/

























































































































































































































































































































































































































































Автор: Фред Дж. Шиффман
Теги: кровообращение кровь патология сердечно-сосудистой системы сердечно-сосудистые заболевания общая терапия внутренние болезни патофизиология
ISBN: 5-7989-0158-0
Год: 2000
Похожие




Текст
Серия Патофизиология
ПАТОФИЗИОЛОГИЯ
КРОВИ
Lippincott's Pathophysiology Series
HEMATOLOGIC
PATHOPHYSIOLOGY
Editor Fred J. Schiffman, MD
Professor of Medicine
Department of Medicine
Brown University School of Medicine
The Miriam Hospital
Providence, Rhode Island
With 16 additional contributors
Lippincott - Raven
PUBLISHERS
Phllmdmlphlm • Nmw York
Фред Дж. Шиффман
ПАТОФИЗИОЛОГИЯ
КРОВИ
Перевод с английского
под редакцией
д-ра мед. наук, проф. Е. Б. Жибурта,
д-ра мед. наук, проф. Ю. Н. Токарева
Под общей редакцией
акад. Ю. В. Наточина
I^binom d8f%& %
^PUBLISHERS ЛГШГАМШЯКШ jum
Москва Санкт-Петербург
2001
УДК 612.1+616.15
ББК 53.53+54.11
Ш65
Перевод с англ.: канд. мед. наук Н. Б. Серебряная, В. И. Соловьев
ШиффманФ. Дж.
Ш65 Патофизиология крови. Пер. с англ.— М.-СПб.: "Издательство БИНОМ"-
"Невский Диалект", 2000.- 448 с, ил.
Монография является кратким руководством по физиологии и патофизиологии крови и системы
кроветворения. В ней представлены современные данные о развитии клеток крови, их
функциональных и морфологических особенностях и свойствах. Рассмотрены нормальные и нарушенные
механизмы гемостаза; патологические состояния, обусловленные недостаточностью костного мозга, и методы
их коррекции; злокачественные опухоли кроветворной ткани; гематологические проявления
ВИЧ-инфекции. В книге подробно излагаются методы обследования, в том числе лабораторные, пациентов с
болезнями крови, а также современные схемы лечения патологии крови и кроветворения.
Для терапевтов, врачей-гематологов и студентов медицинских учебных заведений.
Все права защищены. Никакая часть этой книги не может быть воспроизведена в
любой форме или любыми средствами, электронными или механическими, включая
фотографирование, магнитную запись или иные средства копирования или сохранения информации,
без письменного разрешения издательства.
© Published by arrangement with Lippincott-
Raven Publisher's, Inc., 227 East Washington
Square, Philadelphia, PA 19106-3780 U.S.A.
ISBN 5-7989-0158-0 (Издательство БИНОМ) © "Издательство БИНОМ", "Невский Диалект",
ISBN5-7940-0039-2 (Невский Диалект) 2000
ISBN 0-397-51536-7 (англ.) © Обложка Н. Лозинской, 2000
Оглавление
Авторы 8
Предисловие редакторов перевода 10
Предисловие 11
Благодарности 12
Список сокращений 13
Глава 1. Гемопоэз. Развитие клеток крови (Стивен Дж. Эмерсон) 17
Введение 17
Гемопоэз у эмбриона и плода 19
Гемопоэз во взрослом организме 22
Модель гемопоэза стволовых клеток 34
Клиническое использование гемопоэтических факторов роста 38
Глава 2. Клинический подход к пациентам
с гематологическими проблемами (ФредДж. Шиффман) 43
Глава 3. Эритроциты (Майкл Дж. Роуз, Нэнси Берлинер) 71
Структура и функции эритроцита 71
Клиническая патофизиология анемии 75
Дифференциальная диагностика гипопролиферативной анемии 86
Анемия с ретикулоцитозом 101
Глава 4. Лейкоциты (АланДж. Розмарин) 123
Нейтрофилы 123
Аномалии гранулоцитов 128
Нарушение функции нейтрофилов.. 129
Нарушение локомоции нейтрофилов и захвата ими
микроорганизмов 129
Нарушение бактерицидной активности 132
Нейтрофилия 132
Нейтропения 134
Эозинофилы „ 135
Базофилы 135
Моноциты 136
Лимфоциты 137
В-лимфоциты 138
Образование антител 139
Т-лимфоциты 142
Лимфоцитарные нарушения 145
Глава 5. Тромбоциты (Эрик М. Мазур) 149
Образование и кинетика тромбоцитов 150
Структура тромбоцитов 153
Функции тромбоцитов 154
Лабораторные исследования 156
Тромбоцитопатии .. 161
Тромбоцитопении 167
6 Оглавление
Переливание тромбоцитов 173
Тромбоцитоз 183
Глава 6. Гемостаз и тромбоз (Анджелина К. А. Карвальхо) 191
Нормальный гемостаз 191
Нарушения гемостаза 214
Нарушения тромбообразования 253
Глава 7. Недостаточность костного мозга, апластическая анемия
(Даниель Е. Дани) 283
Приобретенная апластическая анемия 283
Приобретенная парциальная
красноклеточная аплазия 290
Приобретенная амегакариоцитарная тромбоцитопеническая
пурпура 293
Агранулоцитоз и другие нейтропенические синдромы 293
Наследственные синдромы недостаточности костного мозга 301
Глава 8. Трансплантация стволовых клеток
(Митчелл Е. Горовиц, Синтия Е. Данбар) 307
Введение 307
Аллогенная трансплантация 309
Аутологичная трансплантация 327
Глава 9. Злокачественные опухоли кроветворной ткани
(Лоуренс Н. Шульман) 335
Молекулярные основы возникновения злокачественных опухолей
кроветворной ткани 335
Острые лейкозы 336
Миелопролиферативные заболевания 343
Миелодиспластические синдромы 347
Лимфомы 349
Глава 10. Гематологические проявления ВИЧ-инфекции
(Бхарат Рамратнан, Джанти Парамесварап, Тимоти П. Фланиган,
Джеймс А. Хокси) 365
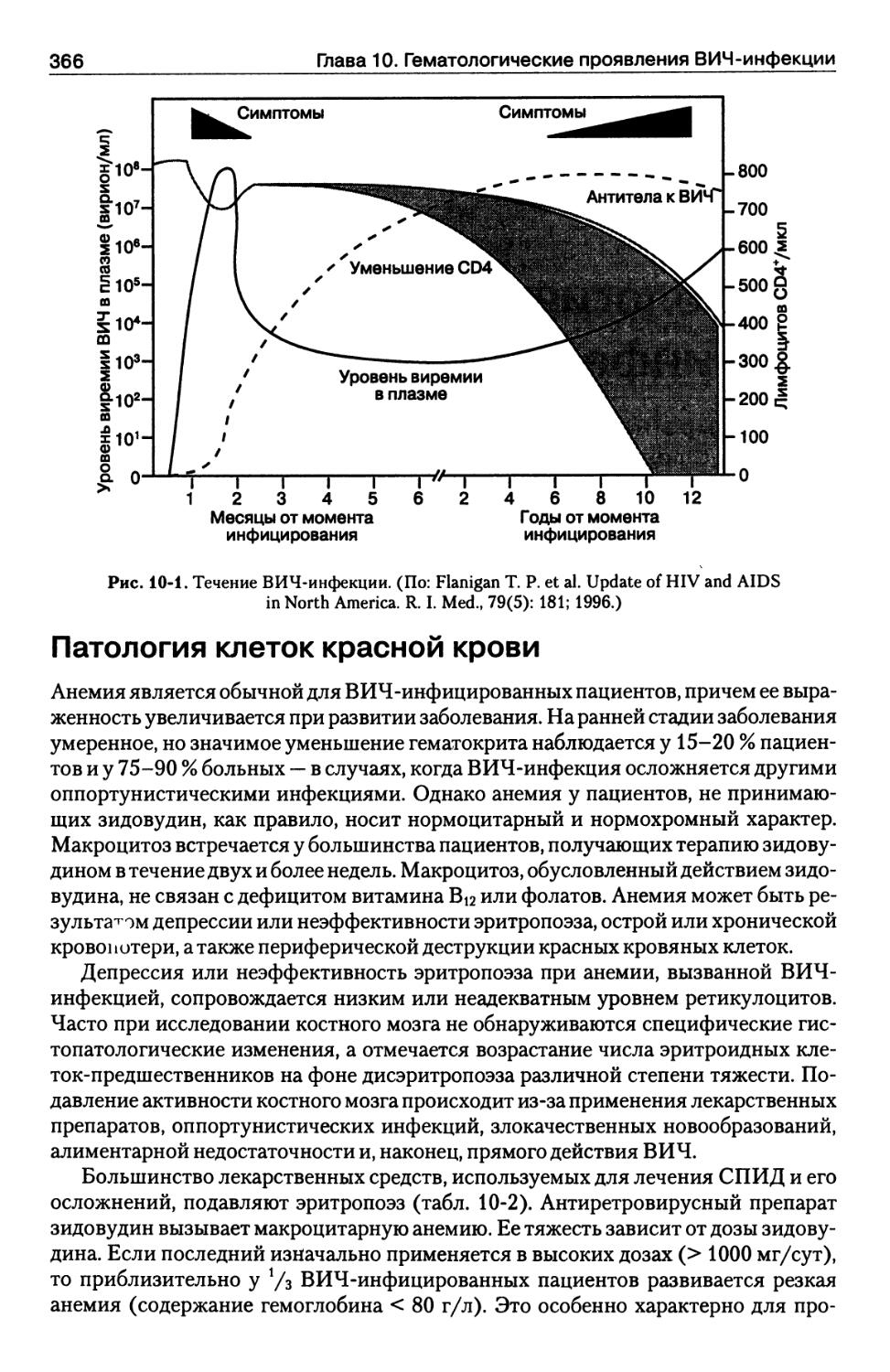
Патология клеток красной крови 366
Патология клеток белой крови 371
Патология тромбоцитов 372
Патология свертывания 374
Злокачественные опухоли кроветворной ткани 375
Глава 11 ♦ Клиническая лабораторная гематология
(Питер Мак-Федран, Натали Ортоли-Дрю) 379
Приложение 1. Эритроцитарные индексы 425
Приложение 2. Время жизни клеток крови 426
Приложение 3. Порфирии 427
Приложение 4. Морфологические характеристики лейкоцитов
(окраска по Райту) 428
Предметный указатель 432
Моему отцу Генри, матери Белл и брату Роберту —
первым постоянным и лучшим учителям
Докторам Солу Фарберу, Самюэлю Тьеру
и Чарльзу Карпентеру — моим наставникам
Джерри, Джошуа, Джессике, Джейку и Джудит —
детям
Авторы
Nancy Berliner, M.D.
(Нэнси Берлинер)
Associate Professor of Medicine
Department of Internal Medicine
Yale University School of Medicine
333 Cedar Street
New Haven, Connecticut 06520-8021
Angelina C. A. Carvalho, M.D.
(Анджелина К. А. Карвапьхо)
Professor of Medicine
Department of Medicine
Brown University School of Medicine
Chief of Hematology
Providence Veterans Administration
Medical Center
830 Chalkstone Avenue
Providence, Rhode Island 02908-4799
Cynthia E. Dunbar, M.D.
(Синтия Е. Данбар)
Senior Investigator
Hematology Branch
National Heart, Lung, and Blood Institute
9000 Rockville Pike
Bethesda, Maryland 20892-1652
Daniel E. Dunn, M.D., Ph.D.
(ДаниельЕ. Данн)
Attending, Hematology and BMT Services
Hematology Branch
National Heart, Lung, and Blood Institute
National Institute of Health
9000 Rockville Pike
Bethesda, Maryland 20892-1652
Stephen G. Emerson, M.D., Ph. D.
(Стивен Дж. Эмерсон)
Francis С. Wood Professor of Medicine
Chif, Hematology-Oncology Division
Department of Medicine
University of Pennsylvania School of
Medicine
3600 Spruce Street
Philadelphia, Pennsylvania 19104-4283
Timothy P. Flanigan, M.D.
(Тимоти П. Фланиган)
Associate Professor of Medicine
Department of Medicine
Brown University School of Medicine
The Miriam Hospital
164 Summit Avenue
Providence, Rhode Island 02906
Mitchell E. Horwitz, M.D.
(Митчелл Е. Горовиц)
Clinical Associate
Department of Hematology
National Heart, Lung, and Blood Institute
National Institute of Health
9000 Rockville Pike
Bethesda, Maryland 20892-1652
James A. Hoxie, M.D.
(Джеймс А. Хокси)
Professor of Medicine
Department of Medicine
University of Pennsylvania School of
Medicine
415 Curie Boulevard
Philadelphia, Pennsylvania 19104
EricM. Mazur, M.D.
(ЭрикМ. Мазур)
Associate Clinical Professor of Medicine
Department of Internal Medicine
Yale University School of Medicine
333 Cedar Street
New Haven, Connecticut 06520-8021
Peter McPhedram, M.D.
(Питер Мак-Федран)
Professor of Laboratory Medicine and
Medicine
Department of Labolatory Medicine
and Internal Medicine
Yale University School of Medicine
Yale-New Haven Hospital
20 York Street
New Haven, Connecticut 06504
Авторы
9
Natalie Ortoli-Drew, B.S,M.T.(A.S.C.R)
(Натали Ортоли-Дрю)
Supervisor of Clinical Hematology
and Lecturer in Laboratory Medicine
Department of Labolatory Medicine
and Clinical Hematology
Yale University School of Medicine
Yale-New Haven Hospital
20 York Street
New Haven, Connecticut 06504
Jayanthi Parameswaran, M.D.
(Джанти Парамесваран)
Attending Physician
Department of Medicine
Newport Hospital/Aquidneck Medical
Associates
Memorial Boulevard
Newport, Rhode Island 02840
Bharat Ram rat nan, M.D.
(Бхарат Рамратнан)
Assistant Instructor of Medicine
Department of Medicine
Brown University School of Medicine
Chief Medical Resident
The Miriam Hospital
164 Summit Avenue
Providence, Rhode Island 02906
Michael G. Rose, M.D.
(Майкл Дж. Роуз)
Post Doctorant Fellow in Hematology
Department of Medicine
Yale University School of Medicine
333 Cedar Street
New Haven, Connecticut 06520-8021
Alan G. Rosmarin, M.D.
(Алан Дж. Розмарин)
Associate Professor of Medicine
Department of Medicine
Brown University School of Medicine
The Miriam Hospital
164 Summit Avenue
Providence, Rhode Island 02906
Fred J. Schiffman, M.D.
(Фред Дж. Шиффман)
Professor of Medicine
Department of Medicine
Brown University School of Medicine
The Miriam Hospital
164 Summit Avenue
Providence, Rhode Island 02906
Lawrence N. Shulman, M.D.
(Лоуренс Н. Шульман)
Associate Professor of Clinical Medicine
Harvard Medical School;
Department of Adult Oncology
Dana-Farber Cancer Institute
44 Binney Street
Boston, Massachusetts 02 115
Предисловие
редакторов перевода
Достижения клинической гематологии последних лет являются ярким примером
прогресса современной биологической и медицинской науки. Возможность
радикального излечения злокачественных опухолей кроветворной ткани, увеличение
продолжительности и улучшение качества жизни пациентов практически при
всех гематологических заболеваниях, профилактика наследственных
заболеваний системы крови обусловлены внедрением в практику здравоохранения
достижений: генетики, цитологии, иммунологии, фармакологии и биотехнологии.
Отечественная медицинская традиция предполагает понимание
патофизиологических процессов как основу диагностики и лечения. Соответственно,
формирование правильного клинического мышления невозможно без знания
механизмов развития и течения заболеваний на молекулярном уровне. Именно
современное представление о патофизиологии системы крови и кроветворения
составляет основную ценность данной книги для российского читателя. Вторым
важным достоинством является сочетание простоты и доступности информации с
ее значительным объемом. Издание может служить как учебным пособием для
студентов, так и,настольной книгой для практических врачей. Иллюстрация
сведений о клинической патофизиологии примерами из врачебной практики
наглядно демонстрирует внедрение в жизнь "медицины, основанной на
доказательствах".
Невозможно в одной компактной монографии охватить весь круг проблем
частной гематологии и гемокомпонентной терапии. В связи с этим можно адресовать
читателя к книге А. Г. Румянцева и В. А. Аграненко "Клиническая трансфузиоло-
гия" (1997) и другим специальным изданиям. К сожалению, современного
руководства по гематологии в России пока нет.
Интегрирующая роль системы крови делает настоящую монографию
полезной не только для гематологов, онкологов, трансфузиологов, но и для
специалистов в разных областях биологии и медицины.
Д-р мед. наук, проф. Е. Б. Жибурт
Д-р мед. наук, проф. Ю. Н. Токарев
Предисловие
Патофизиология крови предназначена для студентов или врачей, желающих
получить общее представление о гематологии. Основу книги составляет изложение
принципов патофизиологии, дополненное описаниями историй болезни и обсуждением
клинических проблем. Авторы считают, что такое представление материала, когда
данные гематологии подкрепляются изучением проблем конкретного пациента,
принесет существенную пользу. Во избежание сухости подачи материала мы
расположили гематологические детали рядом с касающимися их клиническими ситуациями.
Мы надеемся показать читателю, как на почве знаний процессов,
происходящих на молекулярном уровне, базируются клинические достижения. Мы
попытались придать особое значение построению того научного фундамента, на котором
только и возможна успешная клиническая гематологическая практика.
Мы полагаем, что разбор клинических случаев и дискуссии помогут ощутить
весь реализм и драматизм практической гематологии. Иллюстрируя таким
образом нашу книгу, мы хотим не только рассказать о том, чего нельзя забывать, но и
подкрепить теоретические знания и убедить читателя к необходимости
понимания процессов, происходящих на фундаментальном уровне, для того, чтобы
лечение пациентов было как можно более эффективным.
Большинство глав книги организовано по единой схеме: сначала очерк
нормальной физиологии крови, а затем порядок обследования пациента с
сопоставлением научных методов и клинических аспектов.
Читателю будут представлены жизненный цикл, структура и физиология
клеток красной крови в норме и при патологии. Таким же образом рассматриваются
лейкоциты и тромбоциты. Далее следует обсуждение проблем гемостаза,
патологии костного мозга и злокачественных опухолей кроветворной ткани. Каждый
раздел включает клинические иллюстрации и их анализ. Глава "Клиническая
лабораторная гематология" предоставляет возможности для расширения и закрепления
знаний, изложенных в предшествующих главах. Ее табличный формат
обеспечивает быстрый доступ к информации по всем аспектам гематологической
лабораторной диагностики. Приведенные в приложениях общепринятые гематологические
величины осуществляют информационную связь между отдельными главами.
Хотя предприняты определенные усилия по обеспечению однообразия стиля и
объема глав, из уважения к авторам, мы попытались сохранить индивидуальность
и особенности творчества каждого из них. Кроме того, в процессе создания
единого текста мы не устраняли информационные пересечения между главами.
Основной материал каждой из них построен так, что, несмотря на различие тем,
вариантов графического представления и объема, он позволяет ясно и полно раскрыть
особенности сложных проблем гематологии.
Таким образом, наша задача заключается в том, чтобы обеспечить доступный,
исчерпывающий патофизиологический подход к диагностике и терапии
заболеваний крови. Используя истории болезни реальных пациентов и освещая вопросы
оказания помощи, мы надеемся с большей убедительностью показать то, как
знание гематологии можно и должно использовать для лечения пациентов с
гематологическими заболеваниями.
Фред Дж. Шиффман
Благодарности
Я благодарю Элеонору Алоизио за ее выдающееся секретарское и
стилистическое мастерство в подготовке к публикации рукописи этой книги.
Я признателен Ричарду Винтерсу, Делуа Паттерсону и Эмили Гаркави из
издательства Lippincott-Raven Publishers за придание данной работе особенностей,
делающих ее необыкновенно приятной.
Особая моя признательность адресована врачам-исследователям, в чьих
лабораториях я имел честь трудиться: Майклу Фридману, Марко Рабиновичу, Эду
Кадману и Леону Вейссу. Их знания и сила духа помогли мне найти путь, по
которому медицина и жизнь идут рука об руку.
Я ценю замечания и проницательные комментарии Джошуа Шиффмана,
который прочитал всю рукопись как студент-медик и поэт.
Список сокращений
а2-АП — а2-антиплазмин
2,3-ДФГ — дифосфоглицерат
ABVD — Adriamycine (doxorubicine), Bleomycin, Vinblastine, Dacarbazine
CHOP — Cyclophosphamide, Hydroxyldaunomycine, Oncovin (vincristine),
Prednison
DD AVP — 1 -дезамино-8Б-аргинин-вазопрессин
G-SH — глутатион
Hb — гемоглобин
HELPP- — гемолитическая анемия, повышенная активность печеночных
синдром ферментов, низкое содержание тромбоцитов
Ig — иммуноглобулин
МAI — Mycobacterium avium intracellular
МОРР — nitrogen Mustard, Oncomvin [vincristine], Procarbazine, Prednisone
AA — апластическая анемия
АД Б — анемия Даймонда-Блекфана
АИК — аппарат искусственного кровообращения
АКН — абсолютное количество нейтрофилов
АлАТ — аланинаминотрансфераза
АЛГ — антилимфоцитарный глобулин
АПК — антигенпрезентирующие клетки
АПС — активированный протеин С
АсАТ — аспартатаминотрансфераза
AT III — антитромбин III
АТГ — антитимоцитарный глобулин
АТФ — аденозинтрифосфат
АФ — анемия Фанкони
АФ А — антифосфолипидные антитела
АФС — антифосфолипидный синдром
АЧТВ — активированное частичное тромбопластиновое время Q f
БГЛ — большиегранулоцитар/шелимфоциты £&~£о№$>Н1» pUAfaj&HCjfi
БЕ — единица Бетесда
БОЕ-МК — бурстообразующая единица, мегакариоцитарная
БОЭ — бурстообразующая единица, эритроцитарная
БТПХ — болезнь трансплантат против хозяина
ВА — волчаночный антикоагулянт
ВВИГ — внутривенный иммуноглобулин
ВИЧ — вирус иммунодефицита человека
ВК — время кровотечения
ВМК — высокомолекулярный киноген
ВОБ — веноокклюзионная болезнь
ВПГ — вирус простого герпеса
ВФ — внутренний фактор
ВЭБ — вирус Эпштейна- Барр
14
Список сокращений
Г-6-ФДГ — глюкозо-6-фосфатдегидрогеназа
ГА — гемофилия А
ГБГ — гликопротеин, богатый гистидином
ГК — гематокрит
ГКГ — главный комплекс гистосовместимости
Г-КСФ — гранулоцитарный колониестимулирующий фактор
ГМ-КСФ — гранулоцитарный-макрофагальный КСФ
ГП — гликопротеин
ГУ С — гемолитический уремический синдром
ГФ — гепариновый фактор
ГФА(С) - гликофорин А(С)
ГФР — гемопоэтический фактор роста
ДВС — диссеминированное внутрисосудистое свертывание
ДМГ — доброкачественная моноклональная гаммапатия
ДМС — демаркационная мембранная система
ДМСО — диметилсульфоксид
ДНК — дезоксирибонуклеиновая кислота
ДЭБ — диэпоскибутан
ЖДА — железодефицитная анемия
ЖКТ — желудочно-кишечный тракт
ИАП — ингибитор активатора плазминогена
ИИП — идиопатический интерстициальный пульмонит
ИЛ — интерлейкин (ы)
ИМН — инфекционный мононуклеоз
ИПТФ — ингибитор пути тканевого фактора
ИТП — иммунная тромбоцитопеническая пурпура
ИФ А — иммуноферментный анализ
КИЭФ — количественный электроиммунофорез
КОЕ-В — колониеобразующая единица, В-клеточная
КОЕ-Г — колониеобразующая единица, гранулоцитарная
КОЕ-ГМ — колониеобразующие единица, гранулоцитарная-моноцитарная
КОЕ-Т — колониеобразующая единица, Т-клеточная
КОЕ-Э — колониеобразующая единица, эритроцитарная
КППК — клетки-предшественники периферической крови
КСФ — колониестимулирующие факторы
КТ — компьютерная томография
ЛГМ — лимфогранулематоз (болезнь Ходжкина)
ЛДГ — лактатдегидрогеназа
МГНЗ — моноклональная гаммапатия неизвестного значения
МДС — миелодиспластический синдром
М-КСФ — макрофагеальный КСФ
ММС — митомицин С
MHO — международное нормализованное отношение (INR)
МПН — миелопролиферативные нарушения
НАДФН — никотинадениндинуклеотидфосфат
НК — нормальные, "натуральные", киллеры
НПВС — нестероидные противовоспалительные средства
НСТ — нитросиний тетразолий
НХЛ — неходжкинскиелимфомы
OAK — общий анализ крови
Список сокращений
ОАОЛ — общий антиген острого лейкоза
оБТПХ - острая БТПХ
ОКС — открытая канальцевая система
О Л Л — острый лимфобластный лейкоз
О МЛ — острый миел обл астный лейкоз
ОММЛ — острый миеломоноцитарный лейкоз
ОПМЛ — острый промиелоцитарный лейкоз
ПА — пернициозная анемия
ПАЛО — периартериальная оболочка
ПАФС — первичный антифосфолипидный синдром
ПБКА — нарушенная белоклеточная аплазия
ПВ — протромбиновое время
ПГ — простагландин (ПГА2, h и т. д.)
ПДФ — продукты деградации фибрина и фибиногена
ПК — прекалликреин
ПККА — парциальная красноклеточная аплазия
ПМЯЛ — полиморфно-ядерный лейкоцит
ПН-1 — протеаза-нексин-1
ПНГ — пароксизмальная ночная гемоглобинурия
ПНТ — процедура нейтрализации тромбоцитов
ПРФ — продукты расщепления фибриногена
ПС — протеин С
ПЦР — полимерная цепная реакция
ПЭ — протопорфирин эритроцитов
РА — ревматоидный артрит
РИА — радиоиммунный анализ
РИ AT — ристоцетининдуцированная агрегация тромбоцитов
РЭС — ретикуло-эндотелиальная система
С1-И — ингибитор эстеразы С1
СГК — стволовые гемопоэтические клетки
СЗП — свежезамороженная плазма
СКА — серповидно-клеточная анемия
СКВ — системная красная волчанка
СКГ — средняя концентрация гемоглобина в эритроцитах
СНД — совместимый неродственный донор
СОК — средний объем клетки
СОЭ — скорость оседания эритроцитов
СПИД — синдром приобретенного иммунодефицита
ССГ — среднее содержание гемоглобина/число эритроцитов
ТЗ — трийодтиронин
ТАП — тканевый ак-Гиватор плазминогена
ТАФ — тромбоцитактивирующий фактор
ТГВ — тромбоз глубоких вен
ТГФ — тетрагидрофолат
ТДТ — терминальная нуклеотидтрансфераза
ТДЭ — транзиторная детская эритробластопения
ТИФА — твердофазный иммуноферментный анализ
TKI — транскобаламин I
ТКМ — трансплантация костного мозга
ТКР — Т-клеточный рецептор
16
тм
тмп/смк
толк
тпл
тпо
ттп
ТФ
ТФР
ф
ФАТ
ФВ
ФНО-а
ФПА(В)
ФСК
хБТПХ
ХГ
ХЛЛ
ХМЛ
цмв
цпэ
ЦсА
шд
ЭАКК
ЭДТА
ЭК
эквс
эп
ЭФР
— тромбомодулин
— триметоприм/сульфаметоксазол
— тромбоцитопения с отсутствием лучевой кости
— трансплантат против лейкоза
— тромбопоэтин
— тромбическая тромбоцитопеническая пурпура
— тканевый фактор
— трансформирующий фактор роста
— фактор (свертывания крови, например ФШ, Ф1У)
— фактор активации тромбоцитов
— фактор Виллебранда
— фактор некроза опухоли-альфа
— фибринопептид А (В)
— фактор стволовых клеток
— хроническая БТПХ
— хроничский гранулематоз
— хронический лимфолейкоз
— хронический миелолейкоз
— цитомегаловирус
— цинковый протопорфирин (эритроцитов)
— циклоспорин А
— синдром Швахмана-Даймонда-Оски
— е-аминокапроновая кислота
— этилендиаминтетрацетрат
— эндотелиальные клетки
— эндотелиальные клетки венозных синусов
— эритропоэтин
— эндотелиальный фактор релаксации
Глава 1
Гемопоэз.
Развитие клеток крови
Стивен Дж. Эмерсон
Введение
Все клетки, циркулирующие в периферической крови взрослого человека, имеют
костномозговое происхождение и вовлечены в сложный процесс, называемый ге-
мопоэзом. В результате образуются различные типы клеток крови, каждая из
которых обладает уникальными особенностями и определенной
продолжительностью жизни. Главные элементы гемопоэза описываются так называемой моделью
стволовой клетки. На основании этой модели в книге рассмотрены вопросы,
касающиеся гемопоэза в норме и при патологии, а также проблемы терапии
заболеваний системы крови.
Теория стволовой клетки. В отличие от других тканей мезодермального
происхождения, которые, как правило, характеризуются редкой сменой клеточных
популяций, клетки крови постоянно погибают и заменяются новыми (рис. 1-1). При
этом эритроциты циркулируют в крови приблизительно 4 мес, тромбоциты —
около 1 нед, а гранулоциты — менее 10 ч. Подсчитано, что каждый день теряется
1 х Ю11 клеток крови, которые стареют, разрушаются и заменяются на равное
количество новых. Чтобы удовлетворить эту постоянную потребность в новых
клетках, гемопоэз не прерывается в течение всей жизни. Вследствие этого
кроветворные ткани наряду с эпителием желудочно-кишечного тракта, яичками и
эпидермисом являются одними из наиболее митотически активных.
Подтверждение высокой частоты обновления клеток крови привело к созданию теории
стволовых клеток, обеспечивающих развитие и поддержание клеточного состава
крови, или гемопоэз. В этой главе мы подробно рассмотрим данную теорию и ее
применение в клинической практике.
Гемопоэтические факторы роста. Одно из наиболее значительных достижений
в изучении гемопоэза в последнем десятилетии связано с исследованием роли
специфических гемопоэтических полипептидов в регуляции дифференцировки
клеток крови. Эти гемопоэтические гормоны, называемые также гемопоэтически-
ми факторами роста, по-видимому, контролируют все этапы кроветворения. В
настоящее время известны преимущественно ростовые факторы, стимулирующие
стволовые клетки и их производные. Однако были обнаружены и некоторые ин-
гибиторные факторы, которые, как представляется, играют такую же важную
Первичная
стволовая
клетка
Полипотентные
клетки-
предшественники
Унипотентные
клетки-
предшественники
Зрелые
циркулирующие
клетки
Рис. 1-1. "Иерархическая" модель гемопоэза, включающая важнейшие цитокины
Гемопоэз у эмбриона и плода
19
роль в регуляции гемопоэза. Другие гормоны, действие которых первоначально
не связывали с контролем гемопоэза, как теперь стало известно, также влияют
на кроветворение. В этой главе мы кратко опишем роль как стимулирующих, так
и ингибирующих гемопоэтических ростовых факторов.
Гемопоэтическое микроокружение. Термин гемопоэтическое микроокружение%
относится к стромальным элементам органов, в которых происходит гемопоэз.
Микроокружение формируют клеточные и неклеточные элементы,
непосредственно не участвующие в кроветворении, но образующие трехмерный
структурный матрикс, где стволовые клетки и их потомки пролиферируют и
дифференцируются до перемещения в кровоток. Гемопоэтическое микроокружение имеет
первостепенную важность для регуляции развития клеток крови. И стромальные
клетки, и секретируемые ими белки межклеточного матрикса влияют на процесс
гемопоэза столь же существенно, как и растворимые секретируемые гемопоэти-
ческие ростовые факторы. В эту главу включено краткое обсуждение роли гемо-
поэтического микроокружения.
Гемопоэз у эмбриона и плода
Роль желточного мешка. Через некоторое время после оплодотворения яйца (2-
3 нед) возникает эмбриональное кроветворение (рис. 1-2). Первые этапы этого
процесса происходят в желточном мешке, где найдены недифференцированные
клетки, называемые мезобластами, которые мигрируют в него из первичной
полоски эмбриона. Мезобласты имеют высокую митотическую активность и
впоследствии дифференцируются в клетки, называемые первичными эритробластами,
несомненно родственные зрелым кровяным клеткам взрослого человека, а также
первичным эндотелиальным клеткам, образующим сосудистую систему
желточного мешка. В течение нескольких часов после миграции происходит деление
и дифференцировка мезобластов желточного мешка до первичных эритроцитов.
Большинство этих клеток ядросодержащие, некоторые же не имеют ядер. Но все
они синтезируют гемоглобин, что обусловливает красноватый цвет хорошо
различимых кровяных островков желточного мешка.
В кровяных островках найдены также предшественники тромбоцитов, мегака-
риоциты, которые тоже происходят от мезобластов. Другие мезобласты, видимо,
дифференцируются в клетки, называемые гемоцитобластами.
У эмбрионов некоторых млекопитающих описана вторая стадия гемопоэза
в желточном мешке. Она существует и у человеческих эмбрионов, но протекает
не так энергично, как, например, у кролика, эмбриогенез клеток крови которого
наиболее изучен. На второй стадии гемопоэза в желточном мешке гемоцитобла-
сты дифференцируются в окончательные эритробласты, которые впоследствии
синтезируют гемоглобин и становятся окончательными, или вторичными, нор-
мобластами. Последние могут терять свои ядра и становиться окончательными
эритроцитами. В кровяных островках формируются сосудистые каналы,
объединяющиеся в конечном счете в сеть кровеносных сосудов. Эта сеть примитивных
кровеносных сосудов на ранних этапах содержит первичные эритробласты и ге-
моцитобласты, а на более поздних — зрелые эритробласты и эритроциты. К
концу третьей недели эмбрионального развития кролика гемопоэтическая
активность кровяных островков падает и процесс гемопоэза перемещается в печень.
20
Глава 1. Гемопоэз. Развитие клеток крови
Хромосома 16
5' С, а2 a^
^2^2 C2Y2 0282
Гауэр 1 Портлэнд Гауэр 2
А ' 1 '
Эмбрион
Место эритропоэза
Хромосома 11
5'
ш-ш-ш
™sy;a^ Иди ИшгпЯ
6
""[fS$S^ «Г"™"
Р
a2Y2
F
a2Y2 a262
. F Az
Плод
1
Взрослый
1. Пре-a: а», fce2 (Гауэр 1)
2. Ранний а: а2е2 (Гауэр 2)
3. Мало или отсутствует а: &уг (Портлэнд)
4. Мало или отсутствует а: уд (Барт)
5. Присутствует а: а2уг (нормальный Hb F)
6. Мало или отсутствует а (поздний): В4 (Hb H)
7. Нормальный после рождения: a202 (Hb A)
8. 0-варианты Hb А (пример: a2fc Hb S)
9. Нормальный: агбг (Hb А2)
12 18 24 36 36 42 48
Рис. 1-2. (А) Юшстеры гена глобина на хромосомах 16 и И. У эмбриона, плода и взрослого человека
активируются или подавляются разные гены. Различные цепи глобина синтезируются независимо,
а затем объединяются друг с другом, что приводит к образованию нескольких типов гемоглобина. Ген
Y может иметь две последовательности, что приводит к синтезу цепей, отличающихся наличием остатка
глутаминовой кислоты или аланина в позиции 136 (Gy или Ay соответственно). (Цит. по: Hoffbrand A. V.,
Pettit J. E. Essential Hematology, 3rd ed. Cambridge, Mass.: Blackwell Scientific Publishing; 1993.) (Б)
Соотношение стадии развития, локализации гемопоэза и синтеза гемоглобина. Петли соединяют цени,
которые связываются в норме и при патологии. (По: Brown M. S. Fetal and Neonatal Erythropoiesis in
Developmental and Neonatal Hematology. New York: Raven Press; 1988. Из: Handin R. I., Stossel Т. Р,
Lux S. E. (eds.) Blood: Principles and Practice of Hematology. Philadelphia: J. B. Lippincott, 1995.)
Эмбриональная мезенхима. Дополнительную роль в раннем эмбриональном ге-
мопоэзе непосредственно в полости тела играют первичные мезенхимные клетки,
особенно в районе передней прекардиальной мезенхимы. Малая часть мезенхим-
ных клеток развивается в эритробласты, мегак^риоциты, гранулоциты и
фагоцитирующие клетки, аналогичные соответствующем клеткам взрослых. Количество
этих клеток невелико, и больших разрастаний клеток крови, подобных
кроветворным островкам желточного мешка, в мезенхиме полости тела не формируется.
Стволовые клетки, располагающиеся среди этих гемопоэтических клеток (вне
желточного мешка), вероятно, играют главную роль в генерации последующих
поколений гемопоэтических клеток у плода и в постнатальном периоде, хотя
относительный вклад первичных стволовых клеток, находящихся в желточном
мешке и вне его, в более поздний гемопоэз пока не ясен.
Печеночный период эмбрионального гемопоэза. У человека, начиная примерно
со стадии 12 мм эмбриона (возраст 6 нед), гемопоэз постепенно перемещается
Гемопоэз у эмбриона и плода
21
в печень (рис. 1-2). Печень скоро становится основным местом гемопоэза и
является активной в этом отношении до момента рождения. Поскольку энтодермаль-
ные тяжи печени формируются в поперечные перегородки, они сталкиваются
с блуждающими мезенхимными клетками с морфологией лимфоцитов. Эти
маленькие круглые лимфоидные клетки, называемые лимфоцитоидными
блуждающими клетками, впоследствии улавливаются между первичными печеночными
энтодермальными тяжами и эндотелиальными клетками врастающих
капилляров. Они образуют гемоцитобласты, подобные таковым в желточном мешке. Эти
гемоцитобласты вскоре формируют очаги гемопоэза, аналогичные кровяным
островкам желточного мешка, где вторичные эритробласты образуются в больших
количествах. Вторичные эритробласты впоследствии делятся и
дифференцируются в зрелые эритроциты, при этом происходят активация синтеза гемоглобина
и потеря клеточного ядра. Хотя зрелые эритроциты обнаруживаются в печени
эмбриона уже в возрасте 6 нед, в значимом количестве они появляются в
циркуляции гораздо позднее. Таким образом, к четвертому месяцу жизни плода
большинство циркулирующих эритроцитов представлено вторичными зрелыми формами.
Мегакариоциты также, вероятно, образуются из гемоцитобластов в печени
эмбриона и плода. В эмбриональной печени находят гранулоцитарные клетки, но
развиваются они, видимо, не из гемоцитобластов, а непосредственно из блуждающих
лимфоцитоидных клеток.
Эмбриональный костный мозг и миелопоэз. Различные кости у эмбриона
образуются не одновременно. Раньше других — длинные кости добавочного скелета.
Первоначально формируется хрящевая модель каждой кости. Центральное ядро
диафиза впоследствии оссифицируется, и вскоре вслед за врастанием мезенхимных
клеток из периоста развивается область костной резорбции. Процесс движения
мезенхимных клеток сопровождается врастанием внутрь капилляров. Количество
мезенхимных клеток продолжает увеличиваться за счет непрерывного притока
новых клеток, а также делением тех, которые уже находятся внутри недавно
сформировавшейся костномозговой полости. Они нарабатывают неклеточный
материал, или матрикс, заполняющий развивающуюся полость кости. Из этих ранних
костномозговых мезенхимных клеток образуются клетки, морфологически сходные
с гемоцитобластами печени и желточного мешка. Аналогично последним, они
дают начало мегакариоцитам и эритроидным клеткам, а также миелоидным,
включая нейтрофилы, базофилы и эозинофилы. Эмбриональный костный мозг заметно
отличается от центров более раннего развития гемопоэза тем, что образование ми-
елоидных клеток идет здесь особенно энергично и доминирует в гемопоэзе.
Процесс формирования ранних миелоидных клеток, или миелопоэз, начинается в
центральной части костномозговой полости и распространяется оттуда, чтобы
в конечном счете захватить всю полость кости. Эритропоэз в эмбриональном
костном мозге развивается немного позже и в основном смешивается с процессом
миелопоэза, так что среди большинства созревающих клеток миелоидной линии
можно наблюдать малые очаги эритропоэза. После рождения у человека гемопоэз
в печени прекращается, но продолжается в костном мозге всю оставшуюся жизнь.
Гемопоэз в селезенке эмбриона и плода. Последним важнейшим очагом
гемопоэза, который образуется в эмбриональном периоде, является селезенка. Хотя сама
селезенка формируется у человека! намного раньше, циркулирующие гемопоэти-
ческие предшественники начинают заполнять ее примерно на четвертом месяце
22
Глава 1. ГемопоэЗ. Развитие клеток крови
беременности. Вероятно в результате скопления большого объема крови
селезенка плода становится центром гемопоэза до момента рождения, когда селезеночный
эритропоэз постепенно прекращается. В целом миелопоэтическая активность
селезенки эмбриона и плода сравнительно невелика. Позднее, в течение пятого
месяца эмбрионального развития, формируется белая пульпа селезенки. Этот
процесс связан с дифференцировкой мезенхимных клеток, которые группируются
вокруг селезеночных артериол. Образование селезеночных лимфоцитов у
эмбриона полностью пространственно отделено от центров эритропоэза в этом органе.
Другие места гемопоэза у эмбриона и плода. Эмбриональный тимус развивается
как производное третьего жаберного кармана. Тимический эпителий заполняется
блуждающими мезенхимными клетками, которые начинают быстро размножаться
и дифференцироваться в лимфоциты. Одновременно в тимусе формируется
незначительное количество эритроидных и миелоидных клеток, но преобладает процесс
лимфопоэза. Лимфоциты, образующиеся в этом органе, представляют собой
особый класс лимфоцитов со специальной функцией — участие в клеточном
иммунитете. Лимфатические узлы развиваются как разрастания примитивных
лимфатических сосудов, которые вскоре окружаются большим количеством мезенхимных
клеток. Впоследствии эти клетки округляются и становятся похожими по виду
на лимфоциты взрослого. Некоторые из мезенхимных клеток дают начало клеткам
других линий, таких как эритроциты, гранулоциты, мегакариоциты, но это явление
преходящее, поскольку основным процессом в тимусе является лимфопоэз.
Заключение. Во всех гемопоэтических органах эмбриона и плода происходят
тождественные процессы (рис. 1-2). Циркулирующие первичные гемопоэтичес-
кие стволовые клетки расселяются в специфической тканевой нише способом,
который до конца еще не понят. Там они дифференцируются в клетки,
распознаваемые как гемопоэтические предшественники. Эти эмбриональные гемопоэти-
ческие предшественники, вероятно, способны к мультилинейной дифференци-
ровке, но в каждом конкретном месте процесс гемопоэза может быть нацелен
на формирование определенной линии клеток, возможно, под влиянием
локального микроокружения. Различные очаги эмбрионального гемопоэза активны
только на соответствующих этапах развития. За этой активацией следует
программируемая инволюция. Исключение составляет костный мозг, который
сохраняется как основной центр гемопоэза у взрослых. Лимфатические узлы, селезенка,
тимус и другие лимфоидные ткани продолжают выполнять лимфопоэтическую
функцию и у взрослого человека.
Гемопоэз во взрослом организме
С момента рождения развитие первичных полипотентных стволовых клеток и
миелопоэз происходят в костном мозге, в то время как лимфопоэз — в тимусе,
селезенке и лимфатических узлах. При патологии миелопоэз может возобновляться
в селезенке, а также в печени, повторяя стадию развития плода. Главным местом
гемопоэза постепенно, на смену печени и селезенке, становятся костномозговые
полости почти всех костей осевого и добавочного скелета. Вследствие активации
гемопоэза костный мозг приобретает красный цвет, аналогичный цвету крови, что
отражает усиленную продукцию эритроцитов, содержащих гемоглобин.
Костномозговая полость служит местом продукции нелимфоидных клеток крови, в то
Гемопоэз во взрослом организме
23
время как лимфопоэз у взрослого происходит преимущественно в селезенке,
лимфатических узлах, тимусе и лимфоидной ткани, ассоциированной с кишечником,
включая миндалины, аденоиды и пейеровы бляшки. Исследования с
использованием световой микроскопии показали, что костный мозг взрослого человека
составляют эритроидные и миелоидные клетки-предшественники вместе с
рассеянными мегакариоцитами. Имеется также популяция клеток, известных как
"стромальные клетки", которые определяют созревание
клеток-предшественников и высвобождение полностью дифференцированных клеток в кровоток.
Костный мозг
В костном мозге существуют области так называемого гемопоэтического
индуктивного микроокружения, которые обеспечивают продукцию эритроцитов,
лейкоцитов и тромбоцитов (рис. 1-3). Их формируют стромальные клетки (ретику-
Эритробластный
островок
Рис. 1-3. Схематическое изображение поперечного среза костного мозга, показывающее гемопоэти-
ческие и сосудистые компартменты. Аде — адвентициальные клетки (в тексте рассматривается их
роль в регуляции гемопоэза). Мег — мегакариоцит. Эмп обозначает лейкоцит, находящийся в
цитоплазме мегакариоцита,— явление, известное как эмпериполезис. (По: Weiss L. P. Cell and Tissue
Biology, 6th ed. Baltimore, Munich: Urban & Schwarzenberg; 1988.)
24
Глава 1. Гемопоэз. Развитие клеток крови
лярные и барьерные), а также внутрикостные и лимфоидные клетки,
остеобласты, остеокласты, макрофаги и их растворимые ростовые факторы (цитокины).
Они создают и поддерживают "почву" для прорастания "семян" гемопоэтических
стволовых клеток и их потомства. Таким образом, имеется много уязвимых точек
для нарушения гемопоэза.
Сосудистые компартменты костного мозга содержат сосудистые синусы,
которые представляют собой широкие тонкостенные вены. Сосудистые синусы —
доминирующая структура этих компартментов. Клетки крови из гемопоэтических
компартментов входят в синусы (см. ниже), перемещаются от периферии к
центральным венам и в конечном счете попадают в общий кровоток. Артерии
постепенно превращаются в капилляры, которые затем переходят непосредственно
в венозные синусы. В отличие от селезенки циркуляция в данном случае является
"замкнутой" (см. ниже).
Эндотелий сосудистого синуса прилежит к окончатой базальной мембране,
под которой находятся адвентициальные клетки. Это крупные отростчатые стро-
мальные ретикулярные клетки, которые обеспечивают поддержание гемопоэти-
ческого компартмента. Они покрывают и раскрывают эндотелиальные клетки
сосудов, что помогает регулировать проход клеток из гемопоэтического
компартмента к сосудистому. Эти клетки могут превращаться в адипоциты (или
накапливать желатиновый материал) и таким образом контролировать объем
гемопоэтического компартмента.
Гемопоэтические компартменты, в которых группируются клетки,
находящиеся на разных стадиях развития всех трех ростков кроветворения, окружены
венозными пространствами. Здесь же находятся артериальные сосуды и добавочные
клетки. Отношение миелоидных клеток к эритроидным равно приблизительно 3:1.
Развитие эритроцитов происходит в эритробластных островках, которые состоят
из центральных макрофагов, окруженных дифференцирующимися и пролифери-
рующими эритробластами. Такой островок лежит непосредственно напротив
сосудистого синуса, составляющие его клетки располагаются в порядке,
определяемом их зрелостью: ретикулоциты и/или ортохроматофильные пронормобласты
(наиболее дифференцированные эритроидные предшественники) прилегают
непосредственно к эндотелиальным клеткам сосудистых синусов, тогда как ранние
предшественники в большей степени удалены от синусов. Макрофаги
расположены так, чтобы они могли физически взаимодействовать с эритроидными клетками
для обеспечения фагоцитоза ядер и ядерных остатков и поставки цитокинов
развивающимся эритроцитам. Изоляции эритробластных островков способствуют
также барьерные клетки. Через отверстия в эндотелии сосудистых синусов мега-
кариоциты высвобождают цитоплазматические фрагменты (тромбоциты). Кроме
того, в созревании мегакариоцитов и продукции тромбоцитов важную роль могут
играть легкие. Перед выходом в сосудистый синус гранулоциты достигают стадии
превращения в метамиелоциты. Микроворсинки метамиелоцитов отделяют от
базальной поверхности эндотелия адвентициальные клетки для проникновения
последних в просвет сосудистого синуса. Как уже отмечалось, нарушение
гемопоэза может быть обусловлено многими причинами, в том числе физическими,
метаболическими, химическими, инфекционными, воспалительными или
иммунологическими (глава 7).
С возрастом мозговая ткань костей добавочного скелета теряет красноватый
цвет и преобразуется в желтый мозг, что отражает постепенную замену гемопоэ-
Гемопоэз во взрослом организме
25
тической ткани жировой. Уже в молодом возрасте трубчатые кости не содержат
красный костный мозг, поскольку он полностью замещается негемопоэтическим
желтым костным мозгом. Красный костный мозг сохраняется в грудине, ребрах,
позвонках и тазовых костях. Хотя стимул для преобразования красного костного
мозга в желтый неизвестен, в патологических условиях, связанных с усилением
гемопоэза, может происходить нарушение этого процесса и красный костный
мозг обнаруживается в костях, которые обычно не связаны с гемопоэтической
активностью, например в двойных пазухах костей черепа. В таких случаях местами
локализации экстрамедуллярного (внекостномозгового) гемопоэза также могут
быть печень, селезенка и лимфатические узлы. Максимальное распространение
костномозгового кроветворения на все черепные и длинные кости можно
наблюдать у лиц с тяжелой талассемией — болезнью, при которой эритропоэз протекает
необычайно интенсивно в течение всей жизни, что и является сутью данного
заболевания. При талассемии рентгенограмма черепа в области черепных пазух имеет
характерный вид "hair-on-end" ("волосатый череп"). Гиперплазия костного мозга
в костях верхней челюсти приводит к типичному изменению лица: скулы
выдаются и нарушается прикус зубов из-за того, что верхняя челюсть становится
диспропорционально больше, чем нижняя (глава 3).
Селезенка
Железистый шар, спрятанный
за грохочущим дном желудка
под крепкой кривой
диафрагмой, толкаемой
ударами, передаваемыми
сердечным мотором. Она размещена
в шумном углу, по
преданию, в центре
страсти. Маленький эллиптический
мусорный мешок живота, который
поглощает старые клетки, использованную кровь;
возможно, он же — источник
меланхолии, сожаления
о необходимости каннибализма,
вины за излишнюю злость
и постоянное кусание. Это искупается
тем, что маленькая наседка,
кудахча, высиживает
новые клетки, сохраняющие
природу своей матери,
истребляя чужеродные бактериальные частицы,
прибывающие в темную железу.
Фильтр между артерией и веной,
микроскопические канальцы или
26
Глава 1. Гемопоэз. Развитие клеток крови
лужицы крови, маленькие неогороженные моря
хранят тайны глубоко
в красной висцеральной пульпе.
Элис Джонс, M.D. Окленд, Калифорния1
Селезенка размещена в левом верхнем квадранте живота. Она связана с
некоторыми другими органами и имеет почечную, панкреатическую и диафрагмальную
поверхности. У взрослого человека она весит приблизительно 150 г вместе с
небольшими придатками, величиной от горошины до сливы, которые находятся
в желудочно-селезеночной связке, большом сальнике, а также в некоторых других
местах. Хотя в древности рассматриваемый орган представлялся таинственным,
теперь его функция определена. Структура селезенки и характер кровотока
обеспечивают уникальную основу для выполнения многих установленных на
сегодняшний день задач (рис. 1-4). Капсула, состоящая из плотной соединитель-
I Лимфатический узелок
I (содержит зародышевые центры)
Рис. 1-4. Схема строения селезенки (описание тока крови и клеточного распределения см. в тексте).
(Из: Weiss L., Tavassoli M. Anatomical hazards to the passage of erythrocytes through the spleen.
Seminars in Hematology, 7: 732, 1970.)
1 Впервые опубликовано в: JAMA, vol. 267, p. 1454, March 18, 1992, Copyright Alice Jones,
"Anatomy", San Francisco, Bullnettle Press, 1997; с разрешения автора.
Гемопоэз во взрослом организме 27
ной ткани, прорастает, формируя сеть перегородок в ткани селезенки. В отличие
от животных, у человека в капсуле органа есть только небольшая мышца,
способная расширять и сбкращать селезенку. Паренхима называется селезеночной
пульпой, в которой выделяют красную лульпу, состоящую в значительной
степени из селезеночных синусов, и тонкие пластинки ткани — селезеночные тяжи,
находящиеся между синусами. Кластеры лимфоцитов селезенки бывают двух
типов. Одни состоят преимущественно из Т-лимфоцитов (тимусного
происхождения) и вспомогательных клеток и формируют цилиндрическую оболочку,
окружающую центральную артерию. Это так называемая периартериальная
лимфатическая оболочка (ПАЛО). В-лимфоциты (термин "В-клетка" образован от
bursa Fabricius — органа, расположенного в клоаке птиц и необходимого для про-
цессинга и созревания В-клеток; костный мозг человека считают аналогом этого
органа) внутри ПАЛО формируют узелки. ПАЛО центральной артерии
постепенно суживается, переходя в белую пульпу вместе с капиллярами,
соединяющимися непосредственно с венозными синусами. Кровь может изливаться прямо
в красную пульпу, куда клетки свободно просачиваются и попадают в конечном
счете в венозный синус.
Краевая (маргинальная) зона селезеночной пульпы представляет собой
переходную область между красной и белой пульпой. Здесь начинается процесс
фильтрации и сортировки клеток.
Кровоток в селезенке обеспечивает ее функционирование. Кровь поступает
в орган по селезеночной артерии, проходящей через ворота. Селезеночная
артерия разветвляется на трабекулярные артерии, которые в свою очередь делятся на
центральные артерии, расположенные в центре цилиндрических ПАЛО. Как
отмечалось ранее, центральные артерии прямо или косвенно переходят в венозные
синусы. После попадания в селезеночные синусы кровь течет по пульпарным
венам, которые переходят в трабекулярные вены. Из ворот селезенки кровь
выносится по селезеночной вене.
Ток лимфы в селезенке совпадает с направлением венозного потока и
противоположен току артериальной крови, но лимфатическая система селезенки у
человека не так сильно развита, как у животных. Барьерные клетки, описанные
Вейсом как "сильно активизированные, быстро мобилизующиеся блуждающие
фибробластные клетки", являются клетками стромы. Хотя функция барьерных
клеток неизвестна, их центральное расположение предполагает
полифункциональность, включая образование оболочки вокруг кровеносного сосуда,
формирование барьеров между кровью и тканью, концентрацию регуляторных
факторов, изоляцию иммунокомпетентной ткани после запуска иммунного ответа,
отгораживание гемопоэтических колоний, концентрацию темопоэтических
факторов и защиту от паразитов. Подобные клетки представлены в других
гемопоэтических и иммунных тканях, где они могут функционировать так же, как в
селезенке.
Селезенка выполняет много важных функций, часть из которых
непосредственно определяется сложным движением потока крови. В отличие от
лимфоузлов, реагирующих на местный антигенный стимул, получая лимфу, селезенка
тестирует кровь, которая собирается со всего тела, и иммунологически
взаимодействует с ней. Здесь же происходит и "просматривание" плазмы, поскольку
ветви центральных артерий повернуты под прямым углом, что позволяет плазме
просочиться прежде, чем кровь достигает красной пульпы. Различные фильтра-
28
Глава 1. Гемопоэз. Развитие клеток крови
ционные прослойки состоят из ретикулярных клеток и ретикулярных волокон,
а также других типов клеток стромального происхождения, включая макрофаги,
интердигитальные клетки и фолликулярные дендритные клетки. Барьерные
клетки также помогают включать механизм фильтрации. Как уже отмечалось,
периартериальные лимфатические оболочки, прослойки краевой зоны и красная
пульпа служат фильтрами наряду с эндотелиальными клетками венозных
синусов (см. ниже). Это позволяет селезенке распознавать, выбраковывать и удалять
дефектные, старые и изношенные клетки. Включения частиц типа телец Хауэл-
ла-Жолли, телец Гейнца, бактерий, паразитов и гранул железа (табл. 3-3)
удаляются путем "складывания в селезеночную яму". Повторное использование
железа, концентрирование тромбоцитов, удаление эритроцитов, регуляция объема
крови, эмбриональный (и иногда патологический у взрослых) гемопоэз,
иммунные функции — все это элементы комплексной функции селезенки.
На ранних стадиях воспалительного ответа селезенка функционирует и как
первичный бактериальный фильтр или губка. При эпизодах массивной
бактериемии селезенка улавливает бактерии и переваривает их в макрофагах. Эндотели-
альные клетки венозных синусов (ЭКВС) формируют специализированную
ткань, с которой сталкиваются клетки крови и которую они должны успешно
пересечь, покидая губчатую петлю красной пульпы и продвигаясь к селезеночной
вене. ЭКВС имеют уникальные антигенные характеристики и способность
двигаться, что позволяет им тестировать аномальные, старые клетки или клетки,
содержащие бактерии (например, полиморфно-ядерные лейкоциты), паразитов или
простейших (например, эритроциты), по мере перемещения клеток между
пальцеобразными межэндотелиальными расщелинами. Этот физический барьер и се-
теобразная базальная мембрана — плацдарм для межклеточных взаимодействий,
на котором макрофаги взаимодействуют с задержанными клетками и "ищут" на
поверхности и внутри их дефекты и частицы, которые подвергнутся фагоцитозу
(рис. 1-5 и 1-6).
Макрофаги не только поглощают бактерии, но и представляют их
обработанные антигены непосредственно лимфоцитам в селезенке, стимулируя продукцию
специфических антител. Собственно фагоцитоз макрофагов значительно
уменьшает бактериальную нагрузку в кровотоке. Эта функция чрезвычайно важна,
поскольку несколько полисахаридов на поверхности и грамотрицательных, и грам-
положительных бактерий являются мощными системными токсинами. Если их
не изолировать в макрофагах, эти бактериальные антигены до развития
гуморального иммунного ответа могут запускать альтернативный путь активации
комплемента, что приводит к вазодилатации, увеличению проницаемости капилляров и в
конечном счете — к шоку и смерти.
Помимо выполнения функции очень сложного фильтра селезенка служит в
качестве лимфатического "суперузла", в котором в присутствии Т-клеток образуется
большое количество В-клеточных клонов (приблизительно 80 % клеток селезенки
— В-клетки и около 15 % — Т-клетки). Кроме того, главным образом в селезенке
происходит Т-независимое развитие В-клеток, имеющее важное значение для
ответа организма на углеводные антигены, экспрессированные на капсулах бактерий
Streptococcus pneumoniae у Hemophilus influenzae и Neisseriae meningitidis.
Т-клетки и В-клетки взаимодействуют в ПАЛО и лимфатических узелках
внутри ПАЛО. Кластеры антителопродуцирующих клеток, состоящие из
В-клеток, плазматических клеток, хелперных и супрессорных Т-клеток, макрофагов
Гемопоэз во взрослом организме
29
и других вспомогательных клеток, формируют в центре лимфатических узелков
герминативные центры (центры размножения).
Наконец, селезенка выполняет две родственные неиммунные механические
функции. Она служит резервуаром для тромбоцитов, наработанных в костном
мозге. Обычно в селезенке сохраняется только небольшая часть всех тромбоцитов
Наружный ретикулум
периартериальной
лимфатической оболочки
Рис. 1-5. Схема строения селезеночной артерии, покидающей периартериальную лимфатическую
оболочку белой пульпы и входящей в красную пульпу. Показано, что селезеночная артерия входит
в селезеночный тяж и раздваивается между двумя синусами. (Из: Weiss L. The Cells and Tissues of the
Immune System. Englewood Cliffs, N. J.: Prentice-Hall; 1972.)
30
Глава 1. Гемопоэз. Развитие клеток крови
Рис. 1-6. Синус красной пульпы селезенки человека. Палочковидные эндотелиальные клетки
с филаментами, расположенными в трех направлениях, управляют размером межэндотелиальной
щели. Эритроциты и лейкоциты проходят через эндотелиальные клетки благодаря отверстиям
в базальной мембране (описание прохода эритроцита см. в тексте) (Из: Chen L. Т., Weiss L. Am. J.
Anat., 1972; 134: 425.)
организма. Однако при увеличении размера селезенки в ней может находиться
до 90 % всех тромбоцитов. Селезеночные тромбоциты, как представляется,
находятся в состоянии равновесия с пулом циркулирующих тромбоцитов, которые
медленно меняют свою локализацию.
Селезенка задерживает также эритроциты, но этот процесс менее пассивен
и более динамичен. Стареющие, покрытые антителами или поврежденные
эритроциты фильтруются в селезенке, где они либо удаляются, либо частично
восстанавливаются, или "ремоделируются", ЭКВС и селезеночными макрофагами.
Ремоделированные эритроциты могут затем повторно рециркулировать, тогда
как аномальные клетки распознаются селезенкой и быстро удаляются для
последующей переработки. При гипофункции селезенки или ее отсутствии
специальными методами микроскопии обнаруживаются эритроциты с ямками,
щербинками и кратерами. Обычная световая микроскопия позволяет в этих случаях
рассмотреть на мазках крови, окрашенных по Райту, ядерные остатки,
называемые тельцами Хауэлла-Жолли. Следует подчеркнуть, что у пациентов с
дисфункцией или отсутствием селезенки в течение всей жизни существует риск
развития бактериального сепсиса, особенно вызываемого инкапсулированными
бактериями. Кроме того, у них тяжелее протекают паразитарные инфекции, такие
как малярия или бабезиоз.
Гемопоэз во взрослом организме
31
Селезенка увеличивается по ряду причин. Одна из них — функциональная
гиперактивность, называемая гиперспленизмом, часто сопровождается увеличением
органа. Гиперспленизм может характеризоваться "прожорливостью" селезенки
по отношению к собственным клеточным элементам организма, что приводит
к цитопениям. Наблюдаются болевые ощущения из-за расширения или инфаркта
селезенки, а при сдавлении желудка может развиваться преждевременное
чувство насыщения. Существует несколько патофизиологических механизмов
увеличения селезенки, в частности гиперплазия эндотелиальных или иммунных
элементов вследствие инфекционных болезней или нарушений иммунитета.
Расширение селезенки из-за нарушенного селезеночного кровотока происходит
при циррозе печени, тромбозе селезеночной, печеночной или портальной вены.
Первичные или метастатические опухоли, внекостномозговой гемопоэз или
аномальный материал, инфильтрующий селезенку, например при амилоидозе либо
таких болезнях накопления, как болезнь Гоше, гемангиомы или кисты, также
вызывают увеличение селезенки. Решение о хирургическом удалении гиперсплени-
ческой, расширенной, болезненной или кровоточащей селезенки очень
ответственно, так как после спленэктомии у пациента ослабляется иммунитет.
К причинам функционального и анатомического гипоспленизма относятся:
врожденное отсутствие селезенки, спленэктомия, миелофиброз и другие миело-
пролиферативные нарушения, дефекты васкуляризации селезенки, иммунные
или аутоиммунные болезни (волчанка или ревматоидный артрит), целиакия и
воспалительные заболевания кишечника, инвазивные опухоли, системный амило-
идоз, нефротический синдром, мастоцитоз, а также состояние новорожденности.
Проявления гипоспленизма в периферической крови включают транзиторный
тромбоцитоз и наличие телец Хауэлла-Жолли (ядерные остатки в эритроцитах),
а также ямок и расщелин на эритроцитах, образующихся из-за недостаточной
переработки этих клеток в селезенке. При отсутствии селезенки появляются мише-
невидные клетки и акантоциты (табл. 3-3). Как упоминалось ранее, затрудняется
инактивация инкапсулированных бактерий. Нарушается ответ на антигенный
стимул, в том числе вакцинацию некоторыми вакцинами. Кроме того, следствием
асплении является повышенная восприимчивость к паразитарным болезням,
таким как бабезиоз или малярия.
Лимфатические узлы
Лимфатические узлы располагаются по ходу лимфатических сосудов и
представляют собой маленькие овальные или почкообразные образования длиной 0,1-
2,5 см. Они соединены с системой лимфоциркуляции афферентными
лимфатическими сосудами, которые проникают в лимфатический узел в области большой
кривизны, и эфферентными сосудами, которые выходят из ворот (рис. 1-7).
Клапаны в лимфатических сосудах обеспечивают однонаправленный ток лимфы. В
ворота входят артерии, а выходят из них вены. Каждый узел заключен в фиброзную
капсулу, которая распространяется в паренхиму в виде перегородок (трабекул).
Специализированные сети или фильтрационное ложе, составленное из
ретикулярных клеток и волокон, получает Т- и В-лимфоциты из рециркулирующего
лимфатического пула. Т-лимфоциты занимают периферийную область
лимфатических узлов и концентрируются в межфолликулярной зоне (между зонами
первичных и вторичных фолликулов), а также в паракортикальной области.
32
Глава 1. Гемопоэз. Развитие клеток крови
Первичный
В-клеточный
фолликул
(узелок)
Вторичный фолликул
(В-клеточная зона)
с герминативным
центром
(центром размножения)
Паракортикальная
Т-клеточная область
(зона)
Кора
Капсула
Трабекула
Медуллярный
(мозговой)
синус е
Рис. 1-7. Схема строения лимфатического узла. Лимфа попадает в лимфатический узел через
афферентные лимфатические сосуды (А) и покидает лимфатический узел через эфферентные сосуды (Е)
В-клеточные области — это первичные и вторичные фолликулы в кортикальной зоне
лимфатического узла, тогда как Т-клетки концентрируются в паракортикальных областях. Стрелками А и Е
указано направление тока лимфы. (Из: Isselbacher К. J. et al. (eds). Harrison's Principles of Internal
Medicine, 13th ed. New York: McGraw-Hill, 1994.)
Т-клетки в лимфатических узлах — СБ4-хелперного типа (80 %) и CD8-cyn-
рессорного типа (20 %). В-лимфоциты в корковом веществе лимфатического узла
содержатся внутри первичных и вторичных лимфоидных фолликулов (узелков).
Интердигитальные ретикулярные (дендритные клетки) могут быть идентичны
клеткам Лангерганса в эпителии, которые перемещаются в лимфатическую ткань
с накопленными на их поверхности антигенами. ■
Каждый лимфатический узел является агрегатом В-лимфоидных
фолликулов, а в каждом фолликуле происходит экспансия нескольких В-клеточных
клонов. Т-лимфоциты группируются вокруг этих фолликулов, функционируя
совместно с В-клетками. На Т-клетках, непосредственно окружающих фолликулы,
экспрессированы специализированные молекулы межклеточного
взаимодействия, которые служат молекулами адгезии при контакте Т- и В-клеток и
помогают осуществить Т-В-взаимодействие в процессе созревания и секреции антител.
Барьерные клетки фибробластного происхождения объединяются, обозначая
путь крови и участки секвестрации. Макрофаги совместно с барьерными
клетками предотвращают развитие инфекции и участвуют в иммунном ответе.
Афферентные лимфатические сосуды, содержащие лимфу, антигены, лимфоциты
и макрофаги, проникают в субкапсулярное пространство. Лимфа попадает в па-
ракортикальные и медуллярные области, медуллярные синусы и, наконец, в
эфферентные лимфатические сосуды. Артерии доставляют Т-клетки из тимуса
и В-клетки из костного мозга к лимфатическим узлам. В- и Т-клетки входят
внутрь лимфатического узла, проходят через его венулы, где высокие эндотели-
альные клетки распознают лимфоциты и направляют их в лимфатический узел.
Структурный и клеточный состав лимфатических узлов позволяет
взаимодействовать антигену и лимфоцитарным клеткам, которые определяют оптимальный
уровень активации иммунного ответа.
Гемопоэз во взрослом организме
33
Лимфатические узлы могут увеличиваться в размере и при нормальных
условиях, но чаще это происходит при значительно усиленном иммунном ответе,
а также при некоторых патологических состояниях: а) увеличение тока крови
и клеточного состава как части иммунного ответа; б) явные инфекции или
воспаление непосредственно лимфатического узла (лимфаденит); в) захват клеточных
остатков или конечных продуктов метаболизма макрофагами или клетками
накопления при некоторых болезнях накопления; г) вовлечение в первичный
опухолевый процесс или метастазы лимфоретикулярных или солидных опухолей.
Тимус (вилочковая железа)
Тимус находится в переднем средостении. Эта двудольная железа при рождении
весит 10-15 г, быстро увеличивается до 20-40 г и затем ее масса уже не изменяется
значительно. Хотя с возрастом количество лимфоидной ткани постепенно
уменьшается и в железе начинает преобладать жировая ткань, тимус сохраняет
иммунологическую активность. Он закладывается на восьмой неделе жизни эмбриона из
3-го и 4-го жаберных карманов как эпителиальный орган, заполняемый тимоцита-
ми (Т-клетками), которые происходят из костномозговых протимоцитов. Т-кле-
точные маркеры характеризуют стадии развития Т-клеток в тимусе. Железа
подразделяется на дольки капсульными перегородками. В каждой дольке имеются
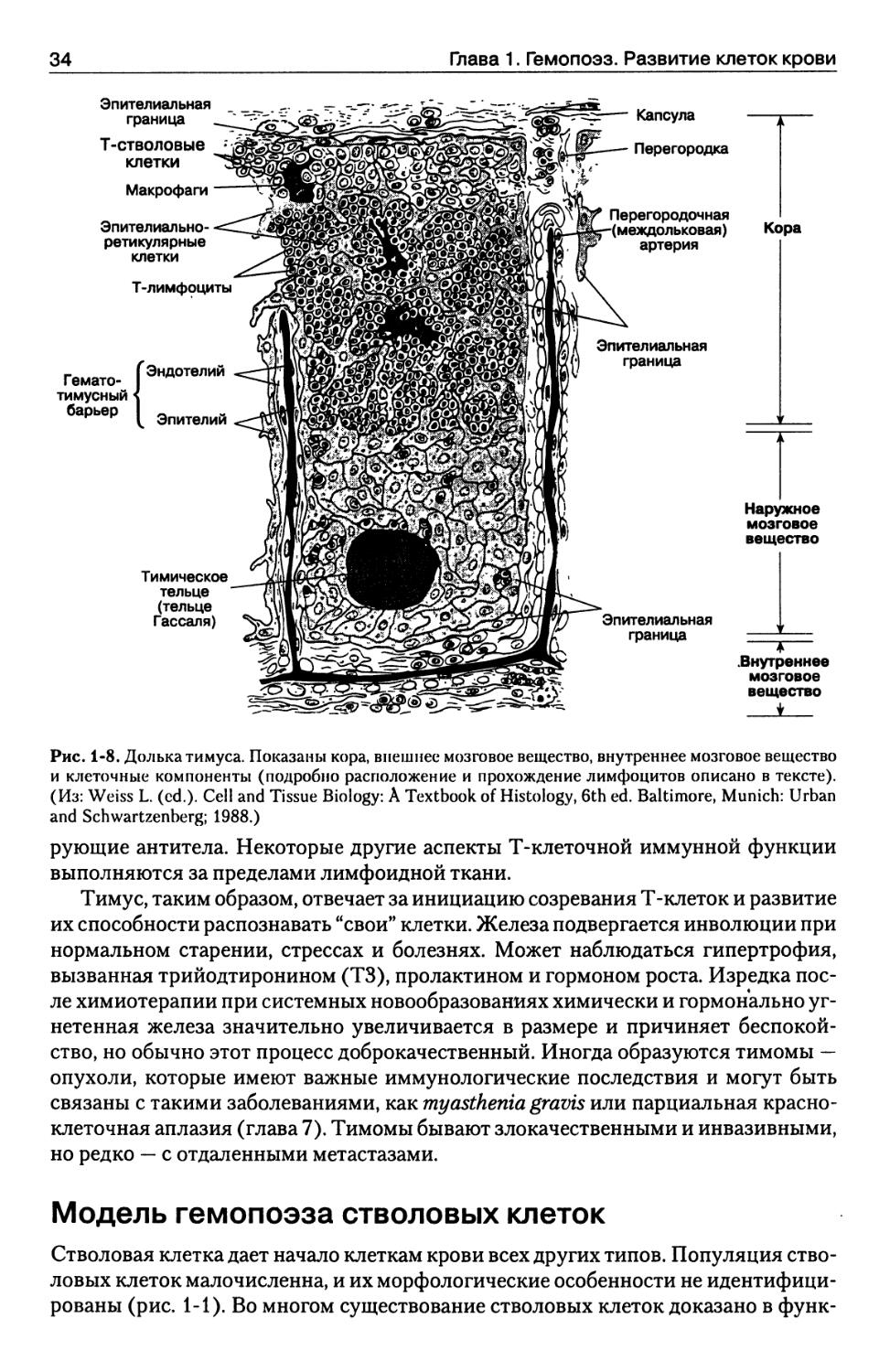
кортикальная и медуллярная зоны (рис. 1-8). Кортикальная зона содержит 95 %
тимических лимфоцитов, а также поддерживающие эпителиальные клетки
(эпителиальные ретикулярные клетки). Протимоциты входят в паренхиму тимуса
высоко в корковом веществе и продвигаются глубже к кортикомедуллярному
переходу, созревая по мере перемещения. Они взаимодействуют со стромальными
клетками (эпителиальными ретикулярными клетками, ретикулярными клетками,
барьерными клетками и макрофагами), которые "обучают" развивающиеся
Т-клетки различать свой и чужеродный антигенный материал.
Мозговое вещество содержит 5 % лимфоцитов тимуса. Это зрелые Т-клетки.
Наиболее широко представленные клеточные элементы — многогранные
эпителиальные клетки, которые могут принимать неправильный кольцевой,
пластинчатый вид с центральным некрозом, кальцификацией и формированием кист.
Такие структуры называются тимическими тельцами, или тельцами Гассаля.
Они представляют собой конечную стадию тимико-медуллярно-эпителиальной
дифференцировки. В кортико-медуллярной зоне или в мозговом веществе
Т-лимфоциты попадают в вены либо лимфатические сосуды и продвигаются к селезенке,
а затем повторно попадают в рециркулирующий пул лимфоцитов.
Приблизительно 95 % лимфоцитов погибают внутри коры тимуса, и только 5 % выходят в
кровоток как иммунокомпетентные клетки (рис. 1-9).
Тимическое развитие Т-клеток происходит преимущественно в детском
и юношеском возрасте. После второго десятилетия жизни тимус в значительной
степени подвергается инволюции, хотя некоторая тимическая активность все еще
имеет место и у взрослого. Некоторые этапы развития Т-клеток в эмбриогенезе,
как представляется, происходят вне тимуса, возможно в лимфатических узлах, но
детали этого процесса неизвестны. Как только лимфоциты дифференцируются
в зрелые Т-клетки, они начинают циркулировать по организму. Их иммунная
функция реализуется в основном в лимфатических узлах, где они индуцируют
созревание В-клеток и превращение последних в плазматические клетки, секрети-
34
Глава 1. Гемопоэз. Развитие клеток крови
Эпителиальная
граница
Т-стволовые
клетки
Макрофаги
Эпителиально-
ретикулярные
Т-лимфоциты
Гемато-
тимусный
барьер
Г Эндотелий
Эпителий
Тимическое
тельце
(тельце
Гассаля)
^с:— Капсула
Перегородка
Перегородочная
(междольковая) Кора
артерия
Эпителиальная
граница
Эпителиальная
граница
Наружное
мозговое
вещество
.Внутреннее
мозговое
вещество
±
Рис. 1-8. Долька тимуса. Показаны кора, внешнее мозговое вещество, внутреннее мозговое вещество
и клеточные компоненты (подробно расположение и прохождение лимфоцитов описано в тексте).
(Из: Weiss L. (cd.). Cell and Tissue Biology: A Textbook of Histology, 6th ed. Baltimore, Munich: Urban
and Schwartzenberg; 1988.)
рующие антитела. Некоторые другие аспекты Т-клеточной иммунной функции
выполняются за пределами лимфоидной ткани.
Тимус, таким образом, отвечает за инициацию созревания Т-клеток и развитие
их способности распознавать "свои" клетки. Железа подвергается инволюции при
нормальном старении, стрессах и болезнях. Может наблюдаться гипертрофия,
вызванная трийодтиронином (ТЗ), пролактином и гормоном роста. Изредка
после химиотерапии при системных новообразованиях химически и гормонально
угнетенная железа значительно увеличивается в размере и причиняет
беспокойство, но обычно этот процесс доброкачественный. Иногда образуются тимомы —
опухоли, которые имеют важные иммунологические последствия и могут быть
связаны с такими заболеваниями, как myasthenia gravis или парциальная красно-
клеточная аплазия (глава 7). Тимомы бывают злокачественными и инвазивными,
но редко — с отдаленными метастазами.
Модель гемопоэза стволовых клеток
Стволовая клетка дает начало клеткам крови всех других типов. Популяция
стволовых клеток малочисленна, и их морфологические особенности не
идентифицированы (рис. 1-1). Во многом существование стволовых клеток доказано в функ-
Модель гемопоэза стволовых клеток
35
Капсула
Перегородка
(трабекула)
Тимоциты
Тимическое тельце
(тельце Гассаля)
,о
О О
Эпителиальная
клетка
Дендритная
клетка
Кора
Мозговое вещество
©
оо
о
Т-лимфоциты
(зрелые)
Периферия
Стволовая
клетка ~
ГС04* fCD4+ fCD4+
J CD4* —► J CD8+ v > \ CD8*
[TCR* [TCRaP \ [TCRc
tap
(низкий)
Общий
предшественник —
пре-Т-клетка
Кар
(высокий)
CD4
CD8+
TCRaP
(высокий)
Предшественник
Перестройка (TCR),
позитивная/негативная
селекция
Функциональные
Т-лимфоциты:
хелперы (Тх)
цитотоксические (Тц)
Рис. 1-9. Структура тимуса. Незрелые тимоциты развиваются в коре по пути к медуллярной
области. Изображены TCRaP-молекулы. (ТСЯуб-клетки встречаются редко). CD — кластер дифферен-
цировки; TCR — Т-клеточный рецептор; ар — аР-молекулы TCR; низкий, высокий обозначают
соответственно низкий и высокий уровни ap-экспрессии. (Из: Nichols W. S., Kipps Т. J. Structure and
function of other lymphoid tissues. In: Beutler E., Lichtman M. A., Coller B. S., Kipps T.J. (eds). Williams
Hematology, 5th ed. New York: McGraw-Hill; 1995, 45.)
циональных исследованиях, показавших способность одиночных клеток
генерировать несколько ростков кроветворения. Таким образом, стволовые клетки в
настоящее время определены не морфологически, а функционально. Точное их
количество неизвестно, поскольку нет единого мнения о том, что считать стволовой
клеткой. Предполагают, что стволовые клетки встречаются с частотой 1 на 106
ядросодержащих костномозговых клеток человека; по более сдержанным
оценкам эта доля составляет 1 на 107.
Для продукции огромного числа гемопоэтических клеток необходимо, чтобы
примерно через день в течение всей жизни костный мозг воспроизводил необхо-
36
Глава 1. Гемопоэз. Развитие клеток крови
димое количество миелокариоцитов; для этого он должен обладать клетками,
которые могут генерировать зрелые клетки непрерывно в больших количествах
(т. е. без потери этой способности). Способность к самообновлению является
ключевой в концепции стволовой клетки. В настоящее время имеются две теории
механизма самообновления. Согласно первой, деление стволовой клетки
асимметрично: из двух произведенных стволовых клеток одна —
недифференцированная, другая — дифференцированная, предназначенная продуцировать зрелые
клетки крови. В соответствии со второй теорией, стволовая клетка при каждом
делении производит или две новые стволовые клетки, или две более зрелые
клетки. Пул стволовых клеток, таким образом, поддерживается не точным
асимметричным делением каждой стволовой клетки, а равновесием между числом
делений, увеличивающих количество стволовых клеток, и делений, связанных с
появлением более зрелых клеток.
В момент, когда стволовая клетка оставляет самообновляющийся пул, чтобы
пополнить дифференцирующийся пул, морфологически она по-прежнему
выглядит как примитивная бластная клетка и сохраняет способность производить
клетки всех линий. С каждым последующим делением дочерние
клетки-предшественники становятся все более ограниченными в их способности к продукции клеток
крови разных линий. Если изолировать клетки-предшественники и разрешить им
размножаться и дифференцироваться, они будут генерировать к. 1етки, которые
в совокупности принадлежат только к одной или нескольким линиям (росткам
гемопоэза). Более дифференцированные клетки-предшественники производят
меньшее количество клеточных линий с меньшим количеством образующихся
клеток. Эти концепции, подтвержденные результатами проводимых в течение
нескольких десятилетий экспериментов in vivo и in vitro, легли в основу
существующей иерархической модели гемопоэза стволовых клеток (рис. 1-1).
Гемопоэтическое микроокружение. Стволовые клетки, если их поместить в
простую питательную среду, погибнут без дифференцировки или деления. Чтобы
поддерживать процесс гемопоэтического самообновления и дифференцировки,
стволовые клетки и их потомство должны находиться в непосредственной
близости от негемопоэтических мезенхимных клеток, называемых стромальными
клетками. Эта гетерогенная группа состоит из фибробластов, эндотелиальных
клеток, остеобластов и адипоцитов, располагающихся на эндостальной
поверхности в костномозговой полости. Гемопоэтические клетки нуждаются в двух тесно
связанных между собой элементах — в растворимых гемопоэтических факторах
роста и мембраносвязанных молекулах присоединения (прикрепления). И тем
и другим их обеспечивают стромальные клетки.
Гемопоэтические факторы роста (ГФР), или колониестимулирующие
факторы (КСФ), являются классом гликопротеиновых гормонов, которые необходимы
для регуляции деления и дифференцировки гемопоэтических клеток. Эти
гормоны требуются для выживания, пролиферации, дифференцировки и
функционирования всех гемопоэтических клеток. Хотя первоначально они были
обнаружены как спонтанно секретируемые продукты Т-клеточных опухолей, теперь
очевидно, что эти гормоны продуцируются в основном стромальными клетками
костного мозга, а также Т-лимфоцитами и моноцитами.
КСФ нарабатывают в двухэтапном процессе. Во-первых, малые количества
определенных КСФ (интерлейкин-6 [ИЛ-6], грацулоцитарно-макрофагальный
Модель гемопоэза стволовых клеток
37
колониестимулирующий фактор [ГМ-КСФ], фактор стволовых клеток [ФСК]
и Flt-3 [Flt-ЗЦ-лиганд) непрерывно продуцируются стромальными клетками
костного мозга, вероятно, в ответ на стимуляцию белками плазмы, тем самым
определяя основной гемопоэз, который поддерживает количество клеток крови в
нормальном диапазоне (рис. 1 -1).
Во-вторых, секреция КСФ значительно возрастает в ответ на инфекцию.
Бактериальные и вирусные продукты активируют моноциты, которые затем сек-
ретируют интерлейкин-1 (ИЛ-1), фактор некроза опухоли-альфа (ФНО-сс), гра-
нулоцитарный колониестимулирующий фактор (Г-КСФ) и собственный макро-
фагальный колониестимулирующий фактор (М-КСФ). Эти продукты в свою
очередь стимулируют дополнительную секрецию КСФ. ИЛ-1, помимо
антигенной стимуляции специфических рецепторов активирует секрецию ГМ-КСФ
и интерлейкина-3 (ИЛ-3) Т-клетки. ИЛ-1 и ФНО-а стимулируют фибробласты
и эндотелиальные клетки стромального микроокружения костного мозга к
увеличению секреции ими ИЛ-6 и ГМ-КСФ, а также к секреции больших количеств
Г-КСФ. Эти гемопоэтические ростовые факторы (цитокины), таким образом,
напрямую увеличивают количество циркулирующих нейтрофилов, моноцитов
и плазматических клеток и активируют эти клетки в процессе их созревания.
Генерация зрелых клеток крови каждой специфической линии регулируется
набором определенных гемопоэтических факторов роста. Хотя наборы гемопоэтичес-
ких факторов роста, стимулирующих специфическое созревание субпопуляций
клеток крови, перекрываются по своим функциям, каждый из них имеет
характерные отличия.
Эритропоэз. Эритроидная дифференцировка на последних стадиях в
значительной степени регулируется эритропоэтином (ЭП) — гликопротеином,
производимым в ответ на тканевую гипоксию в печени плода и почке взрослого. Из
предположительно 18 делений, происходящих в процессе превращения стволовой
клетки в зрелый эритроцит, эритропоэтин существенно стимулирует
заключительные 8-10 делений. Транскрипция гена эритропоэтина в почечных перитубу-
лярных эндотелиальных клетках и гепатоцитах регулируется кислородчувстви-
тельными факторами транскрипции, которые усиливают экспрессию гена при
сокращении доставки 02. Гиперпродукция эритропоэтина, наблюдаемая в
некоторых случаях почечноклеточной карциномы и гепатомы, ведет к развитию эрит-
роидной полицитемии (глава 3).
Предшествующие деления клетки, дающие начало эритропоэтинчувствитель-
ным эритроидным клеткам-предшественникам, в основном независимы от
эритропоэтина. Пролиферация и созревание на этих этапах определяются ГМ-КСФ
и ФСК, которые продуцируются местно в стромальном микроокружении
костного мозга. Кроме того, они могут быть специфически усилены ИЛ-3, секретируе-
мым активированными Т-лимфоцитами.
Гранулоцитопоэз. Нейтропоэз и монопоэз на заключительных стадиях
индуцируются Г-КСФ и М-КСФ соответственно. Ранние деления, в результате
которых полипотентные предшественники становятся коммитированными в
отношении определенных ростков, регулируются синергичными взаимодействиями
ГМ-КСФ, ФСК и ИЛ-3. Как описано ранее, несмотря на постоянный уровень
основной секреции КСФ стромальными фибробластами, выстилающими эндо-
стальную поверхность костного мозга, секреция ГМ-КСФ и Г-КСФ существен-
38
Глава 1. Гемопоэз. Развитие клеток крови
но повышается при воспалении в ответ на продукцию ИЛ-1 и ФНО-а
моноцитами.
В продукции эозинофилов главную индуктивную роль играет интерлейкин-5
(ИЛ-5), в меньшей степени участвуют ИЛ-3 и ГМ-КСФ. Базофилы и тучные
клетки непосредственно стимулируются ФСК и ИЛ-3. В обоих случаях
начальные центростремительные (афферентные) сигналы, которые вызывают
высвобождение этих цитокинов, пока недостаточно поняты.
Мегакариоцитопоэз. Развитие клеток-предшественников мегакариоцитов на
самых ранних стадиях, по-видимому, индуцируется тромбопоэтином (ТПО) в
сочетании с ФСК. ТПО также стимулирует конечные стадии созревания
мегакариоцитов и "отшнуровывание" тромбоцитов. Напротив, интерлейкин-11 (ИЛ-11)
может играть главную роль в "отшнуровке" тромбоцитов при сравнительно
слабом влиянии на образование и развитие самих мегакариоцитов (глава 5).
В-лимфопоэз. Как и в случае миелоидных линий, развитие В-клеток начинается
с дифференцировки полипотентной стволовой клетки в недифференцированные,
но коммитированные предшественники В-клеток. Пролиферация и дифференци-
ровка этих предшественников В-клеток на начальных стадиях индуцируются ин-
терлейкином-7 (ИЛ-7) и ФСК. Как только образовались распознаваемые пре-В-
и В-клетки, происходит их дальнейшая дифференцировка и деление при участии
иммуноглобулинового антигенного рецептора и Fc-рецептора, стимулируемая
растворимым интерлейкином-4 (ИЛ-4) и ИЛ-6. После формирования
плазматических клеток, производящих антитела, ИЛ-6 и ГМ-КСФ стимулируют
дополнительную пролиферацию и секрецию антител.
Т-лимфопоэз. Первоначально пре-Т-клетки подвергаются сложной негативной
и позитивной селекции в тимусе, в процессе которой они "обучаются"
распознавать "свои" и "не свои" (чужеродные) клетки. Возникающие в результате зрелые
Т-клетки являются субъектами антиген- и цитокининдуцированной активации и
экспансии. Стимуляция антигеном в присутствии интерлейкина-2 (ИЛ-2) или
антигеном в сочетании со стимуляцией вспомогательными макрофагами либо
дендритными клетками, которые экспрессируют В7-1 или В7-2, ведет к прямой
активации и CD4+, и'СБ8+ Т-клеток.
Клиническое использование гемопоэтических
факторов роста
После того как были клонированы и изолированы первые человеческие гемопоэ-
тические факторы роста (8 лет назад), успешные доклинические и клинические
испытания способствовали скорому их введению в клиническую практику для
рутинного и экспериментального использования. В настоящее время эритропоэ-
тин, Г-КСФ и ГМ-КСФ уже одобрены FDA к применению у человека, а ИЛ-3,
ФСК, ИЛ-6, ТПО и Flt-3-лиганд проходят первую фазу клинических испытаний.
Эритропоэтин. Анемия при почечной недостаточности — основное состояние,
которое непосредственно отвечает на лечение ЭП. Введение адекватных доз ЭП
3 раза в неделю ведет к быстрому развитию ретикулоцитоза при условии
достаточных запасов железа, фолиевой кислоты и витамина Bi2 у пациента и отсут-
Клиническое использование гемопоэтических факторов роста 39
ствии какого-либо другого источника продолжающегося воспаления, которое
подавляло бы эритропоэз. Главный побочный эффект такого лечения — гипертен-
зия, особенно при значительном повышении уровня гемоглобина. Использование
в медицинской практике терапии ЭП сделало тысячи пациентов,
подвергающихся почечному диализу, независимыми от гемотрансфузий и существенно
улучшило качество их жизни.
Вторую группу состояний, поддающихся терапии ЭП, составляют анемии при
таких хронических состояниях, как рак, СПИД и ревматологические болезни.
Однако страдающим данными заболеваниями требуются большие дозы ЭП, чем
больным с почечными нарушениями, и даже на высокие дозы наблюдаются
различные реакции. В целом, чем выше базальный уровень ЭП, циркулирующего
в плазме пациента, тем меньше эффективность терапии ЭП.
Третьей областью применения терапии ЭП является аутодонорство
эритроцитов у хирургических пациентов. Назначая ЭП в контролируемых условиях,
гематолог может стимулировать умеренный эритроцитоз, который позволяет при
кровопотере избежать развития тяжелой анемии. Таким способом удается
сохранить несколько доз эритроцитов и исключить тем самым необходимость в
переливании аллогенных эритроцитов и ассоциированный с этим риск. Единственным
препятствием к назначению данной терапии являются ограниченные
возможности гематологов и банков крови. Учитывая, что при проведении срочных и
внеплановых операций используется большое количество продуктов крови, без аллоген-
ного донорства и переливания крови вряд ли можно будет обойтись. Однако при
все увеличивающемся числе состояний, требующих поддержки эритроцитами,
доля аутологичного, ЭП-стимулированного донорства будет возрастать.
Гранулоцитарный колониестимулирующий фактор. Когда количество нейтро-
филов в периферической крови снижено, используя высокочувствительные
методы (например, ИФА), можно обнаружить повышенное содержание
циркулирующего Г-КСФ. Однако чтобы стимулировать быстрый гранулоцитопоэз, этого
количества Г-КСФ недостаточно. Дополнительное введение Г-КСФ в виде
фармацевтического препарата позволяет восстановить содержание в крови нейтро-
филов скорее и до более высокого уровня. Вначале терапия Г-КСФ применялась
при нейтропении, развившейся в результате химиотерапии. Подкожное введение
Г-КСФ на первой неделе после химиотерапии способствует сокращению
продолжительности обусловленной ею лейкопении. В контролируемых исследованиях
на сегодняшний день показано, что такое лечение помогает предотвратить
инфицирование больного и его госпитализацию. Единственный существенный
побочный эффект, наблюдаемый при терапии Г-КСФ,^- это боли в костях, которые
возникают у 10-15 % пациентов и могут быть легко купированы анальгетиками.
Предостережение: использование Г-КСФ, позволяющее врачам применять более
высокие дозы химиотерапии, пока не привело к полному излечению и
достоверному увеличению выживаемости.
Фармакологические дозы Г-КСФ также эффективно увеличивают количество
нейтрофилов в крови при некоторых хронических нейтропениях, например
врожденной нейтропении (болезнь Костманна), идиопатической нейтропении и имму-
ноопосредованных нейтропениях (в частности, при Т-лимфопролиферативных
заболеваниях). Однако требуемые в этих случаях дозы Г-КСФ могут быть
существенно выше обычных, а результаты неоднозначны. Кроме того, недавние иссле-
40
Глава 1. Гемопоэз. Развитие клеток крови
дования подтвердили, что примерно у 15 % пациентов с врожденными нейтропе-
ниями, лечившихся Г-КСФ, выявляется вторичный острый миелоидный лейкоз.
Вызвано ли развитие лейкоза стимуляцией аномальных костномозговых клеток
высокими дозами Г-КСФ, неизвестно, но это, безусловно,— основание для
проведения дальнейших исследований.
Гранулоцитарно-макрофагальный колониестимулирующий фактор. ГМ-КСФ
увеличивает, подобно Г-КСФ, количество нейтрофилов in vivo; кроме того,—
моноцитов и эозинофилов, а по сообщениям некоторых авторов,— ретикулоцитов и
тромбоцитов. Применение ГМ-КСФ, обладающего по сравнению с Г-КСФ
значительно более широким спектром действия, одобрено FDA для ускорения
восстановления гемопоэтических функций после трансплантации костного мозга и для
снижения риска развития связанных с ней заболеваний.
Кроме того, использование ГМ-КСФ при профилактике и лечении
выраженных аплазий костного мозга, индуцированных химиотерапией, почти так же
результативно, как и Г-КСФ. Хотя, возможно, в большей степени, чем Г-КСФ,
осложнено побочными эффектами (озноб, лихорадки, сгущение плазмы). Правда,
следует отметить, что на сегодняшний день нет точных данных о сравнении
эффективности и безвредности применения ГМ-КСФ и Г-КСФ.
Одно из новых направлений использования ГМ- и Г-КСФ — мобилизация
клеток-предшественников. При первых клинических испытаниях ГМ-КСФ было
выявлено, что хотя доля клеток-предшественников в костном мозге увеличилась
незначительно, количество циркулирующих клеток-предшественников возросло
весьма существенно — в 50-100 раз выше исходного уровня. Позже сходные
данные были получены для Г-КСФ (то же самое, вероятно, относится и к ИЛ-3, ЭП
и ФСК). Данная технология позволяет сократить число заборов периферической
крови (лейкоцитаферез) для получения трансплантата донорских стволовых
клеток (глава 8). (Собранные стволовые клетки используются впоследствии, чтобы
обеспечить восстановление костного мозга после миелоаблативной терапии, цель
которой — избавить организм от всех неопластических клеток.) Хотя пока остается
много вопросов о причинах и значении такого эффекта ростовых факторов, а также
роли мобилизации клеток-предшественников при трансплантации, применение
гемопоэтических факторов роста для "гемотерапии" имеет обширные перспективы
в трансплантационной медицине грядущего десятилетия.
Избранная литература
Bannister L. Hemolymphoid system. In: Peter L. Williams, ed. Gray's Anatomy.
Edinburgh: Churchill Livingstone; 1995:1400-1442.
Ярко написанный и иллюстрированный раздел во всемирно известной книге по
анатомии, удивляющий глубиной и ясностью изложения функциональной анатомии и
физиологии костного мозга, селезенки, лимфатического узла и тимуса.
Bonilla M. A., Gillio A. P., Ruggiero M. et al. Effects of recombinant human G-CSF on
neutropenia in patients with congenital agranulocytosis. N. Engl.J. Med., 1989; 320:1574.
В этом исследовании наряду с работами Eschbach и соавт. (1987) и Nemunaitis и со-
авт. (1991) продемонстрирована клиническая эффективность ЭП, Г-КСФ и ГМ-
КСФ в онкогематологии. Авторы приводят доказательства того, что, используя
естественные гемопоэтические гормоны в фармакологических дозах, можно
улучшить состояние больного.
Клиническое использование гемопоэтических факторов роста 41
Bowdler A. J. (ed.). The Spleen: Structure, Function and Clinical Significance. New York:
Chapman and Hall Medical, Van Nostrand Rheinhold; 1990.
Исчерпывающий обзор различных аспектов анатомии и физиологии селезенки.
Emerson S. G., Sieff С. A., Wang E. A. et al. Purification of fetal hematopoietic progenitors
and demonstration of recombinant multipotential colony stimulating activity.
J. Clin. Invest., 1985; 76:1286.
Первая публикация, посвященная стволовым гемопоэтическим клеткам человека и
клеткам-предшественникам.
Eschbach J. W., Egrie J. С, Downing M. R. et al. Correction of the anemia of end stage
renal disease with recombinant human erythropoietin. N. Engl. J. Med., 1987; 316:73.
В этом исследовании вслед за Bonilla и соавт. (1989) и Nemunaitis и соавт. (1991)
показаны клинические эффекты ЭП, Г-КСФ и ГМ-КСФ в онкогематологии и впервые
представлены доказательства того, что, используя естественные гемопоэтичес-
кие гормоны в фармакологических дозах, можно улучшить состояние больного.
Ford С. Е., Hamerton j. L., Barnes D. W. H., Loutit J. F. Cytological identification of
radiation chimeras. Nature, 1961; 177:452.
В этом исследовании и работе Till (1961) авторы впервые экспериментально
подтвердили существование иерархической модели стволовой клетки, которая активно
разрабатывалась последние 35 лет.
Guba S. С, Sartor С. I., Gottschalk L. R. et al. Bone marrow stromal fibroblasts secrete IL-6
and GM-CSF in the absence of inflammatory stimulation. Blood, 1992; 80:1190-1198.
В этом исследовании доказана продукция небольших, но значимых количеств ИЛ-6
и ГМ-КСФ нормальными человеческими стромальными фибробластами
костного мозга.
Haynes В. F. Enlargement of lymph nodesNand spleen. In: Isselbacher K.J. et al. (eds). Harrison's
Principles of Internal Medicine, 13th ed. New York: McGraw-Hill; 1994:323-329.
Краткое, но высокопрофессиональное обсуждение нарушений, возникающих в
лимфатических узлах и селезенке.
Henry P. H., Longo D. L. Enlargement of lymph nodes and spleen. In: Fauci A. S. et al.
(eds). Harrison's Principles of Internal Medicine, 14th ed. New York: McGraw-Hill;
1998:345-351.
Современный подход к лечению пациентов с лимфаденопатией и спленомегалией.
Holzer Н., Biehl J., Antin P. et al. Quantal and proliferative cell cycles: how lineages
generate cell diversity and maintain fidelity. Prog. Clin. Biol. Res., 1983; 134:213.
Классическое описание сбалансированной модели репликации стволовой клетки на
примере нормального гемопоэза и таких заболеваний, как недостаточность
костного мозга и лейкоз.
Lemischka I. F., Raulet С. Н., Mulligan R. С. Developmental potential and dynamic
behavior of hematopoietic stem cells. Cell, 1986; 45:917.
Замечательная статья, в которой обсуждаются функциональные возможности
стволовых клеток в течение жизни человека.
Nemunaitis J., Singer J. W., Buckner С D. et al. Long-term follow-up of patients who
received recombinant human granulocyte-macrophage colony stimulating factor
after autologous bone marrow transplantation for lymphoid malignancy. Bone
Marrow Transplant, 1991; 7: 49.
В этом исследовании вслед за Eschbach и соавт. (1987) и Bonilla и соавт. (1989)
показаны клинические эффекты ЭП, Г-КСФ и ГМ-КСФ в онкогематологии, впервые
представлены доказательства того* что, используя естественные гемопоэтичес-
кие гормоны в фармакологических дозах, можно улучшить состояние больного.
Nichols W. S., Kipps T.J. Structure and function of other lymphoid tissues. In: Beutler E.,
Lichtman M. A., Coller B. S., Kipps T.J. (eds). Williams Hematology, 5th ed. New
York: McGraw-Hill; 1995:43-48.
Доступное объяснение физиологии лимфоидной ткани.
Sills R. H. Hyposplenism. In: Pochedly С, Sills R. H., Schwartz A. D. (eds). Disorders of
the Spleen: Pathophysiology and Management. New York: Marcel Dekker; 1989: 99-
144.
В этой статье имеются полезные таблицы, касающиеся гипоспленизма.
Stockman J. A. Splenomegaly: diagnostic overview. In: Pochedly С, Sills R. H., Schwartz
A. D. (eds). Disorders of the Spleen: Pathophysiology and Management. New York:
Marcel Dekker; 1989:217-238.
Хороший обзор причин спленомегалии и возможностей ее диагностики.
Till J. E., McCulloch E. A. Direct measurement of the radiation sensitivity of normal
mouse bone marrow cells. Radiat. Res., 1961; 14:213.
В этом исследовании и работе Ford (1961) впервые экспериментально доказано
существование иерархической модели стволовой клетки, которая активно
разрабатывалась последние 35 лет.
Tsai S., Sief С. A. et al. Stromal cell associated erythroppiesis. Blood, 1986; 67:1418.
Прекрасная статья, показывающая, как эритроидные клетки развиваются на фиб-
робластоидных и моноцитарных прилипающих клетках. Впервые описана
тесная и динамическая физиологическая связь эритроидных клеток с их стромаль-
ным микроокружением в реальном времени.
Weiss L. P. Cell and Tissue Biology: A Textbook of Histology, 6th ed. Baltimore: Urban
and Schwarzenberg; 1988.
Замечательный учебник, отредактированный признанным цитологом и гистологом.
Богато иллюстрирован диаграммами и микрофотографиями, которые
дополняют блестящее изложение материала.
Weiss L. P. Functional organization of hematopoietic tissue. In: Hoffman R. H. et al. (eds).
Hematology: Basic Principles and Practice, 2nd ed. New York: Churchill Livingstone;
1995:193-206.
Современный обзор по анатомии и физиологии гемопоэтической ткани; особое
внимание уделяется кровотоку в гемопоэтических тканях.
Weiss L. P. The structure of hematopoietic tissues. In: Handin R. I., Lux S. E., Stossel T. P.
(eds). Blood: Principles and Practice of Hematology. Philadelphia: J. B. Lippincott;
1995:155-169.
Краткий трактат о структуре, функции, анатомии и физиологии гемопоэтической
ткани, написанный одним из наиболее цитируемых в мире экспертов в этой
области. Подробно обсуждается кровоток в селезенке как основа для обеспечения
множества функций селезенки.
Williams D. A. The stem cell model of hematopoiesis. In: Hoffman R., Benz E., Shattil S.,
Furie В., Cohen H., Silberstein L. eds. Hematology: Basic Principles and Practice.
Baltimore: Williams & Wilkins; 1995:180-92.
Вероятно, лучшее из изданных за последние годы современное всестороннее изложение
вопросов гемопоэза.
Глава 2
Клинический подход
к пациентам
с гематологическими
проблемами
Фред Дж. Шиффман
Пациенты с заболеваниями крови приходят к врачу при появлении тех или иных
симптомов, прямо указывающих на патологию крови, или обращаются по поводу
недомоганий, на самом деле обусловленных гематологическими проблемами.
Например, пациент жалуется врачу на одышку, а при лабораторном исследовании
обнаруживается низкий гематокрит. Это может быть проявлением кардиоваску-
лярной патологии, но такая анемия нуждается в обязательном выявлении ее
этиологии (возможны желудочно-кишечное кровотечение из пептической язвы или
карцинома толстой кишки). Таким образом, анемия (как и большинство других
гематологических проблем) должна служить лишь исходным пунктом для поиска
патофизиологического механизма, лежащего в основе гематологических
признаков или симптомов. Так же, как обычная кожная сыпь может оказаться либо
проявлением местных нарушений, либо следствием глубокого расстройства гомеос-
таза, гематологические синдромы или заболевания могут иметь как вполне
безобидные, так и весьма опасные для жизни причины, которые должны быть
вскрыты и изучены клиницистом.
В таблицах этой главы представлены основные элементы оценки анамнеза
и результаты физикального обследования применительно к заболеваниям крови.
В первых двух колонках перечисляются данные анамнеза и физикального
обследования; в третьей колонке — возможные варианты патофизиологических
объяснений. Там же представлены связи между различными заболеваниями и ключевые
этиологические моменты. (Графическое представление информации по
некоторым разделам таблицы дано в полной и хорошо иллюстрированной монографии
A. Mitus и D. Rosenthal, указанной в списке литературы.)
Когда изучение анамнеза и физикальное обследование завершены, проведены
исследования периферической крови, костного мозга, оценены другие важные
лабораторные данные, появляется возможность создания рабочей диагностической
гипотезы, объединяющей множество сложных переменных. Необходимо
свободное, творческое и в то же время вполне конкретное приближение к формулировке
44 Глава 2. Клинический подход к пациентам с гематологическими проблемами
диагноза, как это прекрасно описано в начальных главах книги W. В. и Е. D. Shelley
(см. список литературы).
Изучение анамнеза, физикальное обследование и наконец лабораторная
диагностика предоставляют врачу все больше и больше информации. Иногда
приходится вернуться к койке больного или в лабораторию для того, чтобы
перепроверить начальные данные. Могут появиться дополнительные, более обстоятельные
или косвенно подтверждающие определенную точку зрения вопросы. Для
координирования все нарастающего объема данных может потребоваться проведение
пропущенных при первичном осмотре компонентов физикального обследования,
равно как и других манипуляций, обусловленных появлением новой
информации. Таким образом, для полного согласования всех данных необходимо, как
правило, неоднократно обследовать пациента. В этом есть свои преимущества, с
другой стороны, бесконечный пересмотр и перепроверка "установившихся фактов"
могут подрывать репутацию врача. Более того, при первичных расспросах врача
пациент невольно "настраивается" на определенные ответы, и повторный сбор
анамнеза часто не дает никаких результатов.
Когда диагноз практически поставлен, понятно стремление врачей не
включать или активно исключать оставшиеся диагностические возможности, если те
или иные данные, полученные впоследствии, не согласуются с рабочей гипотезой
или если терапия, основанная на рабочей гипотезе, не приводит к желаемым
результатам. Такой достаточно распространенный феномен "раннего закрытия"
опасен как возможная причина врачебных ошибок. Будучи уверенным в
недостаточно обоснованной гипотезе, врач может исключить из рассмотрения
следующие возможности:
1. Одновременное развитие нескольких патофизиологических процессов
(например, гемолитической анемии, сопровождающейся падением гематокри-
та с необходимостью постоянных гемотрансфузий, может сопутствовать
потеря крови из желудочно-кишечного тракта).
2. Редко встречающееся проявление обычного заболевания (например, при
нарушении познавательной функции у пациента без признаков анемии
необходимо рассмотреть возможность недостаточности витамина Bi2).
3. Распространенное проявление редко встречающегося заболевания
(например, причиной тромбоцитопении, обнаруживающейся петехиями и
суставными болями, обычно бывает такой коллагеноз, как системная красная
волчанка, но вместе с тем может быть и пурпура Шенлейна-Геноха).
Для более детального обсуждения темы клинического подхода к пациенту и
проникновения в суть диагностической аргументации и стратегии рекомендуем
читателям обратить внимание на весьма информативную книгу "Дифференциальный
диагноз" Barondess и Carpenter (1994).
Гематологи — это врачи, которые видят пациента через окуляр микроскопа.
Это определение можно было бы признать эксцентричным, если бы оно не было
так близко к истине. Согласно ему, любой клиницист мог бы легко стать членом
"гематологического клуба", но в действительности это не так. Конечно, мазок
периферической крови не может полностью заслонить собой пациента. Но тот, кто
тщательно исследовал этот мазок, может свести воедино фрагментарные данные,
сфокусировать неясно сформулированные идеи, поднять на уровень научного
знания интуитивные догадки.
Клинический подход к пациентам с гематологическими проблемами 45
Поскольку для постановки диагноза требуется соблюдение правил получения
и анализа клинической и лабораторной информации, при оценке мазка
периферической крови необходимо тщательно сопоставить морфологические особенности
крови пациента с данными анамнеза и физикального обследования. Иными
словами, возвращение к постели больного или в лабораторию позволяет получить,
прояснить и согласовать новую информацию. Этот процесс стимулируют
результаты микроскопического исследования мазка крови (размеры, форма,
патологические включения эритроцитов, лейкоцитов или тромбоцитов). Например,
обнаружение микросфероцитов может указывать на то, что в основе заболевания
желчного пузыря, отмеченного в истории болезни пациента, лежит
наследственный сфероцитоз. Весьма вероятно, что более детальное исследование анамнеза
поможет обнаружить подобные случаи у родственников, а более внимательный
осмотр области лодыжек пациента позволит убедиться в наличии следов
язвенных поражений, характерных для наследственных сфероцитозов. И наоборот, га-
стрэктомия в анамнезе и небольшие неврологические нарушения могут привести
врача к микроскопу в поисках макроовалоцитов и гиперсегментированных нейт-
рофилов, характерных для дефицита витамина Bi2 — возможной причины
заболевания данного пациента даже при отсутствии анемии.
Все врачи при исследовании мазка периферической крови должны:
1. Разработать подход.
2. Знать, как приготавливается мазок крови (глава 11).
3. Знать, как мазок окрашивается (метиленовый синий окрашивает кислые
структуры в синий цвет; эозин окрашивает основные структуры в красный).
4. Найти поле просмотра, где артефакты минимальны или отсутствуют.
5. Начать с малого увеличения в поисках типичных признаков и наиболее
распространенных дефектов клеток крови (например, агрегация эритроцитов
часто наблюдается при холодовой аллергии; увеличение частоты атипичных
лимфоцитов является маркером хронического лимфолейкоза).
6. Проверять все три ростка кроветворения (эритроциты, лейкоциты,
тромбоциты) одновременно. Будьте дисциплинированны, не рассеивайте
внимание (в главах 3-5 и приложении даны характерные особенности
нормальной и патологической морфологии всех трех ростков кроветворения).
7. Быть наблюдательными.
8. Не пытаться идентифицировать каждую клетку.
9. Просмотреть достаточное количество полей.
10. Обратить внимание на общую конфигурацию, цвет,
консистенцию/внутреннюю структуру, форму, размер.
Врачи-специалисты при исследовании аспирата костного мозга должны:
1. Разработать подход.
2. Знать, как приготавливается мазок костного мозга.
3. Знать, как мазок окрашивается (метиленовый синий окрашивает кислые
структуры в синий цвет; эозин окрашивает основные структуры в красный).
4. Найти поле просмотра, где артефакты минимальны или отсутствуют.
5. Начать с малого увеличения (общая картина, опухолевые клетки, клеточ-
ность, жир, гетерогенность, гранулема, фиброз, лимфома, инфильтраты, ме-
гакариоциты, соотношение миелоидных и эритроидных клеток).
6. Просмотреть достаточное количество полей.
46 Глава 2. Клинический подход к пациентам с гематологическими проблемами
7. Проверять все три ростка кроветворения (эритроидный, миелоидный, мега-
кариоцитарный) одновременно. Будьте дисциплинированны, не
рассеивайте внимание.
8. Быть наблюдательными.
9. Не пытаться идентифицировать каждую клетку.
10. Просмотреть достаточное количество полей на нескольких различных
стеклах.
11. Обратить внимание на дисмиелопоэз, наличие мегалобластов, на
лимфоциты и плазматические клетки, аномальные клетки (например, клетки Гоше).
Более детальное обсуждение направлений исследований и анализа специальных
гематологических тестов не входит в задачу данной главы. Надеемся, что
представленные здесь положения послужат моделью проведения и интерпретации
результатов исследования. Эта тема будет рассмотрена подробнее в главе 11.
Сопоставление в таблице отдельных признаков и симптомов с
гематологическими заболеваниями приведено в соответствии с порядком сбора анамнеза и
проведения физикального обследования. Это должно помочь практикующему
врачу правильно описать, проанализировать, интегрировать и реинтегрировать
специфические признаки и симптомы гематологических заболеваний.
Избранная литература
Barondess J. A., Carpenter С. С. J. Differential Diagnosis. Philadelphia: Lea & Febiger;
1994.
Детально разработанное классическое руководство по всем аспектам
дифференциальной диагностики.
Braverman I, M. Skin signs of systemic disease, 2nd ed. Philadelphia: W. B. Saunders;
1981.
Классическое издание, в котором обсуждаются и хорошо иллюстрируются многие
кожные реакции, рассмотренные в этой главе.
Berger Т. С, Silverman S. Oral and cutaneous manifestations of gastrointestinal disease.
In: Sleisenger M. H., Fordtran J. S. eds. Gastrointestinal disease. Pathophysiology/
Diagnosis/Management, 5th ed. Philadelphia: W. B. Saunders; 1993: 268-285.
В данном издании заслуживает особого внимания таблица по оральным и кожным
проявлениям желудочно-кишечных заболеваний, многие из которых связаны с
гематологическими проблемами.
Degowin E. L., Degowin R. L. Bedside diagnostic examination, 5th ed. New-York:
McGraw-Hill; 1994.
Пятое издание великолепной книги. Очень полезны вступительные главы.
Grover S. A., Barkun A. N., Sackett D. L. Does this patient have splenomegaly? JAMA,
1993;270:2218-2221.
Переработка предыдущей книги этой группы авторов. Посвящена обследованию
больного со спленомегалией.
Henry P. H., Longo D. L. Enlargement of lymph nodes and spleen. In: Fauci A. S. et al. eds.
Harrison's Principles of Internal Medicine, 14th ed. McGraw-Hill; 1998: 345-351.
Современные подходы по обследованию пациента с лимфаденопатией и
спленомегалией. ч .
Клинический подход к пациентам с гематологическими проблемами 47
Mitus A. J., Rosental D. S. History and physical examination of relevance to the
hematologist. In: Handin R. I., Lux S. E., Stossel T. P. eds. Blood: Principles and
Practice of Hematology. Philadelphia: J. B. Lippincott; 1995:3-21.
Хорошо написанная и прекрасно иллюстрированная работа по методам изучения
истории болезни и проведения физикального обследования.
Shelley W. В., Shelley E. D. Advanced dermatologic diagnosis. Philadelphia: W. В.
Saunders; 1992.
Представленный материал далеко выходит за рамки дерматологии. Мастерское
описание методов дерматологической диагностики, диагностических
умозаключений и общей стратегии.
Williams W.J. Approach to the patient. In: Beutler E., Lichtman M. A., Coller B. S., Kipps
T. J. eds. Williams Hematology, 5th ed. New-York: McGraw-Hill; 1995:43-48.
Детальный обзор того, каков должен быть подход к пациенту с гематологическим
заболеванием с точки зрения анамнеза и физикального обследования.
ТАБЛИЦА 2-1. Некоторые признаки и симптомы, а также другие данные, характерные для гематологических заболеваний
Составляющие
анамнеза
Данные/признаки/симптомы
Патофизиологическая значимость
Основная Причины, побудившие пациента обратиться Служат важной исходной точкой проведения дифференциальной
жалоба больного за помощью диагностики
Анамнез данного Корректный (во временном и физиологи-
заболевания
Общий
медицинский
анамнез
ческом смысле) анамнез заболевания ла-
циента с незначительным врачебным
редактированием. Включает максимально
полное описание и оценку основных
симптомов, ощущение пациентом их
серьезности и опасности. Всегда необходимо
отмечать провоцирующие факторы, качество,
локализацию, интенсивность и временные
характеристики симптомов
При изучении анамнеза необходимо
выяснить, какие лекарственные средства
принимал пациент
Включает общие и системные симптомы,
такие как повышение температуры тела,
потливость, озноб, зуд, изменение веса
Лекарственная терапия/препараты
(предписанные врачом, приобретенные без
рецепта или незаконно)
Аллергии
Госпитализации
Позволяет врачу составить первоначальное мнение
относительно заболевания пациента
Прием всякого лекарственного препарата вне зависимости от
времени может стать причиной развития любого заболевания,
особенно гематологического
Важно оценить, как повлияло заболевание пациента на организм
в целом. Многие серьезные гематологические и онкологические
болезни сопровождаются системными признаками
Крайне важные данные при оценке любых гематологических
заболеваний. Прием всякого лекарственного препарата в любое'
время может вызвать заболевание крови. Необходимо отметить
в истории болезни все препараты, применяемые пациентом
без рецепта врача. Весьма важно выявление приема нелегальных
препаратов и пути их введения
Могут оказаться причиной эозинофилии
Сегодняшние гематологические нарушения могут быть
последствием серьезных заболеваний в прошлом. Сведения об имевших
Несчастные случаи/повреждения/травмы
Хирургические операции
Тяжелые заболевания и их лечение
Гинекологический статус: менархе;
характер и продолжительность менструального
цикла; геморрагические или тромботичес-
кие проблемы при выкидышах, абортах,
родах, в перинатальном периоде; менопауза
Детские заболевания
Иммунизации
место госпитализациях или выписки из историй болезни всегда
важны; лабораторные данные нужно оценить в динамике
Данные об этих событиях очень существенны. Особенно серьезны
гематологические и иммунологические последствия спленэкто-
мии. Необходимо расспросить и о возможном наличии инородных
тел в тканях организма, особенно о содержащих свинец
фрагментах ранящих снарядов (могут быть причиной свинцовых
отравлений, сопровождающихся сидеробластной анемией). Состояние
пациента после травмы может дать полезные сведения о гемостазе
Гастрэктомия и илеоэктомия нарушают процесс всасывания
питательных веществ (железа, витамина В12). Синдром укороченной
тонкой кишки также может быть причиной мальабсорбции. То, как
пациент перенес операцию (например, удаление зубов
мудрости), может дать информацию о нарушениях гемостаза
ВИЧ, туберкулез, другие инфекционные заболевания;
заболевания почек, печени, желудочно-кишечного тракта, сердца и легких,
ревматические и неврологические расстройства, равно как и их
лечение, могут оказывать негативное влияние на
гематологический статус
Выявляет проблемы гемостаза. Волчаночный антикоагулянт и
другие антифоефолипидные синдромы могут быть следствием
выкидышей или абортов. Менопауза, вызванная гистерэктомией,
может сочетаться со злокачественными заболеваниями.
Нарушения менструального цикла могут свидетельствовать о патологии
эндокринной системы, имеющей в том числе и гематологические
проявления
Внутрисуставные кровоизлияния или носовые кровотечения
могут указывать на нарушение гемостаза. Инвагинация после
2-летнего возраста может быть связана с наличием пурпуры Шенлей-
на-Геноха
Недостаточная иммунизация приводит иногда к развитию у
взрослых заболеваний, имеющих, в частности, и гематологические
ТАБЛИЦА 2-1. (Продолжение)
Составляющие
анамнеза
Данные/признаки/симптомы
Патофизиологическая значимость
Общий
медицинский
анамнез
(продолжение)
Семейный
медицинский
анамнез
Иммунизации (продолжение)
Трансфузии
Факторы риска ВИЧ-инфекции
Причины смерти или опасных заболеваний
у членов семьи, их возраст при этом,
причины госпитализаций, осложнения лечения
(в частности, кровотечения/тромбозы). По
родословной, которую необходимо
изобразить графически, можно отследить тип
наследования
Этническая принадлежность и страна
происхождения
Текущие заболевания у членов семьи
проявления. В то же время вакцинация пациентов, принимающих
иммунодепрессанты, может иметь различные патологические
последствия (как при введении живой вакцины больным
хроническим лимфолейкозом [ХЛЛ])
Могут вызывать появление аллоантител и гемолитические транс-
фузионные реакции. Существует риск передачи вирусных,
бактериальных или паразитарных заболеваний
Всегда необходимо выяснять: имели ли место парентеральное
применение наркотиков и переливания крови, каковы стиль
жизни и сексуальная ориентация
Многие гематологические заболевания (расстройства,
вызывающие анемию, тромбоцитопению и лейкопению) являются
наследственными, равно как и многие нарушения свертывания крови.
С этой позиции необходимо рассматривать и нарушения тромбо-
образования, если легочная или артериальная эмболии
периодически повторяются у членов семьи или развиваются в раннем
возрасте
Имеют значение для многих гематологических заболеваний
(серповидно-клеточной анемии, талассемии, дефицита глюкозо-6-
фосфатдегидрогеназы). Необходимо помнить, что как женщина,
так и мужчина могут при вступлении в брак или по другим
соображениям изменить фамилию и тем самым скрыть свое этническое
происхождение
Крайне важными могут оказаться данные о заболеваниях
наследственной или инфекционной природы у членов семьи, а также
сведения о бытовых отравлениях. Например, неисправное печное
отопление может явиться причиной отравления угарным газом и,
соответственно, развития эритроцитоза. Его распознавание у па-
Социальный
и личностный
анамнез
Стиль жизни: гомосексуальность, гетеро-
сексуалйность, моногамность, семейный
статус
Профессиональные и экологические
условия
Туризм
Домашние животные
циента может быть облегчено, если эритроцитоз или потеря
сознания обнаружены у членов семьи. И наоборот, наличие
гематологических заболеваний у пациента может положить начало поиску
аналогичных заболеваний у членов его семьи (например,
эритроцитоз при отравлении угарным газом, наследственная патология
свертывания, гемохроматоз, наследственный сфероцитоз)
Все это имеет отношение к диагностике заболеваний,
передаваемых половым путем, особенно ВИЧ
Контакт на рабочем месте или дома с токсинами, пылью, дымом,
газами, химическими реактивами может явиться одним из
основных этиологических факторов развития заболеваний крови.
Установлена четкая связь между воздействием "Оранжевого агента"
(смесь 2,4-дихлор- и 2,4,5-трихлорфеноксиуксусных кислот) и
возникновением лимфом; отравлений свинцом — с анемией,
бензолом — с апластической анемией и лейкозом, тяжелыми
металлами и пестицидами — с различными болезнями крови
Экзотические болезни, особенно паразитарные, могут развиться
во время путешествий. Данные о посещении пациентом
локальных эндемических зон, таких как полуостров Кейп-Код, острова
Нантакет и Мартас Вайньярд, могут навести на мысль о наличии
у него гемолитической анемии, связанной с бабезиозом.
Гематологические заболевания могут возникнуть также вследствие
контактов пациента с теми, кто возвратился из зарубежной поездки,
или употребления им в пищу импортных продуктов. К тому же
некоторые лекарственные препараты, применяемые для лечения
экзотических заболеваний, сами могут стать причиной развития
заболеваний крови (гемолитические или тромбоцитопенические
нарушения, связанные с приемом хинина)
Орнитоз (передаваемый птицами), сальмонеллез (передаваемый
пресмыкающимися) могут иметь гематологические осложнения.
ТАБЛИЦА 2-1. (Продолжение)
Составляющие
анамнеза
Данные/признаки/симптомы
Патофизиологическая значимость
Социальный
и личностный
анамнез
(продолжение)
Домашние животные (продолжение)
Диета
Привычки, хобби, развлечения
Прием лекарственных препаратов (главным
образом, без рецепта)
Употребление алкоголя
У пациентов с удаленной селезенкой иногда наблюдаются
тяжелые заболевания, вызываемые бактериями DF2 (Capnocytophagia
canimorsus) и Eikenella corrodens (передаются собаками).
Болезнь кошачьей царапины, требующая дифференциации с лим-
фомой, также, как и токсоплазмоз, передается кошками
При выяснении общего анамнеза часто остается без внимания то,
что может иметь принципиальное значение в этиологии многих
заболеваний крови. Так, при некоторых диетах может
наблюдаться дефицит железа, фолиерой кислоты, Bi2 и других витаминов
группы В или витамина К. С другой стороны, гипервитаминоз
может повлечь за собой изменение состояния клеток крови и
нарушение гемостаза. Определенные пищевые отклонения, такие как
употребление глины (геофагия) или льда (пагофагия), могут быть
проявлениями недостатка железа
Бег на длинные дистанции, занятия каратэ, интенсивная игра
на барабане способны приводить к развитию гемолитической
анемии напряжения. Заболевания крови могут возникать вследствие
продолжительной работы с клеями, растворителями, красками
Прием всякого лекарственного средства в любое время может
стать причиной гематологических проблем, однако в большей
степени это относится к препаратам определенных фармакоте-
рапевтических групп. Описаны как клеточные нарушения, так и
расстройства свертывания крови. Поэтому при сборе анамнеза
необходимо спрашивать пациента о приеме им лекарств
безрецептурного отпуска
Сбор всестороннего анамнеза врач должен проводить с большим
тактом. Возникновение нарушений свертывания крови, анемии,
тромбоцитопении может быть связано с острым токсическим
Курение
Обзор систем Покровы. КОЖА: цвет, пигментация,
температура, влажность, сыпь, зуд, шелушение,
кровоподтеки, кровоизлияния. ВОЛОСЫ:
цвет, структура, аномальная потеря или
избыток, распределение. НОГТИ: изменения
цвета, ломкость, появление бороздок,
образование впадин,искривление
Лимфатические узлы: увеличение, боль,
нагноение, свищи, локализация
Кости, суставы, мышцы: переломы,
артриты, боль, припухлость, тугоподвижность,
блуждающие симптомы, степень потери
трудоспособности, мышечная слабость,
изнурение или атрофия. Ночные судороги
воздействием алкоголя на костный мозг или хроническим — на
печень (поражение паренхимы, портальная гипертензия). Боли в
области поврежденных регионарных лимфоузлов после
употребления алкоголя встречаются^ пациентов с болезнью Ходжкина
Хорошо известна связь между курением и развитием некоторых
злокачественных опухолей. На фоне последних могут
наблюдаться анемия, эритроцитоз, лейкоцитоз, тромбоцитопения, тромбо-
цитоз, повышенная или пониженная свертываемость крови.
Курение само по себе может вызывать лейкоцитоз
Сухая кожа, сухие и тонкие волосы, ломкие ногти могут быть
признаком железодефицитной анемии. Грубая кожа может
свидетельствовать о микседеме. Зуд возникает после купания у
некоторых пациентов с болезнью Ходжкина, грибовидным микозом и
истинной полицитемией. (Далее описаны и. другие
специфические изменения кожи и ногтей и их патофизиологическая
значимость.)
Подробно описаны в разделе, посвященном физикальному
исследованию пациента.
Боли в костях могут являться признаком их вовлечения в
опухолевый процесс, например множественную миелому или лимфому, а
также признаком метастатического поражения, что
преимущественно характерно для позвоночника. Боли в костях возникают
также при гемоглобинопатиях, особенно талассемии и
серповидно-клеточной анемии. Боли в области грудины могут быть
симптомом лейкоза, в первую очередь хронического миелолейкоза.
Псевдоподагра (хондрокальциноз), обусловленная гемохромато-
зом, может вызывать боли в суставах. Миелопролиферативные
расстройства, высокозлокачественные лимфомы и острые
лейкозы проявляются иногда острой подагрой с гиперурикемией..
Суставы поражаются при ревматоидном артрите и синдроме
Фелти, пурпуре Шенлейна-Геноха и гемофилической артропа-
ТАБЛИЦА 2-1. (Продолжение)
Составляющие
анамнеза
Данные/признаки/симптомы
Патофизиологическая значимость
Обзор систем
(продолжение)
Гемопоэтическая система: анемия (тип,
лечение, чувствительность к терапии), лим-
фоаденопатия, кровотечение (спонтанное,
травматическое, наследственное)
Эндокринная система: анамнез развития,
строение тела, вес. Размер кистей, стоп,
головы и, особенно, изменения их в
подростковом возрасте. Распределение
волосяного покрова. Пигментация кожи. Слабость.
Зоб, экзофтальм, сухость кожи и волос,
непереносимость жары или холода, тремор.
Полифагия, полидипсия, полиурия, глкжо-
зурия. Вторичные половые признаки,
импотенция, бесплодие, лечение.
Диагностирование и терапия эндокринных расстройств
в анмнезе
тии. Боль в левом плече может быть симптомом спленомегалии
или инфаркта селезенки (отраженная боль). Судороги ног в
анамнезе свидетельствуют о возможном употреблении хинина —
причины возникновения тромбоцитопении и анемии
Наличие в анамнезе каких-либо указаний на любое
гематологическое заболевание может прояснить сегодняшние жалобы
пациента
Многие эндокринные заболевания могут иметь
гематологические проявления
Эритроциты. Анемия может сопутствовать пептической язве,
сидрому Золлингера-Эллисона. Нормохромная нормоцитарная
анемия часто сопровождает гипофизарную кахексию. Гипотиреоз
может ассоциироваться с макроцитарной анемией (±
недостаточность витамина В12 или фолиевой кислоты) или, чаще, с нормох-
ромной нормоцитарной анемией. Гипотиреоз,
сопровождающийся нарушением всасывания железа или меноррагиями, способен
приводить к гипохромной микроцитарной анемии. У пациентов с
тяжелым тиреотоксикозом иногда наблюдается нормохромная
нормоцитарная или гипохромная анемия
Примерно у 3 % пациентов с болезнью Грейвса встречается пер-
нициозная анемия с выработкой антител к париетальным
клеткам. Недостаточность надпочечников сопровождается
умеренной нормохромной нормоцитарной анемией. У пациентов с
болезнью Кушинга может возникать умеренный эритроцитоз с
уровнем гемоглобина на 10-20 г/л выше нормы. Примерно у 20 %
пациентов с первичным гиперпаратиреозом выявляется нормо-
Аллергический и иммунологический
анамнез: дерматит, крапивница, ангионевроти-
ческий отек, экзема, сенная лихорадка,
вазомоторный ринит, астма, мигрень, весенний
конъюнктивит. Сенсибилизация к пыли,
пищевым продуктам, перхоти или
лекарственным препаратам. Результаты кожных тестов
в анамнезе. Результаты туберкулиновой и
других реакций. Десенсибилизация,
введение сыворотки, вакцинации и иммунизации
Голова: головные боли, мигрень, травма,
головокружение, обморок, конвульсивные
приступы
Глаза: потеря зрения или цветовая слепота,
диплопия, гемианопсия, травма,
воспаление, использование очков (дата последнего
осмотра окулистом)
хромная нормоцитарная анемия. Железодефицитная анемия
может развиваться при гиперпаратиреозе, осложненном язвенной
болезнью
Лейкоциты. Лейкопения может наблюдаться при гипопитуита-
ризме. Тиреотоксикоз сопровождается лейко- или гранулоцито-
пенией, а лечение метимазолом или пропилтиоурацилом может
вызывать агранулоцитоз. У пациентов с синдромом Кушинга
встречается эозинопения. Лимфоцитоз и эозинофилия могут
обнаруживаться при болезни Аддисона, а лейкоцитоз — при
диабетическом кетоацидозе. Повышенный уровень глюкокортикоидов
становится причиной развития лейко- и тромбоцитоза
Аллергические нарушения могут сопровождаться эозинофилиеи
Подобные симптомы обнаруживаются при анемии или полиците-
мии, поражениях ЦНС при лейкозе и лимфоме, а также при таких
инфекциях, как криптококкоз или туберкулез. Причиной
появления признаков поражения ЦНС может быть внутричерепное
кровоизлияние (паренхиматозное, субарахноидальное, эпидураль-
ное или субдурапьное) на фоне коагулопатии
Тяжелая анемия приводит к ишемии сетчатки или ее инфаркту,
что проявляется потерей остроты зрения или слепотой.
Коагулопатии способны вызывать инфаркт сетчатки (при
гиперкоагуляции) или кровоизлияние (при гипокоагуляции). Диплопия или
выпадение полей зрения могут наблюдаться при лимфомах ЦНС или
ТАБЛИЦА 2-1. (Продолжение)
Составляющие
анамнеза
Данные/признаки/симптомы
Патофизиологическая значимость
Обзор систем
(продолжение)
Глаза (продолжение)
Уши: глухота, звон вушах, головокружение,
выделения из ушей, боль, мастоидит, операции
Нос: острый или хронический ринит,
синусит, выделения, заложенность, носовое
кровотечение
Рот. болезненность органов полости рта,
симптомы заболеваний зубов
Горло: дисфония, боли в горле, тонзиллит,
изменения голоса
Шея: припухлость, гнойные поражения,
увеличение лимфоузлов, зоб, тугоподвиж-
ность или ограниченность движений
других злокачественных новообразованиях, поражающих
паренхиму или черепно-мозговые нервы
Причиной шума, дроби, звона в ушах может быть тяжелая анемия.
Тугоухость встречается у детей с серповидно-клеточной анемией
Носовым кровотечением могут проявляться коагулопатия,
обусловленная нарушениями каскада свертывания крови или
дефектами тромбоцитов, артериовенозные пороки, наблюдающиеся,
например, при синдроме Рандю-Вебера-Ослера. При перници-
озной анемии встречаются обонятельные галлюцинации или
аносмия. Развитию последней способствуют также опухоли,
поражающие ядро обонятельного нерва
Болезненность языка возникает при пернициозной или железо-
дефицитной анемии. Увеличение языка происходит при микседе-
ме или амилоидозе. Инфильтрационные повреждения десен при
остром моноцитарном лейкозе могут сопровождаться
соответствующими болями; язвенные поражения слизистой оболочки
ротовой полости характерны для пациентов с нейтропенией.
Сухость во рту может быть обусловлена гиперкальциемией при
злокачественных опухолях кроветворной ткани, например при
множественной миеломе
Дисфония может развиваться при лимфоретикулезе (вовлечение
в процесс возвратного нерва гортани при распространении
процесса на шейный или верхнегрудной уровень). Боли в горле могут
быть связаны с инфекционным мононуклеозом. Изменения
голоса наблюдаются при ретрофарингеальных гематомах,
обусловленных коагулопатией
Припухлостью в области шеи могут проявляться увеличение
щитовидной железы, лимфаденопатия или синдром верхней полой
вены
Грудные железы: развитие, лактация,
травма, болезненные припухлости, выделения
из сосков, гинекомастия, изменения сосков
Дыхательная система: боль, одышка, шум
при дыхании, диспноэ, ночное диспноэ, ор-
топноэ, кашель, мокрота, кровохарканье,
ночные поты, плеврит, бронхит, туберкулез
(наличие контактов), пневмония, астма,
другие респираторные инфекции
Сердечно-сосудистая система:
сердцебиение, тахикардия, нарушения ритма, боли в
груди, одышка напряжения, пароксизмаль-
ная ночная одышка, ортопноэ, кашель,
цианоз, асцит, отек. Перемежающаяся
хромота, холодные конечности, флебит, посту-
ральная или постоянная гиперемия. Гипер-
тензия, ревматическая атака, хорея,
сифилис, дифтерия. Применение лекарственных
препаратов наперстянки, хинидина,
нитроглицерина, диуретиков и др.
Желудочно-кишечный тракт: аппетит,
изменения веса, дисфагия, тошнота, отрыжка,
метеоризм, абдоминальные боли или
колики, рвота, рвота с кровью, желтуха (боль,
лихорадка, интенсивность,
продолжительность, цвет мочи и стула), стул (цвет,
частота, консистенция, запах, газы, прием
слабительных), геморрой. Изменения в кишечных
отправлениях
Увеличение массы груди может свидетельствовать о первичных
опухолевых процессах или лимфоматозных отложениях,
экстрамедуллярном гемопоэзе или распространении плазмацитомы
Одышка может быть симптомом анемии. Кровохаркание —
признаком коагулопатии, инфекций, связанных с дефектом
иммунитета пациента, или болезни Рандю-Вебера-Ослера,
сопровождающейся артериовенозными пороками. Кашель может
свидетельствовать о повреждении лимфатических узлов
средостения, развитии опухоли или инфекции, протекающей на фоне
нейтропении. Необходимо принять во внимание возможное
вторичное поражение плевры и паренхимы легких
При анемии или полицитемии ухудшается транспорт кислорода и
проявляются различные нарушения сердечно-сосудистой
системы. Гиперкоагуляция способствует возникновению
артериальных или венозных тромбозов и эмболии. Следствием многих
заболеваний сердечно-сосудистой системы и приема
лекарственных препаратов, назначаемых для их лечения,
становятся изменения в системе крови
Анорексия относится к неспецифическим жалобам при многих
системных заболеваниях. Ею страдают больные с лимфомами и
лейкозами. Она может быть проявлением вторичных эффектов
гематологических нарушений, включая гиперкальциемию или
азотемию. Быстрое насыщение нередко указывает на
затруднение эвакуации пищи из желудка или спленомегалию.
Возникновение болей в животе может иметь множество причин:
увеличение печени и селезенки (часто обнаруживается при
гематологических заболеваниях), желудочно-кишечная
непроходимость, ретроперитонеальное кровоизлияние, осложнения
ТАБЛИЦА 2-1. (Продолжение)
Составляющие
анамнеза
Данные/признаки/симптомы
Патофизиологическая значимость
Обзор систем
(продолжение)
Желудочно-кишечный тракт
(продолжение)
Мочеполовая система: цвет мочи, полиурия,
олигурия, никтурия, дизурия, гематурия,
пиурия, задержка мочеиспускания, частота
мочеиспускания, недержание мочи, боль
или колика, выведение камней или песка
Гинекологический анамнез: возраст начала
менструаций, продолжительность цикла,
регулярность, длительность, обильность
менструаций, боли, дисменорея, дата
последних месячных, длительность
предшествующего цикла, характер менопаузы,
постклимактерическое кровотечение
Беременности: количество; наличие
абортов, выкидышей, мертворождений;
хронологическая последовательность; осложнения
беременности. Меры по регулированию
рождаемости
химиотерапии, редкие заболевания крови (например, пурпура
Шенлейна-Геноха или острая перемежающаяся порфирия), а
также многие другие более распространенные заболевания
(например, серповидно-клеточная анемия, при которой боли в животе
являются следствием тромбоза сосудов). При
желудочно-кишечном кровотечении в анамнезе можно предположить развитие
коагулопатии или новообразований ЖКТ. Запор может быть
признаком наличия опухоли ЖКТ или нарушений метаболизма,
например гиперкальциемии. Дисфагия встречается на фоне железоде-
фицитной анемии или сидеропенической дисфагии (синдромы
Паттерсона-Келли и Пламмера-Винсона). Новообразования
пищевода и желудка могут кровоточить и приводить к анемии
Красный цвет мочи может быть признаком кровотечения из моче-
выводящих путей, в основе которого лежит коагулопатия. Вместе
с тем, темно-красноватое окрашивание мочи может указывать на
гемоглобинурию, миоглобинурию, порфиринурию, а также
появляться вследствие употребления некоторых пищевых продуктов
или приема медицинских препаратов. Поэтому при наличии в
анамнезе мочи красного цвета необходимо назначить ее
микроскопическое исследование и другие более специфические
анализы. Как уже отмечалось, гинекологический и акушерский
анамнез может оказаться ключевым с точки зрения выявления и
оценки коагулопатии. Нарушение эрекции или дисфункция мочевого
пузыря служат индикаторами злокачественных заболеваний
крови, затрагивающих нервную систему. Пернициозная анемия
может стать причиной различных нервно-психических расстройств,
связанных, в частности, с нарушениями мочеполовой системы.
Камни в почках вероятны при гиперкальциемии или гиперурике-
мии, развивающихся на фоне злокачественных опухо-
Венерический анамнез: сексуальная
активность; шанкр, бубон, выделения из пениса;
диагностирование и лечение венерических
заболеваний
Нервная система: черепно-мозговые нервы
Психиатрический анамнез
Жизненно
важные
симптомы
Частота и ритмичность пульса
лей системы крови. Постоянная эрекция (приапизм) наблюдается
при лейкозе или серповидно-клеточной анемии
Жалобы на покалывания (парестезия) могут быть признаком
развития пернициозной анемии, злокачественных опухолей
системы крови, амилоидоза, множественной миеломы или
последствием приема лекарственных препаратов для лечения
заболеваний крови (в разделе, посвященном физикальному
обследованию, содержатся некоторые специфические примеры
и описание неврологических симптомов, связанных с
определенными патофизиологическими процессами)
При многих гематологических заболеваниях больные
предъявляют жалобы психиатрического характера. При анемии и полиците-
мии ухудшается транспорт кислорода к головному мозгу.
Нарушения коагуляции могут предшествовать эпидуральному,
субдуральному, субарахноидальному или паренхиматозному
кровоизлияниям с соответствующими нейропсихиатрическими
симптомами. Головной мозг и его оболочки могут быть
инфильтрированы лейкозными или лимфомными клетками. При пернициозной
анемии возможен "мегалобластный психоз" — одно из наименее
изученных ее проявлений. Некоторые типы порфирии приводят
к нервно-психическим расстройствам (острая перемежающаяся
порфирия). Злокачественные заболевания крови могут
сопровождаться гиперкальциемией, вызывающей в свою очередь нервно-
психические проблемы
Тахикардия может быть следствием нарушения транспорта
кислорода при анемии, признаком легочной эмболии как
осложнения гиперкоагуляции, а также симптомом поражения перикарда,
приводящего к сдавлению или тампонаде сердца (кровоизлияние
в околосердечную сумку, вовлечение в опухолевый процесс)
ТАБЛИЦА 2-1. (Продолжение)
Составляющие Данные/признаки/симптомы
Патофизиологическая значимость
анамнеза
Жизненно
важные
симптомы
(продолжение)
Частота дыхания
Температура (оральная/ректальная)
Артериальное давление
Кожа
Тургор, строение, пигментация, цианоз, те-
леангиэктазия, петехии, пурпура, экхимо-
зы, инфекции, повреждения; волосы; ногти;
слизистые оболочки
Тахипноэ может свидетельствовать об анемии, вовлечении
дыхательных путей, плевры или паренхимы легких в опухолевый
процесс при злокачественных заболеваниях крови или легочно-
плевральной инфекции у пациента с иммунодефицитом
Повышение температуры характерно для многих первичных
гематологических заболеваний, в первую очередь злокачественных
опухолей системы крови и миелофиброза. Кроме того,
температура повышается при вторичных процессах, обусловленных
инфекционным или воспалительным осложнением обструктивных
опухолей. Хотя лихорадка иногда развивается при
инфицировании больных с ослабленным иммунитетом, все же подъем
температуры обычно не столь значителен у пациентов, принимающих
кортикостероиды и иммунодепрессанты, а также у пожилых
людей и больных диабетом
Низкое артериальное давление при нарушениях
свертываемости может быть симптомом кровопотери. Кроме того,
артериальное давление может понизиться вследствие заболевания
перикарда (см. выше) или септического шока на фоне ослабленного
иммунитета
ЦВЕТ КОЖИ весьма важен при заболеваниях крови. Бледность
обычно сопутствует анемии. На разжатой кисти ладонные
складки розовые при уровне гемоглобина выше 70 г/л
ТУРГОР, СТРОЕНИЕ. При железодефицитной анемии кожа
сухая, волосы сухие и тонкие, ногти ломкие, ложкообразной формы
(койлонихия). Микседема может проявляться сухостью,
грубостью, чешуйчатостью кожи
ЦВЕТ, ПИГМЕНТАЦИЯ. Желтуха может наблюдаться при
различных гематологических заболеваниях, приводящих к пораже-
нию печени, либо быть следствием гемолиза (непрямой,
несвязанный билирубин). Лимонно-желтый цвет кожи при перници-
озной анемии обусловлен сочетанием желтухи и бледности.
Голубизна склер, светлые или рано поседевшие волосы связаны с
пернициозной анемией. Бронзовая, сероватая пигментация
кожи встречается при гемохроматозе. Периферический цианоз
(кончик носа, уши, пальцы кисти или стопы) наблюдается при
появлении криоглобулинов или Холодовых агглютининов.
Периферический и центральный цианоз (синие губы, язык) — при
сердечно-легочной патологии с содержанием гемоглобина 50 г/л. При
таком состоянии, как полицитемия, общая масса эритроцитов
увеличена, а уровень восстановленного гемоглобина часто более
50 г/л. Явный цианоз развивается при уровне метгемоглобина
15-20 г/л; коричневатый цианоз — при уровне сульфгемоглобина
5 г/л. Красный цвет кожи и различные виды сыпи могут
встречаться при кожных формах Т-клеточных лимфом, хроническом
лимфолейкозе (ХЛЛ), некоторых лимфоцитарных лимфомах и
болезни "трансплантат против хозяина". При отравлении окисью
углерода кожа может быть цианотичной или (реже) вишнево-
красной. Участки гипопигментированной кожи (витилиго)
могут свидетельствовать о появлении антител к меланоцитам —-
одного из аутоиммунных нарушений, с которыми сочетается пер-
нициозная анемия
ПОВРЕЖДЕНИЯ, СЫПИ. Телеангиэктазия может развиваться
при заболевании печени, болезни Рандю-Вебера-Ослера или
синдроме Блюма. Гигантские гемангиомы встречаются при
синдроме Казабаха-Меррит, связанном с диссеминированным
внутрисосудистым свертыванием. Красновато-пурпурные
рельефные повреждения при саркоме Капоши обусловлены ВИЧ-
инфекцией. Петехии являются признаком нарушения структуры
или количества тромбоцитов, патологии кровеносных сосудов
или кожи, васкулита (при цинге обнаруживаются в области про-
ТАБЛИЦА 2-1. (Продолжение)
Составляющие Данные/признаки/симптомы
анамнеза
Кожа
(продолжение)
Патофизиологическая значимость
межности наряду со спиралевидным скручиванием волос). Могут
быть связаны с диспротеинемией. Аналогично происхождение
пурпуры (кроме того, она встречается при менингококковой
септицемии). Пурпура в области рта в виде кровяных пузырьков
(влажная пурпура) — признак крайне опасного снижения уровня
тромбоцитов. Периорбитальная и периректальная пурпуры могут
относиться к симптомам амилоидоза. Экхимоз — проявление
различных нарушений свертывания, бывает специфической
локализации: боковые поверхности туловища или околопупочная
область, что указывает на забрюшинное или внутрибрюшинное
кровоизлияние соответственно. Некротические поражения
кожи являются признаком диссеминированного внутрисосудис-
того свертывания (ДВС) при молниеносной пурпуре, а также
индуцируются криопротеинами, Холодовыми агглютининами, вар-
фарином или гепарином. Хлоромы обнаруживаются в виде
плотной опухоли кожи с инфильтрацией лейкемическими мие-
лобластами. Синдром Свита — фебрильный нейтрофильный
дерматоз, входит в симптомокомплекс острого лейкоза. Кожные
проявления типичны для волосатоклеточного лейкоза, ангиоим-
мунобластной лимфоаденопатии и пигментозной крапивницы.
