/

















































































































































































































































































































































































































































































































































































Текст
В. С. Хайлов,
В. Б. Брандт
ВВЕДЕНИЕ В ТЕХНОЛОГИЮ
ОСНОВНОГО
ОРГАНИЧЕСКОГО СИНТЕЗА
Ь ' Ь О h ИН X Э
ОЗЯБШИХ
Издательство „Xимия“, Ленинградское отделение
1969
УДК 661.7:658.5
В. С. Хайлов, Б. Б. Брандт. Введение в тех-
нологию основного органического синтеза. Изд-во
«Химия». 1969 г., стр. 560, рис. 250, табл. 54.
Книга является первой попыткой обобщен-
ного изложения технологии основного органиче-
ского синтеза с количественным инженерным ана-
лизом основных процессов и производственного
оборудовании. Большое внимание уделяется во-
просам оценки экономичности процессов основ-
ного органического синтеза. Используемый мате-
матический аппарат доступен для рядового инже-
нера и иллюстрируется расчетными примерами.
Кинга представляет собой инженерное руко-
водство по химической технологии и предназна-
чена для инженерно-технических работников
химической и связанных с ней отраслей про-
мышленности. Кроме того, она может служить
пособием для студентов и аспирантов химико-
технологических вузов.
3-14-2
137-68
1
ПРЕДИСЛОВИЕ
Развитие современной химической промышленности и
одной из ее ведущих отраслей — основного органиче-
ского синтеза тесно связано с развитием технологии как науки
о рациональных средствах и способах осуществления производ-
ственных процессов.
Настоящая работа является первой попыткой изложения основ-
ных вопросов технологии промышленного органического синтеза
путем анализа форм связи и взаимозависимости отдельных ста-
дий переработки сырья.
Любая технологическая схема может рассматриваться как
комплекс последовательных химических превращений, каждое из
которых осуществляется в три стадии: 1) подготовительная обра-
ботка исходного вещества; 2) химическое превращение исходного
вещества; 3) выделение и очистка целевого продукта синтеза.
Основой для описания количественной взаимосвязи этих стадий
служит материальный поток, размер и состав которого однозначно
определяются степенью превращения сырья на стадии его химиче-
ского взаимодействия. Такой подход к анализу технологической
схемы облегчает поиск экономического оптимума при исследовании
и проектной разработке процесса.
В соответствии с поставленной в книге задачей в главе I
дается анализ структуры ряда основных показателей экономиче-
ской эффективности химико-технологического процесса.
Для аналитического решения уравнений, полученных при ана-
лизе структуры экономических показателей, необходимо количе-
ственное выражение зависимости выхода продуктов реакции от
степени превращения сырья. Поскольку в химической кинетике
указанная зависимость, как правило, дается в неявном виде, воз-
никла необходимость написания главы II, в которой приведены
основные сведения о кинетике гомогенных и гетерогенных реак-
ций в условиях закрытых и открытых систем. Вопросы термоди-
намики в главе II рассмотрены только в связи с условиями рав-
новесия, определяющими значение предельно достижимой сте-
пени превращения исходного вещества.
Для количественной оценки влияния отдельных стадий пере-
работки сырья на экономические показатели многостадийной
технологической схемы авторы сочли необходимым рассмотреть
некоторые специфические для промышленного органического син-
теза вопросы аппаратурно-технологического оформления процес-
сов подготовительной переработки исходных веществ (глава III),
процессов химического превращения сырья (главы IV и V), а так-
же процессов, связанных с разделением реакционной смеси, вы-
делением и очисткой целевого продукта (глава VI). Учитывая не-
обходимость соблюдения условий безопасности при переработке
горючих продуктов и их смесей, в главе VII рассмотрены основ-
ные вопросы горения газов и меры защиты.
Несмотря на то,- что теплообменные устройства, как правило,
используются на всех стадиях переработки сырья, авторы сочли
возможным рассмотреть их в главе III, хотя это несколько нару-
шает стройность изложения материала.
В главе V реакционная аппаратура сгруппирована по прин-
ципу фазового состава и агрегатного состояния реакционной си-
стемы, что облегчает рассмотрение специфических условий тепло-
и массообмена, а также конструктивных особенностей реакторов.
Сведения о представителях каждой группы реакторов даны в
объеме, необходимом Для их сравнительной оценки при выборе
типа и принципа действия реактора. Читателям, интересующимся
расчетом реакционной аппаратуры, рекомендуется специальная
литература, посвященная этому вопросу. Более подробно в гла-
ве V рассмотрен-тепловой режим и условия устойчивости экзо-
термических процессов.
Приводимые в книге сведения о физико-химических основах
процессов подготовки сырья (глава III) и процессов разделения
реакционных смесей (глава VI) сознательно ограничены в расчете
на инженерную подготовку читателя.
Подход к рассмотрению технологии основного органического
синтеза в данной книге несколько отличается от общепринятого,
поэтому авторы будут благодарны читателям за все критические
замечания и пожелания.
Раздел «Очистка продуктов промышленного синтеза» главы VI
написан канд. хим. наук В. П. Евдаковым. Материал по оптималь-
ному соотношению размеров секций каскада любезно предостав-
лен канд. техн, наук В. Г. Горским, которому авторы выражают
глубокую благодарность за ряд весьма ценных замечаний, ка-
сающихся расположения материала и содержания некоторых глав
книги.
В. Хайлов, Б. Брандт
Глава I
1
ВОПРОСЫ ЭКОНОМИКИ В ТЕХНОЛОГИИ
ОСНОВНОГО ОРГАНИЧЕСКОГО СИНТЕЗА
Конечной целью технологии — науки о рациональных
методах и процессах переработки сырья в продукты
потребления и средства производства — является создание процес-
сов, максимально эффективных по своим технико-экономическим
показателям, т. е. обеспечивающих максимальный выпуск про-
дукции на единицу капитальных вложений и основных производ-
ственных фондов.
Оценка промышленного процесса с помощью критериев эконо-
мической эффективности основана на таких показателях техниче-
ского уровня производства, как себестоимость продукта, удель-
ные капитальные затраты и производительность труда.
Значение каждого из этих показателей определяется не толь-
ко совершенством технологических приемов и способов перера-
ботки сырья, но и химической схемой синтеза продукта.
ПРОМЫШЛЕННЫЙ СИНТЕЗ
НЕКОТОРЫХ ПРОДУКТОВ
К важнейшим задачам экономики химической промыш-
ленности и химической технологии на первом этапе разработки
нового промышленного процесса относится выбор наиболее ра-
ционального пути синтеза целевого продукта из числа возможных
вариантов осуществления этого синтеза на основе различных ви-
дов органического и неорганического сырья.
Решение такой задачи требует количественного сопоставления
технологических и технико-экономических показателей сравнивае-
мых вариантов синтеза; выбор оптимального варианта для проек-
тирования связан с необходимостью дополнительного учета ряда
условий, определяемых местом размещения производства, энер-
гетическими ресурсами и т. п.
Детальное рассмотрение этого сложного вопроса выходит за
рамки настоящей работы. Ниже приводятся лишь результаты
сравнительной оценки на примере некоторых продуктов, способы
промышленного синтеза которых характеризуются многообразием
вариантов.
5
СИНТЕЗ ДИМЕТИЛТЕРЕФТАЛАТА
Диметилтерефталат (ДМТ)—одно из исходных ве-
ществ промышленного производства полиэфирных волокон, из-
вестных под названием дакрон (США), терилен (Англия), лавсан
(СССР). Возможны следующие варианты промышленного син-
теза ДМТ:
Промышленное производство ДМТ, занимающее по темпам
развития за последнее десятилетие одно из первых мест в миро-
вой промышленности основного органического синтеза, базирует-
ся на переработке п-ксилола.
Необходимость расширения сырьевой базы развивающегося -
производства ДМТ вызывает интерес к процессам получения
n-ксилола из смеси изомерных ксилолов, а также к возможности
замены n-ксилола другими, более дешевыми и доступными пред-
ставителями ароматического ряда.
Из опубликованных отечественных и зарубежных данных сле-
дует, что способы переработки n-ксилола мало отличаются один
от другого по расходу основного сырья. Использование азотной
кислоты и соединений серы в качестве окислителей несколько
в
усложняет схему синтеза и требует более тщательной очистки
целевого продукта, чем при проведении процессов, основанных на
применении в качестве окислителя кислорода воздуха. Указанные
соображения, а также сравнительная оценка количественных по-
казателей обусловили преимущественное развитие процесса, осно-
ванного на жидкофазном окислении смеси n-ксилола и метилто-
луилата кислородом воздуха с последующей этерификацией
смеси монометилтерефталата и толуиловой кислоты метиловым
спиртом *.
Представление о расходе сырья и энергетических средств на
1 т продукта при производстве ДМТ указанным способом можно
получить из следующих данных:
Сырье
и-Ксилол (96%-ный), т....... 0,75
Метиловый спирт, т.......... 0,53
Энергетические средства
Пар, Мкал ............ 12,6
Вода, м1 * 3................ 1100
Электроэнергия, квт.ч........ 900
Выбор рациональной схемы переработки смеси изомерных кси-
лолов, играющий основную роль при оценке себестоимости «-кси-
лола, решается на основе тщательного учета потребности про-
мышленности основного органического синтеза в о-ксилолеи этил-
бензоле.
Вопрос о наиболее целесообразной последовательности
процессов окисления и изомеризации, составляющих химическую
схему превращения о- и .м-ксилола в терефталевую кислоту или
ДМТ, до настоящего времени не имеет однозначного решения, и
в промышленной практике известны производства, основанные на
обоих вариантах.
Синтез ДМТ из толуола отличается большим разнообразием
вариантов, однако некоторые из них не выдерживают конкурен-
ции даже при предварительной оценке. Способ синтеза ДМТ,
основанный на реакциях карбонилирования и хлорметилирования
толуола и последующем окислении полученных продуктов с по-
мощью азотной кислоты или перманганата калия, характеризуется
значительным расходом минерального сырья, большим количе-
ством побочных продуктов, требует применения коррозионно-
устойчивых материалов и по технико-экономическим показателям
уступает процессу, осуществляемому через бензойную кислоту.
В настоящее время синтез ДМТ из толуола через бензойную кис-
лоту является одним из наиболее перспективных.
1 В последнее время все большее внимание уделяется процессу одностадий-
ного окисления n-ксрлола в терефталевую кислоту, которая после тщательной
очистки используется как исходный продукт поликонденсации. По технико-эко-
номическим показателям' этот способ выгодно отличается от способа, связанного
с получением днметилтерефталата.
7
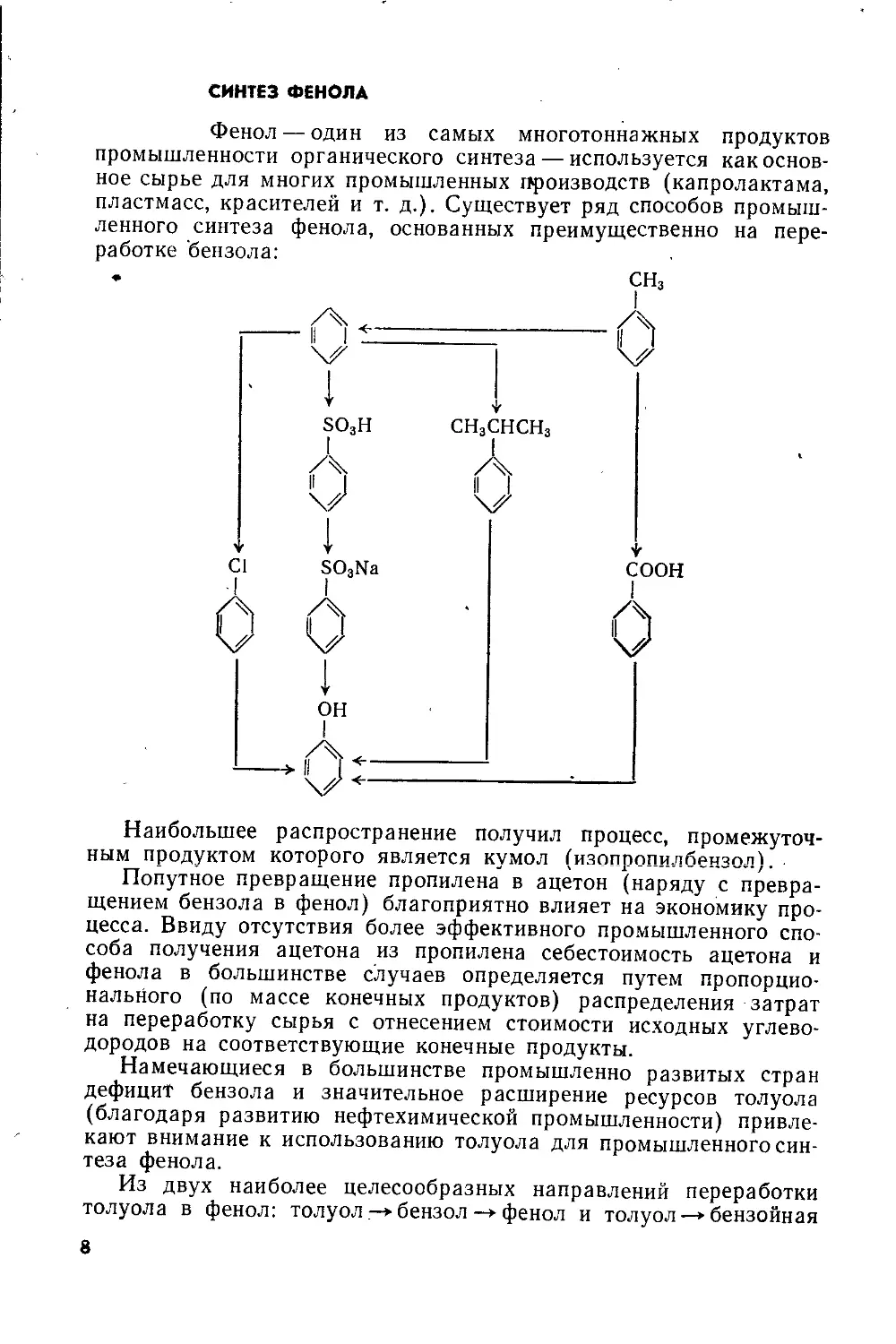
СИНТЕЗ ФЕНОЛА
Фенол — один из самых многотоннажных продуктов
промышленности органического синтеза — используется как основ-
ное сырье для многих промышленных производств (капролактама,
пластмасс, красителей и т. д.). Существует ряд способов промыш-
ленного синтеза фенола, основанных преимущественно на пере-
работке бензола:
. СН3
Наибольшее распространение получил процесс, промежуточ-
ным продуктом которого является кумол (изопропилбензол).
Попутное превращение пропилена в ацетон (наряду с превра-
щением бензола в фенол) благоприятно влияет на экономику про-
цесса. Ввиду отсутствия более эффективного промышленного спо-
соба получения ацетона из пропилена себестоимость ацетона и
фенола в большинстве случаев определяется путем пропорцио-
нального (по массе конечных продуктов) распределения затрат
на переработку сырья с отнесением стоимости исходных углево-
дородов на соответствующие конечные продукты.
Намечающиеся в большинстве промышленно развитых стран
дефицит бензола и значительное расширение ресурсов толуола
(благодаря развитию нефтехимической промышленности) привле-
кают внимание к использованию толуола для промышленного син-
теза фенола.
Из двух наиболее целесообразных направлений переработки
толуола в фенол: толуол —* бензол —> фенол и толуол—» бензойная
8
кислота—> фенол—первое является более освоенным. Второе на-
правление, весьма близкое к первому по технико-экономическим
показателям, имеет все основания для развития при благоприят-
ных условиях в отношении себестоимости толуола и ацетона.
Ниже будет показано (см. стр. 13), что в подобных случаях
(производство одного и того же целевого продукта из определен-
ного вида исходного сырья) влияние цены сырья на себестоимость
продукта различно, в связи с чем отношение себестоимости про-
дукта, получаемого в результате сравниваемых процессов, яв-
ляется функцией себестоимости сырья.
СРАВНИТЕЛЬНАЯ ОЦЕНКА
СПОСОБОВ ПРОМЫШЛЕННОГО СИНТЕЗА
Выбор оптимального варианта схемы промышленного
синтеза является сложной задачей, решаемой путем сравнитель-
ной оценки ряда вариантов.
Показателями технического уровня химического производства
могут служить себестоимость продукта и производительность обо-
рудования, используемого в технологической схеме. Для боль-
шинства процессов промышленности основного органического син-
теза эти показатели находятся в сложной взаимозависимости, что
практически исключает возможность одновременного достижения
оптимальных значений каждого из них и лишает смысла поиск
оптимального режима технологического процесса без учета их со-
вместного влияния на конечный результат.
В качестве обобщающих критериев экономической эффектив-
ности производства, характеризующих его рентабельность, могут
быть использованы [1—4]:
1) отношение чистого дохода предприятия к себестоимости
продукции;
2) показатель приведенных затрат, учитывающий себестои-
мость продукта и удельные капитальные затраты;
3) чистый доход предприятия, отнесенный к объему основных
и оборотных средств предприятия.
Первый из указанных критериев определяется себестоимостью
и достигает своего максимального значения при минимальной се-
бестоимости продукта.
Более полный учет расхода средств, связанных с производ-
ством продукта, дается показателем приведенных затрат (77):
77=Спр + £нК (1-1)
где Сдр — себестоимость единицы продукта; Еп — отраслевой нор-
мативный коэффициент эффективности капитальных затрат; К —
удельные капитальные затраты.
Для химической промышленности коэффициент Еа в среднем
равен 0,2—0,33 [5]. Величина П характеризует химико-технологи-
ческую сторону процесса и его аппаратурное оформление, но
9
недостаточно четко отражает количественную связь между рента-
бельностью производства и его мощностью.
Каждый из рассмотренных критериев экономической эффек-
тивности может использоваться при оценке производств, мощ-
ность которых по выпуску продукта строго регламентирована.
Связь рентабельности производства с размером выпуска про-
дукции находит выражение в чистом доходе предприятия (Д):
Д — (Дпр — Спр) Л1пр (1-2)
где Дпр — цена единицы продукта; Л4пр — количество выпускаемых
единиц продукта в единицу.времени (например, в год).
Из структуры показателя величины чистого дохода следует,
что при заданной цене на продукт наиболее рентабельным яв-
ляется производство, выпускающее максимальное количество это-
го продукта при минимальной его себестоимости. Себестоимость
продукта и производительность оборудования, используемого в
технологической схеме, взаимозависимы, поэтому максимальный
доход не будет соответствовать оптимальным значениям Спр и
Afnp, если находить их, рассматривая доход как функцию только
одной из этих величин. Максимальное значение Д должно нахо-
диться с учетом влияния обеих этих величин на доход.
Поскольку в состав каждого из рассмотренных выше крите-
риев экономической эффективности химического производства в
явном или неявном виде входят два основные показателя техни-
ческого уровня химико-технологического процесса (себестоимость
продукта и удельные капитальные затраты), при рассмотрении
экономических задач технологии основного органического синтеза
в первую очередь заслуживает внимания анализ структуры каж-
дого из этих показателей и выяснение формы их взаимосвязи с
учетом специфики промышленного органического синтеза. Значи-
тельный интерес представляют также условия, которые определяют
производительность оборудования химических производств, оказы-
вающую решающее влияние на величину удельных капитальных
затрат и количество продукта, выпускаемого предприятием.
Для оценки степени влияния себестоимости продукта и удель-
ных капитальных затрат на показатель экономической эффектив-
ности химико-технологического процесса в табл. 1-1 приведены
значения Спр, К и П для ряда производств промышленности основ-
ного органического синтеза.
Из табл. 1-1 следует, что на величину показателя приведенных
затрат решающее влияние оказывает себестоимость продукта, до-
стигающая 80—85% значения П.
В отличие от промышленного органического синтеза структура
показателя приведенных затрат химико-технологических процес-
сов получения неорганических веществ характеризуется заметно
меньшей долей себестоимости продукта при относительно более
высоком значении удельных капитальных затрат. Примером мо-
жет служить крупнотоннажное производство аммиака, где себе-
IQ
Таблица 1-1
Значения С„п, К и П для некоторых производств
lip
промышленности основного органического синтеза
• Продукт Спр. руб 1т X, рубЦт/год) 77. руб!т
Диметилтерефталат Фталевый ангидрид (из на- 610 412 745
фталина) Метиловый спирт (из природ- 440 295 540
кого газа) 200 114 237
стоимость продукта составляет 55% от величины/7 [себестоимость
аммиака 60 руб/т, удельные капитальные затраты 150 pt/б/(т/год)].
Вполне естественно, что при характеристике нового способа
промышленного органического синтеза основным требованием
является снижение себестоимости продукта по сравнению с себе-
стоимостью этого же продукта, получаемого в результате суще-
ствующего . процесса. Если себестоимости целевого продукта,
синтезируемого по новому и существующему способам, обозна-
чить соответственно через Спр и Спр, то конкурентоспособность
нового процесса выразится неравенством:
с
ьпр ипр
(1-3)
Вместе с тем необходимо отметить, что тщательно разрабо-
танная и усовершенствованная в результате длительной эксплуа-
тации технология действующего производства и технология раз-
рабатываемого нового процесса при сравнительной оценке эф-
фективности находятся в неодинаковых условиях. Возможности
нового процесса нередко трудно установить на основе суммы ко-
личественных показателей (расход и стоимость сырья, энергии
и пр.), характеризующих данный этап разработки технологии
синтеза.
Потенциальные возможности нового процесса и способность
(или отсутствие такой способности) его конкурировать с суще-
ствующим эталонным процессом может быть ориентировочно оце-
нена на основе анализа структуры себестоимости целевого про-
дукта. Результаты такого анализа позволяют также установить
целесообразность и направление дальнейших исследований, от-
носящихся к изучению возможностей повышения эффективности
процесса.
Если химико-технологический процесс состоит из ряда после-
довательно осуществляемых стадий, то себестоимость целевого
продукта можно выразить в зависимости от удельного расхода
основного сырья и затрат на его переработку на каждой из этих
стадий.
11
Рис. 1-1. Область значений
расходных коэффициентов,
соответствующих экономи-
ческой целесообразности но-
вого процесса.
Обозначив расход сырья на i-й стадии через v,- (в т/т), стои-
мость (или себестоимость) этого сырья через а{ (в руб1т), за-
траты на переработку сырья в промежуточный или конечный про-
дукт (включая расходы на вспомогательное сырье, материалы,
энергию, зарплату с начислениями и т. д.) через Ь{ (в руб]т},
себестоимость целевого продукта можно представить уравнением:
{[(«iVi + 61) v2 + b2] v3 + b3] v4 + ... = Спр (1-4)
Как следует из уравнения (1-4), себестоимость целевого про-
дукта производства, состоящего из W химических стадий, являет-
ся функцией 1-|-2jV независимых пере-
менных.
Подставив уравнение (1-4) в нера-
венство (1-3), получим:
{[(«1*1 + 6i) *2 + М v3 + 63) v4 + ... < С'р (1-5)
Выражение (1-5) учитывает только
экономику рассматриваемого процесса.
Для определения границ экономических
возможностей нового способа производ-
ства выражение (1-5) должно быть до-
полнено системой неравенств, учитываю-
щих стехиометрические соотношения.
Действительно, расходные коэффициенты
в выражениях (1-4) и (1-5) не могут быть
меньше теоретических (viT), т. е.
V/ > Чц В-6)
Выражение (1-5) и условия (1-6) описывают область потен-
циальных экономических возможностей рассматриваемого про-
цесса.
Если требования конкурентоспособности связаны с наруше-
TJTJOAJ ЛГГ» ТЛТЛ 'ГГ» ГТГ» V а О QTP ПП UfIDfirn Т7ППТ1ОПЛ ттг»тг> О
j x-vx у* w f , iv nviiuuuivnn nvuviv cuvwUu 1 XJCl
не могут быть улучшены за счет уменьшения расходных коэффи-
циентов.
Равенства (1-5) и (1-6), описывающие границы области эко-
номической целесообразности некоторого нового двухстадийного
процесса в предположении, что новый способ отличается от су-
ществующего лишь расходными коэффициентами и v2 (осталь-
ные переменные рассматриваются как параметры), представлены
графически на рис. 1-1. Если линия равной себестоимости, опре-
деляемая равенством (1-5), представляется кривой 1, то область
значений vi и v2, при которых организация производства по
новому способу экономически оправдана, соответствует заштрихо-
ванной площади. В том случае, когда линия равной себестоимо-
сти проходит подобно кривой 2, неравенства (1-5) и (1-6) не
совместны и область экономической целесообразности вообще от-
сутствует.
12
В качестве примера рассмотрим вопрос о целесообразности
организации производства фенола из толуола по новой схеме
СН3 СООН ОН
I I I
при наличии конкурирующего производства фенола из толуола по
существующей схеме:
+н2
-СН4
ОН
I
+ Ог+С3Н6> || + СНзСОСНз
Допустим, что цены на продукты
нефтехимического синтеза (бензол, то-
луол, ксилол) недостаточно стабильны
и можно ожидать изменения стоимости
толуола в довольно широких пределах.
Для исследования влияния стоимости
толуола на результаты сравнительной
оценки рассматриваемых способов про-
изводства фенола неравенство (1-5) сле-
дует написать в развернутом виде:
Рис. 1-2. Область значений
расходных коэффициентов,
обеспечивающих экономич-
ность нового процесса.
(tZjV, + 6]) v2 + b2 < (afv[ + 6]) v2 + b2 (1-7)
Здесь величины, помеченные штрихом,
относятся к существующему процессу,
для которого на основании длительной практики эксплуатации
производства значения ряда величин можно считать стабильными:
Vj=l,27; Ь'г= 15,5 руб/т; V2=l,0; b'2 = 54 руб/т
В данном случае граница области экономических возможно-
стей нового процесса представляет собой поверхность в много-
мерном пространстве. Проведем ее исследование в двухмерном
пространстве, рассматривая остальные независимые переменные
как заданные параметры.
Решив равенство (1-7) относительно Vi и учтя значения Vi, V2,
bi и bi, получим:
Зависимость vi(v2) при различных значениях bi представлена
на рис. 1-2. Переменные щ и Ь2 приняты равными 77 и 30 руб!т
соответственно. Из сопоставления графиков уравнения (1-8) с
теоретическими значениями расходных коэффициентов (viT = 0,75;
13
V2t=1,3), обозначенных на рис. 1-2 прямыми линиями, следует.
что при затратах на переработку
Рис. 1-3. Область значений расходных
коэффициентов по толуолу при окисле-
нии его в бензойную кислоту, обеспечи-
вающих экономичность нового процесса.
в зависимости от цены на толуол.
Рис. 1-4. Область значений расходных
коэффициентов по толуолу при окисле-
нии его в бензойную кислоту, соответ-
ствующих конкурентоспособности ново-
го процесса в зависимости от затрат
на переработку сырья.
толуола в бензойную кислоту,
превышающих 48 руб]т, новый
процесс неконкурентоспособен
ни при каких реально до-
стижимых значениях удельно-
го расхода толуола (область
экономической целесообразно-
сти сводится к нулю). При
условии, что 61 <48 руб/т, су-
ществует область, в которой,
при соответствующем значении
расходных коэффициентов,
рассматриваемый процесс ста-
новится экономически оправ-
данным.
Зависимости Vi(fli) при по-
стоянных значениях vj, Ь\, Ь%м
vi(6i) при постоянных значе-
ниях V2, Qi и 6г> построенные по
уравнению (1-8), изображены
на рис. 1-3 и 1-4 соответствен-
но. Зависимость vi(ai) пред-
ставляет собой семейство ги-
пербол с асимптотой v(v2/v2.
При V2=l,5 экономика сравни-
ваемых процессов не зависит
от цены на толуол, если bi =
=26,3 руб 1т, а £>2=30 руб/т.
Как следует из рассмотрен-
ного примера, независимо от
цены на толуол могут быть
подобраны такие условия осу-
ществления процесса произ-
водства фенола, при которых
новый процесс окажется эко-
номичнее существующего. По-
добный подход к сравнитель-
ной оценке химико-технологи-
ческих процессов полезен уже
на стадии лабораторного изу-
чения нового способа, по-
скольку он позволяет более
целенаправленно проводить
исследования.
Наглядное графическое сопоставление экономики конкурирую-
щих между собой йроизводств, достигаемое описанным методом,
дает возможность определять лишь границы области экономиче-
14
ской целесообразности нового процесса. Определение же опти-
мального варианта осуществления нового процесса требует более
детального рассмотрения экономики производства.
ОСНОВНЫЕ ПОКАЗАТЕЛИ
ХИМИКО-ТЕХНОЛОГИЧЕСКИХ ПРОЦЕССОВ
При оценке нового химико-технологического процесса
в период его лабораторного изучения и при составлении калькуля-
ции себестоимости продукта промышленного производства основ-
ное влияние на технико-экономические показатели оказывают две
величины, характеризующие стадию химического превращения
сырья:
1) степень превращения исходных веществ;
2) выход конечных продуктов (или продукта).
Степень превращения исходного вещества представляет собой
отношение числа молей (или килограммов) этого вещества, всту-
пившего в химическое взаимодействие с другим реагентом или
подвергнутого каким-либо иным химическим превращениям (на-
пример, крекингу, пиролизу), к числу молей (или килограммов)
этого же вещества в питании, поступающем в реакционный ап-
парат или агрегат:
а = п/по
где а — степень превращения, выраженная в долях единицы; п —
число молей (или килограммов) превращенного исходного веще-
ства; ио — число молей (или килограммов) исходного вещества в
питании.
В том случае, когда в химическом превращении участвует не-
сколько исходных веществ, процесс может характеризоваться сте-
пенью превращения каждого из них.
Величина а в общем случае может колебаться от нескольких
сотых до 1 (или от нескольких процентов до 100%), определяя
тем самым количество исходного вещества, подаваемого в реак-
ционный аппарат на единицу этого вещества, вступившего в хими-
ческую реакцию.
Для количественной оценки результатов химического превра-
щения могут быть использованы следующие показатели: 1) выход
продукта в расчете на поданное сырье (выход от теории) и 2) вы-
ход продукта в расчете на превращенное сырье (селективность).
В последнем случае иногда различают понятия «дифференциаль-
ная селективность» (оценка мгновенного состояния процесса) и
«интегральная селективность» (оценка процесса по конечному ре-
зультату).
Так как только второй из показателей (выход продукта в рас-
чете на превращенное сырье) дает возможность для оценки удель-
ного расхода сырья, последующее изложение основывается на
этом показателе, для краткости именуемом «выход продукта».
Выход продукта обычно определяется как отношение числа мо-
лей х данного продукта к числу молей п превращенного сырья
(с учетом стехиометрических соотношений):
р = ± = _£_
п ап0
В общем случае выход продукта может быть отнесен к любому
из исходных веществ. В практике технологии основного органиче-
ского синтеза процесс, как правило, характеризуется выходом ос-
новного продукта из основного (наиболее дорогого, дефицитного)
вида сырья.
Сказанное можно иллюстрировать примером хлорирования бен-
зола. Материальный баланс процесса описывается следующим
уравнением:
20C6He + 15С12 = 8СвН5С1 + 2СвН4С12 + ЗС12 + 10CeH6 + 12НС1
Степени превращения реагентов и выходы продуктов:
В расчете В расчете
на бензол на хлор
Степень превращения................0,5 0,8
Выход:
монохлорбензола................... 0,8 0,667
дихлорбензола................. 0,2 0,333
хлористого водорода............. — 1,0
В том случае, когда целевым продуктом производства является
конечный продукт последовательной реакции (например, дихлор-
бензол в рассмотренной выше реакции) и промежуточное соеди-
нение после его отделения от основного продукта возвращается в
реакционный аппарат, выход целевого продукта соответственно
повышается.
Понятие «выход», являясь характеристикой селективности хи-
мического превращения, может служить также для оценки потерь
сырья или продукта реакции при их переработке на отдельных
стадиях технологической схемы. В этом случае значение выхода
подсчитывается как отношение количества вещества (сырья, про-
дукта реакции), полученного в результате той или иной перера-
ботки, к количеству его, введенному на переработку (степень пре-
вращения равна единице).
Независимо от особенностей отдельных технологических схем
процесс превращения исходного вещества в промежуточный или
конечный продукт химического производства слагается из опреде-
ленных стадий, разнородных по своим технологическим задачам.
Основными стадиями являются:
1) подготовительная обработка сырья перед вступлением его
в химическое взаимодействие;
2) химическое взаимодействие исходных веществ;
3) разделение смеси продуктов химического превращения и
выделение целевого продукта.
16
Кроме того, технологическая схема включает такие стадии, как:
4) дополнительная очистка продукта (если в этом возникает
необходимость);
5) хранение, транспорт и расфасовка готового продукта;
6) очистка сточных вод и отходящих газов, уничтожение от-
ходов.
В зависимости от числа химических стадий синтеза комплекс
основных технологических стадий переработки сырья осуществ-
ляется соответственное число раз.
Первая основная стадия (подготовка сырья) слагается из ряда
единичных операций, характер и аппаратурное оформление кото-
рых определяется свойствами сырья и условиями химического
превращения. К целям таких операций относятся хранение сырья
(свежего и обратного), транспорт по заводской территории, изме-
нение параметров состояния сырья, освобождение от механических
примесей и т. д. (химическая очистка сырья с большими основа-
ниями может быть отнесена ко второй стадии его переработки).
Осуществление всех этих операций связано с расходом различных
видов энергетических средств и сопровождается расходом сырья
в результате неизбежных потерь.
Химическое превращение исходных веществ, осуществляемое
на второй основной стадии, заканчивается получением реакцион-
ной массы, состоящей, как правило, из непревращенного сырья и
\^смеси продуктов реакции. В состав реакционного агрегата входит
Хл^ряд аппаратов, эксплуатация которых требует затраты энергети-
' г\ческих средств, а результат химического превращения характери-
X. зуется расходом сырья на единицу целевого продукта.
На третьей основной стадии смесь продуктов реакции и непре-
^\.вращенного сырья подвергается переработке с целью разделения
\\ее на основные фракции: непревращенное сырье, целевой продукт,
побочные продукты. Целевой продукт часто подвергается допол-
нительной очистке от примесей с применением разнообразных хи-
мических и физико-химических приемов. Все операцйи разделения
реакционной массы и очистка целевого продукта требуют расхода
энергии и сопровождаются расходом сырья и готового продукта
в результате не только механических потерь, но и нежелательных
химических превращений (осмоление, деструкция и т. д.).
Удельный расход сырья на единицу готового продукта химиче-
ского производства слагается из расхода его на всех стадиях тех-
нологической схемы.
На примере технологической схемы, состоящей из трех основ-
ных стадий (подготовка сырья, химическое превращение, разделе-
ние реакционной массы), можно установить характер зависимости
суммарного (технологического) выхода £' продукта от показате-
лей (выходов), характеризующих каждую из стадий.
Схема, изображенная на рис. 1-5, представляет собой упрощен-
ный вариант реального процесса, рассчитанного на полное исполь-
зование одного из исходны^^еДёс^р’. ТНёпрр^^гировавшее сырье
2 В. С. Хайлов, Б. Б. Брандт I '. РлНЧ" '' <
после его выделения из реакционной массы возвращается на пер-
вую стадию, а один из побочных продуктов — на вторую (в реак-
ционный агрегат).
Исходное вещество (в данном случае А), как правило, содер-
жит инертные примеси. Условием стационарности состава мате-
риальных потоков схемы является баланс инертных примесей в
системе:
А0Ха — (Aj — Aj) Х| + Фз — D4) Х3 + (В3 — В4) Х3 +
+ (А3 - 4<) *< + (1 - й) Xs + D4XB
где Ао, Ai, А2, Аз, А4— количества молей исходного вещества А
(или величины потоков сырья в единицу времени); В3, В4— коли-
чества молей продукта В (или величины потоков продукта В в
единицу времени); D3, D4— количества молей продукта D (или
Рис. 1-5. Схема материальных потоков промышленного процесса
с одной химической стадией.
величины потоков продукта D в единицу времени); б — доля по-
тока продукта В, возвращаемая в реакционный агрегат; Хо — на-
чальное содержание инертных примесей в исходном веществе; Хь
Х2, Хз, Х4, Х5, Х6— содержание инертных примесей в материаль-
ных потоках, выводимых из системы.
Если при заданных на каждой стадии параметрах процесса и
заданных размерах выводимых потоков баланс инертных приме-
сей не достигается, концентрация этих примесей в материальных
потоках, стремясь к балансу, повышается или понижается, нару-
шая допустимые пределы. В первом, наиболее часто встречаю-
щемся и иногда опасном случае возникает необходимость сведе-
ния баланса инертных примесей путем удаления их из системы.
Это достигается либо выводом части1 потока сырья в точке, где
концентрация инертных примесей максимальна (на рис. 1-5 такой
18
вывод обозначен пунктирной линией), либо удалением инертных
примесей химическим или иным способом в результате соответст-
вующей обработки потока сырья, возвращаемого на переработку
после разделения реакционной массы.
Указанная выше задача—установление характера зависимости
: р' от выходов на каждой стадии — может быть решена примени-
’ тельно к различным химическим схемам превращения исходного
вещества. Рассмотрим, например, параллельную и последователь-
ную реакции:
А->В; A->D
A->B->D
Промежуточный продукт В последовательной реакции после
выделения его из реакционной массы может рассматриваться как
конечный продукт производства (6=0) или быть использован для
превращения в конечный продукт D путем возврата в реакцион-
ный агрегат (в этом случае 6 = 1).
Приняв условно стехиометрические коэффициенты равными
единице и использовав приведенные на рис. 1-5 обозначения, не-
трудно выразить значения степени превращения сырья в реакци-
онном агрегате и выходов на любой стадии при стационарном
режиме процесса:
выход сырья на стадии предварительной подготовки
„ Аг Aj-
рА.1“ А; = А0 + А4
степень превращения сырья в реакционном агрегате
Аа ~ Аз
А2
выход продукта В на стадии химического превращения
« Вз
рв.2 аА2
выход продукта D на стадии химического превращения
Д3 + дВ4
РО.2= цА2
выход непревращенного сырья на стадии разделения реакционной
массы
Рл,3 = Л4/Л3 <ИЗ>
выход продукта В на стадии разделения реакционной массы
0В,3 = В4/В3 <Н4)
выход продукта D на стадии разделения реакционной массы
₽О,3 = П4/П3 (1‘15)
Совместное решение уравнений (1-9) — (1-15) позволяет исклю-
чить величины, определяющие размер материальных потоков, и
2* 19
(1-9)
(МО)
(I-H)
(1-12)
выразить значения суммарных (технологических) выходов продук-
тов В и D
/10
о' — В* — &3 + ^4 а
Аа До ря-з
в зависимости от безразмерных величин а, рА, ь рА, з, Рв, 2, ₽о, 2-
₽в, з, ₽о, з, характеризующих процесс параллельного и последова-
тельного превращения исходного вещества в условиях схемы,
представленной на рис. 1-5.
При осуществлении параллельной реакции и возврате непре-
вращенного сырья (Д4) на первую стадию технологические вы-
ходы продуктов В и D описываются следующими выражениями:
а' аРл,1Рв,гРв,3
!-^4,3(1-а)
р/ а0Л, 10Ц, 2₽й,3
Р° ^.Льз^-и)
(Мб)
(1-17)
При практически полном отсутствии потерь на первой и тре-
тьей стадиях (рА, i = pA, з = ₽в, з = ₽о, з=1) выходы продуктов будут
равны:
Рв~Рв,2; Рв = Рв,2 = 1 — Рв, 2
Когда в условиях приведенной на рис. 1-5 схемы осуществляется
последовательная реакция и непревращенное вещество и промежу-
точный продукт возвращаются на повторное использование (6=1),
технологические выходы можно выразить следующим образом:
Й-°; Й - , _ °(?- .) (й.,+й. А.,) ,Ы8>
Для этого случая при практически полном отсутствии потерь на
первой и третьей стадиях (0А, i = ₽A, з=₽в, з=₽о, з= 1) выход ко-
нечного продукта D равен единице независимо от степени превра-
щения сырья. При этом выход промежуточного продукта В остается
равным нулю.
Если по каким-нибудь соображениям возврат непревращенного
сырья невозможен или нежелателен, т. е. рА,з = 0 (например, про-
дукт D выпускается как товарный в смеси с веществом А и т. д.),
выход продукта D в расчете на поданное сырье будет описываться
выражением
Рв = “Рл, i₽£>, з (Рв, 2 + Рв, гРв, з) (М9)
из которого следует, что при практически полном отсутствии по-
терь « а.
Если схема, представленная на рис. 1-5, рассчитана на выпуск
обоих продуктов последовательной реакции (при полном исполь-
20
зовании сырья), для выражения технологических выходов про-
дуктов В и D могут быть использованы уравнения (1-16) и (1-17).
В отличие от параллельной реакции одного кинетического по-
рядка, где выход продуктов не зависит от степени превращения
сырья, для последовательной реакции отношение Рд/Рд опреде-.
ляется значением а и при практически полном химическом пре-
вращении исходного вещества Р^->0 и р£ ~>РЛ 1РП З-
Решение системы уравнений (1-9) — (1-15) позволяет также
установить зависимость размера потоков сырья от степени его пре-
вращения в реакционном агрегате:
^0 4" ^4 ^4 О , п о /у \
д — д В = А _____1_______
Л2-Д1рл, 1 з(1 _а)
. _ . Рд. 1Рд. 3 (1 ~ а)
4 ° ’-PaJW1-'»
(1-20)
(1-21)
(1-22)
Отсюда нетрудно получить значение коэффициента рециркуля-
ции (/(л), широко используемого в работах Нагиева [6]:
к _____________1
(Ь23)
Из уравнений (1-20) — (1-23) следует, что при отсутствии по-
терь сырья на первой и третьей стадиях размер потоков исходного
вещества описывается более простыми выражениями:
Д1 = д2 = д0—
4 Л 1 ~а
Д4 — -
К -А-1
До а
Из структуры выражений (1-16) — (1-19) следует, что техноло-
гический выход продуктов не только последовательных, но и па-
раллельных реакций определяется степенью превращения исход-
ного вещества, а также относительной величиной потерь сырья
(Ра, 1, Рд, з) на первой и третьей стадиях и продуктов (Рв. з, Pd, з)
на третьей стадии.
Ясно, что при постоянных условиях механической или физико-
химической переработки любого продукта неизбежные потери
этого продукта как в абсолютном, так и в относительном выраже-
нии находятся в зависимости от размера перерабатываемого по-
тока этого продукта, т. е. от степени превращения сырья. Для
получения точного значения выхода продукта технологического
процесса необходим учет указанной зависимости.
21
При отсутствии данных о форме связи рг-, t(a) и Р,-,з(а) Для
приблизительной оценки значений выхода приемлемым является
допущение независимости рг, i и рг, 3 от величины а в достаточно
узком интервале значений степени превращения.
Величины рл, 1 и Ра,з. как правило, близки к единице (0,97—
0,99); относительные потери продуктов (рв.з, Ро, з) сравнительно
мало зависят от степени превращения сырья при изменении ее в
небольшом интервале.
Принимая Ра, 1 = рА,з=Рв,з = ро, з=0,98, можно оценить значе-
ния выхода продуктов
Рис. 1-6. Зависимость выхо-
дов продуктов параллель-
ной реакции А--------> В,
A—->D (£2/*i=0,33) от
степени превращения исход-
ного сырья в условиях про-
мышлениогопроцесса(Рл ,=
*“₽д, з = ₽в, з = ₽£>, з = •
а
для рассмотренных параллельной и по-
следовательной реакций как функцию
степени превращения исходного веще-
ства А.
В случае параллельной реакции од-
ного кинетического порядка выход про-
дуктов на стадии химического превраще-
ния определяется соотношением констант
скорости реакции
r = k'
РЛ. 2 ki + ki
о **
Р0-2 ki+kt
и не зависит от степени превращения.
Однако выход каждого из этих продуктов
при осуществлении процесса по схеме с
возвратом непревращенного сырья будет
меняться по мере изменения степени пре-
вращения исходного вещества (рис. 1-6),
монотонно возрастая с увеличением а.
Выходы рв> 2, ро, 2 продуктов последо-
вательной реакции зависят от степени
превращения исходного вещества. При отсутствии уравнения, опи-
сывающего функцию pt>2(a), можно воспользоваться эксперимен-
тально найденными данными.
Предположим, что экспериментальные результаты, полученные
при проведении последовательной реакции в аппарате непрерыв-
ного действия с мешалкой (реакторе полного смешения), пред-
ставлены кривой, выражающей связь между степенью превраще-
ния исходного сырья и выходом продукта В (верхняя кривая на
рис. 1-7, а).
Для оценки выхода продукта В при проведении промышлен-
ного процесса в тех же условиях (аппарат непрерывного действия
с мешалкой) по схеме, изображенной на рис. 1-5, можно восполь-
зоваться уравнением (1-16). На рис. 1-7, а даны результаты рас-
чета для двух вариантов значений выходов на первой и третьей
стадиях (средние кривые —для рл, i = Pa, з = рв, з = рв,з=0198, ниж-
ние кривые —для рА, 1 = ₽а,з = ₽в,з = ро, з = 0,95).
22
Кривые ₽g (а) имеют экстремальный характер. Абсцисса точки
экстремума определяется только размером относительных потерь
сырья (Рд, 1, Ра, з). Значение ре в точке экстремума зависит не
только от расхода сырья на первой и третьей стадиях, но и от ве-
личины потерь продукта В при выделении его из реакционной
массы.
С увеличением потерь сырья и продукта В экстремальная точка
перемещается в область больших значений а и сопровождается
уменьшением величины р'. Поиск оптимума может быть основан
на экспериментально установленной зависимости Рв, г(а); задача
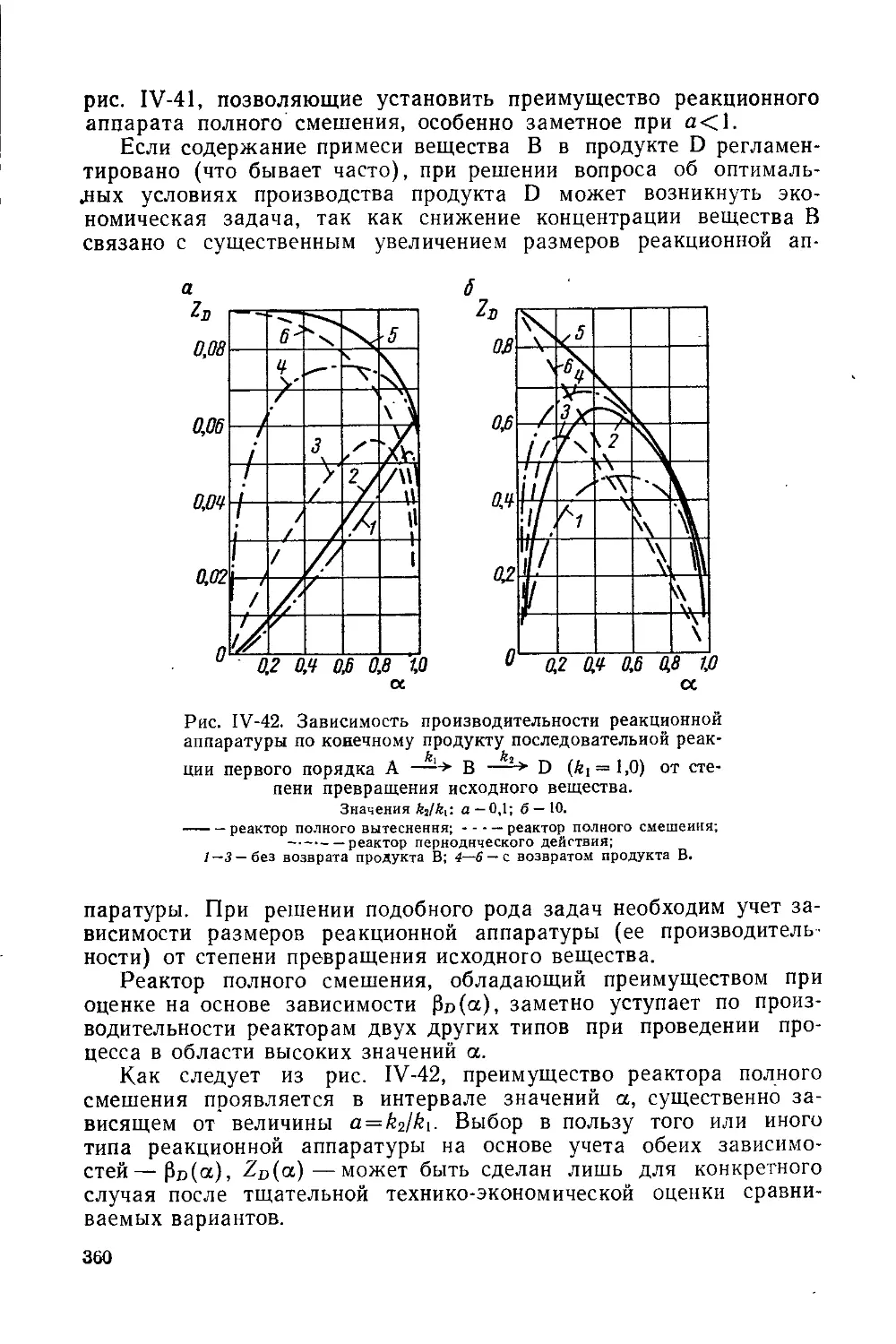
Рис. 1-7. Зависимость выхода промежуточного и конечного
продуктов последовательной реакции А—-* В —D
(й2/А1 — 2) от степени превращения исходного вещества А в ус-
ловиях промышленного процесса (реактор полного смешения).
решается путем построения кривой Pg (а) по точкам с помощью
уравнения (1-16).
Аналитическое решение задачи возможно, если зависимость
Рв,г(а) описывается достаточно простым выражением. В качестве
простейшего примера можно привести результат расчета для по-
следовательной реакции, осуществляемой в аппарате непрерывного
действия с мешалкой, для которого рв, 2=1 — а. В этом случае
„ V1-₽A,1₽A.3-1
«ОПТ — А Я-------------
Ра, >Ра, з
Значения рЛ, i и рЛ, 3 устанавливаются по аналогии с родствей-
ными производствами. При отсутствии таких аналогий для вновь
пускаемого производства произведение РадРа, з определяется экс-
периментально (см. стр. 32).
Выход продукта D, получаемого в тех же условиях, что и про-
дукт В, представлен кривыми на рис. 1-7, б [верхняя кривая иа этом
рисунке получена по кривой Рв, 2 (а) рис. 1-7, а, исходя из равен-
ства рв, 2=1 — Рв, г]-
23
Общий технологический выход продуктов В и D, равный еди-
нице при отсутствии потерь, в реальных условиях схемы выра-
жается уравнением:
0в + Рд ~
Ра, 1а[Рр,2(Рр, з~Рв,з) + Рв,з]
1 ~ Ра, 1Ра, з U — а)
< 1
об
Рис. 1-8. Зависимость выхо-
да конечного продукта по-
следовательной реакции
А—>В—>D от .степени
превращения исходного
сырья при возврате (кри-
вая /) н выводе (кривая 2)
промежуточного продукта.
Когда целевым продуктом является D и процесс проводится
с возвратом исходного вещества А и промежуточного продукта В
на повторное их использование, выход продукта D, равный еди-
нице при отсутствии потерь, в реальных условиях схемы стано-
вится зависимым от степени превраще-
ния сырья. На рис. 1-8 представлены
результаты расчета по уравнению (1-18)
при условии Ра, 1 = Ра, з = Рв,з=Рр, з = 0,98
(кривая /). Для сравнения приведена
также кривая 2 для случая, когда про-
межуточный продукт не возвращается на
стадию химического превращения. Из
рисунка следует, что влияние степени
превращения заметно лишь при низких
значениях а, когда потоки сырья, про-
ходящего первую и третью стадии, зна-
чительны.
Рассмотренные выше закономерности
относились к схеме, которая включает
лишь одну стадию химического превра-
щения сырья. Промышленный синтез ор-
ганических продуктов нередко основан
на процессах, состоящих из нескольких
химических стадий, вследствие чего связь
выхода конечного продукта и показателей отдельных стадий со-
ответственно усложняется.
Рассмотрим два способа промышленного синтеза продукта D
из основного сырья А:
С] С2
А\ : в\
D
. Каждая из написанных выше параллельных реакций протекает с
образованием побочного продукта (Ci, С2, С3), не имеющего ПО'
требительской ценности (отход).
24
Если для упрощения принять, что степень превращения исход-
ного вещества в обоих случаях практически равна единице, то
схемы материальных потоков будут иметь вид, представленный на
рис. 1-9.
Рис. 1-9. Схемы материальных потоков производства продукта D:
а — по первому способу
Выход продукта D для первого и второго случая можно выра-
зить следующим образом:
Рд (I) = Рл, 1 (1)Рв, 2 (1)Рв, 3 (1)Рв, 4 (1)Рд, 2 (1)Рд, 3 (I) (1-24)
Рд (П) = Рл, 1 (П)Рд, 2 (11)Рд. 3 (II) (!'25)
Если принять потери на каждой нехимической стадии равными
2 масс. % от потока сырья, то сравнительная оценка процессов
упрощается:
Рд (I) = О,984РВ_ 2 (1)Рд, 2 (I)
Рд (п)= °>982Рд, 2 (II/
Основным показателем промышленного процесса является вы-
ход продукта из основного вида сырья, поэтому конкурентоспо-
собность рассматриваемых способов производства продукта D воз-
можна при условии:
Рд (I) > Рд (II)
Следствием этого условия является неравенство:
0,982Рв>2(1)Рд,2(1) > Рд, 2 (II)
Допустив, что выход продукта D по второму варианту равен
80% (Рд, 2(П)=0,8), и считая для упрощения, что ₽в, 2® = ₽д, г®, по-
лучим условие конкурентоспособности сравниваемых вариантов-:
РВ.2(1) = РД.2(1)>У
25
Осуществление процесса по первому способу предъявляет
весьма высокие и часто недостижимые требования к условиям про-
ведения химических реакций.
Сравнительная оценка двух рассматриваемых вариантов
процесса получения продукта D приводит к выводу в пользу
второго из них.
Пример 1-1. Экспериментальные результаты, полученные при изуче-
нии реакции хлорирования бензола молекулярным хлором (СвНв -> СвН5С1 ->
->С6Н4С12) в реакторе периодического действия и реакторе непрерывного дей-
ствия, снабженном мощной мешалкой, при 40—50° С, представлены в виде за-
висимостей РСвн5с1 (“) и ₽свн4С12 («) в табл- 1'2-
Т а б л и ц а 1-2 >
Зависимость ₽СбНеС1 и РСбЩС12 от а дли реакции
хлорирования бенэонв молекулярным хлором
а Реактор периодического действия Реактор непре- рывного действия! с мешалкой | а Реактор периодического действия Реактор непре- рывного действия с мешалкой
₽СвН5С1 ₽С6Н4С12 ₽СвН5С1 ₽С6Н4С12 ЭС6Н5С1 ₽СвН4С12 РС6Н5С1 ₽С6Н4С12
0,05 0,994 0,006 0,50 0,952 0,048 0,892 0,108
0,10 0,994 0,006 0,986 0,014 0,60 0,936 0,064 0,846 0,154
0,20 0,980 0,020 0,970 0,030 0 70 0,916 0,084 0,780 0,220
030 0,975 0,025 0,950 0,050 0,80 0,884 0,116 0,675 0,325
0,40 0,967 0,033 0,920 0,080 0,90 0,830 0,170 0,480 0,520
Перед началом технологических разработок требуется обосновать выбор
типа реакционного аппарата (реактор^периодического действия, реактор непре-
рывного действия с интенсивным перемешиванием) и оценить оптимальные зна-
чения степени превращения бензола прн проведении процесса по схеме, изобра-
женной на рис. 1-5 (монохлорбеизол — целевой продукт, т. е. 6=0).
Предполагается, что при разделении моиохлорбензола и смеси дихлорпро-
изводных методом ректификации содержание монохлорбензола в кубовой жид-
кости составляет 10 масс.% и не превышает 0,1 масс.% в дистилляте. Потери
монохлорбензола и смеси дихлорпроизводных на третьей стадии составляют по
1 масс.%, потери бензола на первой и третьей стадиях — по 2 масс.%.
Решение. Обозначив бензол буквой А, монохлорбензол — В и
смесь дихлорпроизводных — D, воспользуемся уравнением (1-16).
При указанных потерях бензола иа первой и третьей стадиях значения
0a,i=Pa,s=O,98. Потери монохлорбензола при его выделении из реакционной
массы, оцениваемые значением pa,s, могут быть подсчитаны, исходя из задан-
ного содержания монохлорбеизола в кубовой жидкости и потерь в результате
испарения.
Отношение числа молей монохлорбензола к числу молей дихлорпроизводных
в кубовой жидкости:
10/112,5
90/147
«0,145
где 112,5; 147—молекулярные массы монохлорбензола и дихлорпроизводных
бензола соответственно,
26
Пренебрегая содержанием дихлорпроизводиых в моиохлорбензоле, выражаем
величину рв,з следующим образом:
В. В-Ъ,]АЫ. ( DA ( ₽п2\
6 „ , = 0,99 = 0,99 ——б = 0,99 1 - 0,145 = 0,99 1 - 0,145 -^=-
в‘3 Вз Вз \ вз/ \ PB.2J
Для удобства интерполирования экспериментальных результатов по дан-
ным табл. 1-2 строим кривые 2(а) для сравниваемых типов реакционных ап-
паратов (рис. 1-10, а).
Рис. 1-10. К примерам 1-1 и 1-2:
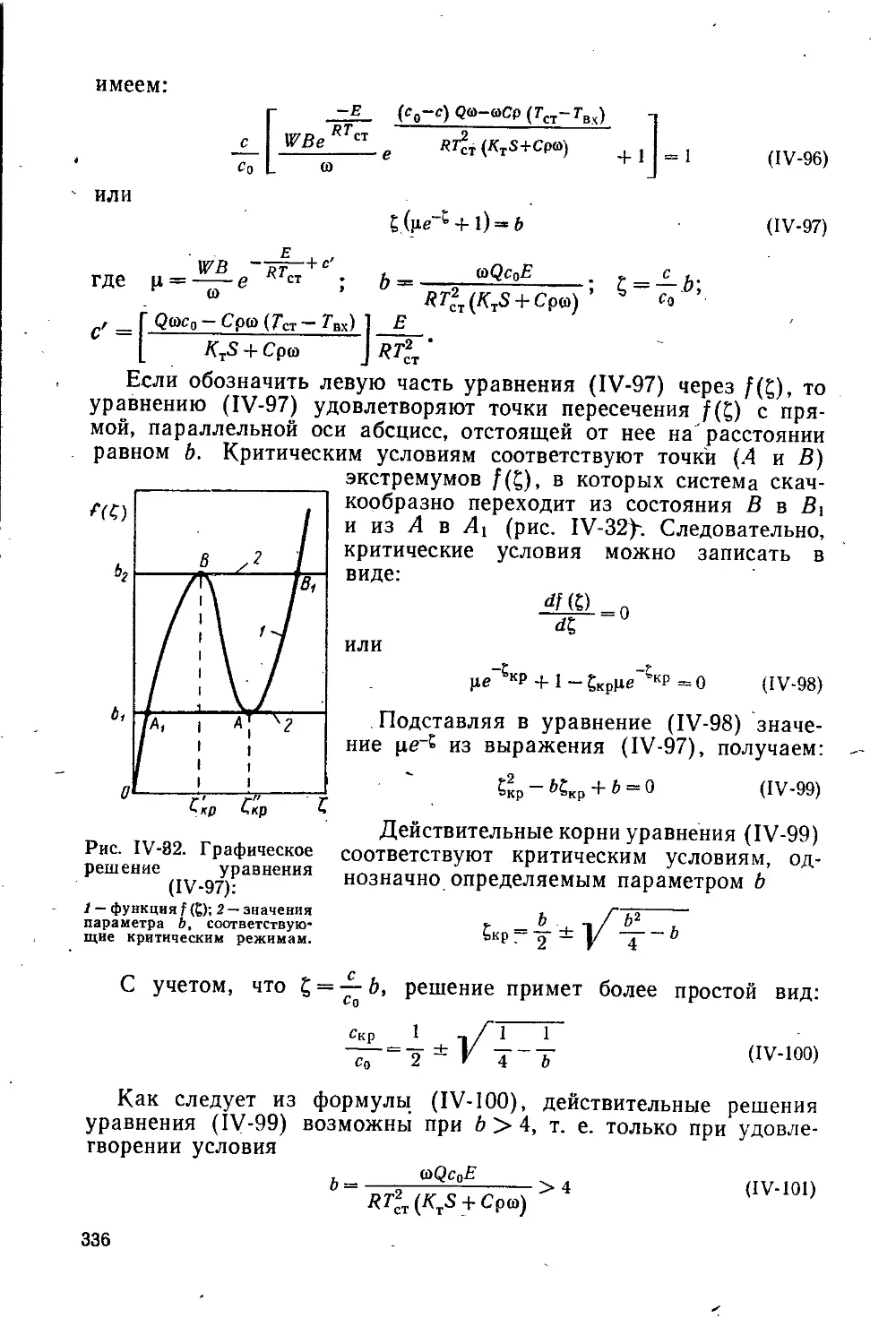
а — зависимость выходов моиохлорбёизола от степени превращения бензола
Номера кривых
₽В,2<°> Эд (а)
Реактор периодического действия....... 1 3; 5
Реактор непрерывного действия с мешал-
кой ...................................... 2 4
5 — зависимость выходов смеси дихлорпроизводиых бензола от степени превращения бензол
Номера кривых
₽D, 2 <“) Эр (а)
Реактор периодического действия....... 1 3
Реактор непрерывного действия с мешал-
кой ...................................... 2 4
Пользуясь уравнением (1-16) и кривыми 1 и 2 рис. 1-10, а, получаем ряд
значений рд (а), сведенных в табл. 1-3.
Из табл. 1-3 и рис. 1-10, а (кривые 3 и 4) видно преимущество реактора
периодического действия: максимальный выход, равный 0,882, достигается при
степени превращения бензола 0,5 (кривая 3).
Для оценки эффекта достигаемого в результате совершенствования техно-
логии и аппаратуры процессов на первой и третьей стадиях, предположим, что
при новой технологии рл,1 = рл,з=0,99.
Повторив расчет для реактора периодического действия, получим зависи-
мость, выражаемую кривой 5 на рис. 1-10, а. Видно, что максимальный выход
(0,915) достигается при степени превращения бензола, равной 0,4.
27
Таблица 1-3
Зависимость $'в от а
а Реактор пери- одического действия Реактор непрерывного действия с мешалкой а Реактор пери- одического действия Реактор непрерыв- ного действия с мешалкой
0,05 0,538 0,40 0,880 0,822
0,10 0,696 0,692 0.50 0,882 0,818
0,20 0,818 0,812 0,60 0,875 0,760
0,30 0,856 0,832 0,70 0,862 0,714
Пример 1-2. Пользуясь исходными данными примера 1-1, обосновать
выбор типа реакционного-аппарата н оценить оптимальную степень превращения
бензола для двух вариантов эксплуатации схемы, приведенной на рис. 1-5:
1) производство смеси дихлорпроизводных бензола состава, указанного в
примере 1-1;
2) производство смеси моно- и дихлорпроизводных бензола состава 3: 1 (со-
отношение молей).
Решение. 1. Для производства смеси дихлорпроизводных бензола
необходимо проведение процесса либо при степени превращения бензола, близ-
кой к единице, либо по схеме, изображенной на рис. 1-5, при возврате моно-
хлорбензола на стадию химического превращения. Учитывая чрезмерно большую
продолжительность реакций первого кинетического порядка при степени превра-
щения, близкой к единице, первый из указанных способов осуществления про-
цесса ие заслуживает рассмотрения. Решение задачи применительно ко второму
способу основано на использовании уравнения (1-18).
Приняв Ра,1 = Рд,з=0,98, Ро,з=0,99, Ро,а(«)—см. пример 1-1, —по уравне-
нию (1-18) подсчитываем значения Рд (а) для сравниваемых типов реакцион-
ного аппарата. Результаты расчета представлены на рис. 1-10,6 (кривые 3 и 4).
Из них следует, что сравниваемые типы реакторов практически равноценны
2. Монохлорбензол и дихлорпроизводные бензола в заданном соотношении
могут быть получены в каждом из сравниваемых типов реакционной аппаратуры
при степени превращения бензола, соответствующей условию рв = ЗРд.
Из используемых в данном случае уравнений (1-16) и (I-J.7) получим:
OZ О О
нв 3 р-8.2Р-8.з
₽D,2₽D,3
При условии, что Pb,3=Pd,3=0,99, имеем:
Рв _ Рв, 2 _ 3
Ро Ро,2
Отсюда Рв,2=0,75; [Jo,2=0,25.
Из рис. 1-10, а следует, что требуемое соотношение достигается в реакторе
периодического действия при степени превращения бензола, равной 0,94, а в ре-
акторе непрерывного действия с интенсивным перемешиванием при а=0,714-
-=-0,72. .Суммарный выход смеси продуктов в каждом из реакторов прн этом
равен:
р Рл,1Рв,за
Рг 1-РЛ(1Рл>з(1-«)
28
Для сравнительной оценки реакционных аппаратов найдем отношение сум-
марного выхода, достигаемого б каждом из них:
Рп.д = «n.A[l-P4.^.3(»-«H.„)] = 0.94 [1-0,9841-0,72)]
₽н.д «н.д[1-₽Л.1₽Л,з(1-«п.д)] 0,72 [1-0,9841-0,94)]
(индекс «п. д» относится к реактору периодического действия, индекс «н. д» —
к реактору непрерывного действия с интенсивным перемешиванием). Получен-
ный результат приводит к выводу в пользу реактора периодического действия.
СТРУКТУРА СЕБЕСТОИМОСТИ ПРОДУКТА
ПРОМЫШЛЕННОГО СИНТЕЗА
Экономическая эффективность химико-технологического
процесса определяется себестоимостью продукта и размером ка-
питальных затрат, связанных с организацией производства этого
продукта.
Себестоимость продукта промышленного производства яв-
ляется денежным выражением стоимости или общественных из-
держек производства продукта за вычетом той части, которая идет
на образование чистого дохода.
В себестоимости продукта находят отражение объем производ-
ства, степень полезного использования сырья и энергетических
Таблица 1-4
Структура себестоимости химической продукции
некоторых отраслей промышленности [7]
Элементы затрат Величина затрат, %
азотная промышлен- ность промышлен- ность основной химии (щелочи, кислоты, соли) аиилиио-кра- сочная промышлен- ность горно-хими- ческая промышлен- ность
Сырье н основные мате- риалы 21,2 54,6 58,2 6,5
Вспомогательные мате- риалы . 6,6 6,0 6,3 11,3
Топливо 7,1 4,2 4,0 12,9
Электроэнергия, пар . . . 24,0 4,2 3,6 4,5
Амортизационные отчисле- НИЯ 13,7 5,6 4,6 8,8
Заработная плата (основ- ная я дополнительная) н отчисления на соци- альное страхование . . . 23,4 22,4 18,3 51,3
Прочие денежные расходы 4,0 3,0 5,0 4,7
Всего: 100,0 100,0 100,0 100,0
29
средств, производительность аппаратуры и оборудования, уро-
вень организации труда и ряд других показателей производствен-
ного процесса.
Затраты предприятия, связанные с производством данного про-
дукта, составляют заводскую себестоимость. Заводская себестои-
мость, дополненная затратами, связанными со сбытом готового
продукта, а также затратами на подготовку кадров предприятия,
проведение научно-исследовательских работ и т. п., образует пол-
ную (коммерческую) себестоимость.
Поскольку технологические вопросы решаются в процессе про-
изводства продукта внутри предприятия и внезаводские затраты
не находятся в прямой связи с процессом производства, для выяв-
ления задач технологии в первую очередь представляет интерес
анализ структуры заводской себестоимости продукта химического
производства.
Структура себестоимости химической продукции некоторых от-
раслей промышленности приведена в табл. 1-4.
Структура себестоимости различных продуктов промышленно-
сти основного органического синтеза дана в табл. 1-5 (приведены
характеристики ряда производств, отличающихся количеством тех-
нологических стадий и условиями переработки сырья).
Т а б л и ц а 1-5
Структура себестоимости различных продуктов
промышленности основного органического синтеза
Элементы затрат Величина затрат, %
нитрил акри- ловой кислоты (из пропилена) фенол (из бензола че- рез кумол) ДМТ (из л-кснлола) бензойная кислота (нз толуола) капролактам н адипино- вая кислота (нз бензола)
Сырье, основ- ные и вспо- могательные материалы 65 58 60 72 60
Энергетичес- кие средства 20 22 15 8 15 '
Амортизаци- онные отчисле- ния .... 6 6 10 9 10
Заработная плата и про- чие денеж- ные расходы 9 14 15 11 15
Всего: 100 100 100 100 100
Из табл. 1-5 следует, что основу себестоимости продукта состав-
ляют энерго-материальные затраты, величина которых опреде-
30
f * - "'Ч
k '
дяется расходом сырья, энергетических средств и размером амор-
тизационных отчислений.
Несмотря на существенное различие технологии производств,
представленных в табл. 1-5, доля энерго-материальных затрат от
себестоимости продукта этих производств в достаточной степени
; постоянна и составляет 85—90%.
СЫРЬЕ И ОСНОВНЫЕ МАТЕРИАЛЫ
Стоимость сырья и основных материалов, расходуе-
мых на единицу готового продукта, составляет, как следует из
табл. 1-5, около 60—70% себестоимости этого продукта.
Ясно, что удельный расход А каждого из видов сырья обратно
пропорционален выходу целевого продукта, рассчитанному по
этому виду сырья, и для многостадийных процессов может быть
описан выражением:
V,- 1
4 = (1‘26)
^прод **
где Мисх, Мпрод — молекулярные массы исходного вещества и го-
тового продукта; v', v" — стехиометрические коэффициенты; ₽' —
выход целевого продукта, отнесенный к данному виду сырья.
Вполне очевидно, что первая статья себестоимости готового
продукта складывается из стоимости отдельных видов сырья и
определяется не только их удельным расходом, но и стоимостью
единицы каждого вида сырья.
Выше было показано, что выход продукта многостадийного
процесса является сложной функцией расходов сырья, промежу-
точных и готового продуктов на каждой технологической стадии.
Значение выхода, характеризующего работу каждого агрегата
или стадии, в свою очередь зависит от комплекса условий проведе-
ния того или иного единичного процесса. Пользуясь обозначе-
ниями, принятыми на рис. 1-5 и в приведенных выше уравнениях,
и рассматривая эту зависимость даже в самом общем виде, мож-
но установить форму связи величины выхода и основных пара-
метров, влияющих на выход продукта.
Потери сырья при проведении операций, составляющих первый
этап его переработки, вообще говоря, зависят от свойств этого
сырья, условий его транспорта, хранения, нагрева или охлажде-
ния, изменения агрегатного состояния, типа и количества аппара-
туры и оборудования, качества их монтажа, квалификации обслу-
живающего персонала и т. п.
При одинаковых условиях переработки одного и того же вида
сырья размер его относительных потерь (1 — ₽д,i), как правило,
связан с величиной потока этого сырья. Общим выражением мо-
жет служить:
Рд. 1 = Ф(Л1- Т’р’ «*-••)
31
где Л] (а)—количество сырья, поступающего в единицу времени
на первый этап переработки; Т, Р, ц, ... — параметры элементар-
ных процессов, составляющих первый этап переработки сырья, и
физико-химические свойства сырья.
По понятным соображениям количественная связь 0А, i и Д1(а)
в общем виде выражена быть не может. Однако для конкретных
условий представляется возможным установить эту форму связи
путем сравнения размера относительных потерь для двух различ-
ных по размеру потоков (при условии, что размер аппаратуры ме-
няется с изменением потока сырья). Например, оценка потерь
жидкости в результате испарения со свободной поверхности в хра-
нилищах либо потерь жидкости (газа) через неплотности в разъем-
ных соединениях аппаратов и трубопроводов для двух случаев
(соответственно 0^ , и 0” j) приводит к выражениям:
потери в результате испарения
/ А1 \»
\ л2 /
потери через неплотности
/ А1 \г
\ л2 /
(у, г— некоторые постоянные величины). Эти выражения позво-
ляют сделать вывод, что для случаев, когда размер аппаратуры и
трубопроводов меняется соответственно изменению размера потока
сырья, величины у и г заключены в пределах 0<#<1 и 0<z<l.
Ясно, что при сохранении размеров аппаратуры и соответствую-
щем увеличении числа агрегатов значение выхода сохраняется:
^=2=0; 0” ,=0*...
При работе с газами или летучими жидкостями в производ-
ственных условиях потери сырья в результате неудовлетворитель-
ной работы систем улавливания (конденсаторы, абсорберы, адсор-
беры) и за счет утечки через неплотности в отдельных случаях
могут быть весьма заметными. Размер этих не поддающихся рас-
чету потерь может быть с достаточной степенью точности установ-
лен в период так называемой «обкатки на рабочих средах», т. е.
на основе баланса сырья, перерабатываемого во всех аппаратах
технологической схемы в рабочих условиях (температура, давле-
ние), но без проведения химической реакции (а=0).
Из уравнения (1-22) при а=0 получим:
Рл. 1Рл.з= л4 + л0
При постоянстве потока сырья (Д4=const) убыль исходного ве-
щества в хранилищах соответствует значению До-
Выход 0{> 2 продукта химической реакции является функцией
ряда параметров процесса, причем внд этой функции зависит от
32
схемы и природы химической реакции и от того, какой из продук-
тов (если образуется ряд продуктов) является целевым.
Для процессов промышленного органического синтеза харак-
терна сложная схема химических реакций, включающая комплекс
параллельно и последовательно протекающих превращений, за-
канчивающихся образованием ряда продуктов. В этих условиях
неизбежна зависимость выхода большинства продуктов от сте-
пени превращения исходного вещества. Зависимость эта обычно
является либо монотонно возрастающей, либо монотонно убываю-
щей;.случаи экстремальной зависимости сравнительно редки (про-
межуточные продукты многостадийной последовательной реакции).
В отличие от простых необратимых и обратимых реакций, где
выход продукта (всегда равный единице) не зависит от парамет-
ров процесса, для сложных реакций изменение параметров при
одной и той же степени превращения может влиять на соотноше-
ние констант скоростей отдельных реакций, обусловливая тем са-
мым зависимость выхода от температуры, давления, каталитиче-
ского и инициирующего воздействия добавок и т. д.
Сложная схема большинства реакций органического синтеза
обусловливает также связь относительной скорости отдельных пре-
вращений и характера изменения концентрации исходного веще-
ства и промежуточных продуктов реакции в процессе ее проведе-
ния, создавая, таким образом, зависимость выхода продукта от
гидродинамического режима в реакционном пространстве, харак-
теризующегося значением критерия Боденштейна (Во).
Общее выражение зависимости выхода продукта от условий
проведения химического процесса можно представить следующим
образом:
Р», г = 'Ф (“ Т> Р, К, Во,...)
где К — катализатор.
Расход сырья и готового продукта на стадиях, связанных с раз-
делением смеси продуктов реакции, вызывается не только механи-
ческими потерями, но и является результатом нежелательных хи-
мических превращений, протекающих при повышении температуры
реакционной массы, каталитическом действии материала аппара-
туры и т. п. и определяемых как свойствами компонентов разде-
ляемой смеси, так и условиями ре разделения. Общим выражением
для выхода на этом этапе может служить:
Рш ~f(G3, T,P,kbk2,...)
где G3(a) =Лз+Вз+£)з—количество реакционной смеси, поступаю-
щее в единицу времени на стадию разделения; Т, Р — параметры
процесса разделения; k\, k2 — кинетические показатели побочных
реакций, протекающих в условиях разделения реакционной смеси.
Кроме рассмотренных выше основных технологических стадий,
расходом сырья (в форме потерь готового продукта) сопрово-
ждается очистка, хранение, расфасовка и транспорт готового про-
дукта, что должно учитываться при определении'значения 0'.
3 В. С. Хайлов, Б. Б Брандт 33
Как правило, выходы на стадиях подготовки сырья и разделе-
ния реакционной смеси значительно превосходят выход на стадии
химического превращения, которым в основном и определяется
удельный расход всех видов сырья.
Степень влияния величин р,-, i и рг-, 3 на выход продуктов в зна-
чительной мере зависит от а. Для количественной оценки этой
зависимости могут служить уравнения (1-16)—(1-18).
ЭНЕРГЕТИЧЕСКИЕ СРЕДСТВА
Все виды энергетических средств, расходуемых на про-
изводство продукта, можно разбить на две группы по степени
связи с технологией промышленного синтеза: 1) расход на перера-
ботку сырья; 2) расход на вспомогательные нужды (отопление,
освещение и вентиляцию производственных помещений, работу
станков механической мастерской и т. д.). Первая из этих групп
является определяющей.
На стадии предварительной обработки сырья энергетические
средства расходуются на перемещение сырья, сжатие газов, на-
грев или охлаждение сырья и перевод его из одного агрегатного
состояния в другое для доведения его параметров до уровня, опре-
деляемого требованиями реакционного агрегата.
Расход тепловой энергии (Q') определяется начальными и ко-
нечными параметрами состояния и свойствами исходного вещества
(теплота фазовых превращений г), а также количеством перера-
батываемого сырья.
Расход механической энергии (Qi) зависит от разности конеч-
ного и начального давлений газообразных продуктов, высоты подъ-
ема жидкостей, а также от количества перерабатываемого сырья.
Общим выражением может служить:
Q( + Ql=f0!, ДГ, ДР,г, С, ...)
где Л1(а) —количество сырья, поступающее в единицу времени на
первый этап переработки; ДТ, ДР, г, С — изменение параметров
процессов и теплофизические константы сырья.
Расход энергетических средств на стадии химического превра-
щения сырья связан, как правило, с поддержанием заданного тем-
пературного режима химического процесса (нагрев, охлаждение)
и с перемешиванием реакционной смеси.
Удельный расход тепловой энергии, расходуемой на поддержа-
ние температуры реакционной смеси в случае проведения экзотер-
мического или эндотермического процесса, определяется величи-
ной теплового эффекта реакции, а стоимость энергетических
средств — температурным уровнем.
Тепловой эффект сложной реакции (сумма тепловых эффектов
отдельных превращений), отнесенный к единице основного целе-
вого продукта, зависит от степени превращения исходного веще-
ства. Количество тепла, подводимого или отводимого от реакцион-
34
ной смеси для поддержания заданного температурного режима
процесса, тоже определяется значением а, так как водяной' экви-
валент реагирующего потока, отнесенный к единице содержаще-
гося в нем основного продукта, находится в обратной зависимости
от степени превращения сырья.
Общим выражением расхода тепловых энергетических средств,
' связанного с поддержанием температурного режима (Q2). может
служить:
где 2 (а) — суммарный тепловой эффект сложной реакции, от-
. несенный к единице основного продукта; С'(а) —отнесенное к еди-
нице основного продукта количество тепла, необходимое для изме-
нения температуры реакционной смеси на 1 град.
Потребление механической энергии, расходуемой на переме-
щение или перемешивание реакционной.смеси (Q2), зависит от гид-
родинамического режима перемешиваемой массы и от ее количе-
ства:
Q2 = 4j(A2, Т, Р, Re, ...)
где Л2(а)—количество сырья, поступающего в единицу времени
в реакционный агрегат; Re — критерий Рейнольдса.
Стадия разделения смеси продуктов реакции и очистки гото-
вого продукта также связана с расходом механической и тепловой
энергии. Величина расхода и параметры тепловых энергетических
средств целиком определяются свойствами разделяемых систем,
схемой и аппаратурой, принятой для осуществления процесса раз-
деления.
Когда реакционная смесь представляет собой смесь жидко-
стей, расход тепла на ректификацию определяется не только коли-
чеством реакционной смеси, но и содержанием в ней непрореаги-
ровавшего сырья (как правило, обладающего более высоким дав-
лением паров, чем продукт синтеза), т. е. степенью превращения
сырья на стадии химического взаимодействия.
Расход энергетических средств на стадии разделения. (Q3) в об-
щем виде можно выразить зависимостью:
Q3 = H>(G3, At, Т, Р, г, С,...)
где G3(a)—количество реакционной смеси, поступающее в еди-
ницу времени на стадию разделения; Д4(а)—количество непре-
вращенного сырья, отделяемого от реакционной смеси в единицу
времени; Т, Р, г, С — параметры процессов разделения и тепло-
физические свойства разделяемых продуктов.
Учитывая расход энергии на дополнительную очистку продукта,
его транспорт и т. д. (Q4), суммарный расход энергетических
средств на единицу готового продукта может быть выражен урав-
нением:
2Q = Q{ + Qi + Q2 + Q2+Q3 + Q4 (1-27)
з*
35
Естественно, что стоимость энергетических средств опреде-
ляется их параметрами и в значительной степени зависит от усло-
вий конкретного производства.
В качестве примера рассмотрим одну из современных много-
стадийных схем производства капролактама, включающую разно-
характерные процессы и аппаратуру. Технологическая схема со-
стоит из пяти основных химических стадий:
1) получение циклогексана (парофазное гидрирование бензола
на сульфидных катализаторах);
2) получение циклогексанона (жидкофазное каталитическое
окисление циклогексана);
3) получение циклогексаноноксима (взаимодействие циклогек-
санона и гйдроксиламина);
4) получение сернокислой соли капролактама (изомеризация
циклогексаноноксима в олеуме);
5) получение капролактама (нейтрализация сернокислой соли
капролактама водным раствором аммиака, выделение и очистка
капролактама). '
Анализ проектной документации позволяет распределить энер-
гетические средства, потребляемые на каждой стадии синтеза, по
основным технологическим стадиям переработки сырья (табл. 1-6).
Таблица \-f>
Распределение энергетических средств,
потребляемых на различных стадиях синтеза капролактама
(в % от суммарной стоимости энергетических средств
на каждой основной стадии)
Несмотря на некоторую условность разграничения этапов, не-
избежно допускаемую при рассмотрении технологической схемы,
порядок величин, приведенных в табл. 1-6, достаточно обоснован
и позволяет установить, что определяющей является последняя
технологическая стадия, при осуществлении которой расходуется
до 96% суммарной стоимости энергетических средств. Расход же
энергетических средств на работу реакционного узла в основном
относительно невелик.
36
АМОРТИЗАЦИОННЫЕ ОТЧИСЛЕНИЯ
Амортизация основных фондов промышленности пред-
ставляет постепенное возмещение их физического и морального
износа в процессе эксплуатации.
Амортизационные отчисления, включаемые в себестоимость
продукции, служат источником финансирования воспроизводства
(ремонт, полная или частичная замена износившихся деталей или
узлов) зданий, сооружений и производственного оборудования.
При составлении калькуляции применяются дифференциро-
ванные нормы амортизации. В зависимости от конструкции зда-
ний и сооружений, материалов и условий эксплуатации аппара-
туры и оборудования размер амортизационных отчислений колеб-
лется в довольно широких пределах и соответствующая статья
затрат представляет собой сумму амортизационных отчислений на
капитальный ремонт и восстановление зданий, сооружений и обо-
рудования, а также других объектов основного и вспомогатель-
ного производственного назначения.
Порядок значений амортизационных отчислений для предприя-
тий промышленности основного органического синтеза можно ви-
деть из данных, приведенных в табл. 1-7.
Таблица 1-7
Амортизационные отчисления для предприятий
промышленности основного органического синтеза (в %] [8]
Группы и виды основных фондов Общая норма амортизацион- ных отчисле- ний В том числе
на капиталь- ный ремонт на полное восстанов- ление
Производственные здания (каменные, бетонные, железобетонные) Машины и оборудование: колониые аппараты для производ- ства синтетического каучука . . 2,5-3,2 1,5-1,7 1,0-1,5
18,1 10,4 7,7
колонные аппараты для производ- ства пластмасс ' 13,0 4,0 9,0
теплообменники для производств синтетического каучука и спирта 13,8 7,1 6,7
красителей и полупродуктов . . 14,1 8,3 5,8
внутрицеховые трубопроводы в производстве синтетического кау- чука и спирта 10,8 5,8 5,0
Применение конструкционных материалов высокой коррозион-
ной устойчивости значительно увеличивает срок службы аппара-
туры, трубопроводов и арматуры. Это обстоятельство, не говоря
уже о защите материальных потоков от загрязнений, вызываемых
коррозией, искупает относительно высокую стоимость таких мате-
риалов, как титан, тантал, ниобий или их сплавы, и замена ими
87
более дешевых и менее коррозионноустойчивых материалов яв-
ляется экономически оправданной, если
“1 > Д1
U-2 0-2
(и — скорость коррозии; а — стоимость материала; индексы 1 и 2
относятся соответственно к дешевому и дорогому материалам).
Естественно, что в нормы амортизационных отчислений при этом
должны быть внесены соответствующие коррективы.
Из анализа структуры основных промышленно-производствен-
ных фондов химической промышленности (табл. 1-8) следует, что
Таблица. 1-8
Структура основных промышленно-производственных фондов
химической промышленности (в %) [9]
Группы основных фондов Отрасли промышленности
аннлино- красочная синтетического каучука пластмасс
Здания и сооружения .... 50 45 50
Производственное н силовое оборудование 40 50 42
Прочие объекты 10 5 8
Всего: - 100 100 । 100
производственное и силовое оборудование оценивается величиной
порядка 40—50% от основного фонда промышленного предприя-
тия. Таким образом, на долю основного производственного и си-
лового оборудования приходится около 75—80% амортизационных
отчислений, включаемых в себестоимость продукции промышлен-
ного предприятия.
Разделение технологической схемы на ряд последовательных
этапов переработки сырья позволяет оценить-долю каждого из них
в суммарной стоимости производственного оборудования и степень
влияния на себестоимость продукта производства, проявляемую
в виде амортизационных отчислений.
Размер затрат на технологическое и силовое оборудование, а
следовательно, и размер амортизационных отчислений в значи-
тельной мере определяется стоимостью аппаратуры, используемой
при подготовке сырья и разделении реакционной смеси. Стоимость
реакционной аппаратуры в большинстве рассмотренных схем не
превышает 50% суммарной стоимости оборудования.
Амортизационные отчисления от затрат на технологическое
оборудование устанавливаются, исходя из стоимости оборудова-
ния и норм амортизации. Размер амортизационных расходов на
технологическую аппаратуру и оборудование определяется стои-
38
мостью этого вида оборудования и его монтажа. Распределение
амортизационных расходов по стадиям технологической схемы р
общем случае вряд ли возможно. Общим выражением может слу-
жить уравнение (1-29)—см. стр. 43 — после учета норм амортиза-
ции, соответствующих типам аппаратуры и оборудования.
ЗАРАБОТНАЯ ПЛАТА
Заработная плата производственного, обслуживающего
и административного персонала химического производства с отчис-
лениями на социальное страхование определяется численностью и
квалификацией работников всех категорий и находится в зави-
симости от сложности технологической схемы, количества машин
и аппаратов, степени механизации и автоматизации технологиче-
ского процесса.
При рассмотрении структуры себестоимости ряда продуктов
промышленного органического синтеза можно заметить, что в оте-
чественных условиях заработная плата производственного персо-
нала редко превышает 2—3% себестоимости продукта.
Общее выражение для описания количественной связи между
численностью и квалификацией персонала и степенью автомати-
зации химического производства найти трудно. Вопрос о необ-
ходимой степени автоматизации химических производств решается
с учетом специфики их технологического и аппаратурного оформ-
ления, при этом имеется в виду наличие оптимума степени авто-
матизации, т. е. такого положения, когда дальнейшее сокращение
количества персонала, обслуживающего технологическую аппа-
ратуру, в результате внедрения средств автоматического управ-
ления сопровождается непропорциональным увеличением расхо-
дов на содержание и обслуживание КИП и предприятие-автомат
становится нерентабельным. Задача об оптимуме автоматизации
химического производства пока не решена.
УДЕЛЬНЫЕ ЭНЕРГО-МАТЕРИАЛЬНЫЕ ЗАТРАТЫ
Удельные энерго-материальные затраты (Ф) на произ-
водство продукта как сумма затрат, связанных с расходом всех ви-
дов сырья и энергетических средств и погашением амортизацион-
ных отчислений, могут быть выражены с помощью уравнений (1-26),
(1-27) и (1-29) следующим образом:
Ф = й,2^(а> ...) + а25<2(а, ...) + йз2 ^(а, •••) (1-23)
где 51, аг — средняя стоимость сырья и энергетических средств со-
ответственно; аз — переходной коэффициент для расчета аморти-
зационных отчислений.
Первое слагаемое в уравнении (1-28) выражает стоимость
сырья, расходуемого на всех стадиях технологической схемы, вто-
рое— стоимость энергетических средств, третье — размер аморти-
зационных отчислений [форма зависимости № (а) дана на стр. 43].
39
Размер энерго-материальных затрат зависит от сочетания ус-
ловий проведения процесса на каждой стадии переработки сырья
и при некотором варианте этого сочетания может приобретать ми-
нимальное значение (см. стр. 49).
СТРУКТУРА КАПИТАЛЬНЫХ ЗАТРАТ
Структура капитальных затрат, связанных с организа-
цией химического производства,.показывает, что на долю объектов
основного производственного назначения в среднем приходится от
50 до 85% всей суммы капитальных затрат (табл. 1-9).
Т а б л и ц а 1-9
Структура капитальных затрат при организации различных
химических производств (в %)
Производство
Объекты ос-
новного
производ-
ственного
назначения:
строитель-
но-мон-
тажные
работы .
оборудо-
вание . .
Объекты
вспомога-
тельного на-
значения . .
53 32 23
42 50 29
5 18 48
48 50
100 100 100 100 100
100 , 100 100 100 100
Даже в самом общем виде трудно выразить зависимость сто-
имости зданий и сооружений от размера и стоимости производ-
ственного и силового оборудования, так как технические решения
при проектировании производственных зданий весьма разнооб’
разны, зависят от ряда внешних условий н в общем имеют тенден-
цию к упрощению. Однако в действительности зависимость эта
имеется, и размер капитальных вложений в объекты основного
производственного назначения тесно связан с размерами и коли-
чеством технологического оборудования.
Относительная величина стоимости аппаратуры и оборудова-
ния каждой из стадий переработки сырья в общей стоимости ап-
паратуры и оборудования технологической схемы в значительной
40
степени зависит от специфических особенностей и условий прове-
дения реакции, требований к чистоте продукта, масштабов про-
изводства и т. п. Анализ структуры стоимости технологической
аппаратуры и оборудования ряда производств основного органи-
ческого синтеза позволяет оценить порядок величин, характери-
зующих относительную стоимость отдельных групп аппаратов и
машин.
Для одностадийных химических схем стоимость аппаратуры
реакционного агрегата (реактор, циркуляционная система, вы-
носные теплообменники и т. д.) составляет 50—70% от общей
стоимости технологического оборудования, если процесс прово-
дится при среднем и высоком давлении; для схем, в которых ре-
акционная аппаратура работает при атмосферном давлении, от-
носительная стоимость реакционного агрегата понижается до 10—
20%. Что же касается реакционного аппарата, т. е. сосуда, разме-
ры которого зависят от скорости реакции, то его относительная
стоимость для процессов, проводимых при среднем и высоком дав-
лении, составляет 40—60% от общей стоимости технологического
оборудования и не превышает 5—10% для процессов, осуществ-
ляемых при атмосферном давлении.
Стоимость аппаратов и машин, используемых для хранения,
транспорта и подготовительной обработки сырья, и Стоимость
аппаратуры, связанной с разделением реакционной смеси и очи-
сткой готового продукта, находятся, как правило, в отношении
1 : (1,5-ь2,0), за исключением тех производств, продукт которых
требует тонкой очистки. В последнем случае схема очистки мо-
жет быть весьма сложной и дорогой.
Для многостадийных схем, включающих разнохарактерные по
природе и условиям осуществления химические и физико-химиче-
ские процессы, подобная характеристика затруднительна, так как
при этом сказывается влияние относительной стоимости аппара-
туры отдельных стадий. Иллюстрацией может служить описанная
выше многостадийная схема промышленного синтеза капролак-
тама из бензола. Каждая стадия превращения бензола в капро-
лактам может рассматриваться как самостоятельная производ-
ственная схема, по которой выпускается продукт различной сте-
пени чистоты. Последнее обстоятельство, а также различие
давлений при проведении реакционного процесса влияют на от-
носительную стоимость отдельных групп аппаратов, используемых
на каждой из стадий. Результаты анализа проектной документа-
ции группы цехов производства капролактама из бензола при-
ведены в табл. 1-10.
Отклонение от средних значений относительной стоимости
оборудования можно видеть на стадии 3 синтеза вследствие ее
специфических особенностей (готовый продукт, получаемый на
этой стадии, передается на последующую стадию синтеза почти
без разделения реакционной смеси) и на стадиях 4—5, связанных
с выпуском продукта высокой степени чистоты.
41
Таблица 1-10
Относительная стоимость оборудования для производства
капролактама из бензола (в %)
• Основные технологические стадии Стадии синтеза По всему производ- ству (ста- дии 1—5)
1 2 3 4—5
Предварительная обработка сырья (хранение, транспорт, нагрев и т. д.) 15 20 50 18 20
Химическое превращение сырья (реактор, циркуляци- онные насосы, теплообмен- ники и т. д.) 65 40 10 9 50
Разделение реакционной сме- си и очистка продукта . . . 20 40 40 73 30
Всего: 100 100 100 ioo 100
Наиболее часто встречающиеся соотношения между стоимо-
стью аппаратуры первой и третьей основных технологических
стадий выдерживаются при рассмотрении всей технологической
схемы (стадии 1—5). Относительно высркое значение стоимости
реакционной аппаратуры (50%) является следствием влияния
стадий 1 и 2 синтеза (реакционные процессы проводятся при вы-
соком и среднем давлении), на которые приходится основная доля
капитальных затрат на технологическое оборудование. Эффект
этого влияния усиливается также положением этих стадий в после-
довательности превращения бензола: размер потока основного
сырья в многостадийной схеме уменьшается от первой к последней
стадии.
Размеры основных видов аппаратуры и оборудования техно-
логической схемы находятся в1 прямой зависимости от величины
потоков перерабатываемого сырья, т. е. от степени его превра-
щения. Общий вид этой зависимости нетрудно установить из чисто
геометрических соображений, основываясь на уравнениях (1-20) —
(1-22):
- /1 \р
Трубопроводы, емкости, теплообменники, насосы 1 — 1 (0<р<И)
Реакционная аппаратура ........................ a4 (q > 1)
Аппаратура для разделения реакционной сме-
си:
ректификационная.......................... (1 — a)s (0 < s 1)
/ 1 V
прочая...................................... 1—1 (0 < t 1)
Относительный (отнесенный к единице готового продукта) раз-
мер (вес, стоимость) аппаратуры и оборудования можно выра-
. ". >
зить следующей зависимостью:
гДе Х> Si’ S2’ S3’ ^ — коэффициенты пропорциональности. Показа-
• тели степени р, q, s, t — положительные величины Они обусловли-
• вают монотонную зависимость каждого слагаемого от а и экстре-
мальный в общем случае характер зависимости №(а).
Относительно малое влияние стоимости реакционной аппара-
туры на суммарную стоимость технологического и силового обо-
рудования указывает на расположение минимума в области вы-
соких значений степени превращения.
В большинстве практических случаев промышленные процес-
сы основного органического синтеза осуществляются в условиях,
когда a<Sl и размер технологического оборудования заметно пре-
восходит возможный минимум.
ЭКОНОМИЧЕСКИЙ ОПТИМУМ z
ТЕХНОЛОГИЧЕСКОГО ПРОЦЕССА
Основной задачей химической технологии является со-
здание промышленных процессов, максимально эффективных по
своим технико-экономическим показателям. Решение этой задачи
состоит в поиске такого сочетания условий промышленного син-
теза экономически важного продукта, которое обеспечивает ми-
нимальное значение показателя приведенных затрат как суммы
себестоимости продукта и удельных капитальных затрат.
Рассмотрение структур себестоимости продукта и удельных
капитальных затрат дает представление о числе и разнообразии
параметров, требующих учета при поиске минимального значения
экономического показателя, а также о сложности взаимозависи-
мости этих параметров.
Сложность решения подобной задачи применительно к техно-
логической схеме современного химического производства уве-
личивается вследствие разнообразия и разнохарактерности про-
цессов и аппаратов, последовательно участвующих в переработке
сырья.
Классический метод поиска экстремального значения функции
многих переменных (заключающийся в составлении и решении
системы уравнений, получаемых в результате определения част-
ных производных функций по каждому из неизвестных перемен-
ных, и приравнивания их нулю) применительно к задаче опти-
мизации Af-стадийной последовательности с М независимыми пе-
ременными на каждой из стадий требует одновременного учета
NM переменных. Сложность взаимных связей между многими не-
зависимыми переменными, не говоря уже о чрезвычайной гро-
моздкости расчетных операций, лишает этот метод практической
ценности при решении задач химической технологии.
43
В последнее время оптимизации сложных производственных
систем уделяется особое внимание. Это привело к созданию
различных способов решения такой задачи, отличающихся по кри-
териям оценки оптимальности и методам [10—16].
В зависимости от объекта оптимизации и сложности исследуе-
мых зависимостей используются принципы линейного [17, 18] и
нелинейного [19—21], геометрического [22, 23] и динамического
[24—26] программирования. На основе этих принципов в настоя-
щее время разработаны методы решения экономических задач
и методы оптимизации единичных (отдельных) процессов, а так-
же последовательностей однотипных процессов и аппаратов хи-
мической технологии (например, каскада секций реакционного
агрегата) [27—29].
При поиске экономического оптимума технологической схемы
с использованием показателя приведенных затрат в качестве кри-
терия оценки оптимальности задача сильно осложняется необхо-
димостью учета одновременного влияния значительно большего
числа еще более разнохарактерных переменных, связанных к
тому же сложной зависимостью при ограничениях на переменные,
налагаемых условиями конкретного производства. Поэтому ме-
тоды, используемые при оптимизации единичных процессов или
последовательностей однотипных аппаратов, значительно услож-
няются и строго аналитический поиск оптимума обычно затруднен.
Трудности, возникающие при использовании чисто аналитиче-
ских методов поиска экономического оптимума многостадийной
технологической схемы, обусловлены в первую очередь невозмож-
ностью сохранения непрерывности функций, описывающих от-
дельные процессы технологической схемы в широком диапазоне
условий их осуществления (с учетом изменения не только техно-
логических параметров, но и аппаратурного оформления).
При разработке схемы нового многостадийного технологиче-
ского процесса, когда количественное описание зависимостей, ха-
рактеризующих каждую из стадий, не является достаточно стро-
гим и точным, постановка вопроса об оптимизации процесса с уче-
том одновременного влияния всех параметров вообще лишена
практического смысла. Речь в этом случае может идти лишь об
оценке оптимальных условий с учетом минимального числа вели-
чин, определяющих экономику всего процесса.
Подход к решению такой задачи требует, по-видимому, огра-
ничения независимых переменных минимальным числом величин,
влияние которых наиболее заметно отражается на экономических
Показателях технологической схемы.
Анализ структуры показателей, определяющих экономическую
эффективность технологического процесса (себестоимости продук-
та и удельных капитальных затрат), позволяет отнести к числу
таких определяющих параметров степень превращения основного
вида сырья на стадии его химического взаимодействия (а). Ис-
пользование а как единственной независимой переменной при за-
44
данной совокупности остальных параметров на каждой из стадий
дает возможность приближенного решения задачи на первом этапе
оптимизации технологического процесса *.
Найденная таким образом область оптимальных значений сте-
пени превращения определяет тип и размеры основной аппара-
туры на каждой из стадий технологической схемы и, сужая тем
самым границы области изменения остальных параметров, облег-
чает возможность использования аналитических методов поиска
оптимума с учетом влияния всех переменных.
Поскольку экономика производств основного органического
синтеза определяется в первую очередь удельным расходом сырья,
наибольшего внимания заслуживает характер зависимости р,,2(а)-
Эта зависимость устанавливается при изучении процесса в допу-
стимо широком диапазоне условий, и для технологической разра-
ботки используется наиболее благоприятный ее вариант. Естест-
венно, что параметры реакционного процесса, соответствующие
выбранной зависимости рг-,2(a), являются основой для аппара-
турного оформления реакционного агрегата.
Для поиска оптимального значения а, соответствующего мини-
муму суммы удельных энерго-материальных затрат (и, следова-
тельно, себестоимости продукта), может служить уравнение (1-28).
Первое слагаемое этого уравнения выражает зависимость расхода
(и стоимости) сырья от степени его превращения в реакционном
аппарате в условиях оптимального варианта экспериментально
установленной зависимости рг-, 2(«) и с учетом потерь сырья и про-
дукта на первой и третьей основных стадиях технологической схемы.
Второе и третье слагаемые учитывают удельный расход и стои-
мость энергетических средств и размеры амортизационных отчис-
лений в зависимости от размера потока перерабатываемого сырья
и комплекса параметров, характеризующих отдельные процессы.
В пределах возможностей того или иного типа аппарата раз-
меры (и стоимость) этого аппарата, а также расход энергии, свя-
занный с эксплуатацией аппарата, являются непрерывными функ-
циями от а. Изменение значения а может привести к необхо-
димости замены одного типа аппарата другим (например, при
увеличении потока сырья может возникнуть необходимость пере-
хода от змеевикового теплообменника к кожухотрубному и т. д.).
Использование нового типа аппарата требует учета присущего
ему характера зависимостей Q(a) и IF(a).
Для выбранного типа аппарата или машины с достаточной
для технических расчетов точностью можно считать, что
а2 Q = а2 Ф («) Д (Л Л г, С, ЬТ, ЬР,...)
a2'^iW^a1'^l<f(a)-x(T,P, Re, ...)
1 Степень превращения сырья как основной параметр технологического про-
цесса при поиске экономического оптимума используется в ряде работ [30, 31],
однако при этом учитывается лишь зависимость размера потоков от а.
45
Полное отсутствие связи между приведенными выше функция-
ми (и право на использование их произведения) строго справед-
ливо лишь для механических и тепловых процессов (транспорт,
нагрев, охлаждение и т. п.). Размер аппаратуры, предназначенной
для разделения реакционной смеси, и расход используемых при
этом энергетических средств находятся в зависимости от состава
реакционной смеси и являются функцией от а. Учитывая, однако,
что изменение размеров таких аппаратов, как, например, ректи-
фикационные и абсорбционные, вызываемое изменением состава
исходной смеси, относительно мало сказывается на суммарной
стоимости аппаратуры и оборудования соответствующих устано-
вок (связанное с этим изменение расхода энергетических средств
учитывается вторым слагаемым), допущение независимости упо-
минаемых выше двух функций для приближенного расчета яв-
ляется оправданным.
Ясно, что расход энергии и размер аппаратуры, на стадиях
подготовки сырья и разделения реакционной смеси меняются с
изменением степени превращения сырья, возрастая с увеличением
размера перерабатываемого потока. Размер реакционной аппара-
туры во всех случаях пропорционален значению а. Относительно
малое влияние стоимости реакционной аппаратуры на суммарный
размер амортизационных отчислений (и удельных капитальных
затрат) дает основание ожидать минимум функции «зЕ№(а) в ин-
тервале высоких значений а.
При заданном значении а удельный расход энергетических
средств и размер амортизационных отчислений для каждого из
аппаратов технологической схемы определяются уровнем оптими-
зации процесса, осуществляемого в этом аппарате. Как будет по-
казано ниже (см. стр. 233) сумма удельных энерго-материальных
затрат и удельных капитальных вложений даже для простейших
процессов является экстремальной функцией специфичных для
данного процесса параметров.
Независимо от степени приближения к оптимиму, характери-
зующему каждый из единичных процессов технологической схемы,
сумма удельных энерго-материальных затрат, связанных с произ-
водством целевого продукта, остается экстремальной функцией
степени превращения сырья.
Большинство реакций промышленного органического синтеза
протекает по сложной схеме, и выход продуктов реакции опреде-
ляется степенью превращения исходного вещества, монотонно убы-
вая или возрастая с увеличением а (экстремальный характер за-
висимости выхода от степени превращения встречается лишь при
проведении многостадийных последовательных реакций, если речь
идет о выходе промежуточного продукта).
Минимум функции Ф(а) имеет место как при монотонном сни-
жении выхода продукта, так и при монотонном его повышении
с увеличением а. В первом случае величина а0Пт в значительной
степени зависит от вида функции р», а(а) и может изменяться в
46
описывается схемой не-
Рис. 1-11. Зависимость вы-
хода продукта В от степени
превращения исходного ве-
щества А:
---------реактор полного сме-
шения; — -------реактор пол-
ного вытеснения.
широких пределах. Во втором случае область монотонного сниже-
ния суммы энерго-материальных затрат по мере увеличения а
определяется относительной стоимостью реакционного агрегата и
чаще всего влияние этой величины становится ощутимым лишь
при 1.
В качестве примера рассмотрим некий технологический про-
цесс, состоящий из трех стадий: 1) подготовка сырья (хранение,
транспорт, нагрев); 2) химическое превращение сырья; 3) разде-
ление реакционной смеси. Пусть реакция
следовательного превращения A->B->D.
экзотермичны. Процесс проводится в
изотермических условиях. Часовое коли-
чество целевого продукта (продукт В)
является заданным.
Зависимость выхода продукта В от
степени превращения исходного веще-
ства А, установленная экспериментально
в интервале температур Т\—Т3, выра-
жается изотермами (рис. 1-11), полу-
ченными в условиях реакционного ап-
парата полного смешения и реактора
полного вытеснения. Нумерация кривых
на рис. 1-11 соответствует температуре
(/,/'—Г,; 2,2'—Тг-, 3,3'—Т3).
Пусть разделение реакционной смеси
условно ограничивается отделением не-
прореагировавшего сырья (возвращае-
мого на повторное использование) про-
стой дистилляцией; выделение продукта
В из смеси его с продуктом D не производится или осуществ-
ляется простым способом, затратами на проведение которого
можно пренебречь.
В качестве предварительного этапа исследования примем не-
кий вариант аппаратурно-технологического оформления процессов
на первой и третьей стадиях, при котором потери сырья и конеч-
ного продукта составляют 2% от величины перерабатываемого по-
тока этих компонентов, т. е. Ра, i = ₽a, з=₽в, з=0,98; полагаем при
этом, что используется реактор полного смешения.
Из рис. 1-11 следует, что в пределах допустимого интервала
температур наиболее выгодная форма зависимости рв, 2(а) для
реактора полного смешения описывается кривой 3, для реакцион-
ного аппарата полного вытеснения — кривой 3'. Вполне понятно,
что для рассматриваемого случая в расчет принимается лишь кри-
вая 3 или 3'.
Пользуясь выражениями (1-20) — (1-22), можно подсчитать
размер потоков на первой и третьей, стадиях (на единицу це-
левого продукта В) и установить размер потерь соответствую-
щих продуктов на этих стадиях (рис. 1-12,а и в), расход сырья
47
в реакционном агрегате (рис. 1-12,6) и, как результат, удельный
расход сырья на всех стадиях его переработки (рис. 1-12,а). Кри-
вые на рис. 1-12, г могут быть также построены по уравнениям
(1-19), (1-26).
Рис. 1-12. Зависимость удельного расхода сырья, энергетических средств и
размера амортизационных отчислений при производстве продукта В от
степени превращения 'исходного вещества А:
---------реактор полного смешения; -----реактор полного вытеснения.
Расход энергии (в условных единицах) на первой и третьей
стадиях нетрудно установить, исходя из размера потоков сырья
и реакционной смеси на этих стадиях (рис. 1-12,6 и ж). Расход
48
энергетических средств (охлаждающего агента) в реакционном
агрегате будет пропорционален количеству превращенного сырья
(рис. 1-12, е). Суммарный удельный расход энергии как функция
степени превращения представлен на рис. 1-12, з.
Оценка зависимости размера амортизационных отчислений от
а может быть произведена лишь в самом общем виде, т. е. абсо-
лютное значение величин определяется размером, конструкцией
и материалом аппаратуры и оборудования, а также нормами
амортизации. Несомненным является
пропорциональность размера аппара-
туры на первой и третьей стадиях ве-
личине потока сырья и реакционной
смеси и, как следствие этого, экстре-
мальный характер зависимости W от
а (рис. 1-12, и и л).
Размер реакционного пространства
по мере увеличеция а возрастает по
законам химической кинетики, и для
продукта В реакции А—>-В > D, про-
водимой в аппарате полного смеше-
ния, зависимость размера реактора
(и пропорциональная ему величина
амортизационных отчислений) выра-
жается кривой на рис. 1-12, к.
При решении конкретных задач
зависимости, приведенные на рис. 1-12,
представляются в денежном выраже-
нии и отражают стоимость всех видов
сырья, энергетических средств, аппа-
ратуры и оборудования.
Построение результирующей кри-
вой производится по кривым, изобра-
женным на рис. 1-12, г, з и м.
В рассматриваемом общем случае
Рис. 1-13. Зависимость удель-
ных энерго-материальных за-
трат при производстве про-
дукта В от степени превраще-
ния исходного вещества А:
---------реактор полного смеше-
ния; ---- — реактор полного
вытеснения.
результирующая кривая
строится на основе учета относительного значения каждой из трех
основных статей, составляющих себестоимость продукта основного
органического синтеза (сырье, энергетические средства, аморти-
зационные отчисления), которые принимаются равными соответ-
ственно 12 : 4 : 1.
Экстремальный характер результирующей кривой на рис. 1-13
вытекает из формы связи зависимостей Л (a), Q(a), W'(a).
Поскольку область оптимального значения а определяется
стоимостью сырья, исследование функций Q(a) и W'(a) может
быть ограничено сравнительно узким интервалом значений а.
Минимальное для рассматриваемого варианта технологической
схемы значение себестоимости продукта достигается при степени
превращения сырья, лежащей в области 0,20—0,22. Работа при
меньшем или большем значении а, по-видимому, нецелесообразна.
4 В. С. Хайлов, Б. Б. Брандт
49
Изменение параметров или аппаратурного оформления любого
из единичных процессов многостадийной схемы требует пересмотра
режима работы всей схемы. Примером может служить изменение
типа (с сохранением параметров режима) реакционного аппарата:
замена реакционного аппарата полного смешения, принятого
для разобранного случая, на реактор, работающий по принципу
аппарата полного вытеснения, требует учета зависимости
₽в,2(a), характерной для реакторов этого типа (см. кривую 3' на
рис. 1-11).
Результатом повторения всех расчетных операций является
серия пунктирных кривых на рис. 1-12, выражающих зависимость
соответствующих показателей процесса на каждой из стадий, и
кривая зависимости Ф(а) на рис. 1-13. Оптимальное значение
степени превращения перемещается в область более высоких зна-
чений а с соответствующим уменьшением значения Ф. После за-
мены реакционного аппарата проведение процесса при прежнем
значении а становится неоправданным.
Совершенствование аппаратурного оформления, оптимизация
режима каждого из единичных процессов, а также улучшение теп-
ловой схемь^либо переход на более дешевые виды сырья, энерге-
тических средств или материала аппаратуры влияют на положе-
ние экстремальной точки результирующей кривой.
Условием максимально полного использования эффекта, до-
стигаемого в результате усовершенствования технологии на лю-
бом этапе многостадийного процесса, является пересмотр режима
работы всего многостадийного процесса и поиск оптимального
значения а, соответствующего новому варианту. Несоблюдение
этого условия снижает экономический эффект оптимизации.
Эксплуатация рассматриваемой технологической схемы после
установки реакционного аппарата полного вытеснения целесооб-
разна при степени превращения 0,3—0,35 (точка А на рис. 1-13);
эффект замены реакционного аппарата заметно снижается, если
значение а остается прежним (точка В на рис. 1-13).
Оптимизация единичного процесса сопровождается изменением
каждого из трех основных показателей (стоимость сырья, энергии,
аппаратуры), причем достижение минимума суммы этих показа-
телей может быть связано с перераспределением их относитель-
ных значений и изменением абсолютных величин, т. е. не исклю-
чается возможность увеличения одних показателей за счет непро-
порционально большого снижения других.
«Местный» эффект оптимизации может быть выражен с
помощью рис. 1-12 при нанесении на горизонтальные строчки (со-
ответственно стадиям схемы) показателей, достигнутых в резуль-
тате усовершенствования того или иного агрегата или аппарата.
Оценка конечного экономического эффекта, а также границ об-
ласти допустимого отклонения от оптимального' режима всего про-
цесса требует учета взаимосвязи оптимизируемой стадии с осталь-
ными на основе зависимости от степени превращения.
50
По рис. 1-13 можно установить, что для рассматриваемого типа
реакции улучшение показателей процессов на стадиях подготов-
ки сырья или разделения реакционной массы сопровождается пе-
ремещением экстремальной точки кривой в область более низкой
степени превращения с соответствующим понижением себестоимо-
сти продукта. Снижение себестоимости, достигаемое при повыше-
нии- выхода продукта в реакционной аппаратуре, вызывает пере-
мещение ctonT в область высоких значений а. '
Из анализа структуры себестоимости ряда продуктов промыш-
ленного органического синтеза (см. табл. 1-5) следует, что наи-
большее влияние на себестоимость продукта оказывает расход
сырья. Повышение степени превращения сырья при сохранении
выхода продукта резко снижает расход энергии, что особенно за-
метно в области низких значений а.
Наименьшее влияние на величину, Фмин оказывает слагаемое,
характеризующее размер реакционной аппаратуры. Увеличение
размеров реакционной аппаратуры, вызываемое необходимостью
повышения значения а, во всех случаях экономически оправды-
вается.
Рассмотрение закономерностей кинетики химических реакций
в открытых и закрытых системах показывает, что форму связи
Р'(а) для данной реакции обусловливает в основном тип реак-
ционного аппарата, который в ряде случаев оказывает решающее
влияние на относительный расход сырья и, следовательно, на се-
бестоимость конечного продукта. Производительность реакцион-
ной аппаратуры, как уже сказано, играет второстепенную роль при
определении основного показателя экономической эффективности
химического производства. Это не следует рассматривать как от-
каз от интенсификации работы реакционной аппаратуры, однако
основная задача технологии состоит в обоснованном выборе типа
реактора, способного обеспечить наиболее выгодную для каждого
случая форму связи р,, 2(а).
Первым этапом поиска экономического оптимума при разра-
ботке нового технологического процесса является сравнительная
оценка вариантов зависимости р,-, 2(а), полученных эксперимен-
тальным или расчетным путем.
Из уравнений (1-16) — (1-18) следует, что оптимальному вари-
анту соответствует зависимость 2(а), при которой заданное
значение степени превращения исходного вещества обеспечивает
получение максимального выхода продукта. Этот принцип срав-
нительной оценки справедлив как при выборе типа реакционного
аппарата, так и при поиске оптимальных условий проведения про-
цесса химического превращения сырья *.
1 При оценке влияния температуры на селективность сложных реакций не-
трудно допустить серьезную ошибку, если при экспериментальном определении
зависимости Р<,2(Г) отсутствует строгий учет влияния температуры на степень
превращения исходного вещества. Примером может служить последовательная
реакция А > В D при £i =/= Е2 (feb k2—константы скорости реакции;
4* S1
При составлении исходных данных для проектирования не сле-
дует, как это нередко практикуется, ограничиваться рекоменда-
цией определенного комплекса значений выхода продукта и со-
• ответствующей ему степени превращения сырья. Нужно рекомен-
довать оптимальную форму зависимости pir 2(«) в аналитическом
или графическом виде. Это облегчает задачу проектировщика при
поиске оптимального значения степени превращения на основе
учета показателей первой и третьей основных технологических
стадий.
Удельные капитальные затраты на технологическое оборудо-
вание являются количественной характеристикой аппаратурного
оформления технологического процесса и составляют заметную
долю показателя приведенных затрат. Уменьшение капитальных
затрат сказывается не только на эффективности химического про-
изводства, но и на сроках выполнения строительно-монтаж-
ных работ — периоде вынужденного омертвления денежных
средств.
Если необходимо организовать новое производство и имеется
возможность выбора технологической схемы из числа известных
схем производства данного продукта, то, естественно, наимень-
шими , удельными капитальными затратами будет характеризо-
ваться схема, состоящая из небольшого числа высокопроизводи-
тельных аппаратов, выполненных из недефицитного и недорогого
материала. Когда рассматривается конкретный технологический
процесс, состав и параметры материальных потоков на входе и
выходе из каждого аппарата и агрегата являются заданными.
Снижение удельных капитальных затрат при разработке аппара-
турного оформления процесса может быть достигнуто в резуль-
тате: 1) использования дешевых конструкционных материалов;
2) совмещения тепловых потоков в одном теплообменном аппа-
рате; 3) интенсификации процессов тепло- и массообмена; 4) со-
кращения длины всех коммуникаций путем правильного размеще-
ния оборудования и пультов управления; 5) расположения обору-
дования на открытых площадках и т. д.
На примере расчета суммы затрат, связанных с работой таких
аппаратов, как теплообменник, и таких агрегатов, как ректифи-
кационная установка для разделения бинарной смеси, можно ви-
деть, что эксплуатационные расходы (стоимость энергетических
средств плюс амортизационные отчисления) достигают своего ми-
нимального значения при удельных капитальных затратах, дале-
ких от возможного в каждом конкретном случае минимума.
£ь Ег — энергии активации). Если Ei>E2, повышение температуры сопрово-
ждается увеличением значений k!t k2 и уменьшением отношения k2)k\. Постоян-
ная продолжительность опытов, проводимых при различной температуре, может
привести к экстремальной зависимости Рв,2(Т’). Вывод об «оптимальной темпе-
ратуре процесса», сделанный на основании положения экстремальной (в рас-
сматриваемом случае — максимум выхода продукта В) точки будет ошибоч-
ным.
52
Оценка упомянутых выше аппаратов и агрегатов показателем,
аналогичным по структуре показателю приведенных затрат П, не-
сколько полнее характеризует экономическую эффективность, од-
нако не вносит существенных корректив в значение определяю-
щего параметра, соответствующего минимуму суммы затрат. Ясно,
что при таком способе оценки оптимальное значение параметра,
по которому производится поиск минимума, перемещается' в об-
ласть меньших значений удельных капитальных затрат.
Если допустить, что каждый из аппаратов и агрегатов техно-
логической схемы удовлетворяет требованию П = ПМ1Ш, и просле-
дить за изменением размера удельных капитальных затрат К,
характеризующих всю технологическую схему (подобную рас-
смотренной выше) при изменении степени превращения сырья,
то можно заметить, что зависимость К (а) описывается кривой,
имеющей минимум при некотором значении а = а0Пт- При этом
удельные капитальные затраты для всей технологической схемы
будут иметь абсолютно минимальное значение. Понятно, что чем
выше относительная стоимость реакционного агрегата, тем мень-
ше значение а0Пт-
Поскольку величины Спр и К, определяющие значение показа-
теля приведенных затрат (n=CBP+EsK), являются функциями
степени превращения сырья, имеющими минимум, естественно, что
зависимость П(а) для технологической схемы также описывается
кривой, имеющей минимум.
Для производств промышленности основного органического
синтеза произведение ЕВК в уравнении показателя приведенных
затрат не превышает 14—15% значения П и определяющей яв-
ляется себестоимость продукта. Вследствие этого минимальное
значение П может соответствовать минимуму удельных капиталь-
ных затрат только в тех редких случаях, когда минимальное зна-
чение себестоимости продукта и минимальное значение удельных
капитальных затрат достигается при одной и той же степени пре-
вращения сырья. Если это условие не выполняется, для достиже-
ния максимальной эффективности технологического процесса со-
блюдение условия минимума удельных капитальных затрат не
обязательно.
Рассмотренный выше подход к поиску минимального значения
себестоимости продукта основного органического синтеза и мини-
мального значения показателя приведенных затрат имеет, как уже
сказано, значение для приближенной оценки показателей опти-
мального режима осуществления многостадийного технологиче-
ского процесса на начальном этапе его разработки.
Изложенные соображения указывают на необходимость изуче-
ния технологии основного органического синтеза как совокупности
последовательных стадий (этапов) переработки сырья, объединяе-
мых зависимостью своих показателей от степени превращения
этого сырья на стадии его химического взаимодействия.
53
Если характер влияния а на расход энергии и размер аппара-
туры, используемой на стадиях предварительной обработки сырья
и разделения реакционной смеси, достаточно ясен, то связь выхода
продукта и степени превращения исходных веществ для различ-
ных реакционных систем и реакционной аппаратуры различных
типов требует тщательного рассмотрения.
Размер реакционной аппаратуры определяется скоростью хи-
мического превращения, являющейся функцией степени превра-
щения сырья.
Изучение технологии основного органического синтеза требует
анализа законов термодинамики и химической кинетики для вы-
яснения вида функции р», г(а) и зависимости скорости образования
отдельных продуктов от степени превращения сырья.
ВЛИЯНИЕ МОЩНОСТИ ПРОИЗВОДСТВА
НА ЕГО ЭКОНОМИЧЕСКИЕ ПОКАЗАТЕЛИ
Значение показателя эффективности химического произ-
водства определяется не только совершенством технологической
схех»ы и степенью приближения режима процессов к оптимальному,
но и в известной степени зависит от мощности производства. Ха-
рактер этой зависимости нетрудно установить из результатов ана-
лиза структуры себестоимости продукта и структуры затрат на
технологическое оборудование.
Расход средств по основным статьям, определяющим себестои-
мость продукта (расход сырья, расход энергетических средств,
амортизационные отчисления), с увеличением мощности производ-
ства при сохранении технологической схемы и режима проведе-
ния основных процессов имеет .тенденцию к сокращению.
В то время как расход сырья на стадии химического превра-
щения практически не зависит от размера производственной ап-
паратуры (если такая зависимость и существует, то в основе ее
лежат более сложные причины, обусловленные отсутствием пол-
ного подобия при переходе от одного масштаба к другому), отно-
сительные потери сырья и готового продукта на других стадиях
технологической схемы, обратно пропорциональные величине пе-
рерабатываемых потоков, уменьшаются с увеличением мощности
производства почти прямолинейно, приводя к снижению удельного
расхода сырья на единицу готового продукта.
Расход тепловых энергетических. средств определяется вели-
чиной тепловых потоков в теплообменной аппаратуре, а также
неизбежными потерями тепла от стенок аппаратов и трубопрово-
дов, вследствие утечки теплоносителей и т. д.
Зависимость величины поверхности аппаратов и трубопроводов
от мощности производства приближенно может быть оценена по
изменению диаметра трубопроводов и аппаратуры, имеющей ци-
линдрическую форму. При условии полной аналогии тепловых
схем сравниваемых производств и одинаковой степени регенера-
54
ции тепла расход тепловой энергии на компенсацию основных
теплопотерь в производстве большей мощности, п<?-видимому, бу-
дет пропорционален j/Afi/jW2 (Afi, М2 — мощности исходного и
укрупненного сравниваемых производств). Аналогично этому ме-
няется также размер удельных капитальных затрат и, следова-
тельно, размер амортизационных отчислений.
Примером могут служить данные, приведенное в ряде работ
[32—34], относящихся к анализу влияния мощности производства
на размер производственных расходов. Из рис. 1-14 следует, что
стоимость продукта во всех случаях снижается с увеличением
мощности производственной установки
(весьма заметный в начале эффект умень-
шается по мере увеличения мощности про-
изводства).
Влияние мощности производства на раз-
мер удельных капитальных затрат также
несомненно, хотя эффект в этом случае за-
висит от возможности укрупнения аппа-
ратуры.
Величина капитальных затрат для ос-
новных категорий химического оборудова-
ния при увеличении его размеров описы-
Рис. 1-14. Зависимость
производствеиных затрат
от мощности производ-
ства:
1 — производство циклогекса-
на; 2 — производство хлори-
стого винила; 3 —производ-
ство циклогексанола.
вается зависимостью:
Ki IМ2J
(1-30)
где /Ci, Кг — удельные капитальные затра-
ты исходного и укрупненного производств,
руб!(т!год)-, М\, М2— мощности произ-
водств, т/год; у=0,3 ч-0,5.
Аналогичные выводы были получены [33] в результате анализа
большого числа вариантов состава технологического оборудова-
ния, разбитого на три группы, различающиеся величиной z в зави-
симости
К2М2 _(М2\г
(1-31)
1) группа аппаратов, стоимость которых пропорциональна мощ-
ности производства (г=0,9ч-1,0);
2) группа аппаратов, возрастание стоимости которых с увели-
чением мощности производства характеризуется значением пока-
зателя степени, равным в среднем 0,6;
3) группа аппаратов, возрастание стоимости которых с увели-
чением мощности производства характеризуется показателем сте-
пени, равным 0—0,4.
При анализе было рассмотрено 28 вариантов состава обору-
дования, в которых доля отдельных групп аппаратов изменялась
от 10 до 80%. Полученное среднее значение z равно 0,5—0,6. Не-
трудно видеть, что у=г—1.
55
или машины
парата
Рис. 1-15. Зависимость удель-
ных капитальных затрат от
мощности промышлэнной уста-
новки.
Выражение (1-31) справедливо в любом диапазоне значений
Л42/Л4ь однако показатель степени является функциёй М%/Мх и
при увеличении мощности производства меняет свое значение,
стремясь в пределе к нулю, а в реальных условиях — к некоторой
постоянной величине, значение которой зависит от возможности
увеличения размеров технологических аппаратов и машин.
Естественно, что при достижении предельных для каждого ап-
ров дальнейшее увеличение мощности
производства связано с необходимо-
стью увеличения числа аппаратов или
агрегатов. Размер удельных капи-
тальных затрат при этом почти не за-
висит от мощности производства.
Зависимость удельных капиталь-
ных затрат от мощности промышлен-
ной установки приведена на рис. 1-15.
Кривая 1 описывает изменение удель-
ных капитальных затрат для некото-
рого производства, характеризующе-
гося значением у=0,5. Принимая
удельные капитальные затраты на ос-
новное технологическое оборудование
для определенной мощности производ-
ства равными единице (точка Л), мож-
но экстраполировать значение К2 для
производств меньшей и большей мощ-
ности.
В последнем случае происходит
уменьшение удельных капитальных
затрат (кривая /), однако если пред-
положить, что все используемые ап-
параты и машины достигли предель-
ных размеров (точка Б), дальнейшее
увеличение мощности производства
практически не будет сопровождаться
рямая 5). Поскольку предельные раз-
меры отдельных групп аппаратов и машин неодинаковы, измене-
ние значения у происходит монотонно (кривая 4).
Изменение (увеличение) предельных размеров машин и аппа-
ратов (прямая 2) позволяет рассчитывать на снижение удельных
капитальных затрат рассматриваемого производства (точка В).
Понятно, что суммарная стоимость аппаратуры и оборудова-
ния основного производственного назначения (равная произведе-
нию удельных капитальных затрат на мощность производства) по
мере увеличения мощности производства растет вначале по урав-
нению (1-31) при z=0,6, а после достижения всеми группами ап-
паратуры предельных размеров значение z приближается к еди-
нице.
изменением значения К2
56
Рис. 1-16. Зависимость суммар-
ной стоимости аппаратуры и
оборудования основного произ-
водственного назначения от
мощности промышленной уста-
новки.
На рис. 1-16 представлена зависимость суммарной стоимости
аппаратуры и оборудования основного производственного назна-
чения от мощности производства. Различие предельных размеров
отдельных групп используемых аппаратов обусловливает посте-
пенное возрастание значения z от 0,6 до ~ 1,0 (пунктирный от-
резок).
Показатель приведенных затрат как функция мощности про-
изводства выражается кривой, имеющей характер, аналогичный
зависимости Спр(Л4пр)- Анализ зави-
симости /7(Л4пр) дает возможность
оценить оптимальные размеры произ-
водственной установки.
Естественно, что абсолютное зна-
чение «оптимальной мощности» стро-
го индивидуально, т. е. справедливо
лишь по отношению к определенному
продукту и определенной технологии
его производства. Переход к другой,
более совершенной технологии сопро-
вождается соответствующим измене-
нием «оптимальной мощности».
В связи с анализом структуры зат-
рат на технологическое оборудование
химического производства уместно
вернуться к вопросу влияния разме-
ров (и стоимости) реактора на пока-
затель проведенных затрат.
Экономика производств, основан-
ных на применении высокого и сред-
него давлений при осуществлении ре-
акционных процессов, и производств, в
которых реакционные аппараты работают при атмосферном или
низком избыточном давлении, в разной степени зависит от раз-
мера и стоимости реактора.
Для многих случаев, когда значения слагаемых в уравнении
показателя приведенных затрат составляют соответственно 0,8—
0,9 и 0,2—0,1, влияние размеров реактора на величину П не пре-
вышает 8—10% Для процессов высокого давления и 2—3% для
процессов низкого и атмосферного давления.
ДОХОД ПРЕДПРИЯТИЯ
Доход предприятия, являющийся мерой роста на-
коплений, которые получаются от сбыта продуктов производства,
для каждого из продуктов описывается приведенным выше выра-
жением (1-2).
Ясно, что при заданных Мщ и ДПр размер дохода зависит
только от себестоимости этого продукта и достигает своего мак-
симального значения при минимальной себестоимости продукта.
57
Если размер используемой технологической аппаратуры до-
пускает возможность изменения производительности в опреде-
Plc. 1-17. Зависимость размера
дохода предприятия от цены на
продукт.
Значения 3:2 —1,2; 2 —1,5;3 —2,0.
ленных пределах (это иногда про-
исходит в результате «запасов»,
предусматриваемых при проектиро-
вании), то мощность производства
становится функцией производи-
тельности аппаратуры и степени
превращения сырья.
Вернемся к примеру на стр. 47,
в котором рассматривалась после-
довательная реакция А -> В -> D,
осуществляемая в непрерывнодей-
ствующем реакционном аппарате
полного смешения (см. рис. 1-11).
Количество продукта, выпускае-
мого производством в год, может
быть выражено как произведение
объема реакционного аппарата V
на его производительность Z:'
Afnp=VZ(a) (1-32)
Пусть производительность реак-
ционного аппарата как функция
степени превращения сырья описы-
вается выражением
Z = R(\ -a)2 (ЬЗЗ)
и цена продукта В установлена, ис-
ходя из минимальной его себестои-
мости:
Цпр — SCnp. мин (а) (1-34)
где R, S — постоянные множители.
Подставив выражения (1-32) —
(1-34) в уравнение (1-2), получим:
Д =• (TZnp £-пр) Л1пр =
= [SCnp. мин - Сдр (a)] VR (1 — а)2 (1-35)
Из структуры уравнения (1-35)
следует, что функция Д(а) имеет
экстремальный характер.
Пусть производство рассчитано
на выпуск продукта В в количе-
стве 1,0 ед./год при себестоимости единицы продукта, дости-
гающей минимального значения (1,0 руб}ед.) при a=0,234-0,25.
Зависимость себестоимости продукта В от степени превраще-
ния сырья можно установить, пользуясь рис. 1-13 и принимая, что
58
сумма энерго-материальных затрат составляет 90% себестоимо-
сти. Полученная таким образом зависимость Спр(а) изображена
на рис. 1-17, а.
Зависимость Л4пр(а), описываемая выражением (1-32), пред-
ставлена на рис. 1-17,6.
Принимая различную цену на продукт В (задаваясь значе- х
нием S) с помощью выражения (1-35) можно выразить зави-
симость Д(а). Для сопоставимости результатов расчетов при
5=1,2; 1,5 и 2,0 выражаем их в виде отношения дохода, получае-
мого при данном значении а (Да), к доходу, соответствующему
минимальной себестоимости продукта В (До)- Результаты расчета
можно видеть на рис. 1-17, в, из которого следует, что.максималь-
ный доход не соответствует минимуму себестоимости и дости-
гается в области значений а, соответствующих увеличенному вы-
пуску продукции.
Левые ветви кривых 1—3 на рис. 1-17, в выражают суммарный
эффект понижения себестоимости продукта и уменьшения произ-
водительности промышленной установки в результате увеличе-
ния а, а правые ветви этих кривых — эффект повышения себе-
стоимости продукта В при одновременном уменьшении произвол
дительности.
Ясно, что увеличение выпуска продукта за счет повышения
его себестоимости, увеличивая доход предприятия, приводит вме-
сте с тем к ухудшению (увеличению значения) показателя при-
веденных затрат П.
Повышение дохода предприятия (при заданной цене на про-
дукт) без изменения себестоимости достигается при увеличении
выпуска продукта в результате укрупнения размеров аппарату-
ры или увеличения числа параллельно работающих агрегатов.
При этом сохраняется оптимальный технологический режим (и
соответствующая ему минимальная себестоимость продукта) и
доход возрастает пропорционально увеличению выпуска продукта.
Значение показателя приведенных затрат при этом сохраняется
(или несколько снижается).
ЛИТЕРАТУРА
1. Сб. «Закон стоимости и его роль при социализме», под ред. Н. А. Цаба-
лова, Госпланиздат, 1959.
2. А. С. Кансон, Экономическая эффективность новой техники, Госполит-
издат, 1959.
3. В. М. Добкин, сб. «Автоматизация химических производств», вып. 3,
изд. Госкомитета Совета Мин. СССР по химии, 1959.
4. Основные методические положения по определению экономической эф-
фективности научно-исследовательских работ, Изд. «Экономика», 1964.
5. Методика определения экономической эффективности внедрения новой
техники, механизация и автоматизация производственных процессов в промыш-
ленности, Изд. АН СССР, 1962.
6. М. Ф. Нагиев, Основы разработки комплексных химических процессов
и проектирование реакторов, Баку, 1961.
7. Н. Н. Некрасов, Экономика химической промышленности, Изд. «Эко-
номика», 1966.
59
8. Нормы амортизационных отчислений по основным фондам народного хо-
зяйства СССР, изд. Госплана СССР, 1961.
9. Н. Н. Калмыков, С. А. В о й с б е й н, Экономика социалистической
химической промышленности, Госхимиздат, 1955, стр. 255.
10. Р. Q. С a n d е, Chem. Proc. Eng., 45, № 7, 380 (1964).
11. А. Р. S h a h b е n d e r i a n, Chem. Process Eng., 45, № 11, 647 (1964).
12. J. E. R. В r a n d e r, Brit. Chem. Eng., 9, № 11, 745 (1964).
13. M. Ф. Нагиев, Азерб. хим. ж., № 3, 64 (1966).
14. D. J. Wilde, Ind. Eng. Chem., 57, № 8, 18 (1965); Chem. Eng. Progr,
61, № 3, 86 (1965).
15. B. A. Hills, Brit. Chem. Eng., 8, № 10, 683 (1963).
16. R. Jackson, Chem. Eng. Sci., 19, № 4, 253 (1964).
17. G. B. D a n t z i g, Linear Programming AND Extentions, Princeton, 1963.
18. С. Л. Зуховицкий, Л. И. Авдеева, Линейное и выпуклое програм-
мирование, Изд. «Наука», 1964.
19. К. Д. Эрроу, Л. Гурвиц, X. У д з а в а, Исследования по линейному
и нелинейному программированию, ИЛ, 1962.
20. Г. П. К ю н ц и, В. Крелле, Нелинейное программирование, Изд. «Ра-
дио», 1965.
21. Ю. М. Ермолаев, Методы решения нелинейных экстремальных задач,
Кибернетика, № 4, 1 (1966).
22. С. Zener, Proc. Natl Acad. Sci., 47, 537 (1961); 48, 518 (1962); 50, 162
(1964).
23. R. J. Duffin, Soc. Indust. Appl. Math., 10, 119 (1962).
24. P. Веллман, Динамическое программирование, ИЛ, 1960.
25. Е. С. В е н т ц е л ь, Элементы динамического программирования, Изд.
«Наука», 1955.
26. Р. Веллман, С. Дрейфус, Прикладные задачи динамического про-
граммирования, Изд. «Мир», 1965.
27. Р. Арис, Оптимальное проектирование химических реакторов, ИЛ, 1963.
28. С. Робертс, Динамическое программирование в процессах химической
технологии и методы управления. Изд. «Мир», 1965.
29. И. И. Иоффе, Л. М. Письмен, Инженерная химия гетерогенного
катализа. Изд. «Химия», 1965, стр. 235.
30. М. Ф. Нагиев, Т. М. М и р д ж а ф а р о в а, Азерб. хим. ж., № 2, 97
(1966).
31. М. Ф. Нагиев, Т. М. Мирджафарова, В. И. Школьников,
Азерб. хим. ж., № 4, 64 (1966).
32. А. В. Woodier, J. W. W о о 1 с о с k, European Chem. News. Large
Plant Supplement, September 10, 7—9 (1965).
33. A. Chauvel, ibid., p. 12, 43. 45.
34. С. M a n t о v a n i, G. Goodchild, ibid., p. 19. 21.
Глава JI
НЕКОТОРЫЕ ВОПРОСЫ
ТЕРМОДИНАМИКИ И КИНЕТИКИ
ХИМИЧЕСКИХ РЕАКЦИЙ
Для отыскания оптимальных решений при разработке
любого технологического процесса целесообразно
рассматривать технологическую схему как совокупность трех по-
следовательных этапов (стадий) переработки исходных веществ:
1) подготовки сырья, 2) химического его превращения и 3) разделе-
ния продуктов реакции,- Аппаратурно-технологическое оформление
этих стадий существенно зависит от степени превращения сырья (а),
достигаемой в реакционном аппарате. Влияние а на расход энергии
и размер аппаратуры стадий подготовки сырья и разделения реак-
ционной смеси обычно достаточно ясно; зависимость же выхода
целевого продукта (0) и скорости его образования (щ) от степени
превращения сырья для разных реакционных систем и различных
типов реакционной аппаратуры имеет более сложный характер.
Вследствие того, что зависимости 0(a) и щ(а) существенным
образом влияют на величину и состав материальных потоков, пе-
рерабатываемых на каждой из стадий технологической схемы,
рассмотрению процессов и аппаратов, характерных для каждой
стадии, предпосылается изложение некоторых вопросов термоди-
намики и кинетики химических реакций.
Реакции, лежащие в основе многих процессов промышленного
органического синтеза, весьма сложны. Число возможных парал-
лельных и последовательных стадий превращения исходного ве-
щества столь велико, что полное кинетическое описание процесса
является практически неразрешимой задачей. Если бы даже такое
описание было выполнено, полученные выражения из-за своей
крайней сложности не имели бы практического смысла при ре-
шении технологических задач.
В подобной ситуации единственно оправданным является опи-
сание кинетического механизма посредством упрощенных модель-
ных схем. Каждая реакция в такой схеме представляет символи-
ческое отображение целой совокупности элементарных процессов,
а каждый промежуточный продукт модельной схемы соответствует
группе веществ, кинетическа'я роль которых одинакова.
61
Такого рода модельные схемы, учитывающие лишь продукты,
доступные аналитическому определению (и достаточно стойкие,
чтобы быть выделенными), могут быть описаны уравнениями фор-
мальной кинетики.
Несмотря на приближенный характер описания сложного про-
цесса модельными схемами, последние имеют важное значение
при поиске экономического оптимума, так как облегчают нахож-
дение оптимальных решений на основе ограниченной информации
в период исследования или проектной разработки нового процес-
са, когда число варьируемых переменных и интервал их возмож-
ных значений намного больше, чем при оптимизации действую-
щего производства.
Анализ механизма и количественных закономерностей процес-
са с помощью модельных схем позволяет установить границы .
области условий, соответствующих желаемому оптимуму. Уточнение
значений отдельных внешних параметров, влияющих на конечный
результат, производится при автоматическом регулировании про-
мышленного процесса (метод «черного ящика») в границах уже
установленной области оптимальных условий.
В химической технологии, очевидно, имеет смысл рассматри-
вать только такие продукты реакции, которые могут быть выде- '
лены из реакционной смеси. С этой точки зрения химические ре-
акции можно классифицировать следующим образом: 1) реакции,
при проведении которых могут быть выделены только конечные
продукты превращения, и 2) реакции, при проведении которых мо-
гут быть выделены не только конечные, но и относительно неустой-
чивые, подвергающиеся дальнейшим химическим превращениям
вещества. Наряду с такой классификацией все реакции можно раз-
делить на две категории: 1) реакции, при проведении которых вы-
ход продуктов не изменяется в течение всего процесса химического
превращения, и 2) реакции, при проведении которых выход про-
дуктов зависит от степени превращения исходного сырья. Таким
образом, имеем:
А->В; А-'ЛЛ—₽¥=f(a)
Wi
---= const;
^- = <р(Сл); ₽=Н«>
₽ = Ф(а)
F
Здесь А — исходное вещество; В, С, D, Е, F — конечные продукты,
которые могут быть выделены из реакционной смеси; сА — кон-
центрация вещества А. Прямыми стрелками обозначены односта-
дийные реакции, волнистыми стрелками — стадии сложных реак-
ций, промежуточные продукты которых весьма неустойчивы и не
могут быть выделены из реакционной смеси.
В случае одностадийной реакции, когда исходное вёщество А
превращается только в конечный продукт, выход последнего бу-
дет равен единице при любых значениях степени превращения ве-
щества. При двух параллельных реакциях, соотношение скоростей
которых постоянно в диапазоне всех возможных значений а, доля
каждого продукта от их суммы будет оставаться постоянной в
течение всего процесса и fj=tf(a). Во всех остальных случаях вы-
ход продуктов будет изменяться по мере протекания реакции.
Зависимость выхода от степени превращения, необходимая для
выбора технологического режима, может .быть получена, если из-
вестно изменение концентрации всех участвующих и образующих-
ся в реакции веществ во времени. Исключив время, в некоторых
случаях можно получить искомую зависимость Р(а). Однако вве-
дение новых переменных а(т) и р(а) в исходные дифференциаль-
ные уравнения быстрее приводит к цели. Для осуществления ука-
занных преобразований концентрации всех веществ, участвующих
и образующихся в реакции, выражают через выход целевого про-
дукта и степень превращения исходного вещества 1 по формулам:
сл = сЛо(1-а)
св =
(II-1)
Здесь'индексом «О» отмечена начальная концентрация исходного
вещества А. Дифференцированием по времени получим выраже-
ния для производных:
dcA da )
dr Са« dr !
dcB da dp da (
, г л a , И- с я -z P I
dr dr da dr J
(П-2)
Подставив выражения (II-l) и (П-2) в исходные дифферен-
циальные уравнения, описывающие изменение концентрации реа-
гентов и продуктов реакции во времени, а также учтя скорость
расходования исходного вещества, получим зависимость 0(a) в
дифференциальной форме.
1 При наличии нескольких исходных веществ концентрации последних долж-
ны быть предварительно выражены через концентрацию одного нз них с по-
мощью стехиометрических соотношений.
63
Так, скорости реакций
L L'
2 viA~> 2 v'/B/
2 V2/B/ + 2 v2i^i “* 2 VnPn
Z-l n = l
(здесь v — стехиометрические коэффициенты; для веществ, не уча-
ствующих в реакции, стехиометрический коэффициент равен нулю)
в общем виде описываются следующей системой дифференциаль-
ных уравнений:
dcAl
~fa~ - - Л (СА,’ СА2' СА3’ • ')
dcB,
—fa~ = fi (CAt> СА1’ %- • • -)~Ь(СА1’ СА2’ сА3.cBfcB2’ СВ3’ •••)
~~fa h(cA,’ СА2’ СА,.СВ,’ СВ2’ сВз’ •••)
Произведя указанные выше преобразования, а также выразив
са., са,, са. и т. д. через сА,, а св,, св,, св и т. д. через св,, получим:
! г ча^/)1 (11.3)
• • 1
dat at at fi (а<)
Дифференциальное уравнение типа (П-З) должно быть допол-
нено областью изменения степени превращения
О sC a sC ак
(ак — конечная степень превращения). Если рассматриваемый про-
цесс или хотя бы одна из его стадий необратимы и если процесс
устойчив, т. е. невозможны критические явления, то ак=1. В про-
тивном случае ак равно либо значению а при равновесии, либо
соответствующему критическому значению степени превращения.
Ниже будут рассмотрены некоторые вопросы термодинамики, свя-
занные с расчетом химического равновесия, а также некоторые
критические явления, возможные при осуществлении промышлен-
ного процесса синтеза.
Из уравнения (П-З) следует, что для получения зависимости
р(а) в явном виде необходимо знать конкретный вид функций fi
и /г- Последний можно получить, зная механизм химических пре-
вращений и кинетические закономерности каждой стадии рассмат-
риваемого процесса.
ХИМИЧЕСКОЕ РАВНОВЕСИЕ
Для проведения инженерных расчетов необходимо вла-
деть методами, позволяющими вычислять равновесный состав про-
дуктов реакции.
