериального давления, покраснением кожи и слизистых оболочек, усиле-
Нарушения периферического крово- и лимфообращения
85
нием обмена веществ и повышением температуры тела. Патологическая
общая артериальная гиперемия возникает в условиях гипертермии при
общем перегревании организма, а также при лихорадке у больных с
инфекционными заболеваниями. Она может быть следствием быстрого падения
барометрического давления (общая вакатная гиперемия).
Местное артериальное полнокровие - наиболее частая форма
артериальной гиперемии. Ее внешними проявлениями являются покраснение,
пульсация мелких артерий, увеличение объёма (припухание) ткани и
повышение температуры в месте усиления артериального кровотока.
Различают следующие виды патологической местной артериальной
гиперемии: ангионевротическая, воспалительная, коллатеральная, постише-
мическая, вакатная и гиперемия на почве артериовенозного свища.
Ангионевротической гиперемией называется местное расширение
артерий, вызываемое нервными импульсами или гуморальными факторами.
Примером может служить гиперемия лица и слизистых оболочек при
некоторых инфекционных заболеваниях, сопровождающихся поражением
симпатической нервной системы.
Воспалительная гиперемия является проявлением компенсаторного
регулирования кровотока при воспалении. Она, в частности, компенсирует
изменения микроциркуляции, возникающие вследствие резкого увеличения
количества функционирующих капилляров, обеспечивая локальное
усиление кровоснабжения в очаге местной воспалительной реакции.
Коллатеральная гиперемия вокруг ишемического очага является
проявлением рефлекторного расширения коллатеральных сосудов при
нарушении кровотока по магистральным артериям. Недостаточность
коллатерального кровообращения в этих случаях способствует развитию инфаркта или
ишемической гангрены.
Гиперемия после ишемии (постишемическая, постанемическая)
развивается в тех случаях, когда быстро ликвидируется причина, вызвавшая
нарушение кровотока по приносящей артерии, т.е. за счет увеличения
артериального кровенаполнения в предварительно ишемизированных тканях. Это
может происходить, например, после быстрого снятия ранее наложенных
жгутов. Частным случаем этого вида гиперемии является так называемая
посткомпрессионная гиперемия, возникающая после временного сдавления
сосудов органа или ткани. Такие условия возникают, например, после
одномоментного извлечения больших объёмов транссудатов и экссудатов из
полостей или удаления крупных опухолевых узлов, сдавливавших
прилежащие артерии. При этом может возникнуть обморок в связи с относительным
полнокровием головного мозга.
Вакатная гиперемия (от лат. vacuus - пустой) развивается при
уменьшении барометрического давления над какой-либо частью тела. Например, при
постановке медицинской банки или разного рода присосок.
Гиперемия на почве артерио-венозного свища - это полнокровие,
возникающее при образовании соустья между артерией и веной. Она может
наблюдаться при повреждении артериальных и венозных сосудов. При этом
артериальная кровь под давлением устремляется в венозное русло, приводя
86
Глава 7
к полнокровию тканей. Этот вид гиперемии может наблюдаться и при
врожденных аневризмах сосудов головного мозга.
Исход артериальной гиперемии может быть различным. В большинстве
случаев она сопровождается усилением обмена веществ и функции органа,
ткани, что является приспособительной реакцией. Однако резкое
расширение атеросклеротически изменённого сосуда может сопровождаться
разрывом его стенки и кровоизлиянием в ткани (например, головного мозга).
Венозное полнокровие - повышенное кровенаполнение венозных сосудов
органа или ткани в результате затруднения оттока крови по венам. Об-
щая венозная гиперемия развивается в ответ на центральную венозную ги-
пертензию при ослаблении функции правого желудочка сердца,
уменьшении присасывающего действия грудной клетки (экссудативный плеврит,
гемоторакс), затруднении кровотока в малом круге кровообращения
вследствие пневмосклероза, эмфиземы лёгких, тромбоэмболии, а также
ослабления функции левого желудочка. Общее венозное полнокровие,
обусловленное левожелудочковой недостаточностью с преимущественным нарушением
кровотока в малом круге кровообращения, характеризуется наиболее
выраженными «застойными» (отёчными, дистрофическими, склеротическими)
изменениями в лёгких. По характеру течения заболеваний сердца общее
венозное полнокровие бывает острым или хроническим. При этом клици-
чески в острых случаях преобладают явления отёка лёгких и дыхательной
недостаточности: обильная пенистая мокрота с примесью крови, влажные
и крупнопузырчатые хрипы, притупление перкуторного звука,
рентгенологически - расширение корней лёгких, а также явления гидроторакса.
Кожный покров и видимые слизистые бледные с акроцианозом. При
хронической левожелудочковой недостаточности на этом фоне прогрессируют
дистрофические и склеротические изменения в лёгких с развитием
синдрома «бурой индурации лёгких». Последний морфологически
характеризуется сочетанием диффузно-очагового склероза и гемосидероза лёгких. При
этом мокрота приобретает «ржавую» окраску за счёт значительного
количества сидерофагов, получивших название «клеток сердечных пороков».
Наиболее информативными показателями, свидетельствующими о нарушении
функции левого желудочка, являются снижение фракции изгнания и рост
давления крови в легочной артерии и левом предсердии.
Общее венозное полнокровие, обусловленное преимущественно
снижением насосной функции правого желудочка (правожелудочковая
недостаточность), характеризуется синюшностью тёплой кожи и видимых слизистых
(«тёплый» цианоз), набуханием шейных вен и резким увеличением размеров
и уплотнением печени, селезёнки (цианотическая индурация), почек (циано-
тическая индурация) и других внутренних органов, а также
распространённым отёком тканей (анасарка) и появлением полостных транссудатов,
особенно заметных в полости брюшины (асцит). На электрокардиограмме, как
правило, обнаруживают признаки перегрузки правых отделов сердца.
Местное венозное полнокровие чаще всего является следствием
образования обтурирующих тромбов (флеботромбоза), сдавления или
прорастания вены опухолью, а также наложения «венозного» жгута. В отдельных
Нарушения периферического крово- и лимфообращения
87
случаях предрасполагающим моментом венозной гиперемии является
конституциональная слабость эластического аппарата вен, недостаточное
развитие и пониженный тонус гладкомышечных элементов их стенок.
Профессии, требующие ежедневного длительного пребывания в вертикальном
положении, способствуют венозной гиперемии в дистальных отделах
нижних конечностей у лиц с конституционно обусловленной
неполноценностью эластических и гладкомышечных элементов стенки венозных сосудов.
Особой разновидностью местного венозного полнокровия считается
коллатеральное венозное полнокровие. Оно возникает, например, при циррозе
печени, а также при тромбозе воротной или печёночной вен (синдром Бад-
да-Киари), следствием которых становится сброс венозной крови,
оттекающей из кишечника, в обход печени через порто-кавальные анастомозы (вены
пищевода, желудка, передней брюшной стенки, таза).
Общая симптоматика при венозной гиперемии связана с уменьшением
объемной скорости кровотока и переполнением сосудов кровью,
гемоглобин которой преимущественно восстановлен. В результате этого ткани
испытывают недостаточность кровоснабжения и гипоксию. Тонкостенные
вены могут сдавливаться в участках резкого повышения гидростатического
тканевого давления, например, в очагах воспаления. Продолжительный
венозный застой сопровождается значительными изменениями элементов
стенки вены, их атрофией. Наряду с этим на участке местной венозной
гиперемии происходит заместительное разрастание соединительной ткани
(склероз). Классическим примером является цирроз печени, вызванный
венозным застоем при недостаточности функции сердца.
МАЛОКРОВИЕ
Малокровие (анемия) - сниженное кровенаполнение сосудов или
обескровливание тела, органов. Различают общее и местное малокровие. Общее
малокровие характеризуется абсолютным уменьшением количества
эритроцитов и снижением содержания гемоглобина в единице объёма крови.
Важнейшими патофизиологическими характеристиками общего
малокровия являются абсолютное уменьшение объёма циркулирующей крови (ги-
поволемия) со снижением венозного возврата, системной гипотонией с
тахикардией и тахипноэ, а также так называемая централизация
кровообращения - перераспределение кровотока преимущественно в жизненно важные
органы (головной мозг, сердце, лёгкие). От истинной анемии следует
отличать гемодилюцию, т. е. разжижение крови за счёт обильного притока
тканевой жидкости в сосудистое русло, наблюдаемое, например, при острой
почечной недостаточности с анурией или после операций с применением
искусственного кровообращения. Подобная псевдоанемия исчезает по мере
того, как ликвидируется вызвавший её «отёк крови». Наоборот, истинная
анемия может маскироваться сгущением крови, например, при обильной
88
Глава 7
рвоте или профузных поносах; при этом вследствие уменьшения объёма
циркулирующей плазмы количество эритроцитов в единице объёма крови
(гематокрит) может оказаться нормальным или даже повышенным.
Целесообразно отличать общее малокровие с абсолютной гиповолемией,
обусловленное кровопотерей, от состояния относительной гиповолемии, когда имеет
место несоответствие объёма циркулирующей крови увеличивающейся
ёмкости сосудистого русла. Такое состояние лежит в основе патогенеза
анафилактического шока и других реакций, проявляющихся развитием коллап-
тоидных (гипотензивных) состояний, в том числе при инфекционно-токси-
ческом коллапсе («септическом шоке»). Относительное уменьшение объёма
циркулирующей крови в последнем случае связывают с генерализованным
расширением резистивных сосудов (артерий небольшого диаметра и арте-
риол) и секвестрацией (выключением из кровотока) в них и в зонах
микроциркуляции значительной части крови. При этом цвет кожных покровов
становится серо-землистым с мраморным рисунком.
Неизбежным следствием абсолютной гиповолемии становятся тяжёлые
генерализованные нарушения микроциркуляции, неэффективная перфузия
тканей и гипоксия. Кожа и видимые слизистые при этом бледнеют. Одним
из механизмов, компенсирующих абсолютную гиповолемию при кровопо-
тере, является выброс в сосудистое русло из интерстициального
пространства жидкости, однако в объёме не более 0,5 л.
Местное малокровие или ишемия (греч. ischo — задерживать + haima -
кровь) -уменьшение кровенаполнения ткани, органа, части тела в
результате недостаточного притока крови. Возникающие при этом
патологические изменения в тканях определяются в конечном счете тяжестью и
длительностью кислородного голодания - гипоксии.
Местное малокровие возникает в случае, когда значительно
увеличивается сопротивление в артериях, приносящих кровь в данную область, и
отсутствует или недостаточен коллатеральный приток крови. В зависимости
от причин и условий возникновения местного малокровия различают
следующие виды ишемии: обструкционную, обтурационную, компрессионную,
ангиоспастическую, перераспределительную и др.
Обструкционной ишемией называется местное малокровие,
возникающее в результате механического разрушения сосудов при травме.
При так называемой обтурационной ишемии препятствием для
нормального кровотока может являться закрытие сосуда эмболом, тромбом, атерос-
клеротической бляшкой и т.д.
Местное компрессионное малокровие может возникнуть в результате
сдавления сосудов органа, например, головного мозга при значительном
повышении внутричерепного давления, или вследствие сдавления сосуда
растущей опухолью. Вариантом этого вида ишемии является наложение
жгута или перевязка артериального сосуда.
Специально выделяют ангиоспастическую (рефлекторную) ишемию,
возникающую вследствие спазма питающей артерии, например, вазоконст-
рикции, обусловленной действием нервных влияний (в т. ч. при болевом
раздражении) или гуморальных факторов, циркулирующих в крови или об-
Нарушения периферического крово- и лимфообращения
89
разующихся внутри сосудистой стенки. Выделяют ишемию, обусловленную
значительным увеличением вязкости крови в мелких сосудах, особенно если
оно сочетается с вазоконстрикцией. Такая ишемия имеет значение в
патогенезе ишемической болезни сердца.
Перераспределительная ишемия возникает в результате
межрегионального, межорганного перераспределения крови. Такова, в частности,
природа обморочных состояний, наблюдаемых при ортостатическом коллапсе, а
также при быстром извлечении значительных объёмов жидкости у больных
с асцитом. Ишемия характеризуется следующими признаками: побледне-
нием ишемизированных тканей, органов или частей тела, снижением
температуры, нарушением чувствительности (онемение, покалывание),
болевым синдромом, уменьшением скорости кровотока и объема органа,
снижением артериального давления на участке артерии, расположенном
ниже препятствия, понижением напряжения кислорода в ишемизирован-
ном участке, уменьшением образования межтканевой жидкости и
снижением тургора тканей, а также дистрофическими и некротическими
изменениями с нарушением функции органа или ткани. Указанные проявления ишемии
зависят от особенностей причины и продолжительности её действия. Так,
малокровие вследствие спазма артерии обычно непродолжительно и не
вызывает особых расстройств. Однако, при длительных спазмах возможно
развитие дистрофических изменений и даже ишемического некроза (инфаркта
или гангрены). Острое обтурационное малокровие почти всегда приводит к
инфаркту. Если же закрытие просвета артерии происходит медленно, то
кровенаполнение может быть сохранено за счёт коллатералей.
ЭМБОЛИИ
Эмболия (греч. embole - вторжение) - патологический процесс,
обусловленный циркуляцией в крови или лимфе различных субстратов (эмбо-
лов), не встречающихся в норме и способных вызвать острую окклюзию
сосуда с нарушением кровоснабжения органа или ткани. Перемещение эм-
болов происходит в основном по трём направлениям:
♦ из венозной системы большого круга кровообращения и правых
отделов сердца - в сосуды малого круга кровообращения;
♦ из лёгочных вен, левой половины сердца и аорты - в артерии
большого круга кровообращения (сердца, мозга, почек, селезёнки,
кишечника, конечностей);
♦ из ветвей портальной системы - в воротную систему.
Иногда, при незаращённом овальном отверстии, наличии дефекта меж-
предсердной или межжелудочковой перегородки со сбросом крови из
правых отделов сердца в левые, может наблюдаться парадоксальная эмболия
большого круга кровообращения, минуя разветвления легочных сосудов.
Исключением также является ретроградная эмболия, когда движение эмбо-
90
Глава 7
ла подчиняется не гемодинамическим законам, а силе тяжести эмбола.
Такая эмболия развивается в крупных венозных стволах при замедлении
кровотока и уменьшении присасывающего действия грудной клетки.
Клинические проявления эмболии определяются её локализацией (малый
или большой круг кровообращения), особенностями ангиоархитектоники, в
частности, состоянием коллатерального кровообращения и его нейрогумо-
ральной регуляции, размером и составом эмболов, их общей массой, темпом
попадания в кровоток, реактивностью организма. Эмболы могут быть
единичными и множественными. В зависимости от величины эмбола выделяют
эмболию крупных сосудов и микроциркуляторную эмболию. Механическая
окклюзия артерии эмболом сопровождается региональной вазоконстрикци-
ей. В результате этого сокращается или полностью перекрывается просвет
артерии и снижается давление крови дистальнее места окклюзии с
последующим развитием сначала ишемии, а затем ишемического некроза
(инфаркта). Следует иметь в виду, что эмболия не может быть сведена только к
простой окклюзии сосуда. Любая эмболия вызывает расстройства
физиологического равновесия в функциональных системах организма. Последние
в значительной мере связаны с рефлекторными явлениями за пределами
участка эмболизации. Так, в случаях эмболии бифуркации лёгочной артерии
возникает пульмокоронарный рефлекс, приводящий к быстрому снижению
системного артериального давления и остановке сердца. В последнее время
установлено, что окклюзия эмболами легочных артериол, где расположено
множество барорецепторов, приводит к выраженным нарушениям системной
гемодинамики в виде коллапсов, вплоть до остановки сердца.
В зависимости от природы эмболов различают следующие виды эмболии:
♦ тромбоэмболия,
♦ жировая,
♦ тканевая (клеточная),
♦ микробная,
♦ воздушная,
♦ газовая,
♦ инородными телами.
Самым распространённым видом эмболии является тромбоэмболия.
Наиболее часто встречается тромбоэмболия малого крута, кровообращения,
так как тромбоз чаще возникает в венах большого круга кровообращения,
особенно системы нижней полой вены (например, глубоких вен голени, вен
параректальной и паравезикальной клетчатки), или в камерах правой
половины сердца. Наиболее частой и опасной являются эмболия лёгочного
ствола, основных ветвей лёгочных артерий и микротромбоэмболия лёгких.
Установлено, в частности, что мелкие тромбоэмболы порядка 40-100 мкм в
диаметре могут вызвать более значительные нарушения лёгочной и
системной гемодинамики, чем крупные эмболы. Источником тромбоэмболии
сосудов большого круга являются тромбы, которые образуются в левой
половине сердца (при эндокардите, аневризме) или в аорте и отходящих от неё
артериях (при атеросклерозе). Эмболия воротной вены, хотя и встречается
реже, чем эмболия малого и большого круга кровообращения, имеет харак-
Нарушения периферического крово- и лимфообращения
91
терную клиническую симптоматику и приводит к тяжёлым гемодинамичес-
ким последствиям. Благодаря большой вместимости портального русла
окклюзия эмболом главного ствола воротной вены приводит к увеличению
кровенаполнения органов брюшной полости (желудка, кишок, селезёнки) и
развитию синдрома портальной гипертензии. В основе общих нарушений
гемодинамики лежит преимущественно уменьшение массы циркулирующей
крови, вызванное скоплением её в портальном русле, что может иметь
танатологическое значение.
В последние несколько десятилетий отмечено учащение
тромбоэмболии, как непосредственных причин смерти. Это объясняют, с одной
стороны, ростом числа травматичных и обширных хирургических вмешательств,
с другой - ухудшением экологической обстановки на планете.
Жировая эмболия - перенос с кровью и окклюзия мелких сосудов
внутренних органов каплями нейтрального жира (липидов). Современные
данные указывают на то, что основным источником жировой эмболии
являются продукты общего нарушения обмена липидов при гипоксии и шоке, когда
хиломикроны плазмы крови агрегируются в крупные жировые капли. В
развитии жировой эмболии свою роль играет снижение способности
эндотелия противостоять эмульгации липидов в сосудистом русле. Кроме этого,
источниками липидов, образующих жировые капли, могут являться
мембранные структуры клеток крови (например, при их повреждении в
аппаратах искусственного кровообращения во время операций на сердце).
Описана жировая эмболия при лечении масляными растворами лекарственных
веществ или в случаях несоблюдения правил введения жировых эмульсий,
применяемых для парентерального лечения.
Необходимо дифференцировать жировую эмболию от тканевой
(клеточной) эмболии липоцитами, которая отмечается при тяжёлых травмах с раз-
мозжением жировой ткани (например, костного мозга, подкожной жировой
клетчатки). Тканевая эмболия - это окклюзия кровеносных сосудов
обрывками собственных тканей и (или) продуктами их распада. Она может
встречаться у рожениц при попадании околоплодных вод в венозные сосуды
матки. Особый вид тканевой эмболии составляет эмболия отделившимися от
основного узла клетками злокачественной опухоли. Следствием
последнего вида эмболии может быть начало роста этой злокачественной опухоли в
месте остановки эмбола.
Микробная эмболия представляет собой тяжёлое осложнение инфекци-
онно-воспалительного процесса практически любой локализации.
Встречается в двух основных клинико-морфологических формах - бактериемии
и сепсисе. Бактериемия служит механизмом диссеминации инфекции из
первичного очага. Окклюзия сосудов микроорганизмами или чаще
фрагментами инфицированного тромбоэмбола, подвергшегося гнойному
расплавлению, приводит к развитию септикопиемии и образованию
вторичных гнойных очагов в различных органах и тканях. Последние два вида
эмболии (опухолевой и микробной) получили особое название.- метастази-
рование. Таким образом, метастазирование - это перенос током крови эм-
болов, содержащих такие элементы, которые способны расти и развиваться
92
Глава 7
на новом месте. Патологическое образование, возникающий на месте
такого переноса, называется метастазом (перемещение местоположения).
Воздушная эмболия (аэроэмболия) развивается в результате попадания
в кровоток пузырьков воздуха. Чаще встречается венозная воздушная
эмболия, которая возникает в момент вдоха при ранениях ярёмных,
подключичных, бедренных вен, а также синусов твёрдой мозговой оболочки, которые
слабо спадаются. Этому в значительной мере способствует близкое к нулю
или отрицательное центральное венозное давление. Описаны редкие
случаи венозной воздушной эмболии при зиянии поверхностных вен матки
после родов и при случайном введении воздуха во время внутривенных
инъекций, особенно при поднятой кверху конечности у лежащего человека.
Артериальная воздушная эмболия большого круга кровообращения может
наблюдаться при некоторых нейрохирургических операциях, проводимых
в положении больного сидя, оперативных вмешательствах на лёгких,
операциях на сердце и аорте с использованием аппарата искусственного
кровообращения, наложении диагностического или лечебного пневмоторакса, а
также при деструктивных процессах или повреждении (ранение, взрывная
ударная волна) лёгкого.
Газовая эмболия возникает при высвобождении растворённых в крови
летучих веществ - газов, обычно азота. Это бывает связано с перепадами
внешнего давления и изменениями способности крови накапливать в растворе азот,
поступающий в лёгкие с вдыхаемым воздухом. В частности, такие условия
могут наступать при быстром подъёме с глубины у кессонных рабочих,
водолазов и моряков-подводников, что иногда случается в аварийных ситуациях.
Такое же явление может наблюдаться у лётчиков при скоростных подъёмах, а
также у членов экипажей авиационных и космических летательных
аппаратов при аварийной разгерметизации кабины. Эмболия инородными телами
(дробью, пулей, осколком снаряда, обрывками одежды при огнестрельных
ранениях и др.) встречается крайне редко. В практике отделений
интенсивной терапии, преимущественно у больных, находящихся в состоянии
психомоторного возбуждения, изредка наблюдается эмболия фрагментами
катетера, введённого в крупную вену, и обломками инъекционных игл. Известны
случаи эмболии кальцинированными фрагментами атеросклеротических
бляшек, попадающих в кровоток при операциях на обызвествлённых клапанах
сердца или при изъязвлении атеросклеротической бляшки.
НАРУШЕНИЯ МИКРОЦИРКУЛЯЦИИ
Нарушения микроциркуляции принадлежат к типовым патологическим
процессам, лежащим в основе многих заболеваний и травм. Расстройства в
системе микроциркуляции можно разделить на 4 большие группы:
нарушения в стенках микрососудов, внутрисосудистые нарушения, внесосудистые
изменения и комбинированные расстройства.
Нарушения периферического крово- и лимфообращения
93
Патологические расстройства на уровне сосудистых стенок микрососудов
выражаются в изменениях формы и расположения эндотелиальных клеток.
Одним из наиболее часто наблюдающихся нарушений этого типа является
повышение проницаемости сосудистой стенки, которые также могут
вызывать прилипание (адгезию) к их поверхности форменных элементов крови,
опухолевых клеток, инородных частиц и др. Проникновение (диапедез)
форменных элементов через стенки микрососудов имеет место после
прилипания соответствующих клеток к эндотелию. Следствием нарушения
целостности при повреждении стенки микрососудов являются микрокровоизлияния.
Внутрисосудистые нарушения микрогемоциркуляции крайне
разнообразны. Среди них чаще всего встречаются изменения реологических свойств
крови, связанные прежде всего с агрегацией (англ, aggregate - соединение
частей) эритроцитов и других форменных элементов крови. Такие внутри-
сосудистые расстройства, как замедление кровотока, тромбоз, эмболия
также в значительной степени зависят от нарушения реологических свойств
крови. Следует отличать агрегацию форменных элементов крови от их
агглютинации. Первый процесс характеризуется обратимостью, в то время как
второй необратим. Крайняя степень выраженности агрегации форменных
элементов крови получил название «сладж» (англ. sludge - тина, густая
грязь, болото). Главным результатом таких изменений является увеличение
вязкости крови вследствие слипания эритроцитов, лейкоцитов и
тромбоцитов. Такое её состояние в значительной степени ухудшает кровоснабжение
тканей через микрососуды и снижает объём циркулирующей крови. В
потоке крови при этом наступает разделение (сепарация) на клетки и плазму.
Ведущая роль в агрегации эритроцитов принадлежит факторам плазмы
крови, в частности высокомолекулярным белкам, таким, как глобулины и,
особенно, фибриноген. Увеличение их содержания, что встречается
нередко при злокачественных опухолях, усиливает агрегацию эритроцитов.
Так как гемостаз является защитной реакцией организма при любом
нарушении целостности сосудистой стенки, такие расстройства
реологических свойств крови встречаются при различных местных повреждениях.
Последствием этих расстройств является замедление кровотока в микроцир-
куляторной системе вплоть до стаза (греч. stasis - стояние), под которым
понимается местная остановка крови в просвете сосудов того или иного
органа, ткани. Стаз может быть вызван уменьшением разности давлений на
протяжении микрососуда и (или) увеличением сопротивления в его
просвете. В зависимости от причин, его вызвавших, различают ишемический,
застойный и истинный капиллярный стаз. При ишемическом стазе градиент
давления в микрососудах уменьшается вследствие значительного
понижения давления в их артериальных отделах, что связано с прекращением
притока крови из более крупных артерий (например, при тромбозе, эмболии,
ангиоспазме и др.). Застойный стаз возникает при уменьшении градиента
давления на протяжении микрососудов вследствие резкого повышения
давления в их венозных отделах (например, при застое крови вследствие
венозной гиперемии, тромбозе более крупных вен, сдавления их опухолью и
др.). Истинный капиллярный стаз связан со значительным первичным увели-
94
Глава 7
чением сопротивления кровотоку в соответствующих сосудах. Причиной
истинного капиллярного стаза является усиленная внутрисосудистая агрегация
эритроцитов. Возникновению стаза может способствовать относительно
высокая концентрация эритроцитов в крови, протекающей по капиллярам. На
развитие и разрешение истинного капиллярного стаза влияют нервные и
гуморальные механизмы. Нервная система воздействует на внутрисосудистую
агрегацию с помощью биологически активных веществ.
Поскольку остановка кровотока в капиллярах при стазе вызывает
прекращение доставки кислорода к соответствующим участкам, проявления
стаза схожи с симптоматикой ишемии. Исход стаза зависит от его
длительности и места возникновения. Кратковременный стаз - явление
обратимое. Если стаз сохраняется в течение длительного времени, происходит
распад тромбоцитов с последующим выпадением фибрина и образованием
тромба.
Одной из частых, распространённых форм патологического тромбооб-
разования в микроциркуляторном русле - синдром диссеминированного
внутрисосудистого свёртывания (ДВС) крови.
В случаях длительного стаза, который охватывает большое количество
капилляров и происходит в тканях, высоко чувствительных к нарушениям
циркуляции крови, может наступать некроз отдельных структурных
элементов органов и тканей. Прежде всего, это относится к центральной нервной
системе, особенно чувствительной к любым нарушениям кровоснабжения.
Внесосудистые тканевые факторы могут влиять на состояние
микроциркуляции. Наиболее выраженное влияние на систему микроциркуляции
оказывают тучные клетки (мастоциты, тканевые базофилы), содержащие в своих
гранулах гистамин, гепарин, серотонин и другие биологически активные
вещества, действующие на микрососуды. Комбинированные расстройства
микроциркуляции, связанные с внутрисосудистыми нарушениями,
изменениями стенки сосудов и внесосудистых компонентов, встречаются
довольно часто. Обычно они представляют собой разные сочетания уже
описанных выше расстройств. Другой тип нарушений микроциркуляции
внесосудистого происхождения обуславливают изменения периваскулярного
транспорта интерстициальной жидкости вместе с растворёнными в ней
веществами, в том числе образования и транспорта лимфы.
НЕДОСТАТОЧНОСТЬ ЛИМФООБРАЩЕНИЯ
Недостаточность лимфообращения - нарушение оттока лимфы.
Проявляется переполнением лимфой лимфатических сосудов и превращением
их в тонкостенные широкие полости (лимфангиэктазии). Недостаточность
лимфообращения находится в тесной патогенетической связи с
нарушениями кровообращения и нередко непосредственно обусловливается ими. В
свою очередь, декомпенсация недостаточности лимфообращения неизбеж-
Нарушения периферического крово- и лимфообращения
95
но приводит к тяжёлым расстройствам микроциркуляции, что проявляется
развитием лимфогенного отёка-лимфедемы. В основу современных
представлений о недостаточности лимфатической системы и её классификации
положено знание механизмов нарушений транссудации и резорбции
межтканевой жидкости. Под недостаточностью лимфатической системы
следует понимать состояние, при котором лимфатические сосуды не выполняют
свою основную функцию - осуществление постоянного и эффективного
дренажа интерстиция. Различают следующие формы недостаточности
лимфообращения:
♦ механическая недостаточность, при которой течение лимфы
затруднено в связи с наличием органических (сдавление опухолью,
рубцом, экстирпация лимфатических узлов и сосудов, облитерация
лимфатических сосудов при их воспалении и др.) или функциональных
причин (повышение давления в магистральных венозных сосудах,
спазм лимфатических сосудов и др.);
♦ динамическая недостаточность, при которой объём транссудации
межтканевой жидкости превышает возможности лимфатической
системы обеспечивать эффективный дренаж межуточной ткани;
♦ резорбционная недостаточность, обусловленная морфофункциональ-
ными изменениями межуточной ткани, накоплением белков и
осаждением их в интерстиции.
Недостаточность лимфообращения может быть общей и местной,
острой и хронической. К развитию острой общей недостаточности
лимфообращения может вести двусторонний тромбоз подключичных вен.
Хроническая общая недостаточность лимфообращения закономерно включается
в проявления общей хронической недостаточности кровообращения
(венозного застоя). Основными клинико-анатомическими проявлениями
недостаточности лимфообращения в острой стадии являются лимфедема,
накопление белков и продуктов их распада в межуточной ткани (слоновость,
хилёзный асцит, хилоторакс), а в хронической - развитие фиброза.
ДИССЕМИНИРОВАННОЕ ВНУТРИСОСУДИСТОЕ СВЕРТЫВАНИЕ
Диссеминированное внутрисосудистое свертывание (ДВС) -
типический патологический процесс, индуцируемый действием множества экзо- и
эндогенных факторов (табл. 7.1). Клиника и патогенез данного
патологического процесса в его варианте, вызванном укусом ядовитой змеи, во
многом верно были описаны известным целителем средневекового Востока
Дисурджани, который описывал ДВС следующим образом: люди умирают
от свертывания крови в сердце и сосудах, а затем из всех отверстий трупа
течет жидкая кровь; более слабый змеиный яд вызывает онемение и лишь
частичное (то есть в тканях, прилегающих к месту укуса - прим. автора)
онемение и свертывание крови.
96
Глава 7
Таблица 7.1
Причины ДВС
Эндогенные
Высвобождение тканевого тромбоп ласти -
на при повреждениях тканей механической,
термической и химической травмой, а
также при воспалительной вторичной
альтерации
Попадание тканевого тромбопластина
при предварительной отслойке плаценты
через ретроплацентарную гематому в
циркулирующую кровь матери
Влияние на систему гемостаза коллагена,
высвободившегося в интерстиции
травмированных, обоженных и воспаленных тканей
Прямая нервная адренергическая
стимуляция сосуцистой стенки и гиперкатехола-
минемия как элементы патологической
стрессорной реакции различного генеза
(травма, кровопотеря, гиповолемия,
оперативное вмешательство и т.д.)
Аутоиммунный механизм развития
заболевания как причина активации системы
комплемента по классическому пути
Системные патологические изменения
сосудистой стенки:
- аутоиммунные
- связанные со злокачественным
клеточным ростом
- вследствие лейкозов и др.
Экзогенные
Инфекции как стимулы системной
реакции, включающей ДВС, вызываемые:
- минингококками,
- стрептококками,
- стафилококками,
- кишечной палочкой,
- возбудителем брюшного тифа,
- риккетсиями
В результате реакции отторжения
трансплантатов
Под влиянием ядов змей, пауков и
ящериц, содержащих экзогенные коагу-
лазы
Вследствие отравлений кислотами
и щелочами
Патологическая активация форменных
элементов крови в результате их контакта
с сорбентом колонок для гемосорбции и
элементами оксигенатора при
искусственном кровообращении
Неэффективное обезболивание при
высокотравматичных вмешательствах как
причина патогенной адренергической
стимуляции сосудистой стенки
Длительно некоррегируемые
гиповолемия и дегидратация у больных с
тяжелыми ранениями и травмами
ДВС - это приобретенная коагулопатия, обусловленная потреблением
факторов свертывания (гиперкоагулемией), активацией фибринолиза,
образованием микротромбов и появлением кровоточивости (гипокоагулемия).
Все эти дисфункции системы гемостаза могут развиваться одновременно
или последовательно. При их последовательном развитии гиперкоагулемия
как патологический сдвиг в системе гемостаза, обусловленный
повышенным потреблением факторов свертывания, приводит к гипокоагулемии. В
этой связи выделяют следующие стадии ДВС как тромбогеморрагического
синдрома:
♦ гиперкоагуляции;
♦ коагулопатии потребления;
♦ дефибринации;
♦ восстановительная или стадия остаточных тромбозов и окклюзии.
В первой стадии гиперкоагуляции и гиперагрегации в организме
происходит массивное и распространенное образование микротромбов. Во
второй и третьей стадиях гипокоагулемия ведет к геморрагиям или к явному
кровотечению. В течение всех стадий происходит обтурация микрососудов
фибриновыми микроэмболами («микросвертками»). Можно считать, что
Нарушения периферического крово- и лимфообращения
97
ДВС как типический патологический процесс в основном развертывается
на уровне микроциркуляторного русла с прижизненным образованием
мельчайших тромбоцитарных и фибриновых тромбов.
Первичным звеном патогенеза ДВС выступает попадание в сосудистое
русло разнообразных активаторов свертывания крови, вызывающих
агрегацию тромбоцитов и образование тромбина в циркулирующей крови. Это в
свою очередь активирует, а значит и истощает свертывающую, калликреин-
кининовую и фибринолитическую ферментные системы крови.
Циркуляция с кровью множества микросгустков и агрегатов форменных элементов
обуславливает распространенную микроэмболизацию в микроциркулятор-
ном русле. Это служит фактором распространенных микрогеморрагий,
микротромбозов, возникновения во внутренних органах микроскопических
очагов некробиотических изменений и воспаления, лишенного биологической
цели. В результате внутренние органы могут потерять критическое число
своих анатомо-функциональных единиц. Таким образом, ДВС выступает
фактором развития синдрома множественной системной недостаточности
у больных после тяжелых ранений и травм, в состоянии сепсиса.
Начальным звеном патогенеза ДВС при септическом шоке служит
повреждение эндотелия микрососудов при бактериемии и модуляция
секреции эйдотелиоцитами биоактивных веществ под влиянием возросшей
действующей концентрации цитокинов в циркулирующей крови.
Систему пара- и аутокринной регуляции микрососудов в структурном
отношении составляют эндотелиоциты мельчайших артериол, секретирую-
щие вещества, препятствующие агрегации тромбоцитов, простагландин 12,
VIII фактор свертывания крови, активатор плазминогена с антикоагулянт-
ной активностью, матричные белки и фибронектин.
Повреждение бактериальными эндотоксинами эндотелия, стимуляция
эндотелиоцитарной секреции в системе пара-аутокринной регуляции
микрососудов приводит к активации фактора Хагемана, высвобождению
тканевых тромбопластинов клеточными элементами микрососудов и
агрегации тромбоцитов. Это в свою очередь активирует факторы свертывания
крови. Активация факторов свертывания происходит тремя путями:
1. Активация внутренней свертывающей системы крови. При этом
варианте развития гиперкоагулемии каскад коагуляции запускает активация
фактора Хагемана. Деструкция сосудистого эндотелия обуславливает прямой
контакт циркулирующей крови с коллагеном соединительной ткани. Это
ведет к трансформации предшественника XII фактора свертывания крови в
активный фермент, индуцирующий алгоритм реакций свертывания крови
путем перевода неактивного XI фактора свертывания в его активную форму.
2. Активация внешней по отношению к циркулирующей крови тканевой
системы свертывания. В результате бактериемической травматизации и
вследствие адренергической стимуляции в ответ на снижение общего
периферического сосудистого сопротивления эндотелиоциты микрососудов при
сепсисе начинают интенсивно высвобождать тромбопластин в
циркулирующую кровь. Тромбопластин, взаимодействуя с кальцием и седьмым
фактором свертывания, образует комплекс, способный к активации фактора X.
98
Глава 7
После этого этапа гиперкоагулемия продолжает развиваться в соответствии
с униформным алгоритмом.
3. Активация калликреиновой системы плазмы. Активированный
фактор Хагемана вызывает в плазме циркулирующей крови интенсивное
образование фермента калликреина из прекалликреина. Калликреин в свою
очередь воздействует на свой главный субстрат, брадикининоген, который
высвобождает нонапептид брадикинин, один из наиболее мощных
естественных вазодилятаторов. У больных в состоянии сепсиса еще до активации
калликреинвой системы под влиянием высокого содержания в крови цито-
кинов общее периферическое сосудистое сопротивление снижено в
результате артериоло-венулярного шунтирования. Расширение сосудов
сопротивления при росте в плазме крови содержания брадикинина приводит на таком
фоне к особенно быстрому и выраженному падению общего
периферического сосудистого сопротивления и трудно купируемой артериальной гипо-
тензии. Адренергическая стимуляция сосудистой стенки при септическом
шоке не в состоянии восстановить общее периферическое сосудистое
сопротивление и артериальное давление, но усиливает интенсивное
высвобождение тромбопластинов эндотелиоцитами. Падение артериального
давления ведет к циркуляторнои гипоксии, повреждающей эндотелий на
периферии, что способствует прямому контакту неактивированного
фактора Хагемана с обнажившимся веществом соединительной ткани. Все это
превращает шок и артериальную гипотензию в самостоятельные факторы
диссеминированного внутрисосудистого свертывания.
Шок и артериальная гипотензия усиливают внутрисосудистое
свертывание, снижая очищение печенью крови от активированных факторов
свертывания.
Диссеминированное внутрисосудистое свертывание при септическом
шоке ведет к отложению фибрина на всем протяжении сосудистого русла,
что приводит к очагам некроза в органах и ускоряет развитие
множественной системной недостаточности, наиболее грозным из элементов которой
является септический респираторный дистресс-синдром взрослых. Биопсий-
ные исследования морфогенеза начальной экссудативной стадии дистресс-
синдрома у септических больных выявляют интенсивное отложение
фибрина вдоль альвеолярных протоков, которые включают остатки погибших
клеток и служат основой для формирования гиалиновых мембран.
Трансформация отложений фибрина в гиалиновые мембраны связана с
повышенным содержанием в легочной паренхиме как ингибиторов урокиназы, так и
антиплазминов. В наибольшей степени в легких возрастает содержание
такого антиплазмина, как ингибитор активатора плазминогена первого типа.
Глава 8
ТРОМБОЗ ГЛУБОКИХ ВЕН
И ТРОМБОЭМБОЛИЯ ЛЕГОЧНОЙ АРТЕРИИ
Тромбоз - это прижизненное формирование и фиксация сгустка
тромбоцитов, фибрина, лейкоцитов и эритроцитов на внутренней поверхности
сосуда, которое препятствует венозному оттоку и (или) поступлению в
ткани артериальной крови, у некоторых больных приводя к ишемии.
Следует отличать внутрисосудистое свертывание крови от тромбоза
(тромбообразования, тромбогенеза). Результат внутрисосудистого
свертывания - образование рыхлых сгустков фибрина, не фиксирующихся на
сосудистой стенке. Тромбогенез завершается формированием на стенке
сосуда плотных отложений крови, относительно прочно связанных не только с
эндотелиоцитами, но и с субэндотелиальными структурами стенки сосуда.
Время от начала внутрисосудистого свертывания до образования на стенке
сосуда плотного сгустка (тромба) составляет около шести часов.
Свертывание крови как защитная реакция, направленная на сохранение
целостности сосудистого русла, во многом представляет собой результат
взаимодействий между (1) эндотелиоцитами, (2) макромолекулами субэн-
дотелиального слоя, (3) тромбоцитами и плазменными факторами
свертывания. Патологические сдвиги в одном из этих трех эффекторов систем
регуляции свертывания крови могут служить пусковым механизмом
тромбогенеза.
Ключевой фермент тромбогенеза и эффектор системной и локальной
регуляции тромбообразования - это тромбин. Тромбин является
катализатором конечных этапов тромбоза (превращение фибрина в
перекрестно-связанный фибрин и активация фактора XIII свертывания крови).
Активированный тромбином фактор XIII стабилизирует сгусток фибрина. Тромбин
представляет собой высокоспецифичную сывороточную протеазу,
обладающую биологической активностью, не связанной со свойствами тромбина
как фермента. Так, относительно моноцитов и нейтрофилов тромбин
выступает в качестве хемоаттрактанта и соответственно индуктора воспаления.
Биологический смысл наличия у тромбина хемоаттрактивных свойств
заключается в запуске воспаления через интенсивный тромбогенез в зоне
первичной альтерации. Воспаление в данном контексте мы рассматриваем как
защитно-патогенную системную реакцию, готовящую условия для
последующего замещения дефектов тканей и органов через клеточную
пролиферацию и ангиогенез. В этой связи становится ясной биологическая
целесообразность свойства тромбина стимулировать клеточную пролиферацию и
ангиогенез.
Прямое повреждение сосудистой стенки всегда вызывает действие
механизмов тромбогенеза. Тромбообразование, особенно в микрососудах,
может начинаться и без первичной альтерации тканей и эндотелия. При этом
100
Глава 8
распространенный тромбогенез в микрососудах может быть элементом
системной воспалительной реакции. Тромбогенез без первичной альтерации
тканей и эндотелиальных клеток индуцируют:
♦ падение тромборезистентности сосудов (см. ниже);
♦ рост высвобождения эндотелиоцитами индукторов тромбогенеза в
результате защитных и одновременно патогенных сдвигов в системах
регуляции организменного и местного уровня;
♦ замедление тока крови и ее патологически высокая вязкость.
Известно, что фактор некроза опухолей (ФНО), чья концентрация в
крови столь значительно возрастает при системной воспалительной реакции,
сепсисе, септическом шоке и в ответ на острую циркуляторную гипоксию,
вызывает образование и высвобождение эндотелием тканевого тромбопла-
стина и фактора активации тромбоцитов. Кроме того, при этом ФНО
снижает образование тромбомодулина и активность других антикоагулянтных
систем. Все это резко повышает тромбогенную активность эндотелиоци-
тов. При состояниях, которые во многом характеризует патологически
интенсивное высвобождение ФНО, возникают выраженные нарушения
периферического кровообращения вплоть до стаза в микрососудах. Все это
служит причиной системного микротромбоза во внутренних органах, который
вызывает воспаление, лишенное биологической цели. Вторичная
альтерация при таком воспалении обуславливает недостаток
структурно-функциональных единиц в органах-эффекторах функций. Через распространенный
и не имеющий защитно-приспособительного значения тромбоз
микрососудов развиваются осложнения тяжелых раневой, травматической болезни,
системной воспалительной реакции, сепсиса, септического и
военно-травматического шока.
Тромбин, играя роль ключевого фермента тромбогенеза, одновременно
представляет собой индуктор активации молекулярных систем
тромборезистентности эндотелия. Так, тромбин не только активирует факторы
свертывания V и VIII, но и взаимодействует с белком наружной клеточной
поверхности эндотелиоцитов тромбомодулином, что ведет к резкой активации
протеина С. Этот белок, перешедший в активированное состояние, инакти-
вирует факторы свертывания V и VIII, что ограничивает образование
фибрина. Связывание тромбомодулином тромбина лишает тромбин
способности превращать фибриноген в нерастворимый фибрин и блокирует реакцию
активации тромбоцитов, а значит и тромбообразование.
Протеин С - это естественный антикоагулянт, представляющий собой
протеолитический фермент, синтез которого в печени проходит с участием
витамина К. Свойствами антикоагулянта белок С обладает лишь в
активированной форме. Так как тромбомодулин, участвующий в активации белка
С, находится на поверхности только интактных эндотелиоцитов, то в
поврежденном участке сосудистой стенки нет тромбомодулина и нет
активной формы белка С. В результате в месте повреждения эндотелия и стенки
сосуда свертывание крови и тромбогенез не испытывают ограничений. На
поверхности прилежащих к локусу альтерации сосудистой стенки
неповрежденных эндотелиальных клеток тромбомодулин есть, что ограничивает
Тромбоз глубоких вен и тромбоэмболия легочной артерии
101
Таблица 8.1
Причины приобретенного дефицита белка С
Побочное действие антикоагулянтов кумаринового ряда
Интенсивная утилизация вследствие диссеминированного внутрисосудистого
свертывания
Недостаточность печени
Респираторный дистресс-синдром взрослых
Послеродовая коагулопатия
Коагулопатия после интраоперационных кровотечений, тромбоз сосудов с
патогенно высоким потреблением белка С
Некоторые острые лейкемии
Миеломная болезнь
Системная красная волчанка и волчаночные иммуноглобулины как антикоагулянты,
циркулирующие с кровью при расстройствах иммунной системы и инфекционных
заболеваниях (синдромы первичного иммунодефицита и др.)
Побочное действие антибиотиков, молекула которых содержит N-метилтиотетразо-
ловое кольцо
Побочные эффекты циклофосфамида, метотрексата
Потери антикоагулянтов с ультрафильтратом при гемодиализе и при нефротичес-
ком синдроме
тромбообразование местом первичного повреждения стенки сосуда.
Приобретенный дефицит белка С (табл. 8.1) может быть причиной
распространенного тромбоза и тромбоэмболии.
Некоторые из ингибиторов протеаз вовлечены в регуляцию активности
тромбина, циркулирующего с кровью. Наиболее мощный из известных
естественных антикоагулянтов такого рода - это антитромбин III, чье
сродство к тромбину усиливает гепарин.
Сосудистая стенка представляет собой не просто локус тромбообразо-
вания. Это эффектор систем регуляции тромбогенеза, самым
непосредственным образом влияющий на скорость и распространенность тромбоза.
Кроме того, сосудистая стенка во многом определяет коагуляционную и
фибринолитическую активность крови и уровень активации кровяных
пластинок. В эндотелии, гладкомышечных и адвентициальных клетках
сосудистой стенки происходит образование широкого спектра биоактивных
веществ и цитокинов активирующих и тормозящих все этапы свертывания
крови, фибринолиза, адгезии и агрегации тромбоцитов. Кроме того, эндоте-
лиоциты обладают свойством активировать и инактивировать плазменные про-
и антикоагулянты на своей поверхности, обращенной в просвет сосуда.
При незатрудненном токе крови в просвете патологически не
измененных сосудов в физиологических условиях эндотелиоциты непрерывно
высвобождают в кровь тромбогенные и атромбогенные вещества. К наиболее
изученным из тромбогенных веществ относят тканевой тромбопластин,
фибронектин, фактор Виллебранда, тромбоксан А, фактор активации
тромбоцитов. Одни из этих веществ вызывают тромбиногенез и образование
фибрина, другие - адгезию и активацию тромбоцитов. Эндотелиоциты и в
меньшей степени другие клетки сосудистой стенки образуют и
высвобождают ряд факторов, которые тормозят свертывание крови, активируют фиб-
102
Глава 8
ринолиз, тормозят агрегацию и адгезию тромбоцитов к эндотелию и субэн-
дотелиальному слою сосудистой стенки. Эти биоактивные вещества
формируют атромбогенный потенциал сосудов, препятствуя избыточному
образованию фибрина на эндотелиальной поверхности и блокируя излишнее
тромбообразование вследствие повреждения сосудистой стенки.
К факторам, противодействующим свертыванию крови, относят белки
С и S. Агрегацию тромбоцитов тормозят простациклин, фактор релаксации
эндотелия, оксид азота. Некоторые из протеогликанов также тормозят
агрегацию тромбоцитов и их адгезию к эндотелию и субэндотелию.
Модуляция под влиянием системных и локальных регуляторных
влияний экспрессии всего спектра биоактивных веществ, формирующих
атромбогенный потенциал сосудов, лежит в основе изменения их
тромборезистентности как причины формирования патогенных предпосылок тромбоге-
неза. Тромборезистентность сосудистой стенки (Петрищев Н.Н.) - это ее
свойство ограничивать процесс тромбообразования зоной повреждения или
в месте локального возрастания тромбогенного потенциала; одна из
причин местного роста тромбогенного потенциала - локальное падение
тромборезистентности. Местное падение тромборезистентности может быть
следствием системной и (или) локальной дизрегуляции баланса между тром-
богенными и атромбогенными свойствами сосудистой стенки. Активация
местных противосвертывающих, антиадгезивных и антиагрегационных
механизмов при повреждении ранее интактных сосудов ограничивает
распространение тромбоза, не препятствуя гемостазу.
Различия в тромбогенном потенциале артерий (артериол) и вен (венул)
нельзя объяснить только разными скоростями кровотока в их просвете.
Не исключено, что венозная стенка, лишенная опиоидных рецепторов,
изолирована от местных и системных влияний опиоидергических стресс-
лимитирующих систем. В результате адренергическая нейрогуморальная
стимуляция, как компонент патологической стрессорной реакции,
присущей болезням и патологическим состояниям, стимулирует образование и
высвобождение эндотелиоцитами тромбогенных факторов, и тромбоплас-
тина в частности. В этой связи длительную патогенную боль можно
рассматривать как фактор венозного тромбоза через патогенную стрессорную
адренергическую нейрогуморальную стимуляцию сосудистой стенки и
эндотелия венозных сосудов.
Ряд болезней и патологических состояний связаны с особо высоким
риском тромбоза глубоких вен нижних конечностей и тромбоэмболии
легочной артерии (табл. 8.2).
Патогенный рост давления в тканях нижних конечностей, окружающих
глубокие вены, предрасполагает к развитию их тромбоза. Этот рост
вызывается отеком после ранений и травм, к нему ведет транспортная и лечебная
иммобилизация после огнестрельных и других переломов костей нижних
конечностей. Замедление кровотока, нарушения текучести крови и цирку-
ляторная гипоксия как следствия застойной сердечной недостаточности,
военно-травматического и гиповолемического шока, а также в результате
обезвоживания, могут индуцировать тромбогенез в глубоких венах нижних
Тромбоз глубоких вен и тромбоэмболия легочной артерии
103
Таблица 8.2
Заболевания и патологические состояния, предрасполагающие к тромбофлебиту
нижних конечностей, тромбоэмболии легочной артерии
Послеоперационные сдвиги системной регуляции, внешнего дыхания и
кровообращения
Раневая и травматическая болезни
Застойная сердечная недостаточность
Нарушения мозгового кровообращения
Тромбоцитоз
Полицитемия как следствие эритроцитоза в результате длительной адаптации к ги-
поксической гипоксии
Серповидно-клеточная анемия
Последствия приема контрацептивов
Беременность
Длительная гиподинамия
Потеря тромборезистентности сосудов вследствие системных сдвигов,
обусловленных злокачественными опухолями
Сепсис и системная воспалительная реакция
Злокачественные опухоли
конечностей. При этом причинами начала тромбообразования выступают
не только падение объемной скорости кровотока и патологически
высокая вязкость крови, но и снижение тромборезистентности эндотелия и
рост его тромбогенности под влиянием адренергической стрессорной
стимуляции сосудистой стенки и роста содержания в циркулирующей крови
цитокинов (ФНО и др.). Частыми источниками эмболов служат тромбы,
локализованные в варикозных расширениях вен нижних конечностей. У
больных с митральными стенозом и недостаточностью местом
тромбообразования может быть расширенное левое предсердие. В этом случае
вероятность тромбообразования повышает мерцательная аритмия,
нарушающая активный выброс крови из левого предсердия в левый желудочек во
время диастолы.
Вследствие полицитемии, которую обуславливает приспособительный
рост содержания эритроцитов в циркулирующей крови в ответ на гипок-
сическую гипоксию у людей в условиях^зысокогорья, падает текучесть
крови. Падение текучести крови в основном обуславливает рост ее
вязкости. Это снижает объемную скорость кровотока в микрососудах и
доставку кислорода к клетке, несмотря на увеличение кислородной емкости
крови. Одновременно циркуляторная гипоксия клеток, которую обуславливает
снижение текучести крови, ведет к системной активации эндотелия
вследствие системной адренергической стимуляции, вызванной циркуляторной
гипоксией клеток. Все это позволяет рассматривать распространенный
тромбоз микрососудов как болезнь длительной адаптации к гипоксичес-
кой гипоксии.
Причинами тромбоза и тромбоэмболии могут быть наследственные и
приобретенные аномалии свертывающей и противосвертывающей систем
104
Глава 8
Таблица 8.3
Патологические сдвиги свертывающей и противосвертывающей систем
как причины тромбоза и тромбоэмболии
Патологически высокая адгезивная способность тромбоцитов
Аномально длительная циркуляция жизнеспособных тромбоцитов с кровью
Патологически высокое содержание в плазме крови V и VII факторов свертывания
крови и антитромбина III
Дефицит в организме белков С и S
Наследственные патогенные особенности строения молекулы фибриногена
Наследственный недостаток функционально активного плазминогена и дефицит
двенадцатого фактора свертывания крови
крови (табл. 8.3) и баланса между реализацией тромбогенного потенциала
и тромборезистентностью сосудистой стенки.
Патогенно высокие концентрации в плазме крови седьмого и пятого
факторов свертывания крови и недостаток белков С и S выявляют у 30 %
больных с неоднократными тромбоэмболиями и у пациентов, ближайшие
родственники которых часто страдают от тромбоза. В этой связи
тромбоэмболии данного генеза у таких больных с большой вероятностью можно
считать наследственно обусловленными.
Волчаночные иммуноглобулины со свойствами антикоагулянтов,
которые циркулируют с кровью не только при системной красной волчанке, но
и при синдроме приобретенного иммунодефицита и других инфекционных
заболеваниях, ведут к тромбозу предположительно через угнетение
образования и высвобождения эндотелиоцитами простациклина.
ПАТОГЕНЕЗ РАССТРОЙСТВ ВНЕШНЕГО ДЫХАНИЯ
ВСЛЕДСТВИЕ ТРОМБОЭМБОЛИИ ЛЕГОЧНОЙ АРТЕРИИ
И ЕЕ ВЕТВЕЙ
Обструкция легочной артерии и ее ветвей вследствие тромбоэмболии
увеличивает альвеолярное мертвое пространство, то есть число
вентилируемых, но не омываемых смешанной венозной кровью альвеол. Это снижает
поглощение кислорода легкими и ведет к артериальной гипоксемии.
Артериальная гипоксемия возникает при эмболии на определенном уровне
легочной артерии и ее ветвей или при значительном распространении
тромбоэмболии ветвей легочной артерии небольшого диаметра в пределах
легочной паренхимы. Артериальная гипоксемия выступает стимулом для
компенсаторной гипервентиляции, которая как защитная реакция
избыточна относительно снижения напряжения кислорода в артериальной крови,
вызвавшего рост минутной альвеолярной вентиляции. В данном случае
гипервентиляция как компенсаторная реакция исходно обречена на
неэффективность, так как рост минутной альвеолярной вентиляции у больных с тром-
Тромбоз глубоких вен и тромбоэмболия легочной артерии
105
Обструкция легочной артерии и ее ветвей
Рост альвеолярного мертвого пространства
Снижение поглощения кислорода легкими
Артериальная гипоксемия
Компенсаторная
гипервентиляция
Увеличение потребления
кислорода организмом
Рост потребления кислорода в системе внешнего дыхания
Схема 8.1. Патогенез артериальной гипоксемии вследствие
тромбоэмболии легочной артерии и ее ветвей
бозом легочной артерии и ее ветвей происходит и в зоне альвеолярного
мертвого пространства. Кроме того, следует учитывать, что если вентиляция
легких растет через увеличение частоты дыханий, то одновременно
нарастает неравномерность вентиляции, которая усиливает артериальную гипок-
семию. Как защитная реакция гипервентиляция всегда имеет избыточный
характер и служит причиной двух патологических сдвигов:
♦ значительное увеличение потребления кислорода всем организмом
через рост потребления кислорода в системе внешнего дыхания,
♦ дыхательный алкалоз.
Напряжение кислорода в артериальной крови представляет собой
прямую функцию отношения поглощения кислорода легкими к его
потреблению организмом. При снижении поглощения кислорода легкими вследствие
роста альвеолярного мертвого пространства и увеличении потребления
кислорода организмом при гипервентиляции напряжение кислорода в
артериальной крови падает, и у больных с тромбоэмболией легочной артерии
обостряется (развивается) артериальная гипоксемия (схема 8.1).
Снижение парциального давления углекислого газа в альвеолах,
находящихся в зоне альвеолярного мертвого пространства, служит одной из
причин их спадения, так как ведет к снижению общего давления смеси газов в
просвете альвеол. Парциальное давление углекислого газа падает, так как в
зоне альвеолярного мертвого пространства некоторое время, до спадения
альвеол и респираторных бронхиол, продолжается вентиляция респиронов
газовой смесью, почти не содержащей углекислого газа. При этом
поступления в альвеолы зоны альвеолярного мертвого пространства углекислого
газа нет в силу блокады на определенном уровне притока смешанной
венозной крови. В результате углекислый газ вымывается из зоны
альвеолярного мертвого пространства, и в ней спадаются альвеолы. Известно, что ин-
106
Глава 8
галяция больным с тромбоэмболией легочной артерии газовых смесей,
обогащенных углекислым газом, подвергает обратному развитию пневмокон-
стрикцию легких, то есть спадение альвеол как причину ателектазирова-
ния легких в результате обтурации легочной артерии и ее ветвей при
тромбоэмболии.
Еще одна из причин пневмоконстрикции вследствие тромбоэмболии
легочной артерии и ее ветвей - это нарушения системы сурфактанта в зоне
альвеолярного мертвого пространства. Их вызывает прекращение
поступления смешанной венозной крови в ее респироны, нарушающее легочную
микроциркуляцию. Расстройства системы сурфактанта прогрессируют
медленно, достигая верхнего предела выраженности через сутки после
тромбоэмболии, когда они начинают приводить к спадению альвеол и ателектазу.
Пневмоконстрикция вследствие тромбоэмболии снижает вентиляцию
альвеолярного мертвого пространства, но не в такой степени, чтобы
подвергнуть обратному развитию артериальную гипоксемию. Нарушения
микроциркуляции в легких вследствие тромбоэмболии легочной артерии вызывают
циркуляторную гипоксию клеток легочной паренхимы в зоне
альвеолярного мертвого пространства. Гипоксия и гипоэргоз клеток легочной ткани в
зоне альвеолярного мертвого пространства ведут к высвобождению ими
целого ряда биоактивных веществ (серотонин, гистамин, фактор некроза
опухолей и др.), которые вызывают некардиогенный отек легких и без
первичной альтерации индуцируют воспаление в зоне альвеолярного мертвого
пространства. Альтерация при воспалении, лишенном биологической цели,
разрушает респироны.
Предположительно, наиболее быстрой из патологических реакций,
приводящих к артериальной гипоксемии, следует считать рефлекторный ней-
рогенный рост сосудистого сопротивления на уровне респиронов,
распространенный далеко за пределы собственно зоны альвеолярного мертвого
пространства. В соответствии с законом асинхронного патологического
реагирования структурно-функциональных единиц эффекторов функций
данная патологическая реакция ведет к патогенной вариабельности вентиляци-
онно-перфузионных отношений респиронов легких, которая вызывает
артериальную гипоксемию.
У некоторых больных ведущим механизмом развития артериальной
гипоксемии следует считать не столько возникновение альвеолярного
мертвого пространства, сколько патологически низкие вентиляционно-перфузи-
онные отношения респиронов и частей легких вне его зоны.
Предположительно их обуславливает микроателектазирование вследствие отрицательных
нейрогуморальных влияний на легочную паренхиму, вызванных
тромбоэмболией легочной артерии.
Если тромбоэмболия ведет к острой циркуляторной гипоксии, то
падение напряжения кислорода в смешанной венозной крови присоединяется к
факторам развития артериальной гипоксемии.
Пневмоконстрикция у больных с тромбоэмболией легочной артерии
через увеличение на уровне всех легких вентиляционно-перфузионного
отношения несколько снижает артериальную гипоксемию. Разрешение тром-
Тромбоз глубоких вен и тромбоэмболия легочной артерии
107
боэмболии, восстанавливая кровоток в зонах пневмоконстрикции, может
обострить артериальную гипоксемию через увеличение фракции неоксиге-
нированной смешанной венозной крови.
ПАТОГЕНЕЗ НАРУШЕНИЙ ЛЕГОЧНОГО И СИСТЕМНОГО
КРОВООБРАЩЕНИЯ, СВЯЗАННЫХ С ТРОМБОЭМБОЛИЕЙ
ЛЕГОЧНОЙ АРТЕРИИ И ЕЕ ВЕТВЕЙ
Первичное звено патогенеза нарушений кровообращения вследствие
тромбоэмболии легочной артерии - это снижение на определенном уровне
системы легочной артерии общей площади поперечного сечения ее
сосудов. В результате растут сосудистое сопротивление в системе легочной
артерии, давление в ней крови и постнагрузка правого желудочка, падают
ударный объем левого желудочка, минутный объем кровообращения и
артериальное давление. Степень выраженности этих патологических
сдвигов определяют:
♦ распространенность и уровень обтурации легочной артерии и ее
ветвей вследствие тромбоэмболии;
♦ степень влияния на системное кровообращение и легочный кровоток
патологических рефлексов, стимулом для которых выступает
тромбоэмболия легочной артерии;
♦ состояние больных перед тромбоэмболией легочной артерии.
Если минутный объем кровообращения не слишком высок, то легочная
артериальная гипертензия в условиях покоя развивается лишь тогда, когда
тромбоэмболия снижает общую площадь поперечного сечения сосудов
определенного уровня системы легочной артерии на 50 % ее максимального
значения. Это отражает высокую податливость сосудов в системе
легочной артерии. Высокая податливость сосудов системы легочной артерии
предотвращает легочную артериальную гипертензию лишь при
определенных размерах тромба. При большей обструкции для поддержания
адекватного легочного кровотока уже необходим рост давления крови в
легочной артерии, и развивается легочная артериальная гипертензия. Если
минутный объем кровообращения находится на высоком уровне (срочные
и устойчивые компенсаторные процессы, лихорадка, сепсис, системная
воспалительная реакция, септический шок, анемия и др.) то легочная
артериальная гипертензия возникает и при меньшей обструкции. При этом
легочная артериальная гипертензия как компенсаторная реакция
направлена на поддержание объемной скорости легочного кровотока адекватной
потребностям организма.
При острой тромбоэмболии легочной артерии и ее ветвей давление
крови в системе легочной артерии растет до 40 мм рт. ст. и выше. Тонкостей-
108
Глава 8
ный правый желудочек не справляется с такой высокой постнагрузкой.
Острая правожелудочковая недостаточность резко снижает преднагрузку
левого желудочка. В результате падают ударный объем левого желудочка,
минутный объем кровообращения и артериальное давление, причем
артериальная гипотензия часто имеет необратимый характер.
Свою роль в развитии легочной артериальной гипертензии могут играть
рефлекторные сдвиги нейрогуморальной регуляции тонуса легочных
сосудов, вызванные тромбоэмболией легочной артерии и ее ветвей.
СВЯЗЬ ПАТОГЕНЕЗА И СИМПТОМОВ ТРОМБОЭМБОЛИИ
ЛЕГОЧНОЙ АРТЕРИИ И ЕЕ ВЕТВЕЙ
Наиболее частым и достоверно коррелирующим с развитием
тромбоэмболии в системе легочной артерии симптомом являются субъективные
ощущения затрудненного дыхания и нехватки воздуха, то есть одышка.
Более 90 % больных с тромбоэмболией легочной артерии и ее ветвей
предъявляют жалобы на одышку, выраженность которой зависит от величины
альвеолярного мертвого пространства. Под одышкой у больного с
тромбоэмболией легочной артерии и ее ветвей следует понимать комплекс
ощущений, отражающих неэффективность компенсаторно-патогенной
гипервентиляции и снижение податливости легких вследствие
тромбоэмболии легочной артерии.
Если тромбоэмболия легочной артерии возникает внезапно и сразу
приводит к легочной артериальной гипертензии (признак высокого уровня или
распространенности обструкции), то одышка особенно выражена и
сопровождается тахипноэ при частоте дыханий в диапазоне 40-50 мин1. Тахип-
ноэ в данном случае - это признак неэффективной компенсаторной
гипервентиляции, имеющей прямо патогенное значение.
Следствием патогенности избыточной гипервентиляции у больных с
тромбоэмболией следует считать респираторный алкалоз, выявляемый
специальными исследованиями одновременно с артериальной гипоксемией.
Пневмоконстрикция вследствие тромбоэмболии легочной артерии и ее
ветвей через ателектазирование приводит к падению звучности дыхания.
Инфаркт легкого как причина пневмонии ведет к возникновению влажных
хрипов, лихорадке, появлению в плевральной полости транссудата и
экссудата, лейкоцитозу и боли вследствие возбуждения ноцицепторов
париетальной плевры. В последующем у небольшой части больных появляется шум
трения плевры. Турбулентное движение крови по обтурированным ветвям
легочной артерии генерирует у небольшой части больных выслушиваемые
над легкими специфические сосудистые шумы. Эти шумы называют
симптомами «приобретенной коарктации легочной артерии».
Тромбоз глубоких вен и тромбоэмболия легочной артерии
109
Психомоторное возбуждение и чувство страха у больных с
тромбоэмболией легочной артерии - это следствие мощного потока афферентации,
который вызывают системная респираторно-циркуляторная гипоксия и обту-
рация сосудов в системе легочной артерии
Тахикардия у больных с тромбоэмболией легочной артерии и ее ветвей
компенсирует низкий ударный объем левого желудочка, который падает
вслед за снижением его конечно-диастолического объема Тахикардия и
гипервентиляция при тромбоэмболии легочной артерии и ее ветвей - это во
многом результат сдвига регуляции по возмущению
Легочная артериальная гипертензия (ЛАГ) как причина роста
постнагрузки правого желудочка и острого легочного сердца увеличивает время
изгнания крови из правого желудочка в легочную артерию, что приводит к
раздвоению второго сердечного тона и появлению выявляемого при аус-
культации «ритма галопа». Второй тон расщепляется не только вследствие
увеличения фазы изгнания правого желудочка, но и в результате быстрого
закрытия створок аортального клапана при небольшом ударном объеме
левого желудочка, который падает из-за низкой преднагрузки ЛАГ вызывает
акцент второго тона над легочной артерией Если обтурация вследствие
тромбоэмболии происходит на уровне главного ствола легочной артерии, то над
легочной артерией выслушивают «царапающий» систолический шум По
мере усиления правожелудочковой недостаточности падают объемная и
линейная скорости кровотока в легочной артерии, что снижает звучность
второго тона и ведет к исчезновению его акцента
ПАТОГЕНЕТИЧЕСКИЕ ПРИНЦИПЫ ПРОФИЛАКТИКИ
ТРОМБОЗА ГЛУБОКИХ ВЕН НИЖНИХ КОНЕЧНОСТЕЙ
И ТРОМБОЭМБОЛИИ ЛЕГОЧНОЙ АРТЕРИИ
У ХИРУРГИЧЕСКИХ БОЛЬНЫХ
Адренергическая системная стимуляция как элемент срочных
компенсаторных процессов активирует эндотелий на организменном уровне, что
усиливает экспрессию тромбогенного потенциала эндотелиоцитов,
повышая высвобождение ими факторов свертывания крови и тромбогенеза, в
частности тромбопластина В этой связи эффективную проводниковую и
центральную анальгезию как средство блокады системной реакции гипер-
коагулемии в афферентном и центральном звеньях следует считать
патогенетическим средством предупреждения тромбоза В эфферентном звене
адренергическую нейрогуморальную стимуляцию стенки вен как фактор
тромбоза в известной мере могут блокировать ганглио- и альфа-адреноли-
тики и другие симпатолитические средства
Артериальная гипоксемия, гипоксия и гипоэргоз на организменном
уровне, ацидоз, снижение объемной и линейной скорости кровотока и рост
вязкости крови вследствие шока и недостаточности кровообращения не только
110
Глава 8
служат стимулами для системной активации тромбогенеза, но и
способствуют тромбообразованию, активируя эндотелиоциты В этой связи всю
систему интенсивной терапии, ориентированной на восстановление и усиление
достдоки кислорода к клетке, анальгезию и гемодилюцию в том числе, можно
считать наиболее эффективной мерой профилактики тромбоэмболических
осложнений ранений, травм и хирургических вмешательств
У больных с генетически детерминированной или приобретенной
высокой предрасположенностью к тромбозу предупреждать и блокировать
гиперкоагулемию и тромбогенез приходится не только воздействуя на
системы регуляции, но и оказывая непосредственное влияние на эффекторы
свертывающей, противосвертывающей систем крови и элементы тромбо-
генного и атромбогенного потенциалов сосудистой стенки Так, после
общехирургических вмешательств на органах груди и живота больным без
тромбоза глубоких вен нижних конечностей и тромбоэмболии легочной
артерии в анамнезе через 2 ч после окончания операции начинают вводить
по 5000 единиц гепарина подкожно через каждые двенадцать часов 3-7
суток послеоперационного периода Кроме того, часто массируют голени и
бедра или применяют специальные пневматические манжеты, которые цир-
кулярно накладывают на бедра, голени и периодически раздувают
Известно, что длительность и интенсивность сокращений произвольных мышц
нижних конечностей - это одна из детерминант линейной и объемной
скоростей кровотока в их венах, которая находится с ними в прямой связи
Падение скорости продвижения крови по сосудам - условие или причина
тромбоза В этой связи индукцию сокращений мышц периодическим сжатием
нижних конечностей манжетами и раздражением при массаже,
выталкивание крови из вен в направлении к легким вследствие роста давления в
окружающих вены тканях при компрессии голеней и бедер следует считать
патогенетически обоснованными средствами предупреждения тромбоза
глубоких вен нижних конечностей
Если у больного в анамнезе есть тромбоз глубоких вен нижних
конечностей или тромбоэмболия легочной артерии, то после обшехирургических
вмешательств на органах груди и живота гепарин подкожно в дозе 5000
единиц вводят чаще, каждые восемь часов
Эндогенный гликозоаминогликан гепарин, воздействуя на антитромбин
сразу же после подкожного введения, вызывает увеличение тромбинового
времени, активированного частичного тромбопластинового времени и в
меньшей степени протромбинового времени Быстрота его действия
представляет собой основное преимущество этого наиболее широко
используемого антикоагулянта Свойство гепарина снижать выраженность тромбо-
генного потенциала и экспрессию тромбогенных свойств эндотелиоцитов
основано не только на усилении гликозоаминогликаном сродства
антитромбина III к тромбину Гепарин как антикоагулянт выступает катализатором
торможения антитромбином III тромбина и активированного десятого
фактора свертывания крови Предотвращая таким образом активацию
тромбином пятого и восьмого факторов свертывания крови, гепарин блокирует
активацию тромбогенезом общего механизма свертывания крови
Тромбоз глубоких вен и тромбоэмболия легочной артерии
111
Таблица 8 4
Принципиальные недостатки гепарина как
антикоагулянта (Hush J, 1991)
Отсутствие достоверной прямой связи между величиной дозы и выраженностью
эффекта
Необходимость частых повторных исследований показателей гемостаза
Нет клинически значимого действия на тромбин, связанный с тромбом
Зависимость действия от активности антитромбина III в плазме крови
Послеоперационные кровотечения как побочный эффект гепарина
Реакции повышенной чувствительности, в которых гепарин играет роль аллергена
Несмотря на широкое использование гепарина в клинической практике,
этот антикоагулянт обладает рядом принципиальных недостатков, которые
в основном связаны с его разносторонним действием на системы регуляции
гемостаза и тромбообразования (табл 8 4), получившего название
«множества точек тормозящего действия» Так, гепарин оказывает мощное
влияние на функции тромбоцитов, инактивируя их У небольшой части больных
введение гепарина в дозах, обычных для профилактики тромбоэмболичес-
ких осложнений критических состояний, ведет к тромбоцитопении Из
побочных эффектов гепарина известны остеопороз, некроз кожи, алопеция и
гипоальдостеронизм, рост проницаемости стенки микрососудов
Наиболее существенным из «фундаментальных» недостатков
гепарина следует считать отсутствие у него влияний на тромбин, связанный с
фибрином в составе сгустка и тромба Этот тромбин продолжает быть
активным как энзим и обуславливает образование фибрина из фибриногена в
крови, омывающей тромб, несмотря на содержание в ней в
физиологических пределах естественных антикоагулянтов антитромбина III и
гепаринового кофактора II Данные антикоагулянты в физиологических
концентрациях снижают активность свободного тромбина, циркулирующего с плазмой
крови Дело в том, что фибрин сгустка одновременно со связыванием
тромбина связывает и эти биоактивные вещества, после чего они теряют
свойства антикоагулянтов Поэтому введение гепарина больным после
индукции тромбогенеза не может остановить тромбообразования, так как не
приостанавливает образование фибрина из фибриногена в крови,
контактирующей с поверхностью тромба Кроме того, показано, что тромбин,
связанный с растворимыми веществами, образующимися при деградации
фибрина, также свободен от угнетающих его активность влияний
антикоагулянтов плазмы крови и гепарина
Если альтерация эндотелия и сосудистой стенки ведет к связыванию
тромбина внеклеточным матриксом, то связанный таким образом
тромбин продолжает быть активным в среде с содержанием антитромбина
III, эквивалентным его физиологической концентрации в плазме крови Все
это проясняет причину ретромбоза венечных аргерий после их
ангиопластики, несмотря на рутинное введение гепарина в больших дозах Травма
сосудистой стенки и десквамация эндотелиоцитов ведут к образованию зна-
112
Глава 8
чительной поверхности для связывания тромбина, который остается
активным, несмотря на достаточное для инактивации свободного тромбина
содержание в плазме антикоагулянтов Недостатки гепарина - это частное
проявление верности одного из общих патогенетических принципов
фармакотерапии, который мы позволим постулировать себе следующим
образом излишние разноуровневые блокада и модуляция регуляции на разном
уровне в результате действия лекарств - это одна из наиболее частых и
фундаментальных причин неэффективности действия средств
фармакотерапии и ее патогенности
В этой связи препаратами, привлекающими все большее внимание в
качестве антикоагулянтов выступают гирудин и его производное гирулог
Гирудин - это наиболее мощный из известных прямых ингибиторов
тромбина, ключевого фермента тромбогенеза В последние десятилетия
специфическая фармакопрофилактика тромбоза и тромболитическая терапия стали
необходимыми элементами системы лечения больных с нестабильной
стенокардией, инфарктом миокарда, после ангиопластики венечных артерий и
интенсивной терапии во многих областях хирургии При этом стало ясно,
что противопоказания к лечебной гипокоагулемии, системные побочные
действия антикоагулянтов и неуправляемость их эффектов делают
невозможной профилактику тромбоза и тромболизис лекарствами у многих
больных Так, ацетилсалициловая кислота, нарушая функционирование простаг-
ландиновой стресс-лимитирующей системы, представленной на периферии
во всех органах и тканях, может у части больных усилить артериальную
гипертензию и вызывать обострения язвенной болезни и бронхиальной
астмы Поэтому для предупреждения гиперкоагулемии и тромбоза
ацетилсалициловую кислоту назначают в относительно небольших дозах (30 мг
внутрь в сутки), стремясь к избирательному эффекту Избирательный
эффект ацетилсалициловой кислоты как антикоагулянта - это торможение
агрегации тромбоцитов через снижение образования тромбоксана А2 при
неугнетенном образовании и высвобождении эндотелиоцитами антиагреганта
простациклина
Варфарин препятствует витамин-К зависимому карбоксилированию в
печени Это снижает активность в плазме крови факторов свертывания,
образование которых в печени идет с участием витамина К Протромбиновое
время у больных, которым вводят варфарин, возрастает через 48 ч от начала
введения препарата В основном варфарин снижает протромбиновое время
через возникновение под влиянием препарата дефицита седьмого фактора
свертывания крови При острых коагулопатиях у хирургических больных
применение варфарина затруднительно из-за относительно медленного
развития гипокоагулемии и длительного последействия
Все это заставляет искать альтернативу гепарину в практике
интенсивной терапии хирургических больных При этом основные преимущества
прямых ингибиторов тромбина гирудина и гирулога представляются
следующим образом
♦ способность ингибировать тромбин, связанный с тромбом и
внеклеточным матриксом,
Тромбоз глубоких вен и тромбоэмболия легочной артерии
113
ВНУТРЕННИЙ
МЕХАНИЗМ
фактор XII
высокомолекулярный кининоген
прекалликреин
фактор XI
фактор IX
фактор 1Ха
+
фактор Villa
фактор X
ВНЕШНИЙ
МЕХАНИЗМ
фактор Vila
+
тканевой фактор
фактор IX
фактор X
ОБЩИЙ
МЕХАНИЗМ
фактор Ха
+
фактор Va
протромбин
тромбин фактор ХШа
фибриноген « ^^ фибрин перекрестно
связанный
Схема 8.2. Внешний, внутренний и общий механизмы свертывания крови (Ленц С, 1995)
♦ дефицит в организме антитромбина III не может обусловить
толерантность к гирудину и гирулогу;
♦ в отличие от гепарина, прямые ингибиторы тромбина не теряют
свойств антикоагулянтов под влиянием высвобождаемого
активированными кровяными пластинками четвертого фактора тромбоцитов.
Внедрение в клиническую практику высокоэффективных антикоагулянтов,
чей эффект достоверно предсказуем, не исключит из лечебной работы
необходимость частой фиксации показателей свертывающей системы крови,
выраженности тромбогенного потенциала сосудов и активности их антисистем.
Протромбиновое время - это временной интервал между
добавлением тканевого тромбопластина (тканевого фактора и фосфолипидов) к ре-
кальцифицированной плазме и образованием в плазме крови сгустка
фибрина. У здоровых людей протромбиновое время находится в пределах 11-14
с. Определение протромбинового времени позволяет судить об
активности внешнего и общего механизмов свертывания крови (схема 8.2). Рост
114
Глава 8
Таблица 8.5
Факторы свертывания крови
I
П
Ш
IV (устарев
ший термин )|
V
VI
vn
VIII
VHI:C
МП: Ag
VniR:RCo
VIE: vWF
DC
X
XI
ХП
ХШ
Фибриноген
Протромбин
Тро мбоп ласти н
Кальций
Проакселерин
Акселерин (устаревший термин, обозначавший активированный пятый
фактор свертывания)
Проконвертин (термин, вышедший из употребления)
Композит, состоящий из различных протеинов с разными свойствами
Низкомолекулярный компонент со свойствами коагулянта, дефицит
которого определяет гипокоагулемию при классической форме
гемофилии
Антигенная детерминанта композитной молекулы
Способствует индуцируемой ристоцетином агрегации тромбоцитов
Фактор Фон Виллебранта, вызывающий адгезию тромбоцитов
Фактор Кристмаса, тканевой тро мбоп ласти н
Фактор Стюарта-Проуэлла
Предшественник тромбопластина плазмы крови
Фактор Хагемана
Фактор Лаки-Лоранда, чья активная форма, воздействуя на фибрин
S, приводит к образованию фибрина I как структурной основы пер-
вичного красного тромба
протромбинового времени - это наиболее чувствительный маркер
дефицитов в крови седьмого и десятого факторов свертывания крови.
Увеличение протромбинового времени могут также обуславливать дефициты
в организме пятого фактора свертывания крови, протромбина и
фибриногена. Протромбиновое время - это критерий состояния общего и
внутреннего механизма свертывания крови на организменном уровне и
дефицитов на системном уровне факторов свертывания крови (табл. 8.5).
Поэтому при тромбогенезе как преимущественно локальном типовом
патологическом процессе оно часто остается в пределах диапазона
нормальных колебаний.
Активированное частичное тромбопластиновое время (АЧТВ) у
здоровых людей колеблется от 22 до 36 с. Оно представляет собой
временной промежуток после добавления фосфолипида как действующей
основы тромбопластина к рекальцифицированной плазме, которую
предварительно инкубируют с инертными частицами, вызывающими
контактную активацию внутреннего механизма свертывания крови.
Увеличение этого времени свидетельствует о снижении в плазме крови
концентраций факторов ее свертывания VIII, IX, XII на 30 % и более от
верхнего предела диапазона нормальных колебаний. АЧТВ как критерий
преимущественно внутреннего механизма свертывания крови часто
остается в нормальных пределах после индукции тромбогенеза у больных
Тромбоз глубоких вен и тромбоэмболия легочной артерии
115
с тромбозом глубоких вен нижних конечностей и тромбоэмболией
легочной артерии.
Тромбиновое время, составляющее в норме от 11 до 18 с, растет при
диссеминированном внутрисосудистом свертывании в стадии гипо-
коагулемии (коагулопатии потребления), гипо- и дисфибриногенемиях.
Этот показатель не отражает интенсивность тромбообразования и
увеличения массы сгустка за счет приращения к нему фибрина в локусе
тромбогенеза, которые определяет активность тромбина, связанного с
фибрином сгустка.
Глава 9
ПАТОФИЗИОЛОГИЯ ИШЕМИЧЕСКОЙ
БОЛЕЗНИ СЕРДЦА
Angina pectoris (стенокардия, грудная жаба) - это длительные тупая боль
или ощущение сдавления, локализованные преимущественно в области
сердца. Зона распространения angina pectoris в данной области сравнима с
площадью поверхности сжатого кулака. Грудная жаба может иррадиировать в
шею, челюстно-лицевую область, в левые руку и предплечье. Нередко angina
pectoris иррадиирует в область живота. Чаще всего грудную жабу вызывают
отрицательный эмоциональный стресс, физическая нагрузка, или она
возникает при употреблении пищи.
Angina pectoris как симптом всегда свидетельствует о неудовлетворенной
потребности сердца в свободной энергии, что чаще всего связано с
обструкцией кровотоку по венечным артериям. Наиболее частая причина
обструкции - это фиксированный стеноз венечной артерии вследствие ее атероскле-
ротических поражений. Если сужения просвета венечных артерий,
обусловленного атеросклерозом, не выявляют, то циркуляторная гипоксия
(ишемия) миокарда может быть следствием других причин (табл. 9.1).
Выделяют грудную жабу (стенокардию), которую вызывает
физическая нагрузка. Физическая нагрузка увеличивает потребность организма в
кислороде и энергопластических субстратах, что повышает их системный
транспорт через увеличение минутного объема кровообращения, которое
Таблица 9.1
Наиболее частые причины стенокардии
Заболевания и патологические состояния, сужающие венечные артерии:
- атеросклероз
-спазм венечных артерий
-васкулит
Врожденные и приобретенные сердечные пороки:
-аортальные стеноз и недостаточность
-митральные стеноз и недостаточность
-стеноз легочной артерии
Кардиомиопатии
Гипертрофия миокарда в стадии декомпенсации
Артериальная гипертензия
Сердечные аритмии (в особенности суправентрикулярная тахикардия)
Генетически обусловленные сужение просвета венечных артерий и гипоксия
миокарда:
-врожденные аномалии сердечных артерий
-цианотические врожденные пороки сердца
- коарктация аорты
Патофизиология ишемической болезни сердца
117
невозможно без адаптивной тахикардии. Тахикардия - это основная
детерминанта роста потребности сердца в кислороде. При физической нагрузке
весьма значителен рост потребности сердца в кислороде под влиянием
увеличения частоты сердечных сокращений. При этом возможность
увеличения доставки 02 кардиомиоцитам у больных ишемической болезнью
сердца ограничена из-за фиксированного атеросклеротическим поражением
стеноза венечных артерий. В результате возникает несоответствие между
потребностью сердца в кислороде и доставкой его кардиомиоцитам,
которое проявляет себя angina pectoris. Грудная жаба при этом выступает не
только признаком, но и звеном патогенеза циркуляторной гипоксии сердца.
У многих больных ишемической болезнью сердца патологическая
реакция на физическую нагрузку в частности заключается в росте общего
периферического сосудистого сопротивления. Это повышает работу левого
желудочка по преодолению его постнагрузки и обуславливает стенокардию
у больных с атеросклеротическим стенозом венечных артерий, у которых
доставка кислорода кардиомиоцитам не может возрасти адекватно росту
потребности сердца в 02. Рост общего периферического сосудистого
сопротивления (ОПСС) оценивают косвенно, ориентируясь на уровень диастоли-
ческого артериального давления, находящийся с ОПСС в прямой связи. Для
каждого больного определяют уровни («пределы») частоты сердечных
сокращений и среднего артериального давления, превышение которых при
физической нагрузке ведет к циркуляторной гипоксии (ишемии) сердца.
Грудная жаба этого типа обычно беспокоит больных в течение 1-5 мин
после прекращения нагрузки. Вторичной по отношению к циркуляторной
гипоксии миокарда кардиомиопатии при этом виде стенокардии не
развивается. Дисфункция сердца, связанная с его циркуляторной гипоксией, в
этом случае возникает только при физической нагрузке.
Степень стенокардии (циркуляторной гипоксии, ишемии сердца),
вызываемой физической нагрузкой, позволяют оценить классификации нью-
йоркской сердечной ассоциации (NYHA) и канадского
сердечно-сосудистого общества (CCS) (табл. 9.2).
Грудная жаба, от которой больные страдают в условиях покоя, - это или
следствие полной обтурации просвета венечных артерий атеросклеротичес-
кими бляшками, или это стенокардия, вызванная спазмом венечной артерии
на протяжении участка, просвет которого сужен вследствие атеросклероза.
Нередко angina pectoris в условиях покоя возникает у больных с аортальным
стенозом. Причиной грудной жабы в данном случае выступает снижение
доставки кардиомиоцитам левого желудочка кислорода и источников
свободной энергии при гипертрофии миокарда в стадии декомпенсации.
В последние годы у больных все чаще выявляют циркуляторную
гипоксию (ишемию) миокарда, которая не проявляется стенокардией, но ведет к
сердечной недостаточности. Первым симптомом сердечной
недостаточности, обусловленной ишемической болезнью сердца без грудной жабы,
выступает повышенная утомляемость, к которой может присоединиться
одышка. Стенокардии у таких больных часто нет среди симптомов болезни и тогда,
когда внезапное развитие кардиогенного шока, отека легких высокого дав-
118
Глава 9
Таблица 9.2
Классификации степени тяжести циркуляторной гипоксии миокарда
(функциональных классов стенокардии)
Степень
I
II
Ш
IV
Классификация
NYHA
Ишемическая болезнь
сердца, не ограничивающая
двигательной акта вн ости
Умеренное ограничение
физической активности,
связанное с груцной жабой.
Обычная повседневная
физическая нагрузка приводит к
утомлению и стенокардии
Значительное ограничение
физической активности. Нет
стенокардии в условиях
покоя
Стенокардия в покое,
нарастающая при увеличении
физической активности
CCS
Нет стенокардии при обычной физической
активности: ходьба, подъем по лестнице. Angina
pectoris при интенсивной и длительной
физической нагрузке
Умеренное ограничение повседневной
физической активности. Ограничение
стенокардией скорости ходьбы вверх по наклонной
плоскости и подъема по лестнице. Стенокардия,
связанная с приемом пищи и при низкой
температуре окружающей среды
Значительное ограничение физической
активности; без стенокардии больной по ровной
поверхности может пройти не более 1-2
кварталов
Любое увеличение физической активности
приводит к стенокардии. Стенокардия может
возникнуть в покое
ления и нарушений сердечного ритма осложняет течение ишемической
болезни сердца Безболевую (немую) циркуляторную гипоксию сердца с
помощью специальных методов исследований выявляют при обследовании
больных, не предъявляющих жалобы на грудную жабу. Эпизоды безболевого
обострения циркуляторной гипоксии (ишемии) миокарда фиксируют у 2,5 %
мужчин среднего возраста без жалоб, свидетельствующих о развитии
сердечной недостаточности, и приступов стенокардии в анамнезе (первый тип
безболевой ишемии миокарда). У 40 % больных ишемической болезнью сердца,
которая проявляет себя грудной жабой, при непрерывной регистрации
электрокардиограммы также периодически выявляют приступы безболевой
ишемии. В этой группе безболевая ишемия указывает на повышенный риск
инфаркта миокарда. Предположительно в основе безболевой ишемии миокарда
лежит генетически обусловленная недостаточность ноцицепторов, стимулом
для которых служит недостаток кислорода в сердце и кардиомиоцитах.
Нестабильная стенокардия - это синдром, который характеризуют:
♦ грудная жаба, возникшая впервые;
♦ angina pectoris в условиях покоя;
♦ стенокардия при минимально интенсивной физической нагрузке;
♦ нарастание частоты, интенсивности и продолжительности приступов
стенокардии.
Морфопатогенез нестабильной стенокардии состоит в разрыве или
расщеплении атеросклеротической бляшки с образованием тромба, чему
способствуют спазм ветви венечной артерии и активация тромбоцитов в ее
просвете.
Патофизиология ишемической болезни сердца
119
У части больных нестабильную стенокардию вызывают: а)
недостаточная доставка кислорода клеткам сердца из-за падения кислородной
емкости крови вследствие анемии; б) рост потребности сердца в кислороде при
преодолении патологически высокой постнагрузки левым желудочком у
больных с артериальной гипертензией. У 10-20 % больных с нестабильной
стенокардией течение ишемической болезни сердца осложняет инфаркт
миокарда. В этой связи нестабильную стенокардию следует рассматривать
как показание к неотложной госпитализации больного.
Спонтанная стенокардия (стенокардия Принцметала) - это
стенокардия, возникающая в условиях покоя в результате спазма венечной
артерии. При стенокардии Принцметала циркуляторная гипоксия
миокарда проявляет себя временным подъемом сегмента S-T
электрокардиограммы без последующего развития инфаркта миокарда. Спазм венечной
артерии чаще возникает в ее суженных атеросклерозом участках
предположительно в силу большей чувствительности пораженной части к
сосудосуживающим влияниям. Если спазм венечной артерии, приводящий
к стенокардии данного вида, происходит в интактных сосудах, то
прогноз более благоприятен. Через циркуляторную гипоксию кардиомио-
цитов спазм венечной артерии может привести к сердечным аритмиям,
вентрикулярным и суправентрикулярным тахикардиям и атриовентри-
кулярной блокаде. У небольшой части больных спазм венечной артерии
при стенокардии Принцметала служит причиной инфаркта миокарда и
внезапной сердечной смерти.
Сила сокращений сердечной мышцы находится в прямой связи с
содержанием ионизированного кальция в цитоплазме кардиомиоцитов ([Са2+]).
Высокие значения [Са2+] индуцируют сокращение кардиомиоцитов, а
снижение концентрации ионизированного кальция в цитозоле ведет к
расслаблению саркомеров миокарда. В конце систолы связанное с утилизацией
свободной энергии (активное) функционирование ионных насосов
перемещает ионизированный кальций обратно из цитозоля в саркоплазматический
ретикулум. В результате снижается [Са2+], и сердечная мышца
расслабляется. Патологический рост отношения потребности сердца в кислороде к
доставке 02 в клетки сердца ведет к дефициту в кардиомиоцитах свободной
энергии, то есть их гипоэргозу. Гипоэргоз кардиомиоцитов снижает
активное возвращение ионизированного кальция в саркоплазматический
ретикулум. Это обуславливает сохраняющееся в диастолу сокращение саркомеров
миокарда желудочков, вызывая их диастолическую жесткость.
Патологическая ригидность стенки желудочков в диастолу, обусловленная гипокси-
ческой контрактурой саркомеров миокарда, повышает давление крови,
которую желудочек содержит в конце диастолы. При этом не растет конечно-
диастолический объем. Это приводит к диастолической левожелудочковой
недостаточности, ко вторичной легочной венозной гипертензии и кардио-
генному отеку легких (крайняя стадия развития левожелудочковой
недостаточности).
Патологический рост соотношения между потребностью сердца в
кислороде и доставкой ему кислорода через гипоэргоз кардиомиоцитов уг-
120
Глава 9
нетает активные сокращение и расслабление желудочков и вызывает сие-
толическую и диастолическую сердечную недостаточность.
Нормальный систолический компонент насосной функции левого
желудочка сердца необходимым условием имеет аэробное биологическое
окисление в кардиомиоцитах выше определенного уровня интенсивности.
Падение интенсивности аэробного биологического окисления вследствие
циркуляторной гипоксии ниже этого уровня через гипоэргоз
сократительных кардиомиоцитов быстро приводит к падению ударного объема и
фракции изгнания левого желудочка сердца, вызывая систолическую сердечную
недостаточность.
Потребность сердца в кислороде находится в прямой связи с величиной
детерминант потребления сердцем кислорода, к которым относят:
♦ частоту сердечных сокращений (ЧСС),
♦ сократимость миокарда,
♦ напряжение стенки желудочков при сокращении.
При каждом сокращении сердце утилизирует относительно
фиксированное количество кислорода. Поэтому потребление сердцем кислорода за
минуту и потребность сердца в кислороде растут при возрастании частоты
пульса. При росте ЧСС в два раза потребность сердца в кислороде растет в
большей степени. Степень роста потребности миокарда в 02 преобладает
над степенью вызвавшего его роста ЧСС, так как увеличение частоты
пульса повышает сократимость сердца. В результате рост потребности сердца в
кислороде начинает определять не только тахикардия, но и увеличение
сократимости.
Напряжение стенки желудочков при сокращении миокарда их стенок
(S) зависит от давления крови в желудочке во время сокращения
(постнагрузка, Р), от размеров камеры сердца (преднагрузка), то есть от ее радиуса
(R) и толщины ее стенки (h). В соответствии с законом Лапласа,
напряжение стенки желудочка представляет собой прямую функцию радиуса
полости желудочка и давления в ней крови. S растет по мере увеличения
давления крови в желудочке в фазу изгнания, а также возрастает при неизменном
давлении в сердечной камере вследствие роста ее радиуса. Если для
лучшего понимания и разумного упрощения форму камеры желудочка сердца
считать шаром, то: S = PR / 2h.
Увеличение давления в сердечной камере в фазу изгнания в два раза
вызывает двухкратный рост потребности ее миокарда в кислороде через
удвоение S. Р - это прямая функция среднего артериального давления.
Поэтому рост среднего АД может повышать соотношение между
потребностью сердца в кислороде и доставкой кардиомиоцитам 02. Такой рост
потребности миокарда в кислороде может служить причиной гипоэргоза
клеток сердца и прогрессирования сердечной недостаточности у больных
ишемической болезнью сердца. Поэтому целью интенсивной терапии
сердечной недостаточности может служить снижение среднего
артериального давления, которое не приводит к опасному падению перфузионного
давления миокарда и через снижение S снижает потребность сердца в
кислороде.
Патофизиология ишемической болезни сердца
121
Увеличение объема камеры сердца в два раза увеличивает ее радиус
всего на 26 %. Поэтому увеличение размеров желудочка само по себе в
меньшей степени влияет на потребность сердца в кислороде, чем рост Р. Тем не
менее, экцентрическая гипертрофия желудочка в стадии декомпенсации
может приводить к большему росту потребности миокарда в 02, чем тот,
который можно ожидать, ориентируясь только на увеличение радиуса
желудочка. Дело в том, что дилатация желудочка через увеличение объема
крови, который сердечная камера содержит в конце диастолы, увеличивает
Р. В результате потребность миокарда в кислороде повышают как
увеличение R, так и рост Р.
Возрастание преднагрузки через увеличение конечно-диастоличесого
давления повышает Р и, увеличивая конечно-диастолический объем крови
в желудочке, обуславливает рост R. В результате рост преднагрузки может
вызвать опасный рост S. Поэтому снижение общего венозного возврата,
которое не приводит к артериальной гипотензии и снижению перфузионно-
го давления миокарда, можно считать целью терапии у больных
ишемической болезнью сердца. Такое снижение общего венозного возврата, снижая
преднагрузку, уменьшает S и потребность сердца в кислороде. В этом в
частности и заключается желательный эффект на сердце нитроглицерина,
применяемого крайне осторожно, дабы не вызвать острой артериальной
гипотензии.
Биологический смысл гипертрофии стенки желудочка заключается в том,
что утолщение стенки снижает ее напряжение и потребность сердца в
кислороде. Гипертрофия миокарда как защитная реакция в ответ на рост
напряжения стенки желудочка в фазу изгнания эффективна лишь до развития
вторичной по отношению к гипертрофии кардиомиопатии, вследствие
которой в клетках сердца падает эффективность биологического окисления.
Кроме того, при вторичной кардиомиопатии такого происхождения гипоэр-
гоз клеток сердца обусловлен снижением доставки к ним кислорода,
которую ограничивает недостаточность капиллярной сети
гипертрофированного миокарда.
Единственный механизм удовлетворения возросшей потребности
сердца в кислороде - это рост объемной скорости кровотока по венечным
артериям.
Объемная скорость кровотока по венечным артериям представляет
собой обратную функцию общего сосудистого сопротивления в системе
венечных артерий (Re). Re в свою очередь зависит от соотношений эффектов
на уровне сосудистой стенки ряда регуляторных влияний.
При росте работы сердца по изгнанию крови из желудочков в нем
происходит накопление молочной кислоты и протонов, расширяющих
венечные артерии небольшого диаметра. Кроме того, вследствие падения
напряжения кислорода в цитозоле и связанного с ним гипоэргоза кардиомиоцитов
происходит усиленный распад аденозинтрифосфата, и в клетках сердца
растет содержание аденозина. Аденозин расширяет венечные артерии, их
мелкие ветви и микрососуды сердца. В результате падение Re увеличивает
доставку кислорода кардиомиоцитам. Следует заметить, что при
122
Глава 9
фиксированном стенозе венечных артерий вследствие атеросклероза этот
механизм ауторегуляции неэффективен, и любой рост потребности сердца
в кислороде опасен развитием циркуляторной гипоксии миокарда, так как
не приводит к снижению Re через аккумуляцию протонов и аденозина.
Стенка венечных артерий и сердечных артериол содержит альфа- и бета-
адренорецепторы. Альфа-адренергическая стимуляция стенки венечных
артерий может приводить к их спазму, несмотря на противодействие
метаболических факторов регуляции Re. Это чаще всего происходит в условиях
покоя, когда действие метаболических факторов можно считать невысоким
по интенсивности. По мере нарастания содержания в клетке протонов и
аденозина, связанного с увеличением работы сердца, действие
метаболических факторов регуляции сопротивления венечных артерий начинает
преобладать над эффектом нервных альфа-адренергических
сосудосуживающих влияний. Нервная альфа-адренергическая стимуляция стенки венечных
артерий как компонент патологической стрессорной реакции может
обуславливать циркуляторную гипоксию миокарда вплоть до ишемии и
цитолиза кардиомиоцитов (стенокардия Принцметала). Наиболее частая причина
необратимых ишемических повреждений миокарда вследствие нейроген-
ной констрикции венечных артерий - спазм сосуда в области его атероскле-
ротического поражения.
Нельзя исключить и значение в развитии спазма венечных артерий
действия на них таких «стрессорных гормонов» как вазопрессин и ангиотензины.
Свою роль в ауторегуляции сопротивления венечных артерий играет
простагландиновая система их эндотелия (тромбоксан, простациклин,
другие производные арахидоновой кислоты). Тромбоксан, повышая тромбо-
генный потенциал микрососудов сердца, вызывает их спазм при инфаркте
миокарда и циркуляторной гипоксии сердца, а действие простациклина
противодействует этим эффектам. Соотношение действий тромбоксана и
простациклина во многом определяет состояние периферического
кровообращения на уровне сердца.
В миокарде человека соотношение между кардиомиоцитами и
капиллярами составляет 1:1. В условиях покоя функционируют 3/5- 4/5 всех
капилляров сердца. Рост потребности сердца в кислороде мобилизует резерв
капилляров, и Re падает. Кроме того, мобилизация резервов капилляров
снижает путь диффузии кислорода из просвета микрососудов к
митохондриям кардиомиоцитов, что увеличивает транспорт кислорода в клетки
сердца. При гипертрофии миокарда в стадии декомпенсации эффективность
данной компенсаторно-приспособительной реакции падает, и рост
потребности сердца в кислороде усиливает циркуляторную гипоксию сердца.
В системе венечных артерий и микроциркуляции сердца есть
множество коллатералей, которые не функционируют в условиях покоя. При
обструкции вследствие атеросклеротического поражения венечной артерии
кровоток по коллатералям предотвращает необратимые ишемические
повреждения миокарда. Дозированная физическая нагрузка, несмотря на
развитие атеросклероза венечных артерий, может увеличить степень
возрастания объемной скорости кровотока по коллатералям в ответ на рост
Патофизиология ишемической болезни сердца
123
соотношения между потребностью сердца в кислороде и доставкой 02 кар-
диомиоцитам.
Re представляет собой линейную функцию вязкости крови.
Обезвоживание повышает вязкость крови через увеличение гематокрита. Если
больным гипертонической болезнью, страдающим от циркуляторной гипоксии
миокарда, назначают мочегонные средства, то существует опасность ятро-
генного снижения доставки кардиомиоцитам кислорода. Его причина -
избыточное мочеотделение, вызывающее эксикоз и рост гематокрита.
Гипотермия повышает вязкость крови и Re, снижает доставку 02 клеткам сердца.
В этой связи искусственную гипотермию у кардиохирургических больных
считают показанием к гемодилюции, снижающей вязкость крови. В остром
периоде тяжелой раневой болезни, гемодилюция, оптимизируя
реологические свойства крови и снижая ее вязкость, повышает доставку кислорода к
кардиомиоцитам сердца, страдающего от острой нейродистрофии.
Снижение вязкости крови под влиянием дезагрегантов (курантил и др.) лежит в
основе их свойства повышать транспорт 02 в клетки сердца больных,
страдающих от его ишемической болезни.
Объемная скорость кровотока по венечным артериям (Vc) - это прямая
функция перфузионного давления миокарда (Рт) и обратная Re: Vc=Pm/Rc,
где Pm - это градиент давлений, который вызывает ток крови в
определенном участке системы венечных артерий. Кровь, протекающая по венечным
артериям, возвращается в системный кровоток в основном через
коронарный синус. Поэтому перфузионное давление миокарда для части сердца,
которой отдает кислород кровь, попадающая в правое предсердие через
коронарный синус, - это различие между диастолическим артериальным
давлением и соответствующим давлением в правом предсердии. Венозная кровь,
оттекающая от субэндокардиального слоя миокарда левого желудочка,
возвращается в системный кровоток последовательно через тебезиевы вены и
полость левого желудочка. Поэтому перфузионным давлением для
миокарда субэндокардиального слоя левого желудочка следует считать градиент
диастолического артериального давления и конечно-диастолического
давления крови в полости левого желудочка.
Кровоток в правом желудочке непрерывен и не прекращается в
диастолу. Кровоснабжение субэндокардиального слоя миокарда левого желудочка
происходит только во время диастолического интервала сердечного цикла.
Артериолы субэндокардиального слоя подвергаются почти полному
сжатию во время систолы под воздействием роста давления крови в левом
желудочке и вследствие особой выраженности укорочения миокардиоцитов
данной части сердечной мышцы во время ее систолического сокращения.
Лишь 15 % объема крови, поступающего по венечным артериям в левый
желудочек за единицу времени, омывает субэндокардиальный слой миокарда
левого желудочка во время систолы. В систолический интервал сердечного
цикла кровь поступает преимущественно в эпикардиальный слой миокарда
левого желудочка. В диастолический интервал вся кровь, поступающая по
венечным артериям в левый желудочек, попадает в субэндокардиальный
слой.
124
Глава 9
Результатом наибольшей среди всех сократительных элементов сердца
интенсивности сокращения кардиомиоцитов субэндокардиального слоя
левого желудочка выступают наибольшие потребность в кислороде и его
потребление в данной части сердца. Поэтому большинство из микрососудов в
субэндокардиальном слое предельно расширены вследствие максимальной
в пределах сердца интенсивности аэробного метаболизма в кардиомиоци-
тах этой локализации. Можно считать, что в субэндокардиальном слое
почти нет резерва роста доставки кислорода с кровью кардиомиоцитам по
микрососудам.
Высокая потребность в кислороде кардиомиоцитов
субэндокардиального слоя левого желудочка, прерывистый характер его кровоснабжения,
а также низкий резерв роста объемной скорости тока артериальной
крови по микрососудам в данной части сердца - все это определяет особую
подверженность субэндокардиального слоя левого желудочка инфаркту
миокарда.
Общая длительность диастолических интервалов за минуту (tD) - это
обратная функция частоты сердечных сокращений. Рост частоты
сердечных сокращений от 60 мин"1 до 100 мин"1 вызывает снижение tD от 34 с/мин
до 27 с/мин и соответственно снижает кровоснабжение
субэндокардиального слоя. Особенно неблагоприятная ситуация складывается тогда, когда
снижение ударного объема левого желудочка как основное звено
патогенеза тяжелой сердечной недостаточности недостаточности служит стимулом
для предельно выраженной тахикардии (схема. 9.1).
Сужение (стеноз) венечных артерий вследствие спазма или атероскле-
ротического поражения через рост сосудистого сопротивления вызывает
снижение доставки кислорода кардиомиоцитам.
Фиксированный стеноз - это сужение просвета венечной артерии,
которое некоторое время остается неизменным вследствие обтурации сосуда
атеросклеротической бляшкой, ареактивности патологически измененной
сосудистой стенки к сосудосуживающим влияниям и ригидности
измененного атеросклерозом участка сосуда.
Динамический стеноз - транзиторное сужение венечной артерии как
результат интенсификации сосудосуживающих регуляторных влияний.
Грудную жабу вследствие динамического стеноза называют вазоспастической
стенокардией (стенокардия Принцметала). Если динамический стеноз
возникает в области атеросклеротической обтурации венечной артерии при
сохраненной или повышенной реактивности ее стенки по отношению к
сосудосуживающим влияниям, то развивается нестабильная стенокардия.
Различают локальный стеноз, захватывающий небольшой участок
венечной артерии, равный по протяженности длиннику атеросклеротической
бляшки обычного размера и более протяженный сегментарный стеноз. В
соответствии с законом Пуазейля, сегментарный стеноз, в большей степени
повышая сосудистое сопротивление, вызывает большее падение объемной
скорости кровотока и доставки кислорода к клеткам сердца.
Снижение диаметра внутреннего поперечного сечения венечной
артерии (просвета) (Dc) на 50 % снижает площадь просвета артерии на 75 %.
Патофизиология ишемической болезни сердца
125
Тахикардия
Увеличение потребности
сердца в 02
Снижение tD
Падение доставки 02 к
миокарду
субэндокардиального слоя
Снижение фракции изгнания
левого желудочка и его
ударного объема
Рост конечно-
диастолического давления
в левом желудочке
Угнетение сократимости
Падение перфузионного давления
субэндокардиального слоя
Схема 9.1. Порочный круг патогенеза тяжелой
сердечной недостаточности
Стеноз такой степени уже проявляет себя стенокардией. Если Dc снижен на
75 %, то площадь просвета падает до 10 % от исходной величины. Такое
сужение приводит к стенокардии покоя. Два структурно не связанных
между собой сужения просвета венечной артерии атеросклеротического
происхождения приводят к двум последовательным падениям динамического
давления крови в просвете артерии, которое может представлять собой звено
патогенеза циркуляторной гипоксии кардиомиоцитов.
Если атеросклеротическая обтурация венечной артерии развивается
медленно, то снижение скорости объемного кровотока компенсирует
возросший ток крови по коллатералям, соединяющим разные венечные артерии
или разные сегменты одной артерии. Компенсаторная интенсификация
коллатерального кровотока не предотвращает циркуляторной гипоксии
миокарда вплоть до ишемии вследствие стеноза крайней степени, при которой
площадь просвета артерии падает до 10 % от исходной.
Степень дисфункции сердца как насоса системы кровообращения,
которую вызывает стеноз венечной артерии, связана с величиной той части
сердца, в которой вследствие сужения венечной артерии развиваются цир-
куляторная гипоксия или ишемия. В этой связи в прогностическом
отношении наиболее неблагоприятным следует признать стеноз левой главной
венечной артерии, когда единичный локальный стеноз приводит к ишемии
значительной части всего сердца. Стеноз ветвей левой главной венечной
артерии приводит к гипоксии миокарда аналогичной по протяженности той,
которую вызывает сужение левой главной венечной артерии. При этом кол-
латераль от стенозированной артерии соединяет ее с полностью обтуриро-
ванной ветвью левой главной венечной артерии (эквивалент стеноза левой
главной венечной артерии).
126
Глава 9
У больных сахарным диабетом доставку кислорода кардиомиоцитам
снижает диабетическая микроангиопатия, которая утолщает стенку
капилляров, препятствуя массопереносу кислорода из их просвета в цитозоль
клеток сердца.
Если строго в патофизиологическом смысле под ишемией понимают
патологическое состояние и некробиотические изменения тканей,
обусловленные полным прекращением их кровоснабжения, то термин «ишемия»,
используемый в клинической практике, означает неадекватное снабжение
кардиомиоцитов кислородом вследствие падения к ним транспорта 02 с
кровью.
Падение объемной скорости кровотока по венечной артерии как
причина циркуляторнои гипоксии кардиомиоцитов выступает инициирующим
моментом ряда порочных кругов пато-танатогенеза. Поэтому одно
восстановление объемной скорости кровотока по венечной артерии (вазодилята-
торы, тромболитическая терапия) не может подвергнуть обратному
развитию нарушения функций и гибель клеток сердца вследствие ишемии.
К механизмам гибели клеток сердца при тяжелой ишемии сердца относят:
♦ накопление продуктов обмена веществ, дальнейшая трансформация
которых на путях метаболизма невозможна вследствие гипоэргоза;
♦ активация мембранных фосфолипаз;
♦ образование свободных радикалов кислорода;
♦ инфильтрация ишемизированного участка сердца активированными
нейтрофилами;
♦ гиперкатехоламинемия в крови, притекающей к кардиомиоцитам по
венечным артериям;
♦ рост концентрации ионизированного кальция в цитоплазме
кардиомиоцитов;
♦ гипоэргоз как следствие недостаточного гликолиза и причина
нарушений ионного состава и отека клеток сердца.
Электрофизиологические свойства и сократительная способность
кардиомиоцитов в основе своей имеют активное (происходящее с
использованием свободной энергии) функционирование системы поддержания
ионного равновесия между клетками сердца и средой их обитания. Уже через
несколько секунд после возникновения ишемии кардиомиоцитов
изменения клеточного метаболизма вызывают нарушения трансмембранного
потенциала и способности клеток к сокращению.
Вследствие гипоэргоза (энергетического голодания) в клетках сердца
компенсаторно активируется анаэробный гликолиз, который не может устранить
недостатка свободной энергии. Гипоэргоз клеток сердца в силу
ограниченности в них запасов аденозинтрифосфата (АТФ) и креатинфосфата развивается
быстро. Данные экспериментов свидетельствуют, что процесс цитолиза
клеток сердца становится необратимым, когда содержание в них АТФ падает до
уровня меньшего, чем 10 % величины нормального содержания.
Сердце для биологического окисления может использовать все из
доступных в организме источников свободной энергии. Натощак, когда
уровень липолиза относительно высок, миокард в основном утилизирует сво-
Патофизиология ишемической болезни сердца
127
бодные жирные кислоты.' Если физическая нагрузка,
военно-травматический шок, сепсис, другие патологические состояния и процессы приводят к
лактатному ацидозу, то основным источником свободной энергии для
кардиомиоцитов становится молочная кислота. У больных, которым
производят внутривенную инфузию раствора глюкозы и инсулина, биологическому
окислению в сердце преимущественно подвергается глюкоза. В этой связи
можно считать, что в силу широкого спектра веществ, циркулирующих с
кровью, которые сердце может использовать в качестве энергетических
субстратов, снижение содержания в плазме крови того или иного источника
свободной энергии не ограничивает потребления свободной энергии миокардом.
Интенсивность и адекватность улавливания и утилизации кардиомио-
цитами свободной энергии в основном зависит от соотношения между их
потребностью в кислороде и доставкой к ним Ог
35 % массы сердечной мышцы составляют митохондрии. Как орган, в
котором особенно высок уровень аэробного метаболизма, сердце обладает
рядом особенностей:
1. Особо высокая активность в кардиомиоцитах энзимов биологического
окисления - необходимое условие нормальной насосной функции сердца. Этот
исключительный среди других органов организма уровень активности
дыхательных ферментов возможен лишь в определенном диапазоне колебаний в
цитозоле и митохондриях кардиомиоцитов напряжения кислорода. При
патологическом снижении напряжения 02 уровень активности дыхательных
ферментов резко падает, и возникает дисфункция сердца как насоса системы
кровообращения, то есть развивается сердечная недостаточность.
2. Функция сердца зависит от определенного уровня аэробного
гликолиза, поддержание которого невозможно без нормального отношения
потребности сердца в кислороде к доставке ему 02. Патологический рост
отношения быстро угнетает аэробный гликолиз. В результате возникает
дефицит свободной энергии в кардиомиоцитах, обуславливающий падение
сократимости и расстройства сердечного ритма.
Когда транспорт кислорода по венечным артериям полностью
удовлетворяет потребность в нем сердца, то концентрации цитрата и аденозинтри-
фосфата в цитозоле кардиомиоцитов достаточно велики для того, чтобы
тормозить активность ключевого фермента анаэробного гликолиза фосфоф-
руктокиназы. Падение напряжения кислорода в цитозоле кардиомиоцитов
ниже определенного уровня ведет к снижению в нем содержания аденозйн-
трифосфата и цитрата и растормаживает анаэробный гликолиз. При
ишемии в строго патофизиологическом смысле, когда аккумуляция лактата и
протонов вследствие прекращения кровотока тормозит в кардиомиоцитах
анаэробный гликолиз, клетки сердца лишаются последнего источника
свободной энергии. Кроме того, свою роль в торможении анаэробного
гликолиза играет обусловленное гипоксией и прекращением кровотока
накопление в кардиомиоцитах восстановленного никотанамидадениндинуклеотида.
Энергетическое голодание клетки и рост содержания протонов в ее
цитозоле тормозят большинство из биохимических реакций на клеточных
мембранах и в клетке, которые протекают при участии ферментов. Основным
128
Глава 9
следствием тотального на клеточном уровне угнетения активности
ферментов выступает нарушение ионного равновесия между клеткой и средой ее
обитания. В частности, прекращается активный АТФ-зависимый транспорт
ионов.
Вызванная энергетическим голоданием блокада активного выведения
натрия из клетки и связанного с ним поступления в клетку калия приводит
к росту содержания натриевого катиона в цитозоле клеток, испытывающих
недостаток кислорода. Кроме того, рост содержания протонов в цитозоле
клеток увеличивает АТФ-независимое выведение из клетки Н+ в обмен на
поступление в нее катиона натрия. Это служит причиной еще большего
увеличения содержания натрия в клетках. Рост содержания натрия ведет к
поступлению в клетки воды из межклеточных пространств. Отек клеток
обостряет их гипоксию, создавая чисто механическое препятствие
периферическому кровообращению в зоне циркуляторной гипоксии.
Одновременно с ростом содержания натрия и развитием клеточного отека
из клеток выходит калий. Предположительно выход калия обусловлен
открытием в результате гипоэргоза АТФ-зависимого мембранного калиевого
канала.
В соответствии с законом поддержания электронейтральности жидких
сред организма выход калия из клеток задерживает в них протоны, что
ускоряет развитие внутриклеточного ацидоза. Ацидоз развивается быстро, в
течение первых 10 с ишемии, одновременно со снижением сократительной
способности ишемизированного участка миокарда.
Увеличение содержания кальция в цитоплазме клеток сердца,
испытывающих острый дефицит кислорода, - это результат снижения скорости
связывания кальция саркоплазматическим ретикулумом вследствие
гипоэргоза кардиомиоцитов. Накопление кальция служит инициируюпщм моментом
трех звеньев патогенеза ишемии клеток сердца, одновременно
выступающих частными механизмами цитолиза кардиомиоцитов:
♦ ишемическая контрактура сократительных кардиомиоцитов как
результат постоянно высокой концентрации ионизированного кальция
в цитоплазме, контрактура ведет к еще большему снижению
кровоснабжения участка сердца, страдающего от циркуляторной гипоксии,
и необратимости некробиотических изменений;
♦ увеличение содержания кальция в митохондриях, которое само по себе
угнетает улавливание свободной энергии в виде АТФ при
биологическом окислении;
♦ активация фосфолипаз, ведущая к деструкции клеточных мембран
через накопление поверхностноактивных лизолецитинов.
Увеличение содержания кальция в цитоплазме ведет к захвату ионов
кальция митохондриями, что увеличивает суммарный положительный
заряд во внутримитохондриальном пространстве. Для уравновешивания
роста положительного заряда в митохондриях в них происходит активный, свя-
занный с потреблением свободной энергии в виде АТФ, процесс
перемещения протонов из митохондрий в цитоплазму. Это еще в большей
степени ограничивает величину свободной энергии, улавливаемой кардио-
Патофизиология ишемической болезни сердца
129
миоцитами в виде АТФ, так как часть АТФ утилизируется для выведения
протонов из митохондрий.
Избыток кальция в цитоплазме ведет к сердечным аритмиям,
снижающим ударный объем левого желудочка. Артериальная гипотензия как
осложнение сердечной недостаточности, прогрессирующей вследствие
аритмий, увеличивает гипоэргоз сердца и кардиомиоцитов, снижая перфузионное
давление миокарда.
Повреждения и дисфункция кардиомиоцитов вследствие ишемии и
последующего возобновления кровотока в ишемизированном участке сердца
во многом связаны с действием на клетки сердца свободных радикалов
кислорода.
Под свободным кислородным радикалом понимают молекулу, которая
содержит непарный электрон на внешней орбите, составляющего ее атома
кислорода. Это лежит в основе высокой способности такой молекулы
вступать в реакции с другими соединениями, то есть проявлять высокую реак-
тогенность. В кардиомиоцитах восстановление кислорода с образованием
молекулы воды при биологическом окислении идет двумя путями:
♦ тетравалентная восстановительная реакция с участием митохондри-
альной цитохромоксидазы без образования каких-либо
промежуточных соединений, в ходе которой 95 % кислорода входят в состав
молекул образуемой воды;
♦ моновалентная реакция, в ходе которой образуются промежуточные
соединения: супероксидные анионы, перекись водорода, гидроксиль-
ный радикал, обладающие непарными электронами на внешней
орбите атомов кислорода.
Супероксидный анион относительно других свободных кислородных
радикалов, образуемых при моновалентной реакции, обладает
относительно низкой реактогенностью. В физиологических условиях в реакции дис-
мутации он трансформируется в перекись водорода. Это более стойкое
соединение с еще меньшей реактогенностью, которое способно
диффундировать на значительное расстояние от места своего образования.
Опасность роста содержания в клетках сердца перекиси водорода состоит в
образовании из нее в реакциях Хабер-Вайса и Фентона гидроксильного
радикала.
Гидроксильный радикал - соединение нестойкое, но он обладает крайне
высокой реактогенностью и активно встраивается в боковые цепи
ненасыщенных жирных кислот, приводя к перекисному окислению липидов.
Выраженность цитотоксичности этих реакций во многом определяется
доступностью в локусе их протекания ионов металлов с переходной
валентностью, выступающих катализаторами образования высоко реактогенно-
го гидроксильного иона.
Ишемия вызывает сдвиги клеточного обмена, способствующие
образованию свободных кислородных радикалов из того молекулярного
кислорода, который клетка содержит после развития ишемии.
В разные периоды ишемии и реперфузии источники образования
свободных кислородных радикалов различны. Например, радикалы образуют-
130
Глава 9
ся в митохондриях, на других мембранных структурах клеток миокарда,
эдотелио- и лейкоцитов. Во время ишемии идет восстановление элементов
электронно-транспортной цепи митохондрий, что приводит к усиленной
утечке электронов из дыхательной цепи, которые, реагируя с
молекулярным кислородом, образуют супероксидные радикалы. Реперфузия
восстанавливает биологическое окисление и улавливание свободной энергии в
митохондриях, но интенсивность тока электронов по
электронно-транспортной цепи при реперфузии невелика из-за недостатка аденозиндифосфата,
что вновь служит причиной образования свободных кислородных
радикалов. Вероятно, что в ранних стадиях ишемии увеличенному образованию
свободных кислородных радикалов противостоит действие супероксиддис-
мутазы (СОД). Активность супероксиддисмутазы выступает обратной
функцией длительности ишемии. В результате низкой активности СОД после
длительной ишемии митохондрии подвергаются все более и более
сильному деструктивному действию свободных кислородных радикалов.
Эндотелиоциты капилляров содержат фермент ксантиндегидрогеназу.
Ишемия ведет к трансформации энзима в оксидазную форму, катализатор
превращения гипоксантина и ксантина в мочевую кислоту. При этом
кислород выступает в роли акцептора электронов. Кислород, поступающий в
ишемизированный участок сердца при реперфузии, восстанавливается
данной ферментной системой с образованием свободных радикалов.
Активированные нейтрофилы генерируют свободные радикалы,
действие которых на мембраны клеток служит одной из причин вторичной
альтерации при воспалении, в том числе и сугубо патогенном, которое не
имеет биологической цели. В результате вторичной альтерации, связанной с
высвобождением свободных кислородных радикалов активированными
полинуклеарами, растет зона ишемического повреждения миокарда.
Неясно, является ли активация нейтрофилов следствием ишемии и реперфузии,
или это результат воспалительной реакции на некробиотические изменения
клеточных элементов сердца.
Образование свободных кислородных радикалов на мембранах кардио-
миоцитов связано с каскадом реакций образования арахидоновой кислоты
и аутокисления катехоламинов. Активация фосфолипаз служит причиной
гысвобождения арахидоната и норадреналина. Аутоокисление
катехоламинов, высвобождаемых ишемизированным миокардом, также служит
источником свободных кислородных радикалов.
Итак, ишемия и реперфузия ведут к массивному образованию
свободных кислородных радикалов в клетках сердца. Свободные кислородные
радикалы вызывают перекисное окисление липидов, выступающего в
качестве основной причины деструкции клеточных мембран,
Предположительно, свободные кислородные радикалы также вызывают конформационные
изменения мембранных протеинов и локализацию ряда белков-ионообмен-
ников. Этим объясняют патологические изменения трансмембранного
транспорта ионов, предшествующие необратимой деструкции мембраны.
При неугнетенных аэробных биологическом окислении и гликолизе,
несмотря на непрерывное образование свободных кислородных радикалов,
Патофизиология ишемической болезни сердца
131
кардиомиоциты нормально функционируют, благодаря равновесию между
системами, генерирующими оксиданты, и защитными антиоксидантными
системами. В сердце антиоксидантные системы представлены ферментами
супероксиддисмутазой (СОД), каталазой и глутатионпероксидазой, а также
другими эндогенными антиоксидантами: витамином Е, аскорбиновой
кислотой и цистеином. Основной антиоксидантной системой кардиомиоцитов
выступает СОД. Этот фермент представляет собой катализатор дисмутации
супероксидных анионов в перекись водорода и молекулярный кислород,
ускоряя скорость этой реакции в 109 раз. Одна из форм СОД содержит медь
и цинк и присутствует в цитоплазме, другая, содержащая марганец,
находится в митохондриях.
В инактивации перекиси водорода как основного источника высокоре-
актогенного и особо цитотоксичного гидроксильного радикала
существенную роль играют ферментные системы каталазы и глутатионпероксидазы,
содержание которой в кардиомиоцитах выше. При ишемии резко падает
концентрация в клетках сердца восстановленной формы глутатиона (ВФГ).
Одновременно гипоэргоз угнетает активное выведение из клетки
окисленной формы глутатиона (ОФГ). В результате в кардиомиоцитах почти
блокируется превращение ВФГ в ОФГ, необходимое для активации
глутатионпероксидазы. Поскольку глутатионпероксидаза удаляет из клеток перекись
водорода, ишемия и гипоэргоз через резкое снижение активности
глутатионпероксидазы ведут к аккумуляции в цитозоле клеток сердца Н202.
Витамин Е как антиоксидант действует в синергизме с аскорбиновой
кислотой, захватывая свободные кислородные радикалы. В силу своей ли-
пофильной природы витамин Е выступает в качестве внутримембранного
антиоксиданта, тогда как витамин С, основной элемент водорастворимой
системы электронного транспорта, осуществляет свое действие в
цитоплазме или во внеклеточной жидкости.
Ишемия угнетает защитные антиоксидантные внутриклеточные
системы. В основном эти патологические сдвиги затрагивают, по-видимому, ми-
тохондриальную супероксиддисмутазу, активность которой вследствие
полного прекращения доставки к клеткам кислорода с кровью падает на 50 %.
После угнетения гипоксическим гипоэргозом антиоксидантных систем
клетки восстановление доставки к ней молекулярного кислорода
предположительно повышает образование свободных кислородных радикалов до
уровня, превышающего нейтрализующую способность митохондриальной
СОД (окислительный стресс). Восстановление кровотока (реперфузия) после
кратковременной ишемии (30-60 мин) не вызывает окислительный стресс
в силу относительной интактности антиоксидантных систем. Реперфузия
после длительной ишемии, когда защитные механизмы более ослаблены,
через окислительный стресс вызывает некробиотические изменения
кардиомиоцитов. Снижение активности СОД вследствие ишемии и реперфузии
специфично для митохондриальной СОД.
Падение активности митохондриальной СОД нарушает функцию
митохондрий и в еще большей степени блокирует улавливание клеткой
свободной энергии. Предположительно вследствие угнетения антиоксидант-
132
Глава 9
ной системы митохондриальной СОД резко растет активность второй по
значимости антиоксидантной системы кардиомиоцитов, системы глута-
тионпероксидазы.
Быстрое восстановление кровообращения в ишемизированном участке
сердца под влиянием тромболитической терапии - это наиболее
эффективный способ восстановления функций, испытавших гипоксию и гипоэргоз
кардиомиоцитов. Однако запоздалая реперфузия, восстанавливающая
доставку кардиомиоцитам с угнетенными антиоксидантными системами
кислорода как субстрата образования свободных кислородных радикалов,
усиливает некробиотические изменения клеток сердца и ускоряет их цитолиз.
Поэтому в настоящее время ведется интенсивный поиск и исследование
средств, увеличивающих антиоксидантную способность клеток сердца.
Ишемия вызывает цитолиз, активируя разрушающие мебраны
кардиомиоцитов фосфолипазы. Эти ферменты катализируют трансформацию
нормальных липидов клеточных мембран в поверхностноактивные
соединения, которые разрушают мембраны. Так, гипоксия и гипоэргоз активируют
фосфолипазу А2, при воздействии которой на лецитин мембран образуется
лизолецитин. Лизолецитин, вступая в соединение с молекулой любой
несвязанной внутриклеточной кислоты (линолевой и др.), образует высоко
поверхностноактивные лизофосфолипидные мицеллы, что служит
причиной деструкции мембран.
Ишемия сердечной мышцы, особенно проявляющая себя стенокардией,
ведет к усилению адренергической нейрогуморальной стимуляции сердца.
В результате происходит избыточная аккумуляция в кардиомиоцитах
циклического аденозинмонофосфата. Это служит причиной устойчивого и
патогенного возрастания содержания в цитозоле ионизированного кальция,
дисфункций и цитолиза клеток сердца.
Гипоксический гипоэргоз ведет к аккумуляции в цитозоле клеток сердца
цитотоксичных продуктов метаболизма. К ним относят
поверхностноактивные липиды, образующиеся из неокисленных экзогенных жирных кислот.
Ишемия, тяжелая циркуляторная гипоксия и гипоксический гипоэргоз
сердца вызывают цитолиз его клеток и через патогенные межклеточные
взаимодействия, составляющие порочный круг патогенеза при ишемии (схема 9.2).
Активированные нейтрофилы в зоне ишемического повреждения
сердца представляют собой клеточный эффектор острого воспаления, во
многом лишенного биологической цели, которое через высвобождение поли-
морфонуклеарами свободных кислородных радикалов и экзоцитоз из
нейтрофилов протеолитических ферментов увеличивает массу
кардиомиоцитов, подвергшихся некробиотическим изменениям. Каскад реакций
свертывания крови и связанная с ним активация системы комплемента по
альтернативному пути, аккумуляция в ишемизированном участке такого
флогогена как лейкотриен В4 служат индукторами воспаления в части
сердца, пораженной гипоксией и гипоэргозом.
Роль патогенных межклеточных взаимодействий в распространении зоны
ишемических повреждений клеток, которые в очаге ишемии и воспаления
во многом осуществляются через высвобождение клетками цитокинов, под-
Патофизиология ишемической болезни сердца
133
Ишемия
Активация полиморфонуклеаров
Активация тромбоцитов
Высвобождение активированными
кровяными пластинками
тромбоксана А2 и фактора
активирующего тромбоциты
Спазм и тромбоз микрососудов
Схема 9.2. Порочный круг межклеточных взаимодействий при ишемии
черкивает следующее определение ишемии миокарда: миокардиальная
ишемия - это гипоксия миокарда, которую характеризуют
♦ интенсивное высвобождение соответствующими клетками в очаге
ишемического повреждения фактора некроза опухолей;
♦ усиленное образование в нем свободных кислородных радикалов;
♦ потеря микрососудами из системы венечных артерий в зоне
ишемического повреждения способности к расширению в ответ на
сосудорасширяющие паракринные влияния со стороны эндотелиоцитов;
♦ некробиотические изменения кардиомиоцитов.
Известно, что рекомбинантный фактор некроза опухолей, не
обладающий биологической активностью своего естественного аналога, но
тормозящий экспрессию его гена, сохраняет реактивность гладкомышечных
элементов сосудистой стенки ветвей венечных артерий по отношению к
вазодилатирующим паракринным сигналам их эндотелиоцитов и снижает
выраженность некробиотических изменений клеток сердца, обычно
вызываемых эндогенным фактором некроза опухолей.
Усиленная адгезия полиморфонуклеаров к эндотелиоцитам в зоне
ишемии представляет собой результат активации нейтрофилов и (или)
эндотелиоцитов. In vitro гипоксия эндотелиоцитов с последующим восстановлением
доставки к ним кислорода приводит к росту прилипания
полиморфонуклеаров к эндотелиальным клеткам, который в основном связан с
функционированием таких адгезивных молекул как Р-селектин, Е-селектин, а также
межклеточная адгезивная молекула-I. Свою роль в адгезии нейтрофилов к
эндотелию играют эндотелиоцитарный интерлейкин-1, фактор активации
тромбоцитов, а также высвобождаемые эндотелиоцитами при ишемии и
реперфузии свободные кислородные радикалы, тромбоксан В2 и
сниженное образование оксида азота. Значение адгезивных молекул в развитии
воспаления в ответ на ишемию подчеркивает торможение воспаления анти-
134
Глава 9
телами, специфичными по отношению к ним. В эксперименте было
показано, что такие антитела снижают выраженность некробиотических
изменений тканей, связанных с ишемией и реперфузией.
Главную цель терапии ишемической болезни сердца, то есть его цирку-
ляторной гипоксии, можно определить следующим образом:
Оптимальное снижение отношения между потребностью сердца в
кислороде и доставкой к его клеткам 0Т которое повышает способность
сердца реагировать усилением насосной функции в ответ нарост
потребления кислорода всем организмом. Иными словами, необходимо так
снизить потребность кардиомиоцитов в кислороде и повысить транспорт 02
в их цитозоль, чтобы сердце отчасти или полностью восстановило
способность повышать выброс крови в аорту и легочную артерию для
удовлетворения возросшей потребности органов и тканей в кислороде,
энергопластических субстратах и агентах гуморальной регуляции.
Данная цель терапии хронической циркуляторной гипоксии миокарда,
обусловленной атеросклерозом, недостижима без строгого следования
общим принципам лечения стенокардии, которые сводятся к устранению
факторов риска атеросклероза. Так, важно прекратить курение и через
применение системы общеоздоровительных мероприятий и антигиперлипопро-
теинемических средств произвести коррекцию гиперхолестеринемии;
существует возможность обратного развития атеросклеротического
поражения сосудов, если содержание холестерина в плазме крови падает
до уровня 1,5-1,8 г/л.
Исторически первыми средствами терапии стенокардии были нитраты,
которые остаются одним из основных средств лечения ишемической
болезни сердца. Одной из положительных сторон действия нитратов следует
считать снижение ими конечно-диастолического давления в левом желудочке
через снижение общего венозного возврата к сердцу. Действуя таким
образом, нитраты повышают перфузионное давление субэндокардиального слоя
миокарда левого желудочка (схема 9.3).
Назначения нитратов и других сосудорасширяющих средств избегают
при стенокардии, обусловленной тяжелым аортальным стенозом. Гипоэр-
гоз клеток сердца, который проявляет себя грудной жабой, у больных с
аортальным стенозом обусловлен не атеросклеротическим поражением
венечных артерий, а прежде всего кардиомиопатией, вторичной по отношению к
концентрической гипертрофии стенок левого желудочка в стадии
декомпенсации. В этой стадии развитие сети капилляров миокарда и его нервных
элементов отстает от роста массы кардиомиоцитов. У больных с
аортальным стенозом в его терминальной стадии нитраты могут через снижение
преднагрузки сердца опасно уменьшить конечно-диастолический объем
левого желудочка. Это резко ограничивает его ударный объем, и без того
уже сниженный вследствие сердечного порока. Поэтому назначение
нитратов больным со стенокардией вследствие аортального стеноза может
привести к трудно купируемой артериальной гипотензии.
Нитраты и вазодилятаторы не применяют при гипертрофической кар-
диомиопатии, так как снижение конечно-диастолического объема левого
Патофизиология ишемической болезни сердца
135
Прямое расслабляющее действие нитратов на
гладкомышечные элементы стенки сосудов
1
Дилатация
емкостных сосудов
(расширение вен)
i
г
Снижение
преднагрузки
ч
Г
t
Расширение артерий,
резистивных сосудов
i
Уменьшение
постнагрузки
ч
г
Снижение потребности
сердца в 02
1
г
т
Дилатация
венечных артерий
▼
Увеличение транспорта 02
по венечным артериям
▼
Увеличение доставки 02
кардиомиоцитам
▼
Устранение гипоэргоза сердца
Купирование стенокардии и нормализация
возбудимости, проводимости и сократимости
Схема 9.3. Положительные стороны фармакодинамики нитратов
при лечении ишемической болезни сердца
желудочка при обструкции его выносящего тракта также может привести к
опасному снижению АД. При кардиогенном шоке дилатация вен и
резистивных сосудов вследствие эффекта нитратов на сосудистую стенку может
привести к необратимой артериальной гипотензии.
Бета-блокаторы - это неоднородная группа препаратов, представителей
которой классифицируют в зависимости от избирательности действия на
бета-один- и бета-два-адренорецепторы. Через снижение интенсивности
стимуляции кардиальных бета-адренорецепторов бета-блокаторы снижают
частоту сердечных сокращений и в меньшей степени уменьшают силу
сокращений саркомеров миокарда. Все это может привести к снижению
потребности сердца в свободной энергии, снизить остроту гипоэргоза кардио-
миоцитов и устранить стенокардию.
В малых дозах бета-один-селективные средства (метопролол и атено-
лол) вызывают бронхоспазм реже, чем бета-блокаторы, лишенные такой
избирательности. Кроме того, в небольших дозах, бета-один-селективные
средства в меньшей степени способствуют прогрессированию
атеросклероза периферических артерий.
136
Глава 9
Блокада кальциевых каналов клеточных
мембран кардиомиоцитов и гладкомышеных
элементов сосудистой стенки
▼ ▼
Расширение сосудов
сопротивления
1
Снижение
сократимости
' Г
Уменьшение потребности
сердца в 02
i
г
▼
Дилатация
венечных артерий
▼
Рост доставки 02 к
кардиомиоцитам
т
Ослабление гипоэргоза
Схема 9.4. Позитивные стороны фармакодинамики
антагонистов кальция
Неизбирательные бета-блокаторы, блокируя возбуждение сосудистых
бета-два-адренорецепторов на периферии, через спазм периферических
артерий небольшого диаметра и связанную с ним модуляцию высвобождения
эндотелиоцитарных цитокинов, могут ускорить развитие атеросклероза.
Необходимо тщательно «титровать» дозу бета-блокатора у каждого
больного, прекращая ее увеличивать, когда частота сердечных сокращений
снизится до 55-60 мин1 в покое и до 90-100 мин1 при нагрузке. Дальнейшее
увеличение дозы, еще более усиливая отрицательный эффект блокады бета-
один-адренорецепторов на частоту генерации импульсов возбуждения
водителем ритма, проводимость и сократимость кардиомиоцитов, может
привести к артериальной гипотензии. В этой связи вполне понятно выделение
таких противопоказаний к назначению бета-блокаторов как тяжелая
сердечная недостаточность и выраженная брадикардия в покое. Следует
заметить, что при использовании бета-адреноблокаторов, обладающих
внутренней симпатомиметической активностью (ацебутолол), не следует
ориентироваться на частоту сердечных сокращений в условиях покоя как на
критерий силы действия препаратов данного класса.
Кроме того, противопоказаниями к назначению бета-один-адрено-
блокаторов служат обструктивные расстройства внешнего дыхания (в том
числе и в анамнезе) и тяжелый облитерирующий атеросклероз
периферических артерий.
Антагонисты кальция - неоднородная группа лекарственных средств, в
различной степени обладающих рядом позитивных эффектов на звенья
патогенеза ишемической болезни сердца (схема 9.4). В основе такого
побочного действия антагонистов кальция как головокружение лежит снижение
Патофизиология ишемической болезни сердца
137
кровотока через соответствующие центры головного мозга, связанное с
уменьшением ударного объема левого желудочка при падении общего
периферического сосудистого сопротивления под влиянием антагонистов кальция. При
этом в результате церебральной ауторегуляции объемной скорости
кровотока, на которую не влияют антагонисты кальция, общее сосудистое
сопротивление на уровне головного мозга не подвергается значительным изменениям.
Все это влечет за собой перераспределение минутного объема
кровообращения с падением объемной скорости кровотока в головном мозге.
Опасным осложнением терапии с использованием антагонистов
кальция выступает артериальная гипотензия, обусловленная снижением
сократимости сердца и общего периферического сосудистого сопротивления.
Периферические отеки как побочный эффект препаратов данной группы
выступают следствием падения линейной скорости кровотока по
капиллярам и возрастанием в них гидростатического давления вследствие
снижения общего периферического сосудистого сопротивления и уменьшения
силы сердечных сокращений.
Антагонисты кальция, которые служат средством выбора при лечении
больных с ишемической болезнью сердца и непереносимостью нитратов и
бета-блокаторов, как мощные коронарные вазодилятаторы особенно
эффективны при стенокардии Принцметала, основное звено патогенеза которой -
спазм венечных артерий.
Глава 10
ГОЛОДАНИЕ
Голодание - патологическое состояние, которое на определенной
стадии развития характеризуют дисфункции всех функциональных систем, а
также дефицит энергии и массы во всех органах эффекторах функций и
клеточных элементах организма. Голодание вызывает низкая относительно
потребностей клеток доставка к ним нутриентов или устойчивый
(патологический) сдвиг метаболизма в сторону катаболических процессов.
Нутриенты - это вещества, которые представляют собой
потенциальные источники свободной энергии, улавливаемой при биологическом
окислении, или субстраты для анаболических процессов. Нутриенты поступают
во внутреннюю среду организма после переработки пищи в ходе
полостного, мембранного пищеварения и всасывания продуктов переработки из
просвета кишечника или в результате расщепления с высвобождением
нутриентов соединений, в виде которых организм аккумулирует источники энергии
и пластический материал (гликоген, триглицериды жировой ткани и др.).
Экзогенное голодание - это следствие полного отсутствия или
недостаточного потребления пищи или результат сниженного поступления в
организм отдельных питательных веществ, витаминов и микроэлементов
(частичное, качественное голодание; белковое, углеводное, витаминное
голодание и т. д.).
Абсолютное голодание - это экзогенное голодание при полном
отсутствии пищи и воды, а полное - это голодание при отсутствии пищи, но с
сохранением питья. При неполном голодании питание недостаточно для
удовлетворения потребностей организма в нутриентах.
Эндогенное голодание - у тяжелых больных чаще всего обусловлено
одновременными нарушениями мембранного, полостного пищеварения,
моторики кишечника и всасывания из его просвета. Кроме того, эндогенное
голодание может быть следствием роста потребности в нутриентах
вследствие повышенных утилизации свободной энергии и потребления
субстратов анаболизма. Рост энерготрат и усиление анаболизма представляют
собой метаболическую основу реакций саногенеза, направленных на
устойчивую компенсацию дисфункций и выздоровление. Так, усиление
синтеза белка в защитных системах (в энтероцитах, иммунокомпетентных
клетках и т. д.) - необходимый элемент системных реакций саногенеза в ответ
на инфекцию, гипоксию, ишемию и некробиотические изменения тканей
вследствие ранений и травм.
Голодание в результате повышенного потребления нутриентов и
низкой относительно потребления доставки нутриентов в клетки называют
ускоренным голоданием.
Патологические сдвиги гомеостазиса, действие патогенных
раздражителей (ноцицептивных и др.) служат стимулами неспецифической защит-
Голодание
139
Таблица. 10.1
Стадии приспособительных изменений обмена веществ
в органах и тканях человека при полном голодании
Орган
Головной
мозг
Печень
Адипоциты
жировой
ткани
Скелетные
мышцы
Через 2-72 ч после начала
полного голодания
Биологическое окисление как
источника свободной энергии только глюкозы
Потребление гепатоцитами
аминокислот, глицерина, свободных жирных
кислот. Глюконеогенез, кетогенез, гли-
когенолиз. Высвобождение клетками
печени глюкозы и кетоновых тел
Липолиз, то есть распад триглицери-
дов до глицерина и свободных жирных
кислот
Биологическое окисление свободных
жирных кислот и кетоновых тел. Про-
теолиз в миоцитах и высвобождение
ими аминокислот
После 72 ч от начала
полного голодания
Биологическое окисление не
только глюкозы, но и
кетоновых тел
Интенсивные глюконеогенез
и кетогенез при почти полном
прекращении гликогенолиза
Липолиз
Окисление свободных жирных
кислот. Протеолиз и
высвобождение аминокислотной смеси
ной стрессорной реакции, биологический смысл которой - это
мобилизация резервов энергии и анаболических субстратов для утилизации в ходе
реакций саногенеза, то есть выздоровления через реакции самого
организма. При стрессорной реакции через активацию нейроэндокринной катабо-
лической системы в ответ на действие патогенных раздражителей сразу
активируются все процессы катаболизма, и нет известной стадийности
использования запасов энергии и пластического материала, характерной для
физиологической адаптации к голоданию (табл. 10.1).
Ранения и травмы, ожоги, сепсис, другие тяжелые болезни и
патологические состояния ведут к длительному действию целого спектра сильных
и неотвратимых раздражителей (патологическая боль, гиповолемия,
артериальная гипоксемия, метаболический ацидоз и др.), которое
представляет собой постоянный и сильный стимул для стрессорной реакции.
В результате мощность катаболической стресс-реакции не спадает и может
стать чрезмерной.
Стрессорное голодание - это патологическое состояние системного
дефицита массы и резервов энергии вследствие потерявшей
биологический смысл и защитное значение катаболической стрессорной реакции. Это
патологическое состояние развивается быстро, несмотря на сохраненное
поступление в клетку нутриентов из внешней среды, что обусловлено
полным угнетением анаболизма или сохранением анаболических процессов
только в виде усиленного белкового синтеза в защитных системах организма.
Устойчивость нарушений гомеостаза как стимулов стресса - это не
единственная причина патогенного характера стресс-реакции у тяжелых
больных. Дело еще в том, что превращение стресс-реакции в звено пато-танато-
генеза происходит в соответствии со вторым принципом компенсации
140
Глава 10
нарушенных функций акад. П.К. Анохина: защитные реакции в ответ на
нарушения функций относительно самих нарушений выражены
значительно сильнее, и поэтому сами часто приводят к дисфункциям,
патологическим состояниям и болезням несостоявшейся компенсации.
ФИЗИОЛОГИЧЕСКАЯ АДАПТАЦИЯ
К ЭКЗОГЕННОМУ ГОЛОДАНИЮ
Физиологическую адаптацию к экзогенному голоданию характеризует
известная стадийность изменений обмена веществ со сменой основных
источников свободной энергии, высвобождаемой при биологическом
окислении и улавливаемых клеткой в виде макроэргов (табл. 10.1). Несмотря на
то, что уже через 24 ч полного голодания организм начинает через
глюконеогенез использовать белки как источники свободной энергии,
энергетическим резервом первой очереди следует считать гликоген и триглицериды
жировой ткани.
Роль основного источника свободной энергии при физиологической
адаптации к голоданию играют триглицериды жировой ткани. У здоровых
людей в виде триглицеридов содержатся 80 % всех энергетических резервов.
У ожиревших триглицериды жировой ткани могут содержать до 95 % орга-
низменного резерва энергии. При потреблении свободной энергии на
уровне в 2000 ккал в сутки здоровые люди живут 30-60 суток после начала
полного голодания. Что касается ожиревших, то описаны случаи почти
безвредного полного лечебного голодания в течение более, чем 250 дней.
Эти наблюдения не должны служить причиной поверхностного отношения
к назначению и проведению лечебного голодания с целью снижения
патологически высокой массы тела. Следует учитывать, что ограниченное
поступление нутриентов во внутреннюю среду - это стимул стрессорной ка-
таболической реакции, включающей протеолиз и глюконеогенез. Поэтому
лечебное голодание может стать стрессорным и привести к белковой
недостаточности и алиментарной дистрофии.
Наиболее мощный регуляторный фактор секреции инсулина - это
концентрация глюкозы в плазме крови. Ее снижение до уровня более низкого,
чем 1 г/л (5,5 ммоль/л), угнетает секрецию инсулина. Кроме того,
высвобождение гормона в кровь падает в ответ на снижение содержания в ней
свободных жирных кислот, аминокислот лейцина и аргинина. Действие
пищи как раздражителя в просвете желудочно-кишечного канала повышает
секрецию гастроинтестинальных гормонов, рост концентрации которых в
крови усиливает секрецию инсулина.
При экзогенном голодании в ответ на снижение концентрации в крови
глюкозы и других нутриентов, вследствие падения секреции
гастроинтестинальных гормонов и под влиянием нервных импульсов из рецепторов
желудка и других органов желудочно-кишечного тракта секреция инсулина
Голодание
141
Таблица 10.2
Влияние гормонов на обмен веществ при голодании и в остром периоде
после тяжелых ранений и травм
Гормон
Инсулин
Адреналин
Глюкагон
Кортизол
Гормон роста
Анаболические процессы
Синтез
гликогена
+
-
-
+/-
0
Липо-
генез
+
0
0
+/-
0
Синтез
белка
+
0
0
-
+
Катаболические процессы
Гликогенолиз
-
+
+
-
-
Глюко-
неогенез
-
+
+
+
+
Липолиз
-
+
?
+
+
Протеолиз
-
-
+
+*
+*
Примечание: «+» - стимуляция; «-» торможение; «О» - отсутствие влияния; «+/-» - стимуляция
при неугнетенной секреции инсулина и торможение при сниженной секреции инсулина или инсу-
линопении; * - эффект реализуется при сниженной секреции инсулина или инсулинопении.
падает до базального уровня и происходит возбуждение пищевого центра.
Возбуждение входящего в него на уровне латеральных ядер гипоталамуса
центра голода активирует симпатический отдел автономной нервной
системы. В результате растет секреция гормонов-антагонистов инсулина.
Изменение соотношения секреции инсулина и гормонов с преимущественно ка-
таболическим действием стимулирует гликогенолиз, липолиз, протеолиз и
глюконеогенез при угнетении гликогенообразования, синтеза жиров и
белков (табл. 10.2).
При неспецифической стрессорной реакции у тяжелых хирургических
больных и раненых через активацию всей автономной нервной системы
усиливается как секреция инсулина, так и выброс в кровь его антагонистов ката-
болических гормонов. Биологический смысл такой реакции - это увеличение
массы нутриентов, доступных для биологического окисления и
анаболических процессов в клетке. Такая доступность невозможна без роста секреции
главного анаболического гормона инсулина, который происходит
одновременно с усилением секреции его антагонистов, катаболических гормонов.
При ускоренном голодании секреция инсулина также растет, что
обеспечивает транспорт в клетки (миоциты скелетных мышц и др.) нутриентов,
поступающих во внутреннюю среду из внешней в количестве,
недостаточном относительно высокой интенсивности биологического окисления и
анаболизма.
Адаптация к полному голоданию состоит в последовательном
использовании энергетических резервов организма. При этом субстраты анаболизма
(аминокислоты и др.) становятся субстратами биологического окисления.
Организм стремится предупредить гипоэргоз (недостаток свободной
энергии) как причину остановки работы мозга, легких и сердца ценой блокады
анаболизма, резко снижая уровень секреции основного эндогенного
анаболика инсулина. Поэтому при физиологической адаптации к полному голо-
данию в отличие от стрессорного и ускоренного голодания секреция
инсулина падает.
142
Глава 10
В первые 2-72 ч полного голодания адаптивные сдвиги обмена веществ
направлены на поддержание концентрации глюкозы в плазме крови на
уровне, превышающем минимально достаточный для функционирования
нейронов и других клеток, в основном в качестве субстрата биологического
окисления использующих глюкозу. Запасы гликогена в печени достаточны
для поддержания нормальной концентрации глюкозы в крови в течение 12-
24 ч. В дальнейшем содержание глюкозы удерживается на минимально
достаточном уровне за счет ее образования печенью из глицерина,
аминокислот и свободных жирных кислот в ходе глюконеогенеза. Поступление
аминокислот и продуктов липолиза в кровь возрастает вследствие
адаптивной интенсификации анаболизма.
При длительном полном голодании, то есть в период после 24 ч от его
начала, только нейроны головного и спинного мозга испояьзуют глюкозу
как энергетический субстрат. Клетки всех других тканей и органов для
биологического окисления утилизируют свободные жирные кислоты и
кетоновые тела (бета-гидроксимасляная и ацетоуксусная кислоты).
Кетоновые тела накапливаются в крови при неспособности печени окислить до
углекислого газа возросшую во внутренней среде массу свободных
жирных кислот, а также вследствие стимуляции кетогенеза глюкагоном. Хотя
кетоновые тела при длительном полном голодании и служат источниками
энергии для мозга, лишь определенный уровень доставки глюкозы к его
нейронам позволяет сохранить их функции и предотвращает цитолиз
клеток нервной системы.
При полном голодании, длящемся более 72 ч, падает выделение азота с
мочой. Это свидетельствует о падении утилизации белка как источника
свободной энергии. Образование глюкозы из аминокислот к этому моменту уже
не может удовлетворить потребность нейронов в свободной энергии.
Компенсация снижения глюконеогенеза из аминокислот возможна в двух
вариантах:
♦ переход на утилизацию других энергетических субстратов;
♦ более интенсивная утилизация печенью длинноцепочечных
аминокислот как субстратов глюконеогенеза.
При снижении интенсивности глюконеогенеза мозг начинает
интенсивнее использовать кетоновые тела. Кроме того, начинается трансформация
кетонов в глюкозу через ацетон, в результате которой образуется 20 %
глюкозы, потребляемой мозгом в период после 72 ч от начала полного голодания.
В ответ на экзогенное полное или тяжелое неполное голодание
возникает системная адаптивная реакция снижения потребления свободной
энергии организмом на 15-20 % от его уровня нормального для здорового
человека в условиях относительного покоя. В этом состоит одно из отличий
физиологической адаптации к полному голоданию от стрессорного и
ускоренного голодания, при которых растут потребление кислорода и
выделение углекислого газа организмом. При стрессорном или ускоренном
голодании рост энерготрат на системном уровне представляет собой защитную
реакцию обеспечения активного саногенеза, которая может превратиться в
звено патогенеза алиментарной дистрофии.
Голодание
143
ПАТОГЕНЕЗ АЛИМЕНТАРНОЙ ДИСТРОФИИ
И СТРЕССОРНОГО ГОЛОДАНИЯ
Алиментарная дистрофия - это болезнь несостоявшейся компенсации
несоответствия меэюду поступлением нутриентов во внутреннюю среду
из внешней и потребностью в них организма. Основное звено патогенеза
алиментарной дистрофии - это блокада той или иной степени экспрессии
генома клеток и всего организма в виде белковых носителей функций из-за
системных гипоэргоза и недостатка субстратов белкового синтеза.
Единственные молекулы, которые синтезируются под прямым
контролем генома клетки, - это белки и рибонуклеиновые кислоты. Белки могут
быть структурными элементами (кератин и коллаген) или играть
функциональную роль (инсулин, фибриноген, альбумины плазмы крови, другие
белковые носители функций и, главное, ферменты, регулирующие клеточный
метаболизм). Именно набор ферментов, который содержит клетка,
определяет ее нормальное функционирование в качестве дифференцированного
структурно-функционального элемента тканей и органов. Недостаток
субстратов белкового синтеза и его торможение ведут к прекращению или
угнетению многочисленных клеточных функций, так как угнетают и
расстраивают экспрессию генома в виде трансляции ферментов. Поэтому потери
резервов аминокислот и белка, снижение во внутренней среде
аминокислот-субстратов синтеза протеинов, прекращение или падение
образования белка на системном уровне как звенья патогенеза ускоренного и стрес-
сорного голодания приводят к распространенным в пределах всего
организма нарушениям функций клеток, органов и тканей (табл. 10.3).
Белковая недостаточность на всех уровнях организма вызывает нарушения
нервной регуляции, системную эндокринопатию, приобретенный
комбинированный иммунодефицит, обуславливает частые инфекционные осложнения,
замедляет регенерацию тканей и заживление ран, через слабость
скелетных мышц задерживают реабилитацию и т. д.
Экспрессию генома в виде белков с различными функциями блокирует
патологическая активация нейроэндокринной катаболической системы у
больных со стрессорным голоданием. При этом нутриенты как источники
свободной энергии и субстраты белкового синтеза поступают во
внутреннюю среду, но не утилизируются из-за торможения анаболизма гормонами
антагонистами инсулина. Чаще всего неотвратимыми стимулами для
активации нейроэндокринной катаболической системы у тяжелых больных
являются патологическая боль, артериальная гипоксемия, гиповолемия, цир-
куляторная гипоксия, гиперцитокинемия. Стрессорное голодание -
необходимый элемент системной воспалительной реакции.
Ускоренное и стрессорное голодание у тяжелых, и в особенности у
хирургических больных, могут развиваться одновременно. При этом
стрессорное голодание представляет собой извращение защитной реакции,
направленной на усиление протеолиза с биологической целью обеспечения
субстратами повышенного анаболизма как основы саногенеза. Кроме того,
144
Глава 10
Таблица 10.3
Патогенез алиментарной дистрофии на органном и клеточном уровнях
Морфопатогенез
Дисфункции, заболевания
и симптомы
Дилатация всех
сердечных камер и истончение
их стенок
Эмфизема, инфаркты
легких, дегенерация эпи-
телиоцитов дыхательных
путей
Атрофия клубочков неф-
ронов, кальцификация
коркового слоя почек,
отек тубулярного эпителия
Дегенеративные
изменения проэритробластов
Атрофия слизистой
оболочки со снижением
вертикального размера энте-
роцитов
Снижение числа гепато-
цитов, перипортальная
аккумуляция жира и
жировая дистрофия печени
Угнетение способности
полиморфо- и мононукле-
аров к хемотаксису.
Снижение содержания
лимфоцитов в крови
вследствие падения содержания
в ней T-хелперов при
увеличении содержания в
крови Т-супрессоров и
натуральных киллеров
Падение сократимости сердца и ударных
объемов желудочков. Застойная лево- и правоже-
лудочковая недостаточность. Толерантность
сердца относительно положительного инотроп-
ного действия лекарственных средств.
Угнетение физиологического автоматизма,
проводимости и возбудимости, которое проявляет себя
брадикардией, увеличением интервала Q-T и
снижением вольтажа на электрокардиограмме
Падение защитной функции эпителия
дыхательных путей. Пневмонии. Снижение
функциональной, остаточной и жизненной емкости
легких. Уменьшение максимально возможного
минутного объема дыхания. Снижение
чувствительности центральных хеморецепторов к ги-
перкапнии и периферических к гипоксемии.
Патологическое снижение коэффициента
использования кислорода.
Снижение скорости клубочковой фильтрации.
Падение реабсорбции натрия как причина по-
лиурии. Метаболический ацидоз. Снижение
образования эритропоэтина, которое вызывает
анемию
Анемия
Полиферментопатия, то есть тотальное
снижение синтеза и высвобождения
энзимов-катализаторов внеклеточного, внутриклеточного и
мембранного пищеварения
Угнетение белкового синтеза и микросомаль-
ного окисления в клетках печени. Нарушения
всех функций печени
Снижение роста клональной экспансии
T-лимфоцитов при выполнении теста
«смешанная культура лимфоцитов». Иммунная анергия.
Частые инфекции
стрессорное голодание - это результат системного угнетения анаболизма
при интенсификации катаболизма.
Ускоренное голодание представляет собой следствие повышенной
утилизации белка по ходу реакций саногенеза при относительно низком по-
Голодание
145
ступлении нутриентов как субстратов анаболизма во внутреннюю среду из
внешней. При одновременном развитии стрессорного и ускоренного
голодания растет утилизация свободной энергии на системном уровне, что
обеспечивает и отражает высокую интенсивность реакций саногенеза.
Следует учитывать, что ряд патологических раздражителей при
условиях достаточно длительного и интенсивного действия приводит к
устойчивой и избыточной активации нейроэндокринной катаболической
системы (НЭКС). Известно, что патологическая боль, артериальная гипоксемия,
гиповолемия и циркуляторная гипоксия, гиперцитокинемия при сепсисе
и системной воспалительной реакции обуславливают патогенную
активацию супрасегментарного звена НЭКС, то есть констелляции нейронов вен-
тромедиальной части гипоталамуса. Активация этих нейронов через ней-
роэндокринные регуляторные связи усиливает выброс гормонов
антагонистов инсулина и вызывает системное преобладание катаболизма
над анаболизмом.
Алиментарная дистрофия и стрессорное голодание приводят к
расстройствам функциональных систем через дефициты белковых
молекулярных эффекторов функций и свободной энергии в исполнительных
органах (табл. 10.3).
Знание суровых реалий жизни нашего общества не позволяет
исключить из числа возможных причин быстрого развития алиментарной
дистрофии одновременного отрицательного психоэмоционального стресса как
стимула устойчивой активации нейроэндокринной катаболической системы
(боевые действия, издевательства, тюрьма, побои и пр.) и неполного или
даже полного голодания.
У больных в состоянии стрессорного голодания вследствие гиперцито-
кинемии (сепсис, системная воспалительная реакция) активация катаболи-
ческих процессов включает и протеолиз, особенно выраженный в
скелетных мышцах. Усиленное высвобождение белка из миоцитов скелетных мышц
ведет к мышечной слабости как причине замедленной реабилитации. При
этом протеолиз в скелетных мышцах имеет защитно-приспособительное
значение, так как обеспечивает аминокислотами как субстратами синтез
структурных белков во внутренних органах, образование белков острой фазы
воспаления, глюконеогенез, а также образование белка как основу
нормальной реакции системы иммунитета на антигенную стимуляцию. Протеолиз
в миоцитах скелетных мышц - это защитная реакция, которая у
тяжелых больных часто теряет свой защитный характер, становясь через сие-
темную дизрегуляцию избыточной относительно вызвавших ее
повреждений и патологических сдвигов гомеостаза.
