/

































































































Автор: Девятых Г.Г. Еллиев Ю.Е.
Теги: химия химические науки химические вещества
ISBN: 5-06-000073-7
Год: 1990




Текст
KWIIII QJIMIIIH IHtal^f,
O'UHOU UUJMPAIA
Ki hi Aii.iHtHii биты кокрицепа
гио гбважс указанного здесь ер*ка
Количество предыдущих выдач
к/*/\ щии:
Арт. КГ 087-03.JS6
Ней» »а 1000 шт. — 8J кав
4 07.90 К т. 1 3 4S30-13 или
П.»»«рп1г* пингу не н1»«1ше •зиаченог* т*р*><иу
Г Г Девятых, Ю Е. Еллиев
ГЛУБОКАЯ
ОЧИСТКА
ВЕЩЕСТВ
ИЗДАНИЕ ВТОРОЕ,
ИСПРАВЛЕННОЕ И ДОПОЛНЕННОЕ
Допущено
Государственным комитетом СССР
по народному образованию
в качестве учебного пособия
для студентов
химических и химика технологических
специальностей
высших учебных заведений
МОСКВА «ВЫСШАЯ ШКОЛА» 1990
Больше химической литературы на
vk.com/chemzone
More chemistry books you can find on
vk.com/chemzone
vk.com/chemzone
М - W г А
ББК 24 5
Д26
УДК 541 1
Рецензенты: кафедра физической химии Mihkoik кого щеудар-
ствепного университета им М В Ломоносова (i.n кдфаддой нроф
Ю А Пентин), д-р хим. наук А А Ефремов (НПО «ИРГЛ»)
Девятых Г. Г., Еллиев Ю. Е.
Д26 Глубокая очистка веществ Учеб пособие для хим ц
хим -технол спец, вузов -2-е изд., испр и доп.— М:
Высш, шк., 1990. — 192 с. ил.
ISBN 5-06-000073-7
Излагаются теоретические аспекты используемых в настоящее время основ-
ных методов глубокой очистки веществ Большое внимание уделяется широко
распространенным дистилляционным н кристаллизационным методам Во втором
издании <I-е — в 1974 г) добавлены новые разделы, посвященные расчету отно
сительноЯ летучести примесей, периодической ректификации, загрязняющему дей
ствию материала аппаратуры при кристаллизационной очистке веществ, глубокой
очистке от взвешенных частиц.
Книга может быть полезна для сотрудников научно исследовательских инсти
тутов н специалистов промышленных предприятий, работающих в области хнмни
веществ высокой чистоты.
д 1707000000(4309000000)—083
001(01)—90
118—89
ББК 24 5
541
И.
Б1БЛ1ОТЕКД
УКРАКНСЬКОГО
Г 7 ?. "iM-Ttx
УЦ1ЕЕ?СИТ^ТУ
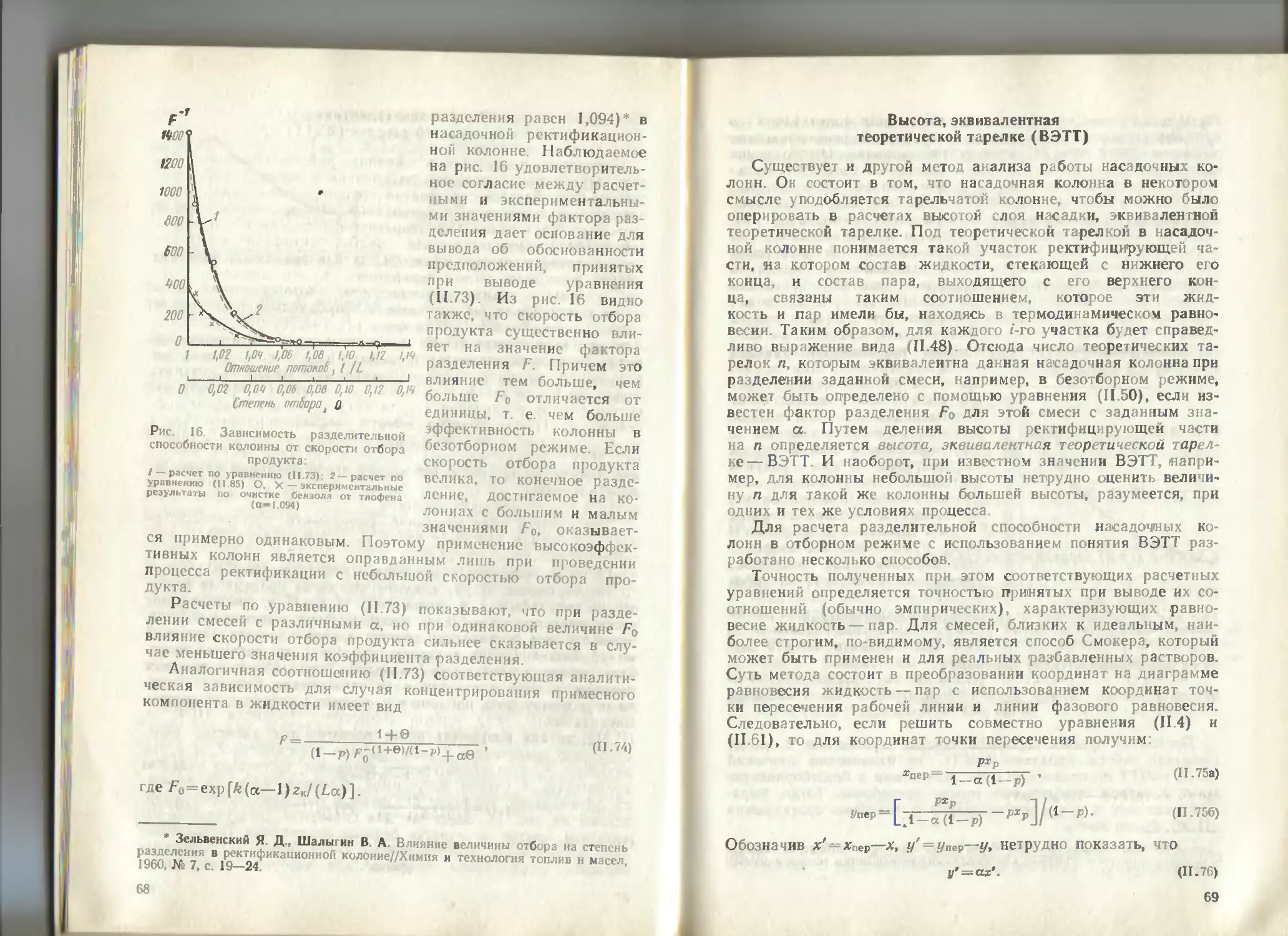
Учебное издание
Девятых Григорий Григорьевич.
Еллиев Юрий Ефремович
"""Глубокая очистка веществ
Зав редакцией С Ф Кондрашкова Редактор 7 С Костян Мл редактор Л С Мокор
кина Художественный редактор £ Д Косырева Художник Н Н Л hi аршин Ггяинче
скнй редактор В М Романова Корректор Р К Косинове
ИБ Ki 7411
Изд К» Хим 866 Сдано в набор 26 06 89 Подл в печать 30 11 89 Форм»! «.".я#1 «.
Бум офс. № 2. Гарнитура литературная Печать офсетная Объем II I и цен. л.
2,01 усл. кр-отт 11.61 уч изд. л. Тираж ЫЮ0 *к> зам 3ft к Цку. 41
Издательство «Высшая школа. 101430. ГСП 4 Псглиниая уя д .44
Набрано в московской типографии К> II I кимитчап! <• « I •
113105 Москва П.птннискяя ул . д I
Отпечатано с готового набора в московской гнпо!рафии М Я I aiiMiwaill i < ' Р
101898 Москва. Центр Хохлит кий игр 7
ISBN 5-06-000073-7
(Ф Дилипа* II । I, <11»п К) Е.
1'1411
ПРЕДИСЛОВИЕ
После выхода первого издания учебного пособия «Глубокая
очистка веще! тв» ( 974 г) исследования в области получения
высокочистых веществ ознаменовались новыми успехами С се-
редины 70-х годов проблема высокочистых веществ оказалась
связанной с развитием волоконной оптики, для которой потре-
бовались новые материалы с низким содержанием примесей.
Возросли требования и к чистоте веществ, используемых в мик-
роэлектронике и полупроводниковой технике В соответствии с
этим актуальной стала проблема максимальной очистки веществ
от примесей, в виде взвешенных частиц субмикронного раз-
мера. Для решения этой задачи был разработан новый ме-
тод очистки — пленочная ректификация с воздействием на пар
температурного градиента (термодистилляция). Большое зна-
чение придается подбору малозагрязняющих конструкционных
материалов и созданию технологических комплексов, которые
исключали бы контакты очищаемого вещества с исходным
сырьем В эту цепочку включают методы аналитического конт-
роля.
Во втором издании шире представлены химические методы»,
глубокой очистки. Добавлены новые разделы, посвященные рас-
чету коэффициента разделения при фазовом равновесии жид-"
кость — пар и твердое тело — жидкость, периодической ректифи-
кации с постоянным и дискретным отбором продукта. Больше
внимания уделено вопросам многократной перегонки и много-
кратной перекристаллизации, загрязняющему действию материа-
ла аппаратуры при кристаллизационной очистке веществ, глу-
бокой очистке от взвешенных частиц Соответственно сокраще-
ны некоторые из рассмотренных в первом издании разделов;
произведены необходимые исправления. При этом общий план
построения книги сохранен прежним: основное внимание в ней,
как и ранее, уделено дистилляционным и кристаллизационным
методам глубокой очистки.
Авторы надеются, что второе издание учебного пособия
«Глубокая очистка веществ» будет полезно не тотько студен-
там, специализирующимся в области химии высокочистых ве-
ществ, но и специалистам работающим в этой области.
Авторы
ВВЕДЕНИЕ
1 Вещества высокой чистоты
Быстро развивающаяся современная техника требует раз-
нообразных материалов с заданными свойствами. Качество та-
ких материалов во многих случаях определяется чистотой ис-
пользуемых при их производстве исходных веществ, т е. нали-
чием в последних различных примесей
То. что содержащиеся в веществе примеси влияют на его
свойства, было замечено задолго до того, как возникла химиче-
ская наука. Наглядный пример в этом отношении представлен в
древней легенде об определении Архимедом содержания золота
в короне сиракузского правителя Гиерона Вопросу чистоты ве-
ществ в свое время большое внимание уделяли и алхимики
М. В Ломоносов указывал на необходимость проведения науч-
ных исследований только с чистыми веществами И уже в нача-
ле XIX в., когда в химии установилось понятие об индивидуаль-
ном веществе как химическом соединении постоянного состава,
стало совершенно очевидным, что многие свойства вещества
действительно определяются степенью его чистоты. Проводив-
шиеся в то время физико-химические исследования, как прави-
ло, требовали очистки веществ не от каких-либо отдельных при-
месей. а от примесей вообще, ибо физико-химические свойства
веществ от природы примесей практически не зависят вследст
вие слабой зависимости от природы веществ сил межмолеку-
лярного взаимодействия, определяющих эти свойства.
В настоящее время тенденция к повышению чистоты веществ
обусловлена возрастающими потребностями науки и техники в
чистых материалах. Особенно резко возросли требования к чис-
тоте ряда веществ в 40—50-е годы в связи с бурным развитием
атомной энергетики и полупроводниковой техники. Оказалось,
что ядерные и электрофизические свойства веществ более «чув-
ствительны» к чистоте, чем физико-химические и, главное, чрез-
вычайно сильно зависят от природы примеси: зависимость ука-
занных свойств от примесей, называемых лимитируемыми, может
быть неизмеримо более высокой, чем от других Тогда же полу-
чил распространение термин «элементы особой чистоты» Так
стали называть простые вещества, подвергнутые очистке до та-
кого остаточного содержания лимитируемых примесей, когда
начинали проявляться те или иные специфические свойства
4
этих веществ Наряду с производством простых особо чистых
веществ появилось и производство сложных веществ особой чис-
тоты Это полупроводниковые соединения диэлектрики, реакти-
вы и т д. Допустимое содержание примесей в веществах осо-
бой чистоты может изменяться в широких пределах. Это зави-
сит от области применения вещества, от сложности технологии
его получения и трудности анализа. Так, содержание ряда при-
месей в полупроводниковых кремнии и германии не должно пре-
вышать 10~9—10 10 %. В то же время даже в наиболее чистых
образцах этих веществ содержание примесей углерода, кислоро-
да и азота может достигать 10~5—10~6 % Это объясняется тем,
что очистка кремния и германия от углерода, кислорода, азота
и определение последних представляют собой наиболее труд-
ную задачу, а главное — в основном не эти примеси лимитируют
полупроводниковые свойства указанных веществ.
Вещества особой чистоты играют исключительно важную
роль во многих областях новой и новейшей техники, развитие
которых определяет темпы и уровень научно-технического про-
гресса, в данном случае для создания принципиально новых
приборов или технологических процессов, которые были бы не-
возможны при недостаточной степени чистоты этого вещества
по лимитирующим примесям. Проблема веществ особой чисто-
ты, таким образом, становится проблемой материаловедения,
от прогресса которого зависит само существование и развитие
практически любой отрасли промышленности.
Особо чистые материалы со строго выдержанным количест-
венным и качественным составом примесей нужны в значитель-
ных количествах, например, в радиоэлектронной и атомной про-
мышленности, волоконной оптике. Допустимый уровень кон-
центрации лимитируемых примесей во многих веществах осо-
бой чистоты оценивается в настоящее время величиной 10-6—
10%. Если при этом суммарное содержание примесей в таких
веществах не превышает 10~3—104 %, то для их обозначения,
по-видимому, более целесообразно использовать термин «высо-
кочистые вещества». Это позволяет рассматривать задачу по-
лучения веществ особой чистоты как один из этапов более об-
щей проблемы получения веществ высокой чистоты в целом.
Вещества высокой чистоты имеют непреходящее самостоя-
тельное значение в сугубо научном плане. Повышение степени
чистоты часто приводило к открытию новых свойств вещества
и новых явлений, т. е к повышению уровня знаний о веществе
как форме существования материи. Имеющиеся в нашем распо-
ряжении материалы высокой чистоты пока представляют собой
лишь слабое приближение к «абсолютно чистому веществу»,
поскольку суммарное число примесных атомов или молекул в
них всего иа пять-шесть порядков меньше числа частиц основ-
ного вещества
5
Вместе с тем высокочистое вещество можно рассматривать
как вещество в экстремальных условиях, ибо состав — такой
же фундаментальный параметр состояния вещества, как темпе-
ратура и давление Подобный подход поддерживает наш опти-
мизм в отношении обнаружения новых свойств при повышении
степени чистоты веществ.
2. Классификация веществ высокой чистоты
До настоящего времени не существует единой международ-
ной классификации химических веществ по степени их чистоты.
Это объясняется разнообразными специфическими требования-
ми, предъявляемыми к высокочистым веществам и материалам
различными отраслями науки и техники.
Чтобы подчеркнуть различие химических веществ по их чис-
тоте, наиболее чистые вещества, применяющиеся при химиче
ском анализе, а также для научных исследований уже в нача-
ле текущего столетия были объединены под общим названием
реактивы, которое часто используется и в настоящее время.
В Советском Союзе эти вещества делятся на четыре категории:
чистые (ч), чистые для анализа (ч. д. а.), химически чистые
(х. ч ) и особо чистые (ос. ч.) Перечень нежелательных приме-
сей и их предельное содержание лимитируются техническими
условиями. Поэтому содержание примесей в двух различных
реактивах одной и той же категории, например «чистый», мо-
жет быть различным и определяется в основном трудностью
освобождения реактива от той или иной примеси, а также пре-
делом обнаружения используемого метода анализа. Отсюда яс-
но, что приведенная классификация реактивов является весьма
условной. То же самое можно сказать и о таких бытующих в
практике определениях степени чистоты вещества, как «спект-
рально чистое», «хроматографически чистое», «криоскопически
чистое», «люминесцентной чистоты» и т д.
Содержание примесей или основного компонента в веществе
выражается в массовых (мае. %), атомных (ат. %) процентах
или в молярных долях. Иногда концентрацию примесей выра-
жают в частях на миллион (ррм), в частях на миллиард (ррв
или ррМ). Однако, к сожалению, при такой маркировке часто
не указывается, какие части (атомы или их масса) имеются в
виду.
С возникновением производства веществ особой чистоты Го-
сударственным научно-техническим комитетом СССР в 1959 г.
была утверждена новая система классификации веществ по
степени их чистоты. По этой классификации все чистые веще-
ства делятся на три класса: А, В, С. Суммарное содержание
примесей в веществе указывается в виде отрицательного деся-
тичного логарифма их атомного или массового процента (раз-
6
личие между этими величинами невелико) и записывается сра-
зу же после буквы, характеризующей класс чистоты. Вещества
с содержанием суммы примесей от 10~’ до КУ"2 % составляют
класс А и обозначаются А!—А2 Вещества с содержанием при-
месей от 10~3 до 10~6 % составляют класс В и обозначаются
ВЗ—В6. И наконец, вещества с содержанием примесей от 10~7
до 10~‘° % составляют класс С и обозначаются С7—СЮ
Указанной системой маркировки предлагалось пользовать-
ся, в первую очередь, при характеристике степени чистоты по-
лупроводниковых веществ. Подобная классификация появилась
и в других странах. В частности, в ФРГ содержание примесей
в веществах особой чистоты указывается в виде индекса чис-
тоты /?—п, где индекс R (Reinheitsgrad) обозначает степень
чистоты, п = —lgc-f-2, с — суммарная концентрация примесей
(в мае. %). Таким образом например, индекс чистоты R— 4
означает, что препарат содержит 99,99 мае. % основного веще-
ства
Выражение чистоты вещества через содержание основного
компонента широко используется в металлургии При этом со-
держание основного компонента принимается равным разности
100 % —%, где Хе, — суммарное содержание определяемых
I :
примесей, %. Так если общее содержание определяемых в ве-
ществе примесей составляет 10“2 %, то говорят, что это веще-
ство имеет чистоту 99,99 %, или, по терминологии металлур-
гов, не выше четырех девяток.
Как можно видеть, в рассмотренных способах выражения
чистоты вещества в качестве определяющего критерия принято
суммарное содержание примесей в веществе. Однако, как уже
отмечалось, количества определимых и действительно содер-
жащихся в веществе примесей могут быть далеко не одинако-
выми. Отсюда становится ясным, что использование указанно-
го критерия в качестве основы для классификации веществ по
степени их чистоты оказалось преждевременным Тем более,
что из-за отсутствия достаточно хорошей базы для проведения
анализов на содержание большого числа примесей требование к
суммарной чистоте вещества выдвигалось не очень настойчиво
Поэтому в 1965 г Комитетом стандартов, мер и измерительных
приборов СССР была введена система классификации, в соот-
ветствии с которой при установлении чистоты вещества контро-
лируется содержание в нем только лимитируемых примесей
Таким образом, предложенная классификация относится не к
высокочнстым веществам вообще, а лишь к веществам особой
чистоты По этой классификации особо чистому веществу при
сваивается определенная марка в зависимости от числа контро-
лируемых в нем примесей и их суммарного содержания Для
веществ, в которых лимитируются только примеси неорганиче
7
ских веществ, марка обозначается буквенным индексом «ос ч»
н следующими за ним двумя числами Первое число показыва-
ет количество лимитируемых примесей, содержание которых
определялось, а второе представляет собой отрицательный де-
сятичный логарифм суммарного процентного содержания этих
примесей (цифры пишутся через тире). Например, запись
«ос. ч. 10—6» характеризует особо чистое вещество, в котором
количественно определены 10 лимитируемых примесей с их сум-
марным содержанием 1-10“6—4-10“6мас %
Для особо чистых веществ, в которых контролируются толь-
ко примеси органических веществ, марка обозначается буквами
«оп» (органические примеси) с числом (через тире), которое
соответствует отрицательному десятичному логарифму суммар-
ного процентного содержания определяемых примесей органи-
ческих веществ, и индексом «ос. ч». Так, марка вещества, в ко-
тором суммарное содержание примесей органических веществ
составляет 2-10-3 %, обозначается «оп — 3 ос. ч». Если же в ве-
ществе контролируются примеси неорганических и органиче-
ских веществ, то при установлении его чистоты необходимо учи-
тывать содержание и тех, и других примесей Например, марка
особо чистого вещества, в котором суммарное содержание опре-
деляемых примесей органических веществ равно 4-Ю-3, а сум-
марное содержание восьми лимитируемых примесей неоргани-
ческих веществ равно 6-10“5 % (или округленно 10“4 %), обо-
значается «оп — 3 ос. ч 8—4».
Нетрудно видеть, что рекомендуемая классификация также
не лишена недостатков. В частности, в ней не нашел отражения
вопрос о содержании примесей, присутствие которых в данном
веществе не определялось, но которые в нем могут находиться.
В то же время этот вопрос имеет принципиальное значение, по-
скольку с расширением наших знаний о свойствах индивидуаль-
ного вещества число лимитируемых по отношению к нему при-
месей, естественно, будет увеличиваться. Необходимо также
принять во внимание и то, что в последние годы растет тен-
денция к повышению спроса на вещества, очищенные от всех
примесей. Отсюда вытекает необходимость разработки такой
классификации веществ по их чистоте, чтобы в ней учитыва-
лись как содержание в веществе лимитируемых примесей, так
и суммарная степень его чистоты. Исследования в этом направ-
лении ведутся п уже имеются работы* в которых рекомендуют
для построения соответствующей классификации использовать
* Девятых Г. Г., Степанов В. М. К вопросу о рациональной классифика-
ции веществ по степени чистоты//В ки.: Получение и анализ чистых веществ.
Изд-во ГГУ, 1978, вып 3, с. 3—7, Девятых Г Г., Каржииа Г. А., Степа-
нов В, М. Понятие абсолютно чистого выцества//В ки.: Химия и мировоззре-
ние М 1986, с. 117—123
8
статистический подход и термодинамические критерии, харак-
теризующие предельно разбавленные растворы, каковыми и яв-
ляются высокочистые вещества.
3. Классификация методов
глубокой очистки веществ
Природа не приготовила для люден ни простых, ни сложных
веществ в особо чистом состоянии Хотя ряд веществ, таких,
как алмаз, кварц, самородное золото и т. д., и встречается в
природе на первый взгляд в чистом виде, но и эти вещества со-
держат разнообразные примеси — одних больше, других мень-
ше. Если мы, например имеем дело с серой самородной, то уже
визуально заметно, что она загрязнена примесями в ней кроме
атомов серы, составляющих основную массу вещества, находят-
ся атомы селена, мышьяка, железа, углерода и других элемен-
те Любое простое или сложное вещество —это смесь многих
веществ, и задача получения индивидуального вещества состо-
ит в выделении из этой смеси основного вещества. При получе-
нии того или иного вещества с помощью химической реакции
примеси, содержащиеся в реагентах, частично переходят в про-
дукты реакции. Кроме того, при этом всегда образуются по-
бочные соединения, загрязняющие получаемое вещество. Таким
образом, получение простых и сложных веществ в высокочис-
том состоянии заключается в глубокой их очистке и освобож-
дении от примесей. Отличие от обычного разделения здесь со-
стоит в том, что при получении вещества высокой чистоты глу-
бина разделения должна быть значительно большей, а материал
стенок аппаратуры не должен в сколько-нибудь заметной степе-
ни загрязнять очищаемое вещество.
Методы, применяемые для глубокой очистки веществ, раз-
нообразны. Выбор метода определяется свойствами очищаемого
вещества и природой отделяемых примесей Хотя теоретически
можно оценить возможности методов, тем не менее в большин-
стве случаев приходится проводить специальные исследования,
дающие возможность выбрать оптимальный метод или найти
метод, позволяющий достигнуть требуемой степени чистоты.
Глубокая очистка вещества потребовала усовершенствова-
ния известных методов разделения смесей, а также разработки
новых методов. Так, например, именно для глубокой очистки
вначале металлов, а затем и других веществ в 50-х годах был
разработан метод зонной перекристаллизации (зонной плавки),
который и в настоящее время широко применяется и в лабора-
ториях, и в промышленности. Совсем недавно предложен метод
термодистилляции, позволяющий производить эффективную
очистку жидкостей от находящихся в них примесей в виде
взвешенных частиц. Развит метод ректификации в режиме
9
эмульгирования. Установлено, что отбор продукта (или кон-
центрата примесей) в ректификации при определенном сочета-
нии параметров процесса целесообразно производить дискрет-
но, а не непрерывно, как это принято. Успешно ведутся иссле-
дования возможностей электродиффузионного метода очистки
металлов
Характерной особенностью применяемых в настоящее вре-
мя методов глубокой очистки является то, что все они основа-
ны на использовании различия в химических или физико-хими-
ческих свойствах разделяемых веществ, т. е в свойствах, обус-
ловленных главным образом строением электронных оболочек
атомов или молекул разделяемых веществ Методы, в основе
которых лежит различие в физических свойствах разделяемых
веществ, например разделение в центрифугах, электромагнит-
ная сепарация, широко используемые для разделения изотопов,
для глубокой очистки веществ применения пока не находят.
Таким образом, все используемые в настоящее время мето-
ды глубокой очистки веществ по своей сути можно объединить
в две группы: химические и физико-химические. Для достиже-
ния требуемой степени чистоты конкретного вещества часто
приходится прибегать к различным сочетаниям этих методов
ГЛАВА I
ХИМИЧЕСКИЕ МЕТОДЫ
глубокой очистки веществ
§ 1 ОБЩАЯ ХАРАКТЕРИСТИКА
Химические методы разделения смесей веществ основаны на
различии в константах равновесия или константах скоростей ре-
акций с участием основного вещества и примесей. Это наиболее
древние методы очистки веществ Например, получение того или
иного металла—это не что иное, как процесс отделения атомов
лого металла от сопутствующих им атомов других элементов,
выделение атомов данного металла из природных смесей, более
пли менее богатых этим металлом. На химических методах раз-
деления смесей основаны классические методы химического ана-
лиза. Эти методы в большинстве своем включают стадию отде-
ления примесей от основного вещества путем перевода их в не-
растворимые соединения с последующим отделением осадка или
стадию отмывки примесей реактивом, в котором основное ве-
щество не растворяется.
В связи с проблемой получения веществ особой чистоты хи-
мические методы стали применяться и для глубокой очистки
веществ. Например, обработка кремния минеральными кисло-
тами позволяет перевести значительную часть содержащихся в
поверхностном слое кремния соединений металлов в раствори-
мые соли, которые затем можно отмыть Таким образом можно
достичь значительного снижения содержания металлов в крем-
нии. При этом, разумеется, используемые реагенты сами долж-
ны иметь высокую степень чистоты во избежание возможного
загрязнения очищаемого вещества.
Хорошо зарекомендовали себя методы связывания примесей
специально подобранными реагентами в такие химические со-
единения, которые сравнительно легко тем или иным способом
(фильтрование, центрифугирование, отгонка и т. д.) отделяются
о1 основного вещества. Так, действуя на водные растворы хло-
ридов и сульфатов некоторых щелочных и щелочноземельных
элементов диэтилдитиокарбаминатом натрия (метод избира-
тельного комплексообразования), можно перевести содержа-
щиеся в этих солях примеси железа, кобальта, меди и некого*
рых других переходных металлов в малорастворимые соедине-
ния типа хелатов по схеме:
11
2(C2H5)2N—С +Mc«*=
^SNa
/Ч
= (С2Н0)2Ь1-С Me С- N(C2II6)t+2Na+
XS / \s^
Эти соединения выпадают в осадок и могут быть отфильтрова-
ны Данным методом удается понизить содержание железа в
указанных солях до 10~5 %, меди — до 10-6 % и т. д.
Другими весьма эффективными химическими методами глу-
бокой очистки веществ являются методы избирательного окис-
ления или восстановления по отношению к очищаемому веще-
ству или примесям. В качестве окислителя используется кисло-
род, галогены, в особенности хлор (метод избирательного хло-
рирования). При использовании кислорода обычно стремятся с
его помощью химически связать и удалить примеси, находя-
щиеся в очищаемом веществе. Но иногда лучшая очистка до-
стигается переводом в оксид основного элемента с последую-
щим его восстановлением
При использовании же в качестве окислителя галогена поч-
ти всегда преследуется цель перевода в галогенид основного
элемента. Полученный галогенид подвергается восстановлению
водородом или термораспаду. В результате протекания соответ-
ствующих химических реакций происходит значительное сни-
жение содержания примесей в очищаемом веществе. Для уве-
личения степени чистоты получаемого материала промежуточ-
ное соединение — галогенид и выделяемый из него элемент под-
вергают дополнительной очистке.
На практике в качестве промежуточных соединений в рас-
сматриваемом галогенидном методе используют летучие галоге-
ниды, под которыми условно подразумевают галогениды, имею-
щие давление насыщенного пара при 500 К более 103 Па, и для
которых разработаны достаточно эффективные методы очистки.
Из рассмотрения свойств галогенидов элементов периодической
системы следует, что возможности галогенидного метода доста-
точно высоки (рис. 1). Действительно, как видно из рис. 1, ле-
тучие галогениды имеют более чем 20 элементов, в то время как
галогенидный метод используется для глубокой очистки лишь
некоторых из них (бор, галлий, олово, мышьяк, сурьма, висмут,
молибден, вольфрам) Расширению возможностей галогенидно-
го метода может способствовать!!более широкое использование
реакций термораспада летучих галогенидов (иодидов). Однако
следует иметь в виду, что при повышенных температурах, обыч-
но характерных для процесса термораспада, возрастает веро-
12
Рис. I Элементы периодической системы, имеющие летучие галогениды
и шесть загрязнения выделяемого элемента материалом anna-
р.i гуры.
Ряд элементов может быть подвергнут глубокой очистке по
< шдующей схеме: перевод в гидрид — очистка гидрида — тер-
мическое разложение гидрида на элемент и водород. Примени-
мость этой схемы в основном определяется тем, обладает ли
нужными свойствами гидрид данного элемента. Поэтому такой
метод очистки следует назвать гибридным методом получения
। юментов особой чистоты.
Для осуществления этого метода необходимо, чтобы интере-
сующий гидрид можно было переводить в жидкое или газооб-
р иное состояние без разложения В противном случае его очи-
< ini становится затруднительной. Ввиду этого твердые солеоб-
рашые гидриды для указанной цели не пригодны, так как они,
। । исключением LiH, разлагаются при температурах ниже тем-
пературы их плавления В то же время гидрид должен иметь не
>чень высокую температуру разложения*, чтобы равновесие
реакции ЭоНь^аЭ-р/гЬНг (где Э — гидридообразующий эле-
мент) было смещено вправо вследствие низкой упругости пара
। н мента при температуре разложения Указанными свойства-
ми обладают простейшие неорганические (летучие) гидриды
• Под температурой разложения условно подразумевается такая темпе-
при которой термическое разложение соединения протекает с замет-
ной скоростью.
13
элементов В, С, Si, Ge, Sn, Р, As, Sb, S, Se, Те. Для гидридно-
го метода характерна высокая селективность, что обусловлива-
ет большую глубину очистки уже в стадии синтеза гидрида.
Но, с другой стороны, гидриды — термически неустойчивые со-
единения, легко распадающиеся на элемент и водород; в осо-
бенности это характерно для гидридов В, Sn, Sb, Те. Тепловой
эффект реакций распада может быть достаточно большим Вы-
деляющаяся теплота повышает температуру системы, что ве-
дет к увеличению скорости реакции термораспада гидрида. Рас-
пад всех гидридов, за исключением сероводорода, селеноводо-
рода и теллуроводорода, протекает с увеличением количества
молей газообразных веществ. Отсюда следует, что термораспад
гидридов может иметь взрывной характер. Необходимо отме-
тить также высокую токсичность гидридов. Все это создает оп-
ределенные трудности в использовании гидридного метода по-
лучения элементов особой чистоты, вследствие чего в промыш-
ленном масштабе он находит ограниченное применение. Тем не
менее исследования по применению гидридного метода ввиду
его больших потенциальных возможностей в отношении чисто-
ты полученного элемента проводятся и в настоящее время.
В литературе имеются сведения о том, что гидридная техноло-
гия является весьма перспективной при получении пленочных
материалов для микроэлектроники. Поэтому интерес к гидрид-
ному методу, к разработке его новых вариантов, несмотря на
отмеченные трудности, очевидно, будет проявляться и в даль-
нейшем
В последнее время все большее значение приобретает метод
получения высокочистых металлов, диэлектрических и полупро-
водниковых материалов через металлоорганические соединения
(МОС). Этот метод по своей структуре адекватен гидридному,
поскольку он осуществляется по аналогичной схеме: синтез
МОС — очистка МОС — термораспад МОС.
Как и в гидридном методе, селективность синтеза МОС поз-
воляет уже при протекании первой стадии процесса достигнуть
весьма существенной очистки металла от сопутствующих при-
месей Синтезированные металлоорганические соединения, как
правило, содержат лишь примеси углеводородов и других МОС
того же металла, но не содержат заметных количеств примесей
других металлов Более полное удаление последних осуществ-
ляется в двух других стадиях. Термораспад МОС протекает при
достаточно низких температурах, что является преимуществом
рассматриваемого метода по сравнению с галогенидным мето-
дом, поскольку при этом снижается вероятность загрязнения
получаемого металла материалом аппаратуры Другим преиму-
ществом данного метода является принципиальная возмож-
ность его более широкого применения, чем галогенидного и гид-
ридного методов, так как летучие МОС известны для большин-
14
ства элементов — металлов периодической системы. Но, к сожа-
лению, выделяющийся при термораспаде ЛЮС металл всегда
|.1Г )язнен углеродом, хотя эффект такого загрязнения можно
та штельмо уменьшить правильным выбором типов МОС, а так-
же использованием специальной обработки получаемого метал-
ла С этой точки зрения применение МОС наиболее целесооб-
разно для получения таких металлов, из которых углерод мо-
жет быть «вымыт» водородом, т. е. для металлов, не образую-
щих соединений с углеродом или карбиды которых могут быть
легко восстановлены водородом. Следует отметить, что реакция
< овместиого термораспада специально подобранных МОС и гид-
рида интересующих элементов лежит в основе одного из совре-
менных методов получения сложных полупроводниковых мате-
риалов химическим осаждением из паровой фазы Имеются
Ы1кже сведения о том, что металлоорганические соединения яв-
ляются перспективным сырьем для плазмохимического синтеза
веществ особой чистоты, осуществляемого в безэлектродной
плазме при высокочастотном (ВЧ) или сверхвысокочастотном
(СВЧ) разрядах. Чистота продукта, получаемого в результате
i.iKoro синтеза, определяется чистотой исходных МОС.
В практике получения высокочистых металлов в виде порош-
ков, пленок и покрытий применяют и так называемый карбо-
нильный метод, в котором в качестве связывающего реагента
используется оксид углерода (II). Образующееся соединение —
карбонил подвергается термораспаду. Метод по существу явля-
сгся двухстадийным, так как специальной очистке синтезируе-
мый карбонил обычно не подвергают ввиду его достаточно вы-
сокой чистоты, обусловленной специфичностью протекающей
химической реакции. Однако получаемый карбонил может быть
агрязнен образующимися при протекании реакции примесями
карбонилов других металлов, близких по свойствам к очищае-
мому и содержащихся в нем. В результате получаемое вещество
будет загрязняться примесями этих металлов, хотя последующая
стадия термораспада карбонила сама по себе является селектив-
ной Термораспад карбонилов в целом протекает при более высо-
ких температурах, чем термораспад гидридов и МОС, и возмож
кость загрязнения материалом аппаратуры здесь больше. Тем
нс менее карбонильный метод с успехом используется при по-
лучении целого ряда чистых металлов, таких, как Fe, Со, NI,
Os Мп, Re, Сг, Mo, W, там, где другие методы оказываются
или неприемлемыми или осуществление которых связано с боль-
шими трудностями.
Из других химических методов глубокой очистки веществ
особого внимания заслуживает метод химических транспортных
реакций Этот метод более подробно будет рассмотрен ниже
Нужно отметить, что химические методы очистки веществ
обычно используются в сочетании с физико-химическими мето-
15
дами (фильтрование, отмывка осадков, выпаривание, дистил-
ляция, экстракция и т. д.) Значительный интерес представля-
ет оценка эффекта разделения, который может быть достигнут
в химических методах глубокой очистки веществ.
§ 2. ОЦЕНКА ПРЕДЕЛЬНЫХ ВОЗМОЖНОСТЕЙ
Пусть протекает обратимая реакция
Ki
ViA-j-v2B у*~ v3C (1.1а)
и параллельная ей реакция
х2
vJA'+vjB Т*- v3C' 1.1 б)
в которых А — основное вещество; А'—примесь, В — химиче-
ский реагент; С, С' — продукты реакции; vi, V2, vs, v/, v/, v3'—
стехиометрические коэффициенты; Ki, Кг — константы равно-
весия.
Как видно из выражений (1.1а) и (1.16), очищаемое вещест-
во А будет связываться с веществом С, а образующееся соеди-
нение С' будет заключать в себе исходную примесь А' Поэто-
му в дальнейшем под содержанием примеси в продуктах подра-
зумевается молярная доля вещества С' в продуктах реакций
(1.1а) и (1.16).
Дробь, в числителе которой находится относительная кон-
центрация примеси в продуктах реакции, а в знаменателе — 6т-
носительиая концентрация примеси в равновесном с продукта-
ми реакции исходном веществе, называется коэффициентом раз-
деления:
х* х
а = _4--: 1-----
1—1* 1—X
где х* — молярная (атомная) доля примеси в продуктах реак-
ции, х— молярная (атомная) доля примеси в равновесном с
продуктами реакции исходном веществе. Коэффициент разделе-
ния характеризует эффективность разделения с помощью дан-
ной химической реакции. Чем больше а отличается от едини-
цы, тем выше эффективность разделения. Если а=1, то разде-
ление отсутствует. Когда концентрация примеси в продуктах ре-
акции ниже, чем в исходном веществе (примесь концентрирует-
ся в остающемся веществе), то за величину а принимают обрат-
ное ей значение, для того чтобы коэффициент разделения всег-
да выражался числом больше единицы. Тахим образом, коэф-
фициент разделения для реакций (1.1а) и (1.16) равен
сС'сА
(1.2)
а =--------,
сА'сС
(1.3)
16
где сл, Се, сХ', Сс — равновесные концентрации веществ А, С,
Л', С', моль/л. Если реакции (I 1а) и (I 16) являются гетероген-
ными, то в выражении (1.3) вместо концентраций веществ сле-
дует подставлять их парциальные давления.
Го определению константы равновесия, для реакций (1.1а)
и (1 16) имеем
Из соотношений (1.3) и (1.4) следует, что
сс с
а = Дк_,_____
а А« V2
(1.5)
Из уравнения (1.5) видно, что коэффициент разделения прямо
пропорционален отношению констант равновесия реакций с уча-
стием примеси и с участием основного вещества
Для оценки порядка величины коэффициента разделения
рассмотрим случай когда стехиометрические коэффициенты в
уравнениях реакций (1.1а) и (I 16) равны единице. Уравнение
(1.5) при этом будет иметь вид
а=-ф-. (1-6)
Л1
IКонстанта равновесия химической реакции может быть рас-
считана с помощью известного уравнения изотермы химической
реакции
AG"
1пК=----(1.7)
111
в котором А6°т — изменение энергии Гиббса системы вследст-
вие протекания в ней химической реакции при температуре Т.
Изменение энергии Гиббса связано с изменениями энталь-
пии ЬН°т и энтропии Д5°т реакционной системы следующим
соотношением
AG«. = Ы1°т — Т bs\ . (1.8)
Из соотношений (1.7) и (18) для реакций (I 1а) и (I 16) имеем
Л'2 Д//Т(2)~Д//Г(1) । Д,?Т(2)—Д,?Т(1)
1П^=-----------пт------+---------Я------- (19>
(индексы «1» и «2» относятся к реакциям, характеризуемым
константами равновесия К\ и Ki соответственно).
В первом приближении разницей в изменениях теплоемко-
сти системы с ростом фТцер&турЫ. пфоцеН]У а также измене-
ниями энтальпии и энтрюпиц^а,£«.Т /озмбж тых фазовых пере
I ДЕГЖ. "'М-ТЕХ J7
I УНИВЕРСИТЕТУ
ходов реагентов и продуктов реакций при температуре выше
стандартной (298 К) можно пренебречь С учетом этого и вы-
ражения (1.6) уравнение (I 9) преобразуется к виду
Д//г<)8(2)—। Л^гвмг)—т «гл
In а ~ -------i-------, I 10)
Г
где ДЯ°2ва и AS°29b — соответствующие изменения энтальпии и
энтропии системы, отнесенные к стандартным условиям; эти ве-
личины для многих простых и сложных веществ приведены в
справочниках.
В качестве конкретного примера оценим возможность очист-
ки теллура от примеси селена гидридным методом:
Ki
Те+Н2 7^ Н2Те (1.11а)
к2
Se+H2 II2Sc I 116)
Используя значения стандартных величин термодинамических
функций для реакций (111а) и (1116)*: ДЯ°29в(н те) =
— 99,7 кДж/моль (23,83 ккал/моль); Д5°298(н2те) =228,8 Дж/
/(моль-К) [54,69 кал/(моль-К)]; А//°298(н2зе) =33,47 кДж моль
(8 ккал/моль); Д5°29в(н2зе) =218,8 Дж/(моль-К) [52,3 кал/
/(моль-К)], из соотношения (I 10) имеем:
прп 7 = 298 К а~10“;
при Т=400 К а ~ 10я;
при Г =1000 К а ~ 103.
Как следует из приведенных результатов расчетов, гидридный
метод очистки теллура от селена должен быть весьма эффек-
тивным
Еще более эффективен гидридный метод очистки олова от
углерода.
Sn-)-2H2 SnII4 (1.12а)
кг
С4-2Щ (X сн« (1.126)
Подставляя в уравнение (1 10) значения А//°298(зпн4) =
= 162,8 кДж/моль (38,9 ккал/моль); ASSgecsnH^ =228,7 Дж/
/(моль К) [54,65 кал/(моль-К)]; ДД°298(сн4> =—74,81 кДж/моль
• Термические константы веществ. Справдчинк/Под ред В П. Глушко.
М 1966, вып 2, 1970, вып 4
18
(- 17,88 ккал/моль); Д50298(сн4 = 186 3 Дж/(моль-К) [44,53кал/
/(моль К)], получаем
при 7 = 298 К а~10,в;
при 7=400 К а~10*®;
при 7=1000 К а~1010.
Из рассмотренных примеров видно, что вклад энтропийного
члена в выражении (1.10), а следовательно, и в выражении
(19) невелик по сравнению с энтальпийным членом. Это поз-
воляет производить расчет коэффициента разделения на осно-
ва ни I лишь сведений о тепловых эффектах протекающих хими-
ческих реакций вида (1 1а) и (1.16), т е если пренебречь вто-
рым членом в правой части уравнения (1 10), будем иметь
1па~—ДЯ,'Я7, (1.13)
I де \Н— разница в тепловых эффектах реакций (116) и
(1.1а). Разница в тепловых эффектах химически?: реакций ред-
ко превышает 400 кДж/моль. Принимая, что ДЯ=—400 кДж/
/моль и подставляя эту величину в соотношение (1.13), полу-
чаем:
а ~ 10*1 при 7 = 1000 К;
а ~ 1071 ирп 7 = 298 К.
И ( приведенной предельной оценки значения а следует, что с
помощью химических реакций содержание примеси в очищае-
мом веществе в принципе можно снизить до ничтожно малой
величины. Разумеется, в каждом конкретном случае нужно под-
бивать соответствующие температурные условия, при которых
скорость интересующих реакций была бы достаточно высока
для достижения в системе равновесия.
Когда очистка вещества от примеси обусловлена различием
и константах скорости химических реакций (1.1а) и (1.16), то
и случае равенства стехиометрических коэффициентов единице
можно записать
X lna = ln^ = - ^+ln-|, (1.14)
। де k — константа скорости прямой реакции; k° —предэкспо-
пеициальный множитель в уравнении для скорости реакции
/—энергия активации реакции при заданной температуре Т;
индексы «1» и «2» относятся к реакциям (1.1а) и (I 16) соот-
ветственно.
К сожалению, необходимых для оценки коэффициента раз-
(еления данных о константах скорости химических реакций в
литературе очень мало. Отсутствуют также и способы их вычис-
ления из молекулярных данных. В общем случае, как следует
19
Рис 2. Схема установки для проведе-
ния транспортной реакции при нали-
чии градиента температур (Т2>Т\):
Tt—температура в зоне прямой реакции;
Т3 — температура в зоне обратной реакции
подобранным реагентом, может
из теории абсолютных скоро-
стей химических реакций, отно-
шение fe2°/^i° может изменять-
ся от 1 до 108. Энергия акти-
вации изменяется примерно в
тех же пределах, что и тепло-
вой эффект реакций. Следова-
тельно, достигаемое химиче-
скими методами разделение,
основанное на различии в кон-
стантах скорости реакций очи-
щаемого вещества и содержа-
щихся в нем примесей с
'ыть так же эффективно, как и
оспованное на различии в константах равновесия этих реакций.
§ 3. МЕТОД ХИМИЧЕСКИХ ТРАНСПОРТНЫХ РЕАКЦИИ
В ряде случаев для глубокой очистки веществ, как уже от-
мечалось, с успехом используются химические транспортные ре-
акции (реакции переноса). Химическими транспортными реак-
циями называют гетерогенные обратимые реакции с участием
газовой фазы, с помощью которых можно осуществить перенос
вещества из одной части системы в другую, если между этими
частями имеет место разность температур или давлений. Обыч-
но для осуществления транспортных реакций используют Си-
стемы с разностью температур. В качестве примера рассмотрим
перепое никеля в виде тетра карбонила никеля (рис. 2) В один
конец стеклянной трубки помещается никель, который необхо-
димо подвергнуть очистке. Из трубки откачивается воздух, пос-
ле чего она заполняется оксидом углерода (II). В холодном
конце трубки (7’1 = 3184-323 К) протекает реакция образования
тетракарбонила никеля по схеме
Ni-HCO —Ni(CO)4 (1.15)
Газообразный тетракарбонил никеля диффундирует в горячий
конец трубки, где под влиянием высокой температуры (Т2 =
=4534-473 К) разлагается по уравнению
Т2
Ni(CO)4 —► Ni-f-4CO 1.16)
Выделяющийся никель оседает на стенках трубки, а освобож-
дающийся оксид углерода (II) диффундирует к холодному кон-
цу трубки. Уравнения реакций (I 15) и (1.16) можно записать
в виде уравнения одной реакции
Т1
Ni + 4CO Ni (СОЦ (1.17)
1'2
20
При температуре Т2 равновесие реакции (I 17) заметно смеще-
но плево по сравнению с тем, что имеет место при температуре
/,. Вследствие этого постепенно весь никель из холодного кон-
ца трубки может быть перенесен в ее горячий конец. Поэтому
тлкие реакции и называются реакциями переноса. Переносимое
вещество при условиях опыта должно быть твердым или жид-
ким и обладать низким давлением пара, а химический реа-
leiiT — в данном случае оксид углерода (II)—и продукт его
взаимодействия с переносимым веществом — тетракарбонил
никеля—должны быть газообразными. Уравнение транспорт-
ной реакции для вещества А можно записать в виде
VjA (тв, >k)4-v2B (г) v3C (г) (1.18а)
и для примесного вещества А' в виде
v'jA' (тв, zK)-f-vjB (г) ч» v3C' (г) (1.186)
где vi, V2, V3, V|Z, V2Z, V3Z — стехиометрические коэффициенты;
Н(г)—реагент; С, Cz — продукты реакций (1.18а) и (1.186) со-
ответственно. Обычно формула транспортируемого вещества за
нисывается в левой части уравнения, а через Т2 обозначают
температуру более горячей части системы (горячей зоны).
Рис. 3. Схема установки для проведения транспорт*
иой реакции при наличии градиента давления
(Р|>Рг):
Pi — давление газа в зоне прямой реакции, — давление
газа в зоне обратной реакции
Равновесие реакции (1.18а) может смещаться не только при
изменении температуры, но и при изменении давления. Если
V3<V2, то при уменьшении давления равновесие реакции
(I 18а) смещается влево. Это приводит к выпадению переноси-
мого вещества в правой части установки (рис. 3). Однако боль-
шой перепад давления, необходимый для заметного смещения
равновесия реакции (I 18а), осуществить трудно. Значительно
проще осуществить это смещение путем создания разности тем-
ператур между зонами. Поэтому двухтемпературный вариант
метода транспортных реакций является более распространен-
ным Направление переноса определяется тепловым эффектом
реакции. Если реакция вещества с заданным реагентом является
экзотермической, то в соответствии с принципом Ле Шателье
21
будет иметь место перенос этого вещества из холодной зоны в
горячую. Если же реакция является эндотермической, то ис-
ходное вещество необходимо помещать в зону с более высокой
температурой; перенос вещества при этом будет происходить из
горячей зоны в холодную.
Возможность переноса вещества с помощью химических ре-
акций была отмечена еще во второй половине предыдущего
столетия на примере реакций
Fe2O3 (тв) + 6НС1 (г) 2FeCl3 (г)4-ЗН2О (г)
Si (тв) -J-SiCl4 (г) 2SiCla (г)
2А1 (тв) + А1С13 (г) ЗА1С1 (г)
Однако практическое использование явлений такого массопере-
носа, получившего название химического транспорта, началось
лишь с ЗО-х годов текущего столетия в связи с необходимо-
стью получения чистых металлов, особенно тугоплавких. Для
этой цели были использованы реакции с участием иодидов ме-
таллов В качестве реагента использовался газообразный иод,
например:
Zr (тв)+212 (г) Zrl4 (г)
Такой метод очистки металлов от примесей получил название
иодидного метода. Он, в частности, применяется в промышлен-
ности для глубокой очистки циркония от примеси гафния. Бы-
ли получены хорошие результаты при очистке этим методом ти-
тана, ванадия, ниобия, вольфрама, тория.
В настоящее время явление химического транспорта успеш-
но используется в целях глубокой очистки ряда веществ, как
простых, так и сложных, а также для получения эпитаксиаль-
ных полупроводниковых пленок и монокристаллов. Реагентами,
с помощью которых осуществляется перевод очищаемого веще-
ства в транспортируемое соединение, помимо указанных выше
оксида углерода (II) и иода служат хлор, бром, галогеноводо-
роды, галогениды Интересно отметить, что при использовании
последних процесс переноса обычно протекает через стадию об-
разования соответствующего субгалогенида, т е соединения с
низшей валентностью В результате перенос вещества в целом
осуществляется за счет реакции диспропорционирования, как
это, например, имеет место в случае очистки элементов III—
IV групп периодической системы:
2Э (тв, ж)-|-ЭНа13 (г) ЗЭНа! (г) (1.19)
где Э = В, Al, Ga, In, Hal = F, Cl, I и соответственно
Э (тв, ж) + ЭНа14 (г) 2ЭНа12 (г) (1.20)
где 3=Si, Ge, Ti, Pb.
Рассмотрим вопрос о скорости переноса очищаемого веще-
ства с помощью химических транспортных реакций. Пусть
ранспортная реакция выражается уравнением (I 18а). Пере-
нос вещества из одной зоны в другую может быть осуществлен
тремя путями: а) потоком газа-реагента с продуктами реакции;
б) молекулярной диффузией газа-реагента и продуктов реак-
ции в) конвективной диффузией газа-реагента н продуктов
реакции.
Перенос вещества потоком газа-реагента
Схема установки приведена на рис. 4. Для осуществления
этого варианта необходимо, чтобы реагент В уже при комнатной
температуре был в газообразном или конденсированном состоя-
нии с высоким давлением пара В последнем случае через си-
стему можно продувать инертный газ, насыщая его предвари-
Зона прямой реакции (I)
Рис. 4 Схема установки для химического транс
порта вещества из зоны прямой реакции (/) в зо-
ну обратной реакции (II) потоком газа оеагеита
тельно парами вещества В. При глубокой очистке этот вариант
обладает тем недостатком, что требует высокой чистоты ре-
агента В, так как при продувании через систему больших ко-
личеств реагента даже с ничтожным содержанием примесей бу-
дет происходить загрязнение очищаемого продукта Для час-
тичного устранения этого недостатка выделяющийся в зоне об-
ратной реакции реагент В вновь перекачивают в первую зону.