При многих заболеваниях желудочно-кишечного тракта,
обусловленных болезнями крови, имеют место оральные и кожные
проявления. (См. работу Berger и Silverman, указанную в списке
литературы.) Кожные инфекции часто связаны с лейкопенией или
патологией лейкоцитов, например при синдроме Чедиака-Хига-
си, хроническом гранулематозе и синдроме Джобса. Кандидиоз
кожи и слизистых может развиваться при недостаточности мие-
лопероксидазы. Некоторые виды повреждений кожи описаны
Череп Общее описание
Глаза Подвижность глазного яблока
Исследование зрачка
Склера и конъюнктива
при гиперэозинофильном синдроме. Язвы нижних конечностей
наблюдаются обычно при серповидно-клеточной анемии и
наследственном сфероцитозе. Для 15 % детей с
серповидно-клеточной анемией характерны краснота, болезненность, неотечная
припухлость всех дистальных отделов конечностей
НОГТИ. Изменения ногтей, особенно койлонихия
(ложкообразные ногти), могут быть признаком дефицита железа
Выпуклости лобной и теменной костей (фронтальный выступ)
и другие нарушения лицевого скелета наблюдаются при
серповидно-клеточной анемии и талассемии, при которых объем
полостей, заполненных костным мозгом, расширяется для
компенсации гемолиза. Подобные проявления могут иметь место при
анемии Фанкони, синдроме Блюма, врожденном сфероцитозе, плаз-
моцитоме и эозинофильных гранулемах
Подвижность глаз может нарушаться при расстройствах
метаболизма, таких как пернициозная анемия или порфирия. Кроме того,
воздействие на глазную моторику оказывают кровоизлияния
и многие злокачественные заболевания крови (прямое давление),
а также паранеопластические состояния или инфекции,
возникновение которых обусловлено ослабленным иммунитетом пациента
Размер зрачка и его рефлексы подвержены влиянию тех же
патофизиологических механизмов, что и подвижность глазного яблока
Бледность конъюнктив может указывать на анемию,
краснота — на полицитемию или синдром верхней полой вены. При
исследовании склер и конъюнктивы может быть выявлена желтуха
Выпячивание — признак гипертиреоза, орбитальной лимфомы,
хлоромы, нейробластомы, экстрамедуллярного гемопоэза.
Маслянистость характерна для болезни Гоше. Другие нарушения со
стороны склер могут указывать на коллагеноз или лимфому.
Неправильные, в форме запятой или спиралеобразные сосуды
конъюнктивы наблюдаются при серповидно-клеточной анемии
ТАБЛИЦА 2-1. (Продолжение)
Составляющие
анамнеза
Данные/признаки/симптомы
Патофизиологическая значимость
Глаза
(продолжение)
Радужка
Сосудистая оболочка
Исследование глазного дна
Кольцо Кайзера-Флейшера, обнаруживаемое с помощью
щелевой лампы,— диагностический признак болезни Вильсона
(проявляющейся, в частности, гемолитической анемией). При ги-
перкальциемии, которая может быть одним из симптомов
злокачественных заболеваний крови, на радужке нередко наблюдается
секторная кератопатия в позиции 3 и 9 часов. Строгая
определенность позиции объяснима: кальций и углекислый газ воздуха
вступают в реакцию с образованием фосфата кальция на
участках, не прикрытых веками. Серые или коричневые кристаллы
могут формироваться в результате отложения парапротеина, что
наблюдается при нарушении гомеостаза плазматических клеток
Увеит — один из симптомов саркоидоза или васкулита
Кровоизлияние в стекловидное тело наблюдается при
врожденном или приобретенном нарушении свертывания.
Кровоизлияние в сетчатку, инфаркт (пятно Рота) и экссудат
встречаются у пациентов с анемией и тромбоцитопенией (или при других
коагулопатиях)
Изменения вен сетчатки имеют место при синдроме
повышенной вязкости (перекрещенные, извитые, похожие на "поезд"
или "связку сарделек"), при серповидно-клеточной анемии
италассемии. Серповидно-клеточная анемия сопровождается
весьма характерными изменениями сосудов, которые могут
прогрессировать, приводя к слепоте. Обнаруживаются также
кровоизлияния в виде черного "солнечного пятна",
оранжево-розовых бляшек и ангиоидных прожилок. При гемоглобинопатии
SC неоваскуляризация принимает форму "гребного винта". Ре-
тинальная патология развивается при серповидно-клеточной
анемии и талассемии, а также при гиперэозинофильном синдро-
Уши
Осмотр, острота слуха
Рот, нос, горло См. "ОБЗОР СИСТЕМ" (выше)
Шея
Лимфатические
узлы
Выявление лимфаденопатии (увеличение
лимфатического узла более 1 см в
диаметре) крайне важно для гематолога. Врач
должен ртветить на следующие ключевые
вопросы: "Является ли пальпируемое
образование лимфатическим узлом?", "Как долго
наблюдается лимфаденопатия?", "Каков
характер увеличенного узла?", "Локальной
или генерализованной является
лимфаденопатия?", "Связана ли она с системными
или локальными симптомами и признака-
ме. При лейкозе могут повреждаться любые ткани глаза путем
прямой инфильтрации лейкозных клеток, а также за счет
вторичных эффектов опухолей или вследствие терапии. Пернициозная
анемия поражает главным образом афферентную зрительную
систему, вызывая оптическую нейропатию и нарушения зрения,
включая нарушение цветоразличения (дисхроматопсия)
Подагрические узлы, обнаруживаемые на ушной раковине,
являются признаком многих гиперпролиферативных
гематологических нарушений. Звон или иные шумы в ушах могут быть связаны
с анемией или истинной полицитемией. Потеря слуха
развивается при серповидно-клеточной анемии
Гладкий мясисто-красный болезненный язык может указывать
на недостаточность витамина В12 или железа. Изъязвления
слизистой — один из признаков нейтропении. Увеличение миндалин
может быть вызвано мононуклеозом или лимфомой. Пурпура с
кровяными пузырями (влажная) — проявление тромбоцитопении
Увеличение щитовидной железы может означать как ее
первичное заболевание, так и вовлечение ее в лимфоматозный
процесс (лимфаденопатия описана в графе "Лимфатические узлы")
Этиологические факторы лимфаденопатии многообразны. К ним
относятся: инфекции; дерматологические, воспалительные
заболевания; нарушения метаболизма, болезни накопления;
патология, связанная с побочным действиемлекарств, а также
первичными или метастатическими эффектами злокачественных
опухолей. Если через 1 -2 месяца диагноз еще не поставлен,
должна быть сделана биопсия лимфатических узлов (глава 1)
О)
ел
ТАБЛИЦА 2-1. (Продолжение)
Составляющие
анамнеза
Данные/признаки/симптомы
Патофизиологическая значимость
Лимфатические ми?", "Связана ли она с необычными эпиде-
узлы
(продолжение)
Грудь
миологическими обстоятельствами?". Не
менее важны данные о болезненности,
явных* признаках воспаления, темпах
развития лимфаденопатии
Грудная клетка Осмотр
Пальпация
Перкуссия
Увеличение груди наблюдается при первичном злокачественном
или лимфоматозном поражении, плазмоцитозе или
экстрамедуллярном гемопоэзе. Гинекомастия у мужчин может быть
связана с атрофией яичек, развивающейся при многих заболеваниях
печени, включая гемохроматоз, может быть признаком
первичной опухоли у мужчин, вторичным проявлением опухолей других
локализаций или побочным эффектом фармакотерапии
Гиперемия и расширенные кровеносные сосуды верхней части
грудной клетки могут быть признаком синдрома верхней полой
вены, возникшего вследствие поражения данного сосуда
опухолью. Болезненность грудины свидетельствует о леикемическои
или лимфоматозной инфильтрации костного мозга в этой области
Болезненность ребер, лопаток, позвоночника или грудины
нередко является признаком леикемическои Глимфоматозной или
карциноматознои инфильтрации костей или костного мозга в этих
областях
Притупление звука так же, как и ощутимая вибрация, может
указывать на заболевания плевры, наблюдаемые при многих
гематологических нарушениях, в том числе и на опухоли лимфоретикуляр-
ной ткани, сопровождающиеся плевральным выпотом. Кроме того,
экссудативный плеврит может развиться в результате вовлечения
в опухолевый процесс органов средостения, включая лимфоаде-
нопатию, и инфицирования пацентов с ослабленным
иммунитетом. Притупление звука также может быть признаком уплотнения
Аускультация
Сердечно-сосу- Частота пульса и артериальное давление
дистая система (см. "ЖИЗНЕННО ВАЖНЫЕ СИМПТОМЫ")
Осмотр груди и пальпация
Шум трения перикарда
Шумы
Аускультативные изменения в форме ослабления или усиления
дыхательных шумов могут свидетельствовать о наличии
плеврального выпота или уплотнения легочной ткани, обусловленных
гематологическими заболеваниями. Выслушиваемые хрипы и
шумы весьма значимы, поскольку часто указывают на наличие
легочного уплотнения еще до появления рентгенологических
признаков. Острый грудной синдром, наблюдаемый при серповидно-
клеточной анемии, этиологически связан с инфекцией или
инфарктом легкого. В свою очередь, к инфаркту легкого может
привести появление эмболов некротизированного костного
мозга, возникающих при гемоглобинопатиях. Эмболия легких может
быть следствием гиперкоагуляции, развивающейся при
недостаточности протеинов С, S или антитромбина III, а также при анти-
фосфолипидном синдроме, Кроме того, эмболия легких и
венозные тромбозы выявляются при гиперкоагуляции, связанной с
некоторыми диссеминированными злокачественными
опухолями. Классические признаки уплотнения встречаются при лимфо-
матозной или лейкемической инфильтрации легочной
паренхимы; кроме того, появление их обусловлено множеством легочных
инфекций (особенно у пациентов с ослабленным иммунитетом),
поражающих плевру и паренхиму легких
Оценка состояния яремной вены. Патология наружйой
яремной вены возникает при синдроме верхней полой вены,
тампонаде сердца или сдавливающем перикардите — осложнении
злокачественных опухолей системы крови
Признаком анемии может быть энергичный сердечный толчок
Заболевания перикарда имеют важное значение для
дифференциальной диагностики гематологических заболеваний (см.
"Оценка состояния яремной вены")
При анемии можно услышать шум потока крови. "Опухолевый
хлопок" характерен для миксомы левого предсердия. S3 и другие
ТАБЛИЦА 2-1. (Продолжение)
Составляющие Данные/признаки/симптомы
анамнеза
Патофизиологическая значимость
Сердечно-сосудистая система
(продолжение)
Шумы (продолжение)
Живот
Осмотр
Аускультация
Перкуссия и пальпация
признаки застойной сердечной недостаточности могут быть
симптомами анемии. Патология правых камер сердца,
вызываемая легочной гипертензией, нередко связана с эритроцитозом и,
особенно, с его вторичными формами. Вовлечение перикарда,-
миокарда или эндокарда в опухолевый процесс приводит к
появлению симптомов перикардита, кардиомиопатии или поражения
клапанов
Вздутие живота может указывать на асцит, обусловленный пери-
тонеальными или печеночными метастазами. Асцит встречается
и при синдроме Бадца-Киари (тромбоз печеночной вены) на
фоне гиперкоагуляции
Венозный рисунок живота и направление венозного
кровотока — признак обструкции воротной или печеночной вен.
Закупорка печеночной или нижней полой вены наблюдается при гиперко-
агуляционном статусе, что особенно характерно для истинной
полицитемии — причине синдрома Бадца-Киари
Экхимозы в боковых отделах (симптом Грея Тернера) и
околопупочные экхимозы (симптом Каллена) возникают при забрюшин-
ном или внутрибрюшном кровоизлиянии соответственно
вследствие нарушения свертывания крови
Уменьшение интенсивности кишечных шумов характерно для
анатомической или функциональной непроходимости кишечника,
обусловленной вазоокклюзивным кризом при
серповидно-клеточной анемии, пернициозной анемии, острой перемежающейся пор-
фирии, гиперкальциемии (связанной со злокачественными
опухолями системы крови)
Заболеваниям крови часто сопутствует гепатомегалия, поэтому
всегда необходимо тщательно проводить перкуссию и пальпацию
Конечности Признаки тромбоза глубоких вен (например,
венозные тяжи и симптомы Хомана)
Признаки артериального тромбоза
Суставы
Мужские Пенис
гениталии
Яички
живота. Ключевым моментом при гематологическом
исследовании живота может стать выявление спленомегалии. Большинство
доступных и информативных методов исследования, включая
перкуссию пространства Траубе и пальпацию живота (от
подреберья до нижних отделов) в положении пациента лежа на спине,
описаны Grover и соавт. (работа указана в разделе "Избранная
литература"). Причины и проявления спленомегалии чрезвычайно
разнообразны, в их основе лежат инфекционные,
воспалительные, застойные, гематологические, злокачественные,
кумулятивные и многие другие патологические нарушения (глава 1)
Артериальный или глубокий венозный тромбоз может
указывать на состояние гиперкоагуляции, поэтому на признаки его
развития необходимо обратить особое внимание при физикальном
обследовании пациентов с системными гематологическими
заболеваниями
Патология суставов различного характера, в том числе при кол-
лагенозах, является весьма важным в гематологии симптомом.
Такое же значение имеет артрит. Кроме того, хондрокальциноз
может быть признаком гемохроматоза, а подагра —
проявлением многих гематологических нарушений, для которых характерно
быстрое клеточное обновление
Приапизм может наблюдаться при лейкозе или серповидно-
клеточной анемии как вторичное поражение вследствие
инфильтрации пещеристых тел аномальными лейкоцитами или
эритроцитами
Атрофия яичек — один из признаков поражения печени при
некоторых заболеваниях системы крови, в частности при гемохро-
матозе. Увеличение яичек может указывать на их вовлечение
в лейкемический или лимфоматозный процесс или на развитие
злокачественной опухоли
ТАБЛИЦА 2-1. (Окончание)
Составляющие
анамнеза
Данные/признаки/симптомы
Патофизиологическая значимость
Женские
гениталии
Прямая кишка
Яичники
Неврологическое
исследование
Психический статус, черепно-мозговые
нервы, чувствительность, моторика,
координация
Лимфоматозная инфильтрация яичников может быть причиной их
увеличения. Маточные кровотечения — возможный признак
коагулопатии
Геморрой может быть симптомом портальной гипертензии,
связанной с поражением печени при многих гематологических
заболеваниях. Патология слизистой включает эпителиальные
новообразования, характерные для ВИЧ-инфекции, а также эк-
странодальные проявления лимфомы Ходжкина. Желудочно-
кишечные кровотечения наблюдаются при лимфоматозном
поражении желудочно-кишечного тракта, других первичных
злокачественных опухолях или коагулопатии
Многие типы неврологических нарушений обусловлены
заболеваниями крови. Нарушения коагуляции (геморрагические или тром-
ботические), злокачественные опухоли системы крови (прямая
инвазия, сдавление или опосредованное действие), метаболические
нарушения (пернициозная анемия или порфирия), инфекционные
осложнения, особенно у пациентов с ослабленным иммунитетом,
могут лежать в основе психиатрических проблем, патологии
черепно-мозговых нервов, расстройств чувствительности,
моторики, координации. Периферические нейропатии нередко
вызываются метаболическими или гематологическими заболеваниями,
такими как пернициозная анемия, порфирия, парапротеинемия и
близкими к ним состояниями (амилоидоз). Использование
лекарственных препаратов для лечения лейкозов и лимфом может
приводить к специфическим неврологическим проблемам. Например,
побочным действием винкристина и цисплатина является
развитие периферической нейропатии. Высокие дозы цитозина араби-
нозида могут стать причиной повреждения клеток Пуркинье в
мозжечке и расстройства координации
Глава 3
Эритроциты
Майкл Дж. Роуз, Нэнси Берлинер
Структура и функции эритроцита
Общие положения
Эритроцит является высокоспециализированной клеткой, основная задача
которой состоит в транспорте кислорода из легких в ткани и двуокиси углерода
(С02) — обратно в легкие. Клетка имеет форму двояковогнутого диска, что
обеспечивает наибольшую площадь поверхности газообмена. Диаметр эритроцита
составляет 8 мкм, однако особенности клеточного скелета и структуры мембраны
позволяют ему претерпевать значительную деформацию и проходить через
капилляры с просветом в 2-3 мкм. Такая способность к деформации обеспечивается
за счет взаимодействия между белками мембраны (сегмент 3 и гликофорин) и
цитоплазмы (спектрин, анкирин и белок 4.1) (рис. 3-1). Дефекты этих белков ведут
к морфологическим и функциональным нарушениям эритроцитов (табл. 3-1).
Зрелый эритроцит не имеет цитоплазматических органелл и ядра и поэтому
Рис. 3-1. Структура мембраны эритроцита. ГФА — гликофорин А; ГФС — гликофорин С. (Из:
Handin R. I., Stossel Т. P., Lux S. G. eds. Blood: Principles and Practice of Hematology, Philadelphia: J. B.
Lippincott, 1995: 1726.)
72
Глава 3. Эритроциты
ТАБЛ ИЦА 3-1. Наследственная патология мембраны эритроцита
Патологическое
состояние
Дефектный белок
Способ наследования
Сфероцитоз
Эллиптоцитоз
Пиропойкилоцитоз
Стоматоцитоз
Спектрин, анкирин
Сегмент 3, белок 4.2
Спектрин
Белок 4.1
Спектрин
Дефект проницаемости Na+
Аутосомно-доминантный
Рецессивный (редкий)
Аутосомно-доминантный
Рецессивный (редкий)
Рецессивный
Аутосомно-доминантный
не способен к синтезу белков и лшщдов, окислительному фосфорилированию
и поддержанию реакций цикла трикарбоновых кислот. Он получает большую
часть энергии через анаэробный путь Эмбдена-Мейергофа и сохраняет ее в виде
АТФ. В, зависимости от степени окислительной стимуляции через гексозомоно-
фосфатный шунт отводится соответствующее количество глюкозы с
образованием восстановленных соединений (глутатион и никотинамидадениндинуклеотид-
фосфат [НАДФН]) (рис. 3-2).
Приблизительно 98 % массы белков цитоплазмы эритроцита составляет
гемоглобин (НЬ), молекула которого связывает и транспортирует кислород.
Гемоглобин представляет собой гетеродимерный тетрамер, состоящий из двух цепей
глобина типа а и двух цепей другого типа (Р, у или б), соединенных с четырьмя
молекулами гема. Гем — это молекула протопорфирина IX, связанная с атомом
железа. Каждый тетрамер гемоглобина может обратимо связывать и
транспортировать не более четырех молекул кислорода.
Гексозомонофосфатный шунт
Г-6-ФДГ
Гликолиз
НАДФ <-
-> НАДФН
СНАД^^
► НАДН '
G-SH ч-
-> G-SS-G
2,3-ДФГ
Рис. 3-2. Пути метаболизма эритроцита. НАД — цикотинамидадениндинуклеотид (НАДН —
восстановленная форма); 2,3-ДФГ — 2,3-дифосфоглицериновая кислота; Г-6-ФДГ — глюкозо-6-фосфатде-
гидрогеназа; НАДФ — иикотинамидадениндинуклеотидфосфат (НАДФН — восстановленная
форма); G-SH, G-SS-G — восстановленная и окисленная формы глутатиона
Структура и функции эритроцита
73
ТАБЛИЦА 3-2. Гемоглобины человека
Типы гемоглобина Состав % содержания
А (основной)
А2 (минорный)
F (фетальный)
Пауэр 1
Гауэр 2
Портлэнд
а2р2
а262
а2у2
^e2
а2е2
&Y2
95-98 % гемоглобина взрослого человека
1,5-3,5 % гемоглобина взрослого человека
0,5-1 % гемоглобина взрослого человека
Эмбриональный гемоглобин
Эмбриональный гемоглобин
Эмбриональный гемоглобин
К нормальным типам гемоглобина относятся: НЬА (а2Рг — основной
гемоглобин взрослого человека), HbF (a2Y2 — фатальный гемоглобин) и НЬА2 (а2Рг —
минорный гемоглобин взрослого) (табл. 3-2). Смена гемоглобина F на гемоглобин А
происходит во время рождения ребенка; к 4-6 месяцу жизни уровень фетального
гемоглобина в крови составляет менее 1 % (рис. 3-3).
Хромосома 16
5' С а2 at 3'
МИД
ИИ ИЛ ИИ
£е2 &Y2 а2ег
,Гауэр 1 Портлэнд Гауэр 2 ,
i
Эмбрион
Место эритропоэза
Хромосома 11
5' е Gy
Eli ■■
a2Y2
Плод
I
Ау б з з
рщ рта щц
||ш| iiiiii iiiili
ишд| ишмш шшжи
a2Y2 Огбг а2Эг
, F А, А ,
I
Взрослый
1. Пре-а: и, &в2 (Гауэр 1)
2. Ранний а: (Дог (Гауэр 2)
3. Мало или отсутствует a: {zYS (Портлэнд)
4. Мало или отсутствует a: Y4 (Барт)
5. Присутствует а: а2у2 (нормальный Hb F)
6. Мало или отсутствует а (поздний): fc (Hb H)
7. Нормальный после рождения: сдог (Hb A)
9.0-варианты НЬА (пример: агЭг Hb S)
9. Нормальный: агб2 (Hb A2)
6 12 18 24 36 36 42 48
Рис. 3-3. (А) Кластеры глобинового гена на хромосомах 16 и И. У эмбриона, плода и взрослого
человека активируются разные гены. Различные цепи глобина синтезируются по отдельности, а
затем объединяются друг с другом и образуют те или иные гемоглобины. Известны два варианта у-це-
пи: с остатком глутаминовой кислоты (Gy) или остатком аланина (Ау) в позиции 136. (Из: Hoffbrand
А. V., Pettit J. E. Essential Hematology, 3rd ed. Cambridge, Mass.: Blackwell Scientific Publishing; 1993.)
(Б) Локализация гемопоэза и синтез гемоглобина в процессе онтогенеза. Петли объединяют
глобины — варианты нормы и патологии. (Из: Brown M. S. Fetal and Neonatal Erythropoesis in Developmental
and Neonatal Hematology. New York: Raven Press; 1988. Из: Handlin R. I., Stossel T. P., Lux S. E. (eds).
Blood: Principles and Practice of Hematology. Philadelphia: J. B. Lippincott, 1995.)
74
Глава 3. Эритроциты
40 80 120
р02ммрт. ст.
Рис. 3-4. Кривая диссоциации оксигемоглобина. Алкалоз, низкая температура и снижение уровня
2,3-ДФГ приводят к сдвигу кривой влево и повышению сродства НЬ к кислороду. Результатом
ацидоза, повышения температуры и/или уровня 2,3-ДФГ является сдвиг кривой вправо и снижение
сродства к кислороду. (Из: Isselbacher K.J. et al., eds. Harrison's Principles of Internal Medicine, 13th ed.
New York: McGraw-Hill, 1994:1719, Fig. 302-4.)
Кривая диссоциации оксигемоглобина иллюстрирует зависимость насыщения
гемоглобина кислородом от напряжения этого газа (рис. 3-4). Сигмоидальная
форма данной кривой обусловлена взаимодействием между субъединицами
тетрамера НЬ. Главным регулятором сродства гемоглобина к 02 является
промежуточный продукт гликолиза — 2,3-Дифосфоглицериновая кислота (2,3-ДФГ).
2,3-ДФГ снижает сродство НЬ к 02, что приводит к сдвигу кривой диссоциации
оксигемоглобина вправо и к усиленному поступлению 02 в ткани. Сдвиг кривой
вправо может быть также вызван увеличением температуры и рС02 или
снижением рН (рис. 3-4). Фетальный гемоглобин слабо связывается с 2,3-ДФГ и поэтому
характеризуется более высокой кислородсвязывающей способностью по
сравнению с НЬ А.
Гены гемоглобина
Каждая хромосома 16 содержит два практически идентичных гена а-глобина,
а хромосома 11 — подобную пару у-генов и по одной копии б- и Р-генов. Глобин
синтезируется только в эритроидных клетках и лишь в период созревания рети-
кулоцитов из пронормоцитов. На каждой стадии развития гены а-глобина
и других глобинов координированно экспрессируются. Это очень важный
момент: поскольку тетрамеры, состоящие из одинаковых цепей НЬ (например, а4
или р4), практически нерастворимы, то для сохранения растворимости тетрамера
НЬ необходим сбалансированный синтез разных цепей. Избыточное образование
одной цепи гемоглобина, происходящее, например, при талассемии, может
привести к преципитации белка в эритроците* повреждению клетки и ее
преждевременной элиминации клетками ретикулоэндотелиальной системы.
Клиническая патофизиология анемии 75
Бурстобразующая Колониеобразующая
эритроидная эритроидная
единица (БОЕ-Э) единица (КОЕ-Э) Нормоциты
Рис. 3-5. Эритропоэз. (Из: Isselbacher К. J. et al., eds. Harrisson's Principles of Internal Medicine} 13th ed.
New York: McGraw-Hill, 1994:1718, Fig. 302-1. Differentiation and morphologic maturation of eryhroid
cells.)
Эритропоэз
Исследования тканевых культур позволили выявить два вида наиболее ранних
эритроидных предшественников. Это бурстобразующая эритроидная единица
(БОЕ-Э) и продукт ее дифференциации — колониеобразующая эритроидная
единица (КОЕ-Э). Последняя дает начало проэритробласту — самому раннему
из эритроидных клеток-предшественников, морфологически различимых в
костном мозге. После 4-5 митотических делений и соответствующих
морфологических изменений проэритробласт становится зрелым безъядерным эритроцитом
(рис. 3-5), который циркулирует в периферической крови в течение 90-120 дней,
после чего удаляется селезенкой и другими структурами ретикулоэндотелиаль-
ной системы.
Фактором роста, необходимым для развития эритроидцых клеток, начиная
со стадии КОЕ-Э, является эритропоэтин — гормон, продуцируемый в перитубу-
лярных клетках почек. Его задача состоит в поддержании необходимого объема
(в соответствии с потребностями организма в кислороде) эритроцитарной массы.
Высвобождение эритропоэтина регулируется специальным механизмом и
зависит от содержания кислорода в тканях почек. >
Клиническая патофизиология анемии
Анемия — это уменьшение эритроцитарной массы. Поскольку объем крови
обычно поддерживается на постоянном уровне, степень анемии можно определить
либо на основании объема эритроцитов, выраженного в процентах по отношению
к общему объему крови (гематокрит [ГК]), либо на основании содержания
гемоглобина в крови. В норме эти показатели различны у мужчин и женщин, посколь-
76
Глава 3. Эритроциты
ку андрогены повышают как секрецию эритропоэтина, так и количество
костномозговых клеток-предшественников. При диагностике анемии необходимо также
учитывать, что на большой высоте над уровнем моря, где напряжение кислорода
ниже обычного, величины показателей красной крови возрастают.
Клинические проявления
Анемия является одним из наиболее важных признаков заболевания, поэтому
причину ее возникновения необходимо определять во всех случаях по объективным
и субъективным симптомам, отражающим также уровень гематокрита и скорость
прогрессирования болезни. У пациентов со слабо или умеренно выраженной
анемией яркая симптоматика часто отсутствует. При быстром развитии анемии
компенсаторные механизмы не успевают развить достаточную мощность:
соответственно, клинические проявления, как правило, значительнее, чем при заболевании
той же степени тяжести, но развивающемся постепенно. Больные с анемией могут
жаловаться на усталость, одышку, сильное сердцебиение и плохую переносимость
физической нагрузки. Возможны головокружения, головная боль или шум в ушах.
Тяжелая анемия вызывает анорексию, расстройство пищеварения,
раздражительность, нарушения сна и затруднение концентрации внимания. У женщин возможно
нарушение менструального цикла, у мужчин — импотенция и/или утрата либидо.
Кроме того, анемия может стать причиной возникновения коронарной
недостаточности и, как следствие, стенокардии и/или инфаркта миокарда.
Бледность — основной симптом анемии, легче всего .ее можно определить
по цвету слизистой ротовой полости, ногтевого ложа, конъюнктивы, складок
ладоней. К другим физикальным признакам анемии относятся тахикардия,
усиленный сердечный толчок, шум потока крови. Анемия, вызванная гемолизом и гемо-
глобинопатиями, может привести к желтухе и спленомегалии (глава 2).
Лабораторные исследования
Мазок периферической крови
Ключевой этап диагностики любого вида анемии — оценка морфологии
эритроцитов (табл. 3-3). При микроангиопатических гемолитических анемиях и
травматическом гемолизе, обусловленном использованием искусственных клапанов
сердца, происходит фрагментация эритроцитов. При аутоиммунном гемолизе
и наследственном сфероцитозе появляются сфероциты. Для заболеваний печени,
талассемии и гемоглобинопатии С характерны мишеневидные эритроциты.
Ядерные эритроциты и эритроциты в форме капли (дакриоциты) обнаруживаются при
миелофиброзе и миелофтизе (следствие опухолевого поражения костного мозга
при карциноме, лимфоме, фиброзе и/или гранулематозе). При малярии и бабези-
озе определяются внутриклеточные паразиты (табл. 3-3).
Исследование лейкоцитов и тромбоцитов в мазке периферической крови
помогает выяснить этиологию анемии. При мегалобластных анемиях наблюдаются
гранулоциты с гиперсегментированными ядрами (табл. 4-1). Анемия может быть
первым проявлением острого или хронического лейкоза, диагностика которых
обычно основывается на обнаружении аномальных лейкоцитов в
периферической крови. Тромбоцитопения и лейкопения указывают на патологию костного
мозга, затрагивающую все три клеточные линии.
Клиническая патофизиология анемии
77
Подсчет ретикулоцитов
Анемия может быть результатом первичного нарушения продукции эритроцитов,
их ускоренного разрушения или острой not ери крови. Для выяснения причин
возникновения анемии важно определить реакцию костного мозга. В норме
примерно 1 % эритроцитов в организме ежедневно заменяется юными клетками,
которые называются ретикулоцитами. Поскольку эти клетки все еще содержат по-
лирибосомальную РНК, их можно определить с помощью специального
окрашивания мазка периферической крови. Краситель Райта, содержащий эозин
и метиленовый синий, окрашивает ретикулоциты в серо-пурпурный цвет. Под
действием более специфических красителей (новый метиленовый синий,
бриллиантовый крезиловый синий) происходит преципитация РНК и полирибосомы
ретикулоцитов проявляются в виде сетчатого (ретикулярного) узора, отсюда и
название этих клеток (табл. 3-3).
Содержание ретикулоцитов обычно выражают как процент от общего числа
клеток красной крови. Данный показатель может повышаться как вследствие
абсолютного увеличения количества ретикулоцитов в крови, так и вследствие
сокращения массы циркулирующих эритроцитов (анемия). Поэтому при анемии
используют нормализованное количество ретикулоцитов, вычисляемое по формуле:
Нормализованное количество ретикулоцитов =
ГК пациента
= % ретикулоцитов х
ГК в норме
Увеличение нормализованного количества ретикулоцитов свидетельствует о
стимуляции костного мозга эритропоэтином, что способствует не только повышению
продукции ретикулоцитов, но и их более раннему поступлению в кровь. В крови
оказываются незрелые ретикулоциты, которые циркулируют там более 1 дня. Срок
зависит от времени, требующегося для созревания ретикулоцитов и утраты ими
РНК. Он коррелирует с уровнем гематокрита: при ГК, равном 45 %, ретикулоцит
проведет в периферической крови только 1 день; при 35 %, 25 % или 15 % — 1,5,2 и
2,5 суток соответственно. Индекс продукции ретикулоцитов позволяет уточнить
время пребывания ретикулоцитов в периферической крови по следующей формуле:
Индекс продукции ретикулоцитов =
_ Нормализованное количество ретикулоцитов
Время созревания ретикулоцитов
Другой способ оценки продукции эритроцитов состоит в подсчете абсолютного
количества ретикулоцитов, равного произведению процентного содержания
ретикулоцитов и числа эритроцитов. В норме эта величина составляет 50 000-
60 000/мкл. Подсчет абсолютного количества ретикулоцитов проводится во
многих лабораториях, и, возможно, он информативнее стандартных показателей.
Если причиной развития анемии являются кровопотеря или деструкция
клеток крови вне костного мозга, то секреция эритропоэтина возрастает и
относительное количество ретикулоцитов поднимается выше нормального уровня (1 %), а
абсолютное число ретикулоцитов превышает величину 100 000/мкл. Отсутствие
соответствующего ретикулоцитоза при анемии указывает на нарушение
продукции эритроцитов в костном мозге из-за недостаточности питания, остановки
созревания и/или заболеваний костного мозга.
ТАБЛИЦА 3-3. Аномалии морфологии эритроцитов и их клиническое значение
Название клетки Внешний вид (микроскопия)
Описание
Клиническая значимость
1.Норма
Эритроцит
Полихромато-
фильная клетка
2. Патология формы и размеров
Макроцит
(круглый)
Круглой формы, 8 мкм в диаметре, бледнее Выявляются у здоровых людей и
в центре при нормохромной анемии
Большая клетка серо-фиолетового цвета;
внешне напоминает матовое стекло (поли-
хромазия/полихроматофилия). Нет
бледного участка в центре. СОК увеличен
Большая клетка. Бледный участок в центре
выражен слабо. СОК увеличен
Присутствуют в норме (1 % от
числа эритроцитов); увеличение
количества свидетельствует о
реакции костного мозга на анемию
Полихромазия обусловлена
наличием в цитоплазме
полирибосом, продуцирующих
гемоглобин. При суправитальном
окрашивании проявляется
сетчатый (ретикулярный) узор,
поэтому такие клетки называют
ретикулоцитами (см. ниже)
Наблюдаются при заболеваниях
печени (особенно вызванных
алкоголем) и после спленэкто-
мии. При виде сбоку клетка
кажется тонкой. Изменено
соотношение лецитин/холестерин
в клеточной мембране
Большая овальная клетка без центрального
бледно окрашенного участка. СОК увеличен
Небольшая клетка (СОК уменьшен); гипо-
хромна при дефиците железа. Усилена
бледность в центре
Выявляются в периферической
крови у пациентов с мегалобла-
стными анемиями, а также при
многих других патологических
состояниях
Обнаруживаются при железоде-
фицитной анемии и талассемии
Может быть микро-, нормо- или макроци-
тарным. Бледная область в центре
отсутствует. Обычен микроцит с уменьшением
СОК и повышением СКГ
Шипы одинаковых размеров,
распределенные равномерно по поверхности
эритроцита
Определяются при
наследственном сфероцитозе или
любой гемолитической анемии,
при которой мембрана
эритроцита удаляется селезенкой или
РЭС, а количество гемоглобина
остается постоянным
• Артефакт
• Уремия
• Недостаточность пируватки-
назы
• Трансфузия крови,
содержащей старые эритроциты
• Рак желудка
• Пептическая язва,
осложненная кровотечением
• Гипофосфатемия
• Гипомагниемия
ТАБЛИЦА 3-3. (Продолжение)
Название клетки Внешний вид (микроскопия)
Описание
Клиническая значимость
Акантоцит
(листоподобная
клетка)
Шпорообразная
клетка
"Надкусанная"
клетка (дегма-
цит)
Пузырчатая
клетка
Выпячивания различной величины,
расположенные на разных расстояниях друг
от друга по поверхности клетки
Клетка выглядит так, будто ее надкусили
Клетка выглядит так, как будто на ее
поверхности имеется пузырек или волдырь
• Абеталипопротеинемия
• Алкогольное поражение печени
• Состояние после удаления
селезенки
• Нарушение всасывания
• Недостаточность Г-6-ФДГ
• Нестабильность гемоглобина
Эти клетки образуются при
удалении телец Гейнца РЭС с
частью мембраны и гемоглобина
•.Иммунная гемолитическая
анемия
Механизм образования неясен
Пойкилоцит
Клетка имеет причудливую форму.
Выявляются фрагменты
Ожоги
Наследственный пойкилоци-
тоз
Миелофиброз
Талассемия
Недостаток железа
Мегалобластная анемия
Миелодисплазия
Клетка овальной или удлиненной формы.
Бледность в центре не видна. Аномалии
мембраны или гемоглобина приводят к
изменению формы клетки
Наследственный эллиптоцитоз
Талассемия
Недостаток железа
Мегалобластная анемия
Чашеобразный эритроцит
Внешний вид клетки является следствием
артефакта окраски по Райту. Если смотреть
на клетку сбоку, то она похожа на две
соединенные мексиканские шляпы
На самом деле клетки похожи на колокол
Клетки похожи на каски, треуголки, осколки
• Наследственный сфероцитоз
• Наследственный стоматоцитоз
• Новообразования
• Алкоголизм
• Цирроз
• Обструктивные заболевания
печени
• Дефекты NaVfO-насоса
мембраны
• Лекарственные препараты
• Заболевания печени
• Гемоглобинопатии С, D и Е
• Талассемия
• Железодефицитная анемия
• Патологическое состояние
после удаления селезенки
Осмотическая резистентность
клеток повышена, что
обусловлено утолщением мембраны
• Микроангиопатическая
гемолитическая анемия любой
этиологии
ТАБЛИЦА 3-3. (Продолжение)
Название клетки Внешний вид (микроскопия)
Описание
Клиническая значимость
Серповидная
клетка (дрепа-
ноцит)
Слезоподобная
клетка (дакрио-
цит)
Клетки похожи на серп или на листья
остролиста
Клетки напоминают каплю или головастика
3. Внутриклеточные включения в эритроциты при окраске по Райту
Ядро ^^m^t Это ортохроматофильный нормобласт.
Последняя стадия перед удалением ядра
РЭС и превращением клетки в ретикулоцит
Тельце Хауэлла-
Жолли
Остаток ядра после удаления его РЭС.
Можно дифференцировать от тромбоцита,
лежащего поверх эритроцита, поскольку
в последнем случае вокруг тромбоцита
наблюдается ореол (см. выше)
• Гемоглобин SS или S в
сочетании с Hb D, С, Мемфис, а также
талассемия
• Миелофиброз
• Миелоидная метаплазия
• Анемия при миелофтизе
(ослабление или нарушение
функции костного мозга при
росте опухоли, гранулеме,
лимфоме или фиброзе)
• Талассемии
Наблюдаются в условиях
выраженного анемического криза.
Обычно при интенсивном
гемолизе или желудочно-кишечном
кровотечении, особенно при ги-
поксемии. Встречаются и при
миелофтизе, когда происходят
лейкоэритробластные
изменения
Выявляются при отсутствии
селезенки, а также при
интенсивном гемолизе и "перегрузке"
РЭС. Кроме того,
обнаруживаются при мегалобластной
анемии
Рассеянные гранулы синего цвета,
связанные с рибосомальной преципитацией;
могут быть довольно крупными
Темно-синие гранулы трехатомного
железа. Если они появляются в ядерных
эритроцитах, то последние называются сидеро-
бластами, а если окружают ядро,—
кольцевыми сидеробластами
Полные или неполные кольца либо
восьмеркообразные фигуры. Могут быть
представлены гранулами
красновато-синего цвета. Образуются из митотических
нитей или ядерной мембраны
Шестиугольные или ромбовидные
кристаллы при SC-гемоглобинопатии. Могут
быть искривлены, неправильной формы,
с более светлой зоной между кристаллами
Интоксикация свинцом или
тяжелыми металлами
Талассемия
Алкогольная интоксикация
Цитотоксическое действие
лекарственных препаратов
Гемолитическая анемия
Сидеробластная анемия
Гипоспленизм
s
i
S
X
CD
О)
20
3
■в"
s
со
S
§
о
-I
г°
ш
I
CD
S
Is
Мегалобластная анемия
С-гемоглобинопатия
00
I со
ТАБЛИЦА 3-3. (Окончание)
Название клетки Внешний вид (микроскопия)
Описание
Клиническая значимость
Включения
при
малярии
Включения
при бабезиозе
Обычно наблюдаются ранние кольцевидные
формы. Они синеватого цвета и могут иметь
на концах красную точку (точки). P. falciparum
распознаются по характерной конфигурации
в виде наушников и бананообразному мак-
рогаметоциту. При инвазии P. vivax и P. ovale
наблюдаются гранулы Шюффнера
Паразиты похожи на малярийный
плазмодий. Можно наблюдать фигуру в виде
"мальтийского креста"
4. Внутриклеточные включения в эритроцитах при суправитальнои окраске
Ретикулоцит Окраска новым метиленовым синим
позволяет увидеть зернистые диффузно-базо-
фильные нитевидные структуры
Тельца Гейнца
После инкубирования с ацетилфенилгид-
разином и окрашивания кристаллическим
фиолетовым гемоглобин денатурируется и
выглядит как синие округлые преципитаты.
В нормальных клетках можно увидеть от
одного до четырех телец Гейнца, а при
патологических состояниях — 5 и более
Малярия, вызываемая
Plasmodium falciparum, vivax, mala-
riae и ovale
Инвазия Babesia microti
В норме составляют около 1 %
от числа эритроцитов;
увеличение содержания
свидетельствует о реакции костного мозга
на анемию
Недостаточность Г-6-ФДГ
Нестабильные гемоглобины
и другие наследственные
гемолитические анемии,
вызванные применением
препаратов-окислителей. Могут
наблюдаться "откусанные"
клетки, образующиеся после
удаления РЭС телец Гейнца
Клиническая патофизиология анемии
85
Средний объем эритроцита
Приблизительно средний объем эритроцита (СОК — средний объем клетки)
можно определить при анализе мазка периферической крови. СОК измеряют
непосредственно с помощью автоматического цитометра или подсчитывают путем
деления величины гематокрита на число эритроцитов (приложение 1). Этот
параметр особенно важен для оценки гипопролиферативных анемий (анемии с
низким содержанием ретикулоцитов), которые подразделяют в соответствии с
величиной СОК на три категории: микроцитарная (СОК < 80 мкм3), нормоцитарная
(80 мкм3 < СОК < 100 мкм3) и макроцитарная (СОК > 100 мкм3) (табл. 3-4).
Еще два показателя: среднее содержание гемоглобина в эритроците (ССГ =
содержание гемоглобина/число эритроцитов) и средняя концентрация
гемоглобина в эритроците (СКГ = гемоглобин/гематокрит) — также определяются с
помощью автоматических цитометров, хотя их информативность не столь высока,
как СОК. ССГ обычно коррелирует с величиной СОК. СКГ понижена при
большинстве микроцитарных анемий, связанных с дефицитом железа, и повышена
при наследственной или аутоиммунной гемолитической анемии.
Если при первичном обследовании больного с анемией обнаружена ретикуло-
цитопения, то следует предположить недостаточность эритропоэза или наличие
гипопролиферативной анемии (табл. 3-4). Исследование костного мозга может
способствовать выявлению миелодиспластического процесса, лейкоза или
инфильтрации костного мозга солидной опухолью (наиболее часто — молочной
железы, предстательной железы или легкого). При исследовании костного мозга ус-
ТАБЛ ИЦА 3-4. Причины развития гипопролиферативной анемии
Микроцитарная
Железодефицитная анемия
Талассемия
Сидеробластная анемия
Отравление свинцом
(Анемия при хронических заболеваниях)
Нормоцитарная
Недостаточность костного мозга (апластическая анемия)
Миелофтиз
Анемия при хронических заболеваниях
Эндокринопатии
Ранняя стадия дефицита железа
"Смешанные" анемии
Макроцитарная
Мегалобластная
Недостаточность витамина В12
Недостаточность фолата
Миелодисплазия
Анемия вследствие приема лекарственных препаратов
Немегалобластная макроцитарная анемия
Ретикулоцитоз
Болезни печени
(Гипотиреоз)
86
Глава 3. Эритроциты
танавливаются также мегалобластные изменения. Инфекции, вызванные такими
возбудителями, как Mycobacterium tuberculosis, Mycobacterium avium intracellular,
цитомегаловирус и гистоплазма, можно диагностировать при культивировании
костномозгового образца или на основании типичной морфологии аспирата или
биоптата костного мозга. С помощью специфического окрашивания удается
подтвердить диагноз дефицита железа, а также продемонстрировать наличие
кольцевых сидеробластов, являющихся одним из признаков миелодисплазии или
врожденной патологии. При оценке анемий, сопровождающихся ретикулоцитозом,
значимость исследования костного мозга невелика, поскольку адекватная
реакция кроветворения на анемию обычно исключает интрамедуллярный дефект
продукции эритроцитов.
Дифференциальная диагностика
гипопролиферативной анемии
Микроцитарные анемии
Дифференциальная диагностика микроцитоза (табл. 3-4) включает железодефи-
цитную анемию, талассемию, отравление свинцом, сидеробластную анемию
и анемию, связанную с хроническим заболеванием. Несмотря на то, что анемия
при хронической патологии может быть микроцитарной, все же гораздо чаще она
носит нормоцитарный характер. Наиболее типичной причиной развития
микроцитоза является дефицит железа.
Железодефицитная анемия
N
Метаболизм железа. В пищевых продуктах железо присутствует или в виде
составной части гема, или как негемовое железо. Хотя железо гема обычно
составляет меньшую долю этого элемента, поступающего с пищей, оно достаточно хорошо
всасывается (в норме примерно 20-30 %). Негемовое железо всасывается хуже
(менее 5 %), а при одновременном нахождении в пище таннинов и фосфатов —
еще меньше. Несколько увеличивает абсорбцию негемового железа аскорбиновая
кислота.
Всасывание железа регулируется главным образом клетками слизистой
оболочки проксимальной части тонкой кишки, но механизм этого процесса
недостаточно понятен. Она возрастает при его дефиците, а также у пациентов с
неэффективным эритропоэзом. В последнем случае при тяжелой форме нарушения
эритропоэза, характерной для некоторых видов талассемии, несмотря на высокий
уровень содержания железа в организме, его всасывание повышено. Вследствие
проводимых в этих условиях переливаний крови часто наступает клинически
значимая перегрузка железом.
Транспорт железа осуществляется с помощью плазменного белка — трансфер-
рина, который связывается со специфическим рецептором мембраны. Комплекс
рецептор-трансферрин-железо поглощается развивающимися эритроцитами,
после чего железо высвобождается, а соединение трансферрин-трансферрино-
вый рецептор завершает цикл на клеточной мембране.
Ферритин является внутриклеточным белком, депонирующим в нетоксичной
форме железо, которое мобилизуется по мере необходимости. Каждая молекула
Дифференциальная диагностика гипопролиферативной анемии
87
ТАБЛИЦА 3-5. Распределение железа
Локализация Содержание (мг/кг)
Гемоглобин 30 (мужчины); 27 (женщины)
Гемсодержащие и не содержащие тема ферменты 2
Миоглобин 5
Ферритин и гемосидерин 13 (мужчины); 6 (женщины)
Всего 50 (мужчины); 40 (женщины)
ферритина может связывать 4500 атомов железа, но в нормальных условиях
содержит примерно половину этого количества. Некоторая часть ферритина
превращается в гемосидерин — нерастворимое в воде соединение, хранящее железо
в большем количестве, но в менее доступной форме. В организме мужчин и
женщин содержится 50 и 40 мг/кг железа соответственно. От 28 до 31 мг/кг железа
входит в состав гемоглобина, 4-5 мг/кг — в состав миоглобина, 12 мг/кг —
ферритина и гемосидерина, а остальные — в состав гемсодержащих и других ферментов
(табл. 3-5). Основными депо железа являются печень (гепатоциты и макрофаги),
костный мозг, селезенка и мышцы.
Внутри клетки специфический Fe-зависимый белок связывается с регулятор-
ным участком на мРНК и координирует экспрессию ферритина, трасферрина
и трансферринового рецептора. При низком внутриклеточном уровне железа он
инициирует синтез трасферринового рецептора и подавляет синтез ферритина.
Этиология железодефицитной анемии. Наиболее распространена железо-
дефицитная анемия (ЖДА). Для большинства мужчин, а также для женщин в
постменопаузе причиной дефицита железа оказывается кровопотеря (табл. 3-6),
обусловленная кровотечением из желудочно-кишечного тракта (ЖКТ). При
обнаружении дефицита железа каждый мужчина, а также женщина в период
постменопаузы должны пройти полное обследование ЖКТ на предмет поиска
возможной патологии, особенно опухолевых поражений. Примерно у 15 % пациентов
причину кровотечения из ЖКТ установить не удается. У женщин детородного
ТАБЛИЦА 3-6. Причины дефицита железа
Кровопотеря
Желудочно-кишечная
При менструации и родах
Легочная (гемофтиз, гемосидероз легких)
Через мочеполовой тракт (заболевание почек, урологическое заболевание,
гемоглобинурия)
Нарушение всасывания
Резекция желудка
Недостаточность поджелудочной железы
Глютеновая энтеропатия и спру
Болезнь Крона и синдром короткого кишечника
Повышение потребности
Быстрый рост (недоношенные новорожденные, дети, подростки)
Беременность и лактация
Бедная железом диета
88
Глава 3. Эритроциты
возраста таковыми в основном являются вагинальное кровотечение,
беременность и лактация. Вследствие менструации ежемесячная потеря железа
составляет около 15 мг. Примерно 900 мг железа теряется в период нормальной
беременности, поскольку транспортируется в плаценту и к плоду, и при родах в результате
родового кровотечения. К более редким причинам потери железа крови относятся
возвратный гемофтиз, пневмосидероз, синдром Гудпасчера и гематурия.
Дефицит железа может развиваться и без потери крови, например в период
интенсивного роста детей потребность в данном элементе превышает его
поступление. Подобная ситуация характерна для новорожденных, особенно
недоношенных, а также для подростков (главным образом девушек в период становления
менструального цикла). Другая причина дефицита железа — нарушение
всасывания, которое наблюдается у пациентов, подвергшихся хирургическому
вмешательству на желудке, или у больных с синдромами кишечной мальабсорбции.
Клинические проявления. Дефицит железа приводит к развитию обычных для
всех анемий объективных и субъективных симптомов: бледность, усиленное
сердцебиение, шум в ушах, головная боль и слабость. К редким, но более
специфическим симптомам относятся: извращение аппетита; странное желание съесть такие
вещества, как крахмал, лед, глина (амилофагия, пагофагия, геофагия); койлони-
хия и голубой цвет склер. Недостаток железа в неэритроидных тканях может
стать причиной глоссита, ангулярного стоматита и эзофагита (глава 2).
Лабораторное обследование. Хотя классическая ЖДА проявляется как гипо-
хромная микроцитарная анемия, у большинства пациентов в начале заболевания
наблюдается нормохромная анемия. Анемия обычно предшествует микроцитозу,
а гипохромия относится к наиболее поздним проявлениям прогрессирующей
ЖДА. Причина сохранения уровня СКГ за счет эритроцитарной массы вплоть
до поздних стадий ЖДА неизвестна. Анемия часто сопровождается реактивным
тромбоцитозом. При тяжелой ЖДА в мазке периферической крови можно
наблюдать микроцитоз с бледными, гипохромными эритроцитами, а также одиночные
удлиненные "карандашеподобные" клетки и мишеневидные клетки.
Запасы железа можно определить непрямым методом, измеряя содержание
ферритина, железа и степень насыщения трансферрина (железосвязывающая
способность) в крови. Печень продуцирует ферритин пропорционально
концентрации доступного железа, и небольшое определенное количество этого белка сек-
ретируется в плазму. Таким образом, содержание ферритина в плазме является
важным показателем запасов железа в организме (концентрация < 12 нг/мл, как
правило, свидетельствует о наличии дефицита железа). Но следует помнить, что
гипотиреоз и дефицит аскорбиновой кислоты также иногда снижают содержание
ферритина в плазме. Кроме того, и нормальный уровень ферритина не исключает
дефицита железа, поскольку ферритин относится к острофазовым реактантам;
его количество может увеличиваться при высокой температуре тела, инфекции,
воспалительном процессе, поражении печени, злокачественных новообразованиях,
гемолизе, неэффективном эритропоэзе. Однако подобные состояния сами по себе
поднимают уровень ферритина до 50-100 нг/мл; при более высоком показателе
дефицит железа обычно исключается.
Запасы железа можно определить и по степени насыщения трансферрина —
соотношению содержания железа в плазме и концентрации трансферрина
(железо/общая железосвязывающая способность х 100 %). В норме это соотношение
Дифференциальная диагностика гипопролиферативной анемии
89
равно по меньшей мере 20 %; дефицит железа уменьшает его, снижая уровень
железа и повышая концентрацию трансферрина в плазме. При хронических
патологиях, таких как инфекционный или воспалительный процесс, злокачественное
новообразование или болезнь печени, уровень железа и железосвязывающая
способность падают, а соотношение между ними сохраняется на уровне выше 20 %
при отсутствии дефицита железа (рис. 3-6).
Определение уровня ферритина и степени насыщения трансферрина —
непрямые методы измерения запасов железа, поэтому окончательный диагноз
дефицита железа можно поставить только после железоспецифической окраски аспирата
или биоптата костного мозга. Наличие в костном мозге железа исключает его
дефицит, а полное отсутствие позволяет поставить диагноз ЖДА.
Терапия. Общий дефицит железа в организме можно определить по следующей
формуле:
Железо (мг) = (НЬ в норме - НЬ больного) х масса тела (кг) х 2,21 + 1000.
Именно такая величина необходима для коррекции анемии и востановления
запасов железа на 1000 мг. Методом выбора является пероральный прием
железосодержащих лекарственных препаратов. Переливаний крови обычно удается
избежать, если у больного не возникла ишемия миокарда или сердечная
недостаточность. Если гемотрайсфузии необходимы пожилым больным с длительной
анемией, их следует назначать с большой осторожностью, поскольку у таких
пациентов обычно увеличен объем плазмы и дальнейшее его увеличение может
привести к сердечной недостаточности.
Перорально железо применяется в виде его солей, как правило, сульфата
железа. Пациенты должны получать 150-200 мг железа/сут за 3-4 приема. Примерно
у 20 % больных развивается понос или запор, которые купируют симптоматичес-
500
Норма Дефицит Поздние Инфекция, Гемо-
железа сроки воспаление, хроматоз,
бере- злока- гемо-
менности чественное сидероз
новообразование
Рис. 3-6. Уровень железа и железосвязывающая способность сыворотки крови при различных
клинических состояниях. (По: Isselbacher K.J. et al. eds. Harrison's Principles of Internal Medicine, 13th ed.
New York: McGraw-Hill, 1994: 1722, Fig. 303-2. Serum iron and total iron binding capacity in various
disorders.)
90
Глава 3. Эритроциты
кими средствами. Признаки раздражения желудка, такие как тошнота и
ощущение дискомфорта в эпигастральной области, минимизируют, принимая
препараты железа в процессе еды или снижая их дозу. Постепенное повышение дозы
часто помогает избежать возникновения этих симптомов.
Парентеральное введение железа показано только пациентам с расстройством
всасывания, небольшой группе пациентов с непереносимостью энтеральных
препаратов, а также больным с хроническими кровотечениями, которые не могут
удовлетворить потребность в железе лишь пероральным путем. Основное
опасное последствие парентеральной терапии — развитие анафилаксии, которая
наблюдается у 1 % больных (чаще у женщин с коллагенозами) и, по-видимому, не
связана с дозой. Несмотря на проводимое лечение, анафилактический шок может
оказаться фатальным.
У больных ЖДА повышение числа ретикулоцитов следует ожидать через 3-
4 дня после начала железотерапии, а пик ретикулоцитоза — на 10-й день. Через
3 недели после начала терапии концентрация гемоглобина возрастает примерно
на 20 г/л. Назначение железосодержащих препаратов обычно продолжается 4-
6 мес или до тех пор, пока концентрация ферритина не превысит 50 нг/мл.
Синдромы талассемии
Талассемия относится к гетерогенной группе наследственных анемий, для
которых характерно нарушение синтеза одной или нескольких субъединиц глобина.
При а- или Р-талассемии снижается или вообще отсутствует синтез
соответственно а- или Р-глобина. Диагноз талассемии основывается на определении
количества гемоглобина и глобиновых фракций с помощью электрофореза (глава 11).
Тяжелые талассемические синдромы связаны с выраженной гемолитической
анемией и диагностируются в начале жизни. Вместе с тем, легкая степень талассемии
вызывает только умеренную микроцитарную анемию и незначительный гемолиз
(или отсутствие такового). Подобные формы талассемии часто неверно
диагностируют как ЖДА из-за низкого СОК.
Наиболее часто талассемии наблюдаются в эндемичных по малярии районах
земного шара (Средиземноморье, Аравийский полуостров, Турция, Иран, Индия,
Юго-Восточная Азия). Полагают, что это обусловлено относительной
устойчивостью к возбудителям малярии эритроцитов гетерозиготных по талассемии лиц.
Р-Талассемия. Р-Талассемия обусловлена рядом мутаций в локусе Р-глобина
на хромосоме 11, которые нарушают синтез Р-глобиновой цепи. Описано более
100 мутаций, приводящих к блокаде разных стадий экспрессии гена, включая
транскрипцию, процессинг мРНК и трансляцию. Промоторные мутации,
ограничивающие транскрипцию мРНК, и мутации, нарушающие сплайсинг мРНК,
обычно снижают синтез Р-цепи (р+-талассемия), в то время как нонсенс-мутации
в зоне кодирования, вызывающие преждевременную остановку синтеза Р-глоби-
новых цепей, приводят к полному отсутствию последних (р°-талассемия).
Патогенез Р-талассемии связан как с неспособностью синтезировать адекватное
количество нормального гемоглобина, так и с наличием относительно
нерастворимых тетрамеров а-цепи, которые образуются из-за недостаточного числа Р-цепей.
Из-за неадекватного синтеза гемоглобина возникает гипохромная микроцитарная
анемия, а вследствие несбалансированного накопления а-глобиновых цепей
образуются а4-тетрамеры, которые преципитируют в развивающихся и зрелых эритро-
Дифференциальная диагностика гипопролиферативной анемии
91
цитах. Появление внутриклеточных преципитатов гемоглобина заставляет ретику-
лоэндотелиальную систему начать их удаление из эритроцитов, в процессе чего
клетки повреждаются, сокращается продолжительность их жизни и как результат
разрушаются интрамедуллярные эритроциты и наблюдается гемолиз. При
тяжелой форме талассемического синдрома гемолиз приводит к интенсивной эритроид-
ной гиперплазии и значительному расширению объема зон кроветворения, что
в свою очередь вызывает аномалии скелета. Кроме того, неэффективный эритропо-
эз (деструкция клеток-предшественников эритроцитов в костном мозге)
индуцирует повышенное всасывание железа, поэтому даже у больных талассемией, не
получавших гемотрансфузии, может развиваться патологическая перегрузка железом.
Клинические синдромы, связанные с р-талассемией, зависят от тяжести
генетического поражения и степени дефицита Р-глобиновых цепей (табл. 3-7).
Большая Р-талассемия, или анемия Кули, обычно возникает в результате гомозигот-
ности по аллелю Р°-талассемии. У больных с промежуточной Р-талассемией
имеет место значительная анемия, однако постоянная трансфузионная терапия
не требуется. Обычно пациенты наследуют две р-талассемические мутации: одну
слабую и одну тяжелую. Малая Р-талассемия возникает как следствие единичной
Р-талассемической мутации только одной хромосомы из пары И. У таких
больных, как правило, наблюдается бессимптомный микроцитоз и гипохромйя, и их
часто неправильно лечат препаратами железа, считая доказанным наличие
дефицита железа. Диагноз Р-талассемии ставят на основании обнаружения анемии,
микроцитоза, относительно большого числа эритроцитов и нормального уровня
железа. При исследовании мазка периферической крови можно обнаружить
причудливой формы эритроциты, ядерные эритроциты, мишеневидные и слезопо-
добные клетки, различные включения (табл. 3-3). При гетерозиготной (3-талассе-
мии все эти изменения могут быть выражены очень слабо. Электрофорез
гемоглобина выявляет компенсаторное повышение уровня гемоглобинов А2 и F.
ТАБЛИЦА 3-7. Синдромы талассемии
Синдром
Генетический дефект
Клинический синдром
а-Талассемия
Скрытый носитель
Слабые признаки
Гемоглобин Н
Водянка плода
/?-Талассемия
Малая
Промежуточная
Большая
a-/aa
a-la- или -/aa
~/a-
--/--
0*Ьа,-мутация
Нет проявлений
Умеренная микроцитарная анемия
Умеренная анемия и гемолиз; нет
необходимости в проведении
трансфузий
Дяжелая анемия, водянка;
внутриутробная гибель плода или
смерть при родах
Умеренная микроцитарная анемия
Сочетание j3°- и /Г-мутаций Не зависит от трансфузии;
перегрузка железом
Гомозигота по )3° Анемия Кули — тяжелый гемолиз,
неэффективный эритропоэз,
зависимость от трансфузии,
аномалии скелета, гепатоспленоме-
галия, перегрузка железом
92
Глава 3. Эритроциты
а-Талассемия. Две почти идентичные копии гена а-глобина находятся на
хромосоме 16. В 80-85 % случаев а-талассемии происходит потеря одного или нескольких
из этих четырех генов. У остальных больных эти гены сохраняются, но не
функционируют. Клинические проявления а-талассемии коррелируют со степенью
нарушения синтеза а-глобиновой цепи (табл. 3-7), однако они обычно выражены слабее,
чем при (3-талассемии. Это объясняется двумя причинами. Во-первых, наличие
четырех а-глобиновых генов способствует образованию адекватного количества
а-цепей до тех пор, пока не утрачиваются 3 или 4 гена. Значительный дисбаланс
гемоглобиновых цепей возникает только в том случае, если поражаются три из
четырех генов. Во-вторых, агрегаты Р-цепей (Р4-тетрамеры образуются при
недостаточности а-цепей) более растворимы, чем а4-тетрамеры, и поэтому даже у больных
с существенно нарушенным синтезом а-глобина при а-талассемии гемолиз гораздо
слабее, а эритропоэз более эффективен, чем при Р-талассемии.
Утрата или дисфункция одного из четырех а-генов клинически незначима
и обычно выявляется только у родителей детей с Н-гемоглобинопатией (см.
ниже). Размер эритроцитов, содержание НЬА2 и F нормальны. У больных с
потерей двух а-глобиновых генов на одной и той же хромосоме (цис-форма, a-thal 1)
или на разных хромосомах (транс-форма, a-thal 2) также отсутствует
симптоматика, но имеют место микроцитоз и гипохромия. У жителей Азии и
Средиземноморья, как правило, встречаются и a-thal 1-, и a-thal 2-варианты, в то время как
у африканцев только a-thal 2 (рис. 3-7).
Утрата или дисфункция трех а-глобиновых генов приводит к Н-гемоглобино-
патии. Гемоглобин Н представляет собой р4-тетрамеры, образующиеся при
избытке р-цепей, и выявляется при электрофорезе в геле как наиболее подвижная
фракция. НЬН преципитирует в основном в зрелых эритроцитах, что сопровождается
умеренной гемолитической анемией и некоторым нарушением эритропоэза.
У пациентов с гомозиготной а-талассемией 1 отсутствуют нормальные а-гло-
биновые гены (поражены все четыре гена, по два на каждой хромосоме) и, таким
образом, не продуцируется функциональный гемоглобин, за исключением
эмбриональной стадии, на которой синтезируются а-подобные £-цепи. Свободный
Р-глобин образует тетрамеры, называемые гемоглобином Барт, с очень высоким
Нормальная хромосома 16
Скрытый носитель
Малая а-тапассемия
a-thal 1 (Азия, Средиземноморье)
a-thal 2 (Африка, Азия, Средиземноморье)
Н-Гемоглобинопатия
Водянка плода (НЬ Барт)
Рис. 3-7. Генотипы больных с синдромами а-талассемии. Овалами обозначены копии гена а-глобина
Дифференциальная диагностика гипопролиферативной анемии 93
сродством к кислороду. Этот гемоглобин не высвобрждает кислород в ткани
плода, из-за чего возникают тканевая асфиксия, отек, застойная сердечная
недостаточность и наблюдается клиническая картина водянки плода. Младенцы обычно
рождаются мертвыми или умирают в первые часы. Гемоглобин Барт наблюдается
почти исключительно у жителей Юго-Восточной Азии, у которых встречается
преимущественно цис-делеция а-глобиновых генов.
Лечение синдромов талассемии. Пациентам с гомозиготной (3-талассемией
(анемия Кули) показано переливание крови для поддержания гемоглобина на уровне
90-100 г/л, необходимом для нормального роста и развития, а также
профилактики деформации скелета. Избыток железа выводят при помощи дефероксами-
на — водорастворимого соединения, которое связывает железо и способствует его
экскреции с мочой. Спленэктомию следует выполнять лишь при развитии реф-
рактерности к гемотрансфузиям и не ранее 5-летнего возраста. Эффективной
может оказаться трансплантация пациенту костного мозга от HLA-совместимых
сибсов. Большинство больных с а-талассемией доживают до взрослого возраста
без гемотрансфузий или спленэктомии, хотя последняя способна помочь
больным с тяжелой анемией. Все пациенты с талассемией должны пройти
генетическое обследование, поскольку в настоящее время возможна антенатальная
диагностика (ДНК-диагностика) большинства талассемических мутаций.
Макроцитарные анемии
Макроцитарные анемии подразделяются на две группы: мегалобластные и немега-
лобластные (табл. 3-8). В первом случае нарушение синтеза ДНК приводит к
аномалии клеточного роста и созревания, что влияет на все делящиеся клетки
организма. Немегалобластный макроцитоз обычно связан с патологией липидов
эритроцитарной мембраны (в то время как другие клеточные линии нормальны)
и наблюдается в основном при заболевании печени и гипотиреозе. Причиной его
развития может быть также злоупотребление алкоголем, причем макроцитоз
продолжается в течение всего жизненного цикла эритроцитов после того, как
произошло воздействие. Увеличение СОК, выявляемое с помощью гематологического
анализатора, наблюдается и у больных с ретикулоцитозом, поскольку ретикуло-
циты крупнее зрелых эритроцитов. Однако это не истинный макроцитоз.
Мегалобластные анемии
Мегалобластные анемии — состояния, которые характеризуются наличием
клеток с определенными морфологическими особенностями: крупные, незрелые
по внешнему виду ядра, окруженные относительно более зрелой цитоплазмой.
С биохимической точки зрения, первичным при этих состояниях является
нарушение синтеза ДНК: клетки прекращают развиваться в S-фазе клеточного цикла
при частичной репликации ДЙК, но не могут завершить процесс деления. В
результате мегалобласты представляют собой крупные клетки и имеют больше
ДНК, чем нормальные. В мегалобластный процесс обычно вовлекаются активно
делящиеся клетки, следствием чего является возникновение
желудочно-кишечной симптоматики. Чаще всего мегалобластоз обусловлен недостатком фолиевой
кислоты или кобаламина (витамин Bt2), а также миелодисплазией (глава 9) и
приемом лекарственных препаратов, ингибирующих синтез ДНК (табл. 3-8).
94
Глава 3. Эритроциты
Недостаточность кобаламина (витамина Bi2). Кобаламин представляет собой
сложную молекулу, состоящую из центрального атома кобальта, связанного с
четырьмя пиррольными кольцами и нуклеотидом. Он продуцируется
микроорганизмами — обитателями корнеплодов и бобовых, и присутствует в мышцах и
паренхиматозных тканях животных, питающихся этими растениями. Человек получает
кобаламин с животной пищей. Общее содержание кобаламина в организме
человека составляет 2-5 мг, а поскольку ежедневная потеря его очень невелика, то в
случае внезапного прекращения поступления запасов достаточно на 2-3 года.
Процесс абсорбции кобаламина сложен. В желудке пепсин отщепляет витамин
от белка. Далее кобаламин переносится к высокоаффинному связывающему
веществу желудочного секрета — транскобаламину I. Транскобаламины I и III
ТАБЛИЦА 3-8. Этиология мегалобластной анемии
Дефицит кобаламина (витамина В12)
Пищевой
Строгие вегетарианцы
Вскормленные грудью дети строгих вегетарианцев
Нарушение всасывания витамина В12
Пернициозная анемия
Частичная или тотальная гастрэктомия
Недостаточность поджелудочной железы
Синдром Золлингера-Эллисона
Чрезмерный бактериальный рост
Заболевания терминального отдела подвздошной кишки (резекция,
шунтирование, болезнь Крона, туберкулез, лимфома)
Паразитирование ленточных червей (Diphyllobothrium latum)
Врожденное отсутствие В12-связывающих компонентов (синдром Иммерсланда-
Грасбека [Immerslund-Grasbeck])
Медикаменты (закись азота)
Дефицит фолиевой кислоты
Недостаточное поступление с пищей
Повышенная потребность
Беременность и лактация
Недоношенность
Гемолиз
Эксфолиативный дерматит
Быстро прогрессирующая злокачественная опухоль
Нарушение всасывания
Спру
Глютеновая энтеропатия
Болезнь Крона
Синдром короткого кишечника
Амилоидоз
Миелодисплазия и эритролейкоз
Лекарственные средства
Антиконвульсанты (фенитоин, карбамазепин)
Ингибиторы дигидрофолатредуктазы (серосодержащие препараты, метотрексат)
Химиотерапия (ингибиторы синтеза ДНК)
Прочие
Дифференциальная диагностика гипопролиферативной анемии 95
(известные также как R-связывающие белки) присутствуют во всех секретах
и плазме. Их функция в этих жидкостях неизвестна, но полагают, что они
участвуют в депонировании кобаламина. Внутренний фактор (ВФ) — другой белок,
с меньшим сродством к кобаламину. Он секретируется париетальными клетками
желудка и поступает в дистальную часть двенадцатиперстной кишки вместе
с протеиновым комплексом кобаламин-транскобаламин I. Затем под действием
панкреатических протеаз R-белок распадается, а кобаламин транспортируется
к ВФ. Комплекс ВФ-кобаламин продвигается в дистальный отдел подвздошной
кишки, где абсорбируется при участии мембраносвязанных рецепторов. В крови
кобаламин связывается с транскобаламином II — транспортным белком, который
осуществляет целенаправленную доставку кобаламина в ткани-мишени по
механизму рецепторно-опосредованного эндоцитоза.
Этиология дефицита кобаламина. Алиментарный дефицит кобаламина —
явление редкое и наблюдается только у больных, длительное время находящихся
на строгой растительной диете, исключающей прием животных продуктов, а
также у вскормленных грудью детей таких вегетарианцев (табл. 3-8).
Наиболее часто дефицит витамина Bi2 возникает вследствие пернициозной
анемии (ПА) — аутоиммунного нарушения, характеризующегося атрофией
париетальных клеток желудка, отсутствием ВФ и секреции соляной кислоты.
Название "пернициозная анемия" было введено Бирмером в 1872 г., однако патогенез
этого заболевания оставался неясным до 1920 г. В настоящее время термин
"пернициозная анемия" используется применительно к состоянию, возникающему
в результате нарушения секреции внутреннего фактора слизистой желудка (ранее
смертельному). ПА обычно обнаруживается у пожилых людей, но может
наблюдаться у лиц любого возраста и расовой принадлежности. Частота заболевания
составляет 25:100 000 человек в год среди людей старше 40 лет. Предполагается
существование генетического компонента, предрасполагающего к ПА, и наличие
связи с другими аутоиммунными заболеваниями, например с болезнью Грейвса,
тиреоидитом Хашимото, витилиго, болезнью Аддисона, гипопаратир'еозом и ги-
погаммаглобулинемией, начавшейся во взрослом возрасте. Более чем у 90 %
пациентов с ПА в сыворотке крови выявляются антитела класса IgG к париетальным
клеткам, однако эти антитела можно также наблюдать у 50 % больных атрофичес-
ким гастритом без ПА. Анти-ВФ-антитела обнаруживают в сыворотке только
у 60 % пациентов с ПА, но они высокоспецифичны для данного заболевания.
Поскольку процесс абсорбции и усвоения кобаламина достаточно сложен,
многие поражения желудочно-кишечного тракта приводят к мальабсорбции и
дефициту кобаламина (табл. 3-8). Дефицит обычно возникает спустя 4-10 лет после
возникновения нарушений абсорбции, так как запасы кобаламина в организме
значительны, а его физиологические потери минимальны. Дефицит кобаламина
может развиться вследствие чрезмерного бактериального роста, поскольку
бактерии разрушают этот витамин. Более редкие причины дефицита включают
врожденные дефекты в молекулах ВФ, рецепторов ВФ или нарушения пострецептор-
ных процессов, а также врожденное отсутствие транскобаламина И. Описаны
редкие случаи врожденных дефектов метаболизма кобаламина.
Дефицит фолиевой кислоты. Фолаты — это витамины группы В, имеющиеся
в листовых овощах (бобовые, салат, шпинат, брокколи), фруктах, грибах,
животных белках. При продолжительной варке фолаты разрушаются, поэтому их луч-
96
Глава 3. Эритроциты
шим пищевым источником являются свежие фрукты и овощи. После попадания
в организм фолаты взаимодействуют со связывающими белками слизистой
тонкого кишечника, где происходит их поглощение и метилирование. Фолиевая
кислота слабо связывается с белками плазмы крови и быстро усваивается клетками.
Ее уровень в сыворотке зависит от диеты и от сохранности энтерогепатической
циркуляции. Резкое прекращение приема фолиевой кислоты в течение трех
недель приводит к ее дефициту. Искусственное дренирование желчи может
вызывать фолиевый дефицит через несколько часов.
Этиология дефицита фолиевой кислоты. Основная причина дефицита
фолиевой кислоты — недостаточное ее потребление. Как правило, это наблюдается
у лиц с нарушенным питанием, например у алкоголиков, наркоманов', пожилых
людей, потребляющих очень мало свежих фруктов и овощей. Дефицит может
также развиваться при быстром обновлении клеток, когда повышенные потребности
организма не успевают удовлетворяться: беременность, острый или хронический
гемолиз, эксфолиативный дерматит; а также у пациентов, находящихся на
гемодиализе, поскольку в ходе этой процедуры происходит потеря витамина.
Нарушение абсорбции фолиевой кислоты наблюдается при таких заболеваниях тонкой
кишки, как глютеновая энтеропатия, спру и болезнь Крона.
Другие причины развития мегалобластной анемии
Лекарственные средства и токсины. Лекарственные средства вызывают мега-
лобластную анемию посредством различных механизмов. Некоторые из них,
например метотрексат, являются сильными ингибиторами дигидрофолатредукта-
зы, другие (пентамидин, триметоприм, триамтерен и пириметамин) —
антагонистами фолиевой кислоты. Такие химиотерапевтические препараты, как
аналоги пурина (азатиоприн, 6-меркаптопурин, 6-тиогуанин), пиримидина
(5-фторурацил, цитозин арабинозид) и другие (прокарбазин, гидроксимочеви-
на) непосредственно ингибируют синтез ДНК. Прием противосудорожных
препаратов (фенитоин, барбитураты) приводит к дефициту этого витамина
посредством еще не изученных механизмов. Метаболизм фолата нарушает алкоголь.
Противовирусные препараты зидовудин и ацикловир вызывают выраженную
мегалобластную анемию, однако механизм ее развития не ясен. Закись азота
окисляет кобаламин и может провоцировать острое мегалобластное состояние,
характеризующееся быстро развивающейся тромбоцитопенией и/или
лейкопенией при отсутствии анемии.
Миелодисплазия и эритролейкоз. Мегалобластный эритропоэз наблюдается
при рефрактерной анемии, приобретенной сидеробластной анемии и других
формах миелодисплазии. Мегалобластоз и созревание эритроцитов причудливой
формы являются признаками эритролейкоза (синдром Ди Гульельмо [Guglielmo]).
Метаболизм витамина Bi2 и фолиевой кислоты. Кобаламин — кофактор двух
внутриклеточных ферментов. В митохондриях он выполняет коферментную
функцию метилмалонил-КоА-мутазы, катализирующей превращение метилма-
лонил-КоА в сукцинил-КоА (рис. 3-8 А). В цитоплазме кобаламин — кофактор
гомоцистеин-метионинметилтрансферазы (метионинсинтазы), которая
катализирует перемещение метальных групп от N-метилтетрагидрофолата к гомоцисте-
ину с образованием метионина (рис. 3-8 Б).
Находясь в клетке, фолат восстанавливается в 5-метилтетрагидрофолат,
отдающий метиловую группу кобаламину в процессе образования метионина из гомо-
Дифференциальная диагностика гипопролиферативной анемии
97
цистеина (рис. 3-8). Соединения фолиевой кислоты играют также важную роль
в синтезе ДНК, являясь донорами одного атома углерода при превращении дезок-
сиуридина в дезокситимидин. Тетрагидрофолат подвергается полиглутаминиро-
ванию; полагают, что этот механизм обеспечивает сохранение фолиевой кислоты
в клетке. Дефицит кобаламина приводит к блокаде метаболизма фолата на этапе
образования метилтетрагидрофолата, в результате чего фолат расходуется на
синтез дезоксиуридина; при этом полиглутаминирование протекает менее
эффективно, что вызывает утечку фолиевой кислоты из клетки (рис. 3-8 Б).
Недостаточность кобаламина приводит к мегалобластозу за счет
функционального дефицита фолата, поэтому дефицит этих двух витаминов невозможно
дифференцировать на основании морфологии крови и костного мозга. Вместе
с тем дефицит только кобаламина вызывает неврологическое заболевание, для
которого характерна пятнистая демиелинизация серого вещества в головном и
спинном мозге и периферических нервах. Причина этого феномена пока не
установлена. Предполагают, что ингибирование метилмалонил-КоА-мутазы
(вследствие недостаточности Bi2, но не фолиевой кислоты) угнетает метаболизм
жирных кислот, содержащих нечетное число атомов углерода, в результате чего
происходит внедрение аномальных жирных кислот в миелин. Эти аномальные
кислоты можно обнаружить при биопсии нервов у больных с дефицитом кобала-
Метилмалонил-КоА ««-
Метилмалонил-
КоА-мутаза
Сукцинил-КоА
Рис. 3-8 А. Метаболизм кобаламина в митохондриях
Метил-
л-ТГФ I
ж ■
Метил-ТГФ
ТГФ
ПолиглугамированныйТГФ I
Клеточная мембрана
Рис. 3-8 Б. Метаболизм фолиевой кислоты и кобаламина в цитоплазме. ТГФ — тетрагидрофолат
4 3ак 313
98
Глава 3. Эритрбциты
мина. Дефицит метионина также может способствовать появлению нейропатии
за счет нарушения йродукции холинсодержащих фосфолипидов.
Симптомы и признаки мегалобластной анемии. При беседе с больным мегало-
бластозом необходимо обратить внимание на особенности его диеты и состояние
ЖКТ. Наличие в анамнезе плохого питания, нарушений всасывания,
алкоголизма дает возможность предполагать дефицит фолиевой кислоты, а хирургическое
вмешательство на желудке, равно как и неврологическое или психическое
заболевание в анамнезе — возможный дефицит кобаламина. Необходимо узнать о
приеме лекарств, особенно противосудорожных, цитостатических средств,
антагонистов фолата и закиси азота. Аутоиммунные нарушения в анамнезе (гипотиреоз,
болезнь Аддисона, витилиго) позволяют предположить пернициозную анемию.
При физикальном обследовании пациента некоторые данные могут помочь
постановке диагноза и явиться ключом к пониманию этиологии. Возможен лимонный
оттенок кожи — следствие сочетания желтухи и бледности. При мегалобластной
анемии часто наблюдаются глоссит и хейлит. У больных с нарушенным питанием
встречаются и другие проявления витаминной недостаточности: остеомаляция,
дерматит, кровотечения и инфекции. При дефиците фолата или кобаламина часто
имеется усиленная пигментация ногтевого ложа и кожных складок. Может
развиться умеренная спленомегалия как результат экстрамедуллярного гемопоэза.
Неврологические признаки и симптомы дефицита кобаламина различны в
зависимости от тяжести патологии. К ранним признакам относится дисфункция
задних рогов спинного мозга с потерей проприоцепции и ощущением вибрации.
Пациенты двигаются с трудом, широко расставляя ноги при ходьбе. Позже у них
развивается поражение пирамидного, спинно-мозжечкового и спиноталамическо-
го трактов, сопровождающееся мышечной слабостью, прогрессирующей спастич-
ностью, гиперрефлексией, ножницеобразной походкой. Возможно также
повреждение периферических нервов с утратой глубоких сухожильных рефлексов,
параличом черепно-мозговых нервов или потерей контроля над сфинктерами.
При длительном дефиците кобаламина возникают деменция и нейропсихическое
заболевание. Неврологическая симптоматика при дефиците кобаламина может
иметь место и без анемии. Результаты некоторых исследований дают основание
предполагать, что чем выше гематокрит, тем тяжелее неврологические
нарушения. Таким образом, по уровню содержания витамина Bt2 можно лишь отчасти
судить о глубине деменции и тяжести нейропсихических симптомов. Клинические
признаки дефицита фолиевой кислоты подобны таковым при дефиците
кобаламина, но неврологическая симптоматика отсутствует; впрочем, некоторые авторы все
же описывают слабо выраженные нейропсихические нарушения.
Диагностика недостаточности фолиевой кислоты и кобаламина. Наличие мак-
роцитоза должно насторожить клинициста в отношении возможного дефицита
фолиевой кислоты и/или кобаламина, особенно если СОК больше 110 мкм3. В то
же время у больных с одновременным дефицитом железа, талассемией и анемией
при хроническом заболевании мегалобластоз может быть без макроцитоза.
Низкий уровень ретикулоцитов, умеренная лейкопения и тромбоцитопения также
указывают на дефицит фолата или кобаламина.
В мазке периферической крови видны макроовалоциты, обнаруживается ани-
зоцитоз, пойкилоцитоз, одиночные ядерные эритроциты и базофильная
зернистость (табл. 3-3). Уникальной особенностью мегалобластной анемии является ги-
Дифференциальная диагностика гипопролиферативной анемии 99
персегментация ядра полиморфно-ядерных клеток, что объясняется нарушением
синтеза ДНК. Наличие даже одной клетки с ядром из 6 долей — верный признак
недостаточности кобаламина или фолата (глава 4,. табл. 1). Тромбоциты имеют
тенденцию увеличиваться в размерах и изменять форму. В костном мозге
наблюдается изобилие клеток при уменьшении количества мегакариоцитов и
появлении аномально крупных предшественников гранулоцитов (гигантские палоч-
коядерные клетки и метамиелоциты). Предшественники эритроцитов также
увеличены в размерах и характеризуются ядерно-цитоплазматической асинхро-
нией (ядро выглядит менее зрелым, чем цитоплазма). У больных
обнаруживаются признаки, свидетельствующие о неэффективности эритропоэза, в сочетании
с непрямой гипербилирубинемией и повышением уровня ЛДГ по причине интра-
медуллярного (костномозгового) лизиса эритроцитов.
Диагноз мегалобластной анемии ставится на основании определения
содержания фолиевой кислоты и кобаламина в крови. Уровень фолата можно измерить
в сыворотке или в эритроцитах периферической крови. Поскольку уровень
фолата в сыворотке отражает недавнее поступление последнего с пищей, то он будет
низким уже через 24-48 ч после прекращения приема пациентом фолиевой
кислоты. Наоборот, у больных с дефицитом фолата можно наблюдать нормализацию
его уровня через несколько часов после принятия пищи, содержащей фолат. Ге-
молизированные эритроциты выделяют фолиевую кислоту в сыворотку, ложно
повышая сывороточный уровень даже при внутриклеточном дефиците фолата.
Уровень эритроцитарного фолата в 30 раз выше сывороточного и обычно
коррелирует с таковым в тканях. Следует иметь в виду, что дефицит кобаламина,
ухудшающий полиглутаминирование фолата, может вызывать утечку последнего из
клеток, что приводит к снижению уровня фолата в эритроцитах при нормальном
уровне в сыворотке.
Содержание кобаламина в сыворотке обычно точно отражает его запасы в
организме. Примерно у 5 % больных с дефицитом кобаламина и неврологическими
и/или гематологическими нарушениями уровень кобаламина в сыворотке
находится на нижней границе нормы. У таких пациентов диагноз можно поставить на
основании измерения содержания в моче метаболитов метилмалоновой кислоты
и гомоцистеина: при дефиците кобаламина показатели будут повышены, что
связано соответственно с метаболической блокадой метилмалонил-КоА-мутазы
и метионинсинтазы (рис. 3-8 А и 3-8 Б).
После постановки диагноза определить этиологию дефицита кобаламина
помогает проба Шиллинга. В ходе пробы перорально вводится радиоактивный коба-
ламин. Затем парентерально — большая доза немеченного витамина с целью
насыщения циркулирующих кобаламинсвязывающих белков транскобаламинов I
и II и обеспечения максимальной экскреции абсорбированного из кишечника
радиоактивного кобаламина с мочой. В суточной порции мочи радиоактивность
должна составлять более 8 % от введенной дозы. Одновременное пероральное
введение меченного другим изотопом кобаламина, связанного с ВФ, позволяет провести
сравнение абсорбции комплекса ВФ-кобаламин с абсорбцией свободного
витамина. Избирательное нарушение абсорбции свободного кобаламина,
компенсированное комплексом ВФ-кобаламин, указывает на пернициозную анемию и
отсутствие ВФ. Если нарушение абсорбции не компенсируется, пробу можно повторить
после назначения курса антибиотиков широкого спектра действия (подавление
избыточного бактериального роста) и последующего применения панкреатичес-
100
Глава 3. Эритроциты
ких ферментов (устранение панкреатической недостаточности). Проба Шиллинга
показательна лишь при тщательном сборе мочи и в настоящее время используется
не столь широко, поскольку выявление наличия анти-ВФ или антител к
париетальным клеткам позволяет поставить диагноз пернициозной анемии и без нее.
Лечение больных с дефицитом фолиевой кислоты и кобаламина. К лечению
следует приступать после определения уровней фолиевой кислоты и кобаламина.
Больные с вторичным в результате пернициозной анемии дефицитом кобаламина
должны получать парентеральную терапию. Она обычно начинается с дозы
1 мг/сут подкожно или 100 мкг внутривенно в течение7 дней, затем 1 мг подкожно
1 раз в месяц. Пациенты с нормальной абсорбцией кобаламина могут получать пе-
роральную заместительную терапию по 1 -5 мг/сут, некоторым больным ПА
показаны более высокие дозы. Данная терапия проводится пациентам, не желающим
получать препарат парентерально. Всем пациентам, принимающим кобаламин,
необходимо назначать и фолат, следя за тем, чтобы не развивался дефицит железа,
поскольку фолат и железо потребляются пролиферирующими тканями. При
тяжелой анемии и мегалобластозе коррекция дефицита кобаламина может привести
к острой гипокалиемии, гиперурикемии и гипофосфатемии в связи со
стремительной активацией клеточной пролиферации и метаболизма ДНК и белков.
Пациентам с дефицитом фолиевой кислоты назначают перорально по 1-5 мг
фолата ежедневно. Поскольку недостаточность кобаламина может привести к
дефициту фолата, еще до начала лечения последним очень важно убедиться в том,
что у больного содержание кобаламина в норме. Хотя при дефиците кобаламина
анемия может корригироваться высокими дозами фолата, это никак не
отражается на неврологической патологии, отсрочка соответствующего лечения которой
способна привести к необратимым изменениям.
Иногда после начала заместительной терапии у больных быстро наступает
значительное улучшение состояния, вплоть до повышения величины гемато-
крита. Количество ретикулоцитов увеличивается через 2-3 дня и достигает пика
на 8-й день. В пределах 1 недели наблюдается повышение уровня гемоглобина и
гематокрита, а полная коррекция анемии — в течение 2-х месяцев.
Неврологические нарушения не всегда обратимы: чем дольше период проявления
неврологических симптомов, тем хуже прогноз. У большинства пациентов максимальное
улучшение наступает через 6-12 месяцев.
Нормоцитарные анемии
Перечень заболеваний, которые необходимо учитывать при дифференциальной
диагностике нормоцитарных анемий, достаточно велик (табл. 3-4). Макроцитар-
ные и микроцитарные анемии обычно нормоцитарны на ранних стадиях процесса.
Кроме того, смешанная анемия, такая как при сочетанном дефиците фолиевой
кислоты и железа, на основании показаний автоматического цитометра может
быть квалифицирована как нормоцитарная. Поэтому для подтверждения
результатов автоматической цитометрии всегда необходимо исследовать мазок
периферической крови. Анемия с нормальным средним объемом клеток наблюдается как
при различных хронических заболеваниях, так и при недостаточности костного
мозга (апластическая анемия и парциальная красноклеточная аплазия [ПККА]).
Анемия, сопровождающая уремию, гипотиреоз, гипертиреоз и другие
эндокринные нарушения, обычно нормоцитарна.
Анемия с ретикулоцитозом
101
Анемия при хронических заболеваниях
Анемия может развиваться у больных с хроническими инфекционными или
воспалительными процессами, злокачественными новообразованиями,
аутоиммунными нарушениями. Она характеризуется относительно низким уровнем эритро-
поэтина, пониженной реакцией костного мозга на эритропоэтин и несколько
сокращенным сроком жизни эритроцитов. У больных отмечается снижение
уровня железа, пониженная железосвязывающая способность при насыщении транс-
феррина обычно больше 10 %. Уровень ферритина, относящегося к белкам острой
фазы, часто повышен. Исследование костного мозга помогает определить
наличие адекватных или увеличенных запасов железа в ретикулоэндотелиальной
системе и сниженное его количество в предшественниках эритроцитов.
Анемия, возникающая при хронической патологии, разрешается при
излечении первичного заболевания. У некоторых больных, главным образом у тех, кто
имеет низкий уровень эритропоэтина, его введение в больших дозах может
корригировать анемию. Это характерно для некоторых злокачественных опухолей
(особенно множественной миеломы), ВИЧ-инфекции, отдельных
воспалительных процессов, например ревматоидного артрита. Эритропоэтин служит
основным терапевтическим средством лечения анемии при почечной недостаточности.
Анемия с ретикулоцитозом
Гемолиз — это преждевременное разрушение эритроцитов в кровеносных сосудах
и/или сосудистых пространствах ретикулоэндотелиальной системы. Признаком
гемолиза является большое число ретикулоцитов, что отражает тенденцию
костного мозга компенсировать уменьшение эритроцитарной массы и понижение кис-
лородтранспортной способности крови. Еще одной причиной ретикулоцитоза
может быть кровопотеря. При обследовании пациента с подозрением на гемолиз
необходимо обратить внимание на такие анамнестические данные, как
перенесенные инфекции, применяемые больным лекарственные средства, воздействие
токсинов, дыма, химических веществ и экстремальных температур, имевшие место
путешествия; иными словами, важно тщательное изучение профессионального
и семейного анамнеза. Этническая и расовая принадлежность пациента может
навести на мысль о специфических наследственных гемоглобинопатиях и энзимо-
патиях. Следует отметить факты переливания крови и ее продуктов. При физи-
кальном обследовании пациента с гемолизом могут обнаруживаться бледность,
желтуха и спленомегалия. Для острого внутрисосудистого гемолиза, который
развивается при посттрансфузионных осложнениях или пароксизмальной
ночной гемоглобинурии, характерны боль в спине и темная моча.„У больных с
тяжелым хроническим гемолизом наблюдаются типичные изменения лица и скелета,
вызываемые усилением гемопоэтической функции костного мозга, подобные
симптомам талассемии. У лиц молодого возраста хронический гемолиз приводит
к образованию пигментных камней в желчном пузыре.
При лабораторном исследовании обнаруживается анемця, которая, если
использовать автоматический цитометр, обычно определяется как макроцитарная,
поскольку ретикулоциты крупнее зрелых эритроцитов. Ключевым этапом
диагностики является исследование мазка периферической крови, так как
морфология эритроцитов часто отражает этиологию деструктивного процесса (табл. 3-3).
102
Глава 3. Эритроциты
У больных гемолитической и мегалобластной анемией повышается уровень
непрямого билирубина и активность лактатдегидрогеназы. Содержание гаптогло-
бина в плазме (гаптоглобин связывает свободный гемоглобин) снижается, так как
после связывания с гемоглобином он быстро выводится из кровообращения
печенью. При иммуноопосредованном гемолизе оказывается положительным прямой
антиглобулиновый тест Кумбса, с помощью которого определяют
иммуноглобулины и комплемент на поверхности эритроцитов. У больных с внутрисосудистым
гемолизом возможны гемоглобинурия, гемосидеринурия и повышение уровня
свободного гемоглобина в плазме.
Гемолитические анемии могут вызываться как аномалиями собственно
эритроцитов, так и другой патологией, быть врожденными или приобретенными
(табл. 3-9 и 3-10).
Гемолитические анемии, вызванные аномалиями эритроцитов
Патология мембраны
Наследственный сфероцитоз — это гетерогенная группа состояний,
обусловленных мутациями в генах, кодирующих мембранные белки цитоскелета эритроцитов.
Болезнь почти всегда наследуется аутосомно-доминантным путем, хотя описаны
и редкие случаи аутосомно-рецессивного наследования. Наболее частым
дефектом при наследственном сфероцитозе являются аномалии спектрина и анкирина,
реже — сегмента 3 или протеина 4.2 (рис. 3-1). У больных наблюдаются
гемолитическая анемия, спленомегалия и сфероцитоз периферической крови. Сфероциты
образуются, когда эритроциты с аномальными структурными белками мембраны
проходят через селезенку, теряя при этом часть клеточной мембраны. В результате
такие эритроциты утрачивают свою двояковогнутую форму и становятся
похожими на мячик. Площадь поверхности клеток уменьшается так же, как и
способность их мембраны к растяжению в гипотоническом растворе. Последнее обстоя-
ТАБЛИЦА 3-9. Классификация гемолитических анемий
Патология эритроцитов
Энзимопатии
Дефицит глюкозо-6-фосфатдегидрогеназы
Дефицит пируваткиназы
Прочие
Гемоглобинопатии
Аномалии мембраны
Наследственный сфероцитоз
Наследственный эллиптоцитоз
Наследственный пиропойкилоцитоз
Наследственный стоматоцитоз
Шпороклеточная анемия
Пароксизмальная ночная гемоглобинурия
Внеэритроцитарная патология
Спленомегалия
Аутоиммунная гемолитическая анемия
Микроангиопатическая гемолитическая анемия
Анемия, индуцированная инфекциями, токсинами
Анемия с ретикулоцитозом
103
ТАБЛИЦА 3-10. Препараты, вызывающие гемолиз при недостаточности Г-6-ФДГ
Антибактериальные препараты
Сульфаниламиды
Нитрофурантоин
Налидиксовая кислота
Противомалярийные средства
Дапсон
Хлорамфеникол
Доксорубицин
Прочие
Аминосалициловая кислота
Фенацетин
Пробенецид
Прокаинамид
Витамины Си К
Аспирин в высоких дозах
тельство объясняет то, что лизис сфероцитов происходит при более высоком
уровне осмолярности среды, чем лизис нормальных эритроцитов. Соответственно,
диагноз ставится на основании определения осмотической резистентности (рис. 3-9).
У большинства больных развивается компенсированная умеренная анемия,
которая нарастает в условиях подавленного ретикулоцитоза, что и происходит во
время инфекций и/или при применении медикаментов, оказывающих супрессив-
ЮОг
80
60
со
S
§
,© 40
Г Наследственный
сфероцитоз
L После спленэктомии
I До спленэктомии N.
\ <
\/.
/;
';
//
\— У . I <'\
/
/
/
/ ;
/ /
/ /
/ /
/ /
11
'i
'/
* X
/ /
* t
\ . Норма
^
■■
/
/
\у/ 1 1 1
20 L
0
0,9 0,8
0,7 0,6 0,5 0,4
Натрия хлорид, г/100 мл
0,3 0,2
Рис. 3-9. Осмотическая резистентность эритоцитов у больного с наследственным сфероцитозом до
и после спленэктомии. (По: Isselbacher K.J. et al. eds. Harrison's Principles of Internal Medicine, 13th ed.
New York: McGraw-Hill, 1994:1539, Fig. 307-1. Osmotic fragility of red cells in hereditary spherocytosis.)
104
Глава 3. Эритроциты
ное действие на костный мозг. При этом часто необходимо назначение фолиевой
кислоты. У многих пациентов при НС в желчном пузыре образуются пигментные
камни, что требует иногда хирургического вмешательства. В более тяжелых
случаях, при выраженной анемии, следует выполнить спленэктомию. Эта процедура
снижает степень гемолиза и увеличивает продолжительность жизни
эритроцитов, купируя анемию.
При наследственном эллиптоцитозе цитоскелетные аномалии локализуются на
участках взаимодействия между цитоплазматическими белками, особенно между
а- и р-спектрином, а также спектрином и протеином 4.1. В результате образуются
эритроциты овальной или эллиптической формы (табл. 3-3) с уменьшенным
сроком жизни. Наследование обычно аутосомно-доминантное, причем у
большинства больных симптомы отсутствуют или имеет место умеренная анемия и спле-
номегалия. Тяжелая форма наследственного элиптоцитоза — наследственный
пиропойкилоцитоз, характеризуется аутосомно-рецессивным способом
наследования, выраженной анемией, наличием фрагментированных эритроцитов, микро-
сфероцитов и эллиптоцитов. Лечение такое же, как и при наследственном сферо-
цитозе. Спленэктомия выполняется только у больных с тяжелой формой анемии.
Третья аномалия эритроцитов — стоматоцитоз. Для него характерна
поперечная щель (стома) в эритроцитах, обнаруживаемая при исследовании мазка
периферической крови (табл. 3-3). Подобное явление наблюдается при редких
наследственных нарушениях катионной проницаемости мембраны, алкоголизме,
новообразованиях, заболеваниях сосудов и желчевыводящих путей, а также
изредка как побочный эффект принимаемых лекарственных средств.
Акантоциты, или шпоровидные клетки, имеют выпячивания мембраны
различного размера, которые легко распознаются при изучении мазка крови. Эти
клетки находят у пациентов с терминальной стадией заболевания печени, а
причина их появления связана с синтезом в этих условиях аномальных липопротеи-
нов плазмы и эритроцитарной мембраны. Акантоциты могут также наблюдаться
у больных с удаленной селезенкой, при крайне неполноценном питании и
различных нарушениях всасывания. Аналогичные морфологические изменения
периферической крови в сочетании с легким гемолизом встречаются у пациентов с абе-
талипопротеинемией — врожденным отсутствием Р-липопротеина. При уремии
эритроциты имеют симметричные выступы меньшего размера и называются
шиловидными клетками, или эхиноцитами.
Пароксизмальная ночная гемоглобинурия
Пароксизмальная ночная гемоглобинурия (ПНГ) — приобретенное заболевание,
при котором клон гёмопоэтических клеток утрачивает способность
синтезировать гликанфосфатидилинозитол — якорь, необходимый для связывания
регуляторов комплемента с эритроцитарной мембраной. При этом виде патологии
эритроциты чрезмерно чувствительны к лизису комплементом. Для диагностики
ПНГ используется выявление аномального лизиса эритроцитов в присутствии
комплемента, активированного с помощью подкисленной сыворотки (тест Хэма
или лизис в гипотонической среде в присутствии сахарозы). В настоящее время
возможна прямая диагностика методом проточной цитометрии, позволяющим
определить отсутствие на эритроцитарной мембране фактора ускорения распада
и ингибитора реактивного лизиса. Эти два белка — важные регуляторы функции
Анемия с ретикулоцитозом
105
комплемента и обычно присоединяются к клеточной мембране с помощью гли-
канфосфатидилинозитола.
ПНГ, которой страдают преимущественно молодые люди, проявляется острыми
приступами боли в спине — следствием врутрисосудистого гемолиза. Поскольку
гемолиз протекает прежде всего внутри сосудов, ретикулоэндотелиальная система
не может полностью захватить освобождаемые при этом гемоглобин и железо, что
приводит к гемоглобинурии и/или гемосидеринурии. Кроме того, возможны тром-
ботическиеЪсложнения (тромбозы глубоких вен, воротной вены, брыжеечной вены
и церебральных вен, синдром Бадда-Киари). Полагают, что причиной подобных
осложнений ПНГ служит агрегация тромбоцитов, вызываемая неконтролируемой
активацией комплемента. Заболевание ассоциируется также с апластической
анемией, острым лейкозом и миелофиброзом. У пациентов наблюдаются нормоцитар-
ная анемия, умеренные тромбоцитопения и лейкопения, низкая активность
лейкоцитарной щелочной фосфатазы. Лечение только поддерживающее. Пациентам
следует назначать гемотрансфузии для подавления пролиферации аномального
клона. Иногда эффективно применение кортикостероидов. Молодым людям
целесообразно проведение трансплантации костного мозга, особенно при
апластической анемии или тяжелых тромботических осложнениях.
Эритроцитарные энзимопатии
Дефицит глюкозо-6-фосфатдегидрогеназы. Наиболее распространенной
патологией метаболизма эритроцитов является дефицит глюкозо-6-фосфатдегидро-
геназы (Г-6-ФДГ) — фермента, восстанавливающего пиридиннуклеотид в гексо-
зомонофосфатном шунте (рис. 3-2). Этот путь окисления углеводов важен для
продукции восстановленного глутатиона, который в свою очередь защищает
сульфгидрильные группы гемоглобина и эритроцитарную мембрану от
окисления. У больных с недостаточностью Г-6-ФДГ проокисидантный эффект
инфекций, ацидоза, определенных лекарственных препаратов и токсинов (табл. 3-10)
приводит к преципитации гемоглобина и внутрисосудистому гемолизу.
Преципитацию гемоглобина в виде эритроцитарных включений (тельца Гейнца) можно
наблюдать при исследовании мазка периферической крови, окрашенного
кристаллическим фиолетовым. Кроме того, в периферической крови можно увидеть
так называемые "надкусанные" эритроциты — следствие удаления селезенкой
части клетки, содержащей включения (табл. 3-3).
Наследование дефицита Г-6-ФДГ сцеплено с полом (с Х-хромосомой),
поэтому у гетерозиготных женщин выраженный гемолиз встречается крайне редко.
Существуют два клинических синдрома: легкая форма (наблюдается у лиц
африканского происхождения) и более тяжелая (выявляется у жителей Азии и
Средиземноморья). Первый тип (африканский) характеризуется небольшим
снижением ферментативной активности, главным образом в старых эритроцитах. Гемолиз
обычно самоограничен, так как активность фермента в ретикулоцитах и юных
эритроцитах нормальна. У больных со вторым типом (средиземноморским)
возможен умеренный хронический гемолиз, однако при действии окислителей
тяжесть гемолиза резко возрастает: по неясным причинам тяжелый гемолиз
развивается также при употреблении этими больными в пищу некоторых сортов бобовых.
Диагноз ставится на основании определения уровня Г-6-ФДГ в крови, однако
у пациентов, только что перенесших острый приступ гемолиза, этот показатель
106
Глава 3. Эритроциты
может соответствовать норме — свидетельство нормальной ферментативной
активности выживших ретикулоцитов и юных эритроцитов. Лечение сводится
к поддерживающей терапии во время острого приступа и исключению
провоцирующих гемолиз средств (или продуктов питания). При хроническом гемолизе
может быть эффективна спленэктомия.
Другие виды энзимопатий. Описана недостаточность большинства ферментов
гликолиза, однако едва ли не самым распространенным нарушением является
дефицит пируваткиназы. Многие виды энзимопатий наследуются аутосомно-рецес-
сивным путем. У больных наблюдается врожденный несфероцитарный гемолиз
различной степени тяжести. Лечение только поддерживающее; в некоторых
случаях показана спленэктомия.
Гемоглобинопатии
Гемоглобинопатии — заболевания, вызванные синтезом аномального гемоглобина.
Подобно талассемии и дефициту Г-6-ФДГ, гемоглобинопатии чаще всего
развиваются у лиц, проживающих в "малярийном поясе", поскольку гетерозиготы
относительно устойчивы к, P. falciparum. Диагноз гемоглобинопатии ставится на
основании результатов электрофореза гемоглобина (глава 11).
Серповидно-клеточная анемия. Серповидно-клеточная анемия (СКА) —
гемоглобинопатия, наиболее распространенная у народов Америки, африканцев,
уроженцев центральной Индии, жителей Средиземноморья и Средней Азии.
Гемоглобин S образуется в результате точечной мутации в гене Р-цепи, приводящей
к замещению в 6-м положении глутаминовой кислоты валином. Дезоксигениро-
ванный гемоглобин S обладает пониженной растворимостью и способен к
полимеризации, приводящей к деформации эритроцитов и гемолизу (табл. 3-3).
Острые проявления СКА. Серповидно-клеточная анемия может вызвать
острую патологию почти любой системы организма (табл. 3-11). Большинство этих
осложнений связано с эпизодическим образованием серповидных клеток,
закупоривающих микрососуды. Болевые (вазоокклюзионные) кризы возникают в том
случае, когда деформированные серповидные эритроциты, взаимодействуя
с тромбоцитами, эндотелием, коагуляционными белками и другими
циркулирующими факторами, вызывают нарушение микроциркуляции. Это является
причиной возникновения тканевой гипоксии, инфарктов и характерных
рецидивирующих болевых приступов (область спины, груди, живота, длинных трубчатых
костей). Вазоокклюзионные кризы часто провоцируются инфекцией, холодом,
физической нагрузкой и эмоциональным стрессом. У детей возможно падение ге-
матокрита вследствие секвестрации крови в селезенке. Рецидивирующие
инфаркты селезенки обусловливают ее некроз и фиброз; у большинства взрослых
пациентов с СКА развивается функциональная аспления. Они обладают повышенной
чувствительностью к различным инфекциям, особенно вызываемым
инкапсулированными бактериями (глава 1).
Средняя продолжительность жизни эритроцитов у больных, гомозиготных
по гемоглобину S, составляет только 17 дней (в норме 120 дней). У больных с СКА
наблюдается хроническая гемолитическая анемия с уровнем гематокрита от 18
до 32 % и ретикулоцитов — около 10 %. Вследствие сокращения срока жизни
эритроцитов больные особенно чувствительны к транзиторной супрессии кост-
Анемия с ретикулоцитозом
107
ного мозга, вызванной инфекцией, например парвовирусом В19 (избирательное
поражение предшественников эритроцитов), пневмококком, сальмонеллой,
вирусом Эпштейна-Барр и др. В этой ситуации может развиться "апластическии
криз" с резким падением гематокрита, содержания гемоглобина и числа ретику-
лоцитов. У больных не исключен некроз костного мозга, сопровождающийся
болями в костях, лихорадкой и картиной лейкоэритробластоза в периферической
крови. Подобное состояние может осложниться эмболией легких агрегатами
клеток костного мозга.
Вазоокклюзионные кризы в жизненно важных органах иногда сопровождают-1
ся серьезными заболеваниями и даже приводят к смерти.
Острый грудной синдром вызван появлением в микрососудистом русле
легких серповидных клеток и характеризуется болью в груди, одышкой, гипоксеми-
ей, лихорадкой и обнаруживаемыми при рентгенологическом исследовании
инфильтратами. Клинические признаки синдрома не отличимы от таковых при
пневмонии, поэтому обязательно назначение антибиотиков (обычно
эритромицина). Это патологическое состояние угрожает жизни пациентов и является
показанием для проведения им заменных трансфузий и интенсивного мониторинга.
Примерно у 25 % больных развиваются острые неврологические нарушения,
в том числе припадки, тромботические и геморрагические инсульты, транзитор-
ные ишемические приступы. Мозговые инсульты — результат закупорки
крупного сосуда, возникают в основном у детей и в 70 % случаев при отсутствии
лечения рецидивируют в течение 3 лет. При инсульте целесообразно проведение
курса заменных трансфузий, значительно снижающих частоту рецидивов.
У взрослых больных с СКА не исключено возникновение острых
геморрагических инсультов в результате неоваскуляризации и образования аневризм сосудов
головного мозга.
ТАБЛИЦА 3-11. Клинические проявления серповидно-клеточной анемии
Органы
Острые проявления
Хронические проявления
Легкие
Мочеполовая
система
Нервная
система
Гепато-били-
арный тракт
Скелет
Глаза
Кожа
Острый грудной синдром
Гематурия
Папиллярный некроз
Приапизм
Тромботический инсульт
Геморрагический инсульт
Припадки
Транзиторные
ишемические приступы
Синдром правого верхнего
квадранта
Вирусный гепатит
Остеомиелит (особенно
вызванный сальмонеллой)
костный инфаркт
Ишемия и геморрагии сетчатки
Отслоение сетчатки
Хроническая гипоксемия
Гипостенурия
Тубулярные дефекты
Хроническая почечная
недостаточность
Заболевания спинного мозга
Неоваскуляризация
с образованием
аневризмы
Холелитиаз
Гепатопатия
Аваскулярный некроз
Рентгенологическая патология
(деформация "рыбий рот")
Пролиферативная ретинопатия
Кожные язвы
108
Глава 3. Эритроциты
Для СКА характерна острая патология мочеполовой системы.
Рецидивирующий приапизм наблюдается более чем у 50 % мужчин с СКА. Приапизм,
продолжающийся несколько часов, следует лечить заменными переливаниями крови
с целью уменьшения эрекции, предотвращения рубцевания и развития
импотенции. Если консервативное лечение неэффективно, можно прибегнуть к
хирургическому вмешательству, обеспечивающему декомпрессию пещеристых тел.
Появление серповидных клеток в мозговом слое почки обусловливает возникновение
гематурии и папиллярного некроза. Вместе с тем, для исключения других причин
(инфекции мочевых путей, гломерулонефрит, опухоли, камни) больных СКА
с гематурией необходимо обязательно обследовать.
Пациенты с СКА склонны к остеомиелиту, который важно дифференцировать
от костных инфарктов. Остеомиелит чаще вызывают сальмонеллы,
стафилококки и энтеробактерии. Бактериологический диагноз должен быть поставлен до
начала лечения.
Хронические проявления СКА. Повторяющиеся эпизоды сосудистой окклюзии
и хронический гемолиз — причина прогрессирующей дисфункции фактически
всех органов (табл. 3-11). Рецидивирующие легочные инсульты обычно приводят
в 40-50-летнем возрасте к слабой или умеренно выраженной гипоксемии и часто
становится причиной дыхательной недостаточности с летальным исходом.
Возникновение ацидоза и гиперосмолярности мозгового слоя почек связано с
образованием серповидных клеток, поэтому у всех больных СКА рано возникает
хроническое заболевание почек. Одной из первых дисфункций почек является
гипостенурия (неспособность концентрировать мочу), обнаруживаемая уже к 10
годам жизни. Нарушение концентрационной способности почек делает больных
с СКА особенно чувствительными к дегидратации. Дефекты канальцев могут
проявляться в виде тубулярного ацидоза и гиперкалиемии. К другим
хроническим осложнениям СКА относятся желчнокаменная болезнь, нарушение функции
печени, ретинопатия и хронические язвы кожи.
Лечение серповидно-клеточной анемии. В основе лечения острой вазоокклюзии
лежит использование жидкостей, кислорода, фолиевой кислоты и наркотических
анальгетиков. Необходимо иметь в виду возможность присоединения внешнего
усиливающего патологию фактора, как правило, инфекции, что требует
соответствующего лечения. При симптоматической анемии и снижении величины гема-
токрита необходимы гемотрансфузии, но при этом гематокрит нельзя увеличивать
более 30 %, поскольку возрастание вязкости крови может ухудшить
микроциркуляцию. Показания для заменных трансфузий: острый грудной синдром с гипоксеми-
ей, инсульт, некроз костного мозга, приапизм, непереносимая боль. Цель обменных
трансфузий заключается в сокращении доли гемоглобина S до уровня меньше 35 %.
Чтобы предотвратить вазоокклюзию и сепсис у больных с функциональной
аспленией, необходимо интенсивное лечение инфекции. У детей опасность
развития сепсиса особенно высока, поэтому обязательна пенициллинопрофилактика
для предотвращения инфицирования Streptococcus pneumoniae. У взрослых чаще
всего пневмония вызывается Mycoplasma pneumoniae, чувствительной к эритромицину.
При подготовке к хирургическому вмешательству проводится инфузионно-
трансфузионная терапия с целью достижения нормоволемии и уровня
гемоглобина 100 г/л — состояния, при котором риск летального исхода во время операции
минимален. Последние рандомизированные исследования, проведенные в
различных медицинских центрах, показали, что этот консервативный режим столь
Анемия с ретикулоцитозом
109
же эффективен для снижения содержания гемоглобина S до уровня менее 30 %,
как и радикальный подход с заменными трансфузиями, при котором чаще
отмечаются трансфузионные осложнения.
Больные должны быть привиты поливалентной пневмококковой вакциной и
ежегодно проходить противогриппозную вакцинацию. К другим общим
профилактическим мерам относятся добавление к рациону фолиевой кислоты и
ежегодная консультация окулиста для своевременного выявления патологии сетчатки.
Сексуально активные женщины должны быть информированы о способах
контрацепции и возможных тяжелых последствиях беременности для матери и плода.
Так, могут развиваться пиелонефрит, инфаркт легкого, пневмония, острый
грудной синдром, послеродовое кровотечение, недоношенность, гибель плода.
Профилактическое использование заменных трансфузий во время беременности —
вопрос спорный, но в случае замедленного внутриутробного роста их следует иметь
в виду. При абортах и выкидышах необходимо тщательное наблюдение за
пациенткой, поскольку подобные состояния могут вызвать острое осложнение СКА.
Одно из последних рандомизированных исследований показало, что гидроксимо-
чевина повышает уровень фетального гемоглобина у больных с СКА и снижает
количество и тяжесть болевых приступов. Более высокая концентрация у-гемоглобина
в эритроцитах предотвращает вазоокклюзионные состояния за счет образования
а2р5у-тетрамеров, которые не подвергаются полимеризации и не вызывают
формирования серповидных клеток. Образованию фетального гемоглобина также
способствует масляная кислота, проходящая в настоящее время клинические испытания.
У большинства больных СКА развивается перегрузка железом, прежде всего
в результате многочисленных трансфузий. При уровне ферритина выше 2500-
3000 нг/мл необходимо начинать лечение дефероксамином.
Работники здравоохранения должны всегда помнить о том, что серповидно-
клеточная анемия — хроническое заболевание, сопровождающееся
рецидивирующими болевыми приступами и угрожающими жизни состояниями, и поэтому
пациентам с СКА чрезвычайно важна моральная поддержка.
Малые признаки СКА. У 8-10 % черного населения Америки, гетерозиготных
по СКА, клинические проявления развиваются редко. Анемия и вазоокклюзионные
приступы, как правило, отсутствуют. Вместе с тем могут развиваться гематурия,
изостенурия, а в редких случаях — патология сетчатки. Лица, относящиеся к этой
категории, должны пройти генетическое обследование в связи с риском развития
гемоглобинопатии у потомства, если у партнера есть один или несколько генов СКА,
гемоглобин С (о нем еще пойдет речь) или (3-талассемия. Возможен пренатальный
диагноз практически всех наиболее часто встречающихся гемоглобинопатии.
Другие гемоглобинопатии. В гемоглобине С глутаминовая кислота замещена
лизином в 6-м положении Р-глобиновой цепи. Эта мутация преобладает у жителей
Западной Африки, а гетерозиготность наблюдается примерно у 2 % афро-
американцев. У больных, гомозиготных по гемоглобину С, обычно выявляют
умеренную гемолитическую анемию, требующую минимального лечения. В
периферической крови обнаруживаются множественные мишеневидные клетки и
складчатые эритроциты; в некоторых из них выявляются внутриклеточные
кристаллы (табл. 3-3). У больных с гемоглобином SC серповидно-клеточный синдром
выражен более слабо, чем у больных с гемоглобином SS. В первом случае
наблюдается тенденция к спленомегалии и чаще встречаются ретинопатии. У больных с ге-
110
Глава 3. Эритроциты
моглобином S и р+-талассемией заболевание также выражено слабее, чем у
гомозигот по гемоглобину S. Однако при серповидно-клеточной Р°-талассемии
нормальный (3-гемоглобин не продуцируется и больные имеют такой же фенотип, как и
пациенты с гемоглобином SS. Гемоглобин Е определяется преимущественно у
жителей Юго-Восточной Азии, особенно Бирмы и Таиланда, и является
результатом мутации гена, кодирующего аминокислоту в 26-м положении Р-цепи.
Симптоматика у гомозигот практически отсутствует, анемия выражена слабо,
обнаруживается микроцитоз и мишеневидные клетки; у гетерозигот — только микроцитоз.
Гемолитическая анемия, вызванная внеэритроцитарной
патологией
Иммунный гемолиз
При иммунном гемолизе причина ускоренной деструкции эритроцитов
заключается в связывании клеточной мембраны с антителами и/или комплементом.
Диагноз ставится на основании результатов прямого антиглобулинового теста Кумб-
са, в ходе которого антитела против иммуноглобулинов и комплемента
смешиваются с эритроцитами пациента. Если на эритроцитарной мембране при-
ч сутствует иммуноглобулин или комплемент, происходит агглютинация клеток.
С помощью непрямого антиглобулинового теста Кумбса в сыворотке крови
пациента выявляют свободно циркулирующие антитела. При иммунной
гемолитической анемии он оказывается положительным у 80 % больных.
Антитела, вызывающие иммунный гемолиз, обычно подразделяются на две
группы: с максимальной активностью при обычной температуре тела (тепловые
антитела — обычно IgG, редко IgA) и с максимальной активностью в холодовой
среде (холодовые антитела — обычно IgM, иногда IgG). Каждая группа антител
ассоциируется с определенными клиническими синдромами (табл. 3-12).
Тепловая иммунная гемолитическая анемия. При этом виде анемии образование
антител может быть первичным (идиопатическим) или вторичным в результате
аутоиммунных заболеваний (системная красная волчанка, антикардиолипино-
вый синдром), лимфопролиферативных нарушений (хронический лимфоцитар-
цый лейкоз, лимфома), приема лекарственных препаратов, новообразований.
Идиопатическая иммунная гемолитическая анемия чаще всего наблюдается
у женщин среднего возраста. Как правило, появляется желтуха, симптомы
анемии, у % больных увеличивается селезенка. Количество ретикулоцитов выше
нормы. В периферической крови обнаруживаются сфероциты, которые
формируются в результате действия макрофагов селезенки на мембрану эритроцитов,
покрытых антителами. Иногда появляются ядерные эритроциты (табл. 3-3).
Отсутствие ретикулоцитов не исключает возникновение гемолиза, так как в редких
случаях антитела направлены также и против ретикулоцитов и/или у больных
может иметь место сопутствующий дефицит фолиевой кислоты или железа.
Однако уровень гаптоглобина обычно низкий, и, если гемолиз внутрисосудистый,
уровень гемоглобина в плазме повышается.
Диагноз ставится на основании результатов прямого антиглобулинового
теста, который выявляет наличие IgG, обычно IgGi и IgG3, на эритроцитарной
мембране. Оба подкласса IgG могут активировать комплемент; иногда комплемент
(C3d) также обнаруживается на мембране эритроцитов.
Анемия с ретикулоцитозом
111
Лечение иммунной гемолитической анемии включает устранение, если это
возможно, основного патологического состояния, вызывающего гемолиз; подавление
образования антител и/или ингибирование уничтожения покрытых антителами
эритроцитов в селезенке и печени. Кортикостероиды, угнетающие активность
макрофагов и блокирующие образование антител, являются для большинства
больных базисным терапевтическим средством. Для подавления антителообразу-
ющего клона используют иммуносупрессию (циклофосфамид, хлорамбуцил, аза-
тиоприн). Некоторым пациентам помогает даназол — синтетический андроген,
ингибирующий функциональную активность макрофагов и, вероятно,
оказывающий иное неспецифическое действие на иммунную систему. Иногда эффективно
внутривенное введение иммуноглобулина, блокирующего активность
макрофагов. При стероидной зависимости и рефрактерности к другим видам лечения
целесообразно выполнение спленэктомии, эффективной примерно у 50 % больных.
Гемолиз, индуцированный лекарственными препаратами. сс-Метилдопа — антиги-
пертёнзивное средство, которое примерно у 20 % пациентов вызывает образование
антител против эритроцитарной мембраны. Однако клинически выраженная
гемолитическая анемия возникает редко. Другие лекарственные препараты
индуцируют выработку антиэритроцитарных антител или в ответ на их связывание
с эритроцитарной мембраной (пенициллин, цефалоспорины и пр.), или путем
формирования тройных иммунных комплексов (хинидин, сульфаниламиды и др.).
Во всех случаях лечение сводится к отмене лекарственного средства (табл. 3-12).
Холодовая иммунная гемолитическая анемия. Преходящий иммунный гемолиз
холодового типа обычно связан с инфекцией, особенно вызываемой Mycoplasma
pneumoniae и вирусом Эпштейна-Барр (ВЭБ). Патологическим
иммуноглобулином является IgM с максимумом активности при температуре 4-18 °С. IgM
активирует комплемент в конечностях, где температура ниже, чем в других частях тела.
Каскад реакций комплемента прерывается, когда эритроциты проходят через
теплое "ядро" тела. В итоге развивается внутрисосудистый гемолиз. Прямой антигло-
булиновый тест обычно подтверждает наличие только СЗЬ на эритроцитарной
мембране, так как IgM отщепляется при циркуляции эритроцитов. Антитела (хо-
лодовой агглютинин), как правило, поликлональны и направлены против
антигенов группы крови I (при инфекциях, вызванных Mycoplasma pneumoniae) или i
(при инфекциях, вызванных ВЭБ). У больных в стадии ремиссии часто
развиваются гемолиз и анемия. Лечение поддерживающее, гемолиз самоограниченный.
Хроническое заболевание с синдромом Холодовых агглютининов нередко
наблюдается у лиц пожилого возраста (50-80 лет) и часто связано с образованием
моноклональных антител IgM и лимфопролиферативными нарушениями
(хронический лимфолейкоз, макроглобулинемия Вальденстрема). Обычно возникает
легкая анемия, но иногда развивается тяжелый внутрисосудистый гемолиз и
почечная недостаточность. Лечение включает устранение переохлаждения;
некоторым пациентам показаны кортикостероиды. Проведение спленэктомии не
рекомендуется, поскольку эритроциты, покрытые СЗЬ, элиминируются в основном
в печени, а не в селезенке.
Пароксизмальная холодовая гемоглобинурия наблюдается в поздних стадиях
сифилиса (антитела Доната-Ландштейнера), а также после перенесенных
вирусных инфекций. Антитела класса IgG реактивны как в холодной среде, так и при
температуре 37 °С и способны активировать комплемент.
112
Глава 3. Эритроциты
ТАБЛИЦА 3-12. Аутоиммунная гемолитическая анемия
Вид
Характерная патология/причины
Класс антител
Тепловая аутоиммунная гемолитическая анемия
Обычно
IgG
Аутоиммунная Коллагеноз
гемолитичес- Заболевания щитовидной железы
кая анемия Инфекции
(АИГА) ХЛЛ
НХЛ
Миелома
Болезнь Ходжкина
Другие новообразования
Гемолитическая анемия, вызванная лекарственными препаратами
Тип 1
Адсорбция
препарата-
гаптена
Тип 2
"Невинный
свидетель";
образование
тройного
иммунного
комплекса
Высокая доза пенициллина
Fab-фрагмент
антитела (IgG)
Хинин, хинидин, фенацетин, хлорпро-
мазин рифампин, изониазид, стибо-
фен, р-аминосалициловая кислота,
гидрохлортиазид, тетрациклин
ТипЗ
Вызванная
препаратом
адсорбция
белков на
эритроцитах
(неиммунная)
Тип 4
Индукция
и связывание
аутоантител
Цефалотин
Метилдопа, леводопа, прокаинамид,
ибупрофен, диклофенак, тиоридизин,
интерферон-а
Ч&
Анемия с ретикулоцитозом
113
Прямой Непрямой
Фиксация антиглобули- антиглобу-
комплемента
Иногда
новый тест
(ПАТ)
IgG
Комплемент
+ +/-
линовый
тест (HAT)
+
Локализация
гемолиза
РЭС Внутри
сосудов
+
Примечания
Типичны микросфероцитоз и
спленомегалия
Лечение:
- иммуносупрессия
- спленэктомия
Редко
+, если
тестируемые
эритроциты
инкубировать с
лекарственным
препаратом
Слабо. Лекарственный препарат
связывается с мембраной эритроцита.
Антитела направлены против
лекарственного препарата
Да
+, если
тестируемые
эритроциты
инкубировать с
лекарственным
препаратом
Антитело и лекарственный
препарат образуют иммунные
комплексы, связывающиеся
неспецифически с эритроцитами, и
активируют комплемент.
Антитело направлено как против
лекарственного препарата, так и
против мембранного белка
Нет
Нет
Лекарство вызывает
неспецифическое связывание белков
плазмы (включая IgG, белки
комплемента, трансферрин,
альбумин и фибриноген) с эритроци-
тарной мембраной. ПАТ
положителен, но гемолиз бывает редко
Антитела направлены против
белков эритроцита, например
Rh, а не против лекарственного
препарата. Положительный ПАТ
наблюдается у 10-20 % больных,
получающих метилдопу, но
гемолиз — только у 2-5 %.
114
Глава 3. Эритроциты
ТАБЛИЦА 3-12. (Продолжение)
Вид Характерная патология/причины Класс антител
Холодовая аутоиммунная гемолитическая анемия
Холодовой Поликлональные антитела: микоплаз- IgM
агглютинин ма (анти-l), ИМН (анти-i), ЦМВ, листе-
риоз, свинка, ПБЭ, сифилис, коллаге-
ноз
Моноклональные антитела: макрогло-
булинемия Вальденстрема, лимфома,
ХЛЛ, саркома Капоши, миелома
Пароксиз- Сифилис, перенесенная вирусная ин- IgG
мальная фекция (корь, свинка и пр.) (антитела Доната-Ландштейнера)
холодовая
гемоглобин-
урия
АИГА — аутоиммунная гемолитическая анемия; РЭС — ретикулоэндотелиальная система;
ХПП — хронический лимфолейкоз; НХЛ — неходжкинская лимфома; ИМН — инфекционный
мононуклеоз; ПБЭ — подострый бактериальный эндокардит; ПАТ — прямой антиглобулиновый
тест; ЦМВ — цитомегаловирус. По: Packman С. Н., Leddy J. P. Drug-related IHA. Из: Beutler E.,
Lichtman M. A., Colles В. S., Kipps T. J. eds. Williams Hematology, 5th ed. New York: McGraw-Hill,
1995:693.
Микроангиопатическая гемолитическая анемия
При микроангиопатической гемолитической анемии гемолиз вызван
травматической фрагментацией эритроцитов при прохождении их через поврежденные
кровеносные сосуды или через искусственный сердечный клапан. Подобные
явления наблюдаются при диссеминированном внутрисосудистом свертывании
(глава 6), тромботцческой тромбоцитопенической пурпуре/гемолитическом
уремическом синдроме, преэклампсии и эклампсии, HELLP-синдроме при
беременности (гемолиз, повышение активности ферментов печени, тромбоцитопения),
метастазирующих опухолях и введении некоторых лекарственных препаратов
(митомицин С, циклоспорин). Диагноз моя^но поставить на основании анализа
мазка периферической крови, на котором обнаруживаются фрагментированные
эритроциты, называемые шистоцитами (табл. 3-3). Лечение базируется на
купировании первичного патологического процесса (главы 5 и 6).
Гемолиз, связанный с инфекцией
Токсины и другие бактериальные продукты могут вызвать прямое поражение
эритроцитарной мембраны и гемолиз, что наблюдается при клостридиальном
сепсисе, сепсисе и/или эндокардите, вызванных стрептококком, стафилококком,
энтерококком. Прямое поражение эритроцитов такими микроорганизмами, как
малярийный плазмодий, бабезия, бартонелла, вызывает тяжелый внутрисосудис-
Анемия с ретикулоцитозом
115
Фиксация
комплемента
Да '
Да
Прямой
антиглобули-
новый тест
(ПАТ)
IgG
+
(на
холоде)
Комплемент
+
+
(во
время
приступа)
Непрямой
антиглобу-
линовый
тест (HAT)
+/-
+
(на холоде)
Локализация
гемолиза
РЭС Внутри
+
(печень и
костный
мозг,
ноне
селезенка)
+
сосудов
+
++
Примечания
IgM и комплемент связываются^
с эритроцитами на периферии
(кисти, стопы)
IgM отщепляется, когда
эритроцит перемещается в более
теплую зону. организма;
комплемент остается. Лечение: эти-
отропное, плазмаферез,
избегать охлаждений
Те же явления, что и при холодо-
вой агглютинации
тый гемолиз. Диагноз легко ставится при обнаружении паразитов в эритроцитах
во время исследования периферической крови (табл. 3-3).
Выявление эритроцитоза
Под эритроцитозом понимают увеличение эритроцитарной массы (в среднем
36 мл/кг у мужчин и 32 мл/кг у женщин). Такое состояние можно заподозрить при
повышении гематокрита и концентрации гемоглобина. Однако эти параметры
зависят от объема крови: если у больного уменьшен объем плазмы, то концентрация
гемоглобина и гематокрит будут расти, а эритроцитарная масса останется неизменной.
Истинное увеличение эритроцитарной массы вызывается или тканевой
гипоксией и избыточной секрецией эритропоэтина, или автономным ростом и
созреванием клеток-предшественников эритроцитов, не зависящим от обычных регуля-
торных процессов (табл. 3-13). Наиболее частой причиной эритроцитоза является
тканевая гипоксия, стимулирующая повышение секреции эритропоэтина.
На большой высоте над уровнем моря, где снижено давление кислорода,
повышение кислородной емкости крови происходит за счет увеличения эритроцитарной
массы. Аналогичный компенсаторный механизм наблюдается у больных с
хронической гипоксемией, развивающейся в результате заболевания легких,
право-левого шунта камер сердца, хронического отравления окисью углерода. Пациенты
' с синдромом ночного апноэ страдают от перемежающейся ночной гипоксемии,
приводящей к эритроцитозу. Поскольку днем у них нормальное р02, такой
диагноз трудно поставить. При наличии гемоглобинов с высоким сродством к
кислороду, которые выделяют его в ткани при более низком, чем в норме, р02, также
развивается эритроцитоз, поскольку имеет место тканевая гипоксия.
Высокий уровень эритропоэтина наблюдается преимущественно при
заболеваниях почек, особенно при почечной кисте и/или гидронефрозе. У 15 % больных
116
Глава 3. Эритроциты
после трансплантации почек эритроцитоз возникает в результате повышенной
секреции эритропоэтина. Некоторые опухоли могут секретировать эритропоэтин
автономно (почечно-клеточная карцинома, печеночно-кдеточная карцинома, ге-
мангиобластома мозжечка, опухоли матки). Повышенный уровень
эритропоэтина иногда выявляется у женщин, получавших андрогены.
Истинная полицитемия представляет собой миелопролиферативное
заболевание, при котором предшественники эритроцитов делятся и созревают
самостоятельно, не подчиняясь регуляции эритропоэтином. Уровень последнего обычно
понижен. Это клональное нарушение, для которого характерна активация всех трех
ростков кроветворения с повышением гематокрита и уровня гемоглобина (глава 9).
При эритроцитозе часто наблюдаются симптомы, связанные с увеличением
объема крови и ее вязкости: головная боль, нарушения зрения, гипертензия,
тромбоз (инсульты, ишемическая болезнь сердца, поражения периферических сосудов).
При обследовании пациента с высоким гематокритом и повышенным уровнем
гемоглобина необходим тщательный сбор анамнеза: пребывание на большой
высоте над уровнем моря, ночное апноэ, курение, воздействие угарного газа, семейный
анамнез (наследственность по высокоаффинным гемоглобинам). Необходимо
исследовать вязкость крови. Для истинной полицитемии характерно увеличение
селезенки. Шумы в сердце или легких могут свидетельствовать о право-левом шунте.
Диагноз эритоцитоза ставится на основании результатов измерения величины
эритроцитарной массы. Ее определяют по степени разведения изотопа, получен-
ТАБЛ И ЦА 3 -13. Дифференциальная диагностика эритроцитоза
Ложный эритроцитоз (снижен объем плазмы)
Обезвоживание организма
Преэклампсия
Гипертензия
Повышенный объем эритроцитарной массы
Гипоксемия
Пребывание на большой высоте над уровнем моря
Хроническое заболевание легких
Синдром ночного апноэ
Право-левый шунт камер сердца (врожденный порок сердца, артерио-венозная
аномалия)
Болезни почек
Гидронефроз
Поликистоз
Трансплантация почек
Опухоли
Почечно-клеточный рак
Печеночно-клеточный рак
Гемангиобластома мозжечка
Другие эритропоэтинсекретирующие опухоли
Эндокринные нарушения
Тиреотоксикоз
Андрогенотерапия
Семейный эритроцитоз
Миелопролиферативные нарушения
Анемия с ретикулоцитозом
117
ные данные выражаются в мл/кг. Однако этот'тест ненадежен. Поэтому, если
предполагается наличие эритроцитоза, диагноз следует подтвердить, несмотря
на нормальную эритроцитарную массу. При диагностике этого заболевания
необходимо также определить артериальное напряжение кислорода, уровень эритро-
поэтина, концентрацию СО и выполнить электрофорез гемоглобина. Необходимо
провести сканирование почек и помнить о возможном развитии синдрома
остановки дыхания во сне.
Лечение должно быть направлено на устранение причины возникновения
патологии (у больных с вторичным эритроцитозом). Для снижения гематокрита
до уровня ниже 45 % необходима флеботомия. Лечение истинной полицитемии
обсуждается в главе 9.
Клинический пример
Мужчина (белый) в возрасте 40 лет пришел на очередной осмотр.
Занимается бегом, иногда принимает аспирин или другие НПВС для купирования
боли в суставах, особенно после 10-километрового пробега. Последняя
пробежка была 5 дней назад. Общий осмотр не выявил никаких
особенностей. Однако анализ кала показал скрытое кровотечение (проба на
определение скрытой крови). Гемоглобин —132 г/л, гематокрит — 38,1 %.
Вопрос 1. Должно ли Вас это насторожить?
Ответ. Да. Хотя потеря крови из ЖКТ может объясняться приемом
лекарственных средств, но несмотря на то, что гематокрит и уровень гемоглобина
только слегка понижены, требуется дальнейшее обследование пациента.
Вопрос 2. Ваши дальнейшие действия?
Ответ. Необходимо тщательно проанализировать мазок периферической
крови и оценить характеристики эритроцитов.
При исследовании периферической крови выявлены анизоцитоз и пойки-
лоцитоз и незначительная полихромазия эритроцитов. Тромбоциты и
лейкоциты без морфологических изменений. СОК — 79 мкм3, ССГ —
26 пг/1 эритроцит, СКГ — 320 г/л эритроцитов.
Вопрос 3. О чем свидетельствуют результаты анализа периферической
крови и что Вы намерены предпринять дальше?
Ответ. Отсутствие выраженной полихромазии указывает на то, что,
несмотря на анемию, отмечается незначительная реакция со стороны костного
мозга, который должен поставлять в кровь большее число юных эритроцитов.
При окрашивании по Райту (обычная окраска мазка периферической
крови) незрелые эритроциты выглядят как крупные клетки без характерной
бледной области в центре и имеют матовую серовато-пурпурную окраску.
Используя другие красители, необходимо провести подсчет ретикулоцитов.
Оказалось, что ретикулоциты составляют 1 %. С помощью приведенных
в этой главе формул было определено, что нормализованное количество
ретикулоцитов — 0,79 %, а индекс продукции ретикулоцитов равен 0,39 %.
Полученные данные подтверждают, что костный мозг слабо реагирует
на анемию. Можно было ожидать, что индекс продукции ретикулоцитов
будет выше нормы. Но выяснилось, что он составляет менее половины
от нормальной величины (1 %), т. е. продукция эритроцитов не адекватна
степени тяжести анемии.
118
Глава 3. Эритроциты
Вопрос 4. Что это может означать?
Ответ. Необходимо выяснить причины низкого гематокрита и
недостаточного образования эритроцитов. На данный момент очевидно развитие
у пациента гипохромной микроцитарной анемии с гипопролифератив-
ной реакцией костного мозга. Обнаружение крови в кале (что следует
подтвердить дополнительными пробами) прежде всего предполагает
кровопотерю из ЖКТ, приводящую к дефициту железа. Недостаток
железа не дает возможности костному мозгу обеспечить компенсаторное
продуцирование эритроцитов. Если бы эритроциты разрушались
(например, за счет гемолиза, опосредованного селезенкой) или происходила их
секвестрация (например, в увеличенной селезенке), то железо должно
было вновь включаться в метаболизм, позволяя нормально
функционировать костному мозгу, продуцирующему эритроциты. С другой
стороны, если создаются биохимические препятствия костному мозгу в
осуществлении компенсации путем дефицита необходимых компонентов
(железо, фолиевая кислота, витамин В12) или структурная блокада за счет
воспаления, развития инфекции, опухоли, лимфомы, фиброза, гранулемы,
то возникает гипопролиферативная реакция, что и наблюдается в
данном случае.
Вопрос 5. Каков следующий этап исследования?
Ответ. Необходимо исследовать несколько образцов кала на скрытую
кровь, а также оценить уровень железа в сыворотке.
Анализы сделаны. Кал коричневого цвета, но пробы на скрытую кровь
положительные.
Содержание ферритина в сыворотке — 2 нг/мл
Содержание железа в сыворотке — 300 мкг/л
Общая связывающая способность железа в сыворотке — 5000 мкг/л
Вопрос 6. Как Вы интерпретируете эти данные?
Ответ. Они подтверждают, что у больного выраженный дефицит железа,
несмотря на незначительно сниженные гематокрит и уровень гемоглобина.
Вопрос 7. Есть необходимость в дальнейших гематологических
исследованиях (например, костного мозга)?
Ответ. Хотя исследование костного мозга могло бы помочь определить
присутствие или отсутствие депонированного железа и
продемонстрировать слабую реактивность костного мозга (например, сопутствующее мега-
лобластное состояние), нет данных (например, наличие микроовалоцитов),
позволяющих предположить наличие вторичной причины данного
состояния. Кроме того, результаты лабораторных тестов характерны для железо-
дефицитной анемии, связанной с желудочно-кишечным кровотечением.
Вопрос 8. Что следует предпринять?
Ответ. Необходимо полное исследование ЖКТ, чтобы выяснить место
кровотечения (исследование верхнего и нижнего отделов ЖКТ).
Результаты:
С помощью рентгенографического исследования обнаружена аденокарци-
нома слепой кишки. Сделана операция, прогноз благоприятный.
Дальнейшей терапии не потребовалось. Через 5 лет после хирургического
вмешательства состояние пациента нормальное.
Анемия с ретикулоцитозом
119
Клинический пример
Женщина в возрасте 60 лет поступила в больницу с анемией. Чувствовала
себя хорошо, за исключением "артрита", в связи с чем принимала на-
проксен в течение 17 месяцев.
За неделю до госпитализации почувствовала одышку, поднявшись всего
на 3 ступеньки крыльца. Соседка обратила внимание на желтизну белков
глаз пациентки. Появление мочи темного цвета встревожило больную, и
она обратилась в клинику. Врач, сделав анализы, обнаружил отклонение
гематокрита от нормы и отправил ее в неотложное отделение госпиталя.
Больная выглядела изнуренной, желтушной. Склеры желтые. На коже и
слизистой рта патологии не обнаружено. Лимфаденопатия
отсутствовала. Сердце и легкие в норме, но селезенка на 3 см ниже уровня левой
реберной дуги. Гемоглобин — 50 г/л, гематокрит — 15,3 %, количество
лейкоцитов и тромбоцитов в норме.
Вопрос 1. Ваши действия?
Ответ. Исследование мазка периферической крови. Тромбоциты и
лейкоциты не изменены. Выражен анизоцитоз и пойкилоцитоз эритроцитов.
Обнаружено большое число сфероцитов и пузырчатых клеток, а также клеток
с признаками полихромазии.
Вопрос 2. Какой тест надо провести, чтобы подтвердить подозрение, что
костный мозг реагирует на низкий гемоглобин и гематокрит?
Ответ. Подсчитать количество ретикулоцитов. Нормализованное
количество ретикулоцитов составило 8 %.
Вопрос 3. Как вы определите характер анемии?
Ответ. Проведены пробы Кумбса: тест прямого определения антител
положительный, а непрямого — отрицательный. Клетки покрыты IgG-антителами.
Вопрос 4. Каков оптимальный терапевтический подход в данном случае?
Ответ. Вполне вероятно, что у больной гемолитическая анемия с
синдромом тепловых агглютининов, возможно, связанная с коллагенозом или
применением напроксена. Прием напроксена следует прекратить. Для
коррекции низкого гематокрита и симптомов анемии необходимы гемотранс-
фузии. Рекомендуются высокие дозы кортикостероидов, которые
постепенно уменьшают после стабилизации гематокрита.
Результаты.
Несмотря на прием высоких доз кортикостероидов, гемолиз продолжался.
Добавление даназола не привело к положительному эффекту. Накануне
плановой спленэктомии проведена вакцинация против Haemophilus
influenzae, Streptococcus pneumoniae и Neisseria meningitidis. Проведение
спленэктомии купировало гемолитический процесс. После операции кор-
тикостероиды отменили. Наступила ремиссия.
Клинический пример
У 39-летнего мужчины при обследовании обнаружен повышенный
гематокрит. Пациент жалуется на периодические головные боли,
неконтролируемую гипертензию, несмотря на прием диуретиков. Пациент—большой
любитель пива, выкуривает несколько пачек сигарет в день. Это довольно тучный
мужчина с гиперемией лица. Артериальное давление —160/97 мм рт. ст.,
120 t Глава 3. Эритроциты
других аномалий при физикальном обследовании не обнаружено. Анализ
крови: лейкоциты — 9900/мкл, нормальная лейкоцитарная формула;
тромбоциты — 489 000/мкл; гемоглобин —184 г/л; гематокрит — 53,7 %.
Вопрос 1. Каков должен быть подход к обследованию этого пациента?
Ответ. Первый диагностический вывод — это истинный, или абсолютный,
а не относительный эритроцитоз. Для определения эритроцитарной массы
и объема плазмы предпочтительно использовать радиологическое
исследование, поскольку у больного есть предрасполагающие факторы как для
развития абсолютного эритроцитоза (курение, которое способствует
образованию карбоксигемоглобина, пониженному поступлению кислорода в ткани,
повышенной секреции эритропоэтина с последующим повышением
продукции эритроцитов), так и относительного эритроцитоза (применение
диуретиков, которые снижают объем плазмы и тем самым повышают гематокрит).
Вопрос 2. Обнаружился абсолютный эритроцитоз (объем эритроцитов
39 мл/кг). Ваши действия?
Ответ. Причин истинного эритроцитоза много. При определении лучшего
диагностического подхода следует учитывать такие факторы, как степень
неотложности, инвазивность, экономичность. В данном случае уже
определено, что повышение гематокрита — реальный факт, но без клинической
симптоматики, поэтому необходимо тщательное обследование. Степень
насыщения кислородом (пульсовая оксиметрия) нормальная. Содержание
карбоскигемоглобина (и это не удивительно) повышено. Источник
угарного газа дома не обнаружен (даже при нормальном содержании
карбоксигемоглобина эритроцитоз может быть индикатором преходящего
воздействия угарного газа). По рекомендации врача больной прекратил курить.
Спустя 1 месяц повторное определение уровня карбоксигемоглобина
показывает, что он нормализовался. Зато теперь гематокрит повысился до 56 %.
Вопрос 3. Каковы дальнейшие диагностические мероприятия?
Ответ. Нужно определить уровень эритропоэтина1. Он чрезвычайно
высок — 94 единицы.
Вопрос 4. Что это означает?
Ответ. Теперь диагноз истинной полицитемии становится менее
вероятным. Это состояние развивается независимо от стимуляции стволовых
эритроцитарных клеток эритропоэтином, и при нем уровень эритропоэтина
низкий. Надо учесть все факторы, вызывающие повышение содержания
эритропоэтина. Дома источника угарного газа нет. Ночное апноэ — еще
одна проблема, последствия которой проявляются в дневное время.
Связанная с ним ночная гипоксемия может являться причиной эритроцитоза,
что следует учесть, особенно если пациент храпит ночью (об этом сообщает
жена). Следует провести исследование характера сна пациента.
Вопрос 5. Исследование не выявило патологии сна. Не обнаружено аномалий
и при поиске гемоглобинов с повышенным сродством к кислороду. Что дальше?
Ответ. Поскольку эритроцитоз и высокий уровень эритропоэтина
наблюдаются при некоторых опухолях, необходимо обследовать пациента в
отношении новообразований: гемангиобластомы мозжечка (что в данном случае
1 Некоторые врачи выполнили бы это исследование ранее.
Анемия с ретикулоцитозом
121
маловероятно из-за отсутствия симптомов), печеночно-клеточной
карциномы, почечно-клеточной карциномы, опухолей матки у женщин.
Вопрос 6. Ваши действия?
Ответ. Проведена компьютерная томография (КТ) живота, и обнаружено
увеличение почки. Повторный анализ мочи показал гематурию (1 месяц назад ее
не было). Биопсия под контролем КТ показала, что причина увеличения — по-
чечно-клеточная карцинома. Отдаленных метастазов не выявлено.
Произведено хирургическое лечение. Поражения лимфатических узлов сосудов нет.
Результаты.
После операции пациент чувствует себя хорошо. Гематокрит
нормализовался. Спустя 4 года после операции рецидивов не наблюдалось, метастазы
отсутствовали.
Избранная литература
Berliner N., Duffy T. P., Ablelson H. Т. Approach to the adult and child with anemia. In:
Hoffman R. H. et al. (eds). Hematology: Basic Principles and Practice, 2nd ed. New
York: Churchill Livingstone, 1995:468-483.
Рассмотрена этиология анемий, представлены критерии группировки анемий по
числу ретикулоцитов и СОК.
Beutler E. The common anemias./ЛМД, 1988; 259:2433-2437.
Подробное описание железодефицитной анемии, талассемии и анемии при
хронических заболеваниях, включая патогенез, клинику и лечение.
Tabbara L. A. Erythropoietin: biology and clinical applications. Arch. Intern. Med., 1993;
153:298-304.
Краткий обзор, посвященный структуре и функции эритропоэтина и его роли в
лечении анемии при почечной недостаточности, злокачественных опухолях и
хронических заболеваниях.
Железодефицитная анемия
Brittenham G. M. Disorders of iron metabolism: iron deficiency and overload. In: Hoffman
R. H. et al. (eds). Hematology: Basic Principles and Practice, 2nd ed. New York:
Churchill Livingstone; 1995:492-522.
Описание метаболизма железа, методов лабораторной оценки дефицита, а также
клинических проявлений и лечения больных с недостатком и избытком железа.
Гемоглобинопатии
Bunn H. F. Sickle hemoglobin and other hemoglobin mutants. In: Stamatoyoannopoulos
G., Nienhuis A. W., Majerus P. W., Varmus H. (eds). The Molecular Basis of Blood
Diseases, 2nd ed. Philadelphia: W. B. Saunders, 1994:207-256.
Обзор молекулярных основ и патофизиологии гемоглобинопатии.
Bunn H. F., Ferget В. G. Hemoglobin: Molecular, Genetic and Clinical Aspects.
Philadelphia: W. B. Saunders; 1986.
Детальное обсуждение структуры и функции гемоглобина и гемоглобинопатии,
включая талассемию и серповидно-клеточную анемию.
Embury S. H.j Hebbel R. P., Mohandas N., Steinberg M. H. Sickle Cell Disease: Basic
Principles and Clinical Practice. New York: Raven Press; 1994.
Рассмотрены все аспекты серповидно-клеточной анемии, включая молекулярные
основы, патофизиологию, клинические проявления и лечение.