64
Устанавливаемые термодинамикой законы справедливы только
для материальных систем, изолированных от внешней среды не-
которой физической или мысленной оболочкой, препятствующей
обмену веществами, но допускающей обмен движением!. Такие
системы обычно называют изолированными. Для краткости при
дальнейшем изложении они будут называться просто системами.
Работа и теплота являются количественными мерами передачи
движения между системами. Первым началом термодинамики яв-
ляется закон сохранения энергии, т. е. закон ее неуничтожимости
как неуничтожимости движения вообще. Теплота (Q), поглощен-
ная системой из окружающей среды или выделенная системой в
окружающую среду,' изменяет внутреннюю энергию (U) системы
и затрачивается на преодоление сопротивлений, препятствующих
изменению состояния системы, т. е. на совершение работы (А):
dU = dQ-dA (П-4)
Первое начало термодинамики, выражая только закон сохра-
нения энергии, не дает никаких, сведений о направлении процес-
сов, протекающих в системе. Для макроскопических систем (пред-
ставляющих собой совокупность большого количества более
мелких подсистем, состояние которых можеъбыть различно) спра-
ведливо утверждение, что процессы во всей системе протекают
только в том направлении, которому соответствует переход систе-
мы в целом из менее вероятного состояния в более вероятное.
В этом заключается сущность второго начала термодинамики.
Обычно в термодинамике пользуются не самой вероятностью
состояния (IT), а некоторой функцией (S), связанной с вероят-
ностью соотношением:
Я. dW
N ’ W (Н"5)
(/? — газовая постоянная; N — число Авогадро). Функцию S на-
зывают энтропией. Связь энтропии с другими термодинамическими
величинами дается уравнением
dS = dQ)T (П-6)
справедливым для обратимых процессов (Т — абсолютная тем-
пература). Для необратимых процессов знак равенства в урав-
нении (П-6) должен быть заменен на знак неравенства.
Для обратимого процесса, проводимого при постоянной тем-
пературе, из уравнений (П-4) и (П-6) получим:
— dA = dU—TdS (П-7)
Работа, совершаемая системой, или работа, совершаемая над си-
стемой, есть сумма работ против сил внешнего давления, поверхно-
стного натяжения, сил электрических, магнитных, химических
1 Под движением здесь и далее подразумевается движение вообще (мо-
лекулярное, механическое, электрическое и т. д.).
5 В. С. Хайлов, Б. Б. Брандт
65
и т. д., препятствующих изменению состояния системы. В инте-
ресующих нас системах работа обычно совершается только про-
тив внешнего давления и химических сил.
Обозначив затрачиваемую на химическую реакцию работу
(т. е. полезную работу) через А', уравнение (П-7) можно пред-
ставить в виде:
dA' = — dU+TdS — PdV (П-8)
(Р — общее давление; V — объем). Полученное выражение, оче-
видно, может служить критерием направления процесса. Действи-
тельно, если tM'>0 (система может быть источником работы), то
химическая реакция может протекать самопроизвольно. Если
dA'=0 (система совершать работу не может), то в системе уста-
новилось химическое равновесие. Если dA'<0, то над системой
надо совершать работу для проведения реакции в желаемом на-
правлении (несамопроизвольный процесс или процесс, протекаю-
щий самопроизвольно в противоположном направлении).
Для химического процесса, проводимого при постоянных тем-
пературе и объеме, выражение (П-8) принимает вид:
. dA'=-dU + TdS (П-9)
ИЛИ
dA'=-d(U-TS) = -dF (П-10)
Функция U—TS = F называется изохорно-изотермическим потен-
циалом или свободной энергией. Интегрирование уравнения (П-9)
с учетом выражения (П-10) дает:
ДГ=Д1/-ГД5 (П-11)
Значит при постоянных Т и V критерием равновесия является из-
менение свободной энергии системы. Реакция протекает самопро-
извольно в желаемом и противоположном направлении при-А^<0
и AF>0 соответственно. Равновесие достигается при ДЁ=0.
Для химического процесса, проводимого при постоянных дав-
лении и температуре, выражение (П-8) можно представить в виде:
dA'=-dl/ + d(TS)-d(PV) (П-12)
ИЛИ
dA'=-d(U + PV-TS) (П-13)
Функцию U + PV = H называют теплосодержанием или эн-
тальпией, а Н—TS = Z — изобарно-изотермическим потенциалом.
Скомбинировав уравнение (П-12) с (П-13) и учтя определения
Н и Z, получим после интегрирования:
(П-14)
Таким образом, при постоянных Р и Т критерием равновесия яв-
ляется изобарно-изотермический потенциал.
Аналогичным образом может быть показано, что для процес-
сов, проводимых при постоянных Р и S и постоянных V и S, кри-
териями равновесия являются соответственно АН и АП.
66
Обычно на практике химические процессы осуществляются при
постоянных Р и Т: Поэтому наиболее часто для расчета равно-
весия приходится использовать изобарно-изотермический потен-
циал. При заданных давлении и температуре по изменению Z мож-
но определить равновесие системы. Состояние равновесия, оче-
видно, будет различным, когда давления, при которых проводится
процесс, тоже различны. Для вычисления равновесия в этом слу-
чае необходимо знать зависимость изобарно-изотермического по-
тенциала от давления. Продифференцируем развернутое выра-
жение Z=U+PV—TS при постоянной температуре:
dZ^dU + PdV+VdP-TdS
Однако dU+Pd.V—TdS = —dA', поэтому
dZ = - dA' + V dP
При равновесии dA' — O, следовательно
dZ^VdP (П-15)
Если и P=const, то dZ=0.
Интегрирование уравнения (П-15) от некоторого начального
состояния до произвольного дает:
р
\Z = f VdP
*0
Для выявления зависимости AZ(P) необходимо указать связь
между V и Р. Так, для одного моля идеального газа V=RT/P и
дг = 7?Г1п^- (П-16)
Аналогичная зависимость для реального газа:
дг=т?Пп-Д (п-17)
/о
где f=yP — летучесть, или исправленное давление (у — коэффи-
циент летучести).
Величина у является однозначной функцией приведенных дав-
ления Рщ> = Р1Рщ> и температуры 7'пр=7’/7’кр. Здесь Ркр и TKV —
критические давление и температура. Зависимость у(Рпр, Гпр)
представлена графически на рис. П-1 (см. также приложения 1,а
и 1,6).
Если за начальное состояние принять такое, при котором ле-
тучесть f0 равна единице, то соответствующее изменение изобарно-
изотермического потенциала/—Z° = RT\nf называют стандартным.
Рассмотрим систему, в которой при постоянных Р и Т осуще-
ствляется обратимая реакция:
',1A1 + v2A2+ ... + v/A/^v*Dj + V2D2+ ... + v*D„ (П-18)
5*
67
Рис. П-1. Зависимость коэффициента активности от приведенных давления и температуры.
При этом для произвольных условий процесса изменение изобарно-
изотермического потенциала во всей системе равно алгебраиче-
ской сумме изменений изобарно-изотермических потенциалов всех
подсистем (компонентов) системы:
AZ = v’zOi + v2ZDi + ••• ~(viza, + v2za2 + •••)
Аналогично для процесса, проводимого в стандартных усло-
виях, т. е. в условиях, когда летучести всех компонентов равны
единице, получим:
AZ° = viZ’O1 + v2Zo2+ ... ~(y1ZAl + v2ZAt + ...)
Изменения изобарно-изотермического потенциала системы при
проведении процесса в произвольных и стандартных условиях:
RZ — \Z = Vj (zDi — ZOi) + v2 (ZO2 — Zo) + ... — Vj (zAi — ZAl)~v2 (za2~ za2) ~
Так как изменение изобарно-изотермического потенциала для
любого компонента равно RTlnf, для всей системы имеем:
AZ-AZ° = RT(y* In + v2 In fDi+ ... -Vjlnf4i-v2lnf42- ...)
f1 p
AZ-AZ° = /?r In D'„ ”~ (11-19)
fV,fV2 ' <
IAt'Ai ‘ •
гДе fa ' fa » • • > fjj > fa > • • • — летучести компонентов системы,
соответствующие любому, в том числе и начальному, моменту
протекания реакции.
Если система находится в равновесии, то AZ=0, a fA, fA, ...
..., fD ,f D , ... соответствуют равновесным значениям летучести.
При этом уравнение (П-19) принимает вид:
— AZ° = 7?rinKf (П-20)
« *
fVlfV2 ...
где Kf = у1 ---------константа равновесия (все значения летуче-
Ж---
стей берутся при равновесном состоянии).
Уравнения (П-19) и (П-20) позволяют рассчитывать равнове-
сие химического процесса. Комбинируя уравнение (П-20) с урав-
нением (II-14), записанным для стандартных условий, получим:
— ДЯ° +rAS° = /?rinKf (П-21)
Таким образом, расчет константы равновесия сводится к вычис-
лению изобарно-изотермического потенциала. Для вычисления по-
следнего можно воспользоваться уравнением (П-14), но ДЯ и AS,
вообще говоря, зависят от температуры.
69
Зависимости Н(Т) и S(T) могут быть выявлены, если извест-
ны теплоемкости всех компонентов системы при различных тем-
пературах. По определению теплоемкость равна:
Ср—$ (П-22)
Из первого начала термодинамики следует, что
dQ = dH + dA'
(dH — dU + PdV; dA = dA' + PdV)
При равновесии dA'=0, следовательно, dQ=dH. Выражая dQ
как CPdT и интегрируя, имеем:
т
H = HQ+^cpdT (П-23)
То
Зависимость S(T) может быть получена при комбинировании
выражения (П-22) с (П-6):
С
dS = ^-dT (П-24)
Интегрирование уравнения (П-24) дает:
г С
S = S0+ J -Z-dT
То
Обычно в качестве начальной температуры (То) выбирают
298°К (стандартная температура).
Подставив выражения для Н(Т) и 5(7) в уравнение (П-14),
записанное для стандартных условий, получим:
д2Н5Х₽0Д-1Хсх)-г(1Х0д-2М=
т Т АС
^AH^-TAS^ J ACpdT-T J -P-dT (П-25)
298 298
где AC Р = 2 (Ср)прод 2(СР)исХ.
Для грубой оценки изменения изобарно-изотермического по-
тенциала при химической реакции можно принять, что теплоем-
кость продуктов реакции равна теплоемкости исходного веще-
ства (первое приближение). Тогда ДСР=0 и
AZ = АН^ — Т Д.$298
Изменения энтальпии и энтропии в стандартных условиях могут
быть вычислены из значений соответствующих величин для реак-
ции образования веществ из элементов — см. приложение II; бо-
лее подробные данные имеются в литературе [1, 2].
70
В качестве второго приближения можно принять, что во время
реакции значение ДСР не изменяется и не равно нулю. Тогда
т т
bZ° = АЯ^8- Т AS^8 + ДСр | dT-b£pT | —
298 298
ИЛИ
AZ = ДЯ298 ~ Т ^*^298 + р 298 — Т In 2gg j
Зависимость теплоемкости от температуры обычно дается в
виде степенного ряда:
С р—а +ЬТ + сТ2+ -^ (П-26)
Разность теплоемкостей продуктов реакции (П-18) и участву-
ющих в ней исходных веществ может быть представлена в виде:
Дс'
ДСр = Да + ДЬТ + ДсТ2 + -^
где
Aa = 2anpoA-SaHCx = v*«B, + v2aB2+ -(vlaZl + v2aZ2+ •••)
АЬ = У Ьпрод 2 ^ИСХ
Ас = 2 слрод — 2 Сисх
Ас ~ 2 спрод — 2 Сисх
Учитывая зависимость ДСР от температуры, получим для из-
менения изобарно-изотермического потенциала при реакции:
Д2° >= Д/^98 - Т AS°298 - Т (ЬаМй + AMI, + ДсМ2 + Дс'М_2) (П-27)
где
т т т т
Л.--4 рг+ jTdT+ [<1Т
298 298 298 298
ГТ ГТ
.. ' Г Т2 JT ! Г т JT >» If f
М2 =—J Т2 dT + J TdT-, М-i =—I + J "уг
298 298 298 298
Значения функций М0(Т), М2(Т) и Л4_2(7') табулированы
(табл. П-1). Этот метод вычисления изобарно-изотермического
потенциала был предложен и разработан Темкиным и Шварцма-
ном [3].
По вычисленной величине стандартного изобарно-изотермиче-
ского потенциала можно определить константу равновесия и зави-
симость ее от температуры. Равновесный состав продуктов реакции
71
Таблица 11-1
Коэффициенты уравнения (11-27) (3]
т м0 Mlf10“3 М2-ю-в М_2-105 т "о Afj-10-3 М2-1(Г6 м_2-ю5
300 0,0000 0,0000 0,0000 0,0000 1700 0,9162 0,5780 0,4424 0,3824
400 0,0392 0,0130 0,0043 0,0364 1800 0,9635 0,6265 0,5005 0,3915
500 0,1133 0,0407 0,149 0,0916 1900 1,0'09 0,6752 0,5619 0,3998
600 0,1962 0,0759 0,0303 0,1423 2000 1,0525 0,7240 0,6265 0,4072
700 0,2794 0,1153 0,0498 0,1853 2100 1,094 0,7730 0,6948 0,4140
800 0,3597 0,1574 0,0733 0,2213 2200 1,134 0,8220 0,7662 0,4203
900 0,4361 0,2012 0,1004 0,2521 2300 1,173 0,8711 0,8411 0,4260
1000 0,5088 0,2463 0,1134 0,2783 2400 1,210 0,9203 0,9192 0,4314
1100 0,5765 0,2922 0,1652 0,2988 2500 1,246 0,9696 1,0008 0,4363
1200 0,6410 0,3389 0,2029 0,3176 2600 1,280 1,0189 1,0856 0,4408
1300 0,7019 0,3860 0,2440 0,3340 2700 1,314 1,0683 1,1738 0,44505
1400 0,7595 0,4336 0,2886 0,34835 2800 1,346 1,1177 1,2654 0,4490
1500 0,8141 0,4814 0,3362 0,3610 2900 1,3775 1,1672 1,3606 0,4527
1600 0,8665 0,5296 0,3877 0,3723 3000 1,408 1,2166 1,4585 0,4562
может быть найден по известному значению константы равнове-
сия следующим образом. Константа равновесия по определению
равна:
*7“
V. V.t
tpfp2 ••
fVifV,
(11-28)
Учитывая, что летучесть компонента пропорциональна его пар-
циальному давлению, уравнение (П-28) можно представить сле-
дующим образом:
V. V2
. Yp.Yp, •••
yM--’
V1 v2
РрхРрг---
Ра,Ра,-
(П-29)
^Кр
(р— парциальное давление компонента). В К,> входят только
исправленные, т. е. подчиняющиеся закону идеальных газов, пар-
циальные давления компонентов. Величину р можно выразить
через количество молей:
Р
р = п---
Иобщ
где п — число молей компонента; Р — общее давление; побщ—
общее число молей компонентов смеси.
Подставив выражение для р в уравнение (П-29), получим:
V. V,
К К П°'П^ ! Р
“ Л „V, „V, ( „
пА1пАг • • • \ лобщ
Если в исходной смеси содержалось по одному молю компонен-
тов А[, Аг и т, д., то в равновесной смеси содержится х моль ком-
72
понента Dj и соответственно (y*2fv*)x моль компонента Dz и т. д.
Следовательно, получим:
/ р ^+'’2+--(vi+v2+-)
(П-30)
где
%6m = * + (v2/vl)x+ ••• +[1-(vl/vl)x]+t1~(v2M)*]+ •••
Пример II-1. 1. Вычислить константу равновесия реакции
С2Н4 + Н2О 5=± С2Н5ОН
при температуре 300° С и постоянном давлении. Расчет произвести при следую-
щих допущениях: сумма теплоемкостей этилена и паров воды равна теплоемко-
сти этилового спирта; теплоемкости не равны, но одинаково зависят от темпе-
ратуры.
Результаты сопоставить с точным значением константы равновесия.
2. Определить зависимость равновесной степени превращения этилена от да-
вления, если состав исходной смеси в молях равен:
Н2О __ Hqi
С2Н4 Лр2
Решение. 1. Константа равновесия (К/) согласно уравнению (11-20)
определяется изменением изобарно-изотермического потенциала при реакции:
Л7°
Таким образом, задача сводится к вычислению значения AZ3.
Стандартные теплоты образования из элементов и энтропии компонентов
реакции (см. приложение II): С2Н4 н2о С2Н5ОН
ЛН°98, кжал!моль . . . . 12,496 —57,798 -56,51
AS29?, кал!(моль • град) .... . . . . 52,48 45,13 66,39
Стандартный (при 7=298° К) тепловой эффект реакции равен разности стан-
дартных теплот образования продукта (этилового спирта) и исходных веществ
(этилена и воды):
ЛН2Э8 = —56,51 — 12,496 + 57,798= — 11,208 ккал!моль
Изменение энтропии при реакции:
AS298 = 66,39 — 52,48 — 45,13= —31,22 к.ал!(моль • град)
Зависимость изменения стандартного изобарно-изотермического потенциала
от температуры в первом приближении:
= ДЯщ8 - Т Л8°э8 = - 11 208 + 31,227
Теплоемкость компонентов реакции при температуре 298° К и коэффициенты
уравнения, аппроксимирующего зависимость теплоемкости от температуры (см.
приложение II):
С2Н4 н2о С2Н5ОН
Ср 298’ кал/(моль • град) . . . . . . . 10,41 8,025 15,4
а . . . 2,08 7,55 2,16
ь • 103 . . . 31,1 1,3525 49,7
с • 108 . . . -10,66 0,8658 -15,53
73
Если теплоемкость не зависит от температуры, то
ДСр 2д8 = 15,4 — 8,025 — 10,41 = — 3,035 кал/(моль град)
и зависимость изменения стандартного изобарно-изотермического потенциала от
температуры (второе приближение) принимает вид:
AZ’ = ДЯ’^ - Т Д5^8 + ЬСр ( Т - 298 - Г In ) =
= - 11 208 + 31,22Т-3,035 (г-298- Tin
\ 29о /
Для определения точной зависимости AZ°(7) при реакции воспользуемся
методом Темкина — Шварцмана. Для этого вычислим изменение коэффициентов
уравнения, апроксимирующего зависимость теплоемкости от температуры:
Да = 2,16 — 2,08 -7,55= -7,47
Дб = (49,7-31,1 - 1,35) • 10-3 = 17,25 • 10"3
Дс = ( — 15,53+ 10,66 + 0,866)-10'6= -5,736-10-6; Дс' = 0
Использовав уравнение (П-27), получим:
AZ = — Т Д3|д8 — Т (\аМ0 + Д6Л1] + ДсЛТ2 + &с'М_<^ =
= - 11 208 + 31,227— Г(т 7,47Л10 + 17,25 • Ю"3^! - 5,736 • 10‘6М2)
При температуре 573° К для изменения стандартного изобарно-изотермиче-
ского потенциала и константы равновесия в первом приближении имеем:
AZZ,3 = 6681 кал'моль-, 1gК, = 698* = — 2,549; Kf *= 2,825 • Ю-3
' I ,УоО • Z,u • □ /и «
Второе приближение дает:
Дг°573 = 6978 кал/моль-, 1g К. = - . Q -- . = - 2,662; К, = 2,178 • 10~3
Для вычисления точного значения AZ373 и Kf воспользуемся табл, II-1. Ин-
терполяцией находим величины Af0, Mi и М2 для температуры 573° К:
Мо = 0,1738; 441 = 0,0664-103; М2 = 0,0262 • 10е
AZ373 = 6855 кал)моль- lg Kf = - —г = - 2,614; Kf = 2,4111 • IO-3
* 1 >УоО * • D/и •
2. Если при установлении равновесия образуется х моль этилового спирта,
то в равновесной смеси будет содержаться «01 — х моль водяного пара и
«ог — х моль этилена. Всего в равновесной смеси будет содержаться (noi — х) +
+ («02 — х)+x=«oi+«o2 — х моль веществ. В соответствии с выражением (П-30)
константу равновесия можно представить следующим образом:
х / Р \1-2
= Ку -------£--------г ----—--------
V («01 - х) («02 - х) \ «01 + «02 - X )
Разрешая это уравнение относительно х, получим:
«о1+«02 у Г («01 +«ог)2 К\Р
Х== 2 ± У 4 KfP + Ky "°‘"°2
Физический смысл имеет только решение со знаком минус перед корнем, так
как содержание этилового спирта в равновесной смеси равно нулю при Я/=0.
74
Степень превращения этилена может быть представлена следующим образом:
И01
П02
Полученное выражение дает искомую зависимость степени превращения от да-
вления.
При высоких температурах и низких давлениях, когда все компоненты ре-
акции ведут себя как идеальные газы, Ку = 1. При низких температурах и вы-
соких давлениях Ку =<f(P,T). Вычислим п₽и температуре 573° К для давле-
ний 10, 100 и 400 атм. Для этого определим приведенные температуры (ТПр)
и давления (РПр) Для всех компонентов реакции. В данном случае значения
Т'пр и Дпр равны:
С2Н4 Н2О С2Н5ОН
ТКр, °К .... 9,7 374,1 243,0
Ркр- атм .... 50,8 218,5 63,1
т 573 ‘пр ~ т J кр .... 59,0 1,53 2,36
Рпр’1~ Ркр .... 0,2 0,046 0,158
р -J00 П₽'2 Ркр . . . . 2 0,46 1,58
р , = ПР- 3 Ркр .... 8 1,84 6,35
Используя рис. П-1 и приведенные в приложении I данные, по найденным
значениям ГПр и РПр определим коэффициенты летучести у Для компонентов
реакции *;
С2Н4 Н2О С2Н5ОН
Yp-ioo
Yp=400
„ г, Yc2H5oh
Таким образом, для константы Ку =------------
YC2H4Yh2O
400 атм имеем соответственно 1, 1,04 и 1,25.
Вычислим степень превращения этилена при
1 0,966 1
1 0,839 1,05
при общем давлении 10, 100 и
пятикратном избытке водяного
пара (По1/ло2=5):
общее давление
10 атм
а = 3 —
9 _ -2’41 :.1о7 ‘ ю • 5 ,0)019
2,41 -10~3 -10-1-1
общее давление
100 атм
а = 3 —
9- _ML_ia-3- ioo-5
2,41 • 10~3 • 100 + 1,04
общее давление
400 атм
а = 3-
9__J.41.H-3.400-5 =
2,41 • 10~3-400+ 1,25
1 Значение у Для этилена определено экстраполяцией соответствующих дан-
ных из приложения I.
76
КИНЕТИКА ХИМИЧЕСКИХ РЕАКЦИЙ
В ЗАКРЫТЫХ СИСТЕМАХ
Обычно при изложении химической кинетики ограничи-
ваются рассмотрением гомогенных реакций. Однако большинство
реакций в промышленности протекает в гетерогенной системе. По-
этому в данном разделе рассматриваются не только гомогенные,
но и гетерогенные реакции. Гомогенными принято называть реак-
ции, компоненты которых полностью смешиваются между собой,
вследствие чего концентрация каждого из них во всех точках
реакционного пространства одинакова. Гетерогенными обычно на-
зывают реакции, компоненты которых не смешиваются или сме-
шиваются лишь ограниченно; особенностью реакционной системы
в этом случае является наличие нескольких фаз и градиента кон-
центраций реагирующих компонентов между фазами
В отличие от разнообразных открытых систем, течение реак-
ции в закрытых системах, т. е. при отсутствии обмена веществом
с окружающим пространством, имеет свои особенности, изуче-
ние которых составляет предмет классической кинетики.
Ниже будут рассмотрены основные закономерности гомоген-
ных и гетерогенных реакций в условиях закрытых систем.
СКОРОСТЬ РЕАКЦИИ
Под скоростью химической реакции обычно понимают
изменение количества вещества (т), происходящее вследствие
химического превращения, в единицу времени (т). Однако при та-
ком определении скорости она зависит не только от самого хими-
ческого процесса, но и от общего количества вещества, подвер-
гаемого химическому превращению. Поэтому обычно рассмат-
ривают скорость химического превращения (ш), отнесенную к
единице объема (V) или поверхности (Г). В случае реакций, про-
водимых в гомогенной среде
1 ат
w ~ V ‘ dx
Для реакций, протекающих на поверхности раздела фаз
1 dm
ТО) = ——- --
F dx
Иногда для описания скорости химического превращения в гете-
рогенной системе изменение количества вещества в единицу вре-
мени относят также к объему одной из фаз.
Если химическое превращение происходит в системе, объем ко-
торой не изменяется, то скорость такого превращения может быть
определена как изменение концентрации (с) вещества в единицу
времени:
(П-31)
(П-32)
1 dm de
ТО) --- . ------ --
V dx dx
(П-ЗЗ)
76
При протекании химической реакции происходит только ка-
чественное изменение вещества, общее же его количество остает-
ся без изменения, поэтому скорость реакции можно измерять
только по изменению количества вещества определенного вида,
т. е. по убыли одного из реагентов или по приросту какого-либо
продукта реакции. Поскольку вещества при химическом превра-
щении взаимодействуют в строго определенных, стехиометрических
соотношениях, при измерениях скорости реакции необходимо учи-
тывать стехиометрические коэффициенты (v).
Так, для реакции
Ог 4- 2Н2 = 2Н2О ,
скорость расходования кислорода вдвое меньше скорости расхо-
дования водорода и образования воды. Скорость же реакции в
данном случае может быть выражена одним из следующих ра-
венств:
_ _ dc°2 = L rfCH2 = 1 . rfcH2o '
W dx 2 dx 2 dx
В общем виде для реакции
V[A! + v2A2 + ... = v*Dj + v2D2 + ... (11-34)
имеем:
1 ^Л1 1 dcA 1 dcDr 1 dcD
w =----------------------= ... =—j--------- —j- ---= ••• (H-35)
V, dx v2 dx V] dx v2 dx
Необходимо отметить, что равенства (И-35) справедливы толь-
ко для простых реакций. Для сложных, протекающих в несколько
ступеней реакций, как будет показано в дальнейшем, между ско-
ростью расходования исходных веществ и скоростью образова-
ния продуктов реакции существует более сложная зависимость.
Величина скорости реакции зависит от условий проведения ре-
акции: температуры, давления, концентрации реагирующих ве-
ществ и т. д.
Химическое взаимодействие между молекулами может про-
исходить только при столкновении их между собой. Так, осу-
ществление реакции (Ш-34) возможно только при одновремен-
ном столкновении vi молекул вещества Ai с V2 молекулами веще-
ства А2 и т. д. Общее число таких столкновений (No) в единице
объема и в единицу времени пропорционально концентрации ре-
агентов в соответствующих степенях:
V . . /V, V2
Л0 • cAtcA2 •
Очевидно, что скорость реакции должна быть прямо пропорцио-
нальна общему числу столкновений No-
W = k$cvA\... (П-36)
где k — коэффициент пропорциональности.
77
Такая зависимость скорости реакции от концентрации реаги-
рующих веществ носит название закона действующих масс, впер-
вые четко сформулированного Гульдбергом и Вааге (см. [4]).
Коэффициент пропорциональности k называют константой скоро-
сти реакции или удельной скоростью реакции. Константа k не яв-
ляется безразмерной величиной. Размерность ее определяется
характером зависимости скорости реакции от концентрации ре-
агентов.
По зависимости скорости реакции от концентрации реагентов
определяется порядок реакции. Суммарный порядок реакции ра-
вен сумме показателей степеней концентраций (vi + v2+ ...). Пока-
казатели vi, V2, ... носят название порядка реакции по данному
Рис. П-2. Энергетические
переходы химической
реакции.
реагенту. Обычно различают реакции пер-
вого, второго и высшего порядков. Экспе-
риментально наблюдаемый порядок реак-
ции нередко оказывается нулевым или
дробным.
В случае простых реакций их порядок
по данному реагенту обычно совпадает со
стехиометрическим коэффициентом того же
реагента. Простые реакции первого поряд-
ка называют мономолекулярными, второ-
го — бимолекулярными и т. д.
Так как скорость реакции является
функцией концентрации реагирующих ве-
ществ, то в том случае, когда хотя бы один
из реагентов является газом, величина w
будет зависеть от давления, причем эта зависимость опреде-
ляется порядком реакции по данному газообразному реагенту. За-
висимость скорости реакции от температуры оказывается более
сложной.
Как было указано выше, протекание химической реакции воз-
можно только при столкновениях молекул реагентов. Возникаю-
щая при сближении молекул сила отталкивания быстро возрастает
при уменьшении расстояния между ними. Химическое взаимодей-
ствие проявляется только на очень близких расстояниях между
молекулами. Вследствие этого в химическую реакцию вступают
только те молекулы, которые обладают энергией (Е) поступатель-
ного движения, обеспечивающей их сближение на достаточно
близкое расстояние. Доля молекул, обладающих энергией, боль-
__________________Е_
шей Е, равна е яг (R — газовая постоянная). Следовательно, к
химической реакции приводят не все No столкновений, а только
часть их {,Noe Rr). Таким образом:
Е
RTAM
е Аг
78
На существование экспоненциальной зависимости между ско-
ростью реакции и температурой впервые указал Вант-Гофф. Если
предположить, следуя Вант-Гоффу, что тепловой эффект реакции
ДЯ есть разность Е<— Е2 некоторых величин энергии (рис. П-2),
то константа равновесия (Ар) может быть представлена в виде:
AS E2-Ei
K^eRe *т
где AS — изменение энтропии в ходе реакции.
Так как Ар представляет собой отношение констант скоростей
прямой и обратной реакции (Kv = ki/k2), константы k\ и k2 должны
Д1 е2
быть пропорциональны величинам е RT и е RT соответственно.
___________________________________Е
Физический смысл зависимости k:: е RT был вскрыт Аррениусом
[5], предположившим, что в реакцию могут вступать только те мо-
лекулы, которые обладают некоторым избытком энергии Е по
сравнению со средним ее значением.
Минимальное значение избыточной энергии молекул, достаточ-
ное для осуществления единичного акта химического превраще-
ния, называют энергией активации, а зависимость константы ско-
рости реакции от температуры
__Е_
k = koe RT (П-37)
(feo—некоторая постоянная величина)—законом Аррениуса. Энер-
гию активации обычно относят к одному молю вещества и выра-
жают в кал/моль. Несмотря на то, что аррениусовская зависимость
скорости реакции от температуры многократно подтверждалась
экспериментально, она не является вполне точной [6, 7].
ГОМОГЕННЫЕ РЕАКЦИИ
Мономолекулярные реакции
Химическое превращение возможно только при столкно-
вениях молекул, обладающих достаточным запасом энергии. Од-
нако это утверждение требует некоторых уточнений, если рас-
сматривается какая-либо мономолекулярная реакция (например,
реакция распада СгНэОСгНб, C2H5ONO или N2O, реакция изоме-
ризации, рацемизации и т. д.). Для протекания таких реакций мо-
лекулы исходного вещества должны обладать запасом энергии,
достаточным для преодоления силы химического взаимодействия
• между отдельными частями молекул. Эту энергию молекулы по-
лучают либо за счет теплового движения при столкновениях (теп-
ловые реакции), либо от внешнего источника, например, при по-
глощении квантов света в фотохимических реакциях. Вследствие
этого при рассмотрении мономолекулярных реакций необходимо
учитывать процесс активации, накладывающий свой отпечаток на
кинетику химического превращения.
79
А + А ч-А 4- А
*2
А* —D-
Так, в случае термического распада вещества А, когда актив-
ные молекулы А* находятся в тепловом равновесии с неактив-
ными молекулами, механизм мономолекулярной реакции может
быть описан следующими уравнениями:
(П-38)
(11-39)
Скорость образования продуктов реакции пропорциональна кон-
центрации активных молекул:
= (J 1-40)
Неизвестная концентрация сА, может быть рассчитана из мате-
риального баланса, составленного для активных молекул:
&СА* о о
= k\CA ~ ^2САСА* ~ ^3СЛ* С1 f‘41)
Так как абсолютное значение концентрации активных молекул
обычно невелико, изменение этой концентрации во времени прак-
тически равно нулю, т. е. dc^jdr = 0. Из условия стационарности
следует, что
_ klcA
Слг ktfA + k3
Отсюда для скорости образования продуктов реакции получим:
dcD____^1^3СА
dx kgA + k3
(П-42)
При протекании мономолекулярной реакции возможны два
крайних случая: когда скорость дезактивации значительно больше
скорости мономолекулярного превращения, что соответствует ус-
ловию k2cA кз, и, наоборот, когда химическое превращение про-
текает значительно быстрее процесса активации (k2cA <^k3). В пер-
вом случае скорость образования продуктов пропорциональна кон-
центрации исходного вещества
dco ktk3
dx k2 Ca
(11-43)
во втором — квадрату концентрации:
- dcD
(II-44)
В соответствии с этим при достаточно низких давлениях все моно-
молекулярные реакции протекают как бимолекулярные.
30
С л *” СА С л х
Учитывая, что —2------= —, уравнения (П-43) и (П-44)
VA VD
можно привести к виду:
^£о_ = _Мз/ _2л_ \
dx k2 \Сл° vD Cf>)
dcD . {-_1аг
dx 1 ул» vd D
Здесь с. — начальная концентрация исходного вещества.
До
Сложные реакции
Обычно при химическом взаимодействии исходные ве-
щества вступают одновременно в несколько простых химических
реакций. В этом случае реакцию называют сложной. Протекание
сложных реакций возможно, если исходные вещества взаимодей-
ствуют между собой различным образом или если первичный про-
дукт реакции является достаточно реакционноспособным соедине-
нием и подвергается вследствие этого дальнейшим химическим
превращениям и т. д. К сложным реакциям можно отнести обра-
тимые, параллельные, последовательные, гомогенные каталитиче-
ские, сопряженные автокаталитические и цепные реакции.
Обратимые реакции. Часто продукты реакции
V|Aj + V2A2 + ... VjD] + V2D2 + ...
способны к химическому взаимодействию, приводящему к обра-
зованию исходных веществ, т. е. наряду с прямой реакцией про-
текает обратная реакция:
v |D[ + V2D2 + ... —> V| А] + V2A2 + ...
Суммарное стехиометрическое уравнение для всего процесса запи-
сывается в виде:
L L*
(П-45)
1 = 1 п=1
Реакции, описываемые уравнением (П-45), называют обра-
тимыми.
Скорость обратимой реакции равна сумме скоростей прямой
(Wi) и обратной (ш>2) реакций:
W = 1И] + w2
или
(П-46)
По мере протекания обратимой реакции и накопления продук-,
тов возрастает скорость обратной реакции. При израсходовании
Q В. С. Хайлов, Б. Б. Брандт 81
некоторого количества исходного вещества скорости прямой и об-
ратной реакции выравниваются
w = a>i + w2~ О
и в системе устанавливается подвижное равновесие. Подставив
в последнее уравнение значения для и w2, имеем:
<п-47)
п—1
ИЛИ
/! V*
, П cd"
TL=!Tj— = (П-48)
! 0Л
Так как показатели степени при концентрациях в выражении
для Лр равны соответствующим стехиометрическим коэффициен-
там, то показатели степени при концентрациях в выражении для
скорости любой сложной обратимой реакции, описываемой урав-
нением вида (П-46), тоже будут совпадать со стехиометрическими
коэффициентами. Если концентрации реагентов совпадают с их
активностями, из уравнения (П-48) следует также, что отношение
предэкспоненциальных множителей констант ki и k2 равно еД8/л,
поскольку тепловой эффект равновесной реакции равен по абсо-
лютной величине разности энергий активации прямой и обратной
реакции.
Для возможности интегрирования уравнения (П-46) необхо-
димо концентрации выразить через концентрацию какого-либо
продукта реакции с помощью стехиометрических соотношений:
Сд .— ~~ СD
—------L = —2__°Д- (11-49)
Здесь и в дальнейшем индексом «О» отмечены начальные концен-
трации.
Так, скорость образования продукта Di в соответствии с урав-
нениями (П-46) и (П-49) можно представить в виде:
Зависимость скорости обратимой реакции от температуры
имеет некоторые особенности. Пусть стехиометрическая смесь ис-
ходных веществ содержит некоторое количество продуктов реак-
ции и система далека от равновесия при температуре Ть Пусть
82
также температура 7\ настолько низка, что скорость расходования
исходных веществ практически равна нулю. При достаточно вы-
сокой температуре Т2, когда исходная смесь является равновес-
ной, скорость реакции тоже будет равна нулю. При температуре,
значение которой заключено между Т\ и Т2, скорость реакции мо-
жет принимать конечные значения и иметь экстремум. Покажем
это аналитически.
Определим сначала температуру Т2=Тт, при которой исходная
смесь становится равновесной и скорость реакции обращается в
нуль, для наиболее общего случая — химического взаимодействия
газов, протекающего с изменением числа молей при постоянном
давлении. От температуры зависит не только константа скорости
таких реакций, но и концентрация реагентов. Зависимость двух
последних величин от температуры описывается функциями раз-
личных классов
Е
. L RT
k = kae ; с =
(с — концентрация; р — парциальное давление компонента; М —
молекулярная масса компонента), вследствие чего оказывается
не всегда возможным подобрать температуру Тт, при которой
данная смесь исходные вещества — продукты реакции оказалась
бы равновесной. Подставив выражения для константы скорости
реакции и концентрации в уравнение (П-47), приведем его к виду:
Л£
е RTm = N1±n
(П-50)
где
Искомым значениям температуры соответствуют действительные
корни уравнения (П-50), равные абсциссам точек пересечения
ЬЕ
графиков <Pi = e RT и ф2 = Лг7’Лп- В зависимости от знака величин
АЕ и Ап возможны четыре случая.
1. Экзотермическая реакция, протекающая с уменьшением чис-
ла молей (АЕ < О, Ап>0). К этому случаю относятся различные
реакции присоединения (например, реакции гидратации олефи-
нов). С ростом температуры от 0 до оо функция qpi убывает от оо
до 1, а <р2 возрастает от 0 до оо, и для всех значений N всегда
существует температура Тт, при которой данная смесь оказывает-
ся равновесной.
6*
83
2. Экзотермическая реакция, протекающая с увеличением числа
молей (Д£<0, Дп<0). К этому случаю относятся наиболее рас-
пространенные реакции—реакции горения. С ростом температуры
от .0 до оо функция ф1 убывает от оо до 1, ф2 также убывает от оо
до 0, причем скорость, с которой убывает ф2, зависит от величины
параметра N. Если ф2 убывает медленнее, чем фь возможны две
точки пересечения графиков. Если ф2 убывает быстрее, чем фЬ
пересечений графиков нет. Следовательно, в этом случае только
смеси некоторых составов могут быть равновесными.
3. Эндотермическая реакция, протекающая с увеличением чис-
ла молей (ДЁ>0, Дп<0). К этому случаю относятся реакции кре-
кинга углеводородов. При увеличении температуры от 0 до оо
функция ф1 также возрастает от 0 до 1, а ф2 убывает от ро до 0.
Графики функций ф1 и ф2 всегда пересекаются, уравнение (П-50)
имеет только действительные корни и для любой смеси исходные
вещества — продукты реакции может быть подобрана температура,
при которой эта смесь оказалась бы равновесной.
4. Экзотермическая реакция, протекающая с уменьшением чис-
ла молей (ДЕ>0, Дп>0). К этому случаю относятся некоторые
реакции гидрирования, а также реакция синтеза углеводородов по
Тропшу—Фишеру. С ростом температуры обе функции ф! ц ф2
возрастают, но с различной скоростью. В зависимости от скорости
их возрастания функции ф] и ф2 могут пересекаться или не иметь
точек пересечения. При этом для смеси исходных веществ и про-
дуктов реакции не всегда существует температура, при которой
эта смесь становилась бы равновесной.
Введем безразмерную температуру:
1
| = AZAnTm (П-51)
Тогда уравнение (П-50) можно представить в виде:
еь^ = £ (П-52)
Уравнение (П-52) содержит только один параметр:
Таким образом, безразмерная температура, при которой ско-
рость обратимой газовой реакции обращается в нуль, является
функцией только параметра Ь. Искомая температура может быть
определена с помощью рис. П-З, на котором представлена зави-
симость £(5). Максимальное значение параметра Ь, при котором
уравнение (П-52) имеет действительные решения, равно 1/е, при
этом £ равно е.
84
Подставив в уравнение (П-51) значение 7VAn из выражения
(П-53), получим:
Т - ЕЕ = Е2~Е1 = Е2-Е1 Пт
(п2-Л1)/?Ь5 (n2-m)fllng ’
ь
0,4 _
0,3
0,2-
0,1
0-
-0,1
-0,2-
-0,3 -
-0,4-
-0,5-
Рис. П-З. Графическое решение
уравнения (П-52).
с
23456783^10
Так, для экзотермической реакции (Е2>Е}), сопровождающей-
ся уменьшением объема (n2 —ni<0), параметр 6<0 и возрастает
по абсолютной величине по мере накопления продуктов, как это
следует из уравнения (П-53).
Из рис. П-З (ветвь АВ) и табл.
П-2 следует, что параметр Ь, изме-
няясь. от 0 до —оо, изменяет g
только от 1 до 0. Следовательно,
температура, при которой дости-
гается равновесие — см. уравнение
(П-54), — убывает для экзотерми-
ческой реакции, протекающей с
уменьшением объема, и возрастает
для эндотермической реакции, сопро-
вождающейся увеличением объема.
Для экзотермической реакции,
протекающей с увеличением объе-
ма, или эндотермической реакции,
сопровождающейся уменьшением объема, параметр Ь>0 и убы-
вает по мере накопления продуктов, если АЕ<0, Ап<0, и возра-
стает, если ДЕ > 0, Ап > 0 — см. уравнение (П-53). В условиях, при
которых параметр Ь>1/е, уравнение (П-52) не имеет действи-
тельных корней, и не существует температур, для которых данный
состав смеси исходных веществ с продуктами реакции оказался
бы равновесным.
в
Таблица П-2
Зависимость | и In от &
ь 5 In 1 ь 1 Ing Ь g Ing
— оо 0 — оо -0,2788 0,8 -0,2231 0,3662 3,0 1,0986
— 23,026 0,1 -2,3026 -0,1171 0,9 -0,1054 0,3465 4,0 1,3863
-8,0470 0,2 -1,6094 | 0 1,0 0 0,3229 5,0 1,6094
-4,0133 0,3 -1,2040 0,1519 1,2 0,1823 0,2986 6,0 1,7918
-2,2901 0,4 -0,9163 0,2703 1,5 0,4055 0,2773 7;0 1,9459
-1,3862 0,5 -0,6931 0,3121 1,7 0,5306 0,2537 8,0 2,0794
-0,8513 0,6 - Q-,5108 0,3465 2,0 0,6931 0,2441 9,0 2,1972
-0,5095 0,7 -0,3567 0,3665 2,5 0,9163 0,2303 10,0 2,3026
Как следует из рис. П-З и табл. П-2 изменению параметра Ь
от 1/е до 0 соответствует изменение g от е до оо (ветвь CD) или
85
от е до 1 (ветвь СА). Так как Тт обратно пропорциональна произ-
ведению bl, температура убывает по мере накопления продуктов
для экзотермической реакции, протекающей с увеличением объ-
ема, т. е. при низких температурах (ветвь CD), и возрастает при
высоких температурах (ветвь СА). Для эндотермической реакции,
протекающей с уменьшением объема, Тт убывает при высоких и
возрастает при низких температурах.
Определим порядок величины Тт при различном содержании
продуктов в исходной смеси и различных значениях АЕ. Пусть
А£«104 кал/моль, ni = 2, п2=\, тогда feO2/&oi~ Ю7 л/моль. Для ис-
ходной смеси, содержащей 1% продуктов, при концентрации ис-
ходного вещества 5-10-2 моль!л имеем b ~ 109, т. е. 1п£~—5 и
7'т«103°К. При меньшем содержании продукта в исходной смеси,
например 10~4% и 10-9%, величина Тт возрастает соответственно
по порядку величины до 2-Ю3 и 104°К. Если АЕ—103 кал/моль, а
содержание продуктов в исходной смеси ~1%, то 7’т«102°К.
Рассмотрим условие экстремума скорости реакции
4^=0
dx
(П-55)
и определим температуру, при которой реакция протекает с мак-
симальной скоростью. Эту температуру будем называть оптималь-
ной, обозначая ее индексом «опт».
Подставив в условие (П-55) выражение для w и продифферен-
цировав, находим:
£а—£,
, ^7ОПТ _ ыт^п. — n2RTnm
опт Et-niRTom
(П-56)
Сопоставление уравнений (П-50) и (П-56) показывает, что условие
экстремума скорости обратимой газовой реакции отличается от
условия, при котором скорость этой реакции обращается в нуль,
на множитель 0) = ^—д£ч-т-. Поскольку всегда 'TOm<Tm и тем-
£1 — tliKlom
пература Тт для смесей с не очейь малым содержанием продукта
по порядку величины равна 103, а Е«И04 кал/моль, /г~1 и R=
= 1,986 кал/ (моль•град), в выражении для <р можно опустить вто-
рые слагаемые и уравнение (П-56) преобразовать в eb^'^h. Здесь
1 1
= = (П-57)
Температура, при которой обратимая газовая реакция, прово-
димая при постоянном давлении, протекает с максимальной ско-
ростью, приближенно равна:
Е2- Е, = E2 — Et
(n2 — ni)Rblh (п2-nd R In g.
(11-58)
Очевидно, что все указанные выше закономерности для Тт
справедливы и для Тоа1..
86
Для реакций, концентрации реагентов которых не зависят от
температуры (для газовых реакций, протекающих без изменения
числа молекул, или реакций в жидкости) условие экстремума ско-
рости (П-56) принимает вид [8]:
Г. v*
Ei-Et ЕД2 11 сй”
е^опт =------2x1----- (П-59)
ЗД)! П СА;
1 = 1 ‘
Из уравнения (П-59) определяется оптимальная температура, т. е.
температура, при которой реакция протекает с максимальной ско-
ростью:
Гопт ------------Ei~Ex L* . • (П-60)
е п 4:
Я 1п + AS + я 1п --------
1 ТТ V/
11 СА.
1 = 1 ‘
При этом учтено, что
R 1п = AS
feoi
Как и ранее, оптимальная температура убывает по мере протека-
ния экзотермической реакции (Д2>Д1) и возрастает в случае эн-
дотермической реакции (Е2<Е\).
Полученная зависимость (П-60) объясняется тем, что в соот-
ветствии с изменяющимся составом реакционной смеси равновес-
ный состав должен меняться таким образом, чтобы поддержива-
лась максимально высокая скорость реакции. Действительно, если
для состава реакционной смеси в некоторый момент времени тем-
пература Т, при которой осуществляется обратимая реакция,
является оптимальной, то по мере дальнейшего накопления про-
дуктов реакция, приближаясь к равновесию, определяемому зна-
чением Т, снижает свою скорость.
Пусть в некоторый момент времени при Т=ТОт соотношение'
продуктов реакции и исходных веществ дано выражением (П-59).
Вычислим температуру, при которой этот состав реакционной
смеси был бы равновесным, т. е. скорость прямой реакции равня-
лась бы скорости обратной реакции:
Е Е
i=l 1 n—i n
Исключив из последнего равенства концентрации реагентов с по-
мощью уравнения (П-59), получим:
Д2-Д1
£*2
87
Отсюда для температуры, при которой данный состав реакцион-
ной смеси был бы равновесным, имеем:
у, ____________Трпт_______
т~ . ,Т R , Е,
+ Л>пт д£ In
Разность Тт — Тот—АТ равна:
,\Т = ЬЕ п Е2
1 । ЕТопт । Е{
Д£ Е2
Таким образом, для увеличения скорости необходимо смещать
равновесие реакции так, чтобы текущий состав реакционной смеси
был всегда достаточно -удален от равновесного. Последнее может
быть достигнуто изменением температуры. Для сильно экзотерми-
ческих реакций в соответствии с правилом Ле-Шателье оптималь-
ная температура должна убывать, а для сильно эндотермических
реакций — возрастать.
Своеобразие обратимых реакций должно проявляться и в том,
что при температурах, больших Тот, должен наблюдаться отрица-
тельный температурный коэффициент скорости реакции.
Параллельные реакции. Если исходные вещества взаи-
модействуют между собой по двум или более простым химическим
реакциям, то последние называют параллельными. Например,
вследствие протекания двух параллельных реакций при присоеди-
нении двуокиси азота к углеводородному радикалу
R. + N = O —► R—N=O
II \
О О
R.+ O=N —► R—О—N = O
II
О
образуются нитросоединение и эфир азотистой кислоты. В общем
виде между i исходными веществами могут протекать ш парал-
лельных реакций, каждая из которых приводит к образованию п
продуктов:
V11A1+v12A2+ ••• +vuA/->v*1DH +v12D124- ... +v*„Dlra
V21A1 + v22A2 + • • • + V2ZA(. -> v21 D21 + Vi2D22 + . . . + V2rtD2ra
vmlA1 + vm2A2 + • • • + VmiA. vml Dm2 + v*m2Dm2 + • • +
88
Скорость образования продуктов каждой из параллельных ре-
акций описывается уравнением (П-36), т. е.
1 dC°H 1 ^12 _ 1 dCg in b. L n
* 1 1 c A,
dx V12 dx dx J. г . i
1 dc^ 21 1 de rj ^22 1 dc^ ^2П L L n ' rV2l
w2 = — • • 1 — ... • — «2 1 1 CA-
V21 dx v22 dx v2n dx J-J i = l . L
(П-61)
1 dcr> um\ 1 dcD um2 1 de L Dmn ь H ''ml
dx ~* • vm2 dx ‘ • * V vmn • . km I I dx J-J. i = l CAt
' Уравнения (П-61) интегрируются в
т реакций — одного порядка; 2) если
трех случаях: 1) если все
выполняются соотношения
k\
W2
wm
km
и 3) если в реакции участвует только одно исходное вещество.
Скорость превращения исходных веществ равна сумме скоро-
стей параллельных реакций, т. е. сумме скоростей образования
продуктов:
rfc m т L
= VmiWm — Vmikm JJ
zn=l Z7Z = 1 1 = 1
Последовательные реакции. Если первичный продукт
химического взаимодействия исходных веществ превращается в
результате одной или нескольких следующих одна за другой ре-
акций в конечное соединение, то такую совокупность реакций на-
зывают консекутивной или последовательной. Продукты, которые
одновременно образуются и расходуются в таком процессе, назы-
вают промежуточными.
Например, при фотонитрозировании циклогексана
NOC1 —> NO + C1
CI + С6Н12 —► СеНп" + НС1
C6Hn. + NO —> CeHnNO
C6HUNO —> C6H10=NOH
первичным продуктом реакции являются атомарный хлор и окись
азота. В результате их взаимодействия с циклогексаном образу-
ются циклогексильные радикалы, хлористый водород и нитрозоцик-
логексан, который затем изомеризуется в циклогексаноноксим.
Приведенные реакции консекутивны, промежуточными продукта-
ми являются NO, С1, СбНц- и CeHnNO.
89
Две последовательные реакции могут протекать по различным
схемам. Например
А —> В + продукты
В —> D +продукты
обе реакции мономолекулярны;
Ai + А2 —► В + продукты
В -—>- D + продукты
первая реакция бимолекулярна, а вторая мономолекулярна;
А, + А2 -—► В + продукты
В + А] —> D + продукты
обе реакции бимолекулярны;
А —> В + продукты
А + В —> D + продукты
первая реакция мономолекулярна, вторая бимолекулярна.
В общем виде две последовательные реакции можно предста-
вить следующей схемой1 *:
2 vliAi 2 Vi/B/
i=l /=1
2 + Zj v2iAi —•> S vnDn
i=i t=i n=i
Если скорости первой и второй реакций обозначить соответст-
венно через Wi и щ2, то с учетом стехиометрических коэффициен-
тов для скорости накопления продуктов и расходования исходных
веществ имеем:
— —----Vjiw'i -г v2iw2
dcB, , ,
~^- = vl!Wl-v2iw2
Исключив Wi и w2, получим соотношение между скоростями ре-
акций; измеренными по различным компонентам:
dcA, dcB, ( V2i ^2/ \ dcD
------- = —---------------L +------------+ —-- (П-62)
dr dr-) vn dr
1 Для веществ, пе участвующих в реакции, стехиометрический коэффициент
равен нулю.
90
Отсюда для стехиометрических соотношений имеем:
, \ V1/ / У>21 < Vii > -г- \
• _(чгч)-^(Ч/-с*/)д v„ v* (С/^
Скорость накопления промежуточного продукта В, равна раз-
ности скоростей его образования и расходования
,lr J*- У' '
П JJ Д 42/ (П-63)
/=1 1=1 /=1
а скорость образования конечного продукта — скорости расходо-
вания В,:
1 dc ь L' ,
п i = l j—l
Интегрированием системы уравнений (П-62) — (П-64) может
быть получено аналитическое выражение для изменения концент-
рации исходных, промежуточных и конечных веществ во времени.
Однако трудности, возникающие при интегрировании нелинейных
уравнений, позволяют довести решение системы уравнений (П-62) —
(11-64) до аналитического выражения только для последователь-
ных реакций первого порядка.
Для преодоления этих математических трудностей Боденштей-
ном [9] был предложен приближенный метод решения *, получив-
ший название метода стационарных концентраций. Заключается
он в следующем. Скорость образования промежуточных продук-
тов убывает вследствие расходования исходных веществ, скорость
же расходования промежуточных продуктов возрастает вследст-
вие накопления последних. В некоторый момент времени (0) ско-
рости образования и расходования промежуточных продуктов
dcB dcB^ dcBl
уравниваются, т. е. производные = ... = и в даль-
нейшем автоматически поддерживаются вблизи нуля. Для вре-
мен, равных или больших 9, можно получить приближенное реше-
ние системы уравнений (П-62) — (П-64), так как дифференциаль-
ное уравнение (П-63) может быть заменено алгебраическим
поскольку 0.
1 Метод Боденштейна, несмотря на появившуюся в последнее время воз-
можность численного интегрирования системы уравнений (П-62) — (П-64) с по-
мощью электронно-счетных машин, не утратил своего значения, так как он при-
водит, хотя и к поиближеиным, но аналитическим решениям.
91
-. - -J
Сопряженные реакции. Часто оказывается, что некото-
рая химическая реакция может протекать только в том случае
если в этой же системе осуществляется другая реакция, которая
как бы индуцирует первую. Реакции в этом случае называют со-
пряженными, а само явление — химической индукцией.
Химическая индукция возможна, если промежуточный продукт
двух последовательных реакций вовлекает в химическое взаимо-
действие вещество, не являющееся исходным для первых двух ре-
акций, т. е, наряду с реакциями
5 vi Л 2 VGB/
i=i /=1
2 V2/BJ + 2 v2iAZ 2 V«Dn
/=1 t=l n=l
протекает реакция
' 2Йв/+2*з^--2(<)Ч
/=1 (=1 П=1
или схематически:
А
к
♦
А'+В—^>D'
к
v
D
Скорости расходования исходных веществ и накопления про-
межуточных и конечных продуктов описываются следующей си-
стемой уравнений:
Точное решение этой системы уравнений может быть получено
только в некоторых частных случаях. Приближенно система ре-
шается методом стационарных концентраций.
92
Гомогенные каталитические реакции. Каталитическими
называются реакции, которые могут протекать или ускоряться в
присутствии какого-либо вещества (катализатора), не расходую-
щегося в данном процессе. Гомогенные каталитические реакции
обычно являются сложными последовательными реакциями, при-
чем катализатор, расходуясь в первой из них, регенерируется в
последней. Часто эти реакции можно представить упрощенной
схемой:
Л + К^>В
В + А'—3-> D + К
где К — катализатор; ku k2, k3— константы скоростей реакций.
Скорости расходования исходных веществ и накопления проме-
жуточных и конечных продуктов в этом случае описываются си-
стемой уравнений:
de .
---7^ = k,C .С„
dx 1 а К
deq de
~dx~ = dx~ = klCACK ~ k2cB ~ k3cA' cs
dcD dcA,
dx dx 3 B A f
Применяя метод стационарных концентраций, можно получить
приближенное выражение для скорости гомогенной каталитиче-
dcR
ской реакции. Из предположения, что = 0, следует:
сл = -*7+*зСл, саск (П-65)
Подставив выражение (П-65) в предыдущее уравнение, получим:
dc A’ ~ dcD = k\k3cAcA'cK
dx dx k2 + k?cA,
(11-66)
Из уравнения (11-66) следует, что скорость гомогенной ката-
литической реакции пропорциональна концентрации катализатора
(в соответствии с тем, что обычно наблюдается на практике).
При стационарности концентрации промежуточного продукта
В в этих реакциях скорости его образования и расходования рав-
ны. Вещество В расходуется в двух параллельных реакциях: за
счет распада на исходные продукты со скоростью k2cB и в реак-
ции с исходным веществом. Скорость последнего процесса равна
&3сл,св. Если распад веществ В протекает существенно медленнее,
чем его превращение в продукт реакции D, т. е.
(II-6 7)
93
то из стационарности концентрации св следует, что скорость об-
разования вещества В (k\CAcK) равна скорости образования про-
дукта D реакции. Этот же результат
^ср ,
dr ~ kicAcK
получается из уравнения (П-66), если пренебречь в знаменателе
первым слагаемым. Если k2 k3cA„ то из уравнения (П-66)
следует:
rfcD
dr k2 caca'ck
Таким образом, гомогенная каталитическая реакция формально
может описываться как реакция первого или второго порядка.
Автокаталитические реакции. Весьма своеобразной ока-
зывается кинетика каталитической реакции, если катализатором
является один из продуктов реакции:
А + К —пК + продукты
где п — количество молей катализатора, образующееся из одного
моля катализатора, вступающего в реакцию *. Скорость такой ре-
акции проходит через максимум. Увеличение скорости реакции в
начальный период (а не уменьшение, как для обычных реакций)
обусловлено возрастанием концентрации катализатора — продукта
реакции. Начаться автокаталитическая реакция может только в
том случае, если система вначале содержала некоторое количество
катализатора (/Со) или если независимо от этой реакции проте-
кает другая, приводящая к образованию катализатора.
В простейшем случае когда последняя реакция — первого по-
рядка, т. е.
А-±->К
для скорости накопления продукта получим уравнение:
dx = ^2СА + МЛ~ 1) САСК (П-68)
Обозначая прирост концентрации катализатора за счет реак-
ций через у и учитывая, что
^ = с№сКо = (сло-сл) («-D
преобразуем уравнение (П-68):
du , , / ц \ [ k2 , \
= k. (п - 1) с.-У-г- -------гг+ СЛ- + У
dr 1 \ п — 1 / \ k-i (П — 1) Ла 1
Цепные реакции. Во многих осуществляемых в промыш-
ленности реакциях, являющихся большей частью сложными па-
раллельно-последовательными реакциями, в качестве промежуточ-
1 Если п=1, реакция является гомогенной каталитической.
94
ного продукта образуются очень реакционноспособные валентно-
ненасыщенные частицы — радикалы или атомы. Если такой про-
межуточный продукт является катализатором реакции, то послед-
нюю называют цепной.
Как уже указывалось, скорость каталитической реакции про-
порциональна концентрации катализатора. Следовательно, для
протекания неразветвленной цепной реакции с достаточной ско-
ростью необходимо, чтобы в системе поддерживалась некоторая
концентрация активных частиц. Поскольку последние могут всту-
пать в различного рода побочные реакции, приводящие к гибели
активных частиц (реакция с материалом стенки аппарата, с при-
месями, реакции рекомбинации), необходимо, чтобы в системе
протекали также реакции, приводящие к возникновению новых
активных частиц. Первую группу реакций
В + материал стенки аппарата —> продукты
В + А —► продукты
В + В —► продукты
называют реакциями обрыва цепи, вторую группу реакций — ре-
акциями зарождения цепи.
Обычная цепная реакция описывается суммарным уравнением,
совпадающим формально с уравнением каталитической или авто-
каталитической реакции:
А + D —► nD —> продукты
где n = consH>l. Если гг = 1 (реакция не оказывает влияния на
концентрацию активных частиц), реакцию называют неразветв-
ленной цепной реакцией. Если п>1, т. е. концентрация активных
частиц возрастает вследствие протекания реакции, последнюю на-
зывают разветвленной цепной реакцией.
Элементарные реакции, приводящие к расходованию исходного
вещества и образованию продуктов реакции, не изменяющие при
этом концентрацию активных частиц, называют реакциями про-
должения цепи.
Например, при взаимодействии хлора с водородом реакциями
продолжения цепи являются:
Н2 + С1 —> НС1 + Н
С12 + Н —> НС1 + С1
Элементарные реакции, приводящие к увеличению концентрации
активных частиц, называют реакциями разветвления цепи. Так,
например, реакция
Н + О2 —► НО + О
является реакцией разветвления цепи, так как атом кислорода
образует две новые активные частицы:
О + н2 —► НО + Н
95
В некоторых случаях разветвления обусловлены реакциями
относительно устойчивого продукта цепной реакции. Так, при окис-
лении углеводородов первичный продукт реакции — гидроперекись
может распадаться по связи О—О с образованием -двух радика-
лов. Такие цепные реакции называют реакциями с вырожденными
разветвлениями.
Скорость неразветвленной цепной реак-
ции. Неразветвленные цепные реакции являются своеобразными
гомогенными каталитическими реакциями. Скорость расходования
исходного вещества в реакциях продолжения цепи, как и для лю-
бой гомогенной каталитической реакции, пропорциональна кон-
центрации катализатора и концентрациям исходных веществ.
Обычно активная частица взаимодействует только с одной молеку-
лой исходного вещества, следовательно, скорость расходования
последнего можно представить следующим образом:
de,
--^ = kCACB (П'69)
где k — константа скорости реакции.
Скорость накопления активных частиц равна разности скоро-
стей их образования (да') и расходования (w") т. е.
Дс„
(П-70)
ат
Из принципа стационарности следует, что
w' = w"
Если активные частицы в основном дезактивируются на стенке
аппарата или на молекулах примеси, то скорость w" пропорцио-
нальна св и из уравнения (П-70) получаем:
с „ = w'lk"
D ’
где k" — константа скорости соответствующего процесса дезакти-
вации.
Если активные частицы дезактивируются в основном за счет
рекомбинации, т. е. скорость w" пропорциональна св, то
Св = К w'lk"
Соответственно уравнение скорости расходования исходного ве-
щества примет вид:
или
de. ,____
----^ = kVw’lk" сА
Так как обычно скорость w' пропорциональна сА, то w" пропор-
циональна либо сА, либо Сд/2.
96
Поскольку в каждом элементарном акте продолжения цепи на
одну молекулу исходного вещества образуется соответствующее
стехиометрическому уравнению количество молекул продукта ре-
акции, при достаточно длинных цепях, когда дезактивация актив-
ных частиц, приводящая к образованию побочных продуктов, про-
f исходит относительно редко, скорость расходования исходного
вещества и скорость образования продукта реакции D связаны
соотношением (11-35), т. е.
1 dcA 1 dcD
Vi dx v2 " dx
Скорость разветвленной цепной реакции.
. В разветвленных цепных процессах скорость расходования исход-
ного вещества за счет реакций продолжения цепи, как и ранее,
пропорциональна концентрациям исходного вещества и активных
частиц. Скорость же накопления последних равна алгебраической
сумме скоростей их образования в реакциях зарождения (ш') и
разветвления цепи (ш"') и скоростей расходования за счет реак-
ций обрыва цепи (ш"). Рассмотрим частный случай, когда w" и
w"' пропорциональны св. Тогда
de „
-^w' + k’"cAcB-k"cB (П-71)
где k'"— константа скорости реакции разветвления.
Интегрированием уравнения (П-71) при начальных условиях
т=0, св=0 получим зависимость концентрации св от времени:
cB = V(e*T-1) (П'72)
где ty — k"'cA— k". При этом обычно пренебрегают малым измене-
нием концентрации исходного вещества по сравнению с измене-
нием концентрации активных частиц.
Из анализа уравнения (П-72) следует, что при ф<0 (т. е. ког-
да скорость дезактивации больше скорости реакций разветвления)
по истечении некоторого времени 1/чр устанавливается стацио-
нарная концентрация сВс[ активных частиц, равная
Учитывая уравнения (П-69) и (П-73), получаем для скорости рас-
ходования исходного вещества формулу
de. kw'c,
_ Л ________А
dx k"-k"'cA
которая переходит в выражение для скорости неразветвленной цеп-
ной реакции при k'"cA k". Если скорость реакции разветвления
цепи больше скорости обрыва (ф>0), концентрация активных' ча-
стиц, а вместе с ней и скорость всего процесса, быстро возрастает
7 В. С. Хайлов, Б, Б. Брандт 97
во времени. Для т> 1/-ф скорость экспоненциально зависит от вре-
мени, как это следует из уравнений (П-69) и (П-72):
Внешне такое увеличение скорости реакции воспринимается как
воспламенение или взрыв. Это явление, обусловленное накопле-
нием в системе активных частиц, получило название цепного вос-
пламенения.
При больших степенях превращения исходного вещества кон-
центрацию последнего нельзя считать постоянной. Для вычисле-
ния скорости расходования исходного вещества необходимо
решить уравнение (П-71) совместно с уравнением
dc,
-^—k'"cAcB (П-74)
Пренебрежем скоростью зарождения активных центров. Разделив
уравнение (П-71) на (П-74), получим:
dc„ k" 1
dc, k'" С.
А Д
Интегрирование последнего уравнения дает зависимость концент-
рации промежуточного продукта от текущей концентрации исход-
ного вещества:
k" с.
СВ ~ САа ~СА~ I*1 (П-75)
А
Поскольку концентрация промежуточного продукта В не может
стать отрицательной величиной, реакция при достижении некото-
рой глубины превращения, описываемой уравнением
k" сАо
САа ~СА = 1п
А
прекратится.
Фотохимические реакции. Реакции, протекающие под
воздействием света, носят название фотохимических. Механизм
этих реакций может быть весьма сложен. Однако общим для них
является активация молекул реагирующего вещества за счет по-
глощения световой энергии (в отличие от процесса активации
обычных темновых реакций, осуществляющихся за счет соударе-
ния молекул). Решающим шагом в разработке кинетики фотохи-
мических реакций является открытие Эйнштейном закона фотохи-
мического эквивалента. Согласно этому закону каждый квант
света при поглощении его веществом вызывает один элементарный
акт химической реакции.
Фотохимическую реакцию можно представить как два последо-
вательных процесса: активация молекулы под действием света
08
(первичный фотохимический процесс, подчиняющийся закону Эйн-
штейна) и следующие за первым процессы дезактивации или хи-
мического превращения активных частиц (вторичные процессы).
Для выяснения особенностей кинетики фотохимических реак-
ций рассмотрим слой газа или жидкости, поглощающий свет опре-
деленной длины волны (X). Концентрация поглощающего веще-
ства во всех сечениях слоя поддерживается постоянной и равной
с (в l/см3). При облучении этого слоя параллельным в направле-
нии х пучком света с длиной волны 7 и интенсивностью I, выра-
жаемой числом фотонов в единицу времени через единицу поверх-
ности сечения слоя, наблюдается ослабление светового потока с
увеличением толщины слоя. Это связано с поглощением фотонов
при движении их через слой. Поглощение фотонов, очевидно, тем
больше, чем более значительная доля поверхности сечения слоя
перекрывается частицами поглощающего вещества и чем интен-
сивней поток фотонов. Если о — эффективное сечение одной ча-
стицы поглощающего вещества (в см2), a s — сечение рассматри-
ваемого слоя, то доля поверхности сечения слоя, перекрываемая
частицами, равна нчст/$ («ч—число частиц в слое).
Число частиц в бесконечно тонком слое dx равно cs dx. Отсюда
для уменьшения интенсивности светового потока получим (закон
Ламберта—Бэра):
dl = — ocldx (И-76)
Уменьшение интенсивности светового потока в слое конечных
(х = /) размеров равно:
М = 10-1
После подстановки значения для /, получаемого интегрированием
уравнения (П-76) с учетом того, что 1 = 10 при х=0, имеем:
М = /0 (1 - ё~ас1) (11-77)
Относя число фотонов, поглощаемых в единицу времени в данном
слое, к объему этого слоя ', в силу закона фотохимического экви-
валента получим скорость возникновения активных частиц ауа (ско-
рость активации):
ша = -^ = -^-(1-е"’с/) (П-78)
В случае цилиндрического слоя, наиболее часто встречающе-
гося на практике, выражение для скорости активации принимает
вид:
и-а = /° 2 /о [1 - еас (П-79)
г ~го
где г0, г — внутренний и внешний радиусы поглощающего слоя.
1 Сечение слоя равно 1 см2.
Т
99
Для источника любой формы скорость активации можно пред-
ставить следующим образом:
= ЯР/о [1 - еос <r<,-r)] (П-80)
где <р—некоторая функция, имеющая размерность см-1, опреде-
ляемая геометрической формой источника света
Уравнение (П-80) может быть выведено, исходя из следующих
соображений. Пусть имеется источник с поверхностью S. Если при
этом расстояние по нормали к поверхности источника равно г (на
его поверхности г = г0), то разность светового потока на входе и
выходе из слоя толщиной dr равно количеству фотонов, погло-
щаемых в этом слое, т. е.
SI - (S + dS) (/ + di) = acIS dr
Производя алгебраические преобразования и опуская - величину
высшего порядка малости, получим:
di J , dS
- — = acdr + -^-
Уравнение следует проинтегрировать, учитывая, что S=So и /=/а
при г=г0. Произведя указанные действия, получим:
SI = I0Sae~ac (г~Го)
Таким образом, суммарное во всем слое ослабление светового по-
тока описывается тем же законом, что и для плоского слоя. Ко-
личество фотонов, поглощаемых в слое толщиной г — г0, равно
S0I0—SI, а скорость активации (количество фотонов, поглощае-
мых единицей объема слоя) равна (5оЛ>—SI)/V (V—объем слоя).
Отсюда, учитывая значение для SI, получим уравнение (П-80) в
следующей форме:
-Wa = 4L/o[l-e’ac(r’r“,l
Нас, однако, интересует увеличение концентрации конечного
продукта Р. Скорость его образования пропорциональна концент-
рации активных частиц сс„, т. е., учитывая стехиометрический ко-
эффициент v реакции, можно записать:
</с„
®p = -^- = vfe2cc» (11-81)
где k2 — константа скорости реакции.
Концентрация активных частиц, вообще говоря, не известна.
Однако она может быть легко установлена, исходя из следующих
соображений. Образовавшиеся в результате первичного фотохи-
1 Для источника сферической формы <р = —-j
г ~го
ИЮ
мического процесса активные частицы (возбужденные молекулы
или продукты диссоциации последних — атомы и радикалы1) мо-
гут вступать в реакцию с невозбужденными молекулами исходного
вещества или растворителя, превращаясь в конечные продукты,
либо дезактивироваться. Изменение концентрации активных ча-
стиц, таким образом, определяется тремя величинами: скоростью
образования этих частиц, скоростью их расходования в химиче-
ской реакции и скоростью их дезактивации.
Очевидно, что начиная с некоторого момента времени скорости
возникновения и расходования активных частиц должны урав-
няться, при этом установится стационарная, не меняющаяся во
времени концентрация активных частиц.
Химическая реакция, следующая за первичным фотохимиче-
ским процессом, в общем виде может быть представлена следую-
щей схемой:
С* + А —► Р + |ЧС*
где С* — активная частица; А — исходное вещество; Р — продукты
реакции; |ч— число активных частиц, образующихся в каждом
элементарном процессе.
При активные частицы расходуются за счет химической
реакции, при g4=l химическая реакция не влияет на концентра-
цию активных частиц, а при > 1 увеличивает количество
последних.
Поскольку дезактивация активных частиц может происходить
как моно-, так и бимолекулярно2, материальный баланс активных
-частиц можно представить следующим образом:
dcc, 2
= wa — fejfc* + An4fe2cc* — fe3c£* = О (П-82)
где wa — скорость активации; kt, ks — константы скоростей процес-
сов дезактивации; k2 — константа скорости химической реакции;
Апч=1ч—1 — прирост числа частиц в каждом элементарном йро-
цессе.
Из полученного уравнения может быть вычислена искомая кон-
центрация активных частиц
kt - \n4k2 Г. 4fe3taa \
-----2k3 V К 1 + (kt - An4fe2)3 ~ 1
1 В зависимости от величины энергии светового кваита и особенностей
строения поглощающей свет молекулы при их взаимодействии может произойти
возбуждение или диссоциация последней.
2 Возникающие в результате фотохимической диссоциации радикалы или
атомы могут дезактивироваться мономолекулярно, например, на стенках сосуда,
и бимолекулярио прн их рекомбинации в объеме. Возбужденные молекулы дез-
активируются либо вследствие излучения кваита света (люминесценция), либо
вследствие рассеивания их избыточной энергии при неудачных столкновениях,
т. е. дезактивируются только моиомолекулярио.
101
и скорость образования продуктов реакции
2«з \ Т \К{ — ш1цК2Г /
Рассмотрим два крайних случая: 1) когда дезактивация про-
исходит только мономолекулярно и 2) когда активные частицы
возникают в основном за счет первичного фотохимического про-
цесса, а расходуются в основном в результате дезактивации, при-
чем дезактивация осуществляется за счет рекомбинации атомов
(радикалов).
В первом случае в уравнении (П-82) можно не учитывать ве-
личину k3c2c*. Концентрация активных частиц и скорость образо-
вания продуктов реакции оказываются пропорциональными скоро-
сти активации:
wa
с* ki — tM4k2
w =Wa =___________yh____A (j _ M
В дальнейшем рассматривается слой, облучаемый параллельным
пучком света. Такое ограничение не оказывает существенного
влияния на конечные результаты.
Во втором случае в уравнении (П-82) можно пренебречь вто-
рым и третьим слагаемым. Концентрация активных частиц, а сле-
довательно, и скорость образования продуктов реакции будут про-
порциональны У wa:
/wa
w = У^га = 1/А- (1 - e~°ct)
УТ3 V I
Из полученных уравнений следует, что скорость образования
продуктов фотохимической реакции нельзя описать уравнениями
формальной кинетики. Однако в некоторых случаях они все же
могут быть использованы.
Если величина ocl велика (достаточно толстый слой или высо-
кая концентрация поглощающего вещества), то значение e~acl мало
по сравнению с единицей и
wa = Ioll
Тогда скорость образования продуктов фотохимической реакции
не зависит от концентрации реагирующих веществ.
Если ocl 1 (тонкий слой, низкая концентрация), то
ё~ас1 « 1 - <scl
и скорость образования продуктов может быть приближенно опи-
сана уравнением реакции первого или половинного порядка. По-
лученные результаты сведены в табл. П-3,
102
Таблица 11-3
Выражения для определения скорости образования
продуктов w фотохимической реакции
Скорость образования продуктов
Условия
<scl < 1
act » 1
vk2alo
kl ~ ^n4k2 * k3cC* kx - \n4k2 °
< -» /~ Gin 1/9
kx-\n4k2^kzcc. ^2УДГС
vfea/p
(k{ — Дгач£2) 1
ГЕТЕРОГЕННЫЕ РЕАКЦИИ
Если участвующие в реакции вещества находятся в раз-
личном агрегатном состоянии, реакцию называют гетерогенной.
Для таких реакций характерным является то, что наряду с хими-
ческим превращением в системе происходит перемещение веще-
ства вследствие различия концентраций реагентов и продуктов ре-
акции в различных точках системы. К гетерогенным относятся все
реакции, протекающие в присутствии твердого катализатора (на-
пример, получение пропаргилового спирта или бутандиола из аце-
тилена и формальдегида в присутствии ацетиленида меди; гид-
рохлорирование ацетилена до хлористого винила, осуществляемое
на сулеме; окисление метилового спирта до формальдегида на се-
ребряном катализаторе; получение метилового спирта из СО и Нг
на различных медных и цинк-хромовых катализаторах; одновре-
менное окисление метана и аммиака воздухом на платиновом кон-
такте в производстве синильной кислоты; окисление нафталина
воздухом на пятиокиси ванадия до фталевого ангидрида; гидриро-
вание бензола на сульфидных катализаторах до циклогексана и
т. д.). К гетерогенным относятся также реакции между газом и
твердым веществом (получение ацетилена из карбида кальция),
все реакции окисления жидких углеводородов воздухом и т. д.
Даже из этого краткого перечисления видно, насколько широко
распространены гетерогенные реакции и какое важное значение
они имеют в промышленности основного органического синтеза.
Следует различать гетерогенные реакции, собственно химиче-
ское превращение в которых осуществляется:
1) на поверхности (точнее вблизи поверхности) твердого веще-
ства, а в жидкости (газе) происходит только транспорт вещества;
2) во всем объеме одной из фаз;
3) во всем объеме реагирующей системы.
Скорость реакций первого, второго и третьего типа можно от-
носить соответственно к единице реакционной поверхности, к еди-
нице объема той фазы, в которой осуществляется химическое пре-
вращение, и к единице объема всей реагирующей системы.
109
Реакции на твердой поверхности
В реакциях, протекающих на твердой поверхности, об-
ласти осуществления химического превращения и переноса реа-
гентов разграничены в пространстве. Из глубины жидкости (газа)
реагенты перемещаются к поверхности, на которой происходит хи-
мическое превращение, а продукты реакции перемещаются в про-
тивоположном направлении, т. е. реакции данного типа представ-
ляют собой три последовательных процесса. Если скорости этих
процессов существенно различны, то может оказаться, что ско-
рость расходования исходного вещества определяется не скоростью
химического превращения, а скоростью переноса реагентов.
В соответствии с этой особенностью для реакций гетерогенного
типа различают так называемые «диффузионную область» проте-
кания реакции, определяемую переносом реагентов, и «кинети-
ческую область», определяемую превращением реагентов на по-
верхности.
Перемещение реагентов к реакционной поверхности существен-
но зависит от скорости (и) движения жидкости (газа) вдоль твер-
дой поверхности^ Если ы=0, перенос к поверхности оЬуществляет-
ся только за счет молекулярной диффузии. При ламинарном
движении потока перенос реагентов к поверхности из глубины
жидкости (по нормали к направлению движения жидкости) также
осуществляется только молекулярной диффузией. Если величина и
достаточно велика, то на направленное движение накладывается
хаотическое движение отдельных небольших объемов жидкости,
затухающее вблизи стенки канала. Такой режим движения назы-
вают турбулентным. Вблизи стенки на расстояниях 6, меньших
lOvmRe'k/u (v® — кинетический коэффициент вязкости жидкости,
см2!сек-, Re — критерий Рейнольдса; и — скорость потока, см/сек),
движение жидкости, по-видимому, ламинарно [10]. На расстояниях
от стенки, превышающих 6, вещество переносится по нормали к
потоку хаотическими пульсациями жидкости, а у стенки в слое
толщиной 6 — молекулярной диффузией.
Удобный для решения инженерных вопросов метод приближен-
ного описания кинетики процессов, протекающих в системе газ
(жидкость)—твердая поверхность разработан Франк-Каменецким
[10]. Метод основан на том, что для каждого момента времени
диффузионный поток q (количество вещества, доставляемого к
единице поверхности стенки в единицу времени) предполагается
пропорциональным разности концентраций этого вещества в глу-
бине жидкости (с) и у стенки (ci), т. е.
<7 = Рд (c-ci)
(рд — константа скорости диффузии). Предполагается также, что
константа скорости диффузии мало зависит от скорости реакции
на стенке.
Рассмотрим допустимость этих предположений на простейших
примерах.
104
Стационарный процесс. Если процесс доставки вещества
к стенке стационарен и осуществляется путем молекулярной диф-
фузии, то на основании первого закона Фика справедливо равен-
ство:
г>/ dc ,
— и -г- = q const
dx
где D' — коэффициент молекулярной диффузии; dcfdx— градиент
концентрации диффундирующего вещества. Отсюда после интегри-
рования, учитывая, что у стенки (толщина слоя жидкости х—0)
концентрация вещества равна Ci, получим:
<7 = — (c-ci) = const
Таким образом, константа скорости диффузии в стационарном
процессе есть отношение коэффициента диффузии к толщине слоя
жидкости, перепад концентрации в кото-
ром равен с — ср
Нестационарный процесс. Рас-
смотрим простейший случай — процесс
нестационарной диффузии в простран-
стве, ограниченном плоскостью, на кото-
рой протекает химическая реакция со
скоростью w. В начальный момент кон-
центрация вещества у стенки равна его
концентрации с0 в глубине жидкости
(газа). Скорость реакции в начальный
момент времени равна kc% и затем убы-
вает по мере расходования реагента.
Диффузионный поток вещества к стенке
в начальный момент равен нулю, так как
нулю равен градиент концентрации. По мере расходования веще-
ства у стенки за счет реакции и увеличения градиента концентра-
ции диффузионный поток возрастает. По истечении некоторого
времени к стенке доставляется почти столько же вещества, сколь-
ко его превращается на стенке. Процесс переходит в квазистацио-
нарный режим (рис. П-4). Для этого режима справедливо при-
ближенное равенство w^q.
Скорость образования продуктов реакции для любого момента
времени можно представить как произведение скорости реакции в
начальный момент времени на некоторую убывающую со временем
от 1 до 0 функцию <р', производная которой при т->оо стремится
к нулю. Например, для реакции первого порядка
w = fec0<p' (11-83)
Вид функции ф' можно получить из условия квазистационар-
ности, принимающего для реакции первого порядка форму
^1 = ₽д (со~<ч)
Рис. П-4. Установление
квазистационарного режи-
ма.
IQ5
Отсюда, исключив неизвестную концентрацию на поверхности, по-
лучим:
= (П-84)
Вследствие уменьшения скорости реакции по мере протекания
процесса функция qf = -д-^\ и константа скорости диффузии так-
Рд "г гс
же убывают во времени.
Зависимость рд(т) можно получить, исходя из следующих со-
ображений. Если константа скорости дйффузии, как и ранее, рав-
на отношению D'lx (х— расстояние, на котором концентрация ве-
щества убывает от Со до Ci), то из закона диффузии Эйнштейна
(среднее смещение частиц за время т пропорционально У D'x)
расстояние, на котором происходит перепад концентраций Со — Сь
пропорционально У D'x и с точностью до постоянного множителя
Сопоставим полученные результаты с точным аналитическим
решением.
Процесс диффузии в неподвижной фазе описывается уравне-
нием
(ц-85)
дх2 дх ' '
решение которого должно удовлетворять условию поставленной
задачи. В начальный момент времени концентрация вещества оди-
накова в объеме и на поверхности:
т = 0 I х = 0; с = с0 )
| х > 0; с = са )
В любой последующий момент времени при условии квазиста-
I дс \
ционарности количество вещества D -д— , доставляемое к по-
\ах >х-0
верхности, равно количеству вещества, исчезающему за счет реак-
ции, а градиент концентрации вещества на бесконечно большом
расстоянии от поверхности равен нулю:
0
x = 0; D'-^- = kcn |
дх I
<3с n I
х о°- —------> 0 |
дх J
(11-87)
Преобразуем уравнение (П-85) по Карсону—Хевисайду:
со оо
dx=p 1
о о
106
После интегрирования, учитывая, что с = с0 при т=0, получим:
7>'-§- = рс-рсо (П-88)
оо
Здесь с = р j ce~pxdx — новая функция, называемая изображением
о
искомой функции; р — новая комплексная переменная. Для удоб-
ства последующих преобразований введем функцию z = c—Со- То-
гда уравнение (П-88) принимает более простой вид:
= (П-89)
Граничные условия для вспомогательного уравнения (П-89) по-
лучаются аналогичным преобразованием условий (П-87) прип=1:
х = 0; D' kz + kca (П-90)
X—>оо; -^-->0 (П-91)
дх
Для того чтобы определить скорость реакции на стенке, до-
статочно наити производную!-^-) =|'д7'1 • Решение уравнения
(П-89), удовлетворяющее условию (П-91), имеет вид
_ -х
z = Ле r D (П-92)
а производная по координате:
— = -Л 1/£
дх А V D'
“X
е
(П-93)
Здесь А — решение уравнения (П-89) для плоскости (х = 0). Зна-
чение константы А находится из условия (П-90). Сопоставляя вы-
ражения (П-90) и (П-93) и учитывая, что zx=o=H, получаем
-г / р k -г k
AVD'~D'A+D' с°
откуда
-т - k/D'
А ----------- ,-г^- Со
и
I -----
х-0 у D'p + k
Окончательно для изображения скорости реакции имеем:
/ дс \
w = D'[ -5— = kc0
\ дх х-0
V~iyp+k
(П-94)
107
Переходя от изображения к оригиналу [11], получаем точное
решение:
Из выражения (П-94) можно получить также приближенное ре-
ч шение уравнения (П-84), а затем сравнить его с точным реше-
нием.
При сопоставлении уравнений (П-83) и (П-94) нетрудно заме-
тить, что для изображения <р' можно записать:
.,=
Ф У D'p + k
Поскольку изображение постоянной величины есть та же посто-
янная величина, изображение для ф', как и сама функция, должно
изменяться от 1 до 0. Предельные значения для изображения ф'
равны 1 и при У D'p k и VD'p k соответственно. Значе-
ния оригинала равны 1 и у jZпри jZ~ > k и jZ-^- < k
соответственно. Полученным предельным значениям ф' удовлетво-
ряет функция Ф':
Из сопоставления полученного и выведенного ранее выражения
для ф' следует, что константа скорости диффузии, как и следовало
ожидать, пропорциональна yZ-у- (коэффициент пропорциональ-
1 \
ности оказывается равным —== .
Ул/
* На рис. П-5 сопоставлены точное и приближенное решение,'
совпадающее между собой при ₽д^£ и 0Д<%. Максимальное от-
клонение приближенного решения не превышает 15,5% абсолют-
ного значения ф' при У^л 0Д=£.
Усложним теперь несколько задачу. Рассмотрим нестационар-
ный процесс диффузии в ограниченном объеме. Очевидно, что до
тех пор пока диффузионные потоки к противоположным стенкам
сосуда, расстояние между которыми равно 21, не оказывают влия-
ния друг на друга, т. е. до момента времени />, когда концентра-
ция в центре сосуда (сг) остается еще равной начальной концент-
рации вещества (с0), задача не отличается от разобранного выше
случая. В дальнейшем изменение движущегося к стенкам диффу-
зионного потока будет происходить только вследствие изменения
108
перепада концентрации (концентрация убывает как на стенке, так
и в центре сосуда) на расстоянии I.
Время О, в течение которого концентрация в центре сосуда ос-
тается равной Со, очевидно, равно времени, в течение которого ча-
стица может переместиться на расстояние /, т. е. 0 с точностью до
множителя порядка единицы должно равняться /2/О'. Константа
скорости диффузии как в стационарных, так и в нестационарных
процессах равна отношению коэффициента диффузии к толщине
Рис. П-5. Диффузия к поверхности, на которой
протекает реакция первого порядка:
1 — точное решение; 2 — приближенное решение; 3 — отно-
сительная ошибка.
слоя, в котором сосредоточен основной перепад концентрации. Та-
ким образом, для времен, меньших l2/D', константа скорости диф-
/D'
— , а С/ = с0; для времен, больших
l2/D', величина рд D'/l и сг убывает со временем.
Для проверки правильности сделанных выводов рассмотрим
задачу аналитически. Задача отличается от предыдущей тем, что
в любой момент времени градиент концентрации поддерживается
равным нулю не на бесконечном удалении от стенки, а на рас-
стоянии /, т. е. второе из условий (П-87) примет вид: _
Л/»
т>'0;х=/;~=0 (П-96)
Применяя преобразование Карсона — Хевисайда и учитывая
граничное условие (П-96), получим изображение скорости образо-
вания продуктов реакции: __
/Z^th/т/
w = kca——------------ (П-97)
VD’p\hiy -fr + k
109
Предельные выражения для w равны
fec0 и
Со / D'p th I
(П-98)
при У D'p th / у 3> k и У D'p th I у k соответственно.
Предельные выражения для самой скорости, очевидно, равны
/ /2 \ Д' t р \
£с0 Iпри т « —гр-1 и с0—•/(*) (при т »
где убывающая со временем безразмерная функция /(т)—ориги-
нал функции I th/y^. По этим предельным выраже-
ниям можно построить два выражения для скорости реакции, яв-
ляющиеся приближенными решениями задачи:
-г • Нт)
w — kca-fy------- (П-99)
-7*-/(T) + fe
w=^'11 kc0-f(x) (П-100)
^T + k
Величина Со‘/(т) в выражении (П-100)—концентрация в центре
сосуда в любой момент времени, больший l2/D.
Выражение (П-99) описывает начальную стадию процесса, вы-
ражение (П-100)—стадию со сформировавшимся профилем кон-
центрации. ____
Разлагая в выражении (П-98) в экспоненциальный
ряд1 и переходя к оригиналу, получаем для /(т):
/——
Нт) = 1/ -7)7-
F Ли Т
оо
1 + 2 (- 1)йе
ь=1
ЬЧ2 -
й'х
При т l2/D' сумма в выражении для f(r) мала по сравнению с
единицей и уравнение (П-99) для скорости реакции принимает вид
совпадающий с решением предыдущей задачи.
th х = 1 + 2 2 (-1)" e~2bx.
110
Более удобное для процесса со сформировавшимся профилем
концентрации (т5?>/2ДУ) выражение f(x) можно получить, если
для перехода к оригиналу воспользоваться изображением в
виде
°° -я»
=2 У е
ь=»о
причем для достаточно больших времен можно ограничиться пер-
вым членом ряда. Следовательно, во второй стадии процесса кон-
станта скорости диффузии постоянна, концентрация же со време-
нем убывает экспоненциально.
Константы скорости диффузии для стадии со сформировав-
шимся профилем концентрации в нестационарных процессах,
протекающих в плоском, цилиндрическом и сферическом сосудах,
приведены в монографии Франк-Каменецкого [10]:
Плоский сосуд...............
л2 D'
4 ’ I
Цилиндрический сосуд 1 . . .
Сферический сосуд..........
Но D'
2 ’ I
л2 D'
3 ’ I
Таким образом, в неподвижной относительно сосуда жидкости
скорость реакции на стенках при условии квазистационарности
процесса равна диффузионному потоку к стенкам. Последний мож-
но представить как произведение разности концентрации в центре
и у стенок сосуда на константу скорости диффузии. При этом кон-
станта скорости диффузии с точностью, достаточной для практи-
ческих расчетов, может приниматься не зависящей от скорости
реакции.
Во всех случаях константа скорости диффузии пропорциональ-
на отношению коэффициента диффузии к толщине слоя, в котором
сосредоточен основной перепад концентраций, т. е. для времен,
меньших Р/D' (Z — радиус сосуда), рд = (перепад концент-
раций сосредоточен в слое толщиной У 2£)'т), а для времен, боль-
ших Р/D', рд: : D'/l, причем коэффициент пропорциональности за-
висит от формы сосуда.
Для процессов, протекающих в жидкости, движущейся вдоль
твердой поверхности, нет оснований полагать, что общие законо-
мерности переноса вещества к поверхности будут, отличаться от
рассмотренных выше, поскольку транспорт вещества из глубины
ламинарного потока к поверхности (по нормали к движению
1 р.0=2,4 — первый корень Бесселевой функции нулевого порядка.
111
жидкости) и через ламинарную пленку при турбулентном движе-
нии 1 осуществляется за счет молекулярной диффузии.
Так, при ламинарном движении жидкости в начальном участке
канала, где диффузионные потоки к противоположным стенкам
еще не взаимодействуют между собой и в центре канала концент-
рация вещества остается еще равной начальной концентрации,
йонстанта скорости диффузии должна зависеть от времени дви-
жения данного элемента жидкости по каналу. Как и ранее
Рд = О'/Л (П-101)
где А — толщина слоя жидкости у стенки, в котором сосредоточен
основной перепад концентрации. Величина А равна среднему рас-
стоянию У2Б'т;, пройденному диффундирующей частицей по на-
правлению к стенке за время т. Если принять, что закон, распре-
деления скоростей в потоке — линейный, то время нахождения
данного элемента жидкости в канале, т. е. время диффузии, можно
представить в виде
где L — путь, пройденный элементом жидкости; и — скорость по-
тока в центре канала; d — диаметр канала.
Исключив время из соотношений (П-101) и (П-102), получим
выражение для толщины слоя, в котором сосредоточен основной
перепад концентрации:
Следовательно, константа скорости диффузии с точностью до мно-
жителя порядка единицы равна
или в безразмерном виде
а _ ~i/~ Уж d
Рд D' ~ V хж ’ D’ ~L
т. е.
3/ J-
№1 = у 2 Re Pr ~-
Здесь й = и/2 — средняя скорость потока; — кинематический ко-
эффициент вязкости жидкости.
Данная задача была аналитически решена в тех же предпо-
ложениях Левеком [12]. Решение, представленное как зависимость
1 Перемещение вещества из глубины потока к ламинарной пленке осуще-
ствляется значительно быстрее, чем через саму пленку. Поэтому основное диф-
фузионное сопротивление оказывается сосредоточенным в этой пленке.
112
между безразмерными величинами, отличается от полученного
выше множителем пропорциональности (1,077 вместо J^)1.
Время, в течение которого частицы диффундирующего веще-
ства из элемента объема, располцженного на оси канала, смеща-
ются в среднем на расстояние, равное радиусу канала (гк), можно
представить следующим образом:
За это время элемент жидкости на оси переместится от начала
канала на расстояние L = u/Q. Исключая из двух последних уравне-
ний. время диффузии и учитывая, что средняя скорость потока
вдвое меньше максимальной, получаем с точностью до множителя
порядка единицы расстояние от начала канала, проходимое жидко-
стью, после которого диффузионные потоки к противоположным
стенкам начинают взаимодействовать между собой и толщина слоя,
в которой сосредоточен весь перепад концентраций, остается по-
стоянной:
£' = rKRePr
На расстояниях, больших L', константа скорости диффузии остает-
ся постоянной и пропорциональной D'Id. Коэффициент пропорцио-
нальности, аналитически вычисленный Нуссельтом, равен 3,659.
Если поток жидкости турбулизован, то рд вследствие недоста-
точной точности в определении толщины ламинарной пленки целе-
сообразно вычислять, используя эмпирические зависимости Nu= '
= f(Re, Рг). Эти зависимости могут быть получены из анализа
данных по физической абсорбции или теплопередаче, так как про-
цессы переноса тепла и вещества, обусловленные хаотическим
движением молекул, подобны. Наиболее часто для вычисления
константы скорости диффузии используется формула Краусольда:
Nu = 0,024 Re018 Рг0'35
Достоинство метода приближенного описания кинетики реак-
ций на твердой поверхности, разработанного Франк-Каменецким,
заключается в том, что этот метод может быть применен не толь-
ко для реакций первого, но и любого другого порядка, а также
для описания кинетики сложных реакций, протекающих на твер-
дой поверхности. Рассмотрим несколько примеров [10].
На поверхности протекает необратимая ре-
акция n-го порядка. Скорость образования продукта реакции
В равна:
1 Коэффициент пропорциональности для среднего значения Nu равен 1,615.
8 В. С- Хайлов, Б. Б. Брандт ИЗ
где с — неизвестная концентрация реагента на поверхности, опре-
деляемая из условия квазистационарности процесса
kctt = ₽д (с0 - с)
Введением безразмерной концентрации £ = с/со условие квазиста-
цйонарности приводится к виду, содержащему только один без-
размерный параметр И — \-п ;
Рдсо
(П-ЮЗ)
При решении полученного алгебраического уравнения удобно при-
менять графический метод, заключающийся в том, что искомая
Рис. П-6. Диффузионная кинетика реакций дробного порядка.
Графическое решение уравнения (П-103)
безразмерная концентрация £ находится как абсцисса точки пере-
1 — С
сечения графиков w = t,n и q=----- (рис. П-6). Из рис. П-6 еле-
Ц
дует, что при уменьшении константы скорости диффузии или уве-
личении константы скорости реакции, т. е. при увеличении пара-
метра ц, концентрация реагента на поверхности приближается к
нулю. Диффузионный поток, а вместе с ним и скорость реакции
114
при этом стремятся к величине рдс0. При увеличении константы
скорости диффузии или уменьшении константы скорости реакции
концентрация реагента на поверхности приближается к его кон-
центрации в объеме, а скорость реакции (и вместе с ней и диффу-
зионный поток) — к величине kcm. В первом случае говорят о диф-
фузионной области протекания реакции, во втором — о кинетиче-
ской области.
Обратимые реакции. Рассмотрим прямую и об-
ратную реакции первого порядка.
Пусть обратная реакция протекает в кинетической "области,
прямая — в диффузионной, тогда с'в св, с'А <С сА (штрихом отме-
чены концентрации на поверхности). Неизвестная концентрация
вещества А на поверхности находится из условия квазистационар-
ности:
₽д1(сЛ-сл) = *1сА-*2сВ
(индекс «1» относится к прямой реакции, индекс «2» — к обратной
реакции). После подстановки значения с'А в выражение для ско-
рости реакции, получим, учитывая, что рд i<Sfei (диффузионная
область)
но отношение констант скорости прямой и обратной реакций есть
константа равновесия. Тогда
w=^ca(i~4^
Реакция может протекать в нужном направлении (с накоплением
продукта В) только в том случае, если максимально полезная ра-
бота Амакс>0
ЛМакс = RT In К - RT In > О
С А
с *^макс
Подставив значение К. — — е RT в выражение для скорости, по-
св
лучаем:
(^макс \
1 ~е RT I
Если система находится вдали от равновесия, то и
Вблизи от равновесия Лмакс<^7?Т. Разлагая экспоненту в ряд и
ограничиваясь первыми двумя членами разложения, получаем,что
скорость реакции пропорциональна максимально полезной работе:
л ЛМакс
ю“Рд1сл-^-
8*
115
Последовательные реакции. Пусть обе реак-
ции— первого порядка:
А —> в —> С
Условия квазистационарности для данной реакции примут вид:
®Л = РД1 (CA-cA) = klcA
WB = Рд2 (СВ ~ св) = ^lcA ~ k2cB
w С = РдЗ (сС — Сс) = ^2СВ
(11-104)
Решая равенства (П-104) относительно с'А, с'в и с'с и подставляя
найденные значения в выражения для скоростей накопления про-
межуточного и конечного продуктов, получаем:
= Мд1СЛ о
В (*1 + ₽Д1)(^ + РД2) Рд2
k^CA 1
с (ki + РдО (кг + 0дг) 2
Нетрудно заметить, что отношение скоростей, т. е. относительный
выход промежуточного и конечного продуктов, равен отношению
константы скорости диффузии промежуточного продукта к кон-
станте скорости его расходования. В диффузионной области, когда
промежуточный продукт медленно отводится от стенки в объем
(Рдг’Сйг), выход его будет существенно ниже выхода конечного
продукта. В кинетической области, когда скорость диффузионного
транспорта вещества В значительно больше скорости его расходо-
вания в реакции на стенке (Рдг^^з), выход промежуточного про-
дукта будет велик по сравнению с выходом продукта С.
Автокаталитические реакции. В этом случае
при увеличении скорости диффузионного транспорта катализирую-
щего продукта реакции от стенки в объем жидкости скорость ре-
акции должна убывать. Покажем это аналитически. Пусть на стен-
ке протекает реакция А+С->2С и сА)»сс. Тогда сА — const и
Определив неизвестную концентрацию продукта реакции на по-
верхности из условий квазистационарности, получим:-
dcc &₽д
dx ~ ₽д-£ Сс
Из этого выражения для скорости накопления продукта следует,
что при увеличении константы скорости диффузии от 0Д=& до
рд3>/г скорость реакции действительно убывает от оо до значения
kcc.
116
Рис. П-7. Конвектив-
ные токи, возникаю-
щие в движущихся
пузыре или капле.
Реакции в слое
Химические реакции, протекающие в системах газ —
жидкость, жидкость — жидкость и между жидкостью (газом) и
слоем твердого пористого катализатора, имеют много общего.
Наиболее существенным для таких реакций является одновремен-
ный с химическим превращением транспорт вещества через по-
верхность раздела фаз (или поверхность слоя катализатора) в глубь
материала.
Для интенсификации процессов, протекающих между газом и
жидкостью или между двумя несмешивающимися жидкостями,
одну из фаз обычно диспергируют в другой.
При этом пузыри газа или капли жидкости
движутся через сплошную фазу. Такое дви-
жение является причиной возникновения
внутри пузырей и капел^ь конвективных то-
ков, направленных вдоль их поверхности
(рис. П-7). Если в жидкости растворены по-
верхностно-активные вещества, блокирующие
поверхности капель, циркуляционные токи
могут в них и не возникать.
Интенсификация процессов, протекающих
в слое пористого катализатора, достигается
тем, что катализатор применяют в виде не-
больших гранул (зерен), заполняющих ап-
парат. Через слой зерен движется газ (жид-
кость). В зависимости от скорости газа и
конструкции аппарата зерна катализатора могут быть либо непо-
движными, либо тем или иным способом перемещаться в аппа-
рате. Внутри зерен газ (жидкость) перемещается только за счет
диффузии, конвективные токи полностью отсутствуют.
Химическое превращение в системе газ — жидкость осуществ-
ляется обычно только в жидкой фазе, в системе жидкость —
жидкость — в одной или обеих фазах, в системе газ — зерно ката-
лизатора^- только внутри последнего. Фазу, в которой протекает
химическая реакция, называют активной.
В подвижных фазах при наличии конвекции основной перепад
концентрации транспортируемого через поверхность раздела ре-
агента сосредоточен в тонком слое вблизи этой поверхности. Вне
слоя устанавливается некоторая средняя по всему объему фазы
концентрация (рис. П-8,а). В зерне катализатора и в каплях, в
которых не происходит циркуляции жидкости, концентрация
транспортируемого реагента убывает с расстоянием от поверхно-
сти по всей глубине слоя (рис. П-8, б).
Если средняя концентрация реагента по всему объему подвиж-
ной фазы, в которой протекает химическая реакция, близка к тер-
модинамически равновесной, так что перепад концентрации в диф-
фузионном слое у поверхности весьма мал, то скорость реакции
117
достигает при данной температуре своего максимального значе-
ния, не зависящего от гидродинамического режима в данной фазе.
При этом процесс протекает во внутренней кинетической области.
Внутренняя кинетическая область в зерне катализатора или капле,
при отсутствии конвекции в последней, достигается тогда, когда
концентрация реагента в глубине зерна или капли близка к кон-
центрации на поверхности. Скорость реакции во внутренней кине-
тической области вдали от поверхности равна скорости реакции
на этой поверхности.
Концентрация транспортируемого через поверхность реагента
во внутренней диффузионной области быстро убывает до нуля в
Расстояние от поверхности
развела фаз
Расстояние от поверхности
катализатора
Рис. П-8. Распределение концентрации при диффузии через поверхность
раздела фаз:
а — диффузия сопровождается конвекцией; б — конвекция отсутствует.
глубине зерна или капли, так что химическая реакция протекает
в основном только в тонком слое у поверхности.
Если скорость реакции в активной фазе или зерне катализа-
тора достаточно велика, весь процесс начинает лимитироваться
скоростью диффузии в неактивной фазе. При этом химическая ре-
акция протекает только на внешней поверхности зерен, капель,
пузырей и процесс становится идентичным с реакциями на твер-
дой поверхности, рассматриваемыми ранее. В этом режиме воз-
можны также две области протекания реакции — кинетическая и
диффузионная, называемые внешними.
В кинетической области (внутренней и внешней) макроскопи-
ческая скорость реакции описывается уравнением истинной хими-
ческой кинетики. Во внешней кинетической области макроскопи-
ческая скорость реакции пропорциональна. поверхности раздела
фаз (поверхности зерен), во внутренней — объему активной фазы.
Во внешней диффузионной области макроскопическая скорость
реакции равна диффузионному потоку к поверхности и пропор-
циональна концентрации реагента в объеме неактивной фазы. Ко-
эффициент пропорциональности (константа скорости диффузии)
118
определяется из решения уравнения диффузии или приближенно
из закона диффузии Эйнштейна.
Слой твердого пористого катализатора. Во внутренней
диффузионной области макроскопическая скорость реакции также
равна диффузионному потоку (q) реагента, транспортируемого че-
рез поверхность раздела фаз (зерен катализатора). Диффузион-
ный поток при отсутствии посторонних возмущений (таких, как
конвекция) и в установившемся режиме, т. е. когда концентрация
более не изменяется со временем, как и для внешней диффузиоН’
ной области, можно представить в виде:
? = Рдсп
где сп — концентрация транспортируемого реагента на поверхно-
сти; рд — константа скорости диффузии.
Величина рд представляет собой отношение коэффициента диф-
фузии D' к расстоянию х, на котором сосредоточен основной пе-
репад концентрации. Это расстояние, очевидно, равно' среднему
смещению частиц при диффузии, т. е.
х::У1Ут (П-105)
Так как молекулы, диффундируя одновременно, подвергаются
химическому превращению, время диффузии не может опреде-
ляться каким-либо внешним процессом, а равняется времени жиз-
ни основной массы молекул, т. е.
(П-106)
Подставляя в соотношение (II-105) значение времени из вы-
ражения (П-106) и учитывая, что ^=D'/x, получаем с точностью
до постоянного множителя порядка единицы для диффузионного
потока реагента через поверхность раздела:
?::сп УЪ'йс"-1
Для вычисления постоянного множителя необходимо проинте-
грировать дифференциальное уравнение
DJ^+^ + ^\ = kcn
\ dx2 dy2 dz2 /
(П-107)
при условии, что на поверхности поддерживается концентрация,
равная сп. Для не очень малых зерен или капель с радиусом, су-
щественно большим толщины диффузионного слоя, в котором в
основном протекает химическая реакция и сосредоточен основной
перепад концентрации, уравнение (П-107) может быть записано
в более простом виде:
D' d2c = kca
U dx* ке
(П-108)
119
Диффузионный поток через поверхность
определяется из решения уравнения (П-108) и граничных усло-
вий
х = 0; с = сп
de п п
х-> оо; —->0; с->0
dx
de
Введением новой переменной = р, зависящей от с, полу-
чаем уравнение с разделяющимися переменными
£>,р-^- = йсп (П-109)
с граничным условием
с = 0; р = 0
Интегрирование уравнения (П-109) дает:
р = = 1/c«+i _L
Р dx V n+l- D'
Отсюда искомый диффузионный поток через поверхность равен
9=Сп1/тттр'Ас"-1 (НПО)
что отличается от найденного ранее выражения множителем
1/ 2
т п 4- 1
Таким образом, макроскопическая' скорость реакции во внут-
ренней диффузионной области в стационарном состоянии и при
отсутствии конвекции описывается уравнением (П-110). Уравне- •
ние (П-110) применимо, если расстояние, на котором сосредоточен
основной перепад концентрации, мало по сравнению с радиусом
капли или зерна катализатора (/?), т. е.
Рассмотренная задача впервые была решена Зельдовичем [13]
и одновременно с ним Тиле [14].
Хемосорбция. Макроскопическая скорость реакции А +
+ В-> продукты во внутренней диффузионной области в системах
газ — жидкость и жидкость — жидкость, когда транспортируемые
реагенты переносятся как диффузией, так .и конвекцией, может
быть найдена из решения уравнений стационарной конвективной
диффузии с источниками для каждого из компонентов реакции.
120
Для случая распределения концентрации вблизи поверхности
уравнения имеют вид:
, д2с , дс,
г\' _____А __А
Л ду2 х дх
дс. _
У ду л °
(П-111)
где их, иу — скорости потока в направлениях осей координат. Ве-
личины их и иу в системе уравнений (П-111) являются некоторыми
функциями координат, удовлетворяющими уравнению непрерыв-
ности:
дих диу
Последнее уравнение позволяет написать для их и иу выражения:
дф . _____йф
ду ’ у дх
(П-112)
где ф — функция тока
Концентрации реагентов несоизмеримы.
Если концентрация одного из реагентов в активной фазе значи-
тельно выше концентрации другого реагента (например, при вза-
имодействий плохо растворимого газа с жидкостью), изменением
количества реагента, находящегося в избытке, можно пренебречь
и считать его концентрацию постоянной. В этом случае макроско-
пическая скорость реакции находится из решения только одного
из уравнений системы (П-111) при условии, что на поверхности
раздела t/=0, концентрация всюду (для любых х) поддерживает-
ся постоянной и равной Со, а по мере удаления от поверхности
в глубь активной фазы концентрация быстро убывает до нуля.
Для интегрирования уравнения конвективной диффузии
D'^-k^ = ux^-+uy^- (П-113)
ду2 дх у ду
как показано Левичем [15], целесообразно в качестве новой пере-
менной выбрать функцию тока, т. е. перейти от функции с(х, у)
к функции с(х, ф). При этом для производных будем иметь:
Г д , .1 Г д , ..1 , дс йф
— с (X, и) = -т— С (х, Ф) + -VT • -^-L-
L дх ]у L дх т _]ф йф дх
дс _ дс йф
ду йф ду
д2с йф й / дс йф\
ду2 ду йф (йф ду)
(П-114)
Подставляя значения производных (П-114) в уравнение
(П-113) и учитывая определения для функции тока (П-112), по-
лучаем:
“•*Gfs") ] = + fec" (П-115)
Цйф \йф/^х \ дх /ф
121
Тангенциальная составляющая скорости потока жидкости
вблизи поверхности раздела фаз газ — жидкость и жидкость —
жидкость различным образом зависит от расстояния до поверхно-
сти (рис. П-9). Составляющая скорости жидкости, движущейся
вдоль поверхности раздела фаз газ — жидкость, не зависит от рас-
стояния до этой поверхности. При движении вдоль поверхности
раздела фаз жидкость—жидкость составляющая скорости убывает
или возрастает от некоторого значения на поверхности, определяе-
мого вязкостями обеих жидкостей, до скорости основного потока
вдали от поверхности.
В тех случаях, когда тангенциальная составляющая скорости
не зависит ни от расстояния до поверхности раздела фаз, ни от
вляющей скорости в жидкости у подвижной
границы раздела фаз.
расстояния вдоль этой поверхности, отношение ф/иж в точности
, дс
равно расстоянию до поверхности раздела фаз, а их — произ-
водной от концентрации по времени. Уравнение (П-113) в этом
случае принимает вид:
D'-Si=^+kcn (п-116)
где т — время, в течение которого данный элемент жидкости дви-
жется вдоль поверхности тела. Это время обычно называют те-
кущим временем контакта.
Уравнение (П-116) описывает процесс, протекающий в жидко-
сти у плоской поверхности раздела фаз газ—жидкость. Если вяз-
кость жидкости в активной фазе не намного больше вязкости
жидкости в неактивной фазе, то уравнение (П-116) описывает
также процесс, протекающий в системе жидкость—жидкость.
Для определения макроскопической скорости реакции во внут-
ренней диффузионной области необходимо вычислить интегриро-
ванием уравнения (П-116) производную дс)ду на поверхности
раздела фаз при следующих начальных и граничных условиях.
Концентрация диффундирующего через поверхность раздела фаз
реагента в приближающемся к этой поверхности элементе жидкости
122
равна нулю. При достижении этим элементом жидкости поверхно-
сти раздела фаз концентрация реагента скачком возрастает до
конечной величины, определяемой термодинамическим равнове-
сием, т. е.
т = О I у> 0; с = 0 ) (11-117)
I у = 0; c = c0J
Концентрация со поддерживается постоянной на поверхности
в течение всего времени контакта. В глубине элемента жидкости,
движущегося вдоль поверхности, концентрация реагента и ее про-
изводная дс/ду быстро убывают до нуля, что можно представить
в виде:
Уравнение (П-116) оказывается линейным и может быть про-
интегрировано только для двух случаев: если реакция протекает
по нулевому или по первому порядку. Преобразуя уравнение
(П-116) по Карсону—Хевисайду, получаем уравнение для изобра-
жения функции:
D'-^^Pc + kcn (П-119)
где с — изображение концентрации реагента; п принимает значе-
ние 0 или 1. Интегрированием уравнения (П-119) получаем изо-
бражение для диффузионных потоков через поверхность раздела
фаз при реакциях нулевого и первого порядка соответственно:
= СП VD'kc~n +
q = са VD'k V р +1
Оригиналы для диффузионных потоков имеют вид1 * * * У:
при п=0
q = сп V D'kc~l (—1= + -1—
\ г ли V Л /
(11-120)
1 Поскольку диффузионный поток реагента через поверхность раздела фаз
не может возрастать со временем, выражение (П-120) имеет смысл для о 1/г.
При о>'/2 поток постоянен и равен своему минимальному значению
q == тV^D'kCn
У л
что отличается от полученного выше стационарного решения множителем Кл/2.
Такое различие связано с некоторыми допущениями, сделанными при реше-
нии. Конечность времени, в течение которого устанавливается стационар-
ный диффузионный поток через поверхность раздела фаз для реакций с га<1,
обусловлена тем, что в указанных реакциях время, необходимое для полного
израсходования реагентов, конечно.
123
при п= 1
q = са V D'kl-Д_ е ” + erf V ю
\У лг>
(П-121)
где x> = xkcn ’ — безразмерное время; ег1]ЛГ = —Д^ f e~z2dz.
Y11 о
Из анализа уравнений (П-120) и (П-121) следует, что в не-
стационарном режиме часть потока реагента, транспортируемого
через поверхность раздела фаз, расходуется на увеличение кон-
центрации реагента в
активной фазе, а дру-
гая часть — на хими-
ческую реакцию. При
и<§;1 первые слагаемые
в уравнениях (И-120)
и (II-121) много боль-
ше вторых (основная
часть потока реагента
расходуется на форми-
рование профиля кон-
центрации) и выраже-
ния для q принимают
вид:
П-10. Изменение скорости хемосорбции
Рис.
вдоль поверхности жидкости в направлении дви-
жения газа (диффузия сопровождается реакцией
нулевого порядка):
1 -^точное решение; 2 — приближенное решение.
q = cn
При и З> 1 профиль кон-
центрации практически
сформирован [вторые
слагаемые в уравне-
ниях (П-120) и (П-121) много больше первых и стремятся к ве-
/* о 1
личине у t J и реагент расходуется только на химическую
реакцию: '
ттг^’1
Указанным предельным значениям для потока удовлетворяет
приближенное решение для реакции любого порядка:
9 « сп К D'kc" 1 (-г^=г + тЛ~у)
\ У лв ~ п + 1 /
(II-122)
Точные и приближенные решения для реакции нулевого и пер-
вого порядка сопоставлены на рис. П-10 и П-11. Приближенные
решения, как это следует из рис. П-10 и П-11, хотя и правильно
отражают ход зависимости <7(0), однако оказываются несколько
завышенными. Ошибка при этом не превышает 50—55% при
124
11-11. Изменение скорости хемосорбции
Рис.
вдоль поверхности жидкости в направлении дви-
жения газа (диффузия сопровождается реакцией
первого порядка):
7 — точное решение; 2— приближенное решение.
. и~1, убывая как в сторону больших, так и в сторону меньших
значений и. Данная задача была подробно рассмотрена в ра-
ботах [16, 17].
Концентрации реагентов соизмеримы. При
хемосорбции хорошо растворимого газа разбавленным раствором
какого-либо реагента (например, абсорбция СОг щелочью, этанол-
аминами и т. д.) или
. при экстракции, сопро-
вождающейся химиче-
ской реакцией, возмо-
жен случай, когда кон-
• центрация транспорти-
руемого через поверх-
ность раздела фаз ре-
агента соизмерима с
концентрацией реаген-
та, растворенного в ак-
тивной фазе. В этом
случае для вычисления
диффузионного потока
через поверхность раз-
дела фаз необходимо
решить
нений
плоской
раздела
систему урав-
(11-111). Для
поверхности
фаз при сла-
бой зависимости их
расстояния до этой
более простой вид:
от
поверхности уравнения (П-111) принимают
, д2с . дс .
°А ду2 дх +kcAcB
д2св_дсв_
в ду2 - дх +kcAcB
(П-123)
(П-124)
Для начальных условий имеем:
т = 0 z/ = 0; сА = сАо; св = сВо
у>0; сд = 0; св = сВо
условия для компонента А аналогичны условиям
Граничные
(П-118):
т>0 y = 0; сА=сА,
дс,
-^-->0
ду
(П-125)
условия для компонента В могут быть однозначно
Граничные
сформулированы на достаточно большом расстоянии от поверх-
Зсо
ности, где св = сВо, а = 0. На поверхности раздела фаз
125 ‘
концентрация реагента В убывает со временем от начального его .
значения до нуля. Поток компонента В к поверхности (qB) в
начальный момент времени равен нулю. Скорость же реакции (w)
на поверхности максимальна. По мере уменьшения св на поверх-
ности скорость реакции убывает, а поток возрастает. После до-
стижения квазистационарного режима, при котором на поверх-
ности
4B~W
диффузионный поток начинает убывать вслед за скоростью хими-
ческой реакции на поверхности.
Система уравнений (П-123) при п = т=\ была решена Киши-
невским [18] в предположении, что диффузионным к поверхности
раздела фаз потоком реагента В, растворенного в активной фазе,
можно пренебречь. Сделанное допущение, очевидно, должно при-
водить к правильным результатам как для очень малых, так и
для очень больших времен, когда диффузионный поток реагента
В к поверхности мал. Для не очень малых и не очень больших г
решение, полученное в работе [18], должно давать максимальные
отклонения от точного решения. Оценить эти отклонения пока не
представляется возможным.
Граничные условия для реагента В имеют вид [18]:
У~* °°; ~>0’ св~* св„
или
(П-126)
Вычитая первое уравнение системы (П-123) из второго и пола-
гая, что D'a = D’b = D', получаем уравнение нестационарной диф-
фузии для разности концентраций реагентов В и А:
где Z = cB—сА-
Уравнение (П-127) с учетом условий (П-124) — (П-126) наи-
более просто решается с использованием преобразования Карсо-
на—Хевисайда. Вспомогательное уравнение для изображения раз-
ности концентрации Z, учитывающее начальные условия (П-124),
является линейным:
D~d^=pZ-PcBt
126
Интегрирование вспомогательного уравнения с учетом граничных
условий
о = 0; Z = с г. ----с.
а в° p-kcAa л»
У dz п
в« dy
дает изображение градиента концентрации реагента А на поверх-
ности:
- =-'«.) - тк ('I128,
(сА и св — изображения концентраций реагентов). При этом было
использовано предположение, что поток реагента к поверхности
I п \
равен нулю, т. е. 1-ч—) =0. Для потока реагента А через по-
\ оу /^=0
верхность раздела фаз имеем:
____ VXv
<?Л = сЛо У VD’kcAo J e“2 da
Г Л 0
или
/1с V ” \
U = + (11-129)
\ Р JVU 6Л0 ' П о /
где и = т£сЛо — безразмерное время.
Первое слагаемое уравнения (П-129) характеризует часть
потока, расходуемую на формирование профиля концентрации,
второе слагаемое — часть потока, расходуемую на химическую
реакцию. При малых значениях и первое слагаемое велико, второе
мало и уравнение (П-129) совпадает по виду с уравнением
(П-122), поскольку при малых значениях и концентрацию реаген-
та В можно считать практически постоянной. Значения функции
X
е-х2 J еа2 _ ф [| 8].
о
х = у^...................... 0 0,2 0,4 0,5 0,6 0,8 1,0 2,0 3,0
<р.......................... 1 0,975 0,898 0,849 0,791 0,665 0,537 0,149 0,070
Стационарный режим при соизмеримости концентраций ре-
агентов невозможен, и с увеличением и поток стремится к нулю
вследствие израсходования реагента В.
Таким образом, для диффузионного потока через поверхность
раздела фаз во внутренней диффузионной области в системах газ—
жидкость и жидкость—жидкость, когда транспортируемые реаген-
ты переносятся как диффузией, так и конвекцией, поверхность
127
раздела фаз — плоская, а массовый поток параллелен последней,
имеем:
q = cAaVD'kc^-f^) (П-130)
где f\v) ~ -р— + у/ —, если концентрация реагента, перво-
начально растворенного в активной фазе, значительно больше
концентрации транспортируемого через поверхность реагента, и
/(v)~ ' +^е-» JL f e^da
У «V % V Л 0J
если концентрации обоих, реагентов соизмеримы и скорость реак-
ции описывается выражением kcAcB-
Диффузионный поток, как следует' из уравнений (11-122) и
(П-129), а также из рис. П-10 и П-11, убывает с увеличением вре-
мени контакта. Очевидно, что макроскопическая скорость реакции
w, отнесенная к единице поверхности раздела фаз, равна среднему
за время контакта (щ) значению диффузионного потока:
ш==саУ D'kcA^
О
Произвольная геометрическая форма по-
верхности раздела фаз. В промышленных аппаратах в ус-
ловиях интенсивного барботажа поверхность раздела фаз искрив-
ляется и может принимать любую произвольную форму. Пока-
жем,, что выражение для диффузионного потока через единицу
поверхности и в этом случае имеет вид уравнения (П-130). Рас-
смотрим случай, когда концентрации реагентов несоизмеримы.
Правая часть уравнения конвективной диффузии (П-113)
представляет собой полную производную от концентрации по вре-
мени, так что уравнение можно переписать следующим образом:
= (П-131)
ду2 dx
Проанализируем решение этого уравнения, удовлетворяющее
условиям (П-117) — (П-118), методом теории подобия. Выберем
произвольные масштабы длины /, времени Ф и концентрации са
и приведем уравнение (П-131) и условия (П-117), (П-118) к без-
размерному виду:
= — (П-132)
/2 <Э|2 п dv
v = 0 I g = 0; /п=1
I g >0; /77 = 0
v>0
g = 0; /77=1
g —> оо; /77 -> 0;
dm
*0
(П-133)
128
(т = с/сп, l=y/l). Поскольку масштабы длины и времени
выбраны произвольно, потребуем, чтобы
О
D'-J = l (П-134)
\ Тогда уравнение (П-132) будет содержать только один параметр
О/гс"-1, представляющий собой отношение масштаба времени О к
характерному времени реакции 0р = —^т-. Производная от без-
размерной концентрации по координате может быть функцией
только тех величин, которые входят в уравнение (П-132) и усло-
вия (П-133), т. е. в, s и 4/йр:
(П-135)
Для поверхности раздела фаз выражение (П-135) примет вид:
[ dm \ , I & \
Переходя к размерным-величинам и учитывая равенство (П-134),
получаем
=-£г-( («. = f(v, -^1 (П-136)
\ ду )у=о I \ ©Р) У D'ft \ *р /
или для диффузионного потока
?=Сп]/
В рассматриваемой задаче надо различать два времени: время
конвекции {>k, т. е. время движения элемента жидкости вдоль по-
верхности тела (йк=Ь/и), и характерное время реакции Фр. Здесь
L — характерный размер тела; и — скорость. Если химическая ре-
акция протекает с большой скоростью, так что то диффу-
зионный поток будет определяться только характерным временем
реакции. Выбирая последнее в качестве масштаба, получим для
диффузионного потока значение
9 = сп 1 (vp)
(dp = т/йр),совпадающее с уравнением (П-130). Макроскопическая
скорость реакции равна среднему за время Ок значению диффу-
зионного потока, т. е.
______*
к о
В пределе, когда скорость реакции очень велика 0к/0р-* оо и
~ w = -сп УD'kcп-1 • const
9 В. С. Хайлов, Б. Б. Брандт
129
что совпадает с точностью до постоянного множителя с соответ-
ствующим выражением для плоской поверхности раздела фаз.
Если скорость реакции мала, так что то процесс опре-
деляется временем движения элемента жидкости вдоль поверх-
ности раздела фаз. Выбирая в качестве масштаба времени
0к = L/и и полагая, что бк/бр = 0, для диффузионного потока
получаем:
1 / О'и ,. .
q = у --fM
где Dk^x/'&k- Макроскопическая скорость реакции при этом равна
J f (vK) dvK = сп у • const
о
что с точностью до постоянного множителя совпадает с соот-
ветствующим выражением для плоской поверхности раздела фаз.
Поскольку предельные при очень больших и очень малых значе-
ниях и выражения для макроскопической скорости реакции отли-
чаются лишь постоянным (порядка единицы) множителем от со-
ответствующих выражений, полученных для плоской поверхности,
то можно ожидать, что в общем случае при любых и выражения
для w у плоской поверхности раздела фаз и у поверхности произ-
вольной формы будут отличаться не более, чем на постоянный
множитель.
ЗАКОНОМЕРНОСТИ
МНОГОСТАДИЙНЫХ РЕАКЦИЙ
В ЗАКРЫТЫХ СИСТЕМАХ
Характерными для большинства процессов промышлен-
ного органического синтеза являются многостадийные (параллель-
ные, последовательные, параллельно-последовательные и т. д.)
превращения исходных веществ, заканчивающиеся образованием
достаточно стойких продуктов, выделяемых из реакционной си-
стемы.
Как указывалось в начале этой главы, для решения техноло-
гических задач необходимым является количественное описание
зависимости выхода каждого из продуктов и скорости их образо-
вания от степени превращения исходного вещества. Выяснение
этих зависимостей сводится к определению связи относительной
и абсолютной скоростей отдельных превращений со степенью пре-
вращения исходного вещества.
Выше было показано, что независимо от природы элементар-
ного акта химического превращения скорость не только простых,
но и большинства сложных реакций (параллельных, последова-
тельных, сопряженных и т. д.), продукты которых не могут быть
130
выделены из реакционной смеси, описывается уравнениями фор-
мальной кинетики.
Применительно к многостадийным реакциям подобное описа-
ние каждой из стадий является, в известной мере, схематичным,
однако это пока единственный надежный путь анализа кинетики
сложных реакционных систем, приводящий к выяснению каче-
ственной и количественной связи р(а) и w(a). В дальнейшем из-
ложении в основном будет использован этот принцип.
ИЗМЕНЕНИЕ КОНЦЕНТРАЦИЙ ИСХОДНЫХ ВЕЩЕСТВ
И ПРОДУКТОВ МНОГОСТАДИЙНЫХ РЕАКЦИЙ
Последовательные многостадийные реакции
Характерные особенности расходования исходных ве-
ществ и накопления продуктов последовательной многостадийной
реакции рассмотрим на примере последовательных односторон-
нйх реакций, кинетический порядок которых на всех стадиях —
первый:
А В —> D — > F
Значения констант скорости реакций могут находиться между
собой в любом соотношении, в том числе могут быть равны друг
другу. Вероятность равенства констант скорости для многостадий-
ных последовательных реакций крайне мала и рассмотрение слу-
чаев й1 = йг оправдано лишь для двухстадийных реакций:
А —В —D
Решение уравнений (П-62) — (П-64) для случаев kx = k2 и
kx=/=k2 приводит к выражению зависимости концентраций исход-
ного вещества (сА), а также промежуточного (св) и конечного
(сс) продуктов от продолжительности процесса:
При k\=k2 = k
c^cke-kx (П-137)
св = cKkxe~kx (П-138)
cD = cAo(l-He-feT-e“feT) (П-139)
при k\^k2
сА = сАе-к^ (П-140)
св=%7^т(^'т-^2Т) (П-141)
где a = k2lki.
Характер кривых, выражающих зависимости (П-137) — (П-142),
позволяет видеть (рис. П-12), что концентрация исходного веще-
ства А монотонно убывает во времени по закону кинетики простых
9*
131
реакций, тогда как изменение концентрации продуктов В и D
во времени описывается более сложной зависимостью.
Экстремальный характер зависимости св{х) указывает на ра-
венство скорости реакции образования промежуточного продукта
А — > В и скорости реакции его расходования В —D, дости-
гаемое в точке, соответствующей максимуму концентрации про-
дукта В.
Изменение концентрации конечного продукта D во времени
выражается S-образной кривой,
характерной для автокаталити-
ческих реакций. Продолжи-
тельность периода автоускоре-
ния (или индукционного пе-
риода), измеряемого отрезком
на оси абсцисс до точки пере-
гиба, так же как и момент
достижения и значение макси-
мума концентрации промежу-
точного продукта В, опреде-
ляются величиной а.
Рассматривая уравнения
(П-141) и (П-142), нетрудно
заметить, что при очень боль-
шом или очень малом значе-
нии а (и соответствующих
значениях kx и k2) эти урав-
нения превращаются в урав-
нения, описывающие односто-
роннюю реакцию первого по-
рядка.
Характер изменения кон-
центрации продуктов во вре-
Рис. П-12. Концентрации продуктов
двух последовательных реакций пер-
вого порядка в различные моменты
от начала процесса:
-------ktlki = 1,0; ....- k2lki = 0,1;
-------42/*1 = 2,0.
мени сохраняется и для более сложной последовательной реакции.
Предположим, что конечный продукт D реакции достаточно
реакционноспособен и при своем превращении приводит к получе-
нию продукта F; допустим также, что реакция D — •> F необра-
тима и имеет первый кинетический порядок.
Изменение концентраций при протекании последовательной
реакции А — •> В — •> D —+ F для случая, когда =А й2 =А &з.
описывается следующими уравнениями:
(П-143)
ш-144)
Г e~k,x e~klX e~k,x ]
Сд== СА?Л L(fe3-*,)№-*>) “ (k3-k2) (кг-kt)+ (k3-k2) (ks-kt) J (IM45)
„ _r Г.________Мзе~*'т , k,k3e~k'x________________
F (k3-kl)(k2-kl)+(k3-k2)(k2-kl) (k3-k2)(k3-ki)J ’
132
Рис. II-13. Концентрации продуктов
трех последовательных реакций первого
порядка при различных глубинах пре-
вращения исходного вещества (&2/&i=2,0,
kilk i = 0,5).
отношения рассмотрим (для про-
Сравнивая уравнения (П-141) и (П-144) нетрудно заметить,
что характер изменения концентрации промежуточного продукта
В остался прежним, тогда как в выражениях концентрации про-
дукта D — см. уравнения (П-142) и (П-145)—появились новые
члены, характеризующие скорость реакции D —F.
Графическое изображение уравнений (П-143) —(П-146), при-
веденное на рис. П-13, позволяет видеть экстремальный характер
кривых св(т) и со(т), а также S-образный характер кривой ск(т)
с еще более ясно выражен-
ным, чем на рис. П-12, индук-
ционным периодом.
Положение точек макси-
мального значения концентра-
ции продукта В и продукта D
во времени соответствует по-
следовательности их образова-
ния по схеме А —В —D
F, а ординаты этих
точек, как и -в рассмотренном
выше случае, определяются ве-
личиной отношения констант
скоростей реакций расходова-
ния и образования этих про-
дуктов (Й2/^1 И Йз/^1).
Количественную зависи-
мость координат точек экстре-
мума от абсолютной величины
констант скоростей реакций
образования и расходования
промежуточных продуктов и и
стоты) на примере реакции, описываемой схемой А—-> В D.
Так как по смыслу явления в точке экстремума скорости обра-
зования и расходования промежуточного продукта достигают ра-
венства, то, приравнивая нулю значение перв'ой производной кон-
центрации продукта В по времени, легко получить значение
т=тМакс, соответствующее экстремальной точке:
для случая ki = k2 = k из уравнения (II-138)
Тмакс — 1/£
для случая k\=f=k2 из уравнения (П-141)
1п а
Тмакс — ~т т~
«2 —
(П-147)
(П-148)
Максимальное значение’ концентрации продукта В легко най-
ти, подставляя значение т = тмакс из уравнений (П-147) и (П-148)
в уравнения (П-138) и (П-141) соответственно.
133
После подстановки имеем:
при k\ = k2
С D
"макс
при k\4=k2
— = 0,368с.
е • А<
Вмакс Сд» а — 1
(П-149)
(П-150)
Из уравнений (П-147) — (11-150)
дината точек экстремума определяются
с, дел. ед
а
Рис. П-14. Концентрации продуктов
двух последовательных реакций пер-
вого порядка при различной глубине
превращения исходного вещества:
---------kzlki = 2,0;...- kzlki = 0,5.
следует, что абсцисса и ор-
абсолютным значением и
отношением констант скорости
реакций А В —-> D —> F и
А —В —D, если ki =/= k2.
В том случае, когда отноше-
ние констант равно единице
{k\/k2 =1), координаты определя-
ются только начальной концент-
рацией исходного продукта и аб-
солютным значением константы
скорости реакции — уравнение
(П-147).
Наряду с рассмотренными
выше зависимостями с(т) боль-
шое значение при выполнении
расчетов, связанных с определе-
нием производительности реак-
ционной аппаратуры, имеют за-
висимости вида с(а).
Связь концентрации исходно-
го вещества, промежуточного и
конечного продуктов со степенью
превоащения исходного веще-
(11-143).
ства легко получить, заменяя т его значением из уравнения
1
т = In (1 — a) k'
После несложных преобразований получаем искомые зависи-
мости для промежуточного и конечного продуктов:
св = % - а "(1 “ а)а] (П-151)
со = сА„-^ТТ а-аП (П-152)
Описываемые уравнениями (П-151) и (П-152) кривые (рис.
П-14) несколько напоминают графическое выражение зависимо-
сти с(т) уравнениями (П-137) — (П-142), по крайней мере харак-
тером кривой Сд(а), имеющей максимум,
134
Приемом, аналогичным использованному выше, нетрудно под-
считать значение степени превращения а, соответствующее мак-
симальной концентрации промежуточного продукта В:
при k\=j=k2
__1
амакс = 1 - а а-‘ (П-153)
при k\=k2 = k
амакс=1-е-‘=0,632 (П-154)
I, при
В, не
и оп-
Уравнение (П-153) показывает, что степень превращения
которой достигается максимальная концентрация продукта 1
зависит от абсолютного значения
ределяется лишь величиной их от-
ношения, а при равенстве кон-
стант значение амаКс равно0,632.
Для трехстадийных последо-
вательных реакций из уравнений
(П-143) — (П-146) можно полу-
чить зависимости с (а):
cB = cA0^ZTTl1-«-(1-“)fl] (1Ь155)
Г 1-а (1—а)а ,
CD CAO«2[(Z,_ 1) (а-1) (Ь-а)(а-1) +
+ /h(1J/g? J (П-156)
(о - а) (Ь - 1)J
, Fl ofesd-q)
CF (6-l)(a-l) +
k3 (l~a)a _ k2(l-a)b J
+ (b - a) (a - 1) (b - a) (b - 1)J U ’
где a = k2/ki; b = k2/ki.
Графическое изображение за-
висимостей (П-155) — (П-157)
приведено на рис. П-15.
Скорость изменения концентрации продуктов последовательной
реакции как функция продолжительности процесса или как функ-
ция степени превращения исходного вещества часто фигурирует
в расчетах реакционной аппаратуры периодического и непрерыв-
ного действия.
Зависимость tw(-v) легко получается из уравнений (П-137) —
(П-142) путем их дифференцирования по т:
при ki=^k2
констант скорости реакции
с,усл.ед.
1,0
09
0,8
0,7
0,6
0,5
0,4
03
03
0,1
° 0,1 0,2 03 0,4 0,5 0,6 0,7 0,8 03 1J3
Рис. П-15. Концентрации продуктов
трех последовательных реакций пер-
вого порядка при различной глубине
превращения исходного вещества
(V*i-2,0; *3/*i - 3,0).
wBac^-^\(ae k2X-e~k'x)
(П-158)
(П-159)
135
При k;=k2 = k
fc(l ~kT)
ekx
WD == CA„
WB = CA<
(11-160)
(11-161)
Более наглядным и удобным для дальнейшего использования
является выражение скорости изменения концентрации продуктов
как функции степени превращения. Эту зависимость легко найти,
заменив значение т в
a
Рис. П-16. Скорости образо-
вания промежуточного и ко-
нечного продуктов двух
последовательных реакций
первого порядка при раз-
личных глубинах превраще-
ния исходного вещества
(W*i“2,0).
уравнениях (П-158)—(П-161) на его зна-
чение из уравнения (П-143). После не-
сложных преобразований имеем:
При kt = k2 = k
= cAofe (! - а) [! + !п (1 - а)] (П-162)
wD = cAk (а— 1) In (1 — а) (П-163)
При &1=#&2
^‘Ч-^^-^-О-а)] (П-164)
wd = са0-^Т U ~ а ~ (1 ~ “)“] (П-165)
Графическое изображение зависимо-
стей (11-162)— (П-165) в координатах
w—а представлено на рис. П-16 для слу-
чая, когда &i = l, ^2=2. Скорость изме-
нения, концентрации продукта В моно-
тонно снижается по мере увеличения
степени превращения исходного вещества, причем наклон кривой
а>в(а) тем больше, чем меньше величина a=kilk\, так что точка
пересечения кривой с осью абсцисс (соответствующая нулевому
значению wB) с увеличением значения а перемещается в область
более низких значений а.
Максимальная скорость образования конечного продукта D
достигается в тот момент, когда прекращается нарастание кон-
центрации продукта В.
Связь между отдельными величинами становится более ясной
при совместном рассмотрении рис. П-14 и П-16. Наблюдаемая
картина не является неожиданной: скорость расходования исход-
ного вещества А (или скорость образования продукта В) убывает
цо закону простых реакций; повышение концентрации продукта
В (т. е. положительное значение наблюдаемой скорости изменения
его концентрации) продолжается до момента, когда убывающая
скорость его образования из вещества А станет равной нарастаю-
щей скорости его расхода в реакции B->D. В этот момент вре-
мени (или при соответствующем значении а) концентрация про-
дукта В достигает своего максимального значения, а скорость
136
изменения его концентрации равна нулю. Этот же момент соот-
ветствует максимальному значению возрастающей скорости обра-
зования продукта D, которая затем снижается по мере уменьше-
ния концентрации продукта В.
Количественная зависимость а (а) может быть найдена как
максимум функции (П-163) или (11-165), когда условно сЛо = 1-
при kx=k2=k
а = 0,632 (П-166)
при k\4=k2
1
а=1—аа-1 (П-167)
Сопоставление полученных
выражений с уравнениями
(П-153) и (П-154) показыва-
ет, что максимальная скорость
изменения концентрации ко-
нечного продукта D достига-
ется в тот момент, когда кон-
центрация промежуточного
продукта В имеет максималь-
Рис. П-17. Скорости образования про-
дуктов трех последовательных реакций
первого порядка при различных глуби-
нах превращения исходного вещества
(fe2/fei =2,0; kt!ki = 3,0).
ное значение.
Несколько более сложный
вид имеют уравнения, связы-
вающие скорость изменения
концентрации промежуточных
и конечного продуктов трех-
стадийной последовательной реакции со степенью превращения
исходного продукта, однако принципиальный характер зависимо-
сти при этом не меняется. Для случая k^k2^=k3, получим:
“’B = cA0-^-j-la<1-a)a-1+aJ (П-168)
, Г а (1 — а)а 1-а 6(1—а)& 1
W г\ — 1^2 I (t, \ ( /L 1 \ / i\ it, \ it, i\l (11*169)
и Ао 1 2 1.(6 — а) (а—1) (Ь — 1)(а— 1) (b — a)(b— 1)J
..Г 1-а (1-а)“ , (1—а)6 1 ,_т
щр = С . 77-777-77 - 77-77---------77 + 77-----------777----77 (II -170)
г Ао 2 зцй — 1) (а — 1) (& — а)(а— 1) (& — a) (b — 1)J
где a = k2ik\\ b = kzlk\.
Ha рис. П-17 в координатах w — а построены кривые, описы-
ваемые уравнениями (П-168) — (П-170) для случая, когда а = 2,
6 = 3 (при Л1 = 1,0). В отличие от кривой wB(a), изображенной на
рис. П-16 (для двухстадийной последовательной реакции), нуле-
вое значение шв не соответствует максимальной скорости измене-
ния концентрации продукта D, так как последняя достигается при
меньшем значении а вследствие расходования продукта D на об-
разование конечного продукта F. Ясно, что абсолютное значение
137
Таблица 11-4
Зависимость концентрации конечных и промежуточных продуктов
от времени и степени превращения исходного вещества
Схема реакции Промежуточн ы й п родукт Конечный продукт
Зависимость от времени
а Г„ —(fel + ^2) Т ЙзТ] а L _ b r. „ —festl
1 lb~laq b-a-1 СВ~СА» 1+а-Ь I a+ 1 I1 -e 1 - e * ?
, 1 h
lF-%e+l U ~ e 1 J
II 1 св~сАа 1-а-Ь [е —№1 + fez) т _ е—fel Т] ь CF~ сАа a + b- 1 I1 - a + b t1 ’ _ e—(62 + кз) t] _ -feltj.
а 1 - 6 [i
^D-^Ao a + b-l I ' a+b (
III а св~сАа ь-а-1 [е—Ui + кг) г _ е—fest] 1 cD~cAa b-a- 1 (6-1) [1 — e~ -(fel + fez) t] _ a (1 _g~fe3T
Зависимость от степени, превращения
Г ь 1 f г b 1 ]
I LB - Mo b - а - 1 Ll - а— (I — а) а+* J 1 з а 1 a О • II Q (a + 1 “Д1' -(1—a)a+1 }
"F-Mo а + 1
II 1 СВ~САО Ь + а-1 [1 - а-(1 — а)а + ь] b CF ~cAa b + a-1 + [1 — (1 — <z)a+b]}
a CD ~cAa b+a-l {a~a + 6 [1 - (1 - a)a+b]}
Г ь I Г b I
III а СВ ~сАо Ь-а-1 [1 — а — (1 — а) 0+1 J 1 CD ~CAO b-a-1 |(6 — 1) a — a l-(l-a)a+1 t
Примечание: a- kzlki, Ь = kzlkt.
ординат и относительное положение на оси абсцисс экстремальных
точек кривых wD(a) и Wp(a) определяются относительным зна-
чением величин а и Ь. При а^>Ь характер кривых toD(a) на
рис. П-16 и П-17 становится практически одинаковым.
Параллельно-последовательные многостадийные реакции
В реальных условиях случаи только последовательных
или только параллельных реакций сравнительно редки и гораздо
чаще встречаются комбинации последовательных и параллельных
реакций, различающихся по числу элементарных стадий и их ки-
нетическому порядку.
Не ставя задачей рассмотрение всех возможных схем такого
рода реакций, ограничимся количественной характеристикой наи-
более простых случаев, схемы которых представлены ниже:
(I)
(П)
(Ш)
Таблица 11-5
Зависимость скорости накопления
промежуточных и конечных продуктов
от степени превращения исходного вещества
Промежуточный продукт
Конечный продукт
II
akt
WB = СА« У.-Г Х
° о — а — 1
Г Ь 1
X Ь(1 — а)й+1 — (а — 1)(1 — а)]
ki
wB = cAol-^x
X [(1 — а) — (а + 6) (1 — а)й+ь]
wD=cAokl 0 “°)
WD = САО f—a -b ~ Я)а+Ь “ 1 * * + “1
WF = СЛО x~-a-b f(1 ~ а)й+Ь ~ 1 + “1
III
aki 1
^D = ^Aob~_a_l X
Г — i г ь
X 1&(1 — а)й+1 - (а+ 1) (1 -а)] х[а6(1-а)а+1 - (а + 1) (1 - 6) (1-а)
Примечание: а = k^lki, Ь - kslki.
139
К подобным схемам приводятся, в частности, процессы, в ко-
торых вторым компонентом, участвующим в химическом взаимо-
действии, является вещество, взятое в большом избытке и сохра-
няющее свою концентрацию практически постоянной.
. Решение системы дифференциальных уравнений, выражающих
скорость изменения концентрации компонентов, дает возможность
вылазить зависимость с(т) для исходного вещества, промежуточ-
ных и конечных продуктов реакций, описанных приведенными вы-
ше схемами.
Изменение концентрации промежуточных и конечных продук-
тов в процессе реакции описывается уравнениями, приведенными
в табл. П-4.
Дифференцированием уравнений, приведенных в табл. П-4, по
времени получена ’зависимость скорости накопления промежуточ-
ных и конечных продуктов от степени превращения исходного ве-
щества (табл. П-5).
ЗАВИСИМОСТЬ ВЫХОДА ПРОДУКТОВ
МНОГОСТАДИЙНЫХ РЕАКЦИЙ
ОТ СТЕПЕНИ ПРЕВРАЩЕНИЯ
ИСХОДНОГО ВЕЩЕСТВА
Для иллюстрации описанного в начале этой главы ме-
тода аналитического определения зависимости 0(a) рассмотрим:
1) три последовательные реакции
I 1
А —В —D —F
2) две параллельные реакции
Пусть скорость первой реакции в обоих случаях пропорцио-
нальна с", скорость второй реакции пропорциональна или с™,
а скорость третьей реакции пропорциональна cpD. Тогда весь про-
цесс химического превращения описывается системой дифферен-
циальных уравнений:
для последовательных реакций
/
de .
__h гп
dX
^В_-Ьсп_ь ст
~ Й1СА Й2СВ
de
UCD ^Ьгт- k.cp
дх r2cB r3cD
(11-171)
140
для параллельных реакций
de.
___4 = ь гп + ь ГШ
dx Я1СА + Я2СА
dcn
___£_ _ Ь гп
dr “ftlCA
(П-172)
Переходя к новым переменным а и 0 и произведя несложные
преобразования, из уравнений (П-171) и (П-172) получаем соот-
ветственно:
d₽„ k2 т „
д ।__£. гт-п___
da ki А» (1—а)"
^ + ЛсР-п_^
da + л» (1 - а)"1
d$B | Рв 1
da а а
₽s+^-4=o
ат
1
----ъ----------= °
l+v-c£"n(l-a)m~n
(П-173)
(П-174)
В уравнения (П-173) и (П-174) входят только два параметра
и у-сР“л, представляющие собой отношение характери-
стических (т. е. при концентрации всех реагентов, равной сЛо) ско-
ростей реакций. Очевидно, что выход продуктов может быть толь-
ко функцией а и параметров -р с^”п и -у- ср~*.
Определим теперь из уравнений (П-173) и (П-174) зависи-
мость 0(a) при различных значениях тип.