Таким образом, общее количество введенного в систему реаген-
та может быть уменьшено.
Количество вещества А, прореагировавшее с веществом В в
первой зоне, в пересчете на моль вводимого в систему вещест-
ва В составляет
rl
Vi ьс
Уз £в ’
где Lb — скорость поступления вещества В в систему (скорость
потока газа), моль/с; L'c— скорость образования вещества С
в первой зоне, моль/с
22
23
Количество вещества А, выводимого из системы потоком га-
за, в пересчете на моль вводимого в систему вещества В равно
/II
v3 Рв ’
где Lc11—скорость вывода вещества С из второй зоны, моль/с.
Следовательно, скорость та переноса вещества А из первой зо-
ны во вторую определяется уравнением
rl г п
т / Vi ьс Vi ьс
—— (г Г j II
v3 (Lc hc >
(1.21)
(1.22)
(1.23)
Pc
(124)
zB *з
Если в первую зону ввести Мв(исх> молей вещества В, то в
результате протекания реакции (I 18а) общее количество мо-
лей газообразных веществ в зоне будет равно
7Уобщ= 7Vb(Hcx)T TVc (1 ^7 j >
гте /Vc — количество молей вещества С.
Из уравнения Менделеева — Клапейрона следует, что
ТУс _ Рс
ТУобщ ^общ
где Рс — парциальное давление пара вещества С; РОбщ — общее
давление в рассматриваемой зоне.
Подставляя соотношение (1.22) в (1.23) и проводя преобра-
зования, получаем
ТУс ____________________.
ТУв(исх) ^общ—РсЦ-^3)1
Из вполне очевидных соображений следует, что
Рс 1Ус
Рв N В(исх)
Из (1.25) с учетом (1.24) имеем
Lc=Pb г-
^общ [_1
(125)
(1.26)
Vs
Vs
Рс
г общ \
При Рс^/’общ. что обычно имеет место на практике, или при
v2=V3 выражение (126) упрощается.
^общ
Так как в системе поддерживается постоянное давление за
счет постоянства скорости вводимого газа-реагента, то очевид-
но, что
(1.27)
Т’общ— Т’н(исх). (1.28)
где Рв(исх) — давление газа-реагента, вводимого в систему.
Подставляя соотношение вида (1.27) для £с[ и Ас1 в уравне-
ние (1.21) и используя выражение (1.28), получаем
nl nil
V» * с
Из уравнения (1.29) видно, что скорость переноса вещества
прямо пропорциональна относительной разности парциальных
давлений пара продукта реакции в первой и второй зонах. Бо-
лее строгое выражение для скорости переноса может быть по-
лучено подстановкой уравнения (I26) в уравнение (1.21)
При переносе вещества из одной зоны в другую оно будет
освобождаться от примесей Одни примеси при этом менее охот
но взаимодейс вуют с реагентом В с образованием летучих про-
дуктов и концентрируются в остатке, другие примеси с реаген-
том В дают более устойчивые продукты, чем основное вещест-
во, и выносятся потоком газа-реагента из прибора. Оценим ве-
личину разделительного эффекта.
Пусть одновременно с реакцией (1.18а) протекает парал-
лельная ей реакция (1.186) Скорость переноса вещества А' из
первой зоны во вторую будет выражаться, по аналогии с урав
пением (1.29), соотношением
, pl pH
У, *С ~^С'
(130>
в котором Р'с , Рс —давление пара вещества С' в первой и
второй зонах Молярная доля х* примеси в продукте, выпадаю-
щем в данный момент во второй зоне, может быть вычислена
из соотношения
Если в системе достигается равновесие, то Рс и Рс могут
быть выражены через константы равновесия реакции (I 18а) и
(1.186). Как и выше, ограничимся рассмотрением характерного
для практики случая, когда Рс<^.РОбщ и Рс <^Po6ui. В соответ
ствии с выражением (1 28) величину РВ(исх> в системе при этом
можно считать постоянной. С учетом последнего, полагая, что
конденсированная фаза гетерогенна и не обладает сколько-
нибудь заметной упругостью пара, имеем
Кр^=Р^, К'р=Р$.
Подстановка значений Рс и Рс из этих равенств в выражение
(1.31) дает
у(у3 (Л#)1'^-^11)17'»
V3V1 (z:J,)1/v3_(^n)i/vs • (1-32)
24
25
Из уравнения (1.32) следует, что концентрация примеси в про-
дукте не зависит от концентрации ее в исходном веществе
Для осуществления переноса с заметной скоростью подбира-
ют такие условия, чтобы Кр'^Кр'1; тогда уравнение (1.32)
упрощается ,
v',v3 ^р1)1^
(133>
Если конденсированная фаза является жидкостью и облада-
ет высокой скоростью выравнивания концентрации, то пар-
циальные давления паров примеси и очищаемого вещества бу-
дут определяться соотношениями:
РА-=Р°и-ху2, Рл=Р\а—х) Vi,
где к — молярная доля примеси; -у2, fi— коэффициенты актив-
ности примеси и основного вещества соответственно. Если, как
и выше, принять, ЧТО Рс'^Робщ, Рс<СРобщ и Рв(исх) = const, то
выражения для констант равновесия будут иметь следующий
вид:
Р$
₽с3
КР=-------------— . (1.35)
(J’a (1 —я) Yi] 1
Подстановка соотношений (1.34), (1 35) в (132) с учетом то-
то, что 1, дает
У',УЯ (Kp)l/V* (PVY2)i':/V3-(^pr)1M
X*~ ЪП (Kj>)1/v4^Yi)V1/V3-(^p)1/V3(^Yi)^/Va ’ (I36>
Из уравнения (1.36) следует, что в данном случае концентра
ция примеси в продукте зависит от ее содержания в исходном
веществе. Когда Кр* »КР“, то уравнение (1.36) принимает бо-
лее простой вид:
, _ v;vs (^pr)1/V3(PW?/V3
VSV1 (kJ ,/V3 (P^Yi)p/V3 ' ‘
Для большей наглядности оценки эффекта очистки примем, что
стехиометрические коэффициенты в уравнениях протекающих
химических реакций (I18а) и (I 186) равны единице и что
конденсированная фаза представляет собой идеальный раствор
Тогда из (I 37) имеем
X* Р°А' к'р
а^~=~Р\1ё~- (L38)
26
Перенос вещества молекулярной диффузией
В этом случае транспортная реакция осуществляется в зам
кнутом сосуде (см. рис. 2). Перенос вещества из одной зоны в
другую происходит за счет диффузии. Преимущество этого ме-
тода состоит в отсутствии необходимости введения в систему
больших количеств реагента В и включения в схему насоса для
перекачки реагента из одной зоны в другую.
Скорость переноса вещества С из зоны прямой реакции в
лону обратной реакции определяется скоростью его диффузии,
которая в соответствии с законом Фика характеризуется соот-
ношением
(1.39)
где тс — скорость переноса вещества С, моль/с; De — коэффи-
циент взаимной диффузии для смеси (С+В), см2/с; S — пло-
щадь поперечного сечения трубки, см2; z—координата вдоль
длины трубки
Без большой погрешности можно принять, что
, . ,г „ы
асе ~ АсС _ Сс~ Сс
dz Az zK
(1-40)
где zK — длина диффузионного участка (длина трубки, соеди
пяющей первую и вторую зоны).
Заменив концентрации на давления в соотношении (1.40)
и подставив их в (I 39), получим
Tc=v&(pc“^cr). (!•«)
где Т — средняя температура диффузионного участка. В соот-
ветствии с выражением (141), скорость переноса вещества А
будет определяться соотношением
т =_11_ ..foS, ,pi_pii)__lL рс^сОбщ j п
А v3 рс> Vs zkPo6u< (Рс рс)> i-42)
где Собщ — суммарная концентрация газа, моль/см3.
f Составив уравнение вида (1.42) для скорости переноса при-
меси А' и используя соотношение тА /та=х*/(1—х*)~х*, бу-
дем иметь
v',v2 D& рс' Р(/
vsvl Dc Я—рИ
(143)
Это уравнение отличается от соответствующего выражения для
«метода потока» [уравнение (131)] только сомножителем
Dc ' Dc, который близок к единице, так как диффузия не чисто
молекулярная, а в значительной степени является конвектив
27
ной Рс и Рс в уравнении (I 43) могут быть заменены на Кр и
Кр' так же, как это было проделано для «метода потока» При
этом получаем выражение
рс,у,у3
ГХ*~ DC^i (Xj,)Vve_ ' (L44>
Если реакция экзотермична, то Кри<Кр1 и, как уже отмеча-
лось, транспорт вещества будет направлен из холодной зоны в
горячую. Могут быть и такие случаи, когда транспорт одного
вещества направлен в одну сторону, а другого — в противопо-
ложную. Например, хром и теллурид хрома (II) в атмосфере
иода переносятся в противоположных направлениях (рис. 5)
вследствие протекания реакций
Сг4-12(г) з=е Сг12(г)
СгТе+12(г) =6* СП2(г) + 1/2Тс2(г)
СгТе
Зона обратной
реакции
Сг ♦ Сг Ге
Зона прямой
реакции
Рис. 5. Схема установки для одиовремеииого прове-
дения двух транспортных реакций (Тз>72>71)
первая из которых является экзотермической, а вторая — эндо-
термической.
Перенос вещества посредством конвекции
Перенос вещества из одной зоны в другую при осуществле-
нии химических транспортных реакций может быть увеличен за
счет конвекции. Если ампулу с очищаемым веществом и газом-
реагентом (например, ампулу, схематично изображенную на
рис. 2) расположить наклонно так, чтобы горячий конец ампу-
лы был обращен к низу, то в ампуле возникнут конвекционные
потоки газа-реагента и продуктов реакции Эффект конвекции
может значительно повысить скорость процесса массопереноса.
Для того чтобы оценить выход транспортной реакции за счет
перемещения газообразных веществ путем конвекции, рассмот-
рим устройство, схематично изображенное на рис. 6 Установка
представляет собой кольцевую трубку длиной z и радиусом г,
которая находится при температуре Ть за исключением отрез-
ка длиной Zr2; последний нагрет до температуры Т? (T2>Ti).
28
Пусть трубка заполнена газом Разница в давлениях, а следо-
вательно, и в плотностях газа находящегося в различных тем-
пературных зонах, и вызывает конвекционное движение газа.
Объемная скорость Loc прохождения газа через трубку дли-
ной z и радиусом г вследствие разницы в давлениях на концах
трубки определяется известным соотношением Пуазейля:
лг’ДР .
£'°б---8тр~~ ’ (145)
в котором т) — вязкость газа, Па-с; АР = 2 98,1 Па;
ini — масса газа в «горячей» части трубки при Т=7’2; "Ъ — мас-
са газа в таком же объеме при Т=1\.
Используя уравнение Менделеева — Клапейрона, можно за-
писать
zT лг2РМ г ! 1 х
----Т(— ’ (L46)
где Zr2—длина «горячей» части трубки; Р— среднее значение
общего давления; М — относительная молекулярная масса.
Рис. 6 Схема установки для химического
транспорта вещества из зоны прямой реак-
ции в зону обратной реакции посредством
конвекции:
Pi, Pt — давление газа при температурах 7\, Т7,
соответственно; z^—участок трубки, находящей-
ся при температуре
Комбинируя уравнения (1.45) и (1.46), получаем.
nr*zT PAf98,l
—
1
71 Тг
НИ И
1
Тг
38,5r«z? PAf ( t
£°б ( 7,
(/ б и 1) относятся к температуре 7,).
Обозначив, как и раньше, скорость поступления газа-реаген-
i.i в горячую зону через LB, имеем
lb - Рв^об.
(1.47)
(1.48)
где pD= ----------плотность газа, моль/см3.
29
Подставим соотношение (148) в вырлжнин (I • ')
г 38,5zT г« 1/ и /I I \ I
LB = ^ВСисх) [ r]zHTl (_г; У? ) I (’/,5,)
Из (I 49) с учетом (I 29^ следует
Сравнивая выражения (150) и (142) и учи шипи t <><>i ношение
(1.28), можно видеть, что если в метоле диффузии скорость
процесса переноса обратно пропорциоп алыц niii.iciiiuo газа-
реагента. то в методе конвекции опа прямо пропорциональна
/-'в(исх). Поэтому диффузионный метод це ie< оо<>р,| то осуществ-
лять при низких давлениях газовой ф»зы, л мен i иопш кипи —
при повышенных. Разделительный эффект и мгюк конвекции
в общем такой же, как и в методе потока, а игройiвис п> за-
грязнения очищаемого вещества примесями, < <* м-pm4uiiimiich в
газе-реагенте, меньше.
Методу химических транспортных реакций iipih-yin.ii те же
недостатки, что и другим химическим мепыпм он пре icт.чнля-
ет собой одноступенчатый процесс разделения и цо»н>му еффек-
тнвен только при отделении примесей, сущие пн ппо оглпчаю-
щихся по свойствам от основного вещее гни Важным преиму-
ществом этого метода является то, что при «ни <»< уннч iii.iciiiih
процесс очистки обычно проводится n .uiMKiiyшм пЛы’ме, что
резко снижает возможность загрязнении очипцшмо!и вещества
окружающей средой.
§4 СРАВНИТЕЛЬНАЯ ХАРАК 11 1ЧК IIIН А
ХИМИЧЕСКИХ И ФИЗИКО ХИМИЧ1 ( KIIX
МЕТОДОВ ОЧИСТКИ ВНИК III
Как было уже показано, химические mcioili очпгпеи ве-
ществ обладают большими возможное i я мп В ш*ко|орых слу-
чаях хороших результатов можно ожидать тяже при очистке
простых веществ от сопутствующих нм примесных «дементов-
аналогов [уравнения (I lla), (I I I6)J В целом же очисти ве-
ществ от близких к нему по свойствам примел ей химическими
методами обычно малоэффективна. Действн ic п.но, шли обра-
титься к периодической системе элементов, то можно иметнть,
что сходные по свойствам элементы имеют и близкую по вели-
чине электроотрицательность. В таблице элекгроотрип.цельно-
сти некоторых из них, например Si и Ge, iinniMiiioi одно место.
Это означает, что различие в энергиях их взаимодействия с ка-
ким-либо третьим элементом не должно бы и. большим, особен-
но при высокой температуре. При более пилкой температуре
30
это различие, возможно, окажется более существенным, но здесь
возникает вопрос о скорости протекания указанных химических
реакций при заданной температуре.
Рассмотрим такой пример. Если пропускать смесь азота и
кислорода через нагреваемую до некоторой определенной тем-
пературы трубку заполненную свежевосстановлениой медью
то в результате протекания химической реакции
2Си Og —* 2CuO
кислород будет связываться медью и задерживаться в колонне.
Таким образом можно достичь глубокой степени очистки азота
от кислорода Если ту же смесь азота с кислородом пропускать
через трубку, заполненную адсорбентом, например активиро-
ванным углем, то будет также наблюдаться разделение газовой
смеси вследствие различной сорбируемости азота и кислорода.
Но в этом случае разделение оказывается менее эффективным,
чем при применении трубки с медью В первом случае разделе-
ние основано на различии в химических силах, действующих
между атомами меди и кислорода, меди и азота. Во втором
случае — на различии в силах межмолекулярного взаимодейст-
вия между углем и кислородом, углем и азотом. Силы межмо-
лекулярного взаимодействия значительно слабее химических
сил. Различие в энергии взаимодействия молекул азота и кис-
лорода с адсорбентом намного меньше, чем в энергии химиче-
ского взаимодействия азота и кислорода с медью Поэтому эф-
фект разделения смеси азота с кислородом в адсорбционном
процессе, который относится к физико-химическим, будет су-
щественно ниже, чем в реакции их химического взаимодейст-
вия с медью Однако несмотря на то что разделительный эф-
фект, имеющий место в физико-химических процессах, обычно
невелик, последние широко используются для глубокой очистки
веществ Это объясняется следующими причинами.
Не всегда удается подобрать химическую реакцию, с по-
мощью которой можно было бы эффективно очистить данное
вещество от всех лимитируемых примесей. Химические методы
не пригодны для очистки веществ от примесей с близкими к ос-
новному веществу свойствами; это обусловлено тем, что до на-
стоящего времени еще не разработаны достаточно удовлетво-
рительные схемы многоступенчатых процессов глубокой очист-
ки веществ с помощью химических реакций Кроме того, хими
ческие методы разделения смесей всегда сопровождаются за-
грязнением выделяемого вещества химическими реагентами.
Физико-химические методы разделения смесей не требуют
химических реагентов для своего осуществления При осуще-
ствлении же химических методов почти всегда возникает про-
блема отделения очищаемого вещества от продуктов реакции.
3!
Физико-химические процессы, в отличие от химических реак-
ций, сравнительно просто можно осуществлять в многоступен-
чатом варианте, в частности с использованием принципа проти-
вотока. Это позволяет многократно умножать разделительный
эффект и тем самым достигать требуемой глубины очистки
Из совокупности используемых в настоящее время физико-
химических методов глубокой очистки веществ ограничимся
рассмотрением теоретических основ дистилляционных и кри-
сталлизационных методов, которые являются наиболее рас-
пространенными в практике получения веществ высокой чисто-
ты. Кратко рассмотрим также основы термодиффузионного ме-
тода, обладающего большими потенциальными возможностями
для повышения степени чистоты веществ, в особенности при
освобождении от примесей в виде взвешенных частиц субмик-
ронного размера
ГЛАВА II
ДИСТИЛЛЯЦИОННЫЕ МЕТОДЫ
Из физико-химических методов разделения смесей веществ
широкое применение как в лабораторной практике, так и в про-
мышленном производстве находят дистилляционные методы.
Во избежание недоразумений следует заметить, что в лите-
ратуре термин «дистилляция» часто используется в более узком
смысле и отождествляется с термином «простая перегонка».
С другой стороны, продукт, получаемый конденсацией пара,
выходящего из верхней части перегонного разделительного ап-
парата независимо от его конструкции, принято называть дис-
тиллятом. Поэтому в дальнейшем мы будем пользоваться тер-
мином «дистилляционные методы» для общего обозначения всех
известных методов очистки веществ с использованием фазово-
го перехода жидкость — пар.
Дистилляционные методы, включающие в себя различные
варианты простой перегонки, ректификации и молекулярной
дистилляции, с успехом используются при получении веществ
особой чистоты. Проблема глубокой очистки веществ потребо-
вала усовершенствования этих методов. Так, здесь особенно на-
глядно проявляется настоятельная задача выбора конструкци-
онного материала для аппаратуры, который не загрязнял бы
очищаемое вещество. Поиск же оптимальных условий осуществ-
ления дистилляционных методов применительно к глубокой очи-
стке веществ непосредственно связан с разработкой вопросов
теории процесса дистилляции разбавленных растворов.
Несмотря на многообразие разработанных к настоящему вре-
мени способов дистилляции, все они основаны на использовании
различия в составах разделяемой жидкой смеси и образующего-
32
ся из нее пара. Это раз. ичи€ можно охарактеризовать величи-
ной относительной летучести отделяемого компонента, называе-
мой в этом случае коэффициентом разделения.
§ 1 КОЭФФИЦИЕНТ РАЗДЕЛЕНИЯ
Относительная летучесть интересующего компонента разде-
ляемой смеси, которая в принципе всегда является многокомпо-
нентной, зависит прежде всего от свойств компонентов смеси.
Учет этой зависимости составляет одну из основных задач тео-
рии и практики разделения смесей. Разумеется, и при глубокой
очистке веществ рабочим объектом также является многоком-
понентная смесь, состоящая из очищаемого вещества и приме-
сей. Однако здесь мы имеем специфическую особенность, кото-
рая заключается в том, что исходное очищаемое вещество со-
держит примеси уже в сравнитечьно небольших количествах.
Обычно для достижения этой цели применяется предваритель-
ная очистка вещества. Таким образом, при глубокой очистке
веществ приходится иметь дело с разбавленными растворами.
В таких растворах содержание каждого из растворенных ве-
ществ (примесей) незначительно по сравнению с содержанием
растворителя (основное вещество) и поэтому взаимным влия-
нием примесей в них можно пренебречь. Следовательно, в этом
случае разделяемую многокомпонентную смесь условно можно
рассматривать как бинарную, состоящую из основного компо-
нента и данной примеси. При этом обычно принимают также,
что в паровой фазе (при невысоких давлениях) ввиду ее боль-
шой разряженности отсутствует взаимодействие не только меж-
ду молекулами примесей, но и между молекулами примесей и
основного компонента, т. е. тем самым постулируется, что обра-
зующийся из жидкости пар представляет собой идеальный газ.
Но даже при указанных упрощающих допущениях установле-
ние зависимости коэффициента разделения от свойств компо-
нентов такой псевдоби парной смеси представляет непростую
задачу.
Понятием «коэффициент разделения» мы уже пользовались
при рассмотрении химических методов глубокой очистки ве-
ществ Применительно к бинарной системе жидкость — пар, обо-
значая через х молярную долю примесного компонента в жид-
кости, а через у — молярную долю этого же компонента в рав-
новесном с жидкостью паре, будем иметь
При этом принципиально неважно, какую из фаз считать в ка-
честве первой, обычно выражение вида (II.1) составляют та-
2 46
33
кнм образом, чтобы коэффициент разделения а был больше
единицы*.
Коэффициент разделения а является важнейшей характери-
стикой однократного, элементарного процесса разделения. Зна-
ние этой величины позволяет определить состав паровой фазы
при известном составе равновесной жидкой фазы (или наобо-
рот). В этом случае выражение (II 1) записывают в виде, раз-
решенном относительно у (или х),
(,Е2)
и называют уравнением кривой фазового равновесия системы
жидкость — пар. Это уравнение является основным при описа-
нии и расчетах дистилляционного процесса
Уравнение (II 1) можно преобразовать и к другому виду;
у—х=(а — l)as(l — у), (II.3)
из которого наглядно видно, что чем больше а отличается от
единицы, тем больше эффект разделения, выражаемый через
разницу (у—х) составов равновесных фаз Поэтому в теории
процессов разделения иногда пользуются величиной (а—1), на-
зываемой коэффициентом обогащения Если коэффициент обо-
гащения равен нулю, то, как следует из выражения (П.З), раз-
деление смеси не происходит, т. е. состав обеих равновесных
фаз будет одинаков. Это имеет место тогда, когда основное ве-
щество образует азеотроп с отделяемой примесью Как было
показано выше, в химических методах очистки коэффициент
разделения обычно намного больше единицы. Поэтому понятие
о коэффициенте обогащения используется лишь в теории физи-
ко-химических методов, в которых величина а часто мало отли-
чается от единицы.
Для предельно разбавленных растворов х-С 1 и t/<C 1, вслед-
ствие чего выражение (II I) упрощается и может быть записа-
но в виде
a=yz. (II 4)
При принятых обозначениях соотношение (II 4) характери-
зует процесс очистки, когда примесь концентрируется в паровой
фазе В том же случае, когда паровая фаза обедняется при-
месью, коэффициент разделения будет выражаться соотноше-
нием
а=х/у. (П-5)
Применение уравнения (II.4) или (II.5) в расчетах определяет-
ся схемой проведения процесса очистки, построение которой за-
висит от природы отделяемой примеси.
* Иногда встречаются и другие обозначения коэффициента разделения,
например- k в кристаллизационных методах очистки у в термодиффузии
34
Использование условий x<gl и и принятие допущения
о постоянстве коэффициента разделения для разбавленных рас-
творов существенно упрощают анализ работы разделительных
аппаратов. При этом многие зависимости, характеризующие
взаимосвязь параметров процесса разделения, принимают прос-
той и удобный для расчетных целей вид.
Знание коэффициента разделения необходимо для оценки
эффективности используемого метода, для вьбора рациональ-
ной схемы и режима процесса, для оценки размеров раздели-
тельной аппаратуры. Иными словами говоря, при разработке
того или иного дистилляционного способа очистки конкретного
вещества в первую очередь необходимо знать коэффициент раз-
деления.
§ 2. ТЕОРЕТИЧЕСКАЯ ОЦЕНКА
КОЭФФИЦИЕНТА РАЗДЕЛЕНИЯ
Как известно, парциальные давления пара каждого из ком-
понентов бинарной! смеси над раствором (в нашем случае ос-
новной компонент — примесь) при заданной температуре Т вы-
ражаются соотношениями
Р1 = РО(1—г) уь
Р2 = .Р&ГЬ,
(И 6)
где р — парциальное давление пара компонента смеси над рас-
твором; -у— коэффициент активности; Р° — давление насыщен-
ного пара индивидуального (чистого) компонента; индексы «1»
и «2» относятся к основному и примесному компонентам соот-
ветственно.
Для идеального газа, с каковым отождествляется равновес-
ный с рассматриваемым раствором пар,
Рг = Ру,
(П.7)
где Р — общее давление пара над раствором
Подставляя (II.7) в (II.6), с учетом (II.1) получаем выра-
жение для коэффициента разделения а через относительную ле-
тучесть примеси
а =
^?Yi
(П.8)
Для разбавленного раствора при х—>0 коэффициент активно-
сти fi—>-1, а коэффициент активности у2—»-const Если при этом
const = 1, то будет справедлив закон Рауля, в соответствии с ко-
торым из выражения (II.8) следует, что
ро
а—ф (П.9)
2
35
Соотношение (II.9) в большинстве случаев качественно пра-
вильно отражает характер перераспределения примеси в кон-
тактирующих паровой и жидкой фазах, т. е. верно указывает
фазу, в которой концентрируется примесь. Для количественной
же интерпретации оно неприменимо, так как по отношению к
реальным растворам условие ^2=1 обычно не выполняется По-
этому количественные расчеты величины а требуют учета откло-
нений поведения реальных растворов от закона Рауля, т. е тре-
буют знания коэффициента активности каждого из компонентов
в растворе, как это видно из выражения (П.8) Но поскольку
для разбавленного раствора очищаемое вещество — примесь
можно принять, что К| = 1, то определение а с помощью выраже-
ния (II.8) сводится, по существу, к определению -уг; значения
Pi° и Р20 или известны, или их измерение не представляет боль-
шой трудности Для оценки величины используют различные
методы. Широко применяются методы ван Лаара, Редлиха —
Кистера, Вильсона и т п., которые на основании эксперимен-
тальных данных по изучению фазового равновесия в системе
жидкость — пар с соизмеримым содержанием компонентов поз-
воляют путем экстраполяции найти величину -у2, а следователь-
но, и величину а для разбавленного раствора
С другой стороны, величина а для разбавленного раствора
может быть найдена и расчетным путем В этом отношении хо
рошо зарекомендовали себя методы, основанные па положениях
статистической термодинамики и использующие либо модели
структуры раствора, либо модели межмолекулярного взаимо-
действия. При использовании последних связь между характе-
ристиками межмолекулярного взаимодействия и величиной а
устанавливается по результатам численных расчетов, для про-
ведения которых с успехом применяется так называемый метод
Монте-Карло, являющийся одним из методов численного экс-
перимента моделирования на ЭВМ поведения изучаемой систе-
мы для заданной модели потенциала межмолекулярного взаи
модействия и дающий практически точные результаты расчета
Поэтому в дальнейшем ограничимся рассмотрением одного из
способов расчета коэффициента разделения а, в основе которо-
го лежит использование метода Монте-Карло.
Для описания межмолекулярного взаимодействия в раство-
рах предложено несколько моделей: из них наиболее часто при
меняют модель Деннарда—Джонса, в соответствии с которой
выражение для потенциала межмолекулярпого взаимодействия
записывается в виде
Ц{дг) = 4ео[(-^-)12—(-2Д.)6], , (П.К)>
где Bi,- — минимальное значение энергии взаимодействия двух
молекул (глубина потенциальной «ямы»): аг]— расстояние меж-
36
ду молекулами, при котором энергия их взаимодействия равна
нулю (эта величина характеризует эффективный размер моле-
кулы); г—расстояние между молекулами; индексы i, j харак-
теризуют тип взаимодействующих молекул.
Для определения е|; и о,; обычно используют комбинацион-
ные правила:
eU= lzE/jew, (Н.П)
o,j«<o,( + ow)/2. (II 12)
Как видно из выражения (11.10), модель Леппарда —Джонса
является моделью парных взаимодействий молеку;, и в соответ-
ствии с ней энергия раствора должна определяться суммарной
энергией таких парных взаимодействий В интересующем нас
случае бинарного раствора индексы i и j будут относится к мо-
лекулам примесного (2) и основного (1) компонентов соответ-
ственно. т. е. в этом случае значения eji и 021 в соответствии с
выражениями (11.11), (1112) будут определяться по парамет-
рам егг, £и и 022, оц для индивидуальных компонентов раствора.
Но, как известно, через эти же параметры с помощью мето-
дов статистической термодинамики (на которых мы здесь оста-
навливаться не будем) могут быть выражены такие характери-
стики индивидуального компонента, как плотность, давление
насыщенного пара, температура тройной и критической точек и
т. и., а также его коэффициент активности и химический по-
тенциал в растворе. Последнее дает возможность выразить раз-
личие между составами равновесных паровой и жидкой фаз с
помощью соотношения*
кт In—= Дцж — Др", (II.13)
X
где k — постоянная Больцмана; Држ и Дцп— разность химиче-
ских потенциалов примесного и основного компонентов в жид-
кой и паровой фазах соответственно при температуре фазового
равновесия Т
С учетом (II 4) соотношение (11.13) удобнее записать в виде
lna = Av«<— Avn- (II 14)
где Дмж = Држ/(&7'), Av" = Дцп/(kT). Расчеты, проведенные ме-
тодом Монте-Карло с использованием потенциала Леннарда-—
Джонса применительно к разбавленному раствору очищаемое
вещество — примесь и при допущении о том, что составляющие
их молекулы являются сферически симметричными, позволили
* Колесников А Н„ Степанов В. М_, Яиьков С. В. Фазовые равновесия
предельно разбавленных лениард-джонсовых растворов//Ж>ри. физ. химии,
1985, т. 59, № I, с. 83—86
37
получить следующее аппроксимирующее выражение для вычис-
ления Дтж-П:
Дуж-п = I _ 12,4%+ЗОХ2 — (19,09 + 35,7Х +45,2Х2) Y +
+ (2,61 + 12.08Х) У2| Л -[3,4Х —90Х2+(23,23 + 71,4Х+136Х2) У—
— (0,87+30,08Х) К2) Л2+(16Х+68Х2 —(23,84 + 09,ЗХ
+ 114Х2)У+(1 27,7Х)У2]Л2, (П.15)
где Х= (022/011) — 1; У = (егг/еп) — 1; Л=1,35—kT ец; а интерва-
лы изменения параметров составляют:
0,84 < O22 <*п С1.2; 0 4 С «22/611 С 2.4; 0.68 < kT/elt <1.0. (II 16)
Результаты вычислений по (II.14) и (11.15) показывают, что в
указанных практически важных областях значений параметров
величина а в большей степени зависит от отношения егг/еи, чем
от отношения 022/оц.
В теории строения вещества в настоящее время достигнуты
большие успехи и, как выше уже отмечалось, некоторые свой-
ства индивидуального вещества можно предсказать, если изве-
стны параметры взаимодействия его молекул между собой, и,
наоборот, эти параметры можно определить исходя из тех же
свойств, если последние известны. Вследствие обычно имеюще-
го место отличия характеристик реального межмолекулярного
взаимодействия от предсказываемых моделью Деннарда —
Джонса определяемые значения его параметров будут зависеть
от выбранного свойства. Совершенно очевидно, что точность
расчета величины а при этом должна быть выше, когда пара-
метры межмолекулярного взаимодействия определяются исхо-
дя из свойств, наиболее близких к коэффициенту разделения по
физическому смыслу — давлению насыщенного пара Р рассмат-
риваемого компонента и его плотности рж в жидком состоянии
па линии насыщения. Соответствующие температурные зависи-
мости, полученные на основании расчетов методом Монте-Кар-
ло можно представить в виде следующих интерполяционных
формул*:
1„ (^-)==__^_з.0421пУ-0,7857-*-
-1п [1,068 0.165Г» —0.207 (Г*)2) + 5,647; (11.17)
р* =1,068 0,165г*—0,207 (Г*)2, (II 18)
в которых T*—kTle, — приведенная температура; рж*=ржо3—
приведения плотность, рж=А/д/Кл; Ад— постоянная Авогадро;
Vi— молярный объем жидкости при температуре Т
♦ Степанов В М. Яиьков С. В., Сенников П. Г. Возможности статистиче-
ских методов расчета коэффициента распределения микропримеси прп фазо-
вом равновесии жидкость — пар//В кн . Получение и анализ веществ особой
чистоты. М 1978, с. 73—76.
38
Таблица III. Данные для расчета коэффицие та разделения
и сравнение полученных результатов с экспериментом
Вещество т. к Р, топп 1'А. см’/моль о 6 1(1111) oul ‘’’“о L> X г> е
Беизол 353 760 95.6 484 5,1 10"» 1,15 1,09
Тиофеи 353 670 84 6 498 4,91 Ю’
Тетрахлорид германия 356 760 124 474 5,54 10"» 2,49 2,00
Трихлорид мышьяка 356 263 89,6 551 5.0610-»
С помощью выражений (11.17) и (11.18) можно рассчитать
параметры е и о для молекул каждого из компонентов системы
в интересующем нас конкретном случае. Действительно, зада-
вая значения Р, Т и V=VA при температуре Т в выражении
(П 17), путем последовательных итераций (обычно с помощью
ЭВМ) можно найти величину е. Подставляя ее значение в вы-
ражение (11.18), нетрудно определить значение величины о.
Поскольку в дистилляционных методах очищаемое вещество на-
ходится при температуре кипения, то в расчетах температуру
равновесия Т системы можно отождествить с температурой ки-
пения первого основного компонента (для заданного давления
пара), так как влияние содержания примесного компонента на
равновесную температуру при этом будет пренебрежимо мало.
При проведении соответствующих вычислений необходимо обра-
щать внимание на то, чтобы значения всех величин в расчетных
соотношениях были выражены в одной системе единиц.
Для большей наглядности оценим коэффициент разделения
в системе бензол — тиофен которая, как уже упоминалось, ис-
пользуется в качестве модельной, и в системе тетрахлорид гер-
мания— трихлорид мышьяка, представляющей собой типичную
систему в практике глубокой очистки веществ. Необходимые
для расчета данные* представлены в табл. II.1. Исходя из этих
данных, с помощью соотношений (II 17) и (II 18) можно найти
параметры межмолекулярного взаимодействия для каждого
* Вайсбергер А., Проскауэр Э., Риддик Дж., Тупс Э. Органические рас-
творители М, 1958, с. 520; Фурман А. А. Неорганические хлориды М 1980
с. 416.
39
из четырех приведенных в табл II. 1 веществ: полученные зна-
чения также представлены в табл. II 1 Если подставить полу-
ченные значения параметров в выражение (11.15), то нетрудно
получить значение Д\ж'", а затем с помощью соотношения
(11.14) найти значение коэффициента разделения, которое, как
видно из таблицы, не (Тчень сильно отличается от эксперимен-
тального значения. Следовательно, рассмотренный способ рас-
чета может быть рекомендован для приближенной оценки ко-
эффициента разделения при фазовом равновесии жидкость —
пар. Следует отметить, что в приведенных примерах рассчитан-
ные коэффициенты разделения меньше единицы, поскольку при-
месный компонент в обеих системах является вышекипящим.
Поэтому для сравнения с экспериментальными величинами в
табл. 11.1 приведены коэффициенты разделения, обратные рас-
считанным по выражениям (11.14) н (11.15).
При отсутствии необходимых для вычислений эксперимен-
тальных данных по температурным зависимостям давления на-
сыщенного пара Р и плотности рж вещества параметры потен-
циала Леппарда — Джонса можно оценить с помощью соотно-
шений:
А ТКР е--1.35, р,,-рО3 = 0,34; (11.19)
АГгр'е =0.68, р,1,(Тр)Л3 = 0,864, (11.20)
где Ткр — критическая температура; ркр — критическая плот-
ность; Лр — температура тройной точки; рж<тР) — плотность
жидкой фазы в тройной точке. Соотношения (П.19) и (11.20)
получены, подобно (II 15) — (II 18), на основании расчетов ме-
тодом Монте Карло. Разумеется точность расчета при этом
будет ниже, так как температуры критической и тройной точек
вещества обычно значительно отличаются от температур, для
которых определяется фазовое равновесие жидкость—пар в
системе, содержащей данное вещество.
§3. ЭКСПЕРИМЕНТАЛЬНЫЕ МЕТОДЫ ОПРЕДЕЛЕНИЯ
КОЭФФИЦИЕНТА РАЗДЕЛЕНИЯ
Существует несколько экспериментальных методов определе-
ния коэффициента разделения, каждый из которых имеет свои
преимущества и недостатки. Рассмотрим некоторые из этих ме
тодов, нашедших применение в практике работы с разбавлен-
ными растворами.
Метод испарения небольших количеств раствора. Г ели в рас
поряжеиии экспериментатора имеется метод анализа, не требу-
ющий больших количеств исследуемого вещества — пробы, то
коэффициент разделения можно определить наиболее простым
способом: путем отгонки небольшого объема пара от большого
40
объема раствора Отводимый пар при этом конденсируется
а конденсат подвергается анализу на содержание компонентов.
Можно полагать, что при испарении небольшой части раствора
состав жидкой фазы практически не изменяется. Аналогично
принимается, что в процессе такого испарения не изменяется и
первоначальный состав пара, т. е. состав отгона-дистиллята не
отличается от состава пара, находящегося над растворителем в
начале процесса испарения. Отсюда, зная составы раствора и
сконденсированного пара, по уравнению (II.4) или (11.5) не-
трудно определить коэффициент разделения.
Метод релеевской дистилляции (рис. 7). Этот метод является
более точным. Отличие его от предыдущего состоит в том, что
отгонке подвергается
большая часть раствора с
целью достижения по воз-
можности более значи-
тельной разницы между
его начальным составом
и составом остающейся в
перегонной колбе жидко-
сти, которая при этом бу-
дет обогащаться высоко-
кипящим компонентом.
Взаимосвязь между эти-
ми величинами и коэффи-
циентом разделения мож-
но установить следующим
образом. Обозначим че
рез N общее число молей
жидкой смеси в некото-
рый момент процесса ис-
Рис. 7. Схема прибора для определения ко-
эффициента разделения методом релеевской
дистилляции:
1 — нагреватель; 2 — теплоизоляция. 3 — мешалка;
4 — перегонная колба; 5— холодильник; 5— при-
емник дистиллята
парения, в который мо-
лярная доля примеси равна х. Принимаем, что в любой момент
процесса испарения количество паровой фазы пренебрежимо ма-
ло по сравнению с количеством жидкой фазы Исходя из этого до-
пущения, можно полагать, что если dN молей жидкости переходит
в пар, то содержание примеси в образовавшемся паре будет
равно г/d V, где у — молярная доля примеси в паре. В результа-
те испарения раствора изменяется и его количество и его со-
став Поэтому уравнение баланса можно записать как
<1 (Л'т) — у &N.
(П 21)
После дифференцирования и разделения переменных получаем
dJV dx
—. (11.22)
Л у — х
Пусть примесью является высококипящий компонент. Тогда,
41
подставив соотношение (II.5) в (1122) и проинтегрировав, полу-
чим
In —а j Inx-f- const. (11.23)
Постоянную интегрирования легко определить из условия х—
= х0 при N=N , гдеЛ;0— число молей исходного раствора, а х0—
молярная доля примеси в этом растворе В результате будем
иметь
x/x0=(V ДГ)(«-‘)/а (П-24)
или
a-*o=(l’o F)(“-,)/a, (II 25а)
так как для разбавленных растворов можно полагать, что
No/N— Vo/V, где Vo — объем жидкости до испарения; V — объ-
ем жидкости в перегонной колбе в заданный момент испарения
Уравнение (П.25а) связывает изменение концентрации рас-
твора с изменением его количества вследствие испарения и с
коэффициентом разделения Это уравнение называется уравне-
нием Релея, так как впервые аналогичное соотношение было по-
лучено О Релеем еще в 1896 г при разделении смеси газов пу-
тем диффузии через пористую перегородку.
Определив концентрацию примеси в растворе и замерпв со-
ответственно его объем в начале и конце испарения, с помощью
формулы (II 25а) можно определить коэффициент разделения
а. С большей точностью величина а может быть найдена из
данных нескольких Опытов графическим путем по углу наклона
прямой линии в координатах lgx~lg V Преимущество графи-
ческого способа заключается в том. что при его использовании
не требуется знание величины Хо, которую иногда трудно опре-
делить с достаточной степенью точности В том случае, когда
примесью является низкокипящий компонент, определение а
производится с помощью формулы
х/х.=(У/У,)«-1, (11.256)
которая вытекает из соотношений (11.22) и (II 4) или из фор-
мулы (II.25а), если в последней взять обратную величину ко-
эффициента разделения
Как следует из вывода формул (II 24) и (II 25а), они спра-
ведливы при следующих условиях: а) состав жидкости должен
быть одинаков (однороден) во всем ее объеме в любой момент
испарения, б) между жидкостью и паром во время испарения
должно иметь место термодинамическое равновесие.
Первое условие осуществляется путем достаточно интенсив
ного перемешивания раствора (например, с помощью мешал-
ки), разумеется, при условиях, исключающих явления брызго-
42
заниженным по
Таким образом, метод
величину так
к нулевой скорости этого
Рнс 8 Зависимось коэффи-
циента разделения от ско-
рости испарения раствора
уноса. Второе — применением медленного испарения Такой про-
цесс отгонки в целом получил название релеевской дистилляции
(отсюда и название метода). Однако поскольку процесс испаре-
ния и при релеевской дистилляции происходит все же с конеч
ной скоростью значение коэффициента разделения, определен-
ное по результатам такой перегонки с помощью формулы
(II.25а) или (II 256), получается несколько
сравнению с равновесным значением,
релеевской дистилляции позволяет определять
называемого эффективного коэффициента разделения аэф Рав-
новесную величину а можно найти путем экстраполяции значе-
ний эффективного коэффициента разделения аЭф, полученных
при различных скоростях испарения,
процесса. Вообще же, как показы-
вает опыт уже в некотором интер
вале достаточно малых, но конеч-
ных скоростей испарения коэффи
циент разделения почти не изменя-
ется (рис 8). Поэтому при опти-
мальном выборе рабочей скорости
процесса в области, где дальнейшее
уменьшение скорости испарения не
вызывает заметного изменения ко-
эффициента разделения, можно по-
лучить эффективный коэффициент
разделения, достаточно близкий к
равновесной величине а.
Максимальное значение скорости
испарения можно оценить
следующим образом В случае высокого вакуума скорость
рения вещества характеризуется уравнением Лэнгмюра
Р
испа-
7.0 =___
,,сп /2л.Ш?Г ’
(11.26)
где Т°исп — скорость испарения в вакууме; Р-~-давление пара
над поверхностью испарения при температуре Т М — относи-
тельная молекулярная масса вещества; Р— молярная газовая
постоянная.
Уравнение (11.26) выведено в предположении, что испаряю-
щееся в ходе процесса вещество полностью отводится в кон-
денсат. Однако даже в условиях вакуума нельзя избежать как
взаимного столкновения испарившихся молекул, так и их
столкновения с молекулами остаточных газов. В результате
таких столкновений часть испарившихся молекул будет воз-
вращаться обратно в исходную фазу. Это приходится учиты-
вать введением коэффициента аккомодации который пред-
ставляет собой долю образующегося конденсата от количества
испаряющегося в единицу времени вещества. Кроме того, жид-
43
кость испаряется в атмосфере собственного пара, находящего-
ся под давлением Р, меньшим, чем равновесное давление Р
соответствующее температуре жидкости. С учетом изложенного
уравнение (П.26) запишется в виде
L ис п — 5 —/ • —
(11.27)
где £1|СП — скорость испарения смеси в атмосфере собственного
пара.
Таким образом, как следует
из уравнения (II.27), максималь-
ная скорость испарения зависит
от величины допустимого рас-
хождения между равновесным
давлением пара Р и давлением
пара Р при данных условиях ис-
парения, от температуры процес-
са и относительной молекуляр-
ной массы испаряемого вещества.
При больших значениях а допус-
тимое расхождение между Р и
Р может быть значительно боль-
ше, чем при значениях а, близ-
ких к единице. К сожалению, не-
определенность численного зна-
чения величины которая может
быть найдена лишь эксперимен-
тально, в большинстве случаев
не позволяет непосредственно
пользоваться уравнением (11.27)
для расчета допустимой скоро-
сти испарения, вследствие чего
она обычно подбирается опыт-
ным путем (см. рис. 8)
Метод релеевской дистилля-
Рис 9 Схема прибора для опреде-
ления коэффициента разделения
циркуляционным методом:
Д—куб (сосуд с исследуемой жидко-
стью) 0 В —приемник конденсата (ди-
стиллята), Сл. Сп — холодильники (ле
вый и правый); / — кран для отбора
проб из куба Л; 2— кран для отбора
проб из приемника В. 3. 6—-счетчики
капель; 4—трубка для соединения с
маностатом или атмосферой; 5 — внут-
ренняя трубка: 7 — нагреватель
ции позволяет с удовлетворитель-
ной точностью определить даже небольшие значения а и поэто-
му он с большим успехом используется при работе со смесями
близких по свойствам веществ. Действительно, если отношение
объемов жидкости в начале и конце испарения достаточно ве-
лико, то отношение л/х0 в уравнениях (11.25а) и (11.256) будет
заметно отличаться от единицы даже при малых а и может
быть определено с достаточной точностью.
Циркуляционный метод. В этом методе испарение жидкости
и контактирование сосуществующих фаз обычно осуществляют
в специальных аппаратах, называемых приборами эффектив-
44
мостью в одну теоретическую ступень (теоретическую тарелку).
В литературе описано много конструкций таких приборов Схе-
ма одного из них приведена на рис 9 Пар, образующийся при
кипении в емкости (кубе) Л, поднимается во внутренней труб-
ке 5 и конденсируется в холодильнике Сл, конденсат стекает в
приемник В, откуда егоизбыток поступает в куб/. Температура
стенок трубки с помощью внешнего нагревателя 7 поддержи-
вается при температуре кипения жидкости во избежание час-
тичной конденсации пара на стенках трубки и связанного с
этим дополнительного разделения компонентов смеси. В тече-
ние опыта нижний конец трубки 5 остается частично погружен-
ным в кипящую жидкость, в результате чего пар из паровой
рубашки не попадает в холодильник Сл, а поступает в холодиль-
ник Сп, где конденсируется; конденсат поступает в куб А. Таким
образом, в приборе имеет место циркуляция жидкости, отсюда и
название метода. Через некоторое время собранный в приемни-
ке дистиллят практически будет иметь состав, отвечающий со-
ставу пара, равновесного с жидкостью в кубе А На основании
результатов анализа проб жидкости из приемника В и куба А
по уравнению (114) или (115) нетрудно найти а Циркуляци-
онный метод дает хорошие результаты, когда величина а ис-
следуемой системы не очень велика. Отмеченное ограничение
обусловлено тем, что в процессе циркуляции парожидкостной
смеси сосуществующие фазы не находятся в термодинамическом
равновесии. При этом особенно заметно составы фаз отличают-
ся от равновесных в системе с большими значениями а вследст-
вие повышенного испарения ннзкокипящего компонента По-
этому для определения коэффициента разделения в таких систе-
мах целесообразно использовать метод статического уравнове-
шивания фаз Циркуляционный метод приводит к неточным ре-
зультатам и тогда, когда коэффициент разделения мало отлича-
ется от единицы, поскольку при этом трудно с удовлетворитель-
ной точностью определить различие в составах фаз, даже если
в распоряжении имеется достаточно чувствительный метод ана-
лиза. В этом случае лучше воспользоваться методом релеевской
дистилляции.
Принцип циркуляции парожидкостной смеси используется
также и в так называемом эбуллиометрическом методе. Он ос-
нован на определении температур кипения смеси с разным со-
держанием составляющих их компонентов при заданном давле-
нии с последующим расчетом по полученным данным коэффи-
циента активности (и коэффициента разделения). Непосред-
ственно для исследования разбавленных растворов этот метод
практического применения не нашел. Однако из полученных с
его помощью значений а для области растворов средних кон-
центраций путем экстраполяции можно оценить коэффициент
разделения в области разбавленных растворов.
45
Метод статического уравновешивания фаз. По этому методу
для установления термодинамического равновесия между фаза-
ми раствор и находящийся над ним пар выдерживают при за-
данной температуре в течение некоторого времени. Затем из
паровой и жидкой фаз отбирают пробы на анализ. По резуль-
татам анализа с помощью уравнения (114) или (115) опреде-
ляется коэффициент разделения.
Дифференциальный метод. Близким к методу статического
уравновешивания фаз является так называемый дифференци-
альный метод, который нашел применение в практике разделе-
ния изотопных смесей, смесей изомеров, смесей гидридов. Он
основан па определении разности в давлении насыщенного па-
Рис. 10. Схема прибора для определения коэффици-
ента разделения дифференциальным методом
1, 2 — отделения сосуда со стандартным н исследуемым об-
разцами веществ соответственно; 3— манометр для опреде-
ления давления лара вещества-стандарта; 4—в — краны,
9— дифференциальный манометр
ра исследуемой бинарной смеси и вещества-стандарта. В каче-
стве вещества-стандарта берется та же смесь, но с другим, по
возможности заметно меньшим содержанием примесного ком-
понента. Поэтому веществом стандартом может быть имеющий-
ся в индивидуальном виде или предварительно выделенный
(очищенный) основной компонент данной бинарной смеси.
Принцип метода легко понять из рис. 10. Сосуд А состоит
из двух отделений 1 и 2 В отделении / помещен образец ве-
щества-стандарта, молярная доля примесного компонента в ко-
тором мала и составляет х'. В отделении 2 находится смесь с
более высоким содержанием примесного компонента х". Оба
отделения соединяются с плечами дифференциального маномет-
ра 9, который позволяет измерять разность давлений пара ДР
46
образцов при заданной температуре; отсюда и название мето-
да— дифференциальный Для точности отсчета обычно исполь-
зуется катетометр С помощью манометра 3 определяется дав-
ление насыщенного пара вещества-стандарта Чтобы поддержи-
вать постоянную температуру в системе, установка термостати-
руется. Выбор тер моста тирующей и манометрической жидко-
стей зависит от условий опыта.
Примем, как и ранее, что для разбавленных растворов коэф-
фициент разделения а не зависит от их концентрации, т. е. ве-
личины 7i и ^2 в выражении (11.8) являются постоянными и,
следовательно, для разбавленных растворов справедлив закон
Генри Тогда при условии установления в системе стационар-
ного состояния, с учетом (П.6) и в соответствии с законом
Дальтона применительно к бинарной смеси будем иметь
Р2-Л?1(1-И4-₽°гТгЛ
где Р|—давление насыщенного пара в отделении 1; Рг—дав-
ление насыщенного пара в отделении 2. Из выражений (П.28)
для случая, когда примесью является низкокнпящий компо-
нент, т. е. с учетом соотношения (II 8), следует, что
1- ^+[(Р°уг)/(^у,)13-»_ 1 +(а-1)^
Pt 1 + (а-1)*'’ * ->
откуда
а-1 (Рг-РдКР^-РгР)- (П.30)
Как правило, Рг мало отличается от Pi, но зато для повыше-
ния точности определения а обычно берется х' <g.x" и поэтому
членом Р2х' по сравнению с членом Р(х" в знаменателе правой
части уравнения (II 30) можно пренебречь. В результате урав-
нение (11.30) запишется как
а 1=ДР Ptx"), (11.31)
где ДР=Р2—Рр
Легко заметить, что уравнение (11.31) будет справедливым и
для случая, когда веществом-стандартом является индивиду-
альный основной компонент; при этом в данном уравнении
По аналогии с выводом уравнения (П.31) нетрудно пока-
зать, что, когда примесью является высококипящий компонент,
выражение для определения коэффициента разделения а будет
иметь вид
(а —1) а = ДР,'(Р1И1 (П.32)
где уже &P = Pt—Р2.
При использовании дифференциального метода необходимо
иметь в виду, что определяемая величина ДР существенно за-
47
висит от наличия в сравниваемых образцах посторонних при-
месей, особенно иизкокипящих. Поэтому соответствующим из
мереииям должна предшествовать тщательная очистка исход-
ных веществ
Известны и другие ме оды определения равновесия жид-
кость— пар, которые используются в отдельных случаях (метод
газожидкостной распределительной хроматографии, метод рек-
тификационной колонны). Однако они находят ограниченное
применение, и па практике в основном используются метод ре-
леевской дистилляции и циркуляционный метод.