122
Weatherhall D. J. The Thalassemias. In: Stamatoyoannopoulos G., Nienhuis A. W.,
Majerus P. W., Varmus H. (eds). The Molecular Basis of Blood Diseases, 2nd ed.
Philadelphia: W. B. Saunders, 1994:157-206.
Обзор молекулярньис механизмов возникновения талассемии, включает раздел,
посвященный эпидемиологии, клиническому течению и лечению.
Гемолитические анемии
Benz E. J. Jr. The erythrocyte membrane and cytoskeleton: structure, function, and
disorders. In: Stamatoyoannopoulos G., Nienhuis A. W., Majerus P. W., Varmus H.
(eds). The Molecular Basis of Blood Diseases, 2nd ed. Philadelphia: W. B. Saunders,
1994:257-292.
Обзор структуры и функции липидов и протеинов мембраны эритроцита, а также их
молекулярных дефектов.
Paglia D. E. Enzymopathies. In: Hoffman R. H. et al. (eds). Hematology: Basic Principles
and Practice, 2nd ed. New York: Churchill Livingstone, 1995: 656-666.
Краткий обзор энзимопатий, вызывающих гемолиз.
Palek J., Kahr К. Е. Mutations of the red blood cell membrane proteins: from clinical
evaluation to detection of the underlying genetic defect. Blood, 1992; (80): 308-330.
Краткий обзор молекулярных дефектов белков мембраны эритроцита и их
клинических проявлений.
Schwartz R. S., Silberstein L. E., Berkman E. M. Autoimmune hemolytic anemias. In:
Hoffman R. H. et al. (eds). Hematology: Basic Principles and Practice, 2nd ed. New
York: Churchill Livingstone, 1995:710-728.
Обзор по иммунологическим механизмам деструкции эритроцитов, клиническим
проявлениям различных типов гемолиза и их лечению. )
Мегалобластные анемии
Anthony А. С. Megaloblastic anemias. In: Hoffman R. H. et al. (eds). Hematology: Basic
Principles and Practice, 2nd ed. New York: Churchill Livingstone; 1995: 552-585.
Подробный анализ метаболизма витамина В12 и фолиевой кислоты, а также
этиологии, клиники и лечения мегалобластных анемий.
Schilling R. F., Williams W.J. Vitamin B12 deficiency: underdiagnosed, overtreated. Hosp.
Pract., 1995; 30(7): 47-52.
Обоснование метода парентеральной терапии пациентов с пернициозной анемией и
неврологическими симптомами. Рассмотрены вопросы проведения лечения при
пограничных уровнях витамина В12-
Toh В. Н., van Driel I. R., Gleeson P. A. Mechanisms of disease: pernicious anemia. New
Engl.J. Med., 337:1441-1448,1997.
Новейшие данные по патогенезу, клинике и лечению пернициозной анемии.
Эритроцитоз
Spivak J. L. Erythrocytosis. In: Hoffman R. H. et al. (eds). Hematology: Basic Principles
and Practice, 2nd ed. New York: Churchill Livingstone, 1995:484-491.
Обсуждение этиологии, дифференциальной диагностики, оценки критериев тяжести
и лечения эритроцитоза.
Глава 4
Лейкоциты
Алан Дж. Розмарин
Клетки "белой крови", или лейкоциты, являются основой антимикробной защиты
организма. В эту разнородную группу "защиты" входят основные эффекторы
иммунных и воспалительных реакций. Участвуя в возникновении и развитии
физиологической воспалительной реакции, лейкоциты играют важную роль и при
патологическом воспалении, например при аутоиммунных заболеваниях.
Термин "лейкоцит" больше относится к внешнему виду клеток (leukos с греч. —
белый) в образце крови после центрифугирования. Лейкоциты представляют собой
гетерогенную группу клеток, которые можно классифицировать по
происхождению (миелоидные или лимфоидные) и по их функции (фагоциты или иммуноци-
ты). В клинической практике лейкоциты обычно группируют в соответствии
с морфологией клеточного ядра (полиморфно-ядерные или мононуклеарные)
или по наличию цитоплазматических включений (гранулоциты).
Нейтрофилы
Нейтрофильные гранулоциты представляют собой самую большую группу
циркулирующих лейкоцитов (рис. 4-1). Термин "нейтрофилъный" описывает
внешний вид цитоплазматических гранул при окрашивании по Райту-Гимзе. Вместе
с эозинофилами и базофилами нейтрофилы относятся к классу гранулоцитов.
В связи с наличием характерного многодолевого (сегментированного) ядра нейт-
рофил называют также полиморфно-ядерным лейкоцитом (ПМЯЛ). Эти
высокоспециализированные клетки мигрируют в очаги инфекции, где они распознают,
захватывают и уничтожают бактерии. Осуществление этой функции возможно
благодаря наличию у нейтрофилов способности к хемотаксису, адгезии,
передвижению и фагоцитозу. У них имеется метаболический аппарат для
продуцирования токсических веществ и ферментов, разрушающих микроорганизмы.
Развитие нейтрофилов
В костном мозге можно наблюдать шесть последовательных морфологических
стадий созревания нейтрофилов: миелобласт, промйелоцит, миелоцит, метамиелоцит,
палочкоядерная и сегментоядерная клетка (рис. 4-2 и приложение 4). Кроме того,
там же имеются более ранние, морфологически не идентифицируемые, коммитиро-
ванные предшественники нейтрофилов: КОЕ-ГМ и КОЕ-Г (рис. 1 -1). Наличие этих
клеток-предшественников доказано способностью костного мозга порождать ком-
митированные миелоидные колонии in vitro и возобновлением гранулопоэза после
экспериментальной или терапевтической трансплантации костного мозга.
124
Глава 4. Лейкоциты
Рис. 4-1. Лейкоциты в мазке периферической крови. (А) Нейтрофил (полиморфно-ядерный
нейтрофил); (Б) эозинофил; (В) базофил; (Г) моноцит; (Д) лимфоцит. (По: Hoffbrand А. V., Pettit J. E.
Essential Hematology, 3rd ed. Cambridge, Massachusetts: Blackwell, 1993: 169.)
Созревание нейтрофилов сопровождается прогрессирующим снижением
размера ядра за счет конденсации хроматина и потери ядрышек. По мере созревания
нейтрофила ядро зазубривается и наконец приобретает характерную
сегментацию. Одновременно происходят изменения и в цитоплазме нейтрофила, где
накапливаются гранулы, содержащие биологические соединения, которые впоследствии
будут играть столь важную роль в защите организма. Первичные (азурофильные)
гранулы — включения синего цвета размером приблизительно 0,3 мкм,
содержащие эластазу и миелопероксидазу. Впервые они появляются на промиелоцитар-
ной стадии; при созревании их количество и интенсивность окрашивания
снижаются. Вторичные (специфические) гранулы, которые содержат лизоцим и другие
протеазы, появляются на стадии миелоцита. Окраска этих вторичных гранул
обусловливает характерный нейтрофильный вид цитоплазмы (рис. 4-2 и
приложение 4).
Митотическая Постмитотическая
стадия (7,5 дней) стадия (6,5 дней)
Миело- Промие- Миелоцит Мета- Палочко- Зрелый
бласт лоцит миелоцит ядерная сегментоядерный
клетка нейтрофил
Рис. 4-2. Созревание нейтрофила. Показана конденсация ядерного хроматина, утрата ядрышек
и развитие первичных и вторичных гранул
Нейтрофилы
125
Продукция нейтрофилов
Нейтрофилы непрерывно поставляются в кожу, слизистые оболочки и другие
периферические ткани. Их ежедневный оборот составляет порядка 100 млрд клеток.
Нейтрофилы обладают уникальной способностью увеличивать свою численность,
когда это необходимо организму, как за счет расширения пула пролиферирующих,
так и за счет "рекрутирования" зрелых клеток. В противоположность
большинству других клеток крови они очень небольшую часть своей жизни проводят
во внутрисосудистом пространстве. Поэтому количество циркулирующих
нейтрофилов не вполне отражает кинетику образования этих клеток (рис. 4-3).
Большую часть своей 15-дневной жизни клетки нейтрофильного ряда
проводят в костном мозге. Именно здесь происходит расширение пула нейтрофильных
предшественников. Митотический, или пролиферативный, пул клеток состоит
из коммитированных миелоидных клеток-предшественников КОЕ-ГМ и КОЕ-Г
и нейтрофильных предшественников вплоть до стадии миелоцита. Расширение
этого костномозгового пула ускоряется воспалительными цитокинами — Г-КСФ
и ГМ-КСФ (рис. 1-1).
На пути к периферическим тканям нейтрофилы проводят примерно 10 ч,
находясь во внутрисосудистом пространстве. Этот внутрисосудистый компартмент
является для них не просто коридором, ведущим от костного мозга к
периферическим тканям: в любой момент времени только половина внутрисосудистых гра-
нулоцитов находится в движении, другая — обратимо прилипает к эндотелиаль-
ной поверхности микрососудистого русла. Эти так называемые пристеночные
клетки представляют собой запасной пул зрелых клеток, которые могут быть
востребованы в случае инфекции или воспаления.
Костный мозг
Митотическая Постмитотическая
стадия и стадия
Рис. 4-3. Пролиферация и дифференцировка нейтрофильных предшественников в костном мозге.
Зрелые нейтрофилы депонируются в костном мозге и сосудах и распределяются по периферичес-
Количество
клеток
126
Глава 4. Лейкоциты
Функция нейтрофилов
Функция нейтрофилов заключается в защите организма от инфекции. Этот процесс
включает хемотаксис, фагоцитоз и уничтожение микроогранизмов. Хемотаксис
предполагает способность к обнаружению и целенаправленному движению по
направлению к микроорганизмам и очагам воспаления. Нейтрофилы имеют
специфические рецепторы для С5а-компонента системы комплемента (вырабатываемого
в классическом или альтернативном путях активации комплемента) и протеаз,
выделяемых при повреждении тканей или при непосредственном бактериальном
воздействии. Кроме того, у нейтрофилов есть рецепторы для N-формилъных пептидов,
выделяемых бактериями и пораженными митохондриями. Они реагируют и на
такие продукты воспаления, шклейкотриенЬТВ4 и фибринопептиды.
Нейтрофилы продвигаются к очагам воспаления по градиенту хемотаксиса.
Движение осуществляется за счет сложного взаимодействия между молекулами
на их поверхности и контррецепторами эндотелиальных тканей. Нейтрофилы
катятся по эндотелиальнои поверхности, замедляя свое движение относительно
скорости потока крови. Это явление инициируется, когда поверхностные
молекулы нейтрофила, известные как L-селектины, взаимодействуют с фукозильными
и сульфатидильными остатками, расположенными на эндотелиальнои
поверхности. Р- и Е-селектины — родственные молекулы, которые экспрессируются эндо-
телиальными клетками в ответ на выработку воспалительных цитокинов и
связываются с молекулами клеточной поверхности нейтрофилов. Затем лейкоциты
экспрессируют адгезивные молекулы интегринового суперсемейства, в частности
$2-интегрины, являющиеся гетеродимерами. Они .состоят из молекулы CD18
и одной из трех молекул CD11. р2-Интегрины связываются с контррецепторами
на эндотелиальных клетках, например ICAM-1 и ICAM-2 (рис. 4-4А).
Локомоторная система, напоминающая сократительный аппарат мышцы,
позволяет нейтрофилу продвигаться по эндотелиальнои поверхности к месту
воспаления. Нити актина взаимодействуют с сократительным белком миозином в
процессе, при котором потребляется АТФ. Медленно двигаясь, нейтрофилы
выпускают псевдоподии в направлении воспалительных хемотаксинов. При этом
процессе белок гелзолин катализирует переход цитоплазматической актиновой
решетки из состояния геля в состояние золя. Потоки цитоплазматического
содержимого устремляются в псевдоподию, за ними следует ядро, форма клетки
изменяется. Микротубулы поддерживают стабильность клетки во время процесса миграции.
Нейтрофилы распознают инородные организмы при помощи рецепторов
к опсонинам (термин взят из греческого языка и означает "подготовиться к
обеду"). Фиксация сывороточного IgG и комплемента на бактериях делает их
распознаваемыми для гранулоцитов. Нейтрофил имеет рецепторы для Fc-фрагмента
молекулы иммуноглобулина и продуктов каскада комплемента. Эти рецепторы
инициируют процессы захвата, поглощения и адгезии инородных объектов.
Нейтрофилы поглощают опсонизированные микроорганизмы с помощью ци-
топлазматических пузырьков, называемых фагосомами. Эти пузырьки
продвигаются от складчатых псевдоподий и сливаются с первичными и вторичными гранулами
за счет энергетически зависимого процесса, во время которого в фагоцитах
происходит взрывная активация гликолиза и гликогенолиза. При дегрануляции клетки
содержимое гранул выбрасывается в фагосому и выделяются ферменты деградации:
лизоцим, кислая и щелочная фосфатазы, эластазаилактоферрин (рис. 4-4Б).
Нейтрофилы
127
Наконец, нейтрофилы разрушают бактерии, метаболизируя кислород с
образованием продуктов, токсичных для поглощенных микроорганизмов. Оксидаз-
ный комплекс, генерирующий эти продукты, состоит из флавин- и гемсодержаще-
гося цитохрома Ь558- В этих реакциях используется восстанавливающий агент
никотинамидадениндинуклеотидфосфат (НАДФН), а стимуляторами их
являются глюкозо-6-фосфатдегидрогеназа и другие ферменты гексозомонофосфатно-
Рис. 4-4. (А) Маргинация и адгезия нейтрофилов опосредованы адгезивными молекулами. Селек-
тины способствуют начальной слабой адгезии к эндотелиальным клеткам сосудов. Прочное
соединение, возникающее за счет лейкоцитарных (32-интегринов, позволяет осуществляться диапедезу и
открывает доступ в ткани за пределами сосуда. (Из: Hillman R. A., Ault К. A. Hematology in Clinical
Practice. New York: McGraw-Hill, 1995: 242.) (Б) Уничтожение бактерий нейтрофилами происходит
«следствие комбинированного действия протеаз в первичных и вторичных гранулах и
деструктивного эффекта токсичных форм кислорода
128
Глава 4. Лейкоциты
го шунта. В результате клетка генерирует супероксид (Ог) и перекись водорода
(Н202), которые выделяются в фагосому для уничтожения бактерий. Лактофер-
рин участвует в образовании свободных гидроксильных радикалов■, а миелоперок-
сидаза, используя галоиды в качестве кофакторов,— в продукции гипохлорной
кислоты (НОС1) и токсичных хлораминов.
Активные формы кислорода токсичны как для микроорганизма, так и для
самой клетки. Но благодаря тому, что они локализованы в фагоцитарной вакуоли,
поражение других компартментов клетки незначительно. К другим механизмам
защиты клетки от воздействия этих продуктов относятся: супероксиддисмутаза,
превращающая супероксид в перекись водорода; каталаза, разрушающая
перекись водорода; восстановленный глутатион, также разрушающий перекись
водорода (и способствующий тем самым регенерации НАДФН, который в
дальнейшем может вновь использоваться для пероксидации).
Наряду с выработкой токсичных активных форм кислорода нейтрофил
обладает и другими механизмами (способами) бактерицидного действия: губительная
для микроорганизмов кислая среда фагосомы; лизоцим, гидролизирующий муко-
пептидную клеточную стенку некоторых бактерий; вырабатываемые в фагосоме
бактерицидные белки (дефензины и перфорины), изменяющие проницаемость
мембран клеток-мишеней.
Аномалии гранулоцитов
Морфологически идентифицируют различные аномалии гранулоцитов
(табл. 4-1). Токсическая зернистость — это наличие в цитоплазме множества азуро-
фильных гранул, что можно наблюдать при тяжелых инфекционных заболеваниях
или воспалении. Тельца Деле — это концентрирующиеся на периферии серовато-
синие цитоплазматические включения, которые также иногда обнаруживаются
при сильной стрессовой реакции. Они представляют собой остатки эндоплазмати-
ческого ретикулума, содержащего рибосомы. Болезнь Альдера-Рейли — это
наследственная аномалия, при которой не нарушаются функции нейтрофилов, но за счет
изменения метаболизма полисахаридов появляются окрашенные в сиреневый цвет
азурофильные гранулы. Синдром Чъдиака-Хигаси — врожденное нарушение, для
которого характерно появление крупных азурофильных гранул в клетках всех мие-
лоидных ростков, гипопигментация глаз и кожи, наличие гигантских меланосом и
ненормально функционирующих тромбоцитов без плотных гранул. Во время
фагоцитоза в гранулоцитах происходит патологическое слияние лизосом; нарушение
хемотаксиса приводит к рецидивированию инфекций. Полагают, что этот синдром
связан с синтезом аномальных микротубулярных белков. Аномалия Мей -Хегглина
— редкое аутосомно-доминантное нарушение, которое проявляется наличием ги-
ганских тромбоцитов и больших базофильных включений в гранулоцитах. У
некоторых больных наблюдается склонность к кровотечению, тромбоцитопения с
количественными или качественными аномалиями тромбоцитов.
Нормальные нейтрофилы имеют ядра с 4-5 долями. Аномалия Пельгера-
Хьюета — врожденное нарушение сементации ядра. Зрелые гранулоциты гете-
розигот имеют только две доли в ядре и называются пенснеобразными клетками,
поскольку напоминают по внешнему виду пенсне. Ядро гомозигот состоит
только из одной доли. Это доброкачественное аутосомно-доминантное заболевание
наблюдается у одного из 6000 человек. Несколько чаще встречается аномалия,
Нарушение локомоции нейтрофилов и захвата ими микроорганизмов 129
называемая псевдо-Пелъгера-Хъюета и представляющая собой приобретенную
патологию ядра. Наблюдается она при миелопролиферативных и миелодисплас-
тических нарушениях, вызванных аномалией созревания ядра.
Избыточная гиперсегментация ядер иногда является следствием
доброкачественного аутосомно-доминантного нарушения. Чаще гиперсегментация
нейтрофилов возникает при мегал областной анемии. Как правило, если гранулоцитов
с пятью (или более) долями свыше 5 %, то констатируют наличие
гиперсегментации. В этих случаях необходимо выявить причину мегалобластоза. К сожалению,
невозможно дифференцировать гиперсегментацию ядер гранулоцитов и
аномальное созревание эритроидных ядер, вызванные недостатком фолиевой
кислоты и витамина Bi2. Однако дефицит витамина Bt2 (но не дефицит фолиевой
кислоты) ассоциируется с неврологическими симптомами, вызванными дегенерацией
задних рогов спинного мозга (глава 3). Гиперсегментация гранулоцитов
наблюдается также при воздействии химиотерапевтических средств, которые нарушают
синтез ДНК, например при использовании антиметаболита гидроксимочевины.
Нарушение функции нейтрофилов
Чтобы защитить организм от микробов, нейтрофилы должны взаимодействовать
с другими видами клеток (табл. 4-2). Поскольку для распознавания и деструкции
бактерий необходима опсонизация последних, синдромы недостаточности антител
и нарушения системы комплемента могут вызывать рецидивирующие бактериемии,
менингит и легочные инфекции. Агаммаглобулинемия или гипогаммаглобулинемия,
а также аномалия СЗ-компонента комплемента предрасполагают к инфекциям,
вызываемым такими патогенами, как стрептококки, пневмококки, гемофильные и си-
негнойные палочки. Эти состояния дефицита защитных звеньев могут быть
врожденными и приобретенными вследствие лимфопролиферативных нарушений,
аутоиммунных заболеваний и иммунокомплексной патологии. Синдром Джоба —
нарушение неизвестной этиологии, при котором рецидивирующие абсцессы и
легочные инфекции связаны с очень высоким содержанием иммуноглобулина Е (IgE).
Нарушение локомоции нейтрофилов
и захвата ими микроорганизмов
Кортикостероиды нарушают способность нейтрофилов мигрировать в очаг
воспаления и поглощать микроорганизмы, однако механизм, посредством которого
это нарушение возникает, неизвестен. Пациенты, получающие большие дозы
стероидов, а также больные с синдромом Кушинга весьма чувствительны к пиоген-
ной инфекции. Нарушение адгезивной способности лейкоцитов — редкая аутосом-
но-рецессивная аномалия, при которой наблюдаются медленное заживление
пуповинной культи, рецидивирующие бактериальные и грибковые инфекции.
При этом нарушении нейтрофилы плохо прилипают к поверхности и не могут
адекватно реагировать на бактерии, покрытые СЗ. В данном случае возможны
различные молекулярные дефекты, но всегда обнаруживается количественная
или качественная аномалия лейкоцитарного интегрина CD18. Дисфункция
актина нейтрофилов — редкое аутосрмно-рецессивное нарушение локомоции
нейтрофилов и их захватывающей способности. Оно связано с дефектом полимеризации
ТАБЛИЦА 4-1. Аномалии лейкоцитов
Название
Микроскопическая картина
Описание
Клиническая значимость
Нормальный
нейтрофильный
лейкоцит(полиморфно-ядерный
лейкоцит — ПМЯЛ)
Токсическая
зернистость
Цитоплазматичес-
кие вакуоли
Тельца Деле
Аномалия
Альдера-Рейли
Диаметр 10-15 мкм; ядро имеет Наблюдается у здоровых людей и при
от 2 до 5 долей, светло-розовые лейкоцитозе
или сиренево-розовые гранулы
Цитоплазма содержит множество
темных азурофильных гранул с
незрелыми мукополисахаридами
Цитоплазма содержит вакуоли
В цитоплазме светло-синие глыбки
различного размера и формы,
представляющие собой РНК из
фрагментов шероховатого эндо-
плазматического ретикулума
Цитоплазма содержит
многочисленные темно-сиреневые гранулы,
состоящие из кислых мукополиса-
харидов
Наблюдается при инфекционных или
воспалительных процессах. Может
сопровождаться присутствием вакуолей и
телец Деле
Наблюдаются при инфекциях. Могут
указывать на дегенерацию ПМЯЛ и
сочетаться с токсической зернистостью
или тельцами Деле. Они иногда
выявляются при аномалии Джордана —
семейной вакуолизации лейкоцитов
Обнаруживаются при инфекционных
состояниях в сочетании с токсической
зернистостью или цитоплазматическими
вакуолями
Наблюдается при генетическом мукопо-
лисахаридозе типа Гурлера, Гунтера и
Мароте-Лами
Цитоплазма содержит
многочисленные крупные слившиеся лизо-
сомальные гранулы. Некоторые
из них имеют гигантский размер и
причудливую форму. Они могут
содержаться также в лимфоцитах
и других клетках крови
Тельца Деле выглядят крупнее,
веретенообразной формы,
отчетливее прокрашиваются, расположены
в цитоплазме более центрально
1-2 % полиморфно-ядерных нейт-
рофилов имеют размер в 2 раза
больше нормы, ядра гиперсегмен-
тированы
Редкое аутосомно-рецессивное
заболевание, проявляющееся наличием
включений в лейкоцитах, частичной ги-
попигментацией глаз и кожи,
повышенной склонностью к кровотечениям
Редкое аутосомно-доминантное
нарушение, для которого характерно
наличие крупных патологических телец Деле
в гранулоцитах и моноцитах; слабо
гранулированные гигантские тромбоциты,
тромбоцитопения
Наблюдается редко, является аутосом-
но-доминантным нарушением
Ядро имеет форму кольца
Наблюдаются при тяжелом алкоголизме
Ядро гипосегментировано и похоже
на пенсне или горошины в случае
наследственных заболеваний; ядро
круглой или продолговатой формы
при приобретенных нарушениях
Ядро имеет более пяти долей, со-
единеных тонкой хроматиновой
нитью
При наследственных нарушениях функция
лейкоцитов нормальна. Приобретенные
могут быть связаны с лейкозом, миелоп-
ролиферативными и миелодиспластичес-
кими нарушениями, влиянием инфекции
или лекарственных препаратов
Характерны для мегалобластной
анемии (редко при бессимптомном ауто-
сомно-доминантном нарушении)
132
Глава 4. Лейкоциты
ТАБ Л И ЦА 4- 2. Нарушение функции нейтрофилов
Нарушенная
функция
Примеры
Этиология
Опсонизация А- или гипогаммаглобулинемия
Локомоция/ Недостаточная адгезия
захват лейкоцитов
Дисфункция актина в нейтрофилах
Синдром Чедиака-Хигаси
Действие кортикостероидов
Киллинг Хронический гранулематоз
Дефицит миелопероксидазы
Врожденная или
приобретенная патология
Врожденная
(дефицит CD18)
Врожденная
Врожденная
Синдром Кушинга или
ятрогенное заболевание
Врожденная
Врожденная
или приобретенная
актина. Для синдрома Чедиака-Хигаси характерны нейтропения, появление
гигантских лизосомальных гранул, которые не могут нормально сливаться с фаго-
сомами, и нарушение хемотаксиса. Нарушение миграции нейтрофилов
определяется в тесте "кожного окна" — методике оценки миграции лейкоцитов in vivo.
Нарушение бактерицидной активности
Хроницеский гранулематоз (ХГ) представляет собой аутосомно-рецессивное или
сцепленное с Х-хромосомой нарушение, при котором нейтрофилы не способны
метаболизировать кислород в супероксид или перекись водорода. В большинстве
случаев заболевание вызвано аномалией оксидаз, генерирующих эти токсичные
формы кислорода. В редких случаях нейтрофилы, не имеющие Г-6-ФДГ, не мета-
болизируют кислород из-за отсутствия НАДФН. Пораженные фагоциты не
убивают микроорганизмы (стафилококки, большинство грамотрицательных энтеро-
бактерий) и грибки, экспрессирующие каталазу, которая разрушает перекись
водорода. У больных наблюдаются рецидивирующие инфекции кожи, костей
и внутренних органов, вызываемые каталаза-позитивными микроорганизмами.
Из-за того, что дефектные нейтрофилы не метаболизируют кислород, активность
гексозомонофосфатного шунта не повышается. Лабораторным тестом для
определения функции фагоцитов (которая нарушена в гранулоцитах при хроническом
гранулематозе) является тест восстановления нитросинего тетразолия (НСТ)
в синий формазан под действием активных форм кислорода.
Недостаточность миелопероксидазы — аутосомно-рецессивное заболевание,
при котором в нейтрофилах отсутствует миелопероксидаза. Нарушения
бактерицидной активности фагоцитов у таких больных менее выражены, чем при
хроническом гранулематозе, и они не столь восприимчивы к инфекции.
Нейтрофилия
Диагноз нейтрофилии обычно ставится в том случае, когда количество
нейтрофилов составляет более 7500/мкл (табл. 4-3). Стойкая нейтрофилия, особенно при
воспалительном процессе с увеличением незрелых циркулирующих миелоидных
Нейтрофилия
133
предшественников ("сдвиг влево"), может быть определена как лейкемоидная
реакция. Причиной нейтрофилии обычно является стимулирование и расширение
миелоидного пролиферирующего ростка в связи с инфекцией, воспалением,
опухолью или эндокринными нарушениями. Острая нейтрофилия обычно
характеризуется увеличением числа циркулирующих гранулоцитов по сравнению с
пристеночными, а также рекрутированием зрелых клеток из костномозгового
резерва. Быстрая мобилизация пристеночного пула может быть вызвана
адреналином, острым стрессом, гипоксией или физической нагрузкой. Мобилизация
костномозгового пула происходит под влиянием кортикостероидов, эндотоксина,
химической интоксикации или хронического стресса.
Реактивные причины нейтрофилии следует отличать от первичных
нарушений гемопоэза, в частности лейкозов. Хронический миелолейкоз (ХМЛ)
представляет собой злокачественную экспансию миелоидных клеток, связанную, как
правило, с наличием филадельфийской хромосомы, которая образуется
вследствие реципрокной транслокации хромосом 9 и 22, приводящей к образованию
химерного продукта bcr/abl. ХМЛ часто сочетается со спленомегалией и тромбо-
цитозом и может сопровождаться пониженной активностью фосфатазы
лейкоцитов. Однако отличить ее от реактивной нейтрофилии на основании анализа мазка
крови и исследования костного мозга трудно. Острый нелимфоцитарный лейкоз
(ОНЛЛ) ассоциируется с прекращением созревания нейтрофилов в костном моз-
ТАБЛ И ЦА 4- 3. Количественные изменения нейтрофилов
Нейтрофилия (> 7500/мкл)
Реактивная
Хроническая инфекция или воспаление
Злокачественная
Острый или хронический лейкоз
Нейтропения (< 1800/мкл) — селективная
Врожденная
Синдром Костманна
Циклическая нейтропения
Доброкачественная (например, расовая)
Инфекционная
Вирусная (например, гепатит, ВИЧ)
Бактериальная (например, милиарный туберкулез)
Лекарственная
Антибиотики
Противосудорожные препараты
Противовоспалительные средства
Антитиреоидные средства
Фенотиазины
Аутоиммунная
Системная красная волчанка (СКВ)
Синдром Фелти (ревматоидный артрит)
Нейтропения как проявление панцитопении
Недостаточность костного мозга
Лейкоз, лимфома, метастазы рака
Апластическая анемия
Пищевые нарушения (например, недостаточность фолата или витамина В12)
Гиперспленизм
134
Глава 4. Лейкоциты
ге и периферической крови. Незрелые миелоидные клетки, или "бласты", могут
содержать тельца Ауэра — продолговатые синего цвета цитоплазматические
включения, которые представляют собой видоизмененные гранулы (глава 9).
Нейтропения
Нейтропения (количество нейтрофилов менее 1800/мкл), как и нейтрофилия,
может быть вызвана временными или постоянными изменениями различных
нейтрофильных пулов и подтверждается несколькими последовательными
анализами крови. Если абсолютное число нейтрофилов составляет более 500/мкл,
обычно существенного нарушения в иммунной защите организма не происходит.
Однако тяжелая нейтропеция (особенно при показателях ниже 200/мкл) всегда
связана с повышенным риском возникновения инфекции, особенно если
снижение уровня нейтрофилов происходит быстро. Тяжелая острая нейтропения может
быть вызвана истощением резерва нейтрофилов в связи с септицемией, высокой
температурой, прострацией. Эта форма нейтропении, особенно связанная с
лихорадочным состоянием, требует быстрой оценки и раннего начала лечения
антибиотиками широкого спектра действия. Хроническое постоянное истощение нейт-
рофильного резерва может быть клинически бессимптомно и проявляться
незначительным повышением температуры, стоматитом, лимфаденопатией и
рецидивирующими абсцессами. Среди здоровых афро-американцев нижняя
граница нормы количества нейтрофилов меньше обычной (1400/мкл), что однако не
повышает риск развития у них инфекций.
Нейтропения иногда вызывается различными лекарственными препаратами,
чаще всего сульфаниламидами, фенотиазинами, антитиреоидными средствами и
анальгетиком фенилбутазоном (табл. 7-5). При многих видах химиотерапии
происходит дозозависимая супрессия гранулоцитообразования; эти препараты могут
быть наиболее частой причиной ятрогенной гранулоцитопении. Содержание гра-
нулоцитов в крови снижается при таких системных нарушениях, как
ревматоидный артрит и системная красная волчанка. В некоторых случаях причиной могут
быть антинейтрофильные антитела, которые участвуют в интрамедуллярной
деструкции созревающих нейтрофилов или влияют на выживание и пролиферацию
предшественников нейтрофилов (глава 7). Псевдонейтропения обусловлена
повышенной маргинацией (краевое стояние) гранулоцитов. Такие временные
сдвиги могут быть вызваны реакциями повышенной чувствительности, виремией,
гемолизом или гемодинамическими изменениями.
Периодическая, или циклическая] нейтропения — аутосомно-доминантное
нарушение, встречающееся с различной частотой, которое впервые проявляется
в младенческом возрасте и сохраняется десятилетиями. Каждые 3-4 недели
у больного развивается нейтропения, продолжающаяся в течение 3-6 дней и
сопровождающаяся высокой температурой, мукозитом, кожными инфекциями.
В это время в костном мозге возникает миелоидная гипоплазия. Хотя причина
неизвестна, полагают, что циклическая нейтропения представляет собой десинхро-
низацию механизмов обратной связи, которые регулируют образование
гранулоцитов и их выход в кровь. У некоторых больных положительные результаты дает
лечение ГМ-КСФ или Г-КСФ. Доброкачественная аутосомная нейтропения
Глассена, наследуемая по доминантному типу, относится к семейным нейтропе-
ниям. Синдром Костманна, напротив, является аутосомно-рецессивным заболе-
Эозинофилы
135
ванием, при котором постоянно наблюдается низкое содержание нейтрофилов.
Многие пациенты умирают от сепсиса в детском возрасте. Врожденные
нарушения метаболизма, например болезнь Гоше, равно как и миелофтиз (например,
вследствие метастазов опухолей), изменяют архитектуру костного мозга и также
способны приводить к нейтропении (глава 7).
Эозинофилы
Эозинофилы имеют двухдольчатое ядро и цитоплазму, заполненную отчетливо
видимыми гранулами, приобретающими красный цвет после окрашивания по Райту -
Гимзе (рис. 4-1). Основные (положительно заряженные) белки этих гранул
окрашиваются в красный цвет из-за их высокого сродства к эозину. Эозинофилы
проходят те же стадии созревания, что и нейтрофилы, однако по причине своей
малочисленности предшественники эозинофилов в костном мозге выявляются
реже (за исключением некоторых патологических состояний). Во время
созревания предшественники эозинофилов аккумулируют в цитоплазме крупные
окрашиваемые в красный цвет гранулы, а не нейтрофильные вторичные гранулы;
их ядро не сегментируется, как у нейтрофилов. Созреванию эозинофилов
способствуют интерлейкины 3 и 5 (рис. 1-1).
Эозинофилы играют особую роль в борьбе с паразитами и контроле аллергии.
Поскольку они редко обнаруживаются в периферической крови, их участие в
защите от бактериальных инфекций неясно. Однако, подобно нейтрофилам,
эозинофилы способны к хемотаксису, фагоцитозу и обладают бактерицидной
активностью. Эозинофильные гранулы содержат особую группу бактерицидных
белков, включая эозинофильный катионный протеин, белковые кристаллы Шар-
ко -Лейдена и эозинофильную пероксидазу.
Эозинофилия (абсолютное количество эозинофилов более 700/мкл) обычно
наблюдается в том случае, когда эозинофилы составляют более 10 % от общего числа
лейкоцитов. Как правило, это вызвано аллергическими или атопическими
состояниями, паразитарной инфекцией, аутоиммунными или воспалительными
процессами, злокачественными опухолями, а также болезнью Ходжкина. Эозинофилию
часто можно наблюдать в сочетании с базофилией при хроническом миелолейкозе.
Характерный синдром острого нелимфоцитзрного лейкоза с наличием необычных
предшественников эозинофилов ассоциируется с аномалией хромосомы 16.
Гиперэозинофилия (количество эозинофилов выше 1500/мкл) может привести
к характерному поражению органов, особенно если она сохраняется на
протяжении многих месяцев. Она может быть вызвана прямой инфильтрацией тканей,
воздействием токсических продуктов кислорода и гранулярных белков. Как
правило, поражаются эндокард и ЦНС. Лечение обычно включает применение кор-
тикостероидов или антиметаболитов, например гидроксимочевины.
Базофилы
Базофилы — самая малочисленная группа циркулирующих гранулоцитов,
составляющая менее 1 % лейкоцитов (рис. 4-1В и приложение 4). В крупных цито-
плазматических гранулах базофилов содержатся сульфатированные или карбок-
силированные кислые белки, такие как гепарин, приобретающие синий цвет при
окрашивании по Райту-Гимзе. Базофилы опосредуют аллергические реакции,
136
Глава 4. Лейкоциты
особенно те из них, которые базируются на IgE-зависимых механизмах. Они
экспрессируют IgE-рецепторы и при соответствующей стимуляции освобождают
гистамин. Не исключено, что базофилы родственны тучным тканевым клеткам,
которые также выделяют вазоактивные вещества, например гистамин, в ответ
на воздействие IgE и антигена.
Если количество базофидов составляет более 150/мкл, ставится диагноз
базофилии. Как и эозинофилия, она может быть связана с острыми реакциями
повышенной чувствительности, например с аллергическими реакциями по типу
крапивницы. Другими причинами базофилии могут быть вирусные заболевания
(ветрянка, грипп), хронические инфекции (туберкулез), воспалительные
процессы (ревматоидный артрит, язвенный колит), дефицит железа, рак. Базофилия
может наблюдаться при миелопролиферативных нарушениях и указывать на бласт-
трансформацию при хроническом миелолейкозе. Острый нелимфоцитарныи
лейкоз при наличии клеток, напоминающих базофилы, ассоциируется с
симптомами, появление которых связано с высвобождением гистамина.
Моноциты
Моноциты циркулируют в периферической крови в виде крупных клеток с
цитоплазмой синего/серого цвета и почкообразным или складчатым ядром,
содержащим нежно-сетчатый хроматин (рис. 4-1Г и приложение 4). Моноциты
являются производными КОЕ-ГМ (общего предшественника для гранулоцитов
и моноцитов) и КОЕ-М (предшественника только моноцитарного ростка).
Моноциты проводят в кровотоке всего около 20 ч, а затем попадают в периферические
ткани, где трансформируются в макрофаги ретикулоэндотелиальной системы
(РЭС). Эти тканевые макрофаги, или гистиоциты, представляют собой крупные
клетки с эксцентрично расположенными ядром и вакуолизированной
цитоплазмой, содержащей многочисленные включения (приложение 4).
Моноциты и макрофаги — долгоживущие клетки, функциональные
особенности которых во многом схожи с таковыми у гранулоцитов. Они более эффективно
захватывают и поглощают микобактерии, грибки и макромолекулы; менее
значима их роль в фагоцитозе пиогенных бактерий. В селезенке макрофаги
ответственны за утилизацию сенсибилизированных и стареющих эритроцитов. Макрофаги
играют важную роль в процессинге и представлении антигенов лимфоцитам в
ходе клеточных и гуморальных иммунных реакций. Продуцирование ими цитоки-
нов и интерлейкинов, интерферонов и компонентов комплемента способствует
координации сложных взаимодействий в интегрированном иммунном ответе.
В норме моноциты составляют от 1 до 10 % циркулирующих лейкоцитов.
Когда количество моноцитов превышает 1000/мкл, можно говорить о моноцитозе,
который наблюдается у пациентов с хроническими инфекциями (туберкулез,
инфекционный эндокардит) или воспалительными процессами (аутоиммунные
заболевания, воспалительные заболевания кишечника). Первичные заболевания
костного мозга (хронический лейкоз или миелопролиферативные состояния)
могут также сопровождаться моноцитозом. Временный моноцитоз часто
предшествует восстановлению уровня циркулирующих нейтрофилов через 1-2 дня после
супрессии костного мозга химиотерапевтическими препаратами. Гистиоцитоз X
связан с неактивными новообразованиями моноцитарно-макрофагальной
системы, при которых происходит инфильтрация патологическими клетками костного
В-лимфоциты
137
мозга, кожи, легких и центральной нервной системы. Истинная гистиоцитарная
лимфома является злокачественной аномалией моноцитов/макрофагов и имеет
много общего с гистиоцитарным медуллярным ретикулезом, для которого
характерен фагоцитоз гематопоэтических клеток мононуклеарными клетками.
Лимфоциты
Лимфоциты — небольшие мононуклеарные клетки (рис. 4-1Д и рис. 4-9А),
координирующие и осуществляющие иммунный ответ за счет продуцирования
воспалительных цитокинов и антигенспецифических связывающих рецепторов.
Лимфоциты подразделяются на две основные категории: В-клетки и Т-клетки —
и несколько менее многочисленных классов, например естественные
("натуральные", нормальные) клетки-киллеры. Подгруппы лимфоцитов отличаются по
месту их образования и эффекторным молекулам, которые они экспрессируют, но
имеют общее свойство — способность опосредовать высокоспецифический
антигенный ответ (рис. 4-5).
Рис. 4-5. Лейкоциты являются производными общей лимфоидной клетки-предшественника.
Созревающие В-лимфоциты экспрессируют специфические антигены CD 19 и CD20; они синтезируют
поверхностные иммуноглобулины (slg), а позднее и цитоплазматические (Ig). Т-лимфоциты
последовательно по мере развития экспрессируют антигены CD2, CD5 и CD7, а также ТДТ на первых
стадиях. Позднее они приобретают Т-клеточный рецептор и временно коэкспрессируют CD4 и CD8,
что происходит до коммитирования или хелперов (С04-позитивных), или супрессоров (CD8).
Процесс созревания НК-клеток менее изучен. (По: Hillman R. A., Ault К. A. Hematology in Clinical Practice.
New York: McGraw-Hill, 1995: 304.)
138
Глава 4. Лейкоциты
В процессе развития плода важнейшими лимфопоэтическими органами
являются желточный мешок, печень и селезенка (рис. 1-3). В постнатальный период
основные лимфоидные органы — костный мозг и вилочковая железа, в которых
происходит деление лимфоцитов до воздействия антигена. Вторичные (или
реактивные) лимфоидные ткани включают лимфатические узлы, селезенку и
лимфоидные образования желудочно-кишечного и дыхательного трактов. Лимфоциты
проходят по кровотоку и через посткапиллярные венулы попадают в селезенку,
лимфатические узлы и другие лимфоидные ткани (глава 1). Они возвращаются
из периферических тканей через эфферентную систему лимфатических сосудов,
впадающих в венозную систему через грудной лимфатический проток.
В-лимфоциты
В-лимфоциты осуществляют экспрессию уникальных антигенных рецепторов —
иммуноглобулинов — и запрограммированы на продукцию их в большом
количестве в ответ на антигенную стимуляцию. В-клетки образуются из стволовых
клеток костного мозга. Термин В-клетки происходит от латинского названия фабри-
циевой сумки {bursa Fabricius) — органа, необходимого для созревания В-клеток
у птиц. Аналогичного органа у человека нет; созревание В-клеток у него
происходит в основном в костном мозге.
Иммунная система содержит большую популяцию отдельных клонов В-лим-
фоцитов. Каждый клон экспрессирует уникальный антигенный рецептор,
идентичный в основном иммуноглобулиновой молекуле, которую он будет
синтезировать. Эти молекулы отличаются друг от друга й связываются только
с ограниченным числом антигенов. Чрезвычайно большое количество
индивидуальных клонов В-лимфоцитов, для каждого из которых характерна определенная,
специфичная молекула иммуноглобулина, обеспечивает удивительное
разнообразие вырабатываемых антител.
В-лимфоциты, образующиеся в костном мозге, иммунологически незрелые,
поскольку они еще не подвергались воздействию антигена (рис. 4-5).
Первоначальные этапы созревания В-клеток(не зависят от антигена. Пре-В-клетка
временно продуцирует терминальную дезоксинуклеотидтрансферазу (ТДТ) и общий
антиген острого лейкоза (ОАОЛ; CD10). Несколько позднее она экспрессирует
характерные поверхностные антигены В-клетки — CD 19 и CD20, и образует инт-
рацитоплазматические ji-цепи иммуноглобулина. Когда В-клетки созревают, они
экспрессируют на своей поверхности целые молекулы антител (slg). Позже эти
иммуноглобулины секретируются. Зрелые В-лимфоциты находятся в основном
в герминативных центрах коры лимфатических узлов и в белой пульпе селезенки.
В-клетки составляют менее 20 % циркулирующих лимфоцитов.
Последующие этапы созревания В-клеток зависят от антигена. С помощью
Т-хелперов и специализированных макрофагов, называемых антигенпрезентиру-
щими клетками, В-клетки, рецепторы которых распознают антиген, пролифери-
руют и созревакж. Образующиеся в результате этих процессов плазматические
клетки продуцируют большое количество иммуноглобулиновых молекул строго
определенной специфичности. Эти клетки имеют характерный внешний вид:
эксцентричное ядро с распределенным по периферии хроматином, базофйльную
цитоплазму и светлую чистую перинуклеарную зону, где располагается активный
комплекс Гольджи (рис. 4-9Г). Другие стимулированные В-лимфоциты становят-
Образование антител
139
ся В-клетками долговременной памяти, сохраняющими информацию о ранее
встречавшемся антигене; они быстро пролиферируют и продуцируют большое
количество иммуноглобулина при повторной встрече с известным антигеном.
Образование антител
Существует 5 основных классов иммуноглобулинов (антител), обозначаемых IgG,
IgA, IgM, IgD и IgE. Наиболее распространены иммуноглобулины класса G,
которые подразделяются на IgGt, IgG2, IgG3 и IgG4. IgA преобладает в секретах ЖКТ и
дыхательных путей и имеет два подтипа. IgD и IgE — минорные группы
иммуноглобулинов, участвующие главным образом в аллергических реакциях и реакциях
гиперчувствительности замедленного типа. IgM полимеризуется, формируя
большие пентамерные структуры, секретируемые зрелыми В-клетками (табл. 4-4).
IgM — это первые антитела, продуцируемые В-лимфоцитами в ответ на
антигенную стимуляцию; по мере созревания В-клетки могут переключаться на синтез
IgG, IgA, IgD или IgE. Такой переход называется переключением изотипа. При
переключении на другой изотип тяжелых цепей структура вариабельных участков
антител, определяющих специфичность распознавания антигена, сохраняется.
Все классы иммуноглобулинов построены по общему плану: две тяжелые цепи
соединены дисульфидными связями с двумя легкими цепями (табл. 4-4). Тяжелые
цепи IgG, IgA, IgM, IgD и IgE называются соответственно гамма (у), альфа (а), мю
(ц), дельта (б) и эпсилон (с). Легкие цепи в любой из молекул называются каппа (х)
или лямбда (К). Легкие и тяжелые цепи имеют константные участки, которые почти
идентичны у антител одного класса. Каждая легкая и тяжелая цепь также имеет
вариабельные участки, обеспечивающие специфичность распознавания антител.
Антитела могут расщепляться протеазами (папаином) на Fc-фрагмент, содержащий
константный участок, и высокоизменчивый Fab-фрагмент (рис. 4^6).
ТАБЛИЦА 4-4. Свойства молекул иммуноглобулинов
Молекулярная масса (кД)
Константа седиментации
Содержание в сыворотке
в норме (г/л)
Имеется в
Фиксация комплемента
Трансплацентарная
передача
Тяжелая цепь
Секреторная форма
IgG
150 000
7S
8-15
сыворотке и внекле
точной жидкости
Обычная
Да
(Yl-4)
Мономерная
IgA
150 000
300 000
400 000
7S
0,9-3,3
- сыворотке и
других жидкостях
(например,
секреты бронхов,
кишечника)
Да
(альтернативный путь)
Нет
а (с^или а2)
Вариабельная
IgM
950 000
19S
0,45-1,50
только
в сыворотке
Обычная и очень
эффективная
Нет
ц
Пентамерная
Из: Hoff brand A. V., Pettit J. E. Essential Hematology, 3rd ed. Cambridge, Massachusetts: Blackwell
Scientific Publication, 1993: 168.
140
Глава 4. Лейкоциты
Разнообразие антител определяется сложным процессом сборки генов,
кодирующих отдельные домены иммуноглобулинов из сегментов, а также
соматическими мутациями в генах. Тяжелая иммуноглобулиновая цепь и легкие х- и Х-цепи
закодированы соответственно в хромосомах 14, 2 и 22. На эмбриональной стадии
развития каждый из этих генов содержит несколько участков: вариабельный (V),
.ответственный за разнообразие (D), соединительный (J) и константный участок
(С), состоящие в свою очередь из множества копий генов (сегментов), которые
кодируют отдельные белковые домены. Во всех тканях, за исключением В-лим-
фоцитов, эти гены остаются1 в конфигурации, характерной для эмбриональной
стадии. Однако в начале развития В-клетки происходят перегруппировки внутри
гена, за счет которых комбинируется по одному из сегментов V-, D- и J-участков
с соседним С-участком. Перегруппировки катализируются рекомбиназами,
активными только в иммунных клетках, а лишние сегменты удаляются (рис. 4-7).
Теоретически рекомбинация сегментов гена иммуноглобулина может
обеспечивать синтез тысяч всевозможных антител. Дальнейшее разнообразие антител
обусловлено действием терминальной дезоксинуклеотидтрансферазы (ТДТ). В проти-
Вариабельный
антигенсвязывающий
участок
Fab-
фрагменты
Рс^фрагмент
b-S-S
f-s-s-tei
||^ Легкая цепь (к или Л)
CL
Расщепление
при помощи
папаина
Тяжелая цепь (ц, а, у. 6 или е)
Рис. 4-6. Молекулы иммуноглобулина представляют собой гетеротетрамеры, состоящие из двух
легких и двух тяжелых цепей, связанных дисульфидными мостиками. Вариабельные участки
обусловливают антигенную специфичность, а Fc-области определяют подкласс иммуноглобулина (IgA,
IgD, IgE, IgG или IgM). (По: Hoffbrand ■ A.V., Pettit J.E. Essential Hematology. Cambridge,
Massachusetts: Blackwell, 1993: 167.)
Образование антител __^ 141_
воположность большинству ДНК-полимераз дезоксинуклеотидтрансфераза
встраивает различное число нуклеотидов в место рекомбинации сегментов имму-
ноглобулинового гена, не требуя шаблона ДНК. Добавленные нуклеотиды
соответственно изменяют последовательность кодирования иммуноглобулина и
опосредуют молекулярную гетерогенность. Аналогичные процессы перегруппировки
характерны и для легких х- или Х-цепей. Перегруппировка приводит к тому, что
промотор, находящийся перед V-участком, под влиянием ускорителя
транскрипции попадает в область между участками J и С. Последующая транскрипция
перегруппированных генов и сплайсинг приводят к образованию мРНК; позже
происходит ее трансляция, то есть синтез функциональных молекул иммуноглобулина.
Поскольку эти рекомбинации происходят на уровне генома, все потомство
В-лимфоцита, прошедшего эту стадию созревания, будет сохранять их, что можно
установить с помощью лабораторных методов. Для характеристики
определенного клона В-лимфоцитов используют амплификацию перегруппированного гена
посредством полимеразной реакции или идентификацию специфической
перегруппировки гена при помощи саузерн блоттинга! Эти методы позволяют
дифференцировать поликлональную пролиферацию лимфоцитов от моноклональ-
ной (и, видимо, злокачественной). Если большая популяция лимфоцитов имеет
одинаковую перегруппировку иммуноглобулиновой молекулы, то это, по всей
вероятности, злокачественные В-клетки.
Иммуноглобулины играют важную роль в защите организма от микробов,
вместе с тем аномальное продуцирование антител свидетельствует о нарушениях
в лимфоидных клетках. Множественная миелома представляет собой, например,
злокачественную пролиферацию В-лимфоцитов, когда клональные клетки миело-
мы секретируют одинаковый иммуноглобулин, обычно IgG, IgA или IgM (глава 9).
Уровень антител, экспрессируемых клетками миеломы, может служить маркером
опухоли. У больных с множественной миеломой возникает иммунодефицит, так
как злокачественные лимфоидные клетки продуцируют большое количество им-
D J
5' —ШШ—'Е—Н/ ■■■■ // И1Ё1Ё ЕШЗ— з«
Vi V„ 123 л 12 3 4 5 6 см
Перестройка ДНК
Перестроенная ДНК \ ШЖМММ
в области гена тяжелой цепи ' V/ 'тг^ивГВГЖ
D J См
Транскрипция и сплайсинг РНК
М-мРНК I И I
V DJ См
Трансляция
ц-цепь в цитоплазме
Рис. 4-7. Перегруппировка ДНК приводит к связыванию отдельных экзонов из участков V, D, J, С.
В результате транскрипции, сплайсинга РНК и последующей трансляции образуется белковая
молекула иммуноглобулина определенного типа
142
Глава 4. Лейкоциты
муноглобулиновых молекул одного типа, ограничивая при этом синтез широкого
диапазона антител, свойственных здоровому организму. Из-за склонности IgM
к полимеризации злокачественные В-клетки, секретируя этот изотип
иммуноглобулина, повышают вязкость крови и вызывают нарушения в ЦНС или системе
кровообращения. При некоторых видах миеломы продуцируется неполный
иммуноглобулин в виде фрагментов легкой цепи (определяются в моче как белок
Бенс-Джонса). Лимфомы и аутоиммунные Заболевания часто ассоциируются
с продуцированием аберрантного иммуноглобулина, а антитела, реагирующие
с антигенами клеток крови, способны вызывать иммунную цитопению.
Т-лимфоциты
Т-лимфоциты играют ключевую роль в клеточном иммунитете.
Сенсибилизированные Т-клетки опосредуют гиперчувствительность замедленного типа,
отторжение аллотрансплантата, болезнь "трансплантат против хозяина", контактную
аллергию, а также иммунитет против опухолей и внутриклеточных паразитов.
Клеточноопосредованный иммунитет обеспечивает уничтожение различных
клеток непосредственно цитотоксическими Т-клетками. Он усиливается под
воздействием цитокинов, которые вырабатываются в результате сложного
взаимодействия Т-клеток и макрофагов. Кроме того, Т-лимфоциты активно и избирательно
участвуют в регуляции пролиферации В-клеток и продукции иммуноглобулинов.
Образовавшись из стволовых клеток костного мозга, Т-клетки обязательно
проходят стадию развития в тимусе, в результате чего генерируются зрелые,
функционально полноценные Т-клетки (глава 1). Незрелые протимоциты мигрируют
из костного мозга и "заселяют" субкапсулярный кортикальный слой селезенки.
В тимусе Т-лимфоциты созревают и приобретают характерные для зрелых Т-клеток
антигенные функции. Они экспрессируют антигены CD2 (рецептор эритроцитов
барана), CD3, CD4, CD7, CD8 и Т-клеточный рецептор. Позже, когда они
становятся клетками-хелперами или супрессорами, они переключаются соответственно
на экспрессию либо CD4, либо CD8. Важнейшая функция селезенки в развитии
Т-клеток состоит в ликвидации аутореактивных Т-клеток, которые способны
распознавать антигены других нормальных клеток организма. В результате отбора
в тимусе происходит элиминация этих клеток посредством ряда сложных
процессов, приводящих к апоптозу, то есть программированной гибели клеток.
Т-лимфоциты экспрессируют на своей поверхности антигенраспознающую
структуру, называемую Т-клеточным рецептором (ТКР), структура которого
соответствует молекуле иммуноглобулина и состоит из двух отдельных
субъединиц. Большинство этих рецепторов состоит из а- и Р-цепей, и лишь немногие
содержат у- и б-цепи. Каждая из а-, (3-, у- и б-цепей ТКР включает V-, D-, J-
и С-участки, которые подвергаются соматическим перегруппировкам под
действием рекомбиназ и ТДТ в ходе процессов, аналогичных происходящим
в В-клетках. ТКР прикрепляется к плазматической мембране клетки, но в
отличие от молекул иммуноглобулинов не секретируется (рис. 4-8).
Для полной активации Т-лимфоцита необходимы антигенпрезентирующие
клетки (АПК), например макрофаги. Они обрабатывают антигены, чтобы
сделать их более иммуногенными, и представляют их Т-клеткам вместе с
рецепторами главного комплекса гистосовместимости (ГКГ). ГКГ-рецепторы неодинаковы
у разных людей (в зависимости от генотипа), но в противоположность иммуно-
Т-лимфоциты
143
глобулинам и ТКР изменчивость ГКГ не является результатом генной
перегруппировки. Данные структуры впервые были описаны как рецепторы,
обусловливающие отторжение тканевых трансплантатов у больных с инородными тканями
или у женщин с многочисленными беременностями. Эти так называемые
антигены лейкоцитов человека (HLA) могут предопределять иммунную реакцию на
некоторые антигены, а также чувствительность к аутоиммунным заболеваниям.
Поскольку Т-клетки не связываются с антигенами, находящимися в растворе
в свободном состоянии, активация Т-клеток зависит от распознавания
рецепторами антигена в сочетании с ауто-ГКГ. Существуют два класса ГКГ-белков: класс I
и класс II. Протеины класса I (HLA-A, -В и -С) опосредуют распознавание
внутриклеточных антигенов, образующихся в результате таких процессов, как, например,
вирусные инфекции. Молекулы класса I, которые стабилизируют взаимодействие
АПК с Т-клетками, связываются молекулами CD8, экспрессирующимися на клет-
ках-супрессорах. Белки класса II (HLA-DP, -DQ и -DR) распознаются СБ4-ан-
тигенэкспрессирующими Т-хелперами. Они имеют особое значение для реакции
на внеклеточные антигены, например белки бактерий. Оба класса ГКГ-молекул
имеют "карман", в котором происходит индикация антигенов, обработанных АПК.
Экспрессия класса I стабильна, тогда как экспрессия класса II (В-клетками и мо-
нонуклеарными фагоцитами) возрастает после воздействия у-интерферона и
других цитокинов. Взаимодействие между АПК и Т-клетками далее стабилизируется
под воздействием СБЗ-комплекса и других лейкоцитарных молекул адгезии
и антирецепторов. Не всем антигенам требуются ГКГ-молекулы для активации
Т-клеток. Так, суперантигены (некоторые вирусные продукты и бактериальные
токсины) непосредственно активируют Т-клетки, возможно, посредством Р-субъе-
диницы рецептора В-клеток.
Т-клетки активируются при связывании антигена с ТКР в окружении ГКГ- и
СЭЗ-молекул. Этот процесс связан с фосфорилированием тирозина, о чем
сигнализируют вторичные мессенджеры — диацилглицерин и инозитолтрифосфат,
а также с активацией протеинкиназы С. Эти события являются
кульминационными в пролиферации клеток и экспрессии интерлейкина 2 (ЙЛ-2) и других
характерных белков. ИЛ-2 принимает существенное участие в аутокринцом механизме
активации Т-клеток, хотя и другие цитокины (включая ИЛ-3, ИЛ-4, ИЛ-6 и
ИЛ-7) также играют важную роль в этом процессе.
Обработанный
антиген
ГКГ класса 1/11
Рис. 4-8. Т-лимфоциты обладают антигенной специфичностью, обусловленной Т-клеточным
рецептором, который они экспрессируют. Т-клетки распознают антигены, представленные
макрофагами и другими антигенпрезентирующими клетками в сочетании с ГКГ-молекулами и CD4- или
С08-контррецептором
144
Глава 4. Лейкоциты
Клетки-хелперы, для которых характерна экспрессия антигена CD4,
составляют большинство циркулирующих лимфоцитов. Их пролиферация и экспрессия
ИЛ-2 после взаимодействия CD4 с молекулами ГКГII класса существенно
повышают экспрессию иммуноглобулинов В-клетками. Хелперы также усиливают
ответ супрессорных Т-клеток после взаимодействия CD8 с антигеном,
представленным в сочетании с молекулами ГКГ класса I антигенпрезентирующими клетками.
Клетки, экспрессирующие CD8, составляют большую часть супрессорных и цито-
токсических Т-клеток. Они уничтожают свои мищени, создавая поры в
клеточных мембранах путем введения перфоринов и других токсических молекул.
Очень сложно дифференцировать В- и Т-лимфоциты, окрашенные по Райту.
Диаметр лимфоцитов в 1-3 раза больше диаметра эритроцитов; их цитоплазма
синего, или серого цвета, а хроматин обнаруживается в виде скоплений. Клетки
с небольшим количеством цитоплазмы описываются как зрелые (малые)
лимфоциты, а с хорошо выраженной цитоплазмой называются большими лимфоцитами.
Другие варианты, или нетипичные лимфоциты, обычно наблюдаются при
вирусных заболеваниях, например инфекционном мононуклеозе. Эти реактивные
клетки имеют моноцитоидные ядра, часто с ядрышками, и обильную синевато-
серую цитоплазму, которая как бы заходит на соседние эритроциты. У больших
гранулярных лимфоцитов значительная часть площаДи клетки представлена
цитоплазмой с четкими красноватыми гранулами (рис. 4-9В).
К распространенным категориям лимфоцитов относятся нулевые
лимфоциты, названные так из-за того, что они не экспрессируют антигены, характерные
Рис. 4-9. Лимфоциты. (А) Малый лимфоцит. (Б) Активированный лимфоцит. (В) Большой
гранулярный лимфоцит. (Г) Плазматическая клетка. (Из: Hoffbrand А. V., Pettit J. E. Essential Hematology.
Cambridge, Mass.: Blackwell, 1993: 161.)
Лимфоцитарные нарушения
145
для В- или Т-клеток. Эта группа клеток включает незрелые лимфоциты, коммити-
рование которых в В- или Т-линию пока еще не произошло. Они экспрессируют
ТДТ — ДНК-полимеразу, участвующую в перегруппировке антигенного рецептора.
Естественные (нормальные, "натуральные") клетки-киллеры (НК) в
противоположность другим категориям лимфоцитов обладают свойством цитотоксич-
ности без предварительной антигенной сенсибилизации. Их способность убивать
не ограничивается ГКГ-антигенами; она не требует перегруппировки рецепторов
Т-клеток или Ig и существенно усиливается под влиянием цнтерферонов.
НК-клетки экспрессируют поверхностный антиген CD16 и морфологически
выглядят как большие гранулярные лимфоциты. Они атакуют аномальные клетки
(поврежденные клетки; клетки, инфицированные вирусом; раковые клетки),
выделяя цитолитические гранулы. НК-клетки способны убивать непосредственно
или за счет антителозависимой клеточно-опосредованной цитотоксичности. В
результате этого антитело, связанное с клеткой-мишенью, привлекает НК-клетки
посредством взаимодействия с их рецепторами для Fc-участка иммуноглобулина.
Лимфоцитарные нарушения
Лимфоцитоз (> 5000/мкл) наблюдается как реакция на острые вирусные
инфекции, хронические инфекции (туберкулез и сифилис), а также может быть связан
с эндокринопатией (тиреотоксикоз). Хронический лимфоцитарный лейкоз
вызывает лимфаденопатию, гепатоспленомегалию и лимфоцитоз. В мазке
периферической крови часто наблюдаются дегенерирующие клетки. Лимфоцитоз может
быть также вызван острым лимфоцитарным лейкозом, некоторыми неходжкинс-
кими лимфомами и волосатоклеточным лейкозом (глава 9).
Инфекционный мононуклеоз — клинический синдром, для которого
характерны повышение температуры тела, фарингит, лимфаденопатия, спленомегалия и
атипичный лимфоцитоз. Возбудителем инфекции является вирус Эпштейяа-
Барр (ВЭБ) — вирус семейства герпеса (имеет двухцепочечную ДНК). ВЭБ
инфицирует В-клетки через поверхностные антигены CD21 и заставляет их пролифери-
ровать. СЭв-экспрессирующие Т-клетки и НК-клетки реагируют на эти
инфицированные клетки и в большом количестве выходят в кровоток и
лимфатические ткани в виде больших лимфоцитов. Аналогичный атипичный лимфоцитоз
наблюдается при инфекциях, вызванных большинством вирусов, микоплазмой,
при туберкулезе и реакциях гиперчувствительности к лекарственным препаратам.
Многие наследственные или приобретенные аномалии лимфоцитов вызывают
иммунодефицита (табл. 4-5). У больных с наследственной сцепленной с Х-хромо-
сомой агаммаглобулинемией Т-клетки функционируют нормально, обеспечивая
отторжение трансплантата и гиперчувствительность, в то время как дисфункция
В-клеток приводит к рецидивирующим гнойным инфекциям. К другим
нарушениям функции В-клеток относятся общий вариабельный иммунодефицит и
изолированная IgA- или IgG-недостаточность. Синдром Ди Георге связан с аплазией
тимуса и нарушением функции Т-клеток. Соответственно, у таких больных
возникает недостаточность клеточного иммунитета, и они становятся необычайно
чувствительны к инфекциям, вызванным вирусами, микобактериями и грибками.
При тяжелом комбинированном иммунодефиците в связи с аномалией аденозин-
дезаминазы или другими нарушениями отсутствуют как В-, так и Т-клетки.
В этих случаях обычно требуется проведение трансплантации костного мозга, что
146
Глава 4. Лейкоциты
позволяет противостоять вирусным, бактериальным и грибковым инфекциям.
К другим редким нарушениям, вызывающим дисфункции В- и Т-клеток,
относится синдром Блюма, атаксия-телеангиэктазия и синдром Вискотта-Олдрича.
Лимфопения (лимфоцитов менее 1000/мкл) наблюдается при тяжелой
недостаточности костного мозга и после его супрессии в результате облучения или
химиотерапии. Нарушения иммунной функции иногда сохраняются в течение нескольких
лет после трансплантации костного мозга, что приводит к частому инфицированию
цитомегаловирусом и вирусом опоясывающего герпеса (глава 8). Поскольку корти-
костероиды нарушают различные звенья лимфопоэза, их применение
сопровождается дисфункцией лимфоцитов и повышенной чувствительностью к инфекции.
Синдром приобретенного иммунодефицита (СПИД) вызывается вирусом
иммунодефицита человека (ВИЧ) (глава 10). ВИЧ — это СБ4-лимфотропный рет-
ровирус, который передается через кровь, ткани или секреты. Чаще всего
заражение происходит при половом контакте, внутривенном введении наркотиков,
переливании зараженной крови или в перинатальный период (передача вируса
от матери плоду). В начальной стадии ВИЧ-инфекции наблюдаются слабовыра-
женные гриппоподобные болезненные состояния. Со временем наступает
прогрессирующая деструкция защитных механизмов организма, прежде всего
за счет истощения СБ4-экспрессирующих клеток. СПИД связан с цитопенией
всех ростков кроветворения, что приводит к тяжелому иммунодефициту. Цито-
пения усугубляется под действием многочисленных антиретробирусных
препаратов и антибиотиков, применяемых для лечения подобных больных. Как
правило, у пациентов возникают инфекции, вызываемые Pneumocystis carinii или
атипичными микобактериями, а также СПИД-энцефалопатии, саркома Капоши,
высокозлокачественные неходжкинские лимфомы или кахексия.
ТАБ Л И ЦА 4- 5. Классификация иммунодефиците
Первичный
Вторичный
В-клеточный
(дефицит антител)
Т-клеточный
Смешанный В-
и Т-клеточный
В-клеточный
(дефицит антител)
Т-клеточный
Т- и В-клеточный
Наследственная Х-сцепленная агаммаглобу-
линемия, приобретенная общая
вариабельная гипогаммаглобулинемия,
изолированный дефицит IgA или IgG
Аплазия тимуса, дефицит пуриннуклеозид-
фосфорилазы
Тяжелый комбинированный иммунодефицит,
вызванный недостаточностью аденозинде-
заминазы или другими причинами
Синдром Блюма
Атаксия-телеангиэктазия
Синдром Вискотта-Олдрича
Миелома
Нефротический синдром, энтеропатия с
потерей белка
СПИД
Болезнь Ходжкина, неходжкинская лимфома
Лекарства: стероиды, циклоспорин, азатио-
принидр.
Хронический лимфоцитарный лейкоз,
состояние после трансплантации костного мозга,
после химиотерапии и облучения
Из: Hoffbrand А. V., Pettit J. E. Essential Hematology. Cambridge, Mass.: Blackwell, 1993:178.
Лимфоцитарные нарушения
147
Клинический пример
65-летняя женщина была доставлена в отделение неотложной помощи
в связи с лихорадкой и ангиной. За 4 месяца до этого лечащим врачом был
поставлен диагноз "повышенной активности" щитовидной железы
(причина обращения к врачу — тахикардия). Больная получала дигоксин и
пропилтиоурацил в дозе 300 мг/день. Она регулярно принимала
лекарства, при этом ежемесячно контролировали содержание лейкоцитов. При
последнем исследовании, выполненном за 2 недели до поступления,
содержание лейкоцитов в крови было нормальным. Исследование функции
щитовидной железы патологии также не выявило. В течение трех дней
до госпитализации отмечались саднящая боль во рту и горле, озноб.
Температура тела (измерение во рту) составила 39,1 °С. При поступлении:
температура 39,4 X, пульс 120 ударов/мин, регулярный. Ротовая полость
и глотка эритематозны. На слизистой оболочке видны язвы диаметром
0,5 см; у некоторых язв в основании — густой экссудат. Иных данных,
позволяющих выяснить причину лихорадочного состояния, при физикаль-
ном обследовании не получено.
В периферической крови показатели гемоглобина, гематокрита и
тромбоцитов находятся в пределах нормы. Однако количество лейкоцитов
существенно снижено: 980/мкл. Лейкоцитарная формула: нейтрофилы — 5 %,
палочкоядерные нейтрофилы — 2 %, лимфоциты — 79 %, моноциты —
10 %, эозинофилы— 4 %.
Вопрос 1. Что следует предпринять?
Ответ. Необходимо провести полное обследование больной для выявления
источника инфекции (посев крови и мазков из зоны ротоглоточных
поражений), незамедлительно начать применение антибиотиков широкого
спектра действия.
Вопрос 2. Как лекарственные препараты, которые принимала больная,
могли повлиять на состояние крови?
Ответ. Теоретически пропилтиоурацил может быть причиной выраженной
лейкопении, однако надо учитывать и другие факторы. Прием
пропилтиоурацил а следует прекратить. Что касается дигоксина, то маловероятно,
чтобы он вызвал нейтропению, к тому же этот препарат, вероятно, необходим
больной для предотвращения фибрилляции предсердий.
Вопрос 3. Что еще необходимо предпринять?
Ответ. Гранулоцитарный колониестимулирующий фактор (Г-КСФ)
сокращает продолжительность нейтропении, но не компенсирует
нейтропению, вызванную лекарственным веществом. Описаны случаи применения
Г-КСФ в подобных ситуациях, когда задействован как иммунный, так и
токсический компонент. Поэтому больной назначен Г-КСФ.
Вопрос 4. Следует ли возобновить прием пропилтиоурацила, но в меньших
дозах, после устранения нейтропении?
Ответ. Нет! Нельзя назначать ни пропилтиоурацил, ни другие
аналогичные препараты (например, метемазол), поскольку они могут
спровоцировать такую же или даже более сильную нейтропеническую реакцию.
Вопрос 5. Мог ли врач этой больной обеспечить лучший контроль
состояния крови и тем самым предотвратить развитие нейтропении?
148
Глава 4. Лейкоциты
Ответ. Нет. Считается, что нейтропения может возникнуть внезапно,
поэтому даже1 частое исследование крови в этих случаях не помогает. Известны
факторы риска развития нейтропении у больных, принимающих метемазол,
а именно: женский пол, возраст старше 40 лет, доза препарата,
превышающая 40 мг в день. Больных следует предупредить, что появление ангины,
боли во рту (частые симптомы при лейкопении), а также повышение
температуры — признаки, о которых нужно незамедлительно сообщать врачу.
Результаты. Температура у больной снизилась до нормальной только через
3 дня, хотя и не было роста бактериальных, вирусных и грибковых культур.
Количество лейкоцитов сохранялось низким в течение 1 недели, а затем
резко повысилось и вернулось к норме. Симптомы исчезли одновременно
с повышением содержания лейкоцитов. Лечение гипертиреоза продолжали
с помощью 1311, больная чувствует себя хорошо.
Избранная литература
Clark E. A., Ledbetter J. A. How В and T cells talk to each other. Nature, 1994; 367:425-428.
Обзор, посвященный молекулярным механизмам взаимодействия В- и Т-лимфоцитов
в ходе иммунного отбета.
Cline M.J. Histiocytes and histiocytosis. Blood, 1994; 84(9): 2840-2853.
Обзор, посвященный вопросам онтогенеза гистиоцитов и клинических нарушений,
связанных с дисфункцией этих клеток.
Miller R. A., Britigan В. Е. The formation and biologic significance of phagocyte-derived
oxidants./. Invest Med., 1995; 43(1): 39-49.
Описание процесса генерации токсических форм кислорода фагоцитами и их роли
в иммунной защите
Moore M. A. S. Expansion of myeloid stem cells in culture. Semin. HematoL, 1995; 32:183-200.
Обзор характеристик стволовых миелоидных клеток, стимулирующих их цитокинов,
а также культуральных систем для выращивания этих клеток.
Orkin S. H. Transcription factors and hematopoietic development./. Biol. Chem., 1995;
270:4955-4958.
Обзор, посвященный роли факторов транскрипции в гемопоэзе и их влиянию на
дифференциацию клеточных линий и коммитирование.
Rosen F. S., Cooper M. D., Wedgewood R. J. P. The primary immunodeficiencies. New
Engl J. Med., 1995; 333 (7): 431-440.
Краткое описание клинических проявлений иммунных нарушений лимфоидных клеток
и последние данные о молекулярной основе этих процессов.
Schwartz R. S. Jumping genes and the immunoglobulin V gene system. New Engl. J. Med.,
1995; 333 (7): 42-44.
Краткое иллюстрированное сообщение, посвященное механизмам, лежащим в основе
перегруппировки генов иммуноглобулинов и обеспечения разнообразия антител.
Spits H., Lanier L. L., Phillips J. H. Development of human T and natural killer cells.
Blood, 1995; 85 (10): 2654-2670.
Обзор по проблеме онтогенеза НК и Т-лимфоцитов.
Stossel Т. P. The machinery of blood cell movements. Blood, 1994; 84 (2): 367-379.
Обстоятельное описание молекулярных основ удивительной способности нейтрофи-
лов к миграции.
Глава 5
Тромбоциты
ЭрикМ. Мазур
Принято считать, что нормальный гемостаз достигается за счет кооперации и
взаимодействия двух самостоятельных систем свертывания крови: гуморальной
(плазменной), состоящей из прокоагулянтных белков (глава 6), и клеточной,
состоящей из тромбоцитов. Конечным результатом активации гуморальной
системы свертывания крови является формирование фибринового сгустка, или
красного тромба, в то время как реакция тромбоцитов, сопровождаемая клеточной
адгезией и агрегацией, приводит к образованию тромбоцитарной пробки, или
белого тромба. Хотя эти две системы свертывания, как правило, рассматриваются
отдельно, следует понимать, что фактически они тесно связаны. Растворимые
факторы свертывания (например, фибриноген и фактор Виллебранда) имеют
большое значение для нормальной функции тромбоцитов, и наоборот,
тромбоциты являются важными поставщиками прокоагулянтных белков и
необходимым катализатором ряда реакций гуморальной системы свертывания крови.
Поэтому в настоящей главе будут рассмотрены в основном собственно
тромбоциты, а также их интегральная роль для системы коагуляции крови и
репарации тканей.
Нестимулированные тромбоциты представляют собой небольшого размера
(диаметр 3,6 ± 0,7 мкм) дискообразной формы безъядерные фрагменты мега-
кариоцитарной цитоплазмы, циркулирующие в периферической крови. В норме
их содержание составляет 150 000-400 000/мкл. Тромбоциты играют важную
роль в воспалительной реакции и в репарации тканей, а также в атерогенезе.
Простота получения нормальных тромбоцитов позволяет использовать их в
экспериментальных моделях для изучения биологических функций клеток:
трансмембранной сигнальной трансдукции, синтеза простагландинов, поглощения и
секреции нейротрансмиттеров. В настоящее время в клинической практике стали
часто прибегать к переливаниям тромбоцитарной массы, что связано с широким
использованием высоких доз цитотоксических препаратов, и трансплантации
костного мозга при злокачественных заболеваниях. Можно надеяться, что
открытие и клонирование тромбопоэтина — физиологического регулятора образования
тромбоцитов, позволит снизить потребность в переливании тромбоцитарной
массы и поможет более детально изучить нормальную функцию этих самых
маленьких клеток крови.
150
Глава 5. Тромбоциты
Образование и кинетика тромбоцитов
Клеточная биология мегакариоцитопоэза
Тромбоциты, наименьшие по размеру форменные элементы крови, образуются
из самых крупных костномозговых клеток — мегакариоцитов. Обнаружено, что
мегакариоциты в.свою очередь являются производными той же полипотентной
гемопоэтической стволовой клетки, из которой образуются эритроциты, нейтро-
филы и моноциты (глава 1). Стволовая клетка, формирование которой
сфокусировалось исключительно на одном из ростков кроветворения (например, мегака-
риоцитарном), называется коммитированнои. Для развития коммитированнои
мегакариоцитарной стволовой клетки характерны четыре почти
последовательные стадии: 1) экспоненциальное увеличение числа коммитированных
стволовых клеток за счет митотического деления; 2) серийная репликация ядра без
деления клеток (т. е. эндоредупликация, или полиплоидизация); 3) созревание
цитоплазмы мегакариоцита; 4) отделение тромбоцитов от мегакариоцита.
Любой из этих четырех процессов может влиять на скорость образования
тромбоцитов и выведения их из костного мозга в периферическую кровь.
Поэтому каждый из них — потенциальное место приложения гомеостатической регу-
ляторной системы, контролирующей количество тромбоцитов. От степени мито-
тической экспансии коммитированнои стволовой клетки зависит, сколько из нее
образуется мегакариоцитов. Поскольку количество коммитированных мегака-
риоцитарных стволовых клеток существенно не меняется с изменением
потребности крови в тромбоцитах, то число митотических делений стволовой клетки,
по всей вероятности, определяет количество сформировавшихся зрелых
мегакариоцитов в костном мозге. Эндоредупликация инициируется только в том
случае, когда развивающийся мегакариоцит утрачивает свою способность к
делению клетки. В ядре продолжается редупликация без деления клетки, и
поскольку это происходит на протяжении нескольких циклов, то окончательная
популяция мегакариоцитов имеет гетерогенный индекс плоидности. Общее
число эндомитотических циклов обычно варьируется между 3 и 6, что соответствует
индексам плоидности мегакариоцита от 8 до 64 п. Хотя целесообразность эндо-
редупликации кариоцитов неясна, вполне очевидно, что существует прямая
зависимость между плоидностью мегакариоцита и количеством тромбоцитов,
которое он может продуцировать. Поэтому при повышении степени эндоредуп-
ликации мегакариоцитов быстро увеличивается продукция тромбоцитов. Как
у экспериментальных животных, так и у человека было обнаружено, что при
значительно повышенной потребности в тромбоцитах происходит сдвиг в
сторону увеличения плоидности мегакариоцитов (составляющий обычно один
дополнительный эндомитотический цикл). Увеличение плоидности представляет
собой раннюю реакцию адаптации к повышенной потребности в тромбоцитах, так
как дополнительный цикл эндоредупликации, возникающий относительно
поздно в процессе формирования мегакариоцита, может усилить образование
тромбоцитов быстрее, чем активация митозов стволовой клетки.
Хотя развивающиеся мегакариоциты приобретают некоторые характерные
мембранные гликопротеины и начинают синтезировать небольшое количество
белков сс-гранул во время эндоредупликации, созревание цитоплазмы
происходит в основном после завершения эндомитоза. На самой ранней постэндомитоти-
Образование и кинетика тромбоцитов
151
ческой стадии при световой микроскопии незрелый мегакариоцит виден как
клетка 6-24 мкм в диаметре с плотным многодолевым ядром, окруженным тонким
кольцом темно-синей цитоплазмы. С помощью электронной микроскопии
выявляется аппарат Гольджи с рудиментарной системой демаркационной мембраны и
редкими а-гранулами (см. далее). Последующее созревание характеризуется
прогрессивно возрастающими ацидофилией, гранулярностью и объемом
цитоплазмы, что можно наблюдать под световым микроскопом. Увеличиваясь в объеме,
цитоплазма заполняется а-гранулами, плотными гранулами, а также обширной
сетью переплетенных мембранных каналов и тубул, которая называется
демаркационной мембранной системой (ДМС). Полностью созревшие мегакариоциты,
размер которых колеблется от 20 до 50 мкм, составляют приблизительно 0,04 %
ядросодержащих костномозговых клеток. Для них характерны объемная розовая
цитоплазма с большим количеством гранул и эксцентрично расположенное
компактное ядро с несколькими долями.
Процесс терминальной стадии тромбоцитопоэза недостаточно изучен. Согласно
одной из гипотез, зрелая ДМС расщепляет цитоплазму мегакариоцита на тром-
боцитарные поля и отдельные тромбоциты образуются за счет растворения
цитоплазмы мегакариоцита в определенных плоскостях демаркации. Позже было
доказано, что формирование тромбоцитов из мегакариоцитов представляет собой
более динамичный процесс. В ответ на некий сигнал мегакариоциты
трансформируются в паукообразные клетки, от которых отходит множество длинных
нитевидных отростков с равномерно расположенными очагами констрикции.
Полагают, что эти отростки, называемые протромбоцитами, входят в костномозговые
синусоиды (рис. 1-3) и там фрагментируются натромббциты,
возможно/благодаря сдвигающей силе кровотока. Хотя терминальная стадия тромбоцитопоэза
затрагивает только наиболее зрелые мегакариоциты, она представляет собой
регулируемый процесс. После резкого повышения периферической потребности
в тромбоцитах незамедлительно выявляется увеличение объема этих клеток, что
отражает изменения в механизме образования тромбоцитов.
Гуморальная регуляция продукции тромбоцитов
В стабильных условиях количество циркулирующих тромбоцитов поддерживается
в определенном контролируемом узком диапазоне. Но при патологических
состояниях, вызывающих ускоренное потребление тромбоцитов, продукция последних
костным мозгом может в 8 раз превышать норму, что происходит за счет
стимуляции образования мегакариоцитов и увеличения их плоидности, размера и скорости
созревания. Ключевые патогенетические факторы тромбоцитопении обычно
связаны с функционированием костного мозга. При возрастании количества
костномозговых мегакариоцитов можно полагать, что тромбоцитопения вызвана
потреблением тромбоцитов в периферической крови или ретикулоэндотелиальной
системе (или секвестрацией селезенкой), соответственно, нет необходимости
в проведении дополнительных клинических исследований.
Давно установлено, что процесс образования тромбоцитов строго
регулируется, но специфический механизм этой регуляции был неясен. Более 30 лет назад
было предположено, что продукция тромбоцитов контролируется
циркулирующим в плазме фактором, концентрация которого увеличивается в ответ на тром-
боцитопению. Впоследствии он получил название тромбопоэтин. Однако как
152
Глава 5. Тромбоциты
специфический цитокин, регулирующий гемопоэз, тромбопоэтин
идентифицировали только в 1994т., когда была определена его аминокислотная
последовательность и его ген был клонирован одновременно исследователями четырех разных
групп. Клонирование существенно облегчилось после того, как обнаружили, что
тромбопоэтин является лигандом для гемопоэтического рецептора с-Мр1,томо-
логичным белку, кодируемому вирусным онкогеном, который индуцирует мие-
лопролиферативное лейкемическое состояние у мышей.
Известно, что тромбопоэтин является стимулятором (специфическим в
отношении линии дифференцировки) развития мегакариоцитов и образования
тромбоцитов. Он действует как на уровне коммитированной мегакариоцитарной
стволовой клетки, так и на уровне развивающегося мегакариоцита (рис. 1-1).
Тромбопоэтин (другие обозначения: тромбопоэтина, Mpl-лиганд, фактор роста и
развития мегакариоцита, мегапоэтин) продуцируется в основном печенью, и
только незначительное количество его мРНК определяется в почках. В
противоположность другим гемопоэтическим цитокинам тромбопоэтин, вероятно, не
регулируется на уровне генной транскрипции. Даже при тяжелой тромбоцитопении,
вызываемой в эксперименте ускоренным потреблением тромбоцитов или ингиби-
рованием их образования, уровень мРНК тромбопоэтина в печени почти не
отличается от нормы. Полагают, что тромбопоэтин продуцируется печенью с
постоянной скоростью и его стимулирующий эффект зависит от количества молекул, не
связанных с рецепторами тромбопоэтина циркулирующих тромбоцитов. Такая
модель требует подтверждения, однако с ее помощью можйо объяснить, почему тром-
боцитопения, вызванная депонированием тромбоцитов в селезенке при
стабильном их содержании, не приводит к компенсаторной стимуляции тромбоцитопоэза.
Было обнаружено, что многие другие гемопоэтические цитокины
стимулируют образование тромбоцитов, но не в той степени или не с той строгой
специфичностью, как тромбопоэтин. Интерлейкин-3 (ИЛ-3), мультилинейный гемопоэти-
ческий фактор роста, стимулирует митотическую экспансию и начальное
развитие мегакариоцитарной коммитированной стволовой клетки, но
незначительно влияет на мегакариоцитарную эндоредупликацию и созревание
цитоплазмы. И наоборот, ИЛ-6 и ИЛ-11 оказывают тромбоцитопоэтическое действие,
стимулируя в умеренной степени постмитотические фазы развития мегакариоцита.
Согласно предварительным данным, ИЛ-6 может опосредовать реактивный
тромбоцитоз, связанный in vivo с отдельными системными воспалительными
заболеваниями (например, ревматоидным артритом). Все три указанные выше ци-
токина проявляют некоторую активность в стимулировании образования
тромбоцитов, что наблюдалось в эксперименте. Однако степень максимальной
тромбоцитостимулирующей активности этих цитокинов in vivo умеренная
(количество тромбоцитов возрастает только в 2-3 раза). Кроме того, они обладают
определенной токсичностью. В литературе нет сведений, которые бы
свидетельствовали о физиологической роли ИЛ-3, ИЛ-6 или ИЛ-11 в регуляции
мегакариоцитопоэза и количества циркулирующих тромбоцитов. Лишь
активность тромбопоэтина плазмы может меняться обратно пропорционально числу
тромбоцитов (по механизму биологической обратной связи).
Благодаря своей значительно большей активности, высокой специфичности и,
по предварительным данным, низкой токсичности, вполне вероятно, что только
тромобопоэтин может использоваться в клинической практике при лечении
тромбоцитопении.
Структура тромбоцитов
153
Структура тромбоцитов
В состоянии покоя тромбоцит представляет собой дискообразную клетку с гладкой
цитоплазматической мембраной, поддерживаемой микротубулиновым кольцом.
Мембрана клетки инвагинирует и соединяется с сетью многочисленных каналов,
так называемой открытой канальцевой системой (ОКС), которые тесно
переплетены внутри тромбоцита. Обнаружено, что центральные каналы этой системы
соединяются с внеклеточным пространством и ОКС экспрессирует те же гликопротеины,
что и клеточная мембрана. Вторая система внутренней оболочки (плотная тубуляр-
ная система), которая, как полагают, образуется из эндоплазматического ретикулу-
ма мегакариоцита, не зависит от ОКС и не соединяется с внеклеточным
пространством. Сократительные микрофиламенты распространяются от субмембранного
пространства по всей цитоплазме тромбоцита и обусловливают изменения его
формы, происходящие во время активации клетки. В цитоплазме неактивированных
тромбоцитов можно обнаружить 4 вида гранул: а-гранулы, плотные гранулы, лизо-
сомы и пероксисомы (рис. 5-1). Наиболее многочисленные а-гранулы содержат
тромбоцитоспецифические и тромбоцитонеспецифические пептиды, участвующие
в механизмах коагуляции, воспаления, иммунитета и репарации и модулирующие
эти процессы. Плотные гранулы, названные так сообразно их внешнему виду под
электронным микроскопом, представляют собой богатое хранилище АДФ и серо-
тонина — веществ, способствующих агрегации тромбоцитов, а также антиагреганта
АТФ и основного кофактора коагуляции Са2+. Лизосомальные гранулы содержат
гидролитические ферменты, а пероксисомы — каталазу (табл. 5-1).
Наружная клеточная оболочка и ОКС усеяны гликопротеинами, играющими
важную роль в адгезии и агрегации тромбоцитов (табл. 5-2). Эти молекулы состоят
из наружных доменов, действующих в качестве рецепторов, которые связываются
с внеклеточными адгезивными гликопротеинами (фибриногеном, коллагеном,
фактором Вилленбранда), и трансмембранных пептидов, которые фиксируют
гликопротеины и опосредуют процессы активации тромбоцитов и изменения
их формы (см. далее). Большинство тромбоцитарных гликопротеинов, за исклю-
Рис. 5-1. Схематическое изображение тромбоцита человека (справа — поперечный разрез). (Из:
Bentfeld-Barker M. Е., Bainton D. F. Identification of primary lysosomes in human megakaryocytes and
platelets Blood, 59: 472-481, 1982.)
154
Глава 5. Тромбоциты
ТАБЛИЦА 5-1. Компоненты о-гранул тромбоцита и их предполагаемые функции
Компонент Функция
Тромбоцитарный фактор роста (ТрФР) Репарация за счет усиления деления
фибробластов
Трансформирующий фактор Репарация ткани
роста Р(ТФР-Р)
Тромбоцитарный фактор 4 (ТФ-4) Нейтрализация гепарина,
воспалительный процесс
Воспаление, репарация ткани
Свертывание, адгезия тромбоцитов
Свертывание, агрегация тромбоцитов
Свертывание
Антикоагулянт
Связывание гормонов, токсинов,
лекарственных препаратов
Иммуноглобулин Иммунитет
чением гликопротеинового комплекса Ib-IX, кодируется генами интегринового
семейства. На тромбоцитарных мембранах имеются рецепторы для
физиологических медиаторов активации тромбоцитов (АДФ, адреналина, серотонина и
тромбоксана А2) и для Fc-фрагмента иммуноглобулинов. Кроме того, на мембране
тромбоцита экспрессированы антигены системы HLA класса I (но не класса II).
Функции тромбоцитов
Тромбоциты выполняют различные функции in vivo: 1) запуск немедленного
гемостаза за счет адгезии и агрегации тромбоцитов, что приводит к формированию
тромбоцитарной пробки; 2) местное выделение вазоконстрикторов для
уменьшения кровотока в пораженном участке; 3) катализ реакций гуморальной системы
свертывания с образованием в конечном счете фибринового сгустка; 4)
инициирование репарации тканей; 5) регулирование местной воспалительной реакции и
иммунитета. Нестимулированные тромбоциты циркулируют в виде гладких дис-
коидных клеток с незначительной метаболической активностью. Такие
тромбоциты не вступают в физиологически значимое взаимодействие с другими
форменными элементами периферической крови или монослоем эндотелиальных
клеток, который выстилает эндоваскулярное пространство.
ТАБ Л И ЦА 5- 2. Тромбоцитарные гликопротеины и их лиганды
Лиганды
Гликопротеин
Первичные Вторичные
ГП llb-llla Фибриноген ФВ, фибронектин, витронектин
ГП Ib-IX ФВ Тромбин
ГП la-lla Коллаген
ГП1с-На Фибронектин Ламинин
(Vila Ламинин
Рецептор витронектина Витронектин Тромбоспондин
/Мромбоглобулин 03-ТГ)
Фактор Виллебранда (ФВ)
Фибриноген
Фактор V
Протеин S
Альбумин
Функции тромбоцитов
155
Физиологическая активация тромбоцитов начинается только тогда, когда
поврежден сосудистый эндотелий и обнажен субэндотелиальный внеклеточный мат-
рикс. При поражении эндотелия воздействию подвергается коллаген, другие белки
внеклеточного матрикса, а также микрофибриллы, фиксирующие большие муль-
тимеры фактора Виллебранда (которые синтезируются и секретируются эндотели-
альными клетками). Рецептор гликопротеина lb (ГП lb) тромбоцитарной
мембраны связывается специфически с фактором Виллебранда (ФВ), который вторично
(другим участком) связывается с ГП ПЬ-Ша тромбоцитарной мембраны.
Субэндотелиальный коллаген связывается с рецептором мембранного ГП 1а. Адгезия
тромбоцитов сама по себе, а также посредством участия в запуске гемостаза, инициирует
процесс активации тромбоцитов, который выражается в существенном изменении
их формы, необратимой секреции содержимого плотных и а-гранул, агрегации
тромбоцитов с образованием гемостатической тромбоцитарной пробки.
Изменение формы представляет собой раннее и обратимое проявление
активации тромбоцитов, которое опосредовано внутриклеточной системой
сократительных микрофиламентов. В тромбоцитарной мембране возникают волны
возбуждения и формируется большое количество коротких нитевидных псевдоподий
или филоподий, продвигающихся по открытой канальцевой системе. В результате
этого процесса значительно увеличивается площадь поверхности
тромбоцитарной мембраны, что необходимо для катализа реакций гуморальной системы
свертывания крови и, возможно, для стабилизации тромбоцитарных агрегатов
(рис. 6-2, 6-4, 6-6, 6-11). Активация тромбоцитов приводит также к конформаци-
онному изменению в ГП ПЬ-Ша, которое способствует связыванию
фибриногена с тромбоцитарной мембраной, что в свою очередь является предпосылкой для
агрегации тромбоцитов. С инициированием активации тромбоцитов
внутриклеточные органеллы сосредотачиваются в центре клетки за счет сокращения мик-
ротубулярного кольца, после чего происходит слияние мембран плотных
и а-гранул друг с другом, с клеточной мембраной тромбоцита и мембранами
ОКС. Это слияние приводит к экзоцитозу содержимого гранул в наружную
микросреду, так как внутренние каналы ОКС соединяются с внеклеточным
пространством. Посредством экзоцитоза белок мембраны а-гранул Р-селектин
(синонимы: GMP-140, PADGEM) перемещается с внутренней поверхности гранул
в ОКС и к наружной оболочке клетки. В это время имеют место такие
биохимические реакции, как синтез тромбоксана А2 из арахидоновой кислоты,
высвобождаемой мембраной плотной тубулярной системы, и синтез тромбоцитак-
тивирующего фактора (ТАФ) — сложной липидной молекулы. Тромбоксан А2
(нестабильный член семейства простагландинов) — сильный проагрегант и вазо-
констриктор, а ТАФ усиливает реакцию агрегации тромбоцитов и является
мощным активатором нейтрофилов. Секретируемые с началом активации
тромбоцитов АДФ, серотонин и тромбоксан А2 в свою очередь активируют
близлежащие тромбоциты. Этот каскад аутоактивации тромбоцитов, синтез тромбоксана
А2 и выделение содержимого гранул приводят к появлению тромбоцитарного
агрегата, прошитого фибриногеновыми мостиками с участием ГП ПЬ-Ша
мембранных рецепторов соседних тромбоцитов. Фибриногеновые мостики
существенно стабилизируются тромбоспондином — другим компонентом а-гранул,
освобождаемым при активации тромбоцитов. Образование продукта
гуморального коагуляционного каскада тромбина усиливает агрегацию тромбоцитов,
поскольку тромбин — еще один мощный ее агонист (рис. 6-15).
156
Глава 5. Тромбоциты
Происходит не только активация тромбоцитов тромбином, но и тромбоциты
в значительной степени способствуют генерированию тромбина, катализируя
и обеспечивая факторы для реакций гуморального коагуляционного каскада
(рис. 6-2, 6-4, 6-6, 6-11). Известно, что мегакариоциты синтезируют и депонируют
в а-гранулах факторы свертывания V, VIII, XIII, ФВ и фибриноген. Эти факторы
свертывания крови выбрасываются в микросреду при активации тромбоцитов и эк-
зоцитозе, что повышает их локальную концентрацию. Внеклеточные адгезивные
гликопротеины (фибронектин, остеонектин и витронектин) т&кже депонируются в
тромбоцитарных а-гранулах и секретируются при активации тромбоцитов.
Тромбоцитарные мембраны играют не менее важную роль в запуске
специфических реакций свертывания. Липопротеин мембраны, так называемый фактор 3,
является значимым катализатором при активации фактора X факторами 1Ха и
VIII и образовании протромбиназы за счет взаимодействия между факторами Ха
и V (рис. 6-4 и 6-6). Кроме того, тромбоцитарная мембрана может существенно
влиять на взаимодействие других растворимых факторов свертывания с целью
достижения оптимальной реактивности. И наоборот, активированные
тромбоциты ограничивают коагуляционный каскад. Мембрана активированного
тромбоцита связывает тромбин и тромбомодулин, компонент а-гранул, которые вместе
вызывают активацию антикоагуляционного протеина С (рис. 6-11). а-Гранулы
к тому же поставляют в микросреду активатор тканевого плазминогена.
Значительно менее изучено участие тромбоцитов в воспалительном процессе
и репарации тканей. Однако вполне очевидно, что тромбоцитарные а-гранулы
поставляют мощные цитокины, тромбоцитарный фактор роста и
трансформирующий фактор роста Р (ТФР-Р) в очаг поврежденной ткани. Тромбоцитарный
фактор роста является сильнодействующим стимулятором пролиферации фиб-
робластов и гладкомышечных клеток, в то время как трансформирующий фактор
обладает как стимулирующими, так и ингибирующими рост свойствами.
Тромбоцитарный фактор 4 и Р-тромбоглобулин — два других тромбоцитоспецифических
белка а-гранул, члены небольшого семейства индуцибельных белков, играющих
определенную роль в коагуляции, воспалительной реакции и клеточном росте.
Наконец, тромбоциты контактируют с системой гуморального иммунитета. Как
указывалось ранее, тромбоцитарные мембраны имеют рецепторы для Fc-участка
молекулы IgG и связывают иммунные комплексы и агрегированный IgG.
Обнаружено, что покоящиеся тромбоциты поглощают иммуноглобулин за счет эндоци-
тоза и хранят его в а-гранулах, чтобы позже секретировать путем экзоцитоза.
Лабораторные исследования
Подсчет тромбоцитов
У здоровых людей количество тромбоцитов в норме обычно достаточно стабильно,
но у различных пациентов может изменяться в пределах 150 000-400 000/мкл.
Средний объем тромбоцитов изменяется обратно пропорционально их
количеству, поэтому циркулирующая тромбоцитарная масса у индивидуума более
постоянна, чем можно было бы ожидать. В настоящее время лабораторное определение
числа тромбоцитов в периферической крови осуществляется автоматически в
разведенной пробе после лизиса эритроцитов. Поскольку этот метод позволяет
установить только количество частиц данного размера и не обеспечивает специфичес-
Лабораторные исследования
157
кого дифференцирования тромбоцитов, то в случае отклонения показателя
от нормы необходимо сделать анализ мазка периферической крови. Количество
тромбоцитов в поле зрения при 100-кратном увеличении умножают на 12 000-
15 000, полученная величина равна их содержанию в 1 мкл. Как артефакт низкое
число тромбоцитов может быть связано с индуцированной ЭДТА агглютинацией
тромбоцитов (так называемой псевдотромбоцитопенией) или сателлитизмом —
слипанием тромбоцитов с плазматическими мембранами нейтрофилов в
результате действия IgG- или IgM-тромбоцитарных агглютининов. Напротив, ложное
завышение количества тромбоцитов может наблюдаться в том случае, если в
периферической крови имеют место небольшие фрагменты нетромбоцитарных клеток
(например, при остром лейкозе, когда циркулирующие бласты могут выделять
малые цитоплазматические пузырьки, а для эритроцитов характерен микросфероци-
тоз). Если есть сомнение в достоверности результатов автоматического подсчета,
необходимо выполнить его вручную. Для подтверждения данных лабораторного
тестирования нужно всегда исследовать мазок периферической крови (глава 2).
Измерение объема тромбоцитов
Внедрение в практику автоматического подсчета клеток крови сделало
доступным определение размера циркулирующих тромбоцитов. Установлено, что объем
тромбоцита коррелирует со средней плоидностью мегакариоцитов и что этот
объем возрастает при ускоренном образовании тромбоцитов. Наличие большого
количества крупных тромбоцитов или мегакариоцитов в мазке периферической
крови больных с тромбоцитопенией считается диагностическим маркером тром-
боцитопении потребления. Однако на результаты автоматического измерения
среднего объема тромбоцитов оказывают влияние такие неклинические
переменные факторы, как температура образца крови, срок его хранения, используемый
антикоагулянт. Поэтому этот вид измерения трудно стандартизировать,
полученные данные следует интерпретировать с осторожностью.
Время кровотечения
Тромбоцитарный гемостаз зависит от двух переменных: количества тромбоцитов
и их функциональной активности. В то время как определение количества
тромбоцитов — простая процедура, оценка функции тромбоцитов — довольно сложная
задача, к тому же на интерпретацию теста оказывают влияние различные сложные
клинические переменные. Несмотря на гетерогенность возможных механизмов и
клинических проявлений тромбоцитарной дисфункции, существует только один
широко применяемый клинический тест для определения функции этих клеток,
а именно — время кровотечения (ВК). ВК соответствует времени между
нанесением небольшой стандартной поверхностной раны на предплечье и образованием
стабильного тромбоцитарного тромба, о чем свидетельствует прекращение
кровотечения. В модифицированном тесте используется лезвие (с пружиной), которым
на ладонной поверхности предплечья наносится рана длиной 1 см и глубиной 1 мм,
а на плече манжеткой нагнетается давление до 40 мм рт. ст. Кровь промокают
на краю раны фильтровальной бумагой каждые 30 с так, чтобы не задеть
образующуюся в центре тромбоцитарную пробку, до остановки кровотечения. В норме ВК
составляет 3-8 мин; так называемое нормализованное ВК — 2-7 мин (рис. 5-2).
158
Глава 5. Тромбоциты
Несмотря на примитивность теста и связанный с ним дискомфорт, он является
методом выбора в тех случаях, когда предполагают дисфункцию тромбоцитов.
При правильно выполняемом ВК-тесте отклонение от нормы наблюдают только
в трех случаях: 1) при тромбоцитопении менее 100 000/мкл; 2) при качественной
дисфункции тромбоцитов любого типа; 3) редко при аномалии микрососудов
(например, амилоидозе). При нормальной функции тромбоцитов и их количестве
10 000-100 000/мкл ВК изменяется линейно в зависимости от количества
тромбоцитов согласно эмпирически полученной формуле:
ВК (мин) = 30,5 - (количество тромбоцитов в 1 мкл)/3850.
(рис. 5-2). При тромбоцитопениях, связанных с увеличением потребления
тромбоцитов (например, иммунная тромбоцитопения), ВК обычно ниже, чем
величина, рассчитанная по этому уравнению, что объясняется избыточным содержанием
фосфолипидов в молодых и более крупных тромбоцитах при данном состоянии.
И наоборот, в некоторых случаях продуктивной тромбоцитопении, когда
нарушается созревание мегакариоцита и тромбоцита (например, миелодисплазия), ВК
будет больше, чем подсчитано по уравнению. Прием одной таблетки аспирина
удлиняет нормальное ВК до 8-10 мин. В то же время аспирин может
непропорционально увеличивать ВК у пациентов со слабой дисфункцией тромбоцитов и
обнаруживать таким образом незначительные качественные нарушения. Так
называемый тест толерантности к аспирину следует применять с большой
осторожностью у пациентов с возможной кровоточивостью, поскольку аспирин
усилит ее. Аспирин необратимо ацетилирует циклооксигеназу тромбоцитов и
поэтому изменяет функцию тромбоцитов на протяжений всего срока их жизни —
приблизительно 8-10 дней. (Другие нестероидные противовоспалительные
средства связываются с этим ферментом обратимо и, следовательно, ингибируют
функции тромбоцитов только в период циркуляции лекарства в крови.)
>60
Время кровотечения (мин)
Рис. 5-2. Зависимость между количеством тромбоцитов и нормализованным ВК в норме. Обратите
внимание, что ВК нечувствительно к числу тромбоцитов, если их > 100 000/мкл, но изменяется
линейно, если их < 100 000/мкл и > 10 000/мкл. (Из: Harker L.A., Philadelphia: F. A. Davis; Hemostasis
manual - 1974: 8.)
Лабораторные исследования
159
Исследование агрегации тромбоцитов
Количественное исследование агрегации тромбоцитов позволяет
дифференцировать качественные нарушения тромбоцитов в отдельные группы. Это простой
лабораторный тест, в котором конгломерация одиночных тромбоцитов в группы или
агрегаты большего размера определяется количественно по степени пропускания
света через тромбоцитосодержащую плазму. Тромбоцитарный агрегометр (рис. 5-3)
состоит из небольшой прозрачной кюветы, в которую помещают пробу тромбоцитов
пациента в стандартной концентрации. Через кювету пропускается свет от
калиброванного источника, поступающий на фотоэлемент. Фотоэлемент связан с
регистрирующим устройством, измеряющим светопрохождение (или оптическую плотность)
с учетом времени. Если тромбоциты представляют собой суспензию одиночных
клеток (т. е. до агрегации), то пропускание света минимально. С увеличением
агрегации плазма, содержащая тромбоциты, становится все светлее, а пропускание света
возрастает. Представить себе это явление можно, сравнивая прозрачность жидких
суспензий, содержащих одну и ту же массу глины или в виде большого количества
мелких частиц, или сконцентрированную в нескольких кусочках.
Агрегацию тромбоцитов in vitro можно инициировать, вводя различные
экзогенные вещества (чаще всего АДФ, адреналин, коллаген, тромбин, арахидоновую
кислоту и ристоцетин) в агрегометрическую кювету. АДФ, адреналин и тромбин
обладают уникальной способностью вызывать агрегацию тромбоцитов
самостоятельно, без обязательной дегрануляции клеток и экзоцитоза. Поэтому в низких
концентрациях эти вещества будут стимулировать только обратимую первичную
Агрегометрия тромбоцитов ~|
• •.;
••а ••
• • ••
Мутная
АДФ
Адреналин
Он
Фотоэлемент
Записывающее}
устройство
%
Прозрачная
1004
10-
% пропускания
света
Время
100-
ю-
1
f Вторичная волна
/первичная
^/ волна
Время
Рис. 5-3. Процесс агрегометрии Тромбоцитов. Значок * означает, что к системе добавлен агонист
агрегации тромбоцитов, например АДФ или адреналин. Образование тромбоцитарных агрегатов из
одиночных тромбоцитов превращает мутную, богатую тромбоцитами плазму в относительно прозрачную
160
Глава 5. Тромбоциты
волну агрегации. При более высоких концентрациях АДФ, адреналина и
тромбина тромбоциты полностью активируются и выделение гранулярнЪго
содержимого наряду с синтезом тромбоксана А2 приводит к несколько отсроченной, но
усиленной вторичной волне агрегации тромбоцитов (рис. 5-4). При еще больших
концентрациях первичная волна агрегаций погружается во вторичную волну и
можно видеть только одну полную волну агрегации.
Хотя коллаген связывается с гликопротеином 1а тромбоцитарной мембраны,
он не вызывает немедленной агрегации тромбоцитов. Однако спустя некоторое
время коллаген индуцирует полную агрегацию в результате внутренней
активации тромбоцитов. Способность арахидоновой кислоты стимулировать агрегацию
зависит от функционирования фермента циклооксигеназы в ходе синтеза про-
стагландинов.
Синтез тромбоксана А2 из арахидоновой кислоты приводит к полной
активации тромбоцитов, экзоцитозу и агрегации.
Уникальность ристоцетина состоит в том, что агрегация под его воздействием
не опосредуется изменением метаболизма тромбоцитов. Аналогичная по
механизму агрегация наблюдается при фиксации тромбоцитов в формалине. Полагают,
что ристоцетин обеспечивает агрегацию путем ассоциации с ГП lb
тромбоцитарной мембраны, активируя эту молекулу для связывания фактора Виллебранда.
Поэтому в присутствии ристоцетина ФВ соединяет тромбоциты в большие агре-
100
О)
00
о
ф
2
С
50
± АДФ (2 ммоль/л)
100
£
о
m
о
ш
S
X
± Адреналин (2 ммоль/л)
100
Q)
00
S 50
S
I
о
о.
С
Коллаген
100
0)
50
о
а
± Ристоцетин
Рис. 5-4. Пример агрегации тромбоцитов здорового человека при использовании агрегометра,
представленного на рис. 5-3. Реакции на введение АДФ, адреналина, коллагена и ристоцетина в
определенных концентрациях (момент добавления указан стрелкой).
Тромбоцитопатии
161
гаты. Если тромбоциты жизнеспособны, индуцированная ристоцетином
агрегация приводит к активации клеток и выделению эндогенных гранул. В
отсутствии фактора Виллебранда активации не происходит.
Тромбоцитопатии
Введение и клинические особенности
Качественные нарушения функции тромбоцитов, имеющие клиническое
значение, могут быть врожденными или приобретенными в результате воздействия
лекарственных средств или вследствие системного заболевания (табл. 5-3). Тем не
менее, тромбоцитарная дисфункция petse обычно не связана с тяжелым спонтан-
ТАБЛИЦА 5-3. Классификация и этиология качественных дефектов тромбоцитов
I. Врожденные нарушения
A. Дефицит/недостаточность мембранных гликопротеинов
1. Тромбастения Гланцмана (болезнь Гланцмана)
2. Синдром Бернара-Сулье
Б. Врожденные аномалии белков плазмы
1. Болезнь Виллебранда
2. Наследственная афибриногенемия
B. Недостаточность гранул
1. Дефицит плотных гранул
2. Дефицит пула а-гранул (синдром серых тромбоцитов)
3. Первичные дефекты высвобождения гранул:
а) дефицит циклооксигеназы
б) дефицит тромбоксансинтетазы
II. Приобретенные нарушения
A. Вторичные дефекты высвобождения гранул
1. Вызванные приемом лекарственных препаратов
а) аспирин и другие НПВС
б) тиклопидин
в) дипиридамол
г) карбенициллин и другие антибиотики (0-лактамы)
д) со-3 жирные кислоты
2. Уремия
3. Врожденный порок сердца с цианозом
Б. Недостаточность гранул
1. Вследствие дисфункции стволовых клеток
а) миелопролиферативные нарушения
б) миелодиспластические синдромы
в) острый лейкоз
2. В связи с частичной активацией
а) диссеминированное внутрисосудистое свертывание (ДВС)
б) тяжелые заболевания сердечных клапанов
в) экстракорпоральное кровообращение
B. Нарушение взаимодействия между тромбоцитарной мембраной и белками
внеклеточного матрикса
1. Парапротеинемия
2. ДВС
3. ИТП (единичные случаи)
162
Глава 5. Тромбоциты
ным геморрагическим диатезом. Сильное кровотечение возникает у таких
больных почти исключительно в связи с хирургическим вмешательством, травмой,
экстракцией зуба и другими видами гемостатического стресса. У больных с
качественной тромбоцитарной дисфункцией легко возникают синяки и
периодические кровотечения из слизистой (например, носовые кровотечения,
кровоточивость десен при чистке зубов, скрытое желудочно-кишечное кровотечение).
Гемартрозы и глубокие кровоизлияния в мягкие ткани характерны для тяжелой
недостаточности одного из гуморальных факторов свертывания и обычно не
связаны с тромбоцитами. Качественную тромбоцитарную дисфункцию следует
подозревать на основании подтверждающих данных анамнеза, патологического
изменения ВК при нормальных значениях АЧТВ, протромбинового и тромбинового
времени, а также нормальном количестве тромбоцитов. Измерение агрегационной
способности тромбоцитов у таких больных обычно позволяет поставить
специфический диагноз. При анамнезе, предполагающем дисфункцию, но нормальном ВК
чрезмерное увеличение последнего после обычной дозы аспирина, введенного пе-
рорально (так называемый тест толерантности к аспирину), позволяет выявить
пациентов, которым показано дальнейшее обследование с использованием агре-
гометрии. Если у больного увеличены ВК и АЧТВ, можно подозревать болезнь
Виллебранда (см. далее).
Врожденные нарушения адгезии тромбоцитов
и тромбоцитарной мембраны
Болезнь Виллебранда — наиболее распространенное наследственное гемофили-
ческое заболевание человека. В различных популяциях ее распространенность
составляет от 1 % до 2 % и более. Поскольку патология вызвана недостаточностью
или качественной аномалией белкового комплекса фактор Виллебранда-фактор
VIII, то это по сути дефект гуморальной системы свертывания, который будет
детально обсуждаться в следующей главе. Однако нормальная концентрация и фун-
кция'ФВ имеет значение и для поддержания функции тромбоцитов. Таким
образом, болезнь Виллебранда считается также нарушением тромбоцитарной
функции. Как отмечалось ранее, ФВ присутствует в плазме и субэндотелиальном
экстрацеллюлярном матриксе и связывается специфически с гликопротеином
тромбоцитарной мембраны ГП lb и вторично с ГП ПЬ-Ша. ФВ играет важную роль
в начальной адгезии тромбоцитов к коллагену и другим белкам внеклеточного мат-
рикса при повреждении субэндотелиальной поверхности, болезнь Виллебранда
может быть результатом двух разных молекулярных дефектов продукции ФВ:
1) количественно низкого синтеза функционально нормального белка ФВ; 2)
количественно нормального синтеза ФВ с нарушенной функцией. Эти два класса
дефектов называются соответственно болезнь Виллебранда типа I и типа ПА. Частота
первого составляет более 70 %, второго — 10-15 % (табл. 6-5 и 6-6).
Функциональную недостаточность ФВ можно определить в лаборатории как ослабленную
способность плазмы больного поддерживать ристоцетининдуцированную агрегацию
тромбоцитов in vitro (рис. 5-4). Эта способность называется активностью
кофактора ристоцетина и является обычно пониженной при болезни Виллебранда типа
I и типа ПА (табл. 6-5 и 6-6). Третий (редкий) вариант заболевания — тип ПВ,
характеризуется повышенным сродством дефектных ФВ-мультимеров с высокой
молекулярной массой к ГП lb, что приводит к специфической адсорбции этих
Тромбоцитопатии
163
мультимеров на тромбоцитарной мембране и в итоге — к недостаточности ФВ
с высокой молекулярной массой в циркулирующей крови, а также к тромбоцито-
пении, по-видимому, из-за внутрисосудистой агрегации тромбоцитов. При этом
редком варианте индуцированная ристоцетином агрегация тромбоцитов (при
низкой дозе ристоцетина) парадоксально повышена (табл. 6-5, рис. 6-20 В, С).
Субъединицы ФВ-мультимера кодируются геном (около 180 килобаз) на конце
короткого плеча хромосомы 12 (рис. 6-23). Болезнь Виллебранда наследуется
аутосомно-доминантным путем. Однако пенетратность гена может варьироваться,
поскольку на уровень циркулирующего фактора Виллебранда влияют многие
другие переменные, включая терапию эстрогенами, беременность, группу крови,
стрессовое состояние. Эти факторы определяют также существенную
клиническую гетерогенность среди пациентов с болезнью Виллебранда даже в пределах
одного типа. Поэтому у больного может изменяться предрасполЬженность к
кровотечению под воздействием клинических факторов и факторов окружающей среды.
Так как ФВ связывается с циркулирующим фактором VIII, стабилизирует его
и предотвращает его катаболизм, то количественные и качественные нарушения
фактора Виллебранда могут привести к снижению уровня прокоагулянтной
активности фактора VIII. Уровень фактора VIII ниже 30-40 % будет влиять на АЧТВ и
нормальный гемостаз. Поэтому у пациентов с БВ могут обнаруживаться
клинически значимые нарушения как гуморальной системы свертывания крови, так и
функции тромбоцитов и, как следствие АЧТВ и ВК. Однако столь низкий уровень
фактора VIII у пациентов с болезнью Виллебранда наблюдается редко (это
характерно для гемофилии А даже умеренной степени).
Клинические проявления заболевания очень разнообразны. Как правило,
обнаруживается склонность к внутрикожным кровоизлияниям, кровоточивость
слизистых оболочек, меноррагия. Предрасположенность к кровотечениям может
возрастать при приеме аспирина или других НПВС. Спонтанные гемартрозы и
кровотечения из мягких тканей нехарактерны,' однако существует высокий риск
возникновения кровотечения при выполнении хирургических и
стоматологических вмешательств. Лечение направлено на повышение уровня ФВ в крови путем
вливания экзогенного, полученного из донорской крови ФВ или за счет
мобилизации эндогенного ФВ из депо. Экзогенный фактор можно вводить в виде
свежезамороженной плазмы, криопреципитата и ряда коммерческих препаратов
концентрата фактора VIII. Поскольку количество ФВ в коммерческих концентратах
зависит от процесса выделения, необходимо индивидуально оценить
эффективность препаратов по замещению ФВ. Использование продуктов крови можно
ограничить с помощью десмопрессина, который мобилизует эндогенный ФВ из эн-
дотелиальных депо. Десмопрессин, синтетический аналог вазопрессина, вводится
внутривенно или интраназально. Он особенно эффективен для больных с БВ
типа I, менее действен при заболевании типа НА и противопоказан для пациентов
с редким типом НВ (глава 6). Так как терапевтический результат применения
десмопрессина у конкретного пациента нельзя предсказать, рекомендуется
выполнение диагностического мониторинга перед его использованием (т. е.
определение после введения десмопрессина протромбинового времени, уровня
фактора VIII, ФВ и кофактора ристоцетина). .В некоторых случаях, особенно
в стоматологической практике, назначение антифибринолитического агента
(е-аминокапроновой или транексамовой кислоты) может быть достаточным для
предотвращения кровотечения.
164
Глава 5. Тромбоциты
Синдром Бернара-Сулье — редкое наследственное нарушение функции
тромбоцитов, характеризующееся дефектом мембранного комплекса ГП lb. Так как ГП
lb является основным рецептором для фактора Виллебранда, у этих больных
клиническая картина напоминает болезнь Виллебранда умеренной тяжести и обычно
проявляется в младенческом или раннем детском возрасте. Функционально, как и
при БВ, нарушается адгезия тромбоцитов к сосудистому субэндотелиальному
матриксу. Хотя агрегация тромбоцитов под действием обычных агонистов
нормальна, индуцированная ристоцетином агрегация существенно нарушена или
вообще отсутствует и не корригируется добавлением нормального фактора
Виллебранда (рис. 6-19). К другим характерным клиническим особенностям относится
тромбоцитопения (от слабой до умеренной) и увеличение объема
циркулирующих тромбоцитов. Склонность к кровотечению может быть достаточно
выраженной и даже приводить к гибели отдельных больных, однако у других пациентов
тяжесть заболевания со временем ослабевает.
Тромбастения Гланцмана. Это редкое врожденное качественное нарушение
тромбоцитов, возникающее в результате отсутствия или дисфункции
мембранного рецептора фибриногена и ГП IIb-Ша. Как и синдром Бернара-Сулье,
тромбастения Гланцмана обычно начинает проявляться в младенческом или раннем
детском возрасте и характеризуется склонностью к внутрикожным кровоизлияниям,
носовыми кровотечениями, кровоточивостью десен и меноррагией. ВК
отличается от нормы, как и все агрегационные пробы, за исключением ристоцетиновой.
При этой серьезной дисфункции тромбоцитов нарушаются связывание
фибриногена с мембраной клетки и в результате — "сшивание" тромбоцитов при участии
фибриногена, что необходимо для их агрегации. У таких больных сохраняется
только ристоцетининдуцированная агрегация, не зависимая от фибриногена и
зависимая исключительно от остающихся в норме при этом заболевании фактора
Виллебранда и мембранного ГП lb (рис. 6-19).
Врожденные заболевания, обусловленные дефектами гранул
тромбоцитов
Эти заболевания представляют собой гетерогенную группу нарушений, для
которых характерны увеличенное ВК, склонность к внутрикожным
кровоизлияниям, умеренно или слабо выраженная склонность к кровотечению при
травме, хирургическом вмешательстве, стоматологических процедурах (табл. 5-3).
Обычно эти заболевания подразделяются на две группы: 1) с абсолютным
дефицитом проагрегантных накопительных гранул (т. е. плотных гранул,
содержащих АДФ, АТФ, кальций и серотонин); 2) с нормальным содержанием
гранул, но нарушенной функцией гранулосекреторного механизма — так
называемым аспириноподобным дефектом (см. далее). В обоих случаях адгезия
тромбоцитов и первичные волны агрегации in vitro с АДФ, адреналином и
тромбином остаются в норме. Однако отсутствует или ослаблена вторичная
волна агрегации из-за того, что тромбоциты не могут высвобождать адекватное
количество эндогенного АДФ и других проагрегантных веществ. Кроме того,
отсутствует или слабо выражена реакция in vitro на коллаген, поскольку для
индуцируемой им агрегации также необходима активная секреция плотных
гранул (рис. 5-1).
Тромбоцитопатии
165
Врожденный дефицит плотных гранул может существовать как
самостоятельное заболевание различной степени тяжести. Известен аутосомно-доминантный
вариант семейного наследования этой патологии. Описаны случаи сцепленного
аутосомно-доминантного наследования дефицита плотных гранул и
альбинизма — синдром Германского-Пудлака. Так как гранулодефицитные тромбоциты
не содержат гранулярного АДФ, диагностическим критерием может быть
повышенное соотношение АТФ : АДФ в тромбоците.
Дефицит ос-гранул, или синдром серых тромбоцитов, клинически
характеризуется умеренным геморрагическим диатезом, умеренной тромбоцитопенией
(60 000-100 000/мкл) и увеличением размера циркулирующих тромбоцитов.
Полагают, что нарушение возникает в результате дефекта "упаковки" продуктов
синтеза развивающихся мегакариоцитов в зарождающиеся а-гранулы. Это положение
подтверждается наблюдаемым повышением уровня циркулирующего тромбогло-
булина и тромбоцитарного фактора 4, а также наличием ретикулинового фиброза
костного мозга, по-видимому, связанного с утечкой тромбоцитарного фактора
роста и трансформирующего фактора роста р в костномозговое микроокружение.
Врожденные дефекты в процессе выделения тромбоцитарных гранул имеют
такую же клиническую картину, как и состояния абсолютного дефицита
накопительного пула. У больных, по-видимому, нарушается связь стимул-реакция
между активацией тромбоцитов и секрецией гранул. При исследовании агрегации
тромбоцитов помимо отсутствия или нарушения вторичной волны наблюдается
измененная реакция на арахидоновую кислоту (метаболический предшественник
мощных проагрегантных простагландиновых эндопероксидов и тромбоксана А2).
Способ наследования этой гетерогенной группы нарушений неизвестен.
Аспирин, который резко увеличивает аномалию ВК у таких пациентов, способен также
повышать склонность к кровотечению. Поэтому проба с аспирином может быть
полезна в дифференциальной диагностике.
Лечение подобных врожденных нарушений обычно неспецифично.
Переливания тромбоцитов с целью временной коррекции геморрагического диатеза
следует применять с осторожностью, учитывая риск развития осложнений после гемо-
трансфузий. Функции тромбоцитов улучшает десмопрессин (механизм действия
пока еще не известен), поэтому назначение данного лекарственного препарата
предпочтительно для профилактики или лечения кровотечения. В некоторых
клинических ситуациях прибегают к криопреципитату, но при его использовании
могут возникать посттрансфузионные осложнения.
Приобретенные тромбоцитопатии
Прием лекарственных препаратов. Приобретенная дисфункция тромбоцитов
может иметь различную этиологию (табл. 5-3). Наиболее характерной причиной
нарушений функции тромбоцитов в клинической практике является прием аспирина или
других НПВС. Аспирин необратимо связывает и инактивирует циклооксигеназу —
основной фермент в биосинтезе простагландинов. Происходят нарушения
внутреннего тромбоцитарного механизма стимул-реакция, что приводит к неспособности
секретировать тромбоцитарные гранулы и в целом — к приобретенному
расстройству секреции. После инактивации тромбоцитарной циклооксигеназы аспирином
тромбоцит остается недееспособным, пока окончательно не покинет сосудистое
русло. Поскольку каждые 5 дней происходит замена примерно 100 000-150000тромбо-
166
Глава 5. Тромбоциты
цитов/мкл, а концентрация 100 000 тромбоцитов/мкл обеспечивает нормальный
гемостаз, то именно стблько дней должно пройти после последнего приема аспирина,
чтобы устранить вызванный им дефект. Поэтому хирургическое вмешательство и
другие инвазивные процедуры следует отложить не менее чем на 5 дней после
лечения препаратом. Другие НПВС тоже оказывают ингибирующее влияние на циклоок-
сигеназу, но их эффект в противоположность аспирину обратим и исчезает при
элиминации этих препаратов из плазмы. Хотя время циркуляции каждого из НПВС
различно и зависит от периода его полувыведения, функция тромбоцитов обычно
нормализуется в пределах 24 ч. Если хирургическое вмешательство абсолютно
необходимо в ситуации, когда больной принимал аспирин или другие нестероидные
противовоспалительные препараты, назначение десмопрессина может временно
улучшить функцию тромбоцитов. При отсутствии эффекта и продолжающемся сильном
кровотечении прибегают к переливанию тромбоцитов.
Уремия. Вызывает различные качественные дефекты тромбоцитов. Хотя ВК
и показатели агрегационной способности тромбоцитов часто отличаются от
нормальных, ни наличие аномалии, ни ее степень не позволяют прогнозировать риск
развития кровотечения. Полагают, что функциональная недостаточность
тромбоцитов, вызванная уремией,— следствие аккумуляции токсических метаболитов
из уремической плазмы. Функцию тромбоцитов может улучшить адекватный
диализ, который в данном случае предпочтителен. Считают также, что дисфункция
тромбоцитов возникает из-за значительной анемии, характерной для
уремических больных. Уменьшение массы циркулирующих эритроцитов позволяет
тромбоцитам проникать в центр просвета кровеносного сосуда, тем самым снижая
произвольное взаимодействие тромбоцитов со стенками сосудов (у здоровых людей
тромбоциты заполняют обычно возникающие пробелы между эндотелиальными
клетками). Повышение уровня гематокрита более чем 30 % за счет трансфузион-
ной терапии или введения эритропоэтина существенно корректирует тромбоци-
тарную дисфункцию. К другим средствам, способствующим восстановлению
функции тромбоцитов при уремии, относятся назначение десмопрессина,
терапия эстрогенами и применение криопреципитата. Переливание тромбоцитарной
массы имеет относительное противопоказание, поскольку введенные тромбоциты
приобретают уремический дефект после контакта с плазмой больного.
Экстракорпоральное кровообращение. При выполнении операций с
экстракорпоральным кровообращением у больных развивается сложная коагулопатия,
компонентом которой является качественная дисфункция тромбоцитов.
Полагают, что она возникает из-за взаимодействия тромбоцитов с поверхностью трубок
аппарата искусственного кровообращения (АИК), что приводит к частичной
активации тромбоцитов и выделению гранул. Одновременно происходит
механическое повреждение тромбоцитов и их мембран элементами АИК. Частично
активированные и/или пораженные тромбоциты возвращаются в кровообращение,
где проявляются их множественные аномалии, включая нарушения реакции сек-
ретирования. Тромбоцитарная дисфункция обычно разрешается в течение
нескольких часов. При клинически значимом кровотечении эффективны
переливания тромбоцитарной массы и введение десмопрессина.
Парапротеинемии. Примерно у !/3 больных IgA-миеломой и макроглобулинемией
Вальденстрема (глава 9) наблюдается сочетанная качественная недостаточность
тромбоцитов. Хотя патогенез этого дефекта неясен, предполагают, что в некоторых
Тромбоцитопении
167
случаях он вызван взаимодействием между парапротеином и гликопротеинами
тромбоцитарных мембран, что препятствует связыванию фибриногена фактора
Виллебранда. В таких случаях эффективным оказывается плазмаферез и другие
меры, направленные на снижение концентрации парапротеина в плазме.
Диссеминированное внутрисосудистое свертывание (ДВС). ДВС может
сопровождаться тромбоцитопатией, что вызвано частичной активацией тромбоцитов
циркулирующим тромбином, а также увеличением количества продуктов
деградации фибрина/фибриногена, конкурирующих с фибриногеном в отношении
рецептора ГП Ilb/IIIa на мембране тромбоцита (глава 6).
Миелопролиферативные и миелодиспластические нарушения. Как показывают
наблюдения, у больных с одним из миелопролиферативных нарушений или мие-
лодиспластическим синдромом обнаруживаются качественные дефекты
тромбоцитов. Поскольку нарушения возникают на уровне гемопоэтических стволовых
клеток, то дисфункция тромбоцитов, вероятно, является результатом нарушения
развития мегакариоцитов и тромбоцитов.
Тромбоцитопении
Введение
Общие данные. Формально под тромбоцитопенией понимают уменьшение
содержания тромбоцитов ниже нормальных пределов 150 000-400 000/мкл.
Однако удовлетворительный гемостаз при хирургических вмешательствах
наблюдается при их количестве 50 000/мкл, и спонтанное кровотечение возникает редко,
пока число тромбоцитов не снизится до 10 000-20 000/мкл и менее. Клинические
признаки тромбоцитопении обычно имеют место только в том случае, когда
количество тромбоцитов составляет < 50 000/мкл. Они включают повышенную
склонность к внутрикожным кровоизлияниям, кровоточивость десен, меноррагию,
возникновение петехий в различных местах. Более тяжелый гемостатический дефект
проявляется носовыми и желудочно-кишечными кровотечениями,
геморрагическими пузырьками на слизистых (влажная пурпура).
Псевдотромбоцитопения. Если лабораторные данные указывают на тромбоцито-
пению, требуется незамедлительная оценка мазка периферической крови. Хотя это
исследование помогает определить патогенез тромбоцитопении, необходимо также
исключить лабораторный артефакт псевдотромбоцитопении. В присутствии ЭДТА,
стандартного антикоагулянта при сборе крови, тромбоциты могут формировать
агрегаты, чрезмерно большие для подсчета с помощью автоматического цитометра.
В других случаях, при наличии тромбоцитарных аутоантител или высокого уровня
тромбоцитосвязанного IgG, тромбоциты могут агглютинировать или образовывать
розетки вокруг циркулирующих нейтрофилов in vitro. При автоматическом
подсчете эти тромбоциты также не учитываются. Псевдотромбоцитопению можно
исключить повторным взятием крови в пробирку с гепарином и новым подсчетом.
Тромбоцитопении распределения и разведения. После регистрации истинной
тромбоцитопении необходимо определить ее этиологию. Лучше всего подойти
к этому вопросу, отнеся тромбоцитопению к одной из 4 основных
патогенетических категорий (табл. 5-4). Классификация по двум из них (тромбоцитопения
распределения и разведения) осуществляется по клинической картине и данным фи-
168
Глава 5. Тромбоциты
зикального обследования пациента. Тромбоцитопения разведения развивается
в связи с сильным кровотечением при возмещении кровопотери растворами
кристаллоидов, плазмой, переливанием эритоцитарной массы. Кровопотеря 5-10
единиц1 с возмещением может привести к тому, что количество тромбоцитов
уменьшится до 20-25 % от исходной величины. Тромбоцитопения распределения
отражает степень секвестрации тромбоцитов в увеличенной селезенке. В норме
примерно 70 % тромбоцитарной массы существуют в виде циркулирующих в
периферической крови тромбоцитов. Остальные 30 % секвестируются в селезенке и
образуют селезеночный тромбоцитарный пул (глава 1). Спленомегалия приводит
к росту тромбоцитарной массы, секвестрированной в селезеночном пуле. При
массивной спленомегалии селезеночный пул составляет до 90 % тромбоцитарной
массы. Поскольку система регуляции тромбоцитопоэза осуществляет контроль за
тромбоцитарной массой, а не концентрацией (см. выше), то массивная
спленомегалия и -секвестрация в селезенке могут вызвать значительную циркуляторную
тромбоцитопению. Если спленомегалия, способствующая развитию умеренной
тромбоцитопении, не всегда бывает клинически очевидной, то увеличение селе-
ТАБЛИЦА 5-4. Патогенетическая классификация тромбоцитопении
Продуктивная
Апластическая анемия
ТАР-синдром
Приобретенная амегакариоцитарная тромбоцитопения
Миелодиспластические синдромы
Острый лейкоз
Миелофтиз
Цитотоксическая химиотерапия
Лучевая терапия
Вызванная приемом лекарственных препаратов (эстрогены, тиазиды)
Обусловленная чрезмерным приемом алкоголя
Дефицит витамина В12 и фолиевой кислоты
Циклическая тромбоцитопения
Пароксизмальная ночная гемоглобинурия
Вирусная инфекция (редко)
Разведения
Массивное кровотечение
Распределения
Спленомегалия
Потребления
Иммунная тромбоцитопеническая пурпура взрослых
Иммунная тромбоцитопеническая пурпура детей
Индуцированная приемом лекарственных препаратов (хинидин, золото, гепарин)
Посттрансфузионная пурпура
Неонатальная аллоиммунная тромбоцитопения
Тромботическая тромбоцитопеническая пурпура/гемолитический уремический
синдром
Диссеминированное внутрисосудистое свертывание
Эклампсия/преэклампсия/НЕНР-синдром
Злокачественные новообразования
1 Примеч. ред. Единицей крови считают стандартную дозу при донорской кроводаче (в России —
400 мл).
Тромбоцитопении
169
зенки, связанное с тяжелой тромбоцитопенией, должен обнаружить даже
малоопытный специалист. Следует отметить, что иммунная тромбоцитопеническая
пурпура (см. далее) развивается при нормальном размере селезенки.
Продуктивная тромбоцитопения и тромбоцитопения потребления. Как только
выяснится, что тромбоцитопения не связана с разведением крови или дефектом
распределения клеток, клиницист должен дифференцировать нарушение
продукции и потребления тромбоцитов. Тромбоцитопения потребления является
результатом ускоренной утилизации тромбоцитов и сокращенного периода
полужизни циркулирующих тромбоцитов без адекватной компенсации их костным
мозгом. Так как костный мозг может увеличивать образование тромбоцитов в 8-
12 раз, то тромбоцитопения не возникнет до тех пор, пока не будет превышена
компенсаторная способность.
Развитие большинства тромбоцитопении потребления обусловлено иммуно-
опосредованной деструкцией тромбоцитов, внутрисосудистой активацией системы
свертывания или распространенным поражением эндотелия (табл. 5-4).
Продуктивная тромбоцитопения возникает, когда костный мозг не в состоянии поставить
тромбоциты в количестве, необходимом для их нормального кругооборота (так как
в норме период жизни циркулирующего тромбоцита составляет 8-10 дней,
костный мозг вынужден замещать в среднем 10-13 % тромбоцитарной массы в день).
Продуктивная тромбоцитопения обычно возникает в связи с недостаточностью ге-
мопоэтических стволовых клеток в результате неоплазии, плохого питания, мие-
лофтиза, замещения костного мозга некровеобразующей тканью (табл. 5-4).
В ответ на усиленную потребность в циркулирующих тромбоцитах здоровый
костный мозг повышает количество мегакариоцитов, их размер, плоидность,
скорость созревания. Продуцируемые при этом тромбоциты, так называемые
стрессовые тромбоциты, часто крупнее и обладают большей гемостатической
эффективностью по сравнению с нормальными. Этим объясняется тот факт, что
у больных с тромбоцитопенией потребления при отсутствии других
отягощающих факторов обычно не развивается угрожающее жизни кровотечение. При
обследовании больного с тромбоцитопенией приведенные ниже клинические
особенности могут помочь клиницисту предположить повышенное потребление
клеток как причину заболевания:
1. Наличие крупных тромбоцитов или мегакариоцитов в мазке
периферической крови.
2. Отсутствие сильного кровотечения.
3. При количестве тромбоцитов в пределах 10 000-100 000/мкл ВК менее
расчетного (рис. 5-2).
4. Наличие сочетанной микроангиопатической гемолитической анемии
(указывающей на возможные ДВС, тромботическую тромбоцитопеническую
пурпуру, метастатическую неоплазию микрососудов легких).
5. Сочетание тромбоцитопении с аутоиммунными заболеваниями у молодых
женщин.
6. Миелопролиферативное заболевание, особенно хронический лимфоцитар-
ный лейкоз без признаков миелофтиза, вызванного неопластическими
лимфоцитами.
7. У детей предшествующая вирусная инфекция.
8. Положительный анализ на ВИЧ.
170
Глава 5. Тромбоциты^
Клинические признаки продуктивной тромбоЦитопении:
1. Тромбоциты нормального размера, значительные кровотечения; ВК равно
или превышает расчетное (рис. 5-2).
2. Лейкоэритробластоз в мазке периферической крови (наличие слезоподоб-
ных ядросодержащих эритроцитов, ранних предшественников лейкоцитов)
свидетельствует о миелофтизе.
3. Циркулирующие бласты подтверждают наличие острого лейкоза.
4. Другие цитопении с умеренным эритроцитарным макроцитозом (или без
него) являются признаком дисфункции стволовых клеток (например, апла-
стическая анемия, миелодиспластический синдром или амегакариоцитар-
ная тромбоцитопения).
5. Гиперсегментация нейтрофилов с выраженным эритроцитарным
макроцитозом характерны для дефицита витамина Bi2 или фолиевой кислоты.
Исследование костного мозга. Поскольку полученные клинические данные и
результаты анализа периферической крови могут оказаться характерными и для
тромбоцитопении потребления, и для продуктивной тромбоцитопении, для четкого
дифференцирования этих состояний необходимо провести исследование костного
мозга. Наличие нормального или повышенного числа мегакариоцитов в костном
мозге является надежным показателем того, что тромбоцитопения по крайней мере
частично обусловлена повышенным потреблением клеток. Единственное важное
исключение из этого правила наблюдается при дефиците витамина Bt2 и дефиците
фолата, при которых мегакариоцитопоэз неэффективен и гибель интрамедуллярно-
го мегакариоцита в костном мозге может произойти до освобождения тромбоцитов.
В этом случае одновременно развиваются выраженная костномозговая мегакарио-
цитарная гиперплазия и продуктивная тромбоцитопения. Однако диагноз обычно
поставить несложно, поскольку имеются мегалобластные изменения в других
клеточных линиях дифференцировки. В некоторых случаях повышенной потребности
в тромбоцитах мегакариоциты костного мозга могугформировать большую
популяцию менее зрелых форм из-за быстрого отделения тромбоцитов от полностью
зрелых мегакариоцитов и апоптоза последних. Такой сдвиг к незрелости часто
неверно расценивают как остановку созревания, которую ошибочно интерпретируют
как показатель нарушения продукции тромбоцитов. При продуктивной
тромбоцитопении обычно значительно уменьшено количество нормальных мегакариоцитов
(или они вообще отсутствуют). Диспластические мегакариоциты можно наблюдать
при дисфункции стволовых клеток (например, миелодисплазии, остром лейкозе,
апластической анемии). При этом диагностически значимые морфологические
аномалии обычно выявляются и в других клетках ростков кроветворения.
Продуктивная тромбоцитопения
Химиотерапия и лучевая терапия. Химиотерапия и лучевая терапия вызывают
поражение делящихся и созревающих клеток, оказывая влияние на репликацию
ДНК и трансляцию соответственно. Так как при остановке продукции
тромбоцитов их содержание в периферической крови линейно сокращается, то самый
низкий уровень тромбоцитов обычно наблюдается через 7-10 дней после мощного
химиотерапевтического воздействия. Что касается лучевой терапии, проводимой
почти всегда в возрастающей дозе в течение нескольких недель, время
максимального снижения уровня тромбоцитов менее предсказуемо. Дозы химиотерапии,
Тромбоцитопении
171
как правило, ограниФны уровнем нейтропении, а не тромбоцитопении.
Исключением являются карбоплатин, оказывающий диспропорциональный эффект
на тромбоциты; нитрозомочевины, способные вызывать тромбоцитопению через
3-6 недель после окончания приема (даже после восстановления уровня
лейкоцитов); бусульфан, который в больших дозах приводит к значительной
тромбоцитопении, продолжающейся в течение нескольких недель или месяцев.
Миелофтиз. Инфильтрация костного мозга злокачественными клетками, фиброз
или гранулема костного мозга могут снизить образование тромбоцитов за счет
вытеснения мегакариоцитов из их обычной гемопоэтической ниши. Этот процесс,
известный как миелофтиз (опустошение костного мозга), как правило,
ассоциируется с лейкоэритробластными изменениями в периферической крови (см. выше),
а также со спленомегалией. Помимо тромбоцитопении, при миелофтизе имеются
и другие аномалии — анемия, сочетающаяся с лейкопенией или лейкоцитозом.
К солидным опухолям, вызывающим миелофтиз, относятся прежде всего рак
легкого, предстательной или молочной, грудной желез, желудка, толстой кишки.
Дисфункция гемопоэтических стволовых клеток. Гемопоэтические стволовые
клетки обладают способностью реплицироваться и дифференцировать
(созревать). Оба процесса необходимы для непрерывного образования зрелых клеток
крови, поступающих в циркуляторное русло. Нарушение одного из этих
процессов может привести к недостаточной продукции клеток крови и вызвать, в
частности, тромбоцитопению. Однако изолированная тромбоцитопения,
обусловленная дисфункцией стволовых клеток, — редкое явление, которое клинически
проявляется как амегакариоцитарная тромбоцитопения. Чаще тромбоцитопения
возникает только как один из гематологических симптомов при патологии
гемопоэтических стволовых клеток, что обусловлено их полипотентностью (глава 7).
Патологию гемопоэтических стволовых клеток можно классифицировать на
основании механизма их дисфункции (табл. 5-5). Полная неспособность к
репликации этих клеток приводит к апластической анемии; репликация без созревания
ведет к острому миелоидному лейкозу, а репликация с нарушениями созревания
является причиной появления одного из миелодиспластических синдромов.
Амегакариоцитарная тромбоцитопения, по-видимому, связана с миелодиспластичес-
кими синдромами, поскольку при ней часто наблюдается эритроцитарный макро-
цитоз (симптом, в данном случае не связанный с мегалобластной анемией,
а обусловленный миелодисплазией), как правило, без эволюции в миелоидный
лейкоз. Другие нарушения функции стволовых клеток, приводящие к
тромбоцитопении, включают пароксизмальную ночную гемоглобинурию (ПНГ),
циклическую тромбоцитопению и врожденную мегакариоцитарную гипоплазию (тромбо-
ТАБЛ ИЦА 5-5. Классификация гематологических заболеваний по механизму
дисфункции стволовых клеток
Функция стволовых клеток
Нарушения
Пролиферация Дифференциация
Отсутствует —
Нормальная Отсутствует
Нормальная Нарушена
Повышена Нормальная
Апластическая анемия
Острый миелоидный лейкоз
Миелодиспластические синдромы
Миелопролиферативное заболевание
172
Глава 5. Тромбоциты
цитопения и синдром отсутствия лучевой кости). Кроме того, изредка развиваются
врожденные тромбоцитопении, которые ассоциируются с рядом
негематологических дефектов (синдром Вискотта-Олдрича, синдром Альпорта). Геморрагический
диатез в сочетании с тромбоцитопенией, вызванной дисфункцией стволовых
клеток, может ухудшаться или за счет качественного функционального дефекта
тромбоцитов вследствие нарушения созревания мегакариоцитов, или за счет
одновременного ДВС, что особенно характерно для промиелоцитарного варианта острого
миелогенного лейкоза. ПНГ, напротив, ассоциируется с повышенной
свертываемостью крови и непровоцированными артериальными и венозными тромбозами.
Тромбоцитопения, связанная с инфекцией. Хотя большинство вирусных
инфекций вызывает тромбоцитопению посредством механизма потребления (см. далее),
некоторые вирусы обладают прямым цитотоксическим действием на костный
мозг и мегакариоциты, что приводит к развитию продуктивной
тромбоцитопении. В настоящее время полагают, что связанная с ВИЧ тромбоцитопения
является продуктивной, по крайней мере частично (глава 10). Об этом свидетельствуют
следующие факты: обнаружено, что ВИЧ непосредственно инфицирует
мегакариоциты; при исследовании костного мозга ВИЧ-инфицированных людей часто
можно наблюдать диспластическое созревание мегакариоцитов; результаты
кинетических исследований продемонстрировали пониженное образование
тромбоцитов у таких пациентов. Парвовирус В19, хоть и имеет сродство к
развивающимся эритроидным клеткам, в редких случаях приводит к общей супрессии костного
мозга и панцитопении. Аналогичным образом инфицирование вирусами
гепатита С и Эпштейна-Барр вызывает продуктивную тромбоцитопению с вирусинду-
цированной апластической анемией.
Мегалобластная анемия. Как обсуждалось выше, дефицит витамина Bi2 или фо-
лиевой кислоты при неэффективном гемопоэзе и изолированном неэффективном
мегакариоцитопоэзе может привести к тяжелой тромбоцитопении. На практике
в клинической картине редко доминирует тромбоцитопеническое кровотечение.
Диагноз обычно ставят на основании данных исследования периферической
крови и костного мозга. В последнем, как правило, обнаруживаются увеличенные ги-
персегментированные мегакариоциты. Соответствующее питание обычно
купирует тромбоцитопению в течение 1-2 недель.
Избирательная супрессия мегакариоцитопоэза, вызванная приемом
лекарственных препаратов и употреблением алкоголя. Хотя на мегакариоциты
отрицательно влияет любой препарат, поражающий полипотентные гемопоэтические
стволовые клетки, некоторые вещества оказывают избирательное действие на
развивающиеся мегакариоциты, прежде всего — этиловый спирт. Хроническое
потребление алкоголя часто ассоциируется со слабой или умеренной тромбоцитопенией,
многофакторной по своей этиологии. По всей вероятности, ее возникновению
способствуют возможный гиперспленизм, пониженное образование тромбопоэтина
в пораженной печени и непосредственное токсическое действие алкоголя на
тромбоциты и мегакариоциты. Значительное потребление алкоголя может
сопровождаться умеренно выраженной тромбоцитопенией и сниженным количеством
костномозговых мегакариоцитов, что указывает на супрессию
мегакариоцитопоэза. В одном из исследований обнаружено, что спирт избирательно воздействует
на развивающиеся мегакариоциты, прекращая их развитие в постмитотической,
Переливание тромбоцитарной массы
173
но предэн^омитотической стадии (т. е. когда клетки после первого эндомитоза
имеют гликопротеины тромбоцитарной оболочки, а специфические тромбоцитар-
ные органеллы отсутствуют). Прекращение потребления алкоголя приводит к
восстановлению количества тромбоцитов в пределах 1-2 недель, что часто
сопровождается временным тромбоцитозом.
Тиазидные диуретики и эстрогены также оказывают у некоторых больных
избирательное, различное по силе супрессивное воздействие на образование
тромбоцитов. Влияние тиазидов обычно слабо выражено и проявляется примерно через
2-3 недели после начала использования препарата, причем только у 25 % больных.
Даже при длительном его приеме тромбоцитопения обычно остается стабильной.
Количество тромбоцитов возвращается к норме, как правило, спустя 2-3 недели
после прекращения приема тиазида. Тромбоцитопенический эффект эстрогенов
точно не описан. Однако имеются сообщения о случаях амегакариоцитарной тром-
боцитопении после продолжительного лечения диэтилстильбэстролом.
Переливание тромбоцитов
Введение аллогенных тромбоцитов, как правило, прекращает кровотечение,
вызванное исключительно тромбоцитопенией. И хотя переливание тромбоцитарной
массы к тому же оказывает положительный эффект при коррекции качественных
тромбоцитарных дефектов, в этих обстоятельствах предпочтительнее прибегать
к менее рискованным терапевтическим мерам (например, введению десмопресси-
на). В 1987 г. на согласительной конференции было высказано предположение, что
не следует переливать тромбоцитарную массу при качественной дисфункции, если
ВК не превышает в 2 раза верхнего предела нормы. Как правило, к этой процедуре
прибегают в двух клцнических ситуациях: 1) с терапевтической целью, если у
больных тромбоцитопения сочетается с опасным для жизни кровотечением; 2) с
профилактической целью у пациентов с высоким риском возникновения тромбоцито-
пенического кровотечения. В обоих случаях нет абсолютных правил определения
показания к переливаниям, однако существуют некоторые общие указания.
Если количество тромбоцитов менее 50 000/мкл и имеет место активное
кровотечение, необходимо предусмотреть возможность переливания
тромбоцитарной массы. Но если количество тромбоцитов более 50 000/мкл, маловероятно, что
основной причиной развития геморрагического синдрома является
тромбоцитопения. При проведении некоторых нейрохирургических процедур считается
необходимым поддерживать количество тромбоцитов на уровне более 100 000/мкл,
но даже в этой ситуации, вероятно, достаточно концентрации циркулирующих
тромбоцитов выше 50 000/мкл. Тромбоцитопения может сопровождаться
качественной дисфункцией тромбоцитов (например, при тромбоцитопении,
связанной с экстракорпоральным кровообращением, или у больных с
тромбоцитопенией, недавно принимавших аспирин). В таких случаях тромбоциты переливают
даже при более высоком их содержании в крови.