Усиленная утилизация субстратов белкового синтеза, высвобождаемых
при стрессорном голодании и защитной реакции интенсификации протео-
лиза, придает аминокислоте глутамину качество условно-незаменимой
(полунезаменимой).
К условно-незаменимым относят аминокислоты, синтезируемые в
организме, дефицит которых во внутренней среде легко развивается и
становится звеном патогенеза при болезнях и патологических состояниях.
146
Глава 10
Содержание глутамина в организме здорового человека велико
относительно содержания других аминокислот. При защитном росте протеолиза у
тяжелых больных межорганный транспорт белкового азота из скелетных
мышц в кишечник и почки осуществляется в основном через высвобождение
глутамина миоцитами и циркуляцию этой аминокислоты с кровью. Глутамин
как субстрат незаменим для синтеза нуклеотидов, белков скелетных мышц,
образования в почках NH3 и глюконеогенеза из аминокислот в гепатоцитах.
Эндогенные резервы глутамина у тяжелых больных при прогрессировании
алиментарной дистрофии истощаются первыми, что обостряет дефицит
энергии и пластических субстратов в ряде органов и клеток. В этой связи
недостаток во внутренней среде глутамина можно считать фактором эндоге-
низации алиментарной дистрофии и звеном патогенеза ее осложнений.
Дефицит глутамина как субстрата синтеза нуклеотидов снижает
эффективность системных компенсаторных и защитных реакций, в основе
которых лежит усиление клеточной пролиферации или клональной экспансии
клеток (воспаление, первичный и вторичный иммунный ответ, заживление
ран). Кроме того, недостаток аминокислоты служит причиной
несостоятельности физиологического механизма предотвращения эндотоксикоза через
частую репликацию энтероцитов кишечника. Репликация энтероцитов
угнетается вследствие дефицита условно-незаменимой аминокислоты и
потому, что глутамин для энтероцитов служит не только пластическим
материалом, но и энергетическим субстратом.
Достаточное выведение протонов во внешнюю среду почками
невозможно без поступления в нефроны с кровью достаточного количества
глутамина. Поэтому дефицит глутамина при ускоренном голодании может быть
одним из звеньев патогенеза метаболического ацидоза.
Известно, что содержание глутамина во внутренней среде организма
находится в обратной связи с абсолютной величиной отрицательного
азотистого баланса. В этой связи становится понятным, почему парентеральное
введение глутамина снижает отрицательный азотистый баланс. Дефицит
глутамина через действие во многом непонятных регуляторных
механизмов интенсифицирует протеолиз. Поэтому парентеральное введение
глутамина снижает потерю массы скелетными мышцами, что облегчает полную
реабилитацию больного.
Через обеспечение одним из необходимых субстратов повышенного
белкового синтеза в иммунокомпетентных клетках и клеточных эффекторах
воспаления парентеральное питание с использованием растворов, содержащих
глутамин, устраняет приобретенный комбинированный иммунодефицит и снижает
частоту инфекционных осложнений у тяжелых хирургических больных.
До недавнего времени не были доступны растворы, содержащие L-глута-
мин. Причиной тому были его низкая растворимость и нестойкость L-глута-
мина при тепловой стерилизации. Использование холодовой стерилизации и
глутаминовых дипептидов, сделало растворы глутамина и его соединений
доступными для клинической практики парентерального питания.
Стрессорное голодание у тяжелых хирургических больных в состоянии
сепсиса и системной воспалительной реакции характеризует качественное
Голодание
147
изменение обмена веществ. Это невозможно без усиленных образования и
выброса катаболических гормонов антагонистов инсулина. Одновременно
растут синтез и секреция гормона роста, глюкагона, катехоламинов и
анаболического гормона инсулина, субстратом синтеза которых является
аминокислота аргинин. У многих больных в состоянии стрессорного
голодания повышен синтез белка и пептидов в элементах иммунной системы, что
обеспечивает клональную экспансию иммунокомпетентных клеток и их
интенсивные при этом межклеточные взаимодействия, реализуемые через
секрецию цитокинов. Для обеспечения реакции системы иммунитета
достаточным белковым синтезом также необходима повышенная утилизация
аргинина в качестве субстрата синтеза протеинов. Поэтому у тяжелых
больных хирургического профиля в состоянии стрессорного и ускоренного
голодания аргинин становится условно-незаменимой аминокислотой.
В этой связи становится понятным, почему добавление аргинина к
смесям для парентерального питания (25 г в сутки) устраняет
комбинированный иммунодефицит как частное проявление алиментарной дистрофии
вследствие стрессорного голодания.
Рост интенсивности катаболизма и торможение анаболизма при таких
состояниях как сепсис, инфекция, тяжелые травматическая и раневая болезнь
ведут к потерям белка. Организм теряет белок в результате усиленного
протеолиза и угнетения синтеза белка вследствие роста катаболизма и падения
анаболизма при сниженном поступлении нутриентов извне во внутреннюю
среду. Регуляторный механизм интенсификации протеолиза и снижения белкового
синтеза после тяжелых ранений, травм, хирургических вмешательств, при
сепсисе и инфекциях - это не только преобладание секреции катаболических
гормонов антагонистов инсулина, но и высвобождение цитокинов-флогоге-
нов (фактор некроза опухолей, интерлейкин-1 и др.). Моно- и другие цитоки-
ны вызывают супрасегментарную активацию нейроэндокринной катаболи-
ческой системы и прямо индуцируют протеолиз в миоцитах скелетных мышц.
Гиподинамия и ацидоз при этом усиливают потери белка.
Системные гипоэргоз и дефицит белка как носителя функций прежде
всего нарушают эффекторы тех систем, где и при адаптивном ограничении
физической активности интенсивно потребление энергии и лабилен
белковый синтез. К ним можно отнести печень, энтероциты и иммунокомпетент-
ные клетки. Даже непрерывное функционирование сердца при ускоренном
голодании не может предотвратить компенсаторный протеолиз в миокарди-
оцитах, и у больных при длительном течении тяжелых заболеваний в
палатах интенсивной терапии выявляют истончение стенок желудочков.
Основные симптомы алиментарной дистрофии - это общее угнетение
восприятия и когнитивных функций, анорексия, кахексия, отеки, асцит, спле-
номегалия и диарея. Угнетение белковообразующей функции печени у
больных алиментарной дистрофией приводит к снижению содержания в крови
альбуминов, что снижает коллоидноосмотическое давление плазмы крови.
В результате на системном уровне происходит перемещение внеклеточной
жидкости из внутрисосудистого сектора в интерстициальный, что
проявляется периферическими отеками и может служить одной из причин асцита.
148
Глава 10
Второй фактор отеков вследствие алиментарной дистрофии - это падение
экскреторной функции почек.
Квашиоркор - это алиментарная дистрофия у детей вследствие
качественного белкового голодания. Заболевание впервые описано у африканских
детей, которые после полноценного питания материнским молоком
переходили на богатую углеводами, но бедную белком диету при кормлении
исключительно кашами из маниоки или сладким картофелем. В результате
наступала задержка роста и умственного развития. Частый симтом кваши-
оркора - это сплено-гепатомегалия как реакция на бактериемию вследствие
нарушений защитных функций энтероцитов и на дистрофию селезенки и
печени. О кваширкоре свидетельствуют характерная сыпь и десквамация
эпидермиса, незаживающие язвы любой локализации на коже и видимых
слизистых оболочках. В переводе с диалекта народности Гиа,
проживающей на территории Ганы, «квашиоркор» означает «болезнь, которую
получает старший ребенок тогда, когда должен родиться младший», то есть с
началом экзогенного качественного белкового голодания.
Маразм (лат. marasmus - истощение) - это алиментарная дистрофия у
детей вследствие неполного голодания. Это возможное опасное следствие
энтеритов и диспепсий раннего детского возраста.
Anorexia nervosa (нервическое угнетение чувства голода) -
алиментарная дистрофия как следствие полного или неполного голодания, которое
развивается в силу внутренних причин. Обычно заболеванием страдают
женщины в возрасте от 10 до 30 лет, находящиеся в состоянии
эмоционального стресса, вызванного безответной любовью или первоначально
избыточной массой тела. Можно предположить, что циркуляция возбуждения
внутри гипоталамо-лимбико-ретикулярных структур лежит в основе
формирования очагов «застойного» возбуждения при эмоциональном стрессе,
захватывающих и вентромедиальное ядро гипоталамуса. Одним из
следствий «застойного» возбуждения на уровне вентромедиального ядра
выступают повышенный уровень катаболизма и почти полное отсутствие
аппетита. Это сочетание обуславливает ускоренное голодание вплоть до
алиментарной дистрофии.
ПАТОГЕНЕТИЧЕСКИЕ ПРИНЦИПЫ
ИСКУССТВЕННОГО ЛЕЧЕБНОГО ПИТАНИЯ
Главными целями интенсивной терапии, ориентированной на
нормализацию утилизации нутриентов и всего метаблизма у тяжелых больных,
следует считать:
♦ выявление и коррекцию предшествующих заболеванию голодания и
торможения анаболических процессов;
♦ оптимизацию состояния обмена у больных и устранение нарушений
водно-солевого обмена;
Голодание
149
♦ снижение смертности и времени, уходящего на выздоровление и
реабилитацию.
При достижении этих целей следует учитывать, что современные способы
энтерального и парентерального искусственного питания без системы
лечения, направленной на предотвращение и снижение патогенной активации ней-
роэндокринной катаболической системы и возобновление необходимого для
саногенеза анаболизма, сами по себе не предотвращают потерь белка. Если
система такой терапии, включающая обезболивание, нейровегетативную
блокаду, способы предотвращения и устранения гипоксемии, инфузии,
трансфузии не достигает своих целей, то продолжают действовать сильные и
неотвратимые раздражители, что активирует нейроэндокринную катаболическую
систему. Длительная активация нейроэндокринной катаболической системы
через преобладание секреции катаболических гормонов надолго угнетает
анаболизм, без которого невозможны выздоровление, компенсация и адаптация. В
этой связи становится ясным, почему больные после тяжелых операций,
ранений и травм и в состоянии сепсиса часто теряют 25-50 г белка в день, несмотря
на смешанное (парентеральное и энтеральное) питание.
Причина потерь белка у таких больных - интенсивное высвобождение
при усиленном протеолизе аминокислот, которые не могут служить
субстратами анаболизма вследствие его угнетения.
Результаты ряда исследований последнего десятилетия убедительно
свидетельствуют, что рост массы тела у тяжелых больных под влиянием
парентерального и искусственного питания не связан с усилением образования
белка. Он в основном обусловлен увеличением массы адипоцитов жировой
ткани и увеличением объема внеклеточной жидкости.
Относительную неэффективность современных способов
искусственного питания в предотвращении патологических последствий стрессорно-
го и ускоренного голодания связывают с низким использованием
аминокислот как субстратов анаболизма при гиподинамии, а также считают
следствием использования для парентерального питания
несбалансированных или неполноценных растворов аминокислот.
Одна из целей энтерального и парентерального питания тяжелых
больных и фармакокоррекции патологического торможения анаболических
процессов - это поддержание должного всасывания из просвета кишечника и
нормальных иммунной и барьерной функций клеточных элементов его
стенки. В настоящее время эту цель достигают в частности через использование
для парентерального питания условно незаменимых аминокислот глюта-
мина и аргинина, введением в составе сред для парентерального и
энтерального питания антиоксидантов, а также использованием таких
стимулирующих анаболизм препаратов как гормон роста.
Гипералиментация - это парентеральное питание, при котором
тяжелому больному в состоянии гиподинамии внутривенно в организм
вводят углеводы, аминокислоты и жиры в дозах, превышающих потребности
здорового человека того же возраста, пола и с аналогичными ростом и
нормальной массой тела в условиях относительного покоя. Общая доза нут-
риентов, вводимая с целью гипералиментации и измеряемая килокалориями,
150
Глава 10
составляет 40 ккал/кг. При этом доза белка находится в пределах 1-1,5 г/кг.
Из общей энергетической ценности нутриентов, инфузируемых с целью ги-
пералиментации, вычитают величину потенциально доступной для клеток
энергии, которую содержит вводимый белок. Оставшуюся часть общей
энергетической ценности на 2/3 покрывают введением углеводов. Суточная доза
воды при гипералиментации в виде полного парентерального питания
составляет 30 мл воды на кг массы тела. Гипералиментация предусматривает
поступление во внутреннюю среду организма адекватной дозы витаминов и
микроэлементов.
В настоящее время показанием к гипералиментации является
коррекция дефицита нутриентов во внутренней среде при болезни Крона,
злокачественных опухолях стенок желудочно-кишечного канала, вследствие
энтерита, обусловленного радиационным поражением, и при синдроме короткой
кишки. Показаниями к гипералиментации в виде полного парентерального
питания также служат болезни и патологические состояния, при которых
для выздоровления или успешной хирургической коррекции необходимо
временное прекращение поступления пищи в просвет
желудочно-кишечного канала: неспецифический колит как причина задержки роста у детей,
кишечные свищи, панкреатит, состояние после операций по поводу диаф-
рагмальных грыж и атрезии пищевода у новорожденных и др.
Гипералиментация считалась необходимым элементом интенсивной
терапии в семидесятых годах и в начале восьмидесятых. Сейчас ясно, что
гипералиментация, особенно проводимая в виде полного парентерального
питания, у многих больных вызывает ряд нарушений обмена веществ:
гипергликемию, нарушения водно-солевого обмена в виде гипофосфатемии,
гипомагниемии и других его патологических сдвигов, азотемию и
дыхательную недостаточность вследствие преобладания образования организмом
углекислого газа над его экскрецией в ходе внешнего дыхания. Азотемию и
дыхательную недостаточность, обусловленные парентеральным питанием,
можно связать с неспособностью организма при стрессорном голодании
использовать вводимые извне нутриенты в качестве субстратов для
анаболизма. В результате они служат субстратами биологического окисления. Это
повышает образование организмом углекислого газа и приводит к азотемии
вследствие окисления больших количеств экзогенных аминокислот. Кроме
того, гипералиментация в виде полного парентерального питания ведет к
жировому перерождению печени и нарушениям ее функций.
Парентеральное питание может быть причиной и фактором развития
инфекций с очагами, отдаленными от места нахождения чрезкожного
внутривенного катетера и локализованных в легких, мочевыводящих путях и
ранах. Полагают, что некоторые компоненты сред для парентерального
питания (липидные эмульсии, получаемые из соевых бобов и цветков
шафрана) обладают прямым иммунодепрессивным действием, которое снижает
резистентность организма и ведет к инфекциям.
Итак в последние годы стало ясным, что полное парентеральное
питание при устойчивом торможении анаболических процессов у больных в
состоянии стрессорного голодания вызывает ряд патологических сдвигов
Голодание
151
метаболизма и служит фактором приобретенного иммунодефицита. В
этой связи общую дозу энергии, белков и жиров, вводимых для гиперали-
ментации тяжелым больным в отделениях интенсивной терапии, в
настоящее время значительно снизили, расширив показания к искусственному эн-
теральному питанию (ИЭП). Это произошло во многом благодаря
совершенствованию техники и нутритивных смесей ИЭП. Современный
подход к проведению искусственного питания характеризуют
сбалансированность смесей нутриентов и более широкое использование именно тех нут-
риентов, потребность в которых организма тяжелых больных особенно
высока (глютамин, аргинин, некоторые липиды). Кроме того, в составе
смесей для парентерального питания сейчас вводят антиоксиданты
необходимые для предотвращения фатальных последствий реперфузионного
синдрома у больных после тяжелых ранений и травм в первые двое суток
посттравматического периода. Главная же тенденция состоит в широком
использовании ИЭП по назогастроинтестинальному зонду, кончик
которого заводят за привратник. В развитых странах в настоящее время
производят различные смеси для энтерального питания, которые содержат
источники свободной энергии в виде среднецепочечных триглицеридов или
углеводов. Аминокислотный состав таких смесей варьирует в зависимости
от специфики болезней и патологических состояний, при которых
применяют ту или иную смесь. Полагают, что страх перед ранним энтеральным
питанием в послеоперационном периоде следует считать проявлением
некоторой косности подхода к лечению тяжелых хирургических больных. Если
энтеральное питание оказывается недостаточным, то прибегают к
парентеральному. Парентеральное питание прекращают сразу после
восстановления функций желудка.
В первые трое суток тяжелых травматической, раневой и ожоговой
болезней и в гиперкатаболическую фазу сепсиса, легко вызвать ятрогенный
патологический рост содержания нутриентов во внутренней среде как
причину иммунодепрессии и нарушений обмена у тяжелых больных. Дело в
том, что в этот период болезней и патологических состояний резко
снижена анаболическая утилизация нутриентов и активирован катаболизм. В
результате больные почти неизбежно теряют белок в ходе системной
защитно-патогенной реакции протеолиза. Восстановление массы белковых
носителей функций происходит относительно медленно, в течение
нескольких недель, начиная с четвертого дня критических состояний. Именно в
этот период искусственное питание как способ улучшения исходов
хирургического лечения эффективно в наибольшей степени. В этой связи
считают, что общая доза потенциальных источников свободной энергии,
измеряемая в килокалориях, в начале проведения искусственного питания
больным в состоянии стрессорного голодания должна составлять 50 %
суточных энерготрат.
Величину суточных энерготрат в килокалориях рассчитывают,
применяя непрямую калориметрию, то есть определение энерготрат, исходя из
фиксируемых у постели больного потребления кислорода организмом и
выделения углекислого газа. При невозможности непрямой калориметрии
152
Глава 10
используют формулу Харриса и Бенедикта, которая позволяет рассчитать
потребность в энергии для условий покоя (ПЭП). ПЭП - это суточная
потребность в свободной энергии у здоровых мужчин и женщин, большую
часть времени суток пребывающих в условиях относительного покоя. Эта
потребность в свободной энергии всегда меньше потребности в ней
тяжелых больных. Поэтому для определения суточной потребности в свободной
энергии у того или иного больного уровень основного обмена умножают на
соответствующий коэффициент (схема 10.1).
Для предотвращения отрицательного азотистого баланса тяжелый
хирургический больной должен за сутки потреблять не менее чем 130 % ПЭП
в виде небелковых источников свободной энергии. При парентеральном
питании этого часто стремятся достичь внутривенным вливанием растворов
глюкозы. Внутривенная инфузия концентрированных растворов глюкозы
через интенсификацию липогенеза и снижение содержания гликогена в
печени нередко ведет к нарушениям функций гепатоцитов (жировая
дистрофия печени). Использование липидов в качестве источников энергии при
парентеральном питании иногда позволяет избежать этого осложнения.
Кроме жировой дистрофии печени, внутривенная инфузия гиперосмо-
ляльных растворов глюкозы часто ведет к глюкозурии, осмотическому
диурезу и нарушениям водно-солевого обмена. Особенно часто это бывает, если
внутривенная инфузия глюкозы происходит слишком быстро и не
сопровождается одновременным введением адекватной дозы инсулина.
Так как глюкозу могут использовать в качестве субстрата
биологического аэробного окисления клетки всех тканей, то внутривенная инфузия
растворов глюкозы может значительно снижать интенсивность глюконеоге-
неза. Тем самым блокируется действие одного из основных механизмов
развития белковой недостаточности у тяжелых хирургических больных.
Внутривенное введение экзогенного инсулина при парентеральном
питании и рост секреции эндогенного инсулина в ответ на ятрогенную
гипергликемию стимулирует липогенез и угнетает липолиз. Это служит еще
одной причиной жировой дистрофии и препятствует утилизации жира в
качестве источника и резерва свободной энергии. Сниженное
использование жира предрасполагает к глюконеогенезу и белковой недостаточности.
Энергетический эквивалент дозы глюкозы при парентеральном
питании должен составлять 4 ккал/г массы тела. Утилизация глюкозы оптимальна
при ее вливании со скоростью 4-5 мг/кг/мин. Вливание глюкозы со
скоростью, превышающей 7 мг/кг/мин, не увеличивает массу глюкозы,
утилизируемой при биологическом окислении. Избыток глюкозы превращается в
глицерин и жирные кислоты, что вызывает жировую дистрофию
внутренних органов, и печени в особенности.
Так как метаболический дыхательный коэффициент при окислении
глюкозы составляет 1,0, то рост уровня ее утилизации в качестве
энергетического субстрата ведет к увеличению продукции организмом С02 и
респираторному ацидозу. Превращение неокисленной глюкозы в липиды еще в
большей степени увеличивает высвобождение двуокиси углерода при
обмене веществ и обостряет респираторный ацидоз. Во многом нежелатель-
Голодание
153
Расчет потребностей в энергии для условий покоя
(ПЭП, ккал/сут)
Т
Женщины
ПЭП=655+9,6хМТ+1,7хР-4,7хВ
т
Мужчины
ПЭП=660+13,7хМТ+5,0хР-6,8хВ
Определение уровня двигательной активности больного
Находится в постели
Обычный уровень
ПЭПх1,2
Расчет суточной потребности в энергии без учета
специфики болезни и состояния больного (ПЭО
I
ПЭПх1,3
Определение специфики
болезни и состояния больного
Определение индивидуальной
суточной потребности больного
в свободной энергии
Лечебное голодание
Состояние после полостных
плановых вмешательств
Сепсис, системная воспалительная
реакция и острый период после
тяжелых ранений травм
Перитонит
Раковая кахексия
Лихорадка
I
ПЭ,хО,85
ПЭ,х1,0-И,2
ПЭ,х1,4-г1,8
ПЭ,х1,2-И,5
ПЭ,х1,1+1,45
I13ixl,0+0,13 на каждый
градус выше 37 °С
Схема 10.1. Расчет суточной потребности тяжелых хирургических
больных в свободной энергии
(МП - масса тела, кг; Р - рост, см; В - возраст)
ные побочные эффекты использования глюкозы для парентерального
питания можно избежать через использование в качестве экзогенных
источников свободной энергии липидных нутриентов. Липидные нутриенты
обычно используют в виде жировых эмульсий. В основном жировые эмульсии
состоят из длинноцепочечных жирных кислот. Энергетический эквивалент
дозы жировой эмульсии при парентеральном питании должен составлять
9 ккал/г массы тела в сутки. Превышение этой дозы длинноцепочечных
жирных кислот угнетает защитные функции нейтрофилов, клеток системы мо-
нонуклеарных фагоцитов, тем самым снижая быстроту очищения крови от
154
Глава 10
Таблица 10.4
Влияние гормона роста на обмен веществ и течение
послеоперационного периода у хирургических больных
Действие гормона на метаболизм
Снижение степени отрицательного азотистого баланса и образования мочевины
Усиление белкового синтеза при уменьшении протеолиза и перемещения организмен-
ного пула аминокислот между органами и тканями
Снижение уровня протеолиза и высвобождения миоцитами глутамина в произвольных
мышцах
Усиление липолиза
Рост потребления кислорода организмом и выделения им углекислого газа на 15-25 %
Задержка в организме калия, фосфатных анионов, магния и натрия
Пререцепторная резистентность по отношению к инсулину, гипергликемия, рост
концентрации в крови неэстерифицированных жирных кислот, ионизированного кальция и ин-
сулиноподобного фактора роста-1
Восстановление нормального распределения воды между клеточным и внеклеточным.
жидкостными секторами
Влияние гормона на заживление ран и некоторые функциональные
системы в послеоперационном периоде
Ускорение заживления ран
Снижение частоты инфекционных осложнений, повышение эффективности первичного
и вторичного иммунного ответа
Рост силы сокращений скелетных мышц (снижение слабости дыхательных мышц и др.)
патогенных бактерий. Поэтому существует верхний предел дозы длинноце-
почечных жирных кислот, составляющий 1,5 г/кг массы тела в сутки. Сред-
нецепочечные триглицериды также используют как экзогенные нутриенты
при гипералиментации. Они не подавляют защитных функций полиморфо-
нуклеаров и мононуклеарных фагоцитов. Так как среднецепочечные
триглицериды не содержат незаменимых жирных кислот, то для
парентерального питания обычно применяют смесь длинно - и среднецепочечных
жирных кислот.
При парентеральном питании анаболическое действие экзогенного
инсулина в основном складывается из усиления захвата глюкозы миоцитами
скелетных мышц. При этом организменный пул аминокислот может
перекачиваться из внутренних органов в произвольные мышцы. Это может
обострять недостаточность внутренних органов как эффекторов функций.
Стрессорное голодание, которое характеризует блокада анаболизма, у
тяжелых больных стремятся подвергнуть обратному развитию за счет
анаболических эффектов препаратов гормона роста (табл 10.4).
Глава 11
ТИПОВЫЕ НАРУШЕНИЯ ЛИПИДНОГО ОБМЕНА
ОЖИРЕНИЕ
Благодаря функционированию еще во многом неизвестных систем
регуляции человек и животные потребляют пищу как источник свободной
энергии в соответствии с уровнем энерготрат в ходе жизнедеятельности.
При этом в виде жира здоровый организм аккумулирует около 0,3 %
энергетической цены всего съеденного и выпитого. Патогенное преобладание
поступления источников свободной энергии нутриентов в организм над
уровнем ее утилизации для функционирования клеток, органов-эффекторов и
функциональных систем ведет к увеличению массы жира, накопленного в
жировой ткани, то есть приводит к ожирению.
Ожирение рассматривают как состояние предболезни, которое
характеризует аномальное увеличение масс1ы тела. Критерием нормальной массы
тела или ее патологических сдвигов выступает индекс массы тела, то есть
отношение массы тела к росту тела в квадрате. Нормальный индекс массы
тела составляет 21 кг/м2.
Ожирение - это патологическое состояние, во многом определяемое
наследственным фактором. Так, у ожиревших родителей часто рождаются
дети, которые потом страдают от ожирения. Тем не менее, ожирение - это
не жестко детерминированный фенотипический признак. Известно, что
избыточное питание в первые три месяца жизни через усиление
дифференциации адипобластов до адипоцитов вызывает ожирение, от которого
человек страдает на всех этапах онтогенеза. Рациональные питание и
двигательный режим могут предотвратить реализацию наследственной
предрасположенности к ожирению. Генетический фактор определяет ожирение
у 70 % всех больных с избыточной массой тела. У 30 % ведущую роль в
развитии ожирения играет сложившийся стереотип питания в семьях, где
вырастают ожиревшие люди. Тут свою роль играет действие внешних
стимулов роста потребления пищи.
За последние двадцать лет число людей с ожирением в развитых
странах возросло на 54 %. При этом на 98 % увеличилось число людей,
страдающих от морбидного ожирения (сверхожирения).
Анатомическая классификация ожирения выделяет два его типа, андро-
идное и гипоидное ожирение.
При первом андроидном типе, более частом у мужчин, жир
аккумулируется в основном в жировых клетках туловища (жировые подушки в области
живота, в подмышечной области и т. д.) и сальника (висцеральные адипоци-
ты). У женщин с андроидным типом ожирения существует прямая связь
между массой тела и концентрацией андрогенов в крови. Абдоминальные и
156
Глава 11
висцеральные адипоциты отличаются от других высокодифференцирован-
ных клеток жировой ткани большей интенсивностью в них липолиза, то есть
распада триглицеридов до глицерина и свободных жирных кислот.
Показатель накопления жира в адипоцитах туловища и висцеральных жировых
клетках - это отношение длины окружности талии к длине окружности
нижней конечности в области верхней трети бедра. Об андроидном ожирении
свидетельствует рост отношения, которое находится в прямой связи с
вероятностью таких заболеваний и патологических состояний как сахарный
диабет, гиперлипидемии и первичная артериальная гипертензия.
Предположительно, генетический фактор развития андроидного ожирения
реализует себя через наследственные нарушения метаболизма андрогенов.
Патогенно высокая концентрация андрогенов в циркулирующей крови может
приводить к нарушениям связывания инсулина и глюконеогенеза в печени,
а также вызывает рецепторную и пострецепторную резистентность к
инсулину. Следует заметить, что аналогичные патологические сдвиги
метаболизма вызывает и рост содержания в плазме крови свободных жирных
кислот.
При гипоидном ожирении, которое в большей степени свойственно
женщинам, жир откладывается в нижней части живота и на бедрах. При гино-
идном ожирении риск атеросклероза меньше, чем при андроидном.
В зависимости от механизма развития ожирения на уровне жировой ткани
выделяют два его вида:
♦ обусловленный ростом числа адипоцитов (гиперплазический),
♦ связанный с увеличением их размеров и содержанием в них жира
(гипертрофический).
Первичное идиопатическое ожирение у больных связано с экспрессией
гена HLA-B18 из главного комплекса тканевой совместимости и возникает
при врожденных заболеваниях, вызывающих ожирение: синдромы Праде-
ра-Вилли, Лоренса-Муна-Бидля и Альстрема.
Сами по себе болезни, без переедания и низкой физической активности,
редко приводят к ожирению. Наиболее часто из заболеваний к ожирению
приводят такие эндокринопатии как синдром Кушинга, гипотиреоз, гипого-
надизм и высокое содержание в крови инсулина (гиперинсулинемия)
вследствие инсулинсекретирующих опухолей.
Морбидное ожирение (сверхожирение) - патологическое состояние, при
котором масса тела взрослого человека больше нормальной на 45 кг. Весьма
затруднительно вызвать обратное развитие морбидного ожирения, так как
снижение массы тела у таких больных ведет к активации в просвете
капилляров жировой ткани липопротенилипазы. Этот фермент служит
катализатором гидролиза триглицеридов, связанных с липопротеинами плазмы крови, и
высвобождения жирных кислот, вновь захватываемых адипоцитами.
Количество свободной энергии, потенциально доступной для
утилизации клетками организма, поступающее во внутреннюю среду вместе с
пищей, регулируют вегетативные центры. Для осуществления регуляции
массы и состава потребляемой пищи в центры по афферентам поступает
информация о состоянии обмена веществ и изменениях внешней среды. Дей-
Типовые нарушения липидного обмена
157
ствие некоторых эндогенных раздражителей (внутренние стимулы),
гипогликемии и др., через возбуждение соответствующих центров усиливает
прием пищи. Побуждает к приему пищи и действие ряда экзогенных
раздражителей, снижение температуры окружающей среды, запах пищи, вид хорошо
сервированного стола и т. д. (внешние стимулы).
Разрушение части нейронов вентромедиального гипоталамуса ведет к
росту потребления пищи экспериментальными животными, увеличению
массы тела и росту содержания в организме триглицеридов и жировой
ткани. Синдром гипоталамического ожирения развивается у больных с
аналогичными повреждениями головного мозга вследствие опухолей
гипоталамуса. Синдром характеризуют: а) усиленное потребление пищи (гиперфа-
гия); б) рост секреции инсулина и гипертрофия островков Лангерганса;
в) снижение двигательной активности; г) эмоциональная неустойчивость,
которая у части больных проявляет себя вспышками аггресивности.
Выделяют две фазы синдрома. В первую динамическую фазу происходит
быстрое увеличение массы тела. Во вторую статическую фазу масса тела
длительное время остается на одном уровне.
Экспериментальная деструкция нейронов гипоталамуса,
локализованных латеральнее его вентромедиального ядра, приводит к
противоположному результату. Животные временно прекращают есть (афазия), и у них
снижается масса тела.
Основной нейромедиатор нервных клеток и окончаний в вентромеди-
альной области гипоталамуса - это дофамин. Через латеральную область
проходят дофаминергические волокна, исходящие из ствола головного
мозга. Повреждение в этой области уменьшает содержание в гипоталамусе
дофамина, что находится в прямой связи со снижением потребления пищи.
Функционирование нейронов как вентро-медиальной, так и латеральной
части гипоталамуса находится под постоянным контролем симпатической
части автономной нервной системы. Симпатические волокна пронизывают
гипоталамус почти во всех направлениях, кроме медио-латерального.
Введение альфа-адреномиметиков в гипоталамус повышает интенсивность
поиска и потребления пищи экспериментальным животным, а инъекция бета-
адреномиметиков прекращает потребление пищи и вызывает поведенческий
эквивалент насыщения. В этой связи выделяют альфа-адренергический
центр голода и бета-адренергический центр насыщения.
Нарушение внутрицентральных отношений на уровне гипоталамуса
приводит к значительным сдвигам в системной регуляции пищеварения и
усвоения нутриентов. Так, повреждение вентромедиальной части через
усиление вагальных регуляторных влияний увеличивает секрецию в просвет
желудка соляной кислоты и усиливает высвобождение инсулина
поджелудочной железой. Нейроны вентромедиальной части гипоталамуса,
повреждение которых ведет к увеличению потребления пищи, рассматривают не
как специализированный центр голода, а как совокупность в
функциональном отношении объединенных супрасегментарных нейронов
симпатической части автономной нервной системы. Их возбуждение вызывает
системную реакцию подготовки к борьбе с угрозой или активному бегству от нее,
158
Глава 11
в частности через установление преобладания катаболических процессов
над анаболическими. Эта совокупность нейронов находится в реципрок-
ных отношениях с парасимпатическими центрами. Снижение уровня
возбуждения симпатических нейронов вентромедиальной локализации
активирует соответствующие парасимпатические центры, локализованные
латеральнее.
Активация парасимпатических центров вызывает системную реакцию
усиления утилизации нутриентов в ходе анаболизма, в частности через рост
потребления пищи, интенсификацию пищеварения и усиление всасывания
из просвета кишечника. Эта реакция у наших предков на пути эволюции не
была столь частой, как у нас, в силу того, что они ели далеко не каждый
день. В наше время в развитых странах легкодоступный полноценный или
даже избыточный суточный рацион - это норма жизни для большинства
населения. Это можно считать одной из причин гипоталамического
ожирения через действие внешних стимулов к потреблению пищи, которое
снижает уровень возбуждения вентромедиальных нейронов и активирует гипо-
таламические нейроны, локализованные латеральнее. Известно, что у
больных, страдающих от ожирения, действие внешних стимулов к
потреблению пищи усиливает его в большей степени, чем у людей с нормальными
массой тела и содержанием жира в организме.
Существуют три главных механизма аккумуляции триглицеридов как
потенциальных источников свободной энергии в организме. Первый
механизм - это увеличение размеров клеток жировой ткани, адипоцитов, по мере
роста содержания в них жиров. Второй - увеличение числа адипоцитов без
изменения их размеров. Третий - это одновременное увеличение как
размеров адипоцитов, так и их числа. Адипоцит - это высокодифференцирован-
ная клетка. Ее мезенхимальная клетка-предшественник - адипобласт. Не
все адипобласты дифференцируются до адипоцитов. Результат развития
части из них - образование клеток, не содержащих жира. В ответ на
увеличение поступления с пищей в организм детей потенциальных источников
энергии растет число адипоцитов, возникших в ходе дифференциации от
адипобластов. В этой связи считают, что переедание в детстве приводит к
трудно устранимому ожирению в зрелом возрасте через увеличение числа
дифференцированных клеток жировой ткани, которое на определенном этапе
онтогенеза становится патологически высоким и постоянным. Переедание
как преобладание поступления в организм источников свободной энергии
над его энерготратами увеличивает размеры адипоцитов. С другой
стороны, увеличение размера адипоцитов усиливает липогенез, то есть
образованию жирных кислот
Интенсивность синтеза жирных кислот как этапа образования жира в
виде триглицеридов, зависит не только от размеров адипоцитов, но и от
соотношения на уровне всего организма секреции и эффектов главного
анаболического гормона инсулина и его гормонов-антагонистов.
Превалирование активности инсулина усиливает липогенез.
Интенсивность липогенеза меньше, когда человек ест часто и немного,
и, наоборот, она больше, когда он ест много и редко.
Типовые нарушения липидного обмена
159
Ожирение служит причиной ряда сдвигов эндокринной регуляции
обмена веществ и представляет собой следствие эндокринопатий. Эндокри-
нопатии при этом могут быть следствием избыточного потребления
источников свободной энергии с пищей. Наиболее частое из нарушений
эндокринной регуляции обмена веществ у больных с ожирением - это
повышенная активность в крови инсулина, гиперинсулинемия. Чем больше
ожирение, тем больше концентрация инсулина в крови утром и натощак.
Гиперинсулинемия у части больных с ожирением приводит к большей
суммарной длительности действия гипогликемии как внутреннего стимула к
потреблению пищи. Кроме того, у больных с ожирением усилена реакция
роста секреции инсулина в ответ на действие ее обычных стимулов
(гипергликемия и др.).
У части больных с ожирением выявляют сниженную секрецию в кровь
гормона роста в ответ на ее обычные стимулы:
♦ вызванное инсулином перемещение глюкозы в клетки, вызывающее
гипогликемию;
♦ рост концентрации в циркулирующей крови аргинина после
внутривенной инфузии раствора аминокислоты.
Как у мужчин, так и у женщин, страдающих от ожирения, в крови
снижается содержание тестостерона и эстрадиола. Концентрация же эстрона
растет. Это происходит вследствие усиления его образования в строме
жировой ткани из андрогенного кортикостероида. Из-за роста содержания в
циркулирующей крови эстрона ожирение у женщин считают фактором
риска злокачественных опухолей матки.
Избыточное потребление с пищей потенциальных источников
свободной энергии для клеток в виде жиров и углеводов приводит к ряду
взаимосвязанных патологических сдвигов обмена веществ и работы
функциональных систем.
Через гипергликемию избыточное поступление нутриентов во
внутреннюю среду ведет к повышенной секреции инсулина. Гиперинсулинемия
вызывает резистентность клеток к эффекту инсулина на рецепторном и по-
стрецепторном уровнях. Кроме того, гиперинсулинемия связана с
гиперплазией инсулинобразующих клеток островков Лангерганса, которая на
определенном этапе своего развития может служить причиной недостаточности
внешнесекреторной функции поджелудочной железы (схема 11.1).
Гиперинсулинемия повышает утилизацию аминокислот как субстратов
белкового синтеза на уровне всего организма. Это в частности вызывает
гиперплазию гладкомышечных элементов стенки сосудов сопротивления.
В результате их просвет сужается, общее периферическое сосудистое
сопротивление растет и возникает артериальная гипертензия. Избыточное
потребление источников свободной энергии с пищей активирует
симпатический отдел автономной нервной системы и повышает секрецию
щитовидной железой триийодтиронина, что вызывает рост потребления
кислорода всем организмом. Вслед за ростом потребления кислорода повышается
минутный объем кровообращения, что вызывает артериальную гипертен-
зию и (или) предрасполагает к ней (схема 11.2).
160
Глава 11
Избыточное поступление нутриентов как источников
свободной энергии с пищей во внутреннюю среду
Гипергликемия
Рост секреции инсулина
Резистентность по отношению
к инсулину на рецепторном и
пострецепторном уровне
Патологическая гиперплазия
инсулинобразующих клеток в
островках Лангерганса как
причина инсулинопении
Неинсулинзависимый сахарный диабет
Схема 11.1. Ожирение как причина и фактор риска
неинсулинзависимого сахарного диабета
Избыточное поступление нутриентов как источников
свободной энергии с пищей во внутреннюю среду
Активация симпатического
отдела автономной нервной
системы
Рост секреции
трийодтиронина
Гиперинсулинемия
Увеличение потребления
кислорода организмом
Гиперплазия гладкомышечных
элементов стенок сосудов
сопротивления
Рост минутного объема
кровообращения
Повышение общего
периферического сосудистого
сопротивления
Артериальная гипертензия
Схема 11.2. Ожирение как фактор риска
и причина артериальной гипертензии
Типовые нарушения липидного обмена
161
Избыточно^ ширеб^ение-слищей источников свободной энергии ведет
к гиперлипидекии и аккумуляции свободной энергии в виде тригтшцеридов
жировой ткани. Это повышает интенсивность обмена холестерина на
системном уровне. В результате растет экскреция холестерина с желчью, что
ведет к образованию камней в просвете желчного пузыря. Поэтому
ожирение считают фактором риска хронического калькулезного холецитита.
ГИПЕРЛИПВДЕМИИ/ГИПЕРЛИПОПРОТЕИНЕМИИ
Гиперлипидемия/гиперлипопротеинемия - патологическое состояние
предболезни или заболевание, связанные сростом содержания в плазме крови
свободных жирных кислот, триглицеридов, холестерина, хиломикронов и
липопротеинов (ЛП).
Существует прямая связь между концентрацией липидов в плазме крови и
вероятностью атеросклероза как причины ишемической болезни сердца.
Полагают, что а) гиперхолестеринемия (патологически высокая концентрация
холестерина в плаще крови) и в меньшей степени рост содержания в плазме
других липидов представляют собой этиологический фактор ишемической болезни
сердца; б) снижение концентраций липидов через диету, физические
упражнения и с использованием средств фармакологической коррекции замедляет
развитие атеросклероза и ишемической болезни сердца. Эта два положения и
составляют липидную гипотезу атеросклероза и ишемической болезни сердца,
аргументами сторонников которой служат следующие факты:
♦ атеросклеротическая бляшка содержит липиды, большинство
которых поступает в нее прямо из липопротеинов, циркулирующих с
плазмой крови;
♦ атеросклеротические поражения сосудов могут быть
воспроизведены в эксперименте при кормлении животных пищей с высоким
содержанием холестерина;
♦ гиперлипидемию всегда выявляют у больных с окончательным
диагнозом атеросклероза;
♦ результаты эпидемиологических исследований свидетельствуют о
высоком риске ишемической болезни сердца как причине летальных
исходов при росте в плазме крови концентрации атерогенных
липопротеинов низкой плотности (ЛПНП) и снижении в ней содержания
антиатерогенных липопротеинов высокой плотности (ЛПВП).
Атерогенными называют те липопротеины, рост концентрации которых
в плазме крови вызывает атеросклероз, и, наоборот, неатерогенные - это
ЛП, не обладающие таким свойством (табл. 11.1).
Выделяют вторичную гиперхолестеринемию, то есть патологический
рост концентрации холестерина в крови вследствие перемежающейся пор-
фирии, блокады желчевыводящих путей, гипотиреоза и беременности.
Вторичную гипертриглицеридемию (рост содержания в крови триглицеридов)
162
Глава 11
Таблица 11.1
Липопротеины и атеросклероз
Липопротеины
Липопротеины низкой плотности
(ЛПНП)
Липопротеины промежуточной
плотности (ЛППП)
Бета-липопротеины очень низкой
плотности (Р-ЛПОНП)
Окисленные ЛПНП (лиганды к
«рецепторам-мусорщикам» наружной
поверхности макрофагов сосудистой стенки)
Липопротеин(а)
Хилом икроны
Липопротеины очень низкой плотности
(ЛПОНП)
Липопротеины высокой плотности
Плотность
fllinAniUlTQUII 4ft
JlnllOlipU IcHHU
1,019-1,063 кг/л
1,006-1,019 кг/л
<1,006 кг/л
1,063-1 Д10 кг/л
Атерогенность (+),
неатерогенность (0) и
антиатерогенность (-)
+
+
+
+
+
0
0(?)
-
обуславливают сахарный диабет, острая алкогольная интоксикация, сепсис,
индуцированный грамотрицательной инфекцией, нарушения синтеза и
аккумуляции гликогена гликогена I типа, а также другие патологические
состояния и болезни.
Семейная гиперлипидемия первого типа - врожденное нарушение ли-
пидного обмена, обусловленное недостаточным расщеплением хиломикро-
нов и липопротеинов очень низкой плотности вследствие низкой
активности катализатора их гидролиза липопротеинлипазы или недостатка активатора
этого фермента, аполипопротеина С-П.
Расщепление хиломикронов с участием липопротеинлипазы
происходит в плазме крови, на поверхности эндотелиальных клеток и адипоцитов.
Низкая активность липопротенинлипазы ведет к снижению очищения
плазмы крови от хиломикронов. Семейная гиперлипидемия первого типа,
обусловленная только низкой активностью липопротеинлипазы без недостатка
аполипопротеина С-П, - это врожденное расстройство липидного обмена,
которое наследуется по аутосомально-рецессивному типу и обусловлено
мутацией 207 остатка в пятом экзоне гена липопротеинлипазы. Его
характеризуют повышенное содержание в плазме крови триглицеридов и
периодические обострения хронического панкреатита, начиная с неонатального
периода. Панкреатит у таких больных вызывает перерождение паренхимы
поджелудочной железы, вызванное высоким содержанием в крови
хиломикронов Низкий относительно нормального уровень гидролиза
хиломикронов и липопротеинов очень низкой плотности в просвете капилляров,
прилежащих к адипоцитам, ведет к росту концентрации хиломикронов в плазме
крови. Кроме того, падает захват триглицеридов адипоцитами. Это ведет к»
росту их концентрации в плазме выше уровня в 4 г/л. Кроме того, рост кон-,
центрации триглицеридов в плазме крови - это следствие увеличения не-
Типовые нарушения липидного обмена
163
ферментативного расщепления в ней хиломикронов, то есть не связанного
с функционированием липопротеинлипазы, а обусловленного ростом
содержания хиломикронов во внутренней среде организма. Высокое содержание
хиломикронов и триглицеридов в плазме крови ведет к ксантоматозу кожи,
непереносимости жирной пищи и гепатоспленомегалии у больных с гипер-
липидемией данного типа. Так как при гиперлипидемии I типа в плазме
растет концентрация неатерогенных липопротеинов очень низкой
плотности и хиломикронов, то атеросклероза у таких больных обычно не
развивается. Следует заметить, что системная красная волчанка ведет к сдвигам
липидного обмена почти идентичным гиперлипидемии первого типа.
Лабораторные исследования плазмы крови больных с гиперлипидемией первого
типа выявляют снижение липолитической активности плазмы крови, не
связанной с действием гепарина как активатора липолиза.
Гиперлипидемия второго типа-это наследственное нарушение
липидного обмена, при котором у родственников или членов одной семьи как фе-
нотипический признак выявляют патологически высокое содержание
холестерина в крови (семейная гиперхолестеринемия). Эту наследуемую по
аутосомально-доминантному типу гиперлипидемию характеризуют
бугорчатый ксантоматоз в области сухожилий, раннее и быстрое развитие
атеросклероза, ишемической болезни сердца и инфаркта миокарда, который
часто служит причиной внезапной сердечной смерти в возрасте от 20 до 50
лет. Ведущим звеном патогенеза гиперхолестеринемии, высокого
содержания в плазме крови атерогенных липопротеинов низкой плотности и
атеросклероза у больных с гиперлипидемией второго типа является или
полное отсутствие рецепторов к липопротеинам низкой плотности на наружной
клеточной поверхности или нарушения их строения и функции вследствие
мутации аллелей генов RbO, Rb- и Rtio. В результате наследуемой
недостаточности рецепторов к ЛПНП возникает гиперхолестеринемия, которая по
механизму положительной обратной связи повышает синтез атерогенных
липопротеинов низкой плотности в печени (схема 11.3).
Если патогенные мутации затронули один из аллелей гена,
кодирующего рецептор к липопротеинам низкой плотности (больные гетерозиготные
по мутантному патогенному гену), то концентрация холестерина в плазме
крови составляет 5-6 г/л. У гомозиготных (патогенный ген содержит два
мутантных аллеля) больных она находится в пределах 3-4 г/л. Выделяют
два вида гиперлипидемии II типа, Па и lib. При а-типе в крови увеличена
концентрация холестерина при нормальных содержаниях в ней
липопротеинов очень низкой плотности и триглицеридов. При b-типе концентрации в
крови липопротеинов очень низкой плотности и триглицеридов повышены.
Изменения обмена веществ, обусловленные злокачественными
опухолями печени, часто приводят к сдвигам содержания в крови липидов,
аналогичным гиперлипидемии На.
Гиперлипидемия третьего типа - это наследуемая по аутосомально-
доминантному типу недостаточность катаболизма атерогенных
липопротеинов промежуточной плотности. Как синдром гиперлипидемию
третьего типа характеризуют ускоренное развитие атеросклероза и частая
164
Глава 11
Наследственно детерминированная
недостаточность ЛПНП-рецепторов
Снижение пиноцитоза комплексов ЛПНП-холестерин
Низкий уровень высвобождения
холестерина в лизосомах
Снижение содержания в клетке
свободного холестерина
Рост концентрации
атерогенных ЛПНП
в плазме крови
Рост активности гидроксиметил-
глютарил-коэнзим А-редуктазы
Рост синтеза холестерина и увеличение высвобождения
холестерина клеткой
Схема 11.3. Патогенез семейной гиперхолестеринемии
тромбоэмболия сосудов из системы венечной артерии, сахарный диабет,
ожирение, гипотиреоз и особо выраженный ксантоматоз. При исследовании
плазмы крови выявляют рост содержания в ней концентраций триглицеридов и
холестерина, которые липопротеины промежуточной плотности переносят
от печени на периферию. О сахарном диабете у таких больных
свидетельствует сниженная толерантность по отношению к глюкозе.
Гиперлипидемия четвертого типа - это наследуемое по аутосомаль-
но-доминантному типу нарушение липидного обмена, которое
характеризует гипертриглицеридемия, то есть аномально высокое содержание
триглицеридов в плазме крови. Это наиболее частое из нарушений липидного
обмена у больных, при котором атеросклероз редко поражает
периферические и венечные артерии. При гиперлипидемии четвертого типа в плазме
крови повышено содержание как триглицеридов, так и липопротеинов очень
низкой плотности. Отдельно выделяют приобретенную гиперлипидемию
четвертого типа, которую вызывают сахарный диабет, уремия, препараты
из группы глюкокортикоидов, а также бета-адреномиметики.
Гиперлипидемия/гиперлипопротеинемия пятого типа - это полиэтио-
логичное и варьирующее по патогенетическим механизмам нарушение
липидного обмена, вследствие которого у части больных возникают
ксантоматоз и панкреатит как следствия патогенно высоких концентраций в плазме
крови липопротеинов очень низкой плотности и хиломикронов.
Типовые нарушения липидного обмена
165
АТЕРОСКЛЕРОЗ
Атеросклероз - отложение в интиме сосудов атерогенных липопроте-
инов низкой плотности (ЛПНП) вследствие взаимодействие гладкомышеч-
ных клеток стенок сосудов с атерогенными липопротеинами при их
высокой концентрации в циркулирующей крови. Атеросклеротическое
перерождение сосудов во многом определяет пролиферация миоцитов сосудистой
стенки, индуцируемая цитокинами тромбоцитов, активированных мононук-
леаров, а также агентами аутокринной регуляции (митогенами) самих глад-
комышечных клеток. Термин атеросклероз описывает изменения
сосудистой стенки при этом системном типовом патологическом процессе. Слово
«атере» (греч.) означает «кашица» и указывает на образование в стенках
сосудов кашицеобразных липидных отложений с их дальнейшим
склерозированием и уплотнением.
По определению Всемирной организации здравоохранения
«Атеросклероз - это вариабельная комбинация изменений внутренней оболочки
(интимы) артерий, включающая накопление липидов, сложных углеводов,
фиброзной ткани, компонентов крови, кальцификацию и сопутствующие
изменения средней оболочки (медии)».
При гистологическом исследовании пораженных атеросклерозом
участков сосудистой стенки выявляют характерное патологическое образование,
атеросклеротическую бляшку (рис. 11.1). Ее формируют липиды,
лейкоциты, гладкомышечные клетки и межклеточное вещество интимы артерий.
Образованию бляшки предшествует адгезия (прилипание) циркулирующих
моноцитов и лимфоцитов к артериальному эндотелию с последующим
локальным накоплением пенистых клеток. Пенистые клетки представляют
собой активированные мононуклеары, видоизмененные в результате
интенсивного эндоцитоза липидов.
До сих пор общественное сознание связывает атеросклероз с
пропитыванием холестерином сосудистой стенки, что верно лишь отчасти.
Холестерин и триглицериды переносятся во внеклеточном пространстве
липопротеинами. Не гиперхолестеринемия (рост концентрации холестерина выше
верхнего предела нормальных колебаний) ведет к атеросклерозу, а
аккумуляция в сосудистой стенке определенных липопротеинов. Некоторые ли-
попротеины можно считать атерогенными, другие неатерогенными, а
третьи антиатерогенными.
Под атерогенными следует понимать липопротеины, которые
проникают в сосудистую стенку, где происходит их эндоцитоз
макрофагами, которые в результате эндоцитоза превращаются в пенистые
клетки. В последующие стадии патологического процесса происходит
отложение аморфного холестерина и его кристаллов в межклеточных
пространствах сосудистой стенки, то есть вне пенистых клеток. При гиперли-
пидемии второго типа, которая часто приводит к атеросклерозу,
пенистые клетки, насыщенные липидами, находят во внутренней оболочке
сосудистой стенки.
166
Глава 11
Тромб
\ Фибрин
Рис. 11.1. Схематическое изображение строения атеросклеротической бляшки,
вызывающей стенокардию
Поверхность макрофагов содержит рецепторы к липопротеинам очень
низкой плотности и непостоянные рецепторы к измененным в результате
окисления липопротеинам низкой плотности («рецепторы-мусорщики»).
Предположительно активация мононуклеаров через связывание этих рецепторов с
окисленными атерогенными липопротеинами или липопротеинами очень
низкой плотности ведет к активации макрофагов, в результате которой они
приобретают способность к эндоцитозу атерогенных липопротеинов.
В ранней стадии атеросклеротического поражения сосудистой стенки
в ней выявляют липидные прожилки и пенистые клетки. Этот начальный
этап формирования атеросклеротической бляшки вызывает активация
непостоянно присутствующих на клеточной поверхности макрофагов
рецепторов к окисленным липопротеинам низкой плотности (ацетил-ЛПНП-
рецепторы, «рецепторы-мусорщики»). Мусорщиками эти рецепторы
называют потому, что они обладают высоким сродством к «мусору» в виде
окисленных ЛПНП. Изменения ЛПНП в результате окисления, которые
обуславливают их особо высокое сродство к рецепторам-мусорщикам
включают:
♦ трансформацию лецитина в составе липопротеинов в лизолецитин;
♦ окисление холестерина;
♦ рост отрицательного заряда и плотности липопротеинов;
♦ снижение содержания в ЛПНП полиненасыщенных жирных кислот.
Распад структурного апопротеина липопротеинов низкой плотности В-
100 с высвобождением гистидина, лизина и пролина. Окисленные липоп-
ротеины служат хемоаттрактантами и активаторами для макрофагов
сосудистой стенки, одновременно являясь для них и объектом эндоцитоза.
Как результат патогенных межклеточных взаимодействий атеросклероз
представляет собой патологическое изменение стенок артерий большого и
среднего диаметра, которые составляют и вызывают локальная аккумуляция
в интиме липидов и макрофагов, миграция и пролиферация гладкомышеч-
Типовые нарушения липидного обмена
167
ных клеток, а также отложения вещества внеклеточного матрикса.
Патогенные межклеточные взаимодействия, приводящие к атеросклерозу,
реализуются не только через действие факторов роста, но и в результате эффектов
цитокинов флогогенов, медиаторов воспаления и экспрессии адгезивных
молекул. Начальным этапом патогенеза атеросклероза как последовательности
межклеточных взаимодействий является адгезия моноцита циркулирующей
крови к эндотелиальным клеткам с их последующей миграцией в интиму.
При этом индукторы атеросклероза окисленные (модифицированные) липоп-
ротеины низкой плотности, воздействуя на лейкоциты циркулирующей
крови и эндотелиальные клетки, вызывают экспрессию на их поверхности
адгезивных молекул. Так, лизофосфатидилхолин - элемент молекулы окисленных
атерогенных липопротеинов - индуцирует экспрессию межклеточной
адгезивной молекулы-1 на поверхности эндотелиальных клеток. Известно, что
атерогенные липопротеины оказывают на эндотелий прямые и непрямые
влияния, повышающие экспрессию на их поверхности эндотелиально-лейкоци-
тарных адгезивных молекул (ЭЛАМ). Непрямой эффект атерогенных
липопротеинов на рост экспрессии ЭЛАМ состоит в стимуляции паракринной
секреции гладкомышечными клетками и мононуклеарными макрофагами
сосудистой стенки. Адгезия лейкоцитов к сосудистой стенке служит первым
этапом их проникновения в интиму. Там активированные моноциты секрети-
руют ряд активаторов эндотелия и хемоаттрактантов, стимулирующих
дальнейшую инфильтрацию очага атеросклеротического повреждения
моноцитами и лимфоцитами. Патогенное функционирование иммунокомпетентных
клеток в качестве эффекторов атеросклероза указывает на участие в развитии
атеросклероза иммунопатологической реакции.
Атеросклероз во многом представляет собой хроническое воспаление
сосудистой стенки, протекающее с преобладанием пролиферативного
компонента, основными клеточными эффекторами которого являются
моноциты циркулирующей крови, мононуклеарные фагоциты субинтимального
слоя, гладкомышечные сосудистые клетки, активированные атерогенны-
ми липопротеинами или в результате межклеточных взаимодействий.
Воспаление в очаге атеросклеротического поражения усиливает
взаимосвязанное с ним локальное свертывание крови, которое могут вызвать
сами атерогенные липопротеины. Так, атерогенный липопротеин(а),
содержит гликопротеин(а), связанный с апопротеином В. Идентичность
структуры липопротеина(а) строению плазминогена может обуславливать внутри-
сосудистый тромбогенез через конкурентное связывание рецепторов к
плазминогену на поверхности эндотелиоцитов.
Таков патогенез индукции атеросклероза через образование прожилок
атерогенных липопротеинов и пенистых макрофагов в сосудистой стенке
как начального этапа образования атеросклеротической бляшки. В нем
особую роль как эффекторы и регуляторы патологического процесса играют
макрофаги сосудистой стенки, которые сами могут индуцировать
окисление ЛПНП через высвобождение свободных кислородных радикалов и (или)
активацию липооксигеназы. Так как макрофаги представляют собой
эффектор единой системы иммунитета организма, то мы вправе предположить
168
Глава 11
роль в развитии атеросклероза нервного эндогенного этиологического
фактора, столь сильно меняющего состояние системы иммунитета, и ее макрофа-
гального звена в частности. Кроме того, патогенный стресс как состояние,
которое характеризует системная интенсификация свободнорадикального
окисления липидов, может повышать уровень окисления ЛПНП и на уровне
сосудистой стенки предрасполагать к атеросклерозу.
Пенистые клетки высвобождают ряд цитокинов, чье действие вызывает
пролиферацию клеточных элементов, и в особенности миоцитов гладкомы-
шечных элементов сосудистой стенки. Кроме того, цитокины
активированных макрофагов активируют эндотелиоциты, что ведет к росту экспрессии
их тромбогенного потенциала. Цитокины пенистых клеток активируют и
нейтрофилы циркулирующей крови, что вызывает воспаление с полимор-
фонуклеарами в качестве его клеточных эффекторов. В результате в составе
атеросклеротической бляшки находят пролиферирующие миоциты
сосудистой стенки, агрегаты активированных тромбоцитов, других форменных
элементов крови, активированные нейтрофилы и нити фибрина. Все это
характеризует атеросклеротическую бляшку как очаг воспаления и локус
тромбоза.
Макрофаги, окисленные липопротеины, ацетил-ЛПНП-рецепторы
играют определяющую роль в индукции атеросклероза. В дальнейшем
процесс образования атеросклеротической бляшки теряет связь с этими
этиологическими факторами, то есть через патогенные межклеточные
взаимодействия происходит его эндогенизация.
Одно из наиболее впечатляющих достижений медицинской генетики
последних десятилетий - выяснение генетической детерминированности
гиперхолестеринемии и высокой концентрации в плазме крови липопроте-
инов низкой плотности как причины атеросклероза. Исследовав 500
больных, выживших после инфаркта миокарда, Гольдштейн, Браун и Мотульс-
кий более, чем в половине случаев выявили семейную гиперхолестеринемию,
семейную гиперлипидемию или комбинированную гиперлипидемию. Было
установлено, что эти расстройства липидного обмена представляли собой
патогенные фенотипические признаки, детерминированные одним геном,
то есть были моногенными наследственными расстройствами липидного
обмена. Последующие исследования клеточных культур фибробластов кожи,
лимфоцитов, и миоцитов аортальной стенки, взятых у гомозиготных и
гетерозиготных по данному гену больных, позволили выявить генетически
детерминированные дефекты связывания молекулярного комплекса
липопротеины низкой плотности-холестерин с наружной клеточной мембраной и
его последующего пиноцитоза.
Взаимодействие молекулярного комплекса холестерин-ЛПНП при
нормальной экспрессии ЛПНП-рецептора на поверхности клеток ведет к пино-
цитозу молекулярного комплекса. После пиноцитоза комплекс
инкорпорируется в лизосомы, где и происходит высвобождение свободного
холестерина. Рост концентрации свободного холестерина в клетке снижает
активность ключевого фермента внутриклеточного синтеза холестерина гид-
роксиметилглютарил-коэнзим А-редуктазы. Наследуемая по аутосомально-
Типовые нарушения липидного обмена
Ъ9
доминантному типу недостаточность ЛПНП-рецепторов ведет к снижению
пиноцитоза комплекса холестерин-ЛПНП и к падению концентрации
свободного холестерина в клетках. В результате низкого содержания
свободного холестерина в клетках с низким содержанием на наружной поверхности
ЛПНП-рецепторов в них высока активность гидроксиметилглютарил-коэн-
зим А-редуктазы. Это ведет к интенсивному образованию холестерина
клетками, его высвобождению во внеклеточное пространство и росту в нем
содержания атерогенных липопротеинов переносчиков холестерина.
Гиперхолестеринемия вызывает атеросклероз не через пропитывание
стенки сосудов холестерином, а через повышение интенсивности образования
печенью и высвобождения ею в кровь атерогенных липопротеинов
(схема 11.3). Кроме того, гиперхолестеринемию при семейной гиперхолестери-
немии и других гиперлипидемиях, связанных с атеросклерозом,
обуславливает и низкий уровень связывания комплекса ЛПНП-холестерин с
ЛПНП-рецепторами наружных клеточных мембран. Гиперхолестеринемия
алиментарного генеза также повышает риск атеросклероза через
увеличение в крови концентрации ЛПНП и других атерогенных липопротеинов,
переносящих холестерин во внеклеточном пространстве. Рост
концентрации ЛПНП повышает массу циркулирующих с кровью продуктов их
окисления, вступающих во взаимодействие с рецепторами-мусорщиками
наружных клеточных мембран макрофагов, то есть повышает вероятность
реализации инициирующего момента атеросклероза.
Глава 12
ВОСПАЛЕНИЕ
ОПРЕДЕЛЕНИЕ И БИОЛОГИЧЕСКОЕ ЗНАЧЕНИЕ
ВОСПАЛЕНИЯ
Воспаление - это системная защитная реакция уничтожения и
элиминации всего чужеродного, которая достигает своей биологической цели в
основном посредством:
♦ активации системы комплемента;
♦ дегрануляции тучных клеток;
♦ роста проницаемости микрососудов и адгезивной способности
эндотелия;
♦ миграции плазмы крови в межклеточные пространства;
♦ адгезии к эндотелиальным клеткам нейтрофилов, моноцитов и
лимфоцитов циркулирующей крови и их выхода в интерстиций;
♦ фагоцитоза, бактерицидного и цитолитического действия
фагоцитов;
♦ расширения, спазма и тромбоза микрососудов;
♦ замещения дефекта тканей через ангиогенез и пролиферацию фиб-
робластов.
Под чужеродным в данном контексте следует понимать не только
микроорганизмы и инородные частицы, попавшие во внутреннюю
среду, но и свои переродившиеся или некробиотически измененные клетки.
Подвергшиеся цитолизу клетки приобретают свойство антигенной
стимуляции системы иммунитета, высвобождая антигены для
взаимодействия с системой иммунитета, в том числе и фрагменты клеточных
мембран. Изменение строения наружной клеточной мембраны
некробиотически измененных клеток обуславливает активацию на них системы
комплемента по альтернативному пути. Активация системы
комплемента вызывает воспаление. Переродившиеся малигнизированные клетки
при взаимодействии с системой иммунитета вызывают ее антигенную
стимуляцию. При этом на их клеточной поверхности появляются
антигены, к которым нет иммунологической толерантности. При
извращении реакций иммуной системы нормальные антигены клеточных
поверхностей приобретают свойство стимулировать систему иммунитета
собственного организма. В результате на поверхности таких нормальных
клеток образуются иммунные комплексы, что через активацию системы
комплемента по классическому пути индуцирует воспаление. В данном
случае клетки не перерождаются, но через потерю иммунной
толерантности организма к их поверхностным антигенам приобретают свойство
чужеродности.
Воспаление
171
В этой связи можно выделить следующие основные причины воспаления:
♦ некробиотические изменения тканей и клеток под влиянием
экзогенных физико-химических факторов',
♦ инвазия чужеродных микроорганизмов или антигенов, иммуногенов
во внутреннюю среду,
♦ малигнизация собственных клеток организма',
♦ потеря иммунологической толерантности по отношению к
антигенам собственного организма.
Достигая главную биологическую цель, то есть уничтожение и
элиминацию всего чужеродного, при реализации острой воспалительной реакции
организм решает промежуточную задачу локализации очага воспаления,
также направленную на достижение конечного приспособительного
результата защитной реакции. Локализуются очаг инфекции, зона некробиотичес-
ких изменений тканей и клон злокачественных клеток. В результате
блокируются распространение инфекции и метастазирование в пределах
внутренней среды. Некробиотически измененные ткани представляют
собой «транзиторный орган внутренней секреции», функционирование
которого имеет исключительно патогенное значение. Поэтому их изоляция
через воспаление также биологически целесообразна. Моноциты и иммуно-
компетентные клетки в очаге воспаления секретируют цитокины (фактор
некроза опухолей, интерлейкин-1 и др.), которые, попадая в системно
циркулирующую кровь, могут обусловить патологическое состояние
системной воспалительной реакции. Системная воспалительная реакция по
дисфункциям функциональных систем и сдвигам гомеостазиса эквивалентна
сепсису. В этой связи одной из целей локализации воспалительной
реакцией ее очага следует считать предотвращение гиперцитокинемии, то есть
патогенно высокой концентрации цитокинов мононуклеарных фагоцитов и
лимфоцитов в циркулирующей крови. Наконец, цитолиз в очаге
воспаления, высвобождая из структуры клеток множество антигенов, создает
возможность антигенной стимуляции системы иммунитета антигенами
собственного организма. Локализация предотвращает системную
аутоиммунную реакцию. Очаг воспаления локализуют спазм, тромбоз и деструкция
микрососудов, их обтурация агрегатами активированных лейкоцитов, сдав-
ление микрососудов вследствие интерстициального отека, а также
компрессия и тромбоз микроскопических лимфатических сосудов.
Воспаление, как и любая защитная реакция организма, избыточна
относительно стимулов ее вызвавших и потому часто трансформируется в
типовой патологический процесс. Локально это проявляется избыточным
повреждением нормальных клеточных элементов при уничтожении и
элиминации всего чужеродного. Функционирование моноцитов и иммуно-
компетентных клеток в очаге воспаления ведет к высвобождению ими в
системную циркуляцию цитокинов, активирующих нейроэндокринную катабо-
лическую систему, что через стрессорное голодание при системной
воспалительной реакции может привести к алиментарной дистрофии. Кроме
того, гиперцитокинемия как следствие несостоятельности локализации
очага воспаления, активирует клеточные эффекторы воспаления в удаленных
172
Глава 12
органах и тканях, вызывая лишенное биологического смысла воспаление как
причину разрушения структурно-функциональных элементов эффекторов
функциональных систем и множественной системной недостаточности.
КОМПОНЕНТЫ И ПРИЗНАКИ ВОСПАЛЕНИЯ
Выделяют три компонента воспаления - альтерацию, экссудацию и
пролиферацию, которые тесно связаны между собой.
Первичная альтерация (лат. alteratio - изменение) - это совокупность
изменений обмена веществ, физико-химических свойств, структуры и
функции клеток и тканей под влиянием этиологического фактора воспаления.
Первичная альтерация как результат взаимодействия этиологического
фактора с организмом сохраняется и служит причиной воспаления и после
прекращения этого взаимодействия. Примером могут служить некробиотичес-
кие изменения тканей вследствие тяжелого ранения, вызывающие
воспаление после воздействия на них ранящего снаряда.
Вторичная альтерация-это совокупность изменений клеток и тканей в
результате самого воспаления. Одна из их причин - это изменения
микроциркуляции в очаге воспаления и прилежащих к нему тканях,
обуславливающие значительное падение напряжения кислорода в центре очага
воспаления. Гипоксия и связанный с ней гипоэргоз клеточных элементов служит
одной из причин цитолиза в очаге воспаления. Через паракринную секрецию
гипоксичными мононуклеарами гипоксия в очаге воспаления стимулирует
клеточную пролиферацию. Клеточная пролиферация замещает дефекты
тканей вследствие первичной и вторичной альтерации при воспалении.
Причиной цитолиза поврежденных первичной альтерацией клеток служит и
активация системы комлемента, предшествующая острой воспалительной
реакции в ее очаге. Цитолиз также вызывают свободные кислородные радикалы и
ферменты, высвобождаемые в очаге воспаления активированными
фагоцитами. Цитолиз служит причиной высокого содержания в межклеточных
пространствах очага воспаления калия, а гипоэргоз повышает содержание в нем
протонов (лактатный метаболический ацидоз типа А).
Коллоидно-осмотическое давление в интерстиции очага воспаления
повышают:
♦ аккумуляция в нем белков плазмы;
♦ выход из клеток калия и сопуствующих ему макромолекулярных
анионов как следствие цитолиза;
♦ тромбоз венул и лимфатических сосудов, препятствующий
выведению осмолей из очага воспаления.
Клеточные эффекторы воспаления становятся таковыми через
изменение своего функционирования, что также составляет альтерацию. Некроби-
отические изменения клеток в очаге воспаления ведут к их дисфункциям,
характеризующим как первичную, так и вторичную альтерацию.
Воспаление
173
Экссудация (лат. exsudatio - выпотевание) - выход жидкой части
плазмы крови в интерстиций и эмиграция из сосудов в межклеточные
пространства лейкоцитов как клеточных эффекторов воспаления.
Результатом экссудации является заполнение интерстициальных пространств и
очага воспаления экссудатом. Транссудат - это жидкость, которая
накапливается в межклеточных пространствах в результате роста
гидростатического давления в микрососудах и (или) увеличения их
проницаемости, не связанных с острой воспалительной реакцией. При воспалении
гидростатическое давление в микрососудах очага воспаления растет в
результате спазма и тромбоза венул, лимфатических микрососудов,
расширения артериол и прекапиллярных сфинктеров. Проницаемость в очаге
воспаления повышает ряд медиаторов воспаления. В результате жидкая
часть плазмы кровы выходит в интерстиций. Отличием экссудата от
транссудата является высокое содержание в нем белка (не менее 30 г/л),
протеолитических энзимов, иммуноглобулинов, лейкоцитов и остатков
тканевых элементов.
Отдельно как элемент экссудации выделяют реакцию роста
проницаемости стенки микрососудов в очаге воспаления. Ее обуславливает
локальный рост содержания флогогенов, повышающих проницаемость стенки
микрососудов в очаге воспаления и вызывающих эмиграцию лейкоцитов.
Раннюю транзиторную реакцию роста проницаемости обуславливает
действие гистамина, лейкотриена Е4, серотонина, брадикинина. Ранняя транзи-
торная реакция в основном затрагивает венулы с диаметром не более, чем
100 мкм. Проницаемость капилляров при этом не меняется. В эту фазу
воспалительной реакции роста проницаемости эндотелиоциты венул
сокращаются, что ведет к образованию между ними зазоров, через которые может
происходить выход жидкой части плазмы крови и эмиграция лейкоцитов в
интерстиций. При немедленной и длительной реакции роста
проницаемости ее в основном вызывает первичная альтерация как следствие действия
экзогенных этиологических факторов механической (травма, ранение),
термической или химической природы. В результате действия
этиологического фактора происходит некроз эндотелиальных клеток на уровне артериол
небольшого диаметра, капилляров и венул, что ведет к стойкому
возрастанию их проницаемости. Отсроченная и стойкая реакция роста
проницаемости микрососудов развивается в очаге воспаления через часы или сутки
от его начала. Она характерна для воспаления, вызванного ожогами,
действием бактериальных токсинов, ультрафиолетовым и ионизирующим
излучением и аллергическими реакциям отсроченного (замедленного) типа.
Одним из ведущих медиаторов этой реакции является медленно
реагирующая субстанция анафилаксии, то есть лейкотриены и полиненасыщенные
жирные кислоты, которые синтезируется из арахидоновой кислоты и
фактора активации тромбоцитов. Медленно реагирующую субстанцию в очаге
воспаления образуют и высвобождают тучные клетки. Стойкий рост
проницаемости микрососудов в очаге воспаления медленно реагирующая
субстанция анафилаксии обуславливает, вызывая протеолиз базальных
мембран микрососудов.
174
Глава 12
Биологический смысл экссудации как компонента воспаления состоит
не только в обеспечении уничтожения чужеродного через его атаку
системой комплемента и активированными фагоцитами из циркулирующей
крови, но и в отграничении очага воспаления через сдавление кровеносных и
лимфатических микрососудов вследствие интерстициального отека, а
также в разведении флогогенов и факторов цитолиза в очаге воспаления для
предотвращения избыточной вторичной альтерации.
Пролиферация (лат. proliferatio - размножение) - интенсификация
деления фибробластов и образования ими стромы соединительной ткани
(коллагеновых структур) в очаге воспаления для замещения дефектов
ткани вследствие первичной и вторичной альтерации. Пролиферацию
завершает инволюция рубца, то есть уничтожение и элиминация лишних
коллагеновых структур. Основные клеточные эффекторы пролиферации -
это активированные мононуклеарные фагоциты, фибробласты и имму-
нокомпетентные клетки. Фибробласты в очаге воспаления образуют и
высвобождают коллаген и энзим коллагеназу, ответственный за
формирование коллагеновых структур стромы соединительной ткани. Кроме
того, они образуют фибронектин, определяющий миграцию,
пролиферацию и адгезию фибробластов. Мононуклеары и лимфоциты секрети-
руют цитокины как стимулирующие, так и подавляющие эти функции
фибробластов. Нейтрофилы как клеточные эффекторы воспаления
влияют на пролиферацию, секретируя стимуляторы пролиферации кейлоны
и антикейлоны.
Покраснение и жар как местные признаки воспаления в основном
связано с артериальной гиперемией и интенсивными окислительными
процессами в очаге воспаления, при которых почти не происходит улавливания
свободной энергии клетками. Припухлость в очаге воспаления - это
следствие артериальной гиперемии и интерстициального отека как результата
экссудации. Боль в очаге воспаления представляет собой результат прямого
и (или) опосредованного действия флогогенов на чувствительные нервные
окончания. Боль в очаге воспаления усиливает ацидоз. Ее могут вызывать
механические раздражения нервных окончаний в результате роста
давления в очаге воспаления. Расстройства функциональных систем, чьи
органы-эффекторы поражены воспалением, как признаки воспаления в
основном связаны с разрушением их структурно-функциональных единиц,
препятствующим системным регуляторным влияниям и вызывающим в
эффекторах функций дефицит массы и энергии.
НЕЙТРОФИЛИЯ И ЛЕЙКОЦИТОЗ
Исследования с использованием нейтрофилов, меченных
радиоактивными изотопами, выявили в крови два пула (англ. pool - букв, общий котел)
нейтрофилов:
Воспаление
175
♦ пул полиморфонуклеаров, циркулирующих с кровью;
♦ пул нейтрофилов краевого стояния.
Каждый пул содержит примерно одинаковое число клеток. Между
пулами происходит постоянный обмен полиморфонуклеарами, то есть они
находятся между собой в состоянии динамического равновесии. Величину
пула полиморфонуклеаров, циркулирующих с кровью можно рассчитать,
умножая число нейтрофилов, которое содержит единица объема крови из
вены или артерии, на величину объема циркулирующей крови.
В начале воспаления пул циркулирующих с кровью нейтрофилов
растет, что в частности представляет собой реакцию на действие
бактериального эндотоксина. К росту пула циркулирующих нейтрофилов приводят
физические упражнения, а также гиперкортизолемия и гиперкатехоламине-
мия, связанные со стрессом. Биологический смысл этой в
филогенетическом отношении древней стрессорной реакции увеличения пула
циркулирующих нейтрофилов - это мобилизация организменного резерва
полиморфонуклеаров для защитной воспалительной реакции в ответ на
возможные повреждения и инвазию патогенных микроорганизмов.
Лейкоцитоз-рост общего содержания лейкоцитов в крови выше 1ЫШ/л.
Выделяют три вида лейкоцитоза: нейтрофилию, лейкемоидную реакцию
и лейкемию (лейкоз).
Нейтрофилия - это рост содержания в крови нейтрофильных
лейкоцитов (нейтрофилов, полиморфонуклеаров) выше верхнего предела его
нормальных колебаний, который составляет 7,2x109/л.
Физиологический лейкоцитоз (нейтрофилия) -реакция здорового
человека на действие того или иного раздражителя или рост содержания
нейтрофилов в крови, не представляющий собой признак или звено патогенеза
болезни. Выделяют следующие виды физиологической нейтрофилии:
♦ идиопатическая или наследственная;
♦ неонатальная;
♦ вызванная высокой температурой окружающей среды или действием
солнечного излучения;
♦ стрессорная, в том числе связанная с рвотой, болью и состоянием
тревожности;
♦ при овуляции, смене фаз менструального цикла и беременности;
♦ в ответ на физическую нагрузку.
Патологический лейкоцитоз (нейтрофилия) - следствие
патологических состояний и болезней, которое особенно часто вызывает инфекция.
Непосредственными причинами нейтрофилии служат:
♦ усиление образования нейтрофилов костным мозгом;
♦ снижение выхода полиморфонуклеаров из крови в ткани;
♦ уменьшение пула маргинальных (пристеночных)
полиморфонуклеаров и увеличение числа нейтрофилов, циркулирующих с кровью, при
неизменном общем содержании в ней нейтрофильных лейкоцитов
(псевдонейтрофилия).
Нейтрофилия у беременных и у больных с судорожными синдромами -
это в основном псевдонейтрофилия.
176
Глава 12
Любой из этих трех механизмов развития нейтрофилии может
действовать одновременно с одним или двумя другими. Так, нейтрофилию
вследствие стрессорного роста содержания кортикостероидов в циркулирующей
крови обуславливают ускоренное высвобождение гранулоцитов костным
мозгом, снижение выхода полиморфонуклеаров в ткани и умеренное увеличение
пула циркулирующих нейтрофилов. Биологический смысл этой реакции
понятен. Это один из элементов неспецифической системной защитной (стрес-
сорной) реакции, в ходе которой организм, готовясь к возможному
повреждению, быстро создает в крови мобильный «отряд» циркулирующих с ней
полиморфонуклеаров. Эти нейтрофилы после активации готовы как
клеточные эффекторы острого воспаления уничтожить патогенные
микроорганизмы и (или) свои омертвевшие ткани и локализовать очаги инфекции.
Наиболее частая причина длительной нейтрофилии - инфекция.
Инфекция как причина острого воспаления вначале увеличивает число
пристеночных нейтрофилов в крови и усиливает их выход в интерстиций. Это
временно снижает пул нейтрофилов, циркулирующих с кровью (псевдонейт-
ропения). Затем следует быстрое увеличение образования нейтрофилов
костным мозгом. При этом скорость образования гранулоцитов начинает
преобладать над интенсивностью их выхода в ткани из сосудистого русла
при воспалении. В результате возникает нейтрофилия. При длительной и
тяжелой инфекции способность костного мозга генерировать полиморфо-
нуклеары падает, что ведет к нейтропении истощения. Нейтропения
истощения при тяжелой гнойной бактериальной инфекции - крайне
неблагоприятный прогностический признак. При нейтропении истощения не только
падает генерация гранулоцитов костным мозгом, но и уменьшается время
их полужизни в циркулирующей крови.
Обычно нейтрофилию вызывают острые бактериальные инфекции
(возбудители: Pneumococcus, Streptococcus, Staphylococcus), а также инвазия в
организм патогенных грибов и вирусов. Некоторые инфекции, как
бактериальные, так и паразитарные, не всегда приводят к нейтрофилии.
Если стимулом для нейтрофилии выступает злокачественный
клеточный рост, то нейтрофилия может приобретать хронический характер. При
серповидноклеточной анемии нейтрофилию выявляют в течение всей
жизни больного.
Острое воспаление любой этиологии и обострение хронического
воспаления могут приводить к нейтрофилии.
Из других наиболее частых патологических состояний и болезней,
вызывающих нейтрофилию, следует выделить уремию, диабетические кетоа-
цидоз, кому, токсемию, связанную с беременностью, желтухой и
алкогольным циррозом печени, отравления ядом змей и насекомых, тяжелыми
металлами, лекарствами и химикалиями.
Лейкемодиная реакция - это патологически интенсивная
политональная пролиферация лейкоцитов, которую вызывает рост секреции колоние-
стимулирующих факторов в ответ на травму, нарушения обмена веществ,
побочное действие лекарств, воспаление и злокачественный клеточный
рост, а также другие патологические состояния и болезни.
Воспаление
177
При лейкемоидной реакции общее содержание лейкоцитов в крови
больше, чем 25хЮ-/л, но меньше, чем 50х109/л. Лейкемоидная реакция - это
результат нормальной реакции костного мозга на усиление секреции колони-
естимулирующих факторов. При лейкемоидной реакции вследствие крайнего
напряжения нормального механизма развития лейкоцитоза через усиление
образования клеток костным мозгом в крови появляются такие незрелые
формы лейкоцитов как метамиелоциты и миелоциты. Общее число палоч-
коядерных нейтрофилов, циркулирующих с кровью, растет и становится
выше, чем 5 % от общего содержания лейкоцитов в крови, что характерно
для длительной лейкемоидной реакции, связанной с очагом бактериальной
гнойной инфекции. Лейкоцитоз при лейкемоидной реакции - это в
основном нейтрофилия. Патогенно интенсивное образование нейтрофилов при
лейкемоидной реакции ведет к появлению их токсической зернистости.
Вакуолизация нейтрофилов при лейкемоидной реакции свидетельствует об
интенсивном фагоцитозе бактерий фагоцитами крови. У больных с
лейкемоидной реакцией в плазме крови определяют повышенную активность
лейкоцитарной щелочной фосфатазы, чего нет при лейкемиях.
Рост числа нейтрофилов, циркулирующих в кровью, вследствие
увеличения при стрессе содержания в крови кортизола и катехоламинов
называют перераспределительной нейтрофилией, то есть обусловленной
увеличением пула циркулирующих нейтрофилов и усилением их выхода из
костного мозга без усиления миелопоэза. Перераспределительная
нейтрофилия, характеризующая начальный этап острой воспалительной
реакции, в дальнейшем по мере активации в очаге воспаления моноцитов
циркулирующей крови, их трансформации в долгоживущие тканевые
макрофаги и после установления тесных межклеточных взаимодействий
между макрофагами в локусе воспаления и находящимися в нем
лимфоцитами сменяется регенераторной нейтрофилией. Регенераторную нейт-
рофилию обуславливает в основном воздействие на миелопоэз в костном
мозге колониестимулирующих факторов мононуклеарных макрофагов и
иммунокомпетентных клеток.
Колониестимулирующие факторы (КСФ) - это гликопротеины, которые
влияют на образование, дифференциацию и функции гранулоцитов и
клеток системы мононуклеарных фагоцитов. Гранулоцитарный КСФ,
кодируемый геном 17 хромосомы, образует эндотелиальные клетки, фибробласты
и макрофаги. Этот КСФ стимулирует образование гранулоцитов.
Одновременно вместе с интерлейкином-3 гранулоцитарный КСФ увеличивает
содержание в циркулирующей крови мегакариоцитов и юных на пути
клеточной дифференциации форм гранулоцитов-макрофагов. Гранулоцитарно-
макрофагальный КСФ, чья структура закодирована в гене 5 хромосомы,
образуют эндотелиальные клетки, фибробласты и фагоциты. Он
стимулирует образование гранулоцитов и макрофагов. Ген КСФ-5 находится в 5
хромосоме. КСФ-5 образуют и секретируют эндотелиоциты, фибробласты и
макрофаги. Его биологическое действие повышает содержание моноцитов
в циркулирующей крови. Интерлейкин-3 называют мульти-КСФ. Его ген
также находится в 5 хромосоме. Интерлейкин 3 образуется Т-лимфоцитами.
178
Глава 12
Он стимулирет образование гранулоцитов, макрофагов, эозинофилов, а также
усиливает пролиферацию тучных клеток.
Появление в циркулирующей крови юных форм нейтрофилов как
правило свидетельствует о сохранении очага гнойной инфекции как локуса
персистирующего воспаления. Более полувека для выявления
инфекционных осложнений хирургических вмешательств используют предложенный
в 1941 году Я.Я. Кальф-Калифом лейкоцитарный индекс итоксикации (ЛИИ):
[(4Ми+ЗЮ+2П+С)(Пл+1)]: [(Л+Мо)(Э+1)],
где Пл, Ми, Ю, П, С, Л, Мо, Э - соответственно процентное содержание
плазматических клеток, миелоцитов, юных, палочкоядерных, сегментоядер-
ных нейтрофилов, лимфоцитов, моноцитов, эозинофилов.
Рост ЛИИ выше 1,1 у хирургических больных обычно свидетельствует
о воспалении, протекающем в очаге острой гнойной инфекции.
ФУНКЦИОНИРОВАНИЕ НЕЙТРОФИЛОВ
КАК КЛЕТОЧНЫХ ЭФФЕКТОРОВ ОСТРОГО ВОСПАЛЕНИЯ
Уже давно известна главная функция полиморфонуклеарных лейкоцитов
(нейтрофилов, полиморфонуклеаров) - поиск, поглощение и уничтожение
через биохимическое расщепление чужеродных веществ, частиц и
микроорганизмов. Полиморфонуклеары как клеточные эффекторы воспаления могут
поглощать и подвергать расщеплению остатки омертвевших клеток
(продукты цитолиза). Функционирование нейтрофилов как эффекторов острого
воспаления может разрушать ткани организма, клетки которых переродились в
такой степени, что система иммунитета уже не признает их «своими».
При ряде критических состояний и типовых патологических процессов
(сепсис, состояние после тяжелых ранений и травм, тяжелая респираторно-
циркуляторная гипоксия, ишемия, распространенные нарушения
периферического кровообращения как причина «полиорганной» недостаточности
и др.) системная адренергическая стимуляция, потерявший защитный
характер интенсивный выброс цитокинов-флогогенов активированными
макрофагами и иммунокомпетентными клетками, активация эндотелиоцитов и
рост экспрессии их тромбогенного потенциала, а также замедление
кровотока на периферии вызывают во многих органах в той или иной степени
выраженное воспаление, не имеющее биологической цели. При воспалении,
лишенном биологической цели, то есть защитно-приспособительного
значения, функционирование нейтрофилов как основного клеточного
эффектора воспаления часто происходит без эндоцитоза, но приводит к
повреждениям тканей и разрушению структурно-функциональных единиц органов.
Все сказанное о нейтрофилах как о клеточных эффекторах острого
воспаления относится и к клеткам, составляющим систему мононуклеарных
фагоцитов, которые также могут выступать в роли эффекторов воспаления.
Воспаление
179
Таблица 12.1
Этапы функционирования нейтрофилов как клеточных
эффекторов острого воспаления
Миграция полиморфонуклеаров
до объекта эндоцитоза
Марги нация
Адгезия нейтрофилов к эндотелио-
цитам
Агрегация
Диапедез
Ориетированная миграция в
соответствии с градиентом
концентрации хемоаттрактантов
Эндоцитоз и уничтожение
объекта
Распознавание
Адгезия
Эндоцитоз
Слияние гранул и вакуоли с образованием
фагосомы и дегрануляция
Респираторный взрыв с одновременным
внутриклеточным уничтожением
микроорганизма и биохимическим расщеплением
объекта фагоцитоза
Экзоцитоз
Функционирование нейтрофилов как клеточных эффекторов острого
воспаления складывается из двух основных этапов (табл. 12.1).
Эффективная воспалительная реакция в ответ на инфицирование и некробиотичес-
кие изменения тканей невозможна без локальной аккумуляции лейкоцитов.
Ключевым этапом аккумуляции лейкоцитов в очаге острой воспалительной
реакции является адгезия (лат. adhaesio - прилипание) лейкоцитов к эндо-
телиальным клеткам. Адгезия зависит от появления и содержания на
поверхности эндотелиальных клеток и нейтрофилов эндотелиально-лейкоцитар-
ных адгезивных молекул (ЭЛАМ). При этом определенная эндотелиально-
лейкоцитарная адгезивная молекула на поверхности эндотелиоцита
представляет собой лиганду к рецептору на наружной мембране нейтрофила в
виде комплементарной ЭЛАМ, и наоборот.
В частности к ЭЛАМ относят селектины. Выделяют два вида
селектинов, L- и Е-селектины, которые присутствуют на плазматической мембране
нейтрофилов и других фагоцитов. Функция селектинов эндотелиоцитов -
это распознавание и связывание углеводных соединений (гликоконьюгатов)
на поверхности нейтрофилов. Вторая разновидность ЭЛАМ на
поверхности нейтрофилов - это сходные по строению с иммуноглобулинами
молекулы (эндотелиальные межклеточные адгезивные молекулы 1 и 2). Общее
название для второго после селектинов вида поверхностных рецепторов
нейтрофила, ответственных за их адгезивные свойства, - интегрины.
Идентифицировано пять видов интегринов. Наибольшее значение для адгезии
как для этапа функционирования нейтрофила в качестве клеточного
эффектора воспаления из интегринов имеют три гетеродимера, состоящие из
общей бета- и трех альфа-цепей.
Рост содержания цитокинов в циркулирующей крови (фактор некроза
опухолей, интерлейкины и др.) вследствие патогенной системной
активации мононуклеарных фагоцитов и иммунокомпетентных клеток при
сепсисе и системной воспалительной реакции (СВР) вызывает экспрессию ЭЛАМ
на поверхности эндотелиальных клеток и на наружной мембране
нейтрофилов и моноцитов. При нарушениях микроциркуляции как неизбежном
180
Глава 12
элементе СВР, тяжелых травматической и раневых болезней, при которых
гиперцитокинемия выступает звеном патогенеза системного
патологического раневого процесса, экпрессия ЭЛАМ приводит к адгезии лейкоцитов
циркулирующей крови к эндотелиальным клеткам. Адгезия активирует эн-
дотелиоциты как клеточные эффекторы воспаления. Они высвобождают
флогогены-хемоаттрактанты и вместе с активированными лейкоцитами
запускают острую воспалительную реакцию в органах и тканях, удаленных
от первичного локуса воспаления. Такое лишенное защитного значения
воспаление приобретает чисто патогенный характер и служит одной из причин
множественной системной недостаточности.
С участием селектинов происходит неокончательная адгезия нейтрофи-
лов к эндотелию. При этом они как бы катятся по поверхности эндотелио-
цита при возвратнопоступательном движении лейкоцитов на внутренней
поверхности стенки сосуда. Интегрины осуществляют «твердое»
прикрепление лейкоцитов к эндотелию. Нарушение адгезивных свойств
фагоцитирующих лейкоцитов, в частности связанное с низкой экспрессией на их
поверхности ЭЛАМ, ведет к неспособности лейкоцитов накапливаться в очаге
воспаления и уничтожать в нем все чужеродное. В результате возникают
частые гнойные инфекции. Это характерно для двух наследственных
заболеваний, при которых недостаточна экспрессия ЭЛАМ на поверхности
лейкоцитов, для синдромов недостаточности адгезии лейкоцитов I и II. При
синдроме недостаточности адгезии I на поверхности лейкоцитов нет или
недостаточно содержание общей бета-цепи интегринов, без чего
невозможна нормальная экспрессия всех гетеродимеров-интегринов на наружной
клеточной поверхности. При синдроме недостаточности адгезии
лейкоцитов II наружная поверхность эндотелиальных клеток не содержит лиганды
к интегринам как к рецепторам наружной поверхности лейкоцитов. Кроме
того, адгезивные свойства лейкоцитов с фагоцитарными функциями
снижают такие генетически детерминированные заболевания фагоцитарной
системы как болезнь накопления гликогена и дефицит специфических
гранул нейтрофилов. Одновременное снижение способности нейтрофилов к
хемотаксису и падение их адгезивной способности происходит вследствие
многих хронических грибковых, вирусных и бактериальных инфекций.
После выхода в интерстиций нейтрофил перемещается в направлении к
большему содержанию в нем медиаторов воспаления, которые называют
хемоаттрактантами. Иными словами, хемотаксис полифорфонуклеара к
объекту эндоцитоза происходит в соответствии с градиентами
концентраций хемоаттрактантов. Хемотаксис - это ориентированное движение
микроорганизмов, растений и животных, а также отдельных клеток под
влиянием химических веществ.
Хемоаттрактанты, кроме свойства обуславливать движение полиморфо-
и мононуклеаров, обладают способностью индуцировать адгезию
нейтрофилов к эндотелию, усиливать их краевое стояние и вызывать агрегацию
полиморфонуклеаров в просвете микрососудов. При воспалении в
значительных массивах некробиотически измененных тканей организм не может
локализовать очаг острой воспалительной реакции. В результате ее медиа-
Воспаление
181
торы-хемоаттрактанты выходят в циркулирующую кровь. Это ведет к
системной адгезии полиморфонуклеаров к эндотелиоцитам, особенно
выраженной на посткапиллярном уровне. Дело в том, что эндотелиальные
клетки, выстилающие просвет венул, обладают среди всех эндотелиоцитов
наибольшей адгезивной способностью. Распространенные в пределах всего
организма адгезия нейтрофилов к эндотелиоцитам и агрегация
полиморфонуклеаров в микрососудах могут быть звеньями патогенеза множественной
системной («полиорганной») недостаточности. Одновременно во многих
органах и тканях под влиянием поступивших из очага воспаления и из
вторично активированных мононуклеаров и иммунокомпетентных клеток в
циркулирующую кровь цитокинов (фактор некроза опухолей, интерлейкин-1 и
др.) растет экспрессия ЭЛАМ на поверхности эндотелиоцитов и
лейкоцитов. У больного с такой реакцией иммунной системы, как это бывает в
первые-третьи сутки после тяжелых травм, во внутренних органах
сохраняются нарушения периферического кровообращения при точечном цитолизе.
Цитолиз ведет к высвобождению в интерстиций циклических нуклеотидов,
обладающих свойствами эндогенных хемоаттрактантов. Все это вызывает
воспаление, направленное на уничтожение собственных омертвевших
клеток и замещение дефектов ткани. Вторичная альтерация при таком во
многом лишенном биологического смысла воспалении разрушает структурно-
функциональные элементы органов эффекторов функций, вызывая
множественную системную недостаточность. Биологический смысл наличия у
циклических нуклеотидов свойств хемоаттрактантов - это индукция ими
острой воспалительной реакции, которая после высвобождения
циклических нуклеотидов погибшими клетками, направлена на уничтожение
организмом собственных нежизнеспособных клеточных элементов.
Функция эндогенных хемоаттрактантов есть и у фракций системы
комплемента, в особенности у С5а. Через хемоаттрактивные свойства своих
фракций система комплемента, атакующая после активации патогенные
микроорганизмы, усиливает неспецифическую защитную реакцию
иммунной системы, вызывая острое воспаление. Если некробиотические
изменения тканей приводят к активации системы комплемента по
альтернативному пути, то фракции системы комплемента как хемоаттрактанты вызывают
острое воспаление, направленное на уничтожение омертвевших клеток и
элиминацию их остатков.
Свойствами хемоаттрактантов обладают и иммуноглобулины,
образующие на поверхности объектов эндоцитоза комплексы антиген-антитело.
Благодаря функционированию хемоаттрактантов, через неспецифическую
воспалительную реакцию усиливается специфический иммунный ответ.
Бактериальный эндотоксин, взаимодействуя с клетками иммунной
системы, усиливает высвобождение ими цитокинов-хемоаттрактантов, что
служит одним из звеньев патогенеза системной воспалительной реакции.
Лизосомальные ферменты, в том числе высвобождаемые
активированными полиморфонуклеарами, при воздействии на сыворотку крови и стро-
му органов и тканей, приводят к образованию в них эндогенных
хемоаттрактантов.
182
Глава 12
Экзогенными хемоаттрактантами являются пептиды бактериального
происхождения, в особенности те, которые содержат Л/-формиловые группы.
Свойствами хемоаттрактантов обладают кинины и активированный
фактор Хагемана. Известно, что повреждение (активация) эндотелия при
первичной альтерации ведет к мгновенной аутоактивации фактора Хагемана
на поверхности эндотелиальных клеток. В дальнейшем в локусе первичной
альтерации происходят связанные друг с другом активация фактора
Хагемана, кининогенез и активация системы комплемента по альтернативному
пути. Таким образом, активация эндотелия вследствие первичной
альтерации приводит к резкому возрастанию содержания хемоаттрактантов в
поврежденных тканях.
После взаимодействия хемоаттрактантов со своими рецепторами на
поверхности нейтрофилов и активированных моноцитов, их хаотическое движение
прекращается. Фагоциты начинают ориентированно перемещаться по
направлению к объекту эндоцитоза в соответствии с градиентами концентраций
хемоаттрактантов, то есть становятся ориентированными. Взаимодействие
хемоаттрактантов со своими рецепторами приводит к гиперполяризации наружной
клеточной мембраны нейтрофилов и увеличивает вхождение в них кальция.
Кроме того, в нейтрофилах возрастает скорость синтеза функционально
активных фосфолипидов и образование циклических нуклеотидов с функциями
вторичных мессенджеров. Как и после адгезии к эндотелиоцитам, при
ориентированной миграции в полиморфонуклеарах растут число и размеры
внутриклеточных органелл (микроскопических канальцев и нитей),
функционирование которых обеспечивает перемещение фагоцита.
Рост содержания в очаге воспаления кортизола тормозит
ориентированный хемотаксис нейтрофилов. Гиперкортизолемия, тормозящая миграцию
ориентированных полиморфонуклеаров, направлена на предотвращение
трансформации воспаления из защитной в патологическую реакцию. Ги-
перкортизолемию в частности обуславливает супрасегментарный эффект
на системы нейроэндокринной регуляции таких медиаторов воспаления как
цитокины фактор некроза опухолей и интерлейкин-1.
Эндоцитоз - это процесс захвата клеткой микроорганизмов, твердых
и жидких частиц через инвагинацию вокруг них части наружной
клеточной мембраны с образованием вакуолей или везикул, которые в
последующем продвигаются внутрь клетки.
Эндоцитоз жидких частиц называют пиноцитозом, а твердых -
фагоцитозом.
Необходимое условие фагоцитоза - это адгезия микроба, или
фагоцитируемой частицы на поверхности нейтрофила или макрофага. Адгезия как
этап в филогенетическом отношении древней воспалительной
неспецифической реакции происходит через взаимодействие углеводных остатков,;
входящих в состав наружной клеточной мембраны микроба и адгезивных;
молекул плазматической мембраны фагоцита. Иными словами, в основе;
неспецифической иммунной адгезии лежит взаимодействие рецепторов,
поверхности фагоцита, которые обладают свойством связываться с
элементами наружной клеточной мембраны, общими для многих микробов.
Воспаление
183
Адгезия активирует актин-миозиновую систему фагоцита, что приводит к
обхвату фагоцитируемой частицы или клетки выпячиваниями наружной
клеточной мембраны, псевдоподиями. По мере присоединения друг к
другу близлежащих рецепторов наружной клеточной мембраны фагоцита
плазматическая мембрана надвигается на фагоцитируемую клетку или
частицу до тех пор, пока частица не будет полностью заключена в
вакуоль, которую называют фагосомой. В течение минуты после образования
фагосомы цитоплазматические гранулы сливаются с ней и изливают в нее
свое содержимое. Заключенный в фагосому микроорганизм подвергается
действию целого ряда бактерицидных механизмов, связанных с
действием содержимого гранул полиморфонуклеаров. Действие этих механизмов
приводит к биохимической деградации частиц, перемещенных в ходе эн-
доцитоза внутрь клетки.
Адгезию нейтрофилов и других фагоцитов к объекту фагоцитоза
ускоряют и усиливают биоактивные молекулы, получившие название опсонинов
(греч. opsonion - снабжение пищей). Опсонинами могут быть
иммуноглобулины. Через синтез и секрецию иммуноглобулинов, которые как антитела
специфичны по отношению к антигенам объектов эндоцитоза, первичный и
(или) вторичный иммунный ответ с участием иммунокомпетентных клеток
повышает эффективность защитной реакции острого воспаления. При
иммунной адгезии относительно стабильный по структуре фрагмент молекулы
иммуноглобулина Fc связывается со своим рецептором на поверхности
фагоцита. Вариабельный по структуре фрагмент молекулы иммуноглобулина Fab
фиксируется на объекте фагоцитоза. Таким образом, осуществляется
связывание активированного полифорфонуклеара с объектом эндоцитоза.
Иммуноглобулины - это специфические опсонины, которые
соединяются с одним определенным антигеном на поверхности объекта
фагоцитоза. Если причиной воспаления служит инвазия в организм бактерий,
поверхностные антигены которых в прошлом уже стимулировали систему
иммунитета, то эффективность защитной реакции острого воспаления
особенно велика. Её повышает специфический вторичный иммунный
ответ, который в частности состоит в быстро нарастающей секреции имму-
нокомпетентными клетками комплементарных к антигенам бактерий им-
муноглобулинов-опсонинов.
Способность нейтрофилов и моноцитов к эндоцитозу снижают
некоторые острые и хронические инфекционные заболевания и аутоиммунные
патологические процессы. Падение способности полиморфонуклеаров и
мононуклеарных фагоцитов к эндоцитозу может быть связано со
снижением содержания иммуноглобулинов-опсонинов в сыворотке крови. Это
обычное следствие врожденного или приобретенного В-клеточного
иммунодефицита. В частности это бывает у больных с хроническим сепсисом, при
котором избыточная стимуляция клональной экспансии
иммунокомпетентных клеток приводит к приобретенному иммунодефициту с недостатком
иммуноглобулинов-опсонинов в сыворотке крови.
Следующим этапом уничтожения чужеродного в ходе острого
воспаления через эндоцитоз является дегрануляция. Она заключается в слиянии
184
Глава 12
фагосомы, содержащей объект эндоцитоза, с лизосомами и в образовании
фаголизосомы. В фаголизосоме происходят уничтожение и деструкция
объекта. Первым в фагосому попадает содержимое специфических
(вторичных гранул), содержащих лизоцим, лактоферрин, белок, связывающий
витамин В12 и другие вещества с прямым или опосредованным
бактерицидным действием. Затем в фагосому попадает содержание первичных
(азурофильных гранул), которые содержат ряд гидролаз.
Уничтожение объектов эндоцитоза, которое в случае фагоцитоза
патогенных микроорганизмов составляют прекращение их
жизнедеятельности и биохимическая деградация, происходит через действие кислородза-
висимых и кислороднезависмых механизмов. При действии кислородза-
висимых механизмов происходит окисление кислорода НАДФ Н-оксидаз-
ной системой, образующей активные формы кислорода, обладающие
сильным бактерицидным действием. Кислороднезависимое уничтожение
объекта фагоцитоза происходит вследствие высокой концентрации
протонов в фаголизосоме, активности в ней гидролаз, а также эффекта
бактерицидных протеинов и пептидов. Среди них своим бактерицидным
действием выделяются дефензины, то есть белки со структурой, насыщенной
аргинином, которые обладают способностью встраиваться в липидный бис-
лой наружной клеточной мембраны, вызывая резкий рост ее
проницаемости в участке внедрения для обычно не мигрирующих через
мембрану осмолей. В результате гипергидратация клеток служит
причиной их цитолиза.
Процесс эндоцитоза и уничтожения его объекта происходит
одновременно и взаимозависимо с респираторным взрывом. Под респираторным
взрывом понимают резкое увеличение потребления кислорода для
образования бактерицидных свободных кислородных радикалов со снижением
напряжения (парциального давления) кислорода в клеточном эффекторе
воспаления, осуществляющем эндоцитоз. При этом значительно снижается
напряжение кислорода в его цитозоле. Стимулом для респираторного
взрыва служит контакт с объектом фагоцитоза или высокие концентрации хе-
моаттрактантов в очаге воспаления. Образование свободных кислородных
радикалов при респираторном взрыве может вызвать цитолиз самого
фагоцита и служит фактором вторичной альтерации, в том числе и потерявшей
приспособительно-защитное значение. Индукция респираторного взрыва
обычно происходит одновременно с дегрануляцией. Начальный этап
респираторного взрыва- это потеря одного электрона молекулярным
кислородом (02), что превращает его в супероксид 02\ Это происходит через
активность связанной с клеточными мембранами оксидазы. Другая основная
инициирующая реакция респираторного взрыва - это соединение протонов,
высвобождаемых при реакции гексозомонофосфатного шунта, с
кислородом, что образует перекись водорода. Образование свободных
кислородных радикалов, характеристика которых дана в главе, посвященной ишеми-
ческой болезни сердца, при респираторном взрыве фагоцитов в очаге
воспаления протекает через спонтанную дисмутацию и с участием
ферментов миелопероксидазы, супероксиддисмутазы и каталазы.
Воспаление
185
КЛЕТОЧНЫЕ ЭФФЕКТОРЫ И МЕДИАТОРЫ ВОСПАЛЕНИЯ
Клеточные эффекторы воспаления (флогогены) - это клетки,
реализация взаимодействий между которыми, вызывает в тканях местную острую
воспалительную реакцию и системные сдвиги регуляции, обмена веществ
и функций, составляющие реакцию острой фазы и системную
воспалительную реакцию.
Медиаторы воспаления - это вещества, обладающие определенными
функциями, которые, отчасти представляя собой агенты ауто- и пара-
кринной регуляции и реализуя межклеточные взаимодействия, способны
вызывать в тканях компоненты острой воспалительной реакции.
Многие из флогогенов (медиаторов воспаления) вызывают компоненты
острой воспалительной реакции, активируя ее основные клеточные
эффекторы, полиморфонуклеарные лейкоциты. Роль нейтрофилов как ключевого
клеточного эффектора острого воспаления особенно видна при острой
воспалительной реакции, вызванной образованием в тканях иммунных
комплексов антиген-антитело. Известно, что воспаление, инициирующим
моментом которого является образование иммунных комплексов, значительно
ослабевает при нейтропении, то есть при снижении содержания
нейтрофилов в циркулирующей крови. Иммунные комплексы запускают воспаление
через нейтрофилы, наружная поверхность которых содержит рецепторы к
относительно стабильной по структуре части молекулы иммуноглобулинов
(Fc-фрагмент). Взаимодействие соответствующих рецепторов наружной
поверхности нейтрофилов с Fc-фрагментом активирует полиморфонуклеа-
ры. Активированные нейтрофилы выделяют массу флогогенов, в частности
обладающих свойством вызывать тромбоз микрососудов. Тромбоз сосудов
через ишемию вызывает быстрый некроз прилежащих к обтурированным
сосудам тканей и формирование полиморфонуклеарных инфильтратов в
очагах их некробиотических изменений.
Эволюционно воспаление формировалось в первую очередь как
неспецифическая защитная реакция уничтожения потенциально патогенных
микроорганизмов после их проникновения во внутреннюю среду организма.
Сам по себе богатый арсенал фагоцитов, фиксированных в тканях и
циркулирующих с кровью, еще не гарантирует надежной защиты организма от
болезнетворных бактерий, попадающих во внутреннюю среду из внешней,
и эффективных уничтожения и элиминации из внутренней среды всего
чужеродного посредством эндоцитоза. Для того, чтобы при воспалении
произошел эндоцитоз, необходимы: а) сближение фагоцита и объекта
фагоцитоза (патогенного микроорганизма в том числе); б) адгезия микроорганизма
на поверхности фагоцита; в) активация плазматической мембраны
фагоцита как инициирующий момент эндоцитоза. Некоторые из патогенных
микроорганизмов в качестве потенциальных объектов эндоцитоза на своей
поверхности содержат лиганды к поверхностным рецепторам нейтрофилов,
другие высвобождают хемоаттрактанты и биоактивные вещества,
активирующие наружную мембрану фагоцита. Если чужеродный потенциальный
186
Глава 12
объект эндоцитоза не обладает такими свойствами, то в дело вступает
система комплемента, функционирование которой реализует три необходимых
условия эндоцитоза при воспалении.
Комплементом называют белковый комплекс, состоящий примерно из 20
протеинов, которые присутствуют в плазме крови и в интерстиции (фракции
системы комплемента). Больше всего в сыворотке крови содержится
фракции системы комплемента СЗ (1,2 г/л). Активация системы комплемента -
это алгоритмизированный процесс, состоящий из ряда последовательных
реакций, продукт каждой из которых служит катализатором последующего
этапа активации (следующей реакции). Поэтому для системы комплемента
характерен быстрый многократно усиленный ответ на действие первичного
стимула ее активации. Это частное проявление общей биологической
закономерности потенциально патогенной избыточности защитных реакций в
сравнении с силой действия стимулов, их вызывающих. Данная закономерность
определяет способность защитных реакций к превращению в звенья
патогенеза болезней, патологических процессов и состояний. Так избыточная и на
уровне всего организма активация системы комплемента является одним из
звеньев патогенеза системной воспалительной реакции, разрушающей
структурно-функциональные единицы эффекторов функциональных систем.
В физиологических условиях в плазме крови и интерстиции происходит
постоянное и очень медленное расщепление СЗ с образованием его
продукта СЗЬ или аналогичной по функциям молекулы. В присутствии ионов
магния СЗЬ, соединяясь с другим элементом системы комплемента фактором
В, образует молекулярный комплекс. В составе молекулярного комплекса
фактор В расщепляется одним из ферментов плазмы крови фактором D. В
результате образуется молекулярный комплекс СЗЬВЬ, представляющий
собой «СЗ-конвертазу», способную расщеплять СЗ на СЗа и СЗЬ. После
образования СЗ-конвертазы возможно быстрое и неограниченное
образование СЗЬ из СЗ, так как отщепление СЗЬ от СЗ - это начальный этап
образования конвертазы. В физиологических условиях этот цикл биохимических
реакций блокируется через замещение в составе конвертазы фактора В
фракцией системы комплемента фактором Н. В результате молекулярный
комплекс становится доступным для взаимодействия с фактором I, инактивиру-
ющим СЗЬ. Инактивированный СЗЬ в дальнейшем расщепляется трипсино-
подобными ферментами.
СЗЬВЬ-конвертаза обладает способностью связываться с углеводными
элементами наружной клеточной мембраны некоторых микроорганизмов.
Фиксация на поверхности микроорганизмов стабилизирует ее, то есть
делает невозможным замещение в ее составе фактора В фактором Н.
Стабилизацию конвертазы усиливает другой белок плазмы крови,
присутствующий в интерстиции, - пропердин. Связанная с поверхностью
микроорганизма конвертаза отщепляет от СЗЬВЬ фракцию СЗЬ. СЗЬ ковалентно
связывается с наружной мембраной микроорганизма. Таким образом, одна
фиксированная на наружной клеточной поверхности СЗЬВЬ-конвертаза
позволяет связаться с микроорганизмом большому количеству молекул СЗЬ.
СЗЬ и ее неактивная форма, фиксированные на поверхности микроорганиз-
Воспаление
187
мое, представляют собой флогогены с функцией опсонинов, которые
обуславливают адгезию микроорганизмов на поверхности фагоцита и актива-
цию плазматической мембраны фагоцита как инициирующий момент эн-
доцитоза. Адгезия фагоцитов и объектов фагоцитоза происходит через
связывание СЗЬ и ее неактивной формы со своими рецепторами на поверхности
полиморфонуклеаров и макрофагов.
С фиксацией множества молекул СЗЬ на поверхности фагоцитов
завершается альтернативный путь активации системы комплемента, который
также называют пропердиновым. Первичная альтерация тканей под
воздействием экзогенного этиологического фактора физико-химической природы
разрушает защитное покрытие наружных клеточных мембран,
преимущественно состоящее из слоя сиаловых кислот. Это делает возможным
альтернативную активацию системы комплемента на поверхности поврежденных
клеток, которая также состоит в стабилизации на их поверхности СЗЬВЬ-
конвертазы с участием пропердина. В результате система комплемента
атакует некробиотически измененные клетки своего организма, что
обуславливает их цитолиз. Поверхность продуктов цитолиза, в том числе фрагментов
плазматической и цитоплазматических мембран, также пригодна для
активации комплемента по пропердиновому пути. Активация системы
комплемента по альтернативному пути в очаге некробиотических изменений в
основном происходит на поверхности поврежденных межклеточных структур,
эритроцитов, попавших после повреждения микрососудов в интерстиции, на
микротромбах и сгустках фибрина, которые все способны связывать СЗЬ,
которая в свою очередь служит субстратом для образования СЗЬВЬ-конвертазы.
Таким образом, в очаге некробиотических изменений, связанных с
первичной альтерацией, происходит почти полная активация всех поступивших в
него неактивных протеинов, составляющих систему комплемента.
Функция системы комплемента состоит не только в атаке
патогенных микроорганизмов и своих переродившихся и ставших чужеродными
клеток. Активация комплемента в ответ на первичную альтерацию или
инфицирование по пропердиновому пути индуцирует острое воспаление как
неспецифическую реакцию уничтожения и элиминации всего чужеродного
в основном через функционирование фагоцитов крови и тканей. При этом
фракции системы комплемента выступают в роли флогогенов.
Специфический иммунный ответ сенсибилизированного определенным
иммуногеном организма также усиливается острой воспалительной
реакцией, которую вызывает активация системы комплемента по более в
филогенетическом отношении молодому классическому пути (был открыт
позже пропердинового).
Индукция активации системы комплемента по классическому пути
состоит в связывании и активации иммунными комплексами
антиген-антитело на поверхности чужеродного объекта фагоцитоза первой фракции
системы комплемента Clq. Эта фракция системы комплемента в небольших
концентрациях постоянно присутствует в плазме крови и интерстиции. Clq -
это молекула, состоящая из коллагеноподобного стержня, который
разветвляется на шесть пептидных цепочек. Каждая из цепочек оканчивается свя-
188
Глава 12
зывающей антитело субъединицей. Такое строение определяет
поливалентность Clq при связывании антител в составе комплексов антиген-антитело
на поверхности чужеродных объектов фагоцитоза. На поверхности объекта
эндоцитоза Clq связывается с одной молекулой иммуноглобулина М или с
двумя молекулами иммуноглобулина G.
Clq, фиксированный относительно стабильными по структуре
фрагментами молекулы иммуноглобулина Fc на поверхности объекта фагоцитоза,
объединяется с двумя другими элементами системы комплемента, ее
субъединицами ClrnClsB молекулярный комплекс, стабилизируемый ионами
кальция. В составе данного молекулярного комплекса субъединица С1 s
приобретает свойство протеолитического фермента. Протеаза Cls расщепляет
фракцию системы комплемента С4. В результате расщепления фракции
системы комплемента С4 образуются два ее фрагмента, больший С4Ь,
обладающий свойствами протеолитического фермента, и меньший С4а. Фракция
системы комплемента С2 образует комплекс с С4Ь. Этот молекулярный
комплекс представляет собой субстрат для Cls. Под влиянием Cls как энзима
из данного субстрата образуется молекулярный комплекс С4Ь2Ь, который
обладает функциями специфичной СЗ-конвертазы.
В результате расщепления фракции СЗ высвобождается ее субъединица
СЗЬ. Молекула СЗЬ, соединяясь с комплексом С4Ь2Ь, превращает его в
фермент, расщепляющий фракцию С5. При альтернативном пути активации
комплемента ферментом, расщепляющим фракцию системы комплемента
С5, является молекулярный комплекс СЗЬВЬ. В дальнейшем активация
системы комплемента происходит одинаково как по классическому, так и по
альтернативному пути. Механизм блокирования активации системы
комплемента по классическому пути состоит в деградации комплекса С4Ь2Ь под
влиянием С4-связывающего белка или вследствие взаимодействия с СЗЬ-
рецепторами клеточной поверхности в присутствии фактора I.
Фракция системы комплемента С5 расщепляется на короткий пептид
С5а и большую часть С5, C5b. C5b фиксируется на объекте фагоцитоза и
последовательно связывает фракции системы комплемента С6, С7 и С8.
Образованный молекулярный комплекс способствует правильной
ориентации одной или более молекул последней фракции системы комплемента
С9. Это обуславливает развертывание молекул С9 и их проникновение в
липидный бислой наружной мембраны атакуемых микроорганизма или
чужеродной клетки. Полимеризация молекул С9 внутри наружной мембраны
образует в ней кольцеобразный «мембраноатакующий комплекс»,
формирующий канал, по которому незатрудненно в соответствии с градиентами
осмотических концентраций мигрируют осмоли и вода. Высокое
коллоидно-осмотическое давление в клетке приводит к поступлению в нее воды и
натрия, что и служит основным механизмом цитолиза.
Основными функциями системы комплемента и его фракций как
молекулярных эффекторов острого воспаления являются:
♦ фиксация на поверхности потенциальных объектов фагоцитоза
(патогенные микроорганизмы и свои ставшие чужеродными клетки и
антигены);
Воспаление
189
♦ создание градиента концентрации хемоаттрактантов между
просветом прилежащих к объекту эндоцитоза микрососудов и
поверхностью объекта;
♦ активация фагоцитов циркулирующей крови; индукция их адгезии к
эндотелиоцитам в локусе воспаления;
♦ рост проницаемости стенок микрососудов для белков плазмы и
форменных элементов крови;
♦ обеспечение эндоцитоза через функционирование фракций системы
комплемента в качестве опсонинов;
♦ индукция воспаления как реакции уничтожения и элиминации всего
чужеродного после связывания в ходе вторичного иммунного
ответа антител с чужеродными антигенами;
♦ воздействие на тучные клетки в качестве стимула для их дегрануля-
ции с высвобождением флогогенов.
СЗа и С5а - небольшие пептиды, отщепляемые при активации
комплемента от своих молекул-предшественниц, представляют собой флогогены,
обладающие рядом важных функций медиаторов острого воспаления. Они
оказывают непосредственное влияние на фагоциты в просвете сосудов,
прилежащих к локусу нахождения объекта фагоцитоза в интерстиции. СЗа и
С5а активируют фагоциты и особенно нейтрофилы, резко повышая
потребление полиморфонуклеарами кислорода, что связано с усиленным
образованием свободных кислородных радикалов.
Как флогогены СЗа и С5а в первую очередь являются хемоаттрактантами
и индукторами адгезии нейтрофилов и активированных моноцитов к
эндотелию в очаге острого воспаления. Их эффект в качестве медиаторов
воспаления, вызывающих адгезию фагоцитов к эндотелиоцитам и выход фагоцитов в
интерстиции, в основном реализуется через экспрессию на поверхности по-
лиморфонуклеаров, моноцитов и эндотелиальных клеток ЭЛАМ.
Кроме того, СЗа и С5а обладают свойством активировать тучные клетки
и базофилы, высвобождающие целый ряд флогогенов. Активация тучной
клетки приводит к высвобождению ею медиаторов воспаления по двум
основным путям:
♦ выброс в интерстиции ранее образованных медиаторов из гранул
тучной клетки;
♦ образование из арахидоновой кислоты и ее метаболитов медиаторов
воспаления и высвобождение новосинтезированных флогогенов во
внеклеточное пространство (табл. 12.2).
Лейкотриены (ЛТ) - это низкомолекулярные биоактивные молекулы,
которые синтезируются и высвобождаются лейкоцитами, макрофагами,
тучными и другими клетками в ответ на антигенную стимуляцию или действие
иных раздражителей. Синтез ЛТ происходит через окисление
арахидоновой кислоты в положении С-5, в результате которого образуется
промежуточное соединение лейкотриен А4 (ЛТА4) Катализатором этой реакции
является 5-липооксигеназа. Удаление из состава ЛТА4 гамма-глютамилового
остатка образует ЛТЭ4. ЛТ04 лишенный гаицина, - это ЛТЕ4. Гидратирова-
ние ЛТА4 приводит к образованию ЛТВ4. Синтез ЛТС4 состоит в добавлении
190
Глава 12
Таблица 12.2
Медиаторы острого воспаления, высвобождаемые в его
очаге тучными клетками
Медиаторы
Функционирование в качестве флогогенов
Образованные тучными клетками до начала острого воспаления
Гистамин I Расширение артериол и спазм венул, увеличение
гидростатического давления в капиллярах, рост проницаемости
микрососудов на уровне венул через сокращение венупярных эндотелиоци-
тов и образование «зазоров»между ними, интерстициальный отек
Гепарин | Предупреждение тромбоза в микрососудах, несмотря на рост
экспрессии тромбогенного потенциала эндотелиоцитов, для
эффективной мобилизации организменного резерва фагоцитов,
комплемента, других клеточных и молекулярных эффекторов о
строго воспаления
Хемотаксис эозинофилов и нейтрофилов
Хемотаксические
факторы
нейтрофилов и эозинофилов
Фактор,
активирующий тромбоциты
Расширение резистивных сосудов с раскрытием артериоло-
венулярных анастомозов (предположительно) и агрегация
тромбоцитов с биологической целью отграничения очага воспаления
через тромбоз, обусловленный падением обменной скорости
кровотока на постартериолярном уровне и функционированием
агрегатов активированных тромбоцитов
Ново синтезированные производные арахидоновой кислоты
Лейкотриены,
образованные по
пути липооксигеназы
Простагландины
(синтез происходит
через активность
ци кл ооксигеназы)
Тромбоксаны
(синтез происходит
с участием цикло-
оксигеназы)
Спазм бронхов. Раскрытие артериоло-венулярных анастомозов и
расширение резистивных сосудов, рост гидростатического
давления крови в капиллярах, увеличение проницаемости
микрососудов для усиления экссудации как компонента воспаления.
Функционирование в качестве хемоаттрактантов
Расширение микрососудов для мобилизации эффекторов
воспаления и экссудации, функционирование в качестве иммуносупрес-
соров, блокирующих индукцию системной иммунной реакции в
ответ на антигенную стимуляцию в очаге воспаления, в том числе
и эндогенными иммуногенами, высвобождаемые вследствие
первичной альтерации и цитолиза (в основном простагландин Е2)
Спазм и тромбоз микрососудов с биологической целью
изоляции очага воспаления от внутренней среды
глютатиона к молекуле ЛТА4. ЛТА4 образуется в больших количествах нейт-
рофилами и тучными клетками в легких больных бронхиальной астмой. ЛТА4
вызывает сокращение гладкомышечньгх элементов стенок бронхов. ЛТВ4 пред-1
ставляет собой агент аутокринной регуляции активированных нейтрофилов,
функционирующих как клеточные эффекторы воспаления в его очаге. ЛТВ4.
стимулирует адгезию и хемотаксис полиморфонуклеаров, то есть обладает
функциями хемоаттрактанта. Кроме того, действие ЛТВ4на нейтрофилы
усиливает экзоцитоз ими протеолитических ферментов, а также синтез и вые- -
вобождение полиморфонуклеарами свободных кислородных радикалов.
Воспаление
191
ЛТС4, JITD4, ЛТЕ4 и фактор активации тромбоцитов составляют мед-
ленно реагирующую субстанцию анафилаксии.
Патогенно высокий уровень образования и секреции лейкотриенов
представляет собой звено патогенеза заболеваний и синдромов, механизм
развития которых во многом составляет воспаление, лишенное биологической цели:
респираторный дистресс-синдром взрослых, аллергический ринит,
бронхиальная астма, ревматоидный артрит, неонатальная легочная гипертензия и др.
Тромбоксаны - это ряд биоактивных веществ, которые образуются из
арахидоновой кислоты циклооксигеназой. Циклооксигеназа образует из
арахидоновой кислоты простагландин G2, который тромбоксансинтетаза
превращает в наиболее функционально активный тромбоксан А2. Синтез и
высвобождение тромбоксанаэндотелиоцитами растет в очаге острого
воспаления в ответ на первичную альтерацию стенки микрососудов. Под
влиянием тромбоксана А2 в просвете микрососудов зоны острого воспаления
происходит агрегация тромбоцитов. При этом предположительно
тромбоксан действует через торможение активности аденилатциклазы кровяных
пластинок. Одновременно тромбоксан вызывает сокращение гладкомышеч-
ных элементов стенок микрососудов. Биологический смысл секреции
тромбоксана эндотелиоцитами при остром воспалении как причины тромбоза
и спазма микрососудов-это отграничение от внутренней среды зоны пер-
винной альтерации, очага инфекции и изоляция от всего организма очага
злокачественного клеточного роста.
В начальный период острого воспаления его основным клеточным
эффектором со свойствами фагоцита является нейтрофил. Функционирование
полиморфонуклеаров как эффекторов острого воспаления не приводит к
значительным системным последствиям и в основном сводится к эндоцитозу,
бактерицидному действию и высвобождению лизосомальных ферментов и
свободных кислородных радикалов, эффект которых во многом
обуславливает вторичную альтерацию. Первые моноциты и лимфоциты из
циркулирующей крови появляются в очаге воспаления через 3-6 ч от его начала.
Максимума инфильтрация очага воспаления моноцитами и лимфоцитами достигает
во вторые-третьи сутки острой воспалительной реакции. За это время
происходят активация моноцитов и лимфоцитов и установление между ними
функциональных связей, реализуемых через секрецию и действие цитокинов.
Одновременно активированные моноциты превращаются в фиксированные
долгоживущие тканевые мононуклеарные макрофаги. На трансформацию
одного вышедшего из циркулирующей крови моноцита или лимфоцита в
активно функционирующий клеточный эффектор воспаления уходит от двух
до трех дней. В это время функциональная активность моноцитов и
лимфоцитов незначительна, и жизнедеятельность клеток сводится
преимущественно к интенсивному изменению экспрессии генома и связанному с ним
усилению белкого синтеза. В результате клетки приобретают функции клеточных
эффекторов воспаления. Можно считать, что за двое-трое суток острого
воспаления в его очаге формируется функционально связанный комплекс
клеточных эффекторов, моноцитов и лимфоцитов, которые высвобождают
множество цитокинов с функциями флогогенов (молекулярных эффекторов
192
Глава 12
воспаления). Кроме того, функциональный комплекс моноцитов и
лимфоцитов секретирует цитокины, через супрасегментарное действие
вызывающие сдвиги системной регуляции обмена веществ.
Активированные лимфоциты в очаге воспаления высвобождают лим-
фокины-цитокины, которые относительно моноцитов в просвете
микрососудов выступают в качестве флогогенов-хемоаттрактантов.
Активированные лимфоциты секретируют фактор, тормозящий миграцию моноцитов,
задерживающий активированные моноциты в очаге воспаления.
Одновременно лимфоциты высвобождают фактор, активирующий моноциты,
действие которого вызывает превращение моноцитов, вышедших в интерсти-
ций, в долгоживущие тканевые макрофаги. Со вторых-третьих суток
воспаления активированный моноцит становится основным фагоцитом,
функционирующим в его очаге. Фагоцитоз моноцитами приводит к
высвобождению в очаге воспаления функционально связанными макрофагами и
лимфоцитами многих цитокинов-флогогенов: простагландина Е2, лейкот-
риенов, фракций комплемента, фибронектина, прокоагулянтов, интерлей-
кинов, фактора некроза опухолей. Как и полиморфонуклеары,
активированные моноциты секретируют лизосомальные ферменты и свободные
кислородные радикалы.
Действие простагландина Е2 и лейкотриенов как
флогогенов-хемоаттрактантов еще больше усиливает инфильтрацию очага воспаления его
клеточными эффекторами. Одновременно простагландин Е2 расширяет
микрососуды в тканях, прилежащих к очагу воспаления, что усиливает экксудацию
и поступление в очаг воспаления флогогенов и клеточных эффекторов
воспаления из системно циркулирующей крови.
Свободные кислородные радикалы, высвобождаемые моноцитами,
воздействуют на секретирующие их макрофаги в качестве агентов паракрин-
ной регуляции. В результате действия свободных кислородных радикалов
активированные моноциты высвобождают флогогены: простагландины,
фракции системы комплемента, лейкотриены и тромбоксаны.
Деструкция межклеточных тканевых элементов и гибель клеток в
результате вторичной альтерации создают в очаге воспаления значительную
поверхность поврежденных межклеточных элементов и продуктов
цитолиза, на которой происходит активация системы комплемента по
альтернативному (пропердиновому) пути. Это служит одной из причин
распространения в тканях зоны вторичной альтерации и увеличения очага воспаления.
Высвобождение клетками макрофагально-лимфоцитарного
функционального комплекса цитокинов со свойствами прокоагулянтов (фактор,
активирующий тромбоциты, тромбоксаны и др.) ведет к образованию зоны
тромбоза микрососудов в самом очаге воспаления и на его периферии. Это
формирует матрицу в виде «фибринового вала», на которой через
клеточную пролиферацию происходит замещение дефекта тканей,
образовавшегося вследствие первичной и вторичной альтерации.
Тромбоз микрососудов на периферии очага воспаления приводит к
падению в нем напряжения кислорода и к гипоксии активированных
макрофагов. Гипоксия служит стимулом для высвобождения макрофагами факто-
Воспаление
193
ров ангиогенеза и роста фибробластов, которые в очаге воспаления
секретируются и активированными тромбоциты. Действие этих цитокинов
со свойствами факторов клеточного роста приводит к образованию новых
капилляров, прорастающих фибриновый вал, а также стимулирует
пролиферацию фибробластов и образование ими коллагена и коллагеназы,
фермента, изменяющего и стабилизирующего структуру новообразованного
коллагена в межклеточном пространстве. В результате формируется фронт
соединительной ткани, который продвигается к центру очага воспаления,
где напряжение кислорода находится на минимальном уровне,
концентрация лактата максимальна, и наиболее интенсивна секреция гипоксичными
макрофагами факторов клеточного роста. Клеточную пролиферацию в
ответ на острое воспаление через паракринные влияния на макрофаги в его
очаге регулируют и Т-лимфоциты.
Полагают, что некоторые из активированных макрофагов в очаге
воспаления функционируют как клетки, обеспечивающие взаимодействие
антигенов из очага воспаления с иммунокомпетентными клетками, то есть как
антиген-презентирующие клетки, индуцирующие первичную или
вторичную специфическую реакцию системы иммунитета. Такие макрофаги
называют активирующими. Другая популяция макрофагов в очаге
воспаления лишена свойств антиген-презентирующих клеток. Эти макрофаги
называют тормозящими. Их функция состоит в очищении очага
воспаления от нежизнеспособных клеточных и интерстициальных элементов, а
также в предотвращении иммуного ответа на стимуляцию собственными
антигенами организма, которые высвободились при остром воспалении и стали
доступными для взаимодействия с системой иммунитета. Предположительно
в предотвращении системной иммуной реакции на стимуляцию «своими»
антигенами из очага воспаления играет роль кахектин (фактор некроза
опухолей) как цитокин с иммуносупрессивными свойствами, высвобождаемый
активированными макрофагами.
Обычно один медиатор воспаления обладает многими функциями.
Предполагают, что находящийся на клеточной поверхности активирующих
макрофагов интерлейкин-1-1 альфа необходим для презентации антигенов Т-
лимфоцитам. Это очевидно происходит посредством миграции
активирующего моноцита с антигеном на его поверхности в регионарный
лимфатический узел. Интерлейкин-1-1 бета, который активированные
макрофаги в качестве флогогена высвобождают в интерстиций очага
воспаления, вызывает экспрессию ЭЛАМ на поверхности эндотелиоцитов и
лейкоцитов, что усиливает задержку в очаге воспаления его клеточных эффекторов
из циркулирующей крови. Таким свойством обладает и фактор некроза
опухолей. Интерлейкин-1-1 бета вызывает экзоцитоз (высвобождение вне
клетки) фашцитами лизосомальных ферментов и свободных кислородных
радикалов фагоцитами, которые подвергают деструкции объекты эндоцитоза
и нежизнеспособные клеточные элементы, что облегчает их фагоцитоз.
Кроме того, интерлейкин-1 в качестве флогогена вызывает дегрануляцию
тучных клеток с высвобождением медиаторов воспаления. Он также
активирует эндотелиоциты, высвобождающие медиаторы воспаления простациклины
194
Глава 12
и тромбоксаны. Биологический смысл секреции простациклинов
активированными эндотелиоцитами состоит в увеличении объемной скорости
кровотока в микрососудах через их расширение для усиления экссудации и
большего поступления клеточных и молекулярных эффекторов воспаления
в его очаг с циркулирующей кровью. Кроме того, простациклины как
антикоагулянты ограничивают тромбоз микрососудов, вызванный тромбокса-
нами и другими флогогенами-прокоагулянтами (фактор некроза опухолей и
т. д.). Фактор активации тромбоцитов повышает экспрессию на
поверхности фагоцитов рецепторов к фракциям системы комплемента со свойствами
хемоаттрактантов. Одновременно он усиливает секрецию эндотелиальны-
ми клетками в очаге воспаления флогогенов-прокоагулянтов.
В физиологических условиях функция эндотелиоцитов состоит в
поддержании текучести крови через торможение коагуляции и противодействие
адгезии к эндотелиальным клеткам лейкоцитов циркулирующей крови. В ответ
на повреждение эндотелия, связанное с инфекцией или действием
экзогенного фактора физико-химической природы, или вследствие воспаления,
вызванного активацией системы комплемента по классическому пути при аутои-
мунных поражениях тканей, эндотелиоциты активируются и становятся
клеточными эффекторами воспаления. При этом эндотелиоциты вызывают
экссудацию и выход лейкоцитов в интерстиций через высвобождение вазоак-
тивных веществ (простагландин 12), цитокинов, в состав молекулы которых
входит липидное основание (фактор активации тромбоцитов и др.),
активирующих лейкоциты, интерлейкинов со свойствами хемоаттрактантов (интер-
лейкин-8 и др.), а также посредством экспрессии на своей поверхности ЭЛАМ.
Повреждение эндотелия при первичной или вторичной альтерации
ведет к моментальной активации на поверхности поврежденных
эндотелиоцитов фактора Хагемана (ФХ). В дальнейшем активация ФХ связана с
образованием плазменного калликреина. Активный ФХ служит катализатором
образования калликреина из прекалликреина. Калликреин - это фермент,
отщепляющий от высокомолекулярного кининогена брадикинин и другие
кинины. Кроме того, активированный фактор Хагемана, связанный с
отрицательно заряженной поверхностью поврежденного эндотелиоцита,
индуцирует каскад реакций внутрисосудистого свертывания крови. Все это
становится возможным при замедлении линейной скорости кровотока в
просвете микрососуда. При нормальных объемной и линейной скорости
кровотока в микрососудах в локусе воспаления не может возникнуть
действующая концентрация флогогенов, достаточная для индуции острого
воспаления. В этой связи становится ясным, что нарушения микроциркуляции
представляют собой один из начальных и необходимых этапов воспаления.
Брадикинин - это один из флогогенов-кининов. Кроме брадикинина к
кининам относят каллидин и метионил-лизил-брадикинин. Кинины
образуются в циркулирующей крови и в тканях из своих неактивных
предшественников кининогенов, синтезируемых в печени, легких, почках, коже,
миокарде и других специализированных тканях. Образование кининов из
кининогенов происходит при участии специфических энзимов калликреи-
нов, которые в основном синтезируются в поджелудочной железе. У кал-
Воспаление
195
ликреинов также есть свои неактивные предшественники прекалликреины.
Кроме активированного фактора Хагемана трансформацию прекалликреи-
нов в калликреины и образование кининов вызывают ацидоз,
неспецифические протеазы (в том числе и лизосомальные ферменты,
высвобождаемые активированными нейтрофилами), фибринолизин и катехоламины.
Снижение степени гиперкатехоламинемии у хирургических больных
эффективной аналгезией и устранением гиповолемии гемодилюцией,
которые, нормализуя микроциркуляцию, подвергают обратному развитию
метаболический ацидоз, уменьшает образование кининов в циркулирующей
крови и тканях. Неэффективная аналгезия у тяжелых хирургических больных
способствует прогрессированию системной воспалительной реакции не
только через гиперкатехоламинемию, но и вызывая метаболический ацидоз
вследствие расстройств микроциркуляции и юкстакапиллярного
шунтирования, связанных с патологической болью. Падение интенсивности
образования кининов может быть фактором нормализации системного и
периферического кровообращения при организменной воспалительной реакции и
септическом шоке.
Эффект кининов как флогогенов сводится к расширению артериол,
росту проницаемости сосудов, что вызывает экссудацию в очаге воспаления.
Кроме того, кинины в афферентном звене обуславливают возникновение
болевых ощущений при воспалении.
ВОСПАЛЕНИЕ И СИСТЕМЫ РЕГУЛЯЦИИ ОРГАНИЗМА
Взаимодействие воспаления как типового патологического процесса с
системами регуляции организма происходит во многом через ноцицептив-
ную афферентацию, которую вызывает взаимодействие флогогенов с
нервными окончаниями в очаге воспаления. При этом ноцицепторы
реагируют на целый ряд стимулов химической природы, вызванных первичной
альтерацией и воспалением. Ноцицептивную афферентацию из очага
воспаления вызывает рост в нем содержания брадикинина, простагландина Е2,
лейкотриена В4 и вещества Р. Эти флогогены вызывают боль в афферентном
звене посредством действия двух основных механизмов:
♦ прямой активации сенсорных нейронов;
♦ непрямой активации нейронов вследствие действия на них факторов,
высвобождаемых симпатическими окончаниями.
Полагают, что высвобождение медиаторов воспаления нервными
окончаниями в тканях вызывает в них острую воспалительную реакцию,
получившую названиенейрогенного воспаления.
В настоящее время уже получен ряд убедительных данных о
существовании генетически детерминированной программы развития воспаления,
информация о которой хранится в нейронах сегментарного уровня. Можно
считать, что в спинном мозге существуют нейрональные сети, чьи функции
196
Глава 12
не исчерпываются только передачей той или иной информации, но
заключаются и в реализации программы воспаления в соответствующем
сегменте на периферии. В этой связи становится понятным часто положительный
эффект блокад местными анальгетиками соответствующих нервных
стволов при воспалении травматической или аутоиммунной этиологии (тендо-
вагиниты, артриты и т. д.). Предположительно, снижая афферентацию в
сегментарные констелляции нейронов, хранящие информацию о
программе воспаления и способные к ее реализации, блокада уменьшает уровень
возбуждения таких физиологических нейрональных интеграции и
блокирует алгоритм воспаления. С учетом значимости супрасегментарного
контроля за функционированием подсистем сегментарной нервной регуляции
можно также предположить, что центральная аналгезия как индуктор
системной антистрессорной реакции нервной системы может менять степень
выраженности системной воспалительной реакции, неизбежно явно или
скрыто вызываемой травматичными оперативными вмешательствами и (или)
тяжелыми ранениями и травмами.
Активация и сенситизация мембран нейронов в очаге воспаления
происходит через взаимодействие их рецепторов со своими лигандами-флогогена-
ми, которое открывает ионные каналы (эффект серотонина), активирует
системы вторичных мессенджеров (действие брадикинина и простагландинов)
или меняет экспрессию генома нейрона. Экспрессия генома нейронов
меняется под влиянием нейронального фактора роста, цитокина, концентрация
которого в очаге воспаления находится на высоком уровне.
Флогогеном, рост концентрации которого в наибольшей степени
ответственен за болевую афферентацию из очага воспаления считают брадики-
нин, который возбуждает ноцицепторы через взаимодействие со своими В1-
и В2-рецепторами. Интерлейкин-1-1 бета усиливает возбуждение и сенси-
тизацию нервных окончаний через усиление экспрессии их В1- и В2 бради-
кининовых рецепторов.
Опиоидные пептиды, синтезируемые и высвобождаемые клетками
иммунной системы в очаге воспаления (бета-эндорфин и др.), взаимодействуя
со своими рецепторами на поверхности чувствительных нервных
окончаний, блокируют ноцицептивную афферентацию из очага воспаления. Тот
факт, что интенсивность боли, первично генерируемой в воспалительном
очаге, падает после введения в него интерлейкина-1-1 бета или кортикотро-
пин-рилизинг-фактора показывает, что эта функция клеток иммунной
системы находится под контролем систем регуляции организма, иммунной и
нейроэндокринной.
Предположительно антиген-презентирующие клетки,
функционирующие в качестве клеточных эффекторов острого воспаления, перемещаясь по
лимфатическим сосудам, обеспечивают контакт антигенов из очага
воспаления с иммунокомпетентными клетками ближайшего регионарного
лимфатического узла. Таким образом происходит антигенная стимуляция иммунной
системы организма, вызывающая специфический первичный или вторичный
иммунный ответ. Специфический иммунный ответ через образование
антител к антигенам из очага воспаления, возникновение иммунных комплексов
Воспаление
197
на поверхности чужеродных объектов, через активацию системы
комплемента по классическому пути и опсонизацию повышает эффективность и силу
воспалительной реакции. При избыточном или извращенном аутоиммунном
характере реагирования системы иммунитета воспаление из защитной
реакции может трансформироваться в типовой патологический процесс.
Нервная и иммунная система представляют собой тесно
взаимодействующие системы регуляции организма, которые обеспечивают восприятие и
анализ информации, поступающей из внешней среды и элементов
организма. Эти задачи нервная и иммунная система решают равным количеством
клеток, способных воспринимать информацию, осуществлять ее процес-
синг и формировать на уровне систем регуляции алгоритм адекватной или
патогенной системной реакции. Установлена возможность модуляции
системной иммунной реакции воздействием на функциональную активность
различных структур головного мозга: гипоталамуса, лимбической системы
и коры. В этой связи можно считать, что воспаление как относительно
неспецифический элемент защитной реакции системы иммунитета и всего
организма находится под контролем нервной системы, в том числе и ее суп-
расегментарного уровня. Отсюда проистекает значимость влияний сугубо
нейротропных воздействий - анестезии и нейровегетативной блокады - на
резистентность больного к хирургической инфекции и на частоту
инфекционных осложнений хирургических вмешательств.
Цитокины, высвобождаемые моноцитами и иммунокомпетентными
клетками в очаге воспаления, пока еще неясным путем влияют на высшие
вегетативные центры, активируя вентромедиальные нейроны гипоталамуса и
всю нейроэндокринную катаболическую систему. Это через усиление
секреции катаболических гормонов антагонистов инсулина приводит к
преобладанию на уровне всего организма катаболических процессов над
анаболическими. При этом анаболизм может тормозиться в такой степени, что
данный супрасегментарный эффект цитокинов становится звеном
патогенеза стрессорного голодания. При развитии кахексии вследствие
длительно текущего воспаления защитная реакция мобилизации резервов
энергопластических субстратов для саногенеза теряет свой биологический смысл,
вызывая в органах эффекторах функций дефицит массы и энергии.
Воспаление вызывает сдвиги системной нейроэндокринной регуляции,
направленные на предотвращение трансформации острой воспалительной
реакции в патологический процесс. Так, гиперкортизолемия, вызываемая
острым воспалением, снижает интенсивность функционирования его
клеточных эффекторов и ограничивает распространение очага воспаления в
пределах здоровых тканей. Это, в частности, происходит вследствие снижения
образования простагландина Е2 через торможение синтеза фосфолипазы А2.
Снижение активности фосфолипазы \ под влияние кортикостероидов в
очаге воспаления уменьшает содержание в нем таких флогогенов как лейкотри-
ены и фактор активации тромбоцитов. Дело в том, что фосфолипаза
^высвобождает арахидоновую кислоту из фосфолипидов клеточных мембран, а
арахидоновая кислота представляет собой субстрат синтеза лейкотриенов.
Глава 13
ЛИХОРАДКА И РЕАКЦИЯ ОСТРОЙ ФАЗЫ
Лихорадка - защитная реакция теплокровных животных, которая
через повышение температуры внутренней среды и цитозоля клеток
усиливает реакцию системы иммунитета в ответ на экзо- и эндогенную
антигенную стимуляцию и действие других стимулов при инфекциях,
злокачественном клеточном росте и некробиотических изменениях
собственных тканей организма. Кроме того, биологическое значение
лихорадки состоит в интенсификации защитных сдвигов обмена веществ
посредством увеличения температуры среды обитания клеток. Реакция системы
иммунитета потенцирует саму себя, повышая температуру внутренней
среды организма высвобождением эндогенных пирогенов. К защитным
изменениям метаболизма при лихорадке, остром воспалении и системной
воспалительной реакции в первую очередь относят усиление протеолиза для
обеспечения субстратами интенсивного белкового синтеза в защитных
системах организма, и в системе иммунитета в том числе.
Реакция системы иммунитета как причина лихорадки может быть
обусловлена:
♦ Бактериальными, вирусными, дрожжевыми и другими инфекциями.
Бактериальные инфекции часто вызывают лихорадку при
высвобождении бактериальных эндотоксинов.
♦ Лекарственными средствами, которые сами могут быть иммуно-
генами или становится таковыми после биотрансформации или
фиксации на поверхности клеток организма больного в качестве
гаптенов.
♦ Некробиотическими изменениями и цитолизом в собственных
тканях организма при ранениях, травмах, ожогах и синдроме
длительного раздавливания. Некробиотические изменения приводят к
действию ряда мощных стимулов острого воспаления и системной
воспалительной реакции, элементом которых и является лихорадка.
♦ Антигенной стимуляцией после озлокачествления клеток организма.
К ней приводит появление на поверхности малигнизированных
клеток неспецифических опухолевых и опухолевых специфических
трансплантационных антигенов.
♦ Аллергией, при которой активация соответствующих клеток
иммунной системы ведет к высвобождению ими эндогенных пирогенов.
Предположительно вызывать лихорадку может гормональный дисбаланс,
в частности нарушения секреции прогестерона.
Все эти факторы возникновения лихорадки традиционно называют
экзогенными пирогенами, что не совсем верно, так как экзогенными часть из
них является лишь для системы иммунитета, а не для всего организма. Так
множественный цитолиз после ранения часто вызывает лихорадку через
Лихорадка и реакция острой фазы
199
высвобождение антигенов, которые система иммунитета не воспринимает
в качестве своих.
Древние называли лихорадку «священной болезнью». Гиппократ
считал что посредством лихорадки организм сжигает попавшие в него
токсины. Эту концепцию лихорадки и в наши дни следует признать
принципиально верной. «Английский Гиппократ» Сиденхэм называл лихорадку
«мощной машиной, которую Природа дает нам в этом мире для борьбы с
врагами».
Современное переосмысление лихорадки как защитной реакции
организма в некоторой мере связано с экспериментами, объектом для которых
служило холоднокровное животное, пустынная ящерица Dipsosaurus
dorsalis. Если до инфицированиия патогенным микроорганизмом животные
хаотически перемещались по клетке, то после него они оставались в ее
самых теплых местах. После инфицирования чаще выживали те ящерицы,
которым была предоставлена возможность свободно передвигаться по клетке
в ее наиболее обогреваемые места. У пойкилотермных животных по
биологическому смыслу аналогична лихорадке активная защитная реакция
перемещения для наибольшего подъема температуры внутренней среды под
воздействием внешней.
Экзогенные пирогены вызывают лихорадку через эффект
веществ-посредников эндогенных пирогенов (ЭП). Один из ЭП идентичен интерлейки-
ну-1 (ИЛ-1). В основном его высвобождают активированные фагоцитозом,
антигенной стимуляцией и цитокинами клеток иммуннной системы
моноциты циркулирующей крови и клетки системы тканевых мононуклеарных
фагоцитов. ИЛ-1 (лейкоцит-активирующий фактор) представляет собой
полипептидный цитокин с молекулярной массой в 11 килодальтон. Кроме
активированных моноцитов и тканевых макрофагов его высвобождают В-
лимфоциты и лимфоциты со свойствами естественных киллеров, микро-
глиальные, мезангиальные клетки, большие гранулярные лимфоциты, ма-
лигнизированные клетки миелодиного ряда при острой миелоидной
лейкемии, озлокачествленные клетки при Ходжкинской лимфоме, мелано-
ме и других злокачественных новообразованиях.
ИЛ-1 как индуктор системной реакции острой фазы при воспалении
действует не только в качестве пирогена, вызывающего лихорадку при
воздействии на центральную нервную систему (ЦНС). ИЛ-1 усиливает нейт-
рофилию, повышая выход полиморфонуклеаров из костного мозга.
Активирующее влияние ИЛ-1 на полиморфонуклеары увеличивает образование
и высвобождение ими коллагеназы, что повышает способность нейтрофи-
лов форсировать базальные мембраны капилляров на пути к объекту
фагоцитоза при воспалении. Кроме того, эффект интерлейкина-1 на
активированные полиморфонуклеары усиливает их дегрануляцию и повышает в ней-
трофилах оксидазную активность и интенсивность реакций гекосозомоно-
фосфатного шунта, что увеличивает экзоцитоз свободных кислородных
радикалов. Это изменение обмена нейтрофилов усиливает их
бактерицидные свойства, повышая способность активированных полифорфонуклеаров
вызывать вторичную альтерацию. Относительно фибробластов и синови-
200
Глава 13
альных клеток ИЛ-1 выступает как фактор клеточного роста, повышая
образование ими коллагена и активатора плазминогена. Усиление клеточной
пролиферации фибробластов под влиянием ИЛ-1 направлено на замещение
дефектов тканей, возникающих вследствие первичной и вторичной
альтерации при воспалении. Действие ИЛ-1 как агента паракринной регуляции
ведет к усилению синтеза и секреции простагландинов-медиаторов
воспаления многими из его клеточных эффекторов.
Влияние ИЛ-1 на ЦНС повышает уровень секреции адренокортикотроп-
ного гормона и гормона роста, что вызывает защитный сдвиг эндокринной
регуляции обмена веществ в сторону преобладания эффектов катаболичес-
ких гормонов-антагонистов инсулина. В результате в крови растет
концентрация энергопластических субстратов, доступных для утилизации в ходе
защитных реакций организма.
Краткое описание функций ИЛ-1 рисует нам этот цитокин как агент
паракринной регуляции, стимулирующий почти все клеточные эффекторы
воспаления, первичного и вторичного иммунного ответов, и пептидный
гормон, вызывающий быструю и качественную перестройку обмена
веществ и термодинамики организма для повышения эффективности его
защитной реакции. Эта реакция происходит при определяющем участии
единых ЦНС и системы иммунитета. Лихорадка - это всего лишь один из
элементов системной реакции воспаления, первичного и вторичного
иммунного ответов, которые во многом индуцируются через действие ИЛ-1.
Воздействие ИЛ-1 на ЦНС как стимулятора секреции адренокортикот-
ропного гормона через увеличение секреции кортизола тормозит
высвобождение интерлейкина-1 клетками организма. Таким образом, происходит
самоограничение системной воспалительной реакции и реакции острой фазы
при воспалении.
ИЛ-1 играет ключевую роль в развитии системной иммунной реакции,
когда активирует Т-лимфоциты хелперы и индукторы. Активация под
воздействием ИЛ-1 этих лимфоцитов ведет к синтезу и высвобождению ими
интерлейкина-2, который выступает относительно Т-лимфоцитов
определенного клона в качестве митогена. Рост частоты деления Т-лимфоцитов
под воздействием интерлейкина-2 усиливается при повышении
температуры среды их обитания и достигает оптимума при температуре тела 39,5 °С.
Такое повышение температуры тела усиливает эффект интерлейкина-2 как
митогена на Т-клетки в 400 раз.
ИЛ-1 снижает содержание железа и цинка в плазме крови. Так как
железо - это необходимый фактор роста для многих микроорганизмов, то
данный эффект эндогенного пирогена противодействует росту патогенных
бактерий на системном уровне. ИЛ-1 повышает синтез белков острой фазы в
печени.
Известно, что одним из механизмов возникновения злокачественной
опухоли из одной малигнизированной или клона озлокачествленных
клеток (промоции) является их выход из-под контроля системой иммунитета
организма. Поэтому промоцию может обуславливать врожденный или
приобретенный комбинированный иммунодефицит. Лихорадка вследствие ле-
Лихорадка и реакция острой фазы
201
небной антигенной стимуляции системы иммунитета (пиротерапия)
всегда сопровождается ростом выброса и системных эффектов цитокинов мо-
нонуклеарных фагоцитов и иммунокомпетентных клеток. Лихорадку
искусственно вызывают для усиления цитокинами атаки лимфоцитов нормальных
киллеров на клетки злокачественной опухоли. В последние десятилетия было
показано, что сочетание пиротерапии с радио- и хемотерапией
злокачественных опухолей может задержать их развитие и препятствовать метастазиро-
ванию. Необходимым условием эффективности пиротерапии является
подъем температуры тела до уровня не меньшего, чем 40 °С.
В этой связи нельзя не вспомнить о токсине Кулея (Coley) (основатель
знаменитой династии американских хирургов и лечащих
врачей-исследователей). В 1893 году, в Нью-Йорке, впервые было показано, что смесь
протеинов бактериального происхождения, в которую в частности входили
энзимы гемолитического стрептококка стрептокиназа и стрептодорназа,
(коктейль Кулея) после парентерального введения через индукцию
системной реакции, о которой свидетельствовала лихорадка, подвергала
некоторые опухоли у некоторых больных обратному развитию. Толчком для
создания и применения коктейля послужили наблюдения Coley регресса
некоторых опухолей головы и шеи после лихорадки, связанной с рожистым
воспалением. Результаты современных исследований антигенной
стимуляции как причины лихорадки и усиления цитолиза злокачественных клеток
показали, что эндогенным пирогеном, секреция которого растет в ответ на
введение коктейля Coley и вызывает лихорадку, в основном является
фактор некроза опухолей.
Следует заметить, что пиротерапия как средство устранения и
замедления развития злокачественных опухолей может быть эффективна далеко не
всегда. Искусственная гипертермия в основном направлена на усиленный
выброс цитокинов, повышающих клональную экспансию активированных
Т-лимфоцитов нормальных киллеров. Но сами Т-лимфоциты-киллеры не
могут уничтожать многие из злокачественных клеток, поверхность
которых не содержит антигенов из главного комплекса тканевой
совместимости, необходимых для распознавания нормальными киллерами их клеток-
мишеней.
Кроме фактора некроза опухолей и ИЛ-1 эндогенным пирогеном может
быть макрофагальный воспалительный белок-1-альфа. Этот эндогенный
пироген обладает способностью связываться с гепарином. Свойство
данного ЭП вызывать лихорадку не связано с активностью циклооксигеназы.
Поэтому лихорадка, обусловленная действием макрофагального
воспалительного белка, не купируется ацетилсалициловой кислотой и другими
нестероидными противовоспалительными средствами, снижающими
активность циклооксигеназы. В частности это относится к лихорадке в ответ на
септицемию, при которой секреция макрофагального воспалительного
белка происходит в ответ на взаимодействие бактериального эндотоксина с
макрофагами циркулирующей крови.
Под влиянием ИЛ-1 через увеличение интерлейкином активности
циклооксигеназы в миоцитах произвольных мышц и образование ими простат-
202
Глава 13
ландина Е, происходит активация протеолиза. Усиление протеолиза
повышает содержание свободных аминокислот в циркулирующей крови как
субстратов для интенсивного белкового синтеза в защитных системах
организма. Рост температуры внутренней среды, связанный с лихорадкой, при этом
усиливает как выработку простагландина Е, так и распад протеинов мышц.
Поэтому при лихорадке больной может потерять до 1 кг массы скелетных
мышц. Протеолиз усиливает гиподинамия, связанная с сонливостью. Ее
обуславливает воздействие ИЛ-1 на нейроны головного мозга,
ответственные за медленный сон.
Взаимодействие эндогенного пирогена интерлейкина-1 на нейроны тер-
морегуляторного центра в преоптической области гипоталамуса вызывает
на периферии резкое повышение теплообразования в мышцах (озноб) с
одновременным снижением теплоотдачи. Снижение теплоотдачи достигается
через спазм кожных сосудов, который в период озноба может проявить себя
побледнением кожных покровов. Характерное ощущение («мороз по коже»)
связывают с сокращением рудиментарных мышечных элементов в области
волосяных фолликулов, в результате которой возникает так называемая
«гусиная кожа». Эта древняя реакция симпатического отдела автономной
нервной системы у млекопитающих снижает отдачу тепла во внешнюю среду
через увеличение прослойки теплого относительно температуры внешней
среды воздуха во вздыбленной шерсти животных.
Вследствие усиления теплообразования температура «ядра» тела при
лихорадке повышается до тех пор, пока не достигнет верхней стабильной
точки, при достижении которой устанавливается равновесие между
теплопродукцией и теплоотдачей. В результате температура крови, омывающей
гипоталамус, становится равной новому установленному уровню.
Полагают, что в гипоталамусе ИЛ-1 усиливает синтез простагландинов
типа Е. Субстратом синтеза простагландинов при этом является арахидоно-
вая кислота мембран клеток, с которыми цитокин взаимодействует на суп-
расегментарном уровне. Известно что один из простагландинов, синтез
которых растет под влиянием ИЛ-1, простагландин Е1, активирует механизмы
теплопродукции и теплоотдачи путем усиления синтеза циклического аде-
нозинмонофосфата в клетках на периферии. Жаропонижающее действие
ацетилсалициловой кислоты и других нестероидных противоспалительных
средств обусловлено их способностью тормозить фермент циклооксигена-
зу, то есть блокировать образование простагландинов Е, и Е2
Препараты на основе гормонов коры надпочечников и эндогенные глю-
кокортикоиды оказывают свое антипиретическое действие, снижая
высвобождение ИЛ-1 клетками системы иммунитета на периферии и
стабилизируя клеточные мембраны с уменьшением высвобождения ими арахидоновой
кислоты в ЦНС.
Кровоизлияние в ткань головного мозга вызывает цитолиз его астроци-
тов, которые при этом высвобождают содержащийся в них интерлейкин-1.
В результате ИЛ-1 вызывает повышение температуры в ответ на
кровоизлияние. Кроме того, ИЛ-1, воздействуя на нейроны, ответственные за
медленный сон, обуславливает сонливость.
Лихорадка и реакция острой фазы
203
Патогенное значение лихорадки состоит в том, что при ее
патологических величине и длительности через избыточный иммунный ответ может
развиться приобретенный комбинированный иммунодефицит. Кроме того,
лихорадка как фактор протеолиза вызывает дистрофию скелетных мышц,
что задерживает реабилитацию больных. Рост температуры тела повышает
потребление кислорода организмом, что у больных со скрытой
недостаточностью внешнего дыхания и низкой насосной функцией сердца может
приводить к артериальной гипоксемии, дыхательному ацидозу и обострять
сердечную недостаточность.
КЛИНИЧЕСКАЯ КЛАССИФИКАЦИЯ ВИДОВ ЛИХОРАДКИ
Нормальной считают температуру тела, не превышающую 37 °С. Под
субфебрильной лихорадкой понимают подъем температуры тела до уровня
не более высокого, чем 38 °С. При фебрильной лихорадке температура тела
находится в диапазоне от 38 до 39 °С. Если температура тела поднялась до
уровня в 39 °С и не выше 41 °С, то ее определяют как тяжелую. Крайне
тяжелая лихорадка, которая сама по себе может быть причиной комы и
терминального состояния, - это подъем температуры тела до уровня более
высокого, чем 41 °С.