Для термодинамической проверки экспериментальных дан-
ных о фазовом равновесии жидкость — пар разработаны спо-
собы, основанные на использовании зависимости того или иного
свойства раствора (коэффициент активности компонентов, тем-
пература кипения и т д.) от его состава Для разбавленных
растворов такого рода зависимости выражены очень слабо,
и поэтому в рассматриваемом случае известные способы термо-
динамической проверки практически не применимы.
Перейдем теперь к рассмотрению основных дистилляцион-
ных методов очистки веществ.
§ 4. ПЕРЕГОНКА
Однократная перегонка
Однократная перегонка как метод очистки оформляется ана-
логично релеевской дистилляции (равновесная перегонка), ко-
торая представляет собой по существу идеальный вариант од-
нократной перегонки.
Если коэффициент обогащения велик, то при испарении рас-
твора первые порции конденсата (дистиллята) в значительной
степени будут освобождены от высококипящего компонента.
Этот компонент, следовательно, будет концентрироваться в ос-
татке, .т. е. в той части раствора, которая остается в перегонной
колбе (см. рис. 7). Аналогичным образом такая простая пере-
гонка позволяет разделить многокомпонентную смесь на фрак-
ции, состоящие в основном из индивидуальных веществ. В соот-
ветствии с этим при разделении обычных смесей можно полу-
чить несколько фракций, заметно отличающихся по своему со-
ставу; поэтому для обозначения такого процесса иногда при-
меняют термин «фракционированная перегонка». Необходимо,
однако, отметить, что последний термин чаще используется как
синоним другого дистилляционного метода — ректификации.
Достоинством метода простой перегонки является, как видно
из рис. 7, простота его осуществления. Примером очистки рас-
сматриваемым методом может служить дистилляция воды.
Соли, содержащиеся в воде, имеют температуру кипения зна-
48
чительно выше, чем у воды, и в процессе дистилляции буду*
концентрироваться в жидкости остающейся в перегонной кол-
бе. В дистилляте же содержание солеи будет ничтожно малым.
Вследствие простоты аппаратурного оформления перегонка
применяется для очистки веществ с высокой температурой кипе-
ния металлов, солей и т. д когда изготовление и эксплуата-
ция более сложной аппаратуры для осуществления других ме-
тодов очистки сталкиваются со значительными трудностями
Когда примесью является низкокипящий компонент про-
дуктом будет жидкость, остающаяся в перегонном кубе. Исхо-
дя из аналогии простой перегонки с релеевской дистилляцией,
содержание примеси в продукте в этом случае можно оценить
по формуле (11.256) Для определения концентрации примеси
•го в отогнанной фракции жидкости Л'о, объем которой равен
Ко, можно принять, что в любой момент времени процесса пере-
гонки образующийся пар непрерывно отводится из отгонной
системы в виде дистиллята. Примем также, что количество
пара по сравнению с количеством жидкости в течение всего
процесса пренебрежимо мало. Указанное условие для простой
перегонки практически выполняется всегда, поскольку даже
при фракционированной перегонке процесс заканчивается, не
доходя до полной отгонки жидкости. С учетом этих допущений
уравнение материального баланса по примесному компоненту
запишется в виде
ЛГото= Л'х+ Л’с^о= Л'*+ (Л^—Л’) xd, (П.ЗЗ)
откуда
*о —*(У/Рр)
*D~
т0-х (N/Ne)
1—N/NO
(П.34)
(11.356)
Подставляя соотношение (II 25) или (11 26) в (11.34), получаем
l-(V/Ve),/e_ i-d-VD/Fe)1^
1- V/Ve 1-(1-VD/VO) ’
когда примесью является высококипящий компонент, и
ственно
1 - (F/Vo)“ _ 1 - (1 - VD/V0)a
Х° i — V/V0 ' 1(1-VD/VO) ’
когда примесью является низкокипящий компонент.
При использовании формул (II 25а), (11.256) и
(11.356) для определения содержания примеси в остающейся
или отогнанной фракциях необходимо, разумеется, предвари-
тельно знать величину а. Поскольку в очищаемом веществе
всегда содержатся и высоко- и низкокипящие примеси практи-
ческий интерес представляет рассмотрение поведения при пере-
гонке хотя бы двух условно выбранных примесных компонен-
(II.35а)
соответ-
49
тов (высоко и иизкокипящие по отношению к основному ком
поненту) Примем, что эти примеси лимитируют свойства основ-
ного компонента и в то же время являются наиболее трудно-
отделимыми с коэффициентами разделения а' и а" соответст-
венно Тогда все другие примеси будут концентрироваться
в первых порциях дистиллята или кубовом остатке. Эти фрак-
ции и являются «хвостовыми».
Пусть объем «хвостовой» фракции дистиллята составляет
Vd<i). Тогда из выражений (11.25а) и (П.256) следует, что кон-
центрации высококипящего компонента х'щц и низкокипящего
компонента x"NW в оставшейся жидкости V%a) До отгона ос-
новной фракции Vd(2) будут составлять соответственно
g'-i
x.v(i)=I’^—*1) а > (11.36а)
xN(l)~xo^— Xi)a *, (11.366)
где 7<i — Vo(i)/Vo, х0', х0" — содержание примесных высоко- и
низкокипящего компонентов в исходном очищаемом веществе.
После отброса первой «хвостовой» фракции в приемник дистил-
лята отгоняется продукт объемом Vd(2)=Vo—Уон)—^М2>. где
V/v(2)— объем второй «хвостовой» фракции, т. е жидкости, ос-
тающейся в перегонном кубе по окончании процесса Величины
л'*(1) и х"^) при этом будут представлять собой как бы исход-
ные концентрации рассматриваемых компонентов соответствен
но, т. е их следует подставить вместо х0 в выражения (П.35а)
и (П.356), которые в этом случае будут характеризовать чисто-
ту продукта С учетом этого после небольших преобразований
получим выражение для концентрации х'о(2) примесного высо-
кокипящего компонента
(II.37a)
и выражение для концентрации x"d(2) примесного нижекипяще-
го компонента в продукте
XIX2) — ХИ (1—К1)а 1 ' (11376)
Простая перегонка иногда используется и для очистки тер-
монестойких и нестойких по отношению к воздуху веществ.
В этих случаях процесс перегонки обычно проводят при пони
женном давлении (перегонка в вакууме) с целью снижения тем-
пературы кипения очищаемого вещества или для удаления воз-
духа из перегонной системы. В таком варианте простая пере-
гонка особенно часто применяется в лабораторной практике
для выделения продуктов реакции из реакционной смеси.
50
Та же цель, которая преследуется перегонкой в вакууме»
в ряде случаев достигается перегонкой с водяным паром Ввод
водяного пара в разделяемую смесь понижает ее температуру
кипения, что создает лучшие условия для отгонки более лету-
чих или термонестойких компонентов смеси Осуществление
этого варианта простой перегонки требует выполнения условий
расслоения дистиллята на отгоняемое вещество и воду. Следо-
вательно, данный способ пригоден лишь для смесей с малой
взаимной растворимостью их компо! ентов и воды в конденси-
рованном состоянии; в противоположном случае возникла бы
проблема разделения дистиллята. Указанному условию обыч-
но удовлетворяют смеси углеводородов, н поэтому перегонка
с водяным паром в основном и используется для их грубого
разделения на отдельные фракции. В принципе вместо водяно-
го пара может быть использован любой подходящий, т. е. хи-
мически инертный по отношению к разделяемой смеси, газ.
Применительно к глубокой очистке вещества однократная
перегонка может быть использована и в качестве предвари-
тельной стадии перед применением многоступенчатых процес-
сов разделения. Для разделения смесей веществ с близкими
температурами кипения однократная перегонка малоэффектив-
на. В этих случаях эффект очистки может быть увеличен путем
частичной дефлегмации (конденсации) поднимающегося из
перегонного куба пара перед его поступлением в конденсатор
и переходом в дистиллят. Это достигается с помощью специ-
альных устройств — дефлегматоров. Образующаяся в результа-
те частичной конденсации пара жидкость — флегма стекает об-
ратно в перегонный куб. Получаемый же при этом дистиллят
будет содержать уже меньше вышекипяшей примеси, чем, на-
пример, в случае простой перегонки.
Многократная перегонка
Существенного повышения чистоты вещества можно достичь
в результате его многократной перегонки с отбросом «хвосто-
вых» фракции в каждом цикле Как уже было показано, со-
держание примесей в продукте первого цикла нетрудно опре-
делить с помощью уравнений (П.37а) и (11.376). Сам цикл
заканчивается сливом из перегонного куба оставшейся жидко-
сти объемом КЛ(2) — второй «хвостовой» фракции Во втором
цикле процесс перегонки повторяется. Продукт первого цикла
загружается в перегонный куб и часть его объемом Ущз) отго-
няется в качестве третьей «хвостовой» фракции Содержание
примесного компонента в остающейся при этом жидкости объ-
емом УЛ(з) можно оценить с помощью выражения вида (П.Зба)
или (П.Збб). Например, в случае высококипящего компонента,
51
как легко показать, соответствующее соотношение запишется
как
1Л’(3) = 1Ь(2)[т^7] = (П.38)
или, с учетом выражения (П.37а),
«'-t j___________________________х>/а' a’-i
хК(3)=х'о^-^ а’ "i-щ (1-*з) . (П.39)
где хз= 1^0(3)/Иод.
Далее, из оставшейся жидкости отгоняется дистиллят объ-
емом К0(4) — продукт второго цикла; в перегонном кубе будем
иметь жидкость объемом I6v(4)— четвертую «хвостовую» фрак-
цию. Концентрация примесного высококипящего компонента
л'о(4) в продукте второго цикла по аналогии с (II.37а) будет
определяться выражением
*D(*)-*M3) 1-Vv(4) гМз) (пл°)
или, подставляя соотношение (11.39) в выражение (11.40),
_«2J 1-х’/«'
a:b«) = Ii(l-’<1) “ Т-Хд (1~ хз) _"Хз • (11.41)
Таким же образом можно получить выражение для концентра-
ции примесного высококипящего компонента в продукте и пос-
ле п циклов. Если в каждом цикле доли «хвостовых» фракций
одинаковы, т. е. если Х| =х2=хз=Х4... =х, то выражение вида
(II.41) применительно к n-му циклу перегонки упрощается и
принимает вид
^b=Iol(l— х)1/а' (1—х‘/сс')]п. (Н.42а)
Аналогично выводится выражение и для определения концен-
трации низкокипящего примесного компонента после п-го цик-
ла перегонки:
хЬ=1оК1—х>“ (1—x“")]n. (11.426)
Выход продукта при этом будет составлять
УдрОЛ = V0(l—x)sn. (11.43)
§ 5 РЕКТИФИКАЦИЯ ОСНОВНЫЕ ПОНЯТИЯ
Процесс ректификации осуществляется в специальных аппа-
ратах, называемых ректификационными колоннами. Простей-
шая схема ректификационной колонны изображена на рис. 11
Пар, образующийся при кипении жидкости в кубе колонны А,
52
поднимается вверх по ректифицирующей части В и попадает
в конденсатор С Конденсат (флегма) стекает вниз по колонне
в куб А. Поднимающийся пар, вступая в контакт со стекаю
щей жидкостью, обедняется высококипящим компонентом. Та-
ким образом, в ректифицирующей части В, которая представ-
ляет собой вертикальный цилиндр, обычно заключающий
в себе устройство для улучшения контакта жидкости и пара,
осуществляется противоток фаз. В результа е между жид-
костью и паром протекает процесс массообмена, т. е. происхо-
дит межфазовое перераспределение компонентов. Па концах
колонны имеет место обращение фаз нар превращается в жид-
fl
дистиллята
СмВ
остатка
Рис. 12. Схема ректификацион-
ной колонны непрерывного
действия с питанием в сере-
дине:
Д — куб (кипятильник); В — ректи-
фицирующая часть; С — конденса-
тор; Хо — концентрация примеси в
исходной смеси
Рис. II. Схема ректификацион
пой колонны периодического
действия:
Л — куб (кипятильник): В — ректи-
фицирующая часть; С —конденса-
тор
кость в конденсаторе, а жидкость — в пар в кубе колонны. Все
эго приводит к умножению элементарного акта разделения,
наблюдаемого при обычном испарении жидкости, т. е. ректи-
фикация в отличие от однократной перегонки является много-
ступенчатым процессом. Чем лучше контакт между жидкостью
и паром в ректифицирующей части, тем выше скорость межфа-
зового массообмена и выше эффект разделения в колонне.
Вследствие межфазового массообмена в процессе ректифика-
ции ннжекипящий компонент концентрируется в верху колон-
ны и в виде дистиллята (конденсата) может оттуда отбирать-
ся в качестве продукта (или, наоборот, в качестве отбрасывае-
53
мой фракции) Для этой цели в конденсаторе осуществляется
деление потока жидкости, образующейся из поступающего
пара, на две части. Одна, меньшая часть отбирается в сборник
дистиллята, а другая, большая часть возвращается в колонну
в противоток пару в виде орошения — флегмы. Отношение ско-
рости орошения к скорости отбора продукта называется флег-
мовым числом. Часто скорость отбора продукта характеризуют
также степенью отбора, которая представляет собой долю по-
тока, отводимого в качестве продукта. Нетрудно показать, что
между этими двумя величинами имеется следующая взаимо-
связь:
(П-44)
где р— степень отбора; Ф — флегмовое число.
Если продукт из колонны не отбирается, то р = 0, а Ф = оо.
При этом говорят, что колонна работает в безотбориом режиме,
или при полной флегме (полном орошении).
При работе колонны в отборном режиме, т. е. при частичном
орошении (р>0, Ф<оо), загруженная в куб колонны смесь
(в случае, когда ее нельзя отождествить с бинарной) может
быть разделена путем непрерывного отбора дистиллята на не-
сколько фракций с различными температурами кипения. В этом
случае термин «ректификация» отождествляется с термином
«фракционированная разгонка». При этом, разумеется, в зави-
симости от интересующего компонента нужным продуктом бу-
дет являться та или иная фракция.
В промышленных условиях ректификация осуществляется
обычно в виде непрерывного процесса (рис. 12). Разделяемую^
смесь — питание — подают в среднюю часть колонны. Низкоки-
пящие компоненты смеси концентрируются при этом в верхней
части колонны, а высококипящие—в нижней части. Местом
ввода питания колонна делится на две секции Секция, в ко-
торой концентрируется интересующий компонент, носит назва-
ние укрепляющей. Другая секция называется исчерпывающей.
С точки зрения теории ректификации каждую из этих секций
можно рассматривать как отдельную колонну. Непрерывно дей-
ствующие колонны иногда еще называют колоннами с от-
крытым циклом.
Таким образом, при работе непрерывно действующей колон-
ны исходная питающая смесь в принципе делится на две фрак-
ции: низкокипящую, которая отбирается из конденсатора ко-
лонны в виде части дистиллята, и высококипящую, которая
отбирается из куба колонны в виде части кубовой
жидкости. Применительно к случаю глубокой очистки од-
на из этих фракций и будет представлять собой ин-
54
гересующии продукт. В лабораторной практике чаще применя-
ются колонны периодического действия, т. е. колонны с закры-
тым циклом работы. Теория же ректификации развивалась в ос-
новном применительно к колоннам с открытым циклом Эта
теория справедлива и для колонн с закрытым циклом, обла-
дающих «бесконечно» большим кубом (при этом соблюдается
условие постоянства состава разделяемой смеси), и удовлетво-
рительно описывает работу колонн с кубом конечных размеров.
По характеру контакта между жидкостью и паром ректифи-
кационные колонны условно можно разделить на следующие
типы.
Тарельчатые колонны. Контакт между жидкостью и паром
в таких колоннах происходит скачкообразно на специальных
горизонтально установленных в различных сечениях ректифици-
рующей части колонны устройствах — тарелках.
Насадочные колонны. К насадочным колоннам обычно от-
носятся колонны, ректифицирующая часть которых заполнена
засыпной (нерегулярной) насадкой. Контакт между жидкостью
и паром здесь осуществляется непрерывно по всей высоте ко-
лонны в ее объеме.
Пленочные колонны. В колоннах этого типа жидкость дви-
жется сверху вниз в виде пленки по поверхности специального
приспособления (регулярная насадка), вводимого в ректифици-
рующую часть для обеспечения большей площади контакта фаз
и их движения по заданному пути. Контакт между жидкостью
и паром при этом происходит на поверхности этой пленки не-
прерывно по всей высоте колонны. Наиболее простой из таких
колонн является колонна с ректифицирующей частью в виде
полой трубки (колонна с орошаемыми стенками).
По специфике работы к колоннам с регулярной насадкой
примыкают роторные колонны, в которых введенное в ректифи-
цирующую часть приспособление для улучшения межфазового
контакта в ходе процесса может вращаться. Но в виду трудно-
стей в их изготовлен-ии и эксплуатации они используются редко;
при глубокой очистке веществ роторные колонны практически
не применяются.
Для установления взаимосвязи между параметрами процес-
са ректификации разбавленных растворов рассмотрим процесс,
протекающий в ректифицирующей части одной из секций не-
прерывно действующей колонны исходя из следующей схемы
(рис. 13).
1. Разделяется бинарная смесь основное вещество—при-
месь. Смесь имеет постоянный состав и со скоростью L непре-
рывно поступает в верхний конец ректифицирующей части.
2. В нижнем конце ректифицирующей части происходит об-
ращение фаз. Образующийся пар поднимается со скоростью I
вверх по колонне и отводится из нее.
55
3 В случае отборного режима часть потока жидкости при
обращении фаз в низу ректифицирующей части с определенной
и постоянной скоростью п отводится в качестве продукта.
С момента начала работы ректификационной колонны до-
стигаемый в ней эффект разделения постепенно увеличивается
до установления в колонне стационарного состояния. Приме-
нительно к рассматриваемой схеме при стационарном состоя-
нии в колонне не происходит изменения составов жидкой и па-
ровой фаз. Следовательно, отношение концентраций примеси
Рис 13. Схема ректификационной
колонны непрерывного действия с
питанием вверху:
L — скорость потока жидкости; I — ско-
рость потока лара; п — скорость отбора
продукта
Рис. 14. Схема верхнего участка
ректифицирующей части тарельча-
той (колпачковой) колонны:
Хо— молярная доля примеси в исходной
смеси, поступающей в колонну; Л|, Л\>.
х3 — молярная доля примеси в жидко-
сти иа I. 2 и 3-Й тарелках соответст-
венно; у1г уз, ул — молярная доля при
.меси в паре над I. 2. и 3 й тарелками
соответственно
и жидкости (или пара) в низу и в верху ректифицирующей
части будет при этом постоянной величиной. Этим отношением
обычно и характеризуют разделительную способность ректифи-
кационной колонны, разумеется, при заданных условиях прове-
дения процесса.
Исходя из задачи глубокой очистки веществ, анализ работы
ректификационной колонны будем проводить для рассмотрен-
ного выше случая, когда содержание примеси (редкого компо-
нента) в разделяемой бинарной смеси незначительно по срав-
нению с содержанием основного вещества, т. е. для случая
5Ъ
разбавленных растворов. При этом можно принять, что такая
бинарная смесь близка к идеальной и, следовательно, скорость
потоков жидкости и пара по высоте колонны практически не
меняется, а количества жидкости и пара в ректифицирующей
части постоянны, т. е. не изменяются в течение процесса ректи-
фикации.
§ 6. РЕКТИФИКАЦИЯ В ТАРЕЛЬЧАТЫХ КОЛОННАХ
В колоннах этого типа, как уже отмечалось, ректифицирую-
щая часть содержит ряд чередующихся специальных уст-
ройств— тарелок для осуществления контакта между стекаю-
щей сверху жидкостью и поступающим снизу паром. В практи-
ке используется много конструкций таких устройств. Схема
действия одного из них — колпачковой тарелки приведена на
рис. 14; в промышленных колоннах тарелка этого типа обычно
содержит несколько паровых патрубков с колпачками.
Как видно из рис. 14, на каждой тарелке колонны происхо-
дит перераспределение компонентов смеси между жидкостью и
паром, т. е на каждой тарелке имеет место межфазовый массо-
обмен. Результатом такого массообмена является скачкообраз-
ное увеличение содержания низкокипящего компонента в паре,
покидающем тарелку, по сравнению с составом пара, поступаю-
щим на эту же тарелку. Соответственно стекающая с тарелки
жидкость имеет более высокую концентрацию высококипящего
компонента по сравнению с его содержанием в жидкости, по-
ступающей на тарелку. При этом находящаяся на тарелке
жидкость и поднимающийся с нее пар имеют тенденцию к ус-
тановлению термодинамического .равновесия между ними. Чем
лучше условия контакта, тем ближе будет соотношение между
составами жидкости и пара на тарелке к равновесному В пре-
дельном случае это соотношение становится равновесным. Та-
релка, соответствующая такому разделению, называется тео-
ретической тарелкой (ТТ) или теоретической ступенью (ТС).
Отсюда легко объясняется факт частого использования термина
«прибор в одну теоретическую тарелку» для обозначения, как
отмечалось выше, некоторых циркуляционных аппаратов при
исследовании равновесия жидкость—пар.
Совершенно очевидно, что достигаемый в ректификацион
ной колонне общий эффект разделения будет выражаться че-
рез совокупность изменений составов жидкости и пара на та-
релках колонны. Первая попытка установления этой взаимосвя-
зи была предпринята еще в конце прошлого столетия Е. Соре-
лем. Суть предложенного им способа состоит в последователь-
ном расчете состава фаз от тарелки к тарелке в ректифицирую-
щей части колонны исходя из соответствующих уравнений ма-
териального и теплового балансов для каждой тарелки и дан-
57
ных по равновесию жидкость—пар разделяемой смеси. Анало-
гичный, но более приближенный подход к решению указанной
задачи был предпринят одновременно Э. Хаусбрандом, который
принял допущение о постоянстве теплот испарения жидкости
на каждой тарелке колонны и исключил из рассмотрения теп-
ловые балансы. ОбаГ способа расчета позволяют вычислить
число теоретических тарелок (ЧТТ), которое требуется для по-
лучения продукта с заданной степенью чистоты, но процедура
соответствующих вычислений является весьма трудоемкой и в
том и в другом способах. Поэтому в дальнейшем метод расчета
«от тарелки к тарелке» подвергся усовершенствованию и упро-
щению. В результате были разработаны эквивалентные ему
графические и графоаналитические методы расчета ЧТТ, более
простые, но требующие геометрических построений, как, напри-
мер, известный метод Мак-Кэба и Тиле, или использования но-
мограмм. При глубокой очистке веществ, когда обычно требу-
ются высокоэффективные колонны, т. е. колонны с большим
числом тарелок, эти методы становятся практически неприем-
лемыми ввиду возникающих трудностей графического представ-
ления, в частности применительно к отборному режиму работы
ректификационной колонны, т. е. для расчета разделительной
способности колонны или для оценки ЧТТ более предпочтитель-
но использование соответствующей аналитической зависимости,
полученной при тех или иных допущениях. Особенно простой и
удобной для практических расчетов эта зависимость становится
при допущении о постоянстве величины коэффициента разделе-
ния а и при характерном для задачи глубокой очистки веществ
условии х<^\,
Это нетрудно показать исходя из материального баланса по
примесному компоненту для отдельной тарелки колонны в со-
ответствии с принятой схемой (см. рис. 13, 14) Когда в колон-
не уже достигнуто стационарное состояние, т. е. дальнейшего
изменения составов жидкости и пара на любой тарелке больше
уже не происходит, можно записать, что
/>о = *!/1-|-пгр, (11.45)
где L—скорость потока жидкости, моль/(см2-с); / — скорость
потока пара, моль/(см2-с); п — скорость отбора продукта,
моль/(см2 с); у\ — молярная доля примеси в ларе, покидаю-
щем первую тарелку; х0—молярная доля примеси в исходной
смеси; хр — молярная доля примеси в отбираемом продукте,
т. е в жидкости, отводимой с последней, л-й тарелки колонны
(счет ведется сверху вниз).
Уравнение материального баланса (11.45) по содержанию
примеси в ректифицирующей части колонны отражает очевид-
ный факт: сколько примеси поступает в единицу времени в рек-
тифицирующую часть колонны с потоком L, столько же приме-
58
си выводится из нее за это время с потоками I и и. П-ри этом
предполагается, что жидкость на тарелке полностью переме-
шивается, т. е ее состав в любой момент времени однороден;
разумеется, это в равной степени относится и к паровой фазе.
Нетрудно видеть, что, с другой стороны, это уравнение харак-
теризует также материальный баланс по примесному компо-
ненту на первой тарелке. Отсюда следует, что уравнение вида
(11.45) будет справедливым для любой t-й тарелки колонны
и его можно записать в общем виде как
Lxi^lyt-i-nxn. (11.46)
Из соотношения (1146) следует, что в безотборном режиме
(п = 0 и, следовательно, L=I) x0 = t/|. Аналогично из (11.46)
следует, что
(И 47)
т. е. в стационарном состоянии и безотборном режиме работы
колонны концентрация примесного компонента в жидкости, сте-
кающей с (i—1)-й тарелки, равна его концентрации в паре,
поступающем с соседней нижележащей (,-й тарелки.
Так как при принятой схеме очистки примесь концентриру-
ется в паровой фазе, то, отождествляя реальную тарелку в ко-
лонне с теоретической и исходя из определения теоретической
тарелки, имеем
— (П.48)
Подстановка (П.47) в (II 48) дает
(П.49)
Составляя п уравнений вида (1149) для t=l, 2,..., п, где
п — число теоретических тарелок в ректифицирующей части
колонны, и перемножая соответственно левые и правые части
этих уравнений, получаем
/?о = ^п/^ = а-п. (П.50)
Соотношение (П.50) представляет собой видоизмененную при-
менительно к ректификации разбавленных растворов форму
широко известного уравнения М Фенске.
Величина Fo называется фактором разделения (степень раз-
деления, фактор фракционирования) в безотборном режиме.
Фактор разделения Fo определяет разделительную способность
колонны. Чем больше Fo отличается от а, тем больше эффект
разделения, достигаемый в ректификационной колонне, по
сравнению с эффектом разделения при обычном испарении жид-
кости. Уравнение (П.50) наглядно отражает многоступенча-
тость процесса ректификации и большую ее эффективность по
отношению к простой перегонке. Так, например, при а=2 со-
59
держание примеси в дистилляте в ходе процесса однократной
перегонки нельзя уменьшить более чем в 2 раза по сравнению
с ее содержанием в исходной смеси. В результате же ректифи-
кации этой смеси на колонне, содержащей всего лишь 10 таре-
лок, следует ожидать в соответствии с уравнением (11.50)
уменьшения содержания примеси в очищаемом веществе при-
мерно на три порядка. В действительности, однако, результат
получается иной, поскольку разделение, достигаемое на реаль-
ной тарелке, всегда меньше теоретически возможного, т е. со-
ставляет лишь долю (50—90%) того разделения, которое соот-
ветствует теоретической тарелке, и характеризуется соотноше-
нием (11.48). Для выражения количественной характеристики
этого различия, которая называется коэффициентом, полезного
действия (КПД) тарелки, используется несколько способов.
Так, по способу Г. Хаузена эффективность реальной тарелки
выражается в виде отношения количества интересующего ком-
понента, перешедшего в единицу времени из одной фазы в дру-
гую на дайной тарелке, к соответствующему его количеству,
которое перешло бы за это же время и в том же направлении
при условии достижения на тарелке фазового равновесия При
расчетах иногда пользуются также величиной условного КПД
Е. Мерфри. Под этой величиной понимается отношение измене-
ния концентрации пара (жидкости) на данной i-й тарелке ио
сравнению с концентрацией пара (жидкости) на предыдущей
(последующей) тарелке к соответствующему изменению, кото-
рое имело бы место, если бы между паром и жидкостью дости-
галось термодинамическое равновесие. Обозначая указанную
величину через Ем'ау, для паровой фазы имеем
. (П.51)
где у,* — равновесная концентрация пара на i-й тарелке. Соот-
ветствующее выражение можно записать и для жидкой фазы.
Поскольку оба способа связаны с некоторыми эксперименталь-
ными неудобствами, касающимися определения состава фаз на
отдельных тарелках, на практике часто выражают КПД тарелок
через КПД колонны, под которым понимается отношение чис-
ла теоретических тарелок п, которым эквивалентна данная ко-
лонна, к числу содержащихся в ней реальных тарелок пд. При-
нимая, что средний КПД тарелок Е равен КПД колонны,
имеем
E=n/nR. (П.52)
Из опубликованных в литературе экспериментальных данных
известно, что КПД тарелок разных конструкций заметно отли-
чаются друг от друга. Причем КПД зависит не только от осо-
бенностей устройства тарелки, но и от условий проведения
60
процесса, и от свойств разделяемой смеси. Для приближенной
оценки КПД предложены различные критериальные уравне-
ния, составленные иа основании соответствующих эмпириче-
ских зависимостей. Достаточно же надежная величина КПД
может быть определена лишь опытным путем, т, е на основа-
нии предварительных опытов. Такие определения обычно про-
водят для безотборного режима работы колонны, чтобы исклю-
чить влияние скорости отбора продукта на величину КПД
Как показывает практика эксплуатации ректификационных
колонн, их разделительная способность в отборном режиме ра-
боты ухудшается по сравнению с безотбориым. Имеющиеся
в литературе результаты проведенных исследований свидетель-
ствуют о том, что зависимость разделительной способности 9т
скорости отбора продукта практически одинакова для колонн
разных типов. Поэтому подробнее отборный режим рассмотрим
в разделе, посвященном анализу работы насадочных колонн,
которые чаще, чем колонны других типов, используются при
ректификационной очистке веществ.
§7 РЕКТИФИКАЦИЯ В НАСАДОЧНЫХ КОЛОННАХ
В насадочных колоннах контакт между движущимися про-
тивотоком жидкостью и паром происходит на насадке, запол-
няющей объем ректифицирующей части. В качестве насадки
используют фарфоровые или стеклянные кольца, отрезки ме-
таллической спирали, тела различной геометрической формы из
проволочной сетки и т д. Использование насадки преследует
цель увеличения площади взаимодействия жидкой и паровой
фаз, что приводит к возрастанию эффекта разделения в колон-
не. При глубокой очистке веществ важную роль играет мате-
риал насадки, который может быть источником загрязнения
очищаемого вещества С целью сведения к минимуму послед-
ствий этого эффекта в качестве материала насадки широко ис-
пользуется кварцевое стекло особой чистоты
Один из путей анализа работы насадочных ректификацион-
ных колонн состоит в решении дифференциальных уравнений,
составленных на основе материального баланса разделяемой
смеси. При составлении таких уравнений учитывается, что мас-
сообмен между жидкостью и паром имеет место по всей высоте
колонны.
Понятие о движущей силе массообмена
Исходя из непрерывного характера процесса межфазового
массообмена по высоте насадочной колонны составим уравне-
ние материального баланса по редкому компоненту (примеси)
для единицы объема слоя насадки. Введем обозначения х —
6!
молярная доля примеси в жидкой фазе в некотором попереч-
ном сечении слоя насадки; z—координата вдоль высоты ко-
лонны; v — скорость межфазового перехода примеси, т е. ко-
личество молей примеси, переходящее из жилкой фазы в паро-
вую (в соответствии с принятой схемой) или, наоборот, из па-
ровой фазы в жидкую (когда продукт отбирается в виде части
дистиллята) в единице объема колонны в единицу времени.
Пусть колонна работает в стационарном состоянии, когда изме-
нения концентрации примеси по высоте колонны со временем
не происходит Примем далее, что движущиеся противотоком
жидкость и пар равномерно распределены по всему сечению
колонны, что скорость движения отдельных частей потоков фаз
одинакова, а продольное перемешивание в них отсутствует. По-
лагаем также, что перенос примеси вдоль высоты колонны за
счет осевой молекулярной диффузии обусловленной концентра-
ционным градиентом, пренебрежимо мал по сравнению с пере-
носом примеси потоками фаз. Тогда, при принятых допущениях,
скорость поступления примеси на единичную площадь сече-
ния z с потоком L составит Lx, а на единичную площадь сече-
ния (z+Дг) колонны — L(x—Дх). Отсюда следует, что
Lx — L(x—&.r) = i?Az (П.53)
или, переходя от приращений к дифференциалам,
£17 = ’- (11.54)
Скорость межфазового перехода компонентов в целом оп-
ределяется механизмом массообмена, поверхностью раздела
фаз, а также степенью отклонения системы жидкость — пар от
равновесного состояния. Действительно, если паровая и жидкая
фазы находятся в термодинамическом равновесии, изменения
их состава не происходит, т. е. скорость перехода примеси (или
равная ей по величине скорость перехода основного вещества)
из одной фазы в другую будет равна нулю. Следовательно, чем
больше состояние системы отклоняется от равновесного состоя-
ния, тем выше в этой системе будет скорость массообмена.
В общем случае, как известно, отклонение системы от рав-
новесия определяется разностью химических потенциалов дан-
ного компонента в обменивающихся фазах. Когда эта разность
равна нулю, система находится в термодинамическом равнове-
сии и перераспределения компонентов между фазами не про-
исходит, т. е. значения химического потенциала данного компо-
нента во всех фазах системы, находящейся в равновесии, рав-
ны между собой Отсюда следует, что мерой отклонения систе-
мы от равновесия может служить также разность между фак-
тическим значением химического потенциала р. данного компо-
нента в одной из фаз системы и тем значением химического
62
потенциала р.*, которое этот компонент имел бы в той же фазе,
если бы система находилась в равновесии Таким образом ско-
рость массообмена можно охарактеризовать соотношением
г~р—[!♦ <11.55)
Линейная зависимость между и и (ц—ц*) соблюдается тем
точнее, чем меньше величина (р—р*)_ Поэтому значения хи-
мического потенциала ц и р* в соотношении (II 55) целесооб-
разно рассматривать применительно к основному компоненту,
для которого разность (р—р*) всегда меньше, чем для примес-
ного компонента. Эту разность обычно относят к паровой фазе,
хотя в принципе она может быть отнесена и к жидкой фазе.
Химический потенциал основного компонента в паровой
фазе (в расчете на моль) связан с концентрацией последнего
известным уравнением
н"= In (1-0 (В-56)
И
рГ=Ро1+лг (В.57)
где у, у*—текущая и равновесная концентрации примесного
компонента в паровой фазе.
Так как скорость переноса примесного компонента v равна
по величине скорости переноса основного компонента, но про-
тивоположна по знаку, то с учетом этого, подставляя выраже-
ния (II56) и (II57) в соотношение (11.55), получаем
v= — к’ ВТ [In (i-у)-In (11.58)
где k' — коэффициент пропорциональности.
Разлагая в ряд функции In (1—у) и in (1—у*) по степеням у
и у* соответственно и отбрасывая члены второй н более высо-
ких степеней, имеем
1п(1—у)~ — у, In (1—у*) ~ — у*.
С учетом этих соотношений из (II 58) следует, что
о—к(у—у*), (11.59)
где k — kRT — константа скорости массообмена, или коэф-
фициент массопередачи а разность (у—у*) называется дви-
жущей силой массообмена (массопередачи) или концентраци-
онным напором.
Понятие движущей силы широко применяется при анализе
массообменных процессов При этом часто весьма важным ока-
зывается соответствующий выбор фазы, через составы которой
выражается движущая сила с учетом явлений, лимитирующих
скорость массообмена. При небольших нагрузках что харак-
терно для процесса глубокой очистки, жидкая фаза в насадоч-
63
jioft колонне обычно распределена в виде непрерывно обнов-
ляющейся тонкой пленки, обволакивающей поверхность эле-
ментов насадки. Так как можно считать, что в тонкой жидкой
пленке однородность состава по ее толщине устанавливается
сравнительно быстро, то массообмен в этом случае будет опре-
деляться диффузией» примесного компонента в паровом про-
странстве: из парового пространства к поверхности жидкой
пленки или от поверхности жидкой пленки в паровое простран-
ство. Скорость же изменения содержания примеси в паровой
фазе в результате диффузии в свою очередь зависит от степе-
ни отклонения состава фазы от равновесного; в состоянии рав-
новесия количество примеси, диффундирующей из жидкости
в пар, равно количеству примеси, диффундирующей из пара
в жидкость, вследствие чего изменения составов фаз при этом
не происходит. Таким образом, при течении жидкости по на-
садке в виде достаточно тонкой пленки предпочтительный выбор
выражения движущей силы процесса ректификации в шасадоч-
ной колонне через концентрации в паровой фазе является впол-
не обоснованным. Константа массообмена k в выражении
(П.59) зависит от свойств системы и условий процесса: k бу-
дет тем больше, чем больше удельная поверхность контакта
фаз и чем выше скорость диффузии (коэффициент диффузии)
примесного компонента в паровой фазе. С другой стороны,
диффузия в паре имеет турбулентный характер, поскольку пар
проходит между элементами насадки по каналам различной
формы и различных направлений. При возрастании линейной
скорости парового потока турбулентность увеличивается, вслед-
ствие чего может происходить значительное изменение величи-
ны k и, следовательно, скорости массообмена в целом.
Фактор разделения в стационарном
состоянии и безотборном режиме
Дифференциальное уравнение материального баланса
(11.54) по примесному компоненту для элемента объема насад-
ки колонны, работающей в стационарном состоянии, в соответ-
ствии с (11.59) и (II.4) будет иметь вид
fix
L——=k(y —az). (11.60)
Концентрация у в уравнении (11.60) может быть выражена
через концентрацию х с помощью уравнения рабочей линии, ко-
торое по форме аналогично соотношению (11.45), но с учетом
непрерывности процесса массообмена в насадочной колонне
запишется как
г=(1-р)у+рхр, (П.61)
где х и у означают соответственно концентрации примеси
в жидкой и паровой фазах в некотором поперечном сечении ко-
<4
_____________________________ J
лонны, хр — концентрацию при 1еси в отбираемом продукте,
p — nlL — степень отбора
Для безотборного режима работы колонны (р=0) из выра-
жения (II.6I) следует, что у=х, т. е. составы жидкости и пара
в одном и том же сечении колонны одинаковы и, следовательно,
неравновесны. Последнее и является причиной наличия движу-
щей силы процесса разделения, о котором говорилось выше.
В соответствии с изложенным из уравнений (II 60) и (II61)
имеем
Лт
L~=^k(x—ax) (11.62)
или, разделяя переменные,
Исходя из принятой схемы процесса и допущения о том, что при
постоянстве параметров процесса (£ = const, a = const) величи-
на k также является постоянной, интегрируем уравнение
(П.63) при следующих граничных условиях:
x=xQ при 2 = 0, Х — Х при Z=ZK, (11.64)
ZK
где гк — полная высота ректифицирующей части: xZ|t— концен-
трация примеси в жидкости в нижнем конце колонны. В ре
зультате получаем
/’=ехр [ — к (а—1) zKfL], (11.65)
где F=xzJxq — фактор разделения
Из уравнения (II.65) видно, что величина Го-1 возрастает
с увеличением длины колонны, коэффициента обогащения
(а—1), константы скорости массообмена k и уменьшается при
увеличении скорости потока жидкости L. Из уравнения (II 65)
следует также, что величина —Info должна обратно пропор-
ционально зависеть от L. Так как k зависит от степени турбу-
лентности парового потока, то в действительности зависимость
—In Fo от L имеет более сложный характер, как это, например,
схематично изображено на рис. 15. На участке б—в зависи-
мость между —InF0 и L соответствует уравнению (П.65), так
как при небольших скоростях движения пара (заметим, что в
рассматриваемом безотборном режиме L — 1) степень турбу-
лентности парового потока слабо зависит от его скорости.
На участке в—г, соответствующем большим скоростям движе-
ния пара, имеет место резкое увеличение турбулентности паро-
вого потока, при этом величина k растет, что приводит к уве
личению значения —1п£0 (при работе в так называемом «пен
ном режиме» или «режиме эмульгирования») На участке а—б
происходит изменение удельной поверхности контакта фаз
3—46
65
Рнс. 15 Зависимость разделитель-
ной способности колонны от ско-
рости потока жидкости
получаем
вследствие того, что при очень
малой скорости жидкого потока
не вся поверхность насадки мо-
жет быть смочена жидкостью.
Следовательно, в этой области k
также не является постоянной
величиной, что приводит к изме-
нению величины —In Fp-
_ Из соотношения (П.63) мож-
4но получить выражение для рас-
пределения примеси по высоте
ректифицирующей части Ис-
пользуя граничное условие х~х0
при г=0 и интегрируя 11.63),
х = хоехр[—к(а — 1) i/L]
или, с учетом (11.65),
(11.66)
Х= X
(11.67)
Таким образом, в логарифмических координатах распределе-
ние примеси по высоте колонны выражается уравнением пря-
мой линии, что подтверждается и экспериментально. На осно-
вании анализа проб жидкости в различных сечениях по высоте
колонны с помощью уравнения (11.67) можно определить вели-
чину Fq или концентрацию примеси в жидкой фазе в нижнем
конце колонны. К этому можно прибегнуть в том случае, ког-
да для очистки применяется высокоэффективная колонна и
концентрация примеси в жидкой фазе на выходе из колонны
может лежать ниже чувствительности используемого метода
анализа.
Влияние скорости отбора продукта
на фактор разделения
Фактор разделения, достигаемый в ректификационной ко-
лонне, как уже отмечалось при рассмотрении колонны тарель-
чатого типа, существенно зависит от того, с какой скоростью
отбирается из колонны конечный продукт. Применительно
к насадочной колонне количественная зависимость между ско-
ростью отбора продукта и фактором разделения может быть
получена следующим путем. Из уравнения рабочей линии
(П.61) следует, что
66
Подставляя выражение (11.68) в (II 60), имеем
dz
х — рхр
—т-------ах
1—Р
(11.69)
или, после разделения переменных
dz к (а— 1)
(1-ав)«+е«р — L(l —р) dг’
(II. 70)
где 0=р/(а—1).
Интегрирование уравнения (II 70) при граничных условиях
(11.64) дает:
Iп (*-««) ^K+6zp____fc(«-l)zK 1—и8
(1—ав z0 + ©zp “ L 1-р 4 ' '
Так как в рассматриваемой нами схеме процесса продукт от-
бирается из нижнего конца колонны, то XzK=xp. С учетом по-
следнего, а также используя соотношение (11.65), из (II 71)
получаем
(1-ав)хг-|-ехр
(1-ав)х0+вхр
(II 72)
Выражение (11.72) после небольших преобразований можно за-
писать в виде
1—ай
Л (1-р)р-(‘-“е)/ч-р)_в ’
(11.73)
где F—Xpfxc — фактор разделения в отборном режиме.
Из уравнения (11.73) следует, что когда продукт из колон-
ны не отбирается, т. е. когда р = 0, то F=F0 и, следовательно,
разделительная способность колонны будет максимальной, ра-
зумеется, при постоянстве других условий процесса. При р=1
из уравнения (1173) получаем, что F=l; это означает отсут-
ствие эффекта разделения в колонне. Таким образом, процессу
очистки должно соответствовать ограничение 0<р<1. Вели-
чина р может быть найдена на основании расчетов по уравне-
нию (11.73)
Результаты соответствующих расчетов для удобства целе-
сообразно представить в виде графика зависимости F от р при
заданных Fo и а; такой график позволяет достаточно надежно
предсказать, с учетом допустимой производительности процес-
са, оптимальную величину р. Примеры подобных графиков при-
ведены на рис. 16. Здесь же для сравнения представлены ре-
зультаты опытов по очистке бензола от тиофена (использова-
лись искусственно приготовленные смеси бензол — тиофен с не-
большим содержанием тиофена, для которых коэффициент
3*
67
F
ПОО''
1200
1000
ООО
ООО
ООО
200
разделения равен 1,094)* в
насадочной ректификацион-
ной колонне. Наблюдаемое
на рис. 16 удовлетворитель-
ное согласие между расчет-
ными и экспериментальны-
ми значениями фактора раз-
деления дает основание для
вывода об обоснованности
предположений, принятых
при выводе уравнения
(11.73) Из рис. 16 видно
также, что скорость отбора
продукта существенно вли-
яет на значение фактора
разделения F. Причем это
влияние тем больше, чем
больше Fo отличается от
единицы, т. е чем больше
эффективность колонны в
безотборном режиме. Если
скорость отбора продукта
велика, то конечное разде-
ление, достигаемое на ко-
лоннах с большим и малым
значениями Fo, оказывает-
ся примерно одинаковым. Поэтому применение высокоэффек-
тивных колонн является оправданным лишь при проведении
процесса ректификации с небольшой скоростью отбора про-
дукта.
Расчеты по уравнению (11.73) показывают, что при разде-
лении смесей с различными а, но при одинаковой величине Fo
влияние скорости отбора продукта сильнее сказывается в слу-
чае меньшего значения коэффициента разделения.
Аналогичная соотношешию (11.73) соответствующая аналити-
ческая зависимость для случая концентрирования примесного
компонента в жидкости имеет вид
о ....
1 1,02 1,04 1,00 1,08 !,Ю 1,12 /,/4
Отношение потоков, I IL
।___।___।___।---1 । —J----1
О 0,02 0,04 0,00 0,08 0,10 0,12 0,14
Степень отвара t о
Рис 16. Зависимость разделительной
способности колонны от скорости отбора
продукта:
/ — расчет по уравнению (11.73): 2—расчет по
уравнению (11.85) О, X — экспериментальные
результаты по очистке бензола от тнофеиа
(а-1.094)
(1—p)Fo(1+e)/(1-J')+<z0 ’
где Е0 = ехр[Л(а—l)zK/(La)].
* Зельвенский Я. Д., Шалыгин В. А. Влияние величины отбора на степень
разделения в ректификационной колоиие//Хнмия и технология топлив н масел
I960, № 7, с. 19—24.
68
Высота, эквивалентная
теоретической тарелке (ВЭТТ)
Существует и другой метод анализа работы насадочных ко-
лонн. Он состоит в том, что насадочная колонна в некотором
смысле уподобляется тарельчатой колонне, чтобы можно было
оперировать в расчетах высотой слоя насадки, эквивалентной
теоретической тарелке. Под теоретической тарелкой в насадоч-
ной колонне понимается такой участок ректифицирующей ча-
сти, на котором состав жидкости, стекающей с нижнего его
конца, и состав пара, выходящего с его верхнего кон-
ца, связаны таким соотношением, которое эти жид-
кость и пар имели бы, находясь в термодинамическом равно-
весии. Таким образом, для каждого (-го участка будет справед-
ливо выражение вида (11.48). Отсюда число теоретических та-
релок п, которым эквивалентна данная насадочная колонна при
разделении заданной смеси, например, в безотборном режиме,
может быть определено с помощью уравнения (П.50), если из-
вестен фактор разделения Fo для этой смеси с заданным зна-
чением а. Путем деления высоты ректифицирующей части
на п определяется высота, эквивалентная теоретической тарел-
ке— ВЭТТ. И наоборот, при известном значении ВЭТТ, /напри-
мер, для колонны небольшой высоты нетрудно оценить величи-
ну п для такой же колонны большей высоты, разумеется, при
одних и тех же условиях процесса.
Для расчета разделительной способности насадочных ко-
лонн в отборном режиме с использованием понятия ВЭТТ раз-
работано несколько способов.
Точность полученных при этом соответствующих расчетных
уравнений определяется точностью принятых при выводе их со-
отношений (обычно эмпирических), характеризующих равно-
весие жидкость — пар. Для смесей, близких к идеальным, наи-
более строгим, по-видимому, является способ Смокера, который
может быть применен и для реальных разбавленных растворов.
Суть метода состоит в преобразовании координат на диаграмме
равновесия жидкость — пар с использованием координат точ-
ки пересечения рабочей линии и линии фазового равновесия.
Следовательно, если решить совместно уравнения (II4) и
(11.61), то для координат точки пересечения получим
РХр
®пер- ’ (И.75а)
= [J-a(l-P) ~Рхр]1 (П-756)
Обозначив х'=хпер—х, у' = Упер—у, нетрудно показать, что
у* = ах*. (П.76)
69
Представим теперь, что слой насадки колонны представляет со-
бой совокупность последовательно расположенных участков
размером ВЭТТ. Тогда для t й ВЭТТ выражение (11.76) запи-
шется как
, р; = ах:, (11.77)
а уравнение (П.46) —в виде
* 1— Р ‘ (П.78)
Подставляя (П.77) в (II 78), получаем
xi-i «(l-Р) • (П.79)
Из соотношения (II.79) для первой теоретической тарелки
(/ = 1) будем иметь
Х’~ х* - сх (1 р) • (П.80)
Соответственно для второй (i=2)
, ^1 • 2 а(1 — р) (а(1—p)J2 ’ (П.81)
для третьей (i = 3)
х' = Х'а X'Q 3 а(1 —р) - [а(1 —Р)]3 (П.82)
и для п-й (i = n) теоретической тарелки
х' — Xq п [а(1 —р)]п" (II.83)
Так как в соответствии с введенными обозначениями
*п/*о==(Р*р/[1 —а(1 —p)J—хп}/{рхр/[1—а(1 —/>)]—х0), a хр = хп,
то из соотношения (11.83) получаем
£=--------1 -CZ0-----
(l-Р) [<х(1-р)|"-е- u
Поскольку процесс глубокой очистки проводится с малой
степенью отбора продукта (р<С1), то изменением значений
ВЭТТ и ЧТТ по сравнению с их значениями в безотборном ре-
жиме в первом приближении можно пренебречь. Тогда, выра-
жая величину п в уравнении (II 84) с помощью соотношения
(11.50), будем иметь
1—ав
(l-p)fo,g (“(‘-PN/tea-e
(11.85)
70
Уравнение (П.85) отличается от уравнения (И.73) лишь
показателем степени при F и поэтому результаты расчетов по
ним не должны заметно различаться Об этом наглядно свиде-
тельствует соответствующая графическая интерпретация, пред-
ставленная на рис. 16. Отсюда следует, что метод анализа ра-
боты насадочных колонн с использованием понятий ВЭТТ и
ЧТТ приводит к удовлетворительным результатам, несмотря на
то, что в насадочных колоннах разделение по высоте колонны
происходит непрерывно, а ие скачками, как это имеет место
в тарельчатых колоннах Таким образом, уравнением (11.84)
можно пользоваться и при расчете эффекта очистки в тарельча-
той колонне, работающей в отборном режиме. Более того, не-
трудно показать, что аналогичное уравнение можно получить
и путем последовательного «перехода» от тарелки к тарелке
исходя из соотношения (11.46). На рис. 17 показано, что харак-
терное для тарельчатой колонны скачкообразное изменение
концентрации примеси от тарел-
ки к тарелке может быть вы-
ражено через плавное изменение
ее по высоте колонны Очевид-
но, чем меньше коэффициент раз-
деления смеси и КПД тарелки,
тем меньше будет отличаться
распределение примеси в тарель-
чатой колонне от распределения
в насадочной колонне.
Через величину ВЭТТ часто
сравнивают разделительную спо-
собность различных насадочных
и пленочных колонн; последние
по характеру контакта фаз близ-
ки к насадочным. Такое сравне-
ние обычно проводят для без-
отборного режима их работы
(£ = /), который, как правило,
является более стабильным. В
(11.50) и (П.65) будем иметь
Рис. 17. Распределение примеси по
высоте ректификационной колонны:
1 — для тарельчатой колонны; 2 — для на-
садочной колонны
этом случае из соотношений
и In g——-(а~^ Zk
Li
откуда
вэтт__——
п к (а — 1) к а — 1"
(11.86)
(11.87)
Таким образом, высота, эквивалентная теоретической тарелке,
возрастает с увеличением нагрузки колонны и уменьшается с
71
увеличением k. Под нагрузкой колонны понимается скорость
прохождения потока одной из фаз: L — нагрузка по жидкости,
I — нагрузка по пару. Чем меньше ВЭТТ, тем больше в целом
эффект разделения в колонне. В соответствии с этим для уве
личения разделительной способности ректификационной колон-
ны при заданной оптимальной нагрузке стремятся подобрать
такую насадку, которая обусловливала бы наибольшую кон-
станту массообмена (коэффициент массопередачи) К сожале-
нию, достаточно строгая теоретическая оценка величины k в на-
стоящее время пока не представляется возможной ввиду слож-
ности процесса межфазового массообмена, который зависит от
характера движения потоков жидкости и пара между элемен-
тами насадки колонны, от поверхности контакта фаз, от
свойств разделяемой смеси и т. д. На первый взгляд кажется,
что для достижения большей величины константы массообме-
на достаточно использовать очень мелкую насадку. Однако
с уменьшением размера элементов насадки увеличивается ее
способность к захвату «неактивной» жидкости в пространстве
между ними, вследствие чего условия межфазового массообме-
на будут ухудшаться. Например, при этом может создаться
большой перепад давлений пара в нижней и верхней частях
колонны, что является крайне нежелательным для процесса
разделения. Поэтому выбор оптимальных размеров элементов
насадки, материала, из которого она изготовляется, ее формы
приходится определять опытным путем. В литературе имеется
много практических рекомендаций по использованию того или
иного типа насадки применительно к различным случаям раз-
деления смесей веществ.