С профилактической целью переливание обычно рекомендуется больным с
продуктивной тромбоцитопенией и количеством тромбоцитов менее 20 000/мкл.
Однако в одном из последних обзоров клинической литературы по этому вопросу
высказывается мнение, что порогом следует считать 5000-10 000/мкл. Порог
5000/мкл относится к клинически стабильным нелихорадящим пациентам, тогда
как порог 10 000/мкл — к больным с факторами риска с точки зрения возможного
174
Глава 5. Тромбоциты
кровотечения: снижающееся количество тромбоцитов; лихорадка и сепсис;
действие антитромбоцитарных препаратов; тромбоцитопения, связанная с лейкозом.
Переливание тромбоцитарной массы редко показано при клиническом
проявлении тромбоцитопении потребления. Тромбоциты, продуцируемые в ответ на
ускоренное потребление, обычно гиперфункциональны, поэтому даже при очень низком
количестве тромбоцитов могут проявляться лишь минимальные геморрагические
осложнения. Более того, из-за короткого периода полужизни циркулирующих
тромбоцитов у таких пациентов введенные тромбоциты оказывают лишь
кратковременное положительное действие. Только в случае активного кровотечения или
при подозрении на сочетанную качественную дисфункцию тромбоцитов при
тромбоцитопении потребления требуется переливание (см. далее). Под 1 единицей
тромбоцитов подразумевается количество тромбоцитов, получаемое из одной дозы
цельной крови, что составляет приблизительно 0,7 X Ю1'1 клеток. Стандартная доза
тромбоцитов, которая может повысить их количество в крови до 30 000-50 000/мкл,
составляет примерно 6 единиц; обычно этого достаточно для достижения гемостаза.
Подсчет тромбоцитов следует проводить в интервале от 10 мин до 1 ч после
переливания, а затем ежедневно с целью мониторирования аллоиммунизации у больного и
поддержания жизнеспособности тромбоцитов. Перелитые тромбоциты обычно
находятся в кровообращении 2-4 дня, после чего может потребоваться повторное
переливание. 20-70 % больных, получающих повторное переливание
тромбоцитарной массы, аллоиммунизированы, поэтому у них не происходит незамедлительного
посттрансфузионного прироста количества тромбоцитов. В этих случаях часто
прибегают к переливаниям HLA-совместимых тромбоцитов от одного донора.
Тромбоцитопения потребления
Этот вид тромбоцитопении является результатом ускоренной утилизации
периферических тромбоцитов при недостаточной компенсации их убыли костным
мозгом. Хотя у таких больных, как правило, наблюдаются петехии и пурпура при
отсутствии тромбоцитопатии, для них не характерна тяжелая геморрагия.
Относительная защита от угрожающего жизни кровотечения обусловлена большей ге-
мостатической эффективностью тромбоцитов, продуцируемых в "стрессовых
условиях" повышенной потребности в циркулирующих тромбоцитах. Поэтому
возникновение тромбоцитопении потребления, независимо от выраженности
количественных параметров, редко опасно для жизни. Терапия направлена на
снижение потребления тромбоцитов ретикулоэндотелиальной системой и
купирование основного заболевания. Переливание тромбоцитов показано редко и
используется только в том случае, когда временное повышение их числа имеет
значение для остановки серьезного кровотечения. Как отмечалось ранее,
увеличенный объем циркулирующих тромбоцитов может указывать на наличие какого-
либо истощающего патологического процесса. Однако для постановки диагноза
обычно необходимо провести аспирацию и биопсию костного мозга, выявляющие
нормальное или повышенное количество мегакариоцитов. Для всех больных
с тромбоцитопенией важно исследование мазка периферической крови — это
позволит правильно диагностировать заболевание и исключить сочетанные микро-
ангиопатические изменения эритроцитов, которые являются показателем тром-
ботической тромбоцитопенической пурпуры. Последняя может представлять
угрозу для жизни пациента, и для ее лечения существует эффективная
специфическая целенаправленная терапия (см. далее).
Переливание тромбоцитарной массы
175
Иммунная тромбоцитопения потребления
Иммунная тромбоцитопеническая пурпура взрослых (хроническая ИТП).
Аутоиммунная тромбоцитопения является типичной формой тромбоцитопении
потребления у взрослых, а ИТП — наиболее характерной формой аутоиммунной
тромбоцитопении. Как правило, ИТП поражает женщин в возрасте 20-30 лет
(соотношение женщины: мужчины составляет 3:1) и представляет чаще всего
изолированную тромбоцитопению умеренной или тяжелой степени. Однако при наличии
других гематологических аномалий необходимо учитывать возможность развития
системного нарушения, компонентом которого может быть ИТП. Обычно ИТП
ассоциируется с системным аутоиммунным заболеванием, например системной
красной волчанкой, и лимфопролиферативными нарушениями, особенно хроническим
лимфоцитарным лейкозом и болезнью Ходжкина. Сочетание ИТП с аутоиммунной
гемолитической анемией получило название синдрома Эванса; в этом случае
следует предположить наличие первичного лимфопролиферативного нарушения.
Как и у больных с тромбоцитопенией, у пациентов с ИТП чаще всего
наблюдаются петехии, пурпура, кровоточивость десен, меноррагия. Начало симптомов
может быть острым или подострым. В противоположность ИТП детского
возраста (см. далее), у взрослых в анамнезе нет эпизодов инфекции. Наличие
геморрагических пузырьков на слизистой оболочке, носовые и желудочно-кишечные
кровотечения менее характерны и являются предвестниками возможного
возникновения сильного кровотечения. Селезенка может чрезвычайно активно
элиминировать тромбоциты, покрытые антителами, однако развитие спленомегалии
не типично для ИТП, поэтому при ее обнаружении врач должен задуматься
о правильности поставленного диагноза.
ИТП возникает в результате сенсибилизации аутореактивными антитромбо-
цитарными антителами, обычно класса IgG, против компонентов тромбоцитарной .
мембраны. Наиболее часто поражается мембранный гликопротеиновый ПЬ-Ша
комплекс, хотя могут быть задействованы и другие мембранные гликопротеины.
Сенсибилизация аутоантителами к тромбоцитам определяется у 90 % больных
с ИТП (на основании выявления у них повышенного количества тромбоцитарно-
го IgG). Однако при измерении уровня связанного с тромбоцитами IgG нередки
артефакты, что затрудняет постановку диагноза. Так, содержание связанного
с тромбоцитами IgG повышается независимо от уровня циркулирующих в плазме
иммуноглобулинов и объема тромбоцитов. Концентрация тромбоцитарного IgG
увеличивается также в присутствии циркулирующих иммунных комплексов.
Сенситизация тромбоцитов IgG приводит к существенному сокращению
времени их жизни вследствие опосредованного Fc-рецепторами фагоцитоза
селезеночными макрофагами. Реактивность IgG по отношению к мембране
развивающихся мегакариоцитов может в некоторой степени нарушать мегакариоцитопоэз:
до 50 % больных с ИТП имеют несколько сниженную тромбопоэтическую
реакцию на ускоренное потребление тромбоцитов. Полагают, что антитромбоцитар-
ные аутоантитела образуются главным образом в селезенке.
Взрослых больных с ИТП госпитализируют только в случае активного
кровотечения или при количестве тромбоцитов менее 10 000-20 000/мкл. С помощью
аспирации и биопсии костного мозга подтверждают предполагаемый диагноз,
после чего можно начинать лечение. В большинстве случаев ежедневный перо-
ральный прием умеренных доз преднизолона дает положительные результаты.
176
Глава 5. Тромбоциты
Однако при кровотечении или повышенном риске его развития клиническая
реакция наступает значительно быстрее после внутривенного введения
иммуноглобулина, высокой дозы метилпреднизолона или их сочетания. При сильном
кровотечении в комплекс неотложной терапии следует включить переливание
тромбоцитарной массы (см. выше).
Считают, что эффект кортикостероидов заключается в ингибировании
взаимодействия между тромбоцитами, покрытыми IgG, и Fc-рецепторами
макрофагов селезенки, что нарушает фагоцитоз. При длительном применении кортико-
стероиды подавляют, по-видимому, и образование антитромбоцитарных
аутоантител. Вводимый внутривенно иммуноглобулин, насыщая Fc-рецепторы
селезеночных макрофагов, также эффективен, что связано со снижением
скорости фагоцитоза тромбоцитов.
Реакция на эти методы лечения наблюдается обычно в пределах нескольких
дней или 1-3 недель. При ее отсутствии следует прибегнуть к альтернативному
методу терапии — спленэктомии. Ответ на внутривенно введенный иммуноглобу-
. лин может быть временным, и фактически во всех случаях для поддержания
необходимого количества тромбоцитов требуется постоянный пероральный прием
преднизолона. Стероиды, как правило, продолжают применять в течение 2-3
месяцев, постепенно уменьшая дозу, поддерживая при этом количество
тромбоцитов на уровне 30 000-50 000/мкл. У 10-20 % больных таким образом достигается
ремиссия и лечение стероидами прекращают. Даже при умеренной хронической
тромбоцитопении можно не принимать стероиды, если течение заболевания
бессимптомно и функция тромбоцитов нормальна. К сожалению, примерно у 80 %
взрослых больных с ИТП возникают рецидивы при уменьшении дозы стероидов
и в конце концов требуется выполнение спленэктомии.
По реакции на кортикостероиды и, вероятно, на внутривенное введение
иммуноглобулина можно прогнозировать эффективность спленэктомии (рис. 5-5).
К сожалению, в случае отрицательного результата после спленэктомии пока не
разработана схема дальнейшей терапии. Однако всегда необходимо учитывать
возможность присутствия остаточной селезеночной ткани (спленоз или
добавочная селезенка). На это могут указывать отсутствие телец Хауэлла-Жолли в мазке
периферической крови или результаты сканирования коллоидной серой,
меченной 99,пТс (глава 3). Некоторым больным с рефрактерной ИТП помогает
постоянный или через день прием стероидов в низкой дозе и периодическая внутривенная
иммуноглобулиновая терапия. В рефрактерных случаях можно назначить дана-
зол, алкалоиды барвинка, а-интерферон и иммуносупрессивные препараты: цик-
лофосфамид, азатиоприн, циклоспорин. Несмотря на неудачи в лечении
рефрактерной ИТП, длительное наблюдение показывает, что большинство больных
чувствует себя неплохо в течение продолжительного времени.
Иммунная тромбоцитопеническая пурпура детей (острая ИТП). Хотя
клинические проявления ИТП у детей такие же, как у взрослых, патогенез и течение болезни
существенно отличаются, что обусловливает разный подход к лечению. Острая
ИТП, как правило, наблюдается у детей в возрасте 2-9 лет с максимальной
частотой в 3-5 лет. Заболеванию в равной степени подвержены мальчики и девочки.
Обычно нарушение возникает после вирусной инфекции (через 1-3 нед). Чаще
всего на фоне хорошего самочувствия внезапно возникают петехии и пурпура. Как
и у взрослых, у детей ИТП проявляется характерной картиной крови (тромбоцито-
пения, отсутствие других цитопений или аномальных циркулирующих клеток кро-
Переливание тромбоцитарной массы
177
ви). При физикальном обследовании не обнаруживаются ни лимфаденопатия, ни
гепатоспленомегалия. Дифференциальная диагностика изолированной тромбоци-
топении у детей значительно проще, чем у взрослых: ИТП составляет более 95 %.
Поэтому необходимость исследования костного мозга для подтверждения диагноза
является предметом дискуссии. При тромбоцитопении у детей следует также
учитывать возможность ВИЧ-инфекции, системной красной волчанки, врожденного'
гуморального иммунодефицита (особенно IgA). Однако при отсутствии
характерных клинических признаков этих заболеваний обычное исследование
периферической крови позволяет установить лишь гуморальный иммунодефицит.
В противоположность ИТП у взрослых, когда основной патогенетический
процесс — выработка антитромбоцитарных аутоантител, острая ИТП у детей
вызвана, как полагают, антителами, направленными против антигенов вирусных
белков. Процесс сенситизации циркулирующих тромбоцитов происходит за счет
адсорбции на их мембране или вирусного антигена (который впоследствии
связывается с антителом), или иммунных комплексов вирус-антитело. Поскольку
антигены вируса непременно элиминируются, острая ИТП является
самоограниченным заболеванием, которое у 80 % пациентов проходит спонтанно в течение
2 месяцев. Спленэктомия показана редко, так как у 95 % больных количество
тромбоцитов достигает приемлемых величин в пределах нескольких месяцев.
У детей по сравнению со взрослыми несколько чаще возникает внутричерепное
кровоизлияние, нередко без признаков кровотечения, что оправдывает лечение
практически всех заболевших с количеством тромбоцитов менее 20 000/мкл.
Рекомендуется парентеральная терапия большими дозами стероидов и/или
внутривенное введение иммуноглобулина. Из-за высокого риска постспленэктомическо-
го сепсиса удаление селезенки у детей относят как можно на бол,ее поздний срок.
Идиопатическая тромбоцитопеническая пурпура
Первоначальное лечение преднизолоном
Спленэктомия 80 %
^Отсутствие
Реакция Щ реакции
80-100% |1 0-20% И 70-100% И 0-30%
S.
— ^^„.Отсуг^иеШн^НОгсутствие]
Реакция Щ реакции щ реакция Щ реакции
60-70% ■30-40%
Рис. 5-5. Варианты исходов хронической идиопатической тромбоцитопенической пурпуры
у взрослых пациентов в зависимости от реакции на лечение преднизолоном и спленэктомию
178
Глава 5. Тромбоциты
Иммунная тромбоцитопения, вызванная лекарственными препаратами.
Развитие этого вида тромбоцитопении обусловлено взаимодействием принимаемого
лекарственного препарата, антител к этому препарату и мембраны тромбоцита.
В результате возникает опсонизация и/или активация тромбоцитов. Поэтому
индуцированная лекарственным средством тромбоцитопения развивается через
2-3 недели после первичного его приема, т. е. во время пика гуморального
иммунного ответа. При повторном воздействии препарата тромбоцитопения может
развиться значительно быстрее из-за присутствия уже наработанных антител к
лекарственному препарату или мобилизации иммунной памяти. Поскольку для
связывания антител с мембраной тромбоцита необходимо наличие препарата,
то время купирования тромбоцитопении определено кинетикой выведения этого
лекарства из организма. Практически в большинстве случаев тромбоцитопения
сохраняется еще несколько дней после элиминации вызвавшего ее препарата.
Наиболее часто тромбоцитопения индуцируется гепарином (нарушение
развивается у 3-5 % пациентов, получавших гепарин в терапевтической дозе).
Обычно это слабая или умеренная тромбоцитопения со средним содержанием
тромбоцитов 50 000/мкл. Однако в отдельных случаях количество тромбоцитов
снижается до 20 000/мкл, а иногда вызываемая гепарином тромбоцитопения
приводит к образованию у больных венозных и артериальных тромбов. Хотя степень
риска возникновения связанного с гепарином тромбоза точно не определена,
очевидно, что фактически у всех пациентов первым признаком этого грозного
осложнения является тромбоцитопения. Учитывая тяжесть течения и высокую
летальность при индуцированной гепарином тромбоцитопении, осложнившейся
тромбозом, рекомендуется мониторинг количества тромбоцитов у всех больных,
получающих гепарин, а при выявлении тромбоцитопении — немедленное
прекращение использования этого лекарственного препарата.
В основе механизма развития гепарининдуцированной тромбоцитопении лежит
связывание специфических антител с комплексом гепарина и одного из компонентов
тромбоцита, который, согласно последним данным, идентифицирован как тромбо-
цитарный фактор 4. В результате Fc-участок молекулы IgG приближается к Fc-pe-
цептору мембраны тромбоцита и как следствие возникают активация и агрегация
тромбоцитов и наблюдается прокоагулянтный эффект. Агрегированные тромбоциты
удаляются ретикулоэндотелиальной системой, что приводит к тромбоцитопении.
Но если активация и агрегация тромбоцитов выражены достаточно сильно, может
иметь место тромбоз. Гепарининдуцированная тромбоцитопения подтверждается
при лабораторном исследовании, когда антитела плазмы больного активируют
нормальные тромбоциты при терапевтической концентрации гепарина (глава 6).
Тромбоцитопению вызывают и другие лекарственные препараты за счет
связывания мембраны тромбоцита со специфическими антителами, обычно класса
IgG. Хотя точный механизм этого процесса неясен, для развития
тромбоцитопении необходимо взаимодействие между лекарством и неким компонентом тром-
боцитарной мембраны, в большинстве случаев — одним из гликопротеинов. Такое
взаимодействие делает гликопротеин, препарат или комплекс гликопротеин-
препарат иммуногенным. Последующее связывание специфических антител
с мембраной тромбоцита в присутствии препарата или опсонизирует тромбоциты
для удаления РЭС, или приводит к активации комплемента с внутрисосудистым
разрушением тромбоцитов. Фактически во всех случаях лекарственной
тромбоцитопении существенно повышается уровень связанного тромбоцитами IgG.
Переливание тромбоцитарной массы
179
У некоторых больных также можно обнаружить связывание тромбоцитами
антител к лекарственному препарату. Однако нередко причиной развития
тромбоцитопении является не сам препарат, а его метаболит, поэтому не всегда
определяются специфические антитела.
Хинин и хинидин — типичные лекарственные препараты, индуцирующие
тромбоцитопению (вероятность возникновения — 1 на 1000 случаев приема).
Клиническое начало тромбоцитопении, вызванной хинином, может быть резким,
а сама тромбоцитопения тяжелой. Из-за того, что многие лекарственные средства
содержат хинин, необходим тщательный сбор анамнеза, чтобы
дифференцировать хинининдуцированную тромбоцитопению от ИТП. Довольно часто
наблюдается тромбоцитопения, вызванная препаратами золота (примерно у 2 %
больных). Предполагается, что существует генетическая предрасположенность
к этому осложнению, так как у большинства пациентов с этим видом
тромбоцитопении выявляется антиген гистосовместимости HLA-DR3. Заболевание
протекает вяло и проходит в пределах нескольких недель, что, возможно, обусловлено
продолжительным периодом полураспада золота, а также тем, что золото
стимулирует образование аутоантител к тромбоцитам. Тромбоцитопению могут также
вызвать дигоксин, рифампицин, сульфаниламиды.
При лечении прежде всего необходимо идентифицировать и удалить
этиологический фактор. Если больной получает лекарства нескольких наименований,
необходимо прекратить прием всех препаратов или заменить их, за исключением
наиболее важных. Особое внимание следует обратить на перечисленные выше
лекарственные средства, вызывающие тромбоцитопению. Большинство
клиницистов полагает, что тяжелую тромбоцитопению, индуцированную лекарственными
препаратами, следует лечить умеренными дозами кортикостероидов.
Рациональной альтернативой может служить и внутривенное введение иммуноглобулина.
При тромбоцитопении, возникшей вследствие приема гепарина, переливание
тромбоцитарной массы не рекомендуется.
Другие причины иммунной тромбоцитопении. Посттрансфузионная пурпура
представляет собой редкую, но тяжелую форму иммунной тромбоцитопении в
результате аллоиммунизации. Обычно это нарушение развивается через 1 неделю
после гемотрансфузии и проявляется тяжелой рефрактерной тромбоцитопенией
и кровотечением. Высокая летальность при посттрансфузионной пурпуре
обусловлена внутричерепными кровоизлияниями. В связи с этим большое значение
имеют ранняя диагностика и рациональная терапия. Как правило, заболевают
повторно родящие женщины или те пациенты, которым ранее переливали кровь или
ее компоненты. Это редкое нарушение встречается у людей с отсутствием тромбо-
цитарного антигена PlA1 (приблизительно 2 % населения) после переливания
продуктов крови, положительных по Р1А1. Происходит выработка эндогенных
антител к Р1А1-антигену, которые разрушают не только аллогенные тромбоциты, но и
собственные тромбоциты больного, отрицательные по PlA1. Причина аутореак-
тивности этих анти-Р1А1-антител неясна. По последним данным, эффективным
оказывается раннее лечение внутривенным введением иммуноглобулина,
которое может спасти жизнь больного.
Аутоиммунная тромбоцитопения новорожденного. Заболевание может
развиваться у младенцев в результате трансплацентарной передачи материнских
антител, вступающих в реакцию с тромбоцитами плода. Полагают, что наиболее ве-
180
Глава 5. Тромбоциты
роятный патогенез заболевания — иммунизация Р1А1-отрицательной матери
тромбоцитами плода, несущими этот антиген. Возможна несовместимость и по другим
антигенам тромбоцита. В противоположность посттрансфузионной пурпуре
данный процесс обычно не затрагивает мать. Ребенок при рождении имеет слабую
тромбоцитопению, которая усиливается в 1-2 недели жизни и исчезает к 4-й
неделе. Лечение рекомендуется только при тяжелой форме нарушения и сводится к
переливанию отмытых тромбоцитов матери и/или к введению иммуноглобулина
внутривенно.
Неиммунная тромбоцитопения потребления
Тромботическая тромбоцитопеническая пурпура (ТТП) и гемолитический
уремический синдром (ГУС). ТТП и ГУС являются острыми тромбоцитопениями
неясной этиологии с потенциально летальным исходом. Оба нарушения
характеризуются острым началом тяжелой неиммунной тромбоцитопении потребления
и микроангиопатической гемолитической анемии (МАГА, или
фрагментационный гемолиз). В связи с вероятностью быстрого летального исхода при этой
патологии (и доступностью относительно простой и достаточно эффективной
терапии) необходимо сразу же исследовать мазки периферической крови у всех
пациентов с тромбоцитопенией на предмет обнаружения признаков возможного
микроангиопатического гемолиза (рис. 3-3). При сочетании тромбоцитопении
потребления с микроангиопатической гемолитической анемией следует
предполагать, что у больного имеют место ТТП или ГУС, пока эти диагнозы не исключены.
Для ТТП и ГУС типичны отложения гиалиновых тромбоцитарных тромбов
в терминальных артериолах и капиллярах. Полагают, что такие тромбы
развиваются в результате спонтанной агрегации тромбоцитов и окклюзии сосудов. Хотя эти
тромбы могут содержать фибриновые полимеры, генерализованная активация
гуморальной системы свертывания крови не наблюдается. Таким образом, этот
синдром легко отличим от диссеминированного внутрисосудистого свертывания
(см. ниже и главу 6). При ТТП, которая в основном обнаруживается у взрослых,
отложение тромбоцитарных тромбов имеет общесистемный характер, вызывая
повреждение микрососудов многих органов за счет микроваскулярной ишемии.
Характерная клиническая "пятерка" признаков ТТП включает тромбоцитопению
потребления, микроангиопатическую гемолитическую анемию, лихорадку,
перемежающиеся неврологические симптомы и почечную недостаточность. Эти
нарушения у разных пациентов встречаются в различных сочетаниях. При ГУС, который
главным образом наблюдается у детей, тромбоцитарные тромбы развиваются
преимущественно в почках, приводя к острой почечной недостаточности — основному
клиническому проявлению заболевания. ТТП и ГУС не всегда клинически
отличаются друг от друга, поэтому их лучше рассматривать как исход одного
патологического процесса. Эти клинические диагнозы обычно не требуют морфологического
подтверждения. Однако при биопсии костного мозга, почек или десен
определяются характерные, но не патогномоничные тромбоцитарные гиалиновые тромбы.
Хотя основным звеном патогенеза при ТТП и ГУС является агрегация
тромбоцитов и окклюзия микрососудов, механизм возникновения агрегации неизвестен.
Есть предположение, что инициирующее событие — повреждение сосудистого
эндотелия с выделением в кровообращение необычайно большого количества
мультимеров ФВ. Как полагают, эти мультимеры спонтанно связываются с ГП1Ь-1Х
Переливание тромбоцитарной массы
181
и ГПНЬ-Ша тромбоцитарной мембраны, приводя к агрегации тромбоцитов и
блокаде микроциркуляции. Другие авторы считают, что агрегация тромбоцитов
стимулируется иными, неэндотелиальными, белками.
Развитию ТТП иногда предшествуют вирусное заболевание, бактериальная
инфекция или иммунизация. Кроме того, ТТП часто наблюдается среди
ВИЧ-инфицированных больных (глава 10). Другие причины ТТП: беременность, применение
пероральных противозачаточных средств, введение некоторых противоопухолевых
препаратов и заболевания соединительной ткани. В редких случаях ТТП —
наследственное нарушение, но в большинстве своем возникает спорадически, без видимых
предпосылок. ГУ С, в противоположность ТТП, в 90 % случаев выявляется у детей,
ему часто предшествует кровавый понос, вызванный Shigella dysenteriae или энтеро-
токсичными Escherichia coli серотипа 0157:Н7. Оба этих микроорганизма ускоряют
развитие ГУ С, образуя токсины, разрушающие эндотелиальные клетки почечных
капилляров, что приводит к попаданию в сосудистое русло значительного
количества мультимеров ФВ с последующей агрегацией тромбоцитов.
Как отмечалось ранее, при тромбоцитопении потребления в сочетании с микро-
ангиопатической гемолитической анемией всегда следует предполагать диагноз
ТТП/ГУ С. Тип тромбоцитопении подтверждается характером аспирата костного
мозга, в котором наблюдается нормальное или повышенное количество мегакари-
оцитов. Микроангиопатическая гемолитическая анемия диагностируется на
основании характерных изменений эритроцитов в мазке периферической крови и
подтверждается наличием очень высокой активности лактатдегидрогеназы в
сыворотке крови, а также другими признаками внутрисосудистого гемолиза (ретику-
лоцитоз, повышенное содержание свободного билирубина, железа в сыворотке,
пониженный уровень гаптоглобина). Диссеминированное внутрисосудистое
свертывание можно исключить при нормальных величинах протромбинового
времени, АЧТВ, фибриногена, продуктов деградации фибрина; а злокачественную ги-
пертензию — на основании нормального или почти нормального АД. Системный
васкулит сложнее дифференцировать от ТТП, однако затруднения в постановке
окончательного диагноза не должны отсрочить терапию предполагаемой ТТП.
Раньше большинство пациентов с ТТП умирали, но с введением в
клиническую практику трансфузий плазмы и терапии плазмообмена они в основном
переживают острую фазу заболевания. Плазмообмен, или переливание плазмы (3-4 л
ежедневно), следует начинать незамедлительно при первом подозрении на ТТП/
ГУС и проводить до появления симптомов улучшения состояния: снижения
активности ЛД Г и увеличения количества тромбоцитов. Иногда терапию
продолжают в течение нескольких недель, поскольку возможны рецидивы после
прекращения плазмообмена. Некоторые врачи дополнительно вводят кортикостероиды,
а в рефрактерных к терапии случаях прибегают к спленэктомии. Переливание
тромбоцитарной массы показано только при активном кровотечении, так как оно
может быть опасно для жизни пациента.
Диссеминированное внутрисосудистое свертывание (ДВС) (см. также главу 6).
Некоторые системные нарушения вызывают активацию гуморальной системы
свертывания, что способствует формированию внутрисосудистого сгустка и
потреблению факторов V и VIII, фибриногена и тромбоцитов. Сочетанная
активация фибринолитических ферментов неизменно приводит к образованию
продуктов деградации фибрина и фибриногена (ПДФ), которые можно определить
182
Глава 5. Тромбоциты
в плазме и которые мешают полимеризации фибрина и нарушают функцию
тромбоцитов. ДВС возникает при тяжелой бактериальной инфекции, гибели
тканей, ишемии в связи с травмой или гипоперфузией, осложнениях беременности и
родов, при некоторых опухолях. В тяжелых случаях из-за внутрисосудистого
отложения фибрина развивается микроангиопатическая гемолитическая анемия.
ДВС следует предполагать при наличии признаков тромбоза или
кровотечения, при пролонгированном ПВ, снижении количества тромбоцитов. АЧТВ при
ДВС более изменчиво, но обычно увеличено. Для ДВС характерны низкий
уровень фибриногена и повышенный уровень ПДФ. Поскольку ДВС — динамичный
процесс, при постановке диагноза данные о снижении количества тромбоцитов
могут иметь болыпее'значение, чем их абсолютные уровни. В редких случаях
требуется количественное определение факторов V и/или VIII.
Лечение ДВС обычно направлено на контроль или ликвидацию основного
нарушения. В острой фазе при необходимости переливают эритроциты,
свежезамороженную плазму, тромбоцитарную массу. Некоторые клиницисты рекомендуют
терапию гепарином в тех случаях, когда преобладающим симптомом является
тромбоз или трансфузионная терапия недостаточна для поддержания гемостаза.
Гепарин можно использовать и в неотложной акушерской практике. При
хроническом ДВС, связанном со злокачественными заболеваниями, необходима
длительная антикоагулянтная терапия с введением гепарина подкожно (глава 6).
Тромбоцитопения потребления вследствие патологии микрососудов. Любое
повреждение сосудистого эндотелия сопровождается адгезией тромбоцитов к суб-
эндотелиальному коллагену, мультимерам ФВ и другим адгезивным гликопроте-
инам экстрацеллюлярного матрикса. При выраженном поражении микрососудов
скорость утилизации тромбоцитов может превысить компенсаторную
способность костного мозга и стать причиной развития неиммунной формы тромбоци-
топении потребления. Такое распространенное патологическое состояние может
возникать в сочетании со злокачественной гипертензией, системным васкулитом
небольших сосудов и отторжением почечного аллотрансплантата. Терапия
направлена на основное заболевание, и по мере его излечения тромбоцитопения
обычно разрешается.
Особая форма тромбоцитопении потребления развивается при
злокачественных заболеваниях с распространением микроскопических субэндотелиальных
метастазов в микрососудистое русло легких, повреждение которого приводит
к микроангиопатической анемии, часто сопровождаемой тромбоцитопенией без
признаков ДВС. Этот синдром сопутствует метастатической аденокарциноме
желудка, хотя может быть связан и с другими метастатическими аденокарциномами,
в частности грудной железы, легких. Дифференцирование с ДВС обязательно.
Тромбоцитопения нередко ассоциируется с осложнениями акушерской
практики — эклампсией и преэклампсией. Показано, что у 20 % больных с преэклампси-
ей и у 40 % пациентов с эклампсией возникает тромбоцитопения. Хотя механизм ее
развития не совсем ясен, некоторые исследователи полагают, что в данном случае
имеет место поражение системного и плацентарного микрососудистого русла.
В большинстве случаев признаки активации гуморальной системы свертывания
минимальны или вообще отсутствуют. После рождения ребенка тромбоцитопения
у матери проходит. Улучшения состояния достигают также при помощи активной
гиготензивной терапии или антитромбоцитарной терапии аспирином.
Переливание тромбоцитарной массы
183
При клиническом варианте преэклампсии тромбоцитопения может
ассоциироваться с микроангиопатическим гемолизом и повышенными показателями
печеночных проб. Это так называемый синдром HELLP (Hemolytic anemia, Elevated
Liver tests, Low Platelets — гемолитическая анемия, повышенные печеночные
пробы, низкое содержание тромбоцитов). Такое сочетание симптомов иногда
предшествует развитию гипертензии и протеинурии, но в остальном клинически не
отличается от преэклампсии. Хотя синдром HELLP необходимо
дифференцировать от преэклампсии, метод их лечения одинаков — удаление плода.
Тромбоцитопения и беременность
Умеренная тромбоцитопения с количеством тромбоцитов 70 000-150 000/мкл
возникает приблизительно у б % практически здоровых беременных женщин.
Такую тромбоцитопению следует рассматривать как несущественную, поскольку
количества тромбоцитов достаточно для обеспечения гемостаза матери, а частота
рождения младенцев с тромбоцитопенией от таких женщин не увеличена.
Поэтому дополнительное обследование не требуется. ИТП у женщин может
проявляться как во время беременности, так и до ее наступления. Из-за теоретически
возможного риска проникновения антитромбоцитарных антител класса IgG матери
через плаценту и развития тяжелой тромбоцитопении плода существуют разные
мнения по поводу лечения таких больных. Беременным (как и не беременным)
с ИТП и потенциально симптоматичной тромбоцитопенией (менее 50 000/мкл)
назначают или кортикостероиды, или иммуноглобулин внутривенно до
увеличения количества тромбоцитов. Однако чаще во время беременности развивается
ремиссия (нередко в результате спленэктомии). Установлено, что у 10-30 % матерей
с ИТП могут родиться дети с количеством тромбоцитов менее 50 000/мкл, но
клинически значимые осложнения у младенца, особенно внутричерепное
кровоизлияние, возникают редко. Некоторые клиницисты рекомендуют оценить количество
тромбоцитов в крови пупочной вены или вены черепа плода, чтобы определить
показания к родам посредством кесарева сечения. Другие авторы предполагают, что
высокий уровень связанного с тромбоцитами IgG помогает выявить
новорожденных с наибольшим риском развития симптоматической тромбоцитопении.
Предлагается также введение преднизона всем матерям с ИТП в последние 3-4 недели
беременности. Но поскольку вероятность возникновения тяжелой неонатальной
тромбоцитопении очень мала даже у детей от матерей с ИТП в анамнезе, а
предложенные подходы пока не признаны достаточно эффективными и экономичными,
то ни один из них не получил широкого распространения.
Тромбоцитоз
Хотя в широком смысле тромбоцитоз можно определить как состояние, при
котором количество тромбоцитов выше нормы (т. е. более 400 000-450 000/мкл),
незначительное увеличение их количества может наблюдаться при различных
болезненных состояниях и без клинических последствий. В данной книге под
тромбоцитозом понимается состояние, когда количество тромбоцитов более
600 000/мкл. С точки зрения патогенеза, тромбоцитоз подразделяют на две
группы: реактивный и первичный. Реактивный тромбоцитоз проявляется как
следствие патологического процесса, который, как полагают, не поражает гемопоэти-
184
Глава 5. Тромбоциты
ческие стволовые клетки, т. е. регуляторные механизмы, контролирующие тром-
боцитопоэз и развитие стволовых клеток, не нарушаются. И наоборот, первичный
тромбоцитоз возникает вследствие дефекта в гемопоэтических стволовых
клетках, что приводит, по крайней мере частично, к автономному гемопоэзу и
нарушению развития стволовых клеток. Таким образом, первичный тромбоцитоз
возникает почти исключительно как осложнение одного из миелопролифера-
тивных нарушений — клональных заболеваний гемопоэтических стволовых
клеток, для которых характерна избыточная пролиферация.
Реактивный тромбоцитоз
Возможные причины реактивного тромбоцитоза многообразны (табл. 5-6).
Патогенетические механизмы, развитие этого заболевания гетерогенны и часто неясны.
Полагают, что постспленэктомический тромбоцитоз отчасти обусловлен утратой
тромбоцитарного пула селезенки, который обычно составляет примерно Уз общего
количества зрелых тромбоцитов. С удалением селезенки тромбоцитарная масса не
изменяется, но увеличивается количество циркулирующих тромбоцитов за счет тех,
которые должны были бы находиться в селезеночном пуле, но не учитывались при
подсчете. Однако после удаления селезенки тромбоцитоз возникает не сразу, а
спустя 1 -3 недели, и степень его часто значительно выше той, которую можно объяснить
мобилизацией селезеночного пула. Поэтому вполне вероятны другие механизмы
развития тромбоцитоза после этого хирургического вмешательства. Во многих
случаях реактивный тромбоцитоз ассоциируется с системными воспалительными
заболеваниями и, как считают, является результатом выделения иммуномодуляторных
цитокинов, которые вторично стимулируют продукцию тромбоцитов. Эта модель
наиболее соответствует состоянию при ревматоидном артрите, когда содержание
интерлейкина-6 (ИЛ-6) в сыворотке крови прямо коррелирует со степенью
тромбоцитоза. ИЛ-6 известен как провоспалительный цитокин, который также
стимулирует полиплоидизацию мегакариоцитов и образование тромбоцитов.
Реактивный тромбоцитоз следует рассматривать как доброкачественное
нарушение. Хотя количество тромбоцитов повышено, их морфология и функция не
изменены. Нормальная агрегация тромбоцитов может помочь при
дифференцировании реактивной и первичной форм тромбоцитоза. При реактивном варианте
ТАБЛИЦА 5-6. Этиология реактивного тромбоцитоза
Спленэктомия
Железодефицитная анемия
Острое кровотечение
Хронические воспаления
(особенно ревматоидный артрит, колит)
Острые и хронические инфекции
(особенно хронические легочные инфекции)
Злокачественные заболевания
(особенно легких, поджелудочной железы, болезнь Ходжкина)
Отказ от алкоголя
Гемолитическая ^немия
Прием лекарственных препаратов (винкристин, адреналин)
Восстановление после тромбоцитопении
(лечение дефицита витамина В12 и фолиевой кислоты)
Тромбоцитоз
185
количество циркулирующих тромбоцитов редко превышает 106/мкл;
значительно большие величины свидетельствуют о первичном нарушении. Не обнаружено
четкой связи между наличием реактивного тромбоцитоза и развитием тромботи-
ческих или геморрагических клинических проявлений; и, следовательно, нет по-
, казаний для лечения ингибиторами функции тромбоцитов и терапии,
направленной на снижение их количества.
Первичный тромбоцитоз
Под этим термином понимают тромбоцитоз, возникающий вследствие клональ-
ного дефекта гемопоэтических стволовых клеток. У большинства больных
первичный тромбоцитоз развивается в сочетании с одним из миелопролиферативных
заболеваний, например хроническим миелогенным лейкозом, полицитемией, эс-
сенциальной тромбоцитемией или идиопатическим миелофиброзом (глава 9).
В редких случаях первичный тромбоцитоз возникает в связи с одним из миело-
диспластических синдромов, особенно Sq-синдромом, а также в связи с идиопати-
ческой кольцевой сидеробластной анемией.
Количество тромбоцитов у больных с первичным тромбоцитозом может быть
различно: от величин ненамного выше нормы до нескольких миллионов в
микролитре. Изменяется также морфология тромбоцитов. Обнаруженные в мазке
периферической крови гигантские тромбоциты и даже фрагменты цитоплазмы мега-
кариоцита свидетельствуют о первичном процессе, хотя эти изменения могут и
отсутствовать. Поскольку первичный тромбоцитоз является результатом
дефекта полипотентных Гемопоэтических стволовых клеток, как правило, наблюдается
изменение гематокрита и/или содержания лейкоцитов. Наличие спленомегалии
также подтверждает первичный процесс. Время кровотечения у больных с
первичным тромбоцитозом варьируется: оно может быть пониженным, нормальным,
пролонгированным. Однако у большинства пациентов обнаруживаются
нарушения функции тромбоцитов, чаще всего агрегации, индуцированной адреналином.
Клинические проявления заболевания в значительной степени связаны
с основным патологическим процессом. При этом возникновение тромбоза и
кровотечения объясняется наличием первичного тромбоцитоза. Характерным
осложнением является тромбоз сосудов необычной локализации (например,
брыжеечные вены, печеночная вена, артерии пальцев). Дигитальная артериальная
ишемия приводит к клинически выраженному синдрому эритромелангии,
болезненной эритеме, отеку пальцев рук. У пожилых людей иногда наблюдаются
приступы ишемии, в том числе сердца. Геморрагические осложнения развиваются
примерно в 2 раза чаще тромботических и локализуются, как правило, в
желудочно-кишечном тракте. Четкая связь между увеличением содержания тромбоцитов
и риском возникновения тромботических или геморрагических нарушений не
прослеживается. Но если тромбоз или геморрагии возникают при первичном
тромбоцитарном нарушении, полагают, что вероятность возникновения
осложнений можно существенно снизить, уменьшив содержание тромбоцитов.
Поскольку у многих пациентов в течение нескольких лет отсутствует
клиническая симптоматика, специфическая терапия, направленная на снижение количества
тромбоцитов, необходима не всегда. В проспективных исследованиях показано, что
к факторам, повышающим риск развития тромбоза, относятся возраст пациента,
продолжительность тромбоцитоза и наличие тромботических эпизодов в анамнезе.
186
Глава 5. Тромбоциты
Уменьшить количество тромбоцитов можно с помощью различных цитотоксичес-
ких алкилирующих агентов, включая бусульфан и мелфалан. Однако назначать эти
препараты следует с осторожностью, особенно молодым больным, из-за возможного
развития лейкоза; предпочтительнее использовать антиметаболит гидрокси-
мочевину. Возможно, еще безопаснее лечение специфическим препаратом,
снижающим количество тромбоцитов,— анагрелидом, который оказывает тромбоцитопе-
ническое действие, прерывая созревание мегакариоцитов. Некоторым больным
цомогает сс-интерферон. Одно из последних клинических исследований
показывает, что введение гидроксимочевины больным с высоким риском эссенциальной
тромбоцитемии существенно уменьшает частоту тромботических осложнений,
окклюзии периферических артерий, поверхностного и глубокого тромбофлебита.
Поскольку терапевтические средства, нарушающие функцию тромбоцитов, могут
способствовать желудочно-кишечному кровотечению, необходимо контролировать
состояние больных, принимающих эти препараты. Аспирин и другие антитромбо-
цитарные средства показаны пациентам, у которых клиническое течение
заболевания осложняется исключительно тромботическими эпизодами.
Клинический пример
Женщина в возрасте 21 года пришла на прием к врачу, поскольку
чувствовала недомогание и небольшое повышение температуры. Неделю
назад, как она полагает, перенесла ОРЗ, после которого появились одышка
при незначительной нагрузке, головокружение, слабость, повышенная
температура тела. При осмотре ректальная температура — 37,9 X, другие
показатели в норме. Лейкоциты — 14 300/мкл, нейтрофилы — 82 %, па-
лочкоядерные — 3 %; лимфоциты — 15 %; гемоглобин — 52 г/л; гематок-
рит — 16 %; тромбоциты — 8000/мкл; биохимический анализ: азот
мочевины крови — 32 мг%, креатинин — 1,7 мг%; ПВ и АЧТВ в пределах
нормы; ЛДГ - 6700 ЕД.
Вопрос 1. О чем свидетельствуют эти данные?
Ответ. У больной лихорадочное состояние, легкая прострация; имеется
анемия, тромбоцитопения, умеренная азотемия. Все это соответствует
диагнозу тромботическая тромбоцитопеническая пурпура (ТТП). Поскольку
существует эффективная специфическая терапия, а фактор времени
чрезвычайно важен, необходимо прежде всего подтвердить диагноз.
Вопрос 2. Что для этого следует сделать?
Ответ. Оценить мазок периферической крови. Лейкоциты — без отклонений.
Количество тромбоцитов снижено, сами они несколько увеличены.
Значительные изменения эритроцитов. Многие клетки имеют форму шлема,
треуголки, можно видеть фрагменты меньшего размера. Подобные шистоциты
характерны для микроангиопатической гемолитической анемии (табл. 3-3).
Вопрос 3. Какова дифференциальная диагностика микроангиопатической
гемолитической анемии?
Ответ. Кроме ТТП (или ГУ С) следует учитывать:
а) патологию со стороны сердца и крупных кровеносных сосудов, что
может нарушать кровоток и вызывать разрушение клеток из-за
механической травмы. Примером может служить синдром Воринга-Блендера, при
котором эритроциты сжимаются (сплющиваются) за счет плохо
функционирующих протезов клапанов сердца;
Тромбоцитоз
187
б) аномалии микрососудов, например их врожденные анастомозы;
в) иммунные повреждения малых сосудов при остром гломерулонефрите,
гипертензии, злокачественных заболеваниях. Они могут
сопровождаться местным внутрисосудистым свертыванием и прокоагулянтными
нарушениями гуморальной свертывающей системы;
г) другие микроангиопатические процессы (отслойка плаценты, промие-
лоцитарный лейкоз, укус змеи);
д) воспалительные процессы: сепсис, панкреатит, перегревание, посттранс-
фузионные реакции.
Для постановки диагноза можно провести исследование на гиалиновые
тромбы (состоящие из агрегатов тромбоцитов, сшитых фибриногеном и
ФВ) в терминальных артёриолах или капиллярах. Биопсия костного мозга
или десны помогает обнаружить подобные изменения в сосудах. В данной
ситуации с учетом анамнеза и данных обследования больной, наличия
тромбоцитопении, анемии, нормальных значений ПВ и АЧТВ наиболее
вероятный диагноз — ТТП. Для достижения максимального эффекта лечение
следует начинать немедленно. ,
Вопрос 4. Что следует предпринять?
Ответ. Терапия выбора—плазмаферез с введением свежезамороженной
плазмы (плазмообмен). Некоторые авторы считают, что патогенез ТТП связан
с попаданием необычных форм ФВ в кровообращение при повреждении эн-
дотелиальных клеток. Эти мультимеры более эффективно, чем другие формы
ФВ, связываются с гликопротеиновыми комплексами на поверхности
тромбоцитов. Такое связывание вызывает усиленную агрегацию тромбоцитов и
механическое повреждение эритроцитов при прохождении "сквозь строй"
агрегатов. Свежезамороженная плазма поставляет фермент, видимо,
терминальную дисульфидредуктазу, который помогает освободиться от гигантских
комплексов ФВ, выделяемых пораженными эндотелиальными клетками.
Результат. Интенсивная терапия помогла больной. Плазмаферез и инфу-
зия донорской свежезамороженной обедненной тромбоцитами плазмы (3-
4 л/день) позволили устранить гемолитическую анемию и тромбоцитопе-
нию, снизить уровень ЛДГ, нормализовать функцию почек и устранить
неврологические изменения.
Клинический пример
Женщина в возрасте 31 года без патологии в анамнезе (за исключением
болей в суставах) обратилась в отделение неотложной помощи. Жалобы
на красные пятна на лодыжках и "кровавые пузырьки" во рту.
Менструация началась на 1 неделю раньше положенного срока. При осмотре
обнаружены геморрагические пузырьки на слизистой оболочке полости рта,
петехии в области бедер и лодыжек. Печень, селезенка и лимфатические
узлы не увеличены. Кровотечение из влагалища. Лейкоциты — 11200/мкл;
гемоглобин — 117 г/л, гематокрит — 35 %, тромбоциты — 2000/мкл.
Вопрос 1. Что надо предпринять?
Ответ. Исследовать периферическую кровь. Морфология эритроцитов и
лейкоцитов без изменений. Тромбоцитов мало, и они большого размера.
Очевидно ускоренное образование тромбоцитов, многие из которых явля-
188
Глава 5. Тромбоциты
ются незрелыми, но после попадания в периферический кровоток активно
потребляются.
Вопрос 2. Что необходимо предпринять для постановки диагноза?
Ответ. Выполнена аспирация костного мозга. Обнаружено нормальное
созревание всех ростков кроветворения, но много базофильных мегакариоци-
тов. Уровень антитромбоцитарных антител не является диагностически
значимым. Поскольку у больной есть признаки патологии суставов, то
после стабилизации состояния следует дифференцировать ревматический
процесс/коллагеноз.
Вопрос 3. Как лечить данную пациентку?
Ответ. Так как на слизистой оболочке ротовой полости имеются
геморрагические пузырьки (влажная пурпура) и выявлено активное маточное
кровотечение, лечение нужно начинать безотлагательно. Большинство
клиницистов предпочитают использование высоких доз преднизолона перорально,
но можно также применить внутривенно кортикостероиды и
иммуноглобулин. Хотя переливание тромбоцитарной массы не всегда приводит к
значимому приросту количества тромбоцитов, но обычно оно быстро
останавливает кровотечение. Среди гематологов нет единого мнения в отношении
приоритета использования данных терапевтических подходов.
Вопрос 4. Лечение привело к остановке кровотечения. Дозу кортикостеро-
идов снизили, но через несколько месяцев количество тромбоцитов у
больной снова упало до опасно низкого уровня. Несмотря на две
дополнительные попытки поддержать количество тромбоцитов на достаточном уровне
высокой дозой преднизолона, а затем медленно ее снизить, количество
тромбоцитов стало < 1000/мкл, появилось больше геморрагических
пузырьков на слизистой оболочке рта и незначительное желудочно-кишечное
кровотечение. Какие терапевтические меры необходимы?
Ответ. Можно выполнить спленэктомию после предварительной
иммунизации вакцинами против Streptococcus pneumoniae, Neisseria meningitidis,
менингококка, Haemophilus influenzae] попытаться использовать даназол или
другие иммуносупрессоры или провести химиотерапию, но эти средства
обычно оставляют как резервные на случай возникновения постспленэкто-
мических проблем.
Результат. Больная хорошо отреагировала на проведенную спленэктомию,
наступила стойкая ремиссия с достаточным содержанием тромбоцитов.
Избранная литература
Ballem P. J., Belzberg A., Devine D. V. et al. Kinetic studies of the mechanism of
thrombocytopenia in patients with human immunodeficiency virus infection. New
Engl.J. Med., 1992; 327:1779-1784.
Интересное исследование, доказывающее, что тромбоцитопения у пациентов с ВИЧ-
инфекцией является результатом как уменьшения продукции тромбоцитов,
так и ускоренного их разрушения. Лечение зидовудином купирует тромбоцито-
пению посредством ускорения продукции тромбоцитов.
Beutler E. Platelet transfusions: the 20,000/uL trigger. Blood, 1993; 81:1411-1413.
Сообщение, подвергающее сомнению необходимость назначения трансфузии
тромбоцитов при снижении их содержания в крови ниже 20 000/мкл. Автор полагает.
Тромбоцитоз
189
что более обоснованным показанием для назначения профилактической
трансфузии тромбоцитов является уровень 5000-10 000/мкл.
Burrows R. FM Kelton J. G. Incidentally detected thrombocytopenia in healthy mothers
and their infants. New Engl J. Med., 1988; 319:142-145.
Проспективная оценка частоты возникновения и последствий тромбоцитопении у
здоровых беременных женщин.
Choi E. S., Nichol J. L., Hokom M. M, Hornkohl А. С, Hunt P. Platelets generated in vitro from
proplatelet-displaying human megakaryocytes are functional. Blood, 1995; 85:402-413.
Исследование in vitro, доказывающее, что развитие тромбоцитов из мегакариоцитов
путем образования промежуточных филаментных цитоплазматических
выростов ограничено стадией протромбоцитов.
Consensus Conference. Platelet transfusion therapy./AMA, 1987; 257:1777-1780.
Несколько устаревшее, но в основном актуальное руководство по трансфузии
тромбоцитов.
Cortelazzo S., Finazzi G., Ruggeri M. et al. Hydroxyurea for patients with essential
thrombocythemia and a high risk of thrombosis. New Engl.J. Med., 1995; 332:1132-1136.
Важное проспективное исследование, демонстрирующее, что цитотоксическая перо-
ральная терапия гидроксимочевиной уменьшает количество тромботических
осложнений у больных с эссенциальной тромбоцитемией и высоким риском
тромбозов. К числу пациентов с высоким риском отнесены те, у которых тромбозы
наблюдались ранее и/или возраст превышает 60 лет.
Cortelazzo S., Viero P., Finazzi G., d'Emilio A., Rodeghiero F., Barbui T. Incidence and
risk factors for thrombotic complications in a historical cohort of 100 patients with
essential thrombocythemia./ Clin. Oncol., 1990; 8: 556-562.
Ретроспективная оценка факторов риска тромботических осложнений у пациентов
с эссенциальной тромбоцитемией. Возраст, предшествующие тромбозы и
длительный анамнез тромбоцитоза определены как независимые факторы риска.
George J. N., Caen J. P., Nurden A. T. Glanzmann's thrombasthenia: the spectrum of
clinical disease. Blood, 1990; 75:1383-1395.
Современный обзор редкого заболевания.
George J. N. et al. Idiopathic thrombocytopenia purpura: a practice guideline developed by
explicit methods for the American Society of Hematology. Blood, 88:3-40,1996.
Всесторонний обзор диагностики и лечения ИТП. См. также редакционную статью
A. Lichtin о процессе разработки руководства по диагностике и лечению.
Gernsheimer Т., Stratton J., Ballem P. J., Slichter S.J. Mechanisms of response to treatment
in autoimmune thrombocytopenic purpura. New Engl.J. Med., 1989; 320:974-980.
Важное научное исследование, посвященное изучению механизмов возникновения
тромбоцитопении при ИТП. Выявлено, что выработка аутоантител к
тромбоцитам так же, как и ускоренное разрушение тромбоцитов в селезенке, приводит
к повреждению мегакариоцитов € костном мозге и уменьшению продукции
тромбоцитов. Кинетика процесса неодинакова у разных пациентов.
Gewirtz A. M., Hoffman R. Transitory hypomegakaryocytic thrombocytopenia:
aetiological association with ethanol abuse and implications regarding regulation of
human megakaryocytopoiesis. Br. J. Haematol, 1986; 62:333-334.
Одно из немногих исследований, в которых рассматривается механизм алкогольной
тромбоцитопении. Показано, что алкоголь приводит к задержке развития
мегакариоцитов на границе митотической и эндомитотической фаз.
190
Глава 5. Тромбоциты
Harker L. A. Hemostasis Manual. Philadelphia: FA Davis; 1974.
Давно изданное, но по-прежнему прекрасное руководство, дающее краткий и
превосходный обзор кинетики мегакариоцитопоэза и продукции тромбоцитов.
Kelton J. G., Smith J. W., Warkentin T. E., Hayward С. Р. М, Denomme G. A.,
Horsewood P. Immunoglobulin G from patients with heparin induced
thrombocytopenia binds to a complex of heparin and platelet factor 4. Blood, 1994;
83:3232-3239.
Описана возможная антигенная мишень для патологических антител у пациентов
с гепарининдуцированной тромбоцитопенией.
Kuter D.]., Beeler D. L., Rosenberg R. D. The purification of megapoietin: a physiological
regulator of megakaryocyte growth and platelet production. Proc. Nat Acad. Set.
USA, 1994;91:11104-11108.
Отчет нескольких научных групп, добившихся успеха в клонировании тромбопоэтина
(в оригинале — мегапоэтин). Авторы предполагают, что состояние тромбопоэ-
тической регуляторной системы зависит от концентрации свободного
тромбопоэтина (не связанного с соответствующими рецепторами тромбоцитов и ме-
гакариоцитов).
Mazur E. M. Megakaryocytes and megakaryocytopoiesis. In: Loscalzo J., Schafer A. I.
(eds). Thrombosis and Hemorrhage. Cambridge, Mass.: Blackwell Scientific
Publishing; 1994:161-194.
Обзор сведений о мегакариоцитах и мегакариоцитопоэзе с точки зрения биохимии и
цитологии. Обсуждение процесса регуляции мегакарицитопоэза частично
устарело в связи с последними достижениями в области клонирования тромбопоэтина.
Metcalf D. Thrombopoietin-atlast. Nature, 1994; 369:519-520.
Краткое описание предпосылок, трудностей, успешного открытия и клонирования
тромбопоэтина.
Picozzi V. J., Roeske W. R„ Creger W. P. Fate of therapy failures in adult idiopathic
thrombocytopenic purpura. Am.]. Med., 1980; 69: 690-694.
Ретроспективное описание ошибок при первичном лечении пациентов с ИТП.
Примечательно, тем не менее, что спустя несколько лет большинство пациентов
чувствовали себя прекрасно.
Souyri M., Vigon I., Penciolelli J. F., Heard J. M., Tambourin P., Wendling F. A putative
truncated receptor gene transduced by the myeloproliferative leukemia virus
immortalizes hematopoietic progenitors. Cell, 1990; 63:1137-1147.
В работе освещена история идентификации онкогена миелолейкоза, приведшей к
открытию тромбопоэтинового рецептора и к успешному клонированию
тромбопоэтина.
Stoll D. В., Blum S., Pasquale D., Murphy S. Thrombocytopenia with decreased
megakaryocytes. Evaluation and prognosis. Ann. Intern. Med., 1981; 94:170-175.
Ретроспективное описание клинической сущности амегакариоцитарной тромбоци-
топении.
Zuker-Franklin D., Cao Y. Megakaryocytes of human immunodeficiency virus-infected
individuals express viral RNA. Proc. Natl Acad. Sci. USA, 1989; 86:5595-5599.
Первая экспериментальная демонстрация прямого поражающего воздействия ВИЧ
на мегакариоциты при использовании метода гибридизации in situ. Это
открытие объясняет причину подавления продукции тромбоцитов, наблюдаемого
у ВИЧ-инфицированных пациентов. Впрочем, результаты данной работы
оспариваются.
Глава 6
Гемостаз и тромбоз
Анджелина К. А. Карвальхо
Нормальный гемостаз
Гемостатический процесс начинается с травмы или разрыва сосуда, а
заканчивается образованием тромбоцито-фибриновой сетки (гемостатическая пробка). Она
служит механическим затвором, предотвращающим дальнейшую кровопотерю, и
очагом для восстановления тканей. Функция гемостатического механизма вклю-/
чает взаимодействие между стенкой сосуда, тромбоцитами, коагуляционными
белками крови и фибринолитической системой (рис. 6-15). Нарушения гемостаза
ведут к серьезным клиническим последствиям. Дисбаланс в одном направлении
может сопровождаться чрезмерным кровотечением, в другом — образованием
тромба.
Сосудистый эндотелий
Потеря крови из интактных сосудов предотвращается сосудистой стенкой. Ее
структурная и функциональная целостность зависит от свойств образующих ее
клеточных компонентов и внеклеточного матрикса, который синтезируют
клетки. Тромбоциты также оказывают трофическое влияние на эндотелий. Все
компоненты сосудистой стенки (эндотелий, субэндотелий, средняя и наружная
оболочки) участвуют в реакции на травму. Эндотелий заслуживает особого внимания
вследствие его активной роли в гемостазе.
Монослой эндотелиальных клеток выстилает базальную мембрану и
составляет первый защитный барьер против различных процессов, включая гемостаз и
тромбоз (рис. 6-1). ЭК поставляют в субэндотелий ряд веществ: компоненты 6а-
зальной мембраны, коллаген, эластин, ламилин, протеазы и их ингибиторы, тром-
боспондин, мукополисахариды, витронектин, фибронектин и фактор Виллебран-
да (ФВ). Эти белки имеют большое значение для межклеточного взаимодействия
и образования диффузионного барьера, который предотвращает попадание крови
из внутрисосудистого пространства во внесосудистое. Кроме того, эндотелиаль-
ные клетки выполняют различные физиологические функции, включая
регулирование реактивности тромбоцитов, контроль за направленной миграцией
лейкоцитов, регулирование активности фактора роста, регулирование текучести крови.
Эндотелиальные клетки также продуцируют вещества, которые секретируются в
просвет сосуда и способствуют текучести крови. К этим веществам относятся:
гликозаминогликаны, комплексы гепарин (гепарин-сульфат)-антитромбин III,
192
Глава 6. Гемостаз и тромбоз
тромбин-тромбомодулин-протеин С, плазминоген-активатор плазмина. Кроме
того, они продуцируют простациклин (ПП2) и эндотелиальный фактор
релаксации (ЭФР) — два возможных ингибитора адгезии и агрегации тромбоцитов. Эти
вещества являются вазодилататорами и действуют синергично.
Прежде всего нормальный эндотелий действует как мощная антикоагулянт-
ная поверхность, которая не активирует белки свертывания крови и не
привлекает к себе клеточные компоненты крови. Но после стимуляции или травмы
эндотелий трансформируется в мощную прокоагулянтную поверхность. Это происходит
за счет синтеза, выделения или привлечения многих прокоагулянтных веществ,
включая тканевый фактор (ТФ), фактор Виллебранда (ФВ), фактор V,
ингибиторы активатора плазминогена (ИАП-1 и ИАП-2), интерлейкин-1 (ИЛ-1), фактор
некроза ткани (ФНТ), эндотелин-1 (вазоконстриктор) (рис. 6-1).
Незамедлительной реакцией на травму сосудистого эндотелия является вазо-
констрикция. Эта преходящая реакция длительностью менее 60 с приводит к
снижению кровотока, что улучшает взаимодействие между тромбоцитами,
факторами свертывания крови и поврежденным участком. Вазоконстрикция как реакция
на небольшую травму может остановить кровотечение, но при обширном
повреждении только снижает кровопотерю, предотвращая тем самым обескровливание.
Продукты эндотелиальной клетки
•Базальная • Эластин • Фибронектин
мембрана • Ламилин • Мукополисахариды
• Коллаген (III, IV) • Витронектин • ФВ
• Микрофибриллы • Ингибиторы протеаз * Протеазы
Субэндотелиальные структуры (секретируемые эндотелиальными клетками)
Продукты поверхности эндотелиальной клетки (секретируемые в кровоток)
Рис. 6-1. Продукты эндотелиальной клетки, секретируемые в субэндотелиальную зону
и в просвет сосуда (сокращения см. в тексте)
Нормальный гемостаз
193
Тромбоциты
Обнажение субэндотелиальных структур в результате повреждения ЭК приводит
к изменению формы тромбоцитов и их адгезии. Адгезия тромбоцитов
опосредована ФВ, который является своеобразным мостиком между гликопротеином lb
мембраны тромбоцита и обнаженными коллагеновыми структурами на месте
травмы. Коллаген (типы I, II, III) субэндотелия и образующийся локально
тромбин заставляют прилипшие тромбоциты секретировать содержимое гранул.
Коллаген и тромбин также стимулируют фосфолипазы тромбоцитарной мембраны,
освобождая арахидоновую кислоту из ее фосфолипидного слоя. Арахидоновая
кислота посредством тромбоцитарной циклооксигеназы превращается в проста-
гландины Н2 и G2 (ПГН2 и nrG2). Последние под воздействием тромбоксансинте-
тазы превращаются в тромбоксан А2 (ТхА2) и другие гидроксижирные кислоты.
Внутриклеточный ТхА2 активирует сократительную реакцию в канальцевой
системе тромбоцитов, за счет чего гранулы продвигаются к центру клетки и
освобождаются быстрее. ТхА2 совместно с выделяемой тромбоцитами АДФ способствует
агрегации циркулирующих тромбоцитов и закрытию участка поражения
тромбоцитарной "пробкой". На периферии этой пробки происходит дезагрегация
тромбоцитов из-за выделения антиагрегатов, в частности простациклина (ПП2), окиси
азота (NO), аденозиндифосфатазы (АДФазы) и других ферментов,
синтезируемых интактными соседними эндотелиальными клетками. Такой первичной
тромбоцитарной пробки достаточно для того, чтобы начался гемостаз и
незамедлительно остановилось развившееся кровотечение, но сама по себе она не в
состоянии поддерживать гемостаз (глава 5).
Свертывание крови
Постоянная гемостатическая пробка формируется при образовании тромбина
посредством активации процесса свертывания крови. Тромбин играет важную роль
в возникновении, росте и локализации гемостатической пробки. Он вызывает
необратимую агрегацию тромбоцитов и отложение фибрина на тромбоцитарных
агрегатах, образующихся в месте сосудистой травмы. Фибрино-тромбоцитарная
сеточка является структурным барьером, предотвращающим дальнейшее
вытекание крови из сосуда, и инициирует процесс репарации ткани.
Свертывающая система крови (рис. 6-2) — это фактически несколько
взаимосвязанных реакций, протекающих при участии протеолитических ферментов.
На каждой стадии данного биологического каскада профермент
(предшественник, зимоген) превращается в соответствующую сериновую протеазу, которая
катализирует превращение следующего профермента в сериновую протеазу. Се-
риновые протеазы гидролизуют пептидные связи в активном центре, основу
которого составляет аминокислота серии. Тринадцать таких белков (факторы
свертывания крови) составляют систему свертывания. Из них семь активируются
до сериновых протеаз (факторы XII, XI, IX, X, II, VII и прекалликреин), три
являются кофакторами этих реакций (факторы V, VIII и кининоген с высокой
молекулярной массой), один — кофактор/рецептор (тканевый фактор, фактор III), еще
один — трансглутаминаза (фактор XIII) и, наконец, фибриноген (фактор I)
является субстратом для образования фибрина, конечного продукта каскада
свертывания крови (табл. 6-1).
194
Глава 6. Гемостаз и тромбоз
Чтобы в результате процесса свертывания крови был достигнут гемостаз,
необходимо сосредоточение циркулирующих факторов свертывания крови в месте
повреждения. Это осуществляется за счет реакций свертывания крови,
происходящих на обнаженном коллагене, тканевом факторе и клеточных мембранах,
включая фосфолипиды тромбоцитарной мембраны. Амплификация местной
Свертывание крови: пути активации
| Внутренний путь |
Белки
контактной
фазы
| Внешний путь |
(Sg^+fviri +П"1
■ Тканевый фактор
4(Ca20/Vlla/ Ш
z
т-г^!^^ 1 Общий путь j
+ |ФПА| + [ФПВ1
Рис. 6-2. Активация системы свертывания крови. Система положительной обратной связи
(амплификация) вызывает усиление начальных реакций. Система отрицательной обратной связи (ингиби-
рование), напротив, ограничивает коагуляцию. Стрелки в виде мелкого пунктира и знака "+"
показывают облегчение процесса; стрелки в виде тире и знака"-" указывают на ингибирование процесса
(сокращения см. в тексте)
Нормальный гемостаз
195
свертывающей реакции является мощным механизмом, необходимым для
образования тромбина. Подсчитано, что одна молекула активированного фактора XII
может генерировать 1 млн молекул тромбина. Основная функция тромбина —
превращать фибриноген в фибрин. Фибрин удерживает агрегаты тромбоцитов
в месте сосудистой травмы и изменяет нестабильную тромбоцитарную пробку
(первичную) в стабильную гемостатическую.
Пути активации свертывания крови
Механизмы активации свертывания крови подразделяют на внешние и
внутренние. Такое деление искусственно, поскольку оно не имеет места in vivo, но данный
подход облегчает интерпретацию лабораторных тестов in vitro.
ТАБ Л И ЦА б-1. Факторы свертывания крови
Фактор Название Синтез Т1/2* Локализация Функция
гена
Хромосома 4 Субстрат
Хромосома 11 Фермент
— Рецептор/
кофактор
Хромосома 1 Кофактор
Хромосома 13 Фермент
Х-хромосома Кофактор
Х-хромосома Фермент
Хромосома 13 Фермент
Хромосома 4 Фермент
Хромосома 5 Фермент
Хромосомы 6,1 Транс-
глутами-
наза
Хромосома 4 Фермент
Хромосома 3 Кофактор
* Полупериод существования.
I
II
III
V
VII
VIII
IX
X
XI
XII
XIII
Фибриноген
Протромбин
Тканевый
фактор
Лабильный
фактор (про-
акцелерин)
Проконвертин
Антигемофиль-
ный фактор
Фактор Крист-
маса (тромбо-
пластин плазмы
Фактор Стюарт-
Прауэра
Гепатоциты
Гепатоциты/
витамин К *
ЭК + многие
другие клетки
Гепатоцит
ЭК/тромбоцить
Гелатоциты/
витамин К
Синусоиды
печени
Гепатоциты/
витамин К
D
Гепатоциты/
витамин К
Предшественник Гепатоцит
плазменного
тромбопластина
Фактор Хагемана Гепатоциты
Фибринстаби-
лизирующий
фактор
Прекалликре-
ин (ПК)
(фактор Флетчера)
Высокомолекулярный кини-
ноген (фактор
Вильямса,
факГепатоциты /
тромбоциты
Гепатоциты
Гепатоциты
тор Фложека или
фактор Фитцже
ральда)
4-5 дней
Здня
—
12-15ч
i
4-7 ч
8-10 ч
1 день
(-24 ч)
2 дня
2-3 дня
1 день
8 дней
196
Глава 6. Гемостаз и тромбоз
Внешний путь. Основным путем активации свертывания крови in vivo считается
внешний путь (рис. 6-3). Компоненты этого пути следующие: тканевый фактор,
(ТФ, ФШ), его ингибитор (ингибитор превращения тканевого фактора, ИПТФ;
рис. 6-12) и плазменный фактор VII (ФУП). ТФ представляет собой внутренний
мембранный гликопротеин, имеющийся во многих клетках, которые находятся
в контакте с кровью; он не поступает в кровь до тех пор, пока не образуются проте-
азы или не произойдет повреждение клетки iv vivo. ТФ функционирует в качестве
кофактора/рецептора, который в присутствии ионов кальция активирует фактор
VII. Фактор VII представляет собой одноцепочечный гликопротеин (Мг 50 000),
образуемый гепатоцитами; в крови циркулирует в виде профермента. Он
содержит 10 остатков у-карбокСиглутаминовой кислоты (Gla-домен), для биосинтеза
которых требуется витамин К.
Активация фактора VII приводит к открытию (обнажению) его активного
серинового центра. Это вызвано прежде всего связыванием ФУН с ТФ/Са2+.
Фактор VII может также активироваться за счет незначительного протеолитичес-
кого действия других сериновых протеаз свертывания (тромбина, ФХИа, Ф1Ха
и ФХа), а также за счет самоактивации, однако самоактивация in vivo идет
незначительно. Комплекс TФ/ФVIIa/Ca2+ действует на два субстрата: ФХ и Ф1Х,—
вследствие чего образуется тромбин.
Внутренний путь. Внутренний путь активации свертывания крови определяется
как коагуляция, инициируемая компонентами, полностью находящимися в
пределах сосудистой системы. In vivo этот путь существует совместно с внешним.
Компоненты внутренней системы: факторы XII, XI, IX, VIII, кофакторы —
высокомолекулярный кининоген (ВМК) и прекалликреин (ПК), а также их ингибиторы
(рис. 6-4>.
Инициация активации ФХП начинается, когда обнажается отрицательно
заряженная поверхность (например, коллаген) в пределах сосудистой стенки; в ре-
Внешний путь активации крови
Тканевый фактор/(^са^)
_ffi-
а'
Фактор IX —>> Фактор IXa \
FVIII \Сра»*) \
®
Фактор X-
-^Ха
Рис. 6-3. Фактор Vila облегчает активацию Ф1Х в системе внутреннего пути и ФХ — в общем
пути. ФУШа и Са2+ необходимы также для осуществления активации ФХ
Нормальный гемостаз
197
зультате самоактивация ФХП приводит к конформационным изменениям
молекулы с раскрытием его активного серинового центра (ФХПа). Наличие
небольшого количества ФХПа вызывает активацию его субстратов: ПК, ВМК и ФХ1
(рис. 6-4). ПК и ФХ1 связываются с активирующей поверхностью посредством
ВМК. Без ВМК активации обоих проферментов не происходит. Связанный ВМК
может расщепляться калликреином (К) или связанным с поверхностью ФХПа и
инициировать взаимную активацию систем ПК-ФХИ. Поскольку ПК и ФХ1
плазмы существуют в бимолекулярных комплексах с ВМК, это облегчает прямую
поставку большого количества ПК и ФХ1 к поверхности для прикрепления к ней,
где активируется ФХП. Затем связанный с поверхностью ФХПа расщепляет ФХ1
до ФХ1а и прекалликреин до калликреина. Механизм взаимной активации ФХИ
и прекалликреина отличается большей быстротой по сравнению с механизмом
самоактивации ФХИ, что обеспечивает многократное усиление системы активации
ФХП. Образовавшийся калликреин превращает ВМК в ВМКа и брадикинин.
В результате этих первых контактных реакций комплексы ВМКа/К и ВМКа/
ФХ1а размещаются вблизи ФХПа, где начинается активация процесса
свертывания крови, фибринолиз и активация комплемента (рис. 6-4).
Калликреин в комплексе с ВМКа недостаточно тесно связан с
поверхностью и выделяется в жидкую фазу, чтобы взаимодействовать с различными
субстратами, включая ФХП, плазминоген, проренин и компонент комплемента С1
(рис. 6-4).
Внутренний путь активации свертывания крови
убрана клетки (тромбоц^^
Рис. 6-4. Самоактивация ФХИ и реципрокная активация ФХН и ПК вызывают мощную активацию
внутреннего пути свертывания с возникновением многих реакций положительной обратной связи
(облегчающие реакции). Конверсия ФХПа в ФХШ за счет К приводит к остановке связанной с
поверхностью коагуляции (подробности см. в тексте)
198
Глава 6. Гемостаз и тромбоз
Калликреин также воздействует на ФХНа, отщепляя фрагмент ФХП (ФХШ),
который сохраняет активный сериновый участок, но утрачивает домен
связывания. Подобное регулирование отрицательной обратной связью с помощью кал-
ликреина "выключает" поверхностносвязанное свертывание. ФХШ в жидкой
фазе может действовать как мощный активатор прекалликреина: превращать
ФУИ в ФУПа, а С1 — в активированный С1. И наоборот, ФХ1а, связанный
с ВМКа, остается тесно прикрепленным к поверхности, где он пространственно
приближает проферменты к ФХНа. С другой стороны, ФХ1а расщепляет ВМК,
нарушая его кофакторную активность, в результате чего ФХ1а отделяется от зоны
поверхностной активации. ФХ1а превращает Ф1Х в Ф1Ха как в жидкой фазе, так
и на тромбоцитарных мембраносвязанных фосфолипидах (рис. 6-4).
Витамин К необходим для пострибосомального карбоксилирования
терминальных остатков глутаминовой кислоты всех витамин К-зависимых факторов
коагуляции, ФХ, Ф1Х, ФУП, ФИ (мнемоник "1972") и двух ингибиторов
факторов свертывания (С и S). Это карбоксилирование способствует присоединению
ионов кальция, что необходимо для освобождения участка, связывающего фосфо-
липид, и активации всех зависящих от витамина К белков.
Хотя активация Ф1Х инициируется или ФХ1а, или ФУПа/ТФ (III) (см.
рис. 6-2), последний считается наиболее важным путем активации свертывания
крови in vivo. Активированный Ф1Х требует наличия кальция и кофактора
(ФУШ) для прикрепления к тромбоцитарному фосфолипиду и превращения
ФХ в ФХа. Фактор VIII действует в качестве мощного ускорителя
завершающей ферментативной реакции.
Фактор УШ„который также называют антигемофилъным фактором, является
единственным полипептидом, синтезируемым различными тканями, включая ЭК
синусоид печени. Он кодируется большим геном (186 кб), расположенным на
конце Х-хромосомы. Анализ кДНК фактора VIII показал наличие двух типов
повторяющихся последовательностей в белке, состоящем из доменов: А1-А2-В-АЗ-
С1-С2 (рис. 6-5). Активация фактора VIII тромбином приводит к образованию
тяжело- и легкоцепочечных фрагментов ФVIII. Домены А локализованы в обоих
фрагментах, а домены С — только легкой цепи. Коагулянтная активность ФVIII
ассоциируется с доменами А и С, но не В (зона соединения), который не обладает
прокоагулянтными свойствами. Молекулярный анализ Ф VIII и другого
кофактора свертывания крови — ФV, выявляет сходство этих двух молекул. ФVIII и ФV
имеют общую структуру (А1-А2-В-АЗ-С1-С2) с гомологией между доменами
А и С, чем и обусловлена коагулянтная активность оббих кофакторов. Зона В
различна у факторов VIII и V, но она не связана с их функцией (рис. 6-5).
ФVIII циркулирует в крови, будучи связанным с ФВ — большим гликопроте-
ином, продуцируемым ЭК и мегакариоцитами. ФВ служит внутрисосудистым
белком-носителем для ФVIII (рис. 6-21). Связывание ФВ с ФVIII (посредством
аминокислотных остатков ФVIII с 1649 по 1689) стабилизирует молекулу ФVIII,
увеличивает ее период полусуществования внутри сосуда и способствует ее
транспорту к месту повреждения. Однако чтобы активированный фактор VIII
мог проявить свою кофакторную активность, он должен отсоединиться от ФВ.
Воздействие тромбина на комплекс ФVIII/ФB приводит к отделению ФVIII
от несущего протеина и расщеплению на тяжелую и легкую цепи, которые важны
для коагулянтной активности фактора VIII. Другая физиологическая роль связи
ФVIII/ФB заключается в способности ФВ повышать концентрацию cf>VIII в ме-
Нормальный гемостаз
199
Структура факторов VIII и V
Тромбин
i Фа*тору,п i L \
NH2
Тромбин
Фактор Ха
1969
Ci С2 ICOOH
Тяжелая цепь
Связывающий
участок
Тромбин
| Фактор V
NH2 Ai
709
Легкая цепь
- Тромбин
1545
А2
В
Ci
С2 ICOOH
Тяжелая цепь
Связывающий
участок
ч/1
Легкая цепь
Рис. 6-5. Гомология, характерная для факторов VIII и V. Тромбин играет важную роль
в активации обоих факторов, расщепляя их в обозначенных участках
сте повреждения сосуда. Поскольку циркулирующий ФВ связывается как с
обнаженными субэндотелиальными тканями, так и со стимулированными
тромбоцитами, он направляет ФУШ в зону поражения, где последний необходим для
ускорения превращения ФХ в ФХа при участии Ф1Ха.
ФУШ может также активироваться под действием ФХа и Ф1Ха в присутствии
фосфолипида, находящегося в форме аниона, и ионов кальция. Эта реакция
протекает значительно медленнее, чем тромбинактивируемая реакция in vitro. Таким
образом, предполагают, что тромбин является основным активатором ФУШ
in vivo.
Общий путь активации свертывания крови
Протром-
биназный
комплекс
Рис. 6-6. ФХа, OVa, фосфолипид и Са2+ (протромбиназный комплекс) превращают ФП
(протромбин) в ФНа (тромбин) (см. рис. 6-2 и 6-5). L — легкая цепь ФУа; Н — тяжелая цепь ФУа
200
Глава 6. Гемостаз и тромбоз
Общий путь. Завершение процесса активации свертывания крови называется
общим путем (рис. 6-6). В этой стадии ФХа связан с OVa на фосфолипидной
поверхности и в присутствии Са2+ (так называемый протромбиназный комплекс)
превращает протромбин (ФИ) в тромбин (ФНа).
Протромбин — наиболее распространенный фактор свертывания крови в
плазме из всех, зависимых от витамина К. Он синтезируется гепатоцитами как пре-
пропептид. Посттрансляционные процессы модифицируют препротромбин в
зрелую молекулу посредством карбоксилирования остатков глутаминовой кислоты
и встраивания углеводных боковых цепей. Зрелая молекула протромбина
содержит 4 домена: первые два связываются с ионами кальция и фосфолипидом;
третий, видимо, также вовлечен в связь с кальцием, а четвертый содержит активный
предшественник сериновой протеазы (рис. 6-7).
Активация протромбина в тромбин может происходить различными путями.
Физиологически наиболее важный путь — за счет действия протромбиназного
комплекса (ФХа + OVa + фосфолипид + Са2+). Протромбиназа расщеляет
протромбин в двух местах (Arg 320 и Arg 271). Преобладающими продуктами
расщепления являются а-тромбин (аминокислотные остатки 272-579) и протромбино-
вый фрагмент 1.2 (аминокислотные остатки 1-271; рис. 6-7). ФХа в присутствии
ионов кальция без OVa способен расщеплять протромбин в тех же точках (в
обратном порядке), но со значительно меньшей скоростью (в 300 000 раз). Сам
тромбин может расщеплять протромбин по аминокислотным остаткам 155 и 284.
Неактивный предшественник фактора V циркулирует в плазме в виде
одиночного полипептида, асимметричного гликопротеина. Он синтезируется в печени
и мегакариоцитах, причем первый впоследствии превращается в ФУ плазмы,
а второй — в тромбоцитарный ФУ.
Взаимодействие ФУ с протромбином обеспечивается с помощью тромбоци-
тарного скелета на участке плазматической мембраны, связывающем все
элементы протромбоцитарного комплекса (рис. 6-6). ФУ служит рецептором для при-
Активация протромбина
Протромбиназа (Ха, Va, Ca2+, фосфолипид)
t т
155 271 284 320 579
ГСа2+;
/ Протромбин
(eg)
271 272 284
+
579
Фрагмент 1.2
а-Тромбин
Рис. 6-7. Протромбиназный комплекс расщепляет протромбин в указанных аминокислотных
сайгах. Фрагмент 1.2 и а-тромбин являются результатом расщепления между аминокислотами 271—
272. Сам тромбин расщепляется в сайтах 155 и 284
Нормальный гемостаз
201
крепления ФХа к активированным тромбоцитам в месте повреждения. ФУа
также связывается с протромбином, облегчая взаимодействие ФХа и протромбина.
Анализ кДНК ФУ показывает, что его доменная структура аналогична структуре
ФУШ, представленной выше (рис. 6-5). Ген ФУ локализован на хромосоме 1. ФУ
превращается в активный кофактор (ФУа) за счет ограниченного протеолиза
тромбином и фактором Ха. Тромбин удаляет домен В, превращая одноцепочеч-
ный полипептид ФУ в двухцепочечный ФУа.
Связывание ФУа с тромбоцитарной фосфолипидной мембраной или
клеточной поверхностью происходит при участий МН2-конца легкой цепи. Оказавшись
на клеточной поверхности, ФУа действует как часть рецептора ФХа. Чтобы этот
связывающий сайт функционировал, ФУа должен содержать интактные легкие
и тяжелые цепи. ФУ осуществляет бинарное взаимодействие с каждым
компонентом протромбиназного комплекса. Затем ФУа, связанный с мембраной,
изменяет конформацию и ориентацию как фермента, так и субстрата. Хотя фосфоли-
пид является подложкой, на которой происходит концентрирование фермента
и субстрата, однако именно ФУа значительно ускоряет конверсию протромбина
в тромбин при участии ФХа (рис. 6-6).
Тромбин вызывает гидролиз фибриногена до фибрина (рис. 6-8). Фибриноген
(Мг 340 000) представляет собой сложный гликопротеин, состоящий из трех пар
неидентичных полипептидных цепей: 2 а-, 2 |3- и 2 у-цепи. Это — продукт 3 генов,
каждый из которых кодирует одну из разновидностей полипептидных цепей.
Гены сгруппированы вместе на длинном плече хромосомы 4. Фибриноген
синтезируется в основном гепатоцитами, но имеется в мегакариоцитах и тромбоцитах.
Его синтез индуцируется повреждением тканей, воспалением, стрессом
(состояниями острой фазы). Фибриноген играет важную роль в нормальном гемостазе,
а также задействован в процессах при нарушенном гемостазе. Таким образом,
кровотечение или тромбоз — обычные проявления или качественных, или
количественных изменений в фибриногене.
Тромбин прежде всего расщепляет аргинин-глициновые связи фибриногена
с образованием двух пептидов (фибринопептид А — ФПА и фибринопептид В —
ФПВ) и мономера фибрина (рис. 6-6). Эти мономеры образуют полимер,
соединяясь бок в бок (фибрин I), и удерживаются рядом водородными связями
(растворимые фибриновые комплексы). Последующий гидролиз этих комплексов
при действии тромбина приводит к выделению ФПВ. Кроме того, тромбин
активирует ФХШ, который в присутствии ионов кальция связывает боковые цепи
полимеров (лизин с глутаминовыми остатками) изопептидными связями. Между
мономерами возникают многочисленные перекрестные связи, создающие сеть
взаимодействующих фибриновых волокон (фибрин II), весьма прочных и
способных удерживать тромбоцитарную массу на месте травмы (рис. 6-8 и 6-15).
Биологические функции тромбина. Помимо образования фибрина, тромбин
активирует тромбоциты, факторы и профакторы коагуляции, действует за
пределами механизма свертывания крови (рис. 6-9). Тромбин активирует фибриноли-
тическую систему, стимулирует ЭК и лейкоциты. Он также вызывает миграцию
лейкоцитов и регулирует тонус сосудов. И наконец, стимулируя рост клеток,
способствует репарации тканей.
Некоторые общие характеристики факторов свертывания крови
представлены в табл. 6-2. Активность факторов I-XIII можно измерить в плазме. Плазма
приготавливается с добавлением связывающего кальций вещества (например,
Воздействие тромбина на фибриноген, фибрин I и ФХШ
1 llpl Тромбин
Фибриноген
01
Фибрин-
мономер
-J la ["+ | фПА | + 1 ФПВ |
Фибрин-полимер
Фибрин I
(растворимый)
ПШП—SWpoiia}-»» @
I ФПВ
Фибрин II
(нерастворимый)
эб:
Сс
=с^.
Фибрино-
пептиды-
Фибрин-
мономер+
Первая фаза ограничивается протеолизом
фибриногена тромбином с образованием
фибрин-мономера и фибринопептидов
Фибрин-полимер
Когда от фибриногена отщепляются сильно
электроотрицательные фибринопептиды,
фибрин-мономеры спонтанно полимеризуются
с образованием фибрин-полимера.
Первоначально связывание происходит
за счет водородных связей
Тромбин, фактор XIII, Са2+
На 3-м этапе фибрин-полимер стабилизируется
ковалентными связями. Для этой фазы требуется
ФХШ, тромбин и ионы кальция.
Рис. 6-8. Виды действия тромбина. Справа представлена молекула фибриногена в форме диаграммы в момент, когда она подвергается превращению в
нерастворимый фибрин. (Рис. справа из: Hirsh J., Braun E. eds. Hemostasis and Thrombosis: A Conceptual Approach, 2nd ed. New York: Churchill Livingston; 1983.)
Нормальный гемостаз
203
Множественные эффекты тромбина
Агрегация
тромбоцитов
| Регуляция тонуса сосудов
Клеточный рост
| Миграция клеток
Высвобождение
эндотелина,
EDRf,PGfe
ИЗ ЭК
Высвобождение
ФВ, ФУ из ЭК
Активация ФУ, I
Ф\Л1,ФХ1иФХ11||
шШ* ——■—*■■
Деградация ФУ, ФУ1И, ФХ11
Образование фибрина |
Эмуляция I
фибринолиза J
Активация
тромбомодулин/
тромбоцитарного нуги
(непрямой фибринолиз) 1
Высвобождение ТхА2
из фибробластов
и тромбоцитов
Высвобождение
лейкотриенов
из лейкоцитов
Рис. 6-9. Функции тромбина не ограничиваются влиянием на фибрин и фибриноген
цитрата натрия) в цельную кровь. Когда кровь в пробирке свертывается в
отсутствии хелатирующего агента или антикоагулянта, образуется сыворотка.
Сыворотка не содержит фибриногена, ФП, OV и ФУШ, поскольку они утилизуются
в процессе образования сгустка. При активации ФУ и ФУШ незначительным
количеством тромбина в пробирке или in vivo их активность быстро исчезает,
поэтому ФУ и ФУШ называются лабильными факторами свертывания.
Регуляция системы свертывания крови
Активация свертывания крови in vivo модулируется рядом регуляторных
механизмов, которые ограничивают реакции местом повреждения и предотвращают
возникновение массивного внутрисосудистого тромбоза. К регулирующим
факторам относят: кровоток и гемодилюцию; клиренс, осуществляемый печенью и
ТАБЛИЦА 6-2. Некоторые общие характеристики факторов свертывания крови
Факторы контактной
фазы
Факторы, зависящие
от витамина К
Другие факторы
XII, XI, ПКиВМК
Стабильны
Са2+-независимы
Присутствуют в сыворотке
Нет кровотечения
при их дефиците,
за исключением XI
X, IX, VII, II
Стабильны
Са2+-зависимы
Все находятся в сыворотке,
за исключением II
Кровотечение
при дефиците
V, VIII, XIII, I
Лабильны^ и VIII)
Са2+-независимы
В сыворотке только XIII
Кровотечение
при дефиците
204
Глава 6. Гемостаз и тромбоз
РЭС; протеолитическое действие тромбина (механизм обратной связи);
ингибиторы сериновых протеаз; фибринолиз.
Кровоток и гемодилюция. При быстром кровотоке происходит разбавление
активных сериновых протеаз и транспорт их в печень для утилизации. Кроме того,
диспергируются и отсоединяются периферические тромбоциты от тромбоцитар-
ных агрегатов, что ограничивает размер растущей гемостатической пробки.
Клиренс печенью и РЭС. Растворимые активные сериновые протеазы инактиви-
руются и удаляются из кровообращения гепатоцитами и ретикулоэндотелиаль-
ными клетками печени (купферовскими клетками) и других органов.
Протеолитический эффект тромбина. Тромбин ускоряет отложение фибрина
на месте повреждения ткани за счет усиления активации факторов XI, V, VIII
(рис. 6-2). Однако тромбин также может и ограничивать гемостаз. Он вызывает
протеолиз и деградацию факторов XI, V, VIII (рис. 6-2), что облегчает их
инактивацию соответствующими ингибиторами и быстрый клиренс. Тромбин
обеспечивает гемостатический контроль, инициируя активацию фибринолитической
системы посредством белка С. Это неизменно приводит к растворению фибрина и
в том числе — за счет стимуляции лейкоцитов (клеточный фибринолиз).
Ингибиторы активных сериновых протеаз. Процесс свертывания крови строго
контролируется присутствующими в плазме белками (ингибиторами), которые
ограничивают выраженность протеолитических реакций и обеспечивают защиту
от тромбообразования.
Главными ингибиторами факторов свертывания крови являются антитромбин III
(AT III), гепариновый кофактор II (ГКII), протеин С (ПС), протеин S (ПБ),
ингибитор пути тканевого фактора (ИПТФ), протеаза нексин-1 (ПН-1), С1-ингибитор,
агантитрипсин (агАТ) и а2-макроглобулин (а2-М). Большинство этих
ингибиторов, за исключением ИПТФ и а2-М, принадлежит к суперсемейству белков,
гомологичных ингибитору (Х1-протеазы,.и называется серпины. Термин "серпин" (serpin)
образован из слов "serineprotease inhibitor" — "ингибитор сериновой протеазы".
Механизм, лежащий в основе действия большинства ингибиторов протеаз,
связан с образованием прочного стехиометрического комплекса с протеазой и
последующим медленным гидролизом ингибитора и быстрым гидролизом слабо
связанного субстрата. У серпинов иной механизм ингибирования — это результат
взаимодействия между субстратсвязывающим участком активированного
фактора свертывания крови и активным центром ингибитора. Вследствие такого
взаимодействия блокируется активный центр фермента (без гидролиза в активном
центре ингибитора) и сериновая протеаза не вступает в протеолитическую
реакцию. Функция ингибиторов in vivo сводится к ограничению активации
свертывания крови за счет быстрого образования комплексов с сериновыми протеазами.
Этот процесс предупреждает системную амплификацию активации свертывания
крови и ограничивает коагуляцию зоной повреждения.»
В соответствии с механизмом действия ингибиторы свертывания крови
подразделяются на группы: 1) серпины (AT III, ГК II, ПН-1, С1-ингибитор, агАТ);
2) кунины (ИПТФ), которые представляют собой белки, гомологичные апроти-
нину (ингибитор панкреатического трипсина); 3) а2-макроглобулин, ингибитор-
"мусорщик" (активный центр сериновой протеазы не участвует в образовании
комплекса с а2-М).
Нормальный гемостаз
205
Серпины
Антитромбин III (AT III) является серпином и основным ингибитором
тромбина, ФХа и Ф1Ха. Он также инактивирует ФХ1а и ФХПа. AT III нейтрализует
тромбин и другие сериновые протеазы посредством ковалентного связывания.
В результате формируется неактивный 1:1 стехиометрический комплекс между
ферментом и ингибитором путем образования связи аргининсодержащего
активного центра AT III и активного серинового центра тромбина (рис. 6-10). Скорость
нейтрализации сериновых протеаз антитромбином III в отсутствии гепарина
(антикоагулянта) невелика и существенно увеличивается в его присутствии (в 1000—
100 000 раз).
AT III представляет собой сс2-гликопротеин (Мг 580 000), синтезирующийся
в печени. Его называют также гепариновым кофактором I. Гепарин имеет два
сайта связывания с AT III, а тромбин — один. Гепарин связывается с лизиновыми
остатками на AT III, что делает аргининовый активный центр более доступным
для активного серинового центра тромбина. Связывание гепарина с AT III
ускоряет образование комплекса тромбин-AT III-гепарин. Ковалентная связь между
активным сериновым центром тромбина и аргининовым сайтом комплекса AT
III-гепарин вызывает инактивацию активной сериновой протеазы.
После образования комплекса между AT III и тромбином гепарин
диссоциирует из комплекса и связывается с другой молекулой AT III, генерируя
множественные циклы инактивации фермента. Нейтрализация активированных форм иных
факторов свертывания крови посредством AT III происходит по аналогичному
механизму, но скорости инактивации различны. Для катализа ингибирования
ФХа достаточно, чтобы гепарин связался только с AT III. Но чтобы катализиро-
| AT 111 ~~~\ [Потенцирование гепарином]
Рис. 6-10. Взаимодействие AT III с факторами Па, Ха и 1Ха без гепарина и с гепарином. Не
представлен фактор ХПа, который также инактивируется AT III. (AT III — антитромбин III; L — места
связывания лизина; * — аргининсодержащий активный центр AT III; сериновая протеаза (Па, Ха,
1Ха); ** — серинсодержащий протеазный центр.)
206
Глава 6. Гемостаз и тромбоз
вать ингибирование тромбина гепарин должен связаться и с AT III, и с тромбином
(ФПа) (рис. 6-10).
Облегчение взаимодействия между AT III и активными ферментами
свертывания крови может быть опосредовано другими мукополисахаридами и гепарин-
сульфатом. Поскольку эти вещества присутствуют в сосудистой стенке,
предполагается, что они играют определенную роль в контроле процесса активации
свертывания крови. Значение AT III как основного модулятора гемостаза
подтверждается наличием тенденции к тромбообразованию у лиц с врожденным или
приобретенным дефицитом AT III.
Гепариновый кофактор II (ТКII) — серпин, ингибирующий тромбин, но не
другие коагуляционные протеазы в присутствии гепарина или дерматан-сульфата.
ГК II секретируется печенью в кровоток, где он циркулирует в течение примерно
2,5 дней, но комплексы ГК П-тромбин выводятся значительно быстрее с участием
рецептора для комплекса серпин-фермент на гепатоцитах. Ингибирующее
воздействие ГК II на тромбин менее выражено по сравнению с таковым у AT III. ГК II
находится преимущественно во внесосудистом пространстве, где локализуется
дерматан-сульфат, и именно здесь может играть решающую роль в ингибировании
тромбина. Тромбину присущи и иные функции, не связанные со свертыванием
крови или активацией тромбоцитов: пролиферация фибробластов и других клеток,
индуцирование хемотаксиса моноцитов, облегчение адгезии нейтрофилов к ЭК,
стимуляция образования простациклина и прочих медиаторов эндОтелиальными
клетками, ограничение повреждения нервных клеток (рис. 6-9). Способность ГК II
блокировать эту деятельность тромбина играет определенную роль в
регулировании процессов заживления ран, воспаления или развития нервной ткани.
Протпеаза нексин-1 (ПН-1) — серпин, еще один вторичный ингибитор
тромбина, предотвращающий его связывание с клеточной поверхностью.
о.г Антитрипсин (at-AT) нейтрализует ФХ1а и активированный протеин С
(АПС).
С1-ингибитор (Cl-И) также является серпином и главным ингибитором се-
риновых ферментов контактной системы. Он нейтрализует 95 % ФХПа и более
50 % всего калликреина, образующегося в системе кровообращения, а при его
дефиците возникает ангионевротический отек. ФХ1а в основном нейтрализуется
arAT и AT III.
Протеин С (ПС) — витамин К-зависимый белок, синтезируемый гепатоцита-
ми. Циркулирует в крови в неактивной форме. Состоит из легкой цепи (с
доменом, содержащим глутаминовую кислоту и двумя доменами, подобными эпидер-
мальному фактору роста) и тяжелой цепи (домен сериновой протеазы).
Протеин С (при участии остатков глутаминовой кислоты) связывается с
поверхностью ЭК посредством кальциевых мостиков.
ПС активируется небольшим количеством тромбина. Эта реакция
значительно ускоряется тромбомодулином (ТМ), поверхностным белком ЭК, который
связывается с тромбином (рис. 6-11). Взаимодействие между тромбином
и тромбомодулином, видимо, опосредовано другим белком — витронектином.
Тромбомодулин обеспечивает 60 % сайтов связывания тромбина на ЭК.
Тромбин в комплексе с тромбомодулином становится антикоагулянтным протеином,
способным активировать сериновую протеазу — ПС. Комплекс тромбин-тром-
бомодулин интернализуется ЭК, где тромбин разрушается, а тромбомодулин
возвращается к поверхности.
Нормальный гемостаз
207
Протеин S (IIS). Активированный протеин С (АПС) в присутствии своего
кофактора — протеина S — расщепляет и инактивирует OVa и ФVilla (рис. 6-11).
nS является витамин К-зависимым белком, который синтезируется гепатоцита-
ми и ЭК. Он связывается с мембраной ЭК и АПС, образуя мембранный
поверхностный комплекс. Поскольку ФУа соединяется с ЭК вблизи ПБ, этот процесс
способен ускорить активацию ПС. АПС расщепляет ФУа в положении Arg506,
a OVIIIa — в районе Arg562. В результате утрачивается их коагулянтная
активность. Так как факторы Villa и Va являются кофакторами для генерирования
соответственно ФХа и тромбина, ПС оказывает антикоагулянтное воздействие
через две взаимодействующие системы на клеточной мембране. Активирован-
Рис. 6-11. Протеин С быстро активируется комплексом ТМ-Па или только Па (медленнее).
Сродство АПС с клеточной мембраной/фосфолипидом усиливается ПБ, а ФУа и OVIIIa становятся
более доступными для расщепления, опосредованного АПС, и инактивации. С4ЬВР связывает и
инактивирует nS. arAT или АПС-И связывают и инактивируют АПС. Поскольку АПС ингибирует
ИАП, то инактивация АПС стимулирует фибринолиз посредством повышенной активности ТАП
208
Глава 6. Гемостаз и тромбоз
ный ПС ингибирует свободный ФУа, но не связанный с ФХа. Однако в
присутствии IIS (кофактор ПС-активации) происходит ингибирование как свободного,
так и связанного ФУ, что усиливает антикоагулянтный эффект АПС.
Активность АПС контролируется собственным циркулирующим плазменным
ингибитором (АПС-И) и (ХгАТ.
Нарушение активации ПС возникает при патологических состояниях,
обусловленных наличием таких факторов, как гипоксия, эндотоксин, ИЛ-1, ФНО-а
и высоким уровнем гомоцистеина. Известно, что все они ускоряют свертывание
крови, индуцируя экспрессию тканевого фактора эндотелиальными клетками
и подавляя транскрипцию тромбомодулина.
Активированный ПС посредством механизма обратной связи подавляет
продукцию ЭК ингибитора активатора плазминогена-1 (ИАП-1), оставляя без
контроля ТАП. Это косвенно стимулирует фибринолитическую систему и усиливает
антикоагулянтную активность АПС (рис. 6-11).
ПС и nS являются важными модуляторами активации свертывания крови,
и их врожденный дефицит связан со склонностью к тяжелым тромботическим
нарушениям. Клиническое значение системы ПС доказывает повышенное тромбо-
образование (тромбофилия) у лиц с врожденной патологией ФУ. Аномалия
молекулы ФУ (т. н. ФУ Лейден) характеризуется резистентностью к инактивации,
связанной с действием АПС. Мутация в гене ФУ (замена гуанина 1691 аденином)
приводит к замещению аргинина глутамином в позиции 506 аминокислотной
последовательности белка. Такая патология ФУ устраняет сайт, по которому
происходит расщепление активированным протеином С, что мешает инактивации ФУ
и способствует возникновению тромбоза.
Кунины
Куниновые ингибиторы представляют собой суперсемейство белков,
гомологичных апротинину, который называют также ингибитором панкреатического
трипсина. Они содержат один или несколько куниновых доменов. Этот домен состоит
из 58 аминокислотных остатков (он значительно меньше серпиновых доменов).
Для кунинов характерна строгая ориентация остатков цистеина. В
противоположность серпинам, структуру которых не определяют дисульфидные связи,
активность кунинов зависит от правильного образования 3-х дисульфидных
мостиков на 1 домен. Из всех ингибиторов сериновых протеаз крови только ингибитор
пути тканевого фактора (ИПТФ) является ингибитором кунинового типа.
ИПТФ — это ФХа-зависимый ингибитор комплекса ФУНа-ТФ (рис. 6-12).
Ингибитор пути тканевого фактора — это гликопротеин (40 кД), состоящий
из кислого аминокислотного остатка, трех куниновых доменов и основной
СООН-концевой области. Ингибиторная активность ИПТФ обусловлена
первым и вторым куниновых доменом. Первый связывается с комплексом ТФ-
ФVila, второй — с ФХа; третий — с липопротеинами и не обладает ингибиторной
активностью. ИПТФ в основном синтезируется эндотелиальными клетками и
в меньшей степени — мононуклеарными клетками и гепатоцитами. ИПТФ
распределен в трех внутрисосудистых пулах: 50-90 % в ЭК, 10-50 % в плазме и
2,5 % — в тромбоцитах. Плазменный пул главным образом связан с
липопротеинами и лишь 5 % циркулируют в свободном состоянии. Однако ингибиторная
активность обусловлена свободным ИПТФ.
Нормальный гемостаз
209
Инактивация посредством ИПТФ происходит в 2 стадии. Сначала ИПТФ
связывается с ФХа и инактивирует его (1:1 стехиометрический комплекс) при
отсутствии ионов кальция; затем комплекс ИПТФ-ФХа связывается с
комплексом ТФ-OVIIa и инактивирует его, образуя кальцийзависимый четвертичный
ингибиторный комплекс (рис. 6-12). Нефракционированный гепарин,
фракционированные гепарины (гепарины с низкой молекулярной массой) и пентозан-по-
лисульфат стимулируют выделение ИПТФ и усиливают его антикоагулянтную
активность. Ускорение ингибирующего действия ИПТФ за счет гепариновых
соединений и аффинности гепарина к ИПТФ расценивается как причина проти-
восвертывающих свойств эндотелия, содержащего молекулы гепаринового
семейства. Ингибируя небольшие количества ТФ, ИПТФ играет важную роль
в поддержании нормального гемостаза.
Фибринолиз. Конечная стадия в репаративном процессе после повреждения
кровеносного сосуда происходит за счет активации фибринолитической системы
(фибринолиза), что приводит к растворению фибриновой пробки и началу
восстановления сосудистой стенки (рис. 6-15). Фибринолиз — основной эндогенный
механизм, предотвращающий тромбообразование. Существуют 2 главных
компонента фибринолиза: фибринолитическая активность плазмы и клеточный
фибринолиз.
Путь тканевого фактора и его инактивация при помощи ингибитора
Четвертичный
ингибиторный
комплекс,
зависимый
отСа2+
Рис. 6-12. Двухфазный процесс инактивации ФХа и OVIIa посредством ИПТФ
(подробное описание в тексте)
210
Глава 6. Гемостаз и тромбоз
Фибринолитическая система плазмы состоит из плазминогена (профермент),
плазмина (фермент), активаторов плазминогена и соответствующих
ингибиторов. Активация фибринолитической системы приводит к образованию
плазмина — мощного протеолитического фермента, обладающего разнообразным
действием in vivo. Предшественник пл&змина — плазминоген, представляет собой
гликопротеин (92 кД), продуцируемый печенью, эозинофилами и почками.
Молекула плазминогена состоит из А-цепи с пятью доменами и В-цепи
(каталитический домен) с N-концевой глутаминовой кислотой (Glu-плазминоген). Период
полусуществования плазминогена составляет 2,2 сут. У несколько деградированной
формы (Lys-плазминоген) более короткий период полусуществования (0,8 сут).
Плазминоген имеет сильное сродство к лизиновым остаткам. Особенности
структуры придают плазминогену способность связываться со свободными лизиновы-
ми остатками фибрина; а2-антиплазмина; гликопротеина, богатого гистидином;
поверхностных рецепторов; внеклеточного матрикса; тромбоспондина и
иммуноглобулинов.
Активация плазминогена. Превращение плазминогена в плазмин катализируется
активаторами плазминогена и строго регулируется различными ингибиторами.
Последние инактивируют как плазмин, так и активаторы плазминогена (рис. 6-13).
Активаторы плазминогена образуются или сосудистой стенкой (внутренняя
активация), или тканями (внешняя активация). Внутренний путь активации
включает активацию белков контактной фазы: ФХИ, ФХ1, ПК, ВМК и калликре-
ина (рис. 6-4). Это важный путь активации плазминогена, но основной — через
ткани (внешняя активация); он происходит в результате действия тканевого
активатора плазминогена (ТАП), выделяемого эндотелиальными клетками. ТАП
также продуцируется другими клетками: моноцитами, мегакариоцитами и мезо-
телиальными клетками.
ТАП представляет собой сериновую протеазу (период полусуществования
приблизительно 4 мин), которая циркулирует в крови, образуя комплекс со
своим ингибитором, и имеет высокое сродство к фибрину. Зависимость ТАП
от фибрина ограничивает образование плазмина зоной аккумуляции фибрина.
ТАП и Glu-плазминоген связываются с фибриновыми нитями. Как только
небольшое количество ТАП и плазминогена соединилось с фибрином,
каталитическое действие ТАП на плазминоген многократно усиливается. Затем
образовавшийся плазмин разлагает фибрин, обнажая новые лизиновые остатки,
с которыми связывается другой активатор плазминогена (одноцепочечная уро-
киназа). Плазмин превращает эту урокиназу в иную форму — активную двухце-
почечную, вызывая дальнейшую трансформацию плазминогена в плазмин и
растворение фибрина (рис. 6-13).
Одноцепочечная урокиназа выявляется в большом количестве в моче. Как
и ТАП, она относится к сериновым протеазам. Основная функция этого фермента
проявляется в тканях и заключается в разрушении внеклеточного матрикса, что
способствует миграции клеток. Урокиназа продуцируется фибробластами,
моноцитами/макрофагами и ЭК. Период полусуществования составляет
приблизительно 7 мин. В отличие от ТАП циркулирует в не связанной с ИАП форме. Она
потенцирует действие ТАП, будучи введенной после (но не до) ТАП. Как ТАП,
так и урокиназа синтезируются в настоящее время методами рекомбинантной
ДНК и используются в качестве лекарственных препаратов.
Нормальный гемостаз
211
Другими активаторами плазминогена (нефизиологическими) являются стреп-
токиназа (продуцируемая гемолитическим стрептококком), антистреплаза
(комплекс человеческого плазминогена и бактериальной стрептокиназы) и стафило-
киназа (продуцируемая Staphylococcus aureus). Первые два — фармакологические
тромболитические средства, применяемые для лечения острого тромбоза.
Основная функция плазмина — расщеплять фибрин и поддерживать сосуды
в открытом состоянии. Однако плазмин разрушает многие другие субстраты,
включая фибриноген, OV, OVIII, ФХ, Ф1Х, ФВ и тромбоцитарные гликопротеи-
ны. Он также активирует компоненты каскада комплемента (С1, СЗа, C3d, C5).
Расщепление плазмином пептидных связей в фибрине и фибриногене
приводит к образованию различных дериватов с меньшей молекулярной массой,
Активация и ингибирование фибринолитической системы
Внутренняя!
активация!
ФХНа, ФХ1а,
ПК, ВМК
| Плазминоген
Нефизиологическая
активация
Стрептокиназа
Стафилокиназа
Антистреплаза
Внешняя
gjcrggaijjjgi
-|П*ГП1е«
| Ингибиторы |
1ИАП-1|
Стимуляторы ИАП-1, которые
приводят к ингибированию
лизиса сгустка
а2-анти плазмин
а2-макроглобулин
а 1-антитрипсин
Антитромбин III
С1-ингибитор
^NH ИАП-1 ИАПТ|
Продукты
деградации
фибриногена
и фибрина
i ±z
Тромбин
Трансформирующий
фактор роста р
Тромбоцитарный фактор роста
Интерлейкин-1
ФНО-а
Инсулиноподобный фактор роста
Глюкокортикоиды
Эндотоксин
Ингибитор ИАП-1, который
приводит к стимулированию
лизиса сгустка
»
v. Активированный протеин С
ФПА(АсИ-16)
ФПВ(ВР1-14)
Фибриноген
Тромбин
Фибрин-мономер
и макромолекулярные комплексы
Фибрин
без перекрестных
сшивок
Плазмин
Продукты деградации
фибриногена
Ао-полярные фрагменты (A.B.C.H)
Ва1-42
X,Y,D,E
Плазмин
Продукты деградации
растворимого фибрина
Ао-полярные фрагменты (А,В,С,Н)
Ва 15-42
X,Y,D,E
Xllla
Перекрестно
сшитый
фибрин
I Плазмин
Продукты деградации
нерастворимого фибрина
Полимеры полярного остатка (dp)
D-димер
у-удимер и другие комплексы
KDD/E), 2 (DY/YD), 3 (YY/DXD) и т. д.
X.Y.D.E
Рис. 6-13. В верхней части рисунка представлена активация и ингибирование фибринолитической
системы. Можно видеть различные сложные взаимодействия (подробное описание и сокращения
см. в тексте). В нижней части изображен процесс трансформации фибриногена в фибрин и их
деградации. ОЦ - одноцепочечная; ДЦ — двухцепочечная. (Из: Beutler E., Lichtman M. A., Coller B. S.,
Kipps T.J. (eds). William's Hematology, 5th ed. New York: McGraw-Hill, 1995.)
212
Глава 6. Гемостаз и тромбоз
а именно продуктов деградации фибрина (фибриногена) (ПДФ) или продуктов
расщепления фибрина (фибриногена) (ПРФ). Самый крупный дериват
называется фрагментом X, который еще сохраняет аргинин-глициновые связи для
дальнейшего действия, осуществляемого тромбином. Фрагмент Y (антитромбин)
меньше, чем X, он задерживает полимеризацию фибрина, действуя как
конкурентный ингибитор тромбина. Два других, меньших по размеру фрагмента, D и Е,
ингибируют агрегацию тромбоцитов (рис. 6-14).
Плазмин в кровотоке (в жидкой фазе) быстро инактивируется естественно
образующимися ингибиторами, но плазмин в фибриновом сгустке (гелевая
фаза) защищен от действия ингибиторов и лизирует фибрин локально. Таким
образом, в физиологических условиях фибринолиз ограничен зоной фибрино-
образования (гелевая фаза), то есть гемостатической пробкой. Однако при
патологических состояниях фибринолиз может стать генерализованным,
охватывая обе фазы плазминообразования (жидкую и гелевую), что приводит к ли-
тическому состоянию (фибринолитическое состояние, активный фибринолиз).
Оно характеризуется образованием избыточного количества продуктов
деградации фибрина (фибриногена) в крови, а также проявляющимся клинически
кровотечением.
Подобно активным сериновым протеазам системы свертывания крови,
функции активаторов плазмина и плазминогена модулируются ингибиторами.
Ингибиторы плазмина: а2-антиплазмин (а2-АП), а2-макроглобулин (а2-М), аг
антитрипсин (агАТ), антитромбин III (ATIII) и ингибитор эстеразы С1 (С1-И).
а2-Антпиплазмин представляет собой серпин и является основным
ингибитором плазмина в крови. Ему присущи 3 основных свойства: быстро ингибировать
плазмин; затруднять присоединение плазминогена к фибрину; образовывать
перекрестные связи с а-цепями фибрина во время фибринообразования. Он
продуцируется печенью. При избыточном образовании плазмина в крови его
нейтрализация происходит в следующей последовательности: а2-антиплазмином,
а2-макроглобулином, at-антитрипсином, AT III и Cl-И. Несмотря на присутствие
различных ингибиторов, участвующих в нейтрализации плазмина in vivo, наслед-
Рис. 6-14. Действие плазмина на фибриноген, другие факторы свертывания и тромбоциты
Нормальный гемостаз
213
ственный дефицит сс2-антиплазмина проявляется сильным кровотечением —
очевидное свидетельство недостаточности контроля активности плазмина другими
ингибиторами.
а2-Макроглобулин — ингибитор плазмина (второй линии) и иных протеаз
(калликреина и ТАП); действует как ингибитор-"мусорщик" (без связывания
со специфическим активным центром).
Ингибиторы активатора плазминогена 1, 2, 3 (ИАП-1; ИАП-2; ИАП-3).
ИАП-1 — основной ингибитор ТАП и двухцепочечной урокиназы, но он не инак-
тивирует одноцепочечную урокиназу. Продуцируется ЭК, клетками гладких
мышц, мегакариоцитами и мезотелиальными клетками; депонируется в
тромбоцитах в неактивной форме и является серпином. Уровень ИАП-1 в крови
регулируется очень точно и возрастает при многих патологических состояниях. Его
продукция (и последующее ингибирование лиЬиса сгустка) стимулируется
тромбином, трансформирующим фактором роста Р, тромбоцитарным фактором
роста, интерлейкином-1, ФНО-а, инсулиноподобным фактором роста, глюкокор-
тикоидами и эндотоксином. Активированный С ингибирует выделенные из ЭК
ИАП и тем самым стимулирует лизис сгустка (рис. 6-11 и 6-12).
Основная функция ИАП-1 — ограничить фибринолитическую активность
местом расположения гемостатической пробки за счет ингибирования ТАП. Это
выполняется легко за счет большего (в молях) содержания его в сосудистой
стенке по сравнению с ТАП. Таким образом, на месте повреждения активированные
тромбоциты выделяют избыточное количество ИАП-1, предотвращая
преждевременный лизис фибрина.
ИАП-2 — основной ингибитор двухцепочечной урокиназы.
С1-ингибитор инактивирует связанный с контактной фазой фибринолиз, в
частности трансформацию одноцепочечной урокиназы в двухцепочечйую.
Гликопротеин, богатый гистидином (ГБГ), является еще одним
конкурентным ингибитором плазминогена. Высокий уровень ИАП-1 и ГБГ в плазме
обусловливает повышенную склонность к тромбозу.
Клеточная фибринолитическая система. Клеточный фибринолиз связан с
лейкоцитами, макрофагами, ЭК и тромбоцитами. Он поддерживает специфическую
активность как местного, так и системного фибринолиза. Лейкоциты
привлекаются в зону отложения фибрина хемостатическими веществами, которые
освобождают тромбоциты, образованием калликреина и продуктами деградации
фибрина. Наряду с влиянием эстераз и других протеаз на разрушенный фибрин
лейкоциты и макрофаги фагоцитируют фибрин и клеточные остатки,
скопившиеся в месте повреждения.
Гемостатическая реакция
Реакция на повреждение сосуда зависит от разнообразных процессов
взаимодействия между сосудистой стенкой, циркулирующими тромбоцитами, факторами
свертывания крови, их ингибиторами и фибринолитической системой (рис. 6-15).
Гемостатический процесс модифицируется посредством положительной и
отрицательной обратной связи, которая поддерживает стимуляцию констрикции
сосудистой стенки и образование комплексов тромбоциты-фибрин, а также
растворение фибрина и релаксацию сосудов, что позволяет вернуться к нормальному
состоянию.
214
Глава 6. Гемостаз и тромбоз
Рис. 6-15. Интеграция растворимых и клеточных факторов, приводящая к образованию
гемостатической пробки и восстановлению сосудистой стенки (ЭТ^-эндотелин)
Нарушения гемостаза
Нарушения механизма нормального гемостаза клинически проявляются или
кровотечением, или тромбозом. Кровотечение способно возникнуть из-за нарушения
любого процесса, задействованного в образовании гемостатической пробки и ее
стабилизации. Тромбоз может быть или следствием чрезмерной активации гемо-
статического механизма, или результатом нарушения его регуляции. Стадии ак-
Нарушения гемостаза
215
тивации свертывания крови, приводящие к образованию агрегатов фибрина и
тромбоцитов, одинаковы как при образовании гемостатической пробки, так и при
образовании тромба. Основное различие — их локализация. Гемостатическая
пробка формируется вне сосуда, а тромб — внутри него.
Нарушения, связанные с кровоточивостью
Патогенез
Патологическая кровоточивость может быть обусловлена дефектом любого
компонента нормального физиологического механизма гемостаза. Дефекты бывают
наследственными или приобретенными и обычно классифицируются в
зависимости от места действия гемост&тического механизма следующим образом: дефекты
сосудов, дефекты тромбоцитов (количественные и качественные), дефекты
факторов свертывания крови, дефекты фибринолиза (чрезмерный фибринолиз).
Дефекты сосудов наследственного характера представляют собой
структурную патологию сосудистой стенки (например, аномалия коллагена);
приобретенные — являются результатом воспалительных или иммунных нарушений,
поражающих кровеносные сосуды. Последние, достаточно часто встречающиеся
в медицинской практике, называют также сосудистыми пурпурами.
Дефекты тромбоцитов — это изменение их количества (меньше 100 000/мкл —
тромбоцитопения) или качества (тромбоцитопатия). Приобретенные нарушения
тромбоцитов наблюдаются значительно чаще, чем врожденные (глава 5).
Дефекты свертывания крови могут быть наследственными или
приобретенными. Последние — значительно более частое явление в клинике. Нарушения
свертывания крови возникают при: 1) абсолютной недостаточности синтеза одного
из факторов свертывания крови; 2) пониженном синтезе фактора (или факторов)
свертывания крови; 3) образовании аномальной молекулы фактора свертывания
крови; 4) чрезмерной деструкции коагуляционных факторов при патологическом
состоянии активации свертывания крови (диссеминированное внутрисосудистое
свертывание) с последующим быстрым клиренсом; 5) нарушении активности
фактора (факторов) свертывания крови за счёт приобретенных циркулирующих
ингибиторов(антител).
Фибринолитические дефекты характеризуются избыточной фибринолитичес-
кой активностью, которая приводит к тяжелому геморрагическому диатезу.
К этим дефектам относятся: 1) наследственный дефицит сс2-антиплазмина,
проявляющийся длительной тяжелой кровоточивостью вторично вследствие
неконтролируемого уровня плазмина в кровообращении; 2) избыточное образование
активаторов плазминогена (например, ТАП и урокиназы); 3) недостаточность
инактивации активаторов плазминогена — вторичное явление, связанное с
отсутствием ингибиторов или нарушением механизма их выведения (например,
болезнь печени). Приобретенные нарушения встречаются значительно чаще, чем
врожденные.
Клинические последствия кровоточивости
Патологическая кровоточивость иногда приводит к заболеванию и даже
летальному исходу. Последний развивается редко, но соматическая патология —
обычное явление и имеет различные клинические формы. Острая кровопотеря сопро-
216
Глава 6. Гемостаз и тромбоз
вождается острой анемией, а та в свою очередь — гипоксией и гиповолемической
циркуляторной недостаточностью. Хроническая кровоточивость вызывает
анемию вследствие возникновения дефицита железа. Массивные кровоизлияния
могут сдавливать жизненно важные органы, в частности глотку, приводя к
асфиксии, или головной мозг, вызывая тяжелые неврологические нарушения.
Кровоизлияния в суставы и мышцы также связаны с опасными последствиями —
возникает деформация суставов, мышечные контрактуры.
Клинические данные
Анамнез. При оценке больных с патологической кровоточивостью сбор анамнеза,
осмотр больного и лабораторное обследование (скрининговые и специфические
тесты) проводятся с учетом понимания основных механизмов нормального
гемостаза. Анамнез (глава 2) должен быть направлен на определение вида
кровоточивости (местная или генерализованная), тяжести и частоты ее проявления (кожа,
слизистые оболочки или кровоизлияние в суставы, мышцы). Очень важно
выявить, возникло кровотечение спонтанно или вследствие приема лекарства,
травмы, удаления зубов или других видов хирургического вмешательства. Семейный
анамнез должен помочь выяснить, является ли патология наследственной, и
определить вид наследования (Х-сцепленное, аутосомно-доминантное или
рецессивное). Установление факта заболевания в семье имеет большое значение для
понимания этиологии нарушения, однако отсутствие в семейном анамнезе случаев
заболевания не исключает возможности развития наследственного
геморрагического диатеза. У больных с наследственной патологией нарушения обычно
проявляются в детстве. Часто это — семейная кровоточивость. В социальном анамнезе
большое значение имеет потребление алкоголя и прием лекарственных
препаратов, особенно аспирина и родственных ему соединений. При изучении
медицинского анамнеза необходимо выявить заболевания, которые могли быть причиной
или условием для развития кровоточивости.
Физикальное обследование. При осмотре больных (глава 2) следует обращать
внимание на вид и локализацию геморрагии (кожа, ротовая полость, глотка, глаза,
суставы). Как правило, обнаруживаются следующие проявления кровоточивости.
Петехии — красные пятна размером с булавочную головку. Они могут быть
рассеяны по коже и слизистым оболочкам в различных частях тела. Эти пятна
образованы внесосудистыми эритроцитами — клетками, которые вышли из сосуда
вследствие повышенной проницаемости сосудистой стенки (сосудистая пурпура)
или патологии тромбоцитов, физиологическая функция которых состоит в
закрытии пробелов между ЭК. Может снижаться количество тромбоцитов либо
нарушаться их функция. Петехии не бледнеют при нажатии на них, так как вышедшие
из сосудов эритроциты не способны проникать в сосуд вновь.
Пурпуры возникают за счет слияния петехии, причиной развития которых
являются те же (указанные выше) факторы.
Экхимоз, или "синяк", представляет собой большой многоцветный плоский
участок кожи с внутри- и подкожным кровоизлиянием. Экхимоз может быть
результатом травмы сосуда, а также возникать у больных с нарушением
функции сосудистой стенки или патологией тромбоцитов. Иногда является
клиническим проявлением наследственных или приобретенных нарушений
свертывания крови.
Нарушения гемостаза
217
Гематома — большое скопление крови, вышедшей из сосудов и инфильтрую-
щей подкожные ткани и мышцы. Кожа над инфильтратом возвышается и
изменяет цвет (синий). Гематома — клиническое проявление наследственного или
приобретенного дефицита фактора (факторов) свертывания крови.
Гемартрозу или кровоизлияние в сустав, связан с тяжелыми дефектами
факторов свертывания крови: недостаточностью фактора VIII (гемофилия А), IX
(гемофилия В), в редких случаях — тяжелым дефицитом фактора XI (гемофилия С)
или дефицитом факторов V, X или II.
Гематурия, или наличие крови в моче, часто наблюдается при гемофилии А
и В, болезни Виллебранда и тяжелом дефиците витамина К. Причиной может
быть местное поражение почек.
Носовые кровотечения обычно вызваны незначительной травмой
расширенных кровеносных сосудов в ноздрях, однако могут быть вторичным явлением,
связанным с нарушением функции тромбоцитов или стенки сосуда либо с
болезнью Виллебранда.
Телеангиэктазия и ангиомы — изменения сосудов, приводящие к появлению
красных пятен или бляшек, которые исчезают при надавливании.
После изучения анамнеза и данных осмотра необходимо провести ряд
обычных лабораторных тестов: пробу на обнаружение крови в кале и моче,
исследование мазка периферической крови, полный клинический анализ крови,
функциональные пробы печени (для исключения заболевания печени), определение азота
мочевины и креатинина в крови (для исключения почечной недостаточности).
После этого выполняются так называемые скрининговые тесты на
кровоточивость, необходимые для характеристики геморрагического диатеза.
Лабораторное исследование. Лабораторная оценка нарушений, связанных с
кровоточивостью, должна касаться каждого компонента гемостатического
механизма: сосудистой стенки, тромбоцитов, факторов свертывания крови.
Лабораторные скрининг-тесты на кровоточивость включают:
1. Определение количества тромбоцитов (подсчет и измерение).
2. Изучение мазка периферической крови.
3. Время кровотечения (ВК), если подозревается качественное нарушение
тромбоцитов или БВ. ,
4. Активированное частичное тромбопластиновое время (АЧТВ).
5. Протромбиновое время (ПВ).
При приобретенных нарушениях, например ДВС> требуется проведение двух
дополнительных проб:
6. Определение содержания фибриногена.
7. Определение содержания продуктов деградации фибрина (ПД Ф) или
уровня D-димера,
1. Подсчет тромбоцитов. Определяется при исследовании образца крови под
микроскопом. Число тромбоцитов, подсчитанное при большом увеличении
в одном поле, умножается на 10 000/мкл. Более точный подсчет проводится
с помощью автоматического счетчика или методом фазовоконтрастной
микроскопии.
2. Изучение мазка крови дает дополнительную информацию о причине
аномального количества тромбоцитов. Так, обнаружение небольшого числа
крупных тромбоцитов при наличии фрагментированных эритроцитов или
218
Глава 6. Гемостаз и тромбоз
шистоцитов (глава 3) свидетельствует о повышенной деструкции
тромбоцитов как причине низкого их количества и освобождения молодых
больших тромбоцитов из костного мозга. Наличие миелобластов одновременно
с низким количеством малых тромбоцитов в периферической крови
указывает на костномозговую инвазию лейкемическими клетками, вызывающую
уменьшение продукции тромбоцитов.
Кровоточивость у больного с тем или иным дефектом тромбоцитов может
быть результатом низкого количества тромбоцитов (тромбоцитопения)
(< 100 000/мкл), нарушения функции тромбоцитов при нормальном их
количестве или обусловлена качественными нарушениями этих клеток.
3. Время кровотечения (ВК) позволяет определить состояние сосудов после
взаимодействия между тромбоцитами и сосудистой стенкой. Оно
измеряется модифицированным методом Айви. После наложения манжетки на
верхнюю часть плеча и создания в ней давления 40 мм рт. ст. делается разрез
на коже сгибательной поверхности предплечья (1x9 мм) с помощью
одноразовой матрицы. ВК — время, необходимое для остановки кровотечения,
в норме оно составляет 3-8,5 мин. ВК стандартизовано для числа
тромбоцитов > 100 000/мкл. Более низкое количество тромбоцитов связано с
прогрессивным увеличением ВК. При первичных нарушениях сосудистой
стенки ВК отклоняется от нормы (например, при сосудистой пурпуре) так же,
как при качественных нарушениях тромбоцитов и БВ.
4. Активированное частичное тромбопластиновое время (АЧТВ, рис. 6-16).
Оно позволяет измерить внутренние факторы свертывания крови (факторы
XII, ПК, ВМК, XI, IX, VIII) и факторы общего пути (X, V, И, I). Для прове-
Реактив (измельченный оксид кремния или каолин —
заменители фосфолипида тромбоцитарной мембраны)
Рис. 6-16. Оценка целостности внутреннего и общего пути по величине АЧТВ.
При наличии нарушений факторов свертывания АЧТВ увеличивается
Нарушения гемостаза
219
дения пробы используются активирующий агент (измельченный оксид
кремния или каолин) — заменитель фосфолипидов мембраны тромбоцита,
кальций и цитратная плазма больного или нормальная плазма. После
добавления активирующего агента к плазме "открывается" активный сериновый
центр фактора XII, что приводит к последующей активации как факторов
свертывания крови внутреннего пути, так и факторов общего пути.
Активатор заменяет фосфолипид тромбоцитарной мембраны, который связывает
активированные факторы IX, X, V, и II для ускорения образования сгустка
в присутствии добавленного кальция. Окончание свертывания
регистрируется в секундах. Величина АЧТВ в норме составляет 25-38 с.
При дефиците факторов свертывания крови XII, ПК, ВМК, XI, IX и VIII, а также
X, V, II и I АЧТВ возрастает.
5. Протромбиновое время (ПВ) (рис. 6-17). Этот тест позволяет оценить
фактор VII (внешний путь коагуляции) и факторы X, V, II и I (факторы общего
пути). В плазму пациента добавляют тканевой фактор и кальций. ТФ
активирует OVII, который, в свою очередь, активирует факторы общего пути
(X, V, Са2+ и фактор II), что приводит к образованию тромбина. Тромбин
трансформирует фибриноген в фибрин. ПВ не учитывает состояние
факторов внутреннего пути свертывания крови. Нормальный диапазон
составляет 10-14 с.
ПВ можно выражать как международное нормализованное отношение (MHO),
которое рассчитывается следующим образом: MHO (INR) = (ПВ больного в се-
кундах/ПВ стандартной плазмы здоровых людей)мин, где МИЧ (ISI) — междуна-
Реактив
(Тканевый фактор)
Рис. 6-17. Тест на ПВ. Определяется целостность внешнего и общего пути.
При нарушении того или другого протромбиновое время увеличивается
220
Глава 6. Гемостаз и тромбоз
родный индекс чувствительности, соотносящий активность тканевого фактора
из животных источников со стандартом тканевого фактора у человека. Величина
MHO используется для контроля за действием антикоагулянтов, вводимых перо-
рально. Использование MHO рекомендовано Всемирной организацией
здравоохранения с целью достижения более точного контроля при лечении
антикоагулянтами:, а также для обеспечения сравнимости межлабораторных данных.
ПВ увеличено у лиц с наследственным дефицитом факторов VII, X, V, II и I
или приобретенным комбинированным дефицитом факторов (дефицит
витамина К или пероральное применение антикоагулянтов).
Интерпретация лабораторных тестов
Увеличение АЧТВ. С помощью скрининга и проб на свертываемость,
проведенных до операции, можно выявить увеличение АЧТВ. В дальнейшем рекомендуется
исследовать тромбоциты, ПВ и ВК (после тщательного анамнеза и общего осмотра
больного). В ходе лабораторного обследования пациента с пролонгированным
АЧТВ, нормальным количеством тромбоцитов, нормальным протромбиновым
временем и временем кровотечения необходимо последовательно выполнить ряд
исследований, интерпретация результатов которых представлена на рис. 6-18
(также рис. 6-2,6-16, 6-17). Особое значение при этом имеет клиническое
определение геморрагического статуса пациента (есть у него кровоточивость или нет).
Если больной впервые обратился за медицинской помощью в связи с
проблемой кровоточивости, необходимо провести подсчет тромбоцитов, определить
АЧТВ, ПВ, ВК после получения данных анамнеза и физикального обследования.
При кровоточивости и пролонгированном А ЧТВ, нормальном количестве
тромбоцитов, нормальных ПВ и ВК тест на измерение АЧТВ выполняют в следующей
смеси: 1 объем нормальной плазмы +. 1 объем плазмы больного — так называемый
1:1 смешанный (микст) АЧТВ-тест. Если в таком варианте теста
пролонгированное АЧТВ корригируется, можно подозревать дефицит факторов VIII, IX или
фактора XI, так как для нормального АЧТВ требуется лишь 30-50 % от их
нормального уровня. И наоборот, если коррекции не происходит, подозревается
наличие ингибитора. Чаще всего это ингибиторы, нейтрализующие активность
факторов VIII или IX, но возможно также присутствие ингибитора активности
фактора XI.
При кровоточивости и пролонгированном АЧТВ, нормальных ПВ и количестве
тромбоцитов, но пролонгированном ВК необходимо рассмотреть причины,
влияющие на состояние внутреннего пути, а также исследовать состояние функции
тромбоцитов (рис. 6-19).
Без кровоточивости с пролонгированным АЧТВ при нормальном количестве
тромбоцитов и коррекции АЧТВ в тесте 1:1 имеет место дефицит одного из
факторов свертывания крови контактной фазы (например, факторов XII, ПК или
ВМК). Без коррекции АЧТВ в тесте 1:1 у таких пациентов, по всей вероятности,
присутствует в плазме волчаночноподобный ингибитор, который ассоциируется
с тромбозом без геморрагических осложнений (рис. 6-18).
Для рассмотрения представленных далее клинических ситуаций
рекомендуется пользоваться рисунками 6-2,6-16 и 6-17, что облегчит понимание того, как
лабораторные данные помогают выявить дефицит определенных факторов или их
ингибиторов.
Нарушения гемостаза
221
Увеличение протромбинового времени (ПВ). При кровоточивости с
пролонгированным ПВ (нормальное АЧТВ, количество тромбоцитов и ВК) можно
предполагать дефицит фактора VII.
Увеличение АЧТВ и ПВ. При кровоточивости с пролонгированными АЧТВ и ПВ
(нормальные тромбоциты и ВК) и с коррекцией в 1:1 микст-тесте вероятно
наличие следующих состояний: 1) дефицит факторов X, V, II или I (факторы общего
пути); 2) дефицит витамина К (недостаточность всех витамин К-зависимых
факторов: X, IX, VII и II); 3) влияние терапии варфарином (также недостаточность
витамин К-зависимых факторов); 4) болезнь печени (пониженный синтез
факторов свертывания крови); 5) диссеминированное внутрисосудистое свертывание
(см. далее); 6) активный фибринолиз (как вторичное явление после терапии или
патологического процесса).
При кровоточивости с пролонгированными АЧТВ и ПВ (нормальные
тромбоциты и нормальное ВК), но без коррекции в 1:1 микст-тесте можно предполагать
наличие ингибитора одного из факторов общего пути. Чаще всего это ингибитор
фактора V, но возможны также ингибиторы факторов X или II.
Патология
Проба
Результат
Клиническое
состояние
Увеличенное АЧТВ
(при нормальном количестве
тромбоцитов, ПВ и ВК)
Кровоточивость
Пробы
Диагностические
заключения
Отсутствие
кровоточивости
OVIII
или Ф1Х
или ФХ1
ФХП
ВМК
ПК
Гемофилии
А
В
С
Недостаточность
ФХП, ВМК
или ПК
Крово- 1
точивость
>
г
Ингибитор 1
OVIII 1
Ингибитор 1
Ф1Х 1
А ,
Г Наличие
ингибитора
OVIII
или ингибитора
| Ф1Хит.д. 1
Отсутствие
кровоточивости
Волчаночный
антикоагулянт
Пробы -dRVTTPNP,
а также АФА и АКА
Наличие волчаночного]
антикоагулянта,
антител
к фосфолипидам
или к кардиолипину
Рис. 6-18. Оценка значения пролонгированного АЧТВ является как лабораторной, так и
клинической процедурой. Первый этап — повторно определить АЧТВ и подтвердить, что оно
действительно увеличено. Второй этап — установить, корригируется ли АЧТВ в 1 : 1 микст-тесте.
Существенное значение для определения схемы дальнейшего обследования имеют такие факторы, как
кровотечения в анамнезе и наличие или отсутствие кровоточивости в текущий момент
(сокращения см. в тексте)
222
Глава 6. Гемостаз и тромбоз
Патология
Проба
Результат
Диагностические
заключения
Пробы
Результат
Диагностические
заключения
Пробы
Результат
Диагностические
заключения
Пролонгированное время кровотечения |
(нормальное количество тромбоцитов и ПВ) |
||АЧТВ
т
| БВ
т
ФВ: антиген |
ФВ:
JOB: антиген
JKP
1РИАТ
1
'
1 БВ 1
1 Тип 1 1
активность
KP
РИАТ
\Г
1 Норма
ЦФВ: антиген
JKP
у^или|РИАТ
1
1 БВ 1
1 Тип 2 1
Норм. АЧТВ
Другие качественные
дефекты тромбоцитов
Определение размера
тромбоцитов в мазке
периферической крови
Нормальный!
размер
Качественные
дефекты
тромбоцитов
Электрофорез
мультимеров
ФВ
Исследование
агрегации
тромбоцитов
Патология
Отсутствие
агрегации
Снижение
агрегации
Подтипы БВ
II типа
Тромбастения
Гланцмана
(дефект ГППЬ/Ша)|
Примечание:
см. также
освобождение
тромбоцитов,
адгезия, ТФЗ
Нарушение
выделения;
низкая АТФ;
нарушение
пула хранения!
ASARx
Большой
размер
Синдром
Бернара-
Сулье
РИАТЭМ I
тромбоцитов I
1РИАТДМ
"внешний вид
швейцарского
сыра" I
Синдром
Бернара-
Сулье
(дефект ГП lb)
Рис. 6-19. При оценке значения пролонгированного ВК прежде всего необходимо подсчитать
количество тромбоцитов и определить ПВ. Если они в пределах нормы, определяют АЧТВ. При
пролонгированном АЧТВ коррекция в 1 : 1 микст-тесте может свидетельствовать о дефиците факторов
свертывания; однако наличие пролонгированного ВК позволяет также предположить патологию
тромбоцитов и/или сосудистой стенки. Очень важно знать состояние кровоточивости больного, но
уместно отметить, что болезнь Виллебранда может протекать на фоне отсутствия геморрагических
проблем (у пациента и членов его семьи). При кажущихся нормальными величинах лабораторных
данных (или незначительных отклонениях от нормы) угрожающее жизни кровотечение может
возникнуть при травме или тяжелом гемостатическом стрессе (ДМ-дефект мембраны, остальные
сокращения в тексте)
Нарушения гемостаза
223
Без кровоточивости с пролонгированием А ЧТВ и ПВ (нормальные
тромбоциты и нормальное ВК), без коррекции в 1:1 микст-тесте обычно причиной
пролонгирования является ингибитор, подобный волчаночному антикоагулянту.
Отсутствие аномалий при проведении обычных скрининговых тестов крови.
Больные с небольшой кровоточивостью в анамнезе и без отклонений при
проведении обычных скрининговых проб (нормальное количество тромбоцитов,
нормальные ВК, ПВ и АЧТВ) могут иметь дефицит ФХШ или а2-антиплазмина. Дефицит
ФХШ удается выявить скрининговым тестом, в котором измеряется
растворимость фибринового сгустка в 5 М мочевине. Фактор XIII образует прочные кова-
лентные связи молекул фибрина-полимера, делая сгусток нерастворимым в
мочевине. Отсутствие ФХШ приводит к растворению фибриновых сгустков
(поддерживаемых водородными связями) в 5 М мочевине.
Специальные пробы для определения факторов свертывания крови. Ниже
перечислены специальные пробы, которые необходимо проводить для определения
причины кровоточивости после детального и полного сбора анамнеза и общего
осмотра больного.
1. Анализы специфического фактора свертывания крови.
2. Пробы на функциональное состояние тромбоцитов (адгезия, агрегация,
секреция, прокоагулянтная активность).
3. Анализ ФВ (количество и функция).
4. Определение.ингибитора (например, определение волчаночного
антикоагулянта или ингибитора специфических факторов VIII или IX).
5. Скрининговые пробы ДВС (ПВ, АЧТВ, количество тромбоцитов, уровень
фибриногена и определение ПДФ или D-димёра).
6. Пробы на определение нарушений фибринолиза.
7. Специфические пробы на активацию свертывания крови.
Анализ специфического фактора свертывания крови. Функциональная актив-
ность факторов свертывания крови определяется в клинических лабораториях
с помощью модифицированных методов АЧТВ и ПВ. Модификация включает
добавление к реакционной смеси плазмы с известным дефицитом
интересующего фактора свертывания крови. Эта плазма называется "субстратом" и содержит
нррмальное количество всех факторов свертывания крови за исключением
одного, содержание которого нужно измерить. Корригирующий эффект плазмы
больного на пролонгированное время плазмы-субстрата сравнивается с
корригирующим эффектом известной концентрации исследуемого фактора в
нормальной плазме (например, фактор VIII -.Д00 %, 75 %, 50 %, 25 %, 10 %, 5 % и 1 %).
Результаты выражают в процентах от активности стандартной нормальной
плазмы; 1 мл смешанной нормальной плазмы равен 100 % активности или
1 ед/мл. В других анализах, применяемых для измерения активности факторов
свертывания крови, используются хромогенные (колориметрические)
субстраты. Последние расщепляются сериновыми протеазами факторов свертывания.
Это позволяет получить кривую титрования и измерить активность конкретного
фактора свертывания крови.
Концентрация белков свертывания крови определяется различными
иммунологическими методами. Это твердофазный иммуноферментный анализ (ТИФА),
радиоиммунныи анализ (РИА) или количественный иммуноэлектрофорез
(КИЭФ).
224
Глава 6. Гемостаз и тромбоз
Пробы на определение функциональной активности тромбоцитов описаны
в главе 5.
Пробы на определение болезни Виллебранда (БВ). Активация ФВ. Активность
ФВ в качестве кофактора ристоцетина измеряется в модифицированном тесте
агрегации тромбоцитов. Ристоцетин используют как агрегант, а лиофилизирован-
ные мембраны тромбоцитов здоровых людей — как источник гликопротеинов.
Ристоцетин связывается с фактором Виллебранда; последний взаимодействует
с ГШЬ и ГППЬ/Ша в мембранах тромбоцитов, вызывая взаимодействие по типу
тромбоцит-тромбоцит (агрегация тромбоцитов). Эффект плазмы больного
(фактор Виллебранда) в тестовой системе сравнивают с эффектом известных
концентраций нормального ФВ. Результаты выражают в процентах активности ФВ
нормальной плазмы (рис. 6-20).
Ристоцетининдуцированная агрегация тромбоцитов (РИАТ) — еще один тест
для определения реакции тромбоцитов на нормальную и субоптимальную кон-
ФВ (концентрация)
методом КИЭФ
|75% 50% 25% 12,5%
^ v ^Тйп1 Тип II
Норма БВ
%
ФВ (активность) (КР)
Ю0-, Норма
75-
50
25
3IIA
БВ Тип I
оД
Ристоцетин
Ристоцетининдуцированная
агрегация тромбоцитов
% 100 НоримИ, 2 мг/мл
75
50
25
Л
! / БВ ИВ
// (0,6 мг/мл)
// БВ Тип 1(1,2 мг/мл)
—
У/^ Норма (0,6 мг мг/мл)
Ристоцетин
Многомерный анализ ФВ
Норма
Тип!
Тип ПА
Тип ИВ
БВ тромбо-
цитарного
типа или
псевдо-БВ
Рис. 6-20. Определение концентрации ФВ методом КИЭФ (А); функциональной активности ФВ
по КР (Б) и РИАТ (В) у здоровых лиц и некоторых больных с БВ. Электрофоретическое разделение
мультимеров ФВ (Г) у здоровых лици больных с БВ. (В — высоко-, С — средне- и Н — низкомолеку-
лярпые фракции ФВ; прочие сокращения см. в тексте)
Нарушения гемостаза
225
центрации ристоцетина. Отличие РИАТ в том, что в первом случае в качестве
источника ФВ суспендируются тромбоциты больного в его плазме (в методе "кофак-
торристоцетина" используются нормальные лиофилизированные тромбоциты).
РИАТ применяется для дифференцирования вариантов патологии ФВ
(аномальный белок ФВ, тип lib) от классической болезни Виллебранда типа I (сниженный
синтез ФВ). Больные с БВ типа ПВ дают полную реакцию на полунормальную
концентрацию ристоцетина (0,6 мг/мл), в то время как для такой же реакции
тромбоцитов у здоровых людей необходимо 1,2 мг/мл. Реакция тромбоцитов на
низкие концентрации ристоцетина уникальна для типа БВ ПВ (рис. 6-20).
Концентрация ФВ (антигенная концентрация ФВ — ФВ: Аг) измеряется
методами количественного электроиммунофореза (КИЭФ), твердофазного иммуно-
ферментного анализа (ТИФА) или радиоиммунного анализа (РИА). На рис. 6-20
представлены данные КИЭФ. При выполнении этой пробы агар с кроличьими
анти-ФВ-антителами помещается на пластиковые предметные стекла. До одной
стороне стекол сделаны ячейки для 10 мл разведенной плазмы (стандарты) или
проб больного. Электрофорез проводится в течение суток. После промывания
и окраски стекол определяют высоту окрашенных столбиков, образуемых преци-
питированными антигеном и антителом, которая прямо пропорциональна
концентрации ФВ. Высота пробы больного сравнивается со стандартом. Это точный
и недорогостоящий метод. ТИФА и РИА (их нет на рисунке) имеют некоторое
преимущество, позволяя анализировать большое количество проб одновременно
и определять незначительную концентрацию ФВ. Разделение фракций ФВ по
молекулярной массе с помощью электрофореза в агаровом геле с додецилсульфа-
том натрия используется для идентификации различных подтипов болезни
Виллебранда (тип ИА-Н), связанных с аномальной структурой белка Виллебранда
(рис. 6-20).
Пробы на определение ингибиторов. Ингибиторы отдельных факторов
свертывания крови (например, факторов VIII, IX и V) определяются
смешиванием разведений плазмы больного с нормальной плазмой и инкубированием
смеси в течение 2 ч при температуре 37 °С. В конце инкубационного периода
в пробах повторно определяют АЧТВ. При отсутствии коррекции в каждом
образце оценивают активность (содержание) интересующего фактора (например,
фактора VIII).
Считается, что образец с остаточной активностью фактора, составляющей
50 % от нормальной активности, содержит одну единицу Бетесда (БЕ) (Bethesda
Unit, BU) ингибитора в 1 мл. Титр ингибитора равен обратной величине
разведения тестируемой плазмы, которая нейтрализует 50 % активности фактора
нормальной плазмы (например, ФVIII). Одна Оксфордская единица ингибитора
(Великобритания) соответствует 1,21 единицы Бетесда (США).
Волчаночный антикоагулянт (ВА) пролонгирует как АЧТВ, так и ПВ; но
в низких титрах оказывает влияние только на АЧТВ. Оценить активность ВА
в плазме можно двумя клиническими тестами: определением времени действия
разведенного яда гадюки Рассела и процедурой нейтрализации тромбоцитов
(ПНТ). В первом тесте реактив (яд гадюки) непосредственно активирует фактор
X. Последовательные разведения яда способствуют образованию небольших
количеств активированного фактора X, в результате чего низкие уровни ВА
проявляют свою активность против протромбиназного комплекса (ФХа + ФVa + Са2+ +
фосфолипид тромбоцита) и увеличивают время свертывания крови.
226
Глава 6. Гемостаз и тромбоз
В процедуре нейтрализации тромбоцитов (ПНТ) используются мембраны
лиофилизированных нормальных тромбоцитов и плазма пациента. ВА в плазме
связывается с Fc-рецепторами мембраны нормальных тромбоцитов. Повторное
определение АЧТВ в супернатанте этой смеси показывает нормализацию
времени свертывания крови. Характеристика ВА может быть получена с помощью
иммунологических методов с применением специфических фосфолипидов,
например сфингомиелина, фосфатидилэтаноламина, фосфатидилхолина, фосфати-
дилинозитола, кардиолипина (КЛ). Другой метод, подтверждающий наличие ВА
в плазме, основан на использовании фосфолипидов с измененной
конфигурацией, например гексагонального фосфатидилэтаноламина.
Антикардиолипиновые антитела (АКА) определяются методом ТИФА с
применением антител против кардиолипина и против человеческих иммуноглобули-
новых антител (IgG и IgM) к кардиолипину. Подобно волчаночной
гиперкоагуляции, их наличие связывают с повышенной свертываемостью крови.
Скрининг-тесты на ДВС. (См. ниже раздел "Тесты на фибринолиз", а также
табл. 6-13.)
Тесты на фибринолиз. Ниже приведены результаты скрининг-тестов,
свидетельствующих о повышенной фибринолитической активности в плазме.
1. Сокращенное время свертывания цельной крови или время лизиса эуглобу-
линовых сгустков по сравнению с нормальным.
2. Повышенный лизис фибрина в чашечном тесте или радиоизотопном
исследовании.
3. Низкое содержание плазминогена, выявленное иммунологическими
методами.
4. Повышенное содержание продуктов деградации фибрина (фибриногена)
(ПДФ) в сыворотке, измеряемое различными иммунологическими методами.
5. Повышенное содержание D-димера в плазме, указывающее на повышенный
фибринолиз и активацию свертывания крови.
При определении ПДФ применяются поликлональные антитела к ПДФ с целью
выявления фибринолиза в сыворотке простым латекс-методом, однако с
помощью этого теста нельзя дифференцировать фибриноген и дериваты фибрина. При
определении D-димера выявляются специфические продукты лизиса фибрина.
(При распаде фибриногена н^ образуется D-димер, так как для этого необходим
перекрестносшитый фибрин.)