Случай 1:две последовательные реакции, вторая из кото-
рых— нулевого порядка (т=0).Такой случай реализуется обычно,
если в процессе, осуществляемом в гетерогенной системе, проме-
жуточный продукт выводится из фазы, в которой протекает ре-
акция. Так, если реакция проводится в жидкости, в которой про-
дукт В весьма плохо растворим, или если продукт В весьма ле-
туч, или если реакция осуществляется в паровой фазе и продукт
В значительно менее летуч, чем вещества А и D, что приводит
к его конденсации, то концентрация продукта В в той фазе, в
которой протекает химическое превращение, поддерживается
постоянной и реакция В -> D протекает как бы по нулевому
порядку.
Для рассматриваемого случая из уравнений (П-173) получим:
-ЁЁ- +10 1 - А a~‘. (П-175)
da ar a fej а« (1 — а)”
Если первая реакция А->В также протекает по нулевому по-
рядку, то выход промежуточного продукта не зависит от степени
141
d0 л т-
превращения исходного вещества, т. е. = 0. 1огда из уравне-
ния (П-175) следует, что выход продукта В постоянен и равен
Когда п>0, интегрирование уравнения (П-175) при условии, что
Св = сЛоар-> сВа, если а=0, дает1:
Co k„ 1 — (1 — а)1""
0=1 + —2S----------?------------------- (II-176)
*14, о-") “
Поскольку выход промежуточного продукта не может быть отри-
цательным, необходимо, чтобы при а>0 удовлетворялось усло-
вие:
1 + Св° fe2 .
еА„« “
или
(1 — а)1-” — 6'(1 — а) + &'+ с'— 1 >0 (11-177)
где
(1 — п.) kic'l Со
----/ ..‘.Л;
*2 СА,
Очевидно, что при некоторых значениях параметров Ь' и с'
уравнение (П-177) имеет решения при ак<1 («к — конечная сте-
пень превращения исходного вещества), т. е. при некоторых зна-
чениях параметров средняя во всей системе концентрация про-
дукта В. становится равной нулю. Физически это означает, что
при достижении степени превращения, близкой к ак, продукт В
в фазе, где не протекает реакция, исчезает. При этом реакция
B->-D перестает протекать как реакция нулевого порядка.
Выход продукта В в начале процесса, т. е. при ,а->0
(если сВй = 0), становится неопределенным, как это следует из
уравнения (П-176). Применив правило Лопиталя, получим:
lim
а->0
0=1----------
(П-178)
Из уравнения (П-178) видно, что при k2 = (1 — п) kvc^, lim 0 = 0.
Следовательно, при выполнении условия
k2 7^ (1 — л) &2СХ
(П-179)
значение 0 равно нулю для любых значений а, так как выход про-
межуточного продукта не может возрастать с увеличением степе-
1 Если реакция А -> В — первого порядка, т. е. п=1, выражение (П-176)
становится неопределенным. Раскрытие неопределенности дает;
*2
*1еА,
! сВа_
“ еА,“
142
ни превращения. Физически это означает, что при выполнении ус-
ловия (П-179) в процессе реакции вообще не происходит образо-
вание новой фазы и реакция B->D не может протекать по нуле-
вому порядку.
Рис. П-18. Зависимость выходов проме-
жуточного ((5g) и конечного ((5В) про-
дуктов двух последовательных реакций
А —В—► D от степени превращения
исходного вещества (вторая реак-
ция — нулевого порядка; п — порядок пер-
„ г \
вой реакции; а =-----— |.
^1 сАо /
Рис. П-19. Зависимость выходов про-
межуточного ((5В) и конечного (0Д)
продуктов двух последовательных
реакций А > В —> D от степени
превращения исходного вещества
(порядок реакции А —> В н В —> D
соответственно равен п н т—1;
= О*
Случай 2: две последовательные реакции, вторая из ко-
торых— первого порядка (т — 1). Уравнение (П-173) для про-
дукта В в этом случае линейно:
. Г &2 1-п 1 । 1 1 д _ 1
da + L ki Аа (1—а)" а]р а
Интегрирование его при начальных условиях св = сЛ(ар—>0 при
а—>0 дает для п = ’/г, 2/3, 3/4 . . . зависимость выхода промежуточ-
ного продукта от степени превращения в виде ряда:
О - + 11 -
_ а'[1-(1-а)1~ге] Г. П , zi(2zi—1)
е L (I -п) а' + (1 — n)2(a')2 'Jj
где а = -
143
Если реакция А->В — нулевого или первого порядка, то зависи-
мость р(а) принимает соответственно более простой вид:'
Л2сА0„
ki 1-е
k2CA„ ’ «
Зависимость выходов промежуточного и конечного продуктов двух
последовательных реакций от степени превращения исходного ве-
щества, если реакция А->В имеет порядок О, V2, 1, 2, а реак-
ция В -► С во всех случаях — первого порядка, представлена на
рис. П-19.
Рис. П-20. Зависимость выходов промежуточного (Рв) и конечного (рв) про-
дуктов двух последовательных реакций А------> В —D от степени превраще-
ния исходного вещества прв различных значениях параметра a = k2c^~n/k1:
в —реакция А-> В — нулевого порядка; б—реакция А-> В—первого порядка; реакция В -* D
в обоих случаях — первого порядка
Как следует из рис. П-19, при одинаковом значении парамет-
ра И2с^~п/к1 выход промежуточного продукта при данном значе-
нии степени превращения исходного вещества тем больше, чем
ниже порядок реакции А->В. Если продукт В образуется по ре-
акции, порядок которой п меньше порядка т реакции расходо-
вания продукта В, то при полном превращении (а = 1) исходного
вещества выход промежуточного продукта не равен нулю. Если
n>m, то рв=0 при а=1. Уменьшение параметра для
данного значения степени превращения исходного вещества при-
водит к увеличению выхода продукта В вследствие уменьшения
скорости его расходования (рис. П-20).
144
Случай 3: две последовательные реакции, вторая из ко-
торых— второго порядка (т = 2). Для этого случая уравнение
(П-173) для продукта В переходит в уравнение Риккати:
4®-=-- —₽ + /?₽2 (П-180)
da а а
где R- сАо (1 _а)П •
Уравнение Риккати, вообще говоря, не интегрируется в квад-
ратурах, т. е. нахождение его решения не может быть сведено к
конечному числу последовательных интегрирований. Однако если
известны два частных решения уравнения Риккати, то общее ре-
шение может быть определено из выражения [19]:
₽ = ₽>+---------------- (II-181)
J?(₽2-₽,)da
1 +CeJ
где С — постоянная, определяемая из начальных условий.
Введением новой переменной, определяемой равенством
1 га
(П-182)
уравнение (П-180) преобразуется в линейное дифференциальное
уравнение второго порядка:
" n i &г 2-п 1 а
Z„----1----2 ~ Т~ сл 2 = О
“ 1—a kt А° (1—а)"
нли
Упг"у + пуп 'z' - az = О
(П-183)
где у = 1 — а; а = с2Гп.
«1 ^0
Если удастся найти два частных линейно независимых реше-
ния уравнения (П-183), связанных с уравнением (П-180) соот-
ношением (II-182), то тем самым окажутся известными и два
частных решения уравнения Риккати. Общее решение уравнения
(П-180) при этом может быть преобразовано к виду
(1 rfz2_______________1_ dzi \
1 dzx , z2 ' da zx ' da |
77 "dT +------~TT]--------- (II’184)
1 Г b — /
22 /
f 1 dz , ,
так как — •-5—aa = In z.
J z da
Рассмотрим теперь несколько частных случаев.
1. Пусть п=0, т. е. реакция А->В протекает по нулевому по-
рядку. Тогда уравнение (П-183) переходит в линейное дифферен-
циальное уравнение с постоянными коэффициентами:
d2z
da2
— az = Q
Ю В. С. Хайлов, Б. Б. Брандт
145
Его общее решение имеет вид:
2 = С1е-/“а + С2/Уа
Подставляя два частных решения zl = e~^aa и z2 — e^aa в
уравнение (П-184), получаем искомую зависимость выхода про-
дукта от степени превращения исходного вещества:
а.. £ ( 2 _ 1
Каа \ 1 + се~2Уаа
(П-185)
Постоянная интегрирования С должна быть выбрана в соот-
ветствии с условиями задачи. В начальный момент времени, ког-
Рис. П-21. Зависимость выходов
промежуточного (Рв) и конечного (Рд)
продуктов двух последовательных
реакций А —> В —D от степени
превращения исходного вещества (по-
рядок реакций А —> В и В —> D
соответственно равен п и т = 2).
да степень превращения исходно-
го вещества А еще равна нулю,
концентрация промежуточного
продукта В также равна нулю.
Но св :: ар, следовательно, по-
стоянная интегрирования может
быть вычислена из условия:
ар~>0 при а->0 (П-186)
Подстановка условия (11-186)
в уравнение (П-185) дает С=1.
Следовательно, выход проме-
жуточного продукта двух после-
довательных реакций, первая из
которых — нулевого порядка, а
вторая — второго, описывается
выражением:
(П-187)
График зависимости р(а), вычисленный по уравнению (П-187)
при j/ -^- сЛо = 1, представлен на рис. П-21.
.2. Пусть га = 2, т. е. реакция А->В протекает по второму по-
рядку. Тогда уравнение (П-183) можно записать следующим об-
разом (уравнение Эйлера):
Y2z"y + 2yz' - az = О (П-188)
Вид общего решения уравнения (П-188) определяется кор-
нями _____
---4 + Т^Т4'”
s—-Ь/F»
146
характеристического уравнения г2 + г—а=0. Поскольку a = k2jk\
всегда больше нуля, корни о и 6 действительны и различны и
общее решение уравнения (II-188) имеет вид:
г = С,у6 + С2уа
Подставляя два частных решения zt = (1 —а)6 и z2 = (l— а)ст
в уравнение (П-184), получаем искомую зависимость р(а):
/-:---1 1/ —Ь
/т+“+4----------------(I,-1S”
“2 г Т+а
1 + С(1 - а) 4 J
Постоянная интегрирования С с учетом условия (П-186) равна:
Таким образом, зависимость выхода промежуточного продукта
двух последовательных реакций второго порядка имеет вид:
fei <1 — «)
fe2a
Уравнение (П-190) при k\ = k2 графически представлено на
рис. П-21. Из сопоставления кривых, данных на рис. П-21, сле-
дуют те же закономерности, которые были описаны выше. Выход
продукта В тем больше, чем меньше порядок реакции образова-
ния В. Если n<m, то рв>0 при а=1.
3. В том случае, если и =4=0 и tv£2, т. е. реакция А->В не ну-
левого и не второго порядка, уравнение (II-183) может бытц све-
дено к уравнению Бесселя [20].
Случай 4: две параллельные реакции разного порядка
(реакция А — > В — n-го порядка; реакция А —D — m-го по-
рядка). Для определения зависимости выхода продукта необхо-
димо проинтегрировать линейное уравнение (II-174). Его общее
решение имеет вид:
oil" da , const
Р~7Г J 1 +a(l — a)m~n + a
)
(11-191)
где ° = I; са7-
10*
147
Зависимость ₽(а), как следует из уравнения (П-191), опреде-
ляется разностью порядков параллельных реакций. Если разность
т—п равна l/2, 1 и 2, то для зависимости, выхода продукта В от
степени превращения исходно-
го вещества А имеем соответ-
ственно:
Рис. 11-22. Зависимость выхода целе-
вого продукта двух параллельных
>В
реакций AI ki ?
от степени превра-
щения исходного вещества при
различных зиачеииях параметра
а = Сд~а и разности т — п = М
(порядок реакций А —► В и А —> D
равен соответственно п и т):
1-М = 0,5; 2-М = 0,1; 3-М = 2.
§ = — / — [1 — (1 — а)1/21—
а I а
1 . 1+а
-----1п ----------jT-
а2 1+а(1-а)1/2
й 1 1 1+0
Р аа 1+а(1-а)
Q ___1 , Каа
6 = —— arctg---------
Каа 1+(1— а) а
Зависимость р(а) для двух
параллельных реакций при раз-
личных значениях параметра а
и разности порядков обеих реак-
ций представлена на рис. П-22.
Выход продукта в случае двух
параллельных реакций, как это
и должно быть, тем больше, чем
выше скорость его образования.
При большем числе реакций,
одновременно протекающих в си-
стеме, возможен более сложный
механизм процесса, представляю-
щий собой различные комбина-
ции рассмотренных выше последовательных и параллельных ре-
акций. Общие закономерности этих сложных процессов будут в
качестве элементов содержать закономерности более простых
(двух параллельных и двух последовательных) реакций.
Рассмотрим простой пример. Пусть в системе протекают три
реакции первого порядка. Это возможно, если концентрация од-
ного из двух реагентов поддерживается постоянной. Из трех ре-
акций могут быть составлены четыре следующие комбинации:
А-^>В—
А —В D
А В —D
А В —D
I . *
(I)
(П)
(Ш)
(IV)
Ц8
Законы изменения выходов промежуточных и конечных про-
дуктов в зависимости от степени превращения исходного веще-
ства представлены в табл. П-6.
Таблица 11-6
Зависимость выходов
промежуточных и конечных продуктов
от степени превращения исходного вещества
дпя систем трех реакций первого порядка
Выход продукта
Схема
В D
F
. а (а — 1)
1 ab(l—a)
(Ь~1)(а—.1)
, Ь(1 —а)а
(b-а) (а—1)
а(1 — а)6
(Ь-аЦЬ-1)
____
(b + a-l) '
х X
I а + b
а (6 + а — 1)
X [1 - а - (1 - а)а+ь]
III
а (b — а — 1)
Г Ь 1
X [1 - а- (1 -а)а+1 J
IV
а (b — а — 1)
Г Ь 1
X L1 — (1 — «) а+ J
Примечание: а = kztki, Ь — kzfk\.
Графически зависимость Р(а) для рассматриваемых четырех
случаев представлена на рис. П-23—П-26.
В первом из рассматриваемых случаев (схема I) выход пер-
вого промежуточного продукта, как и в случае двух последова-
тельных реакций, убывает от 1 до 0 с увеличением а. Выход
149
Рис. П-23. Выходы промежуточных
и конечного продуктов трех последо-
вательных реакций первого порядка
при различной степени превращения
исходного вещества (&2/&1 ~ 2,0;
йз/^3,0).
Рис. П-24. Выходы промежуточного и
конечных продуктов трех параллель-
но-последовательных реакций первого
порядка А ——> В -2~ > D при раз-
L^F
личной степени превращения исход-
ного вещества:
1 — ki/k\ = 1; &з/&1 =1; 2 — &2/&1 = 1;
£3/&1 = Ю.
Рнс. П-25. Выходы промежуточного и
конечных продуктов трех параллель-
но-последовательных реакций перво-
го порядка А —В ~3 * D при раз-
Рис. П-26. Выходы промежуточного и
конечного продуктов трех параллель-
но-последовательных реакций пер-
вого порядка А —-> В — -> D при
личной степени превращения исход-
ного вещества:
l-k2/k} «0,5; fe3/fe] = l;2-fe2/fcl-l, Mh-U
3-kz/ki = 12,5; fc3/fcl - 0,1.
различной степени превращения ис-
ходного вещества:
Z-61/63-2; 2-Ms-l; 3-fe,/fe3-l/3;
4 — £1/63 = 1/5; 5 — £i/&3 = l/Ю; во всех слу-
чаях fei/fe2 = Ь
конечного продукта возрастает при этом от 0 до 1. Выход второго
промежуточного продукта проходит через максимум.
Во втором случае (схема II) выход промежуточного продукта
В, как и должно быть для последовательных реакций, убывает
от 1 до 0 с повышением а. Выходы конечных продуктов D и F
возрастают. При а=1 сумма их выходов равна единице, как для
двух параллельных реакций.
В третьем случае (схема III) сумма выходов веществ В и F
при а->0 равна единице, как для двух параллельных реакций.
Отношение
£в_ _ k2 „п-т
, если порядок реакций
А -> В и А -> F со-
ответственно равен пит. Выход промежуточного продукта В
убывает до 0 при а->1. Выход конечного продукта D возрастает.
Выход продукта F постоянен, если обе параллельные реакции —
одного порядка. При а=1 сумма выходов D и F равна единице,
поскольку оба продукта образуются по параллельным реакциям.
Отношение ро/рк при а=1 равно Рв/рт? при а->0.
В четвертом случае (схема IV) сумма выходов промежуточ-
ного (В) и конечного (D) продуктов при а->0 равна единице, как
fa
для параллельных реакций. Отношение Pb/Pd = если по-
рядок реакций A->D и А->В равен пит соответственно. Выход
промежуточного продукта В убывает до 0 при а-*1. Выход ко-
нечного продукта D возрастает при этом до единицы.
КИНЕТИКА РЕАКЦИИ
В ОТКРЫТЫХ СИСТЕМАХ
Рассмотренные выше закономерности строго справед-
ливы по отношению лишь к закрытым системам, т. е. к реакцион-
ным системам, находящимся в объеме, полностью изолированном
(в смысле материального обмена) от окружающей среды.
Объем закрытой системы сохраняется в течение всего периода
свершения химического процесса, и изменение концентрации реа-
гирующих веществ происходит только в результате их расходова-
ния или образования за счет химической реакции.
Как при осуществлении химических процессов в промышлен-
ных условиях, так и при экспериментальном изучении кинетиче-
ских закономерностей химических реакций весьма часты случаи
использования так называемых «открытых систем», т. е. проведе-
ние процесса в условиях непрерывного ввода в реакционное про-
странство исходных веществ и продуктов реакции при непрерыв-
ном выводе из него непревращенных исходных веществ и продук-
тов реакции.
Параметры (температура Т, давление Р), а также состав
непрерывного потока реагирующих веществ в общем случае мо-
гут быть переменными как во времени, так и по оси потока.
151
Режим, характеризующийся зависимостями
Р = ф(т, L); Т = )(т, L); с/ = ф(т, L)
(т—время, L — длина реакционного пространства) носит назва-
ние нестационарного, так как параметры состояния и состав по-
тока изменяются во времени. При постоянной скорости потока
нестационарный режим наблюдается лишь в период, предшествую-
щий установлению материального и теплового баланса и при из-
менении параметров и состава входящего потока.
При стационарном режиме сохраняется зависимость:
Р = ф(1); T=f(L); С/ = ф(£)
В большинстве практических случаев перепад давления по оси
потока (АР) в пределах реакционного объема исчезающе мал
(особенно при общем высоком давлении), и, пренебрегая значе-
нием АР/L, без большой погрешности можно считать давление в
открытой системе постоянным.
Температура вдоль оси потока может быть либо постоянной
(изотермический режим), либо меняться по различным законам.
Это обстоятельство, а также особенности поля концентраций реа-
гирующих веществ в реакционном объеме, обусловливаемые мас-
сообменом вдоль оси потока, придают своеобразие кинетическим
закономерностям реакций в открытых системах и затрудняют или
полностью исключают возможность описания кинетики реакций
законами, установленными для закрытых систем.
Общие принципы теории химических реакций в потоке впервые
были сформулированы в 1908 г. в работе Боденштейна и Вольгаста
[21], установивших, что закономерности кинетики реакций в потоке
аналогичны законам химической кинетики в закрытой системе
лишь в том случае, когда реакционный объем имеет форму длин-
ной и узкой трубы, т. е. когда перемешивания реагирующих ве-
ществ по оси потока практически не происходит.
В указанной работе рассмотрен и противоположный случай,
когда в реакционном объеме перемешивание и диффузионный об-
мен протекают столь энергично, что полностью исключается кон-
центрационный градиент по оси и сечению потока и концентрация
реагирующего вещества в любом элементарном объеме реакцион-
ного пространства одинакова и равна концентрации его на вы-
ходе. В этих условиях течение химической реакции подчиняется
иным, чем в первом случае, закономерностям. На примере одно-
сторонних реакций первого и второго кинетических порядков
была сформулирована количественная зависимость с(т) для обоих
случаев.
В ряде последующих отечественных и зарубежных работ [22—
31] этот чрезвычайно важный для химической технологии вопрос
обстоятельно разработан.
152
В идеальных условиях полного вытеснения или полного сме-
шения реакционной массы особенность открытых систем (стацио-
нарный режим) характеризуется признаком:
система, полного вытеснения
система полного смешения
Количественной характеристикой гидродинамического режима
открытых систем, определяющей степень приближения любой
открытой системы к условиям идеального режима полного вы-
теснения или полного смешения, служит критерий Боденштей-
на Во:
где U — средняя линейная скорость потока; L — длина реакцион-
ного пространства; D — коэффициент продольного перемешивания.
Для систем полного вытеснения D->0, Во->оо; системы пол-
ного смешения характеризуются значением £)->оо и Во->0.
Открытые системы, для которых 0<£)<оо и оо>Во>0, отно-
сятся к промежуточному типу, наиболее часто встречающемуся в
реальных условиях.
В отличие от закрытых систем, для которых характер изменения
концентрации реагентов (при заданных параметрах) не зависит
от конструкции реакционного аппарата, условия осуществления
и результаты химического процесса в открытой системе сущест-
венно зависят от степени приближения данной системы к систе-
мам идеального режима, т. е. от значения Во.
Степень и характер влияния типа реакционного аппарата не-
прерывного действия на течение реакции неодинаковы по отно-
шению к отдельным группам реакций. В связи с этим целесооб-
разно выяснить зависимости, характерные для систем идеальных
режимов и систем промежуточного типа на примере основных
групп реакций. Для упрощения задачи рассматривается стационар-
ный режим процесса, проводимого в изотермических условиях.
ОТКРЫТЫЕ СИСТЕМЫ ПОЛНОГО ВЫТЕСНЕНИЯ
Такой тип открытых систем характеризуется идеальным
гидродинамическим режимом (Во->оо). Моделью такой системы
может служить труба, для которой L/d->oo.
Выражением скорости реакции в открытой системе полного вы-
теснения является [32, 33]:
пЛ^-^А-кспА (П-192)
153
где Пд^ — начальное число йолей исходного вещества А, вводимое
в систему в единицу времени; s — поперечное сечение реакцион-
ного пространства; и—порядок реакции.
На примере одностадийной необратимой реакции первого по-
рядка
vzA + v;VN = vBB + v;VN
(N — инертное вещество, находящееся в смеси с исходным веще-
ством, va, vb, Vjv — стехиометрические коэффициенты) концентра-
ция исходного вещества А в любой момент времени может быть
описана следующей зависимостью:
са = са0 (1 ~ а) --------------------------------—
VA VA
где Wa, WB, — мольные объемы веществ.
Выражая число молей продукта В и инертного вещества N
в долях от числа молей исходного вещества А, имеем:
1 — а
са “ wA (1 - а) + WBza + WNy
(П-193)
где z=vb/va, X=Vn/va (у>0).
Для газовых реакций WA=WB=WN=RT/P (/? —газовая по-
стоянная), и уравнение (П-193) можно записать следующим об-
разом:
Р(1 —а)
А = [1 _ а (1 - г) + у] RT
(П-194)
Подставляя значение сА из уравнения (II-194) в уравнение
(П-192), получаем:
w = „ = kcn = fe ) __1 ~ 7 _______
А nA*sdL А [1 — а (1 — г) + у] ЛТ
После разделения переменных имеем:
_ks_ I (1 —а)Р
пА, I [1 —а(1 —г) + у]/?Т
(II-195)
Интегрирование уравнения (П-195) в пределах от L=0 до L
и от а=0 до а дает выражение зависимости степени превраще-
ния исходного вещества по длине реакционного объема:
для реакции нулевого порядка
для реакции первого порядка
kPt
t = а(1 — г) — (у + г) 1п(1-а)
До
154
Так как
PsL
~Х'
то предыдущее уравнение может быть запи-
сано в следующем виде:
т = 4- [« (1 - z) - Cv + ?) In (1 - а)]
к
(П-196)
При взаимодействии исходного вещества, свободного от при-
месей (у—0), и в случае, если Va = vb (z=1), выражение (П-196),
принимает структуру уравнения реакции первого порядка для
закрытой системы:
На рис. П-27 показана зависимость a(L) для реакций первого
порядка. Видно, что наклон кривых a.(L) возрастает с уменьшением
значения г, т. е. реакции, идущие
с увеличением объема (z>l), про-
текают значительно медленнее ре-
акций, для которых z<JCl. Прямо-
линейный характер кривой 3 (z =
=0,01) напоминает картину тече-
ния реакции нулевого порядка.
Разбавление исходных веществ
инертным продуктом (или, для не-
обратимых реакций, избыток одно-
го из исходных веществ) наряду с
уменьшением скорости реакции за-
метно меняет характер кривых, вы-
ражающих зависимость a(L): как
видно на рис. П-27, даже равное по
Рис. 11-27. Зависимость a (L) для
системы полного смешения:
/-z = 2; 2 — г-=1; 3-z = 0,01;
-----------у=о-,-------v = 1,0-
объему количество инертных примесей (у=1) нивелирует разницу
между реакциями со столь различным значением z (0,01—2,0), при-
ближая картину их течения к случаю, когда реакция не сопро-
вождается изменением объема (z= 1,0).
Нетрудно представить, что при значении 1 кривые /, 2 и 3
полностью совпадут.
Такая закономерность дает право считать, что во всех случаях
осуществления процесса при малой степени превращения исход-
ного вещества или при проведении процесса в растворе (жидкая
фаза) влияние уменьшения концентрации реагирующих веществ
в результате изменения объема реакционной системы пренебре-
жимо мало.
В реальных условиях (zSgl) при достижении заданной степени
превращения продолжительность реакции в открытой системе пол-
ного вытеснения отличается от продолжительности реакции в
закрытой системе (принимаем у=0).
Из изложенного ясно, что при г=1 открытая система пол-
ного вытеснения может быть уподоблена элементарному объему
155
закрытой системы, перемещающемуся вдоль оси реакционного про-
странства с постоянной скоростью, и тогда все кинетические за-
кономерности закрытой системы сохраняют свою справедливость.
Для газовых реакций при г=#1 учет изложенных выше зависи-
мостей совершенно необходим.
, Пример II-4. Необратимая реакция разложения ацетальдегида
2СН3СНО — * 2СН4 + 2СО
осуществляется при температуре 518° С (А=0,331 л • моль4 • сек~‘) и давлении
Р=1 атм в трубчатом реакторе. Число труб реактора—100 шт., внутренний
диаметр труб — 33 мм, скорость подачи ацетальдегида — 5,0 кг/ч.
Установить зависимость длины трубы реактора от степени превращения
ацетальдегида и характер изменения линейной скорости потока (uK/uH) по
длине трубы для двух случаев:
а) разложение чистого ацетальдегида;
б) разложение ацетальдегида, разбавленного азотом (мольное соотношение
азот: ацетальдегид составляет 1:1). Здесь uH, ин — начальная и конечная ли-
нейные скорости потока.
Решение. 1. Зависимость L(а).
Рассматриваемый случай описывается уравнением (П-195), где п=2; Р=
= 1,0 атм\ 7=273+518=791° К; /?=0,08206 л • атм/(моль • град)
па, = йо^РГзбОО=3116 ’10-4 моль,сек
s = 0,785 • 0,332 = 8,55 • 10-2 дм2
Интегрирование уравнения (П-195) при п=2
0,331-8,55-10-2 Г,, (0,08206 - 791 )2 ГГ 1-а 1
-----------j--- I aL =------------- I I-------------I а а
3,16-10^4 J 1 J Ll+y + (z—l)aj
приводит к выражению:
£ = 47,2[^^-(г + у)2 + 2 (z — l)(z + y) In (1 - a) + (z- I)2 a] (11-197)
Для рассматриваемых в примере случаев выражение (П-197) упрощается:
а) у=0; z=2
Г4<х Т
-р2~-+ 4 In (1 — a) + а (П-198)
б) у=1; z=2
Г 9а Т
£ = 47,2 р7Г^ + 61п(1-а) + а (П-199)
2. Зависимость uK/uH(£). Относительный объем потока реагентов и соот-
ветствующую ему линейную скорость потока можно рассчитать, пользуясь зави-
симостью:
В табл. П-7 приведены результаты расчетов на основе выражений
(П-197) —(П-199) для ряда произвольно взятых значений а, откуда можно
иывести искомую зависимость.
166
Таблица 11-7
Результаты расчетов
Y а L, м ик/“н
0 1 0,13 0,82 1,13
1 J 3,10 1,065
0 1 0,5 18,0 1,50
1 J 24,2 1,25
0 1 0,8 72,0 1,8
1 f 128,0 1,4
Полученные значения могут быть использованы при
и определении размеров реакционного аппарата.
выборе конструкции
ОТКРЫТЫЕ СИСТЕМЫ ПОЛНОГО СМЕШЕНИЯ
Гидродинамический режим реакционной системы пол-
ного смешения характеризуется полным и мгновенным перемеши-
ванием поступающих в реакционное пространство веществ с ве-
ществами, заполняющими этр пространство (D -> оо, Во->0).
Моделью подобной системы может служить сосуд, снабженный
мощной мешалкой, размер и устройство которого (расположение
входа и выхода поступающей и удаляемой смеси, интенсивность
перемешивания) обеспечивают идеальную равномерность состава
в любой точке объема.
При проведении химической реакции в открытой системе пол-
ного смешения одновременно и непрерывно совершаются два яв-
ления, различные по своей природе, но одинаковые по характеру
влияния на величину движущей силы химического процесса.
Одно из этих явлений сопровождается снижением концентра-
ции исходных веществ в результате полного и мгновенного смеше-
ния непрерывно поступающих исходных веществ с содержимым
реакционного объема; другое явление, обусловленное химическим
взаимодействием реагирующих частиц, также сопровождается по-
нижением движущей силы химического процесса. Совокупность
этих двух явлений в условиях постоянного состава и количества
поступающей смеси исходных веществ неизбежно приводит к уста-
новлению состояния реакционной смеси, характеризующегося со-
отношением:
=0; =0
\drjL-Li \dL)t-rl
Период, предшествующий наступлению этого состояния (при вве-
дении системы в работу), а также периоды, в течение которых
157
происходит изменение состава (количества) поступающей смеси
исходных веществ или условий химической реакции (температура,
давление, катализ), характеризуются переменным во времени со-
ставом реакционной массы.
Рассмотрим первое из этих явлений на примере смеси хими-
чески инертных веществ.
Пусть в реакционное пространство системы полного смешения,
заполненное смесью химических инертных продуктов, непрерывно
и с постоянной скоростью вводится смесь этих же продуктов, от-
личная по своему составу от состава смеси, заполняющей реак-
ционное пространство.
Условия полного смешения дают основание исходить из пол-
ного равенства состава смеси в реакционном пространстве и со-
става выходящего потока.
Нетрудно предположить также, что при постоянном объеме
реакционного пространства, полностью заполненного смесью,
состав этой смеси, а следовательно, и состав выходящего потока
будут меняться во времени до установления материального баланса
по любому из веществ, входящих в состав поступающего потока,
что соответствует стационарному состоянию системы.
Примем следующие обозначения: U7P— объем реакционного
пространства; оц— объемная скорость потока на входе в реак-
ционное пространство; иг — объемная скорость потока на выходе
из реакционного пространства; сАо — концентрация вещества А
в поступающем потоке; сА — концентрация вещества А в смеси,
заполняющей реакционное пространство, или, исходя из усло-
вий полного смешения, концентрация вещества А в выходящем
потоке.
При отсутствии химического взаимодействия, постоянстве тем-
пературы и давления можно принять, что cdi = cd2 = c0, и скорость
изменения концентрации вещества А в реакционном пространстве
выразить следующей зависимостью:
Разделение переменных и интегрирование в пределах
т = тн = 0; сл = сЛн
СА = слк
приводит к общему выражению зависимости состава выходящего
потока (или, что то же самое, состава смеси в реакционном про-
странстве) от продолжительности процесса:
Сд С Л Q)
In °-с K~~-WX (П-201)
С д Од И'
Ло Лн Р
158
Рассмотрим два из возможных вариантов проведения про-
цесса:
1) реакционное пространство заполнено смесью, свободной от
вещества А (сА =0)1 концентрация вещества А в входящем по-
токе равна сАо;
2) реакционное пространство заполнено смесью, содержащей
вещество А (сА входящий поток свободен от присутствия ве-
щества А (сЛо = 0).
Подставляя значения с.
Ло
выражение для состава смеси в реакционном пространстве (или
смеси в выходящем потоке) от продол-
жительности процесса для каждого из
рассматриваемых вариантов: '
и с. в уравнение (11-201), получаем
(СОТ
1-е
“К “0
(I I-202)
Рис. 11-28. Вымывание ве-
щества А из реакционного
объема (% = ьо).
от
“uz
сА = сА е Р
лк лн
Из уравнений (П-202) и
рис. П-28 следует, что концентрация ве-
щества А в выходящем потоке (или в
реакционном пространстве) достигает по-
ловинного значения его концентрации
в входящем потоке (вариант 1) или половинного значения его
начальной концентрации в реакционном пространстве (вариант 2)
при -jp—т = 0,7, т. е. после подачи в реакционное пространство
смеси в количестве, равном 70% объема этого пространства.
После подачи в реакционное пространство равного ему по объ-
ему количества смеси концентрация вещества А в этом простран-
стве в первом случае составляет 63,2% его концентрации во вхо-
дящем потоке, а во втором — 36,8% начальной концентрации в
смеси, заполнившей реакционное пространство.
Состав смеси в реакционном пространстве в обоих случаях
становится практически равным составу входящего потока после
подачи в реакционное пространство 6—8-кратного по объему ко-
личества смеси.
Усложняя условия, примем, что вещество А способно к хими-
ческому взаимодействию и превращается в продукт D по законам
реакции n-го порядка:
(11-203)
(П-203) И
7Аа yD
В этом случае изменение концентрации вещества А в реакцион-
ном пространстве ставится в зависимость от дополнительного ус-
ловия и уравнение (П-200) приобретает дополнительный член,
159
(П-204)
выражающий скорость изменения (уменьшения) концентрации
вещества А за счет реакции n-го порядка:
</сл ш
—5— = (сл, - Сл) - kcA
ах wp ' л' А’ л
Рассмотрим уравнение (П-204) применительно к реакции пер-
вого кинетического порядка (п = 1).
Преобразование уравнения (П-204) и замена величин, являю-
щихся постоянными при равномерной и непрерывной подаче ис-
ходных веществ и при T=const
о о , . .
Сл’1Гр=а: Wp+kfab
^ает возможность проинтегрировать по-
лученное выражение:
ч
Рис. П-29. Зависимость
с. (0) и с. (т) для реак-
к к
. * -
ции первого порядка А —> D
(Сло = 1,О; k =1,0).
(П-205)
а — bcA J
тн-о
уравнение (П-205) для
ч
Используем
установления зависимости сА(т) приме-
нительно к двум рассмотренным выше
вариантам начального периода работы
системы полного смешения, приняв, что
в обоих случаях входящий поток содержит исходное вещество А
концентрацией сДд (во втором случае принимается, что сА° — сА у
После интегрирования и соответствующего преобразования по-
лучаем выражение для концентрации исходного вещества А в ре-
акционном пространстве как функцию продолжительности про-
цесса для обоих рассматриваемых случаев:
СА.,
Сак Гр
1+ —
о
(П-206)
(П-207)
Сл I ®р
СА.----1+ — ke
Ак IFn I о
1 +—-k
ш
Из уравнений (П-206) и (П-207) нетрудно видеть, что конеч-
ным результатом обоих вариантов является стационарная кон-
центрация исходного вещества сА . достигаемая при т->оо и рав-
ная
СА,
с - СА°__________________
Лк Wp l+0fe
1 + — k
О
(П-208)
160
реакционной системы
режима, описываемый
открытой
Рис. II-30. Нестационарный режим работы реак-
ционного аппарата полного смешения.
Зависимость с, (0), выражаемая уравнением (П-208), представ-
лена на рис. П-29. Здесь же приведена кривая, описывающая за-
висимость с. (т) для реакции первого порядка в закрытой системе.
По формальному признаку т в уравнении (П-196) и 0 в урав-
нении (П-208) можно рассматривать как величины тождествен-
ные, хотя 0 = ИТр/со является расчетным временем пребывания ре-
акционной массы в реакционном пространстве и в отличие от т
не выражает продолжительность пребывания всех участвующих
в химическом превращении частиц в реакционном пространстве.
Стационарному состо:
предшествует период н<
уравнениями (П-206) и
(П-207). Характер зави-
симости с. (т, 0) можно
видеть на рис. П-30; здесь
в координатах 0—сА по-
казана зависимость, опи-
сываемая уравнением
(П-208), т. е. стационар-
ная концентрация веще-
ства А в выходящем
потоке, достигаемая при
т->оо, а в координатах
сот/IF — с. — условия до-
стижения стационарной
концентрации вещества А
для двух рассматривае-
мых выше случаев, описываемых уравнениями (П-206) и (П-207).
Нетрудно заметить, что время, необходимое для практически пол-
ного приближения к стационарному режиму (т->оо), находится
в зависимости от 0; с точностью, достаточной для инженерных рас-
четов, пределом достижения стационарного режима является
8—10-кратное по объему количество смеси исходных веществ, от-
несенное к объему реакционного пространства (cor/lFp>8 ч- 10).
В качестве примера может быть рассмотрен пусковой режим
реакционного аппарата полного смешения, предназначенного для
проведения реакции первого кинетического порядка.
k
Пример П-5. Односторонняя реакция А —> D первого кинетиче-
ского порядка проводится в реакторе полного смешения при подаче в него с по-
стоянной скоростью раствора, содержащего исходное вещество А концентрацией
сАо. Имеется возможность осуществить пуск реакционного аппарата и вывод
его на заданный стационарный режим (определяемый значением а) по трем
вариантам:
1) реактор заполняется раствором, содержащим вещество А концентрацией
сл0> и непосРеДственно вслед за этим начинается непрерывная подача исходного
раствора;
2) реактор заполняется чистым растворителем;
И В, С. Хайлов, Б. Б. Брандт
161
3) реактор заполняется исходным раствором (концентрация исходного ве-
щества А равна Сдо) и реакция проводится до достижения заданного значения
а, после чего начинается непрерывная подача исходного раствора.
Требуется выбрать режим пускового периода путем сравнения его продол-
жительности по каждому из трех вариантов для а=0,2 'и а=0,8, принимая зна-
чение допустимого отклонения М' 10-3сл .
Решение. Условия достижения стационарного режима при пуске
реактора по вариантам 1 и 2 описываются уравнением (П-206) и (П-207). Ис-
ходя из заданного допустимого отклонения, задача сводится
венств:
к решению нера-
е
(П-209)
«’р -\г-+4)т
k~— е ' Р 7 >ЛГ
<о
Для сравнительной оценки рассматриваемых вариантов
мость т(а) для каждого варианта.
1. Из выражения (П-209) имеем:
<о
W'p
левой частей
® , 1 .
П7_ Т — 1П ВТ
(П-210)
выразим
зависи-
(II-211)
После деления правой и
САК/САО = 1— а получим:
уравнения
(П-208)
на с. и
Ло
замены
Подстановка значения
водит к зависимости т2(а):
О
Fp
— = 0—j-r,----г
<о й(1—а)
из выражения (П-212)
а
(П-212)
в уравнение (П-211) при-
2. Из выражения (П-210)
а 1
k пм-
(П-213)
имеем:
<0 Wpk
X 1П 1 ““ &Х
Wp ©ЛГ
В результате аналогичных преобразований получаем
а . а
T| k п (1 - а) М'
3. Из уравнения для реакций первого порядка в закрытой
Исходя из выражений (П-213)—(П-215) можно оценить
продолжительность пускового периода:
при а=0,2
выражение
для Ti(a):
(П-214)
системе имеем:
(П-215)
относительную
а
т2
1 , 0,2
1п--------- In---------т
(1- а)М' 0,8-10-3
М'
1пг^~
т3 1 —а
т2
. 1
1П---5
10~3
. 1
п 0,8
, 1 ~0,21п103~0,161
а1п -тт;
М
5.521
6,908 0,8
162
при а=0,8
, 0,8
In--------5-
т, 0,2-10"3
т2 In103
т3 1п0,2
т2 0,8 In 103
= 0,29
Нетрудно видеть преимущество варианта 3, наиболее часто применяемого на
практике.
Продолжительность пуска реактора по вариантам 1 и 2 будет одинакова
при условии
а . 1 а . а
Т " ЛГ =1П(1-а)Л4'
а
т. е. при -j—— ~ 1 или при а = 0,5.
Зависимости Т1(а), Т2.(а) и т3(а) во всем диа-
пазоне значений а представлены на рис. П-31
Цг = 1,0).
Стационарное состояние реакционной
системы описывается уравнением (П-204)
при dcA/dx=0:
Са“~ сЛ = kcn. (П-216)
и Л
Левая часть уравнения (П-216) выра-
жает закон изменения концентрации исход-
ного вещества в реакционном объеме как
результат обогащения реакционного объе-
ма исходным веществом за счет входяще-
го потока, а правая часть — скорость рас-
ходования исходного вещества как результат
Рис. П-31. Продолжи-
тельность пускового ре-
жима реактора полного
смешения.
его химического пре-
вращения.
Структура уравнения (П-216) как схема баланса исходного
вещества в реакционном объеме системы полного смешения мо-
жет быть использована для описания баланса продукта превра-
щения исходного вещества А (продукт D):
0
-МСЛ, С°к)
(П-217)
Для реакции A->D первого кинетического порядка (сЛо=1,
сДо = 0), осуществляемой в реакторе полного смешения, зависи-
мость Сд(0), описываемая уравнением (П-217), представлена на
рис. П-32. Для сравнения на рис. П-32 приведена та,кже зависи-
мость cd(t) при проведении той же реакции е аппарате полного
вытеснения.
Л*
163
Характер кинетических зависимостей обратимых реакций в
открытых системах полного смешения можно видеть на примере
реакции
прямое и обратное направление которой описывается кинетически-
ми уравнениями первого порядка. При стационарном режиме урав-
нения (П-216) и (П-217) для этого типа
реакции имеют вид:
Л„~сА , ,
0 k\CA k2CD
0 k2CD klCA
Рис. 11-32. Зависимость
с (0, т) для реакции пер-
вого порядка А —> D.
Отсюда нетрудно найти зависимость
сг(0) как для исходного вещества, так и
для продукта реакции:
_ сл„ О ^2®) сД0^2®
сл i + ikt + k^e
с0о(1 +М) + слЛ®
D l + (ki + k2)&
Соотношение концентраций исходного вещества А и гр
реакции D как функцию расчетного времени пребывания
ционной смеси в реакционном объеме нетрудно выразить
деления уравнения (П-218) на уравнение (П-219):
СА _ cAAl + k2Q) + cD,k2Q
CD cDAX+kiQ) + cAjtfi
(П-218)
(П-219)
(П-220)
продукта
реак-
путем
Когда исходная смесь свободна от примеси продукта реак-
ции, т. е. когда cDo = O, уравнение (П-220) принимает вид:
с А 1 + &20
со ^1®
(П-221)
При 0->-оо соотношение концентраций исходного вещества и про-
дукта обратимой реакции в пределе ’будет равно отношению кон-
стант скорости, т. е. константе равновесия К — kz/k\.
Естественно, что ответственность за возможность достижения
состояния равновесия лежит на 0; при конечном значении 0 в
открытой системе полного смешения могут сколь угодно долго
поддерживаться термодинамически неравновесные концентрации
реагирующих веществ.
Понятно также, что скорость реакции (при условии, 4tocDc = 0)
в открытой системе полного смешения ниже соответствующей ве-
личины для закрытой системы. Иллюстрацией этого может слу*
164
жить рис. П-ЗЗ, где в координатах cD—0 представлены зависи-
мости, выражаемые уравнениями (П-217) и (П-219) для рассмат-
риваемой реакции при k2=~ki = i,0.
Продолжительность пребывания компонентов в реакционном
объеме системы полного смешения, необходимая для достижения
заданного значения св, легко выразить путем
преобразования уравнения (П-219):
kl(cA0~co)~k2cD
Зависимость расчетного времени пребыва-
ния реакционной смеси в аппарате полного
смешения от степени превращения исходного
вещества может быть описана простыми урав-
нениями, если концентрации исходного веще-
ства и продукта реакции выразить через на-
чальную концентрацию исходного вещества
(сЛо), приняв ее равной единице: !
СА = % (! - “ 1 - а
В.усл.еЯ.
Рис. П-ЗЗ. Зависим
мость cD (0) для реак-
ций первого порядка
(fel = fe2 = l,0):
ся = %а = а 2-A^±D.
*2
Эта зависимость приведена в табл. П-8.
Особенность условий, присущих открытым системам полного
смешения (и отличие их от закрытых систем и открытых систем
полного вытеснения), особенно заметна при осуществлении реак-
ций, схема которых включает последовательное превращение од-
ного из реагирующих компонентов. Этот тип реакций, пожалуй,
Таблица 11-8
Зависимости 0 (cD) и 0(a) для различных реакций
Схема реакции Порядок реакции 6 (со) 0(a)
А D 0 О |t) —
А D 1 CD a
kl (CAO ~ cd) ki (1 - a)
fel 1 CD a
A D ks ^1 (cAq ~ cd) ~ ^2CD fej — a (fej + fe2)
2А —> D 2 CD a
k2 (1 — a)2
fe2 2A —2D fej 2 CD a
^2 (CAq “ Cd) ~ ^3C2 k2 (1 — 2a) + (fe2 — &з) «2
}65
наиболее распространенный в промышленном органическом син-
тезе, может быть представлен рядом наиболее типичных схем
А — -> В D (I)
> »• ю ft* о Is- 4- и (П)
А~->В—М) L_5!_^F (III)
А—->-В—->D
I s, t (IV)
каждая из стадий которых может иметь либо первый, либо второй
кинетический порядок.
При стационарном состоянии реакционной системы баланс
каждого из веществ может быть выражен уравнением, аналогич-
ным по структуре уравнению (П-216) или (П-217).
Для схемы I при первом кинетическом порядке каждой из
стадий и при неравенстве констант скорости первой и второй ста-
дий (A2¥=^i) можно написать:
c . — с, — --=k с . e ' a (П-223)
Cn 0 = *1СЛ k2CB (11-224)
~kc 0 K2CB (П-225)
Из уравнения (П-223) нетрудно получить значение сА как
функцию 0:
сл = сл01 + вЙ1 (П-226)
Деля правую и левую части уравнения (П-223) на сА , полу-
чаем:
-5-=й1(1-а) или 6 = (1П-П) (П-227)
Замена 0 в уравнении (П-226) его значением из уравнения
(П-227) дает возможность выразить сА как функцию а:
(П-228)
Приняв, чтосВо = ОиСдо=0, подстановкой в уравнение (П-224)
значения сА из уравнения (П-226) можно выразить концентрацию
промежуточного продукта В как функцию 0. Замена в полученном
выражении 0 на его значение из уравнения (П-227) приводит к
зависимости св(а):
_______М________ «(1 - а) OOQ.
СВ = СЛ. 0 + fej0) (! + akfl) + «Ч - В (П-229)
16$
Х9Т
ор5 * (I - Л + О) (о - [)-гвог Х0Ъ~°:> ар3 °уэ _ дэ (I —Л + о)(о—I) (0-!)% = ^ *ж х г?о + 1 Л - ггР9‘0 + I Х%=Л г?ег х 1 - г*р + I Л X °v3 = аз Чегоуэ = уэ г г a <iT v
(Р-Р<? + I) (П-РР+ D epqp X = аз (р-р<?+ I)(p-pd+ I) (р-1)гр° х%-°з Р —РР+ I Оу„_ д (»-!)» (р— 1)°Ь'э= V3 (I^ei?+ i)(l;?ep +1) х х ^S-±-L.%= J3 ew + iH'gQP+i) х X 'У0+ I . °Ь'Э = <7Э ('^ер + i)(la?e+i) v . z\ X% = ff3 'W+ I Oy? Vj I i [L. Oil « ta Q 4 1 <
о -00+1 0Fj ffj гоо Р — РР + ] ок д (о-1)р (р — I) °¥з - ¥з ('?ep +1) ('?e + t) x &ep X% = ffo ('?ep + i)('?o+i) v ‘w x °¥Э ~ аэ 'W+Ioy., I i a <— a v
(О)Ь (e) порядок | реакции | HHtixead внэхэ
ВиЬяеэс! XHHhHUEed Birtf (»рэ и (0)‘э MiaowMMBeg
6-|| в h и и 9 в 1
Продолжение
Схема реакции Порядок 1 реакции 1 Cf (в) CZ (a,
A -51-> B —D ! -> F 1 1 Ca Ca<> 1 + (kt + k3) 9 CB = CAO X X Qkl t? II II о о X - 1 5 g. 1 3,
'' (1 + 0fe2)[l + (kl + k3)Q] CD = CAO X 4Z 92*1*2 '' (6 + l)(l-a) + aa CD = CAO X У aa2 (& + l)[(fr+l)(l-a) + aa] ab CF CAo 1 + b
(1 + 9/г2) [1 + (ki + k3) 9] 9£3 Cf Ca« l + (*i + fe)9
A —> B —5+> □ L*3 F 1 1 CA CA3 I+0ftj CB = CAO X x 0/г‘ сЛ = сДо(1-а) _ a (1 — a) CB CAO 1 + a(a + b-i) _ a a2 C° % 1 +a(a + Z>- 1). _ ba2 Cf C»o l+a(a + Z>-l)
(1 + 9/si) [1+9 (k3 + /г3)] CD = cAa x xz 92M2
(1 + 9^1) [1 + 9(/г2 + /гз)[ CF = CAO x 4Z e2*!^
'' (1 +9Л,) [1 +9(й2 + й3)]
A В D 1 k3 t 1 1 Ca Ca° l+(ki + k3)Q CB = CAO X x 0/г‘ сЛ = сЛо(1-а) CB = CA3 X x «(!-«) (b + 1) (1 — a) +aa CD = CA3 x xz a [6 -a (b -a)]
z' (1 +9й2) [1 +9(Й! + й3)] CD = cAa X xz 9 [&3 + 6&2 (&1 + &3)1
z' (i + e*2) [i +9 (k, + MJ (b + 1) (1 — a) +aa
Примечали е: a = k2Jkl, b=k3/kv z= 1 + - 1, Y -/(1 - a)2 + 4aa2.""
168
Концентрация конечного продукта D как функция 0 может
быть получена из уравнения (П-225) после подстановки значе-
ния св из уравнения (П-229). Зависимость cD(a) легко выводится
также из баланса продуктов: cD = асДо — св. Окончательно имеем:
ak262 аа2
с° ~ Са« (1 + kfi) (1 + akfi) =Сл»1+аа — а (II 230)
Совместное решение системы уравнений, описывающих реак-
ционные системы II—IV, произведенное аналогичным образом,
приводит к выражению зависимостей концентрации исходного ве-
щества, промежуточного и конечного продуктов как функции 0
и а. Полученные выражения помещены в табл. П-9.
Рис. П-34. Зависимости ct (0) и ct (т) для реакции первого порядка
А —В —D (сЛо = 1,0):
а — fe2/kl == 2; б — ^2/^1 в 0,5;
-----------cz(0);......
Изменение концентрации продуктов последовательной реакции
первого порядка в зависимости от продолжительности пребыва-
ния реакционной массы в аппарате полного смешения приведено
на рис. П-34 (сплошные линии). Здесь же дана зависимость сДт)
для закрытой системы (пунктирные линии).
Характер зависимостей с, (а) можно видеть на рис. П-35, где
приведены также кривые (пунктирные), описывающие картину со-
ответствующей реакции при проведении ее в закрытой системе
(или в открытой системе полного вытеснения).
Изучение рис. П-35 позволяет заметить общую для всех слу-
чаев закономерность: концентрация промежуточного продукта ре-
акции, осуществляемой в закрытой системе, во всем интервале
значений а превышает аналогичную величину в открытой системе
полного смешения. Положение точки экстремума на кривой св(а)
в закрытой системе соответствует более высокому значению сте-
пени превращения исходного вещества, чем в открытой системе.
169
Естественно, что кривая, выражающая изменение концентра-
ции конечного продукта последовательной реакции при проведе-
нии ее в открытой системе, лежит выше кривой, характеризующей
закрытую систему.
Вполне понятно также отсутствие влияния перехода от закры-
той системы к открытой на зависимость ср(а) для продукта па-
раллельной реакции (рис. П-35, г).
Рисунок Схема реакции Кинетн ческнй порядок реакции ks/ki
а I 2
б a-*i_>b-^->d 2 1
в 1 2 3
л A21_^b^D - _>F 1 1 1
д 1 * 1 fea [ 1 1 1
Л А-^В-^О 1 1 2
170
Графический способ расчета состава
реакционной смеси
Расчет состава реакционной смеси при проведении про-
цесса в открытой системе полного смешения требует количествен-
но сформулированных данных о кинетике реакции.
При расчете сложных реакций и реакций высших порядков со-
ставление уравнений кинетики часто бывает связано с значитель-
ными трудностями и неточность, допущенная при определении
кинетических параметров (порядок реакции, константы скорости),
приводит к значительным погрешностям при использовании кине-
тического уравнения для расчета открытых систем.
Для технических расчетов определенное удобство представля-
ют графические методы, допускающие использование эксперимен-
тальных данных.
Из числа известных графических и графо-аналитических мето-
дов [34—36] наиболее удобной является схема расчета, предло-
женная Крыловым и Шабалиным [34], сущность которой сводится
к следующему.
Правая и левая части уравнения (П-217), описывающего ба-
ланс продукта D реакции A->D n-го кинетического порядка в ре-
акторе полного смешения, содержат одно неизвестное—cD (зна-
чения с, = с. и сп = сп заданы) и могут быть выражены раз-
личными по форме функциями от сп . Для общего случая
Ф (ct) -f (Ci) (П-231)
Решение этого уравнения достигается путем нахождения коор-
динат точки пересечения кривых, описываемых левой и правой
частями уравнения.
Примером применения этого метода могут служить односта-
дийные реакции типа:
(v^ = vd)
На рис. П-36 в координатах cD — <p(cD), ?(cd) нанесены зависи-
мости левой и правой частей уравнения (П-231):
f (cd) = k (са„ со)п
При заданном значении 0 прямая, выражающая <p(cD) и при
нулевом значении cD* выходящая из начала координат, пересе-
кает кривые, описываемые выражением f(cn) при разном значе-
нии п в точках 1 и 2, абсциссы которых выражают концентрацию
продукта D в реакционном объеме.
171
Для построения кривых, выражающих функцию f(cD), доста-
точны результаты графического дифференцирования эксперимен-
тальных кривых с©(т), построение которых производится на са-
мом раннем этапе изучения каждого химического процесса.
При достаточно малых значениях Дт, применяемых при графи-
ческом дифференцировании, точность значений f(cD) вполне до-
статочна для технических расчетов.
а
с 0) дел ед.
Рис. П-36. Схема графического расчета состава реак-
ционной смеси в реакторе полного смешения (Сд0= 1,0).
Рисунок Схема реакции ' Кинети- ческий порядок реакции а = ki/ki
а А —D 1
б Q Ь * * 1 1/3
В случае параллельных реакций
->D
.А Lр
соответствующая обработка экспериментальных результатов по-
зволяет получить данные для построения кривых
WF~HCf)
в координатах Ci—f(cD), f(cF).
Задаваясь значением 0, легко получить величину тангенса
наклона прямой, описываемой зависимостями
CD ~ CD CF ~ CF
ё = ф (cz>): ё = ф (ср)
172
При нулевом значении cF и cD прямая, как показано на
рис. П-36, выходит из начала координат и пересекает кривые в
точках 1 и 2 соответственно, давая значения cF и сп для условий
гк
проведения процесса в открытой системе полного смешения.
На рис. П-36 представлена картина тече-
ния реакций первого кинетического порядка
^2/^1 = 1/з; сЛо= 1,0. Видно, что отношение кон-
центрации продуктов F и D в реакционном
объеме сохраняется равным отношению кон-
стант скорости независимо от изменения сте-
пени превращения исходного вещества.
Графический расчет реакций, включаю-
щих последовательное превращение исходного
вещества, удобно рассмотреть на примере по-
следовательной реакции
v .А -> v„B -> vnD -> vPF
Для графического расчета состава продуктов
этой реакции в открытой системе полного
смешения необходимо экспериментально опре-
делить зависимости щ(т) путем постановки
соответствующих опытов (например, в закры-
той системе):
первая серия опытов
v^A->(vBB + vDD + vfF)
вторая серия опытов
Д?в "* (vzjd + VF)
третья серия опытов
vdd
Анализ периодически отбираемых проб
реакционной массы (при проведении экспе-
римента в закрытой системе) и соответст-
вующая обработка результатов анализа по-
зволяют графически выразить зависимости
Св(т); го(т); ск(т). При этом концентрация
смеси продуктов превращения исходного вещества в каждой се-
рии опытов выражается через фиктивную концентрацию первого
V
продукта превращения, например, в первой серии св = сА^А
у А
во второй - CD = Свав И т. д.
Графическое дифференцирование Дсг-/Ат полученных таким
образом кривых дает возможность построить кривые, изображен-
ные на рис. П-37 в соответствующих координатах.
а. ' 7-а
^0 1 J
>| 1 III 1
О 0,2 0,4 0,6 0,8 1,0
сs, у ел ей.
б
0 0,2 Й Ofi 0,8 ~t,0
cD , у еле В.
8
।
I
0 02 0,6 0,8 1,0
J, сг,услед
Рис. П-37. Схема гра-
фического расчета со-
става реакционной
массы в реакторе пол-
ного смешения
(сАй = 1,0; схема реак-
. ki &2 .
ции: А------► В---->
—•>D‘^~*F; поря-
док реакции — первый;
k3: k2: k, = 4: 2: 1).
173
Задаваясь степенью превращения исходного вещества А или
Временем пребывания реагирующей смеси в реакционном объеме
полного смешения, находим точку 1 на кривой рис. П-37, абсцисса
которой соответствует концентрации продукта В при условии, если
дальнейшее превращение его не происходит (максимально воз-
можная концентрация продукта В).
В первом случае (задана степень превращения исходного ве-
щества) точка 1 находится по абсциссе, равной св = сА(а; во втором
случае (задано время пребывания 0) точка 1 находится в резуль-
тате построения прямой О—1, тангенс угла наклона которой к оси
абсцисс равен 1/0. В том случае, когда концентрация продуктов
реакции в исходном веществе равна нулю, прямая, как это пока-
зано на рис. П-37, выходит из начала координат.
Совершенно очевидно, что концентрация продукта D в реакци-
онной смеси не может быть больше концентрации продукта В с
учетом стехиометрических соотношений и возможные пределы ее
v„
изменения (при заданном 0) ограничены значениями О cD^.cB,
' vo
при —— cD = cB скорость образования продукта D равна нулю,
' VD
а максимальное значение скорости достигается при cD=0.
Таким образом, превращение продукта В в продукт D в реак-
ционной системе полного смешения описывается участком кривой
’iwd(Cd), пересекающим ось ординат в точке k2cBn, ось абсцисс —
в точке cD=cB (точка 2).
Построение такой кривой достигается перемещением экспери-
ментально построенной кривой wD(cD) влево по оси абсцисс или
перенесением начала координат вверх по оси ординат, как пока-
зано на рис. П-37, а и б.
Возможная концентрация продукта D (т. е. концентрация, ко-
торая имела бы место при полной химической инертности продук-
та D) определяется как абсцисса точки пересечения прямой 3—4,
параллельной прямой 0—1 (рис. П-37,а), с кривой wD(cD) (точ-
ка 4).
Для определения концентрации продукта F поступают анало-
гичным способом. Значение концентрации продукта F, установлен-
ное описанным выше способом, является максимально возможной
концентрацией этого продукта (по условию продукт F дальней-
шего превращения не претерпевает). »
Концентрации промежуточных продуктов (В и D) в реакцион-
ной смеси определяются как разность между максимально воз
можной их концентрацией и концентрацией продукта их превра-
щения.
Так как конечным продуктом является продукт F, концентра-
ция которого не снижается в результате его взаимодействия, то
первым вычитаемым является концентрация этого продукта.
174
Разность между асбциссой точки 7 (концентрация продукта F)
и точки 4 (максимально возможная концентрация продукта D)
численно равна концентрации продукта D.
Разность между абсциссой точки 4 (максимально возможная
концентрация продукта D) и точки 1 (максимально возможная
концентрация продукта В) численно равна концентрации продук-
та В в условиях открытой системы полного смешения.
Абсцисса точки 1 (с учетом стехиометрических коэффициентов)
выражает величину степени превращения исходного вещества (а).
Длина отрезка оси абсцисс вправо от точки / (с учетом сте-
хиометрических коэффициентов) является выражением концентра-
ции непрореагировавшего исходного вещества Айв относитель-
ных единицах выражает величину 1 — а.
Следует заметить, что перемещение кривых даг-(сг) влево по
оси абсцисс является обязательным во всех случаях, когда поря-
док реакции не равен единице. При п = 1,0 зависимость Wf(Ci) —
прямолинейна и можно ограничиться перемещением начала ко-
ординат в точку, ордината которой равна ординате точки 2
(рис. П-37, б) или 5 (рис. П-37, в).
На рис. 11-37, использованном для иллюстрации применения
графического способа расчета многостадийных последовательных
реакций, выполнен расчет последовательной реакции
А — -> В —D —F
первого порядка, константы скорости отдельных стадий которой
равны &1 = 1,0; &2 = 2,0; йз=4,0 при численном равенстве стехио-
метрических коэффициентов, сЛо=1,0 и а = 0,6.
Результаты графического и аналитического расчетов:
Аналитиче- Графический
ский расчет расчет
Св...................... 0,15 0,15
cD...................... 0,06 0,07
cF...................... 0,39 0,38
а ...................... 0,60 0,60
1—а .................... 0,40 0,40
Примером использования графического метода для расчета по-
следовательных реакций, кинетический порядок которых больше
единицы, может служить двухстадийная последовательная реакция
A->B->D
У
Как и в предыдущем случае, результаты двух серий опытов
А->В
В-> D
проведенных в закрытой системе, наносятся в виде кривых св(т),
Ср(т).
175
Графическое дифференцирование полученных кривых дает воз-
можность построения зависимостей wB(cB) и wD(cD) (рис. П-38).
При заданной степени превращения (а=0,6) строится ломаная
линия 0—1—2—3—4—5 [в отличие от рассмотренного выше слу-
чая реакции первого порядка для
дукта D обязательно смещение
кривой wD(cD) влево по оси абс-
цисс, как показано на нижнем
рис. П-38].
Результаты аналитического
расчета при а = 0,6, &i = l,0, k2 —
определения концентрации про-
ш,
Шц
Cl
^1
0 02 0,0 0,6 0,8 1,0
. । । Се,ушд.
з 5_
0 02 Oft 0,6 0,8 1,0
J с^ум.ед.
Рис. П-38. Схема графи-
ческого расчета соста-
ва реакционной массы
в реакторе полного сме-
шения (сЛо=1,0; схема
реакции: А---->В---->D;
порядок реакции — вто-
рой; Л2/^1 “ 2).
Рис. П-39. Схема графи-
ческого расчета соста-
ва реакционной массы
в реакторе полного сме-
шения (сАо = 1,0; схема
->D
реакции: Ан k ;
порядок реакции — пер-
вый; k4'.k3:k2:ki = 4:3:2:1).
=2,0 дают значения св и cD, практически не отличающиеся
зультатов графического расчета.
Параллельно-последовательные реакции типа
„В-> D
от ре-
при проведении их в открытой системе полного смешения
могут быть рассчитаны графическим методом при наличии
риментальных данных о реакциях
также
экспе-
А<
; в
176
Схема расчета для случая, когда порядок всех реакций — пер-
вый, приведена на рис. П-39.
Рассмотренные выше примеры графического расчета состава
реагирующих компонентов в открытой реакционной системе пол-
ного смешения показывают, что при достаточном масштабе и точ-
ности графических построений полученные результаты практиче-
ски не отличаются от результатов аналитического расчета.
Результаты графического определения состава реакционной
массы ‘могут быть использованы для расчета выхода каждого из
продуктов реакции. Для этого достаточно определить отношение
длины каждого из отрезков, выражающих концентрацию продуктов
реакции в реакционной массе, к длине отрезка, выражающего
степень превращения исходного вещества (рис. П-39). Разумеется,
что при этом необходим учет отношения стехиометрических коэф-
фициентов.
Изучение кинетики химических реакций
с использованием открытых систем
Область применения открытых реакционных систем не ограничивается ис-
пользованием их при организации непрерывнодействующих реакционных аппара-
тов и агрегатов.
Изучение кинетики 'быстротекущих реакций в статических условиях закры-
той системы зачастую бывает связано с значительными экспериментальными
трудностями, особенно ощутимыми, когда объектом исследования является про-
цесс, характеризующийся значительным тепловым эффектом. '
Решение экспериментальных задач значительно упрощается при использо-
вании особенностей открытых реакционных систем. В зависимости от характера
реакции метод исследования может быть основан либо на принципе полного вы-
теснения (проточный метод), либо на принципе полного смешения (безградиент-
ный метод).
Проточный метод основан на пропускании с постоянной скоростью смеси
реагирующих веществ через реакционное пространство (при заданных темпера-
туре и давлении) в условиях, максимально приближающихся к условиям от-
крытой реакционной системы полного вытеснения.
Анализ проб реакционной массы, отбираемых в ряде точек по длине реак-
ционного пространства, позволяет определить изменение состава реакционной
смеси во времени, подсчитываемом из соотношения о = .
Установленная таким образом зависимость с, (9) обрабатывается одним из
известных способов и приводит к получению уравнений скорости.
При использовании проточного метода строго обязательным является мак-
симальное приближение к режиму открытой системы полного вытеснения (ра-
венство скорости потока по сечению, отсутствие продольного перемешивания).
Соблюдению этих условий уделяется серьезное внимание, так как, напри-
мер, отклонение локальной скорости потока газа при проведении гетерогенного
процесса в слое твердого катализатора от средней расчетной ее величины при
малом диаметре реакционного пространства достигает 40%.
Столь же значительным может быть влияние продольного перемешивания,
вносящего дополнительное искажение в результаты эксперимента.
Наибольшие трудности (ограничивающие область применения этого метода)
возникают при изучении кинетики реакций, обладающих большим тепловым эф-
фектом; неравномерность тепловыделения по длине реакционного объема услож-
няет условия теплообмена, необходимого для изотермического режима потока.
Обработка результатов проточного метода, полученных в неизотермических ус-
ловиях. крайне сложна [37].
J2 В. С. Хайлов, Б. Б. Брандт 177
Несмотря на необходимость соблюдения указанных выше условий, проточ-
ный метод находит применение для исследования кинетики гомогенных быстро-
текущих реакций. Использование проточного метода для гетерогенных систем
ограничивается системой газ — твердое тело (каталитические процессы). Изуче-
ние гетерогенных жидкофазных систем проточным методом крайне затрудни-
тельно.
Безграднентный метод основан на использовании открытой реакционной си-
стемы полного смешения [38].
Затруднения, обусловливаемые наличием градиента концентраций и гра-
диента температур (практически неизбежных особенностей проточного метода),
сравнительно легко устраняются в открытой системе полного смешения.
Основным требованием к условиям изучения кинетики безграднентиым ме-
тодом является мгновенное и полное перемешивание непрерывно и с постоянной
скоростью подаваемых в реакционное пространство исходных веществ со смесью
продуктов реакции, заполняющих это пространство.
Практическое отсутствие градиента температур и концентраций в реакцион-
ном объеме достигается при условии, что масса циркулирующих продуктов по
крайней мере на одни порядок превышает количество подаваемых исходных ве-
ществ.
Анализ проб реакционной массы на выходе нз реакционного объема при за-
данном (постоянном во времени) составе исходных веществ дает возможность
описать процесс выражениями
сА - сА
—- = (для расходуемых веществ)
с,-к ~ <7и
----§---=kf(c{^ (для продуктов реакции)
и, устраняя необходимость интегрирования кинетических уравнений, позволяет
непосредственно получать выражение скорости реакции как функцию концентра-
ции реагирующих веществ [39].
Результаты, полученные безграднентиым методом, наиболее удобны для
использования при расчете открытых систем.
Безграднентный метод исследования используется для изучения кинетики
как гомогенных, так н гетерогенных реакций всех фазовых состояний, для кон-
тактно-каталитических процессов, а также при оценке активности катализаторов.
Возможность проведения процесса при высокой степени превращения исход-
ного вещества представляется весьма удобной для изучения кинетики равновес-
ных' реакций и для исследования тормозящего действия некоторых продуктов
превращения.
ОТКРЫТЫЕ СИСТЕМЫ ПРОМЕЖУТОЧНОГО ТИПА
Противоток реагентов
Естественным следствием снижения концентрации исход-
ных веществ по мере их параллельного перемещения по длине ре-
акционного пространства аппарата полного вытеснения является
снижение скорости реакции. Интенсивность тепловыделения (или
поглощения тепла) реагирующего потока, определяемая скоростью
реакции, меняется по длине реакционного пространства, затрудняя
осуществление необходимого теплового режима.
Структура кинетического уравнения реакции v^A + vgB—> vflD
7 VR СЛ \
w = k.c, (1 — а) Со 11-------- a I
‘Ло В°\ % /
178
объясняет сказанное и позволяет вместе с тем видеть возмож-
ность сохранения значения скорости путем увеличения одного из
сомножителей при малом значении другого. Это может быть до-
стигнуто при противотоке реагирующих веществ, рассмотренном
Орочко [40].
Принимая для простоты, что Va=vb=vd=1, допуская постоян-
ство объема реагирующих потоков и рассматривая в качестве ос-
новного компонента исходное вещество А, желаемая степень пре-
вращения которого (ак) задана условиями задачи, нетрудно вы-
разить концентрацию реагирующих веществ в любом стечении
реакционного пространства:
са = са^* 1~^
сВ = сВо-СДо«К + СДоа
Так как значение разности сВо~сЛоак за-
дано условиями процесса и является постоян-
ной величиной (Р'), уравнение скорости мож-
но написать в виде:
а = сл| = ^1-«Г + Ча) (п-232)
Рис. П-40. Зависимость скорости реакции А 4-В—>D
от степени превращения вещества:
/ — прямоток реагентов; 2—6 — противоток реагентов;
кривая . . . . 2 3 4 5 6
ак........... 0,2 0,4 0,6 0,8 0,9
Интегрирование последнего выражения в пределах
т = 0; а — 0
г; а = ак
приводит к уравнению:
1 Со
Т =1п_________________Щ_____
klCAa (1 - «к) + СВО (‘ - «к) (сва - Сд ак)
Ясно, что эффект противотока в максимальной степени прояв-
ляется при стехиометрическом соотношении компонентов и мало
ощутим при большом избытке одного из них.
На рис. П-40 приведены результаты расчета зависимости w(a)
при сЛо = сВо = 1 и kx = 1,0 л • ч/моль. Видно, что в случае противо-
тока скорость реакции характеризуется большей равномерностью
по длине реакционного объема с максимумом значения в сере-
дине процесса.
В большинстве практических случаев весьма трудно осущест-
вить идеальный противоток и более реальным является каскад
секций смешения и секций разделения (рис. П-41).
12*
179
Процесс описывается уравнением (П-232), где значение Р' ме-
няется от секции к секции.
А
B+D
Рис. П-41. Схема ступенчатого противотока реагентов.
Распределение концентраций при ступенчатом противотоке по-
казано в табл. 11-10.
Таблица 11-10
Распределение концентраций при ступенчатом противотоке
СА св
вход выход ВХОД ВЫХОД
Ввод вещества А и вывод не- прореагиро- вавшего веще- ства В . . . Первая секция СА0 САо %(’-“) СВО ~ сАоа1 С о С я СС__ DQ ЛО К СВО ~ сАоак
Вторая секция сЛо(1_а1) СВ о - СА 0ат-2 сва ~ сАоат-1
х-я секция . . . -«i-l) САоО-'М СВО~ сАоат-1 cBa~cAtflm+i-i
(т — 1)-я секция сЛо (1 — am-z) % (!-««-.) СВО ~ сАоа1 СВа~сА0а2
т-я секция . . %(*-%->) Cloo-aj св0 сВо ~ сАо“1
Вывод иепро- реагировавше- го вещества А и ввод веще- ства В ... — СВо —
Ступенчатый подвод реагентов
Выход продуктов параллельных реакций неодинакового
кинетического порядка зависит от степени превращения исходного
вещества. Выход продукта реакции меньшего кинетического по-
рядка увеличивается с понижением концентрации исходного ве-
щества, т. е. оптимальный для этого продукта режим лежит в об-
ласти высоких значений степени превращения.
Сказанное сохраняет справедливость и для параллельных ре-
акций одинакового кинетического порядка (например, второго)
180
при большом избытке одного из реагентов. Последний случай мо-
жет быть иллюстрирован схемой реакций
А + В —•> D (I)
2А—->Е (II)
осуществляемых при большом избытке вещества В (cB~const).
Выход продукта D в этом случае (реакция I — псевдопервого
порядка) может быть повышен при условии поддержания низкой
концентрации вещества А в реакционной системе.
Рассмотрим частный случай схемы реакций (I) и (II), приняв
k2fk} = а.
Выход продукта D за бесконечно малый промежуток времени
периодического процесса или процесса, осуществляемого в аппа-
рате полного вытеснения, выражается зависимостью:
о то = 1 =_____1
Рд ®д + ®£ 1+асд 1+асЛ))(1—а)
Выход продукта D в интервале заданной степени превращения
(ссн=О; ак) можно выразить уравнением:
“к
If da 1 1 + ас л
В„ =— —--------? =------1ПТ-------: (П-233)
ак J 1+асЛо(1-а) ааксЛо 1 + асАо (1 - ак)
а для случая, когда aH=cCi>0:
1 f da 1 1 + асА (1 — a.)
=------- ТЗ------71----Г = “Г----7- 1п ГЧ—-------Ч (П-234)
Di ак~аг J 1+асЛо(1-а) а(ак-аг.)сЛо 1 + acA° (1 - ак) .
а(.
где а, (0<аг<ак)—нижний предел степени превращения в ис-
следуемой области значений а.
Сравним значения pD из уравнений (П-233) и (П-234):
. 1 + асд0(1 ~а»)
Рог п П1+аСЛо(1-ак)
₽Г>» ,п 1+асЛ
(II-235)
₽ог а
Значение р— определяется величиной П — , выражающей
количество точек ввода исходного вещества в реакционный поток
для понижения степени превращения от ак до а,.
Иллюстрацией может служить пример параллельной реакции,
характеризующейся равенством k\=kz (a=l) при степени превра-
щения исходного вещества ак=0,95.
181
Ро (П) и at (П) для параллель-
ной реакции А —> D, 2А —> Е
(ак=0,95).
Результаты расчета по уравнению (П-234) при 0-Са,-С0,95
представлены на рис. П-42. Кривая 2 показывает значение аг-,
определяющее (и определяемое) количество точек ввода исход-
ного вещества. Кривая 3 выражает связь выхода продукта Е
(Рв=1 — рд) и количества точек ввода исходного вещества в ре-
акционный поток и указывает на
монотонное снижение выхода этого
продукта по мере увеличения коли-
чества точек ввода.
Зависимость pD(aK) для одной—
уравнение (П-233)—и пяти—урав-
нение (П-234) — точек ввода исход-
ного вещества А представлена на
рис. П-43.
В ряде работ [37, 40—46] систе-
ма реакций, описываемая схемами
(I) и (II), рассмотрена для случая
-соизмеримых (равных) концентра-
ций реагентов А и В и установле-
ны оптимальные условия ее осуще-
ствления.
Следствием уменьшения концен-
трации исходного вещества В, уча-
ствующего в образовании продукта D, является некоторое сни-
жение выхода этого продукта по сравнению с выходом в разо-
бранном выше примере.
Максимальный эффект достигается при
ступенчатом подводе компонента А в по-
ток компонента В при ак=0,95 и равном
мольном количестве исходных веществ А и
В. Оптимальное распределение компонен-
та В по пяти точкам питания в долях от
потока вещества В соответствует следую-
' щим значениям (по ходу потока): 0,38;
0,24; 0,17; 0,12; 0,09.
Результаты расчетов для пяти точек
питания [46] показывают также, что равен-
ство объемной скорости потоков компонен-
та А, вводимых в реактор, не вносит сколь-
ко-нибудь заметных изменений в значение
выхода продукта D, получаемого при опти-
мальном распределении.
Увеличение количества точек ввода пи-
тания реактора, благоприятствующее по-
Рис. П-43. Зависимость
(ак) для параллельной
реакции А + В-----> D,
2А-^-Е (*2 = *i;
сАо=1>°; % = “nst):
1 — одна точка ввода веще
ства А; 2 — пять точек ввода
вещества А.
вышению выхода продукта D, сопровождается снижением произ-
водительности реакционного пространства. Сравнительная оценка
различных вариантов схемы реакционного узла, предназначенного
для проведения рассмотренной выше параллельной реакции (I, II),
182
при равенстве констант скорости отдельных реакций и степени
превращения исходного вещества А, равной 0,95, приведена в
табл. П-11.
Таблице Н-11
Сравнительная оценка различных вариантов
схемы проведение параллельной реакции
А + В —> D, 2А —> Е
Характеристика П = 1 П = 5 П “ оо
Выход продукта D в расчете на веще- ство А 0,625 0,87 0,90
Производительность реакционного объема (относительные величины) 1,0 0,16 0,10
Для этого случая зависимость от количества точек ввода
питания можно видеть на рис. П-42 (пунктирная кривая).
Ясно, что в реакторе периодического действия низкая концент-
рация исходного вещества достигается при подаче компонента А
в реакционную массу либо непрерывно с заданной скоростью, либо
определенными порциями через равные промежутки времени.
Ступенчатый подвод реагентов используется не только при осу-
ществлении системы параллельных реакций: этот прием удобен
также для регулирования интенсивности тепловыделения при про-
ведении экзотермических процессов.
Каскад реакционных аппаратов
полного смешения
Рассмотренные ранее закономерности открытой реакци-
онной системы полного смешения показывают, что в результате
разбавления исходного вещества продуктами его превращения ве-
личина движущей силы химического процесса уменьшается, вслед-
ствие чего для открытых систем полного смешения характерна
меньшая (по сравнению с закрытой системой или открытой систе-
мой полного вытеснения) скорость реакции по исходному веществу.
Различие между системами полного смешения и полного вы-
теснения количественно выражается критерием Боденштейна, пре-
дельные значения которого характеризуют системы идеальных ре-
жимов.
Промежуточным (между идеальными) режимом характеризу-
ются системы, для которых 0<Во<оо, т. е. коэффициент продоль-
ного перемешивания (D) является положительной конечной вели-
чиной: oo>D>0.
Подобные условия имеют место в реакционном объеме, где
продольное перемешивание несколько усредняет состав реакци-
онной массы, или в серии последовательно соединенных систем
183
полного смешения. В последнем случае в объеме каждой системы
О—>-оо, а на границе систем О—>-0.
Такого рода серия последовательно соединенных систем пол-
ного смешения изображена на рис. П-44, где каждая из секций
представляет собой открытую систему полного смешения при
полном отсутствии химического взаимодействия в потоке между
секциями.
При непрерывной и равномерной подаче исходных веществ в
первую секцию после периода нестационарного режима в каждой
секции устанавливается стационар-
ный режим, описываемый уравне-
нием
1 2.3 т
Рис. П-44. Схема каскада аппа-
ратов полного смешения.
которое позволяет подсчитать кон-
центрацию исходного вещества в
любой т-й секции каскада (сА при заданной начальной или ко-
нечной его концентрации.
Характер зависимостей сА (т, 0(.) можно видеть на примере
реакции первого порядка А—> В:
слт = Т+ОГ = СЛ° (l+Wi+W (1+е^,) (1Ь236)
Аналогичные выражения могут быть получены для реакций
других (кроме нулевого) порядков. Результаты расчета на основе
таких уравнений определяются не только абсолютным значением
01, 02 и т. д„ но и соотношением этих значений.
Как будет показано ниже, вопрос о рациональном соотношении
размеров секций каскада имеет существенное значение при реше-
нии задач, связанных с оценкой производительности реакционной
аппаратуры промежуточного типа.
Соотношение размеров секций каскада. Расчетное вре-
мя пребывания реакционной массы в каждой из секций каскада,
в котором осуществляется простая необратимая реакция n-го по-
рядка (п>0), можно получить из уравнения:
С д , С л t
ег = —-------1- (11-237)
Тогда время пребывания реакционной массы в каскаде из т
секций выражается суммой:
kcnA.
СА ~СА
ят-1 ят
kcA
лт
(П-238)
Для отыскания оптимального соотношения размеров секций
каскада, при котором общее расчетное время пребывания 0s ре-
184
акционной массы минимально, приравниваем нулю частные про-
изводные от 02 по сА (i=l, 2, ..т—1). В результате получим
систему из т— 1 уравнений типа
Эти уравнения позволяют при заданных значениях сА и сА найти
все промежуточные концентраций сА^ от сА до сА и после под-
становки их в уравнение (П-238) определить соответствующее ми-
нимальное значение общего времени пребывания.
В общем случае, однако, задача решается только численно, что
затрудняет анализ характера зависимости оптимального распре-
деления размеров секций каскада от различных параметров. Пред-
ложенный в последнее время [47] изящный способ анализа струк-
туры этой зависимости позволяет решить задачу сравнительно
просто.
Вместо степени превращения исходного вещества в i-й секции
каскада, выражаемой отношением
вводится новая переменная
СА:
Я/ = ---= 1 - at
%-,
(П-240)
которую можно трактовать как степень непревращения исходного
вещества в i-й секции каскада; подобно степени превращения
0<T]j<l.
Уравнение (П-239) в новых переменных будет иметь вид;
1+ге-—Э*- = —!— (1.1,2.....т-1)
п» п"+1
(П-241)
Введение новой переменной величины позволяет сократить чис-
ло «соседних» параметров уравнений (П-239) — сА^ сА? сА^ — до
двух параметров тр и тр+i, характеризующих i-й и (i+ 1)-й аппа-
раты каскада.
Структура тр дает возможность установить дополнительную за-
висимость:
т
ТЦТ12 ... Tlm = JJ1!*
К“1
С л
с
%
(II-242)
185
Текущие концентрации cAi можно выразить через новые пере-
менные с помощью соотношений:
' = Лxn2 • • • 'Hf = И (П-243)
Подставляя выражение (П-243) в (П-237) и (П-238), получаем
уравнения, описывающие время пребывания реакционной массы
в i-й секции каскада и в каскаде в целом:
ег =
4-i~4
с"-1
(П-244)
1
и
(П-245)
Таким образом, расчет каскада с оптимальным распределением
объемов секций (времен пребывания) сводится к определению со-
вокупности параметров т],, которые удовлетворяют условиям
(П-241) и (П-242).
Величины т)ь рассматриваемые как функции исходного пара-
метра Гц, монотонно возрастают при увеличении t]i от 0 до 1. Be-
т
личина П т)«, рассчитанная с помощью уравнения (П-242), в ин-
№=1
тервале от 0 до 1 также является монотонной функцией параметра
т]ь проходящей через начало координат.
Вследствие указанных свойств параметров т], задача построе-
ния каскада с оптимальным распределением объемов секций мо-
жет, по существу, рассматриваться как задача нахождения такого
параметра гц, что последовательность остальных параметров, рас-
считанная с помощью рекурентного соотношения (П-241), отве-
чала бы условию (П-242). Величина гц может быть найдена с ис-
пользованием метода проб и ошибок или определена прямыми
численными методами.
Наряду с этим в данном случае можно применить также про-
стой и наглядный графо-аналитический способ расчета [48], осно-
ванный на использовании графика зависимости (П-241), связы-
вающий между собой коэффициенты неэффективности соседних
реакторов каскада. Для известного порядка реакции построим
график зависимости величины t]i+i от в четырех системах ко-
ординат (Т]1— T)i+1, T|i+1— fli+Z. T)i+2—ТК+З, Т|«+3—Т|«-н)» КОТОрые СОПрЯ-
жены таким образом, что их начала совмещены, а ось абсцисс
последующей системы совпадает с осью ординат предыдущей си-
стемы (рис. П-45). Отложим теперь на оси г), отрезок Оа\, равный
в масштабе начальному значению гц последовательности коэффи-
циентов т]ь которым необходимо задаться. Проведем через точку
186
сц вертикаль до пересечения с первой кривой в точке Через по-
лученную точку проведем горизонталь до пересечения со второй
кривой в точке Ь2. Через эту точку проведем вертикаль до пересе-
чения с третьей кривой в точке Ьз. Через точку Ьз проведем гори-
зонталь до пересечения с четвертой кривой в точке Ьц. Затем че-
рез полученную точку проведем вертикаль до пересечения с пер-
вой кривой и т. д. Отрезки Опь Оа2, Оаз, Ощ и т. д., отсекаемые
Рис. П-45. Схема графического расчета параметров тц-
этой «ломаной» спиралью на осях, равны соответствующим вели-
чинам коэффициентов T)l> 1)2, Чз, Л4, • ; Ч]т-
Ограничившись определением стольких величин этих коэффи-
циентов, сколько секций будет в каскаде, необходимо проверить,
отвечает ли избранная последовательность коэффициентов
желаемой степени превращения исходных реагентов в каскаде
а=1—Л2, т. е. необходимо выяснить соблюдается ли условие
- т
(П-242). Если произведение коэффициентов ф'Пг • • • Лт = 11'Пл
feel
ж
будет больше
(меньше) заданной величины т]2 = —то необхо-
САа
димо повторить расчет, уменьшив (увеличив) исходное значение
начального параметра.
Оптимальное соотношение размера секций каскада может быть
выражено как соотношение времен пребывания реакционной мас-
сы в соседних секциях с помощью уравнения (П-244):
Рис. П-46. Зависимость оптимально-
го соотношения размеров секций
каскада от параметра тц+1.
Z + 1
д 1 - п,+1 п nl’4
°Л-1 к=1
V/+1. /--— ----------------i--------
' -Hi-t-iG—Пг)
1 - Пл-i Пг
П?+1
Подставляя в это выражение
П,-
значение комплекса , наи-
1-Т)г
денное из уравнения (П-241)
1-т)г 1-П?+1
получим искомую зависимость:
= (П-246)
1 Пл-!
Характер полученной зависимости представлен семейством
кривых на рис. П-46. Частные случаи выражения (П-246):
1 + Пл-i + Пл-1
1 + Щ+1
1 ... 1
2 ••• 2
1 + V Пл-! + V П Л1
3 ••• 3
Как видно, величина vj+i, изменяется в пределах от п при
4)1+1 —0 до 1 при T]i+i = l. При п>1 величина v1+i, i монотонно убы-
вает с увеличением Цг+1, при 0<га<1 монотонно возрастает.
Величина т]г-ы, входящая в уравнение (П-246), однозначно за-
висит от значения цг = 1 — а , характеризующего каскад в целом,
188
а также от числа секций каскада т и порядкового номера секции
i+1. При га>1 величина rji+i тем больше, чем больше заданное
значение т)2, т. е. чем ниже заданная степень превращения исход-
ного вещества в последней секции каскада (ат = 1 — т]2), чем боль-
ше число секций каскада и чем выше порядковые номера рассмат-
риваемой пары секций. При 0<п<1 величина rji+I тем больше, чем
меньше заданное значение а, чем больше число секций каскада и
чем ближе рассматриваемая пара секций к началу каскада (чем
меньше i+1).
Таким образом, оптимальное соотношение объемов соседних
секций каскада v,+i,i представляет собой в общем случае не чис-
ло, а функцию п, а, т, i и i+1 и изменяется в пределах от п до
1. При м>1 величина v^+i, г тем меньше, чем ниже конечная сте-
пень превращения исходного вещества, чем больше общее число
секций каскада и чем больше порядковые номера рассматривае-
мой пары секций (т. е. величина v,+i,, убывает по ходу потока ре-
акционной массы). При 0<п<1 величина v,+i, { тем больше, чем
меньше общая степень превращения в каскаде, чем больше число
секций и чем меньше порядковые номера реакций, входящих в
рассматриваемую пару (т. е. в этом случае величина Vi+i, г возра-
стает по ходу потока).
При п = 1 (реакция первого порядка) величина v,+i, г- представ-
ляет собой число, равное единице, т. е. минимальное расчетное
время пребывания реакционной массы в каскаде секций дости-
гается при равновеликих секциях.
Для иллюстрации указанных выше общих закономерностей
приводятся результаты расчетов различных по числу секций кас-
кадов для реакции второго порядка при условии, что конечная
степень превращения исходного вещества во всех случаях равна
ат=1-т12 = 0,77:
Пз 02 V2, 1 — -gj- 03 04 V3, 2 “ "д~ V4, 3 — -д— “г “з
т = 1 . . 0,230 — — — 0,230 — — —
т = 2 . . 0,436 0,528 — — 0,230 1,31 — —
т = 3 . . 0,554 0,619 0,670 — 0,230 1,24 1,20
т = 4 . . 0,631 0,679 0,717 0,748 0,230 1,19 1,17 1,14
Анализируя уравнение (П-246) легко прийти к приближенным
решениям, полученным в работах [24, 27, 28, 36, 49, 50].
В самом деле, когда конечная степень превращения исходного
вещества мала и величина rj2 стремится к единице,..тогда к еди-
нице стремятся также и параметры r)i+i, и уравнение (П-246) име-
ет пределом величину Vj+i, 4 = 1, т. е. соотношение размеров секций
каскада, рассматриваемое в работах [24, 50].
В другом (крайнем) случае, когда конечная степень превра-
щения исходного вещества близка к единице, а величина т]2 стре-
мится к нулю, из уравнения (П-246) получаем vt+i,i = «, т. е.
189
соотношение объемов секций каскада, вытекающее из работ [27,
28,. 36, 49].
На рис. П-47 приведены результаты расчета, выполненного на
вычислительной машине по уравнению (П-239) для реакций вто-
2
рого порядка [51] применительно к каскаду с числом секций 2, 4,
б, дающие возможность видеть зависимость отношения размера
секций от числа секций, порядково-
го номера пары секций и степени
превращения в области высоких
значений а.
Наряду с влиянием относитель-
ного объема секций каскада на ско-
099 0995 09367
оь
4?,
0,9
Рис. П-47. Относитель-
ный объем секций каскада
(схема реакции: А —> D;
порядок реакции — вто-
рой):
1—6 — секции каскада.
рость превращения исходного ве-
щества, выход промежуточного и
конечного продукта последователь-
ной реакции также находится в за-
висимости от значений 0,/0,-ь
Рис. 11-48. Зависимость
Р (0i/02) (схема реакций
А —В -2—> D; поря-
док реакции — первый;
k2Jki =0,5; а = 0,8).
Пусть в каскад, состоящий из двух последовательно включен-
ных секций полного смешения, с постоянной объемной скоростью
подается исходное вещество А, реагирующее по схеме
А—'->В—^->D
описываемой уравнениями реакций первого кинетического порядка.
Задаваясь степенью превращения исходного вещества и изме-
няя отношение объемов секций, можно проследить за изменением
выхода промежуточного и конечного продуктов реакции,
J90
Результаты расчета для случая fe2/fei=0,5 и а=0,80, приведен-
ные на рис. П-48, позволяют видеть, что кривые, выражающие за-
висимость р (62/61), имеют экстремальный характер с точкой экст-
ремума, соответствующей равенству объемов секций. Значения рв
и pD при 02/0i -> 0 и 02/0i -> оо соответствуют условиям реактора
полного смешения.
Наблюдаемая зависимость вполне закономерна, так как ре-
жим, соответствующий минимальному значению 02 в максималь-
ной (для принятого числа секций каскада) степени приближается
к режиму работы аппарата полного вытеснения.
Поиск оптимального соотношения размера секций каскада
оправдывается не только стремлением к увеличению производи-
тельности каскада по исходному веществу реакции, но главным
образом стремлением использовать форму связи 0(a), характери-
зующую реакционный аппарат полного вытеснения.
Влияние числа секций каскада на производительность.
Каскад секций полного смешения является промежуточным зве-
ном между системами идеальных режимов.
Для количественной оценки степени приближения каскада сек-
ций к той или иной идеальной системе может служить значение
критерия Во.
Исследование гидродинамического режима каскада реакторов
полного смешения позволяет выразить [52] значение критерия Во
как функцию числа ступеней каскада *:
2т (т — I) (4т — 3)
В0=-----(2т“1)2---
Зависимость Во(т), почти прямолинейная даже при малом
значении т, при т>100 практически точно прямолинейна и
Во=2ш.
На примере реакции первого порядка степень приближения
каскада равновеликих секций к системам идеального режима
можно оценить величиной отношения продолжительности процесса
в закрытой системе к расчетному времени пребывания реакцион-
ной массы в каскаде секций
три одной и той же степени превращения исходного вещества.
Зависимость т/02(т) выражается семейством кривых, хорошо
иллюстрирующих ход изменения скорости реакции с увеличением
числа секций.
При т —►оо каскад секций становится системой полного вы-
теснения.
1 Зависимость Во(т) может быть выражена более просто: Bo«2(m— 1) [53].
191
Как будет показано ниже, в колонных аппаратах могут быть
созданы условия, соответствующие нецелому числу ступеней пол-
ного смешения, поэтому зависимость r/0s(zn) может выражаться
монотонными кривыми (рис. П-49).
Из рис. П-49 следует также, что эффект, достигаемый увели-
чением числа секций, особенно значителен при переходе от одной
секции к 5—7 секциям. Дальнейшее увеличение числа секций
сопровождается сравнительно небольшим изменением отноше-
ния т/02.
При низкой степени превращения исходного вещества, как это
следует из сопоставления кривых 1 и 3 или 4, высокое значе-
ние т/02 достигается даже при малом числе секций, в то время как
а
° 2 Ч 6 8 10 12 14 16 18 20
т
Рис. П-49. Зависимость Во (т) и т/02 (т, а) (схема
реакции: А—> D; порядок реакции —первый):
а — Во (тУ, б — т/02 (т, а);
кривая .... 7 2 3 4
а............. 0,2 0,5 0,9 0,95
реакция, проводимая с высокой степенью превращения, требует
применения многосекционного каскада.
После рассмотрения вопроса об оптимальном соотношении
расчетного времени пребывания реагирующей смеси в секциях
каскада и вывода в пользу каскада, состоящего из равновеликих
секций, выражения зависимостей, описывающих концентрацию
исходного вещества и продуктов реакции в секциях каскада, не-
сколько упрощаются и для основных типов реакций выглядят сле-
дующим образом.
Простые односторонние реакции (А —D).
Реакции нулевого порядка, скорость которых не зависит от кон-
центрации реагирующего вещества, протекают по одним и тем
же законам в открытых и закрытых реакционных системах. Осу-
ществление их в каскаде секций полного смешения в практических
целях нецелесообразно и рассмотрение закономерностей их ли-
шено смысла.
192
Для реакций первого кинетического порядка в условиях рав-
новеликих секций каскада уравнение (П-236) можно записать
следующим образом:
с, =с. --------------------г (П-248)
Al Al-i 1 +вй, Ло (l+0fei)z
Составление и решение системы уравнений типа (П-236) для
многосекционного каскада приводит к следующим зависимостям
концентрации исходного вещества по секциям:
=_L
СА СА2
сА2 1
СА2 СА>
1
СА
лт-1 _____
г2 = г
.СА С А
лт лт+!
Отсюда легко получить общую зависимость (сЛо= 1):
С • С л , * • •• •С л • С я — С л
А1 л2 Л1 лт
(П-249)
и
с — rilm
CAi~CAm
где i — порядковый номер (начиная с 1-го) секции; т — число
секций в каскаде.
Уравнения (П-249) и (П-250) позволяют определять концен-
трацию (степень превращения) исходного вещества в любой сек-
ции каскада, а также подсчитывать расчетное время пребывания
реагирующей смеси в каждой из секций и в каскаде секций:
(П-250)
02 —
(П-251)
Реакции второго кинетического порядка типа
A-^>D
при установившемся режиме описываются следующими выраже-
ниями:
с4;._1 “ cAt f1 + 0fe2%) <П'252>
или
(П-253)
у1+40/г2сл._1-1
СаГ 20^
Зависимость концентрации в любой секции каскада от кон-
центрации исходного вещества на входе в первую секцию (сл#)
или от его конечной концентрации (сл ) имеет очень сложное, не-
удобное для пользования при практических расчетах выражение.
13 В. С. Хайлов, Б. Б. Брандт 193
Расчет каскада секций для реакции второго порядка произ-
водится последовательным решением уравнений (П-252) и (П-253)
методом последовательного приближения или рассматриваемым
ниже графическим способом.
Обратимые реакции. Зависимости сА (сА ) для различ-
ного типа обратимых реакций представлены в табл. II-12.
Таблица 11-12
Зависимости сА (сл;-) для обратимых реакций
Схема реакции Уравнение скорости
А1 А D *2 klcAi-k2(cA0~cAi')
А1 . 2А D Аг {^icAi + cAt ~ k?cAa
Al 2А s—2D Аг kicAi-k2(cA0~cAl)2
Al,
А 2D Аг klcAi~k2(cA0-cAi)2
1 + 0; I fei + &2
Расчет концентраций исходного вещества по секциям каскада
для обратимых реакций производится последовательным реше-
нием приведенных в табл. П-12 уравнений, начиная с заданной
концентрации.
Параллельные реакции. Процесс в каскаде
равновеликих секций полного смешения, где осуществляется па-
раллельная реакция первого порядка
А
I—-> D
La>f
может быть описан следующим образом:
= сЛг [1+0 (fej + &2)]
САа
СаГ [1 + 0(fe, + fe2)]z
(II-254)
(II-255)
В том случае, когда порядок параллельно текущих превраще-
ний неодинаков (например, реакция АF —второго порядка),
зависимость с(сА ) имеет вид:
[1+0(*1 + *2Ч)] (Н-256)
194
Последовательные реакции. Если в некото-
рых реакциях, рассмотренных выше, концентрация исходного
вещества и продукта его превращения связаны постоянными, не-
зависящими от степени превращения исходного вещества стехио-
метрическими зависимостями и концентрация продукта реакции
может быть определена при использовании уравнений, справедли-
вых для исходного вещества, то для реакций, схема которых вклю-
чает последовательное превращение одного из исходных веществ,
выражение для концентрации промежуточного и конечного про-
дуктов имеет более сложный характер. Рассмотрим этот вопрос
на примере последовательной реакции первого порядка
А —В — -> D
приняв, для простоты, стехиометрические коэффициенты равными
едйнице.
При сЛо = 1,О скорость изменения концентрации исходного ве-
щества может быть выражена через степень его превращения
сА =1— ат. Тогда уравнение (П-223) примет вид:
и
откуда
_ «т-1 + 8^1
т~ 1 + Qki
Скорость изменения концентрации продукта D также можно выра-
зить из условий материального баланса через степень превращения
исходного вещества:
CD CD
~т.9 (П-259)
Делением уравнения (П-259) на уравнение (П-257) получаем:
С Г) с Т\ ~ С Г»
Dm Dm-i . т Dm
--------------- = а—-----------
У-т — ^m-i-----------1
где a=kzlki.
Из уравнения (П-260) нетрудно получить значение
функцию cDm_x, ат и am-i:
О - %) cDm_, + аат (ат - ат-1)
1—dm + a.(dm— dm-i)
(П-257)
(П-258)
(Н-260)
cD как
т
(П-261)
Учитывая, что для рассматриваемого случая (равенство стехио-
метрических коэффициентов) в любой секции каскада
CA + cB + cD== 1
СВт ^ат~ CDm
СВт.Гат-1~СВт_1
13*
195
получаем:
' т/ m m-i
C a = O — С- =-----; ; T----------;--- (11-262)
m Dm 1 — am 4- a (dm ~ ®m-i)
При втором кинетическом порядке каждой из стадий последова-
тельной реакции (сохраняем равенство стехиометрических коэффи-
циентов)
=fe[(1_am)2 (П-263)
и
откуда
аш_1 = ат-0*! (1-ат)2 (П-264)
Скорость изменения концентрации продукта D выразится ана-
логично зависимости (П-259):
(П-265)
(П-266)
(П-267)
имеем:
(П-268)
Деля уравнение (П-265) на (П-263), получаем
— сп — с_ )2
Ozn Рт-\ . д' т
dm ~ ат-1 (1 ~ Я/п)2
откуда нетрудно получить зависимость [50]:
b + Vb2 — 4ес
Сот= 2е
Здесь
е = a (dm — dm-i)
b = 2adm (dm ~ dm-i) + (1 - dm)2
с = аат (ат ~ «m-О + cDm_t (l ~ ат?
Из условий материального баланса (ca'+Zcb+cd= 1)
С Я —— П
Схема расчета и порядок выполнения расчетных операций зави-
сят от состава исходных данных.
Пример П-6. Задано: схема реакции
vBB —> vdD (va = vB = v.D = 1)
кинетический порядок обоих превращений — первый; константы скорости /г> =
=0,004 сек*1, ^2=0,002 се/r1; начальная концентрация исходного вещества
сА = 1,0 кмоль/м3', степень превращения исходного вещества а = 0,956 (сдт =
= 1 — а = 0,044); число секций — 4.
Требуется рассчитать состав реакционной массы по секциям каскада и рас-
четное время пребывания.
196
Решение. 1. Концентрация
кого вещества в секциях каскада:
и степень превращении исход-
1 секция
2 >
3 »
4 »
^.-4:
(0,044)1/4 = 0.455
(0,044)2/4 = 0,209
(0,044)3/4 = 0,094
0,044 (по условию)
сЛг
а, = 1----
1 сАо
1-0,455 = 0,545
1-0,209 = 0,791
1 - 0,094 = 0,906
1 - 0,044 = 0,956
(по условию)
2. Концентрация продукта D в секциях каскада. Расчет ведется по уравне-
нию (П-261) при а = A2/fej = 0,5, Сд0 = 0 последовательно для каждой секции,
начиная с первой.
Результаты расчета:
cD = 0,204 кмоль!м3
cDj = 0,424
сПз = 0,605
cBi — 0,732 „
3. Концентрация продукта В в секциях
каскада. Расчет ведется по уравнению (П-268):
св = — cD = 0,545 — 0,204 = 0,341 кмоль /м3
cDi = 0,791 -0,424 = 0,367
св3 ~ аз ~ cd3 ~ 0>906 — 0,605 = 0,301 „
св = а, - с г, = 0,956 - 0,732 = 0,224
4. Расчетное время пребывания реагирую-
щей смеси в секциях:
в каждой из секций
с, — с. 1 — 0,455
0 = ----— = аааГ а асе = 300 сек
ktcA 0,004 • 0,455
в каскаде секций
02 = тО — 4 • 300 = 1200 сек
Время пребывания реакционной массы в
реакторе периодического действия:
Т= ki 1П 1 —а = 0,004 1П 1-0,956 780 сеК
На рис. П-50 можно видеть общую кар-
тину изменения концентрации продуктов рас-
9,т,с«
Рис. П-50. Зависимости ci (0)
и ci (г) (схеми реакции:
. k\ _. k% _
А----► В —-> D; порядок реак-
ции — первый; fe2/fei = 0,5):
----------реактор полного вытесне-
ния; -----реактор полного сме-
шения; ступенчатые линии —каскад
равновеликих секций;
1—4 — секции каскада.
сматрнваемого процесса в четырехсекционном
каскаде.
Для сравнения полученных результатов с
результатами проведения реакции в закрытой
системе и в аппарате полного смешения на
рис. П-50 приведены кривые (сплошные линии — закрытая система; пунктирные
линии — реактор полного смешения), полученные расчетом для условий рас-
сматриваемого примера.
197
Рис. П-51. Схема графиче-
ского расчета состава реак-
ционной массы в каскаде
секций полного смешения
(схема реакции: А —> D:
порядок реакции — первый).
в исходной смеси (при cDa = v —
Рис. П-52. Схема графического расчета
состава реакционибй Массы в каскаде
секций (две секции) полного смешения
(схема реакции: А —> В —> D; порядок
реакций — первый).
Графический способ расчета каскада секций. Рассмот-
ренный ранее принцип графического расчета состава реакционной
массы в открытых системах полного сме-
шения (см. стр. 171) применим и для
каскада секций полного смешения.
Для односторонней реакции типа А->
—>D независимо от ее кинетического по-
рядка наиболее простой вариант расче-
та основан на заданной начальной или
конечной концентрации исходного веще-
ства, числе секций и расчетной продол-
жительности пребывания реакционной
массы в каждой из секций (значения 0,
т задаются либо исходя из предвари-
тельных расчетов, либо принимаются
на основе аналогий, опытных данных
и т. п.).
В случае, если заданной является на-
чальная концентрация исходного веще-
ства сАо, из точки, лежащей на абсциссе и соответствующей на-
чальному содержанию продукта ~
из начала координат), прово-
дится прямая, тангенс угла
наклона которой коси абсцисс
равен 1/0, до пересечения с
кривой Wd(cd) в точке 1 (рис.
П-51).
Абсцисса точки 1 выража-
ет концентрацию продукта D
в первой секции; смесь тако-
го же состава поступаем во
вторую секцию.
Прямая, выражающая ско-
рость изменения концентрации
продукта D во второй секции
и параллельная прямой 0—1
(01 = 02=0з=. •), проводится
из точки 2, лежащей на оси
абсцисс (так как при cD = cDi2
скорость цу = О), до пересече-
ния с кривой wD(cD).
Абсцисса точки 3 выражает
концентрацию продукта D во
второй секции.
Многократное (соответствен-
но заданному числу секций)
повторение этой операции дает возможность определить конечную
концентрацию продукта D в последней секции каскада [34].
198
В том случав( когда заданным является значение концентрации
продукта D в последней секции каскада, построение производится
справа (из точки 4) налево.
Если заданы начальная и конечная концентрации продукта
и число секций каскада, искомой величиной является расчетное
Рис. П-53. Графический расчет состава реакционной массы
в каскаде секций полного смешения (схема реакции:
А—>В—>D).
время пребывания реакционной массы в каждой из секций. Ре-
шение задачи достигается многократным построением ломаной
линии (как показано на рисунке) до совпадения прямой, соответ-
ствующей последней секции каскада, с точкой на кривой wD(cD),
абсцисса которой равна сп .
ит
Для последовательной реакции А -* В -*• D при заданных сте-
пени превращения исходного вещества и числе секций, поступая,
как указано выше, строят ломаную линию и определяют абсциссы
точек пересечения прямых св/0 с кривой wB(cB), соответствующие
максимально возможной концентрации продукта В.
199
Для определения концентрации продукта D в каждой из секций
точки 2 и 4 (рис. П-52) проектируют на нижнюю ось абсцисс и
перемещают линию wD(cD) влево по этой оси к полученным точ-
кам 2' и 4'. Концентрация продукта D в секциях определяется по-
строением линий 0 — I и 11 — III, которые параллельны прямым
О — /и 2 — 3.
Концентрация продукта В по секциям определяется как раз-
ность между сп и cD для каждой секции соответственно.
Для иллюстрации метода выполнен расчет рассмотренного
выше (см. пример П-6) процесса в четырехсекционном каскаде.
На рис. П-53 можно видеть построение и полученные результаты,
практически равные результатам аналитического расчета.
Аппарат с продольным перемешиванием
Наряду с каскадом секций полного смешения, условия
промежуточного (между идеальными) режима имеют место в реак-
ционном пространстве,, вдоль оси которого непрерывно осуще-
Рис. П-54. Схема аппаратов с циркуляцией
реакционной массы.
Химическая реакция протекает в
ствляется перемешивание
реакционной массы.
Подобные условия со-
здаются в барботажных ап-
паратах, в реакторах с
псевдоожиженным слоем
твердых частиц, а также в
реакционных аппаратах,
снабженных мешалками или
циркуляционными насосами.
Для удобства рассмот-
рения этого сложного во-
проса процесс смешения ис-
ходного вещества со смесью
продуктов реакции можно
представить так, как пока-
зано на рис. П-54,
кольцевом пространстве по
законам, справедливым для аппаратов полного вытеснения; часть
реакционной массы, выходящей из кольцевого пространства и
представляющей смесь непревращенного исходного вещества (сА )
с продуктами его превращения, выводится из реакционного аппа-
рата для дальнейшей переработки. Остальная часть реакционной
массы погружным насосом или установленным снаружи циркуля-
ционным насосом непрерывно подается в нижнюю часть реактора,
где мгновенно и полно смешивается с исходным веществом, не-
прерывно вводимым в нижнюю часть реактора (концентрация
исходного вещества в вводимой смеси — сЛо).
200
Вводя понятие коэффициента кратности циркуляции (гц) как
отношение объема смеси продуктов, возвращаемых в нижнюю
часть аппарата, к объему исходной смеси [40], можно выразить
концентрацию исходного вещества на входе в реакционное про-
странство как функцию коэффициента кратности циркуляции:
гисА + СА,
. = г = ц Л'О
Гц + 1
(П-269)
Крайние пределы возможных значений гц (0-Сгц<оо) соот-
ветствуют условиям идеальных режимов:
'ц=°; члч
гц-*°°; слн^Ч
Промежуточные значения коэффициента кратности циркуляции
характерны для различных вариантов промежуточного режима
А = С А *
Лн Лц
В этих условиях продолжительность превращения исходного
вещества в кольцевом пространстве описывается уравнением, спра-
ведливым для аппарата полного вытеснения в пределах от
Для реакции первого кинетического порядка это выражение
после интегрирования и замены с, его значением имеет вид:
ЛЦ
. г „с . + с .
т ' 1„ Д ~^K А>
Т = — 1П
k (ГЦ+1)СЛК
(П-270)
(П-271)
Принимая сА — 1 и заменяя сА его значением (сА
уравнение (П-270) можно записать следующим образом:
Т = 11П М1-«) + 1
k 1П (гц+1)(1-а)
Средняя скорость реакции в кольцевом пространстве выражает
среднюю скорость превращения исходного вещества от концентра-
ции сЛо до концентрации сА , что дает возможность выразить рас-
четное время пребывания исходного вещества в реакционном ап-
парате:
С л ~~ с А Г-4-1 г (1 — а) + 1
САЯ~САК * (гц+1)(1~й)
(П-272)
201
Степень приближения гидравлического режима работы реак-
ционных аппаратов к работе реакторов идеальных режимов, ха-
рактеризуемая значением критерия Во, может быть оценена также
значением коэффициента кратности циркуляции. Последний спо-
соб оценки в ряде случаев может оказаться более доступным, учи-
тывая возможность расчета значения гц, например, в случае при-
менения схемы рис. 11-54, б.
Пусть реакция А—»В первого кинетического порядка прово-
дится в реакторе с внутренней циркуляцией и в каскаде секций
Рис. П-55. Зависимость га(т) и Во (т):
1 - а = 0,95; 2 - а = 0,80; 3 - а = 0,05.
полного смешения при одинаковых для обоих случаев (и равных
единице) концентрациях исходного вещества (сЛо), одинаковых зна-
чениях с. и расчетного времени пребывания (0х = 0ц). Эти уело-
вия дают основание приравнять выражение (П-251) к выражению
(П-272):
- ‘]-^+ >'" <"-2га>
Результаты числового решения уравнения (П-273) в форме
зависимости гц(ш) для ряда значений а представлены на
рис. П-55.
При малом значении степени превращения эффективность ап-
парата с мешалкой становится практически эквивалентной одной
секции полного смешения уже при гц~ 10 (см. кривую 3).
Для процессов, проводимых при высоком значении а, харак-
терна менее заметная зависимость степени приближения к идеаль-
ному режиму от интенсивности продольного перемешивания. На-
пример, при степени превращения исходного вещества, равной 95%
202
(а=0,95), даже 15-кратное разбавление исходного вещества про-
дуктами реакции не достигает эффекта, соответствующего идеаль-
ному режиму полного смешения.
ВЫХОД ПРОДУКТОВ МНОГОСТАДИЙНЫХ РЕАКЦИЙ
В ОТКРЫТЫХ СИСТЕМАХ
Если выход продуктов одностадийных реакций как в за?
крытой, так и в открытых системах не связан со степенью превра-
щения исходного вещества, то для сложных реакций связь выхода
промежуточного и конечного продуктов с а, отмечающаяся ранее
при проведении их в закрытых системах, сохраняется и в откры-
тых системах.
Системы полного вытеснения
Выход продуктов сложных (параллельных, последова-
тельных, параллельно-последовательных и т. д.) реакций как функ-
ция степени превращения исходного вещества сохраняет форму,
установленную для закрытых систем, при одинаковом кинетиче-
ском порядке отдельных превращений, независимо от соотношения
числа молей исходных веществ и продуктов их превращения.
Для сложных реакций неодинакового кинетического порядка,
осуществляемых в открытых системах вытеснения, разница в
числе исходных веществ и продуктов реакции существенно сказы-
вается на выходе продуктов лишь при высокой степени превраще-
ния исходных веществ и в отсутствие инертных разбавителей
(см. стр. 153).
Системы полного смешения
Рассмотрим кинетику реакций
А -В —D F
(D
(II)
при условии, что критерий Боденштейна (Во) значительно меньше
единицы.
Изменение концентрации исходных веществ и продуктов реак-
ции в зависимости от среднего времени пребывания реакционной
массы в аппарате описывается системой алгебраических урав-
нений:
для реакции (I)
(П-274)
203
для реакции (II)
СЛ,~ СА
о
£ D „
_Д-с А /Л
0 RlcA
При введении переменных р и а системы уравнений
и (П-275) принимают вид:
! ат
а7Г^₽в+₽в-1=о
пр пт
ь₽’Л(Г^^ + ^-“(Г^₽в=О
₽в= 1+а(1— а)т-п
(П-275)
(П-274)
(П-276)
(П-277)
где а = -т2- с’”~п; Ь==^~ ср~п.
«1 Ao ’ k\ Ло
Для выяснения влияния продольного перемешивания реагентов
на выход продуктов реакции в непрерывнодействующих аппаратах
сопоставим уравнения (П-173) и (П-273), (П-174) и (П-277):
для последовательных реакций
' «т т
а^ + ай-^Г₽в+₽в-1 = <>
а7Г^)й^ + ₽в-1 = 0
«Ко <•’ , «" . (1,‘278’
“ da +b (1—а)" +Pd а(1—а)лРв—°
6 (1 _ а)« + ₽£>, ~ а (1-а)Л 0в. = 0
для параллельных реакций
а-Г5. + й-------------= О
da в 1 +а (Д-а)тп (П-279
йв-------------= 0
В‘ Л + а{1-а)т~п
Здесь индексом «1» отмечены выходы продуктов в аппаратах
полного смешения.
Вычитая попарно уравнения (П-278) и (П-279), получим:
а^ + а7Г^^~^) + (₽в-₽В.)“° (П-280
a~d^+b (Г^Г (₽Б-₽S.)+ (рл-ы-а(рв -₽ВJ“° (И-281 ’
а^-+(Р-₽1)“0 (П-282)
204
Выход промежуточного продукта В последовательной реакции (1)
не может возрастать с увеличением степейи превращения. Следо-
dp»
вательно, всегда а < 0 и в уравнении (П-281)
ат
+ (П-283)
Длц удовлетворения неравенства (П-283), учитывая, что а ц_ayi
всегда положительно, необходимо, чтобы — 0В1>О.
Следовательно, выход первого промежуточного продукта по-
следовательных реакций, проходимых в аппаратах полного сме-
шения, всегда меньше, чем в аппаратах полного вытеснения.
Влияние продольного перемешивания реагентов на выход вто-
рого промежуточного продукта становится уже неоднозначным,
как это следует из уравнения (П-281).
Экстремальный характер функции рд(а) позволяет записать
неравенства:
dpc _ g,n (ат _ от\ > п
Таким образом
rtP
откуда следует, что выход продукта D в аппарате полного сме-
шения может быть как меньше, так и больше, чем в аппарате пол-
ного вытеснения, т. е.
Рд
в зависимости от степени превращения.
Для параллельных реакций, в зависимости от кинетических
параметров процесса (величина константы скорости и порядков
реакции), выход целевого продукта может как возрастать с уве-
личением степени превращения, так и понижаться. Следовательно,
производная в уравнении (П-282) может быть как больше,
так и меньше нуля:
p-Pi^O
Зависимость выхода промежуточных и конечных продуктов
более сложных реакций от степени превращения в аппаратах пол-
ного смешения может быть получена при использовании соотно-
шения из уравнений, представленных в табл. П-13.
Для обратимой последовательной реакции первого порядка
205
Таблица 11-13
Зависимость Р.(а) для различных реакций
Схема реакции Порядок реакции ₽(- (а.
А —> В _£*-> D 1 я 1—« й 1 + аа — а я аа Рд 1 + аа — а
А —> В —2> D F 1 й 1-« й 1+аа —а й = аа(1—«) р° (1 + аа— а) (1 + Ьа — а) й aba2 Рр (1 + аа — а) (1 + Ьа — а)
А В —> D 2 я = С1 ~«) (« + У~ 1) 2аа2 о _ 2аа2 — (1 — а) (а + у — 1) рО 2аа2
А —* В —D 1 > F 1 я _ 1 -« Р* (6 + 1) (1 - а) + аа й аа pD (b + 1) [(6 + 1) (1 - а) + аа] ft b pF l + b
д _*!-> В —D / 1—F 1 о 1-« . рв 1 + а(а + /> - 1) аа 1+а (а+ 6 — 1) о Ьа pF 1 + а(а + 6-1)
А в D 1 + 1 *3 1 1 о 1~« рв (6 + 1) (1 — а) + аа b-a(b-a)
PD~ (Ь + 1) (1 — а) + аа
k\ А В D *3 kf Примечание: я “ k?lki, b - „ Al 1—а Б=ТГ— +к" 1 Лз/fej; У"1 Рд = 1 — Рд /(1-а)2 + 4аа2;Е = -^-.1—2- + 4^ + 1 +-^- Лз “ *з «з
206
зависимость сДа) имеет очень сложное и неудобное для пользо-
вания выражение. Более простым является вывод, основанный на
анализе уравнений скорости при условии cBo = z?Do = O:
= ' <n-284>
-^-= kAcD — (k2 + fe3) св (П-285)
Заменяя сА, св и cD их значениями
СА = сАо ~ а)> СВ = сЛоаРв> CD= CA^D
и исходя из того, что Pb+Pd=1, после деления левой и правой
частей уравнений (П-284) и (П-285) на сЛа имеем:
-g- = fej (1 — а) — й3а₽в
а₽я
g- — (1 — <х) + k^a (1 — ₽в) — (fe2 + fe3) afjg
Исключение 0 из обоих уравнений приводит к выражению, свя-
зывающему выход продукта В и степень превращения исходного
вещества:
₽В-₽в(-Г--—- + -Т- + -Т-+1}+-^~£+ф- = ° (П-286)
\ fe3 a k3 k3 ) k3 a k3
Характер зависимостей, приведенных в табл. П-13, можно ви-
деть на рис. П-56.
Для иллюстрации особенностей открытой системы полного сме-
шения на рис. П-56 дана также зависимость рДа) для условий
закрытой системы.
Как следует из рис. П-56 качественный характер зависимо-
сти Pi (а) в открытой системе полного смешения аналогичен ха-
рактеру ранее рассмотренной зависимости для открытых систем.
Количественная разница в выходе промежуточного продукта (для
открытой системы выход снижается) и конечного продукта (по-
вышение выхода) является естественным следствием условий
полного смешения, при которых концентрация промежуточного
продукта в реакционной зоне относительно выше, чем в закрытой
системе. Этим объясняется и разница в величине максимального
выхода продукта D при проведении трехстадийной последователь-
ной реакции в закрытой и открытой системах (см. рис. П-56,б).
Кривая выхода продукта D в открытой системе имеет более кру-
той наклон, чем в закрытой, что и определяет положение экстре-
мальной точки кривой pD(a) на рис. 11-56,6 как по абсолютному
значению выхода, так и по степени превращения, соответствую-
щей достижению этого максимального значения.
Для обратимой последовательной реакции в условиях аппа-
рата полного смешения имеет место предел превращения исход-
ного вещества, определяемый значением ар.
207
a
6
Рис. П-56. Зависимость Р/(а) присАо = 1,О для открытой (штри-
ховые линии) и закрытой (сплошные линии) реакционных систем:
Рисунок Схема реакции Кинети- ческий порядок реакции *2/*l ki/kt
а aA->b-^>d 1 0,1; 2; 10 —
б А В—->D—->F I 2 3 —
в a2l_>b-^>d 2 0,1; 1 — —
г a2l.>b-^->d L-*3_->F 1 1 1 -
д a2l^b^d 1 *з t 1 1 1 -
е A —В — -> D L*3.^f fel „ *2 . 1 1 1 -
ж AT B, D *3 *4 1 1 1 1
Значение ар нетрудно определить, приравнивая нулю скорость
изменения концентрации вещества А и продукта В — уравнения
(П-284) и (П-285)—и решая полученные выражения относи-
тельно а=ар:
а *Ж+1)
где /С' = k\lk3’ К" = — константы равновесия первой и второй
стадии последовательной реакции.
' Для случая, изображенного на рис. П-56, ж (равенство кон-
стант К' = Кр = 1)> равновесная^ степень превращения исходного
вещества составляет:
9
“₽=Ж=0'667
Выход продуктов В и D при достижении равновесной степени
превращения составит:
Рвр = ₽ор = 0-5
Системы промежуточного типа
Занимая промежуточное положение между системами
идеальных режимов по значению критерия Боденштейна, каскад
секций полного смешения и реакторы с продольным перемеши-
ванием характеризуются зависимостью выхода продуктов слож-
ных реакций не только от степени превращения исходных веществ,
но и от интенсивного продольного перемешивания, т. е. от числа
секций каскада т или от кратности циркуляции реакционной
массы Гц. Это можно проследить на примере последовательной
реакции первого порядка А—»В—»-D.
Изменение концентрации промежуточного продукта В по рав-
новеликим секциям каскада описывается системой уравнений:
ся, , ekicA,
СВ' l + 0fe2 + l+0fe2
_ cBt . ®kicA2 CBo , 6*1СЛ , ®klcA2
В* l+0fe2 l+0fe2 (1+0fe2)2 + (1+0fe2)2 + 1+0fe2
„ _ СВ2 , вАг1сЛ, сВа Qk\cAx , QklcA2 0felcA,
в> l+0fe2 l+.0fe2 (l+0fe2)3 + (l+0fe2)3 + (l+0fe2)2 l+0fe2
CAt CA2 cA, ,
+ (i + 0fc2)m’1 (i- Qk1)m~2
, = 1 и заменяя cA^, cA и 6 их соответствую-
Cd
- ” (l+0fe2)m +
Приняв cR = 0, c.
D0
щими выражениями (см. стр. 193)
14 В. С. Хайлов, Б. Б. Брандт
209
нетрудно получить выражение выхода продукта В в потоке, по-
кидающем последнюю секцию каскада:
^ОТ = "ДГ = '—(П-287)
где
V = (l — a)1/m(l — а) + а
При т=\ выход продукта В описывается ранее полученным
уравнением для реактора полного смешения
р =
1 — а + аа
а при m-*Q для выражения выхода продукта В справедливо
уравнение, характеризующее закрытые системы.
При отсутствии количественно сформулированных кинетиче-
ских зависимостей для определения выхода продуктов сложных
реакций в каскаде секций могут быть использованы результаты
графического расчета состава реакционной массы (см. стр. 198).
В качестве иллюстрации в табл. П-14 приводятся результаты
Таблица 11-14
Результаты расчета выходов продуктов
последовательной реакции
Секции СО ' а Выход продуктов В и D при
четырехсекционный каскад открытая система полного смешения закрытая система
В D в D в D
I 0,545 0,626 0,374 0,626 0,374 0,810 0,190
II- 0,791 0,466 0,534 0,342 0,658 0,630 0,370
III 0,906 0,334 0,666 0,172 0,828 0,470 0,530
IV 0,956 0,236 0,764 0,084 0,916 0,347 0,653
расчета выхода продуктов В и D последовательной реакции, рас-
смотренной в примере П-6. Для оценки эффекта, достигаемого
при проведении реакции в каскаде секций, в табл. П-14 приве-
дены результаты расчета для систем идеальных режимов.
Для оценки влияния интенсивности продольного перемешива-
ния на выход продуктов последовательной реакции могут служить
зависимости (П-273) и (П-287). Заменяя т в уравнении (П-287)
его значением т(гц), определяемым графически из уравнения
(П-273), можно получить графическое выражение зависимостей
рв(гц, а) и Рд=1-рв(гц, а).
210
ЛИТЕРАТУРА
1. М. X. К а р а п е т ь я н ц, Химическая термодинамика, Госхимиздат, 1953.
2. F. R. В i с h о w s к у, F. D. Rossini, The Thermochemistry of Chemical
Substances, New York, 1936.
3. А. И. Темкин, Л. А. Шварцман, Усп. хим., 17, № 2 (1936).
4. Л. А. Вайнштейн, Б. М. Яворский, ДАН СССР, 89, 813 (1950).
5. S. А г г h е n i u s, Z. phys. Chem., 4, 226 (1889).
6. В. П. Кондратьев, Кинетика химических газовых реакций, Изд. АН
СССР, 1958.
7. J. R. Hulett, Quart. Revs London Chem. Soc., 18, № 3, 227 (1964).
8. Г. К. Боресков, M. Г. Сл и нько, ЖПХ, 16, 377 (1943).
9. M. В о d e n s t e i n, Z. phys. Chem., 85, 329 (1913).
10. Д. А. Франк-Каменецкий, Диффузия и теплопередача в химиче-
ской кинетике, Изд. «Наука», 1967.
11. В. А. Диткин, П. И. Кузнецов, Справочник по операционному
исчислению, Гостехиздат, 1951.
12. Leveque, Ann. Mines., (12), 13, 201, 305, 381 (1928).
13. Я. Б. Зельдович, ЖФХ, 13, 163 (1939).
14. Т h i е 1 е Е. W., Ind. Eng. Chem., 31, 916 (1939).
15. В. Г. Л е в и ч, Физико-химическая гидродинамика, Физматгиз, 1959.
16. Б. Б. Брандт, Е. Ш. Ш е х т м а н, Д. И П е р а з и ч, Сборник трудов
2-го Всесоюзного совещания по жидкостной экстракции и хемосорбции, Изд.
«Химия», 1966, стр. 68.
17. Б. Б. Брандт, Химия и технология азотных удобрений. Труды ГИАП,
г. XXII, вып. «Процессы и аппараты», 1969.
18. М. X. Кишиневский, Автореф. докт. дисс., МХТИ им. Д. И. Менде-
леева, 1955.
19. Э. Меделунг, Математический аппарат физики, Физматгиз, 1961,
стр. 315.
20. Т. Карме н, М. Био, Математические методы в инженерном деле,
Гостехиздат, 1948, стр. 66.
21. * М Boden stein, К. W о 1 g a s t, Z. phys. Chem., 39, 722 (1907).
22. R. McMullin, M. Weber, Trans. Am. Inst. Chem. Eng., 31, 409
(1935).
23. H. И Кириллов, ЖПХ, 13, 978 (1940).
24. A. H. П л а н о в с к и й, Хим. пром., № 5, 5; № 6, 5 (1944).
25. К. G. Denbigh, М. Hicks, F. М. Page, Trans. Faraday Soc., 44,
№ 307, 479 (1948).
26. J. M. Smith, Chemical Engineering Kinetics, New York, 1956.
27. W. В г о t z, Grunoriss der Chemischen Reactiontechnik, Weinheim, 1958.
28. С. В э й л а с, Химическая кинетика и расчеты промышленных реакторов,
Изд. «Химия», 1967.
29. X. Крамере, К. В е с т е р т е р п, Химические реакторы, расчет и
управление ими, Изд. «Химия», 1967.
30. Р. Арис, Анализ процессов в химических реакторах, Изд. «Химии»,
1967.
31. Я. М. Браннее, Введение в теорию и расчеты химических и нефте-
химических реакторов, Изд. «Химия», 1968.
32. Г. М. П а и ч е н к о в, ЖФХ, 22, 209 (1948).
33. П. М. П а н ч е н к о в, В П . Л е б е д е в, Кинетика и катализ, Изд. МГУ,
1961.
34. К. Н. Крылов, С. Ф. Шабалин, Хим. пром., № 10 (1944).
35. R. W. Jones, Chem. Eng. Progr., 47, 46 (1951).
36. Г. H. Богачев, Хим. пром., № 4, 152 (1962).
37. И. М. Колесников, ЖФХ, 37, № 10, 283 (1963).
38. А. И. Т е м к и н, Кинетика и катализ, 3, № 4, 509 (1962).
39. К- Хартманн, П. В Пас сет, ЖПХ, 37, № 12, 2662 (1964).
40. Д. И. О р о ч к о, Теоретические основы ведения синтеза жидких топлив,
Гостоптехиздат, 1951, стр. 146,
14*
211
41. А. П. Зиновьева, Д И. Орочко, Хим. и технол. топлив и масел,
№ 11 (1959).
42. А. П. Зиновьева, Д. И. Орочко, Кинетика и катализ, 3, № 3, 385
(1962).
43. А. П. Зиновьева, Д. И. Орочко, Хнм. и технол. топлив и масел,
№ 3 (1966).
44. В. Г. Горский, Хим. и технол. топлив и масел, № 12, 34 (1967).
45. I. G. V u s s е, Van de Н. Voether, Chem. Eng. Sci., 14, 90 (1961).
46. В. H. M e s s i k о m m e r, Proc. Int. Seminar Analogue Computation
Applied to the Study of Chem. Proc., Brussels, 1960.
47. В. Г. Горский, А. И. Бояринов, В. А. Гончаров, Тезисы докла-
дов на III Межвузовской конференции по проблеме «Непрерывные процессы и
аппараты производства полимерных материалов», изд. МИХМ, 1967, стр. 5
48. В. Г. Горский, В. Н. Мельников, В. 3. Б р о д с к и й, ЖПХ, 40,
№ 8, 1527 (1967).
49. К- G. D е n b i g п, Trans. Faraday Soc., 40, 352 (1944).
50. A. H. П л а н о в с к и й, Д. А. Гуревич, Аппаратура промышленности
полупродуктов и красителей, Госхимиздат, 1961, стр. 47.
51. К. Хартманн, Б. В. П а с с е т, И. С. П а в л у ш е н к о, ЖПХ, 37, 838
(1964).
52. Р. Т г о m b о u s е, Canie chimique, 84, № 6, 189 (1960).
53. В. В. Д и л ь м а н, Автореф. докт. дисс., МИХМ, 1968.
Ашмор П., Катализ и ингибирование химических реакций, Изд. «Мир»,
1966.
Б еисон С., Осноиы химической кинетики, Изд. «Мир», 1964.
Заградник В., Усп. хим., 30, 1272 (1961).
Захарьевский М. С., Кинетика и катализ, Изд. ЛГУ, 1963.
К и п е р м а н С. Л., Введение в кинетику гетерогенных каталитических реак-
ций, Изд. «Наука», 1964.
Лейдлер К-, Кинетика органических реакций, Изд. «Мир», 1966.
П а и ч е н к о в Г. М., Лебедев В. П., Химическая кинетика и катализ,
Изд. МГУ, 1961.
Рейхсфельд В. О. и др., Кинетика и катализ, 4, № 3 (1963).
Рогидии П. М., Р о г и д н и а Э. Н., Последовательные химические реак-
ции, Изд. АН СССР, 1960.
Р о й т е р В. А., Введение в теорию кинетики и катализа, Изд. АН УССР,
1962.
Темкин М. Н., Кинетика сложных реакций, Труды Всесоюзной конферен-
ции по химическим реакторам, т. IV, 1966.
Химическая кинетика и цепные реакции, Изд. «Наука», 1966.
Эммануэль Н. М., Кнорре Д. Г., Курс химической кинетики, Изд.
«Высшая школа», 1962.
Cher min Н. A. G„ Van D. W. К rev el in D. W., Chem. Eng. Sci., 14, 58
(1961).
J u n g e г s J. C., Cinetique Chemique appliquee, Paris, 1958.
Ju n gers J. C., Sa Ju s L., Rev. Inst, franc. petrole, 20, № 10, 1531 (1965);
Экспресс-инф. Пром, органич. синтез, № 9, изд. ВИНИТИ, 1966.
Schneider Р., Kraus М., Chem. Listy, 55, 1358 (1961),
Глава III
ВСПОМОГАТЕЛЬНЫЕ ПРОЦЕССЫ
И АППАРАТЫ
Химическое производство потребляет разнообразные по
природе и агрегатному состоянию основные и вспо-
могательные исходные вещества. Столь же разнообразны по свой-
ствам и агрегатному состоянию промежуточные продукты про-
мышленного органического синтеза, используемые как исходные
вещества при переработке сырья по многостадийной химической
схеме.
Комплекс процессов и аппаратов, составляющих первую, под-
готовительную, стадию технологической схемы, предназначен для
хранения основных и вспомогательных исходных веществ промыш-
ленного синтеза (или промежуточных продуктов, передаваемых
на последующую переработку), транспорта этих веществ по за-
водской территории и внутри цеха, а также для изменения пара-
метров их состояния перед поступлением в реакционный агрегат.
Подготовительная обработка сырья требует затрат (иногда
значительных) энергетических средств и сопровождается неизбеж-
ным расходом исходных веществ органического синтеза в резуль-
тате разнообразных потерь.
Энерго-материальные затраты, связанные с подготовительной
обработкой сырья, оказывают заметное влияние на экономические •
показатели производства. Снижение удельных энерго-материаль-
ных затрат на этой стадии технологической схемы составляет
одну из задач химической технологии.
ХРАНЕНИЕ И ТРАНСПОРТ СЫРЬЯ И ПРОДУКТОВ
Значительная часть стоимости технологического обору-
дования химического производства приходится на долю аппаратов
и машин, предназначенных для хранения жидких и газообразных
веществ и транспорта их по заводской территории ц в границах
цеха или установки. Для хранения таких веществ используются
различные по устройству и размеру резервуары, а транспорт осу-
ществляется по трубопроводам с помощью разнообразных по
принципу действия и конструкции машин.
213
ХРАНЕНИЕ ЖИДКИХ И ГАЗООБРАЗНЫХ ВЕЩЕСТВ
Хранение жидких и газообразных веществ производится
на складе предприятия или на складах, обслуживающих группу
цехов, а также на промежуточных складах и в цеховых храни-
лищах.
Размеры и устройство заводского склада рассчитаны на бес-
перебойное снабжение предприятия всеми видами основного и
вспомогательного сырья и подсобных материалов и на возмож-
ность единовременного приема партий этого сырья, поступающего
извне. Промежуточные склады, входящие в состав группы цехов,
объединяемых общностью технологического процесса, служат для
хранения сырья, а также для сбора, хранения и распределения
по цехам промежуточных продуктов производства и реакционных
смесей, передаваемых из цеха в цех.
Техническое оснащение складов, предназначенных для хране-
ния жидких веществ, обусловливается способом транспорта жидко-
стей на склад (трубопроводы, железнодорожный или водный
транспорт), а также степенью их горючести и токсичности. Орга-
низация складов жидких веществ является сложной задачей, рас-
сматриваемой в ряде специальных работ [1—3] и регламентируе-
мой особыми нормами и техническими условиями [4].
По степени горючести жидкости разделяются на негорючие,
горючие (температура вспышки>45°С) и легковоспламеняющиеся
(температура вспышки<45°С).
Нормы хранения жидкого сырья на складах и в производст-
венных помещениях установлены с учетом категории горючести
жидкого продукта и способа размещения хранилища. Максималь-
ное количество горючей жидкости, допускаемой к хранению в под-
земных резервуарах, расположенных вне здания, составляет
10 000 м3; в резервуарах, расположенных в производственных по-
мещениях,— до 300 м3.
Для хранения жидкостей используются разнообразные по
форме, размеру и материалу резервуары, которые различаются
по способу их расположения относительно низшей планировочной
отметки производственной площадки на подземные, полуподзем-
ные и наземные, устанавливаемые внутри и вне производственных
помещений.
Резервуары для хранения горючих и легковоспламеняющихся
жидких веществ должны быть несгораемыми, устанавливаться на
несгораемом основании и быть покрытыми изоляцией из негорю-
чего материала.
Наряду с металлическими резервуарами для хранения вязких
органических жидкостей на крупных складах применяются желе-
зобетонные, кирпичные и каменные резервуары, а также резер-
вуары из синтетических материалов [5, 6]. Как правило, неметал-
лические резервуары бывают заглубленными, т. е. подземными
или полуподземными, и только в отдельных случаях применяются
214
наземные неметаллические резервуары. Для хранения нефтепро-
дуктов в неметаллических резервуарах наиболее часто исполь- .
зуются железобетонные сооружения, емкость которых может до-
стигать 30 000 м3.
В промышленности органического синтеза наибольшее распро-
странение получили стальные (из углеродистой и легированных
сталей) резервуары, выполняемые в виде горизонтальных и верти-
кальных цилиндрических сварных сосудов, емкость которых может
достигать 2000 м3, и рассчитанные чаще всего на давление (избы-
точное) до РИЗб=16 ат (емкость до 100 .и3) и температуру до
200° С. Для хранения низкокипящих жидкостей и сжиженных га-
зов находят применение каплевидные и сферические резервуары,
отличающиеся наименьшим удельным расходом металла. Сфери-
ческая форма резервуара позволяет сооружать хранилища значи-
тельных размеров (до 800 .и3), рассчитанные на давление
Ризб=Ю ат.
При хранении на складе и при транспорте жидкостей по тру-
бопроводам неизбежны потери, происходящие в результате утечки
жидкостей из неплотностей в разъемных соединениях и местах
сварки аппаратов и трубопроводов, из сальников насосов, а также
в результате испарения жидкостей со свободной поверхности в
хранилищах и т. д. Эти потери вызывают расход сырья на всех
стадиях его переработки, начиная с поступления на склад пред-
приятия. Относительный размер потерь зависит от величины по-
тока перерабатываемого сырья и во всех случаях определяется
качеством изготовления и монтажа аппаратуры и трубопроводов,
а также культурой обслуживания.
Для оценки порядка величин потерь жидкого сырья в резуль-
тате утечки могут служить данные, относящиеся к нефтепродуктам
(табл. Ш-1).
Таблица 111-1
Потери жидких нефтепродуктов
в результате утечки [1]
Характер утечки Потери, л!месяц
Одна капля в секунду .......................
Капли, временами переходящие в струю........
Струя нефтепродукта прн давлении 1 ат:
диаметр струи 3,2 мм .................
» » 4,8 мм........................
До 130
До 200
До 25 000
До 40 000
Потери жидкостей в результате испарения происходят при на-
полнении и опорожнении цистерн через открытый люк, при запол-
нении резервуаров, газовое пространство которых сообщается с
атмосферой, и т. п. Для хранилищ, расположенных вне зданий,
величина потерь на испарение зависит от климатических условий
215
и времени года; значительное влияние при этом на величину по-
терь оказывает степень заполнения резервуара (табл. Ш-2).
Таблица 111-2
Потерн жндкого сырья в результате нспарёння [1]
Заполнение емкости, % Годовые потери, % от объема резервуара Заполнение емкости, % Годовые потери, % от объема резервуара
в среднем климатиче- ском поясе в южном климатиче- ском поясе в среднем климатиче- ском поясе В южном климатиче- ском поясе
90 0,3 ' 0,4 60 1,6 2,3
80 0,6 0,9 40 3,6 5,2
70 1,0 1,5 20 9,6 13,9
Потери на испарение уменьшаются с увеличением размера
резервуара вследствие большей тепловой инерции последнего и
более благоприятного соотношения площади испарения к объему
жидкости.
Данные о потерях некоторых жидких нефтепродуктов в ре-
зультате испарения приведены в табл. Ш-3.
Таблица 111-3
Предельные нормы потерь нефтепродуктов
прн хранении в закрытых емкостях
(в кг на 1 м2 поверхности испарения в месяц) [2]
Жидкости Южная зона Средняя зона Северная зона
осень — зима весна — лето осень — зима весна w— лето осень — зима весна — лето
Бензин авиационный Керосин осветитель- 1,94 4,99 1,3 4,8 1,37 3,95
ный 0,06 0,22 0,04 0,17 0,04 0,13
Дизельное топливо 0,03 0,11 0,02 0,08 0,02 0,06
В целях уменьшения потерь жидкости на испарение при на-
полнении хранилищ применяются резервуары с пластинами, пла-
вающими на поверхности жидкости внутри цилиндрического кор-
пуса с дышащими крышами, изготовленными из бензостойкой
ткани, пропитанной полимерными материалами. Пластины изго-
тавливаются из металла. Поверхность жидкости покрывается
слоем «пены» из пластмассовых шариков размером до 0,03 мм или
тонкими (толщина 0,3 лл) мембранами из синтетических мате-
риалов. Указанные меры, а также тепловая изоляция или охлаж-
дение резервуара (например, орошение водой) сокращают потери
жидкости на испарение до 10—20% от первоначальной величины,
окупая произведенные затраты.
216
Действенным средством борьбы с потерями жидкостей в ре-
зультате их испарения из хранилищ является применение адсорб-
ционных установок, располагаемых на выводе воздушников в ат-
мосферу. Эти установки в настоящее время используются еще
недостаточно.
Для хранения газа широко используются газгольдеры низкого
или высокого давления [7, 8].
На современных заводах для хранения газа под низким давле-
нием используются преимущественно сухие газгольдеры, обладаю-
щие рядом преимуществ перед мокрыми газгольдерами и допу-
скающие сооружение емкостей значительных размеров (до
600 000 л«3).
В тех случаях, когда имеется возможность сжатия газа, целе-
сообразно использовать газгольдеры высокого давления, выпол-
няемые в виде цилиндрических или сферических стальных сосудов
емкостью до 250 jh3 (Ризб^С^ ат) или 4 jh3 (РИзб= 150-г-160 ат)
и устанавливаемые горизонтально или вертикально группами, ко-
торые объединяются коллектором (газгольдеры высокого давления
150—160 ат укладываются в грунт).
Одним из основных преимуществ газгольдеров ,высокого дав-
ления является относительно низкий удельный расход металла.
Если расход металла на изготовление газгольдеров низкого дав-
ления в зависимости от емкости составляет от 11 до 150 кг/м3,
то удельный расход металла на газгольдер высокого давления на
порядок ниже.
Хранение и транспорт газа также связаны с потерями в ре-
зультате утечки через неплотности. Величина потерь достигает
2% от объема газа в хранилище (сухой газгольдер) и 1% от
потока газа, передаваемого по трубопроводу.
ТРАНСПОРТ ЖИДКИХ И ГАЗООБРАЗНЫХ ВЕЩЕСТВ
Транспорт жидких и газообразных веществ по трубо-
проводам осуществляется с применением разнообразных устройств
и машин, создающих напор, необходимый для преодоления сопро-
тивления трубопровода и давления в сосуде-приемнике.
Для периодического перемещения небольших количеств жидко-
сти используется давление сжатых инертных газов, подаваемых
в газовое пространство сосуда, содержащего транспортируемую
жидкость (так называемое «монтежю»). Этот прием, весьма рас-
пространенный в фармацевтической и анилино-красочной промыш-
ленности [9], иногда может оказаться целесообразным при осуще-
ствлении отдельных операций в крупнотоннажном производстве.
В некоторых случаях давление в монтежю может быть создано
путем подачи воды из промышленного водопровода под уровень
транспортируемой жидкости (при этом неизбежны потери жидко-
сти в виде пара в сбрасываемом из монтежю сжатом , газе или в
виде раствора в воде).
217
Перепад давления между сосудом, в котором хранится транс-
потируемая жидкость, и сосудом-приемником в ряде случаев
создается в результате понижения давления в сосуде-приемнике.
Такой прием находит применение как в периодических, так и в
непрерывных процессах химической технологии, если сосуд или
аппарат, предназначенный для приема жидкости, рассчитан на
работу под вакуумом (питание дистилляционной, ректификацион-
ной и выпарной аппаратуры, работающей под вакуумом).
Использование струйных насосов и эрлифта, простота устрой-
ства которых не окупается значительными потерями перемещае-
мого вещества с потоком инертной жидкости или газа, ограничено.
Подобные приемы целесообразны лишь при использовании энер-
гии материальных потоков, массообмен между которыми не свя-
зан с потерями продуктов, определяющих экономику производства.
Конструкции центробежных насосов, находящих широкое при-
менение в химической промышленности, чрезвычайно разнооб-
разны. Для перемещения агрессивных жидкостей изготавливаются
гуммированные и эмалированные насосы, а также насосы, выпол-
ненные из коррозионноустойчивых металлов (чугун, легированные
стали), фарфора, стекла, углеграфитовых композиций, пластиче-
ских масс. Производительность центробежных насосов (Q) покры-
вает весь диапазон потребностей промышленности основного орга-
нического синтеза, однако зависимость Q(H), характерная для
центробежных насосов, ограничивает возможность создания высо-
кого напора (Я) при относительно малой производительности.
В настоящее время конструкции центробежных насосов совер-
шенствуются [10]. Значительно расширяются также возможности
использования лопастных насосов. Например, насосы черпакового
типа развивают значительно больший (на 50%) напор и обладают
большим к. п. д., чем центробежные насосы тех же размеров и
быстроходности [11], а лабиринтные насосы просты в изготовлении
и обладают относительно высоким напором при малой производи-
тельности.
Большие перспективы в развитии и совершенствовании техни-
ки насосостроения и расширении областей применения центробеж-
ных насосов открывает использование герметичного привода [12].
Насосы с герметичным приводом развивают напор до 300 м столба
жидкости и пригодны для работы при температуре до 400° С под
давлением в контуре до 1000 ат. Отсутствие сальникового уплотне-
ния полностью исключает возможность потерь продукта в резуль-
тате утечки, что делает эти насосы незаменимыми для перемеще-
ния токсичных продуктов вне зависимости от давления в контуре.
Привод насоса осуществляется с применением защитной гильзы,
отделяющей ротор двигателя от статора, снабженного системой
охлаждения.
Отечественной промышленностью намечен широкий план про-
изводства герметических центробежных насосов для работы под
давлением (избыточным) в контуре в пределах 0—25, 25—50 и
2ia
50—350 ат при температуре жидкости от 50 до 400° С. Применение
герметичных центробежных насосов упрощает решение задач пере-
мешивания и циркуляции жидкости в аппарате или контуре под
высоким давлением.