Высота единицы переноса (ВЕП)
Эффективность разделительных аппаратов колонного типа
с непрерывным контактом фаз, к каковым относятся насадоч-
ные и пленочные ректификационные колонны, часто выражают
также через высоту единицы переноса — ВЕП и соответствен-
но через число единиц переноса — ЧЕП. В основе этих харак-
теристик лежит рассмотренное выше понятие о движущей силе
массообмена, обусловливающей перенос вещества в колонне;
отсюда и термин «единица переноса». Высоте единицы переноса
соответствует высота такого участка разделительной части ко-
лонны, для воображаемых концов которого разница в составах
входящего (выходящего) и выходящего (входящего) потоков
одной из фаз равна средней движущей силе на этом участке.
Поскольку применительно к ректификации движущая сила
в принципе может быть представлена в виде разности (у—у*)
или (х*—х), то по отношению к соответствующей разности вы-
соту единицы переноса обозначают как (ВЕП)<^ или (ВЕП)ох-
72
Правильный выбор выражения для движущей силы определяет-
ся скоростью диффузионных явлений в контактирующих фазах.
Когда скорость межфазового массообмена определяется ско-
ростью диффузии в одной из фаз (при определенных условиях
процесса), то говорят, что в этой фазе сосредоточено основное
диффузионное сопротивление массообмену. Когда же диффузи-
онные процессы в обеих фазах протекают с соизмеримой ско-
ростью, то часто возникает необходимость определения «вкла-
да» каждой из фаз в диффузионное сопротивление. В (ряде слу-
чаев это имеет важное значение при конструкционном и техно-
логическом оформлении процесса, например при /разделении
обычных смесей.
В интересующем же нас случае глубокой очистки веществ
ректификацией в насадочных и пленочных колоннах, жидкая
фаза в которых распределена в виде тонкой пленки, можно
полагать, что скорость диффузии в такой пленке достаточно
высока для быстрого выравнивания состава пленки по ее тол-
щине и, следовательно, скорость межфазового массообмена
в колонне будет определяться скоростью диффузии в паре, т. е
основное диффузионное сопротивление массообмену в колоннах
указанных типов при обычно реализуемых условиях будет ока
зывать паровая фаза, и выражение, определяющее ВЕП*, за-
пишется как
Лу = Увх — Увых = (У— У*)ср. (П.88)
Отсюда появляется возможность выразить эффективность ректи-
фикационной колонны через число единиц переноса, подобно
тому как выше она выражалась через число теоретических та-
релок.
Для этой цели составим дифференциальное уравнение мате-
риального баланса по примесному компоненту в паровой фазе,
которое по аналогии с (П.54), с учетом (П.59), будет иметь вид
г-^-=Л(у-у*) (1189)
или, после разделения переменных,
, (11.90)
у — У* he
где Л. —Ijk.
Интегрируя выражение (П.90) при граничных условиях
У = Ув при z = 0, y=yZK при z = zK, (11.91)
* Здесь и в дальнейшем будем пользоваться обозначением ВЕП хотя
в соответствии с общей терминологией в рассматриваемом случае следовало
бы пользоваться обозначением (ВЕП)^.
73
получаем
^ZK
2lt •* <1Ч
= <IL92>
Уо
Как видно из (II.92)f величина 1ге имеет размерность длины.
Далее, поскольку из выражения (11.90) с заменой в нем диф-
ференциалов приращениями следует, что при Az=/ie, Д// =
— (У—У*)ср, то, согласно уравнению (П.88), he в выражении
(П.92) соответствует высоте единицы переноса — ВЕП, а пе —
числу единиц переноса — ЧЕП
Число единиц переноса, которым эквивалентна данная на-
садочная (пленочная) колонна, можно определить графическим
нли аналитическим интегрированием уравнения (11.92) как для
безотборного, так и отборного режима работы. Так, примени-
тельно к безотборному режиму и при выполнении условия
i/*/x = a = const аналитическое интегрирование (11.92) дает
(IL93>
ИЛИ, С учетом ТОГО, ЧТО Угк/Уо = Хгк До = Е0,
Сравнивая соотношения (II 94) и (11.50) или же исходя из
выражения (11.87), с учетом того, что ВЕП = /ге = ///г, получим
следующую взаимосвязь между высотой единицы переноса и
высотой, эквивалентной теоретической тарелке
ВЭТТ=ВЕП (П.95)
Соотношение (11.95) позволяет сделать вывод о том, что в об-
щем случае при одних и тех же условиях ВЭТТ колонны будет
меньше ВЕП, т. е одна теоретическая тарелка будет вызывать
меньшее изменение концентрации, чем одна единица переноса.
Но в расчетах это не дает каких-либо преимуществ ВЕП перед
ВЭТТ. Тем более, что при очистке вещества от трудноудаляе-
мой примеси (примесей), когда (а—1)<1, различие между ве-
личинами ВЭТТ и ВЕП, как следует из соотношения (11.95),
вообще становится несущественным. Поэтому, хотя понятие
ВЕП является теоретически обоснованным, в практике глубо-
кой очистки веществ для сравнивания эффективности ректифи-
кационных колонн обычно используется более наглядная вели-
чина ВЭТТ*.
* Иногда вместо ВЭТТ используют термин БЭТС — высота, эквивалентная
теоретической ступени. Но во избежание путаницы следует заметить, что тео-
ретической ступенью является и единица переноса.
74
§ 8. ВЛИЯНИЕ ЗАГРЯЗНЯЮЩЕГО ДЕЙСТВИЯ
МАТЕРИАЛА АППАРАТУРЫ НА ГЛУБИНУ
ОЧИСТКИ ВЕЩЕСТВ МЕТОДОМ РЕКТИФИКАЦИИ
Так как конечной целью ректификации является получение
продукта заданного состава, то при глубокой очистке веществ
этим методом весьма важной задачей является рассмотрение
вопроса о возможном загрязнении очищаемого вещества мате-
риалом аппаратуры. Эффект загрязнения при этом может быть
обусловлен «вымыванием» нежелательной примеси из материа-
ла аппаратуры, а также химической реакцией материала аппа-
ратуры с очищаемым веществом или какой-либо агрессивной,
но легко отделяемой примесью, содержащейся в очищаемом ве-
ществе. К этим случаям, очевидно, можно отнести и проникно-
вение примеси из окружающей среды через стенки раздели-
тельного аппарата — колонны — и самопроизвольное дисперги-
рование конструкционных материалов при их контакте с очи-
щаемым веществом в ходе процесса.
Поскольку уравнения, выведенные для оценки работы наса-
дочных колонн, применимы и для колонн других типов, рас-
смотрим поставленную задачу применительно к насадочной
колонне, тем более, что поверхность контактного устройства
в ней обычно велика. Установим взаимосвязь между эффектив-
ностью очистки, достигаемой в колонне, работающей в стацио-
нарном состоянии, и скоростью «вымывания» примеси из стенок
и насадки колонны, полагая, что загрязняющая примесь одно-
именна с отделяемой. П/римем, что предел растворимости за-
грязняющей примеси в разделяемой смеси не достигается и что
в течение процесса примесь поступает из материала аппарату-
ры в жидкую фазу равномерно и с постоянной скоростью по
всему объему ректифицирующей части.
Если обозначить через оп скорость поступления примеси
в единицу объема колонны, то, пренебрегая эффектом продоль-
ного перемешивания, для отрезка колонны высотой Дг с еди-
ничным поперечным сечением будет справедливым следующее
уравнение баланса по примесному компоненту:
[£+ («4- Дг)) U+M—(Ь4-«'пг)аг=(₽4-’’п) (11.96)
Из уравнения (11.96) после перехода от приращений к диффе-
ренциалам, пренебрегая членом vndzdx как величиной второго
порядка малости, получаем
+ (11.97)
\ L / QZ
Соответственно запишем уравнение рабочей линии
(L + vnz)x-|-i>n (z>< —z) = (£+«>п2к) (1 —р) р4-(Д + ^п2и) Ргр
75
или
*=(l-p)v+₽*p+—М1-у+р(у-*р)1 + -^(1-х). (11.98)
Так как для процесса глубокой очистки веществ справедливы
соотношения х<С1, р{у—хР)<С1 и, кроме того, есть смысл
(рассматривать задачу только для случая (vnz/L)^l (в против-
ном случае глубокая очистка веществ методом ректификации
становится нецелесообразной), то, пренебрегая этими величи-
нами по сравнению с единицей в уравнениях (11.97) и (11.98),
имеем, с учетом соотношений (II 59) и (П.4),
L (dx/dz) = fc (jr—ссг)4-рп, (11.99)
я —Р^р-М^п/Ь) (zK — z)
у =-------------------• (И. 100)
Фактор разделения в стационарном состоянии
и безотборном режиме
В случае безотбориого режима (р = 0) из соотношения
(11.100) следует, что
I'=a:+JT!L(1-v)- (П*101)
Подставляя (II 101) в (П.99), приходим к линейному диффе-
ренциальному уравнению первого порядка:
-^-+Ро*=Со(*). (П.102)
где Р0=*(а—1)/L, Q0(z) = vn[k(zK—z) +L]/L2.
Решение уравнения в общем виде записывается как
*=ехр( —J podz) [J <2o(z)exp(J Podz)dz+A]. (П.103)
Произведя в (II 103) интегрирование и определяя постоян-
ную А из условия, что при z=0, х=хо, после некоторых преоб-
разований с учетом соотношения (II.65) получаем выражение
для распределения примеси по высоте колонны
[(1-a/ln Fo) (l-</z«) -z/zK], (11.104)
где p = onzK/L(a—1).
Если теперь подставить в (II 104) z=zK и, следовательно,
х=Хгк, то получим выражение для фактора разделения
где Fm = xzJx0—фактор разделения в безотборном режиме при
наличии эффекта загрязнения; Fo—фактор разделения в без-
76
Рис. 18. Распределение примеси по
высоте колонны при наличии загряз-
няющего действия материала аппара-
туры:
/ — расчет ло уравнению (11.67); 2, 4. 5 —
расчет по уравнению (11.104) для значений
vn, моль/(см3 с): I0*M, I0-'0, 1(М соответ
ственно; 3 - расчет по уравнению (11.104)
для vn — 10-10 моль/(смэ-с) при у=х в лю-
бом сечении колонны
отборном режиме при отсут-
ствии эффекта загрязнения.
Из сравнения уравнений
(П.65) и (П.105), а также
(II 67) и (II. 104) видно, что
влияние загрязняющего дей-
ствия материала аппаратуры
должно проявляться тем силь-
нее, чем меньше скорость оро-
шения и коэффициент разде-
ления смеси и чем больше вы-
сота колонны и скорость поступления примеси.
На рис. 18 представлены результаты расчетов по уравне-
ниям (П.67) и (II 104). При этом были использованы следую-
щие величины: £=10-3 моль/(с-см2), гк=100 см; хо=102;
Го=1О~5; а = 2. Из рис. 18 видно, что поступление примеси из
материала колонны даже с незначительной скоростью может
оказать существенное влияние на глубину очистки.
Влияние скорости отбора продукта
иа фактор разделения
Применительно к отборному режиму подстановка (II 100)
в (П.99) приводит к дифференциальному уравнению, аналогич-
ному уравнению (II.102):
^.+ PpX=Qp(z), (11.106)
fc[a(l —р) —11 „ , , гп(1— р) — kpxp + (kvn/L)(zK — z)
W РР ~ L (1 — р) • <?Р(г) - L (1 - р)
Решение уравнения (П.106) можно записать в виде соотноше-
ния, подобного выражению (II 103); постоянная интегрирова-
ния полученного выражения будет определяться из того же гра-
ничного условия, что и при решении уравнения (II.103): х=х0
при z=0 В результате получаем
77
z/z„ f XpeinF0+P[(a-l)(l — p) — In r0]
+ ("ли>
где mp = F0<1-ae)/<'-p); 0s=p/(a—1).
Уравнение (11.107) характеризует распределение примеси по
высоте колонны, работающей в стационарном состоянии и от-
борном режиме.
Введя в (II 107) условие, что х = хр при г—гк, после неко-
торых преобразований получим выражение для фактора раз-
деления
р = F г t_₽_ сс(1~р)5?р,~<)+1,|?В|<
1 L х0 (1 — a6)lnmp
(11.108)
Рис. 19 Зависимость раз-
дельной способности колон-
ны от скорости отбора про-
дукта при наличии загряз-
няющего действия материа-
ла аппаратуры:
1 —расчет по уравнению (11.73);
2. 3, 4 — расчет по уравнению
(II 108) для значений vn,
моль/(см3с): 10-", 10-*°, 10—®
соответственно
меси vn из интересующего
если известны значения Fo,
где Fi—Xplxo — фактор разделения
в отборном режиме при наличии эф-
фекта загрязнения; F—фактор
разделения в отборном режиме при
отсутствии эффекта загрязнения
[см. уравнение (11.73)].
Влияние скорости отбора про-
дукта на фактор разделения графи-
чески иллюстрирует рис. 19. Как
можно видеть из рисунка, даже при
небольших степенях отбора эффект
загрязнения может быть сущест-
венным; при увеличении степени
отбора он постепенно «затухает».
Следовательно, загрязняющее дей-
ствие материала аппаратуры долж-
но проявляться сильнее при малой
скорости отбора продукта. В тоже
время применение высокоэффектив-
ных колонн, необходимых для глу-
бокой очистки, требует небольших
скоростей отбора продукта.
С помощью уравнений (11.104),
(II 105), (II 107), (11.108) можно
оценить скорости «вымывания» при-
материала по результатам опытов,
а, х0.
Предельное значение концентрации примеси
Значительный интерес представляет оценка предельного ми-
нимального значения концентрации примеси, достигаемого в
ректификационной колонне при наличии эффекта загрязнения.
78
Для этой цели соотношение (11.65) с учетом (11.50) запишем
в следующем виде
(IT. 11'3)
Подставив соотношение (П.109) в уравнение (II.105) и приняв,
что ВЭТТ — величина постоянная получаем следующее выра-
жение для FCI при zK->oo:
„ ВЭТТ гпа
Inn Го1=—:----,
, «X, 01 Ina i£(a —1
откуда
ВЭТТ гпа
1 ПП xz = —:------—-------— .
г -.оо In a L (а — 1)
(П.110)
(11.111)
Из выражения (11.111) следует, что при наличии эффекта за-
грязнения имеет место предельное значение концентрации при-
меси на выходе из колонны, которое нельзя снизить за счет
увеличения длины последней. Таким образом, для заданной ве-
личины vn должна существовать оптимальная длина колонны,
дальнейшее увеличение которой по существу не влияет на глу-
бину очистки. Из выражения (II 111) также видно, что пре-
дельное значение концентрации примеси не зависит от ее ис-
ходного содержания в очищаемом веществе. Отсюда следует,
что соотношение (II 111) будет характеризовать и тот случай,
когда примесь в исходном веществе не содержится, а вносится
в него только вследствие загрязняющего действия материала
аппаратуры.
§ 9 ОЦЕНКА ГЛУБИНЫ РЕКТИФИКАЦИОННОЙ
ОЧИСТКИ ТЕРМОНЕСТОЙКИХ ВЕЩЕСТВ
Приведенные выше соотношения, очевидно, должны быть
справедливы и в том случае, когда примесь проникает из внеш-
ней среды через колонну или образуется в результате протека-
ния в течение процесса ректификации той или иной химиче-
ской реакции, например реакции термического разложения
очищаемого вещества. При этом в соответствующие уравнения
вместо ип будет входить скорость проникновения примеси вслед-
ствие химической реакции, отнесенная к единице объема ко-
лонны. Это нетрудно показать на примере очистки термонестой-
ких веществ.
В общем случае для осуществления процесса глубокой очист-
ки термонестойких веществ требуются методы, исключающие
использование таких температур, при которых очищаемое ве-
щество разлагалось бы с заметной скоростью. Последнее обус-
79
ловливает возможность появления ощутимых количеств неже-
лательных примесей. Но если скорость разложения вещества
при его температуре кипения невелика, то для его очистки мо-
жет быть применена и ректификация, особенно если другие ме-
тоды малоэффективны, требуют больших затрат или вообще
неприменимы. Тем более, что температура кипения очищаемого
вещества, а следовательно, и температура процесса очистки
может быть снижена путем уменьшения давления в колонне.
Применительно к поставленной задаче рассмотрим ректи-
фикационную колонну, работающую в стационарном состоянии.
Пусть в процессе ректификации очищаемое вещество А частич-
но разлагается на вещества В, С,... и т. д. по реакции
А v1B+vi!C+..., (И. 112)
где vi=vb/va; V2 = vc/va; va, vb, vc — стехиометрические коэффи-
циенты.
Как уже отмечалось, в разбавленных растворах, с которыми
приходится иметь дело при глубокой очистке веществ, взаим-
ное влияние примесей можно считать незначительным. Следо-
вательно, задачу можно свести к рассмотрению бинарной систе-
мы, состоящей из основного вещества А и образующегося при
его разложении вещества В, одноименного с отделяемой при-
месью. В соответствии с этим запишем уравнение реакции
(11.112) в следующем виде.
A^=v1B+.. . (11.113)
Тогда скорость реакции распада очищаемого вещества при
предположении, что скорость обратной реакции незначительна,
выразится уравнением
dHA/dt=—k1ilA, (II.114)
где ЯА — концентрация компонента А в жидкой фазе в данном
элементе объема колонны (моль/см3) в некоторый момент вре-
мени; — константа скорости прямой реакции.
В соответствии с (11.114) скорость образования примеси В
(скорость поступления примеси в разделяемую смесь) .может
быть выражена соотношением
(1 - хА) (1 -х), (11.115)
где хА — степень разложения вещества А.
Так как паровой захват в колонне всегда значительно мень-
ше жидкостного захвата, то загрязнением вещества А за счет
его частичного разложения в паровой фазе можно пренебречь.
Также пренебрегаем изменениями величин жидкостного захва-
та и скорости потока жидкости в колонне за счет термического
разложения, поскольку рассматривается случай, когда хА<С1-
На основании вышеизложенных допущений и с учетом того,
80
что х<С1, дифференциальное уравнение, описывающее суммар-
ный процесс (маесообмен и термическое разложение) в эле-
менте объема колонны, и уравнение рабочей линии будут
иметь вид:
L-^- = k(y-ax} + vlkllf>fi (П.Н6>
QZ
И
Ix+vi/tiWiK (zK — z) = L (1 — p) y+Lpxp. (II.117>
Решая дифференциальное уравнение (11.116) после подстанов-
ки в него соотношения (11.117) методом, изложенным в преды-
дущем параграфе, с использованием принятых обозначений,
получаем выражение для фактора разделения применительно
к безотборному и отборному режимам соответственно:
^»=[1^7?=1Й“]Го+тЙ)7!ет‘ (ПЛ18)
L 1) Xq j (сс Xq hi г Q
и
Г v,k,T а(1 —р) (1 —m;1) —lnm„ n
(а‘-ГГ.. ]•
где Tm = li^zKIL — полное время прохождения потока жидкости
через колонну.
Сравнивая уравнения (11.118) и (11.119) с соответствующими
уравнениями (11.105) и (11.108), характеризующими влияние
загрязняющего действия материала ректификационной колон-
ны на глубину очистки, можно видеть, что они действительно по
форме идентичны. Это объясняется тем, что как в первом, так
и во втором случае скорость поступления примеси принималась
величиной постоянной, а содержание примеси в разделяемой
смеси по сравнению с содержанием основного очищаемого ве-
щества— пренебрежимо малым. Нетрудно видеть, что сюда
же следует отнести и важный для практики глубокой очистки
веществ случай загрязнения продукта примесью, образующейся
вследствие химической коррозии стенок и контактного устрой-
ства ректификационной колонны. Правда, понятие коррозии
в этом случае приобретает несколько иной смысл, поскольку
заметного разрушения материала колонны здесь не наблюда-
ется даже в течение длительного времени ее эксплуатации,что
обусловлено микроколичеством образующейся примеси. Прак-
тически здесь даже трудно провести различие между этим слу-
чаем и рассмотренным выше случаем «вымывания» примеси из
материала аппаратуры. Однако при установленном факте, что
ректификация сопровождается теми или иными химическими
превращениями, появляется возможность расчета такого про-
цесса хеморектификации исходя из заданных констант скоро-
стей соответствующих химических реакций.
81
§ 10 ПЕРИОДИЧЕСКАЯ РЕКТИФИКАЦИЯ
Выше были рассмотрены закономерности процесса глубокой
очистки веществ в непрерывно действующих колоннах. Однако
поскольку производство веществ особой чистоты в целом явля-
ется малотоннажным,»для их получения обычно используются
ректификационные колонны периодического действия. Харак-
терной особенностью таких колонн является то, что ректифика-
ции в них подвергается определенное, заданное количество ис-
ходной смеси. Перед началом процесса исходная смесь — за-
грузка— помещается в питающую емкость — куб, и колонна
вводится ® рабочий режим. По окончании процесса оставшаяся
в кубе жидкость удаляется, и операция повторяется со следую-
щей загрузкой. В зависимости от природы отделяемых приме-
сей и очищаемого вещества применяют колонны разных конст-
рукций, отличающиеся в основном расположением питающего
куба и соответственно способом отбора примеси или продукта.
Наиболее широкое применение на практике находят колон-
ны с нижним кубом, играющим одновременно и роль кипятиль-
ника (см. рис. 11). Колонны такой конструкции особенно удоб-
ны в случае, когда примесь является высококипящим компо-
нентом; в ходе процесса продукт отбирается в виде дистиллята,
а примесь концентрируется в кубовом остатке. В принципе эти
колонны могут быть использованы и в случае, когда примесью
является низкокипящий компонент: продуктом при этом будет
кубовый остаток. Но здесь имеет место большая вероятность
загрязнения продукта за счет его взаимодействия с материа-
лом аппаратуры (куба) или вследствие протекания химических
превращений, более характерных для высоких температур
(температура кипения), при которых в течение всего процесса
находится очищаемое вещество. Указанные причины возможно-
го загрязнения в значительной мере устранены в конструкциях
колонн с «верхним кубом»—питающим резервуаром, распо-
ложенным в верху колонны непосредственно за конденсатором
или совмещенным с ним. В таких колоннах, которые называют
колоннами с обратным питанием, отбор продукта (или кон-
центрата примеси) производится из зоны обращения фаз, иг-
рающей при этом роль испарителя (куб полного испарения).
Поскольку в очищаемом веществе содержится и низко-
м высококипящие примеси, процесс ректификационной очистки
иногда осуществляют в двухкубовой колонне. Ректификация
при этом происходит в две стадии. Загрузка помещается в ниж-
ний куб, затем колонна вводится в рабочий режим и из верх-
него куба (конденсатора) колонны производится отбор низко-
кипящих примесей до достижения заданного их содержания
в жидкости, находящейся в нижнем кубе, после чего процесс
прекращается. Затем жидкость из нижнего куба перегоняется
82
в верхний, колонна опять вводится в рабочий режим, и из ниж-
него куба, играющего при этом роль куба полного испарения,
производится отбор высококипящих примесей до < пределенной
степени чистоты конечного продукта, отводимого по окончании
второй стадии из верхнего куба.
Существенным недостатком такой двухстадийной ректифи-
кации является ее длительность С целью сокращения времени
ректификационной очистки предложены конструкции колонн со
«средним кубом», расположенным в средней части колонны
(рис. 20). Такое расположение питающего куба позволяет про-
изводить очистку вещества от высоко- и низкокипящих приме-
сей одновременно, т. е в ходе одностадийного процесса.
Отбор продукта илй концентрата примеси в процессе пе-
риодической ректификации приводит к тому, что и количество
и состав загрузки в течение рабочего цикла непрерывно изме-
няются. При этом соответственно изменяются также
жидкой и паровой фаз в колонне на любой ее тарел-
ке в случае тарельчатых колонн или в любом ее се-
чении в случае насадочных и пленочных колонн. Ука-
занная причина и обусловливает то, что при прове-
дении процесса периодической ректификации с по-
стоянной скоростью отбора продукта (постоянное
флегмовое число) состав последнего будет перемен-
ным. Соответственно для того, чтобы в ходе процесса
можно было отбирать продукт постоянного состава,
необходимо изменять скорость его отбора (перемен-
ное флегмовое число). Таким образом, при описании
Рис 20 Схема ректификационной колонны со «средним кубом»:
А — нижний куб (куб полного испарения); Bi, В?—верхняя и нижняя рек
тифицирующие части, С — конденсатор. Е — питающая емкость («сред-
ний куб»)
составы
процесса очистки и в том и в другом вариантах периодической
ректификации следует иметь в виду, что уравнение рабочей ли-
нии, характеризующее материальный баланс в колонне по по-
токам фаз и их составам, здесь уже не будет выражаться урав-
нением прямой линии, как в случае непрерывно действующих
колонн [см. уравнение (11.61)]. Вследствие этого расчеты пе-
риодической ректификации в целом являются более сложными
по сравнению с расчетами непрерывной ректификации
Хотя анализу периодической ректификации посвящено
сравнительно много исследований, достаточно строгие методы
ее расчета по сравнению с методами расчета непрерывной рек-
тификации до настоящего времени еще не разработаны Так,
большинство описаний аналитических зависимостей процесса
относится к случаю пренебрежимо малого жидкостного «захва-
83
та» в ректифицирующей части колонны по сравнению с коли-
чеством жидкости в питающем кубе. Обычно это условие вы-
полняется для промышленных установок. Но когда количество
загрузки относительно невелико, жидкостный захват в ректи-
фицирующей части колонны может оказывать влияние на выход
очищенного вещества,'его чистоту, а также на время проведе-
ния процесса.
С целью установления соответствующих зависимостей рас-
смотрим работу насадочной колонны с нижним питающим ку-
бом (см. рис. 11); полученные соотношения в целом будут
справедливы и для колонн других конструкций, кратко охарак-
теризованных выше. Пусть в начале работы колонны в ее кубе
находится No молей загрузки, в которой молярная доля выше-
кипящей примеси составляет х0- Для равномерного смачивания
насадки жидкостью колонна вначале обычно подвергается «за-
хлебыванию», после чего в ней устанавливается необходимый
тепловой режим, чтобы скорости потоков Ж1идкой и паровой фаз
по колонне были постоянными Избыток жидкости из ректифи-
цирующей части при этом стекает в куб; насадкой «захваты-
вается» (задерживается) лишь некоторое определенное коли-
чество жидкости. Величина жидкостного «захвата» (задержки)
зависит в основном от типа н поверхности насадки, а также от
скорости потоков жидкости и пара в колонне. Затем в течение
некоторого времени (пусковой период) колонна работает в без-
отборном режиме (режим полного орошения) до достижения
в ней стационарного состояния и лишь после этого включается
система отбора части дистиллята. Время пускового периода мо-
жет быть определено расчетным путем. Однако такая оценка
является весьма приближенной и поэтому время пускового пе-
риода определяется экспериментально. Как показали результа-
ты соответствующих исследований, время пускового периода
можно несколько снизить, если с самого начала процесса ко-
лонна будет работать в отборном режиме. Разумеется, отби-
раемый при этом дистиллят по своему составу не будет отве-
чать составу требуемого продукта вплоть до выхода колонны
к заданному стационарному состоянию, и его целесообразно во
избежание потерь исходного вещества отводить в питающий
куб. В результате будем иметь случай стабилизированной рек-
тификации, для которой справедливы закономерности, характе-
ризующие непрерывную ректификацию. Действительно, посколь-
ку при циркуляции жидкость — пар количество вещества в ко-
лонне не изменяется, по достижении стационарного состояния
будет постоянным и состав питания — образующегося в кубе
колонны пара. Совершенно очевидно, что пренебрегая, как
и выше, эффектом продольного перемешивания, уравнение ра-
бочей линии колонны, работающей в стационарном состоянии,
для рассматриваемого случая можно записать в виде
«4
y=(l—p)x+pxD,
(11.120)
где р—л/l— степень отбора; п—скорость отбора дистиллята;
Хи — молярная доля примеси в дистилляте.
По аналогии с (II 54), с учетом (11.59), дифференциальное
уравнение, характеризующее обеднение примесью паровой фазы
в элементарном объеме ректифицирующей части, т. е. при ус-
ловии, что у*=х)а, запишется как
(ПЛ2,)
UZ \ & /
Подставляя соотношение (11.120) в (11.121), после разделения
переменных получаем
______dy , Г. fc(a-l) , ,|т
(1-а©)у + ©zp - I(l-p)a dz- 1 ’
Решение (11.122) с граничными условиями
a)z=0, y=yD; (И123)
о) Z=z„, У=УК
приводит к выражению
(1 —а6)уу+вхр _ fc(ot —t) (1—а»)
(l-a©)yD4-©zD ' ta(l-p) ’ 1 ’ '
где ус, Уы — молярные доли примеси в паре, поступающем в
конденсатор и в ректифицирующую часть колонны соответ-
ственно.
Конденсат, образующийся из пара состава ус, имеет тот же
состав, т. е Хс=ус- Поднимающийся же из куба пар состава
Ун в общем случае отличается от состава xN остающейся кубо-
вой жидкости: в пределе это различие должно достигать вели-
чины а. Но в действительности равновесие между жидкостью
и паром в кубе колонны ввиду конечной скорости испарения не
достигается и поэтому для высокоэффективных колонн, исполь-
зуемых при глубокой очистке веществ, указанным различием
можно пренебречь. С учетом указанного допущения из (II 124)
после преобразования имеем
Г = — =-------!<~Итм Т-----. (И. 125)
где Р.-(^) =е,рГ- 1.
' xn ,'р=о L la д
Соотношение (11.125), характеризующее разделительную спо-
собность колонны периодического действия в процессе стаби-
лизированной ректификации, идентично ранее полученному
выражению (11.73)
Из соотношения (11.122) можно получить также выражение
для описания распределения примеси по высоте колонны, кон-
85
станту интегрирования при этом находят с учетом граничного
условия (II. 123а):
/ 1—аб \ z
1-р г«-е
У= UD--------^^0-----------. (И.126)
Подставив (II 126) в (II 120), получим уравнение, характери-
зующее распределение примеси в жидкой фазе по высоте ко-
лонны:
_/ I-а& \ z
1-Р ' -а©
------Г“^0-------' П127>
С помощью выражения (11.127) и с учетом соотношения
(11.125) нетрудно оценить концентрацию примеси х в жидкост-
ном «захвате» в зависимости от концентрации примеси в кубо-
вой жидкости:
х=х$пр, (11.128)
где пр= [F(l — a© In Fo)—1]/(1—ae)lnF0.
Ректификация с постоянной скоростью отбора продукта
В ходе осуществления этого варианта ректификационной
очистки при непрерывном отборе части дистиллята в виде про-
дукта будет изменяться и мгновенный состав дистиллята Ха и
состав кубовой жидкости Xn. С учетом того, что разделитель-
ная способность колонны при ректификации разбавленных рас-
творов не зависит от концентрации разделяемой смеси, величи-
ны Xd и xn при постоянной заданной скорости отбора продук-
та п в процессе такой квазистационарной ректификации будут
связаны соотношением (II 125) Совершенно очевидно, что и
средняя концентрация примеси в жидкостном «захвате» в тече
ние процесса также будет изменяться Это можно выразить че-
рез изменение величины Хы с помощью соотношения (11.128)
Таким образом, использование соотношений (11.125) и (II.128)
позволяет рассчитать кривую разгонки, которая обычно выра
жается в виде графической зависимости состава дистиллята от
доли отгона. С другой стороны, исходя из кривой разгонки,
нетрудно определить оптимальную скорость отбора продукта
или соответственно время проведения процесса, в которое, ра-
зумеется, не входит время пускового периода.
С этой целью, пренебрегая величиной парового «захвата»
в колонне по сравнению с величиной жидкостного «захвата»,
запишем уравнение материального баланса по примесному ком-
86
поненту для (некоторого момента времени t процесса ректифи-
кации:
NBx0 — Nxn+ Нх+ А^конд^о-Ь Ndxd, (II .129)
где N — количество (моль) кубовой жидкости к моменту вре-
мени /; Н = //Ж5гк — жидкостный «захват» ректифицирующей
части колонны; s — поперечное сечение колонны; М<онд — мгно-
венное количество жидкости в конденсаторе; Лго—количество
отобранного продукта в сборнике дистиллята к моменту време-
ни /; xD— молярная доля примеси в этом продукте. Используя
теорему о среднем значении, имеем
xjd=-~ J z£>dt, (П.130)
отсюда
t
st) ~ J zDdl=ns J xDdt. (II 131)
о о
Подставив (П.131) в (П.129), с учетом того, что N=N0—Н—
--^конд—ND=N0—Н—Л^конд—П st, получим
t
JVqZq = [Nbxn — I{(xn — х) — NKOHa(xN—xD) + astxN] + us J zodt. _ (П.132)
О
Решение уравнения (II. 132) определяется взаимосвязью значе-
ний Xd их со значением xN. Подставляя в уравнение (11.132)
Xd и х из уравнений (II 125) и (11.128), получаем
t
(1 — «1—«2—J ®wdt=z0,
о
(П 133)
где «I = (Н/No) пр, п2 = (^онд/Л^о) (I — F), п3=us/N0, п4=
= (ns/N0)F
Дифференцирование (11.133) дает
(1 — «j —n2—n3t) ---n3x.v4-n4zx = 0 (II.134)
или после разделения переменных,
dz 1 — nL — п2— n3t
Решая дифференциальное уравнение (II 135), получаем
X№const (! — «! — ns—n3t _|. (11.136)
87
После подстановки (П.136) в выражение (II 133) имеем
const=x0/(l—Hi—n2)f и, следовательно, соотношение (11.136)
запишется как
(1 —nt —rig—1
Z,v^»Zq ,, ,F
(1—«1 —
(II 137)
Уравнение (II 137) позволяет установить зависимость соста-
ва продукта от состава кубовой жидкости. Так, из (II 137) и
(11.125) следует, что
Г(1 —«! —и2 —nst)F 1
XD~ хо .р
(1 — «1 — пгу
(11.138)
Если теперь подставить соотношение (11.138) в выражение
(II 130), то можно получить уравнение кривой разгонки:
-Ч^)ЛНЛс2-У',Л- <'1,э9>
Уравнение (II 139) позволяет оценить концентрацию приме-
си в отобранном продукте в любой момент времени при извест-
ных параметрах процесса и величине Nd/N0- Если же, наоборот,
величина xd задана, то с помощью уравнения (11.139) можно
определить выход продукта, допустимую скорость отбора или
время процесса. Так как практически всегда Nkoh^No, то зна-
чением п2 в выражении (II 139) можно пренебречь. Учитывая,
что Nd/Nq= Vd/Vo, из выражения (II 139) получаем
Гр-Г1- ( 1 -"1-. (ц.140)
VD,V0 L \ 1—»1 / J
При H<C/V0, т е когда п^О, уравнение (11.140) упрощается
^“^['-(‘--кЛ- <П141>
При соблюдении указанных условий (п^О, п2—0) уравнение
(11.137) также преобразуется в более простое соотношение
(11.142)
которое по виду аналогично уравнению (11.256), характеризую-
щему релеевскую дистилляцию.
На рис. 21 приведены кривые разгонки, рассчитанные по
уравнениям (II 140) и (П.141) для различных значений а.
Из сравнения кривых следует, что наличие жидкостного «за-
хвата» в ректифицирующей части колонны приводит к сниже-
нию чистоты продукта и это необходимо иметь в виду при вы-
боре типа ректификационной колонны и условий ее работы. Од-
88
нако при небольшом жид-
костном «захвате» (Н<^
<C/Vo), его влиянием в пер-
вом приближении можно
пренебречь, что позволяете
ориентировочных расчетах
пользоваться соотношения-
ми (П.141) и (П.142).
В качестве примера рас-
смотрим часто используе-
мую на практике фракцио-
нированную ректификацию,
при которой отбор дистил-
лята производится фрак-
циями; в простейшем слу-
чае продуктом обычно яв-
ляется средняя фракция
дистиллята Применительно
к задаче глубокой очистки
примем, как и при анализе
процесса многократной пе-
регонки (см. § 4), что в очи-
щаемом веществе содержат-
ся и низко- и высококипя-
Рис. 21. Зависимость относительной кон-
центрации примеси в дистилляте от до-
ли его отгона в процессе ректификации
(F0=103; р-0.05):
/ — а—1,25; 3 — а = 15. 5 —а—2 (кривые /. 3,
5 рассчитаны по уравнению (11.140) для Н№—
—0.1); 2 —а—1.25. 4 —а —1.5; 6 — 0=2 (кривые
2 4 6 рассчитаны по уравнению (II 141)]
щие примеси. В процессе
фракционированной ректификации низкокипящая примесь будет
концентрироваться в дистилляте, поэтому некоторую началь-
ную часть дистиллята следует отбросить как «хвостовую»
фракцию. Концентрация xN^ интересующей примеси, например
яизкокипящей, в жидкости объемом V, находящейся при этом
в перегонном кубе, будет выражаться в соответствии с (П.142)
соотношением
*м>)=*о = z0 (l-V^o/Vo/d)-1, (II 143)
где х0—концентрация примеси в исходной загрузке; Кад—
объем первой «хвостовой» фракции; F(i)=Xo(i)/*jv(i); Xd(d—
мгновенная концентрация низкокипящей примеси в отгоняемой
«хвостовой» фракции.
После удаления первой «хвостовой» фракции в сборник дис-
тиллята отгоняется фракция (продукт). Отбор при этом целе-
сообразно производить с большей скоростью, поскольку основ-
ное количество низкокипящей примеси из очищаемого вещества
уже удалено с первой «хвостовой» фракцией. Если обозначить
объем фракции (продукта) через Ко(2), то, исходя из выражения
(П.141) , можно записать
ID<2) VjmIV С1 f1
(11.144)
89
или, после преобразований с учетом (И.143),
ZD(!)_ I'niil/l’o r Vo / L I Vd4)IV0 I J’
(II 145)
где ход—концентрация интересующей примеси в продукте;
F(2)=Xd(2)Ixn(2), Xd(2) И Хлг(2)—мгновенные концентрации примеси
в дистилляте и кубовой жидкости в данный момент времени
отбора продукта. Оставшаяся в перегонном кубе жидкость яв-
ляясь концентратом высококипящей примеси представляет со-
бой вторую «хвостовую» фракцию.
На рис. 22 представлены
Рис. 22. Зависимость относительной
концентрации примеси в продукте от
его выхода в процессе фракциониро-
ванной ректификации: а=2. p(i)=0,01,
Р<2>=0,1 [кривые рассчитаны по урав-
нению (11.145)]:
/ Vп(1)/Го"0 I и 2 12 для
Го—10’: 3 VD<1)/V0—0,12 и 4 — VD(I)/V0-
—0,1 для Fq — 10-’
примеры кривых зависимости
относительного содержания
примеси (кривые /, 2 для низ-
кокипящей, а кривые 3, 4 для
высококипящей) в продукте
от доли его отгона, рассчитан-
ные по уравнению (11.145).
Используемые в расчетах зна-
чения F(i) (при степени отбора
P(d) и F(2) (при степени отбора
р(21) вычисляли по уравнениям
(II 73) и (11.74) для низко- и
высококипящих примесей со-
ответственно, Из приведенных
кривых видно, что на чистоту
продукта влияет объем пер-
вой «хвостовой» фракции.
Причем это влияние в боль-
шей степени проявляется в от-
ношении содержания нижеки-
пящей примеси- с возрастани-
ем объема «хвостовой» фрак-
ции концентрация указанной
примеси в продукте уменьша-
ется Следует заметить, что ка-
чественно этот вывод вытека-
ет и из общих соображений
Ректификация при постоянном составе продукта
Вернемся к анализу процесса ректификации смеси основное
вещество — вышекипящая примесь; продукт отбирается в виде
дистиллята. В этом случае периодической ректификации урав-
нение материального баланса для некоторого момента времени
с учетом того, что состав отбираемого продукта в ходе процес-
са не изменяется, имеет вид
90
^0*0—(^ Н Л^КСНД----Nd)xN-] I Л^КОИД^о+^и^О (II 146
или
(1-—
^D==^o —--
^конд \ И - АККОНД
ТУо /lN+~7v~a: 7^~XD~Xt
XN~XD
11.147)
Так как скорость отбора продукта можно охарактеризовать со-
отношением nS = dWo/d/, то дифференцированием (11.147) по-
лучим выражение
nS= zN-zjD)» {[*« - 0" -^) ID“^] Т+
+’^-(z№z^)4t} ’ (11.148)
откуда с учетом того, что u=pl, следует
No l^-zD-(H/7V0)(z-zD)]dxN+(H/A'0)(zN-zD)dx
IS J p(xW-zD)»----------------’ (П •149)
^(иач)
где p — степень отбора продукта в заданный момент времени;
-Хл/(наЧ), Хл,(кои)—концентрации примесного компонента в кубовой
жидкости в начале и конце процесса соответственно.
При проведении ректификации рассматриваемым способом
сам процесс должен проводиться с переменной скоростью (сте-
пенью) отбора продукта для поддержания постоянства его со-
става. Поэтому, строго говоря, уравнение (II.128), характери-
зующее взаимосвязь между мгновенными составами кубовой
жидкости xn и жидкостного «захвата» в ректифицирующей ча-
сти колонны х в случае постоянной скорости отбора продукта,
применительно к рассматриваемому случаю будет недействи-
тельным. Однако расчеты показывают, что входящая в это вы-
ражение величина пр в интервале допустимых степеней отбо-
ра р изменяется мало. Так, например, при а=2 и Fo= Ю-3
в интервале р = 0,1—0,005 значение пр изменяется от ~0,18 до
~0,14. Это позволяет использовать уравнение (11.128) для
приближенного выражения х через xN в соотношении (11.149).
Таким образом можно записать, что
(11.150)
Подставляя (II.128) и (11.150) в соотношение (11.149), по-
лучаем
ЛГ0 ’Мною 1о_[1_(Н/ЛГо)(1_п^]>с
IS J
ж?Пнач)
(П.151)
9
Когда жидкостным «захватом» ректифицирующей части колон-
ны можно пренебречь, уравнение (П.151) переходит в извест-
ное уравнение Богарта:
x2V<koh)
_ —др) Г
is J p(*№*d)2 ’
*Мнач)
(И.152)
Уравнения (11.151) и (П.152) позволяют определить время
проведения процесса, например, графическим интегрировани-
ем. С этой целью при известном значении р, соответствующем
началу процесса, когда в колонне уже установился режим ста-
билизированной ректификации, с помощью уравнения (11.125)
задается состав кубовой жидкости х^наЧ) (или, наоборот, при
заданном значении х^нач, находится начальное значение р)
Рассчитав по уравнению (II 125) для ряда последовательно
уменьшающихся значений р соответствующие значения х^
(хо — заданный состав продукта остается постоянным), нетруд-
но построить кривую зависимости подынтегральной функции в
(11.152) от Xn. Площадь под кривой в интервале от х^нач) до
Xw(koh) и предынтегральный множитель дадут искомую величи-
ну t. Сравнение обоих рассмотренных вариантов периодической
ректификации (без учета жидкостного «захвата» в ректифици-
рующей части колонны) при получении продукта одинакового
состава с одинаковым выходом показывает, что осуществление
процесса с постоянной скоростью отбора продукта требует не-
сколько большего времени. Тем не менее на практике этот ва-
риант по существу лишь и используется, что объясняется его
большей технической простотой по отношению к варианту с
переменной скоростью отбора.
Ректификация с дискретным отбором дистиллята
Интересным вариантом периодической ректификации явля-
ется способ, в котором безотборный режим работы колонны
чередуется с периодическим отбором продукта. Отбор при этом
производится из соответствующей емкости («кармана»), свя-
занной с ректифицирующей частью и расположенной выше или
ниже ее, в противоположном от питающего куба конце. Вмести-
мость «кармана» должна быть невелика по сравнению с вмести-
мостью питающего куба, в противном случае будет возрастать
время достижения стационарного состояния, по истечении кото-
рого производится отбор. Для полноты извлечения интересую-
щего вещества цикл такой операции можно повторить несколько
раз в ходе процесса.
Указанный вариант периодической ректификации может
быть использован также для концентрирования примеси и от-
92
бора концентрата. По окончании процесса из питающего куба
отводится продукт. Рассмотрим работу колонны с нижним пи-
тающим кубом. Пусть исходная загрузка составляет No молей,
в которой мольная доля прнмеси (низкокнпящей) равна х0.
После достижения стационарного состояния в колонне, работаю-
щей в безотборном режиме, будет справедливо следующее
уравнение материального баланса (первый цикл):
7VOZO = ^(1)XN^) Влг<^Т-^К0НД1О(1)> И 153)
где М(1), xW(1)—число молей жидкости в кубе и ее состав соот-
ветственно, Н, х<1)—полный жидкостный захват ректифицирую-
щей части и его средний состав; ЛгКонд, Хдщ—число молей жид-
кости в «кармане» конденсатора и ее состав.
В соответствии с уравнениями (11.125) и (II 128) при р=0
и с учетом того, что в паровой фазе концентрируется низкоки-
пящий компонент (примесь), можно записать, что
(11.154)
Fp-1
ln£o •
Так как A\d=7V0—П—Л^коил, то, подставляя
(П 154) — (И 155) в уравнение (II 153), получаем
_____N ого__
— Л^кокд ’
где
^JV(1) = ^0+ Н ( In £ I + ^О^КОНД-
(11.155)
выражения
(П.156)
Первый цикл заканчивается сливом содержимого «кармана»
конденсатора в сборник дистиллята.
В начале второго цикла идет простая перегонка до запол-
нения «кармана» конденсатора жидкостью, после чего в колон-
не, работающей в безотборном режиме, достигается новое ста-
ционарное состояние. Соответствующее уравнение нового ма-
териального баланса будет иметь вид
^охо~ ^(аИ-Л"^) 4" Д'ГО(2) 4 ^конд1^!, (П.157)
или, с учетом выражений (11.154) и (II 155),
г — 1
Л^О ~ (-^0 Н — 21УКопд) Z»V(2) 4" В jn у— xN(2) 4"
4" ^КОНД^?01Л’(2) 4* NКОНД^0^(1)- (11.158)
93
После преобразований (П.158) имеем
Л'вх0 — N кондТozN(i}
^(1)- 2Лконд • (П159)
Нетрудно показать, что
_ TVqIq —/Уконд^о (zn(1)4-J~N(2))
*(3) ^(„-ЗЛГконд
и, следовательно,
т -1
NBXB— -/Уконд^о 2 Z^(i)
------------------------------ЧгХ...............- (П161)
где т — число циклов.
Таким образом, последовательным расчетом по формулам
(11.156), (П 159), (11.160) и т. д нетрудно оценить содержание
примеси в кубовом продукте после каждого цикла.
В интересующем нас случае отбора концентрата примеси
Л'коид^СЛ'о, и поэтому величиной (/пЛ'конд) по сравнению с вели-
чиной можно пренебречь. В результате уравнение (11.161)
преобразуется к виду
771-1
Zjv(m) — ZN(1> —9л'(1) 2 1Л'(«)’ (П.162)
где =Лгкоид/:'о//?у(1). Из выражения (11.162) следует, что
Zjv(2>=z','rii> У‘',г( О1 v(i) 9n(o)> (Ч 1 >3)
lN(3)= 9iv(nTA'(t) — Ул’(|)1;7(2) =
= Ж^1)Н —9W(i) — 9.V(O (!—9.V(1))I XN{1) (! —9N(1)P> (П 164)
= XN(и —’ 9W( ,) — QN(t)xX(2) — !«( , )XN(3) =
— ZW(I) U—GiVu) — 9v(1)(l— УЛ’И))2) =iN(d(I — 9JV(o)3- (11.165)
На основании соотношений (11.163) — (11.165) выражение
(11.162) можно записать в виде
I^(m) = a'iv(l) (1 УА'(1)>т 1 (11.166)
или, с учетом выражений для
~~ Л^о + Н [Fo -l)/ln°/0-11 + ТУдонд^о ?
fl Лкокд^о -.m-l
I Af0 + H[F0-l)/lnF0-ll + JVKOKnF0 J • ЩЛ0°
Полученные выражения будут справедливы и для случая
очистки вещества от высококипящей примеси в колонне с верх-
94
ним кубом. Они могут быть использованы также для оценки со-
става продукта при ректификационной очистке вещества с ку-
бом в средней части.
Необходимо иметь в виду, что при осуществлении рассчиты-
ваемого варианта ректификации общее время работы колонны
относительно времени работы ее по предыдущим вариантам
будет больше, так как по окончании каждого цикла в колонне
должно достигаться стационарное состояние или хотя бы близ-
кое к нему.
§ 11 ВЛИЯНИЕ СПОСОБА ОТБОРА
КОНЦЕНТРАТА ПРИМЕСИ
НА ЭФФЕКТИВНОСТЬ ПРОЦЕССА РЕКТИФИКАЦИИ
Выше, при анализе процесса периодической ректификации
с дискретным отбором концентрата примеси отмечалось, что
этот вариант может быть использован для извлечения примеси
из очищаемого вещества; продуктом при этом будет кубовая
жидкость, например в колонне со средним кубом. Но примесь
из очищаемого вещества можно извлекать и непрерывным отбо-
ром ее концентрата из ректификационной колонны. В связи
с этим представляет интерес сравнение эффективности процесса
ректификации, достигаемой в каждом из указанных способов.
Поскольку важнейшей характеристикой эффективности процес-
са является его движущая сила (или выражаемая через нее
скорость межфазового массообмена), поставленная задача сво-
дится к расчету движущей силы для непрерывного и дискретно-
го способов отбора концентрата примеси с последующим срав-
нением полученных величин.
Рассмотрим непрерывную ректификацию бинарной системы
основное вещество — низкокипящая примесь с содержанием по-
следней хо- Пусть питание колонны поступает снизу в виде пара
состава уо^хо (например, из среднего куба большой емкости),
а сверху, из конденсатора колонны отбирается обогащенный
примесью дистиллят с содержанием примеси хо=уо- В этом
случае уравнение рабочей линии колонны (уравнение мате-
риального баланса по составам фаз в заданном сечении колон
ны, работающей в стационарном состоянии с постоянной ско-
ростью отбора дистиллята) представляет собой соотношение
(11.120). Подставив выражение для х из (П.120) в уравнение
(П.89), с учетом того, что в рассматриваемом случае у*=ах,
получим
(П.168)
или, после разделения переменных,
______dy_______fc(g—1) , „ ,RQ.
(1+е)У-аеУс ~ z(i-p) dz- <п 169)
95
Решение дифференциального уравнения (11.169) при граничном
условии z=0, у—уо дает
1 + 0 \ Z
,-₽Ьк + ае
У—Уо--------""1'-|1 ё---’ (П.170)
где F°=(#Lo^(v)P=o = eX₽ На-,)2г] -Факт°Р раз-
деления колонны в безотборном режиме.
Из выражений (П.170) и (11.120) будем иметь
/i+e \ z
zK+e]/(l+6)- (П.171)
Следует заметить, что при использовании принятого граничного
условия z=zK, у=уо, из (11.170) получим
1 в
У о=Уо------—j------. (И. 172)
П-рИц-^+ае
Выражение (П.172), как и следовало ожидать, идентично вы-
ражению (П.74), характеризующему разделительную способ-
ность ректификационной колонны в отборном режиме с обрат-
ным питанием применительно к концентрированию примеси
в жидкой фазе (примесь — вышекипящий компонент).