В течение суток температура тела здорового человека колеблется.
Минимальную температуру фиксируют в 4-6 ч утра. Максимума температура
тела достигает в 5-7 ч вечера. При лихорадке повышенная температура тела
в течение суток варьирует аналогичным образом. Постоянную лихорадку
характеризуют суточные колебания температуры, не превышающие 1 °С.
Под послабляющей понимают лихорадку, при которой суточные колебания
температуры составляют 1-2 °С. При перемежающейся лихорадке в
течение суток происходит временное снижение температуры до нормального
уровня. При истощающей лихорадке колебания суточной температуры
достигают 3 °С, лихорадка быстро нарастает и быстро снижается. При
длительных септических состояниях и персистирующей системной
воспалительной реакции тяжелую лихорадку у части больных фиксируют по утрам,
а вечером у таких пациентов температура тела может снижаться до
«нормального уровня». Такую лихорадку называют неправильной.
Неправильная лихорадка - это неблагоприятный прогностический признак.
Отдельно выделяют лихорадку неясного происхождения. Лихорадка нет
ясного происхождения - это минимум однократный в течение суток подъем
температуры тела выше 37 °С без видимой причины, который непрерывно
фиксируют в течение не менее чем двух недель. При лихорадке неясного
происхождения данные углубленного клинического обследования и
всесторонних специальных исследований некоторое время не позволяют
окончательно идентифицировать ее генез. В дальнейшем у 30-40 % таких
больных как причину лихорадки выявляют ту или иную инфекцию. У 15-20 %
204
Глава 13
больных с лихорадкой неясной этиологии ее причина - это заболевания
соединительной ткани и обусловленные ими васкулиты. В 20-30 % случаев
лихорадка неясной этиологии у взрослых - это проявление
злокачественного клеточного роста. У детей лихорадку неясной этиологии
злокачественные опухоли вызывают в 10 % случаев. К редким причинам лихорадки
неясного происхождения относят колит и саркоидоз.
Наследственная лихорадка неясного происхождения обычно
обусловлена такими генетически детерминированными заболеваниями и
патологическими состояниями как семейная средиземноморская лихорадка, гипер-
липидемия/гиперлипопротеинемия первого типа и циклическая нейтропения
(периодическое снижение содержания полифорфонуклеаров в
циркулирующей крови до уровня более низкого, чем нижний предел его нормальных
колебаний).
РЕАКЦИЯ ОСТРОЙ ФАЗЫ
При воспалении и типическом раневом патологическом процессе нейт-
рофилы участвуют в реакции первой очереди в ответ на инфекцию и
повреждение. Хотя полиморфонуклеары и способны высвобождать цитоки-
ны, вызывающие реакцию всей системы иммунитета, их функционирование
в очаге воспаления и ране в основном состоит из миграции к объекту
фагоцитоза, эндоцитозу, высвобождению протеаз и свободных кислородных
радикалов, эффект которых на клетки и элементы межклеточных структур
приводит ко вторичной альтерации.
Уже через 3-6 ч после первичной альтерации и (или) инфицирования в
ране и очаге воспаления начинает нарастать содержание моноцитов, Т- и В-
лимфоцитов, что свидетельствует о начале первичной реакции системы
иммунитета, которая является необходимым условием эффективного
вторичного иммунного ответа. Межклеточные взаимодействия между мононуклеар-
ными фагоцитами и иммунокомпетентными клетками в основном
осуществляется через высвобождение цитокинов. Цитокины связываются своими
рецепторами на поверхности иммунокомпетентных клеток и фагоцитов.
Высвобождаемые клетками, функционирующими при воспалении, цитокины не
только обеспечивают интегрирование элементов системы иммунитета для ее
эффективной реакции на инфицирование, повреждение или перерождение
своих тканей. Они вызывают системную реакцию острой фазы. Своего
максимума реакция острой фазы достигает на второй-третий день воспаления
или типического раневого процесса, когда в очаге воспаления начинает
взаимодействовать временный комплекс из тесно в функциональном отношении
связанных активированных моноцитов, тканевых мононуклеарных
фагоцитов и претерпевших бласттрансформацию лимфоцитов.
В основном реакцию острой фазы вызывают ИЛ-1, интерлейкин-6, ин-
терфероны и фактор некроза опухолей (ФИО).
Лихорадка и реакция острой фазы
205
Интерлейкин-6 (ИЛ-6, интерферон бета-два) - это цитокин, масса
молекулы которого составляет 26 килодальтон. Он является фактором роста,
дифференциации, а значит и клональной экспансии В- и Т-лимфоцитов. Тем
самым ИЛ-6 потенцирует вторичный иммунный ответ, эффективность
которого часто представляет собой необходимое условие завершения саноге-
неза при инфекциях, начатого воспалением.
Известно 15 интерферонов, то есть протеинов-иммуномодуляторов,
синтезируемых и секретируемых Т-лимфоцитами, фибробластами и другими
клетками в ответ на стимуляцию антигенами вирусов, бактерий или под
влиянием митогенов.
Многие из интерферонов составляют фактор, активирующий
макрофаги (ФАМ). ФАМ - это группа гетерогенных цитокинов, секретируемых
сенсибилизированными лимфоцитами. Из них как ФАМ наиболее активен
интерферон-гамма. Под влиянием ФАМ усиливаются бактерицидный эффект
макрофагов и их способность вызывать лизис малигнизированных клеток.
Под влиянием ФАМ они увеличиваются в размерах, их скорость миграции
растет, у макрофагов появляются псевдоподии, а в цитоплазме - множество
вакуолей (вакуолизация). Другие интерфероны обладают свойствами
фактора тормозящего макрофаги, то есть замедляют их миграцию, что в
частности связано с увеличением под влиянием интерферонов содержания в
макрофагах циклического аденозинмонофосфата. В различные фазы
воспаления в соответствии с биологической целью этапа воспаления меняется
экспрессия генома Т-лимфоцитов в его очаге. Если на начальном этапе
генетический материал Т-клеток в основном экспрессирует интерфероны,
активирующие фагоциты, то после завершения фагоцитоза в очаге
воспаления и элиминации из него продуктов цитолиза, начинается экспрессия
интерферонов, тормозящих мононуклеарные фагоциты.
Стимулом для системной реакции острой фазы воспаления служат
травматические и раневые повреждения тканей, инфекция, и, что бывает реже,
злокачественный клеточный рост (ходжкинская лимфома, почечная
клеточная карцинома и др.). Реакцию острой фазы в первую очередь составляют
сонливость и гиподинамия, предрасполагающие к защитной мобилизации
аминокислот из белков скелетных мышц. Участие организменной системы
иммунитета в реакции острой фазы проявляет себя нейтрофилией со
сдвигом лейкоцитарной формулы влево и ростом содержания в плазме крови
иммуноглобулинов. Сдвиги эндокринной регуляции метаболизма при
реакции острой фазы приводят к росту содержания в плазме крови глюкозы,
свободных жирных кислот и глицерина, а также к высвобождению в кровь
несбалансированной смеси аминокислот. Превалирование на системном
уровне эффектов катаболических гормонов-антагонистов инсулина
приводит к толерантности по отношению к глюкозе. В плазме крови при реакции
острой фазы падает содержание железа. На уровне печени реакцию острой
фазы в основном составляют усиленный глюконеогенез и синтез белков
острой фазы.
Белки острой фазы - это иммуномодуляторы, протеины с прямым или
опосредованным бактерицидным и (или) бактериостатическим действи-
206
Глава 13
ем, медиаторы воспаления, хемоаттрактанты и неспецифические опсо-
нины, ингибиторы первичной альтерации, синтез которых растет в
печени в острый период воспаления после определенного распространения его
очага в пределах здоровых тканей. К ним относят белки, мигрирующие при
электрофорезе в геле в его области альфа-1 и альфа-2:
альфа-1-антитрипсин, альфа-1 кислый гликопротеин, амилоид А и Р, антитромбин III, С-реак-
тивный белок, ингибитор С-1-эстеразы, фракцию комплемента СЗ, церул-
лоплазмин, трансферрин, гаптоглобулин, оросомукоид, плазминоген.
Рост концентрации белков острой фазы в циркулирующей крови
представляет собой маркер острого воспаления. При этом наиболее чувствительна
к острому воспалению концентрация в плазме крови С-реактивного белка,
которая за первые несколько часов воспаления может возрасти в 10-100 раз.
С-реактивный белок - это полипептид с молекулярной массой в 120 кДа.
Этот белок обладает способностью связываться с полисахариом С на
поверхности Streptococcus pneumoniae. С-реактивный белок активирует
систему комплемента, подавляет функции тромбоцитов и лимфоцитов,
тормозит ретракцию сгустка и стимулирует фагоцитоз нейтрофилами.
Глава 14
КАНЦЕРОГЕНЕЗ
ИЗМЕНЕНИЯ ФЕНОТИПА КЛЕТКИ ПРИ МАЛИГНИЗАЦИИ
В основном злокачественную клетку характеризуют потеря феноти-
пических признаков дифференцированной клетки специализированной
ткани, интенсивная клеточная пролиферация и способность к метастазиро-
ванию.
Многие из малигнизированных клеток обладают фенотипическими
признаками здоровых дифференцированных клеток той специализированной
ткани, из которой произрастают составляемые ими злокачественные
опухоли. Например, в цитозоле клеток злокачественных опухолей щитовидной
железы находятся коллоидсодержащие фолликулы. Чем больше в фенотипе
злокачественных клеток признаков клеточной дифференциации, тем в
меньшей степени прогрессирует и метастазирует злокачественная опухоль (вы-
сокодифференцированные опухоли).
Одним из механизмов приобретения нормальной клеткой фенотипа
злокачественной можно считать блокаду клеточной дифференциации на
определенном ее этапе (частичная дифференциация злокачественных клеток),
когда потенциал ее пролиферации значительно выше, чем у высокодиффе-
ренцированных клеток. Клетка, не достигая зрелости в качестве
структурно-функционального элемента специализированной ткани, начинает
интенсивно делиться, образуя клон потенциально злокачественных или уже
малигнизированных клеток. Так, например, гепатоцеллюлярная карцинома
содержит много альфа-фетопротеина, белка, образуемого эмбриональными
гепатоцитами. Экспрессия генома злокачественной клетки может давать
протеины, которых нет в здоровой специализированной ткани того же
происхождения.
Злокачественная клетка в повышенном количестве образует и
высвобождает цитокины, представляющие собой факторы клеточного роста и
злокачественного перерождения клеток. При этом малигнизированная клетка
теряет свою нормальную реакцию на действие других цитокинов и митоге-
нов. Метастазирующие злокачественные клетки интенсивно
высвобождают протеазы.
Малигнизированная клетка во многом теряет способность к адгезии при
взаимодействии с другими клетками. Трансмембранный потенциал
злокачественной клетки меняется таким образом, что происходит ее
отталкивание от других клеток. В злокачественной клетке падает содержание калия и
кальция. Некоторые из малигнизированных клеток приобретают
способность к эктопической секреции гормонов, а другие секретируют гормоны,
высвобождаемые их клетками-предшественницами на пути малигнизации.
208
Глава 14
К первым можно отнести клетки некоторых злокачественных опухолей
легких, секретирующие гормон околощитовидных желез, а ко вторым клетки
инсулиномы, злокачественной опухоли поджелудочной железы, секретиру-
ющей инсулин.
Малигнизированная клетка лишена многих из тех ее элементов,
которые определяют ее специфичность как структурно-функциональной
единицы органа. Малигнизация миоцитов ведет к потере мышечными клетками
актиновых нитей. Злокачественная клетка, происходящая из энтероцитов,
теряет микроворсинки. У межклеточных соединений между
злокачественными клетками, десмосомами, нет органоспецифичности. Это относится и
к промежуточным нитям. Кроме того, патологические изменения
происходят и в некоторых внутриклеточных органеллах. В частности развивается
отек митохондрий и митохондриальной матрицы.
Обмен злокачественной клетки отличается от метаболизма
доброкачественной. Сдвиги обмена злокачественной клетки ведут к устранению
особенностей ее метаболизма, определяющих специфику клетки как
дифференцированного клеточного тканевого элемента. Это явление получило
название биохимической конвергенции. Метаболизм злокачественной
клетки ориентирован преимущественно на обеспечение частого деления
малигнизированных клеток.
Патологические изменения генома злокачественной клетки проявляют
себя гиперхромазией клеточного ядра, то есть более интенсивной, чем у
доброкачественных клеток, его окраской. При этом часто выявляют марги-
нацию ядра (смещение хроматина на периферию ядра клетки).
Клетки злокачественных опухолей содержат на своей поверхности
антигены, которых нет на плазматической мембране их нормальных клеток-
предшественниц. Один из таких антигенов присутствует на клетках
разнообразных по происхождению злокачественных опухолей (неспецифический
опухолевый антиген, НОА). Лимфоциты с функциями естественных
киллеров могут распознавать НОА и, связываясь с ним на наружной поверхности
злокачественных клеток, вызывают цитолиз малигнизированных клеток без
предварительной иммунизации данным антигеном. Кроме того, наружная
клеточная мембрана злокачественных клеток может содержать
специфичные для данного вида опухоли антигены, которых нет на нормальных
клетках той же ткани (опухоль-специфичные траснплантационные антигены,
ОСТА). После иммунизации экспериментальных животных
нежизнеспособными злокачественными клетками, несущими ОСТА, их организм
приобретает способность противостоять приживлению и размножению
введенных извне живых злокачественных клеток с аналогичными специфичными
антигенами на плазматической мембране. ОСТА не участвуют в озлокаче-
ствлении клетки и представляют собой лишь маркеры ее малигнизации.
В соответствии с иммунопатологической теорией возникновения
злокачественных опухолей, по ходу онтогенеза неоднократно происходит
малигнизация клеток разных тканей. Это не приводит к появлению
злокачественных новообразований, так как озлокачествленные клетки уничтожаются с
преимущественным участием того или иного звена системы иммунитета.
Канцерогенез
209
Ряд мутаций как причина
эквивалентного малигни-
зации клетки изменения ее
генетического материала
Изменения набора
антигенов
наружной клеточной
мембраны
Первичный и
(или) вторичный
иммунодефицит
Выход клетки из-под контроля
системой иммунитета
Неограниченное деление злокачественной клетки и образование
злокачественной опухоли
Схема 14.1. Патогенез выхода злокачественной клетки
из-под иммунного надзора
Иммунодефицит различного генеза может способствовать превращению
первичного клона злокачественных клеток в злокачественную опухоль
(схема 14.1). О роли иммунопатологических сдвигов в развитии опухолей
свидетельствуют: а) лимфоцитарная инфильтрация злокачественных опухолей;
б) частые малигнизация В-лимфоцитов (лимфома) и саркома Калоши у
больных с синдромом приобретенного иммунодефицита; в) высокая частота
злокачественных новообразований, которая у больных с различными видами
иммунодефицита в 200 раз выше, чем у людей без каких-либо
иммунопатологических сдвигов.
Один из механизмов выхода злокачественной клетки из-под иммунного
надзора - это прекращение экспрессии на их поверхности антигенов из
системы антигенов лейкоцитов человека (HLA), экспрессия которых на
наружной клеточной мембране необходима для распознавания лимфоцитами
клеток-мишеней, подлежащих уничтожению иммунной системой.
В частности, содержание антигенов HLA на клеточной поверхности
снижается под воздействием на клетку онкогенных аденовирусов. Поверхность
злокачественных клеток, составляющих нейробластому, почти лишена
антигенов системы HLA.
КАНЦЕРОГЕНЕЗ КАК КОМПЛЕКСНОЕ ИЗМЕНЕНИЕ
ГЕНОТИПА КЛЕТКИ
Канцеризация (канцерогенез, озлокачествление, малигнизация) - ряд
изменений генотипа и фенотипа клетки, которые окончательно придают ей
свойства злокачественной (малигнизированной). При этом признаком
окончательного злокачественного перерождения является приобретение
свойства метастазирования. Канцеризация складывается не только из генотипи-
210
Глава 14
ческих изменений, которые через трансляцию изменяют клеточный
фенотип. Известно, что изменения генетического материала в виде
амплификации генов, транслокации хромосом и утраты хромосомами своих частей (де-
леции), которые in vitro часто вызывают озлокачествление клетки, in vivo
не всегда приводят к малигнизации.
В настоящее время общепринятая схема механизма канцеризации
включает ряд вполне определенных этапов. Инициирующим моментом
канцеризации признают действие на клетку ряда генотоксических канцерогенов
(химические соединения, токсины, ионизирующее излучение и др.), которые
придают геному клетки свойство определять предрасположенность к
патогенной реакции малигнизации на действие внешних и внутренних
факторов или сразу вызывают озлокачествление клетки. Канцерогенез начинают
нарушения генетического материала клетки (альтерации генома) в виде
активации прото-онкогенов (доминирующие альтерации) и (или)
инактивация генов белков супрессоров опухолевого роста, эффект которых на
экспрессию генома клетки противодействует канцерогенезу (гены-супрессоры
клеточного роста). Изменение генома клетки, обуславливающее
канцерогенез, составляют сниженная экспрессия генов белков, подавляющих
клеточную пролиферацию и злокачественный клеточный рост
(гены-супрессоры опухолей), и усиленная экспрессия генов, продукты которых, воздействуя
на геном, усиливают клеточную пролиферацию (прото-онкогены).
Канцерогенез клетки - это во многом многоступенчатый процесс
накопления изменений генома как причины изменения фенотипа клетки
эквивалентного окончательной малигнизации. Полагают, что обычно для
окончательной малигнизации клетки требуется около десяти мутаций генов
различной локализации.
Результаты экспериментов, в основном проведенных во второй
половине XX века, позволили предположить, что изменения фенотипа клетки,
характеризующие ее озлокачествление, представляют собой прямое следствие
изменений или повреждений клеточного генома (генетическая теория
канцерогенеза). Альтернативой генетической теории канцерогенеза выступает
эпигенетическая теория злокачественной трансформации клетки.
Эпигенетическая теория связывает канцерогенез с нарушениями регуляции
клеточного роста, функций клетки и экспрессии генов, составляющих ее
геном, без изменений и повреждений самого генетического материала.
Свидетельством в пользу генетической теории рака является выявление
у веществ и воздействий на клетку (ионизирующие излучения и др.),
которые способны повреждать ДНК и менять ее структуру (мутагены), свойств
канцерогенов. Установлено, что предшественники злокачественных клеток
на пути озлокачествления претерпевают ряд повреждений и изменений
генетического материала, приводящих к образованию мутантных генов.
Экспрессия мутантных генов дает фенотип клетки, определяющий ее
озлокачествление. Эти генетические изменения происходят в клетках органов, где
первично возникают злокачественные опухоли и получили название
соматических мутаций. Тем самым подчеркивается их отличие от мутаций
генетического материала в гаметах, которые передаются по наследству.
Канцерогенез
211
В начале шестидесятых годов экспериментальным путем было
показано, что включение генов ДНК-содержащих и других вирусов (вирусных
онкогенов) в состав генома инфицированной клетки приводило к изменениям
клеточного фенотипа эквивалентным клеточной малигнизации. Длительное
присутствие вирусных онкогенов в составе генома злокачественной клетки
было необходимым условием сохранения ее злокачественности. После
выведения вирусных онкогенов из генетического материала клетки малигни-
зация нередко подвергалась обратному развитию.
Вирусные онкогены, которые содержал геном клеток, малигнизирован-
ных ретровирусами, были относительно небольшими по размерам и
простыми по строению. Это позволило предположительно считать, что
мутация небольшого числа генов из состава генома клетки может обусловить
множество изменений ее фенотипа, приводящих к озлокачествлению. В
шестидестях-семидесятых годах было впервые высказано предположение,
что изменения генетического материала в небольшом числе генов как при-
чина канцерогенеза в первую очередь нарушают реакцию клетки на
системные и местные интегрирующиерегуляторные воздействия, в том
числе и в виде эффектов факторов клеточного роста. Гены, мутации которых
могут быть одним из этапов озлокачествления, назвали онкогенами.
Идентификация онкогенов связана с изучением трансформирующих ретровиру-
сов (ТРВ). ТРВ - это РНК-содержащие вирусы, которые интегрируются в
геном инфицированной клетки и включают ее генетический материал в свой
собственный геном. Иногда это приводит к мутациям генов
инфицированной клетки, которые вирус включает в свой геном. Результатом такой
мутации может быть образование онкогена, то есть гена, экспрессия которого
служит одной из причин озлокачествления клетки.
Разработка технологии клонирования генов в начале восьмидесятых годов
позволила выявить онкогены, играющие роль при озлокачествлении клеток
разной локализации. Один из наиболее хорошо изученных онкогенов EJ/T24
играет роль в озлокачествлении клеток мочевого пузыря (карцинома
мочевого пузыря человека). Это относительно небольшой по размерам онкоген
включает только 6000 пар спаренных оснований ДНК, тогда когда геном клетки
содержит 6 миллиардов таких пар. Тем не менее, онкоген качественно меняет
фенотип клетки, вызывая изменения ее размера, секреции клеткой факторов
роста, нарушение связей клетки с близлежащими клетками, а также ряд
других изменений фенотипа, включающих приобретение способности
образовывать злокачественную опухоль и метастазировать.
Онкоген карциномы мочевого пузыря, выявленный с помощью
использования технологии клонирования генов, происходил из нормального
клеточного гена, строение которого была почти идентично структуре EJ/T24.
Такой нормальный ген, мутация которого приводит к образованию
онкогена, называют npomo-онкогеном. Прото-онкоген - это нормальный ген,
который приобретает свойства онкогена в результате изменения нуклео-
тидной последовательности ДНК.
Прото-онкогены содержит геном многих клеток многих организмов.
Это позволяет считать их играющими важную роль в функционировании
212
Глава 14
клеток и всего организма. Поэтому прото-онкогены остались
незатронутыми мутациями в течение всего длительного пути эволюции. Вовлечение
прото-онкогенов в канцерогенез после их трансформации в онкогены
происходит только в результате относительно редких повреждений
генетического материала клетки. В результате этих повреждений нарушаются
функции прото-онкогенов, что расстраивает систему регуляции клеточного роста.
Для многих прото-онкогенов, мутация которых в составе генома
трансформирующего ретровируса была причиной малигнизации клеток
инфицированной клеточной культуры, была установлена способность
превращаться in vivo в онкогены в результате мутаций, не связанных с действием
вирусов. Так Ha-ras-онкоген (англ. rat's sarcoma - саркома крыс),
превращение которого в онкоген из протоонкогена было связано с эффектом на геном
клеток ретровируса саркомы крыс, по строению своему был почти
идентичен EJ/T24. Оказалось, что прото-онкоген аЫ (англ. mouse Abelson leukemia -
лейкемия мышей Абельсона), впервые идентифицированный как протоон-
коген при исследовании культуры клеток, инфицированной ретровирусом,
вызывающим у мышей лейкемию, участвовал в канцерогенезе при миело-
генных лейкемиях у людей, не инфицированных ретровирусом.
Прото-онкоген туе (англ. avian myelocytomatosis - миеломатоз птиц), который
трансформировался в онкоген после включения в геном соответствующего
ретровируса, был причастен к этиологии многих хронических лейкемий,
возникавших без инфицирования больных ретровирусами (табл. 14.1).
Трансформацию прото-онкогенов в онкогены обуславливают различные
соматические мутации. Мутация, вызывающая рак мочевого пузыря, - это
точечная мутация (генная или точечная мутация - это результат изменения
нуклеотидной последовательности молекулы ДНК в определенном участке
хромосомы, гене), которая касается только одной пары спаренных
оснований двойной спирали ДНК.
Озлокачествление клетки начинается с изменения структуры ДНК под
влиянием способной вызывать изменения нуклеотидной
последовательности ДНК формы канцерогена (реактивной формы канцерогена) или
свободных кислородных радикалов при их усиленном образовании в клетке.
Изменение структуры ДНК служит причиной мутации определенных генов
(прото-онкогенов) и генов-супрессоров опухолей. Экспрессия измененного
генома клетки, содержащего онкогены и инактивированные гены-супрессо-
ры опухолей, патологически меняет клеточный фенотип. В результате
клетка приобретает свойства, характеризующие ее как злокачественную.
Когда у определенного химического вещества выявляют свойство
вступать в реакцию с ДНК, меняя ее структуру и вызывая мутации генов как
причину озлокачествления различных клеток, то его относят к
ДНК-реактивным или генотоксическим канцерогенам. Большинство из
канцерогенов, чье действие вызывает малигнизацию клеток организма человека, -
это генотоксические канцерогены.
Действие некоторых из гормонов (диэтилстильбэстрол, эстрадиол и др.)
ведет к образованию в организме веществ со свойствами генотоксических
канцерогенов.
Канцерогенез
213
Таблица 14.1
Некоторые клеточные онкогены, играющие роль в этиологии злокачественного
клеточного роста у человека
Онкоген
аЫ
eib-B
erb-B-2
HER-2/neu
Ha-ras
Ki-ras
N-ras
myc
N-myc
Ретровирус,
трансформирующий прото-онко-
ген в онкоген
Вирус лейкемии
мышей Абель-
со на
Вирус эритро-
бластоза птиц
Вирус саркомы
мышей Гарвея
Вирус саркомы
мышей Кирстена
Вирус миелома-
тоза птиц
Зл окаче стве иная
опухоль, в развитии
которой играет роль
данный онкоген
Миелогенные
лейкемии
Карцинома молочной
железы, глиобластома
Рак яичников,
молочной железы и желудка
Множество
злокачественных опухолей
Множество
злокачественных опухолей
Множество
злокачественных опухолей
Лимфома
Нейробластома
Локализация
белка продукта
экспрессии
онкогена
Плазматическая
мембрана клеток
крови миелоид-
ного ряда
Плазматическая
мембрана
Нар>окная
клеточная мембрана
Цитоплазматичес-
кие мембраны
Цито
плазматические мембраны
Цитоплазматичес-
кие мембраны
Ядро клетки
Функция
белка
Рецептор ти-
розинкиназы
Рецептор ти-
розинкиназы
Рецептор ти-
розинкиназы
Связывание и
гидролиз гуано-
зинтрифосфата
Связьшание и
гидролиз гуано-
зинтрифосфата
Связьшание и
гидролиз гуано-
зинтрифосфата
Транскрипционный фактор
Транскрипционный фактор
(предположительно)
При обмене веществ в нормальной клетке высвобождаются свободные
кислородные радикалы. В клетке есть эндогенные системы инактивации и
выведения из нее свободных кислородных радикалов, главными
молекулярными эффекторами которых часто являются ферменты (супероксиддис-
мутаза, глютатионпероксидаза и др.). Несмотря на действие эндогенных
защитных систем свободные кислородные радикалы могут изменять
структуру ДНК через образование нуклеотидов, содержащих 8-гидроксидеокси-
гуанин. Особенно велика вероятность таких изменений структуры ДНК в
присутствии ряда канцерогенов химической природы. Работа механизмов
«ремонта» ДНК, которая складывается из экспрессии соответствующих
генов и действия белка эффектора функции, может восстановить структуру
ДНК, устранив из ее состава 8-гидроксидеоксигуанин.
Вещества способные вступать в реакции метилирования и этилирова-
ния с ДНК также могут быть ДНК-реактивными канцерогенами. Под
метилированием в молекулярной биологии понимают присоединение
метиловой группы к цитозиновому остатку как элементу двойной спирали
дезоксирибонуклеиновой кислоты. В результате метилирования гены, со-
214
Глава 14
ставляющие двойную спираль ДНК, становятся неактивными.
Метилирование может быть нормальным процессом регуляции экспрессии
элементов генома. В частности, метилирование служит механизмом клеточной
дифференциации В-лимфоцитов. Метилирование может закрепляться в
генетическом материале гамет и определять фенотипические признаки,
передаваемые по наследству при мейозе (импринтинг, англ.
imprinting-штампование, закрепление информации). Нередко метилирование подвергается
обратному развитию (деметилирование), что меняет экспрессию генома и
функции клетки, в некоторых случаях приводя их в соответствие с
биологической целесообразностью.
Если действие генотоксического канцерогена приводит к
метилированию гена-супрессора опухоли, которое инактивирует этот ген, то такое
изменение генома клетки и связанная с ним модификация фенотипа может
служить одним из этапов канцерогенеза.
Канцерогены, способные вызывать метилирование или этилирование
ДНК ведут к появлению в ее составе 06-алкилгуанинов. В клетках
существует система восстановления структуры ДНК, измененной через
метилирование или этилирование. В результате ее действия, связанного с
активностью фермента метилтрансферазы, каждый моль О^метиловой группы
трансформируется в один моль сульфгидрильного остатка. Исследование
данной эндогенной системы восстановления структуры ДНК привело к
идентификации многих канцерогенов.
Чаще всего одним из этапов канцерогенеза при возникновении разных
злокачественных опухолей человека является инактивация гена-супрессора
опухолей, экспрессия которого дает фосфопротеин р53. Ген р53
локализован на коротком плече семнадцатой хромосомы. Функция данного фосфоп-
ротеина состоит в торможении клеточного роста и блокаде
злокачественной трансформации клетки. Продукт гена р53 блокирует малигнизацию,
задерживая пролиферацию клетки с измененной нормальной нуклеотидной
последовательностью ДНК, что дает системам восстановления нормальной
структуры ДНК достаточное время для достижения своего конечного
полезного присособительного результата. Для этого р53 активирует
транскрипцию генов, кодирующих белки, которые подавляют пролиферацию клетки.
Инактивация гена р53 или предрасполагает к канцерогенезу через
неограниченную пролиферацию или служит одной из причин озлокачествления
клетки в случае потери фосфопротеином вследствие мутации гена свойства
блокировать канцерогенез. Синтез и содержание р53 снижены в клетках
большинства злокачественных опухолей легких, при колоректальном раке
(у 50 % больных) и раке мочевого пузыря. Инактивация гена р53 чаще всего
представляет собой последний этап изменения генетического материала
клетки на пути ее озлокачествления.
При канцерогенезе, ведущем к образованию многих злокачественных
опухолей человека, сначала происходит делеция (потеря, удаление) одного
из аллелей гена р53. Оставшийся аллель (альтернативная форма гена)
претерпевает мутацию. Если делеции одного из аллелей нет, то для
инактивации гена-супрессора клеточного роста необходимы мутации двух аллелей.
Канцерогенез
215
Обусловленная мутацией замена лишь одной аминокислоты в составе р53
может инактивировать его как протеин, подавляющий пролиферацию
клеток и тем самым препятствующий малигнизации. Об инактивации
соответствующего гена-супрессора опухолей свидетельствует возросшая
способность р53 связываться с белком теплового шока hsp70. Мутации гена р53,
играющие роль в канцерогенезе, неоднородны по механизму
возникновения. В частности инактивировать ген могут транзиция1 и изменение
структуры одной пары спаренных азотистых оснований ДНК. Это указывает на
полиэтиологичность мутации гена-супрессора опухолей р53 как одного из
этапов канцерогенеза.
Если онкогенная мутация закрепляется в геноме всего организма и
попадает в генетический материал гамет, то потомство наследует высокую
предрасположенность к политопным злокачественным опухолям, то есть
одну или две инактивированные аллели прото-онкогена.
Синдром Ли-Фраумени - это наследуемое по аутосомально-доминант-
ному типу состояние высокой предрасположенности к злокачественному
клеточному росту различной локализации (опухоли надпочечников,
молочной железы, гортани, легких, мозга, лейкозы и лимфомы), при котором
вероятность озлокачествления клеток велика на любом этапе онтогенеза.
Фибробласты кожи больных с синдромом Ли-Фраумени
высокорезистентны к ионизирующей радиации (характерная особенность клеток,
вступивших на путь канцерогенеза) и содержат в 3-8 раз больше нормального
продукта экспрессии прото-онкогена с-тус. Кроме того, в них
активирован прото-онкоген c-raf. Предрасположенность к канцерогенезу при
синдроме Ли-Фраумени представляет собой результат двух мутаций двух
аллелей гена р53. Один аллель мутирует в ряду предшествующих поколений
предков больного, а второй - по ходу онтогенеза. Аналогично происходит
и двухэтапное изменение генетического материала клеток,
предрасполагающее к возникновению ретинобластомы. Данный механизм
наследования и приобретения предрасположенности к раку и индукции
канцерогенеза получил название «двух последовательных ударов по генетическому
материалу клетки».
Прото-онкогены - это элементы генома клетки, которые кодируют
белки (npomo-онкопротеины), необходимые для нормального
функционирования систем регуляции клеточного роста. Обычно эти гены кодируют белки,
играющие важную роль в регуляции экспрессии генов или в передаче
сигнала геному от цитокинов со свойствами факторов клеточного роста.
Мутация прото-онкогенов ведет к их активации, то есть к приобретению
продуктами их экспрессии свойств онкопротеинов, белков, чье действие на клетку
может привести к малигнизации клетки. В настоящее время известно более
чем 50 прото-онкогенов (табл. 14.1).
1 Транзиция (англ. Transition - переход, перемещение) - точечная мутация,
состоящая в замене одного пурина или пиримидина в составе дезоксирибонуклеино-
вой кислоты другими, занимавшими до транзиции другое место в нуклеотидной
последовательности ДНК.
216
Глава 14
Онкогены составляют разнородную группу «раковых генов», которые,
возникая в результате мутаций прото-онкогенов, обладают способностью
вызывать злокачественное перерождение клетки. Одна из частых причин
мутаций прото-онкогенов - это нарушение нормального
функционирования фермента ДНК-полимеразы. Онкогены могут происходить из генома
содержащих рибонуклеиновую кислоту вирусов или быть элементом
генома нормальной немалигнизированной клетки (прото-онкогены).
Кроме точечных мутаций, механизмом активации прото-онкогенов
могут быть цитогенетические аномалии: транслокация, амплификация генов,
а также потеря хромосомами своих частей (делеция).
Транслокация - это перемещение сегмента одной хромосомы в другую.
Филадельфийская хромосома - это небольшая акроцентрическая
хромосома, которая представляет собой остаток двадцать второй хромосомы после
транслокации дистальной части ее длинного плеча в длинное плечо
девятой. Чаще всего транслокация приводит к образованию филадельфийской
хромосомы в полипотентной стволовой клетке и сохраняется в клетках ми-
елоидной, эритроидной, мегакариоцитарной и лимфоидных линий
клеточной дифференциации. Транслокацию с образованием филадельфийской
хромосомы как причину возникновения онкогена и канцерогенеза
выявляют у 95 % хронической миелоидной лейкемией и у 5-20 % пациентов с
острой лимфоцитарной лейкемией. В результате транслокации ген с-аЫ из
состава двадцать второй хромосомы становится в положение юкстапози-
ции с геном Ъсг девятой, функция которого еще полностью не ясна. Это
ведет к образованию нового гибридного гена, экспрессия которого
завершается образованием онкопротеинаР210*сг/аА/. Онкопротеины,
представляющие собой продукт экспрессии онкогена, образовавшегося при
возникновении из двух генов гена-гибрида (слияние генов) называют слившимися или
спаянными белками (от англ. fusion proteins, что можно перевести как
«белки, образовавшиеся в результате слияния»).
Действие онкопротеинов, приводящее к малигнизации клетки, всегда
можно свести к изменениям или индукции конкретных биохимических
реакций. Так онкопротеин Р21 $>сг/ш предположительно обладает активностью
тирозинкиназы, которая не зависит от функционирования эндогенной
системы регуляции активности данного энзима. Такой же активностью
обладает прото-онкопротеин (продукт экспрессии онкогена src). Тирозинкиназа
осуществляет фосфорилирование остатка тирозина в составе белковых
молекул. Тирозинкиназа фосфорилирует белки, находясь в составе тирозинки-
назного рецептора наружной клеточной мембраны. Через
фосфорилирование осуществляется передача сигнала к интенсификации пролиферации клеток
на уровне взаимодействия ФР со своими рецепторами на поверхности клеток
(эпидермальный и тромбоцитарный факторы клеточного роста и др.).
Амплификация генов (лат. Amplificatio -распространение, увеличение) -
это возникновение в ядре клетки одного или нескольких дубликатов целой
хромосомы, ее частей или отдельных фрагментов генетического
материала. В ходе амплификации прото-онкогены становятся онкогенами как в
результате мутаций и слияний генов, так и вследствие повышенного син-
Канцерогенез
217
теза клеткой продуктов копий амплифицированных прото-онкогенов,
стимулирующих клеточный рост. Морфологически амплификация генов
проявляет себя появлением в ядре хроматиновых телец, не связанных с
хромосомами, или изменением генетического материала, которое характеризует
возникновение гомогенно окрашиваемых участков хромосом. Так,
канцерогенез при нейробластоме отчасти представляет собой результат
амплификации прото-онкогена N-myc.
Делеция, то есть потеря хромосомами части входящего в их состав
генетического материала, служит причиной канцерогенеза, когда эта
часть содержит ген-супрессор опухолей. Потеря одного плеча первой
хромосомы служит одним из этапов многоступенчатого изменения
генетического материала клетки, составляющего канцерогенез при образовании
твердых злокачественных опухолей.
Высокая частота деления клеток предрасполагает к канцерогенезу, так
как снижает время, «отпущенное» для воссстановления нормальной нукле-
отидной последовательности ДНК. Растущий организм более
предрасположен к индукции малигнизации эффектом канцерогенов. Воздействие
радиации при атомной бомбардировке Хиросимы привело к возникновению рака
молочной железы по ходу онтогенеза в четыре раза чаще у тех женщин,
которым в момент взрыва было 10-15 лет (период онтогенеза, в который
наиболее интенсивна пролиферация клеток молочной железы).
ДНК-реактивные канцерогены вступают в прямые или опосредованные
реакции не только с ДНК, но и с другими протеинами организма. Это ведет к
появлению аномальных белков, специфических для того или иного варианта
канцерогенеза (маркеры злокачественных клеточного роста и опухолей).
Одного изменения генетического материала, включающего все этапы
канцерогенеза на уровне генома одной клетки, которое вызывают действие
химических канцерогенов, свободных кислородных радикалов, онкогенных
вирусов и эффект ионизирующих излучений, еще недостаточно для
возникновения из одной малигнизированнои или ряда озлокачествленных клеток
раковой опухоли.
Процесс возникновения выявляемых при обследовании больного клона
(гр. klon-ветвь, побег, отпрыск) злокачественных клеток или раковых
опухолей из одной малигнизированнои или группы озлокачествленных клеток
называют промоцией (англ. promotion, содействие). Экзогенное вещество
или эндогенный агент эндокринной, пара- или аутокринной регуляции, чье
действие на клетки обуславливает промоцию, называют промотором.
Промотор не может вызвать рак без предшествующего многоэтапного и
комплексного изменения генетического материала клетки.
Воздействуя на малигнизированные клетки, промоторы ускоряют их
клональную экспансию как причину возникновения злокачественных
опухолей. Многие из промоторов вызывают промоцию через увеличение
активности фермента протеинкиназы С и снижение в клетке активности
энзимов, относящихся к классу белковых фосфатаз. Во многом действие
промоторов сводится к нарушению межклеточных соединений в области
зазоров между клетками и функциональных связей между ними. Действие
218
Глава 14
Таблица 14.2
Классификация химических соединений со свойствами канцерогенов
Вид канцерогена
А. Гено}поксические
1. Не требующие
активации
2. Становящиеся
канцерогенами после
активации (проканцерогены)
3. Неорганические
канцерогены, не
требующие активации
Б. Эпигенетические
4. Твердые
канцерогены
5. Гормоны
6. Иммунодепрессанты
7. Вещества,
усиливающие образование
свободных кислородных
радикалов (СКР)
8. Цитотоксические
вещества
9. Вещества
предрасполагающие к геноток-
си чес кому действию
ДНК-реактивных
канцерогенов
Механизм действия
Прямая реакция электрофильных
органических соединений с ДНК
Вступает в реакцию с ДНК после
трансформации на путях
метаболизма
Избирательные нарушения
репликации ДНК и систем
восстановления ее нормальной структуры
после мутации
Обычно оказывают действие на
мезенхимальные клетки и ткани;
механизм канцерогенного
действия неизвестен
Системная эндокринопатия как
причина неконтролируемой
клеточной пролиферации
Ускоряют выход
малигнизированных клеток из-под контроля
системы иммунитета
Рост содержания в клетке
свободных кислородных радикалов,
преобладающий над способностью
клетки инактивировать и
элиминировать СКР
Цитолиз как причина воспаления,
усиления пролиферации и роста
содержания СКР в клетке и интер-
стиции
Усиление клеточной
пролиферации, которое уменьшает время,
необходимое для эффективной
работы систем восстановления
структуры ДНК
Пример
Бис(хлорметил) эф ир
Винилхлорид,
бензопирен
Никель,
хром
Полимеры,
используемые для изготовления
современных
упаковочных материалов, фольга
Эстрадиол,
диэтилстильбэстрол
Циклоспорин А, анти-
лимфоцитарная
сыворотка
Клофибрат,
диэтилгексилфталат
Нитрилотриацетат,
четыреххлористый
углерод
Фенолы,
желчные кислоты
промотора как причина промоции должно быть достаточно интенсивным,
непрерывным и длительным. В противном и крайне желательном случае
злокачественная опухоль подвергается обратному развитию.
У мышей, инфицированных вирусом, вызывающим рак молочной
железы, действие эстрогенов ведет к возникновению раковых опухолей. При этом
образование опухоли находится в прямой связи с дозой эстрогенов, а также
частотой и длительностью их парентерального введения. Если мышей не
инфицировать вирусом, то эстрогены, вводимые в той же дозе, таким же
образом и столь же длительно, рака не вызывают. В данном случае
эстрогены выступают в роли промоторов.
Канцерогенез
219
Промоторы могут обладать тканевой специфичностью. Так желчные
кислоты - это промоторы рака толстой кишки, а сахарин натрия может
обуславливать промоцию при раке мочевого пузыря.
Канцерогены - это химические вещества или воздействия на клетки
как экзогенного, так и эндогенного происхождения, обуславливающие
изменения генетического материала клетки при канцерогенезе или
промоцию (табл. 14.2).
Генотоксические или ДНК-реактивные канцерогены - это мутагены,
которые обладают свойством менять нуклеотидную последовательность
ДНК, активируя прото-онкогены или инактивируя гены-супрессоры
опухолей. Канцерогены, чье действие не затрагивает генетический материал
клетки, и которые выступают исключительно в роли промоторов, называют
эпигенетическими канцерогенами. Эпигенетические канцерогены вызывают
злокачественные новообразования и клоны малигнизированных клеток
через свои цитотоксические эффекты, хроническое травмирование тканей, при
эндокринопатиях, приводя к иммунодефициту и другим системным
сдвигам во взаимосодействии функциональных систем, направленном на
поддержание гомеостазиса.
КАНЦЕРОГЕНЕЗ И ФАКТОРЫ КЛЕТОЧНОГО РОСТА
Необходимым условием нормального клеточного роста является не
только достаточное поступление в клетку нутриентов, витаминов и
микроэлементов. Для нормального роста клеток необходима достаточная концентрация в
среде их обитания факторов клеточного роста и его ингибиторов. Наиболее
изученными из общих для многих видов млекопитающих пептидных
факторов роста являются инсулин, трансферрин и эпидермальный фактор роста.
Злокачественные клетки способны к неограниченному делению при крайне
малом содержании в среде их обитания факторов роста (ФР), которые
содержат сыворотка крови и вся внеклеточная жидкость человека. Независимость
роста злокачественных клеток от ФР сыворотки человека связана с
изменениями обмена веществ, которые представляют собой результат изменения
фенотипа клетки на пути малигнизации. Клетка как бы постоянно находится
в состоянии вследствие воздействия на нее факторов роста. Это состояние
персистирует и без взаимодействия факторов роста со своими рецепторами.
В результате клеточный рост злокачественной клетки уже не зависит от
влияния на нее факторов клеточного роста.
Геном злокачественной клетки и синтез белков в ней часто меняется
таким образом, что на пострецепторном уровне постоянно через
экспрессию онкогенов в виде онкопротеинов воспроизводится сигнал к
усилению клеточной пролиферации от факторов роста во внеклеточном
пространстве без взаимодействия ФР со своими рецепторами на наружной
поверхности клеток.
220
Глава 14
Мутация некоторых прото-онкогенов из семейства ras, которая служит
одной из причин возникновения рака легких, прямой кишки и
поджелудочной железы, ведет к озлокачествлению клетки именно таким путем. Ras-
прото-онкогены кодируют внутриклеточные белки, связанные с цитоплаз-
матическими мембранами. Эти белки могут связывать гуанозинтрифосфат
и обладают свойствами энзимов, вызывающих гидролиз
гуанозинтрифосфата (ГТФ) в молекулярном комплексе г<х?-прото-онкопротеин-
гуанозинтрифосфат. Связывание продуктами экспрессии прото-онкогенов
из семейства ras гуанозинтрифосфата представляет собой один из этапов
передачи сигнала к усилению пролиферации клетки, который на уровне
плазматической мембраны передает взаимодействие лиганды в виде
фактора роста с рецептором наружной клеточной поверхности. Взаимодействие
фактора роста, находящегося во внеклеточном пространстве, с
трансмембранным рецепторным белком на поверхности клетки ведет к обратимому
связыванию ras-прото-онкопротеином гуанозинтрифосфата, что служит
следующим этапом передачи сигнала к усилению пролиферации клетки,
пришедшего из внеклеточного пространства. В дальнейшем через
гидролитическую активность ras-прото-онкопротеина ГТФ в составе
молекулярного комплекса ras-прото-онкопротеин-ГТФ превращается в гуанозиндифос-
фат, и передача сигнала прекращается. Онкогенная мутация ras-прото-он-
когена меняет его экспрессию таким образом, что ras-прото-онкопротеин
претерпевает конформационные изменения, становясь онкопротеином. Этот
онкопротеин почти не обладает гидролитической активностью, что
обуславливает необратимый характер его связывания с ГТФ. В результате
нарушений передачи информации на пострецепторном уровне геном клетки
получает постоянный интенсивный сигнал к пролиферации без какого-либо
взаимодействия внеклеточного фактора роста с рецептором наружной
клеточной поверхности.
Независимость деления клеток злокачественных опухолей от влияния
факторов роста, находящихся во внеклеточном пространстве, лишает ма-
лигнизированную клетку свойств элемента целостных ткани или органа.
Дело в том, что межклеточные взаимодействия, интегрирующие клетки для
реализации нормальной работы тканей и органов как эффекторов функций,
во многом осуществляются через регуляторные влияния факторов
клеточного роста. Злокачественный клеточный рост представляет собой
типовой патологический процесс дезинтеграции ткани на автономные
клеточные элементы, утратившие качества структурно-функциональных единиц
ткани, органа и всего организма.
Экспрессия онкогенов может приводить к такой автономии
злокачественных клеток посредством действия четырех основных механизмов.
Первый из них состоит в секреции малигнизированной клеткой митоге-
нов, которые в качестве агентов аутокринной регуляции стимулируют
пролиферацию самой высвобождающей их злокачественной клетки.
Продукты онкогенов могут усиливать аутокринную стимуляцию
пролиферации злокачественных клеток, резко повышая уровень экспрессии
нормальных генов, кодирующих факторы клеточного роста. Транскрипция и
Канцерогенез
221
трансляция активированных прото-онкогенов ведет к неконтролируемой
экспрессии нормальных генов, приобретающих таким образом качество
онкогенов. Среди факторов клеточного роста (ФР), осуществляющих ауток-
ринную стимуляцию клеток злокачественных опухолей, наиболее изучены
альфа-фактор роста опухолей, тромбоцитарный ФР и бета-фактор роста
опухолей.
Второй механизм независимости роста злокачественной клетки от ФР
сыворотки крови - это изменение содержания рецепторов к ФР (ФР-ре-
цепторов) на единице площади поверхности злокачественных клеток или
изменение структуры ФР-рецепторов. Это обуславливает или
повышенную чувствительность клетки к действию на нее митогенов, находящихся
вне клетки, или постоянное возбуждение рецепторов без взаимодействия
с лигандами в виде ФР.
Поверхность клеток агрессивно растущей и метастазирующей опухоли
молочной железы почти полностью покрыта ФР-рецепторами, белковый
элемент которых представляет собой продукт прото-онкогена HER-2/лем.
Наружная мембрана клеток эпидермоидной карциномы (клеточная линия
А-431) содержит аномально много рецепторов к эпидермальному фактору
роста, белковый элемент которых лишен своей определенной части. Эта
аберрация рецептора приводит к тому, что для последующих элементов,
передающих сигнал к клеточному росту до генома, он предстает как бы в
постоянно активированном состоянии.
В культуре доброкачественных клеток некоторых видов
экспериментальных животных (грызуны и др.) размножение большинства клеток
ограничено во времени, то есть в определенном поколении одной клеточной линии
митоз прекращается. В части же клеток продолжают чередоваться фазы
митоза. Эта часть клеток как бы становится бессмертной. В клеточной
культуре нормальных тканей человека через некоторое время прекращается
деление всех клеток. У культуры злокачественных опухолевых клеток
человека потенциал клеточного роста практически неисчерпаем, то есть клетки
всех линий являются бессмертными.
Приобретение клеткой в культуре клеток тканей человека бессмертия
еще не эквивалентно ее малигнизации. Так экспрессия генов из состава
генома некоторых онкогенных вирусов, инфициирующих клетки, ведет к их
бессмертию без других изменений фенотипа, характеризующих
завершенный канцерогенез.
Третий механизм автономии клетки относительно содержания
факторов роста в среде ее обитания связан с элементами системы передачи
сигнала к клеточному росту на цитоплазматическом пострецепторном
уровне. Кроме экспрессии онкогенов из семейства ras (см. выше в данном
разделе), постоянную генерацию сигнала к клеточному росту на
пострецепторном уровне без взаимодействия ФР с ФР-рецепторами
обуславливают продукты онкогенов типа scr.
Четвертый механизм приобретения пролиферации злокачественной
клетки независимости от влияний факторов клеточного роста состоит в
экспрессии онкогенами в качестве онкопротеинов транскрипционных факторов.
222
Глава 14
Транскрипционные факторы (ТФ) - это функционирующие в пределах
ядра клетки протеины (ядерные белки), которые обладают способностью
связываться с ДНК в регуляторных элементах генов. Один
транскрипционный фактор может регулировать транскрипцию или экспрессию многих
генов, взаимодействуя с энзимом РНК-полимеразой. Поэтому мутация
одного гена ТФ через изменение или усиление образования его продукта может
менять весь клеточный фенотип, придавая ему черты малигнизации. Если
на нормальную клетку не действует тот или иной фактор клеточного роста,
то ее ядро почти не содержит соответствующих транскрипционных
факторов. Воздействие фактора роста резко усиливает образование ТФ. В ряде
злокачественных клеток концентрация ТФ в ядре постоянно и аномально
высока, что ведет к их интенсивному делению, не зависящему от влияний
на клетку ФР.
В развитии нейробластомы детского возраста имеет значение
амплификация прото-онкогена N-myc, представляющего собой ген
транскрипционного фактора. Повышенная экспрессия данного прото-онкогена - может
быть одной из причин малигнизации.
МЕТАСТАЗИРОВАНИЕ ЗЛОКАЧЕСТВЕННОЙ КЛЕТКИ
КАК ТИПОВОЙ ПАТОЛОГИЧЕСКИЙ ПРОЦЕСС
Способность злокачественных клеток прорастать в близлежащие ткани
и метастазировать в отдаленные органы часто определяет невозможность
успешного хирургического лечения злокачественной опухоли. Метастази-
рование клеток опухоли - это многоэтапный процесс. Выделяют пять
основных этапов метастазирования:
♦ выход злокачественной клетки из локуса первичной малигнизации и
ее миграция через прилежащие к ней тканевые барьеры (базальные
мембраны и др.);
♦ проникновение злокачественной клетки в просвет кровеносных и (или)
лимфатических сосудов (интравазация) и начало циркуляции
злокачественных клеток с кровью;
♦ приобретение малигнизированными клетками, циркулирующими с
кровью, жизнеспособности, что связано с их выходом из-под
контроля иммунной системы;
♦ выход малигнизированных клеток через базальную мембрану
капилляров в ткани (экстравазация);
♦ внедрение злокачественной клетки в ткань отдаленного от места
первичного озлокачествления органа и возникновение нового очага
злокачественного клеточного роста.
В некоторых случаях злокачественные клетки смешиваются с
нормальными клетками или замещают их, но при этом сохраняется порядок
структурной организации ткани. Например, при возникновении некоторых опу-
Канцерогенез
223
холей молочной железы злокачественные клетки в протоках железы
замещают нормальные клетки на протяжении нескольких миллиметров, но не
пенетрируют базальную мембрану с выходом в прилежащие к ней
межклеточные пространства. При таком распространении злокачественных
клеток в пределах здоровой ткани метастазирование происходит редко (рак
in situ). Вероятность метастазирования при раке молочной железы
особенно высока, когда злокачественные клетки приобретают способность
выходить из протока железы через базальную мембрану в прилежащие к
ней ткани.
Большинство злокачественных новообразований возникает из
эпителиальных клеток, образующих клеточный слой на базальной мембране,
состоящей из плотных протеогликанов. Базальная мембрана обычно
является первым барьером для метастазирования злокачественных клеток.
Малигнизированные клетки часто секретируют энзимы коллагеназу, гепа-
риназу и стромогеназу, разрушающие физический барьер на пути
метастазирования в виде базальной мембраны. Экспрессия генов этих ферментов
может обуславливать приобретение злокачественной клеткой способности
мигрировать через прилежащие к ней тканевые структуры и клеточные слои
в циркулирующую кровь.
После выхода в циркулирующую кровь экранирование поверхности
малигнизированной клетки фиксированными на ней нитями фибрина и
агрегатами тромбоцитов, защищает её от взаимодействия с элементами
системы иммунитета организма. Попадание злокачественных клеток в
циркулирующую кровь еще не означает неизбежности возникновения
метастатического очага злокачественного клеточного роста. Дело в том, что
выживаемость злокачественной клетки в циркулирующей крови
исключительно мала. После интравазации выживает лишь 0,1 % злокачественных
клеток, попавших в циркулирующую кровь
Клетки опухолей разных видов метастазируют преимущественно в
определенные органы. Иногда это связано с особенностями венозного оттока
из органа, где происходит первичная малигнизация. Например, опухоли
толстой кишки в основном метастазируют в печень, в которую
злокачественные клетки поступают с кровью по воротной вене. Во многих случаях
преимущественное метастазирование опухоли определенного вида в
определенный орган не представляется возможным связать с
анатомическими особенностями венозного оттока из органа, где происходит первичная
малигнизация. Это, в частности, относится к нередкому метастазированию
рака желудка в надпочечниковые железы.
Злокачественная клетка, которую покрывают нити фибрина и агрегаты
тромбоцитов, может эмболизировать прекапиллярные артериолы в первом
из органов, в который она поступает с артериальной кровью. В дальнейшем
озлокачествленные клетки часто высвобождаются из состава эмбола и
попадают в просвет посткапиллярных венул, минуя капилляры. Для адгезии
малигнизированной клетки к стенке посткапиллярной венулы необходимо,
чтобы поверхность злокачественной клетки содержала рецепторы к
адгезивным молекулам эндотелия определенного органа, которым предположи-
224
Глава 14
тельно присуща органоспецифичность. Межклеточное взаимодействие ма-
лигнизированной клетки с эндотелиоцитами посткапиллярных венул ведет
к сокращению эндотелиальных клеток. Между эндотелиальными клетками
в результате их сокращения образуются зазоры, что делает возможным
прямое взаимодействие малигнизированных клеток с адгезивными
молекулами субэндотелиальной базальной мембраны.
Фиксация малигнизированной клетки на субэндотелиальной базальной
мембране представляет собой первый этап экстравазации с последующим
внедрением в строму органа, отдаленного от очага первичной
малигнизации. После фиксации к субэндотелиальной мембране злокачественная клетка
высвобождает ряд ферментов, разрушающих ее. Малигнизированные
клетки, которые не обладают способностью, высвобождая энзимы, разрушать
базальную мембрану посткапиллярной венулы, после обратимой адгезии с
мембраной возобновляют циркуляцию с кровью. Некоторые продукты
вызванной гидролазами злокачественной клетки деструкции протеогликанов
базальной мембраны, представляют собой хемоаттрактанты для
малигнизированной клетки. В результате после расщепления базальной мембраны
посткапиллярной венулы гидролазами злокачественной клетки ее инвазия
в строму отдаленного органа происходит хемотаксически.
Инвазия злокачественной клетки в строму органа, отдаленного от очага
первичной малигнизации, зависит от ее способности разрушать через вые-
вобождение ферментов-гидролаз субэндотелиальную мембрану с
расщеплением протеогликанов мембраны до органо-специфичных биоактивных
веществ, обладающих относительно данной малигнизированной клетки
свойствами хемоаттрактантов.
После инвазии злокачественной клетки в строму органа, отдаленного от
локуса первичной малигнизации, возникновение метастаза опухоли может
быть связано с содержанием в органе факторов и ингибиторов клеточного
роста. Установлено, что фактор роста, который содержит паренхима
легких, стимулирует рост клеток аденокарциномы молочной железы,
попадающих в легкие.
Часть II
КЛИНИЧЕСКАЯ
ПАТОФИЗИОЛОГИЯ
ФУНКЦИОНАЛЬНЫХ
СИСТЕМ
Глава 15
ДИЗРЕГУЛЯЦИЯ И ПАТОЛОГИЧЕСКИЕ
ИЗМЕНЕНИЯ ЭФФЕКТОРОВ КАК ПРИЧИНЫ
РАССТРОЙСТВ ФУНКЦИОНАЛЬНЫХ СИСТЕМ
(НОЗОЛОГИЧЕСКИЙ ОЧЕРК)
Необходимым условием роста приспособительных возможностей
через эволюцию является совершенствование механизмов (систем)
регуляции. При этом совершенствование механизмов регуляции в основном
достигается посредством:
а) все большей эволюционной дифференциации клеточных элементов
организма, в ходе которой они становятся потенциальными эффекторами
ряда специфических функций, в том числе и клеточными эффекторами
систем регуляции;
б) все большей специализации передачи информации по
афферентным, эфферентным и другим каналам при одновременном появлении
нескольких систем регуляции, способных к реализации идентичных или
схожих по полезному приспособительному результату реакций;
в) возникновения ряда регуляторных интегрирующих систем
организма (центральная нервная система, система иммунитета и др.)
способных на основе афферентного синтеза оперативно перестраивать аппараты
регуляции и мобилизовать новые эффекторы для достижения полезного
приспособительного результата адекватного изменившимся условиям
существования организма. При этом интегрирующие системы организма
получают в ходе эволюции способность опережающего реагирования в
ответ на действие повреждающего организм или потенциально
патогенного экзо- или эндогенного фактора. В результате опережающего
реагирования алгоритм защитной реакции часто реализуется избыточно
относительно стимула ее вызвавшего, что обуславливает высокий риск
трансформации системных защитных реакций в звенья патогенеза болезней и
патологических состояний. Акад. П.К. Анохин формулирует этот
принцип компенсации нарушенных функций организма следующим образом:
«В то время как силы, отклоняющие функцию от нормы, растут в
арифметической пропорции, силы сопротивления этому отклонению растут в
геометрической пропорции, благодаря чему в нормальных условиях
отклоняющаяся от нормы функция, как правило, возвращается к ее
постоянному уровню».
Кроме того, совершенствование регуляторных механизмов по ходу
эволюции состоит во все большем образовании внутри организма
межсистемных регуляторно-функциональных связей, что обеспечивает
повышение эффективности взаимосодействия функциональных систем при
поддержании гомеостазиса и достижении полезных приспособительных
результатов. В результате защитные реакции при определенной интен-
Д из регуляция и патологические изменения эффекторов как причины расстройств... 227
сивности взаимодействия организма с этиологическим фактором
становятся системными сдвигами регуляции и состояния эффекторов во всех
функциональных системах организма. При потере физиологическими
реакциями гомеостатического смысла и приспособительного значения
(потеря физиологической меры реакции) данная особенность систем
регуляции функций высших организмов определяет организменный характер
патологической реакции. При этом системная патологическая реакция
выступает причиной патологических изменений регуляции и эффекторов
в относительном отдалении от локуса первичного взаимодействия
этиологического фактора и организма.
Все эти особенности и основные механизмы совершенствования
систем регуляции по ходу эволюции определяют возможность
возникновения болезней и патологических состояний, обусловленных
преимущественно нарушениями регуляции, то есть дисфункциями ее
систем. В данном контексте под дисфункцией систем регуляции мы
понимаем расстройство регуляторных систем различных уровней, которое
в первую очередь характеризует устойчивая генерация исполнительно-
го и одновременно тормозящего некоторые функции сигнала к
эффекторам функциональных систем, вызывающая их патологические
изменения, препятствующая достижению конечного полезного
приспособительного результата и расстраивающая регуляцию и всю работу
смежных (интерферирующих, в функциональном и морфологическом
отношении проникающих друг в друга) функциональных систем. Так
как каждому уровню структурно-функциональной организации
присущи свои особенности регуляции и соответствующие регуляторные
механизмы, то на каждом из таких уровней расстройства функциональных
систем имеют свою специфику.
В патологический процесс на клеточном и субклеточном уровнях
нередко оказываются вовлеченными многообразные системы передачи
информации, индуктором каскада реакций которых является
взаимодействие экзо- или эндогенной лиганды со своим рецептором.
Например, патогенные мутации, составляющие комплексное изменение
генома при канцерогенезе, через экспрессию мутантными аллелями онко-
протеинов могут на субклеточном уровне обусловить стойкий и
интенсивный сигнал геному о взаимодействии фактора клеточного роста со
своим рецептором. В данном случае происходит патогенный сдвиг
передачи информации к одной из граней всего возмоэюного спектра силы
и качества потока информации в пределах информационного канала
системы регуляции. Это предрасполагает к озлокачествлению клетки
посредством интенсификации клеточной пролиферации и закрепления
соматических мутаций. Такую модуляцию передачи информации в
регуляторных системах можно считать общей закономерностью дизрегу-
ляции при патологических процессах и болезнях. Следует учитывать,
что при развитии аномалий на субклеточном уровне происходит не
просто постоянная передача информации о взаимодействии лиганды со
своим рецептором. Передача не имеющей биологического смысла ин-
228'
Глава 15
формации происходит одновременно с амплификацией сигнала
эндогенной усилительной системой. Как указывает акад. Г.Н. Крыжа-
новский, такая стойкая и потерявшая физиологическую меру реакции
амплификация передачи информации внутриклеточной усилительной
системой ведет к патологии, то есть обуславливает растормаживание
клетки, ее гиперактивацию и неконтролируемое усиление клеточных
функций. Иными словами, патогенная модуляция передачи информации
в регуляторных системах, приобретение ею в пределах определенного
временного интервала патологических высокой интенсивности и
неизменности качества представляет собой один из универсальных
патогенетических механизмов, действие которых определяет аномалии
клеточного и субклеточного уровней. Позволим себе предположить, что
таким образом мы описали общепатологическую закономерность,
проявляющую себя в патогенезе на всех уровнях
структурно-функциональной организации.
Гиперактивацию в нашем контексте можно определить как
предельно возможную и стойкую мобилизацию клетки в качестве эффектора
функциональной или патологической системы. Гиперактивация
определенного числа клеток расстраивает функциональные системы,
обуславливая:
1. Потерю функциональными системами пластичности, то есть
возможности менять характеристики конечного полезного
приспособительного результата для приспособления, компенсации и саногенеза. Дело в
том, что одну и ту же клетку можно считать локусом эффекторов разных
и альтернативных по виду реализуемой функции функциональных
систем. Мобилизация клетки в качестве эффектора одной физиологической
(патологической) системы на основе реципрокных отношений почти
исключает использование данной гиперактивированной клетки в качестве
эффектора других систем. Вызывая снижение числа доступных
функциональным системам клеточных эффекторов, гиперактивация клеток при
соответствующем распространении данного потенциально патогенного
процесса через падение пластичности функциональных систем снижает
приспособительные возможности организма, а также эффективность
компенсации и саногенеза.
2. Превращение клеток после гиперактивации в источник
патологически интенсивных и устойчивых ауто- паракринных влияний, реализуемых
через секрецию цитокинов.
Гиперактивация клеток как эффекторов функциональных и
патологических систем никогда не происходит изолированно, то есть всегда
через изменение экспрессии генома мобилизует несколько потенциалов
клетки. Мобилизация определенных потенциалов клетки (их
максимальная экспрессия) происходит полностью и удерживается длительно.
В результате гиперактивация клетки не только снижает
приспособительные возможности организма и эффективность компенсации и
саногенеза, но и вызывает ряд сопряженных и усиливающих друг друга
патологических процессов.
Дизрегуляция и патологические изменения эффекторов как причины расстройств... 229
Особенно явно данные общие закономерности возникновения
расстройств функциональных систем проявляют себя при сепсисе,
системной воспалительной реакции и связанных с ними диссеминированном
внутрисосудистом свертывании и множественной системной
недостаточности. Длительная антигенная стимуляция иммуногенами
болезнетворных микроорганизмов и своими аутоантигенами из некробиотически
измененных клеток приводит к гиперактивации клеточных элементов всех
звеньев системы иммунитета организма. В результате на определенном
этапе развития системной защитной иммунной реакции преобладающее
число клеток системы иммунитета, находясь в гиперактивированном
состоянии, начинают высвобождать цитокины с предельной
интенсивностью. Следует заметить, что большинство клеток системы иммунитета
обладает способностью образовывать и высвобождать все известные в
настоящее время цитокины, оказывающие на клетки всех звеньев
системы иммунитета гиперактивирующие ауто- паракринные влияния. В
результате и без циркуляции возбудителя с артериальной кровью, при
отсутствии вторичных очагов гнойной инфекции и, несмотря на
радикальную санацию ее первичного очага, сепсис как типовой патологический
процесс продолжает свое развитие во времени, превратившись в
системную воспалительную реакцию. Системная воспалительная реакция через
гиперцитокинемию вызывает экспрессию тромбогенного потенциала эн-
дотелиоцитов, одновременную с мобилизацией потенциалов эндотели-
альной клетки как эффектора воспаления. Рост на системном уровне
секреции эндотелиальными клетками цитокинов со свойствами прокоагулян-
тов обуславливает диссеминированное внутрисосудистое свертывание
как причину коагулопатии потребления. Образование микросгустков в
циркулирующей крови нарушает периферическое кровообращение во
всех органах и тканях. В результате часть клеток в зоне микротромбоза
впадает в состояние гипоэргоза или даже становится на грань ишемии.
Это обуславливает первичную гипоксическую альтерацию во многих
органах и тканях как причину воспаления и множественной системной
недостаточности. Одновременно патогенное воспаление во многих органах
и тканях усиливается с активацией фактора Хагемана и системы кинино-
генеза при свертывании крови, что активирует систему комплемента по
альтернативному пути.
Данный пример связи разнообразных следствий гиперактивации
клеток и патологических реакций системного уровня представляет собой
одну из иллюстраций проявления еще одной общепатологической
закономерности: гиперактивация определенного числа клеточных эффекторов
функциональных (патологических) систем всегда приводит к
патологическим реакциям системного уровня, которые, в свою очередь, также
вызывают ряд патологических реакций на уровне всего организма.
Частные варианты проявления данной закономерности нередко представляют
собой механизмы эндогенизации патологических процессов.
Под эндогенизацией болезни и патологического процесса мы
понимаем приобретение им свойства генерировать на определенных этапах
230
Глава 15
причины своего дальнейшего развития в виде индукторов патологических
реакций, типовых патологических процессов и действия
патогенетических механизмов.
Эндогенизация не означает перерыва во времени цепочки причинно-
следственных связей, связывающей патологическое состояние в
настоящий момент развития болезни (патологического процесса) с
первопричиной болезни. Первопричина болезни посредством патогенного
изменения структуры и функций может менять функциональное
состояние эффекторов функциональных систем таким образом, что
восстановление физиологических условий их существования, нарушенных
вследствие взаимодействия с этиологическим фактором, представляет собой
фактор эндогенизации патологического процесса. Такое изменение
свойств эффекторов как причину эндогенизации не обязательно
вызывают первые по времени возникновения следствия взаимодействия
организма с этиологическим фактором. Между первопричиной болезни и
патогенным изменением реактивности эффектора по отношению к
восстановлению физиологических условий существования часто
прослеживается не прямая, но достаточно жесткая опосредованная связь
действий причин и возникновения следствий.
При гипертонической болезни, которая развивается параллельно с
атеросклерозом венечных артерий, возвращение артериального давления
в физиологические пределы может снизить повышенное перфузионное
давление субэндокардиального слоя миокарда левого желудочка, что
может обострить его циркуляторную гипоксию и обусловить прогрессиро-
вание сердечной недостаточности.
Взаимодействие организма с этиологическим фактором тяжелой
раневой болезни, действием ранящего снаряда, вызывая гиповолемию и
системные расстройства периферического кровообращения ставит многие из
клеточных элементов организма на край их ишемического цитолиза.
Известно, что повреждения тканей вследствие острой циркуляторной
гипоксии (ишемии) усиливаются восстановлением доставки кислорода в гипок-
сичные ткани (схема 15.1).
Позволив себе перефразировать известное положение Рудольфа Вир-
хова о том, что «болезнь начинается с недостаточности регуляторного
аппарата», можно считать, что нарушения регуляции в функциональных
системах представляют собой необходимое качество болезни и типового
патологического процесса. Если признать критерием эффективности
регуляции ее способность объединить во взаимодействии регуляторные
аппараты и эффекторы различных уровней для достижения конечного
полезного приспособительного результата, то невозможность его достиже^
ния через взаимосодействие функциональных систем следует считать
свидетельством неэффективности регуляции. В этой связи
патологические изменения эффекторов, препятствующие достижению полезного
результата, в том числе и гиперактивацию клеток как результат дизрегуля-
ции на системном, клеточном и субклеточном уровнях, можно считать
причиной неэффективности регуляции.
Дизрегуляция и патологические изменения эффекторов как причины расстройств... 231
Восстановление доставки кислорода с
артериальной кровью в гипоксичные ткани
Снижение образования
в ранее гипоксичных
тканях оксида азота
Рост активности
ксантиноксидазы
Дисфункции на
уровне митохондрий
Рост образования и
выброса клетками
медиаторов воспаления
Накопление
в тканях
реакто генных
метаболитов
кислорода
Активация ядерных
транскрипционных
факторов,
повышающих экспрессию
адгезивных молекул
Активация нейтрофилов и
других лейкоцитов
Рост экспрессии адгезивных
молекул на поверхности эндотелиоцитов
Адгезия лейкоцитов к эндотелиальным клеткам
Высвобождение активированными лейкоцитами и эндоте-
лиоцитами протеаз, реактогенных метаболитов кислорода и
цитокинов, повышающих проницаемость стенки капилляров
Расстройства микроциркуляции и повреждения
тканей и клеток
Схема 15.1. Патогенез альтерации гипоксичных клеток и тканей
после восстановления доставки к ним кислорода
(реактогенные метаболиты кислорода - свободные кислородные радикалы)
Известно, что системные регуляторные влияния при условии
устойчивого сдвига количества и качества информации, передаваемой по
информационным каналам систем регуляции, к одной из граней их
возможного спектра могут менять состояние эффектора таким
образом, что его аномальные изменения становятся звеном патогенеза
болезней и типовых патологических процессов. Например, патогенно
интенсивная адренергическая нервная стимуляция сердца в остром
периоде после массивной кровопотери, обуславливая острую нейродистро-
фию кардиомиоцитов, становится одним из звеньев танатогенеза.
Следует подчеркнуть, что не существует достоверной положительной связи
232
Глава 15
между гиперкатехоламинемией и смертностью вследствие острой ней-
родистрофии миокарда у экспериментальных животных после
массивной кровопотери. Смертность достоверно снижается, если кровопотерю
предварить высокой (в грудном отделе) преганглионарной
симпатической блокадой посредством эпидурального введения раствора местного
анальгетика. Последнее доказывает чисто нервную природу
отрицательных нейротрофических влияний на миокард, стимулами для
которых являются гиповолемия, артериальная гипотензия и циркуляторная
гипоксия.
В данном случае отрицательные адренергические нервные влияния
теряют свой защитно-приспособительный смысл (рост частоты
сердечных сокращений и сократимости сердца для роста минутного объема
кровообращения) и приобретают качество звена патогенеза сердечной
недостаточности. В контексте учебника общей патологии человека акад.
Д.С. Саркисова и соавт. острую нейродистрофию миокарда можно
считать результатом патогенного сдвига антагонистической регуляции
функций в одну из двух возможных крайностей^ то есть максимальной
нейрогенной активации системы аденилатциклазы кардиомиоцитов как
причины тахикардии и роста сократимости. В частности тут проявляет
себя и такая общепатологическая закономерность как наличие у
приспособительных, компенсаторных и защитных реакций потенциала
трансформации в звенья патогенеза болезней и патологических процессов.
Наличие у приспособительных, защитных и компенсаторных реакций
свойства при определенных условиях на определенном этапе развития
болезни и патологического процесса превращаться в звено патогенеза
вполне адекватно известному с древности диалектическому
философскому принципу энантиодромии, который К. Юнг подробно разбирает в
своем ч<Определении терминов». Буквальный перевод с греческого термина
энантиодромия - «бег навстречу», то есть универсальный принцип бытия,
согласно которому все, что есть, переходит в свою противоположность.
Гераклит описывает энантиодромию следующим образом: «Из живого
делается мертвое, а из мертвого живое, из юного старое, а из старого юное,
из бодрствующего спящее, из спящего бодрствующее, поток порождения
и уничтожения никогда не останавливается». «Созидание и разрушение,
разрушение и созидание - вот норма, охватывающая все круги природной
жизни, самые малые и самые великие».
Можно считать, что частный случай энантиодромии, то есть
превращение приспособительных, защитных и компенсаторных реакций в
звенья патогенеза может происходить особенно быстро в силу особенностей
регуляции и структурно-функциональной организации живых систем.
Активный характер реагирования живых систем на результат
взаимодействия этиологического фактора с организмом через модуляцию регуляции и
эффекторов на основе роста утилизации свободной энергии и
пластических субстратов определяет возможность быстрой трансформации
защитных (приспособительных, компенсаторных) реакций в звенья
патогенеза и системные патологические реакции. Представляется возможным
Дизрегуляция и патологические изменения эффекторов как причины расстройств... 233
выделить следующие основные причины возможной трансформации
реакций, составляющих саногенез, в звенья патогенеза.
♦ Реализация защитных реакций обычно происходит через сдвиг
параметров регуляторных влияний и состояния эффекторов в область
значений одной из альтернативных крайностей функционального
состояния {гиперактивация регуляции и эффекторов). Такая
модуляция регуляции и эффекторов при условии ее определенных
длительности и интенсивности, как мы уже пытались показать,
представляет собой один из универсальных патогенетических
механизмов болезней и типовых патологических процессов.
♦ Поддержание гомеостаза осуществляется взаимосодействием
функциональных систем, которые, будучи связаны между собой
множеством функциональных связей, интерферируют на всех уровнях
структурно-функциональной организации. При этом клеточные
элементы систем регуляции и эффекторов одной функциональной
системы представляют собой структурно-функциональные единицы
множества других функциональных систем. В результате
потенциально патогенная гиперактивация систем регуляции и эффекторов,
реализующая защитные физиологические реакции, приобретает
системный характер. Системный характер физиологических
защитных реакций обуславливает не имеющее биологического смысла
вовлечение интерферирующих между собой функциональных
систем в потенциально патогенную гиперактивацию. Системный
характер гиперактивации снижает приспособительные
возможности организма и его резистентность.
♦ Защитная (приспособительная, компенсаторная) реакция организма
на результат своего взаимодействия с этиологическим фактором
всегда является системной и состоит в гиперактивации клеток регу-
ляторного аппарата и других элементов множества
функциональных систем. В частности это обуславливает некоторую степень
избыточной амплификации реакции на результат взаимодействия
организма с этиологическим фактором. В результате защитные
(компенсаторные, приспособительные) реакции всегда избыточны
относительно своих стимулов, в частности относительно степени
патологических сдвигов гомеостаза. Относительная избыточность
компенсаторных реакций служит одним из факторов приобретения ими
патогенного качества через расходование резервов массы и энергии
организма.
Не исключено, что самым ярким примером стремительной
трансформации защитных (компенсаторных), то есть направленных на
предотвращение энтропии системы организма, реакций в патологические
является патогенез кардиогенного шока. Когда вследствие ишемического
цитолиза кардиомиоцитов и их гибернации из синхронного сокращения
стенок левого желудочка выпадает работа целого сегмента, ударный объем
левого желудочка падает в такой степени, что, несмотря на тахикардию,
критически снижается минутный объем кровообращения. В результате
234
Глава 15
возникают циркуляторная гипоксия и артериальная гипотензия, в ответ на
которые растет адренергическая стимуляция сосудистой стенки сосудов
сопротивления, что повышает общее периферическое сосудистое
сопротивление. Рост общего периферического сосудистого сопротивления
(ОПСС) как защитная реакция направлен на поддержание артериального
давления достаточного для сохранения минимума объемной скорости
кровотока через головной мозг, сердце и легкие. Одновременно рост
ОПСС сразу становится звеном патогенеза кардиогенного шока, повышая
постнагрузку сердца. Рост постнагрузки сердца увеличивает работу
миокарда, что усиливает дефицит свободной энергии в зоне циркуляторной
гипоксии, расширяет зону инфаркта и придает необратимость кардиоген-
ному шоку. Одновременно рост системной адренергической стимуляции
посредством возбуждения бета-один-адренорецепторов почечной
паренхимы активирует ренин-ангиотензин-альдостероновый механизм. Это
задерживает в организме натрий и повышает объем внеклеточной
жидкости. Биологический смысл возрастания объема внеклеточной жидкости
состоит в росте преднагрузки сердца для возрастания минутного объема
кровообращения, сниженного из-за падения насосной функции сердца. У
больных в состоянии кардиогенного шока рост преднагрузки, повышая
работу сердца, усиливает гипоэргоз кардиомиоцитов, расширяет зону
инфаркта и ведет к необратимости шока. В данном случае мгновенное
превращение защитных реакций в звенья патогенеза обусловлено
критическим снижением той части клеток основного эффектора защитных
(компенсаторных) реакций (сердца), которую системам регуляции можно
задействовать для реализации компенсаторных реакций.
Если эффектор или совокупность органов-эффекторов
функциональной системы, повреждения и (или) изменения функционального
состояния которых вызывают болезнь, патологическое состояние или
типический патологический процесс, одновременно является эффектором
защитных (компенсаторных) реакций в ответ на расстройства
функциональных систем и нарушения гомеостаза, то защитные реакции сразу
приобретают преимущественно или сугубо патогенный характер.
Позволим себе привести еще один пример действия данной
общепатологической закономерности.
Вариабельность функционального состояния
структурно-функциональных элементов органов-эффекторов функций - универсальное
свойство функциональных и живых систем. Благодаря вариабельности
состояния элементов органов-эффекторов существует возможность
интенсификации функций для экстренной адаптации к изменившимся условиям
существования или срочной компенсации последствий расстройств
функциональных систем и нарушений гомеостаза. Вариабельность позволяет
системам регуляции мобилизовать структурно-функциональные элементы
эффекторов для усиления функций. Одним из способов мобилизации
структурно-функциональных элементов является гиперактивация систем
регуляции и клеток органов-эффекторов. При этом вследствие исходной
вариабельности функционального состояния структурно-функциональ-
Дизрегуляция и патологические изменения эффекторов как причины расстройств... 235
ных элементов (СФЭ) органов-эффекторов гиперактивация приводит к
асинхронному изменению функционального состояния СФЭ. В некоторых
функциональных системах асинхронное изменение состояния СФЭ
приводит к еще большему дефициту конечного полезного
приспособительного результата.
При обострении какого-либо легочного заболевания или гастроэзофаге-
ального рефлюкса до астматического статуса из-за физиологической
вариабельности просвета бронхиол и сократимости гладкомышечных элементов
их стенки, сужение просвета дыхательных путей, по которым вдыхаемая
газовая смесь поступает в респироны, происходит неравномерно. В
результате возникает определенная вариабельность просвета бронхиол, которая в
соответствии с законом Пуазейля обуславливает патогенную
неравномерность сопротивления дыхательных путей респиронов (СФЭ легких).
Патологическая неравномерность сопротивления дыхательных путей вызывает
патологическую вариабельность вентиляционно-перфузионных отношений
респиронов в пределах всех легких и артериальную гипоксемию.
Патологические изменения структуры и функционального состояния основного
органа-эффектора системы внешнего дыхания (легких) вызывают
компенсаторные реакции, эффекторы которых - это все те же легкие и другие
органы системы внешнего дыхания. Одна из таких реакций - это
гипервентиляция, которая при патологически высоком сопротивлении дыхательных
путей приводит к росту потребления кислорода дыхательными мышцами
без роста напряжения кислорода в артериальной крови, которому в
основном препятствует все та же патологическая вариабельность вентиляционно-
перфузионных отношений респиронов. В результате растет потребление
кислорода всем организмом, что снижает напряжение кислорода в
артериальной крови еще в большей степени.
Нарушения регуляции (дизрегуляция) - необходимый атрибут
болезней, патологических состояний и процессов. Обязательное условие
возникновения дизрегуляции и (или) ее причина - это результат
взаимодействия организма с этиологическим фактором и (или) системная на него
реакция. Результатом взаимодействия организма с этиологическим
фактором может быть повреждение или утрата клеточных, молекулярных
элементов систем регуляции или же невозможность их мобилизации в
качестве эффекторов регуляторных систем. Одной из причин такой
невозможности может быть врожденная или приобретенная недостаточность
экспрессии генома клеток. Такова принципиальная схема индукции
болезней, патологических процессов и состояний при прямом повреждении регу-
ляторного аппарата или его патологическом изменении.
Вне зависимости от уровня структурно-функциональной организации,
на котором происходит прямое повреждение или патологическое
изменение функционального состояния эффекторов систем регуляции, они всегда
приводят к системным патологическим реакциям в виде болезней,
патологических состояний и процессов.
Одним из примеров системной патологической реакции в ответ на
недостаток или патогенное изменение молекулярных эффекторов систем
236
Глава 15
регуляции на субклеточном уровне является гиперлипидемия второго
типа. Ведущим звеном патогенеза гиперхолестеринемии, высокого
содержания в плазме крови атерогенных липопротеинов низкой плотности и
атеросклероза при этом виде гиперлипидемии является или полное
врожденное отсутствие рецепторов к липопротеинам низкой плотности на
наружной клеточной поверхности или нарушения их строения вследствие
мутации аллелей генов RbO, Rb-, Rtio.
Нарушения нормальных межклеточных взаимодействий клеточных
эффекторов систем регуляции иммунного ответа как результат
генетических дефектов дифференциации, инфицирования и других аномалий Т- и
В-лимфоцитов, лежат в основе разнообразных первичных и вторичных
иммунодефицитов, а также аутоиммунных заболеваний.
Повреждения центральных супрасегментарных звеньев систем нервной
регуляции при черепно-мозговых ранениях и травмах вызывают целый
спектр патологических состояний и процессов, из которых представляется
целесообразным выделить нейрогенный респираторный дистресс-синдром
взрослых, пневмонию, острую нейродистрофию миокарда и др.
Одним из основных механизмов регуляции функций и образования
функциональных систем служат антагонистические отношения между
системами регуляции на различных уровнях их
структурно-функциональной организации. При этом у каждой из систем регуляции есть своя
антисистема, функционирование которой направлено на альтернативный
по биологическому смыслу (защитно-приспособительному значению)
полезный приспособительный результат. Дефицит массы и энергии в одной
из антисистем регуляции вызывает устойчивый сдвиг соответствующей
составляющей функционального состояния организма к одному из
пределов возможных физиологических колебаний показателей функций. Тем
самым за счет преобладания на уровне систем регуляции, а также
исполнительных органов регуляторных влияний одной направленности
снижаются приспособительные возможности организма (снижение
пластичности конечного полезного приспособительного результата) и возникает
потенциально патогенная гиперактивация функциональных систем.
При одной из самых частых эндокринопатий человека, инсулинзави-
симом сахарном диабете, дефицит массы обладающих нормальными
физиологическими свойствами молекулярных эффекторов регуляторных
антисистем приводит к гиперактивации нейроэндокринной катаболической
системы. В результате эффектам целого спектра гормонов
катаболической системы на организменном уровне не противодействует инсулин как
главный анаболический гормон и главный гормон-антагонист катаболи-
ческих гормонов. Устойчивые блокада анаболизма и интенсификация
катаболизма у больных сахарным диабетом приводят к дефициту массы тела и
вторичному иммунодефициту, что в частности отличает таких пациентов от
больных неинсулинзависимым сахарным диабетом.
Действие различных патогенетических механизмов возникновения
дефицита массы нормальных молекулярных эффекторов функций,
возникающего в одной из регуляторных антисистем, приводит к одному и то-
Дизрегуляция и патологические изменения эффекторов как причины расстройств... 237
му же патологическому результату. Так, инсулинзависимый сахарный
диабет могут обусловить:
♦ связывание эпитопов инсулина с аутоантителами;
♦ генетически детерминированные дефекты строения молекулы
данного полипептидного гормона;
♦ аутоиммунное поражение инсулиновых рецепторов.
Вазоактивный интестинальный полипептид (ВИП) можно считать
молекулярным эффектором системы регуляции, действие которого
направлено на снижение сопротивления дыхательных путей через активацию
системы аденилатциклазы клеток стенок бронхов. Активация
внутриклеточной системы аденилатциклазы не только расслабляет гладкомышеч-
ные элементы дыхательных путей, но и тормозит высвобождение в их
просвет бронхиального секрета. В настоящее время есть определенная
совокупность фактов, которая позволяет считать дефицит ВИП как
молекулярного эффектора регуляторных антисистем причиной развития
бронхиальной астмы у значительного количества больных.
Непосредственное повреждение или патологическое изменение
функционального состояния эффекторов систем регуляции вовсе не является
обязательным условием наличия у дизрегуляции качества ведущего звена
патогенеза. Через формирование патологических систем регуляции в
ответ на результат взаимодействия организма с этиологическим
фактором (причина болезни) дизрегуляция может персистировать и после
элиминации из организма первопричины заболевания.
Если считать необходимым условием патологического состояния
организма его постоянное взаимодействие с этиологическим фактором, то
легко впасть в противоречие с философским законом основания, то есть
принципом детерминизма. Не исключено, что уместным тут будет
обращение к немецкой классической философии в лице Шопенгауэра, который
основные положения закона основания определяет следующим образом:
«Бытие материи- это ее действие; только действуя, наполняет она
пространство, наполняет она время ...Таким образом, причина и действие- в
этом вся сущность материи: ее бытие и есть действие... Какое состояние
должно последовать в это время и на этом месте - вот определение, на
которое только и распространяется законодательная сила причинности».
При возникновении болезни любой из экзо- или эндогенных
этиологических факторов, воздействуя на организм, через активный и
системный характер реагирования его биологической системы, придает
организму в определенный момент времени качественно новое состояние. Это
патологическое состояние качественно отлично от здоровья и работы
функциональных систем в физиологических условиях и само по себе
может обуславливать ряд патологических реакций, которые мы назовем
вторичными. Непосредственной причиной этих реакций выступает
результат взаимодействия организма с этиологическим фактором
(первичная реакция). В момент возникновения вторичных реакций
взаимодействие организма с этиологическим фактором может быть прекращено.
Сохранение взаимодействия организма с этиологическим фактором по ходу
238
Глава 15
болезни и патологического процесса утяжеляет их течение и без
элиминации причин патологических состояний придает аномалиям
необратимость, индуцируя танатогенез. Особенно жестко эта нозологическая
закономерность проявляет себя при инфекционных болезнях. Поэтому поиск
способа лечения больного, в этиопатогенетическом отношении наиболее
приближенного к причине болезни и ориентированного на элиминацию
этиологического фактора, следует признать непреложным
императивом для любого врача у постели любого больного. Тем не менее, следует
признать, что танатогенез может начаться, находясь в связи с
причиной болезни, но после прекращения ее действия. Иными словами,
непосредственные причины физических состояний и танатогенеза могут быть
связаны цепью причинно-следственных отношений с первопричиной
болезни (взаимодействие этиологического фактора и организма), но могут и
возникать под действием причин, не представляющих собой такой
первопричины. В этом и заключается практический смысл концепции эндоге-
низации болезней и патологических процессов. Следствие
(патологическая реакция организма) не возникает из ничего и всегда имеет свою
причину в виде взаимодействия этиологического фактора с организмом. Как
причина патологическая реакция организма в соответствии с
диалектическим законом основания обуславливает появление во времени новых
следствий, которые могут быть и патологическими реакциями.
Примеров тому в патофизиологии несть числа, но наиболее яркий -
патологическое состояние вследствие тяжелых боевых ранений.
Системная воспалительная реакция (СВР) у больных с тяжелыми ранениями и
травмами вторична по отношению к некробиотическим изменениям
тканей, и является особенно частой при той их протяженности, которая
обуславливает невозможность эффективной первичной хирургической
обработки (минно-взрывные ранения и травмы). При этом первопричиной
патологического состояния следует считать взаимодействие организма с
этиологическим фактором, то есть повреждающим ткани механическим
воздействием ранящего снаряда. Вторичная по отношению к
первопричине болезни СВР представляет собой основную причину множественной
системной недостаточности, индуцирующей танатогенез.
Наша попытка описать наиболее общие закономерности дисфункций
функциональных систем на пути развития болезней и патологических
состояний нуждается в некотором итоговом завершении, императивом
которого выступает связь с основными положениями теории
функциональных систем акад. П.К. Анохина.
Функциональная система представляет собой единицу интеграции
целостного организма, которая динамически складывается для достижения
целей его приспособительной деятельности (конечного полезного
приспособительного результата, КППР). Для достижения КППР
функциональная система избирательно объединяет специальные
центрально-периферические образования. Физиологическая суть нормальных
компенсаторных приспособлений к дисфункциям функциональных систем и
нарушениям гомеостаза состоит в том, что каждый этап деятельности орга-
Дизрегуляция и патологические изменения эффекторов как причины расстройств... 239
низма, направленный через работу функциональных систем на
устранение дефекта, может наступить только тогда, когда произошла оценка
предыдущего этапа. Таким образом на каждом отдельном этапе
компенсаторного процесса как необходимого элемента всего комплекса реакций
саногенеза имеется оценка полученного результата для определения
степени его полезности и закрепления функциональной системы
компенсации. Таким образом конечный полезный приспособительный результат
выступает системообразующим фактором.
При болезнях и приспособительных процессах, развивающихся через
дизрегуляцию и патологические изменения эффекторов, конечный
приспособительный результат уже не представляет собой определяющего
фактора формирования совокупностей центрально-периферических
образований, объединенных регуляторными системами организменного
уровня при интерференции клеточных элементов аппаратов регуляции и
органов-эффекторов. Возникают новые центрально-периферические
интеграции, объединение элементов которых лишено биологического смысла и
приводит к исключительно патогенному результату. Речь идет не о
приобретении физиологическими функциональными системами свойства
патологических. Возникают новые и устойчивые во время болезни
(патологического состояния) патогенные и гиперактивированные констелляции
составляющих аппаратов регуляции и эффекторов, информационные
потоки в которых однонаправлены и патогенно интенсивны
(патологические системы). При формировании патологических систем происходит
структурно-функциональная перестройка клеточных элементов аппаратов
регуляции, эффекторов, а также функциональных связей между ними.
При соответствующих интенсивности взаимодействия этиологического
фактора с организмом и его реактивности происходит
структурно-функциональное закрепление патологической системы.
Патологическая система индуцирует формирование на всех уровнях
регуляции и структурно-функциональной организации образование
антисистемы, функционирование которой направлено на достижение целей
саногенетических реакций: элиминацию взаимодействия организма с
этиологическим фактором, блокаду болезней, патологических процессов
и выздоровление. При этом антисистему следует рассматривать как
новую по характеру регуляции и массе эффекторов
центрально-периферическую интеграцию, специфика которой определяется особенностями
болезни и патологического процесса. Формирование антисистем - это
необходимый элемент патологии и цель терапии.
При этом условием эффективности патогенетической терапии
являются устранение дефицита массы и энергии в аппаратах регуляции и
эффекторах антисистемы, формируемой, как и антисистема, на основе
сложившихся центрально-периферических образований. Лишь устранение
дефицита энергии и массы в клетках аппаратов регуляции и эффекторов
антисистем позволяет в ходе интенсивной (превентивной) терапии
тяжелых больных вызвать гиперактивацию антисистем, без которой не
заблокировать патологические процессы и не вызвать выздоровления. Реализа-
240
Глава 15
ция этого принципа терапии способствовала снижению летальности у
раненых, поступивших с поля боя в военно-полевые лечебные учреждения,
на 13 % (Крыжановский Г.Н., Шанин В.Ю., 1993) (схема 15.2).
Рост системного
транспорта кислорода в результате
гемодилюции
Устранение
дефицита энергии и
массы в клетках
антиноцицептив-
ной системы
Устранение первичной
недостаточности тормозных механизмов в
генерации нейронов ГПУВ
Снижение степени
дизрегуляции как
нарушения
межсистемных отношений
ноцицептивной и ан-
тиноцицептивной
систем
Рациональные дозы
анестетика и анальгетика
Проводниковое
обезболивание для блокады
ноцицептивной афферентации
и устранения вторичной
недостаточности
тормозных механизмов в
генерации нейронов ГПУВ
Гиперактивация антино-
цицептивной системы
Схема 15.2. Патогенетические принципы устранения и предупреждения
патологической боли вследствие тяжелых ранений и травм
(ГПУВ - генератор патологически усиленного возбуждения)
Болезни и патологические процессы развиваются по своим законам,
которые качественно отличны от закономерностей работы
функциональных систем в физиологических условиях. Это определяет уникальность
объекта изучения патофизиологии и ее самостоятельность как науки.
Глава 16
КЛИНИЧЕСКАЯ ПАТОФИЗИОЛОГИЯ КОМЫ
И ДРУГИХ НАРУШЕНИЙ СОЗНАНИЯ
Кома (от греческого кота, глубокий сон) - бессознательное состояние,
при котором нет реакции на действие внешних раздражителей. Кома всегда
опасна острой тяжелой артериальной гипоксемией и клинической смертью.
Кома - это показание к реанимации (первая стадия реанимационных
мероприятий А, то есть восстановление и поддержание проходимости
дыхательных путей). Если поддержание сознания - это интегративная функция
всей нервной системы (ЦНС), то кома как тяжелое бессознательное
состояние означает нарушение функциональных связей между подотделами ЦНС,
которое обуславливает дизрегуляцию на системном уровне, в частности как
одну из причин нарушений водно-солевого обмена и терморегуляции.
Ступор (от латинского stupeo, то есть быть или стать
парализованным) - помрачение сознания, при котором у больного ослаблена
реакция на действие внешних раздражителей.
Сознание относят к интегративным функциям ЦНС, которые
непосредственно не связаны с обработкой сенсорных сигналов или регулятор-
ными влияниями на двигательные или вегетативные центры. Структуры
ЦНС ответственные за ее интегративные функции локализованы в двух
отделах конечного мозга, лимбической системе и новой коре (неокор-
текс). Необходимым условием осуществления сознания как интегратив-
ной функции через функционирование соответствующих структур
лимбической системы и неокортекса являются неспецифические
активирующие влияния со стороны ретикулярной формации. Ретикулярная
формация представляет собой скопление нейронов, окруженное нервными
волокнами, находящееся между боковыми и задними рогами спинного
мозга и в центральных отделах ствола вплоть до промежуточного мозга.
Ясно, что кому как бессознательное состояние могут обуславливать:
♦ прямые повреждения отделов мозга, в которых локализованы
структуры конечного мозга и ретикулярная формация, ответственные за
поддержание сознания, в частности вследствие расстройств
мозгового кровообращения, кровоизлияний в ткань мозга и полость
черепа, а также связанные со злокачественным клеточным ростом и
доброкачественными опухолями соответствующей локализации,
гидроцефалией и энцефалитом;
♦ системные гипоксия и нарушения обмена веществ, а также
токсические эффекты лекарств, угнетающие активность нейронов структур
ЦНС, ответственных за поддержание сознания.
Ступор и кома обычно представляют собой следствия поражения двух
полушарий головного мозга одновременно и (или) ствола головного мозга.
Поражения только одного полушария, в частности обусловленные ишеми-
242
Глава 16
ческим инсультом в бассейне средней мозговой артерии, редко служат
причиной ступора или комы. Поражения одного полушария вследствие его
опухоли, кровоизлияния, распространенной ишемии (инсульт), которые
резко увеличивают его объем, приводят к бессознательному состоянию
через сдавление интактного полушария. Объемные поражения мозжечка как
причины сдавления ствола головного мозга также могут вызывать кому.
Тщательное изучение и целенаправленное выявление симптомов
комы позволяют идентифицировать локализацию повреждения структур
ЦНС или определить первопричину комы как не связанную с поражением
структур ЦНС, ответственных за поддержание сознания (табл. 16.1).
Таблица 16.1
Связь симптомов комы и поражений определенных структур ЦНС
Симптом, признак комы,
результат специальной пробы
Средний размер зрачков (диаметр
4-6 мм) при отсутствии реакции
сужения зрачков на свет
Одностороннее расширение
зрачка и отсутствие его реакции на свет
Суженные, но реагирующие на
свет зрачки
Равномерно расширенные и не
реагирующие на свет зрачки
Отклонение глаза кнаружи и
вниз (расходящееся косоглазие)
Отклонение глаза внутрь
Отклонение взора в сторону,
противоположную гемипарезу
Направление взора в сторону
гемипареза
Отрицательная окулоцефальная
проба
Отрицательная окуловестибуляр-
ная проба
Пораженный отдел головного
мозга и причины комы
Средний мозг
Поражение III черепно-мозгового нерва, в
частности вызванное вклинением медиальных отделов
височной доли головного мозга
Диффузное поражение нейронов головного
мозга вследствие системных расстройств обмена
веществ; промежуточный мозг, варолиев мост.
Распространенное угнетение активности церебральных
нейронов из-за токсического действия анестетиков,
транквилизаторов и других нейротропных средств
Тяжелая гипоксическая энцефалопатия,
интоксикация М-холинолитиками, метиловым спиртом и
другими ядами
Поражение III черепно-мозгового нерва (при
отсутствии признаков поражения IV и VI черепно-
мозговых нервов)
Сходящееся косоглазие (поражение VI черепно-
мозгового нерва)
Поражение головного мозга на стороне, в
которую происходит отклонение
Ствол с противоположной стороны
Варолиев мост и средний мозг
Ствол мозга от варолиева моста до среднего
мозга
Известно, что необходимым условием поддержания сознания является
непрерывное поступление в нейроны головного мозга кислорода и
глюкозы. В этой связи императивом обследования больного в коме является
определение того, не являются ли гипоксия и гипогликемия причиной комы.
Клиническая патофизиология комы и других нарушений сознания
243
Кома вследствие гипонатриемии, патологического роста осмоляль-
ности во внеклеточном жидкостном секторе и плазме крови, гиперкап-
нии или энцефалопатии вследствие печеночной и почечной
недостаточности - это результат множества нарушений обмена веществ в нейронах
и астроцитах. При этом возникает гипоэргоз клеток, патологическое
изменение их трансмембранных потенциалов, расстройства
высвобождения и эффектов нейромедиаторов, нередко приводящие к гибели
нейронов. Например, печеночная кома (кома вследствие энцефалопатии,
обусловленной печеночной недостаточностью) повышает
концентрацию аммиака в головном мозге, что нарушает его энергетический обмен
и активный трансмембранный перенос катионов натрия и калия через
наружную мембрану нейронов. Наблюдаемую при печеночной коме
гипертрофию астроцитов предположительно считают следствием
защитной реакции, направленной на обезвреживание аммиака. Полагают, что
гипертрофия астроцитов может быть звеном патогенеза печеночной
комы. Кроме того, считают, что при печеночной коме обмен нейротранс-
миттеров меняется таким образом, что появляются патологические
субстанции способные к конкуренции с нормальными эндогенными лиган-
дами за нейрорецепторы.
При энцефалопатии у больных с уремическим синдромом кому
связывают с повышением проницаемости гематоэнцефалического барьера по
отношению к таким эндогенным токсинам как органические кислоты.
Кроме того, кому у больных с энцефалопатией вследствие хронической
почечной недостаточности в частности считают результатом роста
содержания кальция в головном мозге и повышенной концентрации
фосфатного аниона в спинно-мозговой жидкости. Гипонатриемия вызывает
кому через отек нейронов. Гипернатриемия у больных с комой говорит о
том, что дегидратацию нейронов нельзя исключить из ряда ее причин.
Обычно при коме, связанной с гипернатриемией, осмоляльность
внеклеточной жидкости не меньше, чем 350 мосм/л.
Гиперкапния вызывает кому в основном как причина респираторного
ацидоза.
Многие из лекарственных средств (анестетики, наркотические
анальгетики и др.), побочное действие которых обуславливает кому, вызывают
гипоэргоз и расстраивают трансмембранный потенциал нейронов во всем
головном мозге, что может обусловить ошибочный диагноз
органического поражения больших полушарий и ствола головного мозга.
Кома вследствие и после эпилептических припадков может быть
результатом вторичного по отношению к эпилептическому припадку гипо-
эргоза нейронов. При этом из патогенеза комы у больных эпилепсией
нельзя исключить гиперактивацию антисистемы, чье функционирование
противодействует патологической системе ответственной за реализацию
эпилептического припадка.
У больных в состоянии комы при поступлении в лечебное учреждение
обязательно фиксируют температуру тела. Если температура тела ниже
31 °С, то причиной комы чаще всего является гипотермия. При темпера-
244
Глава 16
туре тела выше 41 °С из причин комы в горно-пустынных климатических
условиях нельзя исключить перегревания.
Движение глаз больного в коме оценивают, выполняя окулоцефалъную
и окуловестибулярную пробы. Окулоцефальная проба - врач быстро
поворачивает голову больного или быстро наклоняет и выпрямляет ее,
наблюдая за положением глаз. При вероятном переломе позвонков шейного
отдела позвоночника окулоцефальная проба противопоказана. При условии ин-
тактности проводящих путей мозга, расположенных в варолиевом мосту и
среднем мозге, глазные яблоки отклоняются в направлении
противоположном движению головы («глаза куклы»), то есть окулоцефальная проба
положительна. Если окулоцефальная проба не выявила каких-либо движений
глазных яблок, то применяют окуловестибулярную пробу. Для
осуществления окуловестибулярной пробы необходимо, чтобы наружные слуховые
проходы были свободно проходимы, а барабанные перепонки как рецеп-
торное поле соответствующих рефлексов интактны. В наружный ушной
проход вливают 10-50 мл ледяной воды, приподнимая голову на 30
градусов. При бессознательном состоянии, если нет поражений ствола мозга от
варолиева моста до среднего мозга, глазные яблоки отклоняются в сторону
раздражаемого слухового прохода (положительная проба).
У больных в состоянии комы существует связь между видом
расстройств внешнего дыхания (его патологическими ритмами) и
локализацией повреждений головного мозга, обусловивших потерю сознания. Эта
связь не всегда достоверна, но, тем не менее, можно выделить следующие
корреляты:
♦ поверхностное (при патологически низком дыхательном объеме),
но ритмичное дыхание обычно свидетельствует о коме вследствие
нарушений обмена веществ или в результате побочного действия
лекарственных средств;
♦ дыхание Куссмауля (частое и глубокое, то есть при повышенном
дыхательном объеме) чаще всего указывает на метаболический
ацидоз (кетоацидоз у больных инсулинзависимым сахарным
диабетом и др.) или характеризует кому вследствие поражений моста и
среднего мозга;
♦ дыхание Чейн-Стокса (дыхание между периодами апноэ с
нарастанием величины дыхательного объема и его последующим
снижением до остановки дыхания) обычно говорит об умеренном
двустороннем поражении больших полушарий или о нарушениях обмена
веществ как о причине комы;
♦ неполноценный вдох, при котором больной как бы судорожно
пытается «проглотить» воздух при крайне сниженном дыхательном
объеме, - это проявление поражения нижних отделов ствола головного
мозга или тяжелых гипоксических диффузных поражениях
церебральных нейронов. В частности такое дыхание «глотающего» типа
можно заметить у больных в состоянии клинической смерти.
Кома может быть и следствием смерти головного мозга, о
наступлении которой говорит ряд ее признаков (табл. 16.2). Для констатации
Клиническая патофизиология комы и других нарушений сознания 245
смерти мозга необходимо наличие всех критериев. При этом признаком
смерти мозга как состояния, наличие которого позволяет рассматривать
пациента в качестве потенциального донора органов, является
температура тела не меньшая, чем 32 °С. Врач может констатировать наступление
смерти мозга только тогда, когда уверен, что кома не является следствием
передозировки уличных наркотиков или побочного эффекта средств,
угнетающих активность нейронов головного мозга.
Таблица 16.2
Гарвардские критерии смерти головного мозга
Полное отсутствие словесного контакта и реакции на действие любого раздражителя
Никаких координированных и судорожных движений. Нет дыхательных движений,
в том числе и его патологических ритмов
Полное угнетение всех рефлексов
Полное отсутствие какой-либо активности на электроэнцефалограмме (плоская
электроэнцефалограмма)
Персистирующий вегетативный статус характеризуют стойкое
угнетение когнитивных функций и потеря способности к высшей нервной
деятельности. Наиболее частые причины статуса у больных - это острые
инфекции, отравления, тяжелые механические травмы, кровопотеря и
электротравма. Больной в персистирующем вегетативном статусе
прикован к постели, он мочится и ходит под себя. При этом он не может
питаться самостоятельно, и ему проводят исскусственное и нередко
смешанное питание (энтеральное по назогастральному зонду и
парентеральное). У таких пациентов обычно нет необходимости в интенсивной
терапии, направленной на устранение расстройств внешнего дыхания и
кровообращения. Это свидетельствует о достаточно сохраненной регуляции
внешнего дыхания и кровообращения. В персистирующий вегетативный
статус больные обычно переходят из состояния комы. При этом глаза
открываются и создается впечатление пробуждения. Больные в статусе
могут зевать, издавать нечленораздельные звуки, а также совершать
беспорядочные, но частично координированные движения. Все это может
сочетаться с признаками распространенного и необратимого поражения
нейронов головного мозга (табл. 16.1). Если за три месяца пациент не
выходит из персистирующего вегетативного статуса, то восстановление
когнитивных функций можно считать почти невозможным.
Глава 17
ДЫХАТЕЛЬНАЯ НЕДОСТАТОЧНОСТЬ
И АРТЕРИАЛЬНАЯ ГИПОКСЕМИЯ
Основная функция системы внешнего дыхания - это поглощение из
внешней среды кислорода и выведение в нее из внутренней среды
углекислого газа.
Дыхательная недостаточность - это состояние организма
вследствие неспособности системы внешнего дыхания обеспечить:
♦ поглощение легкими 02 и уровень его напряжения в артериальной
крови (PaOz), достаточные для адекватного потребностям
организма насыщения гемоглобина кислородом;
♦ выведение из организма углекислого газа, позволяющее организму
удерживать концентрацию протонов во внеклеточной жидкости
и плазме крови ([£t]) в диапазоне нормальных изменений.
Кроме того, под дыхательной недостаточностью следует понимать
неспособность системы внешнего дыхания реагировать ростом
поглощения кислорода легкими и экскреции ими углекислого газа в ответ на
увеличение потребности организма в кислороде (рост потребления
организмом О2) и на возросшую потребность в экскреции углекислого газа
(усиление высвобождения углекислого газа в ходе обмена веществ во
внутреннюю среду организма).
Снижение в артериальной крови напряжения кислорода и рост в ней
напряжения углекислого газа (РаС02) может быть следствием роста
потребления кислорода организмом (ПОг) и образования в нем углекислого
газа при физической нагрузке, лихорадке, сепсисе, при неспецифических
компенсаторных процессах («стресс-реакция»), при тиреотоксикозе, а
также при других патологических состояниях и приспособительных реакциях.
При этом в определенные периоды болезни и патологического процесса
напряжения газов в крови по мере снижения утилизации организмом
свободной энергии возвращаются в «нормальные» пределы. Это не означает
устранения терапией дыхательной недостаточности. Она остается скрытой,
и проявляет себя артериальными гипоксемией и гиперкапнией при
последующих по ходу болезни росте потребления кислорода и образования
углекислого газа.
Легочный газообмен происходит на уровне терминальных
респираторных единиц (ТРЕ) легких. ТРЕ (респирон) - это часть легких, дис-
тальная по отношению к конечной нереспираторной бронхиоле, которая
омывается смешанной венозной кровью, поступающей по ветви легочной
артерии (легочной артериоле), параллельной данной терминальной
нереспираторной бронхиоле. Легочная артериола распадается на
капиллярную сеть, обволакивающую альвеолы и образующую легочные венулы,
находящиеся на периферии ТРЕ. Такое строение ТРЕ в структурном от-
Дыхательная недостаточность и артериальная гипоксемия
247
ношении обеспечивает продвижение смешанной венозной крови строго
из артериол через легочные капилляры в венулы и ее оксигенацию на
уровне альвеол.
Для наиболее полных на уровне одного респирона оксигенации
смешанной венозной крови и освобождения ее от углекислого газа (легочный
газообмен) необходимо наличие и совпадение во времени двух условий:
1. Достаточное поступление кислорода из внешней среды в состав
альвеолярной газовой смеси, то есть определенный уровень минутной
альвеолярной вентиляции (МАВ) дыхательной смесью газов с
достаточным содержанием в ней кислорода. Кроме того, достаточная МАВ
выступает необходимым условием адекватной экскреции углекислого газа в
ходе внешнего дыхания из смешанной венозной крови и альвеолярной
газовой смеси во внешнюю среду.
2. Оптимальное снабжение респирона смешанной венозной кровью.
Если на уровне одного респирона МАВ начинает преобладать над
кровотоком, то есть становится избыточной, то патогенных сдвигов в
напряжении кислорода и углекислого газа в крови, которую содержат
венулы данного респирона, не произойдет. Однако рост МАВ одних респиро-
нов, одной совокупности структурно-функциональных единиц легких,
приводит к падению вентиляции других, а значит и к нарушению в них
легочного газообмена. Причиной нарушений газообмена в легких может
быть и преобладание снабжения смешанной венозной кровью одних рес-
пиронов за счет других.
Дыхательная недостаточность - это всегда следствие нарушений
соответствия МАВ структурно-функциональных единиц легких, их от-
делов и частей, всех легких объемной скорости кровотока, то есть рас-
стройств вентиляционно-перфузионных отношений.
В физиологических условиях всегда существует нормальная
вариабельность вентиляционно-перфузионных отношений респиронов и
отделов легких, которая на уровне всех легких не приводит к дыхательной
недостаточности. При дыхательной недостаточности физиологическая
вариабельность трансформируется в патологическую. В результате на
уровне всей системы внешнего дыхания нарушается легочный газообмен. В
силу большей диффузионной способности углекислого газа нарушения
легочного газообмена чаще сначала приводят к патогенному падению
оксигенации смешанной венозной крови, то есть обуславливают
артериальную гипоксемию.
АРТЕРИАЛЬНАЯ ГИПОКСЕМИЯ
Под артериальной гипоксемией понимают патологически низкий
уровень оксигенации смешанной венозной крови, в результате которого
транспорт кислорода от легких на периферию становится таким
низким, что возникает респираторная гипоксия.
248
Глава 17
Артериальная гипоксемия чаще всего выступает следствием:
♦ нарушений физиологической вариабельности вентиляционно-пер-
фузионных отношений (ВПО) респиронов и отделов легких;
♦ патологического внутрилегочного шунтирования крови справа
налево, то есть роста объема неоксигенированной смешанной
венозной крови, который проходит через легкие в артериальную кровь,
не участвуя в легочном газообмене;
♦ снижения парциального давления кислорода во вдыхаемой газовой
смеси;
♦ падения напряжения кислорода в смешанной венозной крови
(Pv02);
♦ нарушений диффузии газов через альвеоло-капиллярную
мембрану;
♦ низкого МАВ, то есть гиповентиляции.
Степень тяжести артериальной гипоксемии оценивают в зависимости
от уровня напряжения кислорода в артериальной крови (табл. 17.1).
Таблица 17.1
Степени тяжести артериальной гипоксемии
Степень
тяжести
Легкая
Умеренная
Тяжелая
Крайне
тяжелая
РаОг при дыхании атмосферным
воздухом, мм рт.
80-62
61-51
50-40
<40
ст.
Обязательные
лечебные мероприятия
Инсуффляция кислорода при
самостоятельном дыхании больного и
содержании 02 во вдыхаемой газовой смеси
(Fi02) в пределах 30-40 %
Искусственная вентиляция легких при
Fi02 не менее, чем 50 %
Искусственная вентиляция легких
чистым кислородом
ПАТОЛОГИЧЕСКИЕ ВАРИАБЕЛЬНОСТЬ ВЕНТИЛЯЦИОННО-
ПЕРФУЗИОННЫХ ОТНОШЕНИЙ И ШУНТИРОВАНИЕ
СМЕШАННОЙ ВЕНОЗНОЙ КРОВИ В ЛЕГКИХ КАК
ПРИЧИНЫ АРТЕРИАЛЬНОЙ ГИПОКСЕМИИ
Общая площадь легочной мембраны, через которую происходит
диффузия свободных молекул кислорода и углекислого газа, у здоровых
людей колеблется от 160 до 200 м2. Эту площадь составляет сумма
газообменной поверхности (легочной мембраны) около 100 000 респиронов
(ТРЕ) и нескольких миллионов альвеол. Массоперенос кислорода и
углекислого газа из альвеолярной газовой смеси в смешанную венозную кровь
и в обратном направлении моэюет быть достаточным лишь при условии
незатрудненного кровоснабжения определенного числа оптимально вен-
Дыхательная недостаточность и артериальная гипоксемия
249
тилируемых респиронов. Общий объем крови, которую содержат
легочные капилляры, составляет 100 мл. Этот объем крови «растягивается» до
тончайшей пленки на поверхности легочной мембраны в пределах всех
легких, что в структурно-функциональном отношении обеспечивает
наиболее оптимальные условия для нормального легочного газообмена.
Критериями соответствия легочного газообмена потребностям организма
являются парциальные давления и напряжения газов в альвеолярной
газовой смеси, артериальной и смешанной венозной крови (табл. 17.2).
Таблица 17.2
Парциальные давления (напряжения) кислорода и углекислого газа
в альвеолярной газовой смеси, смешанной венозной и артериальной крови
в условиях относительного покоя у здорового молодого человека на уровне моря
Парциальные давления и
напряжения газов
Р02
РС02
Альвеолы
100
40
Смешанная
венозная кровь
40
46
Артериальная
кровь
95
40
Вентиляционно-перфузионное отношение (ВПО) на уровне всех
легких представляет среднее значение ВПО их респиронов, участков
легочной ткани и отделов легких. ВПО всех легких здорового человека
в условиях относительного покоя равное 0,8 может свидетельствовать о
физиологической вариабельности ВПО в пределах всей легочной
паренхимы. При физиологической вариабельности ВПО газообменный
дыхательный коэффициент, отношение выделения углекислого газа к
потреблению кислорода организмом (ПО^) равен метаболическому
дыхательному коэффициенту, то есть отношению образования
углекислого газа в ходе обмена веществ к П02, и они оба находятся на уровне
близком 0,8.
В легких здорового человека нет патологической вариабельности вен-
тиляционно-перфузионных отношений респиронов, участков легочной
паренхимы и отделов легких, несмотря на постоянное действие ряда
факторов физиологической вариабельности ВПО:
♦ большее растяжение нижележащих и периферических отделов
легких на высоте вдоха, обуславливающее преимущественное
распределение дыхательного объема в эти участки легких;
♦ подверженность легочного кровотока влияниям со стороны силы
земного притяжения, которые приводят к преобладанию
кровоснабжения нижележащих респиронов;
♦ зависимость объемной скорости легочного кровотока в различных
участках легких от величины дыхательного объема.
Тогда, когда в результате патологического процесса (пневмония, ге-
мопневмоторакс, крайняя стадия респираторного дистресс-синдрома
взрослых, ушиб легкого, раневой пульмонит и др.) респироны легких в
одинаковой степени теряют и альвеолы и легочные микрососуды, патоло-
250
Глава 17
гической вариабельности ВПО респиронов легких может и не развиться.
При этом артериальной гипоксемии в условиях относительного покоя не
возникает до тех пор, пока легкие не потеряют 70-80 % респиронов.
Если поступление смешанной венозной крови в какой-либо респирон
или часть легких снижается или блокируется, то растет часть
дыхательного объема, которая достигает альвеол, но не участвует в легочном
газообмене. Это снижает поглощение кислорода всеми легкими, что вызывает
падение напряжения кислорода в артериальной крови и артериальную ги-
поксемию. Когда рост вентиляции части альвеол не сопровождается
адекватным возрастанием кровотока по капиллярам, то из альвеолярной
газовой смеси все легкие поглощают меньше кислорода. Это также
увеличивает фракцию дыхательного объема, не задействованную для легочного
газообмена, и снижает поглощение кислорода легкими. Результатом и
того и другого варианта патологической вариабельности ВПО респиронов
может быть артериальная гипоксемия. Если в легких одновременно
возникают два описанных варианта нарушений ВПО, то на уровне всей системы
внешнего дыхания это приводит к снижению физиологической площади
легочной мембраны. Физиологическая площадь легочной мембраны - это
та ее часть, диффузия через которую кислорода и углекислого газа
приводит к полным, то есть ограниченным только градиентами парциальных
давлений (напряжений), поглощению кислорода из альвеолярной газовой
смеси, оксигенации смешанной венозной крови и ее освобождению от
углекислого газа. Снижение физиологической площади легочной мембраны до
определенного уровня приводит к артериальной гипоксемии.
Стеноз, обтурация легочной артерии или ее ветвей, а также
множественная эмболия легочных микрососудов (диссеминированное внутрисо-
судистое свертывание, жировая эмболия, тромбоз микросгустками
перелитой крови, агрегатами активированных тромбоцитов и нейтрофилов
при травматическом шоке и сепсисе) ведут к образованию альвеолярных
мертвых пространств, то есть вентилируемых, но не омываемых
смешанной венозной кровью альвеол. В результате возникает различие между
РаС02 и парциальным давлением углекислого газа в конечной части
выдыхаемого воздуха, которое считают эквивалентным парциальному
давлению углекислого газа в альвеолярной газовой смеси, РАС02. Это
результат полного прекращения в респиронах с только мертвыми
альвеолярными пространствами переноса углекислого газа через легочную
мембрану. Смешанная венозная кровь из таких респиронов по коллатера-
лям устремляется в легочные венулы других ТРЕ, не отдав в
альвеолярную газовую смесь углекислый газ. В результате возрастает
шунтирование смешанной венозной крови в легких, что обуславливает:
♦ снижение различия между напряжением углекислого газа в
смешанной венозной крови и РаС02;
♦ рост различия между РаС02 и РАС02.
Первоначально при синдроме распространенной микроэмболизации в
легких компенсаторная гипервентиляция в ответ на рост РаС02 через
увеличение экскреции углекислого газа предотвращает гиперкапнию, то
Дыхательная недостаточность и артериальная гипоксемия
251
есть патологически высокое РаС02. Если в результате обструкции
легочных микрососудов поглощение кислорода легкими падает в такой
степени, что возникает артериальная гипоксемия, то снижение кислородной
емкости крови и падение в ней РаСЬ оказывают на сердце отрицательное
инотропное действие. Падение сократимости сердца может привести к
снижению минутного объема кровообращения (МОК).
Падение минутного объема кровообращения в данном случае
происходит не только из-за артериальной гипоксемии. МОК снижается
вследствие роста общего легочного сосудистого сопротивления как причины
правожелудочковой сердечной недостаточности. Компенсаторная
гипервентиляция в ответ на артериальную гипоксемию повышает П02, что
ускоряет развитие циркуляторной гипоксии. Циркуляторная гипоксия при
синдроме распространенной микроэмболизации в легких представляет
собой результат легочной артериальной гипертензии, правожелудочковой
сердечной недостаточности и связанного с ними падения МОК при
высокой из-за повышенных энерготрат в системе внешнего дыхания
потребности всего организма в кислороде.
Главными симтомами синдрома распространенной микроэмболизации
в легких являются одышка и тахикардия, которые возникают и
обостряются даже при минимальном увеличении физической нагрузки. Если при
обследовании таких больных над легкими не слышат хрипов,
рентгенологическое исследование не выявляет патологических изменений в легких, а
анамнез не содержит данных, свидетельствующих о предшествующей
явной или скрытой хронической сердечной недостаточности, то следует
заподозрить распространенную эмболизацию микрососудов легких. Когда,
кроме того, есть электрокардиографические признаки гипертрофии
правого желудочка, то развитие синдрома сомнений вызывать не должно.
Распространенная микроэмболизация в легких - это не единственная
причина возникновения в их паренхиме альвеолярных мертвых
пространств. Причиной альвеолярных мертвых пространств служат
патологические изменения легких при эмфиземе, которые через облитерацию
микрососудов блокируют кровоснабжение респиронов.
Если вентиляция респирона полностью прекращается при
сохраненном поступлении в него смешанной венозной крови (ВПО=0), то по
венулам такого респирона начинает оттекать смешанная венозная кровь. Эта
кровь проходит через легкие, не участвуя в легочном газообмене и
составляя объем (величину) истинного патологического внутрилегочного
шунтирования смешанной венозной крови.
Объем истинного патологического внутрилегочного шунтирования
составляет та часть попавшей в легкие смешанной венозной крови, ко-
торая примешивается к артериальной, пройдя по капиллярам и венулам
полностью невентилируемых респиронов.
Если вентиляция респиронов, участков и отделов легких, снижается
без адекватного уменьшения в них объемной скорости кровотока, то
легочный газообмен в них не прекращается, но продолжает оксигениро-
ваться лишь часть поступившей в них смешанной венозной крови. Ос-
252
Глава 17
тавшаяся часть составляет величину физиологического шунтирования,
которая вместе с объемом истинного патологического внутрилегочного
шунтирования составляет у больных величину патологического внутри-
легочного шунтирования смешанной венозной крови.
Обструктивные расстройства альвеолярной вентиляции, вызывая ее
неравномерность, резко увеличивают число респиронов, в которых из-за
низкой относительно снабжения смешанной венозной кровью вентиляции
происходит физиологическое шунтирование смешанной венозной крови.
Таким образом формируется основное звено патогенеза артериальной ги-
поксемии у больных с обструктивными расстройствами внешнего
дыхания вследствие бронхиальной астмы и астматического статуса.
ОБСТРУКТИВНЫЕ И РЕСТРИКТИВНЫЕ РАССТРОЙСТВА
АЛЬВЕОЛЯРНОЙ ВЕНТИЛЯЦИИ
Потеря легкими нормальных респиронов снижает физиологическую
площадь легочной мембраны и через утрату части сосудов из системы
легочной артерии повышает общее легочное сосудистое сопротивление.
В результате поглощение кислорода легкими ограничивается патологи-
чески низким верхним пределом. Такое нарушение альвеолярной
вентиляции и легочного газообмена называют рестрикционным (рестрикцией). О
рестрикционном (лат. restrictio, ограничение) нарушении легочного
газообмена может свидетельствовать частое и поверхностное дыхание.
Рестриктивные расстройства внешнего дыхания - это результат
ограничения способности достижения системой внешнего дыхания
своих конечных полезных приспособительных результатов из-за потери
главным эффектором системы, легкими, определенного числа нормально
функционирующих респиронов.
Сравнительно недавно одной из причин падения поглощения
кислорода легкими считали альвеолярно-капиллярный блок, то есть снижение
массопереноса кислорода в респиронах, связанное с утолщением
легочной мембраны вследствие патологических процессов в интерстиции
легких. Сейчас ясно, что в большинстве случаев хронических заболеваний с
патологическими изменениями в легочном интерстиции падение
поглощения кислорода легкими возникает как рестрикционное нарушение
легочного газообмена.
Обструктивные расстройства внешнего дыхания (обструкция) (лат.
obstructio - преграда, помеха) связаны с аномальным возрастанием
сопротивления продвиэюению смеси газов по дыхательным путям во время
вдоха и (или) выдоха.
Выделяют стабильное обструктивное расстройство внешнего
дыхания, которое выявляют как в фазу вдоха, так и в фазу выдоха.
Стабильную обструкцию обуславливает постоянное сужение верхних
дыхательных путей, связанное со злокачественными опухолями, пороками разви-
Дыхательная недостаточность и артериальная гипоксемия
253
тия и последствиями ранений и травм соответствующей локализации. К
стабильной обструкции приводит и постоянное патогенное сужение
дыхательных путей небольшого диаметра при хроническом обструктивном
бронхите и бронхиальной астме. Лабильное обструктивное расстройство
внешнего дыхания преимущественно выражено или в фазу вдоха или во
время выдоха, то есть может быть инспираторным или экспираторным.
При параличе дыхательных связок или трахеомаляции на
экстраторакальном уровне обструкция в основном или исключительно возникает во
время вдоха, являясь инспираторной. Экспираторная обструкция обычно
происходит на интраторакальном уровне. Такую обструкцию вызывают:
коллапс интраторакальной части трахеи из-за трахеомаляции, спадение
бронхов и бронхиол небольшого диаметра вследствие патологических
изменений легочного интерстиция при эмфиземе.
При бронхиальной астме и хроническом обструктивном бронхите
просвет дыхательных путей небольшого диаметра (от сегментарных
бронхов до терминальных нереспираторных бронхиол) постоянно сужен
вследствие их закупорки секретом, бронхоспазма и отека слизистой
оболочки. Это обуславливает как инспираторные, так и экспираторные об-
структивные расстройства альвеолярной вентиляции. При этом в
результате патологической динамической компрессии дыхательных путей (см.
ниже) экспираторные обструктивные расстройства выражены в большей
степени. Рост линейной скорости газотока в дыхательных путях
небольшого диаметра из-за их сужения, патологическая динамическая компрессия
таких дыхательных путей в фазу выдоха вызывают колебания выдыхаемой
газовой смеси и стенок дыхательных путей, которые при аускультации
воспринимаются как свистящие (в основном экспираторные) хрипы.
При нормальных регуляции функции внешнего дыхания и состоянии
ее эффекторов существует верхний предел объемной скорости газотока
выдыхаемой газовой смеси, выше которого скорость не растет, несмотря
на увеличение силы сокращений дыхательных мышц. Верхний предел
объемной скорости газотока при выдохе обусловлен динамической
компрессией дыхательных путей.
Для осуществления выдоха внутриальвеолярное давление должно
возрасти от атмосферного, которое мы будем считать нулевым. Рост
давления смеси газов в просвете альвеол происходит через увеличение
давления в плевральной полости, а значит и в перибронхиальных
пространствах легких. Статическое давление выдыхаемой газовой смеси
снижается по мере ее продвижения по дыхательным путям. В результате во время
выдоха снижается градиент давлений между интерстицием легких (пе-
рибронхиальные пространства) и просветом дыхательных путей. Как
только где-либо на интраторакальном уровне давление в паренхиме
легких и в перибронхиальных пространствах начинает преобладать над
статическим давлением газовой смеси в просвете дыхательных путей
(градиент давлений становится положительным), то происходит сдавление
(компрессия) дыхательных путей, если только его не предотвращает
ригидность их стенки.
254
Глава 17
Градиент давлений между начальной (А) и конечной (В) точками
определенного сегмента дыхательных путей находится в прямой связи с
сопротивлением данного сегмента. Сужение просвета бронхиол и бронхов
небольшого диаметра повышает сопротивление дыхательных путей
данного уровня, увеличивает соответствующий градиент давлений и через
падение статического давления выдыхаемой газовой смеси (Р2, Р'2,
рис. 17.1) может привести к спадению дыхательных путей.
А
Р.
P,-P2=V,xR,
- В
> Р2
Р,=Р',
А ■
P'i -
v,=v2
P',-P'2=V2xR2
P,-Pa<Pi-P2
P2>P'2 <
Ri<R2
- В
-> p2
Рис. 17.1. Снижение давления в просвете дыхательных путей, обусловленное
ростом их сопротивления в результате сужения просвета
(Pi-Рг - градиент статических давлений в просвете дыхательных путей; R - сопротивление
дыхательных путей; V - объемная скорость продвижения вдыхаемой газовой смеси
по дыхательным путям)
Рост сопротивления дыхательных путей снижает объемную скорость
газотока по дыхательным путям, но линейная скорость продвижения
выдыхаемой газовой смеси по отдельным бронхиолам и бронхам растет
вследствие сужения их просвета. В результате падает статическое
давление газовой смеси в просвете дыхательных путей, что служит еще одним
из механизмов их патологической динамической компрессии, связанной с
патологическим ростом сопротивления дыхательных путей.
Патологическая динамическая компрессия дыхательных путей служит
одной из причин легочной эмфиземы у больных с хроническими обструк-
тивными заболеваниями легких. Эмфизема развивается через постоянную
задержку части выдыхаемой газовой смеси в легких и перераздувание
альвеол после полного спадения дыхательных путей в результате
патологической динамической компрессии. При этом параллельно с развитием
эмфиземы выявляют патологическое возрастание остаточного объема и
функциональной остаточной емкости легких.
Патологическая динамическая компрессия дыхательных путей еще в
большей степени усиливает их сопротивление, что резко повышает
использование свободной энергии для внешнего дыхания и потребление
кислорода дыхательными мышцами. Рост потребления кислорода для внеш-
Дыхательная недостаточность и артериальная гипоксемия
255
него дыхания повышает потребление кислорода всем организмом (П02).
Если при этом нет адекватного возрастания поглощения кислорода
легкими, то возникает артериальная гипоксемия. При обструктивных
расстройствах альвеолярной вентиляции не происходит достаточного для
предотвращения артериальной гипоксемии роста поглощения кислорода
легкими из-за обусловленной обструкцией патологической
вариабельности вентиляционно-перфузионных отношений респиронов, участков и
отделов легких.
Представим две идентичные по всем функциональным
характеристикам структурно-функциональные единицы легких, в которые дыхательная
смесь газов поступает по одному бронху (бронхиоле) (рис. 17.2). Из этого
бронха дыхательная смесь идет в структурно-функциональные единицы
легких по их бронхам (бронхиолам) с одинаковым сопротивлением.
Патогенное сужение просвета дыхательных путей
структурно-функциональных единиц легких происходит неравномерно, то есть происходит
асинхронное реагирование структурно-функциональных единиц
эффектора функций.
Рис. 17.2. Асинхронное сужение просвета дыхательных путей
как причина неравномерности вентиляции при вдохе
(Ri - сопротивление дыхательных путей первой структурно-функциональной единицы легких,
R2 - второй; г - радиус дыхательных путей; V - объемная скорость газотока)
Известно, что при ламинарном характере газотока, который
характеризует продвижение смеси газов по дыхательным путям небольшого
диаметра, снижение радиуса их просвета в два раза увеличивает их
сопротивление в 16 раз. Предположим, что в соответствии с принципом аси-
хроннного реагирования после патогенной бронхоконстрикции (сужения
просвета) радиус просвета бронхиолы одной структурно-функциональной
единицы составляет 90 % радиуса другой. При ламинарном газотоке
сопротивление - это обратная функция четвертой степени радиуса. Поэтому
такое небольшое различие в радиусах приводит к значительному
различию в сопротивлениях, при котором сопротивление бронхиолы одной
256
Глава 17
структурно-функциональной единицы больше сопротивления другой
примерно в полтора раза. При турбулентном газотоке в более крупных по
диаметру дыхательных путях небольшие различия в радиусе просвета
также обуславливают значительные различия в сопротивлениях.
Разные сопротивления бронхиол приводят к разной объемной
скорости тока по ним смеси газов. Время вдоха ограничено. Поэтому часть
структурно-функциональных единиц с более низким сопротивлением
дыхательных путей заполняется вдыхаемой газовой смесью избыточно,
тогда, когда в другие, с высоким сопротивлением, она поступает
недостаточно. Это приведет к снижению в конце фазы вдоха в части альвеол
легких общего давления газовой смеси и парциального давления кислорода.
Снижение РА02 через действие механизма альвеоло-капиллярного
рефлекса (спазм микрососудов прекапиллярного уровня респирона в ответ на
падение парциального давления кислорода в альвеолярной газовой смеси)
ограничивает снабжение смешанной венозной кровью респиронов,
вентиляция которых снижена. Тем не менее, альвеоло-капиллярный рефлекс
не может предотвратить снижения физиологической площади легочной
мембраны, вызванного неравномерностью альвеолярной вентиляции
вследствие ее обструктивных расстройств. Из-за гиповентиляции части
респиронов возрастает шунтирование смешанной венозной крови, и
возникает артериальная гипоксемия.
Обструктивные расстройства альвеолярной вентиляции при
хроническом обструктивном бронхите и бронхиальной астме через увеличение
сопротивления дыхательных путей снижают объемную скорость газотока
по дыхательным путям в фазу выдоха. Рост сопротивления дыхательных
путей приводит к снижению максимальной объемной скорости газотока
при выдохе (пиковая скорость), которая у больных бронхиальной астмой
находится на уровне меньшем, чем 60 л/мин, что может служить
показанием к госпитализации. Падение объемной скорости газотока снижает
объем газовой смеси, выдыхаемой при форсированном выдохе за его
первую секунду (объем форсированного выдоха за первую секунду, ОФВО.
Одновременно, но в меньшей степени, вследствие патологической
динамической компрессии дыхательных путей у больного снижается объем
максимально возможного и быстрого форсированного выдоха
(форсированная жизненная емкость легких, ФЖЕЛ). Это приводит к снижению
экспираторного индекса Тиффно, то есть отношения OOBi/ФЖЕЛ, до
значений меньших, чем 75 %.
Крайняя степень обструктивных расстройств внешнего дыхания - это
полная обтурация (закупорка) крупного бронха с прекращением
вентиляции респиронов отдела легких. При этом альвеоло-капиллярный рефлекс
не прекращает полностью поступление в невентилируемые респироны
смешанной венозной крови. В результате кислород из состава
альвеолярной газовой смеси уходит в смешанную венозную кровь, альвеолы
спадаются, и развивается ателектаз. Истинное патологическое шунтирование
смешанной венозной крови в легких при соответствующей
распространенности ателектаза может быть причиной артериальной гипоксемии.
Дыхательная недостаточность и артериальная гипоксемия
257
НЕДОСТАТОЧНАЯ ВЕНТИЛЯЦИЯ РЕСПИРОНОВ
КАК ПРИЧИНА АРТЕРИАЛЬНОЙ ГИПОКСЕМИИ
Гиповентиляция - это состояние легочного газообмена, которое
характеризуют артериальная гипоксемия и рост в артериальной крови
напряжения углекислого газа вследствие несоответствия патологически
низкой альвеолярной вентиляции объемной скорости тока оксигенируе-
мой смешанной венозной крови.
Артериальная гипоксемия вследствие гиповентиляции обусловлена не
только падением парциального давления кислорода в альвеолах,
вызванного тем, что кислород уходит из необновляемой альвеолярной газовой
смеси в смешанную венозную кровь. Одновременно в альвеолах растет
парциальное давление углекислого газа, что служит еще одним фактором
снижения парциального давления 02 в альвеолярной газовой смеси.
Если нет патологических изменений легких и дыхательных путей, то
гиповентиляция является следствием: нарушений центральной регуляции
внешнего дыхания, паралича или слабости дыхательных мышц
(диафрагмы в особенности). Дыхательная недостаточность при закрытой травме
груди отчасти обусловлена нарушением целостности каркаса грудной
клетки и пневмотораксом. Причины гиповентиляции почти эквивалентны
причинам респираторного ацидоза.
Рестриктивные расстройства внешнего дыхания при респираторном
дистресс-синдроме взрослых и тяжелой пневмонии, снижая
физиологическую площадь легочной мембраны, вызывают респираторный ацидоз и
гиповентиляцию как причину артериальной гипоксемии.
Физиологическая площадь легочной мембраны может быть снижена в результате
аспирации воды при утоплении. Аспирация кислого желудочного
содержимого после регургитации вызывает синдром Мендельсона, патогенез
которого составляет острое воспаление в ответ на первичную альтерацию
вследствие высокого содержания протонов в альвеолах, интерстиции и
клетках респиронов.
Асцит и ожирение снижают дыхательный объем и нарушают
равномерность альвеолярной вентиляции. В результате растет часть
респиронов с вентиляцией, низкой относительно объемной скорости кровотока.
Ожирение предрасполагает к падению физиологической площади
легочной мембраны после полостных оперативных вмешательств не только
через низкую податливость грудной клетки, но и являясь фактором риска
ателектазов в послеоперационном периоде.
Угнетение активности нейронов дыхательного центра как причину
гиповентиляции обуславливают:
♦ побочные эффекты лекарственных средств;
♦ черепно-мозговые ранения и травмы с эпидуральной и (или) субду-
ральной гематомой;
♦ злокачественные опухоли или их метастазы соответствующей
локализации;
♦ абсцессы мозга и менингит;
258
Глава 17
♦ расстройства мозгового кровообращения и их следствия, инфаркт
головного мозга и субарахноидальное кровоизлияние.
Причиной гиповентиляции могут быть нарушения сократительной
способности дыхательных мышц в результате патологических
изменений миоцитов или нарушений регуляции на уровне неиромышечного
синапса. Способность миоцитов произвольных мышц к сокращению
падает при таких заболеваниях как периодический паралич, а также
вследствие выраженных гипокалиемии и гипофосфатемии. Ареактив-
ность скелетных мышц к эфференгации на уровне синапса
обуславливают миастения и такое заболевание как синдром Гийена-Барре. Кроме
того, проведение нервного импульса через синапс к миоцитам
блокируют лекарственные средства: миорелаксанты, антибиотики из группы
аминогликозидов и др.
Наиболее частые в клинической практике причины низкого
парциального давления кислорода во вдыхаемой газовой смеси, вызывающего
артериальную гипоксемию, - это дефекты проведения искусственной
вентиляции легких.
В остром периоде тяжелой раневой болезни циркуляторная гипоксия
приводит к падению напряжения кислорода в смешанной венозной крови.
Одновременно в легких может расти общий объем ее. Повышенное
примешивание к артериальной крови смешанной венозной, напряжение
кислорода в которой находится на патологически низком уровне, служит одной из
причин артериальной гипоксемии после тяжелых ранений и травм.
РЕСПИРАТОРНЫЕ АЦИДОЗ И АЛКАЛОЗ
Респираторный ацидоз - это патологическое состояние вследствие
роста концентрации протонов во внеклеточной эюидкости и крови
([Н*]), обусловленное задержкой в организме углекислого газа из-за
преобладания высвобождения С02 во внутреннюю среду при его
образовании в ходе обмена веществ над выделением углекислого газа во внешнюю
среду при внешнем дыхании. О респираторном ацидозе свидетельствуют
рН артериальной крови на уровне более низком, чем нижний предел
диапазона нормальных колебаний (7,38), при напряжении в ней углекислого
газа большем, чем 43 мм рт. ст.
Ведущее звено патогенеза респираторного ацидоза - это снижение
способности системы внешнего дыхания выделять углекислый газ во
внешнюю среду. Его обуславливают нарушения регуляции в системе
внешнего дыхания и повреждения болезнями и патологическими
процессами ее эффекторов, снижающие через действие тех или иных
механизмов очищение альвеолярной газовой смеси от двуокиси углерода.
Наиболее часто к респираторному ацидозу приводят:
1. Хронические заболевания легких, которые в основном
характеризуют обструктивные расстройства внешнего дыхания. Следует заметить,
Дыхательная недостаточность и артериальная гипоксемия
259
что данные заболевания (бронхиальная астма и хронический обструктив-
ный бронхит) приводят к респираторному ацидозу лишь при крайней
степени обострения обструктивных расстройств альвеолярной вентиляции
(внешнего дыхания), которое в частности происходит при астматическом
статусе. При этом непосредственной причиной респираторного ацидоза
является критическое снижение числа тех респиронов, в которых
вентиляция достаточна для нормального очищения альвеолярных пространств
от углекислого газа. Причиной тому служит патологический рост
сопротивлений дыхательных путей респиронов, частей и отделов легких.
2. Падение силы сокращений дыхательных мышц в результате их
патологических изменений и нарушения передачи возбуждения в нейро-
мышечных синапсах {миастения, действие антидеполяризующих миоре-
лаксантов). Слабость дыхательных мышц как причина респираторного
ацидоза возникает при таких заболеваниях как синдром Гийена-Барре
(острая идиопатическая демиелинизурующая полинейропатия), а также
полимиозит и дерматомиозит. При ботулизме причина респираторного
ацидоза - это падение альвеолярной вентиляции вследствие блокады под
влиянием ботулинического экзотоксина высвобождения ацетилхолина из
пресинаптических нервных окончаний в нервно-мышечном соединении.
Действие аналогичного патогенетического механизма приводит к
респираторному ацидозу у больных с синдромом Итона-Ламберта,
3. Угнетение функциональной активности инспираторных нейронов
дыхательного центра как результат побочного действия наркотических
анальгетиков и других лекарственных средств, расстраивающих
центральную регуляцию внешнего дыхания.
При хроническом респираторном ацидозе, длительность которого
превышает 48 ч, происходит полная мобилизация всех возможных
механизмов компенсации, в результате которой почки начинают предельно
интенсивно образовывать и задерживать в организме бикарбонатный
анион. Поэтому при хроническом респиратором ацидозе в ответ на рост
РаСОг концентрация бикарбонатного аниона в плазме крови растет в
большей степени, и происходит меньшее снижение рН, чем при остром
респираторном ацидозе.
Рост напряжения углекислого газа в артериальной крови до уровня
более высокого, чем 60 мм рт. ст., (тяжелый респираторный ацидоз)
служит абсолютным показанием к проведению искусственной
вентиляции легких. Не следует ждать данных исследования кислотно-основного
состояния и газов крови, если симптомы говорят о тяжелом
респираторном ацидозе. О крайней степени тяжести респираторного ацидоза в
частности свидетельствуют такие его симптомы, как сонливость и
заторможенность. Они связаны с ростом давления спинномозговой
жидкости, обусловленным увеличением мозгового кровотока в ответ на
возрастание напряжения углекислого газа в артериальной крови. По
мере прогрессирования респираторного ацидоза к его симптомам
присоединяется артериальная гипотензия как следствие острой
недостаточности кровообращения (схема 17.1).
260
Глава 17
Угнетение
сократимости рабочего миокарда
Падение ударного
объема левого желудочка
Снижение ударного
объема правого желудочка
Снижение минутного
объема кровообращения
I
Падение общего
периферического сосудистого сопротивления
I
Артериальная гипотензия
Легочная
артериальная
гипертензия
Возрастание концентрации протонов во внутренней среде организма
t
Возрастание РаС02
t
Низкая экскреция углекислого газа
Схема 17.1. Патогенез недостаточности кровообращения вследствие
острого тяжелого респираторного ацидоза
Респираторный алкалоз - это патологическое состояние вследствие
избыточной относительно образования углекислого газа в ходе обмена
веществ экскреции двуокиси углерода при внешнем дыхании. О развитии
респираторного ацидоза свидетельствуют снижение напряжения углекислого
газа в артериальной крови до уровня более низкого, чем 37 мм рт. ст., при
возрастании рН до величин, превышающих 7,42.
Ведущее звено патогенеза респираторного алкалоза - это патогенно
избыточное выведение углекислого газа системой внешнего дыхания.
Метаболический алкалоз вызывается сдвигами регуляции внешнего
дыхания и патологическими изменения ее эффекторов, которые повышают
очищение альвеолярной газовой смеси от двуокиси углерода.
Наиболее частая причина острого респираторного алкалоза - это
неврозы, при которых внутрицентральные отношения и регуляция внешнего
дыхания расстраиваются таким образом, что система внешнего дыхания
начинает избыточно элиминировать СОг- Аномально повышенное
выделение углекислого газа снижает его напряжение в артериальной крови,
что в соответствии с уравнением Гендерсона-Гассельбаха уменьшает
концентрацию протонов во внеклеточной жидкости, то есть вызывает
респираторный алкалоз.
Гипервентиляционный синдром - это результат обострения невроза,
при котором избыточная вентиляция легких обуславливает
респираторный алкалоз. При этом гипервентиляция нарастает параллельно с
возрастанием тревожности. Тревожность (немотивированное беспокойство)
становится предельно выраженной и может трансформироваться сначала в
заторможенность, а затем (у небольшой части больных) в состояние пре-
комы. Прекому характеризует крайняя затрудненность еще возможного в
отличие от комы контакта с пациентом. Падение насосной функции
сердца как причина прекомы у больных с респираторным алкалозом возника-
Дыхательная недостаточность и артериальная гипоксемия
261
ет при росте рН артериальной крови до уровня 7,7 и выше. Респираторный
алкалоз при гипервентиляционном синдроме приводит к снижению
сократительной способности произвольных мышц, которое может обусловить
острую мышечную слабость (лоэюные параличи). Из других жалоб больных
с гипервентиляционным синдромом следует выделить ощущение
затрудненности вдоха, головокружения без обморока, а также онемение рук и ног.
Электроэнцефалографическим эквивалентом синдрома являются
билатеральные синхронные тета-волны, которые сменяются дельта-волнами с
периодическими пиковыми и медленными разрядами.
Отравление салицилатами приводит к респираторному ацидозу через
состояние патологически усиленного возбуждения инспираторных
нейронов дыхательного центра. Кроме того, хронически повышенный
уровень возбуждения инспираторных нейронов может быть следствием
нарушений мозгового кровообращения, опухолей головного мозга,
инфекционных поражений центральной нервной системы, а также возникать в
результате черепно-мозговых ранений и травм.
При синдромах (патологических состояниях) сепсиса и системной
воспалительной реакции респираторный алкалоз - это следствие
устойчивого возбуждения инспираторных нейронов, обусловленного супра-
сегментарным эффектом цитокинов, вызывающих данные синдромы
при циркуляции с кровью в повышенной концентрации (гиперцитоки-
немия).
Артериальная гипоксемия любого происхождения может быть
причиной респираторного алкалоза, который развивается из-за
гипервентиляции в ответ на возбуждение периферических хеморецепторов вследствие
падения напряжения кислорода в артериальной крови. Таков механизм
развития респираторного алкалоза у больных с эмболией легочной
артерии и ее ветвей, пневмонией, бронхиальной астмой и другими легочными
заболеваниями. Кроме того, причиной респираторного алкалоза у
больных с заболеваниями легких является возбуждение соответствующих
рецепторов патологически измененных легочной паренхимы, бронхов и
плевры, которое представляет собой стимул гипервентиляции.
Если у больных с респираторным алкалозом рН артериальной крови
поднимается до уровня более высокого, чем 7,6, то для коррекции
респираторного алкалоза может быть целесообразным дыхание газовыми
смесями, обогащенными углекислым газом.
ПАТОГЕНЕТИЧЕСКИЕ ПРИНЦИПЫ ПРОВЕДЕНИЯ
ИСКУССТВЕННОЙ ВЕНТИЛЯЦИИ ЛЕГКИХ
Искусственная вентиляция легких (ИВЛ) представляет собой крайне
инвазивный элемент интенсивной терапии. Поэтому врачам, которым
приходится прибегать к ИВЛ, необходимо четко представлять ее в
патогенетическом отношении обоснованные цели:
262
Глава 17
L Достижение адекватных потребностям организма оксигенации
смешанной венозной крови и освобождения ее от углекислого газа.
Достижение данной цели ИВЛ предотвращает летальный исход, когда в
результате типических патологических процессов, легочных заболеваний, а
также нарушений регуляции внешнего дыхания (респираторный
дистресс-синдром взрослых, астматический статус, побочный центральный
депрессивный эффект наркотических анальгетиков и др.) поглощение
кислорода легкими и экскреция углекислого газа падают до опасно низкого
уровня. У многих больных с тяжелой артериальной гипоксемией начало
ИВЛ позволяет достичь лишь минимально достаточного транспорта
кислорода от легких на периферию и устранить респираторный ацидоз.
Низкий уровень ?а02 и патологически высокое РаС02 как показания к
ИВЛ (табл. 17.3) свидетельствуют о критическом несоответствии
угнетенной функции внешнего дыхания потребностям организма.
Таблица 17.3
Показатели функции внешнего дыхания и легочного газообмена
как показания к началу ИВЛ
Показатель
Напряжение кислорода в артериальной крови, мм рт. ст.
Напряжение углекислого газа в артериальной крови, мм рт. ст.
Общее (сумма анатомического, физиологического и истинного)
шунтирование смешанной венозной крови, % от минутного
объема кровообращения
Физиологическое мертвое пространство, % дыхательного
объема
Жизненная емкость легких, мл/кг массы тела
Значения, при которых
необходимо начинать ИВЛ
<50
>55
>20
>60
<10
2. Уменьшение потребления кислорода организмом за счет почти
полного исключения или сниэюения уровня потребления свободной энергии при
внешнем дыхании. Достижение данной цели позволяет снизить патогенное
относительно насосной функции сердца возрастание минутного объема
кровообращения. Снижение потребления кислорода организмом,
обусловленное миоплегией при ИВЛ, может положительно сказаться на насосной
функции сердца у больных с тяжелой сердечной недостаточностью. В
остром периоде после тяжелых травм и у больных в других критических
состояниях недостаточное поглощение кислорода легкими служит причиной
респираторно-циркуляторной гипоксии, которую, кроме того,
обуславливает повышенная потребность клеток в 02. К таким состояниям в частности
относят тяжелые травматическую, раневую и ожоговую болезни, состояние
после высокотравматичных оперативных вмешательств, а также сепсис и
системную воспалительную реакцию. В результате преобладания
потребления кислорода организмом (высокой потребности в 02) над его
поглощением легкими и системным транспортом кислорода напряжение кислорода
в артериальной крови падает, то есть развивается артериальная гипоксемия.
Дыхательная недостаточность и артериальная гипоксемия
263
Артериальная гипоксемия в данном случае является результатом
несоответствия поглощения кислорода легкими и системного транспорта
кислорода высокой потребности организма в 02. Кроме того, артериальная
гипоксемия как звено патогенеза респираторно-циркуляторной гипоксии
через снижение насыщения кислородом гемоглобина обостряет кислородное
голодание всего организма. Поэтому снижение потребления кислорода
организмом после исключения работы мышц при внешнем дыхании может
устранить респираторно-циркуляторную гипоксию или снизить степень ее
тяжести. Под респираторно-циркуляторной гипоксией мы понимаем
патологическое состояние вследствие недостаточных поглощения кислорода
легкими и системного транспорта кислорода.
У больных с тяжелыми обструктивными расстройствами внешнего
дыхания (астматический статус), а также в случае критического снижения
физиологической площади легочной мембраны в результате рестриктив-
ных расстройств (респираторный дистресс-синдром взрослых)
артериальная гипоксемия служит стимулом компенсаторной гипервентиляции,
которая вследствие высоких сопротивления дыхательных путей и низкой
податливости легких приводит к особо выраженному росту работы мышц
при внешнем дыхании. В результате в основном потребление кислорода
всем организмом начинает составлять утилизация 02, которая происходит
при биологическом окислении в мышцах, работающих для внешнего
дыхания. У таких больных патогенно высокое потребление кислорода
организмом служит ведущей причиной артериальной гипоксемии, а ИВЛ
устраняет или уменьшает падение Ра02 в основном посредством снижения
потребления кислорода при самостоятельном внешнем дыхании.
Несмотря на снижение работы сердца после устранения артериальной
гипоксемии искусственной вентиляцией легких, эндотрахеальная
интубация и длительная ИВЛ вызывают расстройства системного, легочного и
периферического кровообращения (схема 17.2). Даже при близком к
идеальному соблюдении правил асептики в отделениях интенсивной
терапии, эндотрахеальная интубация и продленная ИВЛ всегда приводят к но-
зокомиальным легочным инфекциям. В этой связи следует рассматривать
ИВЛ как вынужденную временную меру, а одной из задач интенсивной
терапии считать создание предпосылок прекращения длительной
искусственной вентиляции легких.
Длительную ИВЛ часто проводят у больных, находящихся в
состоянии ускоренного голодания, которое характеризует интенсивное
использование резервов протеинов для поддержания белкового синтеза в
защитных системах (регуляции, иммунитета и пр.). Снижение интенсивности
функционирования дыхательных мышц при длительной ИВЛ быстро
переводит их в разряд депо интенсивно мобилизуемых белковых резервов,
то есть служит причиной их дистрофии. Поэтому ИВЛ легче начать, чем
прекратить без вреда для больного. В этой связи полноценное
парентеральное или смешанное питание следует считать средством
предупреждения дистрофии дыхательных мышц вследствие низкого уровня их
работы при длительной ИВЛ.
264
Глава 17
дение рецепторов трахеи и бронхов
i
Рост интраторакального давления
ч
*
Дегочная венозная и артериальная
гипертензия
ч
7
Увеличение постстнагрузки
правого желудочка
н
1"" Щ
Вагальные рефлексы
i
г
Расширение вен
i
г
Снижение
общего венозного
возврата к сердцу
1
Снижение
ударного объема
правого желудочка |
1
г
Падение ударного объема
левого желудочка
3
г
*-
1
г
11 Брадикардия |
W
Падение
сократимости сердца
▼
Падение минутного объема кровообращения
Схема 17.2. Патогенез расстройств кровообращения вследствие ИВЛ
и эндотрахеальной интубации
Быстро наступающую усталость (слабость) дыхательных мышц после
прекращения длительной ИВЛ считают одной из основных причин ее
возобновления. Слабость дыхательных мышц как следствие длительной
ИВЛ- это результат не только атрофии дыхательных мышц, но и
расстройств регуляции дыхательного акта. Нарушения регуляции
предположительно можно связать с длительным извращением афферентации в
системе внешнего дыхания при длительной ИВЛ. Нельзя исключить, что
дизрегуляция дыхательного акта после длительной ИВЛ повышает
потребление кислорода для самостоятельного внешнего дыхания, что
повышает потребление кислорода всем организмом. Рост потребления
кислорода через снижение напряжения кислорода в артериальной крови
обуславливает необходимость возобновления ИВЛ.
Можно считать, что после определенного периода длительной ИВЛ
возобновление естественного внешнего дыхания становится
невозможным вследствие слабости дыхательных мышц. В этой связи становится
ясной необходимость «переходного» режима ИВЛ, при котором работа
дыхательных мышц несколько выше, чем при длительной ИВЛ, но
остается сниженной. Кроме того, у больных с сердечной недостаточностью
иногда полезно ограничить потребление кислорода организмом для
меньшей работы сердца. Рациональным в таких случаях следует признать
Дыхательная недостаточность и артериальная гипоксемия
265
снижение высокого потребления кислорода при внешнем дыхании без
значительного снижения работы дыхательных мышц. Сохранение
определенного уровня работы дыхательных мышц необходимо для
предотвращения их дистрофии вследствие длительной ИВЛ. Таким
«переходным» режимом ИВЛ является вспомогательная искусственная
вентиляция легких. В этом режиме каждая попытка самостоятельного вдоха
запускает подачу вдыхаемой газовой смеси, которую аппарат
искусственной вентиляции легких вдувает в легкие в заданном объеме. При
снижении частоты дыхательных актов (ЧД) ниже определенного предела
аппарат начинает работать как автомат с заданной частотой искусственных
вдохов. Поскольку каждый вдох больного в режиме вспомогательной
вентиляции дополняется или полностью обеспечивается респиратором, то
работа дыхательных мышц невелика, хотя главная дыхательная мышца
диафрагма продолжает сокращаться. Следует заметить, что увеличение
ЧД у больных, которым проводят вспомогательную ИВЛ, через
увеличение минутного объема альвеолярной вентиляции может привести к
респираторному алкалозу.
ИВЛ в режиме прерывистой принудительной вентиляции является
промежуточным этапом на пути к адекватному самостоятельному дыханию
больного после вспомогательной ИВЛ. В таком режиме ИВЛ проводят
больным, у которых самостоятельное дыхание через определенные
промежутки времени ведет к утомлению дыхательных мышц и его следствию
острой дыхательной недостаточности. Этот режим ИВЛ заключается в том,
что через определенные временные интервалы аппарат ИВЛ начинает
периодическую подачу вдыхаемой газовой смеси в легкие больного вне
зависимости от его попыток сделать вдох. Периоды интенсивной работы всех
дыхательных мышц в данном режиме устраняют или предотвращают их
дистрофию. Кроме того, к преимуществам этого режима относят:
♦ меньший риск развития дыхательного алкалоза, так как при данном
режиме избыточная вентиляция легких практически невозможна;
♦ при прерывистой принудительной вентиляции меньше суммарная
длительность периодов положительного (выше атмосферного) ин-
траторакального и интраальвеолярного давления и, соответственно,
меньше отрицательное влияние ИВЛ на кровообращение;
♦ меньшие дозы, а значит меньшие побочное действие и кумуляция седа-
тивных средств, наркотических анальгетиков и миорелаксантов.
Однако в клинической практике эти преимущества периодической
принудительной вентиляции проявляют себя не всегда. Это связывают во-
первых с тем, что аппарат ИВЛ часто начинает искусственную
вентиляцию уже после того, как развились выраженная усталость дыхательных
мышц и артериальная гипоксемия. В результате потребность
дыхательных мышц в кислороде и энергопластических субстратах часто находится
на патологически высоком уровне и остается неудовлетворенной. Это
обостряет дистрофию мышц, участвующих в дыхательном акте, и
тормозит возобновление полностью самостоятельного и адекватного внешнего
дыхания.
266
Глава 17
У больных с диффузными патологическими изменениями легочной
паренхимы (респираторный дистресс-синдром взрослых, пневмония)
одним из звеньев патогенеза крайне тяжелых артериальной гипоксемии и
острой дыхательной недостаточности является спадение респираторных
бронхиол и альвеолярных мешков, выключающее респироны из
легочного газообмена. Для возобновления вентиляции респиронов при ИВЛ
увеличивают соотношение продолжительности фаз вдоха или выдоха от 1:2
до 1:1-4:1, что ведет к раскрытию респираторных бронхиол и
альвеолярных мешков и улучшению легочного газообмена. С одной стороны,
увеличение соотношения повышает вероятность отрицательных влияний
искусственной вентиляции легких на кровообращение, так как
увеличивает суммарную длительность периодов положительного интраторакаль-
ного и интраальвеолярного давлений. С другой стороны, при данном
соотношении фаз вдоха и выдоха снижается максимальное положительное
инспираторное давление, и становится меньшим угнетение насосной
функции сердца вследствие ИВЛ.
При высокочастотной ИВЛ вдыхаемая смесь подается с высокой
частотой (60-3000 мин'1) и с малым дыхательным объемом, составляющим
2-4 мл/кг. Речь в данном случае идет не о вентиляции легких в силу
градиентов давлений между альвеолами и верхними дыхательными путями,
а о продвижении «сотрясаемой» газовой смеси по дыхательным путям до
респиронов подобно тому, как облако аэрозоля заполняет камеру его
генератора. Поэтому считают, что из-за сложности работы с
соответствующим аппаратом ИВЛ и не доказанной клинической эффективности
данный режим ИВЛ может быть рекомендован только по жизненным
показаниям, когда травма дыхательных путей или бронхоплевральный свищ
обуславливают артериальную гипоксемию и неэффективность обычной
ИВЛ в силу невозможности создания градиента давлений между
альвеолами и дыхательными путями.