При определении D-димера применяются моноклональные антитела против
наименьшего продукта деградации (D-димера), возникающего в результате
лизиса перекрестносшитого фибрина (но не фибриногена). Содержание D-димера
измеряется в плазме с использованием латекс-метода или более чувствительного
метода ТИФА. К преимуществам анализа D-димера по сравнению с ПДФ
относятся: выявление D-димера в плазме и измерение специфического плазмин-фиб-
ринового продукта. Последнее свидетельствует об активации свертывания крови
in vivo с активацией тромбина и плазмина, которые вызывают соответственно
перекрестное связывание фибрина и его деградацию D-димерами.
Специфические тесты определения активаций свертывания крови. Тесты на
определение активации свертывания крови in vivo относятся пока к
исследовательским и в большинстве клинических лабораторий не используются. Они включают:
Нарушения гемостаза
227
1) определение фибринопептидов А и В методом РИА; 2) определение про-
тромбинового фрагмента 1,2 методом ТИФА; 3) определение комплекса AT III
с тромбином иммунологическими и хромогенными методами; 4) определение
ПС-активированного пептида, комплексов АПС-АПС-И, комплекса АПС-
<х2-макроглобулин, АПС-аг AT методом ТИФА или другими
иммунологическими методами; 5) выявление Ф1Ха или комплекса 1Ха-АТ III иммунологическими
методами; 6) обнаружение фактора Ха методами РИА или ТИФА.
Наследственные нарушения свертывания крови
Имеются сообщения о наследственном дефиците каждого из факторов
свертывания крови. Наследственный дефицит любого фактора может стать причиной
возникновения геморрагического синдрома, за исключением дефицита фактора XII,
прекалликреина и ВМК. Наиболее характерны 3 наследственных нарушения
свертывания крови: дефицит фактора VIII (гемофилия А), дефицит фактора IX
(гемофилия В) и патология, связанная с фактором Виллебранда (болезнь Вил-
лебранда, БВ). Способ наследования факторов свертывания крови представлен
в табл. 6-3.
Гемофилия А
Гемофилия А (ГА) — наиболее характерный тип гемофилии (составляет 80-
85 % всех случаев гемофилии). В США ГА встречается у 25 из 100 000 мужчин.
Полагают, что в мире ГА в среднем страдают 30-100 из 1 млн человек. Основное
нарушение при ГА — отсутствие или снижение коагулянтной активности
фактора VIII в плазме (VHI:C), хотя молекула этого фактора, как правило,
присутствует.
ФУШ циркулирует в плазме, будучи связанным с фактором Виллебранда,
который стабилизирует его активность и регулирует синтез (рис. 6-21).
Исследования гена фактора VIII (хромосома X) показали множественные делеции и
точечные мутации у кровных родственников.
Наследование и выявление цосительства. Гемофилия наследуется сцеплено
с Х-хромосомой. Сыновья женщин-носителей гена гемофилии имеют шанс его
унаследовать в 50 % случаев (рис. 6-22). У мужчин заболевание проявляется
кровоточивостью. Все сыновья гемофиликов здоровы, в то время как дочери
обязательно являются носителями дефектной хромосомы. Дефект локализуется на
Х-хромосоме, поэтому у мужчин нет нормального аллеля (XhY) и у них
развивается клинически выраженная патологическая кровоточивость. Наоборот, у
женщин-носителей (XhX), которые наследуют нормальный аллельный ген от матери,
обычно нет тенденции к кровоточивости. Коагулянтная активность OVIII у носи-
ТАБЛИЦА 6-3. Способ наследования факторов свертывания крови
Факторы Способ наследования
ФУ\\\ и Ф1Х Сцепленность с Х-хромосомой
ФВ1 и дисфибриногенемии Аутосомно-доминантный
Дефицит других факторов Аутосомно-рецессивный
1 В редких случаях варианты болезни Виллебранда оказываются аутосомно-рецессивными.
228
Глава 6. Гемостаз и тромбоз
Комплекс ФУШ
Место связывания OVIII
Функции ФВ:
1. Адгезия тромбоцитов ]
2. Переносчик ФУШ
Место связывания тромбоцитов
Рис. 6-21. Фактор Виллебранда действует как мост между тромбоцитами и сосудистой стенкой,
несет и транспортирует ФУШ
телей должна составлять 50 % нормы. Тем не менее это не всегда так из-за
широкого диапазона фактора VIII:C (50-200 %) у населения и повышения уровня
ФУШ после физической нагрузки, при беременности, стрессе и различных
других клинических состояниях. Уровень OVIILC позволяет выявить состояние
носительства только у 60 %. Степень выявления удается повысить до 60-90 %, если
дополнительно определить антиген фактора VIII (VIII:Ar) и получить
соотношение 2 : 1 (<DVIII:Ar/OVIII:C). Состояние носительства и пренатальный диагноз
определяются еще более точно с помощью анализа гена ФУШ. Для этого
применяют два метода: определение полиморфизма длин рестрикционных фрагментов
и проведение полимеразной цепной реакции с обратной транскрипцией
Типичное генеалогическое
дерево при гемофилии
Носитель
?■
XY
Ф
XY
Носитель
?
Xй
S
Больной (j
XY
XY
5Нх) Мх]
Носитель Q
(хН или № —Ген гемофилии, сцеплен с Х-хромосомой
Рис. 6-22. Х-связанная передача гена гемофилии, представленная в типичной родословной
Нарушения гемостаза
229
(ОТ-ПЦР). Для каждого метода требуется небольшое количество крови или био-
птата ворсинок хориона, что позволяет поставить пренатальный диагноз
гемофилии на самой ранней стадии беременности (8-12 недель).
Гемофилия А редко встречается у женщин, ею страдают лишь девочки,
рожденные от гемофилика-мужчины и женщины-носителя гемофилии. Еще более
редкие случаи — абсолютная инактивация нормальной Х-хромосомы у
гетерозиготной женщины (носителя) или генетический дефект у женщин только с одной
Х-хромосомой, что наблюдается при синдроме Турнера.
Клиническая тяжесть и проявление кровоточивости. Тяжесть кровоточивости
при гемофилии (А или В) можно предсказать на основании уровня прокоагулянт-
ной активности фактора VIII или IX (OVIIIrC, Ф1Х:С; табл. 6-4).
У больных с уровнем прокоагулянтной активности указанных факторов < 1 %
спонтанные и тяжелые осложнения, связанные с кровотечением, проявляются
в раннем детстве (например, при обрезании). Обычно наблюдаются
кровоизлияния в суставы (гемартроз) и мышцы (гематомы). Они иногда приводят к тяжелым
последствиям, если больной не получит незамедлительного и адекватного
лечения. У больных с умеренной степенью патологии (OVHI:C или Ф1Х:С — 1-5 %
нормы) кровотечение развивается после незначительной травмы или
хирургического вмешательства, а также может быть спонтанным. У больных со слабой
степенью гемофилии (OVIILC или Ф1Х:С — 5-20 %) кровотечение возникает только
после травмы или хирургического вмешательства. Для всех гемофиликов
характерно чрезмерное кровотечение после экстракции зуба. Также часто наблюдаются
гематурия и желудочно-кишечное кровотечение, особенно если есть местное
поражение (язва, полип или воспалительный процесс)."
Посттравматическое или хирургическое кровотечение опасно для жизни при
всех видах гемофилии, включая слабые формы, поскольку приводит к
образованию обширных гематом. Они могут сдавливать нервы или вызывать тяжелую
окклюзию сосудов и гангрену. Даже незначительная травма ротовой полости или
языка способна стать причиной орофарингеального кровотечения и смерти от
удушья. Любая травма головы при слабом виде гемофилии считается опасной для
жизни и требует незамедлительного лечения. Спонтанное внутричерепное
кровотечение не характерно для гемофилии, но если оно происходит, то иногда
приводит к гибели больных с тяжелой гемофилией.
Псевдоопухоли длинных костей, таза и пальцев рук являются результатом
рецидивирующих субпериостальных кровоизлияний. Последние приводят к
деструкции костей, костным новообразованиям и возникновению склонности к
патологическим переломам.
ТАБЛИЦА 6-4. Лабораторные показатели и клинические проявления гемофилии
Снижение Клинические Симптомы кровоточивости
активности до (%) проявления
<1 Тяжелые В раннем детстве, спонтанные
1 -5 Умеренные После незначительной травмы или
хирургического вмешательства; могут быть спонтанными
5-20 Слабые Только после травмы или хирургического
вмешательства
230
Глава 6. Гемостаз и тромбоз
Лабораторная диагностика. Для гемофилии характерны следующие результаты
лабораторных тестов.
1. Пролонгированное АЧТВ, нормальное ПВ и нормальное ВК.
2. 1 :1 микст-тесты АЧТВ — корреляция при нулевом времени и температуре
37 °С после 2-часовой инкубации (выявляет дефицит OVIII и отсутствие
ингибиторов OVIII).
3. Активность OVIII:C — понижена или отсутствует.
4. Концентрация OVIII:Ar — нормальная или пониженная.
Лечение. Общие принципы лечения сводятся к возмещению недостатка фактора
концентратами ФVIII или введению десмопрессина, а также социальной и
психологической поддержке. Большинство больных и члены их семей получают
медицинскую помощь от групп специалистов-профессионалов (в центрах по лечению
гемофилии). Родителям советуют воспитывать детей-гемофиликов в обычном
режиме. Однако избегать травм надо с раннего детства (обкладывать кроватку
подушками, не давать игрушки с острыми углами). Ребенок может заниматься
только неконтактными видами спорта. Необходимо регулярное проведение
профилактической санации зубов. Членов семьи и пациента обучают
поддерживающей терапии в случае появления малейшего признака кровотечения.
Она включает применение местных средств и незамедлительное введение
концентрата OVIII, который должен храниться в холодильнике. Эти мероприятия
снижают частоту возникновения гемартрозов и случаев госпитализации.
Больные со слабой формой гемофилии могут применять десмопрессин (1-дезамино-
SD-аргинин-вазопрессин [DDAVP]) сразу после травмы или геморрагии интра-
назально либо в виде инфузий.
Лечение спонтанного кровотечения требует инфузий концентратов OVIII для
повышения OVIII:C в плазме до 30 % нормы, но при подготовке к обширному
хирургическому вмешательству его необходимо повысить до 100 %, что
обеспечит остановку посттравматического кровотечения. После так называемой
"ударной" (нагрузочной) дозы OVIII-концентрата каждые 12 ч вводят
поддерживающую дозу (половина нагрузочной) до тех пор, пока рана не заживет (7-10 дней).
Определение величины поддерживающей дозы требует частого контроля уровня
OVIILC, по крайней мере через день.
Количество фактора, которое нужно возместить, подсчитывается следующим
образом:
Ударная доза (единицы OVIII) = желаемый уровень OVIII x масса тела (кг).
Поддерживающая доза = половина ударной дозы
(доза OIX подсчитывается так же, но вводится 1 раз в день,
так как биологический период полураспада OIX (примерно 24 ч) больше,
чем фактора VIII (8-12 ч)).
Другие терапевтические принципы, учитываемые при лечении гемофилии.
1. Свести к минимуму вероятность развития хронического синовиита или
прогрессирующей артропатии; лечение острого гемартроза должно
включать неотложную и интенсивную заместительную терапию.
2. Лечение хронической артропатии должно включать физиотерапию,
шинирование суставов на ночь или вытяжение в сочетании с регулярным
введением концентрата фактора.
Нарушения гемостаза
231
3. Лечение хронического синовиита требует интенсивной заместительной
терапии, физиотерапии, а в тяжелых случаях хирургической синовэкто-
мии.
4. Лечение тяжелой артропатии может включать прием анальгетиков и
протезирование сустава.
5. В дополнение к профилактическому осмотру и лечению зубов перед
хирургическим вмешательством необходимо проведение инфузии концентратов
ФУШ или Ф1Х. Применение антифибринолитических препаратов,
например е-аминокапроновой кислоты (ЭАКК) или транексамовой кислоты,
уменьшает необходимое количество концентрата фактора и частоту
инфузии. Большинству больных требуется только 1 инфузия перед
хирургической операцией, но иногда инфузию повторяют после нее.
В настоящее время исследуется новый терапевтический подход к лечению
гемофилии — генная терапия. Ген вносится в человеческие фибробласты (трансфек-
ция), после чего эти клетки трансплантируются мышам, которые начинают
продуцировать человеческие факторы свертывания крови.
Осложнения терапии. У 10-20 % больных гемофилией образуются антитела
(ингибиторы), направленные против вводимого ФУШ. Это осложнение ведет
к трудно купируемому тяжелому кровотечению. У 70 % больных с ингибитором
ФУШ после введения фактора повышается титр ингибитора, который затем
снижается с различной скоростью. Такие пациенты не реагируют на терапию
концентратом человеческого ФУШ, поэтому у них необходимо использовать другие
терапевтические методы остановки кровотечения. У остальных 30 % больных
содержание ингибитора ФУШ в крови остается неизменным или минимально
повышается после инфузии ФУШ. Данные пациенты чувствительны к терапии
высокими дозами концентрата ФУШ.
Больных с низким уровнем ингибитора ФУШ (< 5 БЕ) лечат введением
свиного ФУШ в количестве, достаточном для повышения уровня ФУШ:С в плазме
до 30-50 ед/дл.
Больные с промежуточной величиной содержания ингибитора ФУШ (5-30
БЕ) получают инфузии человеческого Ф1Х, содержащие активированный ФХ
(чтобы обойти ФУШ), как терапию первого выбора, затем следуют или инфузия
свиного ФУШ, или введение очень высокой дозы концентрата человеческого
ФУШ. Если кровотечение не останавливается, проводят интенсивный плазмафе-
рез или экстракорпоральную иммуноадсорбцию.
Больных с высоким уровнем ингибитора ФУШ (> 30 БЕ) сначала лечат плаз-
маферезом, затем инфузиями свиного и человеческого ФУШ. Пациенты, не
чувствительные к такой терапии, иногда реагируют на введение высокоочищенного
концентрата, содержащего активированный ФУП.
Быстрого снижения титра антител удается достигнуть введением
иммуноглобулина. Этот продукт содержит высокую концентрацию антиидиотипических
антител, которые нейтрализуют ингибирующую активность. Эффективны также
иммуносупрессивные агенты, сокращающие продукцию антител, если их
использовать в сочетании с концентратом ФУШ. Однако их нельзя применять у
молодых пациентов, поскольку не исключены канцерогенные побочные эффекты.
В настоящее время для лечения используются также препараты рекомбинантно-
го ФУШ или высокоочищенные концентраты ФУШ.
232
Глава 6. Гемостаз и тромбоз
У лиц, не страдающих гемофилией, появление антител к OVIII может быть
связано с некоторыми заболеваниями, беременностью, приемом лекарственных
препаратов. Они могут также образоваться спонтанно в пожилом возрасте (см.
последующее обсуждение и табл. 6-10).
Помимо появления ингибиторов у больных иногда возникают и другие
проблемы. Осложнения, связанные с множественными трансфузиями концентратов OVIII
или Ф1Х либо других продуктов крови, при гемофилии могут включать такие
инфекции, как гепатиты В и С, инфицирование ВИЧ или цитомегаловирусом.
СПИД — в настоящее время основная причина смерти больных с тяжелой
гемофилией. Для предотвращения заражения вирусом проводится тщательное обследование
доноров, а также процедуры инактивации вирусов в продуктах крови. Кроме того,
существует рекомбинантный человеческий фактор VIII, не зараженный вирусами.
Другим осложнением, характерным для этих больных, является наркомания
в результате частого использования анальгетиков. К тому же и пациенты, и члены
их семей сталкиваются со многими социальными и психологическими
проблемами в связи с пожизненным дорогостоящим лечением (15 000-90 000 долл. США
в год) и снижающим качество жизни заболеванием.
Гемофилия В
Гемофилия В (болезнь Кристмаса) вызвана дефицитом фактора IX,
возникающим в результате Х-сцепленной мутации/делеции в гене Ф1Х. Гемофилия В,
подобно гемофилии А, вызывает кровотечения (гемартроз и гематомы), но частота
их в 5 раз ниже, чем при дефиците OVIII.
У больных с гемофилией В может синтезироваться нефункциональный Ф1Х,
а также не исключено полное отсутствие синтеза молекулы фактора IX.
Лабораторная диагностика. Для постановки диагноза используются следующие
результаты тестирования:
1. АЧТВ пролонгировано, а ПТ и ВК нормально.
2. АЧТВ (1:1 микст) — коррекция при нулевом времени и температуре 37 °С
после 2-часовой инкубации (это указывает на дефицит Ф1Х, а не на наличие
ингибитора Ф1Х).
3. Ф1Х:С — активность снижена или отсутствует.
4. Ф1Х:Аг — концентрация снижена или отсутствует.
Выявление носителя и антенатальную диагностику можно осуществлять
измерением уровня Ф1Х:Аг и Ф1Х:С в плазме. Однако более точным в этом отношении
является генный анализ в препаратах крови или антенатальная хорионбиопсия
с использованием ДНК-типирования и ОТ-ПЦР-метода.
Лечение. Принципы заместительной терапии дефицита Ф1Х аналогичны
таковым при гемофилии А. Для лечения тяжелых кровотечений используются
концентраты Ф1Х, для небольших кровотечений — свежезамороженная плазма.
Осложнения, связанные с лечением концентратом Ф1Х, включают тромбоз
(тромбоз глубоких вен, легочная эмболия, Д ВС) и вирусные инфекции.
Рекомендуется сочетанное назначение гепарина для профилактики тромбоза, особенно при
хирургических операциях. Применяемое в настоящее время осаждение этанолом и имму-
ноаффинная очистка концентратов Ф1Х сводит к минимуму риск возникновения
инфекции. Генная терапия дефицита Ф1Х пока находится на стадии исследования.
Нарушения гемостаза
233
Болезнь Виллебранда
Болезнь Виллебранда (БВ) относится к гетерогенной группе наследственных
(более 20 подвидов) и приобретенных нарушений фактора Виллебранда (ФВ).
Оба типа БВ клинически проявляются кровоизлияниями в кожу и слизистые
оболочек. БВ обусловлена количественной и качественной патологией ФВ.
Биосинтез ФВ. ФВ продуцируется эндотелиальными клетками и мегакариоци-
тами, где он хранится в органеллах. В эндотелиальных клетках фактор
Виллебранда хранится в тельцах Вейбла-Палада, а в тромбоцитах — в а-гранулах.
Знания о синтезе ФВ основаны главным образом на исследованиях ФВ в культуре
эндотелиальных клеток, в которых синтез ФВ инициируется в эндоплазматичес-
ком ретикулуме в виде пре-про-ФВ-молекул, которые с помощью дисульфидной
связи на С-концах образуют про-ФВ-димеры. Димер подвергается гликозилиро-
ванию и процессингу во время прохождения через аппарат Гольджи. При
формировании секреторной гранулы телец Вейбла-Палада про-ФВ-димер проходит
2 важные стадии: образование дисульфидных связей с N-концом с последующей
высокомолекулярной полимеризацией ФВ и отщепление прополипептида
(ФВ:Аг11). Превращение про-ФВ в зрелую форму осуществляется посредством
ферментативного расщепления парных аминокислот под действием фурина,
который отщепляет ФВ:Аг11 от зрелой формы ФВ. Обе формы (ФВ и отщепленный
прополипептид — ФВ:Аг11) хранятся в секреторных гранулах. После стимуляции
и ЭК, и тромбоциты выделяют ФВ и ФВ:Аг11. Функция ФВ:Аг11 неизвестна.
Структура и функция нормального ФВ. Зрелая форма ФВ выполняет две
основные функции в плазме: 1) обеспечивает адгезию тромбоцитов к коллагену
сосудистой стенки; ФВ служит мостом, связываясь с гликопротеинами lb и lib/
Ша (ГШЬ, ГППЬ/Ша) мембраны тромбоцита и дискретными лигандами
коллагена; 2) ФВ стабилизирует молекулу OVIII, увеличивая период ее
полусуществования и транспортируя в места активного образования гемостатической
пробки (рис. 6-21).
ФВ имеет высокую молекулярную массу (примерно 16 млн дальтон), но его
связывающие функции локализованы в отдельных доменах (рис. 6-23). Домен
Фактор Виллебранда
Дефект
OVIII
БВ БВ
Дефект Дефект
ИВ НА
Дефект
ГППЬ/Ша
| Домены ФВ ]
—щ-
Номер аминокислоты 272
Место связывания VIII
449728 9111114
Гепарин ГШЬ
Коллаген Коллаген
1744-1747
ГПНЬ/Ша
Рис. 6-23. Локализация аминокислот и мест связывания для реализации различных функций ФВ
234
Глава 6. Гемостаз и тромбоз
для связывания с OVIII расположен HaN-конце зрелого ФВ в области
аминокислотного остатка 272; места связывания с гликопротеином lb, гепарином и
коллагеном — на аминокислотах 449-728; и дополнительный домен связывания с
коллагеном — на аминокислотах 911-1114. Домен связывания с ГПНЬ/Ша в ФВ
локализован на аминокислотном участке 1744-1747.
Ген ФВ. Аутентичный ген ФВ (178 кб) находится на хромосоме 12, а псевдоген
(98 % гомология с геном ФВ) — на хромосоме 22. Получены данные, что псевдоген
у человека не функционирует. Установлена полная экзон-интронная структура
ФВ, и имеются усовершенствованные диагностические средства для определения
молекулярных дефектов некоторых подтипов БВ, за исключением типа I. Многие
молекулярные мутации в подвидах БВ (например, виды НА и ИВ) расположены
в пределах экзона 28. Последний кодирует (А1- и А2-последовательности)
участок ФВ, который взаимодействует с тромбоцитарным ГШЬ (домен А2), и часть
ФВ, содержащую сайт (сайты) районов полимеризации с N-конца (рис. 6-23).
Известные генетические изменения гена ФВ включают: делеции, вставки,
точечные мутации, альтернативные экзон-интронные сочленения и сайты
преждевременной остановки транскрипции.
Способ наследования. БВ представляет собой наиболее характерный
наследственный геморрагический диатез. Определено, что им страдает примерно 1 %
населения. Заболевание впервые было описано Эриком Виллебрандом в 1926 г.
у жителей Аландских островов (Финляндия). После этого стало известно, что
БВ — наследственное заболевание, передаваемое аутосомно-доминантным путем.
Мужчины и женщины поражаются в равной степени (рис. 6-24). Аутосомно-до-
минантный тип наследования подтвержден многими исследованиями, однако
некоторые варианты БВ (тип III и некоторые случаи типа ИС) наследуются ауто-
сомно-рецессивным путем.
Болезнь Виллебранда
Способ наследования
Слабая
форма БВ
I
С J—|—\^к <— Слабая форма БВ
Норма
| *ч^ *чНорма
Тяжелая форма БВ
Слабая
форма БВ
Рис. 6-24. Пример типичного аутосомио-доминантного наследования, наблюдаемого
при большинстве форм Б В
Нарушения гемостаза
235
Клинические проявления. У больных с БВ проявляются симптомы геморрагии
кожи и слизистых оболочек. Для них характерны носовые кровотечения, экхимоз,
меноррагия, послеродовое кровотечение, гематурия, чрезмерные кровотечения
(в анамнезе) после незначительной травмы или хирургического вмешательства
(тонзиллэктомия, удаление зуба).
Изучение большого числа больных-родственников с БВ показало, что
тяжелые симптомы кровоточивости наблюдались у 65 % фенотипически аномальных
(с лабораторными аномалиями) пациентов с БВ и у 25 % фенотипически
нормальных (без патологических лабораторных признаков) больных. Таким
образом, только данных анамнеза (ранее в сочетании с результатами лабораторных
тестов) недостаточно для обследования членов семей пациентов, страдающих БВ.
Лабораторное выявление БВ осложняется тем, что ФВ — острофазовый белок.
Его уровень в плазме возрастает во время стресса, физической нагрузки,
беременности, при пероральном применении противозачаточных средств, после
хирургического вмешательства. В этих ситуациях диагноз, основанный только на данных
лабораторного исследования, неточен, если вообще его можно поставить. В таких
случаях необходимы генетические маркеры аллельного наследования.
Лабораторная диагностика
Болезнь Виллебранда (тип Г). Результаты скрининг-тестов:
• пролонгированные ВК и АЧТВ;
• нормальное количество тромбоцитов и ПВ (рис. 6-19).
Тесты на ФВ:
• снижен антиген ФВ (ФВ:Аг);
• снижен кофактор ристоцетина (КР);
• снижена ристоцетининдуцированная агрегация тромбоцитов (рис. 6-20А
и тесты на БВ).
Болезнь Виллебранда (тип II). Результаты скрининг-тестов:
• пролонгированные ВК и АЧТВ;
• нормальное количество тромбоцитов и ПВ.
Тесты на ФВ:
• антиген ФВ может быть в той или иной степени снижен;
• КР значительно или в некоторой степени снижено;
• ристоцетининдуцированная агрегация тромбоцитов снижена или нормальна.
При БВ ИВ РИАТ (в половине нормальной концентрации) повышена, а при
других вариантах типа II отсутствует.
Подтверждающий тест: отклонения от нормы результатов электрофоретичес-
кого разделения мультимеров ФВ (рис. 6-2ЙД).
Классификация. БВ классифицируют по величине концентрации ФВ (антигена
ФВ) в плазме, активности КР и мультимерной структуре ФВ. Фактор
Виллебранда представляет собой белок-носитель для ФУШ:С. Поэтому также
снижается уровень ФУШ, что объясняет пролонгирование АЧТВ. Хотя эта
классификация удобна и действенна, однако есть случаи, когда диагноз Б В поставить трудно
из-за больших различий уровня ФВ в популяции (у людей с группой крови 0
уровень ФВ ниже, чем у имеющих группу крови А, В или АВ) и повышения
содержания ФВ при беременности, пероральном приеме противозачаточных средств,
после физической нагрузки, стресса, травмы и хирургического вмешательства.
Во время беременности у больных с тяжелой БВ уровень ФВ нормализуется,
236
Глава 6. Гемостаз и тромбоз
а после родов происходит его быстрое снижение, что может вызвать тяжелое
кровотечение. Соответственно, диагноз БВ во время беременности можно поставить
только на основании генетических маркеров, а не измерений количества ФВ и
определения его функции.
Многочисленные варианты БВ можно подразделить на 2 основных класса:
1) количественные нарушения ФВ (типы I и III), вызванные сниженным
синтезом; 2) качественные нарушения ФВ (типы ПА-Н, тип НВ, тип ИМ, тип IIN),
обусловленные патологической структурой ФВ (тип БВ называется также
классической БВ).
Описание отдельных подтипов представлено в табл. 6-5 и 6-6, а также на
рис. 6-20.
Тромбоцитарный тип БВ, или псевдо-БВ, можно дифференцировать от БВ
типа ПВ по результатам исследований со смешанными нормальными
тромбоцитами и плазмой. Дефект состоит в нераспознанной мутации ГШЬ мембраны
тромбоцита. Это приводит к повышению связывания мембраны тромбоцита и мульти-
меров ФВ с высокой молекулярной массой и их исчезновению из плазмы.
Варианты БВ типа НС-ПН в настоящее время классифицируются как тип НА.
БВ типа ПС являются единственным вариантом типа II, который наследуется
аутосомно-рецессивным путем. Молекулярная основа этих дефектов ФВ неясна.
При БВ типа ИМ дефект состоит в ГШЬ-связывающей петле ФВ, что
приводит к снижению аффинности ФВ к тромбоцитам. Тип ИМ отличается от типа ПА
наличием в плазме высокомолекулярных ФВ-мультимеров.
Тип IIN (вариант Норманди) характеризуется дефектом связывания ФУШ
с ФВ. Диагноз ставится по уровню ФУШ:С, который непропорционально ниже,
чем уровень ФВ; а также по способности ФУШ связываться с ФВ. Молекулярная
ТАБЛИЦА 6-5. Варианты болезни Виллебранда
Подтипы Концентрация КР РИАТ РИАТ (1/2) Мультимеры
антигена ФВ Функция (половина нор-
ФВ мальной
концентрации =
= 0,6 мг/мл)
Тип1 1(20-50%) 1(20-50%) 1 Отсутствует
Тип НА Кварьируется) HI 1 Отсутствует
Тип ИВ ±1 ±1 Норма Повышен
Тип III Отсутствует Отсутствует Отсутствует Отсутствует
БВ ±1 ±1 Норма Повышен
тромбо-
цитарного
типа
(псевдо-БВ)
Нормальные
Отсутствуют
высоко- и
среднемоле-
кулярные
фракции
Утрата
высокомолекулярных
фракций
И 1(или фракции
не выявлены)
Утрата
высокомолекулярных
фракций
Нарушения гемостаза
237
основа дефекта — нераспознанная мутация в N-конце зрелого ФВ, где ФВ
связывается с ФУШ.
Лечение. Лечение определяется тяжестью кровотечения и подтипом БВ.
Препараты выбора: десмопрессин, рекомбинантный концентрат ФВ, криопреципитат и
конъюгированные эстрогены.
Десмопрессин повышает уровень ФВ в плазме, способствуя его выделению
из телец Вейбла-Палада в эндоплазматическом ретикулуме. Препарат
применяется интраназально, а также внутривенно или подкожно. Реакция изменчива и
иногда ослабевает после нескольких доз в результате истощения запасов фактора.
Поэтому рекомендуется до хирургического вмешательства дать тройную дозу,
чтобы определить эффективность препарата. Десмопрессин наиболее
эффективен при БВ типа I, так как у больных продуцируется меньшее количество ФВ-
белка, но его структура нормальная. Лечение десмопрессином при БВ типа II не
рекомендуется, поскольку у больных синтезируется патологический ФВ, не
способный поддерживать адгезию тромбоцитов. Лечение десмопрессином может
быть даже опасно при БВ типа И. Препарат способствует выделению аномальных
мультимеров ФВ, которые вызывают агрегацию тромбоцитов и могут привести к
тромбоцитопении. Десмопрессин неэффективен и при БВ типа III, так как у этих
больных недостаточно депонированного ФВ. Таким пациентам необходима
заместительная терапия ФВ. Побочные действия при лечении препаратом возникают
редко, к ним относятся приливы, головная боль, гипертензия, задержка жидкости.
Эстрогены применяются для лечения незначительных осложнений при типе I.
Эти препараты повышают синтез ФВ.
Основной вид терапии при БВ — замещение фактором Виллебранда. Имеются
два лечебных препарата: криопреципитат и концентрат ФВ. Криопреципитату
ТАБЛИЦА 6-6. Клинические особенности и лечение вариантов БВ
Подтипы Частота Способ
Тип 1
Тип НА
Тип ИВ
Тип III
БВ
тромбо-
цитарно-
готипа
(%)
70-80
10-12
3-5
1-3
0-1
наследования
Аутосомно-
доминантный
Аутосомно-
доминантный
Аутосомно-
доминантный
Аутосомно-
доминантный
Аутосомно-
доминантный
Степень
тяжести
От слабой
до умеренной
От слабой
до умеренной
От слабой
до умеренной
Тяжелая
От слабой
до умеренной
Дефект
гена
Неизвестен
Не
определяется А2-
домен ФВ
Не
определяется А1-
домен ФВ
Делеции;
мутации;
Лечение
Десмопрессин,
эстрогены,
концентрат ФВ,
криопреципитат
Концентрат ФВ,
криопреципитат
Концентрат ФВ
Концентрат ФВ,
криопреципитат
экспрессия
мРНК
Мутации
ГП1Ь
тромбоцитов
Тромбоциты
238
Глава 6. Гемостаз и тромбоз
присущ значительный недостаток — передача вирусных инфекций, поэтому в
настоящее время он заменен концентратом ФВ, содержащим полный спектр муль-
тимеров ФВ, но применение которого связано с минимальным риском передачи
вирусной инфекции. Концентраты ФУШ не содержат интактного ФВ, а поэтому
их не рекомендуется использовать для лечения БВ,
Продолжительность и интенсивность лечения БВ еще не установлены точно,
как при гемофилии. Во время подготовки к серьезному хирургическому
вмешательству необходимо повысить уровень Ф В до 80 % и поддерживать его в течение
трех дней после операции на уровне 40 %, а последующие 5 дней — на уровне 30 %.
1 ед/кг концентрата ФВ повышает уровень в плазме до 24 ед/дл. В редких
случаях у больных с БВ образуются ингибиторы. Тогда возникает сильное
кровотечение и лечение дополняют плазмообменом, экстракорпоральной адсорбцией или
введением у-глобулина.
Во время беременности у больных с БВ типа I нет осложнений, связанных
с кровотечением, поскольку уровень ФВ повышается. После родоразрешения его
уровень быстро падает, соответственно, необходимо применять десмопрессин
или концентрат ФВ. У женщин с БВ типа III или типа НА и ИВ могут возникать
(интенсивные) кровотечения во время беременности, родов и после родов. Им
необходимо вводить концентрат ФВ.
Приобретенная болезнь Виллебранда. Приобретенная БВ — результат
пониженного синтеза ФВ, вторичной аномалии ФВ или выработки аутоантител.
Последние нарушают функцию ФВ или образуют с ним комплексы, тем самым ускоряя
его клиренс. Состояния, связанные с развитием БВ, представлены в табл. 6-7.
Лечение включает устранение основного заболевания и инфузии концентрата ФВ.
•При злокачественных заболеваниях нужно попытаться удалить опухоль или
провести химиотерапию. Больных с аутоиммунными нарушениями лечат
стероидами, иммуносупрессивными средствами и введением у-глобулина. Иногда этого
достаточно для остановки кровотечения. Если кровотечение не прекращается,
следует использовать концентрат ФВ.
Дефицит фактора XI
Дефицит ФХ1 — врожденная патология, наследуемая аутосомно-рецессивным
путем; наблюдается в основном у евреев-ашкенази.
У гетерозигот кровотечения обычно незначительны. У гомозигот с дефицитом
ФХ1 осложнений, связанных с кровоточивостью, мало. Но при травме или
хирургическом вмешательстве не исключено возникновение сильного
кровотечения с формированием гемартроза и гематомы. *
ТАБЛИЦА 6-7. Патологические состояния,
связанные с приобретенной болезнью Виллебранда
Гипертиреоз Пролапс митрального клапана
Опухоль Вильмса Ангиодисплазия
Миелопролиферативные заболевания Легочная гипертензия
Наследственная телеангиэктазия Множественная миелома
Лимфопролиферативные заболевания Аденокарцинома
Острый респираторный дистресс-синдром
Аутоиммунные нарушения (например, системная красная волчанка)
Нарушения гемостаза
239
Лабораторная диагностика. Результаты скрининг-тестов:
1. Пролонгированное АЧТВ; нормальное ПВ; нормальное количество
тромбоцитов; нормальное ВК.
2. АЧТВ 1:1 микст-тесты — коррекция до нормы; дефицит фактора.
3. Уровни ФХ1:С и ФХ1:Аг снижены или равны нулю.
Лечение. Замещение ФХ1 вливанием свежезамороженной плазмы или рекомби-
нантного ФХ1. Перед экстракцией зуба необходимо профилактическое введение
£-аминокапроновой кислоты или транексамовой кислоты (как при гемофилии).
Наследственные дефициты других факторов свертывания крови
Наследственные дефициты других факторов свертывания крови встречаются
редко (менее чем 1 на 1 млн человек). Путь передачи чаще всего аутосомно-рецес-
сивный. Дефицит ФХИ, ПК и ВМК не связан с возникновением кровотечения,
в отличие от дефицита других факторов. У людей с такой патологией выявлено
снижение функциональной активности факторов, уменьшение их содержания
в плазме или увеличение АЧТВ/ПВ.
Лабораторная диагностика
Дефицит ФХП, ПК и ВМК. Результаты скрининг-тестов:
1. Пролонгированное АЧТВ; нормальное ПВ и нормальное количество
тромбоцитов.
2. АЧТВ 1: 1 микст-тесты — коррекция АЧТВ; дефицит фактора.
3. Уровни ФХИ, или ПК, или ВМК снижены либо равны нулю.
Дефицит Ф VII. Результаты скрининг-тестов:
1. Пролонгированное ПВ; нормальное АЧТВ и нормальное количество
тромбоцитов.
2. Уровень фактора VII снижен или отсутствует.
Дефицит факторов V, X, II, I. Результаты скрининг-тестов:
1. Пролонгированные АЧТВ и ПВ; нормальное количество тромбоцитов.
2. АЧТВ и ПВ 1:1 микст-тесты — коррекция АЧТВ и ПВ;
3. Факторы V, X, И, I: уровень снижен или они отсутствуют.
Аномалии фибриногена. Наиболее тяжелой формой аномалии фибриногена,
ведущей к кровотечению, является афибриногенемия. Гипофибриногенемии, как
правило, бессимптомны. При дисфибриногенемиях увеличиваются АЧТВ и ПВ
на фоне нормальной концентрации фибриногена, но сниженной его активности.
Некоторые мутации, связанные с дисфибриногенемией, клинически
проявляются кровотечением, другие — тромбозом или спонтанными абортами. Большинство
дефектов наследуется аутосомно-доминантным путем.
Дефицит фактора XIII. Это редкое заболевание с аутосомно-рецессивным
типом наследования, которое проявляется тяжелым кровотечением, гемартрозом,
гематомами, плохим заживлением ран и спонтанными абортами. Результаты
скрининг-тестов на ПВ, АЧТВ, количество тромбоцитов, ВК и фибриноген в
пределах нормы. Повышенная растворимость кровяного сгустка в 5 М мочевине или
1 % монохлоруксусной кислоте указывает на наличие дефицита ФХШ, что
подтверждается при определении ФХШ:Аг и ФХШ:С.
Лечение включает проведение заместительной терапии свежезамороженной
плазмой.
240
Глава 6. Гемостаз и тромбоз
Приобретенные нарушения факторов свертывания крови
Приобретенные нарушения системы свертывания крови наблюдаются чаще, чем
наследственные, и, как правило, обусловлены множественным дефицитом факторов
свертывания. В основе его возникновения лежат различные механизмы (табл; 6-8).
Дефицит витамина К
Витамин К поступает с пищей (с зелеными овощами) и образуется в организме
за счет бактериального синтеза в кишечнике. Это жирорастворимый витамин,
содержание которого зависит от активности липаз поджелудочной железы, наличия
желчи, всасывающей способности кишечника (поступление в кровообращение) и
степени утилизации гепатоцитами. Дефицит витамина К возникает в результате
неадекватного питания, заболеваний поджелудочной железы, закупорки
желчных путей, нарушенного всасывания, лечения антибиотиками, а также при
лечении антикоагулянтами.
Витамин К необходим в заключительных стадиях синтеза факторов X, IX, VII
и II, ПС и ITS для проявления их биологической активности. Окисление витамина К
в его эпоксидную форму существенно для у-карбоксилирования остатков глутами-
новой кислоты на этих витамин К-зависимых белках. Данный посттрансляционный
процесс включает карбоксилирование у-углерода нескольких остатков глутамино-
вой кислоты на N-конце белковых молекул. Затем у-карбоксилированная глутами-
новая кислота связывается с кальцием и посредством его прикрепляется к фосфоли-
пидным рецепторам клеточных мембран (например, тромбоцитов, ЭК). Данный
процесс способствует активации системы свертывания крови. Во время карбокси-
лирования витамин К окисляется в эпоксид и затем снова восстанавливается в
активную форму редуктазой (рис. 6-25). Перорально принимаемые антикоагулянты
(кумарины) ингибируют восстановление эпоксида витамина К, мешая
эффективному превращению витамина К в его активную ферментную форму, и ограничивают
действие карбоксилазы. У больных, получающих кумарины, образуются
неактивные витамин К-зависимые факторы, а кровь имеет пониженную свертываемость.
У детей и взрослых с недостатком витамина К возникают кровотечения,
которые могут быть сильными и обычно проявляются в виде экхимоза, гематом,
желудочно-кишечных кровотечений, гематурии. Дефицит витамина К следует
предполагать при наличии геморрагического синдрома (сочетанных заболеваний)
и отклонения показателей крови от нормы. N
ТАБЛИЦА 6-8. Приобретенные нарушения свертывания крови
Дефицит витамина К
Мальабсорбция витамина К (целиакия, спру)
Обструкция желчевыводящих путей
Лечение антагонистами витамина К (кумарины)
Лечение антибиотиками
Геморрагическая болезнь новорожденного
Приобретенный дефицит ФХ
Болезнь печени
Массивный трансфузионный синдром
Приобретенные ингибиторы свертывания крови
Диссеминированное внутрисосудистое свертывание (ДВС)
Фибринолитические состояния
Нарушения гемостаза
241
Роль витамина К в образовании активных факторов
свертывания крови (факторы X, IX,Vll и II, ПС, nS)
Неактивный профермент
(факторы X, IX.VII и II, ПС, ПБ)
Активный фермент к
(факторы X, IX,VU и И, dp, HS) I
Остатки
глутаминовой
кислоты
COOH
Восстановленная
форма витамина К
|Витамин К-
эпоксид
COOH
Глутаминовая
кислота, карбо-
ксилированная
в у-позиции
Витамин К-
редуктаза
А0
Кумарины
Активация
свертывания
Рис. 6-25. Способы влияния витамина К на активацию факторов X, IX, VII, II, ПС и IIS
(сокращения см. в тексте)
Результаты скрининг-тестов:
1. Пролонгированные АЧТВ и ПВ.
2. Нормальное количество тромбоцитов.
3. АЧТВ и ПВ 1:1 микст-тесты — коррекция АЧТВ и ПВ.
4. Пониженная активность факторов VII, IX, X, II.
5. Нормальная концентрация антигенов факторов VII, IX, X, И.
Наличие нефункционирующих витамин К-зависимых факторов подтверждает
дефицит витамина К. Клинически диагноз дефицита витамина К можно
поставить на основании положительного эффекта назначения витамина К,
сопровождающегося нормализацией ПВ и АЧТВ.
Лечение включает коррекцию основного нарушения, введение витамина К,
переливание свежезамороженной плазмы при сильном кровотечении. С целью
профилактики витамин К принимают перорально. ПВ отчасти улучшается через
7 ч благодаря восстановлению OVII (T1/2 — 4—7 ч). Витамин К вводят ежедневно
в течение 3 дней, так как другие витамин К-зависимые факторы имеют более
длительный период полусуществования (Ф1Х — около 1 сут; ФХ — 2 сут; ФП —
3 сут). Через 3 дня ПВ и АЧТВ полностью нормализуются.
Геморрагическая болезнь новорожденного
Дефицит витамина К у новорожденного — иной синдром. Геморрагическая болезнь
возникает на 2 -4-й день жизни и обусловлена пониженным синтезом витамин
К-зависимых факторов свертывания крови. Причины дефицита витамина К —
уменьшение его запасов и функциональная незрелость печени, отсутствие бактериально-
242
Глава 6. Гемостаз и тромбоз
го синтеза в кишечнике, низкое количество витамина К в грудном молоке и, редко,
прием лекарственных препаратов матерью (аспирин, фенитоин и др.).
Результаты скрининг-тестов:
1. Пролонгированные АЧТВ и ПВ.
2. Нормальное количество тромбоцитов.
3. АЧТВ и ПВ 1:1 микст-тесты — коррекция АЧТВ и ПВ.
4. Пониженная активность факторов VII, IX, X, И.
Всем детям, за исключением тех, у кого подозревают дефицит Г-6-ФДГ
(опасность возникновения гемолиза эритроцитов [глава 3]), назначают витамин Kt
(фитоменадион). Детям с кровотечением вводят витамин К и переливают
свежезамороженную плазму.
Приобретенный дефицит фактора X
Дефицит фактора X наблюдается при амилоидозе вследствие избирательной
адсорбции ФХ амилоидным белком. Геморрагический синдром у таких пациентов
проявляется экхимозами, гематомами, желудочно-кишечными и мочеполовыми
кровотечениями.
Результаты скрининг-тестов:
1. Пролонгированные АЧТВ и ПВ.
2. Нормальное количество тромбоцитов.
3. АЧТВ и ПВ 1:1 микст-тесты — коррекция АЧТВ и ПВ.
4. Уровни ФХ:С и ФХ:Аг снижены или равны нулю.
Болезни печени
Коагулопатия при болезнях печени ассоциируется с кровоточивостью, в ряде
случаев — тяжелой. Она возникает в результате множественных нарушений
гемостаза (табл. 6-9). Нарушенный синтез витамин К-зависимых факторов в данном
случае опосредован внутрипеченочным холестазом, ухудшением утилизации
витамина К, снижением его всасывания, нарушениями диеты. Снижение синтеза
других факторов, не зависящих от витамина К (факторы V, XI, XII и XIII,
фибриноген), также способствует возникновению кровоточивости. Кроме того, у
пациентов с болезнями печени наблюдается хроническая активация свертывания
крови и фибринолитической системы; регуляторные механизмы (естественные
ингибиторы) последней также нарушены.
ТАБЛИЦА 6-9. Коагулопатия при заболеваниях печени
Пониженный синтез факторов свертывания крови:
i Витамин К-зависимые факторы X, IX, VII, II
| Витамин К-независимые факторы V, XI, XII, I, ПК, ВМК
Активация факторов свертывания крови (диссеминированная внутрисосудистая
коагулопатия)
Дисфибриногенемия
Тромбоцитопения и нарушение функции тромбоцитов
Пониженный синтез и повышение потребления ингибиторов
Повышенный фибринолиз (ГГАП, |а2-антиплазмин)
Нарушения гемостаза
243
Снижается количество тромбоцитов, возникает их дисфункция. Первое
происходит из-за уменьшения продукции костным мозгом. В результате спленомега-
лии увеличивается секвестрация тромбоцитов селезенкой, а в печени и селезенке
усиливается их разрушение. Механизмы, лежащие в основе дисфункции
тромбоцитов, неясны. Предполагают, что при повышенном фибринолизе аномальный
фибриноген, который связывается с мембраной тромбоцитов, нарушает их
реактивность. У некоторых больных возникает дисфибриногенемия (пониженное
содержание сиаловой кислоты в фибриногене), что также обусловливает
возникновение дефекта гемостаза.
Ускоренный фибринолиз — результат повышенного уровня ТАП в плазме
в связи с нарушением печеночного клиренса, отсутствием соответствующего
прироста содержания ингибиторов активатора плазминогена (ИАП) и снижением
синтеза сс2-антиплазмина. В ответ на активацию системы свертывания крови
возникает гиперфибринолиз.
При хронических заболеваниях печени активация свертывания крови, вероятно,
является результатом выделения тканевого фактора некротизированными
клетками, а также нарушения клиренса активированных факторов свертывания
и снижения синтеза основных регуляторных белков (AT III, ПС, ПБ, ГПИ). При
остром и тяжелом поражениях гепатоцитов острая активация наслаивается
на хроническую активацию свертывания. Полагают, что причинами этого
являются: изменение выделительной функции некротизированных клеток, выделяющих
тканевый фактор, другие прокоагулянты, интерлейкины (ФИО, ИЛ-1),
эндотоксины, а также аккумуляция активированных факторов в расширенной портальной
системе с низкой скоростью кровотока. Все это приводит к образованию тромбина
и плазмина за счет активации как свертывания крови, так и фибринолитических
систем. Наличие тромбина и плазмина in vivo способствует тромбозу в микроцир-
куляторном русле и тяжелому нарушению гемостаза, так называемому синдрому
диссеминированного внутрисосудистого свертывания (ДВС).
Характерные показатели лабораторных исследований: пролонгированное ПВ;
несколько увеличенное АЧТВ; количество тромбоцитов — 80 000-120 000/мкл;
низкий уровень фибриногена; очень высокий уровень ПДФ. Специфические
факторы: снижено содержание факторов X, IX, VII, II (витамин К-зависимых);
также — факторов V, XI, XII, КР, НК, XIII и фибриногена. OVIILC нормальный.
ФВ:Аг повышен (реактант острой фазы); плазминоген снижен; AT III, ПС, nS
и а2-антиплазмин снижены; а2-М и ИАП повышены.
Лечение заключается в коррекции дефицита витамина К. Витамин К вводится
парентерально, и степень коррекции ПВ определяется через 6-8 ч.
Заместительная терапия включает трансфузию больших объемов свежезамороженной плазмы
(6-8 доз), которая содержит все факторы свертывания и ингибиторы,
способствующие остановке кровотечения. При сильном кровотечении (во время или после
хирургического вмешательства) для обеспечения гемостаза необходимо вводить
СЗП каждые 6-12 ч. В некоторых случаях требуются дополнительные меры,
например заменные трансфузии, применение концентратов протромбин-комплекса
(активированный ФХ может вызвать ДВС), трансфузии тромбоцитов, введение
концентратов AT III.
Больным с тяжелыми дефектами функции Тромбоцитов можно вводить дес-
мопрессин. Перед удалением зубов пациентам назначают антифибринолитичес-
кие препараты, например ЭАКК.
244
Глава 6. Гемостаз и тромбоз
Подготовка к большой хирургической операции, как правило, включает: 1)
лечение введением витамина К и свежезамороженной плазмы; 2) трансфузии
тромбоцитов для обеспечения их количества на уровне приблизительно 100 000/мкл.
Трансфузии тромбоцитов при подготовке к эндоскопии, торакоцентезу, лапаро-
центезу и люмбальной пункции показаны больным с количеством тромбоцитов
< 50 000/мкл.
Синдром массивных трансфузий
Синдром массивных трансфузий, являющийся приобретенным нарушением
кровоточивости, развивается после переливания больших объемов консервированной
крови. Возникновению геморрагического диатеза способствуют многие факторы.
Тяжесть кровотечения зависит от количества перелитой крови и скорости
переливания, от времени хранения гемокомпонентов, атакже от предшествующей и
сопутствующей патологии.
1. Количество перелитой крови и скорость переливания. Если больные
получают более 10 доз консервированной крови за период менее 24 ч, у них может
возникнуть кровотечение. После переливания 10 доз кр9ви количество
тромбоцитов иногда снижается на 50 %. В консервированной крови
количество тромбоцитов снижается и происходят качественные изменения этих
клеток, поэтому чем больше скорость переливания, тем тяжелее тромбоци-
топения и сильнее кровотечение. Переливание эритоцитарной массы и
плазмозаменяющих растворов (декстранов) сопровождается разведением
тромбоцитов и факторов свертывания крови. Кроме того, декстраны,
покрывая мембраны тромбоцитов, ухудшают функцию этих клеток, что
усугубляет тяжесть кровотечения.
2. Время хранения продуктов крови. После 24-часового хранения при
температуре 4 °С происходит агрегация тромбоцитов и утрата ими своих функций.
Некоторые факторы свертывания крови теряют активность медленно.
Содержание 2-х лабильных факторов, VIII и V, снижается до 80 %
первоначальной величины. Активация факторов свертывания крови,
микроагрегаты тромбоцитов, пораженные или активированные лейкоциты, бактерии,
вирусы, попадая в кровь больного, могут инициировать или усиливать Д ВС.
3. Предшествующие или сопутствующие клинические нарушения. Чрезмерная
потеря крови приводит к снижению числа тромбоцитов, содержания
факторов свертывания и естественных ингибиторов. Гемостатический дефект в
результате массивных трансфузий усиливается, когда нарушается образование
тромбоцитов или факторов свертывания. Это происходит у пациентов с
заболеваниями печени, идиопатической тромбоцитопенией и ДВС в анамнезе.
При лабораторном обследовании выявляются пролонгированные ПВ и АЧТВ
с коррекцией в 1: 1 микст-тесте. Количество тромбоцитов снижено.
Гемостатический дефект предотвращают введением 2-х доз
свежезамороженной плазмы и 5-ти единиц тромбоцитов на каждые 10 или больше доз перелитой
крови. Может потребоваться использование препаратов кальция, поскольку
раствор цитрата, фосфата и глюкозы (декстрозы), применяемый в качестве
антикоагулянта для продуктов крови, способен связывать кальций в крови у пациента и
вызвать гипокальцемию. Лечение кровотечения включает переливание
свежезамороженной плазмы и тромбоцитов (помимо терапии основного заболевания).
Нарушения гемостаза
245
Приобретенные ингибиторы свертывания крови
Приобретенные ингибиторы свертывания крови, известные также как
циркулирующие антикоагулянты, представляют собой антитела, непосредственно инги-
бирующие факторы свертывания крови или их реакции. Ингибиторы могут
появляться в результате трансфузии белков плазмы, например у больных гемофилией,
или возникать спонтанно у пациентов без исходного нарушения гемостатичерких
механизмов. Большинство приобретенных ингибиторов свертывания крови
вызывает кровотечение, а некоторые — тромбоз (волчаночный антикоагулянт/анти-
фосфолипидные антитела).
Приобретенные ингибиторы ФУШ. Ингибиторы ФУШ, образующиеся у
больных гемофилией А, были описаны ранее. Спонтанно возникающие ингибиторы
ФУШ обнаруживаются с равной частотой у мужчин и женщин,
преимущественно в возрасте после 60 лет. У половины пациентов нет сочетанных заболеваний.
У остальных выявляются иммунологические нарушения, коллагенозы,
злокачественные новообразования или лекарственная аллергия. Ингибиторы ФУШ
могут возникать у молодых женщин после родов (примерно в 7 % случаев) без
каких-либо основных заболеваний. Общее спонтанное исчезновение ингибиторов
ФУШ наблюдается у 38 % больных, а летальность в связи с кровотечением
достигает 22 %. Нарушения, вызванные ингибиторами ФУШ, представлены в табл. 6-10.
Ингибитор появляется при рождении первого или второго ребенка. Склонность
к кровотечению становится очевидной сразу же или через 2-5 месяцев.
Клиническое течение различно, но ингибитор исчезает у большинства пациентов
спонтанно через 12-18 месяцев. Причина такой послеродовой реакции неясна.
Подобно ингибиторам, образующимся у гемофиликов, спонтанно
приобретенные ингибиторы ФУШ являются IgG-антителами, которые не фиксируют
комплемент и видоспецифичны. Их эффект зависит от температуры, поэтому
для обнаружения ингибирующих свойств требуется инкубирование в течение 1-
2 ч при 37 °С. Однако есть разница в кинетике их реакции. При лабораторном
обследовании выявляется пролонгированное АЧТВ без коррекции в 1 : 1 микст-
тесте. Снижена активность ФУШ:С, наблюдаются различные уровни
содержания ингибитора ФУШ. В то время как линейная взаимосвязь между
концентрацией ингибитора и количеством инактивированного ФУШ характерна для
страдающих гемофилией; при появлении спонтанных ингибиторов такая связь
отсутствует. У большинства пациентов активность ФУШ в плазме не
определяется. Полагают, что ФВ может частично блокировать эпитопы ФУШ за счет
взаимодействия с ингибитором.
ТАБЛИЦА 6-10. Нарушения и состояния, связанные с ингибиторами 0VIII
Иммунологические Злокачественные Прочие
расстройства заболевания
Системная красная волчанка Моноклональные гаммапатии После родов.
Ревматоидный артрит Лимфопролиферативные
Герпетиформный дерматит нарушения
Воспаление кишечника Миелофиброз
Рассеянный склероз Солидные опухоли
Аллергия к пенициллину (плоскоклеточный рак)
246
Глава 6. Гемостаз и тромбоз
Лечение аналогично описанному для больных гемофилией, имеющих
ингибиторы ФУШ.
Длительное лечение пациентов с основными заболеваниями включает также
иммуносупрессию продукции аутоантител.
Приобретенные ингибиторы ФУ. Они появляются у больных с дефицитом ФУ,
однако это наблюдается редко. В большинстве случаев ингибиторы ФУ
появляются у пожилых пациентов, недавно перенесших хирургическое вмешательство,
лечение антибиотиками или инфекции (туберкулез), а также не имеющих явных
признаков заболевания. Часть таких ингибиторов исчезает спонтанно. Редко
ингибиторы ФУ образуются при миелофиброзе, множественной миеломе, амилои-
дозе, раке прямой кишки. Ингибиторы ФУ относятся к IgG, IgG-IgM или IgG-IgA.
При лабораторном обследовании выявляются пролонгированные ПВ и АЧТВ
без коррекции в 1:1 микст-тесте. Активность ФУ отсутствует или снижена, титр
ингибитора ФУ повышен.
Метод выбора лечения тяжелых кровотечений при наличии ингибитора ФУ —
трансфузии тромбоцитов. Тромбоциты содержат ФУ в сс-гранулах, поэтому
образующийся комплекс тромбоцит-ингибитор ФУ удаляется из кровообращения
РЭС-клетками. ФУ, избежавший воздействия ингибитора, и внутриклеточный
тромбоцитарный ФУ в данной ситуации способны обеспечить гемостаз.
Приобретенные ингибиторы ФП возникают при системной красной волчанке,
циррозе печени и протезировании сердечных клапанов (у больных после
операции). Лабораторное обследование выявляет пролонгированные ПВ и АЧТВ без
коррекции в 1:1 микст-тесте; увеличение тромбинового времени (глава 11) в
связи с действием ингибитора ФП; снижение активности ФП (или ее отсутствие);
высокий титр ингибитора ФИ.
Приобретенные ингибиторы ФУП — это аутоантитела (IgG), становящиеся
причиной тяжелых кровотечений у больных с моноклональной гаммапатией,
тяжелой апластической анемией либо у пациентов без сопутствующей патологии.
При лабораторном обследовании выявляются пролонгированное ПВ без
коррекции в 1 : 1 микст-тесте; АЧТВ нормальное; активность ФУП отсутствует;
повышен уровень ингибитора ФУП.
Приобретенные ингибиторы Ф1Х встречаются чрезвычайно редко: при
системной красной волчанке и после родов. В большинстве случаев исчезают через
1-7 месяцев.
Приобретенные ингибиторы ФХ1 и ФХП также наблюдаются при системной
красной волчанке и лечении хлорпромазином. За исключением больных с
наследственным дефицитом ФХ1, у которых развиваются антитела к ФХ1 после
трансфузии плазмы, у остальных пациентов ингибиторы к факторам контактной фазы
не появляются.
Приобретенные аутоантитела к фибриногену (I), к факторам стабилизации
фибрина (ФХШ) и полимеризации фибрина также наблюдаются при системной
красной волчанке, моноклональной гаммапатии и после лечения изониазидом.
Последний нарушает стабилизацию фибрина (ингибитор ФХШ). Ингибиторы
ФХШ подразделяются на 3 типа: типа I направлены против активации ФХШ, но
не нарушают трансмидазной активности ФХШ; типа II нарушают трансмидаз-
ную активность; типа III препятствуют взаимодействию ФХШ с фибриновым
субстратом. Появление спонтанных ингибиторов ФХШ (фибринстабилизирую-
Нарушения гемостаза
247
щего фактора) может привести к тяжелым кровотечениям, в том числе с
летальным исходом. Лечение направлено на терапию основного заболевания, отмену
этиологически значимых лекарственных препаратов, переливание плазмы.
Приобретенные гепариноподобные антикоагулянтЫу которые являются
спонтанно возникающими ингибиторами этого типа, наблюдаются у больных с плазмоци-
тарными злокачественными новообразованиями, различными опухолями, а также
у пациентов с аденокарциномой или раком предстательной железы, получающих
терапию сурамином. У некоторых больных развиваются тяжелые геморрагические
осложнения. Все ингибиторы в плазме проявляют свойства глюкозаминогликанов
и нейтрализуются протамин сульфатом или гепариназой. При тяжелых
кровотечениях эффективным лечебным средством считается протамин сульфат.
Диссеминированное внутрисосудистое свертывание (ДВС)
Острое ДВС — приобретенное тромбогеморрагическое нарушение, возникающее
в результате чрезмерного образования тромбина и плазмина в периферической
крови. ДВС — всегда явление вторичное, следствие основного патологического
процесса, способствующего активации системы свертывания крови и генерации
тромбина. Разнообразными способами тромбин вызывает распространенное
отложение фибрина в микроциркуляторном русле с потреблением тромбоцитов
(агрегация тромбоцитов) и специфических факторов свертывания крови. Плазмин,
обеспечивающий протеолиз фибрина и факторов свертывания, еще более влияет
на развитие геморрагических осложнений ДВС (рис. 6-26).
Синонимы ДВС — коагулопатия потребления, синдром дефибринирования,
внутрисосудистое свертывание с вторичным фибринолизом,
тромбогеморрагическое нарушение потребления. ДВС возникает при заболеваниях,
способствующих выделению в кровообращение прокоагулянтов, которые вызывают
распространенное поражение эндотелия или стимулируют тромбоциты/макрофаги.
Острое ДВС наблюдается при состояниях, представленных в табл. 6-11. ДВС
может быть хроническим или локальным.
Основные пути патогенеза ДВС (рис. 6-26):
1. Повреждение тканей. Поступление прокоагулянтов (например, тканевого
фактора) в кровообращение вызывает активацию системы свертывания
крови прежде всего за счет активации <DVII. Этот вид повреждений
наблюдается при акушерских осложнениях, злокачественных заболеваниях, после
травмы, хирургических вмешательств, при некрозе печени, внутрисосудис-
том гемолизе, укусах некоторых змей и ряде инфекций (малярии).
2. Повреждение эндотелия способствует обнажению коллагена в субэндоте-
лиальной зоне, что активирует факторы свертывания контактной фазы,
а также тромбоциты и приводит к избыточному образованию тромбина.
Меняются физиологические свойства эндотелия (в основном, от антикоагу-
лянтных к сильным прокоагулянтным), что сопровождается непрерывной
активацией свертывания крови. Обширное повреждение эндотелия может
быть вызвано бактериями и продуктами их жизнедеятельности
(эндотоксином; грамотрицательными бактериями, включая менингококки; грамполо-
жительными бактериями), некоторыми вирусными инфекциями (вирусом
герпеса, который вызывает фульминантную пурпуру), тяжелыми ожогами,
острым поражением легких, нарушениями обмена веществ.
248
Глава 6. Гемостаз и тромбоз
ТАБЛИЦА 6-11. Патологические состояния, связанные с острым
диссеминированным внутрисосудистым свертыванием
Инфекции
Грамотрицательная септицемия
Грамположительная септицемия
Пятнистая лихорадка Скалистых гор
Тяжелая малярия {P. falciparum)
Брюшной тиф
Вирусные инфекции
(фульминантная пурпура)
Хирургическая травма
Экстракорпоральное кровообращение
Размозжение тканей
Черепно-мозговые травмы
Злокачественные заболевания
Острый промиелоцитарный лейкоз
Другие острые лейкозы (ОМЛ, ОММЛ)
Муцинсекретирующая аденокарцинома
Синдром лизиса опухоли, лимфомы
Реакции гиперчувствительности
Трансфузионные реакции
Внутрисосудистый гемолиз
Анафилактический шок
Метаболические нарушения
Гипер/гипотермия
Острая гипоксия
Гипотензия
Акушерские осложнения
Разрыв плаценты
Эмболия околоплодными водами
Эклампсия
Задержка плаценты
Повреждение тканей
Прочие
Острый некроз печени
Змеиный яд
Тяжелые ожоги
Острый респираторный дистресс-
синдром взрослых
ОМЛ — острый миелогенный лейкоз; ОММЛ — острый миеломоноцитарный лейкоз.
3. Поражение тромбоцитов/макрофагов. Непосредственная стимуляция
тромбоцитов, приводящая к образованию внутрисосудистых тромбоцитар-
ных микроагрегатов, также способна провоцировать ДВС. Вирусы,
некоторые бактерии, эндотоксин, иммунные комплексы могут непосредственно
или за счет стимуляции макрофагов (например, фактор активации
тромбоцитов — ФАТ) повреждать тромбоциты.
Активация системы свертывания крови любого происхождения приводит к
чрезмерному образованию тромбина и плазмина, действие которых обусловливают
проявления ДВС, представленные в табл. 6-12.
Патогенез ДВС
Повреждение
эндотелиальной
клетки
(ТФ.ФАТ)
Стимуляция
тромбоцитов
и макрофагов
(АДФ, ФНТ, ФАТ)
Повреждение
ткани
(ТФ)
^ГТ~У
Активация
свертывания крови
Активация фибрино-
литической системы
► Тромбин ^J
► Плазмин^!
Рис. 6-26. Упрощенная схема патогенеза ДВС
Нарушения гемостаза
249
ТАБЛИЦА 6-12. Действие тромбина и плазмина
Тромбин
Плазмин
Образование фибрина -*
снижение Ф1 и ФН
Агрегация тромбоцитов -►
снижение количества тромбоцитов
Активация Ф\/ и OVIII -+
снижение уровня OV и OVII
Активация ФХШ -»•
снижение уровня ФХШ
Активация системы ПС/ПБ -*
снижение уровня ПС/nS
Деградация фибрина -*
значительное повышение ПДФ
Деградация фибриногена —
снижение уровня Ф1
Протеолиз Ф\/ и Ф\ЛП -►
снижение их содержания
Протеолиз ФВ, ФХН, ФХ1 -*
снижение их содержания
Протеолиз ФХШ -*
снижение уровня ФХШ
Активация фибринолитической системы Изменения в ГП тромбоцитарной мембраны
Образование тромбина в ранних стадиях ДВС способствует образованию
больших растворимых комплексов фибрин-фибриноген и развитию фибриновых
микротромбов, которые вызывают обструкцию микроциркуляторного русла
и полиорганную недостаточность. В этой стадии у больных появляются признаки
поражения органов, характеризующиеся гипосенсорными расстройствами,
снижением выделения мочи, ухудшением оксигенирования крови, отклонением
от нормы показателей печеночных проб, изменениями гемостаза.
В более поздних стадиях .ДВС кровотечение вызвано тромбининдуцирован-
ным потреблением тромбоцитов и истощением факторов I, II, V, VIII, XIII, а
также фибринолизом. При продолжающейся активации системы свертывания крови
увеличивается потребление естественных ингибиторов активных сериновых про-
теаз (AT III, ПС, US и TFPI), что приводит к формированию тромбов. Тромбин
также способствует экспрессии тромбомодулина эндотелиальными клетками —
предпосылке активации системы ПС/ПБ (рис. 6-11). До истощения
активированный ПС ингибирует активированные факторы V и VIII, способствуя развитию
геморрагии. Кроме того, АПС подавляет выделение ИАП посредством
отрицательной обратной связи с ЭК, в результате выделение ТАП не контролируется
(рис. 6-11). Повреждение или стимуляция ЭК эндотоксином и другими
медиаторами воспаления усиливает выделение ТАП, тем самым стимулируя
превращение плазминогена в плазмин (рис. 6-13). Поэтому одновременно и
последовательно развиваются тромбоз и склонность к кровотечению.
Образование плазмина при ДВС усугубляет эффект тромбина, приводя
к дальнейшему-снижению содержания факторов V, VIII, фибриногена и ФХШ
и индуцированию процесса лизиса, на что указывают высокие уровни продуктов
деградации фибрина (ПДФ). Протеолиз ФВ и модификация гликопротеиновых
рецепторов мембран тромбоцитов плазмином предотвращают взаимодействие
между тромбоцитами и сосудом и нарушают первичный гемостаз. В последней
стадии ДВС у больных появляется геморрагический некроз тканей и различные
признаки кровотечения.
Клиническая оценка больных с острым ДВС. У больных с острым ДВС
обнаруживаются признаки как микрососудистого тромбоза, так и тяжелого
геморрагического диатеза. Лихорадка наблюдается у 58 %, гипотензия — у 50 %, микроан-
гиопатическая гемолитическая анемия — у 15 % больных.
250
Глава 6. Гемостаз и тромбоз
Признаки тромбов в микрососудистом русле.
Кожа: акроцианоз; синие пальцы ног, ишемия, поверхностная гангрена.
Неврологические: многоочаговые признаки, притупление чувствительности,
бред или кома.
Легочные: дыхательная недостаточность, острый респираторный дистресс-
синдром.
Желудочно-кишечные: стрессовые язвы.
Гематологические: внутрисосудистый гемолиз (фрагментированные
эритроциты), приводящий к гипербилирубинемии (бледно-желтая кожа и склеры).
Почечные: олигурия, азотемия, кортикальный некроз.
Признаки кровотечения при остром ДВС.
Кожа: петехии, пурпура, экхимоз, гематомы, кровотечение в местах венепункции.
Неврологические: внутримозговое кровотечение.
Желудочно-кишечные: массивное кровотечение.
Слизистые оболочки: носовые кровотечения, кровоточивость десен, маточные
кровотечения.
Почечные: гематурия.
Лабораторный диагноз острого ДВС ставят по результатам тестов, позволяющих
определить истощение тромбоцитов, потребление факторов свертывания крови и
вторичный фибринолиз. В табл. 6-13 перечислены исследования, которые обычно
назначают при диагностике ДВС.
При ДВС содержание OV и ФУШ снижено. Эти тесты не информативны для
постановки диагноза ДВС у пациентов с заболеваниями печени. При неосложнен-
ной коагулопатии и заболеваниях печени содержание ФУ (витамин К-независи-
мый фактор свертывания) снижено, поскольку пораженные клетки печеночной
паренхимы его не продуцируют. ФУШ в пределах нормы. Он также не
продуцируется паренхиматозными клетками и относится к острофазовым белкам.
Поэтому его содержание повышается неспецифически при воспалении или болезни
печени, но может быть в пределах нормы или повышено. Если ДВС протекает
на фоне болезни печени, то содержание ФУ и ФУШ значительно снижается или
они не обнаруживаются. AT III, ПС/nS, а2-антиплазмин истощены.
О внутрисосудйстом свертывании свидетельствуют и другие показатели:
протромбиновый фрагмент 1.2; соотношение фибринопептидов А/В; АПС;
комплексы тромбин-АТ III и др. Но эти тесты, как правило, не проводят при
диагностике ДВС.
Исследование мазка периферической крови позволяет выявить
фрагментированные эритроциты (шистоциты). Их наличие ассоциируется с ДВС, однако их
отсутствие не исключает данный синдром.
ТАБЛИЦА 6-13. Скрининг-тесты при остром синдромедиссеминированного
внутрисосудистого свертывания (ДВС)
Показатели ДВС Норма
Количество тромбоцитов < 150 000/мкл 150 000-400 000/мкл
ПВ >15с 12-14с
АЧТВ > 37 с 25-38 с
Фибриноген <150мг% 150-350 мг%
ПДФ >20мкг/мл 2-10мкг/мл
D-димер Положительный Отрицательный
Нарушения гемостаза
251
Принципы лечения острого ДВС
1. Лечение основного заболевания (инфекции или злокачественной опухоли).
2. Поддерживающие мероприятия, например активное устранение гипоксе-
мии и гиповолемии.
3. Лечение гепарином для прекращения активации системы свертывания крови.
4. Заместительная терапия концентратами AT III, ПС, свежезамороженной
\ плазмой, криопреципитатом и тромбоцитами.
Необходимость применения гепарина при ДВС у больных с тяжелым
геморрагическим диатезом признана не всеми специалистами, отчасти из-за отсутствия
понимания причины кровотечения при ДВС (например, индуцированного
тромбином). В некоторых исследованиях обнаружено снижение летальности при
гепариновой терапии ДВС с 90 % до 18 %. Ряду больных с истощением AT III
необходима одновременная инфузия или концентрата AT III, или
свежезамороженной плазмы для замещения кофактора гепарина.
Есть мнение, что назначение СЗП без одновременного введения гепарина
обеспечивает больше субстрата для продолжения реакции и прогрессирования
ДВС (принцип "дрова в костер").
Новые виды терапии, например использование рекомбинантного гирудина,
антител к ФНО, еще требуют клинических испытаний.
Клинические и лабораторные критерии мониторинга реакции на гепарин при
ДВС представлены в табл. 6-14.
Хроническое ДВС
Это состояние возникает в связи с:
1. Задержкой в матке мертвого плода. Повреждение тканей с выделением ТФ
приводит к ДВС.
2. Злокачественной опухолью. Наблюдается в 70 % случаев хронического ДВС.
Опухоли, секретирующие муцин, также образуют и выделяют в кровообра-
ТАБЛИЦА 6-14. Критерии мониторинга реакции на лечение гепарином
при остром ДВС
Клинические критерии Лабораторные тесты
УЛУЧШЕНИЕ
Прекратилось кровотечение Фибриноген увеличился на 40 мг% за 24 ч
Не появились новые пурпуры Уменьшился уровень ПДФ (на 2 разведения за 24 ч)
Исчез акроцианоз ПВ нормализовалось через 24 ч
Количество тромбоцитов возросло через 4-5 дней
ЧАСТИЧНОЕ УЛУЧШЕНИЕ
Прекратилось обильное Только 2 показателя улучшились
кровотечение
Не появились новые пурпуры
Исчез акроцианоз
БЕЗ ИЗМЕНЕНИЙ
Кровотечение продолжается Нет улучшения показателей крови
Сохраняется акроцианоз
УХУДШЕНИЕ
Кровотечение усиливается Все показатели ухудшились
Больной нестабилен
252
Глава 6. Гемостаз и тромбоз
щение прокоагулянты. Идентифицированы 3 вида прокоагулянтов:
тканевый фактор, фермент, превращающий ФХ в ФХа; микровезикулы (фосфо-
липиды), отделяемые от мембран опухолевых клеток и действующие в
гемостазе как заменители прокоагулянтов мембраны тромбоцита.
3. Заболеванием печени. Цирроз печени и некроз (ранее описан в разделе "Коа-
гулопатия при заболеваниях печени") фактически являются одной из форм
хронического Д ВС.
Ограниченное внутрисосудистое свертывание
Возникает при некоторых заболеваниях, связанных с потреблением тромбоцитов
и факторами свертывания крови, в строго определенных ограниченных
анатомических участках. Эти патологические состояния включают: 1) аневризму аорты;
2) гемангиомы — синдром Казабаха-Меритта (доброкачественные опухоли с
извитой массой сосудистых каналов, которые секвестируют и потребляют
тромбоциты и факторы свертывания); 3) заболевания почек (например, при системной
красной волчанке, уремическом синдроме); 4) острый респираторный дистресс-
синдром, если он возник как последствие травмы.
Лабораторные признаки: 1) снижение количества тромбоцитов; 2) повышение
ПДФ или положительный D-димер; З) нормальные или пролонгированные ПВ
и АЧТВ; 4) повышение уровня фибриногена и ФУШ в случае злокачественного
заболевания и понижение при большинстве других патологий. Распознавание со-
четанных клинических нарушений при постановке диагноза хронического или
ограниченного внутрисосудистого свертывания имеет первостепенное значение,
поскольку в этих случаях дает эффект лечение гепарином: улучшаются
клинические и лабораторные показатели у больных со злокачественными
новообразованиями, аневризмой аорты без разрыва и гемангиомами. В случае задержки мертвого
плода после его удаления гемостаз нормализуется.
Приобретенные фибринолитические состояния
Приобретенные нарушения фибринолиза подразделяются на 2 вида: гиперфибри-
нолиз, вызванный избыточной активацией или нарушенным ингибированием.
Данные расстройства фибринолиза могут привести к кровотечению или тромбозу.
Гиперфибринолитические состояния, связанные с кровотечением,
развиваются в результате или избыточной активации, или нарушенного ингибирования
фибринолиза.
Избыточная активация может быть вызвана:
1. Повышенным выделением в кровообращение активаторов плазминогена
(ТАП, урокиназы), что происходит у больных с доброкачественными и
злокачественными заболеваниями простаты (первичный фибринолиз).
2. Нарушением клиренса активаторов фибринолиза (при заболевании печени).
3. Избыточным локальным выделением активатора (при меноррагии, простат-
эктомии).
4. Ятрогенной вторичной активацией вследствие тромболитической терапии.
Нарушенное ингибирование фибринолиза наблюдается при ДВС и болезнях
печени. Может также возникать вторично из-за снижения синтеза плазмина,
образования комплексов плазмина с антиплазмином и/или ингибитором активатора
плазминогена (ИАП).
Нарушения тромбообразования
253
При лабораторном обследовании выявляется нормальное количество
тромбоцитов. Уровень D-димера не повышен при первичном фибринолизе, поскольку
это прежде всего лизис фибриногена или фибрина, не имеющего перекрестных
связей, однако он повышен при вторичном фибринолизе (как и при ДВС), при
котором в плазме происходит расщепление фибрина с перекрестной связью.
Чтобы при этом выделился D-димер, нужно перекрестное связывание фибрина,
индуцируемое тромбином и OVIII. Содержание в крови продуктов расщепления
фибрина (ПДФ) повышается, а фибриногена, плазминогена, ТАП, ИАП, а2-анти-
плазмина, <DV и OVIII — снижается.
Лечение больных с геморрагическим диатезом включает: 1) коррекцию
основного заболевания; 2) антифибринолитическую терапию; 3) переливание
СЗП для возмещения плазминогена, фибриногена и ингибиторов (ИАП, анти-
плазмины*).
Гипофибринолитические состояния, сочетающиеся с тромбозом, обусловлены:
1) нарушенной активацией фибринолиза вследствие неадекватного выделения
ТАП (врожденного или приобретенного), как при остром респираторном
дистресс-синдроме; 2) чрезмерным ингибированием фибринолиза, врожденного (в связи
с избыточной продукцией ингибиторов активатора плазминогена) или
вторичного (вследствие терапии антифибринолитическими средствами [ЭАКК]). Лечение
заключается в использовании тромболитических средств или антикоагулянтов
(гепарин и варфарин).
Нарушения тромбообразования
Определение понятия и состав тромба. Тромб образуется внутрисосудистой
массой фибрина и клеток крови. Относительное соотношение клеток и фибрина
зависит от гемодинамических факторов. Артериальные тромбы, формирующиеся
при высокой скорости потока, состоят в основном из тромбоцитарных агрегатов,
которые удерживаются вместе фибриновыми нитями (белые тромбы). Венозные
тромбы, возникающие в зоне стаза, состоят из большого количества эритроцитов,
фибрина, но содержат относительно немного тромбоцитов (красные тромбы).
Тромбы, образующиеся в зонах медленного и умеренного кровотока, являются
смесью эритроцитов, фибрина и тромбоцитов (смешанные тромбоцитарно-фиб-
риновые тромбы).
Локализация. Тромбы могут формироваться в венах, артериях, сердце или
микрососудах органа.
Судьба тромбов. Тромб подвергается непрерывным структурным изменениям.
Лейкоциты притягиваются к тромбу хемотаксическими факторами,
продуцируемыми кровью (калликреином) в ходе активации тромбоцитов, и аккумулируются
вокруг тромбоцитарных агрегатов в тромбе. Тромбоциты, находящиеся в тромбе,
начинают набухать и разрушаться и в течение 24 ч заменяются фибрином.
Последующая судьба тромба зависит от процессов, которые вызывают или
аккумуляцию тромботического материала, или его растворение. Тромб растворяется фиб-
ринолитическими ферментами (например, плазмином), которые образуются под
влиянием ТАП и урокиназы, выделяемой эндотелиальными клетками, а также
эластазами, продуцируемыми лейкоцитами. Тромб может организовываться,
прикрепляться к стенке сосуда, разрушаться, распространяться или подвергаться
254
Глава 6. Гемостаз и тромбоз
фагоцитозу лейкоцитами. Если тромбогенные факторы превышают мощность
механизмов растворения, тромб продолжает расти.
Клинические последствия тромбообразования. Тромб, образовавшийся в
любой части сосудистой системы, приводит к развитию ишемии (отсутствие
кровоснабжения) за счет окклюзии сосуда или эмболизации и обструкции дистальной
части системы кровообращения.
Окклюзионные и неокклюзионные тромбы. Артериальный тромб остается
у стенки сосуда как внутрисердечный тромб (неокклюзионный), поскольку он
обычно формируется в участках неоднородного и быстрого кровотока. Напротив,
венозный тромб образуется в зонах медленного кровотока и имеет тенденцию
к обструкции кровообращения (окклюзионный).
Внутрисердечный тромб — своеобразная зона непрерывного взаимодействия
тромбоцитов. Свежий тромбоцитарный тромб способен эмболизировать или
продолжать расти с образованием тромбоцитарных слоев различного возраста. Такой
тромб в конце концов внедряется в стенку сосуда, выделяя тромбоцитарный
ростовой фактор, который способствует ускоренной пролиферации фибробластов
и формированию атеросклеротической бляшки. При эмболизации внутрисердеч-
ного тромба иногда возникает опасная для жизни дистальная ишемия и
поражение органов. Как артериальный, так и венозный тромбоз весьма распространены
в клинической практике и относятся к ведущим причинам заболеваемости и
смертности в странах Запада.
Патогенез тромбоза. Более чем 150 лет тому назад Рудольф Вирхов высказал
предположение, что патофизиология тромбоза включает 3 взаимосвязанных
фактора {триада Вирхова): изменения в сосудистой стенке; изменения кровотока;
изменения свертываемости крови. Наличие первых двух факторов неоднократно
подтверждалось, а правомочность третьего только в наше время доказана в ходе
исследований на молекулярном уровне. Подобные изменения свертываемости
крови называют гиперкоагуляционными состояниями. Повышенная
свертываемость крови, известная также как предтромботическое состояние, определяется
как склонность к тромбозу в условиях, которые не привели бы к тромбозу у
здоровых индивидуумов. Ряд наследственных и приобретенных факторов риска
развития тромбоза идентифицированы как гиперкоагуляционные состояния и
ассоциируются с артериальной (табл. 6-15) или венозной тромбоэмболией (табл. 6-16 и
6-17). Термин тромбофилия используется для описания наследственной
склонности к тромбозу. Каковы причины образования тромба? Тромб образуется, когда
нарушается равновесие между тромбогенными факторами и защитными
механизмами гемостаза.
Известны следующие тромбогенные факторы: стимуляция или повреждение
сосудистой стенки, активация тромбоцитов, активация факторов свертывания
крови, ингибирование фибрйнолиза, застой крови (стаз). К защитным
механизмам, препятствующим возникновению тромбоза, относятся: ненарушенная анти-
коагулянтная активность эндотелия, нормальное количество и функция
естественных ингибиторов сериновых протеаз, клиренс активных протеаз
гепатоцитами и РЭС, интактная фибринолитическая система.
Тромбогенные и антикоагулянтные звенья гемостаза были описаны ранее
в этой главе. Развитие тромба проходит те же стадии, за исключением того, что
тромб локализован во внутрисосудистом пространстве. Избыточное количество
Нарушения тромбообразования
255
тромбогенных факторов или недостаточность защитных механизмов — вот
основные причины образования тромба. Хотя патогенез формирования тромба
одинаков в разных участках системы кровообращения, существует значительная
разница в механизмах образования артериального и венозного тромбов.
Артериальный тромбогенез
Одним из основных факторов образования артериального тромба, как и
ускорения атеросклероза, является активация тромбоцитов. Артериальный тромб
формируется преимущественно тромбоцитами и фибрином. Как правило, он
возникает в пораженной сосудистой стенке (наличие атероматозной или изъязвленной
бляшки), где на кровь действуют деформированные или поврежденные ЭК и су-
бэндотелиальные структуры. В результате таких изменений эндотелий лишается
тромборезистентных свойств {антикоагулянтных свойств). В норме эндотелий
способен нейтрализовать активные сериновые протеазы, ингибировать адгезию и
ТАБЛИЦА 6-15. Факторы риска при артериальном тромбогенезе
(приобретенные состояния гиперсвертываемости)
Наследственность1,2
Гомоцистинурия1,2
Мужской пол1
Гипертензия2
Пол (мужчины)2
Сахарный диабет2
Гиперлипидемия2
Курение2
Подагра2
Ожирение2
Пероральный прием контрацептивных средств
Злокачественные заболевания
Болезнь сердца
Повышенный уровень фибриногена
Повышенный уровень OVII
Хирургическая операция
Травма
Миелопролиферативные нарушения (истинная полицитемия и эссенциальная
тромбоцитемия)
Воспалительные васкулопатии
Воспаление кишечника
Пароксизмальная ночная гемогибинурия
Гепаринассоциированная тромбоцитопения и тромбоз
Антифосфолипидный синдром/волчаночный антикоагулянт
Системная красная волчанка
Синдром гипервязкости крови
Нефротический синдром
Инсульт
Беременность/послеродовый период
1 Наследственный фактор риска.
2 Риск развития атеросклероза.
256
Глава 6. Гемостаз и тромбоз
ТАБЛИЦА 6-16. Наследственные причины возникновения тромбофилии
(первичные состояния повышенной свертываемости)
Обычные причины
Частота
венозного
тромбоза
(%)
Редкие причины
Частота
венозного
тромбоза
(%)
Дефицит AT III 1-5
Дефицит ПС 1-9
Дефицит IIS 1-8
АПС-резистентность 20-60
(Ф\/-мутация)
Гипергомоцистеинемия 19
Дисфибриногенемия
Гипо- или дисплазминогенемия
Дефицит кофактора гепарина
Высокий уровень ингибитора
активатора плазминогена (ИАП-1)
Гликопротеин, богатый гистидином (|)
Аномальный тромбомодулин
агрегацию тромбоцитов, обеспечивать поверхность для активации фибринолиза.
При патологических состояниях пораженный эндотелий не препятствует
адгезии, агрегации тромбоцитов, выделению их содержимого и обеспечивает
поверхность для активации свертывания крови.
ТАБЛИЦА 6-17. Факторы риска при венозном тромбозе
(приобретенные и наследственные)
Вторичные, обусловленные стазом
(приобретенные)
Иммобилизация (после операций)
Ожирение
Сердечная недостаточность
Инсульт
Обезвоживание
Повышенная вязкость
(истинная полицитемия)
Беременность
Постинфарктное состояние
Варикозное расширение вен
Вторичные, обусловленные
наследственными нарушениями
гемостаза (наследственные)
Дефицит AT III, ПСилиПБ
АПС-резистентность (ФУ-мутация)
Высокий уровень ИАП
Дефицит кофактора гепарина II
Аномальные фибриноген
и плазминоген
Аномальный тромбомодулин
Высокий уровень гликопротеина,
богатого гистидином
Гипергомоцистеинемия
Вторичные, обусловленные активацией
свертывания крови (приобретенные)
Большая хирургическая операция
Обширная травма
Злокачественное заболевание
Беременность и послеродовый период
Антифосфолипидные антитела/
волчаночный антикоагулянт
Пероральные контрацептивные средства
Эстрогенотерапия
Нефротический синдром
Трансфузии с концентратами Ф1Х
Вторичные, обусловленные нарушением
тромбоцитов (приобретенные)
Гиперреактивные тромбоциты (например,
при миелопролиферативных заболеваниях)
Тромбоцитоз
Пароксизмальная ночная гемоглобинурия
Вторичные, обусловленные разными
факторами (приобретенные)
Возраст
Сепсис
Высокий уровень фибриногена
Высокий уровень OVII
Нарушения тромбообразования
257
Коллаген и тканевый фактор при нарушении целостности стенки сосуда
активируют систему свертывания крови, что сопровождается образованием
тромбина, последний вызывает агрегацию тромбоцитов и образование фибрина.
Другими важными факторами, способствующими агрегации тромбоцитов на месте
тромба, являются стеноз пораженных сосудов и турбулентность потока. ТхА2,
выделяемый из активированных тромбоцитов, и эндотелии-1, продуцируемый
поврежденными эндотелиальными клетками, вызывают вазоконстрикцию, в силу
чего снижается кровоток и происходит дальнейший рост тромбов (рис. 6-15).
Активация тромбином тромбомодулинового ПС/IIS пути ограничивает размеры
тромба за счет косвенной стимуляции фибринолиза (рис. 6-11).
От основного тромба могут отделяться небольшие тромбоцитарно-фибрино-
вые тромбы, которые закупоривают просвет сосуда. Это часто связано с
образованием сгустка в камерах сердца (внутрисердечные тромбы) или в сонных артериях,
что обусловливает нарушение мозгового кровообращения: Церебральная ишемия
может быть преходящей (преходящее нарушение мозгового кровообращения)
или постоянной (инсульт). Тромбоциты внутрисердечного тромба также
выделяют тромбоцитарный фактор роста, промотер роста фибробластов и развития
атеросклероза.
Внутрисердечный тромбоз развивается вследствие поражения клапанов или
эндотелия. Поражение клапанов наблюдается при ревматическом васкулите,
пролапсе митрального клапана, подостром эндокардите, СКВ или вторичном
эндокардите, обусловленном злокачественным заболеванием. Искусственные
клапаны также нередко вызывают тромбоэмболию. Внутрисердечные тромбы могут
образоваться в любом месте поврежденного эндокарда.
В табл. 6-15 представлен список приобретенных факторов риска
артериального тромбогенеза, некоторые из них участвуют и в развитии атеросклероза.
Венозный тромбогенез
Венозные тромбы обычно формируются в участках медленного или нарушенного
кровотока. Тромб возникает в виде небольших отложений, чаще всего в крупных
венозных синусах голени, карманах клапанных пространств глубоких вен голени
и бедра или других поврежденных венах. В месте образования тромб состоит
из тромбоцитов и фибрина, однако по мере увеличения размеров его состав
меняется на эритроциты и фибрин, и возникает закупорка вены.
Венозный тромбогенез отличается от артериального типом нарушения
равновесия между тромбогенным и защитным механизмами. При венозном тромбозе
основные факторы риска — повышенная системная гиперкоагуляция (активация
свертывания крови с нарушенным механизмом ингйбирования) и стаз.
Поражение стенки сосуда необязательно, однако его наличие является способствующим
фактором. Активация тромбоцитов играет второстепенную роль (табл. 6-17).
Иногда основным фактором служит нарушенный фибринолиз. Возвратный
венозный тромбоз наблюдается при ряде наследственных и приобретенных нарушений,
для которых характерна повышенная свертываемость крови (табл. 6-15 и 6-16).
Воспалительная реакция сосудистой стенки на тромб различна. У некоторых
больных развивается минимальная воспалительная реакция без клинических
симптомов. У других — выраженная и проявляется отеком, лейкоцитарной
инфильтрацией, массивной потерей эндотелия. Воспалительный процесс и прокси-
258
Глава 6. Гемостаз и тромбоз
мальная венозная обструкция в сочетании с повышением венозного давления
являются причиной гиперчувствительности, боли, отека у больных с острым
тромбозом глубоких вен.
Судьбы венозных тромбов различны (рис. 6-27). Тромб может
распространяться, подвергаться лизису, организовываться или эмболизировать.
Распространение тром'ба наиболее вероятно в том случае, если сохраняются тромбогенные
стимулы. Лизис тромба зависит от плазматического фибринолиза и
лейкоцитарной эластазы, которые вызывают организацию и/или эмболизацию тромба. Как
правило, клинически значимые легочные эмболии возникают, когда тромбы
образуются в крупных проксимальных венах ног (подколенной, бедренной,
подвздошной), и только иногда при крупных тромбах вен голеней. Однако летальная
легочная эмболия развивается вследствие как проксимальных, так и дистальных
венозных тромбов.
Полный спонтанный лизис тромба происходит редко. Тромб, который не эмбо-
лизирует или не подвергается растворению, будет медленно организовываться и ре-
канализироваться, создавая аномальную поверхность внутри вены, что приводит
к рецидивирующему тромбозу глубоких вен или постфлебитическому синдрому.
К осложнениям венозного тромбоза относятся: 1) легочная эмболия (ЛЭ) с
частотой 20 на 1000 больных (ежегодно от нее умирают 142 000 американцев); 2) по-
стфлебитический синдром, частоту которого трудно определись точно, но
приблизительно он наблюдается у 500 000 американцев. Причина его возникновения —
венозная гипертензия, связанная с деструкцией венозных клапанов, венозным
Рис. 6-27. Возможные исходы венозного тромбоза. (Из: Cortan R. S., Kumar V., Robbins S. L. (eds),
Robbins Pathologic Basis of Disease, 5th ed. Philadelphia: W.B. Saunders, 1996; 108.)
Нарушения тромбообразования
259
рефлюксом или стойкой обструкцией оттока крови. Постфлебитический синдром
проявляется отечностью лодыжки и голени, болью в области икры и лодыжки при
стоянии и.ходьбе. Эти симптомы ослабевают в покое и приподнятом положении
ног. Из-за высокого интракапиллярного давления эритроциты просачиваются
из кровеносных сосудов и железо откладывается в коже, вызывая
гиперпигментацию. Область вокруг лодыжки и голени отечна, с выраженным коллатеральным
кровообращением. Иногда в средней части лодыжки возникают кожные язвы.
Наследственная тромбофилия
Дефицит AT III явился первой распознанной в 1965 г. причиной наследственной
тромбофилии (называемой также состоянием повышенной свертываемости крови).
В начале 1980-х гг. были обнаружены дефициты протеина С (ПС) и протеина S
(nS). На долю этих 3-х наследственных факторов риска приходится менее 15 %
необъяснимых спонтанных и рецидивирующих тромбозов глубоких вен у
молодых людей (табл. 6-16). В 1993 г. резистентность к активированному ПС (в
результате точечной мутации в гене ФV) была выявлена как наиболее частая причина
наследственной тромбофилии (20-60 %). В 1994 г. обнаружено, что еще одним
фактором риска развития венозной тромбофилии является наследственная гипер-
гомоцистеинемия (19 %). В то время как дефицит AT III, ПС, ПБ и резистентность
к АПС — результат одного генного дефекта, гипергомоцистеинемия
продуцируется множественными генными дефектами, поскольку различные гены
осуществляют контроль нескольких ферментов, участвующих в метаболизме гомоцистеина.
К другим редким причинам венозной тромбоэмболии относятся: дисфибрино-
генемия, аномальный рлазминоген, высокий уровень ингибитора активатора
плазминогена-1 и богатого гистидином гликопротеина, аномальный кофактор
гепарина II и аномальный тромбомодулин.
Виды наследственной тромбофилии
Дефицит антитромбина III. AT III представляет собой гликопротеин с одной
цепью, синтезируемый печенью (период полусуществования — 65 ч). Ген AT III
локализован в хромосоме 1. AT III является серпином, который инактивирует
тромбин и другие сериновые протеазй (активированные факторы X, IX, XI, XII;
рис. 6-10).
Дефицит AT III наследуется какаутосомно-доминантноенарушение. Оно
наблюдается у 0,2-0,4 % населения. У больных с венозным тромбозом частота этого
нарушения составляет 5 %. Большинство людей с этим дефектом гетерозиготны,
и уровень AT III в плазме — 40-70 % от нормы. Гомозиготные больные
встречаются очень редко. Тромботические осложнения у лиц с дефицитом AT III составляют
от 15 % до 100 % в различных семьях. Известный типа наследственного дефицита
AT III. Тип I характеризуется уменьшенным количеством AT III и его
дисфункцией (снижен синтез). Молекулярный дефект при типе I связан с делециями гена,
кратковременными короткими вставками или делециями (мутации сдвига рамки)
и единичными заменами нуклеотидов (нонсенс или миссенс-мутации).
При типе II нарушается функция AT III, но его количество нормальное
(нарушенный синтез). Тип II подразделяется на 3 подтипа, которые характеризуются
аномальным активным центром AT III (сайт ингибирования), аномальными
сайтами связывания с гепарином, дефектом сайтов ингибирования гепарина и проте-
• 260
Глава 6. Гемостаз и тромбоз
азы. Молекулярный дефект при типе II вызван миссенс-мутациями,
приводящими к замене аминокислот.
Дефицит протеина С и протеина S. ПС и US — витамин К-зависимые белки,
синтезируемые печенью. Активированный ПС (АПС) ингибирует активированные
факторы V и VIII. Ti/2 ПС равно 7 ч, а у ПБ — значительно больше (42 ч). Тромбин
медленно активирует ПС; однако тромбин, связанный с тромбомодулином (ЭК-
тромбиновый рецептор), ускоряет активацию ПС примерно в 20 000 раз. Ингиби-
рование активированным ПС OVa и OVIIIa ускоряется посредством кофактора
ПБ. ПБ образует мембраносвязанный комплекс с АПС, который облегчает АПС
инактивацию активированных факторов V и VIII (рис. 6-11).
Недостаточность ПС и US наследуется аутосомно-доминантным путем.
Гомозиготный дефицит ПС вызывает неонатальный тромбоз в форме фульминантной
пурпуры. Распространенность гетерозиготного дефицита ПС составляет 0,3-
0,5 % среди населения и 1-9 % у больных с венозным тромбозом. Таким образом,
большинство гетерозиготных пациентов бессимптомны. Однако полагают, что
у 50 % гетерозиготных больных из семей с симптоматическим тромбозом в
анамнезе могут развиться тромботические осложнения.
Частота дефицита US аналогична частоте дефицита ПС у больных с
тромбозом. Частота дефицита nS у населения в целом неизвестна, но экстраполирование
данных от когорты тромботических больных дало возможность подсчитать, что
она составляет 1:33 000. Дефицит как ПС, так и US имеет 2 фенотипа: тип I
(сниженный синтез) и тип II (нарушенный синтез). При дефиците ПС и ПБ типа I
снижены количество и функция антигена в одинаковой степени. При типе II
функция непропорционально снижена, а содержание антигена нормальное.
Синтез ПС контролируется одним геном, локализованным на хромосоме 2,
а синтез US контролируется двумя гомологичными генами (а и Р), которые
находятся на хромосоме 3.
При дефиците ПС типа I мутации в гене (миссенс-мутации) приводят к
преждевременному окончанию синтеза или разрыву формирования вторичной
структуры белковой молекулы с потерей ее стабильности. Делеции и вставки
наблюдаются редко (< 10 %). Дефицит ПС типа II возникает вторично в связи
с миссенс-мутациями, которые появляются в специфических местах, например
домен протеазы и сайт расщепления тромбина, в результате чего возникает
дисфункциональная ингибирующая активность ПС.
Мутаций гена US идентифицировано мало, возможно, из-за большого размера
гена и присутствия псевдогена (Р). Обнаружены большие делеции а-гена, но
преобладающее число дефектов US — это точечные мутации, которые приводят
к преждевременному окончанию синтеза или нарушению вторичной структуры
белковой молекулы.
Резистентность к активации ПС. Резистентность к активации протеина С
является наиболее частой причиной наследственной повышенной способности крови
к свертыванию. Она наблюдается у 20-65 % больных с необъяснимой венозной
тромбоэмболией. Резистентность к активированному ПС связана с дефектом гена
OV, который локализован на хромосоме 1. Поиск генетических дефектов для
объяснения причин такой резистентности включает скрининг мутации гена ФV,
влияющей на сайт расщепления (Arg 506 — Gly 507) или место связывания
активированного ПС с OV (Arg 1865 — Не 1874). Точечная мутация в гене OV, приво-
Нарушения тромбообразования
261
дящая к замещению Arg 506 Gin, оказалась причиной резистентности к АПС-
инактивации фактора Va. Она наблюдалась у 80-100 % обследованных больных с
семейным венозным тромбозом.
Наследственная гипергомоцистеинемия. Гомоцистеин — аминокислота,
образующаяся из метионина. Внутриклеточный метаболизм гомоцистеина (рис. 6-28)
включает реметилирование в метионин и транссульфирование в цистеин. В реме-
тилировании участвуют 3 фермента: метионинсинтаза (кофактором является ко-
баламин), 5,10-метилентетрагидрофолатредуктаза и бетаин-метионинметил-
трансфераза. При транссульфировании гомоцистеин трансформируется
в цистатионин посредством цистатионинсинтазы (кофактором является пири-
доксин). В плазме гомоцистеин окисляется в дисульфиды и смешанные
дисульфиды и находится как в свободной форме, так и связанным с белком (общий
гомоцистеин); нормальная концентрация в плазме составляет 5-16 ммоль/л.
Наследственные дефициты ферментов, обусловленные наследственной гипер-
гомоцистеинемией, включают: 1) гомозиготный дефицит цистатионинсинтазы,
который является наиболее распространенной причиной тяжелой гипергомоцис-
теинемии. Частота ее среди населения составляет от 1 : 200 000 до 1 : 335 000.
У лиц с этой патологией наблюдается высокое содержание гомоцистеина в
плазме (> 100 ммоль/л), клинически проявляющееся ранней артериальной
окклюзией, а также артериальным и венозным тромбозом; 2) гетерозиготный дефицит
цистатионинсинтазы, который вызывает слабую (16-24 ммоль/л) или умеренную
(25-100 ммоль/л) гипергомоцистеинемию. Частота встречаемости у населения
составляет 0,3-1,4 %; 3) у небольшого числа больных с гомозиготным дефицитом
имеют место наследственные дефекты в реметилировании, например дефицит ме-
тилтрансферазы; 4) обычный дефект при реметилировании — появление мутант-
ной формы метилентетрагидрофолатредуктазы, наблюдаемой у 5 % населения.
Приобретенная гипергомоцистеинемия также достаточно широко
распространена и связана с недостаточным поступлением с пищей кобаламина, фолиевой
кислоты или пиридоксина — кофакторов для метаболизма гомоцистеина.
Лекарственные препараты, нарушающие обмен любого из этих кофакторов, также
могут стать причиной повышения содержания гомоцистеина в плазме.
Гипергомоцистеинемия вызывает артериальный и венозный тромбогенез,
нарушая равновесие между антикоагулянтными и тромбогенными свойствами
эндотелия и косвенно влияя на тромбоциты. Исследования in vivo показали, что
гомоцистеин приводит к десквамации эндотелиальных клеток, пролиферации
гладкомышечной ткани и утолщению интимы (патология сосудов).
Исследования in vitro продемонстрировали, что гомоцистеин может вызывать ряд
изменений в функции ЭК. Он снижает экспрессию ЭК тромбомодулина, нарушает ПС-
активацию, ингибирует ТАП, нарушает продуцирование окиси азота и ПП2,
а также подавляет экспрессию гепаран-сульфатов. Все эти изменения вместе
значительно снижают антикоагулянтные свойства эндотелия. Гомоцистеин также
активирует ФV и экспрессию тканевого фактора эндотелиальными клетками (два
важных тромбогенных эффекта). Кроме того, гомоцистеин-тиолактон,
содержание которого увеличивается в плазме больных с гипергомоцистеинемией,
вызывает агрегацию тромбоцитов in vitro и выделение ТхА2. Этот тромбоцитарный
эффект вместе с повреждением эндотелия, вызываемым гомоцистеином, объясняет
механизм артериального тромбогенеза, характерного для гипергомоцистеинемии.
Белки
Реметилирование
- Тетрагидрофолат -<-
5,10-метилен-
тетрагидрофолат
5,10-метилен-
тетрагидрофолат-
редуктаза
Метионин-
синтаза
+
метил-В12
ч
5-метил-
тетрагидрофолат'
Трансметилирование I
о>
ю
-^ Метионин
-► S-аденозилметионин
-► Диметилглицин
Бетаин-гомоцистеин-
метилтрансфераза
Бетаин
ZZZ Гомоцистеин -<-
S-аденозилгомоцистеин
Цистатионин-
бетасинтаза
+ В6
Цистатионин
i
Цистеин
Транссульфирование I
Рис. 6-28. Внутриклеточный метаболизм гомоцистеина. (Из: De Stephano V., Finazzi G., Mannucci P. M. Inherited Thrombophilia:
pathogenesis, clinical syndromes and management. Blood, 1996; 87: 3531-3544.)
0)
I©
\9*
7
\z
3
ICO
Is
H
о
2
o\
о
I CO
Нарушения тромбообразования
263
Слабая и умеренная гипергомоцистеинемия является независимым фактором
риска развития инсульта, инфаркта миокарда, патологии периферических
сосудов и стеноза экстракраниальных сосудов. У молодых людей с венозной
тромбоэмболией частота гипергомоцистеинемии достигает 19 % и в настоящее время
считается фактором риска развития венозного и артериального тромбозов.
Клинические проявления венозного тромбоза не отличаются от проявлений других
тромбофильных состояний. Тромбоз глубоких вен и легочная эмболия,
наблюдаются в 64 %; поверхностные флебиты — в 24 %; тромбоз церебральных или
брыжеечных вен — в 12 % случаев.
Наследственные дефекты фибринолиза. Несколько фибринолитических
дефектов задействованы в патогенезе тромбоза. К ним относятся: гипоплазминогене-
мия, дисплазминогенемия (аномальный плазминоген), снижение выделения
тканевого активатора плазминогена (ТАП) и повышенное содержание ингибитора
активатора плазминогена (ИАП). Все эти изменения приводят к аномальному
или сниженному образованию плазмина и нарушению (неадекватной) фибрино-
литической реакции на формирование фибрина (рис. 6-13).
Наследственное высокое содержание богатого гистидином гликопротеина,
который связывается с плазминогеном и препятствует образованию плазмина,
обнаружено в семьях с симптоматикой тромбоза. Повышенное тромбообразование
у больных с дисфибриногенемией возникает редко, как правило, вторично в связи
с аномалиями фибриногена, приводящими к резистентности, лизису плазмином,
повышенному связыванию с тромбином или патологической полимеризации.
Генетические дефекты при первичных состояниях повышенной способности к
свертыванию недостаточно полно охарактеризованы дефектами ингибиторов,
описанными выше.
Клинические проявления наследственной тромбофилии
Клинические проявления у больных с дефицитом AT III, ПС, IIS и
резистентностью к АПС сходны. Венозная тромбоэмболия наблюдается более чем у 90 %
пациентов. Тромботические осложнения возникают в любом возрасте. Однако
начальный тромботический эпизод обычно проявляется в раннем возрасте. Наиболее
характерное место тромбоза — нижние конечности с развитием в последующем
эмболии легких. Однако иногда наблюдается тромбоз необычной локализации,
например в брыжеечных или церебральных венах, а также не исключен
поверхностный флебит (приблизительно 5 % случаев). По неизвестным пока причинам
поверхностный тромбофлебит чаще возникает у больных с дефицитами ПС и ПБ,
а также резистентностью к АПС, а не при дефиците AT III.
Венозная тромбоэмболия наблюдается у 60-80 % индивидуумов с дефицитом
AT III, ПС или nS, обычно в возрасте до 40 лет. У больных с дефицитом AT III
тромбоз бывает чаще, чем при дефиците ПС и ПБ, а при резистентности к АПС
склонность к тромбозу меньше, чем при дефиците AT III, ПС или US. Хотя
распространенность резистентности к АПС высокая, в большинстве случаев до конца жизни
течение бессимптомное. Однако резистентность к АПС в сочетании с любым
другим дефектом (AT III; ПС или US) повышает вероятность возникновения тромбоза.
Известно, что приобретенные факторы риска развития тромбоза
(хирургические вмешательства, беременность, послеродовый период, иммобилизация)
повышают частоту возникновения тромбоза у лиц с первичной гиперкоагулируемос-
264
Глава 6. Гемостаз и тромбоз
тью. Так, у женщин, гетерозиготных по дефициту AT III, во время беременности
или в послеродовом периоде частота тромбоза составляет 31-44 %; у больных
с дефицитом ПС или US — 10-19 %, а с резистентностью к АПС — 28 %.
Некоторые исследования выявили повышенную частоту тромбоза у больных с
дефицитом AT III, ПС или П5 и тех, кто подвергался абдоминальному хирургическому
вмешательству по поводу опухолей. Эти клинические наблюдения подтверждают
гипотезу о том, что 2 или несколько генных взаимодействий, связанных с про-
тромбоэмболическими мутациями, приводят к наследственной
предрасположенности к тромбозу (гиперкоагуляционное состояние), но тромботическое событие
вызывается приобретенным протромботическим стимулом.
Диагноз наследственной тромбофилии
Первый этап в диагностике наследственной тромбофилии включает детальное
ознакомление с анамнезом больного с учетом всех приобретенных факторов риска
развития тромбоза (системная красная волчанка, злокачественные
новообразования, антифосфолипидные антитела/волчаночный антикоагулянт). Необходимо
выяснить возможные послеоперационные проблемы, случаи внезапной смерти в
семье. Второй этап состоит в определении склонности к тромбозу: на основе
собранной информации составляется схема генеалогического древа.
Отрицательные показатели в семье не исключают тромбофилии, особенно резистентности
к АПС и гипергомоцистеинемии вследствие низкой частоты распространенности
этой патологии. Последний этап — лабораторное исследование.
Когда необходимо проводить лабораторное тестирование на тромбофилию
и кого обследовать? Лабораторную диагностику нужно проводить после того, как
разрешилось острое тромботическое состояние, у стабильных больных, не
получающих антикоагулянты. Тестирование на определение повышенной
свертываемости крови — дорогостоящая процедура. Функциональные исследования на
ингибиторы свертываемости крови, проводимые в острой тромботической стадии
(даже до начала лечения) неточны из-за потребления ингибиторной активности
этих белков во время тромбообразования. Пероральный прием антикоагулянтов
(например, варфарина) подавляет ингибиторы витамин К-зависимых ПС и ПБ
и стимулирует синтез AT III. Поэтому измерение их активности во время лечения
антикоагулянтами дает недостоверные результаты. Лечение гепарином также
влияет на функциональную активность AT III. Следовательно, лучше всего
проводить лабораторную диагностику через 1-2 нед после окончания
поддерживающей терапии антикоагулянтами (продолжительность обычно 3-6 мес).
Проведение лабораторного тестирования дефицита AT III, ПС и ПБ показано
в первую очередь больным в возрасте до 40 лет, у которых в семье были случаи
тромбоза либо этиология венозного тромбоза необъяснима. Необходимо
определить концентрацию и функцию каждого ингибитора. При дефиците US следует
определить уровень свободного и связанного ПБ. Резистентность к АПС и
содержание гомоцистеина нужно определять у любого больного с тромбозом независимо
от его возраста, поскольку распространенность этой патологии велика.
Резистентность к АПС устанавливается по способности плазмы больного противостоять
пролонгированию АЧТВ, вызванному добавлением активированного белка С.
Чувствительность анализа — 85 %, а специфичность — выше 90 %. Точность
повышается при добавлении к тест-системе плазмы с дефицитом ФУ. Только 5-10 %
Нарушения тромбообраэования
265
больных с пограничными результатами, требуют ДНК-анализа гена фактора V.
Это исследование можно проводить уже в ходе лечения антикоагулянтами.
Содержание гомоцистеина в плазме определяют до приема метионина
(внутрь в дозе 0,1 г/кг массы тела) и через 4 ч после. Это позволяет
дифференцировать нормальное и гетерозиготное состояния при дефиците цистатионин-
|3-синтазы. Для лучшего выявления гетерозиготных состояний прибегают к
молекулярному анализу генных мутаций.
Лечение наследственной тромбофилии
Первичная, кратковременная, профилактика. Больные с тромбофилией (в
возрасте до 40 лет) до, в период и после хирургического вмешательства, во время
беременности, после родов (4 нед) должны получать профилактическое лечение —
подкожное введение гепарина. Больные с дефицитом AT III во время беременног
сти из-за высокого риска возникновения тромбоза должны получать или гепарин
низкого молекулярного веса подкожно, или дозированный гепарин также
подкожно (соотношение АЧТВ 1,5-2,0). В ходе родов рекомендуют использовать
концентраты AT III (через день), а затем гепарин подкожно (в течение 4 нед после
родов). Применение концентратов AT III во время беременности не дает какого-
либо, в том числе экономического, преимущества по сравнению с гепарином.
Гепарин с низким молекулярным весом (фракционированный^гепарин) — еще одна
альтернатива, но он дороже нефракционированного.
Вторичная, или долгосрочная, профилактика. Всех больных, за исключением
женщин с тромбофилией, у которых развился первый эпизод тромбоза, лечат
антикоагулянтами перорально (варфарин в течение 6 мес; MHO = 2-3). При
множественных дефектах и рецидивах тромбоза пациенты должны постоянно
принимать антикоагулянты per os.
Лечение острого тромбоза не зависит от наличия наследственного гиперкоагуля-
ционного состояния. Начинают с внутривенного введения
нефракционированного гепарина для поддержания отношения АЧТВ в пределах 1,5-2,5 в течение
7 дней. Как альтернатива, через день после нефракционированного гепарина
больной может перейти на гепарин низкого молекулярного веса. Введение
антикоагулянтов (перорально) начинают через 24-48 ч после начала гепариновой
терапии и совмещают с гепарином, пока не будет достигнут терапевтический
уровень (MHO = 2-3), это обычно требует 3-5 дней.
Больных с гипергомоцистеинемией лечат по поводу тромбоза, как описано
выше. Профилактика пиридоксином, фолиевой кислотой, витамином Bt2 снижает
уровень гомоцистеина, однако неясно, является ли она эффективной мерой
предотвращения дальнейшего артериального или венозного тромбоза.
Приобретенная тромбофилия
Послеоперационный венозный тромбоз
Послеоперационный венозный тромбоз возникает часто у пациентов в пожилом
возрасте, тучных людей; лиц с венозным тромбозом в семейном анамнезе, а также
после операций на брюшной и грудной полостях, головном мозге. Основные
способствующие факторы: активация свертывания крови во время хирургического
266
Глава 6. Гемостаз и тромбоз
вмешательства, послеоперационная гиподинамия и застой крови. Аналогичные
механизмы действуют, как полагают, при застойной сердечной недостаточности,
инфаркте миокарда, варикозном расширении вен.
Злокачественные заболевания
Три вида прокоагулянтов, продуцируемых злокачественными клетками,
активируют или ускоряют свертывание крови и способствуют тромбозу: тканевый
фактор; ферменты, превращающие ФХ в ФХа; и микровезикулы (фосфолипиды),
которые служат подложкой для связывания с активированными факторами
свертывания (заменители мембраны тромбоцита). Источниками этих
прокоагулянтов являются аденокарциномы молочной железы, легких, предстательной
железы, поджелудочной железы, тонкой кишки, при которых высока частота
возникновения венозного тромбоза. Кроме того, при некоторых опухолях
наблюдается неэффективный фибринолиз.
Заболевания крови
При миелопролиферативных нарушениях (МПН) и миелодиспластических
синдромах, пароксизмальной ночной гемоглобинурии (ПНГ), серповидно-клеточной
и гемолитической анемии возникают тромбоэмболии. При МПН гипервязкость
крови, активация тромбоцитов, повреждения эндотелия приводят к активации
свертывания крови и тромбозу. Основная причина смерти у больных с истинной
полицитемией и эссенциальной тромбоцитемией — артериальный или венозный
тромбоз. У больных с ПНГ часто возникает тромбоз печеночных вен или нижней
полой вены (синдром Бадда-Киари), а также портальной и брыжеечных вен
до того, как установлен диагноз ПНГ. У тромбоцитов повышается
чувствительность к активации комплемента и индукторам агрегации. В случае гемолиза АДФ
из гемолизированных эритроцитов индуцирует агрегацию тромбоцитов. Для этих
больных также характерен артериальный тромбоз. Серповидные эритроциты
закупоривают микрососуды, а АДФ из гемолизированных эритроцитов активирует
тромбоциты. Это — два важных фактора, способствующих развитию как
венозного, так и артериального тромбоза при серповидно-клеточной анемии.
Лечение эстрогенами
При лечении высокими дозами эстрогенов часто возникает тромбоэмболия.
Тромботические явления вторичны по отношению к повышенному синтезу
факторов свертывания крови (ФН, VII, VIII, IX, X, фибриноген), снижению фибри-
нолиза, ТАП, функциональной активности AT III и повреждению эндотелия. При
лечении низкими дозами эстрогенов вероятность тромбоэмболии значительно
ниже. Однако пероральный прием противозачаточных препаратов (низкие дозы
эстрогенов) женщинами с первичным повышением свертываемости крови
противопоказан из-за возрастания риска возникновения тромбоза.
Другие приобретенны% заболевания
Развитию венозной тромбоэмболии способствуют нефротйческий синдром,
воспалительные заболевания кишечника, трансплантация костного мозга. При не-
фротическом синдроме больные теряют AT III с мочой, тромбоциты гиперреак-
Нарушения тромбообразования 267
тивны, характерно поражение сосудистой стенки. При воспалении кишечника
тромбозу способствует повреждение эндотелия, приводящее к активации
свертывающей системы крови и тромбоцитов, снижение содержания ПС, AT III и
nS. После аллогенной трансплантации костного мозга у 21 % больных
развивается веноокклюзионная болезнь, 45 % погибают от прогрессирующего
заболевания печени. Веноокклюзионная болезнь является результатом поражения
эндотелия венул и синусов печени облучением и химиотерапевтическими
средствами (глава 8).
Антифосфолипидный синдром/волчаночный антикоагулянт
Определение. Антифосфолипйдные антитела (АФА) — иммуноглобулины (IgG,
IgM, IgA или их сочетание), взаимодействующие с одним или несколькими
отрицательно заряженными фосфолипидами. Для наличия АФА характерно
увеличение АЧТВ и, в меньшей степени, ПВ. Волчаночный антикоагулянт (ВА) —
ингибитор антифосфолипидов, обладающий гепариноподобной активностью.
Клинически АФА и В А проявляются склонностью к развитию венозного и
артериального тромбозов, но не кровотечения!
Отношение АФА к В А. Об АФА впервые стало известно в 1906 г. при
серологической диагностике сифилиса. При использовании реактивов, состоящих из
смеси липидов (кардиолипин, лецитин, холестерин), например VDRL для выявления
сифилиса, у больных с аутоиммунными нарушениями были получены ложнопо-
ложительные результаты. В 1952 г. описаны двое пациентов с системной красной
волчанкой, у которых был обнаружен циркулирующий антикоагулянт (с
гепариноподобной активностью), не связанный с кровотечением. Его назвали волчаноч-
ным антикоагулянтом (ВА) — не совсем точный термин, так как у большинства
больных с В А нет системной красной волчанки.
В 1983 г. был разработан метод радиоиммуного анализа для выявления анти-
кардиолипиновых антител (АКА). и было высказано предположение, что АКА
и ВА — одни и те же антитела. Последующие исследования показали, что у 60 %
больных эти антитела совпадают, тогда как у других пациентов было выявлено
только одно из них. Некоторые больные имеют несколько антител, которые
реагируют с отрицательно заряженными фосфолипидами. Эти антитела могут быть
различного изотипа с различной авидностью.
Присутствие антифосфолипидных антител нередко выявляют при разных
состояниях, например вирусных и бактериальных инфекциях (в частности, СПИД),
аутоиммунных болезнях соединительной ткани (системная красная волчанка,
ревматоидный артрит), злокачественных новообразованиях, приеме
лекарственных средств (прокаинамид, хлорпррмазин, хинидин). АФА обнаруживаются
и при отсутствии основного заболевания. Клиническая связь между ВА и
тромбозом (венозным, артериальным, отслойкой сетчатки) впервые установлена
в 1963 г. у больных системной красной волчанкой. Последующие
многочисленные исследования подтвердили связь между частыми случаями артериального
и венозного тромбоза и АФА/ВА.
Антифосфолипидный синдром (АФС) определяется как наличие
антифосфолипидных антител (ВА или высоких/умеренных уровней антикардиолипиновых
антител класса IgG или IgM) и возникновение венозного или артериального
тромбоза, рецидивирующих выкидышей и тромбоцитопении. Если основное заболевание
268
Глава 6. Гемостаз и тромбоз
не выявлено, считают, что у больного — первичный антифосфолипидный синдром.
Симптомы аналогичны тем, которые наблюдаются при аутоиммунных
заболеваниях соединительной ткани. Частота АФС у женщин и мужчин соотносится как
9 : 1, а при первичном АФС — 2:1. Тромбоцитопения и антинуклеарные антитела
при первичном АФС наблюдаются у 50 % пациентов, а антифосфолипидные
антитела определяются как высокий титр антикардиолипиновых антител класса IgG.
Тромбоз глубоких вен и легочная эмболия — наиболее характерные
проявления поражения вен при АФС, связанном с аутоиммунным заболеванием
соединительной ткани; но также может быть тромбоз необычной локализации, например
почечных и печеночных вен. Артериальный тромбоз способен поражать любую
артерию, особенно венечную, церебральную и периферические. АФС и ПАФС
характеризуются преходящими ишемическими приступами, нарушениями
зрения, в частности временной слепотой, и одиночным или рецидивирующим
церебральным инфарктом. Больным моложе 50 лет с преходящим ишемическим
нарушением или инсультом необходимо проводить тесты на волчаночный
антикоагулянт и антикардиолипиновые антитела. У 50 % пациентов с АФС
наблюдаются васкулит, сыпь и артралгия. К другим проявлениям относятся
мигрени, отслойка сетчатки, патология сердца (поражения эндокарда с дефектом
митрального клапана).
Характеристика В А и АФА. ВА-антиген представляет собой шестиугольный
фосфолипид, требующий плазматического кофактора (возможно, протромбина).
ВА определяется в плазме при исследовании коагуляции с использованием яда
гадюки Рассела и процедуры нейтрализации тромбоцитов. Результаты могут
быть положительными или отрицательными. Антиген антикардиолипиновых
анител представляет собой пластинчатый или мицеллярный фосфолипид,
требующий в качестве кофактора (32-гликопротеин 1. Антифосфолипидные антитела
определяются методом твердофазного иммуноферментного анализа. Результаты
выражаются как низкий, средний или высокий уровень IgG или IgM единицы ан-
тифосфолипидных антител (каждая единица = 1 мг антитела).
Диагноз АФС на основании лабораторных данных ставится при умеренном
или высоком уровне АФА IgG или IgM либо при положительном ВА
(определение 2 раза с интервалом в 12 нед).
Патогенез тромбоза при АФС, Предполагают, что тромбоз, индуцированный
антифосфолипидными антителами, возникает под действием нескольких
механизмов. После воздействия АФА эндотелиальные клетки изменяют свою антико-
агулянтную активность и могут индуцировать сильные тромбогенные эффекты,
такие как сниженные синтез ПП2, экспрессия тромбомодулина и активность
сульфата гепарина; повышенная экспрессия тканевого фактора, нарушение фиб-
ринолиза (в связи со сниженным ТАП и повышенным ИАП), повышение фактора
активации тромбоцитов. Кроме того, обнаружено, что у больных с АФС падает
функциональная активность в плазме AT III, ПС, свободного ПБ, антител
к р2-ГП1 и активируются тромбоциты. Вероятно, все эти изменения вызваны
АФА, направленными против специфических фосфолипидов. Однако в
последнее время обнаружено, что АФА направлены не против фосфолипидов, а против
белка, связанного с фосфолипидом. В 1996 г. Arnout высказал мнение, что
тромбоз при АФС объясняется механизмом "двойного удара". После первоначального
поражения анионные фосфолипиды осаждаются на клетках крови (тромбоцитах,
нейтрофилах), на эндотелиальных клетках или на трофобластах. Эти реактивные
Нарушения тромбообразования
269
фосфолипиды покрываются фосфолипидсвязывающими белками, например
(32-ГП1, или протромбином. Если в крови присутствуют антитела к этим
связанным с поверхностью белкам, то иммуноглобулины концентрируются на
поверхности клетки, связываются с Fc-рецепторами и индуцируют все тромбогенные
изменения, о которых говорилось выше. Эта гипотеза отчасти подтверждается
экспериментальными данными и наличием аналогичных механизмов,
задействованных в гепарининдуцированной тромбоцитопении в сочетании с тромбозом.
Лечение. Есть сведения, что у больных с АФС высок риск развития тромбоза.
С целью профилактики до и после хирургического вмешательства необходимо
подкожное введение гепарина. Беременных женщин лечат подобранной дозой
гепарина, гепарином с низким молекулярным весом или аспирином в сочетании со
стероидами в небольшой дозе. Лечение больных с острым тромбозом и сочетанным АФА
такое же, как при других состояниях гиперкоагуляции с аналогичными тромбоэм-
болическими проявлениями. Гепарин вводится внутривенно, в количестве,
обеспечивающем АЧТВ в 2-2,5 раза выше исходного, а варфарин начинают вводить через
24-48 ч (совместное введение с гепарином продолжают 5 дней). MHO сохраняется
приблизительно на уровне 3,0. Больные с АФС и рецидивирующим тромбозом
получают антикоагулянты в течение всей жизни. Некоторым пациентам с
артериальным и венозным тромбозом в анамнезе назначают варфарин и аспирин.
Антитромботическая терапия
Антикоагулянты применяются для профилактики и лечения как артериального,
так и венозного тромбоза. Наиболее широко в клинической практике
используются гепарин (парентерально) и кумарины (перорально).
Парентерально вводимые антикоагулянты
Гепарин (нефракционированный) — антикоагулянт, состоящий из смеси гликоза-
миногликанов с различной молекулярной массой (5000-30 000). Гепарин
оказывает антикоагулянтное действие за счет связывания AT III и потенцирования
образования комплекса AT III с активными сериновыми протеазами каскада
свертывания крови (тромбин, ФХа, Ф1Ха, ФХ1а, ФХИа). Это комплексообразо-
вание приводит к необратимой инактивации факторов свертывания крови (рис.
6-10). Только небольшая фракция гепарина обладает AT III-связывающей
активностью. Однако гепарин, соединенный с AT III, значительно усиливает ингиби-
торную способность AT III (примерно в 1000 раз). Гепарин активирует
тромбоциты и связывается со многими другими клетками (макрофагами, эндотелиальными
и жировыми клетками) и активирует липазы и систему комплемента;
предотвращает образование и останавливает рост тромбов.
Гепарин метаболизируется печенью и выделяется почками. Он не всасывается
в желудочно-кишечном тракте, поэтому его вводят внутривенно или подкожно.
При дозе 100 ед/кг биологический период полураспада составляет 1 ч, при более
низких дозах его содержание снижается нелинейно. Гепарин прочно связывается
с ЭК и макрофагами (что обусловливает необходимость болюсного введения),
остаточный гепарин выводится медленно почками. Кроме того, гепарин способен
связываться с белками, которые его нейтрализуют (богатый гистидином глико-
протеин, тромбоцитарный фактор 4, витронектин). Высокое содержание этих
белков в крови может вызвать относительную резистентность к препарату.
270
Глава 6. Гемостаз и тромбоз
Фракционированные гепарины имеют низкий молекулярный вес (4000-8000)
и называются гепаринами низкого молекулярного веса. Их действие как
антикоагулянтов обусловлено связыванием с AT III, они ингибируют специфически ФХа,
а не тромбин. Пока неясно, меньше ли эффект гепаринов низкого молекулярного
веса на тромбоциты, чем нефракционированного гепарина.
Введение гепарина и лабораторный мониторинг гепаринотерапии
Гепарин можно вводить внутривенно постоянно или с перерывами. При остром
тромбозе (тромбоз глубоких вен, легочная эмболия, внутрисердечные тромбы
и пр.) лучший способ введения гепарина — непрерывный внутривенный.
При остром венозном тромбозе (тромбозе глубоких вен) лечение начинают
гепариновым болюсом с последующим непрерывным внутривенным введением.
Дозу корригируют каждые 6 ч для поддержания АЧТВ на уровне в 1,5-2,5 раза
выше исходного (терапевтический диапазон). В первые 24 ч для
предотвращения рецидива тромбоза АЧТВ надо сохранять в пределах терапевтического
диапазона.
Пероральный прием антикоагулянтов (например, варфарина) рекомендуется
начинать через 24 ч или 48 ч и сочетать с лечением гепарином в течение 3-5 дней
до тех пор, пока не проявится антитромботический эффект варфарина. Одна
из причин введения кумадина через 24 ч, а не раньше состоит в необходимости
задержки супрессии ПС (ПС является витамин К-зависимым фактором
свертывания, прерывание синтеза которого варфарином приведет к повышению
свертываемости и, возможно, увеличению и распространению тромба).
При острой легочной эмболии объем гепаринового болюса следует увеличить,
так как средний период полусуществования гепарина в зоне эмбола меньше
20 мин. В остальном антикоагулянтная терапия легочной эмболии аналогична
описанной ранее для тромбоза глубоких вен и других венозных тромбов.
Подкожные инъекции гепарина выполняются периодически для профилактики
тромбоэмболии. Этот краткосрочный вид гепариновой терапии не требует
лабораторного мониторинга. Длительное подкожное введение гепарина у больных
с противопоказаниями к пероральному применению антикоагулянтов
эффективно, но может сопровождаться серьезными осложнениями и остеопорозом.
Подобранная доза гепарина вводится подкожно больным с высоким риском, например
беременным женщинам с дефицитом AT III. Мониторинг дозы осуществляется
путем поддержания АЧТВ на уровне отношения 1,5-2,0 (АЧТВ
больного/среднее нормальное АЧТВ).
Гепарины низкого молекулярного веса в настоящее время используются в США
для парентерального введения. Их вводят подкожно 1-2 раза в день в
фиксированной дозе, поскольку они имеют длительный период полусуществования (Зт
18 ч). Терапия этими гепаринами эффективна, не требует лабораторного
мониторинга, но более дорогостоящая, чем лечение нефракционированным гепарином.
Она рекомендуется при тромбоэмболии у онкологических больных с
противопоказаниями к применению варфарина, а также больным, подвергающимся
ортопедическим хирургическим вмешательствам. Ее рассматривают как
первоочередную терапию при тромбозе глубоких вен и легочной эмболии.. Однако гепарины
низкого молекулярного веса в основном ингибируют ФХа, а не тромбин, поэтому
при остром тромбозе может произойти распространение тромба.
Нарушения тромбообразования
271
Осложнения, связанные с лечением гепарином
При чрезмерном угнетении свертываемости крови может возникнуть вторичное
кровотечение вследствие нарушения функции тромбоцитов гепарином или в
результате тромбоцитопении. Кроме того, кровотечение иногда является
результатом потенцирования гепарином причинного дефекта или сочетанного введения
антитромбоцитарных или тромболитических агентов. Поскольку гепарин,
вводимый внутривенно, имеет короткий период полусуществования (1 ч), для того
чтобы остановить кровотечение, вызванное чрезмерной антикоагуляцией, обычно
достаточно прекращения лечения. В тяжелых случаях кровотечения гепарин
можно нейтрализовать протамин сульфатом, однако избыточное количество про-
тамина также может вызвать кровотечение. Гепарины низкого молекулярного
веса нередко дают аналогичные геморрагические осложнения.
Гепарининдуцированная тромбоцитопения наблюдается примерно у 2 %
больных, получающих свиной гепарин, а при использовании гепарина из других
источников частота ее значительно ниже. Данное состояние — это результат иммуно-
опосредованного побочного эффекта гепарина. Появляются антитела против
комплекса гепарина и тромбоцитарного фактора 4, гепаринсвязывающего гликоп-
ротеина, который депонируется в сс-гранулах тромброцитов, но также имеет
рецептор на мембране тромбоцита. Гепарининдуцированный IgG связывается с этими
мембранными комплексами, а затем активирует тромбоциты за счет соединения
с Fc-рецептором. Гепарининдуцированный IgG способен активировать ЭК с
экспрессией тканевого фактора, приводящего к стимуляции свертывания крови и тром-
бообразованию. Лечение заключается в отмене гепарина. Некоторым больным
необходимо введение кортикостероидов или иммуноглобулинов для нормализации
количества тромбоцитов. Тромбоцитарные трансфузии противопоказаны.
Гепарининдуцированная тромбоцитопения и тромбоз наблюдаются менее
чем у 1 % больных с тромбоцитопенией. Данное сочетание обычно наблюдается
после хирургического вмешательства, причем тромбоз может быть
артериальным или венозным. Лечение сводится к незамедлительной отмене гепарина и
использованию альтернативных антикоагулянтов (анкрод, гирулог, гепаринопо-
добные препараты с низкой молекулярной массой). Трансфузии тромбоцитов
противопоказаны.
Гепарининдуцированный остеопороз возникает у больных с долгосрочной гепа-
ринотерапией (более 3 мес) в дозе 20 000 ЕД или больше еженедельно. Это
проблема касается в основном прикованных к постели больных, пожилых людей или
беременных женщин. Следует прибегнуть к лечению кальцием или
использованию гепарин-кальция.
Новые антикоагулянты. Гирудин, вводимый парентерально, является
специфическим ингибитором тромбина (7к-белок), впервые обнаружен в слюнных
железах пиявок (Hirudo medicinalis). Он соединяется с активным сайтом тромбина,
а также с участком, в котором тромбин связывается с фибриногеном. Поэтому
гирудин способен блокировать тромбин, связанный с фибриногеном, а гепарин не
может. Гирудину не требуется кофактор для ингибирования тромбина, он также
не ингибирует другие сериновые протеазы. Гирудин и его синтетический аналог
гирулог являются эффективными антикоагулянтами, которые не влияют
на АЧТВ и не вызывают геморрагических осложнений, связанных с
кровотечением, а также лишены антитромбоцитарного действия.
272
Глава 6. Гемостаз и тромбоз
Анкрод — фермент, получаемый из ямкоголовой (малазийской) змеи. Он
отсоединяет фибринопептид А от фибриногена, не активируя ФХШ, предотвращая
тем самым образование перекрестной связи фибрина. Затем растворимый
фибрин легко расщепляется плазмином. Лечение анкродом вызывает дефибрино-
генемию и увеличивает количество ПДФ без возникновения массивного
кровотечения. Анкрод эффективен при гепарининдуцированной тромбоцитопении
с тромбозом и уже используется в течение нескольких лет в различных странах.
Пероральные антикоагулянты
Эти антикоагулянты являются дериватами кумарина и инданедиона, которые
действуют как антагонисты витамина К. В США наиболее часто используется
натриевая соль варфарина (кумадин).
Варфарин почти всегда вводится перорально, но имеется также препарат для
инъекций. Варфарин представляет собой эквимолярную смесь двух активных
изомеров, S- и R-форм. Изомерический состав вафарина важен в клиническом
отношении, так как S-варфарин в 5 раз более мощный антагонист витамина К, чем
R-форма. Препараты, ингибирующие выведение метаболитов S-формы,
потенцируют антикоагулянтную активность варфарина в большей степени, чем
препараты, ингибирующие клиренс R-формы.
Варфарин быстро абсорбируется желудочно-кишечным трактом, и пиковая
концентрация в крови достигается в пределах 90 мин. В крови он циркулирует
в основном связанным с альбумином и только 3 % находится в свободной форме
в плазме. Но именно этот свободный варфарин биологически активен. Период
полусуществования варфарина составляет в среднем 36-42 ч. Он превращается
печенью в неактивный метаболит, который конъюгируется с глюкуроновой
кислотой и выделяется с мочой и калом.
Препараты, которые мешают связыванию варфарина с альбумином или его
экскреции, также снижают абсорбцию витамина К и нарушают реакцию
организма на варфарин. Следовательно! реакция на препарат зависит от фармакокинети-
ческих факторов (различия в абсорбции или метаболическом клиренсе
варфарина), фармакодинамических факторов (разница в гемостатической реакции),
технических факторов (лабораторные ошибки, плохое взаимодействие между
врачами и больными) и пока еще неявных факторов.
Варфарин и другие кумарины оказывают антикоагулянтное воздействие
за счет нарушения циклической конверсии витамина К и его окисленной
формы — эпоксида витамина К (рис. 6-25). Восстановленный витамин К является
кофактором для пострибосомального у-карбоксилирования остатков глутами-
новой кислоты факторов II, VII, X, IX, ПС, ПБ. у-Карбоксилирование этих
белков приводит к конформационному изменению, которое в присутствии
кальция способствует связыванию с фосфолипидами мембран и тем самым
проявлению биологической активности. Лечение варфарином снижает
функциональную активность этих витамин К-зависимых белков и приводит к
развитию гипокоагуляционного состояния крови. Самое раннее проявление
эффекта варфарина — изменение содержания ФУИ и ПС, поскольку у этих
белков короткий период полусуществования (6 ч), затем — Ф1Х и ФХ и nS
(24-48 ч) и наконец содержания протромбина (72 ч). Основная задача лечения
состоит в снижении количества протромбина для минимизации образования
Нарушения тромбообразования
273
тромбина, поэтому противосвертывающий эффект варфарина не достигается
ранее чем через 3 дня.
Разовая доза варфарина у пожилых больных, женщин и больных с гемостати-
ческими нарушениями должна быть низкой. Для других пациентов допустимо
начинать с более высокой дозы. Ее следует повторять последовательно в течение
3-х дней, а затем отрегулировать в зависимости от ПВ или MHO. Большие дозы,
равные 20 мг, быстро изменяют MHO вследствие выраженной супрессии OVII,
но для проявления антикоагулянтного эффекта in vivo на протромбин требуется
3 дня. Кроме того, большие дозы иногда вызывают существенное снижение
уровня ПС и способствуют возникновению варфарининдуцированного некроза кожи.
Мониторинг пероральной терапии антикоагулянтами
Мониторинг результатов терапии осуществляют по величине ПВ. Тест
стандартизован и называется ПВ/МНО (описан в разделе лабораторных тестов). Его величины
при определении показаний к антикоагулянтной терапии представлены в табл. 6-18.
Продолжительность лечения
Продолжительность краткосрочного лечения составляет 1 мес при подготовке
к кардиоверсии. При тромбозе глубоких вен и легочной эмболии среднее время
лечения 3-6 мес. Долгосрочное лечение длится свыше 12 мес (рецидивирующая
тромбоэмболия). Больные с фибрилляцией предсердий, протезированными
клапанами сердца, ревматическим заболеванием сердца, артериальным
трансплантатом и тромбофилией или АФС нуждаются в постоянном, или пожизненном,
приеме антикоагулянтов.
Осложнения при лечении варфарином
Ведущее осложнение, связанное с приемом варфарина,— кровотечение. Риск его
развития зависит от интенсивности лечения. Если MHO больше 4, риск выше,
чем при 2,0-3,0. Небольшие кровотечения (носовые) лечат местными средствами
ТАБ Л И ЦА 6 -18. Критерии диагностического мониторинга
при пероральном введении антикоагулянтов
Показания MHO
Венозный тромбоз 2,0-3,0
Профилактика (высокий риск возникновения
при хирургической операции)
Лечение тромбоза глубоких вен и легочной эмболии
Артериальный тромбоз 2,0-3,0
Фибрилляция предсердий
Кардиомиопатия
Гипо- и дискинезия
Инфаркт миокарда
Внутрисердечные тромбы
Повреждения сердечных клапанов
Периферические сосудистые заболевания
Механические протезы клапанов сердца 2,5-3,5
Инфаркт миокарда (для профилактики рецидива) 2,5-3,5
Возвратная системная эмболия 2,5-3,5
274
Глава 6. Гемостаз и тромбоз
и снижением дозы варфарина (или его отменой). Массивные кровотечения
(большие гематомы, внутричерепные или ретроперитонеальные кровотечения)
требуют введения свежезамороженной плазмы и/или витамина К (парентерально).
Кровотечение из желудочно-кишечного тракта или мочевыводящих путей
свидетельствует о наличии патологии внутренних органов и требует соответствующей
диагностики.
Синдром багровых пальцев ног, который связан с некрозом кожи,— более
редкое осложнение терапии варфарином. Он возникает через 1-6 недель лечения
и является результатом тромбоза венул или капилляров подкожной жировой
клетчатки (может быть также поражена жировая ткань в области живота и груди).
Синдром характеризуется болью и посинением подошвенной поверхности
большого пальца стопы, второго, третьего и четвертого пальцев. Отмечается связь
между некрозом кожи и дефицитом ПС и IIS. Однако это осложнение иногда
возникает у больных без дефицита ПС и ПБ и может быть вызвано чрезмерными
дозами варфарина. У больных с такими заболеваниями, как васкулит, внутрисер-
дечные тромбы, большие аневризмы брюшного отдела аорты и с активацией
крови синдром багровых пальцев ног может развиться, если начинать лечение
варфарином без предварительной терапии гепарином. Лечение некроза кожи
включает отмену варфарина и внутривенное введение гепарина. Если требуется
длительное лечение антикоагулянтами, а варфарин противопоказан, больным
вводят подкожно гепарин или низкомолекулярный гепарин.
В табл. 6-19 представлены лекарственные препараты и другие факторы,
влияющие на ход лечения варфарином.
Тромболитические препараты вызывают растворение тромбов (табл. 6-20). Они
обычно применяются для лечения острой стадии инфаркта миокарда, а также эф-
ТАБЛ ИЦА 6-19. Факторы, влияющие на лечение варфарином
Потенцирование действия варфарина Ингибирование действия
варфарина
Препараты, снижающие связывание альбу- Препараты, снижающие действие
мином: фенилбутазон, статины, сульфанил- варфарина: барбитураты, рифам-
амиды, клофибрат пин, пенициллин, алкоголь, карба-
мазепин, гризеофульвин
Препараты, ингибирующие метаболический Повышенный синтез факторов свер-
клиренс: дисульфирам, триметоприм-суль- тывания крови: пероральный при-
фаметоксазол, дифенилбутазон, сульфинпи- ем противозачаточных средств
разон, циметидин, омепразол, статины, ами- '
одарон, аллопуринол, трициклические
антидепрессанты, противогрибковые препараты
Препараты, взаимодействующие с витами- Наследственная резистентность
ном К: цефалоспорины (1 и 2-го поколения), к варфарину
салицилаты в высокой дозе
Препараты, влияющие на печеночный рецеп- Беременность
тор: тироксин и хинидин
Потенцирование антикоагулянтного
действия: эритромицин, клофибрат,
анаболические стероиды, патология печени,
повышенная абсорбция витамина К
Нарушения тромбообразования
275
ТАБЛИЦА 6-20. Тромболитическив агенты
Агенты Период полусуществования
(мин)
Стрептокиназа 18-25
Урокиназа 13-20
Тканевый активатор плазминогена (ТАП) 2-6
Активатор плазминогена — одноцепочечная урокиназа 5-8
Анизолированный плазминоген-стрептокиназный 70-90
активированный комплекс (АПСАК, антистрептаза)
фективны для начального лечения острой окклюзии периферических артерий,
массивной легочной эмболии и в некоторых случаях тромбоза глубоких вен.
Клинический эффект тромболитической терапии основан на способности
препаратов генерировать плазмин из плазминогена в системе кровообращения или
в тромбе (рис. 6-13). Плазмин, образующийся в циркуляции, в физиологических
условиях быстро нейтрализуется антиплазмином. Но в ходе тромболитической
терапии ингибиторы интенсивно потребляются, что позволяет свободному плаз-
мину растворять фибрин. Возникает литическое состояние, при котором избыток
плазмина также переваривает фибриноген, факторы свертывания (V, VIII, XII,
ФВ), тромбоспондин и гликопротеины мембраны тромбоцита (ГШЬ, ГПНЬ/Ша).
Связанный с тромбом плазмин не инактивируется антиплазминами и быстро
инициирует лизис тромба.
При однократном введении стрептокиназы, рекомбинантного ТАП или
комплекса АКСАП лабораторный мониторинг необязателен. При лечении легочной
эмболии стрептокиназой или урокиназой по крайней мере 2 раза в день
определяют тромбиновое время, уровень фибриногена и ПДФ, чтобы обеспечить
состояние лизиса.
К осложнениям тромболитической терапии относятся кровотечение и
аллергические реакции. Кровотечение наблюдается у 3-40 % больных, и его частота
возрастает у пациентов, получающих сочетанную терапию антитромбоцитарны-
ми препаратами, блокаторами кальциевых каналов или антикоагулянтами. Оно
возникает в местах прокола сосудов при проведении диагностических или
терапевтических процедур или мониторинга. Неприменение инвазивных процедур
снижает риск возникновения массивного кровотечения до 5 %. Внутримозговое
кровотечение регистрируется у 2 % больных, особенно у пожилых людей,
женщин, пациентов с низкой массой тела, а также при наличии в анамнезе
заболевания сосудов головного мозга и у больных, принимавших одновременно антитром-
боцитарные или антикоагулянтные препараты.
Аллергические реакции возникают при лечении стрептокиназой или АКСАП
в результате предшествующей стрептококковой инфекции. Противопоказания
к проведению тромболитическойдерапии представлены в табл. 6-21.
Клинический пример
22-летняя женщина жалуется на частое появление синяков. Месяц тому
назад у нее возникло несколько синяков в области бедер и плеч при
отсутствии даже незначительной травмы. Со слов больной, были проблемы
с менструацией: "значительно хуже, чем у знакомых'9 — более длитель-
276
Глава 6. Гемостаз и тромбоз
ТАБЛИЦА 6-21. Противопоказания к проведению тромболитической терапии
Абсолютные противопоказания Относительные противопоказания
Недавно перенесенное или Большая хирургическая операция или травма
активное внутреннее кровотечение (произошедшая менее 10 дней тому назад)
Черепно-мозговая травма, хирур- Геморрагический диатез, врожденный или
гическое вмешательство, крово- приобретенный
излияние или инсульт в последние Желудочно-кишечное кровотечение в анамнезе
два месяца Травмирующая сердечно-легочная
Расслоение аневризмы аорты реанимация?
Внутричерепная аневризма Тромбоцитопения
или опухоль Биопсия органа или артериальная пункция,
Внутриспинномозговая опухоль выполненные менее 10 дней назад
Подострый эндокардит или внутрисердечный
тромб в левом желудочке
Неконтролируемая гипертензия
Беременность или роды (менее 10 дней назад)
ные и обильные. В детстве отмечались носовые кровотечения, легко
возникали синяки. Никогда ранее не подвергалась хирургическому
вмешательству, включая удаление зубов. Беременности не было. У ее матери и
младшей сестры столь же легко возникали синяки и кровотечения; отец
здоров. Больная не принимала никаких лекарств, училась на медсестру-
анестезиолога. Не злоупотребляла алкоголем и наркотиками. Факторы
риска ВИЧ-инфекции отсутствовали. При физикальном обследовании
патологии внутренних органов не выявлено. На коже наружной
поверхности бедер и плеч экхимозы различных стадий разрешения диаметром
2-3 см. На слизистых оболочках нет ни петехий, ни пурпуры. Аномалий
головы, ушей, глаз, носа, горла, шеи, легких, сердца не выявлено.
Органы брюшной полости без патологии. Результаты обследования грудных
желез, прямой кишки, гениталий в норме. Неврологическое
обследование патологии не выявило.
Вопрос 1. Какое заболевание можно предположить?
Ответ. Наиболее характерным наследственным нарушением, связанным
с кровоточивостью, является болезнь Вилленбранда (примерно 1 на 1000
человек). Частота гемофилии А составляет 1 йа 10 000 мужчин
(наследование дефектной Х-хромосомы). Развитие симптоматической коагулопатии
у женщин возможно только при наследовании двух аномальных Х-хромо-
сом. Разумеется, что при здоровом отце, но при наличии симптомов у
матери и сестры наиболее вероятным генетическим дефектом коагуляции
у этой женщины является БВ, а не другие наследственные дефекты.
Возможно и приобретенное расстройство свертывания крови, особенно с
учетом значительного утяжеления симптоматики в последнее время. Следует
обязательно изучить семейный анамнез. '
Вопрос 2. Что делать в первую очередь?
Ответ. Лабораторное обследование необходимо начать с определения
ПВ и АЧТВ, что позволит оценить гуморальную систему свертывания
Нарушения тромбообразования
277
крови. Определить количество тромбоцитов, сделать анализ мазка
периферической крови, а затем, возможно, исследовать функцию
тромбоцитов — определить время кровотечения. Аномалии факторов свертывания
крови обычно приводят к возникновению экхимозов и других
геморрагических нарушений. Качественные и количественные нарушения
тромбоцитов вызывают появление петехий и пурпуры, а в ряде случаев —
экхимозов.
Вопрос 3. Оказалось, что ПВ — 11 с; АЧТВ — 34 с (т. е. оба показателя
в норме). Количество тромбоцитов — 345 000/мкл при нормальном мазке
периферической крови. ВК — 8 мин (норма). Что делать дальше? Можно ли
сказать пациентке, что она здорова?
Ответ. Хотя лабораторные показатели без патологических изменений,
АЧТВ и ВК на уровне верхней границы нормы. При болезни Виллебранда
величины показателей могут быть в различные периоды нормальны или
слегка отклоняться от нормы. В контроле гуморальной системы
свертывания крови могут быть задействованы гормональные или другие факторы,
которые приводят к развитию клинических или лабораторных аномалий
(женщину необходимо обследовать в первую-вторую недели ее
менструального цикла — в прелютеиновой фазе). Следовательно, определение
показателей надо повторить в следующем месяце.