Соотношения (П.170) — (П.172) позволяют получить выра-
жение для средней движущей силы процесса ректификации Дун
при постоянной (непрерывной) скорости отбора дистиллята:
*к
1 Г
ДУн = (у — У*)сР=-“— \ (у — ax)dz. (11.173)
ZK J
0
Подстановка (П.170) и (П.171) в (П.173) дает
Лун=
Ур (1—а—р)
zK (1 + 6)
гк
J p-U‘+e)/(i-p)f/lKdz,
о
(П.174)
После интегрирования уравнения (П.174) и использования
(П.172) получим
д„ ц»(°-1) л;(1+е)/(1“р)-1 '
J" lnF0 Г-И+6>/<‘-Р)+ое/(1-р)
(11.175)
В случае дискретного отбора дистиллята в интервале между
двумя последовательными отводами концентрата примеси рек-
тификационная колонна работает в безотборном режиме (р = 0)
и в нестационарном состоянии. По достижении в колонне ста-
•96
пленарного состояния перед удалением очередной порции кон-
центрата примеси ее распределение в жидкой фазе по высоте
колонны будет выражаться в соответствии с соотношением
(11.171) уравнением
z^xdF~Z,^= z0F^Z,Zk\ (11.176)
Следовательно, исходя из принципа геометрического подобия
стационарного и нестационарного профилей концентрации при-
меси в жидкой фазе, приближенно можно записать, что
x=x0F^~z,z"\ (11.177)
где Fq=xdIxo — текущее значение фактора разделения, xd—
содержание примеси в дистилляте (концентрате), образующем-
ся в момент времени /. Такой подход дает возможность пред-
ставить соотношение (II 168) при р=0 применительно к неста-
ционарному процессу в виде
I dy/dz—k(y—ax)
(П.178)
или, с учетом (П.177),
dy/dz—7>i!/ = ^i (z), ' 1-179)
ГДС !/=y(z, t); О‘> pi=-r-> 0i(0=—-р- го' ^K>-
I *
Решение дифференциального уравнения (П 179) с граничным
условием у=уо = хо при z=zK после несложных преобразований
(см. § 8) дает следующее выражение для у:
Z
У—--------------------- аГ0(1п70И0
(а — 1) Info-pin Fo
, (а—1) (In Fo—In Fo) TT ) 1
Fi/(a-i) f o
о J
Из (П 180) и (11.177) следует, что
Г 2
У-ах------------Цг-------- аГ0 (In Fo-1) Fo г« +
(<z — 1) In F04-ln/0 L
, (g—l)(lnF0 —InF0)
’ £i/(tx-l) f0
(11.180)
(II .181)
Отсюда нетрудно получить выражение для средней движущей
силы процесса ректификации Ду в некоторый момент времени
нестационарной работы колонны:
4—46
97
к
~ 1 Г ~ ~
Ау=~ \ (у—ax)az=
•к J
о
Vo(n—1) Г a Fo—1
(a-l)lnFo + lnFc La-1 InF0 (In F«~I) +
+Ja-l)(lnF.-lnF^ (1_f-l/(a-t))J, (П.182)
В период нестационарной работы колонны движущая сила
изменяется, поэтому необходимо произвести ее усреднение и по
интервалу времени между двумя последовательными отводами
концентрата примеси. Следовательно, выражение для средней
движущей силы процесса ректификации при дискретном отборе
дистиллята Ауд запишется как
t2
&Уя $ д^‘
<1
(П.183)
или, с использованием выражения (11.182),
a (Fo-1) (In Fo-1) In F0+(a— 1) (In Fo In Fo) (1 — F“,/(а" *’) In Fo
X—--------------------------—---------—-------------------dr.
[(a— 1) In Fo+ In Fo| In Fo
(11.184)
Подынтегральное выражение в (11.184) через элементарные
функции не выражается и поэтому для нахождения Дуд сле-
дует использовать численные методы расчета.
В соотношении (11.184) верхний предел интегрирования t2
характеризует собой момент времени отбора концентрата при-
меси. Фактор разделения колонны при этом будет составлять
Fo(tz). После «мгновенного» удаления концентрата примеси и
заполнения емкости для концентрирования в конденсаторе ко-
лонны новой порцией конденсата фактор разделения колонны
уменьшится до значения Fo(Л). Это значение будет равно тому
значению фактора, которое достигалось бы в колонне, если бы
колонна с самого начала процесса проработала в нестационар-
ном состоянии и безотборном режиме время Л. Следовательно,
если принять, что «восстановление» значения фактора разделе-
ния после отбора концентрата примеси до первоначального зна-
чения Fo(ti) происходит в соответствии с ходом кинетической
кривой первичного выхода колонны к стационарному состоянию,
то нижний предел интегрирования /, в (П.184) будет равен
Fo(ti) на расчетной кинетической кривой FG~t* Причем, исхо-
* Девятых Г Г., Еллиев Ю. Е. Введение в теорию глубокой очистки ве-
ществ. М, 1981, с. 320.
98
ля из используемой модели идеального вытеснения и принципа
геометрического подобия кинетических кривых, можно записать,
что
г* (G) = F9 («2)(Vh Гконд>/ h . (П.185)
где Vh — объем жидкостного «захвата» в ректифицирующей час-
ти колонны; Иконд — объем отбираемого концентрата примеси
из конденсатора колонны за один цикл.
По уравнениям (11.175) и (П.184) можно определить об-
ласть значений параметров Fo, а и Р, при которых следует про-
водить непрерывный или дискретный отбор дистиллята, пред-
ставляющего собой концентрат примеси. Ясно, что, когда Дун =
= &Ул., преимущества в эффективности ни у первого, ни у вто-
рого способов нет. При тех же значениях параметров Fo, а и Р,
когда Аг/д>Дуц, отбор дистиллята целесообразно осуществлять
дискретно, так как в этом случае эффективность процесса будет
выше. С использованием современной вычислительной техники
проведение соответствующих расчетов не представляет большо-
го труда.
§ 12. РЕКТИФИКАЦИЯ ПОД ПОНИЖЕННЫМ ДАВЛЕНИЕМ
Для успешного осуществления процесса ректификационной
очистки вещества необходимо, чтобы это вещество могло вы-
держать длительное кипячение без заметного разложения. Если
термическая стойкость очищаемого вещества недостаточно вы-
сока, то ректификацию можно проводить под пониженным дав-
лением, поскольку при этом температура кипения разделяемой
смеси основное вещество — примесь будет ниже, как это следует
из зависимости давления насыщенного пара от температуры,
определяемой уравнением Клаузиуса — Клапейрона:
d In Р ЬН
йТ ~ ПТг ’
(11.186)
где Р — давление насыщенного пара; Д77— энтальпия испаре-
ния вещества при температуре Т.
Учитывая, что при кипении под атмосферным давлением
давление насыщенного пара должно быть равно внешнему дав-
лению, и пренебрегая зависимостью Д// от температуры, из
уравнения (П.186) получаем
Р ЬН I 1 1 \
106 — R ( Т То I
или
Т= ДЯ/70 —Н In (105/Р) ’ (ПЛ*
где То—нормальная температура кипения вещества; Т — тем
пература кипения при давлении Р
4*
99
Из уравнения (11.187) видно, что температура кипения мо-
жет быть существенно понижена путем уменьшения давления
над кипящей жидкостью Например, когда нормальная темпе-
ратура кипения жидкости 70 = 500 К (227°С) и, согласно пра-
вилу Трутона, (—&Н/Т0)~88, то из уравнения (11.187) следует,
что если (105/Р) = 1(Р, то 7~300 К (27°C). Разумеется, в дей-
ствительности, хотя бы потому, что величина ДЯ зависит от
температуры, а правило Трутона является приближенным, тем-
пература кипения жидкости будет несколько отличной от рас-
четной величины. Однако такие оценочные расчеты несомненно
представляют интерес, так как позволяют судить о целесообраз-
ности проведения дистилляции под пониженным давлением
в том или ином конкретном случае.
С понижением температуры коэффициент разделения в дис-
тилляционных методах почти всегда повышается. Кроме того,
при пониженном давлении частичная конденсация может быть
избирательной, так как при этом наблюдается высокая скорость
диффузии, обеспечивающая более свободный доступ молекул
примеси к поверхности конденсатора. Поэтому и в кубе и в кон-
денсаторе ректификационной колонны даже при работе с отбо-
ром продукта может иметь место некоторый эффект разделения.
При обычной же ректификации такое разделение по существу
имеет место лишь в кубе колонны. Следовательно, при прове-
дении дистилляционных процессов под пониженным давлением
можно ожидать не только уменьшения вероятности термическо-
го разложения перегоняемой жидкости, но и более высокого эф-
фекта разделения. Следует, однако, иметь в виду, что при
уменьшении давления в колонне изменяются и другие парамет-
ры процесса, такие, как скорости потоков фаз, их количества
и т. д.; это ведет, как показали экспериментальные исследова-
ния, к увеличению ВЭТТ (ВЕП). Таким образом, с понижением
давления в колонне возрастают и коэффициент разделения а и
ВЭТТ. При понижении давления в области давлений, близких
к атмосферному, преобладающим является рост коэффициента
разделения; в результате разделительная способность колонны
увеличивается. По достижении некоторого давления эффект
возрастания ВЗТТ начинает преобладать над эффектом воз-
растания а: разделительная способность колонны начинает
уменьшаться. Следовательно, в каждом конкретном случае раз-
деления той или иной смеси должно иметь место оптимальное
значение давления, при котором в колонне достигается наиболь-
ший фактор разделения.
Понятие оптимального давления позволяет объяснить имею-
щиеся в литературе противоречивые на первый взгляд резуль-
таты, полученные некоторыми исследователями. В отдельных
опытах было найдено, что в некотором интервале изменения
давления эффективность колонны при уменьшении в ней давле-
100
Рис. 23. Схема установки
для молекулярной дистил
ляции
ния практически не изменяется, растет или уменьшается, эти
результаты в целом находятся в согласии с отмеченной выше
зависимостью, имеющей экстремум. Существование оптимально-
го давления позволяет сделать правильный выбор рабочего дав-
ления в каждом интересующем случае ректификационной
очистки.
К сожалению, это не распространяется на ректификацию под
вакуумом (при давлениях ниже 103 Па) ввиду следующих при-
чин. При понижении давления в колонне увеличивается ско-
рость диффузии в паре, так как коэффициент диффузии в газах
обратно пропорционален давлению. Это вызывает улучшение
переноса примеси в паровой фазе Отсюда следует, что, начиная
с некоторого давления, скорость массообмена в ректификацион-
ной колонне будет лимитироваться
диффузией в жидкой фазе и даль-
нейшее уменьшение давления не
будет увеличивать скорость массо-
обмена. Одновременно при пониже-
нии давления увеличивается ско-
рость диффузии в паровой фазе
вдоль оси колонны. В соответствии
с этим вертикальный градиент кон-
центрации в паровой фазе колон-
ны падает и разделение смеси ухуд-
шается. Далее, при понижении дав-
ления в колонне возрастает также
линейная скорость движения пара,
что приводит к резкому увеличению
перепада давления между кубом и
конденсатором колонны, вследствие
чего в кубе не удается поддержи-
вать низкое давление. В результате
лении ниже (1—2) -103 Па обычно становится неэффективной.
§ 13. МОЛЕКУЛЯРНАЯ дистилляция
Очистку веществ, не выдерживающих длительного кипячения
при остаточном давлении ~ (1—1,5) -103 Па, иногда осущест-
вляют дистилляцией и в высоком вакууме, которую обычно на-
зывают молекулярной дистилляцией. При молекулярной дистил-
ляции (рис. 23) равновесие между жидкостью и паром отсутст-
вует. Установлению равновесия между жидкостью и испарив-
шимися молекулами мешает конденсация последних. Можно
считать, что в идеальном случае молекулы испаряющейся жид-
кости проходят расстояние от поверхности жидкости до поверх-
ности конденсатора без столкновения друг с другом или с мо-
лекулами остаточных газов (вследствие столкновения они могут
не попасть в конденсатор) и полностью конденсируются; кон
ректификация при дав
101
денсат отводится в виде продукта. Одним из условий приближе-
ния к этому допущению является создание и поддержание
в разделительном аппарате такого давления, чтобы средняя
длина свободного пробега испарившихся молекул была больше
расстояния между поверхностью испарения и поверхностью кон-
денсации. Вследствие' этого молекулярную дистилляцию обычно
проводят при остаточном давлении —10 1—10~2 Па.
Для оценки эффекта разделения в процессе молекулярной
дистилляции пользуются коэффициентом разделения ам, кото-
рый определяется следующим соотношением:
где Хконд — молярная доля примеси в конденсате; х— молярная
доля примеси в испаряющейся жидкости соответственно в дан-
ный момент времени.
Так как скорость конденсации в процессе молекулярной дис-
тилляции пропорциональна скорости испарения, то полагая, что
при давлении ~10 ’—10~2 Па в дистилляционном пространстве
скорость испарения описывается формулой (II.26) —уравнением
Лэнгмюра, имеем
ЬКОнд=^Р// 2п.МПТ, (11.189)
где Тконд — скорость конденсации вещества, моль/ (см2 • с);
£ — коэффициент пропорциональности, характеризующий по-
правку на столкновение испарившихся молекул с молекулами
остаточных газов, который может быть назван, как и аналогич-
ный коэффициент в уравнении (II.27), коэффициентом аккомо-
дации.
Коэффициент аккомодации показывает, какая доля испарив-
шихся молекул перейдет в конденсат и зависит от остаточного
давления в дистилляционном пространстве, чем ниже остаточ-
ное давление, тем значение £ ближе к единице. Далее, £ зави-
сит от чистоты (г е степени смачиваемости) поверхности кон-
денсатора, который в процессе перегонки обычно покрывается
пленкой жидкого конденсата. Когда поверхность конденсатора
является чистой, коэффициент аккомодации также приближает-
ся к единице.
Поскольку содержание примеси и основного вещества
в жидкой пленке конденсата в данный момент перегонки будет
пропорциональным их соответствующим скоростям конденсации,
то можно записать, что
хКОНд/(1 —гКОНд) — ^конд/£конд- (11.190)
где Ь'конд и £"Конд — скорости конденсации основного вещества
и примеси соответственно.
102
H’iSi И ”2
основного вещества и примеси
у2—коэффициенты активности
Подставляя соотношение (11.189) в (11.190), получаем
жкоид.'('1—^нонд)— (EgP: ^1Р1) 01 191)
где pt, М), gj и р2, Л12, £2 — парциальное давление пара, относи-
тельная молекулярная масса и коэффициент аккомодации основ-
ного вещества и примеси. Так как pi=pi°(l— х)у, и р2 = Рг°-хуг,
то с учетом этих соотношений из (11.191) имеем
жконд / * _ f Mt /n
1 з'конд । 1 х
где Р\° и Р2°— давление пара
при температуре испарения; и
основного вещества и примеси.
Легко заметить, что P20^2/Pt0tt — не что иное, как коэф-
фициент разделения данной смеси при обычной дистилляции
Учитывая это, а также то, что отношение как правило,
мало отличается от единицы, и используя соотношение (II 188),
уравнение (11.192) можно записать в виде
ад/ ~ ^конд/^ — а 11.193)
Из уравнения (11.193) видно, что коэффициент разделения
пропорционален корню квадратному из отношения молекуляр-
ных масс компонентов. Поскольку обычно при Л1|>Л12 а>1
(и, наоборот, при Mt<M2 а<1), разделение, достигаемое при
молекулярной дистилляции, как правило, выше, чем при обыч-
ной перегонке.
Области применения молекулярной дистилляции весьма раз-
нообразны. Этим методом проводят очистку термонестойких или
высококипящих веществ с молекулярной массой 250—1200 (по-
лучение масла для вакуумных насосов и смазочных масел
с незначительным температурным изменением вязкости, очистка
пластификаторов, приготовление витаминов и т. д.). Молекуляр-
ной дистилляцией могут быть разделены изотопные смеси,
а также вещества с одинаковыми парциальными давлениями
паров при температуре разгонки, но с различными относитель-
ными молекулярными массами. Например, молекулярной дис-
тилляцией, как это следует из уравнения (11.193), можно раз-
делять и азеотропные смеси, для которых а=1.
Известно много конструкций аппаратов для осуществления
молекулярной дистилляцией, большинство из которых предназ-
начено для проведения процесса в виде однократного варианта.
Применительно к глубокой очистке веществ разделение, дости-
гаемое при однократном процессе, обычно все же недостаточно.
Поэтому и здесь стремятся к осуществлению процесса с исполь-
зованием принципа противотока фаз и их обращения на концах
соответствующего аппарата колонного типа, как это имеет мес-
то в ректификации.
103
Рис 24 Схема разделительной части
многоступенчатого аппарата для проти-
воточной молекулярной дистилляции
Tt Схема массообменного
устройства одного из таких
колонных аппаратов для
противоточной молекуляр
ной дистилляции «молеку-
лярной ректификации»),
представлена на рис. 24
Оно состоит из ряда чере-
дующихся секций. Образу-
ющийся из жидкости пар
попадает на лопасть конденсатора, конец которой про-
ходит за перегородку следующей секции; конденсат стекает
в жидкость этой секции Избыток жидкости в секции через пе-
регородку стекает в предыдущую секцию. Таким образом в ко-
лонне имеет место противоток пара и жидкости и, следовательно,
основные уравнения, описывающие работу ректификацион-
ных колонн, будут справедливы и для многоступенчатых аппа-
ратов молекулярной дистилляции, т. е. закономерности обыч-
ной ректификации могут быть использованы для расчета проти-
воточной молекулярной дистилляции, если вместо коэффициента
а в соответствующих уравнениях использовать коэффициент сои
ГЛАВА III
КРИСТАЛЛИЗАЦИОННЫЕ МЕТОДЫ
В основе кристаллизационных методов разделения смесей
лежит различие в составах жидкостей (расплав или раствор) и
образующейся из нее твердой фазы (кристаллы). Это различие
максимально, когда жидкая и твердая фазы находятся в термо-
динамическом равновесии Часто оно оказывается существенно
выше, чем различие в составах той же жидкости (расплав) и
равновесного с ней пара. В таких случаях кристаллизационные
методы очистки являются в принципе более предпочтительны-
ми, чем дистилляционные. К достоинствам кристаллизационных
методов следует отнести более низкую температуру процесса
кристаллизации по сравнению с температурой процесса дистил-
ляции. Это особенно важно при очистке термонестойких веществ
и для снижения загрязняющего действия материала аппарату-
ры. Преимуществом кристаллизационных методов очистки явля-
ется также то, что они требуют меньших затрат энергии, чем
дистилляционные методы, так как теплота плавления вещества
существенно ниже теплоты его испарения.
Хотя любая разделяемая смесь является многокомпонентной,
применительно к глубокой очистке веществ оценку эффекта раз-
деления в кристаллизационных методах, как и в дистилляцион-
ных, можно свести к рассмотрению бинарной смеси основное ве-
104
щество—примесь. При кристаллизации веществ из раствора
третий компонент — растворитель — играет роль среды или
сольватирующего агента. Образующиеся при этом сольваты
можно рассматривать как индивидуальные вещества.
Для глубокой очистки веществ кристаллизация из раствора
применяется реже, чем кристаллизация из расплава, поскольку
растворитель всегда загрязняет очищаемое вещество Поэтому
методы очистки веществ кристаллизацией из раствора будут
рассмотрены менее подробно.
§ 1 КРИСТАЛЛИЗАЦИЯ ИЗ РАСПЛАВА
При кристаллизации из расплава различие между соотно-
шением компонентов в жидкой и равновесной с ней твердой фа-
зах можно изобразить графически в виде диаграммы состояния
(диаграммы плавкости). Некоторые типичные формы диаграмм
плавкости приведены на рис 25, 26, 27.
Рис 25. Диаграмма плавкости для
системы образующей непрерыв-
ный ряд твердых растворов
Рис. 26. Диаграмма плавкости для
системы с ограничеииой растворимо-
стью компонентов
На рис. 25 линия у\Х} характеризует составы жидкой и твер-
дой фаз, которые находятся в равновесии при температуре Т\.
В данном примере концентрация компонента В в твердой фазе
ниже чем в жидкой. Если расплав хорошо перемешивается и
скорость диффузии в твердой фазе достаточна для выравнива-
ния состава кристаллов, а охлаждение расплава проводится так
медленно, что равновесие между фазами поддерживается на
протяжении всего процесса охлаждения, например от темпера-
туры Т, до температуры Т2, то у2 и х2 будут обозначать равно-
весные концентрации компонента В в жидкой и твердой фазах
при указанной температуре. По достижении температуры 7\
весь расплав закристаллизуется и, следовательно, средний со-
105
став твердой фазы будет таким же, как и состав исходного рас-
плава, т. е. концентрация компонента В в затвердевшем рас-
плаве будет равна у{.
Если компоненты только частотно смешиваются в твердом
состоянии, то диаграмма плавкости имеет вид, изображенный
на рис 26. Многие Ойнарные системы имеют диаграмму плав-
кости вида, изображенного на рис. 27. Однако в реальных слу-
чаях и эти системы при преобладающем содержании одного из
компонентов, т. е. когда молярная (атомная) доля одного из
компонентов в смеси близка к единице, имеют диаграмму плав-
кости, соответствующую правой или левой части рис. 26. Это
обусловлено тем, что в процессе кристаллизации часть примеси
захватывается кристаллами основного вещества вследствие об-
разования смешенных кристаллов (твердых растворов) или
вследствие адсорбции примесных молекул на гранях растущих
кристаллов.
Зарождение и рост кристаллов
Рассмотрим процесс кристаллизации расплава индивидуаль-
ного вещества, пренебрегая содержащимися в нем примесями.
При охлаждении расплава до температуры плавления соот-
ветствующего ему твердого вещества в нем возникают флуктуа-
ции плотности, которые представляют собой относительно боль-
шие скопления частиц (молекул, атомов или ионов) вещества
с ориентированным расположением, приближенно подобно тому,
как это имеет место в кристаллической решетке Такие скопле-
ния можно рассматривать как некие комплексы, агрегаты или
ассоциаты; их иногда называют дозародышевыми образования-
ми. Но они еще не являются стабильными образованиями- число
частиц в них вследствие теплового движения в расплаве раз-
лично и не постоянно. Сталкиваясь друг с другом, такие конфи-
гурации групп частиц могут укрупняться или распадаться в за-
висимости от соотношения действующих в них межмолекуляр-
ных сил и воздействия на эти частицы молекул расплава. При
дальнейшем понижении температуры расплава, т. е. при его
переохлаждении, преобладающее влияние будет проявлять пер-
вый из указанных эффектов. Размеры образований при этом
в целом будут увеличиваться до некоторой критической величи-
ны. В результате в расплаве начинается образование зароды-
шей кристаллов («критических кластеров»), которые и стано-
вятся центрами кристаллизации. Скорость их образования опре-
деляется заданным переохлаждением расплава. По достижении
определенного переохлаждения расплава после образования
в нем зародышей кристаллов на последних начинается выде-
ление твердой фазы, характеризующееся той или иной скоро-
стью роста образующихся кристаллов. Одновременно может
106
иметь место и ее частичная перекристаллизация, если для этого
имеются соответствующие условия. Возникновение кристаллов
в расплаве и их рост представляют собой две основные стадии
процесса кристаллизации. Главным условием их протекания яв-
ляется то, что исходный расплав должен быть в определенной
степени переохлажден, т. е. движущей силой процесса кристал-
лизации в расплаве является его переохлаждение А Г, под кото-
рым понимается разность равновесной температуры Тр на гра-
нице раздела жидкой и образующейся твердой фаз и темпера-
туры расплава Т. Стадия роста кристаллов также является
сложным процессом. Для объяснения сопутствующих ей явле-
ний предложены различные теории, основанные на тех или иных
постулируемых механизмах роста. Изложение этих теорий выхо-
дит за рамки рассматриваемой задачи глубокой очистки ве-
ществ, ио все же следует отметить наиболее важную их кон-
цепцию об определяющей роли межфазовой границы твердое
тело — жидкость Как из-
вестно, у поверхности ра-
стущего кристалла име-
ется некий переходный
слой, физико-химические
характеристики которого
(теплоемкость, теплопро-
водность, теплота фазо-
вого перехода и т. п.) ме-
няются постепенно от
значений, близких к тем,
которые соответствуют
твердой фазе, до значе-
ний, соответствующих
жидкой фазе. Следова-
тельно, этот переходный
Т
Кршпалш Вещества А ♦ Кристомы ВещестВа В*
эВтектика эВтектика
А &
Рис. 27. Диаграмма плавкости для си-
стемы с простой эвтектикой
слой расплава, особенно прилегающий к поверхности крис-
талла, должен иметь упорядоченную структуру, сходную
с кристаллической, т. е. имеющую связи как ближнего, так и
относительно дальнего порядка. Это обусловлено существова-
нием у поверхности кристалла ненасыщенных связей. Поэтому
образование указанной ориентированной структуры жидкости
в переходном слое и сам рост кристалла можно представить
как процесс насыщения дальнедействующих поверхностных сил
кристалла. Таким образом рост грани кристалла будет зависеть
от структуры ее поверхности (микроскопическое строение по-
верхности в атомно-молекулярном масштабе и ее макроскопиче-
ский рельеф), адекватной структуре прилегающего переходного
слоя расплава. Структурирование жидкости вблизи поверхности
кристалла способствует образованию в этой жидкости отмечен-
ных выше квазикристаллических ассоциатов. Эти дозародыше-
107
вне образования могут ориентированно высаживаться на по-
верхности грани. Достигая критических размеров, они переходят
в дискообразные (двумерные) зародыши. На 'поверхности грани
одновременно могут образоваться несколько зародышей, кото-
рые быстро разрастаются по всей поверхности: возникает кри
сталлический монослой. Теория такого послойного роста хорошо
объясняет рост кристаллов с плоской поверхностью (с «глад-
кими» гранями), но не объясняет рост кристаллов — сфероли-
тов. Структура поверхности кристалла определяется физико-хи-
мическими свойствами кристаллизующегося вещества и усло-
виями кристаллизации. Так, для предсказания строения фронта
кристаллизации, т. е. предсказания о том, будет ли поверхность
растущего кристалла гладкой или шероховатой, можно вос-
пользоваться результатами исследований К Джексона. Он ука-
зал на важную роль в таком предсказании значения энтропии
плавления (5ПЛ) кристаллизующегося вещества и предложил
следующий приближенный критерий (критерий Джексона) если
5пл>2/?, то поверхность кристалла должна быть гладкой, т. е.
в процессе роста кристалл будет иметь ограненную форму;
если же 5Пл<2/?, где /? — универсальная (молярная) газовая
постоянная, то поверхность кристалла будет шероховатой и кри-
сталл при своем росте будет принимать округлую форму. Шеро-
ховатая же поверхность характеризуется наличием большого
числа различных дефектов, в частности выступов и изломов, ко-
торые образуются флуктуационным путем, например вследствие
флуктуации тепловой энергии в процессе реального кристалло-
образования. Эти дефекты имеют локальные ненасыщенные
связи и играют роль активных центров для присоединения
к кристаллу новых молекул из переходного слоя, и в этом слу-
чае для роста кристалла не требуется образование зародышей
на его поверхности, как в случае «гладкой» грани. Таким обра-
зом, изменение скорости роста кристалла с изменением переох-
лаждения в обоих рассматриваемых случаях будет различным,
что будет характеризоваться показателем степени т в выраже-
нии для скорости роста кристалла щ~А7"т. Однако следует за-
метить, что установление характера зависимости скорости рос-
та кристалла от переохлаждения еще не является установлением
механизма роста. Известно, что с изменением переохлаждения
возможно изменение и формы и механизма роста кристалла;
даже рост отдельных кристаллов одного и того же вещества
при фиксированном переохлаждении может происходить по раз-
личным механизмам Все это указывает на сложность явлений,
сопровождающих кристаллизацию, вследствие чего до настоя-
щего времени еще не создана единая теория кристаллообразо-
вания. Важным фактором является то, что хорошо разработан-
ная теория роста идеальных кристаллов, основанная на концеп-
ции двумерного зародышеобразования на их гранях, удачно
108
дополняется теорией роста несовершенных кристаллов, учитываю
щей наличие в последних тех или иных дефектов, к которым
относятся и дислокации.
Влияние примесей
Большое влияние на процесс кристаллообразования в рас-
плаве оказывают различные примеси. Особенно важную роль
в этом отношении играют механические примеси, находящиеся
в расплаве в виде взвешенных частиц микронного и субмикрон-
ного размера и играющие роль «затравки» при образовании за-
родышей. Последнее объясняется тем, что работа образования
зародышей на готовой поверхности (гетерогенное зародышеоб-
разование) меньше, чем работа флуктуативного образования
зародышей (гомогенное зародышеобразование) в объеме рас-
плава. Такое гетерогенное зародышеобразование возможно
лишь, когда расплав является лиофильным по отношению к по-
верхности частицы. Возникающий на ней в этом случае адсорб-
ционный слой вызывает соответствующее структурирование при-
легающего расплава, что приводит к облегчению образования
зародышей на данной поверхности по отношению к зародыше-
образованию в объеме расплава. Вследствие этого начало
кристаллообразования обычно смещается в сторону меньших
переохлаждений по сравнению с тем, что было бы, если бы ис-
ходный расплав был тщательно очищен от взвешенных частиц.
Аналогичное явление имеет место и в случае кристаллизации на
специально вводимых в расплав затравочных кристаллах, что
широко применяется в различных способах выращивания моно-
кристаллов.
Совсем по-иному влияют на процесс кристаллизации раство-
римые примеси. Дело в том, что зародыш кристалла при своем
образовании стремится «оттеснить» инородные примесные мо-
лекулы, что ведет к обогащению этими молекулами слоя рас-
плава, окружающего границы зародыша. По этой причине уча-
стие молекул основного вещества в росте зародыша становится
затруднительным и для достижения зародышем критического
размера уже требуется большее переохлаждение. В присутствии
примеси может изменяться (как правило, уменьшается) и ско-
рость роста кристалла. Это, по-видимому, обусловлено адсорб-
цией примесных молекул на поверхности кристалла. Если
адсорбция происходит на активных местах роста, то такое ло-
кальное «отравление» поверхности кристалла тормозит образо-
вание кристаллического слоя и рост кристалла замедляется
по сравнению с его ростом из чистого расплава. Но, с другой
стороны, адсорбция примесных молекул может приводить
к уменьшению поверхностной энергии кристалла. Это, в свою
очередь, связано с повышением шероховатости поверхности,
109
в результате чего скорость роста кристалла может даже воз-
растать.
При захвате примеси растущим кристаллом она может войти
в узлы кристаллической решетки, образуя твердый раствор за-
мещения, или в междоузлия, образуя твердый раствор внедре-
ния, а также включения, состоящие из взвешенных частиц.
В кристалле при этом будут возникать напряжения или дефор-
мация, являющиеся причиной образования дислокации. Это
является причиной изменения и механизма и скорости роста
кристаллов, что часто сопровождается изменением формы кри-
сталлов (но не типа кристаллической решетки), о чем уже го-
ворилось выше.
На формирование кристаллов влияет и теплообмен образую-
щейся системы жидкость — твердая фаза с окружающей сре-
дой. Так, наличие в расплаве конвекционных потоков будет при-
водить к естественному отводу теплоты, выделяющейся от обра-
зующихся зародышей и растущих кристаллов, или даже к их
переносу в более «холодные» части расплава. Этому способст-
вует и принудительное перемешивание расплава, которое
с целью поддерживания его однородного состава в объеме в хо-
де процесса осуществляется различными способами. Перемеши-
вание приводит также к обнорлению переходного слоя на гра-
нице кристалл — расплав, что обусловливает более благоприят-
ное межфазовое распределение примеси. Кроме того, при этом
снижается эффект возможной агломерации твердой фазы, вслед-
ствие которой несколько соприкасающихся кристалликов могут
срастаться с образованием включений расплава. Наличие таких
включений, разумеется, ухудшает качество конечного продукта.
Таким образом, при использовании кристаллизации для
очистки веществ отделяемые в качестве продукта кристаллы
будут в той или иной степени загрязнены примесями, содержа-
щимися в исходном расплаве, а также поступающими из внеш-
ней среды и из материала разделительной аппаратуры. Захват
примеси, образующейся в процессе кристаллизации твердой фа-
зой, в общем случае принято называть соосаждением Различа-
ют гомогенное и гетерогенное соосаждение. Гомогенное, или ис-
тинное, соосаждение имеет место тогда, когда очищаемое веще-
ство и захватываемая примесь обладают способностью
кристаллизоваться в совместной кристаллической решетке, об-
разуя, как уже отмечалось, твердые растворы замещения или
В1недрения. Эту разновидность соосаждения называют также
сокристаллизацией.
При гетерогенном соосаждении примесь захватывается по-
верхностью кристалла вследствие адсорбции на активных мес-
тах и последующего «зарастания» вновь образующимся слоем
основного вещества. Вариантом гетерогенного соосаждения
является и захват кристаллами примеси, присутствующей в рас-
110
главе в виде взвешенных частиц. В этом случае частица, ока-
завшаяся вблизи поверхности растущего кристалла, как бы
закрывает соответствующий участок этой поверхности от моле-
кул основного вещества. Следовательно, перенос последних
будет осуществляться главным образом посредством диффузии
в тонком слое расплава между частицей и «закрытым» участком
поверхности. У «открытой» же части поверхности преобладает
перенос молекул основного вещества из объема расплава в пе-
реходный слой конвекционными потоками. В результате вокруг
частицы может образоваться полость, которая затем «захлопы-
вается» или, как говорят, растущая грань выклинивается с вра-
станием частицы в кристалл.
Из кратко рассмотренных специфических особенностей кри-
сталлообразования, обусловленных взаимодействием растущих
кристаллов с примесями, следует, что эти особенности сущест-
венно влияют иа процесс, который обычно является неравновес-
ным. Однако при определенных условиях проведения процесса
между жидкой и образующейся твердой фазами может быть до-
стигнуто и равновесие. В этом случае говорят о равновесном
распределении примеси между указанными фазами
Коэффициент разделения (распределения)
Отношение концентраций компонентов в равновесных твер-
дой и жидкой фазах называют коэффициентом разделения, обо-
значая его а:
где х—молярная доля примесного компонента в твердой фазе;
р — молярная доля примесного компонента в жидкой фазе, на-
ходящейся в равновесии с твердой фазой. Коэффициент разде-
ления является важнейшей характеристикой кристаллизацион-
ных методов разделения смесей, как и дистилляционных мето-
дов. Применительно к глубокой очистке веществ, когда и
соотношение (П.1) запишем в более простой форме:
а=х.у. (III.2)
При использовании соотношения (Ш.2) для характеристики
кристаллизационных процессов часто вместо термина «коэффи-
циент разделения» пользуются термином «коэффициент распре-
деления».
Для фазового равновесия твердое тело — жидкость коэффи
циент разделения а, так же как и для фазового равновесия жид
кость — пар, можно оценить расчетным путем. Так, например,
наиболее простое выражение для расчета коэффициента разде-
111
ления а, полученное ван Лааром* при допущении, что сосущест-
вующие твердая и жидкая фазы являются идеальными раство-
рами, имеет вид
In ге - пл(г) Т Упл(г)
ЯТплЬ) Т
(Ш.3>
где ДА7пл<2) и ТПП(.2) — теплота и температура плавления примес-
ного компонента соответственно; Т — температура фазового рав-
новесия, которую для разбавленного раствора можно принять
равной температуре плавления Тпл^ основного компонента.
Однако выражение (III.3) по сравнению с соответствующим
выражением (II.9) для равновесия жидкость — пар во многих
случаях дает качественно неверный результат, т. е. неправильно
предсказывает фазу, в которой должен концентрироваться при-
месный компонент. Это объясняется тем, что для реальной си-
стемы твердое тело — жидкость отклонения от условий, приня-
тых при выводе (III 3), более значительны, чем для системы
жидкость — пар при выводе (II.9). Приближенно эти отклоне-
ния можно учесть с помощью соотношения вида (II 14), кото-
рое в рассматриваемом случае запишется как
1па= Дтж—AvT, (П1.4)
где AvT=Дрт/(&7'), Арт— разность химических потенциалов
примесного и основного компонентов в твердой фазе (твердый
раствор), равновесной с жидкой фазой. Расчеты Av2”, проведен-
ные методом Монте-Карло для разбавленных растворов, взаи-
модействие молекул в которых описывается потенциалом .Ден-
нарда — Джонса, позволили получить для ее оценки следующую
аппроксимирующую формулу**:
AvT= — 3,25X4- 78,75Х24- 82Х3 —
— (11,1234-9,71Х —14,6Х2) У (2,0344-2,21Х) У2,
(Ш.5)
где X и У — обозначают то же, что и в выражении (II. 15) с ана-
логичными интервалами изменения параметров е2г/Еи и Огг/он
потенциала Деннарда — Джонса. Для определения последних
могут быть использованы соотношения (II 20) Поскольку пара-
метры потенциала входят в (III.5) в виде отношений ег2/еп и
022/оц. для удобства расчетов соотношения (11.20) целесообраз-
но преобразовать к виду:
е22/е11 Упл(г)/7’пл(1)> °22/°11 = 1 Ка.(2)/Ка.(1),
где Рд(1) и Уд(2) — молярные объемы основного и примесного
компонентов в жидком состоянии при температуре их плавления
* Шахпароиов М И Введение в молекулярную теорию растворов М.,
1956, с. 508.
** Колесников А. Н„ Степанов В. М., Яньков С. В Фазовые равновесия
предельно разбавленных лениард-джонсовых растворов//Жури физ. химии.
1985, т 59, Ns 1, с 83—86.
112
соответственно. Величину Av* в соотношении (Ш.4) нетрудно
найти с помощью выражения (11.15) как показывают расчеты,
при фазовом равновесии твердое тело — жидкость Av" по срав-
нению с Av* и Avr обычно пренебрежимо мала. Результаты оце-
нок коэффициента разделения на примере систем бензол — тио-
фен и тетрахлорид германия — трихлорид мышьяка (см.
табл. II.1) и соответствующие экспериментальные данные при-
ведены в табл. III.1.
Таблица HI. 1. Данные для расчета коэффициента разделения
при равновесии твердое тело — жидкость
и сравнение полученных результатов с экспериментом
Вещество
Бензол
Тиофен
Тетрахлорид герма-
ния
Трихлорид мышьяка
1,88
1 98
7,66
279
235
224
257
87,2
74,1
106
0,842
0,947
1,5
0,57
0,42'
80,6
1,15
0,913
0,50
0,66
0,50'
по химии и хим. технол. (Горький). 1973. вып. 4(35). с. 53— 54.
•• Аглиулов 1 X Зеляеп И А Зуев М В и др. Г
• Гурьянов А. Н.. Еллиев Ю. Е. О коэффициенте распределения твердая фаза —
расплав для смеси бензол Д- тнофен в области небольших концентраций тиофеиа//То.
СИ П П „1 С и Ар. Глубокая очистка некоторых
групп противоточной кристаллизацией нз расплава//В KI
особой чистоты Горький, 1974, с. 104—111
хлоридов элементов III—V
Получение и анализ веществ
Из табл III.1 следует, что в отличие от уравнения (III.3) со-
отношение (III 4) позволяет достаточно надежно предсказать,
в какой фазе должён концентрироваться примесный компонент
при фазовом равновесии твердое тело — жидкость и ориентиро-
вочно оценить коэффициент разделения.
Коэффициент разделения а нетрудно определить по диа-
грамме плавкости интересующей бинарной системы. Однако для
целей глубокой очистки необходимо знать числовое значение а
в области малых концентраций одного из компонентов, т. е.
нужно знать «угол» диаграмм плавкости (см. рис. 25 и 26)
Соответствующие экспериментальные данные в литературе
обычно отсутствуют, и поэтому на практике определяют коэф-
фициент разделения исходя из результатов соответствующих
опытов по глубокой очистке веществ.
§ 2. НАПРАВЛЕННАЯ КРИСТАЛЛИЗАЦИЯ
Одним из кристаллизационных способов очистки веществ
является метод нормальной направленной кристаллизации. Суть
его заключается в следующем. Пусть сосуд 1 (рис. 28) охлаж-
113
дается таким образом (например, путем медленного ввода сосу-
да в криостат <?), что находящаяся в нем жидкая смесь (основ-
ное вещество—примесь) начинает кристаллизоваться в нижней
части сосуда и образующийся плоский фронт кристаллизации
постепенно передвигается вверх. Примем также, что жидкая
фаза интенсивно перемешивается и, следовательно, жидкость,
остающаяся в сосуде в процессе такой кристаллизации, одно-
родна в любой момент времени, а диффузия в твердой фазе
по сравнению с диффузией в жидкой фазе незначительна
В дальнейшем для краткости при описании рассматриваемого
процесса будем пользоваться термином направленная кристал-
лизация, хотя следует отметить, что в пршципе направленными
являются и зонная перекристаллизация (плавка), и зонное за-
мораживание (затвердевание)
Однократная направленная кристаллизация
I
Рассмотрим однократный процесс. Если коэффи-
циент разделения больше единицы, то образующаяся
в ходе процесса твердая фаза рудет иметь более вы-
сокую концентрацию примеси, чем остающаяся жид-
кость. По мере продвижения фронта кристаллизации
обеднение жидкости примесью будет более заметным
и верхний слой жидкости может быть уже в значи-
тельной степени освобожден от Фримеси. Если же ко-
I
----------- --------- ----------------------------
Рис. 28 Схема прибора для осуществления процесса нормальной
направленной кристаллизации:
1 —• сосуд с расплавом, 2 — мешдлка; 3 — криостат
эффициент разделения меньше единицы, to образующаяся по-
зади фронта кристаллизации твердая фаза будет содержать
примеси меньше, чем жидкость, находящаяся перед фронтом
кристаллизации. Количество примеси в незакристаллизовав-
шемся расплаве в некоторый момент времени протекания про-
цесса равно у (No—NT), где No — общее количество вещества
(основное вещество-t-примесь) в исходном расплаве; NT—ко-
личество закристаллизовавшегося вещества к заданному мо-
менту времени. Через малый промежуток времени еще часть
расплава закристаллизуется, в результате чего количество при-
меси в оставшемся расплаве изменится. Отсюда будет справед-
ливым следующее уравнение материального баланса;
а(У(^-л^)1=Н(Лг,-Агг). (III.6)
Принимая, что составы выделяющейся твердой фазы и
остающегося расплава в любой момент времени процесса отли-
114
чаются в а раз и что коэффициент разделения не •изменяется
с изменением состава фаз в процессе кристаллизации, после
дифференцирования уравнения (III.6) с учетом (III2) полу-
чаем
^-= -(«-!>
У
ЛГО-ЛГТ •
(Ш.7)
Интегрирование (Ш.7) при начальном условии у = уо, когда
Лгт=0, дает
1г — = (а—1) In — -
Уо No
или
K=j/0(l-xr)a-11 (III.8)
где уо — молярная доля примеси в исходном расплаве хг=
= Nt/No — доля закристаллизовавшегося вещества.
С помощью уравнения
(III8) нетрудно определить
коэффициент разделения. С
этой целью уравнение (Ш.8)
с учетом (III.2) и условия
уо=хо можно записать в виде
х=хоа (1—хг)я-1. (III.9)
Взяв после окончания про-
цесса такой однократной нап-
равленной кристаллизации на
анализ пробы из разных сече-
ний сосуда 1 (рис. 28) и по-
строив график зависимости
величину (а—1), которая равна
чающейся прямой линии
как отношение длины
-lg(f-ae,)
Рис 29 Зависимость концентрации
примеси в твердой фазе от доли за-
кристаллизовавшегося вещества
]gx~lg(l—у.т), нетрудно найти
тангенсу угла наклона полу-
(рис. 29). При этом хт выражается
части слитка, в конце которой
отбирается проба, к общей длине слитка (или как соот-
ветствующее отношение объемов) Величину а можно опреде-
лить из отрезка, отсекаемого на оси ординат. Определяемые
указанными способами величины а обычно отличаются друг от
друга. Одной из причин этого различия, по-видимому, является
неодинаковая точность в последовательном определении со-
держания примеси по длине слитка: в той части слитка, где при-
меси меньше, ошибка определения выше. Поэтому на практике
в качестве искомого значения коэффициента разделения иногда
рекомендуют пользоваться усредненным значением, полученным
из обеих определенных величин а
Уравнение вида (Ш.9) для описания процесса однократной
направленной кристаллизации было предложено Г. Гулливером
115
еще в 1922 г Для количественных же расчетов применительно
к задаче очистки веществ это уравнение впервые, по-видимому,
было использовано В. Пфанном*. Поэтому принятые при выводе
этого уравнения упрощающие допущения в литературе обычно
называются пфанновскими допущениями. При проведении про-
цесса направленной кристаллизации для выполнения условия
поддержания однородности состава жидкой фазы обычно
используют механические мешалки. Известны другие способы пе-
ремешивания расплава: наложение поля центробежных сил, воз-
действие электромагнитного поля и ультразвука Если же в хо-
де процесса кристаллизации расплав перемешивается недоста
точно полно, то определенную роль в этом случае уже будет
играть протекающая в нем молекулярная диффузия, скорость
которой недостаточна для выравнивания (состава всего объема
жидкой фазы даже при относительно небольшой скорости кри-
сталлизации. В результате эффективность очистки ухудшается.
Поскольку в целом процесс направленной кристаллизации
протекает с конечной скоростью, то определенный указанным
способом коэффициент разделения будет? представлять собой
так называемый эффективный коэффициент разделения аЭф.
Равновесный коэффициент разделения можно найти, если опы-
ты проводить при различных скоростях движения фронта кри-
сталлизации и затем полученные значения аЭф экстраполировать
к нулевой скорости процесса. Для этой цели обычно используют
следующее соотношение между эффективным коэффициентом
разделения и скоростью кристаллизации:
аЭФ" а+(1-а)ехр(--^ )
(ШЛО)
где а — равновесный коэффициент распределения; цкр — ско-
рость кристаллизации, см/с (в рассматриваемом случае отож-
дествляется со скоростью движения фронта кристаллизации),
Ож—'коэффициент диффузии примеси в жидкой фазе, см2/с;
с» — толщина так называемого диффузионного слоя в жидкой
фазе на границе расплав — твердая фаза, см (лежит обычно
в пределах 0,01—0,001 см) Для удобства вычисления а соотно-
шение (ШЛО) можно записать в виде
Построив график зависимости 1ё((1/аЭф)—1]-~окр, нетрудно
определить а из отрезка, отсекаемого на оси ординат прямой
линией. При этом предполагается, что величина ож не зависит
от окр, что в общем не совсем точно.
♦ Г)фанн В Зонная плавка. М., 1970, с. 367.
116
На точность определения коэффициента разделения методом
направленной кристаллизации существенное влияние оказывает
частичный захват расплава образующейся твердой фазой По-
следнее особенно проявляется при относительно высоких ско-
ростях движения фронта кристаллизации В результате кон-
центрация примеси в пробах, отбираемых последовательно из
различных сечений по длине слитка после окончания процесса,
не будет соответствовать значениям х, задаваемым уравне-
нием (III 9) Это вносит ошибку в оценку коэффициента аЭф,
а следовательно, и равновесного коэффициента разделения
Но при скоростях движения фронта кристаллизации порядка
нескольких миллиметров в час, при которых осуществляется
процесс направленной кристаллизации с целью определения
коэффициента разделения, доля захватываемого расплава то
отношению к единице поверхности раздела фаз обычно невели-
ка. При этом условии захватом расплава можно пренебречь и
пользоваться для расчетов уравнениями (III 9)— III 11)
Направленная кристаллизация используется и в физико-хи-
мическом анализе для построения диаграмм состояния или уточ-
нения их «углов» при работе с разбавленными растворами Так,
определив методом направленной кристаллизации равновес-
ный коэффициент разделения заданной смеси основное веще-
ство — примесь, нетрудно построить для интересующего нас
концентрационного интервала линию солидуса при известной
линии ликвидуса, полученной, например, методом дифферен-
циального термического анализа. При решении вопроса о су-
ществовании области твердых растворов в бинарных системах
с малым содержанием одного из компонентов она даже имеет
преимущество в точности по сравнению с таким классическим
методом, как метод дифференциального термического анализа.
Направленную кристаллизацию применяют и для кристаллиза-
ционного концентрирования примеси при анализе веществ осо-
бой чистоты.
Из сравнения уравнения (Ш.8) с формулами Релея (П.25а)
и (II 256) для разбавленных растворов (гл. II, § 3) можно ви-
деть, что они идентичны по форме. Отсюда следует, что одно-
кратная направленная кристаллизация, как и простая перегон-
ка, достаточно эффективна лишь при очистке веществ от приме-
сей, существенно отличающихся по физико-химическим свойст-
вам от основного вещества. В отношении же отделения приме-
сей с коэффициентами разделения, близкими к единице, одно-
кратная направленная кристаллизация не эффективна. В даль-
нейшем, как и при рассмотрении дистилляционных методов
(см гл II), для удобства изложения теоретических основ кри
сталлизационных методов не будем подчеркивать различия меж-
ду равновесным и эффективным значениями коэффициента раз-
деления, имея в виду, что такое различие существует.
117
Многократная направленная кристаллизация
С целью повышения глубины очистки вещества процесс на-
правленной кристаллизации можно проводить многократно
с отбрасыванием в конце каждого цикла некоторой части рас-
плава, в которой концентрируется примесь. Для оценки эффек-
та очистки, достигаемого при проведении многократной направ-
ленной кристаллизации, примем, что доля незакристаллизовав-
шейся жидкости, отбрасываемой в виде «хвостовой» фракции,
в конце каждого цикла одинакова и составляет хж. С учетом
этого допущения на основании соотношения (III.8) нетрудно
составить уравнение материального баланса для каждого цикла
и всего процесса в целом.
1-й цикл. Количество первой «хвостовой» фракции состав-
ляет NoKk', количество примеси в ней будет равно Л^охж1/ =
= Л^0х0жуо- Следовательно, в кристаллизате останется примеси
Noyo—Л^охаж!/о=Лго(1—хаж)«/о; концентрация примеси в рас-
плавленном кристаллизате по окончании 1-го цикла будет со-
ставлять:
1—х“
(,П12)
2-й цикл. Количество второй «хвостовой» фракции будет
составлять /Уо(1—хж)хж, а количество примесей в ней —
No (1 —Хж) ХжХж“_' [ ( 1 — Хж“) / ( 1 —Хж) ] у0 = No (1 —Хж“) Хж“Уо- ТОГ-
да концентрация примеси в расплаве, образующемся из кри-
сталлизата, будет равна
- / 1 — х“ \2
(т=йг) • (П,13>
Аналогично для n-го цикла будем иметь
/ ! — к“ V1 Г 1—(1—ХГ)“ 1п ,гт1«,ч
^п-уп-уо( j_X)K ) -жо|_ — —J • (ш14>
Выражение (III.14) позволяет оценить чистоту получаемого
продукта в результате проведения и-кратного процесса направ-
ленной кристаллизации с отбросом некоторой постоянной доли
вещества хж = 1—хг в качестве «хвостовой» фракции.
Важнейшим практическим применением нормальной направ-
ленной кристаллизации является производство монокристаллов.
Для выращивания монокристаллов используется много методов.
Например, для получения монокристаллов методом Бриджмена
используют прибор, схематически изображенный на рис. 28.
Схема вытягивания монокристалла по методу Чохральского
представлена на рис. 30. В этом методе затравка кристалличе-
ского вещества в виде небольшого монокристалла вносится
118
в расплав сверху. На границе между затравкой и расплавом
протекает процесс кристаллизации, вследствие чего размер кри-
сталла-затравки увеличивается Для того чтобы фронт кристал-
лизации всегда находился в верхнем слое расплава, растущий
кристалл постепенно поднимают вверх (вытягивают) С целью
перемешивания жидкой фазы кристалл вращают вокруг верти-
кальной оси; вращение кристалла вызывает вращение жидкости
и, как следствие этого, ее перемешивание. При этом, разумеется,
лучшие результаты в любом методе выращивания монокристал-
лов будут достигнуты тогда, когда исходное вещество предвари-
тельно будет очищено от основной массы примесей
§3 ЗОННАЯ ПЕРЕКРИСТАЛЛИЗАЦИЯ (ПЛАВКА)
Рис. 30. Схемы вытя-
гивания монокристал
ла из расплава
При использовании процесса кристаллизации из расплава
для глубокой очистки веществ от трудноудалимых примесей
необходимы методы, которые позволили бы
увеличивать эффект разделения, имеющий
место при однократной кристаллизации К
таким методам относится метод многократ-
ной направленной кристаллизации, но ему
присущ тот же недостаток, что и методу
многократной перегонки: низкий выход
продукта, обусловленный отбрасыванием
«хвостовых» фракций. Более предпочти-
тельным в этом отношении является мно-
гоступенчатый способ кристаллизационной
очистки веществ — метод зонной перекри-
сталлизации, или как его часто называют,
метод зонной плавки. Идея этого метода
состоит в перемещении узкой расплавлен-
ной зоны вдоль твердого образца (рис 31)
Так как диффузионные процессы в твердой фазе всегда про-
текают значительно медленнее, чем в жидкости, то можно счи-
тать, что твердая фаза, попадая в область нагревателя, превра-
щается в расплав практически без изменения состава Если при
этом скорость кристаллизации вещества позади двигающейся
жидкой зоны мала (скорость кристаллизации равна скорости
плавления или скорости передвижения зоны), то соотношение
между концентрациями примеси в расплаве и в выпадающих
из него кристаллах в каждый момент времени можно считать
близкими к равновесному. Поэтому при а<1 и движении рас-
плавленной зоны по слитку слева направо (рис 31) примесь
будет оттесняться в правый конец. Если а>1, то примесь будет
концентрироваться в левом конце слитка.