Так как гипоксемия более опасна, чем токсическое действие
кислорода при кратковременной ИВЛ газовой смесью с высоким содержанием 02,
то ИВЛ у больных с острой дыхательной недостаточностью начинают
при концентрации кислорода во вдыхаемой газовой смеси, составляющей
90-100 %. Затем, исследуя напряжения кислорода и углекислого газа в
артериальной крови и непрерывно проводя пульсоксиметрию,
содержание кислорода во вдыхаемой газовой смеси (Fi02) снижают не более, чем
до 60%, поддерживая напряжение кислорода в артериальной крови на
уровне более высоком, чем 60 мм рт. ст.
При экстренном начале ИВЛ у больного с острой дыхательной
недостаточностью минутный объем дыхания (вентиляции) должен находиться
на уровне 100 мл/кг. Этого обычно достигают при ЧД равной 8-14 мин"1 и
дыхательном объеме, составляющем 10-15 мл/кг. Затем МОД подбирают
так, чтобы рН крови был в нормальных пределах. При этом не
ориентируются на среднестатистическую норму диапазона изменений
напряжения углекислого газа в крови здоровых людей. Так, РаС02 у больных с
хроническими обструктивными расстройствами внешнего дыхания при
Дыхательная недостаточность и артериальная гипоксемия
267
ИВЛ необходимо поддерживать в пределах, соответствующих состоянию
хронической гипервентиляции в ответ на артериальную гипоксемию. К
этому низкому уровню РаСОг больные с хроническими обструктивными
расстройствами вентиляции легких адаптированы через снижение
образования бикарбонатного аниона почками и бикарбонатурию. Почки у
таких пациентов переходят на стабильно отличный от обычного
физиологического уровень синтеза и реабсорбции бикарбонатного аниона.
Поэтому возвращение напряжения углекислого газа в артериальной крови в
обычные «нормальные» пределы может привести к дыхательному
ацидозу, так как почки некоторое время не смогут адекватно образовывать и
задерживать бикарбонатный анион.
Желательно при проведении экстренной ИВЛ использовать
дыхательный объем такой величины, при которой максимальное давление в
дыхательных путях в фазу вдоха не превышает 40-50 см вод. ст. Это
ограничение направлено на предотвращение падения насосной функции сердца
через рост интраальвеолярного давления. Если необходимо увеличить
объем искусственной альвеолярной вентиляции, то целесообразнее
повысить дыхательный объем, чем увеличить ЧД, так как снижение
дыхательного объема может привести к спадению и обтурации респираторных
бронхиол и альвеолярных мешков. Тем самым увеличение ЧД исключит
из участия в легочном газообмене часть респиронов и приведет к
артериальной гипоксемии.
Положительное, то есть превышающее атмосферное, давление в конце
выдоха (ПДКВ) сохраняет и увеличивает физиологическую площадь
легочной мембраны, предотвращая спадение альвеол и расправляя спавшиеся
альвеолы. Поэтому использование ПДКВ, которое больше атмосферного
давления на 3-5 см вод. ст., при обычном режиме ИВЛ позволяет
уменьшить содержание кислорода во вдыхаемой газовой смеси без снижения
РаОг. ПДКВ при ИВЛ опасно падением насосной функции сердца,
обусловленным ростом интраальвеолярного давления. Когда ПДКВ выше
атмосферного давления на 15 см вод. ст., то резко возрастает риск
баротравмы легких и пневмоторакса.
Кроме усталости (быстрого наступления слабости) дыхательных мышц
причинами медленного перехода от длительной ИВЛ к адекватному
самостоятельному внешнему дыханию служат:
♦ хроническая легочная инфекция;
♦ патологически высокая бронхиальная секреция;
♦ бронхоспазм как причина обструктивного нарушения внешнего
дыхания;
♦ снижение минутного объема кровообращения, ограничивающее
поступление смешанной венозной крови к респиронам на уровне всех
легких.
Все эти патологические состояния тем или иным образом снижают
физиологическую площадь легочной мембраны. Так легочная инфекция
через воспаление выключает респироны из участия в легочном
газообмене, что снижает физиологическую площадь легочной мембраны. Эту
268
Глава 17
площадь уменьшает, вызывая патологическую вариабельность вентиля-
ционно-перфузионных отношений, и бронхоспазм, который вызывает
патогенную вариабельность сопротивлений дыхательных путей респиронов,
участков легочной ткани и отделов легких. По аналогичному механизму
снижает площадь физиологической легочной мембраны и вариабельная,
частичная или полная обтурация просвета дыхательных путей
бронхиальным секретом. Низкий минутный объем кровообращения ведет к
недостаточной оксигенации смешанной венозной крови, приводя на уровне
всех легких к низкому относительно вентиляции кровоснабжению
респиронов смешанной венозной кровью, то есть, снижая физиологическую
площадь легочной мембраны. Снижение физиологической площади легочной
мембраны как причина артериальной гипоксемии приводит к
компенсаторной гипервентиляции и ее следствию, быстрому наступлению
усталости дыхательных мышц, еще не окрепших после длительной ИВЛ.
Патологические изменения легких и дыхательных путей как причины
задержки восстановления адекватного самостоятельного дыхания после
длительной ИВЛ обуславливают повышенную работу мышц для
внешнего дыхания не только в результате компенсаторной гипервентиляции в
ответ на снижение физиологической площади легочной мембраны,
вызывающего артериальную гипоксемию. Так, рост сопротивления
дыхательных путей вследствие бронхоспазма, их полной или частичной закупорки
секретом повышает работу дыхательных мышц во много раз, что
обуславливает быстрое развитие их усталости после прекращения
длительной ИВЛ и необходимость ее возобновления вскоре после экстубации.
Следует заметить, что повторная интубация трахеи через вагальные
рефлексы обостряет бронхоспазм, что еще в большей степени задерживает
восстановление незатрудненного самостоятельного дыхания.
Глава 18
БРОНХИАЛЬНАЯ АСТМА
И АСТМАТИЧЕСКИЙ СТАТУС
Бронхиальная астма - это полиэтиологичное заболевание, основное
звено патогенеза которого - обструктивное расстройство внешнего
дыхания вследствие повышенной чувствительности бронхов к спазмирую-
щим их регуляторным влияниям (гиперреактивности) и патогенного
воспаления бронхиачьной стенки.
Астматический статус - обострение бронхиальной астмы и
соответственно обструктивных расстройств внешнего дыхания, которое не
купируют три последовательных ингаляции, приема или введения обычно
эффективных бронхорасширяющих средств (бета-два-адреномиметиков
и др.).
От бронхиальной астмы страдают 3-8 % представителей основной
популяции в развитых странах. Хотя существует множество этиопатогене-
тических и клинических классификаций бронхиальной астмы,
большинство из них выделяет два основных вида заболевания:
♦ аллергическая (атопическая) астма, которая возникает как правило
в детском возрасте и связана с антигенной стимуляцией системы
иммунитета определенными аллергенами. При этом под атопией
понимают аллергическую реакцию со строго специфической
семейной предрасположенностью, которую чаще всего вызывают
аллергены пыльцы, пищевых продуктов и яда насекомых;
♦ неаллергическая астма, патогенез которой в основном составляет
гиперреактивность, и которая впервые чаще развивается у взрослых
больных.
В настоящее время известен целый ряд экспериментальных и
полученных в результате клинических исследований фактов, которые
позволяют считать воспаление определяющим звеном патогенеза
бронхиальной астмы и других аллергических заболеваний дыхательных путей.
Сегодня стало ясным, что как гиперреактивность бронхиальной
стенки, так и ее отечность, утолщение вследствие клеточной пролиферации во
многом связаны с воспалением, обуславливающим высвобождение из
клеток системы иммунитета на уровне стенки бронхов биоактивных
веществ и цитокинов - гистамина, эйкосаноидов (простагландины, лейкот-
риены, тромбоксан), фактора активирующего тромбоциты и др.
Воздействие этих медиаторов реализуется через G-протеины (связующие
рецепторы) с функцией модуляции фенотипа клетки. Активация G-протеинов
вызывает разнообразные реакции системного и органного уровней, в том
числе протекающие в стенке бронхов и легочной паренхиме.
Иммуноглобулины Е как гомоцитотропные антитела, патогенно функционирующие
при развитии бронхиальной астмы, воздействуют через активацию G-
270
Глава 18
протеинов на бета-адренорецепторы бронхиальной стенки, снижая их
чувствительность и обуславливая гиперреактивность. Кроме того,
связанная с анафилаксией активация G-протеинов как звено патогенеза
бронхиальной астмы происходит в легочных эпителиоцитах, в клетках подслизи-
стых желез бронхиальной стенки, в тучных клетках, эозинофилах, поли-
морфонуклеарах, лимфоцитах.
Легко заметить, что большинство из перечисленных тут клеточных
элементов легочной паренхимы представляют собой потенциальные
клеточные эффекторы воспаления, которые становятся таковыми в
активированном состоянии. Активацию G-протеинов клеточных
эффекторов воспаления при бронхиальной астме нельзя с полной уверенностью
считать первичным (ведущим) звеном его патогенеза, так как спектр
биоактивных веществ, агентов системной, а также аутокринной и пара-
кринной регуляции, активирующих данные связующие рецепторы,
весьма широк и включает все те же эйкосаноиды, фракции системы
комплемента, гистамин и др., то есть медиаторы воспаления,
высвобождаемые его активированными клеточными эффекторами. Поэтому не
исключено, что воспаление бронхиальной стенки как причина
бронхиальной астмы может быть скорее не следствием активации G-белков, а его
причиной.
При бронхиальной астме эозинофилы мигрируют в межклеточные
пространства бронхиальной стенки, единица массы которой у больных
бронхиальной астмой содержит эозинофилов в 100 раз больше, чем
единица массы циркулирующей крови здорового человека. Особенно много
эозинофилов содержит подслизистый слой стенки дыхательных путей. В
этой связи бронхиальную астму определяют как хронический десквама-
ционный эозинофильный бронхит. После активации эозинофила,
происходящей через активацию G-протеинов, он высвобождает цитокины и
ферменты, нарушающие работу реснитчатого эпителия, вызывающие
гиперреактивность и усиливающие воспаление. При этом из гранул
эозинофилов в частности высвобождается фактор, активирующий
тромбоциты, который можно рассматривать в качестве медиатора
бронхиальной астмы, так как данный цитокин обладает рядом соответствующих
свойств:
♦ усиливает бронхоспазм;
♦ индуцирует агрегацию тромбоцитов и вызывает микротромбоз;
♦ в качестве агента паракринной регуляции стимулирует выброс эо-
зинофилами главных медиаторов бронхиальной астмы, то есть эй-
косаноидов;
♦ вызывает активацию полиморфонуклеаров и клеток системы моно-
нуклеарных фагоцитов, высвобождающих альфа-фактор некроза
опухолей и интерлейкины, во многом ответственные за прогрессирова-
ние воспаления бронхиальной стенки как причины астмы.
Особая роль эозинофилов в патогенезе бронхиальной астмы
послужила побуждающим мотивом исследования апоптоза эозинофилов у
больных бронхиальной астмой как механизма обратного развития вое-
Бронхиальная астма и астматический статус
271
паления и гиперрекативности. В переводе с греческого апоптоз означает
следующее: без падения, гибели. Тем самым подчеркивается, что
апоптоз - это в известной мере ачгоритмизированный и запускаемый
воздействиями определенного спектра переход клетки в небытие,
остановленный на определенном этапе; в результате такой остановки
клетка не подвергается цитолитической деструкции, но прекращает
свои многообразные, в том числе в определенной ситуации патогенные,
функции, придающие ей свойства клеточного эффектора конкретной
аномалии, болезни и патологического процесса. В морфопатогенетиче-
ском отношении апоптоз характеризует конденсация хроматина и
деградация дезоксирибонуклеиновой кислоты. Апоптоз как причина
блокирования клональной пролиферации представляет собой основной
механизм элиминации кортикальных тимоцитов при выработке
иммунологической толерантности по отношению к аутоантигенам. Кроме того, апо-
потоз - это основной начальный этап гибели клеток эукариотов при
эмбриогенезе и обратном развитии злокачественных опухолей. Апоптоз в
организме человека и млекопитающих индуцируют цитотоксические
лимфоциты-киллеры, натуральные киллеры, лимфотоксины и глюкокор-
тикоиды, полная элиминация интерлейкинов из циркулирующей крови
как индуктор апоптоза лимфоидных клеток в эксперименте, а также мо-
ноклональные антитела к определенным антигенам впадающих в
апоптоз клеток.
Уже давно стало ясным, что эозинофил - это главное действующее
лицо патогенных межклеточных взаимодействий на уровне стенки
бронхов, которые обуславливают бронхиальную астму. Механизмы же выхода
эозинофилов из циркулирующей крови в интерстиций легких и
накопления данных лейкоцитов в бронхиальной стенке стали лишь отчасти
проясняться только сегодня. Рост содержания эозинофилов в стенках бронхов
{тканевая эозинофилия) находится в прямой связи с выраженностью
симптомов бронхиальной астмы тогда, когда болезнь в своем развитии не
достигла крайних степеней тяжести. Активированные эозинофилы
высвобождают ряд биологически активных веществ, ответственных за
гиперреактивность клеточных элементов стенки бронхов относительно
системных и локальных регуляторных влияний, сужающих бронхи (бронхиолы):
гранулярные белки эозинофилов, протеины с высоким содержанием
аргинина, металлоэндопептидазы, метаболиты липидов и др. По мере
обратного развития тканевой эозинофилии исчезает и астма. Известно, что
эозинофилы, циркулирующие с кровью, активируются и выходят в
межклеточные пространства под влиянием целого ряда цитокинов, хемоат-
трактантов и медиаторов воспаления: анафилатоксина, фактора активации
тромбоцитов, лейкотриена В4, интерлейкина-1, -3, -5, -8 и -16, макрофа-
гального воспалительного белка-1 альфа, моноцитарного протеина-хемо-
аттрактанта-2, -3, -4, эотаксина и др. О значении цитокинов в индукции
воспаления стенок дыхательных путей, связанного с тканевой эозинофи-
лией и вызывающего бронхиальную астму, свидетельствует
предотвращение экспериментально вызванного «эозинофильного» воспаления стен-
272
Глава 18
ки бронхов моноклональными антителами к интерлейкину-5. Полагают,
что инициирующим моментом тканевой эозинофилии и воспаления как
непосредственных причин бронхиальной астмы служит высвобождение
цитокинов эпителиоцитами дыхательных путей после модуляции экс-
пресии генома и фенотипа эпителиальных клеток. Цитокины,
высвобождаемые эпителиоцитами, выступают хемоаттрактантами относительно
эозинофилов. Уникальность в этом ряду эотаксина состоит в том, что он
является хемоаттрактантом исключительно для эозинофилов. Глюко-
кортикоиды ослабляют тканевую эозинофилию, восстанавливают
нормальную реактивность бронхов по отношению к регуляторным
влияниям, спазмирующим бронхи. В результате глюкокортикоиды могут
устранить симптомы бронхиальной астмы. Это положительная сторона
действия гормонов коры надпочечников как препаратов для лечения
больных бронхиальной астмой складывается из следующих эффектов
кортикостероидов :
♦ индукция апоптоза эозинофилов;
♦ торможение образования и высвобождения факторов роста и
дифференциации эозинофилов;
♦ подавления синтеза и секреции цитокинов со свойствами
медиаторов воспаления.
Известно, что цитокины со свойствами медиаторов воспаления
(фактор некроза опухолей-альфа и интерлейкин-1-бета), воздействуя на
легочные эпителиоциты определенных клеточных линий, повышают
экспрессию ими эотаксина, что приводит к тканевой эозинофилии и
обуславливает бронхиальную астму. В этой связи хроническое воспаление
(например, при гастроэзофагеальном рефлюксе) как причину гиперци-
токинемии можно считать фактором предрасположенности к
бронхиальной астме.
Как патологическое состояние организма астму в первую очередь
характеризует периодически усиливающаяся обструкция дыхательных
путей, обусловленная как повышенной реактивностью гладкомышечных
элементов бронхиальной стенки к действию стимулов их сокращения, так
и воспалением на уровне стенки бронхов, имеющим чисто патогенное
значение. При этом для бронхиальной астмы характерна крайняя
вариабельность степени тяжести обструктивных расстройств внешнего
дыхания (табл. 18.1) у одного больного в разные периоды болезни и в пределах
всей совокупности больных.
Астматический статус в частности отличается от затянувшегося
приступа бронхиальной астмы тем, что он может быть обострением не только
атопической или неаллергической бронхиальной астмы, но и
представлять собой осложнение хронического обструктивного бронхита,
эмфиземы легких, интерстициального пневмофиброза, а также других
хронических неспецифических заболеваний легких. Чаще всего астматический
статус - это обострение бронхиальной астмы, гастроэзофагеалъного реф-
люкса или является конечным результатом обострения хронического об-
структивного бронхита.
Бронхиальная астма и астматический статус
273
Таблица 18.1
Оценка тяжести бронхиальной астмы
(Зильбер А.П., 1996)
Количество баллов
Напряжение кислорода
в артериальной крови,
мм рт. ст
Цианоз
Дыхательные шумы на
вдохе
Затрудненный выдох
Дренирование мокроты
Участие
вспомогательных мышц
Расстройства
центральной нервной системы
0
70-100
нет
нормальные
нет
нормальное
нет
нет
1
<70
есть при дыхании
воздухом, не
обогащенным кислородом
свистящие хрипы
умеренный
сниженное
умеренное
угнетение или
возбуждение
2
<70 при содержании
кислорода во вдыхаемой
газовой смеси (НОг) не
меньшем, чем 40 %
Есть при Fi02, не
меньшем, чем 40 %
ослабленные
выраженный
отсутствует
резко выраженное
кома
Примечание: бронхиальную астму, при которой тяжесть состояния составляет 8-14 баллов,
можно считать трансформировавшейся в астматический статус; при сумме баллов в диапазоне от
8 до 14 у пациента следует констатировать компенсированную дыхательную недостаточность;
сумма баллов от 3 до 5 свидетельствует о легком приступе бронхиальной астмы.
Острая дыхательная недостаточность при астматическом статусе
часто становится ведущим звеном танатогенеза вследствие:
♦ быстрого прогрессирования обструктивных расстройств
альвеолярной вентиляции;
♦ неравномерности вентиляции и патологической вариабельности
вентиляционно-перфузионных отношений респиронов как причины
артериальной гипоксемии.
Основным звеном патогенеза бронхиальной астмы, согласно его
классической иммунопатологической схемы, является взаимодействие
аллергенов с гомоцитотропными антителами, фиксированными на поверхности
тучных клеток и базофилов. Образование иммунных комплексов на
поверхности тучных клеток и базофилов переводит их в активированное
состояние с последующей дегрануляцией. При дегрануляции базофилы и
тучные клетки высвобождают целый спектр биоактивных веществ,
обладающих бронхоспастическими свойствами. Хронический бронхоспазм у
больных бронхиальной астмой обостряется, и возникают обострение
бронхиальной астмы или астматический статус.
Исследования иммунопатологического звена патогенеза бронхиальной
астмы в последние десятилетия выявили его новые стороны. Их результаты
позволили отдельно выделить иммунопатологическую реакцию поздней
фазы, обуславливающую обострение бронхиальной астмы и астматический
статус. Реакция поздней фазы взаимодействия иммунной системы с
аллергенами возникает при высвобождении гистамина активированными ней-
274
Глава 18
трофилами. Рост действующей концентрации гистамина служит стимулом
для активации тучных клеток и базофилов с их последующей дегра-
нуляцией. Реакция поздней фазы отличается от немедленной реакции
повышенной чувствительности тем, что при ней не происходят образование и
высвобождение простагландина D2. Реакцию поздней фазы вызывает
действие разнообразных раздражителей: холода, высокого содержания во
вдыхаемой газовой смеси озона и других. Данные раздражители реализуют
свои патогенные свойства при взаимодействии с организмом через
активацию связующих рецепторов клеточных эффекторов воспаления
бронхиальной стенки у больных бронхиальной астмой, то есть G-протеинов. Реакция
поздней фазы может возникать и при вирусных инфекциях.
Классическая схема патогенеза бронхиальной астмы- это описание
реакции повышенной чувствительности первого типа, при которой
превалируют обструктивные расстройства альвеолярной вентиляции в
результате высвобождения активированными тучными клетками
низкомолекулярных медиаторов анафилаксии, которые повышают секрецию
железистыми клетками эпителия дыхательных путей и вызывают бронхоспазм:
эозинофильный хемотаксический фактор, гепарин, гистамин, серотонин и
ряд ферментов. У большинства больных анафилактоидные (сходные с
анафилаксией) реакции развиваются и без взаимодействия аллергенов с
иммуноглобулинами Е, фиксированными на базофилах и тучных клетках.
Анафилактоидная реакция - это высвобождение медиаторов
анафилаксии под влиянием факторов неиммунной природы, то есть не аллергенов
и иммуноглобулинов. Ее вызывают влияния на клетки флогогенов,
высвобождаемых в очаге воспаления, а также побочное действие
радиоконтрастных препаратов, вводимых для рентгенологического исследования, и
нестероидных противовоспалительных средств.
Бронхиальная астма не имеет специфического патогенеза, представляя
собой униформный результат многих и разнообразных патологических
процессов, которые на уровне стенки бронхов и бронхиол через свое
взаимодействие разрешаются воспалением. В этой связи бронхиальную астму считают
не нозологической формой, а типовым патологическим процессом.
Для всех больных бронхиальной астмой характерна неспецифическая
гиперреактивность гладкомышечных элементов стенок бронхов. Она
проявляется спазмом бронхов при ингаляции суживающих их веществ
(гистамина и др.) в дозах, обычно не вызывающих бронхоспазм. При
обследовании здоровых людей гиперреактивность бронхов выявляют как
предболезнь в развитии бронхиальной астмы.
Хотя снижение чувствительности бета-два-адренорецепторов
предположительно может иметь значение в патогенезе бронхиальной астмы, у
большинства больных оно является следствием длительного применения
бета-два-адреномиметиков для купирования и предупреждения бронхо-
спазма.
Бета-два-адренорецепторы широко представлены на всем протяжении
дыхательных путей. Из возбуждение через систему аденилатциклазы и
аденозинмонофосфата миоцитов ведет к расслаблению гладкомышечных
Бронхиальная астма и астматический статус
275
элементов бронхиальной стенки и расширению бронхов. Возбуждение
альфа-адренорецепторов дыхательных путей приводит к эффектам
противоположным вызываемым возбуждением бета-два-адренорецепторов. Об
определенной роли возбуждения альфа-адренорецепторов в развитии
бронхиальной астмы свидетельствует эффективность альфа-адренобло-
каторов в ослаблении бронхоспазма у части больных астмой.
Прямая нервная парасимпатическая стимуляция гладких мышц стенок
бронхов вызывает рост содержания в их клетках вторичного мессенджера,
циклического гуанозинмонофосфата. Это тормозит внутриклеточные
эффекты аденозинмонофосфата. Гладкие мышцы бронхов сокращаются, и
развивается бронхоспазм. Обострение бронхоспазма при отрицательном
эмоциональном стрессе обычно связано с нервной парасимпатической
стимуляцией гладкомышечных элементов стенок бронхов. При таком
обострении бронхоспазма эффективны холинолитики.
Из экзогенных токсинов, играющих роль факторов развития
бронхиальной астмы и астматического статуса следует выделить:
♦ аллергены воздуха;
♦ промышленные и бытовые загрязнения воздуха пылью и дымом;
♦ ядовитые примеси во вдыхаемой смеси газов: двуокись серы, озон;
♦ побочное действие нестероидных противовоспалительных средств,
вызывающих сдвиг обмена арахидоновой кислоты в сторону
образования лейкотриенов, то есть медиаторов воспаления с
выраженной бронхоспастической активностью.
В настоящее время ведущим звеном патогенеза бронхиальной астмы у
многих больных все чаще признают врожденные или приобретенные
расстройства обмена арахидоновой кислоты в легких. В результате этих
расстройств в обмене арахидоновой кислоты на уровне легких начинает
преобладать активность фермента 5-липоксигеназы. Это обуславливает рост
содержания в легких производных арахидоновой кислоты с
бронхоспастической активностью (эйкосаноидов): сульфидопептидных
лейкотриенов, лейкотриена В4 и других.
Нестероидные противовоспалительные средства вызывают
бронхиальную астму, обуславливая дефицит молекулярных эффекторов функций в
локальной системе ауторегуляции, противостоящей системным регулятор-
ным влияниям, спазмирующим бронхи. По-видимому, это частный случай
реализации общепатологической закономерности.
Астматический статус у многих больных сопровождается системными
проявлениями массивного высвобождения биоактивных веществ,
обладающих сосудорасширяющим действием и свойством повышать
проницаемость капилляров. Основными признаками и следствиями роста
высвобождения этих веществ являются артериальная гипотензия и
крапивница. У 10 % больных, перенесших астматический статус с такими
особенностями, выявляют все элементы астматической триады:
♦ бронхоспазм и умеренную артериальную гипоксемию еще до
развития астматического статуса;
♦ полипы носа;
276
Глава 18
♦ повышенная чувствительность к нестероидным
противовоспалительным средствам как препаратам, вызывающим бронхоспазм
через нарушение обмена арахидоновой кислоты в легких.
Астматическая триада как фенотипический признак еще пока неясным
путем может передаваться по наследству.
НЕЙРОГЕННЫЕ МЕХАНИЗМЫ ПАТОГЕНЕЗА
БРОНХИАЛЬНОЙ АСТМЫ
Ведущим звеном патогенеза бронхиальной астмы следует считать
воспаление слизистой оболочки дыхательных путей небольшого диаметра.
Воспаление обуславливает спазм гладкомышечных элементов
бронхиальной стенки, который усиливает обструктивные расстройства альвеолярной
вентиляции. В настоящее время есть ряд фактов, полученных в результате
экспериментов, который позволяет предположить, что воспаление как
причина бронхиальной астмы может быть в основном нейрогенным.
Нейрогенное воспаление может происходить во многих тканях, в том
числе в коже и во внутренних органах. Его вызывают аксон-рефлексы,
реализуемые через патогенное функционирование рецепторов нервных С-
волокон (С-рецепторов), ветвящихся в тканях. Возбуждение этих нервных
окончаний вызывает распространение нервных импульсов во всем рецеп-
торном комплексе, что приводит к высвобождению из нервных
окончаний, а возможно из самих нервных волокон, сенсорных нейропептидов.
Действие нейропептидов обуславливает сокращение гладкомышечных
элементов бронхиальной стенки, стимулирует секрецию слизи, а также
расширяет микрососуды и увеличивает их проницаемость. Стимулами
для С-рецепторов являются эффекты на них составляющих табачного
дыма, двуокиси серы, а также капсаицина (острого экстракта
мексиканского перца). Если в эксперименте вызывают деструкцию С-рецепторов
капсаицином или блокируют их местными анальгетиками, то тем самым
элиминируют нейрогенное воспаление вследствие аксон-рефлексов.
Кроме С-рецепторов, в патогенез нейрогенного воспаления через аксон-
рефлексы могут быть вовлечены так называемые быстро адаптирующиеся
рецепторы (БАР). БАР представляют собой окончания нервных волокон,
контактирующих с эпителием и гладкомышечными элементами
бронхиальной стенки.
С-рецепторы и БАР задействованы в патогенезе бронхиальной астмы
не только как рецепторные комплексы, в пределах которых происходит
аксон-рефлекс, обуславливающий нейрогенное воспаление. Их
возбуждение вызывает центральные нервные рефлексы, также представляющие
собой патогенетические механизмы бронхиальной астмы. Центральные
нервные рефлексы, вызывают бронхиальную астму посредством:
♦ Парасимпатической стимуляции бронхиальной стенки, медиатором
которой является ацетилхолин. Парасимпатическая стимуляция вы-
Бронхиальная астма и астматический статус
277
зывает спазм гладкомышечных элементов стенки бронхов.
Эфферентные нервные волокна, по которым проводятся импульсы,
вызывающие спазм гладкомышечных миоцитов, из своих окончаний
высвобождают расширяющие бронхи вазоактивный интестиналь-
г ный полипептид и оксид азота. Таким образом реализуется общий
принцип регуляции - системные регуляторные влияния всегда
неоднозначны по своему конечному эффекту на состояние
эффекторов функций, что предотвращает патогенную гиперактивацию их
структурно-функциональных элементов. При этом одним из общих
механизмов развития болезней и патологических состояний
является однонаправленность системных регуляторных влияний на
состояние эффекторов, в частности связанная с дефицитом
определенных нормальных молекулярных эффекторов регуляторных
систем. Например, полагают, что врожденный дефицит нормального
вазоактивного интестинального полипептида обуславливает
предрасположенность к бронхиальной астме.
♦ Роста секреции бронхиальными железами дыхательных путей
небольшого диаметра в результате их парасимпатической стимуляции.
♦ Расширения микрососудов бронхов небольшого диаметра как
причины утолщения и отека бронхиальной стенки. Предположительно
данный рефлекс реализуется через ослабление симпатической
стимуляции стенки данных бронхов.
♦ Ларингоспазма.
♦ Брадикардии, артериальной гипотензии и угнетения спинальных
рефлексов, в результате возбуждения С-рецепторов.
♦ Кашля как следствия активации БАР.
♦ Апноэ и учащенного поверхностного дыхания вследствие
возбуждения С-рецепторов.
♦ Кроме того, возбуждение С-рецепторов вызывает неприятные
ощущения, связанные с дыханием, что усиливает отрицательный
психоэмоциональный стресс у больных с обострением бронхиальной астмы.
ПАТОГЕНЕЗ АРТЕРИАЛЬНОЙ ГИПОКСЕМИИ
И НАРУШЕНИЙ КИСЛОТНО-ОСНОВНОГО СОСТОЯНИЯ
ПРИ АСТМАТИЧЕСКОМ СТАТУСЕ
В отличие от приступа бронхиальной астмы, в том числе и
затянувшегося, при астматическом статусе крайнюю степень обструктивных
расстройств дыхания обуславливает не предельный спазм бронхиол, а отек,
воспаление их стенок при полной или частичной обтурации просвета
бронхиол вязкой неоткашливаемой мокротой. Полное прекращение
дренирования мокроты из просвета бронхиол через действие его
естественных механизмов знаменует трансформацию затянувшегося приступа
бронхиальной астмы в астматический статус.
278
Глава 18
Нарушение дренирования дыхательных путей при астматическом
статусе на пути к своей предельной степени выраженности проходит три
стадии:
♦ Компенсированная бронхорея, при которой рост образования
мокроты увеличивает ее транспорт реснитчатым эпителием без падения
дренирования мокроты из просвета дыхательных путей.
♦ Декомпенсированная бронхорея. При декомпенсированной бронхо-
рее продукция мокроты может сохраняться на прежнем уровне или
растет, но действие мукоцилиарного механизма уже не приводит к
адекватному дренированию просвета дыхательных путей, что
проявляется постоянным или приступообразным кашлем.
♦ Обструкция. В этой стадии вязкость мокроты возрастает настолько,
что возникает рефлюкс, то есть обратный ток мокроты, который
обуславливает распространенную закупорку дыхательных путей
разного диаметра.
При бронхиальной астме дыхательные пути с небольшим внутренним
диаметром (от субсегментарных бронхов до терминальных бронхиол)
сужены патологическим секретом, бронхоспазмом и отеком слизистой
оболочки. При такой исходной обструкции бронхов любое усиление их
спазма вызывает рост сопротивления дыхательных путей, приводящий к
артериальной гипоксемии и нарушениям кислотно-основного состояния
(схема 18.1). Во время выдоха сужение бронхов увеличивает дополнительная
обструкция из-за скачкообразного падения статического давления смеси
газов в просвете дыхательных путей вследствие увеличения линейной
скорости газотока в суженных бронхах с небольшим внутренним диаметром.
Рост обструкции бронхов с
небольшим внутренним диаметром
Патологическая вариабельность
вентиляционно-перфузионных
отношений респиронов
Полная обтурация бронхов
с небольшим диаметром
+ —
Снижение числа
респиронов, участвующих
в газообмене
Артериальная гипоксемия
Рост потребления
02 организмом
Гиповентиляция
\Дыхательный ацидоз
Гипервентиляция
Дыхательный алкалоз
Увеличение
образования С02
Утомление дыхательных мышц
Схема 18.1. Патогенез артериальной гипоксемии и дыхательных расстройств
кислотно-основного состояния у больных в состоянии астматического статуса
Бронхиальная астма и астматический статус
279
Поэтому сопротивление дыхательных путей особенно возрастает в фазу
выдоха, что ведет к перераздуванию легких и эмфиземе. Перераздувание
легких, а также экспираторная задержка части дыхательного объема в
легких при спадении дыхательных путей с небольшим диаметром во время
выдоха, повышают функциональную остаточную емкость и остаточный
объем легких.
Реакция любых множественных и одинаковых по свойствам
эффекторов функциональных систем никогда не бывает униформной, то есть
не захватывает все эффекторы в одинаковой степени.
Это положение вполне приложимо к типическим патологическим
процессам, одним из которых можно считать бронхиальную астму. Сужение
просвета мелких бронхов при обострении бронхиальной астмы и
астматическом статусе выражено на уровне каждого бронха по-разному, что
приводит к неравномерности вентиляции участков ткани легких, респиронов,
вызывающей патологическую вариабельность их вентиляционно-перфу-
зионных отношений, которая приводит к артериальной гипоксемии.
При астматическом статусе патологическое варьирование вентиляци-
онно-перфузионных отношений респиронов резко возрастает и
развивается артериальная гипоксемия, которая может опасно ограничить транспорт
кислорода от легких на периферию.
Экспираторное перераздувание легких при астматическом статусе
ведет к неуклонному возрастанию функциональной остаточной емкости.
После достижения функциональной остаточной емкостью уровня
расчетного нормального общего объема легких для поддержания прежнего
дыхательного объема требуется больший градиент давлений. Для создания
большего градиента давления требуется рост работы дыхательных мышц.
Работу повышает и увеличение сопротивления дыхательных путей.
Своего пика работа при внешнем дыхании достигает при падении
напряжения кислорода в артериальной крови ниже 70 мм рт. ст., что резко
повышает потребление кислорода организмом.
Несоответствие высокого потребления кислорода организмом
низкому поглощению кислорода легкими вызывает быстрое падение
напряжения кислорода в артериальной крови. Артериальная гипоксемия достигает
столь низкого уровня, что вызывает сердечные аритмии, которые
переходят в фибрилляцию желудочков.
Рост потребления свободной энергии организмом, связанный с
интенсификацией работы при внешнем дыхании, ведет к увеличению
образования клетками углекислого газа. Исключение из легочного газообмена
части респиронов вследствие полной обтурации просвета дыхательных путей
с небольшим внутренним диаметром и потребность в выведении все
большей и большей массы углекислого газа определяют крайнюю степень
гипервентиляции при астматическом статусе. Гипервентиляция служит
еще одним фактором утомления дыхательных мышц, и диафрагмы в том
числе. Утомление захватывает и мышцы, играющие при дыхании роль
вспомогательных. Вследствие выраженных обструктивных расстройств
альвеолярной вентиляции и предельно высокой энергетической цены
280
Глава 18
внешнего дыхания в конечной стадии астматического статуса
альвеолярная вентиляция уже не соответствует образованию углекислого газа, и
развивается респираторный ацидоз.
Респираторный ацидоз приходит на смену респираторному алкалозу
начального периода астматического статуса. В начале развития
астматического статуса респираторный алкалоз является следствием
гипервентиляции в ответ на гипоксемию. Гипервентиляция не устраняет падения
напряжения кислорода в артериальной крови, но ведет к избыточному
выделению углекислого газа.
Вследствие дыхательной гипоксии в мышцах накапливается молочная
кислота, которая выходит в интерстиций и циркулирующую кровь.
Возникает лактатный метаболический ацидоз. Метаболический ацидоз
нарастает очень быстро, так как блокирован основной механизм его
компенсации, увеличение экскреции углекислого газа при дыхании. Ацидоз
вызывает нарушения насосной функции сердца и кровообращения, которые
часто служат причиной развития терминального состояния при
астматическом статусе: угнетение сократительной способности сердца, аритмии,
снижение общего периферического сосудистого сопротивления вплоть до
некупируемой артериальной гипотензии.
В фазу вдоха для поддержания нормальной объемной скорости
газотока по дыхательным путям через рост силы сокращений дыхательных и
вспомогательных мышц патологически растет абсолютная величина
отрицательного давления в плевральной полости. Рост абсолютной
величины отрицательного давления в плевральной полости повышает
присасывающее действие плевральной полости и общий венозный возврат к
сердцу. Рост преднагрузки правого желудочка увеличивает выброс им крови в
легочную артерию, что служит причиной легочной артериальной гипер-
тензии. Легочная артериальная гипертензия снижает фракцию изгнания
правого желудочка, что повышает конечно-систолический объем правого
желудочка и смещает межжелудочковую перегородку влево. Возрастание
абсолютной величины отрицательного давления в плевральной полости
увеличивает градиент давлений, действие которого приходится
преодолевать левому желудочку для выброса крови в аорту. Это снижает конечно-
диастолический и ударный объемы левого желудочка. Падение ударного
объема левого желудочка в фазу вдоха может быть причиной транзитор-
ной артериальной гипотензии {явление парадоксального пульса).
Чем больше длительность астматического статуса, тем больше
вероятность полной закупорки бронхов «пробками», состоящими из вязкого
секрета. Поэтому квалифицированное лечение при астматическом статусе
следует начинать с догоспитального этапа. Терапию астматического
статуса начинают немедленно после выявления двух из факторов риска и
признаков развития астматического статуса (табл. 18.2).
Из спирометрических показателей у большинства больных в
астматическом статусе возможно лишь определение максимальной скорости
газотока при выдохе. При астматическом статусе максимальная
экспираторная скорость газотока снижается до уровня 100 л/мин и ниже, что со-
Бронхиальная астма и астматический статус
281
Таблица 18.2
Факторы риска и признаки астматического статуса
Госпитализация в связи с обострением бронхиальной астмы или астматическим
статусом в анамнезе
Ранее проводившаяся или непрерывная терапия кортикостероидами
Ощущение особой тяжести обострения бронхиальной астмы самим больным
Ареактивность по отношению к ранее успешной терапии средствами
с бронходилатирующим эффектом
Продолжительность обострения бронхиальной астмы более двух часов
Неспособность больного говорить из-за выраженного стридорозного дыхания
и дыхательной недостаточности
Спутанность сознания
Артериальная гипертензия, частота сердечных сокращений выше 11 О/мин
Сердечные аритмии
Цианоз
Активное участие вспомогательных мышц в дыхательном акте
Парадоксальный пульс, превышающий 10 мм рт. ст.
Эмфизема, пневмоторакс
Объем форсированного выдоха за одну секунду меньший, чем 1 л
Острый дыхательный ацидоз или снижение напряжения кислорода в артериальной
крови до 60 мм рт. ст. и ниже при дыхании воздухом, не обогащенным кислородом
ответствует объему форсированного выдоха за одну секунду меньшему,
чем 0,7 л. Прямое определение объема форсированного выдоха за одну
секунду часто невозможно из-за затрудненного дыхания больных.
В начале развития астматического статуса гипервентиляция в ответ на
гипоксемию снижает напряжение углекислого газа в артериальной крови,
возвращение напряжения углекислого газа в пределы
средне-статистической «нормы» следует рассматривать как признак прогрессирования
астматического статуса, при котором система внешнего дыхания начинает
терять способность выделения углекислого газа. Между величиной
снижения форсированного выдоха за одну секунду и величиной напряжения
углекислого газа в артериальной крови при астматическом статусе
существует положительная связь.
Обострение бронхоспазма любой степени всегда приводит к
артериальной гипоксемии в результате увеличения неравномерности вентиляции
респиронов. Тем не менее, напряжение кислорода в артериальной крови
больных в астматическом статусе падает ниже 50 мм рт. ст. только при
его осложнениях: пневмотораксе и острой сердечной недостаточности.
Прогноз при астматическом статусе становится особенно
неблагоприятным, когда развивается метаболический лактатный ацидоз.
При снижении парциального давления кислорода в альвеолах,
захватывающем критическую массу респиронов, возникает
распространенный спазм микрососудов легких (альвеоло-васкулярный рефлекс).
Растет постнагрузка правого желудочка и возникает его дилатация. На
282
Глава 18
электрокардиограмме это проявляется легочным зубцом Р и смещением
электрической оси сердца вправо. Правожелудочковая сердечная
недостаточность может представлять собой весьма патогенетически значимый
механизм падения системного транспорта кислорода у больных
бронхиальной астмой.
ПАТОГЕНЕТИЧЕСКИЕ ПРИНЦИПЫ ИНТЕНСИВНОЙ ТЕРАПИИ
БОЛЬНЫХ С АСТМАТИЧЕСКИМ СТАТУСОМ
Крайняя степень неравномерности вентиляции легких при
астматическом статусе, артериальная гипоксемия и гиперкапния непосредственно
перед началом искусственной вентиляции легких обуславливают риск
опасного обострения гипоксемии при «десинхронизации» больного с
режимом работы аппарата искусственной вентиляции. Разнонаправленный
ток смеси газов при совпадении во времени самостоятельного выдоха с
искусственным вдохом и наоборот резко обостряет неравномерность
вентиляции. Увеличение неравномерности вентиляции ведет к падению
напряжения кислорода в артериальной крови ниже 60 мм рт. ст. При этом
напряжение кислорода в артериальной крови смещается в область
значений, соответствующих «падающей» части кривой диссоциации оксиге-
моглобина. В результате кислородная емкость крови при
«десинхронизации» падает в такой степени, что ее низкий уровень опасно ограничивает
транспорт кислорода от легких на периферию.
В этой связи больным в астматическом статусе, которым проводят
искусственную вентиляцию, показаны седатация и фармакодепрессия
дыхательного центра. Препаратом выбора для достижения седатации и
искусственного апноэ при астматическом статусе считают морфина сульфат, 3-
5 мг которого вводят внутривенно. Хотя морфин и обладает гистамин-
высвобождающим действием, клиническая практика показала, что
препарат не вызывает обострения бронхоспазма при астматическом статусе.
Одна из главных целей искусственной вентиляции легких при
астматическом статусе - снижение потребления кислорода и образования
углекислого газа организмом за счет почти полного исключения утилизации
свободной энергии при внешнем дыхании. Из-за высокого сопротивления
дыхательных путей используют аппараты искусственной вентиляции,
регулируемые по объему. Дыхательный объем при искусственной
вентиляции должен составлять от 6 до 10 мл/кг массы тела больного.
Экспираторное закрытие дыхательных путей у больных бронхиальной астмой
вызывает патологический рост функциональной остаточной емкости,
который прогрессирует при астматическом статусе. Постоянное
положительное давление во время выдоха увеличивает перераздувание легких и
функциональную остаточную емкость. Поэтому у больных с
астматическим статусом не используют режим искусственной вентиляции легких с
постоянным положительным давлением в конце выдоха.
Бронхиальная астма и астматический статус
283
При крайней степени обструктивных расстройств вентиляции альвеол
наиболее сложным моментом искусственной вентиляции легких при
астматическом статусе является оптимальный выбор соотношения времени
вдоха и выдоха. При определении оптимальных значений соотношения
учитывают:
♦ значительное снижение общей объемной скорости газотока в
дыхательных путях при выдохе вследствие их выраженной
обструкции;
♦ необходимость поддержания минутного объема дыхания на уровне
достаточном для устранения гиперкапнии;
♦ желательность увеличения времени вдоха для сведения к минимуму
неравномерности вентиляции вследствие медленного заполнения
дыхательной газовой смесью респиронов с более высоким
сопротивлением дыхательных путей.
Легко заметить, что эти требования к режиму искусственной
вентиляции легких больных с астматическим статусом во многом являются
взаимоисключающими. Достижение «золотой середины», то есть медленных
вдоха и выдоха и достаточной альвеолярной вентиляции, часто возможно
лишь при условии миоплегии, достигаемой внутривенным введением
миорелаксантов, лишенных гистаминвысвобождающего действия: веку-
рониума и панкурониума.
Исключительно высокое давление на высоте вдоха при искусственной
вентиляции может вызвать недостаточность кровообращения из-за
угнетения преднагрузки сердца и увеличения парадоксального пульса.
Поэтому для искусственной вентиляции при астматическом статусе используют
гелиево-кислородные дыхательные газовые смеси, содержащие не менее
50 % кислорода. Высокая текучесть гелиево-кислородных смесей
позволяет вентилировать альвеолы, недоступные вентиляции из-за высокого
сопротивления дыхательных путей при искусственной вентиляции легких
воздухом, обогащенным кислородом. Искусственная вентиляция легких
более текучими газовыми смесями в первую очередь устраняет
артериальную гипоксемию, главный инициирующий момент танатогенеза при
астматическом статусе. Устранение артериальной гипоксемии через
использование более текучих газовых смесей позволяет увеличить время
вдоха и выдоха без усиления респираторной гипоксии, что снижает
максимальное давление в системе аппарат-легкие больного на вдохе. Это
почти исключает явление парадоксального пульса и снижает
отрицательное влияние искусственной вентиляции легких на преднагрузку сердца.
Следует учитывать, что увеличение длительности фаз вдоха и выдоха
уменьшает частоту дыхательных движений и минутный объем дыхания
при искусственной вентиляции легких. Снижение минутного объема
альвеолярной вентиляции при уменьшении частоты дыхательных движений
может привести к меньшей экскреции углекислого газа и респираторному
ацидозу. Несмотря на возрастание в альвеолярной газовой смеси
содержания углекислого газа, кислородная емкость артериальной крови и
транспорт с ней кислорода при искусственной вентиляции легких гелие-
284
Глава 18
во-кислородными смесями растут. Транспорт кислорода от легких на
периферию увеличивает не только лучшая оксигенация смешанной
венозной крови при искусственной вентиляции легких гелиево-кислородными
дыхательными смесями, но и меньший депрессивный эффект на
кровообращение искусственной вентиляции легких при снижении максимального
уровня давления в системе аппарат-легкие больного вследствие
увеличения фаз вдоха и выдоха.
При астматическом статусе у значительной части больных
искусственная вентиляция легких быстро возвращает в нормальные пределы как
напряжение кислорода в артериальной крови, так и напряжение в ней
углекислого газа. Прекращение искусственной вентиляции легких
возможно лишь после обратного развития обострения обструктивных
расстройств альвеолярной вентиляции. Об этом свидетельствуют снижение
максимального давления на вдохе, отрицательное давление на вдохе при
самостоятельном дыхании больного, составляющее не менее, чем
30 см вод. ст., а также жизненная емкость легких на уровне не меньшем,
чем 15 мл/кг массы тела больного. После искусственной вентиляции
легких с перемежающемся положительным давлением в конце вдоха
больному проводят периодическую обязательную искусственную вентиляцию,
постепенно снижая частоту дыхательных движений. Главное при
прекращении искусственной вентиляции легких у больных в астматическом
статусе - убедиться в таком состоянии внешнего дыхания, которое не
приведет к прогрессирующему росту работы при дыхании и повторному
обострению дыхательной недостаточности.
При фармакотерапии у больных с обострением бронхиальной астмы и
астматическим статусом учитывают, что принципы применения бронхо-
дилататоров не меняются по мере трансформации бронхиальной астмы
умеренной выраженности в ее тяжелые формы. Несмотря на то, что
одним из признаков астматического статуса является ареактивность
выраженности обструктивных расстройств вентиляции по отношению к
действию бронходилататоров, препараты с бронхорасширяющим действием
вместе с кортикостероидами составляют краеугольный камень
фармакотерапии больных с обострением тяжелой бронхиальной астмы. Общим
правилом для интенсивной терапии при астматическом статусе является
одновременное применение производных теофиллина и бета-два-адрено-
миметиков в тех максимальных дозах, которые не приводят к побочному
кардиотоксическому действию.
Если при предыдущих обострениях бронхиальной астмы обструктив-
ные расстройства внешнего дыхания удавалось купировать альфа-адрено-
и м-холинолитиками, то данные препараты применяют при
астматическом статусе с учетом побочных эффектов при кумуляции.
Из современных патогенетических средств фармакотерапии больных
бронхиальной астмой и астматическим статусом следует выделить новую
группу препаратов со свойствами антагонистов медиаторов воспаления,
которое на уровне бронхиальной стенки во многом составляет патогенез
бронхиальной астмы. Так, антагонисты 5-липооксигеназы блокируют
Бронхиальная астма и астматический статус
285
синтез в легких из арахидоновои кислоты леикотриенов как медиаторов
воспаления, подвергая обструктивные расстройства внешнего дыхания
обратному развитию. Патогенные эффекты леикотриенов блокируют
антагонисты специфических по отношению к ним клеточных рецепторов.
Кроме того, есть сообщения о положительных результатах использования
в терапии больных бронхиальной астмой и в астматическом статусе он-
тагонистов фактора активации тромбоцитов.
Глава 19
КЛИНИЧЕСКАЯ ПАТОФИЗИОЛОГИЯ
СЕРДЕЧНОЙ НЕДОСТАТОЧНОСТИ
Под застойной сердечной недостаточностью понимают
патологическое состояние организма, которое в первую очередь характеризуют:
♦ Снижение насосной функции сердца.
♦ Дизрегуляция на уровне всего организма как результат потерявших
биологический смысл и ставших патогенными реакций систем ней-
рогуморальной регуляции в ответ на низкий уровень минутного
объема кровообращения (МОК), циркуляторную гипоксию и
связанный с ними системный дефицит свободной энергии.
♦ Вторичные по отношению к низкому МОК и системной дизрегуля-
ции нарушения водно-солевого обмена.
При застойной сердечной недостаточности ударные объемы левого и
правого желудочков снижены в такой степени, что в условиях покоя у
больных выявляют клинические признаки недостаточной насосной
функции левого желудочка (левожелудочковая сердечная недостаточность) и
правожелудочковую сердечную недостаточность, то есть синдром,
элементы которого - это в первую очередь признаки низкой насосной
функции правого желудочка. Сниженная насосная функция сердца у больных с
застойной сердечной недостаточностью обуславливает крайне низкую
переносимость физической нагрузки.
Сердечная недостаточность - патологическое состояние вследствие
неспособности сердца как насоса системы кровообращения адекватно
участвовать в реакции роста МОК на увеличение потребностей
организма в кислороде, энергопластических субстратах и транспорте агентов
гуморальной регуляции. Застойную сердечную недостаточность крайней
степени тяжести характеризует низкая насосная функция сердца как
причина несоответствия МОК этим потребностям в условиях покоя.
Попробуем описать основное звено патогенеза застойной сердечной
недостаточности рядом несложных вычислений.
Ударный объем левого желудочка молодого здорового человека
составляет примерно 60 мл-м"2 (ударный индекс, УИ). Если в условиях покоя
частота сердечных сокращений (ЧСС) у такого субъекта находится на уровне
60/мин, то МОК составляет 3,6 лм" /мин (сердечный индекс, СИ). Если при
интенсивной физической нагрузке ЧСС у этого здорового юноши возрастет
до 180/мин, то СИ поднимется до уровня в 10,8 л-м"?/мин.
У больного с застойной сердечной недостаточностью УИ может
составлять 20 мл-м"2. Лишь рост ЧСС до 120/мин позволяет поддерживать МОК на
минимально достаточном для удовлетворения потребностей организма в
условиях покоя уровне в 2,4 л-м" /мин. При этом у больного есть все симптомы
как лево-, так и правожелудочковой сердечной недостаточности, и почти нет
Клиническая патофизиология сердечной недостаточности
287
переносимости физической нагрузки Дело в том, что основной механизм
адаптивного роста МОК - это тахикардия при относительно фиксированной
величине ударного объема левого желудочка Дальнейший рост ЧСС у
данного больного может привести к снижению УИ через падение сократимости
миокарда, которая падает в частности вследствие снижения суммарной
длительности диастолических интервалов, во время которых артериальная кровь
поступает в наиболее интенсивно работающий субэндокардиальный слой
миокарда левого желудочка Кроме того, недостаток свободной энергии в
кардиомиоцитах возрастет из-за повышения потребности клеток сердца в
кислороде Эту потребность резко повышает тахикардия при ЧСС выше
120/мин у больного с застойной сердечной недостаточностью
ЭТИОПАТОГЕНЕТИЧЕСКАЯ КЛАССИФИКАЦИЯ
ЗАСТОЙНОЙ СЕРДЕЧНОЙ НЕДОСТАТОЧНОСТИ
Выделяют четыре основные детерминанты насосной функции сердца:
♦ преднагрузка;
♦ постнагрузка;
♦ сократимость миокарда;
♦ нормальный сердечный ритм, то есть отсутствие сердечных аритмий.
У больного нет расстройств сердечного ритма, если при генерации
ритма его нормотопическим синоатриалъным водителем и нормальной
скорости распространения фронта возбуждения по проводящей системе
частота сердечных сокращений в условиях покоя находится в пределах
от 60 до 90 мин1. Любая аритмия в той или иной степени снижает
насосную функцию сердца (табл. 19.1).
Оптимальный уровень детерминант насосной функции сердца
проявляет себя в условиях покоя нормальными значениями ряда показателей
сердечной деятельности (табл. 19.2). Циркуляторная гипоксия, гипоэргоз,
гибернация и станнинг сократительных кардиомиоцитов (см. главу,
посвященную инфаркту миокарда) ведут к дискоординации сокращений
сегментов стенок левого желудочка, которая обуславливает падение его
ударного объема.
При дискоординации сокращений некоторые из сегментов стенки
левого желудочка в недостаточной степени смещаются к геометрическому
центру сердечной камеры, что обуславливает снижение фракции изгнания, УИ
и СИ. Критерием нормального изменения конфигурации левого желудочка
при систолическом сокращении является показатель смещения сегмента
стенок желудочка во время систолы. Сначала его измеряют в десятичных
дробях, вычитая из единицы отношение радиуса соответствующего
сегмента в конце систолы к его радиусу в конце диастолы. Затем десятичные
дроби переводят в проценты. При этом за центр окружности принимают
геометрический центр сердечной камеры (рис. 19.1).
288
Глава 19
Таблица J 9.1
Сердечные аритмии как причины падения насосной функции сердца
Расстройства
сердечного ритма
Мерцательная
аритмия
Суправентрикуляр-
ная синусовая
тахикардия
Вентрикулярная
тахикардия
(эктопический водитель ритма
локализован в
желудочках)
Фибрилляция
желудочков
Полная атриовен-
трикулярная блокада
Ведущие звенья патогенеза
сердечной недостаточности
Прекращение активного диас-
толического наполнения левого
желудочка через сокращение
стенок левого предсердия и
патологическая вариабельность
временных диастолических
интервалов
Снижение суммарной
длительности диастолических
интервалов за минуту
Прекращение нормальной
последовательности сокращений
предсердий и желудочков при
снижении суммарной
длительности диастолических
интервалов
Асинхронное сокращение сар-
комеров миокарда левого
желудочка
Полная дискоординация
сокращений предсердий и
желудочков. Урежение сокращений
левого желудочка вследствие
узлового ритма
Основной механизм развития
артериальной гипотензии
Снижение конечно-диастоли-
ческого объема левого желудочка
как результат прекращения
активного наполнения при
сокращении предсердия и уменьшения
некоторой части временных
диастолических интервалов
Падение ударного объема
левого желудочка вследствие:
а) циркуляторной гипоксии кар-
диомиоцитов субэндокардиаль-
ного слоя в результате снижения
времени его снабжения
артериальной кровью; б) падения
суммарной длительности диастоли-
ческого наполнения левого
желудочка
Снижение суммарной
длительности диастолического
наполнения левого желудочка и времени
снабжения его субэндокарди-
ального слоя артериальной
кровью как причины падения
ударного объема левого желудочка и
гипотензии
Прекращение изгнания крови
левым желудочком в аорту
Падение МОК из-за брадикар-
дии, связанной с узловым
ритмом
Фракция изгнания - это отношение ударного объема левого
желудочка к его конечно-диастолическому объему Нормальные пределы
колебаний этого показателя насосной функции сердца - 0,6-0,78 Определение
фракции изгнания позволяет получить информацию только о том, какая
часть конечно-диастолического объема левого желудочка во время
систолы покидает левую сердечную камеру Определение фракции изгнания
полезно при слежении за прогрессированием сердечной недостаточности
у больных Следует помнить, что фракция изгнания может возрасти и без
восстановления терапией сократительной способности сердца. Причиной
роста ФИ у части больных может быть снижение постнагрузки в
результате действия вазодилятаторов.
Клиническая патофизиология сердечной недостаточности
289
Таблица J9.2
Нормальные величины показателей насосной функции сердца
у здоровых людей в условиях относительного покоя
Показатель
Сердечный индекс, л#мин"7м2
Частота сердечных сокращений, мин"1
Ударный индекс, мл/м2
Конечно-диастолический объем левого желудочка, мл/м2
Конечно-систолический объем левого желудочка, мл/м2
Фракция изгнания
Конечно-диастолическое давление крови в левом
желудочке, мм рт. ст.
Максимальная скорость роста давления крови в левом
желудочке во время систолы, мм рт. ст./сек
Смещение сегментов стенок левого желудочка во время
систолы, %
-верхушечного
-переднего
-нижнего
Нормальная величина
3,6
60-90
60
90
30
0,67
<12
1500
30
50
40
В зависимости от патологических изменений детерминант насосной
функции сердца выделяют четыре вида застойной сердечной
недостаточности:
♦ обусловленная ростом преднагрузки;
♦ в результате увеличения постнагрузки;
♦ вызванная падением сократимости;
♦ как результат расстройств сердечного ритма (табл. 19.3).
При застойной сердечной недостаточности, связанной с падением
сократимости рабочего миокарда левого желудочка, два главных
компонента его насосной функции, систолический и диастолический, претерпевают
свои патологические изменения.
Рис. 19.1. Схематическое изображение смещения апикального сегмента
левого желудочка во время систолы (L)
290
Глава 19
Таблица 19.3
Этиопатогенетическая классификация сердечной недостаточности
Патологическое
изменение
детерминанты насосной
функции сердца
Болезни, патологические состояния
и пороки сердца как причины
аномальных сдвигов детерминанты
Ведущие звенья патогенеза
падения насосной функции сердца
Снижение
сократимости
Возрастание пред-
нагрузки
Рост постнагрузки
- Ишемическая болезнь сердца
- Кардиомиопатии
- Миокардит
- Поражение сердца вследствие
хронической алкогольной интоксикации
- Недостаточность митрального
клапана
- Недостаточность аортального
клапана
- Постинфарктный дефект
межжелудочковой перегородки
- Дефект межпредсердной
перегородки
- Аортальный стеноз
- Артериальная гипертензия
- Коарктация аорты
Патологически низкая
способность сердца реагировать
ростом силы сокращений на
рост преднагрузки
Патогенный рост работы
левого желудочка в фазу
изгнания вследствие роста его ко-
нечно-диастолических объема
и давления
Увеличение работы как
правого, так и левого желудочка во
время фазы изгнания
вследствие роста их диастолического
наполнения кровью из-за
шунтирования крови
слева-направо, то есть из левых отделов
сердца в правые (из
артериального звена системного
кровообращения в систему легочной
артерии)', артериальная гипок-
семия в результате
шунтирования крови слева-направо
Шунтирование крови слева-
направо как причина:
а) роста работы правого
желудочка вследствие увеличения
диастолического наполнения;
б) вторичной легочной
артериальной гипертензии
Падение сократимости
вследствие хронического
патологического возрастания уровня
работы левого желудочка при
преодолении повышенного
сопротивления выбросу крови в
аорту
Систолический компонент - это способность желудочка к
сокращению и выбросу крови с преодолением постнагрузки. О нормальном
систолическом компоненте свидетельствует неугнетенная способность
желудочка реагировать возрастанием ударного объема на рост конечно-диас-
толического давления, то есть на растяжение саркомеров миокарда.
Критерий систолического компонента - отношение конечно-систолического
давления крови в желудочке к объему крови, который он содержит в
конце систолы.
Клиническая патофизиология сердечной недостаточности
291
По мере падения сократимости в силу угнетения систолического
компонента происходят рост конечно-систолического давления крови в
желудочке и падение выброса крови из его полости в фазу изгнания.
Это еще в большей степени повышает конечно-систолическое
давление. При нормальных систолическом компоненте и сократимости это
соотношение неизменно в широком диапазоне колебаний постнагрузки
желудочка.
Сердечную недостаточность, обусловленную преимущественно
падением систолического компонента насосной функции левого желудочка,
называют систолической. Чаще всего к систолической сердечной
недостаточности приводят миокардиты, первичные и вторичные кардиомиопа-
тии, в частности обусловленные ишемической болезнью сердца. Крайняя
по тяжести форма систолической сердечной недостаточности - кардио-
генный шок.
Под диастолическим компонентом насосной функции левого
желудочка понимают способность стенки желудочка к расслаблению, а значит
и к диастолическому наполнению кровью. Этот компонент насосной
функции левого желудочка называют способностью «принимать предна-
грузку». Критерием данного компонента является отношение конечно-
диастолического давления крови в желудочке к ее объему, который
камера сердца содержит в конце диастолы, то есть податливость желудочка.
Расслабление сердца в диастолу - это процесс, требующий потребления
свободной энергии кардиомиоцитами. Энергия при этом тратится на
активное возвращение ионизированного кальция из цитозоля кардиомиоци-
тов в саркоплазматический ретикулум. Поэтому циркуляторная гипоксия
клеток сердца у больных его ишемической болезнью угнетает диастоли-
ческий компонент, что обуславливает рост конечно-диастолического
давления при почти неизменном конечно-диастолическом объеме. В
результате возникает вторичная легочная венозная гипертензия, как
инициирующий момент правожелудочковой недостаточности и кардиогенного
отека легких, которые обуславливает диастолическая сердечная
недостаточность.
ПАТОГЕНЕЗ НАРУШЕНИЙ ПРЕДНАГРУЗКИ И ВОДНО-
СОЛЕВОГО ОБМЕНА КАК ПРИЧИН И СЛЕДСТВИЙ
ЗАСТОЙНОЙ СЕРДЕЧНОЙ НЕДОСТАТОЧНОСТИ
Под преднагрузкой часто понимают объем крови который содержит
левый (правый) эюелудочек сердца непосредственно перед началом фазы
изгнания. В строго физиологическом смысле слова под преднагрузкой
следует понимать максимальную степень растяжения саркомеров
миокарда левого желудочка под влиянием его диастолического наполнения
кровью. В соответствии с основным законом сердца Франка-Старлинга-
Штрауба, чем больше преднагрузка как растяжение саркомеров миокарда
292
Глава 19
желудочков, тем больше сила сокращения их стенок в фазу изгнания. В
клинической практике не представляется возможным прямое
определение преднагрузки в физиологическом смысле. Поэтому о ней судят по
косвенным критериям, то есть по клиническим признакам ее нарушений
и изменений, а также определяя величины параметров насосной
функции сердца, которые находятся в связи с преднагрузкой. К ним в
частности относят конечно-диастолические объем и давление крови в левом
желудочке.
При застойной сердечной недостаточности угнетено возрастание силы
сокращений стенок желудочков в ответ на рост преднагрузки при
увеличении диастолического наполнения. В результате у больных с застойной
сердечной недостаточностью при тех же величинах конечно-диастоли-
ческих объема и давления в левом желудочке, что и у здоровых людей,
фиксируют меньшие, чем у них, скорость изгнания крови и УИ.
Снижение УИ ведет к увеличению объема крови, который левый
желудочек содержит в конце диастолы. Кровь задерживается в полости
левого желудочка, и в ней растет конечно-диастолическое давление. Этот
рост давления передается на правое предсердие, что служит ведущим звеном
патогенеза диспноэ (одышки) и кардиогенного отека легких.
Выделяют следующие основные причины патологического возрастания
преднагрузки у больных с застойной сердечной недостаточностью:
♦ активация ренин-ангиотензин-альдостеронового механизма,
ведущая к увеличению содержания натрия в организме, объемов
внеклеточной жидкости и плазмы крови, общего венозного
возврата к сердцу и конечно-диастолического объема крови его
желудочков;
♦ рост интенсивности системной адренергической стимуляции, через
спазм вен увеличивающей общий венозный возврат к сердцу,
который часто выступает элементом неспецифической стрессорной
реакции на циркуляторную гипоксию.
Ведущим (первичным) звеном патогенеза застойной сердечной
недостаточности выступает патологически низкий в условиях покоя МОК
и угнетение реакции его роста в ответ на возрастание потребностей
организма.
Низкий МОК через падение объемной и линейной скорости кровотока
на периферии выключает из системного кровообращения часть внутрисо-
судистого объема. Патологическую секвестрацию и снижение активно
циркулирующей части внутрисосудистого объема (эффективного объема
крови) у больных с застойной сердечной недостаточностью также
обуславливают расстройства микроциркуляции. Нарушения
периферического кровообращения являются причиной избыточного содержания
внеклеточной жидкости в интерстициальном пространстве и патологического
депонирования крови на периферии, связанных с патогенным спазмом
сосудов сопротивления в ответ на снижение МОК.
У многих больных с застойной сердечной недостаточностью рост
действующих концентраций альдостерона и антидиуретического гормона в
Клиническая патофизиология сердечной недостаточности
293
крови может увеличить преднагрузку сердца, повышая общее содержание
в организме натрия и воды. У другой части пациентов снижение
эффективного объема крови исключает патологический рост объема
внеклеточной жидкости из причин возрастания преднагрузки. У таких больных
возрастание конечно-диастолического давления крови в левом желудочке
связано прежде всего со снижением его насосной функции, а
преднагрузку повышает спазм емкостных сосудов (вен).
В этой связи назначение мочегонных средств для улучшения
насосной функции сердца больных с застойной сердечной
недостаточностью не всегда можно признать строго в патогенетическом отношении
обоснованным лечебным воздействием. Длительное использование
диуретиков, эффект которых ведет к повышенной экскреции калия, у
больных с застойной сердечной недостаточностью при отсутствии
должного слежения за концентрацией катиона в плазме крови может
привести к гипокалиемии и ее опасному следствию, сердечным
аритмиям. Сердечные аритмии служат фактором трансформации застойной
хронической сердечной недостаточности в острую, прогноз при
которой у таких пациентов крайне неблагоприятен. Следует заметить, что
позитивный эффект мочегонных при застойной сердечной
недостаточности нельзя свести лишь к увеличению экскреции натрия и воды и
вторичному снижению преднагрузки сердца через уменьшение объема
внеклеточной жидкости. Так, лазикс (фуросемид), положительно влияя
на микроциркуляцию в почках, снижает уровень активации ренин-
ангиотензин-альдостеронового механизма как звена патогенеза
застойной сердечной недостаточности.
В ответ на снижение ударного объема левого желудочка, МОК и
эффективного внутрисосудистого объема через активацию
симпатической части автономной нервной системы происходит
перераспределение МОК Одно из следствий перераспределения МОК- падение
объемной скорости кровотока через почечные клубочки как стимул для
активации секретирующих ренин клеток юкстагломерулярного
аппарата. Активация ренин-ангиотензин-альдостеронового механизма
происходит не только вследствие падения объемной скорости кровотока по
приводящим артериолам клубочков. К нему ведет повышенный
уровень возбуждения бета-один-адренорецепторов в результате усиления
симпатических неирогуморальных регуляторных влияний в ответ на
падение эффективного объема циркулирующей крови и МОК.
Активация ренин-ангиотензин-альдостеронового механизма сразу же из
компенсаторной реакции трансформируется в звено патогенеза застойной
сердечной недостаточности.
Застойная сердечная недостаточность вызывает нарушения водно-
солевого обмена, которые складываются не только из перераспределения
воды между интерстициальным, сосудистым и внутриклеточным
сектором. Она также ведет к перераспределению анионов и катионов между
клетками и внеклеточным пространством вследствие:
♦ увеличения секреции альдостерона;
294
Глава 19
♦ изменения активности натрий-калий-АТФазы скелетных мышц
вследствие возбуждения при системной адренергической
стимуляции бета-два-адренорецепторов;
♦ роста содержания в плазме крови глюкозы в результате системной
адренергической стимуляции.
Несмотря на снижение выраженности симптомов застойной
сердечной недостаточности под влиянием терапии сердечными гликозидами и
диуретиками, у 40 % больных с застойной сердечной недостаточностью
такое «стандартное» лечение, увеличивая МОК и снижая застой крови в
органах на периферии, не устраняет гипонатриемию, гипомагниемию и
гипокалиемию. Данные, полученные при исследовании биоптатов
скелетных мышц, свидетельствуют, что нарушения ионного состава их миоци-
тов персистируют еще чаще. Можно считать, что высокая действующая
концентрация альдостерона в циркулирующей крови ведет к
патологическим изменениям содержания натрия и калия в клетках и вторичным по
отношению к ним изменениям объема клеток. Одной из причин
изменений ионного состава клеток у больных с застойной сердечной
недостаточностью можно считать стимуляцию альдостероном переноса через
плазматическую мембрану натрия в обмен на протоны, то есть активацию
работы главного эффектора системы регуляции объема клетки.
Свою роль в нарушениях водно-солевого обмена у больных с
застойной сердечной недостаточностью играет рост секреции аргинин-вазо-
прессина, который обусловлен:
♦ повышенной чувствительностью реагирующих на растяжение пред-
сердных рецепторов;
♦ высокой концентрацией в крови ангиотензина II.
Высокая действующая концентрация в крови антидиуретического
гормона приводит к гипонатриемии, столь частой у больных с хронической
сердечной недостаточностью.
Патологические изменения ионного состава клеток и их объема у
больных с застойной сердечной недостаточностью могут неожиданно вызывать
сердечные аритмии как причины внезапной сердечной смерти. Следует
заметить, что этиология нарушений ионного состава клеток у больных с
застойной сердечной недостаточностью остается неясной. Были высказаны
предположения, что назначение ингибиторов ангиотензин-превращающего
фермента (каптоприл, анаприл, престариум и др.) может устранить
нарушения ионного состава клетки и ее избыточное наводнение, так как эти
лекарственные средства уменьшают содержание в крови альдостерона и ар-
гинин-вазопрессина. Этот эффект данных препаратов противодействует
вторичному альдостеронизму у больных с хронической сердечной
недостаточностью и через снижение концентрации альдостерона уменьшает
степень гипокалиемии. Результаты прижизненных исследований ионного
состава клеток у больных не подтвердили этого предположения. Эти
нарушения оставались устойчивыми, несмотря на применение ингибиторов
ангиотензин-превращающего фермента. Более того, их продолжали фиксировать
после нормализации концентраций ионов во внеклеточной жидкости.
Клиническая патофизиология сердечной недостаточности
295
ПАТОЛОГИЧЕСКИЕ СДВИГИ ПОСТНАГРУЗКИ
КАК ЗВЕНО ПАТОГЕНЕЗА ЗАСТОЙНОЙ
СЕРДЕЧНОЙ НЕДОСТАТОЧНОСТИ
Под постнагрузкой понимают силу сокращений саркомеров миокарда
при преодолении левым желудочком сопротивления выбросу крови в
аорту. Это сопротивление складывается из аортального импеданса и
общего периферического сосудистого сопротивления (ОПСС).
Рост аортального импеданса, обусловленный изменением
механических свойств стенки аорты вследствие старения и атеросклероза, а также
возрастание общего периферического сосудистого сопротивления в
результате спазма резистивных сосудов, повышают силу сокращений
стенки левого желудочка, необходимую для адекватного потребностям
организма выброса крови в аорту, то есть повышают его постнагрузку.
Увеличение работы сердца, вызванное ростом его постнагрузки, через прогрес-
сирование гипоэргоза кардиомиоцитов может привести к необратимым
падению сократительной способности сердечной мышцы и острой
сердечной недостаточности.
Одно из ведущих звеньев патогенеза застойной сердечной
недостаточности - это стойкий рост постнагрузки, основная причина
которого - длительное патологическое повышение ОПСС.
Рост ОПСС у больных с застойной сердечной недостаточностью - это
результат устойчивого спазма сосудов сопротивления вследствие прямой
нервной симпатической стимуляции гладкомышечных элементов
сосудистой стенки, гиперкатехоламинемии, высокой действующей
концентрации в крови ангиотензинов и аргинин-вазопрессина. Усиление секреции
вазопрессина происходит в результате супрасегментарной стрессорной
активации центральной нервной системы, вследствие роста в
циркулирующей крови концентрации ангиотензина II, а также в ответ на
возбуждение барорецепторов предсердий при росте в них конечно-диастоли-
ческого давления крови. Рост секреции натрийуретического предсердного
пептида, снижающего тонус сосудов сопротивления и повышающего
почечную экскрецию натрия, у данного контингента больных не в
состоянии предотвратить патогенный рост ОПСС и постнагрузки левого
желудочка. Интенсификация нейрогуморальных системных
сосудосуживающих влияний активирует на периферии местные системы ауторегуляции,
функционирование которых в данной ситуации направлено на
предотвращение падения локального кровотока. Так в тканях растут синтез и
высвобождение простагландинов Е2 и 12, чье сосудорасширяющее
действие противостоит системным вазоконстрикторным влияниям и росту
ОПСС как фактору прогрессирования недостаточности сердца. На уровне
почек эти простагландины вместе с ангиотензином II расширяют
приводящие артериолы почечных клубочков, то есть сохраняют скорость клу-
бочковой фильтрации, несмотря на выраженность системных защитно-
патогенных сосудосуживающих влияний. Кроме того, этот эффект
почечной простагландиновои стресс-лимитирующей системы направлен на
296
Глава 19
предотвращение избыточной активации ренин-ангиотензин-альдостеро-
нового механизма вследствие падения объемной скорости кровотока
через клубочки нефронов. Поэтому назначение больным с застойной
сердечной недостаточностью нестероидных противоспалительных средств,
угнетающих синтез простагландинов-вазодилятаторов во всем организме,
может привести к обострению сердечной недостаточности через усиление
активации ренин-ангиотензин-альдостеронового механизма и увеличение
постнагрузки сердца.
ПАТОФИЗИОЛОГИЯ НАРУШЕНИЙ СОКРАТИТЕЛЬНОЙ
СПОСОБНОСТИ СЕРДЦА У БОЛЬНЫХ С ЗАСТОЙНОЙ
СЕРДЕЧНОЙ НЕДОСТАТОЧНОСТЬЮ
Сократимость сердца - это его способность повышать силу
сокращений в ответ на системную и местную нервную адренергическую
стимуляцию, гиперкатехоламинемию и действие средств с полоэюительным
инотропным эффектом (сердечные гликозиды, бета-один-адреномиме-
тики и др.).
Системная нервная адренергическая стимуляция и гиперкатехолами-
немия представляют собой защитную реакцию в ответ на снижение УИ и
СИ, которая у больных с застойной сердечной недостаточностью быстро
приобретает прямо патогенное значение. Этот защитно-патогенный сдвиг
системной регуляции резко повышает потребность кардиомиоцитов в
кислороде и свободной энергии, вызывая:
♦ рост преднагрузки сердца вследствие спазма емкостных сосудов (вен);
♦ повышение частоты сердечных сокращений и сократимости как
детерминант потребности сердца в кислороде;
♦ рост постнагрузки, обусловленный возрастанием ОПСС, который
повышает давление в левом желудочке в фазу изгнания, а значит и
среднюю величину напряжения его стенок в период систолического
сокращения как фактор роста потребности сердца в кислороде (то
есть обостряет гипоэргоз кардиомиоцитов).
Одновременно адренергическая нейрогуморальная стимуляция сердца,
вызывая тахикардию и снижение суммарной длительности диастолических
интервалов за минуту, снижает объем артериальной крови, протекающей
через субэндоардиальный слой миокарда левого желудочка за минуту.
Все это заставляет считать адренергическую нейрогуморальную
стимуляцию сердца фактором прогрессирования сердечной недостаточности,
который действует через усиление гипоэргоза кардиомиоцитов
вследствие усиления несоответствия потребности клеток сердца в кислороде
доставке к ним Ог.
В этой связи использование средств с положительным инотропным
действием, пусть даже действующих избирательно через возбуждение
только бета-один-адренорецепторов, следует считать опасным усилением
Клиническая патофизиология сердечной недостаточности
297
недостатка свободной энергии в кардиомиоцитах и его следствиями,
аритмиями и необратимой сердечной слабостью.
При острой сердечной недостаточности, обусловленной инфарктом
миокарда, вследствие гипоэргоза, гибернации и станнита кардиомиоци-
тов (см. главу, посвященную инфаркту миокарда) в ишемизированной
зоне сердца усиления сокращений саркомеров миокарда в ответ на действие
бета-один-адреномиметиков и других лекарств с положительным ино-
тропным действием не происходит. Такая ареактивность насосной
функции всего сердца возникает при распространении угнетения
сократительной функции кардиомиоцитов в пределах определенной массы рабочего
миокарда. Кроме того, данные многочисленных клинико-патофизиоло-
гических исследований свидетельствуют, что существует верхний предел
роста сократимости под влиянием средств, увеличивающих силу
сердечных сокращений, который не зависит от природы и фармакодинамики
используемого препарата. В такой ситуации положительный результат
терапии обычно связан с коррекцией артериальной гипоксемии,
устранением сердечных аритмий, ацидоза и гиповолемии.
СВЯЗЬ ПАТОГЕНЕЗА ЗАСТОЙНОЙ СЕРДЕЧНОЙ
НЕДОСТАТОЧНОСТИ И СИМПТОМОВ ЛЕВО-
И ПРАВОЖЕЛУДОЧКОВОЙ СЕРДЕЧНОЙ
НЕДОСТАТОЧНОСТИ
Первыми симптомами низкой насосной функции левого желудочка
выступают клинические признаки сердечной недостаточности и жалобы,
связанные с недостаточным транспортом в органы на периферию
кислорода и энергопластических субстратов. Часто первоначальные симптомы
левожелудочковой недостаточности - это повышенная утомляемость и
мышечная слабость. Сниженная объемная скорость кровотока на
периферии, обусловленная низким ударным объемом левого желудочка,
проявляет себя холодными и бледными кожными покровами, что особенно
выражено в области нижних третей голеней. Кожные покровы на ощупь
влажные в результате повышенного потоотделения. Усиление
потоотделения представляет собой следствие системной адренергической
стимуляции в ответ на циркуляторную гипоксию и падение эффективного внут-
рисосудистого объема крови. Время от времени низкий уровень
снабжения артериальной кровью головного мозга проявляет себя
забывчивостью. Из причин забывчивости не следует исключать нарушения внутри-
центральных отношений под влиянием сдвигов в системной афферентации,
вызванных недостаточной насосной функцией левого желудочка и всего
сердца. Гипоэргоз нервных окончаний на периферии ведет к снижению их
чувствительности и проявляет себя жалобами на онемение кожи в дисталь-
ных сегментах конечностей, которое связано и с сердечными отеками. По
мере прогрессирования циркуляторнои гипоксии к симптомам застойной
298
Глава 19
сердечной недостаточности последовательно присоединяются признаки
дисфункций печени, почек и органов системы пищеварения и, наконец,
цианоз кожных покровов и трофические язвы нижних конечностей.
У больных с недостаточной насосной функцией левого желудочка
тахикардия как симптом свидетельствует о компенсаторной реакции,
направленной на удержание МОК на минимально достаточном уровне через рост
частоты сердечных сокращений при низком УИ. Рост ОПСС как следствие ряда
защитно-патогенных сдвигов в регуляции тонуса резистивных сосудов ведет
к снижению пульсового давления, повышая диастолическое артериальное
давление. Кроме того, пульсовое давление падает в результате снижения
систолического артериального давления из-за низкого УИ. Размеры сердца и
степень патологических изменений его границ варьируют в зависимости от
этиологии дисфункций и болезней сердца. У больных с застойной сердечной
недостаточностью выявляют нечеткость, «смазанность» первого тона,
которую предположительно можно связать с ростом объема крови, который
левый желудочек содержит в конце диастолы.
Патологические третий и четвертый тоны - это проявления роста
давлений крови в левом желудочке в фазу диастолического наполнения.
Возникновение третьего тона связано с быстрым заполнением кровью во
время диастолы левого желудочка, содержащего повышенный конечно-
систолический объем крови. В результате роста конечно-систолического
объема левого желудочка его диастолическое заполнение завершается
почти в начале опорожнения предсердия. Причина четвертого тона - это
рост скорости возрастания давления крови в левом желудочке в конце
его диастолического наполнения вследствие высокого конечно-диас-
толического объема желудочка. По мере прогрессирования сердечной
недостаточности вследствие сердечных пороков шумы, обычно
выслушиваемые как их симптомы, теряют свою интенсивность, что связывают
с дилатацией сердечных камер, ростом их объемов и падением силы
сокращений саркомеров миокарда. Так у больного в терминальной стадии
развития митрального стеноза перестает выслушиваться диастолический
шум. Наоборот, систолический шум как свидетельство инфаркта сосоч-
ковых мышц и недостаточности митрального клапана начинают
выслушивать по мере восстановления насосной функции левого желудочка. О
низких ударном объеме левого желудочка и скорости возрастания
давления в крови в левом желудочке в фазу изгнания свидетельствует
слабый и «размытый» сердечный толчок.
Первый признак легочной венозной гипертензии, вторичной по
отношению к недостаточности насосной функции левого желудочка, - это
одышка (диспноэ), то есть ощущение нехватки воздуха, которое не
устраняет произвольное увеличение частоты и глубины дыхания. Ортоп-
ноэ - это одышка в положении лежа, ослабевающая в вертикальном
положении. Одышку в положении лежа обуславливает рост давления в
легочных венах и капиллярах вследствие увеличения под влиянием силы
тяжести общего венозного возврата, с которым «не справляется больное
сердце».
Клиническая патофизиология сердечной недостаточности
299
Патогенез пароксизмального ночного диспноэ во многом можно свести к
обструктивному расстройству внешнего дыхания, которое вызывают:
♦ отек слизистой оболочки бронхов вследствие застоя крови в
бронхиальных венах при легочной венозной гипертензии;
♦ усиление вагальных суживающих бронхи влияний во время ночного
сна.
Тахипноэ у больных с застойной сердечной недостаточностью может
быть следствием артериальной гипоксемии в результате патологического
падения вентиляционно-перфузионного отношения на уровне всех легких
вследствие легочной венозной гипертензии.
По мере прогрессирования легочной венозной гипертензии растет
гидростатическое давление в легочных капиллярах и ультрафильтрация
плазмы в интерстиций легких. Поэтому у больных над легкими
выслушивается все больше звучных влажных хрипов, и ультрафильтрат,
содержащий незначительное количество альбумина, появляется в плевральной
полости. Первоначально хрипы выслушивают над базальными отделами
легких, где в течение суток уровень давления в легочных венах и
капиллярах под влиянием силы тяжести чаще выше, чем в других отделах. При
острой левожелудочковой сердечной недостаточности для рациональной
лечебной коррекции пред- и постнагрузки левого желудочка может
возникнуть необходимость в непрерывном слежении за величинами
давлений крови в левом предсердии, в легочных венах и в легочных
капиллярах, что невозможно без катетеризации легочной артерии. Показаниями
для катетеризации легочной артерии служат:
♦ выраженная и прогрессирующая застойная сердечная
недостаточность;
♦ кардиогенный шок или устойчивая артериальная гипотензия;
♦ нарастающая недостаточность митрального клапана.
Кардиогенный отек легких высокого давления выступает конечным и
часто фатальным результатом вторичной легочной венозной гипертензии
у больных с застойной сердечной недостаточностью.
Недостаточность насосной функции правого желудочка ведет к росту
давления крови в емкостных сосудах, что обуславливает возрастание
гидростатического давления в капиллярах нижних конечностей, печени и
других органов живота. Рост гидростатического давления в микрососудах
данной локализации вызывает интерстициальный отек, нарушающий
периферическое кровообращение и вызывающий отеки и асцит. При этом у
больных набухшие наружные яремные вены заметны не только в
горизонтальном положении. По мере прогрессирования правожелудочковой
недостаточности дилатация правого Желудочка вызывает недостаточность
трехстворчатого клапана, которая проявляет себя волнами «а» и «Ь» на
кривой венозного пульса.
Нарушения периферического кровообращения в печени вызывают ги-
поэргоз и нарушения ионного состава гепатоцитов и других клеток
печени, что приводит к печеночной недостаточности, циррозу и портальной
гипертензии.
300
Глава 19
СЕРДЕЧНАЯ НЕДОСТАТОЧНОСТЬ ПРИ ВЫСОКОМ МОК
Отдельно выделяют сердечную недостаточность при высоком
минутном объеме кровообращения. У больных с такой сердечной недостаточно-
стью МОК выше минутного объема кровообращения у здоровых людей в
условиях покоя, но низок относительно потребностей организма в
транспорте кислорода, энергопластических субстратов или в самом росте МОК.
У части больных с сердечной недостаточностью причина угнетения
насосной функции сердца - патогенно высокий уровень его работы, которую
повышает возрастание внутрисосудистого объема. При сердечной
недостаточности такого происхождения сократительная способность сердечной
мышцы исходно не угнетена, но со временем падает из-за несостоятельности
компенсации. Возникновение несостоятельности компенсации ускоряют
отрицательные нейрогуморальные влияния на сердце.
Потребность в росте МОК как таковом у организма возникает тогда,
когда он представляет собой защитную реакцию, направленную на
поддержание артериального давления, несмотря на низкий уровень общего
периферического сосудистого сопротивления. Кроме того, рост МОК
выступает необходимым условием эффективности системной реакции
компенсации снижения кислородной емкости крови при анемии. Прогресси-
рование сердечной недостаточности такого происхождения может
приводить к синдромам лево- и правожелудочковой недостаточности, несмотря
на МОК, устойчиво превышающий уровень минутного объема
кровообращения нормальный для здоровых людей в условиях покоя.
Выделяют «физиологические стимулы» для устойчивого возрастания
МОК как причины сердечной недостаточности. Их следует рассматривать
и как раздражители, действие которых вызывает патогенную стрессорную
реакцию, обуславливающую нейродистрофию миокарда:
♦ высокие температура и влажность окружающей среды;
♦ физическая нагрузка;
♦ отрицательный психоэмоциональный стресс;
♦ лихорадка;
♦ анемия.
При эссенциальной артериальной гипертензии, которая у
значительной части больных приводит к снижению внутрисосудистого объема,
рост минутного объема кровообращения как звено патогенеза
гипертонической болезни и причина сердечной недостаточности - это не только
следствие нарушений внутрицентральных отношений, но и защитно-
патогенная реакция на снижение объема циркулирующей плазмы.
Образование патологических артерио-венозных сосудистых анастомозов
снижает общее периферическое сосудистое сопротивление (ОПСС).
Снижение ОПСС служит стимулом для предотвращающего артериальную гипотен-
зию значительного и длительного роста МОК, приводящего к вторичной кар-
диомиопатии и застойной сердечной недостаточности при ряде заболеваний:
♦ болезнь Педжетта (образование артерио-венозных анастомозов в
длинных трубчатых костях);
Клиническая патофизиология сердечной недостаточности
301
♦ гломерулонефрит;
♦ гемангиоматоз.
Падение ОПСС вследствие шунтирования крови из артерий в вены при
проведении гемодиализа вызывает рост МОК для удержания артериального
давления в нормальных пределах, который может обострять сердечную
недостаточность у больных с падением экскреторной функции почек. При
алкогольной гепатопатии, связанный с ней дефицит тиамина, снижая ОПСС,
ведет к росту МОК, усиливающему падение сократимости сердца
вследствие хронической алкогольной интоксикации. У больных с тиреотоксикозом
действие на сердце высокой действующей концентрации трийодтиронина в
циркулирующей крови повышает частоту сердечных сокращений и МОК
через увеличение чувствительности бета-один-рецепторов сердца к
адреналину. Одновременно тиреотоксикоз увеличивает потребление кислорода
организмом, что также повышает МОК. Постоянно повышенная работа
сердца по поддержанию МОК на высоком уровне и его избыточная адре-
нергическая стимуляция приводят к миокардиодистрофии и вторичной
кардиомиопатии. МОК на высоком уровне у больных с гипертиреозом
удерживает и компенсаторная реакция роста насосной функции сердца,
направленная на предотвращение артериальной гипотензии, несмотря на
снижение ОПСС под влиянием трийодтиронина.
Сердечная недостаточность при высоком МОК может развиваться как
компенсаторно-патогенная реакция в ответ на возникновение ведущего звена
патогенеза cor pulmonale, легочной артериальной гипертензии.
Редкими причинами сердечной недостаточности при высоком МОК
являются фиброзная дисплазия костей (болезнь Альбрихта) и карциноид-
ный синдром, связанный с серотонин-продуцирующими метастазами
злокачественных опухолей в печень.
ПАТОГЕНЕЗ ОСТРОЙ ЛЕВОЖЕЛУДОЧКОВОЙ
НЕДОСТАТОЧНОСТИ КАК ПРИЧИНЫ
КАРДИОГЕННОГО ОТЕКА ЛЕГКИХ
В зависимости от основного патогенетического механизма отека
легких выделяют два его вида:
♦ кардиогенный отек высокого давления, ведущее звено патогенеза
которого - легочная венозная гипертензия (патологический рост
давления крови в сосудах венозного звена легочной циркуляции)
(рис. 19.2). Легочная венозная гипертензия вторична по отношению
к острой левожелудочковой недостаточности, вызывающей рост
давления крови в левом предсердии {отек высокого давления)',
♦ отек вследствие патологического роста проницаемости стенки
легочных микрососудов и альвеолярного эпителия для жидкой части
плазмы крови и ее белков (некардиогенный отек легких, отек низкого
давления).
302
Глава 19
Рис. 19.2. Основное звено патогенеза кардиогенного отека легких
La - конечно-диастолическое давление крови в левом желудочке;
Pmv- гидростатическое давление крови в легочных капиллярах;
U- скорость ультрафильтрации в интерстиции легких;
L - объемная скорость лимфатического дренирования из легочного интер-
стиция на уровне респиронов
В паренхиме легких, как и во всех других тканях, постоянно
происходит перемещение жидкости из сосудистого сектора в интерстиции. В
физиологических условиях лимфоотток предотвращает избыточное
накопление жидкости в легочном интерстиции. Факторы, определяющие объем
жидкости, который из легочных микрососудов мигрирует в интерстиции,
детерминируют величину этого объема в соответствии с уравнением
Старлинга, описывающим процесс фильтрации жидкости через
полупроницаемую мембрану. В упрощенном виде уравнение выглядит
следующим образом:
Q = К [ (Pmv-Ppmv) - (Mmv - Mpmv) ],
где: Q - это объем жидкости, перемещающийся из сосудов в
интерстиции; коэффициент К в количественном отношении характеризует
проницаемость мембраны; Pmv - гидростатическое давление в просвете
микрососудов, a Ppmv - гидростатическое давление в интерстиции,
прилегающем к стенке микрососудов; Mmv - коллоидно-осмотическое
давление крови, притекающей к легким, a Mpmv - коллоидно-осмотическое
давление в их интерстиции. Q зависит от градиента гидростатических
давлений, «выталкивающего» жидкость из капилляров (Pmv- Ppmv), и
градиента коллоидно-осмотических давлений, удерживающего жидкость
в просвете сосудов (Mmv - Mpmv).
Обычно гидростатическое давление в легочном интерстиции
соответствует давлению в альвеолах, и его можно считать нулевым. Следовательно,
главной силой, вызывающей фильтрацию жидкости в интерстиции легких,
является гидростатическое давление в легочных капиллярах. Величина Pmv
варьирует в пределах легочной ткани, увеличиваясь под влиянием силы
тяжести в базальных отделах легких. Кроме того, гидростатическое давление в
капиллярах легких меняется в зависимости от соотношения сосудистых
сопротивлений на пре- и посткапиллярном уровне сосудистого русла легких. В
клинической практике гидростатическое давление в легочных капиллярах
обычно считают равным давлению в левом предсердии. Более точен расчет-
Клиническая патофизиология сердечной недостаточности
303
ный метод определения Pmv, когда гидростатическое давление в легочных
капиллярах рассчитывают, прибавляя к величине давления в левом
предсердии 0,5 различия между средним давлением в легочной артерии и давлением
в левом предсердии.
Давление «заклинивания» в конечных ветвях легочной артерии,
определяемое после ориентированной катетеризации плавающим катетером
Сван-Ганца, остается наиболее точным критерием как давления в
легочных капиллярах, так и давления в левом предсердии.
Хроническая левожелудочковая застойная сердечная недостаточность
обуславливает вторичную легочную венозную гипертензию, которая
повышает давление в легочной артерии, вызывая легочную артериальную
гипертензию. Вследствие вторичной легочной артериальной гипертензии
ультрафильтрат начинает поступать в легочный интерстиции из просвета
легочных артериол. Поэтому при застойной сердечной недостаточности
по левожелудочковому типу давление в легочной артерии представляет
собой один из факторов миграции жидкости из сосудов легких в их
интерстиции.
В физиологических условиях коллоидно-осмотическое давление в
циркулирующей крови выше коллоидно-осмотического давления в
интерстиции, окружающем легочные капилляры. Только небольшое количество
белка у здоровых людей проникает в интерстиции легких через соединения
между эндотелиоцитами. Градиент между коллоидно-осмотическим
давлением крови в легочных капиллярах и коллоидно-осмотическим давлением
крови в интерстиции легких противостоит гидростатическому давлению в
капиллярах, выталкивающему жидкость из легочных микрососудов.
Анализ уравнения Старлинга позволяет сделать три основных клини-
ко-патофизиологических вывода:
♦ Объем жидкости, который из микрососудов легких перемещается в
их интерстиции,- это прямая функция проницаемости их стенок
для жидкой части плазмы крови и ее белков и различия между
гидростатическим давлением в легочных капиллярах и градиентом
коллоидно-осмотических давлений между просветом микрососудов
и легочным интерстицием.
♦ Падение градиента коллоидно-осмотических давлений до их
выравнивания по обе стороны стенки легочных микрососудов повышает
миграцию жидкости в легочный интерстиции.
♦ Увеличение проницаемости легочных капилляров обладает двумя
однонаправленными эффектами: а) поступление жидкости в
интерстиции возрастает при нормальном уровне гидростатического
давления в легочных капиллярах; б) по мере нарастания миграции
белка плазмы крови в интерстиции падает градиент коллоидно-
осмотических давлений, что еще больше усиливает накопление
ультрафильтрата в интерстиции легочной ткани.
В основном транскапиллярная миграция жидкости в легких
происходит на уровне микрососудов, находящихся в непосредственной близости
к газообменным структурам респирона.
304
Глава 19
Жидкость и белок, которые попадают из сосудов в интерстиций
легких через щели между эндотелиоцитами, сразу не поступают в просвет
альвеол, так как клетки альвеолярного эпителия плотно связаны между
собой. После того, как ультрафильтрат поступает в интерстиций легких
на уровне респиронов, он перемещается в направлении к перибронхиаль-
ным и периваскулярным пространствам, удаленным от альвеол
(экстраальвеолярный интерстиций). Этому способствует преобладание
гидростатического давления в периальвеолярном интерстиций над
гидростатическим давлением в интерстиций экстраальвеолярном. При дренировании
ультрафильтрата из альвеолярного интерстиция перибронхиальные
соединительнотканные футляры функционируют как растяжимые демпферные
емкости. У здорового человека в условиях относительного покоя 10—20 мл
ультрафильтрата каждый час поступает в интерстиций и выводится оттуда
по лимфатическим сосудам.
Если недостаточность насосной функции левого желудочка через
вторичную легочную венозную гипертензию ведет к росту
гидростатического давления в легочных микрососудах, то растет объем
ультрафильтрата, поступающего в интерстиций легких. При умеренном
повышении давления в левом предсердии (от 14 до 20 мм рт. ст.) у
большинства больных выявляют умеренно выраженные одышку и диспноэ. Эта
фаза развития кардиогенного отека легких не рентгенограммах
проявляется затемнениями междолевых перегородок и щелей (линии Керли).
Умеренный подъем давления крови в левом предсердии ведет к отеку
межальвеолярных перегородок, а также к накоплению жидкости в
рыхлой ткани, которую содержат соединительнотканные влагалища сосудов
и бронхов.
При давлении в левом предсердии от 25 до 30 мм рт. ст. поступление
жидкости в интерстиций легких начинает преобладать над лимфооттоком.
В результате отечная жидкость проникает через барьер альвеолярного
эпителия в просвет альвеол и заполняет их. Когда число незаполненных
жидкостью альвеол снижается до критического уровня и ниже,
развивается артериальная гипоксемия, устойчивая по отношению к увеличению
содержания кислорода во вдыхаемой газовой смеси.
При кардиогенном отеке легких высокого давления содержание белка
в лимфе, оттекающей от легких, снижается. Тому причиной служит
разведение белка в интерстиций ультрафильтратом. Так как
проницаемость стенок легочных микрососудов остается неизменной, то белок
плазмы крови не попадает в легочный интерстиций.
Заполнению альвеол жидкостью при кардиогенном отеке
препятствует действие трех механизмов. Во-первых, возрастает лимфатическое
дренирование интерстиция. Кроме того, падение концентрации белка в
легочном интерстиций вызывает рост градиента коллоидно-осмотических
давлений между плазмой крови и интерстициальной жидкостью легких.
Это противодействует фильтрации плазмы в межклеточные пространства.
Наконец, потере легочной тканью воздушности противостоит, задерживая
заполнение жидкостью альвеол, перибронхиальная и периваскулярная ак-
Клиническая патофизиология сердечной недостаточности
305
кумуляция ультрафильтрата в удаленных от альвеол
соединительнотканных пространствах. При кардиогенном отеке легкие содержат до 500 мл
жидкости в виде «манжет», обволакивающих сосуды и бронхи в
экстраальвеолярных интерстициальных пространствах.
При кардиогенном отеке легких объем транссудата в плевральных
полостях находится в достоверной прямой связи с величиной давления в
левом предсердии и степенью «затопления» альвеол всей легочной
паренхимы.
Разрешение отека легких происходит при обратной миграции
ультрафильтрата из альвеол в кровь. Это активный процесс трансмембранного
переноса жидкости, вторичный по отношению к активизации транспорта
катиона натрия из альвеол в интерстиций, а затем и в кровь. Бета-
адреномиметики устраняют отек легких, не просто усиливая насосную
функцию левого желудочка, но и стимулируя трансмембранный активный
перенос натрия. Непрерывная фиксация давления «заклинивания» после
катетеризации конечных ветвей легочной артерии катетером Сван-Ганца
позволяет отдифференцировать отек легких высокого давления от отека
вследствие возрастания проницаемости стенок микрососудов легких. Для
выявления природы отека могут быть информативными результаты эхо-
кардиографического исследования сократимости левого желудочка и его
фракции изгнания.
ПАТОГЕНЕТИЧЕСКИЕ ПРИНЦИПЫ ТЕРАПИИ
КАРДИОГЕННОГО ОТЕКА ЛЕГКИХ
Эффективность терапии отека легких высокого давления зависит от
того, в какой степени удается снизить патологически высокое давление в
левом предсердии, которое повышает гидростатическое давление крови в
легочных микрососудах, выталкивающее жидкость из их просвета в
интерстиций.
Наиболее давно используемые способы лечения отека легких
основаны на снижении преднагрузки сердца через уменьшение общего
венозного у возврата. Так, метод лечения кардиогенного отека Ослера основан на
снижении общего венозного возврата к сердцу в полусидячем положении
больного; кроме того, Ослер рекомендовал накладывать жгуты на
конечности как препятствие для венозного оттока к сердцу.
В течение многих десятилетий морфина сульфат служил
эффективным средством лечения кардиогенного отека легких. Во многом
положительное действие морфина связано со снижением преднагрузки сердца
вследствие дилатации вен на системном уровне. Вазодилататоры прямого
инотропного действия (нитропруссид и др.) снижают гидростатическое
давление в микрососудах не только вследствие венодилатации и
снижения общего венозного возврата, но и в результате увеличения фракции
изгнания левого желудочка. Рост ударного объема левого желудочка обу-
306
Глава 19
словлен снижением общего периферического сопротивления
(постнагрузки) из-за расширения резистивных сосудов.
Мощные диуретики (лазикс и др.), не только снижают патологически
высокий объем внеклеточной жидкости, но и расширяют вены при
внутривенном введении. Такие диуретики снижают гидростатическое давление в
легочных микрососудах, снижая преднагрузку одновременно через
уменьшение внутрисосудистого объема и дилатацию емкостных сосудов.
Средства, обладающие положительным инотропным действием (до-
памин, добутамин, сердечные гликозиды), повышая сократимость
миокарда, снижают давление в левом предсердии.
Глава 20
ПАТОГЕНЕЗ ИНФАРКТА МИОКАРДА
И КАРДИОГЕННОГО ШОКА
Инфаркт миокарда - патологическое состояние сердца и всего
организма, которое развивается вследствие прекращения или резкого падения
объемной скорости кровотока в определенных сегментах стенок
сердечных камер в результате обтурации венечных артерий атеросклеротиче-
скими бляшками и тромбами. В основном патогенные изменения
системной регуляции, расстройства функций и сдвиги гомеостазиса при
инфаркте миокарда вызывают и составляют:
♦ падение насосной функции сердца вплоть до развития кардиогенно-
го шока;
♦ потерявшая приспособительное значение патогенная боль как
стимул защитно-патогенных реакций, которые почти сразу
превращаются в звенья патогенеза острой сердечной недостаточности и кар-
диогенного шока;
♦ прогрессирующая гиперкоагулемия (патологически высокая
свертываемость крови).
Летальность у больных с инфарктом миокарда составляет 30-40 %. В
период реконвалесценции после острого инфаркта миокарда больные
предрасположены к повторным ишемии миокарда и инфарктам,
сердечным аритмиям и внезапной сердечной смерти.
В клинико-патофизиологическом отношении инфаркт миокарда
прежде всего характеризует асинхронное сокращение сегментов стенок
желудочка, пораженного циркуляторной гипоксией. Острое снижение
выброса крови левым желудочком в аорту происходит не столько
вследствие ишемического цитолиза кардиомиоцитов, сколько в результате
обусловленного циркуляторной гипоксией падения сократительной
способности клеток рабочего миокарда. Уже через 15 с после наступления
ишемии (острой циркуляторной гипоксии) клетки сократительного миокарда
жертвуют своей функцией, дабы сохранить жизнеспособность через
ограничение энерготрат в условиях гипоксического гипоэргоза (связанного с
недостатком кислорода дефицита свободной энергии).
Благодаря коллатеральному кровоснабжению в системе венечных
артерий, а также гибернации сердца (см. ниже), не все кардиомиоциты в
зоне инфаркта одинаково страдают от циркуляторной гипоксии. Но все сар-
комеры миокарда в той или иной степени теряют способность к
сокращению. При этом в соответствии с принципом асинхронного реагирования
структурно-функциональных элементов эффекторов функций при
системных патологических реакциях саркомеры миокарда теряют
сократительную способность по-разному. На органном уровне неравномерное
падение силы сокращений саркомеров миокарда приводит к асинхронному
308
Глава 20
сокращению сегментов стенок левого желудочка, которое служит
причиной снижения его ударного объема.
Возникновение или обострение стенокардии - это первый и основной
симптом инфаркта миокарда, но часто боли нет, и лишь специальные
исследования позволяют обнаружить инфаркт, когда к его
ретроспективному выявлению побуждают осложнения (сердечные аритмии, артериальная
гипотензия).
На основании результатов гистопатологических, ангиографических и
ангиоскопических исследований выделяют шесть стадий (вариантов)
морфопатогенеза инфаркта миокарда:
1. Разрастание атероматозной бляшки: холестерин, атерогенные ли-
попротеины все более и более пропитывают субинтиму венечной
артерии; по мере увеличения размеров бляшки, усиления ее фиброза и каль-
цификации она блокирует просвет сосуда.
2. Патологический спазм пораженного атеросклерозом участка
сосудистой стенки, то есть аномально интенсивное сокращение гладкомышечных
элементов измененной атеросклерозом сосудистой стенки в ответ на
действие нейрогенных, паракринных и механических стимулов.
3. Разрыв или повреждение сосудистой стенки в области
атероматозной бляшки вследствие', а) резкого увеличения массы бляшки; б)
дегенерации и гибели эндотелиоцитов из-за инфильтрации макрофагами
сосудистой стенки и секреции ими протеолитических ферментов; в) спазма
артерии, который повреждает эндотелий в области бляшки; г) разрыва
vasa vasorum.
4. Тромбоз. Повреждение эндотелия, связанное с атеросклерозом,
нарушает свойство интимы артерий противостоять тромбообразованию.
Механические повреждения эндотелия обнажают лежащие под ним фиб-
ронектин, коллаген и фактор фон Виллебранда, каждый из которых
активирует тромбоциты. Адгезия активированных тромбоцитов друг к другу
служит инициирующим моментом тромбообразования. Образование
тромба - это не быстрый и прерывистый процесс, но часто приводящий,
несмотря на периодическое разрушение тромба под влиянием кровотока в
артерии, к полной закупорке ее просвета.
5. Спонтанный лизис тромба. После завершения тромбообразования
тканевой активатор плазминогена превращает плазминоген в плазмин, что
ведет к деполимеризации фибрина. Ингибитор активатора плазминогена и
циркулирующий с плазмой крови альфа-два-антиплазмин тормозят лизис
тромба, инактивируя соответственно плазминоген и плазмин. Соотношение
между тромбообразованием вкупе с падением объемной и линейной
скорости кровотока с одной стороны и лизисом тромба вместе с разрушающим
его действием кровотока с другой определяет время образования или
исчезновения тромба в просвете артерии. Частичная оь^клюзия сосуда
тромбом часто проявляет себя нестабильной стенокардией. Полная обтурация
просвета сосуда обычно служит причиной инфаркта миокарда.
6. Ретромбоз, распространение тромба по сосуду и тромбоэмболия.
Соотношение интенсивности процессов тромбообразования и спонтанно-
Патогенез инфаркта миокарда и кардиогенного шока
309
го лизиса тромба подвергается значительным колебаниям. Если конечным
результатом колебаний является устойчивое превалирование тромбообра-
зования, то создаются условия для распространения тромба по сосуду и
тромбоза ветвей венечных артерий дистальнее первичного локуса тром-
богенеза. У 50 % больных инфарктом миокарда полная обтурация
просвета сосуда происходит быстро. У других больных прогрессирование
окклюзии вследствие тромбоза чередуется с разрушением тромба под
влияниями спонтанного лизиса и кровотока. В результате у таких пациентов
нет внезапного появления всех симптомов инфаркта, первыми из которых
могут быть учащение стенокардии и депрессия сегмента S-T
электрокардиограммы.
Морфопатогенез нестабильной стенокардии как патологического
состояния, которое может быстро перейти в инфаркт миокарда, состоит в
распаде пропитанной липидами атеросклеротической бляшки с эрозией ее
фиброзной головки, инфильтрированной моноцитами и макрофагами.
Деструкция атероматозной бляшки служит причиной и создает условия
тромбоза венечной артерии. Патогенетически ориентированными целями
терапии у больных с нестабильной стенокардией являются:
♦ прерывание тромбогенеза для предотвращения инфаркта миокарда;
♦ создание условий и стимуляция эндогенного лизиса тромба.
Хотя данные цели отчасти достижимы при использовании в» качестве
антикоагулянтов ацетилсалициловой кислоты и гепарина, их полностью
достигают, вводя больным такие избирательные антагонисты тромбина
как гирудин и гирулог. Дело в том, что ацетилсалициловая кислота и
гепарин уменьшают массу сердцевины тромба, где почти нет тромбина,
практически не влияя на пристеночную часть, в которой много тромбина,
связанного с фибрином. Более того, гепарин, снижая уровень активации
белка С, способствует образованию тромбина.
Связанную с тромбозом обтурацию венечной артерии выявляют у
90 % больных с острым инфарктом миокарда и (или) тяжёлой ишемией
(острой циркуляторной гипоксией) миокарда, которые приводят к
подъему сегмента S-T электрокардиограммы. Введение современных фибри-
нолитических препаратов лизирует тромб и восстанавливает объемную
скорость кровотока по ранее обтурированной > венечной артерии у
60-90 % больных с инфарктом. При этом эффективный тромболизис не
только предотвращает гибель клеток сердца, но и через возобновление
доставки к ним кислорода подвергает обратному развитию гибернацию
кардиомиоцитов.
Тромболитические средства классифицируют в зависимости от
свойства препаратов избирательно и преимущественно активировать
плазминоген, связанный с фибрином, и в меньшей степени циркулирующий
плазминоген. Неизбирательные средства типа стрептокиназы вызывают
системную активацию плазминогена, которая обуславливает интенсивное
разрушение фибрина во всем сосудистом русле, высвобождение в
циркулирующую кровь продуктов деградации фибрина и низкую активность в
плазме крови ингибитора активности плазмина и альфа-два-антиплаз-
310
Глава 20
мина. Рекомбинантный активатор плазминогена тканевого типа действует
более избирательно и в основном активирует плазминоген, уже связанный с
фибрином, находящимся в составе сгустка.
Показанием к тромболитической терапии служит ишемия (острая
циркуляторная гипоксия), сохраняющаяся более 30 мин, о которой все это
время свидетельствует подъем сегмента S-T на 0,1 мВ и более по крайней
мере в двух отведениях электрокардиограммы при нижней, передней и
боковой локализации инфаркта миокарда или депрессия сегмента в
правых грудных отведениях при инфаркте миокарда задней стенки левого
желудочка. Для уменьшения зоны инфаркта и обратного развития гибер-
нации кардиомиоцитов тромболизис особенно эффективен в первые 4-6 ч
от начала приступа грудной жабы.
Противопоказаниями к тромболитической терапии у больных с
инфарктом миокарда служат патологические состояния и изменения
органов, сопровождающиеся повышенной кровоточивостью и патологические
состояния с высоким риском кровотечений и кровоизлияний, которые
могут возникнуть вследствие тромболизиса:
♦ Недавнее кровотечение вне зависимости от локализации его
источника. Оперативное вмешательство в течение двух недель перед
острым инфарктом миокарда.
♦ Артериальная гипертензия при артериальном давлении выше, чем
200/120 мм рт. ст., как фактор риска субарахноидального
кровоизлияния.
♦ Нарушения мозгового кровообращения в анамнезе как
свидетельство предрасположенности к инсульту.
♦ Длительная реанимация.
♦ Расслаивающая аневризма аорты.
Протяженность зоны ишемической дисфункции кардиомиоцитов и
цитолиза клеток сердца находится в прямой связи со временем
прекращения кровотока по венечной артерии или резкого падения его объемной
скорости. Зона инфаркта миокарда как масса рабочих кардиомиоцитов,
адекватно не участвующих в сокращении желудочков сердца, растет
вследствие индуцированного гипоксией воспаления, а также в результате
аккумуляции в участке сердца, наиболее страдающем от гипоксии,
свободных кислородных радикалов и агентов ауто- паракринной регуляции,
вызывающих гибернацию клеток сердца. Через 20 мин после полного
прекращения кровотока по соответствующим венечным артериям в
подавляющем большинстве кардиомиоцитов субэндокардиального слоя левого
желудочка фиксируют признаки необратимой гибели клеток. Такой же
уровень цитолиза при трансмуральном инфаркте выявляют через 4 ч. Это
связано с тем, что клетки миокарда субэндокардиального слоя до
инфаркта миокарда работают почти максимально интенсивно. Высокий уровень
работы субэндокардиального слоя обуславливает предшествующую
острой гипоксии полную мобилизацию резервов роста доставки кислорода
его кардиомиоцитам, который происходит через локальное снижение
сосудистого сопротивления.
Патогенез инфаркта миокарда и кардиогенного шока
311
Активность креатинкиназы (КК) в плазме крови представляет собой
наиболее известный и широко используемый показатель выраженности
некробиотических изменений кардиомиоцитов при инфаркте и находится
в прямой связи с массой клеток сердца, потерявших свою сократительную
способность вследствие гипоксического цитолиза и гибернации. Из-трех
цитозольных и одного митохондриального изоферментов КК цитозольная
КК-МВ особо специфична для миокарда, и ее патологически высокая
активность в плазме крови выступает достоверным и специфичным
маркером острой циркуляторной гипоксии клеток сердца и ишемического
цитолиза кардиомиоцитов. Так при гипертрофии миокарда в стадии
декомпенсации, при которой потребность клеток сердца в кислороде всегда
остается неудовлетворенной, содержание КК-МВ возрастает в 10 раз
относительно содержания фермента в здоровом сердце.
ГИБЕРНАЦИЯ, СТАННИНГ И ИШЕМИЧЕСКОЕ
ПРЕКОНДИЦИОНИРОВАНИЕ
КАРДИОМИОЦИТОВ
Если после возникновения циркуляторной гипоксии сердца в его
пораженном недостатком кислорода участке продолжает оставаться
высоким отношение потребности клеток сердца в кислороде к доставке 02
кардиомиоцитам, то связанные с гипоксией патологические изменения
могут прогрессировать вплоть до цитолиза. Циркуляторная гипоксия
сердца индуцирует на органном уровне защитную реакцию гибернирую-
щего миокарда (гибернации сердца).
Гибернация (лат. hibernus - зимний, холодный) - искусственно
вызванное состояние замедленной жизнедеятельности организма,
напоминающее зимнюю спячку животных (естественная гибернация).
Под гибернирующим миокардом понимают состояние сердца,
которое характеризует угнетение насосной функции в условиях покоя без
цитолиза кардиомиоцитов, причина которого - снижение объемной
скорости кровотока по венечным артериям. Гибернация резко ограничивает
возможности сердца реагировать ростом выброса крови в аорту левым
желудочком за единицу времени в ответ на рост потребности организма в
кислороде (физическая нагрузка, лихорадка, гипертиреоз и др.).
Состояние гибернирующего миокарда - это результат защитной реакции,
направленной на снижение высокого соотношения между силой
сокращений гипоксичного участка сердечной мышцы и его кровоснабжением, то
есть отношения потребности кардиомиоцитов в свободной энергии к
уровню улавливания клетками сердца свободной энергии при аэробном
биологическом окислении. Таким образом, гибернация задерживает
цитолиз клеток сердца, обусловленный гипоэргозом.
В ответ на снижение объемной скорости кровотока в два раза
происходит снижение вызываемого систолическим сокращением утолщения
312
Глава 20
стенок соответствующего сегмента на 50 %. Так проявляет себя гиберна-
ция сердца как причина угнетения насосной функции левого желудочка.
Кроме того, о гибернации свидетельствует возвращение на исходный
уровень концентрации протонов, креатинфосфата и напряжения
углекислого газа в венозной крови, оттекающей от сердца, через 1-3 ч после
возникновения его циркуляторной гипоксии, приводящей к инфаркту.
Гипокинезия и акинезия сегментов стенки левого желудочка,
вызванная гибернацией сердца, еще не говорят о необратимых
изменениях кардиомиоцитов, в которых при гистопатологическом исследовании
не находят признаков характерной для начальных стадий гипоксиче-
ского гипоэргоза дегенерации. Гибернация сохраняет кардиомиоциты
таким образом, что возобновление кровотока в течение недели после
возникновения ишемии (аорто-коронарное шунтирование, чрезкожная
эндоваскулярная пластика венечной артерии) подвергает обратному
развитию гипо- и акинезию сегментов стенки желудочков. По мере
исчезновения гипокинезии и акинезии сегментов восстанавливается
синхронность их систолического сокращения, растет фракция изгнания
левого желудочка, и восстанавливается способность сердца
реагировать ростом выброса крови в аорту в ответ на увеличение потребностей
органов и тканей.
Можно считать, что в настоящее время не существует широко
доступных достоверных способов определения жизнеспособности
(гибернации) сердечных клеток в асинхронно сокращающихся сегментах
стенки левого желудочка. Лишь комбинация ангиографии, эхокардиографии,
сцинтиграфии и компьютерной томографии сердца при кумуляции в
кардиомиоцитах и элиминации из них радионуклидов позволяет
получить достоверную информацию о степени жизнеспособности гиберни-
рующего миокарда.
Станнинг (англ. stunning - оглушение, ошеломление) миокарда - это
состояние вследствие снижения насосной функции сердца в результате
его циркуляторной гипоксии, которое не подвергается обратному
развитию, несмотря на восстановление объемной скорости кровотока в
испытавших циркуляторную гипоксию сегментах стенок сердечных камер.
Выраженность и длительность станнинга находятся в прямой связи со
степенью и длительностью циркуляторной гипоксии участка сердечной
мышцы. До сих пор неясно, представляет ли собой станнинг сугубо
патологическое состояние миокарда или следствие защитной реакции
гибернации. Существенным отличием станнинга от гибернации выступает то,
что восстановление доставки клеткам сердца кислорода и
энергопластических субстратов не устраняет угнетения насосной функции сердца.
Предположительно в основе развития станнинга лежат образование
свободных кислородных радикалов, нарушения миграции кальция через
клеточные мембраны и низкая эффективность улавливания кардиомиоцита-
ми свободной энергии при биологическом окислении.
Станнинг миокарда может развиться после тромболитической
терапии, когда внутривенное введение стрептокиназы ведет к лизису тром-
Патогенез инфаркта миокарда и кардиогенного шока
313
ба в области стеноза венечной артерии, или после операции аорто-
коронарного шунтирования. Состояние станнинга миокарда может
длиться дни или месяцы. В этих случаях использование средств с
положительным инотропным действием оправдано лишь в том случае,
если угнетение насосной функции желудочка может стать звеном танз-
тогенеза.
В экспериментах у целого ряда видов млекопитающих было показано,
что краткие периоды острой циркуляторной гипоксии (ишемии) сердца
(ишемическое прекондиционирование миокарда) значительно повышают его
устойчивость к длительной ишемии со снижением зоны инфаркта на 80 %
зоны его распространения у животных контрольной группы.
Ишемическое прекондиционирование - наиболее эффективный из
известных у млекопитающих естественных механизмов защиты клеток
миокарда от ишемии. В кардиопротективном эффекте ишемического
прекондиционирования особая роль принадлежит Gi-белкам,
локализованным в плазматической мембране клеток сердца. Эти
трансмембранные белки выступают медиаторами снижения активности аденилатцик-
лазы, которая падает вследствие возбуждения рецепторов к аденозину
Ai и мускариновых Мг-рецепторов. Возбуждение рецепторов этих двух
типов через активацию Gi-белков приводит к активации АТФ-
зависимых калиевых каналов наружных клеточных мембран кардио-
миоцитов, торможению их натриевого трансмембранного канала и
блокирует перенос через мембраны клеток сердца кальция по его каналам
L-типа. Каждый из этих эффектов активации Gi-белков ведет к
снижению утилизации всеми клетками сердца свободной энергии в основном
за счет меньшей работы клеток рабочего миокарда при сокращении.
Предполагают, что активация Gj-белков при ишемии происходит
вследствие связанного с гипоэргозом высвобождения клетками сердца
большого количества молекул аденозина.
Если сердце не подвергать ишемическому прекондиционированию, то
ишемия служит причиной неуклонного снижения уровня активации Gr
белков, то есть их дисфункции, связанной с гипоэргозом. В сердце
экспериментальных животных после ишемического прекондиционирования
возрастает чувствительность Gi-белков к активации соответствующих
рецепторов при ишемии. Устойчивая активация данных трансмембранных
белков в зоне циркуляторной гипоксии сердца, которое прошло через
несколько периодов кратковременной ишемии, не приводящей к цитолизу,
предположительно лежит в основе кардиопротективного эффекта
ишемического прекондиционирования.
Полагают, что данные, полученные при изучении ишемического
прекондиционирования у экспериментальных животных, позволят
экстраполировать их результаты на практику лечения инфаркта миокарда у
больных. Это в известной мере подтверждает предварительное сообщение об
эффективности блокатора распада аденозина акадезина в предупреждении
интраоперационных инфарктов миокарда при аорто-коронарном
шунтировании.
314
Глава 20
ИНФАРКТ МИОКАРДА И КАРДИОГЕННЫЙ ШОК
Кардиогенный шок - это критическое состояние, которое
развивается вследствие острой артериальной гипотензии, обусловленной резким
падением насосной функции левого желудочка. Первичное звено
патогенеза кардиогенного шока - это быстрое снижение ударного объема левого
желудочка, которое приводит к артериальной гипотензии, несмотря на
компенсаторные спазм резистивных сосудов и рост общего
периферического сосудистого сопротивления (ОПСС), направленные на
восстановление артериального давления.
В силу резкого угнетения сократительной способности сердечной
мышцы у больных в состоянии кардиогенного шока невозможно
компенсаторное возрастание минутного объема кровообращения (МОК) в
результате адренергическои неирогуморальнои стимуляции сердца в ответ
на артериальную гипотензию и циркуляторную гипоксию. Кроме
артериальной гипотензии, доставку кислорода клетке при кардиогенном шоке
снижает юкстакапиллярное шунтирование вследствие компенсаторно-
патогенного спазма сосудов сопротивления. Артериальная гипотензия и
снижение кровотока по обменным капиллярам вследствие спазма
мельчайших артерий, артериол и прекапиллярных сфинктеров нарушают
кровоток в органах на периферии и вызывают основные симптомы
кардиогенного шока (табл. 20.1).
Таблица 20.1
Симптомы кардиогенного шока
Симптом
Нарушения сознания
Бледность кожи, холодные и
влажные конечности
Олигурия (< 20 мл/ч)
Артериальная гипотензия
(систолическое артериальное давление
меньше, чем 90 мм рт. ст.)
Причины возникновения
Гипоксия, ацидоз, отрицательный
эмоционально-болевой стресс
Вазоспазм на периферии в ответ на
артериальную гипотензию и циркуляторную гипоксию,
рост потоотделения как результат системной
адренергическои стимуляции
Спазм приводящих артериол нефрона
Падение сократительной способности
сердечной мышцы
Артериальная гипотензия вследствие травматического шока - это
не ведущее звено патогенеза данного патологического состояния, а
следствие несостоятельности компенсации травматического шока, при
котором патологические сдвиги в органах и тканях возникают задолго до
снижения артериального давления. При кардиогенном шоке, наоборот,
артериальная гипотензия сразу лее начинает выступать одним из
основных звеньев патогенеза.
Наиболее частая причина кардиогенного шока у больных - это острый
инфаркт миокарда. Артериальная гипотензия как следствие падения удар-
Патогенез инфаркта миокарда и кардиогенного шока
315
ного объема левого желудочка развивается тогда, когда некробиотиче-
ским изменениям подвергается (в состояние гибернации впадает) более,
чем одна треть массы миокарда левого желудочка. Такие нарушения
сердечного ритма как полная поперечная блокада сердца, другие брадиарит-
мии, политопная желудочковая тахисистолия ведут к дискоординации
сокращений предсердий и желудочков, снижению времени диастолического
наполнения левого желудочка, падению его ударного объема и кардио-
генному шоку. Как диастолическое наполнение левого желудочка, так и
изгнание крови в аорту блокируется тампонадой сердца вследствие кар-
диохирургических оперативных вмешательств, ранений сердца и
инфаркта миокарда.
Прогрессирующее падение сократимости миокарда при кардиогенном
шоке обуславливают:
♦ снижение перфузионного давления миокарда вследствие
артериальной гипотензии;
♦ увеличение несоответствия между доставкой к кардиомиоциту Ог и
его потребностью в кислороде при росте работы сердца, связанном с
возрастанием общего периферического сосудистого сопротивления;
♦ острая дистрофия миокарда под влиянием избыточной адренерги-
ческой нейрогуморальной стимуляции.
Компенсаторные реакции в ответ на артериальную гипотензию и цир-
куляторную гипоксию при кардиогенном шоке почти идентичны таковым
у больных в состоянии травматического или гиповолемического шока. В
частности они включают:
♦ преимущественно нейрогенный спазм вен в результате усиления
симпатических сосудосуживающих влияний;
♦ активацию ренин-ангиотензин-альдостеронового механизма, в том
числе и в результате системной адренергической стимуляции;
♦ компенсаторную аутогемодилюцию, то есть мобилизацию
жидкости из интерстициального сектора в сосудистый вследствие
изменения на системном уровне соотношения между пре- и
посткапиллярным сосудистым сопротивлением.
Биологическая цель данных компенсаторных реакций - поддержание
МОК и артериального давления через рост общего венозного возврата,
задержку в организме натрия и воды, расширение внутрисосудистого
жидкостного сектора и возрастание ОПСС. При кардиогенном шоке эти
защитные реакции увеличивают пред- и постнагрузку, а значит
повышают утилизацию свободной энергии кардиомиоцитами. Рост работы клеток
сократительного миокарда повышает несоответствие между
потребностью сердца в кислороде и доставкой к нему Ог. В результате растет масса
гипоксичного и гибернирующего миокарда, и еще больше падает его
сократимость.
Основная патофизиологическая особенность кардиогенного шока-
это изначально присущие компенсаторным реакциям свойства звеньев
патогенеза, действие которых обуславливает прогрессирование шока и
приобретение им необратимого характера. Кроме того, при кардиогенном
316
Глава 20
шоке поражен основной эффектор компенсаторных реакций,
направленных на поддержание минутного объема кровообращения, - сердце.
Патологическая системная реакция на кардиогенный шок,
увеличивая преднагрузку правого желудочка при угнетенной функции левого,
повышает легочное капиллярное давление заклинивания, которое
начинает превышать коллоидно-осмотическое давление в просвете легочных
капилляров, формируя первичное звено патогенеза кардиогенного отека
легких.
ПАТОГЕНЕТИЧЕСКИЕ ПРИНЦИПЫ ФАРМАКОКОРРЕКЦИИ
НИЗКОЙ НАСОСНОЙ ФУНКЦИИ СЕРДЦА
Главная цель лекарственной терапии у больных с патологически
низким МОК - это повысить транспорт кислорода и энергопластических
субстратов на периферию, но не за счет преходящего роста
сократимости, который в дальнейшем приведет к необратимому падению насосной
функции сердца.
Если артериальная гипотензия, обусловленная низким ударным
объемом левого желудочка, становится ведущим механизмом танатогенеза, то
применение бета-один-адреномиметиков, других средств, повышающих
сократимость, и вазопрессоров следует признать неизбежной и почти
реанимационной лечебной мерой всегда опасной усилением гипоэргоза
сердца и сердечной недостаточности.
Главная цель патогенетической фармакокоррекции низкой насосной
функции сердца часто недостижима без достижения одной или
нескольких частных целей:
♦ роста МОК через увеличение ударного объема левого желудочка за
счет повышения сократимости сердца под влиянием
положительного инотропного действия лекарственных средств;
♦ увеличения доставки кислорода кардиомиоцитам посредством роста
перфузионного давления миокарда и суммарной длительности
периодов кровотока через субэндокардиальный слой миокарда за
минуту, то есть суммарной длительности диастолических интервалов;
♦ уменьшения потребности сердца в кислороде, которое достигают,
снижая: а) частоту сердечных сокращений и б) напряжение стенки
левого желудочка в фазу изгнания (S); S уменьшают без
артериальной гипотензии и падения перфузионного давления миокарда,
осторожно снижая среднее артериальное давление и конечно-диастоли-
ческое давление крови в левом желудочке.
Перфузионное давление субэндокардиального слоя левого желудочка
повышают, вызывая:
♦ увеличение диастолического АД при артериальной гипотензии (вазо-
прессоры, внутривенные инфузии плазмозамещающих средств при
гиповолемии);
Патогенез инфаркта миокарда и кардиогенного шока
317
♦ снижение конечно-диастолического давления крови в левом
желудочке без опасного падения среднего АД (осторожное
использование сосудорасширяющих средств при артериальной нормотензии).
Суммарную длительность диастолических интервалов, во время
которых артериальная кровь попадает в субэндокардиальный слой, повышают,
оптимально снижая частоту сердечных сокращений.
Упрощенный алгоритм достижения частных и главной цели
фармакотерапии у больных с критически низким относительно потребностей
организма МОК можно представить следующим образом:
♦ оптимизация преднагрузки без артериальной гипотензии, которую у
большинства больных достигают снижением общего венозного
возврата к сердцу за счет действия мочегонных и средств,
расширяющих вены (нитраты и др.);
♦ снижение постнагрузки без опасного падения ОПСС и
артериального давления путем эффективной аналгезии (инфаркт миокарда) и
крайне осторожного использования вазодилятаторов;
♦ увеличение вхождения ионизированного кальция в цитозоль кардио-
миоцитов для усиления сокращений саркомеров миокарда, которое
вызывают следующие препараты: а) бета-один-адреномиметики; б)
соли кальция; в) сердечные гликозиды; г) ингибиторы фосфодиэстеразы.
Если падение МОК обусловлено патологическим снижением частоты
сердечных сокращений, то, устраняя брадиаритмию, устраняют и
патологическое снижение МОК.
Наибольшей эффективности лекарственная терапия больных с
патологически низким МОК достигает при условии непрерывного мониторинга
за эффектами лекарственных средств на детерминанты насосной функции
сердца и МОК. Дозу препаратов с положительным инотропным
действием «титруют», ориентируясь на уровень МОК, который непрерывно
фиксируют с использованием метода термодилюции. Постоянное
определение ОПСС позволяет выявить его патологический рост, оптимально
снизить постнагрузку сердца вазодилятаторами, тем самым увеличив
ударный объем левого желудочка.
Катетеризация легочной артерии плавающим катетером Сван-Ганца
позволяет непрерывно фиксировать давления крови в правом предсердии,
правом желудочке, легочной артерии и легочное капиллярное давление
заклинивания (давление заклинивания легочной артерии, легочное давление
заклинивания). При этом одновременно методом термодилюции
производят постоянное определение величины МОК. Кроме того, катетеризация
легочной артерии катетером Сван-Ганца позволяет часто определять
напряжения в артериальной крови кислорода и углекислого газа и ОПСС,
которое у здоровых людей колеблется в пределах от 900 до 1350 дин-см-с'5:
ОПСС = [(среднее АД - ДПП)/МОК]х80,
где: АД - артериальное давление (мм рт. ст.), ДПП - диастолическое давление крови в
правом предсердии (мм рт. ст.), МОК - минутный объем кровообращения (л-мин'1), а 80 -
коэффициент, необходимый для перевода единиц измерения, обычно используемых в
клинической практике, в единицы международной системы.
318
Глава 20
Комментируя табл. 20.2, следует заметить, что рост выше 18 мм рт. ст.
конечно-диастолического давления в левом желудочке, эквивалентным
которому считают легочное давление заклинивания, обычно
свидетельствует о падении насосной функции левого желудочка вследствие
угнетения сократимости и (или) роста постнагрузки. Легочное давление
заклинивания в пределах от 10 до 18 мм рт. ст. говорит о нормальных диасто-
лическом наполнении левого желудочка и изгнании крови в аорту.
Легочное давление заклинивания в тех же пределах при симптомах и признаках
нарушений периферического кровообращения (табл. 20.2) обычно
свидетельствует о падении ударного объема Левого желудочка, снижении МОК
и сердечной недостаточности. Снижение легочного давления
заклинивания до уровня меньшего, чем 10 мм рт. ст., у большинства больных
говорит о низкой преднагрузке левого желудочка, которая падает в результате
снижения общего венозного возврата к сердцу и гиповолемии.
Таблица 20.2
Патогенетические принципы фармакокоррекции острых недостаточности
кровообращения и падения насосной функции сердца
кддлж,
мм рт. ст.
>18
<18
<10
АД
Н
н
п
н
н
п
п
н
п
ПК
н
п
н
н
п
н
п
н
или
п
н
или
П
Основной принцип терапии
Использование вазодилятаторов для снижения высокой
постнагрузки сердца как причины недостаточности кровообращения
Применение сосудорасширяющих средств с аналогичной целью
Одновременное применение вазодилятаторов и средств с
положительным инотропным действием для увеличения МОК через
увеличение сократимости и снижение постнагрузки
Динамическое наблюдение
Применение сосудорасширяющих средств для устранения
нарушений периферического кровообращения
Динамическое наблюдение или применение препаратов с
положительным инотропным действием для устранения скрытого
снижения насосной функции сердца
Применение средств с положительным инотропным действием
для усиления насосной функции сердца и вазодилятаторов, если
они не вызывают артериальной гипотензии и падения перфузи-
онного давления миокарда
Внутривенная инфузия плазмозамещающих растворов с целью
коррекции гиповолемии и для роста преднагрузки сердца
Внутривенная инфузия плазмозамещающих растворов с целью
коррекции гиповолемии и роста преднагрузки сердца
Примечание; КДДЛЖ - конечно-диастолическое давление крови в левом желудочке; Н -
нормальный или патологически высокий уровень АД, а также нормальное состояние периферического
кровообращения, о котором судят, определяя есть или нет признаки нарушений периферического
кровообращения; П - патологически низкие значения показателя или признаки нарушений
периферического кровообращения: холодные и бледные кожные покровы, патологический уровень значений
кожно-ректального температурного градиента, снижение транспорта кислорода на периферию,
выявляемое непрерывной пульсоксиметрией и чрезкожным определением напряжения кислорода в
тканях, падение диуреза; ПК - состояние периферического кровообращения.
Патогенез инфаркта миокарда и кардиогенного шока
319
При инфаркте миокарда снижение легочного давления заклинивания
до уровня меньшего, чем 10 мм рт. ст., артериальная гипотензия, падение
диуреза и устойчивая синусовая тахикардия позволяют предположить,
что существует возможность увеличить МОК путем внутривенных инфу-
зий для увеличения преднагрузки сердца.
Рост конечно-диастолического давления левого желудочка (давления
заклинивания легочной артерии) до уровня более высокого, чем
18 мм рт. ст., одновременный с падением сердечного индекса до уровня
более низкого, чем 2,5 лмин^м"2, несмотря на систолическое
артериальное давление, превышающее 100 мм рт. ст., свидетельствует о
падении насосной функции левого желудочка. При таком падении насосной
функции левого желудочка артериальную гипотензию предотвращает
рост ОПСС, обусловленный спазмом сосудов сопротивления. Рост
ОПСС у таких больных как защитная реакция всегда избыточен
относительно степени патологического сдвига, вызвавшего его, то есть падения
насосной функции левого желудочка.
Защитно-патогенный рост ОПСС как основная причина опасного
роста постнагрузки у больных с инфарктом миокарда расширяет зону
некробиотических изменений кардиомиоцитов. Дело в том, что
патогенный рост постнагрузки повышает работу сердца, увеличивая давление
крови в левом желудочке во время фазы изгнания. Рост энерготрат
сократительным миокардом при росте его работы повышает потребность
кардиомиоцитов в свободной энергии, что ведет к увеличению массы клеток
сердца в состоянии гибернации и некробиотических изменений
вследствие циркуляторной гипоксии.
В этой связи целью применения сосудорасширяющих средств у
больных с недостаточной насосной функцией левого желудочка и падением
МОК вследствие инфаркта миокарда, которые не вызывают
артериальной гипотензии, следует считать исключение патогенного компонента
роста ОПСС и постнагрузки сердца.
Средствами выбора для снижения постнагрузки в остром периоде
инфаркта миокарда служат нитроглицерин и нитропруссид натрия.
Полагают, что целесообразней использовать нитроглицерин, который не
только уменьшает постнагрузку, но и улучшает доставку клеткам сердца
кислорода, снижая сосудистое сопротивление в системе венечных
артерий. Нитропруссид используют, когда инфаркт миокарда развивается
вследствие и при артериальной гипертензии, или когда артериальная ги-
пертензия как осложнение инфаркта миокарда представляет собой
следствие защитно-патогенного роста ОПСС в ответ на падение насосной
функции сердца.
К бета-один-адреномиметикам или другим лекарственным средствам,
обладающим положительным инотропным действием (амринон и т.д.),
прибегают тогда, когда артериальная гипотензия вследствие падения
насосной функции сердца может вызвать терминальное состояние. При этом
следует быть постоянно нацеленным на прекращение применения этих
препаратов, опасно повышающих сократимость сердца и потребность
320
Глава 20
кардиомиоцитов в свободной энергии через увеличение силы сердечных
сокращений.
Все бета-один-адреномиметики обладают определенными эффектами:
♦ Увеличение ударного объема левого желудочка, не связанное с
изменениями пред- и постнагрузки.
♦ Усиление циркуляторной гипоксии сердца за счет роста его
сократимости и частоты сердечных сокращений.
♦ Рост предрасположенности к* возникновению сердечных аритмий
(аритмогенность бета-один-адреномиметиков).
Аритмогенность бета-один-адреномиметиков связана со снижением
под влиянием этих препаратов абсолютной величины порога спонтанной
деполяризации и с прогрессированием гипоэргоза проводящих
кардиомиоцитов вследствие усиления работы сердца в результате возбуждения
бета-один-адренорецепторов.
Необходимое условие эффективности бета-один-адреномиметиков -
это нормоволемия, что диктует необходимость обязательного исключения
дефицитов объема внеклеточной жидкости и циркулирующей крови из
причин артериальной гипотензии у больных с падением насосной
функции сердца.
Диастолическое расслабление сердечной мышцы - это процесс,
который невозможен без потребления свободной энергии кардиомиоцитами.
Его начальный момент- это активное возвращение ионизированного
кальция из цитозоля клеток сердца в саркоплазматический ретикулум.
Циркуляторная гипоксия миокарда через гипоэргоз кардиомиоцитов
тормозит возвращение кальция в саркоплазматический ретикулум. Это
приводит к недостаточному расслаблению саркомеров миокарда и диастоли-
ческой жесткости стенок левого желудочка. Возбуждение бета-один-
адренорецепторов ускоряет снижение содержания ионизированного
кальция в цитозоле кардиомиоцитов, и диастолическая жесткость во многом
подвергается обратному развитию. В результате под влиянием бета-один-
адреномиметиков растет диастолическое наполнение левого желудочка и
возрастает его ударный объем, что может повысить МОК.
Допамин - это предшественник норадреналина на пути его естественного
синтеза. Он представляет собой лиганду специфическую по отношению к
допаминергическим, бета-один- и альфа-один адренорецепторам. Его
системный эффект варьирует в зависимости от дозы. При непрерывной инфузии
препарата в дозах меньших или равных 3 мкг-кг^мин"1 допамин
преимущественно вызывает возбуждение допаминергических рецепторов, что
увеличивает объемную скорость кровотока в почках, скорость клубочковой
фильтрации и повышает экскрецию натрия и воды. В небольших дозах
(<5 мкг-кг'^мин"1) допамин, ослабляя стимуляцию альфа-один-адреноре-
цепторов, устраняет связанные со спазмом резистивных сосудов
нарушения периферического кровообращения и снижает постнагрузку сердца.
Непрерывная инфузия допамина в дозах до 5 мкгкг^мин"1 через
усиление почечных кровотока и экскреции натрия, нормализацию
периферического кровообращения, а также благодаря снижению ОПСС без
Патогенез инфаркта миокарда и кардиогенного шока
321
артериальной гипотензии уменьшает потребность сердца в кислороде и
свободной энергии, что может способствовать устранению сердечной
недостаточности.
В диапазоне доз 5-10 мкгкг^мин"1 допамин выступает в основном как
агонист к бета-один-адренорецепторам и увеличивает сократимость и
МОК без патогенного роста ОПСС. Как и все бета-один-адреномиметики,
препарат, непрерывно инфузируемый внутривенно в таких дозах,
ускоряет образование циклического аденозинмонофосфата из аденозинтрифос-
фата, повышая активность аденилатциклазы кардиомиоцитов. Рост
содержания в цитозоле кардиомиоцитов циклического
аденозинмонофосфата повышает концентрацию в нем ионизированного кальция, что
увеличивает сократимость. Такая фармакотерапия сердечной недостаточности
эффективна при некоторых нарушениях регуляции сердечной
деятельности, в частности связанных с эффектом бета-один-адренолитиков. При
ишемической болезни сердца, когда ограничены резервы возрастания
транспорта кислорода в клетки сердца, рост его потребности в Ог,
вызванный повышением активности аденилатциклазы под влиянием допа-
мина и других бета-один-адреномиметиков, усиливает гипоэргоз сердца и
сердечную недостаточность. В результате возникает необходимость
увеличения доз допамина от «отметки» в 5 мкг-кг^мин'1.
Если непрерывную инфузию допамина применяют в дозах,
превышающих 10 мкг'кг^мин"1, то допамин начинает оказывать действие в
основном как альфа-один-адреномиметик, вызывая спазм сосудов
сопротивления, который позволяет избежать артериальной гипотензии ценой про-
грессирования острой сердечной недостаточности. В основном
необратимость сердечной недостаточности придается устойчивым ростом
постнагрузки как фактором увеличения работы сердца и усиления его гипоэрго-
за. Кроме того, сердце начинает в большей степени испытывать целый
спектр патогенных влияний, связанных с усилением расстройств
периферического кровообращения под влиянием «высоких» доз допамина.
Добутамин - это синтетический катехоламин, преимущественно
оказывающий бета-один-адреномиметическое действие и слабые бета-два- и
альфа-один-адреномиметические эффекты. Тем не менее, слабые бета-
два- и альфа-один-адреномиметические эффекты добутамина
представляют собой «сильную» сторону его фармакодинамики, так как на
периферии приводят через преобладание вазодилятации вследствие стимуляции
бета-два-адренорецепторов над вазоконстрикцией вследствие
возбуждения альфа-один-адренорецепторов к умеренному снижению сосудистого
сопротивления. Умеренное снижение ОПСС под влиянием добутамина
вкупе с увеличением объема крови, который левый желудочек за минуту
выбрасывает в аорту, ведут к росту системного транспорта кислорода.
Если добутамин использовать одновременно с бета-адренолитиками, то
он начинает выступать преимущественно в роли альфа-один-адрено-
миметика и опасно повышает ОПСС. Непрерывная инфузия добутамина
особенно эффективна для повышения МОК и устранения расстройств
периферического кровообращения, если ее сочетать с непрерывным внутри-
322
Глава 20
венным вливанием допамина в дозах меньших, чем 5 мкг-кг^мин'1, при
использовании которых эффект допамина в основном сводится к
расширению сосудов сопротивления. При этом, осуществляя постоянный
мониторинг МОК, артерио-венозного различия по кислороду и потребления
кислорода организмом, дозу добутамина титруют в диапазоне от 2,5 до
40 мкг-кг^мин"1 до достижения оптимального уровня показателей
системного транспорта кислорода.
Допексамин - это бета-два-адреномиметик, который может
возбуждать допаминергические рецепторы. Препарат обладает свойствами
слабого агониста к бета-один-адренорецепторам и тормозит захват норадре-
налина нервными окончаниями, что и лежит в основе его положительного
инотропного действия. Оказывая положительное инотропное действие,
данное лекарственное средство через стимуляцию бета-два-адренорецеп-
торов и домапинергических рецепторов расширяет сосуды сопротивления
и увеличивает объемную скорость кровотока во внутренних органах.
Резкое увеличение частоты сердечных сокращений как основной
детерминанты потребности сердца в кислороде, которое вызывает допексамин,
ограничивает его использование. Поэтому допексамин используют в
течение недолгого времени для устранения острой сердечной
недостаточности у кардиохирургических больных, инфузируя раствор препарата через
внутривенный катетер большого диаметра. Непрерывную инфузию до-
пексамина в 5 % растворе глюкозы начинают в дозе 0,5 мкг-кг'^мин"1.
Если МОК после начала непрерывной инфузии допексамина не растет, то
дозу увеличивают до 1 мкг-кг'^мин"1. Когда такая доза не приносит
желаемого эффекта, то через каждые 15 мин ее увеличивают на 1 мкг-кг'^мин"1
до максимального уровня в 6 мкг-кг^-мин"1.
Длительное и непрерывное возбуждение бета-один-адреномимети-
ками соответствующих рецепторов сердца ведет к а) снижению
содержания в сердце бета-один-адренорецепторов и б) изменениям структуры бе-
та-один-адренорецепторов, которое снижает их чувствительность. Этот
эффект достигает своего максимума уже за 48 ч непрерывной инфузии
допамина, добутамина и других препаратов данной группы. В такой
ситуации прибегают к ингибиторам фосфодиэстеразы, чей положительный
инотропный эффект на сердце в основе своей имеет угнетение фосфо-
диэстеразных ферментов кардиомиоцитов, превращающих циклический
аденозинмонофосфат в метаболит, не обладающий биологическим
действием и свойствами вторичного мессенджера. Внутривенное введение
первого из препаратов данной группы милринона начинают с медленной
инфузии 50 мкг/кг массы тела препарата в течение 10 мин, вслед за
которой следует непрерывная инфузия со скоростью 375-750 мг-кг'^мин*1
в течение 48-72 ч. Максимальная скорость внутривенного введения
другого из препаратов данной группы эноксимона составляет 12,5 мг/мин.
Вначале эноксимон медленно вливают внутривенно в дозе 0,5-1 мг/кг.
Затем через каждые 30 мин внутривенно вводят по 0,5 мг/кг до
положительного эффекта или до достижения уровня суммарной дозы
эноксимона, составляющей 3 мг/кг.
Патогенез инфаркта миокарда и кардиогенного шока
323
Ингибиторы фосфодиэстеразы, резко увеличивая содержание в
клетках сердца ионизированного кальция, предположительно могут вызывать
контрактуру саркомеров миокарда, резко угнетающую систолический и
диастолический компоненты насосной функции желудочков сердца. Это,
как мы полагаем, может быть одним из механизмов развития такого
побочного эффекта как стойкая артериальная гипотензия.
Клинико-патофизиологическими показаниями к применению при кар-
диогенном шоке бета-один-адреномиметиков и вазопрессоров служат
рост конечно-диастолического давления левого желудочка (давления
заклинивания легочной артерии) выше 18 мм рт. ст., падение сердечного
индекса до уровня значений меньших, чем 2,5 л-мин'^м2, и систолическое
АД ниже, чем 100 мм рт. ст., которые свидетельствуют о падении
насосной функции левого желудочка, выступающей причиной артериальной
гипотензии, несмотря на компенсаторный рост ОПСС. При таком крайне
выраженном нарушении сократительной функции левого желудочка
использование лекарств с положительным инотропным действием и
вазопрессоров часто служит лишь средством предотвращения терминального
состояния вследствие артериальной гипотензии и может придать
угнетению сократимости необратимый характер. И те и другие средства
повышают потребность сердца в кислороде, то есть на фоне предшествующего
значительного дефицита свободной энергии в кардиомиоцитах резко
усиливают их гипоэргоз.
КЛИНИКО-ПАТОФИЗИОЛОГИЧЕСКАЯ ХАРАКТЕРИСТИКА
ОСЛОЖНЕНИЙ И ПЕРИОДА РЕАБИЛИТАЦИИ
ИНФАРКТА МИОКАРДА
Циркуляторная гипоксия кардиомиоцитов вследствие атеросклероти-
ческого поражения венечных артерий, инфаркт миокарда, вызывающий
цитолиз и гибернацию клеток сердца как причины снижения его
сократимости, могут приводить к недостаточности митрального клапана
(митральной регургитации).
Митральная регургитация вследствие ишемической болезни сердца
часто имеет преходящий характер. Обычно недостаточность
митрального клапана, вторичная по отношению к циркуляторной гипоксии
миокарда, возникает у больных, страдающих от застойной сердечной
недостаточности вследствие ишемической болезни сердца. У других
пациентов митральная регургитация возникает в результате обострения
гипоксии сердца и подвергается обратному развитию по мере
восстановления доставки кислорода клеткам сердца. Причинами
недостаточности митрального клапана вследствие ишемической болезни сердца
являются:
♦ снижение силы сокращений сосочковых мышц из-за недостаточной
доставки к ним кислорода;
324
Глава 20
♦ расширение кольца митрального клапана, вторичное по отношению
к дилатации полости левого желудочка при застойной сердечной
недостаточности;
♦ разрыв некробиотически измененных сосочковых мышц.
В две сосочковые мышцы левого желудочка кровь поступает по
конечным ветвям передней и задней венечных артерий. Наиболее
удаленные от стенки желудочка (свободные, дистальные) участки сосочковых
мышц постоянно испытывают прямое воздействие внутрижелудочкового
давления. Недостаточный коллатеральный кровоток в обход обтуриро-
ванного участка конечной ветви венечной артерии, кровоснабжающей
дистальный участок сосочковой мышцы, постоянное прямое воздействие
на свободный сегмент мышцы высокого давления крови в левой
сердечной камере у больных с застойной сердечной недостаточностью
обуславливают особую предрасположенность сосочковых мышц к некробиотиче-
ским изменениям в результате циркуляторной гипоксии и ишемии.
Синхронность сокращения сосочковых мышц и рабочего миокарда
левого желудочка представляет собой необходимое условие нормального
функционирования митрального клапана, то есть плотного смыкания его
створок в течение всей систолы. Недостаточная сила сокращений
сосочковых мышц, удерживающих створки клапана в положении плотного
смыкания, служит причиной пролабирования створок клапана в левое
предсердие в конце систолы. Это проявляется патологическим сердечным
шумом, выслушиваемым в конце систолы.
Дилатация левого желудочка у больных с сердечной
недостаточностью смещает сосочковые мышцы вниз и в латеральном направлении от
митрального клапана. В результате смещения систолическое сокращение
сосочковых мышц удерживает створки клапана во время систолы в
полости левого желудочка, препятствуя их смыканию и вызывая
митральную регургитацию. О недостаточности митрального клапана, которая
обусловлена застойной сердечной недостаточностью, свидетельствует
дующий систолический шум, занимающий всю фазу изгнания сердечного
цикла. Если действие сердечных гликозидов уменьшает дилатацию левого
желудочка, то митральная регургитация такого генеза исчезает.
Расширение левого желудочка любого происхождения, в том числе и
не связанное с ишемической болезнью сердца, на определенной стадии
развития приводит к митральной регургитации. Если ишемическую
болезнь сердца осложняет застойная сердечная недостаточность, то
причинами митральной регургитации у таких больных могут быть как гипоксия
сосочковых мышц, так и дилатация полости левого желудочка.
Вторичную легочную венозную гипертензию в результате
связанного с инфарктом миокарда некроза сосочковых мышц левого
желудочка у части больных можно устранить осторожным внутривенным
введением вазодилятаторов (нитроглицерин, нитропруссид натрия),
которые через снижение постнагрузки сердца увеличивают фракцию
изгнания левого желудочка. Бета-один-адреномиметики, повышая силу
сокращений миокарда левого желудочка, также увеличивают объем
Патогенез инфаркта миокарда и кардиогенного шока
325
крови, который он за единицу времени забирает из системы легочной
артерии и выбрасывает в аорту. Рост этого объема ведет к устранению
кардиогенного отека легких.
Из других осложнений инфаркта миокарда следует выделить:
♦ разрыв межжелудочковой перегородки;
♦ разрыв свободной стенки левого желудочка;
♦ аневризму левого желудочка.
Разрыв межжелудочковой перегородки проявляет себя характерным
систолическим шумом, генерируемым турбулентным движением крови
через дефект перегородки из левого желудочка в правый во время
систолы. Максимум звучности шума выявляют при аускультации у левого края
грудины над ее нижней частью (проекция приобретенного дефекта
межжелудочковой перегородки). Шунтирование крови слева направо в
результате разрыва перегородки приводит к росту насыщения кислородом
гемоглобина крови в правом желудочке. Клинико-патофизиологическим
признаком разрыва межжелудочковой перегородки может выступать
превалирование насыщения кислородом гемоглобина крови в правом
желудочке над соответствующим показателем газотранспортной функции
крови в правом предсердии на 5 % и более.
Транзиторного возрастания минутного объема кровообращения при
кардиогенном шоке вследствие постинфарктного разрыва
межжелудочковой перегородки можно достигнуть, снижая преднагрузку левого
желудочка вазодилятаторами (нитроглицерин, нитропруссид натрия) и
увеличивая выброс крови в аорту. В большинстве случаев лишь интрааорталь-
ная контрпульсация позволяет поддерживать минимально достаточное
артериальное давление в период до полной хирургической коррекции
острого приобретенного порока.
Разрыв стенки левого желудочка как осложнение инфаркта миокарда
обычно возникает в течение недели после его развития и в большинстве
случаев приводит к тампонаде сердца. Это осложнение чаще возникает у
больных с артериальной гипертензией, женщин и у пациентов, постоянно
принимающих нестероидные противовоспалительные средства. Лишь пе-
рикардиоцентез и неотложная интрааортальная контрпульсация
позволяют выиграть время до начала хирургического вмешательства.
Клинико-патофизиологическими признаками инфаркта миокарда
правого желудочка выступают падение минутного объема кровообращения до
уровня меньшего, чем 2,5 л-мин'^м"2 при нормальном или несколько
сниженном конечно-диастолическом давлении левого желудочка и диастоли-
ческое давление крови в правом предсердии выше 10 мм рт. ст. Рост
давления крови в правом предсердии во время диастолы - это следствие его
патологически высокой постнагрузки, которую обуславливает возрастание
конечно-систолического объема крови в правом желудочке в результате
недостаточности его насосной функции, связанной с инфарктом миокарда.
Клинические признаки инфаркта миокарда правого желудочка
представляют собой проявления системной венозной гипертензии как
следствия правожелудочковой недостаточности:
326
Глава 20
♦ заметная (усиленная) пульсация яремных вен;
♦ симптом Куссмауля, то есть набухание яремных вен на вдохе;
♦ выявление при аускультации над правыми отделами сердца
третьего и четвертого тонов.
Появление симптома Куссмауля - это результат переполнения кровью
емкостных сосудов в фазу вдоха, когда присасывающее действие
максимально субатмосферного давления в плевральной полости неадекватно
низкой насосной функции правого желудочка. Угнетение насосной
функции правого желудочка как следствие инфаркта его миокарда резко
снижает объем крови, поступающий за единицу времени в легочную
артерию. В результате гидростатическое давление в легочных капиллярах
чаще остается на нормальном уровне или падает, чем растет, как при лево-
желудочковой недостаточности.
Если инфаркт миокарда правого желудочка осложняет артериальная
гипотензия при систолическом артериальном давлении ниже, чем
90 мм рт. ст., то цель патогенетически ориентированной коррекции
снижения АД - это увеличение минутного объема кровообращения через
увеличение преднагрузки левого желудочка. Роста преднагрузки левого
желудочка достигают одновременными внутривенным вливанием плазмозаме-
щающих растворов (увеличение общего венозного возврата к сердцу) и
непрерывной инфузией в вену бета-один-адреномиметиков, направленной на
усиление насосной функции правого желудочка. При этом давление
заклинивания в легочной артерии не должно быть выше 15-18 мм рт. ст.
Больные, выжившие в остром периоде после инфаркта миокарда,
подвержены развитию застойной сердечной недостаточности, прогрессирова-
нию ишемической болезни сердца и циркуляторной гипоксии миокарда, его
повторному инфаркту, сердечным аритмиям, в том числе и как причинам
внезапной сердечной смерти. Вероятность внезапной сердечной смерти,
связанной с инфарктом миокарда, наиболее высока в период первых шести
месяцев после критического обострения циркуляторной гипоксии миокарда,
приведшей к опасному падению насосной функции сердца (инфаркту
миокарда). Преклонный возраст больных выступает фактором риска внезапной
смерти, который не зависит от проводимых лечебно-профилактических и
реабилитационных мероприятий. Наиболее подверженный благоприятным
изменениям под влияниям лечебно-профилактических воздействий фактор
риска внезапной сердечной смерти после инфаркта- это низкая насосная
функция левого желудочка, о которой судят, определяя его фракцию
изгнания или конечно-систолический объем. К маркерам высокого риска
внезапной сердечной смерти в период реабилитации после инфаркта относят:
♦ грудную жабу в условиях покоя;
♦ циркуляторную гипоксию миокарда при дозированной физической
нагрузке, которую выявляют специальными исследованиями и при
отсутствии стенокардии;
♦ усиление обтурации венечных артерий атеросклеротического генеза;
♦ сердечные аритмии при эктопических водителях ритма,
локализованных в желудочках;
Патогенез инфаркта миокарда и кардиогенного шока
327
♦ сниженная вариабельность временных интервалов R-R на
электрокардиограмме как свидетельство нарушений регуляции сердечной
деятельности автономной нервной системой;
♦ курение;
♦ гиперхолестеринемия;
♦ сахарный диабет.
Следует заметить, что, несмотря на высокую информативность
радиоизотопных методов исследования ишемических повреждений сердца,
если они не выявляют признаков дисфункции и цитолиза кардиомиоцитов
при клинических признаках циркуляторной гипоксии миокарда и ее
электрокардиографическом эквиваленте, то у больных в период реабилитации
после инфаркта миокарда риск внезапной сердечной смерти все равно
считают высоким.
Первые три из маркеров высокого риска внезапной сердечной смерти,
связанной с инфарктом миокарда, представляют собой причины и
следствия усиления циркуляторной гипоксии кардиомиоцитов, обусловленной
атеросклеротическим поражением венечных артерий.
Четвертый маркер свидетельствует о тенденции сердца как органа-
эффектора функциональных систем к дезинтеграции вследствие
ишемических повреждений клеточных элементов сердца, участвующих в
регуляции сердечной деятельности на органном уровне, в частности
проводящих кардиомиоцитов.
Выявление пятого маркера говорит о высвобождении сердца из-под
системных регуляторных влияний как предпосылке внезапной сердечной
смерти вследствие атеросклероза и ишемии.
Последние три маркера - это известные факторы риска атеросклероза
и инфаркта миокарда.
Падение насосной функции левого желудочка как первичное звено
патогенеза синдрома малого сердечного выброса в остром периоде
инфаркта миокарда в дальнейшем естественно превращается в фактор риска
внезапной сердечной смерти в период реабилитации. Поэтому, наиболее
значимые из маркеров риска внезапной сердечной смерти в течение шести
месяцев после инфаркта миокарда - это клинические признаки низкой
насосной функции левого желудочка:
♦ влажные хрипы над верхними и средними отделами легких как
симптом легочной венозной гипертензии вторичной по отношению
к падению систолического и диастолического компонентов
насосной функции левого желудочка во время нахождения больного в
отделении кардиологической интенсивной терапии;
♦ падение фракции изгнания левого желудочка в этот период до
уровня меньшего, чем 40 %.
Глава 21
ПАТОГЕНЕЗ НАИБОЛЕЕ ЧАСТЫХ ВИДОВ
АРТЕРИАЛЬНОЙ ГИПЕРТЕНЗИИ
Любой длительный подъем артериального давления выше верхнего
предела нормальных колебаний (табл. 21.1) (артериальная гипертензия)
может быть причиной заболеваний и патологических состояний сердца и
сосудов. Известно, что вероятность развития ишемической болезни
сердца, скрытых или явных сердечной и почечной недостаточности, а также
нарушений мозгового кровообращения у взрослых людей с диастоличе-
ским артериальным давлением (АД) выше 90 мм рт. ст. в два раза выше,
чем при диастолическом АД ниже 80 мм рт. ст.
Артериальная гипертензия - это патологическое состояние
организма взрослых, обусловленное длительным подъемом систолического
АД выше уровня в 140ммрт. ст. и диастолического АД выше
90ммрт. ст.