Для точной дозировки жидкости, необходимой при осуществле-
нии непрерывных процессов, возможности центробежных насосов
оказываются недостаточными, особенно при высоком давлении в
резервуаре-приемнике. В этих случаях подача жидкости осуще-
ствляется с помощью плунжерных насосов, обеспечивающих напор
до 700 ат при скорости подачи от 0,5 до 8—10 м3/ч. Насос выпол-
няется горизонтальным, трехплунжерным, с регулировкой подачи
как путем изменения числа ходов всех трех скалок, так и путем
изменения хода каждой скалки. Плунжерные насосы пригодны
для подачи суспензий и паст, необходимость перемещения которых
возникает при проведении жидкофазных гетерогенно-каталитиче-
ских процессов под повышенным давлением.
Кроме плунжерных насосов, для перемещения суспензий агрес-
сивных жидкостей пригодны одновинтовые насосы, обладающие
производительностью от 0,6 до 40 м3/ч при давлении нагнетания
от 2,5 до 25 ат.
Перемещение газов по заводской территории чаще всего осу-
ществляется центробежными машинами — турбокомпрессорами
(производительность до 150 000 м3!ч, избыточное давление до
9 ат) и турбогазодувками (производительность до 60 000 м31ч, на-
пор до 4500 мм вод. ст.), применяемыми в производствах высших
спиртов, искусственного жидкого топлива, метилового спирта и
аммиака. Конструктивные и эксплуатационные особенности этих
машин рассмотрены в специальных работах [13, 14].
Для перемещения относительно небольших количеств газа
используются ротационные компрессоры разнообразных типов про-
изводительностью до 6000 м31ч при напоре, не превышающем 5 ат.
Перемещение газа в контуре высокого давления производится
с помощью поршневых циркуляционных компрессоров, представ-
ляющих собой одноступенчатую двухцилиндровую машину, рас-
считанную на преодоление сопротивления циркулирующему в си-
стеме потоку газа. Подобные машины, эксплуатируемые в произ-
водстве аммиака, работают под давлением 300—325 ат, имеют
производительность (по сжатому газу) 680 м3/ч и обеспечивают
перепад давления не более 30 ат [15]. Более целесообразной яв-
ляется конструкция многоступенчатого центробежного компрес-
сора. соединенного с электродвигателем, помещенным вместе с
рабочим органом компрессора в сосуд высокого давления. Отсут-
ствие сальника и высокая производительность этой машины вы-,
годно отличают ее от поршневого компрессора.
Перемещение жидких и газообразных продуктов по территории
завода и создание коммуникации аппаратов технологической схемы
осуществляется с помощью трубопроводов, составляющих неотъем-
лемую часть оборудования любого химического производства.
219
Относительная стоимость трубопроводов (включая стоимость фа-
сонных частей, запорной и регулирующей арматуры) в производ-
ствах основного органического синтеза составляет значительную
долю удельных капитальных затрат.
По производственному назначению трубопроводы химического
производства делятся на две основные категории: материальные
трубопроводы и вспомогательные трубопроводы. Первая группа
предназначена для доставки сырья в цех, перемещения промежу-
точных продуктов между аппаратами технологической схемы и
передачи готового продукта на склад. Группа вспомогательных
трубопроводов объединяет паропроводы, водопровод, линии сжа-
тых инертных газов, используемых для вспомогательных целей,
вакуум-трубопроводы, трубопроводы для холодильных растворов
и нагретых теплоносителей, а также разнообразную кайализацион-
ную коммуникацию.
Условия эксплуатации и конструкция трубопровода зависят
от его расположения. В этой связи различают межцеховые и
внутрицеховые трубопроводы.
Диапазон параметров (температура, давление) режима работы
трубопроводов соответствует параметрам технологического режима
процессов, осуществляемых в реакционной аппаратуре и в аппа-
ратуре для разделения реакционных смесей. Требования к мате-
риалу и конструкции трубопровода и арматуры не ограничиваются
учетом действия только температуры и давления: агрессивные и
абразивные свойства потока в ряде случаев оказывают решающее
влияние на выбор материала труб и арматуры, а также на кон-
струкцию трубопровода. При проектировании и эксплуатации
трубопровода необходимо учитывать температурный режим потока,
пределы изменения температуры стенок труб и последствия пуль-
сации, вызываемой работой насосов й компрессоров.
Сведения о гидравлическом расчете трубопроводов приведены
в большом количестве работ; специальные вопросы, относящиеся
к трубопроводам химической и нефтехимической промышленности,
рассмотрены в ряде монографий [16—23].
Современная химическая промышленность располагает широ-
ким ассортиментом труб. Некоторые данные о неметаллических
трубах приведены в табл. Ш-4.
Максимально допустимые температура и давление рабочей
среды находятся в обратной зависимости; количественное соотно-
шение P(t) специфично для каждой группы материалов и зависит
от толщины стенок трубы. В табл. Ш-4 указано давление, отно-
сящееся либо к минимальному диаметру трубы, либо определяе-
мое свойствами материала.
Большим разнообразием отличается сортамент металлических
(особенно стальных) труб, рассчитанных на работу в широком
диапазоне температур (до 1000° С) и давлений (до 700 ат) в усло-
виях нейтральных и агрессивных сред. Трубы, предназначенные
для работы при высокой температуре, выполняются как из угле-
220
Таблица 111-4
Характеристика неметаллических труб
Материал труб Предельные значения
диаметр, мм ₽ИЗб' ат 1, °C
Керамика 25 -300 4 150
Стекло 13-100 7 300
Резина 3-150 15 175
Винипласт 6-150 6 60
Полиэтилен 13-150 12 100
Фаолит 30-300 6 120
Текстолит 25-150 8 80
Стекловолокно 200 Близко к металлическим
Фторопласт 30 15 -
родистых сталей (максимальная температура 425—450°С), так и
из сталей с легирующими добавками хрома, никеля, молибдена,
вольфрама и т. д., устойчивых при температуре до 1000° С в нена-
груженном состоянии.
Для работы под высоким давлением (выше 100 ат) выпус-
кается ряд сортов стальных бесшовных холоднокатанных и горя-
чекатанных труб диаметром до 200 мм из углеродистых и легиро-
ванных сталей, рассчитанных на рабочее давление до 700 ат при
температуре до 500° С.
Совмещение прочности металлических труб с коррозионной
устойчивостью труб из неметаллических материалов достигается
при использовании стальных труб, футерованных резиной, битумом,
шеллаком, полистиролом и другими полимерными материалами.
Выбор материала труб (и аппаратов) основан не только на
учете скорости разрушения материала стенки трубы под дейст-
вием рабочей среды, но и обосновывается соображениями о воз-
можности загрязнения потока компонентами, входящими в состав
материала стенок трубы. При необходимости получения продукта
высокой степени чистоты материал, характеризующийся высокой
коррозионной устойчивостью по отношению к данной рабочей
среде, часто оказывается непригодным, если коррозия приводит
к тому, что содержание в продукте производства элементов, вхо-
дящих в состав материала стенки трубопровода, превосходит до-
пустимые пределы. Когда требованием к чистоте продукта' регла-
ментировано не только количество, но и состав примесей, мате-
риал стенок трубы не должен содержать элементов, нежелательных
при получении чистого продукта.
Все сказанное относительно материала труб в равной степени
относится к материалу прокладок, арматуры и контрольно-изме-
рительных приборов, соприкасающихся с рабочей средой.
В отличие от реакционной и большинства теплообменной аппа-
ратуры, где вследствие малого значения отношения периметра
221
аппарата к площади сечения относительные теплопотери сравни-
тельно невелики, для трубопроводов характерна значительная
величина отношения периметра к площади сечения и, как след-
ствие этого, заметное влияние теплообмена с окружающей средой
на теплосодержание (энтальпию) потока, движущегося по трубо-
проводу.
При стационарном потоке изменение его теплосодержания при
движении по трубопроводу пропорционально отношению длины
трубопровода к его диаметру. Для потоков, параметры которых
далеки от значений, соответствующих фазовому переходу (конден-
сация, кристаллизация), теплообмен с окружающей средой со-
провождается изменением температуры. Для потока пара, паро-
газовой смеси или раствора, близкого к состоянию насыщения,
уменьшение теплосодержания за гчет теплопотерь сопровождается
образованием новой фазы, что в некоторых случаях нежелательно,
а в других недопустимо по условиям дальнейшей переработки.
Увеличение теплосодержания потока легкокипящей жидкости или
сжиженного газа в результате теплообмена трубопровода с окру-
жающей средой, имеющей более высокую температуру, может
привести к повышению давления сверх допустимого. Изменение
температуры по длине потока, сохраняющего свое агрегатное со-
стояние, регламентируется рядом требований (температура холо-
дильного рассола, температура сырья, поступающего в реактор,
и т. п.).
Изложенные соображения, требования условий безопасной ра-
боты обслуживающего персонала, а также учет теплового баланса
производственного помещения вызывают необходимость изоляции
большого числа трубопроводов химического производства.
В настоящее время химическая промышленность располагает
широким ассортиментом теплоизоляционных материалов различ-
ного происхождения и разнообразных механических и теплофизи-
ческих свойств. Подбор изоляционного материала и расчет тол-
щины изоляции проводится исходя либо из допустимых размеров
теплового потока на единицу длины трубопровода, либо из допу-
стимой температуры внешней поверхности слоя изоляции. По-
следняя для горячих трубопроводов ограничена величиной
45—50° С; температура внешней поверхности изоляции трубопро-
водов, предназначенных для транспорта потоков, имеющих низкую
температуру, должна на 5—10 град превышать значение «точки
росы» для окружающего воздуха (во избежание конденсации па-
ров воды).
В отличие от цилиндрических аппаратов значительных разме-
ров (£>из/Папп —* 1,0), когда расчет толщины изоляции с допусти-
мой погрешностью проводится по уравнениям для плоской стенки,
для трубопроводов, особенно при малом их диаметре, необходимо
применение зависимостей для цилиндрической стенки (здесь
Рт. Рятт — диаметры аппарата с изоляцией и без нее).
222
Использование современных теплоизоляционных материалов
позволяет уменьшить величину коэффициента теплопередачи
между потоком и окружающим трубопровод воздухом до мини-
мальных размеров. Однако в ряде случаев это оказывается недо-
статочным. Поэтому для потока продукта, имеющего, например,
высокую температуру плавления, или для легкокристаллизующихся
растворов трубопроводы снабжаются компенсаторами тепловых
потерь, оформляемыми в виде спутников или рубашек, которые
обогреваются горячей водой, паром или высокотемпературными
органическими теплоносителями. В отдельных случаях исполь-
зуется электрообогрев.
Для ответственных материальных трубопроводов предпочти-
тельным является применение рубашек, несмотря на относительно
высокую стоимость и сложность их изготовления, особенно в про-
изводствах, допускающих возможность только кратковременных
остановок.
Естественно, что экономические задачи на определение опти-
мальной толщины слоя изоляции, подобные рассматриваемым
ниже (см. стр. 269), при изоляции трубопроводов зачастую ли-
шены смысла.
К числу важных технических задач относятся: 1) компенсация
температурных деформаций и 2) компенсация вибрации трубопро-
водов.
Первая из этих задач определяется температурным режимом
работы трубы во времени. В тех случаях, когда колебания темпе-
ратуры стенки превышают допустимые пределы, возникает опас-
ность разрушения прямого, закрепленного в двух точках участка
трубы. Предельная разность наивысшей и наинизшей темпера-
туры (А/) не зависит от длины участка трубопровода и опреде-
ляется свойствами материала трубы:
где о — допустимое напряжение на разрыв; Е—модуль упругости
материала трубы; ал — коэффициент линейного расширения. Зна-
чения для стали составляют 36 град, для винипласта 32 град.
Устройства для компенсации деформации трубы, устанавли-
ваемые на прямых участках между жестко закрепленными опо-
рами, различаются по принципу действия и конструктивному вы-
полнению. В химической промышленности наибольшее распростра-
нение получили компенсаторы следующих типов: 1) линзовые;
2) сальниковые; 3) гнутые.
Линзовые компенсаторы устанавливаются главным образом на
стальных парогазопроводах низкого давления. Область их приме-
нения ограничивается диаметром трубопровода и давлением (при
диаметре до 200 мм Ризб = 5-=-8 ат, при диаметре 200 мм Ризб =
= 1,5^-2,0 ат). Компенсирующая способность одной волны не пре-
вышает 10 мм. Общая компенсирующая способность линзового
Д23
устройства не превышает 70—80 мм, так как увеличение числа
волн сверх 7—8 неблагоприятно сказывается на жесткости трубо-
провода.
Сальниковые компенсаторы применяются для стальных трубо-
проводов, а также для трубопроводов из стекла, керамики, тек-
столита и т. д. К достоинствам этого типа компенсирующих уст-
ройств относятся их компактность и высокая компенсирующая
способность (до 300 мм). Применение их ограничено диаметром
труб до 600 мм и давлением до Ризб = 20 ат. Недостатками сальни-
ковых компенсаторов являются необходимость строгой соосности
труб, необходимость наблюдения за плотностью набивки сальника
и периодическая смена набивки.
Гнутые компенсаторы обладают значительной компенсирующей
способностью (до 400 мм) и нетребовательны к строгости соос-
ности труб и постоянству наблюдения. Пределы давления в этом
случае значительно шире, чем у рассмотренных выше типов.
В тех случаях, когда трасса трубопровода предусматривает
естественные повороты, необходимость в установке компенсаторов
отпадает, так как гибкость прямых участков трубопровода, распо-
ложенных под углом друг к другу, обеспечивает возможность са-
мокомпенсации. Этот способ весьма удобен, и, наряду с гнутыми
компенсаторами, самокомпенсация используется при монтаже
трубопроводов высокого давления (исключение составляют трубо-
проводы с рубашками).
Вторая из указанных технических задач возникает при пере-
мещении жидкости (газа) поршневыми или плунжерными насо-
сами, создающими пульсирующий режим скорости (и давления)
потока в трубопроводах. Следствие пульсации давления — вибра-
ция трубопровода — крайне нежелательное явление, вызывающее
нарушение плотности фланцевых соединений, образование трещин
в сварных швах, разрушение изоляции и т. п. Известны случаи
разрушения трубопроводов химических производств, вызванных
некомпенсированной вибрацией [21].
Возможность возникновения вибраций определяется амплиту-
дой пульсации скорости и давления потока; предельная неравно-
мерность оценивается величиной:
Рмакс Рмии q 5
Pep
где Рмакс, Гмин, Гср — максимальное, минимальное и среднее дав-
ления потока.
Компенсация колебаний скорости достигается установкой на
всасывающей и нагнетательной линиях поршневых и плунжерных
машин воздушных или жидкостных компенсаторов и акустических
фильтров. Подробно этот сложный и важный для химического
производства вопрос рассмотрен в работе [21].
Транспорт жидкостей и газов по трубопроводам как одна из
статей расхода, отражающаяся на технико-экономических показа-
224
телях технологического процесса, составляет содержание экономи-
ческой задачи. Расходы на транспорт слагаются из: 1) аморти-
зационных отчислений от стоимости трубопровода, включая стои-
мость арматуры, приборов, сооружения для трассировки и пр.
(Ф1); 2) амортизационных отчислений от стоимости насосов, ком-
прессоров и т. п. (Ф2); 3) стоимости энергии, затрачиваемой на
работу насосов (Фз).
При перемещении вязких жидкостей сопротивление потоку в
значительной степени зависит от температуры жидкости. В связи
с этим нередко возникает необходимость предварительного подо-
грева жидкости и поддержания ее температуры на заданном
уровне на всем протяжении трубопровода. В этом случае необ-
ходим учет расхода энергии, связанного с подогревом (Ф4), а
также затрат на изоляцию трубопровода (Ф5) либо, если изоля-
ция не обеспечивает нужных условий, на подогрев трубопровода
с помощью спутника или рубашки.
Суммарный расход Фз можно выразить следующим образом:
Ф2 = Ф1 + Ф2 + Ф3 + Ф4 + Ф5 (Ш-1)
При заданных часовом количестве перекачиваемой жидкости
(G), длине (L) и профиле трассы трубопровода, а также началь-
ной температуре жидкости (/о) слагаемые уравнения (Ш-1) можно
выразить, пользуясь известными зависимостями:
Ф] = л dLv\
ф- - w Ш' w [‘+* м 4+S Ф»
Ф4=СС(/-/0)<
Ф5 = Ф (Z, <*) V5
где Vp v2, v'— пересчетные коэффициенты для выражения амор-
тизационных отчислений; v', v' — пересчетные коэффициенты для
выражения расходов в денежных единицах; d — диаметр трубо-
провода; t — температура потока; р, С, А,, АР — средние величины,
характеризующие поток жидкости (плотность, теплоемкость, ко-
эффициент трения, перепад давления); g— ускорение силы тяже-
сти; — сумма местных сопротивлений движению потока.
Подставив выражения для Фь Фг, Фз, Ф4 и Ф5 в уравнение
(Ш-1), можно видеть, что при заданном значении L и G для кон-
кретного потока суммарный расход средств определяется значе-
нием двух переменных Фх(Л^).
Практически возможны три случая:
Z = /0 = const; Ф2 (d) = Ф, (d) + Ф2 (d) + Ф3 (d) + Ф5 (d)
d = const; Ф2 (/) = Ф] + Ф2 (/) + Фз (/) + Ф4 (/) + Ф5 (/)
t^(0; d^ const; ®s(d, t) = ®1(d) + ®2(d, 0 + Ф3(й, 0 + Ф4(0 + ф5(й, О
15 В. С. Хайлов, Б. Б. Брандт
225
В первом случае [t=to) увеличение диаметра трубопровода,
сопровождающееся повышением его стоимости (и соответственно
размера амортизационных отчислений Ф1), приводит к снижению
сопротивления трубопровода, в связи с чем уменьшаются значе-
ние Ф2 (возможность использования более дешевых насосов низ-
кого давления) и удельный расход на работу насосов (Фз). При
необходимости сохранения температуры потока в заданных пре-
делах увеличение диаметра трубопровода, по-видимому, повлечет
за собой необходимость увеличения веса изоляции и, как резуль-
тат, увеличение значения Ф5.
Характер зависимости каждого из слагаемых от диаметра тру-
бопровода свидетельствует об экстремальной форме зависимости
Рис. 1П-1. Условия оптимального режима перемещения жидкости по трубо-
проводу:
а — / = £о; б — перемещение вязкой жидкости (d« const).
Фх(^); как показывает анализ конкретных примеров, при неко-
торой величине d=dfmi достигается минимальное значение Фх
(рис. Ш-1,а).
Если диаметр трубопровода является заданной величиной
(d=const), то Ф1 = const и режим перемещения зависит только
от температуры потока в трубопроводе.
Зависимость вязкости от температуры, характерная для жидко-
стей, обусловливает монотонное уменьшение сопротивления с по-
вышением температуры потока, результатом чего является сниже-
ние значений Ф2 и Ф3 (возможность применения более дешевых
насосов низкого давления). Расход тепла на первоначальный по-
догрев жидкости (Ф4) и стоимость изоляции (Ф5) пропорциональны
повышению температуры потока. В случае Фх(?) также имеет
место минимум при некотором значении /=/Опт (рис. Ш-1,б).
Из рассмотрения зависимостей Фх(^) и Фх(?) нетрудно сделать
вывод о наличии минимума функции &z(d, t).
226
Оптимальный режим транспорта вязкой жидкости по трубо-
проводу заданной длины может быть установлен в результате по-
иска минимума функции двух переменных.
Поиск минимума затрат на транспорт жидкости по трубопро-
воду с несколькими ступенями создания напора составляет содер-
жание задачи на распределение, решаемой методом динамиче-
ского программирования [24].
Вопросы экономики, организации и эксплуатации производст-
венных трубопроводов освещены в ряде работ [25—28].
ТЕПЛООБМЕН И ТЕПЛООБМЕННАЯ АППАРАТУРА
При рассмотрении большинства схем производств ос-
новного органического синтеза можно заметить, что второй по
значимости статьей расхода, определяющей себестоимость продук-
та, является расход энергетических средств. Во многих случаях
основная часть расходуемых энергетических средств используется
для нагрева, охлаждения и изменения агрегатного состояния ис-
ходных веществ, промежуточных и конечного продуктов промыш-
ленного синтеза. Естественно, что удельный расход этой части
энергетических средств находится в зависимости от полноты ис-
пользования их теплосодержания (энтальпии).
Анализ спецификаций технологического оборудования показы-
вает, что значительную долю его стоимости составляют теплооб-
менные устройства, используемые на всех этапах переработки
сырья. Основное применение теплообменной аппаратуры связано
с подготовкой сырья и переработкой реакционной массы.
Современные производства основного органического синтеза
оснащены большим количеством теплообменной аппаратуры раз-
личного назначения и типа, работающей в широком диапазоне
температур (от —200-=—250 до 1000—1200° С) и давлений (до
1000—1500 ат) в разнообразных по коррозионной активности
средах.
Как известно, по способу передачи тепла теплообменные уст-
ройства делятся на три основные категории.
1. Рекуперативный теплообмен — теплообмен через стенку, раз-
деляющую потоки теплоносителей. При рекуперативном теплооб-
мене смешение потоков исключается, тепловой и температурный
режимы могут быть как стационарными, так и нестационарными.
2. Теплообмен смешением — непосредственное смешение пото-
ков теплоносителей.
3. Регенеративный теплообмен — передача тепла от более на-
гретого к менее нагретому теплоносителю при участии аккумуля-
тора тепловой энергии. Процесс аккумулирования тепла и пере-
дача его потоку» холодного теплоносителя совершаются попере-
менно, вследствие чего работа регенеративных теплообменников
характеризуется неустановившимся тепловым и температурным
режимами. Необходимость попеременного соприкосновения обоих
15:
227
потоков с аккумулятором тепла создает возможность для частич-
ного смешения потоков.
В химической промышленности в основном применяются реку-
перативные теплообменники и в ряде случаев теплообменники сме-
шения; регенераторы почти не находят применения, так как ча-
стичное смешение потоков теплоносителей, неизбежное при их экс-
плуатации, редко может быть допущено условиями химического
производства. В тех же случаях, когда это допустимо, более це-
лесообразным оказывается применение теплообмена смешением.
ТЕПЛОВЫЕ СХЕМЫ
При разработке технологической схемы химического
производства значительного внимания требует вопрос регенерации
теплосодержания материальных потоков с учетом необходимых
Рис. Ш-2. Тепловая схема уста-
новки:
а—аппаратурная схема; б— схема темпера-
тур тепловых потоков.
для этого теплообменных уст-
ройств. Вопрос этот является
экономической задачей и решает-
ся, исходя из конкретных условий
данного производства. Возмож-
ные варианты тепловой схемы под-
чинены поиску условий минимума
суммарных затрат, связанных с
поддержанием теплового и тем-
пературного режима процесса.
Критерием оценки может слу-
жить или сумма энерго-матери-
альных затратФ, или показатель,
аналогичный по структуре пока-
зателю приведенных затрат П
(см. гл. I).
Условия оптимума по сумме
энерго-материальных затрат, оче-
видно, соответствуют минимуму
функции:
Ф = v'Q + 2 v"F (Ш-2)
где vz— стоимость единицы энер-
гетических средств; Q — расход
энергетических средств, отнесен-
ный к единице готового продукта
(или единице времени); v"— пересчетный коэффициент для опре-
деления амортизационных отчислений; F — размер теплообменной
аппаратуры (поверхность теплообмена).
Иллюстрацией постановки вопроса может служить простой
пример тепловой схемы установки, показанной на рис. Ш-2.
Поток сырья с начальной температурой Л подогревается в теп-
лообменнике / потоком рабочей среды, выходящей из аппарата IV.
228
По выходе из теплообменника / поток сырья с температурой tx
поступает в аппарат II, где нагревается за счет внешнего источ-
ника тепла до температуры t2. Рабочая среда, выходящая из
аппарата IV с температурой t3, охлаждается в теплообменнике I
до температуры ty, после чего поступает в теплообменник III
(охлаждаемый холодильным агентом), где температура его сни-
жается до Ц.
В практических условиях величины t\, tz, t3 и обычно явля-
ются заданными, причем значения их могут колебаться в весьма
широких пределах; понятно, что анализ зависимости (Ш-2) для
случая, когда tz^tz, смысла не имеет.
В качестве аппарата IV может служить как реакционная ап-
паратура, так и аппаратура для разделения реакционной массы.
В общем случае отношение водяных эквивалентов холодного (U7X)
и горячего (№г) потоков Б= Wx/Wr^ 1.
Затраты, связанные с подогревом сырья и охлаждением рабо-
чей среды до заданной температуры (соответственно t2 и i4) яв-
ляются суммой трех слагаемых: затрат на компенсацию износа
теплообменной аппаратуры, стоимости тепловой энергии и стои-
мости энергии, расходуемой на перемещение потоков.
Поскольку последняя величина находится в зависимости от
типа и размера теплообменной аппаратуры и режима ее работы
и, как правило, не является определяющей, анализ зависимости
(Ш-2) без большой погрешности может быть проведен без ее
учета.
Балансы тепла
в аппарате /
W'r (h-ty)=Wx (tx-tt)
в аппарате //
Vi ~ tx) = Qn
в аппарате ///
Wг Vy tt) = Qin
позволяют выразить ty через tx:
ty — t3 — Б (tx —1\)
(Ш-З)
Тогда поверхности теплообмена аппаратов /, II и III можно
описать следующим образом:
. Wx(tx-tD
К. Л/,
Wx (t2 - tx)
W'r [/з-Д(/х-Л)-М
^ш Л^п
229
После подстановки этих значений уравнение (Ш-2) примет
вид:
Рис. Ш-3. Условия опти-
мального варианта тепловой
схемы установки:
1 — стоимость энергетических
средств; 2 — амортизационные от-
числения; 3—суммарные затраты.
ф- 2 V'Q + S V"F ~ vnQn + v"iQin (^) +
+T[<fI(Q+v;?n(/x)+vfIIfnl(Q] = irx<P(^ (ш-4)
Здесь v' —стоимость энергетических средств, руб/кг-, v" = абв —
пересчетный коэффициент для определения амортизационных от-
числений (а —^материалоемкость теплообменников, кг/м2-, б — стои-
мость материала теплообменника, руб/кг-,
в — размер амортизационных отчисле-
ний, доли единицы); K=f(tx)—коэффи-
циент теплопередачи, ккал/(ж2 • ч•град);
т — число часов работы теплообменной
аппаратуры в год; Д/—средняя разность
температур.
Индексы «I», «II» и «III» соответст-
вуют нумерации теплообменников на
схеме.
Выражение (Ш-4) показывает, что
сумма затрат, связанных с эксплуата-
цией тепловой схемы, является функцией
одного параметра, оказывающего про-
тивоположное влияние на величину от-
дельных слагаемых. Такая структура
зависимости Ф(/х) дает основание для
поиска минимального значения Фмин (по
понятным" соображениям возможность
максимума функции исключается).
Ввиду сложности выражения (Ш-4) решение его относительно
(Ммин более удобно производить графически.
Пример III-1. Установка, схема которой представлена на
рис..Ш-2, работает в следующих условиях: ^=<з=200°С; /i=/4=20°C; ai=an =
=аш=40 кг!м2~ 6i=6ii=6ih=2,0 руб/кг-, ei=en=eni=O,l; оборудование дей-
ствует 8000 ч в году; нагревающий агент, применяемый в теплообменнике И, —
водяной пар (/ii=250oС); охлаждающий агент в теплообменнике Ш—вода
(/н=10°С, /„=20° С); стоимость теплоносителей <Эп=2,6руб/т, <Эш=6-10~3 руб/м3;
направление потоков в теплообменниках / и II— противоток; W’I=W’r=
= 10 000 ккалЦч • град).
Требуется приближенно оценить размеры теплообменников I, II и III, со-
ответствующие минимальному размеру суммарных затрат.
Решение. Для приближенной оценки допускаем постоянство
значений коэффициентов теплопередачи в теплообменниках I, II и III и прини-
маем их равными Ki=Kiii=100 ккалЦм2 • ч • град), Кп=200 ккалЦм2 • ч • град).
Пользуясь уравнением (Ш-4), подсчитываем значения каждого из слагае-
мых, задаваясь произвольными значениями температуры потока сырья на вы-
ходе из теплообменника I (tx).
Результаты расчета представлены кривыми 1—3 на рис. III-3.
Как следует из рис. Ш-3, минимум суммарных затрат достигается при тем-
пературе потока на входе в теплообменник II, равной 185—186° С. Оптимальные
условия достигаются при Гг=12,5 м2; Гц=0,125 м2- Fin=l,15 м2.
230
Влияние величины tx в наибольшей степени сказывается на
размерах теплообменника I и расходе тепла в подогревателе II.
Размер теплообменников // и 111 относительно невелик и во всем
диапазоне значений tx меняется сравнительно мало.
Размер капитальных затрат как функция- tx не имеет мини-
мума, монотонно возрастая с увеличением tx за счет теплообмен-
ника I. Суммарный расход тепловой энергии также монотонно по-
нижается с увеличением tx.
Из рис. III-3 следует, что отклонение от оптимального значения
tx резко сказывается на величине суммарных затрат, что особенно
заметно в области высоких значений tx.
Следует добавить, что величина Фмин, относительные размеры
теплообменников (Fi: Fn: Fin) и расход энергии в значительной
степени зависят от стоимости теплообменной аппаратуры и энерге-
тических средств. При использовании более дешевого материала
теплообменной аппаратуры снижается значение Фмин, a tx пере-
мещается в область более низких температур.
Значение (^х)опт, найденное указанным выше способом, позво-
ляет определить размеры теплообменной аппаратуры и расход
энергетических средств.
Величина относится к категории величин, значение кото-
рых задано условиями расчета тепловой схемы, и определяется
степенью превращения а.
При рассмотрении подогрева сырья за счет регенерации тепло-
содержания материальных потоков как составной части первого
этапа переработки сырья (подготовка сырья) можно, пользуясь
ранее установленной зависимостью размера материальных пото-
ков от степени превращения сырья (см. гл. I), записать:
Fx=CMA,(a) = f(a)
где С — средняя теплоемкость материального потока; М — мощ-
ность производства; А] — размер материального потока, кг! кг про-
дукта.
Окончательное выражение функции Ф с достаточной степенью
приближения может быть описано следующим образом:
Ф = /(а)ф(/х)
Экономические задачи возникают не только при разработке
рациональной тепловой схемы,, но и при расчете каждого аппа-
рата и агрегата этой схемы.
Несмотря на то, что в практике проектирования зависимость
(Ш-2) не всегда учитывается должным образом, анализ ее пред-
ставляет интерес, особенно для энергоемких производств и тепло-
обменных установок больших масштабов.
В приведенном выше примере в качестве нагревающего агента
был принят водяной пар, а для охлаждения использована вода.
Обоснование решения отсутствует, хотя для доказательства при-
нятого режима это необходимо.
231
Выбор источника тепловой энергии требует сравнительной
оценки ряда вариантов проведения процесса с использованием
различных (доступных в конкретных условиях) источников теп-
ловой энергии и теплоносителей.
Размер и стоимость теплообменной аппаратуры, определяемые
поверхностью теплообмена, зависят не только от максимально до-
стижимой температуры энергетического средства, но и от его теп-
лофизических свойств, влияющих на величины коэффициентов теп-
лоотдачи и теплопередачи.
Высокая температура дымовых газов обеспечивает достижение
большого значения разности температур, в то же время коэффи-
циент теплопередачи между дымовыми газами и нагреваемой сре-
дой, ограничиваемый малым значением коэффициента теплоотдачи
со стороны газового потока, в редких случаях превышает несколь-
ко десятков калорий (на 1 м2 в час при А?=1 град) и величина
теплового потока оказывается незначительной.
Сравнительная оценка дымовых газов и, например, водяного
пара как энергетических средств (разумеется, с учетом стоимости
теплообменной аппаратуры) может привести к противоположным
выводам в зависимости от конкретных условий.
Теплообмен между двумя потоками теплоносителей при задан-
ной величине и граничных температурах одного из них и заданной
начальной температуре другого также может быть объектом
поиска оптимального варианта режима. Для иллюстрации этого
случая можно рассмотреть простой пример охлаждения данного
потока при заданных начальной и конечной его температурах (за-
дана, как обычно, также начальная температура охлаждающего
агента).
Для противоточной схемы движения потоков уравнение (Ш-2)
может быть написано в следующем виде (расход энергии на пе-
ремещение теплоносителей для простоты примера не учитывается) :
Ф = v'Q + v"F - JFr (t3 - t4) Г У - + ——_X_— in (III-5)
где v'—стоимость охлаждающего агента, руб/кг-, V—абв—
пересчетный коэффициент для определения амортизационных от-
числений (а—материалоемкость теплообменника, кг/м2- б—стои-
мость материала теплообменника, руб/кг-, в — размер амортизаци-
онных отчислений, доли единицы); т—число часов работы тепло-
обменной аппаратуры в год; K=f (tx)—коэффициент теплопередачи,
ккалЦм2 • ч • град); С — средняя теплоемкость охлаждающего аген-
та, ккал/(кг • град)-, tx, Л, t3, — температуры потоков (рис. Ш-4);
W'r — водяной эквивалент охлаждаемого потока.
Структура приведенного выше уравнения, выражающего зави-
симость Ф(Ц, свидетельствует о наличии минимума функции.
Аналитическое выражение значения tx, соответствующее мини-
муму функции, сложно; более удобно графическое решение урав-
нения.
232
Для иллюстрации зависимости (Ш-5) на рис. Ш-4 приведены
результаты расчета теплообменника для конденсации пара, охла-
ждаемого водой.
Абсолютное значение Фмив в значительной степени зависит от
стоимости материала теплообменника и стоимости энергетических
средств; оптимальное значение конечной температуры опреде-
ляется отношением этих величин.
В рассматриваемом случае (рис. Ш-4) замена более дорогого
материала теплообменника на более дешевый сопровождается пе-
ремещением значений tx в область
более высоких температур и соответ-
ствующим увеличением поверхности
теплообмена; аналогичный резуль-
тат дает также повышение стоимо-
сти теплоносителя.
Интервал возможных изменений
значения tx (теплоноситель—вода)
ограничен верхним пределом, опре-
деляемым, как известно, величиной
порядка 55—60° С (опасность отло-
жений накипи), при этом учет зави-
симости (Ш-2) лишен смысла. Од-
нако при отсутствии такого рода
ограничений поиск значения Фмин
необходим и оправдан.
Рис. III-4. Условия оптимального
режима работы конденсатора:
1 — удельная стоимость воды; 2 — амор-
тизационные отчисления при недорогом
металле конденсатора; 3 — амортизацией*
иые отчисления при дорогом металле
конденсатора; 4 — суммарные затраты
при недорогом металле конденсатора;
5— суммарные затраты при дорогом ме-
талле конденсатора.
кг/м2-, 6=2,0 руб) кг-, 8=0,1; д=
ч в году.
Пример III-2. Пары бензо-
ла (G=100 кг/ч) конденсируются в меж-
трубном пространстве кожухотрубного
теплообменника, охлаждаемого артезиан-
ской водой (/н=10°С).
Определить размеры теплообменника и
расход воды, соответствующие оптималь-
ным условиям процесса, при следующих
исходных данных: /КОнд=80°С; теплота
конденсации бензола г=95 ккал/кг-, а—40
=6-10-3 руб!м2; оборудование действует 8000
Решение. 1. Предварительным ориентировочным расчетом
установлено число труб теплообменника (п=50 шт.) и их размер (d=25X
ХЗ мм).
Для подсчета
формулой:
значения K(tx)—уравнение (III-5) — пользуемся
известной
(1/а9 + (б/Л) + (1/а0
ккалЦм2 • ч • град)
Без большой погрешности можно допустить постоянство значений коэффи-
циента теплоотдачи от конденсирующихся паров бензола к стенке труб (а() и
принять его равным 1000 ккалЦм2 ч - град).
Для расчета значений а2 пользуемся уравнением
Nu = 0,15 Re0-33 Рг°>43 Gr0’1 (-Д-V’25
\ Ргст )
рекомендуемым для потока при 10<Re<2000 [29).
283
Для воды последнее уравнение принимает вид:
, и0-33^’1 ( Рг \0,25
а, — В----AQ7 |------1 ккалЦм2 ч • град)
2 rf0;37 у рГст у
температурой потока [значения В
где В — коэффициент, определяемый средней
прн 0,5(1х+1н) представлены интерполяционной
кривой на рнс. Ш-5, а], и —скорость воды в тру-
бах теплообменника
Gr _ 1,07
“ (G-/H)n-1000 - 0,785^-3600 tx - 10 М‘сеК
Д/ср — средняя разность температур в теплооб-
меннике
<х~Ю
Рг — критерий Прандтля при средней [0,5(fx+ 10)J
температуре потока воды; Ргст — критерий
Прандтля при температуре стенкн трубы.
Учитывая, что cq » а2, принимаем значение
РгСт при температуре 65°С (РгСт=2,5).
Поверхность теплообмена кожухотрубного
теплообменника подсчитываем по уравнению:
Q 1000-95
Шр K(lx)A/cp(G)
Задаваясь значениями tx, находим ряд зна-
чений K(tx) и /Ср(1х) и определяем соответ-
ствующие значения F{tx). Результаты расчетов
приведены на рнс. Ш-5, б интерполяционной кри-
вой F(tx).
2. Годовой расход воды GB (теплопотерями
пренебрегаем) подсчитываем по уравнению теп-
лового баланса:
Q-8000 1000-95-8000
° В ~ (tx - /н) Ю00 (tx - 10) 1000
1
С
Рис. Ш-5. Расчет размеров
теплообменника н расхода
охлаждающей воды.
= 7,6 • 105 ——м31год
tx ~ Ю
Задаваясь значениями tx, находим ряд зна-
чений GB(lx)- Результаты расчетов представле-
ны на рис 111-5,6 интерполяционной кривой
GB«x).
Учет стоимости воды (6-IO-3 руб/м3), мате-
риалоемкости теплообменника (40 кг/л2), стои-
мости материала теплообменника (2 руб/кг) поз-
воляет определить размер затрат, связанных с
расходом охлаждающей воды (кривая 1 на
рис. Ш-5, в) и амортизационными отчислениями
(кривая 2), а также оценить значение суммар-
ных Затрат (кривая 3).
Оптимальный режим, соответствующий тем-
пературе воды на выходе из теплообменника, рав-
ной 30° С, достигается при установке теплооб-
менника с 7=6,0 мг и расходе воды GB =
=4,5 -т- 4,6 м3/ч.
234
РЕКУПЕРАТИВНЫЕ ТЕПЛООБМЕННИКИ
Различия в назначении теплообменников, условиях их
работы, природе теплоносителей, свойствах конструкционных ма-
териалов объясняют разнообразие конструктивных вариантов теп-
лообменной аппаратуры.
Литература по конвективному теплообмену огромна. Вопросы
рекуперативного теплообмена обстоятельно рассмотрены в много-
численных руководствах [30—39]. Конструкции теплообменной ап-
паратуры, специфические особенности отдельных ее типов, усло-
вия эксплуатации и методы расчета изложены в ряде отечествен-
ных и зарубежных работ.
Трудно классифицировать теплообменную аппаратуру по одно-
му единственному признаку, поэтому полная характеристика может
быть составлена в результате совокупности ряда признаков, отра-
жающихся на конструкции аппарата:
1) назначение — теплообмен без изменения агрегатного состоя-
ния теплоносителей (холодильники, нагреватели), теплообмен с из-
менением агрегатного состояния одного или обоих теплоносителей
(нагреватели, конденсаторы, испарители);
2) направление тока теплоносителей — прямоток, противоток,
смешанный ток;
3) конструкционный материал — металл, неметаллические мате-
риалы;
4) форма поверхности теплообмена: из труб — змеевик (по-
гружной, орошаемый), кожухотрубные аппараты («труба в трубе»,
пучок труб); из листа — спиральные, пластинчатые, аппараты с
рубашкой.
По конструкционному материалу теплообменники делятся на
металлические и неметаллические.
Кроме обычных углеродистых сталей, за последние годы широ-
кое применение находят высоколегированные аустенитные хромо-
никелевые стали, а также титан, тантал, ниобий, цирконий и их
сплавы.
Наряду с применением коррозионноустойчивых металлов для
изготовления теплообменной аппаратуры используются плавле-
ный кварц, стекло, эмаль и разнообразные углеграфитовые ком-
позиции.
Направление движения теплоносителей почти в каждом типе
теплообменника может быть по необходимости осуществлено по
любому из трех вариантов — противоток, прямоток, смешанный ток.
Изменение разности температур теплоносителей по длине по-
верхности теплообмена при различных относительных значениях
водяного эквивалента при противотоке и параллельном токе пред-
ставлено на рис. Ш-6.
В случае теплообмена без изменения агрегатного состояния
теплоносителей (или теплоносителя) применение противотока
235
позволяет достичь более низкой температуры теплоносителя А и бо-
лее высокой температуры теплоносителя В, чем при параллельном
Рис. Ш-6. Основные варианты изменения температуры теплоносите-
лей (противоток, прямоток).
движении теплоносителей. Большая полнота использования тепло-
содержания встречных материальных потоков, а также большее
значение средней разности температур при противотоке являются
основанием для более частого использования этого варианта.
236
Преимущества параллельного движения теплоносителей можно
установить по рис. Ш-6 (при а[ = а'). При одинаковых начальных
и конечных температурах теплоносителей, независимо от относи-
тельного значения водяных эквивалентов потоков, стенка тепло-
обменной поверхности в случае противотока имеет участки, более
нагретые, чем самый горячий участок стенки в прямоточном теп-
лообменнике. Это обстоятельство имеет большое значение при ре-
шении вопроса о выборе материала теплообменной поверхности,
и в ряде случаев возможен выбор в пользу прямоточного вари-
анта, несмотря на присущие ему недостатки.
Прямоток теплоносителей может быть также единственным
вариантом осуществления процесса в тех случаях, когда задан-
ными параметрами являются поверхность теплообмена и конечные
температуры обоих теплоносителей и нежелателен (или недопу-
стим) перегрев или переохлаждение каждого из потоков.
Не касаясь сложных вопросов теории коррозии, можно счи-
тать, что скорость коррозии прямо пропорциональна температуре
корродирующего материала и понижение температуры теплооб-
менной поверхности со стороны агрессивной среды (/Ст) благо-
приятствует коррозионной устойчивости стенки теплообменника.
Рассматривая известную зависимость
_ «?1 + «2Z2
‘ст---7 7
«1 + «2
нетрудно заметить, что при расчете и конструировании теплооб-
менника, в котором один из теплоносителей является агрессивной
средой для материала теплообменной поверхности, необходимо
использовать указанную закономерность и путем увеличения ко-
эффициента теплоотдачи со стороны холодного потока улучшить
условия работы материала стенки.
Подобный прием в сочетании с параллельным током теплоно-
сителей дает возможность понизить требования к материалу ап-
парата. Иллюстрацией эффективности этого приема может слу-
жить пример, приведенный в работе [33]. Повышением скорости
азотно-водородной смеси в трубах пускового нагревателя (обогре-
ваемого топочным газом) удалось снизить температуру стенок
труб до температуры, близкой к температуре азотно-водородной
смеси, и с успехом использовать для изготовления аппарата трубы
из низколегированной стали.
Схема расчета
рекуперативной теплообменной аппаратуры
Расчет рекуперативной теплообменной аппаратуры сво-
дится к решению вопросов, характер и совокупность которых опре-
деляются поставленной задачей и имеющимися исходными дан-
ными; число искомых величин обычно не превышает четырех.
237
При проектировании или выборе новой аппаратуры задача
сводится к выбору типа теплообменника и определению его раз-
меров, а в ряде случаев также к выбору одного из теплоносителей
и определению его расхода.
При этом возможны два варианта: 1) теплообмен между ма-
териальными потоками и 2) нагрев или охлаждение материального
потока.
В задачу расчетов, связанных с использованием имеющейся
(готовой) теплообменной аппаратуры, когда тип и размеры теп-
лообменника заданы, входит определение конечной температуры
одного или обоих теплоносителей, а также расхода одного из них.
Во всех случаях определяется потеря напора потоком каждого
из теплоносителей.
При выборе новой аппаратуры для нагрева или охлаждения
материального потока в задачу расчета входит определение ряда
конечных величин, число которых превосходит число возможных
уравнений, описывающих процесс. Для сокращения расчетной ра-
боты, вызываемой необходимостью применения метода последова-
тельных приближений, целесообразна следующая очередность вы-
полнения этапов расчета.
1. Составление теплового баланса по основному теплоносителю,
определение количества передаваемого тепла Q (в ккал/ч) и уста-
новление интервала начальной и конечной температур охлаждаю-
щего или нагревающего агента. .
2. Выбор нагревающего или охлаждающего агента (см.
стр. 239), его начальной и конечной температур и определение рас-
хода агента (в кг/ч).
3. Выбор схемы потоков теплоносителей (прямоток, противо-
ток, смешанный перекрестный ток).
4. Определение вредней разности температур.
5. Выбор значения коэффициента теплопередачи, исходя из
природы теплоносителей и предполагаемой конструкции теплооб-
менного аппарата (см. стр. 244).
6. Определение порядка величины поверхности теплообмена
выбираемого аппарата на основе уравнения
7. Выбор типа, конструкции и размеров теплообменного аппа-
рата по существующим каталогам.
8. Уточнение принятого значения К, исходя из размеров кана-
лов для прохода теплоносителей в выбранном типе теплообмен-
ного аппарата, и повторение расчета величины поверхности тепло-
обмена.
Как правило, такая пс'*ледовательность выполнения этапов
расчета облегчает возможность получения выдерживающих про-
верку результатов.
238
Схема расчета, связанного с проектированием нового теплооб-
менного аппарата, более сложна, так как включает расчеты кон-
структивных размеров теплообменника [35].
Зависимости, необходимые для определения значений коэффи-
циентов теплоотдачи при уточнении величины К, приведены в об-
щей и специальной литературе, посвященной процессам тепло-
обмена.
В тех случаях, когда требуется установить пригодность имею-
щегося в наличии теплообменного аппарата для осуществления
процесса нагрева и охлаждения и определить значение ряда ве-
личин, характеризующих режим процесса, расчет целесообразно
основывать на представлении об эффективности теплообменника
и числе единиц переноса тепла [36].
Энергетические средства и теплоносители
Удельный расход энергетических средств является вто-
рой по величине статьей, определяющей себестоимость продукта
химического производства. Значительная часть энергии, расходуе-
мой в химическом производстве, приходится на долю процессов
охлаждения и нагрева рабочих сред.
Тепловая энергия вводится в поток перерабатываемого сырья
на всех стадиях технологического процесса с помощью разнооб-
разных средств и методов, на различном температурном уровне.
За исключением теплоты химических превращений, во всех
других случаях тепло, вводимое в перерабатываемую среду, выво-
дится затем с помощью охлаждающих агентов; количество вво-
димого и выводимого тепла соизмеримы.
Удельный расход нагревающих и охлаждающих агентов в зна-
чительной степени зависит от полноты использования их теплосо-
держания, значение которой в некоторых случаях весьма невелико.
Основными источниками тепловой энергии, применяемой в хи-
мическом производстве, являются жидкое и газообразное топливо,
а также электроэнергия, используемые при рекуперативном тепло-
обмене либо непосредственно, либо с помощью промежуточных
теплоносителей. В зависимости от конечной температуры охлаж-
даемой среды для отвода тепла применяются разнообразные по
природе охлаждающие агенты.
Так как стоимость единицы количества тепла зависит от тем-
пературного уровня, вопрос о выборе теплоносителя, предназна-
чаемого для нагрева или охлаждения раббчей среды, имеет боль-
шое значение при определении стоимости энергетических средств.
В ряде случаев оказывается целесообразным осуществление на-
грева или охлаждения путем последовательного применения ряда
теплоносителей, используемых по мере изменения температуры
рабочей среды.
Теория процессов горения жидких и газообразных топлив, ос-
новы расчета и конструктивного оформления топочных устройств.
239
применяемых в химической и нефтехимической промышленности,
представлены в ряде специальных работ. Столь же значительным
по объему является материал, посвященный использованию элект-
роэнергии для целей нагрева.
Применяемые в химической промышленности нагревающие и
охлаждающие агенты, служащие для транспорта тепловой энергии
между источником и потребителем, весьма разнообразны.
Подвод тепловой энергии к потоку перерабатываемого материа-
ла осуществляется с помощью насыщенного и перегретого пара
(водяной пар, пар органических жидкостей), а также жидких теп-
лоносителей (вода, высокотемпературные органические теплоноси-
тели, силиконовые жидкости).
В качестве охлаждающих агентов используются вода, воздух,
водные растворы солей, кипящие жидкости и т. д.
Жидкое и газообразное топливо. Применение огневого
нагрева в технологии основного органического синтеза — явление
сравнительно редкое, и установки, связанные с использованием
газообразного и жидкого топлива, как правило, не очень велики.
В качестве горючего используются продукты, доступные в ус-
ловиях данного производства: мазут, углеводородные кубовые ос-
татки, генераторный газ, углеводородные газы и т. п.
Низкое значение коэффициента теплоотдачи от дымовых га-
зов к теплообменной поверхности [15—30 ккал!(л2 • ч • град)] при-
водит к необходимости работы при значительной разности темпера-
тур. Оптимальное значение конечной температуры дымовых газов
составляет содержание экономической задачи, аналогичной рас-
смотренной выше (см. стр. 233), так как снижение расхода топ-
лива сопровождается увеличением поверхности теплообмена на-
гревательного устройства (при этом необходимо учитывать, что
условием естественной тяги является температура дымовых газов
не ниже 250° С; при более низкой температуре необходима уста-
новка дымососа).
При противотоке (трубчатые печи) температура уходящих из
печи дымовых газов на 100—150 град превышает температуру по-
ступающего потока сырья.
При необходимости изменения начальной температуры дымо-
вых газов меняется либо коэффициент избытка воздуха (для
трубчатых печей он равен 1,2—1,4), либо осуществляется рецир-
куляция дымовых газов. Последний прием более предпочтителен,
так как при этом уменьшается агрессивное воздействие избытка
кислорода на материал стенок обогреваемой поверхности (при на-
греве потока выше 400° С применяются легированные или специ-
альные жаростойкие стали).
Несмотря на возможность изменения температуры теплоноси-
теля в широком интервале, недостатки, присущие нагреву дымо-
выми газами — низкое значение коэффициента полезного действия
топок (30—40%), трудность регулирования температурного ре-
240
жима и пожароопасность в условиях производств органического
синтеза, — ограничивают его применение.
В химической и нефтехимической промышленности нагрев ды-
мовыми газами осуществляется в трубчатых печах, а также в ав-
токлавах (для установок небольшой производительности).
Теория процесса горения топлив, основы расчета и конструк-
тивное оформление печей изложены, например, в работах [40, 41].
Теплоносители. Насыщенный водяной пар производится
паровыми котельными химических предприятий или ТЭЦ при дав-
лениях не ниже Ризб = 54-6 ат. Для удобства транспорта пар пере-
гревается.
К цехам чаще всего подводится пар одного из двух давлений.
При необходимости использования пара
ния пар высокого давления редуцируется
с одновременным его увлажнением.
Наряду с многочисленными достоин-
, ствами нагрева водяным паром: высокое
значение коэффициента теплоотдачи,теп-
лоты конденсации и степени использо-
вания теплосодержания (до 85%), обес-
печивающими широкое применение это-
го способа в химической промышленно-
сти, нельзя не отметить особенностей
насыщенного водяного пара, затрудняю-
щих в ряде случаев возможность его
применения в качестве нагревающего
агента.
Характер зависимости P(t) (рис. Ш-7) обусловливает резкое
повышение давления пара в области высоких температур. Это об-
стоятельство ограничивает возможность достижения высокой тем-
пературы рабочей среды в отдельных типах теплообменных уст-
ройств при использовании парового нагрева (например, аппараты
с рубашками, кожухотрубные теплообменники с линзовыми ком-
пенсаторами) и предъявляет высокие требования к механической
прочности теплообменной аппаратуры. Кроме того, резкое сниже-
ние значения теплоты конденсации водяного пара в области высо-
ких температур неблагоприятно сказывается на степени использо-
вания теплосодержания пара.
Вода широко используется как теплоноситель для подвода и
отвода тепла. По понятным причинам верхний предел рабочей
температуры воды, лимитируемый давлением ее пара, не отли-
чается от соответствующей характеристики насыщенного водяного
пара. Теплоемкость воды в широком интервале температур ме-
няется незначительно.
Для нагрева или охлаждения в области высоких температур
вода используется в замкнутом, полностью заполненном контуре
тепловой генератор — холодильник при естественной или принуди-
тельной циркуляции.
промежуточного давле-
Рис. Ш-7. Зависимость
Р (I) для насыщенного во-
16 В. С. Хайлов, Б. Б. Брандт
241
При использовании воды в качестве нагревающего или охлаж-
дающего агента в области более низкой температуры (от 10 до
120—150° С) применяется речная вода, вода артезианская, кон-
денсат водяного пара, а также вода оборотных циклов промыш-
ленного предприятия.
Начальная температура артезианской воды независимо от вре-
мени года составляет 10—12° С. Температура воды из оборотного
цикла зависит от времени года и колеблется в пределах 20—30° С.
Температура воды на выходе из теплообменников зависит от
содержания солей и не превышает 40—60° С (за исключением кон-
денсата) во избежание отложения солей на поверхности тепло-
обмена.
В тех случаях, когда попадание воды в охлаждаемый ею ма-
териальный поток по тем или иным соображениям недопустимо
(изменение концентрации реагирующих веществ, течение нежела-
тельных процессов и т. п.), создаются условия, при которых дав-
ление материального потока превышает давление потока охлаж-
дающей воды и при неплотностях в теплообменной поверхности
утечка происходит в сторону потока воды. Для наблюдения за со-
стоянием теплообменного устройства (отсутствие течи) на выходе
потока воды из теплообменника может быть организован перио-
дический или ^непрерывный контроль за содержанием в воде ком-
понентов охлаждаемого материального потока-
Если конечная температура охлаждаемого потока превышает
100° С, целесообразно применение кипящей воды. Высокое значе-
ние коэффициента теплоотдачи от стенки к кипящей воде, повы-
шающееся с увеличением тепловой нагрузки, обеспечивает высо-
кую интенсивность теплообмена со стороны охлаждающего агента.
При необходимости температура кипения воды может быть изме-
нена соответствующим изменением давления. Естественно, что при
охлаждении кипящей водой следует применять конденсат водя-
ного пара.
В последнее время все более часто появляются сообщения о
применении воздуха в качестве охлаждающего агента [42, 43].
Воздушные теплообменники конструируются из труб круглого или
эллиптического сечения, снабженных ребрами и собранными в па-
кеты. Использование антикоррозионных покрытий снижает корро-
зионное действие воздушного потока и дает возможность впрыска
воды в поток охлаждаемого воздуха для увеличения тепловой на-
грузки в жаркое время года.
Варианты конструктивного оформления воздушных теплооб-
менников достаточно разнообразны (теплообменники с пенным
контактом, с кипящим слоем твердых частиц). Для экономии про-
изводственной площади трубные пакеты располагаются по на-
клонным сторонам равностороннего треугольника, в основании ко-
торого помещается вентилятор, а в вершине — входной коллектор.
Воздушные холодильники часто устанавливаются на крышах
зданий и в верхней части ректификационных колонн (последнее
243
относится к числу преимуществ воздушных холодильников перед
водяными, требующими в этом случае большого напора воды).
Подача воздуха осуществляется вентиляторами с напором до
25 мм вод. ст. Известны установки с тепловой нагрузкой до 26X
X 106_ккал/ч, снабженные вентиляторами мощностью до 360 кет
[42, 43].
Применение воздушных холодильников оправдано не только
недостатком воды. Экономические расчеты показывают, что сум-
марные расходы, связанные с применением воздушных холодиль-
ников, часто оказываются ниже затрат, сопровождающих приме-
нение теплообменников, охлаждаемых водой.
Применение воздуха в пенных теплообменниках и в качестве
возможности как хладо-
средства псевдоожижения расширяет <
агента для широкого диапазона тем-
ператур.
Органические и неорганические
жидкости находят все большее при-
менение в технологии органического
синтеза как теплоносители. Наряду с
кипящей водой, в качестве охлаждаю-
щих агентов при температуре выше и
ниже 100° С применяются кипящие
жидкости и сжиженные газы.
При охлаждении и нагреве в обла-
сти высоких температур используют-
ся высокотемпературные органические
теплоносители (дифенил, дифенилок-
сид, даутерм, минеральные масла, ди-
W 360 380 400 428
Рабочая температура,"С
Рис. Ш-8. Длительность рабо-
ты даутерма в зависимости от
рабочей температуры.
толилметан и др.) и кремнийорганические соединения (тетракре-
зилоксисилан, тетракрезилсиликат, тетраксиленоксисилан и др.),
применяемые в жидком или парообразном состоянии, а также в
состоянии кипения. Малое давление пара этих соединений дает
возможность достижения высоких рабочих температур (до 300—
350° С) при относительно низком давлении, выгодно отличая
их от воды. Обладая достаточной устойчивостью в условиях
высокой температуры, такие теплоносители при периодической за-
мене разложившегося продукта могут работать длительное время
(рис. Ш-8).
Несмотря на относительно низкую по сравнению с водой теп-
лоту парообразования даутерма (120 ккал[кг при Р = 1 ат), тепло-
содержание единицы объема пара воды и даутерма при одинако-
вом давлении соизмеримо.
Теплофизические свойства высокотемпературных теплоносите-
лей и аппаратурное оформление установок' связанных с их приме-
нением, приведены в работах [44, 45].
Сравнительная оценка эксплуатационных расходов установок
для нагрева насыщенным водяным паром высокого давления,
установок с использованием электронагрева и установок для
16*
243
нагрева с помощью органических теплоносителей приводит к вы-
воду в пользу последнего способа. Преимущества нагрева с по-
мощью высокотемпературных теплоносителей особенно заметны
для установок большой мощности. Так, для установки с тепло-
вым потоком 2,5 • 105 ккал/ч расходы на эксплуатацию в случае
применения высокотемпературных органических теплоносителей в
два раз ниже, чем при нагреве паром, и почти в четыре раза ниже,
чем при использовании электронагрева [46].
Для работы в области более низких температур в качестве
хладагентов могут применяться углеводороды и спирты, а также
ряд сжиженных газов, используемых при температуре их кипения.
Из числа последних наибольшее применение находят аммиак, про-
пан и этан. Температура кипения сжиженного газа легко регули-
руется соответствующим изменением давления. Парообразный
хладагент, покидающий теплообменник, либо возвращается для
повторного использования после конденсации в специально для
этой цели организованной установке, либо применяется в техноло-
гической схеме.
Водные растворы солей (холодильные рассолы) используются
как охлаждающие агенты в области температур, более низких,
чем температура замерзания воды. В промышленных установках
находят применение водные растворы хлоридов натрия, кальция
и магния. Выбор соли и ее концентрации в растворе зависит от
минимальной температуры холодильной установки.
Растворы хлористого натрия применимы до температуры
—16° С, растворы хлористого кальция — до —50° С.
Коэффициенты теплопередачи и теплоотдачи. Расчетная
часть работы, связанной с выбором или проектированием теплооб-
менной аппаратуры, сводится к решению системы уравнений теп-
лового баланса и теплообмена, причем число неизвестных, как
правило, превышает число возможных уравнений.
Обычно задача решается методом последовательных прибли-
жений путем просчета ряда вариантов, основанных на произволь-
но выбираемых значениях одной из неизвестных величин. К числу
этих неизвестных величин в первую очередь относятся коэффици-
енты теплопередачи или теплоотдачи, являющиеся функцией ско-
рости и физических свойств потока.
Скорость движения теплоносителей, определяемая сечением
каналов теплообменного аппарата, колеблется в очень широких
пределах и без большой погрешности не может быть принята или
установлена до решения вопроса о типе и размерах теплообмен-
ного аппарата.
Для оценки порядка величин поверхности теплообмена и вы-
бора на этом основании типа и конструкции теплообменного ап-
парата представляется целесообразным использование данных о
значениях коэффициента теплопередачи для различных пар тепло-
носителей, установленных экспериментально или найденных в ре-
зультате эксплуатации промышленных аппаратов.
244
Рис. Ш-9. Двухсекционный
змеевиковый теплообменник.
При пользовании указанными в приложениях III—XII значе-
ниями коэффициентов теплопередачи и теплоотдачи следует дове-
рять только порядку величин для последующего уточнения их зна-
чений с учетом конкретных условий проектируемого аппарата.
Основные типы рекуперативных теплообменников
При выборе типа рекуперативного теплообменника для
работы в заданной области условий необходим учет особенностей
каждого из конструктивных вари-
антов.
Змеевиковые теплообменники,
широко применяемые в производст-
вах основного органического син-
теза, оформляются в двух вариан-
тах: 1) погружные спиральные или
плоские змеевики; 2) оросительные
плоские змеевики.
Погружные теплообменники
представляют собой цилиндриче-
скую или плоскую спираль, изготов-
ленную из труб и погруженную в
сосуд, через который проходит один
из теплоносителей; труба змеевика
служит каналом для другого тепло-
носителя (рис. Ш-9). В подавляю-
щем большинстве случаев погруж-
ные змеевиковые теплообменники ис-
пользуются как холодильники или
конденсаторы (охлаждаемый поток
движется по трубам змеевика).
Вследствие большого объема со-
суда скорость движения теплоноси-
теля, омывающего змеевик, относи-
' тельно мала (даже при условии ус-
тановки внутреннего стакана), по-
этому условия теплообмена между
трубой змеевика и охлаждающим
агентом близки к условиям естест-
венной конвекции. Неблагоприят-
ным для теплообмена условием является также то обстоятельство,
что вследствие малой скорости теплоносителя заметно ощущается
влияние конвективных токов в слое теплоносителя, заполняю-
щего внешний сосуд. Результатом этого будет выравнивание тем-
пературы потока и соответственное уменьшение средней разности
температур.
По понятным причинам такие недостатки отсутствуют в случае
обогрева змеевика конденсирующимся паром.
245
Ограничения, налагаемые предельным значением скорости по-
тока в трубах змеевика и диаметром труб (до 75—100 мм), выну-
ждают прибегать к устройству теплообменников с рядом парал-
лельно соединенных змеевиков. Это решение диктуется также
учетом условий теплообмена при подаче в трубы змеевика конден-
сирующегося пара.
В последнем случае ограничивают скорость пара на входе в
змеевик (для водяного пара при охлаждении змеевика водой не
рекомендуется скорость пара, превышающая 30 м/сек) и соблю-
дают предельное отношение длины секции змеевика к диаметру
трубы (L/d).
В зависимости от давления водяного пара при средней разно-
сти температур между паром и водой от 30 до 40 град максималь-
ное отношение L/d равно:
Р, ат.................... 5 3 1,5 0,8 0,5
L/d................... 275 225 175 125 100
При значении средней разности температур, отличной от 30—
40 град, предельные значения L/d следует умножать на поправоч-
ный коэффициент б/У А/ср [29].
При подаче в змеевик конденсирующегося пара ввод следует
делать сверху, а конденсационный горшок ставить как можно ниже
теплообменника, чтобы избежать заполнения нижней части змее-
вика конденсатом.
- Змеевики выполняются из металлических (сталь, чугун) труб,
а также из углеграфитовых композиций и стекла. Для агрессив-
ных сред применяются змеевики, изготовленные из борсиликат-
ного стекла, отличающегося устойчивостью к резкому изменению
температуры, и пригодные для работы под давлением до РЛЗб =
=3,5 ат. Имеются сообщения о применении змеевиков из тефлона.
Недостатки погружного змеевикового теплообменника — гро-
моздкость, значительная металлоемкость — в значительной мере
окупаются рядом бесспорных достоинств; простота конструк-
ции, возможность изготовления на строительной площадке и ма-
лая чувствительность к режиму подачи охлаждающей воды
обеспечивают змеевиковым теплообменникам достаточно широкое
применение.
Оросительные теплообменники. Оросительными называ-
ются теплообменные устройства, в которых охлаждающей средой
является жидкость, свободно стекающая на поверхности теплооб-
менника в виде тонкой пленки.
Оросительные теплообменники могут быть выполнены в самых
разнообразных конструктивных вариантах (цилиндрический змее-
вик, пучок вертикальных труб и т. п.), однако наиболее широко
известны в промышленной практике теплообменные аппараты,
оформленные в виде плоских одно- или многосекционных змееви-
ков с горизонтально расположенными трубами. В качестве охлаж-
246
дающей жидкости используется вода, подаваемая на верхний ряд
(или ряды) труб с помощью специального устройства, обеспечи-
вающего равномерное распределение ее по длине труб и по гори-
зонтальным рядам змеевика. Стекающая с труб вода собирается
в расположенный под теплообменником стальной или бетонный
кювет.
Для оросительного теплообменника характерен многократный
перекрестный ток теплоносителей.
В отличие от погружных змеевиковых теплообменников, которые
могут быть использованы как для нагрева, так и для охлаждения
рабочей среды с применением разнообразных нагревающих и ох-
лаждающих агентов, оросительные теплообменники используются
только в качестве холодильников и конденсаторов.
Основные преимущества оросительных теплообменников перед
погружными заключаются в большем значении коэффициента теп-
лоотдачи от стенки трубы к охлаждающей воде (лимитирующего
коэффициент теплопередачи в погружных теплообменниках), мень-
шем расходе воды вследствие частичного испарения и еще боль-
шей простоте конструкции и доступности для осмотра.
В отличие от погружных теплообменников, в которых скорость
подачи охлаждающей воды может быть без большого ущерба для
режима теплообмена колебаться в весьма широких пределах,
плотность орошения труб оросительного холодильника следует
поддерживать в определенных пределах: при подаче менее 200—
250 кг/(м-ч) необходима очень точная установка оросительных
желобов для равномерного орошения труб по горизонтали и вер-
тикали, а подача воды в количестве, превышающем 1500 кг/(м-ч),
практически бесполезна, так как часть ее сливается мимо нижних
рядов труб.
В одной из последних работ, посвященной изучению теплоот-
дачи со стороны орошения, приводится зависимость коэффициента
теплоотдачи (а') от стенки труб к орошающей их воде от плот-
ности орошения:
а = 122Г0,4 (для s/d =1,7-5- 2,0)
а/ = 5ОГ0,56 (для s/d =1,3)
где T = G/2L — линейная плотность орошения, кг/(м-ч); G — рас-
ход воды, кг/ч\ L — длина верхней трубы; s—шаг; d — диаметр
трубы.
Значение а' мало меняется в зависимости от изменения диа-
метра труб н скорости движения окружающего теплообменник
воздуха (рис. 111-10); отмечается увеличение а' при повышении
температуры охлаждающей воды, объясняемое уменьшением вяз-
кости жидкости и толщины стекающей пленки.
Трубы оросительных теплообменников изготавливаются из раз-
личных металлов, а также из стекла и углеграфитовых компо-
зиций.
347
Известны оросительные холодильники из борсиликатного стек-
ла, применяемые для охлаждения серной кислоты и выгодно отли-
чающиеся по весу и стоимости от свинцовых.
' Углеграфитовые оросительные холодильники изготавливаются
из труб диаметром от 25 до 150 мм с общей поверхностью тепло-
обмена до 360 м2. Соединение труб осуществляется с помощью уг-
испарение
конструк-
наружной
Г, кг/(и ч>
Рис. III-10. Зависимость коэф-
фициента теплоотдачи от стен-
ки трубы оросительного те-
плообменника к воде от линей-
ной плотности орошения:
l-sld=l,7 - 2,0; 2~s/d= 1,3.
аппаратов
леродистого цемента.
К числу основных достоинств оросительных холодильников ОТ-
НОСЯТСЯ в первую очередь небольшой удельный расход охлаждаю-
щей воды (около 50% теплового потока расходуется на
воды), а также простота
ции и легкость очистки
поверхности труб.
Недостатками таких
являются их громоздкость, интен-
сивная коррозия наружной по-
верхности металлических труб от
воздействия кислорода воздуха и
повышенная чувствительность к ре-
жиму подачи воды.
Тонкослойные теплооб-
менные аппараты. Нагрев термиче-
ски нестойких жидкостей часто со-
провождается нежелательными про-
цессами деструкции и осмоления,
приводящими к потере продукта и за-
грязнению поверхности теплообмена,
проведения процесса нагрева в усло-
сводящих к минимуму) возможность
Задача решается путем
виях, исключающих (или
местных перегревов при минимальной продолжительности пребы-
вания продукта в интервале температур, вызывающих его неже-
лательные превращения. Эти условия удовлетворяются в аппара-
тах, где процесс нагрева осуществляется в тонкой пленке при тур-
булентном режиме ее течения.
Подобный режим может быть осуществлен в любом трубчатом
теплообменнике при установке в его трубы вытеснителей, создаю-
щих кольцевую щель (при надлежащей центровке) шириной до
1,0—2,0 мм. В целях сокращения длины кольцевой щели вытесни-
тель может быть использован для подачи в него теплоносите-
ля [37].
Аналогичные условия имеют место в пластинчатых теплооб-
менниках, где волнистый канал благоприятствует турбулизации
потока и конструкция позволяет регулировать продолжительность
пребывания жидкости в зоне высоких температур.
Ряд конструкций теплообменных аппаратов основан на тепло-
обмене между стенкой и тонкой пленкой жидкости.
По направлению движения пленки жидкости тонкопленочные
теплообменные аппараты различаются на теплообменники свспол-
248
зающей и падающей пленкой. Для обработки термолабильных и
вязких жидкостей во многих случаях более предпочтительными
оказываются теплообменники с всползающей пленкой [47].
На рис. Ш-11, а схематически изображен испаритель для вяз-
ких жидкостей. Поверхность нагрева образована трубой, обогре-
ваемой изнутри паром. Видоизменение этого же принципа исполь-
Рис. Ш-11. Некоторые типы пленочных теплообменников.
зовано в конструкции, показанной на рис. 111-11,6. Изменение тол-
щины пленки жидкости при движении ее от центра нагреватель-
ного элемента к его периферии и в обратном направлении, а так-
же при стекании с одного элемента на другой турбулизует поток
и благоприятствует теплообмену. Аппараты таких типов эмалиро-
ваны [48].
На рис. Ш-11, в изображен трубчатый теплообменник. Этот
аппарат может быть использован как испаритель жидкости, по-
даваемой на верхнюю трубную решетку, а также как реактор для
проведения реакций в жидкой фазе со значительным тепловым
эффектом.
Принцип свободно стекающей пленки использован в конструк-
циях теплообменных аппаратов с вращающимися лопастями (рис.
III-11,г).
Поверхность теплообмена, образуемая стенками цилиндри-
ческого или конусообразного корпуса аппарата, обогревается
249
снаружи теплоносителем. По оси аппарата расположен вал ме-
шалки, приводимый в движение приводом, установленным на
крышке аппарата (для аппаратов, работающих под вакуумом, вал
мешалки соединен с герметическим приводом).
Лопасти либо жестко крепятся на валу, либо соединяются шар-
ниром и прижимаются к стенке под влиянием центробежного
ускорения.
Выпариваемая жидкость подается на вращающуюся распреде-
лительную тарелку и после равномерного распределения по пери-
метру стекает по обогреваемой стенке корпуса, турбулизуясь
вращающимися лопастями. Время пребывания жидкости в зоне
нагрева составляет от 3 до 100 сек.
Концентрат удаляется из нижней части аппарата. Пары отво-
дятся из верхней его части. Аппараты с вращающимися лопастя-
ми хорошо зарекомендовали себя при упаривании очень вязких
продуктов.
Коэффициент теплоотдачи от стенки к пленке жидкости зависит
от свойств последней, а также от числа оборотов мешалки и дости-
гает 1200—2000ккал/(м2-ч-град). Тепловая нагрузка при испаре-
нии органических жидкостей составляет 50000—55000 ккал/(м2-ч)
в случае обогрева паром.
Разновидностью тонкослойного теплообменника с ротором яв-
ляется аппарат, в котором свободно стекающая по обогреваемой
стенке пленка жидкости через небольшие интервалы высоты по-
падает на вращающийся диск и вновь отбрасывается к периферии.
Этот вариант конструкции исключает возможность непрерывного
воздействия мешалки на пленку жидкости и не требует высокой
точности при изготовлении и монтаже перемешивающего устрой-
ства [49].
В последнее время такому типу теплообменной аппаратуры
уделяется значительное внимание [50]. Ряд зарубежных фирм
выпускает аппараты с различной величиной поверхности тепло-
обмена (до 21 м2), применяемые в ряде отраслей химической про-
мышленности [37, 51—53] для испарения термолабильных или
очень вязких продуктов, при сгущении пенящихся жидкостей, для
быстрого удаления легкокипящих компонентов (обезвоживание
жиров, глицерина и т. п.), для дистилляции высококипящих про-
дуктов (жирные кислоты, аминокислоты, капролактам и др.).
Конструкции с охлаждаемым ротором используются как рек-
тификационные аппараты.
Имеется сообщение [54] об использовании аппаратов такого
типа в качестве реакторов для' пастообразных и термически не-
стойких веществ.
Кожухотрубные теплообменники. Наибольшее распро-
странение во всех отраслях химической промышленности имеют
кожухотрубные теплообменники, применяемые для систем, меняю-
щих и не меняющих свое агрегатное состояние.
250
Размеры кожухотрубных теплообменников весьма различны
и колеблются от долей квадратного метра до нескольких тысяч
квадратных метров.
Рис. Ш-12. Кожухотрубные теплообменники [38].
Конструкции кожухотрубных теплообменников отличаются
большим разнообразием. Схемы устройства основных типов таких
теплообменников приведены на рис. Ш-12.
Разница между отдельными вариантами конструкций обуслов-
лена различием в подходе к решению некоторых основных во-
просов:
1) приближение площади сечения межтрубного пространства
к площади проходного сечення труб;
2) компенсация температурных деформаций;
3) удобство чистки поверхности теплообмена;
4) снижение металлоемкости аппарата.
251
В кожухотрубных теплообменниках при наиболее часто при-
меняемом расположении труб в трубчатой решетке сечение меж-
трубного пространства в 1,5—1,8 раза превышает проходное сече-
ние труб, вследствие чего скорость потока теплоносителя в меж-
трубном пространстве недостаточна и при неизменном агрегатном
состоянии теплоносителя коэффициент теплопередачи между по-
токами лимитируется коэффициентом теплоотдачи от внешней по-
верхности труб. Решение этого вопроса достигается установкой
перегородок (рис. Ш-12, б), применением многоходовой схемы
движения потока (рис. Ш-12, и), а также применением теплооб-
менника типа «труба в трубе», допускающего к тому же возмож-
ность оребрения труб.
Отсутствие возможности компенсации температурных деформа-
ций в теплообменниках с прямыми трубами жесткой конструкции
(рис. Ш-12, а) ограничивает возможность их применения раз-
ностью температур между корпусом и стенками труб величиной
порядка 45—50 град.
Разность температур между стенкой корпуса и стенками теп-
лопередающих труб в подавляющем большинстве случаев превы-
шает 45—50 град. Вследствие этого возникает проблема компен-
сации температурных напряжений.
Установка линзовых и сальниковых компенсаторов увеличи-
вает допустимую разность температур между стенками корпуса и
труб, однако устройства эти сложны в изготовлении и недоста-
точно надежны в работе в условиях большого давления или боль-
шого перепада давлений между потоками теплоносителей.
Более совершенной является конструкция аппаратов с жестким
креплением одной трубной доски и свободным перемещением труб
в корпусе. Варианты такого решения представлены S- и U-образ-
ными гнутыми трубами (рис. Ш-12, г и д), допускающими воз-
можность значительного их удлинения. Трудность изготовления и
замены труб с разным радиусом изгиба, а- также сложность их
очистки ограничивают применение такого типа теплообменной ап-
паратуры.
Свободными от ряда указанных недостатком являются тепло-
обменники с так называемой «плавающей головкой». Некоторое
удорожание аппарата из-за увеличения диаметра корпуса и уст-
ройства дополнительного днища и крышки опр-авдываются про-
стотой и надежностью его в эксплуатации.
Представляют интерес распространенные в технике глубокого
холода витые теплообменники (рис. III-13). Конструкция этих теп-
лообменников обеспечивает компенсацию температурных дефор-
маций при неподвижных трубных решетках и значительную по-
верхность теплообмена в единице объема аппарата.
Такие теплообменники пригодны только для теплоносителей,
не содержащих твердых примесей и не дающих отложений на
внутренней и наружной поверхностях труб, чистка которых меха-
ническими методами затруднена.
252
Секционные теплообменники, являющиеся разновидностью ко-
жухотрубных, состоят из нескольких последовательно соединен-
ных секций, каждая из которых представляет собой трубчатку с
малым количеством труб, помещенную в корпусе небольшого диа-
метра.
В секционных аппаратах, сравнительно простых по конструк-
ции, достигаются благоприятные для теплообмена условия. Малый
диаметр корпуса облегчает температурную компенсацию с по-
мощью линзовых компенсаторов даже при сравнительно высоком
давлении в межтрубном пространстве. К недостаткам этих аппа-
ратов следует отнести относительно высокую их стоимость (на
единицу теплообменной поверхности) и высокое сопротивление.
Рис. Ш-13. Витые кожухотрубные теплообменники.
Разновидностью секционных теплообменников являются аппа-
раты типа «труба в трубе», представляющие собой наиболее про-
стой вариант этого типа теплообменных устройств. Возможность
подбора диаметра внешней трубы в соответствии с желаемой ско-
ростью потока в кольцевом канале позволяет обеспечить высокое
значение коэффициента теплоотдачи с обеих сторон теплообмен-
ной поверхности, а малый диаметр внешней трубы облегчает про-
ведение процессов под высоким давлением теплоносителя.
Известны [55] теплообменники типа «труба в трубе» с внутрен-
ними трубами из тантала и стекла, помещенными в эмалирован-
ную внешнюю трубу, предназначенную для работы в сильноагрес-
сивных средах.
В случае разборной конструкции теплообменники типа «труба
в трубе»- удобны для чистки внутренней трубной и межтрубной
поверхностей. Естественно, что удельный (на единицу теплообмен-
ной поверхности) расход материала для этого типа, аппаратов
высок.
Для работы под высоким давлением кожухотрубные теплооб-
менники помещаются в поковки высокого давления. Конструкция
аппарата в этом случае рассчитана только на перепад давления
между потоками. ।
253
Конструкционным материалом кожухотрубных теплообменни-
ков чаще всего служит сталь различных марок; для работы с аг-
рессивными средами используются титан и тантал, а также неко-
торые неметаллические материалы. К числу последних следует
отнести углеграфитовые композиции и применяемый в последнее
время тефлон. Известны [55] теплообменники «стекло — титан» с
поверхностью теплообмена 4 и 8 м2. Титановые трубы рассчитаны
на работу под давлением 25 ат при температуре 200° С; корпус из
стекла марки «Пирекс» выдерживает давление до 1,5 ат.
Кожухотрубные теплообменники из графита, аналогичные по
конструкции теплообменникам из металлических материалов, вы-
полняются в кожухе из стали или полностью из углеграфитового
материала. В первом случае важным является компенсация тем-
пературных деформаций, особенно заметных ^следствие большой
разницы значений коэффициентов температурного расширения
графита и металла; в этих целях применяются либо сальниковые
устройства, либо «плавающая головка».
Поверхность теплопередачи углеграфитовых теплообменников
достигает 360 м2, давление в аппарате — до Ризб = 5 ат [56].
Углеграфитовые теплообменники успешно заменяют не только
теплообменники из легированных сталей, но также титановые.
Обследование ряда отечественных производств, использующих уг-
леграфитовые теплообменники [57], свидетельствует о том, что
амортизационные и эксплуатационные расходы, отнесенные к 1 м2
таких теплообменников, в 2—4 раза ниже соответствующих пока-
зателей, характеризующих работу теплообменников из металла.
Ряд зарубежных фирм [58—61] выпускает кожухотрубные теп-
лообменники из тефлона, применяемые в производствах органиче-
ского синтеза (охлаждение ксилола, латексной эмульсии, конден-
сация паров бензола и толуола и т. п.). Незначительная адгезион-
ная способность тефлона позволяет пользоваться трубами малого
диаметра (2,5—6,5 мм). Толщина стенок труб при этом состав-
ляет 0,25—0,3 мм. Несмотря на незначительную теплопроводность
тефлона, коэффициент теплопередачи через тефлоновую перего-
родку указанной толщины достаточно высок и сохраняет свое зна-
чение при длительной работе в условиях, допускающих выпадение
осадков на поверхности теплообмена.
Теплообменники из тефлона рассчитаны на работу при давле-
нии до Ризб=20 ат и температуре 150° С. Такие аппараты компакт-
ны и относительно дешевы.
Аппараты с рубашками и змеевиками. Рубашка — тип
теплообменного устройства, употребляемого главным образом для
реакционного оборудования, — представляет собой сосуд, окру-
жающий корпус аппарата. Кольцевое пространство, образуемое
корпусом аппарата и стенкой рубашки, служит каналом для теп-
лоносителя. В стенке рубашки устанавливаются штуцеры для вво-
да и вывода теплоносителя и для удаления неконденсирующихся
газов.
254
Пар подается в верхнюю часть рубашки, конденсат отводится
снизу. Жидкие теплоносители могут подаваться как снизу, так и
сверху (в последнем случае необходимо устройство гидрозатвора
для заполнения рубашки). При необходимости снижения темпе-
ратуры стенки корпуса аппарата,
обогреваемой паром, последний мо-
жет быть подан в нижнюю часть
рубашки, которая таким образом
будет заполнена конденсатом, отво-
димым от верхнего штуцера.
Рубашки изготавливаются из уг-
леродистых сталей и рассчитыва-
ются на давление, создаваемое теп-
лоносителем.
В зависимости от конструкции
аппарата рубашка либо крепится
на фланцах к корпусу аппарата,
либо приваривается. Для компенса-
ции температурных деформаций
(если в этом есть необходимость)
устанавливаются линзовые или саль-
никовые компенсаторы.
' Корпус аппарата, снабженного
рубашкой, находится под действи-
ем внешнего давления, т. е. в бо-
лее тяжелых условиях, чем стенки
рубашки. Вследствие этого предель-
ным давлением теплоносителя в ру-
башке принято считать РИЗб =
= 8 н- 10 ат.
Применение штампованных ру-
башек, соединяемых с корпусом кон-
тактной сваркой (рис. Ш-14), раз-
гружает корпус аппарата; внешнее
давление при этом может быть по-
вышено до 70—80 ат.
Другим вариантом конструкции,
пригодным для осуществления теп-
лообмена стенки цилиндрического
аппарата с теплоносителем, имею-
Рис. Ш-14. Варочный котел со
штампованной рубашкой:
а —внешний вид аппарата; б—схема
крепления рубашки;
1 — рубашка; 2 — стенка корпуса аппарата.
щим высокое давление, являются змеевики, приваренные к стенке
корпуса. Змеевики выполняются либо из труб, уложенных на мед-
ные прокладки, либо из фасонной стали. Последний вариант про-
ще в изготовлении, обеспечивает хорошую теплопередачу и при-
годен для работы под давлением 50—60 ат.
Тепловой расчет такого типа теплообменных аппаратов редко
приводится в руководствах по теплообмену и изложен в специаль-
ной работе [62].
255
В тех случаях, когда по конструктивным соображениям разме-
щение в аппарате змеевика или трубчатки не представляется воз-
можным, а поверхность рубашки недостаточна, применяют тепло-
обменные элементы, погружаемые в рабочую среду.
Если теплоносителем является жидкость или пар, нагреватель-
ный элемент выполняется в виде так называемой «трубы Фильда».
Теплоноситель вводится по трубке, открытый конец которой рас-
положен вблизи днища трубы Фильда; конденсат пара или отра-
ботанный жидкий теплоноситель выводится через патрубок, рас-
положенный на крышке теплообменного элемента (рис. Ш-15).
Рис. Ш-16. Кольцевой
теплообменник-диффузор.
Рис. Ш-15. Теплообменный
элемент.
Другой конструктивный вариант теплообменного элемента —
кольцевой теплообменник — в некоторых случаях может быть ис-
пользован вместо труб Фильда для обогрева или охлаждения ра-
бочей среды; в аппаратах с мешалками кольцевой теплообменник
может выполнять роль диффузора (рис. Ш-16).
Шнековые теплообменники. Для нагрева или охлажде-
ния твердых сыпучих материалов, а также вязких жидкостей в
некоторых случаях может быть использован шнековый теплооб-
менник, представляющий собой закрытое корыто, в котором вра-
щаются два полых шнека (рис. Ш-17).
Обрабатываемый материал поступает через люк в верхней
крышке аппарата и удаляется через нижний люк. При вращении
шнеков материал в корыте перемещается вдоль его оси и тщатель-
но перемешивается, соприкасаясь в то же время с греющими по-
верхностями корпуса и шнеков.
Теплоноситель может подаваться в рубашку, которой снабжен
корпус аппарата, или в полый вал шнеков.
Спиральные, пластинчатые и блочные теплообменники.
Одно из основных направлений совершенствования и интенсифи-
856
кации теплообменной аппаратуры основано на создании конструк-
ций, характеризующихся большим значением поверхности тепло-
обмена на единицу габаритного объема (компактность) н пригод-
ных для использования в системах с малым коэффициентом теп-
Рис. Ш-17. Шнековый теплообменник:
1 — устройство для подвода теплоносителя; 2 — пустотелые шнеки; 3 — кор-
пус теплообменника; 4 — приводной механизм.
лопередачи. К числу таких типов теплообменной аппаратуры от-
носятся спиральные, пластинчатые и блочные теплообменники.
В спиральных теплообменниках поверхность теплообмена со-
здается металлическими листами, свернутыми по спирали. Обра-
зуемые между листами каналы, имеющие выход в центре и на пе-
риферии цилиндра, ограничиваются с торцов двумя крышками
Пар
Рис. Ш-18. Вертикальный и горизонтальный спиральные теплооб-
менники:
/, 2 —листы; 3 — разделительные перегородки; 4 — крышки;
/, // — теплоносители.
(рис. Ш-18). Спиральные теплообменники горизонтального типа—
противоточные; применяют их при теплообмене между двумя
жидкостями. Теплообменники вертикального типа используются
при теплообмене между паром и жидкостью (теплоносители дви-
жутся либо противотоком, либо перекрестным током).
17 В. С. Хайлов, Б. Б. Брандт 257
Для уменьшения теплопотерь в окружающую среду холодный
теплоноситель рекомендуется пропускать по наружному каналу.
Спиральные теплообменники характеризуются компактностью,
высоким значением коэффициента теплопередачи между потоками
теплоносителей и относительно малым гидравлическим сопротив-
лением. К числу основных их недостатков в первую очередь отно-
сится трудность уплотнения в местах контакта листов с торцовыми
крышками, ограничивающая верхний предел давления теплоноси-
теля величиной порядка Ризб = 3 -> 6 ат-, упоминания о случаях
применения спиральных теплообменников при давлении Рцз5 > 6 ат
крайне редки.
Несмотря на ряд достоинств, спиральные теплообменники име-
ют сравнительно ограниченное применение в промышленности ор-
ганического синтеза.
Конструкция спиральных теплообменников крайне затрудняет
или полностью исключает возможность ремонта поверхности на-
грева.
Величина коэффициента теплопередачи от воды к воде при
скорости движения потоков, равной 1,8—2,2 м/сек, в спиральном
теплообменнике достигает 2400 ккал/(м2 • ч • град).
Предельными показателями по компактности и удельному рас-
ходу металла на единицу поверхности теплообмена обладают теп-
лообменные аппараты, известные под названием пластинчатых
[36, 63, 64].
В настоящее время известен ряд вариантов конструктивного
оформления пластинчатых теплообменников, различающихся по
степени их разборности и по профилю поверхности теплообмена:
разборные пластинчатые, неразборные пластинчато-ребристые и др.
Принцип устройства разборных пластинчатых теплообменников
основан на прохождении потоков двух теплоносителей по обе сто-
роны пластины, входящей в состав пакета, собранного из пластин
аналогично пакету плит и рам рамного фильтрпресса. Направ-
ляющие фиксируют пластины и обеспечивают совмещение отвер-
стий, расположенных по углам пластин, образующих после сборки
пакета коллекторный канал для распределения теплоносителей по
параллельным каналам теплообменного устройства.
Пакет пластин стягивается опорной стойкой и нажимной пли-
той, имеющими отверстия для ввода и вывода теплоносителей.
Уплотнение пластин и коллекторных отверстий достигается с по-
мощью резиновых прокладок, плотно прижимаемых к поверхности
смежных пластин нажимным устройством.
Для увеличения поверхности теплообмена поверхность пла-
стин делается гофрированной с таким расчетом, чтобы зазор ме-
жду двумя смежными пластинами (расстояние 3—6 мм) представ-
лял волнистый канал прямоугольного сечения. Волнистый про-
филь канала сильно турбулизирует поток теплоносителя, так что
турбулентный режим достигается при значениях критерия Рей-
нольдса, не превышающих 180—200.
258
Потоки, между которыми осуществляется теплообмен, движут-
ся по каналам между пластинами, омывая противоположные сто-
роны каждой из них. Способы включения каналов могут быть
самыми разнообразными: параллельными, последовательными,
комбинированными. При параллельном включении каналов тепло-
носитель распределяется одновременно по всем (через один) ка-
налам, при последовательном — проходит поочередно через каж-
дый из этих каналов.
Одна из возможных схем движения теплоносителей в теплооб-
меннике, собранном из 15 пластин, показана на рис. Ш-19.
а — противоток; б — комбинированная схема;
1 — 16 — секции.
Теплоноситель, поступающий слева, проходит (рис. Ш-19, б)
четыре (/, 3—5, 7—9, 11—13, 15) секции, распределяясь в каж-
дой из них по двум параллельным каналам. Поступающий справа
теплоноситель проходит последовательно две секции (10, 12, 14,
16—2, 4, 6, 8), в каждой из которых распределяется по четырем
параллельным каналам.
Широкие возможности комбинирования распределения потока
по каналам позволяет использовать пластинчатые теплообменники
при самом различном соотношении объема потоков (до 1 : 10)..
При обработке жидкостей, склонных к образованию осадков
или пригоранию, можно при соответствующем расположении от-
верстий в плитах последовательным включением каналов сокра-
тить время пребывания жидкости в зоне высоких температур по-
тока, уменьшив тем самым вероятность течения нежелательных
процессов.
17
259
Пластины чаще всего изготавливаются из нержавеющей стали,
которая хорошо полируется и устойчива в отношении коррозии,
из титана, и его сплавов, из медно-никелевых сплавов и т. д.
Поверхность теплообмена одной пластины, составляющая 65—
80% поверхности, соответствующей габаритным размерам, лежит
в пределах от 0,1 до 0,6 м2 при толщине пластины от 1 до 3 мм.
Общая поверхность теплопередачи теплообменников рассмотрен-
ного типа достигает 150—160 м2 (данные 1958 г.).
Предельные давление и температура в пластинчатых теплооб-
менниках составляют соответственно РИзб=15 ат и 200—300° С.
Вопросы теплопередачи и гидравлики пластинчатых теплооб-
менников освещены в ряде работ [36, 56, 63, 64], технические и
эксплуатационные показатели некоторых пластинчатых теплооб-
менников— в обзоре [65].
Выгодные особенности разборных пластинчатых теплообмен-
ников — высокое значение коэффициента теплопередачи при отно-
сительно невысоком сопротивлении, небольшой расход материала
иа единицу поверхности теплообмена, большая компактность, про-
стота разборки для очистки поверхности теплообмена, возмож-
ность осуществления разнообразных вариантов поточности тепло-
носителей— делают этот тип теплообменных аппаратов, пока еще
сравнительно мало распространенный в технологии основного ор-
ганического синтеза, наиболее пригодным для осуществления ряда
новых процессов.
Неразборные пластинчато-ребристые теплообменники имеют
несколько конструктивных вариантов. Принцип их устройства до-
статочно прост: между плоскими листами толщиной 0,5—0,8 мм
помещается дополнительная теплообменная поверхность в форме
гофрированного листа или прутка, изогнутого в виде плоского
змеевика, образующего каналы. Высота ребер, соответствующая
расстоянию между пластинами, колеблется от 4—5 до 12—13 мм.
Пакет таких элементов с необходимым расположением кана-
лов (для прямотока, противотока или перекрестного тока тепло-
носителей) образует теплообменник.
Материал пластин и дополнительной теплообменной поверхно-
сти— алюминий или медь. Соединения листов и ребер произво-
дятся пайкой, осуществляемой по специально разработанной тех-
нологии.
Полная поверхность теплообмена, отнесенная к единице объ-
ема, ограниченного двумя параллельными пластинами, весьма ве-
лика и достигает 1500—1800 м21м3.
Максимальное рабочее давление для известных типов пластин-
чато-ребристых теплообменников не превышает Рпзб=21 ат.
Материалом для изготовления пластинчатых теплообменников,
кроме металла, служат также углеграфитовые композиции. Пла-
стинчатые теплообменники из графита с поверхностью теплооб-
мена до 11,0 м2 применяются дЛя процессов, при проведении ко-
торых оба потока агрессивны,
260
Рис. Ш-20. Круглоблочный графитовый теплообменник.
В химической промышленности распространены также блочные
теплообменники, отличающиеся компактностью, удобством мон-
тажа, чистки и ремонта. Блочные теплообменники (прямоугольные
и круглоблочные) выпускаются с поверхностью теплообмена до
50 ж2 для работы под давлением до РИзб — 6 ат (рис. Ш-20).
Интенсификация конвективного теплообмена
Наряду с интенсификацией теплообменной аппаратуры
путем использования развитых поверхностей и осуществления теп-
лообмена в тонкой пленке, известны и получили некоторое рас-
пространение методы и устройства для повышения интенсивности
конвективного теплообмена турбулизацией потока теплоносителя.
Такими устройствами являются различного рода турбулизи-
рующие вставки, помещаемые в потоке теплоносителя, трубы с
переменным поперечным сечением, устройства для изменения на-
правления потока и т. п.
Возможности этого направления сравнительно ограничены, од-
нако в ряде случаев реализация рассматриваемых ниже приемов
может представить значительный интерес.
Турбулизация потока теплоносителя достигается в результате
помещения в поток вставок, изменяющих направление потока и
создающих дополнительное сопротивление.
В зависимости от конструкции вставки, характера течения и
свойств теплоносителя значение коэффициента теплоотдачи между
стенкой и потоком может быть увеличено от 1,5 до 5 раз по срав-
нению с показателями, характеризующими гладкую трубу.
В качестве турбулизирующих вставок используется ряд уст-
ройств: диафрагмы, кольцевые, спиральные или дисковые вставки
и т. п.
При наличии диафрагмы и вставок переход от ламинарного
режима потока к турбулентному происходит при значительно
меньших значениях критерия Рейнольдса. Так как определяющим
значение а' является термическое сопротивление пограничного
слоя, больший эффект достигается при использовании диафрагм,
чем дисковых вставок, турбулизирующих ядро потока.
Вставки оказывают особенно благоприятное действие в пере-
ходной области режима течения, препятствуя затуханию турбулент-
ности, и позволяют увеличить коэффициент теплопередачи в 2—
3 раза по сравнению с К для гладких труб.
Эффект, достигаемый применением вставок, может быть полу-
чен при заполнении канала зернистым материалом (сферическими
частицами, кольцами и т. п.). Сопоставление влияния размера ча-
стиц насадки на величину коэффициента теплоотдачи и на сопро-
тивление потоку приводит к выводу в пользу относительно крупных
частиц насадки, при которых интенсификация теплообмена сопро-
вождается относительно малым увеличением сопротивления.
262
Трубы с переменным по длине сечением, получаемые соответ-
ствующей обработкой обыкновенных труб, обеспечивают турбу-
лизацию потока и увеличение интенсивности теплообмена. Так же
как в случае турбулизирующих вставок, в трубах с переменным
сечением обеспечивается переход в турбулентную область при
меньшем, чем в гладкой трубе, значении критерия Рейнольдса и
соответствующее увеличение коэффициента теплопередачи. Послед-
нее наблюдается в ламинарной и переходной областях,и мало за-
метно в области интенсивной турбулентности.
Вполне понятно, что эффект изменения коэффициента тепло-
передачи, достигаемый применением вставок и насадок, в значи-
тельной степени зависит от соотношения значений коэффициентов
теплообмена по обеим сторонам теплопередающей поверхности.
При относительно малом значении а' со стороны вставки увеличе-
ние коэффициента теплопередачи за счет действия этого турбули-
зирующего устройства может быть практически равным (в %) уве-
личению значения коэффициента теплообмена, оправдывая тем са-
мым затраты, связанные с изготовлением и применением вставки.
В противоположном случае даже значительное увеличение а' мало
сказывается на величине К.
Целесообразность применения турбулизирующих вставок и на-
садок каждый раз устанавливается на основе результатов сравне-
ния эффекта, достигаемого интенсификацией теплопередачи, с до-
полнительными затратами, вызываемыми увеличением сопротивле-
ния потоку теплоносителя.
Наряду с рассмотренными выше способами интенсификации
теплообмена для любого (жидкого или газообразного) агрегат-
ного состояния потока, при интенсификации теплообмена между
газом и стенкой следует учитывать возможность использования
свойств псевдоожиженного слоя и слоя вспененной жидкости.
Коэффициент теплоотдачи между псевдоожиженным слоем и
стенкой, мало зависящий от свойств газа, оценивается величинами
порядка десятков и сотен кал/(м2 ч • град). Это обстоятельство мо-
жет быть использовано при необходимости интенсификации тепло-
обмена между потоком газа и стенкой: в„ теплообменных аппара-
тах, предназначенных для нагрева или охлаждения газа, газовый
поток, пропущенный через слой зернистого материала при скорости
псевдоожижения последнего, дает возможность увеличить значе-
ние коэффициента теплоотдачи со стороны газа по крайней мере
на один порядок. При выборе такого способа интенсификации теп-
лообмена следует учитывать унос частиц твердой фазы потоком
газа и необходимость установки соответствующей аппаратуры (см.
стр. 427).
Для повышения интенсивности теплообмена со стороны газо*
вого потока следует также иметь в виду возможность использова-
ния свойств барботажного или пенного слоя. Высокая степень дис-
персности газа в слое жидкости или пены и сравнительно большое
значение коэффициента теплоотдачи между барботажным слоем
263
или слоем пеиы и теплообменной поверхностью обеспечивают вы-
сокое значение коэффициента теплопередачи, отнесенного к еди-
нице площади поверхности теплообмена (см. стр. 393).
Устройство пенных теплообменников крайне просто (рис. Ш-21).
Воздух, подаваемый под решетку пенного аппарата, покидает слой
пены при температуре на 2—3 град более низкой, чем темпера-
тура пены, практически полностью на-
сыщенной парами воды. Коэффици-
ент теплопередачи от охлаждаемого
потока к слою пены достигает 300—
380 ккал/{м~ч-град) [66], причем зна-
чение коэффициента теплоотдачи от
стенки теплообменной поверхности к
слою пены сравнительно мало зависит
от скорости воздуха [67].
Сравнение расхода воздуха для
воздушного и пенно-контактного теп-
лообменников свидетельствует о пре-
имуществе последнего [66].
эффективности теплообменной аппара-
Рис. Ш-21. Схема пенного теп-
лообменника.
Основные показатели
туры— компактность, выраженная размером поверхности тепло-
обмена, приходящейся на единицу габаритного объема аппарата
(в л42/л£3), и металлоемкость, выражаемая количеством металла,
отнесенного к единице теплообменной поверхности (в кг/м2),— за-
висят от размера аппарата, улучшаясь с увеличением поверхности
теплообмена.
' Сравнительная характеристика важнейших типов теплообмен-
ной аппаратуры дана в табл. Ш-5.
Таблица II1-5
Сравнительные показатели теплообменников некоторых типов [56]
Теплообменники Пределы применения Компакт- ность, мУ/м^ Металло- емкость, кг!м2
температура, °C давление, ат
Кожухотрубные 800 1500 40 30
Витые От -252 200 70 —
«Труба в трубе» до +50 800 1000 20 150
Змеевиковые погружные 200 1000 15 100
Оросительные 100 100 10 60
Пластинчатые: разборные 150 25 100 25
полуразбориые 200 40 60 40
Пластинчато-ребристые .От -250 32 900-1400 —
Спиральные до +100 200 10 60 50
С рубашкой 300 10 — —-
264
ТЕПЛООБМЕН СМЕШЕНИЕМ
При теплообмене смешением тепло передается от одного
потока к другому в результате непосредственного контакта тепло-
обменивающихся потоков.
Теплообмен смешением применим лишь в тех случаях, когда
условия технологического процесса допускают непосредственный
контакт потоков и неизбежное изменение состава каждого из них.
В зависимости от агрегатного состояния теплоносителей
[газ (пар)—жидкость, газ — газ, жидкость — жидкость, газ —
твердое тело] и их свойств процесс теплообмена смешением может
сопровождаться изменением агрегатного состояния одного из по-
токов или происходить без изменения агрегатного состояния. В по-
следнем случае взаимное направление движения теплоносителей
может быть осуществлено противотоком или параллельным током.
Теплообменники смешения используются для конденсации па-
ров, охлаждения или нагрева газов, твердого зернистого материала
и нагрева агрессивных жидкостей.
Наиболее распространенным типом аппаратов для теплообмена
между газом (паром) и жидкостью являются конденсаторы смеше-
ния и скрубберы.
Основные типы теплообменников смешения
Конденсаторы смешения. Эти аппараты применяются
при конденсации паров воды в вакуум-выпарных установках, а
также для конденсации паров углеводородов при переработке
нефти, однако принцип их действия может быть использован для
значительно более широкого круга процессов.
В конденсаторах смешения пары и охлаждающая жидкость,
диспергированная различными способами (форсунки, орошаемая
насадка, струйный поток и т. д.), приводятся в контакт, в резуль-
тате чего происходит конденсация пара. Конденсат и охлаждаю-
щая жидкость в виде одной или двух фаз, а также несконденси-
рованные газы (наиболее частый случай) непрерывно выводятся
из аппарата.
Конденсаторы смешения, предназначенные для конденсации па-
ров воды под вакуумом, различаются по способу отвода отрабо-
танной воды. В широко известных барометрических конденсаторах
вода отводится через гидрозатвор (барометрическая труба), вы-
сота которого устанавливается с учетом остаточного давления в
аппарате и сопротивления трубы потоку жидкости. Несконденси-
ровавшиеся газы через брызгоуловитель направляются в вакуум-
насос.
Конденсаторы смешения, работающие без гидрозатвора, приме-
няются в тех случаях, когда установка барометрической трубы по
каким-либо соображениям невозможна. Отработанная вода уда-
ляется с помощью насоса,
265
саторы смешения
'3
Рис. Ш-22. Двух-
ступенчатый кон-
денсатор смеше-
ния:
/ — парогазовая смесь;
2 — охлаждающая во-
да; 3 — неконденси-
рующиеся газы;
4 — охлажденный кон-
денсат продукта;
5 — отработанная вода
(60—70° С); 6 — отрабо-
танная вода(30-35° С);
7-конденсат продук-
та.
Удаление несконденсировавшихся газов в обоих случаях про-
изводится с помощью вакуум-насоса из верхней части аппарата.
Взаимное направление потоков воды и парогазовой смеси в кон-
денсаторах смешения может быть параллельным или встречным.
Предпочтительным является противоток.
Конструкция конденсаторов смешения, работающих при атмо-
сферном давлении, не отличается от конструкции абсорбционной
аппаратуры.
По сравнению с рекуперативными теплообменниками конден-
характеризуются значительно меньшей металло-
емкостью, простотой изготовления и небольшими
габаритами. Поскольку конечная температура
охлаждающей воды и конденсата равны, удель-
ный расход воды выше, чем в поверхностных
конденсаторах. Для случая, когда конденсат и
охлаждающая жидкость взаимонерастворимы,
применение двухступенчатого охлаждения
(рис. Ш-22) в значительной степени устраняет
этот недостаток.
Скрубберы. Такие аппараты исполь-
зуются для охлаждения газовых смесей путем
непосредственного контакта с охлаждающей
жидкостью, которая одновременно служит для по-
глощения пыли или тумана и увлажнения газа.
Конструктивно скрубберы оформляются ана-
логично аппаратуре, применяемой для абсорб-
ционных процессов (колонны с насадкой, рас-
пыление жидкости форсункой и т. п.).
Теория процесса, схема расчета и принципы
конструктивного оформления конденсаторов
смешения и скрубберов для охлаждения газа
изложены в работах [68, 69].
Пенные холодильники. Такие аппара-
ты являются эффективными устройствами для
охлаждения газовых и парогазовых смесей. При
охлаждении влажного воздуха водой, когда
происходит конденсация пара, коэффициент теплопередачи, отне-
сенный к единице поверхности решетки (1 м2), в зависимости от
скорости воздуха в свободном сечении решетки и высоты слоя
пены достигает величины 30000—40000 ккал(ч • град) [70].
Нагрев агрессивных жидкостей
При использовании разбавленных водных растворов ми-
неральных кислот нагрев их в рекуперативных теплообменниках
связан с необходимостью применения коррозионноустойчивых теп-
лопроводных материалов. Задача осложняется, если процесс про-
водится при повышенном давлении.
266
Смешение холодной концентрированной кислоты с потоком го-
рячей воды или насыщенного водяного пара в футерованном
неметаллическими материалами аппарате или трубопроводе по-
зволяет сравнительно просто решить этот вопрос. При составле-
нии теплового баланса процесса необходимо учитывать теплоту
разбавления кислоты.
Нагрев газов
При атмосферном и повышенном давлении нагрев
газов может быть осуществлен в теплообменнике смешения. При-
мером может служить нагрев воздуха путем смешения его с ды-
мовыми газами в топках, работающих под давлением (установки
каталитического крекинга, установки для сушки и т. п.). Продукты
горения жидкого или газообразного топлива обычно смешиваются
с воздухом, добавляемым в необходимом соотношении.
Теплообмен смешением между двумя потоками газа проводится
в контактных аппаратах с кипящим слоем катализатора, куда по-
дается газовая смесь при сравнительно низкой температуре.
При смешении парогазовых смесей, имеющих различную тем-
пературу, следует учитывать условия возникновения пересыщен-
ного состояния пара и возможность образования тумана.
Теплообмен смешением
в системе газ — твердое тело
Такой теплообмен осуществляется при охлаждении и
нагреве катализатора в период пуска или остановки контактного
аппарата, а также при охлаждении адсорбентов после регенера-
ции. Процесс охлаждения, осуществляемый при нестационарном
тепловом и температурном режиме, рассчитывается (если в этом
возникает необходимость) по схеме, принятой для расчета регене-
ративных теплообменников.
Стационарный режим теплообмена между газом и твердым те-
лом имеет место"в процессе нагрева или охлаждения потока зер-
нистого материала потоком воздуха или реакционного газа на
установках с движущимся или псевдоожиженным слоем катализа-
тора (см. стр. 412).
ТЕПЛОВАЯ ИЗОЛЯЦИЯ
Удельный расход тепловой энергии зависит не только от
потребления ее на нагрев или охлаждение материальных потоков,
но и от потерь в окружающую среду. Величина этих потерь, отне-
сенная к единице поверхности теплообмена с окружающей средой,
определяется разностью между средней температурой материаль-
ного потока и температурой окружающей среды, а направление
267
теплового потока — отношением средних температур потока и
окружающей среды.
Снижение размера потерь тепла достигается увеличением со-
противления тепловому потоку при уменьшении теплопроводности
стенки в результате применения теплоизоляционных материалов.
Потери тепла нагретой аппаратурой и трубопроводами учиты-
ваются не только при расчете удельного расхода тепло-энергети-
ческих средств, но и при составлении теплового баланса помеще-
ния в связи с проектированием вентиляционных устройств. Верх-
ний предел допустимых пбтерь тепла от нагретой аппаратуры и
трубопроводов диктуется соображениями безопасности. Макси-
мальная температура внешней поверхности изоляции не должна
превышать 55° С.
Теория и техника тепловой изоляции подробно рассмотрены в
работе [71].
При решении задач, связанных с оптимизацией теплоизоля-
ционных устройств аппаратов цилиндрической формы, следует
иметь в виду критические размеры диаметра изоляции
j __ 2ЛИЗ
<*кр-7—
, анар
(“нар ~ коэффициент теплоотдачи от наружной поверхности изоля-
ции в окружающую среду), соответствующего максимуму тепло-
вых потерь с единицы поверхности цилиндрического сосуда, по-
крытого слоем изоляционного материала, коэффициент теплопро-
водности которого равен Я,из.
При увеличении толщины слоя изоляционного материала, по-
крывающего цилиндрический сосуд, поток тепла вначале увели-
чивается и после достижения внешним диаметром изоляции зна-
чения, равного dKp, начинает уменьшаться.
Таблица 111-6
Характеристика некоторых теплоизоляционных материалов [31]
Теплоизоляционный материал Кажущаяся плотность, кг/м3 Допустимая температура, °C Коэффициент теплопроводности, ккал/(м • ч • град)
Асбест 500 0,091 +0,00016
400 ЛЛЛ 0,081 +0,00016
200 0,064 + 0,00014
100 0,052 + 0,00014
Асбоцементные плиты 400 1 45П 0,076 + 0,00011
300 0,068 + 0,00010
Асбестовый картон 1000 450 0,135 + 0,00012
Асбестовый шнур 750 450 0,153 + 0,000084
Войлок шерстяной 300 90 0,04 +0,00017
Пробковые плиты 150-200 — 0,036 + 0,046
268
Рис. Ш-23. Определение оп-
тимальной ТОЛЩИНЫ ИЗОЛЯ-
ЦИИ.
i
' Если среднее значение а'ар принять равным 9—
10 ккал! (м2 • ч • град) и предположить, что минимальный диаметр
промышленного трубопровода составляет 25 мм, группа изоля-
ционных материалов, для которых обязательным является учет
указанной выше зависимости, ограничится значением коэффи-
циента теплопроводности Хиз 0,1 4-0,15 ккал/(м •чград). Коэф-
' фициенты теплопроводности некоторых теплоизоляционных мате-
риалов приведены в табл. Ш-6.
В тех случаях, когда коэффициенты теплопроводности распро-
страненных теплоизоляционных материалов оказываются недоста-
точными, применяется многослойная изо-
ляция, обладающая весьма низким зна-
чением Хиз- Коэффициент теплопроводно-
сти изоляции, изготовленной из тонких
пленок пластических материалов, чере-
дующихся с металлическими экранами
(2—4 слоя на 1 мм толщины слоя изо-
ляции), составляет (24-3)- 10-5'ккал/(мХ
Хч • град) [72].
Ясно, что для материалов, коэффи-
циент теплопроводности которых ниже
0,1 ккал/ (м- ч • град), учет критического
значения диаметра изоляции лишен
практического смысла.
Рассмотренная закономерность имеет
значение не только при решении задач,
направленных на уменьшение тепловых потерь, но также при ре-
шении противоположных задач.
Применение теплоизоляционных материалов связано с необхо-
димостью решения задач на определение минимума суммы затрат
на эксплуатацию теплоизоляционного устройства:
Ф = Q' + Д = f (диз) + <р (биз)
где Q' — стоимость тепловых потерь; А — размер амортизацион-
ных отчислений; 6ИЗ— толщина изоляции.
Графическое изображение зависимости Ф(биз) представлено на
рис. Ш-23. Нахождение значения ФМин должно быть основано на
тщательном учете конкретных условий.
При изоляции аппаратов и трубопроводов, располагаемых вне
производственных помещений, следует иметь в виду возможность
резкого повышения значения Хиз для гигроскопических теплоизоля-
ционных материалов. Необходимо также учитывать теплопотери от
фланцев и арматуры, как правило, не изолируемых.
ЛИТЕРАТУРА
1. Н. М. О лен ев, Хранение нефти, нефтепродуктов и газа, Гостоптех-
издат, 1951.
2. Т. А. Пектемиров, Справочник инженера и техника нефтебаз, Гос-
топтехиздат, 1954.
269
3. В. И. Титов, В. И. Богданов, А. И Макаров, Проектирование и
строительство нефтебаз, Гостоптехиздат, 1953.
4. Нормы и технические условия проектирования складских предприятий и
хозяйств для хранения легковоспламеняющихся и горючих жидкостей (НиТУ
105—56) ТК СМ СССР по делам строительства.
5. Н. М. Оленев, Б. В. Мишин, Неметаллические резервуары для хра-
нения нефти и нефтепродуктов, Гостоптехиздат, 1957.
6. В. С. Грей, Железобетонные резервуары и емкости, Гостоптехиздат,
1956.
7. В. И. К а н д е е в, Е. Ф. Котляр, Газгольдеры, ОНТИ, 1936.
8. А. Б. Ар око в, Газгольдеры, Госхимиздат, 1933.
9. Н. А. Бакланов, Транспортировка жидкостей в химических производ-
ствах, Машгиз, 1962.
10. Новые конструкции центробежных насосов для перекачки нефти, нефте-
продуктов и сжиженных газов, ГОСИНТИ, 1961.
11. Н. К- Переверзин, Новые насосы для химической промышленности,
изд. ЦБТИ, 1961.
12. Герметические центробежные электронасосы в химической промышлен-
ности, изд. НИИТЭИ, 1965.
13. Ю. С. П о д о б у е в, А. П. Селезнев, Теория и расчет осевых и цен-
тробежных компрессоров, Машгиз, 1957.
14. В. Е. Лисичкин, А. М. Горшков, Компрессорные машины, Машгиз,
1957.
15. С. Н. Г а и з, Технологические процессы и оборудование производств
синтез-газа и связанного азота, Изд. ХГУ, Харьков, 1960.
16. Е. А. Иванов, А. В. Шепелев, Е. В. Лялин, Трубопроводы в хи-
мической промышленности, Машгиз, 1963.
17. Н. А. Бакланов, Трубопроводы в химической промышленности, Гос-
химиздат, 1953.
18. Л. А. К У з и е ц о в, Б. В. Р у д о м и и о, Конструирование и расчет трубо-
проводов теплосиловых установок, Машгиз, 1949
19. М. М. Сапожников, Неметаллические напорные трубопроводы, Гос-
стройиздат, 1957.
20. И. Е. Шапиро, Е. Г. Фролова, Стеклянные трубы, Госстройиздат,
1960.
21. П. А. Гладких, С. А. Хачатурян, Вибрация в трубопроводах и
методы ее устранения, Машгиз, 1959.
22. А. А. Лещинский, А. Р. Т о л ч и н с к и й, Основы конструирования
и расчета химической аппаратуры. Справочник, Машгиз, 1963.
23. А. Д. Домашнее, Конструирование и расчет химических аппаратов,
Машгиз, 1961.
24. С. Робертс, Динамическое программирование в процессах химической
технологии и методы управления, Изд. «Мир», 1965.
25. Р. V. R i 1 е у, Chem. a. Process Eng., 47, № 2, 55 (1966).
26. К. G- Е 1 a n g, Oil a. Gas, 64, № 40, 138 (1966).
27. Chem. Process, 29, № 10, 136 (1966).
28. W. H. Holstein, Chem. Eng. Process, 62, № 3, 107 (1966).
29. К- Ф. Павлов, П. Г. P о м а и к о в, А. А. Носков, Примеры и задачи
по курсу процессов и аппаратов химической технологии. Изд. «Химия», 1964.
30. М. А, Михеев, Основы теплопередачи, Госэнергоиздат, 1956.
31. А. А. Кутателадзе, В. М. Боришанский, Справочник по тепло-
передаче, Госэнергоиздат, 1959.
32. В. М. Рам м, Теплообменные аппараты, Госхимиздат, 1950.
33. Г. Хоблер, Теплопередача и теплообменники, Госхимиздат, 1961.
34. Б. Б а л а й к а, К- Сикора, Процессы теплообмена в аппаратах хими-
ческой промышленности, Машгиз, 1962.
35. А. Д. Домашне в, Конструирование и расчет химических аппаратов,
Машгиз, 1961.
36. Ю. В. Петровский, В. Г. Ф а с т о в с к и й, Современные эффективные
теплообменники, Госэнергоиздат, 1962.
270
37. Ф. М. Тарасов, Тонкослойные теплообменные аппараты, Изд. «Ма-
шиностроение», 1964.
38. П. А. Антикайн, М. С. Аронович, А. М. Б а к л а с т о в, Реку-
перативные теплообменные аппараты, Госэнергоиздат, 1962.
39. Р. А. Долинская, ИФЖ, 7, № 9, 127 (1964); 8, № 1, 128 (1965).
40. С. В. Адельсон, Технологический расчет и конструктивное оформле-
ние нефтезаводских печей, Гостоптехиздат, 1952.
41. А. И. С к о б л о, И. А. Трегубова, Н. Н. Егоров, Процессы и аппа-
раты нефтеперерабатывающей и нефтехимической промышленности, Гостоптех-
издат, 1962.
42. J. A. Henle у, Chem. a. Process Eng., 44, № 8 (1963).
43. Н. К a s s a t, Chem. Ingr. Techn., 38, № 9, 987 (1966).
44. А. В. Чечетки н, Высокотемпературные теплоносители, Госэнергоиздат,
1957.
45. Н. П. Долинин, Установки для нагрева химической аппаратуры высо-
котемпературными органическими теплоносителями, Машгиз, 1963.
46. А. М. Lloyd, J. A. Macintosh, W. Z. Р a d u с h, Chem. a. Process
Eng., 46, № 8 (1965); Suppl.: Heat Transfer Survey, p. 445.
47. Chem. Process., 11, № 17, 10 (1965).
48. G. H о c h g e s a n d, Chem. Ingr. Techn., 36, № 12, 1243 (1964).
49. Chem. Eng., 70, № 5, 64, 66 (1963).
50. H. И. Патрикеева, Хим. пром, за рубежом, № II, 58 (1966).
51. А. В. М u t z е n b е г g, Chem. Eng., 79, № 19, 175 (1965).
52. N. Parker, Chem. Eng., 72, № 19, 179 (1965).
53. R. Fischer, Chem. Eng., 72, № 9, 186 (1965)'.
54. R. Barnabos, Chem. Techn., 16, № 11, 649 (1963).
55. Г. M. Векслер, Ю. ЛА. Виноградов, H. M. Самсонов, Хим. и
нефт. маш., № 5, 18 (1966).
56. Л. АА. Коваленко, В. П. К о к о ш а, А. Р. Я с т р е б е н е ц к и й,
Теплообменные аппараты за рубежом, изд. ЦИНТИАМ, 1963.
57. К. А. Бородина, А. В. Горяйнова, Жх'рн. ВХО, 10, № 1, 55
(1965).
58. Chem. Process, 28, № 8, 61. 67 (1965).
59. Chem. Eng. News, 43, № 22, 44 (1965).
60. Chem. Process, 29, № 12, 80 (1966).
61. R. R. Hood, Chem. Eng., 74, № 11, 181 (1967).
62. H. П. Долинин, Хим. пром., № 3, 234 (1960).
63. H. В. Барановский, Пластинчатые теплообменники в пищевой про-
мышленности, Машгиз, 1962.
64. И. И. Ра ц, Конструкция, исследование и расчет пластинчатых тепло-
обменников, Изд. ЦИНТИАМ, 1962.
65. Л М. Коваленко, Вести, техн, и эконом, информ., НИИТЭХИМ,
№ 4, 47 (1959).
66. A. Poll, W. Smith, Chem. Eng., 71, № 22, 111 (1964).
67. S. В r e t s z n e i d e r, W. К a w e c k i, W. D b i e d z i c, Bull. Acad. Polon.
Sci., 10, № 3, 155 (1962); РЖХим, 1963, 1ИЗЗ.
68. H. Al. Ж а в о p о н к о в, Гидравлические основы скрубберного процесса
и теплопередача в скрубберах, Изд. «Сов. наука», 1944.
69. Н. Н. Егоров, Охлаждение газов в скрубберах, Госхимиздат, 1954.
70. М. Е. П о з и н, И. П. Alyx ле но в, Э. Я. Т а р а т, Пенные газоочисти-
тели, теплообменники и абсорберы, Госхимиздат, 1959.
71. А. М. Факторов и ч, Тепловая изоляция, Изд. «Недра», 1966.
72. L. G. М a t s с h, Adv. Gryog. Eng., 7, 413 (1962); РЖХим., 1964, 23Л156.
Глава IV
ПРЕВРАЩЕНИЕ СЫРЬЯ
И АППАРАТУРНОЕ ОФОРМЛЕНИЕ
ХИМИЧЕСКИХ СТАДИЙ ПРОЦЕССОВ
ТЕХНОЛОГИЧЕСКИЕ ОСОБЕННОСТИ
РЕАКЦИОННОЙ АППАРАТУРЫ
[Понятие «реакционная аппаратура» охватывает ком-
। плекс устройств, обеспечивающих создание и под-
держание условий, необходимых для превращения исходного веще-
ства в смесь продуктов химического процесса, параметры которой
позволяют передавать ее на дальнейшую обработку.
Большинство процессов, осуществляемых в реакционной аппа-
ратуре, представляет собой совокупность химических, тепловых
диффузионных и гидромеханических явлений.
Состав технологического оборудования, предназначенного для
осуществления химического процесса, крайне редко ограничивается
одним аппаратом, в котором протекает химическая реакция; обычно
в него входят устройства, предназначенные для использования
тепла реакционной смеси, нагрева поступающего сырья, цирку-
ляции и т. п. В некоторых случаях эти устройства оформлены в
виде отдельных аппаратов, в других — размещены в реакционном
пространстве или в общей обечайке и составляют реакционный
агрегат.
Проектирование реакционной аппаратуры — одна из наиболее
сложных задач, возникающих при создании химического произ-
водства. Успешное ее решение зависит не только от полноты све-
дений о каждом из явлений, составляющих химический процесс,
но и главным образом от полноты сведений о количественной взаи-
мозависимости этих явлений.
При переходе от масштабов лабораторных или стендовых уста-
новок к масштабу производственных агрегатов возникают серьеэ
ные затруднения, если условия подобия модели и проектируемого
реактора, а также условия поперечной равномерности реагирую-
щего потока не соблюдаются. Применение методов физического и
особенно математического моделирования химических процессов
[1—6] открывает пути решения подобных задач, однако современ-
ное состояние теории моделирования и масштабирования пока
ограничивает ее возможности сравнительно простыми объектами.
272
Основные требования к типу и принципу конструкции реакци-
онного агрегата диктуются необходимостью достижения макси-
мального выхода целевого продукта при возможно более высо-
кой степени превращения сырья. Выполнению этого требования
должны быть подчинены другие показатели работы реакционной
аппаратуры:
1) размер реакционного пространства и вспомогательных
устройств, отнесенный к единице готового продукта, получаемого
в единицу времени (производительность реакционного агрегата);
2) автотермичность процесса;
3) возможность автоматического управления процессом;
4) простота изготовления реакционного агрегата, доступность
и низкая стоимость конструкционных материалов.
Для большого числа процессов основного органического синтеза
выход целевого продукта и степень превращения сырья связаны
между собой [0(a)] и оптимальный вариант находится как мини-
мум функции
Ф = На, ₽,...)
где Ф — сумма энерго-материальных затрат на единицу готового
продукта.
Характер зависимости 0(a) обусловлен химической схемой и
природой реакции и является основой для выбора типа и органи-
зационного принципа работы реакционной аппаратуры (периоди-
ческий, непрерывный, полунепрерывный процесс).
Если организационный принцип по тем или иным соображе-
ниям уже задан, указанная зависимость используется при выборе
оптимального значения а.
Производительность реакционной аппаратуры по целевому про-
дукту для реакции всех типов является функцией степени превра-
щения сырья. Так как значение а, как правило, устанавливается
при анализе зависимости Ф(а, 0, ...), то тем самым предопреде-
ляется производительность единицы объема реакционного прост-
ранства.
Степень автотермичности процесса, определяющая не только
удельный расход энергии, но и размер теплообменных устройств
реакционного агрегата, в ряде случаев является о^ним из важ-
нейших показателей совершенства аппаратурного оформления про-
цесса.
Легкость управления процессом, простота конструкции реак-
ционного агрегата, стоимость и доступность конструкционных ма-
териалов не поддаются строго количественной оценке, однако эти
показатели (особенно последний) часто играют значительную роль
при характеристике конструктивных достоинств реакционной ап-
паратуры.
Основными количественными показателями совершенства аппа-
ратурного оформления реакционного процесса при оптимальном
значении 0 в первую очередь могут служить производительность
13 В. С. Хайлов, Б. Б. Брандт
273
по целевому продукту (Z) и расход энергии на единицу готового
продукта.
Многообразием признаков, характеризующих реакционную
аппаратуру, можно объяснить отсутствие до настоящего времени
единого подхода к ее классификации. Часто применяемый вариант
классификации основан на особенностях конструкции реакцион-
ного аппарата, определяющих его производительность. Этот ва-
риант не учитывает организационного принципа действия реакто-
ров и разделяет их на три основные группы:
1) емкостная реакционная аппаратура, производительность ко-
торой определяется только размерами реакционного пространства;
2) теплообменная реакционная аппаратура, производитель-
ность которой зависит от относительного размера теплообменных
устройств;
3) реакционная аппаратура, производительность которой опре-
деляется размером и конструкцией устройств, предназначенных в
основном для интенсификации процессов массообмена.
Каждую из указанных выше групп приходится рассматривать,
подразделяя ее на реакционные аппараты периодического и непре-
рывного действия (последние в свою очередь необходимо разде-
лить по признаку приближения к реакторам идеальных режимов).
Изменчивость взглядов на критерии и принципы классификации
реакционной аппаратуры в ходе развития учения о реакционных
процессах вполне закономерна. Исходя из задач технологии на
современном этапе ее развития, в качестве основного принципа
оценки реакционного аппарата следует, по-видимому, рассматри-
вать характеризующую данный аппарат и экономически наиболее
выгодную при осуществлении определенного процесса форму свя-
зей р(а) и Z(a).
На первом этапе разработки нового технологического процесса,
исходя в основном из масштаба производства, устанавливается
организационный принцип действия реакционной аппаратуры. Не-
которые процессы в промышленных условиях осуществляются
только в непрерывнодействующих аппаратах, так как при исполь-
зовании реакторов периодического действия проведение этих про-
цессов либо нецелесообразно (быстро текущие реакции), либо не-
безопасно. При организации многотоннажного производства, когда
более предпочтительно непрерывное проведение процесса, выбор
группы непрерывнодействующих реакторов проводится на основе
результатов сравнительной оценки реакционных аппаратов идеаль-
ных режимов и аппаратов промежуточного типа (независимо от
фазового состава и состояния реакционной смеси).
Конструктивный принцип устройства реактора определяется
количеством и состоянием фаз реакционной системы. Выбор опти-
мального варианта основан на учете специфических особенностей
перерабатываемой системы. Детали конструкции аппарата уточ-
няются в процессе проведения работ на стендах и опытных уста-
новках.
274
ПЕРИОДИЧЕСКОЕ, НЕПРЕРЫВНОЕ
И ПОЛУНЕПРЕРЫВНОЕ ПРОВЕДЕНИЕ ПРОЦЕССОВ
Проведение любого химического процесса в промышлен-
ных условиях слагается в общем случае из ряда этапов, основ-
ными из которых являются:
1) подготовка сырья перед его химическим превращением (за-
грузка сырья, смешение компонентов, нагрев или охлаждение
смеси, создание давления и т. д.);
2) химическое превращение сырья (подвод или отвод тепла
реакции, перемешивание);
3) подготовка реакционной смеси к выделению целевого про-
дукта (охлаждение смеси, разделение фаз, выгрузка реакционной
массы из аппарата).
Проведение этих стадий во времени в общем случае может
быть либо последовательным, либо одновременным; кроме того,
часть операций может проводиться одновременно, а часть — после-
довательно.
Число вариантов аппаратурного оформления процесса ограни-
чено соображениями целесообразности. В промышленной практике
переработка сырья проводится либо последовательно в одном аппа-
рате, либо одновременно в разных аппаратах, либо одновременно-
последовательно в одном аппарате. В соответствии с этим разли-
чают реакционную аппаратуру периодического, непрерывного и по-
лунепрерывного (полупериодического) действия.
В качестве примера рассмотрим реакцию, протекающую в двух-
фазной жидкой среде при повышенной температуре. Химическое
превращение сырья сопровождается выделением тепла и газооб-
разных продуктов. Процесс проводится в изотермических условиях.
Реакционная масса перед ее дальнейшей переработкой охлаж-
дается до комнатной температуры. Ниже приведены возможные ва-
рианты проведения подобного процесса в промышленном масштабе.
Реакционный агрегат периодического действия
В реакционный агрегат периодического действия в за-
данном соотношении и количестве загружаются исходные веще-
ства. После загрузки сырье перемешивается и нагревается до
определенной температуры, при которой химическое взаимодей-
ствие протекает с необходимой скоростью.
Во время проведения химической реакции, сопровождающейся
выделением тепла и газообразных продуктов, необходимая темпе-
ратура реакционной смеси поддерживается путем отвода реакци-
онного тепла с помощью охлаждающих устройств.
После окончания реакции смесь продуктов охлаждается до за-
данной температуры и передается на дальнейшую переработку.
Для подготовки к следующей операции реактор подвергается ос-
мотру и, если это необходимо, очистке.
18* 275
Продолжительность операции, т. е. время от начала загрузки
исходных веществ предыдущей операции до начала загрузки ис-
ходных веществ последующей операции, является суммой продол-
жительностей отдельных этапов:
Подготовка сырья:
загрузка исходных веществ..............
перемешивание и нагрев.................
Химическое превращение сырья, отвод тепла
реакции, отвод реакционных газов...........
Подготовка реакционной смеси к выделению
продукта:
охлаждение реакционной массы...........
выгрузка реакционной массы . ..........
очистка и осмотр реактора .............
Продолжительность операции.................
Т1П
Вполне естественно, что совокупность аппаратов или устройств,
составляющих реакционный агрегат, должна быть рассчитана на
возможность осуществления каждого из указанных выше этапов,
Рис. IV-1. Реакционный агрегат периодического действия:
а — аппаратурная схема; б — температурный режим процесса; в —тепловой режим
процесса.
т. е. агрегат должен иметь перемешивающее устройство, устройство
для нагревания смеси исходных веществ и устройство для охла-
ждения реакционной смеси (если одно из них не может быть
использбвано для выполнения обеих функций), а реакционный
сосуд должен быть рассчитан на максимальное давление в рабо-
чий период.
Ясно также, что материал всех аппаратов и устройств, находя-
щихся в соприкосновении с реакционной смесью, должен быть
стойким к ее агрессивному воздействию при максимально жестких
условиях.
Схема агрегата, предназначенного для проведения рассматри-
ваемого процесса, приведена на рис. IV-l,a. Графическое изобра-
276
жение температурного и теплового режимов процесса дано соот-
ветственно на рис. 1У-1,бив.
Продолжительность работы каждого из аппаратов или устройств
реакционного агрегата в ходе выполнения операции меньше об-
щей продолжительности операции. Степень (коэффициент) исполь-
зования аппарата или устройства по прямому его назначению вы-
ражается отношением:
(для реакционного пространства
тр Тр
топ Л + Тр
где Л — ti-Vtiji).
Если продолжительность механических (загрузка, выгрузка),
теплофизических (нагрев, охлаждение) и гидромеханических (пе-
ремешивание) процессов может быть сокращена до минимума пу-
тем интенсификации действия соответствующих устройств, то
сокращение продолжительности химической реакции, определяемой
кинетическими особенностями процесса, зачастую связано с зна-
чительными трудностями.
Для медленно текущих реакций (тр>>Л) коэффициент исполь-
зования реакционного пространства приближается к единице, в то
время как коэффициент использования вспомогательных аппара-
тов и устройств является весьма малой величиной.
Параметры процесса при его проведении в агрегате периоди-
ческого действия (температура реакционной смеси, давление в
реакционной зоне) меняются во времени. Так как автоматическое
регулирование нестационарных процессов требует сложной аппа-
ратуры, на практике эксплуатация реакционных агрегатов пе-
риодического действия производится с минимальным использова-
нием автоматики. Поэтому такие агрегаты требуют постоянного
наблюдения.
Реакционный агрегат непрерывного действия
Распределение отдельных этапов каждой операции по
аппаратам, приспособленным для выполнения определенных функ-
ций, позволяет осуществлять эти этапы одновременно и непре-
рывно.
Для рассматриваемого случая схема непрерывнодействующего
агрегата состоит из ряда последовательно (по ходу потока реаги-
рующих веществ) соединенных аппаратов и устройств (рис. IV-2).
Исходные вещества в заданном количестве и соотношении не-
прерывно подаются насосами 1 и 2 в подогреватель 3, откуда,
нагретые до необходимой температуры, они поступают в реакци-
онный аппарат 4.
В реакционном аппарате (на аппаратурной схеме агрегата изо-
бражен непрерывнодействующий аппарат полного смешения) про-
текает химическая реакция при температуре, поддерживаемой на
заданном уровне путем охлаждения реакционной смеси через ру-
башку (или змеевик, погруженный в реакционную смесь). Паро-
газовая смесь непрерывно отводится из аппарата 4 через обрат-
ный холодильник 5. Конденсат (жидкие продукты) возвращаются
в реакционный аппарат, а газообразные продукты направляются
в сепаратор 7.
Реакционная смесь из аппарата 4 при температуре процесса
непрерывно поступает в холодильник 6. Разделение газовой и
Рис. IV-2. Реакционный агрегат непрерывного действия:
а — аппаратурная схема агрегата; б — температурный режим процесса;
1, 2 —насосы; 3 — подогреватель; 4 — реакционный аппарат; 5 —обратный холодильник;
6 — холодильник; 7 — сепаратор.
жидкой фаз, охлажденных до заданной температуры, производится
в сепараторе 7, откуда жидкие и газообразные продукты реакции
направляются на дальнейшую переработку.
Режим работы каждого из аппаратов и устройств строго огра-
ничен выполнением заданных функций, конструкция рассчитана
только на выполнение этих функций, а материал — на определен-
ную среду в строго ограниченных температурных условиях. По-
добный режим позволяет строго специализировать аппаратуру.
Например, теплообменники 3 и 6 (агрессивные свойства нагре-
ваемых или охлаждаемых в них продуктов находятся в прямой
зависимости от температуры потока) могут быть выполнены из
двух последовательно соединенных секций, материал теплообмен-
ной поверхности которых различен (более стойкий материал — в
секции, работающей при высокой температуре).
Параметры процесса (температура, давление) в любой точке
потока реагентов не меняются во времени, и автоматическое под-
278
держание их в заданных пределах достигается применением сра-
внительно несложных приборов.
В условиях работы агрегата непрерывного действия степень ис-
пользования каждого из аппаратов, непрерывно выполняющих при-
сущие ему функции, равна единице.
Реакционный агрегат полунепрерывного действия
В зависимости от конкретных условий процесс может
быть осуществлен при одновременном проведении некоторых ста-
дий.
В рассматриваемом случае один из возможных вариантов та-
кого совмещения может быть следующим: часть исходных веществ
в определенном количестве и соотношении загружается в аппарат
и при перемешивании нагревается до температуры начала быст-
рого взаимодействия, сопровождающегося выделением тепла. При
достижении заданной температуры реакционной смеси начинается
непрерывная подача исходных веществ в аппарат в заданном ко-
личестве и соотношении (при непрерывном перемешивании и охла-
ждении реакционной смеси). После заполнения реакционной зоны
смесью продуктов подача исходных веществ прекращается, реак-
ционная смесь охлаждается до заданной температуры и по оконча-
нии отделения газообразных продуктов передается на дальнейшую
переработку.
Общая продолжительность операции несколько меньше, чем в
агрегате периодического действия, так как некоторые этапы осу-
ществляются одновременно; соответствующим образом повышается
степень использования отдельных устройств такого агрегата.
Подобный прием целесообразно использовать не только для
сокращения продолжительности операции, но и в тех случаях,
когда реакционная система склонна к быстрой спонтанной экзо-
термической реакции (выделение реакционного тепла при этом
можно легко регулировать изменением скорости подачи исходных
веществ).
Схемы агрегатов полунепрерывного и периодического действия
мало различаются между собой. Дополнительным устройством, не-
обходимым для подачи исходных веществ в период непрерывного
питания реакционного аппарата, могут служить насосы.
Возможность автоматического регулирования полунепрерыв-
ного процесса несколько большая, чем при проведении периодиче-
ского процесса, однако полную автоматизацию в этом случае тоже
трудно осуществить.
Как следует из сказанного выше, непрерывные процессы ха-
рактеризуются единством времени протекания отдельных этапов
операции, осуществляемых в разных аппаратах схемы. Для перио-
дических процессов характерно единство места протекания различ-
ных этапов операции при последовательном их свершении во вре-
мени.
279
Непрерывным процессам свойственно также четкое распределе-
ние стадий переработки сырья между отдельными аппаратами и
устройствами реакционного агрегата с соответственным сужением
, функций каждого из них.
К основным достоинствам осуществляемых непрерывно процес-
сов в первую очередь следует отнести:
1) высокую степень использования аппаратуры;
2) возможность совершенствования конструкции аппаратуры
в пределах возлагаемых на нее функций, а также применение бо-
лее дешевых конструкционных материалов (снижение требований
к диапазону условий работы конструкционных материалов);
3) возможность механизации и автоматизации технологического
процесса.
Указанные особенности непрерывных процессов облегчают со-
здание высокопроизводительной аппаратуры и производственных
схем большой мощности.
Необходимо отметить, что высокая степень механизации и авто-
матизации, присущая непрерывным процессам, является в то же
время непременным условием их осуществления.
Характерная особенность схем непрерывных процессов — отно-
сительно высокая доля насосов и коммуникаций, обусловленная
необходимостью непрерывного перемещения материальных по-
токов.
Периодические процессы менее пригодны для использования
при создании промышленных производств большой мощности, од-
нако особенности отдельных химических реакций и экономические
соображения в ряде случаев вынуждают отдавать предпочтение
реакционным агрегатам периодического действия. Основанием для
такого выбора могут быть:
1) малый масштаб производства;
2) специфические свойства реакционной смеси, затрудняющие
непрерывный ее транспорт;
3) малая скорость химической реакции.
Выбор организационного принципа работы реакционной аппа-
ратуры требует тщательного анализа всех особенностей каждого из
сравниваемых вариантов в конкретных условиях.
Сравнительную оценку реакционных аппаратов периодического
и непрерывного действия можно продолжить на основе ранее рас-
смотренных кинетических закономерностей реакций в открытых и
закрытых системах. На примере необратимой реакции A + B—>-D
первого кинетического порядка по каждому из исходных веществ
и параллельной реакции А + B-*D, 2А-> Е можно оценить отно-
сительную скорость превращения исходных веществ в условиях
периодически- и непрерывнодействующих реакционных аппаратов
разных типов (рис. IV-3).
В случаях 1, 2, 4—6 (рис. IV-3) концентрация исходного веще-
ства В практически постоянна (вещество В взято в избытке),
в случаях <?, 7 и 8 реакция проводится при стехиометрическом
280
Рис. IV-3. Основные группы реакционной аппаратуры.
действие
непрерывное
Противоток
Прямоток
Противоток
Прямоток
Полное
смешение
Продольное
перемешивание
Каскад секций
Полное вытеснение
Промежуточный режим
Ндеа льный режим
§ §
8- да
Периодическое
действие
$—
1 м т
II —
—
1 .
U1 ill 11111IIi 11 II 111111II
соотношении исходных веществ А и В. Сравниваемые типы реакто-
ров характеризуются видом зависимости са(т) и ca(L) при одина-
ковом для всех случаев значении начальной (сЛо) и конечной (сА
концентрации исходного вещества А. Заштрихованная площадь
может служить мерой оценки изменения скорости превращения ис-
ходного вещества во времени (т) или по длине (L) реакционного
пространства.
Непрерывнодействующие реакционные агрегаты типа 2 и 4 ха-
рактеризуются гидродинамическим режимом, приближающимся к
идеальному режиму открытых реакционных систем полного вытес-
нения и полного смешения соответственно. Кинетические законо-
мерности реакций первого кинетического порядка, установленные
ранее для открытых реакционных систем, позволяют выразить
изменение концентрации исходного вещества во времени и по
длине реакционного пространства аппаратов этого типа (рис. IV-3:
2, а-, 2, б-, 4, а-, 4, 6).
Схемы непрерывнодействующих реакционных агрегатов, гидро-
динамический режим которых отличается от идеального, представ-
лены на рис. IV-3 в случаях 5—8. В одном из них (5) имеет место
продольное перемешивание, вследствие чего ордината кривой на
рис. IV-3 (5, б) в точке входа в реакционную зону меньше соответ-
ствующего значения ординаты кривых, характеризующих режим
полного вытеснения (рис. IV-3: 2,6; 3,6), но больше значения кон-
центрации в реакторе полного смешения (рис. IV-3: 4, а; 4,6).
Одним из вариантов реакторов промежуточного типа является
каскад секций полного смешения (рис. IV-3, случаи 6 и 7). В слу-
чае 6 исходные вещества А и В движутся прямотоком, в слу-
чае 7—противотоком.
Противоток исходных веществ, осуществляемый в условиях
полного вытеснения (рис. IV-3, случай 3) или в каскаде секций
полного смешения (рис. IV-3, случай 7), оправдан, когда исходные
вещества находятся в стехиометрическом или близком к нему со-
отношении. Движущая сила реакции, определяемая произведе-
нием концентрации компонентов, при этом более равномерна во
времени или по длине реакционного пространства, чем при соот-
ветствующих условиях прямотока исходных веществ.
Для получения продукта D по реакции A + B->D, 2А->Е, один
из продуктов которой (Е) нежелателен, снижение относительной
скорости химического превращения 2А->Е достигается путем
уменьшения концентрации вещества А в реакционной системе при
вводе его в реагирующий поток по мере расходования (рис. IV-3,
случай 8).
Анализируя рис. IV-3, нетрудно заметить, что максимальная
скорость превращения исходных веществ достигается при исполь-
зовании аппаратов периодического действия или непрерывнодей-
ствующих аппаратов полного вытеснения. Наименьшее значение
скорости характерно для непрерывнодействующих реакторов пол-
282
ного смешения, процесс в которых протекает при минимальном
значении концентрации исходных веществ.
Непрерывнодействующие аппараты промежуточного- типа зани-
мают по скорости превращения исходных веществ промежуточное
положение между аппаратами идеальных режимов.
Приведенное выше сравнение основных групп реакционной
аппаратуры справедливо для реакций всех типов (кроме автока-
талитических) при оценке относительной скорости превращения
исходных веществ и относительной скорости образования продукта
одностадийных необратимых реакций.
Средняя скорость образования продуктов сложных реакций,
характеризующая производительность реакционной аппаратуры,
зависит от схемы реакции, целевого продукта и типа реакцион-
ного аппарата.
ДИНАМИКА ТЕХНОЛОГИЧЕСКИХ ПРОЦЕССОВ
При организации непрерывных процессов на тех стадиях
переработки сырья, где состав исходного потока (или потоков)
отличается от состава выходящего потока, наряду с постоянством
таких параметров, как температура и давление, обязательным
условием стационарности процесса является постоянство состава
Рис. IV-4. Характер возмущающего действия по соста-
ву подводимого потока.
поступающих в аппарат потоков. Нарушение этого условия при-
водит к изменению среднего состава выходящего потока, т. е. к на-
рушению регламентированных показателей производства. Это бес-
спорное положение в одинаковой мере справедливо как для про-
цессов, связанных с химическим превращением сырья, так и для
физико-химических и физических процессов.
Изменения выходящего потока будут зависеть от характера и
количественной характеристики возмущающего воздействия (им-
пульса). Основные типы импульсов представлены на рис. IV-4.
Рассмотрим простейший случай, когда в аппарат полного сме-
шения непрерывно и с постоянной скоростью поступает поток
жидкости, не содержащей вещества А, т. е. когда концентрация
вещества А в аппарате и в потоке выходящей жидкости постоянна
и равна нулю. Аппарат работает в стационарном режиме. Внесем
возмущение в состав входящего потока в виде прямоугольного им-
пульса продолжительностью си, введя в поток вещество А так,
чтобы его концентрация была равна сА (амплитуда &и = сл ).
283
В соответствии с ранее установленными зависимостями — см.
уравнение П-202 — изменение концентрации вещества А в аппа-
рате полного смешения и в выходящем потоке в период воздей-
ствия импульса, т. е. в интервале описывается выраже-
нием:
СА = СА 1 - е 6 )
лк о
В момент прекращения воздействия импульса (т=аи) концен-
трация вещества А в аппарате будет равна:
/ _^и \
Ч= %(Т = аи) =ч‘1“е '
Состояние жидкости с момента прекращения импульса (т>аи)
можно описать с помощью уравнения (П-203), если за начальную
концентрацию принять сА = с'д и отсчет времени вести от мо-
мента аи (т'=т — ая), т. е.
т'
т
с. = СА е
лк лк
Из двух последних уравнений нетрудно получить соотношение,
связывающее продолжительность периода вымывания т' с ампли-
Рис. IV-5. Изменение концентра-
ции выходящего потока в резуль-
тате возмущения по составу под-
водимого потока.
тудой возмущающего импульса сЛо,
его длительностью аи, а также с
расчетным временем пребывания
потока в аппарате 0 и составом вы-
ходящего потока:
Сл I )
z n 1 Л0 I , 6 I
т'=т —аи = 01п—-\1— е )
СА
лк
На рис. IV-5 представлена ка-
чественная картина изменения кон-
центрации вещества А в реакторе
при воздействии указанного импуль-
са. Период, в течение которого со-
став выходящего потока отличается
момента начала возмущения. Макси-
от исходного, начинается с
мальное отклонение достигается к моменту окончания действия
импульса. После прекращения воздействия импульса концентра-
ция вещества А убывает по закону вымывания, и при т=оо дости-
гается полное вымывание.