119
Распределение примеси по длине слитка
после одного прохода расплавленной зоны
Если расплавленную зону путем перемещения нагревателя
(рис. 31) продвинуть,, на некоторое расстояние dz вдоль длины
слитка так, чтобы ANr молей твердой фазы с исходной кон-
центрацией х0 расплавилось и такое же количество твердой
фазы выделилось из жидкости на задней границе расплавлен-
ной зоны, то будет справедливо следующее уравнение мате-
риального баланса:
^odA^-p j/G = г<1ДГг4-(^-pdj/) G, (III 15)
где G — количество молей вещества в расплавленной Йоне.
Второй член в левой части уравнения (III 15) представляет
собой содержание примеси в расплавленной зоне в некоторый
момент времени, первый — количество примеси, поступившее
в расплавленную зону после перемещения зоны на расстоя-
ние dz вдоль по длине слитка. При этом продвижении на зад-
ней границе расплавленной зоны выпадает dNT молей твердой
фазы. Количество примеси в
Рис. 31 Схема процесса зонной
перекристаллизации (плавки):
/ — нагреватель; 2 — расплавленная
зона; 3 — очищаемое вещество; 4—
вновь закристаллизовавшееся ве-
щество
не выделяется, концентрация
такая же, как и в исходной тв
ней выражается первым членом
правой части уравнения (III 15)
Второй член в правой части это-
го уравнения характеризует ко-
личество примеси в жидкой зоне
после ее продвижения на рас-
стояние dz
При расплавлении нагревате-
лем самого начального участка
слитка, длина которого равна
длине расплавленной зоны, когда
твердая фаза из расплава еще
примеси в жидкой фазе будет
рдой фазе, т. е.
У о—хо-
(1П.16)
Подставляя соотношения (III.2) и (III. 16) в уравнение (III. 15)
и разделяя переменные, придем к дифференциальному урав-
нению
dy d-Vr
aj/ —!/о ' ‘
(III.17)
После интегрирования уравнения (III.17) с использованием на-
чального условия, что у=уо при yVr=O, имеем:
1 1 юУо—Уо NT
а (а-1)!/0 G
или
ау/Уо— 1 — (1—a) exp (—aNT/G).
(III.18)
120
Обозначая NtIG—U, где U — длина слитка, подвергнутого пере-
кристаллизации, выраженная в длинах расплавленной зоны, и
вновь используя соотношения (1П.2) и (III.16), из (III 18) по-
лучаем
x/Xg—i — (1 — а) ехр (—aU).
(Ill ЛЭ)
Расстояние в длинах зоны
Рис. 32 Распределение примеси по
длине слнтка после одного прохода
расплавленной зоны [кривые рассчи-
таны по уравнениям (111.19) и
(HI 9)]
/ — а-0,1 2 — а-0,5: 3 — а-0.9. 4 — а—10;
5— 0.^2; 6 — а-!,!
Уравнение (III 19) описьнает распределение примеси по длине
слитка вплоть до последнего его участка, равного длине зоны
Для характеристики распределения примеси в последней зоне
это уравнение не применимо, так как длина зоны при этом пе-
рестает быть постоянной и поступление примеси в зону за счет
плавления твердой фазы прекращается Отсюда следует, что
в последней зоне должна идти нормальная направленная кри-
сталлизация. Таким образом,
распределение примеси в по-
следнем участке слитка дли-
ной в одну зону при его за-
твердевании описывается урав-
нением (III.9); распределение
примеси по длине слитка в це-
лом после одного прохода рас-
плавленной зоны будет иметь
вид, изображенный на рис. 32
Так как вывод уравнения
(III.19) основан на тех же до-
пущениях, что и вывод урав-
нения (П1 9), то оно будет
справедливо лишь в случае,
когда содержимое расплавлен-
ной зоны при ее продвижении
по слитку интенсивно переме-
шивается В противном случае
лимитирующее влияние при-
обретает диффузионный мас-
соперенос в объеме расплавленной зоны, в результате чего сни-
жается глубина очистки.
Нетрудно заметить, что, используя результаты опытов по
распределению примеси по длине слитка после одного прохода
расплавленной зоны, можно графическим путем на основании
уравнения (III.19) определить коэффициент разделения Это
позволяет использовать метод зонной перекристаллизации, как
и метод нормальной направленной кристаллизации, в физико-
химических исследованиях, в частности при построении диа-
грамм состояния систем кристаллы — расплав.
Распределение примеси по длине слитка
после нескольких проходов расплавленной зоны
Представленные на рис. 32 расчетные кривые распределения
примеси после прохода зоны указывают на то, что глубина
очистки для большей части слитка (70—80%) будет невелика
даже при коэффициенте разделения, значительно отличающем-
ся от единицы. Максимальное разделение не больше чем
в а раз имеет место лишь в начале исходного образца. После
повторного прохода расплавленной зоны глубина очистки воз-
растает. В результате же многократного прохода зоны можно
добиться значительной глубины очистки и от примесей с коэф-
фициентом разделения, близким к единице. Такой многократный
процесс можно осуществить как путем многократного прохода
вдоль слитка одного нагревателя-плавителя, создающего рас-
плавленную зону, так и путем одновременного перемещения по
слитку системы нагревателей (с чередующимися холодильника-
ми в случае очистки относительно легкоплавких веществ) с воз-
вратно-поступательным движением. Второй путь, разумеется,
приводит к выигрышу во времени.
Пусть число зонных проходов составляет Поскольку
в конце образца длиной в одну зону идет направленная кри-
сталлизация, рассмотрим лишь часть слитка длиной (А/о—G).
Распределение же примеси в кристаллизующейся последней
зонной длине образца будет описываться уравнением вида
(Ш.9), в котором хо = xSk |лг0-с/а, где xsK — концентрация при-
меси в твердой фазе у фронта кристаллизации зоны при ее
Sk-m проходе.
Если обозначить через концентрацию примеси в твер-
дой фазе у фронта кристаллизации при (SK—1)-м проходе зо-
ны, то количество примеси в жидкой зоне при Sk-m проходе,
когда уже перекристаллизовалось NT молей вещества, можно
определить по уравнению
yS}p~ J —J xSA.d7VT, (III.20)
о о
где ysK — концентрация примеси в движущейся жидкой зоне
в мгновенный момент времени при 5к-м ее проходе. Полагая,
что состав жидкой зоны в любой момент времени находится
в равновесии с составом твердой фазы у фронта кристаллиза-
ции, из (III.20) получаем
а
xsK— G
Ny+G N
J xs^dAr—J xSxdNT
- о о
(III.21)
122
или в дифференциальной форме
dxsKlwT а
dJVT ~ С f®SJC-d(N1.+G)“a:SKlxTI-
(III.22)
Уравнение (III.22) представляет собой дифференциальное урав-
нение многопроходной зонной перекристаллизации и лежит
в основе методов расчета распределения примеси по длине под-
вергаемого перекристаллизации слитка вещества, за исключе-
нием его конечного участка, равного длине зоны.
Нетрудно показать, что при одном проходе зоны, т. е. когда
jcsic_i|(wt+g)=Xo, а решение дифференциального урав-
нения (III.22) имеет вид соотношения (III.19). Это, по-видимо-
му, и побудило исследователей при аналитическом решении за-
дачи о распределении примеси по длине слитка после S„-ro про-
хода решение искать в виде
xsK/x0=l — (1— а)ехр(—aU) [z],
(1П.23)
где fz] —функция, сложным образом зависящая от длины зоны
(точнее от количества вещества в зоне), а следовательно, и от
числа зонных длин, укладывающихся по длине слитка, от числа
зонных проходов и от коэффициента разделения Все эти реше-
ния, получаемые при пфанновских допущениях, являются при-
ближенными и плохо описывают распределение примеси в ко-
нечной части слитка вообще, а не только на последнем его
участке длиной в одну зону, где идет направленная кристалли-
зация. При этом даже наиболее простое из уравнений типа
(III.23), полученное К- Милликеном для описания указанной
зависимости, имеет довольно сложный вид
х
2 (SK-/)X
i=i
_к+.у3... 2 . (1П.24)
Однако то, что при расчетах по (III.24) приходится оперировать
разностью величин одного порядка, может привести к ошибкам
в вычислениях, особенно при большом эффекте очистки, когда
из единицы в правой части уравнения (III !4) вычитается вели-
чина, также близкая к единице. Чтобы избежать этого, К. Мил-
ликен предложил при расчетах пользоваться другим уравне-
нием, эквивалентным, по его мнению, уравнению (III.24).
хо а 71
j'=SX+ 1
Х(ае~а)] [U(j —1) + (1-а£7)(/-|-С7)]. (Ш.25)
123
Большая точность достигается в методах численного решения
уравнения (III 22) применительно к заданным параметрам про-
цесса и при определенных граничных условиях (слиток беско-
нечной длины, полубесконечный слиток, слиток конечной длины,,
направленная кристаллизация в конце слитка в одну зону).
Наибольшую известность из них получил метод Хемминга Сущ-
ность его заключается в рассмотрении баланса по примеси для
зоны, движущейся по слитку. При этом условно принимается^
что движение зоны происходит не непрерывно, а как бы малень-
кими скачками Это дает возможность проводить вычисления
в конечных разностях и последовательно рассчитывать измене-
ние содержания примеси по длине слитка от одного прохода
зоны к другому Соответствующие вычисления выполняются
с использованием ЭВМ по составляемой программе: в итоге по-
Рис. 33. Распределение примеси
по длине слитка после пяти прохо-
дов расплавленной зоны (сх=О,1)
лучают данные для построения
искомой концентрационной кри-
вой. Результаты таких расчетов,
для различных сочетаний пара-
метров процесса зонной перекри-
сталлизации представлены, на-
пример, в отмеченной выше ра-
боте В. Пфанна в виде большо-
го набора кривых. Последние по-
зволяют оценить ожидаемую сте-
пень очистки при заданных вы-
ходе продукта и числе проходов
зоны
На рис. 33 для сравнения
приведена кривая 1, рассчитан-
ная по уравнению (III25), и
кривая 2, взятая из работы
В Пфанна. Обе кривые построе-
ны для одних и тех же парамет-
ров процесса, в частности для
а=0,1 Из рисунка следует, что для большей части слитка
(~70%) приближенный расчет концентрационного профиля по
уравнению (Ш.25) хорошо согласуется с точным решением.
Поскольку выход продукта обычно не превышает указанной
величины, то выражением (III 25) можно пользоваться при
оценках глубины очистки методом зонной перекристаллизации.
Распределение примеси по длине слитка
после бесконечно большого числа проходов
расплавленной зоны (конечное распределение)
Сопоставим кривую 1 (см рис 32), характеризующую рас-
пределение примеси по длине слитка после одного прохода рас-
плавленной зоны, и кривую 2 (рис. 33), соответствующую пяти
124
проходам расплавленной зоны. Из этого сопоставления нагляд
но видно, что при увеличении числа проходов зоны на характер
распределения примеси по длине слитка оказывает влияние
явление направленной кристаллизации в его конечной части
Действительно, следует ожидать, что уже при втором проходе
зоны распределение примеси на участке слитка в одну зонную
длину, прилегающем к конечному участку слитка также в одну
зонную длину, будет влиять повышенное содержание примеси
в этом конечном участке,
т е. наклон кривой рас-
пределения примеси на
предпоследнем участке
длиной в одну зону при
втором проходе зоны уве-
личится не только за счет
оттеснения примеси из
начала слитка в его ко-
нец вследствие повторной
перекристаллизации, но и
за счет «обратного отра-
жения» эффекта концент-
рирования примеси от
конца образца. В случае
же пяти проходов этот
эффект будет сказывать-
тя на большей длине
слитка, как это и харак-
теризуется кривой 2 на
рис. 33. На рис. 34, за-
имствованном из моно-
графии В. Пфанна, при-
ведено семейство кривых
распределения примеси,
рассчитанных для коэф-
фициента разделения,
равного 0,5 при различ-
ном числе проходов рас-
плавленной зоны по слит-
Рис. 34. Распределение примесн по длине
слитка прн различном числе проходов рас-
плавленной зоны (рисунок взят из моногра-
фии В. Пфанна)
ку размером в 10 зонных длин. Из рис. 34 видно, что с увели-
чением числа зонных проходов SK наклон кривых распределе-
ния последовательно растет. Однако в результате эффекта «об-
ратного отражения» этот рост становится все меньше и мень-
ше. Характер распределения примеси по слитку (разумеется,
за исключением последнего участка длиной в одну зону) при
этом в полулогарифмических координатах, как показано на
рис. 34, будет приближаться к прямолинейному. В результате
после некоторого числа проходов зоны (в пределе бесконечно
125
большого) через твердый образец дальнейшее изменение рас-
пределения примеси по его длине при последующем проходе
зоны может стать уже неразличимым в пределах точности экс
перимента или расчета. Следовательно, распределение примеси
при этом можно описать уравнением вида
х=Лехр(^г) = 4ехр(ВСГ), (III.26)
где А и В — константы.
Когда достигается такое распределение, то оно может рас-
сматриваться как стационарное, конечное распределение, при
котором происходит перемещение примеси с одинаковой ско-
ростью в прямом и обратном направлениях относительно дви-
жения расплавленной зоны. Знание конечного распределения
примеси по длине слитка очень важно, так как оно позволяет
оценивать предельные возможности зонной перекристаллизации
как метода очистки при заданных условиях проведения про-
цесса.
Для такой оценки необходимо знать значения констант А и
В, которые нетрудно выразить через a, G и No- С этой целью
рассмотрим проход расплавленной зоны через слиток очищае-
мого вещества, в котором уже достигнуто предельное распре-
деление примеси и дальнейшего изменения его состава в за-
данном сечении при проходе расплавленной зоны не происхо-
дит. Количество примеси в движущейся жидкой зоне в этом
случае должно выражаться уравнением
yG= J x&NT. (1П.27)
Nr
Из (Ш.27) и (Ш.2) следует
-f-G
Х=~ ( xANT. (Ш.28)
Or J
NT
Подставив (III.26) в уравнение (Ш.28) и проинтегрировав его,
получим
A exp (BGU) = (enG-1) exp (BGU)
HG
ИЛИ
BG
exp(2?G)—1 ’
(III.29)
Таким образом, константа В в уравнении (III 26) зависит от
величин G и а; при заданных значениях G и а она может быть
определена с помощью соотношения (111.29), например, гра-
фически.
126
Константу А нетрудно определить исходя из следующего
уравнения материального баланса:
Л'о-G с
Лохо= J xdjVT’4- J хсйЛ'тсс)- (III 30)
о О
Член, стоящий в левой части уравнения (III.30), выражает
количество примеси в исходном твердом однородном образце
очищаемого вещества. Первый член в правой части указанного
уравнения характеризует количество примеси после достижения
конечного распределения в слитке от его начала вплоть до по-
следнего участка длиной в одну зону. В этом последнем участ-
ке, как уже отмечалось, идет нормальная направленная кри-
сталлизация. В соответствии с этим второй член в правой части
уравнения (III.30) выражает количество примеси в конечной
части слитка длиной в одну зону, содержащей G молей веще-
ства.
Концентрация примеси в конце закристаллизовавшейся час-
ти слитка перед началом кристаллизации последнего участка
равна Дехр[В(Л/0—G)], как это следует из уравнения (111.26).
Следовательно, состав последней расплавленной зоны с учетом
соотношения (1П.2) должен характеризоваться величиной
1 (Л/а) ехр [В (No—G)]. Нетрудно видеть, что эта величина соот-
ветствует величине у0 в уравнении (III8) для направленной
кристаллизации. Если теперь в этом уравнении заменить пере-
менную у эквивалентной переменной хс/а, то получим
xg = A ехр [В (Лг0—G)) (1 —хт)“- *, (III.31 >
где для рассматриваемого случая v.t=NT{C'IIG\ — количе-
ство вещества в закристаллизовавшейся части последней зоны
от ее начала до заданного сечения.
Интегрирование выражения (III.30) после подстановки
в него соотношений (111.26) и (111.31) дает
Arozo= (A/В) {ехр [В ((V0-G)J—1)+ А ехр [В (,у0—G)J G/a,
откуда
а_______________N Jo____________
[(1 B) + (G/a)] ехр [В(ЛГО—G) -1 В]
или, после преобразования с учетом (III.29),
ДА о
ехр(ВЛ’о) —1 •
(III.32)
III.33)
Таким образом, зная величины G и а, вначале с помощью урав-
нения (III.29) находим константу В, а затем из соотношения
(III.33) —константу А Используя найденные конст иты А и В,
по уравнению (III.26) рассчитывают конечное распределение
12Г
примеси по слитку от его начала до последнего участка в одну
зону. Распределение примеси в конечной части слитка вычисли
ется по уравнению (III.31). Результаты подобных расчетов, как
уже отмечалось, позволяют определить, обычно путем их гра-
фической интерпретации, какую часть слитка можно отобрать
в качестве продукт:'
На основании опытов по определению конечного распреде-
ления примеси по длине образца при зонной перекристаллиза-
ции можно определить и коэффициент разделения а. Для этого
уравнение (III.26) записывается в виде
In x=ln A+BGU (Ш.34)
Следовательно, если данные опытов представить графически
в виде зависимости 1пх~<7, то по наклону получающейся пря-
мой линии легко найти величину BG Подставляя найденное
значение BG в уравнение (III.29), находим а.
Процесс зонной перекристаллизации обычно осуществляется
периодически (см. рис. 31) Известны конструкции аппаратов
и для проведения непрерывной зонной перекристаллизации, но
ввиду технических трудностей их эксплуатации при относитель-
но низкой производительности на практике они используются
редко.
В целом зонная перекристаллизация является очень эффек-
тивным методом глубокой очистки веществ Она позволяет про-
извести очистку веществ до содержания в них отдельных лими-
тирующих примесей на уровне 10-7—10'8 мае. % и ниже. Имен-
но с применением этого метода в настоящее время получают
наиболее чистые вещества, такие, как германий, кремний, оло-
во, алюминий и др. Важнейшей областью использования зонной
перекристаллизации является также производство монокри-
сталлов, в том числе с заданным распределением легирующих
добавок.
§ 4 ЗОННОЕ ЗАМОРАЖИВАНИЕ (ЗАТВЕРДЕВАНИЕ)
Для очистки веществ, являющихся при обычных условиях
жидкостями, определенный интерес представляет метод зонного
замораживания. В этом методе исходное очищаемое вещество,
как и в методе направленной кристаллизации, находится в жид-
ком состоянии Однако оттеснение примеси из одного конца за-
грузки в другой здесь осуществляется не путем ее последова-
тельного направленного затвердевания, а путем прохода через
загрузку отвердевающей зоны определенных размеров. Жид-
кость, попадающая в область воздействия затвердевающей зо-
ны, или, как говорят, зоны замораживания, создаваемой, на-
пример, холодильником с циркулирующим через него хладо-
агентом, кристаллизуется. Позади же движущейся зоны замо-
128
раживания вещество вновь оказывается в жидком состоянии.
По окончании процесса оттесняемая примесь сконцентрируется
в конце образца, в переместившейся сюда зоне замораживания
(при а< 1)
Теперь представим, что подобный образец того же вещества
подвергается направленной кристаллизации. Пусть скорость
движения фронта кристаллизации в обоих процессах одинакова.
Совершенно очевидно, что отбрасывая по окончании каждого из
процессов одно и то же количество концентрата примеси, рав-
ное количеству вещества в зоне замораживания, мы получим
каждым из рассматриваемых методов одинаковое количество
продукта с одинаковым содержанием примеси, т. е. зонное за-
мораживание с одним проходом кристаллизующейся зоны и од-
нократная направленная кристаллизация при одинаковых усло-
виях дают одну и ту же степень очистки, которую, как уже
было показано, можно определить с помощью соотношения
(Ш.12) Отсюда следует, что при многократном повторении про-
цесса замораживания с отбрасыванием после каждого цикла GT
молей концентрата примеси, где GT — количество вещества
кристаллизующейся зоне, соответствующую степень очистки
нетрудно оценить исходя нз выражения (III 14).
Таким образом, казалось бы, что по эффективности очистки
между зонным замораживанием и направленной кристаллиза-
цией никакого различия нет. Однако это справедливо лишь для
рассмотренного варианта метода зонного замораживания. Раз
личие начинает проявляться, если процесс многократного зон-
ного замораживания проводить путем одновременного переме-
щения вдоль образца очищаемого вещества нескольких кристал-
лизующихся зон подобно процессу многократной зонной пере-
кристаллизации, в котором вдоль образца вещества одновре-
менно перемещается несколько расплавленных зон. Ясно, что
при этом эффект очистки вещества за счет зонного заморажи-
вания будет усиливаться эффектом очистки за счет зонной пе-
рекристаллизации, поскольку жидкость, заключенная между
двумя соседними кристаллизующимися зонами, будет играть
роль расплавленной зоны. Последнее позволяет для оценки глу-
бины очистки вещества методом зонного замораживания ис-
пользовать дифференциальное уравнение материального балан-
са вида (III 22). Так, для простейшего случая, когда вдоль очи-
щаемого образца вещества одновременно движутся две кри-
сталлизующиеся зоны, по аналогии с уравнением (III.22) мож-
но записать
<l*2l n+gt а
dJV — ~G~ 1г1^+ст+с—(111.35)
где N— количество молей очищаемого вещества от начала об-
разца до второй кристаллизующейся зоны; Gt — число молей
5—46
129
вещества в кристаллизующейся зоне; G — число молей вещества
в части образца, заключенной между двумя кристаллизующи-
мися зонами. Индексы «1» и «2» относятся к первой и второй
кристаллизующимся зонам соответственно.
Так как при перемещении первой кристаллизующейся зоны
очищаемый образец подвергается направленной кристаллиза-
ции, то с учетом выражения (Ш.9) можно записать
Х11л-+Ст+с=ахо(1-(#+Ог+С)/ЛГо1а"1
Подставляя (III.36) в (III.35), получаем
(1П.36)
а а2 ~ t
G" 1N+GT = *0 — И -(#+Gt+C)/Ne]a~ 1
(1П.37)
Расстопние в длинах зоны
35. Зависимость относитель-
Рис.
ной концентрации примеси в про-
дукте от положения кристалли-
зующейся зоны в процессе зонно-
го затвердевания (а=0,5; N = 8Gr;
G=Gr):
кривая 1 рассчитана по уравнению
(III. 12); кривая 2 рассчитана по урав-
нению (II 1.13); кривая 3 рассчитана по
уравнению (111.37) численным методом
(взята из работы Вольпян А. Е., Кур-
дюмов Г М. — Теор. основы хим. тех-
нол., 1970, т. 4, № 2. с. 281—285)
к сожалению, даже
сматриваемого простейшего слу-
чая одновременного движения по
образцу очищаемого образца
двух затвердевающих зон реше-
ние дифференциального уравне-
ния (1П.35) в аналитическом ви-
де получить не удается, вследст-
вие чего и здесь приходится при-
бегать к численным расчетам.
Полученная на основании таких
расчетов (а = 0,5; #o = 8Gr; 6 =
= Gr) зависимость степени очи-
стки вещества от положения
фронта кристаллизации второй
затвердевающей зоны графиче-
ски представлена на рис. 35
(кривая 3). Здесь же для срав-
нения приведены кривые, рас-
считанные для процессов одно-
кратного (кривая /) и двукрат-
ного (кривая 2) зонного замора-
живания с отбрасыванием Gt мо-
лей концентрата примеси по
окончании каждого цикла. Из
рис. 35 видно, что перемещение
вдоль очищаемого образца одно-
временно двух затвердевающих зон (кривая 3) приводит к су-
щественному повышению эффекта очистки по сравнению с пе-
ремещением одной затвердевающей зоны (кривая /). Эффект
очистки, достигаемый при таком двухзоином замораживании,
выше эффекта очистки при двукратном зонном замораживании
с одной затвердевающей зоной (кривая 2)
для рас-
130
Совершенно очевидно, что процесс двухзонного заморажи-
вания вследствие наложения явления направленной кристалли
зации будет более эффективным, чем процесс зонной перекри-
сталлизации с одним проходом расплавленной зоны Следова-
тельно, при проведении процесса трехзонного замораживания
следует ожидать большей глубины очистки вещества, чем при
осуществлении процесса зонной перекристаллизации с двумя
проходами расплавленной зоны и т. д. Однако с увеличением
числа зонных проходов различие в эффективности обоих про-
цессов будет уменьшаться; при достаточно большом числе про-
ходов затвердевающих зон в процессе зонного замораживания
изменение степени очистки с переходом от 5К проходов
к (SK-|-1) проходам практически станет несущественным. Таким
образом, здесь также можно пользоваться понятием конечного
распределения примеси по образцу, как это делается при ана-
лизе процесса многократной зонной перекристаллизации.
§ 5. ПРОТИВОТОЧНАЯ КРИСТАЛЛИЗАЦИЯ
ИЗ РАСПЛАВА
Рассмотренные выше кристаллизационные методы глубокой
очистки веществ позволяют достичь хороших результатов.
Но им присущи такие недостатки, как малый выход продукта
и длительность проведения процесса очистки в целом. В этом
отношении большими возможностями обладает метод противо-
точной кристаллизации Как метод разделения смесей, он был
предложен почти одновременно с методами зонной перекристал-
лизации (в начале 50-х годов), однако для глубокой очистки
веществ стал использоваться сравнительно недавно. Это объяс-
няется прежде всего трудностями в изготовлении и эксплуата-
ции достаточно эффективных разделительных аппаратов — кри-
сталлизационных колонн, в которых осуществляется противоток
кристаллов и их расплава.
В литературе описано много конструкций кристаллизацион-
ных колонн, отличающихся в основном устройством для созда-
ния противотока фаз. Наиболее эффективны колонны, в кото-
рых транспортируются кристаллы с помощью вращающейся
спирали (шнека), и колонны с движением кристаллов под дей-
ствием силы тяжести. Схема колонны первого типа примени-
тельно к очистке низкоплавких веществ 'приведена на рис. 36.
В нижней части колонны 4 температура поддерживается
выше, а в верхней — ниже температуры плавления смеси Кри-
сталлы, образующиеся в кристаллизаторе 2 колонны (подвод
хладагента к кристаллизатору на рисунке не показан), переме-
щаются с помощью расположенного вдоль направляющего
стержня 5 спирального транспорта — «бесконечного винта» 3
в зону плавления с электронагревателем 7.
5’
131
электромотора (на рисунке
Рис. 36. Схема кристаллизаци-
онной колонны с движением
кристаллов сверху вниз с по-
мощью шнекового транспор-
тера:
1 — холодильник; 2 — крнсталлиза -
тор (медный стержень); 3— шнеко
вый транспортер. 4 — корпус колон-
ны; 5 — направляющий стержень.
6 — теплоизоляция 7 — электрона-
греватель; 8— кран для отбора про-
дукта; 9— питающий резервуар.
10—плунжерный насос
Перемещение кристаллов вызывает вытеснение образующе-
гося расплава в противоположном направлении, что создает
противоток твердой и жидкой фаз. В результате такого процес-
са (применительно к бинарной системе) один из компонентов
разделяемой смеси будет концентрироваться в нижней части
колонны, откуда егб можно отбирать в качестве продукта; дру-
гой компонент будет концентрироваться в верхней части колон-
ны. Вращение шнекового транспортера 3 осуществляется через
редуктор с помощью расположенного над кристаллизатором
не показан). Питание колонны раз-
деляемой жидкой смесью произво-
дится из резервуара 9 насосом 10.
Перед поступлением в колонну
смесь охлаждают до температуры,
близкой к температуре кристалли-
зации, с помощью холодильника 1.
Через кран 8 производится отбор
проб (или продукта) в ходе про-
цесса, а также слив содержимого
колонны по окончании опыта. Вся
колонна заключена в теплоизоля-
ционный кожух 6 из пенопласта, в
котором для наблюдения за про-
цессом кристаллизации сделаны
специальные окна. При необходи-
мости отбор проб расплава из раз-
личных по высоте участков колон-
ны производится с помощью шпри-
ца с длинной иглой. Для этой цели
в направляющем стержне в верти-
кальном направлении вырезана спе-
циальная канавка. Температура в
зоне кристаллизации регулируется
расположенным на кристаллизато-
ре электронагревателем.
Как нетрудно заметить, метод
противоточной кристаллизации в
принципе аналогичен другому про-
тивоточному методу разделения смесей — ректификации. Раз-
деление в кристаллизационной колонне, как и в ректификаци-
онной, основано на различии составов равновесных фаз. При
осуществлении противоточной кристаллизации разделяемая
смесь может также вводиться в середину колонны, с одного
конца которой находится устройство для кристаллизации, а с
другого — устройство для плавления В этом случае кристалли-
зационная колонна по существу будет состоять из двух сек-
ций, которые соответственно можно назвать исчерпывающей и
132
укрепляющей. Вообще для характеристики процесса противо-
точной кристаллизации из расплава часто применяют основные
понятия и термины, применяемые в ректификации, такие, как
флегмовое число, ЧТТ и т. д Отсюда следует, что использова-
ние понятий ЧТТ и ВЭТТ для оценки эффективности кристал
лизационной колонны дает возможность охарактеризовать ее
разделительную способность в безотборном режиме работы со-
отношением вида (П.50) (гл. II, § 6) Для некоторых конст-
рукций кристаллизационных колонн (для колонн небольших
размеров) значения ВЭТТ лежат в пределах 2—3 см Пример-
но такие же значения ВЭТТ имеют и эффективные насадочные
ректификационные колонны.
Основные модели процесса массообмена
в кристаллизационной колонне
Несмотря на кажующуюся простоту метода противоточной
кристаллизации из расплава, осуществляемый в кристаллиза-
ционной колонне процесс разделения имеет довольно сложную
природу. Во-первых, помимо эффекта разделения, имеющего
место при образовании твердой фазы в кристаллизаторе колон-
ны, в общий эффект разделения будет входить и эффект отмыв-
ки кристаллов от захваченной (окклюдированной) жидкости
движущимся противотоком расплавом. Во-вторых, в колонне
идет процесс частичной перекристаллизации подобно тому, как
в ректификационной колонне может иметь место частичные кон-
денсация пара и испарение жидкости непосредственно в ректи-
фицирующей части. И, в-третьих, поскольку движующиеся про-
тивотоком по колонне твердая и жидкая фазы находятся в кон-
такте друг с другом, между ними будет происходить диффузи-
онный массообмен, аналогичный диффузионному массообмену
между жидкостью и паром в ректификации. Одновременно
в кристаллизационной колонне протекают и другие явления,
такие, как, например, изменение среднего размера кристаллов
и доли твердой фазы Все это в целом затрудняет решение за-
дачи оценки общего эффекта разделения в колонне Этим и объ-
ясняется то, что для описания процесса противоточной кри-
сталлизации в литературе предложены различные модели мас-
сообмена, каждая из которых основана на том или ином допу-
щении об основной лимитирующей стадии процесса.
Первая модель, предложенная А Г. Аникиным, исходит из
допущения о том, что в состоянии «предплавления» диффузия
в твердом веществе должна резко возрастать. В соответствии
с этим принимается, что поскольку кристаллическая масса в ко-
лонне, будучи все время в контакте с расплавом, находится при
температуре, близкой к температуре своего плавления, скорость
диффузии в движущихся по колонне кристаллах достаточно вы-
сока для выравнивания их состава. Таким образом, в данной
133
модели принимается, что в любом заданном сечении колонны
перемещающиеся в ней кристаллы имеют однородный состав,
который зависит лишь от координаты вдоль высоты колонны,
а при нестационарной ее работе и от времени. Такая модель
позволяет провести полную аналогию между противоточной
кристаллизацией и ректификацией и использовать для описания
процесса массообмена в кристаллизационной колонне мате-
матический аппарат, который применяется для описания работы
ректификационных колонн. При этом, разумеется, следует при-
нимать во внимание соизмеримость количеств твердой и жид-
кой фаз в кристаллизационной колонне, тогда как в ректифика-
ционной колонне количеством одной фазы (паровой) по сравне-
нию с количеством другой фазы (жидкой) при расчетах прене-
брегают.
В другой модели, разработанной Дж. Пауэрсом, наоборот,
принимается, что скорость диффузии в твердой фазе в процес-
се противоточной кристаллизации, как и в других кристаллиза-
ционных процессах, по сравнению со скоростью диффузии
в жидкой фазе пренебрежимо мала Отсюда следует, что боль-
шое влияние на чистоту получаемого продукта должен оказы-
вать эффект разделения, имеющий место в кристаллизаторе ко-
лонны при образовании твердой фазы. Например, при разделе-
нии смеси, компоненты которой образуют непрерывный ряд
твердых растворов, степень очистки не должна превышать вели-
чины а Поскольку в опытах обычно достигается более высокая
степень очистки. Дж. Пауэрс пришел к выводу, что общий эф-
фект разделения в кристаллизационной колонне, по-видимому,
обусловлен многократной перекристаллизацией кристаллов,
движущихся в противотоке с жидкостью. Важную роль при
этом, по его мнению, играет также и «экстрактивная» отмывка
поднимающимся расплавом движущихся кристаллов от захва-
ченной (окклюдированной) маточной жидкости, загрязненной
примесью.
Сравнивая обе модели, можно отметить, что, будучи основа-
ны на прямо противоположных исходных предпосылках, они
характеризуют предельные случаи при оценке влияния скорости
диффузии в твердой фазе на скорость массообмена в кристал-
лизационной колонне. Влияние размера движущихся кристаллов
па эффект разделения учитывается в уравнениях, характери-
зующих обе модели, лишь косвенно, через соответствующие ко-
эффициенты, выражающие удельную поверхность контакта фаз
при диффузионном массообмене между ними — первая модель,
при теплообмене (перекристаллизация) и «экстрактивной» от-
мывке — вторая модель. Основным недостатком первой модели
является то, что она построена на очень грубом допущении, так
как скорость диффузии в твердых веществах, вероятно, всегда
зна штельно меньше чем в жидкостях. Но, с другой стороны,
134
диффузией в кристаллах, находящихся в колонне при темпера-
туре, близкой к температуре своего плавления, не всегда можно
пренебречь, как принято во второй модели. Это тем более нуж-
но иметь в виду, поскольку образование кристаллов в кристал-
лизаторе колонны происходит в неравновесных условиях, ввиду
чего они имеют большее число дислокаций- в кристаллах с по-
вышенной плотностью дислокаций диффузия протекает значи-
тельно быстрее, чем в более совершенных монокристаллах того
же вещества.
Таким образом, совершенно очевидно, что в целом эффект
очистки, достигаемый в кристаллизационной колонне, зависит
от скорости диффузии примеси в движущихся по колонне кри-
сталлах. Это предположение и лежит в основе модели, согласно
которой лимитирующей стадией процесса массообмена в кри-
сталлизационной колонне является диффу-
зия в твердой фазе. В соответствии с этой
моделью размер кристаллов, составляющих
твердую фазу, играет большую роль в
процессе массообмена. Экспериментальная
проверка показала, что указанная модель
хорошо согласуется с результатами опы-
тов. Поэтому рассмотрим ее подробнее.
Пусть в первом приближении кристал-
лы, движущиеся сверху вниз по колонне с
постоянной скоростью, имеют форму пла-
стинок одинакового размера — толщиной
2П7 Принимается также, что доля твердой
фазы по высоте колонны не изменяется и
Рис. 37. Элемент
объема кристалли-
зационной ко-
лонны
что продольным перемешиванием в колон-
не можно пренебречь. Исходя из принятых
допущений, уравнение материального баланса по примеси для
элемента объема кристаллизационной колонны (рис. 37), ра-
ботающей в стационарном состоянии, можно записать в виде
, дх дгх
(Ш.38)
с граничными условиями
4^1 =о’
OW |lr=o
x(w, 0) = x(w, 0) = хо, x(w, zK) = z2^.
(III.39)
(III.40)
Выражение (III 38) представляет собой видоизмененную за-
пись известного уравнения диффузии. Здесь L — Lt^t, Lt — ско-
рость потока твердой фазы, см3/(см2-с); рг — плотность твердой
фазы, моль/см3; х?— доля твердой фазы в элементе объема
135
колонны, DT — коэффициент диффузии примеси в твердой фазе,
см2/с, х— молярная доля примеси в твердой фазе; z— коор-
дината вдоль высоты колонны, w — координата вдоль полутол-
щины кристалла (с началом в середине кристалла).
Соответственно рассматриваемой схеме процесса, по которой
питание поступает сверху, а продукт отбирается в нижней части
колонны, запишем уравнение материального баланса по при-
месному компоненту в сечении колонны z, которое имеет вид,
аналогичный уравнению (II 82)
х=(1-Р)у+рур, (III 41)
но здесь уже х — средняя концентрация примеси в твердой фазе
в данном сечении колонны; у — концентрация примеси в жид-
кой фазе в этом же сечении колонны; уР— степень отбора про-
дукта.
Если скорость диффузии в твердой фазе лимитирует ско-
рость массообмена, то на границе раздела фаз (u»=U7) должно
иметь место равновесное соотношение между x(W,z) и y(z),
т. е. тогда будет справедливым соотношение вида (Ш2). По-
скольку рассматривается процесс очистки основного вещества
от примеси, кристаллы в низу колонны будут содержать меньше
примеси, чем кристаллы в верху колонны, и, следовательно, бу-
дем иметь, что а< 1 В дальнейшем для удобства будем пользо-
ваться величиной а'= 1/а С учетом этого, используя теорему
о среднем значении для выражения х, из (III 41) получаем
w
-j-p- х(и>, z)dt« = a'(l—p)x(W, z) + pj/p. (III.42)
о
Для установления пусть приближенной, но простой и наглядной
аналитической взаимосвязи параметров процесса противоточной
кристаллизации рассмотрим случай, когда интересующая при-
месь является трудноотделимой, т. е. имеет коэффициент раз-
деления, близкий к единице.
При указанном условии (а'—1)<С1 зависимостью dxfdz от
координаты w вдоль полутолщины кристалла м зжно прене-
бречь. Это позволяет перейти в уравнении (III.38) от частных
производных к обыкновенным. В результате уравнение (III.38)
преобразуется к виду
d2x LT dx
dur Pj-Xy dz '
(III.43)
Интегрирование уравнения (III 43) при граничном усл вии
(III.39) дает
, Ltw dx , .
dx=—-------— dtp. (III.44)
PTxy dz
136
С учетом (III 44) преобразуем левую часть выражения (III 42)
IV W IV
— ( x(w, z)dw = ^£ | x(w, z) w — u?dxj =
о о и
IV
=тИ1|И'- ‘IV—% J .>- /дг^‘. (in«)
0
Подставляя (III.45) в (III.42), получаем
„У*?* z} — pyp (Ш.46)
OZzT’Xy QZ
или, так как ур=хр и полагая, что для значении а', близких
к единице, x(W, z)^x^x(w, z),
-^- = {(l-a'(l-P)P-P^)-^r. (IIL47)
Из выражения (III.47) после разделения переменных будем
иметь
------__£тхт (а,_ 1} dz> (HI.48)
(1- ав') х+в'хр L?w
где 0'=р/(а/—1).
Фактор разделения в стационарном состоянии
и безотборном режиме
Для безотборного режима (0'=О) из уравнения (II 1.48) сле-
дует, что
-^=—dz (1Н.49)
х LtW*
Решая уравнение (III 49) с граничными условиями (III 40), по-
лучаем следующее приближенное выражение для фактора раз-
деления кристаллизационной колонны.
Fo=-^-=exp [- 3PT7r!v*~11 Zk] • (IIL50)
Из сравнения формул (П1.50) и (П.86) следует, что 3DTxrlW2—
величина, аналогичная константе массообмена в процессе рек-
тификации (если скорость потока жидкости при этом также вы-
ражать в объемных единицах), которая лежит в пределах
~ Ю ’2—10 3 с-1. Как уже отмечалось, коэффициент диффузии
в твердых телах обычно мал и составляет 10-12—10-14 см2/с и
ниже. Но вблизи температуры плавления вещества он значи-
тельно выше и может достигать значений ~10~6—10 7 см2/с.
137
Например, при U7= Ю~2 см и хг= 1/3 ЗГт-хг/И72» 10~2—10~3 с~'.
Следовательно, противоточная кристаллизация из расплава мо-
жет быть не менее эффективна, чем ректификация для разде-
ления смесей веществ, например, имеющих молекулярную кри-
сталлическую решетку, и при удалении примеси, кристаллизую-
щейся по границам’зерен в поликристаллах основного веще-
ства.
В случае, когда коэффициент разделения заметно отличается
от единицы, решение дифференциального уравнения (III 38)
усложняется; полученное уравнение для расчета фактора раз-
деления колонны в этом случае имеет вид (см. Приложение):
/0-[1 а (е*))ехр[ — Db (е')], (1П.51)
где е'=(а'—1)/а', D= (DTy.TzK)/LrW2, а коэффициенты а(е')
и b(e') выражаются аппроксимирующими формулами
Выражение (III.51) позволяет при заданных значениях D и а'
оценить разделительную способность кристаллизационной ко-
лонны, работающей в стационарном состоянии и безотборном
режиме. Нетрудно видеть, что для коэффициентов разделения,
близких к единице, уравнение (Ш.51) упрощается и переходит
в выражение (III.50).
Влияние скорости отбора продукта
на фактор разделения
В отборном режиме работы кристаллизационной колонны
часть образующегося из поступающих вниз кристаллов распла-
ва постоянного состава уР=хр выводится из колонны в качестве
продукта. В случае, когда коэффициент разделения мало отли-
чается от единицы, решение дифференциального уравнения
(III.48) с граничными условиями (111.40) и с учетом того, что
в отборном режиме хгк—хр, имеет вид
(1—g'eQzp+e'xp
(1—a'©') z0 + ©'zp
(III.52)
Приняв во внимание выражение (111.50), из соотношения
(III.52) после небольших преобразований получаем
1 —а'©'
(111.53)
где F=xPlxo — фактор разделения кристаллизационной колон-
ны в отборном режиме.
Нетрудно заметить, что уравнение (II 1.53) отличается от
уравнения (II.73), характеризующего разделительную способ-
138
ность насадочной ректификационной колонны в отборном режи-
ме (гл. II, § 7), на величину 1/(1— р) в показателе степени при
Fo. Это объясняется тем, что при выводе (111.53) было принято,
что стадией, лимитирующей скорость массообмена в кристалли
зационной колонне, является диффузия в твердой фазе. При вы-
воде же (11.73) принималось, что скорость массообмена в рек-
тификационной колонне пропорциональна разности текущей и
равновесной концентраций в паровой фазе
При заметном отличии коэффициента разделения от едини-
цы решение дифференциального уравнения (III 38) для отбор-
ного режима работы кристаллизационной колонны, тес учетом
соотношения (П142), приводит к следующему приближенному
выражению для фактора разделения (см Приложение)
F=[l>(e', р] ехр [ — Db (е', p)J, (111.54)
где ° («'. р) = ехр{0,21е'—(1 — е')11,2р/е'4-1,7 (р'е')21);
b (е\ р)=2,47е'-|-е'(1—е')[0,497 — 2,42р;'е*—1,68 (р/е')2].
Как можно видеть, для расчетов по формуле (III 54) необ-
ходимо знание величины D, которая может быть найдена, если
известен коэффициент диффузии примеси в твердой фазе Dr при
рабочих условиях процесса противоточной кристаллизации, т. е
вблизи температуры плавления основного вещества. К сожале-
нию, таких данных в литературе мало. Поэтому можно рекомен-
довать косвенный способ определения D из результатов опытов
по определению разделительной способности кристаллизацион-
ной колонны в безотборном режиме с помощью формулы
(III 51) Во избежание недоразумений следует отметить, что ап-
проксимирующие формулы для а(е', р) и Ь(е', р) в выражении
(III 54) при р = 0 не переходят в соответствующие формулы для
о(е') и Ь(е') в выражении (III 51). Это-обусловлено различием
в аппроксимациях. Но тем не менее результаты вычислений
указанных коэффициентов по приведенным формулам практиче-
ски совпадают.
Распределение примеси
по высоте кристаллизационной колонны
При оценке эффекта разделения смеси веществ многоступен-
чатыми методами, к каковым относится и противоточная кри-
сталлизация, важным является вопрос о распределении компо-
нентов смеси по высоте (длине) разделительного аппарата —
колонны. Знание этой зависимости позволяет предсказать, в ка-
кой степени увеличение высоты колонны будет влиять на ее
разделительную способность и целесообразно ли использование
на практике, для достижения большей глубины очистки, высо-
ких колонн, которые обычно работают хуже, чем короткие
139
Ограничиваясь рассмотрением безотборного режима, при при
нятых выше допущениях, распределение примеси в твердой фазе
по высоте кристаллизационной колонны, можно выразить (см.
Приложение) соотношением
X = ХО [1/а (е')1 ехр [—Db (е') <р], (П1.55)
где коэффициенты а(е')> Ь(е') и D имеют те же значения, что
и в уравнении (III.51); tp=z/zK-
Из соотношения (Ш.55) следует, что при постоянных пара-
метрах процесса противоточной кристаллизации стационарное
распределение примеси в твердой (аналогично и в жидкой) фа-
зе по высоте колонны должно иметь экспоненциальный харак-
тер, что наблюдается и в других противоточных методах глубо-
кой очистки [см. уравнение (11.66)] Однако, как известно, в ре-
альных условиях при перемещении твердой фазы в колонном
аппарате она подвергается частичной перекристаллизации,
вследствие чего размер составляющих ее кристаллов изменяет-
ся. Дело в том, что при своем образовании в зоне кристаллиза-
ции они, по существу, имеют уже неодинаковый размер вслед-
ствие неоднородности температуры переохлажденного расплава
у охлаждаемой поверхности. Выходящая из зоны кристаллиза-
ции такая мелкодисперсная кристаллическая масса обладает
избыточной поверхностной энергией. Следовательно, рассматри-
ваемая система кристаллы — расплав при этом является термо-
динамически неустойчивой, что обусловливает протекание в ней
прежде всего процессов, направленных в сторону уменьшения
поверхностной энергии твердой фазы. Это будет характеризо-
ваться увеличением размера частиц твердой фазы, т. е. сниже-
нием удельной поверхности кристаллов в колонне. В результате
кристаллы при своем движении по колонне должны или укруп-
няться или число их должно уменьшаться. Из имеющихся в ли-
тературе экспериментальных данных следует, что в кристалли
зационной колонне протекают оба эти явления: происходит
плавление мелких и одновременно рост более крупных кристал-
лов, т е в процессе противоточной кристаллизации происходит
увеличение среднего размера движущихся кристаллов.
Поскольку увеличение среднего размера кристаллов приво-
дит к уменьшению величины D в уравнении (III.55), то распре
деление примеси по высоте кристаллизационной колонны долж-
но отклоняться от экспоненциального; разделительная способ
ность колонны при этом будет ухудшаться. Для приближенной
оценки ожидаемого при этом эффекта снижение глубины очист-
ки рассмотрим случай, когда доля твердой фазы и скорость ее
движения по колонне не изменяются, что обычно имеет место в
колоннах со шнековым транспортером кристаллов. При этом
можно принять, что в процессе массовой кристаллизации из рас-
плава, находящегося при температуре, близкой к температуре
140
плавления твердой фазы, увеличение среднего размера кристал-
лов обусловлено плавлением метких и ростом более крупных.
Такой процесс, по-видимому, может протекать лишь тогда ко-
гда переохлаждение расплава по высоте колонны относительно
невелико и флуктуационное образование зародышей кристаллов
и их непосредственный рост из переохлажденного расплава ис-
ключаются. Изменение среднего размера кристаллов в ходе про-
цесса при принятом допущении о постоянстве других параметров
будет приводить к изменению разделительной способности рав-
ных по величине, но разных по высоте участков кристаллизаци-
онной колонны, т. е. к увеличению ВЭТТ Соответствующую за
висимость нетрудно найти, если положить, что уравнение (111.51)
справедливо к некоторому среднему размеру кристаллов твер-
дой фазы в колонне, который будем выражать как
И'ср- ^Н2 И/-К , (III.56)
где ТГо — средний размер (полутолщина) кристаллов в зоне
кристаллизации (z — 0); — средний размер (полутолщина)
кристаллов на входе в зону плавления (z=zK).
Тогда из уравнения (III.51) следует, что
DT ~----nnfo-^ lna (е')1 |уг (111.57)
Ъ (₽/) XTZ„
Аналогично средний размер кристаллов па участке колонны от
зоны кристаллизации до заданного поперечного сечения z выра-
зится соотношением
щср , (Ш.58)
где W — средний размер (полутолщина) кристаллов в сечении
колонны z. Подставив выражения (111.56) — (111.58) в уравнение
(III.55), получаем
*=?<> |а(с')ехр — (In Fo-|-In а (е') + (Ш.59)
Соотношение (III.59) позволяет охарактеризовать распреде-
ление примеси по высоте колонны с учетом изменения среднего
размера кристаллов твердой фазы. Для этого необходимо знать
зависимость 1И от г. Опыты по очистке ряда веществ (бензол,
стильбен, сера, аммиак, трихлорид мышьяка, тетрахюрид крем-
ния) показали, что эта зависимость хорошо описывается выра-
жением
W;= И’о+const i<z (111.60)
или, учитывая, что при z = zK T7=TFK,
W'=Fr0+(TPK-iv0) j/<p. «11.61)
141
После подстановки выражения (11161) в соотношение (111.59)
и логарифмирования получаем следующее уравнение для описа-
ния распределения примеси по высоте кристаллизационной
колонны.
lg*=lg[*o/e(Hl+[lg *о+1е°(И1 Г ———- ——7=т-12. (Ш.62)
L (1- И0/Щк) (I- 2 /<р) J
Более строго влияние изменения среднего размера кристаллов
по высоте колонны на распределение примеси можно учесть
путем решения дифференциального уравнения (111.38) с учетом
(III.61). Однако аналитического выражения соответствующей
зависимости получить при этом не удается, что вынуждает огра-
ничиться лишь численными расчетами.
Сравним экспериментальные данные, полученные при очист-
ке трихлорида мышьяка*, и соответствующую кривую, рассчи-
танную по уравнению (III 62). Наблюдаемое на рис. 38 удовле-
творительное согласие между результатами расчета и опыта
позволяет положительно ответить на вопрос о правомочности
использования понятия среднего размера кристаллов в кристал-
лизационной колонне и соответственно выражений (III.56) —
(111.59) для описания процесса очистки. Это в равной степени
будет относиться и к отборному режиму работы колонны. В этом
случае под входящей в приведенный коэффициент диффузии D
величиной W в уравнении (III.54) следует понимать средний по
высоте колонны размер кристаллов Ц/Ср, а под величиной хг —
среднюю величину доли твердой фазы хср, если имеет место
также и изменение доли твердой фазы по высоте колонны
(рис. 39)**
Необходимо заметить, что поскольку выделяющийся на рас-
тущих кристаллах новый слой твердой фазы содержит меньше
примеси, чем окружающий их расплав, имеющая при этом место
перекристаллизация вносит, с одной стороны, свой положитель-
ный вклад в общий эффект очистки, но с другой — рост кристал-
лов приводит к уменьшению эффекта их очистки за счет диффу-
зионного механизма Если бы последствия этих явлений взаимно
компенсировали друг друга, то следовало бы ожидать согласия
получаемых при этом результатов с теоретической зависимостью
(III 55). Но этого не наблюдается- зависимость lgx~z (см
рис. 38) заметно отличается от линейной. Отсюда можно сделать
* Аглиулов Н X., Зеляев И. А., Фещенко И П и др. Глубокая очистка
летучих хлоридов методом противоточной кристаллизации из расплава//В кн .-
Гидриды, галиды и металлоорганические соединения особой чистоты М 1976.
с. 33- 46.
** Еллиев Ю. Е., Гурьянов А. Н., Девятых Г. Г. Исследование работы
кристаллизационной колонны в отборном режиме при глубокой очистке ве-
ществ//Теор основы хим технол. 1975, т. 9, № 5, с. 773—776
142
Рис. 38. Распределение примеси
по высоте кристаллизационной ко-
лонны [кривая рассчитана по
уравнению (III 62)]:
О — экспериментальные результаты по
очистке трихлорида мышьяка от 1.1.2-
трнхлорэтаиа (а—0,16)
Рнс. 39. Зависимость разделитель-
ной способности кристаллизацион-
ной колонны от скорости отбора
продукта:
кривые /—4 рассчитаны по уравнению
(III 54) с учетом выражений (III.56) и
(III.57); О, Ф. X. А — эксперименталь-
ные результаты по очистке бензола от
тиофена (а—0.42)
вывод, что укрупнение кристаллов в колонне — явление в целом
нежелательное и для достижения большей глубины очистки луч-
ше было бы, если бы движущиеся из зоны кристаллизации в зо-
ну плавления кристаллы не меняли своего размера. Таким об-
разом, следует подбирать такие условия протекания процесса,
чтобы по возможности «затормозить» перекристаллизацию твер-
дой фазы или рост кристаллов за счет окружающего расплава.