Если диастолическое АД ниже 90 мм рт. ст., а систолическое АД
выше 160 мм рт. ст., то такую артериальную гипертензию определяют
как изолированную систолическую артериальную гипертензию.
Долгое время рост общего периферического сосудистого
сопротивления (ОПСС) считали ведущим звеном патогенеза первичной
артериальной гипертензии (эссенциальной гипертензии, гипертонической
болезни) у подавляющего большинства больных. В этой связи подъем
диастолического АД, находящегося в прямой связи с ОПСС,
признавали более неблагоприятным, чем рост систолического артериального
давления. Поэтому степень тяжести артериальной гипертензии
оценивали, ориентируясь на уровень диастолического артериального
давления (табл. 21.2).
В последние десятилетия была выявлена достоверная положительная
связь высокого уровня систолического АД с риском осложнений
артериальной гипертензии и связанных с нею заболеваний. Изолированная
систолическая артериальная гипертензия у больных старше 65 лет особенно
часто приводит к опасным осложнениям и болезням, причем в настоящее
время нет эффективного способа ее коррекции. Поэтому тяжесть
артериальной гипертензии определяют в соответствии с данными исследования
как систолического, так и диастолического АД (табл. 21.3).
При артериальной гипертензии в стадии предболезни
фармакотерапию не применяют, стремясь устранить артериальную гипертензию
рациональным усилением физической активности, диетой, психотерапией и
полноценным отдыхом. Большинство клиницистов-исследователей
сходятся во мнении, что фармакокоррекцию артериальной гипертензии
следует начинать при устойчивом подъеме диастолического АД выше
95 мм рт. ст.
Патогенез наиболее частых видов артериальной гипертензии
329
Таблица 21.1
Верхние пределы нормальных колебаний АД
Возраст
3-5 лет
6-9 лет
10-12 лет
13-15 лет
Взрослые
Систолическое АД,
мм рт. ст.
116
122
126
136
140
Диастол и чес кое АД,
мм рт. ст^
76
78
82
86
90
Таблица 21.2
Классификация тяжести артериальной гипертензии
в зависимости от уровня диастолического АД
Степень
тяжести
I
II
III
Определение степени
тяжести
Незначительная, «мягкая»
Умеренная
Тяжелая
Диастолическое АД,
мм рт. ст.
90-104
105-119
выше 120
Таблица 21.3
Классификация тяжести артериальной гипертензии
Степень тяжести
Артериальная гипертензия в
стадии «предболезни»
Незначительная
Умеренная
Тяжелая
Крайне тяжелая
Систолическое АД,
мм рт.ст.
130-139
140-159
160-179
180-209
Не менее 210
Диастолическое АД,
мм рт. ст.
85-95
90-99
100-109
110-119
Не менее120
Причина длительного и устойчивого подъема АД выше верхнего
предела диапазона нормальных колебаний у 95 % больных в возрасте от 18
до 65 лет обычно остается не вполне ясной. Таких пациентов считают
страдающими от гипертонической болезни, то есть первичной или эссен-
циалъной (лат. essentia - сущность) артериальной гипертензии (АГ). При
эссенциальной АГ длительный и патологический подъем АД
представляет собой первичное звено патогенеза болезни, саму ее сущность, а не
является следствием какого-либо другого заболевания. Вторичная
артериальная гипертензия представляет собой следствие заболеваний и
патологических состояний с вполне ясными этиологией и патогенезом. Среди
всех случаев артериальной гипертензии фиксируют следующую частоту
видов вторичной АГ:
♦ АГ как следствие заболеваний паренхимы почек - 3-4 % ;
♦ почечная сосудистая гипертензия - 0,5-1 %;
330
Глава 21
♦ АГ, связанная с гиперфункцией надпочечников (феохромоцитома,
первичный альдостеронизм и синдром Кушинга) - 0,1-0,3 %;
♦ вторичная АГ в силу других причин - 0,1-0,3 %.
Тяжелая и крайне тяжелая артериальная гипертензия часто является
вторичной относительно заболеваний почек. У 33 % таких больных
выявляют почечную сосудистую (вазоренальную) гипертензию.
У 5 % женщин, постоянно принимающих контрацептивы, содержащие
эстрогены, через 5 лет от момента начала их использования диастолическое
АД становится выше 90 мм рт. ст. Патогенез такой вторичной АГ остается
неясным. Полагают, что эстрогены ускоряют реализацию наследственной
предрасположенности к первичной гипертензии и обостряют скрытые
дисфункции почек, которые становятся причинами вторичной АГ.
Если АГ развивается в препубертатном периоде у детей, то ее
вызывают заболевания с патологическими изменениями паренхимы почек, или
она является вазоренальной. В постпубертатном периоде у детей
артериальная гипертензия в большинстве случаев первична, но частота
вторичной артериальной гипертензии в данный период онтогенеза все равно
остается более высокой, чем у взрослых.
ПАТОГЕНЕЗ ГИПЕРТОНИЧЕСКОЙ БОЛЕЗНИ
Артериальное давление представляет собой прямую функцию
произведения минутного объема кровообращения (МОК) и общего
периферического сосудистого сопротивления (ОПСС). Непосредственными
причинами патогенного роста артериального давления служат рост МОК и
(или) подъем ОПСС. Между МОК и ОПСС существует регуляторная
взаимозависимость, которая часто реализуется по принципу отрицательной
обратной связи. Например, у многих больных с застойной сердечной
недостаточностью действие ингибиторов ангиотензин-превращающего фермента
ведет к росту МОК, снижая общее периферическое сосудистое
сопротивление и увеличивая фракцию изгнания левого желудочка.
Функциональная система, конечным полезным приспособительным
результатом которой является адекватный потребностям организма
уровень АД, на пути достижения своего конечного результата как
промежуточных полезных результатов достигает оптимальной величины МОК и
непатогенного уровня ОПСС, соответствующих потребностям органов,
тканей и клеток на периферии.
Если первичный патогенный сдвиг как этиологический фактор
гипертонической болезни происходит в какой-либо из двух систем, или в
системе поддержания адекватного потребностям организма МОК, или в
системах регуляции, ответственных за оптимальный уровень ОПСС, то
он всегда приводит к изменениям функционирования и дисфункциям
другой системы. Вот почему первоначальные патогенные изменения
регуляции и эффекторов как причина эссенциалъной АГ часто остаются не
Патогенез наиболее частых видов артериальной гипертензии
331
вполне ясными, и патогенез первичной артериальной гипертензии при
возникновении взаимосвязанных патологических изменений МОК и ОПСС
представляет собой уже не специфический механизм болезни, а типовой
эндогенизированный патологический процесс.
У большинства пациентов с эссенциальной АГ ведущим звеном ее
патогенеза следует признать прогрессирующий от транзиторного до
патогенно постоянного повышенный уровень ОПСС. У меньшей части
больных всех возрастных групп эссенциальная АГ представляет собой
следствие не имеющего биологического смысла возрастания МОК. На
этапе развития эссенциальной гипертензии, который можно определить
как стадию предболезни первичной АГ, ее признаком у больных в
возрасте до 30 лет считают отсутствие реакции снижения ОПСС в ответ на
физическую нагрузку. Во время физической нагрузки у людей без
предрасположенности к гипертонической болезни ОПСС снижается для
удовлетворения потребности в росте объемной скорости кровотока на
периферии. У молодых людей с предрасположенностью к
гипертонической болезни ОПСС остается на патологически высоком уровне,
который можно признать нормальным только для условий покоя. При
усилении АГ по мере старения у больных с эссенциальной артериальной ги-
пертензией патологически высокий уровень ОПСС все чаще и чаще
представляет собой основную причину подъема АД. По мере прогресси-
рования гипертонической болезни по ходу онтогенеза, когда АГ
становится постоянно умеренной или тяжелой, в 70 % случаев артериальная
гипертензия представляет собой следствие стойкого аномального
возрастания ОПСС. При этом у пожилых больных МОК в условиях покоя
может быть патологически снижен, и нередко возникает застойная
сердечная недостаточность. В данном случае причиной падения насосной
функции сердца является постоянно повышенная постнагрузка левого
желудочка.
Основные этапы развития гипертонической болезни в соответствии с
ее нейрогенной теорией можно описать следующим образом:
♦ отрицательный психоэмоциональный стресс расстраивает внутри-
центральные отношения таким образом, что на супрасегментном
уровне автономной нервной системы возникает устойчивое и
повышенное возбуждение симпатических центров;
♦ на периферии через нервную симпатическую стимуляцию
сосудистой стенки и гиперкатехоламинемию устойчивое возбуждение
симпатических центров приводит к спазму сосудов сопротивления
и постоянно высокому уровню ОПСС , который обуславливает АГ;
♦ высокая интенсивность и длительность усиленного сокращения
гладкомышечных элементов стенки резистивных сосудов ведут к
росту потребления свободной энергии их миоцитами, что служит
стимулом для гипертрофии последних;
♦ гипертрофия миоцитов стенки сосудов сопротивления служит
одной из причин ее утолщения, которое сужает просвет резистивных
сосудов;
332
Глава 21
♦ сужение сосудов сопротивления придает высокому уровню ОПСС
фиксированный характер и делает АГ необратимой;
♦ когда сужение сосудов сопротивления во всем организме
захватывает в соответствующей мере и приводящие артериолы почечных
нефронов, АГ становится не только нейрогенной и связанной с
гипертрофией стенок сосудов сопротивления, но и почечной
сосудистой артериальной гипертензиещ часто приобретая
злокачественный характер.
Нейрогенная теория как концепция прогрессирования
гипертонической болезни по мере старения вполне адекватна обычному развитию эс-
сенциальной артериальной гипертензии по ходу онтогенеза (схема. 21.1).
Устойчивая активация супрасегментарных симпатических центров как
следствие патогенного отрицательного эмоционального стресса и
злоупотребления спиртным (алкоголь обладает центральным прессорным
действием) повышает АД не только через спазм сосудов сопротивления.
Кроме того, преобладание на системном уровне адренергической
стимуляции вызывает констрикцию емкостных сосудов, что повышает общий
венозный возврат к сердцу и соответственно МОК.
Воздействие на организм факторов
социо-экологической среды
Наследственная
предрасположенность
Эссенциальная артериальная
гипертензия
Возраст больных,
лет
В стадии предболезни
Ранняя
Установившаяся
0-30
20-40
30-50
Неосложненная
w Осложненная
Ускоренное прогрес-
сирование вплоть до
злокачественной АГ
Сердечная
Гипертрофия левого
желудочка
Застойная сердечная
недостаточность
Инфаркт миокарда
Почечная
Нефросклероз
Почечная
недостаточность
Сосудистая
Расслаивающая аневризма
аорты
Мозговая
Нарушения
мозгового
кровообращения
Кровоизлияния
Тромбоз
Схема 21.1. Прогрессирование первичной артериальной гипертензии
по мере старения
Патогенез наиболее частых видов артериальной гипертензии
333
Устойчивая активация симпатического отдела автономной нервной
системы ведет к активации ренин-ангиотензин-альдостеронового
механизма, что еще в большей степени усиливает спазм сосудов
сопротивления и вызывает АГ, задерживая в организме натрий и повышая объем
внеклеточной жидкости. Рост действующей концентрации ангиотензинов
в циркулирующей крови через их супрасегментарное действие
потенцирует активацию и без того уже активированных симпатических центров.
Усиление спазма под влиянием ангиотензинов ускоряет гипертрофию
гладкомышечных элементов резистивных сосудов как причину сужения
их просвета и необратимой (установившейся) АГ.
Высокая активность ренина в плазме крови характерна не для всех
больных с эссенциальной АГ. У 40 % больных с установившейся
первичной АГ активность энзима находится в нормальных пределах или даже
несколько снижена. У таких больных бессолевая диета, снижающая
объем внеклеточной жидкости, обычно приносит хороший результат,
уменьшая тяжесть АГ. При нормальной активности ренина в плазме крови у
таких пациентов выявляют рост секреции альдостерона, снижение
кровотока в почках, задержку в организме натрия и усиленную реакцию сосудов
сопротивления на влияния ангиотензина II как вазоконстриктора. У 10 %
больных с ранней эссенциальной АГ определяют повышенную
активность ренина в плазме крови, но артериальная гипертензия при этом не
является вазоренальной.
Избыточное поступление натрия хлорида в организм с пищей и
напитками повышает МОК, увеличивая содержание натрия в организме как
основную детерминанту объема внеклеточной жидкости и плазмы крови.
Рост секреции аргинин-вазопрессина как элемент патогенного стресса
и следствие активации ренин-ангиотензиновой системы также признают
звеном патогенеза эссенциальной гипертензии.
Системные сдвиги регуляции, которые ведут к артериальной
гипертензии у больных с первичной АГ, приводят к ней, несмотря на активацию
стресс-лимитирующих систем на всех уровнях. Так АГ у пациентов с
гипертонической болезнью развивается, несмотря на усиленную секрецию
предсердного натрийуретического пептида и интенсификацию
функционирования калликреин-кининовой системы. Подавление
стресс-лимитирующих систем различных уровней системными однонаправленнными и
интенсивными патогенными регуляторными влияниями представляет собой
одну из закономерностей развития болезней и патологических состояний.
Известна наследственная предрасположенность к гипертонической
болезни. Если один из родителей страдал от заболевания, то его риск у детей
возрастает в шесть раз в сравнении с риском развития первичной АГ у
людей, родители которых не страдали от эссенциальной гипертензии.
Полагают, что наследственный фактор в развитии гипертонической
болезни реализует себя через генетически детерминированные дефекты
трансмембранного переноса ионов, которые обуславливают рост
содержания ионизированного кальция в цитозоле миоцитов стенки
резистивных сосудов. На уровне почек и всего организма определяемые геноти-
334
Глава 21
пом нарушения трансмембранного переноса ионов ведут к задержке во
внутренней среде натрия, росту преднагрузки сердца и устойчивому
усиленному спазму сосудов сопротивления, которые и служат причинами
АГ. Рост содержания свободного кальция в гладкомышечных элементах
сосудистой стенки повышает степень сокращения и сократительную
способность миоцитов стенки сосудов, что связывают с изменениями
активности переноса кальция через наружную и другие клеточные мембраны
посредством функционирования Са2+-АТФазы. Данная ионная помпа,
которая выводит ионизированный кальций в межклеточные пространства,
представляет собой связанную с кальмодулином Са2+-АТФазу. В
настоящее время ведутся поиски той патогенной мутации, которая может лежать
в основе аномально низкой активности изоформ Са2+-АТФазы как причин
артериальной гипертензии.
Генетический фактор в развитии эссенциальной АГ может реализовать
себя и через патогенную экспрессию генома эндотелиоцитов, при которой
снижено образование и высвобождение ими эндогенных вазодилятаторов
(оксид азота, простациклин, другие простагландины-вазодилятаторы и пр.).
Выявить на молекулярном уровне причину первичной артериальной
гипертензии как полигенной болезни представляется весьма затруднительным.
Также затруднительно определить этиологию на уровне ДНК таких
широкораспространенных мультифакториальных болезней человека как сахарный
диабет, ишемическая болезнь сердца и ожирение. Трудности в попытках
связать особенности определенных локусов генома человека с развитием
гипертонической болезни связаны с отсутствием у болезни фенотипической
специфичности, с генетической гетерогенностью ее этиологии, а также с
постепенностью изменений на молекулярном уровне структурно-функциональной
организации организма, которые приводят к гипертонической болезни.
Полагают, что во многом генетическая предрасположенность к
первичной артериальной гипертензии связана с особенностями строения гена
ангиотензиногена (АТГ). О связи особенностей гена АТГ и более высокой
предрасположенности к эссенциальной гипертензии у больных
гипертонической болезнью, чем у представителей основной популяции, говорят
следующие факты:
♦ достоверная связь первичной артериальной гипертензии, от которой
страдают сиблинги, и повышенной концентрацией у них в крови
ангиотензиногена (маркер повышенной экспресии генов АТГ);
♦ статистически значимая положительная связь между между заменой
метионина в 235 позиции молекулы АТГ на треонин как маркера
экспресии соответствующих аллелей генов АТГ (аллель Т235) и
первичной артериальной гипертензии;
♦ достоверная связь между геном АТГ, содержащим две Т235,
(больные гомозиготные по Т235) с более высокой концентрацией АТГ в
циркулирующей крови. Т235 в настоящее время считают маркером
не только высокой предрасположенности к гипертонической
болезни, но и фактором риска других видов АГ, в том числе и эклампсии
беременных.
Патогенез наиболее частых видов артериальной гипертензии
335
Вторичный ги-
перальдостеро-
низм
Задержка в
организме
натрия
Длительное
действие стимулов
патологического
эмоционального стресса
Избыточное
потребление
натрия
хлорида с пищей и
напитками
Генетически
детерминированное снижение
фильтрационной
поверхности почек по
ходу онтогенеза
Спазм емкостных
сосудов
—ж
Увеличение
сократимости
миокарда
Увеличение объема
внеклеточной жидкости
Возрастание частоты
сердечных
сокращений
Т
Устойчивая активация
симпатического отдела
автономной нервной системы
Рост пред нагрузки
сердца
Увеличение минутного объема
кровообращения
Возникающая по ходу
онтогенеза генетически
детерминированная
недостаточность секреции
эндотелиоцитами
эндогенных вазодилятаторов
Активация ренин-
ангиотензин-альдосте-
ронового механизма
Эссенциальная артериальная
гипертензия
Рост общего периферического
сосудистого сопротивления
Спазм резистивных
сосудов
Гипертрофия стенки
сосудов сопротивления
Наследственная и (или) приобретенная
предрасположенность структуры и
функций наружной клеточной
мембраны миоцитов к реализации вазоконст-
рикторных регуляторных влияний
Генетически
детерминированная или
приобретенная повышенная
секреция факторов
клеточного роста
эндотелиоцитами
Связанная
с
ожирением гипер-
инсулине-
мия
Схема 21.2. Синтетическая схема патогенеза гипертонической болезни
(среднее артериальное давление представляет собой прямую функцию произведения
минутного объема кровообращения и общего периферического сосудистого сопротивления;
причиной интенсификации продукции активированными эндотелиоцитами факторов роста
может быть атеросклероз)
336
Глава 21
Недавно были высказаны предположения о том, что треонин в 235
позиции представляет собой просто маркер другой аномалии на уровне
генетического материала, которая приводит к повышенной экспрессии генов
АТГ. Есть основания полагать, что такая аномалия была идентифицирована
и приводит к замене аденином гуанина в шестой позиции по направлению
транскрипции в области промотера гена. Данная мутация обуславливает
повышенный уровень базальной экспресии генов АТГ. Это повышает
концентрацию АТГ в крови. Увеличенная концентрация АТГ предрасполагает
к росту артериального давления через повышенное образование ангиотен-
зина II при реципрокных системной адренергической стимуляции и
активации ренин-ангиотензин-альдостеронового механизма.
Ожирение предрасполагает к гипертонической болезни. Гиперинсули-
немия у больных с ожирением вызывает гипертрофию миоцитов
сосудистой стенки как причину АГ через усиление вхождения в них аминокислот
и калия. АГ, атеросклероз и гиперлипопротеинемии представляют собой
факторы риска ишемической болезни сердца и инфаркта миокарда, причем
вероятность инфаркта особенно велика, когда нарушения обмена липопро-
теинов выявляют одновременно с АГ. Между нарушениями липидного
обмена, вызывающими атеросклероз, и АГ существует достоверная связь. У
больных с гипертонической болезнью и у родственников пробанда с эссен-
циальной гипертензией выявляют значительное увеличение содержания в
сыворотке крови холестерина, связанного с липопротеинами низкой
плотности, и липопротеинов очень низкой плотности. Это позволило отдельно
выделить синдром семейной дислипидемической АГ.
Факторы риска (условия развития, этиологические факторы) эссенци-
альной АГ хорошо известны: курение, ожирение, алкоголизм,
отрицательный психоэмоциональный стресс, а также избыточное потребление
натрия хлорида с пищей.
Достоверные сведения об этиологии, факторах риска и патогенезе
гипертонической болезни легли в основу интегративной схемы ее
патогенеза (схема 21.2).
ВТОРИЧНАЯ АРТЕРИАЛЬНАЯ ГИПЕРТЕНЗИЯ
Вторичная артериальная гипертензия (табл. 21.4) представляет собой
следствие вполне определенных болезней и патологических состояний.
При этом этиология и ведущие звенья патогенеза болезней, вызывающих
вторичную АГ, обычно являются вполне ясными.
Любое заболевание, повреждающее паренхиму почек, через падение
их экскреторной функции может привести к задержке в организме натрия,
воды и ее следствиям: росту объемов внеклеточной жидкости (ОВнЖ),
плазмы крови и артериальной гипертензии. Кроме того, болезни почек с
патологическими изменениями их паренхимы вызывают артериальную
гипертензию, нарушая кровоснабжение клеток юкстагломерулярного ап-
Патогенез наиболее частых видов артериальной гипертензии
337
Таблица 21.4
Частота видов вторичной артериальной гипертензии
среди всех случаев ЛГу больных
Причины АГ
Заболевания паренхимы почек: инфекционно-
интерстициальные, паренхиматозные, тубулопатии
Сужение просвета почечных артерий (вазоренальная АГ)
Первичный альдостеронизм
Феохром оцитома
Синдром Кушинга
Как результат побочного действия лекарственных средств
АГ в силу других причин
Примерная частота
3-5%
1-4%
0,3%
0,1 %
0,5%
0,5-1 %
0,5-1 %
парата нефронов и тем самым активируя ренин-ангиотензин-альдосте-
роновый механизм. Злокачественная (тяжелая) артериальная гипертензия,
повреждая приводящие артериолы нефронов, снижает скорость клубоч-
ковой фильтрации и усиливает высвобождение ренина в плазму крови. В
этой связи гипертоническую болезнь и длительную тяжелую вторичную
АГ следует рассматривать как заболевание (патологическое состояние),
прогрессирование которого до злокачественной артериальной
гипертензии приводит к эндогенизации АГ как типового патологического процесса
вследствие дисфункций почек. У больных с повреждениями паренхимы
почек вследствие тяжелой и (или) злокачественной АГ особенно трудно
выявить первоначальную причину артериальной гипертензии. Следует
заметить, что тяжелая артериальная гипертензия ускоряет
прогрессирование заболеваний и патологических состояний паренхимы почек, в
особенности обусловленных сахарным диабетом.
По мере падения экскреторной функции почек и связанного с ним роста
объема внеклеточной жидкости все более эффективными способами
снижения уровня АГ становятся бессолевая диета и назначение диуретиков,
влияющих на клетки петли Генле канальцев нефрона. Эти лечебные меры
направлены в первую очередь на снижение ОВнЖ, то есть на основное
звено патогенеза АГ вследствие заболеваний паренхимы почек. Следует
учитывать, что избыточное выделение с мочой натрия в результате действия
мочегонных средств может быть причиной преренальной азотемии. У
некоторых больных с нефропатиями, вызывающими АГ, АД снижается лишь
в результате гемодиализа, то есть искусственного натрийуреза.
Кроме задержки в организме натрия и активации ренин-ангиотензин-
альдостеронового механизма, АГ вследствие заболеваний паренхимы почек
вызывается действием ренопривного патогенетического механизма.
Действие ренопривного механизма развития вторичной АГ - это результат
снижения образования почками агентов гуморальной регуляции, снижающих
ОПСС и АД и предотвращающих избыточную активацию ренин-ангиотен-
зин-альдостеронового механизма: простагландинов Еь Е2, А2, фосфоли-
пидного ингибитора ренина, простациклина 12, каллидина, брадикинина.
338
Глава 21
Почечная сосудистая гипертензия представляет собой следствие
снижения объемной скорости кровотока в обеих почках и микрососудах,
по которым кровь поступает к клеткам юкстагломерулярного аппарата
клубочков нефрона; при этом снижение скорости кровотока
представляет собой стимул активации ренин-ангиотензин-алъдостеронового
механизма.
Снижение перфузионного давления в нефронах как результат сужения
сосудов в системе почечной артерии (табл. 21.5) стимулирует
высвобождение ренина. Ренин служит катализатором образования ангиотензина-1
из ангиотензиногена. Ангиотензин-превращающий фермент,
локализованный на эндотелиальных клетках, - это энзим, образующий ангиотен-
зин II из ангиотензина I. Мощный эндогенный вазоконстриктор ангиотен-
зин II усиливает задержку почками в организме натрия, прямо влияя на
его реабсорбцию в канальцах нефронов и оказывая на нее опосредованное
влияние через увеличение секреции альдостерона.
Таблица 21.5
Причины обструкции-окклюзии почечной артерии
и реноваскулярной АГ
Почечные
♦ Атеросклероз почечной артерии и ее ветвей
♦ Фибромышечная дисплазия стенок почечной
артерии
♦ Опухоли
♦ Эмболия
♦ Васкулит (нодозный периартериит и
системная красная волчанка)
♦ Кистозное перерождение почек
♦ Травма
♦ Гематома внутри фиброзной капсулы почек
Внепочечные
♦ Забрюшинные опухоли
♦ Изменения по типу фиброза тканей,
локализованных ретроперитонеаль-
но (перинефрит, склеродерма)
♦ Околопочечная гематома
Гольдблат в своем классическом опыте после экспериментальной
частичной окклюзии лигатурой общей почечной артерии одной почки («А»)
фиксировал у собак устойчивую артериальную гипертензию и
перерождение второй почки («Б»).
Вследствие частичной обструкции общей почечной артерии «А»
падает кровоснабжение юкстагломерулярных аппаратов данной почки, что
служит стимулом для секреции ренина. Активация ренин-ангиотензино-
вой системы вызывает артериальную гипертензию. Через некоторое
время в паренхиме другой почки возникают изменения, характерные для
злокачественной гипертензии. В их основе лежит склероз почечных арте-
риол (артериолонефросклероз) и артериальных почечных сосудов
небольшого диаметра. Артериолонефросклероз в почке «Б» вызывают
высокие действующие концентрации ангиотензинов и альдостерона. Почка
«А» меньше подвержена воздействию такой эндотоксемии, так как в нее
из-за окклюзии артерии поступает меньше ангиотензинов и альдостерона.
Патогенез наиболее частых видов артериальной гипертензии
339
Артериолонефросклероз в «Б» через снижение кровоснабжения юкстагло-
мерулярных аппаратов повышает высвобождение ренина в кровь
гранулярными клетками «Б». Почечная сосудистая АГ достигает особо
высокого уровня при одновременной максимальной секреции ренина как почкой
«А», так и «Б». По мере развития патологического перерождения почки
«Б» может возникнуть краткий «светлый» промежуток со снижением АД.
Это связано с уменьшением высвобождения в кровь ренина юкстагломе-
рулярными аппаратами «Б» по мере снижения в ней вследствие артерио-
лонефросклероза массы функционально активной почечной паренхимы. В
дальнейшем артериальная гипертензия может возникнуть снова, но уже
как следствие почечной недостаточности.
Почечная сосудистая АГ часто бывает злокачественной, так как в ее
развитии одновременно задействованы все возможные из основных
механизмов подъема АД:
♦ спазм сосудов сопротивления под влиянием высокой действующей
концентрации ангиотензинов в циркулирующей крови;
♦ нейрогенный рост тонуса резистивных сосудов как результат
возбуждения симпатических центров, которые активируются
вследствие супрасегментарного эффекта ангиотензинов;
♦ увеличение преднагрузки сердца в результате роста объема
внеклеточной жидкости, который растет вследствие увеличения
концентрации альдостерона в циркулирующей крови;
♦ рост сократимости сердца и частоты сердечных сокращений из-за
усиления адренергической стимуляции сердца.
Феохромоцитома - это опухоль, состоящая из хромаффинных клеток,
секретирующих катехоламины. В организме хромаффинные клетки
представлены в мозговом веществе надпочечников, в симпатических ганглиях
и в органе Цукерхандля, скоплении хромаффинных клеток, лежащем
спереди бифуркации аорты. В 90 % случаев феохромоцитома локализована в
мозговом веществе надпочечников, являясь у 20 % больных двусторонней
опухолью. Экстраабдоминальные варианты локализации феохромоцито-
мы выявляют редко, у 1-2 % больных. Надпочечниковые феохромоцито-
мы преимущественно секретируют адреналин, тогда когда аналогичные
опухоли другой локализации в основном выделяют норадреналин. За
возникновение опухоли у 6 % больных с феохромоцитомой ответственна
доминирующая аллель гена болезни. В таких случаях феохромоцитома
проявляет себя в раннем возрасте артериальной гипертензией и другими
симптомами. Примерно у 40 % больных с наследственной
предрасположенностью к феохромоцитоме возникновение этого новообразования
выступает элементом синдрома множественной эндокринной неоплазии
второго типа (синдром Сиппла), который, кроме феохромоцитомы,
характеризуют злокачественная медуллярная опухоль щитовидной железы,
аденома или гиперплазия паращитовидных желез и, у небольшой части
больных, синдром Кушинга.
В основном постоянные патологические сдвиги у больных с
феохромоцитомой связаны с хронически повышенным уровнем концентрации кате-
340
Глава 21
холаминов в циркулирующей крови. К ним относят высокое потребление
кислорода организмом со снижением массы тела, избыточное
потоотделение и периодическое возрастание температуры тела до субфебрильной. Ор-
тостатическая артериальная гипотензия у больных с феохромоцитомой -
это результат снижения объема плазмы крови из-за миграции жидкости в
интерстициальный жидкостной сектор из сосудистого под влиянием
изменения соотношения между пре- и посткапиллярным сопротивлением на
системном уровне. Это соотношение меняется вследствие гиперкатехола-
минемии. Кроме того, объем плазмы крови снижается из-за избыточного
диуреза, который повышает гиперкатехоламинемия. К ортостатической ги-
потензии у больных с феохромоцитомой предрасполагает падение
чувствительности адренорецепторов стенки сосудов сопротивления в результате
постоянно повышенного содержания катехоламинов в циркулирующей
крови. Постоянно повышенный уровень адренергической стимуляции
стенки артерий и резистивных сосудов проявляет себя снижением наполнения
пульса и температуры кожных покровов нижних конечностей. Некоторые
пациенты предъявляют жалобы на абдоминальные колики, тошноту и
рвоту. Постоянно повышенный уровень возбуждения бета-один-адрено-
рецепторов в сердце как результат хронической гиперкатехоламинемии
служит причиной концентрической гипертрофии миокарда. По мере про-
грессирования гипертрофии падает доставка кислорода по микрососудам к
кардиомиоцитам, что может быть причиной стенокардии.
Следует заметить, что прежде всего о развитии феохромоцитомы
свидетельствуют периодические пароксизмы АГ, которые сопровождаются
головными болями, сердцебиениями и страхом смерти. Эти приступы
напоминают состояние острого патогенного отрицательного
психоэмоционального стресса (паника и т.д.) или гипогликемии, которая служит
стимулом для предельной симпатической активации. Провоцировать
пароксизмы могут физические упражнения, мочеиспускание, дефекация, пик
полового сношения, инъекции глюкагона или гистамина, анестезия,
действие опиоидов, курение и беременность.
Центральный альфа-два-адреномиметик клонидин (клофелин),
воздействуя на вазомоторный центр головного мозга, снижает интенсивность
симпатической эфферентации на периферию. В результате минутный
объем кровообращения становится меньше, но главный эффект клониди-
на состоит в уменьшении ОПСС и снижении степени тяжести АГ через
ослабление адренергической стимуляции сосудистой стенки. У здоровых
людей и у больных, гиперкатехоламинемия у которых возникает из-за
состояния повышенной тревожности, клонидин снижает концентрацию
катехоламинов в циркулирующей крови, в частности, вследствие
уменьшения симпатической нервной стимуляции мозгового вещества
надпочечников. У больных с феохромоцитомой секреция катехоламинов
«автономна», то есть не зависит от системных регуляторных влияний. Поэтому
после приема клонидина у больного с феохромоцитомой содержание
катехоламинов в плазме крови не снижается. Для выполнения «клонидиновой
пробы» больному в положении лежа назначают 0,3 мг клонидина внутрь,
Патогенез наиболее частых видов артериальной гипертензии
341
предварительно взяв пробу крови для определения концентрации в
плазме свободных катехоламинов. Если через 3 ч после приема клонидина
концентрация свободных катехоламинов в плазме крови меньше, чем
500 пг/мл, то с вероятностью в 97 % можно утверждать, что причина АГ
не связана с феохромоцитомой.
Альдостеронизм (гиперальдостеронизм) - это патологическое
состояние, которое обусловлено повышенной секрецией минералкортикоидов как
причиной максимального уровня реабсорбции натрия в канальцах нефрона,
роста объема внеклеточной жидкости и артериальной гипертензии.
Первичный альдостеронизм - это не следствие повышенной секреции
ренина и роста концентрации ангиотензинов в циркулирующей крови. Его
причина - избыточная и не ограниченная системной регуляцией секреция
альдостерона клетками аденомы надпочечников или обладающего
свойствами минералкортикоида кортизола обеими надпочечниками при их
гипертрофии. Гипертрофия надпочечников приводит к первичному альдостеро-
низму у больных с синдромом Кушинга (см. главу, посвященную
патофизиологии эндокринопатий). Вторичный альдостеронизм- это всегда
конечный результат активации ренин-ангиотензин-альдостеронового
механизма. В частности вторичный альдостеронизм развивается в ответ на
сниженное поступление натрия в организм с пищей для сохранения объема
внеклеточной жидкости посредством активации ренин-ангиотензин-
альдостеронового механизма. Этот механизм активируется, приводя ко
вторичному альдостеронизму, и у больных с застойной сердечной
недостаточностью. Причины активации ренин-ангиотензин-альдостеронового
механизма у больных с застойной сердечной недостаточностью- это адре-
нергическая стимуляция паренхимы почек (возбуждение бета-один-адрено-
рецепторов почек), а также спазм приводящих артериол нефрона.
Диагноз первичного альдостеронизма подтверждает выявление потерь
калия из организма в составе конечной мочи большее, чем 30 ммоль за
сутки, без каких-либо ограничений приема натрия с пищей и без
избыточного потребления с ней и напитками калия. Если после выявления ги-
покалиемии концентрация альдостерона в плазме крови превышает
уровень в 20 нг/дл при низкой активности в ней ренина (меньше 1 нг/мл/ч),
то первичный альдостеронизм у больного почти не вызывает сомнений. О
первичном альдостеронизме свидетельствует содержание альдостерона в
плазме крови, превышающее 6 нг/дл, через 4 ч после внутривенной инфу-
зии 2 л изотоничного по отношению ко внеклеточной жидкости кристал-
лоидного раствора.
Радикальным патогенетическим средством терапии первичного
альдостеронизма, обусловленного аденомой коры надпочечников, служит ее
удаление в ходе хирургического вмешательства. Билатеральная
гиперплазия коры надпочечников как причина особенно высокой активности
минералкортикоидов в плазме крови служит показанием к назначению
антагониста альдостерона спиронолактона и других препаратов данной
группы, которые снижают выраженность нарушений водно-солевого обмена
вследствие первичного альдостеронизма.
342
Глава 21
Сдвиги водно-солевого обмена, аналогичные вызываемым первичным
альдостеронизмом, который приводит к АГ, выявляют у небольшой части
больных с низкой концентрацией альдостерона в плазме крови. Эти
нарушения обычно связаны с низким уровнем активности в организме фермента
11-бета-гидроксистероиддегидрогеназы, энзима, переводящего кортизол,
который связывается с рецептором, специфичным по отношению к мине-
ралкортикоидам, в не обладающий свойствами минералкортикоида
кортизон. Глицирризиковая кислота, которую содержит солодковый корень,
добавляемый к жевательному табаку, снижает активность 11-бета-гидро-
ксистероиддегидрогеназы, что ведет к росту в циркулирующей крови
концентрации кортизола, обладающего свойствами минералкортикоида.
Дефицит в организме фермента, превращающего кортизол в кортизон, может
быть идиопатическим и вызывать АГ, гипокалиемию и метаболический
алкалоз. К повышенной активности минералкортикоидов в плазме крови,
обусловленной ростом содержания в ней кортизола, могут приводить и
другие нарушения обмена кортизола (аномальное восстановление кольца А
молекулы гормона), кроме связанных с данными ферментопатиями.
Относительно недавно было выявлена наследственная болезнь,
основное звено патогенеза которой - избыточное содержание гормонов со
свойствами минералкортикоидов в плазме крови, которое снижается после
инъекции дексаметазона. У таких больных причиной роста содержания
альдостерона в плазме крови, АГ и гипокалиемии выступает генная мутация, в
результате которой ген ключевого фермента синтеза альдостерона альдо-
стерон-синтетазы и ген основного энзима при образовании кортизола 11-
бета-гидроксилазы приобретают во многом идентичные свойства, что
определяет появление в организме «гена-химеры» альдостерон-синтетазы и
11-бета-гидроксилазы. В фенотипе это проявляется появлением
альдостерон-синтетазы, которую обычно содержат клетки поверхностной клубоч-
ковой зоны коры надпочечников, в их средней пучковой зоне. Клетки
пучковой зоны у больных с данным наследственным заболеванием под
влиянием кортикотропина начинают усиленно секретировать не только
кортизол, обладающий свойствами минералкортикоида, но и истинный минерал-
кортикоид альдостерон. Поэтому у таких больных повышена минералкор-
тикоидная активность в циркулирующей крови, которую по механизму
отрицательной обратной связи через рост содержания глюкокортикоидов в
плазме и торможение секреции АКТГ снижает иньекция дексаметазона.
Ко вторичным артериальным гипертензиям относят артериальную ги-
пертензию вследствие беременности. Физиологическим изменением
артериального давления во время беременности следует считать его
умеренное снижение в течение первых двух триместров с возвращением АД к
исходному уровню в ее последний триместр. Следует считать, что у
больной развилась АГ вследствие беременности, если систолическое давление
у ней поднялось от исходного (до беременности) уровня на 30 мм рт. ст., а
диастолическое АД- на 15 мм рт. ст. Если до беременности у больной не
было АГ, то развитие артериальной гипертензии вследствие
беременности следует констатировать при АД выше, чем 140/90 мм рт. ст. Известна
Патогенез наиболее частых видов артериальной гипертензии
343
прямая достоверная связь между уровнем диастолического АД и частотой
гибели плода при АГ вследствие беременности.
Преэклампсия - это синдром, который характеризуют АГ вследствие
беременности, протеинурия и отеки. Отеки обуславливает падение
коллоидно-осмотического давления плазмы крови. Обычно преэклампсия
возникает на двадцатой неделе беременности и чаще всего у молодых
первородящих женщин. У таких женщин после первого развития преэклампсии
во время беременности ее вероятность при последующих беременностях
составляет 25 %. Если преэклампсия трансформируется в эклампсию, то у
больных возникают судороги и кома. При этом энцефалопатия вследствие
АГ может развиться при АД 150/90 мм рт. ст., что указывает на
нарушения ауторегуляции объемной скорости мозгового кровотока у
беременных больных с преэклампсией.
АГ в первый и второй триместры беременности обычно представляет
собой эссенциальную первичную АГ или вторичную АГ другого генеза,
то есть не связанную с состоянием беременности. Следует заметить, что
снижение АД и увеличение скорости клубочковой фильтрации как
нормальные изменения функций в первые два триместра беременности у
части женщин могут снизить тяжесть хронической АГ и скрыть заболевания
паренхимы почек, которые могут проявить себя в качестве причин
тяжелой АГ в последний триместр.
Предположительно в основе развития АГ, связанной с беременностью,
лежит активация ренин-ангиотензинового механизма и снижение на
системном уровне образования и высвобождения простагландинов-вазодила-
таторов при росте синтеза и секреции на уровне всего организма вазокон-
стрикторов тромбоксанов; эти нарушения обмена производных арахидоно-
вой кислоты причиной имеют патогенные сдвиги синтеза стероидов в
плаценте. Роль дисбаланса образования и высвобождения простагландинов в
развитии АГ, связанной с беременностью, подтверждает положительный
результат предотвращения АГ вследствие беременности с использованием
антагониста простагландинов ацетилсалициловой кислоты.
Фармакокоррекцию патогенно высокого уровня АД у больных с
преэклампсией начинают тогда, когда диастолическое АД становится выше
95 мм рт. ст. У 75 % больных госпитализация сама по себе устраняет пре-
эклампсию.
В настоящее время доказано, что центральные альфа-два-адреноми-
метики могут вызывать эмбриопатию. У некоторых новорожденных,
матери которых принимали клонидин для коррекции преэклампсии, сразу
после родов возникали судороги, не связанные с гипоксией и родовой
травмой. В этой связи не представляется возможным рекомендовать
использование клофелина (клонидина) для устранения АГ, вторичной по
отношению к беременности. Бета-адреноблокаторы могут вызывать у
плода и новорожденного брадикардию и гипогликемию;
экспериментальные данные свидетельствуют, что препараты данной группы снижают
устойчивость плода по отношению к гипоксии. Поэтому бета-адренолитики
нельзя считать средствами выбора для устранения АГ вследствие бере-
344
Глава 21
менности. Возможно, что действие альфа- и бета-адренолитика лабетало-
ла провоцирует ретроплацентарные кровоизлияния. Известно, что
действие ингибиторов ангиотензин-превращающего фермента может
обусловить смерть плода. Поэтому эти препараты нельзя использовать для
снижения АД при артериальной гипертензии у беременных.
Беременность создает препятствия общему венозному возврату к
сердцу, что через уменьшение его преднагрузки несколько снижает
уровень минутного объема кровообращения. При этом рост общего
периферического сосудистого сопротивления выступает защитной реакцией,
направленной на предотвращение снижения АД. Рост ОПСС как защитная
реакция патогенно избыточен и приводит к АГ вследствие беременности.
Введение нитропруссида-натрия и ганглиоблокаторов больным с АГ
вследствие беременности снижает спазм резистивных сосудов. Если ди-
латация сосудов сопротивления достигает степени, при которой падение
ОПСС без увеличения МОК может привести к артериальной гипотензии,
то у беременных со сниженной преднагрузкой сердца компенсаторного
роста МОК не происходит, и развивается острая артериальная гипотензия.
Поэтому нитропруссид-натрия и ганглиоблокаторы нельзя использовать
для снижения степени АГ вследствие беременности.
Хотя применение эстрогенов для коррекции эндокринопатий,
связанных с менопаузой, обычно не ведет к АГ, прием эстрогенов в качестве
контрацептивов может вызвать цотенциально патогенный подъем
артериального давления. Через пять лет от начала приема контрацептивов,
содержащих эстрогены, у 5 % принимающих их женщин следует ожидать развития
незначительной АГ. Иногда прием контрацептивов, содержащих
эстрогены, служит причиной вторичной по отношению к АГ нефропатии, в основе
которой лежит повреждение приводящих артериол почечных нефронов их
крайне интенсивным разрушающим сосуды спазмом. Предположительно
причиной АГ вследствие действия эстрогенов в составе контрацептивов
следует считать нарушения синтеза и элиминации ангиотензинов.
АГ КАК ПРИЧИНА НЕВРОЛОГИЧЕСКОЙ НЕСТАБИЛЬНОСТИ
Тяжелая АГ может приводить к периодической ишемии целых
областей головного мозга, обуславливать тромбоз и тромбоэмболию мозговых
сосудов, интракраниальные, субарахноидальные кровоизлияния и
энцефалопатию (гипертензивная энцефалопатия). С другой стороны, сам по
себе рост внутричерепного давления через рефлекс Кушинга вызывает
тяжелую артериальную гипертензию, брадикардию и гипервентиляцию.
Причиной гипертензивной энцефалопатии являются очаговая ишемия
головного мозга и его генерализованный отек. На уровне гематоэнцефали-
ческого барьера мозг защищен от системной АГ, как от причины ее
неврологических осложнений, благодаря спазму мозговых сосудов. Поэтому
патогенно избыточная объемная скорость кровотока в артериях головного
Патогенез наиболее частых видов артериальной гипертензии 345
мозга представляет собой следствие АГ только тогда, когда диастолическое
АД превышает уровень в 120 мм рт. ст. Наиболее велика вероятность
неврологических осложнений АГ при остром подъеме артериального давления
у больных без АГ в анамнезе. Хроническая АГ повышает эффективность
сосудистого механизма защиты мозга от системной артериальной
гипертензии, приводя к гипертрофии артериол и тем самым повышая
эффективность защитной спастической реакции сосудов сопротивления головного
мозга в ответ на подъем системного артериального давления.
В этой связи следует исключительно осторожно подходить к
использованию вазодилятаторов как средств снижения АД у больных с
прогрессирующими неврологическими осложнениями артериальной гипертензии.
Дело в том, что сосудорасширяющие средства, снижая степень защитного
спазма сосудов головного мозга на уровне гематоэнцефалического
барьера, могут предрасполагать к неврологическим осложнениям АГ,
увеличивая мозговой кровоток и внутричерепное давление.
В ауторегуляции объемной скорости мозгового кровотока у больных с
АГ происходит сдвиг в сторону спазма мозговых сосудов сопротивления.
Поэтому резкое падение артериального давления у больных с
прогрессирующими неврологическими осложнениями вследствие АГ не
сопровождается адекватным снижением сосудистого сопротивления на уровне
гематоэнцефалического барьера и приводит к ишемии головного мозга. Так
как быстрое и значительное падение АД у больных с прогрессирующими
неврологическими осложнениями вследствие АГ может усилить ишемию
головного мозга, лечебная тактика при снижении артериального давления
с применением гипотензивных средств аналогична схеме постепенного
снижения АД у больных со злокачественной АГ.
АГ КАК ПРИЧИНА СЕРДЕЧНОЙ НЕДОСТАТОЧНОСТИ
И НЕКОТОРЫЕ ПАТОГЕНЕТИЧЕСКИЕ ПРИНЦИПЫ
ФАРМАКОТЕРАПИИ БОЛЬНЫХ С ТЯЖЕЛОЙ АГ
И ЕЕ ОСЛОЖНЕНИЯМИ
АГ увеличивает циркуляторную гипоксию кардиомиоцитов через
увеличение работы левого желудочка в фазу изгнания и снижение перфузион-
ного давления субэндокардиального слоя миокарда левого желудочка при
увеличении его конечно-диастолического давления. Дистрофию и гипоэр-
гоз клеточных элементов сердца вследствие его ишемической болезни и АГ
усиливает гипертрофия стенок миокарда левого желудочка в стадии
несостоятельности, когда развитие сети микрососудов сердца и его нервных
клеточных элементов отстает от роста массы кардиомиоцитов. Одним из
факторов гипертрофии кардиомиоцитов у больных гипертонической
болезнью следует признать рост экспрессии эндотелина как фактора
клеточного роста кардиомиоцитов эндотелиальными клетками венечных артерий
в ответ на гиперкатехоламинемию и возрастание концентрации в крови ан-
346
Глава 21
гиотензина П. Стимулом для гипертрофии выступает постоянно высокий
уровень энерготрат кардиомиоцитами, связанный с хронически
повышенной постнагрузкой у больных с установившейся эссенциальной АГ. В такой
ситуации патогенетически оправданным является использование средств,
снижающих потребность клеток сердца в кислороде и свободной энергии:
бета-один-адренолитиков и антагонистов кальция. При этом надо помнить
о таких побочных эффектах бета-адреноблокаторов как обострение брон-
хоспазма и об опасном падении силы сердечных сокращений при
использовании некоторых средств из класса блокаторов кальциевых каналов.
Тяжелая АГ может быть причиной острой левожелудочковой
недостаточности и кардиогенного отека легких. При этом патогенетически
обоснованными мерами лечебного воздействия следует считать снижение
преднагрузки левого желудочка через действие морфина сульфата и
диуретиков и уменьшение его постнагрузки посредством эффекта вазодила-
таторов. Успешным средством фармакотерапии по жизненным
показаниям может быть внутривенная инфузия нитроглицерина, расширяющего
вены (снижение преднагрузки), снижающего постнагрузку сердца (дила-
тация артерий) и увеличивающего доставку кислорода кардиомиоцитам
по венечным артериям посредством снижения степени их спазма. Блока-
торы кальциевых каналов также могут быть использованы для терапии
острой сердечной слабости вследствие тяжелой АГ. Эти препараты
увеличивают фракцию изгнания левого желудочка, вызывая расширение
венечных артерий и сосудов сопротивления. В результате потребность кар-
диомиоцитов в свободной энергии снижается двумя путями:
♦ увеличение доставки кислорода кардиомиоцитам вследствие
снижения сосудистого сопротивления в системе венечной артерии;
♦ снижение работы миокарда левого желудочка по преодолению
высокой преднагрузки, которую уменьшает ослабление спазма
сосудов сопротивления.
Нитропруссид натрия - это вазодилататор, который расширяет как
артерии, так и вены, одновременно снижая пред- и постнагрузку. В этой связи
его использование при тяжелой АГ и острой левожелудочковой сердечной
недостаточности представляется вполне патогенетически обоснованным.
Однако при фармакотерапии с использованием непрерывной внутривенной
инфузии нитропруссида натрия у больных с тяжелой АГ и ишемической
болезнью сердца следует учитывать, что препарат относительно венечных
артерий выступает мощным вазодилятатором. При этом нитропруссид
натрия мало влияет на сосудистое сопротивление в области фиксированного
стеноза пораженных атеросклерозом венечных артерий. В других венечных
артериях, в которых нет фиксированного стеноза, препарат вызывает
значительное падение сосудистого сопротивления. Такое усиление
диспропорции сосудистых сопротивлений между венечными артериями ведет к
«обкрадыванию» зоны миокардиальной ишемии, при котором резко падает
поступление в нее артериальной крови.
Мочегонные средства у больных с тяжелой АГ, ишемической
болезнью сердца и острой застойной сердечной недостаточностью следует ис-
Патогенез наиболее частых видов артериальной гипертензии
347
пользовать осторожно, помня о том, что они могут вызвать снижение
объема внеклеточной жидкости и плазмы крови. Это опасно уменьшает
преднагрузку левого желудочка, который у больных с ишемической
болезнью сердца мало податлив вследствие связанной с гипоэргозом
высокой концентрации ионизированного кальция в цитозоле клеток рабочего
миокарда. Поэтому диуретики, снижая преднагрузку сердца, могут
быстро и значительно снизить ударный объем левого желудочка у больных
ишемической болезнью сердца. Кроме того, назначение диуретиков часто
приводит к росту общего периферического сосудистого сопротивления,
который предположительно связывают с увеличением вязкости крови при
эксикозе и усилением активации ренин-ангиотензинового механизма в
ответ на падение объема внеклеточной жидкости.
У больных, перенесших аорто-коронарное шунтирование, любой
подъем АД в ближайшем послеоперационном периоде выше обычного
для них уровня представляет собой состояние, опасное для жизни, так как
может привести к несостоятельности сосудистых анастомозов. Для
устранения АГ в такой период и у таких больных препаратами выбора считают
блокаторы кальциевых каналов, особенно, если они расширяют венечные
артерии и снижают общее периферическое сосудистое сопротивление, не
угнетая сократимости сердца. Одновременно, осторожно, чтобы не
снизить силу сердечных сокращений, применяют и бета-адреноблокаторы,
снижающие частоту сердечных сокращений и сократимость сердца как
детерминанты потребления сердцем кислорода и его потребности в 02.
При расслаивающейся аневризме аорты скорость ее расслоения
вследствие АГ находится в прямой связи со средним артериальным
давлением и скоростью выброса крови в аорту в фазу изгнания. Бета-
адреноблокаторы как средства, снижающие и ту и другую детерминанту
скорости расслоения аорты, представляют собой основной элемент
фармакотерапии по жизненным показаниям, предотвращающей летальный
исход вследствие расслоения аорты в месте ее аневризмы под влиянием
АГ. Отрицательное инотропное действие блокаторов кальциевых каналов,
снижающее скорость выброса крови в аорту во время систолы, лежит в
основе их патогенетически обоснованного применения при расслоении
аневризмы аорты у больных с тяжелой АГ. Кроме того, антагонисты
кальция снижают среднее АД, расширяя сосуды сопротивления, что
также снижает скорость расслоения аорты.
ПАТОГЕНЕТИЧЕСКИЕ ПРИНЦИПЫ ЛЕЧЕНИЯ
ГИПЕРТОНИЧЕСКОГО JCPH3A
У части больных АГ различного генеза приводит к гипертоническому
кризу, который в первую очередь характеризует устойчивый подъем
диастол ического АД выше 120-130 мм рт. ст. Гипертонический криз следует
рассматривать как патологическое состояние, требующее неотложной ин-
348
Глава 21
тенсивнои терапии в специализированных отделениях, если он приводит
к острым или прогрессирующим энцефалопатии, инфаркту головного
мозга или кровоизлиянию в него, ишемии или инфаркту миокарда,
расслоению аневризмы аорты, к острой левожелудочковой сердечной
недостаточности с кардиогенным отеком легких, к острой почечной
недостаточности, эклампсии и микроангиопатической гемолитической анемии.
Чаще всего гипертонический криз представляет собой результат про-
грессирования эссенциальной АГ, но может быть обострением почечной
сосудистой гипертензии, АГ, связанной с заболеванием паренхимы почек,
роста ОПСС вследствие склеродермы и заболеваний соединительной
ткани, поражающих стенку сосудов, побочного эффекта адреномиметиков и
трициклических антидепрессантов. Часто гипертонический криз
развивается у больных, переставших принимать антигипертензивные средства.
Иногда он является элементом системной реакции на токсическое
действие наркотиков у наркоманов. Кроме того, гипертонический криз
выступает следствием высвобождения на сегментарном уровне симпатической
части автономной нервной системы из-под иерархически высших влияний
у больных с травмами спинного мозга. У небольшой части больных
причина гипертонического криза- феохромоцитома. Основное звено морфо-
патогенеза гипертонического криза- это фибриноидный некроз стенок
сосудов сопротивления как результат разрушающего их патогенно
интенсивного спазма. Основным патогенетическим принципом неотложной
фармакотерапии больных в состоянии гипертонического криза выступает
ее ориентированность на основное звено артериальной гипертензии. Так,
при гипертоническом кризе, обусловленном артериальной гипертензией
вследствие патологических изменений паренхимы почек, успех могут
принести мочегонные, действующие на петлю канальцев нефрона.
При проведении интенсивной терапии гипертонического криза
следует по жизненным показаниям добиваться быстрого, в течение минут, и
значительного снижения артериального давления, так как в течение этих
минут АГ может стать причиной необратимых некробиотических
изменений головного мозга, сердца, или нарушить целостность аорты при ее
аневризме. Такую артериальную гипертензию определяют как АГ,
требующую «неотложного» устранения в ближайшие минуты после ее
выявления. АГ, требующую неотложного устранения, следует отличать от АГ
как показания к «немедленной» терапии. Дело в том, что при
необратимых нарушениях периферического кровообращения, которые в основном
характеризуют злокачественную АГ, быстрое и значительное снижение
АД всегда приводит к циркуляторной гипоксии клеток, особенно в почке
и головном мозге. Ее причина - это невозможность без артериальной
гипертензии доставки кислорода в клетки тканей с фиксированно высоким
уровнем сосудистого сопротивления, рост которого обуславливают
нарушения периферического кровообращения вследствие злокачественной
АГ. Поэтому часто у больных с АГ, требующей неотложной терапии, для
предотвращения терминального состояния значительно и быстро
снижают АД, понимая, что тем самым могут обострить гипоксию клеток вслед-
Патогенез наиболее частых видов артериальной гипертензии
349
ствие нарушений периферического кровообращения, связанных со
злокачественной артериальной гипертензией.
К артериальной гипертензии, требующей неотложной терапии,
относятся:
♦ АГ как причина энцефалопатии, в том числе и при эклампсии;
♦ тяжелая АГ у больного с острым инфарктом миокарда;
♦ АГ, приводящая к внутричерепным кровоизлияниям или
расслаивающейся аневризме аорты;
♦ АГ в ближайшем послеоперационном периоде у хирургических
больных.
АГ как показание к немедленной интенсивной терапии, снижение АД
при которой в ближайшие минуты после выявления тяжелой
артериальной гипертензии не является необходимым условием предотвращения
терминального состояния, характеризуют:
♦ ускоренное, в течение часов или нескольких суток, прогрессирова-
ние АГ от незначительной или мягкой до тяжелой;
♦ диастолическое АД выше 140 мм рт. ст.;
♦ АГ, выступающая причиной застойной сердечной недостаточности;
♦ АГ при выявленном прогрессирующем тромбозе мозговых сосудов;
♦ АГ как причина нарастающей потери почками их функционально
активной паренхимы и острой почечной недостаточности;
♦ АГ, вызвавшая носовое кровотечение, окончательной остановки
которого нельзя добиться без снижения АД;
♦ АГ вследствие потребления с пищей тирамина больным,
принимающим ингибиторы моноаминооксидазы;
♦ тяжелая АГ как результат нарастания ишемии почек, как это,
например, бывает при изменениях в тканях, окружающих почку,
вследствие склеродермы;
♦ тяжелая АГ, вызванная действием адреномиметиков;
♦ тяжелая АГ вследствие прекращения действия клонидина.
Больных с АГ, требующей неотложной терапии, помещают в
отделение интенсивной терапии, где им для непрерывной острой регистрации
АД производят катетеризацию артерии. Обычно для устранения АГ,
требующей неотложной терапии, парентерально вводят альфа- и бета-адре-
нолитик лабеталол или антагонист кальция никардипин. Использование
этих средств для устранения гипертонического криза в настоящее время
предпочитают непрерывному внутривенному введению нитропруссида
натрия, которое еще недавно считали средством выбора. Постоянную
внутривенную инфузию нитропруссида для устранения гипертонического
криза начинают со скоростью 0,5 мг/кг/мин, производя непрерывные
слежение за уровнем АД и корректировку дозы гипотензивного средства.
Нитропруссид натрия как миотропное средство, непосредственно
влияющее на степень сокращения гладкомышечных элементов сосудистой
стенки, не влияет на звенья патогенеза АГ, которая обострилась до
гипертонического криза. Поэтому после прекращения его инфузии следует
ожидать тяжелой и злокачественной АГ. С другой стороны, непрерывная или
350
Глава 21
периодическая инфузия препарата может производиться не более 3-5 дней,
так как ведет к кумуляции токсичных продуктов биотрансформации вазо-
дилятатора, цианида и тиоцианата, которая особенно быстро возникает
при почечной недостаточности.
Лабеталол в настоящее время - это единственное средство, обладающее
одновременно свойствами бета- и альфа-адренолитика. При использовании
препарата в небольших дозах его бета-адренолитическое действие
превышает свойства лабеталола как альфа-адреноблокатора в три раза.
Увеличение дозы ведет к усилению альфа-адренолитического эффекта. Когда по
мере увеличения дозы альфа-адренолитическое действие лабеталола
достигает максимума, бета-адренолитическое действие лекарственного средства
выше свойств препарата как альфа-адреноблокатора в шесть раз.
При проведении интенсивной терапии больных в состоянии
гипертонического криза диастолическое АД рекомендуют удерживать в
диапазоне 100-120 мм рт. ст., что позволяет предотвратить терминальное
состояние вследствие тяжелой АГ и исключить выраженные нарушения
транспорта кислорода и энергопластических субстратов к клеткам на
периферии. При выявлении клинических признаков ишемии мозга и (или)
сердца, возникновение которой подтверждают данные специальных
исследований, следует помнить, что у больных с хронической тяжелой АГ ее
может обусловить снижение диастолического АД до уровня меньшего, чем
90 мм рт. ст. В таких случаях целью терапии может быть подъем
диастолического артериального давления в диапазоне 100-120 мм рт. ст.
У большей части больных в состоянии гипертонического криза, он
представляет собой крайнюю стадию прогрессирования гипертонической
болезни, звеном патогенеза которой выступает снижение объема
внеклеточной жидкости и плазмы крови. Поэтому, у пациентов с
гипертоническим кризом как осложнением эссенциальной АГ опасно начинать ИТ с
введения или назначения диуретиков, действие которых может привести к
опасной гиповолемии.
Глава 22
ПОЧЕЧНАЯ НЕДОСТАТОЧНОСТЬ
Острая почечная недостаточность - это патологическое состояние
организма, при котором вследствие быстрого развития олигурии или анурии
во внеклеточной жидкости и клетках стремительно растет содержание
конечных продуктов белкового обмена, протонов, натрия, воды и калия.
Метаболический ацидоз и гиперкалиемия, а также высокая преднагрузка сердца
как следствие роста объема внеклеточной жидкости обуславливают
сердечные аритмии и острую сердечную недостаточность. Расстройства сердечного
ритма, вызывающие резкое падение насосной функции сердца, являются
наиболее частыми непосредственными причинами летального исхода при
острой недостаточности почек. При подостром течении почечной
недостаточности у больных успевает развиться уремический синдром с его
неврологическими, кардиальными и гастроинтестинальными симптомами.
Хроническая почечная недостаточность - это патологическое со-
стояние, которое характеризуют длительные снижение скорости клу-
бочковой фильтрации, падение экскреции почками катионов натрия,
калия и других осмолей во внешнюю среду, метаболический ацидоз,
связанный со снижением ренальной экскреции свободных ионов водорода и
падением образования и реабсорбции бикарбонатного аниона в почках, а
также азотемия и уремический синдром.
Олигурия - патологическое состояние вследствие падения объема
выделяемой мочи взрослого человека с массой тела 70 кг до 400 мл/сутки и
ниже. Анурия (отсутствие выделения мочи) обычно обусловлена
обструкцией мочевыводящих путей. Менее частая причина анурии - это
билатеральная одновременная окклюзия почечных артерии и вены. Быстро
прогрессирующий гломерулонефрит и некроз коркового слоя ночек - наиболее
редкие причины анурии у больных. Поэтому повреждение патологическим
процессом собственно паренхимы почек не следует считать причиной
анурии, пока не будут исключены обструкция мочевыводящих путей или
двусторонняя полная окклюзия главных почечных артерии и вены.
Синдром острой почечной недостаточности полиэтиологичен. Его
обуславливают потеря почками критической массы своих структурно-
функциональных единиц (нефронов) в результате заболеваний,
патологических состояний и процессов или резкое угнетение скорости клубочко-
вой фильтрации в нефронах патологически не измененных почек, часто
связанное со спазмом их приводящих артериол. Спазм приводящих арте-
риол как защитная реакция сохраняется и после устранения своих
стимулов под влиянием лечения.
Преренальная почечная недостаточность - это патологическое
состояние организма из-за дисфункций, связанных с падением объемной
скорости кровотока по приводящим артериолам нефронов. Ее причины эквива-
352
Глава 22
лентны причинам синдрома преренальной азотемии (табл. 22.1).
Патологически высокая вязкость крови снижает скорость клубочковой фильтрации
через рост сопротивления микрососудов на уровне клубочков нефронов.
Таблица 22.1
Наиболее частые причины преренальной азотемии у больных
Снижение внутрисосудистого объема крови в результате:
♦ кровопотери;
♦ потери жидкости из просвета желудочно-кишечного канала (понос, рвота, по назога-
стральному зонду у больных с острой кишечной непроходимостью и перитонитом);
♦ избыточной обьемной скорости мочеотделения (оемотический диурез при сахарном
диабете, других болезнях и патологических состояниях, передозировка мочегонных,
недостаточная секреция альдостерона и кортизола как минералкортикоидов);
♦ несостоятельности адаптации к жаре при нехватке воды;
♦ испарения воды с обширных раневых поверхностей (минно-взрывные ранения,
ожоги);
♦ секвестрации внеклеточной жидкости в некробиотически измененных тканях при
остром панкреатите, синдроме длительного раздавливания, минно-взрывных
ранениях и травмах
Снижение минутного объема кровообращения вследствие:
♦ кардиогенного шока:
♦ застойной сердечной недостаточности;
♦ перикардита и тампонады перикардиальной полости;
♦ эмболии легочной артерии и ее ветвей.
Падение общего периферического сосудистого сопротивления
и расширение вен, которые обусловлены:
♦ анафилактическим шоком;
♦ избыточным действием антигипертензивных средств;
♦ сепсисом и системной воспалительной реакцией как причинами раскрытия артерио-
ло-венулярных анастомозов, артериальной гипотензии и шока;
♦ побочными эффектами лекарственных средств: расширение вен наркотическими
анальгетиками и нитратами; дилатация сосудов сопротивления под влиянием альфа-
один-адренолитиков и др.
Системный спазм сосудов сопротивления, и приводящих артериол
почечных клубочков в том числе как следствие:
♦ травматичного оперативного вмешательства в условиях неэффективной анальгезии;
♦ побочного действия альфа-один-адреномиметиков, в том числе и допамина, непрерывно
инфузируемого внутривенно в дозах больших, чем 10 мкгхкгхмин"1;
♦ нарушения регуляции тонуса приводящих артериол нефронов у больных с гепаторе-
нальным синдромом.
Расстройства местной регуляции тонуса приводящих
артериол нефронов под влиянием:
♦ нестероидных противовоспалительных средств (ацетилсалициловая кислота, ибупрофен,
диклофенак и др.), которые тормозят синтез в почках простагландинов-вазодилятаторов Е2
и 12, угнетая активность циклооксигеназы и липооксигеназы;
♦ ингибиторов ангиотензин-превращающего фермента, снижающих на системном
уровне образование ангиотензина II, который вместе с простагландинами-вазодиля-
таторами противостоит системным сосудосуживающим влияниям на приводящие
артериолы нефронов, вызывая спазм резистивных сосудов другой локализации.
Патологически высокая вязкость крови, которую повышают:
♦ множественная миелома и макроглобулинемия;
♦ полицитемия;
♦ гемоконцентрация в результате дегидратации.
Почечная недостаточность
353
Тромбоз и окклюзия внутрипочечных артерий иного генеза, приводя к
ишемии коркового слоя почек, обуславливают не только преренальную,
но и ренальную почечную недостаточность, вызывая деструкцию
сегментов нефронов вследствие ишемического некроза.
Ренальная (паренхиматозная) почечная недостаточность - это
патологическое состояние, которое развивается вследствие
патологических изменений клеток сегментов нефронов, обусловленных деструкцией
клеток при заболеваниях почечной паренхимы. В основе ренальной
почечной недостаточности лежат гломерулопатии (громерулонефриты) и
острый некроз канальцев нефрона (острый тубулярный некроз).
Постренальная почечная недостаточность - это патологическое
состояние, при котором экскреторная функция почек падает и другие ре-
нальные функции расстраиваются из-за механического препятствия
оттоку мочи по мочевыводящим путям. Постренальная почечная
недостаточность проявляет себя синдромом и патологическим состоянием об-
структивной уропатии. Если при остром пиелонефрите особо выражена
воспалительная альтерация клеток почечных сосочков, то олигурию и
острую почечную недостаточность вызывает поражение сегментов
нефронов (ренальная почечная недостаточность). Чаще при остром
пиелонефрите острая почечная недостаточность по происхождению является
постренальной и возникает вследствие обструкции мочеточников
остатками омертвевших почечных сосочков.
Для клинического течения острой преренальной и ренальной
почечной недостаточности характерны две фазы. В первой фазе олигурия или
анурия сохраняются в течение 2-3 недель. Последующая полиурическая
фаза острой почечной недостаточности обусловлена возобновлением
фильтрации в клубочках нефронов при неполном восстановлении тубу-
лярных функций. В результате реабсорбция натрия и воды в
большинстве нефронов остается сниженной, и больной выделяет много гипоосмо-
лярной мочи.
ПРЕРЕНАЛЬНАЯ АЗОТЕМИЯ
Азотемия - это патологическое состояние организма, обусловленное
аккумуляцией в организме, его внутренней среде и крови конечных
продуктов белкового обмена (азотистых ишаков) в результате
преобладания их образования в ходе обмена веществ над экскрецией азотистых
шлаков почками с мочой.
Азотемия может быть следствием падения скорости клубочковой
фильтрации в нефронах (СКФ) на уровне всех почек, которое снижает очищение
крови и внутренней среды от конечных продуктов белкового обмена. При
этом рост содержания в организме, во внеклеточном секторе и плазме крови
азотистых шлаков у части больных с азотемией обусловлен падением СКФ в
результате системной защитно-патогенной реакции на а) дефицит объема
354
Глава 22
внеклеточной жидкости и гиповолемию; б) патологическое снижение МОК и
циркуляторную гипоксию. Эффекторами данной защитно-патогенной
реакции, приводящей к азотемии, являются нормально функционирующие ин-
тактные нефроны. Такую азотемию называют преренальной.
Патологическая боль вызывает преренальную азотемию и предрасполагает к ней через
потерявший биологический смысл спазм резистивных сосудов всего
организма, и приводящих артериол нефронов в том числе.
Ренальная азотемия - это результат падения СКФ из-за потери
почками определенного числа нормально функционирующих интактных
нефронов.
Постренальная азотемия представляет собой следствие обструк-
тивной уропатии.
Азотемия при нормальной СКФ возникает в результате усиления
образования азотистых шлаков в ходе обмена веществ при нормальном
функционировании почек как органа-эффектора систем выделения. При
этом почки, состоящие из нормального числа интактных нефронов, не
претерпевают патологических изменений и не испытывают извращенных
системных регуляторных влияний, которые обуславливают падение СКФ
как причину азотемии.
СКФ представляет собой прямую функцию числа нормально
функционирующих и патологически не измененных нефронов, то есть в
функциональном и структурном отношениях интактной массы почечной
паренхимы. При большинстве хронических заболеваний почек СКФ
неуклонно снижается из-за все большей потери почками нормальных
нефронов. В клинической практике для оценки степени снижения СКФ как
критерия тяжести заболеваний почек и почечной недостаточности
определяют величины следующих показателей, находящиеся в прямой связи со
скоростью клубочковой фильтрации:
♦ концентрацию азота мочевины в крови (АМК);
♦ концентрацию креатинина в сыворотке крови (КК);
♦ клиренс эндогенного креатинина (КЭК).
Катаболизм аминокислот в печени происходит с образованием
аммиака, который после биохимической трансформации в мочевину
определяют как азот мочевины крови. Азот мочевины крови незатрудненно
проходит через фильтрационные барьеры нефронов. У здорового человека
часть азота мочевины крови, попавшего в состав ультрафильтрата,
(примерно 50 %) затем возвращается во внутреннюю среду, то есть реабсор-
бируется из просвета канальцев. Масса азота мочевины крови, которая
реабсорбируется из просвета канальцев, находится в обратной связи со
скоростью мочеотделения. Поэтому у больных с преренальными почечной
недостаточностью и азотемией, которые в частности характеризует
снижение объема диуреза, АМК растет в большей степени, чем падает
СКФ. В этой связи АМК не всегда можно считать надежным критерием
степени снижения величины СКФ, тем более, что концентрация азота
мочевины в крови может расти и без падения скорости клубочковой
фильтрации (табл. 22.2).
Почечная недостаточность
355
Таблица 22.2
Некоторые из патологических состояний и болезней,
повышающих АМК без изменений СКФ
♦ Как следствие высокого уровня потребления белков с пищей, при котором
повышен катаболизм аминокислот в печени
♦ Кровоизлияние в просвет желудочно-кишечного тракта как причина усиленного
распада белков в просвете кишечника, роста всасывания несбалансированной
смеси аминокислот в кровь и повышенного катаболизма аминокислот в гепатоци-
тах
♦ Некробиотические изменения тканей при травмах и ожогах с распадом белков в
очагах цитолиза
♦ Побочный катаболический эффект экзогенных глюкокортикоидов
♦ Побочный эффект антибиотиков из группы тетрациклинов на белковый обмен
Нормальные пределы колебаний АМК составляют 70-180 мг/л (2,5-
6,4 ммоль/л).
КК представляет собой прямую функцию скорости образования креа-
тинина на системном уровне и находится в обратной связи со скоростью
клубочковой фильтрации во всех почках (табл. 22.3).
Таблица 22.3
Нормальный уровень концентрации креатинина в сыворотке крови
Здоровые люди
Мужчины
Женщины
Концентрация креатинина
6-12 мг/л (53-106 мкмоль/л)
5-11 мг/л (44-97 мкмоль/л)
Креатинин - это продукт, образование которого из креатина миоцитов
не зависит от активности какого-либо фермента. Скорость образования
креатинина в организме здорового человека находится на относительно
постоянном уровне и представляет собой прямую функцию общей массы
мышц организма. Содержание креатинина во внутренней среде
увеличивает повышенное потребление жареного мяса. При обычной и
сбалансированной диете концентрация креатинина в сыворотке крови
представляет собой надежный критерий СКФ.
КК растет без падения СКФ в результате побочного действия
аспирина, спиронолактона и других лекарственных веществ, угнетающих
секрецию креатинина в просвет канальцев нефрона. КК снижается по ходу
онтогенеза параллельно со снижением общей массы скелетных мышц. Если
длительное голодание снижает массу произвольных мышц, то
образование креатинина падает, и концентрация креатинина в сыворотке крови
уменьшается без какого-либо снижения СКФ.
Секреция креатинина в просвет канальцев нефрона в физиологических
условиях минимальна и существенно не влияет на очищение от него
плазмы крови, которое в основном происходит через фильтрацию в
почечных клубочках. Данное вещество незатрудненно фильтруется через
356
Глава 22
гломерулярные барьеры. Креатинин не реабсорбируется из просвета
канальцев. Кроме того, креатинин не подвергается трансформациям в ходе
биохимических реакций. Поэтому клиренс эндогенного креатинина,
который высвобождается во внутреннюю среду в ходе обмена веществ,
представляет собой надежный критерий скорости клубочковой фильтрации
при многих болезнях и патологических состояниях.
Клиренс эндогенного креатинина (КЭК), который в физиологических
условиях почти идентичен СКФ, рассчитывают в соответствии со
следующей формулой:
КЭК=(конц. креатинина в моче)х(объемная скорость мочеотделения)/КК
Нормальный диапазон колебаний КЭК у мужчин составляет 97-
137 мл/мин/1,73 м2 площади тела, а у женщин - 88-128 мл/мин/1,73 м2.
Клиренс эндогенного креатинина падает на 1 мл/мин после каждого
года жизни у людей старше сорока лет. При прогрессировании
хронической почечной недостаточности клиренс креатинина может упасть до
уровня 20 мл/мин/1,73 м2. При этом существенно возрастает секреция
креатинина в просвет канальцев нефронов. В результате КЭК перестает
быть надежным критерием СКФ, так как его начинает определять не
только объемная скорость фильтрации плазмы крови в клубочках, но и
величина массы креатинина, который попадает в конечную мочу через
секрецию в просвет канальцев. При этом КЭК может составлять 140-
150 % СКФ. В такой ситуации средства, угнетающие секрецию
креатинина в просвет канальцев, могут вызвать значительное снижение КЭК.
Преренальную азотемию выявляют у 50 % больных с острой почечной
недостаточностью.
Благодаря функционированию системы местной регуляции объемной
скорости кровотока в нефронах, чьи регуляторные воздействия на
приводящие артериолы нефронов противостоят системным сосудосуживающим
влияниям, объемная скорость кровотока через почечные клубочки и
гидростатическое давление крови в просвете их капилляров могут оставаться
стабильными, несмотря на колебания среднего артериального давления в
диапазоне от 60 до 120 мм рт. ст. Несмотря на столь эффективное
сохранение кровотока в клубочках нефронов системой его местной регуляции,
длительная и значительная артериальная гипотензия как стимул
патогенных системных реакций меняет функциональное состояние почек и
вызывает преренальную азотемию. В основном реакция систем регуляции в
ответ на падение минутного объема кровообращения и артериальную ги-
потензию складывается из усиливающих друг друга повышенного
возбуждения высших симпатических центров, системной адренергической
стимуляции и активации ренин-ангиотензин-альдостеронового
механизма. Одновременно растет секреция антидиуретического гормона. Эти
изменения системной регуляции обуславливают падение объемной
скорости кровотока в корковом слое почек, а также усиление реабсорбции
натрия и снижение водного диуреза. При этом почки являются органом-
эффектором неспецифической защитной реакции, направленной на а) за-
Почечная недостаточность
357
держку натрия и воды для предотвращения дефицита объема
внеклеточной жидкости, гиповолемии и дегидратации; и б) разведение
эндогенных токсинов во внутренней среде. Данная защитная реакция может
быстро становиться патологической, превращаясь в звено патогенеза
острой почечной недостаточности и преренальной азотемии.
Гемолиз как причину острой почечной недостаточности и
преренальной азотемии, возникающих через нарушения микроциркуляции в почках,
вызывают:
♦ переливание несовместимой или инфицированной крови;
♦ гемолитическое действие лекарств и возбудителей инфекционных
заболеваний (малярийный плазмодий, Clostridium perfringens и т.д.);
♦ осмотический гемолиз при внутривенном вливании растворов гипо-
осмоляльных по отношению к плазме крови.
В результате защитно-патогенной реакции ограничения экскреции
почками натрия и воды, которая во многом происходит через спазм
приводящих артериол нефронов и падение СКФ, резко снижается скорость
мочеотделения и растет осмоляльность мочи. Так как у больных с
преренальной острой почечной недостаточностью усилена тубулярная ре-
абсорбция натрия и воды, то вслед за водой и натрием из просвета
канальцев интенсивнее реабсорбируется мочевина. Усиление реабсорбции
мочевины при преренальной почечной недостаточности обуславливает
рост АМК, преобладающий над ростом концентрации креатинина в
сыворотке крови. В результате отношение АМК/КК растет выше 20,0 и
может превысить верхний предел диапазона нормальных колебаний, то
есть 30,0.
У больных с преренальными острой почечной недостаточностью и
азотемией нередко выявляют отрицательный жидкостной баланс, то
есть преобладание потерь жидкости с мочой, при неощутимых потерях,
с потом, с каловыми массами и диарейной жидкостью, при рвоте,
вследствие патологической секвестрации внеклеточной жидкости, при
испарении с раневых и ожоговых поверхностей над поступлением воды во
внутреннюю среду с напитками, пищей, в ходе инфузий и трансфузий, а
также по зонду при искусственном энтеральном питании. Обезвоживание
и гипернатриемия вызывают жажду у большинства больных с
преренальной азотемией.
Жажда и олигурия как симптомы дефицита объема внеклеточной
жидкости у больных с преренальной азотемией сочетаются с ортостати-
ческой артериальной гипотензией, при которой после перевода больного
из горизонтального положения в положение сидя систолическое
артериальное давление снижается на 10 мм рт. ст., а частота сердечных
сокращений возрастает на 10 мин"1. Ортостатическая артериальная гипотензия
у больных с преренальной азотемией является признаком гиповолемии
вследствие снижения объема внеклеточной жидкости.
Гемоконцентрация как следствие обезвоживания у больных с
преренальной азотемией приводит к росту содержания в сыворотке крови
альбуминов и показателя гематокрита.
358
Глава 22
Нормальная реакция почек как эффектора систем выделения на
дефицит объема внеклеточной жидкости и гиповолемию у больных с прере-
нальной азотемией состоит в падении скорости клубочковой фильтрации,
предельной интенсификации канальцевой реабсорбции натрия и
снижении водного диуреза. Данная реакция в основном реализуется через спазм
приводящих артериол нефронов, активацию ренин-ангиотензин-альдо-
стеронового механизма и проявляет себя следующими сдвигами физико-
химических характеристик мочи:
♦ рост удельного веса мочи до уровня, превышающего 1,030;
♦ осмоляльность мочи выше, чем 500 мосм/л;
♦ концентрация натрия в моче более высокая, чем 20 ммоль/л.
В основе гепаторенального синдрома, при котором через спазм
приводящих артериол нефрона возникают острые преренальные почечная
недостаточность и азотемия, лежат вторичные по отношению к
заболеваниям печени и ее патологическим состояниям (цирроз печени, острая
жировая дистрофия печени, печеночная недостаточность, холестатический
синдром, поражения печени вследствие сепсиса, инфекционный гепатит)
снижение объемной скорости кровотока через корковый слой почек, ги-
поволемия и альдостеронизм. У части больных с циррозом печени и гепа-
торенальным синдромом выявляют утолщение субэндотелиальных и ме-
зангиальных сегментов базальной мембраны почечных клубочков и
патологические изменения структуры эпителиальных клеток со слиянием их
ножек, контактирующих с базальной мембраной (цирротический гломе-
рулонефрит).
Терапия, направленная на устранение расстройств системного и
периферического кровообращения и дефицита объема внеклеточной
жидкости, которые снижают скорость клубочковой фильтрации (табл. 22.1),
устраняет преренальную азотемию. Свою позитивную роль тут может
играть элиминация этиологического фактора. Так, отмена нестероидных
противоспалительных средств нередко подвергает преренальную
азотемию обратному развитию. После тяжелых ранений и травм эффективная
анальгезия как средство ослабления спазма приводящих артериол
почечных клубочков представляет собой способ профилактики преренальных
острой почечной недостаточности и азотемии.
ГЛОМЕРУЛОНЕФРОПАТИИ
Гломерулонефропатии (острый гломерулонефрит и др.) - это
болезни, нарушения образования мочи в почках и ренальной экскреции, а так-
же соответствующие им синдромы, связанные с поражением патологи-
ческим процессом клубочков нефронов, которое моэюет быть первичным
(избирательное поражение клубочков) или вторичным, то есть
представлять собой на уровне почек результат каких-либо системных
заболеваний и патологических процессов.
Почечная недостаточность
359
Синдром острого гломерулонефрита характеризуют гематурия, про-
теинурия, эритроцитарные цилиндры в моче, почечная артериальная ги-
пертензия и отеки, обусловленные падением экскреторной функции
почек. Для других гломерулонефропатий характерны прежде всего
выраженная протеинурия, незначительные изменения состава мочевого осадка
при СКФ на нормальном или только умеренно сниженном уровне.
Болезнь минимальных изменений - это гломерулонефропатия, которая
приводит только к нефротическому синдрому. При заболевании
отсутствуют патологические изменения, которые позволяют выявить световая
микроскопия и иммунофлуоресцентные способы исследования. Болезнь
минимальных изменений часто сопутствует таким злокачественным
заболеваниям как лимфогранулематоз и другие лимфомы. Хроническая и
острая почечная недостаточность при данной гломерулонефропатий
развиваются редко.
Фокально-сегментарный гломерулосклероз - идиопатическое
заболевание, в основе которого лежит поражение клубочков. Для него в первую
очередь характерны артериальная гипертензия, почечная недостаточность
и нефротический синдром. Гистологическое исследование у больных с
данной нефропатией позволяет выявить слияние ножек подоцитов и
сегментарный склероз клубочков. Прогрессирование
фокально-сегментарного гломерулосклероза постепенно, в течение 5-10 лет, приводит к
возникновению хронической почечной недостаточности.
Мембранозная гломерулонефропатия обычно проявляет себя нефроти-
ческим синдромом. У некоторой части пациентов фиксируют только один
симптом болезни, протеинурию. Скорость клубочковой фильтрации всех
почек при мембранозной гломерулонефропатий нормальна или снижена
крайне незначительно. Данные гистологического исследования
свидетельствуют об утолщении стенок капилляров клубочков в результате
отложения в субэндотелилальном слое иммуноглобулина G и фракции
комплемента СЗ. У части больных нефропатия носит первичный характер. У
других пациентов она осложняет системные заболевания: а) злокачественные
новообразования; б) системную красную волчанку. Мембранозная
нефропатия - нередкое следствие побочного действия некоторых лекарственных
средств: антибиотиков, препаратов золота. Течение болезни характеризуют
периоды спонтанных ремиссий и обострений. У 20 % больных
гломерулонефропатия приводит к хронической почечной недостаточности. Иногда
течение заболевания осложняет тромбоз почечных вен, который может
служить причиной тромбоэмболии в других органах. Если при
мембранозной гломерулонефропатий внезапно падает скорость клубочковой
фильтрации всех почек, то следует заподозрить тромбоз почечных вен.
Клинические признаки мембранозно-пролиферативного
гломерулонефрита аналогичны симптомам острого гломерулонефрита. Иногда об
этой гломерулопатии свидетельствуют нефротический синдром, а также
бессимптомные гематурия и протеинурия. Основанием для
предположительного диагноза служат сочетание клинических симптомов и низкая
концентрация комплемента в циркулирующей крови. При гистопатологи-
360
Глава 22
ческом исследовании выявляют пролиферацию мезангиальных клеток и
повреждения клубочков субэндотелиальными отложениями (тип I) или
отложения внутри базальных мембран клубочков (тип II).
Идиопатический быстропрогрессирующий гломерулонефрит быстро
приводит к острой почечной недостаточности с олигурией, к массивной
протеинурии, нефротическому синдрому и изменениям в осадке мочи
(гематурия и эритроцитарные цилиндры). Быстро прогрессирующему
гломерулонефриту часто предшествуют заболевания неясной этиологии
по клиническим признакам сходные с вирусной инфекцией.
Болезнь антител к базальным мембранам клубочков в своей морфопа-
тогенетической основе имеет аутоиммунное поражение легких и почек
(синдром Гудпастчера) или одних почек. Диагноз заболевания позволяет
окончательно установить выявление в сыворотке крови антител к
базальным мембранам клубочков (БМК), либо линейные отложения этих
антител вдоль БМК, заметные при микроскопическом исследовании биоптата
почек. У части больных заболевание приводит к олигурии и ренальной
азотемии.
Системная красная волчанка служит причиной поражения почек,
которое приводит к медленно нарастающим ренальной азотемии и
протеинурии или обуславливает быстрое прогрессирование почечной
недостаточности. Исследование биоптата почек позволяет выявить изменения,
характерные для мембранозной гломерулонефропатии. О необратимых
изменениях свидетельствуют микроскопические признаки склероза
клубочков, атрофии канальцев и интерстициального фиброза.
Поражения почек в результате системных васкулитов (гранулематоз
Вегенера и др.) часто приводят к синдрому острого гломерулонефрита,
который может быстро прогрессировать.
Гломерулонефрит вследствие инфекций осложняет бактериальный
эндокардит, абсцессы внутренних органов. Постстрептококковый
гломерулонефрит развивается как осложнение поражения верхних
дыхательных путей или кожи бета-гемолитическим стрептококком.
Под гломерулонефритом в морфопатогенетическом отношении
понимают воспалительный процесс в паренхиме почек, преимущественно
затрагивающий клубочки нефронов. Обычно воспаление в почечных
клубочках - это результат их аутоиммунного поражения с образованием
иммунных комплексов, состоящих из аутоантител и клубочковых антигенов.
Воспаление как причина дисфункций почечных клубочков повреждает их
три основных структурно-функциональных элемента: а) базальную
мембрану; б) мезангий, то есть центральную часть почечного клубочка,
находящуюся между капиллярами; в) эндотелий капилляров почечных
клубочков. Гломерулонефрит может составлять морфопатогенетическую
основу первичных заболеваний почек, при которых первичным локусом
протекания патологического процесса и аномальных изменений клеток
является паренхима почек, или быть вторичным, то есть на уровне почек
представлять собой следствие системных болезней и патологических
процессов.
Почечная недостаточность
361
Больные с гломерулонефритами составляют 5 % всех пациентов,
страдающих от тех или иных заболеваний почек. Из всех больных,
которым показан гемодиализ, у 40 % болезнь почек конечной стадии
развивается вследствие гломерулонефрита.
Как орган-эффектор функциональных систем выделения почки
постоянно контактируют с инородными антигенами. Мезангиальные клетки
почечных клубочков обладают способностью захватывать и перерабатывать
антигены, а также иммунные комплексы антиген-антитело. Когда
способность мезангиальных клеток перерабатывать антигены и имунные
комплексы становится патогенно низкой, то аккумуляция в мезангии антигенов
и комплексов антиген-антитело обуславливает нарушение структуры
почечных клубочков. При этом отложение иммунных комплексов через
активацию системы комплемента по классическому пути и связанное с ней
воспаление служит стимулом для пролиферации мезангиальных клеток.
Эндотелиальные клетки отделяют просвет капилляров клубочков от
мезангия и базальной мембраны клубочков. Слой эндотелиальных клеток,
выстилающий клубочковые капилляры, содержит поры, по которым
антигены больших размеров и даже иммунные комплексы попадают в
мезангии. Это обуславливает возможность субэндотелиальных отложений
иммунных комплексов. Мезангиальные клетки, активированные вследствие
образования субэндотелиальных депозитов иммунных комплексов,
мигрируют к месту их отложения для фагоцитоза антигенов в составе
комплексов антиген-антитело. Этот процесс называют мезангиальной интер-
позщией. В результате мезангиальной интерпозиции наступают
повреждение, деструкция и активация эндотелиоцитов как причины тромбоза,
спазма и разрушения капилляров почечных клубочков.
Хотя базальная мембрана является интегрирующим элементом
сосудистой стенки капилляров нефронов, строение клубочков таково, что она
не отделяет эндотелий от мезангия. Антигены и иммунные комплексы
могут задерживаться базальной мембраной как одним из фильтрационных
барьеров клубочков. Отложение иммунных комплексов на базальной
мембране индуцирует острое воспаление, повреждающее мембрану и
нарушающее клубочковый фильтрационный барьер. Дисфункции мембраны
как фильтрационного барьера вызывают протеинурию. Ряд антигенов
базальной мембраны клубочков идентичен антигенам базальной мембраны
легочных капилляров, что обуславливает одновременное аутоиммунное
поражение почечной паренхимы и легочной ткани при синдроме Гуд-
пастчера.
Эпителиальные клетки почечных клубочков образуют отростки,
которые контактируют с внешним слоем базальной мембраны. Полагают, что
поверхность отростков эпителиальных клеток содержит антигены, с
которыми могут образовывать иммунные комплексы иммуноглобулины G,
которые фильтруются через базальную мембрану. Этот процесс называют
образованием иммунных комплексов in situ. Предположительно
образование иммунных комплексов in situ лежит в основе морфопатогенеза
мембранозного гломерулонефрита.
362
Глава 22
Протеинурия - это ежесуточная экскреция белка с мочой в
количестве большем, чем 150 мг. Выделение белка с мочой большее, чем
3,5 г/сутки, свидетельствует о массивной протеинурии. Ее, как правило,
вызывает повреждение клубочков, вследствие которого белки плазмы
крови, и особенно альбумин, могут свободно попадать из крови
капилляров клубочков в состав ультрафильтрата и конечной мочи.
У здорового человека низкомолекулярные (относительная
молекулярная масса не более 40000) белки сыворотки крови (бета-два-микрогло-
булин, лизоцим и др.) незатрудненно попадают в состав ультрафильтрата и
реабсорбируются настолько эффективно, что с мочой выводится лишь
ничтожное их количество. При заболеваниях, в основе которых лежит
преимущественно повреэвдение канальцев нефрона, реабсорбция этих белков
снижается, и они появляются в составе конечной мочи в большем
количестве, то есть развивается канальцевая протеинурия. При этом экскреция
альбумина почти не возрастает, и протеинурия находится на уровне 1-3 г/сутки.
Канальцевая протеинурия обычно не снижает коллоидно-осмотического
давления плазмы крови, так как содержание альбумина в крови остается на
прежнем уровне. В ответ на потерю с конечной мочой низкомолекулярных
белков сыворотки крови в плазме крови растет концентрация некоторых
белков, миоглобина, белка Бене-Джонса, который предположительно
можно считать димером, состоящим из двух легких цепей, и др.
Если дисфункции на уровне канальцев нефрона обуславливают
преобладание объемной скорости ультрафильтрации и поступления протеинов
сыворотки в просвет канальцев над общей реабсорбционной способностью
почек, то возникает протеинурия, которую называют фильтрационной.
При нормальном функционировании нефронов альбумины и
глобулины поступают в состав ультрафильтрата в незначительном количестве.
Эндотелиоциты почечных капилляров образуют первый барьер на пути
фильтрации альбуминов и глобулинов из просвета канальцев в мочевое
пространство (рис. 22.1). Диаметр пор этого барьера составляет около
100 нм. Клетки крови задерживаются эндотелиальным барьером, но
большинство белков проходит через него. Базальная мембрана почечных
клубочков препятствует попаданию в ультрафильтрат молекул с
относительной молекулярной массой более 100000. Поверхность базальной
мембраны клубочков, непосредственно контактирующая с ультрафильтратом,
соприкасается с отростками висцеральных клеток эпителия клубочков
(подоцитов). Эти отростки формируют множество каналов, по которым
молекулы, прошедшие через эндотелиальный барьер, попадают в
ультрафильтрат. Отрицательно заряженные молекулы, альбумины и др.,
фильтруются по этим каналам с большим трудом, чем нейтральные или
положительно заряженные молекулы. Эту избирательность фильтрации связывают
с действием отрицательно заряженных гликопротеинов, выстилающих
поверхность отростков подоцитов. Так как альбумин заряжен также
отрицательно, то отрицательно заряженные участки молекул гликопротеинов
отталкивают отрицательно заряженные участки молекул альбуминов, что
затрудняет их фильтрацию по каналам базальной мембраны.
Почечная недостаточность
363
«Мочевое пространство»
I
Подоцит
Щель в базальной
мембране
Гликосиало-
протеиновое
покрытие
Эндотелиоцит
Просвет
капилляра
Отверстие между
эндотелиальными
клетками
Рис. 22.1. Схематическое изображение барьера между просветом капилляра
и «мочевым пространством» нефрона
Болезни, при которых разрушаются фильтрационные барьеры
клубочков (гломерулонефриты и др.) ведут к протеинурии, которую называют
клубочковой. Если разрушение эндотелиального фильтрационного
барьера клубочков приводит к появлению в конечной моче значительных
количеств клеток крови, то констатируют возникновение гематурии.
Гематурия вследствие поражения клубочков не является изолированной, то есть
развивается одновременно с клубочковой протеинурией.
Можно предположить, что поражение клубочков, которое в основном
состоит из патологических изменений полианионных гликопротеинов,
выстилающих поверхность отростков подоцитов, будет скорее всего
сопровождаться избирательной потерей с мочой отрицательно заряженных
белков, альбуминов и др. Поражение, захватывающее всю базальную
мембрану клубочков, приводит к появлению в составе конечной мочи
протеинов с молекулами более крупными по размерам и массе, чем
молекулы альбуминов.
В начальной стадии острого гломерулонефрита почки выделяют
незначительное количество концентрированной мочи. Это свидетельство
компенсаторной интенсификации экскреции осмолей малым числом пока
еще интактных нефронов. Гипертензия при остром гломерулонефрите в
первую очередь связана с патологическим возрастанием объема
внеклеточной жидкости и с реакцией на уремию, а не с активацией ренин-
ангиотензин-альдостероновой системы.
Отеки при остром гломерулонефрите обусловлены ростом объема
внеклеточной жидкости. При быстром развитии нефротического
синдрома свою роль в возникновении отеков играет падение
коллоидно-осмотического давления плазмы крови.
364
Глава 22
НЕФРОТИЧЕСКИЙ СИНДРОМ
Нефротический синдром как патологическое состояние организма -
это следствие потерь с мочой белков плазмы крови со средними
размерами молекул и реакции систем поддержания гомеостазиса на снижение
их концентрации в плазме крови
Нефротический синдром возникает в результате сниженной
способности клубочковых фильтрационных барьеров не пропускать в состав
ультрафильтрата и конечной мочи белки плазмы крови с молекулами
промежуточной массы (40-200 килодальтонов). Другие белки с молекулами
большей массы не проходят через клубочковый барьер, несмотря на
значительные повреждения клубочков. Белки, которые теряются с конечной
мочой при нефротическом синдроме, - это альбумины,
иммуноглобулины, белки с функциями факторов свертывания крови, эритропоэтин,
другие гормоны, а также протеины, связывающие и переносящие гормоны в
циркулирующей крови. Потеря данных протеинов и рост синтеза печенью
определенных липопротеинов и белков представляют собой ведущие
звенья патогенеза нефротического синдрома.
Многочисленные клетки, ткани и органы синтезируют и секрети-
руют белки, циркулирующие с кровью. Тем не менее, основную массу
протеинов, циркулирующих с кровью, составляют белки, образуемые
печенью, и именно они теряются с мочой при нефротическом
синдроме. Действие неизвестного регуляторного механизма, стимулами для
которого предположительно являются гипоальбуминемия и (или)
падение онкотического давления плазмы крови, резко повышает синтез
белков печенью в ответ на потери с мочой альбуминов и других белков
при нефротическом синдроме. В результате уже через несколько дней
после возникновения нефротического синдрома возрастает масса
печени. При этом в печени растет как образование белков, теряемых с
мочою (альбумин, трансферрин), так и синтез тех протеинов, которые не
попадают в состав ультрафильтрата почечных клубочков (аполипопро-
теины В, Е, фибриноген и др.). Для усиления образования белков
печенью в гепатоцитах усиливается транскрипция генов ключевых
ферментов синтеза протеинов, то есть возрастает реализация генетической
информации на первом этапе, который состоит в биосинтезе
информационной рибонуклеиновой кислоты на матрице дезоксирибонуклеино-
вой кислоты. На данном транскрипционном уровне растет образование
альбумина, трансферрина и других белков. На посттранскрипционном
уровне, то есть вне зависимости от уровня биосинтеза
соответствующей информационной рибонуклеиновой кислоты, при нефротическом
синдроме увеличивается образование аполипопротеинов В и Е и
других веществ.
В клинико-биохимическом отношении нефротический синдром
характеризуют:
♦ суточные потери белка с мочой, не меньшие, чем 3,5 г/1,73 м2
площади тела;
Почечная недостаточность
365
♦ снижение концентрации альбумина в крови (гипоальбуминемия), о
котором свидетельствует концентрация альбумина в ее
сыворотке меньшая, чем 30,0г/л;
♦ отеки вследствие падения коллоидно-осмотического давления
плазмы крови, обусловленного гипоалъбуминемией (у части больных
могут развиться анасарка и асцит);
♦ рост концентрации в крови холестерина, гиперхолестеринемия, то
есть концентрация холестерина в крови утром натощак большая,
чем 2 г/л.
Состав плазмы крови при нефротическом синдроме претерпевает
существенные изменения. Патологические сдвиги состава плазмы крови
связаны с функциями белков, теряемых с мочой, и природой белков,
концентрация которых в крови растет в ответ на гипоальбуминемию. Онко-
тическое давление плазмы крови у больных с нефротическим синдромом
падает из-за того, что общее число осмолей, которые удерживаются
преимущественно в сосудистом русле, снижается из-за гипоальбуминемии.
При этом число осмолей не восстанавливается ростом концентрации в
плазме крови белков с молекулами по размерам большими, чем молекулы
альбуминов. В результате падения онкотического давления плазмы крови
в соответствии с уравнением Старлинга на системном уровне
увеличивается поступление ультрафильтрата в интерстиций, что вызывает отеки.
Кроме того, отеки возникают как результат увеличения обьема
внеклеточной жидкости. Объем внеклеточной жидкости у больных с
нефротическим синдромом растет из-за ареактивности почек к действию предсерд-
ного натрийуретического пептида, а также в результате патологических
изменений почечной паренхимы при заболеваниях, вызывающих нефро-
тический синдром.
В ответ на гипоальбуминемию растет синтез альбуминов в печени.
Так как одновременно с увеличением образования альбуминов растут их
потери с мочой, то компенсаторная реакция интенсификации синтеза
данных белков не предотвращает гипоальбуминемии и отеков. В
результате, несмотря на возрастание образования альбуминов печенью в четыре
раза, их содержание в плазме крови падает до уровня более низкого, чем
равный 25 % нормального. Хотя назначение больным диеты с высоким
содержанием субстратов синтеза альбуминов увеличивает их образование
в печени, такая диета из-за высокого уровня потерь протеинов с мочой не
предотвращает гипоальбуминемии.
Второй, кроме интенсификации синтеза альбуминов, реакцией
компенсации гипоальбуминемии является торможение их катаболизма.
Потерю некоторых белков с мочой (эритропоэтин, иммуноглобулины
G) организм не может компенсировать путем интенсификации
соответствующего белкового синтеза. В результате нефротический синдром
приводит к анемии (следствие потерь эритропоэтина), приобретенному
иммунодефициту как результату потерь во внешнюю среду иммуноглобулинов
G, а также эндокринопатиям, которые развиваются из-за потерь с мочой
протеинов переносчиков гормонов.
366
Глава 22
Большинство гормонов транспортируется с кровью, будучи
связанными с альбуминами и другими белками-переносчиками. При нефротиче-
ском синдроме с конечной мочой теряются вместе с переносчиками как
андрогены, так и эстрогены, что обуславливает клинически незначимый
гипогонадизм. Потери белков, переносящих с кровью некоторые из
витаминов, могут обусловить соответствующие гиповитаминозы (недостаток
витамина D и др.).
Гипоальбуминемия у больных с нефротическим синдромом служит
стимулом для образования печенью белков с размерами молекул большими, чем
размеры молекул альбуминов, то есть некоторых липопротеинов и ряда
факторов свертывания крови. Одновременно с интенсификацией синтеза и
высвобождения в кровь факторов ее свертывания при нефротическом синдроме
с мочой теряются белки с молекулами относительно небольших размеров,
представляющие собой ингибиторы свертывания (антитромбин-Ш, белки С
и S). Поэтому у больных с нефротическим синдромом развивается гжеркоа-
гулемия, то есть патологическое состояние повышенной свертываемости
крови. Гиперкоагулемию у больных с нефротическим синдромом усиливает
рост концентрации в плазме крови белков, связывающих ингибиторы
свертывания крови, которые не попадают в состав ультрафильтрата, так как
масса их молекул больше, чем масса молекул альбуминов. Рост содержания в
плазме этих белков обуславливает снижение в ней концентрации свободных
ингибиторов свертывания, что усиливает гиперкоагулемию. Увеличение
вязкости крови вследствие гиперлипопротеинемии и сужения внутрисосудисто-
го жидкостного сектора является еще одним звеном патогенеза гиперкоагу-
лемии при нефротическом синдроме.
Гиперкоагулемия у больных с нефротическим синдромом может
вызвать венозный тромбоз. В свое время тромбоз почечных вен считали
считали одной из причин нефротического синдрома. Теперь ясно, что
тромбоз вен осложняет нефротический синдром.
Рост образования печенью липопротеинов при нефротическом
синдроме происходит одновременно с падением их катаболизма, что
предрасполагает к развитию атеросклероза у больных с нефротическим
синдромом. Гиперлипопротеинемия у больных с нефротическим синдромом
обуславливает гиперхолестеринемию. При нефротическом синдроме
происходит накопление в плазме крови липопротеинов с низким
содержанием белка и высоким содержанием холестерина и фосфолипидов.
Нарушения обмена липидов вследствие нефротического синдрома в основном
складываются из торможения катаболизма хиломикронов и
липопротеинов очень низкой плотности, а также связаны с падением захвата адипо-
цитами на периферии продуктов распада триглицеридов, циркулирующих
с кровью. Предположительно данные нарушения липидного обмена у
больных с нефротическим синдромом во многом обусловлены низкой
активностью липопротеинлипазы на поверхности эндотелиоцитов
капилляров жировой ткани. Рост содержания хиломикронов в плазме крови ведет
к возрастанию в ней концентрации продуктов их распада, которые
захватываются активированными макрофагами субэндотелиального слоя сосу-
Почечная недостаточность
367
диетой стенки. Тем самым нефротический синдром индуцирует и (или)
ускоряет развитие атеросклероза. При гиперлипопротеинемии у больных
с нефротическим синдромом в плазме крови преимущественно растет
концентрация атерогенных липопротеинов низкой плотности при падении
в ней содержания липопротеина высокой плотности L2, чье
биологическое действие блокирует атерогенез. Можно считать, что у пациентов с
нефротическим синдромом, концентрации липопротеинов в плазме крови
претерпевают такие патологические сдвиги, что развивается дислипопро-
теинемия, в наибольшей степени предрасполагающая к атеросклерозу.
Данную дислипопротеинемию характеризуют снижение содержания в плазме
крови липопротеина высокой плотности Ьг и возрастание в ней
концентраций липопротеинов низкой плотности, очень низкой плотности,
промежуточной плотности, а также высокоатерогенного липопротеина Lp(a).
Стимулом синтеза липопротеинов у больных с нефротическим
синдромом считают протеинурию. Когда у больных с нефротическим
синдромом под влиянием ингибиторов ангиотензин-превращающего
фермента снижаются потери белка с мочой, то падает концентрация
липопротеинов в плазме крови.
ОСТРЫЙ КАНАЛЬЦЕВЫЙ НЕКРОЗ
Под острым каналъцевым некрозом понимают патологическое со-
стояние, которое характеризует резкое падение скорости клубочковой
фильтрации, связанное с повреэюдением клеток каналъцевого эпителия в
результате недостаточного кровоснабжения почек или прямого
повреждения клеточных элементов канальцев экзо- или эндогенными нефро-
токсинами.
У большинства больных состояние острого канальцевого некроза
характеризуется олигурией при снижении объема мочи, выделяемой за
сутки, до уровня более низкого, чем 500 мл. У меньшей части больных
острый канальцевый некроз приводит к полиурии, связанной с
дисфункциями канальцев нефрона. У больных о развитии острого канальцевого
некроза свидетельствуют возникновение или обострение азотемии, которые
не подвергаются обратному развитию после элиминации действия
этиологического фактора.
Среди всех больных, находящихся на стационарном лечении, острый
канальцевый некроз развивается у 1 %. Особенно высок риск острого
канальцевого некроза у хирургических больных с тяжелыми ранениями и
травмами, с синдромом длительного раздавливания, а также после
экстренных и плановых оперативных вмешательств по поводу
расслаивающейся аневризмы аорты.
Нарушения системного кровообращения как причины острых прере-
нальных почечной недостаточности и азотемии приводят к острому
некрозу почечных канальцев у 50 % больных с данным нарушением структу-
368
Глава 22
ры и функций канальцев нефрона. У 25 % больных с острым канальцевым
некрозом его обуславливает патогенное действие на почки экзогенных
нефротоксинов, а в 20 % случаев - эндогенных. Нередко острый каналь-
цевый некроз представляет собой полиэтиологичное патологическое
состояние. Так при синдроме длительного раздавливания острый канальце-
вый некроз вызывают:
♦ дефицит объема внеклеточной жидкости как причина гиповолемии;
♦ влияние на почки эндогенных токсинов (миоглобин, гемоглобин,
кальций и др.), высвобождаемых в циркулирующую кровь
массивом некробиотически измененных тканей;
♦ патологический спазм приводящих артериол почечных клубочков,
одной из основных причин которого является патогенная боль;
♦ угнетение местной простагландиновой системы регуляции
объемной скорости кровотока в почках вследствие эндотоксемии, что
усиливает спазм приводящих артериол из-за системных
сосудосуживающих влияний.
Экзогенными нефротоксинами, действие которых на почки может
обусловить острый канальцевый некроз, являются:
♦ антибиотики (аминогликозиды, цефалоспорины, тетрациклины, ам-
фотерицин В и др.);
♦ рентгенконтрастные вещества;
♦ тяжелые металлы (ртуть, свинец, мышьяк, висмут);
♦ средства, используемые для хемотерапии злокачественных
опухолей (ципластин, метотрексат, митомицин и др.);
♦ иммунодепрессанты (циклоспорин и др.);
♦ органические растворители (этиленгликоль, ССЦ);
♦ пестициды;
♦ фунгициды.
Эндогенными нефротоксинами, вызывающими острый тубулярный
некроз, могут быть мочевая и щавелевая кислоты.
На уровне клубочков нефронов патогенез острого канальцевого
некроза складывается в основном из четырех его звеньев:
1. Патологически интенсивная фильтрация жидкости из просвета
канальцев в почечный интерстиций, а затем в просвет микрососудов
через поврежденные стенки канальцев. Это звено патогенеза в наибольшей
степени свойственно острому канальцевому некрозу вследствие
воздействия на нефроны нефротоксинов.
2. Обструкция канальцев продуктами деструкции их стенок, которая
часто представляет собой основной механизм развития дисфункций на
уровне нефронов у больных с преренальными острой почечной
недостаточностью и азотемией.
3. Резкое падение объемной скорости кровотока в корковом слог
почки, что предположительно может быть следствием подавления
вследствие эндогенной токсемии (синдром длительного раздавливания и
др.) местной простагландиновой системы регуляции кровотока по
микрососудам почек.
Почечная недостаточность
369
4. Патологический рост проницаемости фильтрационных барьеров в
клубочках.
У подавляющего большинства больных с острым канальцевым
некрозом он не только приводит к острой азотемии но и обуславливает острые
и опасные расстройства водно-солевого обмена.
У большинства больных с острыми почечной недостаточностью и ту-
булярным некрозом выявляют олигурию и рост КК. У некоторых
пациентов, страдающих от поражения почек экзогенными нефротоксинами, в
начальный период острой почечной недостаточности вследствие острого
тубулярного некроза олигурии нет. Анурия у больных с острым тубуляр-
ным некрозом чаще всего бывает следствием обструкции мочевыводящих
путей, билатерального некроза кортикального слоя почек, прекращения
кровотока по почечным артериям или быстро прогрессирующего гломе-
рулонефрита.
У меньшей части больных с острым тубулярным некрозом падение
экскреции почками натрия и воды приводит к артериальной гипертензии,
интерстициальным отекам и вызывает кардиогенный отек легких через
патологическое возрастание преднагрузки сердца. Артериальная гипер-
тензия как причина легочной вторичной артериальной гипертензии,
гипервентиляция (компенсаторная реакция в ответ на метаболический
ацидоз, связанный с острой почечной недостаточностью), обусловленная
ростом объема внеклеточной жидкости сердечная недостаточность, все
это вызывает дыхательную недостаточность у больных с острой почечной
недостаточностью в результате острого тубулярного некроза. Нередкой
причиной сердечных аритмий у больных с острым тубулярным некрозом
является гиперкалиемия. В период длительного восстановления функций
нефронов у больных, перенесших острый тубулярный некроз, может
развиваться уремический синдром с повышенными сонливостью, потерей
аппетита, частыми тошнотой и рвотой.
УРЕМИЧЕСКИЙ СИНДРОМ (УРЕМИЯ)
Сдвиги гомеостаза и дисфункции, составляющие патобиохимический
и клинико-патофизиологический эквиваленты уремического синдрома, не
могут служить непосредственными причинами многих из симптомов
уремии и экстраренальных осложнений хронической почечной
недостаточности (гастроинтестинальные, неврологические симптомы,
перикардит и др.). Известно, что состояние больных с уремическим синдромом
можно улучшить посредством гемодиализа, несмотря на то, что мочевину
добавляют в диализат для удержания ее содержания в плазме крови на
исходном, до гемодиализа, уровне. Это свидетельствует о том, что
высокое содержание малотоксичной мочевины в плазме крови может и не
представлять собой звено патогенеза уремического синдрома. Можно
считать, что не существует прямой связи между степенью патобиохи-
370
Глава 22
мических изменений состава плазмы крови у больных с хронической
почечной недостаточностью (табл. 22.4) и выраэюенностью симптомов
уремии. Тем не менее, гемодиализ, проводимый через определенные
промежутки времени, приводит к одновременным исчезновению или
ослаблению симптомов уремии и снижению АМК и КК. Пересадка почки
также может приводить к одновременным снижению КК и АМК и
частичному или полному устранению экстренальных осложнений хронической
почечной недостаточности.
Таблица 22.4
Патобиохимический эквивалент уремического синдрома
Патобиохимические сдвиги в
плазме крови
Рост содержания креатинина и
фосфатного аниона в сыворотке крови и
возрастание АМК
Рост содержания мочевой кислоты в
сыворотке крови
Низкое содержание во внеклеточной
жидкости и плазме крови бикарбонатного
аниона, почечный каналыдевый ацидоз
Звено патогенеза хронической
почечной недостаточности
Снижение СКФ
Падение СКФ и секреции мочевой
кислоты в просвет канальцев нефронов
Снижение ацидификации мочи вследствие
дисфункций канальцев нефрона, то есть
недостаточная секреция протонов в
просвет канальцев, угнетение образования
эпителиоцитами канальцев аммиака, а
также синтеза бикарбонатного аниона
почками и его тубулярной реабсорбции
К симптомам уремии и экстраренальным осложнениям хронической
почечной недостаточности относят:
♦ анемию как результат гемолиза и угнетения гемопоэза;
♦ гастроинтестинальные симптомы и осложнения: анорексию, икоту,
тошноту, рвоту, язвы желудка, язвенный колит, эрозивные
кровотечения, запор и диаррею, паротит;
♦ неврологические симптомы и осложнения со стороны нервной
системы: периферическая нейропатия, зуд, спазмы мышц, эклампсия,
кома;
♦ остеомаляцию;
♦ отеки;
♦ артериальную гипертензию.
У больных с уремическим синдромом угнетена способность костного
мозга к эритропоэзу, что в частности связывают с утратой почками части
клеток, образующих и секретирующих эритропоэтин. Дефицит эритропо-
этина ведет к развитию нормохромной анемии. При уремии небольшой
гемолиз может вызывать значительную анемию, так как происходит при
угнетении эритропоэза. Цитолиз эритроцитов свежей донорской крови, после
ее переливания реципиентам с уремическим синдромом, происходит в три
раза быстрее, чем у других больных. С другой стороны, эритроциты
пациентов с уремией, выступающих в качестве доноров, по времени сохранения
Почечная недостаточность
371
функции и циркуляции с кровью реципиентов не отличаются от
эритроцитов других доноров. Анемия у больных с уремическим синдромом, часто
являясь первым из его элементов, прогрессирует медленно. Из-за гемолиза ге-
мотрансфузии для коррекции анемии у больных с уремией неэффективны.
Вследствие уремии всасывание кальция из просвета кишки становится
аномально низким вне зависимости от поступления во внутреннюю среду
витамина D и влияния (витамин Э)-гормона на абсорбцию кальция из
просвета кишечника (резистентность кишечного всасывания кальция по
отношению к витамину D). В результате при любых потреблении
витамина с пищей и биологической активности (витамин 0)-гормона
развивается гипокальциемия, которую не может предотвратить компенсаторное
снижение экскреции кальция почками. Кроме как с резистентностью по
отношению к витамину D, нарушения всасывания кальция
предположительно связывают с высоким содержанием фосфатов в стенке кишки.
Высокое содержание фосфатов в кишечной стенке обуславливает связывание
ионизированного кальция с образованием нерастворимого кальция
фосфата еще до того, как свободный кальций в ходе его кишечного
всасывания попадает из просвета кишки в кровь. Это приводит к падению
содержания ионизированного кальция в сыворотке крови.
Низкое содержание в организме ионизированного кальция у больных
с почечной недостаточностью отчасти является следствием соединения
свободного кальция с фосфатным и сульфатным анионами во
внеклеточной жидкости. Содержание этих анионов растет во внеклеточной
жидкости вследствие падения экскреторной функции почек. Интенсивное
образование кальция фосфата во внеклеточной жидкости ведет к внекостной
кальцификации тканей в области суставов и позвоночного столба. Внеко-
стная кальцификация захватывает паренхиму легких, печени и почек.
Соединения кальция откладываются в сосудистой стенке и коже. Оссифика-
ция связок и мышц в области суставов, вызывает боль аналогичную той, от
которой страдают больные подагрой. Поэтому синдром внекостной
кальцификации тканей называют «псевдоподагрой».
Если внекостная кальцификация затрагивает проводящую систему
сердца, то это вызывает сердечные аритмии, которые у части больных
могут привести к летальному исходу.
Низкая концентрация ионизированного кальция в плазме крови
служит стимулом для секреции паратиреоидного гормона. Повышенная
секреция паратиреоидного гормона у больных с хронической почечной
недостаточностью лежит в основе синдрома вторичного гиперпаратиреоза.
Паратиреоидный гормон стимулирует активность остеокластов, что
увеличивает резорбцию кальция и фосфатов из костной ткани. Потеря
костной тканью кальции фосфата, которую усиливает ацидоз вследствие
почечной недостаточности, приводит кренальной остеодистрофищ при
которой образование множественных костных кист обуславливает частые
переломы костей.
У небольшого числа больных высокая секреция паратиреоидного
гормона сохраняется и после устранения почечной недостаточности пересад-
372
Глава 22
кой почки. Это патологическое состояние называют третичным гиперпа-
ратиреозом. Для него характерны остеодистрофия и высокое содержание
ионизированного кальция в плазме крови. Третичный гиперпаратиреоз у
некоторых больных устраняют резекцией паращитовидных желез. В
основе синдрома лежит извращение регуляции секреции гормона
паращитовидных желез, основанное на принципе положительной обратной связи.
При этом секреция гормона продолжает оставаться аномально высокой,
несмотря на снижение концентрации ионизированного кальция в
сыворотке крови.
ОБСТРУКТИВНАЯ УРОПАТИЯ
Патологическое состояние вследствие механического препятствия
выведению мочи и связанной с ним постренальной почечной
недостаточности называют обструктивной уропатией.
Механическое препятствие нормальному выведению мочи может
возникать на уровне мочеиспускательного канала, мочевого пузыря, одного
или двух мочеточников. Его обычно обуславливают:
♦ сдавление опухолью, которая является результатом клеточного
роста в тканях, не составляющих мочевыводящие пути;
♦ сужение их просвета вследствие фиброзных изменений в тканях,
окружающих мочевыводящие пути;
♦ обтурация мочевыводящих путей опухолями, по происхождению
связанными с мочевыводящими путями;
♦ закупорка мочевыводящих путей почечными камнями;
♦ обтурация сгустком крови;
♦ нарушения регуляции тонуса сфинктеров, стенок мочевого пузыря
(нейрогенный пузырь) и мочеточников, связанные с патологической
болью, передозировкой адреномиметиков и действием
криминальных наркотиков.
У мальчиков нередкой причиной обструкции нормальному
мочеиспусканию могут быть задние клапаны мочеиспускательного канала. Более
редкие причины обструкции - клапаны мочеточников, а также уретеро-
целе, то есть мешковидная дилатация терминальной части мочеточника,
выбухающая в просвет мочевого пузыря вследствие врожденного стеноза
устья мочеточника.
Обструкция мочеточников резко повышает давление в мочеточниках,
почечной паренхиме и канальцах нефронов. Затем при длительной
обструкции давление в мочеточниках возвращается на исходный уровень.
Через 15 мин после обструкции мочеточника объемная скорость
кровотока в почке, мочевыведение из которой заблокировано, возрастает на
40-50 %. Этот рост кровотока связан с падением почечного сосудистого
сопротивления. В дальнейшем за несколько дней объемная скорость
кровотока падает до уровня значительно более низкого, чем нормальный. Это
Почечная недостаточность
373
двухфазное изменение кровотока связывают с реакцией роста
образования в паренхиме почек простагландинов-вазодилятаторов (простациклин
и др.), которую сменяет интенсификация синтеза простагландина-вазо-
констриктора тромбоксана.
Под влиянием обструкции падает скорость клубочковой фильтрации,
которую снижают: а) рост давления жидкости в просвете канальцев неф-
рона как причина падения градиента давлений, обуславливающего
фильтрацию в клубочках; б) рост почечного сосудистого сопротивления
вследствие высокого уровня образования в почках простациклина.
Если в течение недели обструкцию мочевыведению устраняют, то
функции почек восстанавливаются полностью. При большей
длительности обструкции возможна потеря почками некоторых из своих функций,
несмотря на возвращение скорости клубочковой фильтрации на исходный
уровень. После прекращения обструкции, которая длилась шесть недель,
скорость клубочковой фильтрации возрастает лишь умеренно, на 10-
20 мл в мин. Длительная обструкция выведению мочи приводит к
несахарному мочеизнурению как результату падения концентрационной
способности почек и ареактивности тубулярных эпителиоцитов к действию
антидиуретического гормона. Кроме того, тубулярный эпителий во
многом лишается своих функций, обеспечивающих нормальные синтез в
почках аммиака и секрецию протонов в просвет канальцев нефрона. В
результате развивается почечный каналъцевый ацидоз.
Прекращение продвижения жидкости по мочевыводящим путям и рост
давления в их просвете, повреждающий защитные иммунные системы ка-
нальцевого эпителия, предрасполагает к инфекциям мочевыводящих путей и
почек, которые могут быть причинами некроза почечных сосочков.
После прекращения обструкции наступает полиурия, то есть
аномально усиленное выделение мочи. Выделяют следующие ее причины:
♦ осмотический диурез в силу необходимости для организма
выделять те осмоли, которые были задержаны во внутренней среде в
результате обструкции выведению мочи;
♦ патологический рост содержания в организме натрия и объема
внеклеточной жидкости как причина интенсификации натрийуреза и
выведения мочи;
♦ падение реабсорбционной способности почек как причина
патологически усиленного натрийуреза;
♦ нефрогенное несахарное мочеизнурение, то есть снижение
физиологической реакции на действие антидиуретического гормона.
Если полиурия как результат обструктивной уропатии приобретает
патологический характер, то она приводит к падению объема
внеклеточной жидкости, гипернатриемии (результат избыточного водного диуреза)
и обуславливает гипокалиемию.
Глава 23
ПАТОФИЗИОЛОГИЯ ПЕЧЕНИ
Печень - это орган-эффектор функциональных систем, конечными
полезными приспособительными результатами которых являются:
♦ нормальный углеводный и липидный обмен;
♦ детоксикация и экскреция эндогенных и экзогенных
метаболитов;
♦ образование и секреция желчи;
♦ уничтожение и элиминация всего чужеродного при прохождении
крови через печень посредством функционирования клеток системы
мононуклеарных фагоцитов, локализованных в печени.
Кроме того, печень и гепатоциты, образуя глутамин, представляют
собой эффекторы системы удержания концентрации протонов во
внеклеточной жидкости и клетках в нормальных пределах. Как локус синтеза
альбумина печень задействована функциональной системой поддержания
нормального коллоидно-осмотического давления плазмы крови.
Ненарушенный синтез коагулянтов в печени - необходимое условие нормальной
свертываемости крови. Это лишь немногие из функций печени и гепато-
цитов, осуществляя которые, они участвуют в функционировании
разнообразных систем.
Недостаточность печени как патологическое состояние всего
организма, обусловленное недостатком нормальных гепатоцитов,
складывается прежде всего из нарушений детоксикации в печени, низкого уровня
синтеза белков в гепатоцитах, а также представляет собой следствие
падения образования желчи и ее секреции в просвет кишечника. Цирроз
печени, снижая общую площадь поперечного сечения печеночных
синусоид, ведет к портальной гипертензии и асциту.
Острая печеночная недостаточность - это следствие быстрой (в
течение нескольких недель или даэюе быстрее) потери печенью 90 или
более процентов нормальных гепатоцитов. При этом часто не
успевает развиться такой частый симптом недостаточности печени как
желтуха.
При острой печеночной недостаточности падает утилизация печенью
метаболитов, образуемых при осуществлении цикла лимонной кислоты,
то есть лактата, пирувата, альфа-кетоглютарата. Эти метаболиты
представляют собой органические кислоты, диссоциирующие во
внеклеточной жидкости. В результате аккумуляции и диссоциации данных кислот
во внутренней среде развивается метаболический ацидоз с увеличенным
анионным пробелом плазмы. Метаболический ацидоз через падение
общего приферического сосудистого сопротивления приводит у больных в
состоянии острой печеночной недостаточности к трудно устраняемой
артериальной гипотензии.
Патофизиология печени
375
ХОЛЕСТАЗ
Холестаз (холестатический синдром) - патологическое состояние
организма вследствие падения объемной скорости продвижения желчи по жел-
чевыводящим путям, которое обуславливает сниэюение секреции желчных
кислот и экскреции билирубина в просвет кишечника и аккумуляцию
составляющих желчи в циркулирующей крови (табл. 23.1).
Таблица 23.1
Патогенез холестатического синдрома
Симптом и данные специальных
исследований
Патогенез
Желтуха
Зуд
Обильный рыхлый стул
Белый или светлый стул. Низкое
содержание уробилиногена в моче
Экхимоз (частые подкожные
кровоизлияния)
Ксантоматоз, то есть кожные
умеренно пигментированные
(желтоватые) разной консистенции (иногда
рыхлые) сходные с папилломами на
толстой ножке образования различной
локализации
Гепатомегалия, рост активности в
крови щелочной фосфатазы, аланин-
и апартатаминотрансферазы,
патологические изменения гепатоцитов и
микроструктуры печени, выявляемые
при микроскопии ее биоптата
Рост концентрации билирубина в крови
вследствие падения его экскреции в просвет кишечника
Аккумуляция в крови желчных кислот из-за
падения их секреции в двенадцатиперстную кишку
Снижение всасывания жиров из просвета
кишечника вследствие низкого содержания желчных
кислот в его просвете. Рост содержания жиров в
кале как причина изменения консистенции и
роста массы кала
Отсутствие или снижение содержания в кале
желчных пигментов и уробилиногена в
результате остановки или снижения объемной скорости
продвижения желчи по желчевыводящим путям
Коагулопатия, обусловленная недостаточным
всасыванием витамина К из просвета кишечника
вследствие холестаза и низкой концентрации
желчных кислот в просвете кишки. Нарушение
синтеза факторов свертывания в печени из-за
вторичных по отношению к холестазу
дисфункций гепатоцитов
Рост концентрации холестерина в плазме крови
после длительного холестаза
Некробиотические изменения клеток печени как
следствие блокады отделения желчи и высокого
давления в желчевыводящих путях. Реактивное
воспаление с клеточной пролиферацией
В плазме крови и печени содержится фермент лецитин-холестерин-
ацилтрансфераза (ЛХАТ), который участвует в превращении свободного
холестерина в его этерифицированную форму. Повышение содержания
свободного холестерина в сыворотке крови при снижении в ней концен-
376
Глава 23
трации этерифицированного холестерина у больных с холестазом
предположительно связано со снижением образования ЛХАТ печенью.
Снижение продукции ЛХАТ связано с появлением в сыворотке крови у больных
с холестазом необычного липопротеина низкой плотности, который
называют липопротеином X.
В клинической практике о холестатическом синдроме чаще всего
свидетельствуют желтуха и рост активности в плазме крови фермента щелочной
фосфатазы, синтез которой в печени растет вследствие холестаза. Если,
кроме того, происходит осветление стула, а из мочи исчезает уробилиноген, то
развитие холестатического синдрома сомнений вызывать не должно.
После выявления у больного холестатического синдрома необходимо
исключить или установить механическое препятствие выведению желчи в
просвет кишечника (обтурация камнями при желчно-каменной болезни,
сдавление или заращение желчевыводящих путей вследствие опухолей
печени и поджелудочной железы, стриктура другого происхождения на
каком-либо уровне выведения желчи в просвет кишечника и др.), которое
может служить показанием к оперативному вмешательству. Известно, что
желчные протоки диаметром более 1 мм обладают исключительной
способностью к растяжению под влиянием роста давления в них желчи,
связанного с холестазом. В результате растяжения канальцев такого диаметра
может возникнуть увеличение объема всей печени. Обычная причина роста
массы печени данного происхождения - это обструкция желчевыводящих
путей, которая может служить показанием к хирургическому лечению.
Приобретенные расстройства активного транспорта анионов через
стенку желчных капилляров - более редкие причины холестаза и крайне
редко приводят к холестатическому синдрому, который сохраняется в
течение нескольких дней, как это бывает при механическом препятствии
выведению желчи в просвет кишечника.
Наиболее частая причина механической обструкции желчевыведению,
требующей хирургической коррекции, - это камень желчного пузыря,
закупоривший общий желчный проток. Чаще всего камень закупоривает проток
в его наиболее узкой и наименее растяжимой части, находящейся в стенке
двенадцатиперстной кишки. В результате происходит дилатация
проксимальной части общего желчного протока, которую можно выявить при
рентгенологическом и ультразвуковом исследовании. Одновременно может расшириться
желчный пузырь. Ультразвуковое исследование и компьютерная томография
печени позволяют выявить характерное для механического препятствия
выведению желчи расширение внутрипеченочных желчных протоков.
У небольшой части больных, и особенно у детей, холестатический
синдром представляет собой результат врожденного дефицита
молекулярных переносчиков желчных кислот на уровне печеночных синусоид и
желчных канальцев (капилляров) или какого-либо другого генетически
детерминированного дефекта анионной помпы стенки желчных
капилляров. Примером могут служить синдромы Байлера и Аладжилля. При
синдроме Аладжилля холестаз сочетается с такими врожденными
аномалиями как деформация дуг позвонков и стеноз легочной артерии.
Патофизиология печени
377
Известно, что эстрогены снижают уровень экскреции как желчных
кислот, так и связанного билирубина. Если беременным назначают препараты
на основе эстрогенов, то может развиться холестатический синдром.
Ведущее звено его патогенеза - это рост под влиянием эстрогенов проницаемости
для желчных кислот стенок желчных канальцев. В результате гепатоцит не
может посредством активного транспорта создать градиент концентрации
желчных кислот между своим цитозолем и просветом желчного канальца.
ЖЕЛТУХА
Желтуха (иктеричностъ) - желтое окрашивание колеи или склер,
обусловленное их пигментацией билирубином при его патологически
высоком содержании в крови (гипербилирубинемия). Прежде всего гиперби-
лирубинемия приводит к иктеричности склер, так как они содержат много
эластина, обладающего высоким сродством к билирубину.
У здорового человека содержание билирубина в плазме крови
варьирует от 3 мг/л (5J мкмоль/л) до 10 мг/л (17 мкмоль/л). Большая часть
билирубина, который в норме содержит кровь, - это неконъюгированный
(несвязанный) билирубин, который в плазме крови находится в соединении с
альбумином. Такой нерастворимый в воде билирубин называют
непрямым, так как в прямую реакцию Ван-ден-Берга в водной среде вовлечен
только водорастворимый прямой (конъюгированный, связанный)
билирубин. Большинство врачей легко выявляют желтуху при осмотре больного,
когда содержание билирубина в крови выше, чем 20-25 мг/л (34-
42 мкмоль/л). По мере нарастания гипербилирубинемии кожа становится
зеленоватой из-за окисления билирубина с образованием биливердина.
Окисление прямого билирубина идет быстрее. Поэтому зеленоватый
оттенок кожа чаще приобретает при преимущественном росте содержания в
плазме крови прямого билирубина.
Сразу же после выявления желтухи или гипербилирубинемии следует
выяснить, содержание какого билирубина преимущественно возросло в
плазме крови. Отсутствие билирубина в моче свидетельствует, что
гипербилирубинемия обусловлена ростом концентрации в плазме крови непрямого
билирубина. Неконъюгированный билирубин, будучи связанным с
альбумином, при нормальной клубочковой фильтрации не попадает в
ультрафильтрат, а значит и в конечную мочу. При гипербилирубинемии, обусловленной
повышенным содержанием в плазме крови неконъюгированного
билирубина, 80-85 % общего содержания билирубина в плазме составляет непрямой
билирубин, что определяют, используя реакцию Ван-ден-Берга. Следует
заметить, что при использовании жидкостной хромотографии эта величина
возрастает до 96 %. Считают, что гипербилирубинемия в основном
обусловлена возрастанием концентрации в крови прямого билирубина, если более
50 % билирубина, который содержит сыворотка, - это прямой билирубин, то
есть моно- и диглюкурониды билирубина.
378
Глава 23
В норме, для того чтобы билирубин поступил в желчь, он должен
быть трансформирован из непрямого билирубина в глюкурониды, то есть
превращен в прямой (связанный) билирубин (рис. 23.1). Секрецию
прямого билирубина в желчь считают активным процессом, торможение
которого может снизить скорость образования конъюгированного билирубина
в печени. Если патологический процесс, вызывая дисфункции гепатоци-
тов, тормозит секрецию глюкуронидов в желчь, то:
♦ снижается секреция билирубина в желчь;
♦ возникает регургитация, то есть обратный выход прямого
(связанного) билирубина из клеток печени в кровоток.
Плазма
крови
Б-Ал-
Гепатоцит
Желчь
г
г\
ЛигаЬадин
-Б 1 *»
Глюкуронил-
трансфераза
БМГ
Глкжуронил-
трансфераза
УДФ-ГК
УДФ
т
^ БДГ
^БМГ
Эндоплазматический ретикулум
Рис. 23.1. Поглощение непрямого (несвязанного, неконъюгированного) билирубина гепато-
цитом, его связывание (трансформация в коньюгированный, связанный) и экскреция в виде
прямого билирубина, то есть глюкуронидов билирубина
(Б-Ал - непрямой билирубин, циркулирующий с кровью вместе с альбумином; Б - билирубин; УДФ-
ГК - уридиндифосфат-глюкуроновая кислота;БДГ - билирубиндиглюкорунид; БМГ - билирубинмоног-
люкуронид).
Желтуху с преобладанием содержания в сыворотке непрямого
билирубина вызывают.
♦ патогенно избыточное образование билирубина;
♦ нарушение поглощения клетками печени непрямого билирубина из
циркулирующей крови;
♦ расстройства связывания билирубина глюкуронидами в гепатоцитах.
Избыточное образование билирубина в организме может быть связано
с гемолизом. Под гемолизом понимают состояние организма, которое
характеризует падение времени циркуляции с кровью эритроцитов в
результате их преждевременного разрушения. При этом эритроциты могут
разрушаться как в силу внешних по отношению к ним причин (аутоиммунное
поражение эритроцитов и др.), так и вследствие причин, кроющихся в
самих красных кровяных клетках (наследственный сфероцитоз и др.)
Нарушение поглощения билирубина печенью, то есть отщепления
пигмента от альбумина и последующее связывание его с лигандином, как
причину желтухи могут вызывать некоторые лекарственные средства
Патофизиология печени
379
(препараты флаваспидиновой кислоты и др.). У некоторых больных с
синдромом Жильбера в основе желтухи также лежит нарушение данной
фазы обмена билирубина в печени.
Расстройства связывания билирубина глюкуронидом могут быть
следствием как приобретенной, так и врожденной недостаточности
активности фермента глюкуронилтрансферазы. Ее активность низка у
нормального плода и здоровых новорожденных, что служит одной из причин
физиологической желтухи между вторым и пятым днями жизни. Умеренное
снижение активности энзима выявляют у части больных с синдромом
Жильбера. Кроме того, низкий уровень активности фермента лежит в
основе врожденной негемолитической желтухи II типа. При врожденной
негемолитической желтухе I типа фермента просто нет в печени.
Приобретенное угнетение активности глюкуронилтрансферазы
билирубина может быть результатом побочного действия некоторых
лекарственных средств (прегнандиола, левомицетина). Заболевания печени,
которые приводят к гибели и дисфункциям гепатоцитов (гепатоцеллюляр-
ные болезни, то есть гепатит и цирроз), также нарушают связывание
билирубина глюкуронидом, но в большей степени вследствие гепатоцел-
люлярных болезней падает способность клеток печени и всего органа к
экскреции билирубина, что обуславливает желтуху с преобладанием в
сыворотке крови прямого билирубина.
Желтуха с преобладанием в сыворотке крови прямого билирубина-
это чаще всего результат нарушения экскреции билирубина печенью и ре-
гургитации прямого билирубина, обусловленных дисфункциями
гепатоцитов, всей печени и (или) холестазом, в том числе и связанным с
механическим препятствием желчевыведению. Так как прямой билирубин
циркулирует с кровью отдельно от альбумина, то он попадает в состав первичной
мочи в ходе клубочковой фильтрации. Билирубин в моче служит
доказательством гипербилирубинемии при преобладании в сыворотке крови
содержания прямого билирубина. Обтурация общего желчного протока через
блокаду перемещения связанного билирубина из гепатоцитов в желчь ведет
к регургитации прямого билирубина и росту его содержания в сыворотке
крови. Гепатоцеллюлярные болезни угнетают экскрецию билирубина,
нарушая функционирование гепатоцитов и снижая число нормальных клеток
печени. Поэтому как при гепатоцеллюлярных болезнях, так и при
механическом препятствии выведению желчи в сыворотке крови растет
содержание прямого билирубина. Для окончательного выявления причины желтухи
применяют другие способы диагностики (ультразвуковой и др.).
Предполагают существование следующих патогенетических
механизмов снижения экскреции прямого билирубина печенью:
♦ разрыв желчных канальцев вследствие гибели клеток,
составляющих их стенки;
♦ обтурация желчных канальцев густой желчью или их сдавление
вследствие отека клеток печени;
♦ сдавление и закупорка холангиол как результат воспалительной
инфильтрации;
380
Глава 23
♦ рост проницаемости наружной клеточной мембраны гепатоцитов;
♦ накопление прямого билирубина в печени и ее клетках, связанное с
нарушением обмена билирубина в клетках печени и снижением
числа нормальных гепатоцитов, как причина пассивной диффузии
прямого билирубина в кровь.
Полная обструкция внепеченочных желчных протоков вызывает
желтуху при преобладающем росте содержания в крови прямого билирубина,
билирубинурию (появление прямого билирубина в моче) и осветление
каловых масс до цвета глины. Так как желчь не поступает в кишечник, то из
кала и мочи полностью исчезает уробилиноген. Общая концентрация
билирубина в сыворотке крови растет до уровня 300-400 мг/л, после чего рост
прекращается. Это можно связать с установлением равновесия между
выведением билирубина с мочой и его трансформацией на путях метаболизма.
Частичная обструкция внепеченочных желчных протоков вызывает
желтуху только тогда, когда давление внутри желчных протоков растет до
величины максимального давления секреции желчи, составляющего
примерно 250 мм рт. ст. Поэтому при частичной обструкции желтуха, били-
бинурия и каловые массы цвета глины встречаются не всегда. Желтуха
может развиться и при более низком давлении, если возникает инфекция
желчных протоков и связанные с ней дисфункции гепатоцитов.
НАРУШЕНИЯ ОБМЕНА ВЕЩЕСТВ КАК РЕЗУЛЬТАТ
ПЕЧЕНОЧНОЙ НЕДОСТАТОЧНОСТИ
Исключительно в печени синтезируются многие из молекулярных
эффекторов функций, без которых невозможна нормальная работа ряда
функциональных систем (табл. 23.2). Поэтому потеря печенью
определенного числа нормально функционирующих гепатоцитов приводит к
специфическому для определенных систем дефициту массы на уровне
эффекторов и к разнообразным системным патологическим сдвигам
вследствие расстройств функциональных систем.
Таблица 23.2
Некоторые из протеинов, синтезируемые исключительно в печени
1. Альбумин
2. Факторы свертывания крови
а. Фибриноген (фактор I)
б. Протромбин (фактор II)
в. Факторы III, V, VII, IX, X, XI
3. Транспортные белки
а. Гаптоглобин
б. Трансферрин
в. Церуллоплазмин
г. Белки с функциями переносчиков гормонов
д. Липопротеины низкой плотности
Патофизиология печени
381
Холестерин - это главная составляющая желчи, а также
предшественник желчных солей и стероидных гормонов на пути их синтеза. Кроме
того, холестерин представляет собой интегрирующий компонент клеточных
мембран, основными структурными элементами которых являются фос-
фолипиды и белки. Гепатоциты поглощают холестерина больше, чем
любые другие клетки организма. Холестерин поглощается гепатоцитами для
синтеза в дифференцированных клетках печени желчных солей, а также
для последующего высвобождения гепатоцитами в кровь холестерина,
связанного с атерогенными липопротеинами низкой плотности. В этой
связи нормальное функциональное состояние гепатоцитов и всей печени
можно считать необходимым условием удержания концентрации
холестерина в сыворотке крови в нормальных пределах.
Одновременно с утилизацией холестерина гепатоциты сами образуют
его как для собственных нужд, так и для удовлетворения потребностей
всего организма. Поэтому содержание холестерина в сыворотке крови
представляет собой результат соотношения синтеза холестерина в печени
и кишечнике и его экскреции печенью вместе с желчью.
Если гепатоцеллюлярные болезни снижают число нормальных
гепатоцитов до определенного уровня, то падение синтеза холестерина в
печени преобладает над снижением его экскреции в просвет кишечника
таким образом, что в сыворотке крови падает концентрация холестерина.
Если внешние по отношению к печени системные расстройства
обмена веществ приводят к гиперхолестеринемии, то есть патологически
высокому содержанию холестерина в крови, то печень начинает выделять с
желчью больше холестерина, и его концентрация в желчном пузыре
растет. Рост содержания холестерина в крови предрасполагает к
формированию камней желчного пузыря.
Широкий спектр белков организма синтезируется исключительно в
рибосомальной сети и грубом эндоплазматическом ретикулуме
гепатоцитов. Кроме того, в печени происходит придание аминокислотной смеси,
циркулирующей с кровью, сбалансированности через трансформацию
аминокислот, находящихся в крови в избытке, в другие, которых
организму не достает. Поэтому печеночную недостаточность можно
рассматривать как патологическое состояние организма, которое характеризует
сниженная резистентность к недостатку в пище незаменимых и условно
незаменимых аминокислот. Необходимым условием адекватности синтеза
белков в печени потребностям всего организма являются:
♦ достаточное число дееспособных гепатоцитов;
♦ нормальная активность в печени ингибиторов белкового синтеза;
♦ достаточное поступление нутриентов во внутреннюю среду из
внешней.
Снижение в плазме крови концентрации альбумина как результат
печеночной недостаточности через падение ее коллоидно-осмотического
давления ведет к интерстициальному отеку. Низкая свертываемость крови
вследствие печеночной недостаточности связана с дефицитами в крови
факторов ее свертывания.
382
Глава 23
Известно, что процесс свертывания крови во многом представляет
собой последовательную активацию его факторов. Активированные формы
факторов свертывания крови инактивируются преимущественно в печени.
Поэтому при печеночной недостаточности раз начавшийся процесс
свертывания крови идет аномально долго, распространенно и интенсивно, так
как его не тормозит инактивация факторов свертывания печенью. Его
результатом может быть коагулопатия потребления, то есть нарушение
свертывания крови вследствие дефицита его факторов, обусловленного
избыточным потреблением при неограниченном свертывании. Коагуло-
патию такого генеза обостряет низкий синтез протеинов с функциями
прокоагулянтов в печени.
НАРУШЕНИЯ СОЗНАНИЯ И ЭНЦЕФАЛОПАТИЯ,
СВЯЗАННЫЕ С ПЕЧЕНОЧНОЙ НЕДОСТАТОЧНОСТЬЮ
Как только у больного с печеночной недостаточностью развиваются
нарушения сознания, прежде всего как причину их возникновения
следует исключить или установить гипогликемию. Гипогликемию вследствие
печеночной недостаточности у большинства больных вызывает угнетение
глюконеогенеза в печени в результате:
♦ острой печеночной недостаточности;
♦ застойной сердечной недостаточности;
♦ алкогольного поражения печени.
Можно считать, что цирроз печени любых этиологии и патогенеза
может привести к гипогликемии (табл. 23.3). У алкоголиков
гипогликемия как причина нарушений сознания возникает после длительного
голодания, когда адаптивная реакция интенсификации глюконеогенеза из-за
недостаточного числа интактных гепатоцитов не может удержать
концентрацию глюкозы в плазме крови на нормальном уровне.
Таблица 23.3
Причины гипогликемии при печеночной недостаточности и циррозе печени
♦ Угнетение глюконеогенеза всей печенью из-за снижения числа функционально
интактных гепатоцитов
♦ Падение содержания гликогена в печени
♦ Угнетение нормальной реакции гепатоцитов и всей печени на эффект глюкагона
как стимулятора глюконеогенеза
♦ Рост содержания в крови инсулина как следствие падения его инактивации
печенью при шунтировании инсулина с кровью из системы воротной вены мимо
печени к сердцу
Энцефалопатия, обусловленная печеночной недостаточностью, - это
патологическое состояние, которое вызывают нарушения обмена в ней-
Патофизиология печени
383
ронах головного мозга и связанные с ними расстройства внутрицен-
тралъных отношений, обусловленные прямым шунтированием токсине-
ских веществ с циркулирующей кровью из просвета кишечника, минуя
печень, в головной мозг.
Источником одного из этих токсических веществ, аммиака, являются
протеины в просвете кишечника. Другими токсическими веществами, чье
действие на нейроны головного мозга может вызвать печеночную
энцефалопатию (табл. 23.4), считают меркаптаны и некоторые жирные кислоты.
Нарушения сознания вплоть до комы у больных с печеночной
недостаточностью и связанной с ней энцефалопатией обостряют: гипокалиемия,
которая увеличивает образование аммиака в почках, артериальная гипок-
семия, гипоксия, седативные средства и наркотические анальгетики (в том
числе и через центральное угнетение внешнего дыхания).
Таблица 23.4
Клинические стадии печеночной энцефалопатии
Стадия
I
II
III
IV
Психический статус
Эйфория или депрессия, замедленные
психические реакции, расстройства сна и речи
Летаргия и более выраженные
психические расстройства
Бессвязная речь, сонливость, возбуждение
Кома при потере реакции на болевые
раздражители
Астерикс
+/-
+
+
-
Электроэнцефалограмма
Чаше нормальная
Аномалии
Отклонения от нормы
Выраженные
аномалии
Примечание: Астерикс («хлопающий тремор») - неритмичные ассимметричные «провалы» при
определенном положении конечностей, головы и позвоночника, когда больной не может
удержать их в данном положении.
Были попытки связать развитие энцефалопатии вследствие
печеночной недостаточности с образованием в печени гепатоцитами с
расстроенными функциями биоактивных аминов, сходных по молекулярной
структуре с норадреналином. Предполагали, что данные биоактивные амины
как лиганды к нормальным рецепторам норадреналина в головном мозге,
взаимодействуя с ними, вызывают нарушения сознания.
ЦИРРОЗ ПЕЧЕНИ, ПОРТАЛЬНАЯ ГИПЕРТЕНЗИЯ И АСЦИТ
Цирроз печени - это определенный комплекс патологических
изменений структуры паренхимы печени, который приводит к разнообразным
расстройствам функциональных систем организма и проявляет себя
рядом связанных с ними симптомов. В основном цирроз составляют
обширный фиброз и образование регенеративных узлов в печени. Их
обуславливают некроз гепатоцитов и разрушение поддерживающей ретикулиновой
384
Глава 23
сети печеночной паренхимы как начальный этап последующего фибрози-
рования. Цирроз следует считать конечной стадией многих хронических
патологических процессов, вызывающих дегенерацию и гибель
гепатоцитов. При этом симптомы цирроза отражают тяжесть патологических
изменений печени, то есть степени потери печенью нормально
функционирующих гепатоцитов, а также роста сосудистого сопротивления на уровне
печеночных синусоид, и часто не связаны с этиологическими факторами
ее заболеваний. Рост сосудистого сопротивления на уровне печени
вследствие цирроза приводит к портальной гипертензии и ее следствиям,
асциту и спленомегалии.
Асцит (гр. askos мех для хранения жидкости), брюшная водянка,
скопление жидкости в брюшной полости. Кроме фильтрации лимфы к
асциту ведет активация ренин-ангиотензин-альдостеронового механизма в
ответ на секвестрацию части крови в брюшной полости, вызывающая
задержку в организме натрия и тем самым повышающая объем
внеклеточной жидкости. Предположительным звеном патогенеза асцита как
следствия роста объема внеклеточной жидкости является снижение инактивации
альдостерона в результате низкого числа нормальных гепатоцитов у
больных с циррозом.
Прогрессирующее снижение числа нормальных гепатоцитов у
больных с циррозом печени проявляет себя большей выраженностью
симптомов печеночной недостаточности.
Чаще всего цирроз печени является следствием вирусного гепатита,
хронической алкогольной интоксикации, длительной механической
обструкции желчевыводящих путей (вторичный билиарный цирроз), а также
гемохроматоза.
Портальная гипертензия - это патологический подъем давления крови в
воротной вене как причина дисфункций печени, связанных с ними
патологических сдвигов обмена веществ, а также перераспределения внеклеточной
жидкости между жидкостными секторами организма.
Давление в воротной вене постоянно меняется вслед за изменениями
давления в брюшной полости. Давление в брюшной полости возрастает
на высоте вдоха и умеренно растет после приема пищи. Поэтому
однократное определение давления крови в воротной вене и венах, по которым
кровь поступает в воротную, недостаточно для выявления ^и&клюжння^
портальной гипертензии.
К постоянной портальной гипертензии приводит сужение (частичная
обтурация) воротной вены или обструкция кровотоку любого другого
уровня на пути крови из воротной вены к сердцу. Если нет патологиче-
крови в воротной вене печеночных синусоид снижает сосудистое
сопротивление, которое кровь преодолевает, проходя через печень. В
результате портальной гипертензии не развивается.
В норме давление крови в воротной вене колеблется от 5 до 10 мм рт. ст.
Его можно определить после пункции селезенки и регистрации давления
в этом органе, которое у большинства больных соответствует давлению в
Патофизиология печени
385
воротной вене. Кроме того, давление крови в воротной вене определяют,
производя катетеризацию пупочной вены. Для определения уровня
обструкции-окклюзии сосудов как причины портальной гипертензии
осуществляют катетеризацию нижней полой вены для заведения кончика
тонкого катетера в одну из печеночных вен. Затем катетер осторожно
продвигают по печеночной вене до прекращения свободного продвижения
катетера по сосудам печени. После этого начинают фиксировать давление
крови, которое называют печеночным давлением заклинивания.
Печеночное давление заклинивания у здоровых людей составляет 4-10 мм рт. ст.
при нормальном давлении в печеночной вене на уровне 2 мм рт. ст. Так
как анастомозы между печеночными синусоидами исключительно
многочисленны, то давление заклинивания отражает давление во всех
синусоидах печени. Если нормальный уровень печеночного давления
заклинивания сочетается с увеличенным давлением в воротной вене, то препятствие
кровотоку находится на пресинусоидальном уровне, а портальную гипер-
тензию вызывают:
♦ тромбоз воротной вены или ее сдавление злокачественными
опухолями;
♦ внутрипеченочное препятствие кровотоку на уровне мелких ветвей
воротной вены, которое обуславливают тромбофлебит вследствие
шистозоматоза, врожденная гипоплазия портальных венул и другие
заболевания.
Кроме того, причиной роста сосудистого сопротивления на
пресинусоидальном уровне, который вызывает портальную гипертензию, может
быть интенсификация эритропоэза в печени. После шести недель
беременности главными центрами эритропоэза плода являются печень и
селезенка. На пятом месяце беременности начинаются кроветворение и
эритропоэз в костном мозге. После рождения роль главного центра
эритропоэза играет костный мозг. Если на организм ребенка действуют стимулы
выброса в кровь эритропоэтина и эритропоэза (хроническая циркулятор-
ная гипоксия, постоянно низкое парциальное давление кислорода в
альвеолах как результат его низких значений во вдыхаемой газовой смеси), то
массы всего костного мозга не хватает для обеспечения компенсаторно-
приспособительной реакции интенсификации образования эритроцитов.
Для ее обеспечения возобновляется эритропоэз в печени. Возникновение
очагов кроветворения на пресинусоидальном уровне в печени
увеличивает его сосудистое сопротивление и повышает давление в воротной вене.
Редкой причиной портальной гипертензии является врожденный
дефект развития воротной вены в виде ее окклюзии.
На уровне синусоид рост сосудистого сопротивления часто
обуславливают жировая дистрофия и амилоидоз печени. К редким причинам роста
сосудистого сопротивления на уровне синусоид относят гипертрофию
эндоплазматического ретикулума гепатоцитов.
На синусоидально-постсинусоидальном уровне наиболее частая
причина портальной гипертензии - цирроз печени. Цирроз печени снижает
площадь общего поперечного сечения ее сосудистого русла на уровне си-
386
Глава 23
нусоид через некроз гепатоцитов как причину обширного фиброза и
образования регенеративных узлов.
Рост сосудистого сопротивления на постсинусоидальном уровне про-
движения крови через печень к сердцу и портальную гипертензию вызывают
идиопатический тромбоз печеночных вен (синдром Бадда-Хиари),
обструкция-окклюзия нижней полой вены, констриктавный перикардит. Самая
частая причина портальной гипертензии вследствие возрастания сосудистого
сопротивления на постсинусоидальном уровне - сердечная недостаточность,
которая через падение насосной функции правого желудочка повышает
давление в нижней полой и воротной венах.
Если после развития портальной гипертензии в силу какой-либо
причины (прием пищи, адаптивно-компенсаторное возрастание минутного
объема кровообращения и др.) происходит рост поступления крови в систему
воротной вены, то для поддержания адекватной объемной скорости
продвижения крови через печень к сердцу растет давление крови в сосудах
органов живота, из которых кровь оттекает в воротную вену. Этот рост
давления происходит посредством перенаполнения данных внутренних
органов кровью. В результате происходит секвестрация части объема
циркулирующей крови в органах брюшной полости, вызывающая гиповолемию.
В ответ на гиповолемию, связанную с портальной гипертензией, через
адренергический спазм приводящих артериол нефрона и прямую нейрогу-
моральную симпатическую стимуляцию юкстагломерулярного аппарата
почек активируется ренин-ангиотензин-альдостероновый механизм. Рост
активности альдостерона в циркулирующей крови увеличивает общее
содержание натрия в организме и объем внеклеточной жидкости. Рост объема
внеклеточной жидкости может увеличить объем циркулирующей крови, но
не повышает резистентность больного к кровопотере. Дело в том, что
увеличение объема циркулирующей крови у больных с портальной
гипертензией приводит к ее большей секвестрации во внутренних органах, кровь из
которых оттекает в воротную вену. Поэтому остается низкой та часть
крови, которая может быть задействована для транспорта кислорода и
энергопластических субстратов на системном уровне.
Портальная гипертензия, сохраняющаяся в течение недель или
месяцев, ведет к расширению коллатералей, по которым кровь продвигается
из внутренних органов в обход воротной вены и печени к сердцу.
Некоторыми направлениями коллатерального кровотока вследствие портальной
гипертензии являются:
♦ прямо из воротной вены вдоль желудка вверх по пищеводу в
направлении к анастомозам с межреберными венами;
♦ по сосудистой сети части селезенки, находящейся между
диафрагмой и задней стенкой полости живота;
♦ по пупочной вене.
Прохождение значительного объема крови в обход печени
обуславливает транзиторный патологический рост концентрации в крови глюкозы
вскоре после приема пищи. Затем концентрация глюкозы снижается
вследствие падения образования гликогена в печени из-за низкого числа нор-
Патофизиология печени
387
мальных гепатоцитов и шунтирования крови, содержащей глюкозу, в обход
клеток печени, образующих гликоген. Кроме того, шунтирование крови в
обход печени служит одной из причин возрастания содержания в крови
аминокислот из-за снижения фракции их пула в циркулирующей крови,
утилизируемой для синтеза белков и глюконеогенеза в гепатоцитах.
Коллатеральный кровоток по венам пищевода при портальной гипер-
тензии обуславливает их характерные расширения, которые называют ва-
рикозами. Вены пищевода локализованы в его подслизистом слое, где
соединительно-тканные образования практически не ограничивают их ди-
латации вследствие портальной гипертензии. Диаметр расширенных вен
пищевода у больных с портальной гипертензией достигает 8 мм.
Варикозные расширения образуются, когда давление в воротной вене
становится большим, чем 20 мм рт. ст. При повышении давления в воротной
вене до уровня более высокого, чем 35 мм рт. ст., возможны разрывы вен
пищевода в местах их варикозных расширений и их следствие, массивная
кровопотеря.
Портальная гипертензия как патологическое состояние аномально
высокого сосудистого сопротивления на пути крови от внутренних органов в
печень и дальше к сердцу вызывает застой крови в соответствующих
органах, который увеличивает их размеры и массу. В частности растут
размеры и масса селезенки, то есть развивается спленомегалия.
Застой крови и патогенно высокое давление крови в органе могут
служить причиной тромбоза ее сосудов и инфаркта селезенки. В
результате застоя крови в селезенке происходит избыточная активация
находящихся в ней клеток системы мононуклеарных фагоцитов организма.
Патогенно избыточное число активированных макрофагов селезенки
обуславливает повышенный уровень деструкции в ней тромбоцитов,
эритроцитов и лейкоцитов. В результате содержание тромбоцитов в
циркулирующей крови может снизиться от нормальной величины, составляющей
190-405* 109/л, до в два раза меньших значений. Общее содержание
лейкоцитов в крови при спленомегалии, связанной с портальной гипертензией,
обычно находится на уровне более низком, чем 3,8109/л. Это в основном
связано с падением числа циркулирующих с кровью полиморфонуклеаров.
Портальная гипертензия может обуславливать тромбоз мезентериаль-
ных сосудов как причину непроходимости кишечника, некроза стенки
кишки и перитонита.
Кроме тромбоза относительно крупных мезентериальных сосудов,
портальная гипертензия служит причиной тромбоза мелких
мезентериальных венул, что препятствует всасыванию из просвета кишечника и
нарушает его моторику. Отчасти нарушения моторики и всасывания у
больных с циррозом и портальной гипертензией могут быть связаны с отеком
стенок кишечника.
Печеночная синусоида - это капилляр, стенки которого обладают
наибольшей проницаемостью среди всех других микрососудов организма.
Около 0,3 % массы крови, поступающей в синусоиды печени, уходит из
их просвета в интерстиций. Величина данного показателя в скелетных
388
Глава 23
мышцах находится на уровне 0,01 %. Любое увеличение
гидростатического давления крови в синусоидах печени повышает ультрафильтрацию
из их просвета и лимфооток из паренхимы печени. При этом лимфоотток
может стать столь интенсивным, что начинается фильтрация лимфы через
капсулу печени в брюшную полость. Причина фильтрации лимфы - это
преобладание образования лимфы над объемной скоростью ее тока по
сосудам в печени как фактор роста гидростатического давления в просвете
лимфатических сосудов. В результате фильтрации лимфы через капсулу
печени в брюшной полости накапливается асцитическая жидкость,
которая содержит белок, образуемый гепатоцитами (около 30 г/л). При этом
концентрация белка в жидкости преобладает над его содержанием в
плазме крови, так как лимфа оттекает из основного локуса синтеза протеинов
в организме, то есть из печени.
Через действие аналогичного механизма портальная гипертензия
повышает формирование лимфы в стенке кишечника и ведет к ее
фильтрации в брюшную полость. Лимфа, оттекающая от стенки кишечника,
содержит в большом количестве липиды, поступившие в лимфу в ходе
кишечного всасывания. Это обуславливает преобладание содержания липи-
дов в асцитическои жидкости над их концентрацией в плазме крови, что
особенно выражено сразу после приема пищи.
Глава 24
ПАТОФИЗИОЛОГИЯ ЖЕЛУДОЧНОЙ СЕКРЕЦИИ,
ГАСТРИТОВ, ЯЗВЕННОЙ БОЛЕЗНИ ЖЕЛУДКА
И ДВЕНАДЦАТИПЕРСТНОЙ КИШКИ
Выделяют три связанные между собой фазы секреции соляной кислоты:
♦ нейрогенную (вагальную);
♦ гастральную, основным медиатором которой является гастрин;
♦ интестинальную.
Нейрогенную фазу индуцирует возбуждение рецепторов коры
головного мозга в ответ на действие таких раздражителей как вид, запах, вкус
пищи, которое активирует нейроны ядра блуждающего нерва.
Эфферентная импульсация по блуждающему нерву на периферии приводит к
высвобождению из постганглионарных парасимпатических нервных
окончаний ацетилхолина, оказывающего воздействие на париетальные клетки
слизистой оболочки желудка, повышающее секрецию ими соляной
кислоты. Кроме того, вагальная стимуляция стенки желудка ведет к
высвобождению гастрина клетками его слизистой оболочки в антральном отделе и
повышает чувствительность париетальных клеток к стимуляции гастри-
ном. Вагальная стимуляция также повышает секрецию главными
клетками, что усиливает образование желудочного сока и повышает содержание
в нем соляной кислоты и пепсина.