В реальных условиях, когда имеется хотя бы небольшая веро-
ятность возмущения по составу входящего потока, не следует рег-
ламентировать содержание примесей, являющихся результатом
возмущения, значением «отсутствие». Необходимо установить до-
пускаемое значение концентрации этих примесей, сообразуясь с
284
возможной их концентрацией во входящем потоке и продолжи-
тельностью возмущающего импульса.
При проведении химической реакции результат возмущений по
составу входящего потока приводит к изменению не только сте-
пени превращения исходного вещества, но и выхода продуктов
сложных реакций. Указанное обстоятельство и необходимость со-
блюдения условий безопасной эксплуатации реакционной аппара-
туры повышают требования к контролю за составом исходных
веществ. Среди разнообразных мер, предотвращающих нарушения
заданного состава, в ряде случаев предусматривается возможность
быстрого прекращения подачи сырья.
Продолжительность возмущающего воздействия практически
равна продолжительности операций, связанных с отбором и ана-
лизом проб на содержание контролируемого вещества. Автомати-
зация этих операций, применение быстрых методов анализа
состава входящего потока и автоматизация операций, прекра-
щающих возмущающее воздействие, являются непременными
условиями осуществления непрерывного процесса в допустимых
пределах отклонения от заданного режима.
ПАРАМЕТРЫ РЕАКЦИОННОГО ПРОЦЕССА
Основная задача выбора режима работы реакционного
аппарата сводится к созданию условий превращения исходного
вещества, обеспечивающих достижение максимального выхода це-
левого продукта. Наряду с катализом и инициированием желаемое
изменение как абсолютной, так и относительной скорости отдель-
ных направлений реакции может быть достигнуто соответствую-
щим изменением ряда условий процесса.
К числу основных регулируемых параметров, влияющих на вы-
ход основного продукта и производительность реакционного аппа-
рата, в первую очередь следует отнести температуру и давление,
а также'концентрационные условия проведения процесса.
Ниже на примере основных типов реакций рассматриваются
некоторые закономерности, используемые при выборе оптималь-
ного режима работы реакционной аппаратуры.
Зависимость выхода продуктов от температуры
Для реакций, в схему которых входят параллельно и по-
следовательно протекающие превращения, изменение температуры
сопровождается изменением не только абсолютной, но и относи-
тельной скорости каждого из этих превращений и, как следствие
этого, соответствующим изменением выхода промежуточных и ко-
нечных продуктов. В практических условиях возможности такого
приема ограничены верхним и нижним пределами эксперимен-
тально установленной области температур, однако в пределах этой
области изменение выхода того или иного продукта в желаемом
285
направлении может быть достигнуто соответствующим изменением
температурного режима.
Рассмотренные в гл. II выражения для Р(а) дают возможность
для оценки характера зависимости f}(T) и Р(ос, Т) исходя из до-
пущения о практически постоянном значении разности энергий
активации отдельных реакций.
Выход продуктов двухстадийной параллельной реакции, описы-
ваемой схемой А —В, А — -> D, при одинаковом кинетическом
порядке реакции в обоих направлениях не зависит от степени пре-
вращения исходного вещества и является функцией только тем-
пературы:
где Zi, Z2 — предэкспоненты; Еь Е2 — энергии активации.
В зависимости от относительного значения Е] и Е2 повышение
температуры сопровождается либо повышением (если Е|>Е2),
либо понижением (если Ei<E2) выхода продукта В.
Иная картина наблюдается при проведении двухстадийной па-
раллельной реакции, если кинетический порядок отдельных пре-
вращений неодинаков. Пусть реакция А — -> В имеет второй, а
реакция А —D первый кинетический порядок, причем ЕГ>Е2 и
сЛо=1,О. Тогда мгновенный выход продукта В (или выход в усло-
виях полного смешения) опишется выражением
„ =_________%(!-«)________=____________________
РВ ,, х z2 (Ei-E2\ , z2 (Et—E2\
%(1-«) + 77expH?H '1-а + 7Гехр (—RT~)
из которого следует, что выход продукта В снижается с увеличе-
нием степени превращения исходного вещества и возрастает при
повышении температуры. Условием сохранения выхода продукта
В на желаемом уровне при заданной степени превращения сырья
(максимальный выход достигается при а—>-0), очевидно, является
повышение температуры по мере увеличения а.
Если процесс описывается схемой, включающей три параллель-
ные реакции одинакового кинетического порядка
А В; А D; А F
то мгновенный выход продукта В можно выразить следующим об-
разом:
Найдя производную по Т и приравняв ее нулю, получаем:
z2 (Е, - Е2) exp £‘ + гз (£1 ~ Ез) ехр = 0
286
Условием экстремума является неравенство £’2>£,1>Ез или
Е2<Е,<£'з.
Температура Тот, соответствующая максимальному выходу
продукта В, зависит от кинетических постоянных [7]:
при Е2>Е1>Е3
Е2-Е3
„при Е2<Е1<Е3
-Г £3 ~ £2
«!
z2
Из приведенных выше уравнений следует, что при Т=Топт до-
стигается максимальный выход продукта В при любой степени
превращения исходного вещества и оптималь-
ным является изотермический режим процес-
са. Выход продуктов D и F с изменением
температуры меняется монотонно (рис. IV-6).
Если кинетический порядок параллельно
протекающих реакций неодинаков, необходи-
мо учитывать зависимость выхода продуктов
от степени превращения исходного вещества.
Предположим, что в рассматриваемой трех-
стадийной параллельной реакции первые две
имеют первый порядок, а последняя (А — >
—-»F) — второй. Выход продукта В при
сА= 1,0 в этом случае будет равен:
Рис. IV-6. Зависи-
мость выхода про-
дуктов параллельной
. ki
реакции А-------> В,
А —> D, А —F от
температуры
(Е2 < £’i < Е3).
'в----------
1 +
Ei — Еп \ , ,, , z3
j + (1 - а) — ехр
Максимальный выход продукта В дости-
гается, если £'2<£'1<£'з или E2>Ei>E3, при-
чем температура, соответствующая этому вы-
ходу, меняется по мере изменения степени
превращения. Для условий полного смешения зависимость
Еопт(а) выглядит следующим образом:
при E2<Ei<E3
Еопт —.
« In
при Е2>Ее>Е3
Е3 — Е2_________
z3 (Е3 — £1) (1 — а)
Z2 (£1 ~’ Е2)
Т'опт —
Е2 — Е3
R.
z3 (El — Е3) (1 - а)
Ясно, что в первом случае соблюдение оптимальной темпера-
турной последовательности связано с необходимостью повышения
287
температуры по мере увеличения степени превращения; во втором
случае необходимо соответственно понижать температуру. Харак-
тер зависимости Т'(а), установленный по значению мгновенного
выхода продуктов и соответствующий условиям смешения, сохра-
няется и в условиях полного вытеснения [8].
Выход продукта В двухстадийной последовательной реакции
первого порядка А В D при заданной степени превраще-
ния исходного вещества монотонно возрастает или монотонно
убывает (в зависимости
Рис. IV-7. Зависимость вы-
хода продукта В по-
следовательной реакции
А----► В----> D от темпе-
ратуры (£] > Е2, Tt < Г2)
для реактора полного выте-
снения (кривые 1, 2) и ре-
актора полного' смешения
(кривые 2').
от отношения Е2)Е\) при изменении тем-
пературы. Так, при Е2<_Е] повышение
температуры сопровождается увеличени-
ем выхода промежуточного продукта В
и соответствующим уменьшением выхо-
да конечного продукта.
При заданном значении выхода про-
дукта В повышение температуры (при
Е2<Е\) позволяет увеличить конечную
степень превращения исходного вещества
(что часто экономически целесообразно)
или компенсировать нежелательный эф-
фект продольного перемешивания, если
процесс проводится в реакторе с цирку-
ляцией реакционной массы (рис. IV-7).
Если схема последовательного пре-
вращения включает три стадии: А —->
—-> В —D — -> F, то в тех случаях,
когда E2>Ei>E2 или Е2<Е\<Е3, суще-
ствует оптимальная температура, при
которой достигается максимальный выход
продукта F. При Е'2<Е1<£'з повышение температуры сопрово-
ждается увеличением выхода продукта В и уменьшением выхода
продукта D аналогично тому, как это имеет место при трехстадий-
ной параллельной реакции (см. рис. IV-6).
Зависимость выхода продуктов последовательно-параллельных
реакций от температуры рассмотрена в работах [7—11].
Последовательно-параллельные реакции характеризуются как
оптимальной температурой, так и оптимальной температурной по-
следовательностью. Например, для промежуточного продукта В
реакционной схемы
А — -> в — > D
*з.> F
(все реакции — первого порядка) существует оптимальная темпе-
ратура
7опт —
Е3 ~ Е2
R In
г3 (Е3 - Е,)
z2 (Ei-E2)
288
тогда как в случае реакционной схемы
а в D А _*>-> в
кг
I—FI
для получения максимального выхода продукта В необходимо со-
блюдение оптимальной температурной последовательности.
Несколько иным является механизм влияния температуры на
выход продуктов сложных реакций, осуществляемых в гетеро-
генной реакционной системе. Иллюстрацией может служить
рассмотренная выше (см. стр. 116) последовательная реакция
А—-> В —-> D, протекающая на поверхности раздела фаз и ха-
рактеризующаяся следующей зависимостью относительной ско-
рости образования промежуточного и конечного продуктов от ки-
нетических и диффузионных констант:
wb t Рд, Д
WD ~ k2
где рв, д — константа скорости диффузии продукта В от поверх-
ности в объем.
Следствием повышения температуры реакционной массы яв-
ляется переход реакции в диффузионную область д) и
существенное повышение относительной скорости образования
конечного продукта, т. е. увеличение его выхода.
Хотя оптимальная температура или оптимальная температур-
ная последовательность соответствуют минимальному удельному
расходу сырья, экономическое обоснование оптимального темпе-
ратурного режима требует учета зависимости Z(T), определяю-
щей размеры и стоимость реакционного аппарата. Этот вопрос
будет рассмотрен ниже.
Зависимость выходе продуктов
от концентрационных условий
Как было показано в гл. II, выход промежуточного про-
дукта реакций, схема которых включает две стадии последова-
тельного превращения исходного вещества, находится в зависи-
мости от степени превращения этого исходного вещества и типа
реакционного аппарата. Достижение высокого выхода промежу-
точного продукта такого рода реакций (в том случае, когда он
является целевым продуктом) в промышленных условиях связано
с необходимостью проведения процесса в реакционных аппаратах
типа реакторов полного вытеснения при низком значении степени
превращения. Последнее условие соответствующим образом от-
ражается на размерах аппаратуры и удельном расходе энергии
на стадиях разделения реакционной смеси.
19 В* С. Хайлов, Б. Б. Брандт 289
В тех случаях, когда свойства промежуточного продукта по-
зволяют выводить его из реакционной зоны и тем самым снижать
скорость его дальнейших превращений, становится возможным
достижение высокого выхода этого продукта при достаточно вы-
сокой степени превращения исходного вещества.
Способ удаления промежуточного продукта из реакционной
системы тесно связан с химическими и физико-химическими свой-
ствами не только рассматриваемого промежуточного продукта, но
и других компонентов реакционной системы; в зависимости от
этого могут быть использованы различные способы, основанные
на процессах экстракции, ректификации, десорбции или кристал-
лизации.
Рассмотрим, например, последовательную реакцию первого
кинетического порядка А —* В —D, протекающую в изотер-
мических условиях в жид-
кой фазе.
Пусть реакционная фаза
(I) находится в постоянном
и тесном контакте с другой
фазой (II), в которой нет
условий для протекания ре-
акции В D, и Пр0.
дукт В распределяется по
законам равновесия между
этими двумя фазами.
Фазой II может быть
либо инертная жидкость,
Продукт
В
Рис. IV-8. Схема реакционного агрегата
с экстракцией продукта В.
обладающая селективной растворимостью по отношению к про-
дукту В, либо инертный газ (если продукт В обладает высокой
относительной летучестью).
Для простоты допустим, что процесс осуществляется в реак-
торе полного смешения и инертной фазой является жидкость, рас-
творяющая только продукт В.
Предполагая практическую неизменность объема реагирующей
фазы и инертной жидкости, значительную (по сравнению со ско-
ростью реакции) скорость массообмена между фазами и постоян-
ное значение коэффициента распределения продукта В (К* =
=Св, n/Св, i = const), нетрудно написать уравнение материаль-
ного баланса относительно продукта В для процесса, схема кото-
рого представлена на рис. IV-8:
Ш1СВ, I + ШП**СВ, I = Г (*1СЛ, I “ k2CB, 1)
(IV-1)
где (or, (од — объемные скорости потоков реакционной и инертной
фаз; ki, ki—константы скорости реакций; W — объем реакционной
фазы в реакторе (предполагается, что среднее время пребывания
6 каждой из фаз в реакторе пропорционально их объемам); сА,
Св, cD — концентрация веществ. Индексы «I» и «II» относятся со-
ответственно к реакционной и инертной фазам.
290
Из уравнения (IV-1) можно выразить концентрацию продукта
В в реакционной фазе на выходе из аппарата:
k\CA, I®7
1= (0, + ®пк* + k2w
Разделив числитель и знаменатель правой части полученного
выражения на кц и обозначив комплекс через Е*, полу-
чим [6]:
klcA. Iе ________fel9_______
C3.J 1 + £* + kjd Ca° (1 + M) (1 + E‘ + A29)
Концентрация конечного продукта D
в реакционной фазе описывается выра-
жением:
„ —Ь- О— - ____________Z?ife202______(TV-91
о, I "'Ло (1 + Л10)(1+£* + *29) '
Заменив в уравнение (IV-2) 0 его
значением
А «
Ml-а)
получим:
аа2
Со. I ~ САО (1 — а) (£*+1) + аа
где a = k2!ki.
Выход конечного продукта D в рас-
сматриваемом случае будет равен:
сп . аа
= 1~~а “ (1 —а) (Е*+1) + аа (IV’3)
Рис. IV-9. Зависимость
р5(а, £ *) для последователь-
« А < Г) ^2
нои реакции А —-> В —-> D
(61 = 62).
Выход промежуточного продукта В описывается следующим
выражением:
(1-а)(Е*+1)
Рв Рд (1-а)(Е*+1) + аа
(IV-4)
Нетрудно заметить, что при К*->0 уравнения (IV-3) и (IV-4)
превращаются в уравнения, описывающие зависимость 0(a) для
однофазной реакционной системы.
Значение экстракционного фактора Е*, зависящее не только
от коэффициента распределения К*, но и от относительного объ-
ема фаз, может быть сколь угодно большим, и в этом случае, не-
зависимо от степени превращения, следует ожидать, что выход
промежуточного продукта будет приближаться к единице.
На рис. IV-9 можно видеть, что даже при сравнительно не-
большом значении Е* выход продукта В может быть увеличен в
несколько раз, особенно при высоком значении а.
19*
291
Примером применения экстракции промежуточного продукта
могут служить работы по получению фурфурола из ксилозы [12].
Выход продуктов параллельных реакций типа
A + B->D, 2А->Е (I)
A->D, 2А->Е _ (II)
в значительной степени зависит от концентрации исходных ве-
ществ в реакционной системе.
При необходимости получения продукта D с максимальным
выходом процесс проводится при низкой концентрации вещества
А. В реакторах периодического действия это условие соблюдается
при равномерной
подаче исходного вещества А после загрузки
всего количества вещества В (схема I)
или после превращения части вещества
А, предварительно загруженной в реак-
тор (схема II).
Непрерывнодействующие аппараты
полного смешения более благоприятны
для достижения указанной цели, чем
реакторы полного вытеснения. При ис-
пользовании последних низкая концент-
рация вещества А поддерживается пу-
тем ввода его в ряде точек по ходу
реакционного потока.
Встественно, что условия, способ-
ствующие повышению выхода продукта
D (низкая концентрация вещества А),
приводят к понижению производитель-
ности реакционного аппарата.
Рис. IV-I0. Зависимость тем-
пературного коэффициента
*f+io/^r от температуры н
энергии активации.
. Значения Е (в ккал/моль): /—40;
2-20; 3—10; 4-5,
Зависимость скорости реакции
от температуры
При оценке влияния темпера-
туры на скорость реакции нередко при-
меняется так называемый «темпера-
турный коэффициент», представляющий
собой отношение констант скорости реак-
отличающихся на 10 град (&r+io/kT).
величины следует иметь в виду её зави-
ции при температурах,
При использовании этой
симость от энергии активации реакции и температуры (рис. IV-10)
In ^г+1°
kT ' РТ(Т+10)
и соблюдать осторожность при экстраполировании эксперимен-
тально установленного значения йт+ю/^т на широкую область
температуры.
292
В случае необратимых реакций, для которых практически ис-
ключена возможность побочных превращений и допустимо изме-
нение температуры в широком интервале, иногда может ока-
заться выгодным температурный режим, обеспечивающий посто-
янство скорости реакции (w) независимо от степени превращения
исходного вещества. Это условие соблюдается при повышении
температуры по мере увеличения степени превращения:
Г =------------£---------
/ А0Сд (1 - а)"
Я In ---------------
\ W
Рис. IV-11. Зависимость констан-
ты макроскопической скорости ге-
терогенной реакции первого по-
рядка от температуры.
Подъем температуры в течение реакции тем более значите-
лен, чем выше порядок реакции (п).
Зависимость макроскопической скорости реакции от темпера-
туры в гетерогенной системе будет несколько иной вследствие су-
щественного различия энергии ак-
тивации химической реакции и диф-
фузии.
При достаточно низкой темпе-
ратуре, когда константа скорости
реакции существенно меньше кон-
станты скорости Диффузии, макро-
скопическая скорость реакции сов-
падает с истинной скоростью хими-
ческого превращения на поверхности
(внешняя кинетическая область).
При достаточно высокой темпера-
туре константа скорости реакции
может оказаться существенно боль-
ше константы скорости диффузии
и макроскопическая скорость будет
определяться скоростью диффузион-
ного переноса (внешняя диффузи-
онная область).
Зависимость константы макроскопической скорости реакции
первого порядка, протекающей на твердой поверхности, от темпе-
ратуры представлена на рис. IV-11. Энергия активации химиче-
ской реакции принята равной 30 ккал!моль, энергия активации
диффузии 2 ккал/моль; предэкспоненциальные множители соот-
ветственно равны 10й и 102.
В области низких температур для макроскопической скорости
наблюдается сильная зависимость от температуры, постепенно осла-
бевающая с увеличением Т (нижняя кривая на рис. IV-11). При
переходе реакции в диффузионную область вследствие повышения
температуры существенными становятся условия диффузионного
переноса, и увеличение макроскопической скорости может быть
достигнуто повышением турбулентности подвижной фазы.
293
Рис. IV-12. Зависимость
равновесной (Гр) и опти-
мальной (Гопт) температур
от степени превращения SO2
иа ванадиевом катализа-
торе.
Переход из диффузионной области в кинетическую при посто-
янной температуре представлен на рис. IV-11—с нижней кривой
(точка /) на верхнюю (точка 2).
При проведении обратимых реакций в адиабатических усло-
виях изменение температуры реакционной системы, происходящее
по мере увеличения степени превращения, неблагоприятно сказы-
вается на положении равновесия и скорости прямого превраще-
ния, так как экзотермическая реакция требует понижения, а эндо-
термическая — повышения температуры.
Зависимость скорости обратимых реакций от температуры
имеет экстремальный характер. Каждому составу равновесной ре-
акционной системы, т. е. каждому зна-
чению степени, превращения исходного
вещества, соответствует присущая дан-
ной реакционной системе равновесная
температура (Тр).
Связь между составом реакционной
смеси и значением температуры, при ко-
торой данному составу соответствует
равновесное состояние реакционной си-
стемы, вытекает из общих термодина-
мических закономерностей. Для экзотер-
мических реакций значение равновесной
степени превращения тем выше, чем
ниже температура реакционной смеси;
для эндотермических реакций эти вели-
чины пропорциональны.
Температура, соответствующая максимальной скорости реак-
ции (Топт), связана с равновесной температурной зависимостью
Д>пт -
Л.
RTp Ех
1 + ~КЁ 1п е2
и по мере изменения степени превращения исходного вещества
следует ожидать изменения значения оптимальной температуры.
При проведении обратимых реакций в потоке по мере увели-
чения степени превращения исходного вещества и соответствую-
щего изменения равновесной температуры, отвечающей каждому
мгновенному составу потока, меняется и значение оптимальной
температуры, снижающейся для экзотермических и повышающейся
для эндотермических реакций.
При проведении каталитических процессов достижение макси-
мальной производительности катализатора требует тщательного
учета рассматриваемой зависимости и - соответствующего плани-
рования температурного режима процесса.
В качестве примера на рис. IV-12 показано изменение равно-
весной температуры реакционной системы по мере протекания
2U
одной из наиболее изученных каталитических экзотермических
реакций
2SO2 + O2 2SO3 + Q
осуществляемой в газовой фазе на ванадиевом катализаторе в
области температур 400—600° С [13].
Температура T0WI, соответствующая максимальной скорости
процесса, отличается от равновесной температуры (Гр) на вели-
чину ДГ
1 + ]П £>_
Д£ Е2
т. е. взаимное расположение равновесной и оптимальной кривых
определяется соотношением между энергиями активации прямой
и обратной реакций.
Учет скорости переноса вещества к внутренней и внешней по-
верхности катализатора несколько меняет количественную сто-
рону рассмотренных выше закономерностей (строго количествен-
но справедливых лишь для кинетической области), не'Нарушая,
однако, принципиального их характера. Эти закономерности не
всегда приводят к выводу о целесообразности температурного ре-
жима, соответствующего максимальной скорости реакции во всем
диапазоне значений а. Для некоторых реакций, обладающих боль-
шой величиной теплового эффекта, максимальная скорость про-
цесса при малом значении степени превращения оказывается столь
значительной, что поддержание температуры на заданном уровне
может стать технически неосуществимым.
В некоторых случаях верхний температурный предел, соот-
ветствующий начальной стадии реакции, лимитируется свойства-
ми катализатора (максимальная рабочая температура), термиче-
ской устойчивостью исходных веществ или продуктов реакции
или опасностью возникновения нежелательных побочных реак-
ций, особенно характерных для многих процессов органического
синтеза.
Учитывая, что скорость большинства реакций при малом зна-
чении степени превращения достаточно велика, часто оказывается
целесообразным проведение начальной стадии процесса при тем-
пературе, значительно отличающейся от оптимальной. При этом
основная часть теплоты реакции используется на подогрев реак-
ционной смеси, и только после достижения значения температуры,
соответствующего кривой оптимальных температур (разумеется, в
пределах, допускаемых свойствами компонентов и химическим
характером реакционной системы), становится целесообразным
соблюдение оптимального температурного режима.
295
Зависимость скорости реакции
от концентрации реагентов
Если скорость реакции первого порядка зависит только
от концентрации исходного вещества, то скорость односторонних
реакций, кинетический порядок которых п>1, определяется также
и соотношением концентрации исходных веществ. В этом нетрудно
убедиться, исследуя на экстремум уравнения скорости реакций
типа A + B->D.
Найдя производную скорости по начальной концентрации од-
ного из исходных веществ и приравняв ее нулю, после соответст-
вующих преобразований можно установить, что максимальной
скорости реакции соответствует стехиометрическое соотношение
концентрации веществ А и В. К тому же результату приводит ис-
следование случая проведения этой реакции в присутствии инерт-
ного вещества. Такой вывод не является неожиданным, так как
избыток одного из компонентов играет роль инертного разбавителя.
Для повышения скорости реакции типа A+B->D при стехио-
метрическом или близком к нему соотношении начальных
концентраций исходных веществ может быть использован рас-
смотренный ранее (см. стр. 178) принцип противотока, эффект
которого особенно заметен при высоком значении степени пре-
вращения.
При противотоке реагентов достигается также значительная
равномерность скорости реакции по длине реакционного про-
странства (для реакторов полного вытеснения) с максимумом при
~0,5ак (ак — конечная степень превращения), что в отдельных
случаях может облегчить поддержание теплового и температур-
ного режима процесса.
Зависимость скорости реакции от давления
Естественно, что вопрос о влиянии давления на ско-
рость химического превращения может рассматриваться только
применительно к газовым системам.
Зависимость равновесного значения степени превращения (сер)
исходного вещества от давления для реакций типа
ВА^=*В + А
или
В2 2В
описывается известным уравнением [14]:
Г К? I0-5
«₽ = [^+р] (IV-5)
где Кр — константа равновесия, выраженная через парциальные
давления; Р — общее давление.
296
570° С
степени
к. IV-13. Изменение скорости реакции
тидрнровання бутилена при ~
зависимости от давления и
Из уравнения (IV-5) следует, что при Р=0, т. е. при практи-
чески полном смещении равновесия вправо (а~1), скорость ре-
акции тоже будет практически равна нулю вследствие малой кон-
центрации реагирующих частиц. Повышение давления газовой
смеси снижает равновесное значение а, соответствующее нулевой
результирующей скорости.
Подобная схема зависимости w(P), напоминающая зависи-
мость w{T), рассмотренную выше, предполагает наличие макси-
мума функции w(P).
Для реакций, протекающих с увеличением объема (реакции
крекинга, дегидрирования и др.), зависимость w(P) описывается
следующим выражением [15]:
w = f(T,P)\\-2^L\ (IV-6)
L лр J
В изотермических условиях 6
уменьшение давления исход- ц;
ного вещества (по мере увели- g
чения степени его превраще- "
ния) сопровождается умень-
шением значения f(P), в то
время как второй сомножитель
в выражении (IV-6) с увели-
чением степени превращения О
возрастает. Для каждого зна-
чения а существует такое дав- Pi
ление, при котором скорость Де
реакции имеет максимальное в ,
г превращения llbl.
значение.
Для реакции дегидрирования углеводородов уравнение (IV-6)
примет вид [15]:
te> = fe, [/>0 (1 -<х)]”.Г1 - (IV’7)
L (1 — а ) Др J
Дегидрирование бутилена при 570° С иллюстрируется рис. IV-13.
Соединяя точки, соответствующие максимальному значению ско-
рости реакции дегидрирования (линия /—/), можно наметить об-
ласть давлений, обеспечивающих максимальную скорость реакции
(пунктиром показаны границы области, внутри которой скорость
реакции больше 0,9 от максимальной).
При проведении процесса дегидрирования бутилена в присут-
ствии водяного пара снижение давления реакционной смеси до-
стигается введением в нее соответствующего количества этого
пара.
Для равновесных реакций, протекающих с уменьшением объ-
ема (реакции гидрирования и др.), очевидны отсутствие макси-
мума зависимости w(P) и целесообразность постепенного повы-
шения давления по мере увеличения степени превращения исход-
ного вещества.
297
ПРОИЗВОДИТЕЛЬНОСТЬ РЕАКЦИОННОЙ АППАРАТУРЫ
Под производительностью реакционного аппарата пони-
мается количество целевого продукта (кг, кмоль), получаемого в
единицу времени (мин, ч) с единицы объема реакционного аппа-
рата (л, м3).
Реакционный агрегат, как правило, представляет собой комп-
лекс устройств, предназначенных для подогрева исходных ве-
ществ, охлаждения реакционной смеси, разделения ее на отдель-
ные фазы и т. п. Эти устройства часто монтируются в одном
сосуде с реакционным пространством, поэтому при определении
производительности реакционного аппарата в понятие «единица
объема» вкладывается условный смысл.
По аналогии с контактными аппаратами и для более правиль-
ного отражения интенсивности химического процесса под объемом
реакционного аппарата при оценке его производительности целе-
сообразно понимать пространство, занятое реагирующей смесью.
Если размеры вспомогательных (например, теплообменных) уст-
ройств для заданных параметров процесса, определяемые тепло-
вым балансом, практически постоянны, то удельный объем, за-
нятый реагирующей смесью и определяемый эффективной ско-
ростью химического превращения, может меняться в широких
пределах.
Производительность реакционного аппарата, отнесенная к ре-
акционному пространству (понятие тождественное с понятием о
средней скорости химического превращения), является основным
критерием оценки интенсивности химического процесса и одним
из показателей при выборе типа и режима работы реакционной
аппаратуры.
Ниже на примере реакций основных типов рассматривается
производительность реакторов периодического действия и непре-
рывнодействующих реакторов.
Реакционные аппараты периодического действия
В реакционных аппаратах этой группы продолжитель-
ность периода (операции), в течение которого происходит образо-
вание конечного продукта, складывается из ряда этапов, основ-
ные из которых указаны на стр. 276.
Степень (коэффициент) использования реакционного аппарата
по прямому его назначению оценивается отношением продолжи-
тельности основного этапа (химическое превращение сырья) к про-
должительности всей операции:
тр
(IV-8)
где <Z7=ti + thi-
Из уравнения IV-8 следует, что при постоянной величине Л
коэффициент использования возрастает с увеличением значения
298
Тр, т. е. в случае проведения в аппарате медленно текущих реак-
ций или при увеличении продолжительности процесса в целях по-
вышения степени превращения исходных веществ В то же время
при совместном рассмотрении уравнения (IV-8) и уравнений, вы-
ражающих зависимость скорости реакций всех кинетических по-
рядков от степени превращения исходных веществ, нетрудно за-
метить, что для одной и той же реакции повышение степени ис-
пользования реакционного аппарата неизбежно сопровождается
снижением его производительности.
Так как показателем интенсивности работы реактора является
его производительность, следует отдать предпочтение этому по-
казателю и рассмотреть характер зависимости его величины от
режима осуществления химического процесса (степени превраще-
ния исходных веществ).
Производительность реакционного аппарата периодического
действия по продукту i в общем виде может быть выражена сле-
дующей зависимостью:
С,- (а) Сг- (а)
Zi = тр (а) Л = Л + тр (а) (I V'9)
где с,к — концентрация продукта i в реакционной массе к концу
процесса, кг/м?.
Анализируя структуру выражения (IV-9), нетрудно прийти к
выводу об экстремальном характере зависимости Z(a) для реак-
ций, порядок которых п>1.
.Действительно, при а->0 концентрация ci (а) также стремит-
ся к нулю; величина Л+тр(а) стремится к Л, так как Л3>тр(а).
Таким образом
lim Zj = О
а = О
При а->1 концентрация продукта стремится к некоторой огра-
ниченной величине, соответствующей а=1. В то же время вели-
чина Л+тр(а) стремится к оо, так как тр->оо. Таким образом
lim Zt = О
а= 1
Естественно, что в некоторой области значений а (0<а<1)
следует ожидать максимума функций Zj(a).
1 Строго говоря, Л не является постоянной величиной в широком интервале
значений а, так как при одних и тех же способах проведения вспомогательных
операций их продолжительность будет зависеть от величины загрузки сырья
на операцию (Л2). В общем случае величина Л является некоторой функцией
степени превращения сырья:
= / 1-₽л> Z'.'d-a)]
299
Простые реакции. Подставляя в уравнение (IV-9) зна-
чения с» (а) для простых необратимых реакций и заменяя тр его
выражением через степень превращения для реакций соответст-
вующих кинетических порядков, получаем зависимость произво-
дительности реакционного аппарата периодического действия от
степени превращения исходных веществ:
реакции первого порядка
7 ka
% —1п(1—а)
(IV-10)
реакции второго порядка
2 fea(l-a)
Ло а(1-ЛА>сЛо)-ЛЙСЛо
(IV-11)
Характер кривых, выражающих зависимости (IV-10) — (IV-11),
эизводительности реак-
ционного аппарата,
значение которой быст-
ро снижается при
уменьшении- или уве-
личении степени пре-
вращения, соответ-
ствующей максиму-
му производительности
(рис. IV-14).
Сопоставляя кри-
вые, соответствующие
различному значению
Л=Ti+Tin, можно за-
метить, что уменьшение
величины Л, достигае-
мое за счет сокраще-
ния продолжительно-
сти подготовительного
и заключительного эта-
пов, а также в резуль-
тате осуществления ря-
да процессов, не свой-
ственных реакционному
аппарату (нагрев ком-
понентов, охлаждение реакционной массы и т. п.), в специально
предназначенной для этого аппаратуре, перемещает положение
максимума кривой Z(a) в область малых значений а. В пределе
при Л —> 0 максимальное значение производительности прибли-
жается к началу координат, достигая значения, свойственного не-
прерывнодействующим аппаратам полного вытеснения.
Для медленно текущих процессов, связанных с использованием
реакционной аппаратуры значительных размеров, необходимо тща-
позволяет видеть наличие максимума
Рис. IV-14. Зависимость ZD(a) для реакционного
аппарата периодического действия (схема реак-
ции: А—->D, САо = 1, k = 1):
в—реакция первого кинетического порядка; б —реакция
второго кинетического порядка.
Значения Л (в усл. ед.): /—0; 2—0,25; 5—0,5; 4—1.
300
тельное исследование зависимости Z(a) и выбор режима, соот-
ветствующего максимально возможной производительности.
Сложные реакции. Для параллельных и последователь-
ных реакций производительность реакционных аппаратов перио-
дического действия выражается уравнением (IV-9) при соответст-
вующих значениях с,к(а) а
и тр (а).
Параллельные реак-
ции, осуществляемые в
реакционных аппаратах
периодического действия,
характеризуются зависи-
мостью Z(a), аналогич-
ной зависимости для про-
стых реакций: производи-
тельность реакционного
аппарата как функция
степени превращения ис-
ходного вещества имеет
экстремальный характер
и зависит от относитель-
ного значения Л.
Производительность ре-
акционной аппаратуры пе-
риодического действия,
предназначенной для осу-
ществления последова-
тельных реакций, можно
рассмотреть на примере
реакции первого кинети-
ческого порядка, проте-
кающей по схеме А —
Рис. IV-15. Зависимость Zi (а) для реакцион-
ного аппарата периодического действия (схема
реакции: А —* В d) при Л = 0,1 (а) и
0,5 (б).
Значения 1 — 0.1; 2 — 0,5; 3 — 5; 4 — 10.
-^->В—->D. Примем, что
k\^=k2. Подставив в фор-
мулу (IV-9) значение
Св (а) и cD(а) из уравне-
ний (П-151), (П-152) и за-
менив значение тр(а) его
выражением для реакций
первого порядка, получим зависимость
производительности реакционного аппарата по продуктам В и D
от степени превращения исходного вещества А при разных значе-
ниях a — и Л:
Z =с k (1-а)а-(1-«)
в сАаа1 (a-l)[ln(l-a)-fcpj]
7 г ь 1 —(1—а)д —aa
(а-1)[1п(1-а)-М1
(IV-12)
(IV-13)
301
Рис. IV-15 показывает, что производительность реакционного
аппарата по каждому продукту проходит экстремальную точку.
Положение этой точки на оси абсцисс определяется величиной Л.
При уменьшении значения Л максимум производительности реак-
тора по каждому продукту перемещается к началу координат, т. е.
в направлении уменьшения степени превращения.
При Л —О, т. е. при переходе к непрерывнодействующим аппа-
ратам полного вытеснения, зависимость ZB(a) выражается моно-
тонно снижающейся кривой, а зависимость ZD(a) продолжает со-
хранять экстремальный характер с перемещением максимума в
область более низкой степени превращения.
Заслуживает внимания влияние значения а на рассматривае-
мую зависимость.
Все реакции, описываемые схемой А — -> В —D, можно раз-
делить на две группы:
1) последовательно протекающие реакции, промежуточный про-
дукт которых обладает большей реакционной способностью, чем
исходное вещество, характеризуются значением а>1;
2) последовательные реакции, основанные на превращении ис-
ходного вещества, реакционная способность которого превышает
реакционную способность промежуточного продукта, характери-
зуются значением а<1.
Для первой группы процессов максимальная производитель-
ность по продукту В лежит в области низких степеней превраще-
ния, а производительность реактора по продукту D достигает мак-
симального значения при степени превращения 50—70%•
Реакционный аппарат, предназначенный для проведения реак-
ций, относящихся ко второй указанной группе, достигает макси-
мальной производительности по продукту В в сравнительно широ-
ком интервале значений степени превращения, тогда как максимум
производительности по продукту D достигается только при высо-
ком значении а.
Реакционные аппараты непрерывного действия
Неодинаковая величина движущей силы, характеризую-
щей условия проведения химического процесса в непрерывнодей-
ствующих реакторах полного вытеснения и полного смешения,
проявляется в структуре уравнений, выражающих производитель-
ность реакционных аппаратов этих типов как функцию степени
превращения исходных веществ.
Реакторы полного вытеснения. Исходя из кинетической
аналогии реакторов периодического действия и непрерывнодей-
ствующих реакторов полного вытеснения, производительность по-
следних может быть описана уравнением (IV-9) при нулевом зна-
чении Л-.
(«)
‘ Тр (а)
302
Подстановка в уравнение (IV-9) соответствующих значений
ct (а) и тр(а) приводит к выражению производительности реакци-
онных аппаратов рассматриваемого типа.
Для реакций, описываемых уравнениями дробного, первого и
второго кинетических порядков, имеем:
Z,- = с. k
I Ао
а
1П -7-!—
1 — а
(п=1)
(IV-14)
Продолжительность одностадийных
0<п<1 или п>1, равна
реакций, порядок которых
<А~п _ А-п 1—11—
= сАо СА 1-п 1 U Ц)
(l-n)fe, (l-n)fej
откуда
с а асл а (1 — п)
в т (а) т (а) " 1 1 —(1—а)1-”
Из выражения (IV-15) следует, что
а -> 0; ZB -> kjc2o
а->1; ZB ->k{ (1 -п) сДд
(IV-15)
т. e. для случая п<1 производительность ре-
актора полного вытеснения при полном пре-
вращении исходного вещества (а—>-!) не об-
ращается в нуль, как это имеет место при
п=1 [см. уравнение (IV-15)] и при и>1
(рис. IV-16), а лишь уменьшается в 1—п раз
по сравнению с максимальной производитель-
ностью, достигаемой при малых значениях а.
Зависимость производительности реакто-
ров полного вытеснения для параллельных
реакций имеет аналогичный характер.
Производительность реакционного аппа-
рата при проведении последовательных реак-
Рис. IV-16. Зависи-
мость ZD (а) для реак-
ционного аппарата
полного вытеснения
(схема реакции:
А —> D):
/ — реакция первого ки-
нетического порядка;
2 —реакция второго кине-
тического порядка.
ций первого кинетического порядка, проте-
кающих по схеме А —В — > D =/= k2), характеризуется урав-
нениями:
_ . (1—а)а—(1—«)
в САО 1 (а — ]) in (1 — а)
7 __ , 1 —(1—а)а —аа
° сл„й1 (а_ 1) in (1 — а)
(IV-16)
(IV-17)
В отличие от реактора периодического действия, производи-
тельность реактора полного вытеснения по продукту В монотонно
снижается с увеличением степени превращения исходного веще-
ства (рис, IV-17, а).
303
Иная картина наблюдается при оценке производительности
реакционного аппарата по продукту D (рис. IV-17, б): так же как
и для реактора периодического действия, в этом случае имеется
максимум функции ZD(a).
Значение оптимальной степени превращения исходного веще-
ства определяется величиной а=й2/&ь и для процессов второй
группы, когда а<1, максимальное значение производительности
реактора практически недостижимо (достигается при почти пол-
ном превращении исходного вещества).
Значении А2/Л1: / — 0,1; 2—10; 3 — 2; ./ — 0,5.
Сравнивая кривые, соответствующие &2/&i = 10 на рис. IV-15 и
IV-I7, нетрудно заметить, что при проведении одной и той же по-
следовательной реакции в реакторе периодического действия и в
иепрерывнодействующем реакторе полного вытеснения произво-
дительность последнего значительно выше и достигается при мень-
шем значении а.
Последовательные реакции автокаталитического характера,
описываемые схемой
Б + А—->К + В (I)
К + Б + А—->К+В (II)
[Б — компонент с практически постоянной концентрацией; К —
продукт реакций (I) и (II), являющийся гомогенным катализа-
тором реакции (II), при этом реакция (I) некаталитическая] при
проведении их в реакторе полного вытеснения характеризуются
иным видом зависимости ZA(a) по сравнению с рассмотренными
выше.
304
Интегрируя уравнение скорости превращения исходного ве«
щества, А
= (1 — а) (£, + = fej (1 — а) (1 + аа)
имеем
“к“°
f da 1 аа +1
т= J fei(l-a)(l+аа) “ fei(l+a) 1-а
ав=о
где а = с, %-. Отсюда
л0 К1
7 Са^ СУ1(1+а)<Х
х . аа +1
1П —:---
1 -а
Зависимость ZA(a) для рассматриваемого
случая описывается кривой, имеющей макси-
мум, положение которого определяется отно-
шением констант скорости реакций (II) и (I).
Реакторы полного смешения. При
подстановке в уравнение (IV-9) Л=0 и за-
мене тр(а) на 0(a) получим общее выраже-
ние для производительности реакционных ап-
паратов полного смешения:
7 Ci (<Х)
0(a)
Производительность реактора полного сме-
шения по продуктам одностадийных необрати-
мых реакций типа А —В, очевидно, равна
скорости реакции в этом аппарате:
С О Сп
ZB = е = = k'cX(1 - a)re (IV’18)
Рис. IV-18. Зависи-
мость ZD (a) для реак-
ционного аппарата
полного смешения
(схема реакции:
A—>D):
1 — реакция первого ки-
нетического порядка;
2 — реакция второго кине-
тического порядка.
Аналогичным образом описывается производительность реак-
тора полного смешения относительно продуктов параллельных ре-
акций одинакового кинетического порядка В А —D:
= ZD = M,(l-«)n (IV-19)
Производительность в данном случае монотонно снижается
(рис. IV-18) с увеличением а при всех значениях п (в том числе
и при га < 1).
Процессы, описываемые уравнением последовательных необра-
тимых реакций, для удобства сопоставления с изложенными выше
закономерностями рассмотрим на примере схемы А —В —D,
приняв порядок реакций первым и ki^k2. 20
20 В- С. Хайлов, Б. Б. Брандт £05
Подставив в уравнение (IV-9) выражения для стационарных
концентраций продуктов В и D (см. стр. 167) и значение 0(a) из
уравнения (П-227), получим:
сд (a) fejl-a)2
ZВ “ 0(a) = CA„ 1 -a(l -а)
cD (a) a^afl- а)
Zo = ПГ(аГ = Сл° l-a(l-a)
Для трехстадийной последовательной реакции
А В D F
(IV-20)
(IV-21)
производительность реакционного аппарата по продуктам В, D и
F можно выразить аналогичным образом.
%
0,8
06
0,4
41
П
п
0,2
О
\ X J \
\ vz \
\ \,з \
0.2 0,4 0,6 0,8 1.0
Рис.
IV-19. Зависимость Z, (а) для реакционного
аппарата
полного смешения (схема
аА->в-^ d).
реакции:
Значения k2lk\: 7—0,5; 2 — 5; 3 — 10.
Графическое изображение зависимостей (IV-20) и (IV-21) пред-
ставлено на рис. IV-19.
Кривые ZB (а) на рис. IV-19 похожи на кривые, изображенные
на рис. IV-17. С увеличением степени превращения исходного ве-
щества производительность реакционного аппарата по продукту В
монотонно снижается, а кривые, выражающие производительность
реактора по продукту D, экстремальны, причем характер влияния
а на величину экстремума кривых ZD(a) и на величину оптималь-
ной степени превращения для реактора полного вытеснения и ре-
актора полного смешения одинаков.
Некоторая особенность характера зависимости ZA(a) наблю-
дается при осуществлении в аппаратах полного смешения группы
306
реакций, схема которых дана на стр. 304. Для этого случая про-
изводительность реакционного аппарата по исходному веществу А
выразится следующим образом:
сл а
k,
где а = с,
л0 fel
Зависимость ZA(a) описывается кривой, имеющей максимум
при степени превращения исходного вещества, равной
1 ki
а — -—
2 2k2cAa
При сА k2 kt максимум производительности достигается при
а=0,5[16]°
Рассмотренная выше зависимость ZD(a) справедлива для слу-
чаев, когда исходное вещество А свободно от примеси промежу-
точного продукта В [см. уравнения (IV-17) и (IV-21)].
В ряде промышленных процессов, описываемых схемой после-
довательной реакции, когда промежуточный продукт не имеет
потребительской ценности, представляется целесообразным его
возврат (после отделения от целевого продукта в смеси с непре-
вращенным исходным веществом) в реакционный аппарат. Ясно,
что производительность реакционного аппарата при этом сущест-
венно изменится.
Стационарный режим в реакторе полного вытеснения дости-
гается при условии равенства средней скорости превращения ис-
ходного вещества и средней скорости образования продукта D
(wA = wd) при соответствующей концентрации продукта В (сВо)
в потоке вещества А, поступающего в реактор.
Приняв, что сАо + сВо = 1 [откуда св = сЛо(1 — е_^1Т)4-сВо — cD],
для скорости изменения концентрации продукта D можно запи-
сать:
= ^2СВ = k2 [СЛО (1 “ е k,X) + cBt> ~ cd]
или
dc
dx + ^2cD = ^2 [СЛ0 (1 ~ е k' ) + св0]
Интегрирование последнего уравнения приводит к зависимости
cd(t):
^ = (’ - %) (1 - j + 'в0(1- (IV-22)
где a = k2/ki. Условие wA = wD соблюдается, если
Ср(т) = сЛо(1-е-/'-т) (IV-23)
Подставив в уравнение (IV-22) значение сп(т) из выражения
(IV-23) и решив полученное уравнение относительно cBq, получим
20*
307
значение св (т), т. е. необходимую концентрацию промежуточ-
ного продукта В ности процесса: в исходной смеси при заданной продолжитель- СВ»=а_1+е-М_ае-М (IV-24)
После подстановки значения сВо(т) в уравнение (IV-22) и за-
мены в полученном выражении и уравнении (IV-24) т его значе-
нием получим выражения для cD(a) и сВо(а)
„ а (а—1) [1 — (1 — а)а) (IV-25)
-о а-а-аЦ-а)"
_ сАо П-а-(1~а)в] (IV-26)
при этом Св» (а- 1) [1 -(1 -а)а] cD (а — 1) [1 — (I — а)в]
Cji° а а — а — а (1 — а)а (IV-27)
Производительность реактора полного вытеснения по продукту
D в рассматриваемом случае опишется выражением:
Z Cn(a) MU-a)n-(l-a)*]
•° т (а) In (1 — а) [а — а — а (1 — а)“] ' '
Для реактора периодического действия, приняв, как и ранее,
продолжительность вспомогательных операций постоянной (Л«
«const), нетрудно выразить значение производительности по про-
дукту D:
сд(«) - аМ*-1)И-(1-«)д]
D х(а)+Л [кхЛ-In (1 - а)] [а - а - а (1 - а)в] ' ’
Ясно, что состав исходной смеси для рассматриваемого про-
цесса может быть получен из приведенных выше выражений
(IV-26) и (IV-27).
В реакционном аппарате полного смешения равенство средней
скорости образования продукта D и средней скорости расходова-
ния вещества А достигается, если
откуда следует, что ^1СА = ^2СВ СВ “ СВ« (IV-30) (IV-31)
Исходя из состава исходной смеси (сЛо + сВа = 1 1), можно запи-
сать: Отсюда 1 — а (IV-32)
сва 1 _ а а
а (IV-33)
САО“ 1 _ аа
аа (IV-34)
CD~ 1-а + а
308
Производительность. реактора полного смешения опишется
уравнением скорости образования продукта D:
Ясно, что при отсутствии потерь продукта В в процессе его
выделения из реакционной смеси выход конечного продукта по-
следовательной реакции
« со(а)
ро асЛо (а)
(IV-36)
равен единице независимо от типа реакционного аппарата и сте-
пени превращения исходного вещества. Это можно ввдеть при
Рис. IV-20. Зависимость ZD (а) и сВо[сАв (а) для последователь-
ной реакции первого порядка А —В > D (fe2/&i=2):
д —производительность реакционного аппарата по конечному продукту D;
б —состав питающего потока в реакторе полного смешения возвратом
продукта В (aR — 0,6).
С возвратом продукта В:
/—реактор полного вытеснения; 2 — реактор полного смешения; 3 — реактор
периодического действия.
Кривые 1 н 2 построены по уравнениям (IV-28), (IV-38) и (IV-35), (IV-39) соот-
ветственно.
Без возврата продуктаВ;
4 — реактор полного вытеснения; 5 —реактор полного смещении.
Кривые 4 и 5 построены по уравнениям (IV-17), (IV-40) и (IV-21), (IV-41) соот-
ветственно.
подстановке в выражение (IV-36) соответствующие значений cD (а)
и сл0<а)-
Для иллюстрации характера зависимости ZD(a) на рис. IV-20,a
приведены соответствующие данные при a=k2]k\=2 для реактора
полного вытеснения (кривая /), реактора полного смешения (кри-
вая 2) и реактора периодического действия (кривая <?). Для со-
309
поставления приведена также зависимость ZD(a) для случая,
когда продукт В не возвращается в реакционный аппарат (кри-
вые 4 и 5).
В отличие от работы реакционного аппарата без возврата про-
межуточного продукта, когда производительность реакторов иде-
ального режима имеет экстремальный характер (кривые 4 и 5) и
выбор в пользу того или иного типа реактора может быть прямо
противоположным в зависимости от значения а, производитель-
ность реакционной аппаратуры при возврате продукта В моно-
тонно снижается с увеличением степени превращения, причем ре-
актор полного вытеснения сохраняет преимущества во всем диа-
пазоне значений а (кривая /).
Рассмотренная выше зависимость ZD(a) для случая возврата
продукта В справедлива при отсутствии потерь этого продукта на
стадии его выделения и может быть использована для техниче-
ских расчетов, если потери продукта В невелики. В случае зна-
чительных потерь размер их может существенно отразиться на
производительности реактора (и, естественно, на выходе продукта D).
Примером может служить реактор полного смешения, рабо-
тающий в условиях, когда выход продукта В на стадии разде-
ления реакционной смеси равен £в, з- Сохраняя прежнее условие
сЛо + сВо=1 и принимая сВо = свРв 3, нетрудно получить значение
2л(а ,0в, з)
Z ________________________________ (IV-37)
° (l-a)(l-pa3) + aa + apBi3(l-a) UV
при этом_
=__________аРв.зС1 ~а)_________
Сво (1-а)(1-рв з) + аа + арв 3(1-а)
В указанных выше условиях выход продукта D в реакторе
(₽d, 2) становится зависимым не только от степени превращения
исходного вещества, но и от выхода промежуточного продукта на
стадии его выделения:
2 = (i-a)(i-pB з) + аа
Выход продукта D описывается соответствующим уравнением,
приведенным в табл. II-13, если продукт В не возвращается в ре-
актор (Рв, з = 0), и приближается к единице в том случае, когда
потери возвращаемого в реактор продукта В отсутствую^ (₽в,з=1).
В заключение необходимо остановиться на режиме пуска ре-
акционного аппарата, предназначенного для работы с возвратом
промежуточного продукта. Ясно, что стационарный режим работы
реакционного аппарата может быть достигнут при соблюдении
необходимого состава исходной смеси, который удобно выразить
как функцию сВо[сАо(а). Отклонение от указанной зависимости
увеличивает длительность пускового периода при нестационарном
(по составу реакционной смеси) режиме.
310
Вид функции сВо/сДо(а) легко может быть найден из соответ-
ствующих, приведенных выше, значений сДо(а) и сВо(а):
для реактора полного вытеснения и реактора периодического дей-
ствия
СВ, 1—а —(1—а)°
(а — 1) [1 — (1 — а)в]
для реактора полного смешения
СВо ... 1 ~а
СЛ, а
(IV-38)
(IV-39)
Смесь исходного вещества А и продукта В необходимого со-
става может быть получена из исходного вещества в результате
кратковременной работы реакционного агрегата без возврата про-
дукта В (при условии отделения продукта D). Вид функции
с„ 1сл (а) легко может быть найден из соответствующих значений
сА{а.) и св(а), рассмотренных в гл. II:
для реактора полного вытеснения и реактора периодического дей-
ствия
= 1-(1-а)а 1 (j v-40)
сд а - 1
лк
для реактора полного смешения
' Ч а
с. 1 — а + аа
лк
(IV-41)
В приведенных выше выражениях смесь состава сд jcA (а),
полученная (после отделения продукта D) в результате работы
реакционного агрегата без возврата продукта В, соответствует
требованиям стационарной работы реакционного агрегата с воз-
вратом продукта В.
Для рассматриваемой последовательной реакции (^/^1 = 2)
выражения (IV-38)— (IV-41) иллюстрированы рис. IV-20, б.
При заданной для работы реакционного агрегата с возвратом
продукта В степени превращения, равной 0,6, состав исходной
смеси без возврата продукта В может быть получен при а=0,32
(для реактора полного вытеснения) или приа=0,25 (для реактора
полного смешения).
Реакторы промежуточного типа. Скорость превраще-
ния исходных веществ в реакционных аппаратах промежуточного
типа (аппараты с мешалками, барботажные реакторы, каскад
секций полного смешения и т. п.) по своему значению, как пра-
вило, занимает промежуточное положение между скоростями пре-
вращения, достигаемыми в реакционных аппаратах идеальных
режимов при одинаковой для сравниваемых случаев величине сте-
пени превращения исходного вещества.
311
Производительность реакторов промежуточного типа удобно
выразить, пользуясь понятием «коэффициента кратности цирку-
ляции» (см. стр. 201).
Для продукта одностадийной необратимой реакции А —D
осуществляемой в аппарате с внутренней циркуляцией, общим
выражением средней скорости (ы) превращения исходного веще-
ства является:
\ Лд Лк
W = 1—2-----2
с
J с"
с А
лк
Интегрирование знаменателя правой части этого уравнения и
замена с. его значением
Лц
_ ГЦСлк %
Слц Гц + 1
приводит к выражению для средней скорости реакций, описывае-
мых уравнениями реакций первого и второго кинетического по-
рядков, т. е. к выражению производительности реактора с про-
дольным перемешиванием:
при п=1
при п=2
Гц + 1
(IV-42)
(IV-43)
Нетрудно заметить, что для крайних значений коэффициента
кратности циркуляции (гц=0, /ц-*оо) уравнения (IV-42) и (IV-43)
превращаются в уравнения, выражающие производительность ре-
акторов полного вытеснения и полного смешения соответственно.
На примере аппарата с мешалкой (см. рис. П-54) можно ви-
деть зависимость производительности от изменения кратности
циркуляции. Пусть в этот реактор непрерывно и с постоянной
скоростью подается смесь, содержащая исходное вещество А кон-
центрацией сА = сАо. В условиях постоянства объема реагирую-
щих веществ из реактора непрерывно и с постоянной скоростью
удаляется готовая смесь, концентрация исходного вещества в ко-
торой составляет сА . Варьирование числа оборотов мешалки в
широких пределах позволяет иметь ряд вариантов промежуточ-
ного режима и наблюдать изменение степени превращения исход-
ного вещества (или производительности реактора).
Приравнивая выражение для производительности реактора к
уравнениям (IV-42) и (IV-43), получаем выражение расчетного
312
циркуляции:
времени пребывания реакционной массы (0) как функцию сте-
пени превращения исходного вещества и кратности
при п= I
при n=2
0 =-Г (Гц + I) fa
k (1 — а) (Гц+ 1)
(IV-44)
= __________Ц(гц+ 1)
kcAa ’ [гц(1 —а) + 1] (1 —а)
(IV-45)
Рис. IV-21. Зависи-
мость ZD (гц) для
реактора с циркуля-
цией реакционной мас-
сы (схема реакции:
A—>D):
1 — реакция первого кв-
иетического порядка;
2 — реакция второго кине-
тического порядка.
При постоянной скорости питания реактора исходной смесью
расчетное время пребывания 0 = const, и из уравнений (IV-44) и
(IV-45) можно установить зависимость степе-
ни превращения исходного вещества (и, сле-
довательно, производительности реактора) от
кратности циркуляции. На рис. IV-21 приве-
дены соответствующие кривые для реакций
первого и второго порядков, позволяющие
видеть, что производительность рассматри-
ваемого реактора (т. е. степень превращения
исходного вещества) падает с увеличением
числа оборотов мешалки, причем угол накло-
на кривых, заметный в интервале значений
Гц от 0 до 8—10, при дальнейшем увеличении
Гц приближается к нулю.
Занимая промежуточное положение меж-
ду реакторами идеальных режимов по скоро-
сти течения простых реакций, реактор с ог-
раниченным продольным перемешиванием
представляет интерес при проведении автока-
талитических реакций, подобных рассмотрен-
ной выше (см. стр. 304).
При малой скорости некаталитической ре-
акции (I) длительный индукционный период
обусловливает низкую производительность реактора полного вы-
теснения при малых значениях а. Условия работы реактора пол-
ного смешения позволяют избежать влияния индукционного пе-
риода, однако эффект разбавления неблагоприятно сказывается
на производительности реактора.
Ограниченное перемешивание реакционной массы открывает
возможность повышения производительности реакционного аппа-
рата [17]. Производительность каскада секций полного смешения
(см. стр. 183) определяется соотношением размера секций и для
реакций первого кинетического порядка достигает своего макси-
мального значения при равновеликом объеме секций (и) незави-
симо от степени превращения исходного вещества и схемы реак-
ции [И].
Для реакций, порядок которых отличается от первого, отно-
шение Ui+i/fi не равно единице и зависит не только от степени
313
превращения, но и от числа секций каскада, порядковых номеров
рассматриваемой пары секций и кинетического порядка реакции
[18]. Однако, как указывалось в гл. II, при малых значениях ко-
нечной степени превращения исходного вещества оптимальное со-
отношение размера секций каскада с практической степенью при-
ближения можно считать равным единице независимо от порядка
реакции.
Если в каскаде секций полного смешения осуществляется од-
ностадийная реакция, порядок которой не равен единице, то при
конечной степени превращения исходного вещества, превышаю-
щей 5—10%, учет зависимости Vi+i,i (а, п, т, i+1, ...) приводит
к выводу о необходимости соблюдения оптимального соотноше-
ния объема секций.
В практических условиях выполнение этого требования часто
вызывает удорожание аппаратуры и усложняет взаимозаменяе-
мость отдельных ее частей. Вместе с тем каскад равновеликих
секций характеризуется большим суммарным объемом аппара-
туры, необходимой для достижения заданной степени превраще-
ния. Выбор оптимального варианта оформления реакционного аг-
регата'в этом случае становится объектом экономического иссле-
дования.
Более сложная инженерная и экономическая задача возникает
при осуществлении реакций, характеризующихся зависимостью
Р(а). В этом случае отклонение от оптимального соотношения
размера секций каскада сопровождается также и изменением вы-
хода целевого продукта, оказывающего существенное влияние на
экономику процесса. Если, например, речь идет о получении про-
межуточного продукта последовательной реакции и отклонение от
оптимального значения Vf+i,i приводит к уменьшению выхода это-
го продукта при сохранении заданной степени превращения, то
замена каскада секций с оптимальным соотношением их объемов
на каскад равновеликих секций должна сопровождаться увеличе-
нием числа этих секций.
Влияние относительного размера секций на производитель-
ность каскада, отнесенную к промежуточному или конечному про-
дуктам последовательной реакции первого порядка, можно устано-
вить на примере каскада, состоящего из двух секций.
При постоянной скорости подачи исходного вещества А в двух-
секционный каскад изменение относительного объема секций со-
провождается изменением выхода промежуточного и конечного
продуктов, а также изменением призводительности каскада отно-
сительно каждого из этих продуктов.
Для последовательной реакции первого порядка при заданной
степени превращения исходного вещества зависимости ZB(v2/vi)
и ZD(y2/vi) представлены графически на рис. IV-22.
Максимальная производительность каскада как по продукту
В, так и по продукту D достигается в равновеликих секциях, со-
314
ответствующих максимально возможному приближению рассмат-
риваемой системы к условиям реактора полного вытеснения.
Изменение производительности каскада равновеликих секций
с изменением числа этих секций (т) для простых и сложных ре-
акций нетрудно выразить, пользуясь ранее установленными зави-
симостями:
для реакции А — -> В первого порядка
с. a k,a
для последовательной реакции А —+ В —D
первого порядка
1+т
7 -Аг И — m ~ 1
в m л» 1 ’ Nm (N — 1)
7 __SlL_
D т Ло (1- аГ1'”1 - 1
Рис. IV-22. Зависи-
мость производитель-
ности двухсекцион-
ного каскада аппара-
тов полного смешения
от соотношения объ-
емов секций (схема
реакции: А---►В---->
—> D; порядок — пер-
вый; k2iki = 0,5).
где рп — выход продукта D в зависимости
от числа секций каскада (см. стр. 210); А =
= (1— a)1/m(l— а) + a (a = k2/ki).
Ясно, что производительность каскада сек-
ций по продукту В монотонно возрастает по
мере увеличения числа секций; производи-
тельность каскада по продукту D при этом
снижается.
При заданных производительности реакционного агрегата и
степени превращения исходного вещества выбор числа секций ка-
скада должен быть основан на учете стоимости реакционного аг-
регата. Решение подобного вопроса в общем виде вряд ли воз-
можно, так как результат выбора зависит от степени влияния
числа секций на удельный расход сырья, а также от конструк-
ции реакционной аппаратуры, условий проведения процесса
(температура, давление), стоимости конструкционных материалов
и Т. Д. t
Критерием оптимальности для случая, когда выход продукта
не зависит от степени превращения исходного вещества и числа
секций каскада (простые реакции, параллельные реакции одина-
кового кинетического порядка), является стоимость секций кас-
када. Если выход целевого продукта (промежуточный и конечный
продукты последовательных реакций) находится в зависимости
от числа секций каскада, критерием оптимальности может слу-
жить показатель, учитывающий стоимость сырья на единицу про-
дукта и удельную (отнесенную к единице продукта) стоимость ре-
акционного агрегата. Структура этого показателя (ГЦ) может
3J5
быть принята аналогичной структуре показателя приведенных
затрат:
Я. = с + Еяк
К
1,6
12
1,0
«б-
&
а=0,40
'.-г 0,60
0.70
—0.90
сс=0,9в
12 3 4 5 6 7 8
т
Рис. IV-23. Зависимость относитель-
ной стоимости каскада равновеликих
секций (К) от числа секций каска-
да (т).
г где С — стоимость сырья, расходуемого на единицу продукта; /< —
стоимость реакционного агрегата, отнесенная к единице продукта;
£н=0,2 4-0,33.
Первый случай можно рассмотреть на примере реакции А->В
первого порядка, предположив, для простоты, что каждая секция
представляет собой цилиндрический сосуд с заданной высотой
(барботажный процесс) и прак-
тически постоянной толщиной
стенок. Стоимость перемешиваю-
щего устройства, коммуникаций
и КИП входит при этом в стои-
мость секции. Используя уравне-
ние (П-249), нетрудно подсчитать
расход металла на изготовление
каскада равновеликих секций для
заданного значения а и выра-
зить зависимость относитель-
ной стоимости реакционного аг-
регата от числа секций каскада
(рис. IV-23).
Для рассматриваемого слу-
чая целесообразность примене-
ния односекционного аппарата
ограничивается степенью превра-
щения 0,60—0,65. С увеличением
степени превращения сильно сказывается эффект полного смеше-
ния и возникает необходимость использования каскада секций.
Вполне естественно, что оптимальное число секций каскада
возрастает по мере увеличения степени превращения. Оптималь-
ные условия для практически предельного случая (а=0,98) до-
стигаются в каскаде из 4—5 секций.
Более сложным является поиск оптимума для второго, чаще
встречающегося варианта, так как оптимальное число секций на-
ходится в зависимости не только от величины отношения стоимо-
сти сырья и стоимости материала аппаратуры, но и от размера
аппаратуры (мощность производства, скорость реакции).
Вследствие относительно высокой стоимости сырья подобный
анализ в технологии основного органического синтеза чаще всего
показывает, что экономический оптимум достигается в многосек-
циоином каскаде, стоимость которого значительно превышает ми-
нимально возможное ее значение. Окончательное решение вопро-
са требует учета таких условий, как усложнение коммуникации и
системы КИП, увеличение площади производственного помещения
и т. п.
316
ОС
Коэффициент производительности. Характер кривых,
выражающих производительность реакционных аппаратов как
функцию степени превращения исходного вещества, дает пред-
ставление об относительной производительности реакторов по про-
дуктам простых й сложных реакций:
На примере простых одностадийных реакций ранее было по-
казано, что движущая сила, а следовательно, и средняя скорость
образования продукта имеет максимальное значение в аппаратах
полного вытеснения. Это обстоятель-
ство, послужившее в ряде работ [12,
15, 16] основанием для принятия за
критерий оценки интенсивность про-
цесса, проводимого в реакторе полного
вытеснения, принимается и в настоя-
щем изложении.
При заданной степени превраще-1
ния исходного вещества отношение
производительности реакторов каждой
группы к производительности реакто-
ра полного вытеснения, численно рав-
ное ч отношению продолжительности
пребывания реакционной массы в ап-
паратах рассматриваемых типов и на-
зываемое «коэффициентом полезного
действия реактора» [19] или «концен-
трационным коэффициентом полезного
количественно оценивать эффективность реакционной аппаратуры.
Общим выражением для этого коэффициента является отноше-
ние:
П, = ->Л = 4н^ = /(“)<1 (IV-46)
°п. с .в
Рис. IV-24. Зависимость т|' (а)
для реакции А —> D:
1 — реакция второго кинетического
порядка; 2 — реакция первого кине-
тического порядка.
действия» [20], позволяет
(индексы «п. в» и «п. с» относятся соответственно к реакторам
полного вытеснения и полного смешения).
Для простых односторонних реакций первого и второго кине-
тического порядка зависимость, выражаемая уравнением (IV-46),
может быть легко найдена при подстановке в него значений Z из
уравнений (IV-18) и (IV-14), (IV-15) соответственно.
’ Полученные таким образом выражения позволяют характери-
зовать реакционные аппараты полного смешения величиной т)'(сь):
при п=1
(IV-47)
U 1 — (X
при п=2
n'=l-« (0<п'<1) (IV-48)
график которой приведен на рис. IV-24.
Удобным выражением зависимости т]'(а), отнесенной к исход-
ному веществу реакций всех типов, является предложенный
317
Орочко [20] способ, основанный на учете кратности внутренней
циркуляции. В соответствии с этим значением ц' для реакций
первого и второго кинетического порядков можно определить с
помощью следующих выражений:
при п = 1
при п = 2
In
1 — а
11 " , J,.1 гц(1-а) + 1
(гц+1)1п (1_а)(Гд+1)
, гц(1— а) + 1
11 Гц + 1
(IV-49)
(IV-50)
При r4=Q значения rf в уравнениях (IV-49) и (IV-50) стано-
вятся равными
Рис. IV-25. Зависи-
мость Дд (а) для реак-
* ~ &2 —
ции А------> В-----> D
первого кинетического
порядка.
Значения /—0,1;
2-0,5; 3 — 5; 4- 10; 5-30;
кривая 6 объяснена в
тексте.
единице; при гц—>- оо из уравнения (IV-50) следует,
что коэффициент —а, т. е. приходит
. к виду уравнения (IV-48).
Производительность реакционного аппа-
рата полного смешения для простой односто-
ронней реакции может быть также сопоста-
влена с производительностью реактора пол-
ного вытеснения при одинаковом для обоих
случаев значении расчетного времени пребы-
вания реагирующих веществ в реакционном
пространстве.
Для последовательных реакций выражения
для т)' по промежуточному продукту В и ко-
нечному продукту D имеют различную
туру:
струк-
г______(1 — а)2 (а — 1) In (1 — а)
’Ib- [1 _а(1 _а)] [(1 -а)«-(1 -а)]
z______аа (1 —а) (а— 1) In (1 —а)
[1 — а (1 — а)] (1 — (1 — а)а — аа]
(IV-51)
(VI-52)
где a=k2jk\.
Графическое изображение зависимости
IV-52 дано на рис. IV-25. Вид кривых не яв-
ляется неожиданным. Относительно высокая
концентрация промежуточного продукта В в
реакторе полного смешения обусловливает
значительную скорость реакции образова-
поэтому производительность реактора полного
ния продукта D,
смешения в определенном интервале значений а превосходит про-
изводительность, достигаемую в реакционном аппарате полного
вытеснения.
Интервал значений а, при котором т}д>1, зависит от величины
отношения констант скорости последовательно текущих реакций.
При т. е. когда скорость процесса образования конеч-
ного продукта лимитируется первой стадией и последовательная
318
реакция может быть приближенно описана уравнением простой
реакции, значение Цд совпадает с значением для реакций типа
A->D (кривая 6 на рис. IV-25).
В тех случаях, когда константа скорости превращения проме-
жуточного продукта относительно мала и k2lk\<§С1, высокая кон-
центрация промежуточного продукта в условиях реакционного
аппарата полного смешения обусловливает высокую скорость об-
разования конечного продукта D, превосходящую среднюю ско-
рость, имеющую место в реакторе полного вытеснения.
Отношение рассматриваемых скоростей определяется степенью
превращения исходного вещества, и при повышении значения а
величина этого отношения снижается.
Естественно, что влияние степени превращения сильнее прояв-
ляется для реакций, характеризующихся большим значением k2lk\.
Уравнение (IV-46) может быть записано как отношение про-
изводительности реакторов разных типов, выраженных по одному
из продуктов сложной реакции (например, продукту i)
Zj,n. с = с,, п, с(«) т (а) = Рг, п. с(«И (а) .
Zi, п. в 0 (а) ’ с/, п. в (а) Pi, п. в («) • 6 (а)
и граничное его условие (ц'< 1) справедливо, если
Pi, п. с (а) = Р/, п. в (а)
что соблюдается лишь в тех случаях, когда выход продукта не
зависит от степени превращения исходного вещества (одностадий-
ные реакции, параллельные реакции одного порядка).
Поскольку реакции, лежащие в основе большинства процессов
промышленного органического синтеза, относятся к группе слож-
ных реакций и характеризуются зависимостью выхода продуктов
от степени превращения исходного вещества, при сравнительной
оценке производительности реакционной аппаратуры, по-видимо-
му, следует пользоваться уравнением (IV-53), частным случаем
которого является уравнение (IV-46).
В заключение следует заметить, что рассмотренные выше за-
висимости Zj(a, Т) могут быть использованы при выборе темпе-
ратурного режима реакционного процесса.
При заданной степени превращения сырья характер зависимо-
сти Zi(T) определяется видом функции рг(7'). Для реакций, ха-
рактеризующихся увеличением выхода целевого продукта с повы-
шением температуры, производительность реакционного аппарата
по этому продукту также монотонно увеличивается при повыше-
нии температуры.
В тех случаях, когда повышение температуры сопровождается
увеличением выхода целевого продукта или функция РДТ) имеет
максимум, производительность реактора (в зависимости от соот-
ношения предэкспоненциальных множителей и разности энергий
активации) либо монотонно меняется, либо проходит через ма-
ксимум.
319
Для первого' случая поиск экономически оптимальной темпе-
ратуры, очевидно, лйшен смысла, так как процесс следует прово-
дить при возможно более высокой температуре.
Если в результате повышения температуры наблюдается умень-
шение выхода целевого продукта или если функция р,(Г) имеет
максимум, поиск экономически целесообразного температурного
режима должен быть основан на учете влияния температуры на
суммарные энерго-материальные затраты. Основанием для подоб-
ного поиска может служить зависимость (см. гл. I):
П (Г) = С + ЕЯК = ф (Г) + Еи • i|> (Г)
где С — удельная (на 1 т продукта) стоимость сырья, расходуе-
мого в реакторе; К—удельная (на 1 т продукта) стоимость ре-
акционного аппарата.
Если учесть, что удельная стоимость сырья играет решающую
роль в образовании экономических показателей процессов основ-
ного органического синтеза, то становится очевидным, что опти-
мальный температурный режим может быть весьма далек от ре-
жима, соответствующего минимальному размеру реактора.
ТЕПЛОВОЙ РЕЖИМ
ЭКЗОТЕРМИЧЕСКИХ РЕАКЦИЙ
Выше было показано, что экономичность “производств
основного органического синтеза существенным образом зависит
от степени превращения сырья в реакторе и обычно существует
такое (оптимальное) значение степени превращения, при котором
себестоимость товарного продукта оказывается минимальной. При
минимизации себестоимости, достигаемой вариацией степени пре-
вращения сырья, иногда в некотором интервале значений а про-
цесс оказывается неустойчивым, самоускоряющимся и практически
нереализуемым. В таком случае на возможность вариации сте-
пени превращения должны быть наложены ограничения, опреде-
ляемые границами области неустойчивых режимов. Неустойчи-
вость процесса химического превращения обусловлена, как пра-
вило, тепловыми факторами: соотношением скоростей выделения
тепла при реакции и отвода его из аппарата.
Если тепло отводится из реакционного пространства со ско-
ростью большей, чем скорость выделения тепла, процесс всегда
устойчив. Когда тепло отводится с меньшей скоростью, реакцион-
ное пространство разогревается, скорость реакции (вследствие
сильной зависимости от температуры) быстро увеличивается, а
вместе с ней повышается и скорость выделения тепла. Это приво-
дит к еще большему увеличению температуры, возрастающей
скачком до максимальной величины. В аппаратах, работающих
периодически и в режиме полного смешения, скачок температуры
происходит во времени, в аппаратах идеального вытеснения — во
времени и вдоль пути движения реакционной массы.
320
ГОМОГЕННЫЕ РЕАКЦИИ
Для. выяснения особенностей теплового режима экзо-
термических реакций рассмотрим наиболее общий случай — гомо-
генную экзотермическую реакцию, протекающую в жидкой фазе
в аппарате полного' смешения с теплоотводом через стенку и за
счет испарения части жидкости. Получаемые при этом результаты
могут быть использованы для аппаратов полного вытеснения, а
также для реакторов периодического действия.
Если за начало отсчета температуры принять температуру по-
даваемого в аппарат сырья и пренебречь изменением теплоемко-
сти при реакции, то тепловой баланс реактора полного смешения
может быть представлен в виде:
Qp~ Qncn ~ Qct ~ Qnp (IV-54)
где С — средняя теплоемкость; р — плотность; W — объем реакто-
ра; Т — температура; т — время; Qp— скорость выделения тепла
при реакции; фисп — скорость отвода тепла при испарении жидко-
сти, Qct — скорости отвода тепла к стенке; Qnp — скорость отвода
тепла с горячими продуктами реакции, выходящими из аппарата. .
Скорость выделения тепла равна произведению скорости ре-
акции (ш), отнесенной ко всему объему реактора, на ее тепловой
эффект Q:
__________________в
Qv=QWw = QWcnBe RT
где с — концентрация реагирующего вещества; п — порядок ре-
акции; В — предэкспоненциальный множитель в константе скоро-
сти реакции; Е — энергия активации; /? — газовая постоянная.
Скорость отвода тепла за счет испарения жидкости равна
произведению скорости испарения на теплоту фазового перехода.
Если степень насыщения газа, покидающего реактор, парами
жидкости равна /, объемная скорость газа, выводимого из аппа-
А^ИСП
рата, (01, а парциальное давление паров р = рое RT (АНИСП —
теплота испарения), то
Qncn = aw ®1
Скорость теплоотвода через стенку реактора пропорциональна
разности температур реакционной зоны и стенки (Тст). а также
поверхности стенки (S):
Qct ~ KtS (Т — Т'ст)
где Кт — коэффициент теплопередачи. Скорость отвода тепла из
аппарата горячими продуктами реакции пропорциональна объем-
ной скорости ввода реагентов (ш), средней теплоемкости и плот-
ности реагирующей смеси, а также разности температур на входе
(Тих) и выходе из реактора:
Qnp= Срш (Т— твх)
21 В. С. Хайлов, Б. Б. Брандт
321
Подставив значения скоростей теплоприхода и теплоотвода в
уравнение (IV-54), получим:
СрГ 4? = WQW ~ ~KtS(T~ Т„) -Сра(Т- Гвх) (IV-55)
Если ю = 0, уравнение (IV-55) описывает процесс в периодиче-
ски работающем реакторе, а также в аппарате полного вытесне-
ния; при отсутствии теплоотвода из зоны реакции (р = 0, /Ст = О,
со = 0)—адиабатический режим.
Адиабатический режим
При адиабатическом режиме все выделяющееся тепло
реакции остается в реагирующей системе. Количество тепла, вы-
делившееся к данному моменту времени за счет химической ре-
акции, а также увеличение температуры в системе пропорциональ-
ны убыли исходного вещества.
Скорость химической реакции убывает при уменьшении кон-
центрации реагентов и возрастает с увеличением температуры.
Вследствие более сильной, чем от концентрации, зависимости w
от температуры скорость адиабатической реакции возрастает со
временем. При достижении некоторой концентрации реагентов
скорость реакции начинает убывать, и химическое превращение
прекращается после полного израсходования одного из реагентов.
Температура при этом достигает своего максимального значе-
ния Тк.
Поскольку зависимость скорости адиабатической реакции от
концентрации реагентов существенно отлична от аналогичной за-
висимости для изотермических реакций, рассмотрим более по-
дробно указанную зависимость на примере адиабатической реак-
ции первого порядка. Уравнение теплового баланса для этого
случая принимает вид:
dT
cP~^ = Qw (IV-56)
В выражение для скорости реакции w входят две зависящие
пт времени переменные: количество прореагировавшего в единице
объема вещества m и температура
___Е_
w = (ca — m)Be RT (IV-57)
(здесь и в дальнейшем индекс «О» относится к начальному со-
стоянию, индекс «к» — к конечному.)
Для исследования уравнения (IV-57) необходимо знать зави-
симость ш(Т’). Так как превращение некоторого количества веще-
ства m происходит адиабатически, выделяющееся при этом тепло
mQ расходуется только на нагрев смеси, т. е.
mQ = Cp(T-Ta) (IV-58)
322
Если т = со, температура принимает значение Т = ТК. Исполь-
зуя уравнение (IV-58), текущую концентрацию реагента можно
представить в виде:
Со - m = с0 (1 - —) = с0 (1 - (IV-59)
\ ^о/ \ * к * о /
Подставляя уравнения (IV-57) и (IV-59) в формулу (IV-56) и
учитывая выражение (IV-58), получаем:
__Е_
^=В(ТК-Т)е RT (IV-60)
Зависимость Т (т) может быть получена при интегрировании
уравнения (IV-60). Однако получение аналитического решения в
замкнутом виде не представляется возможным. Для исследования
этой зависимости воспользуемся приближенным решением, кото-
рое будет приведено ниже. Предварительно определим темпера-
туру Тт, при которой реакция. протекает с максимальной ско-
ростью. Указанная температура определяется из условия
^- = 0
dT
или
(IV-61)
Со dT , ,
поскольку т — -=.-=- • -т—, как это следует из дифференцирова-
Л к 1 Q “Т
ния уравнения (IV-59) по времени. Условие (IV-61) с учетом вы-
dT
ражения для примет вид:
Be
Отсюда
или
Е
Мт
F
—г Гк-тт)-1
RT-m
= о
где Z' — Если энергия активации достаточно велика, так что
A iR ______
удовлетворяется неравенство <С 1, то разлагая 1 + — в
ряд и ограничиваясь тремя членами разложения, имеем:
-Г^=1-2ф. (IV-62)
/к -С
4RT
Уравнение (IV-62) получено в предположении, что 1.
Следовательно, Тт мало отличается от конечной температуры,
21*
323
т. е. скорость реакции возрастает почти до полного израсходова-
ния исходного вещества. Причем температура, соответствующая
максимальной скорости реакции, тем ближе к конечной, чем боль-
ше Е и меньше Тк. Так, если Е=4-104 кал/моль, температура Тт
отличается от Тк на -<40% при конечной температуре 2000° С и
на -<2,5% при 500° С.
Доля прореагировавшего вещества при достижении максималь-
ной скорости реакции может быть вычислена из уравнения (IV-59)
при Т=Тт. Подставив уравнение (IV-62) в выражение (IV-59),
имеем
т , ЯГк
4£(Тк-Г0)
(IV.-63)
т. е. доля израсходованного вещества при достижении температу-
ры, равной Тт, тем ближе к единице, чем больше энергия акти-
вации и адиабатический разогрев смеси и меньше конечная тем-
пература. Так, если £'=2,0-104 кал]моль, 7"к=600°К, 7’о = ЗОО°К,
скорость реакции достигает своего максимального значения при
израсходовании 89% исходного вещества.
Как указывалось, точная зависимость температуры смеси при
адиабатическом течении химической реакции не может быть по-
лучена в замкнутом аналитическом виде. Однако для начальных
стадий процесса, когда Т^Т0, может быть получено приближен-
ное решение [21], дающее также удовлетворительные результаты
при То^Т^Тщ- Разложим показатель экспоненты в уравнении
(IV-60) в ряде в точке Т= То.
и ограничимся для малых значений Д = Т—То двумя первыми
членами разложения, как это предложено Франк-Каменецким [22].
Тогда уравнение (IV-60) примет более простой вид:
_ d (Т- Та) Де » (IV-64)
dr
RT2 Е
где •& = —А = В(ТК — Т0)е RTa. Интегрирование уравнения
(IV-64) дает искомую зависимость:
То-01п(1-у т) (IV-65)
- Точная зависимость Т(т), полученная численным интегрирова-
нием уравнения (VI-60), представлена графически на рис. IV-26.
Пунктирной линией отмечен график приближенного решения,
практически совпадающего с точным только до Т~Тт. Решение
уравнения (IV-65) вследствие сделанных допущений не может от-
ражать плавный переход температуры от Тт до Тк, но поскольку
Тт мало отличается от конечной температуры (если энергия ак-
тивации достаточно велика), уравнение (IV-65) достаточно точно
описывает изменение температуры при адиабатическом протека-
нии реакции.
Приближенная зависимость скорости реакции от времени мо-
жет быть получена дифференцированием уравнения (IV-65) по
времени, т. е.
w — (IV-66)
Выражение (IV-66) имеет смысл для т^Стт-
На рис. IV-27 сопоставлена точная зависимость скорости ре-
акции от времени [численное интегрирование уравнения (IV-60),
Рис. IV-26. Возрастание темпера-
туры при адиабатическом проте-
кании реакции первого порядка
(Е = 104 кал/моль, Тк — 1000° К,
То = ЗОО°К, В = 106 1/сек):
— —точное решение, полученное
численным интегрированием,......—
приближенное решение.
Рис. IV-27. Возрастание скорости адиа-
батической реакции первого порядка
(Е = 104 кал/моль, Тк= 1000° К, То=ЗОО° К,
В = 10е 1/сек):
/ — точное решение, полученное численным ин-
тегрированием; 2 — приближенное решение.
Е = 104 ккал/моль, Тк=1000оК, То=ЗОО°К, В=106. l/сек] с прибли-
женной зависимостью (IV-66). Как следует из рис. IV-27,
уравнение (IV-66) с достаточной степенью точности описывает
скорость химической реакции, проводимой в адиабатических
условиях.
Время, в течение которого превращается основная масса ве-
щества, может быть вычислено из уравнений (IV-62) и (IV-65).
Ю
Обозначив это время через хт, имеем:
Правая часть полученного выражения равна малому отрица-
тельному числу. Действительно, учитывая, что Е (в кал/моль) —
величина порядка 104, а Тк и То (в °К)—порядка 102, имеем:
1п(1-^-тт)«-102
Следовательно
_ Л_
О ЯТ20е RT°
~А ~ ЕВ (Тк — То)
(IV-67)
Для выяснения характерных особенностей зависимости Т’(т)
при адиабатическом проведении реакции определим время те, в
течение которого скорость реакции изменяется в е раз (we), а
также соответствующее этому изменение температуры.. Если при
т=0 w = w0, то we = woe, что возможно, как это следует из уравне-
ния (IV-66), при
О / 1 \
= (IV-68)
Подставляя те в уравнение (IV-65), находим, что увеличению
скорости реакции в е раз соответствует повышение температуры
реагирующей смеси на Ф = RTo/E град, т. е. всего на несколько
градусов, так как Е (в кал/моль)—величина порядка 104, а То
(в °К)—порядка 102. Из сопоставления уравнений (IV-67) и
(IV-68) следует, что время, в течение которого скорость реакции
изменяется в е раз, а температура повышается на в град, равно
приблизительно 60% всего времени процесса. Величину те обычно
называют периодом индукции.
Закономерности адиабатической реакции в аппарате полного
вытеснения, вообще говоря, не отличаются от рассмотренных
выше. При определении длины реакционной трубки необходимо в
случае возрастания температуры учитывать изменение объема га-
зов, если они участвуют или образуются в реакции.
Так, длина реакционной трубки, в которой протекает адиаба-
тическая реакция в потоке газов, может быть определена следую-
щим образом. Если и и coi— соответственно линейная и объемная
скорости газа, a F — площадь поперечного сечения трубки, то
путь dL, пройденный газом за время dx, равен:
dL = и dx = dx
326
Учитывая теперь зависимость (oi(T'), имеем:
nRT
aL — —=— dx
Fp
Интегрирование последнего уравнения с учетом выражения
(IV-65) дает:
г Гт < А < Л Л \ . /. А \1
Л=-тИо+т +
Зная зависимость Т(х) и соотношение между концентрацией и
температурой, можно по заданной степени превращения опреде-
лить время реакции и, соответственно, длину трубки. Обычно ади-
абатические реакции проводят до практически полного превра-
щения исходного сырья. Тогда т=,&/Л и
£ = -^-[То + 0] (IV-69)
Г p/i
Пример 1V-1. Хлорирование углеводорода проводят при атмо-
сферном давлении в адиабатических- условиях. Определить объем реактора про-
изводительности) Z=4 т продукта)*, если температура сырья, вводимого в ап-
парат, равна 400° К, а температура продуктов 800° К. Реакция протекает по
первому кинетическому порядку. Константа скорости реакции равна
18 300
107е RT 1/сек. Массовое отношение реагентов равно пг кг углеводорода!кг
хлора.
Решение. Полагая, что хлоратор может быть- оформлен в
виде аппарата полного вытеснения, воспользуемся для расчета формулой
(IV-69).
Зависимость между числом молей газа, подаваемого в реактор в 1 ч, и
производительностью можно найти, исходя из следующих соображений. Так как
реакция хлорирования
RH + С12 —► RC1 + НС1
не сопровождается увеличением числа молей, равно сумме молей хлора и уг-
леводорода:
= «С1 + ”rh
Производительность Z будет определяться количеством подаваемого в реак-
тор хлора (хлор реагирует полностью):
Z = Псг^С!
где Afci=36,5— молекулярная масса хлора. Массовое отношение реагентов
равно:
м 7 ^RH
"rhMrh "S RH Л1С1
m =-----r;— ----------~---------
"Cl-Mci %
Отсюда
327
Здесь Mrh — молекулярная масса углеводорода. Для объема реакционного
пространства окончательно имеем:
LF=(-ar~+•xf-Vtt(To+^)==
\ ^RH ^С! /
1,986 • 4002 / 1,986 • 4002 \
4- 103 18 300 \400+ 18 300 // т 1 \
“ 3600 °’ 82 18 300 МС1 + 36i5 )
Ю7е *’986’400 (800-400)
= 1640+2,7- Ю-2^ л
V^RH /
Изотермический режим
Изотермический режим протекания экзотермической ре-
акции возможен лишь при определенных условиях теплообмена.
При нарушении этих условий начинается самопроизвольный разо-
грев или охлаждение реакционной смеси и реакция переходит в
нестационарный режим. Критические условия, приводящие к сры-
ву теплового режима, необходимо учитывать при проектировании
реакционных аппаратов и при управлении процессами, протекаю-
щими в реакторах.
Для выяснения критических условий целесообразно преобра-
зовать экспоненты, как это сделано выше — см. уравнения (IV-60)
и (IV-64), — в первых двух слагаемых в правой части уравнения
(IV-55), а в последнее слагаемое ввести температуру стенки ТСТ.
После указанных преобразований уравнение (IV-55) примет бо-
лее простой вид:
^- = Ае* - • (IV-70)
ах Д + Гст
Е А#исп
где Л - Ср с е , с RC(>w е . , l> C(fW т w ,
(О /?Г2
8 ~ (Тег — Твх); & = Т — Гст; = Д//исп •
При изотермическом протекании реакции производную от тем-
пературы по времени можно принять равной нулю. При этом урав-
нение (IV-70) переходит в алгебраическое:
А А
' Ае 9 = С'е01 +РД + 8 (IV-71)
Стационарным режимам соответствуют действительные реше-
ния уравнения (IV-71). При невозможности осуществления ста-
ционарного режима уравнение (IV-71) не имеет действительных
корней.
Проанализируем уравнение (IV-71) и определим критические
условия для некоторых частных случаев, если реакция протекает 328
328
Рис. IV-28. Графическое решение
, In g
уравнения b = —.
по нулевому порядку и последовательно отсутствует теплоотвод
через стенку за счет испарения и за счет вывода из аппарата го-
рячих продуктов реакции.
1. Теплоотвод за счет испарения отсутствует, температура ис- -
ходного сырья равна температуре стенки (С'=0, е = 0). Критиче-
ские условия для этого случая определяются из уравнения
. 4 ПА (IV-72)
Введем новую переменную g = -^-A. При этом уравнение
(IV-72) будет иметь вид:
е6* = £ (IV-73)
где Ь = Зависимость Ь(В), соответствующая уравнению
(IV-73), для различных значений b
Как следует из рисунка, уравнение
(IV-73) имеет действительные кор-
ни при значениях &<1/е. Критиче-
ские условия для рассматриваемо-
го случая могут быть вычислены
из максимального значения пара-
метра
А 1
* = = 7 {IV'74)
при котором уравнение (IV-73) име-
ет еще действительное решение
l = = e (IV-75)
Из сопоставления уравнений
(IV-74) и (IV-75) следует, что мак-
симальный в изотермическом режиме перепад температуры в ре-
акторе (разогрев смеси) равен Ф град, т. е.
Ткр-Т„ = —(IV-76)
Подставляя в уравнение (IV-74) значения для A, D, Ф и учи-
тывая, что порядок реакции — нулевой, получаем:
Е
RT?
(Cp® + KTS)—
£
Выше было показано, что при изменении температуры Ша Фарад
скорость реакции изменяется в е раз. Следовательно, числитель
выражения (IV-77) представляет собой скорость выделения тепла
329
во всем объеме реактора при температуре 7’1;р = Т+й, а знамена-
тель— скорость отвода тепла из реактора при перепаде темпера-
тур Ф град. Таким образом, критические условия перехода от изо-
термического режима к адиабатическому достигаются при равен-
стве скоростей теплоприхода и теплоотвода.
2. Теплоотвод за счет испарения отсутствует (С'=0). В этом
случае уравнение (IV-71) принимает вид:
А
Ае* = ОД + е (IV-78)
Для определения критических условий целесообразно ввести
(D е \ ——
новую переменную ф = 1-^-Д +При этом уравнение (IV-78)
Рис. IV-29. Область изотермиче-
ских режимов эндотермической
реакции нулевого порядка в аппа-
рате полного смешения (теплоот-
вод за счет испарения отсутст-
вует). ,
примет вид:
е6* = ф (IV-79)
А -~
где& = -£ф-е . Уравнение (IV-79)
имеет действительные решения при
Ь^Л/е, т. е.
е
А Dft 1
D^e
(IV-80)
Связь между параметрами в вы-
ражении (IV-80) удобно предста-
еА
вить графически. Обозначив
через ф и преобразовав уравнение
(IV-80), получим:
Л'р < <Р
где Ь, = . Зависимость &1(ф) ^приведена на рис. IV-29. Область
под кривой соответствует значениям параметров и , при
которых возможен изотермический режим. Критическим значе-
ниям параметров соответствуют обе ветви кривой.
Например, если -~f=0,3, изотермический режим возможен
в А
при значениях параметра. от 1,65 до 5,9. Максимальное зна-
чение параметра обеспечивающее изотермическое течение
еА
реакции, равно 1/е, при этом •д^ = е'.
Таким образом, критические режимы возможны при значениях
= и dV-Sl)
330
где tn', n' — постоянные, определяемые из рис. IV-29. Подставляя
выражения для A, D, О, е в уравнение (IV-81) и учитывая, что
реакция — нулевого порядка, получаем:
Е
WBQe КТст е
-----------------5—= «
RTCT
(Срш + KtS)
t,
Сра> (Тст — Твх)-1
" е
(IV-82)
(IV-83)
E
WBQe KTqt e
Выражение (IV-82) представляет собой отношение скорости
выделения тепла при Т = ТЩ) к скорости теплоотвода. При пра-
вильно выбранном тепловом режиме скорость выделения тепла
за счет реакции при, критической температуре должна обеспечи-
вать разогрев сырья от Твх до Т^-ТС1:, как это и следует из выра-
жения (IV-83).
Максимальный разогрев реакционной массы при сохранении
изотермического режима может быть вычислен из уравнения
(IV-79). Подставляя в уравнение (IV-79) максимальное значение
параметра й = 1/е, замечаем, что ф при этом равно е, т. е.
или, учитывая выражение (IV-78), получаем:
Отсюда максимальный разогрев реакционной массы без нару-
шения изотермического режима равен:
Д = О - ~ (I V-84)
Подставляя в формулу (IV-84) выражения для Л, О, е и D,
окончательно имеем:
RT* Срш
Гкр-Гст^^-р—(IV-85)
2- С рй) “Н 1\ уО
3. Теплоотвод осуществляется в основном за счет испарения
части жидкости (7’ст = 7’вх, = Qncn^Qnp) Тепловой баланс
реактора в этом случае можно представить, следующим образом
или в безразмерном виде:
е(Л = д + Гвх (lv.86)
331
Уравнение (1V-86) аналогично уравнению (IV-76), рассмот-
ренному ранее. Изотермические режимы в данном случае воз-
можны при
еС' (O-Oj) , ДГВХ ,
---- =п' и - < т
ДОтО еС
(IV-87)
Учитывая выражения (IV-87), а также значения для А и С',
имеем для критических условий:
ею^вх АЯИСП /Я [-А/^исп - 1)
WQwbxRT^
и
IF QWbxTbx
е®1Рвх/ АЯИСП
(IV-88)
(IV-89)
Здесь рвх— давление пара испаряющейся жидкости при темпера-
туре Т—Твх; wa— скорости», реакции при температуре 7'=7'вх.
Максимальный разогрев, при котором еще возможен изотер-
мический режим, равен:
- 'O'O’i
Л=-тг~4---Т„ (IV-90)
0 — 01^ 7
Подставляя выражения для О и в уравнение (IV-90),имеем: '
00, /?ТВ2Х 1
Гкр_ е-в,= Е АЯИСП (IV-91)
Е * 1
- Из уравнения (IV-91) следует, что изотермические режимы
возможны при т. е. Д/7Исп>£- Если ДНИСП<Е, режим все-
гда нестационарный и близкий к адиабатическому *.
Из значений критических параметров может быть вычислена
возможная температура сырья, подаваемого в реактор (Гвх). По-
сле сопоставления уравнений (IV-88) и (IV-89) получим:
1 Уравнение теплового баланса для неизотермнческого режима (,0т>'0’) мож-
но представить в виде
</А . О Г, С Л(о, о)1
Л Ае Г ^(А+Гвх) е J
а так как -----4- < 0, экспоненциальный член убывает с температурой. Если
От v
1 А (<г-т)
при этом . , =—е т0 —л iл""| "т \— мало по сравнению с
Д+Твх С ^(Д+Твх)
единицей и процесс протекает как адиабатический:
д
dA . О
-7— = Де
</т
332
Концентрация
Рис. IV-30. Скорости расходоиаиия и
намыва реагента в аппарате полного
смешения для реакции нулевого по-
рядка:
/ — скорость расходования реагента при от-
воде из аппарата всего тепла реакции;
2 —скорость расходования реагента при ча-
стичном отводе тепла реакции; 3—скорость
намыва.
Если газ является реагентом, то адиабатические режимы не-
возможны.
Таким образом, выше было установлено, что для реакции ну-
левого порядка, проводимой в аппарате полного смешения, при
различных способах отвода тепла существуют критические усло-
вия, при которых происходит срыв теплового режима; были опреде-
лены также условия, при которых'возможны критические явления.
Очевидно, что критическим условиям соответствуют и опреде-
ленные степени превращения исходного сырья. Это весьма су-
щественно для проектирования
реакционной аппаратуры.
Ранее было показано, что в
аппаратах полного смешения при
стационарном режиме скорость
реакции равна скорости намыва,
т. е. разности скоростей ввода ре-
агентов в аппарат и вывода их с
продуктами реакции. Концентра-
ция исходного вещества в реак-
торе определяется точкой пере-
сечения графиков скорости реак-
ции (расходование) и скорости
намыва (рис. IV-30). Для реак-
ции нулевого порядка, проводи-
мой при постоянной температуре,
скорость не зависит от концен-
трации. Если же отводится толь-
ко часть тепла и температура по
мере протекания реакции повы-
шается, то скорость последней
возрастает экспоненциально при
уменьшении концентрации. При
этом скорость намыва независи-
мо от температуры убывает по
линейному закону. Как следует
из рис. IV-30, максимальное значение концентрации в реакторе,
при котором возможен еще стационарный режим, достигается при
касании обеих кривых. Для определения концентрации исходного
вещества в критическом режиме установим связь между Концент-,
рацией и температурой в реакторе.
В аппарате полного смешения при стационарном режиме ско-
рость реакции равна скофости намыва:
Ww = <а (са — с)
Подставляя скорость намыва в уравнение теплового баланса
dT п
аппарата и учитывая, что при стационарном режиме —
ззз
имеем:
А^ИСП
Qa(ca — c) = ^Poi^JLe КТ -р/fTS (Г—Гст) + Ср® (Т—Твх) (IV-93)
Уравнение (IV-93) дает связь между концентрацией исходного
вещества и температурой в реакторе.
Наиболее простая зависимость с(Т) получается при отсутст-
вии отвода тепла за счет испарения жидкости. Вводя в уравнение
(IV-93) температуру Тк, установившуюся в реакторе при полном
Рис. IV-31. Скорости расходования и намыва реагента в. аппа-
рате идеального смешения для реакции первого порядка:
а —тепловой эффект реакции и энергия активации малы (критические явле-
ния невозможны);
б —большой тепловой эффект реакции и большая энергия активации (за-
штрихована область, в которой возможны два стационарных режима);
1 — скорость расходования реагента при отводе из аппарата всего тепла
реакции; 2—4~ скорости расходования реагента при частичном отводе тепла
из аппарата; 5— скорость намыва реагента; 6 — критическая скорость
иамыва.
израсходовании исходного вещества1 и учитывая, что coi = 0,
имеем:
c = -^(KtS + Cp<o)(Tk-T) (IV-94)
Значения с, соответствующие критическим режимам, могут быть
вычислены из уравнения (IV-93) или (IV-94) после подстановки
соответствующих величин для Др.
Для экзотермических реакций ненулевого порядка в аппара-
тах полного смешения критические явления более сложны, по-
скольку скорость реакции может в этом случае проходить через
максимум (рис. IV-31). Стационарные режимы, соответствующие
точкам пересечения графиков скоростей реакции (расходования)
1 Конечная температура Гк вычисляется из уравнения (IV-93) при с=0.
334
и намыва, могут лежать как до максимума, так и после него.
В точках А и Bi оба графика касаются друг друга. В режимах,
соответствующих этим точкам, при незначительном изменении
условий может произойти, срыв стационарного режима. Стацио-
нарный режим в точке А (низкая температура, скорость реакции
мала) скачкообразно сменяется стационарным режимом в точке
Л1 (высокая температура, большая скорость реакции) при незна-
чительном увеличении скорости подачи сырья (®'р). Если система
находится в верхнем температурном режиме, то при уменьшении
скорости подачи сырья срыв теплового режима произойдет не в
точке Ai (при ®'р), а в точке Bi при меньшей, чем ю'р, скорости
подачи. При этом стационарный режим в точке В] (высокая тем-
пература, большая скорость реакции) скачкообразно сменяется
стационарным режимом в точке В (низкая температура, низкая
скорость реакции). Между точками Лги А, В и Вх возможны два1
стационарных режима. Реализация того или иного режима зави-
сит от области (низкотемпературная или высокотемпературная),
в которой первоначально находилась система.
Впервые на возможность подобного рода критических явлений
было указано Зельдовичем. Подробно они были рассмотрены
Зельдовичем и Зысиным [23, 24]. Проследим указанные законо-
мерности математически. Вначале рассмотрим реакции первого
порядка, ограничившись случаем, когда тепло отводится только
через стенки сосуда.
Концентрация реагирующего вещества в стационарном режиме
определяется, как и ранее, из равенства скорости намыва и ско-
рости реакции:
Е
WcBe RT =®(с0-с)
В безразмерном виде получим:
/ __Е \
е ЛГ+1 =1 (IV-95)
Со Л и /
В уравнении (IV-95) температура не является независимой пе-
ременной, а определяется концентрацией реагирующего вещества
в аппарате. Зависимость Т(с) дается выражением (IV-94). Пре-
образуя экспоненту в уравнении (IV-95) указанным выше методом,
получим:
Т-Т„
Е ______-------Г-Е
е~^Тхе ^сте^сТ
Учитывая, что
KTS + Cpa . ,г ™ , Ср .
—г----(Т — 4—(^ст ~ ^вх) — со~ с
<оу у
1 Третий стационарный режим, соответствующий точке пересечения гра-
фика скорости намыва с участком графика скорости реакции между точками А
и Bi, неустойчив.
335
имеем:
с
са
. (co-c)Qo-e>Cp(T„-TBK)
WBe RTcT е (KTS+Cpa) + j
(0
(IV-96)
ИЛИ
?(pe~s + 1) = Ь
(IV-97)
Е , ,
WB ~~rt~+c
и =----е "’ст :
w ’
Qwcp — Срш {Тот — 7вх)
_ (oQco-E r —JLfc-
J?T*t(KtS + CPw) ’ b co ’
E
KTS + CpW J«TcT‘
Если обозначить левую часть уравнения (IV-97) через f(£), то
уравнению (IV-97) удовлетворяют точки пересечения f(Z) с пря-
мой, параллельной оси абсцисс, отстоящей от нее на' расстоянии
равном Ь. Критическим условиям соответствуют точки (.4 и В)
экстремумов f(£), в которых система скач-
кообразно переходит из состояния В в Bi
и из А в Аг (рис. IV-32)-. Следовательно,
критические условия можно записать в
виде:
где
С
ьг
ь,
Рис. IV-82. Графическое
решение уравнения
(IV-97):
1 — функция f (£); 2 — значения
параметра Ь, соответствую-
щие критическим режимам.
Г' С" t
Скр Скр ч
^- = 0
ас,
или
це Sk₽ + 1 - £крре ’к₽ = О (IV-98)
Подставляя в уравнение (IV-98) значе-
ние из выражения (IV-97), получаем:
^р-6£кр + 6 = 0 (IV-99)
Действительные корни уравнения (IV-99)
соответствуют критическим условиям, од-
нозначно определяемым параметром b
5кр = -у :
-ь
решение примет более простой вид:
1
т
(IV-100)
С учетом, что g = — b,
£()
Скр
Со 2
Как следует из формулы (IV-100), действительные решения
уравнения (IV-99) возможны при b > 4, т. е. только при удовле-
творении условия
6 =>4
^^ct(V + CP®)
(IV-101)
336
возможны критические режимы. Зависимость критических кон-
центраций от параметра Ь представлена на рис. IV-33. Соответ-
ствующие критическим условиям значения параметра р, могут
быть вычислены из уравнения (IV-98):
е?кР
Когда Ь = 4, параметр ц принимает минимальное значение,
равное е2, при котором еще возможны критические явления.
Аналогичным образом могут быть вычислены критические ус-
ловия для реакции любого порядка. Так, если в реакторе полного
смешения осуществляется
реакция, скорость которой
описывается выражением
__________Е
w = cnBe RT, то уравнение
(IV-96) примет вид:
или
—Hl-+ £ = & (IV-102)
ь
где pi = р.с£-1. Критическим
условиям, как и ранее, со-
ответствуют экстремумы
функции
Рис. IV-33. Значения концентраций реаген-
та, при которых возможны срывы тепло-
вого режима в аппарате полного смеше-
ния, при различных условиях теплоот-
Т. е.
*р(Е)
d(t>)
£"ре ^КР + 1 = о
(IV-103)
вода:
/ — граница, разделяющая-верхние (над кривой)
и нижние (под кривой) температурные режимы;
2 —критические условия для пористого зерна ката-
лизатора реакции нулевого порядка (точкам пере-
сечения кривых соответствуют минимальные ско-
рости теплоотвода, при которых еще возможны
критические явления; п — порядок реакции).
ь
1пе Е из
(IV-99):
Подставляя в уравнение (IV-103) значение для -Лл-
ьп 1
выражения (IV-102), получаем уравнение, аналогичное
Отсюда
^р-(Ь + л-1)£кр+лЬ = 0
(IV-104)
Ь + n-l 62-2(л+1) 6 + (n-I)2
2 ~ V 4
(IV-105)
Критическим условиям соответствуют действительные корни
уравнения JIV-104). Как следует из формулы (IV-105), £кр при-
нимает действительные значения, если подкоренное выражение
22 В* С. Хайлов, Б. Б. Брандт
337
больше или равно нулю, т. е.
&2 —2(п + 1) ь + (п-1)2>0
Таким образом, критические условия для реакции любого по-
рядка возможны при b >(Уп + 1)2.
Пример IV-2. Определить минимальную поверхность теплооб-
мена, обеспечивающую изотермический режим работы нитратора производи-
тельностью Z кг продукта/ч, если известно, что реакция протекает по первому
порядку. Тепловой эффект реакции Q ккал/кг нитруемого сырья, энергия акти-
вации 5 ккал!моль, коэффициент теплопередачи Лт ккал/(мг • ч •град), тепло-
емкость продуктов С ккал/(кг-град). Процесс необходимо проводить при тем-
пературе 273° К- Максимальное отклонение температуры не должно превышать
13,5 град. (Принять, что теплоотвод за счет испарения отсутствует, а соотно-
шение между нитрующей смесью и нитруемым веществом равно m кг/кг.)
Решение. Реактор для проведения непрерывного процесса
нитрования можно рассматривать как аппарат полного смешения. Поэтому для
расчета воспользуемся формулой (IV-101) и рис. IV-33. Имеем:
wQc0£
RT*T (KtS + Срш)
Выразим производительность нитратора через со, с0, р и т. Если обозначить
индексами «1» и «2» величины, относящиеся соответственно к нитруемому агенту
и нитрующей смеси, то
®1Р1 °>1Р1 . „ 0>1Р1 + Ш2Р2
т —-------; Со=------; Р =--------------
ш2р2 ш ш
Z
Исключая величины с индексом «2» и учитывая, что ~ = ®iPi> получаем:
Z т
--= ШСо = рш---—-
а т + 1
Здесь а — степень превращения нитруемого сырья. Выражение для параметра b
можно представить в виде:
ZQ
RT? I т+\
а—^\К^ +----
Е \ ат
Отсюда
S Q__________т + 1 с
Z '' оу2 аКтт
LV ' СТ
аЬКг—=—
С
В полученной расчетной формуле минимальному значению поверхности теп-
лообмена соответствует знак равенства. При этом достигаются критические зна-
чения степени превращения нитруемого вещества. Связь между акр=1 —
— (скр/со) и параметром Ь определяется с помощью рис. IV-33. В расчетную
формулу входит неизвестная температура стенки (Уст). Полагая, что темпера-
тура реагентов, вводимых в аппарат, совпадает с ТСт, получим из уравнения
(IV-93) выражение для разогрева смеси:
г Т Q® (ср-с)
ст Кт5 + Срш
338
Сопоставляя это выражение с выражением для параметра Ь и учитывая, что
максимальный разогрев смеси (с сохранением изотермического режима) дости-
гается в критических условиях, т. е. с = скр, имеем:
/ с \ RT^
\ с0 /
Связь между величинами 1 — (скр/с0) и b определяется с помощью рис. IV-33.
Так, если 6 = 4, то скр/со = 0,5 и
Т-Тст = 2-^-
Отсюда температура стенки равна:
Если принять, что допустимое регламентом повышение температуры дол-
жно обеспечивать хотя бы двухкратный запас надежности, то изотермический
режим должен сохраняться при повышении температуры на 27 град, т. е. до
300° К. Температура стенок охлаждающих поверхностей, необходимая для под-
держания изотермических условий, при этом должна быть равна:
Т
СТ
5000
4-2
/ 5000^+ 50ОТ зоо = 250оК
Г 16-4 2-2
ГЕТЕРОГЕННЫЕ РЕАКЦИИ
Тепловой режим гетерогенных экзотермических реакций
определяется тремя процессами: переносом реагентов, химической
реакцией и отводом тепла. Тепловой режим гомогенных реакций
в аппаратах полного смешения, как было показано выше, также
определяется подводом реагентов (скоростью намыва), скоростью
реакции и отводом тепла. Очевидно, что в условиях, когда тран-
спорт реагентов в гетерогенной реакции не зависит непосредствен-
но от скорости реакции (например, реакции на твердой поверх-
ности), т. е. когда процесс доставки реагентов в гетерогенной ре-
акции подобен процессу доставки реагентов в аппарат полного
смешения, в обоих указанных случаях должны наблюдаться два
устойчивых температурных режима — высокотемпературный и
низкотемпературный. Переход от одного режима к другому дол-
жен происходить скачкообразно при незначительном изменении
условий осуществления процесса, причем эти условия должны
зависеть от направления перехода — от низкотемпературного ре-
жима к высокотемпературному или наоборот.
Реакция на твердой поверхности
Рассмотрим вначале тепловой режим экзотермической
гетерогенной реакции произвольного порядка, протекающей на
твердой поверхности (например, на поверхности плавленого ка-
тализатора). Подобная задача впервые была решена Франк-Ка-
менецким [22], объяснившим основные закономерности, наблюдае-
мые в таком процессе.
22*
339
Очевидно, что -в стационарном режиме скорости теплоприхода
и теплоотвода должны быть равны друг другу. Скорость тепло-
прихода (Q+=wQ, где Q — тепловой эффект реакции) при низких
температурах, как и скорость реакции (w), зависит от Т по за-
кону Аррениуса. При высоких температурах скорость реакции
переходит в диффузионный режим и Q+ лишь незначительно за-
висит от Т. Скорость теплоотвода является линейной функцией
температуры [Q_=KT(7’—То), где То — температура газа, Кт —
коэффициент теплопередачи, Т — температура поверхности].
Стационарным режимам соответствуют точки пересечения гра-
фиков функций
Рис. IV-34. Стационарные тепловые режимы
поверхности плавленого катализатора:
4 —скорости охлаждения поверхности в различных
условиях; 5 — скорость разогрева поверхности.
Q+ и Q-. На рис. IV-34 сопоставлены графики
скоростей теплоприхода
и теплоотвода при раз-
личных значениях коэф-
фициента теплопередачи
Кт- Если условия тепло-
передачи хорошие, значе-
ние Кт велико и возмож-
но только одно пересече-
ние кривые Q+ и Q_ — в
точке А. При уменьшении
значения Кт (например,
за счет уменьшения ско-
рости газового потока)
кривая теплоотвода будет
поворачиваться по часо-
вой стрелке,вправо, а точ-
ка пересечения, соответ-
ствующая устойчивому
режиму, перемещается
вверх по кривой теплоприхода. Скорость газового потока, при ко-
торой кривые Q+ и Q- касаются в точке А2, является критической,
поскольку дальнейшее даже незначительное уменьшение скоро-
сти газового потока приведет к тому, что устойчивым становится
режим, соответствующий точке В2. Температура поверхности
катализатора при этом возрастает скачкообразно от ТА до Тв.
Дальнейшее уменьшение Кт приведет лишь к монотонному уве-
личению температуры поверхности. Если поверхность катализа-
тора первоначально нагрета до температуры большей, чем Тв,
то при увеличении скорости газового потока, т. е. при увели-
чении Кт, устойчивыми будут режимы, соответствующие точкам
пересечения кривой Q_ с верхней ветвью (В2—В]) кривой Q+.
Срыв верхнего теплового режима произойдет в точке В[. Режим,
соответствующий точке Вь сменится режимом в точке Аь Темпе-
ратура при этом скачкообразно изменится от Тв до ТА. Как сле-
дует из рис. IV-34, условия перехода от иижнего температурного
режима к верхнему не совпадают с условиями перехода от верх-
него температурного режима к нижнему.
340
Рассмотрим данную задачу аналитически и выясним условия,
соответствующие критическим режимам, а также температуру по-
верхности в верхнем и нижнем температурном режиме. В стацио-
нарном состоянии скорость доставки реагентов к поверхности
должна равняться скорости химической реакции, а скорость тепло-
отвода— скорости теплоприхода, т. е.
Е
₽м(с0-с) = Ве RTcn (IV-106)
Е
Кт (Т- То) = QBe~ *т сп (IV-107)
Здесь 0м — коэффициент массопередачи. Исключив из обоих урав-
____________Е_
нений Be R-Tcn, получим связь между температурой и концентра-
цией на поверхности катализатора:
₽м(со-с) = ^-(7’-7’о) (IV-108)
Уравнение (IV-108) позволяет определить температуру поверх-
ности как в нижнем, так и в верхнем температурных режимах.
В нижнем температурном режиме реакция протекает в кинетиче-
ской области, концентрация реагента на поверхности близка к кон-
центрации его в объеме. Из уравнения (IV-108) следует, что при
этом /’«Го, т. е. температура поверхности катализатора мало от-
личается от температуры газа.
В верхнем температурном режиме реакция протекает в диффу-
зионной области, так что с<4Ссо. Для температуры на поверхности
в этом случае имеем:
-QPmC° + Го (IV-109)
Ат
Значения коэффициентов массопередачи и теплопередачи могут
быть вычислены из соответствующих критериальных уравнений:
Nuo = -lT = ^e’ Рго)
*ч=-¥-^-рд)
где б — характерный размер системы; Л — коэффициент теплопро-
водности (индексами «£>» и «?,» отмечены величины, относящиеся
к диффузии и теплопередаче соответственно). Для газов коэффи-
циент диффузии обычно равен коэффициенту температуропровод-
ности и = cjj-, а следовательно, Рго=Ргх и Nud=Nux. Из равенства
критериев Nu следует, что
(IV-ПО)
Ат ^Р
где С — теплоемкость; р — плотность. После подстановки отноше-
ния коэффициентов масса- и теплопередачи в уравнение (IV-109)
341
замечаем, что температура на поверхности в верхнем температур-
ном режиме равна температуре при адиабатическом протекании
реакции (Та):
m CqQ гр
Га = ТТ 0
Для определения условий, при которых происходит срыв теп-
лового режима, подставим
или
значение Т—То из уравнения (IV-108)
в выражение (IV-106), предваритель-
но преобразовав экспоненту. После
указанных преобразований получим:
Е М
„ , КТ Ктд (е“ с}
0м(со-с) = Ве °е
сп
-/!------------1-------------и.
О 5 10
Ь"
Рис. IV-35. Критические усло-
вия «воспламенения» и поту-
хания поверхности плавлено-
го катализатора (п — порядок
реакции; прямая 1 для п = 0;
верхние ветви кривых 3 (для
п = 1) и 4 (для п = 2) соот-
ветствуют «воспламенению»;
2 — «воспламенение» для реак-
где£ = &" —; b
со
Мсо. А
----, tr =
Е ’
п-1 PmQco то
= — Be °
0м
Если для газа коэффициент диффу-
зии равен коэффициенту температуро-
проводности, то
Т -т сп~' —
b" = m 0, = Be * °
v Рм
ции нулевого порядка в зерне
пористого катализатора). Уравнение (IV-111) совпадает по
форме с уравнением (IV-102) в зада-
че о тепловом режиме аппарата полного смещения. Критические
условия удовлетворяют уравнениям:
Н = И
(IV-111)
f b" + n- 1 п/(У7)2 - 2 (п + 1) У7 + (п-1)2
£кр= 2 * V 4
(IV-112)
(IV-U3)
Критическим условиям на нижнем температурном режиме соответ-
ствует знак плюс, на верхнем— знак минус.
Из последнего уравнения следует, как и ранее, что критические
режимы возможны, если удовлетворяется неравенство:
(IV-114)
Если критические значения концентраций, даваемых уравнени-
ем (IV-113), поддаются непосредственному измерению в задаче
342
б тепловом режиме аппарата полного смешения, то задание кри-
тической концентрации на поверхности катализатора не имеет
практической ценности. Критические условия целесообразно пред-
ставить в виде зависимости |лкр(6"), которая получается из урав-
нения (IV-112) после подстановки в нее значений £кр из уравнения
(IV-113):
CD <ь")
е к₽
Ркр-т ~ -
L $кр (Ь")
(IV-115)
i«
Для реакции первого порядка зависимость цкр(6")
принимает
вид:
Икр —
Графики зависимости цкр(6") для реакции нулевого, первого
и второго порядка представлены на рис. IV-35.
Минимальное значение цкр, при котором еще возможны крити-
ческие явления, находится из уравнения (IV-115) при (Ь")2—
—2(п+1)Ь"+(п~ 1)2=0:
_______еп+Уп______
(п + Щ)п-п(п + ГпГ'
(IV-116)
Реакция на пористом зерне
Рассмотрим теперь задачу о тепловом режиме экзотер-
мической реакции в пористом зерне катализатора. Газ диффунди-
рует в глубь зерна, одновременно реагируя. Выделяющееся при
проведении реакции тепло отводится за счет теплопроводности из
глубины зерна к его поверхности.
Если тепло реакции не успевает отводиться, температура в
зерне быстро повышается. Возрастает скорость реакции, а вместе
с ней и скорость теплоприхода. Скорость расходования реагента
за счет реакции оказывается больше скорости доставки его через
поверхность зерна, и концентрация в глубине зерна быстро убы-
вает. При некоторой низкой концентрации и соответственно высо-
кой температуре скорость реакции опять оказывается равной ско-
рости диффузии, возросшей за счет увеличения перепада концен-
трации в зерне. При этом устанавливается стационарный режим,
аналогичный верхнему температурному режиму в аппарате пол-
ного смешения (см. стр. 334). Реакция из внутренней кинетиче-
ской области переходит во внутреннюю диффузионную область,
т. е. при плохом теплоотводе, как и для экзотермической реакции
на поверхности плавленого (непористого) катализатора, стацио-
нарным оказывается высокотемпературный режим в диффузионной
343
области, а при хорошем теплоотводе — низкотемпературный ре-
жим в кинетической области. При переходе от нижнего режима
к верхнему происходит нечто подобное тепловому взрыву. Задача
оказывается, таким образом, подобной задаче о тепловом вос-
пламенении с чисто кондуктивным теплоотводом, решенной Франк-
Каменецким [22], и отличается от последней только тем, что
наряду с переносом тепла происходит одновременный подвод
реагентов.
Решенная выше задача (см. стр. 339—343) позволяет полно-
стью описать процесс, протекающий нй поверхности плавленого
катализатора, так как тепловой и материальный балансы процес-
са можно представить в виде алгебраических уравнений. Если ре-
акция протекает внутри зерна, то процесс транспорта реагентов
и отвод тепла из зерна катализатора описывается системой диф-
ференциальных уравнений1:
__Е_
Х9ф + Qcae *т = О (IV-117)
__Е_
^эф-§- + сЛе RT (IV-118)
где х — расстояние по нормали к поверхности тела; ХЭф и ОЭф —эф-
фективные коэффициенты теплопроводности и диффузии соответ-
ственно.
Уравнения (IV-117) и (IV-118) надо решить для следующих
граничных условий:
на поверхности зерна
х = L; с = с0, Т = То
в центре зерна
Исключив из уравнений (IV-117) и (IV-118) скорость реакции,
получим:
Лэф <РТ <Рс
(IV-119)
Интегрирование уравнения (IV-119) дает связь между темпе-
ратурой и концентрацией в каждой точке зерна катализатора:
T-To = ^-Q(co-c) (IV-120)
Лэф
Введем в уравнение (IV-120) отношения D'^/d' и ЛЭф/Л. Тогда
при равенстве истинного коэффициента диффузии коэффициенту
температуропроводности получим, что во внутренней диффузион-
1 Здесь будет рассмотрена задача, описывающая процесс в неограниченной
пористой пластине.
344
ной области разогрев зерна катализатора пропорционален адиа-
батическому разогреву:
К D'.
Та-Тл = ------(IV-121)
Здесь индексами «ц», «О» и «а» отмечены температуры в центре
и на поверхности зерна и температура, достигаемая при адиаба-
тическом проведении реакции. Из уравнения (IV-121) следует, что
- » ^э<Ь _
температура катализатора меньше адиабатической, если >
или больше адиабатической, если •
Для решения системы уравнений (IV-117) — (IV-118) преобра-
зуем экспоненту в уравнении (IV-118), как в предыдущей задаче.
Подставив затем значение Т—То из уравнения (IV-120), получим:
$ = № (IV-122)
. с .. °эф<? . 6 х .
где ^-b- b = ^c0-, g = T,
Если D'rsx, то
2 п ! Т*~Т° Т°
та-т0 ы'эф вь2с“~' #
О ЧфД п'фб"-1
Граничные условия примут вид:
5 = 1; 5 = 6
Введем, следуя Франк-Каменецкому [22], в граничное условие
в качестве параметра значение безразмерной концентрации в
центре зерна (£ц). Тогда для общего решения уравнения (IV-122)
будем иметь:
5 = Н5. и. Ь5ц)
В центре зерна (g=l) решение принимает вид:
Сп = «1 (щ Ь)
или
ц = и(5ц, Ь)
Если И(£ц) не монотонная функция, то критическим значениям
(см. рис. IV-31) отвечают условия экстремума
345
Первый интеграл уравнения (IV-122) равен:
' |-=/2ЛУ7^7
где f = e^v (£); v (£) = С + 2 п (п - 1)... (п - k + 1) £
Введем новую переменную q>=flfn- Тогда уравнение (IV-123)
примет вид:
(1V-123)
.'n-k
/2ц/ц q> У l-q>F (<p)
(IV-124)
где F (<р) = -^- • — 1. Для граничных условий имеем:
e~bv (6)
g=l; q> =-j--~ = Фо
/ц
Интегрированием уравнения (IV-124) получим для центра зер-
на катализатора:
’ f-.-A------------= „(Sb)
Пц / фУ1-фНф)
'Для реакции нулевого порядка имеем
У 2g = е ^£ц1п
У~£- Фа -1
У1 — <Ро + 1
или, учитывая, что <р0=г_Л (где Д = &—— максимальная безраз-
мерная разность концентраций реагента в зерне катализатора):
____ __д_
У2(хе~6 = 2е 1 2 arth У 1 — е-Л
Срыв теплового режима возможен, если
(IV-125)
= о
2 arth
Отсюда получаем трансцендентное уравнение для определения
критической разности концентрации реагента в зерне:
1 1
—. - = cth —,
/1_е-Лкр У1-е-Дкр
(IV-126)
Уравнение (IV-126) имеет единственное действительное реше-
ние при - . = = 1,2. Отсюда минимальная разность между
V 1 - е к₽
безразмерной концентрацией на поверхности и в центре зерна ка-
тализатора, при которой еще возможны критические режимы,
равна:
Дкр -1.2 (IV-127)
Дкр в
346
Учитывая равенство (IV-120), дающее связь между концен-
трационным и температурным полем в зерне катализатора, заме-
т — т
чаем, что А = “ » °'• Следовательно, минимальный разогрев зер-
на, при котором еще возможны критические явления, равен:
#7*
Гц—То= 1,2——
Критическое значение' параметра це-ь = 0,88 получается из
уравнения (IV-125) после подстановки Дкр= 1,2.
Итак, срыв теплового режима для реакции нулевого порядка
возможен, если
Е
,,С-Ъ _ BL2QE
Для реакций с порядком, отличным от нулевого, задача сво-
дится к численному интегрированию уравнений (IV-117) и
(IV-118).
Сопоставим условия, при которых достигаются критические
режимы для экзотермической реакции нулевого порядка, проте-
кающей в аппарате полного смешения, на поверхности непори-
стого (плавленого) катализатора и в зерне пористого катализа-
тора. Будем сопоставлять зависимости (y-j = f(b) и цкр(&). Для
первых двух случаев указанные зависимости имеют вид:
(—'I =1—(IV-128)
\ С0 /Кр Ь
И
1п цкр = 6—1 , (IV-129)
Для реакций в зерне катализатора зависимость I —) = f(b)
\ Со/Кр
получается из соотношения (IV-127):
=1-^- - (IV-130)
\ СО Др b
Зависимость цкр(&) Для критических условий в зерне катали-
затора получается из уравнений (IV-125) и (IV-126). Исключив
из обоих уравнений гиперболические функции, получим:
'____ ДКР .
V2це~ь = 2е 2 г
/1-Ар
Отсюда
In ркр =6 —0,87 (IV-131)
На рис. IV-33 и IV-35 сопоставлены графики уравнений
(IV-128) и (IV-130), (IV-1.29) и (IV-131). Удовлетворительное
совпадение указанных зависимостей для реакции нулевого
порядка указывает на глубокую аналогию между тремя
347
рассмотренными процессами и позволяет, по-видимому, исполь-
зовать решения уравнений (IV-106) и (IV-107) для описания
критических явлений в зерне катализатора при произвольной
кинётике реакции.
Пример IV-3. Окисление спирта воздухом проводят на медном
непористом катализаторе в адиабатических условиях при начальной темпера-
туре 500° К. Продукты за счет тепла реакции разогреваются до 900° К. Извест-
но, что кинетика изотермической реакции описывается уравнением:
12 000
= 20,9с *rc2_^L_
ах мл • сек.
где с — концентрация спирта. Соотношение между реагентами, подаваемыми в
реактор:
2 моль спирта
т = -гц--------------
. 4,8 моль воздуха
Определить: 1) область давлений, в которой возможно проведение адиабати-
ческой реакции; 2) максимальное значение степени превращения, возможное в
этом процессе. Принять, что коэффициент массопередачн рм=72 м/ч, а коэффи-
циенты переноса равны между собой.
Решение. 1. Для решения воспользуемся уравнением (IV-115)
или рис. IV-35. Имеем:
тт 2Т.
СаВе~~ _ _
а к" 4*кр го о
L ькр 1° ) J
скр b" — I п/(Г)2-66" + 1 . Т-Т RT2
где5кр = -^= — ±]/----------------------4-----;
п ьп 900 — 500
Параметр о равен } Э86 500ад2 qqq' = 9’7- По ₽ис- IV-35 Для реакции вто-
рого порядка находим, что критические режимы возможны при
lgl,3<u <lgl,85
или
(900-1000)12 000
20< со2О,9е 1-986-5002
Отсюда 0,21 < с0 <7,45.
Для того чтобы перейти от
лению воспользуемся мольным
парциальных давлений (р):
9,7 • 72/3600
начальной концентрации спирта к общему дав-
отношением реагентов, равным отношению их
Рвозд
(индекс «еп» — относится к спирту, индекс «возд» — к воздуху). Таким образом,
общее давление (Р) равно
или, учитывая связь между парциальным давлением спирта и его концентра-
цией:
RToco ( 1 \ 0,082-500 „.
Р ‘ —г;—---------hl = ---------3,4с0
А1сп \ tn } 32
345
Следовательно, возможные технологические режимы лежат в пределах 0,9 «С
С Р С 1,2 ат.
2. Максимально возможному значению степени превращения соответствует
минимальное значение £нр:
£кр = ~— Ь" = (1 — амакс) Ъ"
Сй
или
„ Ь"-1 n Г (Ь")2 - Ы>" + 1 9,7-1 1 /9,72 - 6 • 9,7 + 1
= — 2------У 4 ' 2 V 4 “
Отсюда аМакс равно:
Омаке — 1 ду — 0,86
СРАВНИТЕЛЬНАЯ ОЦЕНКА
РЕАКЦИОННОЙ АППАРАТУРЫ
Вопрос о выборе реакционной аппаратуры возникает на
самом раннем этапе разработки основ новой технологии.
Предварительная оценка отдельных групп реакционных аппа-
ратов в общем случае может быть сделана исходя из схемы реак-
ции, вне зависимости от фазового состава и агрегатного состоя-
ния реакционной массы. Учет специфических особенностей пере-
рабатываемых продуктов, свойств реакционной смеси и масштабов
производства дает возможность для выбора типа реактора, в наи-
большей степени удовлетворяющего этим особенностям. Выбран-
ный вариант в дальнейшем становится объектом детального экс-
периментального изучения для уточнения деталей конструкции,
отдельных параметров технологического режима и т. д.
Сказанное справедливо для всех групп реакций, однако осо-
бенно важное значение это имеет при осуществлении сложных
реакций, выход продуктов которых зависит от степени превраще-
. ния исходного вещества.
В зависимости от того, какой из продуктов такого рода реак-
ций является целевым, требования к типу реакционного аппарата
могут быть диаметрально противоположными.
Для освещения основных вопросов, встречающихся при выборе
типа реакционной аппаратуры, ниже рассматриваются две груп-4
пы сложных реакций: параллельные и последовательные.
ПАРАЛЛЕЛЬНЫЕ РЕАКЦИИ
Ряд процессов промышленного органического синтеза
упрощенно может быть описан как система параллельных реак-
ций различного кинетического порядка:
АD (я=1), А —Е (я = 1) (I)
А D (я = 1), 2А — -> Е (я = 2) (II)
А+В D (я = 2), 2Д — * Б (П = 2) (И!)
349
Для реакций типа (I) выход продуктов не зависит от степени
превращения исходного вещества. Выбор типа реакционного ап-
парата в этом случае производится путем сравнительной оценки
производительности основных типов реакционной аппаратуры при
заданном режиме (рис. IV-36).
Производительность реакционного аппарата полного вытесне-
ния во всем интервале значений а выше производительности ре-
Рис. IV-36. Зависимости
ZD(a) и ZE (а) для па-
раллельной реакции
A^>D, А—->Е
(*i = k2):
--------- реактор полного
вытеснения;....—реактор
полного смешения;--- —
реактор периодического дей-
ствия.
акторов других типов. Производительность
реактора полного смешения превосходит
производительность реакционного аппара-
та периодического действия только в ин-
тервале малых значений а. При необхо-
димости проведения процесса при высокой
степени превращения в ряде случаев пред-
почтительнее оказывается аппарат перио-
дического действия, преимущества которо-
го особенно заметны при осуществлении
процессов, протекающих с малой скоростью
(небольшое значение Л).
Выход продуктов реакций типа (II) и
(III) зависит от степени превращения ос-
новного исходного вещества (А), участвую-
щего в параллельных превращениях раз-
ного кинетического порядка.
Ранее (см. стр. 182) было показано, что
выход продукта D монотонно повышается
с уменьшением концентрации исходного
вещества А. При проведении процесса в
реакторе периодического действия поддер-
жание минимальной концентрации исход-
ного вещества в реакционной массе дости-
гается путем подачи его в предварительно
загруженную в аппарат смесь продуктов
реакции либо непрерывной струей с по-
стоянной скоростью, либо порциями через
равные промежутки времени. Ясно, что в
этом случае производительность реакционного аппарата тем мень-
ше, чем выше желаемый выход продукта.
Варианты осуществления реакций типа (II) и (III) в реакци-
онной аппаратуре непрерывного действия более разнообразны,
чем в случае осуществления реакций типа (I).
На рис. IV-37 приведены результаты расчета [12], выполненно-
го применительно к реакциям типа (III) при условии ki = k2, рав-
ном мольном количестве исходных веществ А и В и конечной сте-
пени превращения вещества А ак = 0,95.
Наибольший выход продукта D достигается в том случае, ког-
да компонент А подается в поток компонента В с постепенно
уменьшающейся объемной скоростью; при этом производитель-
350
ность реакционного аппарата относительно невелика (рис. IV-37, а).
Наименьший выход продукта D и наибольшая средняя ско-
рость превращения компонента А характеризуют аппарат пол-
ного вытеснения (рис. IV-37, е).
Реакционные агрегаты промежуточного типа, состоящие из
каскада секций полного вытеснения (рис. IV-37, б, в) или полного
смешения (рис. IV-37,г), позволяют получать выход продукта D,
близкий к максимально достижимому. Следует отметить, что рас-
пределение компонента А по точкам питания каскада секций не
оказывает заметного влияния на выход продукта D.
Рис. IV-37. Варианты осуществления реакции
А+В—->D, 2А—-> Е (Z>i = Z>2, исходные моль-
ные количества веществ А и В равны, ак = 0,95): ц
Выход продукта D Производи- тельность реактора (относитель- ные значения) в
а — идеализированный труб- чатый реактор спопереч-
ным потоком, с^4 = const 0,9 0,1 г
б — оптимальное распределе-
ние А 0,875 0,16
в — ввод А равными долями 0,866 0,16
г — то же д — единичный кубовый реак- 0,856 0,12 д
тор . 0,82 0,06
е — единичный трубчатый
реактор 0,625 1.0
0,1 0,2 0,2 0,2 0,2
0,2 0,2 0,2 0,2 0,2
В аппарате полного смешения (рис. IV-37, д) выход продукта
D достаточно высок вследствие низкой- концентрации компонента
А, однако по этой же причине производительность реактора от-
носительно мала.
ПОСЛЕДОВАТЕЛЬНЫЕ РЕАКЦИИ
Для простоты рассмотрения принимаем, что реакция
описывается схемой:
А —В —D
(кинетический порядок обеих реакций — первый, k2/ki = a^=i).
Такие реакции целесообразно разделить на две группы по
значению величины а:
1) реакции, промежуточный продукт которых обладает боль-
шей реакционной способностью, чем исходное вещество (п>1);
2) реакции, реакционная способность исходного вещества ко-
торых превышает реакционную способность промежуточного про-
дукта (а<1).
К первой из этих групп можно отнести многие процессы техноло-
гии основного органического синтеза, основанные на реакциях
351
замещения (хлорирование, нитрование, сульфирование предельных
И ароматических углеводородов), окисление диал^илпроизводных
бензола, этерификация дикарбоновых кислот и т. п? Вторая, столь
же многочисленная группа процессов связана с реакциями окис-
ления углеводородов.
В случае производств большой мощности, использующих быст-
ротекущие реакции, более высокая производительность непрерыв-
нодействующих реакционных аппаратов по сравнению с аппара-
тами периодического действия приводит, как правило, к выводу
о преимуществе первых из них, и задача обычно сводится к срав-
нительной оценке реакторов полного смешения и полного вытес-
нения.
В тех случаях, когда объектом исследования является медлен-
но текущий процесс, возможность интенсификации которого ог-
раничена, в числе возможных вариантов его аппаратурного оформ-
ления необходимо учитывать и реакционную .аппаратуру перио-
дического действия, отказ от использования которой должен быть
тщательно обоснован. Можно привести немало примеров, когда
вследствие специфических свойств реакционной массы вопрос ре-
шается в пользу реактора периодического действия даже для
сравнительно быстро текущих реакций (несмотря на проигрыш в.
производительности).
Вопрос об оптимальном типе реакционного аппарата необхо-
димо рассматривать отдельно для продукта В и продукта D, каж-
дый из которых в различных случаях может быть целевым. При
этом для простоты полагаем, что другой продукт является отхо-
дом производства, не имеющим потребительской ценности (при-
нятое упрощение не всегда справедливо).
Продукт В
Анализ зависимости Ф(а) позволяет найти значение а,
соответствующее минимальному значению Ф — суммарных энерго-
материальных затрат на единицу готового продукта. Учитывая,
что вид зависимости Ф(а) для реакторов полного смешения от-
личается от вида той же зависимости для реакторов периодиче-
ского действия и реакторов полного вытеснения, выбор опти-
мального значения а необходимо основывать на сопоставлении
функции Ф(а) для аппаратов всех указанных типов.
Оптимальное значение а зависит от величины а и условий раз-
деления продуктов реакции и может лежать в весьма широких
пределах.
Для группы процессов, в которых а^>1, оптимальное значение
а лежит в области малых величин, в то время как для второй
группы (а<$С1) величина а, соответствующая оптимальным усло-
виям, может достигать значений, близких к единице (рис. IV-38,
“опт и “опт соответственно),
352
Независимо от значения а, сравнительная оценка реакторов
полного смешения и реакторов полного вытеснения (или аппара-
тов периодического действия), основанная на сопоставлении вы-
хода продукта В при одной и той
же степени превращения, приводит
к однозначному выводу в пользу
последней группы аппаратов.
В некоторых случаях представ-
ляется целесообразным использова-
ние реакторов различных групп, по-
следовательно располагаемых по
мере возрастания степени превра-
щения исходного вещества. Есте-
ственно, что в подобных случаях ре-
актор полного смешения следует ис-
пользовать в начальной стадии про-
цесса, так как при малом значении
степени превращения выход про-
дукта В в аппарате полного смеше-
ния мало отличается от выхода, до-
стигаемого в реакторе полного вы-
теснения.
Производительность реакционных
Рис. IV-38. Зависимость 0g (а) для
реакторов полного вытеснения
(------) при а = 0,1 и а =10.
аппаратов всех типов опре-
деляется степенью превращения исходного вещества и в значи-
тельной степени зависит от величины а. Зависимость ZB(a) для
Рис. IV-39. Зависимость ZB (а) для реакторов пол-
ного вытеснения (----------), полного смешения
(......) и периодического действия (— — —).
значений а, равных 0,1 и 10 для аппаратов периодического дей-
ствия и непрерывнодействующих реакторов обоих идеальных ре-
жимов представлена на рис. IV-39. Во всех случаях, независимо
23 В- С. Хайлов, Б. Б. Брандт
353
от степени превращения исходного вещества, более производи-
тельным является реакционный аппарат полного вытеснения.
Преимущество непрерывнодействующего аппарата полного
смешения перед реакционным аппаратом периодического действия
проявляется только в области малых значений а, причем протя-
женность этой области зависит от значения а и степени исполь-
зования реакционного объема ц (см. стр. 298).
Для медленно текущих реакций, при осуществлении которых
в реакторах периодического действия продолжительность вспомо-
гательных операций относительно мала, целесообразность приме-
нения реакторов полного смешения для рассматриваемых значе-
ний а ограничивается величиной а=0,3 (точки 1 иЗна рис. IV-39).
Процесс, основанный на относительно быстро текущей реакции
(значение Л велико), целесообразно проводить в реакционном ап-
парате периодического действия лишь в тех случаях,' когда опти-
мальное значение а превышает 0,5 (точки 2, 4 на рис. IV-39).
Из рис. IV-39 следует также, что область применения реак-
ционного аппарата полного смешения расширяется с уменьшением
значения а и преимущества его проявляются при осуществлении
быстро текущих реакций. Реакторы периодического действия ока-
зываются более производительными в случае медленно текущих
реакций в области высоких значений а.
Ниже рассматриваются промышленные процессы, основанные
на последовательно протекающих превращениях исходного веще-
ства, характеризующиеся различным значением а.
Хлорирование бензола. Многотоннажное производство
хлорбензола основано на жидкофазном каталитическом хлориро-
вании бензола молекулярным хлором.
Процесс жидкофазного хлорирования бензола достаточно сло-
жен, и однозначная трактовка механизма реакции до настоящего
времени вызывает затруднения [25].
Первичным продуктом взаимодействия бензола с хлором в при-
сутствии катализатора (хлорное железо) являемся некое промежу-
точное соединение, превращение которого заканчивается образова-
нием монохлорбензола. Взаимодействие монохлорбензола с хлором
(протекающее, по-видимому, по аналогичному механизму) приводит
к образованию изомерных дихлорпроизводных (орто-, мета-, пара-)
и относительно небольшого количества трихлорзамещенных.
Превращение бензола в условиях жидкофазного хлорирования
может быть представлено следующей упрощенной схемой:
С6Н6 С6Н5С1 С6Н4С12 —3-> С6Н3С13
Установлено [26], что реакции описываются кинетическими
уравнениями реакций первого порядка (по бензолу и монохлор-
бензолу) при соотношении констант скорости k2/kl = a=0,\ 18-е-
-е-0,120 (при 25°С).
Сравнительная оценка зависимости выхода монохлорбензола
от степени превращения бензола для аппаратов полного смеше-
354
ния и полного вытеснения приводит к естественному выводу в
пользу последнего типа.
Условия, близкие к условиям аппарата полного вытеснения,
могут быть созданы при проведении химико-технологического про-
цесса либо в каскаде барботажных аппаратов [27], либо в наса-
дочной эмульгационной колонне при параллельном токе жидкости
и газа [28].
В первом случае число секций каскада устанавливается на
основе ранее рассмотренных закономерностей, причем следует
иметь в виду необходимость строгого распределения хлора по сек-
циям каскада в соответствии с принятым соотношением объемов
секций.
При равновеликих секциях четырехсекционного каскада рас-
пределение хлора по секциям (при а = 0,4) должно соответство-
вать соотношению 1 : 0,8 : 0,6 : 0,5. Следствием отклонения от этого
соотношения является увеличение относительного количества ди-
хлорпроизводных.
При проведении процесса в области температур 35—40° С па-
рогазовая смесь не обеспечивает отвода тепла реакции и соблю-
дение температурного режима связано с необходимостью разме-
щения теплообменных устройств в барботажном слое.
Проведение процесса по второму варианту (насадочная эмуль-
гационная колонна) более целесообразно при температуре, близ-
кой к температуре кипения реакционной смеси. Степень превра-
щения бензола и температура реакционной смеси при этом
связаны прямой зависимостью. Избыточное теплосодержание
(энтальпия) парогазовой смеси и жидкой фазы реакционной
массы, равное в сумме тепловому эффекту процесса, избавляет
от необходимости отвода тепла из зоны реакции.
Повышение температуры процесса хлорирования сопровож-
дается увеличением значения а, т. е. относительным (при задан-
ном значении а) снижением выхода монохлорбензола. Сохранение
выхода хлорбензола при повышении температуры процесса хлори-
рования требует соответствующего уменьшения степени превра-
щения бензола.
Сопоставление выхода хлорбензола и смеси дихлорпроизвод-
ных, достигаемого в однотипных реакторах при различной тем-
пературе, без учета степени превращения бензола лишено смысла.
Условием нормальной работы реакционного аппарата типа
эмульгационной колонны является такая скорость подачи хлора,
при которой эффект продольного перемешивания имеет минималь-
ное значение.
Как следует из результатов испытания производственного ре-
актора (табл. IV-1 и рис. IV-40), при малой скорости подачи хло-
ра продольное перемешивание реагентов столь значительно, что
условия работы насадочной эмульгационной колонны прибли-
жаются к условиям, характеризующим реакционный аппарат
полного смешения.
23*
355
Таблица IV-1
Результаты испытания реактора
типа эмульгационной колонны (сс=О,3| [29]
Скорость подачи хлора, кг(ч на 1 л реакционного объема Содержание бензола в реакционной смеси, масс. % Массовое отношение моно- хлорбензол : дихлорпроиз- водиые ₽В, 2
0,4 65-66 16,2 95,2
0,6 65-66 25,0 97,0
0,9 65-66 26,3 97,4
Окисление циклогексана. Одним из современных спо-
собов производства капролактама и адипиновой кислоты является
процесс, основанный на переработке циклогексана по схеме, пер-
вой стадией которой является жидкофаз-
Рис. IV-40. Зависимость
выхода монохлорбензола
от гидродинамического
режима потока реакцион-
ной массы [29]:
Скорость подачи хлора (в кг{ч
на 1 л реакционного объема):
/ _ о,4; 2 - 0,9.
ное каталитическое окисление циклогексана.
Процесс превращения циклогексана в
условиях жидкофазного окисления обстоя-
тельно изучен многими исследователями и
освещен в литературе [30, 31].
Согласно современным представлениям,
превращение циклогексана в процессе его
жидкофазного окисления может быть вы-
ражено следующей схемой:
Н
/\I__q УП > Дикарбоновые
кислоты
I /1=0 ——* Продукты
осмоления
ОН
В кинетической области течения реакции скорость превраще-
ния циклогексана определяется скоростью взаимодействия его
молекулы с перекисным радикалом, приводящего через ряд про-
межуточных стадий к образованию циклогексанола и циклогекса-
нона. Молекула циклогексанона, взаимодействуя с перекисным
радикалом, приводит через ряд промежуточных превращений к
адипиновой кислоте и другим кислородсодержащим продуктам.
Пренебрегая реакциями циклогексанола и вырожденным раз-
ветвлением цепи (допуская заведомую погрешность), можно рас-
356
сматривать реакции III и V как определяющие скорость превра-
щения циклогексана и циклогексанона и описать процесс окисле-
ния упрощенной модельной схемой [31]:
А + Б — -> В (1)
Б + В D (2)
где А — циклогексан; Б — радикал ROO-; В — смесь циклогек-
санола и циклогексанона; D — продукты глубокого окисления
циклогексана.
Разность значений энергии активации реакций (1) и (2) оце-
нивается величиной порядка 5,0 ккал)моль [30].
Скорость изменения концентрации смеси циклогексанола и
циклогексанона (продукт В) описывается уравнением:
dcB
= ^1СА СБ ~ ^2СВСБ
Для определения выхода продукта В система реакций (1) и
(2) может быть представлена схемой последовательной реакции
первого порядка А В —D, не учитывающей участия ра-
дикала Б в реакции.
Характер зависимости а (Г) указывает на целесообразность
проведения процесса при максимально возможной температуре.
Справедливость этого вывода подтверждается опытными данны-
ми [31]. В условиях жидкофазного процесса оптимальная темпе-
ратура равна 180—185° С.
Сопоставление зависимости рв(а) для режимов полного сме-
шения и полного вытеснения приводит к выводу в пользу реак-
торов полного вытеснения (или реакционных аппаратов периоди-
ческого действия).