Однако таких схем процесса противоточной кристаллизации по-
ка не разработано. Но тем не менее одна из практических реко-
мендаций интенсификации процесса глубокой очистки веществ
противоточной кристаллизацией из расплава, исходя из изло-
женного, очевидна — это последовательное дробление (диспер-
гирование) растущих кристаллов в определенных сечениях по
высоте колонны. Хорошие перспективы в этом отношении имеет
ультразвуковое воздействие.
143
§6 ВЛИЯНИЕ ЗАГРЯЗНЯЮЩЕГО ДЕЙСТВИЯ
МАТЕРИАЛА АППАРАТУРЫ НА ГЛУБИНУ
ОЧИСТКИ ВЕЩЕСТВ
КРИСТАЛЛИЗАЦИОННЫМИ МЕТОДАМИ
Как известно, кристаллизация из расплава используется для
очистки многих веществ, в том числе и таких тугоплавких, как
кремний, германий, различных металлов и солей. Однако высо-
кая температура процесса увеличивает вероятность взаимодей-
ствия очищаемого вещества с материалом разделительной ап-
паратуры, что приводит к загрязнению этого вещества. Напри-
мер, в процессах зонной очистки и выращивании монокристал-
лов германия он долго находится в расплавленном состоянии
при температуре ~ 1000 °C в контакте с контейнером (лодоч-
кой). Хотя контейнер обычно изготавливают нз графита высо-
кой чистоты, тем не менее оказывается, что в ходе процесса
имеет место переход некоторых примесей, содержащихся в гра-
фите, в германий. Следовательно, задача подбора подходящих
конструкционных материалов в подобных случаях приобретает
важное значение. С целью выработки рекомендаций по повы-
шению их качества или замены представляет интерес оценка
загрязняющего действия этих материалов. Рассмотрим кратко
некоторые оценки загрязнения очищаемого вещества примесью,
одноименной с отделяемой.
Направленная кристаллизация
В процессе нормальной направленной кристаллизации (в
дальнейшем, как и выше, для краткости будем называть ее на-
правленной) расплав находится в контакте с поверхностью кон-
тейнера площадью S, которая является переменной. Примем,
что скорость поступления примеси v„ с единицы поверхности
контакта постоянна. Тогда для малого промежутка времени
процесса d(, за который закристаллизуется dNT молей вещест-
ва, будет справедливо следующее уравнение материального ба-
ланса:
у (NB— Nr)+vnS df = zdArT+(A'o—A'T—d?VT)(y-pdy). (111.63)
Подставляя выражение (HI 2) в (III 63) и пренебрегая членом
dydNr как величиной второго порядка малости, получаем
(1— a)J/d^ + vnSdi = (^ — ,WT)dy. (III.64)
Пусть контейнер расположен вертикально и имеет форму ци-
линдра длиной zK и радиуса г0; следовательно, можно записать,
что го=У$/л, где S — площадь поперечного сечения контейне-
144
ра Тогда площадь контакта расплава с контейнером будет вы-
ражаться соотношением
5 = 2nr0(zK—z) = 2 /^(zh-s), (III.65)
где z— координата вдоль высоты (длины) контейнера. Но по-
скольку SzKpK=N0 и SzpT = NT и если принять что рж^рг, то
zK—z-(No—NT)/(Sp>«). С учетом этого из (III 65) следует, что
5 = [(2 |^75/рж1 (М—^т)- ПН-66)
Малый промежуток времени d( выразим через количество за-
кристаллизовавшегося вещества dWr, скорость кристаллизации
икр и перемещение фронта кристаллизации на расстояние dz с
помощью соотношений nKpdf=dz и SdzpT=dNr, из которых
имеем
dIVт = vKP5pT dt (III.67)
Подстановка (III 66) и (III 67) в (III 64) дает
дау+г»-?. О"-68»
где Р=(а—l)/(No—NT)', <^=2гп/(лго3ЦкРржрг)
Решение дифференциального уравнения (III 68) имеет вид»
подобный соотношению (11.179):
у=ехр ( — J Р dN? ) [ J (Q ехР J PANt ) йЛтЧ-Л |. (III.69)
Проинтегрировав уравнение (III.69) и определив постоянную
интегрирования А из граничного условия у = уо при ZVr=O, по-
лучаем
/ NT\a-i QN0 I NT CM /. NT\ 0
мг) +2=^(1—nJ nJ’ (IIL7U?
Так как уо=Хо, то, используя соотношение (III.2), из выраже-
ния (III.70) будем иметь
х=(ахо+^)(1-хг)»-1—g^(l-xT). (111.71)
Из соотношения (III 71) нетрудно получить выражение для рас-
чета содержания примеси х в отбираемом продукте-
(Ш72’
где хж — доля отбрасываемой по окончании процесса «хвосто-
вой» фракции (концентрат примеси). Как можно видеть, при
отсутствии эффекта загрязнения, т. е. при (7=0, выражения
(III.71) и (III.72) переходят в уравнения (III.9) и (111.12) со-
145
ответственно. Выражения (III.70)—(Ш.72) могут быть исполь-
зованы для расчета глубины очистки при многократном повто-
рении процесса направленной кристаллизации.
Зонная перекристаллизация
В процессе зонной перекристаллизации длина расплавленной
зоны го не изменяется (за исключением начала слитка) и поэто-
му площадь ее контакта с контейнером Sc будет также посто-
янной Отсюда, обозначая, как и выше, скорость поступления
примеси в расплавленную зону из материала аппаратуры через
г?п. уравнение материального баланса применительно к первому
проходу расплавленной зоны можно записать в виде
yG4-z0<bVT-(-uriSc = x dArT+ (у-f-dy) G (III.73)
или, с учетом соотношений (Ш.2) и (III.61),
(Д<,-«У+--Р°с5 )<LVr = Gdy. (Ш.74)
Полагая, что и в рассматриваемом случае контейнер представ-
ляет собой вертикально расположенный цилиндр радиуса г0,
легко показать, что 5с = 2б/(г0рж), и таким образом, уравнение
(III.74) можно записать как
dy AJVt
z<>—ay + bn ~ G ’
(111.75)
где b„= (2гпб)/(лго3Пкрржрг). Нетрудно видеть, что выражение
для Ьп в уравнении (III.75) лишь на множитель G отличается
от выражения Q в уравнении (III 68)
После интегрирования уравнения (III.75) с граничным усло-
вием у—уо' при NT=0 получим
In —аУ~ЬЬп _ ___ _
^0—°4/o4-bn G
(111.76)
Использование граничного условия означает, что в нача е про-
цесса перед выделением твердой фазы из расплавленной зоны
часть исходного образца очищаемого вещества длиной в одну
зону находится в жидком состоянии и имеет концентрацию при-
меси уо'. Таким образом, величина у0' учитывает загрязняющее
действие материала контейнера в период движения фронта
плавления от начала слитка до создания полной расплавленной
зоны Так как эта величина отличается от величины t/o = xo.
характеризующей случай, когда загрязняющее действие мате-
риала контейнера отсутствует, то, вводя для отражения указан-
ного различия коэффициент kn, можно записать, что yo'=kayo —
14 6
= knxo. Тогда с учетом этого и выражения (Ш.2) соотношение
(III 76) запишется в виде
х0—X—Ьп=(х0—аЛ‘пх„-|-bD)exp ( — aU) (III 77)
или
x=z0[l + (afcn — 1)ехр( — аС)Ц-Ьп[1— ехр( —аР)] (III.78)
Когда ип=0, то 6п=0, а уо'=уо, т. е fen=l, и уравнение (III 78)
переходит в известное выражение (III. 19)
Коэффициент kn нетрудно оценить следующим образом. Ко-
личество примеси в 1-й зоне при отсутствии эффекта загрязне-
ния будет составлять no = yoG молей. За период времени обра-
зования этой зоны tc при наличии эффекта загрязнения в нее
поступит из стенок контейнера дополнительно п„ молей приме
си и, следовательно, будет справедливо уравнение баланса
По + пп = уо'(С + пп). Отсюда получаем
Уо (СЦ- П„) Ид-р Пп ЦГ уд,
y0G — п0
ИЛИ
‘”=(*+х)/(,+^)- <|П-80’
Для оценки коэффициента kn необходимо знать лп. Если обо-
значить через Sg площадь контакта расплава со стенками кон-
тейнера при образовании 1-й расплавленной зоны, то будем
иметь
*G
^п~ d/.
о
(III.81)
Так как 5с = 2лгог, a 2=uKpt где г —часть длины 1-й расплав-
ленной зоны, образовавшейся к моменту времени t, то из (III 81)
следует, что
пп = лгоипркр^. (III.82)
Но, с другой стороны, время образования 1-й расплавленной
зоны связано с ее длиной выражением /с=гс/цкр Длину же
зоны нетрудно выразить через количество вещества в зоне как
2с = О/(лг02Рж) Исходя из этих выражений соотношение (III 82)
можно преобразовать к виду
(III.83)
Подставляя (III 83) в (III 80) и учитывая, что n0=y0G, полу-
чаем
Лп=(1-1
___________) / I1 л_)
П’о^рРжУо II \ ЛГ^нрР^ /
(III.84)
147
Как можно видеть, коэффициент kn в целом зависит от тех же
параметров процесса, что и величина Ьп.
При многократном проходе расплавленной зоны распределе-
ние примеси по длине слитка будет так же изменяться в сторо-
ну снижения концентрационного профиля, но в меньшей степе-
ни, чем при отсутствии эффекта загрязнения. Это обусловлено
тем, что если каждый последующий проход расплавленной зо-
ны вносит все меньший вклад в глубину очистки за счет пере-
кристаллизации, то сопутствующий ему эффект загрязнения
остается практически неизменным. Отсюда следует, что в том
периодическом варианте, в каком обычно осуществляется зон-
ная перекристаллизация, и при наличии эффекта загрязнения
с постоянной скоростью поступления примеси v„ нельзя ожидать
достижения предельного распределения примеси по длине слит-
ка, подобного тому, которое имеет место при отсутствии эффек-
та загрязнения.
Противоточная кристаллизация
При наличии эффекта загрязнения в процессе противоточной
кристаллизации примесь из стенок колонны, а также с поверх-
ности транспортирующего или перемешивающего устройств по-
ступает в объем расплава и распределяется по нему. В резуль-
тате массообмена загрязняемого таким образом расплава с кри
сталлами как за счет диффузии, так и за счет частичной
перекристаллизации глубина очистки кристаллов будет ухуд-
шаться. Соответственно уравнение материального баланса по
примесному компоненту для сечения z кристаллизационной ко-
лонны, работающей в стационарном состоянии, при постоянной
скорости поступления примеси цп в единицу объема колонны
можно записать в виде
= г)] у — <’п(«к—4+пУр (Ш.85)
ИЛИ, ПОСКОЛЬКУ VnZKCl И VnZ-^l,
z=(l — p)y+pyp — (vn/L) (zK—z), (III.86)
где р — степень отбора; L = LTpT.
Ограничимся рассмотрением безотбориого режима работы
колонны (р = 0). Тогда при (а.'—1)«1 из уравнения (III 86)
с учетом выражения (III 45) применительно к среднему размеру
(полутолщине) кристаллов №Ср получаем
«+-Х?Г°
или, используя соотношение (III.50),
_ InFp - In рп .
dz z1( zK L (a' — 1)' K
(III.88)
148
Дифференциальное уравнение (Ш.88) является уравнением то-
го же типа, что и уравнения (11.102) и (III 68). Нетрудно пока-
зать, что его решение с определением константы интегрирова-
ния из граничного условия г = 0, х=х<> приводит к соотноше-
нию вида
x = x0Fz/lK+r-.Vn,ZK Г (1—(1-Г*/гк) -I. (Ill.89)
“о ' L (а — 1)L\ InZ-e/4 » 2к J
Из соотношения (III.89)
тора разделения колонны
можно получить выражение для фак-
F 01 %О'-
^пгк Fo (1 I*1 ^о) 1
Проводя в (Ш.90) преобразования, аналогичные тем, что ис-
пользовались при выводе (II I11), получим выражение для
оценки предельного значения концентрации примеси в очищае-
мом веществе на выходе из колонны
"п
,. _ _ ВЭТТ 1>п ..патт 'Ъ
Ina' L(a'-l) -ВЭТТ L(a'-l)’
к
(111.91)
Как можно видеть, выражение (Ш.91) мало отличается от ана-
логичного выражения (II 111), полученного применительно к
процессу ректификационной очистки. Однако ввиду своего весь-
ма приближенного характера выражение (Ш.91) отражает, по
существу, взаимосвязь параметров процесса очистки при нали-
чии эффекта загрязняющего действия материала разделитель-
ной аппаратуры лишь качественно. Количественную оценку
можно провести на основании числовых расчетов по (111.38)
и (111.86).
§ 7. КРИСТАЛЛИЗАЦИЯ ИЗ РАСТВОРА
Кристаллизация из раствора как метод разделения и очист-
ки веществ находит широкое применение. Особенно успешно
этот метод используется для разделения смесей солей, так как
при этом в большинстве случаев в качестве дешевого раствори-
теля может быть использована вода, а растворимость солей в
воде обычно существенно меняется с изменением температуры.
Последнее дает возможность последовательного выделения из
раствора фракций кристаллов, содержащих в основном тот или
иной интересующий компонент Каждая из этих фракций затем
может быть подвергнута перекристаллизации с целью удаления
находящихся в лей нежелательных примесей других веществ.
Для очистки ряда веществ с этой точки зрения хорошими раст-
ворителями являются различные спирты и эфиры, ацетон, бен-
зол, сероуглерод и т. д. В нефтеперерабатывающей промышлен-
ности в целях избирательной растворимости отдельных
149
компонентов разделяемой смеси иногда используются комбини-
рованные растворители.
При кристаллизации из раствора процесс образования твер-
дой фазы связан с выделением в нее преимущественно раство-
ренного вещества (веществ). Наоборот, при кристаллизации из
расплава в твердую фазу в основном переходит растворитель,
роль которого играет основное вещество, находящееся в рас-
плавленном состоянии. Поэтому в отличие от кристаллизации
из расплава кристаллизация из раствора обычно проводится
при сравнительно невысоких температурах. В этом отношении
кристаллизация из раствора обладает преимуществом, так как
с ее помощью можно производить очистку большего числа тер-
монестойких или тугоплавких веществ. Но, с другой стороны,
наличие растворителя осложняет процесс очистки, так как в
растворе могут происходить процессы сольватации (гидратации)
или сольволиза (гидролиза) растворенного вещества. Кроме
того, исходный растворитель должен быть уже сам по себе до-
статочно чистым во избежание попадания в ходе процесса не-
желательных примесей в кристаллы очищаемого вещества.
Кристаллизация из раствора, как и кристаллизация из рас-
плава,— сложный процесс, представляющий собой совокупность
нескольких последовательно и параллельно протекающих ста-
дий. Основными из них также являются стадии зарождения
кристаллов и их роста. Но движущей силой процесса при этом
будет пересыщение раствора, под которым понимается избыточ-
ная концентрация содержащегося в растворе вещества сверх
его растворимости при заданной температуре в рассматривае-
мом растворителе. Причем оказывается, что образование цент-
ров кристаллизации и рост кристаллов в растворе имеет мест»
лишь при определенном его пересыщении, т. е. используя для
характеристики пересыщенных растворов понятие степень пере-
сыщения 1п^=Уп1у«, где уп и уи— концентрации растворенного
вещества в пересыщенном и насыщенном растворах, можно
утверждать, что образование центров кристаллизации не будет
происходить не только при уп^1, но и в некотором интервале
7п>1 Если же степень пересыщения раствора достаточно вы-
сока, то после возникновения в нем кристаллических зародышей
на последних начинается выделение твердой фазы—массовая
кристаллизация.
Способы осуществления процесса
Так как растворимость большинства твердых веществ умень-
шается с понижением температуры, то необходимого пересыще-
ния, обусловливающего выделение кристаллов, можно достиг-
нуть охлаждением исходных горячих растворов. Этот вариант
методов кристаллизации из растворов получил название изо-
гидрической кристаллизации-, при его осуществлении количест-
во
во растворителя в растворе практически остается постоянным.
Разновидностью этого варианта является кристаллизация вы-
мораживанием, которая осуществляется охлаждением растворов
до температур ниже О °C Этот способ используется для концент-
рирования термолабильных веществ и выделения их из смесей,
для обессоливания воды, для очистки органических раствори
телей.
Перевод исходного раствора в пересыщенное состояние мож-
но осуществить и за счет частичного удаления растворитезя
выпариванием раствора. Такой вариант получил название изо-
термической кристаллизации, поскольку после достижения со-
стояния насыщения выпаривание раствора в этом случае про-
исходит при постоянной температуре.
Выбор оптимального варианта кристаллизации из раствора
зависит от характера изменения растворимости вещества от
температуры. На практике иногда приходится комбинировать
оба варианта, как это, например, имеет место в методах ваку-
умной кристаллизации, в которых вследствие откачки раство-
рителя одновременно происходит понижение температуры раст-
вора: раствор становится пересыщенным и из него выделяется
твердая фаза Преимуществом такого комбинирования является
то, что криста злизация здесь происходит в целом в объеме
раствора, а не на охлаждаемой поверхности, как это обычно
бывает при изогидрической кристаллизации, и при более низкой
температуре по сравнению с изотермической кристаллизацией
в отдельности, что очень важно с точки зрения глубокой очист-
ки термонестойких веществ
Существуют и другие способы выделения твердой фазы из
раствора, например путем добавления в раствор какого-либо
специально подобранного вещества, которое снижает раствори-
мость выделяемого вещества; этот способ получил название
высаливания. При проведении так называемой аддуктивной
кристаллизации в исходный раствор вводится реагент, образу-
ющий с выделяемым веществом менее растворимое комтекс-
ное соединение — аддукт Здесь мы имеем пример проведения
процесса кристаллизации в сочетании с химической реакцией
Для полноты извлечения вещества из раствора процесс иногда
осуществляют в противоточном варианте: раствор подается в
один конец колонного аппарата, а реагент вводится в другой
конец этого аппарата. Кристаллы полученного аддукта отфильт-
ровывают и подвергают разложению и очистке (термораспад с
последующей перекристаллизацией выделяемого вещества из
специально подобранного растворителя, перегонка с водяным
паром и т. д ). Способ комплексообразования применяется и для
химического связывания примесей в соединения, легко отделяе-
мые от основного вещества; образование осадка при ^том не
обязательно.
151
Указанные способы могут быть использованы в качестве
предварительной стадии концентрирования вещества перед его
последующей глубокой очисткой.
Как следует из изложенного, процесс кристаллизации из
растворов характеризуется типичным фазовым превращением.
В этом смысле принципиальной разницы между кристаллизаци-
ей из растворов и кристаллизацией из расплавов нет, хотя каж-
дый из этих процессов имеет свои существенные особенности.
Коэффициент разделения (сокристаллизации)
Разделительный эффект при кристаллизации из раствора
обусловлен различием составов образующейся твердой фазы и
остающегося раствора, который называют маточным. Рассмат-
ривая в общем случае систему основное вещество — примесь —
растворитель как трехкомпонентную, это различие графически
можно представить диаграммой фазового состояния в виде рав-
ностороннего треугольника, строящегося при условии постоян-
ного давления (треугольник Розебома). Чаще, однако, для ха-
рактеристики указанного различия пользуются аналитическим
выражением, вид которого определяется способом выражения
состава фаз и условиями соосаждения (равновесное или нерав-
новесное соосаждение, сокристаллизации или адсорбция и т. д.).
Для процесса кристаллизации из раствора, в котором дости-
гается термодинамическое равновесие между твердой (кристал-
лы) и жидкой (маточный раствор) фазами, будет справедлив,
как и для других фазовых процессов разделения, закон распре-
деления Бертло — Нернста. В соответствии с этим законом
применительно к системе основное вещество — примесь можно
записать, что
^-:^- = Ркр, (Щ.92>
где аж=аисх—ат\ Ьж = ЬКСх—Ьт; аНСх, Ьисх— количества примеси
и основного вещества в исходном растворе; ат, Ъ-т — соответст-
венно количества примеси и основного вещества, перешедших
в твердую фазу, DKP — так называемый коэффициент сокристал-
лизации. Формула (III.92) характеризует гомогенное соосажде-
ние для случая, когда конечные состояния вещества внутри кри-
сталла равновесны с конечными состояниями вещества в раст-
воре; последнее может быть достигнуто несколькими способами.
Соотношение (III 92) нетрудно привести к виду
(П1.92а>
или, в случае малого содержания примеси в очищаемом ве-
ществе,
“ = dkp> (П1.92б>
152
где х—молярная доля примеси в выделившихся кристаллах
очищаемого вещества; у — молярная доля примеси в очищае-
мом веществе, оставшемся в маточном растворе (молярная до-
ля примеси в «солевой» части маточного раствора) Из сравне-
ния уравнений (111.92а) с (III 1) и (III.926) с (III.2) следует,
что коэффициент сокристаллизации Дкр в уравнениях (111.92а)
и (III.926) тождествен коэффициенту разделения а в уравнени-
ях (III.1) и (1П.2) В соответствии с этим в дальнейшем для
характеристики процесса кристаллизации из раствора, так же
как и для кристаллизации из расплава, будем пользоваться
термином «коэффициент разделения», обозначая последний
через а.
Для определения коэффициента разделения применительно
к процессу кристаллизации из раствора предложен ряд мето
дик, в основном отличающихся характером установления равно
весия между выделяющимися кристаллами и маточным раство
ром. Коэффициент разделения при этом находится на основании
данных анализа содержания основного вещества и примеси в
исходном и маточном растворах с помощью соотношения
(Ш.92) При выражении концентраций через молярные доли
для этой цели более удобным является выражение (Ш.92а)
или (Ш.926). Коэффициент разделения может быть также оп-
ределен и методом направленной кристаллизации раствора.
В реальном процессе массовой кристаллизации, протекаю-
щем с конечной скоростью, состав образующихся кристаллов
обычно все же получается неоднородным так как в ходе про-
цесса раствор непрерывно обогащается (или обедняется) при-
месями. Даже при относительно невысокой скорости кристалли
зации и достаточно интенсивном перемешивании жидкой фазы
в равновесии с маточным раствором в лучшем случае будет на-
ходиться лишь поверхностный слой кристаллов. Распределение
примеси в кристаллах конечного продукта в этом случае описы-
вается известной логарифмической зависимостью Дернера —
Хоскинса
]g-^_=alg-±^ , (Ш.93)
“исх °исх
которую удобно представить как
= (1—(1П-94)
L аиСХ Оцсх J ' "» '
Нетрудно показать, что соотношение (III.94) с учетом того,
что содержание примеси в кристаллах и маточном растворе не-
значительно по сравнению с соответствующим содержанием
основного вещества, можно записать в виде уравнения (1II.3)
или (III9) Отсюда, используя ранее принятые обозначения,
153
(III.97)
(Ш.98)
(III.99)
получаем следующее выражение для средней концентрации при-
меси в кристаллах:
S=-g-l1 -а-хтП, (шед
где t/о — молярная доля примеси в солевой части исходного
раствора; хт — доля закристаллизованного вещества. Если
ввести обозначение а=х/у, то из (III.8) и (III.95) будем иметь
сс=[(1—хт)~а—. (I11.9C)
хт
Иногда для характеристики глубины очистки вещества в ре-
зультате такого однократного процесса массовой кристаллиза-
ции пользуются термином кратность очистки (коэффициент
очистки), под которой понимается отношение концентраций при-
меси в исходном очищаемом веществе (х0, уо) и в выделившей-
ся твердой фазе х. Обозначая это отношение через Ккр, на ос-
новании уравнения материального баланса для примеси
хо (аисх + &исх) = (ятЧ-Ь-г) (апсх—ат"|-5исх — 5Т) у
и исходя из условий аИсх<^исх, ат<^Ьт, получим
v _ 1— хт (1 —а)
Лкр-------=-----
а
или, с учетом выражения (II 1.96),
к Хт
К₽ 1—(1 —хт)“
Легко заметить, что соотношения (III.99) и (Ш.95) выражают
одну и ту же зависимость. Когда а>1, т. е. когда примесь кон-
центрируется в твердой фазе, в качестве продукта отбирается
маточный раствор, из которого очищаемое вещество выделяется
путем отгонки растворителя. Кратность очистки при этом рас-
считывается как обратная величина от значения Ккр.
Фракционированная (дробная) кристаллизация
Уже в результате однократного кристаллизационного со-
осаждения иногда удается достичь весьма заметного эффекта
разделения. Однако в большинстве случаев эффект разделения
бывает невелик из-за включения маточного раствора в трещины
и поры образующихся кристаллов, смачивания поверхности
кристаллов маточным раствором, адсорбционного захвата при-
меси твердой фазой н т. д. Степень загрязнения кристаллов
примесью в принципе можно уменьшить путем промывки про-
дукта или последующей его перекристаллизации. Промывка
кристаллического продукта свежим растворителем (иногда для
154
этой цели применяется насыщенный раствор кристаллизуемого
вещества) не всегда дает хорошие результаты. Маточный раст-
вор, включенный в кристаллы твердой фазы, в целом не может
быть удален из них промывкой; для его удаления требуется
перекристаллизация выделенной твердой фазы. Многократное
повторение процесса кристаллизации позволяет, хотя и с не-
большим выходом, достичь высокой степени чистоты продукта.
Кри многократной перекристаллизации происходит очистка ос-
новного вещества от примесей, попадающих в кристаллы как
за счет гомогенного, так и за счет гетерогенного соосаждения.
Существует несколько вариантов многократной перекристалли-
зации с возвратом и без возврата в цикл маточных растворов.
Одним из таких вариантов является метод фракционированной
(дробной) кристаллизации, разработанной еще в 1899—1907гг.
Рис. 40. Схема дробной кристаллизации
М. Склодовской-Кюри и использованный ею для выделения хло-
рида радия из смеси солей, получающейся при переработке
урановой смоляной руды. Суть этого метода схематично пред-
ставлена на рис. 40.
По этой схеме исходный раствор путем кристаллизации раз-
деляется на две фракции: кристаллы и маточный раствор (пер-
вая ступень кристаллизации). В соответствии с законом р опре-
деления обе полученные фракции будут иметь состав, отличный
от состава исходного раствора. Каждая из этих фракций япя-
логичным образом делится на две новые фракции, в результате
чего получается четыре фракции (вторая ступень кристаллиза-
ции). Процесс такого фракционирования (дробления) может
быть продолжен, пока не будет достигнута необходимая (гспень
155
очистки интересующего вещества от примеси. Если примесь
концентрируется в маточных растворах, то конечным продуктом
будут являться полученные в результате процесса кристаллы
(на схеме слева)
Для оценки эффекта очистки будем исходить из материаль-
ного баланса па ступенях разделения. Для первой ступени кри-
сталлизации уравнения материального баланса процесса име-
ют вид:
^оУо — ^тМх1 +
(III.100)
где ^o — общее число молей растворенного вещества (основное
вещество 4- примесь) в исходном растворе; Л\.(1)— общее число
молей вещества в кристаллах; — общее число молей ве-
щества в маточном растворе; у0— молярная доля примеси в
исходном веществе; yi — молярная доля примеси в веществе,
находящемся в маточном растворе; xt — молярная доля приме-
си в кристаллах. Принимая, что Xi/y\=a, из системы уравнений
(III 100) получаем
_ Уо (I 4~61) _ аУо (1~Ь ^1)
1 l-|-a0i ’ 1 14-а©!
(111.101)
где
о _ Т<1)
* Лж(1) •
Аналогичные уравнения материального баланса можно записать
н для второй ступени кристаллизации:
ЛГт(1)л:1= ^т(2)г»+ ^(2)^2’
^(l)^ ЛГт(2) + ЛГж(2)>
ЛГж( i )У1 — Wt( 2 )х2 + ^ж( 2)2/»’
^ж( 1) ~ ^т(2) 4~ Л\к(2)’
откуда следует, что
,, _ !/1 (14-62) r azJl+Oj) .
—1+^еГ’ **=--------------------
_ Д1 (14~ 6а)
14- а62 ’ 1
^(2)
”2—"iv7 •
Аж(2)
J/2
1 4- сс02
_ «yi (1 + е2)
2 1 + аб'
(III.102)
(III.103)
нетрудно вывести
для после-
соотношения
кристаллизации. Как легко видеть, рассмат-
тде ,
7уж(2)
Соответствующие
дующих ступеней
риваемая схема фракционированной кристаллизации представ-
ляет собой схему многоступенчатого кристаллизационного
каскада
и
156
Условием построения рационального каскада является тре-
бование минимума числа фракций при заданном числе ступеней.
Д ля этого процесс должен проводиться таким образом, чтобы
отдельные промежуточные фракции можно бы то объединить,
как это схематично показано на рис. 40 Совершенно очевидно,
что такое смешивание промежуточных фракций наиболее целе-
сообразно в том случае, когда нх состав одинаков Это дости-
гается лишь тогда, когда отношение количества вещества, вы-
павшего в виде кристаллов, к количеству вещества, оставшемуся
в маточном растворе, сохраняется постоянным на всех ступе-
нях. Как видно из соотношений (III 103), для того чтобы со-
блюдалось равенство концентраций у/ и хг в двух промежу-
точных фракциях второй ступени, необходимо равенство вели-
чин 02 и ©г'. С учетом указанного условия минимальное число
фракций после первой кристаллизации будет равно двум, после
второй — трем, после третьей — четырем; соответственно после
n-й ступени оно будет равно п+ 1.
Если 0|=02=©з = - =0п, из соотношений (III 101) и
(III 103) для второй ступени каскада имеем:
г2/у2 = аа
Уг . / 14-61 \2
f/o I 14-а©1 /
аналогично для последней, д-й ступени:
хп'Уп = ап,
Уп I 1 + 61 V
Уо \ 1 + а©! /
(III.104)
(III. 105)
(Ш.106)
(III.107)
Подставляя соотношение (III 107) в (Ш.106), с учетом того,
что уо =хо, получаем:
Xn L 1+<х61 J
Из выражений (III 106) — (III. 108) следует, что методом дроб-
ной кристаллизации можно достичь большой глубины очистки.
Однако он имеет существенный недостаток — низкую произво-
дительность. Кроме того, этот метод включает в себя значи-
тельное число побочных операций: отделение кристаллов от ма-
точных растворов, добавление к кристаллам растворителя, сме-
шивание промежуточных фракций, упаривание маточных раст-
воров и т. п.; помимо того, что эти операции трудоемки, при их
осуществлении могут происходить потери вещества и загрязне-
ние продукта.
157
Противоточная кристаллизация из раствора
Другими вариантами фракционированной кристаллизации
являются метод последовательной кристаллизации твердой фа-
зы с возвратом в цикл маточных растворов н метод многократ-
ной кристаллизаций маточного раствора с возвратом в цикл
твердой фазы. Если примесь концентрируется в жидкой фазе,
то следует использовать первый из указанных методов Если же
примесь концентрируется в твердой фазе, то целесообразнее
применять второй метод. Существенное преимущество этих ме-
тодов перед методом дробной кристаллизации заключается в
том, что при их осуществлении может быть использован прин-
цип противотока, обусловливающий большую производитель-
ность.
h.yt //»:
Рис. 41. Схема каскада для противоточной кристаллизации из раствора:
/, 2, 3, .... I — ступени каскада; п — число ступеней каскада
На рис. 41 приведена простейшая схема противоточного кри-
сталлизационного каскада с рециркуляцией маточных растворов.
Исходя из материального баланса по примесному компоненту,
для каждой ступени каскада, по достижении стационарного со-
стояния, будем иметь.
b;Xi4-Zi+2(/i+2 = Z,i4.1Xi+14-Zi+iJ/i+1,
где i—номер ступени каскада; L—скорость потока твердой
фазы: I — скорость потока очищаемого вещества (основное ве-
щество + примесь), находящегося в растворе. Принимая усло-
вие постоянства потоков твердой и жидкой фаз от ступени, не
158
трудно показать, что система уравнений (III 109) преобразуется
к виду:
где хр = хп—молярная доля примеси в продукте; п — скорость
отбора кристаллического продукта. Из системы уравнений
(ШЛЮ) получаем
yi+x-xt = -^-. (ПИН)
Для случая безотборного режима, т. е. когда п = 0, выражение
(Ш.111) существенно упрощается:
(III.Н2)
При допущении, что выходящие с каждой ступени твердая и
жидкая фазы находятся в термодинамическом равновесии, т. е.
когда %i+i = «!/<+!, из соотношения (III 112) получаем
xt^axt. (III.113)
Если теперь составить уравнение вида (III 113) для каждой
ступени каскада и соответственно перемножить левые и правые
части этих уравнений, то будем иметь
xn = a"-ix1. (III.114)
Так как Xi/y0=a и уо=*о, то
zn/*0=an.
(III. 115)
Выражение (III 115) аналогично выражению (11.50), характе-
ризующему разделительную способность ректификационной та-
рельчатой колонны в безотборном режиме (гл. II, § 6). Следо-
вательно, в рассматриваемой схеме кристаллизационного кас-
када каждая его ступень эквивалентна тарелке ректификацион-
ной колонны. Отсюда следует, что уравнения, описывающие
работу ректификационных колонн, будут справедливы и для
работы кристаллизационного каскада В частности, с помощью
соотношения (II.85) можно оценить разделительную способ-
ность рассматриваемого кристаллизационного каскада в отбор-
ном режиме. Поскольку кристаллизация из раствора осуществ-
ляется при температуре, обычно весьма далекой от температуры
плавления очищаемого вещества, следует ожидать, что эффект
диффузионного массообмена между движущимися кристаллами
и раствором в ходе процесса будет ничтожно мал. Поэтому до-
стигаемая в процессе противоточной кристаллизации из раство-
ра глубина очистки обусловлена в основном эффектом перекри
сталлизации и, таким образом, здесь возникает задача создания
и поддержания благоприятных для его проявления условий
159
ГЛАВА IV
МЕТОД ТЕРМОДИФФУЗИИ
Разделение веществ методом термодиффузии основано на воз-
никновении градиента концентраций в смеси веществ под влия-
нием градиента температуры (например, один конец запаянной
трубки, в которой находится смесь,нагревается, а другой охлаж-
дается) Само явление термодиффузии применительно к водным
растворам солей было открыто еще в середине прошлого века
Этим явлением объясняется также и обнаруженный в то же
время термоэлектрг еский эффект в твердых телах, в частности
в металлах, проявляющийся при наложении на них температур-
ного поля. Возможность протекания термической диффузии в
смесях газов вначале
а затем подтверждена
и
Рис. 42. Схема термоднффузнонной
установки, свободной от конвекции
(T2>Tt)
была предсказана теоретически (1911г.),
экспериментально (1917 г )
Хотя метод термодиффузии
до настоящего времени и не
нашел столь широкого приме-
нения, как дистилляционные и
кристаллизационные методы,
тем не менее он используется
в качестве одного из эффек-
тивных методов разделения га-
зовых смесей. В последнее вре
мя проявляется все больший
интерес к жидкостной термо-
диффузии с целью использова-
ния ее для очистки органиче
ских растворителей
§ 1. ТЕРМОДИФФУЗИЯ В ГАЗОВОЙ фазе
Коэффициент разделения установки,
свободной от конвекции
Рассмотрим систему, состоящую из двух сосудов, находя-
щихся соответственно при температурах 7\ и Тг и соединенных
между собой трубкой (рис. 42) Пусть эта система заполнена
смесью двух газов (основное вещество и примесь) Разность
температур между сосудами вызывает термическую диффузию
газов, скорость которой выражается следующим уравнением
LT = —р{Рт!Т) grad (Г)= — pDT51n T/dz, (IV 1)
в котором Lr — скорость термической диффузии примеси, т. е
количество примеси, переносимое через единицу сечения соеди-
нительной трубки в единицу времени; р — плотность газовой
160
смеси; DT — коэффициент пропорциональности, называемый ко-
эффициентом термодиффузии; Т — температура, К г—коорди-
ната вдоль соединительной трубки.
В результате процесса термодиффузии концентрация приме-
си в одном из сосудов будет возрастать, что приводит к возник-
новению концентрационного градиента вдоль соединительной
трубки. Последний вызывает обычную концентрационную диф-
фузию, которая стремится выровнять состав смеси во всем объ-
еме системы, вследствие чего примесь переносится по соедини-
тельной трубке в обратном направлении Скорость этой кон-
центрационной диффузии будет тем выше, чем больше разница
в концентрациях примеси в сосудах 1 и 2, и в соответствии с
законом Фика может быть выражена соотношением
Ld— —pD2 grad (х) = —pP2 dx/dz, (IV 2)
где Ld — скорость концентрационной (молекулярной) диффузии
примеси, т. е. количество примеси, переносимое через единицу
сечения соединительной трубки в единицу времени; D2 — коэф
фициент диффузии примеси; х— молярная доля примеси в дан-
ном сечении соединительной трубки.
При установлении в системе равновесия, т. е. при Ld=Lt,
из выражений (IV 1) и (IV.2) следует, что
O2dx Brdln7. (IV.3)
Отношение коэффициента термодиффузии к коэффициенту мо-
лекулярной диффузии называют термодиффузионным отноше-
нием kT:
кт ~ D?/D2 = а.тх (i—х), (IV.4)
где ат — постоянная термодиффузии, которую в первом прибли-
жении можно считать не зависящей от концентрации и темпе-
ратуры. Из выражения (IV.4) для рассматриваемого случая
небольших концентраций примеси имеем
DT= Ь2атх. (IV.5)
Так как d In Т=6Т/Т, то с учетом этого соотношения и выра-
жения (IV.5) из уравнения (IV.3) следует
dx d7 .......
— = «T-s-. (IV.6>
X 1
Интегрируя уравнение (IV.6) при граничных условиях
х=хх прп Т— 7\,
х—х2 при Т=Г2,
получаем
= exp ( ат In -Ь- ) (IV.7)
G—46
161
или
?
(£Г
(IV.8)
где q — x.2/x\ — коэффициент разделения*
Постоянная термодиффузии
Из соотношения (IV.8) следует, что коэффициент разделения
при термодиффузии без конвекции нетрудно определить, если
известна постоянная термодиффузии аг (термодиффузионный
фактор), зависящая от природы компонента смеси. С некото-
рым приближением ат может быть найдена расчетным путем
исходя из положений молекулярной теории смеси газов с нало-
женным на эту смесь температурным градиентом. Для проведе-
ния соответствующих расчетов требуется знание характера меж-
молекулярного взаимодействия в заданной газовой смеси.
Теоретическое вычисление постоянной термоднффузии возможно
лишь при использовании той или иной модели межмочекуляр-
ного взаимодействия. Выбор модели определяется требуемой
точностью оценки величины ат и связанными с ней расчетными
трудностями. Соответствующие вычисления существенно упро-
щаются для смесей изотопов.
Модель твердых упругих шаров В этой модели молекулы
представляются в виде твердых шаров диаметром О| (молекулы
основного вещества) и о2 (молекулы примесного вещества).
Принимается, что взаимное притяжение между такими молеку-
лами отсутствует, а отталкивание имеет место лишь при непо-
средственном столкновении молекул, когда их центры находятся
на расстоянии 01.2= (oi + o2)/2 Следовательно, для этой модели
можно записать, что
/м (г) ~ 1 rx, (IV.9)
где /м(г) — сила взаимодействия между двумя молекулами,
центры которых находятся на расстоянии г. В этом случае, если
принять О|=^о2, что характерно для изотопных мочекул, выра-
жение для постоянной термодиффузии имеет вид
105 Мг— .V,
°'т~ 118 ЛГг+Л/1 ’
(IV.10)
где Mi и —относительные молекулярные массы разделяемых
веществ.
* В работах по термодиффузионному разделению через ат (или а) обоз-
начается постоянная термодиффузии, поэтому коэффициент разделения обозна
чается буквой q.
162
Модель твердых шаров (сфер) является наиболее прибли-
женной из всех используемых в расчетах моделей и в реальных
случаях, по-видимому, применима лишь для описания свойств
газов при достаточно высоких температурах, когда взаимное
притяжение молекул действительно становится несущественным
Модель точечных центров отталкивания (инверсионная мо-
дель). По этой модели молекулы отождествляются с точечными
центрами сил отталкивания. Здесь, как и в предыдущей модели,
взаимным притяжением молекул пренебрегают, но учитывают
их взаимное отталкивание, описываемое соотношением
fM (Г) - l/rv. (IV 11)
Показатель v в соотношении (IV 11) представляет собой неко-
торое положительное число и является мерой «сжимаемости»
молекул. Действительно, как можно видеть, прн v->oo инвер-
сионная модель переходит в модель твердых шаров. Таким об-
разом, при больших значениях у будем иметь дело с «жесткими»
молекулами, а при низких значениях у — с «мягкими» Исходя
из данной модели, получено следующее выражение для ат'.
105 ЛГ2 Л/ v 5 , . . /IV 191
Кг==~Й8 М2+^1 ~^1 /( Ь ( ’
где f(v)—функция, сложным образом зависящая от у Величи-
на v может быть определена исходя из данных о свойствах га-
зовой смеси, таких, как вязкость, теплопроводность, диффузия,
применяя для описания этих свойств рассматриваемую инвер-
сионную модель. Для этой цели обычно используют температур-
ную зависимость коэффициента вязкости Так, еще Дж. Релеем
в 1900 г. было установлено, что эта зависимость для газов
имеет вид
И ~ Тп, (IV 13)
где т] — коэффициент вязкости; п — некоторое положительное
число.
С другой стороны, из молекулярно-кинетической теории га-
зов следует, что для молекул, взаимодействие которых описы-
вается инверсионной моделью,
2
Т)- /ГТ"’-1 .
Сопоставляя (IV. 13) и (IV 14), получаем, что
v-f-3
П~2{у-Ц
(IV 14)
(1V.15)
или
2Й+3
У=-----
2л—1
(IV.16)
6
163
Определяя п из уравнения (IV 13), для удобства записанного
в логарифмической форме 1g т] ~ lg Т, из соотношения (IV. 16)
находим v. Как показали такие определения, для большинства
газов значения v лежат примерно в интервале 9—15, т. е. мо-
дель твердых упругих шаров, для которых v-*oo, в целом не
отвечает действительности. Для некоторых газов значения v
оказываются или меньше 9 или больше 15. Для удобства рас-
четов по формуле (IV 12) входящая в нее функция f(v) прота-
булирована- для значений v, изменяющихся от 3 до оо, функция
f(v) изменяется от 0 807 до 1, т е незначительно. Таким обра-
зом, величина ат, рассчитываемая по формуле (IV 12), в основ
ном определяется относительными молекулярными массами
компонентов разделяемой смеси и множителем (v—5)/(v—I).
Легко видеть, что при v = 5, ат=0, откуда следует, что в смесях
газов, для которых ?м(г) ~1/г5, явления термодиффузии не
должно наблюдаться Этим и объясняется известный вывод
Дж. Максвелла о невозможности протекания термической диф-
фузии в газах вообще; им был рассмотрен случай с v=5
Как следует из формулы (IV 12), при v<5 величина ат ста-
новится отрицательной (возможность изменения знака ат и обу-
словливает термин «инверсионная модель»). Это означает, что
обеднение газовой смеси примесью в указанном случае проис-
ходит в противоположной температурной зоне, т. е. если v>5,
обеднение примесью газовой смеси должно происходить в зоне
с более высокой температурой При v<5 обеднение примесью
будет происходить в зоне с более низкой температурой.
Следует заметить, что результаты расчета ат с использова-
нием инверсионной модели, так же как и при использовании
модели твердых упругих шаров, все же обычно заметно отлича-
ются от результатов соответствующих экспериментальных опре-
делений. Это вынуждает исследователей прибегать к другим,
более сложным моделям, учитывающим и взаимное отталкива-
ние и притяжение молекул.
Модель Леннарда — Джонса Из известных в литературе
моделей, предложенных для описания межмолекулярного взаи-
модействия, наиболее часто используется модель, в которой
постулируется, что сила притяжения между молекулами, как и
сила их взаимного отталкивания, описывается степенной зави-
симостью. Следовательно, результирующую силу межмолекуляр-
ного взаимодействия в этом случае можно выразить с помощью
соотношения
fM(r)=ar~V°-br~\ (IV17)
где постоянные а и va относятся к силе отталкивания, постоян-
ные b и Vb— к силе притяжения.
Соотношение вида (IV 17) с v/, = 3 и произвольном va, но при
условии, что va>3, впервые было использовано для объяснения
164
температурной вязкости газов. Позднее было найдено, что луч-
шее согласие дают результаты расчета с гЛ=13 и vt = 7, в част-
ности при исследовании различных свойств газов, состоящих из
неполярных молекул. В соответствующих расчетах обычно ис-
пользуют выражение для так называемого потенциала межмо-
лекулярного взаимодействия, вид которого определяется исходя
из той или иной принятой модели. Значение потенциала взаимо-
действия при этом входит в сложные интегральные соотношения,
характеризующие движение молекул и их отклонение при столк-
новении (интегралы столкновения) Эти соотношения, в свою
очередь, позволяют выразить в общем виде зависимость ат от
свойств исследуемой системы, состава, температуры и т д
Применительно к бинарной системе выражение для потенци-
альной энергии «|,2 парного взаимодействия молекул первого
и второго компонентов смеси в рамках рассматриваемой модели
при va=13 и Vb = 7 характеризуется соотношением Леннарда—
Джонса [см уравнение (II 10)]
которое известно в литературе под названием «потенциал 6—42».
Сама же модель взаимного межмолекулярного отталкивания и
притяжения, характеризуемая соотношением (IV.18), получила
название модели Леннарда — Джонса.
Вычисление ат с использованием зависимости (IV 18) пред-
ставляет собой трудоемкую процедуру, которая облегчается ис-
пользованием табулированных значений интегралов столкнове-
ния. Как можно видеть, при этом необходимо знать величины
fii.2 и О|.2, которые могут быть определены на основании экспе-
риментальных данных о свойствах интересующей газовой смеси
или составляющих ее компонентов. Для этой цели, как и при
определении v в инверсионной модели, удобно пользоваться
данными о температурной зависимости коэффициента вязкости.
Методики таких определений изложены в ряде работ. При этом,
если указанные постоянные для каждого компонента смеси из-
вестны, то для бинарных смесей этих газов значения ei.2 и о,,г
можно найти с помощью комбинационных правил [см выраже-
ния (II.II) и (11.12)].
Для расчета ат (или kT) используют и другие выражения
для потенциальной функции, причем в ряде случаев они дают
лучшее согласие с опытом, чем потенциал Леннарда — Джонса.
В целом же, результаты вычислений ат по сложным моделям
все же не дают достаточно хорошего соответствия с эксперимен-
том, особенно для многоатомных газов. Вследствие этого на
практике обычно прибегают к экспериментальному опреле пению
этой величины
165
Для экспериментального определения постоянной термодиф
фузии ат используют несколько способов. Наибольшее распрост-
ранение получил метод двух сосудов (двухбаллонный метод),
суть которого поясняется рис 42. После установления в системе
равновесия, что определяется из предварительных опытов, из
сосудов 1 и 2 отбирают смеси газов и анализируют их состав.
По результатам анализа с помощью уравнения (IV.8) опреде-
ляют ат.
Следует напомнить, что при выводе этого уравнения прини-
малось, что ат от содержания компонентов в смеси и от темпе-
ратуры последней не зависит Опыт показывает, что в общем
случае ат зависит от указанных параметров; это предсказыва-
ется и теорией Но если концентрационную зависимость ат для
рассматриваемой задачи разделения смеси газов с малым со-
держанием одного из компонентов во внимание можно не при-
нимать, то изменение аг с температурой может быть весьма
существенным, особенно при большом различии величин Т и
Т2, с чем приходится сталкиваться, когда требуется достичь
относительно большой величины q.
В общем виде температурная зависимость ат может быть
охарактеризована эмпирическим уравнением
а-т-А-? , (1V 19)
где А и В — константы.
Но при определении постоянной термодиффузии методом
двух сосудов мы имеем дело не с конкретной температурой Т,
а с интервалом температур Т\—Т2. Здесь возникает вопрос,
к какой температуре следует отнести определяемую эксперимен-
тально величину ат. Часто ее относят к средней температуре
Т= i/2(7’i + 7'2), а иногда к Т=УТ\Т2. Однако лучшее приближе-
ние можно получить, если исходить из того, что определяемая
методом двух сосудов постоянная термодиффузии является
средней величиной ат в температурной области между 1\ и Т2
С учетом этого можно записать, что
где Т — средняя температура в интервале температур 1\—Т2.
Таким образом, определяемая в опыте величина ат отож
дествляется здесь с действительной величиной ат при средней
температуре Г, находящейся в интервале 7,—Т2.
Значение Т нетрудно выразить через значения Т\ и Т2 исхо-
дя из соотношений (IV.6), (IV.19) и (IV 20) С учетом того, что
1CG
постоянная термодиффузии является функцией температуры,
интегрирование уравнения (IV.6) дает
т,
1п-^=Лаг4^. (IV.21)
xi J 1
Г.
Но, с другой стороны, пользуясь понятием среднего значения
постоянной термодиффузии ат в интервале температур Т,—Т2,
из (IV.21) имеем
1п-^ = аг1п-Ь-. (IV 22)
Х1 j
Приравняв выражения (IV21) и (IV22), получим
Т2
аг=$ «г 4Г/1П УТ' (IV 23)
г,
Подставляя уравнение (IV 19) в (IV23), после интегрирования
будем иметь
"Т”А-В Wn~£r.)- <IVM>
Из сравнения выражений (IV.24) и (IV.20) следует, что
Т=--Т^- in A. (iv.25)
Вообще говоря, для определения коэффициента разделения,
а следовательно, и постоянной термодиффузии методом двух
сосудов достаточно знать концентрацию примеси лишь в одном
из сосудов при установлении в системе равновесия, если извест-
но содержание примеси Хо в исходной смеси газов. Это нетруд-
но показать исходя из условий равенства скоростей массообме-
на в рассматриваемом процессе термодиффузии, согласно кото-
рому количество молей примеси, переносимых в один из сосудов,
равно количеству молей основного вещества, переносимого в
другой сосуд: суммарное количество молей газа в сосуде 1
и N2 в сосуде 2 при этом будет постоянным Тогда, пренебрегая
количеством газа в соединительной трубке по сравнению с коли-
чеством газа в сосудах 1 и 2, можно записать, что
N^N^N^l+N^. (IV.2G)
Если теперь принять, что рассматриваемая смесь газов близка
по свойствам к идеальному газу, то, обозначая через Vi и V2
объемы сосудов 1 и 2 соответственно, из выражения (IV.26)
после некоторых преобразований получаем
,,1+(i^)(1+l£.), |,v'27>
167
когда определение концентрации примеси производится в пробе,
отбираемой из сосуда /, или
’ (IV28)
Ч \ \ * 1-* 2 /
когда определение Концентрации примеси производится в пробе,
отобранной из сосуда 2.
Если достигаемый в опыте эффект разделения незначителен,
то для повышения точности определения коэффициента разделе-
ния, а следовательно, и постоянной термодиффузии прибегают к
многократному повторению процесса. С этой целью после уста-
новления в системе равновесия газ из одного сосуда откачивают
и снова добиваются равновесия в системе (соответственно сна-
чала при закрытом кране, а затем при открытом кране на сое
динительной трубке). Далее газ из указанного сосуда вновь от-
качивают и т. д., в результате оставшийся газ существенно
обогащается одним из компонентов Разумеется, после каждой
такой операции общее давление газа в системе будет умень
шаться. Однако полагая, что коэффициент разделения от дав-
ления не зависит (по крайней мере при давлениях ниже
(1—2)-105 Па), нетрудно показать, что, например, когда газ
откачивается из сосуда 2 (см. рис. 42), по завершении первой
операции будем иметь
9
*i(D
1 + -Ы1
(IV.29)
где X](|) — концентрация прнмеси в сосуде 1 при установлении
в системе вторичного равновесия.
Подставляя значение х, из (IV.27) в (IV.29), получаем
х1(1) = а:0
г
(IV.30)
9+ ^172 V2T1
Соответственно после n-й операции (не считая операции по
предварительной откачке системы и загрузке исходной смеси)
ж1(п) —
/ i+ViWr, у-м
\ 94-Pi7,2.'F27’1 I
(IV.31)
откуда
1 + QWVzTt) [ 1 - (х1М!хе}Ч(^) ]
(*l(n)/x0),/("+l)
(IV.32)
где Хцп) — концентрация примеси в сосуде / после n-й операции.