Операция стволовой ваготомии направлена на устранение
преобладания кислотно-протеолитических и деструктивных относительно
слизистой оболочки желудка и всей его стенки свойств эюелудочного сока над
защитной по отношению к ним функцией слизистой оболочки. В
результате снижения интенсивности эфферентации по блуждающему нерву
вследствие ваготомии падают как объем желудочной секреции, так и
концентрация протонов в желудочном соке. Особенно этот эффект ваготомии
выражен у больных с язвой двенадцатиперстной кишки, необходимым условием
возникновения которой является гиперсекреция соляной кислоты
париетальными клетками желудка. Следует заметить, что и после ваготомии
париетальные клетки желудка сохраняют способность усиливать секрецию
соляной кислоты в ответ на прямую стимуляцию гастрином и гистамином.
Это во многом обуславливает возможность повторного возникновения
язвы желудка и двенадцатиперстной кишки после ваготомии.
Сдвиги внутрицентральных отношений и эмоционального статуса в
виде длительных повышенной тревожности, эмоционального эквивалента
вовлеченности в межличностный конфликт (гнев, враждебность)
приводят к росту объемной скорости кровотока в слизистой оболочке желудка
и усиливают секрецию соляной кислоты париетальными клетками
слизистой оболочки стенки желудка. Одновременно растет кровоснабжение
мышечных элементов стенки желудка. Хроническое повышение уровня
390
Глава 24
секреции соляной кислоты при таких патогенных сдвигах внутрицен-
тральных отношений как причин нарушений эмоционального статуса
может быть фактором развития пептической язвы. Чаще при этом
развивается пептическая язва двенадцатиперстной кишки, и реже - желудка.
Длительные депрессия и страх ведут к падению кровоснабжения
слизистой оболочки и угнетают секрецию соляной кислоты ее клетками, а
также снижают уровень кровоснабжения мышечных элементов.
Снижение кровоснабжения всей стенки желудка и ее слизистой оболочки может
быть причиной падения защитной функции слизистой оболочки, что
ведет к образованию язвы желудка (чаще) и двенадцатиперстной кишки.
Известно, что раздражение чревных нервов усиливает выделение
щелочного секрета с низким содержанием пепсина железами слизистой
оболочки стенки желудка в области привратника.
Основным медиатором гастральной фазы желудочной секреции
является гастрин. Его секреция растет как вследствие нервных регуляторных
влияний (вагальная стимуляция), так и в результате действия
механических стимулов, то есть растяжения желудка при поступлении в него пищи
и жидкостей. К росту секреции гастрина приводит воздействие на железы
желудка ряда пептидов, некоторых аминокислот, а также циркулирующих
с кровью адреналина и пептида бомбезина. Тормозят секрецию гастрина
эфферентная импульсация по тормозящим волокнам блуждающего нерва,
действие некоторых гуморальных факторов, а также падение
концентрации протонов в просвете желудка и желудочном соке. Наиболее
изученными из гуморальных факторов торможения секреции гастрина (агентов
паракринной регуляции, тормозящих секрецию гастрина) являются
следующие регуляторные пептиды: секретин, вазоактивный интестинальный
пептид, гастральный тормозящий пептид, соматостатин, кальцитонин и
глюкагон.
Гастрин представляет собой наиболее изученный гуморальный
стимулятор секреции соляной кислоты париетальными клетками слизистой
оболочки стенки желудка. Гастрин содержится в цитоплазматических
секреторных гранулах гастриновых клеток (клетки-G), которые в
одиночку или небольшими скоплениями залегают между другими
эпителиальными клетками, в основном в средних или глубоких частях антральных
пилорических желез. Основная форма гастрина - это гастрингептадека-
пептид (G-17), состоящий из семнадцати аминокислот. G-17 составляет
90 % гастрина, который содержит слизистая оболочка стенки желудка в
его антральном отделе.
Гастрин оказывает ряд регуляторных эффектов, затрагивающих
многие эффекторы функций. На уровне желудка этот ряд включает
значительный рост секреции соляной кислоты париетальными клетками, слабое
или умеренное повышение секреции пепсина, усиление секреции
внутреннего фактора Кастла, рост кровоснабжения стенки желудка,
интенсификацию моторики желудка и действие в качестве фактора клеточного
роста на эпителий слизистой оболочки дна желудка. К системным
эффектам гастрина в первую очередь следует отнести стимуляцию секреции
Патофизиология желудочной секреции...
391
ионов и воды поджелудочной железой, печенью, а также усиление
секреции в просвет кишечника панкреатических ферментов. Гастрин
стимулирует секрецию инсулина и кальцитонина, повышает тонус нижнего
пищеводного сфинктера, усиливает моторику кишечника и вызывает
сокращения гладкомышечных элементов стенок желчного пузыря. Кроме того,
гастрин угнетает всасывание глюкозы, ионов и воды из просвета
кишечника. Гастрин расслабляет пилорический сфинктер желудка, илеоцекальный
сфинктер и сфинктер Одди. Высвобождая гастрин, желудок
функционирует как орган-эффектор системы регуляции пищеварения.
У больных с язвой двенадцатиперстной кишки концентрация гастрина
в циркулирующей крови утром натощак не отличается от его содержания
в крови здоровых людей. У пациентов с язвой желудка утром и натощак
фиксируют умеренный подъем концентрации гастрина в крови. У
больных с низким уровнем секреции соляной кислоты в просвет желудка
(меньше 10 мэкв/ч) выявляют обратную связь между концентрацией
гастрина в циркулирующей крови и уровнем секреции соляной кислоты. Это
связано с отсутствием торможения высвобождения гастрина слизистой
оболочкой антрального отдела желудка под влиянием роста концентрации
протонов в его просвете.
Рост содержания в плазме крови гастрина до уровня более высокого,
чем 1 пг/л, характеризует ряд патологических состояний. При синдроме
Золлингера-Эллисона гипергастринемия (патологически высокая
концентрация гастрина в циркулирующей крови) связана не с гиперсекрецией
гастрина, а выступает результатом повышенного образования гастрина
клетками опухоли поджелудочной железы (гастринома).
У части больных с язвенной болезнью и язвой двенадцатиперстной
кишки причиной как гипергастринемии, так и роста секреции соляной
кислоты в просвет желудка является гиперплазия антральных G-клеток.
Перерастяжение желудка при его заполнении пищей и жидкостью также
обуславливает рост уровня секреции гастрина. У небольшой части
хирургических больных после резекции желудка по способу Бильрот II в
результате дефекта операции оставляют часть антрального отдела, которая в
результате гастроэнтеростомии оказывается соединенной с афферентной
дуоденальной петлей. Растяжение оставшей части антрального отдела
вследствие поступления в него щелочных желчи и секрета
поджелудочной железы ведет к гипергастринемии. При введении секретина больным
с гиперплазией G-клеток в антральном отделе, а также пациентам с ги-
пергастринемией вследствие дефекта резекции желудка по способу
Бильрот II секреция гастрина падает. У больных с гастриномой введение
секретина повышает образование гастрина ее клетками.
У пациентов с пернициозной анемией, обусловленной аутоиммунным
поражением слизистой оболочки желудка, гипергастринемия
представляет собой результат низкого содержания в желудочном соке протонов как
стимула повышенной секреции гастрина G-клетками антрального отдела
и их гиперплазии. У больных с атрофическим аутоиммунным гастритом и
нормальным всасыванием витамина B]2 из просвета кишки механизм раз-
392
Глава 24
вития гипергастринемии аналогичен, то есть гипергастринемия связана с
ахлоргидрией (см. ниже).
Простой (неаутоиммунный) атрофический гастрит не приводит к
росту содержания гастрина в циркулирующей крови. Отсутствие реакции на
ахлоргидрию в виде гипергастринемии у таких больных связано с
патологическим изменением слизистой оболочки желудка по типу атрофии со
снижением числа G-клеток в его антральном отделе.
Почки и тощая кишка - это основные органы-эффекторы системы
инактивации и элиминации гастрина. Поэтому хроническая почечная
недостаточность и состояние после резекции тонкой кишки могут
обуславливать гипергастринемию и быть причиной роста деструктивной
активности желудочного содержимого.
При аномально повышенной секреции гормона паращитовидных
желез в крови растет концентрация кальция, что служит стимулом для
секреции гастрина слизистой оболочкой антрального отдела и может быть
причиной гипергастринемии. Феохромоцитома как причина гиперадрена-
линемии через стимуляцию секреции G-клетками приводит к росту
содержания гастрина в циркулирующей крови.
Роль гистамина как стимулятора секреции соляной кислоты
париетальными клетками была переосмыслена после синтеза антагонистов гис-
таминовых Н2-рецепторов, находящихся в зоне париетальных клеток
желудка. Эти препараты (циметидин, ранитидин и др.) подавляют как ис-
ходную (базалъную, то есть до приема пищи и в условиях относительного
покоя) секрецию соляной кислоты, так и ее интенсификацию в ответ на
воздействие гастрина, гистамина, а также вследствие гипогликемии и
раздражения блуждающего нерва. Гистамин представляет собой основной
медиатор желудочной секреции, который действует на париетальные
клетки вместе с гастрином и холинергическими веществами.
Полагают, что наружная поверхность париетальных клеток содержит
рецепторы к ацетилхолину, гастрину, а также гистамину. Вероятно,
блокада Н2-гистаминовых рецепторов их антагонистами нарушает
взаимодействие между всеми данными рецепторами, что понижает секрецию
соляной кислоты париетальными клетками через снижение
чувствительности обкладочной клетки ко всему спектру стимуляторов выведения в
просвет желудка протонов и хлоридных анионов.
Уровень секреции соляной кислоты обкладочными клетками желудка
связан с концентрацией в циркулирующей крови гипофизарных, надпо-
чечниковых гормонов и гормонов щитовидной и паращитовидных желез.
При полном прекращении или недостаточном уровне их высвобождения в
циркулирующую кровь падает уровень желудочной секреции.
Нормальная регенерация эпителиоцитов желудка предположительно зависит от
секреции гормона роста аденогипофизом, который, кроме того, во многом
детерминирует секреторные функции слизистой оболочки желудка. Инте-
стинальная фаза желудочной секреции вызывается прохождением
частично гидролизированной пищи в тонкую кишку, которое ведет к
высвобождению ее стенкой гастрина и других гормонов. Патогенно интенсивное
Патофизиология желудочной секреции...
393
высвобождение гастрина слизистой оболочки тонкой кишки в начальный
период интестинальной фазы секреции можно считать одним из
механизмов возникновения язвы двенадцатиперстной кишки. После операции по
поводу язвы желудка по способу Бильрот I секреция гастрина G-клетками
желудка уже не может быть причиной избыточной секреции соляной
кислоты, и гипергастринемия как фактор рецидива язвы представляет собой
результат извращения интестинальной фазы желудочной секреции.
Сдвиги внутрицентральных отношений в виде эмоционального
эквивалента отчаяния, подавленности, невротические расстройства по типу
депрессии угнетают желудочную секрецию через эфферентную импуль-
сацию по волокнам блуждающего и чревных нервов.
Выделяют два главных механизма местной регуляции секреции гастрина:
♦ Действие всех стимуляторов секреции гастрина тормозится в
результате роста концентрации протонов непосредственно в межклеточных
пространствах вокруг G-клетки. Угнетение секреции гастрина G-клет-
кой происходит при снижении рН желудочного сока до уровня 1,0.
♦ Попадание в просвет тонкой кишки кислого и с высоким
содержанием липидов содержимого желудка приводит к высвобождению
его стенкой энтерогастронов, которые тормозят как секрецию
гастрина G-клетками, так и снижают стимулирующее действие гастрина
на париетальные клетки. Наиболее изученным из энтерогастронов
является секретин.
Уровень максимальной секреции соляной кислоты париетальными
клетками желудка при ее стимуляции пентагастрином зависит от общего
числа обкладочных клеток в слизистой оболочке желудка, которое у
здоровых мужчин находится на уровне одного миллиарда. У больных с
язвой двенадцатиперстной кишки это число возрастает в 1,5-2 раза.
Причина такого различия точно не известна. Представляется, что ее
можно связать с полигенным наследованием предрасположенности к
язвенной болезни.
Патологические сдвиги желудочной секреции обычно составляют
изменения ее объема и (или) концентрации протонов в желудочном соке.
Показателем секреции соляной кислоты слизистой оболочкой желудка в
его просвет является количество миллиграмэквивалентов протонов,
которое поступает за единицу времени в просвет желудка. Этот показатель
представляет собой произведение объема секрета, поступающего в
просвет желудка, и концентрации в нем свободных ионов водорода. У
больных язвенной болезнью желудка обычно нет достоверной связи между
интенсивностью секреции протонов и выраженностью патологических
изменений стенки полого органа. Известно только то, что сниженная
секреция протонов может быть одним из признаков атрофического гастрита.
У здоровых людей концентрация свободных ионов водорода в
желудочном соке варьирует весьма значительно, от ахлоргидрии (состояние
крайне сниженной секреции соляной кислоты в просвет желудка при рН
желудочного сока на уровне 8,0) до высоких значений содержания протонов
в желудочном соке (рН = 1,0). Клиническое обследование больных обыч-
394
Глава 24
но не выявляет достоверной связи между уровнем активной секреции
протонов и выраженностью симптомов болезни. Можно только считать,
что при постоянной ахлоргидрии вероятность длительного существования
не закрывающихся язв желудка невелика. Полное отсутствие секреции в
просвет желудка (achylia gastrica) встречается редко. У больных почти
всегда выявляют секрецию того или иного компонента желудочного сока
в просвет органа. В этой связи становится ясным, что предпочтительней
использовать термины «ахлоргидрия» и «анацидность».
Анацидность - рН содержимого желудка на уровне более высоком,
чем 6,0, во время наибольшей интенсивности стимуляции пентагастри-
ном секреции соляной кислоты париетальными клетками.
Анацидность - это признак сугубо патологических изменений
функций желудка, который свидетельствует о значительной потере его
слизистой оболочкой париетальных клеток, секретирующих соляную кислоту.
Анацидность могут обусловить рак желудка, аутоиммунный атрофиче-
ский гастрит, при котором иммунная система больного образует аутоан-
титела к аутоантигенам париетальных клеток. Атрофический
аутоиммунный гастрит характеризует последовательная потеря содержимым
желудка сначала соляной кислоты, затем пепсина и, наконец, внутреннего
фактора Кастла. Поэтому анацидность всегда означает высокую вероятность
развития у больного пернициозной анемии.
Следует заметить, что в течение последних десятилетий были
пересмотрены первоначальные представления о нормах желудочной
кислотности. При этом было установлено, что «разнообразие показателей
секреции нельзя втиснуть в узкие рамки границ нормы и деятельность
секреторного аппарата желудка соответствует определенным типологическим
особенностям» (Фролькис А.В.). Среди здоровых людей, у которых не
выявляют признаков гастрита и язвенной болезни, уровень секреции
соляной кислоты в просвет желудка варьирует от состояния идиопатиче-
ской желудочной гиперсекреции, при котором часовой дебит базальной
секреции в 5 раз превышает средние показатели у представителей
основной части популяции, до другой крайности, ахлоргидрии.
Стойкое повышение уровня секреции соляной кислоты в просвет
желудка характеризует такие заболевания как язвенная болезнь с
локализацией язвы в двенадцатиперстной кишке, синдром Золлингера-Эллисона,
гиперплазия G-клеток в антральном отделе желудка, а также
патологическое состояние желудочной секреции в результате дефекта резекции
желудка в виде оставленной части антрального отдела, переполняемой
секретом из просвета приводящей дуоденальной петли.
Образование язвы желудка и двенадцатиперстной кишки представ-
ляет собой результат полиэтиологичного преобладания кислотно-про-
теолитической деструктивной относительно слизистой оболочки
желудка и его стенки активности желудочного сока над защитной
относительно такой активности функцией слизистой оболочки.
Выделяют три уровня защиты слизистой оболочки желудка и всей его
стенки от деструктивных влияний со стороны желудочного сока (рис. 24.1):
Патофизиология желудочной секреции...
395
Эпителиоциты поверхностного слоя слизистой оболочки секрети-
руют слизь и бикарбонатные анионы, тем самым создавая градиент
концентрации протонов между желудочным соком и поверхностью
эпителиальной клетки, непосредственно над которой рН слизи
почти не отличается от рН внеклеточной жидкости и жидкой части
плазмы.
Известно, что апикальная часть плазматической мембраны эпите-
лиоцита поверхностного слоя слизистой оболочки желудка мало
проницаема для протонов, что предотвращает их обратную
диффузию в цитозоль эпителиоцитов. Защитная реакция снижения
электропроводимости апикальной мембраны предотвращает ее
деструкцию, несмотря на снижение рН среды, непосредственно
контактирующей с мембраной, до 2,0.
Кроме того, эпителиоциты слизистой оболочки стенки желудка
обладают способностью выводить протоны через свою базолатераль-
ную мембрану в обмен на катионы натрия и бикарбонатные анионы.
Секреция слизи
эпителиоцитами
желудка
Активное
выведение протонов через
базолатеральную
мембрану эпите-
лиоцита
Действие простаг-
ландинов-
вазодилататоров и
факторов
клеточного роста
Активность пепсина и высокая
концентрация протонов в
желудочном содержимом
♦
ш.
■<*<
н+
Вымывание протонов из
стенки желудка через пе-^
риферическое
кровообращение
Секреция
бикарбонатных
анионов
эпителиоцитами желудка
Снижение
электропроводности апикальной
мембраны эпителиоци-
' та при возрастании
концентрации протонов
в непосредственной
близости от нее
>
Рис. 24.1. Защитные механизмы слизистой оболочки желудка
Нормальная репликация эпителиоцитов слизистой желудка
представляет собой необходимое условие ее нормальной защитной функции.
Ненарушенная микроциркуляция - это необходимое условие эффективности
такого защитного механизма как выведение протонов из стенки желудка,
после того, как они в патогенно высокой концентрации преодолевают за-
396
Глава 24
щитный барьер слизистой в местах нарушения его целостности. При этом
нарушения целостности слизистой оболочки могут быть результатами
недостаточно быстрой репликации эпителиоцитов.
Некоторые простагландины, образуемые в стенке желудка, повышают
ее защитную функцию через рост объемной скорости тока крови по
микрососудам, а также как стимуляторы секреции слизи и бикарбонатного
аниона эпителиоцитами.
ПАТОГЕНЕЗ ГАСТРИТОВ
Острый гастрит как патологическое изменение слизистой оболочки
желудка и его стенки - это в первую очередь воспаление слизистой
оболочки, которое моэ/сет быть распространенным в пределах всего органа
или захватывать какой-либо из его отделов.
Наиболее частой причиной гастрита является побочное действие
лекарственных средств и ядов (нестероидные противовоспалительные
средства, алкоголь), которые, снижая защитную функцию слизистой
оболочки, ведут к обратному поступлению пепсина и протонов из состава
желудочного содержимого в слизистую оболочку желудка и его стенку.
Поступление в желудок сильных кислот (серной, соляной) и
щелочей, а также растворов таких веществ как формальдегид и тринитрофе-
нол через некробиотические изменения стенки желудка приводят к ее
воспалению, происходящему и в слизистой оболочке (коррозивный
гастрит).
Патологическая централизация кровообращения в остром периоде
после тяжелых ранений, травм, ожогов, которую усиливает и поддерживает
длительная патогенная боль, через юкстакапиллярное шунтирование
(прохождение крови мимо обменных капилляров) в желудке снижает
защитную функцию слизистой оболочки, что также может быть причиной
острого гастрита.
Редкая причина острого гастрита - это бактериальная инвазия в
стенку желудка (флегмонозный гастрит). Чаще всего стенку желудка
инфицирует стрептококк, реже - стафилококки, Escherichia coli и некоторые из
видов Proteus.
Лишь у 30 % больных острый гастрит проявляет себя такими
симптомами как ощущение жжения и боль в эпигастральной области, тошнота и
рвота. У небольшой части пациентов острый гастрит приводит к
кровотечениям из поврежденных первичной альтерацией и самим воспалением
сосудов стенки желудка (геморрагический эрозивный гастрит).
Эндоскопическое исследование у больных с острым гастритом
выявляет петехиальные кровоизлияния в слизистую оболочку, ее отечность, а
также различные степени деструкции вплоть до поверхностных
изъязвлений. Эндоскопия - это окончательный этап выявления острого гастрита у
больных.
Патофизиология желудочной секреции...
397
Патогенетические принципы терапии острого гастрита включают:
♦ устранение воздействия, снижающего защитную функцию
слизистой оболочки (прекращение приема или введения нестероидных
противовоспалительных средств, отказ от спиртного) или прямо
повреждающего стенку желудка (промывание желудка щелочными
растворами при отравлении сильными кислотами и т.д.);
♦ применение антацидных средств, и в частности блокаторов Нг-
гистаминовых рецепторов, для снижения деструктивных
относительно слизистой оболочки желудка и всей его стенки свойств
желудочного содержимого.
При геморрагическом гастрите в связи с гиповолемией и
патологическими реакциями на кровопотерю может возникнуть необходимость
инфузий и трансфузий. При этом для прекращения кровотечения из
стенки желудка в его просвет рН желудочного содержимого
необходимо поддерживать на уровне более высоком, чем 3,5. Кровотечение в
связи с геморрагическим гастритом иногда может служить показанием
к оперативному вмешательству с высокой послеоперационной
летальностью.
Хронический гастрит в морфопатогенетическом отношении
характеризует лимфоцитарная инфильтрация мышечного слоя слизистой
оболочки желудка. При этом лимфоциты в основном инфильтрируют
часть мышечного слоя, непосредственно контактирующую со слизистой
оболочкой желудка.
Наиболее частыми причинами хронического гастрита являются
постоянное угнетение алкоголем защитной функции слизистой оболочки
желудка, ее повреждения ионизирующим излучением, а также
аутоиммунное поражение через образование аутоантител к антигенам поверхности
эпителиоцитов желудка.
При хроническом гастрите типа А аутоантитела образуются к
антигенам поверхности париетальных клеток слизистой оболочки. Данный
вид гастрита характеризует высокий уровень секреции гастрина и его
концентрации в циркулирующей крови в результате постоянного
действия такого стимула секреции G-клетками как ахлоргидрия, возникающая
вследствие аутоиммунного поражения обкладочных клеток. При
хроническом гастрите типа А поражения слизистой оболочки желудка в
основном локализованы в области дна и тела желудка. Пернициозная анемия
при данном типе хронического гастрита связана с недостаточным
образованием в слизистой оболочке желудка внутреннего фактора Кастла,
которое падает из-за ее аутоиммунного поражения.
Патологические изменения слизистой оболочки желудка при
хроническом гастрите типа В происходят преимущественно в антральном
отделе, то есть в меньшей степени затрагивают секретирующие соляную
кислоту клетки слизистой оболочки желудка. У некоторых пациентов с
данным типом хронического гастрита в крови выявляют аутоантитела к G-
клеткам. Поражение секретирующих гастрин клеток слизистой оболочки
при хроническом гастрите типа В снижает уровень его секреции. Поэтому
398
Глава 24
у пациентов с таким типом гастрита уровень высвобождения соляной
кислоты в просвет желудка обычно не повышен, и чаще находится на
нормальном уровне.
Хронический гастрит того и другого типов нередко протекает
бессимптомно. Как и многие аутоимммунные болезни хронический гастрит типа
А сочетается с другими заболеваниями аутоиммунной природы,
гипотиреозом, связанным с аутоиммунным поражением щитовидной железы,
инсулинзависимым сахарным диабетом и др.
За 10-20 лет своего течения и тот и другой типы гастрита приводят к
тяжелой атрофии слизистой оболочки желудка и пернициозной анемии.
Оба типа гастрита - это факторы риска язв желудка и
двенадцатиперстной кишки, полипов и рака желудка, но рак чаще возникает у больных с
гастритом типа В.
Под эозинофильным гастроэнтеритом понимают инфильтрацию эо-
зинофилами антрального отдела желудка и (или) тонкого кишечника.
Такая инфильтрация, которую считают следствием неясных
иммунопатологических сдвигов, приводит к утолщению стенок желудка в антральном
отделе, что приводит к обструкции вхождению пищи в желудок из
пищевода. У больных с таким редким видом гастрита часто выявляют рост
содержания эозинофилов в циркулирующей крови. Под влиянием терапии
кортикостероидами эозинофильный гастроэнтерит может подвергнуться
обратному развитию через апоптоз эозинофилов.
Гиперплазию желудка в морфопатогенетическом отношении
характеризует, прежде всего, значительное увеличение складок слизистой
оболочки желудка. Исследование биоптата выявляет гиперплазию
(увеличение числа клеток до величины большей, чем обычная) клеток слизистой
оболочки желудка, а также (у некоторых больных) ее инфильтрацию
активированными клеточными эффекторами воспаления со свойствами
фагоцитов и лимфоцитами. Гиперплазию желудка вызывают:
♦ болезнь Менетрие;
♦ гиперсекреторная гастропатия;
♦ гастринома (синдром Золлингера-Эллисона).
Для болезни Менетрие типичны грубые извитые складки слизистой
оболочки с гиперплазией слизистых клеток и уменьшением числа
париетальных и главных клеток (зимогенные клетки, секретирующие пепсин).
Гиперплазия эпителиоцитов желудка сопровождается образованием
зазоров между ними, что обуславливает потери альбумина через
патологически измененную слизистую оболочку в просвет желудка. Снижение про-
теолитической активности желудочного сока как причина недостаточного
частичного переваривание пищи в желудке, а также рост коллоидно-
осмотического давления содержимого желудка, поступающего в
двенадцатиперстную кишку, из-за миграции в просвет желудка альбумина
вызывают осмотическую диарею у пациентов с болезнью Менетрие. Часто
единственным симптомом болезни является боль, локализованная в эпи-
гастральной области. Гипоальбуминемия как следствие болезни может
обусловить интерстициальные отеки.
Патофизиология желудочной секреции...
399
ЭТИОЛОГИЯ И ПАТОГЕНЕЗ ЯЗВЕННОЙ БОЛЕЗНИ
Язвенная болезнь (болезнь пептической язвы) - совокупность
заболеваний, которые в морфопатогенетическом отношении объединяет
образование под влиянием высокой концентрации свободных ионов водорода в
просвете полых органов желудочно-кишечного тракта и протеолитиче-
ской активности их содержимого (пепсин) язв слизистой оболочки и под-
слизистого слоя (пептические язвы), которые способны обуславливать
ряд вторичных изменений макроструктуры органов, определенные
дисфункции в системе пищеварения, а также сдвиги регуляции и гомеоста-
зиса на уровне всего организма.
Чаще всего пептические язвы возникают в желудке и проксимальной
части двенадцатиперстной кишки. Реже они поражают слизистую оболочку
нижней трети пищевода и тонкой кишки. Язвы нижней трети пищевода,
как правило, представляют собой следствие рефлюксэзофагита. В тонкой
кишке пептическое изъязвление слизистой оболочки может происходить в
дивертикуле Меккеля, слизистая оболочка которого содержит клетки гаст-
рального эпителия способные к секреции соляной кислоты и пепсина.
Пептические язвы реже выявляют у женщин, чем у мужчин. Язвы
двенадцатиперстной кишки возникают в десять раз чаще, чем язвы желудка и
гораздо раньше по ходу онтогенеза (в среднем на 10 лет). Максимума риск
трансформации предрасположенности к язвенной болезни в образование
язвы двенадцатиперстной кишки достигает в 40 лет (50 лет при язвенной
болезни с пептической язвой в желудке). Риск повторного развития язвы
двенадцатиперстной кишки после ее закрытия под влиянием терапии составляет
80 %. Вероятность возникновения язвы двенадцатиперстной кишки у
больных с пептической язвой желудка - 33 %.
Хроническая интоксикация ядами, которые содержит табачный дым,
ведет к образованию пептической язвы желудка и двенадцатиперстной
кишки через действие ряда патогенетических механизмов:
♦ снижение экзокринной функции поджелудочной железы как
причина падения выведения ее щелочного секрета в двенадцатиперстную
кишку и относительно низкой концентрации бикарбонатных
анионов в ее просвете;
♦ увеличение выброса кислого желудочного содержимого в
двенадцатиперстную кишку вследствие падения тонуса пилорического
сфинктера;
♦ рост секреции главными клетками пепсиногена I, о котором
свидетельствует рост его концентрации в сыворотке крови.
Известно, что риск возникновения пептической язвы у больных
ревматоидным артритом, регулярно принимающих нестероидные
противовоспалительные средства, составляет 20-30 %. Нестероидные
противовоспалительные средства (аспирин, ибупрофен, вольтарен и др.) угнетают
синтез простагландинов в стенке желудка, тормозя в ней активность цик-
лооксигеназы и липооксигеназы. В результате падает защитная функция
слизистой оболочки стенки желудка, во многом определяемая образова-
400
Глава 24
нием и функционированием в ней простагландинов вазодилататоров с
функциями факторов клеточного роста.
Кортикостероиды, используемые для длительной фармакотерапии, как
блокаторы анаболизма и катаболические гормоны антагонисты инсулина
тормозят репаративные процессы в желудочном эпителии, что снижает его
защитную функцию. Предположительно через рост чувствительности ад-
ренорецепторов сосудистого русла к эндогенным катехоламинам
кортикостероиды усиливают спазм резистивных сосудов и прекапиллярных
сфинктеров в стенке желудка, что увеличивает объем юкстакапиллярного
шунтирования и снижает защитную функцию слизистой оболочки желудка.
Если у больных язвенной болезнью с локализацией язвы в
двенадцатиперстной кишке уровень базальной секреции соляной кислоты обкла-
дочными клетками и интенсивность ее выброса париетальными клетками
после стимуляции пентагастрином обычно умеренно или значительно
повышены, то у пациентов с пептической язвой желудка эти показатели
находятся на нормальном уровне или даже несколько снижены. Это
заставляет предположить более значимую роль в патогенезе пептической язвы
желудка падения защитной функции слизистой оболочки.
Концентрация гастрина в сыворотке крови у больных с пептической
язвой желудка как натощак, так и после приема пищи несколько повышена.
При пептической язве двенадцатиперстной кишки она аномально
возрастает после приема пищи, а натощак находится на нормальном уровне.
Язвенная болезнь с пептическими язвами желудка и
двенадцатиперстной кишки - это полигенное заболевание. Поэтому для родственников
пробанда первой степени вероятность страдать от пептической язвы в три
раза выше, чем у представителей основной группы популяции.
Гастринома как фактор роста кислотно-деструктивной активности
желудочного сока ведет к образованию пептических язв. При этом синдром
Золлингера-Эллисона может быть элементом синдрома множественной
эндокринной неоплазии. Хронический гастрит типа В может привести к язве
желудка, так как снижает защитную функцию его слизистой оболочки.
Образование в ходе раннего органогенеза легких и органов системы
пищеварения из одного источника, кишки, а также прямая связь систем
внешнего дыхания и пищеварения с внешней средой возможно
предопределяют частое одновременное возникновение у больных хронического
обструктивного бронхита (бронхиальной астмы) и пептических язв
желудка и двенадцатиперстной кишки.
Как явствует из содержащегося в данной главе краткого очерка
физиологии и патофизологиии желудочной секреции патогенные сдвиги
психофизиологического профиля личности могут обусловить
возникновение пептических язв желудка и двенадцатиперстной кишки.
Характерной боли эпигастральной локализации, которую
обуславливает действие протонов в высокой концентрации на лишенную защитной
функции слизистой оболочки стенку желудка в области язвы, нет у 25 %
больных с язвенной болезнью. Так как у пациентов с пептической язвой
двенадцатиперстной кишки ее образование - это прежде всего результат
Патофизиология желудочной секреции...
401
повышенного уровня секреции протонов париетальными клетками, то
прием пищи у таких больных снижает интенсивность боли. Дело в том,
что пища экранирует открытый подслизистый слой в области язвы от
действия протонов, содержание которых в желудочном соке при пептиче-
ских язвах двенадцатиперстной кишки обычно повышено.
Полагают, что свою роль в образовании пептической язвы желудка
может играть инфицирование просвета желудка спиралевидными грамот-
рицателъными бактериями, обитающими в области привратника (пило-
рический геликобактер). Данные бактерии часто обнаруживают в биопта-
те слизистой оболочки желудка при гастрите, язвенной болезни желудка и
при не связанной с пептической язвой диспепсии. Доказана роль данной
инфекции в развитии хронического гастрита. Роль спиралевидных
бактерий в развитии пептической язвы желудка до сих пор не выяснена.
Считают, что препараты висмута в сочетании с амоксиллином и метронида-
золом у части больных могут подавлять рост данных бактерий как
причину хронического гастрита.
Инфицирование просвета желудка Helicobacter pylori (Нр) и контакт
данного микроорганизма с клеточными элементами иммунной системы,
локализованными в слизистой оболочке желудка и его стенке, в
настоящее время признают этиологическими факторами, причиной пептических
язв желудка и двенадцатиперстной кишки или звеньями патогенеза
данных заболеваний. Наличие в просвете желудка Нр достоверно связано с
развитием хронического гастрита, пептических язв, аденокарциномы
желудка, а также лимфоидной тканевой лимфомы желудка. При этом нет
достоверной связи между инфицированием просвета желудка
определенным клоном Нр и развитием определенного заболевания желудка. Так у
членов одной семьи при нахождении в желудке представителей одной
линии Нр развиваются различные заболевания желудка. В данном случае
мы имеем дело с реализацией одной из нозологических закономерностей,
которую представляется возможным сформулировать следующим
образом: индукция развития определенной болезни в силу действия ее
причины, взаимодействия этиологического фактора с организмом, не всегда
находится в жесткой связи с этиологическим фактором и часто
преимущественно определяется системной реактивностью и характером
реагирования на определенном уровне структурно-функциональной
организации. Иными словами, результат взаимодействия Нр с клеточными
элементами слизистой оболочки желудка всегда непредсказуем, но всегда
приводит к изменению экспрессии генома клеток стенки желудка,
относящихся к иммунной системе. Инфицирование Нр просвета желудка
усиливает экспрессию на поверхности эпителиоцитов желудка молекул
второго класса из главного комплекса тканевой совместимости, а также таких
молекул с функцией стимуляторов локальных гастральных Т-лимфоцитов
как В7-1 и В7-2 (костимулирующие молекулы). В результате гастральные
Т-лимфоциты атакуют эпителиоциты слизистой желудка, что
обуславливает образование пептической язвы. Локальная активация Т-лимфоцитов в
стенке желудка повышает в ней содержание цитокинов со свойствами фак-
402
Глава 24
торов клеточного роста, что в частности может предрасполагать к
неконтролируемой клеточной пролиферации и малигнизации клеток.
Основной патогенетический принцип терапии, направленной на
закрытие (эпителизацию) пептической язвы - это восстановление
нормального равновесия между кислотно-протеолитическими свойствами
желудочного сока и защитной функцией ее слизистой оболочки.
Блокаторы Н2-гистаминовых рецепторов, ослабляя действие
основного медиатора желудочной секреции гистамина на париетальные клетки
слизистой оболочки желудка, снижают секрецию в просвет желудка
протонов и хлоридных анионов, тем самым уменьшая деструктивные
свойства желудочного сока. Один из препаратов данной группы, циметидин,
обладает такими побочными эффектами как импотенция, гинекомастия и
олигоспермия.
Сукральфтат представляет собой защищающее слизистую оболочку
желудка средство, которое, вступая во взаимодействие с белками некро-
биотически измененных клеток в области пептической язвы, формирует
на месте язвы экранирующий слой, который защищает стенку желудка от
деструкции под влиянием высокой концентрации протонов, пепсина и
желчных кислот.
Прием внутрь антацидов, то есть препаратов, содержащих слабые
кислоты и их соли, анионы которых связывают протоны в просвете
желудка, снижает концентрацию свободных ионов водорода в просвете желудка
и деструктивные свойства желудочного сока. Общим недостатком
известных антацидов является поступление вместе с ними в просвет
желудка и кишечника чужеродных осмолей, которые через изменение
осмотических концентраций в просвете кишечника и специфическое действие
вызывают дисфункции системы пищеварения, диарею и патологические
сдвиги ионного состава внутренней среды. Так, например, гидроокись
алюминия связывает фосфат в просвете тонкого кишечника, что может
обусловить гипофосфатемию (патологически низкое содержание
фосфатного аниона в плазме крови).
Ингибиторы /Г- КА-АТФазы (омепразол и др.), действуя на уровне
плазматической мембраны, быстро и значительно уменьшают
секрецию соляной кислоты, тем самым обуславливая эпителизацию
пептической язвы.
Следует заметить, что современные средства фармакотерапии
язвенной болезни желудка в основном действуют на уровне клеточных
эффекторов патологических процессов, чьи дисфункции вызывают образование
пептических язв. При этом большинство из них может и не затронуть те
нарушения системной регуляции и внутрицентральных отношений,
которые на уровнях желудка и двенадцатиперстной кишки приводят к
образованию пептических язв. В этой связи при лечении больных с пептически-
ми язвами желудка и двенадцатиперстной кишки особое внимание
следует уделять диете, режиму, нетрадиционным методам лечения,
психотерапии, рассматривая их как средства устранения расстройств
внутрицентральных отношений и патологических сдвигов системной регуляции.
Патофизиология желудочной секреции...
403
До сих пор не получено убедительных доказательств того, что жидкая
щадящая диета способствует заживлению язвы. Больные просто должны
избегать тех продуктов, которые вызывают у них диспепсию. Поздний
ужин в данном случае безусловно вреден, так как он стимулирует
секрецию соляной кислоты в ночное время, когда больной спит и не может
принять антацид.
Глава 25
ПАТОГЕНЕЗ ДИСФУНКЦИЙ СИСТЕМЫ ПИЩЕВАРЕНИЯ
НА УРОВНЕ КИШЕЧНИКА
СИНДРОМ НЕДОСТАТОЧНОГО
ВСАСЫВАНИЯ
Неадекватно низкое относительно потребностей организма
всасывание нутриентов из просвета кишечника вызывают:
♦ расстройства любого вида и этапа пищеварения;
♦ патологические изменения органов-эффекторов пищеварительной
системы.
Симптомокомплекс, вызываемый синдромом недостаточного
всасывания как патологическим состоянием всего организма, весьма
вариабелен. При этом специфичные симптомы могут быть проявлениями
недостаточного всасывания определенных нутриентов (табл. 25.1).
Абсорбцию липидов из просвета кишечника снижают:
1. Падение секреции ферментов поджелудочной железы в
двенадцатиперстную кишку вследствие: а) хронического панкреатита; б) рака
поджелудочной железы; в) фиброза поджелудочной железы, связанного с
ее кистозным перерождением.
2. Дефицит связанных желчных солей в просвете кишечника в
результате холестаза.
3. Деконъюгация желчных солей в кишечнике при падении скорости
пассажа его содержимого. Падение скорости пассажа как причину де-
конъюгации обуславливают: а) слепые петли кишечника,
образовавшиеся в результате оперативных вмешательств на органах живота (после
резекции желудка по способу Бильрота II и др.); б) дивертикул тощей
кишки сам по себе или в сочетании со свищом между тощей и толстой
кишкой; в) расстройства моторики кишечника, в частности
обусловленные нарушениями водно-солевого обмена, склеродермой и другими
причинами низкой сократительной способности гладкомышечных
элементов стенки кишки.
4. Недостаточное перемешивание желудочного содержимого с
желчными солями и панкреатическими ферментами как результат
операций на желудке, и особенно его субтотальной резекции по способу
Бильрота И. После такого вмешательства на желудке одной из причин
недостаточного всасывания может быть стаз в приводящей петле кишки
как причина патогенной интенсификации бактериального роста в ее
просвете.
К недостаточному всасыванию нутриентов могут приводить болезни,
в основе которых лежат патологические изменения стенки тонкого
кишечника:
Патогенез дисфункций системы пищеварения на уровне кишечника
405
Таблица 25.1
Связь симптомов и патогенеза синдрома недостаточного всасывания
как патологического состояния всего организма
Симптом или
проявления синдрома
Ведущие звенья патогенеза
Повышенная
утомляемость, мышечная слабость
и падение массы тела
Понос
Метеоризм
Аменорея, снижение
либидо
Глосситы, хейлоз,
стоматит
Макроцитарные и железо-
дефицитные анемии
Пурпура и экхимоз
Боли в костях
и остеопороз
Тетания и парестезии
Нейропатия на уровне
периферических нервов
Ксерофтальмия и ночная
слепота
Фолликулярный
гиперкератоз и дерматиты
Экзема
Никтурия
Артериальная
гипотензия
Азотемия
Снижение поступления во внутреннюю среду организма
нутриентов как источников свободной энергии.
Компенсаторная активация катаболических процессов в ответ на
голодание, связанное с синдромом недостаточного всасывания.
Анемия. Гипокалиемия как одно из следствий диареи и
причина увеличения трансмембранного потенциала клеток
скелетных мышц и падения возбудимости миоцитов
Нарушение всасывания воды из просвета кишечника как
результат роста содержания в нем невсасываемых осмолей из-за
недостаточности полостного и пристеночного пищеварения
Ферментация не всосавшихся углеводов под влиянием эшимов
бактерий, находящихся в просвете кишечника
Снижение поступления во внутреннюю среду
энергопластических субстратов как причина вторичного гипопитуитаризма
Дефицит в организме железа, витамина В12, фолатов и
других витаминов
Снижение всасывания витамина В12, фолиевой кислоты и
железа
Недостаточное всасывание витамина К. Патологически
низкая концентрация тромбина в крови как результат
эндогенного голодания
Белковое голодание с нарушением процессов образования
костной ткани и остеопорозом. Падение всасывания кальция
Снижение содержания в организме кальция* и магния
Дефицит в организме витамина В]2 и тиамина
Снижение всасывания из просвета кишечника витамина А
Низкое поступление во внутреннюю среду витамина А, других
витаминов, цинка, а также незаменимых жирных кислот
Патогенез неизвестен
Низкая скорость всасывания воды из просвета кишечника и
гипокалиемия
Низкое всасывание воды и ионов из просвета кишечника как
причина гиповолемии. Алиментарная дистрофия вследствие
эндогенного голодания
Снижение скорости клубочковой фильтрации вследствие
падения минутного объема кровообращения, связанного с гипо-
волемией и алиментарной дистрофией. Рост катаболизма в
ответ на эндогенное голодание
Целиакия (нетропическая спру). Хотя этиология и патогенез
заболевания еще не ясны полностью, можно считать, что болезнь представляет
собой следствие повышенной чувствительности к глютену, который
содержат зерна злаков, употребляемых в пищу человеком. Целиакия - это
406
Глава 25
полигенное заболевание. Исследование биоптатов тонкой кишки, взятых
у родственников пробанда первой степени, выявляет у 10-15 % из них
патологические изменения слизистой оболочки, характерные для целиакии.
Кроме того, о мультифакториальном характере наследования заболевания
свидетельствует тот факт, что в организме 60-90 % больных
нетропической спру определяют антигены клеточной поверхности В8 и Dw3 из
системы антигенов лейкоцитов человека.
Энзимопатическая гипотеза трактует целиакию (глютеновую
болезнь) как интестинальную энзимопатию. Глютен - это белковый
компонент клейковины, склеивающего компонента зерен пшеницы, ржи,
ячменя, овса. Одна из фракций глютена глиадин может оказывать токсическое
действие на слизистую оболочку тонкой кишки, причем токсична для
кишки только одна из 50 фракций самого глиадина. В основе энзимопати-
ческой гипотезы лежит предположение, что целиакию обуславливает
врожденный дефицит специфической пептидазы, расщепляющей
токсическую фракцию глиадина.
Суть иммунопатологической гипотезы глютеновой болезни сводится
к тому, что ее вызывает патологическая реакция системы иммунитета на
глютен. Об иммунопатологической природе заболевания свидетельствует
циркуляция с кровью больных антител к глютену. Кроме того,
патологические изменения слизистой оболочки кишки вследствие целиакии
характеризуются признаками аутоиммунного поражения, то есть ростом
содержания в слизистой оболочке тучных, плазматических клеток, а также
эозинофилов.
Согласно синтетической теории целиакии нерасщепленный
вследствие энзимопатии глютен взаимодействует с клетками системы мононук-
леарных фагоцитов в подслизистом слое стенки кишки, что приводит к их
активации как начальному моменту аутоиммунного поражения.
Болезнь Уиппла -это редкое заболевание, которое в первую очередь
проявляет себя спутанностью сознания, ослаблением памяти, такими
признаками поражения черепно-мозговых нервов как нистагм и
офтальмоплегия, общей слабостью, болями в суставах и в области живота, диареей
и прогрессирующим снижением массы тела.
Расширение лимфатических сосудов брыжейки при болезни Уиппла
обычно сочетается с нарушениями всасывания из просвета кишечника. У
больных данным заболеванием падает абсорбция ксилозы в кишечнике. У
части больных голодание из-за нарушения всасывания, обусловленного
болезнью Уиппла, приводит к анемии и гипоальбуминемии. Одной из
причин снижения концентрации альбумина в плазме крови можно считать
потери белка в интерстиций стенки кишки и ее просвет из сосудов
кишечной стенки. Возможно расширение сосудов стенки кишечника
связано с локальной гиперцитокинемией как следствием хронической
активации мононуклеарных фагоцитов подслизистого слоя.
При болезни Уиппла в слизистой оболочке стенки кишки выявляют
специфические для нее макрофаги, цитозоль которых содержит характер- \
ные крупные гранулы. При электронной микроскопии в макрофагах вы-
Патогенез дисфункций системы пищеварения на уровне кишечника 407
являют характерные тельца размером 0,3-2,5 мкм с ультраструктурой
микроорганизмов.
Предположительно основное звено патогенеза болезни Уиппла - это
потерявшее защитное значение воспаление кишечной стенки, основным
клеточным эффектором которого является ее мононуклеарный фагоцит,
активированный при фагоцитозе еще неизвестного возбудителя
инфекционного заболевания после его инвазии в стенку кишки
Эозинофильный гастроэнтерит - это заболевание, в основе
которого лежат патологические изменения желудка, тонкой и толстой кишки
еще пока неясной этиологии. В морфопатогенетическом отношении
болезнь характеризуют рост содержания эозинофилов в циркулирующей
крови и инфильтрация эозинофилами стенок желудка и кишечника.
Предположительно болезнь представляет собой следствие
иммунопатологических сдвигов, о чем свидетельствует ее частое сочетание с
бронхиальной астмой, пищевой аллергией, аллергическим ринитом, а также
аутоиммунным гастритом. У некоторых больных идентификация аллергена
и его элиминация из потребляемой пищи приводит к устойчивой
ремиссии. Не исключено, что по своему патогенезу данное заболевание почти
аналогично бронхиальной астме как эозинофильному десквамационному
бронхиту. Очевидно, что при этих двух заболеваниях эозинофил является
клеточным эффектором основного звена их патогенеза, эозинофильного
воспаления.
Первичный или вторичный синило ид оз подслизистого слоя стенки
тонкого кишечника также может быть причиной падения кишечного
всасывания. При этом связанное с амилоидозом угнетение моторики
тонкого кишечника, предрасполагает к патологическому росту
кишечной флоры в его просвете, что еще в большей степени угнетает
всасывание нутриентов.
Болезнь Крона (региональный энтерит) - это хроническое
воспалительное заболевание стенок органов, составляющих пищеварительный
канал, которое обуславливает их патологические изменения на любом
уровне от ротовой полости до анального отверстия. У 50 % больных
патологические изменения стенок захватывают подвздошную и часть
толстой кишки. Чаще заболевание возникает в возрастном диапазоне от 12 до
30 или в возрасте около 50 лет.
Региональный энтерит - это полигенное заболевание. Известно, что
вероятность страдать от болезни выше у родственников пробанда, чем у
других представителей популяции. Мужчины болеют чаще, чем
женщины. Чаще всего болезнь возникает у мужчин еврейской национальности.
Неоднократно роль возбудителя болезни Крона приписывалась
патогенным микроорганизмам (РНК-содержащим вирусам и др.).
Есть основания считать региональный энтерит аутоиммунным
заболеванием, так как у больных были выявлены патологические: а) рост числа
определенных Т-лимфоцитов; б) клоны Т-клеток; в) функции Т-лимфо-
цитов. Об аутоиммунном характере заболевания свидетельствует его
частое сочетание с аутоиммунным артритом (у 10-12 % больных).
408
Глава 25
Патологические изменения стенки кишки при болезни Крона включают:
♦ утолщение стенки вследствие воспаления, захватывающего все ее
слои;
♦ увеличенные мезентериальные лимфатические узлы;
♦ гранулемы стенки кишки (в 50 % случаев);
♦ глубокие извитые или линейные изъязвления стенки кишки как
причина образования свищей и стриктур;
♦ чередующиеся участки нормальной и патологически измененной
слизистой оболочки кишки.
Боль в области живота у пациентов с болезнью Крона можно описать как
кишечные колики. Кишечные колики у пациентов связаны с обструкцией
пассажу кишечного содержимого вследствие утолщения стенки
патологически измененной кишки со снижением ее просвета и возникновением стриктур.
Из других симптомов болезни следует выделить лихорадку, потерю
веса, сильно выраженную общую слабость, анорексию и диарею.
Клиническое течение болезни характеризует чередование ремиссий и обострений.
Пернициозная анемия при болезни Крона связана с низкой абсорбцией
витамина BJ2 патологически измененной подвздошной кишкой. У части
больных нормохромная нормоцитарная анемия сочетается с задержкой
роста и представляет собой следствие голодания как результата
недостаточного всасывания нутриентов.
Нередко патологические изменения полых органов при болезни Крона
захватывают стенки желчевыводящих путей, что может быть причиной
жирового перерождения печени, перихолангита, неспецифического
гепатита, цирроза или склерозирующего холангита. Это повышает риск
образования камней желчного пузыря, а у некоторой части пациентов даже
приводит к циррозу печени.
Воспалительные изменения кишечника на определенном уровне у
части пациентов с болезнью Крона приводят к блокаде выведения мочи
по мочеточникам и обструктивной уропатии. При болезни Крона из-за
усиленного цитолиза эпителиоцитов полых органов
желудочно-кишечного тракта растет выведение мочевой кислоты с мочой и образование
почечных камней соответствующего химического состава.
До сих пор полностью неясен патогенез такого проявления болезни
Крона как узловатая эритема, из возможных механизмов возникновения
которой нельзя исключить аутоиммунное поражение кожи.
Опасным осложнением болезни Крона является перфорация стенок
полых органов и кровотечение из сосудов, поврежденных при
образовании язв стенки кишки.
Одной из причин низкого относительно потребностей организма
всасывания нутриентов может быть недостаточная абсорбционная площадь
внутренней поверхности кишечника. Снижение абсорбционной площади
до неадекватно низкого уровня - это чаще всего следствие обширной ре-'
зекции тонкого кишечника, которую по жизненным показаниям
предпринимают в связи с болезнью Крона или при ишемическом некрозе
кишечника, вызванном тромбозом мезентериальных сосудов (синдром короткой
Патогенез дисфункций системы пищеварения на уровне кишечника 409
петли). Резекция 50 % тонкой кишки не приводит к синдрому
недостаточного всасывания при условиях структурно-функциональной
сохранности оставшейся части и гладкого течения послеоперационного периода.
Резекция 40-50 % тонкой кишки обычно хорошо переносится больными
при сохранении проксимального отдела двенадцатиперстной кишки, дис-
тального отдела подвздошной кишки и илеоцекального клапана. Резекция
подвздошной кишки и только одного илеоцекального клапана может
приводить к выраженной диарее и нарушениям всасывания при удалении
менее 30 % кишки.
Гипогликемия как результат синдрома недостаточного всасывания
стимулирует секрецию соляной кислоты париетальными клетками
эпителия стенки желудка. Кроме того, транзиторную гиперсекрецию соляной
кислоты клетками желудка можно считать следствием частичного
выпадения тонкой кишки как эффектора эндокринных регуляторных систем из
регуляции желудочной секреции.
Расстройства кишечного всасывания вследствие резекции
проксимальной части тонкой кишки в основном состоят из снижения
абсорбции кальция, фолиевой кислоты и железа. Если удаляется подвздошная
кишка, то в первую очередь падает всасывание желчных кислот и
витамина Ви.
При синдроме короткой петли, в частности вызванном удалением
значительной части подвздошной кишки, желчные кислоты начинают
попадать в толстую кишку, что может быть причиной поноса.
Синдром короткой кишки характеризует связанная с качественным
голоданием и парентеральным питанием жировая дистрофия печени, та
или иная степень ее недостаточности, а также образование оксалатных
почечных камней.
Интестинальная лимфангиэктазия - состояние расширения
лимфатических сосудов стенок кишечника, брыжейки и роста давления в них
лимфы вторичное по отношению к врожденному сужению
лимфатических сосудов на определенном уровне, системным, локальным болезням и
патологическим процессам, которые через возникновение препятствия
оттоку лимфы от кишечника снижают кишечное всасывание.
Интестинальная лимфангиэктазия может быть вторичной по отношению к
туберкулезу кишечника, болезни Уиппла, раневым и травматическим
повреждениям кишечника (кровоизлияние в брыжейку и др.); ее могут
обуславливать злокачественные опухоли соответствующей локализации, а также
репаративное или реактивное образование фиброзных тканей в ретропе-
ритонеальном пространстве.
Интестинальная лимфома - плотное злокачественное
новообразование стенок кишечника и брыжейки, состоящее из малигнизированных
клеточных элементов лимфатической или ретикулоэндотелиальной ткани,
которые кажутся незрелыми или напоминают лимфоциты,
плазматические клетки или гистиоциты. Интестинальная лимфома создает
механическое препятствие лимофоттоку из кишечника, особенно препятствуя ему
при лимфоматозе, то есть при множественных, широко распространенных
410
Глава 25
зонах кишечника и брыжейки, пораженных лимфомой. В результате у
больных с лимфомами и лимфоматозом стенок кишечника и его
брыжейки резко падает кишечное всасывание и развивается эндогенное
голодание. При распространении лимфоматоза за пределы полости брюшины
причиной обструкции лимфооттоку из кишечника становится
механическое ему препятствие, обусловленное образованием увеличенных
лимфатических узлов в ретроперитонеальном пространстве. Интестинальную
лимфому и лимфоматоз в клиническом отношении характеризуют
хроническая лихорадка и периодические кратковременные обострения
синдрома недостаточного всасывания как патологического состояния всего
организма.
При радиационном энтерите, состоянии после гастрэктомии
(субтотальной резекции желудка), а также у больных с инсулинзависимым
сахарным диабетом патогенез синдрома недостаточного всасывания
характеризует одновременное действие нескольких патогенетических
механизмов.
Лучевое поражение кишечника (радиационный энтерит) при
лучевой терапии злокачественных опухолей соответствующей локализации
обуславливает стриктуры кишки, фиброзное перерождение ее стенок и
брыжейки, а также интестинальную лимфангиэктазию. Это вызывает
синдром недостаточного всасывания, ведущее звено патогенеза которого
составляют нарушения микроциркуляции в стенке кишки и брыжейке и
потеря ими кровеносных и лимфатических микрососудов.
Причинами синдрома недостаточного всасывания у больных с
инсулинзависимым сахарным диабетом являются:
♦ нарушения иннервации стенки кишки, связанные с диабетической
нейропатией на уровне периферических симпатических и
парасимпатических нервов и вызывающие нарушения моторики
кишечника;
♦ обусловленный угнетением моторики кишечника патогенно
интенсивный бактериальный рост в его просвете;
♦ недостаточность экзокринной функции поджелудочной железы,
особенно частая при вторичном сахарном диабете.
Нарушения кишечного всасывания липидов может обусловить абета-
липопротеинемия (синдром Бассена-Корнцвейга), представляющая
собой результат патогенной мутации в гене аполипротеина В. Данная
мутация состоит в потере геном четырех пар нуклеотидов, что образует кодон,
хранящий информацию о прекращении синтеза аполипопротеина,
который локализован по ходу транскрипции в цепи нуклеотидной
последовательности гораздо проксимальнее нормального стоп-кодона. В результате
геном экспрессирует аномальный «усеченный аполипопротеин». Эту
моногенную болезнь, наследуемую по аутосомно-рецессивному типу,
характеризуют отсутствие в плазме крови липопротеинов с плотностью ниже
1,063 (липопротеинов низкой плотности), акантоцитоз, пигментная
дегенерация клетчатки, а также атаксия, то есть неспособность к
координации произвольных движений. Акантоцитоз - редкое состояние, при кото-
Патогенез дисфункций системы пищеварения на уровне кишечника
411
ром большинство эритроцитов относятся к акантоцитам, то есть красным
кровяным клеткам с множественными шиловидными выростами. В
плазме больных с абеталипопротеинемией фиксируют патологически низкий
уровень концентрации холестерина и триглицеридов. Недостаток в
организме холестерина и липидов как субстратов синтеза фосфолипидов
клеточных мембран предположительно можно считать причиной акантоци-
тоза. Гиполипопротеинемию со снижением в плазме крови
концентрации липопротеинов низкой плотности следует связать с аномалиями
синтеза аполипопротеина В. Если больные абеталипопротеинемией
гомозиготны по гену болезни, то гипохолестеринемия (патологически
низкое содержание холестерина в плазме крови) особенно выражена. Так
как нормальный аполипопротеин В необходим для образования хило-
микронов в интестинальных клетках (необходимый этап кишечной
абсорбции липидов), то у больных с абеталипопротеинемией падает
всасывание липидов из просвета кишки. Липиды при этом задерживаются
энтероцитами, в цитозоле которых при исследовании биоптатов
выявляют липидные включения.
Патологические изменения структуры стенки кишечника и
дисфункции ее клеточных элементов, связанные с бактериальными, вирусными и
паразитарными инфекциями, также могут вызывать синдром
недостаточного всасывания как патологическое состояние всего организма. Так
вирусные и бактериальные энтериты вызывают преходящее угнетение
кишечного всасывания через снижение активности дисахаридазы,
расщепляющей дисахариды в просвете кишки и на поверхности энтероцитов, и
(или) прямые повреждения интестинальных клеток. Тропическое спру -
эндемическая болезнь кишечника с поносами и стеатореей вследствие
недостаточной кишечной абсорбции липидов. Стеаторея- выделение
большого количества жира с испражнениями. Полагают, что
тропическая спру представляет собой инфекционное заболевание неизвестной
этиологии. Кроме того, причиной недостаточного всасывания может
быть инвазия в просвет кишечника кривоголовок, то есть кровососущих
нематод семейства Ancyclostomatidae, ленточных червей (кишечные
паразитические черви, относящиеся к классу Cestoidea), а также
паразитарная инвазия нематодой Strongyloides stercoralis (угрица кишечная) и
другими нематодами.
Хроническая циркуляторная гипоксия кишки при одновременной ате-
росклеротической обтурации верхней и нижней брыжеечных артерий
приводит к недостаточному всасыванию. Кроме обычных симптомов
недостаточного всасывания, об атеросклерозе мезентериачьных артерий
освидетельствуют понос с кровью в диарейной жидкости и острая боль в
области живота после приема пищи.
Врожденные и приобретенные иммунодефщиты, в основе которых
лежат дисфункции В-лимфоцитов, могут приводить к синдрому
недостаточного всасывания, особенно если они обуславливают недостаточное
{Образование сывороточных иммуноглобулинов А и G, а также интести-
нального иммуноглобулина А, функционирующего в качестве элемента
412
Глава 25
защитной системы слизистой оболочки кишечной стенки,
предотвращающей попадание патогенных бактерий и чужеродных антигенов из
просвета кишечника во внутреннюю среду. В результате
иммунодефицита как следствия гипоиммуноглобулинемии (патологически низкого
содержания иммуноглобулинов в сыворотке крови и внутренней среде), а
также недостатка интестинального иммуноглбулина А патогенные
бактерии и чужеродные иммуногены форсируют энтероцитарный барьер, что
вызывает гиперплазию иммунокомпетентных клеток, локализованных в
брыжеечных лимфатических узлах. Из-за гиперплазии клеток
лимфатических узлов блокируется отток лимфы от кишечной стенки, развиваются
лимфангиэктазия и синдром недостаточного кишечного всасывания.
ДИАРЕЯ
Каждые сутки в просвет желудочно-кишечного канала поступают 9 л
жидкости. Из этого количества общий объем выпитых жидкостей
составляет в среднем только 2 л. Оставшиеся 7 л - это общий объем поступающих в
просвет желудочно-кишечного канала секретов слюнных и желудочных
желез, желчи, секретов поджелудочной железы и желез кишечника,
которые формируют среду, необходимую для нормальных внутриполостного и
пристеночного пищеварения. В физиологических условиях большая часть
жидкости, поступающей за сутки в просвет кишечника, всасывается в его
верхних отделах. Около 1 л жидкости, которая содержит непереваренные
остатки пищи и продукты цитолиза энтероцитов, через илеоцекальный
клапан попадает в толстую кишку. Одна из функций толстой кишки
состоит в превращении поступающей из подвздошной кишки жидкости в
плотные каловые массы, которые поступают в прямую кишку для экскреции
через дефекацию. В результате снижения общей абсорбционной
поверхности стенок кишечника (состояния после удаления части кишечника и др.),
патологических изменений его стенок (хронические воспалительные
заболевания стенок кишечника и др.), появления в просвете кишечника нереаб-
сорбируемых осмолей, в том числе и вследствие недостаточного
пищеварения, а также из-за ускорения пассажа кишечного содержимого падают
абсорбция и реабсорбция жидкости из просвета кишечника (табл. 25.2).
Падение абсорбции и реабсорбции ведет к дефициту объема внеклеточной
жидкости, обезвоживанию и диарее (поносу).
Диарея - учащенное опорожнение кишечника, при котором
фекальные массы имеют жидкую консистенцию.
Классическим примером секреторной диареи является понос при
холере. При холере перегрузка толстой кишки жидким содержимым,
поступающим из тонкой кишки, приводит к частому жидкому стулу, то есть
диарее. В данном случае перегрузка толстой кишки жидкостью и
относительная недостаточность абсорбции в толстой кишке представляют собой
результат дисф икций на уровне тонкой кишки. Диарея при холере -это
Патогенез дисфункций системы пищеварения на уровне кишечника
413
Таблица 25.2
Патогенетическая классификация диареи
Тип диареи
Секреторная
Экссудативная
Как результат
низкого
кишечного всасывания
♦ осмотическая
♦ из-за потери
части
кишечника
♦ вследствие
расстройств
моторики
кишечника
Ведущее звено
патогенеза
Рост секреции
натриевого катиона в
просвет кишечника
как причина
возрастания в нем общего
количества осмолей.
Рост общего
количества осмолей
повышает количество
фекальных масс через
их разжижение
Воспаление стенок
кишечника как
причина низкого
кишечного всасывания
Рост общего
количества осмолей в
просвете кишечника как
следствие появления
в нем неабсорбируе-
мых молекул. Такие
невсасываемые
осмоли в частности могут
появляться в просвете
кишечника из-за
недостаточности
пищеварения
Снижение площади
абсорбционной
поверхности
Снижение времени
кишечной абсорбции
Характеристика
фекальных масс
Светлые, жидкие. Общая
осмоляльность жидкости в
просвете кишечника
примерно равняется общей
сумме осмотических
концентраций в нем
натриевого и калиевого
катионов, умноженной на два
(свидетельство отсутствия
в просвете кишечника не-
абсорбируемых осмолей)
Гнойные. Содержат поли-
морфонуклеары и кровь
(свидетельство
изъязвления стенок кишечника)
Общая осмоляльность
жидкости в просвете
кишечника больше, чем
сумма общей
осмотической концентрации
катионов натрия и калия,
умноженной на два
(свидетельство появления в
просвете кишечника не-
абсорбируемых осмолей)
Изменчивый
Изменчивый
Болезни и патологические
состояния, вызывающие
диарею данного типа
♦ Холера
♦ VIPoMa, гастринома
(опухоли поджелудочной
железы, клетки которых
секретируют вазоактив-
ный интестинальный
пептид и гастрин)
♦ Энтеропатия
(расстройство функций кишечника
вследствие
непереносимости желчных кислот)
♦ Язвенный колит
♦ Шигеллез
♦ Амебиаз
♦ Хронический панкреатит,
вызывающий
недостаточность внутриполост-
ного пищеварения
♦ Врожденный дефицит
лактазы
♦ Состояние вследствие
действия слабительных
средств, содержащих
катионы магния
♦ Состояние после
резекции более 50 % тонкого
кишечника
♦ Патологическое
состояние в результате свища
между желудком и
толстой кишкой
♦ Гипертиреоз
♦ Синдром раздраженной
кишки
результат нарушений массопереноса ионов и воды через стенку
кишечника на уровне тонкой кишки без воспаления ее стенок и заметных при
световой и электронной микроскопии аномалий энтероцитов.
414
Глава 25
При острой холере у взрослого человека секреция жидкости стенками
тонкой кишки в ее просвет в среднем составляет 1-2 л в час. Это изоосмо-
ляльная по отношению к внеклеточной жидкость, которая почти не
содержит белка. Основной катион секретируемой жидкости - натриевый, а
анионы - хлоридный и бикарбонатный. Ионный состав жидкости,
секретируемой в просвет кишечника при холере, одинаков в тощей и
подвздошной кишке. Возбудитель холеры (холерный вибрион) не
инфицирует энтероциты, а присоединяясь к их наружной поверхности,
высвобождает энтеротоксин, активирующий систему аденилатциклазы клеток
слизистой оболочки кишечника. В результате воздействия холерного
экзотоксина на систему аденилатциклазы она устойчиво активируется, что
обуславливает повышенную секрецию натриевого катиона в просвет
тонкой кишки. Рост содержания катиона натрия в просвете кишечника
удерживает в нем воду. В результате в организме падает содержание натрия и
воды. Дефицит общего содержания в организме натрия обуславливает
дефицит объема внеклеточной жидкости при гипернатриемии вследствие
потерь свободной воды с жидкими испражнениями. Дефицит объема
внеклеточной жидкости у больных холерой, может быть причиной
необратимой артериальной гипотензии.
Следует заметить, что активация системы аденилатциклазы эпителио-
цитов не приводит к падению всасывания глюкозы из просвета
кишечника и вторичной по отношению к нему абсорбции натрия. Вот почему
больным холерой показано обильное питье сладкой воды. Это вызывает
рост абсорбции глюкозы и всасывания натрия из просвета кишки.
Такие патогенные и условно патогенные возбудители болезней как
кишечная палочка, возбудители дизентерии, газовой гангрены также
повышают секрецию ионов в просвет кишечника клетками стенки тонкой
кишки. Но только при инфицировании холерным вибрионом объем
жидких испражнений, выделяемых за сутки, составляет 12-14 л.
При некоторых видах диареи сугубо патогененетическая терапия
оказывается безуспешной, и действенным может быть лечение, в основном
направленное на первопричину болезни и первичные звенья ее
патогенеза. Речь идет о поносах вследствие эндокринопатий. Диарея вследствие
эндокринопатий, часто являясь гиперсекреторной, своей
непосредственной причиной имеет активацию системы аденилатциклазы эпителиоцитов
тонкой кишки.
Повышение чувствительности адренорецепторов стенки кишки и
подавление в ней активности фермента моноаминооксидазы, разрушающего
норадреналин, вследствие гипертиреоза (см. главу, посвященную эндок-
ринопатиям) усиливает моторику всего кишечника и толстой кишки в
частности, а также по-видимому усиливает секрецию в просвет кишечника
энтероцитами натриевого катиона через активацию системы аденилат-
цитклазы. Все это обуславливает понос, который устраняет только
хирургическая или терапевтическая коррекция гипертиреоза.
Некоторые из опухолей мозгового слоя щитовидной железы образуют
и высвобождают простагландины, способные стимулировать моторику
Патогенез дисфункций системы пищеварения на уровне кишечника
415
кишечника и секрецию ионов в просвет кишки энтероцитами. Полагают,
что патогенный выброс простагландинов данными опухолями приводит к
диарее. Хирургическое удаление опухолей мозгового слоя надпочечников
прекращает диарею.
Вернер и Моррисон в 1958 г. впервые описали синдром, который в
основном составляли диарея как причина выраженного обезвоживания и
гипокалиемии, ахлоргидрия (см. главу, посвященную патофизиологии
желудочной секреции), а также аденома поджелудочной железы. В
1973 г. из крови пациентов с данным синдромом был выделен вазоак-
тивный интестинальный полипептид (ВИП), высвобождаемый
соответствующими клетками аденомы поджелудочной железы, патогенно
интенсивное действие которого служило причиной синдрома (синдром
панкреатической холеры).
ВИП, относясь к семейству пептидов секретина-глюкагона, состоит из
двадцати восьми аминокислотных остатков. ВИП содержат нервные
волокна и окончания гладких мышц, стенок кровеносных сосудов, а также
желез слизистой оболочки крупных бронхов. Известно, что пептид
стимулирует систему аденилатциклазы, вызывая выраженное системное
расширение сосудов сопротивления, стимулируя внешнесекреторную
функцию поджелудочной железы и секрецию интестинальными
энтероцитами, угнетая высвобождение соляной кислоты обкладочными
клетками желудка, повышая гликогенолиз, обуславливая дилатацию бронхов, а
также снижая выброс макромолекул железами верхних отделов
дыхательных путей. Полагают, что дефицит ВИП может быть фактором
развития бронхиальной астмы. Основными звеньями патогенеза диареи при
синдроме панкреатической холеры являются:
♦ рост секреции катиона натрия в просвет кишечника в результате
активации системы аденилатциклазы при высокой действующей
концентрации ВИП в циркулирующей крови;
♦ увеличение объема жидкости, поступающего в кишечник,
вследствие роста панкреатической секреции.
Не исключено, что одной из причин гипокалиемии у больных с
синдромом панкреатической холеры является рост секреции калия
бокаловидными клетками слизистой оболочки кишечника.
У части больных с синдромом панкреатической холеры ее
обуславливают другие опухоли поджелудочной железы, а не VIPoMa. В таких
случаях медиаторами диареи являются панкреатический полипептид и про-
стагландин Ег.
Гастринома - это опухоль, произрастающая из дельта-клеток
поджелудочной железы и продуцирующая гастрин.
Диарею вследствие гастриномы связывают с ростом концентрации в
циркулирующей крови «минигастрина», состоящего из четырнадцати
аминокислотных остатков. Полагают, что этот гастрин из всех видов
гормона в наибольшей степени обладает способностью стимулировать
секрецию ионов в просвет кишечника его эпителиоцитами через активацию
системы аденилатциклазы клеток слизистой оболочки стенок кишечника.
416
Глава 25
Осмотическая диарея - это следствие наличия в просвете
кишечника невсасываемых осмолей, которое служит причиной задержки в
нем как воды, так и других всасываемых осмолей, всасывание которых
зависит от всасывания воды.
Примером осмотической диареи может служить диарея вследствие
дефицита в просвете кишечника и на поверхности эпителиоцитов лакта-
зы. О данном генезе диареи свидетельствует прекращение поноса через
48-72 ч полного голодания. При осмотической диарее общая осмоляль-
ность кишечного содержимого всегда и значительно преобладает над
суммой осмотических концентраций натрия и калия, умноженной на два.
Это указывает на присутствие в просвете кишечника неабсорбируемых
осмолей (катионов магния, фосфатного аниона, продуктов неполного
пищеварения при его недостаточности и др.).
Недостаточность дисахаридаз ведет к хронической осмотической
диарее, так как дефицит данных энзимов в просвете кишки и на поверхности
энтероцитов обуславливает недостаточное расщепление нереабсорбируе-
мых из просвета кишечника дисахаридов до абсорбируемых
моносахаридов. В результате растет содержание осмолей в просвете кишечника и
толстой кишки в частности, что снижает реабсорбцию из ее просвета
воды и вызывает осмотическую диарею.
Врожденные дефициты таких ферментов из класса дисахаридаз как су-
краза и изомальтаза, от которых страдают 0,2 % представителей основной
популяции, приводят к осмотической диарее в раннем детском возрасте.
Дефицит лактазы в просвете кишечника также приводит к
осмотической диарее и связанному с ней синдрому пониженного всасывания. Лакта-
за (бета-ё-галактозидаза) - это фермент, катализирующий расщепление
лактозы на глюкозу и галактозу, а также расщепление бета-с!-галактозидов
и галактотрансферазные реакции. Частота лактазного дефицита различна у
отдельных народов. В Европе, особенно в ее северо-западных районах,
данная энзимопатия встречается сравнительно редко, а в ряде районов Азии,
Африки она наблюдается значительно чаще. В некоторых случаях его
частота среди отдельных этнических групп достигает 100 %. В настоящее
время можно считать, что способность расщеплять лактозу у взрослых
является врожденным свойством организма, передающимся по наследству.
Первичная недостаточность лактазы и связанный с ней синдром
недостаточного всасывания (первичная лактозная малабсорбция) как моногенная болезнь
передается по аутосомно-рецессивному типу при высокой пенетрантности
гена. Ген, кодирующий лактазу, локализован во второй хромосоме.
Основным клинически проявлением недостаточности активности
лактазы у взрослых являются поносы и рези после приема в пищу молока.
Поносы вызваны тем, что нерасщепленная лактоза попадает в дистальные
отделы тонкой кишки и в толстую кишку, где происходит ее
бактериальная фрагментация с образованием органических кислот (в основном
молочной и уксусной) и углекислого газа. В результате рост осмоляльности
кишечного содержимого приводит к уменьшению абсорбции воды и
возрастанию ее перемещения в просвет кишки.
Патогенез дисфункций системы пищеварения на уровне кишечника 417
Патологически интенсивная или низкая кишечная моторика приводит
к диарее посредством действия ряда патогенетических механизмов:
♦ При слишком быстрой перистальтике тонкой кишки растет
поступление воды, растворенных в ней катионов и анионов, а также осмо-
лей, представляющих собой продукты неполного переваривания
пищи, в толстую кишку.
♦ Замедление перистальтики через патогенно интенсивный
бактериальный рост в просвете кишечника приводит к деконъюгации
желчных кислот в кишечнике и прекращению их абсорбции как причине
диареи.
♦ Патогенно интенсивная моторика толстой кишки может
обуславливать снижение абсорбции в ней жидкости и осмолей вследствие
снижения времени контакта содержимого данного отдела
кишечника с его абсорбционной поверхностью.
Диарея у больных с диабетическими нейро- и микроангиопатиями,
с синдромом раздраженной кишки, в состоянии вследствие ваготомии,
а также из-за повышенной секреции серотонина, гастрина и вазоактив-
ного интестинального пептида злокачественными и
доброкачественными опухолями - это понос, связанный с нарушениями кишечной
моторики.
Диарея может быть следствием синдрома раздраженной кишкщ то
есть патологического состояния организма, обусловленного нарушениями
моторики кишечника. Известны три клинических варианта данного
синдрома. Первый из них называют спастическим колитом. Больные со
спастическим колитом жалуются на постоянные боли в области живота и
запоры. Прием пищи как стимул для регуляторных влияний,
усиливающих моторику толстой кишки, может вызвать боли в области живота
через 1-1,5 ч после еды. При втором варианте синдрома больные страдают
от перемежающейся диареи и часто не испытывают каких-либо болей в
животе (первичная диарея). Объем жидкого стула при первичной диарее
не превышает 500 мл/сутки. Третий вариант характеризуют поносы,
которые сменяют запоры, и боли в области живота.
Можно считать, что патофизиологическую основу синдрома
составляет патологически измененная реактивность местных (на уровне
стенки кишки) систем нервной регуляции кишечной моторики и
соответствующих гладкомышечных элементов по отношению к системным регу-
ляторным влияниям, усиливающим двигательную активность толстой
кишки. Известно, что у пациентов с любым из вариантов клинического
течения синдрома эффекты холиномиметиков и холецистокинина, а также
отрицательный психоэмоциональный стресс усиливают двигательную
активность толстой кишки в большей степени, чем у здоровых людей. У
больных со спастическим колитом период покоя между сокращениями
стенок кишки удлинен, а сила сокращений кишечной стенки больше, чем
при других вариантах синдрома, что проявляет себя сочетанием запоров и
болей. При первичной диарее период между сокращениями укорочен, что
обуславливает понос.
418
Глава 25
Исследования миоэлектрической активности стенки толстой кишки у
больных с синдромом раздраженной кишки показали, что у 40 % таких
пациентов по стенке кишки 3 раза в минуту проходит волна возбуждения,
состоящая из его медленных волн. Такую высокую частоту прохождения
медленных волн выявляют у 10 % здоровых людей. После приема пищи
пик миоэлектрической активности в стенке толстой кишки у здоровых
людей фиксируют через 40 мин после приема пищи, а у больных с
синдромом раздраженной кишки через 70-90 мин. Это позволило
предположить, что падение сократимости гладкомышечных элементов кишечной
стенки и усиление системных и местных регуляторных влияний,
стимулирующих моторику, составляют основные звенья патогенеза синдрома
раздраженной кишки.
Диарея - это весьма полиэтиологичные синдром и патологическое
состояние организма. При определении способа устранения диареи
следует помнить, что ее конечными патогенетическими механизмами часто
являются рост поступления жидкости в толстую кишку при транзите
кишечного содержимого, снижение абсорбционной поверхности толстой
кишки, а также ускоренное прохождение по ней кишечного
содержимого. Поэтому нередко для устранения диареи у больных с синдромом
раздраженной кишки эффективными оказываются средства, содержащие
вещества, связывающие воду и уплотняющие каловые массы в просвете
толстой кишки. Таким образом можно устранить обезвоживание и
нарушения солевого обмена у пациентов, страдающих от данного
заболевания. Это частный пример эндогенизации патологического процесса,
при которой звенья патогенеза в известной мере теряют связь с
первопричиной заболевания, а патогенетическая и совершеннно неэтиотроп-
ная терапия позволяют существенно улучшить состояние больных.
Более того, у части больных такая терапия полностью устраняет синдром
раздраженной кишки. Можно предположить, что снижение объема
свободной жидкости и увеличение времени для всасывания воды в толстой
кишке при уплотнении каловых масс устраняет патогенный характер
афферентации из рецепторов стенки кишки как фактор дизрегуляции,
обуславливающей эндогенизацию локального типового патологического
процесса диареи.
Диарея вследствие влияний желчных кислот на клетки, составляющие
стенки кишечника, представляет собой вариант поноса, при котором нет
патологических изменений клеточных элементов толстой кишки. Когда
концентрация связанных и несвязанных дигидроксижелчных кислот в
просвете толстой кишки возрастает до определенного уровня, желчные
кислоты стимулируют секрецию натрия и воды ее энтероцитами в
просвет кишечника. В результате через рост содержания в просвете толстой
кишки натрия и воды возникает диарея.
Расстройства циркуляции желчных кислот между кишечником и
печенью вызывают диарею посредством действия ряда патогенетических
механизмов. Если циркуляция желчных кислот блокируется при холеста-
тическом синдроме (смотри главу, посвященную патофизиологии пече-
Патогенез дисфункций системы пищеварения на уровне кишечника 41£
ни), то снижение концентрации желчных кислот в просвете кишечника
приводит к падению всасывания липидов и стеаторее. Стеаторея (гр.
стеатос - сало + гр. реуматос - истечение) - выделение большого
количества жира с испражнениями. В результате падения всасывания
липидов из просвета кишечника в нем растет общая концентрация осмолей,
что обуславливает диарею.
При стазе кишечного содержимого в тонкой кишке, который в
частности может быть обусловлен последствиями некоторых хирургических
вмешательств на желудке, в просвете кишечника происходит деконьюга-
ция желчных кислот. Непосредственной причиной деконьюгации при
этом выступает патогенный бактериальный рост в просвете кишки. В
результате деконьюгации в просвете кишки желчных кислот падает
образование мицелл и снижается кишечное всасывание жиров.
Когда расстройства циркуляции желчных кислот (ЖК) между
кишечником и печенью представляют собой результат недостаточной
абсорбции желчных кислот из просвета подвздошной кишки, в просвет толстой
кишки поступает патогенно повышенное количество ЖК. У части
больных недостаточное всасывание желчных кислот из просвета подвздошной
кишки может быть следствием ее хирургической резекции. Рост
концентрации в содержимом толстой кишки коньюгированных и деконьюгиро-
ванных желчных кислот до уровня 3-5 ммоль/л вызывает значительное
возрастание секреции натрия и воды в просвет кишки через активацию
системы аденилатциклазы энтероцитов. При этом вторично возрастает
поступление из внеклеточного сектора в просвет толстой кишки
калиевого катиона и бикарбонатного аниона. Потери калия вызывают гипокалие-
мию, а потери бикарбонатного аниона приводят к метаболическому
алкалозу. Кроме того, в данном случае одним из патогенетических
механизмов развития диареи является рост осмоляльности кишечного
содержимого в толстой кишке из-за подъема в нем содержания желчных кислот.
По-видимому, одним из патогенетических механизмов диареи
вследствие роста содержания в просвете толстой кишки желчных кислот
является вторичное по отношению к нему угнетение активного всасывания
натрия кишечными эпителиоцитами.
Известно, что диарею у части больных с нарушенной циркуляцией
желчных кислот между кишечником и печенью можно устранить
назначением внутрь препаратов на основе определенного вида резины, который
обладает свойством связывать желчные кислоты. При этом следует
опасаться побочного эффекта таких средств, обладающих, кроме того,
свойством связывать в просвете кишечника липиды и вызывать стеаторею.
Изменения стенки толстой кишки, обусловленные ее хроническим или
острым воспалением (бактериальная дизентерия, амебиаз, болезнь Крона,
язвенный колит и др.), также являются причиной диареи.
Непосредственные причины диареи в данном случае - это снижение абсорбционной
поверхности толстой кишки вследствие патологических воспалительных
инфильтративно-пролиферативных изменений ее стенок, а также
ускорение транзита кишечного содержимого по толстой кишке как результат
420
Глава 25
роста уровня возбуждения соответствующих нейронов локальной
системы нервной регуляции моторики кишки.
В физиологических условиях в толстой кишке происходят абсорбция
катиона натрия и хлоридного аниона, вторичное по отношению к ней
всасывание воды, абсорбция калиевого катиона и бикарбонатного аниона, а
также секреция К+ и НСОз". Так как в толстой кишке постоянно
происходит разнонаправленная миграция катионов калия и натрия, то часто
выявить первичное нарушение трансмуральной миграции одного из
катионов как причину диареи не представляется возможным. Кроме того,
практически невозможно установить, что привело к росту содержания в
кишечном содержимом того или другого катиона (аниона), угнетение его
абсорбции или рост секреции ионов в просвет кишечника его энтероци-
тами. Есть только две нозологические формы, диарею при которых
представляется возможным связать исключительно с нарушениями
перемещения через стенку кишки определенных ионов, врожденная хлоридорея и
ворсинчатая аденома толстой кишки.
Хлоридорея - заболевание или патологическое состояние, которые в
первую очередь характеризуют водянистый стул, гипохлоремия
(патологическое снижение концентрации хлоридного аниона во внеклеточной
жидкости и жидкой части плазмы крови), гипокалиемия (патологическое
снижение содержания во внеклеточной жидкости и жидкой части плазмы
крови калия) и метаболический ацидоз.
Хлоридорею как следствие заболеваний, вызывающих патологические
изменения стенки кишки, называют приобретенной. Приобретенная
хлоридорея обычно развивается у пациентов, страдающих заболеваниями
толстой кишки, которые вызывают понос и гипокалиемию. Полагают, что
дефицит калия влияет на проницаемость стенки кишки таким образом,
что прекращается трансмуральный обмен хлоридного аниона на бикарбо-
натный. В результате в просвете толстой кишки возрастает содержание
хлоридного аниона. Рост содержания в просвете кишки хлоридного
аниона происходит одновременно с падением его содержания во
внеклеточном секторе и клетках. Одновременное возрастание содержания в них
бикарбонатного аниона приводит к метаболическому алкалозу.
Гипокалиемия при хлоридорее - это отчасти и результат задержки хлоридными
анионами катионов калия в просвете кишки.
Врожденная хлоридорея наследуется по ^утосомно-рецессивному
типу. Данную моногенную болезнь связывают с патогенной мутацией на
уровне 7q31.
Механизмы переноса хлоридного аниона через стенку кишки
многообразны. Значительная часть хлоридного аниона всасывается из просвета
кишечника пассивно. При этом абсорбция СГ вторична по отношению к
активному всасывания натрия. Кроме того, есть основания полагать, что
существуют и активные механизмы всасывания СГ: сопряженный
транспорт катиона натрия и хлоридного аниона, активный обмен хлоридного
аниона на бикарбонатный. Вероятно при врожденной хлоридорее
нарушены именно эти механизмы.
Патогенез дисфункций системы пищеварения на уровне кишечника 421^
При врожденной хлоридорее рост концентрации хлоридного аниона
в просвете кишки приводит к увеличению в ней содержания натриевого
катиона.
Ворсинчатая аденома - доброкачественная опухоль толстой кишки с
наиболее частой локализацией в ректосигмоидном отделе. Складки
ворсинчатой аденомы покрыты множеством бокаловидных клеток, обильно
высвобождающих секрет. Так как в секрете бокаловидных клеток
аденомы повышено содержание катиона калия (140 ммоль/л), то у больных
развивается гипокалиемия. Если ворсинчатая аденома локализована крани-
альнее ректосигмоидного отдела, то диарея может и не развиться. Дело в
том, что в данном случае часть секрета реабсорбируется по ходу
продвижения кишечного содержимого по прямой кишке. Основной
патогенетический механизм возникновения диареи вследствие ворсинчатой
аденомы - рост массы содержимого толстой кишки вследствие гиперсекреции
бокаловидными клетками опухоли.
ПАТОГЕНЕЗ ОСТРОЙ КИШЕЧНОЙ
НЕПРОХОДИМОСТИ
Острая кишечная непроходимость (илеус1) - это патологическое
состояние вследствие быстрого прекращения пассажа кишечного
содержимого в аборальном направлении, которое в первую очередь
характеризуют стремительное нарастание обезвоживания и дефицита объема
внеклеточной жидкости, экзо- и эндогенная интоксикация вследствие
потери частью стенок кишечника своей барьерной функции и патогенно
интенсивной антигенной стимуляции системы иммунитета в кишечнике
и непосредственно связанных с ним органах брюшной полости, а также
вторичные по отношению к ним расстройства системного и
периферического кровообращения.
Острую кишечную непроходимость может обуславливать действие
факторов как механической (механическая непроходимость)', так и
немеханической природы. Сужение просвета кишечника из-за действия
факторов механической природы чаще всего обуславливают:
♦ спайки брюшной полости;
♦ наружные и внутренние грыжи.
В таких случаях механическая непроходимость - это результат внеки-
шечного сдавления кишки.
При дивертикулах стенок кишечника, его раковых опухолях и
регионарном энтерите причины механической непроходимости связаны с
патологическими изменениями стенок кишечника.
Илеус (от греческого эйлеос - закупорка кишечника) - нарушение прохождения
содержимого по кишечнику.
422
Глава 25
Если кишечная непроходимость развивается в результате инвагинации
(лат. in, в, во внутрь + vagina, ножны), то ее причина - патологическое
изменение положения частей стенок кишечника относительно друг друга,
при котором происходит впячивание или захождение одной части в
просвет кишечника.
Кроме того, механическая непроходимость может быть связана с
патогенным изменением кишечного содержимого, то есть вызвана
закупоркой просвета кишечника желчными или каловыми камнями.
Причиной непроходимости на уровне тонкого кишечника в 70-75 %
случаев являются спайки брюшной полости, то есть чаще всего
непроходимость на уровне данного сегмента является спаечной (один из наиболее
распространенных видов механической непроходимости). Еще чаще
механической является непроходимость на уровне толстой кишки, которую
в 90 % вызывают ее раковые опухоли, инвагинация и дивертикулы.
Кишечник обладает способностью относительно свободного
перемещения в брюшной полости. Эта способность обуславливает возможность
реакций объема кишечника и его моторики в ответ на изменения массы и
состава кишечного содержимого. Способность кишечника к свободному
перемещению во многом определяется состоянием фиксирующего
аппарата, состоящего из брыжейки, париетальной брюшины и связок.
Врожденные и приобретенные изменения фиксирующего аппарата
предрасполагают к механической кишечной непроходимости через ограничение
свободы перемещения кишечника.
Наиболее частая причина ограничения свободного перемещения
кишечника в брюшной полости и нарушений его моторики как причин
механической кишечной непроходимости - это спайки брюшной полости.
Особенно часто механическую кишечную непроходимость вызывают
изолированные межкишечные, кишечно-париетальные или париетально-
сальниковые сращения, которые обуславливают странгуляцию (от лат.
strangulo - душить) подвижных сегментов кишечника, то есть чреватое
ишемией сдавление его стенок.
Известно, что тощая, подвздошная, сигмовидная кишка, а также
поперечно-ободочная кишка как сегменты кишечника, обладающие
максимальной подвижностью в брюшной полости, являются теми его
участками, на уровне которых механическая кишечная непроходимость
вследствие спаек и спаечной болезни развивается наиболее часто.
Спаечный процесс представляет собой защитную реакцию,
направленную на отграничение:
♦ очагов инфекции, воспаления и связанной с ними альтерации
тканей;
♦ участков повреждения висцеральной брюшины;
♦ инфицированных инородных тел и кишечного содержимого,
попадающих в полости живота и брюшины при огнестрельных и
других проникающих ранениях живота, а также при оперативных
вмешательствах и вследствие перфораций и пролежней полых
органов.
Патогенез дисфункций системы пищеварения на уровне кишечника 423
Как и всякая другая защитная реакция спаечный процесс
потенциально патогенен. Патогенность спаечного процесса в частности реализует
себя, когда спайки брюшной полости приводят к механической
непроходимости.
Образование спаек - это частный случай пролиферативного
компонента воспаления на уровне брюшины, который отличает высокая
реактивность соединительнотканных клеток брюшины по отношению к цито-
кинам со свойствами факторов клеточного роста, которые в очаге
воспаления брюшины высвобождаются активированными мононуклеарными
фагоцитами, другими клеточными эффекторами воспаления и клетками
системы иммунитета.
Воспаление при образовании спаек брюшной полости служит
стимулом нервных реакций супрасегментарного уровня, приводящих к
избыточной адренергической стимуляции стенок кишечника, вызывающей
паралитическую непроходимость. Кроме того, воспаление - это местный
процесс, меняющий функционирование локальной нервной системы
регуляции кишечной моторики и клеток кишечника, относящихся к системе
захвата и декарбоксилирования предшественников аминов на пути их
синтеза (англ. amine precursor uptake and decarboxylation system, APUD
system). Таким образом воспаление меняет соотношение системных и
локальных регуляторных влияний на кишечник таким образом, что
вызывает снижение его двигательной и секреторной активности. Поэтому многие
из спаек не вызывают механической кишечной непроходимости, так как
при их образовании через воспаление снижается моторика кишечника и
секреция его эпителиоцитами.
Особенно часто образование спаек служит причиной механической
кишечной непроходимости в том случае, когда оно связано с
формированием обширного гнойника. При этом в очаге гнойной инфекции особенно
велика концентрация цитокинов со свойствами факторов клеточного
роста, что обуславливает максимальную интенсивность процесса
образования спаек. Перитонит как причина системных патологических сдвигов
нервной регуляции, нарушений водно-солевого обмена и расстройств
периферического кровообращения блокирует пассаж кишечного
содержимого не только через образование спаек брюшной полости, но и
обуславливая развитие и паралитической непроходимости (см. ниже).
У большинства больных после завершения воспаления как защитной
реакции спайки брюшной полости подвергаются обратному развитию. У
части пациентов происходит сложное морфологическое преобразование
спаек, в ходе которых развивается фиброз париетальной и висцеральной
брюшины, который нередко распространяется на мышечную оболочку,
подслизистую основу кишечной стенки и на брыжейку, повреждая их
сосуды и интрамуральный нервный аппарат соответствующего отдела
кишечника. При этом степень выраженности фиброза брюшины не
соответствует степени выраженности первичной альтерации, вызвавшей
воспаление. В таком случае говорят о возникновении «спаечной болезни»,
патогенез которой состоит в выходе межклеточных отношений, определяю-
424
Глава 25
щих и стимулирующих пролиферацию соединительнотканных клеток
брюшины, из-под контроля систем их местной и системной регуляции.
При кишечной непроходимости вследствие спаечной болезни действует и
паралитический механизм возникновения илеуса. Паралитическая
динамическая непроходимость (см. ниже) в данном случае - это результат
повреждения фиброзом мышечной оболочки, интрамурального нервного
аппарата кишечника и связанной с фиброзным перерождением
облитерации сосудов брыжейки. При спаечной болезни кишечная непроходимость
нередко вызывается грубым рубцовым сморщиванием и укорочением
брыжейки или рубцовой деформацией кишечной стенки.
Другая группа причин механической кишечной непроходимости -
злокачественные и доброкачественные опухоли кишечника, а также
опухоли других органов живота и забрюшинного пространства,
обуславливающие механическую обструкцию пассажу кишечного содержимого.
Механизмы возникновения кишечной непроходимости вследствие
полипоза кишечника имеют некоторые особенности. Так полипы на ножке,
расположенные в наиболее подвижных отделах кишечника, могут
вызвать инвагинацию, представляющую собой сочетание обтурации и
странгуляции.
Острую кишечную непроходимость, которая развивается в результате
действия факторов немеханической природы называют адинамической
или паралитической, то есть преимущественно связанной не с
механическим препятствием пассажу кишечного содержимого, а представляющей
собой следствие расстройств кишечной моторики.
При попадании на париетальную брюшину кишечного содержимого,
крови и инородных тел угнетение кишечной моторики также является
рефлекторной нервной реакцией, которая предположительно может
происходить при определяющем участии нервных сплетений брюшины,
брыжейки и локальной системы нервной регуляции кишечной моторики
без вовлечения в рефлекс сегментарного и супрасегментарного регуля-
торных аппаратов.
Одна из частых причин адинамической острой кишечной
непроходимости - это патологическая боль в условиях неадекватного
обезболивания у больных после высокотравматичных хирургических
вмешательств. При этом боль выступает стимулом системной адренергической
стимуляции, которая на уровне стенок кишечника приводит к
гиперполяризации гладкомышечных клеток кишечной стенки и тем самым
угнетает моторику кишечника. Патологические изменения
трансмембранного потенциала гладкомышечных клеток стенок кишечника как причины
острой кишечной непроходимости могут быть следствием гипо- или ги-
перкалиемии (соответственно патологически низкого и патологически
высокого содержания калия во внеклеточном жидкостном секторе и
плазме крови). Аномальные изменения стенок кишечника при его
воспалительных и некоторых системных заболеваниях (склеродерма и др.)
также могут быть причиной адинамической кишечной непроходимости.
Из других причин паралитической кишечной непроходимости следует
Патогенез дисфункций системы пищеварения на уровне кишечника 425
выделить тромбоз и атеросклеротическое поражение мезентериальных
сосудов, которые приводят к циркуляторной гипоксии кишечной стенки
и через гипоэргоз ее гладкомышечных элементов вызывают острую
кишечную непроходимость.
В острый период тяжелых травматической и раневых болезней
защитно-патогенная централизация кровообращения приводит к циркуляторной
гипоксии клеточных элементов кишечной стенки, вызывая адинамиче-
скую непроходимость.
Адинамическую непроходимость при злокачественных опухолях
кишечника могут обусловить патологические изменения его стенок,
связанные со злокачественным клеточным ростом.
При развитии непроходимости кишечника прежде всего рефлекторно
подавляется непропульсивная перистальтика, обуславливающая
перемешивание пищевого комка с пищеварительными соками в просвете
кишечника. В основном же нарушения моторики кишечника сводятся к
угнетению пропульсивной перистальтики, то есть волнообразно
распространяющихся по кишке сокращений циркулярных мышц, которым обычно
предшествует волна расслабления. Угнетение пропульсивной
перистальтики связано с блокадой распространения мигрирующих миоэлектриче-
ских комплексов в каудальном направлении из-за механического
препятствия пассажу кишечного содержимого. Кроме того, угнетение
перистальтики представляет собой следствие нервных реакций супрасегмен-
тарного, сегментарного и локального уровней, которые через
преобладание адренергической стимуляции клеточных элементов кишечной стенки
и местной нервной системы регуляции кишечной моторики угнетают и
расстраивают двигательную активность кишечника. Следует заметить,
что критическое состояние вследствие кишечной непроходимости служит
стимулом активации всей вегетативной нервной системы. В результате
растет интенсивность влияний на стенку кишки не только симпатических,
но и парасимпатических регуляторных влияний. Поэтому при сохранении
механического препятствия пассажу усиление парасимпатических
регуляторных влияний может обуславливать антиперистальтику, которую
сменяет угнетение моторики вследствие преобладания эффектов
симпатических регуляторных влияний и циркуляторной гипоксии нейронов
локальной системы нервной регуляции кишечной моторики и
гладкомышечных клеток стенок кишечника.
При послеоперационной адинамической кишечной непроходимости
вследствие преобладания на уровне кишечной стенки адренергических
регуляторных влияний (неустраненная патологическая боль, гиповолемия
и циркуляторная гипоксия, артериальная гипоксемия) активность
расположенного в двенадцатиперстной кишке водителя ритма «медленных
волн» перистальтики сохраняется полностью. При этом миграции
«медленных волн» возбуждения и сокращения в аборальном направлении не
происходит из-за стойкой деполяризации гладкомышечных клеток
кишечной стенки в результате ее патологически интенсивной
адренергической стимуляции. Кроме того, распространение волны возбуждения-
426
Глава 25
сокращения прекращается вследствие обусловленного циркуляторной
гипоксией гипоэргоза кишечных миоцитов и нейронов местной системы
нервной регуляции кишечной моторики.
По мере разрушения острой кишечной непроходимостью экосистемы
микрофлоры кишечника к звеньям патогенеза острой кишечной
непроходимости присоединяется еще один элемент, высвобождение токсинов
алло- и аутохтонными микроорганизмами как в просвете кишечника, так и в
самой кишечной стенке. Данные бактериальные токсины расстраивают
локальную нервную регуляцию кишечной моторики и угнетают гладко-
мышечные миоциты.
Снижение двигательной активности кишечника приводит к угнетению
полостного и пристеночного пищеварения через снижение
ферментативной секреции железистого аппарата.
Угнетение пищеварения при острой кишечной непроходимости
причиной своей имеет структурные и ультраструктурные изменения в
слизистой оболочке тонкой кишки при острой кишечной
непроходимости. В основном такие патологические изменения энтероцитов связаны
с циркуляторной гипоксией. Их составляют истончение слизистой
оболочки, уплощение энтероцитов, повреждение щеточной каемки.
Электронная микроскопия при этом выявляет первичное поражение глико-
каликса щеточной каемки. В частности это свидетельствует о
несостоятельности защитных механизмов на уровне стенки кишки, так как
микроворсинки щеточной каемки в обычных условиях защищают
слизистую оболочку от воздействия патогенных веществ со стороны
просвета кишки и предотвращают адгезию микроорганизмов из просвета к
энтероцитам.
В результате в просвете кишечника аккумулируются молекулы,
которые представляют собой невсасываемые осмоли. Это задерживает
жидкость в просвете кишечника и обуславливает эндогенное голодание.
Кроме того, рост содержания осмолей в просвете кишечника происходит в
результате дисбаланса экосистемы кишечной микрофлоры. Дисбаланс
приводит к неконтролируемому размножению микроорганизмов в
просвете кишечника, продуктами жизнедеятельности которых являются
невсасываемые полипептиды.
Рост общего количества осмолей в части кишечника проксимальной
относительно обструкции пассажу кишечного содержимого в частности
обусловлен прекращением продвижения кишечного содержимого по
кишечнику, что исключает его всасывание в части кишечника,
расположенной относительно места обструкции каудально.
В результате расстройств кишечного всасывания при острой
кишечной непроходимости во внутреннюю среду организма за сутки не
поступают 8 л жидкости, и еще 4 л теряются в стенки кишечника из-за
происходящих в них нарушений микроциркуляции и интерстициального
отека. Связывание в просвете кишечника больших количеств жидкости
приводит к резкому сужению внеклеточного жидкостного сектора и
нарушениям ионного состава клетки, вторичным по отношению к гипово-
Патогенез дисфункций системы пищеварения на уровне кишечника 427
лемии и циркуляторной гипоксии. Тем не менее, следует знать, что
быстрая инфузия нескольких литров плазмозамещающих растворов изото-
ничных по отношению к внеклеточной жидкости, несмотря на ее
крайнюю необходимость для предотвращения танатогенеза при острой
кишечной непроходимости, чревата опасной ятрогенией. Дело в том, что
такая инфузия через частичное восстановление периферического кро-
овообращения в стенках кишечника увеличивает концентрацию цитоки-
нов со свойствами медиаторов чисто патогенного воспаления в крови,
циркулирующей системно, «вымывая» цитокины из гипоксичных стенок
кишечника. Это приводит к системной гиперцитокинемии,
множественной системной недостаточности и обуславливает некардиогенный отек
легких и РДСВ.
Расстройства микроциркуляции в стенке кишки вызываются
следующими основными причинами:
♦ гиповолемией как причиной расстройств системного
кровообращения первичных по отношению к нарушениям периферического
кровообращения в стенке кишки;
♦ спазмом сосудов сопротивления органов брюшной полости в ответ
на гиповолемию и циркуляторную гипоксию;
♦ локальным воспалением на уровне стенок кишечника как
следствием взаимодействия клеток системы иммунитета в кишечной стенке
с чужеродными антигенами и объектами фагоцитоза из просвета
кишки и активации клеточных эффекторов воспаления
циркуляторной гипоксией.
Кроме того, стаз в микрососудах кишечной стенки представляет собой
следствие патогенного роста давления в просвете кишечника, связанного
с увеличением массы и объема его содержимого как в части
проксимальной относительно места обструкции, так и в части кишечника каудальной
по отношению к нему. Дело в том, что системные расстройства
микроциркуляции, нарушения моторики, пищеварения вследствие острой
кишечной непроходимости распространяются и на каудальную часть
кишечника, что нарушает в нем всасывание и повышает давление
кишечного содержимого.
Секвестрация части организменного пула бикарбонатного аниона в
просвете кишечника при острой кишечной непроходимости может быть
столь значительной, что обуславливает метаболический ацидоз. Потери
желудочного содержимого при острой кишечной непроходимости могут
обусловить метаболический алкалоз.
Просвет кишечника здорового человека содержит несколько
миллионов смертельных доз бактерий и токсинов, которые могли бы
вызвать быструю смерть, если бы их попаданию во внутреннюю среду
организма не препятствовал защитный барьер слизистой оболочки
кишечной стенки, В основном конечный полезный приспособительный
результат данной защитной системы состоит в предотвращении
патогенного бактериального роста в просвете кишки и адгезии бактерий из
просвета к энтероцитам, в том числе и как инициирующего момента сие-
428
Глава 25
темной иммунной реакции. Основными элементами данной защитной
системы являются:
♦ Гликокаликс энтероцита и плотные соединения между клетками
слизистой оболочки кишечника.
♦ Секреция слизи клетками кишечных стенок.
♦ Иммуноглобулины А, которые содержатся в желчи и кишечной
слизи. Образование с участием иммуноглобулинов А в слизи,
секретах, попадающих в просвет кишечника, иммунных комплексов на
поверхности чужеродных бактерий и иммуногенов предотвращает
их адгезию к энтероцитам как начальный этап проникновения
патогенных микроорганизмов и токсинов во внутреннюю среду и
индукцию патологических иммунных реакций.
♦ Воздействие на экзогенные бактерии высокой концентрации
протонов в желудочном содержимом и его протеолитической активности
(главный защитный бактерицидный механизм на уровне
желудочно-кишечного канала).
♦ Конкурентное ингибирование роста потенциально патогенных
микроорганизмов в просвете кишки бактериями комменсалами (лат.
сом - вместе + mensa - стол), которые в физиологических условиях
находятся с организмом человека во взаимоотношениях, при
которых один организм извлекает пользу из другого, и другой организм
не повреждается. В более каудальных отделах кишечника (толстой
кишке) как элемент защитной системы функционируют
комменсалы, являющихся облигатными анаэробами, бактериальный рост
которых происходит при низких парциальных давлениях и
напряжениях кислорода.
♦ Вещества, обладающие неспецифической бактерицидной
активностью, которые образуют и секретируют клетки слизистой оболочки
кишечника: лизоцим, лактоферрин и др.
♦ Постоянный процесс распознавания антигенов из просвета
кишечника и периодическая (при инвазии во внутреннюю среду
чужеродных антигенов) индукция первичного и (или) вторичного
иммунного ответа иммунокомпетентными клетками местных
лимфатических узлов кишечной стенки и региональных узлов
брыжейки. Тут особую роль играют М-клетки, покрывающие
пейеровы бляшки.
В физиологических условиях просвет кишечника заселен аутохтон-
ными (нечужеродными) микроорганизмами, видовой состав которых
варьирует в зависимости от диеты и периода онтогенеза. Видовой состав
аутохтонных микроорганизмов относительно стабилен в том или ином
отделе желудочно-кишечного канала. Аллохтонные (чужеродные)
микроорганизмы также присутствуют во всех его отделах. При нормальном
пищеварении, когда состав аутохтонной флоры относительно постоянен,
данная экосистема сообществ чужеродных и нечужеродных
микроорганизмов находится в динамическом равновесии, не обладая патогенными
свойствами.
Патогенез дисфункций системы пищеварения на уровне кишечника 429
Острая кишечная непроходимость подвергает экосистему кишечной
флоры деструкции, обуславливая резкий дисбаланс между
составляющими ее сообществами микроорганизмов. Во-первых, блокада транзита
кишечного содержимого приводит к неконтролируемому системой
иммунитета организма бактериальному росту в просвете кишечника. Во вторых,
расстройства кишечной моторики приводят к миграции аутохтонной для
дистальных отделов кишечника флоры в просвет проксимальных отделов,
для которых она является чужеродной. В третьих, циркуляторная
гипоксия кишечной стенки приводит к быстрому размножению в кишечнике
анаэробной и в основном неспорообразующей микрофлоры, которая
начинает доминировать над другими сообществами микроорганизмов в
пораженных циркуляторной гипоксией отделах кишечника.
Через циркуляторную гипоксию клеток защитного барьера слизистой
оболочки кишечника и дисбаланс экосистемы кишечной микрофлоры
острая кишечная непроходимость резко усиливает взаимодействие
клеток системы иммунитета на уровне кишечника и интерферирующих с
ним органов-эффекторов с чужеродными микроорганизмами как с
источниками иммуногенов. В результате в кишечной стенке происходит
активация мононуклеарных фагоцитов как антиген-презентирующих
клеток, а межклеточные взаимодействия с участием иммунокомпетентных
клеток индуцируют системную иммунную реакцию. При этом данная
локальная и одновременно системная иммунная реакция обречена на
избыточность, так как в физиологических условиях не происходит такого
интенсивного контакта бактерий из просвета кишечника с его энтероцитами.
Особую интенсивность такого контакта как инициирующего момента
защитно-патогенной иммунной реакции обуславливают:
♦ недостаточность секреции в просвет кишечника связывающих
микроорганизмы иммуноглобулинов А в результате
неограниченного размножения аллохтоннных микроорганизмов в просвете
кишечника;
♦ недостаточность секреции иммуноглобулинов А и кишечной слизи,
предотвращающих контакт микроорганизмов из просвета
кишечника с его энтероцитами, как результат гипоэргоза клеточных
элементов стенок кишечника.
В результате защитно-патогенной иммунной реакции при сохранении
нарушений периферического кровообращения в кишечнике он становится
локусом исключительно интенсивного накопления цитокинов (альфа-
фактор некроза опухолей, ряд интерлейкинов и др.), которые, попадая в
системное кровообращение в патологически высокой концентрации,
обуславливают на системном уровне чисто патогенное воспаление,
разрушающее структурно-функциональные элементы органов-эффекторов и
вызывающее множественную системную недостаточность. Таким
образом патогенез критического состояния, вызванного острой кишечной
непроходимостью, начинают составлять гиперцитокинемия и системная
воспалительная реакция вплоть до эндотоксического шока, развитие
которых могут резко ускорить хирургическое устранение механической
430
Глава 25
причины циркуляторнои гипоксии кишечной стенки и гемодилюция
внутривенными инфузиями. При этом гемодилюция обостряет системную
воспалительную реакцию, обуславливая активацию мононуклеарных
фагоцитов кишечной стенки по механизму реперфузионного повреждения
через действие свободных кислородных радикалов. Свой вклад в
индукцию системной воспалительной реакции при острой кишечной
непроходимости вносит и гипоксическая первичная альтерация клеточных
элементов кишечных стенок.
Глава 26
ПАТОГЕНЕЗ ОСТРОГО И ХРОНИЧЕСКОГО
ПАНКРЕАТИТОВ
Панкреатит, воспаление поджелудочной железы, бывает острым и
хроническим. В большинстве случаев (85-90 %) острый панкреатит
подвергается обратному развитию без хирургического лечения и интенсивной
терапии. Если острое воспаление поджелудочной железы приводит к
распространенной альтерации стенок микрососудов поджелудочной железы, то
кровь выходит в интерстиций органа со всеми своими форменными
элементами. В таком случае говорят о развитии геморрагического панкреатита.
Наиболее распространенной гипотезой патогенеза острого
панкреатита является теория самопереваривания (аутодигестии) поджелудочной
железы. Согласно данной гипотезе, протеолитические ферменты железы
(трипсиноген, хемотрипсиноген, проэластаза и неактивная форма фосфо-
липазы А) активируются в поджелудочной железе, а не в просвете
кишечника. Факторами активации панкреатических протеаз в самой железе
считают эндо- и экзотоксины, вирусные инфекции, ишемию, травму
железы и другие этиологические факторы острого панкреатита (табл. 26.1).
Активированные протеазы теперь уже сами выступают факторами
активации проферментов. Активированные проферменты поджелудочной
железы, и трипсин в особенности, переваривают клеточные мембраны и
вызывают протеолиз, что приводит к эмиграции крови в интерстиций
(геморрагический панкреатит), обуславливает отек, а также коагуляционный
и жировой некроз стромы железы и ее дифференцированных клеток.
Деструкция клеток железы обуславливает еще большее высвобождение в
ней активированных ферментов, что усиливает аутодигестию. Через ау-
тодигестию поврежденная железа становится источником массивного
высвобождения кининов-вазодилятаторов. Аутодигестия железы приводит к
ее первичной альтерации как индуктору воспаления в железе. Во многом
через вторичную альтерацию при воспалении аутодигестия выходит за
пределы железы и повреждает прилежащие ткани. Распространенный
очаг воспаления при панкреатите служит причиной системной
воспалительной реакции и септического шока.
У большей части больных с острым панкреатитом он представляет собой
осложнение желчекаменной болезни. Полагают, что естественное
сообщение между общим желчным и панкреатическим протоками, а также обту-
рация общего желчного протока желчным камнем на уровне стенки
двенадцатиперстной кишки служат причинами заброса желчи в панкреатические
протоки как инициирующего момента острого панкреатита у пациентов с
хроническим калькулезным холециститом. Смесь желчи и секрета
поджелудочной железы, воздействуя на ее паренхиму, индуцирует щтолиз ее
клеточных элементов и стромы (аутолиз, аутодигестия поджелудочной железы).
432
Глава 26
Таблица 26.1
Причины острого панкреатита
♦ Желчекаменная болезнь как с обтурацией общего желчного протока камнями, так и
без нее
♦ Травма железы при огнестрельных проникающих ранениях живота и при его
закрытых травмах
♦ Повреждения железы при плановых хирургических вмешательствах
♦ Пенетрация пептической язвы желудка в железу
♦ Гиперлипопротеинемия (семейный дефицит липопротеинлипазы и гиперлипопротеи-
немия пятого типа)
♦ Гиперкальциемия
♦ Наследственный острый панкреатит как в сочетании с аминоацидурией, так и без нее
♦ Побочное действие лекарств
♦ Свинка
♦ Коксакивирус типа В
♦ Обтурация атеросклеротическими бляшками панкреатодуоденальных артерий
♦ Циркуляторная гипоксия железы вследствие системного васкулита (системная
красная волчанка, узелковый периартериит)
♦ Тромбоз при тромбоцитопенической тромбогемолитической пурпуре
♦ Болезнь Бехчета
♦ Опухоли поджелудочной железы
♦ Метастазы в железу злокачественных опухолей другой локализации
♦ Опухоли поджелудочной железы
♦ Глистные инвазии
♦ Отравление ядом скорпиона
♦ Гипотермия
♦ Идиопатический панкреатит
Результаты многочисленных экспериментов, клинических
исследований и аутопсий, позволяют считать ответственными за аутолиз
поджелудочной железы такие компоненты желчи как желчные соли и лецитин.
Фосфолипаза А - это тот фермент секрета поджелудочной железы,
активность которого представляется возможным связать с приобретением
смеси желчи и панкреатического секрета свойства вызывать аутолиз железы.
Фосфолипаза А отщепляет от лецитина одну из жирных кислот,
превращая лецитин в лизолецитин. Лизолецитин является эндогенным токсином,
действие которого на ткань поджелудочной железы в эксперименте
приводит к ее некрозу в морфопатогенетическом отношении схожему с
некрозом железы при остром панкреатите у человека. Клеточные мембраны
ткани поджелудочной железы также в избытке содержат лецитин как
субстрат образования лизолецитина. После заброса желчи в панкреатические
протоки и ее смешивания с панкреатическим секретом в образовавшейся
смеси через активность трипсина образуется неактивная форма фосфоли-
пазы А. Ее активируют желчные кислоты. Активированная фосфолипаза
А образует лизолецитин из лецитина, который содержат желчь и
мембраны клеток поджелудочной железы. Кроме того, в индукции аутолиза иг-
Патогенез острого и хронического панкреатитов
433
рает роль активация зимогенов (класс ферментов, к которым относится
фосфолипаза А) под влиянием трипсина, активность которого в железе
растет в результате прекращения нормального оттока панкреатического
секрета по вирзунгову протоку. Рост активности фосфолипазы А
увеличивает образование лизолецитина. Лизолецитин разрушает клеточные
мембраны, активируя зимогены и фосфолипазу А. Таким образом
замыкается порочный круг патогенеза. В результате концентрация
лизолецитина достигает патогенно высокого уровня в большей части массы
поджелудочной железы.
Другим механизмом развития панкреонекроза при остром
панкреатите считают заброс содержимого двенадцатиперстной кишки в
панкреатический проток через зияющее отверстие Фатерова сосочка после
повреждения его гладкомышечных элементов конкрементом,
«прорвавшимся» в просвет двенадцатиперстной кишки.
Заброс содержимого двенадцатиперстной кишки, содержащего энтеро-
киназу, в панкреатические протоки, ведет к активации неактивной формы
трипсина в их просвете. В результате активность панкреатического
секреторного ингибитора трипсина становится недостаточной относительно
массы активированного трипсина в протоках железы. Это обуславливает
аутодигестию поджелудочной железы через активность трипсина. Есть
предположения об определяющей роли дефицита активности
панкреатического секреторного ингибитора трипсина в развитии острого панкреатита.
Одним из звеньев патогенеза острого панкреатита является
обструкция оттоку секрета по панкреатическому протоку, которая сама по себе
приводит к выходу ферментов поджелудочной железы в виде зимогенных
гранул в интерстиций паренхимы железы, после чего панкреатические
ферменты появляются в циркулирующей плазме крови. Обструкция сама
по себе в эксперименте не вызывает геморрагического панкреатита,
обуславливая жировое перерождение железы. Известно, что прекращение
кровотока по панкреатикодуоденальной артерии в течение пятнадцати
минут или аналогичной длительности блокада венозного оттока могут
приводить к острому панкреатиту, возникновение которого не
представляется возможным связать только с ишемическим некрозом железы.
Очевидно, ишемия индуцирует аутолиз железы через действие во многом
неясных патогенетических механизмов. При этом локальное истощение
ингибиторов протеаз плазмы крови (альфа-два-макроглобулин и др.)
приводит к резкому возрастанию активности ферментов железы и приводит к
скачкообразному ускорению аутолиза.
При обструкции панкреатических протоков ферменты поджелудочной
железы появляются в циркулирующей крови после абсорбции в просвет
лимфатических сосудов. Известно, что сеть лимфатических сосудов
желчного пузыря, общего желчного протока и сеть лимфатических сосудов
панкреатической железы сообщаются между собою. Таким образом флогоге-
ны, образовавшиеся в стенке желчного пузыря при хроническом кальку-
лезном холецистите, могут попадать в интерстиций поджелудочной железы
и вызывать острый панкреатит. Полагают, что таков механизм возникнове-
434
Глава 26
ния острого панкреатита у больных с большими конкрементами в просвете
пузыря, но без обтурации камнями общего желчного протока.
Росту активности ферментов поджелудочной железы в
циркулирующей крови препятствует их связывание ингибиторами протеаз плазмы
крови (альфа-два-макроглобулин, альфа-один-антитрипсин, альфа-два-
макроглобулин и др.). Это биоактивные вещества, циркулирующие с
кровью, действие которых состоит в угнетении активности протеолитических
ферментов. Если связывание протеаз с альфа-один-антитрипсином
приводит к необратимой потере ферментами своих свойств катализаторов
соответствующих реакций, то альфа-два-макроглобулин таким качеством не
обладает. Трипсин, связанный с ним, сохраняет способность активировать
трипсиноген, а также индуцировать образование сгустка фибрина из
фибриногена, одновременно вызывая фибринолиз.
Следует заметить, что в настоящее время нет общепринятой схемы
патогенеза острого панкреатита вследствие заброса желчи и содержимого
двенадцатиперстной кишки в панкреатический проток. Тем не менее,
следует ориентироваться на схему патогенеза аутодигестии, с которой в
основном согласны большинство врачей-исследователей (схема 26.1).
Заброс желчи и содержимого двенадцатиперстной
кишки в панкреатические протоки
1
Трансформация трипсиногена в
трипсин в панкреатических протоках
и интерстиции железы
1
г
^
т
Появление желчных солей в
просвете желчных протоков |
1
г
Активация трипсином зимогенов и фосфолипазы А
i
к
у
г
Образование лизолецитина
1
*
Деструкция клеточных мембран
Аутолиз
i. |
1 1
л
i
Отек и нарушения
мш
:роцир
ку
ляции
Схема 26.1. Патогенез аутодигестии поджелудочной железы вследствие попадания
желчи и содержимого двенадцатиперстной кишки в панкреатический проток
Гиперкальциемия как следствие избыточной секреции паратгормона
может обусловить острый панкреатит. Кроме того, гиперкальциемия как
причина острого панкреатита может быть следствием множественной
миеломы и саркоидоза.
Патогенез острого панкреатита вследствие гиперкальциемии до сих пор
остается неясным. Одно время считали, что в данном случае патогенез
аутодигестии складывается из прямой активации ионизированным кальцием про-
Патогенез острого и хронического панкреатитов
435
теаз поджелудочной железы. Известно, что одна из многочисленных
функций свободных ионов кальция заключается в стабилизации трипсина после
активации соответствующего профермента и предотвращении деструкции
трипсина через его собственную протеолитическую активность.
Предположительно, гиперкальциемия вызывает преципитацию белков в
панкреатических протоках, что ведет к панкреатиту через механическое препятствие
оттоку панкреатического секрета в вирзунгов проток. Может быть, что
панкреатит вследствие гиперкальциемии связан с ростом секреции гастрина G-
клетками слизистой оболочки желудка как стимулятора панкреатической
секреции под влиянием роста содержания кальция в интерстиции стенки
желудка вследствие гиперкальциемии.
При остром панкреатите у 25 % больных развивается желтуха, которая
у 10% больных является следствием обтурации или сдавления общего
желчного протока. Только у 2-3 % пациентов с острым панкреатитом
выявляют обструкцию желчевыведению и желтуху вследствие закупорки
желчевыводящих путей желчными камнями. В остальных случаях
обструкция как причина желтухи связана со сдавлением общего желчного
протока отекшей головкой поджелудочной железы. У 15 % больных
желтуха при остром панкреатите выступает результатом дисфункций гепато-
цитов, не связанных с обструкцией желчеотделению.
У 30 % больных с острым панкреатитом концентрация кальция в
сыворотке крови падает до уровня более низкого, чем 85 мг/л (нижний
предел нормальных колебаний). Содержание кальция начинает снижаться в
первые-вторые сутки после возникновения острого панкреатита. Своего
максимума снижение концентрации кальция в сыворотке крови у больных
острым панкреатитом достигает в пятые-седьмые сутки. Снижение общей
концентрации кальция в сыворотке является плохим прогностическим
признаком. Не исключено, что падение содержания кальция связано со
снижением содержания в плазме крови альбуминов, которые связывают
ионизированный кальций и переносят его, циркулируя с плазмой крови.
Падение концентрации альбуминов обуславливают вторичная по
отношению к острому панкреатиту дисфункция гепатоцитов, а также выход
альбуминов в интерстиции при воспалении в тканях, некробиотически
измененных при остром панкреатите. Аутопсийные исследования у больных с
острым панкреатитом выявляют отложения кальция в области некроза и
аутолиза железы. Очевидно, гипокальциемия у больных острым
панкреатитом не приводит к значительному падению содержания в сыворотке
крови свободного ионизированного кальция, так как у большинства
больных с острым панкреатитом не выявляют его клинических проявлений
(тетания и др.). Рост концентрации кальция в сыворотке крови не
представляется возможным связать с ростом содержания в крови глюкагона и
кальцитонина у больных острым панкреатитом, так как клинико-пато-
физиологические исследования не выявили между ними какой-либо
достоверной корреляции. Предположительно при остром панкреатите
угнетена реакция роста секреции паратгормона в ответ на падение содержания
в сыворотке крови ионизированного кальция.
436
Глава 26
Одним из грозных осложнений острого панкреатита является
формирование ложной кисты поджелудочной железы. Ложная киста
поджелудочной железы заполнена жидкостью, содержащей продукты цитолиза.
Ее стенки могут быть выполнены листками брюшины или фиброзной
ложной капсулой. Ложная киста - это результат воздействия
активированных протеаз поджелудочной железы на ткани, прилежащие к данному
органу, после выхода в них трипсина и других протеолитических
панкреатических ферментов через разрывы в капсуле железы. Ложная киста
может образоваться в ретроперитонеалбном пространстве или
формируется между листками брюшины. Ложную кисту следует отличать от
истинной ретенционной кисты поджелудочной железы, которая
представляет собой результат обтурации панкреатических протоков и роста
давления панкреатического секрета. Инфицирование ложных и ретенционных
кист поджелудочной железы ведет к формированию абсцессов
соответствующей локализации.
80 % ложных и истинных кист поджелудочной железы подвергаются
обратному развитию в течение двух недель. В противном случае киста
начинает проявлять себя характерной опоясывающей болью и приводит к
осложнениям:
♦ желтуха вследствие блокады продвижения желчи по желчевыводя-
щим путям (сдавление общего желчного протока);
♦ тромбоз воротной и селезеночной вен;
♦ блокада выхода содержимого из желудка в двенадцатиперстную
кишку (высокая кишечная непроходимость);
♦ разрыв кисты с эвакуацией ее содержимого в полость брюшины;
♦ наиболее грозное осложнение, то есть кровотечение из
поврежденного эрозией сосуда стенок кисты.
Разрыв кисты с эвакуацией ее содержимого в полость брюшины
может обусловить быстрое развитие необратимого шока, ведущими
звеньями патогенеза которого являются нарастание токсемии через всасывание
флогогенов с поверхности брюшины, а также вагальные рефлексы,
вызываемые возбуждением чувствительных нервных окончаний перитонеаль-
ного рецепторного поля.
Постоянное поступление содержимого кисты в полость брюшины
обуславливает так называемый панкреатический асцит.
Ложная и ретенционная киста могут пенетрировать через диафрагму в
плевральную полость, что в частности может привести к абсцессу
плевральной полости.
Полное устранение причины острого панкреатита как осложнения
хронического калькулезного холецитита, конкрементов в просвете желчного
пузыря, служит необходимым условием восстановления у большинства
больных нормальных функций поджелудочной железы через гипертрофию
и гиперплазию ее клеточных элементов. Следует заметить, что
восстановление железы как органа-эффектора систем регуляции и пищеварения
происходит у большинства больных за 1-3 месяца и приводит к полной рекон-
валесценции больных с острым панкреатитом вследствие желчекаменной
Патогенез острого и хронического панкреатитов
437
болезни. Это диктует необходимость высокой хирургической активности
при выявлении конкрементов в просвете желчного пузыря.
Патогенез распространенного жирового некроза вследствие острого
панкреатита до сих пор остается неясным. Были высказаны
предположения, что выброс активированных липаз и фосполипазы А в системно
циркулирующую кровь приводит на периферии к распаду триглицеридов и
фосфолипидов. При этом игнорировали то, что для обеспечения
взаимодействия липаз с триглицеридами необходимо воздействие на них
эмульгаторов. Некоторыми из чисто предположительных звеньев патогенеза
распространенного жирового некроза у больных с острым панкреатитом
являются:
♦ перенос липаз, активированных в поджелудочной железе, на
периферию внутри гранул лейкоцитов;
♦ активация липаз на периферии гипотетическим фактором их
активации, высвобождаемым железой при остром панкреатите.
Причиной панкреатического асцита обычно является разрыв капсулы
воспаленной железы, когда ее содержимое попадает в полость брюшины.
Известно, что перитонеальный диализ как средство элиминации с
поверхности брюшины флогогенов-цитокинов в качестве эндогенных
токсинов, а также как блокада вагальных рефлексов в афферентном звене
(устранение механического раздражения чувствительных нервных окончаний пе-
ритонеального рецепторного поля) подвергает системные расстройства
кровообращения у больных с острым панкреатитом обратному развитию.
У 60 % больных с острым панкреатитом развивается явная или
скрытая дыхательная недостаточность. У 20 % больных возникает острая
почечная недостаточность. Острая почечная недостаточность у больных с
острым панкреатитом обычно представляет собой следствие критического
несоответствия объема циркулирующей крови емкости сосудистого
русла. При этом опасное увеличение емкости сосудистого русла связывают с
ростом содержания в циркулирующей крови цитокинов-флогогенов
(альфа-фактор некроза опухолей, интерлейкины и др.), раскрывающих арте-
риоло-венулярные анастомомозы, дилатирующих артериолы и
снижающих общее периферическое сосудистое сопротивление, кининов-вазоди-
лятаторов, фракций системы комплемента, расширяющих микрососуды и
других иммунорегуляторов и медиаторов воспаления в качестве
эндогенных токсинов.
При достижении вторичной альтерацией при остром панкреатите
соответствующей протяженности острый панкреатит через гиперцитоки-
нию становится причиной системной воспалительной реакции и
множественной системной недостаточности, а также может приводить к
септическому шоку, нарушения системного кровообращения при котором
обостряет плазмопотеря в массив тканей, некробиотически измененных
как в самой железе, так и в ретроперитонеальном пространстве.
Множественная системная недостаточность у больных острым панкреатитом
часто проявляет себя в первую очередь респираторным
дистресс-синдромом взрослых.
438
Глава 26
ЭТИОЛОГИЯ И ПАТОГЕНЕЗ
ХРОНИЧЕСКОГО ПАНКРЕАТИТА
Примерно у 90 % больных хроническим панкреатитом выявляют
хронический алкоголизм, при котором больные пьют спиртное каждый день.
Следует заметить, что у многих из таких пациентов нет заметных
деградации личности и социальной дезадаптации.
Механизм патогенного влияния постоянно повышенной концентрации
этилового спирта в крови на железу остается неясным. Согласно одной из
известных гипотез патогенеза хронического панкреатита, болезнь
является следствием преципитации белков и липидов панкреатического секрета
в протоках поджелудочной железы под влиянием повышенной
концентрации этанола во внутренней среде. Полагают, что преципитация
вызывает обтурацию панкреатических протоков как одну из непосредственных
причин хронического панкреатита.
Не исключено, что патогенез хронического панкреатита вследствие
хронического алкоголизма, связан с нарушением обмена лактоферрина
вследствие постоянной интоксикации этанолом. Лактоферрин - это
протеин, который содержит молоко женщины, широкий спектр экзокринных
секретов, а также специфические гранулы полиморфонуклеаров.
Молекулы лактоферрина содержат атомы железа. У больных хроническим
панкреатитом содержание лактоферрина в просвете панкреатических
протоков резко возрастает. Известно, что концентрация лактоферрина в
панкреатическом секрете увеличивается после роста секреции холецистоки-
нина. В этой связи можно предположить, что хронический алкоголизм как
причина системной дизрегуляции расстраивает взаимодействие между
органами-эффекторами системы пищеварения, которое осуществляется
через секрецию полипептидных гормонов. Маркером такого расстройства
систем локальной регуляции является рост концентрации лактоферрина в
панкреатическом секрете. Возможно, что лактоферрин, находящийся в
составе панкреатического секрета, высвобождают ацинарные клетки
поджелудочной железы.
Анализ белковых преципитатов, которые закупоривают
панкреатические протоки, выявляет в их составе белки, которых нет в нормальном
панкреатическом секрете. К ним в частности относят панкреолитин,
обладающий повышенным сродством к ионизированному кальцию. В панкреолити-
не высоко содержание глутамина и аспаргиновой кислоты.
Полагают, что у части больных с хроническими панкреатитом и
алкоголизмом, патологические изменения поджелудочной железы - это не
только следствие постоянной интоксикации этанолом, но результат гипер-
триглицеридемии, то есть наиболее частой формы гиперлипопротинемии
(гиперлипопротеинемия четвертого типа). Известно, что в эксперименте
панкреатит можно вызвать перфузией железы раствором с высоким
содержанием триглицеридов. Предположительно интоксикация алкоголем и
потребление пищи с высоким содержанием жиров резко повышают
концентрацию триглицероидов в крови и без того уже высокую вследствие гипер-
Патогенез острого и хронического панкреатитов
439
липопротеинемии четвертого типа. Рост содержания триглицеридов в
крови служит причиной панкреатита. Предположительно гипертриглицериде-
мия приводит к панкреатиту через высвобождение жирных кислот.
Хроническая гиперкальциемия, то есть патогенно высокое содержание
кальция во внеклеточной жидкости и циркулирующей плазме крови,
может приводить к хроническому панкреатиту. Длительную гиперкальцие-
мию, вызывающую хронический панкреатит, обуславливают:
♦ гиперпаратиреоз (патологическое состояние вследствие избыточной
секреции паратгормона околощитовидными железами);
♦ отравление витамином D;
♦ молочно-щелочной синдром, то есть обратимое на ранней стадии
нефрозоподобное патологическое изменение структуры и функций
почек вследствие продолжительной терапии больных с пептиче-
скими язвами желудка и двенадцатиперстной кишки антацидами и
назначением внутрь больших количеств молока.
При хронической гиперкальциемии морфопатогенез хронического
панкреатита во многом складывается из кальцификации железы. У
больных хроническим панкреатитом вследствие хронического отравления
этанолом общая концентрация кальция в панкреатическом секрете
повышена. В частности общую концентрацию кальция в панкреатическом
секрете составляет содержание в нем комплексных соединений, в состав
которых входит СаС03. Не исключено, что у больных хроническим
панкреатитом способность панкреатического секрета растворять
комплексные соединения, содержащие СаСОз, становится низкой относительно
содержания данной соли в панкреатическом секрете, что приводит к ее
преципитации, вызывающей и преципитацию белков. Повышенная
концентрация кальция в поджелудочной железе рассматривается как один из
причинных факторов ее кальциноза у больных с хроническим
панкреатитом. Кроме того, кальциноз связывают с эндогенным белковым
голоданием вследствие недостаточной концентрации протеолитических ферментов
в просвете кишечника.
Наследственный хронический панкреатит начинает проявлять себя
соответствующими симптомами в возрастном диапазоне от 11 месяцев до
61 года. Чаше всего наследственный хронический панкреатит впервые
возникает в возрасте 11 лет. Диагноз данного заболевания обычно
становится окончательным в возрасте 31 года. Это наследственное заболевание
наследуется по аутосомно-доминантному типу при неполной пенетрант-
ности соответствующего гена. Пенетрантность (лат. penetrare -
проникать) - частота проявления в популяции того или иного гена. Для
больных наследственным хроническим панкреатитом характерна высокая
предрасположенность к идиопатической кальцификации железы и ее
злокачественной опухоли.
Голодание, и в особенности качественное белковое, - это не столько
причина хронического панкреатита, сколько фактор развития атрофии
ацинарных клеток железы и падения ее экзокринной функции. Аутопсия у
больных квашиоркором, кроме атрофии ацинарных клеток, выявляет
440
Глава 26
расширение панкреатических протоков, очаги фиброзного перерождения
железы, локусы минимально выраженного воспаления и потерю железой
определенного числа островков Лангерганса. По мере прогрессирования
недостаточности экзокринной функции поджелудочной железы в
панкреатическом секрете неуклонно снижается содержание проферментов
протеаз, а в паренхиме железы падает содержание зимогенных гранул,
несмотря на неизменность массы секрета и содержания в нем бикарбо-
натных анионов. В некоторых регионах мира (Индонезия и др.)
качественное белковое голодание через действие пока неизвестных
патогенетических механизмов вызывает кальцификацию поджелудочной железы при
хроническом панкреатите.
Предположения о том, что хронический панкреатит представляет
собой следствие обструкции выведению в просвет двенадцатиперстной
кишки желчи и панкреатического секрета, не нашли своего
подтверждения результатами клинико-физиологических исследований.
Синдром недостаточного кишечного всасывания у больных с
хроническим панкреатитом в частности характеризует то, что низкое кишечное
всасывание вследствие хронического воспаления поджелудочной железы
не вызывает клинически значимой гипокальциемии и вторичной по
отношению к ней остеомаляции. Это отличает падение кишечного
всасывания данного генеза от снижения всасывания нутриентов из просвета
кишечника при целиакии, спру и других хронических заболеваниях
кишечной стенки. Кроме того, у пациентов с хроническим панкреатитом как
причиной низкого кишечного всасывания обычно не развивается анемия со
снижением концентрации гемоглобина в циркулирующей крови до уровня
более низкого, чем 100 г/л. Дело в том, что при хроническом панкреатите
обычно нет снижения абсорбционной поверхности кишечника, основной
причины эндогенного голодания при кишечной мальабсорбции (синдроме
недостаточного кишечного всасывания), вызывающей анемию и гипокаль-
циемию. Хронический панкреатит при своем медленном прогрессировании
(в течение нескольких лет) обуславливает нарастающее падение
активности панкреатических ферментов в просвете поджелудочной железы.
Падение активности амилолитических ферментов приводит к росту
содержания в просвете кишечника крахмала и других неабсорбируемых
углеводов (сахаридов). Это повышает количество осмолей в кишечном
содержимом, увеличивает его массу, снижает всасывание воды и ведет к
диарее.
Повышенная активность панкреатической амилазы в сыворотке
крови достоверно свидетельствует о развитии острого панкреатита, а также
о хроническом панкреатите и его обострении только тогда, когда
уровень активности фермента превышает верхнюю границу нормы в два-
три раза.
Недостаточная активность в просвете кишечника липолитических
панкреатических ферментов липазы, фосфолипазы А и холестеринэстера-
зы через снижение кишечного всасывания жиров и недостаточность ли-
полиза в просвете кишки приводит к диарее и стеаторее.
Патогенез острого и хронического панкреатитов
441
Низкая активность в просвете кишечника у больных хроническим
панкреатитом эндопептидаз, экзопептидаз и эластазы служит причиной
белкового голодания, может приводить к анемии, а также через
увеличение содержания в просвете кишки неабсорбируемых осмолей вызывает
осмотическую диарею. При этом недостаточность эндопептидаз,
обуславливая патологически низкое всасывание витамина В^, может приводить и
к пернициозной анемии.
Через 6-12 лет своего течения хронический панкреатит у одной трети
больных приводит к появлению классической триады, которую
составляют сахарный диабет, кальциноз поджелудочной железы и стеаторея.
Вторичный сахарный диабет у больных с хроническим панкреатитом - это
следствие потери поджелудочной железой определенного числа инсулин-
продуцирующих клеток в результате воспалительной альтерации и
реактивного фиброза.
Глава 27
САХАРНЫЙ ДИАБЕТ
Сахарный диабет в первую очередь характеризуют гипергликемия,
то есть патологически высокое содержание глюкозы в крови, и другие
нарушения обмена веществ, связанные с патологически низкими
секрецией инсулина, концентрацией нормального гормона в циркулирующей крови
или представляющие собой следствие недостаточности или отсутствия
нормальной реакции клеток-мишеней на действие гормона-инсулина. Как
патологическое состояние всего организма сахарный диабет в основном
составляют расстройства обмена веществ, в том числе и вторичные
относительно гипергликемии, патологические изменения микрососудов
(причины ретино- и нефропатии), ускоренный атеросклероз артерий, а также
нейропатия на уровне периферических соматических нервов,
симпатических и парасимпатических нервных проводников и ганглиев.
Выделяют два типа сахарного диабета (табл. 27.1). От сахарного
диабета! типа страдают 10 % больных сахарным диабетом как первого, так
и второго типа. Сахарный диабет первого типа называют инсулинзависи-
мым не только потому, что больным для устранения гипергликемии
необходимо парентеральное введение экзогенного инсулина. Такая
необходимость может возникнуть и при лечении больных с неинсулинзависимым
сахарным диабетом. Дело в том, что без периодического введения
инсулина больным сахарным диабетом I типа у них развивается
диабетический кетоацидоз.
Если инсулинзависимый сахарный диабет возникает в результате
почти полного отсутствия секреции инсулина, то причина неинсулин-
зависимого сахарного диабета - это частично сниженная секреция
инсулина и (или) резистентность по отношению к инсулину, то есть
отсутствие нормальной системной реакции на высвобождение гормона
инсулинпродуцирующими клетками островков Лангерганса
поджелудочной железы.
Длительное и экстремальное по силе действие неотвратимых
раздражителей в качестве стимулов стресса (послеоперационный период в
условиях неэффективной анальгезии, состояние вследствие тяжелых ранений
и травм, персистирующий отрицательный психоэмоциональный стресс,
вызванный безработицей и нищетой, и др.) обуславливает длительную и
патогенную активацию симпатического отдела автономной нервной
системы и нейроэндокринной катаболической системы. Эти сдвиги
регуляции через нейрогенное снижение секреции инсулина и устойчивое
преобладание на системном уровне эффектов катаболических гормонов
антагонистов инсулина может трансформировать сахарный диабет II типа в
инсулинзависимый, что служит показанием к парентеральному введению
инсулина.
Сахарный диабет
443
Таблица 27.1
Тип сахарного
диабета
Частота в основной
популяции
Возраст больных при
возникновении
заболевания
Наследственная пред-
расположенность
Генетические маркеры
предрасположенности к
заболеванию
Аутоиммунный
механизм развития болезни
Конституция больных
Нарушения обмена
веществ
Основной элемент
терапии
Типы сахарного диабета
Первый (I)
инсулинзависимый
0,2-0,5 %, частота среди
мужчин и женщин одинакова
Меньше 25 лет
Частота инсулинзависимого
сахарного диабета у родственников
пробанда первой степени
меньше, чем 10 %; вероятность
болезни у гомозиготного близнеца
(брата, сестры пробанда)
составляет 50 %
Гены (антигены) из системы
генов (антигенов) лейкоцитов
человека В8, В15, DR3, DR4
У части больных с кровью
циркулируют аутоантитела к
антигенам инсулинпродуцирующих
клеток островков Лангерганса и
другим аутоантигенам
Масса тела часто снижена,
низкое общее содержание жировой
ткани и триглицеридов в теле
Тенденция к развитию
метаболического кетоацидоза;
патологически низкая секреция
инсулина
Инсулин, вводимый
парентерально
Второй (II)
неинсулинзависимый
2-4 %, женщины
болеют чаще, чем мужчины
Больше 40 лет
Частота заболевания у
родственников пробанда
первой степени больше,
чем 20 %; вероятность
болезни у гомозиготного
близнеца находится на
уровне 90-100%
Нет
Нет
У 80 % больных
выявляют ожирение при массе
тела, превышающей 115 %
идеальной расчетной
Метаболический кетоаци-
доз не развивается;
секреция инсулина может быть
повышенной, нормальной
или патологически низкой
Диета и физическая
активность для снижения
массы тела
Вторичный сахарный диабет - это патологическое возрастание
концентрации глюкозы в плазме крови, обусловленное не сахарным
диабетом, а другим патологическим состоянием.
Вторичный сахарный диабет может быть следствием патогенно
избыточного питания, особенно с высоким потреблением углеводов.
Вероятность гипогликемии выше, если переедание происходит одновременно со
снижением физической активности.
Снижение образования эндогенного инсулина как причину
вторичного сахарного диабета вызывают:
♦ разрушение инсулин-продуцирующих клеток в результате
побочного действия таких лекарственных средств как стрептозоцин и
пентамидин;
444
Глава 27
♦ обратимое угнетение секреции инсулина под влиянием
лекарственных средств (диазоксид, диуретики из группы тиазидов, фенитоин);
♦ гипокалиемия и (или) гипомагниемия.
Кроме того, вторичный сахарный диабет может возникать в
результате тяжелых и хронических инфекций, воспаления, тяжелой ишемии и
инфаркта миокарда, травмы, хирургических вмешательств, отрицательного
эмоционального стресса как причин избыточно патогенной активации
нейроэндокринной катаболической системы, приводящей к
превалированию на системном уровне эффектов катаболических гормонов
антагонистов инсулина. В данном случае вторичный сахарный диабет является
следствием патологической резистентности к эффекту инсулина на
уровне экспрессии генома клеток.
Беременность и побочное действие препаратов на основе эстрогенов, в
том числе и пероральных контрацептивов, также могут приводить ко
вторичному сахарному диабету через развитие патологической резистентности
к действию инсулина на клетки и ткани. Патологическая резистентность по
отношению к действию инсулина может быть результатом побочного
действия адреномиметиков и препаратов на основе никотиновой кислоты.
ЭТИОЛОГИЯ САХАРНОГО ДИАБЕТА
Можно считать, что этиология сахарного диабета до сих пор остается
неясной. Тем не менее, в зависимости от этиологии каждый тип сахарного
диабета как отдельную нозологическую форму можно определить
следующим образом:
Сахарный диабет первого типа - это мулътифакториалъная
(полигенная) болезнь, которая, часто являясь аутоиммунным заболеванием,
может быть связана с генетическими маркерами из системы антигенов
лейкоцитов человека, другими аутоиммунными болезнями; инсулинзави-
симый сахарный диабет - это во многом результат взаимодействия
между собой элементов генома, приводящего к инсулинопении.
Сахарный диабет второго типа - полигенное заболевание, во многом
зависящее от кумуляции в организме последствий взаимодействия с
внешними этиологическими факторами. К таким последствиям следует
отнести длительное избыточное поступление в организм источников
свободной энергии вместе с нутриентами, гиподинамию, а таксисе
хронический отрицательный психоэмоциональный стресс.
Можно предположить, что взаимодействие элементов генома,
обуславливающее предрасположенность к сахарному диабету второго типа,
детерминирует и высокую предрасположенность к ряду
распространенных полигенных болезней: первичной артериальной гипертензии,
атеросклерозу, ишемической болезни сердца, а также ожирению.
Неинсулинзависимый сахарный диабет нельзя считать полигенной
болезнью, обусловленной исключительно патологической резистентностью
Сахарный диабет
445
по отношению к инсулину. Об этом свидетельствуют результаты последних
экспериментальных исследований роли генетического фактора в развитии
сахарного диабета второго типа, объектом которых стали две линии
трансгенных мышей. У животных первой линии в геноме отсутствовал ген
первого субстрата инсулинового рецептора, а у мышей второй линии
генетический материал не содержал гена глюкокиназы бета-клеток островков Лан-
герганса, от активности которого зависит синтез инсулина. И у той и у
другой линии мышей не было патологической толерантности по отношению к
глюкозе. Она появилась у потомства мышей двух линий, генотип которого
определял патологическую резистентность клеток-эффекторов по
отношению к действию инсулина (отсутствие гена первого субстрата
инсулинового рецептора) и обуславливал недостаточность секреции инсулина в ответ
на рост концентрации глюкозы во внеклеточной жидкости и крови через
низкую активность глюкокиназы бета-клеток.
Результаты данного эксперимента еще раз подтвердили то, что неин-
сулинзависимый сахарный диабет является полигенной болезнью,
наследственная предрасположенность при которой складывается из
генетически детерминированных патологической резистентности по отношению
к инсулину и недостаточности секреции инсулина. При этом нельзя
исключить из патогенеза неинсулинзависимого сахарного диабета
следующего порочного круга: раз возникнув, гипергликемия сама по себе
становится фактором прогрессирования возрастания концентрации глюкозы в
крови через угнетение секреции инсулина и усиление патологической
резистентности по отношению к действию гормона.
ПАТОЛОГИЧЕСКАЯ РЕЗИСТЕНТНОСТЬ
ПО ОТНОШЕНИЮ К ИНСУЛИНУ
Резистентность по отношению к инсулину характеризует отсутствие
нормальной реакции снижения концентрации глюкозы в крови в ответ на
рост содержания в ней инсулина. Выделяют три уровня резистентности
по отношению к инсулину:
♦ пререцепторный,
♦ рецепторный,
♦ пострецепторный.
Резистентность по отношению к инсулину на пререцепторном уровне
обуславливают:
♦ патологические изменения гормона,
♦ взаимодействие инсулина в качестве носителя аутоантигенов с ау-
тоантителами,
♦ выброс клетками поджелудочной железы в кровь проинсулина
вместо инсулина,
♦ блокада нормального взаимодействия гормона со своим рецептором
на поверхности клеток.
446
Глава 27
При хронической почечной недостаточности и уремическом синдроме
через действие механизма неспецифического антагонизма расстраивается
взаимодействие нормального инсулина со своим нормальным
рецептором. Одновременно патологически низкое содержание ионизированного
кальция во внеклеточной жидкости и жидкой части плазмы крови как
элемент уремического синдрома увеличивает выброс в кровь проинсули-
на, который не обладает свойствами главного анаболического гормона,
вместо инсулина. Таков патогенез пререцепторной резистентности по
отношению к инсулину у больных с уремическим синдромом.
Пререцепторная резистентность по отношению к инсулину
составляет ведущее звено патогенеза сахарного диабета, который возникает
одновременно с проявлениями повышенной секреции андрогенов (опухоли
яичников и их поликистоз, а также гипертекоз, то есть диффузная
гиперплазия текоцитов граафова фолликула) после полового созревания у
девочек. Данный синдром получил название гиперандрогенной рези-
стентности по отношению к инсулину. У больных с
гиперандрогенной резистентностью по отношению к инсулину гормон инактивируется
вследствие связывания с его эпитопами паратопов аутоантител.
У части больных пререцепторная резистентность по отношению к
инсулину представляет собой моногенную болезнь, при которой патогенная
мутация, закрепившаяся в ряду поколений, обуславливает врожденный
патогенный дефект строения молекулы инсулина.
Резистентность по отношению к инсулину на рецепторном уровне
может быть следствием:
♦ низкого уровня связывания гормона со своими рецепторами из-за
врожденных аномалий инсулиновых рецепторов (ряд редких
моногенных заболеваний),
ф низкой экспрессии инсулиновых рецепторов на поверхности
соответствующих клеток (во многом детерминируемое генотипом
патологическое состояние систем регуляции обмена веществ),
♦ аутоиммунного поражения рецепторов.
Инсулиновый рецептор представляет собой мембранный гетеродимер,
состоящий из альфа- и бета-цепей, который после соединения с
инсулином приобретает активность тирозинкиназы. Врожденный дефицит
инсулиновых рецепторов может быть результатом патогенных мутаций:
♦ изменения последовательности пар нуклеотидов дезоксирибонук-
леиновой кислоты, приводящего к потере рецептором домена
тирозинкиназы;
♦ точечной мутации как причины отсутствия в рецепторе локуса
связывания аденозинтрифосфата и др.
У больных с синдромом гиперандрогенного инсулинрезистентного
черного акантоза аутоиммунное поражение инсулиновых рецепторов-
это ведущее звено патогенеза сахарного диабета, действие которого у
части пациенток с данным синдромом не приводит к явному сахарному
диабету. Синдром выявляют у 5 % женщин с гирсутизмом, то есть
излишним оволосением тела. При данном синдроме нередкими являются
Сахарный диабет
447
кистозное перерождение яичников, а также патологически высокие
концентрации в крови тестостерона и андростендиона наряду с черным акан-
тозом. Черный акантоз (acanthosis nigricans) - дерматоз в виде
бархатистых бородавчатых разрастаний и гиперпигментации кожи,
подмышечных впадин, шеи, анальной области, паха.
Инсулин- это полипептидный гормон, составляющие который
аминокислотные цепи, связаны между собой дисульфидной связью. Инсулин
как главный анаболический гормон и антагонист катаболических
гормонов стимулирует экспрессию генов глицеральдегид-3-фосфат дегидроге-
назы, глюкокиназы, альфа-амилазы и тормозит экспрессию гена
ключевого фермента глюконеогенеза фосфоенолпируваткарбоксикиназы.
Механизм действия инсулина как гормона, модулирующего экспрессию генов
клетки, неизвестен, но ясно, что инсулин действует через систему
белковых сериновых киназ (ферментов, катализирующих превращение
профермента в фермент), то есть киназы ацетил-коэнзим А-карбоксилазы,
киназы клеточного микроканальцевого протеина-2, протеинкиназы С и
других киназ. В этой связи становится ясным, что принципиально
возможно извращение действия инсулина на геном клеток на различных
уровнях функционирования той или иной киназы как причина пострецеп-
торной резистентности по отношению к инсулину.
При неинсулинзависимом сахарном диабете пострецепторная
резистентность по отношению к инсулину может быть ведущим звеном
патогенеза. При этом аномалия реакции генома клеток на системном уровне
по отношению к инсулину в основном складывается из нарушений
синтеза гликогена в миоцитах скелетных мышц, то есть главного механизма
аккумуляции избытка глюкозы в организме как у больных сахарным
диабетом, так и в физиологических условиях. Предположительно свою роль в
снижении реакции усиления образования гликогена в миоцитах скелетных
мышц в ответ на действие инсулина может играть извращение его действия
на белковые фосфатазы первого типа, активность которых в клетке во
многом определяет уровень образования в ней гликогена. Инсулин активирует
данные белковые фосфатазы через фосфорилирование (дефосфорилирова-
ние) определенных сериновых аминокислотных остатков.
Нарушенная толерантность по отношению к глюкозе - это
патологическое состояние, при котором концентрация глюкозы в сыворотке
крови выше, чем у здоровых людей, но ниже, чем у больных сахарным
диабетом.
При выявлении сахарного диабета нередко определяющее значение
имеет тест толерантности (переносимости) глюкозы. Исходным
моментом теста является определение концентрации глюкозы в сыворотке
крови через два часа после приема пищи или приема стандартной дозы
глюкозы (75 г). О патологически низкой толерантности глюкозы
свидетельствует концентрация глюкозы в сыворотке крови через два часа после
приема пищи (стандартной дозы глюкозы) не меньшая, чем 140 мг/дл.
Содержание глюкозы в сыворотке не меньшее, чем 200 мг/дл, - это
признак развития сахарного диабета. Предпочтительней исследовать концен-
448
Глава 27
трацию глюкозы в сыворотке крови после приема пищи, чем определять
ее в пробе крови, взятой у больных утром, до завтрака. Дело в том, что по
мере развития у больных явного сахарного диабета способность
концентрации глюкозы в сыворотке крови возвращаться в нормальные пределы
через два часа после приема пищи теряется быстрее, чем возникает
гипергликемия утром и натощак.
После соблюдения больным в течение трех дней нормальной
диеты, при которой больной потребляет с пищей минимум 150 мг
углеводов в день, пациент принимает внутрь 75 г глюкозы. Для выполнения
теста (табл. 27.2) исследуется концентрация глюкозы в крови до
приема стандартной дозы глюкозы, а затем через 30, 60, 90 и 120 мин после
приема.
Таблица 27.2
Тест толерантности глюкозы
Результаты
теста
Нормальная реакция
содержания глюкозы
в сыворотке крови
Нарушенная
толерантность по
отношению к глюкозе
Сахарный диабет
Концентрация
глюкозы в
сыворотке крови
утром, натощак,
мг/дл
< 115
115-139
> 140
Максимальная
концентрация глюкозы в сыворотке
крови через 30,60 и 90 мин
после приема стандартной
дозы углевода, мг/дл
<200
>200
>200
Величина
концентрации глюкозы
через 120 мин после
приема стандартной
дозы, мг/дл
< 140
140-199
>200
ПАТОГЕНЕЗ ГИПЕРГЛИКЕМИИ И ДРУГИХ НАРУШЕНИЙ
ОБМЕНА ВЕЩЕСТВ У БОЛЬНЫХ
САХАРНЫМ ДИАБЕТОМ
Концентрация свободной глюкозы в клетках значительно выше ее
содержания во внеклеточном секторе. Известно, что скорость переноса
глюкозы через наружную клеточную мембрану мио- и адипоцитов
представляет собой детерминанту интенсивности фосфорилирования глюкозы
в клетках. D-глюкоза и другие сахара проникают в клетки путем
облегченной диффузии, опосредованной переносчиком. Инсулин усиливает
трансмембранный перенос глюкозы, увеличивая число переносчиков,
активно функционирующих на уровне плазматической мембраны.
Недостаточная концентрация нормального инсулина в циркулирующей крови,
нарушения взаимодействия гормона со своим рецептором приводят к
снижению числа переносчиков глюкозы, функционирующих на уровне
плазматической мембраны. В результате падает транспорт глюкозы в клетки
Сахарный диабет
449
жировой и мышечной ткани, что повышает содержание глюкозы во
внеклеточном пространстве и обуславливает гипергликемию.
Инсулин не усиливает облегченной диффузии глюкозы в гепатоци-
ты, но увеличивает приток глюкозы в дифференцированные клетки
печени через рост активности в них глюкокиназы, одного из ключевых
ферментов гликолиза, превращающего глюкозу в глюкозо-6-фосфат.
Инсулин, стимулирующий экспрессию гена данного энзима, повышает
содержание глюкокиназы в гепатоцитах, что интенсифицирует гликолиз
и снижает содержание свободной глюкозы в клетках печени. В
результате растет свободная диффузия глюкозы из внеклеточного сектора в
гепатоциты. При инсулинопении или патологической резистентности по
отношению к инсулину на уровне гепатоцитов в них падает фосфорили-
рование глюкозы. В результате растет содержание свободной глюкозы в
дифференцированных клетках печени и падает ее свободная диффузия в
гепатоциты. Это повышает содержание глюкозы во внеклеточном
секторе и служит одним из факторов гипергликемии у больных сахарным
диабетом. ч
Кроме того, инсулин усиливает интенсивность гликолиза в печени и
других органах вследствие того, что повышает активность в них других
его ферментов, фосфофруктокиназы и пируваткиназы. Одновременно
инсулин подавляет в гепатоцитах активность глюкозо-6-фосфатазы, что
повышает концентрацию в них глюкозо-6-фосфата. В результате действия
инсулина глюкоза удерживается в клетках печени, так как их наружная
мембрана непроницаема для глюкозо-6-фосфата. Снижение содержания
инсулина в крови и (или) патологическая резистентность по отношению к
инсулину через повышение активности глюкозо-6-фосфатазы снижают
удержание глюкозы в гепатоцитах в виде глюкозо-6-фосфата. Это может
быть одним из факторов гипергликемии.
Инсулин, повышая активность гликогенсинтетазы, усиливает
образование гликогена на системном уровне. Поэтому инсулинопения и
патологическая резистентность по отношению к инсулину снижают утилизацию
глюкозы из внеклеточного сектора для синтеза гликогена и тем самым
обуславливают гипергликемию.
Под влиянием инсулина происходит избирательное ингибирование
транскрипции гена, кодирующего фосфоенолпируваткарбоксикиназу,
ключевой фермент глюконеогенеза. В результате инсулинопении или
патологической резистентности к инсулину глюконеогенез
растормаживается, что повышает высвобождение глюкозы печенью и ведет к
гипергликемии.
Итак, патогенез гипергликемии у больных сахарным диабетом весьма
сложен и не сводится только к низкой облегченной диффузии глюкозы
через наружную клеточную мембрану клеток мышечной и жировой ткани
(схема. 27.1). Свою роль в развитии гипергликемии у пациентов с
сахарным диабетом играют превалирующие на системном уровне эффекты ка-
таболических гормонов антагонистов инсулина в виде торможения
синтеза гликогена и усиления гликогенолиза.
450
Глава 27
Инсулинопения или патологическая
резистентность по отношению к инсулину
Низкое антикатаболиче-
ское действие инсулина
Снижение
облегченной диффузии
глюкозы через
наружную клеточную
мембрану клеток
мышечной ткани и
адипоцитов
т
Падение
интенсивности фосфорилирова-
ния глюкозы в гепа-
тоцитах; снижение
утилизации глюкозы
из внеклеточного
сектора для гликолиза
Снижение
синтеза
гликогена
Интенсификация
глюконео-
генеза
Распад
гликогена
Гипергликемия
Схема 27.1. Патогенез гипергликемии у больных сахарным диабетом
Инсулинопения и патологическая резистентность по отношению к
инсулину через снижение анаболического эффекта инсулина и его антиката-
болического действия обуславливают стойкое падение процессов
анаболизма, в том числе липогенеза и белкового синтеза. Одновременно в
мышечные и другие клетки вследствие недостаточного системного эффекта
инсулина падает транспорт аминокислот, катионов калия и кальция, нук-
леозидов и органического фосфата как субстратов анаболизма. Снижение
белкового синтеза у больных сахарным диабетом захватывает и систему
иммунитета, что служит одной из причин иммунодефицита, вторичного по
отношению к сахарному диабету. Падение анаболизма и связанная с ним
интенсификация липолиза и распада белков у больных инсулинзависимым
сахарным диабетом, возникшим в детском возрасте, может обусловить
задержку роста тела. После возникновения инсулинзависимого сахарного
диабета у взрослых болезнь приводит к снижению массы тела.
Предположительно инсулин влияет на образование атерогенных ли-
попротеинов или снижает очищение от них плазмы крови. Очевидно
падение этого эффекта гормона у больных сахарным диабетом приводит к
ускоренному атеросклерозу.
Морфопатогенез атеросклероза у больных сахарным диабетом не
отличается от патогенетических механизмов атеросклеротических
патологических измененеий сосудистой стенки у пациентов, не страдающих от
сахарного диабета. И в том и в другом случае патогенные межклеточные
взаимодействия в основном складываются из аномального изменения
функционального состояния эндотелиоцитов, адгезии к ним
активированных мононуклеарных фагоцитов сосудистой стенки, за которой следует
прилипание агрегатов активированных тромбоцитов. Агрегаты этих
активированных клеток становятся источником цитокинов со свойствами фак-
Сахарный диабет
451
торов клеточного роста, что обуславливает пролиферацию гладкомышеч-
ных миоцитов сосудистой стенки как этапа формирования атеросклеро-
тической бляшки. Многие из факторов риска атеросклероза, гиперхоле-
стеринемия, патогенно низкая концентрация в крови липопротеинов
высокой плотности, курение и артериальная гипертензия одновременно
представляют собой факторы риска сахарного диабета второго типа.