При оценке значений степени превращения циклогексана в ре-
акторе полного вытеснения необходим учет условий эксплуатации
реакционного аппарата в составе промышленной технологической
схемы. Для этого можно воспользоваться уравнением (1-16). Пос-
ле подстановки в это уравнение значения рв, 2(а) для условий
полного вытеснения получим:
, X [1 — а — (1 — а)°]
Рв [1 —Г (1 —а)] (а —1)
где Х=₽а, 1₽в,з; У=₽а, 1Ра,з-
Для нахождения значения степени превращения, соответствую-
щей максимальному выходу продукта В в условиях эксплуатации
технологической схемы, найдем производную выхода продукта В
по степени превращения исходного вещества реакции и прирав-
няем ее нулю. Тогда
d$'B Х[1 — К (1 — а)] [а (1 — а)0-1 - 1]-Х[1 - а - (1 - а)а] Y
1а ~ [1-Г (1-а)Д2 (а—I)2 =0
357
В результате преобразований получим:
а (1 - а)а-1 - 1 = У (а - 1) (1 - а)а
Последнее выражение показывает, что искомое значение сте-
пени превращения зависит от условий организации технологиче-
ского процесса и эксплуатации оборудования — потерь циклогек-
сана на^ стадиях 1 и 3 технологической схемы (0а,ь 'Ра,з=5/).
Значение аОпт уменьшается при снижении потерь циклогексана
на стадиях 1 и 3 и заметно увеличивается при увеличении потерь.
В пределах экспериментально установленного значения
=а=5^-6 при допущении потерь циклогексана в размере 1% от
перерабатываемого потока значение Оопт оценивается величиной
порядка 0,06—0,07. Увеличение потерь сырья на стадиях 1 и 3
технологической схемы соответственно повышает значение а0Тп-
При поиске экономического оптимума технологического про-
цесса необходим учет затрат, связанных с разделением оксидата.
Такой учет приводит к необходимости некоторого (сравнительно
небольшого) увеличения а0Тп-
Для установленного выше значения аОТп=0,06 выход смеси
циклогексанола и циклогексанона в реакторе полного вытеснения
составит (см. табл. П-6)
„ 1 г, xai 1-0,06-(1-0,Об)6 по„_
Рв-2 <х(а—1) [1 “ (1 а) 1 0,06(6-1) =0,835
в то время как в условиях полного смешения выход смеси указан-
ных выше продуктов не будет превышать
о _ 1 -а . 1 -0.06 = 0 72-
Рв,2 1-а-аа 1-0,06 + 6 - 0,06 ’
Реализация режима полного вытеснения для реакционного ап-
парата промышленных масштабов может быть обеспечена приме-
нением барботажных аппаратов, секционированных тарелками
либо установленных каскадом. При современных масштабах про-
изводства капролактама, когда мощность реакционного агрегата
составляет 2—3 т/ч (считая на смесь циклогексанола и циклогек-
санона), наиболее оправданным типом реакционного аппарата яв-
ляется каскад барботажных реакторов.
Расчет необходимого числа секций каскада может быть вы-
полнен на основе ранее (см. стр. 183) рассмотренных зависимо-
стей. Поскольку реакция окисления имеет автокаталитический ха-
рактер, для сохранения необходимого отношения Aotj: Лаг : Лаз • • •
по секциям каскада размер их должен убывать по ходу реакцион-
ного потока. Экономически оправданное число аппаратов равно 4—6.
Выход смеси циклогексанола и циклогексанона в каскаде из
трех и шести секций составит соответственно 0,78 и 0,80.
При установке барботажных реакторов с ограниченным про-
дольным перемешиванием условия каскада из шести секций пол-
358
ного смешения могут быть обеспечены в 3—4 последовательно
соединенных аппаратах.
Решение подобной задачи для конкретных условий требует
применения расчетной техники.
Продукт D
При сравнительной оценке различных типов реакцион-
ной аппаратуры, предназначенной для выпуска в качестве товар-
ного конечного продукта последовательной реакции, необходимо
учитывать конкретные условия производства, различая два воз-
можных случая:
Рис. IV-41. Зависимость выхода конеч-
ного продукта последовательной реакции
первого порядка А —•> В D от
степени превращения исходного веще-
ства (kijki = а).
Значениям
Без возврата
продукта В
С возвратом
продукта В
Кривые
теЧ£к
и о 3 я
а; О 2
0,1
10
о,1
10
0,1
10
0,1
10
а
Рд 1 3
I 4
{ 6
О I- я
2
и О 3 я
1) отделение промежуточного продукта от конечного крайне
затруднительно и сопровождается значительными потерями обоих
продуктов;
2) промежуточный продукт сравнительно легко отделяется от
конечного с помощью обычных промышленных методов и без за-
труднений может быть возвращен в реакционный аппарат.
В первом случае целесообразно проводить процесс при макси-
мально возможной степени превращения исходного вещества для
получения реакционной смеси с минимальным содержанием про-
межуточного продукта. Отделение продукта В может быть про-
ведено с помощью современных способов отделения малых коли-
честв примесей. При этом, естественно, продукт В чаще всего
теряется, а стоимость очистки конечного продукта существенно
повышается с увеличением содержания в нем вещества В.
Содержание продукта В в выходящей из реактора смеси будет
зависеть от степени превращения сырья и от типа реакционного
аппарата. Иллюстрацией могут служить кривые 1—4 на
359
рис. IV-41, позволяющие установить преимущество реакционного
аппарата полного смешения, особенно заметное при а<1.
Если содержание примеси вещества В в продукте D регламен-
тировано (что бывает часто), при решении вопроса об оптималь-
ных условиях производства продукта D может возникнуть эко-
номическая задача, так как снижение концентрации вещества В
связано с существенным увеличением размеров реакционной ап-
Рис. IV-42. Зависимость производительности реакционной
аппаратуры по конечному продукту последовательной реак-
ции первого порядка А —В —D = 1,0) от сте-
пени превращения исходного вещества.
Значения k^kx‘. а —0,1; б — 10.
-----реактор полного вытеснения; - - - — реактор полного смешения;
----------------— реактор периодического действия;
1—3 — без возврата продукта В; 4—6 — с возвратом продукта В.
паратуры. При решении подобного рода задач необходим учет за-
висимости размеров реакционной аппаратуры (ее производитель-
ности) от степени превращения исходного вещества.
Реактор полного смешения, обладающий преимуществом при
оценке на основе зависимости 0л(а), заметно уступает по произ-
водительности реакторам двух других типов при проведении про-
цесса в области высоких значений а.
Как следует из рис. IV-42, преимущество реактора полного
смешения проявляется в интервале значений а, существенно за-
висящем от величины a = k2lki. Выбор в пользу того или иного
типа реакционной аппаратуры на основе учета обеих зависимо-
стей— Ро(сс), 2д(а)—может быть сделан лишь для конкретного
случая после тщательной технико-экономической оценки сравни-
ваемых вариантов.
360
Иногда возникает необходимость выпуска в качестве товар-
ного продукта смеси веществ D и В заданного состава. Предпо-
ложим, что желательным является продукт состава 0,5 мол. доли
D и 0,5 мол. доли В. По кривым 1—4 на рис. IV-41 нетрудно сде-
лать вывод в пользу реактора полного вытеснения, обеспечиваю-
щего достижение заданного выхода продукта D (рл=0,5) при
большей, чем в реакторе полного смешения, степени превращения
исходного вещества.
Производительность реакционной аппаратуры, выраженная от-
носительно смеси продуктов D и В, без труда может быть полу-
чена из уравнений (IV-16), (IV-17), (IV-20) и (IV-21):
для реакторов полного вытеснения
ZB+D ==ZB + ZD’= саР\ 1П(1 _а)
для реакторов полного смешения
ZB+D ZB + ZD = CA^i (1 — а)
Для рассматриваемого примера по кривым на рис. IV-41 по-
лучим значения а, указанные в табл. IV-2.
Таблица IV-2
Значения а при Рл=0,5 и различных а
Кривые на рис. IV-41 Значения а Значения a
реактор полного вытеснения реактор полного смещения
1, 2 0,1 0,98 0,9
3, 4 10,0 0,15 0,1
Приведенные выше зависимости ZB+D(a) дают возможность
оценить производительность сравниваемых типов реакционного
аппарата по величине коэффициента производительности
’Ib+o ~
ZB+D (п. с)
7
B+D (п. в)
(l-a) 1
------ In ——
Полученные результаты
а..................................0,1 10
T1B+D .............................°>4 °’975
свидетельствуют о значительном преимуществе реакционного ап-
парата полного вытеснения, особенно в том случае, когда а<1.
Во втором случае, нередко встречающемся в практике про-
мышленного органического синтеза, возврат промежуточного про-
дукта В в реакционный аппарат обеспечивает возможность до-
стижения высокого выхода продукта D в широком диапазоне
36J
значений а. Ясно, что вид зависимости Рд(а) определяется раз-
мером потерь сырья, промежуточного и конечного продуктов на
всех стадиях технологической схемы (см. гл. I).
Как следует из рис. IV-41, вид функции p^(a) при одинаковом
размере потерь мало зависит от типа реакционного аппарата и
значения а, в результате чего сравнительная оценка реакцион-
ной аппаратуры может быть основана лишь на учете ее произво-
дительности.
Для рассматриваемой последовательной реакции зависимость
ZD(a) представлена на рис. IV-42 (кривые 4, 5, 6). Преимуще-
ство реакционного аппарата полного вытеснения вполне очевидно.
Размер реакционного аппарата полного вытеснения рассчи-
тывается исходя из оптимального значения а, устанавливаемого
на основе схемы поиска экономического оптимума, приведенной
в гл. I.
Ниже в качестве примера рассматривается промышленный про-
цесс получения бензойной кислоты из толуола.
Окисление толуола. В связи со значительным расшире-
нием ресурсов толуола, ставшего доступным для промышленно-
сти основного органического синтеза в результате развития неф-
техимической промышленности, большое значение приобретают
процессы, основанные на его переработке.
К числу одной из перспективных схем этого направления от-
носится группа синтезов (е-капролактам, фенол, терефталевая
кислота), первой стадией которых является процесс получения
бензойной кислоты жидкофазным каталитическим окислением
толуола.
Современные сведения о механизме процесса жидкофазного
окисления толуола позволяют описать химические превращения
следующей схемой: >.
О- ОН
I I
Для инженерной оценки способа получения бензойной кислоты
правомерен упрощенный вариант схемы, не учитывающий ряда
362
промежуточных реакций:
£>t
с6н5сн3 —-> со, со2> нсоон
I-----*?-> X С6Н5СООН
где X — промежуточные продукты окисления молекулы толуола
(бензиловый спирт, бензальдегид, гидроперекись), способные к
превращению в бензойную кислоту.
Исследование макрокинетических закономерностей процесса
в условиях барботажа (воздух — толуол) дает возможность опи-
сать скорость расходования толуола уравне-
нием:
_ Атол = . , ,0,5
Й1 \СО2) стол
Полагая, что скорость всех реакций в уп-
рощенном варианте схемы может быть описа-
на уравнениями реакций первого кинетиче-
ского порядка по толуолу, зависимость выхо-
да бензойной кислоты от степени превраще-
ния толуола можно выразить уравнением:
а
Рис. IV-43. Зависи-
мость Ро_ 2 (а) для
процесса окисления
толуола при темпе-
ратуре 180—190° С
(Рй 2 — выход бензой-
ной кислоты).
Кривая построена по урав-
нению (IV-132); точка-
1
______________________1___________________ 1 — (1 — <Х)
0,2 ( । ^1 \ f _ fel _ j \ / fea _ fei _ j\ а
\ fe3 fe3 / \ fe2 fea / \ fe2 fea /
Входящие в это уравнение отношения кажу-
щихся констант скоростей реакций на основа-
нии результатов исследований могут быть
оценены величинами следующего порядка:
k3lk2^ 124-13.
ми отмечены эксперимен-
тальные данные.
Подстановка численных значений отношений констант скоро-
стей реакций в последнее уравнение приводит к зависимости
₽о,2« ‘ [1 — (1 — а)11,36]
(IV-132)
с удовлетворительной степенью приближения описывающей экспе-
риментальные данные (рис. IV-43).
Так же как и в других аналогичных случаях, удовлетворитель-
ное совпадение экспериментальных результатов с расчетной за-
висимостью рв(а) не может служить основанием для выяснения
механизма реакции, и выражение (IV-132) справедливо лишь для
целей интерполяции.
Как было показано ранее, суммарный выход конечного про-
дукта последовательной реакции (Р£) при возврате промежуточ-
ного продукта на стадию химического превращения практически
не зависит от типа реакционного аппарата и, являясь функцией
степени превращения сырья, определяется величиной потерь
сырья и продуктов на первой и третьей основных стадиях техно-
логической схемы.
363
Задаваясь значениями ₽А. ь ₽а, з, рв, з. Ро, з и используя выра-
жение (IV-132), с помощью уравнения (1-18) можно описать за-
висимость рд (а) для рассматриваемого процесса (рис. IV-44).
Учет энергетических затрат и амортизационных отчислений
дает возможность установить значение оптимальной степени пре-
вращения толуола. Поскольку влияние размера реакционной ап-
Рис. IV-44. Зависи-
мость (а) для про-
цесса окисления то-
луола при темпе-
ратуре 180—190° С
(Рд — выход бензой-
ной кислоты):
1 — в соответствии с урав-
нением (IV-132); 2 —в со-
ответствии с уравиеинем
(1-18).
паратуры заметно проявляется лишь при вы-
соких степенях превращения, можно предпо-
ложить (без учета особенностей процесса),
что оптимальное значение а лежит в этой об-
ласти.
Особенности жидкофазного каталитиче-
ского окисления толуола проявляются в виде
почти прямой зависимости предельно дости-
жимой степени превращения углеводорода
от количества катализатора (соли метал-
лов переменной валентности). Поскольку
стоимость катализатора относительно вы-
сока, существует зависимость удельной стои-
мости сырья и вспомогательных материалов
(толуол + катализатор) от степени превра-
щения толуола, имеющая экстремальный ха-
рактер.
Минимум удельной стоимости толуола и
катализатора соответствует значениям степе-
ни превращения в интервале 0,5—0,6.
Сравнительная оценка производительности
реакционной аппаратуры приводит к выводу
в пользу реакционных аппаратов полного вытеснения, а также
реакторов периодического действия.
При проведении процесса в непрерывнодействующих реакто-
рах барботажного типа необходимо использовать каскад аппа-
ратов или, для производств небольшой мощности, колонны с та-
релками. Для определения экономически целесообразного числа
секций каскада или числа тарелок колонны необходим поиск ми-
нимума стоимости реакционного агрегата на основе ранее рас-
смотренных зависимостей.
Экономическая задача, связанная с обоснованием степени пре-
вращения толуола и выбора типа реакционного аппарата при про-
изводстве бензойной кислоты, значительно усложняется, если воз-
никает необходимость выделения бензальдегида и бензилового
спирта в виде товарных фракций.
Сопоставление потребности в бензойной кислоте и промежуточ-
ных продуктах окисления толуола (бензальдегид и бензиловый
спирт), а также сопоставление их цены дает возможность для
обоснования степени превращения толуола и выбора типа реак-
ционного аппарата, соответствующих экономическому оптимуму
производства указанных продуктов.
364
ЛИТЕРАТУРА
I. Я. М. Бра инее, Подобие и моделирование в химической и нефтехими-
ческой технологии, Гостоптехиздат, 1961.
2. Г. К. Боресков, М. Г. С л и н ь к о, Вести. АН СССР, № 10, 29 (1961).
3. М. Г. Слинько, Кинетика и катализ, 3, 481 (1962).
4. М. Г. Слинько, Кинетика и катализ, 1, 153 (I960).
5. М. Г. Слинько, сб. «Моделирование и оптимизация каталитических
процессов», Изд. «Наука» 1965.
6. Г. К. Боресков, М. Г. Слинько, Хим. пром., № 1, 22 (1964).
7. F. Horn, 7. Elektrochem., 65, 209 (1961).
8. К. Г. Д е н б и г, Теория химических реакторов, Изд. «Наука», 1968,
стр. 134.
9. Р. Арис, Оптимальное проектирование химических реакторов, ИЛ, 1963,
стр. 92, 167.
10. К. G. Denbigh, Chem. Eng. Sci., 8, 125 (1958).
11. И. И. Иоффе, Л. М. Письмен, Инженерная химия гетерогенного
катализа. Изд. «Химия», 1965.
12. Х. Крамере, К. Вестертерп, Химические реакторы. Расчет и
управление ими, Изд. «Химия», 1967.
13. Г. К. Боресков, Катализ в производстве серной кислоты, Госхим-
издат, 1954.
14. Э. А. М э л ь в и н - X ь ю з, Физическая химия, кн. 2, ИЛ, 1962, стр. 871.
15. М. Г. Слинько, И. Я. Т ю р я е в, Ю. И. Кузнецов, Хим. пром.,
№ 4, 253 (1962).
16. Г. К. Во р е с к о в, М. Г. С л и н ь к о,' Хим. пром., № 1, 22 (1964).
17. А. П. Зиновьева, Д. И. Орочко, Кинетика и катализ, 3, № 3,
385 (1962).
18. В. Г. Горский, А. И. Бояринов, В. А. Гончаров, Тезисы докла-
дов на III Межвузовской конференции по проблеме «Непрерывные процессы и
аппараты производства полимерных материалов», изд. МИХМ, 1967.
19. А. И. П л а н о в с к и й, Хим. пром., № 5, 5; № 6, 5 (1944).
20. Д. И. Орочко, Теоретические основы ведения синтезов жидких топлив,
Гостоптехиздат, 1951.
21. Я. Б. Зельдович, В. В. Воеводский, Тепловой взрыв и распро-
странение пламени в газах, изд. Моск. мех. ин-та, 1947.
22. Д. А. Фраик-Каменецкий, Диффузия и теплопередача в химиче-
ской кинетике, Изд. «Наука», 1967.
23. Я. Б. 3 е л ь д о в и ч, ЖЭТФ, 11, 493 (1941).
24. Я. Б. Зельдович, Ю. А. 3 ы с и н, ЖЭТФ, 11, 501 (1941).
25. Н. Н. Ворожцов, Основы синтеза промежуточных продуктов и кра-
сителей, Госхимиздат, 1950, стр. 210.
26. F. В о u г i о и, Compt. rend., 990, 1115, 1319 (1920); Ann. chim., 14, 215
(1920).
27. H H. Ворожцов, Г. Б. Зильберман, В. М. Григорьев, ЖПХ,
8, 872 (1935).
28. В. В. К а ф а р о в, Основы массопередачи, Госхимиздат, 1962.
29. Б. Е. Б е р к м а н, Промышленный синтез хлорбензола, Госхимиздат, 1957.
30. И. В. Березин, Е. Т. Денисов, Н. М. Эмануэль, Окисление
циклогексана, Изд. МГУ, 1962.
31. М. С. Фурман и др:, Производство циклогексанона и адипиновой кис-
лоты окислением циклогексана, Изд. «Химия», 1967.
Вильямс Т. Дж., Проектирование химико-технологических процессов мето-
дами системотехники, Изд. «Химия», 1967.
Иоффе И. И., Письмен Л. М., Инженерная химия гетерогенного ката-
лиза, Изд. «Химия», 1965.
Слинько М. Г., Кинетика и катализ, 3, № 3, 460 (1962).
Bil ous О., «Amundsen N., Am. Inst. Chem. Eng. J., 1, № 4, 513 (1955).
Reg enass W., Aris R., Chem. Eng. Sci., 20, Ns 1, 60 (1965).
365
Глава V
ТИПЫ РЕАКЦИОННЫХ АППАРАТОВ
|_Г онструктивные особенности реакционного аппарата
определяются в первую очередь, фазовым составом
и агрегатным состоянием компонентов реакционной смеси. Реак-
торы, предназначенные для проведения гомогенных процессов,
обычно более просты. по устройству, чем аппараты, в которых в
аналогичных условиях осуществляются гетерогенные процессы.
Ниже рассматриваются принципы устройства реакционной ап-
паратуры, применяемой в промышленности основного органиче-
ского синтеза для проведения гетерогенных процессов в систе-
мах газ (жидкость) — жидкость, газ—твердое тело и жидкость—
твердое тело.
РЕАКТОРЫ ДЛЯ ПРОВЕДЕНИЯ ПРОЦЕССОВ
В СИСТЕМЕ ГАЗ —ЖИДКОСТЬ
В зависимости от температуры, давления, соотношения
объемов фаз, теплового эффекта реакции и специфических свойств
реакционной массы применяются различные по конструкции ре-
акционные аппараты: барботажные, пенные, пленочные. В бар-
ботажных и пенных аппаратах газ диспергируется в жидкости
специальным устройством и в виде отдельных пузырей движется
через слой жидкости. Аппараты, в которых линейная скорость
газа w, отнесенная к площади полного их сечения, меньше пре-
дельной скорости всплывания одиночных пузырей (и0), обычно
называют барботажными. К пенным аппаратам относят такие,
в которых w>u0. В аппаратах пленочного типа газ сплошной фа-
зой движется через аппарат, жидкость же, благодаря специаль-
ным устройствам, стекает в виде тонкой пленки по твердым по-
верхностям.
Барботажные аппараты являются наиболее распространен-
ным типом реакционного оборудования рассматриваемой группы
и широко используются в промышленности. Обычно их разделяют
на аппараты: 1) с диспергированием газа через отверстия; 2) с
диспергированием газа в насадке и 3) с механическим дисперги-
рованием газа.
366
ГИДРОДИНАМИКА барботажного слоя
Для выяснения особенностей гидродинамики барботаж-
ного слоя рассмотрим вначале более простой процесс — образо-
вание пузырей из одиночного отверстия.
Образование и движение одиночных пузырей
При малых скоростях газового потока над отверстием
периодически образуются пузыри одинакового размера, всплы-
вающие с одной и той же скоростью на одинаковом расстоянии
друг от друга. Увеличение скорости газового потока приводит к
возрастанию частоты образования пузырей. Их размеры и ско-
рость всплывания остаются прежними, уменьшается лишь рас-
стояние между ними. Дальнейшее возрастание скорости газового
потока приводит к такому режиму образования пузырей, когда
онй, соприкасаясь, движутся цепочкой. При этом над отверстием
образуется газовый факел, близкий по форме к вытянутому эл-
липсоиду вращения, из верхней части которого непрерывно гене-
рируется цепочка пузырей. Так как расстояние между центрами
двух соседних пузырей в цепочке не может быть меньше их диа-
метра, увеличение расхода газа возможно либо за счет увеличе-
ния диаметра пузырей, либо за счет повышения скорости движе-
ния пузырей.
Таким образом, при барботаже возможны два режима обра-
зования пузырей: 1) режим свободного всплывания, когда разме-
ры пузырей не зависят от объемной скорости газа в отверстии
(V), и 2) режим цепочечного движения, когда размеры и скорость
всплывания пузырей зависят от V.
Объем образующихся пузырей в первом режиме может быть
определен из равенства подъемной силы (vApg), действующей на
пузырь, и силы, удерживающей его на краю отверстия nod. Тогда
лМ
о = ——
Apg
Здесь v — объем пузыря; Др — разность плотностей газа и жидко-
сти; g — ускорение силы тяжести; d— диаметр отверстия; а — по-
верхностное натяжение; б — диаметр пузыря.
Диаметр пузырей, образующихся во втором режиме, может
быть вычислен [1] на оснований следующих соображений. Объем
газа V, выходящий из отверстия в единицу времени, должен рав-
няться объему пузырей, образующихся в единицу времени. По-
скольку пузыри движутся цепочкой, соприкасаясь между собой,
частота образования этих пузырей равна отношению скорости их
движения (и) к диаметру б. Такрм образом
у = 2L лз Л.
6 6
367
Отсюда
V л и
На поверхности раздела фаз в рассматриваемой системе
жидкость может двигаться с практически любой скоростью. Дви-
жение жидкости в направлении, нормальном к поверхности раз-
дела фаз, затухает по мере приближения к этой поверхности
вследствие действия сил поверхностного натяжения. Подвижность
жидкости на поверхности пузыря приводит к тому, что сопротив-
ление, испытываемое пузырем при его всплывании, в 1,5 раза
меньше, чем при движении твердых шариков с той же скоростью.
Скорость падения твердых шариков при значениях критерия Рей-
нольдса порядка единицы подчиняется закону Стокса:
2Apgr2 '
Ст 9ц
где г — радиус; р.— вязкость. Скорость свободного всплывания
пузырей в 1,5 раза больше рассчитанной по закону Стокса:
и = 1,5«Ст
Скорость обтекающей пузырь жидкости на его поверхности
равна
3 • д
«пов= - и sin О
где 0 — угол, отсчитываемый от направления движения. Среднее
значение скорости «П0в отличается от скорости движения пузыря
в 3/л раза.
Таким образом, скорость всплывания пузырей возрастает про-
порционально квадрату их диаметра. Однако для пузырей с диа-
метром порядка 2 мм вычисленная скорость оказывается завы-
шенной. Снижение фактической скорости всплывания по сравне-
нию со скоростью, рассчитанной по закону Стокса, объясняется
деформацией пузырей вследствие сопротивления, оказываемого
средой. Пузырь сплющивается в направлении своего движения,
приближаясь по форме к сплющенному эллипсоиду вращения.
При этом пузырь начинает всплывать по зигзагообразной траек-
тории, так что малая ось эллипсоида совпадает с направлением
движения. В результате деформации пузыря миделево сечение1
его возрастает, сила сопротивления оказывается больше, чем для
недеформированного пузыря, а скорость всплывания меньше рас-
четной.
Зависимость скорости всплывания пузыря от его объема мо-
жет быть получена, исходя из следующих соображений. Для про-
стоты последующих выкладок вместо сферического пузыря будем
1 Миделево сечение — максимальное сечение тела в поверхности, нормальной
к направлению движения.
368
рассматривать пузырь, имеющий цилиндрическую форму с пло-
щадью основания s. Тогда для недеформированного пузыря его
высота h равна диаметру основания 6. Вследствие сопротивления
среды перед лобовой и задней частями пузыря возникает пере-
пад давления ДР, равный, по закону Бернулли, ри2/2. Эта раз-
ность давлений сплющивает пузырь (общая его поверхность воз-
растает). Очевидно, что затрачиваемая на деформацию работа
(&Ps'dh) приводит к соответствующему увеличению поверхност-
ной энергии пузыря adS.
Выражая изменение поверхности пузыря dS через его объем и
высоту, получим: .
. п ,, zw 2v \ ,,
kPs dh = и I/ —:--rv I dh
\ V h h2
В форме,
имеем:
разрешенной относительно v, учитывая, что s = vlh,
л h3 о2
(2<у — кР h)2
(V-1)
Высота деформированного пузыря определяется сопротивле-
нием среды и может быть вычислена из условия равенства силы
сопротивления и подъемной силы:
К' = Лр Sv (V-2)
где К' — коэффициент сопротивления.
Исключая h из уравнения (V-1) и (V-2) и учитывая выраже-
ние для ДР, получаем зависимость скорости всплывания пузыря
от его объема:
( %'Р V 2 “s
\2APg/ /2о. К'р2 \2
\ 4Apg 1
или
К'сг2 и6
Ар gp ( So Apg
\ К'р2
(V-3)
Из уравнения (V-З) следует, что при неограниченном возра-
стании объема пузыря, скорость его всплывания стремится к пре-
делу ______
»=i/C-8gApg
V к'р2
совпадающему с точностью до множителя порядка единицы с вы-
ражением, полученным Франк-Каменецким [2].
Для пузырей небольшого объема, скорость всплывания кото-
рых значительно меньше предельной, уравнение (V-З) переходит в
V =
32 Up g У
24 В. С. Хайлов, Б. Б. Брандт
(V-4)
369
Коэффициент сопротивления движению небольших пузырей об-
ратно пропорционален критерию Рейнольдса (ирд/р). Подставляя
в уравнение (V-4) выражение для К' и учитывая, что v : : б3, по-
лучаем для скорости всплывания недеформированных пузырей
и = й Ар^— (V-5)
и
где О — коэффициент пропорциональности.
Экспериментальная зависимость u(v) представлена нарис. V-1,
из которого следует, что при увеличении объема пузырей ско-
рость их всплывания при-
ближается к предельному
значению ы=35 см/сек.
При и==5-10-3 см3 (диа-
метр равновеликой сфе-
ры дэкв~0»12 см) наблю-
дается излом кривой, об-
условленный, очевидно,
изменением характера
обтекания пузыря жидко-
стью. Скорость всплыва-
Рис. V-1. Зависимость скорости всплывания .
пузырей от их величины:
1 — глицерин — воздух [3]; 2 — вода — воздух [3, 4];
3 — вода — воздух (этилен, двуокись углерода) [5]:
4 — вода — воздух (6J; 5 —рассчитано для воды.
ния пузырей, объем кото-
рых превышает 35-10-3
см3 (<5Экв~О,3 см), может
быть рассчитана по урав-
нению (V-3), если К'=
=0,85. Скорость всплывания пузырей, значения бЭКв Для которых
меньше 0,12 см, может быть рассчитана по уравнению (V-5) при
#=0,02.
Диспергирование газа через перфорированные тарелки
В процессе диспергирования газа через перфорирован-
ную тарелку при малых скоростях его подачи происходит обра-
зование пузырей только через несколько отверстий. При доста-
точно больших скоростях подачи газа, соответствующих началу
нормального режима работы аппарата, газ диспергируется одно-
временно через все отверстия решетки. В этом случае автомати-
чески осуществляется такой режим движения пузырей вблизи,та-
релки, при котором каждый последующий пузырь в данном от-
верстии образуется непосредственно после предыдущего. Тогда
для частоты образования пузырей в отверстии имеем:
и
V 6
ИЛИ
— И, = «д2 (V-6)
Л
370
где Уд — объемная скорость газа в отверстии; v — объем одного
пузыря.
Если при образовании пузырей из единичного отверстия не на-
кладывается никаких ограничений на увеличение диаметра пу-
зыря при возрастании Уд, то при диспергировании газа через пер-
форированную тарелку максимальное значение диаметра пузыря
не может превысить расстояния между центрами соседних отвер-
стий (d'). Вследствие этого при достаточно больших скоростях
подачи газа, диспергируемого через перфорированную тарелку,
6=sd'=const и скорость движения пузырей линейно завиЬит от
Уд. Принимая за начало отсчета скорость и0 (соответствующую
минимальному значению УД=У“), при которой Ь-d', а также
учитывая, что линейная скорость газа в свободном сечении ко-
лонны (да) равна -?-Уд (tn — число отверстий; SK — площадь се-
чения колонны), получаем из уравнения (V-6):
6 5/ 6S ) AZ -Л
л (а )2 т \ л (а У т )
„ У наиболее часто применяемых перфорированных тарелок от-
ношение диаметра к расстоянию между соседними отверстиями
велико, так что (d')2m^S. При этом угловой коэффициент в за-
висимости u(w) для барботажного режима обычно порядка 6/я «2.
В случае очень больших скоростей газа при образовании ячеи-
стой пены вместо множителя 6/я в уравнении (V-7) появится мно-
житель 1. При малых скоростях газового потока, когда 6<$Cd',
скорость всплывания пузырей постоянна и не зависит от w, диа-
метр же пузырей возрастает пропорционально ^w.
Анализ экспериментальных данных по определению задержки
газа при барботаже в системе вода—воздух подтверждает суще-
ствование линейной зависимости между скоростью движения пу-
зырей через слой жидкости и скоростью подачи газа в аппарат.
Скорость всплывания пузырей вычисляется, исходя из следую-
щих соображений. Если т — среднее время всплывания пузырь-
ков, то количество газа, поданного в аппарат за это время, равно
задержке газа, т. е. количеству газа, диспергированного в жидко-
сти:
ТУ = nv
где п — число пузырьков в слое; v — средний объем пузыря.
Подставив в последнее равенство выражения для среднего вре-
мени всплывания пузырей 1т = —— 1, скорости подачи газа
(У = и/S) и объема диспергированного газа (nv = S\H), получим:
ДЯ _ w
Но + ДЯ й
Здесь Но — начальная высота слоя жидкости в аппарате; ДЯ—-
изменение высоты слоя жидкости; й — средняя скорость движе-
ния пузырей.
24’
371
Отношение „ ц- = ф обычно называют газонаполнением
пот &Н
или газосодержанием. Авторы монографии [7] указывают, что
определяемое методом у-просвечивания газонаполнение вблизи
барботера мало отличается от газонаполнения в основном слое
жидкости. Поэтому можно полагать, что скорость движения пу-
зырей вблизи барботера не сильно отличается от скорости их дви-
жения в основной части слоя,
Рис. V-2. Зависимость средней ско-
рости всплывания пузырей при дис-
пергировании газа через перфориро-
ванную тарелку от скорости газа
в свободном сечении аппарата.
т. е. и~и.
Зависимость <р(ш) дается эм-
пирическим уравнением [8]:
1
Ф = 1 — е
Здесь
0,2 / Рж \0-2
.__________и0 \ рг ,1______
* / w \0,95 / \0.75
1+о’о875 Ы та
где «о — предельная скорость
всплывания одиночных пузырей;
рж, рг — плотность жидкости и
газа соответственно.
Пересчитанные нами экспери-
ментальные данные [8] для зави-
симости газонаполнения от скоро-
сти подачи газа в колоннах диа-
метром D — 50 4- 300 мм, с шагом
отверстий в перфорированной ре-
шетке соответственно от 6 до
20 мм и высотой слоя //о=50-г-
-=-1000jhjh представлены в координатах и—w на рис. V-2, из ко-
торого следует, что зависимость u(w) линейна уже для w =
=0,1 м!сек. Угловой коэффициент равен 1,6—2,2. Следовательно,
средний диаметр пузырей в барботажном слое можно принимать
равным расстоянию между соседними отверстиями перфорирован-
ной тарелки, которое является единственным характерным разме-
ром системы. Аналогичным образом диаметры пузырей в барбо-
тажном аппарате с насадкой равны характерному размеру гранул.
Влияние размеров аппарата и условий барботажа на величину
коэффициента продольного перемешивания (ОпР) изучалось в ра-
боте [9]. Величина DnP для полых барботажных колонн прямо про-
порциональна их диаметру и практически не зависит ни от вы-
соты газо-жидкостного слоя, ни от диаметра применяемых на
практике (1—3 мм) отверстий барботера. Коэффициент продоль-
ного перемешивания возрастает линейно с увеличением скорости
жидкости w', если аг/>4-е-5 см!сек. Для вычисления Dnp в полых
барботажных колоннах предложены эмпирические зависимости:
372
при линейной скорости жидкости ю'<4-^5 см!сек
, . Г / 42) \2/з1
D' =/ 4,2+1,67и0 —
*р L \ Wo / J
при о/ > 4 ч- 5 см/сек
г /Г 12)' / 12) \2/з1
D' =d' 4,2+12,1-г^—- —
пр L 1 — ф \ «о / J
Коэффициент продольного перемешивания в насадочных бар-
ботажных колоннах имеет максимум при w — б см/сек (w — линей-
ная скорость газа, отнесенная к площади свободного от насадки
сечения аппарата).
Выше было показано, что скорость всплывания одиночных пу-
зырей в жидкости («о) вследствие их деформации стремится к
постоянному значению, равному для воды 20—30 см/сек. Скорость
всплывания пузырей относительно сосуда при диспергировании
их через перфорированную тарелку больше и0 и линейно возра-
стает со скоростью подачи газа. Последнее, очевидно, является
следствием возникающих в барботажном слое конвективных то-
ков жидкости. На наличие конвективных токов жидкости при ин-
тенсивном барботаже указывалось в литературе ранее [10]. Это
явление используется также в барботажных аппаратах с диффу-
зором для создания более регулярного тока жидкости (в диффу-
зоре— вверх, между стенкой аппарата и диффузором — вниз).
Среднее значение скорости U\ восходящего потока жидкости
равно разности между скоростью всплывания пузырей
сивном барботаже и предельной скорости всплывания
пузырей, т. е.
при интен-
одиночных
U] = М"— «о
или, учитывая, что U~2w + u0-,
ut ~ 2w
Если среднее время пребывания жидких реагентов
равно т, то для кратности циркуляции (гц) имеем:
2а>т . .
га « (! -ф)
Но
Используя выражения для <р, можно привести последнее урав-
нение к следующему виду:
2т ш + м0
Гц Но W 2w + «о
Следует отметить, что кратность циркуляции в барботажном
аппарате лишь незначительно зависит от его размеров. Действи-
тельно, если <й и V — объемные скорости жидкости и газа соот-
ветственно, то т=//о5/(в, w=V/S и
V
„ v ~s+“°
Гц ~ 2 <0 . V
2 + Uq
в аппарате
(V-8)
373
Из уравнения (V-8) следует, что кратность циркуляции опре-
деляется в основном отношением объемных скоростей подачи газа
и жидкости. Однако V/o) пропорционально отношению степени пре-
вращения жидкого реагента
AV рж
<х = v---—
ш рг
к степени использования объема газа £=Д7/V, поэтому
ra«2v-^(l-<p)-^ (V-9)
Ь Рг
Здесь v — стихометрический коэффициент; AV — объем газа, всту-
пающего в реакцию.
Как и следовало ожидать, кратность циркуляции тем выше,
чем больше степень превращения жидкости и меньше доля газа,
расходуемого на реакцию. Выражение (V-9) дает возможность
оценивать режим работы барботажных аппаратов. Так, в барбо-
тажном режиме кратность циркуляции — порядка 10 даже при
полном использовании объема газа, поскольку обычно а«>0,01,
Рж/рг~Ю3, а ф~0, Г. Вследствие высокой кратности циркуляции в
барботажном режиме такие аппараты с достаточной степенью
приближения можно считать аппаратами полного смешения. При
интенсивном пенном режиме в аппаратах с насадкой газонаполне-
ние близко к 1, так что рассматриваемые аппараты при небольших
степенях превращения могут приближаться к аппаратам полного
вытеснения.
АППАРАТЫ С ДИСПЕРГИРОВАНИЕМ ГАЗА
ЧЕРЕЗ ОТВЕРСТИЯ
К достоинствам аппаратов с диспергированием газа че-
рез отверстия можно отнести возможность осуществления процес-
са в широком диапазоне соотношений объемов фаз, возможность
отвода или подвода тепла непосредственно от барботажного слоя,
простоту конструкции и достаточно высокую производительность.
Особенность аппаратов такого типа, ограничивающая применение
их в абсорбционной технике, — значительное сопротивление, оп-
ределяемое высотой столба жидкости, не является решающим при
использовании этих аппаратов для проведения химических про-
цессов, особенно в тех случаях, когда химический процесс про-
водится при повышенном давлении.
В зависимости от схемы химического превращения и требова-
ний, предъявляемых к процессу, проводимому в барботажном
слое, тип барботажного реактора по интенсивности продольного
перемешивания жидкой фазы может приближенно соответствовать
реакторам идеальных режимов.
Ниже -кратко рассматриваются принципиальные схемы барбо-
тажных аппаратов.
374
Полное перемешивание
Полное перемешивание жидкой фазы реакционной смеси,
что нередко бывает одним из основных условий проведения про-
цесса, достигается при использовании односекционного аппарата,
устройство которого обеспечивает интенсивное вертикальное пере-
мешивание реакционной смеси.
Продольное перемешивание достигается диспергированием
газа, вводимого в нижнюю часть аппарата, а также механиче-
ским перемешиванием.
Изопропилбензол
Воздух
Рис. V-З. Реакторы типа «эрлифт», используемые при окислении изопропилбен-
зола.
Отработанный,
воздух
На рис. V-З приведены некоторые варианты барботажных ре-
акторов, основанные на использовании эффекта «эрлифта».
Полное вытеснение
Полное вытеснение жидкой фазы с практической полно-
той достигается в эмульгационных апаратах, для которых ф близ-
ко к 1. Необходимая степень приближения, к режиму полного вы-
теснения достигается в каскаде секций. Степень полноты переме-
шивания в отдельной секции при этом несущественна.
Конструктивное оформление каскада секций может быть осу-
ществлено по одному из вариантов в зависимости от выбранной
схемы взаимного перемещения потоков газа и жидкости (рис. V-4).
При прямотоке и противотоке жидкости и газа используются
аппараты типа тарельчатых колонн или каскад секций.
375
На рис. V-5 показан реакционный аппарат для сернокислотной
гидратации олефинов (реакционная колонна для получения этил-
сульфатов) .
Газ, поступающий через барботер в нижнюю часть колонны,
барботирует на каждой тарелке (всего 20 тарелок) через слой
жидкости, подаваемой на верхнюю тарелку, и последовательно
проходит весь каскад тарелок. Высота слоя жидкости на тарелке,
поддерживаемая переливной перегородкой, составляет 800—900 мм.
Для отвода реакционного тепла на каждой тарелке ниже уровня
переливной перегородки помещены охлаждающие элементы в виде
пучка U-образных труб.
Смесь алкилсульфатов выводится из нижней части аппарата,
а отработанный газ (обычно применяется фракция состава эти-
лен— этан, пропилен — пропан) —из верхней.
Если необходимо провести процесс с прямотоком газа и жидко-
сти, можно использовать реакционные аппараты аналогичной кон-
Раз
Жидкость
Рис. V-4. Схема взаимного переме-
щения потоков газа и жидкости
в реакторе.
струкции при условии полного
их заполнения жидкой фазой.
Ряд соображений, основанных
на учете специфических особен-
ностей реакционной системы ино-
гда вынуждает отдавать пред-
почтение каскаду секций, вклю-
ченных последовательно по току
жидкости и газа. Противоток и прямоток при этом осуществляют-
ся простым изменением коммуникации секций.
Число секций устанавливается расчетом. Размер секций в боль-
шинстве случаев принимается одинаковым, хотя для реакций, ки-
нетический порядок которых отличается от нулевого, количества
тепла (особенно при прямотоке жидкости и газа), выделяющиеся
в отдельных секциях, могут значительно отличаться одно от дру-
гого. Более благоприятные в этом смысле условия возможны при
противотоке.
В том случае, когда необходим вывод побочных продуктов ре-
акции из каждой секции, парогазовая смесь, покидающая секцию,
направляется в холодильник-конденсатор, а газовая смесь после
конденсатора поступает в следующую по ходу газа секцию.
На рис. V-6 показаны схемы прямотока (а) и противотока (б)
жидкости и газа в серии последовательно соединенных секций
барботажного аппарата.
Эффективность работы многосекционного барботажного реак-
тора как аппарата для поглощения реакционноспособного компо-
нента газовой смеси, когда парциальное давление газового ком-
понента над раствором исчезающе мало, не зависит от взаимного
направления потоков газа и жидкости.
Выбор того или иного варианта определяется, исходя, напри-
мер, из равномерности тепловыделения по секциям каскада, дав-
ления паров легколетучего компонента жидкой фазы в последней
376
Рис. V-5. Реакционная колонна для получения этилсульфатов:
/ — корпус аппарата; 2 —футеровка; 3 — насадка; 4—люк для осмотра холодильника; 5 — лаз;
б —отверстие для термопары; 7 —спускной штуцер; 8- измерители уровня; 5 —барботер;
10 — переливные перегородки; // — колпачковые тарелки.
по ходу газа секции, условий удаления легколетучих побочных
продуктов и т. п.
Общие рекомендации по этому вопросу затруднительны, и ре-
шение его, как правило, основывается на тщательном учете спе-
цифических особенностей рассматриваемой системы.
Рис. V-6. Последовательно соединенные секции барботажного аппарата:
а —прямоток; б — противоток;
-----жидкость;-----газ.
Ряд недостатков схем, основанных на последовательном про-
хождении потоком газа секций каскада, можно избежать, приме-
няя перекрестный ток жидкости и газа,
Рис. V-7. Перекрестный ток
жидкости и газа в последо-
вательно соединенных сек-
циях барботажного аппа-
рата:
— — жидкость; - — — — газ.
также большая степень
при котором свежая газовая смесь по-
дается в каждую секцию, а жидкость
последовательно проходит все секции
каскада (рис. V-7).
Аппаратура для работы в пенном ре-
жиме несколько отличается от барбо-
тажных аппаратов. Так, вследствие боль-
шой скорости газового потока в пенных
аппаратах (0,3—1 м/сек в расчете на
свободное сечение) газонаполнение мо-
жет достигать величины 0,8—0,85 и бо-
лее. Конструктивно эти аппараты обыч-
но оформляют в виде тарельчатых ко-
лонн (тарелки либо провальные, либо
с переливными трубами). Конструктив-
ные особенности пенных аппаратов, а
газонаполнения, достигаемая в пенном
режиме, приближают эти аппараты к реакторам полного вытес-
нения.
При проведении реакции между газом и жидкостью последняя
обычно является избыточным по отношению к стехиометрическому
составу компонентом. Поэтому скорость реакции определяется, с
одной стороны, концентрацией газа, растворенного в жидкости, а
с другой, — скоростью его ввода в реактор. Поскольку в пенных
аппаратах нагрузка по газу в 8—10 раз больше, чем в барботаж-
ных реакторах, скорость образования продуктов во всем объеме
378
рабочей зоны аппарата также в 8—10 раз больше скорости обра-
зования продукта при работе в барботажном режиме. Производи-
тельность же реакционного аппарата, работающего в пенном ре-
жиме, т. е. количество продукта, образующегося в единице реак-
ционного пространства, мало отличается от производительности
барботажных аппаратов, вследствие значительно большего (в
8—-10 раз) вспенивания жидкости.
Большая сложность конструкции по сравнению с барботаж-
ными аппаратами, а также ограниченный диапазон нагрузок по
газу и жидкости привели к тому, что пенные реакторы не нашли
пока широкого применения.
БАРБОТАЖНЫЕ АППАРАТЫ С НАСАДКОЙ
Для осуществления барботажного процесса в односек-
ционном аппарате при высоком значении критерия Во может быть
использована насадочная колонна с гидродинамическим режимом
работы, близким к точке инверсии.
В ректификационной технике насадочная колонна, процесс в
которой осуществляется в условиях непрерывности потока жидкой
фазы и дисперсного состояния паровой (газовой) фазы, носит на-
звание эмульгационной.
При осуществлении прямотока жидкость подается в нижнюю
часть аппарата, а жидкая часть реакционной смеси отводится из
верхней его части после отделения от парогазовой смеси. В случае
противотока подача жидкости производится в верхнюю часть ко-
лонны, отвод реакционной смеси происходит из нижней ее части
через гидрозатвор. Газ в обоих случаях подается в нижнюю часть
аппарата.
Предельно допустимые скорости газа и жидкости могут быть
оценены на основе закономерностей, установленных для насадоч-
ных ректификационных и абсорбционных колонн в точке инверсии:
, /^иае Рг Рж') ГМ0'25 /Рг\°’125
1g =0,22- 1,751— —)
' ё^нас Рж Рв ! \ G/ \ рж/
где дао— линейная скорость газа в свободном сечении колонны,
м/сек-, рг, рж — плотность газа и жидкости, кг/м3-, р,в, цж— вяз-
кость воды и жидкости, спз; L, G — массовая скорость жидкости
и газа, кг/(л12-ч); пнас — удельная поверхность насадки, ж2/л13;
Гнас — свободное сечение насадки, л2/л<2; g — ускорение силы тя-
жести, мР/сек.
Потеря напора (ДР) на единицу высоты барботажного слоя
(Z) в точке инверсии при соотношении L/G>10, может быть вы-
ражена следующей зависимостью:
Примером использования эмульгационой колонны для осуще-
ствления процесса при прямотоке газа и жидкости может служить
379
процесс жидкофазного хлорирования бензола газообразным хло-
ром. Аппарат для хлорирования (рис. V-8) представляет собой
стальную колонну, футерованную диабазовой плиткой и запол-
ненную смесью стальных и керамических колец. В верхней части
колонны расположена сепара-
ционная камера и штуцер для
вывода жидкой части реакци-
онной массы после отделения
ее от парогазовой смеси. В
нижнюю часть аппарата под-
водятся бензол и хлор.
Наличие насадки и бли-
зость гидродинамического ре-
жима к точке инверсии исклю-
чают необходимость устройства
барботера и создания других
условий для диспергирования
потока газа. Опорная решет-
ка для слоя насадки выполня-
ется из колосников с расстоя-
нием между ними, несколько
меньшим, чем диаметр колец
насадки.
Скорость газа подсчитыва-
ется, исходя из соотношения
L/G, устанавливаемого для
рассматриваемого'случая сте-
пенью превращения бензола
(0,3—0,35) и степенью исполь-
зования хлора, и составляет
0,4—0,45 м/сек-.
При суммарном тепловом
эффекте процесса, равном в
среднем 30,0 ккал/моль, избы-
ток тепла на 1 кг бензола, по-
даваемого в реактор, достига-
ет 75—77 ккал.
Проведение процесса при
температуре, близкой к темпе-
ре кипения бензола, позволяет
осуществить отвод тепла за
счет избыточного теплосодержания (энтальпии) парогазовой сме-
си, покидающей барботажный слой.
Температуру реакционной массы легко подсчитать, зная ее со-
став (бензол 65—68%, хлорбензол 32—33%, смесь дихлорпроиз-
водных бензола 1 — 1,5%) и количество реакционных газов (опре-
деляется по составу реакционной массы, исходя из степени пре-
вращения хлора и атмосферного давления),
380
Объем парогазовой смеси в верхней части барботажного слоя
увеличивается приблизительно в 2,5—3 раза. Скорость парогазо-
вой смеси, превышающая 1 м!сек, соответствует пенному режиму.
Уменьшение скорости газа, даже при наличии насадки, в зна-
чительной мере сказывается на интенсивности продольного пере-
мешивания, о чем можно судить по изменению выхода хлорбен-
зола (степень превращения бензола во всех опытах постоянна и
равна 27—28%):
Скорость газа, м!сек............. 0,2
Массовое соотношение хлорбен-
зол : дихлорбензол................16,2
Выход хлорбензола..................96,0
0,3 0,45
25,0 26,3
97,0 97,5
Рассматривая графическую зависимость Р(а) (рис. IV-40) для
последовательной реакции при &2/&i=0,12 можно видеть, что при
оптимальной скорости га-
за (0,45 м/сек) гидроди-
намический режим рабо-
ты аппарата практически
соответствует условиям
полного вытеснения (точ-
ка 1). Уменьшение ско-
рости газового потока
обусловливает увеличе-
ние интенсивности про-
дольного перемешивания,
приближая режим к ус-
ловиям полного переме-
шивания.
Для осуществления
процессов в системе газ—
жидкость в отдельных
случаях (вязкие, термо-
лабильные жидкости) мо-
жет представить интерес
применение аппаратуры,
принцип действия кото-
рой основан на соприкос-
новении газа с тонкой
пленкой жидкости, стека-
ющей по стенке аппарата
Рис. V-9. Схема роторного реактора:
1 — сепаратор; 2 — рубашки.
или насадки.
Для указанных целей могут найти применение некоторые кон-
струкции рассмотренных ранее тонкопленочных теплообменников
(см. стр. 348), а также абсорбционных и ректификационных ап-
паратов (см. стр. 497) со свободно стекающей пленкой жидкости
и пленкой, турбулизируемой мешалкой.
Так, в работе [11] рассматривается аппарат, предназначенный
для проведения процессов в системе газ — жидкость и жидкость —
381
жидкость (рис. V-9). Отношение высот кбнической и цилиндриче-
ской частей реактора составляет (1,5-т-2):1, угол конусности верх-
ней части равен 15°.
На верхней части вертикального вала жестко закреплены ло-
пасти, образующие со стенками аппарата зазор, равный 2 мм;
лопасти нижней части закреплены на валу шарнирно.
В отличие от цилиндрических аппаратов аналогичной конст-
рукции количество удерживаемой жидкости зависит от скорости
ее подачи, вязкости и числа оборотов мешалки. Расчетное время
пребывания жидкости в аппарате, равное отношению количества
удерживаемой жидкости к объемной скорости подачи, зависит от
высоты конической части аппарата и значительно возрастает с
увеличением числа оборотов перемешивающего устройства (осо-
бенно резко при 1000 об!мин), а при 1200—1500об/мин составляет
в зависимости от вязкости жидкости от 5—8 сек (для 1 спз) до
30 сек (для 13—15 спз).
Коэффициент теплопередачи тоже в значительной степени за-
висит от числа оборотов и при 1000 об/мин достигает 2000—
2400 ккал/ (м2 • ч •град).
АППАРАТЫ С МЕШАЛКАМИ
Для проведения процессов в системе газ—жидкость не-
редко используются аппараты, в которых диспергирование газа в
жидкости достигается за счет энергии движущегося рабочего ор-
гана мешалки.
Применение мешалок для системы газ — жидкость, оправды-
вается в тех случаях, когда относительное количество газа неве-
лико (линейная скорость газа по сечению аппарата значительно
меньше 0,1 м/сек) и использование аппаратов барботажного типа
нецелесообразно вследствие малой интенсивности процесса. Верх-
ний предел применимости мешалок определяется также скоростью
газового потока и при наступлении режима захлебывания (т. е.
режима, при котором газ не диспергируется в жидкости, а обте-
кая мешалку, поднимается вдоль ее вала) механическое переме-
шивание становится излишним. Для маловязких жидкостей эта
скорость составляет ~0,1 м/сек.
По характеру движения рабочего органа мешалки можно под-
разделить на вращающиеся и вибрирующие.
Введение газа под уровень жидкости осуществляется либо с
помощью барботера, помещаемого под мешалку, либо через вал
мешалки, либо в результате всасывающего действия самой ме-
шалки.
Интенсивность работы мешалок в системе газ — жидкость оце-
нивается величиной поверхности фазового контакта (в м2/м3) или
средней величиной коэффициента массопередачи, отнесенным к
единице объема перемешиваемой жидкости [в моль газа/(м3 ч)].
Внешним проявлением эффекта перемешивания является также
382
степень газонаполнения газо-жидкостной смеси и размер пузырей
газа.
Наиболее часто применяемые типы мешалок (турбинная, про-
пеллерная, многолопастная дисковая, лопастная, диспергирующая
и другие) описаны в общих и специальных руководствах по пере-
мешиванию [10, 12—16]. В этих работах освещены вопросы теории
перемешивания, рассмотрены конструктивные варианты оформле-
ния рабочего органа мешалок и даны количественные зависимо-
сти, необходимые для расчета основных размеров мешалок и рас-
хода энергии на перемешивание.
Так как при создании реакционного аппарата для проведения
процесса в системе газ—жидкость основным требованием являет-
ся достижение максимальной степени диспергирования газа в
жидкости, а расход энергии не играет решающей роли, в настоя-
щем разделе кратко рассматриваются основные типы мешалок и
дается сравнительная оценка интенсивности их действия.
Вращающиеся мешалки
Для диспергирования газа в жидкости применяются
различные типы вращающихся мешалок, среди которых, по мне-
нию большинства исследователей, первое место принадлежит тур-
бинной мешалке; диспергирующая, пропеллерная и лопастная ме-
шалки при перемешивании системы газ—жидкость менее эффек-
тивны.
При использовании вращающихся мешалок различных типов
отражательные перегородки оказывают благоприятное действие
на диспергирование газа и установка трех-четырех перегородок
шириной около 0,1 диаметра сосуда почти удваивает эффект пе-
ремешивания.
Интенсивность перемешивания (поверхность фазового контак-
та) при прочих равных условиях (скорость подачи газа, высота
слоя жидкости и т. д.) в значительной степени зависит от пери-
ферийной скорости мешалки, а также от скорости подаваемого
газа и в общем виде выражается уравнением:
где SK— поверхность фазового контакта, м2/м3; пм — число обо-
ротов мешалки, об!сек\ dM — диаметр мешалки, м; Уг — скорость
газа, м1сек\ а, Ь — константы, определяемые конструкцией ме-
шалки.
Значения показателей а и b мало отличаются от единицы [17]:
Турбинная мешалка........... 1,1 0,75—1,1
Дисково-лопастная мешалка .... 0,7—0,9 1,0
На рис. V-10 приведены результаты испытаний турбинной и
дисково-лопастной мешалок в сосудах диаметром £> = 2504-585 мм
383
при различных скоростях газового потока (отношение диаметра
мешалки к диаметру сосуда равно 0,4).
Рис. V-10. Зависимость поверх-
ности фазового контакта от пери-
ферийной скорости мешалки [17].
Скорость газа (в л{ч)\ 7 — 6,1; 2—12,2;
3-27,5;
-----дисково-лопастна я м ешалка;
—_—турбинная мешалка.
Рис. V-11. Зависимость степени газонапол-
нении (/), поверхности фазового контакта (2)
и среднего диаметра пузырей (3) от пери-
ферийной скорости мешалки.
Зависимость степени газонаполнения, поверхности фазового
контакта и диаметра пузырей газа от периферийной скорости тур-
Рис. V-12. Зависимость
поверхности контакта фаз
от отношения диаметра тур-
бинной мешалки к диаметру
аппарата.
бинной мешалки при dM/Z) = 0,4 и скоро-
сти газа 42 м/ч представлена на рис.
V-11. Степень газонаполнения, увеличи-
ваясь почти прямолинейно с возраста-
нием периферийной скорости мешалки,
достигает для каждого случая (в зави-
симости от свойств жидкости) предель-
ного значения, и дальнейшее увеличение
поверхности фазового контакта обусло-
влено уменьшением размера пузырей
газа.
Результаты испытания турбинной ме-
шалки в сосуде диаметром 1,5 м при по-
стоянном значении мощности, потребля-
емой мешалкой, дают возможность сде-
лать вывод, что наиболее экономичной
является мешалка, отношение диаметра
которой к диаметру сосуда не превы-
шает 0,15—0,20 (рис. V-12). В других
работах указанное отношение рекомендуется брать в пределах
0,2—0,4.
Исследование времени пребывания газа в газо-жидкостных
контакторах [18], снабженных турбинной мешалкой, показывает,
что нагрузка аппарата по газу мало влияет на распределение его
384
по времени пребывания и при отношении высоты слоя жидкости
к диаметру аппарата, приблизительно равном единице, с практи-
ческой точностью можно считать перемешивание газа с жидкостью
полным (соответствующим одной ступени аппарата полного сме-
шения), а состав газа на выходе из аппарата равным составу газа
в газо-жидкостной смеси.
Мощность, потребляемая мешалкой в системе газ—'жидкость,
меньше мощности, потребляемой такой же мешалкой при пере-
мешивании одной жидкости.
Самовсасывающие мешалки
Рис. V-13. Самовсасываю-
щая мешалка:
1 — статор; 2 — вал; 3 — труба-диф-
фузор; - 4 — гребная лопасть;
5 — лопатки.
В тех случаях, когда процесс проводится с участием
газа, не содержащего инертных примесей, т. е. когда разбавление
свежего газа отработанным не сказывается на величине движу-
щей силы процесса, целесообразно применять самовсасывающие
мешалки различных конструкций. Прин-
цип действия таких мешалок понятен из
рис. V-13.
Засасывание газа по полому валу ме-
шалок происходит благодаря разреже-
нию, возникающему на периферии вра-
щающейся лопасти. В кольцевую щель,
образованную трубой-диффузором и ста-
тором, также засасывается жидкость.
Сравнительная оценка эффективно-
сти ряда конструкций мешалок для си-
стемы газ — жидкость показывает, что
самовсасывающая мешалка при относи-
тельно невысоком значении коэффици-
ента массопередачи (даже по сравнению
с лопастной и дисковой мешалками) вы-
годно отличается от других типов по-
добных устройств относительно малым
расходом энергии при одной и той же
степени увеличения коэффициента мас-
сопередачи.
Недостатком самовсасывающих ме-
шалок, аналогичных изображенной на
рис. V-13, ограничивающим область и масштабы их применения,
является быстрое снижение объема засасываемого газа с увели-
чением глубины погружения мешалки.
К самовсасывающим мешалкам может быть отнесена мешалка
типа «гребной винт», несмотря на то, что принцип ее действия не-
сколько иной, чем у мешалок, аналогичных изображенной на
рис. V-13.
Гребной винт укрепляется на коротком валу в верхней части
цилиндрического диффузора реактора (рис. V-14). Над винтом
25 В. С. Хайлов, Б. Б. Брандт
385
Рис. V-14. Герметичный реактор с экранированным электродвигателем:
а — разрез по вертикали; б — мешалка с направляющим аппаратом и диффузором;
/ — асинхронный двигатель; 2 — экранирующая гильза; 3 — статор; 4 — мешалка; 5 —направляю-
щий аппарат; б —диффузор; 7 — теплообменная камера; 8 — вертикальные ребра;
диаметр ступицы dCT = 0,4dM; ширина лопасти мешалки b = 0,4dM; толщина лопасти мешалки
О —0,02du; радиус кривизны направляющей лопатки rK-0,3dM; высота направляющих лопаток
Л — бм — диаметр мешалки).
расположен направляющий аппарат, преобразующий кинетиче-
скую энергию вращающегося потока жидкости в статическую
энергию, необходимую для преодоления сопротивления циркуля-
ционного контура. Отражатель, установленный над направляю-
щим аппаратом, является одной из важнейших составных частей
перемешивающего устройства.
Характеристикой работы гребного винта служит аэродинами-
ческое число Рейнольдса Е [19]:
1 ub d2n. У4,85 + т]2р2
Е = -==---- 0,0057 » —----—
70
где vH; — кинематический коэффициент вязкости, м^сек-, и — ско-
рость эквивалентного элемента винта относительно жидкости,
м]сек\ b — средняя ширина лопасти (/> = 0,4 dB), м\ dB — наружный
диаметр винта, м; т]в — гидравлический к. п. д. винта; р — шаговое
отношение винта (7/B/dB); Нв — шаг винта, м.
Оптимальное значение Е лежит в пределах 4000—6000. При
Е<1000 гребной винт работает как лопастная (или пропеллер-
ная) мешалка, при Е^>6000 в перемешиваемой жидкости возни-
кают явления кавитации.
Результаты исследований работы гребного винта [19] с различ-
ным значением шагового отношения показывают, что наиболее
эффективен гребной винт с р = 2=4 (при этом т]в = 0,44-0,6), снаб-
женный направляющим аппаратом из шести ребер, загнутых на-
встречу вращения винта.
Интенсивность перемешивания оценивается значением гидрав-
лического критерия Рейнольдса потока жидкости в кольцевом
пространстве, образуемом корпусом аппарата и диффузором.
Возможности использования гребного винта как перемешиваю-
щего устройства представлены в табл. V-1. •
Таблица V-1
»
Возможности использования мешалок типа «гребного винта»
Re Перемешивание тонких суспензий Перемешивание грубых суспензий и взаимо- нерастворимых жидкостей Перемешивание системы газ — жидкость
2 000-10 000 +
10 000-40 000 + +
40 000 + + +
К положительным особенностям гребного винта относится не-
значительная величина пускового момента, так как винт воспри-
нимает нагрузку лишь при достижении большого числа оборотов,
25* 387
а в момент пуска его к. п. д. и потребляемая мощность весьма не-
значительны (рис. V-15).
Вращение вала мешалок осуществляется с помощью привода
(механического, гидравлического, электрического), расположен-
ного вне аппарата и отделенного от реакционного пространства
сальниковым устройством.
При длительной и непрерывной работе быстроходной мешалки
(характерно для перемешивания системы газ — жидкость) реак-
ционного аппарата, процесс в котором проводится под высоким
избыточным давлением, сальниковое уплотнение может оказаться
Рис. Vt15. Гидравлический к. п. д. винта с различ-
ным шаговым отношением р:
а — зависимость от числа оборотов нм (модельная жидкость —
гексан, V2o = O,6 ест); б — зависимость от шагового отношения
р при пи, равном 1200 и 3000 об/мин»
причиной неполадок. Наряду с неизбежной' утечкой газа, эксплуа-
тация сальникового устройства связана с расходом (иногда зна-
чительным) энергии, а также с быстрым износом вала мешалки и
уплотняющих частей сальника.
Из уравнения, выражающего расход мощности на трение в
сальнике
W = пыг2Р (e°Ms - 1) 10~3 кет
(г — радиус вала мешалки; s — толщина кольца набивки саль-
ника; I—длина набивки сальника; Р—давление внутри аппарата,
ат) следует, что при значительной мощности, потребляемой ме-
шалками промышленных аппаратов, и высоком давлении внутри
аппарата расход энергии на границе в сальнике (а также тепло-
выделение и износ сальника и вала) может быть весьма значи-
тельным и в определенных условиях сальниковое уплотнение
практически неосуществимо.
Из числа возможных способов вращения вала мешалки без
применения сальникового уплотнения (гидравлический привод,
помещенный внутри аппарата, электромагнитный привод, экрани-
388
Рис. V-16. Экранирован-
ный электродвигатель (мощ-
ность 50—65 кет, т] = 0,74,
п = 2910 об/мин, избыточное
давление 300 ат, экранирующая
гильза разгружена противо-
давлением) [19]:
/ — вал; 2 — статор; 3 —ротор;
4 — опорный подшипник.
рованный электродвигатель) наибольший практический интерес
представляет способ, основанный на использовании экранирован-
ного асинхронного двигателя [19, 20].
Экранированный двигатель, гарантирующий полную герметич-
ность сосуда и -обеспечивающий возможность работы при темпе-
ратуре до 500° С при практически неограниченно высоких давле-
нии в аппарате и числе оборотов мешалки, имеет все основания
для широкого внедрения в техноло-
гию основного органического син-
теза.
Пониженные по сравнению с нор-
мальным асинхронным двигателем
значения cos ср и к. п.д. экранирован-
ного двигателя возмещаются отсутст-
вием механических потерь в сальнике
и указанными выше преимуществами
его эксплуатации.
Экранированный двигатель с п =
= 2900 об/мин показан на рис. V-16.
Ротор двигателя насажен на верти-
кальный вал, вращающийся в подшип-
никах. Статор помещен в масляную
ванну, для охлаждения которой мо-
жет быть использовано жидкое сырье,
подаваемое в реакционный аппарат.
Корпус масляной ванны рассчитан
на рабочее давление реакционного
пространства и в случае разрыва
экранирующей гильзы способен
предотвратить выброс реакционной
массы.
Статор отделен от ротора экрани-
рующей гильзой, изготовляемой из
аустенитной стали. Толщина стенок
гильзы рассчитывается на давление,
равное давлению в реакционном про-
странстве.
Для защиты подшипников от кор-
розионного воздействия агрессивных
газов и паров, заполняющих реакционное пространство, часть све-
жего газа вводится в верхнюю часть экранирующей гильзы.
Для смазки подшипников и их охлаждения может служить
(если это допустимо) жидкость, подаваемая в реакционный
аппарат.
Применение экранированного двигателя и высокое число обо-
ротов, связанное с отсутствием сальника, позволяет применять
быстроходные мешалки различных типов для работы под давле-
нием и при глубоком вакууме.
389
Вибрационные мешалки
Для диспергирования газа в жидкости наиболее часто
применяются вращающиеся мешалки. В тех же целях иногда ис-
пользуются вибрационные мешалки, рабочий орган которых со-
вершает периодическое возвратно-поступательное движение.
При использовании для питания вибраторов переменного тока
обычной частоты рабочий орган мешалки может совершать 50 ко-
лебаний в секунду при небольшой амплитуде колебаний.
Энергетически такие мешалки очень экономичны и пригодны
для перемешивания сложных гетерогенных систем (например,
жидкость—жидкость—газ, жидкость—твердое—газ и т. п.). При-
менение их в аппаратах, работающих под давлением, более удоб-
но, чем вращающихся мешалок с сальниковым уплотнением, так
как уплотнение вала, движущегося в вертикальном направлении,
достигается легче.
Время, необходимое для диспергирования газа при использо-
вании вибрационных мешалок, значительно ниже, чем в случае
применения вращающихся мешалок.
В конструктивном отношении размер мешалок ограничивается
длиной вала, которая не превышает 2,5 м [10]. Применяются виб-
рационные мешалки в сосудах емкостью до 3 м3.
ТЕПЛОВОЙ БАЛАНС РЕАКТОРА
Отвод тепла химических и физико-химических процес-
сов, протекающих в жидкой фазе, производится двумя способами,
при раздельном или одновременном их использовании:
1) за счет избыточного теплосодержания (энтальпии) щарога-
зовой смеси, покидающей барботажный слой;
2) путем теплообмена реакционной массы и охлаждающего
агента в теплообменных устройствах различной конструкции.
Первый способ весьма эффективен, когда давление паров
жидкой фазы барботажного слоя при температуре процесса до-
статочно велико, и мало пригоден для жидкостей, обладающих
низким давлением паров.
Рассмотрим общий случай одновременного отвода тепла реак-
ции как за счет испарения части жидких продуктов, так и через
теплообменные поверхности непосредственно из реакционной зоны.
В стационарном режиме работы аппарата тепловой эффект реак-
ции и количество тепла, вносимого исходным сырьем, равны ко-
личеству тепла, отводимого из аппарата. Если Qi и Q2 — теплосо-
держания жидкого и газообразного реагентов соответственно, Q3—
тепло, отводимое или подводимое тепловым агентом, Q4 — тепло,
отводимое из конденсатора, и Q$— теплосодержание продуктов
реакции, то тепловой эффект процесса Q равен:
+- Q *= Qi + Q? + <2з ~ Qi ~ Qs
390
Тепло, отводимое из конденсатора, равно теплу, уносимому па-
рами жидкости и не вступившими в реакцию газами, т. е.
Q4 = Mini ДНИСП + G2C2T (V-10)
где rii — количество молей жидкости, испаряющихся в единицу
времени; — молекулярная масса жидкости; Д77Исп— теплота
испарения жидкости; G2—массовая скорость непрореагировав-
шего газа (в том числе и инертного); С? — теплоемкость газа.
Принимая, что для паров жидко-
сти справедливы законы идеальных
газов, имеем для количества молей
жидкости, испаряющихся в единицу
времени:
где pi — парциальное давление паров;
Р — общее давление в аппарате.
Учитывая уравнения (V-10) и
(V-11), тепловой баланс аппарата
можно представить в следующем виде:
+• Q — Q3 + GjCjTg-l- GgCgTg + G2C2Tq —
Рис. V-17. Тепло, отводимое
(или подводимое) теплоносите-
лем от реакционной массы в
барботажном реакторе при раз-
личных давлениях.
- (G. + G2) C,T- G2-^P(p-Z^) “ G^T
ИЛИ
B + AQ3 = -~—— (V-12)
‘ Pi
±0—0] (С5Т— С]?)]) — g2(c5t— C^Tq)— G2C2(T — Tq) .
ГДе D — “"" «г—Г77 ................. I
г Mi ДЛГцсп
я М9
А — G хц&н—» иг—массовая скорость газа, поглощаемого
жидкостью; Сг — теплоемкость газа, поглощаемого жидкостью.
Если во время проведения реакции меняется давление, то из-
меняется количество паров, уносимых газом в конденсатор, и для
поддержания постоянной температуры в реакторе необходимо
увеличить или уменьшить отвод тепла непосредственно из реак-
ционной зоны через теплообменные поверхности. Первое слагаемое
левой части выражения (V-12) состоит лишь из физических кон-
стант и параметров процесса и при Г=const является, как и пар-
циальное давление паров, постоянной величиной.
График зависимости Qs(P) представляет собой равносторон-
нюю гиперболу, смещенную относительно оси абсцисс на величину
В/А и относительно оси ординат на величину pi (рис. V-17).
Если В>0, что всегда выполняется для сильно экзотермической
реакции при не слишком высоких температурах, Q3 обращается в
391
нуль при некотором значении Р. Аппарат при этом давлении ра-
ботает автотермично. Очевидно, что автотермический режим прин-
ципиально может быть достигнут только при В/А < 0. Чем меньше
по абсолютной величине отношение В[А, тем при большем давле-
нии этот режим достигается.
Полагая в выражении (V-12) фз=0 и решая относительно Р,
получаем искомую зависимость Р(Т) для автотермического ре-
жима:
или
так как
-выше процесс окисления
Рис. V-18. Теплопроизводитель-
ность реактора окисления то-
луола при различных давле-
ниях.
Р=(т+1)Р/
Д]
Р = const (-4-+ 11 е ‘
\ О /
А"исп
Pl = const • е
Примером подобного расчета может служить рассмотренный
толуола при 200° С. На рис. V-18 в
координатах Р—Q3 приведены резуль-
таты расчета для двух вариантов
схемы реакционного агрегата. Значе-
ние Q3 выражено в долях от тепло-
вого эффекта процесса окисления
(Q= 162 ккал/моль), Р — ъ атмосферах,
Автотермичный режим достигается
при давлении ~ 10 ат. При снижении
давления ниже -10 ат резко возраста-
ет дефицит тепла, что сцязано с необ-
ходимостью подогрева исходных ком-
понентов или непрерывного подвода;
тепла к реакционной массе.
Увеличение давления выше 10 ат
уменьшает теплосодержание парогазо-
вой смеси и приводит к необходимо-
сти отвода тепла из жидкой фазы. Ха-
рактер правой части кривой дает возможность обосновать верх-
ний предел повышения давления. Для рассматриваемого случая
увеличение давления выше 18—20 ат вряд ли целесообразно.
Уменьшение количества тепла, передаваемого в конденсаторе-
холодильнике, сопровождается соответствующим уменьшением по-
верхности теплообмена этого аппарата и расхода охлаждаемого
агента.
Несмотря на то, что тепловая схема барботажного реактора в
общем случае позволяет с высокой степенью полноты использовать
тепло реакции, следует иметь в виду, что эффективность работы
теплообменных устройств будет более высокой при отводе тепла
реакции непосредственно из реакционной мдссы. ,В дальнейшем
392
будет показано, что коэффициент теплоотдачи от барботажного
слоя значительно превышает соответствующую характеристику
процесса теплообмена парогазовой смеси с охлаждающей поверх-
ностью.
Рассмотренные выше чисто энергетические соображения, при-
водящие к выводу в пользу режима работы при максимально
высоком давлении парогазовой смеси, следует учитывать при вы-
боре параметров процесса в реакционном аппарате и составлении
тепловой схемы производства. Однако окончательное решение
этого вопроса требует тщательного учета всех последствий, свя-
занных с работой при более высоком давлении.
В ряде случаев некоторые продукты побочных реакций, про-
текающих в барботажном-слое, обладают ингибирующим дейст-
вием по отношению к основному процессу или оказывают неже-
лательное влияние на катализатор. Удобным способом удаления
этих продуктов из реакционной системы является очистка конден-
сата (образующегося в конденсаторе-холодильнике и, как пра-
вило, представляющего собой непревращенное исходное вещество)
перед возвращением его в реакционный аппарат.
Удаление воды достигается путем сепарации водного слоя кон-
денсата, а примесей кислого характера — путем промывки орга-
нической части конденсата щелочными агентами. Для осуществ-
ления такой промывки иногда применяется конденсатор смеше-
ния, орошаемый водным раствором щелочи.
С повышением давления количество примесей, удаляемое с
парогазовой смесью, соответствующим образом снижается и
верхний предел повышения давления в некоторых случаях мо-
жет основываться на балансе скоростей образования и удале-
ния примесей.
ТЕПЛООБМЕН В ГАЗО-ЖИДКОСТНОМ СЛОЕ
Теплообмен между газо-жидкостной системой, образую-
щейся при прохождении газа через слой жидкости, и элементом
теплообменной поверхности относится к наиболее сложным про-
цессам передачи тепла. Изучению закономерностей этого явления
посвящено значительное количество исследований [21—24].
Эффект действия пузыря газа, проходящего через слой жидко-
сти, который граничит с теплообменной поверхностью, сводится к
турбулизации прилегающей к поверхности жидкости, следствием
чего является увеличение коэффициента теплоотдачи от жидкости
к стенке.
Повышение степени турбулентности и уменьшение толщины
пристенного слоя жидкости имеет своим пределом режим, при
котором дальнейшее увеличение объемной скорости газового по-
тока более не изменяет условий теплообмена, а толщина пристен-
ного слоя жидкости достигает своего минимального значения и
перестает зависеть от скорости подачи газа. Этому соответствует
пенный режим.
393
В области значении газоиаполнения <р, характерных для бар-
ботажного режима (ш<0,154-0,2 м/сек), величина коэффициента
Рис. V-19. Коэффициенты тепло-
отдачи при барботаже газа через
жидкость при температуре погра-
ничного слоя:
1 — вода (ЗГ С); 2 — вода (16,6° С); 3 — 44%
водный раствор глицерина (15,2° С);
4 — этиловый спирт (14,5° С); 5 — 68% вод-
ный раствор глицерина (14,2° С).
теплоотдачи как функция свойств
жидкости и скорости газового по-
тока описывается уравнением:
Nu = 0,25 (Ga • Рг)°>33ф°’2
где Nu, Ga, Рг—критерии Нуссель-
ма, Галилея и Прандтля соответст-
венно. При пенном режиме
Nu = const (Ga • Рг)0,33
Для интерполяции эксперимен-
тальных значений коэффициента
теплоотдачи при барботажном ре-
жиме может служить зависимость:
а' = const • ш0,22
Характер зависимости а'=/(ш) представлен на рис, V-19, из
которого следует, что в пенном режиме а' определяется только
физическими свойствами жидкости.
В тех случаях, когда в жидкой фазе барботажного слоя содер-
жатся твердые частицы (образование нерастворимых продуктов
реакции),значение средней вели-
чины коэффициента теплоотдачи
от барботажного слоя теплооб-
менной поверхности повышается
с увеличением содержания твер-
дой фазы в жидкости (рис. V-20).
Естественно, что подобная зако-
номерность соблюдается лишь
при отсутствии адгезии твердых
частиц с поверхностью теплооб
мена.
Высокое значение коэффици-
ента теплоотдачи от барботаж-
ного слоя к теплообменной
поверхности (а') определяет ве-
личину коэффициента теплопере-
дачи только в тех случаях, когда
а' <С а', что возможно, например,
Рис. V-20. Коэффициенты теплоот-
дачи при барботаже газа через
жидкость с различным содержанием
твердых взвешенных частиц (в %):
/ — 40; 2 — 20; 3—10; 4 — 5; 5 — 0 (чистая
вода).
при использовании в качестве охлаждающего агента кипящей
воды [коэффициент теплоотдачи от теплообменной поверхности к
охлаждающему агенту =20004-20 000 ккал/(м2 ч • град)].
Форма теплообменной поверхности барботажных аппаратов
может быть различной в зависимости от величины теплового эф-
394
фекта и специфических свойств реакционной массы. Наиболее ча-
сто применяются рубашки (сплошные и с секционным охлажде-
нием по высоте аппарата), змеевики и трубчатки.
РЕАКТОРЫ ДЛЯ ПРОВЕДЕНИЯ ПРОЦЕССОВ
В СИСТЕМЕ ГАЗ —ТВЕРДОЕ ТЕЛО
Процессы в системе газ — твердое тело широко исполь-
зуются в химической промышленности. Наиболее важные и инте-
ресные из них — гетерогенные каталитические процессы.
Гетерогенный катализ — один из основных методов химиче-
ской технологии. Значительная часть известных в настоящее
время гетерогенных каталитических процессов приходится на долю
промышленности органич.еского синтеза. Осуществляемые в - про-
мышленности газофазные гетерогенные каталитические процессы
характеризуются рядом признаков, совокупность которых опреде-
ляет разнообразие вариантов реакционной аппаратуры.
Известен ряд принципов подхода к классификации процессов
гетерогенного катализа в газовой фазе и аппаратуры, предназна-
ченной для проведения этих процессов [25—27].
По степени подвижности частиц различают неподвижный, дви-
жущийся, а также псевдоожиженный или кипящий слой катали-
затора. Процессы каждой из указанных групп различаются по
тепловому режиму (адиабатический, политропический) и способу
поддержания температуры в заданных пределах.
Заметное влияние на схему реакционного агрегата и конструк-
цию аппаратуры оказывают способ и условия восстановления ак-
тивности катализатора.
В зависимости от гидродинамического режима газового потока
условия течения реакции могут приближаться к условиям прове-
дения процессов в реакторах идеальных режимов или реакторах
промежуточного типа.
КАТАЛИЗАТОРЫ
Несмотря на огромное количество работ в области ге-
терогенного катализа и большие успехи в разработке теории ка-
талитических процессов, проблема получения катализатора с за-
ранее заданными свойствами до настоящего времени не может
считаться решенной и основным путем подбора катализатора для
нового процесса или улучшения свойств известных промышленных
катализаторов является путь максимального использования ана-
логий и эмпирических приемов.
Количество известных в настоящее время катализаторов ог-
ромно. Возможности каждого из катализаторов не исчерпываются
имеющимися сведениями об использовании его в условиях того
или иного производства. При разработке новых каталитических
процессов имеется широкая возможность применения готовых ка-
тализаторов.
395
В качестве катализаторов могут использоваться индивидуаль-
ные металлы или их соединения (окислы, соли). Однако многие
катализаторы имеют сложный состав (смешанные катализаторы)
и представляют собой смесь, твердый раствор или молекулярное
соединение индивидуальных веществ, расширяющих область дей-
ствия катализатора, повышающих его активность (промоторы)
или пассивирующих действие каталитических ядов. Большое рас-
пространение получили также катализаторы, нанесенные на ве-
щества, увеличивающие поверхность катализатора, повышающие
механическую прочность зерна или улучшающие его теплофизиче-
'ские свойства. В некоторых случаях такие вещества (носители)
обладают самостоятельным каталитическим действием, В каче-
стве носителей применяются пемза, силикагель, асбест, керамика,
каолин, активированный уголь и т. п.
По способу приготовления и химической природе катализа-
торы, независимо от их назначения, можно разделить следующим
образом {26]:
1) осажденные катализаторы (солевые, окисные, кислотно-ос-
новные) : монолитные гели, таблетированные и порошкообразные,
формованные;
2) катализаторы на носителях: импрегнированные (солевые,
окисные, кислотно-основные, металлические), зерненые, таблети-
рованные, формованные;
3) природные катализаторы (силикаты и алюмосиликаты);
4) плавленые катализаторы (металлические, окисные), сетки,
проволочные спирали, таблетки;
5) скелетные катализаторы (металлические);
6) органические катализаторы (ионообменные смолы).
Катализаторы многотоннажных производств органического син-
. Теза производятся на специальных фабриках в сформованном
виде и с регламентированными показателями.
Наиболее часто катализатор выпускается в виде цилиндров
размером от 5 до 15 мм (диаметр равен высоте) или сферических
частиц диаметром от 0,3—0,5 до 5—10 мм.
Основными показателями, характеризующими катализатор
и определяющими возможность и условия его применения, яв-
ляются:
1) активность катализатора и продолжительность периода по-
стоянной активности;
2) границы температурного режима работы;
3) физико-механические и теплофизические свойства;
4) каталитические яды, отравляющие данный катализатор
(природа и допустимая концентрация).
Активность катализатора определяется по экспериментально
установленным изотермам и выражается либо количеством исход-
ного вещества или продукта реакции, получаемым в единицу вре-
мени с единицы насыпного объема частиц катализатора, либо так
называемой «объемной скоростью», т. е. объемом газа (приведен-
396
ного к нормальным условиям), подаваемого в единицу времени на
единицу насыпного объема катализатора. Оба эти показателя
аналогичны, и значение их для одного и *гого же процесса опреде-
ляется степенью превращения исходного вещества.
Активность промышленных катализаторов измеряется экспе-
риментально в проточной системе в условиях полного смешения
или полного вытеснения [27, 28] с соблюдением подобия гидроди-
намического режима потока в модели и в промышленном аппарате.
В условиях промышленного производства активность катали-
затора меняется в процессе его эксплуатации, в связи с чем раз-
личают следующие периоды работы катализатора: так называе-
мого «созревания»; постоянной активности; прогрессирующего па-
дения активности; восстановления активности катализатора.
Во многих случаях активность свежеприготовленного катали-
затора весьма низка и достигает своего нормального значения
лишь по истечении некоторого периода пребывания катализатора
в условиях, близких к условиям процесса, или в условиях специ-
альной обработки. В этот период процесс проводится по специ-
ально устанавливаемому для каждого случая режиму с постепен-
но повышающейся нагрузкой.
Продолжительность периода постоянной активности катализа-
тора зависит от специфических особенностей реакционной систе-
мы, соблюдения условий эксплуатации и может достигать не-
скольких лет для одних и не превышать нескольких минут для
других катализаторов. Продолжительность периода постоянной
активности является весьма важной технологической характери-
стикой процесса, во многом определяющей аппаратурное оформ-
ление и схему реакционного агрегата.
Причины дезактивации катализатора (или так называемого
«отравления»), наступающей даже при самом строгом соблюде-
нии условий технологического режима, разнообразны и зависят от
свойств катализатора, а также от состава и свойств реагирующей
смеси.
К числу общих, не специфичных для данного катализатора
причин дезактивации следует отнести блокировку каталитической
поверхности отложениями смолистых и углеподобных продуктов,
часто наблюдаемую в процессах органического синтеза. В резуль-
тате отложений выключаются отдельные участки активной по-
верхности зерна катализатора и забиваются устья пор, что при-
водит к снижению активности, особенно заметному для пористых
катализаторов.
Снижение активности катализатора может быть вызвано и
механической деформацией зерна в результате резких колебаний
температуры, давления газовой смеси, механического давления на
зерно, трения частиц о стенки аппарата, эрозии, спекания под дей-
ствием высокой температуры и т. п.
Другая категория причин дезактивации катализатора связана с
изменением структуры каталитической поверхности под действием
397
примесей, специфичных для данного катализатора (каталитиче-
ских ядов), содержащихся в перерабатываемом сырье и способных
образовывать с катализаторами химические соединения.
Результат совместного действия указанных явлений выражает-
ся в падении активности катализатора, т. е. уменьшении степени
превращения сырья при постоянной скорости его подачи.
Угол наклона кривой, выражающей падение каталитической
активности во времени, может быть значительно уменьшен путем
постепенного подъема температуры процесса. Количественная за-
висимость /(т) устанавливается экспериментально.
Увеличение активности частично дезактивированного катали-
затора путем повышения температуры — явление необратимое:
при снижении температуры до первоначального значения актив-
ность катализатора оказывается ниже исходной.
Период постоянной активности катализатора в промышленных
I условиях заканчивается при достижении нижнего допустимого
предела объемной скорости, а также верхнего предела темпера-
туры процесса, после чего дальнейшая эксплуатация катализатора
становится нецелесообразной. Затем катализатор подвергается
обработке для восстановления активности.
Отравление катализатора принято считать обратимым, если
каталитическая активность восстанавливается в результате обра-
ботки поверхности реагентом, способным к устранению причин
дезактивации. При необратимом отравлении катализатора тре-
буется его замена.
Способы восстановления активности довольно разнообразны и
зависят от природы катализатора и причин его дезактивации.
Углеродистые и смолистые отложения-удаляются в результате
пропускания через слой дезактивированного катализатора потока
горячего воздуха или кислорода (платиновые, алюмосиликатные,
цинк-хромовые и другие катализаторы). Активность хромовых ка-
тализаторов дегидроциклизации восстанавливается при пропуска-
нии через слой катализатора потока водорода при 500° С. Для
медных катализаторов температура восстановления водородом не
превышает 200° С.
Некоторые никелевые и платиновые катализаторы, потерявшие
активность, восстанавливаются окислительно-восстановительным
методом путем последовательной обработки их горячим воздухом
при 150° С, а затем водородом.
Активность сплавных катализаторов, применяемых в процес-
сах гидрирования, восстанавливается при обработке их водным
раствором щелочи (растворяющей слой окиси алюминия) с по-
следующей промывкой водой и сушкой.
Продолжительность и режим периода восстановления актив-
ности устанавливается опытным путем.
s' Схема и аппаратура реакционного узла контактно-каталитиче-
ских процессов должна предусматривать возможность осуществ-
ления всех операций, связанных с восстановлением активности
' 398
катализатора. Период восстановления активности необходимо учи-
тывать при оценке среднечасовой производительности катализа-
тора (Окат):
_ Дкатт1
°кат — Z '_1_ -7“
?! -г ?2
где Ti, 12 — продолжительность периодов постоянной активности и
восстановления активности катализатора соответственно.
Определение продолжительности работы катализатора между
двумя периодами восстановления его активности—продолжитель-
ности соответствуют оптимальным показателям процесса, — со-
ставляет одну из экономических задач химической технологии [29].
Периодическое восстановление активности катализатора, уст-
раняющее причины дезактивации, не останавливает процесса раз-
рушения активной поверхности и механической структуры зерна
катализатора, а лишь отдаляет момент наступления полной (не-
обратимой) потери каталитической активности.
После прекращения работы катализатора реакционный аппа-
рат выключается и производится выгрузка отработанной контакт-
ной массы и замена ее свежим катализатором.
Отработанная контактная масса перерабатывается на катали-
заторной фабрике с целью регенерации наиболее ценных элемен-
тов для производства новых партий катализатора.
Потери катализатора в результате механического разрушения
и уноса пыли (особенно значительные при использовании движу-
щегося и псевдоожиженного слоев катализатора), а также при
его регенерации на катализаторных фабриках относятся к еди-
нице выпускаемого продукта. Стоимость расходуемого таким об-
разом катализатора фигурирует в расходе сырья на единицу го-
тового продукта и учитывается при составлении калькуляции.
Требования к условиям эксплуатации катализатора включают
целый ряд ограничений (температурные границы, качественный и
количественный составы примесей и т. д.), которые необходимо
строго соблюдать.
К числу таких условий относятся допустимые скорости изме-
нения температуры и давления в реакционном пространстве, осо-
бенно в период пуска и остановки оборудования. Резкое измене-
ние температуры или давления может вызвать механическое раз-
рушение зерен катализатора. В целях предотвращения этого
повышение давления производится при соблюдении заданного зна-
чения АР/Ат на всем протяжении периода подъема или спуска
давления; изменение температуры также требует соблюдения за-
данной скорости AZ/Ar.
Следует отметить, что вопрос о необходимой для каждого кон-
кретного процесса активности катализатора требует учета ряда
обстоятельств. Например, использование высокоактивных катали-
заторов позволяет проводить реакцию в мягких условиях (темпе-
ратура, давление), что упрощает аппаратурное оформление и по-
вышает селективность процесса. Вместе с тем, высокоактивные
399
катализаторы, как правило, характеризуются повышенной чувст-
вительностью к присутствию в сырье каталитических ядов и при-
менение их связано с необходимостью' тщательной очистки сырья.
Для решения указанного вопроса необходимо сопоставить эконо-
мические показатели сравниваемых вариантов.
Взаимодействие газообразных реагентов с частицами твердого
катализатора осуществляется в аппаратуре, различающейся в за-
висимости от степени подвижности катализатора, параметров про-
цесса (температура, давление) и способов поддержания теплового
и температурного режимов.
Выбор теплового и температурного режимов гетерогенного ка- .
-талитического процесса и типа реакционного аппарата основан
на тщательном учете всех кинетических и термодинамических
особенностей основной реакции.
При рассмотрении основных типов реакционной аппаратуры,
предназначенной для проведения контактно-каталитических про-
, цессов, целесообразно исходить из классификации реакторов по
степени подвижности катализатора (неподвижный слой, псевдо-
ожиженное состояние, движущийся слой).
АППАРАТЫ С НЕПОДВИЖНЫМ СЛОЕМ КАТАЛИЗАТОРА
Большинство гетерогенных каталитических процессов в
системе газ—твердое тело, используемых в промышленности ос-
новного органического синтеза, осуществляется в неподвижном
слое катализатора. Естественно, применение реактора с таким
слоем оправдано лишь при значительной продолжительности пе-
риода постоянной активности катализатора.
Выбор типа контактного аппарата с неподвижным слоем ка-
тализатора основан в первую очередь на учете температурного
режима процесса. Конструктивное оформление реактора опреде-
ляется величиной и знаком теплового эффекта реакции.
Адиабатический режим
Вопрос о конструкции реакционного, аппарата наиболее
просто решается для экзотермических процессов, режим проведе-
ния которых допускает существование градиента температуры по
высоте слоя катализатора, а величина теплового эффекта реакции
позволяет осуществлять процесс без отвода тепла в пределах мак-
симально допускаемой температуры:
где биакс — максимально допустимая температура, определяемая
свойствами катализатора и компонентов реакционной смеси;
tK — конечная температура реакционной смеси; ^ — начальная
температура реакционной смеси («температура зажигания»); ак—
Конечная степень превращения исходного вещества; Q — тепло-
400
вой эффект реакции; Г—1 среднее значение теплоемкости реак-
ционной смеси, отнесенное к молю исходного вещества.
Расчет размеров реактора, наполненного зернистым катали-
затором, аналогичен расчету реактора полного вытеснения. За-
дача расчета сводится к определению объема слоя катализато-
ра, необходимого для достижения заданной степени превращения
исходного вещества при заданных температурном режиме и да-
влении.
Если на основании опытных данных
водительность катализатора, его объем
известна удельная произ-
(W, в л) подсчитывается
по уравнению:
W — Z/a
где Z — производитель-
ность реактора по це-
левому* продукту, кг!ч-,
а—активность катали-
затора в условиях (тем-
пература, давление,
степень превращения
исходного вещества)
процесса, отнесенная к
единице массы целево-
го продукта, кг/(ч‘Л).
Высота слоя ката-
лизатора вычисляется,
Рис. V-21. Однослойные контактные аппараты.
исходя из скорости га-
зового потока, определяемой экспериментально или устанавли-
ваемой из допустимого гидравлического сопротивления слоя ка-
тализатора. Газовый поток обычно направляется сверху вниз так,
чтобы при этом исключалась возможность разрыхления и исти-
рания катализатора.
Трудность равномерного распределения потока газа по сече-
нию слоя катализатора ограничивает диаметр контактного аппа-
рата величиной порядка 1,5—2,0 м.
Поперечная равномерность газового потока, вводимого в слой
катализатора, достигается путем установки направляющих лопа-
ток, дополнительных сопротивлений и других подобных устройств’
на пути струи газа, входящего в реакционный аппарат через шту-
цер, сечение которого существенно отличается от сечения слоя [30].
Гидравлическое сопротивление слоя катализатора определяет-
ся по известным зависимостям для сухой (неорошаемой) насадки.
Адиабатический режим каталитического процесса реализуется
в однослойных и многослойных реакционных аппаратах.
Однослойные аппараты. Такие аппараты просты по
устройству и представляют собой сосуды, слой катализатора в ко-
торых имеет форму сетки (рис. V-21,a) или состоит из таблети-
рованных частиц (рис. V-21,6).
2Q В. С. Хайлов, Б. Б. Брандт
401
На рис. V-21,e схематически изображен контактный цилиндри-
ческий аппарат относительно небольшого диаметра. В аппарате
достигайтся условия работы катализатора при высокой объемной
и небольшой линейной скоростях газа. Этот тип контактных ап-
паратов, осваиваемый в технологии синтеза аммиака [31, 32], мо-
жет представлять интерес при осуществлении процессов промыш-
ленного органического синтеза.
Реакторы, подобные изображенному на рис. V-21,a, исполь-
зуются для проведения процессов, протекающих во внешней диф-
фузионной области и характеризующихся слабой зависимостью
избирательности от температуры. К этой категории процессов от-
носятся промышленный синтез синильной кислоты из метана, ам-
миака и кислорода, окисление метилового спирта до формальде-
гида, изопропилового спирта до ацетона, бутилового спирта до
масляного альдегида и т. п.
Температура поверхности непористого зерна катализатора
устанавливается из условия равенства количества тепла, выде-
ляющегося (поглощающегося) в результате химической реакции,
количеству тепла, отводимого (подводимого) газовым потоком.
Характерным для реакций, протекающих во внешней диффузион-
ной области, является значительное отличие температуры поверх-
ности зерен катализатора от температуры газового потока и воз-
можность подачи газовой смеси при низкой температуре (на пред-
варительно разогретый катализатор).
В однослойных аппаратах проводятся эндотермические и экзо-
термические процессы, протекающие также и в кинетической об-
ласти (реакции дегидрирования углеводородов, гидратация эти-
лена и др.). Близость температур газового потока и поверхности
зерна катализатора в этом случае вызывает необходимость со-
блюдения соответствующей температуры газового потока на входе
в слой катализатора (так называемой «температуры зажигания»).
При небольшой высоте слоя катализатора повышаются требо-
вания к поперечной равномерности скорости потока газа. Несо-
блюдение этого условия вызывает местные перегревы контакт-
ного слоя.
Для реакций, характеризующихся высоким значением тепло-
вого эффекта и проводимых в аппаратах без теплообмена с внеш-
ней средой, возникает вопрос о способах увеличения среднего зна-
чения теплоемкости.
Добавка инертного газа или избыточного количества одного
из исходных веществ (для которого не требуется достижение
высокой степени превращения) к реакционной смеси, поступаю-
щей в контактный аппарат, сопровождается увеличением объема
газовой смеси, уменьшением парциального давления основного
исходного вещества и соответствующим снижением скорости
реакции.
Относительное количество разбавителя в исходной газовой
смеси для реакций, допускающих малый перепад температуры по
402
высоте слоя катализатора, может быть очень значительным, анё-1
желательные последствия — весьма ощутимыми.
При добавке «разбавителя» к исходной газовой смеси в слу-
чае процесса, текущего в кинетической области (необходимость
соблюдения «температуры зажигания»), его действие как аккуму-
лятора тепла реакции ограничено допускаемым градиентом тем-
пературы в слое катализатора
, QaK С"
С'
где п' — число молей разбавителя, приходящееся на 1 моль реа-
гирующего вещества; С', С" — мольные теплоемкости разбавителя
и реагирующего вещества соот-
ветственно, ккал! (моль•град).
Многослойные аппа-
раты. Введение разбавителя
в поток реагирующего газа
по длине слоя катализатора
позволяет снизить началь-
ную температуру разбавителя
на любую величину и соот-
ветственно уменьшить его
относительное количество
(рис. V-22). Введение свежей
реакционной смеси в поток
газа между слоями катализа-
тора сильно сказывается на
Рис. V-22. Схема установки многослой-
ного контактного аппарата.
увеличении относительного объема катализатора, так как при
этом почти на всем протяжении реакции исходное вещество разба-
влено продуктами. Более целесообразно вводить между слоями
катализатора один из компонентов реакции, конечная степень пре
вращения которого не является определяющей.
При решении вопроса о распределении потока холодного газа
между слоями катализатора и об относительной высоте этих
слоев в общем случае можно рассматривать два основных ва-
рианта:
1) разные по высоте слои катализатора и убывающее по ходу
реакционной смеси количество холодного компонента (h2 = h3 —
=...=hm- WA_2>WA,3>...>WA,m)-,
2) равномерное распределение холодного компонента между
слоями катализатора и возрастающая по ходу реакционной смеси
высота слоев катализатора (h2<h3<.. .<hm\ WA 2« WA 3«s...«
Возможно также большое число промежуточных вариантов,
когда h2^h3^.. s4=hm и W А, 2=kW А, 3=£.. .=f=W At т.
Особого внимания заслуживает первый по ходу газа слой ка-
тализатора, так как высокая скорость реакции на начальном ее
26'
403
этапе вынуждает ограничить высоту слоя очень малой величиной,
что затрудняет возможность создания условий для равномерной
скорости газа по сечению слоя катализатора. Вместе с тем уве-
личение высоты слоя вызывает необходимость резкого увеличе-
ния количества избыточного компонента. Выйти из этого положе-
ния помогут приемы, замедляющие скорость реакции на первом
ее этапе:
Рис. V-23. Варианты распределения потока холод-
ного газа между слоями катализатора в многослой-
ном контактном аппарате.
а) использование малоактивного (отработанного или разбавлен-
ного инертным материалом) катализатора для первого слоя;
б) добавка к реакционной массе продукта реакции (в случае
обратимых реакций).
Оба рассмотренные выше варианта распределения потока хо-
лодного газа между слоями катализатора в многослойном кон-
тактном аппарате представлены графически на рис. V-23.
Основная часть задачи уменьшения количества циркулирую-
щего газа сводится к созданию условий, позволяющих снизить
степень превращения на первой полке аппарата. Например, для
случая гидрирования бензола на сульфидных катализаторах
(/к=340°С; /п=280°С) количество водорода в циркулирующем
газе, приходящееся на каждый моль превращенного бензола на
404
Рис. V-24. Изменение темпе-
ратуры газа на входе в слой
катализатора в многослой-
ном контактном аппарате.
первой полке, в 34() _ 28q »5,5 раза превышает соответствующее
количество водорода для остальных полок.
В случае реакций, проводимых до степени превращения, да-
лекой от состояния равновесия, или практически неравновесных
реакций для сохранения скорости реакции температура газового
потока повышается от слоя к слою. При заданной максимальной
температуре задача решается путем повышения температуры га-
зового потока на входе в очередной слой.
Применительно к двум рассмотренным выше вариантам рас-
пределения потока холодного газа изменение температур на вхо-
де в очередной слой можно видеть на
рис. V-24.
Равновесные экзотермические реак-
ции требуют соблюдения условий опти-
мального распределения температуры по
слоям катализатора (см. стр. 294). В ус-
ловиях многослойного контактного аппа-
рата кривые равновесия (а следователь-
но, и кривые оптимальной температуры)
смещаются вверх по оси ординат по
мере увеличения номера слоя катали-
затора по ходу потока газа; темпера-
тура потока газа на выходе из очеред-
ного слоя должна соответственно сни-
жаться.
Наиболее выгодное для достижения
минимального объема катализатора распределение температуры
и степени превращения исходного вещества между слоями долж-
но отвечать минимуму функции
i=m
- 2
Решение системы уравнений, описывающих условия достиже-
ния минимального значения общего объема катализатора при
заданных начальных и конечных условиях (степень превращения
исходного вещества, начальная концентрация), представляет зна-
чительные трудности. Так, расчет оптимального режима пяти-
слойного контактного аппарата, предназначенного для проведе-
ния равновесной экзотермической реакции при оптимальном тем-
пературном режиме, поддерживаемом подачей холодного газа
между слоями катализатора, требует использования 31 уравне-
ния [34,35]. Для решения столь сложной системы уравнений не-
обходимы вычислительные машины.
Многослойные реакционные аппараты без внутреннего тепло-
обмена отличаются простотой конструкции и сравнительной лег-
костью регулирования температурного режима. К недостаткам
их следует отнести несколько пониженную производительность
495
катализатора, а также необходимость перемещения большего колй-
чества газа и связанный с этим повышенный расход энергии.
Применение многослойных контактных аппаратов целесообраз-
но в тех случаях, когда оптимальная скорость реакции является
функцией не только температуры, но и давления (например, рас-
смотренный ранее случай эндотермических равновесных реакций,
протекающих с увеличением объема). Снижение парциального
реагирующих компонентов по мере повышения степени
давления
Рис. V-25. Многослойный
контактный аппарат с тепло-
обменом между слоями.
превращения достигается за счет ступен-
чатого разбавления основного потока
потоком инертного газа-теплоносителя.
Содержание тепла в газовом потоке,
покидающем однослойные и многослой-
ные контактные аппараты, бывает весь-
ма значительным, поэтому непременной
составной частью реакционного агре-
гата является теплообменник, исполь-
зуемый для нагрева исходной смеси
(см. рис. V-22).
Недостатки, связанные с разбавлени-
ем реагирующего потока инертным га-
зом в многослойных реакторах, устра-
няются при размещении между слоями
катализатора теплообменных устройств
(рис. V-25).
При достаточно высокой температу-
ре процесса использование в качестве
хладагента кипящей воды дает возмож-
ность получения технологического пара. Усложнение конструкции
реактора при размещении в нем теплообменных устройств в не-
которых случаях окупается стоимостью получаемой при этом теп-
ловой энергии. Естественно, что этот вариант целесообразен лишь
для экзотермических процессов, характеризующихся высоким теп-
ловым эффектом.
Оптимальные условия процесса, осуществляемого в последо-
вательности адиабатических реакторов полного вытеснения, при-
менительно к экзотермическим и эндотермическим реакциям рас-
смотрены в ряде работ [26, 33, 36].
Теплообмен в неподвижном слое катализатора
Аппараты, предназначенные для проведения процессов
в неподвижном слое катализатора при теплообмене с внешней сре-
дой, различаются принципом отвода тепла и конструкцией тепло-
обменных устройств.
Для проведения экзотермических процессов наибольшее рас-
пространение получили кожухотрубные теплообменники, внутри
труб которых (реже — между труб) размещен слой катализатора,
406
а межтрубное пространство заполнено охлаждающим агентом
(рис. V-26). В качестве хладагента используется поток реакцион-
ного газа, кипящие жидкости или высокотемпературные тепло-
носители.
Рис. V-26. Контактные аппараты с теплообменом от слоя катализатора.
I — исходная смесь; II — продукты реакции; /// — теплоноситель.
В реакторах, предназначаемых для экзотермических процессов,
тепловой эффект реакции может быть использован для нагрева
исходной смеси (рис. V-26, в—д). Поток исходной смеси и
реакционный поток в реакторах, пред-
ставленных на рис. V-26, в, г, движутся па-
раллельно, в реакторе, изображенном на
рис. V-26, д, — противотоком.
Отвод тепла с помощью кипящих жид-
костей происходит при практически по-
стоянной (если не учитывать влияние гид-
ростатического эффекта) температуре хлад-
агента и обеспечивает поддержание изо-
термического режима лишь для реакций
нулевого порядка. Профиль температур
потока газа в слое катализатора для реак-
ции первого порядка характеризуется мак-
симумом в начальной зоне движения по-
тока (рис. V-27). Реагирующий поток сна-
Рис. V-27. Профиль тем-
ператур в неподвижном
слое катализатора при
постоянной температуре
хладоагента.
а затем, когда ско-
чала разогревается за счет тепла реакции,
рость тепловыделения становится меньше скорости отвода тепла,
температура потока газа падает, приближаясь к температуре теп-
лоносителя. В точке температурного максимума имеется опасность
оплавления катализатора. На выходе из слоя катализатора ско-
рость реакции снижается ниже допустимого предела.
Профиль температур потока зависит от кинетического порядка
реакции и определяется активностью катализатора по высоте слоя.
В результате более быстрой утомляемости катализатора первых
4Q7.
по ходу газа слоев в реальных условиях максимум температурной
кривой выражен несколько менее резко.
Большее соответствие скорости тепловыделения и скорости от-
вода тепла достигается при прямотоке реагирующей смеси и теп-
лоносителя, не меняющего своего агрегатного состояния.
Тепловой баланс единицы объема потока теплоносителя (про-
текающего в межтрубном пространстве диаметром d со скоростью
о>с), характеризующегося теплоемкостью Со [в ккал/(м3•град)]
и нагреваемого стенкой трубы, имеющей постоянную температуру
Рис. V-28. Теплоотвод
от слоя катализатора при
секционированном тепло-
обменном устройстве:
1—3— номера секций.
t, приводит к следующему выражению
плотности теплоотвода qT [26]:
AKTL
<7т--0 с (V-13)
где А — коэффициент, зависящий от рас-
положения труб; Кт — коэффициент тепло-
передачи от потока к теплоносителю.
Выражение для скорости тепловыделе-
ния реагирующего потока (q) зависит от
кинетического порядка реакции и прип=1
имеет вид:
kb
q = QCoe wp (V-14)
где Q — тепловой эффект реакции; k — константа скорости реак-
ции; —скорость движения реагирующего потока.
Для строго изотермического режима функции (V-13) и (V-14)
должны тождественно совпадать, и из условий равенства предэкс-
понентов и показателей степени может быть установлена зави-
симость скорости движения и начальной температуры теплоноси-
теля от кинетических и термохимических показателей химической
реакции в потоке:
ЛКтЛРр
= (V'I6)
Условия, налагаемые зависимостями (V-15) и (V-16), не
всегда могут быть реализованы. В сложных случаях цель может
быть достигнута ири использовании ректификационного тепло-
обменника [37] или с помощью секционированного теплообмена.
В последнем случае изменение температуры теплоносителя по
ходу реакционного потока легко поддается регулированию
(рис. V-28).
Тепловой расчет реакторов, изображенных на рис. V-26, б—д,
значительно усложняется (размер и свойства потока хладагента
заданы) [38—40].
408
Наряду с поддержанием заданного температурного профиля
по высоте слоя, условием нормальной работы катализатора яв-
ляется соблюдение заданного предела температур в поперечном
сечении слоя. Рассматривая заключенный в трубку слой катали-
затора как твердое тело с равномерно распределенными источ-
никами тепла, нетрудно прийти к выводу о зависимости разности
температур между центром и периферийными слоями цилиндри-
ческого тела от его радиуса. Эта зависимость выражается урав-
нением: _____
г = 2т/ (V-17)
где Д/— допустимая разность температур между центром и пе-
риферией цилиндрического слоя катализатора; q' — удельное теп-
ловыделение слоя катализатора; Ха—эквивалентная (по радиусу)
теплопроводность слоя зернистых частиц при прохождении через
них потока газа.
Значение Хэ определяется свойствами и режимом газового по-
тока и выражается зависимостью:
Лэ = Zr (10,5 + В Rer Ргг) + Хизл (V-18)
где Хг— теплопроводность газа; В — коэффициент, зависящий от
формы зерна катализатора (для сферических частиц В=0,076,
для цилиндрических В = 0,114).
При высокой температуре процесса необходим учет измене-
ния (увеличения) значения Ха в результате теплового излучения
между частицами и потоком газа (ЛИЗл)- Значение колеблется
в широких пределах (табл. V-2).
Т а бл и ц a V-2
Значения Аэ [41]
Катализатор Температура, °C Скорость газа, м/сек Кэ- ккалКм • ч град)
Ванадиевый катализатор (производ- ство серной кислоты) 500 0,35 0,57
Серебряный катализатор (производ- ство окиси этилена) 220 0,55 0,73
220 1,65 1,40
Катализатор синтеза метилового спирта (Р — 250 ат) 350 0,12 23,03
Диаметр труб реакционного аппарата ограничен величиной
порядка 50—75 мм. Для процессов, характеризующихся большим
тепловым эффектом, реактор выполняется из труб диаметром
20—25 мм. Естественно, что в аппаратах большой производитель-
ности число труб бывает очень велико.
409
Для нормальной работы трубчатого реактора большое зна-
чение имеет равномерность распределения потока газа по труб-
кам. С этой целью количества катализатора, загружаемого в каж-
дую трубку, а также сопротивления слоя каждой трубки потоку
газа должны быть одинаковыми. В некоторых случаях трубки
снабжаются дополнительными сопротивлениями (установка ди-
афрагм), одинаковыми по величине и существенно превышающими
сопротивление слоя катализатора.
При оценке реакционных аппаратов с неподвижным слоем ка-
тализатора их можно рассматривать с достаточной для практики
точностью как аппараты полного вытеснения.
Несмотря на широкое распространение гетерогенных катали-
тических процессов, проводимых в неподвижном слое катализа-
тора, недостатки, присущие этому методу, в ряде случаев играют
заметную роль при решении задач интенсификации каталитиче-
ских процессов. Основные недостатки неподвижного слоя:
1) необходимость периодического восстановления активности
катализатора и снижение степени использования реакционной ап-
паратуры; кроме того, применение активных катализаторов по-
вышает требования к чистоте исходных веществ;
2) недостаточная полнота использования внутренней поверх-
ности катализатора, ограниченная невозможностью применения
зерен катализатора малых размеров;
3) затруднения, возникающие при осуществлении процессов с
высоким тепловым эффектом (производительность, единицы объ-
ема катализатора в этом случае ограничивается интенсивностью
теплообмена внутри зерен катализатора, эффективной теплопро-
водностью зернистого слоя и интенсивностью теплообмена между
слоями катализатора и поверхностью теплообмена).
Первый из указанных недостатков устраняется при осуществ-
лении процессов в движущемся слое.
АППАРАТЫ С ДВИЖУЩИМСЯ СЛОЕМ КАТАЛИЗАТОРА
Использование достоинств неподвижного слоя (аппарат
полного вытеснения) для катализаторов с малой продолжитель-
ностью периода постоянной активности достигается при проведе-
нии процесса в движущемся слое.
Катализатор для установок с движущимся слоем готовится в
виде таблетированных или чаще сферических частиц размером
3—5 мм.
Движение слоя частиц катализатора в реакционном аппарате
и регенераторе осуществляется под влиянием силы тяжести. Для
уменьшения истирания катализатора и абразивного износа стенок
аппаратуры скорость перемещения слоя в реакционном аппарате
и регенераторе не превышает 0,25—0,8 см/сек..
Установка с движущимся слоем катализатора состоит из трех
основных частей: 1) реакционного аппарата; 2) регенератора и
410
I 3) системы катализаторопроводов, обеспечивающих непрерывную
' циркуляцию катализатора между реактором и регенератором.
В зависимости от взаимного расположения реактора и регене-
ратора по высоте различают две основные системы (схемы) цир-
' куляции катализатора (рис. V-29).
Схема типа «опрокинутая восьмерка» и более новая схема ти-
па «нулевой контур» (терминология, принятая в литературе по
Рис. V-29. Схемы установок с движущимся слоем катали-
затора:
а — типа «опрокинутая восьмерка» (двукратный подъем катализатора);
б —типа «нулевой контур» (однократный подъем катализатора);
У —исходная смесь; II — смесь продуктов реакции; III — воздух;
IV — отработанный газ;
1 — бункер; 2 — напорный трубопровод; 3 —реактор; 4—регенератор;
5, 6 — подъемники катализатора.
промышленному нефтехимическому синтезу) предусматривают по-
следовательное прохождение слоя катализатора через реакцион/
ный аппарат и регенератор.
Циркуляция катализатора между реакционным аппаратом и
регенератором осуществляется с помощью ковшевых подъемни-
ков либо пневмоподъемников. Последние обладают рядом пре-
имуществ (возможность увеличения кратности циркуляции, сни-
жение расхода тепла на подогрев сырья, поступающего в реак-
тор), которые обеспечивают их более широкое использование.
В схеме с двукратным подъемом катализатора реактор и ре-
генератор, а также пневмоподъемники, предназначенные для
подъема отработанного и регенерированного катализатора,
411
расположены на одной высоте. Катализатор непрерывно вводится
в оба аппарата сверху и удаляется из нижней их части.
Взаимное перемещение катализатора и перерабатываемого
сырья может осуществляться как противотоком, так и прямото-
ком (на рис. V-29 показан прямоток, обладающий в данном слу-
чае рядом преимуществ).
Прямоточные реакторы, в отличие от противоточных, пригодны
для переработки не только газообразных, но и жидких исходных
Рис. V-30. Прямоточный реактор для
каталитического -крекинга жидких
углеводородов:
/, 2, 6, 7— штуцеры; 3 —труба для ввода
жидкого сырья в реактор; 4, 5 —трубки;
8 — трубная решетка.
веществ и обеспечивают возмож-
ность использования избыточного
содержания тепла в регенериро-
ванном катализаторе 1 для нагре-
ва газообразного (и испарения
жидкого) сырья2, поступающего
в реакционное'пространство.
При использовании схемы с
однократным подъемом катали-
затора реактор должен быть
расположен под регенератором.
Регенерированный катализатор
последовательно проходит через
бункер Л напорный трубопро-
вод 2, реактор 3 и регенератор 4.
Из регенератора катализатор
поступает в загрузочное устрой-
ство пневмоподъемника, откуда
потоком газа подается в бункер 1.
В обоих случаях потоки сырья
и регенерированного катализато-
ра при выходе в рабочую зону
реактора равномерно распреде-
ляются по его поперечному сече-
нию так, чтобы уровень слоя ка-
тализатора был горизонтальным.
Для равномерного питания ре-
актора регенерированным ката-
лизатором (особенно, если реак-
давлении) напорный трубопровод,
тор работает при повышенном
соединяющий бункер с реактором, должен иметь соответствующую
высоту. Во избежание проникновения реакционных газов в бункер,
в верхнюю зону реактора подается инертный газ или водяной пар.
Прямоточный реактор, предназначенный для каталитического
крекинга жидких углеводородов, схематично представлен на
рис. V-30.
1 Как правило, температура регенерации превышает рабочую температуру
реакционного процесса.
2 Это особенно необходимо при проведении эндотермических процессов.
412
Регенерированный катализатор подается в реактор через шту-
цер 1, парообразное сырье — через штуцер 2, а жидкое сырье —
через трубу 3. Слой катализатора движется параллельно потоку
реагирующих газов.
Устройство для подачи катализатора, имеющее несколько ва-
риантов конструктивного оформления [42], рассчитано на равно-
мерное распределение катализатора по сечению аппарата. Жидкое
сырье разбрызгивается соплом, помещенным на конце трубы 3,
и, перемешиваясь с частицами катализатора, падающими из рас-
пределительного устройства, нагревается и испаряется.
Трубки 4 предназначены для отвода газообразных продуктов
реакции, трубки 5 — для вывода катализатора.
Газообразные продукты, проходят через отверстия в трубах,
защищенные от попадания катализатора щитками, и выводятся
в сборную камеру под трубной решеткой, соединенную с штуце-
ром 6.
Катализатор, осыпающийся по переточным трубкам 5, обраба-
тывается водяным паром или инертным газом для освобождения
от основной части адсорбированных продуктов реакции. Парооб-
разные продукты, удаляемые из отработанного катализатора, по-
ступают в сборную камеру и присоединяются к основному потоку
продуктов реакции, а отработанный катализатор удаляется через
штуцер 7.
Если реактор установки каталитического крекинга углеводо-
родов представляет собой аппарат для проведения эндотермиче-
ских процессов, то устройство регенератора, рассчитанное на осу-
ществление процессов, характеризующихся высоким тепловым
эффектом экзотермических реакций окисления, дает основание
рассматривать его как реакционный аппарат для осуществления
экзотермических реакций.
В зависимости от величины суммарного теплового эффекта,
определяемого в условиях каталитического крекинга количеством
кокса, отложившегося на зернах катализатора, и допустимой тем-
пературы нагрева катализатора регенератор снабжается зональ-
ными охлаждающими устройствами, выполненными из гладких
труб 1 диаметром 50—60 мм, расположенными с просветом, доста-
точным для свободного передвижения катализатора (не менее
50—60 мм).
Коэффициент теплопередачи от горячего слоя движущегося
катализатора к охлаждающей воде в трубах оценивается величи-
ной порядка 80 ккал/(м2 • ч•град).
Аппарат для регенерации катализатора с зональным охлажде-
нием аналогичен по конструкции многослойному аппарату с непод-
вижным катализатором. Отличием является устройство для равно-
мерного распределения' потока катализатора, располагающееся
1 Ребристые трубы легко забиваются катадизаторной мелочью.
413
в верхней части аппарата, и устройство для выравнивания скоро-
сти движения слоя катализатора по сечению аппарата.
Реактор и регенератор выполняются в виде цилиндрических
или прямоугольных сосудов с футеровкой стенок в случае высо-
кой температуры процесса. Размеры аппаратов, применяемых в
установках каталитического крекинга, весьма значительны (диа-
метр 3—4 м, высота 30—40 м).
Гидравлическое сопротивление движущегося слоя катализато-
ра рассчитывается по тем же закономерностям, что и для непо-
движного слоя.
Для возмещения потерь и поддержания активности катализа-
тора на необходимом уровне, циркулирующий поток катализатора
пополняется свежим катализатором, вследствие чего активность
катализатора, поступающего в реактор, является некоей средней
величиной (иногда называемой «равновесной активностью»), зна-
чение которой определяется относительным количеством свежего
катализатора, вводимого в систему.
АППАРАТЫ С ПСЕВДООЖИЖЕННЫМ СЛОЕМ КАТАЛИЗАТОРА
Движущийся зернистый слой практически свободен от
затруднений, возникающих при использовании неподвижного ка-
тализатора с малой продолжительностью периода постоянной
активности. Однако остальные особенности, присущие неподвиж-
ному слою (ограниченная возможность использования внутрен-
ней поверхности зерен, неблагоприятные условия для теплообмена
внутри слоя и на границе между слоем катализатора и тепло-
обменной поверхностью), полностью сохраняются в движущемся
слое катализатора.
Система газ — твердое тело в состоянии псевдоожижения ли-
шена указанных недостатков, что дает возможность использовать
ее при проведении процессов, лимитируемых интенсивностью мас-
со- и теплообмена.
Интерес, вызванный первыми работами в псевдоожиженном и
кипящем слоях, проявился в большом количестве исследований
этой сложной системы. Накопленный к настоящему времени ма-
териал внес серьезные коррективы в первоначальное представле-
ние о псевдоожиженном слое как об универсальном и почти ли-
шенном недостатков способе проведения гетерогенных каталити-
ческих реакций.
Современные сведения о гидродинамике псевдоожиженного
слоя твердых частиц в потоке газа, а также закономерностях мас-
со- и теплообмена в этом слое облегчают оценку его возможно-
стей, достоинств и недостатков применительно к каждому конкрет-
ному процессу. Несмотря на неблагоприятные для отдельных
случаев особенности, псевдоожиженное состояние катализатора
представляет большой интерес для ряда промышленных процес-
сов органического синтеза и заслуживает самого серьезного вни-
мания при разработке новых технологических схем,
414
& Закономерности гидродинамики, массо- и теплообмена в псев-
t доожиженном слое, подробно освещенные в специальных работах
и монографиях [43—47], кратко рассматриваются ниже.
Рис. V-31. Влияние ско-
рости газа на высоту и
сопротивление слоя ка-
тализатора.
Гидродинамика псевдоожиженного слоя
Переход неподвижного слоя твердых частиц (через ко-
торый снизу вверх проходит ток газа или жидкости) в псевдо-
ожиженное состояние соответствует равенству перепада давления
(АР) потока газа по высоте слоя с весом
этого слоя (бел), приходящимся на еди-
ницу площади его поперечного сечения
(бел):
ДР = <?сл/-$сл (V-19)
В состоянии псевдоожижения скорость
газа в каналах, окружающих частицу, рав-
на скорости витания частицы иВИт в по-
токе газа.
При повышении скорости газового по-
тока в свободном сечении аппарата сече-
ние каналов, окружающих частицу-, увели-
чивается (иВит=const), внешним проявле-
нием чего является возрастание объема
(и порозности) слоя при сохранении по-
стоянства сопротивления (рис. V-31).
Дальнейшим увеличением скорости га-
зового потока достигается состояние, при
котором
^ВИТ = Иг (V-20)
(иг — скорость газового потока в свобод-
ном сечении аппарата), т. е. начинается
разрушение псевдоожиженного слоя в ре-
зультате уноса частиц.
Характеристикой плотности упаковки слоя частиц служит от-
ношение объема пустот к объему слоя (порозность слоя)
или приблизительно
e = l_-p££L (V_.22)
Ртв
где №сл, №тв — объемы слоя и твердых частиц; рсл, ртв — плотно-
сти слоя и твердых частиц.
В некоторых случаях в качестве характеристики употребляет-
ся так называемая «степень расширения слоя» (К), т. е. отноше-
ние объема 1ГСЛ (или высоты ЯСл) псевдоожиженного слоя к
объему №0 (высоте Яо) неподвижного слоя:
?<==^л=^л_>1 (V-23)
w Q Л о
415
Связь этих двух характеристик выражается простой зависи-
мостью:
К = ? ~ 8° (V-24)
1 8сл
где со» есл — порозности неподвижного и псевдоожиженного слоев
(для монодисперсных сферических частиц порбзность неподвиж-
ного слоя равна 0,38—0,42).
Зависимость порозности псевдоожиженного слоя от гидроди-
намических условий его существования выражается уравнением
[48]:
/ 18 Re+ 0,36 Re2 \0,21
8сл”( Аг )
Из структуры зависимости (V-19). и рис. V-31, где участок
кривой 0—а представляет собой зависимость сопротивления не-
подвижного слоя от скорости газа,.нетрудно заключить, что при
скорости газа через неподвижный слой, превышающей скорость
начала его псевдоожижения (и'р), сопротивление неподвижного
слоя частиц будет превосходить сопротивление этого же слоя в
состоянии псевдоожижения.
Важнейшими параметрами процесса, играющими основную
роль при расчетах аппаратуры, являются значения и'кр и и" :
1) и'р, называемая начальной критической скоростью псевдо-
ожижения, представляет собой скорость газового потока в момент
перехода неподвижного слоя в состояние псевдоожижения;
2)/ы'р—скорость уноса, соответствующая началу разрушения
псевдоожиженного слоя и равенству скорости потока в свободном
сечении со скоростью витания.
Изучению зависимости начальной критической скорости и'кр
от условий начала псевдоожижения посвящено большое количе-
ство работ. Предложено свыше 50 уравнений для расчета вели-
чины ы'р, отличающихся принципом подхода к физической сущ-
ности явления и структурой.
Обобщение огромного материала, относящегося к этому во-
просу, приводит к простой зависимости [49]:
при ламинарном режиме (Аг<10)
<р = 0,01з«вит (V-25)
при турбулентном режиме (Аг>106)
«'р = 0,1175«вит (V-26)
Интерполяционное уравнение для выражения значения »'р/«внт
во всем диапазоне значений критерия Архимеда имеет вид:
"ко °>1046
кр =01175_______________
«B„T 1 + 0,00373 Аг0,6
416
Это уравнение может быть использовано для определения на-
чальной критической скорости псевдоожижения как для сфериче-
ских частиц, так и для частиц неправильной формы (на основа-
нии экспериментально определенной скорости витания).
Для оценки порядка величины скорости витания в зависимости
от- размера частиц может служить рис. V-32, справедливый для
частиц алюмосиликатного катализатора
(ро=2О8О кг/м3) в потоке воздуха при
20°С и 760 мм рт. ст.).
Значения начальной критической ско-
рости псевдоожижения для некоторых про-
мышленных катализаторов (эксперимен-
тальные данные) представлены в табл. V-3.
Отношение рабочей скорости потока к
начальной критической скорости (число
псевдоожижения Kw) лежит в пределах
1,5—2,0. При /С„=5-^6 начинается харак-
теризующийся барботажем пузырей газа и
канальными проскоками режим кипения,
который ухудшает контакт газа с зернами
катализатора и сопровождается сильными
Диаметр частиц, мкм
Рис. V-32. Зависимость
скорости витания от раз-
мера частиц [50].
колебаниями сопротивления слоя.
Псевдоожиженный слой твердых частиц может без нарушения
своей структуры перемещаться вдоль оси сосуда или трубопро-
вода как под действием силы тяжести, так и в результате раз-
ности плотностей отдельных объемов слоя. Последнее обстоятель-
ство используется для транспорта псевдоожиженного слоя ката-
лизатора в вертикальном (снизу вверх) направлении подобно
циркуляции жидкости, кипящей в вертикальных трубах.
Таблица V-3
Значения и'кр для некоторых промышленных катализаторов
Диаметр частиц, мм Температура, °C Давление, мм рт. ст. “кр- м/сек
Катализатор прямого Синтеза НАК 0,45 20 760 0,09
Катализатор окисления SO2 3,50 500 760 1,5
Катализатор синтеза аммиака 5 470 300 ат 0,22
Катализатор синтеза метило- вого спирта 6 360 250 ат 0,45
При проведении каталитических процессов под давлением сле-
дует иметь в виду появление дополнительной зависимости, опре-
деляющей значение начальной скорости псевдоожижения [51]. На
рис. V-33 можно видеть, что для частиц катализатора малого раз-
мера повышение давления почти не сказывается на величине
27 В. С. Хайлов, Б. Б, Брандт
417
критической скорости псевдоожижения (кривая 1 на рис. V-33, а)
в широком интервале давлений. Начальная критическая скорость
для частиц большего размера понижается с повышением давле-
ния, причем довольно резко при значениях давления до 50—100 ат.
Влияние давления особенно заметно на изменении величины
скорости уноса (а"р). Отношение и''р/и'кр с увеличением давления
значительно уменьшается, сужая тем самым область скоростей,
соответствующих режиму псевдоожижения и ограничивая воз-
можность уменьшения размера зерен катализатора (ср. кривые
1 и 4 на рис. V-33,б).
Повышение температуры и связанное с этим увеличение вяз-
кости газа сопровождается уменьшением значения критической
скорости псевдоожижения [44].
Рис. V-33. Зависимость и' (а) и и" /и' (б) от да-
кр ' ' кр/ кр ' ‘
вления при 15° С.
Размер частиц (в jhjh): 7 — 0,1; 2 — 0,38; 3 — 1,0; 4—1,5.
Как указывалось выше, число псевдоожижения в практических
условиях значительно превышает единицу и гидродинамическое
состояние псевдоожиженного слоя катализатора заметно отли-
чается от состояния, соответствующего критической скорости (ра-
венство весового градиента перепаду давления). На практике
псевдоожижение почти всегда сопровождается барботажем пу-
зырей газа, а также канальными и поршневыми проскоками га-
зовых струй (режим кипящего слоя).
Режим кипящего слоя, в отличие от идеального состояния
псевдоожижения, характеризуется барботажем. Барботаж газа
через псевдожидкость сопровождается некоторым повышением
перепада давления вследствие трения пузырей газа о твердые ча-
стицы. Умеренный барботаж газа не оказывает влияния на ско-
рость наступления состояния псевдоожижения, так как явление
барботажа возможно лишь при скорости, превышающей крити-
ческую.
Отклонение от режима идеального псевдоожижения наблю-
дается при канальных проскоках газовых струй. Происходящее
418
при этом уменьшение перепада давления (ниже значения АР,
соответствующего весовому градиенту) является следствием того,
что основная масса газового потока проходит через образовав-
шиеся каналы, в то время как скорость газа в зернистом слое
меньше критической скорости псевдоожижения.
Так как каналообразование неблагоприятно влияет иа вели-
чину контакта газовой и твердой фаз, большое значение имеет
оценка доли газового потока, который не участвует в процессе
псевдоожижения. Предложен ряд зависимостей для оценки доли
газового потока, участвующего в проскоке [52, 53]. Анализ этих
зависимостей указывает, что повышению коэффициента эффек-
тивности (т. е. уменьшению относительного количества газа, про-
ходящего через слой в виде струй и пузырей) благоприятствуют
увеличение высоты слоя, уменьшение скорости газа, а также по-
вышение степени дисперсности зернистого материала.
Последнее условие оказывает решающее влияние на «качество»
псевдоожижения, так как добавка мелких частиц резко уменьшает
вязкость слоя (повышает его текучесть). Опыт эксплуатации уста-
новок каталитического крекинга дает представление об опти-
мальном гранулометрическом составе зернистого материала
(табл. V-4).
Таблица V-4
Оптимальный гранулометрический состав зернистого материала,
используемого в установках каталитического крекинга [54]
Скорость газа в свободном сечении, м!сек «Качество» псевдоожижения (или текучесть) Гранулометрический состав, % Унос, кг!м^> газа
0—40 мкм 40—80 мкм 80 мкм
0,61 Хорошее 25 36 39 2,80
Неудовлетво- рительное 6 31 63 5,45
0,73 Хорошее 25 36 39 4,80
Неудовлетво- рительное 6 31 63 8,65
«Качество» псевдоожижения, т. е. степень приближения ре-
жима кипящего слоя к идеальному режиму псевдоожижения, ска-
зывается не только на эффективности системы с точки зрения
условий массообмена, но и на уносе частиц газовым потоком.
Последнее явление относится к числу основных недостатков ки-
пящего слоя, так как в промышленных условиях это связано с
потерями катализатора и изменением гранулометрического со-
става, что приводит к необходимости установки пылеулавливаю-
щих устройств.
Для' каталитических процессов в кипящем слое применяются
катализаторы, удовлетворяющие требованиям не только химиче-
ского характера (активность, избирательность), но и повышенным
27*
419
требованиям к механической прочности в условиях псевдоожиже- )
ния и псевдокипения. Эти требования вызваны необходимостью
сохранения гранулометрического состава и уменьшения абразив- >
ного действия катализатора на материал аппаратуры и трубо-
проводов.
Кроме прочности материала, на устойчивость зерна катализа-
тора против истирающего действия влияет форма частицы. Наи-
более устойчивы зерна сферической формы. Для установок ката- 1
литического крекинга используется микросферический катализа- '
тор с размером частиц 10—150 мкм, обладающий, по сравнению
с пылевидным катализатором, меньшим измельчением (соответ- 1
ственно уменьшенным расходом) и меньшим абразивным дей-
ствием на стенки аппаратуры и трубопроводов.
Расход катализатора в результате истирания зависит от его
механических свойств и ряда условий эксплуатации (скорость по-
токов в аппаратуре и трубопроводах, скорость изменения темпе-
ратуры зерен при нагреве и охлаждении и т. д.) и для разных
процессов оценивается величиной порядка 1,5—2,0% в месяц.
Характеризуя реакционные аппараты с кипящим слоем ката-
лизатора интенсивностью внутриреакционной циркуляции, боль-
шинство авторов приходит к выводу о практической полноте пе-
ремешивания твердой фазы, особенно для случаев, когда отно-
шение диаметра сосуда к высоте слоя достаточно велико. Что
же касается вертикального перемешивания газовой фазы, то в
условиях однородного псевдоожиженного слоя, в котором отсут-
ствуют пузыри барботирующего газа (иг ~ последний совер-
шает в основном поступательное движение и перемешивание его
по вертикали невелико.
Режим кипящего слоя («г » м'р), сопровождающийся барбо-
тажем газовых пузырей, вызывает усиленное движение твердых
частиц сверху вниз и заметное вертикальное перемешивание газа.
Для характеристики интенсивности перемешивания предлагается
[55] коэффициент продольного перемешивания Оп.п, зависимость
которого от скорости газа и линейных размеров слоя дается сле-
дующим уравнением:
“г ~ и'кп
Дп.п = /о е р ' (V-27)
Исследование влияния давления на интенсивность перемеши-
вания в кипящем слое показывает, что при повышении давления
газа перемешивание твердых частиц значительно улучшается, в
то время как интенсивность смешения газовой фазы становится
меньше [56].
Структура кипящего слоя и перемешивание твердой и газовой
фаз в нем в значительной степени зависят от конструкции распре-
делительных (дутьевых) решеток, оценка действия которых про-
изводится, исходя из равномерности поля скоростей газа в попе-
речном сечении слоя.
420
Судя по имеющимся данным [57, 58], рядом достоинств обла-
дают так называемые «вихревые» решетки, в которых отверстия
расположены под углом к плоскости решетки. При этом газо-
вый поток совершает не только поступательное, но и вращатель-
ное движение, исключая возможность скопления частиц в про-
межутках между отверстиями. ПодЬбные решетки рекомендуется
применять для кипящего слоя с большой степенью расширения
при значительной его высоте (Н=0,75 4-5,0 D).
По данным ряда исследований [59, 60], для устойчивости ки-
пящего слоя живое сечение отверстий дутьевой решетки (фреш)
не должно превышать 2—2,5% ее сечения. Следует, однако, иметь
в виду, что в зависимости от особенностей зернистого слоя, ха-
рактера процесса и принципа конструкции реактора пределы зна-
чений фреш могут быть значительно шире. Так, наряду с дутьевыми
решетками, рассчитанными на проход только газа (скорость по-
тока газа в отверстиях дутьевых решеток достигает 12—15 м!сек),
живое сечение отверстий в провальных тарелках, применяемых
как при прямотоке, так и при противотоке газа и твердого мате-
риала, достигает 25% при диаметре отверстий 4—10 мм [61].
Теплообмен в псевдоожиженном слое
Интенсивный турбулентный режим движения газа и
твердых частиц в псевдоожиженном слое катализатора в верти-
кальном и радиальном направлениях резко меняет величину эф-
фективной теплопроводности, характеризующую неподвижный
слой зернистого материала.
Величина ?.э зависит от степени расширения слоя и диаметра
частиц [62]:
Функция имеет максимум при = 1,5—1,75 и значение 2.э
при этой степени расширения слоя для частиц песка размером
0,1 мм составляет 170 ккал/(м •ч•град). При большем значении
Кю величина 7.э уменьшается, приближаясь к теплопроводности
газового потока.
Вследствие разной плотности потоков твердых частиц у на-
ружной стенк^ и в центре псевдоожиженного слоя (поток газа
движется предпочтительно в центральной части слоя) влияние
скорости газового потока на величину коэффициента теплоотдачи
от псевдоожиженного слоя к поверхности теплообмена различно
в центре и в периферийных слоях потока. Установлено также, что
интенсивность теплообмена имеет максимальное значение в сред-
ней по высоте части псевдоожиженного слоя, что следует иметь
в виду при размещении теплообменных устройств.
Значение коэффициента теплоотдачи от кипящего слоя к стен-
ке змеевика, погруженного в слой, или к стенке сосуда более чем
424
на один порядок превосходит соответствующую величину для
псевдоожиженного слоя [54].
Отмеченные выше особенности псевдоожиженного состояния
катализатора благоприятны для процессов, протекающих в области
внутренней диффузии, для экзотермических процессов, обладаю-
щих высоким тепловым эффектом и проводящихся в узком интер-
вале температур, а также для процессов, характеризующихся ма-
лой цродолжительностью периода постоянной активности катали-
затора и связанных с необходимостью частой его регенерации.
Конструктивное оформление реакторов
с псевдоожиженным споем
При проведении каталитических реакций в псевдоожи-
женном и кипящем слоях значительно повышаются требования
к механической прочности зерен катализатора и неизбежно воз-
никают задачи, связанные с улавливанием пылевидных частиц
катализатора в результате его истирания.
Основной особенностью каталитических процессов в кипящем
слое является эффект продольного перемешивания твердой и га-
зовой фаз и поперечной неравномерности скорости газового по-
тока в слое.
Наблюдаемое во всех случаях снижение степени превращения
исходного вещества при переходе от неподвижного слоя катали-
затора к кипящему слою этого же катализатора является резуль-
татом не только перемещения части газа в направлении, противо-
положном направлению движения реакционного потока, но и
следствием проскока газа в виде пузырей и струй, сопровождаю-
щих режим псевдокипения. Суммарный результат этих двух яв-
лений и характер зависимости скорости превращения исходного
вещества от степени его превращения подобны результату, дости-
гаемому при продольном перемешивании реакционной массы. Од-
носекционные реакционные аппараты с кипящим слоем практи-
чески могут быть отнесены к категории реакторов промежуточ-
ного типа и характеризуются числом эквивалентных псевдосекций
полного смешения.
Экспериментально установленная [63] зависимость числа псев-
досекций (т) по газовой фазе от числа псевдоожижения (Kw) и
размеров зернистого слоя описывается уравнением
(Но — начальная высота зернистого слоя; D — диаметр аппарата),
справедливым при 0,5-</7о/0-<4.
Внешняя аналогия псевдокипящего слоя и аппарата полного
смешения не дает, однако, основания для перенесения на поток
газа в кипящем слое всех особенностей идеального режима пол-
422
ного смешения (возможность раздельного ввода компонентов, пол-
ная равномерность поля температур и т. п.).
Схема и аппаратурное оформление реакционного агрегата за-
висят от продолжительности периода постоянной активности ка-
тализатора и конечной степени превращения сырья. Примером
использования относительно устойчивых, не требующих частой
регенерации катализаторов может служить процесс парофазного
окисления нафталина (рис. V-34).
Воздух, подогретый в змеевике, установленном над поверхно-
стью кипящего слоя ванадиевого катализатора, поступает в ис-
паритель нафталина 1,
откуда парогазовая
смесь подается под
дутьевую решетку ре-
акционного аппарата 2.
В зоне кипящего слоя
катализатора располо-
жено теплообменное
устройство, охлаждае-
мое водой, подача ко-
торой автоматически
регулируется в зависи-
мости от температуры
слоя. Реакционная
смесь, частично охлаж-
денная при прохожде-
нии через подогрева-
тель воздуха 3, посту-
Смесь продуктов
Рис. V-34. Схема установки для парофазного
окисления нафталина:
/ — испаритель нафталина; 2 — реакционный аппарат;
3 — подогреватель воздуха; -/ — секционный фильтр.
пает в зону сепарации,
в верхней части кото-
рой расположен сек-
ционный фильтр 4, из-
готовленный из пористой керамики. Периодическая продувка сек-
ций фильтра обратным током воздуха обеспечивает их длительную
и надежную работу. Потери катализатора составляют 58—59 г на
1000 кг фталевого ангидрида.
Текучесть псевдоожиженного слоя позволяет легко осущест-
вить транспорт катализатора и дает возможность непрерывного
вывода отработанного катализатора и непрерывной подачи ре-
генерированного катализатора в реакционный аппарат. Схема ре-
акционного агрегата в этом случае аналогична рассмотренной
выше схеме агрегата с движущимся слоем и состоит из реак-
ционного аппарата, регенератора и системы транспортных
устройств.
Отработанный катализатор, выводимый из реакционного ап-
парата, направляется на регенерацию, осуществляемую также в
кипящем слое. В зависимости от причин дезактивации катализа-
тора восстановление активности достигается путем обработки его
423
газообразным окислителем или восстановителем (воздух, водо-
род), используемым в качестве псевдоожижающей среды.
Регенерированный катализатор непрерывно выводится из ре-
генератора и возвращается в реакционный аппарат.
Для эндотермических процессов (например, дегидрирования
углеводородов) тепло катализатора, возвращаемого после реге-
нерации, используется для испарения и нагревания исходного
сырья (температура регенерации, как правило, выше температуры
в реакционном аппарате).
Ввиду того, что технология и аппаратура процессов в кипя-
щем слое наиболее разработаны применительно к процессам ка-
Рис. V-35. Схема реакторно-
регенераторного блока кре-
кинг-установки (модель IV)
[42]:
1 — реактор; 2 — регенератор;
3 — циклоны; 4 — стояки; 5, 7—
трубопроводы; 6 — распредели-
тельные устройства.
талитического крекинга углеводородов,
на рис. V-35 приведена принципиальная
схема реакторно-регенераторного блока
крекинг-установки.
Парообразное сырье смешивается с
горячим регенерированным катализато-
ром в трубопроводе 5 и подается в ре-
актор 1 через распределительное устрой-
ство 6. Продукты реакции после отде-
ления от пылевидных частиц катализа-
тора в системе циклонов 3, выводятся
через верхний штуцер, а пылевидный
катализатор по стояку 4 возвращается
под уровень кипящего слоя.
Отработанный катализатор постоян-
но стекает по трубопроводу 7 и направ-
ляется в регенератор 2. Интенсивность
циркуляции катализатора между реак-
тором и регенератором регулируется
изменением разности плотности потоков путем соответствую-
щего изменения подачи воздуха в верхний участок трубопро-
вода 7.
В регенераторе 2 проводится удаление дезактивирующих от-
ложений на зернах катализатора (в рассматриваемом случае —
выжигание в токе воздуха), после чего восстановивший свою ак-
тивность катализатор через стояк трубопровода 5 возвращается
в реактор.
Для отвода тепла реакций, протекающих в реакторе и регене-
раторе, в кипящем слое катализатора могут быть размещены
теплообменные устройства.
Реакционные аппараты (и аналогичные им по принципу устрой-
ства— регенераторы) оформляются в виде цилиндрических со-
судов 1 с различным отношением высоты к диаметру (для реак-
торов каталитического крекинга HfD = 1,4-т-4,0). При проведении
1 В последнее время все большее внимание привлекают реакторы конической
формы.
424
/ ,
высокотемпературных процессов стенки аппарата футеруются
кирпичом или диабазовой плиткой.
Конструкция дутьевых решеток выполняется с учетом дости-
жения максимально полной равномерности скорости потока газа
по сечению аппарата.
Наряду с достоинствами кипящего слоя, позволяющими пол-
ностью использовать внутреннюю поверхность зерен катализатора,
ему присущи особенности реакционных аппаратов полного сме-
шения. Последнее обстоятельство необходимо учитывать при про-
ведении реакций с заданной высокой степенью превращения ис-
ходного вещества.
Уменьшение эффекта продольного перемешивания газового
потока внутри слоя и ликвидация последствий прорыва струй и
пузырей через кипящий слой достигается последовательным по
ходу газа расположением отдельных секций, разделенных дырча-
тыми решетками. Перегородки, разделяющие слои, исключают
возможность перемещения катализатора и газовой фазы между
секциями в направлении, противоположном реакционному пото-
ку, способствуют усреднению состава газового потока над кипя-
щим слоем- (смешение струй и пузырей непревращенного газа с
основным потоком) и перераспределению его на входе в следую-
щий слой.
Способы секционирования кипящего слоя весьма разнообраз-
ны. При выборе соответствующего варианта необходимо учиты-
вать сменяемость катализатора, а также, какое взаимное направ-
ление потока газа и катализатора является наиболее целесооб-
разным.
Из большого числа возможных схем многослойных реакторов
с кипящим слоем [64, 65] на рис. V-36 даны основные.
В реакторах с несменяемой загрузкой катализатора
(рис. V-36, а) слой катализатора разделяется по высоте проваль-
ными тарелками, между которыми, в толще кипящего слоя, раз-
мещаются теплообменные устройства.
Реакторы с параллельным движением твердой и газовой фаз
представлены на рис. V-36, б, в. В первом случае газовый поток
вводится только в нижнюю часть аппарата и проходит все секции
параллельно движению катализатора. Во втором случае газооб-
разные компоненты (или один из них) вводятся в каждую из
секций, смешиваясь с основным потоком. Секции отделяются одна
от другой провальными тарелками. Теплообменные устройства
размещены в каждой секции.
Противоток газа и катализатора • осуществляется в каскаде
секций, снабженных переточными трубами или отделенных одна
от другой провальными тарелками. Газовая фаза поступает либо
только под нижнюю тарелку (рис. V-36,а), либо часть ее вводит-
ся под нижнюю тарелку, а другая часть распределяется по сек-
циям (рис. V-36, в, д).
425
Температурный режим процесса, проводимого в секциониро-
ванном кипящем слое, поддерживается путем размещения в
каждой секции теплообменных устройств. В отличие от непод-
вижного слоя катализатора теплообменник помещается внутри
кипящего слоя. Регулирование подачи охлаждающего агента обес-
печивает поддержание заданной температуры слоя и позволяет
осуществлять необходимый режим изменения температуры походу
газового потока.
По аналогии с многослойными реакторами неподвижного слоя
катализатора регулирование температуры газового потока при
Рис. V-36. Схемы многослойных реакторов с кипящим слоем:
Г —газ; Т — твердая фаза.
проведении экзотермических реакций может осуществляться пу-
тем ввода холодного потока газа, как это показано на
рис. V-36, в, д.
Распределение катализатора по секциям производится в со-
ответствии с принципами расчета размера секций полного сме-
шения (см. стр. 183).