Вопрос 4. После повторного исследования АЧТВ — 39 с; ВК — 9,5 мин. Что
делать дальше?
Ответ. Обратить внимание на повышенное АЧТВ, выполнить 1 : 1
микст-тест (плазма больной + нормальная плазма). Это скорригирует
дефицит фактора, если в плазме больной нет ингибитора. У пациентки
АЧТВ действительно снизилось до 27 с. Учитывая повышенные ВК и
АЧТВ, можно поставить диагноз болезни Виллебранда (дефицит
системы свертывания плюс дефект тромбоцитов) и провести скрининг-тесты
наБВ.
В дальнейшем получены следующие результаты: OVIII:C — 50 % (норма
50-180 %); КР — 50 % (норма 50—150 %); антиген фактора Виллебранда —
50% (норма 50-180%).
Вопрос 5. Это окончательный диагноз? Надо ли предпринять что-нибудь
еще?
Ответ. Результаты обследования полностью соответствуют БВ, но
необходимо определить ее подтип, так как лечение десмопрессином может
привести у больных с типом ИВ к потреблению тромбоцитов и, следовательно,
к ухудшению состояния системы свертывания крови. У больных с типом I,
наиболее частым, таких осложнений при лечении десмопрессином нет.
Вопрос 6. Как можно определить подтип?
Ответ. Выполнить исследование методом вестерн-блот на мультимеры
Виллебранда. У данной больной мультимеры оказались в качественном
отношении нормальны, но их количество снижено, что позволило
поставить диагноз БВ типа I. Теперь необходимо сообщить всю информацию
больной.
Наблюдение. Через 1 год после постановки диагноза был успешно удален
зуб мудрости с профилактическим использованием десмопрессина.
Больная чувствует себя хорошо.
278
Глава 6. Гемостаз и тромбоз
Клинический пример
Женщина в возрасте 23 лет обратилась за неотложной помощью в связи
с отеком и болью в области левой икры. Ранее была здорова, не
переносила каких-либо серьезных заболеваний, требовавших госпитализации.
Ежедневно принимала противозачаточные средства. За три дня до
обращения почувствовала дискомфорт в области левой икры, которая затем
стала болезненной, отекшей и покраснела. Также появилась боль в бедре.
У больной не было ни одной беременности; не курит, не употребляет
алкоголь и обезболивающие лекарственные препараты. Факторов риска
ВИЧ-инфекции не выявлено. Работает клерком. В последнее время не
было иммобилизации конечностей, и больная не совершала
туристических поездок. Ее дядя скончался в возрасте 37 лет после хирургического
вмешательства. При осмотре выявлены отечность, эритема, гипертермия,
левая икра болезненна. Венозных тяжей не обнаружено, но имеются
признаки отека. Паховые лимфатические узлы не увеличены,
безболезненные, мягкие. Показатели лабораторного скрининга в норме. При
ультразвуковом исследовании выявлен тромбоз глубоких вен икры и бедра.
Начали терапию гепарином и спустя 24 ч — варфарином.
Вопрос 1. Надо ли обследовать больную на предмет гиперкоагуляционного
состояния?
Ответ. Да. Семейный анамнез и, по-видимому» неспровоцированный
тромбоз глубоких вен могут быть признаками наследственного
гиперкоагуляционного состояния, индуцированного приемом противозачаточных средств.
Вопрос 2. Какие надо провести исследования?
Ответ. Массовый скрининг предрасположенности к гиперкоагуляции
дорог, однако во многих лабораториях в настоящее время выполняют весьма
экономичные исследования, позволяющие выявить наиболее
распространенные аномалии, обусловливающие гиперкоагуляционное состояние.
Тесты на дефицит антитромбина III, ПС, ПБ, увеличение уровня ВА и других
антифосфолипидных антител, резистентности к. активированному ПС и
повышения гомоцистеина.
Вопрос 3. Когда их лучше всего проводить?
Ответ. Кровь надо взять до начала лечения гепарином или варфарином, так
как гепарин изменяет содержание антитромбина III, а варфарин — ПС и
nS. В связи с активным образованием сгустка в ряде: тестов могут
получиться неверные результаты. Поэтому необходимо завершить курс терапии
и через 2 недели после этого провести тестирование. Функциональные
пробы в скрининг-тесте на активированный протеин С выявили низкое
содержание этого белка. Впоследствии обнаружена мутация фактора V Лейден.
Вопрос 4. Что следует рекомендовать больной?
Ответ. У больной имеется предрасположенность к повышению
свертываемости крови, обусловленной выявленным нарушением. Ей не желательно
принимать противозачаточные средства, содержащие эстроген, так как он
индуцирует гиперкоагуляционное состояние. Кроме того, в случае
иммобилизации конечностей, например при хирургических вмешательствах,
необходима профилактика антикоагулянтами. Пока нельзя установить, должна
ли больная принимать антикоагулянты всю жизнь, если у нее нет других
Нарушения тромбообразования
279
факторов риска. Проведение антикоагулянтной терапии при состоянии
с высоким уровнем эстрогенов, например при беременности, может
оказаться необходимым. Желательно провести скрининг-тесты у ее
родственников, чтобы также дать им рекомендации.
Наблюдение. Больная сообщила родственникам о своей болезни и убедила
их пройти тестирование. После обсуждения с врачом она решила прекратить
прием противозачаточных препаратов, но не согласилась лечиться
антикоагулянтами и решила вернуться к этому вопросу в случае беременности.
Избранная литература
Нормальный гемостаз
Colman W. R., Marder V. J., Salzman E. W., Hirsh J. Overview of hemostasis. In: Colman
R. W., Hirsh J., Marder V. J., Salzman E. W. (eds). Hemostasis and Thrombosis: Basic
Principles and Clinical Practice, 3rd ed. Philadelphia: J. B. Lippincott; 1994:3-18.
Один из наиболее всеобъемлющих обзоров по гемостазу, имеющихся в литературе.
Esmon С. Т. The roles of protein C and thrombomodulin in the regulation of blood
coagulation./. Biol Chem., 1989; 264:4743-4749.
В статье впервые описана роль протеина С и тромбомодулина в системе
свертывания крови.
Furie W. A., Furie В. С. Molecular basis of vitamin K-dependent gamma carboxylation.
N. Engl.J. Med., 1990; 75:1753-1762.
Детальное описание биохимии витамин KJ3aeucuMbix факторов свертывания крови.
Hawiger J. Adhesive interactions of platelets and their blockade. Ann. NY Acad. Sci., 1991;
614:270-278.
В статье рассмотрены механизмы адгезии тромбоцитов в процессах
физиологического гемостаза и тромбоза. Кроме того, обсуждается возможность использования
методов ингибирования адгезии тромбоцитов в клинической практике.
Ruggeri Z. M., Ware J. The structure and function of vWf. Thromb. Haemost., 1992; 67:
594.
Классическая статья по биохимии, функциям и лабораторной оценке фактора Вил-
лебранда.
Salvesen G., Pizzo S. V. Proteinase inhibitors : a-macroglobins, serpins, and kunins. In:
Colman R. W., Hirsh J., Marder V. J., Salzman E. W. (eds). Hemostasis and
Thrombosis: Basic Principles and Clinical Practice, 3rd ed. Philadelphia: J. B.
Lippincott; 1994:241-258.
Лучший обзор по данной теме.
Shen L., Dahlback B. Factor V and protein S as synergistic cofactors to activated protein С
in degradation of factor Villa./. Biol. Chem., 1994; 269:18735-18738.
Оригинальный обзор, содержащий много информации.
Патология гемостаза и тромбоз
Arnout J. The pathogenesis of the antiphospholipid syndrome: a hypothesis based on the
parallelisms with heparin-induced thrombocytopenia. Thromb. Haemost., 1996; 75:
536-541.
Обзорная статья дискуссионного характера.
280
Глава 6. Гемостаз и тромбоз
Asherson R. A., Khamashta M. A., Gil A. et al. The primary antiphospholipid syndrome:
major clinical and serological features. Medicine, 1989; 68:366-374.
Хороший обзор по антифосфолипидному синдрому.
Dahlback В. Resistance to activated protein, the Arg 506 to Gin mutation in the factor V
gene, and venous thrombosis. Thromb. HaemosL, 1995; 73:739-742.
Великолепная статья, посвященная дефекту фактора Упри тромбозе.
Dahlback В., Hildebrand В. Inherited resistance to activated protein С is corrected by
anticoagulant cofactor activity found to be a property of factor V. Proc. Natl. Acad.
Sci. USA, 1994; 91:1396-1400.
Оригинальная статья, в которой описан механизм резистентности к протеину С.
De Stefano V., Finazzi G.f Mannucci P. M. Inherited thrombophilia: pathogenesis, clinical
syndromes, and management. Blood, 1996; 87:3531-3544.
Информативный обзор источников литературы.
Feinstein D. I. Acquired disorders of hemostasis. In: Colman R. W., Hirsh J., Marder V. J.,
Salzman E. W. (eds). Hemostasis and Thrombosis: Basic Principles and Clinical
Practice, 3rd ed. Philadelphia: J. B. Lippincott; 1994:881-905.
Глава посвящена всем формам приобретенных нарушений гемостаза и их лечению.
Fourth АССР Consensus Conference on Antithrombotic Therapy. Dalen J. E., Hirsh J.
(eds). Chest, (supplement), 1995; 108: 225-521.
Обзор методик проведения антитромботической терапии.
Geerts W. H., Jay R. M., Darmon J.-K. et al. A comparison of low dose heparin with low
molecular weight heparin as prophylaxis against venous thromboembolism after
major trauma. N Engl. J. Med., 1996; 335:701-707.
Обзор современных данных.
Greengard J. S., Sun X., Xu X., Fernandez J. A., Griffin J. H., Evatt B. Activated protein С
resistance caused by Arg 506 Gin mutation in FVa. Lancet, 1994; 343:1361-1362.
Прекрасная работа, демонстрирующая причину резистентности к протеину С.
Нагкег L. A., Slichter S. J., Scott С. R., Ross R. Homocysteinemia: vascular injury and
arterial thrombosis. N. Engl. J. Med., 1974; 291: 537-543.
Hoyer L. W., Hemophilia A. N. Engl.J. Med., 1994; 330:38-47.
Обзор, написанный одним из самых известных исследователей биохимии фактора VIII
и посвященный диагностике и лечению дефицита этого белка, а также
взаимоотношениям ФУШ и ФВ.
Joist H.J. Hemostatic abnormalities in liver disease. In: Colman R. W., Hirsh J., Marder
V.j Salzman E. W. (eds). Hemostasis and Thrombosis: Basic Principles and Clinical
Practice, 3rd ed. Philadelphia: J. B. Lippincott; 1994:906-920.
Обзор нарушений гемостаза при заболеваниях печени.
Marder V. J., Feinstein D. J., Francis C. W.f Colman R. W. Consumptive thrombo-
hemorrhagic disorders. In: Colman R. W., Hirsh J., Marder V., Salzman E. W. (eds).
Hemostasis and Thrombosis: Basic Principles and Clinical Practice, 3rd ed.
Philadelphia: J. B. Lippincott; 1994:1023-1063.
Наиболее всеобъемлющий обзор по данной теме.
Schaffer A. I. Hypercoagulable states : molecular genetics to clinical practice. Lancet,
1994;344:1739-1742.
Краткая, но информативная статья.
Нарушения тромбообразования
281
Scott J. P., Montgomery R. R. Therapy of von Willebrand disease. Semin. Thromb. Hemost,
1993;19:37-47.
Великолепный обзор диагностики и терапии болезни Виллебранда.
Triplett D. A. Laboratory diagnosis of lupus anticoagulant. Semin. Thromb. Hemost., 1990;
16:182-191.
Исчерпывающий обзор по лабораторной диагностике волчаночного антикоагулянта.
Triplett D. A. Antiphospholipid antibodies and thrombosis: a consequence, coincidence or
cause? Arch. Pathol. Lab. Med., 1993; 117:78-88;
Полный обзор с многочисленными идеями автора, касающимися данной проблемы.
Wetz J. I. Low molecular weight heparin. N. Engl. J. Med., 1997; 337:688.
Обзор последних данных по механизмам действия и рекомендации по лечению острого
тромбоза низкомолекулярным гепарином.
Глава 7
Недостаточность
костного мозга,
апластическая анемия
Даниель Е. Данн
Цитопении периферической крови (анемия, лейкопения или тромбоцитопения)
являются следствием ускоренной гибели, депонирования или разрушения зрелых
циркулирующих клеток крови либо следствием нарушения созревания клеток
в результате токсического воздействия на их источник — костный мозг. В данной
главе будут рассмотрены состояния, относящиеся только ко второй категории,
называемые синдромами недостаточности костного мозга. Механизм этого
нарушения может быть первичным (при врожденной или наследственной анемии),
вторичным, обусловленным внешними по отношению к костному мозгу
причинами (при аутоиммунных синдромах, анемиях медикаментозной или вирусной
природы), но чаще всего его этиология неизвестна. В главе делается попытка
систематизировать эти синдромы на основе их клинических проявлений и/или этиологии
(когда она известна).
Приобретенная апластическая анемия
Приобретенная апластическая анемия,(АА) характеризуется панцитопенией
периферической крови и аплазией ("сниженной клеточностыо") костного мозга.
В этом случае гемопоэтическая ткань замещается жировой. АА обычно идиопати-
ческая: ее развитие может быть обусловлено другими состояниями, но причинно-
следственные связи пока не установлены. Наличие клональных нарушений
(таких как злокачественные новообразования или миелодиспластический
синдром — МДС), которые нарушают гемопоэз, исключает диагноз АА.
Эпидемиология и этиология
Частота встречаемости
1. 2 на 106 в Западной Европе (и, возможно, в США).
2. В 2-3 раза больше на Дальнем Востоке.
Эпидемиология. Наибольшая частота заболеваемости отмечена у пациентов в
период ранней зрелости (молодости) и пожилом возрасте. Никакой расовой или
половой предрасположенности не выявлено.
284 Глава 7. Недостаточность костного мозга, апластическая анемия
Этиология
1. Химические агенты. Часто пациенты с нераспознанной АА до проведения
им каких-либо гематологических исследований получали антибиотики и
нестероидные противовоспалительные средства (НПВС); поэтому
причинно-следственные связи между приемом этих препаратов и развитием
идентифицированной впоследствии АА установить трудно. Однако применение
некоторых лекарств и химических агентов (большинство из которых
характеризуется наличием бензольного кольца), по-видимому, значительно
повышает риск заболевания АА:
а) бензол. Причинная связь вероятна. Представляют опасность также
родственные ароматические углеводороды (тринитротолуол) и
инсектициды;
б) хлорамфеникол. НПВС (особенно индометацин и фенилбутазон), соли
золота, сульфаниламиды, некоторые нейролептики, антитиреоидные
препараты, противосудорожные средства (например, дифенилгидантоин
и карбамазепин);
в) цитотоксические химиотерапевтические агенты и ионизирующее
облучение приводят к панцитопении и снижению клеточности костного мозга.
2. Некоторые вирусные инфекции иногда сопровождаются гипопластически-
ми цитопениями:
а) инфекционный мононуклеоз, вызванный вирусом Эпштейна-Барр (ВЭБ);
б) гепатит (ни А, ни В, ни С);
в) ВИЧ;
г) парвовирус В19 вызывает изолированное подавление эритроидного
ростка, транзиторный апластический криз или парциальную красноклеточ-
ную аплазию (см. соответствующий раздел).
3. Иммунные заболевания:
а) болезнь "трансплантат против хозяина" (БТПХ): гемопоэтические
предшественники являются мишенями для аллореактивных лимфоцитов
донора, например при трансфузиях;
б) эозинофильный фасцит;
в) гипоиммуноглобулинемия;
г) тимома и карцинома тимуса.
4. Пароксизмальная ночная гемоглобинурия (ПНГ).
5. Беременность.
6. Идиопатическая АА. В большинстве случаев развитие АА не связано с
какими-либо этиологическими факторами.
Историческая справка
В дотрансфузионную эру нелеченая апластическая анемия обычно заканчивалась
смертью больного в течение 6 месяцев. Благодаря развитию трансфузионной
медицины пациенты, не реагирующие на медикаментозную терапию, получили шанс
продления жизни на несколько лет. При этом у них может сформироваться реф-
рактерность (невосприимчивость) к аллогенным тромбоцитам вследствие
появления аллоантител. В конечном счете при наличии постоянной нейтропении
повышается риск возникновения бактериальных или, чаще, грибковых
заболеваний. Вероятность спонтанных ремиссий очень мала.
Приобретенная апластическая анемия
285
Патофизиология
Результаты многочисленных лабораторных и клинических исследований
свидетельствуют о стойком количественном дефиците наиболее примитивных
гемопоэтических стволовых клеток у больных АА. В большинстве случаев
медикаментозной и идиопатической АА подавление костного мозга опосредовано
иммунными механизмами. В костном мозге и периферической крови больных АА
часто находят активированные цитотоксические Т-лимфоциты (CD8+DR+).
Данные клетки способны производить как интерферон (ИФ-у), так и фактор некроза
опухоли (ФНО-а) — антипролиферативные цитокины, угнетающие гемопоэз
за счет индукции апоптоза в гемопоэтических клетках-мишенях. Этим можно
объяснить безрезультатность трансплантации костного мозга от сингенного
донора (идентичного близнеца без АА). Иногда иммуносупрессивные препараты,
обычно назначаемые как альтернатива облучению до трансплантации, вызывали
ремиссию даже без пересадки костного мозга.
Диагноз
Симптомы и признаки. Типичными проявлениями панцитопении являются
кровоточивость, кровоподтеки, инфекции, вялость, учащение дыхания и др.
Необъяснимые симптомы (снижение веса, зуд) и признаки (лимфаденопатия,
гепатоспленомегалия) должны побудить к поиску иной причины развития ци-
топении.
Степень тяжести. Тяжелую апластическую анемию диагностируют по
сниженной клеточности костного мозга и по наличию двух из следующих трех
критериев.
1. Абсолютное количество ретикулоцитов < 40 000/мкл.
2. Абсолютное количество нейтрофилов < 500/мкл.
3. Количество тромбоцитов < 20 000/мкл.
При очень тяжелой апластической анемии (количество нейтрофилов < 200/мкл)
прогноз значительно ухудшается.
Умеренная апластическая анемия — это гипопластическая панцитопения, не
соответствующая строго критериям, приведенным выше.
Лабораторные исследования
1. В дополнение к стандартным биохимическим исследованиям (значительно
возросшие показатели функциональных проб печени, например, наводят
на мысль о синдроме гепатита/аплазии), а также определению содержания
фолата и витамина Bt2 все пациенты должны быть обследованы на ВИЧ.
Для выявления пароксизмальной ночной гемоглобинурии (ПНГ)
выполняют тест Хэма.
2. Цитогенетические исследования.
3. В зависимости от клинической картины целесообразно выполнить один или
оба следующих теста на наличие:
а) парвовируса В19 — при изолированном поражении эритроидного ростка;
б) анемии Фанкони (АФ) — иногда у лиц среднего возраста (< 50 лет) тест
положителен, несмотря на отсутствие характерных скелетных аномалий.
286
Глава 7. Недостаточность костного мозга, апластическая анемия
Исследование костного мозга
1. Обязательно выполнение биопсии костного мозга, потому что в мазках
аспирата невозможно количественно оценить цитоз (клеточность).
Стандартный критерий АА — цитоз в биоптате менее 25 %; хотя поражение может
быть очаговым. Негемопоэтическая составляющая костного мозга в
основном представлена жировыми клетками. Иногда присутствуют лимфоциты,
плазматические клетки и макрофаги. Если лимфоциты организованы в па-
ратрабекулярные фолликулы, следует предположить развитие лимфомы.
Ретикулин не повышен.
2. В аспиратах костного мозга количество гемопоэтических клеток
незначительное или они отсутствуют. Часто обнаруживается дисплазия эритроид-
ного ростка; выявленная дисплазия гранулоцитарного или мегакариоцитар-
ного ростка подтверждает диагноз миелодиспластического синдрома.
Дифференциальный диагноз (панцитопении с низким цитозом)
1. Наследственные апластические анемии: анемия Фанкони и др.
2. Миелодиспластический синдром — гипоцеллюлярный вариант (табл. 7-1).
3. Алейкемический лейкоз — острый миелобластный лейкоз.
4. Острый лимфоцитарный лейкоз.
5. Лимфома.
6. Волосатоклеточный лейкоз.
7. Инфекции (микобактерии, легионелла, лихорадка Ку).
8. Другие причины (голодание, гипотиреоз).
ТАБЛИЦА 7-1. Морфология костного мозга при миелодисплазии
и апластичвской анемии
Показатели
Миелодисплазия
Апластическая анемия
Цитоз (клеточность)
Эритропоэз
Мегалобластный
Дизэритропоэтический
Дефекты созревания
Кольцевые сидеробласты
Миелопоэз
Преимущественно
моноцитарный
Преимущественно
среднемиелоидный
Повышенный бластоз
Мегакариоциты
Атипичная морфология
Обычно нормальный,
повышенный
Очень часто
Очень часто
Часто
Часто
Очень часто
Очень часто
Имеется
Очень часто
Сниженный
Часто
Редко
Не определяются
Не определяются
Редко
Редко
Не определяется
Не определяется
При некоторых миелодиспластических синдромах обнаруживаются кольцевые сидеробласты
и миелобласты. Выявляются дизэритропоэтические предшественники эритроцитов
необычной формы с множественными или неправильными ядрами. Мегакариоциты могут иметь
дефект ядерной полиплоидизации и увеличение межъядерных пространств; они также бывают
малыми, всего лишь с несколькими ядрами и специфической зернистостью.
Приобретенная апластическая анемия
287
Лечение
Задержка начала лечения больных А А абсолютно не оправдана и лишь увеличивает
риск возникновения инфекций и осложнений при гемотрансфузиях. Необходимо
быстро принять решение о проведении иммуносупрессивной терапии или
трансплантации костного мозга и сразу же приступить к выполнению. Сопутствующие
аплазии неотложные состояния (например, системная инфекция или гепатит)
в большинстве случаев не являются основанием для отсрочки начала терапии.
Трансплантация костного мозга (глава 8). Трансплантация костного мозга
(ТКМ) от HLA-совместймого сибса корригирует два важнейших звена патогенеза
при АА: аутоиммунитет и дефект гемопоэтических стволовых клеток. При этом
возникает риск развития новой патологии — болезни "трансплантат против
хозяина" (БТПХ). При ТКМ следует помнить, что для АА характерно увеличение
частоты отторжения трансплантата. По-видимому, это обусловлено усилением
аутоиммунных реакций реципиента именно против пересаживаемого органа —
костного мозга. Попытки борьбы с одним заболеванием часто ведут к обострению
второго. Таким образом, если для ТКМ используются гемопоэтические
стволовые клетки, очищенные от лимфоцитов донора, то снижается вероятность
развития БТПХ, но повышается риск отторжения/несостоятельности трансплантата.
Важный фактор прогноза отторжения — количество гемотрансфузий,
проведенных до трансплантации: введение более 40 доз тромбоцитов значительно
повышает риск отторжения трансплантата. Следовательно, решение о пересадке должно
быть принято как можно быстрее после диагностирования АА и выполнено
незамедлительно. Важный прогностический фактор для БТПХ — возраст больного:
вероятность возникновения острой или хронической БТПХ у молодых пациентов
(< 20 лет) — приблизительно 25 %; у пациентов старшего возраста — больше.
Стандартная схема подготовки пациента к ТКМ включает введение высокой
дозы циклофосфамида в течение 4-х дней. Общее облучение тела уменьшает
вероятность отторжения трансплантата, но увеличивает риск развития БТПХ и,
возможно, поздних осложнений, таких как злокачественные опухоли.
Долгосрочная выживаемость больных с АА младше 40 лет, получивших трансплантат
от HLA-совместимого сибса, составляет в различных учреждениях от 60 %
до 90 %, но значительно снижается с увеличением возраста пациента.
Иммуносупрессия. ТКМ от HLA-совместимого сибса доступна лишь немногим
пациентам. За последние 20 лет был достигнут значительный прогресс в
иммуносупрессивной терапии: на сегодняшний день при введении стандартной дозы ан-
титимоцитарного глобулина (АТГ) (в Европе антилимфоцитарного глобулина —
АЛ Г) в сочетании с циклоспорином А (ЦсА) можно ожидать ремиссии
приблизительно у 70 % больных независимо от их возраста. Трехлетняя выживаемость для
пациентов после иммуносупрессии соответствует примерно таковой после ТКМ.
Возможно, более длительная выживаемость будет выявлена после обработки
результатов иммуносупрессивной терапии больных АА препаратами последнего
поколения. Оба компонента терапии режима АТГ + ЦсА обладают значительной,
но корригируемой токсичностью. В течение первых 2-х недель для профилактики
тяжелой сывороточной болезни, возникающей в ответ на введение АТГ/АЛГ,
назначается метилпреднизолон в низкой дозе. При тяжелой артралгии и других
осложнениях дозу стероида можно временно увеличить, не забывая, однако, сни-
288 Глава 7. Недостаточность костного мозга, апластическая анемия
зить ее при первой же возможности. Вследствие приема циклоспорина иногда
развиваются почечная недостаточность, гипертензия, гипертрофия десен,
гипертрихоз, миалгия и артралгия; в этом случае дозу препарата уменьшают до 200-
400 нг/мл. В период назначения ЦсА необходимо проводить профилактику
пневмонии, вызываемой Pneumocystis carinii; для этого часто используют ингаляции
пентамидина, позволяющего избежать потенциального миелосупрессивного
действия триметоприма-сульфаметоксазола или дапсона. Длительные заболевания
вследствие назначения АТГ/АЛГ или ЦсА редки.
Сравнение результатов ТКМ и иммуносупрессии. Пациенты младше 20 лет
лучше переносят ТКМ, поэтому для них при наличии HLA-совместимого донора-
сибса предпочтителен данный метод лечения. Для пациентов старше 20 лет оба
терапевтических подхода имеют как преимущества, так и недостатки. Летальные
исходы при ТКМ возникают у них в основном вследствие развития БТПХ и чаще
случаются в течение 2-х лет после трансплантации. Побочные эффекты иммуно-
супрессивной терапии проявляются медленнее: поздние осложнения включают
рецидив (например, в ответ на второй курс иммуносупрессии) и развитие кло-
нальных нарушений типа ПНГ, миелодиспластического синдрома и истинного
лейкоза. Выздоровление больных АА после иммуносупрессии, в отличие от ТКМ,
в большинстве случаев неполное, с частыми бессимптомными цитопениями.
Заслуживают внимания результаты клинического исследования, в котором
небольшой группе пациентов (14 человек) сначала проводили иммуносупрессию,
оказавшуюся неэффективной, а затем — ТКМ. Несмотря на значительную транс-
фузионную нагрузку ко времени трансплантации, явных недостатков такой
тактики лечения не обнаружено.
Поддерживающее лечение
1. Гемотрансфузия. Поскольку многие больные АА нуждаются в обширной
трансфузионной поддержке, важно свести к минимуму возможность
развития у них при каждой гемотрансфузии аллосенсибилизации, что в свою
очередь уменьшит вероятность отторжения трансплантата при проведении
таким пациентам ТКМ (родственной или неродственной). Основной метод
профилактики аллоиммунизации — фильтрация крови и ее компонентов
через устройства, задерживающие лейкоциты. Во избежание
сенсибилизации не рекомендуется при выполнении гемотрансфузии кандидатам
на ТКМ от донора-сибса использовать кровь, взятую от их родственников.
ЦМВ-негативные больные АА, которым будет проводиться ТКМ, должны
получать ЦМВ-отрицательные компоненты крови (или, если это
недостижимо — обедненные лейкоцитами). Компоненты крови, назначаемые
пациенту в ходе ТКМ, необходимо также облучить, чтобы предотвратить
развитие посттрансфузонной БТПХ.
а. Трансфузии эритроцитов. Переливания эритроцитов при АА проводят
с целью обеспечения такого уровня гемоглобина, при котором пациент не
будет страдать от анемии. У больных старшего возраста, особенно при
наличии или возможном развитии заболеваний сердца и легких,
содержание гемоглобина должно быть приблизительно 90-100 г/л. Нет никаких
доказательств того, что анемия ускоряет эритропоэз.
б. Переливания тромбоцитов. Угрожающим жизни осложнением у
пациентов с тяжелой тромбоцитопенией является кровоизлияние в мозг. Из-за
Приобретенная апластическая анемия
289
отсутствия надежных предвестников его развития больным апластичес-
кой анемией обычно назначают профилактические трансфузии
тромбоцитов, если их количество в крови падает ниже установленного порога
(на сегодняшний день — 10 000/мкл). Показаниями для трансфузий
тромбоцитов являются также быстро распространяющаяся пурпура,
кровоизлияния в слизистую щеки или обширные кровоизлияния в сетчатку
глаза. У больных с продолжающимися кровотечениями необходимо
поддерживать содержание тромбоцитов с помощью их переливания на
уровне более 50 000/мкл. При этом содержание гемоглобина должно быть
больше 100 г/л, что способствует достижению оптимального гемостаза
(глава 5). Использование концентратов тромбоцитов (заготовленных
методом афереза от одного донора) уменьшает аллосенсибилизацию.
2. Профилактика пленение инфекционных осложнений. При нейтропенической
лихорадке необходимо сразу же, еще до идентификации источника или
разрешения нейтропении, назначить антибиотики широкого спектра действия.
При затяжной лихорадке, особенно иммуносупрессированным пациентам,
необходимо провести исследования на наличие грибковых возбудителей.
Возможность использования неабсорбируемых антибиотиков в лечении
пациентов с апластической анемией трактуется неоднозначно. Больные с
тяжелой нейтропенией отличаются от пациентов с вторичной нейтропенией,
развивающейся после химиотерапии. У последних почти всегда
наблюдается повреждение слизистой ЖКТ.
3. Факторы роста. Недостаточность факторов роста не является причиной
возникновения апластической анемии; следовательно, они (прежде всего Г-
и ГМ-КСФ) используются в основном для предотвращения развития
инфекций, угрожающих жизни, а не с целью увеличения количества нейтро-
филов у бессимптомных больных. Далеко не все пациенты реагируют на
терапию факторами роста. Эффективность же введения таким больным
тромбопоэтина еще предстоит выявить в клинических испытаниях.
4. Применение хелаторов железа. Риск повреждения внутренних органов,
особенно сердца, вследствие чрезмерного поступления железа повышен у
пациентов, получивших около 100 доз эритроцитов. Терапию хелаторами можно
начинать после приема 50 доз эритроцитов. В настоящее время
рекомендуется подкожное введение дефероксамина.
Возможности лечения рефрактерной анемии
1. Андрогены. Случаи исчезновения потребности в трансфузиях эритроцитов
после терапии андрогенами послужили основанием для проведения
клинических испытаний препаратов этого ряда у рефрактерных пациентов.
Целесообразно назначение 3-месячного курса пробного лечения. Наиболее часто
используются даназол или декадураболин. Из побочных эффектов следует
выделить гепатотоксичность, проявления которой варьируются от
обратимого нарушения показателей функциональных проб печени до пелиозиса
(заполненные кровью кисты) и опухолей печени.
2. Ростовые факторы. Как обсуждалось ранее, назначение ростовых факторов
или их комбинации вряд ли способно уменьшить проявления
апластической анемии. Поэтому не следует ради проведения пробного лечения
факторами роста откладывать иммуносупрессивную терапию или ТКМ.
ЮЗак. 313
290
Глава 7. Недостаточность костного мозга, апластическая анемия
3. Совместимая неродственная ТКМ. Риск возникновения БТПХ — главного
осложнения родственных трансплантаций — многократно увеличивается
при неродственных трансплантациях. Поэтому долгосрочная
выживаемость у детей составляет лишь 50 %, а у пациентов более старшего
возраста — еще меньше.
Конституциональные гипопластические яанцитопенические синдромы будут
рассмотрены далее в этой главе. Другие синдромы недостаточности костного
мозга являются изолированными цитопениями (их классификация представлена
в табл. 7-2).
Приобретенная парциальная
красноклеточная аплазия
Парциальная красноклеточная аплазия (ПККА) проявляется анемией, ретикуло-
цитопенией и отсутствием костномозговых предшественников. ПККА по
клиническим признакам и патофизиологическим механизмам во многом сходна с апла-
стической анемией.
Эпидемиология и этиология
Встречаемость. Редко (имеются сообщения только о нескольких сотнях случаев).
Эпидемиология. Жейщины предрасположены больше, чем мужчины — 2:1.
Средний возраст начала заболевания — около 60 лет.
Этиология. Среди многочисленных причин развития цитопении чаще всего
упоминается тимома. Несмотря на преобладание подобных сообщений, в
действительности доля сопровождаемых тимомой ПККА, вероятно, низка. К другим при-
ТАБЛ ИЦА 7-2. Классификация изолированных цитопении
Приобретенные цитопении Наследственные цитопении
Парциальная красноклеточная аплазия (ПККА)
Идиопатическая Анемия Даймонда-Блэкфана
Лекарственная, индуцированная токсинами (АДБ)
Иммунная
При тимоме
Транзиторная детская эритробластопения (ТДЭ)
Нейтропения
Идиопатическая Синдром Костманна (СК)
Лекарственная, индуцированная токсинами Синдром Швахмана-
Даймонда-Оски (ШД)
Ретикулярный дисгенез
Тромбоцитопения
Идиопатическая амегакариоцитарная
Лекарственная, индуцированная токсинами Тромбоцитопения с отсутствием
лучевой кости (ТОЛК)
Приобретенная парциальная красноклеточная аплазия
291
чинам относятся злокачественные опухоли лимфоидной ткани, хронический мие-
лоидный лейкоз (ХМЛ), миелодиспластический синдром, миелофиброз,
сосудистые коллагенозы, беременность, паранеопластические синдромы, вирусы и
действие лекарственных препаратов. Перечень лекарств, применение которых
вызывает ПККА, сходен с таковым для АА, но более ограничен. Причинная связь
между приемом дифенилгидантоина и возникновением ПККА была установлена
после регистрации рецидива симптомов заболевания у пациента в результате
повторного назначения данного препарата. Однако, как и при АА, большинство
случаев ПККА являются идиопатическими.
Патофизиология
Наиболее понятен механизм селективной красноклеточной аплазии на фоне пер-
систирующей инфекции парвовируса В19. Организм в состоянии хронической
врожденной (синдром Незелоффа), ятрогенной (химиотерапия) или
приобретенной (СПИД) иммунодепрессии не способен элиминировать цитотоксический
вирус В19. Наличие тропности вируса к предшественникам эритроидного ряда
приводит к селективному угнетению эритропоэза. Механизмы поражения костного
мозга при ПККА, не связанных с В19, включают как гуморальную, так и
клеточную иммунную элиминацию гемопоэтических клеток эритроидного ростка,
находящихся на разных стадиях развития.
Диагностика
Отличительными для ПККА признаками являются анемия, ретикулоцитопения
и изолированный дефицит эритробластов в костном мозге. Иногда в малых
количествах обнаруживаются проэритробласты аномально гигантских размеров (про-
нормобласты с диаметром вдвое больше, чем у типичного пронормобласта, с
наличием ядерных включений, цитоплазматических пузырьков или без них). Это
подтверждает инфицирование парвовирусом В19. Мегакариоцитарный и грану-
лоцитарный ростки сохраняются без изменений. Лимфоциты распределяются
диффузно или образуют небольшие агрегаты. В отличие от апластической анемии
общий цитоз не изменен.
Дополнительное обследование должно включать тестирование на наличие
вируса В19 (отметим, что присутствие антител класса IgG к вирусу В19 не является
доказательством активного вирусного процесса), сероконверсию (по антителам
класса IgM) и компьютерную томографию средостеция для обнаружения
возможной тимомы.
Дифференциальный диагноз
1. Наследственная ПККА: анемия Даймонда-Блэкфана (АДБ).
2. Неиммунная гидроцефалия плода: внутриутробное инфицирование
парвовирусом В19.
3. Транзиторные синдромы:
а) транзиторная детская эритробластопения (ТДЭ);
б) транзиторный апластический гемолитический криз. У пациентов с
гемолитической анемией при острой инфекции вирусом В19
ретикулоцитопения способна возникнуть раньше, чем будет достигнут достаточный уро-
292 Глава 7. Недостаточность костного мозга, апластическая анемия
вень вируснейтрализующих антител. Инфицирование здоровых лиц пар-
вовирусом В19 хотя и может вызвать преходящую ретикулоцитопению,
редко привлекает внимание врачей, поскольку продолжительность
циркуляции эритроцитов сравнима по времени с развитием адекватного
иммунного ответа.
Лечение
Необходимо прекратить прием лекарств, воздействие которых увеличивает риск
развития цитопении. При выявлении новообразований назначаются
противоопухолевые средства с минимальными системными эффектами. Если ПККА
сохраняется после исключения всех возможных этиологических факторов, проводится
лечение, как при аутоиммунной ПККА.
Парвовирус В19. Эффективно назначение иммуноглобулинов внутривенно,
поскольку они содержат нейтрализующие антитела.
Тимома. Проводится хирургическое лечение. Если оно безрезультатно, пациент
должен получать терапию как при аутоиммунной ПККА.
Аутоиммунная ПККА. Назначается поэтапная иммуносупрессивная терапия до
достижения ремиссии или до исчерпывания терапевтических возможностей.
Лечение начинают с наиболее щадящих (низкотоксичных) режимов.
1. Преднизолон.
2. Азатиоприн или циклофосфамид (перорально) ± преднизолон; постепенно
увеличивать дозы азатиоприна или циклофосфамида, до тех пор пока:
а) количество ретикулоцитов не возрастет (ремиссия);
б) количество лейкоцитов не снизится менее 2000/мкл;
в) количество тромбоцитов не снизится менее 80 000/мкл.
3. Антитимоцитарный глобулин + преднизолон; при отсутствии эффекта
может быть назначен второй курс АТГ.
4. Циклоспорин ± преднизолон.
Стандартный курс терапии продолжается 4-8 недель. Самый ранний индикатор
ответа — изменение количества ретикулоцитов. Должен проводиться
тщательный мониторинг возможных токсических эффектов применяемых лекарственных
средств, дозы которых после достижения ремиссии нужно медленно снижать
до полной отмены. При рефрактерности пациента используют андрогены, плаз-
маферез, внутривенные препараты IgG, лимфоцитоферез и в конечном счете
спленэктомию. Пациентам, зависимым от постоянных трансфузий эритроцитов,
со временем потребуется проведение терапии хелатирующими препаратами (де-
фероксамин). Их начинают вводить после переливания приблизительно 50 доз.
Прогноз
В конце концов большинство пациентов становятся независимыми от
трансфузий либо спонтанно (приблизительно 15 %), либо после иммуносупрессивной
терапии (примерно 2/3 случаев). Впоследствии у 50 % пациентов развивается
рецидив; из них около 80 % реагируют на второй курс иммуносупрессии. Средний
срок выживания пациентов с приобретенной ПККА составляет 14 лет. Трансфор-
Агранулоцитоз и другие нейтропенические синдромы
293
мация ПККА в другие заболевания, такие как апластическая анемия или лейкоз,
происходит редко, однако в одном исследовании было зарегистрировано, что у 2
из 58 больных развился острый миелобластный лейкоз.
Приобретенная амегакариоцитарная
тромбоцитопеническая пурпура
В соответствии с названием этого редко встречающегося заболевания (имеются
сообщения приблизительно о 30 случаях), при нем поражается мегакариоцитар-
ный росток гемопоэза. При исследовании костного мозга выявляется, что на фоне
нормальных количества и морфологии миелоидных и эритроидных
предшественников отсутствуют мегакариоциты. Цитоз нормальный. Сочетанные состояния:
сосудистые коллагенозы, гепатит, вирусные инфекции (включая ВИЧ) и
применение лекарственных средств (оксифенбутазон). Для обнаружения Sq-синдрома
необходимо провести цитогенетические исследования. Пациенты реагируют на
лечение антитимоцитарным глобулином, циклофосфамидом, циклоспорином А,
литием, андрогенами и преднизолоном.
Агранулоцитоз и другие нейтропенические
синдромы
Изолированная нейтропения, обусловленная дефицитом гранулоцитарных
предшественников в костном мозге, может быть приобретенной или врожденной.
У взрослых в основном встречается приобретенный агранулоцитоз, чаще всего
индуцированный лекарственными препаратами. У детей главным образом
диагностируются наследственные рецессивные состояния, преимущественно
наследуемые по аутосомному типу или в сцеплении с Х-хромосомой. Основным
симптомом заболевания, как правило, является боль во рту или горле. Типичный
мукозит или афтозный стоматит обусловлен нарушением процесса участия нейт-
рофилов (полиморфно-ядерных лейкоцитов) в физиологической защите и
жизнедеятельности слизистых оболочек. При многих из этих синдромов разрушение
клеток гранулоцитарного ряда часто происходит и в костном мозге, и в
периферической крови, поэтому, строго говоря, некоторые из указанных состояний не
соответствуют полностью критериям синдромов.поражения костного мозга. Они
рассмотрены в данной главе для полноты картины. Изолированные нейтропении
могут быть классифицированы в соответствии с диагностической схемой,
представленной в табл. 7-3. Отдельные типы нейтропении обсуждаются ниже.
Приобретенная нейтропения
Индуцированная лекарствами
1. Эпидемиология. Это редкий синдром, наблюдаемый у пожилых пациентов
(> 50 лет), обусловленный, вероятно, повышением частоты приема и
разнообразием используемых в этой популяции лекарственных препаратов;
соотношение женщин и мужчин составляет 2:1.
2. Этиология/патофизиология. Применяемые лекарственные препараты
могут вызывать нейтропению посредством действия иммунных механизмов
294 Глава 7. Недостаточность костного мозга, апластическая анемия
ТАБЛИЦА 7-3. Дифференциальный диагноз изолированной нейтропении
Вторичная нейтропения
Нейтропения, индуцированная лекарственными препаратами
Нейтропения, индуцированная токсинами
Вторичная по отношению к специфическим инфекционным агентам
Вирусы, включая вирусы гепатитов, вирус Эпштейна-Барр, ВИЧ-1
Бактерии, особенно возбудитель брюшного тифа
Риккетсии
Простейшие
Вторичная по отношению к генерализованной инфекции
Сепсис
Диссеминированный туберкулез
Аутоиммунные сосудистые коллагенозы
Системная красная волчанка
Синдром Фелти
Голодание и квашиоркор
Алкоголизм
"Нормальные" варианты
Этническая и семейная нейтропения
Доброкачественная хроническая нейтропения
Первичные гематологические заболевания
Аутоиммунная нейтропения1
Изоиммунная нейтропения
Парциальная белоклеточная аплазия1
Лимфоцитоз с преобладанием больших гранулярных лимфоцитовД-у-лимфопро-
лиферативное заболевание1
* Миелодисплазия
Состояние после химиотерапии, облучения1
Циклическая нейтропения1
Агранулоцитоз1
Хроническая идиопатическая нейтропения
Наследственные/врожденные состояния
Циклическая нейтропения
Младенческий агранулоцитоз (синдром Костманна)
Синдром Швахмана-Даймонда-Оски
Ретикулярный дисгенез
Врожденный дискератоз (синдром Зинссера-Коле-Энгмана)
Синдром Чедиака-Хигаси
Миелокахексия/нейтропения с тетраплоидными лейкоцитами
1 Состояния, при которых абсолютное количество нейтрофилов обычно < 500/мкл.
или прямого повреждения предшественников гранулоцитов в костном
мозге. В табл. 7-4 представлены некоторые особенности этих двух
механизмов. Иммунная природа нейтропении была доказана в начале XX в.
при обнаружении рецидива данного состояния у вылеченного пациента.
Агранулоцитоз и другие нейтропенические синдромы
295
ТАБЛИЦА 7-4. Иммунный и токсический агранулоцитоз
Иммунный
Токсический
Препарат, вызывающий
агранулоцитоз
Время от начала приема
Клинические
проявления
Рецидивы
Лабораторные
показатели
Амидопирин
От нескольких дней до
недель
Острое, часто
молниеносное появление
симптоматики
Повторно вызывается
малыми тест-дозами
Выявление лейкоагглю-
тининов, положительные
тесты на другие антитела
Фенотиазин
От недель до месяцев
Часто бессимптомен
или начало незаметно
После латентного
периода, требуются большие
дозы
Признаки прямой или
опосредованной
метаболитами цитотоксичности
После повторного назначения амидопирина — препарата, вызвавшего ней-
тропению, количество нейтрофилов резко снизилось в течение нескольких
часов. Подобный результат был получен после введения здоровому
добровольцу сыворотки от пациента с такой нейтропенией. Перечень
лекарственных препаратов, наиболее часто вызывающих нейтропению,
аналогичен таковому при апластической анемии и включает производные
амидопирина, антитиреоидные средства, макролиды, прокаинамид, фено-
тиазины, р-лактамы и НПВС (табл. 7-5).
3. Диагноз. Внезапное развитие нейтропении при приеме лекарственного
препарата с установленным супрессивным действием на гранулоцитопоэз в
основном является достаточным основанием для постановки диагноза;
дополнительно необходимо выполнить исследование костного мозга.
4. Коррекция. Восстановление гранулоцитопоэза может происходить от 2-х до
21 и более дней после прекращения приема лекарственного препарата. Его
можно ускорить ростовыми факторами, такими как Г- или ГМ-КСФ.
5. Прогноз. Несмотря на оптимальное лечение, у 10 % пациентов может
наступить летальный исход. Препарат, вызвавший нейтропению (в некоторых
случаях и близкие по механизму действия препараты, например пропилтио-
урацил и метимазол), нельзя назначать повторно после восстановления
гранулоцитопоэза.
Аутоиммунная нейтропения
1. Эпидемиология. В отличие от агранулоцитоза, вызванного принимаемым
лекарственным препаратом, аутоиммунная нейтропения развивается в
основном у младенцев и детей. У пациентов более старшего возраста она
может наблюдаться одновременно с аутоиммунными нарушениями, такими
как системная красная волчанка (СКВ) или ревматоидный артрит (РА).
Данный вид нейтропении — хроническое состояние, которое
обнаруживается только при длительном течении.
296
Глава 7. Недостаточность костного мозга, апластическая анемия
ТАБЛИЦА 7-5.
Препарат
Лекарственные препараты, вызывающие агранулоцитоз
(поданным Международного исследования апластической
анемии и агранулоцитоза)
RR
Избыточный риск
Цинепазид
Сульфасалазин
Антитиреоидные препараты
Макролиды
Прокаинамид
Априндин
Дипирон
Котримоксазол
Теналидин
Карбамазепин
Гликозиды наперстянки
Индометацин
Троксерутин
Производные сульфонилмочевины
Кортикостероиды
Бутазоны
Дипиридамол
Р-Лактамы
Пропранолол
Салицилаты
оо
оо
97
54
-50
-49
16
16
-16
11
2,5-9,9
6,6
6,0
4,5
4,1
3,9
3,8
2,8
2,5
2,0
5,3
6,7
3,1
2,7
0,6
1J
2,4
0,6
,1-0,3
0,4
0,3 ,
0,2
*
0,2
0,2
0,2
0,1
0,06
RR = оценка многовариантного относительного риска.
Избыточный риск выражается как число случаев на 106 пациентов, принимающих
лекарственный препарат в течение 1 нед. Чтобы вычислить избыточный риск в контролируемых
исследованиях, используют следующие показатели: общая встречаемость (I), относительный риск
(RR), доля установленных случаев (Рв), этиологическая фракция (EF), которая определяется
как (RR — I/RR) Ре.
Шаг 1. Вычислить базовую встречаемость: lB = I х (1 - EF).
Шаг 2. Вычислить общую встречаемость среди установленных случаев: lT = lB x RR.
Шаг 3. Вычислить избыточную встречаемость среди установленных случаев: lE = lT - 1в или
[(RR) х | х (1 - EF)] - [I (1 - EF)] или (RR -1) х | х (1 - EF).
* Избыточный риск не рассчитывался из-за отсутствия установленного контроля или, как
в случае кортикостероидов, из-за сложной причинно-следственной связи при диагнозе
ревматоидный артрит.
2. Этиология (табл. 7-6). Лекарственные препараты не являются
этиологическим фактором. У некоторых пациентов определяются антинейтро-
фильные антитела (к антигенам NA1, NA2, NB1, ND1, ND2 и т. д.), но их
нельзя причислить к диагностическим показателям. Присутствие
антител к зрелым клеткам крови — признак аутоиммунного процесса,
направленного прежде всего против зрелых гранулоцитов. При этом могут
также нарушаться последние стадии созревания гранулоцитов в костном
мозге.
Агранулоцитоз и другие нейтропенические синдромы
297
ТАБЛИЦА 7-6. Нейтропения, опосредованная гуморальными иммунными
механизмами
Агранулоцитоз
Парциальная белоклеточная аплазия
Аутоиммунная нейтропения
Синдром Фелти
Системная красная волчанка
3. Диагноз:
а) симптомы. Наряду с возможным отсутствием симптоматики пациенты
иногда страдают от повторных инфекций кожи, среднего отита,
инфекций верхних отделов респираторного тракта, диареи или приступов
лихорадки неясного генеза;
б) физикальное обследование. Гепатоспленомегалия отмечается только у-50 %
пациентов;
в) исследования костного мозга выявляют нормальный или повышенный ци-
тоз с остановкой созревания на поздних стадиях. Предшественники нейт-
рофилов кажутся "замороженными" на характерной стадии созревания,
поскольку представители более поздних стадий созревания отсутствуют.
Можно наблюдать гистиоцитарный фагоцитоз миелоидных клеток.
4. Лечение. Многие пациенты выздоравливают без лечения. Следовательно,
решение о его начале основывается на тяжести симптоматики. Успешно
использовались кортикостероиды и внутривенные препараты IgG. Спленэк-
томия чаще всего сопровождается лишь кратковременным разрешением
нейтропении (об исключениях еще будет сказано), однако иногда создает
риск развития сепсиса. У пациентов с аутоиммунными заболеваниями
(например, СКВ или РА) могут быть эффективны цитотоксические препараты.
5. Особые клинические проблемы:
а) первичная селезеночная нейтропения. Повышенная клбточность
костного мозга с избытком ранних миелоидных элементов; есть реакция на спле-
нэктомию;
б) синдром Фелти. Нейтропения, спленомегалия и ревматоидный артрит
(выраженная миелоидная гипоплазия встречается редко). Может быть
эффективно назначение метотрексата или проведение спленэктомии; при
этом у У4 пациентов нет реакции на спленэктомию, а еще у х/ъ пациентов
будут возникать рецидивы нейтропении.
Изоиммунная нейтропения. Это гранулоцитарный эквивалент
резус-гемолитической болезни новорожденных. Мать сенсибилизируется к аллоантигенам нейт-
рофилов отца и вырабатывает антитела к гранулоцитам плода, проходящие через
плаценту.
1. Встречаемость. Приблизительно 1 на 500 живых новорожденных.
2. Диагностика. Поскольку сепсис у младенцев сам по себе может приводить
к нейтропении, иногда трудно дифференцировать причину от следствия.
Характерной чертой синдрома является блокада миелопоэза на поздней
стадии. Антинейтрофильные антитела, специфичные к антигенам нейтрофи-
лов отца, обнаруживаются в сыворотке крови матери и ребенка.
298 Глава 7. Недостаточность костного мозга, апластическая анемия
3. Дифференциальный диагноз. Если пациент не выздоравливает, необходимо
рассмотреть возможность наличия врожденных заболеваний.
4. Лечение. Большинство младенцев выздоравливают в течение 12-20 недель
без возникновения у них каких-либо опасных инфекций, для
предотвращения развития которых назначают стандартную антибиотикотерапию.
Безопасность и/или эффективность КСФ при этом синдроме не оценивалась.
Парциальная белоклеточная аплазия (ПБКА). Очень редко встречающийся
синдром, иногда ассоциированный с тимомой, иногда возникающий значительно
позже после ее удаления. В костном мозге фактически отсутствуют все миелоид-
ные элементы. Выявлено наличие гуморальных и клеточных ингибиторов мие-
лопоэза in vitro. Имеющуюся тимому надо удалять. Пациенты, у которых тимома
не обнаружена, реагируют на проведение тимэктомии, спленэктомии, плазмафе-
реза, назначение внутривенных препаратов IgG, циклофосфамида, азатиоприна и
циклоспорина.
Нейтропения с лимфоцитозом с преобладанием больших гранулярных
лимфоцитов (БГЛ) (Ту-лимфоцитоз). Часто у больных выявляется спленомегалия,
а в некоторых случаях — сочетание с ревматоидным артритом, что составляет
синдром Фелти. В мазке периферической крови наблюдаются большие гранулярные
лимфоциты (рис. 4-9В). При исследовании костного мозга отмечается блокада
миелоидного ростка. Описаны три подтипа:
1. Т-БГЛ-лейкоз (CD3+). При изучении перегруппировки Т-клеточного
рецептора это заболевание определено как клональное нарушение Т-клеток с под-
острым течением. У пациентов с повторяющимися инфекциями иногда
эффективно использование кортикостероидов, Г- и ГМ-КСФ. Перспективным
лечением считается назначение метотрексата и циклоспорина в низких дозах.
2. НК-БГЛ-лейкоз (CD3"). Клональность определена наличием однородной
цитогенетической аномалии. Течение острое, и только 2 из 11 пациентов
дали положительную реакцию на комбинированную химиотерапию.
Большинство пациентов умерли в течение нескольких месяцев после появления
симптомов заболевания.
3. НК-БГЛ-лимфоцитоз (CD3+ или CD3"). На основе анализа полиморфизма
Х-хромосом у этих пациентов выявлена полшслональная пролиферация
БГЛ, что позволяет предположить реактивный характер процесса.
Клиническое течение очень медленное/и иногда лечение не требуется.
Хроническая идиопатическая нейтропения. В соответствии с названием это
диагноз исключения. Количество нейтрофилов обычно в пределах 200-500/мкл.
В отличие, например, от агранулоцитоза, индуцированного лекарственным
препаратом, стимуляция гидрокортизоном выявляет резервы костномозговых клеток.
При исследовании костного мозга в основном обнаруживается блокада поздних
стадий созревания. Использовать Г-КСФ, кортикостероиды, цитотоксические
агенты и проводить спленэктомию следует только у пациентов с частыми
инфекционными осложнениями.
Нейтропенические состояния. Это нарушения, характеризующиеся
незначительным уменьшением содержания предшественников гранулоцитов в костном
мозге (по определению они не относятся к синдромам недостаточности костного
мозга, но обсуждаются здесь, чтобы дать полное представление о нейтропениях).
Агранулоцитоз и другие нейтропенические синдромы
299
1. Доброкачественная семейная лейкопения. Состояние с аутосомно-доми-
нантным типом наследования и, как следует из названия, не связанное с
увеличением риска инфицирования. Количество нейтрофилов в пределах
2100-2600/мкл. Костный мозг нормоклеточный. Повышенная частота
встречаемости зарегистрирована на Ближнем и Среднем Востоке, а также
у американских и африканских негров. Лечения этого состояния, не
являющегося болезнью, не требуется.
2. Хроническая (доброкачественная) нейтропения младенцев и детей. Диагноз
часто ставится только ретроспективно. Она обычно проявляется на первом
или втором году жизни, как правило, на основании подсчета лейкоцитарной
формулы крови при инфекции, которая отнюдь не всегда связана с нейтро-
пенией. Количество нейтрофилов при рождении, если оно было определено,
нормальное. Костный мозг с нормальным или повышенным цитозом, его
морфология в норме или отмечается угнетение созревания миелоидных
предшественников, как правило, на поздних стадиях. Могут выявляться
небольшие оральные, вагинальные или ректальные язвы. Инфекции
протекают легко и хорошо поддаются лечению антибиотиками. Фактически все
пациенты выздоравливают к 4-летнему возрасту. При несоответствии этим
критериям доброкачественности целесообразно провести дальнейшее
обследование. Количество нейтрофилов можно увеличить назначением кор-
тикостероидов, внутривенных препаратов IgG и Г-КСФ, однако, учитывая
спонтанное разрешение заболевания, эти препараты не следует
использовать постоянно, а лишь при выявлении инфекций.
3. (Пост-) Инфекционная нейтропения. Полиэтйологический процесс, чаще
встречающийся при сепсисе новорожденных, пожилых или истощенных
пациентов. В основном легко распознается на основании клинических данных.
Клеточность костного мозга обычно повышена, может быть блокада
созревания клеток миелоидного ростка на поздних стадиях. Кроме обычных
бактерий, наличие которых обусловлено сепсисом, могут выявляться вирусы
ветряной оспы, кори, краснухи, ВЭБ, ЦМВ, гепатита А и В, гриппа, а также
микобактерии, риккетсии и бруцеллы.
4. Нарушения обмена веществ. Несмотря на нормальный миелопоэз,
нейтропения может развиваться при кетоацидозе, связанном с гипергликемией,
ацидурией (оротовая кислота, метилмалонат) и гиперглицинурией, а также
при гликогенозах типа lb. Кроме того, у некоторых пациентов
обнаруживаются дефекты функции нейтрофилов. Инфекции иногда протекают тяжело.
Для лечения с успехом используются колониестимулирующие факторы.
Наследственные/врожденные нейтропении
Циклическая нейтропения. Возвратная нейтропения, возникающая с
периодичностью 15-35 дней (классически — каждый 21-й день).
1. Проявления. Возвратные лихорадки или инфекционные заболевания у
ребенка отражают степень тяжести нейтропении. Такие проявления
заболевания у взрослых могут быть связаны с увеличением содержания БГЛ.
2. Наследование. Аутосомно-доминантный тип с различной выраженностью.
3. Сочетанные проявления. Возможны циклические колебания содержания
моноцитов, лимфоцитов, ретикулоцитов и тромбоцитов.
300 Глава 7. Недостаточность костного мозга, апластическая анемия
4. Диагностика. Дважды в неделю необходимо определять количество нейтро-
филов по крайней мере в течение 8 недель.
5. Исследование костного мозга. Миелоидная гипоплазия, соответствующая
степени тяжести нейтропении.
6. Лечение. Терапия Г-КСФ корригирует нейтропению и уменьшает частоту
развития инфекций, но не влияет на циклические изменения содержания
других элементов крови.
7. Прогноз. Тяжесть клинических проявлений имеет тенденцию к
уменьшению по мере увеличения возраста пациента.
Синдром Костманна — агранулоцитоз новорожденных
1. Проявления. Тяжелые возвратные гнойные инфекции, в основном в течение
первых 6 месяцев жизни.
2. Наследование. Аутосомно-рецессивное.
3. Сочетанные проявления. Иногда замедление умственного развития,
микроцефалия, катаракта или низкорослость.
4. Исследование костного мозга. Миелоидная гипоплазия с остановкой на
стадии промиелоцита.
5. Лечение. Достаточно обещающей является терапия Г-КСФ.
6. Лейкоз. У 1-2 % пациентов развиваются миелоидные лейкозы (до
внедрения Г-КСФ в клиническую практику).
Синдром Швахмана-Даймонда-Оски. Это комбинация нейтропении с
недостаточностью поджелудочной железы. Гемопоэтические проявления могут также
включать анемию (15%) или тромбоцитопению (9 %'), которые иногда даже
предшествуют развитию нейтропении.
1. Проявления. Повторные инфекции со стеатореей в первые годы жизни. Сте-
аторея может быть скрытой и определяться только при исследовании кала
на содержание жира.
2. Наследование. Аутосомно-рецессивное.
3. Сочетанные проявления. Низкорослость (60 %), задержка умственного
развития (15 %), метафизарный дизостоз (30 %), миокардиальный фиброз.
4. Диагностика. Должна быть доказана экзокринная недостаточность
поджелудочной железы. Необходимо определить хлориды пота, чтобы исключить
муковисцидоз.
5. Исследование костного мозга. Нормальная или уменьшенная клеточность
и миелоидное созревание; возможна гиперплазия эритроидного ростка.
6. Лечение:
а) нейтропения поддается терапии Г-КСФ, но препарат должен быть
зарезервирован для пациентов с тяжелыми или повторными хроническими
инфекциями;
б) анемия может корригироваться при назначении преднизолона или анд-
рогенов;
в) недостаточность поджелудочной железы компенсируется
заместительной терапией ферментами.
7. Прогноз. Примерно у 25 % пациентов развивается панцитопения, а у 5 % —
лейкоз. Средний возраст смерти пациентов с синдромом
Швахмана-Даймонда-Оски примерно 35 лет, но у 25 % больных с панцитопенией он меньше
почти в 2 раза, а у 5 % пациентов с развившимся лейкозом — меньше 10 лет.
Наследственные синдромы недостаточности костного мозга 301
Ретикулярный дисгенез. Ретикулярный дисгенез состоит из триады: 1)
врожденный агранулоцитоз, 2) лимфопения и 3) отсутствие клеточного и гуморального
иммунитета. Наследование рецессивное, возможно, сцепленное с Х-хромосомой.
У младенцев развиваются инфекции на первом или втором месяце жизни.
Выявляется отсутствие лимфатических узлов, миндалин и тимуса; иногда имеют место
анемия и тромбоцитопения. Костный мозг малоклеточный, с отсутствием миело-
идных и лимфоидных элементов. Могут быть признаки депрессии или дисплазии
эритроидного ростка. В некоторых случаях эффективна трансплантация
костного мозга, но только если она выполнена до развития серьезных инфекций. В
противном случае неизбежен летальный исход в течение 4 месяцев.
Врожденный дискератоз (синдром Зинссера-Коля-Энгмана). Определяется
триадой: 1) сетчатая гиперпигментация лица, шеи и плеч; 2) лейкоплакия
слизистых оболочек и 3) дистрофия ногтей. Были выявлены аутосомно-доминантный и
рецессивный типы наследования, но преобладающим является тип наследования,
сцепленный с Х-хромосомой. У 5-15 % пациентов развивается сквамозно-клеточ-
ный рак или аденокарцинома с преобладающей локализацией в ротоглотке и
ЖКТ. На втором десятилетии жизни приблизительно у половины пациентов
обнаруживается нейтропения или АА. Возможна некоторая коррекция цитопении
андрогенами, часто при назначении их в комбинации с преднизолоном. После
трансплантации костного мозга отмечается высокая частота рецидивов в раннем
посттрансплантационном периоде.
Синдром Чедиака-Хигаси. Отсутствие пигментации кожи и радужной оболочки
(альбинизм), прогрессирующие неврологические нарушения и гигантские цито-
плазматические гранулы в лейкоцитах — отличительные признаки этого аутосом-
но-рецессивного заболевания. Характерные гранулы заметны во многих клетках,
включая клетки гранулоцитарного ряда, и предположительно связаны с
дефектом гранулопоэза, объясняющим тяжелую нейтропению при данном синдроме.
Может возникнуть панцитопения. Эффективна ТКМ (глава 4).
Миелокахексия/нейтропения с тетраплоидными лейкоцитами. Этот синдром
в основном наблюдается у детей и проявляется тяжелыми возвратными
инфекциями. Количество двуядерных клеток гранулоцитарного ряда увеличивается
с каждой последующей стадией созревания настолько, что 2/3 этих клеток и
фактически все зрелые нейтрофилы тетраплоидны. Ранние предшественники —
нормальные. Лишь немногие тетраплоидные клетки попадают в кровоток, что
и объясняет возникновение нейтропении. У таких пациентов зарегистрированы
многочисленные аномалии функций нейтрофилов. Подтверждена
эффективность терапии Г-КСФ.
Наследственные синдромы
недостаточности костного мозга
Наследственные синдромы недостаточности костного мозга почти всегда
обнаруживаются в детском возрасте симптомами, связанными с цитопенией, или
характерными физическими аномалиями. Недостаточность костного мозга, часто
скрытая в начальном периоде, к моменту распознавания может проявляться как
угнетение одного или нескольких ростков кроветворения.
302 Глава 7. Недостаточность костного мозга, апластическая анемия
Анемия Фанкони. Классический фенотип анемии Фанкони имеет следующие
признаки: семейная апластическая анемия (аутосомно-рецессивный тип
наследования), низкорослость, отсутствие либо гипоплазия большого пальца или
лучевой кости, микроцефалия и гипопигментированные пятна ("кофе с молоком").
Диагностическая значимость совокупности данных признаков уступает тесту
определения кластогениндуцированных разрывов хромосом in vitro. Спектр
клинических проявлений может также включать гипертелоризм, недоразвитие половых
желез, различные аномалии скелета, глухоту, дефекты ЖКТ и
сердечно-сосудистой системы. С недавних пор почти треть пациентов с анемией Фанкони
составляют лица без очевидной физической патологии, поэтому у всех пациентов
моложе 30 лет, имеющих цитопении и малоклеточный костный мозг, необходимо
проводить исследования хромосомных аберраций, вызываемых диэпоксибутаном
(ДЭБ) или митомицином С (ММС). Эти агенты, вероятно, усиливают нарушение
репарации ДНК, которое является следствием генетического дефекта при данном
аутосомно-рецессивном заболевании.
Гематологические проявления при анемии Фанкони могут варьироваться от
нормального количества клеток крови с небольшим повышением уровня
гемоглобина F до тяжелой панцитопении с полной зависимостью больного от гемотранс-
фузий. Аналогично в костном мозге может наблюдаться снижение клеточности от
умеренной до сильной степени. Первая линия поддерживающей терапии — анд-
рогены, которые эффективны приблизительно у 50 % пациентов. Вначале обычно
реагирует эритроидный росток (в течение 1-2 мес), затем гранулоцитарный;
последним повышается содержание тромбоцитов, что наименее надежно и может
наблюдаться только через год. У большинства пациентов после прекращения
введения андрогенов возникают рецидивы. Трансплантация костного мозга от
подходящего совместимого донора является в настоящее время единственным
радикальным методом терапии, при котором удается добиться 5-летнего выживания
2/з пациентов. Обычная высокодозная химиотерапия, назначаемая перед
трансплантацией костного мозга, должна быть ограничена из-за чувствительности
к ней гемопоэтических клеток при анемии Фанкони (глава 8). В будущем может
оказаться полезной генная терапия аутологичного костного мозга.
Пациенты без цитопении имеют значительный риск развития
новообразований, включая лейкоз (обычно миелоидный), предлейкоз и карциному (рак) (в
частности, сквамозный и гепатоцеллюлярный). Общая частота возникновения
злокачественных новообразований приближается к 20 %. После трансплантации
костного мозга часто образуются эпителиальные опухоли, хотя они не всегда
угрожают жизни пациента.
Анемия Даймонда-Блэкфана. Изолированную врожденную недостаточность
эритропоэза называют анемией Даймонда-Блэкфана (АДБ).
Диагностическими критериями являются: 1) ретикулоцитоления с макроцитарной или нормо-
цитарной анемией; 2) нормальная клеточность костного мозга с
изолированной эритроидной гипоплазией и 3) нормальное или близкое к нормальному
количество лейкоцитов и тромбоцитов. В большинстве случаев АДБ
развивалась спорадически, хотя в различных семьях зарегистрированы
аутосомно-рецессивный и аутосомно-доминантный типы наследования. Пациент обычно
попадает под медицинское наблюдение в течение первых 6 месяцев жизни
и только изредка — после 1 года. Физические аномалии выявляются почти
Наследственные синдромы недостаточности костного мозга
303
у 25 % пациентов (в зависимости от тщательности обследования).
Лекарственное средство первого ряда ■*- преднизолон. Эффективен более чем у 50 % детей,
однако преимущественно при длительном использовании, что обусловливает
развитие обычных для этого препарата побочных действий. Рефрактерным
пациентам показана долгосрочная трансфузионная поддержка с
соответствующей продолжительной терапией дефероксамином для выведения избытков
железа. У 15-20 % всех больных АДБ возникают спонтанные ремиссии.
Отдаленные осложнения включают перегрузку железом, сепсис и миелоидные
лейкозы, которые могут оказаться смертельными.
Транзиторная детская эритробластопения. В соответствии с названием транзи-
торная детская эритробластопения (ТДЭ) подразумевает резкое, но временное
подавление эритропоэза. Такое угнетение — отличительный признак ТДЭ,
однако окончательный диагноз часто ставится ретроспективно. Вероятной причиной
заболевания является вирусная инфекция клеток эритроидного ростка, хотя тест
на обнаружение парвовируса редко бывает положительным. Проявления
синдрома включают изолированную анемию, ретикулоцитопению и обнаруживаемую
при исследовании костного мозга глубокую гипоплазию эритроидного ростка,
т. е. аналогичны таковым при АДБ. Как и при АДБ, миелоидный и мегакариоци-
тарный ростки существенно не повреждены. Различия между двумя синдромами
устанавливают на основании клинических данных. Первые признаки проявления
ТДЭ обычно наблюдаются у детей старше 2 лет. Наличие в анамнезе вирусных
инфекций подтверждает диагноз ТДЭ (хотя, конечно, у детей с АДБ также
развиваются вирусные инфекции, иногда непосредственно перед появлением
симптомов). В течение острой, ретикулоцитопенической, фазы ТДЭ маркеры
напряжения эритропоэза (стрессорного эритропоэза) вообще отсутствуют, что является
диагностическим критерием; иными словами, обнаружение данных маркеров
в этой фазе свидетельствует о развитии АДБ. К маркерам напряжения
эритроцита относятся увеличение среднего объема эритроцитов, уровня гемоглобина F
и уровня антигена I. Однако эти маркеры могут выявляться в течение
восстановительного периода, что затрудняет дифференциальный диагноз ТДЭ и АДБ; но
нормализация количества ретикулоцитов у пациента, не получавшего стероидов,
позволяет исключить диагноз АДБ. Таким образом, если лечащий врач имеет
серьезные основания предполагать у пациента ТДЭ, то больного можно просто
наблюдать. При постановке этого диагноза восстановление произойдет в течение
1-2 месяцев. Пациенты часто удовлетворительно переносят снижение уровня
гемоглобина до 50 г/л, не нуждаясь в трансфузионной поддержке.
Тромбоцитопения с отсутствием лучевых костей. Характерная физическая
аномалия при этом редко встречающемся синдроме — отсутствие лучевых костей
(двустороннее) при наличии больших пальцев (при анемии Фанкони большие
пальцы отсутствуют). Заболевание обнаруживается у младенцев в первые
недели жизни на основании выраженной физической аномалии или из-за
геморрагических проявлений. Характер наследования аутосомно-рецессивный. При
исследовании костного мозга, наряду с нормальным миело- и эритропоэзом,
регистрируется отсутствие, гипоплазия или незрелость развивающихся ,мегака-
риоцитов. Анемия с ретикулоцитозом может возникнуть вследствие кровавой
диареи, связанной с потреблением коровьего молока. Соответственно,
исключение из рациона пациента этого продукта приводит к улучшению его состояния.
304 Глава 7. Недостаточность костного мозга, апластическая анемия
Нередко наблюдается лейкемоидная реакция с резко увеличенным количеством
лейкоцитов. Эффективны лишь переливания тромбоцитов. Если пациенты
переживают первый год, что происходит в большинстве случаев, долгосрочный
прогноз положителен, при этом количество тромбоцитов обычно стабильно
превышает 100 000/мкл.
Амегакариоцитарная тромбоцитопения. Амегакариоцитарная
тромбоцитопения — гипомегакариоцитарный аналог анемии Даймонда-Блэкфана.
Лучевые кости — в наличии; физические аномалии — лишь в качестве
исключения. Тромбоцитопения у младенца в основном проявляется в течение первых
нескольких месяцев жизни, при этом в костном мозге наблюдается
селективное угнетение мегакариоцитов. Характер наследования рецессивный, в
некоторых случаях аутосомный, в других — сцепленный с Х-хромосомой.
Благотворное влияние на течение заболевания оказывают кортикостероиды и/или
андрогены, хотя средний срок выживания составляет только 5 лет. У многих
пациентов развивается апластическая анемия. Эффективна трансплантация
костного мозга.
Клинический пример
Мужчина в возрасте 28 лет, почувствовав слабость, пришел на прием к
терапевту. За несколько недель до появления этого ощущения у пациента
отмечались головные боли, недомогание, миалгия, лихорадка и озноб. За
два-три дня до визита к врачу появилась диффузная яркая макуло-папу-
лезная сыпь и боли в суставах. Уже около 1 недели пациент ощущает
слабость и утомляемость, а в последние несколько дней — постоянную
одышку при обычной физической нагрузке, без лихорадки, потливости или
озноба. При физикальном обследовании выявлена бледность, не
связанная с каким-либо острым состоянием. Частота дыхания составляет
22/мин, пульс регулярный с частотой 100/мин. Температура тела
нормальная. Выраженная бледность кожи и слизистых. Сыпи нет, лимфаде-
нопатия не выявлена. Сердце и легкие без патологии. Живот мягкий.
Конечности не изменены. При ректальном обследовании — стул коричневый,
без скрытой крови. Нервная система без изменений. В моче отсутствуют
эритроциты и билирубин. Данные клинического анализа крови:
гемоглобин — 50 г/л, гематокритное число —15 % при среднем объеме эритроцита
93 мкм3; лейкоциты — 4500/мкл, лейкоцитарная формула в норме;
количество тромбоцитов — 320 000/мкл. В мазке периферической крови внешний
вид тромбоцитов и лейкоцитов нормальный. Окраска и форма
эритроцитов — без отклонения от нормы. Полихромазия не выявлена.
Вопрос 1. Каков следующий этап обследования?
Ответ. Детально расспросить пациента с целью выявить признаки
возможного кровотечения из желудочно-кишечного, мочеполового тракта или
слизистых. Выяснить, была ли у пациента темная моча; это позволит
определить, имелись ли в прошлом гемолитические эпизоды с бийирубинурией.
В данном случае никакой новой информации получено не было.
Вопрос 2. Что выполнено далее?
Ответ. Определено количество ретикулоцитов, а также активность ЛДГ,
содержание гаптоглобина, билирубина в крови и гемосидерина в моче. Пол-
Наследственные синдромы недостаточности костного мозга 305
ное отсутствие ретикулоцитов. Другие исследования не выявили никаких
аномалий.
Вопрос 3. Как интерпретировать результаты обследования?
Ответ. Костный мозг не реагирует адекватно на анемию, так как нет
продукции молодых клеток (ретикулоцитов). Признаков гемолиза при
лабораторном обследовании не обнаружено. Нормальные показатели активности
ЛДГ и содержания билирубина и гаптоглобина указывают на то, что в
последнее время не было каких-либо гемолитических эпизодов. Нормальный
уровень гемосидерина в моче свидетельствует об отсутствии в ближайшем
анамнезе больного гемолиза (в противном случае гемосидерин
откладывался бы в клетках почечных канальцев, которые при слущивании привели бы
к появлению в моче железосодержащих пигментов).
Вопрос 4. Каков следующий шаг?
Ответ. Выполнено исследование костного мозга. Цитоз оказался
нормальным. Предшественники тромбоцитов и лейкоцитов в границах нормы.
Однако выявлено очень мало всех видов предшественников эритроцитов.
Обнаружено несколько гигантских пронормобластов, которые содержали
внутриядерные включения свободного аморфного розового материала.
Положительными оказались дополнительный анализ крови на наличие
генома парвовируса В19 (анализ ДНК методом дот-блот-гибридизации) и тест
на ВИЧ.
Вопрос 5. Каков вероятный диагноз?
Ответ. Больной, скорее всего, инфицирован парвовирусом В19 на фоне
ВИЧ-инфекции.
Вопрос 6. Как лечить анемию у этого пациента?
Ответ. Пациенту было проведено переливание эритроцитов. Вводился
также внутривенный иммуноглобулин.
Продолжение наблюдения
У пациента были определены количество СБ4+-клеток и вирусная
нагрузка. Начато соответствующее лечение ВИЧ-инфекции. В течение
следующих 2-х недель развился ретикулоцитоз и временно повысилось гематок-
ритное число. Однако для сохранения его на приемлемом уровне было
назначено постоянное введение иммуноглобулина.
Избранная литература
Alter В. P. (ed.). Perinatal Hematology. New York: Churchill Livingstone, 1989.
Эта монография включает исчерпывающие сведения по эпидемиологии, генетике,
истории, а также обзор клинических наблюдений, методов лабораторной
диагностики и лечения синдромов недостаточности костного мозга, в основном
конституциональных, которые развиваются на ранних этапах жизни.
Hoffman R. H. et al. (eds). Hematology: Basic Principles and Practice, 2nd ed. New York:
Churchill Livingstone, 1995.
Фундаментальный учебник по гематологии, в котором обсуждаются фактически все
синдромы недостаточности костного мозга, представленные в данной главе.
Приводятся полезные диагностические и терапевтические схемы, хотя они не
являются единственно возможными.
306
Schroeder-Kurth T. M., Auerbach A. D.f Obe G. (eds). Fanconi Anemia: Clinical,
Cytogenetic, and Experimental Aspects. New York: Springer- Verlag; 1989.
В книге представлен полный спектр клинических вариантов анемии Фанкони (АФ)
с рассмотрением анатомических аномалий, примеров наследования, возраста
манифестации заболевания, вариантов лечения. Однако в ней отсутствуют
последние данные о молекулярных основах АФ и применении смягченных
режимов трансплантации.
Young N. S., Alter В. Р'. Aplastic Anemia, Acquired and Inherited. Philadelphia: W. B.
Saunders; 1994.
Это полное руководство, в котором много сведений о патофизиологии и молекулярных
механизмах синдромов недостаточности костного мозга, а также рассмотрена
возможность использования большого количества современных терапевтических
средств, таких как факторы роста, при лечении АА.
Young N., Keisu M. (eds). Drug related blood dyscrasias workshop. Eur. J. Haematol
(Supplement 60), 1996; 57. A
Монография в основном касается вопросов приобретенной апластической анемии
(АА). Обсуждается клиника, эпидемиология, патофизиология и терапия
медикаментозной и идиопатической АА. Подробно обсуждаются возможные
механизмы индуцированной бензолом АА (классическая модель аплазии, развитие
которой связано с приемом лекарственных препаратов), а также фармакология
повреждений, индуцированных лекарствами.
Глава 8
Трансплантация
стволовых клеток
Митчелл Е. Горовиц, Синтия Е. Данбар
Введение
История
Пятьдесят лет назад выявленные у пациентов, подвергшихся облучению в
Хиросиме и Нагасаки, нарушения системы крови стимулировали поиски возможных
радиопротекторов костного мозга. В 1950-1960-е гг. ученые обнаружили, что ге-
мопоэз может быть полностью восстановлен внутривенным вливанием клеток
костного мозга. Клиницисты быстро осознали преимущества трансплантации
костного мозга (ТКМ) для терапии врожденных и приобретенных синдромов
недостаточности костного мозга, а также спасения пациентов от смерти в результате
миелоаблативного облучения и химиотерапии, используемых при лечении рака.
В первых исследованиях аллогенная трансплантация (пересадка костного
мозга от донора реципиенту для постоянной замены всех гемопоэтических
клеток) предлагалась как альтернатива аутологичной трансплантации (реинфузия
пациенту его собственных, предварительно сохраненных клеток после миелоаб-
лативной химио- или радиотерапии). Однако тогда при аллогенных
трансплантациях не учитывались в полной мере иммунологические барьеры,
приводящие к отторжению реципиентом костного мозга донора и развитию болезни
"трансплантат против хозяина" (БТПХ) — реакции иммунологически
компетентных лимфоцитов донора против тканей реципиента. Идентификация системы
HLA (человеческих лейкоцитарных антигенов) в середине 1960-х гг. позволила
клиницистам сделать попытку ТКМ у сибсов в совместимых парах
"донор-реципиент". Нобелевский лауреат Е. Donnell Thomas направил исследования в область
поиска оптимальных миелоаблативных режимов лучевой и химиотерапии, а
также фармакологических методов для предотвращения БТПХ. К концу 1970-х гг.
аллогедная ТКМ превратилась из практически экспериментальной и, как
правило, опасной процедуры в эффективный метод терапии апластической анемии, им-
мунодефицитных состояний и многих форм лейкозов — заболеваний, которые
ранее считались неизлечимыми.
В настоящее время, несмотря на разработку и использование мощных иммуно-
супрессивных препаратов, таких как циклоспорин, и широкого спектра
антимикробных средств, выживаемость больных после аллогенных ТКМ незначительно
308
Глава 8. Трансплантация стволовых клеток
изменилась по сравнению с началом 1980-х гг1. Частично это объясняется тем,
что процедуру ТКМ стали шире применять у пациентов с наиболее
неблагоприятным прогнозом. Впечатляющие результаты от увеличения вводимой дозы КМ
при лейкозах и лимфомах, разработка улучшенных режимов кондиционирования
повторно привлекли внимание специалистов к аутологичным трансплантациям.
Ежегодно, начиная с 1990 г., аутологичных трансплантаций выполняется больше,
чем аллогенных.
Обзор концепций и процедур трансплантации
Как описано в главе 1, стволовые гемопоэтические клетки (СГК) способны
полностью восстановить гемопоэтическую и лимфоидную системы облученного
реципиента. Теоретически этого можно достигнуть при помощи одной клетки, но
в клинической практике быстрое и стабильное приживление гарантирует
переливание по крайней мере 1-3 х Ю8 ядросодержащих клеток костного мозга на 1 кг
массы тела реципиента. Лишь мизерная доля (меньше чем 1 на 100 000)
ядросодержащих клеток костного мозга являются СГК. Они морфологически
неотличимы от лимфоцитов, но имеют характерный состав поверхностных белков,
включая CD34\ HLA класса 1Г и CD38". На них нет линейных маркеров, таких как
CD4, поверхностные иммуноглобулины и гликофорин.
Клетки костного мозга для трансплантации аспирируют из задних гребней
подвздошных костей полыми иглами, многократно вводя их в полости,
заполненные костным мозгом. Обычно эта процедура выполняется под общей анестезией
в операционной. Всего собирается 15-20 мл жидкого костного мозга из расчета
на 1 кг массы тела реципиента. Перед последующей обработкой или
непосредственно перед переливанием реципиенту проводится фильтрация, чтобы удалить
фрагменты кости и клеточные конгломераты. Взятие костного мозга переносится
хорошо, опасность для донора, как правило, связана с общей анестезией и очень
редко — со значительным местным кровотечением или повреждением нерва из-за
неправильного положения иглы.
В течение последних 5-6 лет были обнаружены и внедрены в клиническую
практику альтернативные источники СГК. Введение онкологическим пациентам
таких цитокинов, как Г-КСФ и ГМ-КСФ, изолировано или их применение
после высокодозной химиотерапии стимулирует большое число первичных клеток
к миграции из костного мозга в кровоток, из которого они могут быть легко
собраны посредством одной или нескольких процедур афереза. Полученная таким
образом "мобилизованная" периферическая кровь почти полностью заменила
костный мозг как источник клеток для аутологичных трансплантаций. Считается, что
она содержит действительно долгоживущие репопулирующие СГК. К
преимуществам этого метода относятся легкость сбора, более быстрое приживление после
трансплантации и потенциально меньшая опухолевая контаминация. Совсем
недавно были начаты исследования по применению клеток-предшественников пе-
Примеч. ред. При использовании HLA-генотипирования годовую выживаемость реципиентов
неродственного костного мозга удалось повысить до 70-80 % (против 10-20 % в парах
донор-реципиент, подобранных без исследования генотипа но HLA-антигенам II класса). При этом выживание
пациентов, получивших костный мозг от HLA-идентичных родственников, составило только 40 %
за такой же период. (Hansen J. Development of registries of HLA-typed volunteer marrow donors. Tissue
Antigens. - 1996, 47: 460.)
Аллогенная трансплантация
309
риферической крови (КППК), полученных от здоровых доноров-сибсов, которым
вводился Г-КСФ. Их результаты обнадеживают.
. Третий потенциальный источник СГК — пуповинная кровь, содержащая, как
представляется, большую концентрацию примитивных клеток, чем костный мозг.
До 250 мл крови можно получить при родах без ущерба для матери или
новорожденного. Создание банков пуповинной крови расширяет возможности
неродственной аллогенной трансплантации, правда, собираемая кровь может не
содержать необходимого для взрослых реципиентов количества СГК.
Процедура трансплантации заключается в инфузии клеточного концентрата
через широкий в/в катетер. Даже медики бывают удивлены, узнав, что клетки
практически не вводятся повторно в костномозговые полости и что
трансплантации костного мозга выполняются врачами-интернами или педиатрами, которые
специализировались по гематологии или онкологии, а не хирургами. Раствор ди-
метилсульфоксида (ДМСО), используемый для криоконсервации аутологичного
КМ или КППК, может немедленно вызывать умеренную легочную и системные
токсические реакции, тогда как реинфузии свежих аллогенных компонентов не
имеют таких последствий. Механизмы "хоуминга" (миграции) СГК в полости
костного мозга пока плохо изучены, но известно, что в течение 10-25 дней
начинается продукция первых нейтрофилов, затем тромбоцитов и эритроцитов. Эти
процессы подтверждают приживление донорских стволовых клеток.
Аллогенная трансплантация
Обзор
Перечень приобретенных и генетических болезней, при которых используется
аллогенная трансплантация, все возрастает (табл. 8-1). Из-за высокой
заболеваемости и смертности, наблюдаемой при этом виде лечения, большинство
трансплантаций проводится пациентам с приобретенными гематологическими
заболеваниями, угрожающими жизни, прежде всего лейкозами или апластической
ТАБЛИЦА 8-1. Применение аллогенной трансплантации
Приобретенные болезни
Острый миелолейкоз
Хронический миелолейкоз
Апластическая анемия
Миелодиспластический синдром
Множественная миелома1
Острый лимфобластный лейкоз взрослых1
Лимфома1
Генетические болезни
Талассемия
Врожденные иммунодефицитные состояния
Анемия Фанкони
Серповидно-клеточная анемия1
Болезни накопления1
1 В настоящее время показания к проведению аллогенной трансплантации точно не определены.
310
Глава 8. Трансплантация стволовых клеток
анемией. Но поскольку и заболеваемость, и смертность при трансплантации
постепенно снижаются, данная процедура становится все более обычной для
пациентов с хронической несмертельной врожденной патологией, например
серповидно-клеточной анемией. К потенциальным преимуществам аллогенной
Трансплантации по сравнению с аутологичной при злокачественных новообразованиях
относят обеспечение реципиента гемопоэтическими стволовыми клетками от
здорового донора, что уменьшает риск реинфузии жизнеспособных опухолевых
клеток, которые способствуют возникновению рецидива заболевания. При
некоторых опухолях, особенно хроническом миелолейкозе, чрезвычайно важно
иммунологическое противоопухолевое действие донорских клеток.
Выбор донора
Совместимость по белкам (антигенам) главного комплекса гистосовместимости
(ГКГ) — основа для выбора подходящего аллогенного донора. У человека эти
антигены называются системой человеческих лейкоцитарных антигенов (human
leukocyte antigens — HLA). Они представлены фактически на всех ядросодержа-
щих клетках в организме. Эти белки связывают и презентируют пептиды других
белков Т-клеткам; ГКГ-зависимые взаимодействия являются центральными
почти во всех типах иммунных реакций (глава 4). Чужеродные HLA-белки
распознаются и могут вызывать мощный иммунный ответ. При аллогенной
трансплантации эффекторное звено иммунной системы (Т- и В-клетки) замещается
клетками донора. Это же относится к моноцитам и макрофагам, которые
являются важными в презентации чужеродных антигенов лимфоидным клеткам.
Наличие общих HLA-антигенов у донора и реципиента уменьшает риск немедленного
отторжения трансплантата лимфоцитами реципиента, а также риск распознавания
и "нападения" на ткани реципиента (хозяина) лимфоцитами, полученными от
донора. Последний процесс называют болезнью "трансплантат против хозяина".
Наиболее часто аллогенными донорами становятся HLA-совместимые сибсы.
Таковыми считаются сибсы, имеющие одинаковые HLA-антигены I класса А, В
и С, а также HLA-антигены II класса DR, DQ и DP. Так как все гены HLA-белков
локализованы на коротком плече 6-й хромосомы и каждый сибс получает по
одной хромосоме (гаплотип) с комплексом HLA-генов от каждого родителя, сибсы
будут HLA-идентичными с 25 % вероятностью. Трансплантат от полностью
HLA-совместимого сибса имеет очень низкий риск отторжения, однако может
развиться БТПХ из-за различий между донором и реципиентом по недостаточно
изученным малым антигенам гистосовместимости, не найденным на 6-й
хромосоме. При сингенных трансплантациях, когда донор и реципиент являются
однояйцовыми близнецами, не существует угрозы отторжения трансплантата или
возникновения БТПХ.
Только 25-30 % пациентов, нуждающихся в аллогенной трансплантации,
имеют HLA-идентичных сибсов. У остальных пациентов используют трансплантат от
частично совместимого родителя, иного родственника или HLA-совместимого
неродственного донора, соотнося риск развития БТПХ и других
посттрансплантационных осложнений с потенциальными выгодами от проведения ТКМ. За
последние 10 лет были созданы большие регистры HLA-типированных лиц,
желающих пожертвовать свой костный мозг для неродственных реципиентов, что
позволяет значительно увеличить количество трансплантаций от совместимых
Аллогенная трансплантация
311
неродственных доноров (СНД). В США внедрение национальной программы
донорства костного мозга позволило накопить сведения более чем об 1 млн
потенциальных доноров. К сожалению, только 60 % пациентов находят совместимые пары
через регистры. Иногда на поиски донора уходит 3-6 месяцев, из-за чего
трансплантация становится невозможной. Особенно трудно найти донора для
пациентов-неевропеоидов из-за низкого представительства некоторых этнических групп
в регистрах и их существенного генетического многообразия. Долгосрочная
выживаемость после трансплантаций от СНД или частично совместимых
родственников по сравнению с трансплантациями от совместимых сибсов значительно
ниже вследствие более высокой встречаемости отторжения трансплантата,
развития инфекций и БТПХ, особенно у взрослых пациентов.
Подготовка к трансплантации
Как только показания для аллогенной трансплантации установлены и найден
подходящий донор, начинается предоперационная подготовка пациента. Ее цели
зависят от основного заболевания. Однако в любом случае режим подготовки
(кондиционирования) должен так супрессировать иммунитет реципиента, чтобы
предотвратить отторжение аллотрансплантата, а также подавить костный мозг
реципиента, что позволит прижиться стволовым клеткам донора. При наличии
злокачественных новообразований нужно обеспечить максимальное
уничтожение клеток опухоли при ограниченной токсичности по отношению к другим
органам (не костному мозгу). При доброкачественных заболеваниях для иммуносуп-
рессии используют циклофосфамид в сочетании с антитимоцитарным
глобулином, общее облучение лимфоидной ткани или комбинацию сравнительно
низкой дозы бусульфана для миелосупрессии и циклофосфамида для иммуно-
супрессии. Такие режимы относительно безопасны для больных талассемией
и апластической анемией (при использовании полностью совместимого донора-
сибса смертность, связанная с трансплантацией, составляет у молодых пациентов
только 5-15 %).
При подготовке к трансплантации пациентов со злокачественными
новообразованиями обычно используется общее облучение тела в комбинации с приемом
цитотоксического агента (или агентов). Положительные эффекты данной
процедуры обусловлены антинеопластическим воздействием на организм в целом
и сильной миело- и иммуносупрессией. Циклофосфамид — лекарственный
препарат, наиболее часто назначаемый при общем облучении тела. Для усиления
противоопухолевого действия применяют также этопозид, цитозинарабинозид и
мелфалан. Комбинации циклофосфамида и бусульфана могут служить
альтернативой схемам, включающим общее облучение тела.
Получение стволовых клеток донора и трансплантация
Для успешного и длительного приживления необходимо адекватное количество
СПС. Трансплантаты, содержащие очень большое количество таких CD34+
клеток, имеют более низкую частоту отторжения трансплантата и развития ранних
посттрансплантационных инфекционных осложнений. Процедура взятия
костного мозга безопасна, частота осложнений у здоровых доноров не превышает
0,1 % (их возникновение обычно связано с последствиями общей анестезии).
312
Глава 8. Трансплантация стволовых клеток
Доноры госпитализируются в большинстве случаев на 1 сутки после процедуры,
а возвращение к нормальному образу жизни и плановой работе происходит
в течение 1-2 недель. В настоящее время в аллогенных трансплантациях
преимущественно используют костный мозг донора как источник для СГК, но в
перспективе, вероятно, его заменят трансплантаты донорских КППК. Для
получения КППК донорам вводят Г-КСФ или другие цитокины подкожно в течение
4-6 дней, после чего клетки периферической крови собирают путем афереза, что
позволяет получить для трансплантации клеток CD34+ в 3-10 раз больше, чем
из костного мозга. Увеличивается также частота трансплантаций пуповинной
крови; ее банки могут оказаться ценным источником неродственных донорских
стволовых клеток, если будет доказано, что эти трансплантации безопасны и
эффективны для взрослых и детей.
Техника аллогенной трансплантации
Ранее аллогенные трансплантации выполнялись методом инфузии в
периферическую вену необработанного костного мозга, который впоследствии стали
фильтровать для профилактики тромботических осложнений, вызываемых крупными
частицами. Поскольку показания для аллогенных трансплантаций расширяются
и увеличивается риск возникновения осложнений, становится общепринятым
дотрансплантационное манипулирование с трансплантатом. При
несовместимости эритроцитов донора и реципиента из трансплантата удаляются донорские
эритроциты и/или сыворотка, чтобы предотвратить непосредственный лизис
эритроцитов донора или реципиента изогемагглютининами. Т-клеточное
истощение костного мозга уменьшает частоту развития БТПХ, но его применение имеет
некоторые ограничения. Когда используется костный мозг, обедненный Т-лим-
фоцитами, нарушение функции трансплантата или его отторжение происходят
чаще. Из-за отсроченного иммунного восстановления чаще развиваются
инфекционные осложнения, например цитомегаловируснаяболезнь. Кроме того,
стараясь уменьшить риск возникновения БТПХ, можно свести на нет эффект
"трансплантат против лейкоза" (ТПЛ), что приведет к увеличению частоты рецидивов
лейкозного процесса, особенно при миелолейкозах. Для преодоления этих
проблем активно испытываются такие усовершенствования, как определение
оптимальной степени истощения Т-клеток или возврат Т-клеток после приживления
трансплантата и восстановления организма после иммуносупрессии. Уменьшить
вероятность развития БТПХ без отмены эффекта ТПЛ может также истощение
отдельных субпопуляций Т-клеток, например CD8+, но не CD4+. У пациентов
с доброкачественными заболеваниями истощение Т-клеток, предотвращающее
отторжение трансплантата, становится все более предпочтительным при
использовании адекватного кондиционирования.
График проведения трансплантации
Материалы этого раздела могут служить руководством по ведению пациентов,
подвергающихся трансплантации. В нем хронологически изложена
последовательность ожидаемых событий и осложнений, связанных с этой процедурой
(рис. 8-1). День 0 всегда определяется вливанием аутологичных или аллогенных
СГК пациенту. До приживления характер (виды) осложнений у пациентов при
Аллогенная трансплантация
313
аутологичных и аллогенных трансплантациях очень сходен. После приживления
реципиенты аутологичного трансплантата имеют меньше проблем, и
последующие разделы касаются только реципиентов аллогенного трансплантата.
От подготовительной терапии к приживлению
Профилактика инфекции. С момента начала подготовительной терапии
наиболее частыми являются инфекционные осложнения. Количество лейкоцитов
в крови приближается к самому низкому уровню в день 0 или вскоре после него,
поэтому данный период рассматривается как критический по восприимчивости
к бактериальным и грибковым инфекциям. Даже при нормализации количества
нейтрофилов пациенты имеют глубокую иммунодепрессию, особенно после алло-
генной трансплантации. В некоторых случаях назначение антибиотиков и
асептика окружающей среды эффективно уменьшают частоту развития инфекционных
осложнений. Боксы, оборудованные вентиляционными фильтрами с
повышенным давлением и ламинарным током воздуха, теперь имеются в большинстве
отделений трансплантации. Они эффективны для удаления большинства
переносимых воздухом бактериальных частиц и грибковых спор.
В качестве антибактериальных профилактических средств обычно используют
энтеральные фторхинолоны. Плохо адсорбируемый хинолон норфлоксацин
можно назначать для деконтаминации кишечника. В основе профилактики грибко-
Мукозит
Тошнота/рвота
> Диарея
Веноокклюзионная
болезнь
Острая БТПХ
Хроническая БТПХ
Бактериальные/
грибковые
инфекции
Цитомегаловирусная
инфекция
Интерстициальный
пульмонит
Другие вирусные
инфекции
i i
'День ["*
•^трансплантации'
i I.
Приблизительно
день приживления '
i
_1_
I
_1_
J L_
_L
JL
-10 -5 0 5 10 15 20 25 30 35 40 45 50
-Ih
J_
_L
J L
_l_
_L_
J
50 150200250 300350400
Режим
кондиционирования
Дни
Рис. 8-1. Время появления осложнений при трансплантации стволовых клеток. До приживления
у пациентов, подвергающихся аутологичной и аллогенной трансплантации, осложнения сходны.
После приживления осложнения в основном проявляются у реципиентов аллогенных
трансплантатов. Полосы ограничивают периоды возникновения каждого осложнения (усредненные данные).
Пациенты с тяжелой болезнью "трансплантат против хозяина" имеют намного более длительную
иммуносупрессию, у них иногда развиваются серьезные бактериальные, грибковые и вирусные
инфекции даже после периода наибольшего риска, показанного на рисунке
314
Глава 8. Трансплантация стволовых клеток
вых инфекций лежит терапия флуконазолом. Показана эффективность флукона-
зола в уменьшении риска возникновения как инвазивных, так и поверхностных
кандидозов. Однако Aspergillus и некоторые стойкие штаммы Candida
нечувствительны к нему и по-прежнему вызывают осложнения. В большинстве
медицинских центров прекращают бактериальную и грибковую профилактику после
приживления нейтрофильного ростка. Реципиентам при аллогенных и аутологичных
трансплантациях в течение 3 месяцев или длительнее проводится также
профилактика инфекций, вызываемых Pneumocystis carinii. Используется или триметоп-
рим-сульфаметоксазол перорально 1 раз в неделю, или аэрозоль пентамидина
ежемесячно. Антивирусные мероприятия направлены на вирусы группы герпеса:
вирус простого герпеса (ВПГ), вирус Varicella zoster и цитомегаловирус (ЦМВ).
Реактивация ВПГ и V. zoster предотвращается внутривенным введением низкой
дозы ацикловира. Оптимальный режим профилактики ЦМВ все еще в процессе
разработки. Если и донор, и реципиент ЦМВ-серонегативны, необходимо
предотвратить развитие первичной инфекции, для чего используют ЦМВ-негативные
или профильтрованные компоненты крови. Серопозитивным реципиентам для
предупреждения реактивации назначают ацикловир в высокой дозе. Кроме того,
доказано, что профилактику ЦМВ можно обеспечить еженедельным
внутривенным введением у-глобулина как серопозитивным пациентам, так и серонегатив-
ным, получившим трансплантат от серопозитивного донора. У пациентов с
высоким риском развития ЦМВ-инфекции рассматривалась также возможность
применения ганцикловира, хотя наличие у него миелосупрессивного действия
препятствует широкому профилактическому использованию этого препарата.
Инфекционные осложнения. Несмотря на оптимальные профилактические
меры, инфекционные осложнения являются главной причиной заболеваемости и
смертности при трансплантации стволовых клеток. Клиницисты обычно
расценивают лихорадку как первый признак развития инфекции. Вероятно,
значительная доля эпизодов лихорадки у больных в период трансплантации является не
результатом активной или распознаваемой инфекции, а следствием мукозита,
БТПХ или других воспалений. В любом случае возникновение лихорадки у им-
мунокомпрометированного пациента не следует оставлять без внимания.
Бактерии. Бактериальные инфекции преобладают в дотрансплантационном
периоде не только потому, что пациент имеет глубокую нейтропению, но и в связи
с появлением новых входных ворот для инфекции при поражении кожи, мембран
слизистых оболочек и поверхности ЖКТ из-за воздействия лекарственных
препаратов, применяемых для кондиционирования. Бактериемия наблюдается
у 15-50 % пациентов. Источник ее, как правило, эндогенный, происходящий из
желудочно-кишечного тракта или от установленных постоянно катетеров.
Доминирует грамположйтельная бактериемия, вероятно, в результате использования в
профилактических целях антибиотиков с грамотрицательной активностью. В
некоторых центрах пациенту сразу, как только у него развивается нейтропения,
назначают цефтазидим или вводят внутривенно иной антибиотик широкого
спектра действия, тогда как в других лечение начинают лишь при первых признаках
лихорадки. При возникновении нейтропенической лихорадки часто также
назначают ванкомицин.
Иные распространенные локализации бактериальной инфекции в
дотрансплантационном периоде — кожа, легкие, синусы и ЖКТ. Еженедельная скринин-
Аллогенная трансплантация
315
говая рентгенография или компьютерная томография (КТ) грудной полости и
синусов могут быть полезны для раннего обнаружения очагов инфекции. Тифлит —
скоротечная (молниеносная) некротизирующая инфекция стенки слепой кишки,
вызываемая грамотрицательными микроорганизмами, разновидностями кандида
и/или клостридий. Пациенты обычно жалуются на боль в животе, тошноту и
рвоту. Если ультразвуковое исследование или КТ-сканирование позволяют
предположить утолщение или воспаление кишечной стенки, может понадобиться
хирургическое вмешательство. Смертность при тифлите составляет от 50 % до 100 %.
Нередко возникает бактериальная пневмония, но типичные долевые (крупозные)
инфильтраты встречаются очень редко. Диагностика диффузных инфильтратов
в дотрансплантационном периоде может быть затруднена, поскольку
дифференциальный диагноз нужно проводить между перегрузкой жидкостью, легочным
кровоизлиянием, ранними эффектами облучения и ЦМВ-пульмонитом.
Пациентам с лихорадкой и легочным инфильтратом часто назначают эмпирическое
лечение антибиотиками широкого спектра действия, чтобы обеспечить двойную анти-
грамотрицательную терапию и антигрибковую защиту. Амфотерицин обычно
вводят до получения результатов исследования бронхоальвеолярного лаважа,
который в большинстве случаев бывает неинформативным.
Грибки. Почти у 30 % реципиентов в дотрансплантационном периоде
развиваются грибковые инфекции. Чаще всего удается выделить разновидности Candida,
прежде всего albicans, и Aspergillus. Идентифицировать грибковую инфекцию
бывает трудно. Когда эпизоды возвратной лихорадки длятся более 3-7 дней,
несмотря на применение антибиотиков широкого спектра действия, должна быть
назначена эмпирическая антигрибковая терапия амфотерицином, даже у пациентов,
уже получающих флуконазол профилактически. Кандидозы проявляются в виде
эритематозных пятнистых кожных высыпаний, фунгемии или висцеральной
инфекции. Повышенная активность печеночных ферментов, особенно щелочной
фосфатазы, и типичные повреждения печени при КТ- или при
магнитно-резонансном исследовании (МРИ) позволяют заподозрить развитие гепатоспленического
кандидоза. Лечение инвазивных кандидозных инфекций в
дотрансплантационном периоде относительно неэффективно, и персистирующая инфекция является
очень плохим прогностическим признаком. Поверхностные инфекции слизистой
оболочки полости рта и пищевода можно предотвратить, соблюдая правила
гигиены ротовой полости, профилактическим приемом флуконазола и антигрибковых
средств в таблетках, антисептическими полосканиями полости рта.
Пневмония, вызываемая Aspergillus pneumoniae,— наиболее частое и грозное
грибковое осложнение. Причиной развития инфекции является ингаляция спор
Aspergillus, которые попадают в воздух, ускользая от вентиляторных фильтров,
или их предсуществующая колонизация у пациента. Aspergillus может также
вызывать воспалительный процесс в синусах, коже или ЦНС. Диагностика аспер-
гиллезной инфекции иногда достаточно трудна: характерные для нее узелковые
или полостные проявления и/или специфический ареол, который представляет
собой зону размягчения окружающих легочных масс, определяемую при
просмотре КТ, нередко отсутствуют. Единственное средство подтверждения
диагноза — биопсия поврежденной ткани, но у компрометированных пациентов с
низким количеством тромбоцитов проведение этой процедуры не всегда оправдано.
Если есть серьезные основания подозревать аспергиллез, должно быть назначено
лечение высокими дозами амфотерицина.
316
Глава 8. Трансплантация стволовых клеток
Вирусные инфекции. В дотрансплантационном периоде вирусные инфекции
развиваются реже, чем после трансплантации, по крайней мере, у реципиентов
аллогенных трансплантатов. Реактивация вируса простого герпеса (ВПГ)
происходит у 85 % пациентов, которым не назначался ацикловир с профилактической
целью. Стоматиты с гингивитами — наиболее частое проявление
ВПГ-1-инфекции, эти болезненные повреждения ограничивают прием пищи через рот.
Они также являются входными воротами для бактериальной и грибковой
суперинфекции. К другим потенциально опасным вирусным агентам относятся
аденовирус и респираторно-синцитиальный вирус. Очень опасна также ранняя
ЦМВ-инфекция или ее реактивация, но пик ее встречаемости отмечается в
посттрансплантационном периоде.
Желудочно-кишечные осложнения. Химиотерапия высокими дозами
препаратов, общее облучение тела и прием таких преператов, как имипенем, ганцикловир
и амфотерицин, могут вызывать тяжелую тошноту и рвоту. Мукозит развивается
через 5-14 дней после начала режима кондиционирования и связан с периодом
глубокой нейтропении. Быстро делящиеся клетки, которые выстилают ЖКТ,
повреждаются лекарственными средствами, используемыми в предоперационной
подготовке, а последующая суперинфекция язв бактериями, грибками, ВПГ или
ЦМВ усугубляет нарушение. Заживление этих повреждений происходит не ранее
повышения уровня нейтрофилов. Клинические признаки включают боль, тошноту,
рвоту и диарею. При очень тяжелых мукозитах не исключено поражение
воздухоносных путей из-за слущивания эпителия и кровоизлияний в периларингеальной
ткани. Любой пациент, у которого заподозрено развитие этого осложнения,
должен быть помещен в стационар, где есть условия для немедленного выполнения
трахеостомии. Адекватное обезболивание — наиболее важный компонент в
лечении мукозитов; при прогрессировании симптомов от умеренных к тяжелым для
пациента нужно подобрать индивидуальную дозу анальгетиков. Средства для
полоскания полости рта, например хлоргексидины, способны уменьшить развитие
суперинфекции и должны использоваться в период нейтропении. Поскольку
прием пищи через рот затруднен из-за описанных вторичных осложнений, постоянно
регулируется объем парентерального питания.
Воспаление и слущивание слизистой оболочки ЖКТ нарушает всасывание
в толстом и тонком отделах кишечника. Хотя это наиболее частая причина
возникновения диареи, клиницисты должны помнить, что одновременно могут
развиваться и другие, более тяжелые, осложнения. Распространенной является
инфекция Clostridium difficile, приводящая к псевдомембранозному колиту даже без
длительного применения антибиотиков широкого спектра. Скрининг токсина
Clostridium difficile выполняется при резком увеличении объема стула, особенно
если он кровянистый и сопровождается лихорадкой.
Нарушения функции печени. В дотрансплантационном периоде всегда имеет
место умеренное повышение активности ферментов печени и содержания
билирубина. Причины его множественны: действие принимаемых лекарственных
препаратов, общий холестаз, обусловленный парентеральным питанием,
инфекции. Более серьезной является печеночная веноокклюзионная болезнь, часто
заканчивающаяся смертью во время кондиционирования. Она встречается у 10-
20 % реципиентов аллогенных и аутологичных трансплантатов. Триада
симптомов веноокклюзионной болезни, обычно развивающихся в период от 7 до
Аллогенная трансплантация
317
14 суток, включает гепатомегалию, желтуху и асцит или необъяснимое
увеличение массы тела. Кроме того, есть сведения о возникновении боли в правом
верхнем квадранте живота, напоминающей таковую при "остром животе". Тяжесть
симптомов варьируется. Веноокклюзионную болезнь нужно дифференцировать
от БТПХ либо гепатита, обусловленного инфекцией или действием
принимаемых лекарственных препаратов. Увеличение уровня билирубина при отсутствии
значительного повышения активности трансаминаз и щелочной фосфатазы
позволяет отличить ранние стадии заболеваниями* этих и других осложнений. При.
необходимости для подтверждения диагноза может выполняться трансвенозное
измерение давления в печеночной вене или трансвенозная биопсия печени, но на
практике эти процедуры проводятся редко. Хотя патогенез веноокклюзионной
болезни неясен, при гистологическом исследовании всегда отмечают
повреждение эндотелия венул печени и тромбоз. Выявляются снижение уровня
эндогенных антикоагулянтов, например протеина С, и изменения количественных
соотношений мультимеров фактора Виллебранда (глава 6). Большинство этих
нарушений, по-видимому, вторичны по отношению к повреждению печени.
Лечение веноокклюзионной болезни, за исключением поддерживающей
терапии, не стандартизовано. Пациентам с подозрением на данное заболевание
необходимо следить за массой тела и, по возможности, не допускать ее увеличения,
ограничивая потребление натрия и жидкости или применяя форсированный
диурез; В некоторых исследованиях была показана целесообразность использования
антикоагулянтов или пополнения резервов эндогенных антикоагулянтов, однако
полученные результаты противоречивы, тем более что существует очевидный
риск при назначении антикоагулянтов этим пациентам. Смертность по разным
сообщениям достигает 50 %, но фактически может быть ниже, так как многие
умеренно выраженные случаи скорее всего не учитывались. Плохой прогноз связан
с развитием полиорганной недостаточности, особенно гепаторенального
синдрома. Развитие веноокклюзионной болезни более вероятно при очень интенсивных
режимах кондиционирования, у реципиентов пожилого возраста, а также при
наличии дотрансплантационных повреждений печени любой этиологии.
Поддерживающая терапия. Переливание компонентов крови до периода
приживления неизбежно. В среднем реципиенту аллогенного трансплантата
требуется до 10-15 доз эритроцитсодержащих компонентов и до 150 доз тромбоцитов.
Реципиенты аутологичного трансплантата обычно менее зависимы от поддержки
компонентами крови. Из-за глубокой иммуносупрессии должны
предприниматься шаги для предотвращения посттрансфузионной БТПХ. Этот вид БТПХ
развивается, когда жизнеспособные лимфоциты в компонентах крови реагируют с
антигенами реципиента. Фильтры, удаляющие до 99 % лейкоцитов, и облучение
гемокомпонентов инактивируют остающиеся лейкоциты. Если и донор КМ, и
реципиент ЦМВ-негативны, необходимо предотвратить первичное ЦМВ-инфици-
рование через компоненты крови путем фильтрации через лейкоцитарные
фильтры. Стандартной практикой в большинстве трансплантационных центров
является обеспечение уровня гемоглобина от 80 до 100 г/л.
У многих реципиентов затруднена поддержка тромбоцитоконцентратами из-за
дотрансплантационной аллосенсибилизации, развившейся после
предшествующих трансфузий. В отличие от эритроцитов тромбоциты несут на себе HLA-анти-
гены I класса и являются чрезвычайно иммуногенными. Переливание по крайней
318
Глава 8. Трансплантация стволовых клеток
мере частично HLA-совместимых тромбоцитов способно решить проблему
увеличения их количества у реципиента. Рефрактерность к тромбоцитам может быть
обусловлена также гиперспленизмом, лихорадкой и сепсисом. Считается, что
у пациентов с уровнем тромбоцитов менее 5000/мкл повышен риск развития
спонтанных кровотечений. Клиническая практика свидетельствует, что
содержание тромбоцитов менее 5000/мкл является показанием для переливания.
Активное кровотечение, лихорадка или использование инвазивных методов
мониторинга — серьезные основания для повышения количества тромбоцитов.
Использование мобилизованных периферических клеток крови вместо
костного мозга при аутолбгичных трансплантациях сокращает время приживления
настолько, что лечение цитокинами (Г-КСФ или ГМ-КСФ), стимулирующими
миелопоэз, не приводит к его дальнейшему уменьшению. Продолжающиеся
исследования результатов применения новых цитокинов, таких как тромбопоэтин
иинтерлейкин-11, позволят установить их роль в ускорении восстановления
тромбоцитов. Пока не разработаны общие принципы использования гемопоэти-
ческих факторов роста (ГФР) после аллогенных трансплантаций. Очевидно, что
высокое содержание эндогенных факторов роста в течение дотрансплантацион-
ного периода прогностически благоприятно. Некоторые клиницисты
настороженно относятся к использованию ГФР у пациентов с миелолейкозами из-за
теоретической возможности стимуляции злокачественного клона, однако на
практике не зарегистрировано увеличения частоты рецидивов миелолейкоза при
использовании ГФР. Несмотря на сомнительную эффективность применения
ГФР в раннем посттрансплантационном периоде, их назначение в ситуации
отсроченного приживления общепринято.
Приживление. Существуют большие индивидуальные различия по времени
приживления трансплантатов, но признаки донорского гемопоэза у реципиентов
после аллогенных трансплантаций костного мозга обычно замечают на 12-23
сутки. При аутологичных трансплантациях КППК восстановление нейтрофилов
в периферической крови реципиентов может происходить уже на 7-9 сутки.
Первыми обычно появляются в периферической крови моноциты из
восстановленного костного мозга, потом нейтрофилы, а затем тромбоциты и эритроциты.
Возможных причий отсроченного приживления много. Среди них отторжение
трансплантата у аллогенных реципиентов и низкое число СГК в донорском или
аутологичном трансплантате. Т-клеточное истощение, элиминация опухолевых
клеток, подавление инфекций или медикаментозная супрессия костного мозга
такими препаратами, как ганцикловир, также могут задерживать или угнетать
приживление. Гемопоэтические цитокины, по-видимому, способны ускорять
восстановление, но в некоторых случаях необходима повторная трансплантация
либо донорских КППК, либо дополнительного количества костного мозга.
Приживление через 100 дней
Острая болезнь "трансплантат против хозяина"
Патофизиология. Когда иммунокомпетентные Т-лимфоциты, полученные
от донора, реагируют против антигенов, экспрессированных на тканях
реципиента, развивается БТПХ. Сроки возникновения и клиническая картина БТПХ
достаточно вариабельны и зависят от многих факторов, часть из которых еще плохо
изучена. Совокупность признаков и симптомов, обычно отмечаемых в первые
Аллогенная трансплантация
319
100 дней, характеризует острую БТПХ (оБТПХ), тогда как клинические
проявления после сотого дня очень разнообразны и составляют синдром хронической
БТПХ. При отсутствии адекватной профилактики БТПХ ее первые симптомы
могут обнаруживаться в течение 1-2 недель после инфузии донорских клеток.
Встречаемость. БТПХ наблюдается в той или иной степени почти у 80 %
реципиентов аллогенных трансплантатов. Факторы, предрасполагающие к
возникновению оБТПХ, указаны в табл. 8-2. Наиболее значимый прогностический
фактор развития оБТПХ — HLA-несовместимость. Возникновение БТПХ более
вероятно, когда донорами являются многорожавшие женщины из-за их HLA-ал-
лоиммунизации в течение беременностей или родов. Риск возрастания частоты
БТПХ увеличивается у пациентов старше 30 лет. Значительный возраст донора
также может быть фактором риска. Многие случаи достаточно серьезной
оБТПХ при совместимых неродственных трансплантациях можно объяснить
полиморфизмом HLA-белков. Инициировать возникновение или усиливать
проявления оБТПХ могут инфекции и воспаление из-за высвобождения цитокинов
и стимуляции донорских лимфоцитов к реагированию против антигенов в месте
воспаления.
Клинические проявления. Клинические признаки оБТПХ отмечаются прежде
всего в коже, печени и ЖКТ. При оБТПХ могут повреждаться одна или несколько
систем органов: одновременное развитие всех характерных для оБТПХ
симптомов наблюдается редко. Обычно первым признаком оБТПХ является макуло-па-
пулезная кожная сыпь на сгибательных поверхностях конечностей, а также на
лице, шее и туловище. При тяжелой оБТПХ сыпь становится сливной и может
прогрессировать до образования пузырей или десквамации. С помощью биопсии
кожи можно отличить оБТПХ от реакций на прием лекарственных препаратов и
вирусных экзантем.
Помимо кожной сыпи, как правило, возникают желудочно-кишечные
нарушения, в том числе водянистая обильная диарея, которая в тяжелых случаях может
быть кровянистой; а также спастическая брюшная боль и кишечная
непроходимость. Диагноз оБТПХ ЖКТ ставится после тщательного анализа времени ее
начала и других возможных причин развития симптомов. Ее дифференцируют от
остаточных эффектов кондиционирования; инфицирования Clostridium difficile,
ЦМВ, энтеральными патогенными микроорганизмами или побочного действия
ТАБЛИЦА 8-2. Факторы риска острой болезни "трансплантат против хозяина"
Реципиент
Увеличение возраста
Инфекция ДНК-содержащими вирусами (ВПГ, вирус Varicella zoster, ЦМВ)
Интенсивная предтрансплантационная подготовка
Низкая доза клеток-предшественников
Донор
Предшествующие трансфузии
Предшествующие беременности
Увеличение возраста
Совместимость донора и реципиента
Несоответствие по полу
HLA-A-, -В-, -.DR-несовместимость
Неродственный донор
320
Глава 8. Трансплантация стволовых клеток
принимаемых лекарственных препаратов. С этой целью используется эндоскопия
с биопсией.
О развитии "печеночной" симптоматики оБТПХ свидетельствуют
лабораторные признаки холестаза, который является результатом агрессии цитотокси-
ческих Т-клеток преимущественно в отношении малых междолевых и краевых
желчных протоков. Хотя преобладает повышение содержания билирубина и
активности щелочной фосфатазы, нередко в патологический процесс вовлекается
паренхима печени с увеличением активности АлАТ и АсАТ. Дифференциальный
диагноз оБТПХ печени проводят с вирусным гепатитом, ВОБ и токсическими
побочными эффектами приема лекарственных препаратов (клиническую
картину может искажать гепатотоксичность циклоспорина). Иногда для постановки
диагноза требуется выполнение биопсии печени.
Профилактика БТПХ. Всем реципиентам аллогенных (но не аутологичных
или сингенных) трансплантатов проводят профилактику БТПХ. Наиболее
эффективны два метода: удаление Т-лимфоцитов из донорских клеток или
назначение реципиенту иммуносупрессивных препаратов (обычно циклоспорина, мето-
трексата и преднизолона или других кортикостероидов). Комбинированная
терапия по меньшей мере двумя препаратами показана при использовании
трансплантатов, не истощенных Т-клетками, однако назначить два или три
препарата — устанавливается индивидуально. Профилактически в большинстве случаев
применяют циклоспорин, который вводят до или во время вливания костного
мозга (день 0). Не реже 1 раза в неделю следует определять содержание препарата
в плазме крови и при необходимости для поддержания соответствующего
терапевтического уровня корригировать дозу. Профилактика БТПХ должна
продолжаться по крайней мере в течение 3-6 месяцев после трансплантации. Деконта-
минация ЖКТ антибиотиками и очистка воздуха при помощи фильтров также
снижают риск развития БТПХ, уменьшая частоту возникновения инфекционных
эпизодов.
Лечение. Лечебная стратегия при оБТПХ строится с учетом профилактических
мероприятий с использованием иммуносупрессивных препаратов. Для лечения
БТПХ применяют те же средства, что и для ее профилактики. Решение о начале
терапии принимают на основании степени выраженности органоспецифических
признаков БТПХ (табл. 8-3). Нет необходимости лечить изолированное умеренное
повреждение кожи при БТПХ, но при любых нарушениях ЖКТ или поражении
кожи второй и более степени следует активно воздействовать на патологический
процесс. В основном иммуносупрессивные агенты назуначаются поэтапно в
соответствии с клинической реакцией на их введение.
Обычно вместе с циклоспорином используют метилпреднизолон в высокой
дозе, которую снижают на протяжении нескольких недель с того момента, как
симптомы и признаки оБТПХ ослабевают. При рефрактерной к стероидам БТПХ
прогноз очень плохой, и при тяжелой (III или IV степень) оБТПХ, если нет
положительной реакции на применение высоких доз стероидов в течение 1 недели,
должны быть назначены препараты второй линии: антитимоцитарный глобулин
или экспериментальные иммуносупрессоры. Пациентам со значительным
вовлечением в патологический процесс ЖКТ требуется тщательная регуляция баланса
жидкости и электролитов. Необходимо определить степень повреждения
(сохранности) кишечника и по мере уменьшения диареи увеличивать потребление
пациентом пищи per os. Для лечения спазмов ЖКТ могут применяться болеутоляю-
Аллогенная трансплантация
321
щие лекарственные средства. Лекарственные препараты против диареи, такие как
лоперамид или октреотид, используются ограниченно.
Прогноз. Прогноз развития оБТПХ во многом зависит от ее клинической
степени. При IV степени смертность, несмотря на агрессивную иммуносупрессию,
составляет почти 100 % и чаще обусловлена возникающими из-за подавления
иммунитета суперинфекциями, чем прямым поражением органа или оБТПХ.
Имеющие место иммунные дисфункции являются как непосредственным следствием
оБТПХ, так и результатом лечения. Подавление костного мозга с последующим
увеличением потребности в трансфузиях, особенно тромбоцитов, характерно для
тяжелой БТПХ. У пациентов с тяжелой оБТПХ выше вероятность развития
хронической БТПХ.
Цитомегаловирус. В общей популяции 50-70 % индивидуумов являются серо-
позитивными (носителями антител) по цитомегаловирусу (ЦМВ), что указывает
на предшествующую первичную ЦМВ-инфекцию и латентное "носительство"
вируса в небольшом количестве лейкоцитов или других клеток. При аллогенной
трансплантации глубокая иммуносупрессия сопровождается реактивацией ЦМВ
почти у 70 % реципиентов (в случаях, когда серопозитивные или серонегативные
реципиенты получают клетки от серопозитивного донора). У реципиентов ауто-
логичных клеток такое нарушение наблюдается значительно реже.
Клинические проявления. Посттрансплантационная реактивация ЦМВ может
быть бессимптомной и обнаруживаться при рутинном скрининге активной
вирусной репликации. Реактивация ЦМВ с клинической симптоматикой называется
ЦМВ-болезнью и наблюдается приблизительно у 25-35 % реципиентов аллоген-
ных трансплантатов. Пик манифестации приходится на 50-60-е сутки, но ЦМВ-
болезнь развивается и в иные сроки, особенно у пациентов с предсуществующей
глубокой иммунодепрессией или текущей БТПХ. ЦМВ-болезнь иногда
проявляется как типичная вирусная инфекция с лихорадкой и ознобом, недомоганием
ТАБЛИЦА 8-3. Органоспецифическая классификация острой БТПХ
Тяжесть
повреждения
1
2
3
4
Клиническая
степень
I
II
III
IV
Кожа
Макуло-папулезная сыпь
< 25 % поверхности тела
Макуло-папулезная сыпь
25-50 % поверхности тела
Генерализованная
эритродермия
Генерализованная
эритродермия с пузырями и дес-
квамацией
Печень
(билирубин)
2-3 мг/дл
> 3-6 мг/дл
> 6-15 мг/дл
> 15 мг/дл
Кишечный тракт
500-1000 мл жидкого
стула/сут
1000-1500 мл жидкого
стула/сут
> 1500 мл жидкого
стула/сут
Тяжелые
абдоминальные боли с
непроходимостью или без нее
V
Уровень повреждения
Кожа
1 или 2
1-3
2илиЗ
2-4
Печень
0
1
2илиЗ
2-4
жкт
0
1
2илиЗ
2-4
322
Глава 8. Трансплантация стволовых клеток
и миалгией. Вне зависимости от того, повреждаются ли ранние гемопоэтические
клетки-предшественники или активируется высвобождение ингибиторных цито-
кинов, обычно усугубляется цитопения. Инфекция может быть органоспецифич-
ной, поражая легкие в форме интерстициального пульмонита или ЖКТ в виде
диффузных интерстициальных изъязвлений, сопровождаемых диареей или
болью в животе. Часто отмечается повышение активности печеночных ферментов,
тогда как ЦМВ-ретинит наблюдается редко.
Интерстициальный пульмонит, ассоциированный с ЦМВ,— наиболее частое и
наиболее грозное проявление ЦМВ-болезни. По разным оценкам, он развивается
у 15-30 % реципиентов аллогенных трансплантатов, причем в 90 % случаев
приводит к их гибели. У больных отмечается лихорадка, диспноэ и сухой кашель с ги-
поксемией при всех формах заболевания, кроме самых легких.
Рентгенологическая картина вариабельна, наиболее типичны диффузные интерстициальные
инфильтраты. Из-за сложности трактовки рентгенологических данных возникает
необходимость в широкой дифференциальной диагностике. Следует рассмотреть
возможность развития диффузного альвеолярного кровоизлияния, легочного
отека, идиопатического интерстициального пульмонита и острого
респираторного дистресс-синдрома или инфекции другой этиологии.
Диагноз. Современные диагностические методы ускоряют процесс
распознавания заболевания и тем самым способствуют предупреждению угрожающих жизни
осложнений ЦМВ-инфекции. Обнаружение реплицирующегося ЦМВ в
лейкоцитах периферической крови или быстрое культивирование вируса из биологических
жидкостей позволяет назначить лечение до значительного повреждения органа и
развития выраженной ЦМВ-болезни. Общепринято обследовать пациентов 1 или
2 раза в неделю в раннем периоде после приживления. Определение ЦМВ-специ-
фических антигенов в лейкоцитах крови — быстрый и чувствительный метод
ранней диагностики инфекции. Таковым является и метод "shell-vial",
обнаруживающий ЦМВ при 24-48-часовом выращивании в культуре по экспрессии вирусных
антигенов в монослое индикаторных клеток, культивируемых совместно с
лейкоцитами больного. Значимость получения положительных результатов в культуре
мочи или соскоба из зева не ясна. Положительные культуры крови или легочного
лаважа, так же как и положительный тест на выявление антигена в крови,
указывают на необходимость начала лечения. Если все неинвазивные тесты отрицательны,
но на 30-100 сутки после трансплантации у пациента появляются легочные
инфильтраты, нужно выполнить диагностическую бронхоскопию и получить бронхо-
альвеолярный лаваж (БАЛ). Из-за высокой частоты возникновения и смертности
от ЦМВ-пульмонитов целесообразно начинать эмпирическое анти-ЦМВ-лечение
даже при отрицательных результатах культуральных исследований.
Лечение. При обнаружении ЦМВ-носительства или клинических проявлений
болезни обычно назначают ганцикловир в комбинации с очень высокими дозами
внутривенных иммуноглобулинов (ВВИГ). Как правило, ежедневно
ганцикловир вводят по крайней мере 10 дней, а затем устанавливают поддерживающий
режим (3-4 раза в нед) вплоть до 100-120-го дня. Во время терапии ганцикловиром
почти всегда происходит супрессия костного мозга, поэтому пациенты с ранней
реактивацией или незначительным количеством пересаженных клеток не
переносят ее. Эффективной альтернативой является фоскарнет, который незначительно
подавляет костный мозг, однако обладает выраженной токсичностью в
отношении вируса. При уже развившемся поражении легких лечение практически безре-
Аллогенная трансплантация
323
зультатно, а при необходимости интубации выживаемость пациентов с пульмони-
том не превышает 1-2 %.
Другие инфекции. Реципиент с недавно прижившимся аллогенным
трансплантатом остается иммунокомпрометированным и восприимчивым ко многим
микроорганизмам, перечисленным в разделе, посвященном периоду до
приживления. Необходимость продолжения профилактики БТПХ обусловлена
остаточным мукозитом и оБТПХ разных степеней. Длительное использование
катетеров (в течение 100 дней) способствует частому возникновению грамполо-
жительных бактериальных инфекций. При адекватной профилактике
инфицирование Pneumocystis carinii происходит редко. То же относится и к токсоплазмо-
зу. Необходимо учитывать возможность реактивации туберкулеза при развитии
легочных инфильтратов у больных с положительными туберкулиновыми
пробами в анамнезе или проживающих в регионе высокого риска. У пациентов со
значительной иммунодепрессией из-за неродственной трансплантации или не
полностью идентичного трансплантата, сильного Т-клеточного истощения или
длительной интенсивной терапии оБТПХ велика вероятность развития лим-
фопролиферативных состояний, ассоциированных с вирусом Эпштейна-Барр.
Идиопатический интерстициальный пульмонит. Когда диффузные интерстици-
альные легочные инфильтраты развиваются без видимой причины, их этиология
считается идиопатической. Идиопатический интерстициальный пульмонит
(ИИП) возникает у 5-10 % реципиентов аллогенных трансплантатов. ИИП
клинически не отличим от ЦМВ-ассоциированного пульмонита, и ранее, возможно,
некоторые случаи ЦМВ-пульмонита, которые современными методами могли бы
быть правильно диагностированы, относили к ИИП. Повреждение легких при
ИИП скорее всего является следствие^ побочных токсических эффектов
лекарственных препаратов и мероприятий, применяемых при подготовке к
трансплантации, особенно ООТ и бусульфана. При ИИП предпочтительна
поддерживающая терапия; некоторые пациенты реагируют на высокие дозы стероидов.
После 100-го дня
Хроническая болезнь "трансплантат против хозяина". Хотя в соответствии
с принятой классификацией хроническая болезнь "трансплантат против хозяина"
(хБТПХ) проявляется после 100-го дня от момента аллогенной трансплантации,
некоторые признаки этого синдрома могут отмечаться и ранее. Наиболее часто
(но не обязательно) хБТПХ развивается из оБТПХ. Другими факторами риска
хБТПХ являются высокая степень несовместимости донора и реципиента и
значительный возраст реципиента.
Патофизиология. Патофизиологически хБТПХ отличается от оБТПХ и имеет
сходство с аутоимунными заболеваниями, поражающими соединительную ткань.
Механизмы ее до конца не поняты. Известно, что избыточно активированные при
хБТПХ Т-клетки запускают системную воспалительную реакцию с
последующим отложением коллагена. Диагностирование хБТПХ не вызывает сложностей.
При атипичных проявлениях заболевание можно распознать с помощью биопсии
кожи или печени.
Клинические проявления. Клинические особенности и классификация хБТПХ
представлены в табл. 8-4. У 80 % пациентов отмечается поражение кожи: красный
324
Глава 8. Трансплантация стволовых клеток
ТАБЛИЦА 8-4. Классификация хронической БТПХ
Тип болезни Распространенность поражения
Ограниченная Ограниченное поражение кожи и/или дисфункция печени
Обширная Генерализованное поражение кожи
(распространенная) Ограниченное поражение кожи-или дисфункция печени плюс
любое из следующих проявлений:
- хронический агрессивный гепатит, некроз соединительной
ткани или цирроз
- сухость конъюнктивальных оболочек глаз
- поражение слюнных желез
- повреждение других органов-мишеней
плоский лишай с папуло-десквамозным дерматитом, бляшками и витилиго. Если
не контролировать процесс, повреждение кожи при хБТПХ прогрессирует до
склеродермоидного уплотнения, контрактур суставов и хронических кожных
изъязвлений. Также характерны сухость слизистых оболочек рта и глаз, поэтому
для раннего обнаружения хБТПХ и предотвращения нарушения зрения важны
частые офтальмологические осмотры с проведением теста Ширмера на слезооб-
разование. При хБТПХ может возникнуть дисфункция печени с появлением
хронической гипербилирубинемии, зуда и признаков мальабсорбции.
Встречаются и другие, менее специфические, желудочно-кишечные симптомы,
такие как анорексия, тошнота, потеря веса, преждевременное насыщение и
затруднения при глотании. Одна из наиболее серьезных форм хБТПХ — рестриктив-
ная легочная болезнь, поэтому периодически определяются показатели функции
внешнего дыхания. Сама по себе хБТПХ, как и ее лечение иммуносупрессивными
препаратами, ослабляет иммунную систему и, соотвественно, отсрочивает
восстановление иммунитета. Пациенты с хБ1ГПХ, которым назначены стероиды или
циклоспорин после 100-го дня, должны и далее получать препараты для
профилактики Pneumocystis carinii, ВПГ и даже кандидозного эзофагита.
Лечение. Иммуносупрессивные препараты, используемые для лечения или
профилактики оБТПХ, назначаются и при хБТПХ. При болезни ограниченного
типа часто эффективен преднизолон. При более обширной или рефрактерной
хБТПХ дополнительно может применяться циклоспорин, а также азатиоприн
и талидомид. PUVA (псорален и облучение ультрафиолетом А) — эффективный
способ лечения рефрактерной кожной формы хБТПХ.
Восстановление иммунитета. В раннем посттрансплантационном периоде у
реципиента могут проявляться признаки клеточного и гуморального иммунитета
донора. Это, вероятно, связано с передачей зрелых донорских Т- и В-лимфоцитов.
По мере исчезновения они заменяются незрелыми лимфоцитами,
происходящими от пересаженных стволовых гемопоэтических клеток. Поскольку
костномозговые донорские Т-лимфоциты повторно "обучаются" в присутствии антигенов
реципиента, формируется толерантность. Восстановление гуморального и
клеточного иммунитета у реципиента трансплантата во многом зависит от наличия
хБТПХ. У пациентов без признаков хБТПХ Т-клеточный иммунитет
восстанавливается через 4-6 месяцев после трансплантации. Для формирования
гуморального иммунитета требуется наличие связи между антигенспецифичными Т-лим-
Аллогенная трансплантация
325
фоцитами и антителопродуцирующими В-лимфоцитами. У реципиентов,
получивших костный мозг без элиминации Т-лимфоцитов, нормальная продукция
антител восстанавливается к 7-9-му месяцу. Нарушения, возникающие при
активно текущей БТПХ, и применяемые для ее лечения лекарственные средства
задерживают или предотвращают восстановление иммунитета.
Другие поздние осложнения трансплантации (табл. 8-5). Основные факторы
риска связаны с подготовкой к трансплантации, вторичным повреждением
тканей при БТПХ или хронической иммуносупрессией у аллогенных реципиентов.
Многие пациенты находятся в репродуктивном возрасте, поэтому очень важен
вопрос об их фертильности. Нормализация функции яичников отмечается у 15 %
женщин в постпубертатном возрасте, а восстановление нормальной функции
яичек — у 25 % мужчин. В наибольшей степени на фертильность влияет доза
облучения во время предимплантационной подготовки. Общее облучение тела в
высокой дозе чаще назначают пациентам со злокачественными заболеваниями;
вероятность восстановления фертильности в этой группе пациентов обычно меньше.
Иногда наблюдается поздний гипотиреоз и дефицит гормона роста, который
особенно важен для детей. Раннее назначение синтетического гормона ускоряет
рост костей и предотвращает возникновение карликовости. Наиболее частое
офтальмологическое осложнение — катаракта в результате облучения тела и/или
продолжительной глюкокортикоидной терапии. Любая другая глазная патология,
включая рубцевание или перфорацию роговицы, может быть следствием хБТПХ.
Глюкокортикоидная терапия, назначаемая при БТПХ, может приводить к
повреждению костей. Наиболее серьезное осложнение — несосудистый некроз
кости. При необходимости продолжения глюкокортикоидной терапии весьма
вероятно развитие генерализованного остеопороза.
ТАБ Л И ЦА 8 - 5. Поздние осложнения аллогенной трансплантации
Легочные
Инфекция
Хронические заболевания легких
Эндокринные
Бесплодие
Преждевременная менопауза
Гипотиреоз
Офтальмологические
Синдром сухости конъюнктивальной оболочки
Катаракта
Оральные
Синдром сухости слизистых оболочек
Болезни зубов
Болезни периодонта
Скелетно-мышечные
Несосудистый некроз
Остеопороз (при хронической стероидной терапии)
Общие
Вторичные новообразования
Аутоиммунные явления
Психологические эффекты
326
Глава 8. Трансплантация стволовых клеток
Нередко проведение ООТ и возникновение БТПХ становятся причиной
дисфункции слюнных желез, вызывая синдром сухости слизистых оболочек полости
рта (сухость во рту, или ксеростомия) и как следствие поражение зубов и
инфицирование полости рта. Таким больным необходимо и пред-, и
посттрансплантационное лечение зубов опытным стоматологом.
После аллогенной или аутологичной трансплантации повышен риск развития
новых опухолей. Режим кондиционирования может провоцировать канцерогенез
в результате длительного иммунодефицита с пониженным противоопухолевым
иммунным надзором. В Центре по исследованию рака имени Фреда Хатчинсона
в Сиэтле наблюдали 4294 пациентов после аутологичных или аллогенных
трансплантаций и выявили 82 пациента с вторичными новообразованиями. В
основном это были лимфопролиферативные заболевания, многие из которых связаны
с ВЭБ. Более чем у 10 % реципиентов аутологичных трансплантатов, живущих
длительное время, развивается вторичная миелодисплазия или лейкоз,
по-видимому, вследствие дотрансплантационного повреждения остаточных стволовых
клеток костного мозга.
Эффективность аллогенной трансплантации. Результат аллогенной
трансплантации зависит от основного заболевания, характеристик реципиента и донора.
При остром миелобластном лейкозе (ОМЛ) аллогенная трансплантация
наиболее эффективна в период ремиссии. Если она выполняется при первой ремиссии,
долгосрочная выживаемость достигает 40-70 %. При ТКМ во время рецидива или
в течение последующих ремиссий выживаемость снижается до 20-40 % из-за
увеличения частоты рецидивов. Смертность, связанная с проведением
трансплантации, возрастает также у пациентов с заболеваниями на более поздних стадиях
развития. Только 20-25 % больных ОМЛ, получающих химиотерапию, можно
вылечить, и лишь 20-40 % пациентов с рецидивом после стандартной
химиотерапии могут быть в дальнейшем подвергнуты аллогенной трансплантации. Выбор
времени для трансплантации четко не определен. Общей тенденцией является
. идентификация пациентов с высоким риском рецидива, находящихся в стадии
первой ремиссии, для проведения им немедленной трансплантации.
От 50 % до 75 % больных хроническим миелолейкозом (ХМЛ) живут
длительное время и могут быть излечены аллогенной трансплантацией от HLA-совмести-
мого родственного донора, выполненной в течение хронической фазы; таким
образом, она показана всем пациентам моложе 60 лет при наличии HLA-совместимого
родственного донора. Результаты трансплантации в фазе акселерации или во
время бластного криза гораздо менее благоприятны, но у этих пациентов ожидаемая
продолжительность жизни при любой другой терапии также очень коротка.
Для более точного определения роли аллогенной трансплантации в лечении
других онкогематологических заболеваний, например множественной миеломы,
лимфомы, острого и хронического лимфоцитарного лейкоза и миелодиспласти-
ческих синдромов, необходимы крупномасштабные клинические испытания.
Аллогенная трансплантация показана при лечении смертельных иммунодефи-
цитных состояний: тяжелого комбинированного иммунодефицита (глава 4),
синдрома Вискотта-Олдрича (глава 5), синдрома Чедиака-Хигаси (главы 4 и 7).
Пациентам с тяжелой приобретенной апластической анемией в возрасте до 30 лет
при наличии совместимого донора-сибса должна быть проведена трансплантация
в качестве начальной терапии. Спорной остается проблема выбора между транс-
Аутологичная трансплантация
327
плантацией и иммуносупрессией для пожилых пациентов (глава 7). Пациентам
с большой талассемией при отсутствии повреждений внутренних органов
вследствие перегрузки железом и наличии совместимого донора-сибса трансплантация
должна быть, вероятно, проведена в возрасте до 10 лет (глава 3).
Аутологичная трансплантация
Обзор
Подавление костного мозга относится к проявлениям дозозависимой
токсичности многих химиотерапевтических средств и облучения. Дозы многих из этих
агентов можно значительно увеличить, если сохранить при этом стволовые
кроветворные клетки. Условие успеха аутологичной трансплантации — дозозависимый
ответ опухоли на химиотерапию и/или облучение. Это означает, что, используя
повышенные дозы противоопухолевых агентов при сохранении СГК, можно
уничтожить большинство опухолевых клеток без существенного роста
заболеваемости и смертности, ассоциированных с лечением. Поскольку при аутологичной
трансплантации не заменяют всю гемопоэтическую и иммунную системы
таковыми от другого индивидуума, многие из осложнений, которые наблюдаются после
аллогенной трансплантации, не развиваются (например, отторжение
трансплантата, болезнь "трансплантат против хозяина" и инфекционные осложнения,
связанные с длительным подавлением Т-клеточного иммунитета). Имеются две
причины, по которым злокачественные заболевания чаще рецидивируют после
аутотрансплантации, чем после аллотрансплантации. Это — отсутствие эффекта
"трансплантат против опухоли" и вероятность реинфузии клеток опухоли с
трансплантатом.
Получение стволовых гемопоэтических клеток
До 1990 г. основным источником СГК для огромного большинства аутологичных
трансплантаций был костный мозг. В ряде случаев возникали трудности при
заготовке костного мозга у онкологических пациентов, получавших мощную терапию;
часто требовались 4 или более недель, чтобы поддержать гемопоэз. В течение
этого длительного периода цитопении повышается частота инфекционных
осложнений и кровотечений, а смертность, связанная с трансплантацией, превышает 10 %.
В последнее время мобилизованные КППК почти полностью заменили костный
мозг как источник стволовых клеток для аутологичной трансплантации.
Открытие того факта, что количество циркулирующих примитивных клеток можно
повысить в 100 и более раз при помощи гемопоэтических цитокинов (с
химиотерапией или без нее), уменьшило число аферезов, необходимых для заготовки
адекватного количества трансплантата. Кроме сравнительной легкости отбора,
эти трансплантаты имеют тенденцию приживаться гораздо быстрее с
восстановлением гемопоэза уже на 9-14-е сутки. Столь скорое восстановление уменьшило
заболеваемость и смертность, ив некоторых цецтрах эти процедуры выполняют
уже в амбулаторных условиях (при отсутствии у пациентов нейтропенической
лихорадки или другого осложнения). Верхний возрастной предел для пациентов
при аутологичной трансплантации повысился; сообщается о хороших
результатах даже у больных, достигших 70 лет.
328
Глава 8. Трансплантация стволовых клеток
Техника аутотрансплантации
После сбора аутологичный КМ или КППК иногда обрабатывают с целью очистки
трансплантата от остаточных клеток опухоли. Для этого используются методы
селективного устранения опухолевых клеток: противоопухолевые моноклональ-
ные антитела, цитотоксические агенты, а также физическое разделение,
основанное на различиях в размере или плотности клеток. Кроме того, используется
позитивная селекция гемопоэтических предшественников и стволовых клеток.
Необходимость очистки трансплантатов должна быть подтверждена в больших
рандомизированных и контролируемых клинических исследованиях. При
использовании чувствительных молекулярных и иммуногистохимических методов
часто среди клеток КМ или КППК удается обнаружить опухолевые клетки, даже
если морфологически образцы свободны от них. Это справедливо и для солидных
опухолей, например рака молочной железы. Логично ожидать, что реинфузия
злокачественных клеток приведет к рецидиву, поэтому желательно удаление
клеток опухоли из трансплантата. Однако процесс приживления опухолевых клеток
после трансплантации, по-видимому, относительно неэффективен. В
экспериментах на животных и в некоторых клинических исследованиях получены
свидетельства того, что наиболее вероятным первичным источником рецидива
является остаточная опухолевая ткань. В то же время изучение генетических маркеров
с использованием вирусных векторов для постоянной метки аутологичных транс:
плантатов показали, что по крайней мере некоторые рецидивы опухоли при
детском лейкозе и нейробластоме происходят из трансплантата.
Имеются две главные практические проблемы обработки аутотрансплантата.
Во-первых, недостаток знания о биологии опухолевых и стволовых клеток
затрудняет выбор оптимального метода очистки нормальных гемопоэтических элементов
от клеток опухоли. Во-вторых, многие подходы к очистке, особенно с применением
цитотоксических агентов, таких как 4НС или ИЛ-2, по-видимому, чреваты
повреждением гемопоэтических клеток и замедлением приживления, увеличивая
посттрансплантационную заболеваемость и смертность. Совсем недавно усилия
исследователей сфокусировались на процедуре позитивной селекции. Вместо попыток
разрушить или физически удалить опухолевые клетки из трансплантата,
выполняется иммунологическая селекция СГК с использованием антител, которые
связывают их поверхностные белки, например CD34, отсутствующие на большинстве
опухолевых клеток. Трансплантаты КМ и КППК, отобранных таким образом,
несмотря на то, что они содержат только 1 -2 % клеток от их первоначального
количества, полностью очищены от опухолевых клеток и быстро приживаются.
Предимплантационная подготовка и проведение
трансплантации
После обработки трансплантат криоконсервируют, а пациента подготавливают
в режиме кондиционирования миелоаблативными дозами химио- или лучевой
терапии. Выбор режима основан на действии специфических агентов против
конкретной опухоли. При этом доза, вызывающая миелотоксический эффект, не
должна патологически влиять на другие органы. Например, цисплатин относительно
редко используют при аутологичных трансплантациях, потому что доза,
вызывающая поражение почек, меньше миелотоксической. После завершения предимп-
Аутологичная трансплантация
329
лантационной подготовки, полной метаболизации и выведения активных форм
химиотерапевтических агентов трансплантаты КМ или КППК размораживают и
реинфузируют внутривенно.
Последующий период до приживления аналогичен аллогенной
трансплантации (раздел "Аллогенная трансплантация"). Как только приживление
произошло, самочувствие пациентов быстро улучшается и через 6-12 недель они могут
вернуться к активной жизни. В отличие от аллогенной трансплантации у
реципиентов аутологичных трансплантатов отсутствуют такие поздние осложнения, как
оБТПХ, вирусные или грибковые инфекции. Отсроченные токсические
поражения, например катаракта, дисфункция щитовидной железы и бесплодие, могут
быть следствием общего облучения тела. Недавние исследования показали, что
почти у 10 % пациентов через несколько лет после трансплантации развивается
вторичная миелодисплазия.
Применение и эффективность
Цели аутотрансплантации различны в зависимости от характера
новообразования и клинической ситуации. При острых лейкозах и некоторых лимфомах
у большинства пациентов наблюдалось исцеление или, по крайней мере, очень
длительная ремиссия. При множественной миеломе после проведения
аутотрансплантации не удается добиться излечения, но результаты рандомизированного
исследования подтверждают, что эта процедура обеспечивает достоверно более
длинные интервалы, свободные от проявлений заболевания, чем стандартная
химиотерапия. При раке молочной железы у пациентов с IV стадией метастазирова-
ния (широко распространенные висцеральные или костные метастазы)
выздоровление после ТКМ маловероятно. Возможно, при раке молочной железы
аутотрансплантация перспективна как метод интенсификации
консолидирующей терапии у пациентов со II или III стадией болезни (метастазы в
лимфатических узлах ± большая первичная опухоль).
Важно осознавать, что практически каждая прикладная программа
аутотрансплантации противоречива. Пока выполнено мало рандомизированных
контролируемых испытаний, в которых аутотрансплантация сравнивается со стандартной
терапией. Сопоставление же с "традиционными" методами контроля чревато
неверной трактовкой в связи с усовершенствованиями в поддерживающем лечении,
произошедшими за это время, и различием подходов к выбору пациентов.