Факторы риска атеросклероза, связанные исключительно с сахарным
диабетом, - это гипергликемия, гиперинсулинемия (патологически высокая
концентрация инсулина в крови у больных сахарным диабетом второго
типа, преимущественно обусловленным патологической резистентностью
по отношению к инсулину), а также гипертриглицеридемия.
ПАТОГЕНЕЗ ДИАБЕТИЧЕСКОЙ МИКРОАНГИОПАТИИ
В настоящее время не вызывает сомнений, что именно гипергликемия
является основной причиной патологических изменений сосудов у
больных с длительным инсулинзависимым сахарным диабетом. Тем не менее,
патогенез диабетической ангиопатии вследствие гипергликемии остается
неясным. Так как глюкоза и ее метаболиты выступают субстратами на
различных путях метаболизма, то ангиопатический эффект
гипергликемии реализуется несомненно через действие множества патогенетических
механизмов. В последние десятилетия были предложены четыре
теоретических модели действия таких механизмов:
♦ связанный с активностью альдозоредуктазы (сорбитоловый путь);
♦ через изменение активности протеинкиназы С;
♦ неферментного гликолизирования;
♦ в результате нарушений окислительно-восстановительного
потенциала тканей.
В большинстве тканей организма глюкоза может трансформироваться
в сорбитол через активность альдозоредуктазы. Из-за высокой константы
Михаэлиса1 альдозоредуктазы для глюкозы уровень синтеза сорбитола
при низкой концентрации углевода в клетках невысок. При
гипергликемии растет содержание глюкозы в клетках, в которые углевод проникает
путем обычной, а не облегченной диффузии, усиливаемой инсулином. В
результате в клетках усиливается образование сорбитола. После
образования сорбитола возможна его метаболизация, связанная с активностью
сорбитолдегидрогеназы, в результате которой образуются фруктоза и
восстановленный никотинамидадениндинуклеотид (NADH). Однако пре-
1 Константа Михаэлиса - концентрация субстрата, при которой скорость реакции,
катализатором которой является данный фермент, составляет половину максимальной (в
данном случае субстрат - это глюкоза, фермент - альдозоредуктаза, а реакция - это
образование сорбитола из глюкозы).
452
Глава 27
вращение сорбитола во фруктозу и NADH идет медленно, и сорбитол
накапливается в клетках. Рост содержания сорбитола в клетках (в частности в
нейронах) приводит к их отеку вследствие миграции воды из
внеклеточного сектора в сторону большей осмотической концентрации сорбитола.
Предполагают, что накопление сорбитола в клетках хрусталика глаза
больного инсулинзависимым сахарным диабетом приводит к
образованию у таких пациентов диабетической катаракты.
Так как возрастание концентрации сорбитола в нейронах и клетках
мышечной ткани не может привести к их значительному отеку и
обусловить цитолиз, то аккумуляцию сорбитола нельзя признать ведущим
механизмом диабетических ангио- и нейропатий.
Полагают, что связанный с инсулинопенией рост в клетках и тканях
концентрации свободной глюкозы и сорбитола может быть причиной
увеличения содержания в них миоинозитола как причины диабетической
ангиопатии. Это может быть только предположением, ибо известно, что
рост концентрации миоинозитола вследствие инсулинопении,
гипергликемии и высокой активности альдозоредуктазы преимущественно
происходит в клетках нервной ткани. В эксперименте ингибиторы
альдозоредуктазы снижают содержание миоинозитола только в нейронах.
Одновременно с ростом содержания в нейронах миоинозитола в них снижается
активность натрий-калий-АТФазы. Полагают, что снижение активности
натрий-калий-АТФазы также представляет собой следствие накопления в
нейронах сорбитола, так как ее активность восстанавливается под
влиянием ингибиторов активности альдозоредуктазы.
Были получены обнадеживающие результаты экспериментального
изучения ингибиторов альдозоредуктазы как средств коррекции
диабетических ангио- и нейропатий. Тем не менее, клинические испытания не
принесли ожидаемых результатов, которые бы свидетельствовали о
возможности предупреждения диабетических ретино- и нейропатий через
действие ингибиторов альдозоредуктазы.
Не исключено, что гипергликемия вызывает диабетическую микроан-
гиопатию через патологические изменения обмена фосфолипидов и
активности протеинкиназы С. Некоторые из фосфолипидов и данный
фермент, функционируя в качестве агентов системной и паракринной
регуляции, оказывают на функциональное состояние сосудов и его
характеристики (проницаемость сосудистой стенки, сократимость ее гладкомышеч-
ных элементов, экспрессия тромбогенного потенциала эндотелиоцитов,
объемная скорость кровотока, внутренняя секреция, синтез составляющих
базальной мембраны) широкий спектр, в том числе и разнонаправленных
влияний. Иными словами, данные системы регуляции клеток на уровне
сосудистой стенки контролируют те функции ее клеточных элементов,
расстройства которых и служат причинами диабетической ангиопатии.
Предположительно повышение активности протеинкиназы С,
вызывающее патогенную разностороннюю модуляцию функционального
состояния микрососудов, может быть ведущим звеном патогенеза
диабетической микроангиопатии. В пользу этого предположения говорит факт вы-
Сахарный диабет
453
сокой активности протеинкиназы С в сетчатой оболочке глаз пациентов,
страдающих от инсулинзависимого сахарного диабета и диабетической
ретинопатии; кроме того, известно, что у таких пациентов активность
данного фермента аномально высока в стенке аорты, кардиомиоцитах, а
также в почечных клубочках. Экспериментальные исследования
сахарного диабета показали, что инсулинопения и гипергликемия ведут к росту
активности протеинкиназы С в стенках сосудов сетчатой оболочки глаза,
аорты, почечных клубочков и венечных артерий. Одновременно с ростом
активности энзима в клеточных элементах сосудистой стенки определяли
возрастание уровня содержания диацилглицерола.
Это позволяет предположить, что инсулинопения и гипергликемия
повышают активность протеинкиназы через увеличение содержания в
клетках диацилглицерола, одна из функций которого - это усиление
перемещения протеинкиназы С из неактивного пула фермента в цитозоле в
его активный мембранный пул. Полагают, что изменения активности
протеинкиназы С, связанные с сахарным диабетом и гипергликемией,
происходят преимущественно в сосудистой стенке, где вследствие
инсулинзависимого сахарного диабета растет активность специфической для
сосудистой стенки бета-И-протеинкиназы С. В основном рост активности
протеинкиназы С меняет функциональное состояние сосудистой стенки
посредством модуляции экспрессии генов эндотелиальных и других клеток
сосудистой стенки.
Данные, полученные при экспериментальном исследовании влияний
гипергликемии на обмен веществ, свидетельствуют, что рост активности
протеинкиназы С вследствие гипергликемии не связан с изменением осмо-
ляльности внеклеточной жидкости и со сдвигами в обмене сорбитола.
Обусловленные гипергликемией рост содержания диацилглицерола и
активности протеинкиназы С при инсулинзависимом сахарном диабете
возникают не сразу и не сразу подвергаются обратному развитию после
устранения гипергликемии. Тут мы имеем дело еще с одним проявлением
общей для развития патологических процессов закономерности, то есть
тенденции к эндогенизсщии - относительной потери связи источников их
развития с первичным причинным фактором. Причина эндогенизации в
данном случае состоит в том, что протеинкиназа С, комплексно меняя
экспрессию генома клетки формирует патологическую систему
взаимодействия элементов генетического материала клетки как причины перси-
стирования диабетической ангиопатии (это всего лишь наше
предположение - В.Ю. Шанин).
Полагают, что гипергликемия вызывает патогенные межклеточные
взаимодействия как причину диабетической ангиопатии через
образование соединений на основе ковалентных связей глюкозы с ингредиентами
клеточных мембран и (или) циркулирующими с кровью липопротеинами
и белками. Процесс образования таких соединений называют
неферментным гликолизированием. Через соединение глюкозы ковалентными
связями с аминогруппами протеинов клеточных мембран, ядерных белков и
дезоксирибонуклеиновой кислоты происходит образование так называв-
454
Глава 27
мых транзиторных промежуточных продуктов неферментного гликоли-
зирования. Эта реакция достигает своего равновесия за несколько недель.
В дальнейшем промежуточные продукты неферментного гликолизирова-
ния служат субстратами для образования конечных продуктов
неферментного гликолизирования. С образованием конечных продуктов
неферментного гликолизирования реакция становится необратимой. Конечные
продукты неферментного гликолизирования могут обуславливать
патогенную модуляцию экспрессии генома клеток сосудистой стенки как
причину диабетической микроангиопатии. Кроме того, они служат
субстратами образования свободных кислородных радикалов, которые
повреждают наиболее в функциональном отношении активные фосфолипиды
клеточных мембран и еще в большей степени стабилизируют конечные
продукты неферментного гликолизирования.
Конечные продукты неферментного гликолизирования образуются во
внеклеточном пространстве через соединение глюкозы ковалентными
связями с липопротеинами низкой плотности, альбумином и
гемоглобином. Образование конечных продуктов неферментного гликолизирования
на поверхности клеточных мембран меняет их механические свойства,
повышая жесткость мембраны. В результате плазматическая мембрана
эритроцитов становится более ригидной, что снижает деформируемость
эритроцита как необходимое условие нормальной микроциркуляции
(незатрудненного прохождения эритроцитов по микрососудам и
капиллярам). Эритроциты с патологически низкой вследствие гипергликемии
и неферментного гликолизирования деформируемостью при
прохождении по микрососудам активируют эндотелиальные клетки через
усиленное трение о часть их наружной мембраны, обращенной в просвет
микрососуда. Активация эндотелиоцитов служит первым этапом
патогенных межклеточных взаимодействий, приводящих к диабетической
микроангиопатии.
Кроме того, звеном патогенеза диабетической микроангиопатии
является утолщение базальной мембраны капилляров в результате
образования входящим в ее состав коллагеном и глюкозой конечных продуктов
неферментного гликолизирования, при котором молекулярные цепочки,
составляющие коллаген, соединяются между собой попереречными
связями в виде молекул глюкозы. В результате базальная мембрана не только
утолщается. В ней падает содержание протеогликанов, а
функционирование интегриновых рецепторов сосудистой стенки меняется таким
образом, что возникают дисфункции всех ее клеточных элементов.
Интенсивность образования конечных продуктов неферментного
гликолизирования представляет собой прямую функцию длительности
гипергликемии и степени патологического возрастания концентрации
глюкозы в циркулирующей крови.
При длительной гипергликемии конечные продукты неферментного
гликолизирования (КПНфГ) начинают циркулировать с кровью и
фиксируются на наружной поверхности ее мононуклеарных фагоцитов,
которая содержит рецепторы специфические по отношению к КПНфГ. В
Сахарный диабет
455
результате происходит системная активация мононуклеарных
фагоцитов, циркулирующих с кровью. Активированные мононуклеары
начинают высвобождать такие цитокины как фактор некроза опухолей, ин-
терлейкин-1, что приводит к гиперцитокинемии. Гиперцитокинемия
через системные активацию эндотелиоцитов и рост экспрессии их тромбо-
генного потенциала вызывает диабетическую микроангиопатию на
уровне всего организма.
Если атеросклероз считать результатом хронического воспаления
сосудистой стенки, основным клеточным эффектором которого являются
активированные клетки системы мононуклеарных фагоцитов, то гиперци-
токинемию вследствие активации мононуклеаров в результате
воздействия на них КПНфГ можно предположительно признать фактором
ускорения атеросклероза у больных сахарным диабетом и диабетической микро-
ангиопатией.
Длительная гипергликемия и неферментное гликолизирование
приводят к образованию связей в виде молекул глюкозы, связанных с
аминогруппами, между цепочками нуклеотидов дезоксирибонуклеиновой
кислоты (ДНК). Это вызывает дисфункции генетического материала
эндотелиоцитов, обусловленные изменением строения двойной спирали ДНК,
что предположительно можно считать еще одним фактором
диабетической микроангиопатии.
В эксперименте было показано, что инфузия растворов, содержащих
конечные продукты неферментного гликолизирования, вызывает
характерные для диабетической микроангиопатии утолщение базальной
мембраны микрососудов и патологические изменения сократимости гладко-
мышечных элементов сосудистой стенки. При этом ослабляется
физиологическая реакция расширения сосудов в ответ на действие такого
медиатора как оксид азота. В этой связи были сделаны предположения о
механизме ускоренного развития тяжелой первичной артериальной гипертен-
зии у больных сахарным диабетом и длительной гипергликемией,
который состоит в потере гладкомышечными клетками сосудистой стенки
нормальной реактивности по отношению к расширяющему сосуды
действию оксида азота.
Обусловленный гипергликемией рост утилизации глюкозы как
субстрата гликолиза или образования полиола увеличивает отношение
содержания в клетке восстановленной формы никотинамидадениндинукле-
отида к его окисленной форме (NADH/NAD). Предполагают, что рост
содержания в клетке NADH представляет собой следствие интенсификации
обмена сорбитола и утилизации глюкозы в реакциях пентозного шунта,
что в частности приводит к образованию 1,3-дифосфоглицерата. Рост
NADH/NAD патогенно влияет на клеточные функции через
патологические изменения а) синтеза диацилглицерола, б) функционирования систем
восстановления структуры дезоксирибонуклеиновой кислоты и в)
окисления жирных кислот. В эксперименте было продемонстрировано обратное
развитие диабетической микроангиопатии под влиянием пирувата как
метаболита, снижающего NADH/NAD.
456
Глава 27
Самым ранним из гистопатологических сдвигов при диабетической
ретинопатии является потеря капиллярами сетчатой оболочки своих
перицитов. За несколько лет хронической гипергликемии соотношение
между эндотелиальными клетками и перицитами ретинальных
микрососудов может снизиться от нормального 1:1 до патологического 1:10.
Кроме того, диабетическую микроангиопатию на уровне сетчатой
оболочки глаза характеризуют расширение капилляров, утолщение их ба-
зальной мембраны, рост проницаемости и возникновение капиллярных
микроаневризм. Все эти патологические изменения капилляров сетчатой
оболочки можно связать с утратой нормальных межклеточных
взаимодействий между перицитами и эндотелиальными клетками. Известно,
что гипергликемия угнетает клеточный рост перицитов. Перициты
оказывают на эндотелиальные клетки регуляторные влияния, которые
служат необходимым условием нормальной экспрессии эндотелиоцитами
цитокинов. В результате потери перицитов экспрессия генома и
клеточный рост эндотелиоцитов претерпевают патологические изменения. Это
обуславливает потерю эндотелиоцитами способности обеспечивать
нормальный кровоток в капиллярах. В результате часть капилляров
оказывается обтурированными микротромбами и появляются капилляры, не
содержащие крови («капилляры-призраки»). Вторичная микроциркуля-
торная гипоксия приводит к локальному росту концентрации
соответствующих факторов клеточного роста, что обуславливает разрастание
капилляров как причину пролиферативной диабетической микроан-
гиопатии на уровне сетчатой оболочки глаза (пролиферативная
ретинопатия).
Пролиферативную ретинопатию характеризует прорастание новообра-
зуемых капилляров в стекловидное тело. Новые капилляры
неполноценны. Число окружающих их перицитов резко снижено. В результате стенка
капилляров теряет свои нормальные свойства барьера между кровью и
интерстицием, что служит причиной множества кровоизлияний в
сетчатую оболочку, ее отслойки и потери зрения. Из факторов клеточного
роста и цитокинов, вовлеченных в патогенез пролиферативной ретинопатии,
следует выделить следующие: основные и кислые факторы роста фибро-
бластов, инсулиноподобный фактор роста I, бета-трансформирующий
фактор клеточного роста, а также сосудистый эндотелиальный фактор
клеточного роста.
Гипертрофия мезангиальных клеток почечных клубочков у больных
сахарным диабетом во многом составляет диабетическую нефропатию.
В результате пролиферации клеток мезангия клубочки теряют
фильтрационную поверхность, формируемую эндотелиальными и
эпителиальными клетками. При этом утолщается базальная мембрана и теряются
эндотелиальные и эпителиальные клетки. Все это завершается
склерозом почечных клубочков и чреватым танатогенезом угнетением
экскреторных функций почек. При диабетической нефропатии в клубочках
растет содержание таких факторов клеточного роста, как альфа-фактор
некроза опухолей, бета-трансформирующий фактор клеточного роста,
Сахарный диабет
457
фактор клеточного роста тромбоцитов, а также основной фактор роста
фиброластов.
Развитие диабетической нефропатии и гипертрофию мезангиальных
клеток связывают с ростом содержания в циркулирующей крови
ангиотензина И. При этом полигенную наследственную предрасположенность
к развитию диабетической нефропатии у больных сахарным диабетом
связывают с полиморфизмом генов ангиотензинпревращающего
фермента и ангиотензиногена. Максимальный уровень риска диабетической
нефропатии и соответственно предрасположенности к ней у больных
сахарным диабетом фиксируют тогда, когда ген
ангиотензинпревращающего фермента содержит два аллеля D, а ген ангиотензиногена
составляют два одинаковых аллеля, экспрессия которых дает молекулу
ангиотензиногена, в которой на 235 позиции находится треонин. Кроме того,
свою лепту в предрасположенность к диабетической нефропатии вносит
полиморфизм гена рецептора ангиотензина II, который
предположительно содержит наружная мембрана клеток мезангия. Если вспомнить о
том, что определенная системная гиперактивация эндотелиальных
клеток, участвующих в образовании ангиотензина II, во многом составляет
патогенез осложнений сахарного диабета и, то можно считать
диабетическую микроангиопатию инициирующим моментом роста образования
ангиотензина II, который происходит особенно интенсивно при
соответствующем полиморфизме генов.
Образование конечных продуктов неферментного гликолизирования
миелина предположительно лежит в основе диабетической нейропатии,
которая в начале развития захватывает тонкие нервные волокна. Если
оценивать вагальные регуляторные влияния на сердце, фиксируя
вариабельность частоты сердечных сокращений при глубоком дыхании и
ориентируясь на возрастную норму, то у большинства больных инсулинзави-
симым сахарным диабетом можно выявить признаки блокады вагальных
регуляторных влияний на сердце, которая приводит к меньшей
вариабельности. С возрастом вариабельность падает. Снижение
вариабельности частоты сердечных сокращений при глубоком дыхании у больных ин-
сулинзависимым сахарным диабетом происходит в несколько раз
быстрее. Данные, полученные при исследовании вариабельности частоты
сердечных сокращений при глубоком дыхании свидетельствуют, что
диабетическая нейропатия развивается у большинства из них, но только у
небольшой части таких пациентов она приводит к заметным и тяжелым
последствиям нарушений вегетативной регуляции функций:
♦ ощущению жара и боли в конечностях;
♦ патологически интенсивному потоотделению;
♦ задержке мочеиспускания;
♦ расстройствам сердечной проводимости со снижением интервала
QT, сердечным аритмиям;
♦ диареям и запорам;
♦ угнетению моторики желудка как причины застоя желудочного
содержимого и рвоты.
458
Глава 27
ДИАБЕТИЧЕСКИЕ КОМА И КЕТОАЦИДОЗ
Ведущее звено кетоацидоза - это недостаточная секреция инсулина,
то есть его высвобождение, низкое относительно уровня секреции
гормонов-антагонистов. Это обуславливает усиление липолиза и рост
содержания в крови свободных жирных кислот. Патогенно высокая
интенсивность окисления жирных кислот в печени как следствие их высокой
концентрации в циркулирующей плазме крови ведет к повышенному
образованию печенью так называемых кетоновых тел (ацетона, ацетоацетата и
бета-гидроксибутирата), молекулы которых содержат карбониловую
группу С=0. Одно из кетоновых тел, бета-гидроксибутират - это гидро-
ксильная кислота. У больного инсулинзависимым сахарным диабетом
скорость высвобождения печенью кетоновых тел может быть очень
высокой, достигая уровня в 100ммоль/ч. У больных с кетоацидозом как
осложнением сахарного диабета {диабетический кетоацидоз) выявляют ке-
тонемию (появление кетоновых тел в плазме крови), кетонурию
(кетоновые тела в конечной моче), гиперлипидемию, гипергликемию и глюкозу-
рию. Глюкозурия как причина осмотического диуреза вызывает поли-
урию, в свою очередь приводящую к дефициту объема внеклеточной
жидкости и гипокалиемии.
У пациента с инсулинзависимым сахарным диабетом при
гипергликемии многие клетки испытывают дефицит глюкозы как источника
свободной энергии, что образно определяют как «голодание среди изобилия».
При этом альтернативным источником энергии могут выступать
продукты неполного метаболизма липидов, кетоновые тела. В этой связи
интенсификацию образования кетоновых тел печенью следует рассматривать
как защитную реакцию, которая всегда избыточна относительно стимула,
ее обусловившего. Как защитно-патогенная реакция усиление кетогенеза
не устраняет системный дефицит свободной энергии, а через
метаболический ацидоз усиливает его.
В усилении кетоза как причины метаболического ацидоза при
увеличенном АПП играет роль действие гормона-антагониста инсулина глюка-
гона, который усиливает кетогенез. Глюкагон усиливает кетогенез,
увеличивая в печени содержание малонилкоэнзима А, повышая активность
карнитинацилтрансферазы I и вызывая перемещение жирных кислот из
цитозоля гепатоцитов в их митохондрии, которые через бета-окисление
превращают жирные кислоты в кетоновые тела.
Рост концентрации протонов в жидкой части притекающей к сердцу
крови угнетает его сократимость при снижении чувствительности адре-
норецепторов сосудистого русла к эндогенным вазопрессорам-кате-
холаминам. При этом на периферии стремительно нарастающий
кетоацидоз вызывает дилатацию сосудов сопротивления. Все это приводит к
опасной артериальной гипотензии, основными звеньями патогенеза
которой являются:
♦ снижение минутного объема кровообращения через падение
сократимости, ударного объема левого желудочка и его фракции изгнания;
Сахарный диабет
459
♦ снижение общего периферического сосудистого сопротивления.
Артериальную гипотензию у больного с диабетическим кетоацидозом
усиливают дефицит объема внеклеточной жидкости и гипокалиемия,
вызывающая сердечные аритмии. В этой связи диабетический кетоацидоз
следует признать критическим состоянием, которое без неотложной
коррекции может привести к летальному исходу.
Диабетический кетоацидоз у молодых больных иногда знаменует
собой развитие сахарного диабета. У больных с диабетическим
кетоацидозом гипергликемия связана не только с инсулинопениеи, но и выступает
следствием падения скорости клубочковой фильтрации в ответ на
дефицит объема внеклеточной жидкости, обусловленный полиурией.
Снижение концентрации бикарбонатного аниона в плазме крови до уровня
меньшего, чем 10 ммоль/л, обычно сопровождается выраженной
компенсаторной гипервентиляцией при глубоких и частых вдохах через равные
временные интервалы (дыхание Куссмауля).
По мере роста содержания в плазме крови глюкозы и протонов
нарушения сознания прогрессируют от его умеренного угнетения до комы, в
патогенезе которой, кроме артериальной гипотензии и ацидоза, роль звена
патогенеза предположительно может играть реакция нейронов головного
мозга на патологическое возрастание осмоляльности внеклеточной
жидкости. Если причина диабетического кетоацидоза - это нерегулярное
введение инсулина, то диабетический кетоацидоз обычно развивается через
12-24 ч после введения последней дозы. Оперативное вмешательство,
травма и другие патологические состояния как причины и следствия
патогенного стресса с интенсивным выбросом в кровь гормонов
антагонистов инсулина ускоряют развитие диабетического кетоацидоза.
Если диабетический кетоацидоз развивается медленнее, то к звеньям
патогенеза недостаточности кровообращения вследствие сахарного
диабета и метаболического кетоацидоза присоединяется падение ОВнЖ,
обусловленное полиурией, вызванной осмотическим диурезом. В таком
случае артериальная гипотензия особенно выражена и опасна.
У большинства больных инсулинозависимым сахарным диабетом
концентрация калия во внеклеточной жидкости и в жидкой части плазмы
крови повышена вследствие инсулинопении, при которой недостаток
инсулина служит причиной недостаточного вхождения калия в клетки. Если
течение болезни осложняет развитие диабетической нефропатии, то
гиперкалиемия - это результат не только недостаточной секреции инсулина,
но и падения экскреторной функции почек. Кроме того, выход калия из
клеток как причину гиперкалиемии обуславливает преобладание на
системном уровне катаболических процессов над анаболическими
вследствие превалирования в организме эффектов катаболических гормонов
антагонистов инсулина. Гиперкалиемия через сердечные аритмии обостряет
недостаточность сердца, вызванную метаболическим кетоацидозом.
Если развитие диабетического кетоацидоза идет медленней, то у части
больных успевают развиться гиповолемия и гипокалиемия как следствия
полиурии. Второй механизм развития гипокалиемии у больных с диабе-
460
Глава 27
тическим кетоацидозом - это действие на почки высокой действующей
концентрации минералкортикоидов, секреция которых растет в ответ на
снижение объема внеклеточной жидкости.
У больных с диабетическим кетоацидозом часто выявляют гипонат-
риемию, которую вызывают:
♦ рост концентрации глюкозы во внеклеточной жидкости как фактор
перемещения воды из клеток во внеклеточный жидкостной сектор.
Вода выходит из тех клеток, в которые глюкоза проникает только
при нормальном действии на них инсулина и ненарушенной
клеточной реакции на эффект гормона;
♦ потери натрия и воды организмом вследствие полиурии, которые
восполняются только растворами гипоосмоляльными относительно
внеклеточной жидкости.
Гипонатриемия может быть звеном патогенеза комы вследствие
обострения сахарного диабета. Причина гипонатриемии - это снижение
почечного клиренса свободной воды из-за диабетической нефропатии. Если
во внеклеточном пространстве концентрация катиона натрия падает, то
вода устремляется в церебральные нейроны, что ведет к их отеку.
Диабетическая кома - это потеря сознания вследствие обострения
инсулинзависимого сахарного диабета. Диабетическую кому вызывают:
♦ метаболический кетоацидоз, который всегда выступает звеном
патогенеза диабетической комы;
♦ артериальная гипотензия вследствие падения сократимости сердца,
общего периферического сосудистого сопротивления и гиповоле-
мии, связанной с полиурией;
♦ гипер- или гипокалиемия;
♦ патологическая реакция нейронов головного мозга на гиперосмо-
ляльность, обусловленную гипергликемией;
♦ гипонатриемия.
У диабетической комы нет механизма развития, который можно
признать универсальным. Патогенез комы вследствие обострения
инсулинзависимого сахарного диабета всегда конкретен и индивидуален. Так, у
больного с диабетической нефропатией сердечная недостаточность в
основном связана с диабетическим кетоацидозом и гиперкалиемией как
следствием инсулинопении или (и) резистентности к инсулину и
снижения экскреторной функции почек. Если больной обезвожен вследствие
полиурии, то сердечная недостаточность может быть обусловлена не
только кетоацидозом и гиповолемией, но и гипокалиемией, вызывающей
нарушения сердечного ритма.
Первоочередная цель интенсивной терапии больных в состоянии
диабетической комы - это прекращение кетогенеза. Кетогенез
приостанавливают введением инсулина. Сначала внутривенно вводят 5-10 единиц
инсулина, после чего начинают непрерывную внутривенную инфузию его изоос-
моляльного по отношению к плазме раствора со скоростью 2-10 единиц в
час. Несмотря на то, что действие инсулина сразу прекращает липолиз,
перестройка эндокринного звена регуляции обмена веществ через антиката-
Сахарный диабет
461
болическое и анаболическое действия инсулина происходит не сразу, так
как экзогенный инсулин не устраняет мгновенно превалирования на
системном уровне эффекта катаболических гормонов антагонистов инсулина.
Поэтому из-за персистирования кетогенеза, несмотря на непрерывную ин-
фузию раствора инсулина, концентрация бикарбоната во внеклеточной
жидкости остается неизменной длительное время после начала введения
гормона. Анионный пробел плазмы возвращается в нормальные пределы в
течение 8-10 ч после начала терапии инсулином, что свидетельствует об
остановке кетогенеза.
Обычно артериальная гипотензия при систолическом артериальном
давлении меньшем, чем 90 мм рт. ст., у больных с диабетическим кетоа-
цидозом свидетельствует о дефиците ОВнЖ в результате полиурии.
Поэтому таким больным начинают внутривенную инфузию изоосмоляльных
по отношению ко внеклеточной жидкости растворов, каждые полчаса
вливая в вену 1000 мл, пока систолическое артериальное давление не
возрастет до уровня в 100 мм рт. ст. Если уровень гипергликемии под
влиянием терапии инсулином и вследствие гемодилюции становится ниже,
чем 250 мг/дл, то к инфузируемым растворам для предотвращения ятро-
генной гипогликемии добавляют глюкозу в адекватной дозе.
Если гипокалиемия у больного с диабетическим кетоацидозом
обусловлена потерями калия вследствие полиурии, то при повышенном
диурезе содержание калия в моче больше, чем 20 ммоль/л. При интенсивной
терапии таких больных в каждый литр инфузируемых изоосмолярных
растворов добавляют 20-40 ммоль калия. Если содержание калия в
плазме крови больше, чем 5 ммоль/л, то калий к растворам не добавляют.
Несмотря на гиперкалиемию, повышенное содержание калия в моче и поли-
урия служат показаниями для добавления 20 ммоль калия в каждый литр
вливаемых растворов.
Глава 28
ПАТОФИЗИОЛОГИЯ НАИБОЛЕЕ ЧАСТЫХ
ЭНДОКРИНОПАТИЙ
Термин «гормон» (гр. hormao - привожу в движение, побуждаю)
ввели в 1906 г Старлинг и Бейлис при описании функций секретина и
гастрина. Согласно Старлингу и Бейлису, гормоны - это химические
вещества, которые определенная группа клеток высвобождает в
циркулирующую кровь для специфического в целях поддержания го-
меостазиса эффекта (эффектов) на другую (другие) группу клеток;
при этом группы клеток, на которые воздействует гормон могут
отличаться друг от друга. Хаксли (1935) дал классическое
определение гормонов, которое можно признать верным и в настоящее
время: гормоны - это молекулы, чья основная функция - это перенос
информации от одних групп клеток другим для удовлетворения по-
требностей всего организма.
Как системное интегрирующее регуляторное влияние секрецию
гормонов и их эффекты на функции клеток нарушают:
♦ расстройства синтеза и хранения гормонов в головном мозге и
органах внутренней секреции;
♦ нарушение переноса гормонов до клеток-эффекторов с кровью и в
межклеточных пространствах;
♦ расстройства взаимодействия гормонов со своими клеточными
рецепторами, в том числе локализованными на наружной клеточной
мембране;
♦ блокада или извращение на пострецепторном уровне передачи
информации, посылаемой клетке через секрецию гормона;
♦ невозможность нормальной реакции клетки на получение
информационного сигнала вследствие дефицита в ней массы и энергии,
патогенных мутаций генетического материала или устойчивых
модуляций его экспрессии (гиперактивация клетки).
Все это в той или иной мере расстраивает системные интегрирующие
регуляторные влияния, приводит к болезням, угнетает резистентность
организма при взаимодействии с этиологическими факторами заболеваний и
снижает его приспособительные возможности.
Эндокринное звено систем регуляции организма находится под
постоянным контролем и находится в непрерывной взаимосвязи с высшими
нервными центрами, в том числе на уровнях коры больших полушарий,
подкорковых структур и гипоталамуса. Поэтому нарушения внутрицен-
тральных отношений на любом уровне центральной нервной системы
могут вызывать патогенные сдвиги эндокринной регуляции.
Патофизиология наиболее частых эндокринопатий
463
Известно, что неврозы, психозы, хронический отрицательный
эмоциональный стресс, вызывающий длительное патологическое состояние
повышенной тревожности, нарушают функции половых желез,
щитовидной железы и вызывают другие эндокринопатий. Почти всегда в
патогенез эндокринопатий такого происхождения вовлечены лимбическая
система и гипоталамические центры.
Непосредственной причиной расстройств эндокринного звена
систем регуляции организма на уровне гипоталамуса и гипофиза является
недостаточное или избыточное образование либеринов, статинов,
тропных гормонов, что приводит к нарушениям функций
периферических эндокринных желез. При этом могут возникать нарушения
регуляции секреции гормонов в соответствии с принципом отрицательной
обратной связи, в результате которых изменения концентрации
гормонов периферических желез в циркулирующей крови не оказывают
нормального влияния на секрецию рилизинг-факторов и тропинов
(гормонов аденогипофиза).
Падение роста секреции гормона периферической железы в ответ на
действие тропина аденогипофиза может быть обусловлено
патологическими изменениями клеток периферической железы. При аномальных
изменениях периферических желез секреция их гормонов может
длительно находится на патологически низком уровне, что обуславливает
хроническое возрастание секреции тропина. Так как спектр эффектов
тропинов не ограничивается лишь влиянием на секрецию гормонов
периферических желез, то избыточное высвобождение тропных гормонов
аденогипофиза может быть причиной болезней, расстройств гомеостази-
са и обмена веществ.
Эндокринопатий могут быть следствием патологически высокого
уровня секреции тропина, несмотря на нормальные секрецию и
концентрацию в циркулирующей крови гормона периферической железы, что
также представляет собой нарушение регуляции секреции гормонов по
принципу обратной связи.
Возникновение опухолей периферических желез, их гиперплазия
обуславливают соответствующие расстройства эндокринной регуляции
при патологически низком уровне секреции тропинов аденогипофизом.
Аномалии эндокринного звена системной регуляции могут
представлять собой результат патологических мутаций генов
соответствующих гормонов, а также протеинов составляющих их рецепторы,
выступают результатом изменений генетического материала,
кодирующего ферменты, участвующие в синтезе и катаболизме гормонов, в
том числе и находящихся друг с другом в антагонистических
отношениях. Генетический фактор предрасположенности к расстройствам
эндокринного звена системной регуляции может реализовать себя на
полигенной основе, как это происходит при неинсулинзависимом
сахарном диабете.
464
Глава 28
РАССТРОЙСТВА РЕГУЛЯЦИИ С УЧАСТИЕМ
ГОРМОНОВ АДЕНОГИПОФИЗА
Под гипопитуитаризмом понимают ряд патологических состояний,
болезней и синдромов, которые обусловлены патологически низким
уровнем секреции гормонов аденогипофиза.
Наиболее частые причины сочетанной недостаточности секреции
гормонов аденогипофиза - это опухоли гипофиза и его ножки, а также
цитолиз клеток гипофиза вследствие циркуляторной гипоксии
(ишемии), обусловленной послеродовым кровотечением. У большинства
больных с сочетанной недостаточностью секреции гормонов
аденогипофиза происходит постепенное замещение его нормальных клеток
элементами растущих краниофарингиом или не высвобождающей гормоны
хромофобной аденомы.
Синдром Шихана, основное звено патогенеза которого - это гипопи-
туитаризм, развивается в результате циркуляторной гипоксии гипофиза
из-за артериальной гипотензии вследствие кровопотери во время родов.
Дело в том, что во время беременности происходит гиперплазия клеток,
составляющих гипофиз, что в основном связано с увеличением числа
клеток, секретирующих пролактин (лактотропный гормон). При этом
гиперплазия не сопровождается адекватным расширением гипофизарной мик-
росудистой сети. Поэтому клетки гипофиза становятся особо
чувствительными к циркуляторной гипоксии вследствие кровопотери при родах.
Послеродовый ишемический некроз гипофиза возникает только
вследствие массивной кровопотери. Симптомы недостаточности
аденогипофиза вследствие послеродового кровотечения могут возникать сразу после
кровопотери или через годы после родов в зависимости от
распространенности и степени ишемических повреждений гипофизарных клеток.
Первой падает секреция гонадотропинов, что у женщин проявляет себя
аменореей. В дальнейшем снижается секреция тиреотропина и возникают
признаки недостаточной функции щитовидной железы. Последней
снижается секреция кортикотропина.
Хирургические вмешательства, состоящие в удалении опухолей
гипофиза, как причины гибели клеток гипоталамуса-гипофиза могут
приводить к гипопитуитаризму.
Редкими причинами гипопитуитаризма являются саркоидоз, гемохро-
матоз, туберкулез, сифилис, грибковые инфекции и др.
Дефицит гормона роста при гипопитуитаризме вызывает задержку
роста у детей, но не приводит к клинически значимым сдвигам регуляции
и гомеостазиса у взрослых.
Дефицит секреции гонадотропинов (лютеинизирующего и фоллику-
лостимулирующего гормонов) у больных с гипопитуитаризмом вызывает
аменорею (прекращение менструаций с потерей детородной функции),
атрофию половых органов у женщин и импотенцию с падением полового
влечения у мужчин. Если одновременное снижение секреции адренокор-
тикотропного гормона (кортикотропина) обуславливает падение образо-
Патофизиология наиболее частых эндокринопатий
465
вания и секреции надпочечниками андрогенов, то исчезают волосы на
лобке и в подмышечных впадинах, что особенно часто бывает и более
выражено у женщин с гипопитуитаризмом.
Дефицит тиреотропного гормона (тиреотропина) ведет к гипотиреозу.
Недостаточная секреция кортикотропина может быть причиной
недостаточности надпочечников, которая отличается от надпочечниковои
недостаточности, связанной с первичным поражением самих надпочечни-
ковых желез {первичная недостаточность надпочечников). При
вторичной недостаточности надпочечников вследствие падения секреции
кортикотропина нет характерной пигментации кожи, столь свойственной
первичной недостаточности. Дело в том, что при первичной
недостаточности надпочечников пигментация кожи и слизистых оболочек связана с
высокой действующей концентрацией в крови кортикотропина, уровень
которой повышается из-за низкой концентрации в крови кортизола. При
вторичной недостаточности надпочечников из-за гипопитуитаризма
концентрация кортикотропина в крови снижена, и гиперпигментации не
возникает.
При вторичной недостаточности надпочечников, связанной с
гипопитуитаризмом, суммарная концентрация всех минералкортикоидов в
циркулирующей крови падает, так как в ней из-за низкого уровня секреции
кортикотропина снижается содержание кортизола, обладающего
свойствами минералкортикоида. Однако это не приводит к снижению общего
содержания в организме натрия, гипокалии и гипокалиемии, так как
сохраняется способность надпочечников реагировать ростом секреции аль-
достерона в ответ на соответствующие влияния других, кроме гипотала-
мо-гипофизарно-надпочечниковой, систем регуляции, в частности ренин-
ангиотензин-альдостероновой. При первичной недостаточности
надпочечников секреция всех минералкортикоидов стойко и значительно
угнетена, что служит причиной падения содержания в организме натрия,
дефицита объема внеклеточной жидкости, гиповолемии и гиперкалиемии.
Дефицит секреции пролактина может быть причиной недостаточной
лактации у женщин с синдромом Шихана в период после родов.
При медленной, но неуклонно прогрессирующей деструкции
гипофиза-гипоталамуса, прежде всего, наступает снижение секреции гормона
роста и гонадотропинов. Затем падает секреция тиреотропного гормона,
и, наконец, снижается уровень секреции кортикотропина.
Падение секреции гормона роста человека (соматотропина) и
гонадотропинов при нормальной секреции других гипофизарных гормонов у
детей обуславливает задержку роста тела и замедленное половое
созревание. Изолированное (то есть при нормальной секреции других гормонов)
снижение секреции тиреотропина и кортикотропина у больных с
гипопитуитаризмом бывает крайне редко.
Заподозрить снижение секреции всех гормонов передней доли
Гипофиза (пангипопитуитаризм) следует при симптомах сочетанной
недостаточности щитовидной железы, надпочечников и половых желез (гипого-
надизм).
466
Глава 28
Пангипопитуитаризм - снижение секреции всех гормонов аденоги-
пофиза, известное под названием синдрома Симмонда. Сам Симмонд
считал, что все пациенты с недостаточной секрецией гормонов аденоги-
пофиза страдают от эндогенного голодания и назвал описанный им
синдром кахексией Симмонда. Ретроспективный анализ позволил
установить, что Симмонд описал не больных с пангипопитуитаризмом, а
пациентов с anorexia nervosa. У больных, описанных Симмондом как
страдающих от пангипопитуитаризма, расстройство внутрицентральных
отношений приводило к изменению личности, анорексии (отсутствию
аппетита) и частой рвоте.
Так как вследствие гипофункции щитовидной железы падает
потребность организма в источниках свободной энергии и субстратах
анаболизма, то организм может относительно легко переносить сочетанную
недостаточность секреции гормонов аденогипофиза при заместительной
терапии препаратами на основе кортикостероидов.
У детей с пангипопитуитаризмом недостаточная секреция гормона
роста и тиреотропина обуславливает задержку роста и умственного
развития. Недостаточность когнитивных функций у детей связана в
основном с недостаточной секрецией гормонов щитовидной железы.
У мужчин, страдающих от сочетанной недостаточности секреции
гормонов аденогипофиза, падает половое влечение, возникает импотенция,
уменьшаются яички и исчезают некоторые из вторичных половых
признаков, в частности волосы на лобке. Одновременно с падением либидо и
инволюцией яичек появляются патологические изменения кожи. Она белеет,
истончается, становится сухой и похожей на пергамент. Содержание 17-
гидрокси- и 17-кетостероидов в моче у больных с пангипопитуитаризмом
снижено. Концентрация кортизола в плазме крови (диапазон нормальных
колебаний - 0,22-0,68 мкмоль/л) также патологически низка. При этом у
больных нет нормальной реакции роста содержания кортизола в плазме
крови как элемента стресса в частности, вызываемого гипогликемией,
лихорадкой. Это снижает резистентность организма к инфекциям,
хирургическому стрессу и патологическому стрессу тяжелых ранений и травм.
Снижение секреции гормонов аденогипофиза может быть следствием
нарушений образования и секреции рилизинг-факторов в гипоталамусе.
Например, недостаточное влияние гонадотропин-рилизинг гормона на
соответствующие клетки аденогипофиза, связанное с недостаточностью
гипоталамуса, приводит к аменорее у женщин и прекращению образования
спермы у мужчин.
Акромегалия (гр. akron - конечность + гр. megas или megalu -
большой) - чрезмерный непропорциональный рост конечностей (кистей рук,
стоп) и костей лица вследствие нарушений эндокринной регуляции роста
тела человека с участием соматотропина, секретируемого
соответствующими клетками аденогипофиза.
Наиболее частая причина акромегалии - это патогенно избыточная
секреция соматотропина клетками эозинофильной или хромофобной
аденомы аденогипофиза.
Патофизиология наиболее частых эндокринопатий
467
Избыточная секреция соматотропина вызывает патологические
изменения костей, мягких тканей и метаболических процессов. В частности
патогенно избыточное действие соматотропина как гормона антагониста
инсулина у части больных может обуславливать патологическую толерантность
по отношению к глюкозе и неинсулинзависимый сахарный диабет.
У детей в период до полового созревания избыточная секреция
соматотропина увеличивает линейные размеры длинных трубчатых костей,
что приводит к гигантизму. После полового созревания и окостенения
эпифизов длинных костей такой эффект соматотропина в повышенной
действующей концентрации становится невозможным.
Акромегалия может быть результатом не только избыточной секреции
гормона роста человека клетками аденомы гипофиза. Ее первопричиной у
части больных является избыточное образование соматолиберина. Рост
образования соматолиберина обычно связан с гиполатамической гамар-
томой, его высвобождают некоторые опухоли поджелудочной железы,
стенок бронхов и кишечника. Избыточные образование и секреция
соматолиберина вызывают гиперплазию соматотрофов аденогипофиза, что
может быть принято за аденому гипофиза. Установить истину позволяет
определение концентрации соматолиберина в циркулирующей крови.
Обычно больные приобретают характерный акромегалический облик
в период онтогенеза от тридцати до пятидесяти лет. При этом черты лица
становятся грубыми, возникают прогнатия и неправильный прикус,
надбровные дуги утолщаются, а также увеличиваются масса и размеры языка
(макроглоссия). У больных с акромегалией увеличивается плотность
костной ткани. Гипертрофия костей в области суставов и увеличение
суставных хрящей обуславливают артропатии и остеохондроз. У части
больных выявляют запястный синдром, то есть боли и парестезии в руке в
области разветвления срединного нерва. Непосредственная причина
запястного синдрома - сдавление срединного нерва волокнами сухожилий
мышц-сгибателей, масса которых возрастает при акромегалии.
Гипертрофия сердца у больных с акромегалией может быть причиной
застойной сердечной недостаточности.
Транссфеноидальное удаление гипофизарной аденомы признается
наиболее эффективным способом коррекции акромегалии. Одним из
критериев ее эффектиности является снижение концентрации
соматотропина в циркулирующей крови утром и натощак до уровня более
низкого, чем 5 нг/мл.
ПАТОГЕНЕЗ ЭНДОКРИНОПАТИЙ
КАК ПРИЧИН РАССТРОЙСТВ РОСТА ТЕЛА ЧЕЛОВЕКА
Расстройства образования, синтеза и секреции гормонов
аденогипофиза, их дизрегуляция, извращение эффектов гормонов на свои
эффекторы, могут приводить к нарушениям роста тела.
468
Глава 28
Выделяют следующие основные условия нормального роста:
♦ адекватное поступление во внутреннюю среду из внешней нутриен-
тов, то есть, углеводов, аминокислот (незаменимых в том числе),
жирных кислот, а также витаминов;
♦ ненарушенные синтез, хранение в депо и секреция гормона роста
(соматотропина);
♦ нормальная эндокринная функция щитовидной железы;
♦ ненарушенная эндокринная функция поджелудочной железы,
нормальная секреция тестостерона и эстрогенов;
♦ нерасстроенное функционирование системы синтеза и секреции ин-
сулино-подобного фактора роста I (ИФР-1), ее нормальная реакция
на взаимодействие с соматотропином.
В наибольшей степени в эндокринном контроле за ростом тела
задействована гипоталамо-гипофизарная система регуляции роста, эффектором
которой является система синтеза и секреции ИФР-1 (соматомедина С)
(табл. 28.1).
Таблица 28.1
Гипоталамо-гипофизарная система регуляции роста тела
Место синтеза
и наименование гормона
Гипоталамус
Соматолиберин
Соматостатин
Гипофиз
Соматотроггин
Печень и в меньшей степени клетки
гих тканей
ИФР-1
дру-
Действие гормона
Повышает транскрипцию генов
соматотропина и усиливает его секрецию
Тормозит секрецию соматотропина
Усиливает образование ИФР-1. Катабо-
лический гормон антагонист инсулина
Стимулирует рост костей и клеточную
пролиферацию
Регуляция роста тела млекопитающих в основном осуществляется
через секрецию соматотропина. Гормон роста, молекула которого состоит
из 191 аминокислот, представляет собой конечный результат экспрессии
пяти генов, связанных между собой и локализованных в семнадцатой
хромосоме. Его образуют и секретируют клетки аденогипофиза (сомато-
трофы). Гипоталамические полипептиды соматолиберин и соматостатин,
секретируемые в портальную систему гипофиза, взаимодействуют со
своими рецепторами на уровне аденогипофиза. Соматотропин
высвобождается в кровь через некоторые промежутки времени («пульсирующая
секреция»). Секреция соматотропина определяется в основном
колебаниями уровня секреции соматостатина. Частота выброса соматостатина
возрастает во сне, при ожирении и во время голодания. В этой связи
ожирение и недостаточное питание можно считать факторами задержки роста
Патофизиология наиболее частых эндокринопатий
469
тела. Факт роста частоты выброса соматостатина во сне ставит под
сомнение народную мудрость: «человек растет, когда спит». Общее время
секреции соматотропина возрастает при увеличении поступления в
гипофиз соматолиберина, достигая максимума вследствие усиленного
образования соматолиберина клетками опухоли (гамартомы) соответствующей
локализации или его внутривенного введения в определенной дозе. Свою
пока неясную роль в секреции соматотропина играют эстрогены и
гормоны щитовидной железы. Наиболее частая причина патогенно избыточной
секреции соматотропина - это аденома гипофиза.
Гормон роста может прямо стимулировать клеточную пролиферацию.
Об этом говорит рост экспрессии прото-онкогена с-тус под влиянием
соматотропина (СТ). В основном свойство СТ стимулировать рост тела
реализуется через действие ИФР-I. ИФР-1 (соматомедин С) - это
полипептидный фактор роста, который активирует хондроциты и клеточную
пролиферацию различных эпителиоцитов. Рост содержания ИФР-I в
циркулирующей крови подавляет транскрипцию генов соматотропина в сома-
тотрофах, что представляет собой пример регуляции по принципу
обратной отрицательной связи. Соматомедин С достигает клеток как с
циркулирующей кровью, так и по межклеточным пространствам.
Карликовость (наносомия - от греч. nemos - карлик и soma - тело,
системная дисплазия) - патологически низкая длина тела, нижним
пределом нормальных колебаний которой считают уровень в 140 см. У 35 %
больных с наносомией она представляет собой следствие семейной
болезни. У 25 % больных с системной дисплазией - это идиопатическая
наносомия. Нередко (10 % от всей совокупности случаев наносомии)
карликовость выступает результатом нарушений гипоталамо-гипофизарной
системы регуляции роста тела, а расстройства секреции гормонов
щитовидной железы приводят к наносомии у 10 % больных с системной
дисплазией. У 10 % больных с наносомией рост тела замедляется вследствие
врожденной аплазии половых желез. В остальных случаях наносомия
представляет собой следствие разнообразных причин. Всего известно
55 патологических состояний и болезней (часто моногенных), которые
приводят к наносомии. Выделяют летальную наносомию, которая
возникает как признак и следствие болезней, приводящих к преждевременному
(в детском возрасте) летальному исходу. Примером может служить
системная дисплазия вследствие ахондроплазии у больных, генотип которых
содержит гомозиготный ген данного заболевания с двумя патогенными
аллелями. Наносомия вследствие расстройств эндокринной регуляции
роста тела к летальной не относится (нелетальная наносомия).
Наносомию вследствие врожденной недостаточной секреции аденоги-
пофизом соматотропина называют гипофизарной.
Задержка роста у детей в период онтогенеза до полового созревания
может быть признаком недостаточности секреции всех гормонов адено-
гипофиза, тропных по отношению к периферическим железам внутренней
секреции, то есть свидетельствовать о неизбирательной недостаточности
секреции аденогипофизом. После наступления периода полового созрева-
470
Глава 28
ния еще одним симптомом неизбирательной недостаточности аденогипо-
физа становится задержка появления или недостаточная выраженность
вторичных половых признаков. После достижения больным возраста, в
котором обычно наступает половое созревание, наносомия в сочетании с
задержкой появления или недостаточной выраженностью вторичных
половых признаков чаще всего свидетельствуют о неизбирательной
недостаточности аденогипофиза, при которой нарушена секреция ряда его
гормонов. Такой синдром называют гипофизарным инфантилизмом.
Наиболее частая причина недостаточности аденогипофиза у больных с
гипофизарным инфантилизмом - это опухоли соответствующей локализации.
Если у больных не выявляют каких-либо органических поражений
аденогипофиза, то гипофизарный инфантилизм и неизбирательную
недостаточность гипофиза определяют как идиопатические.
Карликовость, при которой половое созревание наступает в срок, а
вторичные половые признаки выражены вполне достаточно, может
быть признаком расстройств гипофизарно-гипоталамической системы
регуляции роста тела или свидетельствовать о нарушениях
взаимодействия этой системы с системой синтеза и секреции ИФР-1. В частности
такую наносомию обуславливает избирательная недостаточность
аденогипофиза, то есть в данном случае - низкий уровень образования и (или)
секреции аденогипофизом соматотропина при нормальном
высвобождении в кровь гонадотропинов и других тропных гормонов гипофиза
(избирательная недостаточность секреции соматотрофами).
Гипофизарную наносомию как результат избирательной
недостаточности секреции соматотропного гормона (табл. 28.2) можно
предотвратить лечением с использованием препаратов на основе гормона роста.
После системной дисплазии вторым основным симптомом гипофизарной
наносомии является периодическая гипогликемия. Ее причина -
выпадение действия соматотропина как катаболического гормона антагониста
инсулина из совокупного влияния на обмен веществ всего спектра ката-
болических гормонов.
Таблица 28.2
Гипофизарная наносомия
Ведущее звено патогенеза Наследственная избирательная недостаточность
секреции соматотропина
Диагностический тест Исследование реакции роста секреции
соматотропина в ответ на гипогликемию, вызванную
внутривенным введением инсулина
Результат теста Отсутствие роста секреции соматотропина
Основные симптомы Карликовость и периодическая гипогликемия,
иногда приводящая к потере сознания
Патогенетическая терапия Введение лекарственных средств на основе
гормона роста в период онтогенеза до полового
созревания, то есть до закрытия (окостенения)
эпифизов костей
Патофизиология наиболее частых эндокринопатий
471
Стимулами для секреции соматотропина являются гипогликемия, рост
содержания в циркулирующей крови аргинина и действие препарата L-
дофа. Для оценки способности аденогипофиза секретировать гормон
роста у больных вызывают гипогликемию внутривенным введением
инсулина, вводят внутривенно аргинина моногидрохлорид или назначают per os
L-дофа. Затем через определенные промежутки времени берут пробы
крови для определения концентрации в ней гормона роста, которая
натощак в крови у здоровых людей меньше, чем 8 мкг/л.
Наносомия или задержка роста могут быть следствиями
приобретенной недостаточности секреции гормона роста. Если нормальный рост
тела резко или постепенно замедляется на определенном этапе
онтогенеза до полового созревания, то наиболее частая причина нарушения
роста- это приобретенная недостаточность секреции соматотропина,
что следует связать прежде всего с новообразованиями
соответствующей локализации.
Опухоли ножки гипофиза (краниофарингиома) и самого гипофиза,
особенно его хромофобная аденома, замещают нормальную гипофизар-
ную ткань. У большинства больных детей это приводит к
последовательно возникающим эндокринопатиям. Сначала возникает недостаточность
секреции соматотропина, затем падает секреция гонадотропинов, потом
тиреотропина, и, наконец, кортикотропина. В результате возникает
приобретенная неизбирательная недостаточность секреции гормонов
аденогипофиза, в частности приводящая к гипофизарному инфантилизму. При
дифференциальной диагностике отличить приобретенную гипосекрецию
соматотропина от замедленного полового созревания, помогает тест,
используемый при выявлении гипофизарной наносомии (табл. 28.2). В ответ
на гипогликемию у больных с замедленным половым созреванием в
крови растет концентрация соматотропина, чего нет при гипофизарном
инфантилизме.
Карликовость Ларона была впервые им описана на основании
фактов, полученных при обследовании евреев-карликов в восточном Израиле.
У пациентов, обследованных Лароном, были явными симптомы
гипофизарной наносомии, но концентрация соматотропина в сыворотке была
нормальной. Как и у больных с гипофизарной наносомией у евреев-
карликов, обследованных Лароном, была снижена концентрация в
циркулирующей крови и тканях ИФР-I (у здоровых людей концентрация ИФР-1
в сыворотке крови натощак по утрам не превышает 2500 единиц на литр
крови). Лечение таких больных с использованием препаратов на основе
соматотропина не принесло положительных результатов. В дальнейшем
было установлено, что у части больных с наносомией Ларона задержка
роста была обусловлена наследственным нарушением синтеза молекулы
соматотропина, в результате которой гормон роста уже не мог повышать
синтез ИФР-I в печени и клетках других тканей {наносомия Ларона
первого типа). Несмотря на генетически детерминированную аномалию
синтеза соматотропина (СТ), молекула соматотропина сохраняла свои
эпитопы, с которыми взаимодействовали моноклональные и меченные
472
Глава 28
радиоактивными изотопами антитела, используемые для
иммунодиагностики гипофизарной наносомии. Поэтому у таких больных определяли
«нормальную» концентрацию ненормального СТ в сыворотке крови.
Ведущее звено наносомии Ларона второго типа - это
наследственное расстройство системы синтеза ИФР-I, вследствие которого
биологически активный нормальный соматотропин не повышает образование со-
матомедина С.
ПАТОГЕНЕЗ ГИПО- И ГИПЕРТИРЕОЗА
Гипотиреоз - патологическое состояние вследствие низкого уровня
секреции гормонов щитовидной железы и связанной с ним
недостаточности нормального действия гормонов на клетки, ткани, органы и
организм в целом.
Так как проявления гипотиреоза (табл. 28.3) аналогичны многим
признакам других болезней, то при обследовании больных гипотиреоз
нередко остается незамеченным.
Первичный гипотиреоз возникает в результате заболеваний самой
щитовидной железы. Первичный гипотиреоз может быть осложнением
лечения больных с тиреотоксикозом радиоактивным йодом, операций на
щитовидной железе, влияния на щитовидную железу ионизирующих
излучений (лучевая терапия при лимфогранулематозе в области шеи), а
также у части больных представляет собой побочный эффект йод-содер-
жащих препаратов.
В ряде развитых стран наиболее частой причиной гипотиреоза
является хронический аутоиммунный лимфоцитарный тиреоидит (болезнь
Хашимото), который у женщин возникает чаще, чем у мужчин. При
болезни Хашимото равномерное увеличение щитовидной железы едва
заметно, а с кровью больных циркулируют аутоантитела к аутоантигенам
тиреоглобулина и микросомной фракции железы.
Болезнь Хашимото как причина первичного гипотиреоза нередко
развивается одновременно с аутоиммунным поражением коры
надпочечников, обуславливающим недостаточность секреции и эффектов ее
гормонов (аутоиммунный полигландулярный синдром).
Вторичный гипотиреоз - это следствие нарушения секреции тирео-
тропного гормона (ТТГ) аденогипофизом. Чаще всего у больных
недостаточность секреции ТТГ, вызывающая гипотиреоз, развивается вследствие
хирургических вмешательств на гипофизе или является результатом
возникновения его опухолей. Вторичный гипотиреоз часто сочетается с
недостаточной секрецией других гормонов аденогипофиза, адренокортико-
тропного и прочих.
Определить вид гипотиреоза (первичный или вторичный) позроляет
исследование содержания в сыворотке крови ТТГ и тироксина (Т4).
Низкая концентрация Т4 при росте содержания в сыворотке ТТГ свидетель-
Патофизиология наиболее частых эндокринопатий
473
Таблица 28.3.
Связь симптомов гипотиреоза со звеньями его патогенеза как со следствиями низкого
системного действия гормонов щитовидной железы
Симптом
Звено патогенеза
Слабость, сонливость,
повышенная утомляемость
Сухость кожи
Брадикардия, снижение
минутного объема
кровообращения вплоть до вызывающей
кому артериальной гипотензии
Утолщение волос
Утолщение век, «надутое
лицо» и «отекшие» конечности
Непереносимость холода
Запор
Роста массы тела
Изменение тембра голоса
Избыточные и иррегулярные
менструальные кровотечения
Неполная глухота
Сниженный уровень активности психических
процессов и низкая возбудимость центральной нервной
системы
Низкий уровень адренергической стимуляции
потовых желез
Системное падение чувствительности адренорецеп-
торов
Связанное с гипотиреозом извращение белкового
синтеза
Накопление в интерстиции мукополисахаридов,
обусловленное расстройствами образования белков и
дефектами развития вследствие гипотиреоза (микседемах)
Невозможность интенсификации теплопродукции из-
за угнетенного аэробного биологического окисления
Системные гипоэргоз, недостаточность анаболизма и
падение возбудимости нейронов на уровне кишечника
Угнетение липолиза и аккумуляция триглицеридов,
связанные с гипотиреозом
Утолщение голосовых связок, вызванное микседемой
Патогенез не ясен. Результат низкой возбудимости
нервных центров, гипоэргоза, угнетения анаболизма
Дефекты развития при кретинизме, падение
возбудимости соответствующих нейронов и рецепторов
ствует о том, что в соответствии принципом регуляции по обратной
отрицательной связи снижение образования и высвобождения Т4 служит
стимулом для роста секреции ТТГ аденогипофизом. В этом случае
гипотиреоз определяют как первичный. Когда при гипотиреозе снижена
концентрация в сыворотке ТТГ, или в том случае, если, несмотря на
гипотиреоз, концентрация ТТГ находится в диапазоне среднестатистической
нормы, снижение функции щитовидной железы является вторичным
гипотиреозом.
При неявном субклиническом гипотиреозе, то есть при минимальных
клинических проявлениях или отсутствии симптомов недостаточности
функции щитовидной железы, концентрация Т4 может находиться в пре-
1 Микседема (гр. микса - слизь + эйдема - опухоль, отек) - резко выраженная форма
гипотиреоза, которая проявляется отеком кожи, языка, сухостью кожи, выпадением волос,
замедленным пульсом, низким кровяным давлением, общей вялостью, сонливостью,
снижением памяти и умственных способностей.
474
Глава 28
делах нормальных колебаний. При этом уровень содержания ТТГ в
сыворотке повышен, что предположительно можно связать с реакцией роста
секреции ТТГ аденогипофизом в ответ на неадекватное потребностям
организма действие гормонов щитовидной железы. У таких больных в
патогенетическом отношении может быть оправданным назначение
препаратов щитовидной железы для воссстановления на системном уровне
нормальной интенсивности действия тиреоидных гормонов (заместительная
терапия).
Более редкие причины гипотиреоза - это генетически
детерминированная гипоплазия щитовидной железы (врожденный атиреоз),
наследственные нарушения синтеза ее гормонов, связанные с отсутствием
нормальной экспрессии генов определенных ферментов или ее
недостаточностью, врожденная или приобретенная пониженная чувствительность
клеток и тканей к действию гормонов, а также низкое поступление йода как
субстрата синтеза гормонов щитовидной железы из внешней среды во
внутреннюю. Последняя причина гипотиреоза свойственна только
«бедным кварталам» мирового сообщества, то есть может быть и в нашем
удерживаемом за чертой бедности многострадальном Отечестве.
Гипотиреоз можно считать патологическим состоянием,
обусловленным дефицитом в циркулирующей крови и всем организме свободных
гормонов щитовидной железы. Известно, что гормоны щитовидной
железы трийодтиронин (Тз) и тироксин связываются с ядерными рецепторами
клеток-мишеней. Сродство тиреоидных гормонов к ядерным рецепторам
высоко. При этом сродство к Тз в десять раз превышает сродство к Т4.
Несмотря на это, можно считать, что специфической активностью обладают
оба гормона. Кроме того, тиреоидные гормоны взаимодействуют с
низкоаффинными по отношению к ним связывающими цитоплазматическими
участками клеток мишеней. Эти протеины, скорее всего, не аналогичны
белкам ядерных рецепторов и служат для удержания гормонов в
непосредственной близости от истинных рецепторов.
Основное воздействие гормонов щитовидной железы на обмен
веществ - это увеличение потребления кислорода и улавливания клетками
свободной энергии в результате усиления биологического окисления.
Поэтому потребление кислорода в условиях относительного покоя у
больных с гипотиреозом находится на патологически низком уровне. Данный
эффект гипотиреоза наблюдается во всех клетках, тканях и органах,
кроме головного мозга, клеток системы мононуклеарных фагоцитов и гонад.
Таким образом, эволюция отчасти сохранила не зависящими от
возможного гипотиреоза энергетический обмен на супрасегментарном уровне
системной регуляции, в ключевом звене системы иммунитета, а также
обеспечение свободной энергией репродуктивной функции. Тем не менее,
дефицит массы в эффекторах системы эндокринной регуляции обмена
веществ (дефицит гормонов щитовидной железы) приводит к дефициту
свободной энергии (гипоэргозу) на системном уровне. Мы считаем это
одним из проявлений действия общей закономерности развития болезни и
патологического процесса вследствие дизрегуляции, - через дефицит
Патофизиология наиболее частых эндокринопатий
475
массы и энергии в системах регуляции к дефициту массы и энергии на
уровне всего организма.
Системный гипоэргоз и падение возбудимости нервных центров
вследствие гипотиреоза проявляет себя такими характерными
симптомами недостаточной функции щитовидной железы как повышенная
утомляемость, сонливость, а также замедление речи и падение когнитивных
функций. Нарушения внутрицентральных отношений вследствие
гипотиреоза - это результат замедленного умственного развития больных с
гипотиреозом, а также падения интенсивности неспецифической афферен-
тации, обусловленного системным гипоэргозом.
Большая часть свободной энергии, утилизируемой клеткой,
используется для работы Na+/ К+-АТФазного насоса. Гормоны щитовидной железы
повышают эффективность работы этого насоса, увеличивая количество
составляющих его элементов. Так как практически все клетки обладают
таким насосом и реагируют на тиреоидные гормоны, то к системным
эффектам тиреоидных гормонов относится повышение эффективности
работы данного механизма активного трансмембранного переноса ионов. Это
происходит посредством роста улавливания клетками свободной энергии
и через увеличение числа единиц Ыа+/К+-АТФазного насоса.
Гормоны щитовидной железы усиливают чувствительность адрено-
рецепторов сердца, сосудов и других эффекторов функций. При этом в
сравнении с другими регуляторными влияниями адренергическая
стимуляция возрастает в наибольшей степени, так как одновременно
гормоны подавляют активность фермента моноаминооксидазы,
разрушающей симпатический медиатор норадреналин. Гипотиреоз, снижая
интенсивность адренергической стимуляции эффекторов системы
кровообращения, приводит к снижению минутного объема кровообращения
(МОК) и брадикардии в условиях относительного покоя. Другая причина
низких величин минутного объема кровообращения - это сниженный
уровень потребления кислорода как детерминанты МОК. Снижение
адренергической стимуляции потовых желез проявляет себя характерной
сухостью колеи.
Гипотиреоидная (миксематозная) кома - редкое осложнение
гипотиреоза, которое в основном складывается из следующих дисфункций и
нарушений гомеостазиса:
♦ Гиповентиляция как результат падения образования углекислого
газа, которую усугубляет центральное гипопноэ из-за гипоэргоза
нейронов дыхательного центра. Поэтому гиповентиляция при миксема-
тозной коме может быть причиной артериальной гипоксемии.
♦ Артериальная гипотензия как следствие снижения МОК и
гипоэргоза нейронов сосудодвигательного центра, а также падения
чувствительности адренорецепторов сердца и сосудистой стенки.
♦ Гипотермия в результате падения интенсивности биологического
окисления на системном уровне.
Запор как характерный симптом гипотиреоза вероятно обусловлен
системным гипоэргозом и может быть результатом расстройств внутри-
476
Глава 28
центральных отношений вследствие падения функции щитовидной
железы.
Гормоны щитовидной железы, как и кортикостероиды, индуцируют
белковый синтез, активируя механизм транскрипции генов. Это основной
механизм, посредством действия которого влияние Тз на клетки усиливает
общий синтез белка и обеспечивает положительный азотистый баланс.
Поэтому гипотиреоз нередко вызывает отрицательный азотистый баланс.
Тиреоидные гормоны и глюкокортикоиды, повышают уровень
транскрипции гена гормона роста человека (соматотропина). Поэтому развитие
гипотиреоза в детском возрасте может быть причиной задержки роста
тела. Тиреоидные гормоны стимулируют синтез белка на системном уровне
не только через усиление экспрессии гена соматотропина. Они усиливают
синтез белка, модулируя функционирование других элементов
генетического материала клеток и повышая проницаемость плазматической
мембраны для аминокислот. В этой связи гипотиреоз можно считать
патологическим состоянием, которое характеризует угнетение белкового
синтеза как причина задержки умственного развития и роста тела детей с
гипотиреозом. Связанная с гипотиреозом невозможность быстрой
интенсификации белкового синтеза в иммунокомпетентных клетках может
служить причиной дизрегуляции специфического иммунного ответа и
приобретенного иммунодефицита вследствие дисфункций как Т-, так и
В-клеток.
Одним из эффектов тиреоидных гормонов на метаболизм является
усиление липолиза и окисления жирных кислот с падением уровня их
содержания в циркулирующей крови. Низкая интенсивность липолиза у
больных с гипотиреозом приводит к аккумуляции жира в организме, что
обуславливает патологическое возрастание массы тела. Рост массы тела
чаще выражен умеренно, что связано с анорексией (результат падения
возбудимости нервной системы и трат свободной энергии организмом) и
низким уровнем белкового синтеза у больных с гипотиреозом.
Гормоны щитовидной железы - важные эффекторы систем регуляции
развития по ходу онтогенеза. Поэтому гипотиреоз у плодов или
новорожденных приводит к кретинизму (фр. cretin, тупица), то есть сочетанию
множественных дефектов развития и необратимой задержки нормального
становления ментальных и когнитивных функций. Для большинства
больных с кретинизмом вследствие гипотиреоза характерна микседема.
Патологическое состояние организма вследствие патогенно
избыточной секреции гормонов щитовидной железы называют гипертиреозом
(табл. 28.4). Под тиреотоксикозом понимают гипертиреоз крайней
степени тяжести.
Болезнь Грейвса (диффузный токсический зоб) обусловлена
аутоиммунным поражением щитовидной железы и является самой частой
причиной тиреотоксикоза. Примерно у 40 % больных диффузным
токсическим зобом наблюдают офтальмопатию, которую нередко
характеризуют периорбитальный отек, коньюктивит, экзофтальм, а также
поражение внеглазных мышц.
Патофизиология наиболее частых эндокринопатий
477
Таблица 28.4
Гипертиреоз как синдром.
Связь симптомов и звеньев патогенеза
Симптом
Расстройства сна,
раздражительность, непроизвольные
дрожательные движения
Снижение массы тела при
хорошем аппетите и доступности пищи
Избыточное потение, в том
числе в условиях покоя и при
комфортной температуре внешней
среды: непереносимость роста
температуры окружающей среды
Тахикардия, рост минутного
объема кровообращения в
условиях покоя, возрастание пульсового
давления
Мышечная слабость. Больной
помогает себе руками, когда встает
со стула (симптом табурета)
Диарея
Аменорея
Звено патогенеза
(результат усиленного действия тиреоидных гормонов)
Повышение возбудимости центральной нервной
системы и активация психических процессов
Рост потребления кислорода организмом и
утилизации свободной энергии на системном уровне.
Усиление липолиза
Рост теплопродукции в организме, вторичный по
отношению к интенсификации аэробного
биологического окисления на системном уровне Рост адре-
нергической стимуляции потовых желез вследствие
повышения чувствительности адренорецепторов и
угнетения активности фермента (моноаминооксида-
за), разрушающего норадреналин
Рост чувствительности адренорецепторов на
уровне сердца. Возрастание потребления кислорода
организмом
Усиление вхождения в миоциты калия при росте
выхода из них натрия как причина патологических
изменений трансмембранного потенциала и падения
возбудимости
Патогенно избыточная активация системы адени-
латциклазы энтероцитов
Патогенез не ясен. Вторична по отношению к
изменению состояния всех клеток организма из-за
повышенной концентрации тиреоидных гормонов в
циркулирующей крови
Болезнь Грейвса может иметь семейный характер. От данного
заболевания чаще страдают пожилые женщины. Офтальмопатия при болезни
Грейвса также является семейной. Ее патогенез неясен. Офтальмопатию
связывают с присутствием в крови больных так называемого экзофталь-
мического фактора, возможно имеющего гипофизарное происхождение.
Симптом Грефе - при опускании глазного яблока веко двигается
толчками, неравномерною - у больных болезнью Грейвса обусловлен
экзофтальмом и поражением внеглазных мышц при офтальмопатии.
Одиночные или множественные токсические аденомы щитовидной железы
также могут вызывать тиреотоксикоз через не контролируемую
системной регуляцией патогенно избыточную секрецию тиреоидных гормонов.
В данном случае причину тиреотоксикоза может элиминировать лишь
хирургическое удаление аденомы или лечение радиоактивным йодом.
Тиреотоксикоз может быть ятрогенным, то есть обусловленным
приемом внутрь или парентеральным введением препаратов, содержащих йод,
которые приводят к возрастанию уровня синтеза тиреоидных гормонов.
478
Глава 28
Риск тиреотоксикоза как осложнения действия на организм йод-содержа-
щих препаратов особенно велик у больных с многоузловым,
эндемическим зобом и болезнью Грейвса.
Тиреоидит, обуславливая высвобождение тиреоидных гормонов из
воспаленной и болезненной железы, может быть причиной
тиреотоксикоза. Предположительно тиреоидит, вызывающий тиреотоксикоз, связан с
вирусной инфекцией.
Бета-один-адреноблокаторы как средства фармакотерапии при тирео-
токсическом кризе снижают уровень возбуждения соответствующих ад-
ренорецепторов на уровне сердца, тем самым снижая тахикардию как
фактор риска сердечных аритмий у больных с предельно выраженным
гипертиреозом.
Кроме того, данные средства снижают тяжесть тиреотоксикоза,
тормозя превращение тироксина в трийодтиронин, то есть в наиболее
активный из тиреоидных гормонов.
Антагонисты кальциевых каналов снижают возбудимость рабочих и
проводящих кардиомиоцитов, которая растет вследствие роста
возбудимости адренорецепторов на уровне сердца.
Йод-содержащие препараты как средства терапии больных в
состоянии тиреотоксикоза снижают концентрацию тироксина в крови,
угнетая высвобождение тиреоидных гормонов из железы в
циркулирующую кровь.
ПАТОГЕНЕЗ РАССТРОЙСТВ СЕКРЕЦИИ
ГОРМОНОВ НАДПОЧЕЧНИКОВ
Одной из причин недостаточной секреции гормонов коры
надпочечников является поражение ткани железы, вызванное туберкулезом.
Связанный с туберкулезом цитолиз клеток железы как причина
недостаточной секреции гормонов коры надпочечников достигает соответствующей
распространенности через 10-20 лет после первичного инфицирования.
Ведущая в развитых странах причина хронической недостаточности
секреции гормонов коры надпочечников (болезни Аддисона) (табл. 28.5) -
это аутоиммунное поражение надпочечниковой ткани, вызывающее идио-
патическую недостаточность надпочечников. Первичное звено
патогенеза идиопатической недостаточности надпочечников- это локальное
воспаление, которое индуцируется образованием иммунных комплексов
аутоантиген-аутоантитело через активацию системы комплемента по
классическому пути.
Острую недостаточность секреции и эффектов гормонов коры
надпочечников может обусловить хирургическое вмешательство на обеих над-
почечниковых железах, которое предпринимают в связи с болезнью Ку-
шинга или для удаления метастазов злокачественных опухолей легких в
надпочечники. Другая ятрогенная причина недостаточности надпочечни-
Патофизиология наиболее частых эндокринопатий
479
Таблица 28.5
Связь симптомов болезни Аддисона с патогенезом
недостаточности надпочечников
Симптом
Гиперпигментация («бронзовый цвет
кожи»), наиболее выраженная на
открытых участках кожи, видимых слизистых
оболочках, в области кожных складок и
шрамов
Артериальная гипотензия, в том
числе и ортостатическая
Гипогликемия
Слабость и повышенная
утомляемость, сонливость и спутанность
сознания
Анорексия, тошнота и рвота
Звено патогенеза
Рост образования меланоцитстимулирующе-
го гормона, связанный с усилением секреции
кортикотропина в ответ на недостаточную
секрецию кортизола
Снижение пермиссивного действия глюкокор-
тикоидов на адренергическую стимуляцию
эффекторов системы кровообращения
Снижение интенсивности глюконеогенеза в
результате падения действующей концентрации
глюкокортикоидов во внутренней среде
Гипоэргоз вследствие гипогликемии, а также
низкой способности системы кровообращения
повышать минутный объем кровообращения в
ответ на рост потребностей организма.
Артериальная, в том числе и ортостатическая, гипотензия
Изменение внутрицентральных отношений
вследствие гипогликемического гипоэргоза,
скрытой или явной недостаточности
кровообращения
ков - угнетение секреции адренокортикотропного гормона в соответствии
с принципом регуляции по механизму отрицательной обратной связи при
длительном и частом введении во внутреннюю среду кортикостероидов
как лекарственных средств. Такое ятрогенное угнетение секреции
кортикотропина может быть стойким, сохраняясь как причина недостаточности
надпочечников в течение одного года и более.
К редким причинам недостаточности секреции кортикостероидов
относят амилоидоз, грибковые инфекции, двухстороннее кровоизлияние в
надпочечники как побочный эффект лекарственных средств, снижающих
свертываемость крови, а также метастазирование в надпочечники
злокачественных опухолей. Синдром приобретенного иммунодефицита у
части больных приводит к недостаточности надпочечников через их
поражение вследствие цитомегаловирусных и других инфекций.
Под вторичной недостаточностью коры надпочечников
понимают снижение секреции глюкокортикоидов, связанное с гипопитуита-
ризмом.
Нормальные секреция глюкокортикоидов и содержание гормонов
коры надпочечников в циркулирующей крови - необходимое условие
поддержания нормальных, то есть соответствующих потребностям
организма, артериального давления и минутного объема кровообращения. Глю-
кокортикоиды не оказывают прямого физиологического действия на
эффекторы системы кровообращения, но требуются для проявления эффекта
480
Глава 28
на артериолы и сердце катехоламинов и нервной адренергической
стимуляции, то есть оказывают пермиссивное (англ. permissive - дозволяющий,
позволяющий, разрешающий) действие.
Сниженное пермиссивное действие глюкокортикоидов в основном
детерминирует низкую переносимость пациентов с болезнью Аддисона
неспецифической стрессорной, любой защитной и патологической
реакций, которые характеризует возрастание минутного объема
кровообращения, обеспечивающее рост улавливания и потребления клетками
свободной энергии, а также доставку к ним энергетических и пластических
субстратов:
♦ неспецифическая стрессорная приспособительная и компенсаторная
реакция;
♦ состояние после ранений, травм и хирургических вмешательств,
системная реакция на кровопотерю;
♦ сепсис, системная воспалительная реакция и др.
Кроме того, следует учитывать, что усиление секреции кортизола
представляет собой необходимый элемент неспецифической
стрессорной реакции на уровне эндокринного звена регуляции обмена веществ и
кровообращения. При этом увеличение концентрации кортизола во
внеклеточной жидкости и крови через усиление катаболизма и пермиссив-
ного действия глюкокортикоидов на сердце повышает доставку
энергопластических субстратов клеткам при защитно-приспособительном
системном перераспределении энергопластических субстратов,
составляющем суть стресса. В результате стрессорной интенсификации секреции
глюкокортикоидов у пациентов с болезнью Аддисона окончательно
падает уровень секреции гормонов коры надпочечников и развивается ад-
реналовая кома. Звенья патогенеза адреналовой комы в основном
представляют собой следствия низкого пермиссивного действия
глюкокортикоидов на сердце и сосуды, а также недостаточной активности во
внеклеточной жидкости и циркулирующей крови минералкортикоидов. Из
звеньев патогенеза адреналовой комы и недостаточности надпочечников
нельзя исключить гипогликемии, патогенез которой в основном
складывается из неограниченного действием кортизола эффекта инсулина, что
на системном уровне приводит к патологическому преобладанию
анаболизма (в частности росту утилизации глюкозы для гликолиза) и
торможению катаболизма. В результате падение глюконеогенеза, рост
образования гликогена и утилизации глюкозы при гликолизе снижают
концентрацию глюкозы в крови в такой степени, что гипогликемия может быть
ведущей причиной комы у больных с синдромом Аддисона.
При болезни Аддисона и идиопатической недостаточности
надпочечников снижение уровня секреции глюкокортикоидов представляет собой
результат воспаления и цитолиза клеток железы, вызванных туберкулезом
или аутоиммунным поражением надпочечников. Причина недостаточной
секреции нормальных гормонов коры надпочечников у больных с адрено-
генитальным синдромом - врожденный дефицит активности ключевых
ферментов синтеза кортизола, 21-гидроксилазы или 11-бета-гидрокси-
Патофизиология наиболее частых эндокринопатий
481
лазы. Адреногенитальный синдром - это моногенное заболевание,
наследуемое по аутосомно-рецессивному типу. В результате генетически
детерминированного дефицита соответствующих энзимов надпочечники не
обладают адекватной потребностям организма способностью
синтезировать нормальный кортизол. Падение концентрации гормона во
внеклеточной жидкости и плазме крови в соответствии с принципом регуляции
по отрицательной обратной связи служит стимулом для роста секреции
аденогипофизом кортикотропина. Рост секреции кортикотропина
обуславливает гиперплазию надпочечников. Под влиянием высокой
действующей концентрации кортикотропина надпочечники интенсивно
образуют и высвобождают аномальные стероиды, обладающие биологической
активностью андрогенов. В результате у женщин с адреногенитальным
синдромом развивается вирилизм. Вирилизм (лат. virilis, мужской)-
наличие у женщин вторичных мужских половых признаков, то есть гирсу-
тизма, характерного для мужчин строения скелета и произвольных
мышц, массивного клитора, а также низкого тембра голоса. Вирилизм у
больных девочек иногда можно выявить сразу после рождения, и он
прогрессирует в первые годы жизни. Под влиянием высокой концентрации
во внутренней среде аномальных стероидов со свойствами андрогенов
рост тела в первые годы жизни ускорен. В дальнейшем по ходу
онтогенеза рост тела детей с адреногенитальным синдромом замедляется, что
связано с преждевременным (до полового) созревания окостенением в
области эпифизов длинных трубчатых костей. Рост содержания
стероидов со свойствами андрогенов в циркулирующей крови в соответствии с
принципом регуляции по механизму отрицательной обратной связи
ведет к падению секреции аденогипофизом гонадотропинов. В результате
у больных девочек не происходит становления нормального
менструального цикла.
Хотя у мальчиков с адреногенитальным синдромом некоторые из
вторичных половых признаков появляются раньше, чем у здоровых детей,
половое созревание у них полноценным считать нельзя. Дело в том, что
дефицит секреции гонадотропинов обуславливает недоразвитие половых
желез и неполноценный сперматогенез, несмотря на вторичные половые
признаки и наступление соответствующего периода онтогенеза у
подростков мужского пола с адреногенитальным синдромом.
Патогенетическим средством терапии и предупреждения дефектов
развития у больных с адреногенитальным синдромом служит введение им
препаратов на основе глюкокортикоидов (например, 25-50 мг кортизона в
сутки). Рост концентрации экзогенных кортикостероидов во внутренней
среде и плазме крови в соответствии с принципом регуляции по
механизму отрицательной обратной связи снижает уровень секреции адренокор-
тикотропного гормона, тем самым уменьшая концентрацию аномальных
стероидов со свойствами андрогенов в плазме крови. Это возвращает
секрецию гонадотропинов на уровень адекватный для нормального полового
1 Гирсугизм - (лат., hirsutus - волосатый) - мужской тип оволосения у женщин.
482
Глава 28
созревания. В результате у больных девочек вирилизм и гирсутизм
подвергаются обратному развитию, и происходит становление нормального
менструального цикла. У больных мальчиков и подростков
заместительная терапия кортикостероидами посредством действия аналогичного
механизма обуславливает нормальное половое созревание, то есть
формирование образующих и секретирующих тестостерон клеток Лейдига и
выводящих сперму канальцев яичек.
Врожденный дефицит активности 21-гидроксилазы как причина адре-
ногенитального синдрома может приводить к низкому относительно
потребностей систем регуляции и организма синтезу корой надпочечников
альдостерона. Дефицит альдостерона обуславливает потерю организмом
натрия. Снижение содержания натрия в организме ведет к падению
объема внеклеточной жидкости. Снижение объема внеклеточной жидкости
может быть причиной летального исхода у новорожденных, если только
верный диагноз не диктует необходимости экстренной коррекции
дефицита объема внеклеточной жидкости путем внутривенной инфузии изо-
осмоляльных относительно внеклеточной жидкости растворов. При
дефиците альдостерона проводят заместительную терапию, вводя больным
препараты, обладающие свойствами минералкортикоидов.
При адреногенитальном синдроме, обусловленном генетически
детерминированным дефицитом активности 11-гидроксилазы, стимуляция
коры надпочечников высокой действующей концентрацией кортикотро-
пина приводит к усиленному синтезу деоксикортикостерона,
обладающего свойствами минералкортикоида. Рост активности минералкортикоидов
во внутренней среде (частный случай вторичного гиперальдостеронизма)
повышает содержание в организме натрия, что увеличивает объем
внеклеточной жидкости и вызывает артериальную гипертензию.
Одновременно в силу усиленного влияния минералкортикоидов на почки растет
экскреция с мочой калия и развивается гипокалиемия.
В основе синдрома Кушинга лежат избыточная секреция и
действие на клетки-эффекторы глюкокортикоидов. Избыток глюкокортикои-
дов как катаболических гормонов антагонистов инсулина через сдвиг
соотношения между анаболизмом и катаболизмом в сторону последнего
вызывает нарушения развития и деградацию соединительной ткани. В
результате синдром характеризуют: а) повышенная ломкость сосудистой
стенки; б) снижение массы скелетных мышц как причина мышечной
слабости; в) остеопороз. В основном системный остеопороз захватывает
позвонки различных отделов. Повышенная ломкость сосудистой стенки
проявляет себя частыми и массивными подкожными кровоизлияниями,
причинами которых могут быть незначительные травмы. Больные с
синдромом Кушинга страдают от ожирения, при котором жир в основном
аккумулируется в подкожной жировой клетчатке живота, спины, над-
плечий и ягодиц. Своеобразный характер ожирения связан с тем, что;
глюкокортикоиды стимулируют липолиз в одних частях тела (конечно- \
сти) и липогенез в других (лицо и туловище). Из-за увеличения массы \
адипоцитов в области лица, у больных с синдромом Кушинга оно при-j
Патофизиология наиболее частых эндокринопатий
483
обретает характерную «лунообразную» форму. При этом обычно всегда
есть гиперемия кожи лица. Вследствие перерастяжения кожи в областях
аккумуляции жира и в результате системной деградации
соединительной ткани на коже больных появляются характерные красные (почти
фиолетовые) полосы.
Известно, что глюкокортикоиды способствуют повышению
образования и высвобождения глюкозы печенью:
♦ вызывая рост скорости глюконеогенеза;
♦ усиливая через интенсификацию протеолиза высвобождение
аминокислот в качестве субстратов глюконеогенеза из мышечной и
лимфоидной ткани;
♦ оказывая пермиссивный эффект, то есть как бы «позволяя» другим
катаболическим гормонам оказывать свое действие с максимальной
эффективностью.
Все это приводит к гипергликемии, которую усиливает эффект
глюкокортикоидов как антагонистов инсулина. В результате у больных с
синдромом Кушинга выявляют патологически низкую толерантность по
отношению к глюкозе.
Как катаболические гормоны антагонисты инсулина
глюкокортикоиды в избыточной действующей концентрации повышают липолиз и
концентрацию свободных жирных кислот в плазме крови, вызывая
незначительный метаболический ацидоз, который возникает вследствие роста
образования кетоновых тел.
Известно, что адекватные потребностям организма секреция и
эффекты гормонов коры надпочечников представляют собой необходимое
условие нормальных высшей нервной деятельности и развития личности. У
больных с синдромом Кушинга избыточная секреция глюкокортикоидов
приводит к периодическому деструктивному поведению, которое
сменяют периоды депрессии. Патологические сдвиги высшей нервной
деятельности столь выражены, что служат причиной неправильного диагноза
эндогенной депрессии или шизофрении у больных с синдромом Кушинга.
Если синдром Кушинга развивается у детей, то антианаболическое
действие кортизола приостанавливает рост тела.
Выделяют гипоталамо-гипофизарный синдром Кушинга, который
характеризует двухсторонняя гиперплазия коры надпочечников. У 70 %
больных с синдромом Кушинга (пациентов с гипоталамо-гипофизарным
синдромом Кушинга) дизрегуляция на уровне гипоталамических центров
приводит к извращению регуляции секреции глюкокортикоидов в
соответствии с принципом «отрицательной обратной связи». В результате
секреция кортикотропина аденогипофизом не тормозится при
концентрации кортизола в крови, которая в четыре-пять раз больше нормальной.
Патогенетическим средством терапии гипоталамо-гипофизарного
синдрома Кушинга выступает хирургическое вмешательство с целью
двустороннего удаления гиперплазированной железы (билатеральная адре-
налэктомия). После операции патологические следствия избыточной
секреции глюкокортикоидов во многом подвергаются обратному развитию.
484
Глава 28
Падение концентрации кортизола в крови предельно повышает
секрецию кортикотропина. В результате возрастает образование меланоцит-
стимулирующего гормона, что, как и при болезни Аддисона, может
обусловить темную пигментацию («бронзовый оттенок») кожи. Больным с
удаленными надпочечниками показана заместительная терапия
препаратами на основе глюкокортикоидов. В последующем больные,
перенесшие данное оперативное вмешательство, приобретают определенный
опыт, и в предверии стресса той или иной природы сами увеличивают
дозу кортикостероидов.
У части больных после билатеральной адреналэктомии постоянно
предельно повышенный уровень секреции кортикотропина служит
причиной образования вторичной базофильной аденомы гипофиза.
У 20 % больных с синдромом Кушинга его причиной является
первичная базофильная аденома гипофиза. В таких случаях предпринимают
оперативное вмешательство на гипофизе.
У 10 % больных с синдромом Кушинга его причина - это аденома
коры надпочечников, продуцирующая патогенно избыточное количество
глюкокортикоидов при подавлении секреции кортикотропина из-за
высокой концентрации кортизола в крови. После введения таким больным
препаратов кортикостероидов в определенной и высокой дозе секреция
глюкокортикоидов не падает, как это бывает при гипоталамо-гипофи-
зарном синдроме Кушинга. После операции удаления аденомы
существует опасность острой надпочечниковой недостаточности, то есть адренало-
вого криза. Дело в том, что из-за подавления секреции кортикотропина
вследствие патогенно высокой секреции кортизола аденомой, другая и не
содержащая аденомы железа атрофируется, причем атрофия часто имеет
необратимый характер.
Синдром Кона (первичный гиперальдостеронизм) - это следствие
образования опухоли надпочечников, клетки которой обладает
избирательной способностью секреции альдостерона. Избыточно патогенное
образование альдостерона опухолью через рост реабсорбции натрия из
просвета канальцев нефронов увеличивает общее содержание натрия в
организме, а значит повышает объем внеклеточной жидкости. Расширение
внеклеточного жидкостного сектора ведет к отекам и росту преднагрузки
сердца как причинам увеличения минутного объема кровообращения и
артериальной гипертензии.
Одновременно с ростом реабсорбции натрия растут экскреция
почками калия и протонов, а также образование и реабсорбция бикарбонатных
анионов. В результате расстройства водно-солевого обмена и кислотно-
основного при синдроме Кона, кроме увеличения объема внеклеточной
жидкости, составляют гипокалиемия и метаболический алкалоз. Гипока-
лиемия вызывает мышечную слабость, сердечные аритмии, а также
падение концентрационной способности почек. Падение концентрационной
способности почек в результате гипокалиемии повышает объем
выделяемой мочи и служит причиной никгпурии, то есть частого мочеиспускания
по ночам, препятствующего нормальному сну.
Патофизиология наиболее частых эндокринопатий
485
ПАТОГЕНЕЗ ГИПЕР-, ГИПОКАЛЬЦИЕМИИ
И РАССТРОЙСТВ СЕКРЕЦИИ ПАРАЩИТОВИДНЫХ ЖЕЛЕЗ
Ионы кальция как эффекторы систем регуляции вовлечены в
осуществление важнейших функций, оказывая влияние на проведение
возбуждения через нейромышечный синапс, сокращение скелетных мышц и
миокарда, являясь фактором свертывания крови, обеспечивая целостность
мембран и трансмембранный перенос ионов, индуцируя секрецию ряда
гормонов и нейромедиаторов, а также осуществляя на внутриклеточном
уровне действие многих агентов эндокринной регуляции. Нормальное
протекание этих процессов обеспечивается тем, что концентрация
ионизированного кальция в плазме крови удерживается в узких нормальных
пределах. Так как кальций участвует в регуляции важнейших функций, а
диапазон нормальных колебаний его концентрации в плазме крови весьма
узок, то толерантность организма к патологическим сдвигам
концентрации кальция в плазме крови исключительно низка.
Адекватная потребностям организма концентрация катиона кальция и
фосфатного аниона во внеклеточной жидкости представляет собой
необходимое условие ненарушенной минерализации костей.
Очень низкая концентрация ионизированного кальция в цитозоле
клеток поддерживается ионными насосами, встроенными в клеточные
мембраны. Так как градиент концентраций свободных ионов кальция между
цитозолем клеток и интерстицием исключительно высок, то даже
небольшие сдвиги проницаемости наружной клеточной мембраны приводят
ко значительным изменениями внутриклеточной концентрации Са2+.
Интенсивность поступления свободных ионов кальция в клетку
определяется как его содержанием в межклеточных пространствах, так и
характером влияний гормонов и медиаторов. Таким образом, нормальное
осуществление клеточных функций на молекулярном уровне
структурно-функциональной организации критически зависит от постоянства
внеклеточной концентрации кальция. Поэтому небольшие сдвиги
содержания ионизированного кальция во внеклеточном секторе
обуславливают системные патологические изменения состояний клеточных
эффекторов функций, расстройства функциональных систем и нарушения
гомеостазиса.
Нормальная общая концентрация кальция в сыворотке крови
варьирует в пределах от 8,8 до 10,4 мг/100 мл (2,2-2,6 ммоль/л). Ее составляют:
а) кальций, связанный с белками плазмы крови (40 % общей
концентрации кальция); б) кальций, циркулирующий с кровью в комплексе1 с цит-
ратным, сульфатным, лактатным и фосфатным анионами (15 %); в)
ионизированный кальций, то есть фракция общей концентрации кальция в
сыворотке, обладающая наибольшей биологической активностью (45 %).
Комплекс - относительно стабильная комбинация двух или более ионов либо других
соединений, образующих структуру, не содержащую ковалентных связей.
486
Глава 28
Содержание ионизированного кальция в сыворотке определяют сразу
после забора пробы крови с использованием ионообменных электродов и в
анаэробных условиях (отсутствие в среде протекания реакции свободного
кислорода).
Общее содержание кальция в сыворотке зависит от концентрации в
ней альбумина. Снижение содержания альбумина в сыворотке на 10 г/л
приводит к падению общего содержания в ней кальция на 0,8 мг/100 мл.
Перемещение воды из сосудистого жидкостного сектора в интерстици-
альный как следствие портальной гипертензии, застойной сердечной
недостаточности, гиперцитокинемии и системной воспалительной реакции,
других патологических состояний и типовых патологических процессов у
части больных через гемоконцентрацию вызывает рост общего
содержания кальция в сыворотке. Концентрация кальция растет только в том
случае, если патологическое возрастание проницаемости стенки
микрососудов не приводит к одновременному выходу альбумина из плазмы крови в
интерстиций. Так в частности происходит на определенной и критической
стадии системной воспалительной реакции, вызывающей множественную
системную недостаточность. Вместе с альбумином в межклеточные
пространства из плазмы крови уходит и кальций. В результате, несмотря на
гемоконцентрацию, общее содержание кальция в сыворотке остается
неизменным.
Связывание кальция альбуминами сыворотки крови - это обратная
функция концентрации в ней протонов. Снижение рН крови на 0,1
повышает концентрацию ионизированного кальция в сыворотке на 0,12 мг/100 мл.
Паратиреоидный гормон увеличивает концентрацию ионизированного
кальция в плазме крови, в том числе и снижая уровень связывания
кальция ее альбуминами.
Переход наших предков на пути эволюции из водной среды обитания
в наземную, где кальций относительно дефицитен, во многом зависел от
развития сложной физиологической системы, конечный полезный
приспособительный результат которой - это удержание концентрации
кальция во внутренней среде в пределах диапазона нормальных колебаний. В
качестве агентов эндокринной регуляции в функционировании данной
системы участвуют три гормона - паратиреоидный, кальцитриол, то есть
(витамин D)-ropMOH, и кальцитонин.
При падении концентрации ионизированного кальция в плазме крови
до уровня более низкого, чем 1,1 ммоль/л, растет секреция паратиреоид-
ного гормона (ПТГ). ПТГ восстанавливает нормальную концентрацию
кальция во внеклеточной жидкости и сыворотке крови через прямое
влияние на кости, почки и опосредованное действие на слизистую
оболочку кишечника (стимуляция синтеза кальцитрирла). ПТГ увеличивает
скорость вымывания из костей как органических, так и неорганических,
компонентов, что обеспечивает перемещение ионизированного кальция
из костей в плазму крови. Гормон снижает экскрецию почками кальция,
что также повышает его содержание во внеклеточной жидкости и плазме^
Посредством стимуляции синтеза кальцитриола ПТГ увеличивает эффек-.
Патофизиология наиболее частых эндокринопатий
487
тивность всасывания Са2+ в кишечнике и тем самым поднимает уровень
содержания кальция во внутренней среде. Быстрее всего реализуется
эффект ПТГ на почки, но в основном уровень концентрации кальция
повышается вследствие влияния гормона на кости. Таким образом, за счет
вещества кости предотвращается развитие гипокалъциемии.
Для переноса катиона кальция и фосфатного аниона через слизистую
оболочку кишечника необходимы:
♦ захват и перенос ионов через мембрану щеточной каемки и
микроворсинок;
♦ транспорт ионов через обращенную в просвет кишечника часть
плазматической мембраны эпителиоцита;
♦ перенос ионов через базолатеральную часть наружной мембраны
эпителиоцита в интерстиций.
Очевидно, что кальцитриол активирует перенос кальция на одном или
более из этих трех этапов.
Если не происходит интенсивных изменений жесткости скелета в
ответ на возрастание или снижение нагрузки, то костная ткань находится в
состоянии динамического равновесия, которое характеризует
сбалансированность ее образования и резорбции. При этом костная ткань служит
огромным резервуаром кальция, который можно оперативно использовать
для удержания в нормальных пределах концентрации кальция во
внутренней среде. Около 1 % кальция скелета составляет его легкообмени-
вающийся пул. Еще 1 % содержит периостальное пространство. ПТГ
стимулирует транспорт кальция из периостального пространства через
мембрану, отделяющую внеклеточную жидкость от периостальной.
Усиливает данный вид трансмембранного переноса ионов и кальцитриол. Кальци-
тонин способен предотвращать этот эффект и уменьшает резорбцию
костной ткани.
ГИПЕРКАЛЬЦИЕМИЯ
Гиперкалъциемия - это патологическое состояние всего организма,
обусловленное патогенным ростом общего содержания во внеклеточной
жидкости кальция, который приводит к возрастанию общей концентрации
кальция в сыворотке крови до уровня более высокого, чем 10,5 мг/100 мл
(2,63ммолъ/л), и (или) увеличению содержания в сыворотке крови
ионизированного кальция до величины большей, чем 4,8мг/100мл (1,2ммоль/л).
Частота развития гиперкальциемий среди представителей основной
популяции варьирует от 0,05 до 0,6 %. В 70-80 % случаев гиперкальциемию
обуславливают злокачественные опухоли и первичный гиперпаратиреоз.
После внутривенной инфузии определенной массы Са2+ его
концентрация в сыворотке крови возвращается в нормальные пределы после
экскреции почками 10-15 % введенной массы кальция. Остальная часть
перемещается в кости скелета. В этой связи одной из функций костей следу-
488
Глава 28
ет считать функцию депо кальция, которое организм использует для
быстрой аккумуляции Са2+, предотвращающей патогенное возрастание
содержания ионизированного кальция в плазме (сыворотке) крови. Поэтому
гиперкальциемию следует рассматривать как результат дисбаланса между
интенсивностью функционирования остеобластов (действия остеобласти-
ческих факторов) и остеокластов (влияния факторов остеолиза) при
преобладании активности последних.
У 90 % больных с гиперкальциемией ее вызывают:
♦ первичный гиперпаратиреоз;
♦ злокачественные опухоли;
♦ гранулематозные заболевания, которые характеризует образование
множества гранулем (саркоидоз, туберкулез, гистоплазмоз).
При гранулематозных заболеваниях гиперкальциемия - это следствие
повышенного образования на системном уровне (витамин Б)-гормона
мононуклеарными фагоцитами, активированными в области гранулем.
У части больных гиперкальциемия представляет собой следствие
побочного действия диуретиков из группы тиазидов. Тиазиды обостряют
гиперкальциемию у больных с первичным гиперпаратиреозом, усиливая
реабсорбцию кальция в проксимальных и начальных отделах дистальных
канальцев нефрона.
Гиперкальциемия может быть одной из составляющих синдрома ги-
пертиреоза. Гиперкальциемия вследствие гипертиреоза обусловлена
усилением остеолитической активности под влиянием тиреоидных гормонов,
циркулирующих с кровью в повышенной концентрации. Кроме того,
известно, что гипертиреоз вызывает снижение секреции паратгормона, что
ведет к гипокальциемии.
Гиперкальциемия у больных с гипотиреозом - это результат
системного угнетения анаболических процессов в организме на уровне костной
ткани.
Отравление витамином D вызывает ятрогенную гиперкальциемию,
наиболее частую при многократном гемодиализе и у женщин, которым
витамин назначают для лечения остеомаляции.
У больных с хронической почечной недостаточностью и уремическим
синдромом гиперкальциемия выступает следствием вторичного и
третичного гиперпаратиреоза (гиперпаратиреоидизма) (см. главу, посвященную
патофизиологии почек).
Первичный гиперпаратиреоз - это следствие выхода из под контроля
системной регуляции секреции ПТГ одной или более паращитовидными
железами. Вторичный гиперпаратиреоз - патологическое состояние
вследствие роста секреции ПТГ в ответ на снижение концентрации
кальция во внеклеточной жидкости и плазме крови, обусловленное
хроническими заболеваниями почек (хронической почечной недостаточностью),
синдромом низкого кишечного всасывания и другими патологическими
состояниями, снижающим» содержание Са2+ во внеклеточной жидкости.
Как защитная реакция возрастание секреции ПТГ избыточно
относительно стимулов, его вызвавших. В результате реакция становится патоген-
Патофизиология наиболее частых эндокринопатий
489
ной, то есть не возвращает концентрацию кальция в плазме крови в
диапазон нормальных изменений, а вызывает гиперкальциемию.
Причины первичного гиперпаратиреоза - одна или более аденом па-
ращитовидных желез (90 % случаев первичного гиперпаратиреоза),
гиперплазия всех четырех желез (около 10 %), карцинома паращитовидной
железы (менее 1 %). Если первичный гиперпаратиреоз представляет
собой семейное заболевание, то он нередко сочетается с развитием
новообразований в других железах внутренней секреции, чаще всего с
опухолями гипофиза, щитовидной железы и надпочечников {синдром
множественной эндокринной неоплазии). Чаще первичный гиперпаратиреоз
развивается после менопаузы у пожилых женщин.
Ведущее звено патогенеза первичного гиперпаратиреоза - гиперкаль-
циемия. Многообразие функций, в регуляцию которых в качестве
эффекторов регуляггорных систем вовлечены ионы кальция, определяет
широкий спектр симптомов гиперкальциемии. Как синдром гиперкальциемию
составляют тошнота, анорексия, рвота, та или иная степень угнетения
высшей нервной деятельности и сознания вплоть до ступора и комы,
повышенная утомляемость, мышечная слабость, кальцификация роговицы,
пептические язвы желудка (результат избыточной секреции обкладочны-
ми клетками эпителия желудка, стимулом которой является повышенная
концентрация во внеклеточной жидкости ионов кальция), остеопения
(снижение плотности кости и уменьшение костной массы из-за
недостаточного образования остеоидной ткани), «барабанные палочки»
(состояние, приводящее к увеличению кончиков пальцев рук и ног, а также
блеску ногтей и их изогнутости по длиннику), боли в суставах,
кальцификация различных тканей, которая в почках может приводить к полиурии,
укорочение интервалов Q-T на электрокардиограмме.
Псевдогиперпаратиреоз (гиперкальциемия злокачественных
заболеваний) у 50 % больных связан с патологическим возрастанием
концентрации во внеклеточной жидкости и плазме крови пептида (протеина)
подобного паратиреоидному гормону (ПТГ-подобный пептид).
Концентрация ПТГ-подобного пептида в плазме крови находится на уровне более
низком, чем 2 пмоля/л, то есть меньше, чем 210"12 ммоль/л. У больных с
явным синдромом содержание ПТГ-подобного пептида в плазме крови
нередко больше, чем 20,9 ммоль/л. Следует заметить, что ПТГ-подобный
пептид, по-видимому, представляет собой нормальный молекулярный
эффектор системной эндокринной регуляции. Об этом свидетельствует
его высокая концентрация в женском молоке. ПТГ-подобный пептид
вырабатывают и высвобождают клетки карцином легких, молочной железы,
почек, яичников, клоны озлокачествленных клеток при миеломе, лимфо-
ме и саркоме.
Гиперсекреция ПТГ-подобного пептида - не единственное звено
патогенеза синдрома гиперкальциемии вследствие злокачественных опухолей
и заболеваний. Если малигнизация служит стимулом для системной
иммунной реакции, то повышенная секреция цитокинов лимфоцитами
активирует клетки системы мононуклеарных фагоцитов. В результате растет
490
Глава 28
образование кальцитриола, то есть (витамин 0)-гормона, на уровне всего
организма, что связано с его интенсивной продукцией активированными
мононуклеарами и вызывает гиперкальциемию. Более редкое звено
патогенеза синдрома - это прямая резорбция кальция в кровь из
злокачественной опухоли с высоким содержанием кальция в составляющих ее тканях.
Кроме того, гиперкальциемия у больных со злокачественными опухолями
может быть следствием патогенно избыточного образования простаглан-
дина Е2, повышающего резорбцию кальция из кости через усиление
функционирования остеокластов. Гиперкальциемию вследствие
злокачественных опухолей связывают с увеличенным образованием таких цито-
кинов как фактор активации остеокластов, интерлейкин-1, а также альфа-
и бета факторы некроза опухолей.
ГИПОКАЛЬЦИЕМИЯ
Гипокалъциемыя - это снижение концентрации кальция во
внеклеточной жидкости и жидкой части плазмы крови, о котором
свидетельствует падение общей концентрации кальция в сыворотке крови до
уровня более низкого, чем 8,5 мг/дл (2,13 ммоль/л) при нормальной
концентрации в ней альбумина.
Снижение концентрации в сыворотке крови альбумина как протеина
плазмы крови, связывающего и переносящего кальций, приводит к
снижению общей концентрации кальция в сыворотке без изменения ее
наиболее биологически активной составляющей, то есть содержания в
сыворотке крови ионизированного кальция. Гипокальциемию вследствие ги-
поальбуминемии называют псевдогипокалъциемией.
Низкое содержание магния во внеклеточной жидкости и жидкой части
плазмы крови приводит к гипокальциемии через падение секреции пара-
тгормона и снижение физиологической реакции костной ткани на
действие гормона паращитовидных желез.
Острый респираторный алкалоз увеличивает связывание кальция
альбумином, снижая концентрацию ионизированного кальция в сыворотке крови.
Дефицит витамина D обуславливает гипокальциемию через падение
кишечного всасывания кальция. Следует заметить, что снижение
кишечного всасывания любого происхождения может приводить к
гипокальциемии. У больных с хронической печеночной недостаточностью и
низким числом нормальных гепатоцитов гипокальциемия может быть
следствием падения синтеза в печени 25-гидроксивитамина D как субстрата
образования в почках 1,25-дигидроксивитамина D, то есть (витамин D)-
гормона, повышающего кишечное всасывание ионизированного кальция.
Хроническая почечная недостаточность как состояние, в основе которого
лежит потеря почками значительного числа своих нормальных клеточных
элементов, сопровождается падением синтеза (витамин Э)-гормона, что у
части больных обуславливает гипокальциемию. У больных с нефротиче-
Патофизиология наиболее частых эндокринопатий
491
ским синдромом потери 25-гидроксивитамина D с мочой ведут к гипо-
кальциемии. Гипокальциемию вследствие потерь 25-гидроксивитамина D
может обусловить и блокада его физиологической циркуляции между
просветом кишечника в печень и обратно при различных болезнях
кишечника, приводящих к падению кишечного всасывания.
Дефицит секреции гормона паращитовидных желез (гипопаратиреоз)
как причина гипокальциемии чаще всего является следствием удаления
желез во время операций на щитовидной железе, ее амилоидоза или
может быть идиопатическим, сочетаясь с аплазией вилочковой железы
(синдром DiGeorge).
Псевдогипопаратиреоз (болезнь Альбрихта) - патологическое
состояние, которую характеризуют гипокальциемия и аномальное содержание
во внеклеточной жидкости и жидкой части плазмы крови фосфатного
аниона из-за патологической резистентности соответствующих клеток
мишеней по отношению к действию на них гормона паращитовидных
желез. В результате постоянной стимуляции секреции паращитовидных
желез гипокальциемией у всех пациентов повышена концентрация пара-
тгормона в крови, а у части больных выявляют гипертрофию
паращитовидных желез. Ареактивность клеток-мишеней по отношению к действию
паратгормона приводит к низкой активности остеокластов и снижает
выделение фосфата почками.
Псевдогипопаратиреоз первого типа встречается чаще и как
моногенная болезнь наследуется по аутосомно-доминантному механизму или
вместе с Х-хромосомой. В его основе лежит отсутствие физиологической
реакции активации системы аденилатциклазы. Так как
псевдогипопаратиреоз наследуется вместе с Х-хромосомой, то частота данной моногенной
болезни у женщин выше, чем у мужчин. Как синдром этот тип псевдоги-
попаратиреоза характеризуют задержка умственного развития, круглое
лицо, аномалии развития зубов и др. При псевдогипопаратиреозе второго
типа нарушена реакция клетки на рост содержания в ее цитозоле
циклического аденозинмонофосфата. При втором типе псевдогипопаратиреоза
туловище и конечности укорочены, лицо круглое. Кроме того, у больных
выявляют брахидактилию (короткопалость), тетанию (перемежающиеся
судороги), а также локусы остеопении (деминерализации костей).
Если больному без псевдогипопаратиреоза парентерально вводят
экзогенный паратгормон, то реакция системы аденилатциклазы тубулярных
эпителиоцитах в почках приводит к росту концентрации циклического
аденозинмонофосфата в конечной моче. У пациента с псевдогипопарати-
реоом этого не происходит.
У части больных гипокальциемия - это следствие связывания кальция
во внеклеточном пространстве фосфатными анионами, которые патогенно
избыточно высвобождаются во внеклеточный сектор при реперфузии
некробиотически измененных мышц у больных с синдромом длительного
раздавливания. Синдром лизиса злокачественных опухолей вследствие
высвобождения внутриклеточных фосфатных анионов погибающими
злокачественными клетками может включать гипокальциемию. Кроме того,
492
Глава 28
рост концентрации фосфатных анионов во внеклеточном секторе как
причина гипокальциемии может быть следствием хронической почечной
недостаточности с крайне выраженным падением экскреции фосфатного
аниона почками.
Когда метастазирование опухолей, клеточные элементы которых
обладают остеобластической активностью (рак предстательной железы и
др.), достигает определенной степени распространенности, то опухоль и
ее метастазы начинают задерживать столько кальция, что развивается ги-
покальциемия.
Кальцификация тканей в ретроперитонеальном пространстве, некро-
биотически измененных вторичной альтерацией при остром панкреатите,
также может быть причиной гипокальциемии.
Гипокальциемию как синдром составляют проявления повышенной
возбудимости нейронов, миоцитов произвольных мышц и облегченного
проведения возбуждения через нейромышечные синапсы вследствие
низкого содержания кальция в межклеточных пространствах и в синаптиче-
ской щели, а также из-за роста там содержания фосфатного аниона:
♦ симптом Хвостека, то есть сокращение мышц лица в ответ на удар
молоточком в области прохождения лицевого нерва при тетании,
одностороннем спазме мышц;
♦ симптом Труссо - тоническая судорога кисти в ответ на сдавление
в области плеча как симптом скрытой тетании;
♦ судороги;
♦ болезненные непроизвольные сокращения отдельных мышц;
♦ фотофобия и др.
Снижение концентрации ионизированного кальция может быть
следствием массивного переливания донорской крови, в результате которого
свободные ионы кальция связываются во внеклеточном секторе
экзогенными цитратными анионами. В данном случае особенно опасно такое
патологическое следствие гипокальциемии как артериальная гипотензия.
Гипокальциемия через снижение содержания кальция в цитозоле
рабочих и проводящих кардиомиоцитов приводит к падению сократимости
сердца, застойной сердечной недостаточности, артериальной гипотензии,
увеличению интервала Q-T, нарушениям внутрижелудочковой
проводимости, а также ареактивности сердца по отношению к действию
сердечных гликозидов.
ПАТОГЕНЕЗ ГИПОГОНАДИЗМА У МУЖЧИН
Как синдром гипогонадизм у мужчин характеризуют бесплодие,
потеря полового влечения, импотенция, снижение массы скелетных мышц,
а также уменьшение количества андроген-зависимых волос.
В основе гипогонадизма лежат нарушения образования
сперматозоидов в семявыносящих канальцах и (или) расстройства секреции тестосте-
Патофизиология наиболее частых эндокринопатий
493
рона клетками Лейдига. При этом дисфункции на уровне семявыносящих
канальцев обуславливают бесплодие, а недостаточная секреция
тестостерона служит причиной дефектов развития и недостаточной выраженности
вторичных половых признаков.
У мужских половых желез (яичек, семенников) есть две главные
функции: а) синтез и секреция мужских половых гормонов; б)
сперматогенез, то есть образование и развитие сперматозоидов. При этом
достаточный уровень секреции тестостерона представляет собой необходимое
условие нормальных либидо и половой потенции мужчины. Если
образующие тестостерон клетки Лейдига относительно резистентны по
отношению к механическим повреждениям и вторичной альтерации при
воспалении, то образующий сперматозоиды канальцевый эпителий
исключительно чувствителен к таким повреждающим клетки воздействиям. Браки,
не приносящие потомства, могут быть следствием повреждения каналь-
цевого эпителия и расстройств сперматогенеза, которые чаще всего
связаны с крипторхизмом, воспалением того или иного генеза, а также
травмой семенников. У таких больных мужчин, страдающих от потери
способности к зачатию, объем яичек находится в нормальных пределах (12-
25 мл), а сохраненная функция клеток Лейдига обуславливает
нормальные либидо и способность к половому сношению. При потере обеих
семенников (кастрация) или их повреждении, разрушающем не только
канальцевый эпителий но и клетки Лейдига, больной перестает
испытывать половое влечение и становится импотентом.
Недостаточность тестикулярных функций, возникшая в период
онтогенеза до полового созревания, приводит к недостаточным развитию
половых органов и выраженности вторичных половых признаков.
Половой член и яички у больных с недостаточностью тестикулярной функции,
возникшей до периода полового созревания, остаются маленькими
несмотря на то, что данный период уже должен наступить. При этом в
семявыносящих канальцах отсутствуют сперматозоиды. На лице и других
частях тела почти незаметно волос. Голос остается высоким, его «ломки»,
связанной с половым созреванием, не происходит. Так как недостаточная
секреция тестостерона обуславливает задержку эпифизарного закрытия,
то больные приобретают характерный облик евнуха, при котором сразу
заметны непропорционально длинные руки и ноги.
Первыми симптомами недостаточности тестикулярных функций,
возникшей после полового созревания, выступают потеря полового
влечения и половое бессилие. Затем к ним присоединяется постепенное
обратное развитие вторичных половых признаков, в частности замедление
роста волос на лице и других частях тела, а также снижение массы скелетных
мышц.
Если гипогонадизм представляет собой следствие недостаточной
секреции гонадотропинов, то его называют гипогонадотропным. Такой
гипогонадизм - это результат недостаточной секреции лютеинизирующего и
фолликулостимулирующего гормонов как причины патологически низкой
секреции тестостерона, вызывающей синдром гипогонадизма (евнухои-
494
Глава 28
дизма). В частности от гипогонадотропного гипогонадизма страдают
больные с синдромом Кальмана, при котором выявляют недоразвитие
обонятельных долей головного мозга, отсутствие обоняния (аносмию), а
также врожденное расщепление неба. В основе гипогонадотропного
гипогонадизма при синдроме Кальмана лежит врожденная недостаточность
(отсутствие) синтеза гонадолиберина в гипоталамусе.
Если возраст больного с гипогонадизмом меньше, чем 20 лет, то
вывод об эндокринопатии как о причине задержки полового созревания
может быть преждевременным. Дело в том, что в отдельных случаях
половое созревание завершается в возрасте 20 лет.
Одной из причин гипогонадотропного гипогонадизма является панги-
попитуитаризм.
Гипергонадотропный гипогонадизм - это патологическое состояние,
для которого в первую очередь характерно недоразвитие или
приобретенное патологическое изменение яичек, в результате которого
возникает дефицит тестостерона, обуславливающий постоянную
гиперсекрецию фолликулостимулирующего и лютеинизирующего гормонов.
Наиболее частая причина гипергонадотропного гипогонадизма - это
врожденное недоразвитие яичек при синдроме Кляйнфельтера (карио-
тип 47, XXY у 80 % больных). Яички у больных с синдромом
недоразвиты. Их длина не превышает 2 см. При синдроме Кляйнфельтера
происходит гиалинизация семявыносящих канальцев и азооспермия (отсутствие в
сперме подвижных сперматозоидов). Не у всех пациентов с синдромом
Кляйнфельтера клетки Лейдига теряют функцию образования
тестостерона. Поэтому евнухоидизм не всегда является составляющей синдрома. У
всех пациентов выявляют гинекомастию и повышенную секрецию
фолликулостимулирующего и лютеинизирующего гормонов. Секреция гона-
дотропинов у части больных с синдромом повышена, несмотря на
достаточную секрецию тестостерона. Недостаточное развитие когнитивных
функций выявляют у 25 % больных.
Врожденное недоразвитие яичек также может быть причиной
гипергонадотропного гипогонадизма. При этом яичек не находят ни в мошонке
ни в паховых каналах. Потеря яичек происходит после 7-14 недель
беременности. Отсутствие до этого срока в организме плода тестикулярных
гормонов приводит к формированию у больных женского фенотипа.
Орхит при свинке приводит к деструкции зародышевых клеток как
причине азооспермии. При этом секреция тестостерона обычно не
претерпевает патологических изменений.
Крипторхизм может быть причиной гипергонадотропного
гипогонадизма, так как повреждает яички через их постоянную травматизацию,
связанную со сдавлением и растяжением.
У больных с синдромом миотонической дистрофии
(наследственное заболевание) также выявляют гипергонадотропный гипогонадизм
вследствие атрофии яичников. Кроме того, синдром характеризуют мио-
тония (затруднение расслабления мышц после их сильного сокращения) и
катаракты.
Патофизиология наиболее частых эндокринопатий
495
Из других причин гипергонадотропного гипогонадизма у мужчин
следует выделить лучевую терапию злокачественных опухолей
соответствующей локализации, осложнения хирургических вмешательств,
гонорейный орхит, а также травму яичников.
ПАТОГЕНЕЗ ГИПОГОНАДИЗМА У ЖЕНЩИН
Эндокринопатий, обуславливающие нарушения репродуктивной
функции у женщин, обычно приводят к аменорее, то есть к отсутствию или
аномальному прекращению месячных.
Первичная аменорея - это аменорея у женщин старше восемнадцати
лет при наличии признаков полового созревания и отсутствии в анамнезе
хотя бы одной менструации.
Вторичная аменорея - аменорея при наличии менструаций в анамнезе.
Синдром Тернера - это следствие первичного гипогонадизма у
больных с женским фенотипом, который характеризуют недостаточное
развитие гонад, аменорея, задержка роста тела, а также недостаточная
выраженность вторичных половых признаков. Кроме того, при данном синдроме
выявляют разнообразные врожденные дефекты строения тела, в частности
коарктацию аорты, а также перепончатость шеи, то есть врожденный
дефект ее строения, при которой смежные структуры области шеи
соединяются между собой широкой полосой ткани, состоящей из низкодифферен-
цированных клеточных элементов. Обращают на себя внимание большие и
как бы распластанные на поверхности груди соски молочных желез, а
также родимые пятна у таких больных. В первые недели жизни при синдроме
выявляют отеки нижних и верхних конечностей, связанные с
дисфункциями лимфатических сосудов. У больных с данным синдромом, как и в
других случаях гипергонадотропного гипогонадизма, высокий уровень
секреции гонадотропинов сочетается с низкой концентрацией метаболитов
эстрогенов в моче. Чаще всего синдром Тернера, который выявляют у одного
из десяти тысяч младенцев, вызывается кариотипом 45,X.
Известно, что у больных с синдромом Тернера часто развивается
аутоиммунный тиреоидит Хашимото. Еще чаще у таких больных в
циркулирующей крови выявляют аутоантитела к аутоантигенам тиреоглобу-
лина. При этом у родителей пробанда частота циркуляции с кровью ауто-
антител к тиреоидным аутоантигенам выше, чем у представителей
основной популяции. Это заставляет предположить наличие еще непонятной
связи между данной предрасположенностью к аутоиммунным
заболеваниям у родителей и синдромом Тернера у детей.
У меньшинства больных с синдромом Тернера общее число хромосом
является нормальным, и нормальна одна половая Х-хромосома.
Структура другой Х-хромосомы аномальна. Предполагают, что она представляют
собой результат патогенного деления хромосомы в точке центромеры, в
результате которого образуются две аномальные хромосомы. При этом
496
Глава 28
деструкции не подвергается только аномальная хромосома (изохромосо-
ма), которую составляют два длинных плеча Х-хромосомы. Кроме того,
Х-изохромосома образуется в результате транслокации, то есть
структурного изменения хромосом в результате обмена двумя сегментами между
негомологичными хромосомами. У части больных с синдромом Тернера
образование Х-изохромосомы может быть следствием дупликации, то
есть хромосомной аберрации, возникающей в результате неравного крос-
синговера или обмена сегментами между двумя гомологичными
хромосомами. При дупликации одна хромосома теряет небольшой сегмент, а
другая получает его. Хромосома, получившая сегмент, дуплицируется, а
ее гомолог подвергается делеции, то есть удаляется из генома. У части
больных с синдромом Тернера одна Х-хромосома нормальна, а вторая
представляет собой продукт делеции. Делеция - регистрируемая цитоге-
нетически, либо методами молекулярной генетики потеря части
генетического материала.
У части больных с синдромом Тернера среди соматических клеток
выделяют клеточные линии, которые отличаются друг от друга карио-
типом. Известны следующие варианты сочетаний кариотипов клеточных
линий у больных с данным синдромом: 45,Х и 46,ХХ; 45,Х,46,ХХ и
47,ХХХ (три клеточные линии) и др. Синдром Тернера у больных с
такими сочетаниями кариотипов клеточных линий определяют как
вариант данного синдрома. При всех вариантах синдрома у больных есть
задержка роста тела и недостаточное развитие половых органов. Данное
явление получило название мозаицизма кариотиров клеточных линий и
послужило причиной гипотезы патогенеза синдрома, апологеты
которой считают, что хромосомная аберрация возникает не в результате
неразъединения половых хромосом при образовании гамет как
причины аномального набора хромосом в диплоидных клетках (зиготах), а
является результатом хромосомных аберраций, возникших после
образования зигот.
Синдром тестикулярной феминизации выявляют у мужчин с карио-
типом 46,XY. Так как у таких больных гениталии представляют собой
внешние женские половые органы почти нормального строения, то их
воспитывают как девочек. В основе синдрома лежит патологическая аре-
активность клеток-мишеней по отношению к действию андрогенов. Во
время внутриутробного развития таких больных их яички образуют и
секретируют тестостерон, но так как нормальная реакция вольфовых
каналов и тканей гениталий на действие тестостерона отсутствует, то
дифференциация внешних половых органов идет по женскому варианту. При
этом яички плода не теряют способности секретировать фактор
тормозящий развитие мюллеровых протоков, из которых у женщин образуются
часть влагалища и маточные трубы. В результате из соответствующей
структуры эмбриона и плода не развиваются фаллопиевы трубы, матка и
верхняя часть влагалища.
В основе синдрома резистентных яичников (частный случай гиперго-
надотропного гипергонадизма) лежит угнетение или отсутствие физиоло-
Патофизиология наиболее частых эндокринопатий
497
гических реакций яичников на действие гонадотропинов, которое может
быть результатом аутоиммунного поражения женских гонад.
Гипогонадотропный гипогонадизм у женщин, то есть дисфункции,
патологические изменения и (или) недоразвитие женских половых органов
и желез при низкой секреции гонадотропинов, может быть результатом
пангипопитуитаризма вследствие деструктивных поражений
соответствующих клеточных элементов гипоталамуса и (или) гипофиза. В данном
случае гипогонадотропный гипогонадизм может приводить как к
первичной, так и ко вторичной аменорее в зависимости от того, в какой период
онтогенеза развивается пангипопитуитаризм. Патологически низкий
уровень секреции только гонадотропинов без падения уровня секреции
других гормонов аденогипофиза как причина гипогонадотропного гипогона-
дизма обычно связан с дефектом синтеза гипоталамического гонадолибе-
рина, что при синдроме Кальмана нередко сочетается с аносмией.
Задержку полового созревания у девочек можно подозревать тогда,
когда менструальный цикл не устанавливается до шестнадцатилетнего
возраста. При этом нередко сбор анамнеза позволяет выявить задержку
полового созревания у матери. В таком случае вероятность того, что
половое созревание еще наступит, достаточно велика.
Наиболее частая причина вторичной аменореи - это гипоталамическая
(психогенная, функциональная, идиопатическая) аменорея, при которой
временная потеря способности к деторождению может быть связана с
отрицательным эмоциональным стрессом (у части больных психогенный
характер патогенеза аменореи не подтверждается). Полагают, что у
некоторых больных данный вид аменореи связан с нарушениями секреции го-
надолиберина.
Как половое созревание женщины, так и нормальный менструальный
цикл возможны лишь в том случае, если масса тела женщины превышает
определенный уровень. Если масса тела падает ниже этого уровня в
результате голодания, то возникает аменорея.
Известно, что у 50 % балерин интенсивные физические нагрузки
приводят к аменорее, патогенез которой состоит по-видимому в нарушениях
эндокринной регуляции детородной функции на уровне гипоталамуса.
Нередко аменорея, как первичная, так и вторичная, составляет ряд
синдромов в основе которых в частности лежит патогенно избыточная
секреция андрогенов (инсулин-резистентного черного акантоза и др.,
смотри главу, посвященную сахарному диабету). При этом ряде
синдромом нарушения овуляции приводят к поликистозу яичников, избыток
андрогенов к гирсутизму (через рост числа андроген-зависимых волос), и
нередкими являются аутоиммунные поражения, в частности приводящие
к сахарному диабету.
Глава 29
РЕАКЦИИ ПОВЫШЕННОЙ ЧУВСТВИТЕЛЬНОСТИ
Реакция системы иммунитета в ответ на антигенную стимуляцию
может обусловить патологические изменения органов и тканей своего
организма.
РЕАКЦИИ ПОВЫШЕННОЙ ЧУВСТВИТЕЛЬНОСТИ
ПЕРВОГО ТИПА
Реакции повышенной чувствительности первого типа (анафилаксия,
гр. ana - вновь + aphylaxis беззащитность) - это следствие
образования гомоцитотропных антител (иммуноглобулины G и Е), Fc-фрагмент
которых представляет собой лиганду к Fc-рецептору наружной
поверхности тучных клеток и базофилов. В результате гомоцитотроп-
ные антитела фиксируются на поверхности данных клеток.
Экзогенный антиген, вызывающий образование гомоцитотропных антител,
называют аллергеном.
Аллерген вступает во взаимодействие с фиксированными на
клеточной поверхности гомоцитотропными антителами, что активирует базо-
филы и тучные клетки. Активированные тучные клетки и базофилы
высвобождают биоактивные вещества, медиаторы анафилаксии (рис. 29.1).
Системное действие медиаторов анафилаксии складывается из роста
проницаемости эндотелия капилляров, интерстициального отека,
сокращения гладкомышечных элементов стенок бронхов и увеличения
секреции клетками слизистых оболочек. Анафилаксия во многом составляет
патогенез таких болезней и патологических состояний как
аллергическая бронхиальная астма, аллергические риниты, крапивница,
анафилактический шок.
Общая (генерализованная) анафилаксия - это аллергическая реакция
первого типа, которая обуславливает возникновение критического
шокового состояния (анафилактического шока) в течение нескольких минут
после взаимодействия аллергена и антитела.
Местная анафилаксия - это патологическое изменение отдельных
органов или тканей (бронхиальное дерево, желудочно-кишечный тракт,
слизистая оболочка носа, кожа) в результате реакции повышенной
чувствительности I типа. В эксперименте эти изменения можно воспроизвести,
вводя в ткани сыворотку крови сенсибилизированного донора,
содержащую гомоцитотропные антитела. Последующее введение в ткани
аллергена ведет к высвобождению из тучных клеток медиаторов анафилаксии и
локальному отеку.
Реакции повышенной чувствительности
499
эпитоп антигена
Fc-фрагмент молекулы
иммуноглобулина Е Д,, J^ ^ паратоп иммуноглобулина
Fc-рецептор наружной
клеточной поверхности
♦ гистамин ^~ ^ — .
. лейкотриены ^<**. ' N поверхность тучной клетки
♦ фактор активации тромбоцитов
♦ хемотаксические факторы эозинофилов и нейтрофилов
♦ др. медиаторы анафилаксии
Рис. 29.1. Образование «мостика» из иммуноглобулинов Е
и носителя антигенных детерминант как причина дегрануляции тучных клеток
К гомоцитотропным антителам относят иммуноглобулины G и Е. При
некоторых обстоятельствах в анафилактическую реакцию оказываются
вовлеченными иммуноглобулины А и М. Иммуноглобулины G начинают
участвовать в анафилактической реакции через 10-14 дней после
сенсибилизации. Иммуноглобулины Е проявляют себя как гомоцитотропные
антитела гораздо раньше.
Гомоцитотропные антитела обладают способностью фиксации на
клеточной поверхности определенных дифференцированных клеток организма
человека. Это определяет возможность пассивной сенсибилизации как
механизма выработки способности к анафилаксии. При пассивной
сенсибилизации переливанием донорской плазмы, содержащей в качестве гомоцито-
тропных антител иммуноглобулины G, латентный период до выработки
способности к анафилаксии составляет 3-6 ч. Такая сенсибилизация
неустойчива и подвергается обратному развитию за 12-24 ч, в течение которых
иммуноглобулины G отсоединяются от клеток способных к образованию и
высвобождению медиаторов анафилаксии. Пассивная сенсибилизация
трансфузией плазмы сенсибилизированного донора, которая в качестве го-
моцитотропных антител содержит иммуноглобулины Е, по латентному
периоду не отличается от пассивной сенсибилизации переливанием
донорской плазмы с гомоцитотропными антителами из класса
иммуноглобулинов А, но более устойчива и становится максимально выраженной в
течение 24-72 ч после трансфузии. Поэтому анафилаксия, которая развивается
через 48-72 ч после сенсибилизации, связана с фиксацией на поверхности
тучных клеток и базофилов только одного вида антител, принадлежащих
классу иммуноглобулинов Е. Дело в том, что к этому времени на
поверхности тучных клеток остаются фиксированными только эти
иммуноглобулины. Продолжительность устойчивой фиксации иммуноглобулинов Е на
поверхности тучных клеток и базофилов составляет 6 недель.
Базофилы и тучные клетки обладают особо выраженным сродством по
отношению к иммуноглобулинам Е. Это в частности проявляется тем, что
500
Глава 29
данные гомоцитотропные антитела плотно усеивают наружную
поверхность клеток, образующих и высвобождающих медиаторы анафилаксии.
После ингаляции, инъекции аллергена или после его попадания в
организм с пищей и питьем для развития анафилаксии необходима его
миграция к сенсибилизированным клеткам через многочисленные барьеры.
Аллерген, соединяясь с двумя антиген-связывающими участками
иммуноглобулинов Е на наружной клеточной мембране, образует «мостик»,
деформирующий наружную клеточную мембрану (рис. 29.1). Растяжение
плазматической мембраны сенсибилизированных клеток запускает каскад
биохимических реакций, приводящий к высвобождению медиаторов анафилаксии.
Быстрее всего высвобождение медиаторов анафилаксии из
сенсибилизированных клеток вызывает умеренный избыток аллергена вблизи
наружной клеточной поверхности. Значительный избыток аллергена
ослабляет высвобождение медиаторов, так как приводит к большей частоте
связывания одного аллергена с одним иммуноглобулином, то есть к
меньшему образованию «мостиков» на поверхности тучной клетки или
базофила. Аллерген определенных размеров, в наибольшей степени
подходящих для его связывания двумя гомоцитотропными антителами и
образования мостика, быстрее вызывает более сильную анафилаксию.
Локальная анафилаксия быстрее возникает в тех тканях, где быстрее
накапливается аллерген. Так при ингаляционном пути попадания
аллергена во внутреннюю среду аллерген прежде всего накапливается в
слизистых оболочках носа и дыхательных путей. Там аллерген
взаимодействует с гомоцитотропными антителами, фиксированными на поверхности
тучных клеток, что приводит к высвобождению медиаторов анафилаксии.
В результате растет секреция эпителия дыхательных путей, развивается
его отек и возникает спазм бронхов. Все это расстраивает альвеолярную
вентиляцию по обструктивному типу. Такова классическая схема
патогенеза аллергической бронхиальной астмы.
При энтеральном пути попадания в организм аллергена первыми
медиаторы анафилаксии высвобождают тучные клетки подлизистого слоя
желудочно-кишечного тракта. Это стимулирует моторику желудка и
кишечника. Симптомами анафилаксии, преимущественно происходящей в
желудке и кишечнике, являются кишечные колики и диспепсия,
ощущения тяжести в животе и его вздутия. Локальная анафилаксия, патогенез
которой зависит от пути попадания в организм аллергена, всегда
приводит к системной реакции той или иной степени выраженности.
О реакции поздней фазы и анафилактоидной реакции читатель может
узнать из главы, посвященной патогенезу бронхиальной астмы.
Представлятеся возможным выделить следующие патогенетически
обоснованные принципы профилактики и терапии анафилаксии:
♦ Предупреждение контакта с потенциальными аллергенами (не
кормить новорожденных коровьим молоком, избегать контактов с
кошками, собаками, попугаями и др.).
♦ Десенсибилизация посредством неоднократного введения
очищенного аллергена, которая предположительно происходит через а) индук-
Реакции повышенной чувствительности
501
цию и стимуляцию выработки иммуноглобулинов G, которые,
связывая аллерген, предотвращают дегрануляцию тучных клеток и б)
выработку иммунологической толерантности по отношению к
аллергену на уровне Т-лимфоцитов, в частности посредством образования
клона соответствующих Т-супрессоров.
♦ Угнетение продукции гомоцитотропных антител гаптенами,
связанными с тимус-независимыми носителями.
♦ Использование экзогенных лиганд, которые, связываясь с
рецепторами наружной клеточной поверхности тучных клеток,
предотвращают их дегрануляцию.
При анафилактическом шоке массивное высвобождение бронхоконст-
рикторов и вазодилятаторов тучными клетками ведет к обструктивным
расстройствам внешнего дыхания, к которым быстро присоединяется
расширение емкостных и резистивных сосудов как причина острой и
приводящей к терминальному состоянию артериальной гипотензии. В этой
связи рациональными патогенетическими принципами терапии
анафилактического шока следует считать:
♦ Использование прямых адреномиметиков, которые через
возбуждение бета-один-, бета-два и альфа-один-адренорецепторов вызывают
рост частоты сердечных сокращений, повышают их силу,
расширяют бронхи, устраняя обструктивное расстройство альвеолярной
вентиляции, а также обуславливают спазм ^ен (рост преднагрузки
сердца) и сужение рестриктивных сосудов.
♦ Внутривенное введение массивной дозы препаратов на основе
гормонов коры надпочечников; эти средства за счет своего еще не
вполне ясного эффекта снижают интенсивность дегрануляции
тучных клеток как причины анафилактического шока. Кроме того,
препараты из ряда кортикостероидов, оказывая на адренорецепторы
пермиссивное действие, повышают чувствительность адренорецеп-
торов сосудистого русла и бронхов к действию адреномиметиков,
что повышает эффективность использования прямых
адреномиметиков для устранения анафилактического шока.
♦ Инфузия плазмозамещающих растворов, мобилизующих жидкость из
интерстициального сектора и клеток для роста преднагрузки сердца с
целью экстренного повышения минутного объема кровообращения.
При этом нельзя инфузировать внутривенно те растворы,
действующее начало которых является аллергеном (декстраны и др.).
РЕАКЦИИ ПОВЫШЕННОЙ ЧУВСТВИТЕЛЬНОСТИ ВТОРОГО ТИПА
(ГУМОРАЛЬНЫЕ ЦИТОТОКСИЧЕСКИЕ РЕАКЦИИ)
Основу реакций повышенной чувствительности второго типа
составляет гибель клеток, связанная с образованием на их поверхности
комплексов антиген- антитело. Такие клетки могут разрушаться в результате
502
Глава 29
межклеточных взаимодействий с мононуклеарными фагоцитами, наружная
поверхность которых содержит рецепторы к Fc-фрагментам
иммуноглобулинов G и соответствующей части молекулы фракции системы комлемента
СЗЬ. При этом иммуноглобулины G и фракция системы комплемента
функционируют в качестве опсонинов (см. главу, посвященную
воспалению). Кроме того, при реакциях повышенной чувствительности второго
типа клетки разрушаются в результате активации системы комплемента по
классическому пути, начальным этапом которой является образование на
поверхности атакуемой клетки комплекса антиген-антитело. Клетки с
фиксированными на их поверхности иммуноглобулинами G при реакциях
повышенной чувствительности второго типа могут также уничтожаться через
взаимодействие с нейтрофилами, а также К-клетками.
Одним из клеточных эффекторов реакций повышенной
чувствительности второго типа являются К-клетки (киллеры). Это большие
гранулярные лимфоциты, через функционирование которых происходит антитело-
зависимая и не связанная с активацией системы комплемента реакция
цитолиза клеток, покрытых иммуноглобулинами G. В основе цитолитиче-
ского действия К-клеток лежит внедрение в наружную клеточную
мембрану атакуемой клетки белкового полимера киллеров перфорина. Пер-
форин содержат «маленькие темные органеллы» киллеров.
Полимеризацию субстратов образования перфорина внутри киллеров и перфорацию
полимером их плазматической мембраны предотвращает белок протек-
тин, который содержат К-клетки. В результате перфорации наружной
клеточной мембраны атакуемой клетки перфорином в нее из
внеклеточного сектора устремляются катионы натрия и вода. Отек клетки и рост
давления в ней инициируют неконтролируемый процесс клеточной
гибели. К-клетки участвуют в уничтожении и элиминации патогенных
микроорганизмов, а также малигнизированных и инфицированных клеток,
принадлежащих организму больного.
Реакции повышенной чувствительности второго типа во многом
составляют патогенез системной реакции в ответ на переливание крови
несовместимой с организмом реципиента. Они обуславливают связаннное с
генерацией соответствующих иммуноглобулинов В-лимфоцитами реципиента
разрушение пересаженных тканей. При гемолитической болезни
новорожденных эритроциты младенца разрушаются после фиксации на
поверхности красных кровяных клеток антител матери к эритроцитарным резус-
антигенам ребенка. При аутоиммунных заболеваниях, приводящих к
гемолитической анемии и разрушению базальных мембран почечных
клубочков, также действует механизм реакций повышенной чувствительности
второго типа. Когда молекулы лекарственных средств или продукты их
биотрансформации присоединяются к поверхности эритроцитов или
тромбоцитов в качестве гаптенов, то антигенная стимуляция системы
иммунитета приводит к гуморальной цитотоксической реакции, которая
обуславливает гемолиз и разрушейие кровяных пластинок. Таково ведущее звено
патогенеза такой ятрогенной болезни как гемолитическая анемия,
представляющей собой побочный эффект фенацетина и других лекарственных средств.
Реакции повышенной чувствительности
503
РЕАКЦИИ ПОВЫШЕННОЙ ЧУВСТВИТЕЛЬНОСТИ
ТРЕТЬЕГО ТИПА (ОБРАЗОВАНИЕ ИММУННЫХ КОМПЛЕКСОВ)
Аллергические реакции третьего типа обусловлены циркуляцией с
кровью и накоплением во внеклеточных пространствах иммунных
комплексов антиген-антитело. Отложение значительного количества
иммунных комплексов во внеклеточных пространствах тканей ведет к быстрой и
интенсивной активации в них системы комплемента по классическому
пути. При активации системы комплемента высвобождаются ее фракции
со свойствами хемоаттрактантов и медиаторов воспаления (см. главу,
посвященную воспалению). Это ведет к выходу в ткани из кровяного русла
полиморфо- и мононуклеаров, а также лимфоцитов в качестве клеточных
эффекторов воспаления. Реализация алгоритма воспаления как сугубо
патогенной реакции повреждает ткани и разрушает органы-эффекторы. В
частности в местах отложения иммунных комплексов происходит
агрегация тромбоцитов с образованием микротромбов и выделением вазоактив-
ных аминов. Кроме патогенного воспаления, повреждения тканей
обуславливает так называемый реактивный лизис, когда активированные
фракции системы комплемента С5-С7, вступая в соприкосновение с
поверхностью соседней клетки, связывают фракции системы комплемента
С8 и С9, активация которых служит причиной цитолиза.
В местах преципитации иммунных комплексов активация мононукле-
арных фагоцитов крови и тканей весьма устойчива, так как после эндоци-
тоза мононуклеарам не сразу удается лизировать комплексы антиген-
антитело.
При высоком уровне содержания антител в сыворотке крови
преципитат комплексов антиген-антитело образуется в месте проникновения
антигена во внутреннюю среду организма. Давно известна кожная реакция,
в основе которой лежит действие данного механизма
гиперчувствительности (реакция Артюса). Морис Артюс обнаружил, что внутрикожное
введение растворимого антигена кроликам с высоким содержанием в
крови антител, образующих преципитаты вместе с данным антигеном,
вызывает эритему кожи и ее отек, которые достигают максимума через 3-8 ч.
В последующем отек и эритема подвергаются обратному развитию.
Реакции повышенной чувствительности третьего типа возникают в
результате длительной стимуляции системы иммунитета избытком
антигена. Длительную стимуляцию системы иммунитета антигеном
обуславливают хронические инфекции или потеря иммунологической
толерантности к аутоантигенам. Длительная стимуляция системы иммунитета
аллергеном при его небольшом избытке относительно массы антител и числа
их паратопов может привести к циркуляции с кровью и отложению
комплексов антиген-антитело в тканях.
Судьба растворимых и циркулирующих с кровью иммунных
комплексов во многом связана с активацией системы комплемента по
классическому пути. При этом небольшие растворимые комплексы связывают
фракции системы комплемента СЗЬ. В результате блокируется реакция
504
Глава 29
преципитации комплексов и их отложение в тканях, которая происходит
через взаимодействие фрагментов Fc иммуноглобулинов, входящих в
состав комплексов антиген-антитело. Небольшие растворимые
иммунные комплексы, связанные с фракцией комплемента СЗЬ,
присоединяются к поверхности эритроцитов, на которой имеются рецепторы к
данной фракции системы комплемента. После попадания растворимых
иммунных комплексов в селезенку вместе с эритроцитами они
подвергаются там лизису через функционирование мононуклеаров селезенки.
При относительной недостаточности системы комплемента растворимые
иммунные комплексы накапливаются в тканях, в сосудах, коже и
почках, что обуславливает их патогенное воспаление.
Иммуннокомплексные болезни - это группа заболеваний, которые
объединяет связь с комплексами антиген-антитело, циркулирующими с
кровью. При небольшом относительном преобладании массы и числа
эпитопов антигена над числом и массой циркулирующих антител
иммунные комплексы откладываются в микрососудах органов с
наибольшей объемной скоростью кровотока (почки и др. органы,
«фильтрующие» кровь при системном кровообращении). При иммуннокомплекс-
ных заболеваниях отложение комплексов антиген-антитело в тканях
приводит к активации системы комплемента по классическому пути, что
определяет рост концентрации хемоаттрактантов в месте отложения
комплексов и реализацию в данном локусе алгоритма сугубо
патогенного воспаления. Реализация алгоритма воспаления приводит в месте
тканевых повреждений к фагоцитозу нейтрофилами и мононуклеарами, де-
грануляции тучных клеток и базофилов циркулирующей крови, а также
обуславливает активацию Т-лимфоцитов-супрессоров (антиген
кластерной дифференциации CD8) через их межклеточные взаимодействия с
антиген-презентирующими клетками. В местах тканевых отложений
иммунных комплексов организм пытается подвергнуть их деструкции и
элиминации через функционирование иммуноцитов, поверхность
которых содержит рецепторы к Fc-фрагменту соответствующих
иммуноглобулинов и фракции системы комплемента СЗ. У всех иммунокомплекс-
ных болезней латентный период весьма значителен. Дело в том, что
необходимым условием развития болезней данного вида является
приобретение системой иммунитета способности вырабатывать антитела,
образующие иммунные комплексы, в результате длительной стимуляции
системы соответствующим антигеном. В патогенезе иммунокомплекс-
ных заболеваний критическое значение имеет размер иммунных
комплексов. Большие по размерам комплексы нерастворимы и быстро
разрушаются и элиминируются через реакцию системы иммунитета.
Небольшие комплексы циркулируют с кровью, не преципитируя в тканях и
не вызывая реакции системы иммунитета.
Поражения различных тканей при иммунокомплексных болезнях в
частности приводят к воспалению суставов, гломерулонефриту,
миокардиту, поражениям кожи, которые могут обуславливать язвы, пурпуру и
крапивницу. Воспаление селезенки может привести к перисплениту. При от-
Реакции повышенной чувствительности
505
ложении иммунных комплексов в микрососудах ишемия клеток
обуславливает цитолиз. Гломерулонефрит вследствие иммунокомплексного
поражения почек как синдром складывается из артериальной гипертензии,
олигурии, гематурии и почечных отеков. Миокардит вызывает застойную
сердечную недостаточность, которую обостряет недостаточная экскреция
натрия и воды почками при гломерулонефрите. Системная иммунная
реакция при иммунно-комплексных заболеваниях приводит к спленомега-
лии, лихорадке, а также обуславливает лимфаденопатию и эозинофилию.
Интенсивный расход фракций системы комплемента при ее активации в
местах отложения иммунных комплексов обуславливает их
патологически низкое содержание данных фракций в сыворотке крови.
Иммунокомплексный механизм повреждений тканей во многом
составляет патогенез таких болезней как системная красная волчанка, узелковый
периартериит, склеродерма, ревматоидный артрит, анкилозирующий
спондилит, болезнь Бехчета, болезни Рейтера и Вегенера, синдром Сегрена, а
также других заболеваний. К иммунокомплексным поражениям тканей и
органов ведет длительная стимуляция системы иммунитета у больных
вирусным гепатитом типа В, инфекционным мононуклеозом, цитомегалови-
русной инфекцией, подострым склерозирующим панэнцефалитом,
лихорадкой Денге, инфекционным менингитом, гонореей, сифилисом и
проказой. Длительную стимуляцию системы иммунитета как причину иммуно-
комплексных болезней может обусловить злокачественный клеточный
рост. Кроме того, иммунокомплексные заболевания могут быть следствием
хронических воспалительных заболеваний легких и кишечника.
Морфопатоенез рассеянного склероза также отчасти составляется
иммунокомплексным воспалением в соответствующих отделах центральной
нервной системы. Первичный билиарный цирроз, синдром Шанляйн-
Геноха, тромбоцитопеническая пурпура - это также во многом
иммунокомплексные заболевания. Следует заметить, что данный список иммуно-
комплексных болезней является далеко не полным.
РЕАКЦИИ ПОВЫШЕННОЙ ЧУВСТВИТЕЛЬНОСТИ ЧЕТВЕРТОГО
ТИПА (РЕАКЦИИ ЗАМЕДЛЕННОЙ ГИПЕРЧУВСТВИТЕЛЬНОСТИ,
ПАТОЛОГИЧЕСКИЕ ИММУННЫЕ РЕАКЦИИ,
ОПОСРЕДОВАННЫЕ КЛЕТКАМИ)
Реакции этого типа обусловлены патогенными межклеточными
взаимодействиями сенсибилизированных Т-хелперов, цитотоксических Т-
лимфоцитов (Т-киллеров) и активированных клеток системы мононукле-
арных фагоцитов, вызванных длительной стимуляцией системы
иммунитета бактериальными антигенами, при которой возникает относительная
недостаточность способности организма элиминировать из внутренней
среды бактериальные возбудители инфекционных заболеваний. Данные
реакции повышенной чувствительности обуславливают туберкулезные
506
Глава 29
каверны легких, их казеозный некроз и общую интоксикацию у пациентов
с туберкулезом. Кожный грануломатоз при туберкулезе и проказе в мор-
фопатогенетическом отношении во многом составляется реакциями
повышенной чувствительности четвертого типа.
Наиболее известный пример реакции повышенной чувствительности
четвертого типа - это реакция Манту, развивающаяся в месте внутрикож-
ного введения туберкулина больному, организм и система которого
сенсибилизированы к антигенам микобактерий. В результате реакции
образуется плотная гиперемированная папула с некрозом в центре, которая
появляется только через несколько часов (замедленно) после внутрикож-
ного введения туберкулина. Формирование папулы начинается с выхода
из сосудистого русла в межклеточные пространства мононуклеарных
фагоцитов циркулирующей крови. Одновременно начинается эмиграция из
сосудистого русла полифорфонуклеаров. Затем инфильтрация нейтрофи-
лами спадает, и инфильтрат начинает преимущественно состоять из
лимфоцитов и мононуклеарных фагоцитов. Этим реакция Манту отличается
от реакции Артюса, при которой в месте поражения накапливаются
преимущественно полиморфонуклеарные лейкоциты.
При реакциях повышенной чувствительности четвертого типа
длительная стимуляция антигенами сенсибилизированных лимфоцитов
приводит в местах патологических изменений тканей к избыточно
интенсивному и длительному высвобождению Т-хелперами цитокинов.
Интенсивный выброс цитокинов в локусах тканевых повреждений обуславливает
гиперактивацию находящихся там клеток системы мононуклеарных
фагоцитов, многие из которых в гиперактивированном состоянии образуют
тяжи эпителиоидных клеток, а некоторые сливаются между собой с
образованием гигантских клеток. Макрофаги, на поверхности которых
экспонированы бактериальные и вирусные антигены, могут уничтожаться
через функционирование Т-киллеров (натуральных киллеров).
Реакция повышенной чувствительности четвертого типа индуцируется
распознаванием чужеродного бактериального антигена
сенсибилизированными по отношению к нему Т-хелперами. Необходимое условие
распознавания - взаимодействие индукторов с антигенами,
экспонированными на поверхности антиген-презентирующих клеток после эндоцитоза и
переработки мононуклеарными фагоцитами чужеродных иммуногенов.
Еще одно необходимое условие - экспонирование антигенов в комплексе
с молекулами I класса из главного комплекса тканевой совместимости.
После распознавания антигена сенсибилизированные хелперы
высвобождают цитокины и, в частности интерлейкин-2, активирующий
натуральные киллеры и мононуклеарные фагоциты. Активированные мононукле-
арные фагоциты высвобождают протеолитические ферменты и свободные
кислородные радикалы, что повреждает ткани.
Натуральные киллеры составляют 75 % тех больших больших
гранулярных лимфоцитов, которые в свою очередь составляют 3-5 % общего
числа лейкоцитов циркулирующей крови. Натуральные киллеры
характеризует высокое отношение массы цитоплазмы к массе клеточного ядра в
Реакции повышенной чувствительности
507
ряду других лимфоцитов. Данные лимфоциты обладают способностью
уничтожать различные клетки, в том числе претерпевшие вследствие
инфицирования вирусами трансформацию фибробласты, клетки твердых
опухолей и малигнизированные клетки миелоидного ряда,
микроорганизмы, а также незрелые клеточные формы, принадлежащие костному мозгу
и вилочковой железе. Под воздействием интерлейкина-2, который
высвобождают Т-хелперы, натуральные киллеры активируются для
высвобождения содержимого эозинофильных гранул (первичных лизосом) и
интерферона, которые повышают число атакующих натуральных киллеров,
их цитотоксичность и скорость цитолиза атакуемых клеток и
микроорганизмов. Цитолиз клеток под воздействием цитотоксических лимфоцитов
происходит не быстро (в среднем за 18 ч) и связан с устойчивой
активацией протеинкиназы С киллеров. Механизм цитолиза через
функционирование Т-лимфоцитов состоит в действии на атакуемые клетки
образуемого и высвобождаемого киллерами белка перфорина, который после
внедрения в наружную клеточную мембрану атакуемой клетки делает ее в
соответствующем участке проницаемой дли натрия и воды. Известно, что
активация натуральных киллеров связана с ростом экспрессии гена онко-
протеина K-ras озлокачествленных клеток.
Хроническую гранулему как следствие реакций повышенной
чувствительности четвертого типа характеризуют локальное и отграниченное
скопление в месте повреждения ткани размножающихся лимфоцитов и
фибробластов, а также явления некроза. Реакции повышенной
чувствительности четвертого типа обуславливают кожную сыпь при оспе, кори и
лихорадке, а также сыпь, вызванную вирусом простого герпеса. При этом
патологические изменения кожи - это в основном результат уничтожения
цитотоксическими лимфоцитами клеток, зараженных вирусами. Кроме
того, реакции повышенной чувствительности четвертого типа составляют
патогенез таких инфекционных заболеваний как кандидоз, дерматомикоз,
кокцидиомикоз и гистоплазмоз. При паразитарных инфекциях (лейшма-
ниоз и шистозоматоз ) в качестве аллергенов при реакциях IV типа
выступают ферменты, высвобождающиеся из яиц паразитов, которые
откладываются в печеночных капиллярах.
При контактном дерматите антиген проникает в организм через
эпидермис, где его поглощают и перерабатывают клетки Лангерганса,
относящиеся к системе мононуклеарных фагоцитов. В дальнейшем клетки
Лангерганса мигрируют в лимфатические узлы, где представляют антиген
Т-лимфоцитам, что инициирует иммунную реакцию с участием Т-клеток.
При контактном дерматите первичное звено патогенеза складывается из
того, что чужеродные вещества способные связываться с компонентами
организма (предположительно с молекулами плазматической мембраны
клеток Лангерганса) образуют новые антигены, антигенная стимуляция
которыми и обуславливает гиперчувствительность замедленного типа.
Морфопатогенез контактного дерматита в основном составляет
инфильтрация мононуклеарами, которая достигает максимального уровня через
12-15 ч, приводит к отеку эпидермиса и его дефектам в виде пузырьков.
508
Глава 29
Контактный дерматит может обуславливать попадание в организм
некоторых химических соединений (пикрилхлорид, хроматы, парафенила-
мин), которые в частности являются компонентами красителей для волос,
лечебных мазей, неомицина. Кроме того, контактный дерматит вызывает
попадание в кожу солей никеля, образующихся из материала никелевых
застежек ювелирных изделий.
Реакции гиперчувствительности второго типа составляют патогенез
отсроченных реакций повышенной чувствительности в ответ на укусы
насекомых.
Глава 30
ИММУНОДЕФИЦИТЫ
Иммунодефицит - патологическое состояние высокой
предрасположенности к инфекции и связанным с ней заболеваниям вследствие недостаточности
системной защитной иммунной реакции в ответ на антигенную стимуляцию,
обусловленной дефектами развития или приобретенными расстройствами в
одном или нескольких элементах системы иммунитета организма.
Выделяют шесть основных видов иммунодефицитов:
♦ обусловленный нарушением образования антител, то есть
дисфункцией продуцирующих антитела В-лимфоцитов;
♦ вызванный расстройствами клеточного звена иммунной системы, то
есть нарушением функционирования Т-лимфоцитов;
♦ комбинированные иммунодефицита как результат одновременных
расстройств функций В- и Т-лимфоцитов;
♦ вследствие недостаточного фагоцитоза;
♦ в результате нарушений в системе комплемента.
Первичный иммунодефицит вызывают генетически детерминированные
дефекты гуморального и клеточного звена иммунной системы. Вторичный
(приобретенный) иммунодефицит -это следствие целого ряда
патологических состояний и заболеваний, первопричина которых не состоит в
расстройствах и патологических изменениях эффекторов иммунной системы. Кроме
того, вторичный иммунодефицит могут вызывать побочные эффекты
лекарственных средств и лечебных воздействий. Например, у больных со
злокачественными опухолями вторичный иммунодефицит могут обусловить
эффекты цитотоксических средств и ионизирующих излучений. Ко
вторичному иммунодефициту приводят нарушения всасывания нутриентов из
просвета кишечника, нефротический синдром как причина потерь
иммуноглобулинов с мочой, множественная миелома с макроглобулинемией, при
которых увеличен синтез аномальных иммуноглобулинов, лишенных свойств
антител, сниженное образование нормальных иммуноглобулинов при
голодании, а также многие другие заболевания и патологические процессы.
ПАТОГЕНЕЗ НАИБОЛЕЕ ИЗУЧЕННЫХ ВРОЖДЕННЫХ
ИММУНОДЕФИЦИТОВ НА УРОВНЕ
ИММУНОКОМПЕТЕНТНЫХ КЛЕТОК
Расстройства дифференциации общего предшественника лимфоидных
клеток приводят к блокаде образования Т- и В-лимфоцитов, что
обуславливает тяжелую недостаточность системы иммунитета реакции в ответ на ее
стимуляцию антигенами (табл. ЗОЛ.). При этом из организменной реакции
510
Глава 30
Таблица 30.1
Врожденные иммунодефициты
Иммунодефицита в результате недостаточности и расстройств
реакции на антигенную стимуляцию на уровне иммунекомпетентных клеток
Тип клеток
Некоторые из нозологических форм
Стволовые клетки
В-лимфоциты (50%
всех первичных
иммунодефицитов)
T-лимфоциты (10%
всех врожденных
иммунодефицитов)
В- и Т-лимфоциты
(30% первичных
иммунодефицитов)
Ретикулярный дисгенез, тяжелый комбинированный дефицит с
аплазией вилочковой железы, частый вариабельный
иммунодефицит при задержке роста тела, агамма-глобулинемия, связанная с
Х-хромосомой
Болезнь Брутона и другие виды врожденной гипогамма-глобули-
немии, иногда сочетающиеся с тимомой, частый вариабельный
иммунодефицит
Болезнь Незелофа, синдром ДиДжорджи, дефицит нуклеозид-
фосфорилазы
Синдром Вискотта-Олдрича, иммунодефицит при атаксии -
телеанги эктазии
Нарушения на уровне молекулярных иммунных систем
и не специфических клеток системы иммунитета
Эффектор системы
иммунитета
Некоторые из нозологических форм и
патологических состояний
Нейтрофильные
лейкоциты
Полиморфо- и
мононуклеары
Комплемент
Нейтропения, синдром ленивых лейкоцитов
Сниженная бактерицидная активность фагоцитов при хронических
гранулематозных заболеваниях, дефиците миелопероксидазы,
синдроме Чедиака-Хигаси (нейтрофилы и мононуклеары с
дефектными лизосомами, недостаточной способностью к хемотаксису и
высвобождению бактерицидных веществ), дефиците активности
гл юк 030-6-фосфат-дегидрогеназы
Ангионевротический отек (аномалии на уровне ингибитора
С1-эстеразы), системная красная волчанка
системы иммунитета выпадает работа клеточных и молекулярных
эффекторов систем ее регуляции (Т-индукторы, Т-супрессоры и Т-хелперы, а
также их цитокины) и клеток, через активацию которых происходит
уничтожение и элиминация чужеродных иммуногенов и антигенов (В-клетки,
плазматические клетки, Т-лимфоциты натуральные киллеры и цитотокси-
ческие Т-лимфоциты). Кроме того, при тяжелом иммунодефиците
вследствие недостаточности стволовых клеток неэффективен эндоцитоз моно- и
полиморфонуклеарами, что отчасти связано с критическим дефицитом
иммуноглобулинов в качестве опсонинов.