Расстояние между тарелками противоточного реакционного
аппарата зависит от высоты слоя катализатора на тарелке, числа
псевдоожижения и диаметра тарелки. Для описания указанной
зависимости рекомендуется следующее выражение [66]:
//т= (V-28)
где Н?— расстояние между тарелками, м; Нсл — высота слоя, м;
Kw — число псевдоожижения; £)т — диаметр тарелки, м.
Уравнение (V-27) выведено, исходя из допускаемого уноса
частиц с тарелки на тарелку, равного 5—7%.
426
Размеры переточных устройств определяются с учетом скоро-
сти движения материала, принимаемой равной 0,05—0,1 м!сек.
Кипящий слой катализатора, нашедший широкое применение
в промышленности нефтехимического синтеза, в несколько мень-
ших масштабах используется в технологии основного органиче-
ского синтеза. Из числа гетерогенных каталитических процессов,
проводимых в кипящем слое, известны: окисление нафталина, вос-
становление нитробензола в анилин, синтез окиси этилена [65, 67],
синтез акрилонитрила из пропилена, аммиака и кислорода [65,
68], процессы хлорирования низших алканов [65], синтез метил-
хлорсиланов [69], получение малеинового ангидрида окислением
бутана [65, 70] и др.
Кипящий слой находит применение в технологии органиче-
ского синтеза не только при проведении каталитических процес-
сов, но и при перемешивании сыпучих материалов [71], а также
при сушке твердых материалов [72] и растворов [73].
Непременной частью установок для проведения каталитиче-
ских процессов в кипящем слое катализатора являются устрой-
ства для улавливания пылевидных частиц катализатора. В этих
целях применяют систему ступенчатых циклонов, фильтры из
стеклоткани, фильтры из пористой керамики, металлокерамиче-
ские фильтры.
МЕХАНИЧЕСКИЕ СПОСОБЫ СОЗДАНИЯ
КИПЯЩЕГО СЛОЯ
Необходимость создания значительной скоро-
сти газа или жидкости для перевода слоя твердых частиц в со-
стояние псевдоожижения и псевдокипения в ряде случаев может
стать причиной затруднений при осуществлении процесса.
Псевдоожиженное состояние зернистого слоя, практически не
зависящее от скорости газового потока, может быть достигнуто
при замене аэродинамического воздействия механическим. Такие
механические приемы относительно новы, и опыт их практиче-
ского использования отсутствует. Однако, учитывая все возра-
стающее многообразие процессов органического синтеза, они мо-
гут представлять интерес. Ниже кратко рассмотрены принципы,
положенные в основу этих механических приемов.
Виброкипящий слой
Псевдоожиженное состояние зернистого слоя может
быть достигнуто путем воздействия на него вибрирующей пло-
щадки, которая поддерживает слой или соприкасается с ним.
Вибрация вызывает уменьшение коэффициента трения между
частйцами и при определенных параметрах (частота и амплитуда
колебаний) значение эффективного коэффициента трения прибли-
жается к нулю.
427
Состояние вибрирующего слоя, характеризующееся постоян-
ством соприкосновения частиц с вибрирующей площадкой, назы-
вается состоянием вибропсевдоожижения. Начало отрыва частиц
от вибрирующей площадки соответствует критической точке пе-
рехода слоя в состояние виброкипения [74, 75].
Критическая точка перехода слоя в состояние виброкипения
для любого сыпучего материала соответствует равенству ускоре-
ния вибрации площадки ускорению силы тяжести. Характерная
особенность вибрирующего слоя — независимость точки перехода
состояния вибропсевдоожижения к состоянию виброкипения от
размера и плотности частиц слоя. При питании электродвигателя
вибрирующей площадки током обычной частоты (50 гц) ампли-
туда колебаний‘площадки не превышает 0,12—0,13 мм.
Эти особенности виброкипящего слоя выгодно отличают его
от аэродинамического кипящего слоя и открывают широкие воз-
можности для разнообразных режимов работы независимо от
скорости газа и гранулометрического состава частиц.
Приводом перемешивающего механизма может служить лри-
вод, используемый при работе вибрационных машин, широко при-
меняемых в строительстве [76, 77].
г
Псевдоожижение перемешиванием
Значительный интерес представляет возможность пе-
ревода слоя пылевидного материала в псевдоожиженное состоя-
ние путем механического перемешивания.
Исследование этого вопроса показывает [78], что при враще-
нии в среде сыпучего материала системы лопастей с определен-
ной скоростью материал переходит в псевдоожиженное состояние,
аналогичное состоянию, которое достигается аэродинамическим
методом. Внутренняя циркуляция частиц начинается при окруж-
ной скорости ы0Кр лопасти мешалки (лопастная мешалка с углом
наклона лопасти 45°), равной 4—5 м/сек, а при uOKp=5-i-8 м/сек
наступает состояние псевдоожижения и объем слоя увеличивает-
ся на 10—15%.
Продольное перемешивание частиц при этом довольно интен-
сивное, хотя и уступает аэродинамическому псевдоожиженному
слою. В отличие от последнего, движение частиц в периферийных
слоях происходит снизу вверх.
Условия образования псевдоожиженного слоя механическим
методом приведены в табл. V-5.
Экспериментально установлено, что значение окружной скоро-
сти, при которой наступает состояние псевдоожижения в значи-
тельной степени (почти пропорционально) зависит от отношения
высоты слоя к ширине лопасти.
Для каждого материала и конструкции рабочего органа имеет-
ся своя предельная высота слоя над лопастью (Дел), выше кото-
рой материал не переходит й псевдоожиженное состояние неза-
428
Таблица V-5
Условия образования
псевдоожиженного слоя механическим методом
Материал Насыпная плотность, г/смР Влажность, % Пределы дисперсности частиц, мк Окружная скорость лопасти, м!сек
Каолин 0,52 1,33 0-70 5-5,5
Цемент .......... 1,17 0,68 0-50 5-6
Тальк 0,85 0,11 0-60 5-6
Краситель Ж-1 0,185 — 0-50 5,5-6
Краситель Ж-2 0,54 — 0,50 5,5-6
Краситель П-1 0,3 — 0,50 5-5,5
висимо от окружной скорости движущейся лопасти. Для радиаль-
ной прямоугольной лопасти с углом наклона 45° предельная вы-
сота слоя определяется шириной лопасти и зависит от насыпной
плотности перемешиваемого материала, уменьшаясь с ее увели-
чением.
При необходимости псевдоожижения слоя, высота которого
превышает предельную, требуется установка нескольких лопастей
с расстоянием h' между ними по вертикали Н'сл (h' « 0,8//£л),
При этом, однако, окружная скорость, необходимая для псевдо-
ожижения, увеличивается на 20—30%, в связи с-чем для не-
прерывнодействующей установки более целесообразно использо-
вать многосекционный аппарат с высотой слоя в каждой секции
0,8 - 0,85Ясл.
В процессе перемешивания происходит изменение структуры
зернистого слоя, выражающееся в изменении насыпной плотно-
сти и расхода мощности на перемешивание.
Состояние псевдоожижения, характеризующееся уменьшением
насыпной плотности, требует минимального расхода энергии на
перемешивание.
Максимальный (соответствующий периоду уплотнения слоя)
расход мощности на перемешивание выражается следующим об-
разом [79]:
* = С'рХ^л13 (&л sin в)0’82 Я’-л04 (V-29)
где С' — коэффициент сопротивления [для каолина и талька С'
(6--8)10-3, для цемента С' = 19- 10~3, для кварцевого песка С' =
= 56-10~3]; рн — насыпная плотность материала в начале пере-
мешивания, кг^’м3; щл— угловая скорость вращения лопасти, сект1;
Ьл, L — ширина и длина лопасти, м; 0 — угол наклона лопасти;
Нел — высота слоя материала, м.
Рассмотренный выше прием псевдоожижения может найти
применение при осуществлении каталитических процессов в тех
случаях, когда объемная скорость потока газа недостаточна для
аэродинамического псевдоожижения слоя катализатора.
429
РЕАКТОРЫ ДЛЯ ПРОВЕДЕНИЯ ПРОЦЕССОВ
В СИСТЕМЕ ЖИДКОСТЬ — ТВЕРДОЕ ТЕЛО
В качестве реакционной среды система жидкость —
твердое тело в практике промышленного органического синтеза
встречается сравнительно редко; гораздо более часты случаи трех-
фазной системы жидкость — твердое тело — газ, в которой твер-
дая фаза является катализатором взаимодействия жидких и га-
зообразных реагентов.
Иной, чем в газовой фазе, механизм- процесса, протекающего
на твердой поверхности катализатора в жидкой фазе, обусловли-
вает значительную скорость реакции. Так, для одних и тех же
конкретных реакций при проведении их в газовой и жидкой фазах
с применением близких по составу катализаторов активность ка-
тализатора в последнем случае на 1—2 порядка выше; при этом
температура процесса на 100—200 град ниже, чем в первом слу-
чае [26]. Эта особенность гетерогенного катализа в жидкой фазе,
благоприятная для проведения процессов с участием термола-
бильных веществ, а также возможность отвода тепла экзотерми-
ческих реакций за счет испарения легколетучих компонентов
реакционной смеси делают жидкофазные каталитические про-
цессы в отдельных случаях более предпочтительными, чем газо-
фазные.
В промышленной практике известен ряд процессов гетероген-
ного катализа в жидкой фазе с участием газообразного реаген-
та. При гидрировании жиров, непредельных и ароматических уг-
леводородов, восстановлении нитро- и нитрильных групп цикло-
алифатических, алифатических и жирноароматических соединений
процесс проводится в жидкой фазе, контактируемой с водородом.
Известны случаи окисления жирных и жирноароматических угле-
водородов кислородом воздуха в условиях жидкофазного гетеро-
генного катализа. Все эти процессы относятся к таким, скорость
которых определяется не только диффузией реагирующих ве-
ществ к поверхности частицы катализатора, но также скоростью
массообмена между газовой и жидкой фазами и дуффузией ча-
стиц растворенного газа в жидкой фазе. Поскольку диффузия ча-
стиц жидкой фазы к поверхности зерен катализатора гораздо
менее затруднена, скорость процесса может быть лимитирована
скоростью транспорта частиц из газовой фазы к поверхности ка-
тализатора через поверхность раздела газовой и жидкой фаз.
При достаточно высокой скорости реакции, определяемой тем-
пературой процесса и концентрацией реагирующего компонента
(с), малое значение коэффициента диффузии (£))' исходных ве-
ществ и продуктов реакции, характерное для жидкостей, приво-
дит к тому, что эффективная глубина проникновения
kcn-1
430
(k— константа скорости реакции, п — порядок реакции) стано-
вится соизмеримой с диаметром пор зерен катализатора (/э мень-
ше или ~d0) и течение процесса по отношению к поверхности
катализатора характеризуется закономерностями явлений на
внешней поверхности.
Рассмотрение простейшего случая, когда кинетика реакции на
поверхности частиц катализатора удовлетворяет первому порядку
и эффективная константа скорости fe3, отнесенная к единице объ-
ема жидкости с определенным содержанием твердых
тализатора, выражается зависимостью
*Э = * + ₽д'
приводит к выражению скорости реакции, отнесенной
объема жидкой фазы:
de = k3D"FK
Нт D"FK + k3 C°
частиц ка-
(V-30)
к единице
(V-31)
где N4 — число дисперсных частиц в единице объема жидкости;
б —средняя поверхность дисперсной фазы; k — истинная констан-
та скорости реакции на поверхности дисперсной частицы; рд—кон-
станта скорости диффузии к поверхности частицы катализатора;
D" — отношение коэффициента диффузии D' к толщине приведен-
ной пленки на границе раздела газ — жидкость; FK— поверхность
контакта газовой и жидкой фаз, отнесенная к единице объема
жидкой фазы; с0 — концентрация газообразного компонента на
границе диффузионного слоя с жидкой фазой.
Из сопоставления уравнений (V-30) и (V-31) видно, что ско-
рость реакции пропорциональна удельной поверхности контакта
газовой и жидкой фаз и в пределах некоторого значения N4 за-
висит от этой концентрации твердых частиц катализатора.
Повышение концентрации катализатора при постоянстве зна-
чения D"FK способствует увеличению скорости реакции лишь до
известного предела, выше которого влияние концентрации ката-
лизатора становится мало заметным и скорость стремится к пре-
дельному постоянному значению.
Величина k3 пропорциональна степени измельчения частиц ка-
тализатора (N°), однако по мере уменьшения размеров частиц
уменьшается и скорость ее витания, т. е. скорость перемещения
относительно потока жидкости, создаваемого мешалкой или бар-
ботажем. Следствием этого может быть уменьшение скорости кон-
вективной диффузии вблизи частицы. Увеличение общего диффу-
зионного сопротивления способствует переходу процесса из кине-
тической области в диффузионную.
Увеличение степени дисперсности газа, т. е. значения D'FK, со-
провождается повышением скорости процесса только в области
диффузии в объеме, не меняя условий диффузии к поверхности.
Таким образом, для увеличения скорости жидкофазного гетеро-
генного каталитического процесса необходимо одновременное
431
изменение условий диффузии в объеме и диффузии к поверхности
катализатора; интенсификация процесса связана с необходимо-
стью увеличения количества и максимального диспергирования
катализатора при одновременном увеличении поверхности фазо-
вого контакта между газом и жидкостью.
Возможные варианты аппаратурного оформления жидкофаз-
ных гетерогенных каталитических процессов сводятся к двум
группам, которые различаются по способу диспергирования фаз:
Рис. V-37. Схема реакционного агрегата для гетерогенного
катализа в жидкой фазе:
7 —насос; 2 —настовый насос; 3 — аппарат для приготовления суспензии;
4 — подогреватель жидкого сырья; 5 — реакционный аппарат; 6 — сепара-
тор высокого давления; 7 — конденсатор пара; 8 — циркуляционный газо-
вый насос; 9, 10 — дросселирующие устройства; // —сепаратор среднего
давления; 12 — сепаратор низкого давления.
1) катализатор суспендирован в жидкости;
2) катализатор находится в виде сплошного слоя в потоке
жидкости или орошается ею.
Первая группа реакторов может быть представлена аппарата-
ми периодического и непрерывного действия, в которых в слое
жидкости диспергированы газ и частицы катализатора. В неко-
торых случаях суспендирование катализатора осуществляется в
результате продольного перемешивания жидкости в условиях
барботажного режима, достигаемого при пропускании реакцион-
ного газа.
Скорость газа в свободном сечении аппарата, необходимая для
равномерного распределения катализатора по высоте барботаж-
ного слоя, зависит от разности плотностей твердой и жидкой фаз
(устанавливаемой экспериментально) и, как правило, превышает
скорость, соответствующую стехиометрическим соотношениям.
Организация схемы, подобной изображенной на рис. V-37, тре-
бует -установки циркуляционных насосов и создания условий, ис-
432
I
1
ключающих возможность конденсации паров жидкой фазы в ком-
муникациях.
Более полное диспергирование газа и равномерное распределе-
ние твердой фазы по реакционному объему достигается при ис-
пользовании мешалок. Одной из наиболее эффективных является
мешалка типа «гребной винт» (см. рис. V-14), которая может ра-
ботать без сальникового уплотнения при высоком давлении в ап-
парате. Использование мешалок такого типа, обеспечивающее вы-
сокую степень дисперсности газа в жидкости и равномерность
распределения катализатора, исключает необходимость системы
внешней циркуляции газа, что значительно упрощает и удешев-
ляет схему реакционного агрегата.
При непрерывной работе реакционного агрегата питание ре-
актора катализатором производится с помощью пастовых насосов,
обеспечивающих достаточно точную и равномерную подачу ката-
лизатора, суспендированного в жидком исходном веществе, при
любом избыточном давлении.
Прн проведении процесса под давлением поток реакционной
массы, выходящей из реактора, проходит через дросселирующие
устройства и поступает в систему сепараторов для отделения де-
сорбированного газа от жидких продуктов. В ряде случаев дрос-
селирование проводится в две ступени с отбором из сепараторов,
устанавливаемых после каждой ступени дросселирования, паро-
газовой смеси необходимого давления и соответствующего состава
(см. рис. V-37).
Присутствие в реакционной массе твердых частиц весьма услож-
няет работу дросселирующих устройств вследствие значительной
эрозии. В качестве материала для седла и запорной иглы дрос-
селирующего вентиля используются специальные материалы (кар-
бид вольфрама и др.). Практикой промышленности искусствен-
ного жидкого топлива установлено, что для надежности целесо-
образна установка нескольких параллельно соединенных групп
дросселирующих вентилей, включаемых в работу периодически
(иногда автоматически).
Для дросселирования жидкого потока используется также со-
противление, создаваемое каналом соответствующей длины. Дли-
на и формй (спиральная, винтообразная) канала позволяют со-
здавать необходимое сопротивление при скорости потока в канале
меньшей, чем скорость в кольцевом зазоре между седлом и иглой
дросселирующего вентиля.
Интенсивное продольное перемешивание в реакторах без ме-
шалок дает возможность отнести их к категории аппаратов про-
межуточного типа с большим значением коэффициента кратно-
сти циркуляции. Реакторы, снабженные интенсивно работающими
мешалками, практически приближаются к аппаратам полного
смешения. Особенности, присущие этим категориям реакционной
аппаратуры, могут быть использованы при разработке схемы ре-
акционного агрегата.
28 В. С. Хайло», Б. Б. Брандт
433
Применение суспендированного катализатора позволяет осу-
ществлять процессы, катализируемые активными, но быстро те-
ряющими активность катализаторами; непрерывный вывод отра-
ботанного контакта и замена его новым дает возможность под-
держивать каталитическую активность на постоянном уровне.
Выделение катализатора из реакционной массы с целью возврата
его в реакционный аппарат или для передачи на регенерацию —
достаточно сложная задача, решаемая с применением способов и
аппаратуры, предназначенной для разделения систем жидкость—
твердое тело (см. стр. 463). В некоторых случаях задача разделе-
ния реакционной массы усложняется требованием предохранения
катализатора от соприкосновения с воздухом, что бывает либо
нежелательно, либо недопустимо (пирофорность катализатора);
при этом аппаратура для выделения работает в атмосфере инерт-
ного газа.
Возврат катализатора для повторного использования может
быть осуществлен также в реакторе, снабженном быстроходной
мешалкой, на валу которой (выше уровня жидкости) расположен
центробежный отстойный сепаратор. Суспензия катализатора в
реакционной массе делится в сепараторе на две фракции. Концен-
трат возвращается в реакционное пространство, а фугат, освобо-
жденный от основной части катализатора, выводится из реакци-
онного аппарата.
При использовании неподвижно расположенного катализатора
для каталитических реакций в жидкой фазе возможны два вари-
анта проведения процесса: 1) при орошении слоя катализатора
жидкостью в атмосфере газообразного компонента и 2) в устрой-
стве типа эмульгационной колонны.
При первом варианте слой зерен катализатора орошается
жидкостью при противотоке или прямотоке газа. Второй вариант
также может быть осуществлен при обоих способах взаимного
перемещения фаз.
Примером первого варианта может служить процесс гидриро-
вания бензола на сплавных катализаторах — слой зерен катализа-
тора орощается бензолом, водород движется параллельным током.
Второй вариант оформления процесса можно рассмотреть на
примере гидрирования адиподинитрила в гексаметилендиамин [80].
В нижнюю часть реактора, заполненного зернами катализатора
(кобальт на окиси алюминия) диаметром 4—10 мм. подаются ади-
подинитрил, жидкий аммиак и водород. Процесс проводится при
температуре 100—125° С и давлении в системе 200 ат.
Жидкофазное хлорирование бензола (гомогенная каталитиче-
ская реакция) также может служить примером аппаратурного
оформления процессов в системе жидкость — твердое тело — газ.
Насадка колонны, состоящая из железных колец и являющаяся
поставщиком катализатора гомогенной каталитической реакции
между бензолом и хлором, омывается потоками бензола и хлора,
подаваемыми снизу вверх.
434
Теплота экзотермических реакций в обоих рассмотренных слу-
чаях отводится парогазовой смесью в результате испарения
жидких компонентов реакционной массы (аммиак и бензол).
Реакционные аппараты с неподвижным слоем катализатора по
признаку изменения концентрации реагирующих компонентов в
реакционном объеме в зависимости от гидравлического режима
движения газа и жидкости могут быть отнесены либо к одной,
либо к другой группе реакторов идеального режима. Реакторы с
орошаемой насадкой практически могут рассматриваться как ре-
акторы полного вытеснения. Скорость газового потока в реакто-
рах, заполненных жидкостью, определяет близость режима к ус-
ловиям полного вытеснения или полного смешения (см. стр. 356).
Для сравнительной оценки обеих групп реакционных аппара-
тов может быть использован процесс синтеза гексаметиленди-
амина из адиподинитрила, протекающий по схеме
NC(CH2)4CN H2N(CH2)5CN H2N(CH2)6NH2
(И (2) (3)
в присутствии твердого катализатора.
При синтезе гексаметилендиамина из аминокапронитрила в
диффузионной области можно рассчитывать на получение высо-
кого выхода гексаметилендиамина независимо от степени превра-
щения адиподинитрила, так как из уравнения [81]
Рз, д
w2 k2
(р2, д — константа скорости диффузии продукта 2) следует, что
при д скорость образования гексаметилендиамина значи-
тельно превосходит скорость образования аминокапронитрила
Понятно, что понижение температуры будет благопри-
ятно сказываться на выходе промежуточного продукта реакции.
При проведении этого процесса в кинетической области высокий
выход гексаметилендиамина может быть получен лишь при а->1,0.
При выборе режима проведения процесса в реакционном аппа-
рате необходим учет условий и затрат, связанных с разделением
реакционной массы. Если реакционную смесь разделяют методом
ректификации и при этом наблюдается значительное осмоление
адиподинитрила, удельный расход сырья ставится в зависимость
от содержания адиподинитрила в реакционной массе и с точки
зрения экономики значения а<?С1,0 нежелательны.
Сравнительная оценка производительности реакционного ап-
парата при проведении процесса в диффузионной или в кинети-
ческой областях дает все основания для предпочтения последнего
режима и выбора реакционного аппарата с максимально интен-
сивным перемешиванием реакционной массы.
Такой метод обязывает к созданию условий для внешнекине-
тического режима процесса и, в частности, приводит <к необходи-
мости выбора оптимального размера частиц катализатора (при
28*
435
излишне высокой ^степени дисперсности катализатора возможен
переход в диффузионную область).
При использовании реакционной аппаратуры с интенсивным
перемешиванием реакционной массы для достижения высокой сте-
пени превращения исходного вещества возникает вопрос о произ-
водительности реакционного агрегата. Сравнительная оценка про-
изводительности реакционных аппаратов полного вытеснения, пол-
ного смешения и периодического действия при а-> 1 • свидетель-
ствует о преимуществе аппаратов первой группы, а в рассматри-
ваемом случае — о необходимости использования каскада аппара-
тов с мешалками.
РЕАКТОРЫ ДЛЯ ФОТОХИМИЧЕСКИХ РЕАКЦИИ
Впервые фотохимическая реакция в промышленном мас-
штабе была осуществлена для сульфохлорирования парафиновых
углеводородов. Сейчас известны фотохимические процессы суль-
фоокисления, хлорирования и нитрозирования парафинов и других
веществ, освоенные промышленностью. Внедрение в промышлен-
ную практику фотохимических процессов, имеющих часто неоспо-
римые преимущества перед термическими реакциями, сдержи-
вается в основном отсутствием достаточно мощных источников
излучения. Преимущество фотохимического инициирования ре-
акции, при котором концентрация активных частиц и скорость
реакции не зависят от температуры, а определяются только ин-
тенсивностью излучения, заключается в том, что при фотохими-
ческом процессе в значительной степени предотвращаются
различные побочные реакции, протекающие при повышенной тем-
пературе. Фотохимическое инициирование цепных реакций устра-
няет период индукции.
К недостаткам фотохимических процессов следует отнести
ббльшие, по сравнению с процессами, основанными на термиче-
ском или химическом инициировании, капитальные затраты и эк-
сплуатационные расходы.
При выборе метода инициирования реакции (способа актива-
ции) необходимо наряду с химическими факторами, такими как
селективность процесса, учитывать энергетические затраты. Так,
при фотохимическом инициировании энергия непрерывно подво-
дится к реагирующей системе в течение всего процесса, т. е. ко-
личество затрачиваемой энергии возрастает с увеличением сте-
пени превращения сырья, в то время как при термическом или
химическом инициировании энергия (тепловая или химическая,
измеряемая количеством инициатора) вводится в систему однаж-
ды. Таким образом, энергия, затрачиваемая при фотохимическом
инициировании, начиная от некоторого значения степени превра-
щения, может быть больше затрат энергии, которые были бы
возможны при термическом (химическом) инициировании. Рас-
смотрим этот вопрос более подробно.
436
Пусть слой вещества толщиной I с сечением, равным единице,
облучается световым источником, мощность которого 2V, в течение
времени т и при этом в слое протекает реакция:
А2 + fry 2А • А • + В —-* продукт
Концентрация вещества А2 поддерживается постоянной, кон-
центрация вещества В убывает со временем. Тогда затраты энер-
гии на облучение в расчете на единицу продукта составят;
Nx
I [В]н сфкв
где а — степень превращения вещества В к моменту времени т;
[В]н—начальная концентрация вещества В; ркв — квантовый вы-
ход.
Затраты энергии при тепловом инициировании этой же реак-
ции в том же слое вещества равны количеству тепла, сообщае-
мому исходному веществу для того, чтобы нагреть его до темпе-
ратуры, при которой концентрация активных частиц (А-) равна
этой концентрации при фотохимическом инициировании. Очевид-
но, что в расчет должно приниматься только то количество тепла,
которое необходимо для первоначального нагревания реакционной
массы. Количество тепла, выделяющееся (поглощающееся) при
реакции, одинаково при обоих способах проведения процесса.
Концентрацию активных частиц можно определить из равен-
ства скоростей инициирования и дезактивации. Если дезактива-
ция протекает за счет рекомбинации активных частиц, их концен-
трация равна ___
|А-1-/%
где А/ — скорость инициирования, равная /о// при полном погло-
щении света; /о—максимальная интенсивность светового потока
в слое; k\ — константа скорости рекомбинации. Искомая темпера-
тура определяется из значения константы равновесия для про-
цесса:
А2 2А •
Если Q — тепловой эффект реакции, равный при диссоциации
молекулы А2 энергии связи А—А, то константа равновесия может
быть представлена в виде:
О
к, — fA* Р zo . . ~ RT
[А2] lkt [А2] • •
Отсюда
у __________________
1-^0.1П----------
Q [А2]
Здесь Ко—константа равновесия при температуре То, при которой
осуществляется фотохимическое инициирование.
437
Количество тепла, необходимого для нагревания 1 г реакци-
онной массы до температуры, при которой скорость термического
инициирования равна скорости фотохимического инициирования,
можно представить выражением
\ Q IK'fa [А2] /
где С — массовая теплоемкость.
Таким образом, условие экономичности (по расходу энергии)
фотохимического инициирования по сравнению с термическим при-
нимает вид:
Nx т /________________1_______\
/[В]наркв^ 0 L 1д /р
\ Q Wo% [А2] )
Стоящая под знаком логарифма интенсивность светового по-
тока может быть выражена через мощность лампы. Если коэф-
фициент полезного действия светильника, т. е. отношение коли-
чества энергии света (активного по отношению к данной химиче-
ской реакции), поставляемое светильником в зону реакции, ко
всей затрачиваемой им энергии обозначить через цСв, а рабочую
поверхность светильника через 5СВ, то
, Л^св
1 о — “о-
ось
Учитывая, что время реакции, определяемое кинетикой процес-
са, есть некоторая функция степени превращения сырья и заменяя
/о предыдущим выражением, для условия экономичности фотохи-
мического инициирования окончательно имеем:
ЛГ • т (с) /___________1___________\
I [В]н а₽кв ° j-*!» 1П ^св
\ Q [А2] /
Как следует из полученного выражения для данного светиль-
ника, правая часть равенства является постоянной величиной,
левая же зависит от достигаемой при реакции степени превраще-
ния исходного сырья. Следовательно, степень превращения, на-
ряду с указанными выше факторами, может также определять це-
лесообразность выбора того или иного способа инициирования.
Поскольку при осуществлении фотохимического процесса за-
трачивается световая энергия, целесообразно учитывать степень
использования света, определяемую как отношение поглощенной
световой энергии ко всей затраченной энергии света, т. е.
aCB = -^-«l-e-acZ (V-32)
'о
и квантовый выход продуктов реакции, определяемый как отно-
шение числа молей образовавшегося продукта реакции к количе-
438
ству поглощенной световой энергии или как отношение скорости
образования продуктов реакции к скорости активации (см. стр. 102):
wnp vk2 Г-. / 4k3wa
₽кв = —(fei-«fe2) У И--7Т--------yp-l (V-33)
Wa 2йз^а г \К\’—Пк2Г
Практический интерес, например, для конструирования аппа-
рата, выбора оптимальных условий реакции, представляет выяс-
нение зависимости квантового выхода продуктов реакции и ско-
рости их образования от степени использования света.
Поскольку Ясв зависит от концентрации с поглощающего реа-
гента и толщины поглощающего слоя, рассмотрим два случая,
когда концентрация постоянна и асв зависит только от /, и
наоборот, когда толщина слоя постоянна и меняется только
концентрация.
ЗАВИСИМОСТЬ СКОРОСТИ РЕАКЦИИ
ОТ СТЕПЕНИ ИСПОЛЬЗОВАНИЯ СВЕТА
Освещенность данного сечения слоя убывает при уда-
лении его от источника света. Следовательно, средняя освещен-
ность всего слоя убывает с увеличением его толщины. Если в еди-
нице объема количество мо-
лекул, поглощающих свет,
остается постоянным, ско-
рость активации последних
должна при этом, очевидно,
также убывать. Если же в
некотором слое увеличивать
концентрацию поглощающе-
го свет вещества, то будет
возрастать и количество по-
глощаемого света, т. е. в
данном слое число актив-
ных частиц, возникающих
в единицу времени, будет
увеличиваться и скорость
активации повысится.
Степень использования
Рис. V-38. Зависимость безразмерной ско-
рости активации от степени использования
света.
света возрастает с увеличе-
нием толщины слоя и концентрации поглощающего вещества.
Скорость образования продуктов реакции тем больше, чем боль-
ше скорость активации. Следовательно, скорость образования
продуктов реакции должна убывать с увеличением степени ис-
пользования света при c = const, и наоборот, должна возрастать
с повышением асв при / = const.
Действительно, скорость активации при с—const можно пред-
ставить в виде
1 — е~ас1
ша = аС/0--~ —
439
или
“'а = то/оьГ!Т£5Т^ (V-33)
In (1 — aCB)
Из уравнения (V-33) следует, что при аСв = 1 величина wa = 0.
Значение toa при асв—>-0 можно получить, переходя к пределу и
применяя правило Лопиталя:
lim w0 = crc/0
“св"*0
Во втором случае, когда / = const
/о Zl „~Ос1\ Л> _
ОУа--~ ^св
т. е. величина wa пропорциональна степени использования света.
Безразмерная скорость активации (фс = при с = const и
<рг = -“/ при Z=const), т. е. скорость активации, отнесенная к
*0
своему максимальному значению, в зависимости от степени ис-
пользования света представлена на рис. V-38.
ЗАВИСИМОСТЬ КВАНТОВОГО ВЫХОДА
ОТ СТЕПЕНИ ИСПОЛЬЗОВАНИЯ СВЕТА
Квантовый выход, как следует из его определения, при
условии стационарности (П-82) концентрации активных частиц,
может быть представлен в следующем виде:
ПУпО
Ркв — ~(ПРИ
И’пр + ^да
Wnp .
РкВ = ~ (при £ч = 1)
“’да
Ркв = ™<при
ПУ да — ^пр
где о>да — скорость дезактивации активных частиц. Скорость об-
разования продуктов реакции (Юцр) пропорциональна концентра-
ции активных частиц. Скорость дезактивации активных частиц
пропорциональна либо первой степени, либо квадрату их концен-
трации. Концентрация же активных частиц, очевидно, тем боль-
ше, чем больше скорость активации. Поэтому квантовый выход
либо не должен зависеть от скорости активации, либо должен
убывать с возрастанием последней.
Скорость активации, как было показано выше, убывает с уве-
личением степени использования света при c = const и возрастает
с увеличением асв при Z=const. Следовательно, квантовый выход
при увеличении асв должен возрастать при c=const и убывать
при I—const.
440
Максимальное значение квантового выхода может быть полу-
чено из уравнения (V-33) при переходе к пределу при w& -> О
о макс ___________
Ркв kt-(Z4-l)k2
Из полученного выражения следует, что при е<1 величина
Р”вКС —порядка единицы или меньше ее, поскольку стехиомет-
рический коэффициент реакции v обычно не превосходит 2—3.
При g4> 1 квантовый выход может при-
нимать любые значения, определяемые
отношением констант скоростей химиче-
ской реакции и дезактивации.
Зависимость квантового выхода от
скорости активации удобнее представить
в безразмерном виде. Для этого отнесем
квантовый выход- к его максимальному
значению. После такой замены уравне-
ние (V-32), связывающее квантовый вы-
ход со скоростью активации,, примет наи-
более простой вид:
Рис. V-39. Зависимость кван-
тового выхода от скорости
активации.
Здесь | ;
м ъ амакс *
“кв
на рис. V-39.
_ 2fe3te?a
(fel ~ nk$
Зависимость g(<p) представлена
Зависимость безразмерной скорости активации от степени ис-
пользования света вместе с зависимостью безразмерного кванто-
вого выхода от величины ф позволяют выбирать в каждом кон-
кретном случае оптимальные условия осуществления процесса.
КОНСТРУКТИВНОЕ ОФОРМЛЕНИЕ
ФОТОХИМИЧЕСКИХ РЕАКТОРОВ
Фотохимические процессы обычно применяют для реак-
ций, протекающих в растворе. Чаще всего один из реагентов на-
ходится в газообразном состоянии. Обычно этот же реагент ак-
тивируется светом. В реактор вводится либо раствор газа, пред-
варительно приготовленный в сатураторе, либо отдельно жидкость
и газ, так что растворение газа сопровождается одновременно
протекающей химической реакцией. Иногда оба способа ввода
реагентов совмещаются. Предварительное насыщение газом це-
лесообразно в том случае, если газ достаточно хорошо растворим
в жидком реагенте. При низкой растворимости газа очень важно,
чтобы поверхность раздела газ — жидкость была как можно боль-
шей, а в жидкости, непосредственно соприкасающейся с колбой
светильника, поддерживалась достаточная концентрация раство-
ренного газа. Наилучшие выходы можно получить тогда, когда
441
поднимающиеся в жидкости пузырьки газа будут проходить как
можно ближе к стенке вертикального светильника. Последнее
легко осуществить, установив под каждым из светильников со-
осно с ним кольцевой барботер, диаметр которого совпадает с
диаметром колбы светильника.
Фотохимические реакции большей частью проводятся при тем-
пературе, близкой к комнатной. Выделяющееся при реакции тепло
отводится через развитые теплообменные поверхности, размещае-
мые как внутри реактора, так и вне его (выносной холодильник).
Использование различного рода змеевиков, трубок Фильда и дру-
гих подобных устройств менее целесообразно, так как они затруд-
няют создание равномерной освещенности реакционного простран-
ства. Применение выносных холодильников позволяет одновременно
осуществлять достаточно интенсивное перемешивание реакционной
массы в реакторе за счет ее циркуляции.
Если продукты реакции нерастворимы в исходном сырье, то
накопление их в реакционной зоне нежелательно и для удаления
этих продуктов реакционная масса непрерывно циркулирует че-
рез отстойник или какое-либо сепарирующее устройство [82].
Фотохимические реакторы обычно оформляют в виде верти-
кально установленных колонн или горизонтально расположенных
аппаратов. Для предотвращения коррозии аппараты футеруют
пластиком и цветными металлами. В некоторых случаях внутрен-
ние части аппарата покрывают бакелитовым лаком или эмалью.
Последнее менее целесообразно вследствие сложности устранения
дефектов и повреждений антикоррозионного слоя.
Светильники обычно равномерно распределяют по всей длине
аппарата, вводя их через предусматриваемые для этой цели люки.
Светильники представляют собой колбы из стекла прозрачного
для активных в данной реакции лучей. В колбы помещают источ-
ники излучения. Для охлаждения источников излучения через кол-
бу циркулирует хладоагент. При этом с охлаждающей жидкостью
отводится более 80% энергии, потребляемой лампой. В некоторых
случаях в качестве хладоагента применяют специальные рас-
творы, используемые одновременно и как светофильтры. В верти-
кальных аппаратах светильники располагают горизонтально, рав-
номерно по всей высоте аппарата. Иногда против каждого све-
тильника предусматривают смотровой люк для возможности
наблюдения за работой ламп. В горизонтально расположенные ап-
параты светильники вводят через верхнюю крышку аппарата, что
-значительно облегчает обслуживание ламп. Это обстоятельство
весьма существенно для процессов с низким квантовым выходом,
так как реакторы для таких процессов снабжаются по необходи-
мости большим количеством светильников. На рис. V-40 показано
крепление светильника на крышке промышленного реактора для
фотонитрозирования циклогексана. Такие светильники располага-
ются на крышке в три ряда и жестко укрепляются на металличе-
ских фланцах, Конструкция реактора обеспечивает удобный до-
442
ступ к каждому из 24 светильников, что позволяет быстро устра-
нять различного рода неполадки, неизбежно возникающие при эк-
сплуатации подобного рода аппаратов.
В качестве источников излучения применяют в зависимости от
требуемых длин волн лампы ртутные, накаливания или люминес-
центные. Эффективность источника излучения определяется глав-
ным образом спектральным составом излучения и общей мощ-
ностью источника. Источники излучения подбираются таким об-
разом, чтобы максимум излучаемой энергии приходился на
длины волн, при которых спектральная чувствительность реакции
Рис. V-40. Крепление светильника на крышке про-
мышленного реактора для фотонитрозирования
циклогексана н люк для наблюдения за работой
светильника.
максимальна. При этом достигается минимальный расход элек-
троэнергии на единицу продукта.
Так расход электроэнергии при фотохимическом нитрозирова-
нии циклогексана хлористым нитрозилом [83] с использованием
ламп накаливания составляет 17—21 квт-ч на 1 кг циклогекса-
ноноксима. При использовании люминесцентных ламп с люмино-
фором MgWO4 расход электроэнергии равен только 6—9 квт-ч
на 1 кг циклогексаноноксима. Такое различие в расходе электро-
энергии связано со спектральной чувствительностью реакции, име-
ющей максимум при Z,=520- 10~3 мкм, и спектром соответствую-
щего источника света (максимум излучения для люминесцентной
лампы 525-10~3 мкм, для лампы накаливания ~900-10"3 мкм).
На рис. V-41 спектральная чувствительность реакции [83]
<^~^> + NOCl —<^~\=NOH-HC1
сопоставлена со спектром излучения люминофора MgWO4 и лам-
пы накаливания.
443
При низком квантовом выходе, однако, более существенным
может оказаться абсолютное значение мощности источника, при-
ходящейся на излучение с необходимыми длинами волн и опреде-
Рис. V-41. Спектральная чувствительность реакции фотонитрозиро-
вания циклогексана (а), спектры излучения люминофора MgWO4 (б)
и лампа накаливании (в).
ляющей производительность реактора и особенности его конструк-
тивного оформления.
Светофильтры, используемые в светильниках, должны обеспе-
чивать отсутствие в спектре излучения, вызывающего побочные
Рис. V-42. Установка для фотохло-
рирования жидких парафинов [84]:
1 — реактор; 2 — обратный холодильник;
3 — мешалка; 4 — питание ртутной лампы;
5 —циркуляционные трубы; 6 — стеклян-
ный реакционный сосуд; 7— ртутная
лампа.
реакции и различные нежелатель-
ные явления (например, налипание
или пригорание смолы к колбе све-
тильника).
В качестве светильников, как
указывалось выше, могут исполь-
зоваться растворы, поглощающие
свет с нежелательными длинами
волн. Причем эти растворы исполь-
зуют одновременно и как хладо-
агент для отвода тепла, выделяемо-
го лампой. В некоторых случаях
можно ограничиться специальными
сортами стекла для изготовления
колбы светильника. Так, при суль-
фохлорировании парафинов исполь-
зование кварца для обеспечения
жаропрочности колбы светильника
приводит к быстрому почернению
ее внешних стенок, так как коротко-
волновое излучение (Л,<250-10-3
мкм), не поглощаемое кварцем, вы-
зывает, по-видимому, крекинг и полимеризацию продуктов реак-
ции. Последнее устраняется заменой охлаждающей воды 50%
водным раствором нитрата калия. Тот же эффект может быть до-
444
Отходящие газы
Рис. V-43. Хлоратор для получения гексахлорцикло-
гексана [85]:
/ — предохранительная мембрана; 2 — охлаждающие трубы;
3 — рубашка; 4 — светильники; 5 — уравнительная трубка; б — тер-
мопары.
стигнут при использовании для изготовления колбы тугоплавкого
стекла [84].
Конструктивное оформление фотохимических реакторов, как
следует из изложенного выше, может быть весьма разнообразным.
Например, хлорирование жидких углеводородов парафинового ряда
445
может осуществляться в реакторе, представленном на рис. V-42.
Хлор подается в аппарат через барботер, изготовленный из свин-
ца. Аппарат из освинцованного чугуна или керамики, снабжен ме-
шалкой и обратным холодильником. Углеводород насыщается хло-
ром и отводится по трубке, в которой размещен светильник с
ртутной лампой. При движении раствора по циркуляционной тру-
бе происходит фотохимическое хлорирование. Циркуляции рас-
твора способствует пропеллерная мешалка, устанавливаемая в
нижней части циркуляционной трубы. Образующийся при реак-
ции хлористый водород выводится из верхней части обратного
холодильника.
Хлорирование бензола до гексахлорциклогексана проводится в
вертикальном аппарате колонного типа (рис. V-43), освинцован-
ном изнутри для защиты от коррозии и предотвращения катали-
тического воздействия железа, способствующего реакции замеще-
ния [85]. Для облучения реакционного пространства аппарат снаб-
жен ртутнокварцевыми лампами, расположенными горизонтально
в колбах из тугоплавкого стекла. Бензол и хлор вводятся проти-
вотоком. Выделяющееся при реакции тепло (—48 ккал!моль) от-
водится через рубашку и охлаждающие элементы, размещенные
параллельно оси аппарата. На уровень жидкости подается азот
для предотвращения возможности образования взрывоопасных
концентраций хлора с парами бензола.
В качестве реакционного сосуда при сульфохлорировании уг-
леводорода парафинового ряда [84] применяются стальные колон-
ны, футерованные полихлорвиниловой смолой или покрытые ба-
келитовым лаком. Ртутные лампы помещаются в горизонтальных
защитных трубках по всей высоте реакционной колонны. Защит-
ные трубки, изготовленные из йенского стекла, являются одновре-
менно светофильтрами. Трубки вставляются в аппарат через спе-
циальные штуцеры. На каждую защитную трубку приходится не-
сколько ламп (чаще всего по 6 штук) мощность по 75 вт.
Большие агрегаты для сульфохлорирования, например, колон-
ны диаметром 2 и высотой 4 м, имеют 16 защитных трубок, в
каждой из которых 6 ламп, т. е. 96 ламп по 75 вт, общей мощ-
ностью — 7200 вт. В таких агрегатах за один прием перераба-
тывается 10 000 л жидкости. Отвод тепла, выделяющегося при
реакции и работе ламп ( — 30 ккал! моль), осуществляют циркуля-
цией реакционной смеси через холодильник при помощи насоса.
Этим одновременно разрешается задача перемешивания реакци-
онной жидкости.
ЛИТЕРАТУРА
1. D. W. WanKrevelen, Chem. Eng. Progr., 46, 29 (1950).
2. Д. А. Франк-Каменецкий, Труды НИИ-1 НКАП, № 7. 1946.
3. Т. Bryn, Forschung auf dem Gebiete des Ingenierwesens, 4, № 1, 27
(1933).
4. А. В. Городецкая, ЖФХ, 23, № 1, 71 (1949).
5. F. H. Garner, D. Hammerton, Chem. Eng. Sci., 3, 1 (1954).
446
6. К. Н. Шабалин, С. Ф. Крылов, В. И. Оборин, Хим. пром., 16,
№ 10 (1939).
7. С. С. Кутателадзе, М. А. Стыркович, Гидравлика газо-жид-
костных систем, Госэнергоиздат, 1958.
8. М. Б. Айзен бу д, В. В. Д ильм ан, Хим. пром., № 3, 199 (1961).
9. Т. А. Ж и л я е в а, Автореф. канд. дисс., ГИАП, 1965.
10. 3. Ш т е р б а ч е к, П. Т а у с к, Перемешивание в химической промыш-
ленности, Госхимиздат, 1963.
11. В. Barnabas, U. Au г el, Экспресс-информ., № 8, реф. 39 (1964).
12. А. Г. Касаткин, Основные процессы и аппараты химической техно-
логии, Госхимиздат, 1960.
13. В. В. Каф а ров, Перемешивание в жидких средах, Госхимиздат, 1961.
14. N. Н. Parker, Chem. Eng., 71, № 12, 165 (1964).
15. S. Scarlett, Chem. a. Process Eng., 45, № 11, 612 (1964).
16. J. Y. О 1 d s h u e, Ind. a. Eng. Chem., 58, № 11, 50 (1966).
17. losida Fumitake, Miura loshikary, Ind. Eng. Chem. Process
Desing a. Developm., 2, № 4, 263 (1963).
18. J. Hahnhart, H, Kramers, K. R. Westerterp, Chem. Eng. Sci.,
18, № 8, 503 (1963).
19. H. E. Вишневский, H. П. Г л у x а н о в, И. С. Ковалев, Аппа-
ратура высокого давления с герметическим приводом, Машгиз, 1960.
20. Отраслевая нормаль НИИХИММАШа ОН26-01-9-65. Реакторы с гермети-
ческим приводом, Изд. «Машиностроение», 1965.
21. В. И. Соколов, А. Д. С а л а м а х и н, ЖПХ, 35, № 5, 1022 (1962).
22. К. Н. Шабалин, Н. Г. Бляхер, Хим. пром., № 11, 141 (1945).
23. А. Г. Касаткин, Д. М. Попов, Ю. В. Аксельрод, Хим. пром.,
№ 7, 662 (1959).
24. М. Е. Позин, И. П. Мух л ено в, К. С. Тумаркина, 3. Я. Т а-
р а т, Пенный способ обработки газов и жидкостей, Госхимиздат, 1955.
25. Г. К- Боресков, М. Г. Слинько, Хим. пром., № 3, 193 (1960).
26. И. И. Иоффе, Л. М. Письме н, Инженерная химия гетерогенного
катализа. Изд. «Химия», 1965.
27. Г. К. Боресков, Кинетика и катализ, 3, № 4, 470 (1962).
28. М. И. Тем к и н, Кинетика и катализ, 3, № 4, 509 (1962).
29. Ю. Н. Тюрин, Проблемы оптимального планирования проектирования
и управления производством, Изд. МГУ, 1963, стр. 152.
30. И. Е. И д е л ь ч и к, Справочник по гидравлическим сопротивлениям, Гос-
энергоиздат, 1960.
31. Chem. Eng., 72, № 22, 81 (1965).
32. Chem. Week, 98, № 9, 97 (1966).
33. Г. К. Боресков, Катализ в производстве серной кислоты, Госхим-
издат, 1954.
34. М. Г. Слинько, В. С, Бесков, Хим. пром., № 12, 826 (1961),
35. В. С. Б е с к о в и др., Хим. пром., № 10, 721 (1963).
36. Р. Арис, Оптимальное проектирование химических реакторов, ИЛ, 1963.
37. В. В. Фальковский, Автореф. докт. дисс., МФХИ им. Карпова 1961.
38. В. Л. Волков, ЖХП, № 4—5 (1950).
39. И. П. Сидоров, Труды ГИАП, т. VI, 1956.
40. Ю. А. Соколинский, Труды МИХМ, т. XXVI, 1964, стр. 161.
41. Г. К. Боресков, М. Г. Слинько, Хим. пром., № 6, 321 (1957).
42. Б. И. Бондаренко, Установки катали! ического крекинга, Гостоптех-
издат, 1959.
43. Я. Б е р а н е к, Д. Сокол, Техника псевдоожижения, Гостоптехиздат,
1962.
44. Н. М. Боцеловский, Т. X. Мелик-Ахназаров, Псевдоожижение
в химической технологии, ГОСИНТИ, 1960.
45. А. Н. П л а и о в с к и й, Д. И. О р о ч к о, П. И. Лукьянов, Процессы
в кипящем слое, ВИНИТИ, 1959.
46. Н. И. Сыромятников, В. Ф. Волков, Процессы в кипящем слое,
Госметаллургиздат, 1959.
447
47. М. Лева, Псевдоожижение, Гостоптехиздат, 1961.
48. В. Д. Гор'шков, Р. Б. Розенбаум, О. М. Тодес, Изв. высш. шк.
Сер. «Нефть и газ», К» 1 (1958).
49. П. Г. Р о м а н к о в, Н. Б. Р а ш к о в с к а я, А. Д. Г о л ь ц и к е р, ЖПХ,
37, № 3, 615 (1964).
50. М. И. Фридланд, А. И. Скобл о, Хим. пром., Xs 5 (1960).
51. Е. А. Казакова, Р. 3. Хит ер ер, Хим. пром., Xs И, 798 (1962).
52. И. Г. Мартюшин, Труды МИХМ, 13, 1957, стр. 145.
53. П. С о л ч а н и, Хим. пром., Xs 4, 267 (1961).
54. Сб. «Процессы в кипящем слое», Гостоптехиздат, 1958, стр. 173.
55. К П. Лавровский, А. А. Розенталь, Хим. и технол. топлива,
№ 3 (1956).
56. В. С. Альтшулер, Г. П. Сеченов, Процессы в кипящем слое под
давлением, Изд. АН СССР, 1963, стр. 80, 94.
57. В. С. Альтшулер, Г. П. Сеченов, Труды Ин-та горючих ископае-
мых АН СССР, т. 11, 1959.
58. В. Я. Кругляков, Автореф. канд. дисс., Ин-т нефти им. Губкина,
1951.
59. В. Л. Г у р в и ч, Е. В. С л ю д о в и ч, Каталитический крекинг-флюид за
рубежом, ГОСИНТИ, 1960.
60. Н. И. Сыромятников, За экономию топлива, Xs 2 (1951).
61. И. Г. М а р т ю ш и н, В. Н. Головин, Труды МИХМ, т. 26, 1964,
стр. 23.
62. А. К. Б о н д а р е в а, ДАН СССР, 115, Xs 4 (1957).
63. С. Э. Л я и д е р с, Автореф. канд. дисс., МИХМ, 1966.
64. Д. И. Орочко, П. X. М е л и х - А х н а з а р о й, Г. Н. Полубояр и-
нов, Хим. и техиол. топлив и масел, X» 12 (1957).
65. А. Я. Авербух, А. И. В и т в и ц к и й, И. П. М у х л е н о в, Применение
кипящего слоя в химической промышленности, ч. I, Изд. «Знание», 1965.
66. Л. А. Акопян, Хим. пром., Xs 5, 94 (1955).
67. Хим. пром, за рубежом, 7, 20 (1964).
68. П. У. Ш е р в у д, Инженер-нефтяник, 34, Xs 10, 50 (1962).
69. Ю. А. Андрианов, С. А. Голубков и др., ЖПХ, 32, X» 1, 20
(1959).
70. Нефтехим. синтез за рубежом, Xs 10, 23 (1963).
71. А. С. С у щен ко, Применение кипящего слоя в химической промышлен-
ности, ч. I, изд. общества «Знание», 1965.
72. В. А. С е б а л л о, Н. Б. Р а ш к о в с к а я, П. Г. Р о м а н к о в, Там же,
стр. 82.
73. В. Е. Бабенко, П. Г. Р о м а и к о в, Н. Б. Р а ш к о в с к а я, Там же,
стр. 73.
74. Н. И. Сыромятников, Л. К- Басакова, Ю. Н. Шиманский,
Тепло- и массообмен в кипящем слое, Изд. «Химия», 1967, стр. 136.
75. В. А. Членов, Н. В. Михайлов, Хим. пром., Xs 12, 910 (1964);
ДАН СССР, 154, Xs 3, 703 (1964) .
76. Д. Д. Барки н, Виброметод в строительстве, Госстройиздат, 1959.
77. И. Ф. Г о н ч а р е в н ч, П. Д. Сергеев, Вибрационные машины в строи-
тельстве, Машгиз, 1963.
78. А. М. Л а с т о в ц е в, А. М. X в а л ь н о в, Ю. И. Макаров, Хим. пром.,
Xs 11, 815 (1962).
79. А. М. Ластовцев, А. М. Хвальнов, Труды МИХМ, т. 19, 1959,
стр. 125.
80. С. 3. В а н ю ш и и а, М. С. В и л е с о в а, Г. А. Чистякова, Хим.
пром., Xs 4, 205 (1958).
81. Д. А. Франк-Каменецкий, Диффузия и теплопередача в хими-
ческой кинетике, Изд. «Наука», 1967.
82. К. С. В у л ь ф с о н, ЖФХ, 39, Xs 6, 1531 (1965).
83. А. А. Артемьев, А. А. Стрельцова, Е. В. Генкина,
К. С. Вульфсон, Хим. наука и пром., 3, Xs 5, 629 (1958).
448
84. Ф. Аз ин гер, Химия и технология парафиновых углеводородов, Гос-
топтехиздат, 1959.
85. И. И. Юкельсон, Технология основного органического синтеза, Гос-
химиздат, 1958, стр. 240.
Аз бель Д. С., Гидродинамика барботажных процессов, Хим. пром., № 11
(1962).
А з б е л ь Д. С., Хим. пром., № 9, 49 (1964).
Аэро в М. Э., Тодес О. М., Гидравлические и тепловые основы работы
аппаратов со стационарным и кипящим зерновым слоем, Изд. «Химия», 1968.
Б а т у н е р Л. М., Процессы и аппараты органического синтеза и биохими-
ческой технологии, Изд. «Химия», 1966.
Бесков С. Д., Технохимические расчеты, Изд. «Высшая школа», 1966.
В э й л а с С., Химическая кинетика и расчеты промышленных реакторов,
Изд. «Химия», 1967.
Г е л ь п е р и н Н. И., А й н ш т е й н В. Г., Кваша В. Б., Основы техники
псевдоожижения, Изд. «Химия», 1967.
За броде кий С. С., Гидродинамика и теплообмен в псевдоожиженном
слое, Госэнергоиздат, 1963.
Разу.мо и И. М., Псевдоожижение и пневмотранспорт сыпучих материалов,
Изд. «Химия», 1964.
Рейхс фельд В. О., Ер ко в а Л. Н., Оборудование производств основ-
ного органического синтеза и синтетических каучуков, Изд. «Химия», 1965.
Применение кипящего слоя в народном хозяйстве СССР. Материалы Все-
союзного семинара, проведенного в сентябре — октябре 1963 г. на ВДНХ, изд.
ЦНИИ информ, и техн, экономия, иссл. цв. мет., 1965.
К б 1 b е 1 Н., Erdole-Z., 80, № 10 (1964).
Kolbel Н., Langemann Н., Chem. Techn., 13, № 7—8, 394 (1961),
Kolbel H., Siemes W., Chem. Umschau, Fette, 37, № 24, 746 (1957).
S m 11 h J. M., Chemical Engineering Kinetics, New York, 1956.
Van Krevelen D. W., Hoftyzer P, J., Chem. Eng. Sci., 2, № 4
(1953).
29 В. С. Хайлов, Б, Б. Брандт
Глава VI
РАЗДЕЛЕНИЕ И ОЧИСТКА ПРОДУКТОВ
ПРОМЫШЛЕННОГО ОРГАНИЧЕСКОГО
СИНТЕЗА
Заключительный этап переработки сырья в химиче-
ском производстве и на каждой из стадий техноло-
гической схемы связан, как правило, с разделением сложных сме-
сей разнообразных продуктов, образующихся в результате хими-
ческого превращения исходных веществ, т. е. с выделением
непревращенного сырья, разделением продуктов реакции, выделе-
нием и очисткой целевого продукта производства. Все эти опера-
ции в большинстве случаев требуют сложного аппаратурного
оформления, сопровождаются значительным расходом энергетиче-
ских средств и потерей сырья, промежуточных и основного про-
дуктов.
Методы и аппаратура, применяемые для разделения сложных
смесей, которые получаются в условиях производств основного ор-
ганического синтеза, чрезвычайно разнообразны и для решения
технологических задач часто используется комплекс различных
приемов и методов. Общие рекомендации по этому вопросу вряд
ли возможны, так как выбор способа и аппаратурного оформле-
ния отдельных процессов разделения определяется фазовым со-
ставом и состоянием разделяемой смеси, свойствами разделяемых
компонентов и масштабами производства.
Обширная отечественная и зарубежная литература, относя-
щаяся к процессам и аппаратам химической технологии, в основ-
ной своей части посвящена вопросам разделения. Она освещает
общие закономерности процессов разделения гетерогенных и го-
могенных систем и принципы аппаратурного оформления этих
процессов [1—5].
Для решения задач технологии основного органического син-
теза при использовании общих принципов осуществления процес-
сов разделения необходим учет специфических свойств разделяе-
мых продуктов, что в ряде случаев вносит существенные коррек-
тивы в условия проведения и аппаратурное оформление процессов.
Расширение номенклатуры продуктов промышленного органи-
ческого синтеза и характерное для последнего времени повышение
450
требований к их чистоте вызывают необходимость привлечения
ряда новых приемов и методов разделения и очистки продуктов.
В настоящей работе нет необходимости излагать общие прин-
ципы и закономерности процессов разделения гетерогенных и го-
могенных систем и подробно останавливаться на общеизвестных
методах и аппаратуре, применяемых для разделения. Учитывая,
однако, роль этой стадии в технологии и экономике химического
производства, ниже кратко рассматриваются аппаратура и ме-
тоды, применяемые и могущие найти применение в технологии ос-
новного органического синтеза.
На первой стадии переработки многофазной реакционной мас-
сы обычно производится разделение фаз. Отделенные одна от
другой фазы при необходимости подвергаются обработке для
разделения каждой из них на составляющие компоненты или сме-
си этих компонентов заданного состава.
Вопрос, связанный с выбором критерия эффективности и па-
раметров оптимизации промышленных процессов разделения, весь-
ма сложен [6]. В зависимости от состава и свойств разделяемой
смеси, методов и целей процесса разделения, а также в зависимо-
сти от относительной стоимости сырья, энергетических средств и
целевого продукта меняется комплекс критериев эффективности,
применяемых для оценки процесса. Известны технологические,
термодинамические, кинетические, статические, аппаратурные и
экономические критерии эффективности [6]. Наиболее полная и
универсальная характеристика процесса разделения дается эко-
номическими критериями.
Учитывая особенность структуры экономических показателей
эффективности процессов промышленного органического синтеза,
можно утверждать, что основная роль при оценке процессов раз-
деления продуктов органического синтеза принадлежит техноло-
гическим, термодинамическим и, в конечном счете, экономическим
критериям.
Расход сырья на стадии разделения характеризуется отноше-
нием количества продукта, полученного в результате разделения
реакционной смеси, к его количеству, содержавшемуся в смеси,
поступающей на разделение (Рг, з)—см. гл. I. В общем случае
значение Рт, з выражает суммарный эффект, складывающийся из
прямых потерь продукта i (испарение, утечка, нежелательные хи-
мические превращения и т. д.) и «потерь» этого продукта в ре-
зультате распределения его между основной (целевой) и побоч-
ными фракциями. Чаще всего продукт i, содержащийся в побоч-
ных фракциях, утрачивается (кубовые остатки, хвостовые газы
и т. д.).
Рассмотрим простейший случай разделения реакционной сме-
си, состоящей из двух компонентов — непревращенного сырья и
продукта реакции. Пусть смесь, поступающая на разделение в ко-
личестве G (в кг^н), содержит целевой продукт (например, не-
превращенно? сырье) в количестве Хо (в долях единицы) и
29*
451
подлежит разделению на фракции Pi и Р2 (в кг/ч) состава Xi и
Хг, соответственно (по целевому продукту).
При отсутствии прямых потерь баланс целевого продукта мож-
но выразить следующим образом:
GX0 = P1XI + (G-P1)X2
Отсюда
Р1 Хр — Х2
G = Xt-X2
Выход продукта i на стадии разделения реакционной массы
будет равен
а _ Рi-^i _ -^i (*°
PZ'3 GXp Х0(Х,-Х2)
Поскольку состав разделяемой смеси зависит от степени пре-
вращения исходных веществ, выход продукта i на стадии разде-
ления является функцией не только состава фракций, но и сте-
пени превращения сырьр:
_ %! [Хр(а)-Х2]
pz’3
Для заданного состава фракций (Х), Х2) выход целевого про-
дукта существенно повышается при увеличении его концентрации
в исходной смеси (Хо). При заданной степени превращения сырья
(Хо = const) выход целевого продукта. повышается при снижении
его концентрации в обедненной фракции (Х2) тем значительнее,
чем ниже концентрация целевого продукта в исходной смеси. По-
вышение концентрации целевого продукта в обогащенной смеси
(Xi) сопровождается снижением выхода этого продукта. Напри-
мер, при ректификационном разделении смеси следует стремиться
к максимальному обеднению кубового остатка целевым продук-
том, не повышая без необходимости концентрацию его в дистил-
ляте. Для сохранения выхода целевого продукта при повышении
его концентрации в дистилляте необходимо соответственно сни-
жать его концентрацию в кубовом остатке.
Термодинамические критерии эффективности используются для
характеристики процессов разделения газов, смешивающихся
жидкостей и изотопов по удельному расходу энергетических
средств на единицу целевого продукта путем сравнения его с ми*
нимально необходимой затратой энергии (t/):
— Т Д5ра'зд
где Т — температура; АЗразд—изменение энтропии при разделении.
Как было сказано, наиболее полная и универсальная оценка
процесса разделения основана на учете энерго-материальных за-
трат, отнесенных к единице целевого продукта и удельных капи-
тальных затрат. Так как размер удельных энерго-материальных
затрат складывается из стоимости сырья (потери сырья или про*
453
дукта, выраженного в расходе сырья), энергетических средств и
амортизационных отчислений
Ф = Л + Q + W
повышение степени извлечения целевого продукта сопровождает-
ся уменьшением удельного расхода сырья, но требует большего
расхода энергетических средств и, как правило, более совершен-
ной и сложной аппаратуры. Для удельных энерго-материальных
затрат как функции степени извлечения продукта существует ми-
нимум. Аналогичный характер имеет зависимость показателя при-
веденных затрат от степени извлечения целевого продукта из ре-
акционной смеси.
РАЗДЕЛЕНИЕ ГЕТЕРОГЕННЫХ СИСТЕМ
В зависимости от числа и состояния фаз при разделе-
нии многофазных систем используются различные методы и аппа-
ратура. Двухфазные системы наиболее часто встречающиеся в
практических условиях промышленного органического синтеза, мо-
гут быть представлены четырьмя группами смесей: жидкость—газ,
жидкость—жидкость, жидкость—твердое тело и твердое тело—газ.
СИСТЕМЫ ЖИДКОСТЬ — ГАЗ
Необходимость разделения систем жидкость — газ воз-
никает в технологии основного органического синтеза в самых
разнообразных случаях. Методы и аппаратура, применяемые для
разделения, зависят от структуры этой системы и свойств фаз.
Различают две основные группы структур системы:
1) газ диспергирован в жидкости (г./ж.);
2) жидкость диспергирована в газе (ж./г.).
К первой из этих групп относятся дисперсные системы, кото-
рые образованы множеством пузырьков газа или пара, разделен-
ных тонкими пленками жидкости (пены). Чаще всего дисперсной
фазой является газ (так называемые «влажные» пены).
Пены образуются при барботаже газа через слой жидкости,
при перемешивании жидкости и газа мешалкой и при десорбции
газа из слоя жидкости.
Устойчивость пен, т. е. время самопроизвольного разрушения
столба пены на половину его начальной высоты, и наибольшая до-
стижимая высота столба пены зависят от содержания в жидкости
поверхностно-активных веществ (мыла, спирты, жирные кислоты,
высокомолекулярные вещества, фенолы, белки и т. д.), облегчаю-
щих диспергирование газа в жидкости и повышающих устойчи-
вость пленок жидкости между пузырьками газа. Чистые жидко-
сти не образуют пен. Устойчивость пен в присутствии поверхност-
но-активных веществ весьма велика и достигает многих десятков
часов. Нежелательное образование устойчивых пен часто вносит
серьезные осложнения в технологический процесс и расстраивает
работу аппаратуры технологической схемы.
Для разрушения пег. в условиях промышленного производства
применяются различные способы, которые можно разделить на
несколько основных групп:
1) применение химических пеногасителей;
2) применение теплофизических методов;
3) применение механических методов.
Разрушение пен методами первой группы основано на дейст-
вии сильных поверхностно-активных веществ, очень мало раство-
римых в жидкости и быстро растекающихся по ее поверхности в
Рис. VI-1. Уст-
ройство для
разрушения пе-
ны механиче-
ским способом.
виде мономолекулярных пленок. Вытесняя пенооб-
разующие вещества из поверхностного слоя плен-
ки, пеногасящие добавки на несколько порядков
снижают устойчивость пен.
В качестве пеногасящих добавок применяются
средние гомологи спиртов (например, октиловый
спирт), полиамиды жирных кислот, жидкие жир-
ные кислоты и их глицериды, силиконовые масла
и др. Количество пеногасящих добавок, вводимых
в жидкую фазу составляет 10~5—10-3%.
Теплофизические способы основаны на разру-
шении жидкостной пленки, окружающей пузырь-
ки газа, под действием неравномерного нагрева или
в результате внутреннего давления нагреваемого
газа.
Освещение поверхности пены инфракрасными
лучами вызывает разрушение верхнего слоя пены
и препятствует росту высоты ее столба. Анало-
гичный результат достигается расположением над верхней по-
верхностью столба пены решетки или змеевика из труб, обогре-
ваемых паром или газом. Нужный эффект может быть достигнут
при обдувании поверхности пены струей водяного пара (если это
допускается по условиям производства).
Механические способы разрушения относительно мало устой-
чивых пен основаны на применении отбойников или комбинации
отбойников и сетчатых перегородок, установленных на пути дви-
жения пены и вызывающих резкое изменение давления газа в пу-
зырьках пены (рис. VI-1).
В условиях, допускающих потерю абсорбированного газа, эф-
фективным является способ, основанный на использовании эжек-
тирующего потока газа. Увлекаемый струей газа поток пены на-
правляется на отбойник, где в результате мгновенного сжатия пу-
зырьков газа происходит дезаэрация пены [7].
Известен ряд способов [8—10] разрушения пен с помощью уль-
тразвука. При частотах, превышающих 5—6 кгц, и соответствую-
щей интенсивности ультразвукового поля происходит разрушение
даже относительно стойких пен в результате акустического дав-
454
Допустимый
уровень
подъема пены
Рис. VI-2. Устройство для раз-
рушения пены с помощью
ультразвука:
1 — генератор ультразвука; 2 — дат-
чик импульса.
ления на пузырьки газа. По расходу газа и скорости разрушения
пен самого различного типа наиболее эффективными являются
аэродинамические генераторы. Генератор ультразвука (аэро- или
гидродинамический) может работать непрерывно или автомати-
чески включаться по достижении слоем пены заданной высоты.
Устройство для разрушения пены этим методом представлено на
рис. VI-2.
К второй группе относятся туманы и капельно-жидкие системы.
Они часто образуются в промышленных условиях в результате
ряда различных по своей природе процессов, вызывают серьезные
нарушения технологического режима и сопровождаются потерей
продукта.
Туман представляет собой систему
жидкость—газ, в которой дисперсной
фазой является жидкость. Размер
частиц (капель) жидкости в тумане
колеблется в пределах до 2—3 мкм.
Образование тумана является ре-
зультатом объемной конденсации пе-
ресыщенного пара и происходит при
определенном значении степени пе-
ресыщения s [11]:
S = P/PHac (VI-1)
где Р — давление пара в системе;
Риас — давление насыщенного пара
при данной температуре.
Критическое значение s, при котором наступает конденсация
пересыщенного пара, зависит от ряда причин: 1) наличия пыли
или капель жидкости в паре или парогазовой смеси; 2) наличия
ионов газа в паре или парогазовой смеси.
В среде, свободной от присутствия пыли или капель жидкости
и не содержащей ионов газа, образование тумана может происхо-
дить лишь при высокой степени пересыщения; присутствие цент-
ров конденсации (частицы твердого или жидкого вещества) или
ионов газа снижает значение критической степени пересыщения,
приближая его к единице. Критическое пересыщение снижается
при повышении температуры.
В условиях-промышленного синтеза вероятность переработки
паровой или парогазовой системы, полностью свободной от при-
сутствия второй фазы или ионизированных молекул, крайне мала
и образование тумана может происходить при степени пересыще-
ния, близкой к единице.
Разрушение тумана, независимо от его происхождения, оказы-
вается сложной задачей, поэтому более целесообразно предупре-
ждать образование тумана, т. е. создавать условия, при которых
степень пересыщения пара в системе не достигает критического
значения.
455
Пусть состояние пересыщения наступает при конденсации
пара из паровой или парогазовой смеси при соприкосновении этих
систем с холодной поверхностью или при излучении ими тепла в
окружающую среду.
Степень пересыщения пара для первого случая описывается
простой зависимостью:
( Т-Т2 рх-р2 р2
\ Г] Т2 ) Рнас Рнас
(VI-2)
где Т, Т{ — температура
цесса; Т2 — температура
парогазовой смеси в конце и начале про-
поверхности конденсации; — давление
Температура,
охлаждающего
агента,°C
Рис. VI-З. Зависимость со-
става парогазовой смеси от
температуры охлаждающего
агента:
1 — содержание пара; 2 — содер-
жание тумана в отфильтрованном
воздухе; 3 —содержание тумана
в неотфильтрованиом воздухе.
пара в парогазовой смеси в начале про-
цесса; Р2— давление пара у поверхно-
сти конденсации; Рнас — давление насы-
щенного пара; 6?» I—I ' (D'— коэф-
фициент диффузии; а — коэффициент
температуропроводности).
Уравнение (VI-2) показывает, что с
повышением температуры поверхности
конденсации Т2 степень пересыщения па-
ра снижается и при соответствующем ее
значении (меньше критического) может
быть исключена возможность образова-
ния тумана. Эта закономерность должна
учитываться как при конденсации пара
из паровой или парогазовой смеси, так
и при «вымораживании» парообразных
продуктов из парогазовых смесей. Для
иллюстрации последнего случая может
служить рис. VI-З, на котором дана за-
висимость состава парогазовой смеси
этиловый спирт — воздух, покидающей
теплообменник, охлаждаемый агентом
с температурой Т2.
Для отфильтрованного воздуха образование тумана начинает-
ся при —33° С и дальнейшее понижение температуры поверхно-
сти охлаждения сопровождается увеличением туманообразования
(при Т2——-80° С около 70% этилового спирта находится в тума-
нообразном состоянии).
Если воздух не отфильтрован (кривая 3 на рис. VI-З) конден-
сация в объеме наступает уже при s~ 1,0, т. е. при более высокой
температуре. Понижение температуры поверхности охлаждения
также вызывает образование тумана, количество которого при со-
ответствующих температурах выше, чем для случая с отфильтро-
ванным воздухом.
С целью предупреждения образования тумана при конденса-
ции и вымораживании используют повышение температуры по*
456
верхности охлаждения (соблюдение этого условия связано с уве-
личением площади теплообмена). Вероятность образования ту-
мана может быть уменьшена при заполнении газовых каналов
холодильника насадкой, поверхность которой служит местом кон-
денсации. Рекомендуется также орошение насадки потоком хо-
лодной жидкости.
Пусть состояние пересыщения возникает при смешении паро-
газовых смесей с газами или парогазовыми смесями, имеющими
разную температуру. Величина пересыщения зависит от содержа-
ния пара в смешиваемых потоках и от их относительной темпе-
ратуры: чем больше разность температур между потоками, тем
выше возникающее при этом пересыщение. Мерой предупрежде-
ния образования тумана в этом случае может служить выравни-
вание температуры смешиваемых потоков.
Адиабатическое расширение парогазовых смесей сопровождает-
ся понижением температуры смеси и, как следствие этого, воз-
никновением пересыщенного состояния/пара. Это явление может
иметь место при дросселировании горячих жидкостей, содержа-
щих растворенные газы. Охлаждение жидкости перед дросселиро-
ванием снижает давление пара и возможность образования пере-
сыщенного состояния пара уменьшается.
Пересыщенное состояние пара может возникнуть в результате
химической реакции газообразных исходных веществ в объеме и
при механическом распылении жидкости с помощью форсунок.
Меры предупреждения образования тумана в этих случаях за-
труднительны.
Разрушение образовавшегося тумана производится с помощью
ряда методов, среди которых наиболее эффективный основан на
применении осаждения капель в электрическом поле (см. стр. 473).
Задача разделения систем газ (пар)—жидкость не ограничивает-
ся предупреждением образования или разрушения образовавшихся
туманов или пен. В выпарных аппаратах, абсорбционных и ректи-
фикационных колоннах и при десорбции газа из жидкости обра-
зуются системы, представляющие собой взвесь капель жидкости в
потоке газа. При размере капель, превышающих 3—4 мкм, отделе-
ние их от газового потока может быть осуществлено рядом меха-
нических приемов.
Расчет аппаратуры для разделения систем газ—жидкость до
настоящего времени не разработан (за исключением циклонов)
и рекомендации основаны на результатах сравнительной оценки
вариантов конструкций сепарирующих устройств. Выбору типа
сепараторов жидкости посвящены работы [12—14]. По принципу
действия сепараторы можно подразделить на три основные груп-
пы: инерционные, мелкопоточные (фильтрующие, жалюзийные) и
центробежные (рис. V-4).
Широко применяются центробежные сепараторы, эффективные
для отделения пара от капель жидкости в выпарных аппаратах.
Жалюзийные и фильтрующие отбойники жидкости, выполненные
457
из уголков или разнообразных насадок (кольца Рашига, сетки
различного типа и размера) эффективны при размере капель
>5 мкм. Для отделения более мелких капель применяются сепара-
торы-фильтры.
Сравнительная оценка ряда типов сепараторов для очистки
вторичного пара показывает [12] наибольшую эффективность цик-
лонных сепараторов.
Для сепарации жидкости из потока газа под высоким давле-
нием (до 800 ат) рекомендуется также сепаратор центробежного
Рис. VI-4. Типы сепараторов.
типа, эффективность разделения которого достигает 99,5% даже
при относительно малой разнице плотностей жидкости и сжатого
газа [14].
Сепараторы, а также фильтры, применяемые для очистки газа
от капель жидкости (мыло) в условиях высокого давления, рас-
смотрены в работах, посвященных аппаратуре и технологии свя-
занного азота [15].
СИСТЕМЫ ЖИДКОСТЬ — жидкость
Одна из наиболее часто встречающихся в практике про-
мышленного органического синтеза гетерогенных дисперсных си-
стем представляет собой смесь двух практически нерастворимых
жидкостей (эмульсии).
Число таких систем чрезвычайно велико. В большинстве слу-
чаев одной из фаз является вода либо водные растворы. Эмульсии
образуются при конденсации смеси паров взаимнонерастворимых
жидкостей, при охлаждении раствора жидкостей, обладающих ог-
458
раниченной растворимостью, при конденсации паров в конденса-
торах смешения, а также при проведении различных процессов,
связанных с перемешиванием нерастворимых жидкостей.
Известны специальные способы приготовления эмульсий (ме-
шалки, механические и ультразвуковые гомогенизаторы и т. д.).
Эмульсии характеризуются соотношением дисперсной и диспер-
сионной фаз, размером частиц дисперсной фазы и агрегативной
устойчивостью [мерой устойчивости эмульсии является средняя
скорость самопроизвольного разрушения (в сл/сек)].
Различают разбавленные эмульсии (концентрация дисперсной
фазы до 1,0%), концентрированные эмульсии (70—80%) и эмуль-
сии средней концентрации.
Устойчивость эмульсий — наиболее важная характеристика при
решении вопросов разделения — зависит от многих причин [16].
В разбавленных эмульсиях возможность коалесценции частиц
крайне мала вследствие малой вероятности столкновения частиц.
Такие эмульсии могут быть весьма устойчивы даже при высоком
значении поверхностного натяжения на межфазной границе. До-
статочно концентрированные эмульсии при прочих равных усло-
виях всегда оказываются менее устойчивыми.
Системы, характеризующиеся большим значением поверхност-
ного натяжения на границе раздела фаз, не способны к образова-
нию стойких эмульсий.
Устойчивость концентрированных эмульсий обусловлена при-
сутствием в системе эмульгаторов веществ, способных стабилизи-
ровать адсорбционную оболочку на границе раздела фаз и, тем
самым, затруднять коалесценцию капель дисперсной фазы.
Эмульгирующим действием обладают поверхностно-активные
вещества (мыла, спирты, сульфированные углеводороды, высоко-
молекулярные соединения и т. д.), а также высокодисперсные
твердые вещества (глинозем, углекислый кальций, сажа, различ-
ные сорта глин, кизельгур и др.).
Устойчивость эмульсий зависит не только от эффективности
эмульгатора, но и от его концентрации, природы дисперсной и
дисперсионной фаз, значения pH и других факторов. Результаты,
полученные при использовании эмульгатора для одной системы,
не могут быть непосредственно перенесены на другую систему без
экспериментальной проверки.
Прочность адсорбционного слоя, стабилизированного эмульга-
торами, меняется с течением времени, а устойчивость одних эмуль-
сий при хранении увеличивается, других — уменьшается.
Возникновение устойчивых эмульсий в промышленной прак-
тике может быть причиной серьезных нарушений технологиче-
ского режима работы аппаратуры и оборудования и сопровож-
даться потерями сырья и продуктов производства.
Подобно тому, как не существует общих правил для приготов-
ления различных по природе эмульсий, практически не существует
и общих рекомендаций для разрушения любого вида эмульсий.
459
Все известные методы разрушения эмульсий можно разделить
на несколько групп по методу воздействия на систему: тепловые,
физико-механические, химические и электрические.
Тепловое воздействие (нагрев) сопровождается понижением
вязкости и плотности фаз и изменением поверхностного натяже-
ния между ними. В ряде случаев оно дает необходимый эффект
Рис. VI-5. Электродегидратор:
/ — корпус; 2 — проходные изоляторы; 3 — под-
весные изоляторы; 4 — шины электроподводки;
5 — электроды; б —распределитель; 7 —подогре-
ватель; 8 — водомерное стекло; 9 —трубопровод
для вывода обезвоженного продукта; 10 — по-
плавковый выключатель напряжения; //—вто-
ричные обмотки трансформаторов; 12 — трубо-
провод для подачн эмульсин.
при разрушении нестойких
эмульсий. Режим нагрева дол-
жен быть установлен экспери-
ментально во избежание не-
желательных побочных явле-
ний (вспенивание).
Из физико-механических
способов разрушения эмуль-
сий заслуживает внимания ме-
тод фильтрования, основанный
на избирательном смачивании
фазами пористых веществ
фильтровального слоя (песок,
кизельгур, глины, толченая
окись алюминия, ткани и т.п.).
Этот метод в ряде случаев
дает нужный эффект. Для
повышения эффективности
фильтрования процесс целе-
сообразно проводить при по-
вышенной температуре.
Эффективным способом
разрушения эмульсий являет-
ся также отстаивание капель
дисперсной фазы в поле цент-
робежных сил при использо-
вании центробежных сепара-
торов, применяемых в ряде
отраслей промышленности
(пищевой, нефтяной и т. д.).
Теория процесса, конструкция и принцип действия жидкостных
сепараторов и трубчатых сверхцентрифуг, широко применяемых в
промышленной практике, изложены в многочисленных специаль-
ных руководствах и работах [17—21].
Высокое значение фактора разделения (1500—1600) и незна-
чительная высота слоя сепарируемой жидкости, проходящего ме-
жду тарелками пакета (0,3—1,0 мм), позволяет разделять эмуль-
сии даже с малой разностью плотностей фаз. В современных
сепараторах интенсивность разделения эмульсий в 10б раз превос-
ходит соответствующую характеристику отстойников [17].
Выпускаемые отечественной и зарубежной промышленностью
жидкостные сепараторы рассчитаны на широкий диапазон произ-
460
А
Рис. VI-6. Схема устройства сепара-
тора-отстойника.
водительности (от 100 до 5000 л/ч). Предел сепарации (радиус
предельно малой выделяемой сепаратором частицы), зависящий
от разности плотностей фаз и угловой скорости вращения ротора,
составляет 0,2—0,9 мкм, например, для отработанного смазочного
масла 0,3—0,7 мкм, для молока 0,4—0,9 мкм, для латекса 0,2—
0,3 мкм.
Незначительные габариты жидкостных сепараторов, высокие
производительность и разделительная эффективность дают воз-
можность широко использовать эти аппараты в химической про-
мышленности. Обычно расход энергии на работу сепаратора от-
носительно невелик (0,4—0,7 кет • ч/м3).
Необходимость разрушения эмульсии типа В/М (вода/масло)
возникает при обезвоживании органических жидкостей. Процесс
может быть осуществлен путем
воздействия на эмульсию поля
переменного тока высокого на-
пряжения. В этих условиях кап-
ли воды заряжаются и переме-
щаются по направлению силовых
линий. Интенсивное движение
заряженных капель жидкости
способствует их столкновению и
коагуляции. Крупные капли лег-
ко осаждаются под действием
силы тяжести. Для увеличения электропроводности воды к эмуль-
сии добавляют электролит.
Указанный процесс нашел широкое применение при обезво-
живании нефтей и осуществляется на установках значительных
масштабов [22]. Рабочее напряжение тока на промышленных ус-
тановках достигает 20—22,0 кв. Процесс проводится при темпе-
ратуре 60—90° С.
Один из вариантов конструкции основного аппарата установки
для обезвоживания нефтей приведен на рис. VI-5. Электродегид-
ратор представляет собой цилиндрический сосуд (диаметром до
3500 мм), в верхней части которого расположены вводы для элек-
тродов 5 — концентрических колец из полосового алюминия, за-
крепленных на рамах на расстоянии 100—150 мм друг от друга
(рабочий градиент электрического поля равен 500—3000 в/см).
Эмульсия поступает в аппарат по трубопроводу 12 и через рас-
пределитель 6 в пространство между электродами. Обезвоженный
продукт выводится из верхней части аппарата. Вода собирается
в нижней части аппарата и удаляется через нижний штуцер.
В нефтяной промышленности обезвоживанию подвергаются
эмульсии, содержащие 15—20% воды. Степень обезвоживания до-
стигает 99%, расход энергии при этом составляет0,15—0,20 квт-ч/т.
Разделение жидкостей, взаимонерастворимых и неспособных к
образованию стойких эмульсий, производится в отстойниках, ис-
пользуемых в технологии основного органического синтеза наряду
461
Легкая
жидкость
Рис. VI-7. Одноярусный отстойняк для разделения в тонком слое:
/—кожух; 2 — внутренняя камера; 3 —наружные камеры; -/ — трубки, соединяю-
щие наружные камеры; 5 —листы; 6 — лаз.
Рис. VI-8. Многополочный отстойник для разделения в тонком слое:
1 — распределитель; 2 — кожух; 3-наклонные перегородки; 4-воадушник.
с более совершенными методами разделения такого рода систем
(см. выше). Принцип действия и устройство этих аппаратов про-
сты и общеизвестны. Некоторым преимуществом обладает сепа-
ратор-отстойник, схема которого приведена на рис. VI-6. Более
эффективно разделение жидкостей в аппаратах, представленных
на рис. VI-7 и VI-8.
СИСТЕМЫ ЖИДКОСТЬ — ТВЕРДОЕ ТЕЛО
При осуществлении химических процессов, промежуточ-
ные или конечные продукты которых являются твердыми вещест-
вами, нередки случаи образования разнообразных по структуре
взвесей (грубые взвеси, тонкие взвеси, мути).
В зависимости от концентрации твердой фазы, степени дисперс-
ности и структуры твердых частиц (кристаллические, аморфные,
коллоидные осадки), а также в зависимости от специфических
свойств каждой из фаз, для разделения взвесей в технологии
основного органического синтеза применяются разнообразные спо-
собы и аппаратура.
При выборе способа разделения взвесей, помимо указанных
выше условий, решающую роль играют: 1) требования к полноте
разделения фаз (содержание твердого вещества в жидкости, вла-
госодержание осадка); 2) необходимость промывки осадка; 3) ток-
сические свойства компонентов взвеси (требования к герметично-
сти аппаратуры); 4) температура процесса.
По принципу действия аппаратура, используемая для разделе-
ния взвесей, делится на две основные группы — отстойно-осади-
тельную и фильтровальную.
Отстойники
Отстойники (сгустители) периодического или непрерыв-
ного действия, широко используемые в технологии минеральных
веществ, сравнительно редко применяются в технологии основного
органического синтеза (главным образом, только на установках
для очистки сточных вод).
Значительно более эффективны аппараты, в которых разделе-
ние взвесей происходит в поле центробежных сил (гидроциклоны,
центрифуги отстойного типа).
Для выделения из взвесей частиц, обладающих ферромагнит-
ными свойствами, в некоторых случаях (например, при разделе-
нии реакционной массы, содержащей металлический катализатор)
с успехом может быть использована сепарация в магнитном поле.
Электромагнитный сепаратор представляет собой колонну, изго-
товленную из немагнитной стали, заполненную насадкой из сталь-
ных колец (углеродистая сталь) и окруженную соленоидом, пи-
таемым постоянным током. Разделяемую взвесь пропускают через
насадку со скоростью 2,0—2,5 м/сек. После окончания рабо-
чего периода (несколько часов) магнитное поле снимается и
463
Рис. VI-9. Мультигидроциклон:
7—труба для подачи суспензии; 2t 3 —решетки; 4 — патру-
бок для удаления слива; 5 —патрубок для удаления сгу-
щенной суспензии.
металлические частицы смываются с насадки потоком соответ-
ствующей жидкости. Аппарат такой конструкции был использован
для отделения катализатора (кобальт на кизельгуре) в произ-
водстве бутиловых спиртов методом оксосинтеза [23].
Гидроциклоны. Эти аппараты, широко применяемые в
качестве классификаторов взвесей [24, 25], еще недостаточно ис-
пользуются в промышленности основного органического синтеза,
хотя их возможности при сгущении малоконцентрированных сус-
пензий (1—2%) пред-
ставляют несомненный
интерес.
Малые габариты и
простота устройства
(отсутствие движущих-
ся частей) гидроцикло-
нов позволяют приме-
нять разнообразные
конструкционные мате-
риалы (фарфор, спла-
вы титана и т. д.) и
дают возможность ис-
пользовать эти аппа-
раты для работы в ши-
роком интервале тем-
ператур (до 300—
350° С) и давлений (до
200 ат) в условиях аг-
рессивной среды.
При сравнительно
небольшом напоре, не-
обходимом для созда-
ния скорости потока
(0,5—3,0 ат), фактор
разделения F, характеризующий гидроциклон, соизмерим с ана-
логичной характеристикой отстойных центрифуг (£=1000-^1200),
вследствие чего гидроциклон пригоден не только для класси-
фикации, но и для разделения взвесей с размером частиц 15—
100 мкм.
Диаметр гидроциклона, определяющий его производительность,
по понятным соображениям, не может быть значительным, поэтому
производительность аппарата сравнительно невелика.
В целях увеличения производительности установки гидроци-
клоны соединяют в батареи (мультигидроциклон), как это пока-
зано на рис. VI-9.
Отстойные центрифуги. Отстойные центрифуги состав-
ляют более разнообразную по конструктивному оформлению груп-
пу аппаратов, используемых при разделении взвесей в поле цент^
робежных сил,
464
Сведения о теории и практике центрифугирования изложены в
многочисленных отечественных и зарубежных работах и объеди-
нены в монографиях, сборниках и обзорах [18, 26—28].
Для осветления низкоконцентрированных (<1%) и высокодис-
персных (d<0,01 мкм) взвесей применяются тарельчатые и ка-
мерные сепараторы, а также трубчатые суперцентрифуги с ручной
выгрузкой осадка.
Удаление осадка из ротора пока не может быть автоматизи-
ровано, вследствие чего значение индекса производительности со-
временных трубчатых центрифуг не превышает 2500 м2 [28].
Фильтровальная аппаратура
Разнообразная по типу и конструктивным особенностям
фильтровальная аппаратура, применяемая в химической промыш-
ленности, может быть сгруппирована по принципу преодоления
сопротивления фильтрующей перегородки и осадка:
1) пониженное давление под фильтрующей перегородкой;
2) повышенное давление над фильтрующей перегородкой.
К первой группе относятся многочисленные конструкции ва-
куум-фильтров [29]. Вторая группа объединяет аппараты, в кото-
рых повышенное давление над фильтровальной перегородкой обес-
печивается либо за счет напора потока разделяемой суспензии
(создаваемого давлением газа или с помощью насоса), либо в
результате наложения поля центробежных сил (фильтрующие
центрифуги).
Аппаратура первой и второй групп изготавливается примени-
тельно к работе как в периодическом, так и в непрерывном ре-
жиме.
При выборе типа фильтровальной аппаратуры учитываются не
только приведенные выше условия, но и давление паров жидкой
фазы в условиях фильтрования. Для жидкостей, обладающих вы-
соким давлением пара при температуре фильтрования, примене-
ние вакуум-фильтров исключается. \
Вакуум-фильтры. Периодически действующие вакуум-
фильтры в многотоннажных производствах основного органиче-
ского синтеза применяются крайне редко (только для проведения
отдельных операций подсобного характера).
Современная тенденция отечественной и зарубежной техники
фильтростроения направлена на создание крупной, непрерывно-
действующей высокопроизводительной, автоматически управляе-
мой фильтровальной аппаратуры широкое применение корро-
зионностойких металлических (сплавы титана, ниобия и др.) и
синтетических материалов расширяет возможности разделения
агрессивных сред.
Большое распространение получили барабанные вакуум-филь-
тры с наружной поверхностью фильтрования. Площадь поверхно-
сти фильтрования герметизированных отечественных барабанных
30 В. С. Хайлов, Б. 5. Брандт
465
фильтров, предназначенных для депарафинизации минераль-
ных масел (рис. VI-Ю), достигает 75 м2 [30]. Наибольшая вели-
чина поверхности барабанного
фильтра, выпускаемого иностран-
ными фирмами равна 100 м2 [31].
Ступенчатое регулирование
скорости вращения барабана в
настоящее время заменено плав-
ной регулировкой, облегчающей
эксплуатацию аппарата.
Забивание ткани барабанного
фильтра осадком, приводящее к
постепенному снижению его про-
изводительности, устранено в кон-
струкции фильтра фирмы «Рото-
бэлт», благодаря непрерывной ре-
генерации сходящей с барабана
фильтровальной ткани(рис. VI-11).
Производительность фильтра этой
конструкции более чем в 4 раза
превышает производительность
Рис. VI-10. Барабанный герметичный
фильтр.
обычного барабанного фильтра.
Разделение взвесей с неоднородным размером частиц произво-
дится на ленточных вакуум-фильтрах. Совпадение направлений
движения частиц при фильтровании и осаждении, простота конст-
рукции, хорошее разделение фильтрата и промывных вод, относи-
тельно высокая производительность, возможность работы со сло-
ем осадка небольшой толщины, удобство промывки осадка — от-
носятся к достоинствам ленточных вакуум-
фильтров. Большая производственная пло-
щадь, занимаемая фильтром, непригодность
его для переработки продуктов, разрушаю-
щих резину (материал ленты фильтра), а
также для веществ, плохо смачивающих
резину и металл, являются недостатками
Рис. VI-11. Схема бара-
банного фильтра со схо-
дящим полотном:
/ — нож; 2 — разгрузочный
валик; 3—промывочный ро-
лик; 4 — натяжной ролик;
5 — фильтрова ль ное полотно;
6 — барабан.
конструкции такого типа аппаратов.
Площадь поверхности фильтрования оте-
чественных ленточных фильтров составляет
1,6—10 м2 (намечается дальнейшее ее уве-
личение).
Из наливных вакуум-фильтров непре-
рывного действия заслуживает внимания
карусельный фильтр, широко используе-
мый в производстве фосфорной кислоты и на гидрометаллургиче-
ских предприятиях. Отечественной промышленностью выпускаются
карусельные вакуум-фильтры с поверхностью фильтрования до 50 .и2.
Для получения осадков с пониженным влагосодержанием за
рубежом разработан ряд конструкций вакуум-фильтров, позволяю-
466
щих отжимать под значительным давлением (до 20 ат) осадок,
предварительно промытый и просушенный под вакуумом [31].
Фильтры, работающие под давлением. Широко извест-
ные фильтрпрессы периодического действия находят применение в
промышленности основного органического синтеза лишь при
Рис. VI-12. Непрерывнодействующий автоматический фильтрпресс системы
УкрНИЙХИММАШ (поверхность фильтрования 25,0 я2).
выполнении операций подсобного характера. Наиболее распрост-
раненная величина фильтровальной поверхности одной рамы
составляет 1,0 м2, число рам в фильтрпрессе — от 8 до 68, мак-
симальное давление не превышает 10 ат.
Недостатки фильтров такого типа: периодичность работы, руч-
ная выгрузка осадка, потери продукта, длительность и трудоем-
кость подготовительных операций (сборка и разборка фильтр-
пресса). Совершенствование конструкции фильтрпресса направ-
лено на механизацию перемещения рам и плит и выгрузки осадка.
30*
467
Наибольший интерес из конструкций фильтров такого типа
представляет отечественная конструкция (УкрНИИХИММАШ)
непрерывнодействующего автоматического фильтрпресса (рис.
VI-12), обеспечивающая возможность промывки и отжима осадка
под давлением до 15 ат. Выгрузка осадка и регенерация фильт-
ровальной ткани производится непрерывно, с автоматическим
управлением режима процесса. Фильтры такой конструкции вы-
пускаются с поверхностью фильтрования 0,5; 2,5 и 25,0 м2.
Сравнительная характеристика автоматического и рамного
фильтрпрессов на примере разделения ряда мелкодисперсных сус-
пензий приведена в табл. VI-1.
Таблица VI-1
Сравнительнав характеристика автоматического (УкрНИИХИММАШ]
и рамного фильтрпрессов [32]
Взвесь Фильтрпресс Производительность по сухому веществу, кг/(м* * ч) Влажность осадка, %
Краситель прямой Автоматический 18 34-40
черный-3 Рамиый 3 50-55
Дибензатроиил Автоматический 3,5 65
Рамиый 0,15 65
К новинкам в области барабанных фильтров относится бара-
банный фильтр, работающий под давлением [33]. Фильтр приго-
ден для работы с продуктами, требующими полной герметизации,
а также для фильтрования тонкодисперсных взвесей^ и дает воз-
можность значительного увеличения удельного расхода промы-
вочной жидкости (в 5—15 раз превышающего аналогичную вели-
чину, достигаемую на барабанных вакуум-фильтрах). Производи-
тельность фильтров под давлением в 1,5—2 раза превосходит
производительность вакуум-фильтров при соответственно более
низкой влажности осадка.
Для разделения или сгущения взвесей, содержащих неболь-
шое количество твердой фазы, применяются патронные фильтры
с патронами из проволочной спирали „(навитой на цилиндриче-
ский стержень из пористой керамики), фильтровальной ткани, а
также с намывным слоем вспомогательного фильтровального ве-
щества. Поверхность фильтрования патронных фильтров дости-
гает 60—70 м2. Фильтр работает периодически. Осадок смывают
водой и удаляют в виде сгущенной суспензии.
Патронные фильтры компактны и герметичны, удобны в экс-
плуатации. Отсутствие возможности для осмотра фильтровальной
поверхности во время работы фильтра является одним из суще-
ственных недостатков этой конструкции.
Аналогичной по идее является конструкция фильтра, предна-
значенного для осветления жидкостей, содержащих небольшое
468
количество твердой фазы. Поверхность фильтрования образуется
горизонтальными дисками, покрытыми
фильтровального вещества и на-
саженными на полый вал, который
может вращаться от привода, рас-
положенного на крышке аппарата
(рис. VI-13). После окончания цик-
ла фильтрования осадок смывает-
ся обратным током промывной
жидкости и в результате вращения
мешалки удаляется с поверхности
фильтрования (известны конструк-
ции фильтров с применением вибра-
ционного способа удаления осадка).
Разделение или сгущение взве-
сей с небольшой концентрацией
твердой фазы за рубежом произво-
дится с помощью листовых (пла-
стинчатых) фильтров. Поверхность
фильтрования пластинчатых фильт-
ров, выпускаемых за рубежом, до-
стигает 180 м2. Отечественной про-
мышленностью изготавливаются
листовые фильтры с поверхностью
фильтрования 6 и 44 м2. Намечен
выпуск фильтров с поверхностью
до 200 м2 [31].
Листовые фильтры изготавли-
ваются в вертикальном и горизон-
тальном исполнении. Вертикальные
фильтры используются преимуще-
ственно для осветления жидкостей,
горизонтальные — для разделения
взвесей невысокой концентрации.
Более высокая удельная поверх-
ность фильтрования (в м2 на еди-
ницу объема), меньшие размеры
занимаемой площади и удобства
выгрузки осадка являются бес-
спорными преимуществами верти-
слоем вспомогательного
Рис. VI-13. Листовой фильтр.
кальных листовых фильтров перед
горизонтальными [32—34]. Все ука-
занные фильтры действуют перио-
дически. Непрерывнодействующие аппараты типа листового
фильтра могут быть представлены дисковым фильтром, работаю-
щим под давлением. Наличие в неподвижной головке трех зон
фильтрования позволяет (благодаря постепенно понижающемуся
противодавлению в зонах фильтрования) предотвратить проскок
469
твердой фазы в фильтрат в начальной стадии рабочего цикла.
Вследствие избыточного давления в зоне сброса осадка (четвер-
тая зона распределительной головки) осадок непрерывно уда-
ляется с поверхности фильтрования, и сгущенная взвесь может
непрерывно выводиться из аппарата [31].
РАЗДЕЛЕНИЕ СИСТЕМ ТВЕРДОЕ ТЕЛО — ГАЗ
Необходимость выделения из газа взвешенных в нем
твердых частиц пыли возникает при удалении из газа примесей,
затрудняющих последующую его обработку, при выделении целе-
вого продукта, содержащегося в газе в пылеобразном состоянии,
Рис. VI-14. Схема
установки для
очистки газа от
пыли:
/ — конический жалю-
зийный пылеулови-
тель; 2 — циклон.
а также при очистке газа от примесей, загряз-
няющих атмосферный воздух (для выхлопных
газов).
Способы и аппаратура для очистки газов от
пыли довольно разнообразны. Некоторые из них
используются для очистки газа от диспергиро-
ванной жидкости и по принципу действия могут
быть сгруппированы следующим образом:
Осаждение твердых частиц ( Жалюзийные золоуловители
под действием ннерцион- < Циклоны и мультициклоны
ных сил I Ротационные пылеуловители
Фильтрование твердых час-
тиц
Очистка газов промывкой
Рукавные фильтры
Керамические фильтры
Масляные (висциновые)
фильтры
Скрубберы (оросительные,
колпачковые)
Скрубберы центробежные
Скоростной турбулентный
пылеуловитель
Осаждение частиц в поле I Электрофильтры
электростатических сил ( F
В зависимости от фракционного состава и
концентрации пыли и требований к степени очи-
стки газа, схема установки может включать по-
следовательную переработку запыленного газа
различными способами.
К первой группе аппаратов относятся жалюзийные золоуло-
вители, пригодные для удаления частиц размером более 25 мкм
и часто используемые совместно с циклонами (рис. VI-14), а так-
же широко известные циклоны и батарейные циклоны (мульти-
циклоны), принцип действия которых, конструкция и расчет осве-
щены в ряде общих [1—5] и специальных [35—38] работ.
Эффективность действия некоторых типов отечественных цик-
лонов может быть иллюстрирована следующими данными:
Диаметр частиц пыли, мкм .5 10 20
Степень очистки, %........... 41—86 70—90 96—99,8
470
Рис. VI-15. Схема аппарата
для очистки газа от пыли:
1 — вращающееся сопло; 2 —рукав-
ные фильтры.
Степень очистки запыленного газа в циклоне зависит от склон-
ности частиц пыли к слипанию.
Из числа аппаратов первой группы наиболее эффективны ро-
тационные пылеуловители, совмещающие функции вентилятора и
аппарата для очистки газа от пыли. Такие пылеуловители обла-
дают значительной производительностью и обеспечивают высокую
степень очистки газа от частиц размером 5—8 мкм.
Вторая группа аппаратов представлена различного рода филь-
трами, фильтровальная перегородка которых выполнена из слоев
гравия, песка, специальных сортов картона, пористой бумаги, тка-
ней, пористой керамики и т. д. Некоторые из этих фильтроваль-
ных перегородок не регенерируются и
стижении предельной величины со-
противления. В других случаях уда-
ление отфильтрованных частиц пыли
производится периодически путем
встряхивания (рукавные фильтры)
или пропускания обратного тока га-
за. Степень очистки газа от пыли, до-
стигаемая при фильтровании, доходит
до 99,0—99,5%, размер удаляемых ча-
стиц— до 2 мк.
Характерным для этой группы ап-
паратов требованием является ниж-
ниц предел температуры фильтруемо-
го газа — на 10—15 град выше точки
росы паров, содержащихся в газе.
Верхний предел температуры и сте-
пень агрессивности газов определяют-
ся материалом фильтровальной перегородки. Применение синте-
тических тканей и особенно тканей из стекловолокна расширяет
верхний предел работы рукавных фильтров до 300—350° С [39].
Представляет интерес совмещение принципа действия фильт-
ровального устройства (рукавный фильтр) и циклона в одном
аппарате [40]. Запыленный газ вводится тангенциально в цилин-
дрический корпус аппарата (имеющий коническое днище, пе-
реходящее в пылесборник), вследствие чего более тяжелые
частицы отбрасываются к периферии и, как в циклонах, попа-
дают в пылесборник, а более мелкие — улавливаются в рукавных
фильтрах.
Фильтровальная ткань очищается током очищенного газа, пе-
родически направляемого вращающимся соплом внутрь каждого
рукава (рис. VI-15).
Для тонкой очистки газа от пыли применяются фильтры, из-
готовленные из пористой керамики (поролитовые фильтры) или
пористого металла. Поверхность фильтрования оформляется в
виде труб с закрытым концом, монтируемых в трубной доске, как
в патронном фильтре.
471
Вода
. Газ 8
J циклон
Рис. VI-16. Схема скоростного пыле-
уловителя.
Для повышения эффекта пылеулавливания фильтры с насад-
кой из крупных материалов (керамические или металлические
кольца, гофрированная стальная сетка и т. д.) смачиваются высо-
ковязким минеральным (висциновым) маслом (смесь машинного
масла, глицерина и каустической соды). Степень очистки газа в
масляных фильтрах достигает 99% при сравнительно небольшом
сопротивлении потоку газа.
Аппараты, действие которых основано на смачивании частиц
пыли жидкостью и удалении пыли потоком этой жидкости, ис-
пользуются в тех случаях, когда по условиям процесса допускается
увлажнение газа (для орошения аппаратов в большинстве слу-
чаев применяется вода); в то же время этот прием может слу-
жить для охлаждения и осушки
влажных газов.
Наиболее простым вариантом
конструкции таких аппаратов
является насадочная колонна с
противотоком газа и жидкости.
Однако степень очистки газа в
этой колонне не превышает 80—
90%. Более эффективны колпач-
ковые колонны и колонны с про-
вальными тарелками, в которых взаимодействие газа с жидкостью
осуществляется в слое пены [41].
В пенных аппаратах улавливается 97—98% пыли диаметром
частиц >5 мкм и примерно 75—80% частиц размером <5 мкм.
При сравнительно высокой степени очистки по удельной (от-
несенной к 1000 м3 газа), стоимости переработки газа улавлива-
ние пыли в пенных газоочистителях является одним из наиболее
экономичных способов разделения системы газ — твердое тело.
Высокая степень очистки достигается в мокром циклоне
(ЦС-ВТИ), объединяющим действие центробежного осадителя с
промывкой газа. Частицы пыли, отбрасываемые центробежной си-
лой потока газа (вводимого по касательной к стенке корпуса ап-
парата), смываются пленкой воды, орошающей стенки. Повыше-
ние эффекта очистки обусловливается отсутствием уноса пыли от
стенок в результате местных завихрений, происходящих в сухих
циклонах.
Для характеристики эффективности действия аппарата ЦС-ВТИ,
распространенного в промышленности, могут служить следующие
данные [42]:
Диаметр частиц пыли,
мкм............... 1-9 10-14 15-25 25-35 35-45 45-60
Степень очистки, % . . 70—80 80—92 88—94 91—95,5 93—96,6 94—98
Скоростной турбулентный пылеуловитель (рис. VI-16), отли-
чающийся малыми габаритами, основан на взаимодействии ча-
стиц пыли с жидкостью, вводимой в поток газа и тонко диспер-
472
гируемой в диффузоре. Коагуляция капель жидкости с частицами
пыли повышает эффективность циклона, устанавливаемого по ходу
потока газа (размер отделяемых частиц пыли достигает 0,5—
1,5 мкм). Недостаток такого аппарата — значительные затраты
энергии на подачу газа.
При очистке газа в аппаратах этой группы неизбежно приходится
решать вопрос о переработке сточных вод, загрязненных частица-
ми пыли и газообразными, растворимыми в воде веществами.
Наиболее эффективным способом очистки газа от пыли (и от
тумана) является осаждение частиц твердого вещества в поле
электрического тока (электрофильтры). Этот вид очистки нахо-
дит широкое применение в химической промышленности. Теории
процесса, способам расчета и конструкции электрофильтров по-
священ ряд работ [43, 44].
Принципы действия электрофильтра таковы, что достигаемая
степень очистки газа является не только вопросом техники, но и
экономики (расход электроэнергии, относительно высокие капита-
ловложения). Степень очистки, достигаемая с помощью электро-
фильтров, превышает 99,5%. Размер отделяемых частиц пыли оце-
нивается величиной порядка 0,003 мкм.
РАЗДЕЛЕНИЕ ГОМОГЕННЫХ СИСТЕМ
После разделения фаз, составляющих реакционную
смесь, разделение каждой из этих фаз на компоненты или смеси
этих компонентов заданного состава представляет самостоятель-
ную и, пожалуй, более сложную задачу.
В зависимости от фазового состояния разделяемой смеси за-
дача решается различными способами (с применением разнооб-
разной по принципу действия и конструктивному оформлению ап-
паратуры): Кристаллизация и перекристаллиза-
Смеси твердых продуктов ция из растворов Зонная плавка Сублимация (возгонка) Парциальная конденсация
Газовые или парогазовые смеси Ректификация Абсорбция Адсорбция Диффузия через пористые или непо- ристые мембраны Кристаллизация Дистилляция, ректификация
Смеси жидких продуктов Экстракция Диффузия через непорнстые мем- браны . Адсорбция Выпаривание
Растворы твердых продук- тов Кристаллизация Экстракция Диализ, электродиализ Адсорбция
473
Основная часть перечисленных процессов имеет широкое при-
менение в технологии основного органического синтеза, другая
часть используется в иных отраслях химической промышлен-
ности либо находится в стадии изучения и применяется в малых
масштабах.
ЭКСТРАКЦИЯ
Экстракция широко используется в ряде отраслей хи-
мической промышленности и в промышленном органическом син-
тезе [1—5, 45—47]. В соответствии с общей тенденцией разработка
экстракционной аппаратуры преследует цель создания высокопро-
изводительных и эффективных конструкций непрерывного дейст-
вия (в подавляющем большинстве случаев).
Сравнительная оценка основных показателей процесса экст-
ракции (чистота рафината, удельный расход экстрагента, число
ступеней экстракции) приводит к выводу в пользу противотока
фаз, применение которого в большинстве случаев оказывается
более целесообразным.
В современных экстракционных установках находят примене-
ние как ступенчатые экстракторы (смеситель-отстойник), так и
дифференциально-контактные (колонны с равномерным потоком
фаз). Вывод в пользу того или иного типа аппарата должен де-
латься на основе результатов сравнительной оценки их показате-
лей применительно к конкретным условиям и задачам.
В зависимости от назначения стадии разделения экстрагент
либо регенерируется для повторного использования, либо направ-
ляется на дальнейшую переработку в смеси с экстрагированным
продуктом.
В первом случае количество экстрагента не является задан-
ным (во втором оно задано), в состав схемы входит установка для
регенерации экстрагента (ректификация, экстракция) и значение
отношения экстрагент : рафинат определяет экономические показа-
тели всей стадии разделения.
Затраты, связанные с работой установок для экстракции с ре-
генерацией экстрагента, складываются из расхода на амортизаци-
онные отчисления, пропорциональные размеру экстракционной
аппаратуры и оборудования для регенерации экстрагента, стоимо-
сти энергетических средств (особенно заметных при ректифика-
ционном методе регенерации экстрагента), а также стоимости
экстрагента вследствие неизбежных потерь при его переработке.
Оптимальный режим работы стадии экстракционного разде-
ления, соответствующий, очевидно, минимуму суммы указанных
выше затрат, находится для каждого конкретного случая как
функция количества экстрагента, отнесенного к количеству рафи-
ната.
Характер зависимости суммы затрат от соотношения экстр-
агент: рафинат понятен из рис. VI-17.
474
4
Кривая 1, выражающая размер амортизационных отчислений,
при минимальном количестве экстрагента (бесконечно большое
число ступеней экстракции) и максимальном значении его отно-
сительного количества (количество рафината стремится к нулю)
стремится к бесконечности. Кривая 2, выражающая стоимость
энергетических средств, имеет
монотонный характер, стре-
мясь к бесконечности при мак-
симальном количестве экстр-
агента (количество рафината
равно нулю).
Масса рафината
Рис. VI-17. Условия опти-
мального режима работы
экстракционной установки:
/ — амортизационные отчисления
от стоимости аппаратуры и обо-
рудования; 2 —стоимость энерге-
тических средств; 3 —сумма экс-
плуатационных расходов.
Рис. VI-18. Схемы экстракционных
установок:
а — одноступенчатая прямоточная экстрак-
ция; б — многоступенчатая (две ступени)
прямоточная экстракция; в — многоступен-
чатая (две ступени) противоточная экстрак-
ция;
I — смеситель; II — отстойник; 1 — исходный
раствор; 2 — рафинат; 3 — экстрагент;
4 — экстракт.
Суммарная кривая 3 имеет минимум, соответствующий опти-
мальному режиму стадии разделения.
В ступенчатых экстракторах, оформленных в виде батарей че-
редующихся смесителей и отстойников (рис. VI-18), установлен-
ных каскадом, смесительные устройства представляют собой или
центробежные насосы с мешалками, или струйные смесители раз-
нообразных конструкций (рис. VI-19).
Экстракторы типа «смеситель-отстойник», представляющие со-
бой горизонтальные или вертикальные аппараты со смесительны-
ми и отстойными зонами, разнообразны по вариантам конструк-
ций. На рис. VI-20 представлен экстрактор, применяемый при
экстракции капролактама из водных растворов. Камера смешения
475
оформлена в виде сосуда с погружным насосом. Исходный рас-
твор и экстрагент движутся противотоком. Аппарат отличается
значительными габаритами и размещается вне производственного
помещения.
Рис. VI-19. Схемы конструкции струйных смесителей:
А, Б —компоненты эмульсин; А + Б— эмульсия.
Известен ряд конструкций горизонтальных аппаратов с гори-
зонтально расположенным валом и насаженными на нем лопа-
стями. Камеры смешения и отстаивания разделены неподвижными
перегородками.
Конструктивные варианты вертикальных экстракторов более
многочисленны. Некоторые из-них приведены на рис. VI-21.
Рис. VI-20. Горизонтальный смесительно-отстойный экстрактор:
/ — переливная труба для подачи раствора в камеру смешения; 3 —патрубок для подачи
смеси в отстойную камеру; 3 —труба для перетока экстрагента.
По способу диспергирования жидкостей колонные экстракторы
можно разделить на две группы: 1) аппараты, в которых осуще-
ствлено диспергирование одной из фаз в сплошном потоке другой
в результате разделения дисперсной фазы на струи, капли или
пленку (рис. VI-21,a—з) и 2) аппараты, в которых диспергиро-
вание достигается при использовании перемешивающих устройств
(рис. VI-21.U—р).
В наиболее простых по устройству, пригодных для работы
со взвесями и удобных для очистки распылительных колоннад
476
Рис. VI-21. Конструктивные варианты колонных экстракторов.
Л - легкая фаза; Т - тяжелая фаза.
(рис. VI-21, а) удерживающая способность относительно мала.
В связи с этим, а также вследствие продольного перемешивания
сплошной фазы массообмен на единицу объема менее интенсивен,
чем у аппаратов других типов.
Уменьшение продольного перемешивания достигается устрой-
ством поперечных перегородок различной конструкции, которые
служат для образования пленки и дробления на капли дисперс-
ной фазы (рис. VI-21,6—г). Эффективность экстракторов такого
типа невелика: число единиц переноса на одну тарелку не превы-
шает 0,05—0,10.
Насадочные экстракторы (рис. VI-21,6) заполняются насад-
кой, аналогичной насадке абсорбционных аппаратов (кольца Ра-
шига, стальные спирали, куски кокса, деревянные рейки и т. д.),
укладываемой слоями по высоте, превышающей диаметр колонны
в 2—10 раз. Предпочтительной оказывается насадка, составлен-
ная из элементов малых размеров. Гидродинамические условия в
насадочных колоннах зависят от смачиваемости материала на-
садки каждой из фаз. Диспергированная фаза может иметь фор-
му капель или пленки. Удерживающая способность и эффектив-
ность насадочных колонн выше, чем распылительных, но предель-
ная производительность их несколько ниже.
Близкими по эффективности к насадочным колоннам являются
экстракторы с перфорированными тарелками (рис. VI-21,e,ж),
дисперсная фаза в которых многократно дробится при прохож-
дении через отверстия тарелок (диаметр отверстий 2—10 мм, пло-
щадь отверстий составляет ~10% площади поперечного сечения
колонны). В зависимости от того, какая из фаз диспергируется,
соответственно меняется расположение переливных патрубков или
закраин (на рис. VI-21,e диспергирована тяжелая фаза). Вариант
устройства для диспергирования тяжелой фазы представлен на
рис. VI-21,3. В отличие от распылительных и насадочных аппара-
тов экстракторы с перфорированными тарелками менее пригодны
для работы со взвесями.
Группа аппаратов, в которых для диспергирования фаз приме-
няются мешалки, приведена на рис. VI-21, и — о.
Представленные на рис. VI-21,—м аппараты относятся к ка-
тегории смесительно-отстойных экстракторов. Зоной отстаивания
в первом из них служит слой насадки высотой от 0,3 до 0,5 м,
выполненной из колец Рашига или редкоплетеной сетки. В зоне
смешения расположена либо турбинная (рис. VI-21,k), либо ло-
пастная (рис. VI-21, л) мешалка.
Известны экстракторы изображенного на рис. VI-21,o типа, в
которых пространство между мешалками заполнено вертикально
расположенными листами, образующими прямолинейные или кон-
центрические каналы. Жидкость, более хорошо смачивающая ма-
териал насадки, течет по поверхности листа, а вторая жидкость
образует слой между пластинами,
478
Экстракторы, представленные на рис. VI-21,/c— о, отличаются
относительным размером и формой мешалки и неподвижно укреп-
ленных на корпусе аппарата пластин. Роторно-дисковый экстрак-
тор (рис. VI-21,ju) характеризуется относительно высокой произво-
дительностью и высокой интенсивностью процесса массопередачи.
Примером может служить опыт промышленной эксплуата-
ции роторно-дисковых экстракторов в производстве капролакта-
ма [46].
Вертикальные экстракторы с мешалками более компактны, чем
соответствующие им по эффективности горизонтальные смеситель-
но-отстойные аппараты, и характеризуются меньшим расходом
энергии.
Интенсификация массообмена между фазами с помощью пуль-
сации смеси нашла свое отражение и в экстракционных процес-
сах. Пульсационный режим в' экстракционном аппарате создается
путем включения в систему сплошной фазы поршневого или плун-
жерного насоса, присоединенного по одной линии (вторая — за-
глушена) при снятом клапане на этой линии. Частота двойных
ходов насоса соответствует частоте пульсации.
Колебательное движение столба жидкости в экстракторе мо-
жет быть достигнуто также в результате соответствующего ре-
жима работы мешалки, снабженной тарелками и работающей от
привода, сообщающего возвратно-поступательные движения валу
мешалки. Частота пульсаций лежит в широком диапазоне (800—
30 000), амплитуда колебаний составляет 3—12 лии.
Эффект пульсации использован в распылительных и насадоч-
ных колоннах, а также в колоннах с перфорированными тарел-
ками (рис. VI-21,n, р).
Частота и амплитуда, оптимальные для каждой системы, зави-
сят от свойств перерабатываемых жидкостей, однако эффект пуль-
сации неизменно выражается в заметной интенсификации массо-
обмена между фазами. Наряду с этим, а также с возможностью
переработки взвесей (пульсация способствует осаждению твердых
частиц), пульсационный режим способствует эмульгированию и
связан с расходом энергии на работу насоса. Оценка экономиче-
ского эффекта, достигаемого в результате перевода экстракцион-
ного аппарата на пульсационный режим работы, требует сравне-
ния экономических показателей обоих режимов.
В последние годы промышленное применение находят центро-
бежные экстракторы, в которых осуществляется многократно че-
редующиеся процессы смешения и сепарации фаз. На рис. VI-22
изображен один из таких аппаратов — трехступенчатый экстрак-
тор системы «Люва», работающий по принципу противотока.
Экстрагент С поступает по каналу, расположенному в непо-
движном валу 2, в последнюю (третью) ступень, где смешивается
с раствором R2 (в рассматриваемом случае — тяжелая жидкость),
выходящим из второй ступени. Смесь обеих жидкостей разделяет-
ся в периферийной зоне вращающегося корпуса 1 (число оборотов
479
до 3500 об!мин), в результате чего более тяжелая фаза (рафинат
R3) выводится из периферийного слоя, а экстракт (Е3) — из слоя,
имеющего меньший радиус. Экстрагент направляется по соответ-
ствующему каналу вала 2 во вторую ступень экстрактора, оформ-
Третья
ступень
Вторая
ступень
ПерВая
ступень
Рис. VI-22. Центробежный трехступен-
чатый экстрактор системы Люва:
1 — вращающийся корпус; 2 — неподвижный вал
с каналами; 3, 5 — направляющие перегородки;
4 —вращающиеся перегородки; 6 — камеры раз-
деления; 7 — труба для тяжелой жидкости;
в —труба для легкой жидкости; 9, 10 — вход и
выход легкой жидкости, 11, 12 —вход, и выход
тяжелой жидкости;
S — исходный раствор; С — экстрагент; Е — экс-
тракт; R — рафинат*
ленную аналогичным образом,
а рафинат выводится из ап-
парата.
Исходный раствор S по-
дается в первую ступень экс-
трактора, где смешивается с
экстрактом, прошедшим вто-
рую ступень, и, после сепара-
ции, экстракт Ei отводится из
аппарата, а раствор Ri на-
правляется во вторую сту-
пень аппарата.
Перемещение экстракта и
рафината от ступени к ступе-
ни происходит под действием
поля центробежных сил, со-
здаваемого вращающимся ро-
тором аппарата. Регулирова-
ние положения плоскостей
раздела фаз в камерах рас*
слоения дает возможность ис-
пользовать аппарат в широ-
ком диапазоне соотношения
объемов взаимодействующих
жидкостей.
Центробежные экстракто-
ры такого типа изготавли-
ваются со значительным коли-
честном ступеней для пере-
работки смеси жидкостей на
производительность ~5 м3/ч.
Они удобны для разборки и
очистки от твердых осадков.
Сокращенное время кон-
такта фаз, возможность пере-
работки жидкостей с неболь-
шой разницей плотностей (затруднительная в аппаратах, где
сепарация основана на гравитационном разделении), малые габа-
риты относятся к числу несомненных достоинств таких экстракто-
ров и в определенных условиях окупают их относительно высокую
стоимость.
Другой принцип взаимодействия потоков использован в цент-
робежном экстракторе, изображенном на рис. VI-23. Основным
элементом аппарата является свернутый спиралью лист, надетый
480
на вал, имеющий внутренние каналы. Принцип действия аппарата,
понятный из рисунка, обеспечивает противоток фаз, смешение ко-
торых происходит при истечении тяжелой фазы через перегород-
ку; сепарация осуществляется под действием центробежной силы.
жидкости
Рис. VI-23. Центробежный
экстрактор Подбильняка:
а —разрез; б — принципиальная схе-
ма движения жидкости;
/ — барабан; 2 — вал; 3— перфориро-
ванная перегородка (перфорация по
всей перегородке не "показана);
4 — каналы.
Аппараты такого типа отличаются высокой эффективностью
разделения, дают возможность перерабатывать легко эмульги-
рующиеся жидкости при коротком времени контакта фаз, отлича-
ются малыми габаритами. Несмотря на относительно высокую
стоимость центробежных экстракторов, достоинства таких аппа-
ратов в ряде случаев делают их более предпочтительными перед
экстракторами других типов.
Экстрагенты, применяемые для извлечения, весьма разнооб-
разны. Выбор экстрагента определяется спецификой производства
и должен быть основан нЗ сравнительной оценке ряда вариантов.
31 В. С. Хайлов, Б. Б. Брандт 4g[
В технологии основного органического синтеза процесс экс-
тракции используется в производстве фенола по методу Рашига
(экстракция фенола из водного раствора бензолом), глицерина из
пропилена (экстракция окрашивающих продуктов из водного рас-
твора глицерина изооктаном), в производстве ДДТ (экстракция
гексахлорана хлорбензолом), капролактама (экстракция капро-
лактама из водного раствора трихлорэтиленом и экстракция кап-
ролактама из раствора в трихлорэтилене водой) и в ряде других
производств.
Выбор типа экстракционной аппаратуры, в наибольшей степе-
ни удовлетворяющей условиям и задачам процесса (подобно ана-
логичной задаче для других типов химической аппаратуры), ре-
шается путем сравнительной оценки суммы показателей, прису-
щих каждому из вариантов конструктивного оформления. Он
основан на учете общей нагрузки, числа теоретических тарелок,
физических свойств исходного раствора и экстрагента (разность
Плотностей, склонность к эмульгированию, наличие взвешенных
частиц, агрессивные свойства и т. д.), соотношения объема фаз и
прочих соображениях (габариты аппаратуры, легкость очистки,
металлоемкость и др.).
Относительная количественная оценка каждого из известных
типов экстракционных аппаратов применительно к приведенным
выше условиям весьма детально рассмотрена в монографии [45].
Наибольшей эффективностью обладают аппараты с использо-
ванием для контактирования фаз механической энергии (механи-
ческое перемешивание, пульсация), однако в ряде случаев оказы-
вается целесообразным применять экстракционную аппаратуру
типа насадочных или полых колонн.
В качестве примера можно привести результаты сравнитель-
ной оценки [45] насадочного экстрактора, колонны с перфориро-
ванными тарелками (рис. VI-21,e), роторно-дискового экстрактора
(рис. У1-21,л), колонны с насадкой при механической пульсации,
каскада типа «насос-отстойник», центробежного экстрактора Под-
бильняка (рис. VI-23) для экстракции фенола из водного раствора
концентрации 10 г/л—50 кг/л бутилацетатом при общей нагрузке
15 м3/ч, объемном соотношении экстрагент: исходный раствор,
равном 0,1, и небольшом содержании твердых частиц в исходном
растворе. При расчетном количестве теоретических ступеней экс-
тракции, равном четырем, наиболее целесообразным оказалось
применение насадочной колонны с пульсацией, колонны с перфо-
рированными тарелками или колонны с насадкой.
Большие габаритные размеры систем типа «смеситель—отстой-
ник» и относительно высокая стоимость центробежного экстрак-
тора в рассматриваемом случае оказались решающими для от-
клонения этих экстракторов. Естественно, что выбор, не ограни-
ченный площадью производственного помещения, при высокой
стоимости перерабатываемого продукта может привести к заклю-
чению в пользу других типов экстракционной аппаратуры.
482
КРИСТАЛЛИЗАЦИЯ
Под кристаллизацией обычно понимают образование
твердой фазы из растворов, расплавов или парогазовой фазы, про-
исходящее при пересыщении исходной фазы по отношению к твер-
дому веществу в результате понижения температуры системы или
повышения концентрации компонента, образующего твердую фазу.
В зависимости от характера изменения растворимости твер-
дого вещества с температурой для создания пересыщенных рас-
творов используют либо охлаждение (кристаллизация из распла-
вов, кристаллизация веществ, растворимость которых существенно
уменьшается при понижении температуры), либо выпаривание ча-
сти растворителя (кристаллизация веществ, растворимость кото-
рых мало изменяется с понижением температуры), либо одно-
временное охлаждение и испарение растворителя. Состояние
пересыщения достигается также уменьшением растворимости в
результате изменения свойств растворителя путем введения треть-
его компонента (высаливание).
Размер кристаллов твердой фазы определяется соотношением
скорости образования 'и роста кристаллических зародышей. В за-
висимости от режима достижения пересыщенного состояния си-
стемы (скорость охлаждения, скорость удаления растворителя, пе-
ремешивание) могут быть получены кристаллы самых различных
размеров. Кристаллизация из переохлажденных растворов,’ когда
скорость образования кристаллических зародышей существенно
превосходит скорость роста кристаллов, благоприятствует полу-
чению мелкокристаллических осадков. Наличие в системе готовой
поверхности раздела фаз (стенки сосуда, суспендированные кри-
сталлы) облегчает образование кристаллических зародышей и
препятствует достижению пересыщенного состояния.
В технологии органического синтеза процесс кристаллизации
используется для выделения твердых продуктов из растворов и
расплавов (производство моно- и дикарбоновых кислот, гексахло-
рана, дихлорпроизводных бензола, диметилтерефталата, капро-
лактама й т. д.), из парогазовой фазы (выделение фталевого
ангидрида), а также при разделении смеси твердых веществ.
В последнем случае многократным повторением процессов рас-
творения и кристаллизации достигается весьма полное разделение
компонентов твердой фазы; этот прием, называемый перекристал-
лизацией, используется как один из способов очистки твердых
продуктов.
Вопрос об оптимальном размере кристаллов продукта, выде-
ляемого из раствора методом кристаллизации, в каждом отдель-
ном случае требует экспериментального изучения, так как в зави-
симости от желаемой чистоты продукта и условий его дальнейшей
переработки требования к величине кристаллов могут быть совер-
шенно противоположными. Крупнокристаллический осадок легче
фильтруется, промывается и высушивается, однако чистота
31*
483
кристаллов может снижаться из-за возможности сокристаллиза-
ции. Мелкие кристаллы сильно затрудняют процесс фильтрования и
отстаивания, промывка мелкокристаллического осадка также свя-
зана с затруднениями, однако включения маточного раствора или
примесей в мелких кристаллах менее вероятны. Во всех случаях
предпочтительным является осадок максимально однородного гра-
нулометрического состава (осадок, состоящий из кристаллов раз-
Рис. VI-24. Схемы одностадийной (а) и двух-
. стадийной (б) перекристаллизации из раст-
воров:
ного размера, при филь-
тровании имеет склон-
ность к сжатию).
Наряду с переохлаж-
дением раствора причи-
ной образования мелких
кристаллов при выделе-
нии органических продук-
тов могут быть поверхно-
стно-активные вещества
[48, 49]. Адсорбция содер-
жащихся в растворе по-
верхностно-активных ве-
ществ на поверхности
кристаллических зароды-
шей препятствует их ро-
сту и способствует обра-
зованию мелких, трудно
фильтрующихся кристал-
лов. В качестве примера
1,3— аппараты для растворения; 2, 4 — кристаллиза-
торы (и аппаратура для разделения);
А, Б — разделяемые компоненты; В — растворитель;
А*, Б* — продукты после разделения (содержащие при-
месь второго компонента разделяемой смеси); Мь
М2 —маточные растворы.
может служить процесс
выделения алифатиче-
ских дикарбоновых кис-
лот (адипиновой, глута-
ровой и янтарной) из
водных растворов, получающихся при окислении циклогексанола
и кислородсодержащих производных циклогексана, азотной кисло-
той. Водорастворимые нитропроизводные в значительной степени
затрудняют процесс кристаллизации. х
Понижение температуры раствора и удаление части раствори-
теля осуществляется либо попеременно, либо одновременно. В пер-
вом случае маточный раствор циркулирует между кристаллиза-
тором, фильтром и выпарным аппаратом. Во втором случае
понижение температуры раствора происходит в результате испа-
рения растворителя непосредственно из кристаллизующегося
раствора.
Схемы установок кристаллизации и особенно перекристалли-
зации из растворов довольно разнообразны. Простейшая схема
перекристаллизации (рис. VI-24, а) предусматривает вывод при-
меси Б в составе маточного раствора уже после первой кристал-
лизации. При двухстадийной перекристаллизации (рис. VI-24,6)
484
схема усложняется и растворитель В, вводимый в аппарат для
растворения 1 в виде маточного раствора М2 (получаемого после
отделения кристаллов продукта А в результате второй кристалли-
зации), подается на растворение разделяемой смеси в аппарат 3.
Примесь выводится из цикла в составе маточного раствора Л11
после отделения кристаллов, полученных при первой кристаллизации.
> Естественно, продукт А и вещество Б, получаемые в результате
кристаллизации, содержат примесь другого компонента. При не-
обходимости дополнительной очистки продукт А подвергается
дальнейшей перекристаллизации.
С вертикальной
мешалкой
С горизонтальной
мешалкой
Охлаждаемые
вальцы
Рис. VI-25. Основные типы кристаллизаторов.
F — поверхность охлаждения; V — емкость аппарата; Кт — средиее значение коэффициента
теплопередачи.
Аппараты, предназначенные для кристаллизации из раствора
и из расплава, обладают значительной поверхностью теплообмена
и устройством для перемешивания кристаллизующегося раствора.
Некоторые типы аппаратов этой группы приведены на рис. VI-25.
Аппараты с вертикальной мешалкой, в которых отвод тепла
осуществляется через стенки корпуса, змеевик или охлаждающую
поверхность мешалки, используются как периодически- и непре-
рывнодействующие кристаллизаторы. В первом случае интенсив-
ность теплообмена через поверхность охлаждения выше, так как
образующийся на поверхности охлаждения слой кристаллов рас-
творяется во вновь подаваемом горячем растворе.
При проведении непрерывных процессов находят применение
горизонтально расположенные кристаллизаторы, мешалка кото-
рых служит для очистки поверхности теплообмена и перемещения
кристаллов вдоль оси аппарата,
485
При кристаллизации из вязких и высококонцентрированных
растворов, а также из расплава применяются охлаждаемые валь-
цы (рис. VI-25, а, б) с различным способом подачи исходной смеси.
Рис. VI-26. Схема многоступенчатой вакуум-кристаллизационной установки:
/ — кристаллизаторы; 2 — переточные трубы; 3 — поверхностные конденсаторы.
Кристаллизация при, одновременном охлаждении и упарива-
нии раствора проводится в
Рис. VI-27. Схема установки для
получения крупных кристаллов:
1 — испарительная камера; 2 — труба для
насыщенного раствора; 3 — кристаллиза-
тор; 4 — трубы для вывода кристаллов;
5 — кипятильник.
сталлизатора 3, откуда он
серии последовательно соединенных
кристаллизаторов (рис. VI-26), в
которых происходит испарение рас-
творителя и понижение температу-
ры при переходе раствора из ап-
парата в аппарат в результате
понижения давления от первого к
последнему аппарату. С увеличением
числа последовательно соединенных
ступеней кристаллизации увеличи-
вается размер кристаллов вслед-
ствие понижения степени пересы-
щения в каждой из ступеней.
Для получения крупных одно-
родных кристаллов используется
кристаллизатор с принудительной
циркуляцией слабо пересыщенного
раствора через слой взвешенных
кристаллов (рис. VI-27). Раствор
нагревается в теплообменнике 5 и
поступает в испарительную камеру
1. Вторичный пар удаляется на кон-
денсацию, а раствор опускается по
трубе 2 под решетчатое дно при-
поднимается, проходя слой кристал-
лов. При соответствующей скорости потока раствора в слое круп-
ные кристаллы по мере их роста собираются в нижней части
слоя и по трубе 4 выводятся из кристаллизатора.
486
Выделение твердых продуктов из растворов проводится также
в распылительных сушилках и в сушилках с применением кипя-
щего слоя частиц продукта, выделяемого из раствора. В послед-
нем случае псевдоожижающей средой является горячий газ или
воздух. Подача раствора в кипящий слой позволяет получать
материал в гранулированном виде.
В технологии органического синтеза кристаллизация произво-
дится из водных растворов и из растворов в разнообразных орга-
нических растворителях. Процесс кристаллизации и перекристал-
лизации, являясь достаточно эффективным способом очистки
продуктов, связан с относительно высоким удельным расходом
энергии и применение его оправдано лишь для условий, исклю-
чающих возможность использования другого способа очистки.
РЕКТИФИКАЦИЯ И АБСОРБЦИЯ
Процессы ректификации и абсорбции, различные по за-
дачам, решаемым ими в технологии органического синтеза, и по
физико-химической сущности составляющих их явлений, имеют
много общего в принципах конструкции аппаратуры, предназна-
чаемой в обоих случаях для создания контакта между потоком
газа и жидкости.
Современные ректификационные и абсорбционные установки
нефтехимической промышленности и промышленности основного
органического синтеза представляют собой сложный комплекс ап-
паратов и машин, в ряде случаев выделяемый в самостоятельный
цех химического завода.
Последний период времени характеризуется обилием работ,
посвященных теории и технике ректификации и абсорбции. Общей
является тенденция к повышению интенсивности и эффективности
аппаратуры, углубленному изучению кинетики процесса и уточне-
нию его математического описания. Характерно также внимание
к экономической стороне проблемы и поиск оптимальных вариан-
тов решения задачи.
Ректификационному разделению подвергаются многокомпо-
нентные смеси сложного состава, различающиеся по температуре
кипения легких и тяжелых ключевых компонентов, их относитель-
ной летучести, агрессивным и токсическим свойствам.
В зависимости от состава и свойств компонентов и задач, стоя-
щих перед ректификационным разделением, процесс проводится
под вакуумом или под избыточным давлением с использованием
способности отдельных компонентов смеси к образованию азео-
тропных систем.
Производительность и размер аппаратуры ректификационных
установок достигает значительных величин. Материал аппаратуры
весьма разнообразен.
Необходимость получения фракций узкого состава, характерная
для большинства процессов технологии основного органического
487
синтеза, накладывает отпечаток на схему ректификационных уста-
новок, которые в большинстве случаев организуются с сохране-
нием известного принципа зависимости числа колонн (и) от числа
компонентов (Л^):
n = JV-l
При разделении многокомпонентных смесей, содержащих вы-
сококипящие смолистые примеси, последовательность выделения
компонентов от низкокипящего к высококипящему не соблюдает-
ся, так как длительное пребывание смолистых веществ, а также
Рис. VI-28. Схема ректифика-
ционной установки для разде-
ления смеси, содержащей смо-
листый продукт D:
1—3 — ректификационные колоииы.
содержащихся в разделяемой смеси
остатков катализатора способствует
осмолению отдельных компонентов
смеси.
В подобных случаях целесообраз-
ным является освобождение разделя-
емой смеси от высококипящей фрак-
ции на первой стадии разделения в
условиях кратковременного нагрева.
Для этой цели применяются ротор-
ные тонкопленочные испарители,уста-
навливаемые в начале каскада рек-
тификационных устройств или для
уменьшения нагрузки на испаритель—
после отделения легкокипящего ком-
понента (рис. VI-28).
Небольшая разделительная спо-
собность роторных испарителей доста-
точна для отделения фракции смоли-
стых высококипящих веществ от сме-
си низкокипящих продуктов, поступающих в виде смеси паров на
разделение в систему ректификационных колонн.
Для разделения смесей продуктов, кипящих при низкой или
высокой температуре, термически нестойких продуктов, а также
для разделения смесей близкокипящих компонентов используется
ряд приемов:
1) ректификация при повышенном давлении или под вакуумом;
2) ректификация в токе водяного пара;
3) азеотропная ректификация;
4) экстрактивная ректификация.
При разделении смеси, содержащей низкокипящие вещества
(например, сжиженные газы), получение дистиллята-жидкости свя-
зано с необходимостью применения низких температур для конден-
сации паров, выходящих из колонны. Возможность использования
доступных и дешевых хладоагентов (вода) требует повышения
температуры конденсации паров дистиллята, что достигается со-
ответствующим повышением давления этих паров. Значение избы-
точного давления в ректификационной колонне устанавливается с
488
учетом влияния этого давления на конструктивные размеры ап-
паратуры (толщина стенок, площадь сечения отверстий для про-
хода пара), а также с учетом изменения (как правило, уменьше-
ния) относительной летучести разделяемых компонентов и соот-
ветственного уменьшения разделяющей способности колонны.
Ректификация термически нестойких продуктов, обладающих
низким давлением паров, приводит к необходимости осуществле-
ния процесса при пониженном давлении. Проведение ректифика-
ции под вакуумом сказывается на значении коэффициента отно-
сительной летучести разделяемых компонентов и в большинстве
случаев облегчает процесс разделения.
Последнее обстоятельство используется при разделении смесей
продуктов с близкой температурой кипения, ректификация кото-
рых при атмосферном давлении требует применения колонн с
большим числом теоретических тарелок. Благоприятным является
характер зависимости числа теоретических ступеней разделения
от значения коэффициента относительной летучести: необходимое
число тарелок при заданной четкости разделения резко уменьша-
ется при увеличении коэффициента относительной летучести от
1,03 до 1,2; в области значений ал (ал — коэффициент относи-
тельной летучести, равный отношению давлений паров разделяе-
мых компонентов при данной температуре), превышающих 1,8—
2,0, эффект становится мало заметным.
Ректификация под вакуумом предъявляет серьезные требова-
ния к схеме и устройству ректификационной аппаратуры. Харак-
тер зависимости Р(Т) в области низкого давления делает весьма
заметным влияние сопротивления насадки или тарелок ректифи-
кационной колонны и столба жидкости в кубе колонны на темпе-
ратуру кипения компонентов.
Конструкция вакуумных колонн рассчитана на минимальное
сопротивление току пара.
Наряду с применением вакуума, для разделения высококипя-
щих продуктов, не растворимых в воде, применяется ректифика-
ция в токе водяного пара. Этот прием удобен при разделении
смеси, отличающейся высоким значением коэффициента относи-
тельной летучести и высоким давлением паров низкокипящего
компонента. Проведение ректификации такой смеси под вакуумом
(для понижения температуры кипения вещества в кубе) может
привести к значительным потерям низкокипящего продукта, кон-
денсируемого при пониженном давлении.
Схема установки для ректификации в токе водяного пара при-
ведена на рис. VI-29. Конденсат из теплообменника 2, представ-
ляющий собой эмульсию органической фазы и воды, направляется
в аппарат 3, откуда органическая фаза поступает на орошение
колонны /ив холодильник дистиллята, а вода — в парогене-
ратор 4.
Учитывая присутствие в дистилляте примесей высококипящего
компонента, способного к осмолению при длительном нагревании
489
в парогенераторе, для полноты разделения фаз в качестве аппа-
рата 3 целесообразно использовать центробежный сепаратор.
Для предотвращения осмоления теплообменных устройств па-
рогенератора 4 необходимо постоянное или периодическое удале-
ние части воды из парогенератора и замена ее свежей водой.
Увеличение коэффициента относительной летучести, необходи-
мое при разделении смесей близкокипящих компонентов, достига-
ется путем введения в
Загрязненная
вода
Рис. VI-29. Схема установки
для ректификации в токе
водяного пара:
1 — ректификационная колонна;
2 — теплообменник; 3 — аппарат
для разделения; 4 — парогенера-
тор.
систему дополнительного агента, характе-
ризующегося. различной растворимостью
в продуктах, которые составляют разде-
ляемую смесь, этот агент неодинаково
влияет на значение коэффициентов ак-
тивности в уравнении для коэффициен-
та относительной летучести
где у1, у2 — коэффициенты активности
разделяемых компонентов, характери-
зующие степень отклонения их от зако-
нов идеальных растворов.
В зависимости от летучести разде-
ляющего агента по отношению к летуче-
сти разделяемых компонентов разли-
чают азеотропную и экстрактивную рек-
тификации.
В первом случае в результате ректи-
фикации основная часть разделяющего
агента, обладающего высоким давлением
паров, удаляется вместе с дистиллятом,
а из куба колонны выводится высоко-
кипящая фракция, содержащая примесь
разделяющего агента. При осуществле-
нии экстрактивной ректификации тяжело-
кипящий разделяющий агент, вводимый
в верхнюю часть колонны, с практической полнотой выводится с
кубовой жидкостью.
В промышленных схемах в состав установок для азеотропной
или экстрактивной ректификации входит аппаратура, предназна-
ченная для регенерации разделяющего агента из дистиллята и
кубовой жидкости.
Азеотропная ректификация используется для разделения сме-
сей, содержащих относительно малое количество легколетучего
компонента, в то время как близкокипящие смеси, содержащие
небольшое количество тяжелокипящих примесей, целесообразно
подвергать экстрактивной ректификации.
Энергетически более экономичен процесс экстрактивной рек-
тификации, при проведении которого дополнительный расход теп-
490
ла в ректификационной колонне (не считая расхода тепла на ре-
генерацию разделяющего агента) связан лишь с нагревом разде-
ляющего агента, в то время как при азеотропной ректификации
дополнительное тепло расходуется на испарение и создание флег-
мы, содержащей разделяющий агент.
Схемы установки для экстрактивной ректификации приведены
на рис. VI-30. Тарелки, расположенные выше точки ввода разде-
ляющего агента, практически исключают возможность его присут-
ствия в дистилляте. Стадия регенерации разделяющего агента (в'
колонне 2) рассчитана
на переработку толь-
ко кубовой жидкости.
Схемы абсорбцион-
ных установок менее
разнообразны, и, как
правило, состоят из ап-
паратуры, предназна-
ченной для осуществ-
ления абсорбционного
процесса, и аппарату-
ры для процесса, свя-
занного с регенерацией
абсорбента (ректифи-
кация, экстракция).
Аппаратура, приме-
няемая для ректифи-
кации и абсорбции,по
конструктивному при-
знаку имеет много об-
Рис. VI-30. Схема установки для экстрактивной
ректификации:
/ — колонна экстрактивной ректификации; 2 —ректифика-
ционная колонна для регенерации разделяющего агента;
3 — конденсаторы; 4 — холодильники; 5, 6 — нагреватели;
А, Б — компоненты разделяемой смеси; В— разделяющий
агент.
щего и в некоторых
случаях имеется возможность взаимозамены. Специфические осо-
бенности каждого из процессов ограничивают возможность пол-
ной унификации.
Применяемые в ректификационной и абсорбционной технике
аппараты различаются по конструктивному оформлению устройств
для гомогенизации газо-жидкостной смеси. Наиболее распростра-
нены тарельчатые колонны с различными вариантами конструк-
ции тарелок (колпачковые, ситчатые, провальные) и насадочные
с разнообразной формой насадочных тел, удельной их поверхно-
сти и сопротивлением потоку газа (пара). Применяются также
аппараты, в которых контакт жидкой и газовой сред осуществ-
ляется при диспергировании жидкости механическими устройст-
вами.
Тарельчатые аппараты, используемые в ректификационных и
абсорбционных установках, достигают значительных размеров
(диаметр до 7—8 м).
Относительно высокое гидравлическое сопротивление колпач-
ковых тарельчатых колонн ограничивает их применение при
491
ректификации под атмосферным давлением. В абсорбционных уста-
новках эта особенность имеет меньшее значение. Среди различных
вариантов конструкций колпачковых тарелок максимальная ско-
рость газовой фазы в свободном сечении колонны достигается при
использовании тарелок с малым диаметром колпачков (40—60мм).
В группе тарельчатых колонн к числу новых типов с направ-
ленным движением жидкости под действием тока газа относятся
получившие признание тарелки типа «Унифлюкс» (рис. VI-31). Та-
кие тарелки более просты по устройству, чем колпачковые [50, 51],
обладают меньшим гидравлическим сопротивлением и по эффек-
Рис. VI-31. Схема устрой-
ства тарелки типа «Уни-
флюкс».
тивности не уступают колпачко-
вым тарелкам.
Влияние нагрузки по газовой
фазе, сильно^сказывающейся на
эффективности большинства та-
рельчатых колонн, менее замет-
но при использовании балластных
и клапанных тарелок (рис. VI-32).
Переменное сечение каналов для
прохода газовой фазы, регули-
im
Рис. VI-32. Схема устрой-
ства клапанной тарелки.
руемое скоростным напором потока, сохраняет постоянство ско-
рости газового потока в отверстиях колпачков даже при значи-
тельных колебаниях нагрузки аппарата по газовой фазе (отноше-
ние максимальной нагрузки к минимальной равно 7—9).
Другой принцип поддержания постоянства скорости газа в бар-
ботажных отверстиях использован в комбинированных ситчато-
колпачковых тарелках (рис. VI-33). Газ проходит через патрубок
между глухой и перфорированной тарелками и барботирует через
слой жидкости над перфорированной тарелкой. В зависимости от
размера потока газа или пара рабочая площадь перфорированной
тарелки меняется. Постоянный, независимый от плотности оро-
шения уровень жидкости на тарелке позволяет размещать на ней
теплообменные устройства — змеевики, трубчатки [52].