Страховые компании используют очень противоречивые критерии, касающиеся
покрытия расходов на эти дорогостоящие процедуры (50 000-150 000 долл. США), что
обусловлено политическими или законодательными моментами. В следующие
5 лет, вероятно, удастся выяснить, оправдано ли огромное увеличение частоты
аутотрансплантации в различных клинических ситуациях, после чего
разработчики стратегии и экономисты оценят их экономическую обоснованность. Для
этого по возможности все пациенты, подвергающиеся аутологичной
трансплантации, должны быть зарегистрированы в специальных клинических исследованиях
с надежным наблюдением и быстрым предоставлением результатов.
Клинический пример
Мужчина в возрасте 35 лет, страдающий хроническим миелолейкозом
(ХМЛ), направлен на аллогенную трансплантацию костного мозга. Его
330
Глава 8. Трансплантация стволовых клеток
младший брат определен как совместимый по всем шести HLA-антиге-
нам. Дотрансплантационное тестирование выявило предшествующее
ЦМВ-инфицирование и пациента, и донора.
Пациент помещен в отделение трансплантологии. Для профилактики
бактериальной и грибковой инфекций назначены норфлоксацин и флукона-
зол. Начато также внутривенное введение ацикловира для
предупреждения реактивации ВПГ и ЦМВ. Высокие дозы циклофосфамида введены
через катетер центральной вены в 8-7-й ДНИ до трансплантации. Затем
проведено ООТ в дозе 1200 рад в ДНИ 6-1. В процессе
кондиционирования пациент жалуется только на умеренную непостоянную тошноту и
сонливость. Для предотвращения развития БТПХ назначены циклоспорин и
меторексат.
В ДЕНЬ 0 у донора под общей анестезией заготавливают костный мозг.
Днем позже часть донорского костного мозга вводят внутривенно через
центральный венозный катетер реципиенту.
В ДЕНЬ 4 после трансплантации пациент жалуется на дисфагию, диарею
и тошноту. При физикальном обследовании выявлены покраснение и
некоторая отечность слизистых полости рта; афты или изъязвления не
обнаружены. В связи с развитием панцитопении требуется переливание
облученных и обедненных лейкоцитами компонентов крови, чтобы
поддержать количество тромбоцитов больше 10 000/мкл и уровень
гемоглобина выше 80 г/л.
ВДЕНЬ 10 оральная температура 38,8 °С. При физикальном обследовании
отмечены только выявленные ранее изменения в полости рта. В ДЕНЬ 14
обнаружен положительный баланс жидкости с увеличением массы тела
на 2 кг в течение 2-х предшествующих суток. Данные физикального
обследования: иктеричность склер, болезненность в правом верхнем
квадранте живота и смещаемое притупление перкуторного звука,
свидетельствующие об асците. Концентрация циклоспорина в крови — в
терапевтическом диапазоне. Ограничено потребление жидкости, и назначен
калийсберегающий диуретик альдактон. Повышенный вес и гипербили-
рубинемия на прежних уровнях.
В ДЕНЬ 22 обнаружены значимые изменения в крови: увеличилось
количество лейкоцитов (до 500/мкл), преимущественно за счет моноцитов
и незрелых нейтрофилов. Следующие 6 дней характеризуются
устойчивым увеличением в крови числа лейкоцитов и уменьшением потребности
в трансфузиях. Одновременно отмечается значительное улучшение
самочувствия пациента. Пики интермиттирующей лихорадки исчезают.
Начинает разрешаться дисфагия, соответственно возрастает потребление
пищи через рот. Диарея все еще присутствует, уменьшаясь, однако, по
частоте и объему. В ДЕНЬ 28 абсолютное количество нейтрофилов в
крови достигает 1000/мкл, и прием антибиотиков прекращают. Пациент
выписан из больницы. Он продолжает принимать только циклоспорин для
предотвращения развития БТПХ и триметоприм/сульфаметоксазол
3 раза в неделю для профилактики пневмоцистной пневмонии.
Пациент находится под тщательным амбулаторным наблюдением. В ДЕНЬ
50 по результатам еженедельного скрининга отмечена реактивация ЦМВ
(положительный ЦМВ-антиген). Пациент отрицает наличие лихорадки, ча-
Аутологичная трансплантация
331
стота дыхания не увеличена, нет диареи или дисфагии, количество клеток
крови остается устойчивым. Рентгенограмма грудной клетки по-прежнему
без патологии. Начаты инфузии ганцикловира по 2 раза в день и
продолжаются еженедельные инфузии ВВИГ для восстановления иммунной
системы. Через 1 неделю ЦМВ-антиген в крови не обнаруживается. Доза
ганцикловира через 10 дней снижена до поддерживающей (3 введения в неделю).
В ДЕНЬ 120 после трансплантации выявлено повышение активности
АлАТ, АсАТ и щелочной фосфатазы в крови. Отмечена эритематозная
кожная сыпь на спине пациента. Назначен перорально преднизрлон для
лечения предполагаемой хБТПХ, и ее признаки медленно разрешаются. ,
Постепенно дозу преднизолона снижают, вплоть до отмены.
1 ГОД ПОСЛЕ ТРАНСПЛАНТАЦИИ. Пациент чувствует себя хорошо.
Он не принимает лекарственные препараты и возвратился к работе.
Количество клеток крови нормальное, при исследовании костного мозга
признаки остаточного лейкоза не выявляются.
Вопрос 1. Являются ли диарея, тошнота и десквамация эпителия
слизистых оболочек, описанные на 4-й день после трансплантации, характерными
для мукозита? Как лучше всего лечить мукозит?
Ответ. Чтобы контролировать боль, используют наркотическую аналге-
зию. Для профилактики суперинфекции можно назначить
антисептические полоскания рта хлоргексидином. Необходимо начать проведение
парентерального питания, поскольку пероральное потребление снижается.
Диарея, развитие которой связано с мукозитом, обычно нечувствительна
к терапии лекарственными препаратами. Нужен тщательный контроль за
балансом жидкости для предотвращения дегидратации и электролитного
дисбаланса.
Вопрос 2. У пациента развивается лихорадка на 10-й день. Каковы
потенциальные источники инфекции?
Ответ. В этот период после трансплантации костного мозга нередки
бактериальные инфекции. Наиболее вероятные их источники — ЖКТ, легкие,
синусы, кожа и катетер центральной вены. Часто источник инфекции не
выявляется.
Вопрос 3. Как бороться с лихорадкой?
Ответ. Следует очень серьезно относиться к любому эпизоду лихорадки,
возникающему у пациента при нейтропении. Необходимо немедленно
начать эмпирическое антибактериальное лечение антибиотиками широкого
спектра действия, например цефтазидимом. Провести тщательный
функциональный анализ всех систем и физикальное обследование с акцентом
на выявление наиболее частых источников инфекции, перечисленных
выше. Выполняют посев крови на гемокультуру, анализ мочи и
рентгенограмму грудной клетки. Если лихорадка сохраняется более 3-4 дней,
несмотря на адекватное антибактериальное лечение, эмпирически назначают
противогрибковые препараты (амфотерицин).
Вопрос 4. С чем проводится дифференциальный диагноз симптомов,
описанных в день 14?
Ответ. У пациента, вероятно, развилась умеренная форма веноокклюзион-
ной болезни печени. Дифференциальный диагноз проводится с токсичес-
332
Глава 8. Трансплантация стволовых клеток
ким медикаментозным гепатитом, например вследствие лечения
циклоспорином, инфекционным гепатитом либо печеночной формой оБТПХ. ДЕНЬ
14 — еще слишком ранний срок для возникновения БТПХ, к тому же это
патологическое состояние обычно не осложняется развитием асцита,
болями в правом верхнем квадранте живота и увеличением веса.
Вопрос 5. Каковы потенциальные осложнения реактивации ЦМВ?
Ответ. Наиболее грозным осложнением реактивации ЦМВ является
ЦМВ-ассоциированный интерстициальный пульмонит. Также мишенями
ЦМВ-инфекции могут быть печень и ЖКТ. ЦМВ-инфекция, как и ган-
цикловир, который обычно используется при ее лечении, часто вызывает
подавление костного мозга. В этой ситуации для увеличения в крови
количества нейтрофилов можно использовать гранулоцитарный колониестиму-
лирующий фактор. В качестве альтернативы ганцикловиру назначают фос-
карнет.
Вопрос 6. Существует ли вероятность рецидива хБТПХ у этого пациента?
Ответ. Благоприятные прогностические факторы, определяемые у этого
больного, включающие ограниченную степень заболевания, отсутствие
значительных проявлений оБТПХ в раннем посттрансплантационном периоде, начало
хБТПХ после ДНЯ 100 и полную адекватную реакцию на назначенную
терапию, позволяют предположить, что у него не возникнет рецидива хБТПХ.
Избранная литература
Almici С, Carlo-Stella С, Wagner J. Е., Rizzoli V. Umbilical cord blood as a source of
hematopoietic stem cells: from research to clinical application. Haematologica, 1995;
80:473-479.
В этом обзоре рассматривается возможность использования стволовых клеток из
пуповинной крови (СКПК) как источника донорских клеток при
трансплантациях; выделены преимущества и ограничения применения СКПК.
Appelbaum F. R., Clift R., Radich J., Anasetti C, Buckner С D. Bone marrow
transplantation for chronic myelogenous leukemia. Semin. Oncol, 1995; 22: 405-
411.
В данном обзоре суммированы результаты трансплантации костного мозга при
лечении хронического миелолейкоза (ХМЛ). Сравнивается эффективность
трансплантаций от неполностью совместимых родственных доноров и совместимых
неродственных доноров у больных ХМЛ.
Appelbaum F. R., Fisher L. D., Thomas E. D. Chemotherapy versus marrow
transplantation for adults with acute nonlymphocytic leukemia: a five-year follow-
up. Blood, 1988; 72:179-184.
В настоящей работе приводятся результаты проспективного исследования,
предпринятого в одном медицинском учреждении, показывающие преимущества
трансплантаций аллогенного костного мозга перед стандартной
химиотерапией при лечении острого нелимфоцитарного лейкоза. Подчеркивается важность
возраста и прогноза опухоли при выборе лечения.
Barrett J., Treleaven J. Bone Marrow Transplantation in Practice. New York: Churchill
Livingstone; 1992.
Иллюстрированный клиническими примерами учебник, в котором рассматриваются
и аутологичная, и аллогенная трансплантации костного мозга. В первом разделе
Аутологичная трансплантация
333
книги обсуждается роль трансплантации костного мозга в лечении
злокачественных и незлокачественных заболеваний. Во втором разделе
рассматриваются практические аспекты этой процедуры.
Cheson B.D. Chemotherapy and bone marrow transplantation for myelodysplastic
syndromes. Semin. Oncol, 1992; 19:85-94.
Обзор публикаций по трансплантации костного мозга при миелодиспластическом
синдроме. Хотя ТКМ — единственный вид лечения миелодисплазии, дающий
надежду на выздоровление, связанная с трансплантацией смертность и
вероятность рецидива выше, чем при первично выявленном лейкозе. Возможно, это
обусловлено увеличением возраста и длительным предшествующим лечением
пациентов с миелодисплазией.
Fennelly D., Vahdat L., Schneider J. et al. High-intensity chemotherapy with peripheral
blood progenitor cell support. Semin. Oncol., 1994; 21: 21-25.
Инфузия клеток-предшественников периферической крови (КППК) является
эффективным средством, облегчающим проведение высокодозной химиотерапии.
Сообщаются данные из Мемориального центра рака Sloan-Kettering о значительно
более коротком периоде нейтропении при поддержке КППК после курса
химиотерапии, состоящего из карбоплатина, этопозида и циклофосфамида. В
отдельном исследовании КППК использовались для облегчения переносимости двух
быстрых циклических курсов высокодозной химиотерапии.
FerraraJ. L. M., Deeg H.J. Graft-versus-host disease. N. Engl.J. Med., 1991; 324:667-674.
Обзорная статья, в которой подробно рассмотрены острая и хроническая болезни
«трансплантат против хозяина». Изложены детали клинической картины,
патофизиологии, подходов к профилактике и лечению, а также гипотезы
относительно роли продукции цитокинов в этиологии острой БТПХ.
Lu L,. Shen R. N., Broxmeyer H. E. Stem cells from bone marrow, umbilical cord blood and
peripheral blood for clinical application: current status and future application. Crit.
Rev. Oncol. Hematol., 1996; 22: 61-78.
В работе представлена информация о потенциальных возможностях использования
пуповинной крови. Рассматриваются также перспективы генной инженерии
стволовых кроветворных клеток для лечения генетических заболеваний.
Miller A. M. Hematopoietic growth factors in autologous bone marrow transplantation.
Semin. Oncol, 1993; 20:88-95.
В статье дан обзор опубликованных сведений о применении Г-КСФ, ГМ-КСФ и эрит-
ропоэтина при проведении аутологичной трансплантации стволовых клеток.
Использование Г-КСФ или ГМ-КСФ значительно сокращает период
нейтропении. Хорошо представлены полезные эффекты эритропоэтина.
Momin F., Chandrasekar P. H. Antimicrobial prophylaxis in bone marrow transplantation.
Ann. Intern. Med., 1995; 123:205-215.
Исчерпывающий обзор многочисленных профилактических режимов противомикроб-
ной терапии.
Purdy M. H., Shpall E. J. The role and methodology for purging tumor from autologous
bone marrow and peripheral blood progenitor cells. Med. Oncol, 1994; 11:47-51.
В работе обобщены данные об опухолевой контаминации стволовых клеток, взятых
у больных раком. Для очистки стволовых клеток от опухолевых применяли хими-
отерапевтический агент 4-НС и позитивную селекцию стволовых клеток с
использованием антигена CD34.
334
Sable C. A., Donowitz G. R. Infections in bone marrow transplant recipients. Clin Infect.
Dis., 1994; 18:273-284.
Превосходный обзор инфекционных осложнений, возникающих в ходе трансплантации
костного мозга и после нее.
Shulman Н. М., Hinterberger W. Hepatic veno-occlusive disease: liver toxicity syndrome
after bone marrow transplantation. Bone Marrow Transplant, 1992; 10:197-214.
Показана необходимость изучения причин ВОВ и обобщены современные подходы,
используемые для контроля этого осложнения, часто приводящего к смертельному
исходу.
Storb R., Anasetti С, Appelbaum F. et al. Marrow transplantation for severe aplastic
anemia and thalassemia major. Semin. Hematol, 1991; 28:235-239.
Планируя трансплантацию при незлокачественных заболеваниях, необходимо
тщательно взвешивать соотношение риск/польза. Это особенно актуально в случае
талассемии. В обзоре обсуждаются и другие проблемы.
Wingard J. R. Viral infections in leukemia and bone marrow transplant patients. Leukemia
and Lymphoma, 1993; 11:115-125.
Основное внимание в обзоре уделено герпетической и цитомегаловирусной инфекции.
Определены методы их лечения.
Глава 9
Злокачественные опухоли
кроветворной ткани
Лоуренс Н. Шульман
Молекулярные основы возникновения
злокачественных опухолей кроветворной ткани
Все болезни, описанные в этой главе,— результат злокачественного
преобразования гемопоэтических клеток, приводящего к их клональному нерегулируемому
росту. Как показано на рис. 9-1, все гемопоэтические клетки происходят из
стволовых клеток, которые в норме самореплицируются, обеспечивая непрерывную
продукцию клеток крови в течение всей жизни человека, а также
дифференцируются в зрелые специализированные клетки, необходимые для функционирования
организма. Тщательно сбалансированный и регулируемый рост клеток обычно
сопровождается образованием соответствующего количества зрелых потомков.
При этом первичные стволовые клетки не истощаются, и существует
достаточный резерв возможностей для адекватного ответа на такой стресс, как инфекции
или кровотечение (глава 1).
Следует отметить, что созревшие клетки крови: эритроциты, лейкоциты и
тромбоциты — запрограммированы на клеточную смерть, тогда как стволовые
клетки должны сохраняться для поддержания жизни организма. Эта
закономерность нарушается при злокачественных опухолях системы крови. Так, при мие-
лопролиферативных заболеваниях усиливается самовозобновление ранних
стволовых клеток с поддержанием нормального уровня дифференцировки в целом и
интенсификацией дифференцировки некоторых ростков кроветворения
(например, эритроидного ростка при истинной полицитемии). При остром миелобласт-
ном лейкозе бласты самовозобновляются, но не дифференцируются и,
следовательно, бластные формы накапливаются без образования из них зрелых потомков.
Хромосомные повреждения гемопоэтических стволовых клеток вызывают
аномальный и клональный рост. Количество идентифицированных хромосомных
аномалий, связанных с болезнями кроветворного аппарата, увеличивается. Их
обнаружение, например выявление bcr-abl-транслокацйи t(9;22) при хроническом
миелолейкозе или t(15;17) при остром промиелоцитарном лейкозе, позволяет
объяснить нарушение клеточного роста и злокачественную трансформацию клеток.
Вероятно, в будущем мы будем лучше понимать молекулярные механизмы развития
подобных заболеваний и использовать эти знания для их профилактики и лечения.
336 Глава 9. Злокачественные опухоли кроветворной ткани
f*\ ^ Стволовая клетка ^ у^Ш 1
Л^Ь \ Т-лимфоцитов ^^*/
^ _ ш m _ ^ я ССМ-хелперная клетка
Лимфоидная
стволовая
клетка __ ^
С08-супрессорная клетка
Плазматическая
клетка
Стволовая клетка
В-лимфоцитов
€ь€Ь€ь °о°
Стволовая Эритро-
клетка бласт
эритроцитов
о °
fhfbfbfb •••••■
оиднаяЧ Стволовая Мега-
•ловая \ клетка мега- кариоцит
етка \ кариоцитов
Эритроциты
Тромбоциты
Нейтрофилы
Стволовая Миело-
клетка бласт
гранулоцитов
Рис. 9-1. Упрощенная схема миелоидного и лимфоидного ростков кроветворения
Лейкозы подразделяются на острые и хронические. Острые лейкозы
характеризуются продукцией незрелых бластных клеток без созревания, что приводит
к быстрой замене нормальных гемопоэтических элементов костного мозга. Могут
повреждаться миелоидный или лимфоидный ростки кроветворения. Без лечения
острые лейкозы быстро завершаются гибелью пациента из-за нейтропении,
способствующей возникновению инфекции, и тромбоцитопении, вызывающей
кровотечения. Хронические лейкозы при отсутствии лечения развиваются менее
стремительно. При хроническом миелолейкозе повышена пролиферация миело-
идных клеток-предшественников, однако дифференцировка до зрелых клеток
(эритроциты, лейкоциты и тромбоциты) сохраняется. Хронический лимфолейкоз
по сути дела представляет собой вялотекущую неходжкинскую лимфому с
вовлечением периферической крови. Он будет обсуждаться в разделе этой главы,
посвященном лимфомам.
Острые лейкозы
Патофизиология и классификация. К острым лейкозам относятся острый мие-
лобластный лейкоз (ОМЛ) и острый лимфобластный лейкоз (ОЛЛ). Обе болезни
характеризуются злокачественной и клональной пролиферацией незрелых
бластных предшественников, которые в значительной степени потеряли дифференци-
ровочный потенциал. Проявления каждого из этих заболеваний обусловлены
способностью лейкозных бластов замещать нормальные элементы костного моз-
Острые лейкозы
337
га, что ведет к панцитопении, а также инфильтрировать другие ткани, например
десны и мозговые оболочки.
Острый миелобластный лейкоз, ОМЛ объединяет группу болезней,
характеризующихся пролиферацией злокачественных бластов миелоидного ряда. К ним
относятся миелобласты, промиелобласты, монобласты, эритробласты и мегака-
риобласты. Все эти клетки — потомство общей миелоидной стволовой клетки, и
в норме они самовозобновляются и дифференцируются в функционально зрелые
клетки крови: эритроциты, нейтрофилы, моноциты и тромбоциты (рис. 9-1).
Поскольку понятие миелоидный иногда используют, чтобы обозначить только нейт-
рофильную линию, в прошлом эту группу лейкозов называли острыми нелимфо-
цитарными лейкозами, но в настоящее время предпочтительно использовать
термин "острый миелобластный лейкоз". Специалистами
франко-американо-британской (FAB) рабочей группы предложена классификация ОМЛ, приведенная
в табл. 9-1 и 9-2. Подтипы Ml, M2 и МЗ представляют различные стадии диффе-
ренцировкй миелобластов. МО ОМЛ можно определить по миелоидной природе
поверхностных маркеров клетки, например CD33, однако в бластах при этом
заболевании не продуцируются миелоидные ферменты, из-за чего гистохимическое
окрашивание на миелопероксидазу отрицательно. В подтипах М1-МЗ
постепенно возрастает степень первичной грануляции так, что подтип МЗ представляет
собой острый промиелоцитарный лейкоз. М4-М5 ОМЛ — это монобластные
лейкозы с миелоидными элементами или без них соответственно. Мб ОМЛ — эритро-
лейкоз, а М7 — мегакариобластный лейкоз. Как показано в табл. 9-2, подтипы
ОМЛ различаются по морфологии, гистохимическому окрашиванию, кариотипу
и иммунофенотипу патологических клеток. Перечисленные методы помогают
также отличать ОМЛ от ОЛЛ при неоднозначной морфологии клеток.
Для развития ОМЛ достаточно одной клетки, повреждение хромосом
которой приводит к экспансии злокачественного клона со сниженной способностью
к дифференцировке до зрелых потомков. Из-за потребности в саморепликации и
воспроизводстве клеток в течение всей жизни человека бласты не
запрограммированы на умирание. Зрелые потомки (эритроциты, нейтрофилы, тромбоциты)
имеют конечную продолжительность жизни, и их гибель предопределена. При ОМЛ
бласты продолжают реплицироваться без последующей дифференцировки и,
следовательно, накапливаются в костном мозге и других тканях. Дефектные бласты
ТАБЛИЦА 9-1. Франко-американо-британская (FAB) классификация острого
миелоидного лейкоза
МО: Острый недифференцированный лейкоз
М1: Острый миелобластный лейкоз без дифференцирования
М2: Острый миелобластный лейкоз с формированием некоторых гранул
МЗ: Острый промиелобластный лейкоз (промиелоцитарный1)
М4: Острый миеломоноцитарный лейкоз (миеломонобластный1)
М5: Острый моноцитарный лейкоз (монобластный1)
Мб: Эритролейкоз
М7: Мегакариобластный лейкоз
По: Bennett J. M., Catovsky D., Daniel M.-T. et. al. Proposals for the classification of the acute
leukemias. Br. J. Haematol., 1976; 33: 329-331.
1 Названия, принятые в России.
338
Глава 9. Злокачественные опухоли кроветворной ткани
ТАБЛИЦА 9-2. Гистохимические, иммунофенотипические и хромосомные
характеристики измененных клеток при острых лейкозах
Заболевание Гистохимия
Иммуно-
фенотип
Хромосомные
аномалии
ОМЛ, МО
ОМЛ, М1.2
ОМЛ, МЗ
ОМЛ, М4
ОМЛ, М5
ОМЛ, Мб
ОМЛ, М7
ОЛЛ, Т-кле-
точный тип
ОЛЛ, пре-В-
клеточный тип
Все тесты отрицательны
Миелопероксидаза
Судан черный
Миелопероксидаза
Судан черный
Миелопероксидаза
Неспецифическая эстераза
Неспецифическая эстераза
PAS
Антиген фактора VIII
PAS
PAS
CD13, CD33, CD34
CD13, CD33 t(8;21)
CD13, CD33 t(15;17)
CD13, CD33,
CD11b, CD14 Inv16
CD11b, CD14
CD13, CD33 5q-,7q-
CD13, CD33, CD41
CD3, CD5, CD7
CD10, CD19 t(9;22)
развиваются не быстрее нормальных, но из-за отсутствия дифференцировки они
накапливаются, вытесняя неповрежденные элементы костного мозга, что
приводит к дефициту эритроцитов, зрелых лейкоцитов и тромбоцитов.
В большинстве случаев причина возникновения ОМЛ неизвестна, однако
идентифицирован ряд этиологических факторов. Слабым, но достоверным лей-
козогеном является радиация. Лейкозы вызывают и некоторые химиотерапевти-
ческие агенты, такие как ал^илирующие вещества и ингибиторы топоизомеразы И,
а также растворители, например бензол. Кроме того, ОМЛ может развиваться как
следствие предшествующих гематологических нарушений: истинной полиците-
мии, миелоидной метаплазии, пароксцзмальной ночной гемоглобинурии и мие-
лодисплазии.
С лейкозными б ластами ассоциированы определенные хромосомные
аномалии, играющие важную роль в лейкозогенезе. Идентификация этих хромосомных
аберраций имеет прогностическое значение. Некоторые из них являются патогно-
моничными для конкретного подтипа ОМЛ. Так, t(15;17) всегда обнаруживают
при МЗ — остром промиелоцитарном лейкозе (ОПМЛ). Наличие некоторых
аномалий, таких как транслокация t(8;21) или инверсия в хромосоме 16, предвещает
хороший прогноз. Другие, например делеции длинного плеча хромосом 5 и 7,—
маркеры неблагоприятного прогноза. Выяснение молекулярных процессов,
связанных с подобными транслокациями и делецйями, несомненно, значительно
приблизит к пониманию патогенеза ОМЛ и, возможно, повлияет на терапию, что
уже имеет место при МЗ (см. далее). Все пациенты с недавно диагностированным
ОМЛ должны пройти цитогенетическое исследование лейкозных бластов,
полученных из костного мозга или периферической крови.
Острый лимфобластный лейкоз. ОЛЛ могут быть пре-В- или Т-клеточного типа
(в главе 4 рассмотрена физиология лимфоидной ткани). В обоих случаях
продуцируются незрелые лимфобласты, которые в дальнейшем не дифференцируются
до зрелых лимфоидных потомков. Существует две классификации ОЛЛ:
морфологическая (FAB) и иммунологическая (табл. 9-2 и 9-3). Классификация FAB не яв-
Острые лейкозы 339
, 5
ТАБЛ И ЦА 9- 3. Классификация острого лимфобластного лейкоза
Пре-В-клеточный (нулевые клетки) (L1 - или 1.2-морфология1)
75 % случаев: la + (HLA-DR), В4 + (CD19), CALLA + (CD10)
Т-клеточный (L1- или 1.2-морфология1)
20 % случаев: Leu 1 + (CD5), Leu 9 + (CD7)
В-клеточный (1.3-морфология1)
5% случаев: la+, представлены поверхностные иммуноглобулины. ЦЗ-вариант
(FAB-классификация) имеет очень неблагоприятный прогноз, подобен или
идентичен неходжкинской лимфоме из малых нерасщепленных клеток (лимфома Бер-
китта). У пациентов с этой патологией обычно используют методы терапии, при-
менямЫе для лечения лимфомы из малых нерасщепленных клеток, а не ОЛЛ
1 FAB-морфология для ОЛЛ.
L1. Относительно небольшой размер клетки, регулярная форма ядра, высокое ядерно-цито-
плазматическое соотношение, ядрышки незаметные.
L2. Гетерогенный размер клетки, ядерно-цитоплазматическое соотношение ниже.
L3. Большие гомогенные клетки, ядра с тонкими нитями хроматина, ядрышки видны четко,
базофильная цитоплазма с отчетливыми вакуолями.
ляется широко используемой и не отражает иммунологическую природу подтипов
ОЛЛ, что очень важно. Приведем пример. Известно, что морфологически лимфо-
бластные клетки варианта L3 часто выявляются при таком близком к ОЛЛ
заболевании, как неходжкинская лимфома из малых нерасщепленных клеток, или лимфома
Беркитта. Однако терапия последней отличается от лечения ОЛЛ. Для
дифференциации этих заболеваний очень полезна иммунологическая классификация.
Приблизительно у 70 % пациентов с ОЛЛ выявляются лимфобласты пре-В-кле-
тпочного типа. Такие заболевания в прошлом определялись как ОЛЛ с "нулевыми
клетками" из-за отсутствия иммуноглобулинов на клеточной поверхности. Лишь
после изучения иммунофенотипа и исследования генома удалось
дифференцировать ранние В-клетки. Теперь известно, что эти бласты несут ранний В-клеточный
антиген CD 19 и имеют мутации в генах иммуноглобулинов, что подтверждает их
принадлежность к В-клеточному ростку. В остальных случаях развитие ОЛЛ
связано с Т-лимфобластами (Т-клеточный тип), у которых обнаруживаются ранние
антигены Т-клеток и аномальные гены Т-клеточного рецептора.
Как уже отмечалось, для ОЛЛ, в прошлом определявшегося как истинный
В-клеточный, морфологически относящегося к варианту L3, характерны клетки, которые
находятся на более поздней стадии дифференцировки В-клеток с экспрессией
поверхностных иммуноглобулинов и CD20. Это заболевание теперь
классифицировано как неходжкинская лимфома из малых нерасщепленных клеток (лимфома
Беркитта). Типичные для него клетки часто содержат хромосомную транслокацию
t(8;14), а программы лечения отличаются от использующихся при ОЛЛ.
С ОЛЛ ассоциированы также хромосомные аномалии. Наиболее важной из
них является транслокация t(9;22), известная как филадельфийская хромосома.
Первоначально она была описана как характерная для хронического миелолейко-
за. При этой транслокации ген аЫ перемещается из хромосомы 9 в точку разрыва
в кластерной области (breakpoint cluster region — bcr) хромосомы 22. В результате
синтезируется одна из двух bcr-abl аномальных (аберрантных) протеинкиназ —
р210 или р190. Тип р210 чаще выявляется при хроническом миелолейкозе, а тип
р190 — при ОЛЛ. Бласты, содержащие такую аномалию, встречаются у 5 % боль-
340
Глава 9. Злокачественные опухоли кроветворной ткани
ных ОЛЛ детей и более чем у 30 % взрослых, страдающих этим недугом. Данное
явление имеет клиническое значение в силу неблагоприятного прогноза для
пациентов с ОЛЛ, бласты которых несут подобную транслокацию. Важно знать, что
стандартные цитогенетические метафазы трудно получить на клетках ОЛЛ,
поскольку часто они не делятся в культуре ткани. Для идентификации bcr-abl-
транслокации можно использовать молекулярные исследования, которые
должны стать обязательными для всех пациентов с впервые диагностированным ОЛЛ.
Диагноз. Для диагностики ОМЛ или ОЛЛ требуется идентификация лейкозных
бластов в периферической крови или в костном мозге. Даже при нахождении
бластов в периферической крови, чтобы подтвердить лейкозную инфильтрацию,
всегда выполняется исследование костного мозга. Наличие в нем более чем 30 %
бластов является стандартным критерием диагностики ОМЛ (в отличие от мие-
лодиспластических синдромов, обсуждаемых далее).
Часто бласты могут быть идентифицированы по принадлежности к миелоид-
ному или лимфоидному ряду в соответствии с их морфологическими
характеристиками. Миелобласты больше, чем лимфобласты, имеют очень тонкие нити
ядерного хроматина и крупные выделяющиеся ядрышки. Лимфобласты меньше по
размеру, с меньшим количеством цитоплазмы и менее заметными ядрышками.
Нередко бласты у пациента невозможно четко отнести к миелоидной или лимфо-
идной линии и требуется дальнейшее исследование. Практически во всех случаях
диагноз может быть подтвержден с помощью цитохимического окрашивания и
иммунофенотипического анализа (табл. 9-2). Миедоидные бласты типа М1-М4
окрашиваются в тесте на миелопероксидазу, а бласты типа М4-М5 — на
неспецифическую эстеразу. Некоторые бласты типа Мб проявляют PAS-реакцию, а М7 —
реакцию на антиген фактора VIII. Бласты при ОЛЛ не окрашиваются в тесте на
миелопероксидазу или неспецифическую эстеразу, но иногда в них определяются
большие PAS-положительные гранулы. При иммунофенотипировании
принадлежность к миелоидным бластам устанавливается по наличию CD33 и других
миелоидных антигенов, а к лимфоидным — по CD19 и CD10 (CALLA). В трудных
случаях на завершающих стадиях обследования пациента для постановки
окончательного диагноза должно выполняться цитогенетическое исследование. Для
получения цитогенетических данных часто требуется значительное время.
Клинические особенности. Основное проявление острого лейкоза — панцитопе-
ния. У пациентов отмечаются бледность, усталость или учащение дыхания,
вторичные по отношению к анемии. Тромбоцитопения может приводить к
кровотечению, а нейтропения — к инфекции. Часто симптомы слабо выражены и их
трудно отделить от общих жалоб, таких как усталость и признаки затяжной
вирусной инфекции. Ситуацию проясняет клинический анализ крови. Почти всегда
обнаруживаются анемия и тромбоцитопения. Содержание лейкоцитов в крови
может быть высоким или низким, но лейкоцитарная формула, как правило,
аномальна, с уменьшением числа нейтрофилов. Обычно в периферической крови
присутствуют циркулирующие бласты, хотя и при ОМЛ, и при ОЛЛ количество
лейкоцитов в крови может быть низким, а бласты не всегда выявляются. В этом
случае только исследование костного мозга может подтвердить диагноз острого
лейкоза. При наличии циркулирующих бластов в периферической крови обычно
можно быстро выполнить морфологический, гистохимический и иммунофеноти-
пический анализы.
Острые лейкозы
341
Терапия острого миелоидного лейкоза. При отсутствий лечения пациенты с
ОМЛ быстро погибают на фоне неконтролируемой инфекции или кровотечения.
Базисное лечение состоит в назначении интенсивной химиотерапии, приводящей
к гипоплазии костного мозга и освобождению не только от неповрежденных блас-
тов, но и от нормальных клеток, которые должны обеспечивать восстановление
нормальных ростков кроветворения. Наиболее часто используют режимы,
включающие комбинацию антрациклиновых препаратов (даунорубицин или идаруби-
цин) и Ara-С (цитарабин). При таком лечении приблизительно у 65 % пациентов
болезнь переходит в ремиссию, причйм чаще у молодых (примерно у 80 % тех,
кому от 20 до 30 лет, и только у 50 % больных старше 60 лет). Следует различать
ремиссию и выздоровление. Ремиссия лишь отражает отсутствие видимых лей-
козных бластов; и если после ее индукции лечение прекратить, очень небольшое
число пациентов останутся в состоянии ремиссии и действительно будут
вылечены. Проведение постремиссионной интенсивной терапии увеличивает
длительность ремиссии и частоту излечения. Однако даже при назначении интенсивной
консолидирующей терапии (терапии усиления) только 30-40 % пациентов
излечиваются при помощи стандартных химиотерапевтических средств. Улучшить
показатели лечения может как аутологичная, так и аллогенная трансплантация
костного мозга (ТКМ), если выполнять ее во время первой ремиссии (см. рис. 9-2 и
главу 8). В одном из клинических исследований у пациентов была достигнута
ремиссия при использовании интенсивного режима, включающего прием даунору-
бицина в течение 3 дней и инфузии Ara-С в течение 7 дней (стандартная
индукционная терапия), после чего дополнительный 3-дневный курс Ara-С в высокой дозе.
Частота полных ремиссий в этом исследовании составила 89 %. Пациенты,
достигшие состояния ремиссии, подверглись аллогенной трансплантации при наличии
HLA-совместимого сибса или аутологичной трансплантации, если такового не
было. Полученные данные подтверждают, что и аллогенная, и аутологичная
трансплантации увеличивают общую выживаемость пациентов: при использова-
Аллогенная ТКМ
Л Аутологичная ТКМ
Интенсивная терапия
Годы
Рис. 9-2. Выживаемость с полным выздоровлением пациентов с острым миелоидным лейкозом,
получивших индукционную интенсивную химиотерапию с последующей консолидирующей
стандартной химиотерапией, аутологичной или аллогенной ТКМ. Обратите внимание на преимущество
использования ТКМ. (Из: Zittoun R. A. et al. Autologous or allogeneic bone marrow transplantation
compared with intensive chemotherapy in acute myelogenous leukemia. N. Engl. J. Med., 1995; 332: 217.)
342
Глава 9. Злокачественные опухоли кроветворной ткани
нии этих способов лечения 55 % больных остаются в состоянии долгосрочной
ремиссии. Для пациентов с рецидивом ОМЛ аллогенная трансплантация —
единственный потенциально курабельный вид терапии (глЬва 8).
Острый промиелоцитарный лейкоз (ОПМЛ — подтип МЗ) уникален тем, что
по существу во всех случаях обнаруживается транслокация t(15;17). В этой
транслокации участвует а-ген рецептора ретиноевои кислоты, расположенный на
хромосоме 17. Показано, что пероральное назначение транс-изомера ретиноевои
кислоты (all-trans retinoid acid — ATRA) может стимулировать дифференцировку
ОПМЛ-клеток и приводить к полной гистологической и цитогенетической
ремиссии. К сожалению, ремиссия не является длительной, кроме того, необходимо
проведение стандартной химиотерапии. Однако установление того, что лечение
ATRA (однозначно связанное с наблюдаемой аномалией гена рецептора
ретиноевои кислоты) вызывает дифференцировку и подавление злокачественного клона,
стало ключевым компонентом современной терапии этого заболевания.
Лечение острого лимфобластного лейкоза. Выбор терапии для пациентов с ОЛЛ
зависит от их возраста и цитогенетических характеристик лейкозных бластов.
Дети с ОЛЛ, чьи лейкозные бласты не содержат филадельфийскую хромосому
t(9;22), могут лечиться по программам, включающим доксорубицин, винкристин,
преднизолон и L-аспарагиназу; полная ремиссия достигается в 90-95 % случаев.
Длительная поддерживающая терапия проводится в течение 2 лет, включая
профилактическое лечение центральной нервной системы; при э?ом 80-90 % детей
остаются в сострянии ремиссии и, вероятно, излечиваются. При наличии
филадельфийской хромосомы у детей с ОЛЛ прогноз чрезвычайно неблагоприятен;
такие пациенты должны лечиться по более интенсивным программам терапии,
по возможности включающим ТКМ (глава 8).
Для взрослых больных ОЛЛ терапевтические режимы, используемые у детей,
неэффективны: необходимо намного более интенсивное лечение; и даже в этом
случае только 35-50 % пациентов излечиваются при помощи стандартной
химиотерапии. Выживание зависит от возраста больного. Как показано на рис. 9-3, наиболее
обнадеживающие данные получены у пациентов до 30 лет, тогда как у пациентов
<30лет(п = 87)
Рис. 9-3. Выживаемость взрослых пациентов с острым лимфобластным лейкозом после
интенсивной химиотерапии. Обратите внимание на то, что у молодых пациентов выживаемость выше по
сравнению с больными старших возрастов. (Из: Larson R. A., Dodge R. К., Burns С. P. et al. A five-drug
remission induction regimen with intensive consolidation for adults with acute lymphoblastic leukemia:
Cancer and Leukemia Group В study 8811. Blood, 1995; 85: 2025.)
Миелопролиферативные заболевания
343
старше 60 лет показатели выживаемости существенно снижены. Взрослые больные
с Т-клеточным ОЛЛ имеют несколько лучший прогноз, чем при В-клеточных ОЛЛ
(рис. 9-4). Как подчеркивалось ранее, пациентов с истинно В-клеточными ОЛЛ
(L3) лечат полротоколам терапии неходжкинских лимфом из малых нерасщеплен-
ных клеток (лимфома Беркитта). У взрослых пациентов с ОЛЛ, как и у детей, при
наличии филадельфийской хромосомы чрезвычайно неблагоприятный прогноз, и
если есть совместимый донор, им нужно проводить ТКМ во время первой ремиссии
(глава 8). Это также справедливо для больных, чьи лейкозные бласты имеют
другие, менее часто встречающиеся хромосомные аномалии, ассоциированные с
плохим прогнозом. У взрослых, как и у детей, профилактика осложнений со стороны
центральной нервной системы необходима при любом режиме лечения.
Миелопролиферативные заболевания
Патофизиология и классификация. Миелопролиферативные заболевания (как и
острые лейкозы) — группа болезней, характеризующихся клональным и
аномальным ростом. При миелопролиферативных заболеваниях, однако, наряду с
повышенной репликацией клеток-предшественников сохраняется и их дифференци-
ровка. Следовательно, продукция зрелых потомков продолжается, и в начале
развития данной патологии цитопения обычно не наблюдается. При разных
миелопролиферативных заболеваниях пролиферативный толчок испытывают
различные ростки кроветворения. Продукция эритроцитов наиболее существенно
увеличивается при истинной полицитемии, тромбоцитов — при эссенциальной
тромбоцитемии, гранулоцитов — при хроническом миелолейкозе (ХМЛ), а мега-
кариоцитов — при миелоидной метаплазии.
Часто определяется спленомегалия, так как селезенка становится местом
экстрамедуллярного (внекостномозгового) кроветворения. Относительные
изменения количества клеток крови и другие важные клинические дифференциально-
диагностические признаки приведены в табл. 9-4.
Все эти заболевания представляют собой клональные злокачественные
процессы, возникающие вследствие поражения ранней стволовой клетки с ВОВЛече-
ТМуСп* 8)
Т(п = 31)
Рис. 9-4. Выживаемость взрослых пациентов с острым лимфобластным лейкозом, получивших
интенсивную химиотерапию, в зависимости от иммунофенотипических показателей. (Из: Larson R. А.,
Dodge R. К., Burns С. P. et al. A five-drug remission induction regimen with intensive consolidation for
adults with acute lymphoblastic leukemia: Cancer and Leukemia Group В study 8811. Blood, 1995; 85:2025.)
344
Глава 9. Злокачественные опухоли кроветворной ткани
ТАБЛИЦА 9-4. Миелопролиферативные заболевания
Истинная
полицитемия
Эссенциальная
тромбоцитемия
Миелоидная
метаплазия
ХМЛ
+++
+/-
+/-
-
Заболевание Эритро- Лейко- Тромбо- Фиброз Сплено- ЩФЛ Ph1
циты циты циты мегалия
++ ++ +/- + |
+/- +++ +/- +/- H/t
++ +++ +++ +++ н/t
+++ +++ +++ +++ I +
ЩФЛ — щелочная фосфатаза лейкоцитов, Ph1 — филадельфийская хромосома, Т — повышение,
1 — снижение, Н —- нормальное значение.
нием в каждом случае клеток всех трех ростков кроветворения. Доказано, что при
ХМЛ злокачественной является очень ранняя стволовая клетка, сохраняющая
способность к дифференцировке как по лимфоидному, так и по миелоидному
типу. Это заключение базируется на том, что когда ХМЛ прогрессирует к фазе
бластного криза, у Уз пациентов бласты имеют лимфоидный фенотип.
Диагностика. Диагноз миелопролиферативного нарушения подтверждается
аномальным количеством клеток крови или спленомегалией, выявляемой при физи-
кальном осмотре. Если у пациента обнаруживается полицитемия с повышенным
гематокритом, врач должен провести дифференциальную диагностику между
истинной полицитемией (миелопролиферативное нарушение) и вторичной поли-
цитемией, обусловленной аномальным гемоглобином, опухолями,
продуцирующими эритропоэтин, или другими нарушениями, например врожденными
пороками сердца, приводящими к хронической гипоксии (табл. 9-5 и глава 3).
Повышенная клеточность костного мозга — неспецифический признак, не
включенный в диагностические критерии истинной полицитемии (табл. 9-6).
ТАБЛИЦА 9-5. Дифференциальный диагноз полицитемии
Первичная полицитемия
Истинная полицитемия
Вторичная полицитемия
Врожденные цианотические пороки сердца
Злокачественные новообразования
Печеночно-клеточный рак
Почечно-клеточный рак
Болезни легких с гипооксигенацией
Высокоаффинный гемоглобин
Низкое парциальное давление кислорода,
пребывание в высокогорной местности
Поликистоз почек
Псевдополицитемия
Полицитемия напряжения
Сокращение внутрисосудистого объема
Миелопролиферативные заболевания
345
ТАБЛИЦА 9-6. Диагностические критерии истинной полицитемии1
Главные критерии
Увеличение массы эритроцитов
Спленомегалия
Нормальное насыщение кислородом
Малые критерии
Тромбоцитоз > 400 000/мкл
Лейкоцитоз > 12 000/мкл
Повышение активности щелочной фосфатазы лейкоцитов
Повышение уровня витамина В12или В12-связывающего белка в сыворотке крови
По: Berlin N. I. Diagnosis and classification of the polycythemias. Sem. Hematol., 1975; 12: 339.
1 Для постановки диагноза необходимо наличие всех трех главных критериев или двух главных
и двух малых критериев.
Диагностика эссенциальной тромбоцитемии начинается с попытки исключить
другие заболевания, при которых наблюдается увеличение числа тромбоцитов,
особенно вторичный, или "реактивный", тромбоцитоз, обсуждаемый в главе 5.
При наличии дефицита железа у пациентов с истинной прлицитемией иногда
выявляется не эритроцитоз, а тромбоцитоз. У некоторых пациентов изолированный
тромбоцитоз может также быть признаком развития ХМЛ, однако отсутствие
у них филадельфийской хромосомы в большинстве случаев исключает данное
заболевание. Эссенциальный тромбоцитоз необходимо дифференцировать от
реактивного тромбоцитоза, который наиболее часто служит проявлением
негематологических новообразований, инфекций и дефицита железа. При эссенциальном
тромбоцитозе костный мозг может иметь нормальное или повышенное
содержание клеток. Количество мегакариоцитов также часто бывает больше нормы, что
сопряжено с увеличением размера клеток и аномалией их морфологии.
Миелоидная метаплазия характеризуется фиброзом костного мозга, возможно
вызванным аномальным ростом мегакариоцитов с высвобождением факторов
роста фибробластов и экстрамедуллярным гемопоэзом, в первую очередь в
селезенке, которая может увеличиваться до огромных размеров. Весьма характерна
анемия. Количество лейкоцитов в крови может быть повышенным (в начале
заболевания) или пониженным (на поздних стадиях).
Хронический миелолейкоз — злокачественное клональное нарушение,
характеризующееся наличием филадельфийской хромосомы t(9;22), которая является
продуктом транслокации гена аЫ хромосомы 9 в точку разрыва в кластерной
области хромосомы 22. Выделяются две четко различаемые клинические фазы
заболевания: хроническая фаза с увеличением продукции зрелых гранулоцитов
и тромбоцитов, а также умеренной анемией и фаза властной трансформации, когда
ранние властные предшественники теряют способность к дифференцировке и
болезнь принимает фенотип острого лейкоза миелоидного или лимфоидного типа.
Клинические особенности и терапия. Клинические особенности и терапия мие-
лопролиферативных заболеваний весьма различаются и будут рассматриваться
по отдельности.
Истинная полицитемия характеризуется увеличением массы эритроцитов
с повышенным гематокритом и количеством лейкоцитов и, в большинстве случа-
346
Глава 9. Злокачественные опухоли кроветворной ткани
ев, тромбоцитозом (табл. 9-5 и 9-6). Важнейшее проявление болезни —
повышенная вязкость крови с возникающими в результате тромбозом и/или
кровоизлиянием вследствие сгущения крови, закупорки мелких сосудов (сладж-синдром),
а также аномальной функции тромбоцитов. Наличие высокого гематокрита
объясняет причину возникновения многих из признаков повышенной вязкости,
как и некоторых тромботических эпизодов. Гематокрит, следовательно,
необходимо поддерживать на уровне ниже 45 %, что легко достигается кровопусканием.
При повторных венепункциях образуется дефицит железа, вследствие чего
уменьшается продукция красных клеток. Однако даже при надежном контроле
гематокрита могут развиваться тромбозы и кровоизлияния, приводящие к
осложнениям. Частично это связано с аномалиями тромбоцитов, возникновение
которых способно вызвать спонтанные тромбозы и/или кровоизлияния. Лечение
такими антинеопластическими препаратами, как хлорамбуцил, радиоактивный
фосфор или гидроксимочевина, уменьшает количество тромбоцитов в крови
и снижает риск развития тромбоза и кровоизлияния, нередко исключая
необходимость в кровопусканиях. К сожалению, использование хлорамбуцила и
радиоактивного фосфора увеличивает вероятность трансформации заболевания в
острый миелоидный лейкоз.
У пациентов, получавших только кровопускания, трансформация истинной
полицитемии в ОМЛ происходит обычно в 1-3 % случаев. Неясно, увеличивается
ли риск подобного преобразования при лечении гидроксимочевиной. У
некоторых пациентов усиление костномозгового фиброза приводит к фазе "выгорания"
истинной полицитемии и, возможно, к панцитопении. .
Эссенциальная тромбоцитемия ассоциирована с Очень большим количеством
тромбоцитов и может проявляться тромбозом или кровоизлиянием.
Значительная анемия у пациентов обычно не возникает, иногда имеет место лейкоцитоз.
Лечение, как правило, не проводится, поскольку, несмотря на высокий уровень
тромбоцитов, симптоматика отсутствует. В других случаях развиваются
угрожающие жизни тромбоз и/или кровотечение. Оптимальный режим терапии эссен-
циальной тромбоцитемии не разработан. У некоторых больных успешно
применяется аспирин, у других — гидроксимочевина.
Иногда, несмотря на лечение, продолжаются серьезные тромботические
эпизоды. В проводимом рандомизированном исследовании всех больных эссенци-
альной тромбоЦитемией разделили на 2 группы, в одной из которых пациенты
получали лечение гидроксимочевиной, а в другой — только наблюдение. При
использовании гидроксимочевины частота возникновения тромбозов значительно
уменьшилась. Однако из-за опасности того, что долгосрочное лечение
гидроксимочевиной может быть лейкозогенным, а также из-за вариабельности
заболевания подход к лечению пациентов должен быть индивидуальным.
Миелоидная метаплазия. Миелоидная метаплазия — болезнь с гетерогенными
проявлениями, обычно характеризующаяся отчетливым фиброзом костного
мозга и массивной спленомегалией, вызванной экстрамедуллярным гемопоэзом.
Пациенты часто испытывают значительный дискомфорт из-за спленомегалии, а
также гиперметаболических нарушений. Анемия бывает тяжелой, а количество
лейкоцитов в крови может увеличиваться или уменьшаться. Вследствие
имеющей место нейтропении возможно развитие инфекции. Количество тромбоцитов
также увеличено или снижено; при выраженном снижении вероятна повышенная
кровоточивость. Схема лечения миелоидной метаплазии пока еще плохо разрабо-
Миелодиспластические синдромы
347
тана. Лечение состоит прежде всего из поддерживающей трансфузионной
терапии и приема антибиотиков. Эффективно использование гидроксимочевины и
других антинеопластических агентов, но они могут спровоцировать
возникновение тяжелой панцитопении. Спленэктомия также способна приводить к панцито-
пении, а в некоторых случаях — к развитию экстрамедуллярного гемопоэза в
печени, вызывающего значительную дисфункцию последней. Излечение пациента
молодого возраста, имеющего донора костного мозга, может быть достигнуто при
помощи аллогенной ТКМ. Если после трансплантации фиброз костного мозга
исчезает, это доказывает, что он был результатом действия ростовых факторов,
производимых аномальными гемопоэтическими клонами.
Хронический миелолейкоз. У больных ХМЛ, как правило, отмечается
хроническая фаза заболевания, когда имеют место лейкоцитоз, тромбоцитоз, анемия, спле-
номегалия и симптомы гиперметаболизма. В хронической фазе продуцируются
зрелые нейтрофилы и функционально полноценные тромбоциты. В данном
случае эффективно применение гидроксимочевины или а-интерферона. Вследствие
уменьшения количества лейкоцитов и тромбоцитов, а также выраженности спле-
номегалии и симптомов гиперметаболизма пациенты чувствуют себя достаточно
хорошо. Лечение гидроксимочевиной не устраняет лейкозный клон, а лишь
контролирует пролиферацию. Цитогенетический анализ костного мозга показывает,
что все клетки, даже при нормальном их количестве, содержат филадельфийскую
хромосому. Лечение а-интерферонрм иногда подавляет лейкозный клон так, что
на стандартных цитогенетических метафазах не находят клеток с
филадельфийской хромосомой. Есть предположение, что лечение а-интерфероном уменьшает
частоту трансформации хронической фазы в бластный криз.
К сожалению, обычно при лечении гидроксимочевиной или а-интерфероном
неизбежен прогресс в сторону "властной трансформации", когда лейкозные
предшественники теряют способность к дифференцировке. У 2/3 пациентов в костном
мозге происходит их замена на миелобласты, а у Уз пациентов — на лимфобласты.
Трансформацию бластов чрезвычайно трудно контролировать, и она, как
правило, заканчивается фатально в течение нескольких месяцев.
Единственным потенциально эффективным методом терапии ХМЛ является
аллогенная ТКМ. Этот метод лечения наиболее результативен, когда
используется в начале заболевания, и менее — если используется во время бластного криза
(рис. 9-5). Всем пациентам моложе 60 лет, имеющим совместимого донора
костного мозга, должна быть проведена аллогенная ТКМ в относительно ранние сроки
развития заболевания. Анемия, связанная с этим заболеванием, называется
рефрактерной, поскольку она не поддается никакой стандартной терапии. Иногда
отмечаются аномалии метаболизма железа с появлением кольцевых сидеробластов.
Миелодиспластические синдромы
Патофизиология и классификация. Миелодиспластические синдромы —
гетерогенная группа нарушений, характеризующихся аномальным ростом миелоидных
компонентов костного мозга. Аналогично ОМЛ и миелопролиферативным
заболеваниям при этих состояниях первично повреждается ранняя стволовая клетка.
Соответственно, миелодиспластические синдромы также относятся к клональ-
ным новообразованиям. В большинстве случаев снижается продукция грануло-
348
Глава 9. Злокачественные опухоли кроветворной ткани
Р< 0,0001
< 12 мес. Д-Т, гидроксимочевина (п = 208)
ч-\ >12мес. Д-Т, гидроксимочевина (п = 84)
"ТХ.
~\
< 12 мес. Д-Т, бусульфан (п = 62)
> 12 мес. Д-Т, бусульфан (п = 96)
1 1 1—
12 24 36 48
Продолжительность жизни (мес)
I
60
Рис. 9-5. Выживаемость пациентов с хроническим миелолейкозом, получавших терапию гидроксимо-
чевиной или бусульфаном с последующей ТКМ. Обратите внимание, что при использовании гидро-
ксимочевины результаты у пациентов лучше, чем при лечении бусульфаном; при раннем начале
лечения выживаемость больных также выше. (Goldman J. M. et al. Choice of pretransplant treatment and
timing of transplants for chronic myelogenous leukemia in chronic phase. Blood, 1993; 82: 2235-2238.)
цитов, эритроцитов и тромбоцитов, что приводит к развитию панцитопении.
Количество лейкоцитов в крови может повышаться с увеличением числа зрелых, но
диспластических нейтрофилов. Часто нейтрофилы качественно аномальны и
плохо функционируют как антибактериальные и антигрибковые клетки.
Эти заболевания иногда относят к рефрактерным анемиям, так как никакая
стандартная терапия, например восполнение запасов железа, витамина Bt2 или
фолата, анемию не корригирует.
Учитывая вариабельность миелодиспластических синдромов, специалисты
FAB-группы разработали их классификацию, которая представлена в табл. 9-7.
Можно заметить, что более продвинутые стадии болезни характеризуются
увеличением процента бластов в костном мозге. Это отражает трансформацию в острый
миелоидный лейкоз, что в конечном счете и происходит у многих пациентов с мие-
лодисплазией. В клетках костного мозга пациентов с миелодисплазиями часто
выявляются клональные хромосомные аномалии. Со временем в этих клетках
развиваются дополнительные хромосомные аберрации, что ведет к дальнейшей потере
их способности дифференцироваться в зрелые клетки и увеличивает число миело-
бластов до уровня, соответствующего ОМЛ. Для этой популяции больных
характерны аномалии длинного плеча хромосом 5,7 и 20, а также трисомия по 8-й
хромосоме. Миелодисплазия, как известно, часто развивается после лечения
алкилирующими лекарственными средствами, облучения, воздействия
растворителей, а также в результате других первичных гематологических нарушений, таких
как истинная полицитемия, но в большинстве случаев конкретный этиологический
фактор установить не удается.
Клинические особенности и терапия. Развитие клинических проявлений
является следствием панцитопении и включает признаки анемии, кровоточивости из-за
Лимфомы
349
ТАБЛИЦА 9-7. FAB-классификация рефрактерных анемий (миелодисплазий)
РА: Рефрактерная анемия (< 5 % бластов в костном мозге)
РАс: Рефрактерная анемия с кольцевыми сидеробластами
(< 5 % бластов в костном мозге)
РАПБ: Рефрактерная анемия с повышенным содержанием бластов
' (5-10 % бластов в костном мозге)
РАПБ-Т: Рефрактерная анемия с повышенным содержанием бластов
в стадии трансформации (10-30 % бластов в костном мозге)
Из: Bennett J. M., Catovsky D., Daniel M-T. et al. Proposals for the classification of the
myelodysplasfic syndromes. Br. J. Haematol., 1982; 51: 189-199.
тромбоцитопении и инфекции вследствие нейтропении. Даже если уровень нейт-
рофилов в крови не очень низкий, возникновение инфекции — частая проблема,
так как нейтрофилы этих пациентов, как правило, функционально
неполноценны. Спленомегалия отмечается редко.
Для поддержки этих пациентов необходимо проведение трансфузии
эритроцитов и тромбоцитов, а также назначение антибиотиков. Изредка анемию можно
ограничить, используя высокие дозы пиридоксина. "Дифференцировочные
агенты", такие как ретиноиды и низкие дозы Ar^-С, при этом заболевании не
оказывают значительного эффекта. Применение гемопоэтических факторов роста,
например Г-КСФ и эритропоэтина, дает переменный успех. У некоторых пациентов эти
цитокины снижают потребность в трансфузиях или количество инфекционных
эпизодов, но болезнь неизбежно прогрессирует, и контролировать ее становится
все труднее. Использовали и противолейкозную терапию, особенно при
трансформации в явный ОМЛ, но, к сожалению, частота ремиссий была низкой и
многие пациенты умирали в течение затяжного периода гипоплазии, возникающего
после проведения такой терапии.
В лечении больных миелодисплазией можно использовать аллогенную ТКМ,
но в большинстве своем больные слишком стары для проведения этой процедуры
или не имеют приемлемого донора.
Лимфомы
Лимфомы — заболевания, при которых происходит неопластическая
трансформация клеток, находящихся преимущественно в лимфоидных тканях.
Злокачественные лимфомы подразделяют на болезнь Ходжкина (или
лимфогранулематоз — ЛГМ) и неходжкинские лимфомы. Оба типа лимфом обычно
характеризуются злокачественным ростом лимфоузлов и селезенки, хотя могут
быть задействованы и экстранодальные ткани. Источник опухоли при болезни
Ходжкина остается неизвестным, а неходжкинские лимфомы возникают из
нормальных лимфоидных клеток, которые подверглись злокачественной
трансформации. Болезнь Ходжкина и неходжкинские лимфомы необходимо
дифференцировать, так как течение патологического процесса, прогноз и терапия этих двух
групп заболеваний отличаются. Неходжкинские лимфомы, хотя и выделяются
в единую группу, весьма гетерогенны.
350
Глава 9. Злокачественные опухоли кроветворной ткани
Болезнь Ходжкина (лимфогранулематоз)
Болезнь Ходжкина (ЛГМ) — заболевание неизвестной этиологии со
злокачественными клетками Рида-Штернберга, источник происхождения которых не
выяснен; тем не менее принадлежит к злокачественным нарушениям, лечение
которых наиболее успешно. ЛГМ был одним из первых онкологических
заболеваний, при которых с помощью комбинированной химиотерапии удалось добиться
положительных результатов даже на поздних стадиях. Клетка Рида-Штернберга
не имеет явного нормального двойника в процессе онтогенеза лейкоцитов, как это
было показано при других лимфопролиферативных нарушениях (например, пре-
В-клетки при ОЛЛ). Кроме того, клетки Рида-Штернберга присутствуют в
пораженных лимфоузлах в весьма малых количествах. Они окружены большим
числом доброкачественных поликлональных Т-клеток, которые, возможно,
реактивны по отношению к злокачественной клетке. Существуют четыре
гистологических подтипа ЛГМ: с преобладанием лимфоцитов, нодулярный склероз,
смешанно-клеточный и с истощением лимфоцитов. Наиболее распространен,
особенно у пациентов молодого возраста, нодулярный склероз. ЛГМ — болезнь
прежде всего молодых людей, обычно из семей с высоким
социально-экономическим статусом. Уже долгое время пытаются выделить вирусный этиологический
агент ЛГМ. Приблизительно у 25 % пациентов в ядрах клеток Рида-Штернберга
был найден вирус Эпштейна-Барр, но значение этого феномена неизвестно,
и происхождение болезни остается в значительной степени невыясненным.
Клинические особенности и лечение. ЛГМ обычно проявляется поражением
шейных, надключичных или медиастинальных лимфатических узлов. Реже
первым вовлеченным участком являются тазовые лимфатические узлы. ЛГМ
постепенно распространяется на лимфатические узлы смежных областей. В конечном
счете поражаются печень, костный мозг и другие экстранодальные зоны.
Возможно бессимптомное увеличение лимфатических узлов шеи или наличие
симптомов, связанных с медиастинальной аденопатией. При прогрессировании болезни
развиваются так называемые В-симптомы, включающие лихорадку, ночные поты
и потерю веса.
Когда при биопсии подтверждается диагноз ЛГМ, необходимо установить
стадию болезни на основании данных КТ-сканирования груди, живота и таза, а
также биопсии костного мозга. Система стадирования, которая наиболее часто
используется у пациентов с ЛГМ, приведена в табл. 9-8. Выяснение стадии
заболевания чрезвычайно важно для больных ЛГМ, так как определяет характер
проводимой терапии. Пациенты, у которых в процесс вовлечены только
лимфатические узлы выше диафрагмы, излечиваются в большинстве случаев одной
лучевой терапией, а те, у кого развивается рецидив,— химиотерапией "спасения". Если
в процесс вовлечены лимфатические узлы с обеих сторон диафрагмы (III стадия
заболевания), максимальной вероятности излечения удается добиться лишь с
применением химиотерапии.
При минимальных проявлениях болезни и если радиологически установлено
ограниченное вовлечение только лимфатических узлов выше диафрагмы
(средостение, область шеи и т. д.),.ставится вопрос о проведении диагностической лапа-
ротомии для уточнения стадии заболевания. Приблизительно у 30 % пациентов,
у которых после радиологического анализа определена II стадия заболевания,
при этом выявляются дополнительные проявления лимфомы. Наиболее частым
Лимфомы
351
ТАБЛИЦА 9-8. Стадии лимфом
Стадия I: Вовлечение одной группы лимфатических узлов с любой стороны
диафрагмы; непосредственное поражение ограниченной области или
наличие одного экстранодального очага, являющегося единственным
проявлением заболевания
Стадия II: Вовлечение 2-х или более групп лимфатических узлов по одну сторону
диафрагмы; может вовлекаться селезенка, если некоторые группы
лимфатических узлов расположены ниже диафрагмы
Стадия III: Вовлечение групп лимфатических узлов с обеих сторон диафрагмы;
может вовлекаться селезенка
Стадия IV: Вовлечение экстранодальных участков, таких как костный мозг или печень
А: Отсутствие симптомов
В: Лихорадка, ночные поты, потеря веса (> 10 % массы тела) ± кожный зуд
Из: Carbone P. P. el al. Report of fhe committee on Hodgkin's disease staging. Cancer Res., 1971; 31:1860.
местом скрытого поражения ниже диафрагмы является селезенка, однако это
почти всегда пропускается при радиологическом обследовании. Альтернативой ла-
паротомии считается лечение химиотерапевтическими средствами, которое
проводится при диссеминированной форме болезни.
Применение только лучевой терапии сопровождается излечением
большинства пациентов с четко локализованным заболеванием, без большой опухолевой
массы. Когда болезнь прогрессировала до стадии III или IV, особенно при экстра-
нодальном распространении (вовлечение костного мозга), основным методом
лечения становится комбинированная химиотерапия, обеспечивающая излечение
у большинства пациентов. В 1960-х гг. так называемая терапия МОРР (nitrogen
Mustard, vincristine [Oncovin®], Procarbazine, Prednisone) позволяла вылечивать
приблизительного % пациентов с III—IV стадиями заболевания. Это было
большое достижение, но терапия была сопряжена с рядом побочных эффектов, в
частности подавлением функции костного мозга. Лечение сопровождалось развитием
долгосрочных осложнений, включая стерильность у всех мужчин и многих
женщин, а также миелодисплазией и ОМЛ. Схема ABVD (doxorubicin [Adriamycin®],
Bleomycin, Vinblastine и Dacarbazine) была разработана в 1970-х гг. и, как
известно, позволяет достигать лучших результатов, чем МОРР. Кроме того, она не
приводит к стерилизации и не осложняется развитием миелодисплазии или ОМЛ.
У некоторых пациентов отмечается токсическое поражение легких блеомицином
или сердца доксорубицином, но эти осложнения редки.
Явление возникновения "вторичных опухолей" после терапии ЛГМ находится
в центре внимания исследователей (рис. 9-6). Как упоминалось, миелодисплазия
и ОМЛ отмечаются у 3-5 % пациентов, получавших химиотерапию МОРР,
причем частота этих осложнений выше у больных старше 40-летнегб возраста.
Индуцированный химиотерапией ОМЛ обычно возникает через 2-9 лет после
лечения, после этого срока риск его развития равен нулю. Лучевая терапия
предрасполагает к развитию опухолей в облученных областях, включая рак
легких, ЖКТ, саркомы и рак молочной железы. Эти солидные опухоли имеют
тенденцию к появлению через 10-20 лет после лечения, и их частота со временем
увеличивается. В настоящее время проводятся исследования по разработке таких
352
Глава 9. Злокачественные опухоли кроветворной ткани
20-
154
104
о.
ш
ш
54
Все опухоли (17,6 %)
Солидные опухоли (13,2 %)
Лейкозы (3,3 %)
Лимфомы(1,6%)
111111111111111111111
■ ■ i«■' i ■
8 10
Время (годы)
12
14
16
18
Рис. 9-6. Развитие вторичных онкологических заболеваний у пациентов, лечившихся но поводу болезни
Ходжкина в Стэнфорде. Отмечается, что риск развития острого миелоидного лейкоза наибольший в
период между 3 и 9 годами, а для солидных опухолей он продолжает увеличиваться со временем. (Tucker
М. A. et al. Risk of second cancers after treatment for Hodgkmin's disease. N. Engl. J. Med., 1988; 318: 76.)
режимов терапии, которые способны поддерживать высокую частоту излечения
от ЛГМ при снижении долгосрочной заболеваемости и смертности, особенно от
вторичных онкологических процессов.
Неходжкинские лимфомы
Неходжкинские лимфомы обладают широким спектром гистологических,
иммунологических и клинических характеристик. Все они характеризуются клональ-
ным размножением пролиферациями лимфоидных клеток типа В или Т.
Патофизиология и классификация. Этиология большинства случаев неходж-
кинских лимфом неизвестна. Некоторые этиологические агенты, например вирус
Эпштейна-Барр, были идентифицированы при ряде лимфом у иммунодефицит-
ных пациентов с ВИЧ-инфекцией или у пациентов, получавших иммуносупрес-
сивную терапию после трансплантации солидного органа. Аномальные лимфоид-
ные клетки способны циркулировать в периферической крови; общим признаком
неходжкинских лимфом является гематогенная диссеминация. Способы
распространения среди различных типов лимфом вариабельны, и пролиферативные
толчки заболевания также неодинаковы. Некоторые лимфомы медленно
прогрессируют, а другие растут чрезвычайно быстро.
Изучение неходжкинских лимфом затруднено из-за отсутствия устоявшейся
номенклатуры. В настоящее время используют несколько классификаций, что
еще более увеличивает неопределенность. Чаще всего употребляют рабочую
классификацию, базирующуюся на рекомендациях симпозиума, поддержанного
Национальным институтом рака США. Она была опубликована в 1982 г. и
представлена в табл. 9-9.
Лимфомы
353
ТАБЛИЦА 9-9. Рабочая классификация неходжкинскихлимфом (НХЛ)
Низкая степень злокачественности
НХЛ из малых лимфоцитов (ХЛЛ) (диффузная, хорошо дифференцированная)
Плазмоцитоидная лимфоцитарная НХЛ (Вапьденстрёма)
Фолликулярная НХЛ из малых расщепленных клеток
Фолликулярная смешанно-клеточная НХЛ
Промежуточная степень злокачественности
Фолликулярная крупноклеточная НХЛ
Диффузная НХЛ из малых расщепленных клеток
Диффузная смешанно-клеточная НХЛ
Диффузная крупноклеточная НХЛ
Высокая степень злокачественности
Иммунобластная НХЛ
Лимфобластная НХЛ
НХЛ из малых нерасщепленных клеток (типа Беркитта и неберкиттовского типа)
Из: The Non-Hodgkin's Lymphoma Pathologic Classification Project. National Cancer Institute
sponsored study of classifications of non-Hodgkin's lymphoma: summary and description of a
working formulation for clinical usage. Cancer, 1982; 49:112.
Эту классификацию удобно использовать, и она получила широкое
распространение, хотя не отражает современного понимания иммунологической сущности
заболевания. В ней не различают Т- или В-клеточные типы заболевания. Кроме
того, многие лимфомы, такие как грибовидный микоз или множественная миело-
ма, в нее не включены. Недавно предложена новая классификация, которая делит
лимфомы на Т- и В-клеточные. Она значительно полнее "Рабочей
классификации", но и намного сложнее. Эта номенклатура представлена в табл. 9-10. (The
Revised European-American Classification of Lymphoid Neoplasms [REAL].)
Клинические особенности и лечение. Обычно неходжкинские лимфомы
поражают лимфатические узлы, но в ряде случаев выявляется и иногда может быть
единственным проявлением болезни экстранодальная локализация. Полное описание
всех лимфом не входит в задачу этой главы, хотя будет рассмотрено несколько
примеров.
Фолликулярная лимфома из малых расщепленных клеток — вялотекущая лим-
фома низкой степени злокачественности, одна из наиболее частых форм (табл. 9-9).
У пациентов не отмечаются нарушения функции органов (например, закупорка
уретры), часто их можно длительно наблюдать без назначения терапии. Когда из-
за появления симптомов или нарушения функции органа требуется лечение,
используют относительно мягкие режимы химиотерапии, нередко с хорошим
результатом. При этом заболевании эффективно индуцируют ремиссию алкилиру-
ющие агенты и аналоги пуриновых оснований, такие как флударабин. К
сожалению, их применение не приводит к полному излечению и лимфома со временем
рецидивирует. В настоящее время исследуются возможности высокодозной
химиотерапии и ТКМ, хотя создается впечатление, что у большинства больных
развиваются рецидивы после трансплантации. Меньшая часть пациентов
излечиваются этой терапией, но необходимо более длительное время наблюдения, чтобы
верно понять роль ТКМ при лечении этого заболевания.
354
Глава 9. Злокачественные опухоли кроветворной ткани
ТАБЛИЦА 9-10. Пересмотренная классификация неходжкинскихлимфом
В-клеточные
1. НХЛ из предшественников В-клеток
а. Пре-В-клеточный лимфобластный лейкоз/лимфома
2. В-клеточные опухоли из клеток с "периферическим" фенотипом
а. В-клеточный ХЛЛ/мелкоклеточная лимфоцитарная лимфома
б. Лимфоплазмоцитоидная лимфома/иммуноцитома
в. Лимфома из мантийных клеток
г. Лимфома из клеток фолликулярных центров, фолликулярная
- Из малых клеток, смешанная из малых и больших клеток, из больших клеток
- Диффузная, преимущественно из малых клеток
д. В-клеточная НХЛ маргинальной зоны
- Экстранодальная (MALT ± моноцитоидная)
- Нодальная (± моноцитоидная)
- Селезеночная (± ворсинчатые лимфоциты)
е. Волосатоклеточный лейкоз
ж. Плазмоцитома/миелома
з. Диффузная крупноклеточная В-клеточная лимфома
Подтип: первичная медиастинальная В-клеточная
и. Лимфома Беркитта
Т-клеточные
1. НХЛ из предшественников Т-клеток
а. Т-лимфобластная лимфома/лейкоз
2. Т-клеточная НХЛ из клеток с "периферическим" фенотипом и NK-клеточные
опухоли
а. Т-клеточный ХЛЛ/лролимфоцитарный лейкоз
б. Крупногранулярный лимфоцитарный лейкоз (Т-клеточный и NK-клеточный)
в. Грибовидный микоз/синдром Сезари
г. Т-клеточная лимфома с "периферическим" фенотипом
- Смешанная НХЛ из средних и больших клеток, из больших клеток, лимфо-
эпителиоидная
д. Ангиоиммунобластная Т-клеточная лимфома (AILD)
е. Ангиоцентрическая лимфома
ж. Кишечная Т-клеточная лимфома (± энтеропатия)
з. Т-клеточная лимфома/лейкоз взрослых (HTLV-1)
и. Анапластическая крупноклеточная лимфома (CD30*), Т- и нулевого клеточного
типа
Из: Harris N. el al. A revised European-American classification of lymphoid neoplasms: a proposal
from the International Study Group. Blood, 1995; 84:1361.
Другая частая разновидность лимфом — диффузная крупноклеточная
лимфома. В противоположность фолликулярной лимфоме из малых расщепленных
клеток крупноклеточная лимфома характеризуется ускоренным ростом и без
лечения быстро приводит к фатальному исходу. С другой стороны, приблизительно
50 % пациентов излечиваются при помощи комбинированной химиотерапии.
Один из наиболее часто используемых режимов — CHOP (cyclophosphamide,
Лимфомы
355
hydroxyldaunomycin [doxorubicin], vincristine/ [Oncovin®], prednisone). Эта схема
хорошо переносится и так же эффективна для большинства пациентов, как и
более сложные и интенсивные. Установлены пять неблагоприятных
прогностических факторов, позволяющих предсказать развитие рецидива у этих пациентов
после интенсивного лечения: возраст больше 60 лет, III/IV стадия заболевания,
повышенная активность ЛДГ, плохое общее состояние и экстранодальное
вовлечение. Большинство этих факторов риска присутствует уже во время постановки
диагноза. У пациентов с меньшей вероятностью рецидива лечение более
эффективно. Пациентам с плохим прогнозом могут быть полезны более интенсивные,
исследовательские, режимы лечения.
Хронический лимфолейкоз (ХЛЛ) — один из видов лимфом низкой степени
злокачественности, соответствующий по "Рабочей классификации" хорошо
дифференцированной лимфоме. У большинства пациентов заболевание
прогрессирует медленно. Однако оно не излечивается стандартной химиотерапией, а роль
ТКМ при ХЛЛ не установлена. Пациенты с ХЛЛ обычно имеют значительное
вовлечение в процесс костного мозга, в конечном счете у них развиваются лимфа-
денопатия и спленомегалия. По мере усиления поражения костного мозга
развиваются анемия и тромбоцитопения. Они возникают также из-за продукции ауто-
антител, которые вызывают иммунологическое разрушение этих клеток крови,
что является частым осложнением у больных ХЛЛ. Выживаемость пациентов
с ХЛЛ тесно связана со стадией болезни (табл. 9-11).
Терапия ХЛЛ обычно включает прием пероральных алкилирующих средств
или аналогов пуриновых оснований, таких как флударабин. В начале лечения эти
препараты эффективны, но со временем к ним формируется резистентность.
Лечение ХЛЛ начинают только при развитии соответствующей симптрматики:
значительной лимфаденопатии, анемии или тромбоцитопении. Раннее начало
терапии ХЛЛ не увеличивает продолжительность жизни и может даже снижать
выживаемость из-за высокой токсичности применяемых препаратов.
Наиболее частой причиной смерти является инфекция, обусловленная
сочетанием недостаточной продукции антител, характерной для многих пациентов
с ХЛЛ, и нейтропении, возникающей вследствие замещения ткани костного мозга
лимфомой или подавления функции костного мозга химиотерапевтическими
агентами.
Пациенты с ХЛЛ имеют также склонность к развитию аутоиммунных
заболеваний, включая аутоиммунную гемолитическую анемию и иммунную тромбоци-
топеническую пурпуру (ИТП).
ТАБЛИЦА 9-11. Стадии хронического лимфолейкоза по К. Rai
Стадия Продолжительность
жизни1
Стадия 0: только лимфоцитоз > 150 мес
Стадия 1: лимфоцитоз и лимфаденопатия 100 мес
Стадия 2: сплено- или гепатомегалия 71 мес
Стадия 3: анемия, гемоглобин < 110 г/л, гематокрит < 33 % 19 мес
Стадия 4: тромбоцитопения, тромбоциты < 100 000/мкл 19 мес
1 После установления диагноза.
Из: Rai К. R. et al. Clinical staging of chronic lymphocytic leukemia. Blood, 1975; 46: 219.
356 Глава 9. Злокачественные опухоли кроветворной ткани
Множественная миелома — злокачественное новообразование из
плазматических клеток, которые являются конечным продуктом дифференцировки В-клеток
и в норме обычно производят антитела. Для этого заболевания характерна
экспансия клона злокачественных плазматических клеток в костном мозге, которая
приводит к проявлениям болезни, представленным в табл. 9-12. Продуцируемые
злокачественными клетками моноклональные иммуноглобулины
идентифицируются при электрофорезе сывороточных белков как острый пик в у-области,
а также могут быть охарактеризованы и количественно определены с помощью
моноклональных антител при использовании методов иммунофиксации или им-
муноэлектрофореза. Обычно моноклональный белок принадлежит к классу IgG,
но это может быть и IgA; реже злокачественные клетки производят только легкие
цепи иммуноглобулинов. Когда выявляются моноклональные антитела типа IgM,
болезнь определяется как макроглобулинемия Вальденстрёма, имеющая
клинические особенности, отличающие ее от множественной миеломы. Как и при ХЛЛ,
несмотря на высокие уровни моноклонального белка, пациенты с миеломой не
вырабатывают специфических антител при антигенной стимуляции, и этот
дефект определяет развитие инфекций — основной причины заболеваемости и
смертности при множественной миеломе. Кроме того, миеломные клетки производят
"фактор, активирующий остеокласты", который вызывает резорбцию костной
ткани и остеопороз, литические повреждения кости и, как следствие —
патологические переломы. В результате действия моноклонального протеина на
почечные канальцы, особенно при избытке легких цепей, очень часто возникает
почечная недостаточность. Обнаруживается также гиперкальциемия, усугубляющая
почечную недостаточность.
ТАБЛИЦА 9-12. Клинические характеристики множественной миеломы
1. Моноклональный иммуноглобулин
а. IgG в 70 % случаев
б. IgA в 20 % случаев
в. Легкие цепи только в 5 % случаев
2. Повреждения костей
а. Остеопороз и литические повреждения кости из-за усиленной резорбции
кости
б. Частые патологические переломы, особенно позвоночника
3. Почечные нарушения
а. Миеломные белки (легкие цепи) накапливаются в почечных канальцах и реаб-
сорбируются клетками почечных канальцев, вызывая их повреждение
б. Гиперкальциемия может вызывать повреждение почек
в. При миеломе может присутствовать амилоид, вызывающий повреждение
клубочков
4. Инфекция
а. Пациенты с миеломой плохо продуцируют антитела при стимуляции
антигеном, что ведет к повышению риска развития инфекций, особенно
инкапсулированными бактериями
б. Заполнение костного мозга миеломными клетками может приводить
к нейтропении
в. Химиотерапия, используемая для лечения миеломы, может вызывать
нейтропению
Лимфомы
357
Диагноз миеломы ставится на основании идентификации моноклонального
иммуноглобулина в сыворотке крови или моче и выявления увеличенного
количества моноклональных плазматических клеток в костном мозге.
Миелома — неизлечимая болезнь, но назначение алкилирующих агентов,
например мелфалана, в сочетании с преднизолоном приводит к частичной ремиссии
почти у половины больных. Средний срок жизни (после установления диагноза)
не превышает тем не менее 24 месяца, и даже использование интенсивной
комплексной (шесть препаратов) химиотерапии не улучшает результатов лечения
(рис. 9-7). Исследуются возможности применения ТКМ, и хотя окончательная
эффективность этого метода лечения еще не известна, очевидно, что у
большинства пациентов после трансплантации болезнь рецидивирует.
Клинический пример
26-летний мужчина обнаружил опухоль в области шеи слева. Он
обратился к врачу, который не выявил у пациента болезненных симптомов и
какого-либо специфического заболевания, но обратил внимание на передний
шейный лимфатический узел слева размером 3X2 см, эластичный,
фиксированный. При биопсии выявлен ЛГМ, нодулярный склероз.
Определен перечень необходимых диагностических тестов.
Вопрос 1. Каков следующий этап диагностического процесса?
Ответ. После проведения стандартного анализа крови и мочи необходима
рентгенограмма грудной клетки для поиска медиастинальной аденопатии
и легочных паренхиматозных нарушений. Если аномалии будут выявлены,
то КТ-исследование грудной полости позволит уточнить диагноз. Допол-
v«uu1 i 1 1 1 1 1 1 1 1 1 1 1
О 6 12 18 24 30 36
Продолжительность ремиссии (месяцы)
Рис. 9-7. Продолжительность жизни (после установления диагноза) больных множественной мие-
ломой, получавших мелфалан с преднизолоном (МП) или интенсивную комплексную терапию
(КоТ). Мелфалан с преднизолоном обеспечивает такой же срок жизни при меньших токсических
эффектах. (Hjorth M. et al. Initial treatment in multiple myeloma: No advantage of multidrug
chemotherapy over melphalan-prednisone. Br. J. Haematol., 1990; 74: 185.)
358 Глава 9. Злокачественные опухоли кроветворной ткани
нительно необходимо КТ-сканирование живота и таза для определения
степени увеличения лимфатических узлов в этих областях.
Вопрос 2. При рентгеновском исследовании грудной клетки и КТ
обнаружено образование приблизительно 3 см в диаметре в области корня левого
легкого. Стандартный анализ крови, мочи и КТ-обследование живота и
таза не выявили никаких аномалий. Нужно ли начинать лечение этого
пациента?
Ответ. Из-за возможности распространения палогического процесса ниже
диафрагмы, особенно на селезенку (что бывает в 30 % случаев), многие
рекомендовали бы проведение лапаротомии для выявления "скрытого"
процесса в брюшной полости. Этот вид агрессивного обследования ("стадиро-
вания") выполняют только при условии, что оно может изменить схему
планируемой терапии, а терапия должна привести к изменению
результатов (обычными критериями эффективности являются общая
выживаемость и излечение от болезни). Если стадирование не показало
распространения болезни ниже диафрагмы, тогда можно проводить только лучевую
терапию. При обнаружении лимфомы необходимо рассмотреть различные
радиотерапевтические и/или химиотерапевтические подходы.
Если из-за неполностью проведенного стадирования субдиафрагмальная
лимфома была первоначально не выявлена и выбрана менее интенсивная
схема терапии, еще не все потеряно. Даже если произошел рецидив, можно
"подкрепить" пациента дополнительной терапией и обеспечить ему
долгосрочную выживаемость. Однако большинство специалистов предпочитают
гипердиагностику и, соответственно, проведение максимально
интенсивного лечения, которое минимизирует риск возникновения рецидива и
осложнений.
У данного пациента при одном стандартном диагностическом
вмешательстве необходимо выполнить лапаротомию и спленэктомию. Если никакого
проявления ЛГМ ниже диафрагмы не будет найдено, следует назначить
лучевую терапию на поверхностные (подкожные) и верхние парааорталь-
ные лимфатические узлы.
Продолжение наблюдения
При КТ брюшной полости и таза не выявлено признаков лимфаденопатии.
Сканирование галлием показало увеличенную активность в области
средостения. Пациент получил вакцины против Haemophilus influenzae,
Streptococcus pneumoniae, Neisseria meningitidis. Затем была выполнена
диагностическая лапаротомия и спленэктомия, при этом никаких признаков
заболевания не найдено. Проведена лучевая терапия на поверхностные и па-
рааортальные области. Пациент находится в состоянии ремиссии в течение
5 лет после курса лечения.
Хотя этот пациент, очевидно, был вылечен, в дальнейшем требуется
всестороннее наблюдение для определения последствий болезни и лечения,
которые могут развиваться спустя многие годы. К ним относят гипотиреоз или
карциному щитовидной железы после лучевой терапии и постспленэкто-
мический сепсис, вызванный инкапсулированными микроорганизмами,
упомянутыми ранее (несмотря на иммунизации). Вызывать проблемы
могут также другие бактерии, малярийный плазмодий и бабезия. Кроме того,
увеличивается вероятность развития вторичных новообразований, особен-
Лимфомы
359
но в облученных областях, через 5,10 и 15 лет после лучевой терапии.
Молодые женщины особенно предрасположены к развитию рака молочной
железы после облучения.
Клинический пример
77-летний мужчина пришел на прием из-за плохого анализа крови. Он
хорошо себя чувствовал в последнее время, хотя 7 лет назад перенес аорто-
коронарное шунтирование из-за стенокардии, резистентной к
лекарственной терапии. В настоящее время принимает только блокатор
кальциевых каналов и чувствует себя прекрасно. При ежегодном
обследовании 2 недели назад у него обнаружено повышение СОЭ и содержания
общего белка в плазме крови. Последующий электрофорез
сывороточных белков показал моноклональный "пик" в области у-глобулинов при
их концентрации 26 г/л (нормальное содержание у-глобулина — 7-
17 г/л); уровень других иммуноглобулинов был в пределах нормы.
Необходимо рассмотреть возможность диагноза множественной миеломы.
Пациент встревожен, особенно потому, что от этой болезни недавно умер
его друг, у которого были переломы костей, кровотечения и инфекции.
При физикальном осмотре пациент выглядит здоровым, но испуганным.
Каких-либо аномалий, кроме хорошо зажившего рубца от срединного
разреза грудной клетки, не найдено.
Вопрос 1. Может ли у этого пациента быть множественная миелома?
Ответ. Постановка диагноза множественной миеломы возможна только
в том случае, если имеющиеся данные соответствуют нескольким
критериям. У данного пациента, как мы надеемся, пик в у-области (моноклональная
парапротеинемия) указывает на то, что он страдает только
доброкачественной моноклональной гаммапатией (ДМГ), известной также как
моноклональная гаммапатия неизвестного значения (МГНЗ). Дальнейшие
действия должны быть направлены на классификацию его заболевания.
Вопрос 2. Каковы следующие диагностические шаги?
Ответ. Анализ крови, выполненный за 2 недели до осмотра, показал, что
кроме высокого уровня белка все биохимические параметры сыворотки
нормальны, включая уровень кальция. Это хорошо, потому что отсутствие
анемии, азотемии или гиперкальциемии является благоприятным
признаком для исключения множественной миеломы. Назначается анализ мочи с
определением наличия легких цепей (белков Бене-Джонса), даже если
общий скрининговый тест определения белка в моче будет отрицательным.
Легкие цепи не найдены. Предпринята дальнейшая попытка
охарактеризовать аномальный сывороточный белок. Иммуноглобулин принадлежит к
классу IgG, при этом в сыворотке имеется лишь небольшое количество
свободных легких цепей, но все подтипа X.
Вопрос 3. Нужно ли еще что-либо делать?
Ответ. Да. Чтобы полностью успокоить пациента (и, возможно, сначала
убедить самого себя), необходимо провести рентгенографию черепа и
длинных костей. При множественной миеломе можно обнаружить диффузное
истощение костной ткани или, чаще, повреждения в виде пробоин (кости
напоминают швейцарский сыр).
360 Глава 9. Злокачественные опухоли кроветворной ткани
В отличие от некоторых других новообразований костной ткани, при
которых сканирование кости (с использованием радиоактивного технеция)
позволяет выявить дефекты, не заметные на простых рентгенограммах, при
множественной миеломе сканирование кости обычно дает отрицательный
результат ("холодные" образования) даже там, где имеется патология. Это
происходит из-за того, что в отсутствии остеобластной активности на фоне
преобладания остеокластной активности при большинстве форм миеломы
не идет дроцесс неоваскуляризации. Поскольку основой для поглощения
радиоактивного технеция служат новые кровеносные сосуды, связанные
с опухолью (это приводит к положительному ["горячему"] результату при
сканировании кости), при этой методике изображения кости выглядят
нормальными даже при наличии костных плазмоцитом. Хотя на обычных
рентгенограммах видны повреждения в виде пробоин.
Вопрос 4. На рентгенограммах не выявлено никаких нарушений. Можно
ли сообщить пациенту теперь, что у него нет множественной миеломы?
Ответ. Чтобы исключать присутствие "тлеющей множественной миеломы",
при который болезнь, как правило, ограничена только костным мозгом,
необходимо выполнить аспирацию и биопсию костного мозга. Эти
исследования показали, что костный мозг не изменен, то есть содержит меньше 5 %
плазматических клеток.
Вопрос 5. Каков вывод?
Ответ. Хорошее самочувствие пациента в настоящее время и нормальные
показатели клинического анализа крови, ее химического состава,
рентгенограммы костей и аспирата костного мозга при относительно низком уровне
парапротеина и неизмененном уровне других иммуноглобулинов
убедительно подтверждают наличие в данном случае доброкачественной моно-
клональной гаммапатии. Кроме того, запись в истории болезни, сделанная
при проведении хирургического вмешательства 7 лет назад, показывает,
что содержание белка в сыворотке было повышено уже в то время, что
свидетельствует об отсутствии прогрессирования заболевания.
Продолжение наблюдения
После 3-х лет наблюдения у пациента не выявлено изменения уровня
иммуноглобулинов и каких-либо клинических признаков развития
множественной миеломы. Приблизительно 25-30 % пациентов с
доброкачественной моноклональной гаммапатией или моноклональной гаммапатией
неизвестного значения прогрессируют до развития полной картины
множественной миеломы в течение 20 лет, но этот пациент, по-видимому, пока
может просто находиться под наблюдением. Однако ему следует обращать
особое внимание на появление болей в костях, которые могут
свидетельствовать об остеолитическом повреждении, и отслеживать результаты
каждого электрофоретического исследования сывороточных белков, которые
могли бы быть первым индикатором прогрессирования болезни.
Клинический пример
78-летняя женщина посещает врача для планового обследования. Она
чувствует себя хорошо, но врач обнаруживает несколько эластичных
мягких лимфатических узлов размером 1 X 2 см в области шеи слева
Лимфомы
361
и в правой подмышечной впадине. В клиническом анализе крови:
лейкоцитов 26 000/мкл, 69 % лимфоцитов, 13 % нейтрофилов, 10 % моноцитов,
5 % атипичных лимфоцитов и 3 % эозинофилов. Гемоглобин — 128 г/л,
гематокритное число — 37,5 %, количество тромбоцитов — 353 000/мкл.
Вопрос 1. Каков вероятный диагноз?
Ответ. Картина сопоставима с хроническим лимфолейкозом (ХЛЛ).
Вопрос 2. Что должно быть выполнено далее?
Ответ. Диагноз может быть подтвержден исследованием иммунофенотипи-
ческих маркеров методом проточной цитометрии и цитогенетическим
анализом, но сначала необходимо исследовать мазок периферической крови.
Вопрос 3. Просмотр мазка периферической крови показал типичные черты
хронического лимфолейкоза. Что они включают?
Ответ. Красный ряд клеток морфологически не изменен. Нет полихрома-
зии, что позволяет предполагать отсутствие повышения количества рети-
кулоцитов. (Если полихромазия и повышение количества ретикулоцитов
выявлены при анемии, это позволяет предположить активную потерю
крови или гемолиз с последующей компенсаторной реакцией костного мозга.)
Морфология тромбоцитов нормальна. В мазке обнаружено несколько
нейтрофилов. Лимфоциты находятся в изобилии, большая их часть
представлена малыми и зрелыми клетками. Присутствует несколько клеток с
нечеткими очертаниями (хрупкие лимфоциты при хроническом лимфолейкцзе
способны разрушаться в процессе приготовления мазка крови).
Вопрос 4. Какой иммунофенотипический профиль является типичным для
хронического лимфолейкоза?
Ответ. Большинство маркеров относится к В-клеточным, но имеется
двойная экспрессия В-клеточных антигенов CD 19, CD20, CD21, CD24 с Т-кле-
точным антигеном CD5 на клетках, что обычно является диагностическим
признаком В-клеточного ХЛЛ. Особенно важна для диагностики коэксп-
рессия CD20 и CD5. В большинстве случаев на поверхности клетки
выявляются моноклональные иммуноглобулины. Иногда обнаруживаются
сывороточные парапротеины и/или гипогаммаглобулинемия.
Вопрос 5. Какие события в течении заболевания можно прогнозировать?
Ответ. Повышение восприимчивости к инфекции, аутоиммунную тромбо-
цитопению или гемолитическую анемию, прогрессирующую лимфаденопа-
тию и спленомегалию. Не исключен переход в более агрессивную форму
лимфомы.
Вопрос 6. Когда пациентка должна начать лечиться по поводу ХЛЛ?
Ответ. Каждая из проблем, упомянутых в пункте 5, является поводом для
начала терапии. Кроме того, причиной для начала медикаментозной или
лучевой терапии могут служить значительный лейкоцитоз или органные
нарушения, обусловленные лимфаденопатией.
Продолжение наблюдения
С гематологической точки зрения состояние пациентки не внушает
опасений. Хотя имеется умеренный лейкоцитоз, не отмечено ни одного из
грозных осложнений хронического лимфолейкоза, включая трансформацию в
агрессивную крупноклеточную лимфому. К сожалению, через 5 лет после
постановки диагноза больная погибла в автомобильной катастрофе.
362
Глава 9. Злокачественные опухоли кроветворной ткани
Избранная литература
Canellos G. P. et al. Chemotherapy of advanced Hodgkin's disease with MOPP, ABVD, or
MOPP alternating with ABVD. N. Engl J. Med., 1992:327:1478.
Результаты большого мультицентрового обследования пациентов с Л ГМ,
лечившихся с использованием трех наиболее распространенных режимов химиотерапии.
ABVD, оказавшийся наиболее эффективным при минимальной токсичности,
признан терапией выбора для этих пациентов.
Canellos G. P., Lister Т. Н., Sklar J. L. The Lymphomas. Philadelphia: W. B. Saunders,
1998.
Современный всесторонний обзор по лимфомам.
Clavell L. A. et al. Four-agent induction and intensive asparaginase therapy for the
treatment of childhood acute lymphoblastic leukemia. N. Engl. J. Med., 1986; 315:
657.
В статье проанализированы результаты лечения острого лимфобластного лейкоза
у детей. Показано, что использование комбинированной химиотерапии
существенно повышает вероятность излечения.
Fisher R. I. et al. Comparison of a standard regimen (CHOP) with three intensive
chemotherapy regimens for advanced non-Hodgkin's lymphoma. N. Engl. J. Med.,
1993; 328:1002.
В статье сравнивается СНОР-химиотерапия с более сложными и токсичными
режимами лечения пациентов с крупноклеточной лимфомой. СНОР-терапия столь
же эффективна, как и другие варианты лечения, но переносится лучше.
Foon К. A., Rai К. R., Gale R. P. Chronic lymphocytic leukemia: New insights into biology
and therapy. Ann. Intern. Med., 1990; 113:525.
Великолепный обзор по биологии, патофизиологии и лечению ХЛЛ.
French Cooperative Group on Chronic Lymphocytic Leukemia. Effects of chlorambucil
and therapeutic decision in initial forms of chronic lymphocytic leukemia (Stage A):
Results of a randomized clinical trial on 612 patients. Blood, 1990; 75:1414.
Результаты большого рандомизированного исследования показывают, что лечение
ХЛЛ на ранних стадиях заболевания не повышает выживаемости.
Целесообразно начинать его только тогда, когда болезнь прогрессирует и появляются
симптомы или органные нарушения.
Gahrton et al. Allogeneic bone marrow transplantation in multiple myeloma. N Engl. J.
Med., 1991; 325:1267.
Исследование результатов трансплантации костного мозга у пациентов с
множественной миеломой показывает, что этот метод имеет ограниченное
применение у таких больных.
Harris N. et al. A revised European-American classification of lymphoid neoplasms:
a proposal from the International Study Group. Blood, 1995; 84:1361.
Представлена пересмотренная и достаточно полная классификация
злокачественных лимфом, которая нашла широкое признание.
Hjorth M. et al. Initial treatment in multiple myeloma: No advantage of multidrug
chemotherapy over melphalan-prednisone. Br.J. Haematol., 1990; 74:185.
Показано, что интенсивная химиотерапия не обеспечивает никаких преимуществ
перед имягким" мелфаланом и преднизолоном при лечении больных миеломой.
Лимфомы
363
International Non-Hodgkin's Lymphoma Prognostic Factors Project. A predictive model
for aggressive non-Hodgkin's lymphoma. N Engl. J. Med., 1993; 329:987.
Детализированы прогностические факторы у пациентов с крупноклеточной лимфо-
мой, что является важным достижением в понимании сути этой болезни.
Прогнозирование имеет большое практическое значение для лечения таких
пациентов.
Kaplan H. S. Hodgkin's Disease: Biology, treatment, prognosis. Blood, 1981; 57: 813.
Обзор по биологии и лечению болезни Ходжкина, написанный Henry Kaplan, который
произвел революцию в подходах к лечению этой болезни.
Kyle R. A. Multiple myeloma: Review of 869 cases. Mayo Clin. Proc, 1975; 50:29.
Хотя этой работе уже 20 лет, она остается одним из лучших описаний
множественной миеломы, очень хорошо детализирующим клинические характеристики.
Larson R. A., Dodge R. К., Burns С. P. et al. A five-drug remission induction regimen with
intensive consolidation for adults with acute lymphoblastic leukemia: Cancer and
Leukemia Group В study 8811. Blood, 1995; 85: 2025.
Обобщены результаты исследования по лечению острого лимфобластного лейкоза
у взрослых. Показано, что при использовании интенсивной химиотерапии
значительная доля пациентов может быть излечена. Это главное достижение в
лечении ОЛЛ, давшее новое понимание результатов лечения с учетом специфических
подгрупп этой болезни.
Longo D. L. et al. Twenty years of MOPP therapy for Hodgkin's Disease./. Clin. Oncol,
1986; 4:1295.
MOPP была первой схемой комбинированной химиотерапии, позволившей излечивать
распространенные злокачественные новообразования. Результаты
долгосрочного исследования подтвердили факт излечения почти у половины пациентов.
Mayer R. J., Davis R. В., Schiffer С. A. et al. Intensive postremission chemotherapy in
adults with acute myeloid leukemia. N. Engl. J. Med., 1994; 331: 896.
Обследование более 1000 пациентов показало, что интенсивная постремиссионная
химиотерапия улучшает результаты лечения пациентов с ОМЛ.
Mitus A.J., Miller K.B., Schenkein D.P. et al. Improved survival for patients with acute
myelogenous leukemia./ Clin. Oncol., 1995; 13:560.
Показано, что интенсивная терапия для достижения ремиссии с последующей ауто-
логичной или аллогенной трансплантацией костного мозга повышает
выживаемость пациентов с ОМЛ.
Rai К. R. et al. Clinical staging of chronic lymphocytic leukemia. Blood, 1975; 46: 219.
В статье подробно обсуждаются вопросы биологии ХЛЛ и прогноза у пациентов
с этой болезнью.
The Non-Hodgkin's Lymphoma Pathologic Classification Project. National Cancer
Institute sponsored study of classifications of non-Hodgkin's lymphoma: summary and
description of a working formulation for clinical usage. Cancer, 1982; 49: 2112.
Все еще наиболее широко используемая классификация лимфом, дополняемая в
настоящее время пересмотренной классификацией Harris N. и соавторов.
Tucker M. A. et al. Risk of second cancers after treatment for Hodgkin's disease. N. Engl. J.
Med., 1988; 318:76.
Большинство пациентов с болезнью Ходжкина теперь излечиваются и живут
достаточно долго, но они могут страдать от побочных эффектов терапии. Ихимио-,
364
и лучевая терапия являются мутагенными и вызывают лейкоз и солидные
опухоли у перенесших эту болезнь. Представлен отчет о 1500 пациентах с подробным
анализом эффектов современного лечения.
Warrell Jr. R. P. et al. Differentiation therapy of acute promyelocytic leukemia with
tretinoin (All-trans-retinoic acid). N. Engl J. Med., 1991; 324:1385.
Одно из первых указаний на способность ATRA вызвать дифференцировку клеток
острого промиелоцитарного лейкоза, что ведет к клинической ремиссии. Это
также наиболее наглядный пример связи между хромосомной аномалией и
специфическим вмешательством, нацеленным на ее коррекцию.
Zittoun R. et al. Alternating v repeated postremission treatment in adult acute
myelogenous leukemia: A randomized phase III study (AML6) of the EORTC
Leukemia Cooperative Study. Blood, 1989; 73:896.
Результаты крупного исследования по сравнению стандартной химиотерапии с алло-
генной и аутологичной трансплантацией костного мозга у пациентов с ОМЛ.
Показано, что дополнительное интенсивное лечение, которое является частью
процедуры трансплантации, способно повышать выживаемость.
Глава 10
Гематологические проявления
ВИЧ-инфекции
Бхарат Рамратнан, Джанти Парамесваран,
Тимоти П. Фланиган, Джеймс А. Хокси
Синдром приобретенного иммунодефицита (СПИД), вызываемый вирусом
иммунодефицита человека (ВИЧ), впервые был зарегистрирован в 1983 г. Несмотря
на то, что ВИЧ, принадлежащий к группе ретровирусов, выделяется из многих
жидких сред организма, распространение заболевания может происходить только
при половом (гомо- или гетеросексуальном) контакте, парентерально
(трансфузия зараженной крови и ее продуктов или внутривенное введение наркотиков)
и вертикально (от зараженной матери к ребенку). По приблизительным оценкам,
на сегодняшний день этим вирусом инфицированы более 20 млн человек по всему
миру. В США ежегодно инфицируется около 40 тыс. человек. В 1996 г. СПИД
стал основной причиной смерти мужчин в возрасте от 25 до 44 лет.
Отдельные фазы ВИЧ-инфекции определяются в соответствии с лабораторными
и клиническими данными (рис. 10-1). Механизмы, с помощью которых
ВИЧ-инфекция приводит к состоянию иммунной недостаточности, продолжают
интенсивно исследоваться и, по-видимому, являются отражением комплексного
взаимодействия между вирусом и его носителем. ВИЧ способен поражать множество клеток,
однако максимальные разрушения он производит в СВ4-лимфоцитах и макрофагах.
Именно количественные и качественные патологические изменения СБ4-лимфо-
цитов представляются центральным звеном развития иммунной дисфункции при
СПИД. Предполагаются 2 механизма истощения этих клеток: прямое уничтожение
и разрушение, опосредованное клеточным или гуморальным ответом на
ВИЧ-инфицирование. Задолго до того, как СБ4-клетки гибнут, они теряют свою
способность к стимуляции эффективного иммунного ответа на ВИЧ и другие инфекции.
Гематологические проявления ВИЧ-инфекции обширны и включают анемию,
лейкопению, тромбоцитопению и нарушения свертываемости. Патогенетические
механизмы разнообразны и могут содержать следующие звенья:
- прямое поражение клеток костного мозга или других клеток, необходимых
для гемопоэза;
- нарушение регуляции иммунной системы, приводящее к разрушению или
угнетению гемопоэтических клеток и продукции аномальных иммуноглобулинов;
- осложнения антиретровирусной терапии;
- вторичные осложнения при оппортунистических инфекциях, малигнизации
и/или лечении этих заболеваний (табл. 10-1,10-2).
366
Глава 10. Гематологические проявления ВИЧ-инфекции
Месяцы от момента Годы от момента
инфицирования инфицирования
Рис. 10-1. Течение ВИЧ-инфекции. (По: Flanigan Т. P. et al. Update of HIV and AIDS
in North America. R. I. Med., 79(5): 181; 1996.)
Патология клеток красной крови
Анемия является обычной для ВИЧ-инфицированных пациентов, причем ее
выраженность увеличивается при развитии заболевания. На ранней стадии заболевания
умеренное, но значимое уменьшение гематокрита наблюдается у 15-20 %
пациентов и у 75-90 % больных — в случаях, когда ВИЧ-инфекция осложняется другими
оппортунистическими инфекциями. Однако анемия у пациентов, не
принимающих зидовудин, как правило, носит нормоцитарный и нормохромный характер.
Макроцитоз встречается у большинства пациентов, получающих терапию зидову-
дином в течение двух и более недель. Макроцитоз, обусловленный действием зидо-
вудина, не связан с дефицитом витамина Bt2 или фолатов. Анемия может быть
результатом депрессии или неэффективности эритропоэза, острой или хронической
крово1 ютери, а также периферической деструкции красных кровяных клеток.
Депрессия или неэффективность эритропоэза при анемии, вызванной ВИЧ-
инфекцией, сопровождается низким или неадекватным уровнем ретикулоцитов.
Часто при исследовании костного мозга не обнаруживаются специфические гис-
топатологические изменения, а отмечается возрастание числа эритроидных
клеток-предшественников на фоне дисэритропоэза различной степени тяжести.
Подавление активности костного мозга происходит из-за применения лекарственных
препаратов, оппортунистических инфекций, злокачественных новообразований,
алиментарной недостаточности и, наконец, прямого действия ВИЧ.
Большинство лекарственных средств, используемых для лечения СПИД и его
осложнений, подавляют эритропоэз (табл. 10-2). Антиретровирусный препарат
зидовудин вызывает макроцитарную анемию. Ее тяжесть зависит от дозы зидову-
дина. Если последний изначально применяется в высоких дозах (> 1000 мг/сут),
то приблизительно у Уз ВИЧ-инфицированных пациентов развивается резкая
анемия (содержание гемоглобина < 80 г/л). Это особенно характерно для про-
Патология клеток фасной крови
367
ТАБЛИЦА 10-1. Гематологические проявления некоторых ВИЧ-ассоциироранных
инфекций
Нозологическая Постановка диагноза
форма
Гематологические Лечение
и клинические
проявления
Вирусные
Парвовирус
В19
Поражение
предшественников эритроцитов
и симптомы
тяжелой анемии
Количество рети-
кулоцитов равно 0
Назначение
внутривенных
препаратов
иммуноглобулина
Оптимизация
антивирусной
терапии
Цитомегало-
вирус
Выявление антител IgM к
В19 (могут отсутствовать
в прогрессирующей
стадии заболевания)
Выявление наличия
вирусной ДНК методом ПЦР
в сыворотке крови или
костном мозге
Выявление парциальной
красноклеточной аплазии и
наличия гигантских пронор-
моцитов в костном мозге
Выявление внутриядерных Цитопения, колит, Применение ган-
включений в клетках кост- зрительные рас- цикловира или
ного мозга или гастроин- стройства фоскарнета
тестинальном биоптате
Исследование сетчатки
Бактериальная Выделение культуры из
Mycobacterium костного мозга либо изо-
avium лированной гемокультуры
intracellulare и/или визуализация
кислотоустойчивых
организмов в биоптате костного
мозга
Диссеминирован-
ная инфекция при
CD4<50
Инфильтрация
костного мозга с
признаками анемии и
нейтропении
Цитопения
на фоне
инфильтрации костного
мозга при диссе-
минированном
заболевании
Грибковые Выделение культур из
различных сред организма,
включая аспираты
костного мозга, и/или
выявление возбудителя в
биоптате костного мозга при
соответствующем
гистохимическом окрашивании
Протозойные Выделение культур из Цитопении на фо
Leishmania крови, костного мозга, пе- не гиперсплениз
species чени, лимфатических уз
лов или селезенки
Гистохимическое иссле
дование
Комбинированное лечение
активной формы
заболевания
с
использованием макролидных
антибиотиков
(кларитромицин
или азитромицин)
Применение
антигрибковых
средств, включая
триазолы (флу-
коназол, итрако-
назол), или
амфотерицина В
Сочетание
противопарази-
ма, инфильтрации тарных средств и
костного мозга и амфотерицина В
возможного
аутоиммунного
процесса
368
Глава 10. Гематологические проявления ВИЧ-инфекции
ТАБЛИЦА 10-2.
Гематологические осложнения при использовании препаратов,
применяемых для лечения ВИЧ-инфицированных пациентов
Лекарственные препараты
Гематологические
осложнения
Оптимальная терапия
Антиретровирусные средства
Зидовудин (АЗТ) Анемия
и макроцитоз
Антивирусные средства
Ацикловир (герпетические
вирусные инфекции)
Фоскарнет (ЦМВ)
Ганцикловир (ЦМВ)
Интерферон (гепатит С,
саркома Калоши,
остроконечные кандиломы)
Миелосупрессия при
приеме в высоких дозах
Анемия и гранулоцито-
пения
Обычно нейтропения
(могут также
наблюдаться анемия и тром-
боцитопения)
Нейтропения, анемия
или тромбоцитопения
Антипротозойные средства
Примахин (ППК)
Триметоприм/сульфамет-
оксазол (профилактика
и лечение ППК)
Дапсон (профилактика
и лечение ППК)
Пириметамин
(токсоплазмоз)
Пентамидин (ППК)
Гемолитическая анемия
на фоне
недостаточности Г-6-ФДГ
Миелосупрессия
Дозозависимый гемолиз
у пациентов с
недостаточностью Г-6-ФДГ или
без нее, нейтропения
Мегалобластная
анемия, тромбоцитопения,
нейтропения
Анемия
Антибактериальные/микобактериальные средства
Рифабутин
(профилактика MAI)
Рифампин (МТБ)
Нейтропения, редко
тромбоцитопения
Тромбоцитопения при
высокодозной интер-
миттирующей терапии,
Отмена препарата
Уменьшение дозы
Назначение эритропоэтина
Трансфузионная терапия
Отмена препарата при
тяжелых осложнениях
Назначение Г-КСФ
Отмена препарата
Замена препарата
(ганцикловир)
Назначение Г-КСФ
Отмена препарата
Замена препарата
(фоскарнет)
Отмена препарата при
тяжелых осложнениях
Использование
альтернативного препарата
Использование
альтернативного препарата
Использование
альтернативного препарата
Назначение фолиновой
кислоты (НЕ фолиевой
кислоты) для профилактики
нейтропении
Рассмотрение
возможности замены препарата
Использование
альтернативного препарата из
группы макролидных
антибиотиков
Использование
альтернативного препарата
противотуберкулезного ряда
Патология клеток красной крови'
369
ТАБЛИЦА 10-2. (Продолжение)
Лекарственные препараты Гематологические Оптимальная терапия
осложнения t
транзиторная анемия,
гемолитическая анемия
и лейкопения
Антигрибковые средства
Амфотерицин В (при диссе- Нормохромная, нормо- Внимательное наблюдение
минированной или резис- цитарная анемия Рассмотрение возможнос-
тентной грибковой инфек- ти замещения препарата
ции) триазоловыми средствами
или липосомальными
композициями
ЦМВ — цитомегаловирус; ППК — интерстициальная пневмония, вызываемая Pneumocystis
carinii; MAI — Mycobacterium avium intracellulars; Г-КСФ — гранулоцитарный колониестимули-
рующий фактор; МТБ — микобактерия туберкулеза.
грессирующей стадии развития ВИЧ-инфекции с уменьшением числа CD4-ioie-
ток (< 200/мкл). Однако при приеме обычно рекомендуемых доз (600 мг/сут)
тяжелая анемия наблюдается реже.
Многие ассоциирующиеся с ВИЧ оппортунистические инфекции могут
подавлять эритропоэз при инфильтрации костного мозга (например, Mycobacterium
avium intracellular [MAI]) или прямом цитопатичёском воздействии на гемопоэ-
тические клетки-предшественники (например, парвовирус В19). MAI является
нетуберкулезной микобактерией, ставшей распространенным
оппортунистическим патогеном у пациентов со СПИД, вторым по частоте после Pneumocystis
carinii. MAI-инфекция участвует в патогенезе тяжелого иммунодефицита
(обычно ниже 50 СБ4-клеток/мкл) и, как правило, сопровождается лихорадкой,
потерей веса, глубокой анемией, болями в животе и диареей. Гемокультуры
позитивны у большинства пациентов, а сам возбудитель может быть визуализирован
в биоптатах костного мозга при кислотоустойчивом окрашивании мазков.
Применение профилактических лечебных средств (азитромицин или рифабутин)
уменьшает частоту возникновения MAI-инфекции приблизительно на 50 %. Для
лечения диссеминированной инфекции используются комбинации антибиотиков
макролидного ряда (кларитромицин или азитромицин). Однако MAI-инфекция
обычно персистирует и реакция на проводимое лечение незначительна.
Цитопатическое воздействие парвовируса В19 на эритрридные
клетки-предшественники костного мозга приводит к тяжелым апластическим кризам у
пациентов с серповидно-клеточцой анемией или другими гемоглобинопатиями.
Данная инфекция среди популяции в целом достаточно обычное явление; избавление
от вируса зависит только от эффективности выработки антител к главному белку
капсида вируса. Именно потому, что у пациентов с прогрессирующей стадией
ВИЧ-инфекции репродукция нейтрализирующих антител зачастую неадекватна,
парвовирусная инфекция может персистировать и являться причиной развития
не только тяжелой гипопластической анемии, но даже парциальной краснокле-
точной аплазии (глава 7). Внутривенное введение иммуноглобулина повышает
уровень гемоглобина и уменьшает виремию путем пассивного восстановления
370
Глава 10. Гематологические проявления ВИЧ-инфекции
необходимого количества нейтрализирующих антител. Для некоторых пациентов
может потребоваться довольно длительный курс лечения иммуноглобулином.
По-видимому, нарастание периферической деструкции эритроцитов может
осуществляться в результате реализации как чисто иммунных, так и неиммунных
механизмов. Почти у всех ВИЧ-инфицированных пациентов наблюдается поли-
клональная гипергаммаглобулинемия, а серологические исследования выявляют
пациентов (5-20 %) с антиэритроцитарными антителами и положительным
прямым антиглобулиновым тестом (Кумбса). Однако клиническая значимость этих
данных пока неясна, так как иммунная гемолитическая анемия для таких
пациентов нехарактерна. Недостаточность Г-6-ФДГ ВИЧ-позитивных пациентов может
рассматриваться как фактор риска в отношении гемолиза при использовании
препаратов оксидантов (дапсон, примахин), назначаемых для профилактики или
лечения оппортунистических инфекций. На фоне тромботической тромбоцитопе-
нической пурпуры, являющейся осложнением ВИЧ-инфекции, может
наблюдаться тяжелая микроангиопатическая гемолитическая анемия.
Необходимо подчеркнуть, что, поскольку инфекции (цитомегаловирусные
колиты) или процессы малигнизации (саркома Калоши, лимфосаркома), связанные
с ВИЧ-инфицированием, часто сопровождаются острой или хронической крово-
потерей из желудочно-кишечного тракта, для адекватной оценки анемии
необходимо проводить исследование кала на скрытую кровь.
Таким образом, диагностика анемии включает подробный сбор анамнеза, фи-
зикальное обследование и лабораторные тесты. Всегда важно учитывать поли-
этиологичность анемии. Полнота анализа последней может быть гарантирована
при соблюдении следующих условий:
- учет вероятности острой кровопотери: физикальное обследование, включая
ортостатические пробы, исследование органов брюшной полости и кала на
скрытую кровь;
- исследование периферической крови (табл. 3-3) для выявления макроцитов
(характерно для пациентов, получающих зидовудин), шистоцитов (указание
на ТТП или ДВС), сфероцитов (вероятность иммунного гемолиза), а также
полихромазии, соотносимой с уровнем ретикулоцитов (правда, нужно
признать, что у ВИЧ-инфицированных пациентов число ретикулоцитов часто
не отражает степень анемии);
- полная количественная оценка крови, включая подсчет ретикулоцитов; кроме
того, при подозрении на М AI-инфекцию и сокращении числа СБ4-клеток
(менее 100) — выделение изолированных гемокультур; при признаках гемолиза —
анализы на ЛДГ, гаптоглобин и непрямой билирубин; при алиментарной
недостаточности — проверка уровня фолиевой кислоты в эритроцитах и витамина
В12 (уменьшение уровней железа, витамина Bt2 или фолата не обязательно
усиливает ВИЧ-опосредованную хроническую анемию, однако обычно проводят
исследование этих потенциально обратимых^тиологических факторов);
- анализ проводимой лекарственной терапии; например, хорошо известно, что
анемия может быть осложнением терапии зидовудином (для ее коррекции
отменяют или уменьшают дозы препарата, назначают поддерживающие
трансфузии или эритропоэтин); кроме того, иногда обратимую анемию
вызывает прием таких препаратов, как триметоприм/сульфаметоксазол,
дапсон, изониазид, а также химиотерапевтических средств, используемых в
терапии ВИЧ-ассоциированных опухолей (табл. 10-2);
Патология клеток белой крови
371
- исследование костного мозга для исключения инфильтративных процессов
злокачественного (неходжкинская лимфома и лимфома Ходжкина) или
инфекционного (MAI и парвовирус В19) характера; в частности, присутствие
в исследуемом материале гигантских пронормобластов с ядерными и цито-
плазматическими нарушениями характерно для инфекции, вызываемой пар-
вовирусом В19 (глава 7);
- анализ уровня сывороточного эритропоэтина: при данном показателе ниже
500 ЕД/л необходимо оценить целесообразность дополнения терапии эрйт-
, ропоэтином. Подкожное введение эритропоэтина у ВИЧ-инфицированных
лиц, как свидетельствуют исследования, значительно уменьшает
Выраженность анемии и снижает потребность в переливании крови.
Нужно лечить пациента, а не приводить к норме количестведные параметры
крови. Прибегать к переливанию крови следует лишь в тех случаях, когда налицо
признаки выраженной анемии, указывающие на существенное ухудшение
физиологических функций (снижение толерантности к физической нагрузке, одышка,
стенокардия, головокружение, ортостатические нарушения или застойная
сердечная недостаточность).
Патология клеток белой крови
Лейкопения является обычным следствием ВИЧ-инфекции и наблюдается у 75 %
больных СПИД. Так как применяемые лекарственные препараты,
сопутствующие инфекционные и опухолевые осложнения сами по себе способны вызывать
лейкопению, выбор терапевтической стратегии зачастую крайне затруднен.
Лейкопения может свидетельствовать о поражении как лимфоидного, так и миелоид-
ного ростков кроветворения. Как уже отмечалось, прогрессирующее уменьшение
абсолютного и относительного числа Т-лимфоцитов CD4 — характерная
иммунологическая патология при ВИЧ-инфекции. Снижение уровня CD4 меньше
200/мкл служит важным прогностическим индикатором риска развития тяжелых
оппортунистических инфекций.
К частым осложнениям приема сильнодействующих препаратов,
используемых для лечения ВИЧ, ВИЧ-ассоциированных инфекций или новообразований,
относится гранулоцитопения (табл. 10-2). Однако даже если причина
осложнений кроется лишь в применении данных лекарственных препаратов, при
принятии решения важно учесть риск последствий отмены лекарств, столь
необходимых для лечения других клинических проявлений СПИД. Подавлять миелопоэз
могут диссеминированные инфекции, опухоли или их терапия. Механизмы
прямого воздействия ВИЧ на костный мозг еще только изучаются, и вопрос о том,
инфицированы ли ВИЧ миелоидные клетки-предшественники, пока остается
открытым. Хотя антитела к антигенам нейтрофилов обнаруживаются у 32 %
больных СПИД, присутствие этих антител не коррелирует с наличием или
отсутствием нейтропении. Наряду с уменьшением абсолютного числа клеток были
замечены диспластические изменения миелоидных клеток периферической
крови и костного мозга, а также функциональные дефекты, касающиеся способности
гранулоцитов к фагоцитозу и уничтожению внутриклеточных микроорганизмов.
При первичном обследовании ВИЧ-инфицированного пациента с
лейкопенией необходимо сосредоточить усилия на поиске вторичных и потенциально устра-
372
Глава 10. Гематологические проявления ВИЧ-инфекции
нимых причин миелосупрессии. В частности, важно оценить миелотоксичность
принимаемых пациентом лекарственных препаратов и обсудить возможность их
отмены или замены. Применение некоторых препаратов, таких как ганцикловир
и пириметамин, должно сопровождаться постоянным наблюдением за числом ней-
трофилов (табл. 10-2). Пациентам с пониженным содержанием CD4 (< 200/мкл),
а также с признаками и симптомами системной инфекции должно быть назначено
исследование костного мозга для исключения микобактериальной или грибковой
инвазии либо неопластической инфильтрации костного мозга (например, неходж-
скинская лимфома). Гистохимическое исследование костного мозга дает
возможность диагностировать грибковую и микобактериальную флору и предоставляет
ценную клиническую информацию задолго до выделения положительных гемо-
культур.
В работах некоторых авторов был описан опыт использования миелоидных
колониестимулирующих факторов для лечения ВИЧ-ассоциированной
лейкопении. Быстрое повышение уровня нейтрофилов, моноцитов и эозинофилов в
периферической крови наблюдалось после внутривенного и подкожного применения
ГМ- и Г-КСФ и зависело от дозы введенного препарата. Первоначально считали,
что достигаемый лечебный эффект будет кратковременным. Однако
последующие исследования продемонстрировали, что длительный курс подкожного
введения этих препаратов может приводить к стойкому (от нескольких недель до
нескольких месяцев) повышению числа гранулоцитов. Колониестимулирующие
факторы могут успешно применяться в целях коррекции уровня лейкоцитов у
пациентов со вторичной медикаментозной нейтропенией (например, ганцикловир
при ЦМВ-инфекции или химиотерапевтические препараты при ВИЧ-ассоцииро-
ванных лимфомах). Хотя назначение ГМ- или Г-КСФ повышает толерантность
пациентов к миелосупрессивным терапевтическим средствам, остается неясным
его влияние на репликацию ВИЧ и ВИЧ-специфическую терапию. Исследования
in vitro подтверждают, что использование ГМ-КСФ может увеличивать вирусную
нагрузку, но клиническое значение этих данных пока непонятно.
Оценку эффективности и воздействия гемопоэтических факторов роста на
качество жизни, вирусную нагрузку и выживаемость ВИЧ-инфицированных
пациентов предстоит дать в ходе дальнейших научных изысканий.
Патология тромбоцитов
В отличие от анемии и лейкопении, которые обычно наблюдаются при прогрессиро-
вании ВИЧ-инфекции, тромбоцитопения может встречаться на любой стадии
развития ВИЧ-инфекции и зачастую является ее первым гематологическим
проявлением. По данным отчетов, тромбоцитопения характерна для 3-8 % серопозитивных
пациентов с бессимптомной стадией заболевания и для 30-45 % пациентов со
СПИД. Степень тромбоцитопении обычно находится в пределах от весьма
умеренной до средней при абсолютных значениях от 40 000/мкл до 100 000/мкл.
Встречаются случаи и значительного снижения уровня тромбоцитов — меньше 10 000/мкл.
При исследовании механизмов тромбоцитопении у ВИЧ-инфицированных
пациентов было обнаружено снижение продолжительности жизни тромбоцитов,
изменение цитокинов и факторов роста и наличие (у некоторых пациентов)
циркулирующих и связанных с тромбоцитами иммунных комплексов и антитромбо-
цитарнщх антител. В отдельных работах показано также, что одной из причин
Патология тромбоцитов
373
недостаточности тромбоцитообразования может быть чувствительность мегака-
риоцитов к ВИЧ-инфекции.
Терапевтические возможности коррекции тромбоцитопении у
ВИЧ-инфицированных пациентов разнообразны и включают антиретровирусную терапию,
внутривенное введение у-глобулина, назначение кортикостероидов и спленэкто-
мию. Вместе с тем, подобные методы нередко дают лишь частичный эффект, и ни
одна терапевтическая схема не является универсальной для всех пациентов.
Лечение показано, если число тромбоцитов менее 50 000/мкл, а также при
выраженном кровотечении. Особые формы терапии будут описаны ниже, а основные
подходы к лечению тромбоцитопении у ВИЧ-инфицированных пациентов
представлены на рис. 10-2.
Значительное
кровотечение
Безотлагательное,
включающее один
или несколько
следующих
пунктов
Кортикостероиды
(в высоких
или низких
дозах),
lg в/в, дапсон,
интерферон
При прекращении
кровотечений
см. Отсутствие
симптомов
Отсутствие
симптомов
Уровень тромбоцитов
> 20 000 J
^внимательное
наблюдение
Оптимизация
комбинированной
антиретровирусной
терапии,
вт. ч?с включением
зидовудина
Продолжающееся
сильное
кровотечение
Спленэктомия
и последующее
внутривенное
введение
иммуноглобулина
Уровень тромбоцитов
< 20000
Контроль
частоты
возможных
кровотечений
Применение
низких
доз стероидов
Рис. 10-2. Алгоритм терапии ВИЧ-ассоциированной тромбоцитопении
374
Глава 10. Гематологические проявления ВИЧ-инфекции
Зидовудин. Применение зидовудина может повысить уровень тромбоцитов
приблизительно у половины пациентов с ВИЧ-ассоциированной тромбоцитопе-
нией. Хотя механизм его действия не ясен, однако весьма вероятно, что зидовудин
повышает продукцию тромбоцитов, подавленную ВИЧ, поскольку последний,
как уже упоминалось, выбирает в качестве одной из своих мишеней мегакариоци-
ты. В литературе описано успешное использование и других антиретровирусных
препаратов для лечения тромбоцитопении при ВИЧ-инфекции.
Кортикостероиды. Лечение низкими дозами кортикостероидов может
повысить число тромбоцитов у 40-80 % пациентов, однако длительные ремиссии
встречаются нечасто (10-20 %). Недавние исследования показали, что
краткосрочные курсы терапии высокими дозами дексаметазона (10 мг перорально
четыре раза в сутки, 2-4 дня) повышают уровень тромбоцитов у
ВИЧ-инфицированных пациентов с клинически выраженными кровотечениями и тяжелой
тромбоцитопенией, не поддающимися лечению никакими другими средствами,
включая зидовудин. Несмотря на это, очевидной помехой для использования кор-
тикостероидных препаратов у ВИЧ-инфицированных пациентов является их
способность вызывать дополнительную иммуносупрессию.
Внутривенное введение у-глобулина приводит к быстрому повышению уровня
тромбоцитов (как при классической иммунной тромбоцитопенической пурпуре
ИТП) у большинства (около 90 %) пациентов и является методом выбора в рамках
неотложной помощи или в качестве поддерживающей терапии перед инвазивными
процедурами. В типичном случае эффект непродолжителен и сохраняется в
течение нескольких дней или недель. Лечение с применением анти-Ш1(Б)-иммуногло-
булина также может привести к кратковременному, а иногда и более длительному
повышению уровня тромбоцитов. Это происходит в основном за счет ускорения
удаления ретикуло-эндотелиальной системой эритроцитов, покрытых IgG.
Спленэктомия. Хотя более чем у 90 % пациентов спленэктомия приводит
к нормализации уровня тромбоцитов, однако необходимо учесть те
потенциальные осложнения, которые могут сопровождать эту инвазивную процедуру, и
рассмотреть любую возможность применения более щадящих методов лечения.
К другим вариантам лечения (различным по своей эффективности) можно
отнести облучение селезенки, назначение дапсона, даназола и а-интерферона.
Тромботическая тромбоцитопеническая пурпура (ТТП) является
потенциально фатальным осложнением ВИЧ-инфекции, связанным с развитием тромбо-
цитарных микротромбов в микрососудистом русле. У пациентов наблюдаются
тромбоцитопения, лихорадка, неврологическая симптоматика, микроангиопати-
ческая гемолитическая анемия, почечная патология и пурпура. Патогенез ТТП
у ВИЧ-инфицированных пациентов не изучен. Известно лишь, что аналогичный
синдром у пациентов, не зараженных ВИЧ, развивается чаще всего молниеносно,
поэтому требуется неотложное и энергичное вмешательство с привлечением
плазмафереза (с инфузиями свежезамороженной плазмы или без них), анти-
тромбоцитарных агентов, кортикостероидов или винкристина (глава 5).
Патология свертывания
Антифосфолипидные антитела, обнаруживаемые у 20-70 %
ВИЧ-инфицированных пациентов, часто проявляют свою активность как волчаночные
антикоагулянты (например, вызывая увеличение активированного частичного тромбоплас-
Злокачественные опухоли кроветворной ткани 375
тинового времени [АЧТВ], которое не корригируется последующим
смешиванием с нормальной плазмой и инкубацией при 37 °С). Несмотря на удлинение
АЧТВ, волчаночный антикоагулянт не предрасполагает к кровотечениям и не
является противопоказанием к проведению инвазивных процедур (глава 6). Если
у ВИЧ-отрицательных пациентов волчаночный антикоагулянт может быть
причиной повышенной частоты возникновения тромботических осложнений, то
у ВИЧ-положительных подобная корреляция встречается крайне редко.
Приобретенная недостаточность протеина S встречается у 17-73 %
ВИЧ-инфицированных пациентов и, в отличие от волчаночных антикоагулянтов, иногда
предрасполагает к тромботическим осложнениям. По данным трех
опубликованных отчетов, в 8 из 159 случаев наблюдался низкий уровень содержания в крови
протеина S и сопутствующие этому тромбоэмболические осложнения. Механизм
развития недостаточности протеина S у данных пациентов остался
невыясненным. Однако предполагается, что он связан либо со снижением синтеза белка,
либо с нарушением функции эндотелиальных клеток.
Злокачественные опухоли кроветворной ткани
Некоторые злокачественные опухоли, например неходжкинская лимфома
(НХЛ) и саркома Капоши, более характерны для ВИЧ-инфицированных,
нежели для общей популяции. Рак шейки матки и плосковргеточный рак заднего
прохода (у гомосексуальных мужчин) также чаще развиваются при наличии ВИЧ-
инфекции.
Из всех форм злокачественных опухолей кроветворной ткани НХЛ является,
пожалуй, самой распространенной патологией и встречается в 200 раз чаще
у ВИЧ-инфицированных людей, чем в общей популяции. В отличие от
оппортунистических инфекций СПИД-ассоциированные лимфомы возникают у пациентов
с умеренным уменьшением числа CD4-клеток. В то же время НХЛ, как правило,
относится к довольно поздним осложнениям ВИЧ-инфекции. При обследовании
пациентов обычно выявляется экстранодулярная форма заболевания: с
наибольшей частотой в процесс вовлекаются желудочно-кишечный тракт, костный мозг
и центральная нервная система. Эффективность тех или иных режимов
химиотерапии — в пределах от 24 % до 56 %, но средняя выживаемость, как правило,
незначительна — всего 4-7 мес. Как и для НХЛ, для болезни Ходжкина характерны
повышенная частота развития в период прогрессирования (III и IV стадии), а
также экстранодулярная форма.
Первичная лимфома ЦНС — позднее осложнение ВИЧ-инфекции, имеющее
тенденцию развиваться у пациентов с уровнем CD4-ioieTOK ниже 50 клеток/мкл.
Лучевая терапия может привести к клиническому улучшению, но
продолжительность жизни большинства пациентов остается незначительной, поскольку они
умирают от оппортунистических инфекций.
Лечение СПИД-ассоциированных лимфом сложно в силу иммуносупрессив-
ного воздействия, оказываемого как самим ВИЧ, так и часто тяжело переносимой
системной химиотерапией. Подобные лимфомы характеризуются высокой
злокачественностью и неблагоприятной гистологической картиной. Эффект от
введения низких или средних доз химиотерапевтических препаратов сопоставим с тем,
который наблюдается при стандартных дозах химиотерапии. Хотя использование
гемопоэтических факторов роста может улучшить переносимость химиотерапии,
376
Глава 10. Гематологические проявления ВИЧ-инфекции
остается неясным, влияют ли эти препараты на выживаемость пациентов.
Вероятно, в будущем способы терапии и профилактики ВИЧ-ассоциированных
новообразований будут развиваться на основе объединенных стратегий,
ориентированных на нейтрализацию иммуносупрессивного эффекта ВИЧ-инфекции.
Клинический пример
ВИЧ-инфицированная женщина в возрасте 32 лет поступила в приемное
отделение с жалобами на головокружение и одышку, продолжающиеся
в течение 1 месяца. В анамнезе отмечено значительное кровотечение
из желудочно-кишечного тракта на фоне эндоскопически
подтвержденного гастрита, но при отсутствии каких-либо указаний на выраженную
анемию. За восемь лет до данного обращения был установлен диагноз ВИЧ-
инфекции (инфицирование, обусловленное парентеральным введением
наркотиков). В числе прочих лекарственных препаратов больной были
назначены зидовудин, ЗТС, триметоприм/сульфаметоксазол (ТМП/СМК).
Уровень CD4 — 90 клеток/мкл. Пациентка выглядела бледной и весьма
болезненной. Оральная температура 37 Х. Артериальное давление 120/80
мм рт. ст., пульс ритмичный, 80 уд/мин. Данные объективного
обследования: бледность, безжелтушность склер, живот не напряжен, печень и
селезенка не увеличены, лимфаденопатии нет. Кровь в кале отсутствует.
Данные первичных лабораторных исследований: гемоглобин — 69 г/л;
гематокритное число — 22,8 %; уровень лейкоцитов — 1200/мкл; сегменто-
ядерные нейтрофилы — 60 %; палочкоядерные нейтрофилы — 2 %;
моноциты — 18 %; лимфоциты — 15 %; эозинофилы — 5 %. Уровень
тромбоцитов — 140 000/мкл. Средний объем эритроцитов — 116 мкм\
Вопрос 1. Каковы Ваши первые действия?
Ответ. Необходимо провести оценку стабильности гемодинамики и
убедиться в том, что как в положении стоя, так и в положении сидя изменения
пульса и артериального давления не указывают на наличие ортостатичес-
ких нарушений.
Вопрос 2. Каковы, по Вашему мнению, главные причины развития анемии
и лейкопении у этой пациентки?
Ответ. Недостаточность пролиферативных процессов костного мозга
вследствие инфекций (парвовирус В19, MAI), злокачественной
инфильтрации (лимфома или саркома Капоши) и приема лекарственных
препаратов (зидовудин, ТМП/СМК). Вероятно, поражены эритроцитарные и
лейкоцитарные клетки-предшественники.
Деструкция клеток красной и белой крови в результате запуска
аутоиммунных процессов или побочного действия лекарственных препаратов.
Потеря крови. Причиной возникновения анемии в данном случае может
являться острая или хроническая кровопотеря.
Вопрос 3. Что Вы предполагаете делать дальше?
Ответ. Необходимо тщательно исследовать мазок периферической крови.
Это прежде всего касается полихромазии эритроцитов. (Отсутствие поли-
хромазии свидетельствует о слабой реактивности эритропоэза.) Наличие
сфероцитов может указывать на иммунную деструкцию; фрагментов —
на микроангиопатическую гемолитическую анемию. Макроциты встреча-
Злокачественные опухоли кроветворной ткани ^ 377
ются при весьма различных состояниях, а микроциты или гипохромия —
при недостаточности железа (табл. 3-3). Внимательно следует отнестись и к
патологическим изменениям лейкоцитов и тромбоцитов.
Вопрос 4. Каковы результаты исследования мазка периферической крови?
Ответ. При исследовании мазка периферической крови были выявлены
макроциты и небольшое число полихроматофильных клеток. Не
обнаружено ни микроцитов, ни бледных эритроцитов, ни шистоцитов. Лейкоциты —
в небольшом количестве, но их формула в норме. Гиперсегментированные
нейтрофилы отсутствуют. Уровень тромбоцитов умеренно снижен.
Вопрос 5. Какие другие лабораторные исследования необходимо провести?
Ответ. Можно определить уровни витамина Bi2 и фолатов, активность ЛДГ
в сыворотке, содержание непрямого билирубина, гаптоглобина и провести
тест Кумбса. Все они оказались в норме. Хотя, как это уже было
установлено, содержание ретикулоцитов снизилось до 0.
Вопрос 6. На чем теперь следует заострить внимание?
Ответ. Поскольку проведенные исследования не выявили ни полихромато-
филии (полихромазии), ни ретикулоцитоза, ни каких-либо иных значимых
нарушений, представляется, что наиболее вероятной причиной развития
анемии, а равно и лейкопении у этой пациентки является недостаточная
продуктивность костного мозга.
Вопрос 7. Каковы Ваши дальнейшие действия?
Ответ. Необходимо провести исследование костного мозга с целью
выявления алиментарной недостаточности, инфекций и инфильтрационных
процессов. Такое исследование было выполнено и показало, что при нормальной
клеточности число эритроидных клеток-предшественников недостаточно.
Гигантские пронормобласты отсутствовали. Уровень лейкоцитарных
клеток-предшественников и мегакариоцитов в норме. Парвовирус В19 в
костном мозге и периферической крови не обнаружен. Анализ биоптата костного
мозга не выявил признаков злокачественной инфильтрации.
Вопрос 8. Необходимы ли какие-либо дополнительные исследования?
Ответ. Да. Уровень эритропоэтина (ЭП) восстановился до 27 ME, что
подтвердило эффективность введения экзогенного ЭП. Поэтому было решено
отменить зидовудин и ТМП/СМК и продолжить лечение с помощью ЭП.
Поскольку зидовудин и ТМП/СМК из-за своего супрессорного (и еще
до конца не понятого) воздействия на костный мозг могут способствовать
развитию анемии и лейкопении, необходимо внимательное наблюдение
за каждым пациентом, принимающим подобные препараты.
В дальнейшем пациентка получала другие антиретровирусные препараты
и препараты для профилактики пневмоцистной пневмонии. Уровень гема-
токрита и лейкоцитов нормализовался и стабилизировался. Инъекции ЭП
были отменены. В течение 10 месяце^ после выписки из госпиталя больная
чувствует себя хорошо.
Избранная литература
Aboulafia D. M., Mitsuyasu R. Т. Hematologic abnormalities in AIDS. Hematol Oncol
Clin. North Am., 1991; 5:195-215.
Клинический обзор гематологических проявлений ВИЧ-инфекции.
Canellos G. P., Lister T. A., Sklar J. L. The lymphomas. Philadelphia: W. B. Saunders, 1998.
В соответствующих разделах книги достаточно полно описаны
ВИЧ-ассоциированные лимфомы.
Carpenter С. С. J., Fischl М. A., Hammer S. M. et al. Antiretroviral therapy for HIV
infection in 1997 Updated Recommendatoins of the International AIDS-Society —
USA panel./ЛМЛ, 277:1962-1969,1997.
Современное руководство по использованию антиретровирусных препаратов при
лечении ВИЧ-инфекции.
Centers for Disease Control. First 500000 AIDS cases. U. S. 1995. MMWR, 1995,849-851.
Обзор эпидемиологических данных по ВИЧ-инфекции в США.
Coyle Т. Е. Hematologic complications of human immunodeficiency virus infection and
the acquired immunodeficiency syndrom. Med. Clin. North Am., 81:449-470,1997.
Краткий обзор современных данных.
Fauci A. S., Pantaleo G., Stanley S., Weissman D. Immunopathogenic mechanisms of HIV
infection. Ann. Intern. Med., 1996; 124: 654-663.
Качественный обзор, посвященный патогенезу ВИЧ-инфекции.
Forsyth P. A., DeAngelis L. M. Biology and management of AIDS-associated primary
CNS lymphomas. Hematol. Oncol. Clin. North Am., 1996; 10:1125-1134.
Краткий обзор по биологии и лечению ВИЧ-ассоциированных лимфом ЦНС.
Glaspy J. A., Chap L. The clinical application of recombinant erythropoietin in the HIV-
infected patient. Hematol. Oncol. Clin. North Am., 1994; 8:945-957.
Клинический обзор применения рекомбинантного эритропоэтина при лечении ВИЧ-
инфекции.
Glatt A. E., Anand A. Thrombocytopenia in patients infected with human
immunodeficiency virus: treatment update. Clin. Infect. Dis., 1995; 21:415-423.
Хорошо написанный обзор по лечению ВИЧ-ассоциированной тромбоцитопении.
Hoxie J. A. Hematologic manifestations of AIDS In: Hoffman R., Benz E. J., Shattil S. J.,
Furie В., Cohen H. J., Silberstein L. E., eds. Hematology: Basic Principles and Practice.
New York: Churchill Livingstone; 1995:2171-2200.
Детальное описание гематологической патофизиологии при ВИЧ-инфекции.
Knowles D. M. Etiology and pathogenesis of AIDS-related non-Hodgkin's lymphoma.
Hematol. Oncol. Clin. North Am., 1996; 10:1081-1109.
Обзор НХЛ при СПИД.
Koch M. A., Volberding P. A., Lagakos S. W. et al. toxic effects of zidovudine in
asymptomatic human immunodeficiency virus-infected individuals with CD4 cell
counts of 0,50 x 109/L or less. Arch. Intern. Med., 1992; 152:1992.
Проспективное исследование зидовудина, наглядно демонстрирующее его гематоток-
сические эффекты.
Ramratnam В., Parameswaran J., Elliot В. et al. Short course dexamethasone for
thrombocytopenia in AIDS. Am. J. Med., 1996; 100:117-118.
Обоснование целесообразности стероидной терапии для лечения ВИЧ-ассоциирован-
ной тромбоцитопении, резистентной к зидовудину.
Scadden D. Т. The clinical applications of colony-stimulating factors in acquired
immunodeficincy syndrome. Semin. Hematol, 1992; 29:33-37.
Принципы использования КСФ при СПИД.
Глава 11
Клиническая лабораторная
гематология
Питер Мак-Федран, Натали Ортоли-Дрю
Лабораторная гематология представлена в данной главе серией таблиц:
Таблица 11-1. Рутинные гематологические лабораторные исследования.
Таблица 11-2. Лабораторные исследования при анемии.
Таблица 11 -3. Исследования для диагностики дефицита железа.
Таблица 11-4. Тесты для определения аномальных гемоглобинов.
Таблица 11 -5. Основные исследования при диагностике лейкоза/лимфомы
и миелопролиферативных заболеваний.
Таблица 11-6. Исследование системы свертывания крови (кроме ПВ, АЧТВ).
Таблица 11 -7. Тесты для оценки функции тромбоцитов и тромбоцитопении.
Таблица 11-8. Тесты для выявления гиперкоагуляции.
В таблицах все тесты охарактеризованы по единой схеме: приведено название и
описание, нормальные значения у взрослых (в обычно используемых единицах),
основные цели, метод, приблизительная стоимость и объем работы (в клиничес1
ком госпитале на 800 коек в 1997 г.), а также проблемы, возникающие при
проведении теста. Большинство представленных тестов используется при
гематологическом обследовании больных различного профиля, а также может быть полезно
при обследовании гематологических больных.
Другие исследования, проводимые в лабораториях иного профиля, не
рассматриваются здесь подробно, но упомянуты.
Далее приведены комментарии к таблицам.
Таблица 11-1. Рутинные гематологические лабораторные исследования
(глава 3). Понятие "Общий анализ крови" (OAK) имеет различные значения. В
данном случае под OAK подразумевается совокупность тестов по определению
количества эритроцитов и лейкоцитов, а также размера эритроцитов и их
распределения в соответствии с размером. В нашей лаборатории в целях
оптимизации производственной нагрузки подсчет количества тромбоцитов не
входит в OAK, однако, если при подсчете клеток выявляется аномальное
количество тромбоцитов, это исследование проводится дополнительно. Иной подход
к автоматизированному определению лейкоцитарной формулы. Не называя
это частью OAK, мы проводим его без дополнительной оплаты, если не заказы-
380
Глава 11. Клиническая лабораторная гематология
валось определение лейкоцитарной формулы вручную. Мы расцениваем
автоматизированный подсчет нейтрофилов/гранулоцитов, лимфоцитов и эозино-
филов как эквивалентный мануальному тесту. Точное количество моноцитов
редко бывает важным, тем более что прибор определяет количество мононук-
леаров.
Автоматизированные счетчики клеток обычно предлагают больше эритроци-
тарных индексов, чем перечислено в таблице (или в наших лабораторных
бланках). Мы считаем, что определение среднего клеточного объема эритроцита (СОК)
полезно для дифференциальной диагностики анемии: высокий СОК позволяет
заподозрить ретикулоцитоз, алкогольное поражение печени, недостаточность
фолиевой кислоты, витамина В12 или последствия терапии некоторыми
препаратами. Низкий СОК предполагает недостаточность железа, талассемию, анемию
в результате хронического воспаления и некоторые другие заболевания.
Аномальное распределение эритроцитов по их размеру — более чувствительное, но
менее специфичное исследование при анемиях.
Автоматические счетчики позволяют устанавливать и другие параметры
эритроцитов, включаемые в OAK большинством лабораторий: количество
эритроцитов в 1 микролитре; среднее содержание гемоглобина в эритроците (ССГ),
которое рассчитывают путем деления содержания гемоглобина на количество
эритроцитов; средняя концентрация гемоглобина в эритроцитах (СКГ), которая
характеризует концентрацию гемоглобина в расчете на единицу объема эритро-
цитарной массы (приложение 1). Многие лаборатории включают все эти
показатели в бланки анализов, компьютерные сети и при ответах по телефону. СКГ
может использоваться для контроля качества лабораторных показателей.
Ни один из перечисленных показателей (эритроциты, ССГ, СКГ) в настоящее
время не имеет самостоятельного значения при оценке анемии. Существуют
3 коэффициента пересчета гемоглобина в гематокрит (и обратно), что несколько
избыточно, но некоторые клиницисты опираются на показатели гемоглобина,
тогда как другие — на гематокрит. Иногда обычное соотношение нарушается из-
за значительного гемолиза.
В дополнение к автоматизированному подсчету лейкоцитарной формулы
лаборант может выполнить традиционный подсчет типов лейкоцитов в
окрашенных мазках крови, так называемый ручной дифференциальный анализ
крови. Далеко не все пациенты нуждаются в его проведении, поскольку
автоматизированное дифференциальное определение столь же или более эффективно
для подсчета клеток крови основных типов, прежде всего
нейтрофилов/гранулоцитов, лимфоцитов, эозинофилов, а также определения абсолютного
количества нейтрофилов (АКН). (Автоматизированный анализ предпочтителен для
определения АКН, потому что лабораторный компьютер легко вычисляет его
умножением количества лейкоцитов на % гранулоцитов.) Ручной анализ
необходим, чтобы убедиться в наличии клеточных аномалий (например, бластов,
атипичных лимфоцитов, миелоцитов, метамиелоцитов и клеток Сезари),
внутриклеточных включений (в частности, при малярии) и многих других
необычных клеток (главы 3 и 4). Морфология эритроцитов, включая
морфологические изменения, связанные с гемолизом, может быть хорошо оценена только при
проведении мануального исследования (глава 3). При ручном определении
лейкоцитарной формулы возможно также выявление особенностей сегмента-
381
ции ядер в нейтрофилах. Увеличение количества клеток с несегментированным
ядром (палочкоядерных) ранее расценивалось многими клиницистами как
свидетельство в пользу бактериальной инфекции, хотя современная литература не
подтверждает особой чувствительности или специфичности этого показателя
при бактериальной инвазии. Лейкоцитоз или гранулоцитоз являются столь же
предварительными показателями инфекции, но их определение менее
трудоемко. Гранулоцитоз или нейтрофилез лишь подтверждают диагноз при
инфекциях. Снижение количества гранулоцитов или нейтрофилов называется грануло-
цитопенией или нейтропенией и свидетельствует о высоком риске развития
инфекций у пациента, особенно если АКН менее 500 клеток/мкл. Лимфоцитоз
у взрослых заставляет заподозрить хронический лимфолейкоз. Эозинофилия
характерна для многих патологических состояний, включая аллергию,
некоторые заболевания легких (например, астма), болезни кожи, паразитарные
инвазии и некоторые злокачественные заболевания, в том числе синдром гиперэози-
нофилии. Однако многие вариации лейкоцитарной формулы с трудом
поддаются толкованию, например изменение количества моноцитов или эози-
нопения.