У некоторых больных недостаточность дифференциации клеток
предшественниц лимфоцитов является результатом недостаточной активности
аденозиндезаминазы. У 50% таких пациентов к положительному
результату приводит переливание нормальных эритроцитов, содержащих данный
фермент. При длительном течении заболевания, когда иммунодефицит
сопровождается атрофией эпителия вилочковой железы, требуется
дополнительное лечение экстрактами нормальной вилочковой железы (тимозин и
Иммунодефициты
511
др.). Тяжелый комбинированный иммунодефицит, вызванный полным
отсутствием предшественников не только иммунокомпетентных, но и всех
миелоидных клеток, получил название ретикулярного дисгенеза.
Заболевание приводит к смерти вскоре после рождения.
У детей с недостаточностью стволовых клеток нормальная реактивность
иммунной системы может быть восстановлена трансплантацией гистосов-
местимого с организмом донора костного мозга от его брата или сестры.
В-лимфоцит -это иммунокомпетентная клетка, образуемая сумкой
Фабрициуса, органом, локализация которого у млекопитающих до сих пор не
выяснена. В-лимфоциты составляют 30 % всех циркулирующих с кровью
лимфоцитов и сконцентрированы в фолликулярной зоне лимфатических
узлов. В-лимфоциты образуют иммуноглобулины, таким образом участвуя:
♦ в системной защитной реакции, направленной против патогенных
микроорганизмов;
♦ в иммунном надзоре за клетками своего организма, то есть в
цитолизе потенциально злокачественных клеток;
♦ в антитело-зависимой клеточной реакции цитотоксичности и
реакциях повышенной чувствительности.
Как антитело-продуцирующие клетки В-лимфоциты образуют
комплексы антиген-антитело. При образовании комплексов на поверхности
объектов фагоцитоза иммуноглобулины начинают функционировать в качестве
опсонинов, что повышает эффективность защитной реакции фагоцитоза.
Образование на поверхности объектов фагоцитоза комплексов
антиген-антитело активирует систему комплемента по классическому пути.
Активация системы комплемента служит инициирующим моментом воспаления.
Поэтому недостаточность образования В-клетками иммуноглобулинов
приводит к недостаточности воспаления как защитной реакции, направленной
на элиминацию носителей чужеродных антигенов. В-лимфоциты, секрети-
рующие широкий спектр цитокинов, - необходимый эффектор системы
регуляции системной иммунной реакции. В клетках-предшественниках В-лим-
фоцитов есть цитоплазматические иммуноглобулины. При этом поверхность
клеток-предшественников иммуноглобулинов не содержит. Маркерами
зрелости В-лимфоцитов являются антигены их наружной клеточной
поверхности и цитоплазмы. Зрелые В-лимфоциты несут на наружной поверхности
иммуноглобулины и рецепторы фракций системы комплемента. Антигены-
маркеры зрелости (нормальной окончательной дифференциации)
В-лимфоцитов -это CD9, CD 10, CD 19, CD20, CD24, Fc-рецептор, В1, ВА-1, В4 и 1а.
Связанная с Х-хромосомой агамма-глобулинемия (болезнь Брутона) -
первичный иммунодефицит, в основе которого лежит задержка
нормальной дифференциации В-лимфоцитов. Болезнь Брутона встречается только
у мужчин. Вследствие такой задержки у больного младенца нет своих
зрелых В-лимфоцитов и эндогенных гамма-глобулинов плазмы крови.
Заболевание начинает проявлять себя с шестимесячного возраста, в котором из
плазмы крови ребенка элиминируются иммуноглобулины матери. В
результате иммунодефицита, несмотря на нормальные свойства Т-лимфоцитов, у
больного возникают очаги гнойной инфекции разной локализации. Имму-
512
Глава 30
нодефицит усиливается вследствие синдрома нарушенного кишечного
всасывания, обычно связанного с лямблиозом. В частности, синдром
составляют полиартрит, системный коллагеноз, вызывающий распространенный
васкулит, а также дерматомиозит, который у части больных приводит к
летальному исходу.
Транзиторная гипогамма-глобулинемия - иммунодефицит, схожий в
своих проявлениях с болезнью Брутона и обусловленный замедленным
становлением иммунной системы у новорожденных. В результате после
элиминации иммуноглобулинов матери из плазмы ребенка в ней некоторое время
нет зрелых В-лимфоцитов и достаточной концентрации гамма-глобулинов.
Связанная с полом гипогаммаглобулинемия при нормальном или
повышенном содержании иммуноглобулинов М в плазме крови. Этот первичный
иммунодефицит характеризуют дефицит иммуноглобулинов G и А при
нормальной или повышенной концентрации в плазме крови
иммуноглобулинов М. У больных в плазме крови выявляют В-клетки, способные
образовывать и нести на своей поверхности иммуноглобулины М. Грубых
дисфункций Т-лимфоцитов нет. В основе синдрома лежит задержка
трансформации продуцирующих иммуноглобулины М В-клеток в более зрелую
форму В-лимфоцитов, образующую иммуноглобулины G. Большинство
больных - женщины. Синдром часто возникает одновременно с нейтропе-
нией, тромбоцитопенией, гемолитической анемией и В-клеточной лимфо-
мой. Приобретенный иммунодефицит с аналогичным патогенезом
вызывают краснуха, недостаток транскобаламина II. Врожденный и наследуемый
по аутосомно-рецессивному типу недостаток в организме транскобаламина
II (транспортный белок, вовлеченный в метаболизм витамина В12), может
приводить к полному отсутствию в плазме крови гамма-глобулинов, агам-
маглобулинемии. Врожденный недостаток витамина В12 ведет к мегалобла-
стической анемии, гранулоцитопении и тромбоцитопении, снижающим
общую резистентность организма, что вкупе с иммунодефицитом делает
прогноз особенно неблагоприятным.
Тяжелый комбинированный иммунодефицит - весьма вариабельное в
своих клинических проявлениях врожденное иммунопатологическое состояние,
которое наследуется по аутосомно-рецессивному типу. Кроме того,
наследование тяжелого комбинированного иммунодефицита может быть связано с
Х-хромосомой. В Соединенных Штатах этот иммунодефицит чаще
выявляют у черных младенцев в возрасте до пяти месяцев. В основе тяжелого
комбинированного иммунодефицита лежат дисфункции как В-, так и
Т-лимфоцитов. Исследование иммунологического статуса больных выявляет:
♦ сниженную концентрацию иммуноглобулинов в плазме крови или их
полное в ней отсутствие;
♦ нарушение клеточного звена иммунитета, которое проявляет себя
полным угнетением реакции интенсификации митоза Т-клеток в
ответ на действие специфических для них митогенов (фитогемагглю-
тинина и др.);
♦ снижение содержания в крови как В-, так и Т-лимфоцитов;
♦ угнетения образования интерлейкина-2 клетками иммунной системы.
Иммунодефициты
513
Тяжелый комбинированный иммунодефицит вызывают:
♦ наследственная недостаточность энзима аденозиндеаминазы;
♦ врожденный дефицит энзима пуриннуклеозидфосфорилазы;
♦ генетически детерминированный дефект белков, которые, связываясь
с дезоксирибонуклеиновой кислотой, определяют экспрессию генов
из главного комплекса тканевой совместимости (система антигенов
лейкоцитов человека).
Кроме того, тяжелый комбинированный иммунодефицит считают
вариантом болезни Незелофа.
Клинические проявления тяжелого комбинированного
иммунодефицита складываются из сыпи, похожей на сыпь у больных корью, а также
частых легочных инфекций (Candida, Pneumocystis carinii, вирус Эпштейн-Бар-
ра, возбудитель ветряной оспы и др.). Нередкой является гиперпигментация
кожи.
Частый вариабельный иммунодефицит (англ. - common variable
immunodeficiency) - это наследуемое по аутосомно-рецессивному типу
нарушение реакции системы иммунитета организма в ответ на антигенную
стимуляцию, которое характеризует снижение концентрации в плазме
крови иммуноглобулинов большинства изотипов. При вариабельном
иммунодефиците клетки-предшественники В-лимфоцитов циркулируют с кровью,
но не дифференцируются до зрелых форм. Кроме того, у больных
выявляют дефекты развития Т-клеток, которые не служат первопричиной
расстройств клеточного звена иммунитета. Частым этот вид врожденного
комбинированного иммунодефицита называют потому, что его частота среди
всех видов первичного иммунодефицита высока и составляет 20-90
случаев на миллион населения.
Заболевания и синдромы, связанные с частым вариабельным
иммунодефицитом, начинают выявлять с двенадцатилетнего возраста. Их спектр
широк. В этой связи это иммунопатологическое состояние получило
название вариабельного. Чаще всего больные с вариабельным
иммунодефицитом страдают от различных бактериальных инфекций, вызывающих отиты,
синуситы, пневмонии. Нередко их беспокоят поносы неизвестного
происхождения, которые выступают основным симптомом синдрома
нарушенного кишечного всасывания у таких пациентов. Синдром нарушенного
кишечного всасывания часто сопровождается энтеритом, ахлоргидрией и ведет к
пернициозной анемии. Синдром нарушенного кишечного всасывания у
больных вызывается и холестазом той или иной степени, в частности,
связанным с лямблиозом.
Основное звено патогенеза вариабельного иммунодефицита - это
блокирование дифференцировки клеток-предшественников В-лимфоцитов до
зрелых форм (рис. ЗОЛ). Предположительно одним из этиологических
факторов вариабельного иммунодефицита считают вирус Эпштейна-Барра. Его
патогенные эффекты могут резко угнетать образование иммуноглобулинов
зрелыми В-лимфоцитами, нарушать гликозилирование антител или
воздействовать на Т-лимфоциты таким образом, что они начинают подавлять
синтез иммуноглобулинов.
514
Глава 30
Fc-рецептор СЗ-рецептор
Стволовая клетка-пред- Недифференцированный Лимфоцит с маркерами
шественница иммуно- лимфоцит с поверхностными клеточной дифференциации
компетентных клеток иммуноглобулинами на клеточной поверхности
Рис. 30.1. Принципиальная схема дифференциации В-лимфоцитов
Исследование плазмы крови больных с вариабельным частым
иммунодефицитом позволяет выявить значительную гипогаммаглобулинемию. Из
результатов специальных исследований больных с данным
иммунодефицитом можно выделить снижение активности 5-нуклеотидазы в лимфоцитах
пациентов с данным видом иммунодефицита. У таких больных риск
развития рака желудка в 50 раз выше, чем у представителей основной популяции.
Активность этого фермента снижена в лимфоцитах у 70 % больных с
частым вариабельным иммунодефицитом. Падение секреции гастрина под
влиянием бомбезина после инъекции пептида является маркером частого
комбинированного иммунодефицита у больных с высоким риском рака желудка.
Врожденные иммунодефицита, связанные с расстройствами системной
иммунной реакции исключительно на уровне Т-лимфоцитов, у больных с
недостаточной реакцией системы иммунитета в ответ на антигенную
стимуляцию встречаются весьма редко. Врожденные дефекты свойств
Т-лимфоцитов почти всегда приводят к недостаточному образованию антител В-
лимфоцитами. Дело в том, что большинство белковых антигенов и гапте-
нов обладают свойством стимулировать систему иммунитета для
образования антител лишь при условии ненарушенного функционального
состояния как Т-лимфоцитов, так и В-клеток. Полное отсутствие в
организме нормальных Т-лимфоцитов лишает его систему иммунитета способности
реагировать образованием антител в ответ на стимуляцию Т-лимфоцит-за-
висимыми антигенами. В этой связи можно считать, что вне зависимости от
первопричины недостаточной реакции системы иммунитета в виде
избирательного дефекта В- или Т-лимфоцитов, иммунодефицит на уровне имму-
нокомпетентных клеток всегда отчасти является комбинированным, то есть
его патогенез состоит из дисфункций иммунной системы как на уровне Т-
клеток, так и В-лимфоцитов.
Т-лимфоциты участвуют в защитных реакциях, направленных на
элиминацию из организма возбудителей внутриклеточных инфекций. Каждая
Т-клетка имеет специфический рецептор, благодаря которому и распознает
антиген во взаимодействии с мононуклеаром. Т-лимфоцит распознает
антигены возбудителей внутриклеточных инфекций в комплексе с
молекулами из главного комплекса тканевой совместимости на поверхности анти-
Иммунодефициту
515
ген-презентирующей клетки. В результате образования комплекса,
состоящего из Т-клеточного рецептора и антигена, происходит индукция клональ-
ной экспансии лимфоцитов с образованием эффекторных клеток и клеток
памяти, что обеспечивает специфическую реакцию приобретенного
иммунитета.
Т-хелперы распознают антигены возбудителей внутриклеточных
инфекций, экспонированные на поверхности мононуклеарных фагоцитов в
комплексе с молекулами второго класса из главного комплекса тканевой
совместимости (ГКТС). После распознавания Т-хелперы секретируют цитокины,
и в частности гамма-интерферон. Через секрецию цитокинов хелперы
действуют как клеточные эффекторы регуляции системной иммунной реакции.
В результате их действия в данном качестве активируются цитотоксические
лимфоциты и натуральные киллеры, которые уничтожают
инфицированные клетки. Цитотоксические Т-лимфоциты распознают антигены
возбудителей внутриклеточных инфекций, которые экспонированы на
поверхности атакуемых клеток в комплексе с гликопротеинами из первого класса
молекул ГКТС.
Поэтому недостаточность системной иммунной реакции, связанная
преимущественно с дисфункциями системы иммунитета на уровне Т-лимфо-
цитов, приводит к частым болезням, в основе которых лежат вирусные,
бактериальные и паразитарные внутриклеточные инфекции.
Диагноз врожденной аплазии вилочковой железы (синдромы ДиДжорд-
жи, Незелофа) становится ясным вскоре после рождения, так как синдром
как патологическое состояние всего организма включает врожденные
пороки сердца и первичный гипопаратиреоз. В результате гипопаратиреоза у
таких больных уже через 24 часа после рождения развивается гипокальци-
емия как причина тетании, то есть перемежающихся судорог. Синдром ДиД-
жорджи представляет собой результат нарушения эмбрионального
развития на двенадцатой неделе беременности, когда не происходит нормального
формирования вилочковой и паращитовидных желез из третьего и
четвертого парных выпячиваний эмбриональной глоточной эктодермы. У многих
больных нет полной аплазии железы; она лишь частична при нередкой
аномальной локализации. Из-за недоразвития вилочковой железы стволовые
клетки не могут дифференцироваться в Т-лимфоциты, и «тимус-зависимые»
элементы лимфоидной ткани организма характеризует пониженное
содержание лимфоцитов. В результате у больных выявляют патологически
низкое содержание Т-лимфоцитов в циркулирующей крови. Если больные
выживают, несмотря на сердечную недостаточность вследствие врожденного
порока сердца, то причиной летального исхода служат осложнения
соответствующих инфекционных заболеваний. Дело в том, что у таких больных
практически нет нормальной реакции клеточного иммунитета, хотя их
иммунная система способна уничтожать возбудителей бактериальных
инфекций. Система иммунитета у больных с синдромами ДиДжорджи и
Незелофа обладает способностью продуцировать антитела в ответ на стимуляцию
антигенами, но эта реакция ослаблена, так как системную иммунную
реакцию не обеспечивает должным образом взаимодействие Т- и В-лимфоци-
516
Глава 30
тов. У больных с аплазией вилочковой железы «... иммунореактивность
восстанавливается при трансплантации тимуса новорожденных, но, если
трансплантат подобран неудачно, он отторгается "неблагодарными" клетками
хозяина, созреванию которых сам же и способствовал» (Ройт).
Синдром Вискота-Олдрича относится к первичным иммунодефицитам
вследствие выпадения из системной иммунной реакции нормальной
работы как Т-, так и В-лимфоцитов. В частности, синдром характеризует тром-
боцитопения, которую у части больных выявляют сразу после рождения.
Наследование синдрома связано с полом. Его возникновение определяет
патогенный рецессивный аллель гена болезни. Первопричина болезни
состоит в патологическом изменении функционального состояния мононук-
леарных фагоцитов, которое определяет невозможность индукции
системной иммунной реакции через взаимодействие антигенпрезентирующей
клетки и Т-лимфоцита. Дело в том, что мононуклеарные фагоциты у
больных с синдромом не обладают способностью переработки для
представления иммуноцитам полисахаридных антигенов. В результате система
иммунитета не обладает способностью вырабатывать антитела, комплементарные
по отношению к полисахаридным антигенам некоторых патогенных
микроорганизмов {Hemophilus influenzae, пневмококки и др.). В данном случае
первопричина иммунодефицита на уровне мононуклеарных фагоцитов
определяет вторичную специфическую недостаточность антителообразования
В-клетками. Если сначала по ходу онтогенеза исследование функций имму-
нокомпетентных клеток in vitro не выявляет аномалий, то затем они
возникают. Больные с синдромом часто страдают от повышенной
кровоточивости, связанной с тромбоцитопенией, подвержены малигнизации клеток
лимфоретикулоэндотелиальной системы и частым инфекционным
заболеваниям, которые вызывают соответствующие возбудители.
Иммунодефицит у больных с атаксией-телеангиэктазией. Атаксия -
неспособность координации мышц при произвольных движениях. Телеанги-
эктазия - локальное расширение капилляров и мелких концевых артерий.
Атаксия-телеангиэктазия представляет собой моногенную болезнь,
наследуемую по аутосомно-рецессивному типу. Кроме атаксии и телеангиэкта-
зии болезнь характеризуют частые или хронические инфекции. Примерно
у 80% больных в сыворотке крови и секретах почти нет
иммуноглобулинов А. У большинства пациентов патологически снижена не только
концентрация иммуноглобулинов А в секретах и сыворотке, но и содержание в них
иммуноглобулинов G. Болезнь в морфопатогенетическом отношении
характеризуют аутоантитела к иммуноглобулинам А, гипоплазия вилочковой
железы, лимфопения, а также сниженная реакция Т-лимфоцитов в ответ на
эффект митогенов. Предположительно основным звеном патогенеза
иммунодефицита у больных с атаксией-телеангиэктазией можно считать
недостаточные влияния со стороны Т-лимфоцитов на развитие В-клеток или
дефицит их регуляторных влияний в качестве супрессоров.
Иммунодефициты как следствия врожденного недостатка
активности аденозиндезаминазы и нуклеозидфосфорилазы. Если врожденный
дефицит активности аденозиндеаминазы служит причиной иммунодефицита
Иммунодефициту
517
вследствие патологического изменения функционального состояния как Т-,
так и В-лимфоцитов, то наследственный недостаток активности нуклеозид-
фосфорилазы обуславливает иммунодефицит преимущественно на уровне
Т-клеток.
Первичные одновременные нарушения дифференциации как В-, так и
Т-лимфоцитов, приводящие к врожденному комбинированному
иммунодефициту, связаны с наследуемым по аутосомно-рецессивному типу
недостатком фермента аденозиндеаминазы. Ген энзима локализован на
длинном плече 22 аутосомы. Сам фермент представляет собой молекулу с массой
в 38 килодальтон. Дефицит энзима ведет к накоплению в клетках аденози-
на, деоксиаденозина, аденозинтрифосфата, S-аденозилгомоцистеина и
деоксиаденозинтрифосфорной кислоты. Деоксиаденозинтрифосфат - это
мощный естественный ингибитор фермента дифосфатредуктазы, который
участвует в синтезе пуринов. Рост в клетках концентрации аденозина
угнетает внутриклеточное метилирование дезоксирибонуклеиновой
кислоты (ДНК), что служит причиной цитолиза. Кроме того, врожденный
недостаток аденозиндезаминазы угнетает синтез S-аденозилметионина,
который является донатором метальных групп для ДНК. Деоксиаденозин-
трифосфорная кислота (дАТФ) при ее накоплении в клетках тормозит
активность рибонуклеотидредуктазы, без достаточного уровня которой не
происходит фазы S цикла клеточного роста. Это ведет к прекращению
синтеза ДНК и включению дАТФ в полиаденилированную
рибонуклеиновую кислоту (РНК), что угнетает синтез РНК и разделение ДНК на
отдельные нити. В результате не происходит дифференциации
клеток-предшественников В- и Т-лимфоцитов. Врожденный дефицит аденозиндеаминазы
вызывает тяжелый комбинированный иммунодефицит у 40% больных с
этим синдромом.
Иммунодефицит, обусловленный недостатком аденозиндезаминазы, чаще
всего проявляет себя кандидозом полости рта, трудно устранимым поносом,
задержкой роста и другими инфекциями и дефектами развития. Общее число
лимфоцитов в крови находится на уровне более низком, чем 0,5х109/л.
Особенно мало в крови Т-лимфоцитов. Лимфопения (патологически низкое
содержание лимфоцитов в крови) приводит к эозинофилии. В моче и
сыворотке крови больных повышено содержание аденозина и деоксиаденозина.
Врожденный дефицит активности нуклеозидфосфорилазы
наследуется по аутосомно-кодоминантному типу. Расстройства пуринового обмена у
больных с иммунодефицитом обуславливают аккумуляцию в иммуноком-
петентных клетках деоксигуанозинтрифосфата, что приводит к
иммунодефициту через торможение в них активности рибонуклеотидредуктазы.
Вследствие низкой активности рибонуклеотидредуктазы блокируется деление
клеток и преимущественно на уровне Т-клеток возникает иммунодефицит.
Данный иммунодефицит приводит к частым инфекциям легких, мочевыво-
дящих путей. Нередко иммунодефициту сопутствуют аутоиммунная
гемолитическая анемия и гипоплазия клеточных элементов костного мозга. В
циркулирующей крови выявляют патологически низкое содержание
Т-клеток, в моче и сыворотке крови аномально высокое содержание мочевой кис-
518
Глава 30
лоты (признак цитолиза неспособных к клональной экспансиии иммуно-
компетентных клеток). Кроме того, в сыворотке крови возрастает
концентрация свободных инозина и гуанозина.
НЕДОСТАТОЧНОСТЬ ЗАЩИТНОЙ РЕАКЦИИ ФАГОЦИТОЗА
КАК ПРИЧИНА ИММУНОДЕФИЦИТА
Хронический грануломатоз обуславливает неспособность моно- и по-
лиморфонуклеаров синтезировать активные кислородные радикалы, что
связано с дефектом строения цитохрома Ь24Р который обычно активируется
параллельно с активацией фагоцитов. В результате недостаточного
образования свободных кислородных радикалов в активированных фагоцитах
объекты фагоцитоза не уничтожаются через действие свободных
кислородных радикалов. Известно, что многие патогенные микроорганизмы
содержат каталазу, инактивирующую перекись водорода. После фагоцитоза
таких бактерий нейтрофилами с недостаточной активностью глюкозо-6-фос-
фатдегидрогеназы, в фагосомах вообще не образуются активные соединения
кислорода, из действия которых во многом состоит бактерицидный эффект
фагоцитов. В результате не наступает конечный этап фагоцитоза, т. е.
уничтожение патогенных микроорганизмов.
При болезни Чедиака-Хигаси нарушения структуры и функционального
состояния лизосом обуславливают недостаточность защитной реакции
фагоцитоза, что приводит к частым гнойным инфекциям.
При редком врожденном заболевании недостаточности миелоперокси-
дазы фагоцитов недостаточная резистентность больных служит причиной
частых кандидозов.
При «синдроме ленивых нейтрофилов» патогенез в основном
составляется низкой чувствительностью полиморфонуклеаров по отношению к хе-
моаттрактантам.
ВРОЖДЕННЫЕ ИММУНОДЕФИЦИТЫ КАК РЕЗУЛЬТАТ
НАСЛЕДСТВЕННЫХ РАССТРОЙСТВ СИСТЕМЫ
КОМПЛЕМЕНТА
Представляется возможным выделить три основных вида наследственных
расстройств системы комплемента как причин первичного иммунодефицита:
♦ Дефекты ингибиторов системы комплемента, которые на уровне
всего организма обуславливают избыточный уровень активации
системы комплемента как причины патогенно избыточного потребления
ее фракций.
Иммунодефициты
519
♦ Генетически детерминированные дефекты строения фракций
системы комплемента.
♦ Наследственный абсолютный дефицит определенных фракций
системы комплемента.
Наследственная ангиоэдема (ангионевротический отек) обусловлен
дефектом синтеза ингибитора С1-эстеразы. В результате пациенты страдают
от периодических обострений ангионевротического отека, в основе
которых лежат пароксизмы неограниченной активации системы комплемента.
При этом чаще всего патологически изменяется структура верхних
дыхательных путей (ангионевротический отек гортани может быть причиной
асфиксии), органов желудочно-кишечного тракта. Причина состоит в том,
что в данных локусах соприкосновения внутренней среды организма с
внешней через соответствующие барьеры наиболее интенсивно первичное
взаимодействие экзогенных антигенов с системой иммунитета организма.
Есть два вида наследственного ангионевротического отека. Первый
обусловлен полным отсутствием экспрессии гена ингибитора С1 эстеразы. При
другом соответствующий ген экспрессирует ингибитор с патологически
измененной структурой.
У больных с иммунодефицитом, обусловленным недостатком фракции
системы комплемента СЗ, часто возникают гнойные инфекции. Дефицит
фракции системы комплемента СЗ приводит к недостаточности иммунной
адгезии фагоцитирующих клеток через низкое содержание нормальной СЗ в
качестве опсонина на поверхности объекта фагоцитоза. Кроме того, дефицит
СЗ приводит к недостаточно полной активации системы комплемента как
защитной реакции, направленной на уничтожение бактериальных
возбудителей. При этом недостаточность активации системы комплемента сама по себе
представляет звено патогенеза иммунодефицита. Механизмы развития
наследственного дефицита СЗ различны. Дефицит данной фракции системы
комплемента может быть обусловлен недостаточным синтезом СЗ и отсутствием
экспрессии ее инактиватора (причина избыточного катаболизма СЗ).
Патогенетически обоснованной терапией больных с иммунодефицитом вследствие
недостатка фракции системы комплемента СЗ является переливание плазмы
крови доноров без дефицита данной фракции.
Семейная дисфункция фракции комплемента С5 в частности
характеризует больных с синдромом Лейнера, при котором больные страдают от
системного себорейного дерматита, тяжелого поноса, и частых бактериальных
инфекций при инвазии во внутреннюю среду преимущественно грам-нега-
тивных микроорганизмов. У пациентов, страдающих от системной красной
волчанки, выявляют наследственный дефицит фракции системы
комплемента С2, который наследуется по аутосомно-рецессивному типу.
Глава 31
АУТОИММУННЫЙ МЕХАНИЗМ
РАЗВИТИЯ БОЛЕЗНЕЙ
В основе аутоиммунного механизма возникновения и развития
болезней лежит потеря организмом способности распознавать нормальные
ткани, клетки и антигены (аутоантигены) в качестве принадлежащих
организму больного. В результате возникает реакция системы иммунитета,
повреждающая нормальные аутоантигены, клетки, ткани и вызывающая
аутоимунные болезни.
Некоторые иммунные реакции в основном возникают через
образование иммунных комплексов (комплексов антиген-антитело). Основное
звено патогенеза других - это атака нормальных клеток организма больного
его Т-лимфоцитами. Тем не менее, в любую аутоиммунную реакцию в той
или иной степени обычно оказываются вовлеченными все клеточные и
молекулярные эффекторы системы иммунитета. При большинстве
аутоиммунных болезней система иммунитета начинает вырабатывать антитела к ауто-
антигенам (аутоантитела);
Клеточными элементами системы иммунитета, взаимодействия между
которыми обеспечивают иммунологическую толерантность, то есть
состояние ареактивности системы иммунитета по отношению к аутоантигенам,
являются клетки системы мононуклеарных фагоцитов, Т- и В-лимфоциты,
а также антиген-презентирующие В-клетки костного мозга. При этом
определяющую роль в поддержании иммунологической толерантности играет
состояние ареактивности (иммунологической толерантности) Т- и В
лимфоцитов (рис. 31.1)
Многие из аутоантигенов (тиреоглобулин, гормоны белковой природы,
растворенные в жидкой части плазмы крови аутоантигены наружных
клеточных мембран и др.) в физиологических условиях циркулируют с
кровью в небольшой концентрации. При этом толерантность к ним
поддерживается только через ареактивность Т-клеток, а не посредством
толерантности В-лимфоцитов. Известно, что Т-клетки способны приобретать
толерантность под влиянием интенсивной стимуляции массивными дозами
антигенов. В-лимфоциты этим свойством обладают в значительно
меньшей мере. Даже толерантные В-клетки в ответ на стимуляцию аутоантиге-
нами могут начать вырабатывать аутоантитела с низкой аффинностью
(сродством) к данным нормальным антигенам организма больного. Все это
определяет возможность непосредственной стимуляции аутоантигенами в
патологических условиях В-клеток в силу их меньшей, чем у тимоцитов,
спосо'бности приобретать толерантность. Такую стимуляцию называют
антигенной стимуляцией в обход толерантных Т-лимфоцитов-хелперов.
Необходимое условие иммунологической толерантности по отношению к
аутоантигену - это его взаимодействие с иммунокомпетентными клетками.
Аутоиммунные механизмы развития болезней
521
Аутоантигены
Ареактивная
(толерантная)
Т-клетка
Толерантная В-клетка или
не испытывающая влияний
Т-лимфоцитов хелперов
нетолерантная В-клетка
Стимуляция Т-клетки
аутоантигенами
*
К
Отсутствие влияний Т-
хелпера на В-клетку
Нет выработки
аутоантител
Рис. 31.1. Принципиальная схема поддержания
иммунологической толерантности
Поэтому к аутоантигенам, существующим в клетках и органах
изолированно от системы иммунитета, не может быть иммунологической ареактивно-
сти. Отграниченность внутриклеточных и внутриорганных аутоантигенов
от взаимодействия с иммунокомпетентными клетками часто не бывает
полной. Например, такой внутриорганный аутоантиген как тиреоглобулин
циркулирует с кровью в минимальной концентрации, и к нему у здоровых
людей выявляют иммунологическую толерантность. Тем не менее, она
исчезает при соответствующем аутоиммунном поражении щитовидной
железы. Сами по себе аутоиммунные болезни приводят к высвобождению
пораженными клетками изолированных от системы иммунитета
аутоантигенов в циркулирующую кровь, причем часто высвобождаются именно те
аутоантигены, к которым и вырабатываются аутоантитела. Это не
подвергает аутоиммунную болезнь обратному развитию. Например, при
системной красной волчанке в кровь из погибших клеток высвобождаются
именно те аутоантигены (фрагменты дезоксирибонуклеиновой кислоты), к
которым вырабатываются аутоантитела. В результате постоянного
взаимодействия данных ядерных аутоантигенов с системой иммунитета во внутренней
среде иммунологическая толерантность к ним не восстанавливается, а
системная красная волчанка не исчезает.
Одним из патогенетических механизмов, действие которых приводит к
аутоиммунным заболеваниям, можно считать патогенную стимуляцию
системы иммунитета вследствие появления по ходу онтогенеза в структуре
аутоантигена новых детерминант, к которым нет иммунологической
толерантности. Это вызывает образование аутоантител, так как ареактивные Т-
лимфоциты в результате взаимодействия с измененным аутоантигеном как
носителем новых детерминант начинают оказывать на В-клетки хелперные
влияния. Следует заметить, что при подавляющем большинстве известных
сегодня аутоиммунных болезней с помощью современных методов
исследования не удается выявить каких-либо изменений структуры аутоантиге-
522
Глава 31
на, к которому в результате дизрегуляции иммунологической
толерантности система иммунитета вырабатывает антитела.
Не исключено, что побочные эффекты некоторых лекарственных
средств, приводящие к аутоиммунным болезням, связаны с тем, что
препарат, его молекулярная составляющая или продукт их биологической
трансформации фиксируются на поверхности (в структуре) аутоантигенов в
качестве гаптенов или меняют их структуру с появлением новых эпитопов, к
которым нет иммунологической ареактивности Т-клеток. Обычно при
таком побочном эффекте лекарственных средств не происходит
формирование нового эпитопа на аутоантигене, с которым взаимодействует Т-лимфо-
цит. Можно, кроме того, предположить, что лекарственное средство или
продукты его биологической трансформации изменяют какую-либо
независимую молекулу, участвующую в иммунологическом узнавании.
Полагают, что аутоиммунная гемолитическая анемия как ятрогенная
болезнь вследствие побочного эффекта альфа-метилдофа возникает в
результате изменения структуры клеточной поверхности эритроцита под
влиянием препарата. В результате участки клеточной поверхности
эритроцитов начинают нести детерминанты, которые вступают в прямое
взаимодействие с В-клетками, способными к распознаванию антигена Rh.
Системное действие изониазида приводит к образованию антител к
элементам клеточного ядра (антинуклеарные аутоантитела), что
обуславливает аутоиммунный артрит, который сохраняется и после отмены препарата.
В данном случае первичное звено патогенеза аутоимунной болезни
закрепляется в виде изменения строения аутоантигена или независимой
молекулы, участвующей в иммунологическом распознавании.
Из других ятрогенных аутоиммунных болезней, связанных с действием
лекарственных препаратов, следует выделить системную красную
волчанку у больных как осложнение лечения новокаинамидом, а также
миастению в результате побочного эффекта пенициллинов.
Специфичность антител и иммунокомпетентных клеток
определенного идиотипа по отношению к соответствующей детерминанте (эпитопу) не
абсолютна. Поэтому стимуляция системы иммунитета чужеродными
антигенами всегда в определенной степени связана с риском аутоиммунной
реакции. При возникновении аутоиммунной реакции такого генеза
детерминанта, принадлежащая какому-либо чужеродному перекрестно
реагирующему антигену, после попадания во внутреннюю среду организма больного
через стимуляцию его системы иммунитета вызывает образование аутоан-
тител. Напомним, что перекрестно реагирующий антиген при
взаимодействии с системой иммунитета индуцирует образование антител,
способных образовывать иммунные комплексы с детерминантами других
антигенов (одна антигенная детерминанта перекрестно реагирует с детерминантой
другого антигена). Таков в основном патогенез аутоиммунного энцефалита
как осложнения парентерального введения антирабической вакцины,
содержащей разнообразные антигены ткани головного мозга. Некоторые из
антигенов болезнетворных микроорганизмов содержат детерминанты,
перекрестно реагирующие с детерминантами нормальных аутоантигенов. В
Аутоиммунные механизмы развития болезней
523
результате взаимодействие таких чужеродных антигенов бактериального и
другого происхождения с системой иммунитета больного приводит к
образованию аутоантител и аутоиммунной болезни. Так, при ревматизме
антитела к стрептококку являются аутоантителами, взаимодействующими с
нормальными аутоантигенами сердечной мышцы. У 50 % детей, страдающих
от ревматической хореи, то есть от связанного с ревматизмом поражения
нервной ткани {хорея Сиденхема), с кровью циркулируют аутоантитела,
реагирующие как с нейронами, так и с мембранами стрептококка.
Известно, что аутоантитела, взаимодействующие с нормальными аутоантигенами
слизистой оболочки толстой кишки у больных неспецифическим язвенным
колитом, перекрестно реагируют с антигенами Escherichia coli.
Под сопряженным распознаванием в данном контексте следует
понимать реакцию системы иммунитета, при которой патогенно измененный
компонент клеточной мембраны вызывает реакцию системы иммунитета с
аналогичными нормальными участками клеточной мембраны, в том числе
и других клеток. Компонентом, способствующим реакции системы
иммунитета на нормальную антигенную детерминанту, является
модифицированный под влиянием лекарственных средств или вирусов участок
клеточной мембраны. В частности, препараты и продукты их биологической
трансформации меняют структуру участков клеточной мембраны как гап-
тены, а вирусные антигены оказываются встроенными в плазматические
мембраны. Не исключено, что таков патогенез аутоиммунной
гемолитической анемии через образование Холодовых агглютининов к групповому
антигену крови I после инфицирования организма больного Mycoplasma
pneumonia.
При большинстве аутоиммунных заболеваний аутоантитела
относятся к одному или в основном к одному идиотипу. Идиотип - это
антигенная детерминанта, придающая уникальность молекуле
иммуноглобулина. Антитела одного идиотипа образуются В-лимфоцитами определенного
клона. Идиотипы есть и у Т-лимфоцитов. Идиотип Т-клеток - это
антигенная детерминанта изменчивой по структуре части рецептора
Т-лимфоцита, которая определяет специфичность иммунокомпетентной клетки
по отношению к определенному антигену. При системной иммунной
реакции в ответ на антигенную стимуляцию образуются антитела к идиоти-
пам собственных иммуноглобулинов (идиотип-антиидиотипические
взаимодействия). Идиотип-антиидиотипические взаимодействия можно
считать механизмом регуляции реакции системы иммунитета в ответ на
антигенную стимуляцию. Часто встречающиеся идиотипы, присущие
антителам разной специфичности (перекрестно реагирующие идиотипы),
служат эпитопами для паратопов антиидиотипических антител. Идиотип-
антиидиотипические взаимодействия происходят не только на уровне
иммуноглобулинов, но и присущи Т-лимфоцитам хелперам и супрессо-
рам. Лимфоциты могут специфически реагировать с идиотипами
рецепторов других лимфоцитов, что определяют как сетевые взаимодействия.
Сетевое взаимодействие представляет собой один из механизмов
регуляции системной иммунной реакции.
524
Глава 31
Идиотипы определенных антител могут перекрестно реагировать с
идиотипами Т- и В-клеток. В результате образуются антитела
специфичные не только по отношению к идиотипам данных антител, но и к идиоти-
пам определенных иммуноцитов. При этом посредством перекрестного
реагирования происходит активация иммуноцитов и система иммунитета
может начать уничтожать и элиминировать антигены, которые не
участвовали в индукции иммунного ответа.
Выделяют следующие основные механизмы возникновения аутоимун-
ных поражений, действие которых связано с идиотип-антиидиотипически-
ми взаимодействиями.
♦ Перекрестная реакция микробного антигена с идиотипом аутореак-
тивного Т-лимфоцита.
♦ Образование антимикробных антител с идиотипом идентичным иди-
отипу аутореактивных Т-лимфоцитов. Образование таких
антимикробных антител вызывает продукцию соответствующих антиидио-
типических иммуноглобулинов, что активирует аутореактивные
Т-клетки, несущие идиотип общий с идиотипом антимикробных
антител.
♦ Образование антимикробных антител, которые являются антиидио-
типическими антителами к идиотипам аутореактивных
иммуноглобулинов.
♦ Образование антител к вирусу приводит к образованию антидиоти-
пических антител к рецепторам клеточной поверхности, с которыми
избирательно связывается данный вирус. Такие рецепторы могут
быть эффекторами систем регуляции разных уровней структурно-
функциональной организации организма. Их соединение с антииди-
отипическими аутоантителами обуславливает дизрегуляцию. В этой
связи уместно напомнить, что бета-адренорецепторы представляют
собой рецепторы к определенным вирусам, а вирус бешенства - это
лиганда к ацетилхолиновым рецепторам.
Т-хелперы специфичные к идиотипу иммуноглобулинового рецептора
Т-лимфоцитов, вступая в сетевые идиотип-антиидиотипические, могут
активировать иммуноциты, несущие рецептор с данным идиотипом. Это
служит одним из механизмов системной иммунной реакции в ответ на
антигенную стимуляцию. Если какой-либо экзогенный чужеродный антиген
вызывает образование антител, несущих идиотип, перекрестно
реагирующий с рецепторами аутореактивных Т- и В-рецепторов, то образование ан-
тиидиоитпических антител активирует данные иммуноциты.
Активированные иммуноциты вызывают аутоиммунную реакцию.
Стимуляция системы иммунитета чужеродными антигенами через
идиотип-антиидиотипические взаимодействия может приводить к образованию
аутоантител к гормонам. При этом специфичные по отношению к данным
антиидиотипическим антителам иммуноглобулины могут быть
комплементарными по отношению к рецепторам гормона. Это может нарушить
взаимодействие гормона со своим рецептором. Не исключено, что появление в
сыворотке крови больного сахарным диабетом антител к инсулину и его
Аутоиммунные механизмы развития болезней
525
рецепторам, представляет собой следствие патогенных идиотип-антиидио-
типических взаимодействий. Обходная активация аутореактивных Т-индук-
торов, то есть активация данных Т-лимфоцитов не в результате
стимуляции системы иммунитета специфическими для Т-индукторов антигенами,
в большинстве случаев не может быть устойчивой, так как иммунная
система обладает способностью тормозить продукцию аутоантител через
соответствующую реакцию Т-супрессоров. В этой связи можно считать, что
любые воздействия, снижающие активность и число Т-супрессоров,
одновременно повышают продукцию аутоантител. Предположительно в
угнетении Т-супрессоров и во вторичной по отношению к нему аутоиммунной
реакции может играть роль патогенно высокая активность Т-контрсупрес-
соров. Кроме того, есть основанные на экспериментальных данных
предположения, что причиной недостаточного числа Т-супрессоров и
аутоиммунной реакции может быть врожденно низкая экспрессия на клеточной
поверхности молекул 1-Е как стимула образования Т-супрессоров. Можно
считать, что одним из гипотетических звеньев патогенеза аутоиммунных
болезней является недостаточная экспрессия генов молекул второго класса
из главного комплекса тканевой совместимости, обуславливающая
недостаточное образование Т-супрессоров.
Не исключено, что такое свойство В-лимфоцитов с поверхностным
маркером Ly 1 как относительная ареактивность по отношению к влияниям Т-
супрессоров может быть причиной некоторых аутоиммунных реакций.
Недостаточные регуляторные влияния со стороны Т-клеток на другие
клеточные эффекторы системной иммунной реакции могут быть звеном
патогенеза системной красной волчанки. Ряд иммунопатологических
сдвигов у таких пациентов говорит в пользу такого взгляда на патогенез
данного аутоиммунного заболевания:
♦ В-лимфоциты больных системной красной волчанкой продуцируют
гораздо больше иммуноглобулинов, чем В-клетки здоровых людей;
♦ у пациентов почти нет неспецифических Т-супрессоров,
пролиферацию которых стимулирует конканавалин А;
♦ у больных системной красной волчанкой снижено содержание несущих
Fc гамма-рецепторы Т-лимфоцитов, которые подавляют клеточную
пролиферацию лимфоцитов в ответ на действие митогена фитолакки.
Существует прямая связь между степенью тяжести системных и
локальных аномалий у больных системной красной волчанкой и выраженностью
данных иммунопатологических сдвигов.
При поддержании нормальной иммунологической толерантности по
отношению к аутоантигенам они экспрессируются геномом клеток на
наружной клеточной мембране в сочетании с молекулами первого класса из
главного комплекса тканевой совместимости. Это обуславливает
невозможность взаимодействия аутоантигенов с аутореактивными Т-лимфоцитами
индукторами. Полагают, что при угнетении экспрессии
соответствующими генами молекул первого класса из главного комплекса тканевой
совместимости присутствие на клеточной поверхности аутоантигенов в
комплексе с молекулами второго класса комплекса может вызвать системную
526
Глава 31
Аутореактивный
Т-лимфоцит
индуктор
Поверхностный
рецептор
индуктора
Нет распознавания
аутоантигена
аутореактивным
индуктором
Аутоантиген
Клетка-
мишень
Есть
распознавание
аутоантигена
Молекула
первого класса
из главного
комплекса
тканевой
совместимости
Молекула
второго класса
из главного
комплекса
тканевой
совместимости
Рис. 31.2. Аутоиммунная реакция как результат недостаточной экспрессии
молекул первого класса из главного комплекса молекул тканевой
совместимости и экспрессии молекул второго класса
аутоиммунную реакцию (рис. 31.2). Известно, что в клетках щитовидной
железы больных аутоиммунным тиреоидитом идет интенсивная
экспрессия молекул второго класса из главного комплекса тканевой
совместимости. При сахарном диабете на поверхности бета-клеток поджелудочной
железы также выявляют повышенную экспрессию молекул второго класса из
главного комплекса тканевой совместимости. Не исключено, что
повышенная экспрессия молекул второго класса при соответствующих
аутоиммунных заболеваниях представляет собой результат активации
соответствующих Т-индукторов, которые через действие гамма-интерферона вызывают
экспрессию молекул второго класса на клеточной поверхности, тем самым
делая клетку более подверженной разрушению в результате аутоиммунной
реакции.
Рост частоты аутоиммунных заболеваний по ходу старения очевидно
связан с инволюцией вилочковой железы и лимфоидной ткани как
причины недостаточного поддержания иммунологической толерантности по
отношению к аутоантигенам. По-видимому, большая частота аутоиммунных
заболеваний у женщин связана с влияниями эстрогенов.
Глава 32
АНЕМИИ
Анемия - состояние вследствие снижения числа циркулирующих
эритроцитов.
У женщин об анемии свидетельствует содержание гемоглобина в
крови (НЬ) меньшее, чем 120 г/л и гематокрит (Ht) ниже 36 %. У мужчин
возникновение анемии констатируют при НЬ < 140 г/л и Ht < 42 %.
НЬ не всегда отражает число циркулирующих эритроцитов. После
острой кровопотери НЬ может оставаться в нормальных пределах при
дефиците циркулирующих эритроцитов, обусловленном снижением объема
циркулирующей крови (ОЦК). При беременности НЬ снижен вследствие
увеличения объема плазмы крови при нормальном числе эритроцитов,
циркулирующих с кровью.
Клинические признаки гемической гипоксии, связанной с падением
кислородной емкости крови вследствие снижения числа циркулирующих
эритроцитов, возникают при НЬ меньшем, чем 70 г/л. О тяжелой анемии
говорят бледность кожных покровов и тахикардия как механизм
поддержания через рост минутного объема кровообращения адекватного
транспорта кислорода с кровью, несмотря на ее низкую кислородную емкость.
Содержание ретикулоцитов в крови отражает интенсивность
образования эритроцитов, то есть является критерием реакции костного мозга на
анемию. Содержание ретикулоцитов обычно измеряют в процентах от
общего числа эритроцитов, которое содержит единица объема крови. Ре-
тикулоцитарный индекс (РИ) - показатель соответствия реакции
усиления образования новых эритроцитов костным мозгом тяжести анемии:
РИ = 0,5 х (содержание ретикулоцитов х Ht больного/нормальный Ht).
РИ, превышающий уровень в 2-3 %, свидетельствует об адекватной
реакции интенсификации эритропоэза в ответ на анемию. Меньшая
величина говорит об угнетении образования эритроцитов костным мозгом как
о причине анемии.
Определение величины среднего эритроцитарного объема
используется для того, чтобы отнести анемию у больного к одной из трех
совокупностей: а) микроцитарные; б) нормоцитарные; в) макроцитарные. Нор-
моцитарную анемию характеризует нормальный объем эритроцитов, при
микроцитарной анемии он снижен, а при макроцитарной повышен.
Нормальный диапазон колебаний среднего эритроцитарного объема
составляет 80-98 мкм3.
Анемия при определенном и индивидуальном для каждого пациента
уровне концентрации гемоглобина в крови через снижение ее
кислородной емкости вызывает гемическую гипоксию. Гемическая гипоксия
служит стимулом ряда защитных реакций, направленных на оптимизацию и
528
Глава 32
рост системного транспорта кислорода (схема 32.1). Если
компенсаторные реакции в ответ на анемию оказываются несостоятельными, то
посредством неирогуморальнои адренергическои стимуляции сосудов
сопротивления и прекапиллярных сфинктеров происходит
перераспределение минутного объема кровообращения (МОК), направленное на
поддержание нормального уровня доставки кислорода в мозг, к сердцу и легким.
При этом в частности падает объемная скорость кровотока в почках.
Нормальный
системный
транспорт
кислорода
Действие этиологического
фактора анемии
Адекватный
потребностям
организма
минутный объем
кровообращения
Системный транспорт
кислорода, не
соответствующий
потребностям организма
Достаточная
концентрация
гемоглобина в
крови
Низкий
относительно
потребностей
организма МОК
Оптимальный уровень
различия в содержании
кислорода между
артериальной и венозной
кровью (авР02)
Аномально
низкая
концентрация гемоглобина
в крови
Прежний
уровень
авР02
Гемическая гипоксия
Нейрогуморальная
адренергическая
стимуляция сердца
Рост синтеза и
высвобождения в кровь
эритропоэтина
Увеличение
содержания 2,3-дифосфогли-
церата в эритроцитах
X
Метаболический лактат-
ный ацидоз
Рост минутного
объема
кровообращения
Увеличение
концентрации гемоглобина в
циркулирующей крови
ЗЕ
Возрастание
авР02
I
Увеличение системного транспорта кислорода
Схема 32.1. Компенсация гемической гипоксии, связанной с анемией
Гемоглобин, составляющий 95 % массы эритроцита, в основном
состоит из глобина и содержащего железо компонента гема-протопорфири-
на. Любое расстройство синтеза гемоглобина и (или) обмена железа
может привести к анемии, которую характеризует низкое содержание гемо-
Анемии
529
глобина в красных кровяных клетках. Как правило, при анемиях такого
происхождения возникают гипохромия (низкое содержание гемоглобина в
эритроцитах) и микроцитоз (снижение размеров красных кровяных
клеток). Выделяют четыре основных вида анемий, возникающих вследствие
нарушений синтеза гемоглобина и обмена железа:
1. Железодефицитные анемии.
2. Анемии как следствия хронических воспалительных заболеваний.
3. Сидеробластные анемии.
4. Таллассемии.
АНЕМИИ, ВЫЗВАННЫЕ НАРУШЕНИЯМИ СИНТЕЗА
ГЕМОГЛОБИНА И ОБМЕНА ЖЕЛЕЗА
Железодефицитные анемии. Наиболее частая причина дефицита в
организме железа - это кровопотеря, в результате которой поступление
железа в организм с пищей становится низким относительно уровня его
утилизации при образовании эритроцитов. В частности к железодефицитным
анемиям могут приводить: кровоизлияния из сосудов, поврежденных при
образовании пептических язв желудка и двенадцатиперстной кишки,
менструальная кровопотеря. Иногда у новорожденных и детей утилизация
железа для эритропоэза преобладает над его поступлением в организм,
что без какой-либо кровопотери вызывает железодефицитную анемию.
Анемии вследствие хронических воспалительных процессов. У
больных с длительно текущими (более одного месяца) заболеваниями,
патогенез которых во многом составляет хроническое воспаление, обычно
развивается легко или умеренно выраженная анемия. При этом тяжесть
анемии находится в прямой связи с продолжительностью и выраженностью
воспалительного процесса. Болезни, которые наиболее часто приводят к
анемии такого происхождения, - это подострый бактериальный
эндокардит, остеомиелит, абсцесс легкого, туберкулез и пиелонефрит. При
аутоиммунных болезнях на поверхности клеток пораженных ткани, органа
образуются иммунные комплексы аутоантитело-аутоантиген. Это ведет к
активации системы комплемента по классическому пути как
инициирующему моменту воспаления, повреждающего ткани и органы больного.
Поэтому многие из аутоиммунных болезней следует считать заболеваниями,
которые во многом характеризует выраженное хроническое воспаление.
Чаще всего из аутоиммунных болезней к анемии вследствие
хронического воспаления приводит ревматоидный артрит.
Одной из причин анемии у больных со злокачественными
новообразованиями является связанное с ними хроническое воспаление.
Непосредственными причинами анемии, обусловленной хроническим
воспалением, в частности являются:
1. Угнетение образования эритроцитов костным мозгом как результат
его длительной стимуляции цитокинами (колониестимулирующими фак-
530
Глава 32
торами), образуемыми и высвобождаемыми клеточными эффекторами
хронического воспаления.
2. Несостоятельность компенсации снижения продолжительности
жизни эритроцитов в крови.
При анемиях вследствие хронического воспаления снижение
содержания железа в эритробластах является следствием нарушения его
доставки к развивающимся эритроидным клеткам в костном мозге.
Недостаток железа в эритроидных клетках приводит к гипохромии и микроцитозу
эритроцитов. Дефицит железа, доступного для синтеза гемоглобина, ведет
к росту содержания в эритроцитах протопорфирина. Массу железа,
доступную для эритропоэза, несмотря на его нормальное содержание в
организме, снижает избыточная системная активация мононуклеарных
фагоцитов, а также увеличение их числа {гиперплазия). В результате
гиперплазии и гиперактивации в системе мононуклеарных фагоцитов происходит
избыточный захват железа активированными мононуклеарами с
повышенной способностью поглощать данный микроэлемент. Повышенная
способность мононуклеаров поглощать железо во многом связана с
высокой концентрацией в циркулирующей крови интерлейкина-1, которая
растет вследствие хронического воспаления. Под действием интерлейкина-1,
циркулирующего с кровью и находящегося в межклеточных
пространствах в повышенной концентрации, нейтрофилы всего организма
интенсивно высвобождают лактоферрин. Этот протеин связывает свободное
железо, высвобождаемое при деструкции отмирающих красных кровяных
клеток, и в повышенных количествах транспортирует его к мононуклеа-
рам, которые захватывают и удерживают данный микроэлемент. В
результате развивается умеренное угнетение эритропоэза, обусловленное
снижением доступности железа дли образования эритроидных клеток.
Предположительно одним из звеньев патогенеза анемий из-за
хронического воспаления можно считать избыточную деструкцию эритроцитов
как результат гиперактивации и гиперплазии в системе мононуклеарных
фагоцитов. О ней свидетельствует укорочение жизни почти нормальных
эритроцитов, патологические изменения которых сводятся к сниженному
содержанию железа и росту содержания протопорфирина.
Сидеробластные анемии. Анемии такого рода связаны с нарушениями
синтеза тема как компонента гемоглобина. Нарушения синтеза
гемоглобина при сидеробластных анемиях характеризует накопление железа в
митохондриях, локализованных вокруг ядра аномальных эритроидных
клеток (сидеробластов). Данные клетки называют «окольцованными»,
так как внутриклеточные депозиты железа формируют вокруг ядра клетки
контур, напоминающий кольцо. Нарушения синтеза гема у больных с си-
деробластными анемиями служат причиной гипохромии и микроцитоза.
Выделяют два основных вида сидеробластных анемий:
1. Наследственная сидеробластная анемия представляет собой
моногенное заболевание, передача которого от родителей больному связана с
Х-хромосомой или наследуется по аутосомно-рецессивному типу.
Предположительно наследственную сидеробластную анемию вызывает врож-
Анемии
531
денный дефицит активности фермента синтетазы гамма-аминолевулино-
вой кислоты (ключевой фермент первого этапа синтеза порфиринов).
Угнетение активности энзима может быть первичным или является
следствием врожденного нарушения метаболизма ее эссенциального кофактора,
пиридоксаль-5 '-фосфата.
2. Приобретенные сидеробластные анемии возникают чаще, чем
наследственные. Приобретенные сидеробластные анемии могут быть
результатом побочного действия лекарств (изониазид и др). Кроме того, они
могут быть идиопатическими.
Нарушение утилизации железа для образования гема при сидеробла-
стных анемиях проявляет себя ростом содержания его ионов в сыворотке
крови, а также возрастанием в ней концентрации ферритина.
Талассемия - это моногенное заболевание, в основе которого лежит
угнетение синтеза одной из полимерных цепей, составляющих молекулу
глобина. В зависимости от вида цепи, синтез которой снижен у больного,
талассемию относят к одной из трех основных групп:
1. Альфа-талассемии. Эти заболевания вызывает делеция (удаление)
из генома организма генов альфа-глобина. Существуют четыре таких
гена. В зависимости от того, какой ген потерян геномом, сидеробластная
анемия варьирует по степени тяжести от незначительной и без каких-либо
заметных клинических проявлений до тяжелой, которая обуславливает
гибель плода в утробе матери.
2. Бета-талассемии, которые обуславливает отсутствие или
дисфункция соответствующего гена. При дисфункции гена его
транскрипция происходит, но приводит к образованию аномальной РНК. Кроме
того, дисфункция гена может состоять и в сниженном образовании
нормальной РНК. Геном содержит два различных гена бета-глобина.
Поэтому существуют два вида бета-талассемий. При более тяжелом виде
бета-талассемии (анемия Кулея) ее симптомы выявляют уже в детстве.
Обычно в тридцатилетнем возрасте, несмотря на гемотрансфузии,
наступает летальный исход. При менее тяжелой бета-талассемии
показаний к гемотрансфузиям нет, и анемия не ограничивает
продолжительность жизни.
При исследовании мазка крови кроме гипохромии и микроцитоза у
больных с талассемиями выявляют пойкилоцитоз, то есть
патологическую вариабельность формы эритроцитов.
МАКРОЦИТАРНЫЕ АНЕМИИ
Макроцитарные анемии характеризует циркуляция с кровью
аномальных эритроцитов, диаметр которых превышает 100 мкм. Существуют три
основных механизма развития макроцитарных анемий:
1. Усиление нормального эритропоэза. Ретикулоциты и юные формы
эритроцитов по размерам превосходят нормальные красные кровяные
532
Глава 32
клетки. Компенсаторная интенсификация эритропоэза (в ответ на крово-
потерю, хроническую гипоксию и др.) может исчерпать резервы роста
образования эритроцитов, что приводит к макроцитарной анемии с ростом
содержания ретикулоцитов в циркулирующей крови.
2. Увеличение площади наружной мембраны эритроцита. Гиперлипи-
демия (рост содержания липидов в плазме крови) через адсорбцию липи-
дов на поверхности эритроцитов увеличивает размеры красных кровяных
клеток и может обуславливать макроцитоз (появление в циркулирующей
крови эритроцитов с диаметром, превышающим 100 мкм). При этом из-
за патологических изменений эритроцитов падает время их циркуляции с
кровью и развивается макроцитарная анемия.
3. Нарушения синтеза ДНК при пролиферации эритроидных клеток.
Мегалобластные анемии - болезни, которые характеризуют
макроцитоз и прочие патологические изменения клеток костного мозга и других
быстро делящихся клеток, связанные с нарушениями синтеза дезоксири-
бонуклеиновой кислоты.
У 90 % больных с мегалобластной анемией ее обуславливают
дефициты в организме витамина Bi2 и фолиевой кислоты. У других больных ме-
галобластная анемия - это следствие побочного действия лекарственных
веществ, таких как а) препараты, содержащие серу, б) метотрексат и гид-
роксимочевина.
Связанный с белком витамин В^ (цианкобаламин, внешний фактор
анемии Кастла) содержится в продуктах животного происхождения - в
мясе, сырах, печени, яйцах и др. Под влиянием кулинарной обработки и
протеолитических ферментов, секретируемых в просвет желудка,
цианкобаламин освобождается от белка. Незначительная часть свободного
витамина Вп (примерно 1 %) всасывается в кровь из просвета желудка.
Свободный витамин В и соединяется с образуемым и секретируемым
париетальными клетками эпителия желудка гликопротеином {внутренний
фактор анемии Кастла). Комплекс, образованный в результате соединения
витамина и гликопротеина, связывается со специфическими рецепторами
эпителиоцитов средней и каудальной части подвздошной кишки. Это
представляет собой необходимое условие поступления цианкобаламина в
циркулирующую кровь из просвета кишечника. В основном витамин
депонируется в печени. Запасы витамина Вп в организме велики и
составляют 2-5 мг при суточной потребности во внешнем факторе на уровне в
1 мкг. Поэтому недостаток витамина вследствие значительного снижения
его поступления в просвет желудочно-кишечного канала или нарушений
его усвоения развивается медленно, в течение 3-6 лет.
Выделяют следующие главные причины недостатка в организме
витамина Bi2:
1. Недостаточное поступление витамина в организм с пищей, что в
развитых странах бывает крайне редко, и служит причиной
мегалобластной анемии у ортодоксальных вегетарианцев.
2. Нарушения секреции в просвет желудка внутреннего фактора
анемии.
Анемии
533
Это наиболее частая причина мегалобластной {пернициозной) анемии
у больных. При пернициозной (лат. pemiciosus - гибельный, опасный)
анемии аутоиммунное поражение обкладочных клеток снижает
высвобождение в просвет желудка соляной кислоты и внутреннего фактора. Пер-
нициозную анемию можно считать заболеванием аутоимунной природы,
так как у 60 % больных в плазме крови выявляют аутоантитела к
антигенам эпителиоцитов, секретирующих внутренний фактор, и (или) к самому
внутреннему фактору. Пернициозная анемия может быть следствием
снижения числа клеток желудочного эпителия, секретирующих
внутренний фактор, обусловленного удалением части или всего желудка. У
небольшой части больных пернициозная анемия - это результат
наследуемой по аутосомно-рецессивному типу неспособности эпителиоцитов
желудка синтезировать внутренний фактор.
3. Недостаточное всасывание витамина из просвета подвздошной
кишки. К нему приводит снижение общей площади внутренней
поверхности подвздошной кишки после ее резекции. Воспаление стенки кишки
снижает в функциональном отношении интактную часть этой площади.
Кишечные дисбактериоз и гельминтозы вызывают пернициозную анемию
через усиленное потребление внешнего фактора микроорганизмами и
гельминтами в просвете кишки. Дисбактериоз как причина пернициозной
анемии особенно часто развивается в слепых петлях кишки.
Пернициозная анемия может быть одним из звеньев патогенеза синдрома
недостаточного кишечного всасывания, который как причину пернициозной
анемии в частности вызывает снижение внешнесекреторной функции
поджелудочной железы.
Коферментная форма витамина Bi2, метилкобаламин, необходим для
нормального эритробластического кроветворения. Тетрагидрофолиевая
кислота, которая образуется при участии метилкобаламина, - это
субстрат для синтеза 5,10-метилтетрагидрофолиевой кислоты
(коферментная форма фолиевой кислоты). Эта кислота участвует в образовании ти-
мидин-фосфата. Тимидин-фосфат - необходим для образования ДНК
эритрокариоцитами и другими быстро делящимися клетками костного
мозга. Недостаток тимидин-фосфата нарушает синтез ДНК, что
вызывает патологические изменения ее структуры. В результате эритрокарио-
циты увеличиваются в размерах. Такие клетки получили название ме-
галобластов (гр. megas - большой + гр. blastos - росток) и мегалоцитов
и напоминают эритрокариоциты и мегалоциты эмбриона человека.
Продолжительность жизни мегалоцитов невелика, и большая их часть
(до 50 %) преждевременно разрушается в костном мозге, что ведет к
анемии.
Основными патоморфологическими признаками мегалобластов
являются нечетко структурированное «открытое» клеточное ядро и зрелые
цитоплазматические внутриклеточные органеллы, которых нет у
нормальных экитроцитов. Неэффективный эритропоэз и усиленный интраме-
дуллярный гемолиз при мегалобластных анемиях приводят к росту
активности в сыворотке крови лактатдегидрогеназы.
534
Глава 32
НОРМОХРОМНЫЕ НОРМОЦИТАРНЫЕ АНЕМИИ
Под нормохромными нормоцитарными анемиями понимают
совокупность болезней и патологических состояний, которые, в первую очередь,
характеризует анемия при нормальных размерах, массе, строении
красных кровяных клеток и содержании гемоглобина в эритроцитах.
Различные нормохромные нормоцитарные анемии не связаны между
собой общими для них звеньями патогенеза. Выделяют два основных
вида нормохромных нормоцитарных анемий:
1. Анемии со сниженной способностью к интенсификации эритропоэза.
2. Анемии при сохраненной способности организма к усилению
образования эритроцитов.
При анемиях, вызванных сниженным образованием эритроцитов, РИ
находится на патологически низком уровне. К ним относят гипо- или
апластическую, а также миелофтизную анемии. Кроме того, к этому виду
снижения числа циркулирующих с кровью эритроцитов и концентрации в
ней гемоглобина принадлежат анемии вследствие сниженной секреции
эритропоэтина и другие анемии, связанные с падением пролиферативной
активности клеток костного мозга.
Гипо- апластические анемии - это группа анемий, обусловленных
первичными нарушениями функций костного мозга, при которых в нем снижается
образование и содержание клеток предшественников эритроцитов на пути
эритропоэза (стволовых клеток). Термин «апластическая анемия» следует
употреблять только в том случае, если из-за низкого содержания в костном
мозге всех видов стволовых клеток возникают анемия, снижение содержания
в циркулирующей крови нейтрофилов (нейтропения) и тромбоцитов (тром-
бощтопения). Предполагают, что апластическая анемия развивается
вследствие разрушения или патологического изменения полипотентной стволовой
клетки, что затрагивает все клеточные линии гемопоэза.
Примером наследственной апластической анемии может служить
анемия Фанконщ которая наследуется по аутосомно-рецессивному типу.
Клинические проявления анемии Фанкони у больных выявляют уже в
детском возрасте. Как наследственное заболевание анемия Фанкони
сочетается с такими врожденными дефектами развития как гипоплазия почек,
отсутствие первых пальцев кистей и др. В основе болезни лежит
врожденная дисфункция внутриядерных систем репарации ДНК. В результате
соматические мутации легко закрепляются в клеточных клонах, что
может служить причиной хромосомных аберраций, в том числе и как
причин угнетения гемопоэза.
У некоторых больных, которым предстоит пересадка костного мозга
в связи с апластической анемией, после подавления иммунитета,
вызываемого лекарственными средствами со свойствами иммунодепрессан-
тов, анемия во многом подвергается обратному развитию. Это
свидетельствует об аутоиммуном механизме развития апластической
анемии, который в основном составляет патогенное функционирование Т-
лимфоцитов.
Анемии
535
Апластическая анемия может быть следствием побочного эффекта
лекарственных средств и действия токсинов. Кроветворную функцию
костного мозга в определенных дозах подавляют иммунодепрессанты и
противоопухолевые средства, которые влияют на гемопоэз одновременно с
разрушающей злокачественные клетки ионизирующей радиацией. К таким
препаратам относятся антагонисты фолиевой кислоты, алкилирующие
средства, антрациклины, производные нитрозомочевины, а также аналоги
пуриновых и пиримидиновых оснований. Воздействие на костный мозг
ионизирующей радиации может быть причиной необратимой аплазии.
Загрязнение среды обитания человека, и в особенности питьевой
воды, производными бензола через действие данного токсического вещества
на костный мозг также может вызывать обратимую апластическую
анемию, которую в некоторых случаях характеризует макроцитоз.
Такой антибиотик как левомицетин (хлорамфеникол) оказывает на
костный мозг двоякое токсическое действие, которое находится в прямой
связи с суточной дозой и длительностью применения препарата. Во-
первых, побочный эффект антибиотика тормозит пролиферативную
активность и угнетает способность к дифференциации клонов эритровдных
клеток предшественников нормальных эритроцитов. Во-вторых
(значительно реже), левомицетин оказывает аналогичное воздействие на клоны
клеток предшественников гранулоцитов и мегакариоцитов. При этом
падение эритропоэза проявляет себя снижением концентрации в сыворотке
крови железа и содержания ретикулоцитов в циркулирующей крови.
У некоторых больных апластическая анемия как результат побочного
эффекта хлорамфеникола протекает по типу идиосинкразии с быстрым
развитием панцитопении и высоким риском летального исхода.
Следует заметить, что установление связи апластической анемии с тем
или иным лекарственным средством затруднено, так как больные часто
принимают несколько препаратов.
Апластическая анемия может осложнять течение вирусных
заболеваний (вирусный гепатит, кроме его видов А и В, инвазия в организм и его
клетки вируса Эпстайна-Барра, вирусные респираторные заболевания и
др.). Парвовирусы вызывают апластическую анемию, а также
избирательно инфицируют эритробласты, что служит причиной гемолиза,
обостряющего анемию.
Миелофтизная анемия - это следствие инфильтрации костного мозга
опухолями, гранулемами и замещения клеток костного мозга,
участвующих в гемопоэзе, соединительной тканью (миелофиброз). При этом
инфильтрирующие костный мозг клетки могут быть местными по
отношению к костному мозгу или заносятся в него как метастазы твердых
злокачественных опухолей. Чаще всего в костный мозг метастазируют клетки
злокачественных опухолей молочной, предстательной и вилочковой
железы, желудка и легких.
Гранулематозное перерождение костного мозга отмечается при далеко
зашедшем костном туберкулезе. Первичные нарушения накопления липи-
дов (болезни Гоше и Ниманна-Пика) в костном мозге через его жировое
536
Глава 32
перерождение вызывают снижение числа клеток, участвующих в гемопо-
эзе, и обуславливают миелофтизную анемию.
Полагают, что анемия и тромбоцитопения как наиболее частые
следствия патогенной клеточной инфильтрации костного мозга обусловлены
не только простым заполнением пространства костного мозга клетками,
не участвующими в гемопоэзе. Дело еще в том, что патогенная клеточная
инфильтрация костного мозга изменяет в нем микроциркуляцию таким
образом, что в циркулирующую кровь выходят незрелые клетки, недолго
живущие в циркулирующей крови.
Анемия вследствие сниженного образования и высвобождения в кровь
эритропоэтина развивается у больных с хронической почечной
недостаточностью в результате падения образования гормона почками,
постепенно теряющими свои нормально функционирующие клеточные элементы.
Анемии при сохраненной способности организма интенсифицировать
эритропоэз могут быть постгеморрагическими. Если у больного в
состоянии после кровопотери (пациенты с пептическими язвами желудка,
больные после высокотравматичных оперативных вмешательств и др.)
резервы железа в организме достаточны для компенсаторной
интенсификации образования эритроцитов, то о ней свидетельствует рост
содержания ретикулоцитов в циркулирующей крови.
Второй вид анемий с сохраненной способностью к интенсификации
эритропоэза - это гемолитические анемии. Гемолитические анемии
характеризует снижение продолжительности жизни эритроцитов в
циркулирующей крови. Известно два основных пути преждевременной гибели
эритроцитов. Во-первых, они могут разрушаться в кровеносном русле с
высвобождением продуктов цитолиза непосредственно в плазму. Внутри-
сосудистый гемолиз может быть обусловлен механической травмой
эритроцитов, активацией на их поверхности системы комплемента, фиксации
ее фракций на наружной клеточной мембране красной кровяной клетки, а
также воздействием экзогенных токсинов. Второй из двух основных путей
гемолиза - это поглощение эритроцитов макрофагами в печени и селезенке
(элименты системы мононуклеарных фагоцитов), в которых они
разрушаются, а продукты цитолиза подвергаются лизису через активность протео-
литических ферментов фагоцитирующих клеток. Клетки системы
мононуклеарных фагоцитов захватывают эритроциты при двух условиях:
♦ изменение поверхностных свойств эритроцитов, в частности
связанное с фиксацией на них иммуноглобулинов со свойствами ауто-
антител;
♦ снижение деформируемости красных кровяных клеток, в том числе
и в результате врожденных аномалий их строения.
Дискоидная форма эритроцитов способствует их способности
деформироваться для прохождения по микрососудам, поскольку площадь
поверхности нормального дискоцита (нормального эритроцита) на 60-70 %
превышает уровень, минимально достаточный для того, чтобы в клетке
поместилось ее содержимое. Способность эритроцита к изменениям
конфигурации обеспечивают:
Анемии
537
♦ вязкоэластичные свойства его мембран;
♦ высокое отношение площади поверхности нормального эритроцита
к его массе;.
♦ способность агрегации молекул гемоглобина в каком-либо локусе
клетки. Нарушения какого-либо из данных свойств эритроцита
может быть причиной гемолиза.
В ответ на снижение длительности жизни эритроцитов в
циркулирующей крови у больных с гемолитическими анемиями происходит
компенсаторная интенсификация эритропоэза с ростом содержания ретику-
лоцитов в циркулирующей крови и гиперплазией эритроидных клеток в
костном мозге. Усиленная на системном уровне деструкция эритроцитов
приводит к росту в сыворотке крови активности лактатдегидрогеназы и
снижению в ней концентрации гаптоглобина. Усиленное разрушение
эритроцитов может быть причиной роста содержания в плазме крови
несвязанного билирубина и желтухи. Каждый из сотен видов гемолитической
анемии можно отнести к одному из двух типов:
♦ Гемолитическая анемия как результат влияний факторов внешних
по отношению к эритроцитам.
♦ Гемолитическая анемия вследствие причин, присущих самим
эритроцитам.
Гемолитическая анемия, развившаяся под влиянием факторов внешних
по отношению к эритроцитам, может быть следствием образования ауто-
антител к аутоантигенам поверхности красных кровяных клеток. Ауто-
антитела относительно эритроцитов и клеток системы мононуклеарных
фагоцитов выступают в качестве опсонинов. Образование комплексов аутоан-
титело-аутоантиген на поверхности красной кровяной клетки активирует
на ее поверхности систему комплемента по классическому пути, что может
быть одной из причин деструкции эритроцитов и гемолитической анемии.
Окончательно выявить аутоиммунную природу гемолитической анемии
позволяет тест Кумбса. Тест Кумбса дает информацию о фиксации на
поверхности эритроцитов аутоантител и фракций системы комплемента
(иммуноглобулины из класса G, фракция системы комплемента СЗ), которые
как опсонины ведут к усиленной деструкции эритроцитов клетками
системы мононуклеарных фагоцитов и вызывают гемолитическую анемию
(сывороточные протеины-аутоопсонины). В основе теста лежит способность
антител, полученных при иммунизации животных сывороточными про-
теинами-аутоопсонинами, агглютинировать эритроциты больного с
гемолитической анемией при условии фиксации протеинов-аутоопсонинов на
их поверхности. При этом способность сывороток крови животных,
содержащих антитела к протеинам-аутоопсонинам, вызывать агглютинацию
эритроцитов с аутоопсонинами на поверхности обуславливает
положительную прямую пробу Кумбса. Положительная прямая проба Кумбса
свидетельствует об аутоиммунной природе гемолитической анемии.
Тепловые антитела - это иммуноглобулины, наиболее интенсивно
реагирующие с антигеном при температуре тела и относящиеся к классу G
иммуноглобулинов. Эти антитела вызывают гемолиз как собственных
538
Глава 32
эритроцитов больного, так и практически неповрежденных красных
кровяных клеток переливаемой крови. Синдром анемии, связанный с тепловыми
антителами, называют аутоиммунной гемолитической анемией. Так как в
последние годы выявлен ряд лекарств, вызывающих данный синдром, то
основное внимание стали уделять внешнему этиологическому фактору
гемолитической анемии, возникающей через образование аутоантител.
Поэтому стали чаще применять термин иммуногемолитическая анемия.
При иммуногемолитической анемии аутоопсонины вызывают
взаимодействие эритроцитов с клетками системы мононуклеарных фагоцитов
человека, которое придает пораженным красным кровяным клеткам
шарообразную форму, то есть ведет к образованию сфероцитов. Это
снижает нормальную деформируемость красных кровяных клеток как
необходимое условие их быстрого прохождения через микрососуды селезенки,
то есть предрасполагает к деструкции эритроцитов вследствие
патогенного функционирования мононуклеарных фагоцитов.
При наиболее тяжелой форме иммуногемолитической анемии
происходит молниеносный массивный гемолиз как причина появления в
циркулирующей крови свободного гемоглобина (гемоглобинемия) и гемоглоби-
нурии, то есть свободного гемоглобина в конечной моче, а также острой
почечной недостаточности.
При синдроме холодовой агглютинации гемолитическую анемию
обуславливает рост образования системой иммунитета иммуноглобулинов со
свойствами аутоантител к аутоантигенам поверхности эритроцитов
больного (аутоантигены I, i, Pr, Gd, Sda). Наиболее интенсивно агглютинация
эритроцитов под влиянием данных иммуноглобулинов, фиксированных на
поверхности красных кровяных клеток, происходит при температуре
окружающей клетки среды на уровне в 4 °С. Поэтому эти иммуноглобулины
называют Холодовыми аутоантителами (иммуноглобулины класса М). В
низких титрах, меньших, чем 1:32, холодовые аутоантитела присутствуют во
внеклеточной жидкости и жидкой части плазмы крови здоровых людей. Их
образование иммунной системой возрастает при таких инфекциях как ми-
коплазмоз, цитомегаловирусная инфекция, инфекция вирусом Эпстайна-
Барра, трипаносомоз и малярия. Образование Холодовых аутоантител
достигает пика через 2-3 недели инфекционного заболевания и не приводит к
каким-либо патологическим последствиям, если только не вызывает гемолиза.
Массивное высвобождение в циркулирующую кровь Холодовых
антител как причину гемолитической анемии обуславливают: а)лимфома;
б) поликлональная пролиферация иммунокомпетентных клеток в ответ на
стимуляцию иммунной системы антигенами Mycoplasma pneumoniae и
при инфекционном мононуклеозе; в) аденовирусные инфекции и другие
иммунопатологические состояния.
У большинства больных агглютинация эритроцитов Холодовыми
агглютининами наступает при температуре циркулирующей крови 32 °С.
Следует заметить, что большинство Холодовых аутоантител оказывают
минимальное повреждающие действие или вообще не влияют на
продолжительность жизни эритроцитов.
Анемии
539
Гемолитическую анемию, развивающуюся под влиянием факторов
внешних по отношению к эритроцитам, могут вызывать токсические
влияния на красные кровяные клетки со стороны возбудителей
инфекционных заболеваний, и малярии в частности.
Гиперлипопротеинемии через адсорбцию липопротеинов на
поверхности эритроцита снижают его деформируемость, что может обуславливать
незначительную гемолитическую анемию. Из-за адсорбции липопротеинов
поверхностью красных кровяных клеток они становятся как бы
утыканными микроскопическими шипами, что послужило причиной такого названия
данного вида гемолитической анемии как «шпороклеточная». Шпорокле-
точная анемия осложняет течение хронической печеночной
недостаточности как причины гиперлипидемии/гиперлипопротеинемии.
Тромбоз и расстройства функции микрососудов могут быть причинами
деструкции эритроцитов в их просвете и обуславливать гемолитическую
анемию. Например, при диссеминированном внутрисосудистом
свертывании отложения фибрина в просвете микрососудов повышают трение
эритроцитов о сосудистую стенку, что служит фактором гемолиза и анемии.
Причиной гемолиза может быть и деструкция эритроцитов при их
трении о поверхности искусственных клапанов сердца.
В основе гемолитических анемий вследствие причин присущих самим
красным кровяным клеткам лежат различные врожденные нарушения
структуры и функций эритроцитов, которые определяют их особую
предрасположенность к преждевременной деструкции после начала
циркуляции с кровью.
Нарушение функции плазматической мембраны эритроцита как
причина гемолитической анемии в частности обуславливает такую болезнь
как наследственный сфероцитоз. При наследственном сфероцитозе
генетически детерминированное расстройство функционирования активного
переноса натрия и калия через наружную клеточную мембрану служит
причиной отека красных кровяных клеток. В результате эритроциты
приобретают шарообразную форму (сфероцитоз), то есть становятся сферо-
цитами. Сфероциты характеризуют небольшие относительно макроцитов
размеры, отсутствие бледной окраски центральной части клетки, которую
наблюдают у нормальных эритроцитов, а также содержание гемоглобина
более высокое, чем его концентрация в патологически не измененных
красных кровяных клетках. Нарушения содержания натрия и воды в
эритроцитах, связанные с наследственным сфероцитозом, обуславливают их
особую подверженность деструкции в селезенке через функционирование
локализованных там клеток системы мононуклеарных фагоцитов.
Поэтому спленэктомия (удаление селезенки) часто полностью подвергает
гемолитическую анемию данного происхождения обратному развитию,
несмотря на сохранение сфероцитоза.
Наследственные нарушения структуры гемоглобина
(гемоглобинопатии), которых известно уже более 250 разновидностей, - это следствия
точечных мутаций, которые приводят к замене одной аминокислоты в
глобиновой цепи гемоглобина другой. В результате растет жесткость мо-
540
Глава 32
лекулы гемоглобина, что снижает деформируемость эритроцитов, тем
самым приводя к гемолитической анемии. Рост ригидности молекулы
гемоглобина - это наиболее частая причина гемолитической анемии
вследствие гемоглобинопатии. Примером гемолитической анемии, связанной с
гемоглобинопатиями, может быть серповидно-клеточная анемия, при
которой эритроциты содержат аномальный гемоглобин S. При других гемо-
глобинопатиях эритроциты содержат патологические гемоглобины С, О и
SC. Для серповидноклеточной анемии характерны периодические
гемолитические кризы с острой болью различной локализации, которую
обуславливают нарушения периферического кровообращения, связанные с
закупоркой микрососудов продуктами гемолиза.
Генетически детерминированные аномалии на уровне цитозоля
эритроцитов и внутриклеточных ферментов красных кровяных клеток также
могут быть причинами врожденных гемолитических анемий. Красная
кровяная клетка выходит из костного мозга в циркулирующую кровь без ядра
и митохондрий. Лишенный генома зрелый эритроцит в течение 120 дней
циркуляции с кровью в своем функционировании полностью зависит от
того набора ферментов, который содержится в нем сразу после выхода в
циркулирующую кровь. Поэтому любой наследственный дефицит
внутриклеточных ферментов эритроцита снижает жизнеспособность красных
кровяных клеток и может приводить к гемолитической анемии.
Нередко причиной гемолитической анемии служит врожденный
дефицит активности глюкозо-6-фосфатдегидрогеназы. Известно уже
более 150 разновидностей данной энзимопатии, наследование которой
связано с Х-хромосомой. В результате наследственного дефицита активности
фермента красные кровяные клетки становятся особенно подверженными
повреждению свободными кислородными радикалами. Причина такого
патологического изменения свойств эритроцитов состоит в особо
быстром развитии их гипоэргоза вследствие гипоксии из-за невозможности
компенсаторной активации анаэробного гликолиза, что связано с
дефицитом активности его ключевого фермента глюкозо-6-фосфатдегидроге-
назы. Это ведет к гемолитической анемии при некоторых инфекциях и в
результате побочного действия лекарственных средств (хинин и др.).
Другой вид энзимопатии в эритроцитах, обуславливающий
гемолитическую анемию, - это дефицит активности пируваткиназы, который
наследуется по аутосомно-рецессивному типу.
Глава 33
ПАТОФИЗИОЛОГИЯ РАССТРОЙСТВ
ФУНКЦИОНАЛЬНЫХ СИСТЕМ И НАРУШЕНИЙ
ГОМЕОСТАЗА В ОСТРОМ ПЕРИОДЕ ПОСЛЕ
ТЯЖЕЛЫХ БОЕВЫХ РАНЕНИЙ И ПРИ РАЗВИТИИ
ВОЕННО-ТРАВМАТИЧЕСКОГО ШОКА
«Легендарный орел советской военной медицины» начальник
Главного военно-санитарного управления Красной Армии генерал-полковник
медицинской службы Ефим Иванович Смирнов в 1944 году пришел к
следующему выводу: «... нас не удовлетворяет ни существующий подход
к статистике шока, ни результаты работы по профилактике и лечению
шоковых состояний у раненых. Чтобы быть правильно понятым, я
оговариваюсь, что это не следует понимать как заявление, которое делается
людьми, взявшими себе за правило никогда не довольствоваться
достигнутыми успехами... В результате нашей работы не достигнуто тех
успехов, которых можно было бы ожидать, учитывая уровень современной
медицины и давность изучения разбираемой проблемы». С тех лет
прошло уже более полувека. За это время произошла подлинная революция в
теории и практике лечения больных с тяжелой раневой болезнью.
Качественное изменение теоретических представлений о патогенезе раневой
болезни у раненых в состоянии военно-травматического шока и
патогенетических подходов к терапии при данном критическом состоянии в
основном обусловили:
♦ Осознание представлений о военно-травматическом шоке как о
состоянии, которое вызывает и характеризует недостаток объема
циркулирующей крови (гиповолемия) как причина падения системного
транспорта кислорода и стимул ряда защитных реакций, быстро
превращающихся в звенья патогенеза.
♦ Выяснение общего патогенеза нарушений периферического
кровообращения и патогенных межклеточных взаимодействий как
причин расстройств микроциркуляции, гипоксии клеток, а также
нехватки в них свободной энергии (гипоэргоза) и субстратов
метаболизма.
♦ Выявление роли военно-травматического шока как инициирующего
момента системной воспалительной реакции, разрушающей
структурно-функциональные элементы эффекторов функций и
вызывающей множественную системную недостаточность. При этом
военно-травматический шок стали рассматривать как индуктор
развития респираторного дистресс-синдрома взрослых, гиперцитокине-
мии и других патологических состояний организма и его систем,
осложняющих тяжелую раневую болезнь.
542
Глава 33
♦ Определение военно-травматического шока как причины иммунной
анергии, обуславливающей инфекционные осложнения, которые
являются ведущей причиной летальных исходов в отдаленном
периоде тяжелой раневой болезни.
♦ Разработка патогенетических принципов инфузионно-трансфузион-
ной терапии как способа коррекции гиповолемии, нормализации
микроциркуляции, устранения гипоэргоза и голодания клеток, а
также блокады трансформации защитных реакций в звенья
патогенеза тяжелой раневой болезни.
♦ Применение в военно-полевых условиях современных способов
анальгезии, анестезиологического пособия и интенсивной терапии,
разработанных с учетом особенностей патогенеза
военно-травматического шока и тяжелой раневой болезни.
♦ Внедрение в клиническую практику подхода к анальгезии как к
способу нормализации системной регуляции и активации стресс-
лимитирующих систем всех уровней.
Несомненно, что в последние десятилетия экстраполяция
патогенетических принципов терапии, выработанных на основании фактов,
полученных в эксперименте и в ходе клинических исследований, на широкую
практику лечения больных в состоянии тяжелого военно-травматического
шока принесла снижение летальности, частоты осложнений острого и
отдаленного периодов тяжелой раневой болезни. Тем не менее, следует
заметить, что в нашей стране это было в основном связано с внедрением
современных и адекватных патогенезу тяжелой раневой болезни способов
анестезиологического пособия, интенсивной терапии и хирургического
лечения на этапе пусть даже временно развернутого, но стационара. Это
заставляет еще раз вспомнить Ефима Ивановича Смирнова (1944):
«... оказание помощи на поле боя и своевременный вынос раненых -
основная проблема военной медицины. Поэтому крайне важно и
необходимо всесторонне и подлинно освещать и тщательно анализировать все
то, что относится ко всякого рода затруднениям и помехам в этом деле.»
В этой связи представляется необходимым предварить изложение
материала, касающегося военно-травматического шока с клинико-патофи-
зиологической точки зрения, результатами теоретической разработки
реаниматологической помощи тяжелым раненым на поле боя и во время
выноса.
Основными элементами реаниматологической помощи на
догоспитальном этапе следует считать:
♦ Временную или при возможности окончательную остановку
кровотечения.
♦ Восстановление проходимости дыхательных путей.
♦ Обезболивание и транспортную иммобилизацию.
♦ Гемодилюцию, достигаемую через инфузии гипо-, изо- и гиперос-
моляльных относительно внеклеточной жидкости растворов.
♦ Пункцию полости перикарда, медиастинотомию при развитии
тампонады сердца и эмфиземы средостения.
Патофизиология расстройств функциональных систем и нарушений гомеостаза... 543
♦ Устранение напряженного пенвмоторакса пункцией и
дренированием плевральной полости.
Известно, что реаниматологическая помощь на поле боя и во время
выноса существенно улучшает исходы лечения раненых на последующих
этапах медицинской эвакуации. Особенно она важна при оказании
доврачебной и первой врачебной помощи в условиях боевых действий с
использованием высокоточного оружия. В современной войне 67 %
санитарных потерь составят раненые на поле боя, 40 % которых смогут
выжить в период до поступления на этап квалифицированной медицинской
помощи лишь при условии оказания адекватной реаниматологической
помощи на месте ранения, во время выноса и эвакуации. При оказании
медицинской помощи в условиях современной и особенно локальной
войны для обеспечения эффективной реаниматологической помощи на
поле боя, во время выноса и эвакуации этапы доврачебной и врачебной
помощи целесообразно усиливать специально оснащенными
реанимационными бригадами. Врачи и средние медицинские работники, входящие в
их состав, должны пройти специализацию в области хирургии и
анестезиологии-реаниматологии. Так как настоящее сообщение в основном
посвящено теоретическому аспекту проблемы военно-травматического
шока, то в этой связи, коснувшись организации помощи раненым в
состоянии шока на войне, следует еще раз обратиться к Е.И. Смирнову (1944):
«Если вы скажете, что эти соображения недостаточно научны, что
дескать, это вопрос организационного порядка, я с вами не соглашусь и
готов убеждать любого, что это и есть подлинная военно-медицинская
наука. И только в такой постановке этого сложного вопроса - залог нашего
успеха в деле борьбы с шоком».
ПРИНЦИПЫ УСТРАНЕНИЯ ПАТОЛОГИЧЕСКОЙ БОЛИ
И ПРОВЕДЕНИЯ АНЕСТЕЗИОЛОГИЧЕСКОГО ПОСОБИЯ У
БОЛЬНЫХ С ТЯЖЕЛОЙ РАНЕВОЙ БОЛЕЗНЬЮ
Первой на ранение реагирует нервная система. Патологическая
реакция центральной нервной системы (ЦНС) расстраивает функциональные
системы и повреждает эффекторы функций еще до того, как свой вклад в
патогенез военно-травматического шока и раневой болезни внесут гипер-
цитокинемия и эндотоксикоз. Тяжелое ранение - это всегда причина
образования новых патогенных интеграции нейронов в ЦНС, которые
имеют прямо патогенное значение. Ноцицептивная афферентация из гипок-
сичных вследствие нарушений микроциркуляции тканей подавляет анти-
ноцицептивную систему, предрасполагая к образованию генераторов
патологически усиленного возбуждения (ГПУВ). ГПУВ представляет собой
патогенную интеграцию нейронов, которая определяет возникновение в
разных отделах ЦНС и ноцицептивной системы патологических алгиче-
ских систем (Крыжановский Г.Н., 1993) и придает боли свойства типово-
544
Глава 33
го патологического процесса, развитие которого в некоторой мере
свободно от действия первопричины раневой болезни. В данном контексте
под первопричиной раневой болезни мы понимаем взаимодействие
организма с тяжелым ранением как с комплексом специфических патогенных
раздражений.
Условием и причиной формирования ГПУВ служит недостаточность
тормозных механизмов в принадлежащей ему популяции нейронов. Сразу
после тяжелого ранения возникают как первичная, так и вторичная
недостаточность тормозных механизмов в популяции нейронов ГПУВ.
Первичное нарушение тормозных механизмов обусловлено недостатком
свободной энергии в нейронах, а вторичное вызывается функциональным
преобладанием на Системном уровне ноцицептивной афферентации из
поврежденных тканей. В значительной массе гипоксичных вследствие
раневой болезни тканях, когда расстройства микроциркуляции выходят
далеко за пределы собственно раневых повреждений, продолжают
функционировать ноцицепторы, устойчивые к гипоксии. В них рождается особо
патогенная центростремительная импульсация, которая в сочетании с кровопо-
терей через расстройство внутрицентральных отношений расстраивает
доставку кислорода клетке на системном уровне таким образом, что
расстройства гомеостаза становятся более выраженными, чем нарушения
постоянства внутренней среды, обусловленные только кровопотерей.
Тяжелое ранение всегда служит причиной и создает условия
структурно-функциональной организации патологических систем, которые
формируются во всех отделах ЦНС где представлены образования,
проводящие и усиливающие ноцицептивную афферентацию. В
патологические системы оказываются вовлеченными и те образования центральной
нервной системы, где локализованы центральные звенья компонентов
боли, сложного явления, имеющего у тяжелых раненых сугубо патогенный
характер.
Особенностью патологических систем в различных отделах ЦНС при
тяжелых ранениях является их обязательный выход на периферию с
критическими повреждениями эффекторов функций, которые могут
составить основное звено танатогенеза. Так связанный с патологической болью
нейрогенный спазм легочных микрососудов создает отправную точку
развития респираторного дистресс-синдрома взрослых. Избыточная
нервная симпатическая стимуляция сердца, стимулом которой является
патологическая боль, обуславливает острую кардиальную нейродистрофию.
Спазм приводящих артериол нефрона при неустраненной патологической
боли опасно снижает скорость клубочковой фильтрации, предрасполагая
к острой преренальной почечной недостаточности. Известно, что после
минно-врывных ранений, с которыми мы так часто встречались в
афганскую кампанию, нейродистрофические расстройства, во многом
определяемые патологической болью, быстро охватывали как все отделы ЦНС,
так и периферические нервные образования.
Системная нейродистрофия вследствие тяжелых ранений и травм не
является фатальной. Комплексная превентивная (интенсивная) патогене-
Патофизиология расстройств функциональных систем и нарушений гомеостаза... 545
тическая терапия предупреждает стабилизацию патологических систем,
то есть необратимый характер раневой болезни в ее остром и отдаленном
периодах. Один из ее базисных компонентов - это чисто нейротропное
воздействие, эффективная многоуровневая аналгезия.
Общим свойством для всех видов анестезии является способность к
функциональной дезинтеграции ЦНС. Дезинтеграция системных
отношений, их автономизация являются универсальными биологическими
закономерностями, выступая существенными признаками патологии. В
этой связи дезинтегративный эффект анестезии следует рассматривать
как патогенное действие, вызывающее вторичную недостаточность
тормозных механизмов. Поэтому неизбирательную с системным
дезинтегрирующим и депрессивным эффектами анестезию следует считать
воздействием, облегчающим формирование в ЦНС патологических
систем, повреждающих эффекторы функций на периферии. Это служит
причиной выбора для анестезии при тяжелой раневой болезни кетамина.
Данный неингаляционный анестетик минимально дезинтегрирует ЦНС
и потенцирует системное действие наркотических анальгетиков.
Анестезия кетамином в настоящее время признана наиболее
соответствующей военно-полевым условиям.
Кетаминовую анестезию индуцируют медленным внутривенным
введением 2 мг/кг массы тела анестетика. Одновременно с введением в
анестезию кетамином исходного уровня центральной анальгезии достигают
внутривенным введением 7,5 мкг/кг массы тела фентанила в одном
шприце с раствором кетамина.
Центральное обезболивание фентанилом вызывают после
предшествующего сужения входа в антиноцицептивную систему тем или иным
способом блокады нервных стволов местным анальгетиком. Анальгезию
поддерживают и усиливают перед наиболее травматичными моментами
вмешательства внутривенным введением 100 мкг фентанила.
Поддерживающую дозу фентанила вводят каждые 15-30 мин операции и анестезии.
В одном шприце с кетамином и фентанилом вводят 4 мг ардуана, антиде-
поляризующего миорелаксанта без опасных ганглиоблокирующего и гис-
таминвысвобождающего эффектов.
Блокады нервных стволов производят после устранения гемодилю-
цией как элементом стандартизированной предоперационной
подготовки критической гиповолемии, эксикоза и мобилизации в ходе гемоди-
люции эндогенных резервов альбумина. Производят блокады
бедренного и седалищного нервов из двух доступов, межреберную блокаду (при
ранениях живота - билатеральную), блокаду плечевого сплетения. С
учетом повышенной чувствительности тяжелых раненых к резорбтив-
ному действию местных анальгетиков более 240 мг лидокаина, триме-
каина одномоментно не вводят. Бокируя нервные стволы, ограничивают
афферентный вход в ноцицептивную систему, создавая условия
устойчивой активации антиноцептивной системы, в значительной мере
устраняя вторичную недостаточность тормозных механизмов в популяции
ГПУВ.
546
Глава 33
ПАТОГЕНЕЗ РЕСПИРАТОРНОГО ДИСТРЕСС-СИНДРОМА
КАК ОСЛОЖНЕНИЯ ВОЕННО-ТРАВМАТИЧЕСКОГО ШОКА
И ОСТРОГО ПЕРИОДА ТЯЖЕЛОЙ РАНЕВОЙ БОЛЕЗНИ
В соответствии с уравнением Старлинга патологический рост
проницаемости стенки микрососудов резко увеличивает массу жидкости и
белка, которая из просвета сосудов мигрирует в интерстиции. При некардио-
генном отеке легких, основное звено патогенеза которого - аномальное
увеличение проницаемости стенки микрососудов легких для белка и
воды, в легочном интерстиции резко возрастает содержание протеинов
плазмы крови.
Некардиогенный отек легких как синдром не эквивалентен респираторному
дистресс-синдрому взрослых (РДСВ). Это звено патогенеза РДСВ.
Респираторный дистресс-синдром взрослых в клиническом и клини-
ко-патофизиологическом отношении характеризуют: а) артериальная ги-
поксемия, которую не устраняет обогащение вдыхаемой газовой смеси
кислородом до 50 объемных процентов {рефрактерная гипоксемия);
б) двусторонние «пушистые», «хлопьевидные» затемнения легочных
полей на рентгенограмме; в) падение статической податливости легких,
которое проявляет себя ростом давления в системе аппарат ИВЛ-легкие
больного на вдохе.
В основе морфопатогенеза респираторного дистресс-синдрома лежат:
♦ активация эндотелиальных клеток легочных микрососудов с ростом
их адгезивной способности;
♦ повышение проницаемости стенки микрососудов легких;
♦ рост экспрессии тромбогенного потенциала эндотелиоцитов и
сосудистой стенки легочных микрососудов, вызывающий
микротромбоз, распространенный в пределах всей легочной паренхимы;
♦ адгезия активированных нейтрофилов к активированным легочным
эндотелиоцитам как инициирующий момент острого воспаления,
лишенного биологической цели.
Индуктором патогенного острого воспаления, которое без адекватной
интенсивной терапии может разрушить большинство
структурно-функциональных единиц легких (терминальных респираторных единиц, рее-
пиронов) и привести к РДСВ, является военно-травматический шок как
результат взаимодействия типовых патологических процессов в остром
периоде тяжелой раневой болезни. Потеря легкими определенного числа
структурно-функциональных единиц и дисфункции оставшихся респиро-
нов у больных тяжелой раневой болезнью неизбежны и вызывают
артериальную гипоксемию. При достижении этими патологическими
изменениями эффектора функции внешнего дыхания определенной критической
степени возникает РДСВ. Задача анестезиологического пособия и
интенсивной терапии - блокировать типический патологический процесс
потери легкими нормальных респиронов и необратимой деструкции
терминальных респираторных единиц, индуцированный
военно-травматическим шоком. Этой цели анестезия и интенсивная терапия достигают через
Патофизиология расстройств функциональных систем и нарушений гомеостаза... 547
ослабление действия факторов осложнений военно-травматического
шока, нормализуя системную регуляцию и устраняя дефицит массы и
энергий на уровне эффекторов функций.
Респираторный дистресс-синдром - это типический патологический
процесс снижения до критического уровня и ниже числа нормальных респи-
ронов в результате отрицательных неирогуморальных влияний, патогенных
межклеточных взаимодействий и тромбоза легочных микрососудов.
Причина артериальной гипоксемии у больных с РДСВ - это рестриктивное
расстройство внешнего дыхания.
Разрушение респиронов у больных с РДСВ резко снижает
статическую податливость легких. В результате для обеспечения роста силы
сокращений дыхательных мышц растет утилизация свободной энергии в
системе внешнего дыхания, что повышает потребление кислорода всем
организмом. Рост потребления кислорода всем организмом обостряет
артериальную гипоксемию. Падение напряжения кислорода в артериальной
крови приводит к еще более интенсивной гипервентиляции, что
повышает потребление кислорода всем организмом и усиливает артериальную
гипоксемию. Так на системном уровне завершается один из порочных
кругов патогенеза РДСВ, определяющий неизбежность ИВ Л при этом
критическом нарушении легочного газообмена.
О том, что РДСВ представляет собой типический патологический
процесс, который через эндогенизацию на определенном этапе развития
теряет специфичность, связанную с конкретной его причиной, говорит
широта спектра заболеваний и патологических состояний, у части
больных вызывающих дистресс-синдром. РДСВ вызывают сепсис, травма,
острый панкреатит, нарушения регуляции после черепно-мозговых
ранений и травм, жировая эмболия, аспирация желудочного содержимого,
острая горная болезнь как следствие острой гипоксической гипоксии,
токсичное действие наркотиков и лекарственных средств (героин,
барбитураты, нестероидные противоспалительные средства), отравления
никотином и другими ядами табачного дыма, кислородом, NO2, CL2, NH3,
фосгеном. К РДСВ предрасполагают массивные гемотрансфузии, диссемини-
рованное внутрисосудистое свертывание, состояние после
искусственного кровообращения, эклампсия беременных и эмболия околоплодными
водами. Через патогенную реакцию системы иммунитета и прямое
повреждающее ткани действие развитие РДСВ индуцируют возбудители
инфекционных заболеваний, вирусы, бактерии, микобактерии,
возбудитель туберкулеза, Pneumocystis carinii и др. Следует заметить, что этот
список нельзя считать полным.
Уже в первые минуты после тяжелой механической травмы ноцицеп-
тивная афферентация активирует симпатический отдел вегетативной
нервной системы. При этом возрастает уровень возбуждения
симпатических центров, представленных в преоптической области гипоталамуса. На
периферии симпатическая стимуляция ведет к патогенному спазму
легочных микрососудов. Рост сопротивления кровотоку сразу дистальнее
легочных капилляров вызывает стаз в легочных микрососудах, их тромбоз и
548
Глава 33
отек легочной мембраны. Сразу после тяжелого ранения отрицательные
нервные влияния на легочную паренхиму закладывают основу развития
посттравматической дыхательной недостаточности и ее крайней стадии,
респираторного дистресс-синдрома взрослых.
Патогенная системная адренергическая стимуляция вызывает РДСВ
не только через спазм легочных микрососудов и связанные с ним
расстройства периферического кровообращения в легких. В течение часа
после кровопотери и тяжелой механической травмы в легких растет
экспрессия генов цитокинов со свойствами медиаторов воспаления. Данные
цитокины высвобождаются клетками системы мононуклеарных
фагоцитов, которые находятся в легких (интрапаренхимальные и альвеолярные
макрофаги). В легочных мононуклеарах растет транскрипция с генов
таких цитокинов как интерлейкин-1 (бета и альфа), альфа-фактор некроза
опухолей. При этом не происходит транскрипции с аналогичных генов
мононуклеарных фагоцитов, циркулирующих с кровькь Можно считать,
что в течение часа после массивной кровопотери и тяжелой механической
травмы в легких значительно возрастает содержание медиаторов
лишенного биологического смысла патогенного воспаления. Это заставляет
предположительно считать причиной такой «локальной гиперцитокине-
мии» расстройства регуляции нервной системой, первой реагирующей на
массивную кровопотерю и тяжелую травму. Так известно, что
возбуждение альфа-адренорецепторов наружной клеточной поверхности
мононуклеарных фагоцитов легких активирует в их клеточном ядре
транскрипционный фактор кВ, а также транскрипционный фактор, активируемый
ростом содержания в клетке циклического аденозинмонофосфата. Данные
транскрипционные факторы связываются с геномом легочных
мононуклеарных фагоцитов в области промотеров генов цитокинов-медиаторов
воспаления. Возбуждение бета-адренорецепторов наружной поверхности
мононуклеаров тормозит экспрессию данных цитокинов через активацию
системы аденилатциклазы, но рост транскрипции в результате
возбуждения альфа-адренорецепторов преобладает, и выброс медиаторов
воспаления растет. Можно считать, что патогенные отрицательные нервные
влияния на легочную паренхиму и гиперкатехоламинемия в остром
периоде после тяжелых ранений и травм посредством возбуждения альфа-
адренорецепторов мононуклеарных фагоцитов легких вызывает в
легочной паренхиме воспаление, лишенное биологической цели.
Последствия патологической централизации кровообращения и
гипоксии тканей и клеток длительное время препятствуют снабжению
клетки кислородом и энергопластическими субстратами, несмотря на
окончательную остановку кровотечения и достаточные инфузии и трансфузии.
Микроциркуляторую гипоксию тканей усугубляет патологическая боль,
вызывающая устойчивый спазм резистивных сосудов и юкстакапилляр-
ное шунтирование. Гипоксия и восстановление микроциркуляции как
причина реперфузионного синдрома через образование свободных
кислородных радикалов активируют макрофаги и моноциты, которые в
активированном состоянии начинают высвобождать цитокины (интерлейкин-1,
Патофизиология расстройств функциональных систем и нарушений гомеостаза... 549
фактор некроза опухолей и др.), активирующие гранулоциты.
Одновременно интерлейкин-1 и фактор некроза опухолей повышают
проницаемость и адгезивность эндотелиоцитов легочных микрососудов.
Активированные нейтрофилы после адгезии к эндотелию легочных капилляров
становятся источником свободных кислородных радикалов и
гуморальных медиаторов (простаноидов). Все эти агенты гуморального звена
патогенеза РДСВ повышают проницаемость эндотелия легочных
капилляров, угнетая лимфоотток из интерстиция легких. В результате возникает
гипергидратация интерстиция легких.
Следует заметить, что фиксация лейкоцитов и других клеток крови на
эндотелиоцитах легочных микрососудов увеличивают вариабельность
вентиляционно-перфузионных отношений структурно-функциональных
единиц легких, респиронов еще до того, как после тяжелых ранений и
травм развиваются выраженная острая дыхательная недостаточность или
ее крайняя стадия, дистресс-синдром.
Успехи практической реаниматологии в шестидесятых-семидесятых
годах позволили предотвратить смерть многих раненых и пострадавших в
остром периоде. Это не привело к значительному снижению смертности
после тяжелых ранений и травм, так как большинство больных погибало
в отдаленном периоде от дыхательной недостаточности. Аутопсийные
исследования выявили повреждение легочного капиллярного эндотелия с
цитолизом и деструкцией межклеточных соединений, миграцией
форменных элементов крови, плазменных белков в легочную мембрану, а за-
тем и в просвет альвеол. Легкие были тяжелыми (их масса составляла от
800 до 2000 г), застойными, влажными, с множественными мелкими
кровоизлияниями и гиалиновыми мембранами. Часто наблюдали опеченение
легочной ткани. Разные патологи по-разному назвали это состояние:
«застойный ателектаз», «синдром просачивания через капилляры»,
«геморрагическое влажное легкое», «синдром скрытой гипоксемии»,
«прогрессирующее затемнение легочной ткани на рентгенограммах», «синдром
посттравматической дыхательной недостаточности», «травматическое
влажное легкое», «гипоксическая гипервентиляция», «легкие Да Нанга».
Еще в то время стало ясно, что нужен один термин, наиболее отвечающий
этиологии и патогенезу этой крайней стадии посттравматической
дыхательной недостаточности.
Наибольшее распространение получил термин респираторный
дистресс-синдром взрослых (РДСВ), adult respiratory distress syndrome
(ARDS), впервые примененный в 1967 г. рядом исследователей
клиницистов Старого и Нового Света. Тем не менее, весь спектр наименований
синдрома сам по себе ярко рисует его патогенез и клиническую картину.
Так термин «синдром просачивания через капилляры» сразу говорит о не-
кардиогенном отеке легких как о звене патогенеза РДСВ. «Синдром
скрытой гипоксемии» напоминает о признаке продромы РДСВ,
артериальной гипоксемии без остальных проявлений дыхательной
недостаточности. Артериальную гипоксемию можно связать с патологической
вариабельностью вентиляционно-перфузионных отношений респиронов.
550
Глава 33
«Нарастающее затемнение легочных полей на рентгенограммах»
происходит вследствие интерстициального отека легких и заполнения альвеол
плазмой и форменными элементами крови после деструкции респиронов
из-за патогенного воспаления.
Респираторный дистресс-синдром взрослых после ранений и травм -
это острое расстройство легочного газообмена с угрожающей жизни
тяжелой артериальной гипоксемией в результате деструкции вследствие
патогенных межклеточных взаимодействий структурно-функциональных
единиц легких. К нему предрасполагают системные циркуляторная
гипоксия и воспалительная реакция, а также нейрогенный спазм легочных
микрососудов при патологической боли. Рост тромбогенного потенциала
легочных эндотелиоцитову вызванный патологическим стрессом тяжелых
ранений травм, и коагулопатия, неизбежно присущая тяжелым
травматической и раневой болезням, служат причинами распространенного
тромбоза микрососудов легких как звена морфопатогенеза РДСВ.
Адгезия активированных гранулоцитов и других клеток крови к эндо-
телиоцитам как ранняя стадия общей воспалительной реакции в остром
периоде после тяжелых ранений и травм возникает не только в легких.
Она носит генерализованный характер и выступает одной из ведущих
причин полиорганной (множественной системной) недостаточности.
Спустя несколько минут после смерти у людей, погибших от ранений и
травм, в легких выявляют признаки полиморфонуклеарной
инфильтрации, лейкоцитарный стаз и патологический рост проницаемости
микрососудов. Таким образом патогенные межклеточные взаимодействия
детерминируют начало развития дисфункций внутренних органов как
эффекторов функциональных систем. При этом в первую очередь индуцируется
развитие дыхательной недостаточности.
К эффекторам патогенных клеточных реакций, выступающих причиной
крайней стадии респираторного дистресс-синдрома, относятся нейтрофилы,
а также мононуклеарные клетки: лимфоциты разной зрелости и активности,
моноциты, другие мононуклеарные фагоциты и их производные.
Системные гипоксические активация и повреждение эндотелия, его
деструкция под влиянием распространенной эмболизации микрососудов,
разрушение эндотелия в результате прямых механических воздействий -
все это сопровождается моментальной аутоактивацией контактного
фактора свертывания, фактора Хагемана (HF). Дальнейшие этапы активации
HF зависят от образования пламенного калликреина. Активный фактор
Хагемана запускает кининогенез и систему комплемента, а через
активацию плазминогена активирует фибринолиз. Под действием активного HF
включается каскадный механизм внутрисосудисого свертывания крови.
Активация системы комплемента и каскадного механизма свертывания
крови приобретает системный характер и на уровне легочных
микрососудов служит индуктором воспаления, лишенного биологической цели.
Патогенное воспаление разрушает структурно-функциональные единицы
легких и вызывает артериальную гипоксимию. Гиперкоагулемия в мик-
роциркуляторном русле легких ведет к распространенной микротромбо-
Патофизиология расстройств функциональных систем и нарушений гомеостаза... 551^
эмболии, вызывающей дальнейшее падение напряжения кислорода в
артериальной крови.
Все эти процессы становятся возможными при замедлении кровотока в
результате гиповолемии и спазма легочных венул. При нормальном токе
крови плазменные медиаторы ранней фазы респираторного
дистресс-синдрома, повышающие проницаемость легочного эндотелия и индуцирующие
адгезию к нему активированных гранулоцитов, не могут сосредоточиться
на достаточно коротком участке легочного микроциркуляторного русла и
тем самым подготовить почву для последующих этапов чисто патогенного
воспаления. Следовательно, нарушения микроциркуляции представляют
собой необходимую предпосылку респираторного дистресс-синдрома и
являются одним из его наиболее ранних патогенетических механизмов.
Растворимые фракции комплемента не только повышают
проницаемость эндотелия легочных микрососудов. Они активируют фагоциты
крови, взаимодействуя со специфическими рецепторами клеточной
поверхности последних. Растворимые фракции комплемента,
активирующие нейтрофилы, образуются под действием протеаз, высвобождаемых
активированными полиморфонуклеарами.
Через систему комплемента к клеточным взаимодействиям,
составляющим патогенез дистресс-синдрома присоединяются тучные клетки. Они
выделяют целый спектр гуморальных «медиаторов шока» и цитокинов-
флогогенов, включая гистамин, серотонин, фактор активации тромбоцитов,
медленно реагирующую субстанцию анафилаксии и эозинофильный хемо-
таксический фактор. Часть этих веществ активирует нейтрофилы.
Следует учитывать, что под действием плазменных медиаторов
ранней фазы РДСВ (фракции системы комплемента, цитокины, кинины) не
только повышается проницаемость эндотелия легочных микрососудов, но
и резко возрастает его чувствительность к повреждению полиморфонук-
леарными лейкоцитами.
Артериальный конец легочного капилляра выстилают эндотелиоциты
первого типа, формирующие упругую выстилку, отделяющую
циркулирующие лейкоциты, тромбоциты и плазму от подлежащих структур. Тут
прилипание полиморфонуклеаров выражено меньше, или его почти не
наблюдают. Внутренняя же поверхность венул выполнена эндотелиоци-
тами второго типа, которые в ответ на действие плазменных медиаторов
РДСВ при снижении скорости кровотока сокращаются и приобретают
высокую адгезивность. В результате сокращения между эндотелиоцитами
второго типа образуются щели, по которым усиленно фильтруется плазма
с белками и проходят лейкоциты. Развивается генерализованный некар-
диогенный интерстициальный отек легких с инфильтрацией
полиморфонуклеарами легочной мембраны.
Мононуклеарные фагоциты в ишемизированных после тяжелой
травмы тканях выделяют фактор некроза опухолей, интерлейкин-1 и другие
цитокины, которые, попадая в системную циркуляцию, повышают
адгезивность эндотелиоцитов легочных венул к уже активированным
системной циркуляторной гипоксией гранулоцитам. Кроме того, под влиянием
552
Глава 33
интерлейкина-1 усиливается образование в эндотелиоцитах тромбоген-
ных агентов, что создает дополнительные условия для фиксации
лейкоцитов в местах локализации микротромбов.
В регуляции адгезивности эндотелиоцитов основную роль играют их
собственные продукты простациклин и циклический аденозинмонофос-
фат, синтез которых возрастает в эндотелии под влиянием адренергиче-
ской стимуляции. В этой связи аналгезию как афферентную блокаду ад-
ренергической активации следует считать средством предупреждения
респираторного дистресс-синдрома взрослых путем снижения
выраженности адгезии нейтрофилов к эндотелию легочных венул.
После фиксации лейкоцитов к эндотелию развивается стойкое
повышение проницаемости сосудистой стенки. Проницаемость особенно резко
возрастает, если эндотелиоцит не только сокращается, но и повреждается
веществами, высвобождаемыми активированными лейкоцитами.
Активированные полиморфонуклеары выделяют как высокоактивные лизосомаль-
ные протеазы катепсины и эластазу, так и биоокислители (супероксид СЬ,
Н202 и их производные). Эластаза отслаивает эндотелиоцит от базальной
мембраны, а оксиданты запускают перекисное окисление липидов в эндо-
телиальных мембранах. Биоокислители разрушают ингибиторы протеаз и
способствуют образованию простагландинов и других биоактивных
веществ, повышающих проницаемость стенки капилляров. Известно, что
чувствительность к повреждающему действию оксидантов наиболее выражена
у эндотелия из системы легочных артерий, что во многом определяет роль
легких как органа-мишени тяжелых травматической и раневых болезней.
Альвеолоциты менее чувствительны к действию биооксидантов, чем
легочные эндотелиоциты. Это объясняет развитие альвеолярного отека
лишь в терминальной стадии РДСВ, которой предшествуют рефрактерная
гипоксемия и интерстициальный отек легких с проникновением в
легочную мембрану лейкоцитов и других клеток крови. Более устойчивые к
повреждению свободными кислородными радикалами альвеолоциты
некоторое время препятствуют миграции плазмы и форменных элементов
крови из легочного интерстиция в просвет альвеол.
В целом следует отметить, что ранняя фаза повышения
проницаемости стенки легочных микрососудов связана преимущественно с гиперци-
токинемией. Последующее же стойкое увеличение проницаемости
микрососудов, деструкция эндотелия легочной мембраны с миграцией клеток
крови вплоть до альеол зависят от контактных взаимодействий
нейтрофилов с эндотелиоцитами. Гуморальные и клеточные факторы патогенеза
действуют на фоне предшествующего нейрогенного стаза в легочных
микрососудах, создающего отправную точку развития РДСВ.
Дыхательную недостаточность и дистресс-синдром обостряют диссе-
минированное внутрисосудистое свертывание, жировая эмболия,
неизбежно в той или иной степени присущие тяжелой раневой болезни. Они
вызывают распространенную микроэмболизацию в легких с ростом
альвеолярного мертвого пространства, которую в начальных стадиях острой
дыхательной недостаточности позволяет уловить лишь фиксация разли-
Патофизиология расстройств функциональных систем и нарушений гомеостаза... 553
чий между парциальным давлением углекислого газа в конечной части
выдыхаемого воздуха и его напряжением в артериальной крови.
Частота респираторного дистресс-синдрома после геморрагического
шока, тяжелых травм и при сепсисе составляет приблизительно 20 %. С учетом
многочисленных противоречивых описаний синдрома, а также тесной связи
клинических признаков дистресс-синдрома с патогенезом представляется
целесообразным привести описание клинической картины РДСВ.
Гипоксемия и патогенная афферентация при дистресс-синдроме
вызывают возбуждение и ступор с частыми галлюцинациями.
Стремительное падение насыщения кислородом гемоглобина в артериальной крови
обуславливает быстрое возникновение цианоза кожных покровов.
Диспноэ при дистресс-синдроме имеет как первичный характер, то
есть связано с гипоксическим повреждением регуляции дыхания, так и
вторичный, в результате генерализованного интерстициально-альвеоляр-
ного отека легких. Тахипноэ и гипервентиляция (возрастание минутного
объема дыхания) сочетаются со свистящими хрипами и тахикардией.
Для дистресс-синдрома характерна рефрактерная артериальная
гипоксемия, то есть напряжение кислорода в артериальной крови менее 55 мм. рт. ст.
при концентрации кислорода во вдыхаемой смеси более 50 %.
Рентгенография выявляет диффузную альвеолярную инфильтрацию - двустороннее
«хлопьевидное» затемнение легочных полей с тенденцией к расширению
легочных инфильтратов.
Весь симптомокомплекс РДСВ у раненых развивается в течение двух суток
после ранений и травм по мере реализации патогенных межклеточных
взаимодействий в легочной паренхиме. Но уже в первые часы после любого тяжелого
ранения есть явная или скрытая острая дыхательная недостаточность
(начальная стадия дистресс-синдрома), которая без патогенетической интенсивной
(превентивной) терапии (табл. 33.1) может перейти в дистресс-синдром.
К клиническим признакам начальной стадии респираторного
дистресс-синдрома взрослых в первые сутки раневой болезни относят
беспокойство, тахипноэ, жесткое дыхание, единичные сухие и влажные хрипы,
гипоксемию, устраняемую оксигенотерапией, усиление легочного
рисунка на рентгенограмме.
Гипоксемия при РДСВ служит стимулом для компенсаторной
гипервентиляции, которая не устраняет снижения напряжения кислорода в
артериальной крови, но при ненарушенной диффузии ССЬ в легких вызывает
респираторный алкалоз. При респираторном дистресс-синдроме взрослых снижается
податливость и растет функциональная остаточная емкость легких. Быстрый
рост сопротивления дыхательных путей связан вначале с повышением
вязкостного дыхательного сопротивления. В конечных стадиях дистресс-синдрома
податливость снижена в основном из-за диффузного микроателектазирования
вследствие разрушения системы сурфактанта под влиянием биооксидантов.
Альвеолярный отек легких обуславливает особенно резкое возрастание
податливости. Искусственная вентиляция легких при низкой податливости
легких часто приводит к спонтанному пневмотораксу, что заставляет
рекомендовать профилактическое дренирование плевральной полости.
554
Глава 33
Таблица 33.1.
Патогенетическая терапия ОДНу тяжелых раненых и больных
Звено патогенеза
Нейрогенный спазм легочных
сосудов сопротивления под влиянием
адренергической стимуляции
Сокращение эндотелиоцитов
легочных венул с образованием пор
эндотелия, рост проницаемости
стенок венул и легочных капилляров
Интерстициальный отек легких,
микроателектазирование
Адгезия активированных грануло-
цитов к эндотелию легочных венул
Элемент терапии
Эффективная комбинирования (проводниковая
и центральная) аналгезия, симпатическая блокада
без ятрогенной гипотензии
Гемодилюция для устранения циркуляторной
гипоксии как основного фактора повышенного
образования плазменных медиаторов
дистресс-синдрома и для снижения их действующих концентраций
Повышение внутриальвеолярного давления в
ходе дыхания с постоянным положительным
давлением в конце выдоха (ПДКВ), снижение
градиента между гидростатическим давлением в
легочном капилляре и давлением в просвете альвеол
для снижения ультрафильтрации
Улучшение текучести крови для ускорения
кровотока в легочных микрососудах при
повышенном общем легоч ном сосудистом сопротивлении
посредством: гемодилюции с внутривенным
вливанием повышающих текучесть крови плазмоза-
мещающих растворов декстрана и гидроксиэтил-
крахмала
ПАТОГЕНЕЗ РАСТРОЙСТВ СИСТЕМНОГО
И ПЕРИФЕРИЧЕСКОГО КРОВООБРАЩЕНИЯ
В ОСТРОМ ПЕРИОДЕ ТЯЖЕЛОЙ РАНЕВОЙ БОЛЕЗНИ
Вариабельность реакции на кровопотерю так велика, что потеря 25 %
ОЦК вначале может не сопровождаться артериальной гипотензией или
даже тахикардией. Поэтому в условиях локального конфликта, то есть
относительно небольших сроков эвакуации всех раненых с
проникающими ранениями груди и живота, минно-взрывными ранениями сегментов
конечностей, ранениями крупных, как артериальных, так и венозных
сосудов, следует считать находящимися в шоке, который может и не успеть
привести к снижению артериального давления. Для предотвращения
необратимого шока у таких раненых как можно раньше следует начинать
стандартизированную превентивную терапию (табл. 33.2), которую часто
проводят в порядке предоперационной подготовки во время первичного
обследования и вводной анестезии.
Раннюю, в том числе и на догоспитальном этапе, инфузионную
терапию можно считать средством предупреждения инфекционных
осложнений ранений вследствие угнетения иммунитета. Потеря 10%
внутрисосудистого объема крови, не приводя к значительным
изменениям артериального давления и частоты сердечных сокращений, вызы-
Патофизиология расстройств функциональных систем и нарушений гомеостаза... 555
вает 30 %-ное снижение кровотока через толстую кишку с таким же
падением доставки к ней кислорода. Значительный дефицит кровотока
через толстую кишку и другие отделы желудочно-кишечного тракта при
отсутствии гипотензии,тахикардии и опасного снижения диуреза
неизбежно отрицательно сказывается на печеночном кровотоке, 70 %
которого обеспечивается поступлением крови из воротной вены. При этом,
несмотря на относительную устойчивость паренхимы печени к
гипоксии, возникают гепатоцеллюлярные расстройства и связанные с ними
дисфункции локализованных в печени клеток системы мононуклеарных
фагоцитов. В этой связи раннюю профилактику гиповолемии гемоди-
люцией следует считать эффективным средством предупреждения
угнетения иммунитета.
Известно, что начальный этап патогенеза военно-травматического шока
во многом составляется патологической реакцией нервной системы на
тяжелое ранение. В ответ на травму, гиповолемию и циркуляторную
гипоксию тканей активируется вегетативная нервная система. В частности
происходит растормаживание симпатической части сосудо-двигательного
центра, которое на периферии ведет к спазму вен, артериол и прекапиллярных
сфинктеров.
Таблица 33.2
Патогенетическая терапия расстройств кровообращения
в остром периоде после тяжелых ранений
Цель терапии
Регидратация внеклеточной жидкости для нормализации
интерстиция как среды обитания клеток. Устранение
клеточного обезвоживания
Устранение дефицита объема внеклеточной жидкости,
повышение способности крови доставлять кислород клетке,
устранение юкстакапиллярного шунтирования как причины мик-
роциркуляторной гипоксии внутренних органов и связанной с
ней иммунодепрессии, снижение действующих концентраций
биоактивных веществ и цитокинов, мобилизация эритроцитов
для системного кровообращения
Уменьшение степени гиповолемии; профилактика коагулопа-
тии; устранение отека интерстициального пространства;
улучшение текучести крови для увеличения доставки кислорода и
энергопластических субстратов клетке, а также для
предупреждения патологических системных активации эндотелиоцитов,
роста их адгезивной способности и экспрессии тромбогенного
потенциала эндотелиальных клеток
Защита клеточных мембран от деструкции под влиянием
факторов, связанных с циркуляторной гипоксией (липидная
триада, свободные кислородные радикалы)
Способ достижения,
средство
Инфузия 1,2 л 5 %
раствора глюкозы
Вливание 1,2 л раствора
Рингера
Инфузия 0,4 л поли-
глюкина, 0,4 л реополиг-
люкина или 0,5 л 10 %
раствора гидроксиэтил-
крахмала
Внутривенное введение
600 мг преднизолона
Первой по времени реакцией, компенсирующей кровопотерю, следует
считать спазм емкостных сосудов, вен. Веноконстрикция, снижающая
емкость венозного резервуара, мобилизует для циркуляции 1 л крови. По-
556
Глава 33
этому до восполнения сниженного внутрисосудистого объема опасно
стремиться нормализовать микроциркуляцию симпатолитическими
средствами. Артериальная гипотензия может стать необратимой, когда ятро-
генная дилатация вен вызовет критическое несоответствие ОЦК емкости
сосудистого русла.
Артериальное давление - это конечный полезный приспособительный
результат насосной и транспортной, демпферной (обеспечивающей
относительную равномерность кровотока в аорте и крупных артериях в систолу и
диастолу) подсистем кровообращения. Эффекторы этой системы, сердце и
артерии, вместе с емкостной, то есть венозной, системой образуют
макроциркуляцию. Конечный результат макроциркуляции - поддержание
должных артериального давления и объема циркулирующей крови. Венозная,
емкостная система, содержащая 75-80 % всей крови, богата адренореце-
прецепторами, но лишена опиоидных рецепторов. Это позволяет венозной
системе быть эффектором первой очереди аварийной компенсации крово-
потери, легко снижать свой объем под влиянием адренергической
стимуляции. При этом емкостная система играет роль переменной емкости для
сердца и около пятисот миллионов артериальных сосудов от артерий
крупного диаметра до метартериол. Система макроциркуляции всячески
стремится к поддержанию своего полезного результата - должного
артериального давления, что спасает жизнь тогда, когда дефицит массы в системе
макроциркуляции еще не восполнен инфузиями и трансфузиями.
Второй по времени компенсаторной реакцией в ответ на кровопотерю
выступает спазм артериол и прекапиллярных сфинктеров, направленный
на сохранение артериального давления, достаточного для поддержания
адекватной объемной скорости кровотока в артериях головного мозга,
сердца и легких.
Системный спазм артериол и прекапиллярных сфинктеров
обуславливает общее ускорение тока крови в обменных капиллярах, что снижает в
них гидростатическое давление. Внеклеточная жидкость из интерстициаль-
ного сектора устремляется в капилляры. Если только ранению не
предшествовала дегидратация, то внутрисосудистый объем возрастает на 500-
700 мл. Таким образом достигается компенсаторная аутогемодилюция.
Возникающее вследствие роста различия пре- и посткапиллярного
сосудистых сопротивлений увеличение сосудистого жидкостного сектора снижает
содержание воды в интерстиции. Можно считать, что сразу после тяжелых
ранения и травмы среда обитания клеток организма изменяется по
гипертоническому типу. Это служит одним из показаний к внутривеннрому
вливанию в остром периоде тяжелой травматической болезни гипоосмолярных
относительно внеклеточной жидкости растворов.
Микроциркуляторная гипоксия как следствие избыточного
компенсаторного спазма резистивных сосудов сразу обуславливает
генерализованные гипоксические повреждения клеток, главный фактор танатогенеза
при тяжелых травматической и раневых болезнях.
Расстройства доставки кислорода клетке в остром периоде после
тяжелых ранений и травм являются главной причиной возникновения энер-
Патофизиология расстройств функциональных систем и нарушений гомеостаза... 557
гетического дефицита на клеточном уровне. Энергетический дефицит
возрастает вследствие разобщения окисления и фосфорилирования под
влиянием избыточной системной адренергической стимуляции в первые
двое суток после тяжелых ранений и травм.
Кислородная недостаточность уже в первые часы после тяжелого
ранения ведет к усилению перекисного окисления липидов с истощением
системы антиоксидантной защиты клетки. Перекисное окисление
липидов очень быстро, в первые 30-40 мин от момента травмы, а не в течение
1-2 суток, как это считали раньше, повреждает клеточные мембраны с
изменением содержания в них наиболее функционально активных фос-
фолипидов (работы школы Г.Н. Крыжановского).
В результате деструкции клеточных мембран и энергетического
дефицита на клеточном уровне расстраивается функционирование системы
вторичного мессенджера, аденозинмонофосфата, что критически
расстраивает эндокринную регуляцию клеточных функций. Почти
полностью прекращается активный транспорт натрия и калия через клеточную
мембрану. Натрий проникает в клетку из интерстициального
пространства. Вслед за натрием в клетку перемещается вода, что ограничивает
возможности компенсаторной гемодилюции и усугубляет гиповолемию.
Клеточный отек вслед за деструкцией мембран завершает цикл цитолиза.
Перекисное окисление липидов усиливают биооксиданты,
высвобождаемые активированными гипоксией гранулоцитами после их адгезии к
эндотелию капилляров. При этом адгезия гранулоцитов как источников
свободных кислородных радикалов приобретает характер системного
расстройства, нормальных межклеточных отношений и одного из
ведущих звеньев патогенеза расстройств периферического кровообращения.
Распространенная адгезия фагоцитов крови к эндотелиоцитам придает
необратимость капиллярному стазу и разрушает микрососуды.
Деструкция лизосомальных мембран служит причиной
высвобождения лизосомальных ферментов. При попадании протеолитических
энзимов из лизосом в системную циркуляцию в плазме ускоряется
образование вазоактивных веществ, наиболее изученными из которых являются
гистамин и брадикинин. Эти агенты вкупе с кислыми анаэробными
метаболитами вызывают стойкий паралич прекапиллярных сфинктеров.
Общее периферическое сосудистое сопротивление критически падает, и
артериальная гипотензия становится необратимой.
Парез артериол и прекапиллярных сфинктеров наступает несколько
раньше дилатации более устойчивого к ацидозу посткапиллярного
сфинктера. Возникает выраженное различие в пре- и посткапиллярных
сосудистых сопротивлениях, которое обуславливает патологическое
депонирование крови в микрососудах, усиливает адгезию лейкоцитов к
эндотелию, повышает гидростатическое давление в капиллярах. Рост
капиллярного гидростатического давления служит причиной
перемещения внеклеточной жидкости из капилляров в интерстиций. Интерстици-
альный отек тканей возникает также вследствие повышения
проницаемости капилляров эндотелия под влиянием гиперцитокинемии и грану-
558
Глава 33
лоцитарной адгезии. Снижение объема циркулирующей плазмы
обостряет гиповолемию.
Нарушения микроциркуляции в остром периоде тяжелой раневой болезни
усугубляет диссеминированное внутрисосудистое свертывание (ДВС).
Первоначально под влиянием активации как симпатического, так и
парасимпатического отделов автономной нервной системы гемокоагуля-
ция возрастает на 25-50 %. Такое укорочение времени свертывания
происходит за счет снижения самой продолжительной фазы гемокоагуля-
ции - образования протромбиназы.
Гиперадреналинемия ведет к повышенному высвобождению из стенок
сосудов естественных антикоагулянтов и активаторов фибринолиза, но
определяющим для развития ДВС-синдрома является высвобождение из
сосудистой стенки более мощного фактора свертывания, тромбопластина,
который в крови превращается в тканевую протромбиназу.
Кроме того, адреналин в крови активирует фактор Хагемана с ростом
образования кровяной протромбиназы.
Гиперадреналинемия вызывает устойчивую гиперактивацию тканевых
липаз, что усиливает липолиз и обуславливает рост концентрации в крови
свободных жирных кислот, обладающих тромбопластической
активностью. Фосфолипиды, высвобождаемые при гиперкатехоламинемии из
эритроцитов, также ускоряют самую продолжительную фазу гемокоагу-
ляции - образования протромбиназы.
Первую фазу ДВС-синдрома, гиперкоагулемию> также вызывает
активация в ответ на ранение и травму парасимпатического отдела
автономной нервной системы. Парасимпатические центробежные влияния по
блуждающему нерву приводят к высвобождению из стенок сосудов
веществ, индуцирующих свертывание крови.
Гиперкоагулемия, связанная с повышенным потреблением факторов
свертывания, всегда первична по отношению к коагулопатии
потребления, второй фазе ДВС-синдрома. Если учесть, что стимулом
генерализованной адренергической стимуляции (детерминанта ДВС сразу после
тяжелых ранений), служат патологическая боль и гиповолемия, то ранние
эффективная аналгезия и коррекция дефицитов объемов циркулирующей
крови и внеклеточной жидкости выступают основными мерами
предупреждения коагулопатии у больных тяжелой раневой болезнью.
Патогенетически обоснованная стандартизированная терапия (табл. 33.2),
которую следует проводить уже на догоспитальном этапе, нормализует
состав и структуру жидкостных секторов внеклеточного пространства,
снижает патогенно высокую действующую концентрацию биоактивных
веществ и цитокинов, улучшает текучесть крови. Последний из эффектов
гемодилюции особенно важен, так как ДВС всегда есть в остром периоде
после тяжелых ранений. Следует заметить, что коагулопатия вначале не
проявляет себя клинически и не сказывается на данных исследования
кислотно-основного состояния крови, но сразу нарушает доставку
кислорода клетке, так как распространенный тромбоз артериол усиливает
патологическую централизацию кровообращения: Это ведет к гипоксии тканей,
Патофизиология расстройств функциональных систем и нарушений гомеостаза... 559
которая возникает при нормальных напряжениях газов в крови. В
последующем реактивный гиперфибринолиз разрушает микротромбы с
повышенным потреблением фибриногена и тромбоцитов. Коагулопатия
потребления провоцирует кровотечения, которые особенно трудно
остановить при ранениях внутренних органов. Если аналгезия и гемодилюция,
вливание реоактивных плазмозамещающих растворов предотвращают
ДВС и вторичную коагулопатию потребления, то стандартизированную
патогенетическую терапию на догоспитальном этапе и в
предоперационном периоде можно считать способами снижения частоты коагулопатиче-
ских кровотечений после тяжелых ранений.
При лечении тяжелых ранений и травм следует исходить из того, что
тяжелым травматической и раневой болезни всегда присуща сердечная
недостаточность.
При патологическом стрессе тяжелого ранения возбуждение высших
вегетативных центров детерминирует стресс-реакцию, которая уже в
первые часы после травмы вызывает выраженные стрессорные повреждения
миокарда. Их быстрое развитие обусловлено тем, что тяжелое ранение
рождает целый спектр необычайно сильных и неотвратимых раздражителей -
патогенную боль, гиповолемию, артериальную гипоксемию и гиперкап-
нию, метаболический ацидоз. Эти критические нарушения гомеостазиса
сохраняются долго, их интенсивность как составляющих стимула стресса
длительное время остается высокой, несмотря на интенсивную терапию.
Непосредственной причиной стрессорного повреждения миокарда
является избыточная концентрация катехоламинов в миокарде, что связано
в первую очередь с прямой нервной адренергической стимуляцией
сердечной мышцы.
На рубеже 60-х и 70-х годов в экспериментах было показано, что ад-
ренолитические воздействия предупреждают стрессорные повреждения
миокарда при шоке. Равнозначные положительные влияния на функции
миокардиоцитов при геморрагическом шоке были выявлены у
хирургической денервации сердца, адреналэктомии и адренолитиков. При
массивной кровопотере альфа-адреноблокаторы замедляли снижение субэндо-
кардиального кровотока, а бета-адренолитики уменьшали несоответствие
потребления сердцем кислорода его работе. Был сделан вывод, что
выраженность стрессорных повреждений сердца находится в прямой связи со
степенью гиперкатехоламинемии. На основании последних
экспериментальных данных этот вывод подвергнут коррекции.
Эпидуральная блокада на уровне верхних грудных сегментов повышала
выживаемость собак при геморрагическом шоке. При этом не наблюдали
снижения концентрации в крови адреналина, которое при длительной
эпидуральной блокаде обусловлено блокадой местным анальгетиком
симпатических эфферент, иннервирующих надпочечники. Положительное влияние
десимпатизации сердца вследствие эпидуральной блокады на летальность
после массивной кровопотери не было связано со снижением роста
концентрации катехоламинов в крови. Это свидетельствовало о преимущественно
нервном адренергическом механизме острой миокардиопатии.
560
Глава 33
Уже в первые минуты после тяжелого ранения есть все предпосылки
избыточной концентрации катехоламинов в миокарде и чрезмерной
активации аденилатциклазы. Это ведет к действию факторов липидной триады,
повреждающих мембраны кардиомиоцита вплоть до цитолиза. К ним
относят активацию фосфолипаз, липаз, перекисного окисления липидов, детер-
гентного действия лизофосфатидов и жирных кислот. Кроме того, под
влиянием повышенной действующей концентрации катехоламинов в
миокарде растет вхождение ионизированного кальция в кардиомиоциты,
истощается резерв и прекращается обновление гликогена. Под влиянием
факторов липидной триады, вследствие уменьшения резерва и обновления
гликогена происходит лабилизация лизосом, усугубляющая деструкцию и
дисфункции клеточных мембран с нарушением функционирования
кальциевого насоса, повышением проницаемости сарколеммы и саркоплазматическо-
го ретикулума. Лабилизация лизосом окончательно индуцирует эти
нарушения, но предрасполагают к ним липидная триада, увеличение вхождения
кальция и уменьшение резерва гликогена. Избыток кальция в саркоплазме
активирует фосфолипазы, протеазы, вызывает контрактуры миофибрилл и
распад дезоксирибонуклеиновой кислоты (ДНК) в миокарде.
В результате действия всех факторов патогенеза острых стрессорных
повреждений миокарда падает растяжимость и сократительная
способность сердечной мышцы. Падение растяжимости миокарда во многом
извращает действие закона Франка-Старлинга-Штрауба. Волокна сердечной
мышцы во время диастолического наполнения увеличивают свою длину
незначительно. Сердце уже не в состоянии отреагировать адекватным
возрастанием ударного объема желудочков в ответ на рост общего
венозного возврата (преднагрузки) в ходе инфузионнно-трансфузионной
терапии, что может привести к кардиогенному отеку легких.
Свою роль в развитии диффузного некробиоза сердечной мышцы
играет замедление кровотока по венечным артериям при падении
диастолического давления в аорте. Развиваются вторичные по отношению к снижению
скорости коронарного кровотока повышение вязкости крови и адгезия гра-
нулоцитов к эндотелию капилляров миокарда. Снижение текучести крови
нарушает ее реологические свойства, что повышает степень микроциркуля-
торной гипоксии сердечной мышцы. Высвобождение активированными
лейкоцитами биооксидантов после адгезии повреждает не только
эндотелий сердечных капилляров, но и мембраны кардиомиоцитов.
По значимости в генезе посттравматической сердечной
недостаточности факторы ее развития можно расположить следующим образом:
♦ симпатические прямые нервные влияния на сердечную мышцу с
повышенным высвобождением норадреналина из нервных окончаний;
♦ снижение соотношения кровотока в эндокарде и эпикарде,
усугубление несоответствия доставки кислорода к сердцу работе миокарда;
♦ избыточная тахикардия с нарушениями кровотока по венечным
артериям;
♦ гиперкатехоламинемия с преобладанием роста концентрации в
крови адреналина.
Патофизиология расстройств функциональных систем и нарушений гомеостаза... 561^
Из других причин недостаточности сердца после ранений и травм
ведущее значение имеют гипоксемия и респираторный ацидоз.
Ацидоз, который всегда сопутствует острому периоду тяжелой
травматической болезни, обладает широким спектром отрицательных кардио-
тропных влияний. Ацидоз нарушает функционирование сердца, угнетая
диастолическую деполяризацию, снижая порог фибрилляции желудочков,
оказывая отрицательное инотропное действие, расстраивая автономную
регуляцию сердца, а также извращая его реактивность по отношению к
катехоламинам. Известно, что депрессия миокарда более выражена при
респираторном, чем при метаболическом ацидозе.
При присоединении к патогенезу военно-травматического шока кар-
диогенного компонента цель интенсивной терапии становится особенно
трудно достижимой. При кардиогенном шоке в остром периоде тяжелых
травматической и раневых болезней анальгезия, инфузии и трансфузии в
силу невозможности быстрого устранения сердечной недостаточности не
могут нормализовать доставку кислорода к клетке. В этой связи кардио-
генный шок у раненых следует признать состоянием с наиболее
выраженной и длительной циркуляторной гипоксией.
Наиболее частая причина кардиогенного шока при раневой болезни -
это ушибы сердца при закрытой травме груди, причем перелом грудины
всегда приводит к ушибу сердца.
Если патогенетическая терапия в предоперационном периоде (табл. 33.3)
не стабилизирует АД на уровне выше 90 мм рт. ст., а при дополнительном
обследовании не удается выявить признаки продолжающегося кровотечения,
то следует заподозрить или неустраненный гемо-пневмоторакс или ушиб,
ранение сердца, высокую вероятность которых при ранениях и травмах груди,
торакоабдоминальных ранениях следует всегда иметь в виду. Клиническая
картина ушиба, ранения сердца складывается из жесткого дыхания и
влажных хрипов как следствий застоя в малом круге кровообращения, глухих
сердечных тонов и тахикардии, высокого центрального венозного давления с
набуханием шейных вен. Вольтаж электрокардиограммы снижен при
депрессии сегмента ST в грудных отведениях.
Минутный объем кровообращения при кардиогенном шоке остается
низким, несмотря на компенсаторные реакции кровообращения,
индуцируемые адренергической стимуляцией. Из-за падения сократимости
сердечной мышцы артериальное давление падает, несмотря на спазм ре-
зистивных сосудов на периферии. Снижение диастолического давления
ведет к падению перфузионного давления миокарда и коронарного
кровотока.
Вследствие компенсаторных веноконстрикции и аутогемодилюции, а
также в ходе инфузионной терапии у раненых в состоянии кардиогенного
шока возрастает общий венозный возврат к правым камерам сердца,
которые страдают от дистрофии, ишемических и механических
повреждений в гораздо меньшей степени, чем миокард левого желудочка. При этом
некоторое время ударный объем правого желудочка может преобладать
над ударным объемом левого.
562
Глава 33
Таблица 33.3
Патогенетическая терапия кардиогенного шока у раненых
Звено патогенеза
Повреждения и расстройства
функций сердца и кардиомиоцитов
Элемент интенсивной
терапии
Патологическая
боль, избыточное
длительное
возбуждение адренергиче-
ской и гипофиз-ад-
реналовой систем
Артериальная
гипоксемия
Рост действующей на кардиомио-
циты концентрации катехоламинов,
вторичная по отношению к гипер-
катехоламинемии в миокарде
активация аденилатциклазы, увеличение
вхождения в клетки сердца
ионизированного кальция, истощение
резервов гликогена в клетках сердца,
распространенные некробиотиче-
ские изменения миокарда
Падение сократимости сердца,
аритмии
Метаболический
ацидоз
Миокардиодист-
рофия, мехнические
и метаболические
повреждения сердца
Угнетение деполяризации в
диастолу и спонтанной активности
синоатриального водителя ритма,
снижение порога фибрилляции
желудочков, ареактивность по
отношению к катехоламинам,
отрицательное инотропное действие
Устойчивое падение
сократимости миокарда
Эффективная центральная и
проводниковая анальгезия,
умеренная симпатическая блокада,
гемодилюция, блокаторы
кальциевых каналов, глюкозо-инсу-
лин-калиевые растворы
Ингаляции кислорода,
дыхание с постоянным
положительным давлением в конце выдоха
для устранения ателектазирова-
ния и мобилизации невентили-
руемых респиронов;
гемодилюция для улучшения оксигенации
крови при нарушениях
легочной микроциркуляции
Анальгезия и нейровегета-
тивная блокада, гемодилюция,
снижающие выраженность юк-
стакапиллярного шунтирования
Сердечные гликозиды,
препараты с избирательным
положительным инотропным
действием
Приток крови в легкие превышает отток, растет гидростатическое
давление в легочных капиллярах. Это проявляется возрастанием
легочного капиллярного давления заклинивания, которое фиксируют, обтурируя
кончиком катетера Сван-Ганца конечные ветви легочной артерии.
При кардиогенном шоке у раненых гидростатическое давление в
легочных капиллярах особенно быстро начинает превалировать над
коллоидно-осмотическим давлением плазмы крови, так как его, кроме
сердечной недостаточности, повышают спазм легочных микрососудов,
замедление легочного кровотока и тромбоз микрососудов в легких.
В результате капиллярного легочного стаза и влияний цитокинов в
высокой действующей концентрации на легочную мембрану уже в
первые часы после травмы патологически возрастает проницаемость
легочных капилляров, а свободнорадикальные продукты перекисного
окисления липидов и другие гуморальные медиаторы шока угнетают
лимфатическое дренирование интерстиция легких. В результате подъем гидроста-
Патофизиология расстройств функциональных систем и нарушений гомеостаза... 563
тического давления в легочном капилляре резко увеличивает интерстици-
альный отек легких с падением их диффузионной способности для
кислорода. Гипоксемия вскоре становится рефрактерной, то есть ее
невозможно устранить повышением концентрации кислорода во вдыхаемой
газовой смеси. Под влиянием респираторной гипоксии нарастает миокарди-
альная депрессия, и кардиогенный шок становится необратимым.
Следует подчеркнуть, что терапия кардиогенного шока при тяжелых
ранениях не может быть только кардиотропной (табл. 33.3). Например,
применение непрерывных инфузий инотропных средств (допамин, добут-
рекс) может принести лишь транзиторный эффект, если ее не сочетать с
эффективной респираторной терапией.
Содержание
ПРЕДИСЛОВИЕ 5
Часть I
Типовые патологические процессы
ГЛАВА 1. БОЛЕЗНЬ И ТИПОВОЙ ПАТОЛОГИЧЕСКИЙ ПРОЦЕСС 8
ГЛАВА 2. НАСЛЕДСТВЕННЫЙ ФАКТОР В ВОЗНИКНОВЕНИИ
И РАЗВИТИИ ЗАБОЛЕВАНИЙ 12
Мутации как причины наследственных заболеваний 12
Моногенные и полигенные болезни 14
Закон частоты генов в популяции и наследование моногенных
болезней 18
Сцепленное с полом наследование моногенной болезни 19
Непрерывная изменчивость предрасположенности к полигенной
болезни как фенотипического признака в популяции 21
Генный комплекс лейкоцитарных антигенов человека и система
антигенов главного комплекса тканевой совместимости
как маркеры предрасположенности к болезням 23
Хромосомные мутации (изменения числа
и макроструктуры хромосом) 26
ГЛАВА 3. ГИПОКСИЯ (проф. Шанин В.Ю., с.н.с. КропотовСЛ.) 29
ГЛАВА 4. НАРУШЕНИЯ ОБМЕНА ВОДЫ И НАТРИЯ 39
Гипонатриемия 42
Гипернатриемия 50
ГЛАВА 5. НАРУШЕНИЯ ОБМЕНА КАЛИЯ 55
ГЛАВА 6. НАРУШЕНИЯ КИСЛОТНО-ОСНОВНОГО СОСТОЯНИЯ 64
Метаболический ацидоз 67
Метаболический ацидоз, связанный с дисфункциями
на уровне сегментов нефрона 71
Метаболический алкалоз 79
ГЛАВА 7. НАРУШЕНИЯ ПЕРИФЕРИЧЕСКОГО КРОВО-
И ЛИМФООБРАЩЕНИЯ (проф. Шанин В.Ю., д.м.н. Тимофеев КВ.) 84
Полнокровие 84
Малокровие 87
Эмболия 89
Нарушения микроциркуляции 92
Недостаточность лимфообращения 94
Диссеминированное внутрисосудистое свертывание 95
ГЛАВА 8. ТРОМБОЗ ГЛУБОКИХ ВЕН
И ТРОМБОЭМБОЛИЯ ЛЕГОЧНОЙ АРТЕРИИ 99
Патогенез расстройств внешнего дыхания вследствие
тромбоэмболии легочной артерии и ее ветвей 104
Патогенез нарушений легочного и системного кровообращения,
связанных с тромбоэмболией легочной артерии и ее ветвей 107
Связь патогенеза и симптомов тромбоэмболии
легочной артерии и ее ветвей 108
Патогенетические принципы профилактики тромбоза глубоких вен
нижних конечностей и тромбоэмболии легочной артерии
у хирургических больных 109
ГЛАВА 9. ПАТОФИЗИОЛОГИЯ ИШЕМИЧЕСКОЙ БОЛЕЗНИ СЕРДЦА 116
ГЛАВА 10. ГОЛОДАНИЕ 138
Физиологическая адаптация к экзогенному голоданию 140
Патогенез алиментарной дистрофии и стрессорного голодания 143
Патогенетические принципы искусственного лечебного питания 148
ГЛАВА 11. ТИПОВЫЕ НАРУШЕНИЯ ЛИПИДНОГО ОБМЕНА 155
Ожирение 155
Гиперлипидемии/гиперлипопротеинемии 161
Атеросклероз 165
ГЛАВА 12. ВОСПАЛЕНИЕ , 170
Определение и биологическое значение воспаления 170
Компоненты и признаки воспаления 172
Нейтрофилия и лейкоцитоз 174
Функционирование нейтрофилов как клеточных эффекторов
острого воспаления 178
Клеточные эффекторы и медиаторы воспаления 185
Воспаление и системы регуляции организма 195
ГЛАВА 13. ЛИХОРАДКА И РЕАКЦИЯ ОСТРОЙ ФАЗЫ 198
Клиническая классификация видов лихорадки 203
Реакция острой фазы 204
ГЛАВА 14. КАНЦЕРОГЕНЕЗ 207
Изменение фенотипа клетки при малигнизации 207
Канцерогенез как комплексное изменение генотипа клетки 209
Канцерогенез и факторы клеточного роста 219
Метастазирование злокачественной клетки
как типовой патологический процесс 222
Часть II
Клиническая патофизиология
функциональных систем
ГЛАВА 15. ДИЗРЕГУЛЯЦИЯ И ПАТОЛОГИЧЕСКИЕ ИЗМЕНЕНИЯ ЭФФЕКТОРОВ
КАК ПРИЧИНЫ РАССТРОЙСТВ ФУНКЦИОНАЛЬНЫХ СИСТЕМ
(НОЗОЛОГИЧЕСКИЙ ОЧЕРК) , 226
ГЛАВА 16. КЛИНИЧЕСКАЯ ПАТОФИЗИОЛОГИЯ КОМЫ И ДРУГИХ
НАРУШЕНИЙ СОЗНАНИЯ 241
ГЛАВА 17. ДЫХАТЕЛЬНАЯ НЕДОСТАТОЧНОСТЬ
И АРТЕРИАЛЬНАЯ ГИПОКСЕМИЯ 246
Артериальная гипоксемия 247
Патологические вариабельность вентиляционно-перфузионных
отношений и шунтирование смешанной венозной крови
в легких как причины артериальной гипоксемии 248
Обструктивные и рестриктивные расстройства
альвеолярной вентиляции 252
Недостаточная вентиляция респиронов как причина
артериальной гипоксемии 257
Респираторные ацидоз и алкалоз 258
Патогенетические принципы проведения
искусственной вентиляции легких 261
ГЛАВА 18. БРОНХИАЛЬНАЯ АСТМА И АСТМАТИЧЕСКИЙ СТАТУС 269
Нейрогенные механизмы патогенеза бронхиальной астмы 276
Патогенез артериальной гипоксемии и нарушений
кислотно-основного состояния при астматическом статусе 277
Патогенетические принципы интенсивной терапии больных
с астматическим статусом 282
ГЛАВА 19. КЛИНИЧЕСКАЯ ПАТОФИЗИОЛОГИЯ СЕРДЕЧНОЙ НЕДОСТАТОЧНОСТИ
(чл.-корр. РАМН Шевченко ЮМ, проф. Шанин В.Ю.) 286
Этиопатогенетическая классификация застойной
сердечной недостаточности 287
Патогенез нарушений преднагрузки и водно-солевого обмена
как причин и следствий застойной сердечной недостаточности ....291
Патологические сдвиги постнагрузки как звено патогенеза
застойной сердечной недостаточности 295
Патофизиология нарушений сократительной способности
сердца у больных с застойной сердечной недостаточностью 296
Связь патогенеза застойной сердечной недостаточности
и симптомов лево- и правожелудочковой
сердечной недостаточности 297
Сердечная недостаточность при высоком МОК 300
Патогенез острой левожелудочковой недостаточности
как причины кардиогенного отека легких 301
Патогенетические принципы терапии кардиогенного
отека легких 305
ГЛАВА 20. ПАТОГЕНЕЗ ИНФАРКТА МИОКАРДА
И КАРДИОГЕННОГО ШОКА 307
Гибернация, станнинг и ишемическое прекондиционирование
кардиомиоцитов 311
Инфаркт миокарда и кардиогенный шок 314
Патогенетические принципы фармакокоррекции
низкой насосной функции сердца 316
Клинико-патофизиологическая характеристика осложнений
и периода реабилитации инфаркта миокарда 323
ГЛАВА 21. ПАТОГЕНЕЗ НАИБОЛЕЕ ЧАСТЫХ ВИДОВ
АРТЕРИАЛЬНОЙ ГИПЕРТЕНЗИИ (проф. Шанин В.Ю., Лютое В.В.) 328
Патогенез гипертонической болезни 330
Вторичная артериальная гипертензия 336
АГ как причина неврологической нестабильности 344
АГ как причина сердечной недостаточности и некоторые
патогенетические принципы фармакотерапии больных
с тяжелой АГ и ее осложнениями 345
Патогенетические принципы лечения гипертонического криза 347
ГЛАВА 22. ПОЧЕЧНАЯ НЕДОСТАТОЧНОСТЬ 351
Преренальная азотемия 353
Гломерулонефропатии 358
Нефротический синдром 364
Острый канальцевый некроз 367
Уремический синдром (уремия) 369
Обструктивная уропатия 372
ГЛАВА 23. ПАТОФИЗИОЛОГИЯ ПЕЧЕНИ 374
Холестаз 375
Желтуха 377
Нарушения обмена веществ как результат печеночной
недостаточности 380
Нарушения сознания и энцефалопатия,
связанные с печеночной недостаточностью 382
Цирроз печени, портальная гипертензия и асцит 383
ГЛАВА 24. ПАТОФИЗИОЛОГИЯ ЖЕЛУДОЧНОЙ СЕКРЕЦИИ, ГАСТРИТОВ,
ЯЗВЕННОЙ БОЛЕЗНИ ЖЕЛУДКА И ДВЕНАДЦАТИПЕРСТНОЙ КИШКИ .. 389
Патогенез гастритов 396
Этиология и патогенез язвенной болезни 399
ГЛАВА 25. ПАТОГЕНЕЗ ДИСФУНКЦИЙ СИСТЕМЫ ПИЩЕВАРЕНИЯ
НА УРОВНЕ КИШЕЧНИКА 404
Синдром недостаточного всасывания 404
Диарея 412
Патогенез острой кишечной непроходимости 421
ГЛАВА 26. ПАТОГЕНЕЗ ОСТРОГО И ХРОНИЧЕСКОГО ПАНКРЕАТИТОВ 431
Этиология и патогенез хронического панкреатита 438
ГЛАВА 27. САХАРНЫЙ ДИАБЕТ 442
Этиология сахарного диабета 444
Патологическая резистентность по отношению к инсулину 445
Патогенез гипергликемии и других нарушений обмена веществ
у больных сахарным диабетом 448
Патогенез диабетической микроангиопатии 451
Диабетические кома и кетоацидоз 458
ГЛАВА 28. ПАТОФИЗИОЛОГИЯ НАИБОЛЕЕ ЧАСТЫХ
ЭНДОКРИНОПАТИЙ (проф. Шанин В.Ю., с.н.с. Кропотов СП.) 462
Расстройства регуляции с участием гормонов аденогипофиза 464
Патогенез эндокринопатий как причин расстройств
роста тела человека 467
Патогенез гипо- и гипертиреоза 472
Патогенез расстройств секреции гормонов надпочечников 478
Патогенез гипер-, гипокальциемии и расстройств секреции
паращитовидных желез 485
Гиперкальциемия 487
Гипокальциемия 490
Патогенез гипогонадизма у мужчин 492
Патогенез гипогонадизма у женщин 495
ГЛАВА 29. РЕАКЦИИ ПОВЫШЕННОЙ ЧУВСТВИТЕЛЬНОСТИ 498
Реакции повышенной чувствительности первого типа 498
Реакции повышенной чувствительности второго типа
(гуморальные цитотоксические реакции) 501
Реакции повышенной чувствительности третьего типа
(образование иммунных комплексов) 503
Реакции повышенной чувствительности четвертого типа
(реакции замедленной гиперчувствительности,
патологические иммунные реакции,
опосредованные клетками) 505
ГЛАВА 30. ИММУНОДЕФИЦИТЫ 509
Патогенез наиболее изученных врожденных иммунодефицитов
на уровне иммунокомпетентных клеток 509
Недостаточность защитной реакции фагоцитоза
как причина иммунодефицита 518
Врожденные иммунодефицита как результат
наследственных расстройств системы комплемента 518
ГЛАВА 31. АУТОИММУННЫЙ МЕХАНИЗМ РАЗВИТИЯ БОЛЕЗНЕЙ 520
ГЛАВА 32. АНЕМИИ 527
Анемии, вызванные нарушениями синтеза гемоглобина
и обмена железа 529
Макроцитарные анемии 531
Нормохромные нормоцитарные анемии 534
ГЛАВА 33. ПАТОФИЗИОЛОГИЯ РАССТРОЙСТВ ФУНКЦИОНАЛЬНЫХ СИСТЕМ
И НАРУШЕНИЙ ГОМЕОСТАЗА В ОСТРОМ ПЕРИОДЕ ПОСЛЕ
ТЯЖЕЛЫХ БОЕВЫХ РАНЕНИЙ И ПРИ РАЗВИТИИ ВОЕННО-
ТРАВМАТИЧЕСКОГО ШОКА 541
Принципы устранения патологической боли и проведения
анестезиологического пособия у больных
с тяжелой раневой болезнью 543
Патогенез респираторного дистресс-синдрома как осложнения
военно-травматического шока и острого периода
тяжелой раневой болезни 546
Патогенез расстройств системного и периферического
кровообращения в остром периоде тяжелой раневой болезни 554
Всеволод Юрьевич ШАНИН
КЛИНИЧЕСКАЯ ПАТОФИЗИОЛОГИЯ
(Под редакцией Ю. Л. Шевченко)
Ответственный за выпуск Бровко А. В.
Технический редактор Иванова О. Е.
Компьютерная верстка Кропотова ТА.
ЛР Ni 071099 от 09.11.94. Подписано в печать 03.04.98. Формат 70xl00l/i6.
Гарнитура Тайме. Печать офсетная. Усл.-печ. л. 46,1.
Тираж 7000 экз. Заказ 73.
Издательство «Специальная Литература» при участии ТОО «Мифрил».
198052, Санкт-Петербург, Измайловский пр., 29
Отпечатано с готовых диапозитивов
в ордена Трудового Красного Знамени ГП «Техническая книга»
Комитета Российской Федерации по печати
198052, Санкт-Петербург, Измайловский пр., 29