Весьма интересным является то, что определенные рассматри-
ваемым двухбаллонным методом значения ат для паров целого
ряда бинарных смесей органических веществ оказываются по
абсолютной величине на 1—2 порядка выше, чем обычные зна-
чения ат для бинарных смесей газов. Одна из причин такого
различия, по-видимому, зак.вючается в пространственном строе-
1G8
нии молекул органических веществ, которые в парах могут на-
ходиться в виде ассоциатов Наблюдаемые аномально высокие
значения ат для паров смесей органических веществ дают воз-
можность использовать термодиффузию в качестве эффектив-
ного метода глубокой очистки органических растворителей,
в частности, от примесей, образующих с растворителем азео-
тропную смесь
§ 2. ТЕРМОДИФФУЗИОННЫЕ КОЛОННЫ
Разделение смеси, достигаемое за счет непосредственного
использования однократной термодиффузии, обычно очень мало,
и поэтому величины q всегда близки к единице. Этим и объяс-
няется тот факт, что термодиффузионный метод разделения
смесей по существу не находил практического применения до
тех пор, пока для умножения эффекта разделения не был при
менен принцип противотока. Суть противоточного варианта тер-
модиффузионного метода, осуществляемого в вертикальных
аппаратах — термодиффузиоиных колоннах, можно пояснить с
помощью схемы, представленной на рис. 43 Разделяемая смесь
находится между двумя стенками, одна из которых имеет низ-
Рис. 43. Схема движения конвек-
ционных потоков вдоль «горячен»
и «холодной» стенок термодиффу-
зионной колонны:
В — ширина стенки; 2W — расстояние
между стенками
Рис. 44. Схема термодиффузнон-
иой колонны с нагреваемой прово-
локой:
/ — корпус колонны («холодная> стен-
ка), 2—нагреваемая проволока («го-
рячая» стенка). 4 — нижний конец; 5 —
спираль для натяжения проволоки; 6 —
питающая емкость 3. 1 — краны для
отбора проб и фракций газа
169
кую температуру 7\ (холодная стенка), другая — более высокую
температуру Т2 (горячая стенка). Различие в температурах
стенок приводит к возникновению конвективных потоков газа
в вертикальном направлении вдоль этих стенок: «холодный»
поток, движущийся вниз, играет роль «тяжелой фазы», а «го-
рячий» поток, двияфщнйся вверх, — роль «легкой фазы» Такой
противоток позволяет многократно умножать термодиффузион-
ный эффект разделения, имеющий место в горизонтальном на-
правлении между холодной и горячей стенками. В результате
один из компонентов смеси (в случае бинарной смеси) будет
концентрироваться в одном конце колонны, а второй — в дру-
гом ее конце; из соответствующего конца колонны можно отби-
рать интересующий продукт.
Типы термодиффузионных колонн
Термодиффузионные колонны конструктивно обычно выпол-
няются в виде двух типов: а) колонны с нагреваемой проволо-
кой, б) колонны типа коаксиальных цилиндров. Каждый из этих
типов имеет свои преимущества и недостатки.
Колонны с нагреваемой проволокой. Принципиальная схе-
ма конструкции одной из таких колонн приведена на рис. 44
Колонна представляет собой закрытую с обоих концов верти-
кальную трубку 1 (обычно стеклянную), окруженную холодиль-
ником, по которому циркулирует хладоагент (водопроводная
вода). Охлаждаемая поверхность трубки служит холодной стен-
кой. По оси трубки проходит проволока 2, нагреваемая элек-
трическим током, которая играет роль «горячей» стенки; прово-
лока натягивается с помощью спирали 5, которая компенсирует
тепловое расширение проволоки. Горячий газ, окружающий
проволоку, поднимается в верх трубки, вдоль стенки трубки
движется вниз холодный поток газа. Вследствие этого в трубке
имеет место противоток с образованием потоков на концах 4
и 6. Под влиянием разности температур легкие молекулы из
холодного потока диффундируют в горячий поток, а тяжелые
молекулы — в обратном направлении. Следовательно, между
потоками происходит массообмен, в результате чего процесс
разделения становится многоступенчатым; однократный эффект
разделения умножается подобно тому, как это имеет место в
других противоточных процессах. Краны 7 и 3 служат для вво-
да разделяемой смеси и для отбора продукта. Диаметр трубки
обычно составляет 7—12 мм, а диаметр проволоки — 0,3—0,5 мм.
Преимуществом таких колонн является их конструкционная
простота. Именно с помощью такого типа колонн в 1938 г.
К Клузиусу и Г Диккелю впервые удалось применить принцип
противотока к термодиффузионному разделению смесей водоро-
да и углекислого газа, гелия и брома, для концентрирования
170
кислорода в воздухе, тяжелого изотопа 22Ne в обычном неоне,
изотопа 37С1 в хлороводороде. Используя систему колонн общей
длиной около 36 м, они достигли очень хорошего разделения
НЭ5С1 и Н37С1 в обычном хлороводороде (в верхней части ко-
лонны— 96 % Н35С1, в иижней части колонны — 99,4 % Н37С1).
Полученный в этих опытах фактор разделения (~4000) позво-
лил оценить значение ВЭТТ, которое оказалось равным ~-5 см
(при <7 = 1,01).
Основным недостатком колонн подобного типа является то,
Теплоноситель
II
Рис. 45. Схема термолиф-
фузиониой колонны типа
коаксиальных цилинд-
ров-
1. 5 — краны для отбора
проб и фракций газа. 2 —
внутренний цилиндр; 3— за-
зор; 4— внешний цилиндр
что в них нельзя разделять термически нестойкие вещества. Для
того чтобы производительность таких ко-
лонн была более или менее удовлетвори-
тельной, температуру проволоки прихо-
дится поддерживать равной 800—1000 °C,
термически нестойкие вещества при этом
разлагаются на поверхности проволоки
Колонна типа коаксиальных цилинд-
ров (рис. 45). Диаметр внутреннего ци-
линдра 2 обычно равен 20—80 мм, зазор
между внешней стенкой внутреннего ци-
линдра и внутренней стенкой внешнего
цилиндра составляет 0,25—0,5 мм. Через
краны 1 и 5, соединенные с зазором, про-
изводится ввод смеси и отбор продукта.
Преимущество такого типа колонн в том,
что они обладают большей производи-
тельностью, чем колонны с нагреваемой
проволокой, при малой разности темпе-
ратур между холодной и горячей стенка- Хладагент
ми, роль которых играют соответствую-
щие поверхности цилиндров. Это позво-
ляет эксплуатировать их при сравнитель-
но низких температурах; последнее осо-
бенно ценно тогда, когда разделяемые
вещества не обладают высокой термиче-
ской стойкостью Однако колонны типа
коаксиальных цилиндров для разделения
смесей газов применяются редко ввиду
значительных технических трудностей в
их изготовлении и монтаже.
При анализе работы термодиффузиопных колонн обычно
пользуются феноменологической теорией, которая исходит из
указанного выше факта, что градиент температур в колонне
вызывает градиент концентраций. Работу термодиффузнонной
колонны в принципе можно сравнить с работой ректификацион-
ной колонны. Основное различие, не считая различия в физиче-
ской природе процессов в обоих случаях, состоит в том что
171
Рис. 46. Схема участка термодиффу-
зионной колонны, состоящей из двух
параллельных пластин
массообмен в первом случае
происходит между восходящим
и нисходящим конвекционны-
ми потоками газа, а не между
жидкостью и паром, как во
втором случае
Разработанная к настояще-
му времени теория термодиф-
фузионного разделения в ко-
лонне в математическом опи
сании сложна. Однако наряду
со строгим изложением
К. Джонсом и В Ферри* пред-
ложено и элементарное изло-
жение сущности процессов,
протекающих в колонне. Это
изложение приводит к более наглядным результатам, отличаю-
щимся от результатов строгой теории только числовыми коэф-
фициентами. Рассмотрим его основные положения.
Скорость движения газа в колонне
В соответствии со схемой, предложенной на рис. 45, выделим
участок термодиффузионной колонны, состоящей из двух парал-
лельных пластин (рис. 46), находящихся при температуре Т\ и
Т2 соответственно. Скорость течения газа, заключенного между
стенками, определяется следующим уравнением:
Л 2 Г Ар
11 “dr^ Р,€'—а7=0, (iv.33)
где т] — коэффициент вязкости; L — объемная скорость газа;
рг — плотность газа; Р — давление; г, z— координаты вдоль
сечения и вдоль длины колонны соответственно.
Обозначая через рг среднюю плотность газа в данном попе-
речном сечении и учитывая, что dP/dz=—prg, из уравнения
(IV.33) получаем
Т) -д;5- = (Рг—Рг) g. (IV.34)
Далее примем, что теплопроводность газа не зависит от темпе-
ратуры, т. е.
dT/dr=const, (IV.35)
* Джонс К., Ферри В. Разделение изотопов методом термодиффузии. М.,
1947, с. 168
172
а плотность является линейной функцией Т
рг=рг(1-^^), (IV 36)
где 7,= ,/2(7'|4-7'г)—средняя температура
Из рис. 46 с учетом соотношения (IV.35) следует, что
Г = Т+-^г, (IV.37)
где 2W — расстояние между холодной и горячей стенками;
ДТ=Т2—Г,.
Подстановка выражения (IV 37) в соотношение (IV.36) дает
Р, РГ(‘-2ЙГ^). (1V.3S)
С учетом (IV 38) из (IV 34) имеем
После интегрирования уравнения (IV 39) получаем
“ \т
Л=-Т^Г’Т+Л1Г+Л” |,v-40)
где Ai и Аг — константы интегрирования.
Для определения этих констант используем граничные усло-
вия L = 0 при r=W и L = 0 при г = 0, в результате получаем, что
= , (IV.41)
121] т
а Л2 = 0 Подстановка значений Л, и Аг в уравнение (IV 40)
дает
<ive)
С помощью уравнения (IV 42) находим выражение для средней
скорости движения холодного и горячего потоков газа
VV
о
VV
Ь«1г=4г (
И J
о
7£4^-^-(И/1-г2)<1г
12т]РИ т
(IV.43)
откуда, полагая, что рг^рг,
г PrgW2 АГ
481] т
(1V.44)
173
Вычисление скорости переноса
примеси вдоль по колонне
Пусть примесь концентрируется в «холодном» потоке. Тогда
выражение для скорости переноса примеси Т| в нижней части
колонны можно записать в виде
Т1=(х—у) pLWB, (IV.45)
где р — плотность газа, моль/см3, х—средняя концентрация
(молярная доля) примеси в некотором сечении колонны z в хо-
лодном потоке газа, идущем в низ колонны, у — средняя кон-
центрация примеси в этом же сечении колонны в горячем пото-
ке газа, идущем в верх колонны; В — ширина стенок (глубина
щели). Скорость переноса примесн тг в верх колонны за счет
молекулярной диффузии будет выражаться соотношением
т2= И'ВрР, g + WBpD2 . (I V.46)
Отсюда полагая, что dy/dz=dxldz, выражение для результи-
рующего переноса будет иметь вид
—° Их
T=T1-T2=pL(z—V) WB-2WBpD2— . (IV.47)
nz
Вычислим теперь скорость переноса примеси тз из горячего по-
тока в холодный-
тз = Ст—Qd> (IV.48)
где Q-i = WBpDiarx(d In Ttdr)—скорость перехода примеси из
горячего потока в холодный за счет термодиффузии; Qd=
= WBpD? (дх/дг) — скорость перехода примеси из холодного
потока в горячий за счет молекулярной (концентрационной)
диффузии. Используя приближения
д In Т 1 ДГ дх х — у
дг~ ~ У ~2W ' ~ W '
получаем результирующую скорость переноса примеси из горя-
чего потока в холодный:
т3 = IVBpDjarx . (I V.49)
2WT И7
Конвективная скорость переноса т< выражается соотношением
— Лд-
T4 = IV2Bp£ (IV,50)
Приравнивая правые части уравнений (IV.49) и (IV.50),
получаем
ДГ
X—и=ЛтХ——
27
Zip2 дх
D2 dz
(IV 51)
174
Подставив выражение (IV 51) в (IV 47) и заменив в первом
приближении дх./дг на dx/dz, будем иметь
т= WBpLaTx -W^Bp -Lf- ~ -2WBpDt . (IV.52)
£,1 L/2 tlZ
С учетом выражения (IV 44) уравнение (IV 52) преобразуем к
следующему виду:
dx
T = Z/x-(A'c+Kd)-^- . (IV.53)
где
//=^f^Z£(4L)2; (IV.54)
96ij \ т /
р?ррИ”В / ДТ \2
Ас“ / (
Kd = 2WBpDz. (IV.56)
Уравнение (IV 53) выражает зависимость скорости переноса
примеси вдоль колонны от параметров колонны и разделяемой
смеси. Величины Н, Кс и Kd для данной смеси и данной колон-
ны являются постоянными, характеризующими скорость пере-
носа примеси вдоль колонны, и поэтому обычно называются
коэффициентами переноса.
Соответствующие выражения для коэффициентов переноса,
полученные для колонн с нагреваемой проволокой, при более
строгом рассмотрении задачи имеют вид:
Ad = 2n (pD2)Ti r^d (4; '• 57 ) • (IV.5C.a)
где л — радиус колонны; г2 — радиус проволоки; ft, kt, kd—
сложные функции, которые в каждом конкретном случае могут
быть найдены с помощью методов численного интегрирования.
Из приведенных соотношений наглядно проявляется качест-
венная аналогия между приближенными и точными выражения-
ми для коэффициентов переноса.
Фактор разделения в стационарном состоянии
и безотборном режиме
Опыт показывает, что асимметрия стенок колонны приводит
к паразитному перемешиванию газовой смеси и соответственно
к ухудшению разделения. Это перемешивание hmcci конвектив-
ную природу и может быть учтено введением и уравнение
175
(IV 53) коэффициента Кг, аналогичного Кс В результате урав-
нение (IV.53) принимает следующий вид:
Я о*
т=Ях4-(Лс + Кр+К(|)-з? . (IV.57)
В стационарном сострянии и безотборном режиме работы ко-
лонны т = 0. В этом случае из уравнения (IV.57) имеем
dz Н ,
dz.
-г-- к Л.К -л_к — uV-58)
Интегрирование (IV.58) при граничных условиях х=х0 при z = 0
и x=xZk при z=zK дает
До=ехр[
II
(IV.59)
Kc|-Kp+Kd
где Fo—Xzk/xo — фактор разделения; zK — длина колонны. Из
уравнения (IV.59) видно, что, как и в других противоточных
процессах, фактор разделения, достигаемый в термодиффузион-
ной колонне, экспоненциально зависит от ее длины.
Вводя понятие теоретической тарелки как участка колонны,
на концах которого отношение составов газовой смеси в одной
из «фаз» (например, в холодном потоке) отвечает коэффициен-
ту разделения, из уравнения (IV.59) нетрудно получить выра-
жение для ВЭТТ
ВЭТТ=(Хс + + Л 'llIn 9. (iv.GO)
К сожалению, теоретический расчет ВЭТТ в данном случае за-
труднен тем, что в выражение (IV.60) входит коэффициент па-
разитного перемешивания Кр, который может быть определен
только опытным путем; он зависит от того, насколько хорошо
изготовлена колонна. Кроме того, вычисления коэффициентов
переноса И, Кс и Ка в лучшем случае дают лишь весьма при-
ближенные значения этих величин. Таким образом, в целом
здесь сталкиваются с теми же трудностями, что и при теорети-
ческом расчете ВЭТТ для насадочных ректификационных ко-
лонн. Количественную проверку рассматриваемой элементарной
теории термодиффузионного разделения смесей нетрудно про-
извести на основании определения зависимости разделительной
способности колонны от давления находящейся в ней смеси га-
зов. Из кинетической теории газов известно, что коэффициент
вязкости от давления не зависит (в пределах обычно исполь-
зуемых давлений), a pr, р~Р, D2~P~' В соответствии с этим
из соотношений (IV.54) и (IV 55) следует, что Н~Р2, КР,
~РА, и поэтому уравнение (IV.59) можно записать в таком
виде:
lnF° [( к"
где Ан и В к — константы.
176
(IV .61)
(IV.62)
(IV.63)
(IV.64)
Из выражения (IV.61) следует, что зависимость фактора
разделения термодиффузионной колонны от давления должна
проходить через экстремум, т. е при некотором давлении Рогт
разделительная способность колонны будет максимальной
(рис 47). Таким образом, можно записать, что
р =^о(тах)
' опт
И
<1 111 Fo _0
аР 1р-^опт
С учетом условия (IV63) из (IV 61) следует
Однако если коэффициент переноса Ка приближенно нетрудно
I вычислить с помощью соотношения (IV.56), то константу В к
можно рассчитать лишь при условии, когда эффектом паразит-
ного перемешивания в термодиффузионной колонне можно пре-
небречь. Так как это условие практически никогда не выполня-
ется, то константу В* определяют по экспериментальным дан-
I ным. Для этого выражение (IV.61) удобнее записать в виде
Строя график зависимости 1/Р2 (In Fo) ~ 1/Р4, находим величи-
ну Ка/Кн как тангенс угла наклона получающейся прямой ли-
нии, а величину Вк/Ан— как отрезок на оси ординат. Вычислив
Ка по соотношению (IV.56), определяем Ан, а затем и В к- При
определении Гэпт можно и не прибегать к вычислениям Ка и Вк
в отдельности; для этой цели достаточно знать отношение
Ка/Вк, которое легко находится из экспериментально определяе-
мых величин Ka/Ан и В к/Ан В качестве примера на рис. 47
представлены экспериментальные данные* по разделению изо-
топов кремния термодиффузией моносилана в металлической
колонне типа коаксиальных цилиндров. Приведенные на этом
рисунке кривые получены путем обработки результатов опытов
методом наименьших квадратов с использованием зависимости
(IV.65) Кривая /, относящаяся к системе 28SiH4—3 SiH4, опи-
сывается формулой
In Fo = O,134P2/ (14-0.0847Р1), (I V.G6)
а кривая 2, относящаяся к системе 28SiH4—29SiH4, — формулой
In Fo=0,0645P2/ (1 + 0,0759Р‘), (IV.67)
где Р — давление моносилана.
* Девятых Г. Г.. Борисов Г. К. Разделение изотопов кремния в мопосила-
не методом термодиффузии//Докл. АН СССР. 1963, т. 149, № 6, с. 1293—
1294.
177
Рис. 47. Экспериментальные зави-
симости разделительной способно-
сти термодиффузионпой колонны
от давления:
J — данные для системы Я5|Н«—X'SIH,
[кривая описывается формулой (IV 66)1.
2 — данные для системы MSiH,—!*SiH4
[кривая описывается формулой (IV 67)]
Исходя из (IV 66)—(IV.67) и
(IV.64), легко найти, что опти-
мальное давление моносилана в
проведенных опытах для первой
системы составляло 1,87-10® Па,
а для второй системы—1,93х
X 105 Па. Экспериментальное
подтверждение характера теоре-
тически предсказываемой зависи-
мости термодиффузионной колон-
ны от давления находящегося в
ней газа является убедительным
доказательством достоверности
рассмотренной модели процесса
разделения. Для повышения раз-
делительной способности термо-
диффузионных колонн в литера-
туре предлагаются различные
усовершенствования к их конст-
рукциям. Так, для уменьшения паразитного перемешивания ре-
комендуют в ходе процесса осуществлять вращение внешней
или внутренней трубки колонны (типа коаксиальных цилиндров)
или обеих одновременно. С целью снижения конвективного пе-
ремешивания в зазор между трубками иногда вводят перегород-
ки (шайбы). Установлено также, что интенсивность конвектив-
ного перемешивания в колонне заметно снижается, если зазор
между трубками заполнен насадкой Но и при этом требование
об устранении возможной ацентричности нагреваемой и охлаж-
даемой стенок остается жестким, поскольку она является основ-
ной причиной возникновения нежелательного паразитного пере-
мешивания в колонне.
§ 3. ТЕРМОДИФФУЗИЯ В ЖИДКОЙ ФАЗЕ
Хотя явление термодиффузпи в жидкостях было открыто
значительно раньше, чем в газах, использование его для разде-
ления жидких смесей долго не находило практического приме-
нения И лишь после работ К. Клузиуса, Г Коршинга и К Бир-
на стало ясно, что при использовании принципа противотока
жидкостная термодиффузия как метод разделения смесей обла-
дает потенциально высокой эффективностью. Начиная с 1940 г.
были предприняты исследования применимости этого метода
для разделения изотопов урана в жидком гексафториде урана,
в результате которых был разработан промышленный способ
получения концентрата изотопа урана-235. Метод жидкостной
термодиффузии оказался вполне копкурентноспособным по срав-
178
нению с другими методами разделения изотопных смесей. Обна-
деживающие результаты были получены также при использова-
нии этого метода для разделения нензотопиых смесей Одновре-
менно с разработкой и усовершенствованием конструкций
разделительных аппаратов развивалась и теория жидкостной
термодиффузии В результате были предложены различные ме-
тоды расчета жидкостных термодиффузиониых колонн. По ана-
логии с работой термодиффузноиной колонны для разделения
газовой смеси, при разделении жидкой смеси перенос интере-
сующего компонента по высоте колонны в безотборном режиме
будет также описываться уравнением вида (IV.53) или (IV 57).
Соответствующие коэффициенты переноса при этом будут опре-
деляться точностью используемых приближений в рассматривае-
мой модели массообмена. Поскольку в жидкой смеси, п.тходя-
щейся в колонне, конвективная скорость переноса супкчгневно
выше скорости переноса за счет молекулярной диффузии, го
можно принять, что в случае жидкостной термоднффу нт
Kd^Kc. С учетом этого, выражение для фактора pm к iciinw
колонны, работающей в стационарном состоянии, utijty'iciio
в виде
1пГ0 = 252 “гТ^<»Л-н
1
i+KpIKc ’
(iv.n«>
где т] — вязкость жидкости; £>2(ж)— коэффициент днффу нт
примесного компонента в жидкости; рг=—држ/с!Г — к мпгрн
турный коэффициент плотности; IV — половина рясегоиини м< щ
ду «холодной» и «горячей» стенками
Известны попытки теоретического расчета пщчошнтЙ irp
модиффузии для жидких смесей. Однако cootboici нушиин' мг
тодики расчета имеют еще меньшую точность, «им пин i>>i ipihmi*
методики расчета для газовых смесей. Поэтому и гич I. при*
дится прибегать к экспериментальным определениям II 6<оп.
шинстве случаев при этом определяют не аг, а ш* шчппу Л, -
= ат/Т, которую называют коэффициентом Соре Нр - нПпнк
установка для определения коэффициента Copi пр> нЧн >
собой заполненный исследуемой смесью цнлпппр, lu'imu ппн1
рого закрыты днищем и крышкой, изготовленными hi и i|
водящего материала, обычно из металла Олин шнн и ни ш ip.i
охлаждается до температуры Т\, а другой 1 и ренлф и * i । м
пературы Т2 Таким образом, днище и крышка ни ни i ......•
роль «холодной» и «горячей» стенок соответ нении II ы и ши
нием градиента температур в цилиндре (ячейке) и чини" i i
диент концентрации, обусловленный протекающими нр'« «м imh
термической и концентрационной диффузии 1л» р пиния
конвективного перемешивания в растворе п < < «шпи нишки
устанавливают пористую перегородку По ципнинин ранпнне-
179
сия в системе из раствора вблизи «холодной» и «горячей» сте-
нок отбирают пробы на анализ, и с помощью формулы
?т = —(IV.69)
определяют коэффициент Соре. Формула (IV.69) вытекает из
выражения (IV 6) при допущении, что величина Sr в темпера-
турной области Ti—Т2 и в области составов х{—х2 является
постоянной.
Равновесие в такой термодиффузионной ячейке достигается
за время ~ (5-е-6)/г, где tr— (4/л2){W2/D2). Так как в жидкос-
тях коэффициент диффузии порядка 10~5 см2/с, то даже
при 1^ = 5 см равновесие в ячейке установится не менее чем че-
рез месяц. Отсюда ясно, что для снижения времени установле-
ния равновесия размер ячейки должен быть по возможности
минимальным. Но, к сожалению, при этом возрастает ошибка
в определении разности составов раствора на концах ячейки,
что, в свою очередь, сказывается на точности определения ве-
личины Sr. Поэтому оптимальный размер ячейки обычно под-
бирают опытным путем
Анализ имеющихся в литературе опытных данных о величи-
нах постоянной термодиффузии для жидкостей показывает, что
последняя обычно существенно выше, чем постоянная термодиф-
фузии в газах, вследствие чего жидкостная термодиффузия как
метод разделения смесей в принципе имеет преимущество перед
термодиффузией в газах.
Постоянная термодиффузии (или коэффициент Соре) может
быть определена также и методом колонны, по результатам
опытов как в стационарном, так и в нестационарном состояниях.
Знание этой величины позволяет судить о возможностях термо-
диффузии для глубокой очистки жидкостей от примесей, а так-
же производить ориентировочный расчет разделительное™ ап-
паратуры.
В практике разделения смесей термодиффузией используют
колонны плоского типа (в простейшем случае это две плиты с
зазором между ними) или типа коаксиальных цилиндров. Что-
бы свести к минимуму эффект конвективного перемешивания
жидкости в колонне, здесь иногда, как и в колоннах для разде-
ления газовых смесей, зазор между «холодной» и «горячен»
стенками заполняется насадкой. Для повышения эффекта раз-
деления вместо одной колонны можно использовать каскад
колонн.
Основной недостаток жидкостной термодиффузии, как и тер-
модиффузии в газах, — низкая производительность. Этот недо-
статок в целом, к сожалению, не удается устранить, например,
путем увеличения сечения колонны или зазора между «холод-
ной» и «горячей» стенками, поскольку при этом резко возраста-
ло
ет ВЭТТ и термодиффузионный метод лишается своего главного
преимущества — эффективности Это преимущество метода в
отдельных случаях, в частности при очистке от тех примесей,
которые плохо удаляются другими методами может превали-
ровать над его низкой производительностью, т е термодиффу-
зия с успехом может быть использована и как самостоятельный
метод очистки, и как стадия доочистки в комбинированных ме-
тодах, когда не ставится задача получения больших количеств
очищенного вещества, но требуется его высокая степень ин ючц.
§ 4. ТЕРМОДИСТИЛЛЯЦИЯ
В последнее время в практике глубокой очистки н< цкш
успешно применяют комбинированный способ, получивший на
звание метода термодистилляции. В этом методе термоднффу
зия осуществляется в сочетании с ректификацией в колонном
аппарате типа коаксиальных цилиндров Процесс разделении и
такой термодистнлляционпой колонне протекает в ycioinin'
сосуществования движущихся противотоком жидкости и ппрн
При этом на пар налагается температурное поле, подобно т< му,
как это реализуется в рассмотренной выше термодиффузш пн >rt
колонне для разделения смеси газов. Роль «холодной» cieniut
играет поверхность внутренней трубки (цилиндра), темпер.iiypn
которой Т1 путем циркуляции хладоагента поддержи u.iric и ран
ной температуре конденсации пара или несколько ниже В
процесса по этой стенке движется в виде тонкой пленки <кн i
кость, образующаяся в конденсаторе колонны. Темперniу| а /
подбирается таким образом, чтобы на «холодной» crriim in
происходило дополнительной конденсации пара, кои1Ч»ктп[>ук>
щего с жидкостью. Горячей стенкой является поверхшнч ь hii.hi
ней трубки, которая обогревается до заданной темпер и iyp>i /
В результате общий эффект разделения в колонне бу ,< i иПу.
словлен как явлением термодиффузии в паре, так к рсннфиЛ,
цией вследствие массообмена между стекающей по *Х'>.1п41ЙМИ
стенке жидкостью и поднимающимся в зазоре меж iy ipyf»»- <ми
потоком пара.
Для расчета глубины очистки рассмотрим работу
колонного аппарата с питанием, поступающим иниу вилонпи
(например, колонна периодического действия с шжннм муб н)
Пусть примесью является высококипящий компот- ।( ри
ражение для скорости массообмена процесса терм' ни hi i чини
в колонне, работающей в стационарном состоянии, Мотип N
писать как
(IV ’И)
QZ
где v — составляющая скорости массообмепл ЛуЛМ ♦nil и
нч
ректификацией, a vT — составляющая скорости массообмена,
обусловленная термодиффузией.
Выражение для составляющей о нетрудно получить исходя
из решения уравнения (II 121). Из этого решения следует, что
искомое выражение можно представить в виде
(!/-£/») = * (р-- , (IV.71)
где в случае пленочной колонны типа полой трубки радиусом г0
с орошаемыми стенками (46/ti)D2f>/r02. Аналогичный вид
для v получается и в случае рассматриваемой «щелевой» ко-
лонны с одной орошаемой стенкой, но здесь (,2O/52)Z)2p/w2,
где w — зазор между трубками.
Зависимость составляющей vT от параметров процесса легко
находится на основании соотношений (IV 49) и (IV 50), опери-
руя в них полной величиной зазора w=2W между «горячей» и
«холодной» стенками колонны, так как второй фазой в колонне
является жидкость, а не «холодный» поток пара. С учетом это-
го и принимая, как и выше [см уравнения (IV 47) и (IV.52)],
что dx/dz=dy/dz = 6y/dz, нз отношений (IV 49) и (IV.50) по-
лучаем
ат7)2рД7’ /Др (г—у)
vT= т -------х----, (IV.72)
Ты* w*
где Vt=lAy/Az\ l=Lp.
В безотборном режиме работы колонны средняя концентра-
ция примеси в жидкой фазе х равна средней концентрации при-
меси в паровой фазе у в одном и том же сечении колонны г.
При этом условии из (IV.72) следует, что
ат-РгрАГ
«'т = —-------У, (I V.73)
а выражение (IV 71) для v соответственно преобразуется к виду
Подставляя (IV.73) и (IV.74) в (IV.70) и разделяя переменные,
получаем
А=Г I j,. (IV.75)
У L la lTu)2 J
Интегрирование в (IV.75) при граничных условиях (11.123) дает
i„p *(а-1) _ , атГ)2рЛТ _
- In Fo = —-----2к + 2K, (I V.76>
ITw*
где Fo—yo/yN — фактор разделения колонны; yD — концентра-
ция примеси в паре, поступающем из колонны в дистиллят;
182
Уи~Угк—концентрация примеси в паре (питании), поступаю-
щем в низ разделительной части колонны из ее куба. Уравнение
(IV 76) позволяет сделать вывод о том, что сочетание термо-
диффузии с ректификацией в процессе термодистилляции в об-
щем случае должно приводить к увеличению глубины очистки
по сравнению с той, которая имела бы место при проведении
термодиффузии или ректификации в отдельности.
Метод термодистилляции оказался весьма эффективным ме
тодом глубокой очистки ряда веществ от содержащихся в них
примесей в виде мельчайших взвешенных частиц субмикронного
размера (~10-1—10-3 мкм) Такие частицы могут иметь раз-
личную природу, обусловленную их происхождением (химиче-
ские реакции термораспада или гидролиза, диспергирование
конструкционных материалов, окружающая среда и т д.), они
практически присутствуют во всех веществах — газообразных,
жидких и твердых. Установлено, например, что взвешенные час-
тицы, находящиеся в летучих неорганических гидридах и хло-
ридах, на основе которых получают некоторые материалы для
полупроводниковой техники и волоконной оптики, состоят в
основном из оксидов различных элементов Внося существенный
вклад в суммарное содержание примесей, взвешенные частицы
оказывают отрицательное влияние на электрофизические и он
тические свойства этих материалов
Исследования показали, что если для очистки веществ ог
взвешенных частиц относительно больших размеров (>0,1 мкм)
часто достаточно простой перегонки, то в отношении взвешен
ных частиц субмикронных размеров малоэффективной oica.'iuii.i
ется даже ректификация. Применение дтя удаления таких чйс
тиц кристаллизационных методов также показало их низкую
эффективность. Это объясняется существенно меньшей днффу
зионной подвижностью взвешенных частиц по сравнению с по-
движностью молекул. Хорошие результаты по очистке oi n ine
шейных частиц достигаются при использовании mcio.i.i <|oi.ii.t
рации Однако его применение ограничено вследсптс пмеиншто
при этом место заметного загрязнения очищаем!и н<чце< inn
материалом фильтра, в частности по причине химический иди
термической нестойкости последнего В методе же н рмо ннчп.1
ляции загрязняющее действие материала pai.u ни» и.ной .шни
ратуры если и проявляется, то в значительно мепыппй < i u.-iiii
Оценку глубины разделения при использонпннн Mrruiiu гер-
модистилляции для очистки веществ <и и нт
можно провести на основании уравнения (IV 7<1), •< ш | мат-
ривать движущийся в ходе процесса очистки н< ыщм ни щдя-
ционной колонне пар как некий газ — тик и t ihuuki ый i .и .'1о-
рентца. Такой газ представляет собой смей, ivikii* и 'ihMMbix
молекул, причем масса и размер ежих м<> и ц\ । ин . меньше
массы и размера тяжелых молекул < > г < • н-гч и >м инн пннные
183
21г/иг, град/см
Рис. 48 Зависимость разде-
лительной способности тер-
модистилляционной колон-
ны от градиента темпера-
туры:
А О — экспериментальные ре-
зультаты по очистке изопропило-
вого спирта от взвешенных ча
стиц германия по данным спект-
рального анализа и лазерной
ультрамнкроскопии соответст-
венно
частицы с тяжелыми молекулами и
исходя соответственно из уравнения
(IV.76), можно сделать вывод, что
при очистке и от взвешенных частиц
зависимость разделительной способ-
ности термодистилляционной колон-
ны от температурного градиента в
координатах In Fo—АГ fw должна
иметь линейный характер. Это под-
тверждается представленными на
рис. 48 результатами опытов* по
очистке изопропилового спирта от
искусственно введенной в него при-
меси в виде мельчайших частиц гер-
мания со средним размером
0,08 мкм Из рис. 48 видно также,
что приведенная на нем прямая ли-
ния практически исходит из начала
координат. Это указывает на то, что
в соответствие с уравнением (IV 76)
эффект очистки от взвешенных час-
тиц за счет ректификации в термо-
дистилляционной колонне незначи-
телен Аналогичные данные были
получены и в опытах по очистке веществ от взвешенных частиц
непосредственно методом ректификации. Это, как уже отмеча-
лось, объясняется малой скоростью диффузии таких частиц по
сравнению со скоростью диффузии молекул в контактирующих
паровой и жидкой фазах
Таким образом основной вклад в эффект очистки веществ от
взвешенных частиц методом термодистилляции обуслов теи тер-
модиффузией (термофорезом) этих частиц в движущемся паро-
вом потоке, находящемся в температурном поле, т. е за счет
температурного градиента в термодиститляционной колонне
происходит направленное перемещение взвешенных частиц от
«горячей» стеики к «холодной». Роль же стекающей по ней жид-
кой пленки в этом случае сводится в основном к захвату пере-
местившихся к ней частиц и переносу их в кубовую жидкость,
где эти частицы и концентрируются.
* Девятых Г Г., Воротыицев В М. Дроздов П Н. Глубокая очистка ве-
ществ от взвешенных частиц методом пленочной ректификации с воздействи-
ем на пар температурного поля//Докл. АН СССР. 1980, т 252, Ns 3, с. 671—
673.
ПРИЛОЖЕНИЕ
ВЫВОД УРАВНЕНИЯ СТАЦИОНАРНОЮ
МАССООБМЕНА В КРИСТАЛЛИЗАЦИОННОЙ КОЛОННЕ
Рассмотрим работу противоточной кристаллизационной колонны и его
цнонарном состоянии Введем безразмерные координаты
где W— линейный размер кристаллов твердой фазы (пел)! пинии) *
полная длина разделительной части колонны.
Используя соотношения (1), уравнение материальною О.> iiito >ыи < к
мента объема кристаллизационной колонны [см. урлннени<> (111 .1) | можно
записать в виде
да: д2а:
(2)
приведенный коэффициент дпффу >ии (* пи ни..
пишем граничные условия [см соотношения (111.39) и {111-10)1
дх
ди
I =0,
lu-0
х (и, О) = а-о
Поскольку рассматривается процесс очистки ochohiioIO »>тнс• • и> • > при
меси, то при заданной схеме процесса кристаллы н tuny । . . .> i hi >>
держать меньше примеси, чем кристаллы в верху колонны и, > и ,п » . > <•>
но, имеет место случай, когда а<1. В дальнейшем i <»i 6>»м
оперировать величиной а'=1/а. Таким образом с- нн> ......... (>• <> ы.*> рных
координат (I) уравнение рабочей линии [уравнение (III Г. | > . и . >
1
J a:(u, <j>) dw = a' (1 —р) z (1, <р)- /• J « (к 1'1» ><)
Безотбориый режим. Применительно к б > iu»> н HI »>
уравнения (4) следует, что
1
J х(и,
о
Решение уравнения (2) будем искать в виде*
____________
* Батунер Л М., Позин М Е. Матеман • ........ «нмичнтй i«x
•1нке. М., 1960 с. 321
185
x (u, <p) = e“v<1’ (Л cos Pu-f-B sin 0u), (6)
где A, B, p, 7 — постоянные.
Подставим (6) в (2); после дифференцирования будем иметь
— ye-1’4’ (A cos Pu-|-B sin рц) = — Ррге-То (Л cos Pu-|- В sin Pu)
или *
y=DP2. (7)
Из граничного условия (3) следует, что в выражении (6) В=0. С учетом
последнего и соотношения (7) выражение (6) принимает более простой вид
х (и, q>)==e-I,₽ 4cosPu (8)
Подстановка уравнения (8) в (5) дает
cos ₽• (9)
Трансцендентное уравнение (9) имеет бесконечное множество корней рк-
Следовательно, решение уравнения (8) в общем случае будет иметь вид
х (и, <р)= 2 А«е ^^cos рки, (10)
А—О
в котором значения рк в соответствии с соотношением (9) будут определять-
ся характеристическим уравнением
tgPK = a'pK (11)
и могут быть найдены, например, графическим путем.
Для определения коэффициентов Ак используем граничное условие (3);
прн этом нз выражения (10) следует, что
2 OkCOsPku = 1, (12)
к=о
где ак=Ак/хо. Далее, исходя из (12) и нз требования минимума* величины
1 оо /= $ [(2 °kCOS₽km)—1] du, (13) 0 к0
получим 1 С» [(2 UkCOsPku)—1 cos0judu=O 0 j=0
или 1 DO J 2 °к cosPKu cosf5ju du= J cosPjudu. (14) 0 3-0 0
Из соотношения (14), поскольку
1 cos cos ₽ F = — [cos (Pk—pj) и + cos (Рк + P j) и J,
* Бронштейн М Н., Семендяев К. А Справочник по математике. М., 1959,
с. 572
186
можно получить систему линейных уравнений для определения значений а*
(а следовательно, и Лк):
S_ sin Bj
°кйкТ = 2 —pj ,
7 = 0
sin(PK—Pj) , sin(pK+Pj)
Ph-Pj Pk+Pj
, । sin 2PK
1+ 2РИ
На основании вышеизложенного выражение для
шется в виде
при j ф к,
(15)
при ; = к
фактора разделения запи-
J X {и, 1) du У 2 Аке cosPnudu
0__________ . о ио__________________
^0 Z0
2 vui ()6)
гК
к-0
Формула (16) позволяет производить расчет фактора разделения кристал-
лизационной колонны при заданных значениях а' и D. Однако такие рас-
четы, включающие в себя предварительное определение значений ак из си-
стемы уравнений (15), без использования вычислительной машины крайне
трудоемки. Поэтому весьма интересным представляется отыскание на осно-
ве соотношения (16) хотя бы н более приближенной, но зато более удоб-
ной для вычислений аналитической зависимости С этой целью на цифро-
вой вычислительной машине ЭЦВМ М-20 были проведены численные рас-
четы по уравиениям (11) и (15) для сравнительно большого иитервала зна
чений а' (1 01—100) Эти расчеты показали, что ряд, стоящий в правой ча-
сти соотношения (16), быстро сходится, причем определяющей величиной
по существу является первый член ряда Например.
1) а'= 1,05,
Fo = O,991e-°>142D+O,OO47e_2o-3D+O,O016e-59’8r>+(17)
2) а'= 4,
Fo=0,857e-1194D4- 0,07e~2,17I)+0,0025e~ei’2D+- - (18)
Таким образом как следует нз численных примеров (17) н (18), для оцен-
ки величины фактора разделения в выражении (16) можно ограничиться
одним первым членом ряда, т. е.
Пд2 sin Вл
Го~аое-™<>—(19)
или в более удобной для расчета форме
Fo ~ (а (в') е^'))-», (20)
где а(е') =₽о/(До sin рс), Ь(е')=р02.
Рассчитанные с помощью ЭЦВМ М-20 по уравиениям (11) и (15) зна-
чения ро, ав н с(е') представлены в табл. 1. Отсюда необходимые для вы-
числений в каждом конкретном случае величины а(е') и б(е') нетрудно
найти, например, графическим путем. С другой стороны, на основании дан-
ных табл 1 значения коэффициентов а(е') и б(е') в выражении (20) в за-
висимости от а' хорошо аппроксимируются формулами:
187
Таблица 1 Значения коэффициентов ₽о, а0 а
а* Во а0 а
1,01 > 0,1722 1,003 1,002
1,03 0,2947 1,009 1,006
1,05 0,3762 1,015 1,009
1,07 0,4401 1,020 1,013
1,10 0,5175 1,028 1,018
1,15 0,6175 1,040 1,026
1,20 0,6954 1,050 1,033
1,40 0,8999 1,085 1,058
1,60 1,022 1,111 1,078
2,0 1,166 1,146 1,107
3 1,324 1,191 1,146
4 1,393 1.212 1,168
6 1,457 1,233 1,189
8 1,487 1,243 1,200
10 1,504 1,250 1,207
20 1,538 1,261 1,220
100 1,564 1,271 1,231
Ъ (е') = е' [з- (з—(22>
в которых е'— (а'—1)/а'.
Вычисления по соотношению (20) с учетом (21) и (22), показали вполне
удовлетворительное согласие с соответствующими расчетами по уравнению
(16), проведенными на ЭЦВМ. Таким образом, заменяя а' на 1/а в выраже-
ниях (21) и (22), из соотношения (20) можно получить расчетную формулу
для интересующего случая а<1.
На основе изложенного можно определить распределение примеси
в твердой фазе по высоте кристаллизационной колонны, т. е. найти зависи-
мость х(ф). Для этой цели в уравнении (10) ограничимся одним первым
членом ряда так же, как это было сделано при выводе соотношения (20)
Используя при этом теорему о среднем значении, будем иметь
х(ф) ~ J -4oe-D₽o4> cospoudu (23)
о
или. после интегрирования, поскольку A0=floX0,
х (<р) ~ х0 (aeD& <F)~l, (24)
где а и ро2 соответственно характеризуются выражениями (21) н (22).
Отборный режим. Применительно к отборному режиму решение диффе-
ренциального уравнения (2), используя граничное условие (3), будет иметь
вид
х(и, ф)=2 А.е cos + const. (25)
ко
Подставляя соотношение (25) в уравнение рабочей линии (4), после неко-
торых преобразований получим следующее выражение:
— 00 —о₽?ц> 03 —
х(и, ф) — 2 Аке к cosPku—а'в' £ Аке kcosPk, (26)
к—о к—о
188
в котором в'=р/(а —1), а значения определяются из характеристическо-
го уравнения
1кРи-а'(1 -р)Рк. (27)
При р=0 соотношения (26) к (27) переходят в соответствующие выраже
ния (10) и (11) для безотборщно режим.)
Коэффициенты Лк и 11Ы1>.1Ж<>>|>»| {2(1) могут быть найдены тем же спо-
собом, что и в выражении (10). i е с учетом граничного условия (3), ис-
ходя нз требования минимума шынчниы
^=$[(2 2 r~« T7Pkcos Рк)~ 1] dw. (28)
0 к—0 и—1>
При этом получшчеи < » п'Мл .iiicflp.i....-скнх уравнений, которые можно ре-
шить с помощью иы'шгло।елш)пП м.опины
Из соотионим111Й (’.’II) и (#/> можно получить выражение д я фактора
разделения
I
I t (II , I) till со
I а'Н' -оаЗ sinBK
Л i„ I -Р 2 “к* рк , 0)
к=0
где я»- Лк/х |.
Расчеты ио форм) и> 174) проиеденныс с помощью вычислительной ма-
шины БЭ М <1 ii(ih>io<in ,»ii р»л. > ।нищий в правой части форм 1ы быст
ро сходится и otiplAMUMMU нилнннной является первый член ря а. Поэт<
Му ПрНбЛ11Ж<Ч1НО МОЯ1ЛП »1|11Н> ин., Ч1О
I • <»' «лн1пР0 nft2
или
p)eDh<e’P’)-\ (II)
где p)< J 1 /,(e'’ p) = ₽§-
Il ikioim) 1ШМ1«.н1. Ч1О для большинства практн> fin
ных । iv no (ii« e^KII ............ a(r',p) близка к единице Г > )му
ч ине / I|IHIV4<MU* Й HII «ыряжсиия (31), будет в ос ином
де яты « 1«^ь '• м последнюю можно н йти
р KTipiK иг>«> юл* ipamuioi i*h
ЛИТЕРАТУРА
Введение
Сажнн Н. П. Вещества высокой чистоты в науке и технике. — М.: Зна-
ние, 1969. 63 с.
Финкельштейн Д. Н Чистота вещества. — М._ Атомиздат, 1975. 224 с.
Девятых Г. Г. О создании банка данных по высокочистым веществам//
Вести. АН СССР. 1982, Ns 7. с. 25—31.
Глава I
Шефер Г. Химические транспортные реакции. — М.: Мир, 1964. 189 с.
Степнк Б. Д., Горштейн И. Г., Блюм Г. 3. и др. Методы получения особо
чистых неорганических веществ.—Л.: Химия, 1969. 480 с.
Разуваев Г А., Грибов Б. Г., Домрачев Г А. и др Металлоорганические
соедииеиия в электронике.—М.: Наука, 1972. 479 с
Девятых Г Г., Зорин А. Д Летучие неорганические гидриды особой чи-
стоты — М : Наука, 1974. 208 с.
Глава II
Роз А., Роз Е. Теория//Перегонка. — М.: ИЛ, 1954. С. 5—153.
Розен А. М. Теория разделения изотопов в колоннах. — М.: Атомиздат,
1960. 439 с.
Зельвенский Я. Д., Титов А. А., Шалыгин В А. Ректификация разбавлен-
ных растворов —Л.: Химия, 1974 216 с
Крель Э. Руководство по лабораторной перегонке. — М Химия, 1980
520 с.
Глава 111
Херингтон Е. Зонная плавка органических веществ —М.- Мир, 1965.
260 с
Вигдоровнч В. Н. Очистка металлов и полупроводников кристаллизаци-
ей.— М Металлургия, 1969.269 с.
Степин Б. Д., Горштейн И. Г. Блюм Г. 3. и др Методы получения особо
чистых неорганических веществ. — Л.: Химия, 1969 480 с.
Пфанн В. Зонная плавка. — М Мир, 1970. 336 с.
Гельперин Н И., Носов Г. А. Основы техники кристаллизации распла-
вов.— М Химия, 1975 351 с
Девятых Г Г., Еллиев Ю. Е. Введение в теорию глубокой очистки ве-
ществ.— М.: Наука, 1981 320 с
Глава IV
Грю К 3., Иббс Т. Л Термическая диффузия в газах —М- Гостехтео-
ретиздат, 1956 184 с
Розеи А М. Теория разделения изотопов в колоииах.—М.: Атомиздат,
1960. 439 с.
Рабинович Г Д., Гуревич Р Я., Боброва Г. И. Термодиффузиоииое раз-
деление жидких смесей. — Минск: Наука и техника, 1971. 243 с.
Девятых Г Г., Воротынцев В М Дроздов П Н. Глубокая очистка ве-
ществ от взвешенных частиц методом нлеиочиой ректификации с воздействи-
ем на пар температурного поля//Докл АН СССР, 1980, т. 252, № 3, с. 671—673.
ОГЛАВЛЕНИЕ
Предисловие .... .......................
Введение..................................
1. Вещества высокой чистоты .......................
2. Классификация веществ высокой чистоты
3. Классификация методов глубокой очистки веществ
Г лава I Химические методы глубокой очистки веществ
Общая характеристика...................................
Оценка предельных возможностей.................
Метод химических транспортных реакций
Перенос вещества потоком газа-реагента
Перенос вещества молекулярной диффузней
Перенос вещества посредством конвекции
Сравнительная характеристика химических и физнко
ских методов очистки веществ ......
11. Дистилляционные методы
Коэффициент разделения...........................
Теоретическая оценка коэффициента разделения
Экспериментальные методы определения коэффициента раз
деления ..... ..........................
Перегонка...........................................
Однократная перегонка......................
Многократная перегонка .........
Ректификация Основные понятия ......................
Ректификация в тарельчатых колоннах ................
Ректификация в насадочных колоннах..................
§
§
§
§
2.
3.
4
Г лава
§
§
§
1.
2.
3
химиче
§
4
§
§
§
5
6.
7 . ________________________________________
Понятие о движущей силе массообмена.....................
Фактор разделения в стационарном состоянии и безотборнои
режиме...............................................
Влияние скорости отбора продукта иа фактор разделения
Высота, эквивалентная теоретической тарелке {ВЭТТ)
Высота единицы переноса (ВЕП) . .................
§ 8. Влияние загрязняющего действия материала аппаратуры на
глубину очистки веществ методом ректификации
Фактор разделения в стационарном состоянии и безотборном
режиме .....
Влияние скорости отбора продукта на фактор разделения
Предельное значение концентрации примеси ....
§ 9. Оценка глубины ректификационной очистки термоиестойкнх
веществ ...
I
I
I
•»
и
II
II
10
2(1
23
27
28
30
32
33
35
40
48
48
51
52
57
61
61
64
66
69
72
75
§ 10. Периодическая ректификация.........................
Ректификация с постоянной скоростью отбора продукта
Ректификация при постоянном составе продукта
Ректификация с дискретным отбором дистиллята
§ И Влияние способа отбора концентрата примеси на эффеитнн
пасть процесса ректификации
§ 12 1 гкти<|>11кпц11я под пониженным давлением
§ 11, М'1Лек\л«рния дистилляция ...
78
79
82
86
90
92
95
49
101
191
Глава III Кристаллизационные методы .................. . 104
§ 1. Кристаллизация из расплава . 105
Зарождение и рост кристаллов......................... . . 106
Влияние примесей............................................109
Коэффициент разделения (распределения)......................111
§ 2. Направленная кристаллизация.......................... . ИЗ
Однократная направленная кристаллизация.....................114
Многократная направленная кристаллизация .... 118
§ 3. Зонная перекристаллизация (плавка)........................119
Распределение примеси по длине слитка после одного прохода
расплавленной зоны ................................... ... 120
Распределение примеси по длине слнтка после нескольких про-
ходов расплавленной зоны....................................122
Распределение примеси по длине слитка после бесконечно боль-
шого числа проходов расплавленной зоны (конечное распреде-
ление) . ...........................................124
§ 4 Зонное замораживание (затвердевание) .............128
§ 5 Противоточная кристаллизация из расплава ... 131
Основные модели процесса массообмена в кристаллизационной
колонне.....................................................133
Фактор разделения в стационарном состоянии и безотборном
режиме ... ...................... 137
Влияние скорости отбора продукта на фактор разделения 138
Распределение примеси по высоте кристаллизационной колонны 139
§ 6. Влияние загрязняющего действия материала аппаратуры на
глубину очистки веществ кристаллизационными методами 144
Направленная кристаллизация .... .144
Зонная перекристаллизация ...................... . . 146
Противоточная кристаллизация .............................. 148
§ 7. Кристаллизация из раствора 149
Способы осуществления процесса..............................150
Коэффициент разделения (сокрнсталлнзации) 152
Фракционированная (дробная) кристаллизация 154
Противоточная кристаллизация из раствора .................. 158
Глава IV Метод термодиффузии . . 160
§ 1. Термодиффузия в газовой фазе...............................160
Коэффициент разделения установки свободной от конвекции 160
Постоянная термодиффузин....................................162
§ 2. Термодпффузиоиные колонны 169
Типы термодиффузнонных колонн 170
Скорость движения газа в колонне ...........................172
Вычисление скорости переноса примеси вдоль по колонне 174
Фактор разделения в стационарном состоянии и безотборном
режиме......................................................175
§ 3. Термоднффузия в жидкой фазе 178
§ 4 Термолистялляция 181
Приложение Вывод уравнения стационарного массообмена в кристал-
лизационной колонне...................................... . 185
-Литература.........................................................190