Определение количества тромбоцитов стало широко доступным во многих
лабораториях в 1980-е гг. после усовершенствования фильтрации растворителя
в гематологических счетчиках. Несмотря на стандартизацию оценки заметного
повышения и снижения количества тромбоцитов (глава 5), проблематичной
осталась трактовка пограничных значений, отмечающихся у большого количества
пациентов. Часто возникают сомнения, нужно ли этих пациентов обследовать далее
или достаточно просто наблюдать за ними (например, беременная женщина i
количеством тромбоцитов 125 000/мкл).
Протромбиновое время и активированное частичное тромбопластиновое
время (ПВ и АЧТВ) имеют самостоятельное значение в мониторинге антикоагу-
лянтной терапии (глава 6). Иногда они применяются для "скрининга" пациентов
перед проведением агрессивной процедуры при отсутствии возможности сбора
анамнеза или при наличии эпизодов кровоточивости в прошлом. Они также
предназначены для обследования пациентов с недиагностированными
нарушениями гемостаза. Поскольку почти все факторы свертывания продуцируются
в печени, и ПВ, и АЧТВ будут увеличены при тяжелых заболеваниях печени.
(В этом случае лучше ориентироваться на ПВ, поскольку диапазон разброса
этого показателя у здоровых лиц намного уже.) Желательно знать
чувствительность этих тестов при дефиците определенных факторов свертывания, хотя
большинство лабораторий не имеет такой информации. Например, какая
степень недостаточности фактора VIII приведет к удлинению АЧТВ выше нормы?
(В нашей лаборатории подъем АЧТВ регистрируется при снижении уровня
фактора VIII ниже чем на 25 %.)
Количество ретикулоцитов является ключевым показателем при оценке
анемии, позволяющим дифференцировать наличие или отсутствие адекватного
ответа со стороны костного мозга (глава 3). Но надо помнить, что одинаковое
абсолютное количество ретикулоцитов в микролитре составит различную долю
от разного общего количества эритроцитов. При тяжелой анемии здоровые почки
увеличивают продукцию эритропоэтина, что ведет к выходу ретикулоцитов
из костного мозга в кровь. Эти два эффекта могут создавать иллюзию реакции ко-
382
Глава 11. Клиническая лабораторная гематология
стйого мозга, когда на самом деле ее нет, поэтому некоторые лаборатории
определяют нормализованное количество ретикулоцитов, используя формулы,
представленные в главе 3. Другие лаборатории обходят эти сложности, ориентируясь
только на абсолютное количество ретикулоцитов (глава 3).
Скорость оседания эритроцитов (СОЭ) — наиболее доступный и самый
дешевый тест из многих других для определения реактантов "острой фазы". СОЭ
возрастает при увеличении уровня сывороточного глобулина или фибриногена
в плазме. Эти белки покрывают эритроциты, которые обычно несут на
поверхности отрицательные заряды, так называемый дзета-потенциал. Белковое
покрытие нарушает обычное взаимоотталкивание эритроцитов, из-за чего они легко
связываются друг с другом в форме монетных столбиков. Конгломераты клеток
приобретают критическую массу, плотность которой превышает обычную,
определяющую плавучесть эритроцитов. В итоге клетки быстро падают к основанию
капилляра, что и является причиной увеличения СОЭ. Это неспецифический
тест, но некоторые считают его полезным для выявления скрытых форм
заболеваний и для определения активности хронических воспалительных состояний,
таких как ревматоидный артрит. Его проведение особенно важно для
диагностики и мониторинга преходящего артрита, а также выявления костных метастазов
при распространении лимфогранулематоза. Увеличение СОЭ отмечают у
пожилых пациентов и больных с анемией. Это тест иногда дает ложно низкие
значения у пациентов с серповидно-клеточной анемией, трихинеллезом, при
застойной сердечной недостаточности, диссеминированном внутрисосудистом
свертывании и при низком уровне фибриногена — всех состояниях, при которых
изменяется белковое покрытие эритроцитов, их взаимодействие и/или
взаиморасположение.
Таблица 11-2. Лабораторные исследования при анемии (кроме OAK, подсчета
ретикулоцитов, исследования костного мозга). Протопорфирин эритроцитов
(ПЭ) не связан непосредственно с наличием анемии. Этот тест получил
распространение при идентификации отравления свинцом у детей. С появлением в
последнее время простых методов определения микроколичеств свинца ПЭ реже
используется в этих целях. При дефиците железа, как и при отравлении свинцом,
угнетается синтез гема, что и способствует увеличению ПЭ (обычно определяют
свободный или цинковый протопорфирин — СПЭ или ЦПЭ), хотя этот тест не
очень чувствителен к небольшому (начальному) дефициту железа. В обзоре 1989
года показано, что приблизительно у четверти пациентов с дефицитом железа
уровень ЦПЭ в цельной крови выше 1000 мкг/л. Эти значения более высокие, чем
при каком-либо другом типе анемии, поэтому тест можно использовать для
дифференциальной диагностики анемий.
Витамин В12 и фолиевая кислота определяются при диагностике анемии,
особенно макроцитарного типа, а также для оценки мальабсорбции. Определение
уровня витамина В12 также используют при обследовании пациентов с нейропа-
тией и деменцией. Концентрация фолиевой кислоты в эритроцита*
преимущественно отражает ее содержание в тканях, а уровень фолата в сыворотке в первую
очередь связан с характером принятой недавно пищи. Следовательно, при
диагностике нарушений эритропоэза, обуслрвленных дефицитом фолата, более
существенно определение низкого уровня фолиевой кислоты в эритроцитах. У алкого-
383
ликов с макроцитарной анемией часто встречаются недостаток фолата и
заболевания печени Каждое из этих состояний может быть причиной появления
аномально больших клеток (табл. 3-3). Сам по себе прием алкоголя, как свидетельствуют
данные автоматического анализа крови, ведет к увеличению среднего объема
эритроцитов.
Сывороточный гаптоглобину по-видимому, связывается с гемоглобином,
высвобождаемым при внутрисосудистом гемолизе. В этом случае, а также при
интенсивном внесосудистом гемолизе он не обнаруживается (содержание нулевое)
в сыворотке крови. Отсутствие гаптоглобина в этих ситуациях часто
сопровождается повышением активности ЛДГ (из лизированных эритроцитов) и уровня
непрямого билирубина. Гаптоглобин — реактант острой фазы, содержание которого
резко увеличивается у пациентов с воспалительными процессами.
Эритропоэтин (ЭП) определяют относительно недавно, и пока
информативность этого показателя в дифференциальной диагностике анемии и эритроцитоза
не полностью ясна. Неожиданно низкий уровень ЭП у анемичного больного
указывает на нарушение почечного ЭП-ответа на анемию. В этой ситуации
исследование содержания ЭП может быть более чувствительным тестом на снижение
функции почек, чем определение сывороточного креатинина, и более удобным,
чем оценка клиренса креатинина. С другой стороны, высокое содержание ЭП при
анемии указывает на неспособность костномозговых эритроидных
предшественников реагировать на гипоксическое состояние организма. При эритроцитозе
низкое содержание ЭП свидетельствует о нарушении функции костного мозга,
истинной полицитемии; высокое значение — признак гипоксии, наличия ЭП-сек-
ретирующей опухоли или выраженной анемии.
Определение активности глюкозо-6-фосфатдегидрогеназы (Г-6-ФДГ)
помогает объяснить происхождение гемолиза у пациентов-выходцев из
Средиземноморья или Африки. Гемолиз может быть острым (связан со стрессом при остром
заболевании или с приемом лекарств, таких как противомалярийные средства или
препараты серы) или хроническим и компенсированным. Проверка на дефицит
Г-6-ФДГ также должна быть проведена перед назначением некоторых
потенциально гемолизирующих лекарств, например дапсона и БАЛ. Необходимо
учитывать, что причиной гемолитических состояний бывают и другие, возможно
врожденные, эритроцитарные энзимопении. Второй по частоте эритроцитарной
ферментопатией является дефицит пируваткиназы.
Из тестов на пароксизмальную ночную гемоглобинурию (ПНГ) мануальное
определение сахарозного гемолиза является чрезвычайно простым для
выполнения и столь же чувствительным, как и сложный, отнимающий много времени
тест Хэма или более дорогостоящее определение с помощью проточного цито-
метра отсутствия на эритроцитах пациента CD55, фактора, ускоряющего распад
(ФУР). В большинстве лабораторий лишь изредка регистрируются
положительные результаты какого-либо из этих тестов.
Исследование осмотической резистентности, когда выявляется заметное
увеличение чувствительности эритроцитов пациента к осмотическому лизису,
является золотым стандартом для диагностики наследственного сфероцитоза.
Этот тест чувствителен и при других, более редких, врожденных
гемолитических состояниях, обусловленных аномалиями цитоскелета (стоматоцитоз,
некоторые случаи эллиптоцйтоза/овалоцитоза). Снижение осмотической резистент-
384
Глава 11. Клиническая лабораторная гематология
ности обусловлено тем, что эритроциты имеют слишком тонкую мембрану или
избыточную поверхность мембраны (например, при дефиците железа), и также
наблюдается при состояниях, сопровождающихся присутствием большого
количества мишеневидных эритроцитов (например, гемоглобинопатии или тяжелые
заболевания печени).
При обнаружении низкого уровня гемоглобина традиционно исследуют
концентрацию свободного гемоглобина в сыворотке или плазме крови для оценки
степени внутрисосудистого гемолиза. Главная проблема состоит в том, что
стандартная техника взятия крови и транспортировка образца обычно вызывают
гемолиз in vitro. При необходимости этого исследования кровь следует забирать
в шприц с иглой "бабочка". В пробирку для транспортировки кровь выпускают
на стенку по каплям. После заполнения пробирку закрывают пробкой, которую
фиксируют. Кровь нужно осторожно отнести в лабораторию, где также должны
быть приняты меры по недопущению тряски. Иначе положительный ответ
(повышенный уровень сывороточного гемоглобина) не будет поддаваться
интерпретации.
Гемосидеринурияу или обнаружение "гранул железа" в осадке мочи, является
убедительной демонстрацией хронического внутрисосудистого гемолиза. Это
особенно иллюстративно, если железосодержащие гранулы замечены в
спущенных клетках почечных канальцев.
Таблица 11-3. Исследования для диагностики дефицита железа (глава 3).
Среди множества тестов для выявления недостаточности железа ни один не
является полностью удовлетворительным. Не составляет труда диагностировать
дефицит железа у пациента при гематокрите 25 %, СОК 70 мм3, гипохромии в мазке
крови и содержании ферритина 10 нг/мл. Однако СОК на ранних этапах
недостаточности железа не изменяется, при субъективной оценке мазка возможны
артефакты, а содержание ферритина возрастает (по крайней мере, до
сомнительных значений) при лихорадке или воспалении. Исследование костного мозга
обычно не проводится (из-за болезненности процедуры и высокой стоимости),
и к тсУму же его результаты не вполне специфичны. Костный мозг может быть
истощен вследствие иной причины, нежели железодефицйтная анемия. Если
диагноз недостаточности железа поставлен и проводится лечение, то очень важно
отслеживать содержание гемоглобина или величину гематокрита. Обязательной
является оценка состояния желудочно-кишечного тракта с определением крови
в кале, а также эндоскопией или рентгенологическим исследованием (с
бариевым контрастом) не только у мужчин, но и у женщин, поскольку они теряют
железо не только при менструациях. Дефицит железа у взрослых почти всегда
свидетельствует о потере крови из желудочно-кишечного или урогенитального
тракта. П]3и наличии лихорадки у пациента показателям железа в сыворотке
крови нельзя доверять, поскольку лихорадка может маскироваться под железо-
дефицитную анемию. Показатель содержания железа может изменяться в
течение дня, особенно при воспалении.
Таблица 11-4. Тесты для определения аномальных гемоглобинов (глава 3).
Имеются сотни структурных вариантов гемоглобина f-Халичие многих из них не
вызывает каких-либо симптомов. Гемоглобины, которые могут вызвать или
вызывают проблемы у пациента, можно классифицировать следующим образом. Это
385
гемоглобины, определяющие серповидную форму эритроцитов; гемоглобины,
вызывающие эритроцитоз вследствие высокого сродства к кислороду;
гемоглобины, вызывающие цианоз из-за метгемоглобинемии; нестабильные
гемоглобины, приводящие к гемолизу; гемоглобины С и Е, которые вызывают спленоме-
галию, умеренный гемолиз и микроцитоз. Кроме того, следует упомянуть
талассемию, которая возникает из-за дефицита продукции какой-либо глобино-
вой цепи, что определяет раннюю смертность у гомозигот и развитие микроцитар-
ной анемии у гетерозигот.
Многие из клинически значимых гемоглобинопатии подтверждаются
аномальным уровнем гемоглобина и гематркрита (низкий или реже — высокий),
микроцитозом, обнаружением мишеневидных эритроцитов в мазке крови или
совокупностью этих признаков. Основное и необходимое исследование в
идентификации и оценке большинства гемоглобинопатии — электрофорез гемоглобина
в щелочной среде (обычно рН 8,4 или 8,6). При этом обычно выявляется
аномальная полоска гемоглобина, соответствующая клинически значимой
гемоглобинопатии.
При гемоглобинопатиях с эритроцитозом обычно не определяется
аномальная электрофоретическая подвижность и, как правило, необходимо определить
сродство гемоглобина к кислороду ("р50", или р02, при котором гемоглобин
насыщается кислородом на 50 %). При гемоглобинопатиях других типов
алгоритм диагностики зависит от клинической картины и позиции аномальных
полосок, выявленных при электрофорезе, которые сравниваются с образцами
известных нормальных и вариантных гемоглобинов (например, искусственная
смесь гемоглобинов A, F, S и С). Так, если выявлена полоса, характерная для
гемоглобина S, необходимо сделать еще несколько специальных исследований,
таких как электрофорез гемоглобина в кислой среде (рН 6,2), чтобы
подтвердить серповидно-клеточную анемию. В кислой среде гемоглобин S будет по-
прежнему давать отдельную полосу, в то время как обычно мигрирующие
с ним в других электрофоретических системах гемоглобины (D, Gph) такой
полосы не дают. Альтернативу электрофорезу составляют такие
функциональные исследования, как 1) тест "появления серпа" в метабисульфите натрия
(поиск эритроцитов, изменивших форму в гипоксическом растворе в
присутствии сильного восстановителя) или 2) тест растворимости гемоглобина
в фосфатном буфере (гемоглобин А и большинство других растворимы, а
гемоглобин S — нет).
Приводящий к цианозу метгемоглобин обнаруживается или подтверждается
при спектроскопическом исследовании образца. Нестабильные гемоглобины
определяются электрофоретически и/или при суправитальном окрашивании
на тельца Гейнца. (З-Талассемии идентифицируются при определении
гемоглобинов А2 и F, а-талассемии — при наличии доминантного наследственного микроци-
тоза у пациентов без (3-талассемии (или недостаточности железа) либо при
обнаружении быстро мигрирующего гемоглобина Н. Гемоглобины С и Е встречаются
наиболее часто и легко отличимы от других видов гемоглобина при
электрофорезе в щелочной и кислой среде.
-Л- :■
Таблица 11-5. Основные исследования в диагностике лейкоза/лимфомы и мие-
лопролиферативных заболеваний (глава 9). Первоначально лейкозы выявляют-
386
Глава 11. Клиническая лабораторная гематология
ся при рутинном просмотре мазков периферической крови и костного мозга,
окрашенных по Райту. Идентификация типа острого лейкоза может быть проведена
с помощью проточной цитометрии при использовании моноклональных антител
к поверхностным антигенам определенных линий. Проточная цитометрия
необходима для подтверждения лимфобластного лейкоза. Идентификация миелобла-
стных и моноцитарных лейкозов, а также эритролейкозов может быть проведена
при использовании более дешевых методов цитохимического окрашивания
клеток. Окрашивание помогает дифференцировать острые лейкозы и в соответствии
с FAB-классификацией, что бывает полезно для установления прогноза
заболевания (глава 9). Использование и проточной цитометрии, и цитохимии обычно
излишне, но в ряде случаев может принести пользу.
Часто помогает идентифицировать тип острого лейкоза определение
концентрации лизоцима в сыворотке крови или моче. Уровень лизоцима повышен при
вариантах ОМ Л, особенно при острых моноцитарных лейкозах, но не при лим-
фобластном лейкозе. В таблице перечислены и другие исследования, проведение
которых полезно при различных лейкозах. Цитогенетические исследования
хромосомного аппарата особенно помогают в идентификации промиелоцитарного
лейкоза, лейкоза — лимфомы Беркитта, хронического миелолейкоза, а также
в установлении некоторых прогностически благоприятных и неблагоприятных
подтипов миелобластного лейкоза.
Далее приведены некоторые типы хромосомных аномалий, существенных для
диагностики и прогнозирования течения лейкозов.
Тип лейкоза Кариотип
ХМЛ Филадельфийская хромосома,
или транслокация
9-22(t9;22)(q34;qll)
Острый промие- Транслокация 15-17
лоцитарный (tl5; 17) (q22; ql2)
Лейкоз —лим- Транслокация 8-14
фома Беркитта (t8; 14) (q24; q32)
ОМЛ
Инверсия 16 (р13; q22)
Транслокация 8-21 (q22; q22)
Трисомия 8, делеция 5 или 7
Значение
Диагностика ХМЛ
Диагностика острого
промиелоцитарного
лейкоза
Диагностика лимфомы
Беркитта
Благоприятный прогноз
Неблагоприятный прогноз
Таблица 11-6. Исследование системы свертывания крови (глава 6). Выбор
методов исследования системы свертывания крови обычно продиктован клинической
ситуацией (наличие кровотечения или кровотечение в анамнезе) или
результатами скрининговых коагуляционных тестов, ПВ и АЧТВ. Первичное обследование
при кровоточивости пациента включает ПВ, АЧТВ, определение количества
тромбоцитов и времени кровотечения. Если увеличены ПВ и АЧТВ, то после
проведения подтверждающих повторных анализов следует исследовать функцию
печени, назначить прием витамина К и выполнить другие тесты "панели ДВС":
определение уровня фибриногена, ПДФ (продуктов деградации фибрина) или
387
D-димера. Если удлинено только АЧТВ, то обычно сперва повторяют этот тест и/
или, если образец крови получен от пациента внутривенным катетером,
освобождаются от гепарина in vitro (в нашей лаборатории используют полибрен). Если
удлинение АЧТВ подтверждается как не связанное с гепарином, то плазму
пациента смешивают с нормальной плазмой, после чего исследование повторяют. Если
при соотношении образцов плазмы 1:1 удается добиться коррекции, то это
указывает на недостаточность факторов свертывания (VIII, IX, XI или XII).
Невозможность коррекции АЧТВ при добавлении равного объема нормальной плазмы
указывает на присутствие ингибитора: обычно "волчаночного антикоагулянта",
который иногда вызывает гиперкоагуляционный синдром, или антител к
факторам свертывания, например к фактору VIII. Алгоритмы диагностики
"волчаночного антикоагулянта" и ингибиторов факторов свертывания хорошо разработаны
(рис. 6-18 и 6-19).
Таблица 11-7. Тесты для оценки функции тромбоцитов и тромбоцитопении
(глава 5). У пациентов с дисфункцией тромбоцитов обычно имеется умеренная
кровоточивость. Тромбоциты участвуют в начальном, или "первичном",
гемостазе, прилипая к поврежденным краям сосудов и друг к другу при помощи
плазменного фактора Виллебранда (ФВ) и фибриногена. Известно, что
кровоточивость, обусловленная дисфункцией тромбоцитов, обычно выявляется при
определении длительности кровотечения. Наиболее вероятными причинами
увеличения времени кровотечения (если тромбоцитопения исключена)
являются прием некоторых лекарственных препаратов, особенно аспирина и других
НПВС, а также почечная недостаточность. Если эти факторы исключены,
необходимо исключить влияние другие лекарственных препаратов, которые реже
приводят к этому виду осложнений, например высоких доз препаратов пени-
циллинового ряда и нитропруссида натрия (ниприд). Врожденные дефекты
тромбоцитов, например недостаточность метаболизма этих клеток, приводят
к нарушению их агрегации либо из-за отсутствия АДФ, либо из-за
недостаточности или дисфункции циклооксигеназы, катализирующей синтез тромбоксана,
который участвует в агрегации тромбоцитов. Наследственная недостаточность
специфических тромбоцитарных поверхностных гликопротеинов проявляется
в виде редких рецессивных нарушений функции тромбоцитов: тромбастении
Гланцмана и синдрома Бернара-Сулье. Функция тромбоцитов также
нарушается при снижении уровня фактора Виллебранда и, несколько реже, при
недостатке фибриногена.
Сниженное количество тромбоцитов, или тромбоцитопения, встречается
достаточно часто. Причинами развития этого состояния могут быть лекарственные
препараты, перенесенные инфекции, а также аутоиммунная, или идиопатическая,
тромбоцитопеническая пурпура (ИТП). Тромбоцитопения может быть первым
проявлением гемобластозов, но в таких случаях она редко бывает изолированной,
то есть без анемии или лейкопении.
На этиологическое значение лекарственных препаратов и перенесенных
инфекций в развитии тромбоцитопении могут указывать анамнестические
данные. Прием любых препаратов, начатый в последние 2 месяца и
продолжающийся до настоящего времени, заставляет заподозрить их связь с тромбоцито-
пенией (даже если препарат принимается в течение более длительного срока).
388
Глава 11. Клиническая лабораторная гематология
Причиной тромбоцитопении могут быть и острые инфекции, начавшиеся од-
ной-двумя неделями ранее. Если причины снижения количества тромбоцитов
нуждаются в уточнении, целесообразно выяснить, не увеличилась ли
численность тромбоцитарных клеток-предшественников (мегакариоцитов) в
аспирате костного мозга, что может подтвердить процесс усиленного разрушения
тромбоцитов. Можно также определить наличие антитромбоцитарных
антител, количество незрелых тромбоцитов и выполнить серийный анализ
количества тромбоцитов в течение двух недель при терапии кортикостероидами или
внутривенным углобулином.
К недостаткам исследования костного мозга, несмотря на значительную
информативность, относят болезненность, трудоемкость и высокую
стоимость. Большинство тестов для выявления антитромбоцитарных антител
недостаточно чувствительно и специфично, хотя имеются сообщения, что для
ИТП в 70-80 % случаев характерны IgG- и IgM-антитела, специфически
связывающиеся с гликопротеинами тромбоцитов. Обычно количество
"тромбоцитарных ретикулоцитов" (крупные незрелые тромбоциты) составляет менее
10 % клеток. Если их количество достигает 40-50 %, то это подтверждает
высокую скорость продукции и распада тромбоцитов. Указанные исследования,
особенно наиболее инвазивные из них, становятся чрезвычайно важными,
если снижение количества тромбоцитов представляет серьезную угрозу для
жизни и здоровья.
Тромбоцитопения, развивающаяся в период стационарного лечения,
представляет собой особый случай. Как правило, она является следствием приема
миелосупрессивных химиотерапевтических средств или идиосинкразии к
препарату. Наиболее часто причиной развития тромбоцитопении становится
антикоагулянт гепарин. В нескольких тестах можно выявить антитела к тромбо-
цитарно-гепариноврму комплексу. Мы проводим исследование агрегации
тромбоцитов с гепарином, которое чувствительно только в 50 % случаев, но
весьма специфично. Другой важной причиной развившейся в стационаре
тромбоцитопении является бактериальный сепсис. Тромбоцитопения — более
ранний маркер сепсиса, чем повышенное потребление факторов свертывания
или ДВС, определяемые по увеличению ПВ или низкой концентрации
фибриногена.
Таблица 11-8. Тесты для выявления гиперкоагуляции (глава 6). В настоящее
время такое гематологическое обследование проводится у лиц со склонностью
к венозным тромбозам, у больных с венозными тромбозами, особенно в
молодом возрасте без каких-либо известных факторов риска (ожирение, травма,
застой крови), и обязательно у больных в моменты тромботических осложнений
или при неблагоприятном семейном анамнезе. В этих ситуациях необходимо
оценить резистентность активированного протеина С или уровень фактора V
Лейден, а также дефицит естественных антикоагулянтов (антитромбина III,
протеина С, протеина S). На результаты данных анализов влияют прием
антикоагулянтов, заболевания печени и, возможно, ДВС и активный процесс
свертывания крови. Чрезвычайно важен выбор времени исследования. Гомоцис-
теинемию/урию в последнее время также относят к перечню маркеров
гиперкоагуляции.
389
Если тромботическое состояние является приобретенным и/или протекает
с поражением артерий и центральной нервной системы, то целесообразно
провести поиск антифосфолипидных антител и волчаночного антикоагулянта
с использованием АЧТВ, времени свертывания с ядом гадюки Рассела, а также
антикардиолипиновых антител. Пожилые пациенты с венозными и/или
артериальными поражениями могут иметь "синдром Труссо", или тромбоз, как
признак злокачественного новообразования, который иногда является одним
из наиболее ранних симптомов заболевания.
К другим причинам гиперкоагуляции относятся изменения фибринолитичес-
кой системы, что также можно проверить. Дефицит эндогенного тканевого
активатора плазминогена (ТАП) или избыток естественного ингибитора ТАП
(ИТАП) являются двумя из возможных причин гиперкоагуляции. Дефицит
плазминогена сам по себе тоже может быть причиной гиперкоагуляционного
синдрома.
ТАБ Л И ЦА 11 -1. Рутинные гематологические лабораторные исследования
Нормальные
Тест величины
(для взрослых)
Основное
предназначение
Метод
Количество
анализов,
стоимость1
Возможные проблемы
Общий анализ
крови (OAK),
включая
показатели красной
крови
Гемоглобин:
мужчины
женщины
беременные
Гематокрит:
мужчины
женщины
беременные
Средний объем
клетки (СОК)
Распределение
диаметра
эритроцитов
Количество
лейкоцитов (КЛ)
Дифференциальный
анализ
(автоматический)
135-175 г/л
120-160 г/л
> 100 г/л
41-53%
36-46%
>30%
80-95 мкм3
13±1%
4000-10 000/мкл
См.
"Дифференциальный
анализ
(ручной)"
Трактовка
пониженного и повышенного
количества клеток
крови: анемия, полиците-
мия; лейкопения,
лейкоцитоз. СОК и
распределение диаметра
эритроцитов могут
служить основой для
дифференцирования
анемий
Подсчет гранулоци-
тов, лимфоцитов,
моноцитов и эозинофи-
лов — выявление
аномального
распределения. Обнаружение
признаков бластных
клеток, ядерных эрит-
роидных клеток,
атипичных лимфоцитов
Автоматический
подсчет клеток (вместе со
взятием крови,
разведением и анализом
одного образца крови
занимает около минуты)
на основе импедансно-
го, лазерного и/или
радиочастотного
методов. Дублируется
ручными методами,
включая гемоглобиномет-
рию, спектрофотомет-
рическое определение
цианметгемоглобина,
измерение гематокри-
та, подсчет клеток
крови в камере и
определение лейкоцитарной
формулы
Инструментарий OAK;
простейшая
дифференциальная
диагностика, основанная на
анализе 10 000 клеток.
> 100 в день
(10-20 USD)
> 100 в день
(часть OAK)
Критическим пунктом (что
справедливо в отношении
любого другого
лабораторного исследования)
является корректная
идентификация (регистрация) образца
крови.
При взятии крови из
внутривенного катетера
следует учесть разведение крови
инфузионной жидкостью.
Частичное свертывание
крови приводит к
уменьшению количества клеток,
особенно тромбоцитов. Хо-
лодовые агглютинины
могут быть причиной макро-
цитоза и ложной анемии.
Аномальные клетки не
идентифицируются
Подсчет
абсолютного
количества нейтро-
филов (АКН)
по формуле:
КЛ х % грану-
лоцитов/100
Дифференциальный
анализ (ручной)
Сегментоядер-
ные нейтрофилы
Палочкоядер-
ные нейтрофилы
Лимфоциты
Моноциты
Эозинофилы
Базофилы
Атипичные
лимфоциты
Метамиелоциты
Прочие
Морфология
эритроцитов
при окраске
мазка крови
по Райту
1800-7000/мкл
(значения
характерны для
90 % взрослых)
38-71 %
0-10
14-46
2-15
0-5
0-2
0-7
0-1
отсутствуют
Нормальные
размер, цвет,
форма,
расположение
эритроцитов в
мазке; отсутствие
включений
Диагностика опасной
нейтропении: < 5Q0
клеток/мкл. АКН
обычно придают особое
значение
Поиск признаков
бактериальной или
вирусной инфекции,
аллергии и лейкоза.
Иногда имеет
решающее диагностическое
значение
Поиск причин анемии:
воспаление, дефицит,
гемолиз и пр.
Автоматическое
дифференцирование (им-
педансный, лазерный
или радиочастотный
метод)
Каплю крови
размазывают по предметному
стеклу и окрашивают по
Райту. Классификация
лейкоцитов происходит
при исследовании
мазка под микроскопом.
Наряду с этим
оценивается количество
лейкоцитов и тромбоцитов, а
также
морфологические особенности
эритроцитов
Просмотр мазка крови
на предмет поиска
клеток с аномальными
размерами, цветом,
формой, включениями,
межклеточным
соединением
> 100 в день
(часть OAK)
Если не используется
гематологический счетчик,
подсчитывают вручную
> 100 в день
(10-20 USD)
> 100 вдень
(часть диф-
ференци-
ального
анализа)
Занимает много времени,
поэтомудорог. Несмотря на
то, что увеличение доли па-
лочкоядерных нейтрофи-
лов не является
специфическим маркером
бактериальной инфекции,
достаточно часто используется
клиницистами
Интерпретация полученных
данных достаточно
субъективна. Возможны
многочисленные артефакты при
приготовлении мазка и его
окрашивании.
ТАБЛИЦА 11-1. (Продолжение)
Нормальные
Тест величины
(для взрослых)
Основное
предназначение
Метод
Количество
анализов,
стоимость
Возможные проблемы
Подсчет
тромбоцитов
150-400 тыс
клеток/мкл
Протромбино-
вое время
(ПВ)
13±1 с
Часто используется
при скрининге, хотя
это и не обязательно.
Более необходим при
поиске причины
кровоизлияний,
связанных со значительной
тромбоцитопенией,
или (реже) тромбозов
И ПВ, и АЧТВ
используются для контроля
антикоагулянтной
терапии (ПВ при
введении варфарина,
АЧТВ — гепарина);
для
предварительной оценки состояния
гемостаза; для поиска
причин
кровоточивости при гемофилии. ПВ
также используется
при оценке состояния
пациентов,
страдающих тяжелыми
заболеваниями печени
Автоматический
гематологический анализатор
крови; как запасной
вариант — счетная камера
или оценка мазка крови
(умножив среднее число
тромбоцитов в поле
зрения микроскопа при
увеличении х 1000 на
13 000, получаем число
тромбоцитов в 1 мкл)
Смешиваются декапь-
цифицированная
плазма пациента, хлорид
кальция и тромбоплас-
тин или частичный
тромбопластин; с
помощью
автоматического определителя
времени образования
сгустка оценивается
изменение вязкости. То же
самое можно
выполнить и ручным методом
> 100 вдень
(< 10 USD)
> 100 вдень
(10-20 USD)
В прошлом причиной
ошибок могли быть
находящиеся в растворителе частицы.
В настоящее время
причиной ложного заключения о
наличии тромбоцитопении
могут являться
микросгустки в пробирке
Используют различные
активаторы ПВ, поэтому
чувствительность метода
варьируется. Сравнить ПВ
различных активаторов
помогает международное
нормализованное отношение
(MHO). При использовании
этого метода ПВ пациента
соотносится с
контрольным ПВ. Это отношение
корригируется в
соответствии с чувствительностью
тромбопластина,
используемого в лаборатории,
которую определяют при
помощи международного
индекса чувствительности
Активированное частичное
тромбоплас-
тиновое время
(АЧТВ)
Количество
ретикулоцитов
Скорость
оседания
эритроцитов
(Westergren)
25-38 с
0,6-2,7 % от
количества
эритроцитов.
Процент
ретикулоцитов иногда
переводится в
"индекс
ретикулоцитов" (глава 3)
< 20 мм/ч
Определение типа
анемии: гипо- или ги-
перпролиферативная.
Типер" включает как
пациентов с
кровотечениями, так и
пациентов с гемолизом
Скрининг
заболеваний (особенно
воспалительного
характера). Воспаление
приводит к повышению
уровней фибриногена
и глобулинов
сыворотки, что в свою
очередь способствует
соединению
эритроцитов в виде монетных
столбиков и их
быстрому осаждению
И
автоматизированный, и ручной методы
основаны на
РНК-специфической окраске
мазка крови (новый ме-
тиленовый синий)
Измеряется скорость
оседания клеток крови
в калиброванном
капилляре в течение часа
(МИЧ). MHO позволяет
стандартизировать ПВ
> 100 в день Широкий диапазон нор-
(20-50 USD) мальных значений;
вариации активатора
обусловливают сложность создания
АЧТВ-МНО; могут
возникать ошибки при попадании
в образец крови гепарина
10-100 Ручное исследование до-
в день вольно утомительно и, пло-
(10-20 USD) хо воспроизводимо.
Автоматический анализ
целесообразно проводить при
большом количестве
образцов
10-100 Полученный результат не-
вдень специфичен. Осаждае-
(10-20 USD) мость старой крови (> 4
часов до проведения
анализа) имеет тенденцию
к уменьшению
13десь и в следующих таблицах количество анализов и стоимость тестов даны на примере 800-коечного клинического госпиталя, 1997.
ТАБЛИЦА 11 -2. Лабораторные исследования при анемии (кроме OAK, подсчета ретикулоцитов, исследования костного мозга)
Тест
Нормальные
величины
Основное
предназначение
Метод
Количество
анализов,
стоимость
Возможные проблемы
Протопорфи-
рин
эритроцитов (ПЭ)
Предшественник тема. Он
также известен
как "свободный
протопорфирин
эритроцитов"
(СПЭ). В интак-
тных
эритроцитах
протопорфирин
соединяется с цинком
(ЦПЭ)
Витамин В12
100-280 мкг/л
200-1200
пкг/мл
сыворотки
или плазмы
Исследование
применяется при
отравлении свинцом,
дефиците железа у детей,
подростков и
взрослых
Исследование
дефицита витамина В12 как
возможной причины
анемии, особенно
макроцитарного типа,
а также
неврологических симптомов (ней-
ропатия, деменция).
Обычной причиной
является нарушение
всасывания В12
Содержание
экстрагированного ПЭ
измеряется с помощью флюо-
риметра; быстрое
измерение уровня ЦПЭ
можно провести на
проточном флюориметре,
работая с цельной
кровью
Часто используется
радиоиммунный анализ
(РИА): В12 из сыворотки
пациентов конкурирует
с меченым В12 за сайты
связывающего
вещества, например
внутреннего фактора Кастла
10-100
вдень
(10-20 USD)
1-10 вдень
(20-50 USD)
Уровень ЦПЭ умеренно
повышен при анемии,
связанной с воспалительными
заболеваниями,
сопровождающимися дефицитом
железа. Билирубинемия
может оказаться причиной
ложноположительных
результатов
Исследование имеет
ограниченное диагностическое
значение, но не в
отношении макроцитоза, при
котором оно весьма
информативно, если исключены аль-
тернатиные причины
макроцитоза: применение АЗТ,
заболевания печени, дефицит
фолата, ретикулоциоз и др.
Фолиевая
кислота
в эритроцитах
(предпочтительнее) и
в сыворотке
Гаптоглобин
(белок
сыворотки,
связывающий
свободный
гемоглобин; этот
комплекс
разрушается РЭС)
Эритропоэтин
(гормон,
вырабатываемый
почками и
стимулирующий
эритропоэз в
костном мозге)
> 160нг/мл
> 3 нг/мл
200-2000 мкг/л
гемоглобинсвя-
зывающая
способность
(в большинстве
лабораторий
измеряется
уровень общего
гаптоглобина)
8-21 мЕ/мл
Исследование
дефицита фолата как
причины анемии (как
правило, макроцитарной),
особенно при
недостатке питания у
алкоголиков и нарушениях
всасывания
Исследование
достаточно чувствительно
при гемолизе,
особенно внутрисосудис-
том; гаптоглобин яв-
ляе1-ся также
острофазовым белком
Оценка анемии у
пациентов с нарушением
функции почек и поли-
цитемией (уровень ЭП
в этих случаях снижен).
Часто показатель
повышен при анемиях,
обусловленных
недостаточностью костного
мозга, и
ассоциируется с умеренным мак-
роцитозом
РИА: фолиевая
кислота эритроцитов
пациента конкурирует с фо-
латом, меченным
изотопом, за
соответствующие сайты на белке
молока
1-10 вдень
(20-50 USD)
Сыворотка пациента
смешивается с
избыточным количеством
гемоглобина;
несвязанный гемоглобин
разрушается: уровень
связанного (защищенного
от разрушения)
гемоглобина измеряется
химическим способом
РИА;иммунофермент-
ный анализ (ИФА)
1-10 вдень
(20-50 USD)
1-10 вдень
(50-100
USD)
Уровень фолата в
эритроцитах перелитой крови
сохраняется, что может
создавать ошибочное
представление о нормальном
значении этого показателя
после недавней
трансфузии. Концентрация фолие-
вой кислоты в сыворотке
отражает особенности
диеты последних двух недель
Может быть полезным,
однако преимущественно
выполняется серийно.
Чувствительность и
специфичность метода для
различных типов гемолитических
состояний пока
окончательно не установлена
Значимость для
диагностики анемий определена еще
не в полной мере
ТАБЛИЦА 11 -2. (Продолжение)
Тест
Нормальные
величины
Основное
предназначение
Метод
Количество
анализов,
стоимость
Возможные проблемы
Глкжозо-6-
фосфатде-
гидрогеназа
(Г-6-ФДГ)
Тест на паро-
ксизмальную
ночную гемо-
глобинурию
(ПНГ)
Тест Хэма
Тест
на сахарозный
гемолиз
6-11 МЕ/г
гемоглобина
Нормальные
эритроциты не
поддаются
гемолизу в
тестовых растворах.
При ПНГ
эритроциты гемоли-
зируются
Оценка
эпизодической или устойчивой
гемолитической
анемии, особенно у афро-
американцев и
жителей
Средиземноморья
Определение/подтверждение редкой
причины анемии, панцитопе-
нии, флебитов
В качестве аналитичес- < 1 в день
кой системы служит (120-150
смесь реагентов, явля- USD)
ющихся субстратом для
Г-6-ФДГ гемолизата;
вырабатываемый в
реакции НАДФН
определяют спектрофотомет-
рически по изменению
поглощения при 340 нм.
Доступны и скрининго-
вые тесты
Тест Хэма: эритроциты < 1 в нед
пациента подвергаются (50-100
воздействию компле- USD)
мента (нормальная
сыворотка) в
подкисленном растворе с низкой
ионной силой;
оценивается гемолиз.
Тест на сахарозный ге- < 1 в нед
молиз: кровь пациента (< 10 USD)
помещают в раствор
сахара; оценивается
гемолиз
При африканском типе
дефицита ретикулоцитоз
может имитировать
нормальный результат. Дефицит
Г-б-ФДГ трудно выявить у
гетерозигот
Тест Хэма достаточно
трудоемок; относительная
чувствительность обоих тестов
пока не известна. ПНГ —
редкое заболевание. За год
работы лаборатории может
быть не сделано ни одного
анализа.
Предпочтительнее может оказаться
проточная цитометрия,
позволяющая выявить фактор,
ускоряющий распад (CD55)
Осмотическая
резистентность
эритроцитов (ОРЭ)
(чувствительность
эритроцитов к
гемолизу в серии
гипотонических
растворов)
Свободный ге-
моглобин
плазмы или
сыворотки
Свежие
эритроциты
подвергаются лизису
в растворе
с
концентрацией соли между
0,55 и 0,35%;
эритроциты
после инкубации в
течение 24 ч
при 37 °С
разрушаются при
концентрации
растворов
между 0,7 и 0,3 %
< 40 мг/л
Определение
специфических причин
гемолитической анемии:
сфероцитоз
(приобретенный или
врожденный), гемолитический
эллиптоцитоз и стома-
тоцитоз
Эритроциты пациента
помещаются в
растворы соли понижающихся
концентраций и
наблюдается гемолиз
< 1 в нед
(50-100
USD)
Диагностика внутри-
сосудистого гемолиза
< 1 в нед
(10-20 USD)
Плазма или сыворотка
пациента смешивается
с реагентами,
дающими окрашивание в
присутствии пероксидаз-
ной активности
гемоглобина (например,
0-дианизидином)
Гемосидерин в Никогда не на- Диагностика свежего, Осадок мочи, дающий <1внед
моче
(железосодержащие
гранулы в моче,
особенно в
составе клеток слу-
щенного
почечного эпителия)
блюдается в су-
пернатанте
центрифугата
мочи
обычно персистирую-
щего внутрисосудис-
того гемолиза
реакцию образования
берлинской лазури,
подвергается
микроскопическому
исследованию в целях
обнаружения
железосодержащих гранул
(10-20 USD)
Окрининговые
исследования мазка крови не
позволяют выявить сфероцитоз.
Этот тест (ОРЭ), хотя и
является ручным и трудоемким,
все же более специфичен
Стандартные методики
взятия и транспортировки
образца крови по
пневматическим трубкам могут
вызвать гемолиз in vitro и
повлиять на результаты
теста. Тест бесполезен, когда
не предприняты
специальные меры при взятии и
транспортировке крови
Пробу мочи необходимо
собирать в тщательно
промытый стеклянный сосуд.
Тест всегда положителен
при хроническом внутрисо-
судистом гемолизе
ТАБЛИЦА 11-2. (Окончание)
Тест
Нормальные
величины
Основное
г едназначение
Метод
Количество
анализов,
стоимость
Возможные проблемы
Пируват-
киназа
(скрининговое
определение
активности в
эритроцитах)
Основные
ферменты
эритроцитов
Нормальная
активность — ис-
чезновение
флюоресценции (скрининго-
вый тест Бетле-
ра)
Диагностика
врожденной
недостаточности ПК, являющейся
причиной
гемолитической анемии
Гемолизат крови
пациента смешивают с
реагентами,
устраняющими флюоресценцию
НАДФН в присутствии
ПК
Нормальная ак- Диагностика причин Большое разнообразие
тивность около врожденной гемоли- специфических мето-
15 ферментов тической анемии дов; эти тесты доступ-
эритроцитов ны для
специализированных лабораторий
< 1 в нед Скрининговый тест, выпол-
(50-100 няемый эпизодически и не
USD) обладающий высокой
чувствительностью; при
подозрительных результатах
следует провести
исследование полностью
< 1 в нед Большинство этих тестов
(100 USD) проводится редко, поэтому
они не используются в
небольших лабораториях
Для оценки анемии также могут оказаться важными следующие тесты: креатинин, ЛДГ, билирубин, гормоны щитовидной железы; железо
сыворотки, железосвязывающая способность, ферритин (табл. 11 -3); тест Кумбса, титр холодового агглютинина, метилмалоновая кислота в моче,
антитела к парвовирусу, реже — выживаемость эритроцитов, меченых 51Сг (глава 3).
ТАБЛИЦА 11-3. Исследования для диагностики дефицита железа
Тест
Нормальные
величины
Основное
предназначение
Метод
Количество
анализов,
стоимость
Возможные проблемы
СОК
(табл. 11-1 и
приложение 1)
80-95 мкм3
Распределение диаметра
эритроцитов
(РДЭ)
Морфология
эритроцита
(табл. 11-1;
глава 3)
13±1%
Нормохромные
нормоцитарные
эритроциты;
например,
центральная
бледная область
должна
занимать < Уз
клеточного
диаметра
Один из критериев
начального определения
типа анемии: макро-
цитарная, нормоци-
тарная, микроцитар-
ная
Определение анизо-
цитоза, т. е. вариаций
в размерах
эритроцитов -~
Отклонения от
нормальной формы
эритроцита выявляются
при разных анемиях;
например,
расширенная центральная
бледность, длинные эллип-
тоциты (карандаше-
образные формы) в
сочетании с тромбо-
цитозом характерны
для дефицита железа
Импедансный метод и
метод рассеивания
лазерного пучка
("узкоугольный") являются
автоматизированными
методами диагностики;
в прошлом
рассчитывался на основе гема-
токрита и количества
эритроцитов
Как для СОК:
импедансный метод или метод
рассеивания лазерного
пучка
Капля крови
помещается на предметное
стекло, размазывается и
окрашивается по Райту.
Морфология
эритроцитов оценивается под
микроскопом
> 100 в день
(часть OAK)
см. СОК
> 100 в день
(часть
дифференциального
анализа)
Чувствительность СОК при
результате < 78 для
дефицита железа составляет
около 50 %;
специфичность — около 60 % внутри
нашей популяции (так же
определяются талассемия,
анемия при хронических
заболеваниях)
Увеличение РДЭ не
является чувствителым или
специфичным по отношению
к дефициту железа (спорно)
Оценка гипохромии
субъективна; могут мешать
артефакты
ТАБЛИЦА 11 -3. (Продолжение)
Тест
Нормальные
величины
Основное
предназначение
Метод
Количество
анализов,
стоимость
Возможные проблемы
Кровь в кале
Hemoccult®
и пр.
Протопорфи-
рин
эритроцитов (ПЭ)
(табл.11-2)
Железо
сыворотки
Железосвязы-
вющая
способность
сыворотки (ЖСС)
% насыщения
Отрицательный
(кровь не
обнаружена; стул
коричневый)
100-280 мкг/л
цельной крови
Железо
500-1800 мкг/л
ЖСС 2500-
4500 мкг/л
20-40 %
Диагностика потери
крови из ЖКТ,
являющейся обычной
причиной дефицита
железа. (Дефицит
железа у мужчин обычно
связан с потерей
крови через ЖКТ и
требует проведения
эндоскопии и/или
сканирования, не взирая на
результаты данного
исследования.)
Применяется для
выявления отравления
свинцом и дефицита
железа
Определение
дефицита железа (низкий
уровень железа, высокая
ЖСС, низкий %
насыщения) или
перегрузки железом (высокий
уровень железа,
высокий % насыщения)
Образец кала /юмеща-
ется на бумагу, импрег-
нированную гваяковой
смолой (например,
Hemoccult), и
обрабатывается перекисью.
Синее окрашивание
указывает на
присутствие крови
(активность пероксидазы
крови)
Экстракция ПЭ и флюо-
риметрия; ЦПЭ
измеряется проточным
флюори метром
Автоматический
инструментальный анализ;
определение по
изменению цвета после
добавления хромогена
Феррозим
Часть
общего
физикаль-
ного
обследования:
>100вдень;
может
выполняться
у постели
больного, а
не в
лаборатории
10-100
вдень
(10-20 USD)
1-10 вдень
(10-20 USD)
Тест очень чувствителен;
реакция может быть
инициирована растительной пе-
роксидазой. Кровотечение
из ЖКТ часто носит интер-
миттирующий характер
Уровень умеренно
повышен при анемии, связанной
с хроническими
заболеваниями. Заметное
повышение у незначительного
числа пациентов с дефицитом
железа. Значительный
разброс результатов
Уровень железа сыворотки
и % насыщения постепенно
снижаются у пациентов с
лихорадкой или
воспалением; уровень железа
сыворотки колеблется в
пределах суток; ЖСС повышается
при беременности
Исследование
ЖКТ
для
определения места
кровотечения:
ЭГДС и коло-
носкопия
Ферритин
сыворотки
Исследование
эффективности
проводимого лечения
(через 1-2 нед
или 1 мес после
начала
назначения препаратов
железа
сопоставляется
количество
введенного железа
с гематокритом
и количеством
ретикулоцитов)
Не обнаружены
источники
кровотечения: нет
язв или эрозий,
полипов и рака,
артериовеноз-
ной патологии
12-300 нг/мл
(существуют
некоторые
половые
различия, возможно
объясняющие
более частое
развитие
дефицита железа у
женщин)
Ни гематокрит,
ни количество
ретикулоцитов
не повышаются,
если у пациента
сохраняется
дефицит
железа
Нахождение источни- Фиброскопия верхних
ка кровотечения, яв- и/или нижних отделов
ляющегося причиной ЖКТ, проводимая гаст-
дефицита железа роэнтерологом или
хирургом
Диагностика
дефицита железа и
перегрузки железом. Этот тест
менее лабилен, чем
тест на железо
сыворотки (см.
"Проблемы")
Подтверждение
диагноза железодефицит-
ной анемии по мере ее
коррекции
РИА; альтернативным
является метод "IMX-
сэндвич" с ИФА на
заключительном этапе
325 мг FeS04 3 раза в
день в капсулах за 1 час
до еды. Можно
использовать также глюконат
железа, декстран
железа в/в
1-10 в день Инвазивный, неприятный,
(более 1000 дорогостоящий метод; не
USD) проводится до тех пор, пока
не установлено, что именно
дефицит железа является
причиной анемии
1-10 вдень Повышается при воспале-
(50-100 ниях (но незначительно,
USD) обычно < 50 нг/мл, если у
пациента действительно
имеется дефицит железа);
существенное повышение
при некрозе печени
1 -10 в день Повышение гематокрита во
(20-50 время лечения железом
USD*) может произойти и из-за
других факторов, таких,
например, как лечение
лихорадочного состояния или
ранений. Равно и
причинные факторы могут
сдерживать появление ретику-
лоцитоза даже в случае
наличия дефицита железа
ТАБЛИЦА 11-3. (Окончание)
Тест
Нормальные
величины
Основное
предназначение
Метод
Количество
анализов,
стоимость
Возможные проблемы
Аспират
и/или
биоптат
костного
мозга;
окраска
на железо
В норме железо
присутствует в
умеренном
количестве в
гистиоцитах
костного мозга,
большинство из
которых
разрушается во
время взятия
пробы, оставляя
гранулы
различных
размеров
Подтверждение
наличия запасов железа
(обычно
рассматривается в качестве
определяющего теста). У
пациента не
развивается железодефицит-
ная анемия, пока
запасы железа в
костном мозге не
исчерпаны. Наличие железа в
костном мозге в
любой форме
свидетельствует о том, что
дефицит железа не
является причиной анемии
Окрашивания
берлинской лазурью.
Стандартное гистологическое
окрашивание
< 1 в день
(выполняется при
исследовании
аспирата и/или
биоптата
костного
мозга без
отдельной
оплаты)
Исследование костного
мозга — болезненный и
дорогостоящий метод.
Встречаются ложноположитель-
ные и ложноотрицательные
результаты.
Чувствительность и специфичность не
идеальны
* Стоимость медикаментов и последующих измерений гематокрита и уровня ретикулоцитов.
ТАБЛИЦА 11 -4. Тесты для определения аномальных гемоглобинов
Тест
Нормальные
величины
Основное
предназначение
Метод
Количество
анализов,
стоимость
Возможные проблемы
Электрофорез
гемоглобина
щелочная
среда (рН 8,6),
ацетатцеллю-
лозный, агароз-
ный или акри-
ламидный гель
Электрофорез
гемоглобина
кислая среда
(РН6.2),
агарозный гель
Гемоглобины А,
F и А2 обычно
наблюдаются
именно в таком
порядке в виде
отдельных
полос
Обнаруживаются гемоглобины
F, А и А2.
Диагностика
гемоглобинопатии, способных
вызывать гемолиз,
цианоз, полицитемию
2-й этап
исследования аномальных
гемоглобинов после
щелочного
электрофореза. Отделение HbS от
D и G; С от Е и О Араб.
Выявление отличий
между AS и SS у
новорожденных
Эритроциты
отмываются в солевом растворе,
подвергаются лизису в
среде вода-толуол;
гемоглобин отделяется от
клеточных мембран в
ходе
центрифугирования, наносится на гель,
помещенный в
буферный раствор, и
подвергается воздействию
постоянного
электрического поля в течение 20 и
более минут.
Гемоглобины перемещаются по
направлению к
положительному электроду.
Полосы измеряются с
помощью денситометра
Гемолизат,
приготовленный, как это было
описано выше,
наносится на гель агарозы и
подвергается
воздействию электрического
поля. Гемоглобины в
кислой среде
перемещаются к
отрицательному полюсу
1-10 вдень
(<10USD)
< 1 в день
(20-50 USD)
Если появляются полосы,
свидетельствующие о
присутствии аномального НЬ,
необходимо дальнейшее
исследование (включая
функциональные тесты).
Проблемы могут
возникнуть в том случае, когда
обнаруживается несколько
гемоглобинопатии, многие
из которых клинически
незначимы
В нашем госпитале
групповое тестирование
(отсроченное); точная
идентификация HbS и НЬС
достигается лишь после двукратного
тестирования
ТАБЛИЦА 11 -4. (Продолжение)
Тест
Нормальные
величины
Основное
предназначение
Метод
Количество
анализов,
стоимость
Возможные проблемы
Количественное
определение НЬА2
с помощью
метода
микроколонок
Количественное
определение HbS
с помощью
метода
микроколонок
Определение
серповидных
клеток
< 3,8 %
от общего
гемоглобина
"Нормой"
является отсутствие
HbS; у
пациентов с серповид-
но-клеточной
- анемией гемо-
трансфузии
обычно
проводятся до тех пор,
пока HbS < 50 %
Нет
серповидных клеток
Повышение уровня
НЬА2 — один из
характерных признаков р-та-
лассемии
Отслеживание уровня
HbS у пациентов с
серповидно-клеточной
анемией, получающих
гемотрансфузии
Определение
серповидных эритроцитов
(HbS; реже — НЬС-
Гарлем)
Подготовленная
колонка устанавливается
вертикально. НЬ
наносится сверху и затем
последовательно нали-
ваютбуферы; собирают
фракции гемоглобина;
уровни НЬ определяют,
используя раствор
Драбкина; вычисляют
процент НЬА2
Аналогично методу
определения НЬА2
Эритроциты
смешиваются с сильным
окислителем —■ метабисуль-
фитом натрия, и
просматриваются время от
времени (закрытые на 1
час покровным
стеклом) на предмет
появления характерной
формы
< 1 в день
(20-50 USD)
< 1 вдень
(50-100
USD)
2 в неделю
(стоимость
не
определена)
Умеренно трудоемкое
ручное исследование.
Недостаток гемоглобина
свидетельствует о микроцитозе
нежелезодефицитной
природы (дефицит железа
вызывает ложнонизкие
уровни НЬА2). Вместе с НЬА2
элюируется НЬС
При колоночном способе
воспроизводимость
результатов хуже, нежели при
применении электрофореза
Ложноположительные и
ложноотрицательные
результаты обычны до тех пор,
пока не будет наработан
опыт использования этого
метода. Эхиноциты в
длительно хранившихся
образцах крови могут создавать
путаницу. В случае
положительного результата необ-
Фетальный
гемоглобин
Окрашивание
эритроцитов
с фетальным
гемоглобином
по Клейхауэ-
ру-Бетке
< 2 % у детей в
возрасте
старше 6 месяцев
У взрослых нет
фетальных
эритроцитов
(яркое
окрашивание), это
признак
талассемии
Диагностика большой
и малой талассемии,
включая р-б-типы и
наследственное персис-
тирование фетального
НЬ. Наблюдение за
пациентами с
серповидно-клеточной
анемией, получающими гид-
роксимочевину
(уровень HbF может
увеличиваться до 25 %)
Определение
фетальных эритроцитов в
крови матери;
идентификация эритроцитов
в амниотической
жидкости; изучение
распределения
фетального НЬ среди
эритроцитов. Используется для
RhoD-имуноглобулина
дозирования после
родов
резус-несовместимого плода и для
диагностики
наследственного персисти-
рования фетального
гемоглобина (НПФГ)
Разведенный и
обработанный KCN гемолизат
смешивается с 1/15 (от
объема) 1,2 н NaOH, что
вызывает денатурацию
нефетальных гемогло-
бинов; для
преципитации используют
сульфат аммония; раствор
подвергают
фильтрации. Количество
гемоглобина, осажденного
на фильтре,
сравнивается с количеством
гемоглобина в исходном
материале
Подготовка мазка
крови; НЬ взрослого
избирательно удаляется с
помощью цитратного
буфера. Оставшийся НЬ
окрашивается. В случае
Rh-несовместимости
подсчитывается
фетальный НЬ,
содержащийся в эритроцитах.
При НПФГ оценивается
распределение
фетального НЬ в эритроцитах
< 1 в день
(10-20 USD)
< 1 в день
(10-20 USD)
ходимо дополнительное
контрольное исследование
Необходимо тщательно
соблюдать концентрацию
NaOH и время
Субъективная оценка
распределения количества
фетального НЬ; материалы для
исследования и буфер
должны быть
свежеприготовленными. Необходим
тщательный контроль. Оценка
распределения довольно
утомительна. Ретикулоци-
тоз обусловливает ложно-
позитивный результат
ТАБЛИЦА 11-4. (Окончание)
Тест
Нормальные
величины
Основное
предназначение
Метод
Количество
анализов,
стоимость
Возможные проблемы
Тельца Гейнца,
суправиталь-
ное
окрашивание
эритроцитов
Включения
НЬН, супра-
витальное
окрашивание
Тесты на
нестабильный
гемоглобин:
изопропа-
нольная
или тепловая
преципитация
НЬ
Нет телец
Гейнца
Нет включений
Преципитация
отсутствует;
тестируемый су-
пернатант
содержит тот же
НЬ, что и
необработанный ге-
молизат (в тех
же количествах)
Поиск нестабильных
гемоглобинов
(например, НЬ Кёльн) или
механизмов
окислительного гемолиза
при обнаружении
денатурированных телец
НЬ в эритроцитах
Подтверждение
наличия включений НЬН
(или НЬ Барт)
Поиск одного из 50
гемоглобинов,
являющихся причиной тран-
зиторной анемии при
эпизодическом
гемолизе (см. также тест на
определение телец
Гейнца)
Эритроциты смешива- < 1 в нед
ют с кристаллическим (<10USD)
фиолетовым в
пробирке и инкубируют. Затем
проверяют на наличие
телец Гейнца.
Положительный контроль
производится при
обработке нормальных
эритроцитов ацетил-
фенилгидразином
Эритроциты смешива- < 1 в нед
ют с красителем брил- (< 10-20
лиантовым крезиловым USD)
голубым, инкубируют,
проверяют на наличие
характерных включений
Гемолизат смешивают с < 1 в нед
изопропанолом или ин- (< 20-50
кубируют при 50 вС в те- USD)
чение 3 часов;
оценивают преципитацию НЬ.
Обработанный
гемолизат центрифугируют;
содержание НЬ в супер-
натанте сранивают с
базовым уровнем
Для интерпретации
результатов, равно как и для
проведения контроля,
необходим соответствующий
опыт. Ретикулоцитоз
обусловливает ложнопозитив-
ный результат
Позитивные результаты в
нашей лаборатории
достаточно редки; возможны
проблемы с
интерпретацией результатов
Положительные результаты
редки; строма эритроцитов
может давать запутанную
картину нестабильных НЬ
Сродство
гемоглобина
к кислороду
"р50"
Гемоглобин
насыщен 02
на 50 %
при р02> равном
26 ± 1 мм рт. ст.
Определение
аномальных
гемоглобинов с высоким или
низким сродством к
кислороду,
вызывающих полицитемию или
выраженную анемию
соответственно
Гемолизат, цельный или
разделенный хромато-
графически на
соответствующие фракции
гемоглобина,
выдерживают при различных
уровнях 02. Насыщение НЬ
02 определяют для
каждого уровня р02
< 1 в нед
(стоимость
не
определена)
Слишком редкое
исследование для того, чтобы быть
легко выполнимым
Прочие исследования: изоэлектрическое фокусирование гемоглобинов, Sickledex* — определение серповидного гемоглобина, окончательная
идентификация аномальных гемоглобинов с определением аминокислотных последовательностей, пренатальная диагностика с помощью
ДНК-анализа.
ТАБЛИЦА 11-5. Основные исследования в диагностике лейкоза/лимфомы и миелопролиферативных заболеваний
Тест
Нормальные
величины
Основное
предназначение
Метод
Количество
анализов,
стоимость
Возможные проблемы
Проточная ци-
тометрия
Перестройки
генов Т- и
В-клеток:
Т-клеточный
рецептор или
ген
иммуноглобулина
Нормальные и
аномальные
клетки крови
имеют
характерный набор
поверхностных
антигенов, с
помощью которых
и возможна
идентификация
клеток, их типи-
рование и, если
необходимо,
разделение
Только паттерны
клеток-предшественников.
Отсутствие
аномальных полос,
свидетельствующих о новых
клонах
Диагностика и
идентификация лейкоза и
лимфомы, включая их
подтипы. Диагностика
остаточных явлений
после проведенного
лечения. Оценка
степени восстановления
костного мозга после
трансплантации. Ти-
пирование клеток CD4
и CD8 при
ВИЧ-инфекции и CD34 в
стволовых клетках при
трансплантации костного
мозга. Диагностика
врожденных иммуно-
дефицитов
Диагностика
лимфомы и других лимфоид-
ных новообразований
в крови и костном
мозге
Мононуклеарные клет- 10-100
ки собираются из об- вдень
разцов крови или кост- (более
ного мозга с помощью 100 USD)
сепарации в градиенте
плотности фиколл-ги-
пак или электронным
способом после лизиса
эритроцитов.
Впоследствии они помечаются
одним или несколькими
флюоресцентными мо-
ноклональными
антителами (МКА) и через них
пропускают лазерный
луч. Рассеиваясь, свет
попадает на датчики
флюоресценции, что и
позволяет фиксировать
клетки с различными
поверхностными
антигенами
ДНК экстрагируется из < 1 внед
клеток крови или кост- (более
ного мозга, вырезается 100 USD)
с помощью
стандартного набора из трех эндо-
нуклеаз, разделяется в
геле и оценивается на
наличие дополнитель-
Дорогостоящие реактивы;
серьезное ограничение в
использовании количества
МКА на исследование.
Интерпретация требует опыта
и может быть субъективной
Это новая технология,
требующая дорогостоящего
набора реактивов.
Полученные данные о
генетически измененном клоне не
дают абсолютную
уверенность в диагнозе
новообразования
Лизоцим
(сыворотки
или мочи)
Аспирация
костного мозга
(часто
проводится в
сочетании с биопсией
костного мозга,
проточной ци-
тометрией,
специальным
цитохимическим
окрашиванием,
хромосомным
анализом,
выделением культур)
Сыворотка:
7-14 мкг/мл
Моча: 0-2,5
Полуколичественная
оценка клеточности,
мегакариоци-
тов (3-5 в
центре поля),
соотношения мие-
лоидных и эрит-
роидных клеток
(3-4 : 1 у
взрослых), %
лимфоцитов (< 15), %
плазматических
клеток (< 3).
Полнота
созревания миелоид-
ных и эритроид-
ных клеток.
Наличие железа,
но отсутствие
кольцевых си-
деробластов
Увеличение уровня у
пациента с лейкозом
указывает на нелим-
фоцитарный тип;
значительное
увеличение — на моноцитар-
ный лейкоз. Подъем
уровня наблюдается
также при
туберкулезе, саркоидозе и
болезни Крона
Поиск причин
аномального количества
клеток крови,
доброкачественных и
злокачественных
заболеваний. Изредка можно
определить
начальные стадии
злокачественных процессов,
несмотря на
нормальное количество
клеток; использование
для выделения
культур
ных полос по
сравнению с контролем
Суспензию лиофилизи-
рованных бактерий
("micrococcus lysodek-
tlcus") смешивают с
сывороткой крови или
мочой пациента и
наблюдают степень
осветления по сравнению со
стандартным лизоци-
мом яичного белка
Пробу костного мозга
забирают с помощью
шприца, содержащего
1 % раствор анти
коагулянта ЭДТА; готовят
мазок и окрашивают
красителями Райта-Гимзе
и берлинской глазурью
(раздельно).
Микроскопическое
исследование проводится
гематологом или
патологом
< 1 в нед
(50-100
USD)
1-10 в день
(более
100 USD)
Надежный метод,
ограниченный в своем
применении. Не всегда наблюдается
повышение при ОМЛ
Дискомфорт пациента
может помешать получить
хороший образец; требуется
оперативное искусство.
Критическим пунктом
метода является
приготовление мазка и его
окрашивание. Стоимость полного
анализа может превышать
2000 USD
ТАБЛИЦА 11 -5. (Продолжение)
Тест
Нормальные
величины
Основное
предназначение
Метод
Количество
анализов,
стоимость
Возможные проблемы
Цитохимическое
окрашивание
Для выявления
острого
лейкоза:
Пероксидаза:
Судан В, ШИК,
Эстераза
моноцитов
(например,
а-нафтил-
бутират-
эстераза)
Для волосато-
клеточного
лейкоза:
кислая фосфа-
таза с тартра-
том
Для мастоцито-
за:
толуидин
голубой
Окрашивание
неитрофилов и
их
предшественников
вплоть до про-
миелоцитов.
ШИК также
окрашивает мега-
кариоциты и
тромбоциты.
Окрашивание
моноцитов дает
возможность
увидеть как
моноциты, так и
гистиоциты
костного мозга
Все ядросодер-
жащие клетки
окрашиваются
на кислую фос-
фатазу, окраска
пропадает в
присутствии
тартрата
Изредка
встречаются масто-
циты
Если имеются
соответственно
окрашенные бластные клетки,
можно
идентифицировать тип лейкоза
Диагностика волоса-
токлеточного лейкоза
Диагностика мастоци-
тоза
Мазки костного мозга и
периферической крови
окрашивают разными
методами
(исследование периферической
крови часто
используется при уверенности в
наличии интересующих
врача клеток)
Изофермент кислой
фосфатазы в волосатых
клетках
"сопротивляется" обесцвечиванию
тартратом
Мастоциты костного
мозга окрашиваются в
уникальный пурпурный
цвет
< 1 в день
(20-50 USD)
< 1 в нед
(20-50 USD)
< 1 в нед
(20-50 USD)
При лимфобластном
лейкозе часто окрашивание не
дает результата (в 50%
случаев). Чувствительность
метода связана с числом
бластных клеток.
Интерпретация субъективна
Моноциты могут
окрашиваться также положительно
Мастоциты часто
встречаются и при других
состояниях, к примеру при аплас-
тической анемии
Для ХМЛ и по-
лицитемии:
щелочная фосфа-
таза
лейкоцитов (ЩФЛ)
Способность
связывать
свободный
витамин В12
("транско-
баламины")
Окрашивание
нейтрофилов и
палочкоядер-
ных клеток
периферической
крови; при
подсчете по Kaplow
на 100
нейтрофилов
приходится 13-130
баллов
600-1600 пкг/мл
сыворотки (75-
90 % транско-
баламин II; "R",
или "быстрый",
связывает 9-
25%)
Поддержка диагноза
ХМЛ (низкий уровень
ЩФЛ) и истинной по-
лицитемии (высокий
уровень ЩФЛ)
Оценка уровней важна
для миелопролифера-
тивных заболеваний,
особенно ХМЛ и
истинной полицитемии
Окрашивают мазок
периферической крови,
степень окрашивания
цитоплазмы оценивают
под микроскопом
(полуколичественно 1-4+).
< 1 в день
(20-50 USD)
Сыворотку смешивают
с избытком меченного
изотопом витамина В12;
связанный В12
изолируют и измеряют его
уровень; возможно
фракционирование на ТК I
(повышен при ХМЛ), ТК
II (физиологически
важен) и R-транскобала-
мины (повышен при
истинной полицитемии)
Ограниченные
возможности: использование при ХМЛ
и истинной полицитемии
< 1 в нед
(50-100
USD)
Нечастое исследование,
требующее к тому же
внимания к реактивам и
хорошего навыка. В
значительной степени заменимо
другими методами
is
о
I
ho
го
н
S
S
S
к
Прочие методы: биопсия костного мозга, хромосомное кариотипирование.
ТАБЛИЦА 11 -6. Исследование системы свертывания крови (кроме ПВ, АЧТВ)
Тест
Нормальные
величины
Основное
предназначение
Метод
Количество
анализов,
стоимость
Возможные проблемы
Фибриноген
(субстрат
для каскада
свертывания)
Тромбиновое
время
(свертывание
субстрата
при
непосредственном
взаимодействии с
ферментом)
1,5-3,5 г/л,
повышен во время
беременности и
при
воспалительных
заболеваниях
Подбирается; в
нашей
лаборатории 20 ± 5 с
Скрининг диссемини-
рованного внутрисосу-
дистого свертывания
(ДВС); проверка
достоверности пробы, если
ПВ/АЧТВ стабильно
пролонгированы у
пациентов. Кроме того,
используется для
контроля антикоагулянт-
ной терапии с
использованием Ancrod
(змеиного яда)
Определение
эффекта гепарина,
аномального фибриногена или
продуктов
расщепления фибрина; "лити-
ческое состояние" в
фибринолитической
терапии. Можно
использовать для
контроля устранения
антикоагуляции гепарином
Производная от ПВ
плотность сгустка;
оценивается скорость
образования сгустка или
проводится тест по
методу Клаусса,
основанный на измерении
тромбинового времени
Время образования
сгустка цельной или
разбавленной плазмы
пациента после
добавления разбавленного
бычьего тромбина
10-100 в
день(20-50
USD)
< 1 вдень
(20-50 USD)
Нормальная величина не
исключает ДВС, хотя капля
фибриногена может
оказаться полезной.
Присутствие в пробе
гепарина; ПДФ определяются
и другими методами. Дис-
фибриногенемия редка
и обычно незначима.
Рептилазное Подбирается Диагностика диспро- Время образования
время подобно тром- теинемии; особенно сгустка плазмы пациен-
(заменяет биновому вре- полезен у гепаринизи- та с использованием
тромбиновое мени: 20 ± 5 с рованных пациентов; змеиного яда (прямое
время) гепарин не влияет на воздействие на фибри-
результат ноген)
< 1 в нед Потребность в тесте неве-
(50-100 лика; реагент дорогостоя-
USD) щий; расфасован для
массовых исследований;
поэтому тест выполняется
редко
Продукты
расщепления, или
деградации,
фибрина (ПРФ
или ПДФ),
фрагменты D и Е
(тест
Thrombo-Wellco®)
D-димер
(фрагмент
фибрина,
образовавшийся при
лизисе
фибрина плазмином)
Факторы
свертывания
(II, V, VI, VIII, IX,
X, XI, XII)
< 10мкг/мл
Скрининг ДВС, фиб-
ринолиза
< 0,5 мкг/мл
50-150%
(для
большинства факторов)
50-200 % (VIII)
Скрининг ДВС; кроме
того полезен при
диагностике венозного
тромбоза
Оценка врожденных и
приобретенных
нарушений свертывания
крови
Сыворотка, полученная
из свернувшейся
крови, в специальной
камере смешивается с
частицами латекса,
покрытыми антителами к
фибрину или
фибриногену. Определяется
преципитация
Частицы латекса,
покрытые антителами к D-
димеру, осаждаются на
стеклянной плашке,
если в плазме
присутствует D-димер
Образование сгустка,
установленное по времени
появления
соответствующих изменений вязкости
плазмы с дефицитом
фактора свертывания;
"субстрат"
корригируется или не корригируется
плазмой пациента как
источника тестируемого
фактора свертывания
(автоматическое или
ручное исследование). Если
(например) субстрат для
определения дефицита
фактора VIII образует
сгусток с прежней
скоростью после добавления
плазмы пациента, это
означает наличие дефицита
факторами
1-10 в день Повышено содержание при
(50-100 заболеваниях печени, ак-
USD) тивном тромбозе; иногда
дает ложноположительный
результат, если кровь не
образует сгусток; необходимо
использование
специальной пробирки
< 1 в день Преимущества метода пе-
(20-50 USD) ред тестом на ПДФ не
установлены. Чувствительность
и специфичность при ДВС и
тромбозе не определены
1-10 в день Для проведения официаль-
(50-100 ного исследования необхо-
USD) димы весьма
дорогостоящие коммерческие тест-
системы (оборудование,
субстраты, активаторы).
Интерпретация результатов
требует опыта. , Факторы
плазмы, получаемые из
некоторых лабораторий,
зачастую нестабильны и
неудовлетворительной чистоты
ТАБЛИЦА 11-6. (Продолжение)
Тест
Нормальные
величины
Основное
предназначение
Метод
Количество
анализов,
стоимость
Возможные проблемы
Фактор
свертывания XIII
скрининг-тест
Сгусток
сохраняется в
течение суток
Активность 50-200 %
фактора Вил- от среднего
лебранда (или нормального
активность ко- пула (рис. 6-20)
фактора рис-
тоцетина)
Антиген ФВ
Тоже
Мультимеры
фактора
Виллебранда
Разделение
мультимеров по
молекулярной
массе
электрофорезом в геле
с додецилсуль-
фатом натрия
(ДСН) (рис. 6-20)
Определение
дефицита фактора XIII или
ингибитора ФХЦ1
Диагностика болезни
Виллебранда, при
которой наблюдаемая
активность менее 50 %
Тоже
Дифференцирование
различных типов БВ; в
особенности
диагностика БВ типа lib, при
которой обычное
лечение десмопресси-
ном может быть
противопоказано
Определение стабиль- < 1 в нед
ности сгустка плазмы (50-100
в 5 М растворе моче- USD)
вины.
Сравнение агрегации < 1 в день
отмытых тромбоцитов в (50-100
присутствии ристоце- USD)
тина в растворе
стандартной плазмы с
агрегацией в плазме
пациента
Laurell rocket: определе- < 1 в день
ние локализации преци- (более
питата, образованного 100 USD)
антигеном ФВ плазмы
пациента, при миграции
в агарозном геле,
содержащем анти-ФВ-антите-
ла. Преципитат
образуется в зоне равновесия
ДСН-гельэлектрофо- < 1 в нед
рез с последующим (более
анти-ФВ изотопным ве- 100 USD)
стерн-блотом или ИФА
(табл. 6-5 и 6-6)
Редкое нарушение
Точность подобной
диагностики БВ неизвестна.
Уровень активности ФВ в 40-
50 % обычен и труден для
интерпретации.
Содержание ФВ изменяется в
разное время и может зависеть
от группы крови пациента
См. выше. Тест на антиген
может оказаться
излишним. И антиген, и
активность ФВ могут быть
маркерами острофазовых
реакций. Диапазон
содержания антигена зависит от
группы крови
Референтный
(контрольный) тест (длительный срок
получения результатов);
проводится редко
Микст-тесты,
АЧТВ или ПВ
Титр
ингибитора фактора
свертывания
(обычно
антитела
к фактору VIII)
Определение
гепарина
(анти-ФХа)
Выполняют,
если АЧТВ или
ПВ аномальны
Ингибитор
отсутствует; если
он есть, его
активность
измеряется в
единицах Бетесда
Отсутствует; ан-
тикоагуляцион-
ная терапия
проводится до
уровня 0,2-0,4 Е/мл
обычным не-
фракциониро-
ванным
гепарином; выше —
низкомолекулярным гепарином
Исследование
производится независимо
от того, АЧТВ или ПВ
пролонги ровано
вследствие
недостаточности факторов
свертывания или
присутствия ингибиторов
свертывания
Определение
содержания ингибитора
помогает выбрать
необходимое лечение.
Низкий уровень
ингибитора может быть
"перекрыт" концентратом
фактора свертывания.
Высокий уровнень
необходимо "обойти",
если у пациента
кровоточивость
Контроль гепариновой
антикоагуляции для
пациентов, которым
требуются
неожиданно большие дозы
гепарина для удлинения
АЧТВ, пациентов с вол-
чаночным
антикоагулянтом и пациентов,
получающих
низкомолекулярный гепарин
Оценивается
образование сгустка (АЧТВ или
ПВ) в 1 + 1 смеси
плазмы пациента со
стандартной плазмой. Если
стандартная плазма
корригирует
пролонгированное АЧТВ (или
ПВ), имеется
соответствующий дефицит
Плазма пациента
серийно разводится
стандартной плазмой до тех
пор, пока уровень
исследуемого фактора не
составит 50 %
< 1 вдень
(20-50 USD)
Фактор Ха
смешивается с хромогенным
субстратом, дающим
окрашивание; гепарин
плазмы пациента
препятствует окрашиванию
< 1 в нед
(более
100 USD)
< 1 в нед
(более 20-
50 USD)
Пролонгирование АЧТВ под
воздействием ингибитора
может частично
корректироваться стандартной
плазмой
Длительно выполняемый
метод. Дает ошибочные
результаты, если пациент
получает лечение
Более труден для
выполнения, дорогостоящий, менее
стабильные результаты,
чем при определении АЧТВ.
Стандартная кривая
должна быть построена с тем
типом гепарина, который
получает пациент
ТАБЛИЦА 11-6. (Окончание)
Тест
Нормальные
величины
Основное
предназначение
Метод
Количество
анализов,
стоимость
Возможные проблемы
Время лизиса
эуглобулино-
вых сгустков
(стабильность
сгустка
во фракции
плазмы
пациента)
Антиплазмин
("альфа-2и);
эндогенный
ингибитор
фибринолиза
Сгусток
сохраняется
в течение 3 ч
50-150%
от нормы
Определение фибри-
нолитической
активности плазмы
пациента: в ходе фибриноли-
тической терапии
(стрептокиназа, уро-
киназа, ТАП), при ДВС
или "первичном фиб-
ринолизе" время
лизиса эуглобулина
сокращается
Диагностика
дефицита, являющегося
причиной
кровоточивости; выявление
необычны* причин
кровоточивости на фоне
нормальных
результатов других
исследований
Эуглобулины пациента
преципитируются и
отмываются раствором
до освобождения от
эндогенных ингибиторов
фибринолиза; после
чего оценивается лизис
Плазмин лизирует
искусственный хромоген-
ный субстрат с
образованием окрашивания.
Стандартная плазма и
плазма пациента
сравниваются по
способности ингибировать эту
реакцию
< 1 в нед
(20-50 USD)
< 1 в нед
(50-100
USD)
В этой ситуации желательно
быстро получить результат,
что невозможно
осуществить описанным методом.
Тромбоэластография,
выполняемая для диагностики
фибринолиза, дает
результат значительно быстрее
Проводится редко
ТАБЛИЦА 11-7.
Тест
Тесты для оценки функции тромбоцитов и тромбоцитопении (кроме исследований морфологии тромбоцитов,
костного мозга, определения креатинина, фактора Виллебранда и др.)
Нормальные
величины
Основное
предназначение
Метод
Количество
анализов,
стоимость
Возможные проблемы
Время
кровотечения,
поАйви,
Simplate®
(в качестве
альтернативного метода
используется
коммерческая
система
Surgicutt)
Гепарининду-
цированная
агрегация
тромбоцитов
< 9 мин
(некоторые авторы
приводят более
короткое время)
< 25 %
просветления
обогащенной
тромбоцитами
плазмы (ОТП)
донора после
добавления плазмы
пациента и
гепарина
Скрининговый метод
для диагностики
нарушения функции
тромбоцитов
Определение наличия
у тромбоцитов
антител к гепарину, что
встречается
примерно у 5 % пациентов,
получающих гепарин;
они могут стать
причиной тромбоцитопении
с тромбозом или без
него (ГИТ или ГИТТ)
Царапина или разрез
5 мм длиной и 1 мм
глубиной производится на
ладонной поверхности
предплечья, прокси-
мальнее накладывается
манжета, в которой
создается давление
40 мм рт. ст.
Определяется время полной
остановки кровотечения
Смесь ОТП здорового
донора, бедной
тромбоцитами плазмы
пациента и гепарина:
оценивается степень агрегации
тромбоцитов донора
1 -10 в день Зависит от количества
(20-50 USD) тромбоцитов, температуры
окружающей среды и
состояния кожи пациента,
техники скарификации, прибора
для измерения давления.
Не позволяет точно
прогнозировать кровотечение во
время хирургической
операции
< 1 вдень Низкочувствительный ме-
(более тод, дающий результат
100 USD) лишь у 50 % пациентов с
ГИТ. Тесты на
высвобождение серотонина или АТФ
являются, вероятно, более
чувствительными и
специфичными, а также более
доступными и точными
ТАБЛИЦА 11-7. (Продолжение)
Тест
Нормальные
величины
Основное
предназначение
Метод
Количество
анализов,
стоимость
Возможные проблемы
Агрегация
тромбоцитов
(с коллагеном,
АДФ,
адреналином,
ристоцетином,
арахидоновой
кислотой)
Тромбоксан В2
(сывороточный)
(стабильный
конечный
продукт
метаболизма
арахидоновой
кислоты)
Серотонин
(серотонин
крови; главным
образом
в плотных
гранулах
тромбоцитов)
> 50 %
просветления ОТП
пациента
после
добавления любого
из реактивов
300-700 нг/мл
сыворотки
80-180 нг/мл
крови
Определение наличия
и некоторых
особенностей дисфункции
тромбоцитов
Кровь пациента центри- <1 вдень
фугируют при малых (более
оборотах для приготов- 100 USD)
ления обогащенной
тромбоцитами плазмы
(ОТП); добавляется
соответствующее количество
активатора агрегации;
оценивается агрегация
(по увеличению
светового потока,
пропускаемого через раствор)
Оценка метаболизма Радиоиммунный ана- < 1 вдень
арахидоновой кисло- лиз (РИА) тромбоксана (стоимость
ты; позволяет устано- В2 в сыворотке свернув- неопреде-
вить недостаточность шейся крови пациента лена)
ферментов (циклоок-
сигеназы, тромбок-
сансинтетазы) или
дисфункцию
вследствие лекарственной
терапии
Диагностика наруше- Жидкостная хромато- < 1 в день
ний пула накопления графия под высоким (стоимость
в тромбоцитах (дефи- давлением (ЖХВД) неопреде-
цит плотных гранул) лена)
Требуется
соответствующая аппаратура и
достаточно времени. Результаты
неспецифичны.
Чувствительность и специфичность по
отношению к дисфункции
тромбоцитов не известны
Не является широко
распространенным методом.
Оборудование для РИА
весьма дорого
Используется редко;
увеличивается при приеме
препаратов, ингибирующих
обратный захват серотони-
на, хотя гемостаз может
быть ненарушен (Прозак,
Золофт)
Тест на анти-
тромбоцитар-
ные антитела
Качественное
непрямое
определение
антитромбоци-
тарных
иммуноглобулинов
класса IgG и IgM
в сыворотке
Антитела
к глико-
протеинам
мембраны
тромбоцита:
тест MAIPA,
прямой или
непрямой
Антитромбо-
цитарные
антитела
прямой или
непрямой метод
Ретикулоциты
тромбоцитар-
ного ряда
(аналогичны
ретикулоцитам
эритроцитов)
Нет (видимой)
флюоресценции
Нет антител
Определение антител,
способных вызывать
иммунную тромбоци-
топению (ИТП)
Нет антител
<5%
Дифференциальная
диагностика гипер- и
гипопролифератив-
ных тромбоцитопений
Флюоресцентная
микроскопия: отмытые
донорские тромбоциты
помещаются в
сыворотку пациента, после
чего оценивается
флюоресценция анти-lgG и
анти-lgM
- Вестерн-блот:
нормальные гликопротеи-
ны мембраны
тромбоцитов разделяются в
геле, подвергаются
воздействию плазмы или
сыворотки пациента;
добавляемые антитела
класса IgG и IgM служат
в качестве индикатора
Проточная цитометрия
тромбоцитов донора,
смешанных с
сывороткой или тромбоцитами
пациента; оценка
флюоресценции анти-lgG
Проточная цитометрия:
ОТП пациента сначала
смешивается с
флюоресцентным
красителем (оранжевый тиа-
зол), имеющим
аффинитет к РНК
< 1 в нед
(20-50 и
более USD)
< 1 вдень
(более 100
' USD)
< 1 в нед
(более 100
USD)
< 1 в нед
(более 100
USD)
Чувствительность и
специфичность этих тестов
неизвестна. Методики в
различных учреждениях
недостаточно стандартизированы.
Низкая
производительность. Неясна пригодность
методики при ВИЧ-ИТП
Медленно выполняемый и
редко используемый
метод, однако достойный
внимания и широкого
внедрения
ТАБЛИЦА 11-7. (Окончание)
Тест
Нормальные
величины
Основное
предназначение
Метод
Количество
анализов,
стоимость
Возможные проблемы
Морфология
тромбоцитов
(часть ручного
дифференциального
анализа)
Нормальное
строение:тромбоциты
выглядят как гроздь
пурпурных или
красных гранул.
Конгломераты
< 2 мкм в
диаметре
Диагностика
синдромов: серых
тромбоцитов, Бернара-Сулье и
Мей-Хегглина. Серые
тромбоциты выглядят
бледными,
голубовато-синими и без а-гра-
нул при электронной
микроскопии.
Остальные — гигантского
размера и в
уменьшенном количестве
Анализ тромбоцитов в
мазке крови,
окрашенном по Райту.
См. "Диф- Световая микроскопия не
ференци- считается чувствительным
альный методом. Как правило,
анализ, тромбоциты с нарушенной
ручной" функцией выглядят так же,
как и нормальные
Прочие методы включают: высвобождение 14С-серотонина из тромбоцитов при гепарининдуцированной тромбоцитопении (ГИТ); потребление
протромбина; турникетный тест; тромбоцитарный фактор 4, р-тромбоглобулин поверхностного антигена тромбоцита PLA-1 и антитела к PLA-1.
ТАБЛИЦА 11 -8. Тесты для выявления гиперкоагуляции (кроме исследований АЧТВ, ТВ, микст-тестов)
Тест
Нормальные
величины
Основное
предназначение
Метод
Количество
анализов,
стоимость
Возможные проблемы
Волчаночный
антикоагулянт
(ВА):
коррекция
фосфолипидом
пролонгированного АЧТВ
Антитромбин III
(естественный
антикоагулянт,
ингибирующий
тромбин,
фактор Хаи др.;
активизируется
экзогенным
гепарином и
эндогенными
гепаранами)
Протеин С
(естественный
антикоагулянт,
который при
активации
понижает активность
факторов
свертывания V и VIII)
Нормальное
АЧТВ и время
образования
,сгустка при
добавлении яда
гадюки
Рассела; онднако не
исключает ан-
тикардиолипи-
новые антитела
70-130%
70-130(
Подтверждение
наличия волчаночного
антикоагулянта и анти-
фосфолипидных
антител, обычно связанных
с тромбозом
Диагностика
недостаточности или
дисфункции AT III
В плазму пациента с
пролонгированным
АЧТВ добавляется
плазма с повышенным
количеством фосфоли-
пидов и оценивается
сокращение
пролонгированного АЧТВ
Плазма пациента
используется как анти-
ФХа после смешивания
плазмы с Ха, гепарином
и хромогенным
субстратом
Диагностика недоста- Активированный проте-
точности или дисфун- ин С пациента преобра-
кции протеина С зует хромогенный
субстрат
1-10 вдень
(50-100
USD)
Одна из многих причин
удлинения АЧТВ; увеличение
АЧТВ не помогает выявить
все
< 1 вдень
(100 USD)
< 1 вдень Нарушается (уменьшается)
(100 USD) при заболеваниях печени,
применении
антикоагулянтов, ДВС, тромбозе
ТАБЛИЦА 11-8. (Продолжение)
Тест
Нормальные
величины
Основное
предназначение
Метод
Количество
анализов,
стоимость
Возможные проблемы
Протеин S
(I1S), общий
и свободный
(естественный
антикоагулянт,
кофактор
протеина С; часть
его связана с
комплементом,
часть свободна)
Резистентность к
протеину С, или
резистентность к
активированному
протеину С
(обычно
препятствует
образованию фактора
V Лейден;
последний
идентифицируется с
помощью ПЦР
ДНК пациента)
Антикардио-
липиновые
антитела (АКА)
70-130%
Отношение
> 2,3; 2,0-2,3 -
пограничные
значения
Меньше, чем
в контроле
Диагностика недоста- Преципитация антигена < 1 в день Нарушается (уменьшается)
точности свободного методом Laurell rocket
протеина S до и после связывания
I1S полиэтиленглико-
лем. Гель содержит
антитела к F1S
Диагностика
наиболее частых причин
наследственных
венозных тромбоэмболи-
ческих заболеваний
Диагностика антифос-
фолипидного
синдрома; пациенты с волча-
ночным
антикоагулянтом обычно имеют АКА
Измерение АЧТВ у
пациента выполняется до
и после добавления
активированного
протеина С. АЧТВ после
добавления АПС
увеличивается вдвое или еще
больше. В противном
случае имет место
резистентность к АПС
РИА или ИФА
(более
100 USD)
< 1 вдень
(более
100 USD)
1-10 вдень
(более
100 USD)
при заболеваниях печени,
применении
антикоагулянтов, ДВС, тромбозе
Требует добавления
плазмы с дефицитом фактора V
для функционирования в
присутствии
антикоагулянтов. Это важнейшее из
условий пока не везде
достаточно оценено
Уровни стабильны; после
перенесенных инфекций
содержание АКА обычно
постепенно снижается
("доброкачественны")
Плазминоген
(предшественник плазмина)
Активатор
плазминогена
(АП или ТАП)
(эндогенный)
80-120%
Средняя
величина ТАП
0,3 МЕ/мл;
уровень
увеличивается вдвое
после венозной
компресии
(90 мм рт. ст. в
течение 10 мин)
Диагностика недоста- Плазминоген плазмы
точности или дисфун- активируется стрепто-
кции киназой; плазмин
оказывает воздействие на
хромогенный субстрат
Диагностика
недостаточной реакции
уровня ТАП плазмы на
венозную компрессию;
помогает выявить
нарушение нормального
фибринолиза
Плазма пациента
используется в качестве
источника ТАП для
превращения
плазминогена в плазмин, который,
в свою очередь,
действует на хромогенный
субстрат
< 1 в день Недавно перенесенная
(50 USD) стрептококковая инфекция
может оказаться причиной
появления антистрептоки-
назных антител, которые (в
данном методе) имитируют
отсутствие плазминогена
< 1 в нед Не относится к рутинным
(более методам. Значимость ис-
100 USD) следования принята не
всеми специалистами
Ингибитор
активатора
плазминогена
(ИАП)
< 10 условных
единиц/мл
Диагностика избытка
VIАП, что предполагает
нарушение
нормального фибринолиза
Урокиназа
смешивается с плазмой пациента,
а затем подвергается
воздействию
плазминогена.
Высвободившийся плазмин
смешивается с хромогеном. У
пациентов с
повышенным уровнем ИАП
высвобождается меньше
плазмина
< 1 в нед Острофазовый реактант:
(более воспаление приводит к по-
100 USD) вышению содержания
Кроме того, следует обратить внимание на то, что гематокрит и количество тромбоцитов не повышаются; следует иметь ввиду синдром Труссо
(связанный с раком венозный или артериальный тромбоз) у пациентов старше 50 лет.
Приложение 1
Эритроцитарные индексы
Индекс
Формула
Нормальные значения
1 Средний объем
эритроцита (СОК)
Среднее содержание
гемоглобина
в эритроците (ССГ)
Средняя концентрация
гемоглобина
в эритроцитах (СКГ)
гематокрит (л/л) х 1000
1 кол-во эритроцитов (х 1012/л)
1 гемоглобин (г/л)
1 кол-во эритроцитов (х 1012/л)
1 гемоглобин (г/л)
гематокрит (л/л)
80-95 мкм3
25,4-34,6 пг/клетка
310-360 г/л
концентрата
эритроцитов
1 Измеряется непосредственно автоматическим счетчиком.
Приложение 2
Время жизни клеток крови
Клетки
Эритроциты
Лейкоциты (гранулоциты)
Тромбоциты
Моноциты
Время существования в компартменте
Костный мозг
7,5 дней
14 дней
5 дней
55 ч
Кровь
120 дней
< 1 дня
10 дней
12ч
Ткани
1-2 дня
Приложение 3
Порфирии
При порфириях в первую очередь поражаются нервная система и кожа. Эта группа
метаболических заболеваний, при которых имеются дефекты ферментов
биосинтеза гема, характеризуется избыточным накоплением и экскрецией порфиринов и их
предшественников. Клинические проявления порфирии могут варьироваться от
достаточно ярких до стертых, часто оставаясь непонятными даже для опытных
клиницистов. При многих типах порфирии имеются гематологические нарушения.
На схеме показан путь синтеза гема и указаны места ферментативных
дефектов при различных порфириях.
Глицин + Сукцинил-КоА
I
6-Аминолевулиновая кислота
I
Порфобилиноген
I
(Тетрапирролметан)
Неферментный
Дефект
фермента
Заболевание
АЛК-дегидраза
Порфобилиноген-
дезаминаза
Уропорфириноген I
i
Копропорфириноген I
Уропорфириноген III
j
Копропорфириноген III
I
Протопорфириноген IX
Протопорфирин IX
^-Fe2+
Гем
Уропорфириноген III-
синтаза
(косинтаза)
Уропорфириноген-
декарооксилаза
Копрорфирино-
геноксидаза
Протопорфирино-
геноксидаза
Феррохелатаза
Дефицит
АЛК-дегидразы
Острая
перемежающаяся
порфирия
Врожденная
эритропоэтическая
порфирия
Поздняя кожная
порфирия; гепато-
эритропоэтическая
порфирия
Наследственная
копропорфирия
Смешанная
порфирия
Протопорфирия
Избранная литература
Bloomer J. R., Bonkovsky H. L. The porphyrias. Disease-a-Month, 1984; 3-56.
Desnick R. J., Anderson К. Е. Heme biosynthesis and its disorders: The porphyrias and
endoblastic anemias. In: Hoffman R., Benz E.J. Jr., Shattil S.J. et al. (eds). Hematology.
Basic Principles and Practice, 2nd ed. Churchill-Livingstone; 1995:350-367.
Морфологические
характеристики лейкоцитов
(окраска по Райту)
Тип клеток
Ядро
Размер, Положение Форма Цвет
мкм
1. Гранулоциты:
Миел облает1 10-18
Промиелоцит 12-20
Миелоцит 12-18
Метамиелоцит 10-18
"Юный" или па- 10-16
лочкоядерный
Полиморфно- 10-15
ядерный нейтро-
фил
Полиморфно- 10-15
ядерный эозйно-
фил
Полиморфно- 10-15
ядерный базо-
фил
Эксцентричное или
центральное
Эксцентричное или
центральное
Эксцентричное
Центральное
или
эксцентричное
Центральное
или
эксцентричное
Центральное
или
эксцентричное
Центральное
или
эксцентричное
Центральное
Круглое или
овальное
Круглое или
овальное
Овальное или
с небольшой
выемкой
Толстое
подСветлое
красно-
фиолетовое
Светлое
красно-
фиолетовое
Красно-
фиолетовое
Светло-
ковообразное фиолетовое
или с выемкой
Палочка
одинаковой
толщины
2-5 или
более
сегментов
2-3 сегмента
2-3 сегмента
Светло-
фиолетовое
Темно-
фиолетовое
Сине-
фиолетовое
Сине-
фиолетовое
Приложение 4
Хроматин
Очень нежная
сетка
Нежная
ячеистая сетка, но
нити слегка
утолщены
Тонкий,но
становится
более грубым
Ясно различим
кислый и
щелочной хроматин
Ясно различим
кислый и
щелочной хроматин
Скорее грубый
Грубый
Грубый,
перекрывающийся
с гранулами
Ядерная
мембрана
Очень
тонкая
Тонкая
Неотчетливая
Присутствует
Присутствует
Присутствует
Присутствует
Присутствует
рышки
2-5
2-5
Относительное
количество
Скудное
Умеренное
Редко Умерен-
Нет
Нет
Нет
Нет
Нет
ное
Обильное
Обильное
Обильное
Обильное
Обильное
Цитоплазма
Цвет
Голубая
Голубая
Голубо-
вато-
розо-
вая
Розовая
Розовая
но-розовая
Розовая
Бледно-
розовая
Четкая
пери-
нуклеар
Гранулы
1-
ная зона
Нет
Нет
Нет
Нет
Нет
Нет
Нет
Нет
Нет
Первичные (азу-
ро*-, эозино- или
базофильные)
Первичные + в
нейтрофилах
вторичные (спе-
цифические)
гранулы
Нейтро-,
эозино- или
базофильные
Нейтро-,
эозино- или
базофильные
Четкие, розовые
или
розово-фиолетовые
Крупные, грубые,
однородные по
размерам,
темно-красные,
многочисленные
Крупные, грубые,
однородные,
сине-голубые
430
Приложение 4. Морфологические характеристики лейкоцитов
Ядро
Тип клеток
2. Лимфоциты
z^^^\ Лимфобласт1
Ш
Ш^\ "Зрелый"
^^Р' лимфоцит
Размер,
мкм
10-18
7-18
Положение
Эксцентричное или
центральное
Эксцентричное
Форма
Круглое или
овальное
Круглое или
с небольшой
выемкой
Цвет
Светлое
красно-фиолетовое
Темное
синевато-
красное
§
3. Промоноцит1 12-20
12-20
Макрофаг1 15-80
Эксцентрич- Круглое или Бледное
ное или цент- овальное сине-
ральное с умеренной голубое
выемкой
Эксцентрич- Круглое или
ное или цент- овальное,
ральное с выемкой
или
седловидное
Центральное Удлиненное,
зубчатое или
овальное
Бледное
сине-
фиолетовое
Бледное
сине-
фиолетовое
1У здоровых лиц эти клетки локализуются в костном мозге (обычно не встречаются в мазках
периферической крови). Из: Lea, Febriges. Wintrobe's 8th ed.: Clinical hematology, 1981.
(окраска по Райту) {Продолжение)
431
Хроматин
Умеренно
грубая
неравномерность
структуры
Глыбки
среднего или крупного
размера либо
пикнотический
Нежный
сетчатый, клубочко-
образный или
кружевной
Нежный
сетчатый, клубочко-
образный или
кружевной
Губчатый
Ядерная
мембрана
Довольно
плотная
Плотная
Имеется
Имеется
Различимая,
рышки
1-2
Нет
1-2
Нет
Нет
Относительное
количество
Скудное
Скудное
или
обильное
Умеренное
Изобилие
Обычно
изобилие
Цитоплазма
Цвет
Светло-
голубая
но-голубая,
темно-
голубая
или
изредка
но-розовая
Сероватая
или
мутно- .
голубая
Сероватая
или
мутно-
голубая
Темная
но-голубая
Четкая
пери-
нуклеар-
ная зона
Имеется
Имеется,
если
топлазма
темная
Нет
Нет
Нет
Гранулы
Нет
Нет или мало,
азурофильные
'
Мало, четкие,
сиреневые или
красноватые
Множество,
четкие,
сиреневые или
красновато-голубые
Многочисленные, умеренно
грубые
азурофильные грану-
лы и вакуоли
Предметный
указатель
агАнтитрипсин, 206
агАнтитрипсин, 206
а2-Антиплазмин, 212-213
а2-Антиплазмин, 212-213
а2-Макроглобулин, 212-213
а-Гранулы, 153
Р-Тромбоглобулин, 156-157
Р-Тромбоглобулин, 156-157
В-клетки, 25,138-139
В-лимфоидные фолликулы, 33
В-лимфопоэз, 38
В-лимфоциты, 138-139р2-Интегрины, 126-
127
CD55 фактор ускорения распада, 394
IgA дефицит, 145
^Амиелома, 166-167
IgA, 139
IgD, 139
IgE, 139
IgG, 139
IgG- дефицит, 145
IgM, 139
L-селектины, 126-127
Mycobacterium avium intracellulare, 367, 369-
370
N-формил-пептиды, 126-127
Р-селектины, 126-127,155
A
Абеталипопротеинемия, 104-105
Абсолютное количество нейтрофилов, 381,
385 !
Автоматический подсчет клеток, 385
Агаммаглобулинемия, 12J9-130
Агранулоцитоз. См. также Лекарственная
нейтропения, 293,295-296
детский (синдром Костманна), 300
иммунный и токсический, 295
Агрегация тромбоцитов пятью агентами, 414
Агрегация тромбоцитов, 160
Агрегометрия тромбоцитов, 159-162
Адгезия тромбоцитов
врожденная патология, 161-165
фактор Виллебранда, 233-234
Аденокарцинома слепой кишки, 117-120
Аденокарцинома слепой кишки, 118-120
Адреналин, 160
АДФ, 160
Акантоциты, 79-80
Активаторы плазминогена, 210-213
определение, 420-422
Активация свертывания крови. См. также
Свертывание крови
пути, 195-203
регуляция, 203-213
Активированное частичное тромбопластино-
вое время (АЧТВ), 217-220,382,386
Активность ристоцетинового кофактора,
163-164
Актин, 126-127
Алкоголь, 52
тромбоцитопения вследствие, 172-173
Аллергия, 46
история, 54
Аллогенная трансплантация стволовых
клеток, 307-326
Аллоиммунная тромбоцитопения
новорожденного, 179-180
Альфа-2 тест, 409-410
Альфа-гранулы, 144,153
Альфа-метилдопа, 111
Амегакариоцитарнаятромбоцитопеническая
пурпура, приобретенная, 292-293
Амегакариоцитарная тромбоцитопения,
170-172,304
Амфотерицин В, 368
Анамнез
медицинский семейный, 49
настоящего заболевания, 48
семейный анамнез, 46-48
социальный и индивидуальный, 49-52
Ангиомы, 217
Анемия Даймонда-Блэкфана, 302
Анемия Кули, 93
Анемия при хронических заболеваниях,
100-102
Анемия при хроническом заболевании, 101-
105
Анемия с ретикулоцитозом, 101-105
Анемия Фанкони, 301-302
Анемия(и), 53,75-115
Предметный указатель
433
апластическая, приобретенная, 283-290
гемолитическая, 101-105
гипопролиферативная, 85-102
железодефицитная анемия, 86-90
иммунная гемолитическая, 109-113,
114-115,120
клинические проявления, 75-77
Кули, 93
лабораторные исследования, 76-86
определение, 75
пернициозная дефицит кобаламина, 96-97
с ретикулоцитозом, 101-105
Анкрод, 270-272
Аномалия Мейя-Хегглина, 129,131
Аномалия Олдера-Рейля, 129-130
Анорексия, 56-57
Аносмия, 54
Антигемофильный фактор, 195-200
Антиген волчаночного антикоагулянта, 268-
270
Антигенпрезентирующие клетки, 143
Антикардиолипиновые антитела, 225
тестирование, 421-422
Антикоагулянтная поверхность, 192
Антикоагулянты пероральные, 271-273
мониторинг, 269-273
схема применения, 273
Антикоагулянты, 269-270
Антикоакулянты свойства, 255, 257
Антителозависимая клеточная цитотоксич-
ность, 145
Антитромбин 111 (АТШ, кофактор гепарина
1), 204-206
дефицит, 259-260
определение, 417,421
Антифосфолипидные антитела, 267
у ВИЧ-инфицированных, 374
Антифосфолипидный синдром, 267
тромбофилия вследствие, 267-270
Аплазия, парциальная белоклеточная крови,
297
Апластическая анемия приобретенная, 283-
290
Апластическая анемия средней тяжести, 285
Апластическая анемия средней тяжести, 285
Апластическая анемия, тяжелая, 285
Арахидоновая кислота, 160-162
Артериальный тромб, 253,255,257
Артериальный тромбогенез, 255
факторы риска, 254-257
Артериальный тромбоз, 68
Аспират костного мозга, 393-395
при лейкозе/лимфоме, 403-404
при миелопролиферативных
заболеваниях, 403-404
Аспирин,166
Атаксия-телеангиэктазия, 145-146
Аутоиммунная гемолитическая анемия, 109,
112-114
Аутологичная донация, эритроцитов, 39-40
Ацикловир, 368
Б
Бабезиоз, 84
Бадда-Киари, синдром, 104-105,265
Базофилия, 136
Базофилы, 124,135-136
Базофильная зернистость, 82
Барьерные клетки
в лимфатических узлах, 33
в селезенке, 26
функция, 27-28
Белая пульпа, 26
Белок Бене-Джонса, 142
Беременность
обзор системный, 56-57
тромбоцитопения при, 183-184
Бетесда единица, 225
Болезни обмена, нейтропения от, 299-300
Болезнь "трансплантат против хозяина", 307
определение, 310
острая, 318-321
хроническая, 323-324
Болезнь Виллебранда, 161-164, 233-239,
275-278
тестирование, 223-225
Болезнь Гоше, 135
Болезнь Кристмаса, 232-234
Болезнь перикарда, 67
Болезнь Ходжкина, 351-352
клинические признаки и лечение, 351-352
клинический пример, 357-359
Боль в животе, 56-57
Боль в левом плече, 53
Бурстобразующая единица, эритроидная,
74-75 У
В
Вагинальное кровотечение, 69
Варфарин, 271-275
Венерологический анамнез, 58
Венозный тромбоз
послеоперационный, 265-266
тромбогенез, 255-258
факторы риска, 256
434
Предметный указатель
Венозный тромбогенез, 255,257-259
Веноокклюзионная болезнь после
трансплантации, 316-318
Вилочковая железа, 21,33-34
Вирус Эпштейна-Барр, 145
Витальные признаки, 59
Витамин В, 95-96
Витамин В12 (кобаламин), 388,391-392
дефицит, 93-100
Витамин К, 197-198
активация факторов свертывания, 240-241
дефицит, 239-242,248
Витилиго, 60
Витронектин, 206
ВИЧ-инфекция, 145-147,365-378
гематологические осложнения
лекарственной терапии, 368
клинический пример, 375-378
сниженное количество ретикулоцитов,
клинический пример, 304-305
стадии инфекции, 365-366
факторы риска, 48
Внешний путь, 196-197
Внутренний путь, 196-200
Внутривенный гаммаглобулин, 374
Внутрисердечный тромб, 253-255,257
Внутрисосудистое свертывание диссемини-
рованное (ДВС), 247-252
Воздействие окружающей среды, 49
Возраст, 24
Волосы, 52
Волчаночный антикоагулянт, 225,267
тест на, 417,421-423
тромбофилия, 267-270
Время кровотечения, 157-159
Айви тест, 412-414
Время лизиса эуглобулина, 409-410
Время при разведенной яде гадюки Рассела,
225
Время хранения продуктов крови, 244
Врожденная патология мембран
тромбоцитов, 161-165
Врожденная патолргия пула хранения
тромбоцитов, 164-165
Врожденная патология пула хранения, 164—
165
Врожденные нарушения пула хранения,
164-165
Врожденный дефицит плотных гранул, 164-
165
Врожденный дефицит плотных гранул, 164-
165
Врожденный дискератоз, 300
г
Гамма-глобулин для внутривенного
введения, 374
Ганцикловир, 368
Гаптоглобин сыворотки, 388,391
Гастрэктомия, 48
Гексагональная фаза РЕ, 225
Гексозомонофосфат, 129-130
Гелзолин, 126-127
Гемартроз, 217
при гемофилии А, 230
Гематокрит, 380,385
Гематологические проблемы, клинический
подход, 43-70
Гематомы, 217
при гемофилии А, 230
Гематопоэз, обзор, 53
Гематурия, 217
Гемоглобин(ы), 71-73,380,385
выявление аномалий, 394-397
гемоглобин Н, включения, 397-400
гемоглобин S, количество, 396-397
гемоглобин SC, 114
гемоглобин Е, 114
гемоглобин С кристаллический, 83
гемоглобине, 114
гены, 74
железо в, 81
количественное определение
гемоглобина А2,396-398
кривая диссоциации кислорода, 74
нестабильный, 397^399
свободный в сыворотке или плазме, 390,394
среднее содержание в клетке, 77,385,425
фетальный, 399
Гемоглобинопатии, 106-107,112-114
Гемолиз, 101-102. См. также Анемия с рети-
кулоцитозом
Гемолитические анемии, 101-105. См. также
Анемия с ретикулоцитозом
микроангиопатическая, 114-115,186-187
Гемолитический уремический синдром
(ГУСХ 179-182
Гемопоэз желточного мешка, 19-21
Гемопоэз, 17-40
в миелоидных/лимфоидных компарт-
ментах, 335-336
Гемопоэз, 20-25
Гемопоэтическая дисфункция стволовой
клетки, 170-172
Гемопоэтические факторы роста, 18-19,34-40
Геморрагическая анемия, 101—102
Предметный указатель
435
Геморрагическая болезнь новорожденных,
242
Геморрагические нарушения, 214-254
Геморрагические пятна Сальмона, 64
Геморрой, 69
Гемосидеринурия, 390,394-395
Гемостаз, 145, 191-254. См. также
Тромбоциты в
нормальный, 191-213
Гемостатическая пробка, 191 '
Гемофилия А
лабораторная диагностика, 230-232
лечение, 230-232
наследование и определение состояния
носительства, 227-230
тяжесть течения и геморрагические
проявления, 230
фактора VIII комплекс, 226-229
Гемофилия В, 232-234
Гемоцитобласты, эмбриональная печень, 20-
21
Генетика гематологических заболеваний, 49
Гены глобина, 19
Гепарин
введение и мониторинг, 269-270
иммунная тромбоцитопения, 179
осложнения, 270
продолжительность лечения, 273
свойства, 268-270
Гепарининдуцированная тромбоцитопения
и тромбоз, 270
Гепарининдуцированный остеопороз, 270,
382-383
Гепариноподобные антикоагулянты,
приобретенные, 247
Гепарин-тромбоцитарная агрегация, 414
Гепатомегалия, 68
Гигантские гемангиомы, 60
Гидроксильные радикалы, 126-127
Гинекомастия, 64-65
Гиперкоагуляционные состояния, 254-255
Гиперпролиферативные анемии, 85-102. См.
также Анемии, гиперпролиферативные
Гиперсегментированныеполиморфонуклеа-
ры, 129,131
Гиперспленизм, 31-32
Гиперфибринолитические состояния, 252
Гиперэозинофилия, 135
Гипогаммаглобулинемия, 129-130
приобретенная вариабельная, 145-146
Гипосегментированные полиморфонуклеа-
ры, 129,130
Гипоспленизм, 31-32
Гипотензия, 59
Гипофибринолитические состояния с
тромбозом, 253-254
Гипохлорная кислота, 120,127-128
Гирудин,270
Гистидинобогащенный гликопротеин, 214
Гистиоцитоз X, 136
Главный комплекс гистосовместимости, 143
при аллогенной трансплантации
стволовых клеток, 310-311
Глаза, 54,62-64
Гликанфосфатидилинозитол, 104
Глобин, 74
Глобулин,139-140
Глотка, 55-56
Глутатион, восстановленный, 127
Глюкозо-6-фосфатдегидрогеназа, 104-105
дефицит, 102-104
Голова, 54
Гормональные контрацептивы, 278-279
Гранулоцитарно-макрофагальныйколоние-
стимулирующий фактор, 37-38
клиническое использование, 39-40
Гранулоцитарные клетки эмбриональной
печени, 20-21
Гранулоцитарныйколониестимулирующий
фактор, 37
, клиническое использование, 39-40
Гранулоцитоз, 385
Гранулоцитопения, 53,385. См. также Нейт-
ропения при СПИД, 370-372
Гранулоцитопоэз, 37-38
Гранулоциты
определение, 123
патология, 128-131
Гранулы сидерина, 83
Гранулы, тромбоциты, 153
Грануляция, токсическая, 128-129
Грибковая инфекция связанная со СПИД,
367
Грудная клетка, 64-66
Д
Дакроцит, 82
Дапсон, 368
ДВС-синдром, 181-183,244,247-251
патогенез, 239-244,248
поражение тромбоцитов, 166-167
Дегмацит, 79-80
Десмопрессин
при болезни Виллебранда, 236-237,277
при гемофилии А, 230
436
Предметный указатель
Десны, болезненность, 55-56
Детская иммунная тромбоцитопеническая
пурпура, 176-177
Детские болезни, 48
Детский агранулоцитоз (синдром Костман-
на), 300
Дефекты свертывания крови, 214-254
Дефензины, 128
Дефицит железа, организм в целом, 89
Дефицит миелопероксидазы, 129-130,132—
133
Дефицит накопления альфа-пула, 164-165
Дефицит фактора X, приобретенный, 242-
243
Дефицит факторов свертывания,
наследственность, 238-240
дефицит, 239-240
Дзета-силы, 386-387
Диета, 50-51
Дисфагия, 56
Дисфункция нейтрофильного актина, 129-
130
Дисфункция эрекции, 58
Дисхроматопсия, 63-64
Дифференциальная диагностика, 43-70
Диффузная крупноклеточная лимфома,
354-356
Доброкачественная гаммапатия, 359-360
Доброкачественная нейтропения детей, 298-
299
Доброкачественная семейная лейкопения,
298
Дрепаноцит, 82
Дыхательная система, 55-56
Е
Единица тромбоцитарная, 174
Е-селектины, 126-127
ж
Железо сыворотки, 393-395
Железодефицитная анемия, 86-90
клинический пример, 117-120
Железосвязывающая способность, 89,393
Желтуха, 60
Желудочно-кишечное кровотечение, 56-57,
69
Желудочно-кишечный тракт, 56-57
Живот
обследование, 67
перкуссия, пальпация, 68
з
Заболевание печени, коагулопатия, 242-244
Загрудинная боль, 52
Запор, 56-57
Зидовудин (АЗТ)
гематологический эффект, 366,368-371
клинический пример, 376-378
при СПИД-ассоциированной тромбоци-
топении, 373
Злокачественные заболевания системы
крови, 335-361
при ВИЧ-инфекции, 374
Злокачественные новообразования
гематологические, 335-361. См. также
Злокачественные опухоли опухоли
системы крови
тромбофилия от, 265
Золото, иммунная тромбоцитопения
вследствие, 179-180
Зубчатая клетка, 79
и
Идиопатическая интерстициальная
пневмония, 323
Идиопатическая тромбоцитопеническая
пурпура, 175-179
Изменения вены сетчатки, 63-64
Илеоэктомия, 48
Иммунизация, 49
Иммунная тромбоцитопеническая пурпура
взрослых (хроническая ИТП), 175-179
Иммунная тромбоцитопения потребления,
175-180. См. также Тромбоцитопения,
потребления иммунная
Иммунное восстановление, 324
Иммунные гемолитические анемии, 114-115
лекарственная, 109-110
теплового типа, 111,120-121
холодового типа, 112-113
Иммунодефицит, общий вариабельный, 145
Иммунодефициты, 148. См. также
специфические нарушения
Иммунологический анамнез, 54
Иммуносупрессия при приобретенной апла-
стической анемии, 287
и ТКМ, 287-289
Ингибитор волчаночного антикоагулянта,
225
Ингибитор пути тканевого фактора, 195-
198,207-208
Ингибитор реактивного лизиса мембран,
104-105
Предметный указатель
437
Ингибитор С 1-эстеразы
Ингибиторы
нарушения связанные с, 245
приобретенные, 245-247
Ингибиторы активатора плазминогена, 212—
214
Ингибиторы плазмина, 212-213
Ингибиторы сериновых протеаз
кунины, 208-209
протеин С, 207, 211-212
серпины, 205-206
Ингибиторы фактора II, приобретенные, 246
Инжиниринг аллотрансплантата, 311-313
Интегрины, 126-127
Интерлейкин-11,152-153
Интерлейкин-2, 38
Интерлейкин-3,152-153
Интерлейкин-4, 38
Интерлейкин-5, 38
Интерлейкин-6,38,152-153
Интерлейкин-7,38,152-153
Интерферон, 368
Инфаркт легкого, 65-66
Инфекции с низким числом ретикулоцитов,
304-305
Инфекционная нейтропения, 299
Инфекция(и), 48
гемолиз, 114-115
кожа, 61-62
при аллогенной трансплантации
стволовых клеток, 313-315
тромбоцитопения, 172
Исследование генов иммуноглобулинов,
401-402
Исследование костного мозга
при анемии, 85-86
при приобретенной АА, 399-402
при тромбоцитопении, 169-177
Исследование мазка крови, 217-218
Исследование функции тромбоцитов, 412-
416,421
Исследования гиперкоагуляции, 417-423
Истинная полицитемия, 116, 345
диагностика, 345-346
клиника и лечение, 346-347
к
Калликреин, 197-198
Камни почек, 58
Катал аза, 127
Кашель, 55-56
Кининоген высокомолекулярный, 195-196
Кислая фосфатаза, 403
Кишечные шумы, ослабленные, 67
Классификация Rai хронического лимфо-
бластного лейкоза, 356
Классификация рабочей франко-американо-
британской группы острого миелогенного
лейкоза, 337
рефрактерной анемии (миелодисплазии),
349-351
Клейхауэр-Бетке, 399-400
Клетка Барр, 79-80,104-105
Клетка-мишень
активация, 143-144
в тимусе, 33-34
Т-клетка(и), 82
Клетки Белла, 81
Клетки белой крови, 123-147
аномальные, 127-129,370-372
базофилы, 136-137
В-лимфоциты, 138
продукция антител, 139-141
Т-лимфоциты, 137-144
эозинофилы, 135
Клетки крови
время воспроизведения, 426
клеточный цикл, 17
развитие, гемопоэз, 17-40
клетки стромы, 22-23
Клетки-предшественники, 34-36
Клетки-хелперы, 144
Клеточная фибринолитическая система, 213
Клиническая лабораторная гематология,
379-423
Клинический подход к гематологическим
проблемам,43-70
клинический пример, 117-120
Кобаламин (витамин В12), 93-96
Кодоцит. 81
Кожа, 52,59-62
Кожный кандидоз, 61-62
Койлонихия, 61-62
Коллаген, и агрегации тромбоцитов, 160
Колониеобразующая эритроидная единица
(КОЕ-Э), 74-75
Колониестимулирующие факторы (КСФ),
34-38
Кольцевидные ядра лейкоцитов, 129,131
Кольцевые сидеробласты, 83
Кольцо Каузера-Флейшера, 62
Кольцо Кебота, 83
Компартменты гемопоэза, 23-25
Комплекс стрептокиназного активатора
плазминогена, 210,275
438
Предметный указатель
Кондиционирование, предтрансплантационное
при трансплантации аллогенных
стволовых клеток, 310-311
при трансплантации аутологичных
стволовых клеток, 329
Контроль рождаемости, 58
Конъюнктива, 62
Кортикостероиды, 129-130,374
Костномозговая полость, 22-23
Кость(и), 52-53
Кофактор гепарина 1,205-206
Кофактор гепарина II, 205
Краевая зона, 25-26
Краевая мембранная система, 151
Красная пульпа, 25-26,31
Кривая диссоциации кислорода, 73-74
Кристаллы Шарко-Лейдена, 135
Кровоизлияние в сетчатку, 63-64
Кровоизлияние в стекловидное тело, 63-64
Кровопотеря
анемия, 101-102
клинический случай, 117-120
Кровотечение, 215-216
желудочно-кишечное, 56,69
оценка, 393-394
Кровяное давление, осмотр кожи, 59
Кровяные островки, 20-21
Крупногранулярный лимфоцитоз (Т), 298
Крупногранулярный лимфоцитоз, 298
Кумадин. См. Варфарин
Кумарины, 271-272
Кунины, 208-209
Курение, 52
л
Лабораторная гематология, клиническая,
379-423
тесты на анемию, 388-390
исследования свертывания, 405-413
определение аномального гемоглобина,
394-400
исследования гиперкоагуляции, 417-423
определение дефицита железа, 392-393
исследования при лейкозах, лимфомах и
миелопролиферативных заболеваниях,
396-397,401-402
функция тромбоцитов и тромбоцитопе-
ния, 412-416,421-422
Лабораторная диагностика анемии, 388-390
Лабораторное обследование при
кровотечении, 217-226
Лабораторные исследования при дефиците
железа, 392-393
Лактоферрин, 126-127
Легочная эмболия, 65-66,259
Лейкоз
лабораторные исследования, 396,401-404
острый и хронический, 336
Лейкопения, 53
доброкачественная семейная, 298
клинический пример, 147-148
при СПИД, 370-372
Лейкотриен LTB4,126-127
Лейкоцитоз, 53,385
Лейкоциты
морфологическая характеристика (при
окраске по Райту), 428-431
определение, 123
Лекарственно индуцированная иммунная
тромбоцитопения, 178-180
Лекарство(а), 48, 50-51. См. также
отдельные препараты
поражение тромбоцитов, 165
тромбоцитопения, 171-173
Лентовидная кератопатия, 62
Лечение железом, 393
Лизосомы, 152-153
Лизоцим, 126-127
определение, 401-402
Лимфатические сосуды, 31-32
Лимфатические узлы, 52,64-65
гемопоэз, 31-33
клетки, 31-33
развитие, 22-23
увеличение, 33
функции, 33
Лимфобласты, 431
Лимфома Беркитта, 412-413
Лимфомы, 142,350-357
болезнь Ходжкина, 351-352
лабораторные исследования, 396,401-403
неходжкинская лимфома, 355-363
СП ИД-ассоциированные, 375
стадирование, 351
Лимфопения, 147
Лимфопоэз
в эмбриональном тимусе, 21
иерархическая модель, 18
у взрослых, 22-23
Лимфоцитоз, 53
Лимфоциты, 124,136-137,146
нарушения, 146-147
типы, 144
Лихорадка, 59
Локализованное внутрисосудистое
свертывание, 252
Предметный указатель
439
м
Мазок крови. См. Мазок периферической
крови
Мазок периферической крови, 217-218
общий осмотр, 44-45
при анемии, 76,79-84
Макроглобулинемия Валъденстрема, 166-
167,256
Макроовалоцит, 78
Макрофаг(и), 136-137
в лимфатических узлах, 33
окраска по Райту, 431
повреждения при ДВС-синдроме, 247
Макрофагальные колониестимулирующие
факторы, 37
Макроцитарная анемия, 86,93-100. См.
также Мегалобластная анемия
Макроциты, 78
Мальабсорбция, 56-57
Малярия, 84
Мегакариопоэз, 38
Мегакариоцитопоэз
клеточная биология, 150-151
лекарственная и алкоголь-ассоцирован-
ная супрессия, 172-173
Мегакариоциты, 150
в островках желточного мешка, 20-21
в эмбриональной печени, 20
в эмбриональном костном мозге, 21
тромбоциты и, 155-156
Мегалобластные анемии, 93-100
тромбоцитопения от, 171-172
Мембранные нарушения и гемолитическая
анемия, 101-105
Менструальный анамнез, 56-57
Метамиелоциты, 429-430
Механизм защиты от тромбоза, 256
Миелобласт, 429-430
Миелодисплазия, 96-97
и апластическая анемия, 286
Миелодиспластические синдромы, 265-266,
349-351
патология тромбоцитов, 166-167
тромбоцитопения, 170-172
Миелоидная метаплазия, 345
диагностика, 346
клиника и лечение, 347-348
Миелоидный росток, 336
Миелокахексия/нейтропения с тетраплоид-
ными лейкоцитами, 301-302
Миелопероксидаза, 126-127
Миелопоэз, 21-23
Миелопролиферативные заболевания, 265-
266,345-350
лабораторная диагностика, 396,401-404
патология тромбоцитов, 166-167
Миелофтиз, 170-171
Микроангиопатическая гемолитическая
анемия, 114-115
Микроокружение в гемопоэзе, 18,36-37
Микроцит, 78
Микроцитарные анемии, 85-93
Миозин, 126-127
Множественная миелома, 142,355-357
Модель гемопоэза стволовой клетки, 17-19,
34-38
Молочные железы, 64-65
Моноклональная гаммопатия неизвестной
этиологии, 359-360
Мононуклеоз, инфекционный, 145
Моноцит(ы), 124,136
время воспроизведения, 427
окраска по Райту, 431
Моноцитоз, 136-137
Морфология эритроцитов, 392
окраска по Райту, 381
Моча красная, 56-57
Мочеполовая система, 56-57
Мышцы, 52-53
н
"Надкушенная" клетка, 79-80
Нарушения гемостаза, 214-254
Нарушения различения света, 63-64
Нарушения свертывания крови,
врожденные, 131,226-240
Нарушения свертывания крови,
приобретенные, 239-255
Наследственная гигантская нейтрофилия,
129,131
Наследственная гипергомоцистеинемия,
240,261-263
Наследственные дефициты факторов
свертывания, 227-229
Наследственный пиропойкилоцитоз, 103-
104
Наследственный сфероцитоз, 101-104
Наследственный эллиптоцитоз, 103-104
Натуральные киллеры (НК), 145
Неврологическое обследование, 69,132-135
Недостаточность адгезии лейкоцитов, 129-
130
Неиммунная, не связанная с потреблением
тромбоцитопения, 179-183
Нейропатия зрительного нерва, 63-64
440
Нейтропения наследственная/врожденная,
300-302
Нейтропения приобретенная, 292-299
Нейтропения с тетраплоидными
лейкоцитами, 301-302
Нейтропения, 132-135
вследствие метаболических нарушений,
299-300
дифференциальная диагностика, 294
инфекционная, 299
Нейтрофилез, 132-134
наследственный гигантский, 129,131
Нейтрофилы, 123-134
Некротическая кожа, 61-62
Нервная система, 58
Нестероидные противовоспалительные
средства, 165
Неходжкинская лимфома, 352-357
при ВИЧ-инфекции, 376
Низкоклеточная панцитопения, 286
Нитросиний тетразолий, 129-130
НК-БГЛ-лейкоз, 298
Нормобласты, 20-21
Нормоцитарная анемия, 85,86,101-103
Носовое кровотечение, 54,217
Нулевые клетки, 144
о
Обследование гениталий, 68-69
Обследование зрачка, 62
Обследование конечностей, 68
Обследование яремной вены, 65-66
Обследование/пальпация стенки грудной
клетки, 65-66
Общее облучение тела, предтрансплантаци-
онное, 311-312
Общий анализ крови, 379-380,385
Общий антиген острого лейкоза (CALLA;
CD10), 139
Общий вариабельный иммунодефицит, 145
Общий путь, 198-204
Овалоциты, 81
Одноцепочечный урокиназный активатор
плазминогена, 210,275
Одышка, 55-56
Окраска по Райту, 429-430
Окрашивание эритроцитов на фетальный
гемоглобин, 399
Оксфордская ингибиторная единица, 225
Определение D-димера, 126,405-406,412
Определение антигена Виллебранда, 408-
409
Предметный указатель
Определение антиплазмина, 409-410
Определение антител к гликопротеинам
мембран тромбоцитов, 415-416
Определение антител к тромбоцитам, 415
Определение антитромбоцитарного IgG,
IgM, 415-416
Определение антитромбоцитарных антител,
415-416
Определение антифактора Ха, 409-411
Определение гепарина, 409
Определение осмотической устойчивости,
389,344-345
Определение резистентности С-белка, 420-
421
Определение факторов свертывания, 408-409
Остеокластактивирующий фактор, 356-357
Острая иммунная тромбоцитопеническая
пурпура (ИТП), 178-179
Острый грудной синдром, 107
Острый лейкоз, 336-345
острый лимфобластный лейкоз, 337-344
острый миелогенный лейкоз (ОМЛ),
336-337,341-343
Острый лимфобластный лейкоз, 337-341
выживаемость, 342-345
терапия, 342-345
Острый лимфоцитарный лейкоз, 145. См.
также Острый лимфобластный лейкоз
Острый миелогенный лейкоз (ОМЛ), 336-337
лечение, 342
миелобласты, 340
хромосомные перестройки, 412-413
Острый нелимфоцитарный лейкоз, 134
Острый промиелоцитарный лейкоз, 342
хромосомные перестройки, 412-413
Отбор доноров для аллогенной
трансплантации стволовых клеток, 310-311
Ответственный за связывание железа белок,
86-87
Открытая канальцевая система, 153
Охриплость, 55-56
Оценка желудочно-кишечного кровотече-.
ния, 393-395
Оценка пула железа, 89
п
Парапротеинемии, 166-167
Парвовирус В19,367-369
Парестезии, 58
Пароксизмальная ночная гемоглобинурия,
104-105,265-266
исследование, 389,391-392,394
Предметный указатель
441
Парциальная белоклеточная аплазия, 297
Парциальная красноклеточная аплазия,
289-293
Патология микрососудов, тромбоцитопения
потребления,183
Патология суставов, 68
Пельгера-Хьюэта аномалия, 129,131
Первичный антифосфолипидный синдром,
267
Первичный тромбоцитоз, 185-187
Перекись водорода, 126-127
Переливание крови, 48
количество и частота, 244
Переливание тромбоцитов
клиническое использование, 173-174
тромбоцитопения, 173-174
Периартериальная лимфатическая оболочка,
25-26
Пернициозная анемия, 53
вследствии дефицита кобаламина, 95-96
Пероксидаза, 403-404
Пероксисомы, 153
Перфорины, 127-128
Петехии, 61-62,217
Пигментация кожи, 59-60
Пиропойкилоцитоз, 72
Пируваткиназа, 391-392
Плазматические клетки,
Плазмин, 210-213
действие, 249
Плазминоген, 210
действие, 210-213
определение, 420-421
Плод, гемопоэз, 19-23
Плотная гранула, 152-153
Поверхностная оболочка, 52
Повреждение тканей вследствие ДВС, 247
Повреждение эндотелия, ДВС вследствие,
247
Повреждения тромбоцитов при
искусственном кровообращении, 166-167
Повреждения, 60
Повышение свертываемости крови, 258
при приеме оральных контрацептивов,
278-279
Поглощение железа, 86-87
Подагра, 52,68
тофус, 63
Подагрическое отложение солей, 63-64
Подсчет ретикулоцитов, 384,386,391-392
при анемии, 76-77
сниженное при СПИДе, клинический
пример, 304-305
Подсчет тромбоцитов, 158,217-218,381,386
время кровотечения, 157
при тромбоцитопении, 167-168
Подсчет эритроцитов, 385
Пойкилоцит, 81
Показатели эритроцитов, 425
Полимеризация фибрина, приобретенная,
246-247
Полиморфно-ядерный базофил, 429-430
Полиморфно-ядерный нейтрофил, 429-430
Полиморфно-ядерный эозинофил, 429-430
Полихроматофильные клетки, 78
Поражение психики, 58
Порфйрии, 427
после аллогенной трансплантации
стволовых клеток, 317-318
Послеоперационный венозный тромбоз, 265
Постинфекционная нейтропения, 299
Посттрансфузионная пурпура, 179-180
Потеря слуха, 63-64
Почечноклеточная карцинома, 119-121
Прекалликреин, 195-198
Препарат серповидных клеток, 396-398
Претромботическое состояние, 254
Преходящая эритробластопения детей, 303
Приапизм, 58,68
Приживление
при аллогенной трансплантации
стволовых клеток, 317-318
через день, 126-127,318-323
при аутологичной трансплантации
стволовых клеток, 329
Признак Грея Тернера, 67
Признак Куллена, 68
Признаки серповидных клеток, 114
Примахин, 368
Применения железа, 89-90
Приобретенная амегакариоцитарная тромбо-
цитопеническая пурпура, 292-293
Приобретенная апластическая анемия (АА),
283-290
Приобретенная непостоянная гипогаммагло-
булинемия, 149
Приобретенная парциальная
красноклеточная аплазия, 289-293
Приобретенная
амегакариоцитарная тромбоцитопеничес-
кая пурпура, 293-305
апластическая анемия, 283-290
нейтропения, 292-300
См. также Нейтропения, приобретенная
парциальная красноклеточная аплазия,
289-293
442
Предметный указатель
См. также Парциальная красноклеточная
аплазия, приобретенная
наследственные (врожденные) амегакариоцитарная
тромбоцитопения
анемия Даймонда-Блэкфана, 302-304
анемия Фанкони, 301-302
нейтропения,
наследственная/врожденная, 300-302
тромбоцитопения с отсутствием лучевой
кости, 303-304
транзиторная эритробластопения детей,
303
Приобретенные антитела к
стабилизированному фибрину,246-247
Проблемы гемостаза, 48
Продуктивная тромбоцитопения, 167-173
Продукты деградации фибрина, 226
Продукты распада фибрина (ЦРФ), 405-409
Продукция антител, 139-141
Прокоагулянтная поверхность, 192
Промиелоцит, 432
Промоноцит, 431
Проптоз, 61
Протеазанексин-1,206
Протеин S, 206-214
дефицит, 260-261,376
определение, 417-421
Протеин С, 205-207,211
дефицит, 260
определение, 417-418,421
Протопорфирин эритроцитов, 388,391-392
Проточная цитометрия, 396,401
Протромбиназа, 199-201
Протромбиновое время, 218-220, 383, 386-
387
Протромбиновый индекс, 218-219
Протромбоциты, 151
Профессиональные факторы, 49
Процедура нейтрализации тромбоцитов, 225
Псевдоаномалия Пельгера-Хьюэта, 129
Псевдотромбоцитопения, 167-168
Психиатрический анамнез, 58
Психический статус, 69
Пузырчатая клетка, 79-80
Пурпура посттрансфузионная, 179-180
Пурпуры, 61-62,217
Пятно Рота, 63-64
Р
Радужная оболочка, 63
Раннее закрытие, 44
Раннее насыщение, 56-57
Распределение железа, 86-87
Распределение эритроцитов по диаметру,
380,385,392
Реактивный тромбоцитоз, 183-185,345
Резистентность активации протеина С, 261
Рептилазное время, 405-407
Ретикулоцит, 84
Ретикулоцитоз при анемии, 101-115
Ретикулярный дисгенез, 300
Рида-Штернберга клетки, 351-352
Ристоцетин, 160-162
Ристоцетининдуцированная агрегация
тромбоцитов, 224-225
Рифабутин, 380
Рифампин, 380
Роговица, 62
Ротовая полость, 55-56
ксеростомия
с
С1-ингибитор, 206
Саркома Калоши, 60
Сахарозный гемолиз, 389,391,394-395
Свертывание крови, 193-214
пути активации свертывания, 195-204
регуляция активации свертывания, 203-
213
Свободный эритроцитарный
протопорфирин, 388,391-392
Селезенка, 24-32
Селезеночная артерия, 30
Селектины, 126-127
Сердечнососудистая система, 55-56,65-67
Сердечные шумы, 67
Серотонин,415
Серпины, 205-207
Серповидная клетка, 82
Серповидно-клеточная анемия, 106-107,
112-114
Синдром HELLP, 183
Синдром Бернара-Сулье, 163-165
Синдром Блума, 145-146
Синдром Висотта-Олдрича, 145-146
Синдром Германского-Пудлака, 164-165
Синдром Джоба, 129
Синдром Ди Георге, 145
Синдром короткой кишки, 48
Синдром Костманна (детский агранулоци-
тоз), 300
Синдром Кушинга, 129-130
Синдром "ладонь-стопа", 61-62
Синдром массивных трансфузий, 244-245
Предметный указатель
443
Синдром приобретенного иммунодефицита,
145-147
Синдром Свита, 60-61
Синдром Цинссера-Коля-Энгмана
(врожденный дискератоз), 300
Синдром Чедиака-Хигаси, 130г-131,300
Синдром Швахмана-Даймонда-Оски, 300
Синдромы недостаточности костного мозга,
283-305
Система комплемента, 126-127
Системы свертывания, 149
Склера, 61
Скорость оседания эритроцитов, 384, 386-
387,391
Скрининг пируваткиназы, 390
Скрининг XIII фактора свертывания, 408-409
Скрытая кровь в кале, 392,394-395
Слабительные средства, 327-329
Слизистокожный кандидоз, 62
Смешанное исследование, АЧТВ и ПВ, 409-
411
Совместимый неродственный донор
трансплантата, 311
Сосудистые дефекты, 214-216
Сосудистый компартмент, 23
Сосудистый эндотелий в гемостазе, 191-192
Спленомегалия, 68
Спленоэктомия, 374
Среднее содержания гемоглобина, 77,385,425
Средний объем клетки, 380,385,392,394,425
Средний объем тромбоцитов, 156-157
Средняя концентрация гемоглобина в
клетке, 77,385,425
Сродство гемоглобина к кислороду р50,396,
399-400
Стаз, 258
Стафилокиназа, 210
Стволовая клетка гемопоэза
определение, 308
дисфункция, 170-172
заготовка
при аллогенной трансплантациии
стволовых клеток, 311-313
при аутологичной трансплантации
стволовых клеток, 327
источники, 308
Стволовая клетка, 18,34-36
Стоматоцит, 81
Стоматоцитоз, 73,104
Стрептокиназа, 210,273-275
Стромальные клетки, 22-23,34-36
Судороги голеней, 53
Супероксиддисмутаза, 127
Суправитальное окрашивание эритроцитов,
397-400
Суправитальное окрашивание, включения в
эритроцитах, 84
Суставы, 52-53
геморрагии, 48
Сфероцит, 79
Сфероцитоз, 73
Сыпи, 60
т
Т- лимфопоэз, 38
Талассемические синдромы, 90-93
Талассемия большая, 24-25
Тахикардия, 59
Тахипноэ, 59
Тела Хауэлла-Жолли, 31,82
Телеангиэктазия, 60,217
Тельца Гассаля, 34
Тельца Деле, 129-130
Тельце Гейнца, 84,397-400
Тельце Паппенгеймера, 83
Терминальная деокситрансфераза, 139-141
Тест Хэма, 389,394
Тестикулярная атрофия, 69
Тесты свертывания, 405-412
Тесты тромбоцитопении, 412-416
Тимические тельца, 34
Тимомы, 34
Тимоциты, 33-34
Тиннит, 63-64
Титры ингибиторов факторов свертывания,
409-410
Тканевой активатор плазминогена, 210,273-
274
Тканевой фактор, 195-196,273-274
Т-клеточные генные перестройки,
исследование, 401 -402
Т-клеточный рецептор
исследование, 142-143
Т-лимфобластный крупноклеточный лейкоз,
298
Т-лимфоцитоз, 298
Т-лимфоциты, 137-138,142-145
Токсическая зернистость, 128-130
Толуидин, голубой, 403
Транскобаламины, 94-95,403-404
Трансплантация костного мозга, 307
при остром миелогенном лейкозе,
при приобретенной АА, 341-344
Трансплантация стволовых клеток, 307-332
аллогенная, 307-326
аутологичная, 326-330
444
Предметный указатель
Трансферрин, 86-87
Трансформирующий фактор роста-(3,155-157
Трансфузии, 48
количество и частота, 244
Триада Вирхова, 254
Триметоприм/сульфаметоксазол,
гематологические эффекты, 368
клинический пример, 376-378
Тромб, 253-259
Тромбастения Гланцмана, 164-165
Тромбин
действие, 249
биологические функции, 200-204
образование, 225
протеолитические эффекты, 203-205
Тромбиновое время, 405-407
Тромбо-Веллко тест, 405-407
Тромбогенез, 254-257
Тромбогенные факторы, 256
Тромбоз
гипофибринолитические состояния,
253-254
Тромбоз глубоких вен, 68,258
Тромбозы, 253-276
антитромботическая терапия, 269-272
артериальный тромбогенез, 255
гепарининдуцированный, 270
окклюзивный и неокклюзивный тромбы,
253-255
патогенез, 254-256
венозный послеоперационный, 265
факторы риска, 254-256
приобретенная тромбофилия, 265-270
врожденная тромбофилия, 259-266
Тромбоксан А2,155-156
Тромбоксан В2, сывороточный, 414
Тромболитические агенты, 275-276
Тромбомодулин, 205-206
Тромбопоэтин, 30,149,151-152
Тромботическая тромбоцитопеническая
пурпура, 179-182
при СПИД, 374
клинический пример, 186-188
Тромбофилия, 207, 256
приобретенная, 265-270
наследственная, 259-266
Тромбоцит(ы), 149-188
активация, 154-156
дефекты, 215-226
функция,154-158
время воспроизведения, 426
повреждения вследствие ДВС-синдрома,
247
лабораторное исследование, 156-162
продукция и кинетика, 150-153
в селезенке, 29
структура, 152-154
Тромбоцитактивирующий фактор, 155
Тромбоцитарнный фактор роста, 155-157,
255
Тромбоцитарно-ретикулоцитарныйтест,
415
Тромбоцитарные нарушения
при ВИЧ-инфекции, 372-374
тромбоцитопения, 167-183,372-374
тромбоцитоз, 183-187
Тромбоцитарные нарушения,
качественные, 161-167
приобретенные, 165-167
Тромбоцитарный фактор 3,155
Тромбоцитемия
эссенциальная, 345-348
Тромбоцитоз, 53,183-187
Тромбоцитопения Гланцмана, 164-165
Тромбоцитопения потребления, 167-170
вследствие патологии микрососудов, 183
Тромбоцитопения при химиотерапии, 170—
171
Тромбоцитопения разведения, 167-169
Тромбоцитопения распределения, 167-168
Тромбоцитопения с отсутствием лучевой
кости, 302-303
Тромбоцитопения, 167-184
при СПИД, 372-374
амегакариоцитарная, 304
гепарининдуцированная, 270
вследствие инфекции, 421-422
вследствие лекарственной терапии, 412-
413,421
Тромоцитопения вследствие лучевой
терапии, 170
Тяжелая апластическая анемия, 285
Тяжелый комбинированный
иммунодефицит, 145-147
У
Увеит, 63-64
Увеличение щитовидной железы, 64-65
Уремическое поражение тромбоцитов, 166
Урокиназа,210,275
Уши, 54,63
Ф
Фагосомы, 126-127
Фаза бластной трансформации, 346-347
Предметный указатель
445
Фактор IX, 195-198
приобретенные ингибиторы, 245
Фактор V Лейден, 207
ФакторУ, 197-201
приобретенные ингибиторы, 246
Фактор VIII, 195-200
Фактор Виллебранда (ФВ), 161-164
активность, 408-409
биосинтез, 233-234
концентрация, 224-225
мультимерный тест, 408-409
структура и функция, 233-234
Фактор стволовой клетки, 37-38
Фактор XI, 195-198
приобретенные ингибиторы, 246
дефицит, 238
Фактор XII, 195
приобретенные ингибиторы, 246
Фактор XIII
приобретенные антитела, 246-247
дефицит, 239-240
скрининг тест, 408-409
Фактор, ускоряющий разрушение, 104-105
ФакторУН, 195-198
приобретенные ингибиторы, 246
Факторы роста, гемопоэтические, 18-19,34-
40
Факторы свертывания крови, 223
Факторы свертывания, 149,195-196
Факторы свертывания. См. факторы
коагуляции
Ферритин, 86-87,393-395
Фетальный гемоглобин, 399-400
Фибриноген
аномалии, 239-240
приобретенные антитела к, 246-247
определение, 405,412
Фибринолиз,209,214
нарушения ингибирования, 252
дефекты, 215-216
чрезмерная активация, 252
активация плазмиогена, 210-213
тесты, 225-226
Фибринолитическая система плазмы, 210
Фибринолитические состояния,
приобретенные, 252-254
Филадельфийская хромосома, 337
Фолаты
дефицит, 95-96
диагностика, 98-99
лечение, 99-100
Фолиевая кислота, 388,391
метаболизм, 96-98
Фолликулярная мелкоклеточная лимфома,
353-354
Фоскарнет, 368
Фрагментированные клетки, 82
Фрагменты D и Е, 405-406
Фундоскопия, 63-64
х
Хинидин, 179
Хинин, 179
Хлорамины, 126-127
Хлорома, 61-62
Хондрокальциноз, 68
Хроническая доброкачественная нейтропе-
ния детей, 298-299
Хроническая иммунная тромбоцитопеничес-
кая пурпура, 175-179
Хронический гранулематоз, 129-130
Хронический лимфоцитарный лейкоз, 145-
146,335-336
клинический пример, 360-361
Хронический миелогенный лейкоз, 134,345,
349-350
трансплантация алллогенных стволовых
клеток, 329-332
хромосомные перестройки, 412
клиника и лечение, 347-350
диагностика, 346-347
Хрупкость, 394-395
Х-сцепленная агаммаглобулинемия, 145—
147
Ц
Цианоз, 60
Циклическая нейтропения, 299-300
Цинк-протопорфирин, 388,391
Цитокины, 37. См. также Гемопоэтические
факторы роста в гемопоэзе и лимфопоэзе,
18-19
гемопоэтические, 152-153
Цитомегаловирус при трансплантации алло-
генных стволовых клеток, 321-323
клинический пример, 329-332
клинические проявления, 322
лечение, 322-323
Цитомегаловирус, 367
Цитоплазматические вакуоли, 129-130
Цитохимические признаки острого лейкоза,
403-404
ч
Череп, 62
446
Предметный указатель
ш
Шенлейна- Геноха пурпура, 48
Шистоцит, 82
Шлемовидная клетка, 82
Шпоровидные клетки, 79-80,104-105
Шум трения перикарда, 67
Щ
Щелочная фосфатаза лейкоцитов, 403-404
Щелочная фосфатаза, 126-127
э
Экхимоз, 61-62, 67,217
Эластазы, 126-127
Электроферез гемоглобина, 396-398
Эллиптоцит, 81
Эллиптоцитоз, 72
Эмбриональная мезенхима, 20-21
Эмбриональное формирование кости, 21
Эмбриональный гемопоэз, 20-23
Эмбриональный печеночный гемопоэз, 19-
20
Эндокринная система, 53
Эндотелиальные клетки венозного синуса,
27-28
Эндотелиальные клетки, 191-192
примитивные, 19
продукты, 192
Эндотелий, 23
Энзимопатии эритроцитов, 102-103
Эозинофилия, 135,385-387
Эозинофилы, 124,135
Эритробластные островки, 23-24
Эритробластопения детей, преходящая, 303
Эритробласты, примитивные, 19
Эритропоэз, 37,74-75
Эритропоэтин, 37-40,75,389,391
Эритроцитарная ферментная панель, 390
Эритроцитарные энзимопатии, 102-106
Эритроцитоз, 114-117,391
клинический пример, 119-121
Эритроциты, 71-122
анемия, 75-115. См. также Анемия
клинические примеры, 117-121
развитие, 24
время воспроизведения, 426
при ВИЧ-инфекции, 366,369-371
наследственные аномалии мембраны, 71-
72
в селезенке, 29,31
структура и функция, 71 -74
Эритроциты, 71 -74
Эссенциальная тромбоцитопения, 345-348
Эстераза моноцитов, 403-404
Эстрогенотерапия, 265-266
Этничность, 49
Эхиноцит, 79-80,104-105
Я
Язык, 55-56,63-64
Яички, 69
Яичники, 69
Научно-учебное издание
Фред Дж. Шиффман
Патофизиология крови
Главный редактор Я. Я. Новиков
Редактор Я. Ю. Фролова
"Издательство БИНОМ"
Москва, 103473, Краснопролетарская, 16
Лицензия на издательскую деятельность
серия ЛР № 065249 от 26.06.97
Издательство "Невский Диалект"
Санкт-Петербург, 195220, Гражданский пр., 14
Лицензия на издательскую деятельность
серия ЛР, № 065012 от 18.02.97
Подписано в печать 20.12.99 Формат 70x100 %}.
Бумага газетная. Печать офсетная.
Гарнитура PetersburgC.
Усл. печ. л. 28,2. Доп. тираж 3000 экз. Заказ № 313
Отпечатано с оригинал-макета
в Академической типографии "Наука" РАН
199034, Санкт-Петербург, 9 линия, 16.