Ситчатые тарелки с переливом просты по устройству, обла-
дают высоким к. п.д. и допускают значительную скорость пара
при сравнительно малом сопротивлении. Они непригодны для пе-
реработки загрязненных твердыми примесями жидкостей (забив-
ка отверстий в тарелках) и вследствие присущих им особенностей
гидравлики используются в непрерывнодействующих установках
492
при строго регулярном режиме работы (кратковременное прекра-
щение подачи питания или подачи пара нарушает распределение
жидкости по тарелкам).
Рис. VI-33. Схема устройства сетчато-колпачковых тарелок.
Последнее условие является также особенностью тарелок про-
вального типа, для нормальной работы которых обязателен гид-
равлический баланс между потоком пара и жидкости. Необходи-
мость поддержания этого баланса сужает диапазон допустимых
нагрузок.
Рядом достоинств, по сравнению с кол-
пачковыми тарелками, обладают тарелки
конструкции ГИПРОНЕФТЕМАШа (рис.
VI-34), устойчиво работающие в диапазоне
скоростей пара (газа) до 2,6—2,8 м/сек
при соотношении рабочей скорости к ми-
нимальной до 5—8.
Провальные тарелки находят большое
применение в абсорбционной и ректифи-
кационной технике. К числу новых кон-
Рис. VI-34. Схема устрой-
ства тарелок системы
ГИПРОНЕФТЕМАШа.
струкций тарелок провального типа относятся гофрированные та-
релки, характеризующиеся большим диапазоном устойчивости ре-
жима при переменной нагрузке и значительно (в 2—3 раза)
меньшим уносом жидкости [53].
Разновидностью тарелок провального типа являются трубча-
то-решетчатые тарелки, совмещающие функции провальных та-
релок с теплообменным устройством и оформляемые в виде пучка
493
too
Рис. VI-36. Варианты охла-
ждающих устройств в аб-
сорбционных колоннах.
параллельных труб, собранных в коллектор, либо в виде спираль*
но изогнутого змеевика (рис. VI-35).
Трубчато-решетчатые тарелки обладают высокой пропускной
при относительно малой величине гидравлического
[54, 55] и используются как в ректификацион-
способностью
сопротивления
ных, так и в абсорбционных ко-
лоннах.
В последнем случае высокая
интенсивность теплообмена поз-
воляет поддерживать изотерми-
ческий режим процесса, характе-
ризующегося большим тепловы-
делением (абсорбция паров из
парогазовой смеси). При сравне-
нии с другими вариантами ох-
лаждающих устройс!в (рис.
VI-36) трубчато-решетчатые та-
релки имеют преимущество в
простоте устройства.
Для переработки агрессивных
сред представляет интерес та-
рельчатая колонна, выполненная
из керамики (рис. VI-37). Ин-
жектирование газом жидкости
обеспечивает высокоразвитую
поверхность фазового контакта
[56].
Группа насадочных колонн
совершенствуется путем созда-
ния новых типов насадок с повы-
шенно удельной поверхностью
при относительно малом гидрав-
лическом сопротивлении. Извест-
Рис. VI-37. Керамическая тарельча-
тая колонна.
но большое количество насадок
с телами различной формы, из-
готовленными из металла, угле-
графитовых композиций и синтетических материалов с гладкой
и шероховатой поверхностями [57].
Основная причина, ограничивающая применение насадочных
колонн в вакуум-ректификационных установках — высокое гидрав-
лическое сопротивление насадки, соответствующее одной ВЭТТ,
устранена в плоско-параллельной насадке (вертикальные пакеты
из плоских или волнистых металлических листов или полотен из
стеклоткани, устанавливаемых на расстоянии 5—15 мм друг от
друга) [58]. При эффективности, приблизительно равной эффек-
тивности насадки из колец Рашита, плоско-параллельная насадка
характеризуется большой пропускной способностью и малым
гидравлическим сопротивлением. В случае разделения смеси
495
циклогексанол—циклогексанон—высококипящие продукты [59] в
колонне с плоско-параллельной насадкой при остаточном давле-
нии наверху колонны, равном 60 мм рт. ст., сопротивление насадки
высотой ~20 м (число единиц переноса 15, флегмовое число 3,5)
не превышает 25—40 мм рт. ст.
Значительное внимание уделяется равномерности орошения
насадки, оказывающей большое влияние на эффективность рабо-
ты насадочных орошаемых колонн (в том
Рис. VI-38. Скруббер с
псевдоожиженным слоем
насадки.
числе и колонн с плоскопараллельной
насадкой). Результаты сравнительного
испытания ряда устройств [60] свиде-
тельствуют о преимуществах форсунок
и отбойно-разбрызгивающих приспособ-
лений.
Представляют интерес показатели,
получаемые при осуществлении абсорб-
ции в слое псевдоожиженной насадки из
полых металлических или выполненных
из синтетических материалов шаров.
Высокие рабочие скорости газа (до
5—6 м/сек) и высокое значение плотно-
сти орошения [до 150 ж3/(ж2-ч)] указы-
вают на перспективность этого способа,
уже находящего практическое примене-
ние [61,62]. Скруббер с псевдоожиженным
слоем насадки показан на рис. VI-38.
Из разнообразных вариантов конст-
рукции колонн для ректификации под
вакуумом, предложенных в последнее
время, наибольшего внимания заслужи-
вают роторные колонны.
Принцип действия одной из конструк-
ций таких колонн состоит в том, что
жидкость, находящаяся на тарелке, раз-
брызгивается с помощью соосных воро-
нок (рис. VI-39), укрепленных на вра-
щающемся валу, установленном по оси
колонны. Капли жидкости пересекают
движущийся снизу вверх поток пара и орошают цилиндрическую
стенку, выполненную из металлической сетки.
Перепад давления в роторно-разбрызгивающей колонне при-
мерно на один порядок ниже, чем в равной ей по эффективности
тарельчатой колонне. Диаметр промышленных аппаратов такого
типа достигает 1400 мм [63]. Недопустимость чрезмерного удли-
нения вала ротора ограничивает высоту колонн размером, эквива-
лентным 15 ступеням разделения.
В роторно-пленочной колонне вращающийся вал, снабженный
лопастями, охлаждается, а стенки колонны имеют обогрев. Кон-
496
денсат, образующийся на охлаждаемой поверхности вращающего-
ся вала, отбрасывается к периферии лопастями, одновременно
турбулизирующими поток пара (рис. VI-40). Эффективность про-
цесса в колонне определяется перепадом температуры между стен-
кой колонны и стенкой ротора, т. е. интенсивностью потока брызг
конденсата от центра к периферии. Флегма стекает по стенкам
колонны, и равномерность смачивания стенок также влияет на
эффективность разделения. Наибольшая высота известных ротор-
но-пленочных колонн достигает 6 м, диаметр — до 860 мм [63].
Преимуществом роторной колонны при переработке термола-
бильных жидкостей является малое время их пребывания в зоне
Рис. VI-39. Устройство для рас-
пыления жидкости в роторных
колоннах.
Рис. VI-40. Схема устрой-
ства роторно-пленочной ко-
лонны.
нагрева и практическая независимость эффективности от нагруз-
ки. Наличие ротора, усложняющего конструкцию аппарата и огра-
ничивающего его высоту, относится к числу основных недостатков
роторных ректификационных колонн.
В дополнение к рассмотренным выше тарельчатым и насадоч-
ным аппаратам, используемым при проведении ректификационных
и абсорбционных процессов, следует кратко остановиться на ряде
конструкций, находящих применение в абсорбционной технике [64].
Особого внимания заслуживают пленочные, барботажные и рас-
пыливающие абсорберы.
К пленочным абсорберам принадлежат аппараты со стекаю-
щей пленкой (трубчатые абсорберы, абсорберы с плоско-парал-
лельной насадкой) и аппараты с восходящим движением пленки.
В первых двух конструкциях при относительно небольшой ско-
рости газового потока (4—5 м/сек) процесс может быть осуществ-
лен как при параллельном движении газа и жидкости, так и при
их противотоке. В обоих случаях создаются условия, практически
соответствующие режиму полного вытеснения для каждой из фаз.
Восходящее движение пленки, достигаемое в результате зна-
чительной скорости (до 40 м/сек) движущегося снизу вверх газо-
вого потока, характеризуется высокой скоростью массообмена
32 В- С. Хайлов, Б. Б. Брандт 497
между фазами. Естественно, что абсорберы, действие которых ос-
новано на этом принципе, пригодны для случаев, когда желатель-
ным является параллельный ток газа и жидкости; условия про-
тивотока создаются в многоступенчатых аппаратах с восходящей
пленкой жидкости [64].
Трубчатые пленочные аппараты со стекающей пленкой (см.
стр. 248) и аппараты с восходящей пленкой жидкости характери-
зуются весьма высоким значением удельной поверхности теплооб-
мена и пригодны для проведения процессов, связанных с подво-
дом или отводом значительного количества тепла.
Для случаев, когда абсорбент представляет собой суспензию
или когда суспензия образуется в результате взаимодействия газа
и жидкости, рассматриваемые выше типы пленочных аппаратов
малопригодны и задача решается путем использования механи*
ческих пленочных абсорберов с горизонтально расположенным ва-
лом, который снабжен дисками или пакетами дисков. Низкое гид-
равлическое сопротивление и возможность работы при малом ко-
личестве абсорбента выгодно отличают этот тип абсорбционной
аппаратуры, однако наличие движущихся частей и связанную с
этим высокую стоимость эксплуатации следует отнести к числу
серьезных недостатков таких аппаратов.
Абсорберы барботажного типа весьма разнообразны по кон-
структивному оформлению. Кроме рассмотренных выше тарельча-
тых аппаратов, используемых в ректификационной и в абсорбци-
онной технике, для проведения процессов абсорбции находят
применение аппараты со сплошным барботажным слоем и с меха-
ническим перемешиванием барботажного слоя.
Интенсивное продольное перемешивание жидкой фазы в полых
цилиндрических аппаратах со сплошным барботажным слоем
столь значительно снижает движущую силу процесса, что, не-
смотря на предельную простоту устройства этих аппаратов, при-
менение их целесообразно лишь при осуществлении процессов,
характеризующихся незначительным равновесным давлением пара
или газа над жидкостью (хемосорбция).
Уменьшение продольного перемешивания в сплошном барбо-
тажном слое достигается установкой по высоте аппарата тарелок
провального типа или заполнением аппарата насадкой.
Механическое перемешивание барботажного слоя в крупных
абсорбционных установках используется сравнительно редко. При-
менение абсорберов с перемешиванием барботажного слоя оправ-
дано при малых значениях отношения газ: жидкость и в тех слу-
чаях, когда абсорбент содержит взвешенные твердые частицы. Ап-
параты с механическим перемешиванием для системы газ —
жидкость находят применение при проведении химических про-
цессов (см. стр. 382).
В распиливающих абсорберах поверхность межфазовово кон-
такта создается в результате диспергирования жидкости в поток
газа. По принципу работы устройств для диспергирования жидко-
498
с. V1-41), отличающиеся
Газ
Рис. VI-41. Схема устройства абсор-
бера с распылением абсорбента.
сти известные конструкции абсорберов, используемые в химиче-
ской промышленности, делятся на три группы [64]:
1) аппараты, в которых диспергирование осуществлено с по-
мощью форсунок;
2) абсорберы, диспергирование в которых проводится с по-
мощью вращающихся механизмов;
3) аппараты с диспергированием жидкости струей газа.
Распиливающие абсорберы находят сравнительно небольшое
применение в промышленных установках, хотя для хорошо рас-
творимых газов аппараты этого :
простотой конструкции, дают хо-
рошие результаты [65] при отно-
сительно малых эксплуатацион-
ных расходах,
типа абсорбционной
аппаратуры основан на резуль-
татах сравнительной технико-
экономической оценки ряда ва-
риантов аппаратурно-технологи-
ческого оформления процесса с
учетом конкретных условий. Срав-
нительная характеристика основ-
ных групп абсорбционной аппара-
туры, приведенная в табл. VI-2,
облегчает решение этой задачи.
Наряду с совершенствованием
известных способов и улучшением
конструкций ректификационной и
абсорбционной аппаратуры, в ка-
честве средств интенсификации
массообмена в процессах абсорб-
ции и ректификации применяют
пульсацию, высокочастотные ко-
лебания и ультразвук. Использование этих средств, пока не выхо-
дящее за рамки лабораторных исследований, свидетельствует о
возможности значительного прогресса в области интенсификации
промышленных процессов массообмена.
Выбор схемы и режима работы ректификационных и абсорб-
ционных установок составляет одну из важных экономических за-
дач химической технологии, так как эти стадии технологической
схемы в большинстве случаев являются ответственными за энер-
гетические показатели производства и оказывают решающее влия-
ние на удельные капитальные затраты.
Вопросам оптимального проектирования ректификационных
установок в настоящее время уделяется все большее внимание
[66]. Простейшей задачей этого типа является поиск коэффициен-
та избытка флегмы, соответствующего минимальному значению
показателя приведенных затрат (см. гл. I). Задача решается
32*
499
Таблица VI-2
Сравнительная характеристика основных групп
абсорбционной аппаратуры [64]
Пленочные абсорберы Насадоч- ные аб- сорберы Барботажные абсорберы Распыли- вающие абсорберы
Показатели о 3 СС «с л «с «с о X о S X S 3 д $ X X 2S X S 3 5 = « s S о X X X X О СС Si « о 3 S X X S о £ 4) СС X СП
§ ° ° * go 5 ° £ X к * « g я з я Si о X X s X £ я S X СС ф См
S S h S 5 о 4> = «з § ч х S <и <0 X л <У X Л ф с >,
к сс И о « £ X £ О £ X а сс ss- о s £ а р
н о (J я о * О 5- о н О 5- О X о а а и
Возможность осуществления противотока в одной сту- пени + + + + + X
Возможность достижения в од- ном аппарате числа единиц переноса илн числа ступеней: <2 + + + + + + + + + + + + +
2-5 + + X + + + + + + — X X X
5-10 — + X + + + + + + — — — —•
>10 — + — X X + + + + — — — —
Возможность работы с соот- ношением Кж/Уг: <0,001 + + + + + . +
0,001-0,005 + + — X + + + + + — — + +
0,005-0,02 + + — + + + + + + — + + +
> 0,02 — — — + + X — X X + '+ — +
Осуществимость внутреннего отвода тепла + + — — — + + + + — — —
Низкое гидравлическое сопро- тивление + + X + — — — — — + — +
Возможность широкого изме- нения нагрузок по газу и жидкости + + X + + X + X — + X — +
Большое время пребывания жидкости — — + X + + X — + — — +
Возможность работы при на- личии загрязнений X X — — X — + + + + X X +
Количество перерабатывае- мого газа, м/ч'. 1 000 + + + + X + X + + + X + +
1 000-10 000 + + + + + X + + + — + + —
10 000-100 000 X + + + + — + + + — + + X
Простота конструкции .... X X — + + X — X + — + + —
Возможность работы в агрес- сивных средах X X X + + + X X X X + + X
Примечание. + —соответствие требованию; X — частичное соответствие; - не-
соответствие.
500
упрощенными аналитическими или графическими методами либо
с применением ЭВМ.
Более сложен поиск условий экономического оптимума, осно-
ванный на учете нескольких независимых переменных (например,
коэффициента избытка флегмы и давления в колонне).
Необходимость проведения подобного рода работ вполне оче-
видна, так как достигаемый при этом эффект весьма значителен:
отмечается [65], что значение показателя приведенных затрат в ре-
зультате оптимизации процесса по флегмовому числу снижается
на 20—30%.
Рассмотрим процесс разделения бинар-
ной смеси заданного состава при заданном
составе дистиллята и кубового остатка.
Принимая наиболее простую схему
установки (рис. VI-42), состоящей из наса-
дочной колонны 1, конденсатора 2, охлаж-
даемого водой, и куба 3, нагреваемого на-
сыщенным водяным паром, можно выра-
зить суммарный расход, связанный с эк-
сплуатацией установки следующим образом:
Ф = v[Gj + VjWj + v3F2 + V4Q2 + v3F3 + VgQ3 (VI-3)
Рис. VI-42. Упрощенная
схема ректификационной
установки:
1 — колонна; 2 — конденсатор;
3-куб.
где — пересчетные коэффициенты для
выражения стоимости энергетических
средств и размера, амортизационных отчис-
лений в рублях на 1 т продукта (дистил-
лят или кубовая жидкость) или в единицу
времени; F2, F3 — поверхности теплообмена конденсатора и кубо-
вой части колонны; Q2, Q3 — расход энергетических средств (вода.
пар) в конденсаторе и кубовой части колонны (в ккал на приня-
тую выше единицу); G\— вес ректификационной колонны; 11%—
объем насадки ректификационной колонны.
При заданном составе дистиллята режим работы конденса-
тора может быть оптимизирован, исходя из минимума суммы за-
трат, как функция температуры охлаждающей воды на выходе ib
аппарата (см. стр. 233)
при любой нагрузке по пару.
Значение + v'Q2, как слагаемое в уравнении (VI-З) опре-
деляется количеством пара, т. е. является функцией флегмового
числа R.
Из теплового баланса установки следует, что размер поверх-
ности теплообмена и расход пара на обогрев кубовой части ко-
лонны 1 пропорционален R. При заданном давлении в колонне
и постоянной скорости пара диаметр колонны пропорционален
,(1+/?)0,5, в то время как число теоретических тарелок и.
501
следовательно, высота насадки колонны уменьшается с увеличе-
нием флегмового числа по сложной зависимости.
Поиск минимального значения энерго-материальных затрат по
уравнению
Ф = ((Я)
требует учета значений v', v', v', отражающих конкретные
условия организацйи и эксплуатации установки. Значение /?опт за-
висит от относительной стоимости
Рис. VI-43. Условия оптимального
режима работы ректификациоииой
установки:
J, 2,3 —размер амортизационных отчис-
лений для ректификационной колонны,
конденсатора н кубовой части колонны
соответственно; 4 — удельная стоимость
энергетических средств; 5 —суммарные
энерго-материальные затраты
материала аппаратуры и энергети-
ческих средств.
Характер изменения отдельных
слагаемых уравнения (VI-З) с из-
менением избытка флегмы (^?//?мин)
можно видеть на рис. VI-43.
Минимум амортизационных рас-
ходов лежит в области значений
избытка флегмы, приближающихся
к часто рекомендуемым (1,3—1,4),
однако стоимость энергетических
средств, пропорциональная умень-
шению флегмового числа, переме-
щает экстремальную точку в об-
ласть нйзких значений. R тем в
большей степени, чем выше отно-
сительная стоимость энергетических
средств.
Значение избытка флегмы, соот-
ветствующее минимуму суммар-
ных затрат, приближается к еди-
нице, составляя величину порядка
1,05—1,1.
Зависимость Ф(7?) применитель-
но к разделению пропан-пропиле-
новой фракции (пропилен 60%,
пропан 40%) при давлении 12,2 ат
в тарелочной колонне исследована
с применением электронной цифро-
вой машины «Урал» [66].
Результаты расчетов свидетельствуют, что при заданных усло-
виях и принятых для расчета значениях стоимости пара и воды
избыток флегмы составляет 1,09.
Минимальное значение амортизационных расходов лежит в
области более высоких значений R. Последнее обстоятельство со-
ответствует общей закономерности экономики технологической ’
схемы, отмечавшейся ранее.
Показатели работы ректификационной установки, в основу
проекта которой положены произвольно установленные значения •
4-02
коэффициента избытка флегмы, могут значительно отличаться от
их возможных оптимальных значений.
При поиске оптимальных условий процессов ректификации
представляет интерес анализ влияния давления в колонне на раз-
мер суммарных затрат, связанных с эксплуатацией ректификаци-
онной установки.
Увеличение значения ВЭТТ и коэф-
фициента разделения, наблюдаемое при
снижении давления, открывает возмож-
ность достижения минимального объема
насадки при определенном для. каждой
конкретной системы давлении в колон-
не [67—70]. Вполне понятно, что поиск
давления, соответствующего минимуму
суммарных затрат, не может ограни-
чиваться учетом только изменения раз-
меров колонны, необходима также
оценка изменения размера теплообмен-
ных устройств и расхода энергетических
средств.
Изменение давления, сопровождаю-
щееся соответствующим изменением тем-
пературы в верхней и нижней частях ко-
лонны, существенно сказывается на
разности температур в кубе и конден-
саторе и на величине поверхности теп-
лообмена этих частей установки. Тща-
тельного учета требует также расход
энергетических средств (теплоносители,
хладоагенты) и их параметры, изме-
няющиеся при изменении давления в ко-
лонне.
Наряду с флегмовым числом, давле-
ние в ректификационной установке мо-
жет служить основой при оптимизации
ректификационных процессов.
Для разделения многокомпонентной
смеси, как правило, используется систе-
ма последовательно соединенных двух
секционных колонн с выделением на
каждой из них одного компонента или фракции. Ключевыми ком-
понентами разделяемой смеси являются при этом соседние по
летучести продукты, составляющие смесь.
В зависимости от свойств продуктов и условий ректификации
последовательность выделения компонентов может быть различ-
ной (рис. VI-44,а и б). Число двухсекционных колонн в схемах,
представленных на рис. VI-44, а и б, на единицу меньше числа
разделяемых компонентов или фракций. При разделении смесей,
Рис. VI-44. Схемы устано-
вок для отделения трехком-
понентной смеси.
503
кипящих в широком интервале температур, указанные выше схе-
мы дают возможность применения различного давления в каждой
из колонн.
Термодинамически оптимальный вариант схемы ректификации
многокомпонентной смеси отличается двумя основными особенно-
стями [71]: ключевыми компонентами в каждой двухсекционной
колонне являются не соседние, а крайние по летучести компо-
ненты и тепловые потоки всех двухсекционных колонн связаны
между собой. Пример такой схемы, применительно к разделению
трехкомпонентной смеси дан на рис. VI-44, в. Использование по-
добной схемы для ректификационного разделения смеси хлорпро-
изводных метана на три фракции (состав смеси приведен в табл.
VI-З) позволяет существенно сократить расход пара и воды и
уменьшить количество теплообменных устройств в сравнении со
схемой, показанной на рис. VI-44, а.
Таблица VI-3
Состав смеси хлорпроизводных метана
Компоненты •Состав исходной смеси, масс. % Состав фракций, масс. %
АВ АВС вс
СН2С12 (А) 17,05 74,3 0,00132 —
СНС13 (В) 62,06 25,7 99,9287 7,65
СС14 (С) 20,35 — 0,0700 92,35
Результаты расчетов, выполненных для двух вариантов разде-
ления указанной выше смеси [72], приведены в табл. VI-4.
Таблица VI-4
Расчетные данные для двух вариантов разделения смеси
хлорпроизводных метана
Показатели Схема установки
рнс. VI-44, а рис. VI-44, б
Суммарное количество испаряемой жидкости, моль/100 моль сырья ............ 579 255
Суммарное количество сконденсированного пара, моль/100 моль сырья 500 237
Число теоретических тарелок 112 145
Число кипятильников и дефлегматоров 4 2
Особенность схем со связанными тепловыми потоками — прак-
тически постоянное давление во всех частях и аппаратах схемы.
Это обстоятельство затрудняет разделение смесей, кипящих в ши-
504
роком интервале температур, и ограничивает возможную область
применения схем со связанными тепловыми потоками разделением
смесей близко кипящих продуктов.
АДСОРБЦИЯ
Адсорбционные методы находят применение главным
образом при регенерации паров органических веществ из паро-
газовых смесей, а также для очистки жидких продуктов.
В качестве промышленных адсорбентов наиболее распростра-
нены силикагель и активированный уголь, применяемые в виде
зерен размером от 1 до 5—7 мм.
Отдельные сорта силикагеля и активированного угля разли-
чаются размером пор (мелкопористые, крупнопористые) и физи-
ко-механическими свойствами, в зависимости от чего определяют-
ся области их применения. х
Основные свойства некоторых промышленных адсорбентов при-
ведены в табл. VI-5.
Таблица VI-5
Основные свойства силикагеля и активированного угля
Показатели Силикагель Активирован- ные угли
мелко- пористый крупно- пористый
Насыпная плотность, г/см? Объем пор, см3)г Радиус пор, А Удельная поверхность, л2/г 0,8 0,28 5-30 450-500 0,5 0,9 70-100 270-350 0,2-0,6 < 70 600-1700
Значительная гидрофильность силикагеля (количество воды,
поглощаемой силикагелем, достигает 50% от его массы; равно-
весное влагосодержание силикагеля в воздухе при 60% относи-
тельной влажности доходит до 24%), снижающая его адсорбци-
онную способность по отношению к органическим веществам,
делает силикагель в ряде случаев мало пригодным при рекупе-
рации паров органических жидкостей; для осушки газов чаще
всего применяется силикагель.
Активированный уголь гидрофобен и предпочтителен как ад-
сорбент органических веществ. К числу недостатков активирован-
ного угля относится его горючесть, ограничивающая температуру
десорбции (в газовой среде, содержащей кислород) величиной
200° С (для силикагеля максимально допустимая температура
равна 300°С и выше).
В большинстве промышленных установок используется перио-
дически действующая аппаратура, попеременно включаемая в
поток перерабатываемой парогазовой смеси. Различают ряд
505
вариантов осуществления процесса, имеющего стадии адсорбции,
десорбции, сушки и охлаждения адсорбента: применяется четы-
рехфазный способ, при котором указанные стадии осуществляются
последовательно, и двухфазный, при котором процесс адсорбции
проводится одновременно с охлаждением и высушиванием адсор-
бента (после регенерации его горячей паровоздушной смесью)-
Преимущества двухфазного способа [73] делают его более целе-
сообразным.
Регенерация активированного угля производится путем обра-
ботки его водяным паром (при четырехфазном режиме работы)
или смесью воздуха и водяного пара (при двухфазном режиме
работы). Регенерация силикагеля осуществляется путем нагрева
его до 300° С.
Представляет интерес применяемый для разделения углеводо-
родных газов процесс адсорбции в движущемся слое активиро-
ванного угля (гиперсорбция). Все стадии цикла осуществляются
одновременно в разных по ходу потока адсорбента зонах аппа-
рата при противотоке газа и адсорбента. Возможность непрерыв-
ного вывода части адсорбента для реактивации (обработка угля
паром при более высокой, чем на стадии десорбции температуре)
способствует сохранению активности адсорбента при длительной
работе установки. Этот способ разделения углеводородных газов
отличается высокой производительностью адсорбента. Работа ус-
тановки может быть полностью автоматизирована; стационарный
режим процесса облегчает возможность поддержания оптималь-
ных условий и обеспечивает высокую степень разделения компо-
нентов газовой смеси.
Преимущества движущегося слоя используются также приме-
нительно к силикагелю, регенерация которого осуществляется пу-
тем нагрева.
Схема и аппаратура установок для адсорбции газов и паров
описана в общих [1—5] и специальных [73, 74] работах.
Процессу адсорбции в кипящем слое присущи все достоинства
и недостатки этой системы. Интенсивное продольное перемешива-
ние зерен адсорбента в кипящем слое неблагоприятно сказывается
на величине движущей силы процесса. Равномерное распределе-
ние зерен, насыщенных адсорбированным компонентом, по высоте
слоя может быть причиной десорбции в верхних зонах кипящего
слоя и обогащения потока выходящего газа адсорбированным
компонентом.
Указанное обстоятельство, снижающее степень очистки газо-
вой смеси, а также повышенные требования к механической проч-
ности зерен адсорбента являются недостатками этого метода. Не-
желательный эффект продольного перемешивания устраняется
при использовании многослойных аппаратов.
Селективность адсорбентов по отношению к отдельным ком-
понентам смесей используется и при разделении жидких продук-
тов.
506
В качестве адсорбентов применяются крупнозернистые сорта
активированного угля и силикагеля, а также отбеливающие гли-
ны— флоридины и бентониты. Первые из этих глин обладают до-
статочной. адсорбционной активностью и перед применением под-
вергаются лишь термической активации (прокаливание при 150—
400°С для удаления адсорбированной воды); вторые нуждаются в
кислотной активации — обработке разбавленной соляной или сер-
ной кислотой при нагревании.
Адсорбционные свойства отбеливающих глин объясняются не
только величиной удельной поверхности пор (колеблющейся в пре-
делах от 10 до 200 л2/г), но и химическими свойствами этой по-
верхности.
Отбеливающие земли используются при очистке смазочных и
растительных масел, водных растворов карбоновых кислот (на-
пример, в производстве адипиновой кислоты), в пищевой и фар-
мацевтической промышленности.
В зависимости от масштаба производства очистка с помощью
активированного угля производится или в периодически Действую-
щих аппаратах с мешалками, где мелко раздробленный уголь
суспендируется в очищаемой жидкости или в непрерывнодей-
ствующих системах, представляющих собой либо каскад колонн,
заполненных слоем угля, либо колонны с движущимся (или пуль-
сирующим) слоем угля [75, 76].
Многоколонные системы, используемые при необходимости до-
стижения высокой степени очистки, дают возможность повышения
степени использования адсорбента.
Число последовательно или параллельно работающих аппара-
тов многоколонной системы, продолжительность работы каждого
из них (до момента выключения для регенерации или перегрузки-
адсорбента), а также удельный расход адсорбента зависят от со-
держания примесей в исходном растворе и заданной степени его
очистки и определяются экспериментально установленной емкостью
угля.
Кроме установок с неподвижным слоем угля, используются
также установки с движущимся или пульсирующим слоем. Этот
вариант дает возможность осуществлять непрерывный вывод от-
работанного адсорбента и непрерывную его регенерацию.
После отделения от очищенной жидкости (фильтрование, цен-
трифугирование) адсорбент может быть регенерирован с помощью
экстракции или прокаливания. Первоначальная активность обыч-
но не восстанавливается, и в большинстве случаев регенерация
оказывается экономически нецелесообразной, а отработанные от-
беливающие земли после обжига используются как сырье в кир-
пичном и цементном производствах.
Ряд задач технологии современной химической промышленно-
сти не может быть решен без учета возможностей гетерогенного
ионного обмена, находящего все более широкую область приме-
нения [77, 78].
507
В качестве ионообменных сорбентов применяются вещества
минерального происхождения (алюмосиликаты, силикаты, гидрат
окиси алюминия и др.), а также синтетические высокомолекуляр-
ные соединения с сетчатой или пространственной структурой,
По типу ионогенных групп иониты разделяют на нераствори-
мые кислоты—катиониты и нерастворимые основания—аниониты.
По степени ионизации иониты делятся на сильно- и слабокис-
лотные катиониты и сильно- и слабоосновные аниониты.
Наибольшее значение в промышленности имеют полимерные
иониты, отличающиеся высокой поглотительной способностью, ме-
ханической прочностью и химической устойчивостью [79].
Извлечение ионов из раствора осуществляется путем фильтро-
вания через слой гранулированного ионита. Для извлечения со-
лей из растворов применяют последовательно соединенные слои,
одни из которых заполнены катионитом, другие анионитом. В слое,
состоящем из катионита, происходит присоединение катионов соли
ц вытеснение водорода; в анионитовой колонне анионы соли вы-
тесняют гидроксильные группы анионита.
Ионный обмен происходит в эквивалентных количествах и в
большинстве случаев — обратим. Кинетика гетерогенного ионного
обмена имеет диффузионный характер (зависимость от размера
зерен, режима потока).
При достижении предельного для каждого ионита значения
емкости (в мг-экв!г) производится регенерация ионообменной
смолы путем промывки катионитового слоя раствором кислоты, а
анионитового слоя — водным раствором щелочи.
В промышленном органическом синтезе ионообменные процес-
сы находят применение для очистки продуктов от следовых коли-
честв примесей. Примером может служить очистка адиподинитри-
ла и капролактама [80, 81]. Заслуживают внимания работы, по-
священные поиску экономического оптимума промышленных
ионообменных процессов [82]. Ионный обмен позволяет отделить
катионы от анионов, истинные электролиты от коллоидов, элект-
ролиты от неэлектролитов.
Процессы конденсации, абсорбции и адсорбции как способы
промышленного разделения газовых и парогазовых смесей могут
оцениваться по степени полноты разделения смеси компонентов в
конкретных условиях. Исходя йз физико-химических зависимостей,
присущих каждому из этих процессов, нетрудно прийти к выводу
о целесообразной последовательности их применения для любого
начального и конечного состава разделяемой системы:
Конденсация —> абсорбция —> адсорбция
При заданных начальном и конечном составах разделяемой
смеси энерго-материальные затраты, связанные с осуществлением
каждого из указанных выше процессов, являются функцией со-
става разделяемой смеси и состава ее после разделения на каждой
из последовательно осуществляемых стадий.
608
Предположим, что разделению подвергается заданное часовое
количество парогазовой смеси состава Б + А при заданном на-
чальном содержании компонента Б (сБ ) и конечном его содержа-
нии (сБ ) путем последовательного использования процессов кон-
денсации, абсорбции и адсорбции.
Очевидно, что в конкретных условиях
практические границы целесообразности при-
менения каждого из способов разделения,оп-
ределяются концентрацией компонента Б на
выходе из соответствующего аппарата (при
достижении которой движущая сила процес-
са стремится к нулю, а размеры аппарата
безгранично увеличиваются). В пределах этих
границ суммарные энерго-материальные зат-
раты монотонно меняются с изменением кон-
центрации компонента Б на входе или на вы-
ходе потока разделяемой смеси в соответ-
ствующей стадии.
Если рассматривать схемы двухстадийно-
го разделения парогазовой смеси (конденса-
ция — абсорбция; абсорбция — адсорбция), то
суммарные энерго-материальные затраты как
функция содержания в смеси компонента Б
между стадиями имеют минимальное значе-
ние при некотором оптимальном для каждых
конкретных условий значении сд (1—2) и сд(2-з)
(рис. VI-45).
В том случае, когда разделение смеси осу-
ществляется с применением процессов конден-
сации, абсорбции и адсорбции подобная за-
дача сводится к нахождению минимума функ-
ции двух переменных. Решение такой за-
дачи требует строго количественного описа-
ния ЗавИСИМОСТИ Ф (f£(i-2), Сд (2-3))-
При отсутствии данных для подобного опи-
сания область оптимальных значений сб п-2)
и Сб (2-з) может быть установлена графо-аналитическим методом
путем нахождения минимума суммы оптимальных значений энер-
го-материальных затрат для каждой пары стадий на основе про-
извольно задаваемых значений концентрации компонента Б на
выходе из стадии конденсации или абсорбции.
Абсорбция | Адсорбция
Сбц ^Б(г-З) СБК
Рис. VI-45. Оптималь-
ные условия разделе-
ния парогазовой сме-
си:
1 — расходы на стадии
конденсации; 2 — расходы
на стадии абсорбции;
3 — расходы на стадии ад-
сорбции; 4, 5 —суммар-
ный расход средств.
СПЕЦИАЛЬНЫЕ МЕТОДЫ РАЗДЕЛЕНИЯ
При разделении сложных смесей продуктов промыш-
ленного органического синтеза или при необходимости очистки
промежуточных или конечных продуктов от примесей в ряде
509
случаев возможности таких процессов как ректификация, абсорб-
ция или десорбция оказываются либо недостаточными, либо тех-
нологически нецелесообразными.
Решение подобного рода задач может быть основано на ис-
пользовании ряда специальных методов разделения:
1) зонная плавка или зонная кристаллизация;
2) адсорбция на молекулярных ситах;
3) диализ и электродиализ.
Некоторые из этих методов применяются в ряде отраслей про-
мышленности, другие — на лабораторных установках.
Расширение номенклатуры продуктов промышленного органи-
ческого синтеза и повышение требований к чистоте продуктов хи-
мического производства ставят вопрос о необходимости привлече-
ния в технологию основного органического синтеза новых приемов
и методов, среди которых новые методы разделения представляют
значительный интерес.
ЗОННАЯ ПЛАВКА
Зонная плавка, т.е. метод получения продуктов высокой
степени чистоты путем многократной перекристаллизации из рас-
плава, применяется при очистке ряда металлов.
Метод заключается в перемещении легкоплавкого и тяжело-
плавкого компонентов смеси к противоположным концам колон-
ны, заполненной разделяемой смесью. Этот процесс осуществляет-
ся благодаря перемещению вдоль оси колонны зоны нагрева.
Применение зонной плавки пока ограничивается масштабами
небольших установок периодического действия [83, 84]. Намечает-
ся тенденция промышленного использования этого метода [84].
АДСОРБЦИЯ НА МОЛЕКУЛЯРНЫХ СИТАХ
Разделение смеси газообразных продуктов может быть
осуществлено с помощью молекулярных сит [74, 85]. Молекуляр-
ные сита, получаемые при дегидратировании цеолита или его мо-
дификаций (природных или искусственно приготовленных), при
пропускании через них смеси газообразных веществ сорбируют
молекулы, размеры которых соответствуют размерам пор кри-
сталлов материала сита (3—5А). Молекулярные сита задержи-
вают преимущественно молекулы полярных, ненасыщенных и аро-
матических соединений и способны поглощать влагу из газов даже
при сравнительно высоких температурах, когда другие адсорбен-
ты теряют способность выполнять функции осушителей.
Использование сорбционных свойств молекулярных сит позво-
ляет успешно решать задачи очистки от примесей и сушки газо-
образных и жидких продуктов. Применение этого метода дает
возможность понижать содержание примесей до 1-10~4% (очистка
этилена от СОг, удаление влаги из жидких углеводородов и т. д.).
Высокая сорбционная селективность дегидратированных цео-
литов может быть использована при разделении азеотропных си-
510
стем (метиловый спирт — ацетон, этиловый спирт — толуол; серо-
углерод— ацетон и др.).
Схема установки состоит из нескольких параллельно включен-
ных и наполненных адсорбентом аппаратов, попеременно находя-
щихся в стадии абсорбции и десорбции.
Регенерация молекулярных сит производится либо при повы-
шении температуры (максимально допустимая температура 500—
600°С), либо при пропускании неадсорбируемого газа.
Свойствами, аналогичными свойствам цеолитов, обладают по-
ристые стекла, имеющие значительные перспективы для исполь-
зования.
пропускной спо-
Обогащенная
смесь
Рис. VI-46. Схема установки
для разделения смеси изо-
пропиловый спирт — этиловый
спирт — вода:
/ — ректификационная колонна;
2 — конденсатор; 3 — холодильник;
4 — диффузионная камера.
РАЗДЕЛЕНИЕ С ИСПОЛЬЗОВАНИЕМ МЕМБРАН
Для разделения газовых смесей могут быть использова-
ны пористые мембраны, изготовленные из пористого стекла. Имею-
щиеся данные свидетельствуют о зна
собности разделительных установок
такого типа, однако селективность
разделения при этом сравнительно
невелика [74].
Значительной селективности сле-
дует ожидать от применения непори-
стых мембран [86—91]. Механизм про-
хождения веществ через непористые
мембраны принципиально отличен от
механизма, свойственного пористым
мембранам. Для мембран, изготов-
ленных из органических материалов,
процесс состоит из трех последова-
тельных стадий: 1) растворения ве-
щества в материале мембраны; 2) диф-
фузии вещества по направлению к про-
тивоположной поверхности мембраны
и 3) перехода продиффундировавшего
вещества с поверхности мембраны.
Характеристикой вещества мембраны является растворимость
в нем компонентов разделяемой смеси и скорость селективной
диффузионной проницаемости целевого компонента. Выбор мем-
браны диктуется природой разделяемой смеси и должен быть ос-
нован на результатах экспериментального исследования.
В большинстве работ, посвященных изучению этого явления, в
качестве материала мембран использованы высокомолекулярные
соединения (резина, каучук, полистирол, хлоропрен, полиэтилен и
др.). Для практического применения пригодны пленки толщиной
до 25 мм. Производительность мембраны сравнительно невелика
и не превышает нескольких килограммов вещества в 1 ч с 1 м2
поверхности мембраны [74, 92].
511
Схема установки для обезвоживания смеси изопропилового и
этилового спиртов приведена на рис. VI-46. Азеотропная смесь
изопропиловый спирт — этиловый спирт —вода, получаемая в ре-
зультате ректификации и содержа-
Рис. VI-47. Аппарат для диффу-
зионного разделения газовой
смеси (трубы из полиэтилена).
щая воду в количестве >0,5%, по-
ступает в диффузионную камеру.
Обогащенный водой поток направ-
ляется в исходную систему, обезво-
женная смесь (содержание воды
<0,5%) представляет собой гото-
вый продукт. Устройство аппарата,
предназначенного для разделения
легких углеводородов (материал
пленки—полиэтилен), показано на
рис. VI-47.
Непористые мембраны пригод-
ны для разделения как газообраз-
ных, так и жидких близкокипящих
продуктов.
В ряде случаев определенный интерес может представить воз-
можность освобождения коллоидных растворов и растворов высо-
комолекулярных соедине-
ний от истинно раствори-
мых в них низкомолеку-
лярных соединений при
помощи полупроницае-
мых мембран (диализ).
В качестве мембран
при разделении органиче-
ских веществ используют-
ся пленки из нитро- и
ацетилцеллюлозы, поли-
этилена, полипропилена,
поливинилхлорида, поли-
этилентерефталата и дру-
гих веществ.
В промышленном мас-
штабе диализ применяет-
ся для утилизации соды
из отходов производства
вискозы, для очистки же-
Перемеш,аюш,ии поток
поток
латины, а также при из- Рис. VI-48. Установка для электродиализа
готовлении некоторых с селективными мембранами:
фармацевтических пре- /, 2~ ячейки; А —анион; К+— катион.
паратов [93].
Для ускорения диффузии ионов при диализе на обрабатывае-
мый раствор накладывается электрическое поле постоянного тока
(электродиализ). Процесс проводится в многоячейковых агрега-
512
тах, обладающих достаточно высоким электрическим к. п. д.
(рис. VI-48}.
Электрический ток, проходя в поперечном направлении через
ячейки, вызывает движение анионов к аноду, а катионов к катоду.
Анионные и катионные мембраны концентрируют соответствующие
ионы в ячейках /, из которых продукты удаляются потоком
жидкости А. Поток, питающий аппарат, поступает в ячейки 2, от-
куда он выходит освобожденный от части растворенных в нем
низкомолекулярных веществ.
Процесс электродиализа применяется при обессоливании мор-
ской воды. В технологии основного органического синтеза элект-
родиализ используется при производстве себациновой кислоты [93].
ОЧИСТКА ПРОДУКТОВ
ПРОМЫШЛЕННОГО ОРГАНИЧЕСКОГО СИНТЕЗА
В последние годы проблема получения и анализа чи-
стых веществ стала одной из важнейших в современной химии и
технологии. Постановка различных химических, биологических и
других экспериментов, а также создание процессов получения ле-
карственных веществ, красителей и даже таких многотоннажных
продуктов, как мономеры, должны проводиться с веществами вы-
сокой чистоты. Степень чистоты исходных веществ и конечных
продуктов, используемых в различных областях народного хозяй-
ства, оказывает огромное влияние на эффективность и произво-
дительность процессов.
Примеси, находящиеся в веществах, можно разделить на две
категории: 1) примеси, генетически связанные с веществом и 2)
примеси, проникшие в вещество в результате небрежного хране-
ния (загрязнения из атмосферы, тары и другие) или за счет кор-
розии либо эрозии на различных стадиях технологической схемы.
Примеси, генетически связанные с веществом, могут быть двух
видов: 1) примеси, происходящие из сырья, и 2) побочные про-
дукты технологической переработки.
Естественно, что выделение и изучение состава примесей яв-
ляется неотъемлемой частью общей проблемы получения чистых
веществ.
Некоторые примеси, как правило, механические загрязнения,
можно различить под лупой, другие — отделяются и определяются
просеиванием, перекристаллизацией, перегонкой, титрованием и
т. д. Для получения чистых веществ на современном уровне эти
методы уже оказываются неудовлетворительными и будут менять-
ся по мере совершенствования техники очистки и повышения чув-
ствительности и точности методов анализа.
Получение веществ особой чистоты стало особо необходимо в .
связи с развитием атомной, ракетной и .электронной техники. По-
требовались образцы графита, урана, бериллия, германия и дру-
гих веществ, содержащие не более 10~6% примесей.
33 В, С, Хайлов, Б. Б, Брандт. ' 513
Важнейшее значение для- развития химии в целом имеет вы-
пуск чистых химических реактивов, номенклатура которых исчис-
ляется тысячами наименований.
Бурное развитие химии в технологии полимеров во многом
объясняется тем, что для их получения использовались мономеры
высокой степени чистоты.
От наличия той или иной примеси в мономере при его полиме-
ризации может происходить, с одной стороны, «сшивание» макро-
молекул и образование трехмерной структуры (в результате та-
кого вмешательства посторонних веществ меняются основные
свойства полимера — текучесть, жесткость, растворимость и т. д.);
с другой стороны, частица постороннего вещества может оборвать
рост цепи макромолекулы за счет реакции с активным центром.
В результате резко снижается молекулярная масса полимера и
ухудшаются многие его ценные качества. Наличие вредных при-
месей может привести к увеличению доли изделий второго и тре-
тьего сортов и, как следствие, к резкому повышению себестоимо-
сти выпускаемой продукции в целом. В то же время часто необ-
ходимо сознательно вносить определенные примеси в мономеры
для регулирования процесса полимеризации. Например, при до-
бавке определенного количества уксусной и аминокапроновой кис-
лот получают поликапрамид с заданной молекулярной массой.
Наконец, без изучения состава примесей, пусть даже в микро-
количествах, невозможно подходить к выяснению механизма раз-
личных реакций.
Борьба за чистоту веществ определяется самой сущностью лю-
бого нового технологического процесса. Она оказывает большое
влияние на проведение процесса в оптимальном режиме, комп-
лексном использовании сырья и оборудования, исключает кор-
розию и побочные процессы. Например, процесс получения бен-
зойной кислоты прямым окислением толуола требует более высо-
кокачественного сырья, чем многостадийные процессы.
В последние годы во всем мире осуществляется пересмотр
стандартов на различные промышленные изделия, направленный
на дальнейшее снижение предельно допустимого содержания при-
месей.
Без знания состава примесей невозможно разработать рента-
бельный способ очистки и разделения многих промышленных ре-
акционных смесей. Как правило, чем сложнее продукт и техноло-
гический процесс его получения, тем больше химических и техно-
логических стадий, а следовательно, больше побочных реакций и
образующихся примесей.
Для получения товарного продукта необходимого качества
можно использовать очистку конечного продукта, но можно также
применять кондиционирование исходного сырья и промежуточных
продуктов по стадиям.
Опыт показывает, что наиболее успешных результатов удается
достичь при очистке продуктов по всем стадиям, включая более
Б14.
тщательную предварительную очистку основного химического
сырья (серной и азотной кислоты, бензола, толуола, природных
газов и т. д.).
На практике обычно ограничиваются очисткой лишь на двух-
трех стадиях процесса, включая конечную, причем основой для
выбора схемы очистки служит экономика процесса в целом.
Выбор метода очистки в большой мере зависит от тоннажа
продукта. Сложная и многостадийная очистка оказывается пол-
ностью оправданной по отношению к продукту процесса, масштаб
которого исчисляется десятками тысяч тонн в год, но совершенно
не приемлема для процесса, рассчитанного на десятки тонн или
сотни килограммов.
Разумеется, сказанное справедливо в общем случае и не отно-
сится к таким процессам, которые связаны с получением неболь-
ших количеств дорогостоящих продуктов особой степени чистоты,
например, к производству полупроводниковых материалов или ле-
карственных препаратов.
Одним из факторов, сильно затрудняющих очистку продуктов,
является большое разнообразие примесей в них как по количест-
венному, так и по качественному составу, включая различия в хи-
мической природе ш структуре. Образование большого числа при-
месей зависит не только от различных химических и технологиче-
ских схем получения одного и того же продукта, но и от различий
в режиме проведения одного и того же процесса. Часто образова-
нию примесей способствуют отдельные нарушения режима кото-
рых по тем или иным причинам еще не удается избежать: коле-
бания температуры, соотношения реагентов, время пребывания в
реакционной зоне и т. д. Отсюда вытекают два требования, обяза-
тельных при разработке промышленного способа очистки какого-
либо органического продукта.
Во-первых, такой способ должен разрабатываться строго при-
менительно к продукту, который производится по химической и
технологической схеме, работающей в данном режиме, т. е. при-
менительно к продукту данного конкретного завода.
Во-вторых, разработанный способ очистки может быть эффек-
тивным лишь при условии, что колебания технологического режи-
ма схемы синтеза сведены до минимума, при котором состав при-
месей в исходном сырье, а также в продукте, поступающем на
очистку, остается постоянным.
Так как в каждом продукте обычно содержится целый ряд
примесей, резко отличающихся по химической природе и физиче-
ским свойствам, решить задачу очистки с помощью применения
какого-либо одного вида обработки не удается. Для этой цели
всегда приходится прибегать к использованию комбинации из не-
скольких видов обработки продукта. Выбор этой комбинации дол-
жен осуществляться на основе знания природы и состава приме-
сей. Однако такие сведения, как правило, отсутствуют, и решение
задач, связанных с очистцой, осуществляется путем эксперимента
33*
515
в каждом отдельном случае, т. е. путем эмпирического подбора
такой комбинации видов обработки, которая позволяла бы полу-
чить продукт нужной кондиции.
Немаловажную роль в создании промышленной схемы получе-
ния веществ высокой степени чистоты играет общая обстановка в
цехах и лабораториях, где вещества могут загрязняться в резуль-
тате контакта с оборудованием и окружающей средой. Чистота
веществ во многом зависит так же от общей и химической куль-
туры обслуживающего персонала. Современный уровень химической
и физической науки не всегда, к сожалению, позволяет оценить чи-
стоту вещества путем прямого анализа состава взятого образца,
хотя этот путь получения сведений об истинной степени чистоты
вещества в принципе наиболее достоверен. Поэтому в настоящее
время во всем мире ведутся интенсивные разработки методов об-
наружения ультрамалых количеств примесей прямым путем.
В большинстве случаев используют косвенную оценку степени
чистоты путем измерения одной или нескольких физических кон-
стант (например, температуру плавления, кипения, электропро-
водности) или, отделяя сумму примесей, родственных по химиче-
ским свойствам (определения летучих оснований, зольности и
т. д.). В последние годы приобрел огромное значение анализ ор-
ганических примесей методом газо-жидкостной хроматографии,
позволяющий разделять вещества на колонках с эффективностью
в десятки и сотни тысяч теоретических тарелок.
В некоторых случаях удается сначала сконцентрировать при-
меси (например, зонной плавкой и зонным вымораживанием, эк-
стракцией, перекристаллизацией), а затем подвергнуть концент-
рат разделению и анализу полученных компонентов.
Более подробно указанные вопросы могут быть рассмотрены и
проиллюстрированы на примере капролактама, мировое производ-
ство Которого в настоящее время приближается к миллиону тонн.
Ниже (стр. 517) приведены все известные схемы получения
капролактама.
Сопоставление этих схем показывает, что все они базируются
главным образом на бензоле и предусматривают синтез разных
промежуточных продуктов. Совершенно очевидно, что капролак-
там, получаемый по каждой из приведенных схем, будет содер-
жать, во-первых, примеси, приносимые с бензолом и другими
продуктами, общими для всех способов (такими как серная кис-
лота, водород, азот, аммиак), а также возникающие на стадиях,
общих для всех методов (перегруппировка циклогексаноноксима в
капролактам), и, во-вторых, примеси, связанные с превращения-
ми, присущими только данной схеме. Таким образом, капролак-
таму, полученному различными способами, сопутствуют разные
суммы примесей. В ГОСТ, лимитирующий качественные характе-
ристики выпускаемого мономера, заложены следующие главные
показатели:
1) перманганатное число капролактама;
516
2) содержание летучих оснований;
3) окраска 50% водного раствора;
4) температура застывания.
Под перманганатным числом понимают восстановительную
способность 1 % водного раствора капролактама по отношению к
водному раствору перманганата калия. Поскольку сам капролак-
там является веществом, устойчивым к окислению в данных усло-
виях, разрушение перманганата происходит за счет примесей,
способных окисляться. Степень чистоты продукта характеризуется
временем сохранения растворов окраски перманганата.
Летучие основания определяются как сумма веществ, отгоняю-
щихся с водяным паром из щелочного раствора капролактама и
способных связывать серную кислоту.
Окраска 50% водного раствора определяется путем сравнения
с эталонными растворами, приготовленными из специальных ре-
активов.
Вполне понятно, что каждый из трех перечисленных пока-
зателей носит абстрактный характер. Нетрудно, например,
517
представить себе наличие такой примеси, которая, не обладая
восстановительными свойствами, не сказывается на перманганат-
ном числе, не отгоняется с водяным паром и не способствует
-окрашиванию водного раствора капролактама, но, в то же время,
может оказаться крайне вредной при дальнейшей переработке ка-
пролактама. И наоборот, в продукте может присутствовать незна-
чительная примесь, которая обладает способностью ухудшать
один из трех, или даже все три приведенные выше показателя,
но не оказывает ощутимого вредного влияния на синтез полимера
и изделия из него. В то же время, согласно ГОСТу, качество ка-
пролактама, содержащего первую примесь, будет выше, чем каче-
ство капролактама, содержащего вторую примесь. В этих усло-
виях, не представляется возможным найти сколько-нибудь зако-
номерную связь между качеством полимера и качеством мономера,
определяемым по показателям ГОСТа.
Таким образом, ГОСТ в вопросе очистки капролактама обла-
дает, по меньшей мере, двумя серьезными недостатками: во-пер-
вых, приводит к искусственному отождествлению капролактама,
получаемого по разным химическим и технологическим схемам и
ввиду этого представляющего собой разные системы с различ-
ными свойствами: во-вторых, совершенно не вскрывает какую-
либо связь между приведенными в нем прказателями и качеством
поликапрамида.
Естественный и правильный выход из положения, казалось бы,
состоит в том, чтобы оценивать качество капролактама по содер-
жанию в нем конкретных примесей, предварительно изучив влия-
ние каждой из них на свойства получаемого полимера и конечных
изделий. Над этим работают сейчас многие исследователи, однако
задача эта весьма трудна, так как современный технический уро-
вень предъявляет очень высокие требования к выпускаемым мо-
номерам. Содержание примесей в капролактаме, подлежащем
очистке, не превышает 1 -10-2—1 • 10~3%. Прямое качественное и
количественное определение большинства примесей, содержа-
щихся в таких концентрациях, невозможно ни одним из суще-
ствующих методов. В связи с этим задача выделения, идентифи-
кации и количественной оценки примесей к капролактаму привела
к необходимости предварительного концентрирования примесей,
что также требует значительных усилий.
Таким образом, разработка методов очистки капролактама на
основе томного знания качественного и количественного составов
примесей и учета их свойств является делом будущего. В ожида-
нии того, когда это станет возможным, приходится разрабатывать
методы очистки путем эмпирического подбора различных видов
обработки капролактама. Оценка полученных результатов произ-
водится определением показателей ГОСТа в обработанном про-
дукте, которые при всех указанных выше недостатках в общем
случае дают относительную характеристику чистоты продукта.
Использование такого метода оценки принято в Советском Союзе
518
и за рубежом, так как до настоящего времени других критериев
чистоты капролактама не существует.
Эмпирическим путем найдено и предложено большое количе-
ство различных методов очистки капролактама. Подавляющее
большинство методов описано в патентной литературе. Обилие и
разнообразие предложенных методов подтверждают невозмож-
ность единого универсального промышленного способа очистки.
Следует говорить лишь о способе очистки капролактама, полу-
чаемого по данной схеме, на данном заводе.
Все предложенные методы могут быть разделены на три груп-
пы: 1) физические методы; 2) химические методы; 3) физические
методы в сочетании с химическими.
К числу физических методов относятся такие, как сорбция, эк-
стракция, ректификация, перекристаллизация. К группе химиче-
ских принадлежат методы, предусматривающие обработку капро-
лактама окислителями, восстановителями, кислотами, щелочами,
аминами, альдегидами и т. д. Для этой цели используются, на-
пример, водород, кислород воздуха, перманганат калия, гипохло-
рит натрия, бромная вода, серная кислота, олеум, уксусный ан-
гидрид, едкий натр, уротропин, формальдегид и другие вещества.
Разнообразие примесей содержащихся в капролактаме, за-
ставляет применять различные методы очистки.
Физические методы очистки, оказывая положительное влияние
на качественные характеристики капролактама, во многих слу-
чаях, однако, оказываются недостаточно эффективными. Кроме
того, осуществление их нередко связано со значительными труд-
ностями в аппаратурном оформлении и требует больших энерго-
затрат.'
Применение химических методов почти всегда благотворно
влияет на качественные показатели капролактама, не вызывает
серьезных затруднений при реализации в промышленном масшта-
бе, но таит в себе опасность маскировки примесей, т. е. превраще-
ния одних примесей в другие, которые не сказываясь на таких
показателях, как перманганатное число, содержание летучих ос-
нований, окраска, температура застывания, способны ухудшить
качество полимера и изделий из него.
Наиболее оптимальные варианты очистки капролактама осно-
ваны на использовании комбинации физических методов с хими-
ческими. Эмпирический характер поисков при разработке спо-
собов очистки не означает полного отсутствия ориентирующих
факторов. Так, низкое перманганатное число капролактама, для
которого необходимо разработать метод очистки, указывает на
присутствие вторичных и первичных, аминов, карбонильных соеди-
нений, нитрозосоединений, или оксимов. Повышенное содержание
летучих оснований дает повод Считать, что в качестве примесей
капролактам содержит различные амины и некоторые непредель-
ные соединения, способные реагировать с серной кислотой. Ухуд-
шенный показатель окраски свидетельствует о наличии в качестве
519
Капролактам ^Капролактам
из толуола из анилина
Рис. VI-49. Схема очистки капролактама.
520
1
Чистота продукта I
примесей соединений, содержащих хромофорные группы. Однако
такие данные не могут служить основанием для однозначного вы-
бора способа очистки капролактама, и многие вопросы решаются
путем простого подбора. В отдельных случаях технологическая
схема очистки оказывается сложнее схемы самого синтеза капро-
лактама. Примером может служить очистка капролактама, полу-
чаемого из толуола и анилина (рис. VI-49).
Получение продукта промышленного органического синтеза
высокой степени чистоты связано с значительными энерго-мате-
риальными затратами, размер1 которых экспоненциально возра-
стает по мере уменьшения концентрации
примесей.
Количественная оценка влияния чисто-
ты очищаемого продукта (продукт /) на
показатели последующих стадий его пере-
работки в общем виде невозможна, однако
не вызывает сомнения, что критерием оцен-
ки чистоты продукта / могут служить та-
кие показатели, как продолжительность
работы катализатора, используемого при
переработке продукта I в продукт //, вы-
ход продукта II, синтезируемого на основе
продукта /, или количество изделий перво-
го и второго сорта (различающихся по по-
требительской стоимости), получаемых при
переработке продукта I.
Ясно, что затраты, связанные с полу-
чением продукта // (продукт промышлен-
ного органического синтеза или изделие)
жаютСя по мере повышения чистоты этого продукта.
Характер зависимости размера затрат на очистку продукта I
и размера затрат на переработку продукта / в продукт // от
степени чистоты продукта I — различны; сумма затрат на очист-
ку продукта / и получение продукта или изделия // как функция
чистоты продукта / имеет минимум.
Установление степени чистоты продукта /, соответствующей
минимальному значению суммарных затрат на стадиях 1 и 2 (т. е.
себестоимости продукта II)
1 2
Продукт / (сырой) —> продукт I (чистый) —* продукт II
Рис VI-50. Оптимальная
степень очистки про-
. дукта Г.
/ — затраты на очистку про-
дукта; 2 — затраты на. полу-
чение продукта II; 3 — сум-
марные затраты.
из продукта Г, сни-
является содержанием одной из важнейших экономических задач
технологии основного органического синтеза:
Аналитическое решение такой задачи, вряд ли возможно, если
учитывать разнообразие продуктов промышленного синтеза, спо-
собов их очистки, путей их переработки и потребительского ха-^
рактера конечных продуктов или изделий, однако в каждом от-
дельном случае анализ экономических зависимостей, представ-
ленных на рис. VJ-50, должен обязательно проводиться. Такие
521
зависимости являются единственным основанием для требований
к чистоте продукта / со стороны его потребителей.
Пример VI-1. Смесь, находящаяся при температуре кипения и со-
стоящая из низкокипящего компонента А (масс.%) и высококипящего компо-
нента В (10 масс.%), разделяется на ректификационной установке, снабженной
высокоэффективной насадочной колонной заданной высоты (//=10 я). Дистил-
лят, полученный в результате ректификации, направляется в реакционный аппа-
рат, где в избытке компонента С, протекает реакция, описываемая схемой:
А + С — > N (I)
А + С — > М (II)
Некаталнтическая реакция (I) приводит к образованию основного про-
дукта А. Катализатором гомогенно-каталитической реакции (II) служит веще-
ство В, являющееся примесью к веществу А.
Требуется приближенно оценить концентрацию вещества В в дистилляте,
соответствующую минимальному значению суммы энерго-материальных затрат,
связанных с производством единицы продукта N из вещества А, находящегося
в смеси с В.
Решение. 1. Энерго-материальные затраты, отнесенные к
100%-ному продукту А, выражаются зависимостью (VI-3):
ф* = TZ7-----(v<Gi + v2ri + V3f2 + + V5K3 + v'6Q3)
1 В, D
где св, d — содержание примеси вещества В в дистилляте, масс. доли.
Пользуясь известными уравнениями теплового баланса процесса ректифи-
кации, нетрудно видеть, что все слагаемые написанного выше выражения яв-
ляются функцией флегмового числа:
G j = лЯ др У£>о/(0,785ип) К/? + 1 = а (сд о)
W =HDv-^-(R+l) = 6<p(c )
F27Dr xW(/?+1)“e,₽(c* о)
<?2 = Dr t -Г + ° = гф (СВ. d)
Fz = Dr етг(/? ++ 0 (zo ~zd) = еФ(св. о) + ж
<?з = 4? lDr W + D + ° ('о - 'о)] = 3<₽ (св, о) + «
где d — диаметр ректификационной колонны, я; д — толщина стенок колонны, я;
v — средний удельный объем смеси паров в колонне, я3/кг\ г — теплота конден-
сации паров дистиллята, ккал/кг-, — средняя разность температур, град\
tK, ta — конечная и начальная температуры, °C; К — коэффициент теплопередачи,
ккал/{я"1 ч-град); io, ‘о — теплосодержание (энтальпия) кубового остатка и
флегмы, ккал/кг-, О — массовый расход кубового остатка, кг/ч; ип — скорость
пара в колонне, я!сек-, &ia — изменение энтальпии греющего пара в кубовой ча-
сти колонны, ккал/кг-, р — плотность материала стенок колонны, кг)я\
Для упрощения расчета принимаем следующие допущения: 1) независимость
Ki, Кг, Д/з, Мг от значения флегмового числа; 2) при относительно малой кон-
центрации вещества В в исходной смеси и высокой его концентрации в кубовой
жидкости изменение значений св1 о (содержания вещества В в кубовом остатке)
522
и О в результате изменения состава дистиллята пренебрежимо мало, вследствие
чего
I — iD ~ const; сА 0 = 0,01 = const
3) зависимость значения флегмового числа от состава дистиллята установ-
лена экспериментально и выражается кривой на рис. VI-51,a.
Основываясь на сделанных допущениях, приведенное выше выражение для
Ф1 можно переписать следующим образом:
Ф, = —-1------{v{a )/"q> (св + v' бф (св D) +
1 СВ, D
+ ^вф (св д) + <гф (СВ< д) +
+ v5 [еф (св D + ж] + Vg [зф (св д) + к]
2 . Стоимость сырья (вещество А в составе
исходной смеси), расходуемого на получение про-
дукта N (продукт М—отход):
где р — стоимость единицы вещества А в исход-
ной смеси, руб/т-, МА, MN — молекулярные мас-
сы веществ А и N; рл—выход вещества А в
результате ректификации; p.v—выход вещества
N на стадии химического превращения.
Выход вещества А на стадии ректификации
определяется рядом условий (содержание в кубо-
вом остатке, потери при циркуляции потока флег-
мы и т. п.). Упрощенно принимаем, что Pa(cb,d):
(1-0,01 cBtD) (0,9-0,01)
Ь “ Хо (%,-%2) “ 0,9 [(1-0,01 д)-0,01]
Учитывая, что компонент С вводится в реак-
ционный аппарат в большом избытке, реакции
(I) и (II) можно описывать уравнениями реак-
ций первого порядка:
о 1______________1
PjV 1+(W*1) ' 1 + [ф(Сд,д)]/й,
Для гомогенно-каталитической реакции (II)
значение константы скорости пропорционально
величине с я, d и следовательно:
ф2=р------------------------
mn$a(cb,d)$n(.cb,d)
Решение задачи сводится к нахождению минимума функции:
ф1 + ф2 = Ксв,о)
В рассматриваемом случае аналитическое решение затруднено из-за отсут-
ствия описания ф(св, о) и ф(св, о). Задача решается путем численного расчета
ряда значений (Ф1+Ф2) (св, D).
В рассматриваемом случае сумма Ф1 + Ф2 зависит от абсолютного размера
потоков на стадии ректификации, и для расчета принимается массовый расход
продукта А, равный 1000 кг/ч.
Для числового расчета принимаем: 6 = 0,01 м; «п = 0,5 м/сек:,
9=0,4 м?/кг\ Л4д=60; Л1л=90; г=100 ккал/кг-, Д/г=40 град-, Д/2=50 град-.
Рис. VI-51. Определение оп-
тимальной степени очистки
вещества А (к примеру VI-1).
523
Кг=200 ккал! (м2-ч- град); Кз=500 ккал/(м2-ч-град)' материалоемкость тепло-
обменников 50 кг/м2-, число часов работы установки в год 8000; нормы аморти-
зации 0,1; стоимость материала аппаратуры 1,0 руб/кг-, стоимость пара 3,0 руб/т;
стоимость воды 0,006 руб/м2-, стоимость вещества А 100 руб/т\ ki=0,03 ч-1, fe3=cB, в-
Результаты числового расчета, основанного на сделанных допущениях, при-
веденные на рис. VI-51,6, показывают, что оптимальное значение св, в лежит
в области 0,28—0,30%. Это соответствует значению R=12-=-15.
Из структуры выражения (Ф1+Ф2) (св, о) следует также, что повышение
стоимости исходного вещества А сопровождаетси повышением требований к сте-
пени его чистоты от примеси вещества В.
Для получения точного значения Св, в необходим детальный расчет ряда
вариантов режима ректификационного процесса в области значения Л от 11
до 15.
При поиске экономически оптимальной степени очистки продукта промыш-
ленного синтеза следует учитывать ограничения, налагаемые специфическими
особенностями процесса. В том случае, когда эта граница лежит влево от экстре-
мальной точки кривой (Ф1+Ф2) (св, о), целесообразным является режим очистки,
удовлетворяющий ограничительным требованиям независимо от экономических
соображений. Если граница требований к чистоте продукта расположена вправо
от точки экстремума, необходим тщательный анализ процесса и поиск эконо-
мического оптимума.
ЛИТЕРАТУРА
1. А. Г. Касаткин, Процессы и аппараты химической технологии, Гос- ,
химиздат, 1960.
2. Я. Циборовский, Процессы химической технологии, Госхимиздат,
1958.
3. А. Н. П л а н о в с к и й, В. М. Р а м м, С. 3. Каган, Процессы и аппа-
раты химической технологии, Госхимиздат, 1968. ,
4. А. Н. П л а н о в с к и й, П. И. Николаев, Процессы и аппараты хими-
ческой и нефтехимической технологии, Гостоптехиздат, 1960.
5. А. И. С к о б л о, И. А. Трегубова, Н. Н. Егоров, Процессы и аппа-
раты нефтеперерабатывающей и нефтехимической промышленности, Гостоптех-
издат, 1962.
6. Л. А. Барский, И. Н. Плаксин, Критерий оптимизации раздели-
тельных процессов, Изд. «Наука», 1967.
7. Б. Ш. Арутюнян, В. М. Борисов и др., Хим. пром., № 2, 146
(1962).
8. R. М. В a u с h е г, Chem. Eng., 68, № 20, 83 (1961).
9. R. М. Ваис he г, А, Н. Wether, Chem. Process, 25, № 21, 24 (1962).
10. В. А. Носов, Ультразвук в химической промышленности, Гостехиздат
УССР, 1963.
11. А. Г. Амелин, Теоретические основы образования туманов в химиче-
ских производствах, Госхимиздат, 1961.
12. А. И. С к о б л о, И. А. Александров, Новости нефтяной и газовой
техники, ГОСИНТИ, 1962, стр. 44.
13. Е. М. Ковалев, Хим. и нефт. маш., № 1, 9 (1965).
14. W. Со о ply, Chem. Eng., 69, № 3, 118, 120, 123 (1962).
15. A. Scheiman, Hydrocarbon Proc. a. Petrol. Ref., 42, № 10, 165 (1963).
16. В. Клейтон, Эмульсии, их теория и техническое применение, ИЛ, 1950.
17. Г. Н. Бреме р, Жидкостные сепараторы, Изд. «Машиностроение», 1964.
18. В. И. Соколов, Современные промышленные центрифуги, Машгиз,
1961.
19. 3. Г. Гатаулии, Новейшее оборудование для химической промышлен-
ности, Изд. «Машиностроение», 1965.
20. И. А. Александров, А. И. С к о б л о, Изв. вузов. Нефть н газ, № 5
(1960).
21. Фильтры и центрифуги, Стандартгиз, 1961,
524
22. К. А. Чефранов, Электрообезвоживание и электрообессоливание неф-
тей, Гостоптехиздат, 1948.
23. И. П. Сидоров, М. И. Силич, А. Н. Воробьев, О. Г. Кац-
нельсон, сб. «Техническая и экономическая информация». Азотная промыш-
ленность, 1969.
24. А. И. П о в а р о в, Гидроциклоны, Госгортехиздат, 1961.
25. В. А. О л е в с к и й, Конструкция и расчет механических классификаторов
и гидроцнклонов, Госгортехиздат, 1960.
26. Сб. «Центрифугирование в СССР», Машгиз, 1963.
27. М. М. Васильев, Хим. маш., № 1, 14 (1964).
28. В. И. С о к о л о в, Журн. ВХО, 10, № 2, 34 (1965).
29. В. А. Жужиков, Фильтрование, Изд. «Химия», 1967.
30. Б. Н. Борисоглебский, Хим. маш., № 4, 1 (1963).
31. Н. В. Ш панов, Жури. ВХО, 10, № 1, 143 (1965).
32. А. Ф. Сороченко, М. Я. Мешенгиссер, Хим. маш., № 1, 7
(1959).
33. Фильтростроение за рубежом, Машгиз, 1963.
34. Б. Н. Белы некий, Хим. маш., № 4, 6 (1959).
35, Циклоны НИОГАЗ. Каталог, № 22А, Госхимиздат, 1961.
36. Батарейные циклоны. Каталог, № 23А, Госхимиздат, 1961.
37. Г. М. Гордон, И. Л. П е й с а х о в. Пылеулавливание и очистка газов,
Госметаллургиздат, 1958.
38. Н. Г. 3 а л о г и н, С. М. Шухер, Очистка дымовых газов, 1954.
39. Г. М. Гордон, И. А. Аладжалов, Газоочистка рукавными фильт-
рами в цветной металлургии, Госметаллургиздат, 1956.
40. Brit. Chem. Eng., 10, № 2, 136 (1965).
41. М. Е. Позин, И. П. Мух ленов, Э. Я. Тар ат, Пенные газоочисти-
тели, теплообменники и абсорберы, Госхимиздат, 1959.
42. А. П. Андрианов, Краткая химическая энциклопедия, т. 1, Изд. «Сов.
энциклопедия», 1961, стр. 743.
43. В. Н. Ужав, Очистка промышленных газов электрофильтрами, Изд.
«Химия», 1967.
44. Б. М. Ш н е е р с о н, Электрическая очистка газов, Госметаллургиздат,
1950.
45. 3. 3 ю л к о в с к и й, Жидкостная экстракция в химической промышлен-
ности, Госхимиздат, 1963.
46. С. Э. Каган, В. Г. Труханов, П. А. Костин, Е. Н. Кудрявцев,
сб. «Процессы жидкостной экстракции и хемосорбции», Изд. «Химия», 1966,
стр. 220.
47. Л. Альдере, Жидкостная экстракция, ИЛ, 1962.
48. Н. Marten, Е. Lothar, Chem. Techn., 17, № 6, 335 Д1965).
49. М. С. Ге р ш а н о в а, Журн. ВХО, 10, № 1, 51 (1965).
50. В. И. Ш е й н м а н, Ю. С. Коган, М. А. Александров, Хнм. маш ,
№ 3, 15 (1963).
51. А. Г. Касаткин, Ю. И. Дытнерский и др., Хим. пром., № 7,
534 (1963).
52. - Brit. Chem. Eng., 9, № 9, 619 (1964).
53. Ю. И. Дытнерский, М. А. Александров и др., Хим. пром.,
№ 1,70 (1964).
54. М. Э. Аэро в, Т. А. Быстрова и др., Хим. пром., № 1, 62 (1960).
55. М. Э. Аэров, Е. П. Доровских, Хим. пром.>№ 2, 92 (1957).
56. Ю. В. Поплавский, В. Г. Капитальный, Хим. маш., № 3, 11
(1964).
57. Н. И. Гельперин, В. 3. Гришко, Журн. ВХО, 10, № 1, 26 (1965).
58. В. А. Мал юс о в Н. М. Жаворонков и др., Хим. пром., № 7 519
(1962).
59. В. М. Олевский, В. Р. Р учинский, Хим. пром., № 1, 57 (1961).
60. Л. В. Бирюкова, В. Г. Овчаренко и др., Хим. пром., № 6 464
(1963).
61. Chem, Eng., 66, 106 (1959).
525
62. H. R. Douglas, J. W. Snider, G. H. Tomlinson, Chem. Eng.
Progr., 59, № 12, 85 (1963).
63. L. Reich le, R. Billet, Ind. Eng. Chem., 57, № 4, 52 (1965).
64. В. M. P а м м, Абсорбция газов, Изд. «Химия», 1966.
65. А. Н. Терновская, Хим. пром., № 7, 50 (1962).
66. В. И. Горбушин, В. М. Платонов, Н. П. Федоренко, Хим.
пром., № 4, 273 (1962); В. М. Платонов, Б. Г. Берге, Разделение многоком-
понентных смесей, Изд. «Химия», 1965.
67. Я. Д. 3 е л ь в е н с к и й, А. А. Титов, В. А. Ш а л ы г н н, Хим. пром.,
№ 2, 116 (1963).
68. А. А. Ефремов, Я. Д. 3 е л ь в е н с к и й, Хим. пром., № 3, 201 (1964).
69. А. А. Титов, Я. Д. 3 е л ь в е н с к и й, В А. Шалыгин, Хим. пром.,
№ 6, 425 (1964).
70. Я. Д. 3 е л ь в е н с к и й, Д. А. Николаев, В. А. Шалыгин,
В. С. Татаринский, Хим. пром., № 5, 42 (1966).
71. Ф. Б. Пет л юк, В. М. Платонов, Д. М. Славинский, Хим. пром.,
№ 3, 206 (1965).
72. Ф. Б. Пет л юк, В. М. Платонов, В. С. Аветян, Хим. пром., № 11,
865 (1966).
73. Е. Н. С ер пионов а, Промышленная адсорбция газов и паров, Гос-
химнздат, 1956.
74. В. А. Соколов, Новые методы разделения легких углеводородов, Гос-
топтехиздат, 1961.
75. Н. J. Fornwall, R. A. Hutchins, Chem. Eng., 73, № 10, 155 (1966).
76. J. В. Allen, R. S. Joyce, R. H. К a s c h, J. Water Pollut. Control
Federat., 39, № 2, 217 (1967).
77. Г. Осборн, Синтетические ионообмеиники, Изд. «Мир», 1964.
78. Р. Грнссбах, Теория и практика иоииого обмена, ИЛ, 1963.
79. К- М. С о л д а д з е, А. Б. Пашков, В. С. Титов, Ионообменные вы-
сокомолекулярные соединения, Госхимиздат, 1960.
80. М е 1 п h о 1 d Т. Е., Chem. Progress, 29, № 13, 35, 36, 40 (1966).
81. Г. М. Вайнштейн, В. Р. Р у ч и н с к и й, Хим. пром., № 8, 571 (1967).
82. D. Е. Downing, Chem. Eng., 72, № 25, 170, 172 (1965).
83. J. E. Powers, Ind. Chemist, 39, № 10, 541 (1963).
84. В. H. Вигдорович, Teop. основы хим. технол., 1, № 2, 186 (1967).
85. В. А. Соколов, Н. С. Т о р о ч е ш н и к о в, Молекулярные сита и их
применение, Изд. «Химия», 1964.
86. В. В. Кузнецов, В. А. Ма люсов, Хим. пром., № 5, 39 (1962).
87. В. В. Кузнецов, В. А. Ма люсов, Хим. пром., 8, 54 (1963).
88. К- К u m m е г 1 е, Chem. Ingr. Techn., 36, № 10 (1964).
89. Чей-И-Чу, сб. «Новейшие достижения нефтехимии и нефтеперера-
ботки», т. V—VI, Изд. «Химия», 1965, стр. 73.
90. В. Г. Горский, Р. Г. Кочаров, В. 3. Бродский, Ииж.-физ. жури.,
13, № 3, 314 (1967).
91. J. W. С а г t е г, Brit. Chem. Eng., 9, № 8, 523 (1964).
92. А. Г. Касаткин, Ю. И. Д ы т н е р с к и й, Труды МХТИ им. Д. И. Мен-
делеева, вып. 5, 1963, стр. 156.
93. D, Steven, Chem. Eng., 71, № 25, 155 (1964).
Глава VII
ТЕХНИКА ВЗРЫВОБЕЗОПАСНОСТИ
[Лсточниками взрывоопасности промышленных произ-
' водств могут быть конденсированные вещества
(жидкие и твердые — окись этилена, гидроперекись кумола, си-
нильная кислота, полинитросоединения, взрывчатые вещества
и т. д.), газы и парогазовые смеси. При взрыве конденсированных
веществ разрушения обычно большие, чем при взрыве газов. Од-
нако, как показывает опыт эксплуатации промышленных пред-
приятий, чаще аварии связаны именно со взрывами газовых и па-
рогазовых смесей. Это обусловлено, с одной стороны, тем, что
образование взрывоопасных газовых (парогазовых) смесей проис-
ходит часто в производствах, конечные и промежуточные продукты
которых вообще не взрывоопасны, и, с другой стороны, недоста-
точным вниманием, уделяемым обеспечению взрывобезопасности
таких производств по сравнению с производствами конденсиро-
ванных взрывоопасных веществ. Вследствие этого ниже будут рас-
смотрены только взрывоопасные газовые и парогазовые смеси.
Под «горением» обычно понимают процесс окисления одного
вещества (горючего) другим (окислителем), при котором выде-
ляется большое количество тепла, следствием чего является разо-
грев продуктов реакции до высокой температуры, сопровождаю-
щийся свечением этих продуктов. Подобный эффект могут вызы-
вать не только реакции окисления, но и другие, достаточно
экзотермические реакции, например реакции хлорирования и фто-
рирования углеводородов, а также распада некоторых эндотерми-
ческих веществ1 (ацетилена, закиси азота, озона, газообразной
окиси этилена, гидразина и т. д.).
Сгорание газа в замкнутом сосуде сопровождается повышением
давления, зависящего от реакции. Обычно давление возрастает в
8—12 раз. Быстрое повышение («скачок») давления, являющееся
результатом горения газа в замкнутом сосуде, воспринимается как
1 Эндотермические вещества — это такие вещества, при распаде которых
происходит выделение тепла.
527
взрыв, особенно если оболочка сосуда при этом подвергается раз-
рушению.
Таким образом,, взрывоопасными являются не горючее и окис-
литель, а их смеси и эндотермические вещества.
ОСНОВНЫЕ ПОНЯТИЯ ТЕОРИИ ГОРЕНИЯ ГАЗОВ
НОРМАЛЬНАЯ СКОРОСТЬ ПЛАМЕНИ
Реакция, начавшаяся от внешнего импульса в некоторой
точке горючей смеси, распространяется по всему объему. При этом
реакция протекает только в тонком (<1 мм) слое, отделяющем про-
дукты реакции от несгоревшей части смеси и перемещающемся по
нормали к его поверхности в сторону исходной смеси. Происходит
послойное сгорание. Скорость перемещения пламени в неподвиж-
ной смеси по нормали к его поверхности называют нормальной
скоростью. Она является физико-химической характеристикой
данной смеси. Практически наблюдаемые скорости горения зави-
сят от степени турбулентности газового потока и при искривлении
и значительном увеличении поверхности фронта пламени могут
быть значительно больше нормальной скорости горения.
Перемещение фронта пламени по нормали к своей поверхности
происходит вследствие того, что выделяющееся при горении тепло
от горячих продуктов реакции за счет теплопроводности пере-
дается исходной смеси, диффундирующей в противоположном
направлении. Нормальная скорость пламени определяется из сов-
местного решения уравнений теплопроводности и диффузии в пла-
мени. Приближенное решение этой системы уравнений на основе
теории горения, разработанной Зельдовичем и Франк-Каменецким,
дано в работах [1—5]. Нормальная скорость пламени ип может
быть выражена -зависимостью:
(VII-1)
где р — постоянная, для п = 0 и 1 равная 2 и для п = 2 равная 4;
п — порядок реакции, Хь — теплопроводность, СР — теплоемкость;
р — плотность, Q — тепловой эффект реакции; В ехр (—E/RTb)—
константа скорости реакции; Е — энергия активации; R— газовая
постоянная; с0 — концентрация недостающего по отношению к
стехиометрии компонента смеси. Индексы «0> и «&» относятся со-
ответственно к исходному состоянию и состоянию при адиабати-
ческой температуре реакции Ть.
Как следует из уравнения (VII-1), величина ип практически
экспоненциально зависит от адиабатической температуры горения,
которая определяется составом исходной смеси. Адиабатическая
температура, максимальная для стехиометрической смеси, убы-
вает как при увеличении горючего или окислителя сверх стехио-
628
метрического количества, так и при разбавлении смеси инертным
(не участвующим в реакции) газом. Соответственно убывает и
нормальная скорость пламени.
КОНЦЕНТРАЦИОННЫЕ ПРЕДЕЛЫ РАСПРОСТРАНЕНИЯ
ПЛАМЕНИ (ПРЕДЕЛЫ ВЗРЫВАЕМОСТИ)
При малых скоростях пламени вследствие охлаждения
продуктов сгорания возникает поток тепла от фронта пламени в
сторону сгоревшей смеси. В результате этого, а также вследствие
отдачи части тепла во внешнее пространство (стенки сосуда) из
фронта пламени (например, излучением) температура в пламени
оказывается меньшей, чем при адиабатическом сгорании. Сниже-
ние температуры горения ниже адиабатической приводит к еще
большему уменьшению скорости пламени, что вызывает в свою
очередь увеличение тепловых потерь. В результате прогрессивного
охлаждения зоны реакции происходит затухание пламени.
Скорость теплоотдачи во внешнее пространство с единицы по-
верхности пламени (Qi) равна произведению средней скорости
теплоотдачи из единицы объема (Ф) на ширину фронта пла-
мени (б). Поток тепла через единицу поверхности фронта пламени
охлаждающимся продуктам реакции (Q2) равен величине QI( т. е.
Q2 = Q) = 6a>
(VI1-2)
Из теории горения известно, что
б = — (а = — коэффициент
температуропроводности). Тепловой баланс реакции в пламени
с учетом теплопотерь Qi и Q2 может быть записан следующим
образом:
ubnfibmCp{Tbm 7'о) + 2(21
(VII-3)
где wioQ — количество тепла, выделяемое при сгорании 1 г сме-
си. Индекс «О» относится к исходной смеси, индекс «Ьт» — к
продуктам при неадиабатическом сгорании.
Сопоставляя уравнение (VII-3) с балансом тепла без учета
теплопотерь
m0Q~cp(Tb-T0) (vn-4)
и подставляя значение Qi из выражения (VII-2), получаем:
Тьт = Ть—--аФ-2 - (VII-5)
CpPbmubm
Для скорости распространения пламени теория дает:
Е
ubm ~ COnst ’ е 2RTbm
34 В. С. Хайлов, Б. Б. Брандт
(VII-6)
529
Вследствие того, что Tbm не сильно
принять, что TbmTb Т2Ь. Тогда
^b~^bm
----2~Е
U. ** Ubme 2^Tb = ui
о bm j
отличается от Ть, можно
А
—2
lbme
(VII-7)
Уравнение (VII-5) выражает зависимость фактически достигаемой
в пламени температуры от скорости пламени, которая в свою оче-
редь зависит от ТЬт по уравнению (VII-6).
Стационарный режим горения возможен в таких- условиях, ко-
гда уравнения (VII-5) и (VII-6) имеют действительные решения.
Критический режим, соответствующий концентрационному пре-
делу распространения пламени, достигается при равенстве про-
изводных dUbmldTbm Для уравнений (VII-5) и (VII-6). Отсюда
' ть-тьт=~/ (VII-8) *.
Подставляя формулу (VII-8) в уравнение (VII-7), получаем ми-
нимальное значение скорости пламени
= (VII-9)
При котором еще возможно стационарное горение.
Таким образом, горение разбавленной смеси становится воз-
можным в том случае, когда фактически достигаемая в пламени
температура снижается по сравнению с адиабатической на вели-
чину RTb/E- Скорость пламени при этом в Уе раз меньше ско-
рости, вычисляемой для той же смеси при адиабатическом ее сго-
рании.
Подставляя в уравнение (VII-7) значение Ть — Тът из выраже-
ния (VII-5), получаем зависимость между адиабатической и фак-
тически достигаемой скоростью пламени
аФЕ
С„р. и?
и. = и.е pbmbm b
о от
которая, как это было показано выше, в предельном случае пере-
ходит в зависимость
-I/O
ub = ubme
Следовательно, критические условия осуществляются при опреде-
ленном, равном единице, значении параметра А:
. 2аФЕ 2QE ..... ...
Л =-------5----5- -----2т (VII-10)
Ср9ЬтиЬт%ТЬ е%Ср9ьиЪТЪ
В работе [6] было показано, что величины, составляющие без-
размерный комплекс (VII-10), не зависят на нижнем концентра-
ционном пределе распространения пламени от химического строе-
530
ния топлива и, кроме энергии активации, однозначно определяются
коэффициентом избытка окислителя
г-экв окислителя
г-экв восстановителя
и долей I инертных компонентов в смеси, т. е. А = f(a, I),
Безразмерный комплекс А наиболее сильно зависит от состава
смеси (а, /) через адиабатическую температуру горения, так как
тепловой эффект сго-
рания углеводорода
является линейной
функцией числа угле-
родных атомов в мо-
лекуле углеводорода, а
Е
ub:-.e'2RTb. Энергия
же активации в пла-
мени углеводородов (и
их производных) оди-
накова вследствие то-
го, что реакцией в высо-
котемпературной зоне
пламени, определяю-
щей скорость распро-
странения последнего
[7], является взаимо-
Рис. VII-1. Критические значения коэффициентов
избытка окислителя смесей некоторых углеводо-
родов и окиси углерода с закисью азота и азо-
том [10]:
/ — метан; 2 —пропан; 3 —пропилен; 4 — циклопропан;
5 —диэтнловый эфир; б —бутан; 7 —окись - углерода;
8 — П-КС.ИЛОЛ; 9 — циклогексан.
действие СО с кисло-
родом. Реакция в пла-
мени бедных топливо-
воздушных смесей про-
текает двустадийно: в
зоне более низких тем-
ператур углеводород
окисляется до СО, в высокотемпературной зоне СО догорает [8,9].
Так как на пределе распространения пламени А = 1, соответствую-
щие этому пределу значения коэффициента избытка окислителя аКр
являются однозначной функцией содержания инертных компонен-
тов в смеси, причем эта зависимость является универсальной, не
зависящей от химического состава топлива. В качестве иллюстра-
ции на рис. VII-1 сопоставлены границы областей поджигания
для смесей некоторых углеводородов и СО с закисью азота и
азотом.
При описании концентрационных пределов в виде зависимости
акр(/) верхнему концентрационному пределу распространения пла-
мени (избыток горючего) соответствуют значения акр на нижней
ветви кривой, ограничивающей область распространения пламени;
нижнему концентрационному пределу распространения пламени
34*
531
(избыток окислителя) соответствуют значения а[(р на верхней
ветви кривой.
Зависимость концентрационных пределов распространения пла-
мени от давления и температуры может быть выяснена при рас-
смотрении безразмерного комплекса (VII-10). Поскольку нормаль-
ная скорость распространения пламе-
ни экспоненциально зависит от тем-
Е
пературы горения, т. е. ub:: е 2RTb, без-
„ . 2аФЕ
размерный комплекс А =-----------=-
наиболее сильно зависит от состава
смеси (а, 7) через адиабатическую
температуру горения. Последняя, как
Рис. VII-3. Зависимость
критических значений со-
держания кислорода в бо-
гатых горючих смесях ме-
тана с кислородом от да-
вления [11].
Рис. VII-2. Область распро-
странения пламени при раз-
личных давлениях.
указывалось выше, имеет максимум для смеси, близкой к стехио-
метрической. Рассмотрим случай, когда Тъ не зависит от давления
и определяется только составом исходной смеси. Тогда безразмер-
ный комплекс А можно разложить на множители, один из которых
зависит только от давления, а второй — только от температуры
Е
горения. Учитывая, что Ф:: РтТ3ь5, к::Т°ь’5, (up)2 iRTb , по-
лучаем:
V (Р) • g (Ть) = const
Е
где V (Р) = Pn~m, g (Ть) = Ть<п+2)е 2RTt>. Значение т, вычисляемое
из данных Хоттеля для СО2 (доля тепла, излучаемого СО2, со-
ставляет 0,9 всего теряемого тепла), равно 5/э~0,5. Следовательно,
для реакций, порядок которых более 0,5, величина У(Р) возрастает
53?
с увеличением давления. Критические условия при этом дости-
гаются при более низком, чем прежде, значении g(Tb), т. е. при
более низкой адиабатической температуре горения, что объясняет
Рис. VII-4. Критическое да-
вление взрывного распада
закиси азота при разбавле-
нии ее азотом [6]:
О — горит; X — не горнт.
Рис. VII-5. Зависимость кри-
тических значений коэффи-
циента избытка окислителя для
богатых горючим смесей ме-
тай — закись азота — окись азо-
та — азот от давления (доля
окиси азота в сумме окислите-
лей равна 0,715) [12]:
О — горнт; X — не горит.
общеизвестный факт расширения области распространения пла-
мени с увеличением давления (рис. VII-2). В качестве примеров
можно привести зависимости для смесей СН4+О2, N2O + N2 и
CH4+NO+N2O + N2 (рис. VII-3—VII-5).
Рис. VII-6. Влияние давления на пределы распространения пла-
мени [13].
Наблюдаемое в некоторых случаях аномальное изменение
пределов распространения пламени при увеличении давления
(рис. VI1-6) связано, очевидно, с уменьшением порядка реакции
до значений <0,5.
533
ГОРЕНИЕ В ТРУБАХ РАЗЛИЧНОГО ДИАМЕТРА
Скорость распространения пламени для смеси данного
состава понижается, как показал опыт, с уменьшением диаметра
трубы (сосуда),. При этом теплопотери обусловлены в основном
теплопроводностью и количество тепла, теряемого единицей объ- ,
ема газа в секунду, может быть выражено в виде [2]
ф KATbm-T0)F
W
или, учитывая, что для цилиндрических труб, диаметр которых d,
коэффициент теплопередачи Кт равен 8X/d, а отношение поверх-
Рис. VII-7. Зависимость
критических значений диа-
метра сосуда от состава
горючей смеси.
F 4
ности к объему трубы = у
ф = -^(7’6т-Г0) (VII-11) '
Подставив уравнение (VII-1I) в уравне-
ние (VII-10), для критического диамет-
ра получим:
Так как -уД <С 1, а > 1, окончатель-
но имеем:
(VII-12)
Из уравнения (VII-12) следует, что с увеличением температуры
горения, т. е. при переходе к более быстро горящим смесям, кри-
Рис. VII-8. Зависимость предельного давления взрывного и
детонационного распада ацетилена от внутреннего диаметра
трубопровода [14]:
/ — взрывной распад; 2— детонация.
тическое значение диаметра экспоненциально убывает, достигая
своего минимума для смесей, близких к' стехиометрическим
534
(рис. VII-7). Критическое значение диаметра убывает также с
увеличением давления исходной смеси, так как а::l/Р, а скорость
распространения пламени для смесей, температура горения кото,-
рых не зависит от давления, пропорциональна Р2 . Следователь-
но, dKp:: Р 2.
Экспериментально зависимость dKV(P) изучена для ацетилена
(рис. VII-8). Завышенное значение углового коэффициента: 1,7
вместо единицы (порядок реакции распада ацетилена в пламени
~2), связано с тем, что температура горения ацетилена несколько
возрастает с увеличением давления.
САМОВОСПЛАМЕНЕНИЕ И ПОДЖИГАНИЕ ГОРЮЧЕЙ СМЕСИ
Горючая газовая смесь не представляет собой непо-
средственной опасности. Она только потенциально опасна. При
надлежащем обращении с такого рода смесями и применении эф-
фективных мер безопасности возможна даже организация крупных
промышленных производств (например, производство и перера-
ботка ацетилена, окиси этилена и т. д.) и безаварийная их экс-
плуатация.
Одним из важных условий обеспечения безопасности при ра-
боте с горючими смесями является исключение возможности
возникновения условий загорания. Загорание горючей смеси воз-
можно при нагревании ее выше некоторой температуры, называе-
мой температурой самовоспламенения, либо при внесении в холод-
ную горючую смесь сильно нагретого источника (искра, нагретые
детали аппаратуры и т. д.).
Самовоспламенение
Самовоспламенение горючей смеси, т. е. самопроизволь-
ный разогрев до появления пламени, происходит в таких условиях,
когда скорость освобождения тепла (Q+) при реакции превышает
скорость отвода его к стенкам сосуда (Q_) [15]. Скорость освобож-
дения тепла, как и скорость реакции, экспоненциально зависит от
температуры, скорость же отвода тепла линейно зависит от по-
следней, т. е.
__Ег
Q^ — vQw— QvBe
Q_-KtS(T-Tct)
Здесь v — объем сосуда; 3 — поверхность сосуда; Q — тепловой
эффект реакции; w — скорость реакции; В = Вос£ [Во — предэкс-
поненциальный множитель в константе скорости реакции; с0 —
концентрация исходного вещества (недостающий по отношению к
стехиометрии компонент); п — порядок реакции]; — коэффи-
циент теплопередачи; Т, Тст — средние температуры газа и стенки
соответственно.
535
Зависимости Q+(T) и Q-(T) представлены на рис. VII-9. Ста-
ционарному режиму соответствуют точки пересечения обоих гра-
фиков (например, в точке /), так как незначительное смещение
системы в сторону более высоких температур приведет к преоб-
ладанию теплоотвода над теплоприходом и охлаждению системы.
Аналогично смещение системы влево от положения теплового рав-
Рис. VII-9. Зависимость ско-
ростей теплоприхода и
теплоотвода от температуры
в сосуде:
кривая линия — скорость тепло-
прихода; прямые линии — ско-
рости теплоотвода.
новесия приведет к преобладанию теп-
лоприхода над теплоотводом, так что
система самопроизвольно вернется в
точку 1. Точка 2 соответствует неустой-
чивому равновесию. Если графики Q+(7')
и Q-(T) не пересекаются, то теплоприход
при всех температурах больше теплоот-
вода и реакция протекает с автоускоре-
нием до полного выгорания — происхо-
дит самовоспламенение.
Критическими условиями самовос-
пламенения является касание графиков
функций Q+ и Q-. Математически это
условие запишется в виде:
dQ+ dQ_
~dF~4r
Q+ = Q_
(VII-13)
(VII-14)
Подставляя в соотношение (VII-13) и
в равенство (VII-14) значения Q+ и Q_,
имеем:
Е
QvB-^-e ^=^8 (VII-15)
Е
QvBe *Гк₽ -KtS(TKp-TCT)
Совместное решение двух последних уравнений дает:
Отсюда
4г1р-гкр+гст-о
7к₽ 2R V И 1 £ /
(VI1-16) ,
Максимальный разогрев газа, при сохранении стационарного
режима, вычисляется из уравнения (VII-16) разложением подко-
ренного выражения в ряд (все члены ряда после третьего отбра-
сываются):
RT2
Гкр Т’ст + &— (VII-17)
536
Как следует из уравнения (VII-17), предвзрывной разогрев
газа равен характеристическому интервалу температуры, т. е. та-
кому интервалу изменения температуры, при котором скорость
реакции возрастает в е раз. Так как энергия активации реакции
в пламени (в кал/моль) обычно имеет порядок 104, а ТС1: (в ° К)
порядок 102, предвзрывной разогрев обычно мал по сравнению с
температурой стенки.
В уравнение (VII-15) входит значение не поддающейся измере-
нию температуры газа. Критические условия, при которых возможно
Рис. VII-10. Область самовоспламе-
нения метано-воздушных смесей [16].
Температура
самовоспламенения, °C
Рис. VII-11. Зависимость тем-
пературы самовоспламенения
чистого ацетилена от давле-
ния [17].
самовоспламенение горючей смеси, получаются из этого уравне-
ния, если в предэкспоненте принять ТК^ТС^. Аналогичная замена
в экспоненте приведет к появлению в числителе уравнения (VII-15)
дополнительного множителя, равного е. Таким образом
Е
QvB0CgEe rtct
-------е
KtSRT2ct
(VII-18)
Из уравнения (VII-18) следует, что температура самовоспла-
менения убывает с увеличением размеров сосуда (v/S) и по мере
приближения концентрации недостающего компонента к стехио-
метрическому количеству. Аналогичным образом температура са-
мовоспламенения зависит от давления (Р) газа, поскольку с0:: Р.
В качестве примеров на рис. VII-10 приведены температуры
самовоспламенения различных метано-воздушных смесей, а на
рис. VII-H—зависимость температуры самовоспламенения ацети-
лена от давления.
537
Поджигание горючей смеси
Если до высокой температуры нагреты только некото-
рые участки стенок сосуда или в газе движется сильно нагретая
частица, то условия зажигания газа, очевидно, должны быть более
жесткими, чем аналогичные условия при температуре, равномерной
по всей поверхности сосуда. Температура горячего участка стенки
(частицы) при поджигании должна быть более высокой, чем Тст
при самовоспламенении. Вследствие неопределенности условий
теплоотвода при неодинаковом нагреве стенок сосуда в качестве
характерной температуры для данной смеси обычно выбирают наи-
более низкую, соответствующую температуре самовоспламенения.
ДЕТОНАЦИЯ
Явление детонации открыто в конце прошлого века и в
связи с сильным разрушающим действием достаточно хорошо из-
учено. Детонация возникает вследствие автоускорения пламени за
Содержание пропана,”/.
Рис. VII-12. Концентрационные пределы дето-
нации пропано-кнслородных смесей [18].
счет турбулизации.
При этом впереди
фронта пламени фор-
мируется ударная вол-
на (волна сжатия).
Сжатие в ударной вол-
не происходит адиаба-
тически. Газ в зоне
сжатия оказывается
сильно разогретым,
так что создаются ус-
ловия, благоприятные
для его реагирования.
Газ самовоспламеняет-
ся, и ударная волна
сопровождается вол-
ной горения. Обе эти
волны образуют вме-
сте детонационную
волну. Скорость распространения детонационной волны в сотни
раз больше нормальной скорости пламени и обычно равна не-
скольким километрам в секунду. Давление в детонационной волне
может превышать исходное в 20—30 раз, а при отражении от
препятствия — в 40—60 раз.
Скорость распространения детонации ил вычисляется по фор-
муле: ____
-
Ср (Г.) !
где е— степень сжатия газа; у = тт ;т , (СР, Cv — теплоемкости ,
D
при постоянных давлении и объеме); г = (R— газовая по- '
538
стоянная; М — молекулярная масса продуктов реакции); Та — мак-
симальная температура в детонационной волне. Очевидно, что тем-
пература Td является однозначной функцией состава исходной
смеси и максимальна для смеси стехиометрического состава. Со-
ответственно и скорость детонации убывает по мере приближения
к смесям предельного состава (рис. VII-12).
Пределы детонации несколько уже пределов распространения
пламени и достигаются при удовлетворении равенства [7]:
2С у Е
a'b'Ru.e Ru2.
-----— е =1
CVE
где а' — коэффициент, характеризующий потери при распростра-
нении детонационной волны, обусловленные гидравлическим со-
противлением трубы, по которой распространяется детонация, и
турбулентным теплообменом; Ь' — величина, зависящая от состава
смеси и кинетики реакции.
ВЫЯВЛЕНИЕ ОПАСНЫХ РЕЖИМОВ РАБОТЫ
ТЕХНОЛОГИЧЕСКОГО ОБОРУДОВАНИЯ
И ЗАЩИТНЫЕ УСТРОЙСТВА
При технологических операциях состав газовой (паро-
газовой) смеси претерпевает различные изменения вследствие хи-
мических превращений, конденсации паров или испарения контак-
тирующих с газом жидкостей, смешения нескольких газовых
потоков и т. д. Наиболее действенными мерами обеспечения без-
опасности производства является своевременное выявление
возможности образования горючих смесей. Такое выявление це-
лесообразно начинать на стадии лабораторной разработки техно-
логического процесса, что позволяет осуществлять более высоко-
качественное проектирование промышленного производства.
Любой состав газовой (парогазовой) смеси однозначно харак-
теризуется некоторой точкой на плоскости в координатах а—/.
Выявление технологических операций или режимов, при осуществ-
лении которых возможен взрыв, может быть достигнуто путем
изображения изменения состава смеси на плоскости а — I (тех-
нологический процесс представляется некоторой кривой) и сопо-
ставлением полученных результатов с соответствующими преде-
лами взрываемости. При правильно разработанной технологии
график процесса не должен находиться внутри или проходить че-
рез концентрационную область распространения пламени.
Иногда не удается избежать образования в технологическом
процессе горючих смесей. В этом случае необходимо принимать
меры, препятствующие возникновению и распространению по обо-
рудованию и коммуникациям случайно возникшего в одном ме-
сте пламени.
539
Одним из важных условий обеспечения безопасности при ра-
боте с горючими смесями является исключение возможности воз-
никновения условий их загорания. С этой целью должны быть
приняты надлежащие меры для устранения зарядов статического
электричества (заземление коммуникаций и аппаратуры, громоза-
щита и т. д.). Стенки трубопроводов и аппаратов не должны быть
нагреты выше температуры самовоспламенения. Сжатие газа мо-
жет производиться как
компрессорами, однако
поршневыми компрессорами, так и трубо-
степень сжатия должна выбираться такой,
чтобы газ не нагревался выше его тем-
пературы самовоспламенения. Кроме то-
го, необходимо исключить возникновение
искр, вызываемое трением (ударом) в
движущихся частях машин и аппара-
тов.
Для предотвращения распростране-
ния по коммуникациям и аппаратуре
случайно возникшего пламени приме-
няются огнепреградители и разнообраз-
ные отсекатели, а для предотвращения
аппаратов от разрушения — разрывные
мембраны.
Огнепреградители представляют со-
бой насадочные, иногда орошаемые
инертной жидкостью колонны, устанав-
ливаемые вдоль магистральных трубо-
Рис. VII-13. «Мокрый» огне- проводов, а также у входа и выхода из
преградитель: цеха, газгольдера и т. д. Действие огне-
1- насадка из колец Рашига; ПрегрЭДИТвЛеЙ ОСНОВЭНО НЙ СуЖеНИИ КОН-
2 — влагоотделитель (кольца Ра- 11 „ - J
шига). центрационнои области распростране-
ния пламени в узких каналах, приводя-
щем к его потуханию в насадке огнепреградителя. В некоторых
случаях трубопроводы и коммуникации (а иногда и аппаратуру)
заполняют насадкой или пучками тонких трубок.
Вместо огнепреградителей можно использовать также быстро-
действующие отсекатели (водяные затворы, обратные клапаны
и т. д.). Действие таких отсекателей основано на том, что в ре-
зультате повышения давления в коммуникациях вследствие воз-
никшего пламени перекрывается клапан, препятствующий распро-
странению взрыва навстречу потоку газа.
Огнепреградитель и водяной затвор закрытого типа представ-
лены соответственно на рис. VII-13 и VII-14.
Разрывные мембраны применяют главным образом для пред-
отвращения разрушения аппаратуры при возникновении взрыва.
Их сочетают с установкой огнепреградителей и отсекателей. Обыч-
но разрывные мембраны устанавливают на повороте трубопрово-
дов под углом 90° друг к другу (рис. VII-15). Диаметры мембран
не должны быть меньше диаметра трубопровода. Снаружи мем-
540
брани необходимо защитить от механических повреждений. Иног-
да для предотвращения аварий вследствие случайного разрушения
мембраны (например, за счет коррозии) устанавливают две мем-
браны лбследовательно, соединяя пространство между ними с
С2Н2
Рис. VII-14. Водяной затвор закрытого типа (пропускная спо-
собность 35 м31ч).
манометром. Разрушение внутренней мембраны фиксируется по
показанию манометра.
Для защиты людей от травм, возможных при разрыве мем-
браны, на некотором расстоянии перед ней устанавливают предо-
хранительный щит.
541
Для подбора мембран с одинаковыми механическими свойст-
вами предложен ряд приспособлений. Испытания на одном из
таких приспособлений [19] (рис. VII-16) проводят следующим об-
разом. Пластину, неподвижно зажатую между двумя фланцами,
подвергают в течение определенного времени гидравлическому дав-
Рис. VI1-15. Схема установ-
ки разрывных мембран на
повороте трубопровода.
лению, превышающему предел текуче-
сти. О механических свойствах пласти-
ны судят по ее стреле прогиба, измеряе-
мой высотой h подъема штифта. Такая
пластина устанавливается выпуклостью
во внутрь трубопровода.
Пример VII-1. По трубопроводу при
давлении 1,2 ат транспортируется горючая
смесь — воздух, содержащий 1% паров углеводо-
рода. Известно, что адиабатическая температура
горения смеси равна 2000° К, нормальная ско-
рость распространения пламени 41 см'/сек. Из-
вестно также, что энергия активации при горении
углеводородо-воздушной смеси составляет 25—
37 ккал!моль [20]. Для предотвращения разруше-
ния аппаратуры при случайном загорании смеси необходимо установить огне-
преградитель. Определить размеры гранул насадки, необходимой для заполнения
огиепреградителя (диаметр гранул принять втрое большим диаметра пламегася-
щих каналов).
Рис. VII-16. Приспособление для испытания разрывных
мембран повышенной надёжности [19].
Решение. Максимальный (критический) диаметр пламегасящих
каналов оценим по уравнению (VII-12). Подставляя в него значения
* МР _
а Срр ’ Р RT ’ СрМ~ср
получим расчетное уравнение:
дкр = 760 -^5 УЁГЬ
UbCPb
Здесь М — молекулярная масса, Ср, Ср — мольная и массовая теплоемкости
при постоянном давлении.
542
Скорость движения пламени относительно горячих продуктов реакции свя-
зана с нормальной скоростью пламени соотношением
“Л = “Л
или
иЬ = ип 'Г' = 41 = 280 см!сек
и 1 q ХУО
Так как физические константы азота и кислорода близки между собой, а содер-
жание горючего мало, примем, что теплопроводность смеси совпадает с тепло-
проводностью воздуха, а теплоемкость этой смеси — с теплоемкостью азота.
Теплопроводность воздуха при различных температурах:
t ’С.............................. О 100 200 300 400 500 600 700 800 900 1000
Т, °К........................... 273 373 473 573 673 773 873 973 1073 1173 1273
Z.-105, кал!(см-сек-град) . .. 5,83 7,60 9,40 10,98 12,43 13,70 14,87 16,02 17,22 18,20 19,25
Для экстраполяции теплопроводности воспользуемся формулой Сатерленда;
2 =2 273 +С, I Т V/2
z Л273 Г+С, \ 273 /
Для вычисления постоянной Сатерленда Ci, воспользуемся приведенными
выше данными:
А- /273\3/а
~ Г-273
р __ ^273 \ •* /_________
1 / 273\3/2
1-
19,25 / 273 \3/2
wM 1273~273 22?
,т , . 19,25 / 273 у/2
Латз / 1 ~ 5,83 \ 1273/
Вычисления для теплопроводности при 7=2000° К дают /2000=25,6-10~5 кал/(ему.
X сек • град).
Теплоемкость при 2000° К вычисляем по уравнению (11-26), используя спра-
вочные данные для азота:
Ср 2000 = 6,6 + 1,02 • IO'3 • 2000 = 8,7 кал/(моль • град)
Оценка для критического диаметра пламегасящих каналов дает при
£=25 ккал/моль-.
пе с 1 л—5
«кр = 760 25 000 •2000 = °’47 сх
При £=37 ккал/моль 6Кр=0,57 см. Максимальный размер гранул насадки ра-
вен 1,4 — 1,7 см.
Пример VI1-2. Для некоторой горючей смеси (7\ = 15ОО°К, йь =
=200 см!сек) установлено, что диаметр пламегасящих каналов при давлении
1,2 ат равен 0,8 см.
Определить диаметр насадки огнепреградителя, работающего при давлении
5 ат, для горючей смеси, содержащей те же компоненты, но в другом соотноше-
нии, так что Гд(=2000°К, «(,=4800 см!сек. Соотношение между диаметром
пламегасящих каналов и насадки принять таким же, как в предыдущем
примере. Теплопроводность смеси при 1500° и 2000° К равна 21,6-10~5 и
25,6-10-6 кал!(см сек • град), теплоемкость 8,2 н 8,7 ккал/(моль град) соответ-
ственно.
Решение. Из уравнения (VII-12) имеем
или ____ ____________________________________
А . ЧсРгР1иЬ, , _ПЙ 25,6-10-5-8,2-1,2-200 , /2000
Ч1СРРм0 У Tt> 21,6-10 5-8,7-5-480 И 1500
543
ЛИТЕРАТУРА
I. Я. Б. Зельдович, Д. А. Ф р а н к - К а м е н е ц к и й, ЖФХ, 12, 100
(1938).
2. Я. Б. Зельдович, В. В. Воеводский, Тепловой взрыв и распро-
странение пламени в газах, изд. Моск. мех. ин-та, 1947.
3. Я. Б. 3 е л ь д о в и ч, ЖФХ, 22, 27 (1948).
4. Я. Б. Зельдович, Н. Н. Семенов, Журн. эксперим. теор. физ., 10,
1116, 1427 (1940).
5. Н. Н. С е м е н о в, Усп. физ. наук, 24, 433 (1940).
6. Б. Б. Брандт, Автореф. канд. дисс., МХТИ им. Д. И. Менделеева,
1962.
7. Я. Б. Зельдович, Теория горения и детонации газов, Изд. АН СССР,
1944.
8. R. F г i е d m a n n, Е. В u г k е, J. Chem. Phys., 22, 824 (1954).
9. R. F r i e d m a n n, J, А. С у p h e r s, J. Chem. Phys., 23, 1875 (1955).
10. Б. Б. Брандт, Л. А. Матов, А. И. Розловский, В. С. Хайлов,
Хим. пром., № 5, 419 (1960).
И. С. М. Cooper, Р. J. .W i е z е v i с h, Ind. Eng. Chem., 21, 1210 (1929).
12. Б. Б. Брандт, А. И. Розловский, В. С. Хайлов, Хим. пром.,
Xs 4, 279 (1965).
13. D. М. Newitt, D. Т. A. Town end, Sci. Petrol., 4, 2884 (1939).
14. H. S a r g e n t, Chem. Eng., 64, Xs 2, 250 (1957).
15. H. H. Семенов, Цепные реакции, Госхимиздат, 1934.
16. Н. В. Соловьев, П. И. Ермилов, Н. А. С трельчу к, Основы
техники безопасности и противопожарной техники в химической промышленно-
сти, Госхимиздат, 1960.
17. И. И. С т р и ж е в с к и й, А. А. Бородулин, Б. Л. К ан ер, Охрана
химических предприятий от пожаров и взрывов, изд. НИИТЭХИМ, 1961.
18. J. Breton, Ann. off. nat. des combust, liquides, 11, 487 (1936).
19. G. N i t s c h k e, Chem. Ind. Techn., 31, Xs 8, 511 (1959).
20. Б. В. Канторович, Введение в теорию горения и газификации твер-
дого топлива, Госметаллургиздат, 1960.
СООТНОШЕНИЕ ЕДИНИЦ МЕЖДУНАРОДНОЙ СИСТЕМЫ
ЕДИНИЦ (СИ] С ЕДИНИЦАМИ ДРУГИХ СИСТЕМ
Величина Единицы измерения Коэффициент для приведе- ния к едини- цам СИ
в различных системах в СИ
Вес (сила тяжести) Вязкости коэффициент динамический кинематический Давление Диффузии коэффициент Длина Масса Натяжение поверхностное Объем Плотность Площадь Работа, энергия, количество теплоты Теплоемкость удельная Теплоотдачи я теплопере- дачи коэффициенты Теплопроводности коэффициент Теплота удельная (фазово- го преобразования) Энтальпия удельная Энтропия удельная кгс дин кгс сек!м* пз спз ст сст кгс/см2 (или ат) кгс/м2 мм рт. ст. мм вод. ст. дин/см2 атм бар см2/сек м2/ч см мм мкм к г кгс/м дин/см (или эрг/см2) дм2 см3 л г/см3 см2 кгс • м кет • ч ккал ккал/(кг град) ккал/(м2 • ч град) ккал/(м • ч • град) ккал/кг ккал/кг ккал/(кг • град) } Н | н-сек/м2 } м2/сек н/м2 | м2/сек м кг 1 н/м f (или дж/м2) | м3 кг/м3 м2 | дж дж/(кг • град) вт/(м2 град) вт/(м • град) дж/кг дж/кг дж/(кг град) 9,81 10"5 9,81 10-з 10- 10 3 10"’ 9,81 • 104 9,81 133,3 9,81 КГ1 10,1 • 104 105 10"' 277,8 • 10’® 10=з 10 в <в 10-з 10 • ’ 9,81 >Сз 10.® 1О’з 10з 10 9,81 3,6- 106 4187 4187 1,163 1,163 4187 4187 4187
35 В. С. Хайлов. Б. Б. Брандт
545
ПРИЛОЖЕНИЯ
ПРИЛОЖЕНИЕ I, а
Значения упри 7^?=! -=-3,5*
Значения Y при различных ТПр
гпр 1.0 1.1 1,2 1.3 1,4 1,5 1.6 1,7 1.8 ‘ 2,0 2,2 2.4 2,7 3,0 3,5
0 1,000 1,000 1,000 1,000 1,000 1,000 1,000 1,000 1,000 1,000 1,000 1,000 1,000 1,000 1,000
1 0,612 0,735 0,814 0,870 0,906 0,926 0,948 0,956 0,964 0,976 0,990 1,000 1,006 1,010 1,014
2 0,385 0,560 0,668 0,760 0,824 0,822 0,898 0,914 0,930 0,956 0,980 1,000 1,012 1,020 1,028
3 0,288 0,435 0,560 0,668 0,748 0,806 0,854 0,880 0,902 0,940 0,974 1,000 1,020 1,032 1,046
4 0,248 0,370 0,494 0,602 0,690 0,764 0,824 0,858 0,882 0,930 0,972 1,000 1,030 1,048 1,062
5 0,226 0,338 0,464 0,566 0,654 0,736 0,802 0,842 0,866 0,922 0,972 1,008 1,042 1,062 1,080
6 0,210 0,318 0,442 0,544 0,634 0,720 0,788 0,834 0,860 0,920 0,978 1,014 1,052 1 074 1,098
7 0,202 0,310 0,430 0,532 0,626 0,710 0,780 0,832 0,860 0,926 0,988 1,026 1,068 1,092 1,112
8 0,200 0,308 0,428 0,528 0,624 0,712 0,784 0,834 0,868 0,934 1,000 1,040 1,086 1,110 1,136
9 0,200 0,310 0,430 0,532 0,630 0,720 0,792 0,840 0,878 0,948 1,014 1,058 1,106 1,130 1,158
10 0,202 0,312 0,434 0,542 0,640 0,730 0,806 0,852 0,890 0,964 1,034 1,076 1,128 1,153 1,180
И — — 0,460 0,552 0,654 0,746 0,810 0,866 0,908 0,982 1,054 1,100 1,152 1,174 1,204
12 — — 0,474 0,566 0,668 0,760 0,834 0 884 0,928 1,008 1,078 1,126 1,174 1,198 1,226
13 — — 0,490 0,582 0,686 0,778 0,852 0,906 0,952 1,014 1,106 1,152 1,202 1,222 1,250
14 — — 0,510 0,598 0,706 0,798 0,874 0,930 0,978 1,066 1,134 1,180 1,228 1,248 1,280
15 — — 0,532 0,620 0,728 0,826 0,902 0,958 1,006 1,100 1,166 1,214 1,256 1,280 1.310
16 — 0,554 0,646 0,758 0,854 0,934 0,996 1,036. 1,114 1,198 1,240 1,290 1,310 1,340
17 — — 0,554 0,672 0,786 0,890 0,970 1,026 1,072 1,172 1,230 1,274 1,322 1,342 1,368
18 — — 0,578 0,706 0,824 0,930 1,006 1,066 1,110 1,208 1,270 1,310 1,354 1,374 1,402
19 — — 0,604 0,738 0,860 ' 0,970 1,050 1,106 1,150 1,248 1,308 1,358 1,392 1,414 1,434
20 — — 0,628 0,768 0,894 1,006 1,088 1,142 1,180 1,288 1,340 1,386 1,342 1,442 1,468
21 —. —- — — —. — — — 1,328 1,406 1,418 1,472 1,476 1,504
22 — — — — — — — — — 1,366 1,426 1,466 1,514 1,522 1,534
• М. X. Кар а петь яи ц, Химическая термодинамика, Госхимиздат, 1953.
ПРИЛОЖЕНИЕ I, б
Значения? при Гпр^З.5ч-35
рпр Значения Y при различных ГПр 35
3,5 5 6 7 8 9 10 12 15 16 18 20 22 25 30
0 1,000 1,000 1,000 1,000 1,000 1,000 1,000 1,000 1,000 1,000 1,000 1,000 1,000 1,000 1,000 1,000
5 1,080 1,076 1,071 1,063 1,055 1,057 1,048 1,043 1,038 1,036 1,030 1,028 1,024 1,019 1,015 1,012
10 1,180 1,167 1,152 1,135 1,120 1,117 1,102 1,088 1,072 1,070 1,061 1,052 1,048 1,039 1,031 1,028
15 1,310 1,274 1,244 1,214 1,194 1,181 1,160 1,136 1,110 1,108 1,087 1,080 1,072 1/958 1,045 1,042
20 1,468 1,402 1,346 1,302 1,274 1,248 1,210 1,182 1,152 1,148 1,127 1,110 1,100 1,082 1,060 1,054
25 1,540 1,450 1,398 1,356 1,318 1,284 1.434 1,192 1,188 1,158 1,142 1,128 1,106 1,084 1,070
30 1,686 1,570 1,502 1,444 1,392 1,352 1,292 1,234 1,228 1,192 1,176 1,156 1,130 1,106 1,086
35 . 1,868 1,708 1,612 1,534 1,470 1,424 1,350 1,284 1,270 1,228 1,208 1,184 1,160 1,126 1,104
40 2,028 1,854 1,728 1,630 1,554 1,492 1,410 1,328 1,312 1,266 1,240 1,212 1,178 1,146 1,118
45 2,228 2,018 1,850 1,736 1,644 1,570 1,470 1,380 1,354 1,306 1,274 1,242 1,202 1,168 1,134
50 2,450 2,190 1,986 1,850 1,744 1,654 1,534 1,432 1,400 1,346 1,308 1,272 1,228 1,188 1,152
55 2,694 2,372 2,126 1,968 1,844 1,740 1,598 1,486 1,448 1,388 1,342 1,302 1,252 1,208 1,168
60 2,966 2,570 2,274 2,098 1,952 1,828 1,664 1,546 1,500 1,432 1,380 1,334 1,278 1,230 1,182
65 . — — 1,602 1,552 1,476 1,416 1,368 1,306 1,252 1,196
70 — 1,662 1,608 1,526 1,454 1,380 1,332 1,272 1,214
75 — — 1,728 1,668 1,590 1,494 1,438 1,362 1,292 1,238
80 . 1,794 1,728 1,622 1,538 1,472 1,390 1,314 1,248
85 — 1,862 1,790 1,672 1,582 1,512 1,426 1,338 1,268
90 — 1,930 1,862 1,726 1,626 1,548 1,456 1,360 1,288
95 . . — 2,002 1,912 1,774 1,668 1,590 1,490 1,380 1,308
100 — — — — — — — — 2,070 1,978 1,828 1,712 1,628 1,528 1,402 1,328
М. X. Кар а п е тьянц, Химическая термодинамика, Госхимиздат, 1953.
СИ
ПРИЛОЖЕНИЕ II
Теплоемкости [в кал)(моль • град)], стандартные теплоты образования, изобарно-изотермические потенциалы
и энтропии некоторых веществ в газообразном состоянии *
Вещество Теплоемкость ДЯ298' ккал моль az298, ккал моль S298’ кал моль град
Коэффициенту уравнения Ср = о + ЬТ + сГ2 + (с7т2) рекомен- дуемый темпера- турный интервал, °К веро- ятная ошиб- ка, % СР,298
а ъ • 103 с 10-6 с 10б
Хлор С12
Фтор F2
Водород Н2
Кислород О2
Хлористый водород НС1
Фтористый водород HF
Вода Н2О
Неорганические вещества
8,82 0,06 -0,68 — 298-3000 — 8,11 0 0 53,286
8,29 0,44 -0,80 — 273-2000 1,0 7,52 0 0 48,6
6,9469 -0,1999 — 0,4808 300-1500 0,49 6,892 0 0 31,211 лл лл
8,643 0,202 -1,030 — 298-1500 0,13 1 7 Л( 7 л л
8,186 0,594 -2,972 — 600-3000 0,5 | * / и и 4V,UU
6,34 1,10 0,26 — 298-2000 0,5 6,96 -22,063 -22,769 44,17
6,43 0,82 — — 273-2000 0,5 6,95 -64,2 -64,7 41,47
7,17 2,56 0,08 — 298-2500 0,5 8,025 -57,798 -54,636 45,106
7,55 ** 1,3525** — 0,8658** — — — — — 45,13 **
Органические вещества
Метан СН4 3,422 17,845 — -4,165 291-1500 0,71 8,536 -17,889 -12,140 44,50
Ацетилен С2Н2 12,13 3,84 -2,46 — 298-2000 1 10,50 54,190 50,000 48,00
Omw ТТЛ., Г* Т-Т 1 2,706 29,160 — -9,059 291-1500 1,46 10,41 12,498 16,295 52,45
стилен '-'2^14 1 2,08** 31,1 ** — -10,66** — ' — — 12,496 ** —. 52,48**
Этан С2Нв 1,375 41,852 -13,827 291-1000 0,76 12,59 -20,236 -7,860 54,85
Пропилен С3Нв 2,974 45,024 -11,376 270-510 0,16 15,27 4,880 14,99 63,8
Пропан СгНв 0,410 64,71 — -22,582 298-1500 0,73 17,57 -24,82 -5,61 64,51
Бутан С4Н10 4,357 72,552 — — 22,145 298-1500 1,74 23,61 -29,810 -3,750 74,10
Изобутан С4Ню 2,296 82,407 — -38,792 298-1500 0,54 23,14 -31,450 -4,300 70,42
Пентан C5Hi2 3,140 100,523 — -35,560 298- 1500 0,32 29,30 -35,000 -1,96 83,27
Бензол C6H8 Циклогексан CeH12 -5,04 -7,701 95,63 125,675 — -40,6 -41,584 298-1500 2,66 19,54 25,40 19,82 -29,43 30,850 7,59 64,457 71,28
Толуол C7H8 4,74 113,46 — -46,7 — — 24,80 11,95 29,23 76,42
Фенилэтилеи (стирол) -3,13 130,4 — -52,9 — — 29,18 35,11 51,1 82,48
CsHio
о-Ксилол C8H10 4,603 104,476 — -33,616 298-1500 2,63 31,85 4,54 29,18 84,31
я-Ксилол С8Н10 1,846 108,594 — -35,200 298—1500 2,84 30,32 4,29 28,95 84,23
Метиловый спирт СН4О о , ТТ f~\ 4,88 24,78 — -5,889 300-700 — 10,8 -48,08 38,69 56,8
J 3,578 1 2,16** 49,847 16,991 300—1000 0,28 17,59 . —56,24 —40,3 67,4
сЭтиловыи спирт С2Н8О 49,7 ** — -15,53** — 15,4 ** —56,510** 66,39**
Пропиловый спирт —0,62 74,67 — 25,22 — — 34,9 —62,5 —40,9 46,1
С3Н8О
Изопропиловый спирт — — — — — — —64,2 —41,91 73,2
С3Н8О
Диэтиловый эфир — — — — —• — — —45,60 —28,1 —
с4н10о
Муравьиный альдегид 4,498 13,953 — —3,730 291—1500 1,90 8,45 —27,7 —26,3 52,6
СН2О
Уксусный альдегид 7,422 29,029 — -8,742 298—1500 2,00 15,0 —39,76 —31,96 63,5
С2Н4О
Ацетон С3Н6О 5,371 48,227 — — 15,182 298—1500 1,74 18,38 —51,79 —36,5 72,7
Муравьиная кислота 7,33 21,32 — -8,255 300—700 — 12,96 —86,67 —80,24 58,81
СН2О2
Уксусная кислота 5,20 46,16 — — 18,35 300—700 — 17,3 — 104,3 —91,2 70,1
С2Н4О2
Хлороформ СНС13 7,052 35,598 — —21,686 273—773 0,69 15,63 —24 — 16 70,62
Трифторметан (фреон-2,3) CHF3 3,616 18,239 — —2,035 298—600 0,89 12,68 — •—• 53,50
Фторхлорметан CH2FC1 4,292 27,025 — — 10,605 250—600 0,57 11,37 — — —
Дихлорметан СН2С12 — — — — — — 12,28 —21 — 14 64,68
Дифторметан CH2F2 4,203 21,633 — 4,088 250—600 0,95 10,24 —— — —
Хлористый метил СН3С1 3,562 22,998 — -7,541 273—773 0,64 9.75 — 19,6 — 14,0 55,97
Фтористый метил CH3F —- — —• — — — 8,95 — — 53,30
Хлористый этил С2Н5С1 — — — — — — 15 —25,1 — 12,7 65,90
сл • M. X. Карапетьянц, Химическая термодинамика, Госхимиздат, 1953.
S **F. R. Blchowsky, F. D. Rossiny, The Thermochemistry of Chemical Substances, New York, 1936,
ПРИЛОЖЕНИЕ III
Коэффициенты теплопередачи от жидкости к охлаждающей воде
в сосуде, снабженном змеевиком и мешалкой*
Количество мешалок —2
Охлаждаемая жидкость Число оборотов мешалки, об/мин Темпера тура жидкости, °C Средняя разность температур, град Скорость воды в змеевике, м/сек. Кт. ккал
м* • ч • град
Вода 182 50 22,8 2,6 1940
240 50 10,1 0,216 535
22,1 2,3 1790
Толуол 182 70 40,3 1,4 810
240 50 12,7 0,215 405
21,4 1,07 880
26,3 1,9 945
Изопропиловый спирт 182 50 28 2,45, 780
Этиленгликоль 182 50 13,0 0,106 21-0
18,6 0,4 488
28,0 1,23 632
20,5 1,82 760
Минеральное масло 182 70 41,7 0,385 213
51.4 1,64 258
Глицерин 182 50 14,6 0,106 177
34,5 2,58 365
240 50 15,0 0,215 315
28,3 1,34 425
30,1 1,87 485
Процессы
теплообмена в аппаратах химической про-
•Б. Балайка, К. Сикора,
мышлеииости, Машгиз, 1962.
ПРИЛОЖЕНИЕ IV
Коэффициенты теплопередачи от насыщенного водяного паре
к жидкости, нагреваемой в сосуде, снабженном рубашкой
и мешалкой*
Нагреваемая жидкость ' Количество мешалок Число оборотов мешалки, об/мин Темпера- тура жидкости, °C Разность темпера- тур, град Кт. ккал
м% • ч •град
2 182 80 17 27 70 1080 1380 880
Вода 2 240 50 6 12 37 910 1260 1100
1 182 50 7 10 22 990 1085 910
•Б. Балайка, К- Сикора. Процессы теплообмена в аппаратах химической про-
мышленности, Машгиз, 1962.
550
Продолжение приложения IV
Нагреваемая жидкость Количество мешалок Число оборотов мешалки, об!мин Темпера- тура жидкости, °C Разность темпера- тур, град Кт, ккал
м2 • ч •град
2 182 71 18 880
50 820
Толуол 2 240 50 7 26 740 1030
1 182 50 6 1000
15 680
2 182 50 7 625
Изопропиловый спирт 21 23 890 950
2 182 50 5 1050
14 1080
Этиленгликоль 20 910
1 182 50 7 880
19 760
2 182 70 25 278
Минеральное масло 40 240
2 240 70 17 400
2 107 50 17 240
Г лицерин 30 298
2 240 50 16 303
36 495
ПРИЛОЖЕНИЕ V
Коэффициенты теплоотдачи [в ккалЦм1 • ч • град) ]
при кипении воды (1 атм]
Тепловая нагрузка, ккалКм* • ч) Котел с гори- зонтальным днищем Трубы диаме- тром 25—50 мм и длиной 2 м при естествен- ной циркуляции Тепловая нагрузка, ккал[(м2 • ч) Котел с гори- зонтальным днищем Трубы диаме- тром 25—50 jkjk и длиной 2 jk при естествен- ной циркуляции
13 570 2 049 2 682 1 162 750 11 960 14 390
27 120 3 240 4150 189 800 13 420
54 230 5 362 6 590 217 000 15120
81 300 7318 8 780 244 100 16 590
108 600 8 780 10 730 271 200 17 560
135 700 10 490 12 680
551
ПРИЛОЖЕНИЕ VI
Коэффициенты теплопередачи [в ккалЦм2 ч • град) ]
при выпаривании воды в трубах
Трубы Разность температур, град
6 10 14 18 22 26 34
Стальные новые 1 500 1 800 2 100 2 300 2 600 3 200
Стальные заржавленные . . . 1 200 1 300 1 400 1 500 1 600 1 650 1750
Медные чистые — 3 600 4 900 6 300 7 500 8 300 10 800
ПРИЛОЖЕНИЕ VII
Коэффициенты теплопередачи [в ккал/(м2 - ч • град)]
в выпарных аппаратах с подвесной греющей камерой
и в аппаратах типа «Роберт» при дистилляции воды
Разность температур, град Температура кипения воды, °C
50 60 75 100
ю - 586 927 1498 2035
20 1015 1513 2074 2562
30 1318 1806 2391 2830
40 1562 — —
50 1781 — ‘— —
ПРИЛОЖЕНИЕ VIII
Ориентировочные значения коэффициентов теплопередачи*
Вид теплообмена Вынужденное движение Свободное движение
вт ккал вт ккал
л2•град • ч град м2•град № ♦ ч •град
От газа к газу (при обыч- ных давлениях) .... 10—40 10—30 4—12 3—10
От газа к жидкости (газо- вые холодильники) . . . 10—60 10—50 6—20 5—15
От конденсирующегося во- дяного пара к газу (воз- духоподогреватели) . . . 10—60 10—50 6—12 5—10
От жидкости к жидкости (вода) 800—1700 700—1500 140—340 120—300
От жидкости к жидкости (масло) 120—270 100—250 30—60 25—50
От конденсирующего водя- ного пара к воде (подо- греватели) 800—3500 700—3000 300—1200 250—1000
• К, Ф, Пав лов, П. Г. Р о м а и к о в, А. А. Н о с к о в, Примеры п задачи по курсу
процессов и аппаратов химической технологии, Изд. «Химия*, 1964.
552.
Продолжение приложения VIII
Вид теплообмена Вынужденное движение Свободное движение
вт ккал вт ккал
м2.град м2 . ч •град м2 •град м2 • ч •град
От конденсирующегося во- дяного пара к органиче- ским жидкостям (подо- греватели) 120—340 100—300 60—170 50—150
От конденсирующегося па- ра органических веществ к воде (конденсаторы) 340—870 300—750 230—460 200—400
От конденсирующегося во- дяного пара к кипящей жидкости (испарители) . — — 30—3500 250—3000
ПРИЛОЖЕНИЕ IX
Коэффициенты теплопередачи [е кал/(мг • ч • град)]
для различных теплоносителей *
Теплоноситель Естественная циркуляция Незначительная искусственная циркуляция Интенсивная искусственная циркуляция
Воздух — воздух 3—4 7,5—20 30-40
Воздух — вода 6—8 15—40 58—78
Воздух — конденсирующийся пар . . 6—8 15—40 60-80
Вода — вода 98—283 370—1000 1430—2500
Вода — конденсирующийся пар . . . 190—524 690-1860 2630—4650
Вода — масло 33—54 88—179 255—508
ПРИЛОЖЕНИЕ X
Коэффициенты теплопередачи для гладкой трубы *
Теплоносители
Кт,
кал/(м2 • ч • град)
Конденсация
Водяной пар (Рабс=1 ат) —вода.......................
Насыщенный пар органических веществ (7>абс=1 ат) —вода
Насыщенный пар органических веществ (-Рабс~ 1 ат) с не-
конденсирующнмнся примесями — вода...................
Пар органических веществ (атмосферное давление, значи-
тельная часть неконденснрующнхся примесей) — вода . . .
Пар органических веществ (Рабс = 1 аг, значительная часть
неконденснрующнхся примесей) — вода.................
Углеводороды с низкой температурой кипения — вода . . .
Углеводороды с высокой температурой кипения (Рабс= 1 ат)—
вода ...............................................
1700—3700
488—976
245—580
100—400
50—250
390—980
49—150
’ В. Балайка, К. Снкора, Процессы теплообмена в аппаратах химической промыш-
ленности, Машгиз, 1962,
553
Продолжение приложения X
Теплоносители
*т-
кал/(м% • ч • град)
Нагрев
Пар —вода...........................................
Пар —легкое масло...................................
Пар — тяжелое масло.................................
Пар — органический раствор..........................
Пар — газ...........................................
ВОТ — газ...........................................
ВОТ — тяжелое масло ................................
Кипение
Пар — вода........................................
Пар — органический раствор.............................
1220—3650
245—730
49—390
488-976
25-250
20-200
40-300
1700-3650
490—980
ПРИЛОЖЕНИЕ XI
Кипение жидкости в вертикальной трубе
при естественной циркуляции*
Тепловая нагрузка, ккалКм^ • ч) Температура жидкости, °C разность температур, град Замеренный коэф* фицнент тепло- отдачи, ккалКм* • ч •град)
ВХОД ВЫХОД
Вода
16 640 ' 100,4 99,1 5,56 2 990
35200 89,6 98,8 6,85 5 106
91 200 99,3 98,6 10,85 8 440
143 300 98,8 98,3 13,66 10 430
Изопропиловый спирт
92 500 81,8 81,2 7,7 1 210
28 400 80,7 80,3 11,6 2 560
50050 80,7 80,3 15,3 3 320
74 600 80,7 79,8 18,6 4 030
Бутиловый спирт
14 580 116,3 115,7 7,9 1 840
26 260 116,8 115,9 9,3 2 820
14 220 115,6 115,6 11,9 3 540
59 900 116,8 115,6 14,9 4 030
Четыреххлористый углерод
50 400 76,7 74,7 9,5 537
22 080 75,2 74,7 19,8 1 120
35 250 74,1 73,4 22,6 1560
47300 74,2 73,2 26,5 1 780
• Б. Ба лайка, К. Сикора, Процессы теплообмена а аппаратах химической про-
мышленности, МашГиз, 1962.
554
Продолжение приложения XI
Тепловая нагрузка, ккал1(м2 • ч) Температура жидкости, °C Разность температур, град Замеренный коэф* фнцнент тепло- отдачи, ккал/(м2 • ч • град)
ВХОД ВЫХОД
35% водный раствор углекислого натрия
17 520 116,4 105,4 8,3 2 120
28 080 116,4 105,5 10,15 2 780
45 700 114,8 104,0 12,35 3 700
53 900 115,2 104,6 13,50 3 990
50% водный раствор углекислого натрия
16 500 117,3 114,8 8,35 1935
24 700 115,9 114,6 10,8 2 300
51 400 112,4 112,7 14,3 3510
59 900 112,6 111,8 16,2 3 710
ПРИЛОЖЕНИЕ XII
Теплообмен в псевдоожиженном слое
Система Размер частиц, мм Температура, °C Давле- ние, атм Напра- вление тепло- обмена а', ккал мЛ ч •град
твердое тело газ СЛОЯ стенкн
Стеклян- ные шарики Воздух 0,07 147- 239 227—268 1 От стенки К слою 100—520
Карбо- рунд Светиль- ный газ 0,075 - 0,11 21—26 13—15 1 От слоя к стенке 73—1030
Песок Воздух 0,04—0,18 Конден- сирован- ный водя- ной пар 4,6 От стенки к слою 390
Графит Воздух 0,260 31-63 15—56 От слоя к стенке 19-600
ОГЛАВЛЕНИЕ
Предисловие ............................... ...................... 3
Глава 1
Вопросы экономики
в технологии основного органического синтеза
Промышленный синтез некоторых продуктов............................... 5
Синтез диметилтерефталата..........................................6
Синтез фенола......................................................8
Сравнительная оценка способов промышленного синтеза....................9
Основные показатели химнко-технологических процессов..................15
Структура себестоимости продукта промышленного синтеза................29
Сырье и основные материалы........................................31
Энергетические средства ......................................... 34
Амортизационные отчисления........................................37
Заработная плата ................................................ 39
Удельные энерго-материальные затраты..............................39
Структура капитальных затрат..........................................40
Экономический оптимум технологического процесса.......................43
Влияние мощности производства на его экономические показатели .... 54
Доход предприятия.....................................................57
Литература ....................................................... ... 59
Глава II
Некоторые вопросы термодинамики
и кинетики химических реакций
Химическое равновесие..................................................64
Кинетика химических реакций в закрытых системах.......................76
Скорость реакции ................................................ 76
Гомогенные реакции.................................................79
Мономолекулярные реакции.........................................79
Сложные реакции...................................................81
Гетерогенные реакции..............................................103
Реакции на твердой поверхности .................................. 104
Реакции в слое...................................................117
Закономерности многостадийных реакций в закрытых системах.............130
Изменение концентраций исходных веществ н продуктов многостадий-
ных реакций ............... ..... ............................... 131
556
Последовательные многостадийные реакции........................131
Параллельно-последовательные многостадийные реакции............139
Зависимость выхода продуктов многостадийных реакций от степени превра-
щения исходного вещества.............................................140
Кинетика реакций в открытых системах.................................151
Открытые системы полного вытеснения..............................153
Открытые системы полного смешения................................157
Графический способ расчета состава реакционной смеси ......... 171
Изучение кинетики химических реакций с использованием открытых
систем..........................................................177
Открытые системы промежуточного типа.............................178
Противоток реагентов ......................................... 178
Ступенчатый подвод реагентов...................................180
Каскад реакционных аппаратов полного смешения..................183
Аппарат с продольным перемешиванием............................200
Выход продуктов многостадийных реакций в открытых системах . . . 203
Системы полного вытеснения.....................................203
Системы полного смешения.......................................203
Системы промежуточного типа....................................209
Литература...........................................................211
Глава III
Вспомогательные процессы и аппараты
Хранение и транспорт <;ырья и продуктов...............................213
Хранение жидких и газообразных веществ............................214
Транспорт жидких и газообразных веществ...........................217
Теплообмен н теплообменная аппаратура ............................... 227
Тепловые схемы....................................................228
Рекуперативные теплообменники ................................... 235
Схема расчета рекуперативной теплообменной аппаратуры...........237
Энергетические средства и теплоносители.........................239
Основные типы рекуперативных теплообменников....................245
Интенсификация конвективного теплообмена........................262
Теплообмен смешением .............................................265
Основные типы теплообменников смешения..........................265
Нагрев агрессивных жидкостей....................................266
Нагрев газов....................................................267
Теплообмен смешением в системе газ—твердое тело.................267
Тепловая изоляция ............................................... 257
Литература ...........................................................269
Глава IV
Превращение сырья и аппаратурное оформление
химических стадий процессов
Технологические особенности реакционной аппаратуры................... 272
Периодическое, непрерывное и полунепрерывное проведение процессов . 275
Реакционный агрегат периодического действия......................275
Реакционный агрегат непрерывного действия........................277
Реакционный агрегат полунепрерывного действия . .................279
Динамика технологических процессов.................................283
Параметры реакционного процесса....................................285
Зависимость выхода продуктов от температуры......................285
Зависимость выходе продуктов от концентрационных условий . • . 289
557
Зависимость скорости реакции от температуры ......... 292
Зависимость скорости реакции от концентрации реагентов..........296
Зависимость скорости реакции от давления ...................... 296
Производительность реакционной аппаратуры.........................298
Реакционные аппараты периодического действия....................298
Реакционные аппараты непрерывного действия......................302
Тепловой режим экзотермических реакций................................320
Гомогенные реакции................................................321
Адиабатический режим............................................322
Изотермический режим............................................328
Гетерогенные реакции..............................................339
Реакция на твердой поверхности................................ 339
Реакция на пористом зерне.......................................343
Сравнительная оценка реакционной аппаратуры ......................... 349
Параллельные реакции...................................... . . . 349
Последовательные реакции..........................................351
Продукт В.......................................................352
Продукт D.......................................................359
Литература ...........................................................365
Глава V
Типы реакционных аппаратов
Реакторы для проведения процессов в системе газ — жидкость......366
Гидродинамика барботажного слоя.............................367
Образование и движение одиночных пузырей...................367
Диспергирование газа через перфорированные тарелки.........370
Аппараты с диспергированием газа через отверстия............374
Полное перемешивание.......................................375
Полное вытеснение..........................................375
Барботажные аппараты с насадкой............................ 379
Аппараты с мешалками........................................382
Вращающиеся мешалки........................................383
Самовсасывающие мешалки....................................385
Вибрационные мешалки.......................................390
Тепловой баланс реактора .................................. 390
Теплообмен в газо-жидкостном слое...........................393
Реакторы для проведения процессов в системе газ—твердое тело .... 395
Катализаторы ...............................................395
Аппараты с неподвижным слоем катализатора...................400
Адиабатический режим.......................................400
Теплообмен в неподвижном слое катализатора.................406
Аппараты с движущимся слоем катализатора.................. 410
Аппараты с псевдоожиженным слоем катализатора...............414
Гидродинамика псевдоожиженного слоя........................415
Теплообмен в псевдоожиженном слое..........................421
Конструктивное оформление реакторов с псевдоожиженным слоем . . 422
Механические способы создания кипящего слоя.................427
Виброкипящий слой ...................
Псевдоожижение перемешиванием ............................428
Реакторы для проведения процессов в системе жидкость — твердое тело . . 430
Реакторы для фотохимических реакций.............................436
Зависимость скорости реакции от степени использования света .... 439
Зависимость квантового выхода от степени использования света . . . 440
Конструктивное оформление фотохимических реакторов..........441
Литература . .......................................*...........446
668
Глава VI
Разделение и очистка продуктов
- промышленного органического синтеза
Разделение гетерогенных систем........................................453
Системы жидкость —газ.............................................453
Системы жидкость — жидкость.......................................458
Системы жидкость — твердое тело . ................................463
Отстойники ..............................................- . . . 463
Фильтровальная аппаратура ..................................... 465
Разделение систем твердое тело — газ..............................470
Разделение гомогенных систем..........................................473
Экстракция .......................................................474
Кристаллизация ............................................483
Ректификация и абсорбция.........................................487
Адсорбция ..................................................... 505
Специальные методы разделения.......................................509
Зонная плавка ............................................. 510
Адсорбция на молекулярных ситах.................................510
Разделение с использованием мембран.............................511
Очистка продуктов промышленного органического синтеза..............513
Литература ..........................................................524
Глава VII
Техника взрывобезопасное™
Основные понятия теории горения газов . . ......................... 528
Нормальная скорость пламени.....................................528
Концентрационные пределы распространения пламени (пределы взрывае-
мости) ......................................................... 529
Горение в трубах различного диаметра .......................... 534
Самовоспламенение и поджигание горючей смеси....................535
Самовоспламенение ............................................ 535
Поджигание горючей смеси.......................................538
Детонация ......................................................538
Выявление опасных режимов работы технологического оборудования и за-
щитные устройства...................................................539
Литература .........................................................544
Соотношение единиц международной системы единиц [СИ] с единицами дру-
гих систем.................’........................................545
Приложения .........................................................546
Виктор Сергеевич Хайлов,
Борис Борисович Брандт
ВВЕДЕНИЕ В ТЕХНОЛОГИЮ
ОСНОВНОГО ОРГАНИЧЕСКОГО СИНТЕЗА
Издательство „Химия", Ленинградское отделение
Невский пр., 28
Редактор В. Г. Горский
Техн, редактор 3. Е. Маркова
Корректоры: Л. А. Л ю б о в и ч, Н. Я. Шубина
Переплет художника С. В. Гайковича
Сдано в набор 30/1 1969 г.
Подписано к печати 23/IV 1969 г.
Бумага типогр. № 2. Формат 60X90 x/ie
Уч.-изд. л. 37,65. Печ. л. 35. М-40754. Тираж 11 000 экз.
Цена 2 р. 06 к. Заказ 54,
Ленинградская типография Хе 2 имени Евгении Соколовой
Главполиграфпрома Комитета по печати при
Совете Министров СССР. Измайловский пр.» 29.